|
8)
|
в Част Б, се добавят следните глави:
„Б.63 СКРИНИНГОВО ИЗПИТВАНЕ ЗА ТОКСИЧНОСТ ЗА РЕПРОДУКЦИЯТА/РАЗВИВАЩИЯ СЕ ОРГАНИЗЪМ
ВЪВЕДЕНИЕ
|
1.
|
Настоящият метод за изпитване е еквивалентен на насоките за изпитване на ОИСР (ТG) 421 (2016). Насоките на ОИСР за изпитване на химикали се преразглеждат периодично в светлината на научния прогрес. Оригиналните насоки за скринингово изпитване 421 бяха приети през 1995 г., на базата на протокол за „Предварително скринингово изпитване за токсичност за репродукцията“, обсъден на две експертни срещи в Лондон през 1990 г. (1) и в Токио през 1992 г. (2).
|
|
2.
|
Този метод за изпитване бе актуализиран с крайни точки, относими към вещества, нарушаващи функцията на ендокринната система, като последващо действие към дейност с висок приоритет, инициирана в ОИСР през 1998 г. за ревизиране на съществуващите насоки за изпитвания и за разработване на нови насоки за изпитвания за скрининг и изпитване на веществата с потенциал да нарушават функцията на ендокринната система (3). TG 407 на ОИСР (28-дневно изследване за токсичност с повтаряща се орална доза при гризачи, Глава Б.7 от настоящото приложение) например, бяха подобрени през 2008 г. с параметри, подходящи за откриване на въздействие на изпитваните химикали върху ендокринната система. Целта при актуализиране на TG 421 бе да се включат някои крайни точки, относими към вещества, нарушаващи функцията на ендокринната система в насоките за скринингови изпитвания (TG), при които периодите на експозиция обхващат някои от чувствителните периоди по време на развитието на организма (пред- или ранните постнатални периоди).
|
|
3.
|
Избраните допълнителни крайни точки относими към вещества, нарушаващи функцията на ендокринната система, също част от TG 443 (Разширено изследване за токсичност за репродукцията в едно поколение, Глава Б.56 от настоящото приложение), бяха включени в TG 421 на базата на проучване на осъществимостта разглеждащо научни и технически въпроси, свързани с тяхното включване, както и възможните адаптации на плана на изпитването, необходими за тяхното включване (4).
|
|
4.
|
Настоящият метод за изпитване е проектиран да генерира ограничена информация относно ефектите на изпитвания химикал върху мъжката и женската репродуктивна ефективност като например, функция на половите жлези, поведение при чифтосване, зачеване, развитие на заченатия организъм и раждане. Той не е алтернативен на, нито замества съществуващите методи за изпитване Б.31, Б.34, Б.35 или Б.56.
|
ПЪРВОНАЧАЛНИ СЪОБРАЖЕНИЯ
|
5.
|
Настоящият метод за скрининг изпитване може да се използва за предоставяне на първоначална информация за възможните ефекти върху репродукцията и/или развитието, в ранния етап на оценяване на токсикологичните свойства на химикали, или за химикали, които пораждат безпокойство. Той може да се използва и като част от набор от първоначални скрининг изпитвания за съществуващи химикали, за които има малко или липсва токсикологична информация, като проучване за определяне на диапазона на дозата за по-обширни проучвания върху репродукцията/развитието, или когато се счете за уместно по други причини. При провеждане на проучването следва да се спазват ръководните принципи и съображения, очертани в Ръководство на ОИСР № 19 относно признаването, оценката и употребата на клинични признаци като хуманни крайни точки за експериментални животни, използвани за оценяване на безопасността (5).
|
|
6.
|
Настоящият метод за изпитване не предоставя пълна информация по всички аспекти на репродукцията и развитието на организма. По-конкретно, той предлага единствено ограничени средства за откриване на постнатални прояви на пренатална експозиция, или ефекти, които може да бъдат индуцирани по време на постнатална експозиция. Наред с другите причини, поради относително малкия брой животни в групите на определена доза, избирателността на крайните точки и кратката продължителност на проучването, този метод няма да предостави доказателство за категорични твърдения за липса на ефекти. Нещо повече, при отсъствието на данни от други изследвания за токсичност на репродукцията/развиващия се организъм, положителните резултати са полезни за първоначалната оценка на опасността и допринасят за вземането на решения по отношение на необходимостта и времето за допълнителни изпитвания.
|
|
7.
|
Резултатите, получени от свързаните с жлезите с вътрешна секреция параметри, следва да се разглеждат в контекста на „Концептуалната рамка на ОИСР за изпитване и оценка на химикали, нарушаващи функциите на ендокринната система“ (6). В тази Концептуална рамка подобрените TG 421 на ОИСР се съдържат в ниво 4 като in vivo изследване, предоставящо данни за неблагоприятните ефекти върху крайните точки, относими към ендокринната система. Все пак, един ендокринен сигнал сам по себе си не може да се разглежда като достатъчно доказателство, че изпитваният химикал е вещество, нарушаващо функциите на ендокринната система.
|
|
8.
|
Настоящият метод за изпитване предполага орален прием на изпитвания химикал. Ако се използват други пътища на експозиция може да се налагат изменения на метода.
|
|
9.
|
Преди използването на метода за изпитване по отношение на смес с цел генериране на данни за планирана регулаторна цел, следва да се разгледа въпросът дали той може да даде адекватни резултати за тази цел и, ако е така, защо. Подобни съображения не са необходими, когато има регулаторно изискване за изпитване на сместа.
|
|
10.
|
Определенията, които са използвани, са дадени в допълнение 1.
|
ПРИНЦИП НА ИЗПИТВАНЕТО
|
11.
|
Изпитваният химикал се прилага в нарастващи дози на няколко групи от мъжки и женски индивиди. Мъжките следва да бъдат дозирани в продължение на най-малко четири седмици до и включително деня, преди планираното им умъртвяване (това включва минимум две седмици преди чифтосване, по време на периода на чифтосване и приблизително, две седмици след чифтосване). Предвид ограничения период на дозиране на мъжките преди чифтосване, плодовитостта може да не бъде особено чувствителен показател за тестикуларна токсичност. Ето защо, детайлно хистологично изследване на тестисите е от съществено значение. Комбинацията от период на дозиране преди чифтосване от две седмици и последващи наблюдения на чифтосването/плодовитостта с цялостния период на дозиране от минимум четири седмици, последван от детайлни хистопатологични изследвания на мъжките полови жлези, се разглежда като достатъчен за да могат да бъдат определени голяма част от ефектите върху мъжката плодовитост и сперматогенеза.
|
|
12.
|
Женските следва да бъдат дозирани през целия период на проучването. Това включва две седмици преди чифтосване (като целта е да се обхванат най-малко два пълни естрални цикъла), променливото време до зачеване, продължителността на бременността и най-малко тринадесет дни след раждането, до и включително деня, преди планираното им умъртвяване.
|
|
13.
|
Продължителността на проучването, след аклиматизацията и оценката на естралния цикъл преди дозиране, зависи от женската ефективност и е приблизително 63 дни, [най-малко 14 дни преди чифтосването, (до) 14 дни за чифтосване, 22 дни бременност, 13 дни лактация].
|
|
14.
|
По време на периода на прилагане, животните се наблюдават внимателно и ежедневно за признаци на токсичност. Животните, които умират или са умъртвени по време на периода на изпитването, се аутопсират, а преживелите животни се умъртвяват и се аутопсират при приключването на изпитването.
|
ОПИСАНИЕ НА МЕТОДА
Избор на животински вид
|
15.
|
Настоящият метод за изпитване е предназначен за употреба с плъхове. Ако параметрите, определени в рамките на този метод за изпитване, се изследват при друг вид гризачи, следва да бъде представена детайлна обосновка. Плъховете са единственият биологичен вид, използван в рамките на международната валидационна програма за откриване на вещества, нарушаващи функциите на ендокринната система в TG 407 на ОИСР (съответстващи на Глава Б.7 от настоящото приложение). Не трябва да се използват породи с ниска плодовитост или породи, за които е характерна висока честота на вродените аномалии. Трябва да се използват здрави, нераждали животни, които не са били подлагани на предишни експериментални процедури. Тестовите животни следва да се охаракеризират по вид, порода, пол, тегло и възраст. При започване на изследването измененията в теглото на използваните животни следва да бъде минимално и да не надвишава 20 % от средното за всеки пол тегло. Когато изследването се провежда като предварително до дългосрочно проучване или проучване на цяло поколение, за предпочитане е в двете изследвания да се използват животни от една и съща порода и източник.
|
Условия на отглеждане и хранене
|
16.
|
Всички процедури трябва да са в съответствие с местните стандарти за полагане на грижи за лабораторни животни. Температурата в стаята на опитните животни трябва да бъде 22 °C (± 3 °). Въпреки че относителната влажност трябва да е най-малко 30 % и за предпочитане да не превишава 70 %, освен по време на почистване на помещението, целта следва да е 50—60 %. Осветлението трябва да бъде изкуствено, като последователността е 12 часа светлина, 12 часа тъмнина. За храненето могат да се използват обичайните лабораторни хранителни режими с неограничен достъп до вода за пиене. Изборът на хранителен режим може да бъде повлиян от нуждата да се осигури подходящо смесване с изпитван химикал, когато прилагането му е по настоящия метод.
|
|
17.
|
Животните се настаняват групово, в малки групи от един и същи пол; животните могат да се настаняват отделно, ако това е научно обосновано. При групово настаняване в клетки не следва да има повече от пет животни в клетка. Чифтосването трябва да се извърши в клетки, подходящи за тази цел. Бременните женски следва да бъдат настанявани поотделно и да им бъдат предоставяни материали за изграждане на гнездо. Кърмещите женски се настаняват поотделно заедно с потомството си.
|
|
18.
|
Храната следва да бъде редовно анализирана за наличието на замърсители. Проба от хранителния режим трябва да се съхранява до окончателното изготвяне на доклада.
|
Подготовка на опитните животни
|
19.
|
Здрави млади полово зрели животни се определят на произволен принцип за контролните и опитните групи. Клетките трябва да бъдат подредени по такъв начин, че възможните ефекти в резултат на местоположението на клетката да бъдат сведени до минимум. Животните се идентифицират по уникален начин и се настаняват в клетките им най-малко пет дни преди началото на проучването, за да им се осигури време за аклиматизиране към лабораторните условия.
|
Приготвяне на дозите
|
20.
|
Препоръчително е изпитваният химикал да се прилага орално, освен ако други пътища на приложение не бъдат определени като по-подходящи. При избор на оралния път, изпитваният химикал обикновено се прилага с помощта на сонда; въпреки това, друга възможност за приложение на изпитваните химикали е чрез хранителната диета или питейната вода.
|
|
21.
|
Когато е необходимо, изпитваният химикал е в разтвор или в суспензия в подходящ носител. Препоръчва се, когато е възможно, най-напред да се прецени дали може се използва воден разтвор/суспензия, след това — разтвор/емулсия в масло (напр. царевично олио), а след това — разтваряне в други носители. При работа с носители, различни от вода, трябва да бъдат известни токсичните характеристики на съответния носител. Трябва да се определят стабилността и хомогенността на изпитвания химикал в носителя.
|
ПРОЦЕДУРА
Брой и пол на животните
|
22.
|
Препоръчително е в началото всяка група да е с най-малко 10 мъжки и 12-13 женски индивида. Женските се оценяват преди експозицията за естрална цикличност и животните, които не проявят типичните 4-5-дневни цикли не се включват в проучването; това е причината за препоръчителния допълнителен брой женски, с цел да се отделят по 10 женски в група. Освен в случай на ясно изразени токсични ефекти, в резултат от това се очаква да има най-малко 8 бременни женски в група, което обикновено е минималния приемлив брой бременни женски на група. Целта е да се постигнат достатъчно бременности и потомство, за да се гарантира значима оценка на потенциала на изпитвания химикал да оказва влияние върху плодовитостта, бременността, поведението на майката и кърменето, и растежа, и развитието на поколение F1 от зачатието до ден 13 след раждането.
|
Дозиране
|
23.
|
Обикновено се използват поне три опитни групи и една контролна. Нивата на дозиране може да се базират на информацията от изпитванията за остра токсичност или на резултатите от изследванията с повторна доза. С изключение на третиране с изпитвания химикал, животните в контролната група трябва да бъдат отглеждани по идентичен начин на този при животните от изпитваната група. Ако за прилагане на изпитвания химикал се използва носител, контролната група трябва да получава най-голямото използвано количество носител.
|
|
24.
|
Нивата на дозиране следва да бъдат подбрани така, че да вземат отчитат всички съществуващи и налични данни за токсичност и (токсико-) кинетика. Следва да се вземе предвид и факта, че може да съществуват различия в чувствителността на бременни и не-бременни животни. Целта при избор на най-високото ниво на доза е индуциране на токсични ефекти, но не смърт или силно страдание. След това следва да се избере последователност с намаляващи нива на дозиране с цел демонстриране на всеки отклик, свързан с дозирането и достигане на най-ниското ниво на дозиране, при което не се наблюдават неблагоприятни ефекти (NOAEL). За определяне на последователно намаляващите нива на доза в повечето случаи е най-подходящо всяка доза да намалява два до четири пъти спрямо най-близката по-висока доза, а добавянето на допълнителна четвърта изпитвана група често е за предпочитане пред използването на твърде големи интервали между дозите (напр. намаление повече от 10 пъти).
|
|
25.
|
При наличие на наблюдения за обща токсичност (напр. намалено телесно тегло, ефекти върху черния дроб, сърцето, белите дробове или бъбреците и др.) или други промени, които може да не бъдат токсични реакции (напр. намален прием на храна, уголемяване на черния дроб), наблюдаваните ефекти върху ендокринни чувствителни крайни точки следва да се тълкуват предпазливо.
|
Гранично изпитване
|
26.
|
Ако орално изследване с ниво на една доза от най-малко 1 000 mg/kg телесно тегло/ден или при приложение с храната или питейната вода с доза, която е еквивалентен процент в храната или питейната вода, като се използват процедурите, описани в настоящото изследване, не доведе до забележими токсични ефекти и, ако въз основа на данните от структурно свързани вещества не се очаква токсичност, тогава пълно изследване с използване на няколко нива на дозиране може да не се счита за необходимо. При лимитиращото изпитване може да се приложи и по-висока орална доза, когато това се налага във връзка с нивото на експозиция за населението. За другите видове приложения, като например инхалация или дермално приложение, физикохимичните свойства на изпитваните химикали често предполагат използване на максимално постижимата концентрация.
|
Прилагане на дозите
|
27.
|
Животните се дозират с изпитвания химикал в продължение на 7 дни в седмицата. Когато изпитваният химикал се прилага чрез хранене през сонда, това следва да става чрез еднократна доза за животното през стомашна тръба или подходяща интубационна канюла. Максималният обем течност, който може да бъде въведен еднократно, зависи от телесното тегло на изпитваното животно. Обемът не следва да превишава 1 ml/100 g т.м., освен в случаите, когато се прилагат водни разтвори. Тогава обемът може да достигне 2 ml/100 g т.м. С изключени на дразнещите или корозивни изпитвани химикали, които обикновено водят до засилени ефекти при по-високи концентрации, промените в изпитвания обем следва да бъдат сведени до минимум чрез коригиране на концентрацията, за да се гарантира постоянен обем при всички нива на дозиране.
|
|
28.
|
За изпитван химикал, прилаган с храната или питейната вода, е важно да се гарантира, че количествата на приложения изпитван химикал не пречат на нормалния хранителен или воден баланс. Когато изпитваният химикал се прилага чрез хранителен режим, могат да бъдат използвани постоянна хранителна концентрация (ррm) или постоянно ниво на доза спрямо телесното тегло на животното; използваната алтернатива следва да бъде описана. За изпитван химикал, прилаган с помощта на сонда, дозата следва да бъде прилагана в сходни моменти всеки ден, и коригирана най-малко веднъж седмично, за да се поддържа постоянно ниво на дозиране, предвид телесното тегло на животното.
|
Експериментален график
|
29.
|
Дозирането на двата пола следва да започне най-малко 2 седмици преди чифтосване, след като животните са аклиматизирани в продължение на най-малко пет дни и женските са били скринирани за нормален естрален цикъл (в рамките на 2-седмичния период преди третиране). Изследването следва да бъде планирано по такъв начин, че оценката на естралния цикъл да започне скоро след като животните навършат пълна полова зрялост. Това може леко да варира за различните породи плъхове в различни лаборатории, напр. плъхове Sprague Dawley - 10-седмична възраст, плъхове Wistar - около 12-седмична възраст. Майките и потомството следва да бъдат умъртвени на ден 13 след раждането, или скоро след това. Денят на раждането (тоест, когато раждането завърши) се определя като ден 0 след раждането. Женските, които не показват признаци за копулация се умъртвяват 24-26 дни след последния ден от периода за чифтосване. Дозирането продължава за два пола по време на периода за чифтосване. Мъжките следва да бъдат допълнително дозирани след периода за чифтосване, най-малкото докато завърши минималния общ период за дозиране от 28 дни. След това се умъртвяват или друга възможност е да се запазят и да продължат да бъдат дозирани за евентуално провеждане на второ чифтосване, ако това се счете за уместно.
|
|
30.
|
Ежедневното дозиране на женските майки следва да продължи през цялата бременност и най-малко до и включително ден 13 след раждането или до деня преди пожертването им. За изследвания, при които изпитваният химикал се прилага чрез инхалация или по дермален път, дозирането следва да продължи най-малко до и включително 19 ден от бременността, и да се възобнови възможно най-скоро и не по-късно от 4 ден след раждането (PND).
|
|
31.
|
Схема на експерименталния график е дадена в допълнение 2.
|
Процедура на чифтосването
|
32.
|
В настоящото изследване обикновено следва да се използва чифтосване 1:1 (един мъжки с една женска). Изключения могат да възникнат в случай на непредвидена смърт на мъжките. Женските индивиди следва да бъдат поставени заедно с мъжкия, докато не бъде забелязано доказателство за копулация или до изтичането на две седмици. Всяка сутрин женските следва да бъдат преглеждани за наличие на сперма или вагинална запушалка. Ден 0 от бременността се определя като деня, в който е потвърдено доказателството за чифтосване (откриване на вагинална запушалка или сперма). Когато чифтосването е неуспешно, може да се извърши повторно чифтосване на женските животни с мъжки животни с доказан фертилитет от същата опитна група.
|
Брой на животните в котилото
|
33.
|
На ден 4 след раждането, броят на животните във всяко котило може да се коригира като се елиминират излишните новородени чрез произволна селекция, за да се постигне, доколкото е възможно, по четири или пет новородени от един пол в котило, в зависимост от нормалния брой на животните в котилото за използваната порода плъхове. От две от излишните новородени следва да се вземат кръвни проби, които да се обединят и използват за определяне на серумните нивата на T4. Селективно елиминиране на новородени, напр. въз основа на телесното тегло, или разстоянието между ануса и половите органи (РАПО) не е уместно. Когато броят на мъжките или женските новородени не позволява отделяне по четири или пет от един пол във всяко котило, е приемливо частично коригиране (например, шест мъжки и четири женски). Няма да се елиминират новородени, когато броят на животните в котилото падне под целта за бракуване (8 или 10 новородени/котило). Ако има само едно новородено над целта за бракуване, само едно новородено ще бъде бракувано и използвано за вземане на кръв за евентуални оценки на серумните нива на T4.
|
|
34.
|
Ако броят на животните в котилото не се коригира, на ден 4 след раждането се пожертват две новородени на котило и се вземат кръвни проби за измерване на серумните концентрации на тироидния хормон. Ако е възможно, двете новородени на котило, следва да бъдат женски, с цел да се резервират мъжки новородени за оценка на запазването на зърната на млечните жлези, с изключение на случая, когато при премахването на тези новородени не остават женски за оценяване при прекратяване на изпитването. Когато броят на животните в котилото падне под 8 или 10 новородени/котило (в зависимост от нормалния брой на животните в котилото при използваната порода плъхове), няма да се елиминират новородени. Ако има само едно новородено над нормалния брой на животните в котилото, само едно новородено ще бъде елиминирано и използвано за вземане на кръв за евентуални оценки на серумните нива на T4.
|
Интравитални наблюдения
Клинични наблюдения
|
35.
|
По време на периода на изпитване следва да бъдат извършвани общи клинични наблюдения минимум веднъж на ден и по-често, когато бъдат забелязани признаци на токсичност. За предпочитане е те да се правят по едно и също време всеки ден, като се има предвид пиковия период за поява на очакваните ефекти след дозиране. Следва да се записват относимите промени в поведението, признаци за трудно или удължено раждане, както и всички признаци за токсичност, включително смъртност. Тези записи следва да включват часа на започване, степента и продължителността на признаците за токсичност.
|
Телесно тегло и консумация на храна/вода
|
36.
|
Мъжките и женските следва да се претеглят на първия ден от дозирането, и минимум всяка седмица след това, както и при прекратяване на изследването. По време на бременност, женските следва да се претеглят на дни 0, 7, 14 и 20, и в рамките на 24 часа след раждането (ден 0 или 1 след раждането) и минимум на ден 4 и 13 след раждането. Тези наблюдения трябва да се описват индивидуално за всяко полово зряло животно.
|
|
37.
|
По време на периода преди чифтосването, бременността и кърменето, консумацията на храна следва да се измерва най-малко веднъж седмично. Измерването на консумацията на храна по време на чифтосване е незадължително. Консумацията на вода по време на тези периоди също следва да се измерва, когато изпитваният химикал се прилага с питейната вода.
|
Естрални цикли
|
38.
|
Естралните цикли следва да бъдат наблюдавани преди да започне третирането, за да се подберат за изследването женски с редовна цикличност (вж. точка 22). Вагиналните натривки следва също да се наблюдават ежедневно от началото на периода на третиране до потвърждение за чифтосване. Ако има притеснения за ефекти на остър стрес, които биха могли да променят естралните цикли с началото на дозирането, лабораториите могат да изложат тестовите животни за 2 седмици, след което да вземат вагинални натривки ежедневно и да следят естралния цикъл в продължение на минимум две седмици по време на периода преди чифтосване, като наблюдението да продължи и през периода на чифтосване, докато бъде открито потвърждение за чифтосване. Вземането на клетки от влагалището/цервикса следва да бъде внимателно, за да се избегне увреждане на лигавицата, което може индуцира псевдобременност (7) (8).
|
Показатели за потомството
|
39.
|
Продължителността на бременността следва да бъде записвана и изчислявана от ден 0 от бременността. Всяко котило следва да бъде прегледано възможно най-скоро след раждането, за да се установи броя и пола на новородените, броя на мъртвородените, живородените, изтърсаците (новородени, които са значително по-малки от съответните контролни новородени) и наличието на макроскопски аномалии.
|
|
40.
|
Живите новородени следва да се преброят и да им се определи пола, а котилата да се претеглят в рамките на 24 маса след раждането (ден 0 или 1 след раждане) и поне на ден 4 и 13 след раждането. В допълнение към наблюденията, описани в точка 35, следва за се запише всяко аномално поведение на потомството.
|
|
41.
|
РАПО разстоянието следва да се измери на същия ден след раждането между PND 0 до PND 4. Телесното тегло на новороденото следва да бъде измерено в деня на измерване на РАПО, и стойността на РАПО следва да бъде нормализирана към размера на новороденото, за предпочитане към кубичния корен от телесното тегло (9). Броят на зърната/ареолите на гърдите на мъжките новородени следва да се преброи на PND 12 или 13, както се препоръчва в GD 151 на ОИСР (10).
|
Клинична биохимия
|
42.
|
От определено място се вземат кръвни проби на базата на следния график:
|
—
|
от поне две новородени на котило, на ден 4 след раждането, ако броят на новородените го позволява (вж. точки 33-34)
|
|
—
|
от всички майки и поне от две новородени на котило при прекратяването на ден 13, и
|
|
—
|
от всички зрели мъжки, при прекратяването,
|
|
Всички кръвни проби се съхраняват при подходящи условия. Кръвните проби от новородените на ден 13 и от зрелите мъжки се оценяват за серумни нива на тироидните хормони (T4). Ако е уместно се прави допълнителна оценка на T4 в кръвните проби от самките и новородените на ден 4. Също ако е уместно, може да се извърши незадължително измерване на други хормони. Кръвта на новородените може да се обедини по котила за анализите на тироидния хормон. За предпочитане е тироидните хормони (T4 и TSH) да се измерват като „общи“.
|
43.
|
Следните фактори могат да повлияят на варирането и на абсолютните концентрации при определянето на хормони:
|
—
|
времето на умъртвяване, поради дневните колебания в концентрацията на хормони
|
|
—
|
методът на умъртвяване, за да се избегне ненужен стрес за животните, който може да повлияе на концентрациите на хормони
|
|
—
|
комплектите за изпитване за определяне на хормони, които могат да се различават по стандартните си криви.
|
|
|
44.
|
Пробите от плазма, специално предназначени за определяне на хормони, следва да бъдат получени в сравними периоди през деня. Числовите стойности, получени при анализа на концентрации на хормони, се различават при употребата на различни търговски комплекти за анализ.
|
Патология
Макроскопска аутопсия
|
45.
|
В момента на умъртвяване или смърт по време на изследването, следва да се извърши макроскопски оглед на възрастните животни за всякакви аномалии или патологични изменения. Особено внимание при огледа трябва да се обърне на органите на половата система. Броят на местата за имплантация следва да бъде записан. Вагиналните натривки следва да бъдат изследвани на сутринта в деня на аутопсия, за да се определи стадия на естралния цикъл и да се даде възможност за корелация с хистопатологичното изследване на яйчниците.
|
|
46.
|
Тестисите и епидидимите, както и простатата и семенните мехурчета с коагулиращите жлези като цяло трябва да бъдат изрязани от всички мъжки зрели животни и почистени от прилежащите тъкани, според необходимостта, и трябва да се претеглят в мокро състояние възможно най-скоро след дисекцията, за да не се допусне изсъхването им. В допълнение, опционалното тегло на органите може да включва мускула повдигач на ануса плюс булбокавернозния мускулен комплекс, жлезите на Каупър и главичката на пениса при мъжките и двойката яйчници (мокро тегло) и матката (включително маточната шийка) при женските; ако са включени, тези тегла следва да бъдат взети възможно най-скоро след дисекцията.
|
|
47.
|
Мъртвите новородени и новородените, умъртвени на ден 13 след раждането или скоро след това, трябва най-малко да бъдат внимателно огледани външно за макроскопски аномалии. Особено внимание следва да се обърне външните репродуктивни полови органи, които да се огледат за признаци на променено развитие. На ден 13 трябва да се запази щитовидната жлеза от 1 мъжко и 1 женско новородено на котило.
|
|
48.
|
Яйчниците, тестисите, допълнителните полови органи (матката и шийката на матката, епидидимите, простатата, семенните мехурчета плюс коагулиращите жлези), щитовидната жлеза и всички органи показващи макроскопски лезии на всички възрастни животни следва да бъдат запазени. Фиксация във формалин не се препоръчва за рутинно изследване на тестиси и епидидими. Приемлив метод за тези тъкани е използването на фиксаж на Буен или модифициран Дейвидсън (11). Бялото покривало (tunica albuginea) може внимателно да се пробие на неголяма дълбочина в двата края на органа с помощта на игла, за да се позволи бързо проникване на фиксатора.
|
Хистопатология
|
49.
|
Подробно хистопатологично изследване следва да бъде направено на яйчниците, тестисите и епидидимите (със специален акцент върху етапите на сперматогенеза и хистопатология на интерстициалната тестикуларна клетъчна структура) на животните от групата, третирана с най-висока доза и контролната група. Другите запазени органи, включително щитовидната жлеза от новородени и възрастни животни могат да бъдат изследвани, когато е необходимо. Теглото на щитовидната жлеза може да се установи след фиксация. Почистването също следва да се осъществи много внимателно и само след фиксиране, за да се избегне увреждане на тъканите. Увреждането на тъканите може да постави под съмнение хистопатологичния анализ. Изследванията следва да обхванат и животните от групи с други нива на дозата, ако бъдат забелязани промени в групата, третирана с най-висока доза. В Указанията за хистопатология (11) се съдържа допълнителна подробна информация относно дисекцията, фиксирането, разрязването и хистопатологията на ендокринните тъкани.
|
ДАННИ И ПРОТОКОЛИРАНЕ
Данни
|
50.
|
Следва да бъдат осигурени индивидуални данни за всяко животно. Допълнително, всички данни следва да бъдат обобщени в таблична форма, показвайки за всяка изпитвана група броя на животните в началото на изпитването, броя на животните открити мъртви по време на изпитването или умъртвени по хуманни причини, времето на смъртта или хуманното умъртвяване, броя на плодовитите животни, броя на бременните женски, броя на животните показващи признаци на токсичност, описание на наблюдаваните признаци на токсичност, включително времето на началото, продължителността и сериозността на всички токсични ефекти, видовете хистопатологични изменения и всички относими данни за котилото. Табличен формат на обобщено протоколиране, който се е доказал като особено полезен за оценката на ефекта върху репродукцията /развитието, е даден в допълнение 3.
|
|
51.
|
Поради ограничените размери на изследването, статистически анализи във формата на изпитвания за „значимост“ са с ограничена стойност за много крайни точки, и особено за крайните точки на репродукцията. Ако се използват статистически анализи, тогава избраният метод следва да бъде подходящ за разпределението на изследваната променлива и да бъде избран преди началото на изследването. Статистическият анализ на РЕПО и запазването на зърната на млечните жлези трябва да бъде базиран на индивидуални данни за новороденото, като взема под внимание ефектите на котилото. Където е уместно, котилото е единицата за анализ. Статистическият анализ на телесното тегло на новороденото трябва да бъде базиран на индивидуални данни за новороденото, като взема под внимание броя на животните в котилото. Поради малкия размер на групите, използването на контролни данни от предходни периоди (напр. за брой на животните в котилото), когато са налични, могат също да бъдат полезни като спомагателно средство за интерпретиране на изследването.
|
Оценка на резултатите
|
52.
|
Констатациите на това изследване за токсичност следва да бъдат оценени като се вземат предвид наблюдаваните ефекти, аутопсията и микроскопските находки. Оценката включва връзката между дозата на изпитвания химикал и наличието или отсъствието, разпространението и сериозността на аномалиите, включително макроскопски лезии, идентифицирани прицелни органи, безплодие, клинични аномалии, засегната ефективност на репродукцията и на котилото, промени в телесното тегло, ефекти върху смъртността и всякакви други токсични ефекти.
|
|
53.
|
Поради краткия период на третиране на мъжките индивиди, хистопатологичното изследване на тестисите и епидидимите следва да се разглеждат заедно с данните за плодовитост, когато се оценяват ефектите върху мъжката репродуктивност. Използването на контролни данни от предходни периоди за репродукцията/развитието на организма (напр., относно броя на животните в котилото, РЕПО, запазване на зърната на млечните жлези, серумни нива на T4), когато са налични, могат също да бъдат полезни като спомагателно средство за интерпретиране на изследването.
|
|
54.
|
За контрол на качеството се предлага да се събират контролни данни за минали периоди и да се изчисляват коефициенти на вариация за числовите данни, особено за параметрите, свързани с откриване на нарушители на функциите на ендокринната система. Тези данни могат да бъдат използвани за сравнение когато се извършва оценка на актуалните изследвания.
|
Протокол от изпитването
|
55.
|
Протоколът от изпитването следва да включва следната информация:
|
|
Изпитван химикал:
|
—
|
източник, номер на партидата, крайна дата за употреба, ако има такава
|
|
—
|
стабилност на изпитвания химикал, ако е известна.
|
|
|
|
Вещество с една съставка:
|
—
|
външен вид, разтворимост във вода и допълнителни относими физични и химични свойства;
|
|
—
|
химична идентификация, като например наименование по IUPAC или CAS, CAS номер, код SMILES или InChI, структурна формула, чистота, химична идентичност на онечистванията, доколкото е целесъобразно и практически осъществимо, и т.н.
|
|
|
|
Вещество с повече съставки, UVCB и смеси:
|
—
|
определени, доколкото е възможно, с химичната си идентичност (вж. по-горе), количествения състав и относимите физични и химични свойства на съставките.
|
|
|
|
Носител (когато се използва такъв):
|
—
|
обосновка за избора на носител, когато е различен от вода.
|
|
|
|
Изпитвани животни:
|
—
|
брой, възраст и пол на животните;
|
|
—
|
източник за доставка на животните, условия на отглеждане, хранителен режим и т.н.;
|
|
—
|
индивидуално тегло на животните в началото на изпитването.
|
|
—
|
обосновка при използване на видове, различни от плъх.
|
|
|
|
Условия на изпитването:
|
—
|
обосновка за избора на нивото на доза;
|
|
—
|
подробна информация за това в какъв вид е изпитваният химикал/хранителната смес, постигнати концентрации, стабилност и хомогенност на сместа;
|
|
—
|
подробна информация относно прилагането на изпитвания химикал;
|
|
—
|
превръщане от концентрация на изпитвания химикал в храна/питейна вода (ppm) в действителна доза (mg на kg телесно тегло дневно), ако е приложимо,
|
|
—
|
подробности относно качество на храната и водата;
|
|
—
|
подробно описание на процедурата за рандомизация при подбора на новородени за бракуване, ако се прилага бракуване.
|
|
|
|
Резултати:
|
—
|
телесно тегло/промени в телесното тегло;
|
|
—
|
консумация на храна и консумация на вода, ако има налични данни;
|
|
—
|
данни за токсичната реакция по пол и доза, включително плодовитост, бременност и всякакви други признаци на токсичност;
|
|
—
|
продължителност на бременността;
|
|
—
|
токсични или други ефекти върху репродукцията, потомството, постнаталния растеж и др.;
|
|
—
|
природа, сила и продължителност на клиничните наблюдения (независимо обратими или необратими),
|
|
—
|
брой на възрастните женски с нормален или аномален естрален цикъл и продължителност на цикъла;
|
|
—
|
брой на живородените и пост-имплантационни загуби;
|
|
—
|
данни за телесното тегло на новороденото
|
|
—
|
РЕПО на всички новородени (и телесно тегло в деня на измерване на РЕПО)
|
|
—
|
запазване на зърната на млечните жлези при мъжките новородени,
|
|
—
|
нива на тириодния хормон, новородени на ден 13 и възрастни мъжки (и самки и новородени на ден 4, ако е измерван)
|
|
—
|
брой новородени с видими макроскопски аномалии, максоскопска оценка на външните полови органи, брой на изтърсаците;
|
|
—
|
време на смъртта в процеса на изпитването или дали животните са преживели до края на изпитването;
|
|
—
|
брой на имплантациите, брой на животните в котилото и тегло на котилото към момента на записване;
|
|
—
|
телесно тегло в момента на пожертване и данни за тегло на органи за животните родители;
|
|
—
|
находки при аутопсията;
|
|
—
|
подробно описание на хистопатологичните находки;
|
|
—
|
данни за абсорбция (ако има);
|
|
—
|
статистическа обработка на резултатите, ако е подходящо.
|
|
Обсъждане на резултатите.
Заключения.
|
Обяснение на резултатите
|
56.
|
Изследването предоставя оценки на токсичността на репродукцията/развиващия се организъм, свързана с приложението на повторени дози (вж. точки 5 и 6). Може да представи указание за необходимостта от провеждане на допълнителни разследвания и предоставя насоки за планиране на последващи изпитвания. Следва да се консултирате с Ръководството на ОИСР 43 за помощ при тълкуването на резултатите, свързани с репродукцията и развитието на организма (12). Ръководство № 106 на ОИСР относно Хистологичната оценка на ендокринните и репродуктивни изпитвания на гризачи (11) предоставя информация относно подготовката и оценката на (ендокринните) органи и вагиналните натривки, които могат да бъдат полезни за настоящите TG.
|
ЛИТЕРАТУРА
|
(1)
|
OECD (1990). Room Document No 1 for the 14th Joint Meeting of the Chemicals Group and Management Committee. Available upon request at Organisation for Economic and Cooperation and Development, Paris.
|
|
(2)
|
OECD (1992). Chairman’s Report of the ad hoc Expert Meeting on Reproductive Toxicity Screening Methods, Tokyo, 27th-29th October, 1992. Available Upon Request at Organisation for Economic Cooperation and Development, Paris.
|
|
(3)
|
OECD (1998). Report of the First Meeting of the OECD Endocrine Disrupter Testing and Assessment (EDTA) Task Force, 10th-11th March 1998. Available Upon Request at Organisation for Economic Cooperation and Development, Paris.
|
|
(4)
|
OECD (2015). Feasibility Study for Minor Enhancements of TG 421/422 with ED Relevant Endpoints. Environment, Health and Safety Publications, Series on Testing and Assessment (No 217), Organisation for Economic Cooperation and Development, Paris.
|
|
(5)
|
OECD (2000). Guidance Document on the Recognition, Assessment, and Use of Clinical Signs as Humane Endpoints for Experimental Animals Used in Safety Evaluations. Series on Testing and Assessment, (No 19), Organisation for Economic Cooperation and Development,.Paris.
|
|
(6)
|
OECD (2011). Guidance Document on Standardised Test Guidelines for Evaluating Chemicals for Endocrine Disruption. Environment, Health and Safety Publications, Series on Testing and Assessment(No 150), Organisation for Economic Cooperation and Development, Paris.
|
|
(7)
|
Goldman, J.M., Murr A.S., Buckalew A.R., Ferrell J.M. and Cooper R.L. (2007). The Rodent Estrous Cycle: Characterization of Vaginal Cytology and its Utility in Toxicological Studies, Birth Defects Research, Part B, 80 (2), 84-97.
|
|
(8)
|
Sadleir R.M.F.S (1979). Cycles and Seasons, in Auston C.R. and Short R.V. (eds.), Reproduction in Mammals: I. Germ Cells and Fertilization, Cambridge, New York.
|
|
(9)
|
Gallavan R.H. Jr, Holson J.F., Stump D.G., Knapp J.F. and Reynolds V.L.(1999). Interpreting the Toxicologic Significance of Alterations in Anogenital Distance: Potential for Confounding Effects of Progeny Body Weights, Reproductive Toxicology, 13: 383-390.
|
|
(10)
|
OECD (2013). Guidance Document in Support of the Test Guideline on the Extended One Generation Reproductive Toxicity Study. Environment, Health and Safety Publications, Series on Testing and Assessment (No 151), Organisation for Economic Cooperation and Development, Paris.
|
|
(11)
|
OECD (2009). Guidance Document for Histologic Evaluation of Endocrine and Reproductive Tests in Rodents. Environment, Health and Safety Publications, Series on Testing and Assessment (No106), Organisation for Economic Cooperation and Development, Paris.
|
|
(12)
|
OECD (2008). Guidance Document on Mammalian Reproductive Toxicity Testing and Assessment. Environment, Health and Safety Publications, Series on Testing and Assessment (No 43), Organisation for Economic Cooperation and Development, Paris.
|
Допълнение 1
ОПРЕДЕЛЕНИЯ (ВИЖТЕ И GD 150 НА ОИСР (6))
Андрогенност е способността на даден химикал да действа като естествен андрогенен хормон (напр. тестостерон) в организма на бозайник.
Антиандрогенност е способността на даден химикал да потиска действието на естествен андрогенен хормон (напр. тестостерон) в организма на бозайник.
Антиестрогенност е способността на даден химикал да потиска действието на естествен естрогенен хормон (напр. естрадиол 17ß) в организма на бозайник.
Антитиреоидна активност е способността на даден химикал да потиска действието на естествен тиреоиден хормон (напр. T3) в организма на бозайник.
Химикал означава вещество или смес.
Токсичност за развиващия се организъм: проявата на репродуктивна токсичност, представляваща пред-, пери- пост-натални, структурни или функционални разстройства в потомството.
Дозировка е общ термин, обхващащ дозата, нейната честота и продължителност на дозиране.
Доза е количеството приложен изпитван химикал. Дозата се изразява като маса на изпитвания химикал за единица телесно тегло на изпитваното животно на ден (напр. mg на kg телесно тегло дневно), или като постоянна концентрация в храната.
Явна токсичност е общ термин, описващ ясните признаци на токсичност след прилагане на изпитвания химикал. Те следва да бъдат достатъчни за оценка на опасността и следва да бъдат такива, че при увеличаване на прилаганата доза да се очаква развитие на признаци на силна токсичност и вероятна смърт.
Увреждане на плодовитостта представлява разстройства на мъжките или женските възпроизводителни функции или способности.
Токсичност при майката: неблагоприятни ефекти върху бременните женски, възникващи или специфично (пряк ефект), или неспецифично (непряк ефект).
NOAEL е съкращение за нивото, при което не се наблюдават неблагоприятни ефекти. Това е най-високото ниво на доза, при което не се наблюдават свързани с третирането неблагоприятни находки в резултат от третирането.
Естрогенност е способността на даден химикал да действа като естествен естрогенен хормон (напр. естрадиол 17ß) в организма на бозайник.
Репродуктивна токсичност представлява вредни ефекти върху потомството и/или увреждане на мъжките и женските възпроизводителни функции или способности.
Изпитван химикал е всяко вещество или смес, изпитвано(а) с използване на настоящия метод за изпитване.
Тиреоидна активност е способността на даден химикал да действа като естествен тиреоиден хормон (напр. T3) в организма на бозайник.
Валидиране е научна процедура, предназначена за характеризиране на оперативните изисквания и ограничения на даден метод за изпитване и за доказване на неговата надеждност и относимост за определена цел.
Допълнение 2
СХЕМА НА ЕКСПЕРИМЕНТАЛНИЯ ГРАФИК, УКАЗВАЩА МАКСИМАЛНАТА ПРОДЪЛЖИТЕЛНОСТ НА ИЗСЛЕДВАНЕТО, НА БАЗАТА НА ПЪЛЕН 14-ДНЕВЕН ПЕРИОД НА ЧИФТОСВАНЕ
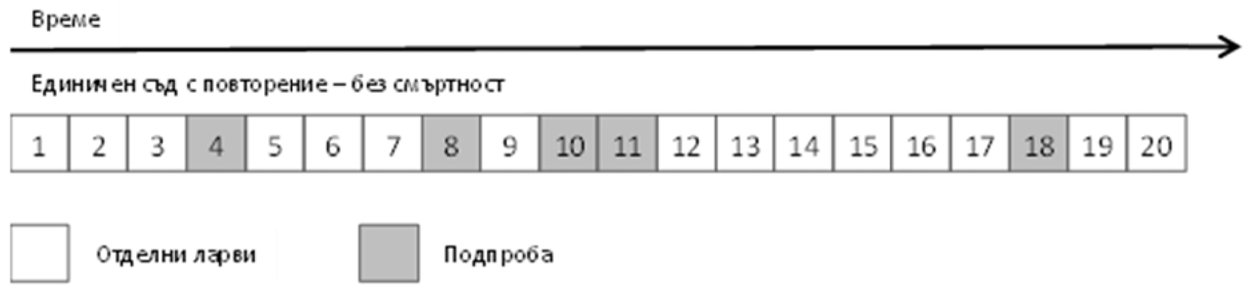
Допълнение 3
ОБОБЩЕН ДОКЛАД В ТАБЛИЧЕН ВИД НА ЕФЕКТИТЕ ВЪРХУ РЕПРОДУКЦИЯТА/РАЗВИТИЕТО
|
КОНСТАТАЦИИ И ОЦЕНКИ
|
Стойности
|
|
|
|
Дозировка (единици)
|
0 (контрол)
|
…
|
…
|
…
|
…
|
|
Стартирани двойки (N)
|
|
|
|
|
|
|
Естрален цикъл (поне средна продължителност и честота на нередовни цикли)
|
|
|
|
|
|
|
Женски, показващи потвърждение за копулация (N)
|
|
|
|
|
|
|
Женски, постигнали забременяване (N)
|
|
|
|
|
|
|
Дни от зачатието 1 - 5 (N)
|
|
|
|
|
|
|
Дни от зачатието 6 -… (21) (N)
|
|
|
|
|
|
|
Бременност ≤ 21 дни (N)
|
|
|
|
|
|
|
Бременност = 22 дни (N)
|
|
|
|
|
|
|
Бременност ≥ 23 дни (N)
|
|
|
|
|
|
|
Самки с живородени малки (N)
|
|
|
|
|
|
|
Самки с живи малки на 4 ден след раждането (N)
|
|
|
|
|
|
|
Импланти/самка (средно)
|
|
|
|
|
|
|
Живи новородени/самка при раждането (средно)
|
|
|
|
|
|
|
Живи новородени/самка на 4 ден (средно)
|
|
|
|
|
|
|
Съотношение на половете (м/ж) при раждането (средно)
|
|
|
|
|
|
|
Съотношение на половете (м/ж) на 4 ден (средно)
|
|
|
|
|
|
|
Тегло на животните в котилото при раждането (средно)
|
|
|
|
|
|
|
Тегло на животните в котилото на 4 ден (средно)
|
|
|
|
|
|
|
Тегло на новороденото при раждането (средно)
|
|
|
|
|
|
|
Тегло на новороденото към момента на измерване на РЕПО (средно мъжки, средно женски)
|
|
|
|
|
|
|
РЕПО на новороденото на същия постнатален ден, раждане — ден 4 (средно мъжки, средно женски, отбелязване PND)
|
|
|
|
|
|
|
Тегло на новороденото на 4 ден (средно)
|
|
|
|
|
|
|
Запазване на зърната на млечните жлези при мъжките новородени на ден 13 (средно)
|
|
|
|
|
|
|
Тегло на новороденото на 13 ден (средно)
|
|
|
|
|
|
|
|
|
НОВОРОДЕНИ С АНОМАЛИИ
|
|
Самки с 0
|
|
|
|
|
|
|
Самки с 1
|
|
|
|
|
|
|
Самки с ≥ 2
|
|
|
|
|
|
|
|
|
ЗАГУБА НА ПОТОМСТВО
|
|
|
|
Пре-натално/след имплантация (имплантации минус живородени)
|
|
Женски с 0
|
|
|
|
|
|
|
Женски с 1
|
|
|
|
|
|
|
Женски с 2
|
|
|
|
|
|
|
Женски с ≥ 3
|
|
|
|
|
|
|
|
|
Постнатално (живородени минус живи на постнатален ден 13)
|
|
Женски с 0
|
|
|
|
|
|
|
Женски с 1
|
|
|
|
|
|
|
Женски с 2
|
|
|
|
|
|
|
Женски с ≥ 3
|
|
|
|
|
|
B.64 КОМБИНИРАНО ТОКСИКОЛОГИЧНО ИЗСЛЕДВАНЕ С ПОВТАРЯЩА СЕ ДОЗА СЪС СКРИНИНГ ИЗПИТВАНЕ ЗА ТОКСИЧНОСТ ЗА РЕПРОДУКЦИЯТА/РАЗВИВАЩИЯ СЕ ОРГАНИЗЪМ
ВЪВЕДЕНИЕ
|
1.
|
Настоящият метод за изпитване е еквивалентен на насоките за изпитване на ОИСР (ТG) 422 (2016). Насоките на ОИСР за изпитване на химикали се преразглеждат периодично в светлината на научния прогрес. Оригиналните насоки за скрининг изпитване 422 бяха приети през 1996 г., на базата на протокол за „Комбинирано скрининг изпитване с повтаряща се доза и за токсичност за репродукцията/развиващия се организъм“, обсъден на две експертни срещи в Лондон през 1990 г. (1) и в Токио през 1992 г. (2).
|
|
2.
|
Настоящият метод за изпитване съчетава частта за скрининг за токсичност за репродукцията/развиващия се организъм, която се основава на опита, придобит от страните членки при използването на оригиналния метод върху съществуващи химикали, произвеждани в големи количества и в проучвателни изпитвания с вещества за положителна контрола (3) (4), и частта за токсичност с повтаряща се доза, в съответствие с насоките за изпитване на ОИСР 407 (28-дневно изследване за токсичност при гризачи с повтаряща се орална доза, съответстващо на Глава Б.7 от настоящото Приложение).
|
|
3.
|
Този метод за изпитване бе актуализиран с крайни точки, относими към вещества, нарушаващи функцията на ендокринната система, като последващо действие към дейност с висок приоритет, инициирана в ОИСР през 1998 г. за ревизиране на съществуващите насоки за изпитвания и за разработване на нови насоки за изпитвания за скрининг и изпитване на веществата с потенциал да нарушават функцията на ендокринната система (5). В този контекст TG 407 (съответстващи на глава Б.7 от настоящото приложение) бяха подобрени през 2008 г. с подходящи параметри за откриване на ендокринна активност на изпитваните химикали. Целта при актуализиране на TG 422 бе да се включат някои крайни точки, относими към вещества, нарушаващи функцията на ендокринната система в насоките за скринингови изпитвания (TG), при които периодите на експозиция обхващат някои от чувствителните периоди по време на развитието на организма (пред- или ранните постнатални периоди).
|
|
4.
|
Избраните допълнителни крайни точки, относими към вещества, нарушаващи функцията на ендокринната система, също част от TG 443 (Разширено изследване за токсичност за репродукцията в едно поколение, съответстващо на глава Б.56 от настоящото приложение), бяха включени в TG 422 на базата на проучване на осъществимостта, разглеждащо научни и технически въпроси, свързани с тяхното включване, както и възможните адаптации на плана на изпитването, необходими за тяхното включване (6).
|
|
5.
|
Настоящият метод за изпитване е проектиран да генерира ограничена информация относно ефектите на изпитвания химикал върху действието на мъжката и женската репродуктивна система, напр. функция на половите жлези, поведение при чифтосване, зачеване, развитие на заченатия организъм и раждане. Той не е алтернативен на съществуващите методи за изпитване Б.31, Б.34, Б.35 или Б.56, нито ги замества.
|
ПЪРВОНАЧАЛНИ СЪОБРАЖЕНИЯ
|
6.
|
При оценката и оценяването на токсичните характеристики на изпитван химикал след получаване на първоначалната информация за токсичността от изпитването за остра токсичност може да се извърши определяне на оралната токсичност с използване на повтаряща се доза. Това проучване предоставя информация за възможните опасности за здравето, които е вероятно да възникнат при повторна експозиция за относително ограничен период от време. Методът се състои от базово проучване за токсичност с повтаряща се доза, което може да се използва за химикали, за които 90-дневно изследване не е гарантирано (напр. когато произведеното количество не превишава определени граници) или като предварително изследване към дългосрочно проучване. При провеждане на проучването следва да се спазват ръководните принципи и съображения, очертани в Ръководство на ОИСР № 19 относно признаването, оценката и употребата на клинични признаци като хуманни крайни точки за експериментални животни, използвани за оценяване на безопасността (7).
|
|
7.
|
Освен това то включва и скринингово изпитване за токсичност за репродукцията/развиващия се организъм и поради това може да бъде използвано за осигуряване на първоначална информация за възможните ефекти върху мъжката и женската репродуктивна способност, като например функция на половите жлези, поведение при чифтосване, зачеване, развитие на зародиша и раждане, или в ранен етап на оценка на токсикологичните свойства на изпитваните химикали, или за изпитваните химикали, които пораждат безпокойство. Настоящият метод за изпитване не предоставя пълна информация по всички аспекти на репродукцията и развитието на организма. По-конкретно, той предлага единствено ограничени средства за откриване на постнатални прояви на пренатална експозиция, или ефекти, които може да бъдат индуцирани по време на постнатална експозиция. Наред с другите причини, поради избирателността на крайните точки и кратката продължителност на проучването, този метод няма да предостави доказателство за категорични твърдения за липса на ефекти върху репродукцията/развитието. Нещо повече, при отсъствието на данни от други изследвания за токсичност за репродукцията/развиващия се организъм, положителните резултати са полезни за първоначалната оценка на опасността и допринасят за вземането на решения по отношение на необходимостта и графика за допълнителни изпитвания.
|
|
8.
|
Резултатите, получени от свързаните с жлезите с вътрешна секреция параметри, следва да се разглеждат в контекста на „Концептуалната рамка на ОИСР за изпитване и оценка на химикали, нарушаващи функциите на ендокринната система“ (8). В тази Концептуална рамка подобрените TG 422 на ОИСР се съдържат в ниво 4 като изследване in vivo, предоставящо данни за неблагоприятните ефекти върху крайните точки, относими към ендокринната система. Все пак, един ендокринен сигнал сам по себе си не може да се разглежда като достатъчно доказателство, че изпитваният химикал е вещество, нарушаващо функциите на ендокринната система.
|
|
9.
|
Методът на изпитване набляга и върху неврологичните ефекти като специфична крайна точка, а акцентът е поставен върху необходимостта от внимателни клинични наблюдения на животните с оглед получаване на възможно най-пълна информация. Методът следва да идентифицира химикали с невротоксичен потенциал и може да служи за основание за допълнително задълбочено проучване в тази посока. В допълнение, методът може да предоставя и базови указания за имунологичните ефекти.
|
|
10.
|
При отсъствието на данни от други изследвания за системна токсичност, токсичност за репродукцията/развиващия се организъм, невротоксичност и/или имунотоксичност, положителните резултати са полезни за първоначалната оценка на опасността и допринасят за вземането на решения по отношение на необходимостта и графика за допълнителни изпитвания. Изпитването може да е особено полезно като част от Информационния набор от скрининг информация (SIDS) на ОИСР за оценка на съществуващите химикали, за които съществува малко или никаква информация и може да служи като алтернатива на провеждането на две отделни изпитвания за токсичност с повтаряща се доза (ОИСР TG 407, съответстващи на глава Б.7 от настоящото приложение) и токсичност за репродукцията/развиващия се организъм (ОИСР TG 421, съответстващи на глава Б.63 от настоящото приложение), респективно. Може да се използва и като проучване за определяне на обхвата на дозата за по-обхватни проучвания по отношение на репродукцията/развиващия се организъм или когато се приема за уместно по други причини.
|
|
11.
|
Като цяло се смята, че съществуват различия в чувствителността между бременни и небременни животни. Следователно в настоящото комбинирано изпитване може да е по-сложно да се определят нива на дози, които да са адекватни за оценяване както на общата системна токсичност, така и на специфичната токсичност за репродукцията/развиващия се организъм, отколкото когато се провеждат индивидуални изпитвания поотделно. Нещо повече, тълкуването на резултатите от изпитването по отношение на общата системна токсичност може да бъде по-трудно отколкото когато се провежда отделно проучване с повтаряща се доза, особено когато не се оценяват серумните и хистопатологичните параметри по едно и също време в проучването. Поради тази техническа усложненост, при изпълнение на подобно комбинирано скринингово изпитване се изисква значителен опит в изпитванията за токсичност. От друга страна, освен малкия брой участващи животни, комбинираното изпитване може да предложи по-добри средства за разграничаване на преките ефекти върху репродукцията/развитието от ефектите, които са вторични спрямо други (системни) ефекти.
|
|
12.
|
При това изпитване, периодът на дозиране е по-дълъг от този при конвенционалното 28-дневно проучване с повтаряща се доза. В него обаче се използват по-малко животни от всеки пол в група, отколкото в ситуацията, при която в допълнение към скрининговото изпитване за токсичност за репродукцията/развиващия се организъм се провежда конвенционално 28-дневно проучване с повтаряща се доза.
|
|
13.
|
Настоящият метод за изпитване предполага орален прием на изпитвания химикал. Ако се използват други пътища на експозиция, може да се наложат изменения на метода.
|
|
14.
|
Преди използването на метода за изпитване по отношение на смес с цел генериране на данни за планирана регулаторна цел, следва да се разгледа въпросът дали той може да даде адекватни резултати за тази цел и, ако е така, защо. Подобни съображения не са необходими, когато има регулаторно изискване за изпитване на сместа.
|
|
15.
|
Използваните оределения са дадени в допълнение 1.
|
ПРИНЦИП НА ИЗПИТВАНЕТО
|
16.
|
Изпитваният химикал се прилага в нарастващи дози на няколко групи от мъжки и женски индивиди. Мъжките следва да бъдат дозирани в продължение на най-малко четири седмици включително до деня преди планираното им умъртвяване и (това включва минимум две седмици преди чифтосване, по време на периода на чифтосване и приблизително две седмици след чифтосване). Предвид ограничения период на дозиране на мъжките преди чифтосване, плодовитостта може да не бъде особено чувствителен показател за тестикуларна токсичност. Ето защо, детайлно хистологично изследване на тестисите е от съществено значение. Комбинацията от период на дозиране преди чифтосване от две седмици и последващи наблюдения на чифтосването/плодовитостта с цялостния периодна дозиране от минимум четири седмици, последван от детайлни хистопатологични изследвания на мъжките полови жлези, се разглежда като достатъчен, за да могат да бъдат определени голяма част от ефектите върху мъжката плодовитост и сперматогенеза.
|
|
17.
|
Женските следва да бъдат дозирани през целия период на проучването. Това включва две седмици преди чифтосване (като целта е да се обхванат най-малко два пълни естрални цикъла), променливото време до зачеване, продължителността на бременността и най-малко тринадесет дни след раждането, включително деня преди планираното им умъртвяване.
|
|
18.
|
Продължителността на проучването, след аклиматизацията и оценката на естралния цикъл преди дозиране, зависи от функционирането на женската полова система и е приблизително 63 дни, [най-малко 14 дни преди чифтосването, (до) 14 дни за чифтосване, 22 дни бременност, 13 дни лактация].
|
|
19.
|
По време на периода на прилагане, животните се наблюдават внимателно и ежедневно за признаци на токсичност. Животните, които умират или са умъртвени по време на изпитването, се аутопсират, а преживелите животни се умъртвяват и се аутопсират при приключването на изпитването.
|
ОПИСАНИЕ НА МЕТОДА
Избор на животински вид
|
20.
|
Настоящият метод за изпитване е предназначен за употреба с плъхове. Ако параметрите, определени в рамките на тези TG 422, се изследват при друг вид гризачи, следва да бъде представена детайлна обосновка. Плъховете са единственият биологичен вид, използван в рамките на международната валидационна програма за откриване на вещества, нарушаващи функциите на ендокринната система в TG 407. Не трябва да се използват породи с ниска плодовитост или породи, за които е характерна висока честота на вродените аномалии. Трябва да се използват здрави, нераждали животни, които не са били подлагани на предишни експериментални процедури. Тестовите животни следва да се харакеризират по вид, порода, пол, тегло и възраст. При започване на изследването разликите в теглото на използваните животни следва да бъдат минимални и да не надвишава 20 % от средното за всеки пол тегло. Когато изследването се провежда като предварително до дългосрочно проучване или проучване на цяло поколение, за предпочитане е в двете изследвания да се използват животни от една и съща порода и източник.
|
Условия на отглеждане и хранене
|
21.
|
Всички процедури трябва да са в съответствие с местните стандарти за полагане на грижи за лабораторни животни. Температурата в стаята на опитните животни трябва да бъде 22 °C (± 3 °C). Относителната влажност трябва да е най-малко 30 % и за предпочитане да не превишава 70 %, освен по време на почистване на помещението. Осветлението трябва да бъде изкуствено, като последователността е 12 часа светлина, 12 часа тъмнина. За храненето могат да се използват обичайните лабораторни хранителни режими с неограничен достъп до вода за пиене. Изборът на хранителен режим може да бъде повлиян от нуждата да се осигури подходящо смесване с изпитван химикал, когато прилагането му е по настоящия метод.
|
|
22.
|
Животните се настаняват групово, в малки групи от един и същи пол; животните могат да се настаняват отделно, ако това е научно обосновано. При групово настаняване в клетки не следва да има повече от пет животни в клетка. Чифтосването трябва да се извърши в клетки, подходящи за тази цел. Бременните женски следва да бъдат настанявани поотделно и да им бъдат предоставяни материали за изграждане на гнездо. Кърмещите женски се настаняват поотделно заедно с потомството си.
|
|
23.
|
Храната следва да бъде редовно анализирана за наличието на замърсители. Проба от хранителния режим трябва да се съхранява до окончателното изготвяне на доклада.
|
Подготовка на опитните животни
|
24.
|
Здрави, млади, полово зрели животни се определят на произволен принцип за контролните и опитните групи. Клетките трябва да бъдат подредени по такъв начин, че възможните ефекти в резултат на местоположението на клетката да бъдат сведени до минимум. Животните се идентифицират по уникален начин и се настаняват в клетките им най-малко пет дни преди началото на проучването, за да им се осигури време за аклиматизиране към лабораторните условия.
|
Приготвяне на дозите
|
25.
|
Препоръчително е изпитваният химикал да се прилага орално, освен ако други пътища на приложение не бъдат определени като по-подходящи. При избор на оралния път, изпитваният химикал обикновено се прилага с помощта на сонда; въпреки това, друга възможност за приложение на изпитваните химикали е също чрез хранителната диета или питейната вода.
|
|
26.
|
Когато е необходимо, изпитваният химикал е в разтвор или в суспензия в подходящ носител. Препоръчва се, когато е възможно, най-напред да се прецени дали може се използва воден разтвор/суспензия, след това да се прецени за разтвор/емулсия в масло (напр. царевично олио), а след това — за разтваряне в други носители. При работа с не-водни носители, трябва да бъдат известни токсичните характеристики на съответния носител. Трябва да се определят стабилността и хомогенността на изпитвания химикал в носителя.
|
ПРОЦЕДУРА
Брой и пол на животните
|
27.
|
Препоръчително е в началото всяка група да е с най-малко 10 мъжки и 12-13 женски индивида. Женските се оценяват преди експозицията за естрална цикличност и животните, които не проявят типичните 4-5-дневни цикли, не се включват в проучването; това е причината за препоръчителния допълнителен брой женски, с цел да се отделят по 10 женски в група. Освен в случай на ясно изразени токсични ефекти, в резултат от това се очаква да има най-малко 8 бременни женски в група, което обикновено е минималният приемлив брой бременни женски на група. Целта е да се осигурят достатъчно бременности и потомство, за да се гарантира значима оценка на потенциала на изпитвания химикал да оказва влияние върху плодовитостта, бременността, поведението на майката и това на малките при кърмене, растежа и развитието на поколение F1 от зачеването до ден 13 след раждането. Ако междувременно се планира убиване на животни преди завършването на изследването, броят се увеличава в зависимост от броя на животните, които е планирано да бъдат убити. Следва да бъде разгледана възможността за допълнителна сателитна група от по пет животни от всеки пол в контролната група и в групата, третирана с най-високата доза за наблюдение на обратимостта, устойчивостта или забавеното възникване на системните токсични ефекти, в продължение на най-малко 14 дни след третиране. Животните от сателитните групи няма да бъдат чифтосвани и следователно, не се използват за оценяване на токсичност за репродукцията/развитието.
|
Дозиране
|
28.
|
Обикновено се използват поне три опитни групи и една контролна. Ако няма налични подходящи данни за общата токсичност, може да се проведе проучване за определяне на диапазона на дозата (с животни от същата порода и източник за набавяне), което да помогне при определянето на дозите, които да бъдат използвани. С изключение на третиране с изпитвания химикал, животните в контролната група трябва да бъдат отглеждани по идентичен начин на този при животните от изпитваната група. Ако за прилагане на изпитвания химикал се използва носител, контролната група трябва да получава най-голямото използвано количество носител.
|
|
29.
|
Нивата на дозиране следва да бъдат подбрани така, че да се отчитат всички съществуващи и налични данни за токсичност и (токсико-) кинетика. Следва да се вземе предвид и фактът, че може да съществуват различия в чувствителността на бременни и небременни животни. Целта при избор на най-високото ниво на доза е индуциране на токсични ефекти, но не смърт или явно страдание. След това следва да се избере последователност с намаляващи нива на дозиране с цел демонстриране на всеки отклик, свързан с дозирането и достигане на най-ниското ниво на дозиране без неблагоприятни ефекти. В повечето случаи е най-подходящо всяка доза да намалява два до четири пъти спрямо най-близката по-висока доза, а добавянето на допълнителна четвърта изпитвана група често е за предпочитане пред използването на твърде големи интервали между дозите (напр. намаление повече от 10 пъти).
|
|
30.
|
При наличие на наблюдения за обща токсичност (напр. намалено телесно тегло, въздействие върху черния дроб, сърцето, белите дробове или бъбреците и др.) или други промени, които може да не бъдат токсични реакции (напр. намален прием на храна, уголемяване на черния дроб), наблюдаваните ефекти върху ендокринни чувствителни крайни точки следва да се тълкуват предпазливо.
|
Гранично изпитване
|
31.
|
Ако изследване с орално третиране с ниво на еднократна доза от най-малко 1 000 mg/kg телесно тегло/ден или при приложение с храната с доза, която е еквивалентен процент, в храната или питейната вода (на базата на определеното телесно тегло), като се използват процедурите, описани в настоящото изследване, не доведе до забележими токсични ефекти и, ако въз основа на данните от структурно свързани вещества не се очаква токсичност, тогава може да не се смята за необходимо провеждането на пълно изследване с използване на няколко нива на дозиране. Изпитването при пределна концентрация се прилага, с изключение на случаи, при които експозиция на хора подсказва нуждата от използване на по-високо ниво на доза. За другите видове приложения, като например инхалация или дермално приложение, физикохимичните свойства на изпитваните химикали често налагат използване на максимално постижимата експозиция.
|
Прилагане на дозите
|
32.
|
Животните се дозират с изпитвания химикал в продължение на 7 дни в седмицата. Когато изпитваният химикал се прилага чрез хранене през сонда, това следва да става чрез еднократна доза за животното през стомашна тръба или подходяща интубационна канюла. Максималният обем течност, който може да бъде въведен еднократно, зависиот телесното тегло на изпитваното животно. Обемът не следва да превишава 1 ml/100 g т.м., освен в случаите, когато се прилагат водни разтвори. Тогава обемът може да достигне 2 ml/100 g т.м. С изключени на дразнещите или корозивни изпитвани химикали, които обикновено водят до засилени ефекти при по-високи концентрации, промените в изпитвания обем следва да бъдат сведени до минимум чрез коригиране на концентрацията, за да се гарантира постоянен обем при всички нива на дозиране.
|
|
33.
|
За изпитвани химикали, прилагани с храната или питейната вода, е важно да се гарантира, че количествата на приложения изпитван химикал не пречат на нормалния хранителен или воден баланс. Когато изпитваният химикал се прилага чрез хранителен режим, могат да бъдат използвани постоянна хранителна концентрация (ррm) или постоянно ниво на доза спрямо телесното тегло на животното; използваната алтернатива следва да бъде описана. За изпитван химикал, прилаган с помощта на сонда, дозата следва да бъде прилагана в сходни моменти всеки ден, и коригирана най-малко веднъж седмично, за да се поддържа постоянно ниво на дозиране, предвид телесното тегло на животното. Когато комбинирано проучване се използва като предварително за дългосрочно или пълно проучване за токсичност за репродукцията, при двете проучвания следва да се използва сходна хранителна диета.
|
Експериментален график
|
34.
|
Дозирането на двата пола следва да започне 2 седмици преди чифтосване, след като животните са аклиматизирани в продължение на най-малко пет дни и женските са били проверени за нормален естрален цикъл (в рамките на 2-седмичния период преди третирането). Изследването следва да бъде планирано по такъв начин, че оценката на естралния цикъл да започне скоро след като животните навършат пълна полова зрялост. Това може леко да варира за различните породи плъхове в различни лаборатории, напр. плъхове Sprague Dawley - 10-седмична възраст, плъхове Wistar - около 12-седмична възраст. Майките и потомството следва да бъдат умъртвени на ден 13 след раждането, или скоро след това. За да се гарантира, че самките няма да приемат храна за през нощта преди вземането на кръв (ако тази опция е предпочитана), не е задължително самките и тяхното потомство да бъдат умъртвявани в един и същи ден. Денят на раждането (тоест, когато раждането завърши) се определя като ден 0 след раждането. Женските, които не показват признаци за копулация се умъртвяват 24-26 дни след последния ден от периода за чифтосване. Дозирането продължава за двата пола по време на периода за чифтосване. Мъжките следва да бъдат допълнително дозирани след периода за чифтосване, най-малкото докато завърши минималния общ период за дозиране от 28 дни. След това се умъртвяват или друга възможност е да се запазят и да продължат да бъдат дозирани за евентуално провеждане на второ чифтосване, ако това се сметне за уместно.
|
|
35.
|
Ежедневното дозиране на женските майки следва да продължи през цялата бременност и най-малко до ден 13 след раждането включително, или до деня преди умъртвяването им. При проучвания, в които изпитваният химикал се прилага чрез инхалация или по кожен път, дозирането следва да бъде продължено най-малко до ден 19 от бременността включително, като то следва да бъде възобновено възможно най-скоро и не по-късно от постнатален ден (PND) 4.
|
|
36.
|
Животните в сателитните групи, планирани за контролни наблюдения, ако са включени, не се чифтосват. Те следва да бъдат задържани за поне още 14 дни след първото планирано умъртвяване на самките, без третиране, за да се открие забавеното възникване или устойчивост на токсичните ефекти, или възстановяване от тях.
|
|
37.
|
Схема на експерименталния график е дадена в допълнение 2.
|
Естрални цикли
|
38.
|
Естралните цикли следва да бъдат наблюдавани преди да започне третирането, за да се подберат за изследването женски с редовна цикличност (вж. точка 27). Вагиналните натривки следва също да се наблюдават ежедневно от началото на периода на третиране до потвърждение за чифтосване. Ако има притеснения за ефекти на остър стрес, които биха могли да променят естралните цикли с началото на дозирането, лабораториите могат да изложат тестовите животни за 2 седмици, след което да вземат вагинални натривки ежедневно и да следят естралния цикъл в продължение на минимум две седмици по време на периода преди чифтосване, като наблюдението да продължи и през периода на чифтосване, докато бъде открито потвърждение за чифтосване. Вземането на клетки от влагалището/цервикса следва да бъде внимателно, за да се избегне увреждане на лигавицата, което може индуцира псевдобременност (8) (9).
|
Процедура на чифтосването
|
39.
|
В настоящото изследване обикновено следва да се използва чифтосване 1:1 (един мъжки с една женска). Изключения могат да възникнат в случай на непредвидена смърт на мъжките. Женските индивиди следва да бъдат поставени заедно с мъжкия, докато не бъде забелязано доказателство за копулация или до изтичането на две седмици. Всяка сутрин женските следва да бъдат преглеждани за наличие на сперма или вагинална запушалка. Ден 0 от бременността се определя като деня, в който е потвърдено доказателството за чифтосване (откриване на вагинална запушалка или сперма). Когато чифтосването е било неуспешно, може да се извърши повторно чифтосване на женските животни с мъжки животни с доказан фертилитет от същата опитна група.
|
Брой на животните в котилото
|
40.
|
На ден 4 след раждането, броят на животните във всяко котило може да се коригира, като се елиминират излишните новородени чрез произволна селекция, за да се постигне, доколкото е възможно, по четири или пет новородени от един пол в котило, в зависимост от нормалния брой на животните в котилото за използваната порода плъхове. От две от излишните новородени следва да бъдат взети кръвни проби, които да бъдат, обединени и използвани за определяне на серумните нива на T4. Селективно елиминиране на новородени, напр. въз основа на телесното тегло, или разстоянието между ануса и половите органи (РАПО) не е уместно. Когато броят на мъжките или женските новородени не позволява отделяне по четири или пет от един пол във всяко котило, е приемливо частично коригиране (например, шест мъжки и четири женски). Не се елиминират новородени, когато броят на животните в котилото падне под целта за бракуване (8 или 10 новородени/котило). Ако има само едно новородено над целта за бракуване, само едно новородено ще бъде бракувано и използвано за вземане на кръв за евентуални оценки на серумните нива на T4.
|
|
41.
|
Ако броят на животните в котилото не се коригира, на ден 4 след раждането се умъртвяват две новородени на котило и се вземат кръвни проби за измерване на серумните концентрации на тироидния хормон. Ако е възможно, двете новородени на котило следва да бъдат женски, с цел да се резервират мъжки новородени за оценка на запазването на зърната на млечните жлези, с изключение на случая, когато при премахването на тези новородени не остават женски за оценяване при прекратяване на изпитването. Когато броят на животните в котилото падне под 8 или 10 новородени/котило (в зависимост от нормалния брой на животните в котилото при използваната порода плъхове), не се елиминират новородени. Ако има само едно новородено над нормалния брой на животните в котилото, само едно новородено ще бъде елиминирано и използвано за вземане на кръв за евентуални оценки на серумните нива на T4.
|
Наблюдения
|
42.
|
Общи клинични наблюдения следва да бъдат извършвани поне един път дневно, за предпочитане по едно и също време всеки ден и като се има предвид пиковият период за поява на очакваните ефекти след дозиране. Здравословното състояние на животните следва да се записва. Поне два пъти дневно всички животни се наблюдават за заболяемост и смъртност.
|
|
43.
|
Веднъж преди първата експозиция (с цел да са възможни сравнения в рамките на изпитвания обект) и най-малко веднъж седмично след това следва да бъдат правени детайлни клинични наблюдения на всички родители от животните. Тези наблюдения следва да се правят извън клетката, в която живеят, на стандартно място и за предпочитане по едно и също време на деня. Наблюденията следва да бъдат внимателно записвани; за предпочитане, като се използват системи за оценяване, изрично определени от изпитващата лаборатория. Следва да се положат усилия, за да се гарантира, че колебанията в условията на изпитването са минимални, както и че наблюденията се извършват за предпочитане от хора, неинформирани за изпитването. Отбелязваните признаци следва да включват промяна в кожата, козината, очите, лигавиците, поява на секреция и екскреция, както и автономна активност (напр. сълзене, пилоерекция, промяна в големината на зениците, необичаен начин на дишане), без да се ограничават до това. Следва да се записван и промени в походката, стойката и реакцията при манипулиране, както и наличието на клонични или тонични движения, стереотипи (напр. прекомерно близане на козината, повтарящо се движение в кръг), затруднено или продължително раждане или странно поведение (напр. само-осакатяване, вървене назад) (10).
|
|
44.
|
В даден момент по време на проучването следва да се извърши оценка на сензорната реактивност на стимули от различно естество (напр. слухови, визуални и проприоцептивни стимули) (8) (9) (11), оценка на силата на захват(12) и оценка на моторната активност (13) на пет мъжки и пет женски животни, произволно избрани от всяка група. Допълнителни уточнения по процедурите, които могат да бъдат следвани, са дадени в съответните препратки. Въпреки всичко могат да се използват и алтернативни процедури, различни от тези, към които се препраща. При мъжките, тези функционални наблюдения следва да се правят към края на техния период на дозиране, малко преди планираното им умъртвяване, но преди вземането на кръв за хематологични или клинико-химични изследвания (вж. точки 53-56, включително бележката под линия 1). Женските следва да бъдат в сходно физиологично състояние по време на тези функционални изпитвания и за предпочитане е да бъдат изпитани веднъж по време на последната седмица кърмене (напр., LD 6-13), малко преди планираното им умъртвяване. Доколкото е възможно, времето за разделяне на самките от новородените трябва да се сведе до минимум.
|
|
45.
|
Функционалните наблюдения, извършени веднъж към края на проучването, може да бъдат пропуснати, когато проучването се провежда като предварително проучване към последващо субхронично (90-дневно) или дългосрочно проучване. В този случай функционалните наблюдения следва да бъдат включени в това последващо изследване. От друга страна, наличието на данни от функционалните наблюдения от проучването с повтаряща се доза може да подобри способността за избор на нива на дозата за последващото субхронично или дългосрочно проучване.
|
|
46.
|
По изключение, функционалните наблюдения могат също да се избегнат за групи, които иначе показват признаци на токсичност в размер, който значително би попречил на извършването на функционалното изпитване.
|
|
47.
|
Продължителността на бременността следва да бъде записвана и изчислявана от ден 0 от бременността. Всяко котило следва да бъде прегледано възможно най-скоро след раждането, за да се установи броят и полът на новородените, броят на мъртвородените, живородените, изтърсаците (новородени, които са значително по-малки от съответните контролни новородени) и наличието на макроскопски аномалии.
|
|
48.
|
Живите новородени следва да се преброят и да се определи полът им, а котилата да се претеглят в рамките на 24 часа след раждането (ден 0 или 1 след раждане) и поне на ден 4 и ден 13 след раждането. В допълнение към наблюденията на животните родители (вж. точки 43 и 44) следва за се запише всяко аномално поведение на потомството.
|
|
49.
|
РАПО разстоянието следва да се измери на същия ден след раждането между PND 0 до PND 4. Телесното тегло на новороденото следва да бъде измерено в деня на измерване на РАПО, и стойността на РАПО следва да бъде нормализирана към размера на новороденото, за предпочитане към кубичния корен от телесното тегло (14). Броят на зърната/ареолите на гърдите на мъжките новородени следва да се преброи на PND 12 или 13, както се препоръчва в GD 151 на ОИСР (15).
|
Телесно тегло и консумация на храна/вода
|
50.
|
Мъжките и женските следва да се претеглят на първия ден от дозирането, и минимум всяка седмица след това, както и при прекратяване на изследването. По време на бременност, женските следва да се претеглят на дни 0, 7, 14 и 20, и в рамките на 24 часа след раждането (ден 0 или 1 след раждането) и минимум на ден 4 и ден 13 след раждането. Тези наблюдения трябва да се описват индивидуално за всяко полово зряло животно.
|
|
51.
|
По време на периода преди чифтосването, бременността и кърменето, консумацията на храна следва да се измерва най-малко веднъж седмично. Измерването на консумацията на храна по време на чифтосване е незадължително. Консумацията на вода по време на тези периоди също следва да се измерва, когато изпитваният химикал се прилага с тази среда.
|
Хематология
|
52.
|
Веднъж по време на проучването следва да се правят следните хематоложки изследвания на пет мъжки и пет женски животни, избрани на произволен принцип от всяка група: хематокрит, концентрации на хемоглобин, брой на еритроцитите, ретикулоцити, общ и диференциален брой на левкоцити, брой на тромбоцитите, както и измерване на времето на съсирване/потенциала за съсирване на кръвта. Други определяния, които следва да се извършат, ако изпитваният химикал или неговите предполагаеми метаболити имат, или се предполага че имат, окислителни свойства, включват концентрация на метхемоглобин и телца на Heinz.
|
|
53.
|
Кръвните проби следва да бъдат вземани от предварително определено място. Женските следва да бъдат в сходно физиологично състояние по време на вземането на проби. За да се избегнат практически трудности, свързани с различното начало на бременността, вземането на кръв от женските може да се прави в края на периода преди чифтосване като алтернатива на вземането на кръв непосредствено преди процедурата за евтаназия на животните или като част от нея. За предпочитане е кръвните проби от мъжките да се вземат непосредствено преди процедурата за евтаназия на животните или като част от нея. Като алтернатива, вземането на кръв от мъжките може да се прави и в края на периода преди чифтосване, когато този момент е бил предпочетен и за женските.
|
|
54.
|
Кръвните проби следва да се съхраняват при подходящи условия.
|
Клинична биохимия
|
55.
|
Клинични биохимични изследвания за проучване на основните токсични ефекти върху тъканите и по-специално, ефектите върху бъбреците и черния дроб, следва да се извършват върху кръвни проби, получени от подбрани пет мъжки и пет женски животни от всяка група. Препоръчва се една нощ гладуване на животните преди вземане на кръвни проби (22). Изследванията на плазма или серум следва да включват натрий, калий, глюкоза, общ холестерол, урея, креатинин, общ протеин и албумин, най-малко два ензима, показателни за хепатоцелуларни ефекти (като аланин аминотрансфераза, аспартат аминотрансфераза и сорбитол дехидрогеназа) и жлъчни киселини. Измерванията за допълнителни ензими (от чернодробен и друг произход) и билирубин могат да дадат полезна информация при определени обстоятелства.
|
|
56.
|
От определено място се вземат кръвни проби на базата на следния график:
|
—
|
от поне две новородени на котило, на ден 4 след раждането, ако броят на новородените го позволява (вж. точки 40-41)
|
|
—
|
от всички майки и поне от две новородени на котило при прекратяването на ден 13, и
|
|
—
|
от всички зрели мъжки, при прекратяването
|
Всички кръвни проби се съхраняват при подходящи условия. Кръвните проби от новородените на ден 13 и от зрелите мъжки се оценяват за серумни нива на тироидните хормони (T4). Ако е уместно се прави допълнителна оценка на T4 в кръвните проби от самките и новородените на ден 4. Също ако е уместно, може да се извърши незадължително измерване на други хормони. Кръвта на новородените може да се обедини по котила за анализите на тироидния хормон. За предпочитане е тироидните хормони (T4 и TSH) да се измерват като „общи“.
|
|
57.
|
По избор, през последната седмица от проучването могат да се проведат следните изследвания на урината от пет мъжки от всяка група, избрани на произволен принцип, като се използва събиране на урината по часове; външен вид, количество, осмолалитет и относително тегло, рН, протеини, глюкоза и кръв/кръвни клетки.
|
|
58.
|
Като допълнение следва да се предвидят изследвания на серумни маркери за основните увреждания на тъканите Други изследвания, които трябва да се проведат, ако известните свойства на изпитвания химикал може или има подозрения, че засягат свързаните метаболитни профили, включват калций, фосфат, триглицериди и глюкоза на гладно, специфични хормони, метхемоглобин и холинестераза. Те трябва да бъдат идентифицирани за всеки отделен случай.
|
|
59.
|
Следните фактори могат да повлияят на варирането и на абсолютните концентрации при определянето на хормони:
|
—
|
времето на умъртвяване, поради дневните колебания в концентрацията на хормони
|
|
—
|
методът на умъртвяване, за да се избегне ненужен стрес за животните, който може да повлияе на концентрациите на хормони
|
|
—
|
комплектите за изпитване за определяне на хормони, които могат да се различават по стандартните си криви.
|
|
|
60.
|
Пробите от плазма, специално предназначени за определяне на хормони, следва да бъдат получени в сравними периоди през деня. Числовите стойности, получени при анализа на концентрации на хормони, се различават при употребата на различни търговски комплекти за анализ.
|
|
61.
|
Ако базовите данни за минали периоди са недостатъчни, следва да се разгледа възможност за определяне на хематологичните и клиничните биохимични променливи преди началото на дозирането или за предпочитане в набор от животни, които не са включени в опитните групи За женските данните трябва да бъдат от кърмещи животни.
|
ПАТОЛОГИЯ
Макроскопска аутопсия
|
62.
|
Всички възрастни животни, подложени на изследване, са обект на цялостна, подробна макроскопска аутопсия, която включва внимателно изследване на външната повърхност на тялото, всички отвори и черепната, гръдната и коремната кухини и тяхното съдържание Особено внимание при огледа трябва да се обърне на органите на половата система. Броят на местата за имплантация следва да бъде записан. Вагиналните натривки следва да бъдат изследвани в деня на аутопсията, за да се определи стадият на естралния цикъл и да се даде възможност за корелация с хистопатологичното изследване на женските репродуктивни органи.
|
|
63.
|
Тестисите и епидидимите, както и простатата и семенните мехурчета с коагулиращите жлези като цяло трябва да бъдат изрязани от всички мъжки зрели животни и почистени от прилежащите тъкани, според необходимостта, и трябва да се претеглят в мокро състояние възможно най-скоро след дисекцията, за да не се допусне изсъхването им. В допълнение, опционалното тегло на органите може да включва мускула повдигач на ануса плюс булбокавернозния мускулен комплекс, жлезите на Каупър и главичката на пениса при мъжките и двойката яйчници (мокро тегло) и матката (включително маточната шийка) при женските; ако са включени, тези тегла следва да бъдат взети възможно най-скоро след дисекцията. Яйчниците, тестисите, епидидимите, допълнителните полови органи и всички органи, показващи макроскопски лезии при всички възрастни животни следва да бъдат запазени.
|
|
64.
|
От всички възрастни мъжки и женски и от едно мъжко и едно женско новородено на 13 дни от всяко котило следва да се запазят щитовидните жлези в най-подходящата среда за фиксиране за предвиденото последващо хистопатологично изследване. Теглото на щитовидната жлеза може да се установи след фиксиране. Почистването също следва да се осъществи много внимателно и само след фиксиране, за да се избегне увреждане на тъканите. Увреждането на тъканите може да постави под съмнение хистопатологичния анализ. Кръвните проби следва да се вземат от определено място, точно преди или като част от процедурата за евтаназия на животните и да се съхраняват при подходящи условия (вж. точка 56).
|
|
65.
|
В допълнение, от най-малко пет възрастни мъжки и женски, произволно избрани от всяка група (освен онези, които са намерени агонизиращи и/или са евтаназирани преди прекратяване на изследването), следва да се изрежат черният дроб, бъбреците, надбъбречните жлези, тимусът, далакът, мозъкът и сърцето от всички прилежащи тъкани, според необходимостта, и да се претеглят в мокро състояние възможно най-скоро след дисекцията, за да не се допусне изсъхването им. Следните тъкани следва да бъдат запазени в най-подходящата среда за фиксиране, както за типа тъкан, така и за предвиденото последващо хистопатологично изследване: всички макроскопски лезии, мозъкът (представителни области, включително главният мозък, малкият мозък и моста), гръбначният мозък, очите, стомахът, тънките и дебелите черва (включително Пайеровите плаки), черният дроб, бъбреците, надбъбречните жлези, далакът, сърцето, тимусът, трахеята и белите дробове (запазени чрез инфлация с фиксатор, след което потопени), половите жлези (тестиси и яйчници), допълнителните полови органи (матка и маточна шийка, епидидими, простата, семенни мехурчета плюс коагулиращи жлези), вагина, пикочен мехур, лимфни възли (освен разположеният най-близо до средата на тялото дрениращ възел следва да бъде взет и друг лимфен възел, в съответствие с опитана лабораторията (16)), периферен нерв (седалищен или тибиален) за предпочитане в непосредствена близост до мускула, скелетен мускул и кост с костен мозък (срез от костен мозък или, като алтернатива, прясно фиксиран костномозъчен аспират). Препоръчва се тестисите да бъдат фиксирани чрез потапяне във фиксатор на Буен или модифициран фиксатор на Дейвидсън (16) (17) (18); фиксиране във формалин не се препоръчва за тези тъкани. Бялото покривало (tunica albuginea) може внимателно да се пробие на неголяма дълбочина в двата края на органа с помощта на игла, за да се позволи бързо проникване на фиксатора. Клиничните и другите сведения могат да посочат необходимостта от изследвания на допълнителни тъкани. Също така всички органи, които на основата на познатите свойства на изпитвания химикал се считат за вероятни прицелни органи, също следва да бъдат консервирани.
|
|
66.
|
Следните тъкани могат да предоставят ценни показания за свързани с ендокринната система последици: полови жлези (яйчници и тестиси), допълнителни полови органи (матка, включително маточна шийка, епидидими, семенни мехурчета плюс коагулиращи жлези, простата, локализирана дорзолатерално и вентрално), влагалището, хипофиза, мъжка млечна жлеза и надбъбречна жлеза. Промените в мъжките млечни жлези не са достатъчно добре документирани, но този параметър може да бъде особено чувствителен към вещества с естрогенно действие. Наблюдението на органи/тъкани, които не са изброени в точка 65, е по избор.
|
|
67.
|
Мъртвите новородени и новородените, умъртвени на ден 13 след раждането или скоро след това, трябва най-малко да бъдат внимателно огледани външно за макроскопски аномалии. Особено внимание следва да се обърне външните репродуктивни полови органи, които да се огледат за признаци на променено развитие.
|
Хистопатология
|
68.
|
На запазените органи и тъкани от избраните животни от контролната група и от групите с най-висока доза (като специално се наблегне върху стадиите на сперматогенеза в мъжките полови жлези и хистопатологично изследване на интерстициалната тестикуларна клетъчна структура) следва да се направят пълни хистопатологични изследвания. Щитовидни жлези от новородени и от останалите възрастни животни може да бъдат изследвани, когато е необходимо. Тези изследвания следва да обхванат и животните от групи с други нива на дозата, ако бъдат забелязани промени в групата, третирана с най-висока доза. В Указанията за хистопатология (10) се съдържа допълнителна подробна информация относно дисекцията, фиксирането, разрязването и хистопатологията на ендокринните тъкани.
|
|
69.
|
Изследват се всички общи наранявания. Прицелните органи от групи, третирани с други дози, особено от групи, за които се твърди, че показват NOAEL, следва да бъдат изследвани, за да се установят нивата на NOAEL.
|
|
70.
|
Когато се използва сателитна група, хистопатология следва да се извършва на тъкани и органи, за които е установено, че показват ефект при третираните групи.
|
ДАННИ И ПРОТОКОЛИРАНЕ
Данни
|
71.
|
Следва да бъдат осигурени индивидуални данни за всяко животно. Допълнително, всички данни следва да бъдат обобщени в таблична форма, като за всяка изпитвана група се посочва броят на животните в началото на изпитването, броят на животните, открити мъртви по време на изпитването или евтаназирани, времето на смъртта или евтаназията, броят на плодовитите животни, броят на бременните женски, броят на животните, показващи признаци на токсичност, описание на наблюдаваните признаци на токсичност, включително времето на началото, продължителността и сериозността на всички токсични ефекти, видовете хистопатологични изменения и всички относими данни за котилото. Табличен формат на обобщено протоколиране, който се е доказал като особено полезен за оценката на ефектите върху репродукцията /развитието, е даден в допълнение 3.
|
|
72.
|
Когато е възможно, цифровите резултати следва да бъдат оценени с помощта на подходящ и общоприет статистически метод. Чрез сравнения на въздействието в рамките на даден обхват на дозиране следва да се избегне използването на множество t-изпитвания. Статистическите методи следва да бъдат избрани при планирането на изследването. Статистическият анализ на РЕПО и запазването на зърната на млечните жлези трябва да бъде базиран на индивидуални данни за новороденото, като взема под внимание ефектите на котилото. Където е уместно, котилото е единицата за анализ. Статистическият анализ на телесното тегло на новороденото трябва да бъде базиран на индивидуални данни за новороденото, като взема под внимание броя на животните в котилото. Поради ограничените размери на изследването, статистически анализи във формата на изпитвания за „значимост“ са с ограничена стойност за много крайни точки, и особено за крайните точки на репродукцията. Някои от най-широко използваните методи, особено параметричните изпитвания за измерване на централна тенденция, са неподходящи. Ако се използват статистически анализи, тогава избраният метод следва да бъде подходящ за разпределението на изследваната променлива и да бъде избран преди началото на изследването.
|
Оценка на резултатите
|
73.
|
Констатациите на това изследване за токсичност следва да бъдат оценени, като се вземат предвид наблюдаваните ефекти, аутопсията и микроскопските находки. Оценката включва връзката между дозата на изпитвания химикал и наличието или отсъствието, разпространението и сериозността на аномалиите, включително макроскопски лезии, идентифицирани прицелни органи, безплодие, клинични аномалии, засегната ефективност на репродукцията и на котилото, промени в телесното тегло, ефекти върху смъртността и всякакви други токсични ефекти.
|
|
74.
|
Поради краткия период на третиране на мъжките индивиди, хистопатологичното изследване на тестисите и епидидимите следва да се разглежда заедно с данните за плодовитост, когато се оценяват ефектите върху мъжката репродуктивна система. Използването на контролни данни от предходни периоди за репродукцията/развитието на организма (напр. относно броя на животните в котилото, РЕПО, запазване на зърната на млечните жлези, серумни нива на T4), когато са налични, могат също да бъдат полезни като спомагателно средство за интерпретиране на изследването.
|
|
75.
|
За контрол на качеството се предлага да се събират контролни данни за минали периоди и да се изчисляват коефициенти на вариация за числовите данни, особено за параметрите, свързани с откриване на нарушители на функциите на ендокринната система. Тези данни могат да бъдат използвани за сравнение когато се извършва оценка на актуалните изследвания.
|
Протокол от изпитването
|
76.
|
Протоколът от изпитването следва да включва следната информация:
|
|
Изпитван химикал:
|
—
|
източник, номер на партидата, крайна дата за употреба, ако има такава
|
|
—
|
стабилност на изпитвания химикал, ако е известна.
|
|
|
|
Вещество с една съставка:
|
—
|
външен вид, разтворимост във вода и допълнителни относими физични и химични свойства;
|
|
—
|
химична идентификация, като например наименование по IUPAC или CAS, CAS номер, код SMILES или InChI, структурна формула, чистота, химична идентичност на онечистванията, доколкото е целесъобразно и практически осъществимо, и т.н.
|
|
|
|
Вещество с повече съставки, UVCB и смеси:
|
—
|
определени, доколкото е възможно, с химичната си идентичност (вж. по-горе), количествения състав и относимите физични и химични свойства на съставките.
|
|
|
|
Носител (когато се използва такъв):
|
—
|
обосновка за избора на носител, когато е различен от вода.
|
|
|
|
Изпитвани животни:
|
—
|
брой, възраст и пол на животните;
|
|
—
|
източник за доставка на животните, условия на отглеждане, хранителен режим и т.н.;
|
|
—
|
индивидуално тегло на животните в началото на изпитването.
|
|
—
|
обосновка при използване на видове, различни от плъх.
|
|
|
|
Условия на изпитването:
|
—
|
обосновка за избора на нивото на доза;
|
|
—
|
подробна информация за това в какъв вид е изпитваният химикал/хранителната смес, постигната концентрация, стабилност и хомогенност на сместа;
|
|
—
|
подробна информация относно прилагането на изпитвания химикал;
|
|
—
|
превръщане от концентрация на изпитвания химикал в храна/питейна вода (ppm) в действителна доза (mg на kg телесно тегло дневно), ако е приложимо,
|
|
—
|
подробности относно качество на храната и водата;
|
|
—
|
подробно описание на процедурата за рандомизация при подбора на новородени за бракуване, ако се прилага бракуване.
|
|
|
|
Резултати:
|
—
|
телесно тегло/промени в телесното тегло;
|
|
—
|
консумация на храна и консумация на вода, ако е приложимо;
|
|
—
|
данни за токсичната реакция по пол и доза, включително относно плодовитостта и бременността, както и всякакви други признаци на
|
|
—
|
продължителност на бременността;
|
|
—
|
токсични или други ефекти върху репродукцията, потомството, постнаталния растеж и др.;
|
|
—
|
природа, сила и продължителност на клиничните наблюдения (независимо обратими или необратими), оценки на сензорната активност, силата на захвата и двигателната активност,
|
|
—
|
хематологични изпитвания с относими базови стойности,
|
|
—
|
клинични биохимични изпитвания с относими базови стойности,
|
|
—
|
брой на възрастните женски с нормален или аномален естрален цикъл и продължителност на цикъла;
|
|
—
|
брой на живородените и постимплантационни загуби;
|
|
—
|
брой на новородените с макроскопски видими аномалии; макроскопска оценка на външните полови органи, брой на изтърсаците;
|
|
—
|
време на смъртта в процеса на изпитването или дали животните са преживели до края на изпитването;
|
|
—
|
брой на имплантациите, брой на животните в котилото и тегло на котилото към момента на записване;
|
|
—
|
данни за телесното тегло на новороденото
|
|
—
|
РЕПО на всички новородени (и телесно тегло в деня на измерване на РЕПО)
|
|
—
|
запазване на зърната на млечните жлези при мъжките новородени,
|
|
—
|
нива на тиродния хормон, новородени на ден 13 и възрастни мъжки (и самки и новородени на ден 4, ако е измерван)
|
|
—
|
телесно тегло в момента на умътрвяване и данни за тегло на органи за животните родители;
|
|
—
|
находки при аутопсията;
|
|
—
|
подробно описание на хистопатологичните находки;
|
|
—
|
данни за абсорбция (ако има);
|
|
—
|
статистическа обработка на резултатите, ако е подходящо.
|
|
|
|
Обсъждане на резултатите.
|
|
Тълкуване на резултатите
|
77.
|
Проучването предоставя оценки на токсичността за репродукцията/развиващия се организъм, свързана с приложението на повторени дози. По-конкретно, тъй като акцентът е поставен както върху общата токсичност, така и върху крайните точки за токсичност за репродукцията/развиващия се организъм, резултатите от изследването позволяват разграничаване на ефектите върху репродукцията/развиващия се организъм, възникващи при липса на обща токсичност, и онези, които се изразяват само на нива, които са токсични и за животните родители (вж. точки 7-11). То може да представи указание за необходимостта от провеждане на допълнителни изследвания и да предоставя насоки за планиране на следващи изпитвания. Неодходима е консултация с Ръководството на ОИСР 43 за помощ при тълкуването на резултатите, свързани с репродукцията и развитието на организма (19). Ръководство 106 на ОИСР относно „Хистологичната оценка на ендокринните и репродуктивните изпитвания на гризачи“ (16) предоставя информация относно подготовката и оценката на (ендокринните) органи и вагиналните натривки, които могат да бъдат полезни за настоящия метод за изпитване.
|
ЛИТЕРАТУРА
|
(1)
|
OECD (1990). Room Document No 1 for the 14th Joint Meeting of the Chemicals Group and Management Committee. Available upon request at Organisation for Economic Cooperation and Development, Paris
|
|
(2)
|
OECD (1992). Chairman’s Report of the ad hoc Expert Meeting on Reproductive Toxicity Screening Methods, Tokyo, 27th-29th October, 1992. Available upon request at Organisation for Economic Cooperation and Development, Paris
|
|
(3)
|
Mitsumori K., Kodama Y., Uchida O., Takada K., Saito M. Naito K., Tanaka S., Kurokawa Y., Usami, M., Kawashima K., Yasuhara K., Toyoda K., Onodera H., Furukawa F., Takahashi M. and Hayashi Y. (1994). Confirmation Study, Using Nitro-Benzene, of the Combined Repeat Dose and Reproductive/ Developmental Toxicity Test Protocol Proposed by the Organization for Economic Cooperation and Development (OECD). J. Toxicol, Sci., 19, 141-149.
|
|
(4)
|
Tanaka S., Kawashima K., Naito K., Usami M., Nakadate M., Imaida K., Takahashi M., Hayashi Y., Kurokawa Y. and Tobe M. (1992). Combined Repeat Dose and Reproductive/Developmental Toxicity Screening Test (OECD): Familiarization Using Cyclophosphamide. Fundam. Appl. Toxicol., 18, 89-95.
|
|
(5)
|
OECD (1998). Report of the First Meeting of the OECD Endocrine Disrupter Testing and Assessment (EDTA) Task Force, 10th-11th March 1998, Available upon request at Organisation for Economic Cooperation and Development, Paris
|
|
(6)
|
OECD (2015). Feasibility Study for Minor Enhancements of TG 421/422 with ED Relevant Endpoints. Environment, Health and Safety Publications, Series on Testing and Assessment (No 217), Organisation for Economic Cooperation and Development, Paris.
|
|
(7)
|
OECD (2000). Guidance Document on the Recognition, Assessment, and Use of Clinical Signs as Humane Endpoints for Experimental Animals Used in Safety Evaluations, Environment, Health and Safety Publications, Series on Testing and Assessment, (No 19), Organisation for Economic Cooperation and Development, Paris.
|
|
(8)
|
Goldman J.M., Murr A.S., Buckalew A.R., Ferrell J.M.and Cooper R.L. (2007). The Rodent Estrous Cycle: Characterization of Vaginal Cytology and its Utility in Toxicological Studies, Birth Defects Research, Part B, 80 (2), 84-97.
|
|
(9)
|
Sadleir R.M.F.S. (1979). Cycles and Seasons, in Auston C.R. and Short R.V. (Eds.), Reproduction in Mammals: I. Germ Cells and Fertilization, Cambridge, New York.
|
|
(10)
|
IPCS (1986). Principles and Methods for the Assessment of Neurotoxicity Associated with Exposure to Chemicals. Environmental Health Criteria Document (No 60).
|
|
(11)
|
Moser V.C., McDaniel K.M. and Phillips P.M. (1991). Rat Strain and Stock Comparisons Using a Functional Observational Battery: Baseline Values and Effects of Amitraz. Toxicol. Appl. Pharmacol., 108, 267-283.
|
|
(12)
|
Meyer O.A., Tilson H.A., Byrd W.C. and Riley M.T. (1979). A Method for the Routine Assessment of Fore- and Hindlimb Grip Strength of Rats and Mice. Neurobehav. Toxicol., 1, 233-236.
|
|
(13)
|
Crofton K.M., Howard J.L., Moser V.C., Gill M.W., Reiter L.W., Tilson H.A., MacPhail R.C. (1991). Interlaboratory Comparison of Motor Activity Experiments: Implication for Neurotoxicological Assessments. Neurotoxicol. Teratol. 13, 599-609.
|
|
(14)
|
Gallavan R.H. Jr, J.F. Holson, D.G. Stump, J.F. Knapp and V.L. Reynolds. (1999). „Interpreting the Toxicologic Significance of Alterations in Anogenital Distance: Potential for Confounding Effects of Progeny Body Weights“, Reproductive Toxicology, 13: 383-390.
|
|
(15)
|
OECD (2013). Guidance Document in Support of the Test Guideline on the Extended One Generation Reproductive Toxicity Study. Environment, Health and Safety Publications, Series on Testing and Assessment (No 151). Organisation for Economic Cooperation and Development, Paris.
|
|
(16)
|
OECD (2009).Guidance Document for Histologic Evaluation of Endocrine and Reproductive Tests in Rodents. Environment, Health and Safety Publications, Series on Testing and Assessment (No. 106) Organisation for Economic Cooperation and Development, Paris.
|
|
(17)
|
Hess RA and Moore BJ. (1993). Histological Methods for the Evaluation of the Testis. In: Methods in Reproductive Toxicology, Chapin RE and Heindel JJ (Eds.). Academic Press: San Diego, CA, pp. 52-85.
|
|
(18)
|
Latendresse JR, Warbrittion AR, Jonassen H, Creasy DM. (2002). Fixation of Testes and Eyes Using a Modified Davidson’s Fluid: Comparison with Bouin’s Fluid and Conventional Davidson’s fluid. Toxicol. Pathol. 30, 524-533.
|
|
(19)
|
OECD (2008). Guidance Document on Mammalian Reproductive Toxicity Testing and Assessment. Environment, Health and Safety Publications, Series on Testing and Assessment (No 43), Organisation for Economic Cooperation and Development, Paris.
|
|
(20)
|
OECD (2011), Guidance Document on Standardised Test Guidelines for Evaluating Chemicals for Endocrine Disruption (No 150), Organisation for Economic Cooperation and Development, Paris.
|
Допълнение 1
ОПРЕДЕЛЕНИЯ (ВЖ. И (20) ОИСР GD 150)
Андрогенност е способността на даден химикал да действа като естествен андрогенен хормон (напр. тестостерон) в организма на бозайник.
Антиандрогенност е способността на даден химикал да потиска действието на естествен андрогенен хормон (напр. тестостерон) в организма на бозайник.
Антиестрогенност е способността на даден химикал да потиска действието на естествен естрогенен хормон (напр. естрадиол 17ß) в организма на бозайник.
Антитиреоидна активност е способността на даден химикал да потиска действието на естествен тиреоиден хормон (напр. T3) в организма на бозайник.
Химикал означава вещество или смес.
Токсичност за развиващия се организъм: проявата на репродуктивна токсичност, представляваща пред-, пери- или постнатални, структурни или функционални разстройства в потомството.
Доза е количеството приложен изпитван химикал. Дозата се изразява като маса на изпитвания химикал за единица телесно тегло на изпитваното животно на ден (напр. mg на kg телесно тегло дневно), или като постоянна концентрация в храната.
Дозировка е общ термин, обхващащ дозата, нейната честота и продължителност на дозиране.
Очевидна токсичност е общ термин, описващ ясните признаци на токсичност след прилагане на изпитвания химикал. Те следва да бъдат достатъчни за оценка на опасността и следва да бъдат такива, че при увеличаване на прилаганата доза да се очаква развитие на признаци на силна токсичност и вероятна смърт.
Увреждане на плодовитостта представлява разстройства на мъжките или женските възпроизводителни функции или способности.
Токсичност при майката: неблагоприятни ефекти върху бременните женски, възникващи или специфично (пряк ефект), или неспецифично (непряк ефект) и свързани със състоянието на бременност.
NOAEL е съкращение за нивото, при което не се наблюдават неблагоприятни ефекти. Това е най-високото ниво на доза, при което не се наблюдават свързани с третирането неблагоприятни находки в резултат от третирането.
Естрогенност е способността на даден химикал да действа като естествен естрогенен хормон (напр. естрадиол 17ß) в организма на бозайник.
Репродуктивна токсичност представлява вредни ефекти върху потомството и/или увреждане на мъжките и женските възпроизводителни функции или способности.
Изпитван химикал е всяко вещество или смес, изпитвано(а) с използване на настоящия метод за изпитване.
Тиреоидна активност е способността на даден химикал да действа като естествен тиреоиден хормон (напр. T3) в организма на бозайник.
Валидиране е научна процедура, предназначена за характеризиране на оперативните изисквания и ограничения на даден метод за изпитване и за доказване на неговата надеждност и относимост за определена цел.
Допълнение 2
СХЕМА НА ЕКСПЕРИМЕНТАЛНИЯ ГРАФИК, УКАЗВАЩА МАКСИМАЛНАТА ПРОДЪЛЖИТЕЛНОСТ НА ИЗСЛЕДВАНЕТО, НА БАЗАТА НА ПЪЛЕН 14-ДНЕВЕН ПЕРИОД НА ЧИФТОСВАНЕ


Допълнение 3
ОБОБЩЕН ДОКЛАД В ТАБЛИЧЕН ВИД НА ЕФЕКТИТЕ ВЪРХУ РЕПРОДУКЦИЯТА/РАЗВИТИЕТО
|
КОНСТАТАЦИИ И ОЦЕНКИ
|
Стойности
|
|
Дозировка (единици)…….
|
0 (контрол)
|
…
|
…
|
…
|
…
|
|
Стартирани двойки (N)
|
|
|
|
|
|
|
Естрален цикъл (поне средна продължителност и честота на нередовни цикли)
|
|
|
|
|
|
|
Женски, показващи потвърждение за копулация (N)
|
|
|
|
|
|
|
Женски, постигнали забременяване (N)
|
|
|
|
|
|
|
Дни от зачеването 1 - 5 (N)
|
|
|
|
|
|
|
Дни от зачеването 6 -… (23) (N)
|
|
|
|
|
|
|
Бременност≤21 дни (N)
|
|
|
|
|
|
|
Бременност = 22 дни (N)
|
|
|
|
|
|
|
Бременност ≥ 23 дни (N)
|
|
|
|
|
|
|
Самки с живородени малки (N)
|
|
|
|
|
|
|
Самки с живи малки на 4 ден след раждането (N)
|
|
|
|
|
|
|
Импланти/самка (средно)
|
|
|
|
|
|
|
Живи новородени/самка при раждането (средно)
|
|
|
|
|
|
|
Живи новородени/самка на 4 ден (средно)
|
|
|
|
|
|
|
Съотношение на половете (м/ж) при раждането (средно)
|
|
|
|
|
|
|
Съотношение на половете (м/ж) на 4 ден (средно)
|
|
|
|
|
|
|
Тегло на животните в котилото при раждането (средно)
|
|
|
|
|
|
|
Тегло на животните в котилото на 4 ден (средно)
|
|
|
|
|
|
|
Тегло на новороденото при раждането (средно)
|
|
|
|
|
|
|
Тегло на новороденото към момента на измерване на РЕПО (средно мъжки, средно женски)
|
|
|
|
|
|
|
РЕПО на новороденото на същия постнатален ден, раждане — ден 4 (средно мъжки, средно женски, отбелязване PND)
|
|
|
|
|
|
|
Тегло на новороденото на 4 ден (средно)
|
|
|
|
|
|
|
Тегло на новороденото на 13 ден (средно)
|
|
|
|
|
|
|
Запазване на зърната на млечните жлези при мъжките новородени на ден 13 (средно)
|
|
|
|
|
|
|
НОВОРОДЕНИ С АНОМАЛИИ
|
|
Самки с 0
|
|
|
|
|
|
|
Самки с 1
|
|
|
|
|
|
|
Самки с ≥ 2
|
|
|
|
|
|
|
ЗАГУБА НА ПОТОМСТВО
|
|
Пренатално (имплантации минус живородени)
|
|
Женски с 0
|
|
|
|
|
|
|
Женски с 1
|
|
|
|
|
|
|
Женски с 2
|
|
|
|
|
|
|
Женски с ≥ 3
|
|
|
|
|
|
|
Постнатално (живородени минус живи на постнатален ден 13)
|
|
Женски с 0
|
|
|
|
|
|
|
Женски с 1
|
|
|
|
|
|
|
Женски с 2
|
|
|
|
|
|
|
Женски с ≥ 3
|
|
|
|
|
|
Б.65
IN VITRO МЕТОД ЗА ИЗПИТВАНЕ С МЕМБРАННА БАРИЕРА ЗА КОРОЗИВНО ДЕЙСТВИЕ ВЪРХУ КОЖАТА
ВЪВЕДЕНИЕ
|
1.
|
Настоящият метод за изпитване е еквивалентен на насоките за изпитване на ОИСР (ТG) 435 (2015). Корозивното действие върху кожата се разбира като причиняване на необратимо увреждане на кожата, проявяващо се като видима некроза през епидермиса и в дермата, настъпило след прилагане на изпитван химикал [както е дефиниран от Глобалната хармонизирана система за класификация и етикетиране на химикали (GHS) на Обединените нации (ООН) (1) и Регламент на Европейския съюз (ЕС) 1272/2008 относно класификацията, етикетирането и опаковането на вещества и смеси (CLP) (24) Настоящият метод за изпитване, еквивалентен на актуализираните насоки за изпитване 435 на ОИСР, предоставя in vitro метод за изпитване с мембранната бариера, който може да се използва за идентифициране на корозивни химикали. Методът за изпитване използва изкуствена мембрана, проектирана да реагира на корозивни химикали по начин, подобен на животинска кожа in situ.
|
|
2.
|
Корозията на кожата традиционно се оценява чрез прилагане на изпитвания химикал върху кожата на живи животни и оценяване на степента на увреждане на тъканта след фиксиран период от време (2). Освен настоящия метод за изпитване, са приети множество други методи за изпитване in vitro като алтернативи (3)(4) на стандартната in vivo процедура с кожа от заек (глава Б.4 от настоящото приложение, еквивалентна на TG 404 на ОИСР), използвана за идентифициране на корозивни химикали (2). Стратегия на GHS на ООН за поетапно изпитване и оценяване за оценката и класификацията на корозията на кожата и Ръководството на ОИСР относно Интегрираните подходи за изпитване и оценка (IATA) за кожно дразнене/корозия препоръчват употребата на валидирани и приети in vitro методи за изпитване в съответствие с модули 3 и 4 (1)(5). IATA описва няколко модула, които обединяват източници на информация и инструменти за анализ, и i) предоставя насоки за това как да се интегрират и използват съществуващите данни за изпитвания и различни от тези за изпитвания за оценка на потенциала на химикалите за дразнене на кожата и за корозивно действие върху кожата, и ii) предлага подход, когато е необходимо по-нататъшно изпитване, включително когато са определени отрицателни резултати (5). При този модулен подход, положителните резултати от in vitro методите за изпитване могат да се използват за класифициране на даден химикал като корозивен, без да е необходимо изпитване върху животни, като така се намалява и рафинира употребата на животни и предотвратяването на болката и стреса, които биха могли да възникнат за животните, ако се използват за тази цел.
|
|
3.
|
Изследванията за валидиране са завършени за модела in vitro на мембранна бариера, който е наличен в търговската мрежа под името Corrositex® (6)(7)(8) и показват цялостна точност на прогнозиране на корозия на кожата от 79 % (128/163), чувствителност от 85 % (76/89), и специфичност от 70 % (52/74) за база данни от 163 вещества и смеси (7). На базата на неговата потвърдена валидност, този валидиран референтен метод (VRM) се препоръчва за употреба като част от стратегията за поетапно изпитване за оценяване на потенциала за опасност от корозивното действие върху кожата на химикалите (5)(7). Преди in vitro модел на мембранна бариера да може да бъде използван за регулаторни цели, следва да се определи неговата надеждност, относимост (точност) и ограничения за предложената употреба, за да се гарантира, че той е сходен с тези на VRM (9), в съответствие с предварително дефинираните Стандарти за ефективност (СЕ) (10). Взаимното приемане на данните от ОИСР се гарантира само след като всеки предложен нов или актуализиран метод, съответстващ на СЕ, е бил разгледан и включен в еквивалентните насоки за изпитване на ОИСР. Понастоящем само един in vitro метод е обхванат от насоки за изпитване 435 на ОИСР и този метод за изпитване е наличният в търговската мрежа модел Corrositex®.
|
|
4.
|
Други методи за изпитване на корозивно дейсвие върху кожата са базирани на употребата на реконструирана човешка кожа (ОИСР TG 431) (3) и изолирана кожа от плъх (ОИСР TG 430) (4). Настоящите насоки за изпитване осигуряват и подкатегоризация на корозивни химикали в трите подкатегории корозивно действие съгласно GHS на ООН и трите групи опаковки при транспорт на ООН по отношение на опасността от корозивно действие върху кожата. Настоящите насоки за изпитване бяха първоначално приети през 2006 г. и актуализирани през 2015 г., за да включват отпратка към Ръководството за IATA и актуализация на списъка на опитните вещества.
|
ОПРЕДЕЛЕНИЯ
|
5.
|
Използваните определения са дадени в допълнението.
|
ПЪРВОНАЧАЛНИ СЪОБРАЖЕНИЯ И ОГРАНИЧЕНИЯ
|
6.
|
Изпитването, описано в настоящия метод за изпитване позволява идентификация и подкатегоризация на корозивни изпитвани химикали, съгласно GHS на ООН/CLP (таблица 1). В допълнение, подобен метод за изпитване може да се използва за вземане на решения относно наличието или отсъствието на корозивно действие на специфични класове химикали, напр. органични и неорганични киселини, киселинни производни (25) и основи за целите на изпитвания при транспортиране (7)(11)(12). Настоящият метод за изпитване описва обща процедура, сходна с валидирания референтен метод за изпитване (7). Тъй като този метод за изпитване не осигурява адекватна информация за дразненето накожата, следва да се отбележи, че МИ Б.46 (еквивалентен на TG 439 на ОИСР) специално разглежда въздействието върху здравето - кожно дразнене in vitro(13). За пълна оценка на локалното въздействие върху кожата след еднократна дермална експозиция следва да се направи справка с Ръководството на ОИСР относно Интегрираните подходи за изпитване и оценка (5).
Таблица 1
Категория и подкатегории на вещества с корозивно действие върху кожата по GHS на ООН (26)
|
Категория корозивно действие (категория 1) (за органи, които не използват подкатегории)
|
Подкатегории за вещества с потенциално корозивно действие (26) (за органи, използващи подкатегории, включително Регламент CLP)
|
Корозивно за ≥ 1 от 3 животни
|
|
Експозиция
|
Наблюдение
|
|
Корозивен
|
Корозивно от подкатегория 1A
|
≤ 3 минути
|
≤ 1 час
|
|
Корозивно от подкатегория 1B
|
> 3 минути / ≤ 1 час
|
≤ 14 дни
|
|
Корозивно от подкатегория 1C
|
> 1 час / ≤ 4 часа
|
≤ 14 дни
|
|
|
7.
|
Ограничение на валидирания референтен метод (7) е, че много некорозивни химикали и някои корозивни химикали не изпълняват изискванията за изпитване, въз основа на резултатите от първоначалното изпитване за съвместимост (вж. точка 13). Водните разтвори на химикали с pH от порядъка на 4,5 до 8,5 често не отговарят на изискванията за изпитване; Все пак, 85 % от химикалите, изпитани в този обхват на pH, са били некорозивни при изпитвания с животни (7). In vitro методът за изпитване с мембранна бариера може да се използва за изпитване на твърди вещества (разтворими или неразтворими във вода), течности (във водни или неводни разтвори) и емулсии. Въпреки това, изпитвани химикали, които не предизвикват откриваема промяна в изпитването за съвместимост (т.е. промяна в цвета в Системата за откриване на химикали (CDS) на валидирания референтен метод за изпитване) не могат да бъдат изпитвани по метод за изпитване с мембранна бариера и следва да бъдат изпитвани чрез други методи за изпитване.
|
ПРИНЦИП НА ИЗПИТВАНЕТО
|
8.
|
Системата за изпитване обхваща два компонента: синтетична макромолекулна био-бариера и система за откриване на химикали (CDS); с помощта на CDS, този метод за изпитване открива увреждане на мембранната бариера, предизвикано от корозивни изпитвани химикали след прилагане на изпитвания химикал върху повърхността на синтетичната макромолекулна мембранна бариера (7), вероятно по същия(същите) механизъм(механизми) за корозия, който(които) действат и на живата кожа.
|
|
9.
|
Преминаването през мембранната бариера (или пробива) може да се измери чрез множество процедури или чрез CDS, включително чрез промяна в цвета на индикаторен оцветител за рН или промяна на друго свойство на индикаторния разтвор под бариерата.
|
|
10.
|
Мембранната бариера следва да бъде определена като валидна, т.е. относима и надеждна с оглед на своето предназначение. Това включва гарантиране, че различните препарати са сходни по отношение на бариерните свойства, напр. могат да осигурят постоянна бариера срещу некорозивни химикали, могат да категоризират корозивните свойства на химикалите в рамките на различните подкатегории на корозивност, съгласно GHS на ООН (1). Определената класификация е базирана на времето, което е необходимо на даден химикал да премине през мембранната бариера до индикаторния разтвор.
|
ДОКАЗВАНЕ НА КОМПЕТЕНТНОСТ
|
11.
|
Преди рутинната употреба на in vitro метода за изпитване с мембранна бариера, който спазва изискванията за този метод за изпитване, лабораториите следва да демонстрират техническа компетентност чрез правилно класифициране на дванадесетте опитни вещества, препоръчани в таблица 2. При ситуации, в които дадено вписано вещество е неналично или когато е обосновано, може да се използва друго вещество, за което има налични адекватни in vivo и in vitro справочни данни (напр. от списъка на референтните химикали (10)), при условие че се прилагат същите критерии за избор, които са описани в таблица 1.
Таблица 2
Вещества за изпитване на пригодност
(27)
|
Вещество (28)
|
CASRN
|
Химичен клас
|
Подкатегория in Vivo по GHS на ООН (29)
|
Подкатегория in Vitro по GHS на ООН (29)
|
|
Борен трифлуорид дихидрат
|
13 319 -75-0
|
Неорганични киселини
|
1А
|
1А
|
|
Азотна киселина
|
7 697 -37-2
|
Неорганични киселини
|
1А
|
1А
|
|
Фосфорен пентахлорид
|
10 026 -13-8
|
Прекурсори на неорганични киселини
|
1А
|
1А
|
|
Валерил хлорид
|
638-29-9
|
Киселинни хлориди
|
1B
|
1B
|
|
Натриев хидроксид
|
1 310 -73-2
|
Неорганични основи
|
1B
|
1B
|
|
1-(2-аминоетил) пиперазин
|
140-31-8
|
Алифатни амини
|
1B
|
1B
|
|
Бензенсулфонил хлорид
|
98-09-9
|
Киселинни хлориди
|
1C
|
1C
|
|
N,N-Диметил бензиламин
|
103-83-3
|
Анилини
|
1C
|
1C
|
|
Тетраетиленпентамин
|
112-57-2
|
Алифатни амини
|
1C
|
1C
|
|
Евгенол
|
97-53-0
|
Феноли
|
NC
|
NC
|
|
Нонил акрилат
|
2 664 -55-3
|
Акрилати/метакрилати
|
NC
|
NC
|
|
Натриев бикарбонат
|
144-55-8
|
Неорганични соли
|
NC
|
NC
|
|
ПРОЦЕДУРА
|
12.
|
Следващите точки описват компонентите и процедурите на метода за изпитване на изкуствена мембранна бариера за оценка на корозивност (7)(15), на базата на текущия VRM, т.е. наличния в търговската мрежа Corrositex®. Мембранната бариера и решенията за съвместимост/индикатор и категоризация могат да бъдат конструирани, подготвени и набавени от търговската мрежа, какъвто е случая с VRM Corrositex®. Наличен е примерен протокол на метод за изпитване за валидирания референтен метод за изпитване (7). Изпитването следва да бъде извършено при околна температура (17-25 °C), а компонентите следва да отговарят на следните условия.
|
Изпитване за съвместимост на изпитвания химикал
|
13.
|
Преди извършване на изпитването на мембранната бариера, се прави изпитване за съвместимост, за да се определи дали изпитвания химикал може да се открива от CDS. Ако CDS не открие изпитвания химикал, методът за изпитване на мембранната бариера не е подходящ за оценяване на потенциалната корозивност на този конкретен изпитван химикал и следва да се използва различен метод за изпитване. CDS системата и условията на експозиция, използвани за изпитването за съвместимост следва да отразяват експозицията в последващото изпитване на мембранната бариера.
|
Изпитване за категоризация на времевата рамка на изпитвания химикал
|
14.
|
Ако е подходящо за метода на изпитване, изпитван химикал, който е бил квалифициран чрез изпитването за съвместимост, следва да бъде подложен на изпитване за категоризация на времевата рамка, т.е. скринингово изпитване за разграничаване на слаби и силни киселини или основи. Например, в рамките на валидирания референтен метод за изпитване, изпитването за категоризация на времевата рамка се използва, за да покаже коя от двете времеви рамки следва да бъде използвана на базата на това дали е открит значителен резерв от киселина или основа. За определяне на корозивността и подкатегорията на кожна корозивност съгласно GHS на ООН следва на бъдат използвани две различни времеви рамки за пробив, въз основа на киселинния или алкалния резерв на изпитвания химикал.
|
КОМПОНЕНТИ НА МЕТОДА ЗА ИЗПИТВАНЕ НА МЕМБРАННАТА БАРИЕРА
Мембранна бариера
|
15.
|
Мембранната бариера се състои от два компонента: протеинов макромолекулен гел във воден разтвор и пропусклива опорна мембрана. Протеиновият гел следва да бъде непропусклив за течности и твърди вещества, но може да бъде подложен на корозия и в резултат от това да стане пропусклив. Напълно изградената мембранна бариера следва да се съхранява при предварително определени условия, които доказано изключват влошаване на качествата на гела, напр. изсушаване, растеж на микроорганизми, изместване, напукване, които биха влошили ефективността му. Следва да се определи приемливият срок за съхранение и подготвените мембранни бариери да не се използват след този срок.
|
|
16.
|
Пропускливата опорна мембрана осигурява механична опора за протеиновия гел по време на процеса на образуване на гела и на експозиция на изпитвания химикал. Опорната мембрана следва да предотвратява провисване или изместване на гела и да пропуска лесно всички изпитвани химикали.
|
|
17.
|
Протеиновият гел, съставен от протеин, напр. кератин, колаген или смеси от протеини, образуващи гелова матрица, служи като цел за изпитвания химикал. Протеиновият материал се поставя по повърхността на опорната мембрана и се оставя да се превърне в гел, преди да се постави мембранната бариера над индикаторния разтвор. Протеиновият гел следва да бъде с равномерна дебелина и плътност по цялата си площ, и да няма въздушни мехури или дефекти, които биха повлияли върху функционалната му цялост.
|
Система за откриване на химикали (CDS)
|
18.
|
Индикаторният разтвор, който е същият разтвор, използван за изпитването за съвместимост, следва да реагира на присъствието на изпитван химикал. Следва да се използва индикаторен оцветител за pH или комбинация от оцветители, напр. крезолово червено и метил-оранж, които показват промяна в цвета, в отговор на наличието на изпитвания химикал. Измервателната система може да бъде визуална или електронна.
|
|
19.
|
Системите за откриване, които са разработени за откриване на преминаването на изпитвания химикал през бариерната мембрана, следва да бъдат оценявани по тяхната относимост и надеждност, за да се докаже обхватът от химикали, които може да бъдат открити, и количествените лимити на откриване.
|
ЕФЕКТИВНОСТ НА ИЗПИТВАНЕТО
Сглобяване на компонентите на метода за изпитване
|
20.
|
Мембранната бариера се позиционира в стъкленица (или тръба), съдържаща индикаторния разтвор така, че опорната мембрана да е в пълен контакт с индикаторния разтвор и да няма наличие на въздушни мехури. Трябва да се внимава, за да не се наруши целостта на бариерата.
|
Прилагане на изпитвания химикал
|
21.
|
Внимателно се нанася подходящо количество от изпитвания химикал, напр. 500 μl течност или 500 mg фино разпрашено твърдо вещество (7) върху горната повърхност на мембранната бариера и се разстила равномерно. За всеки изпитван химикал се подготвя подходящ брой повторения, напр. четири (7), както и съответния брой контроли (вж. точки 23 до 25). Времето за прилагане на изпитвания химикал върху мембраната се записва. За да се гарантира, че кратките времена за корозия се записват точно, времената за прилагане на изпитвания химикал по стъклениците на повторенията се подреждат едно след друго.
|
Измерване на проникванията през мембранната бариера
|
22.
|
Всяка стъкленица се наблюдава по подходящ начин и времето на първата промяна в индикаторния разтвор, т.е. проникването през бариерата, се записва, като се определя изтеклото време между приложението и проникването през мембранната бариера.
|
Контроли
|
23.
|
При изпитвания, които включват използването на носител или разтворител с изпитвания химикал, носителят или разтворителят следва да бъдат съвместими със системата на мембранната бариера, т.е. да не променят нито целостта на системата на мембранната бариера, нито корозивността на изпитвания химикал. Когато е приложимо, контролът на разтворителя (или носителя) следва да се изпитват паралелно с изпитвания химикал, за да се докаже съвместимостта на разтворителя със системата на мембранната бариера.
|
|
24.
|
Положителен (корозивен) контрол с междинна корозивна дейност, напр. 110 ± 15 mg натриев хидроксид (подкатегория корозивни вещества 1B по GHS на ООН) (7), следва да се изпитва паралелно с изпитвания химикал, за да се оцени дали системата за изпитване работи по приемлив начин. Втора положителна контрола, която е от същия химичен клас като изпитвания химикал, може да бъде полезна за оценяване на относителния потенциал за корозивност на корозивен изпитван химикал. Следва да се избере(изберат) положителен(-ни) контрол(и), който (които) е/са междинни в своята корозивност (напр. под-категория 1B по GHS на ООН), за да се открият промените във времето за проникване, което може да бъде неприемливо дълго или по-кратко от установената референтна стойност, чрез което се посочва, че системата за изпитване не функционира правилно. За целта, използването на крайно корозивни (подкатегория 1A по GHS на ООН) или некорозивни химикали се ограничава. Корозивен химикал от подкатегория 1B по GHS на ООН би позволил откриване на твърде бързо или твърде бавно време за преминаване. Може да се използва слабокорозивен химикал (подкатегория 1C по GHS на ООН) като положителна контрола за измерване на способността на метода за изпитване да разграничава неизменно слабокорозивните от некорозивните химикали. Независимо от използвания подход, следва да се разработи приемлив диапазон на отклика на положителната контрола на базата на обхвата от стойности на преминаване от предходни периоди за използваните положителни контроли, като например средните ± 2-3 стандартни отклонения. Във всяко проучване, точното време на пробив следва да бъде определено за положителната контрола, така че отклоненията извън приемливия обхват да могат да бъдат открити.
|
|
25.
|
Паралелно с изпитвания химикал следва да бъде изпитан и отрицателнха (некорозивна) контрола, напр. 10 % лимонена киселина, 6 % пропионова киселина (7) като още една мярка за качествен контрол, за доказване на функционалната цялост на мембранната бариера.
|
Критерии за приемливост на изследването
|
26.
|
Съгласно установените времеви параметри за всяка от подкатегориите корозивност на GHS на ООН, изтеклото време (в минути) между приложението на изпитвания химикал върху мембранната бариера и преминаването на бариерата се използва за прогнозиране на корозивността на изпитвания химикал. За да се разглежда едно изследване като приемливо, паралелната положителна контрола следва да дава очакваното време на реакция, т.е., проникване (напр. 8-16 минути време на пробив за натриев хидроксид, ако се използва като положителна контрола), паралелната отрицателна контрола не следва да бъде корозивна, а когато е включена, паралелната контрола на разтворителя не следва нито да бъде корозивна, нито да променя корозивния потенциал на изпитвания химикал. Преди рутинна употреба на метод, който се придържа към настоящия метод за изпитване, лабораториите следва да докажат техническата си компетентност, като използват дванадесетте вещества, препоръчани в таблица 2. За новите вариантни методи („me-too“), разработени съгласно настоящия метод за изпитване, които са структурно и функционално сходни с валидирания референтен метод (14), предварително определените стандарти за ефективност следва да бъдат използвани за доказване на надеждността и точността на новия метод преди употребата му за редовни изпитвания (10).
|
Тълкуване на резултатите и класификация за корозивност на изпитваните химикали
|
27.
|
Времето (в минути), изтекло между прилагането на изпитвания химикал върху мембранната бариера и проникването през бариерата се използва за класифициране на изпитвания химикал в подкатегориите на корозивни вещества по GHS на ООН (1) и, ако е приложимо, в опаковъчна група на ООН (16). Граничните времеви стойности за всяка от трите корозивни подкатегории се определят за всеки предложен метод за изпитване. Окончателните решения относно граничните времена следва да вземат под внимание нуждата от намаляване до минимум на опасността от подценяване при класифициране на корозивно вещество (т.е. неверни отрицателни резултати). В настоящите насоки за изпитване следва да се използват граничните времена на Corrositex® както са описани в таблица 3, тъй като това е единственият метод за изпитване, който в момента отговаря на насоките за изпитване (7).
Таблица 3
Corrositex® модел за предвиждане
|
Средно време за пробив (в мин.)
|
Предвиждане за GHS на ООН (32)
|
|
Изпитвани химикали от категория 1 (30) (определени чрез изпитването за категоризация на метода)
|
Изпитвани химикали от категория 2 (31) (определени чрез изпитването за категоризация на метода)
|
|
0–3 мин.
|
0–3 мин.
|
Корозивен Незадължителна подкатегория 1A
|
|
> 3 до 60 min.
|
> 3 до 30 min.
|
Корозивен Незадължителна подкатегория 1B
|
|
> 60 до 240 min.
|
> 30 до 60 min.
|
Корозивен Незадължителна подкатегория 1C
|
|
> 240 мин.
|
> 60 мин.
|
Некорозивен
|
|
ДАННИ И ПРОТОКОЛИРАНЕ
Данни
|
28.
|
Времето (в минути), което е изтекло между приложение и проникването през бариерата на изпитвания химикал и положителната(те) контрола(и) следва да бъде протоколирано в таблична форма като отделни данни за повторение, както и като средна стойност ± стандартното отклонение за всяко изпитване.
|
Протокол от изпитването
|
29.
|
Протоколът от изпитването следва да включва следната информация:
|
|
Изпитван химикал и контролни вещества:
|
—
|
Вещество с една съставка: химична идентификация, като например наименование по IUPAC или CAS, CAS номер, код SMILES или InChI, структурна формула, чистота, химична идентичност на онечистванията, доколкото е целесъобразно и практически осъществимо, и т.н.;
|
|
—
|
Вещество с повече съставки, UVCB и смес: характеризирано, доколкото е възможно, с химичната си идентичност (вж. по-горе), количествения състав и относимите физични и химични свойства на съставките;
|
|
—
|
Външен вид, разтворимост във вода и допълнителни относими физикохимични свойства;
|
|
—
|
източник, номер на партидата, при наличност;
|
|
—
|
Обработка на изпитвания химикал/веществото за контроли преди изпитването, ако се прилага (напр. затопляне, смилане);
|
|
—
|
стабилност на изпитвания химикал, крайна дата за употреба или дата за повторен анализ, ако е известна;
|
|
|
|
Носител:
|
—
|
химично идентифициране, концентрация (когато е необходимо), използван обем;
|
|
—
|
обосноваване на избора на носител.
|
|
|
|
Използван модел in vitro на мембранна бариера и протокол, включително доказана точност и надеждност
|
|
|
Условия на изпитване:
|
—
|
Описание на използваната опитна постановка и подготвителни процедури;
|
|
—
|
източник и състав на използваната мембранна бариера in vitro;
|
|
—
|
състав и свойства на индикаторния разтвор;
|
|
—
|
количества на изпитвания химикал и контролните вещества;
|
|
—
|
описание и обосновка на изпитването за категоризация на времевата рамка;
|
|
—
|
описание на приложените критерии за оценка и класификация;
|
|
—
|
доказване на компетентност на изпълнение на метода за изпитване преди редовна употреба чрез изпитване на химикалите за изпитване за пригодност.
|
|
|
|
Резултати:
|
—
|
представяне в табличен вид на отделните необработени данни от индивидуалното изпитване и контролните проби за всяко повторение;
|
|
—
|
описание на други наблюдавани ефекти;
|
|
—
|
получената класификация по отношение на използвания модел за прогнозиране/критериите за вземане на решение.
|
|
Обсъждане на резултатите
Заключения
|
ЛИТЕРАТУРА
|
(1)
|
United Nations (UN) (2013). Globally Harmonized System of Classification and Labelling of Chemicals (GHS), Fifth Revised Edition, UN New York and Geneva, 2013. Available at: http://www.unece.org/trans/danger/publi/ghs/ghs_rev05/05files_e.html.
|
|
(2)
|
Глава Б.4 от настоящото приложение, Остро дразнещо/корозивно действие върху кожата.
|
|
(3)
|
Глава Б.40bis от настоящото Приложение, In Vitro корозивно действие върху кожата: Метод за изпитване с реконструиран човешки епидермис (RhE).
|
|
(4)
|
Глава Б.40 настоящото приложение, In Vitro корозивно действие върху кожата: Транскутанно електрическо съпротивление (TER).
|
|
(5)
|
OECD (2015). Guidance Document on Integrated Approaches to Testing and Assessment of Skin Irritation/Corrosion. Environment, Health and Safety Publications, Series on Testing and Assessment, (No 203). Organisation for Economic Cooperation and Development, Paris.
|
|
(6)
|
Fentem, J.H., Archer, G.E.B., Balls, M., Botham, P.A., Curren, R.D., Earl, L.K., Esdaile, D.J., Holzhutter, H.-G. and Liebsch, M. (1998). The ECVAM International Validation Study on In Vitro Tests for Skin Corrosivity. 2. Results and Evaluation by the Management Team. Toxicology In Vitro 12, 483-524.
|
|
(7)
|
ICCVAM (1999). Corrositex®. An In Vitro Test Method for Assessing Dermal Corrosivity Potential of Chemicals. The Results of an Independent Peer Review Evaluation Coordinated by ICCVAM, NTP and NICEATM. NIEHS, NIH Publication (No 99-4495.)
|
|
(8)
|
Gordon V.C., Harvell J.D. and Maibach H.I. (1994). Dermal Corrosion, the Corrositex® System: A DOT Accepted Method to Predict Corrosivity Potential of Test Materials. In vitro Skin Toxicology-Irritation, Phototoxicity, Sensitization. Alternative Methods in Toxicology 10, 37-45.
|
|
(9)
|
OECD (2005). Guidance Document on the Validation and International Acceptance of New or Updated Test Methods for Hazard Assessment. Environmental, Health and Safety Publications. Series on testing and Assessment (No 34).
|
|
(10)
|
OECD (2014). Performance Standards for the Assessment of Proposed Similar or Modified In Vitro Membrane Barrier Test Method for Skin Corrosion in Relation to TG 435. Organisation for Economic Cooperation and Development, Paris. Available at: http://www.oecd.org/chemicalsafety/testing/PerfStand-TG430-June14.pdf.
|
|
(11)
|
ECVAM (2001). Statement on the Application of the CORROSITEX® Assay for Skin Corrosivity Testing. 15th Meeting of ECVAM Scientific Advisory Committee (ESAC), Ispra, Italy. ATLA 29, 96-97.
|
|
(12)
|
U.S. DOT (2002). Exemption DOT-E-10904 (Fifth Revision). (September 20, 2002). Washington, D.C., U.S. DOT.
|
|
(13)
|
Chapter B.46 of this Annex, In Vitro Skin Irritation: Метод за изпитване на реконструиран човешки епидермис. ICCVAM (2004). ICCVAM Recommended Performance Standards for In Vitro Test Methods for Skin Corrosion. NIEHS, NIH Publication No 04-4510. Available at: http://www.ntp.niehs.nih.gov/iccvam/docs/dermal_docs/ps/ps044510.pdf.
|
|
(14)
|
U.S. EPA (1996). Method 1120, Dermal Corrosion. Available at: http://www.epa.gov/osw/hazard/testmethods/sw846/pdfs/1120.pdf.
|
|
(15)
|
United Nations (UN) (2013). UN Recommendations on the Transport of Dangerous Goods, Model Regulations, 18th Revised Edition (Part, Chapter 2.8), UN, 2013. Available at: http://www.unece.org/fileadmin/DAM/trans/danger/publi/unrec/rev18/English/Rev18_Volume1_Part2.pdf.
|
Допълнение
ОПРЕДЕЛЕНИЯ
Точност
: степента на близост между резултатите от метода за изпитване и приетите референтни стойности. Това е мярка за ефективността на метода за изпитване и един от аспектите на неговата приложимост. Терминът често се използва взаимозаменяемо с термина „съответствие“, за да се обозначи делът на верните резултати при даден метод за изпитване (9).
Химикал
: Вещество или смес.
Система за откриване на химикали (CDS)
: Визуална или електронна система за измерване с индикаторен разтвор, която реагира на присъствието на изпитван химикал, напр. чрез промяна в индикаторния оцветител за pH, или комбинация от оцветители, които показват промяна в цвета като реакция на наличието на изпитвания химикал, или чрез други видове химични или електрохимични реакции.
Съответствие
: Това е мярка за ефективността на метода за изпитване за методи, които дават категоричен резултат и е един от аспектите на неговата относимост. Терминът понякога се използва взаимозаменяемо с термина „точност“ и се определя като делът на всички изпитани химикали, които са правилно класифицирани като положителни или отрицателни. Съответствието зависи в голяма степен от преобладаването на положителните контроли сред видовете на текущо изпитвания химикал (9).
GHS (Глобална хармонизирана система за класифициране и етикетиране на химикали)
: система, предлагаща класифициране на химикали (вещества и смеси) според стандартизирани видове и степени на физическа, здравна и екологична опасност и разглеждаща съответни съобщителни елементи, като например пиктограми, сигнални думи, предупреждения за опасност, препоръки за безопасност и информационни листове за безопасност, така че те да съобщават информация за тяхното неблагоприятно въздействие с оглед защита на хората (включително работодатели, работещи, служители в транспорта, потребители и аварийни служители) и на околната среда (1).
IATA
: Интегриран подход към изпитването и оценката.
Смес
: смес или разтвор, съставен от две или повече вещества.
Вещество с една съставка
: Вещество, определено от неговия количествен състав, в който съдържанието на една основна съставка е най-малко 80 % (w/w).
Вещество с повече съставки
: Вещество, определено от неговия количествен състав, в който повече от една основна съставка се съдържа в концентрация ≥ 10 % (w/w) и < 80 % (w/w). Веществото с повече съставки е резултат от производствен процес. Разликата между смес и вещество с повече съставки е, че дадена смес е получена чрез смесване на две или повече вещества без химична реакция. Веществото с повече съставки е резултат от химична реакция.
NC
: Некорозивен.
Стандарти за ефективност
: стандарти въз основа на валидиран метод за изпитване, осигуряващи основата за оценка на сравнимостта на предложен метод за изпитване, който по своя механизъм и функционално е сходен с валидирания метод. Включени са i) съществени компоненти на метода за изпитване; ii) минимален списък на референтни химикали, подбрани между химикалите, използвани за демонстриране на приемливата ефективност на валидирания метод за изпитване; и iii) сходните нива за точност и надеждност въз основа на получените за валидирания метод за изпитване, които предложеният метод за изпитване следва да демонстрира при оценяването чрез използване на минималния списък на референтните химикали (9).
Относимост
: Описание на взаимовръзката между метода на изпитване и ефекта, представляващ интерес, и дали тя е значима и полезна за определена цел. Това е степента, в която изпитването правилно измерва или прогнозира биологичния ефект, представляващ интерес. Относимостта включва разглеждането на точността (съответствието) на метода за изпитване (9).
Надеждност
: мярка за степента, в която даден метод за изпитване може да се приложи възпроизводимо в една и съща лаборатория и в различни лаборатории по различно време, при използване на един и същи протокол. Определя се, като се изчислява вътрешно- и междулабораторната възпроизводимост (9).
Чувствителност
: Относителният дял на всички положителни/активни химикали, които са класифицирани правилно чрез метода за изпитване. Това е мярка за точността на метода за изпитване, който предоставя категорични резултати и е важен фактор при оценката на относимостта на метода за изпитване (9).
Корозивно действие върху кожата in vivo
: Причиняването на необратимо увреждане на кожата; а именно видима некроза през епидермиса и в дермата след прилагане на изпитван химикал в течение на не повече от четири часа. Характерните прояви на корозивно действие включват язви, кървене, кървави струпеи, а до края на 14-дневния период на наблюдение се появява обезцветяване, предизвикано от избледняване на кожата, области с пълна алопеция и цикатрикси. Хистопатологията трябва да се вземе под внимание при оценката на спорните увреждания.
Специфичност
: Относителният дял на всички отрицателни/неактивни химикали, които са класифицирани правилно чрез метода за изпитване. Това е мярка за точността на метода за изпитване, която предоставя категорични резултати и е важен фактор при оценката на метода за изпитване (9).
Вещество
: Химически елемент и неговите съединения в естествено състояние или получени чрез производствен процес, включително всякакви добавки, необходими за запазване на стабилността му, както и всякакви онечиствания, произтичащи от използваната технология, но с изключение на разтворители, които могат да бъдат отделени, без да се засегне стабилността на веществото или да се промени съставът му.
Изпитван химикал
: Всяко вещество или смес, изпитвано(а) с използване на настоящия метод за изпитване.
UVCB
: Вещества с неизвестен или променлив състав, сложни продукти от реакции или биологични материали.
Б.66
IN VITRO ИЗСЛЕДВАНИЯ ЗА ТРАНСАКТИВАЦИЯ ЧРЕЗ СТАБИЛНО ТРАНСФЕКТИРАНЕ С ЦЕЛ ОТКРИВАНЕ НА АГОНИСТИ И АНТАГОНИСТИ НА ЕСТРОГЕННИ РЕЦЕПТОРИ
ОБЩО ВЪВЕДЕНИЕ
Насоки за изпитване на ОИСР, базирани на ефективност
|
1.
|
Настоящият метод за изпитване е еквивалентен на насоките за изпитване на ОИСР (ТG) 455 (2016). TG 455 са насоки за изпитване базирани на ефективност (PBTG), описващи методиката за In Vitro изследвания за трансактивация чрез стабилно трансфектиране с цел откриване на агонисти и антагонисти на естрогенни рецептори (ER TA изследвания). Състоят се от няколко механистично и функционално сходни методи за изпитване за идентифициране на агонисти и антагонисти на естрогенни рецептори (т.е. ERα, и/или ERβ) и следва да улесняват разработването на нови подобни или модифицирани методи за изпитване в съответствие с принципите за валидиране, установени в Ръководството на ОИСР относно валидирането и международното приемане на нови или актуализирани методи за изпитване за оценка на опасности (1). Напълно валидираните референтни методи за изпитване (допълнение 2 и допълнение 3), които осигуряват основата за настоящите PBTG, са:
|
—
|
Изследване чрез стабилно трансфектиране TA (STTA) (2), използващо клетъчната линия (h) ERα-HeLa-9903; както и
|
|
—
|
изследването VM7Luc ER TA (3), използващо клетъчната линия VM7Luc4E2 (33), която преобладаващо експресира hERα с известно допълнение от hERβ(4)(5).
|
За разработването и валидирането на подобни изследвания за същата крайна точка на опасност следва да се използват наличните стандарти за ефективност (СЕ) (6) (7). Те позволяват навременно изменение на PBTG 455 така, че да могат да се добавят нови подобни изследвания към актуализираните PBTG; въпреки това обаче, подобни изследвания се добавят едва след като бъдат прегледани от ОИСР и бъде сметнато, че отговарят на стандартите за ефективност. Всички изследвания, включени в TG 455, могат да се използват, за да се покрият изискванията на държавите за резултати от изпитвания относно трансактивация на естрогенни рецептори, като същевременно се възползват от предимствата на Взаимното приемане на данни.
|
Контекст и принципи на изследванията, включени в настоящия метод за изпитване
|
2.
|
През 1998 г. ОИСР инициира дейност с висок приоритет за преразглеждане на съществуващите и разработване на нови насоки за изпитване при провеждане на скрининг и изпитване на химикали, които имат потенциала да нарушават функциите на ендокринната система. Концептуалната рамка (КР) на ОИСР за изпитване и оценяване на химикали, които имат потенциала да нарушават функциите на ендокринната система бе преразгледана през 2012 г. Оригиналната и преразгледаната КР са включени в приложенията към Ръководството на ОИСР относно стандартизираните насоки за изпитване за оценка на химикали, нарушаващи функциите на ендокринната система (8). КР обхваща пет нива, като всяко ниво отговаря на различно ниво на биологична комплексност. ER изследванията за трансактивиране (TA), описани в настоящия метод за изпитване, са от ниво 2, което включва „изследвания in vitro, осигуряващи данни за избран(и) ендокринен(ни) механизъм(-ми)/път(-ища)“. Този метод за изпитване е за изпитвания in vitro за трансактивиране (TA), предвидени за идентифициране на агонисти и антагонисти на естрогенни рецептори (ER).
|
|
3.
|
Взаимодействието на естрогените с ER може да окаже въздействие върху транскрипцията на естроген-контролираните гени, които могат да причинят индуциране или инхибиране на клетъчни процеси, включително тези, които са необходими за клетъчната пролиферация, нормалното развитие на зародиша и репродуктивната функция (9)(10)(11). Смущението на функцията на нормалните естрогенни системи може да причини евентуални неблагоприятни ефекти върху нормалното развитие (онтогенеза), репродуктивното здраве и целостта на репродуктивната система.
|
|
4.
|
In vitro TA изследванията са базирани на пряко или непряко взаимодействие на веществата със специфичен рецептор, който регулира транскрипцията на продуктите на репортерния ген. Такива изследвания са широко използвани за оценяване на генната експресия, регулирана от специфични нуклеарни рецептори, като например ER (12) (13) (14) (15) (16). Те са предложени за откриване на естрогенна трансактивация, регулирана от ER (17) (18) (19). Има най-малко два основни под-типа нуклеарни ER, α и β, които са кодирани от определени гени. Съответните протеини има различни биологични функции, както и различно тъканно разпределение и лиганд-свързващ афинитет (20)(21)(22)(23)(24)(25)(26). Нуклеарният ERα медиира класическия естрогенен отклик (27)(28)(29)(30), и поради това повечето модели, които се разработват в момента за измерване на активирането или инхибирането на ER, са специфични за ERα. Изследванията се използват за идентифициране на химикали, които активират (или потискат) ER след свързване с лиганда, след което лиганд-рецепторният комплекс се свързва със специфичните елементи на отклика на ДНК и трансактивира репортерен ген, което води до повишена клетъчна експресия на маркирания протеин. В тези изследвания могат да се използват различни отговори на репортерите. При системите, базирани налуцифераза, луциферазният ензим трансформира луцифериновия субстрат до био-луминесцентен продукт, който може да се измери количествено с помощта на луминометър. Други примери за често срещани репортерни гени са флуоресцентния протеин и генът LacZ, който кодира β-галактосидазата, ензим, който преобразува безцветния субстрат X-гал (5- бромо-4-хлоро-индолил-галактопиранозид) в син продукт, който може да се определи количествено с помощта на спектрофотометър. Тези репортери могат да бъдат измерени бързо и евтино с тестови китове, налични в търговската мрежа.
|
|
5.
|
Валидационните изпитвания на STTA и изследванията VM7Luc TA доказаха тяхната относимост и надеждност за предвидената цел (3)(4)(5)(30). Стандартите за ефективност за базираните на луминесценция ER TA изследвания, използващи клетъчни линии от гръдна тъкан, са включени в Доклада за оценка на метода за изпитване на ICCVAM относно метода за изпитване LUMI-CELL® ER (VM7Luc ER TA): In Vitro изследване за идентифициране на агонистично и антагонистично действие на химикали върху човешки естрогенен рецептор (3). Тези стандарти за ефективност са модифицирани, за да бъдат приложими както за STTA, така и за VM7Luc TA изследванията (2).
|
|
6.
|
Използваните определения и съкращения в рамките на настоящия метод за изпитване, са описани в допълнение 1.
|
Обхват и ограничения, свързани с TA изследванията
|
7.
|
Тези изследвания се предлагат за целите на скрининговите изпитвания и определянето на приоритети, но могат да предоставят и механистична информация, която може да се използва при подход, при който се отчита значимостта на доказателствения материал. Те се занимават с TA, предизвикана от химично свързване с ER в in vitro система. Поради това резултатите не бива да бъдат директно есктраполирани към комплексното сигнализиране и регулиране на здравата ендокринна система in vivo.
|
|
8.
|
Медиираната от от ER ТА се разглежда като един от ключовите механизми за нарушение на функциите на ендокринната система (ED), въпреки че има и други механизми, чрез които може да възникне ED, включително i) взаимодействия с други рецептори и ензимни системи в рамките на ендокринната система, ii) синтез на хормони, iii) метаболитно активиране и/или деактивиране на хормони, iv) разпределение на хормони в прицелните тъкани и v) клиърънс на хормони от организма. Нито едно от изследванията съгласно този метод за изпитване не се занимава с тези режими на действие.
|
|
9.
|
Настоящият метод за изпитване се занимава със способността на химикалите да активират (т.е. да действат като агонисти) и да потискат (т.е. да действат като антагонисти) транскрипция, зависеща от ER. Някои химикали, които могат по начин, зависещ от типа на клетките, да проявяват както агонистична, така и антагонистична активност, са известни като селективни модулатори на естрогенните рецептори (СМЕР). Химикалите, които дават отрицателен резултат в тези изследвания, биха могли да бъдат оценени в рамките на изследване за свързване на ER, преди да се заключи, че този химикал не се свързва с рецептора. В допълнение, вероятно е изследванията само да дадат информация за действието на родителската молекула, като се имат предвид ограничените способности за метаболизъм на in vitro клетъчните системи. Като се има предвид, че по време на валидацията са използвани само единични вещества, приложимостта върху изпитвани смеси не е разглеждана. Независимо от това методът на изпитване е теоретично приложим за изпитването на вещества с повече съставки и на смеси. Преди използването на метода на изпитване по отношение на вещества с повече съставки, UVCB или смес, с цел генериране на данни за предвидената регулаторна цел, следва да се разгледа въпросът дали той може да даде адекватни резултати за тази цел и, ако е така, защо. Подобни съображения не са необходими, когато има регулаторно изискване за изпитване на сместа.
|
|
10.
|
За информационни цели, таблица 1 предоставя резултатите от изпитване на агонисти за 34-те вещества, които бяха изпитани в рамките на двата напълно валидирани референтни методи на изпитване, описани в настоящия метод за изпитване. Въз основа на публикуваните протоколи 26 от тези вещества се класифицират като определени агонисти на ER, а 8 са отрицателни, включително при изследвания in vitro за свързване на ER и TA, и/или утеротрофичното изследване (2)(3)(18)(31)(32)(33)(34). Таблица 2 предоставя резултатите от изпитване на антагонисти за 15-те вещества, които бяха изпитани в рамките на двата напълно валидирани референтни методи на изпитване, описани в настоящия метод за изпитване. От тези вещества, въз основа на публикуваните протоколи, 4 се класифицират като определени/предполагаеми антагонисти на ER и 10 са отрицателни, включително при изследвания in vitro за свързване на ER и изследване за TA (2)(3)(18)(31). Като се имат предвид данните, обобщени в таблица 1 и таблица 2, бе постигнато 100 % съгласие между двата референтни метода на изпитване относно класифицирането на всички вещества, с изключение на едно вещество (Мифепристон) за изследването с антагонисти, като всяко вещество бе правилно класифицирано като агонист/антагонист на ER или като отрицателно. Допълнителна информация за тази група химикали, както и за допълнителните химикали, изпитвани в рамките на изследванията STTA и VM7Luc ER TA по време на изследванията за валидиране, е предоставена в Стандартите за ефективност за ERTA (6)(7), допълнение 2 (таблици 1, 2 и 3).
|
Таблица 1
Общ преглед на резултатите от изследванията STTA и VM7Luc ER TA за вещества, изпитани в двете изследвания с агонисти и класифицирани като агонисти на ER (POS) или отрицателни (NEG)
|
|
Вещество
|
CAS №
|
Изследване (35) STTA
|
Изследване (36) VM7Luc ER TA
|
Източник на данните за класифицирането (38)
|
|
ER TA Дейност
|
Стойност PC10 (M)
|
Стойност PC50
(2) (M)
|
Активност ER TA
|
Стойност EC50
(2), (37) (M)
|
Други ER TA (34)
|
ER Свързващи вещества
|
Утеротрофично
|
|
1
|
17ß-естрадиол (1)
|
50-28-2
|
POS
|
< 1,00 × 10-11
|
< 1,00 × 10-11
|
POS
|
5,63 × 10-12
|
POS (227/227)
|
POS
|
POS
|
|
2
|
17α-естрадиол (1)
|
57-91-0
|
POS
|
7,24 × 10-11
|
6,44 × 10-10
|
POS
|
1,40 × 10-9
|
POS(11/11)
|
POS
|
POS
|
|
3
|
17α-етинил естрадиол (1)
|
57-63-6
|
POS
|
< 1,00 × 10-11
|
< 1,00 × 10-11
|
POS
|
7,31 × 10-12
|
POS(22/22)
|
POS
|
POS
|
|
4
|
17β-тренболон
|
10161-33-8
|
POS
|
1,78 × 10-8
|
2,73 × 10-7
|
POS
|
4,20 × 10-8
|
POS (2/2)
|
NT
|
NT
|
|
5
|
19-нортестостерон (1)
|
434-22-0
|
POS
|
9,64 × 10-9
|
2,71 × 10-7
|
POS
|
1,80 × 10-6
|
POS(4/4)
|
POS
|
POS
|
|
6
|
4-кумилфенол (1)
|
599-64-4
|
POS
|
1,49 × 10-7
|
1,60 × 10-6
|
POS
|
3,20 × 10-7
|
POS(5/5)
|
POS
|
NT
|
|
7
|
4-трет-октилфенол (1)
|
140-66-9
|
POS
|
1,85 × 10-9
|
7,37 × 10-8
|
POS
|
3,19 × 10-8
|
POS(21/24)
|
POS
|
POS
|
|
8
|
Апигенин (1)
|
520-36-5
|
POS
|
1,31 × 10-7
|
5,71 × 10-7
|
POS
|
1,60 × 10-6
|
POS(26/26)
|
POS
|
NT
|
|
9
|
Атразин (1)
|
1912-24-9
|
NEG
|
—
|
—
|
NEG
|
—
|
NEG (30/30)
|
NEG
|
NT
|
|
10
|
Бисфенол A (1)
|
80-05-7
|
POS
|
2,02 × 10-8
|
2,94 × 10-7
|
POS
|
5,33 × 10-7
|
POS (65/65)
|
POS
|
POS
|
|
11
|
Бисфенол Б (1)
|
77-40-7
|
POS
|
2,36 × 10-8
|
2,11 × 10-7
|
POS
|
1,95 × 10-7
|
POS(6/6)
|
POS
|
POS
|
|
12
|
Бутилбензил фталат (1)
|
85-68-7
|
POS
|
1,14 × 10-6
|
4,11 × 10-6
|
POS
|
1,98 × 10-6
|
POS(12/14)
|
POS
|
NEG
|
|
13
|
Кортикостерон (1)
|
50-22-6
|
NEG
|
—
|
—
|
NEG
|
—
|
NEG(6/6)
|
NEG
|
NT
|
|
14
|
Куместрол (1)
|
479-13-0
|
POS
|
1,23 × 10-9
|
2,00 × 10-8
|
POS
|
1,32 × 10-7
|
POS(30/30)
|
POS
|
NT
|
|
15
|
Дайдзеин (1)
|
486-66-8
|
POS
|
1,76 × 10-8
|
1,51 × 10-7
|
POS
|
7,95 × 10-7
|
POS(39/39)
|
POS
|
POS
|
|
16
|
Диетилстилбестрол (1)
|
56-53-1
|
POS
|
< 1,00 × 10-11
|
2,04 × 10-11
|
POS
|
3,34 × 10-11
|
POS(42/42)
|
POS
|
NT
|
|
17
|
Ди-n-бутил фталат
|
84-74-2
|
POS
|
4,09 × 10-6
|
|
POS
|
4,09 × 10-6
|
POS(6/11)
|
POS
|
NEG
|
|
18
|
Етил парабен
|
120-47-8
|
POS
|
5,00 × 10-6
|
(няма PC50)
|
POS
|
2,48 × 10-5
|
POS
|
|
NT
|
|
19
|
Естрон (1)
|
53-16-7
|
POS
|
3,02 × 10-11
|
5,88 × 10-10
|
POS
|
2,34 × 10-10
|
POS(26/28)
|
POS
|
POS
|
|
20
|
Генистеин (1)
|
446-72-0
|
POS
|
2,24 × 10-9
|
2,45 × 10-8
|
POS
|
2,71 × 10-7
|
POS(100/102)
|
POS
|
POS
|
|
21
|
Халоперидол
|
52-86-8
|
NEG
|
—
|
—
|
NEG
|
—
|
NEG (2/2)
|
NEG
|
NT
|
|
22
|
Кемпферол (1)
|
520-18-3
|
POS
|
1,36 × 10-7
|
1,21 × 10-6
|
POS
|
3,99 × 10-6
|
POS(23/23)
|
POS
|
NT
|
|
23
|
Кепон (1)
|
143-50-0
|
POS
|
7,11 × 10-7
|
7,68 × 10-6
|
POS
|
4,91 × 10-7
|
POS(14/18)
|
POS
|
NT
|
|
24
|
Кетоконазол
|
65277-42-1
|
NEG
|
—
|
—
|
NEG
|
—
|
NEG (2/2)
|
NEG
|
NT
|
|
25
|
Линурон (1)
|
330-55-2
|
NEG
|
—
|
—
|
NEG
|
—
|
NEG (8/8)
|
NEG
|
NT
|
|
26
|
мезо-Хексестрол (1)
|
84-16-2
|
POS
|
< 1,00 × 10-11
|
2,75 × 10-11
|
POS
|
1,65 × 10-11
|
POS(4/4)
|
POS
|
NT
|
|
27
|
Метил тестостерон (1)
|
58-18-4
|
POS
|
1,73 × 10-7
|
4,11 × 10-6
|
POS
|
2,68 × 10-6
|
POS(5/6)
|
POS
|
NT
|
|
28
|
Морин
|
480-16-0
|
POS
|
5,43 × 10-7
|
4,16 × 10-6
|
POS
|
2,37 × 10-6
|
POS(2/2)
|
POS
|
NT
|
|
29
|
Норетинодрел (1)
|
68-23-5
|
POS
|
1,11 × 10-11
|
1,50 × 10-9
|
POS
|
9,39 × 10-10
|
POS(5/5)
|
POS
|
NT
|
|
30
|
p,p’-Метоксихлор (1)
|
72-43-5
|
POS
|
1,23 × 10-6
|
(няма PC50) (2)
|
POS
|
1,92 × 10-6
|
POS(24/27)
|
POS
|
POS
|
|
31
|
Фенобарбитал (1)
|
57-30-7
|
NEG
|
—
|
—
|
NEG
|
—
|
NEG(2/2)
|
NEG
|
NT
|
|
32
|
Резерпин
|
50-55-5
|
NEG
|
—
|
—
|
NEG
|
—
|
NEG(4/4)
|
NEG
|
NT
|
|
33
|
Спиронолактон (1)
|
52-01-7
|
NEG
|
—
|
—
|
NEG
|
—
|
NEG(4/4)
|
NEG
|
NT
|
|
34
|
Тестостерон
|
58-22-0
|
POS
|
2,82 × 10-8
|
9,78 × 10-6
|
POS
|
1,75 × 10-5
|
POS(5/10)
|
POS
|
NT
|
|
Съкращения: CASRN = Номер по Служба за химични индекси CAS; M = моларна; EC50 = половината от максимално ефективната концентрация на изпитваното вещество; NEG = отрицателен; POS = положителен; NT = неизпитан; PC10 (и PC50) = концентрацията на изпитваното вещество, при което реакцията е 10 % (или 50 % за PC50) от реакцията, предизвикана от положителната контрола (E2, 1nM) във всяка плака.
|
Таблица 2
Сравнение на резултатите от изследванията STTA и VM7Luc ER TA за вещества, изпитани в двете изследвания с антагонисти и класифицирани като антагонисти на ER (POS) или отрицателни (NEG)
|
|
Вещество (3)
|
CAS №
|
Изследване (40) ER STTA
|
Изследване (41) VM7Luc ER TA
|
ER STTA кандидат ефекти (43)
|
ICCVAM (44) Консенсусна класификация
|
MeSH (45) Химичен клас
|
Клас на продукта (46)
|
|
ER TA Дейност
|
IC50 стойност (39) (M)
|
ER TA Дейност
|
IC50 стойност (39), (42) (M)
|
|
1
|
4-хидрокситамоксифен
|
68047-06-3
|
POS
|
3,97 × 10-9
|
POS
|
2,08 × 10-7
|
умерено POS
|
POS
|
Въглеводород (цикличен)
|
Фармацевтични продукти
|
|
2
|
Дибензо[a.h]антрацен
|
53-70-3
|
POS
|
Няма IC50
|
POS
|
Няма IC50
|
POS
|
PP
|
Полициклично съединение
|
Лабораторен химикал, природен продукт
|
|
3
|
Мифепристон
|
84371-65-3
|
POS
|
5,61 × 10-6
|
NEG
|
—
|
слабо POS
|
NEG
|
Стероид
|
Фармацевтични продукти
|
|
4
|
Ралоксифен HCl
|
82640-04-8
|
POS
|
7,86 × 10-10
|
POS
|
1,19 × 10-9
|
умерено POS
|
POS
|
Въглеводород (цикличен)
|
Фармацевтични продукти
|
|
5
|
Тамоксифен
|
10540-29-1
|
POS
|
4,91 × 10-7
|
POS
|
8,17 × 10-7
|
POS
|
POS
|
Въглеводород (цикличен)
|
Фармацевтични продукти
|
|
6
|
17β-естрадиол
|
50-28-2
|
NEG
|
—
|
NEG
|
—
|
PN
|
PN
|
Стероид
|
Фармацевтичен продукт, ветеринарен агент
|
|
7
|
Апигенин
|
520-36-5
|
NEG
|
—
|
NEG
|
—
|
NEG
|
NEG
|
Хетероциклично съединение
|
Оцветител, природен продукт, междинен фармацевтичен продукт
|
|
8
|
Атразин
|
1912-24-9
|
NEG
|
—
|
NEG
|
—
|
NEG
|
PN
|
Хетероциклично съединение
|
Хербицид
|
|
9
|
Ди-n-бутилфталат
|
84-74-2
|
NEG
|
—
|
NEG
|
—
|
NEG
|
NEG
|
Естер, Фталова киселина
|
Козметична съставка, индустриален химикал, пластификатор
|
|
10
|
Фенаримол
|
60168-88-9
|
NEG
|
—
|
NEG
|
—
|
неизпитан
|
PN
|
Хетероциклено съединение, пиримидин
|
Фунгицид
|
|
11
|
Флавон
|
525-82-6
|
NEG
|
—
|
NEG
|
—
|
PN
|
PN
|
Флавоноид, хетероциклено съединение
|
Природен продукт, фармацевтичен продукт
|
|
12
|
Флутамид
|
13311-84-7
|
NEG
|
—
|
NEG
|
—
|
NEG
|
PN
|
Амид
|
Фармацевтичен продукт, ветеринарен агент
|
|
13
|
Генищайн
|
446-72-0
|
NEG
|
—
|
NEG
|
—
|
PN
|
NEG
|
Флавоноид, хетероциклено съединение
|
Природен продукт, фармацевтичен продукт
|
|
14
|
p-n-нонилфенол
|
104-40-5
|
NEG
|
—
|
NEG
|
—
|
неизпитан
|
NEG
|
Фенол
|
Междинен химикал
|
|
15
|
Ресвератрол
|
501-36-0
|
NEG
|
—
|
NEG
|
—
|
PN
|
NEG
|
Въглеводород (цикличен)
|
Природен продукт
|
|
Съкращения: CASRN = Номер по Служба за химични индекси CAS; M = моларна; IC50 = половината от максималната предизвикваща потискане концентрация на изпитваното вещество; NEG = отрицателен; PN = приема се за отрицателен; POS = положителен; PP = приема се за положителен.
|
КОМПОНЕНТИ НА ER TA ИЗСЛЕДВАНЕТО
Съществени компоненти на изследването
|
11.
|
Настоящият метод за изпитване се прилага за изследвания, използващи стабилно трансфектиран или ендогенен ERα рецептор и модел на стабилно трансфектиран репортерен ген при контрола на един или повече елементи за естрогенен отклик; въпреки това могат да бъдат налични и други рецептори, напр. ERβ. Това са съществените компоненти на изследването.
|
Контроли
|
12.
|
Следва да се опише основата на предложените паралелни референтни еталони за всяко изследване с агонисти и антагонисти. Паралелните контроли (отрицателни, на разтворител и положителни), според случая служат за индикация, че изследването се изпълнява съгласно условията за изпитване и осигурява база за сравнение между отделните експерименти; те обикновено са част от критериите за приемливост за даден експеримент (1).
|
Стандартни процедури за контрол на качеството
|
13.
|
Стандартните процедури за контрол на качеството следва да се изпълняват, както е описано за всяко изследване, за да се гарантира, че клетъчната линия остава стабилна при множество преминавания, остава свободна от микоплазма (т.е. без бактериално замърсяване) и съхранява способността си да предоставя очакваните ER-медиирани отговори във времето. Клетъчните линии следва да бъдат допълнително проверени за правилната им идентичност, както и за други замърсители (напр. гъбички, дрожди и вируси).
|
Доказване на пригодността на лабораторията
|
14.
|
Преди да изпитва непознати химикали с някое от изследванията съгласно настоящия метод за изпитване, всяка лаборатория следва да докаже пригодността си да използва изследването. За да докаже пригодност, всяка лаборатория следва да изпита 14-те вещества за изпитване за пригодност, изброени в таблица 3, в изследване с агонисти и 10-те вещества за изследване за пригодност от таблица 4 в изследването с антагонисти. Това изпитване за пригодност ще потвърди и способността за реагиране на изпитваната система. Списъкът на веществата за изпитване за пригодност е подмножество на референтните вещества, посочени в Стандартите за ефективност за ER TA изследванията (6). Тези вещества могат да се намерят в търговската мрежа, представляват класове химикали, които обикновено се асоциират с активността на агонисти или антгагонисти на ER, проявяват подходящ диапазон на потентност, очаквана за агонисти/антгагонисти ER (т.е. силни до слаби) и включват отрицателни контроли. Изпитването на веществата за изпитване за пригодност следва да се повтори най-малко два пъти, в различни дни. Пригодността се доказва чрез правилното класифициране (положително/отрицателно) на всяко опитно вещество. Изпитването за пригодност следва да бъде повторено от всеки техник, когато се запознава с изследванията. В зависимост от типа на клетките, някои от тези опитни вещества може да се държат като SERM (селективни естроген-рецепторни модулатори) и да проявяват активност както на агонисти, така и на антагонисти. Въпреки това обаче, опитните вещества се класифицират в таблици 3 и 4 по тяхната известна преобладаваща активност, която следва да се използва за оценяване на пригодността.
|
|
15.
|
За да докаже ефективността си и за целите на контрол на качеството, всяка лаборатория следва да събере база данни от агонисти и антагонисти от предходни периоди с референтния еталон (напр. 17β-естрадиол и тамоксифен), химикали за положителни и отрицателни контроли и данни за контрол на разтворителя (напр. DMSO). Като начало, базата данни следва да бъде генерирана от най-малко 10 независими серии с агонисти (напр. 17β-естрадиол) и 10 независими серии с антагонисти (напр. тамоксифен). Резултатите от бъдещите анализи на тези референтни еталони и контролите на разтворителите следва да бъдат добавени, за да се разшири базата данни и да се осигури последователност и ефективност на биологичното изследване от лабораторията във времето.
|
Таблица 3
Списък на (14) вещества за изследване за пригодност в изследване с агонисти
(47)
|
No
(54)
|
Вещество
|
CAS №
|
Очаквана реакция (48)
|
STTA изследване
|
VM7Luc ER TA изследване
|
Химичен клас (52) по MeSH
|
Продуктов клас (53)
|
|
PC10 стойност (M) (49)
|
PC50 стойност (M) (49)
|
Диапазон на конц. при изпитване (M)
|
VM7Luc EC50 стойност (M) (50)
|
Най-висока конц. за определянето на обхвата (M) (51)
|
|
14
|
Диетилстилбестрол
|
56-53-1
|
POS
|
< 1,00 × 10-11
|
2,04 × 10-11
|
10-14 – 10-8
|
3,34 × 10-11
|
3,73 × 10-4
|
Въглеводород (цикличен)
|
Фармацевтичен продукт, ветеринарен агент
|
|
12
|
17α-естрадиол
|
57-91-0
|
POS
|
4,27 × 10-11
|
6,44 × 10-10
|
10-11 – 10-5
|
1,40 × 10-9
|
3,67 × 10-3
|
Стероид
|
Фармацевтичен продукт, ветеринарен агент
|
|
15
|
мезо-хексестрол
|
84-16-2
|
POS
|
< 1,00 × 10-11
|
2,75 × 10-11
|
10-11 – 10-5
|
1,65 × 10-11
|
3,70 × 10-3
|
Въглеводород (цикличен), фенол
|
Фармацевтичен продукт, ветеринарен агент
|
|
11
|
4-трет-октилфенол
|
140-66-9
|
POS
|
1,85 × 10-9
|
7,37 × 10-8
|
10-11 – 10-5
|
3,19 × 10-8
|
4,85 × 10-3
|
Фенол
|
Междинен химикал
|
|
9
|
Генищайн
|
446-72-0
|
POS
|
2,24 × 10-9
|
2,45 × 10-8
|
10-11 – 10-5
|
2,71 × 10-7
|
3,70 × 10-4
|
Флавоноид, хетероциклено съединение
|
Природен продукт, фармацевтичен продукт
|
|
6
|
Бисфенол A
|
80-05-7
|
POS
|
2,02 × 10-8
|
2,94 × 10-7
|
10-11 – 10-5
|
5,33 × 10-7
|
4,38 × 10-3
|
Фенол
|
Междинен химикал
|
|
2
|
Кемпферол
|
520-18-3
|
POS
|
1,36 × 10-7
|
1,21 × 10-6
|
10-11 – 10-5
|
3,99 × 10-6
|
3,49 × 10-3
|
Флавоноид, хетероциклено съединение
|
Природен продукт
|
|
3
|
Бутилбензил фталат
|
85-68-7
|
POS
|
1,14 × 10-6
|
4,11 × 10-6
|
10-11 – 10-5
|
1,98 × 10-6
|
3,20 × 10-4
|
Карбоксилова киселина, естер, фталова киселина
|
Пластификатор, индустриален химикал
|
|
4
|
p,p’- Метоксихлор
|
72-43-5
|
POS
|
1,23 × 10-6
|
—
|
10-11 – 10-5
|
1,92 × 10-6
|
2,89 × 10-3
|
Въглеводород (халогениран)
|
Пестицид, ветеринарен агент
|
|
1
|
Етил парабен
|
120-47-8
|
POS
|
5,00 × 10-6
|
—
|
10-11 – 10-5
|
2,48 × 10-5
|
6,02 × 10-3
|
Карбоксилна киселина, фенол
|
Фармацевтичен продукт, Консервант
|
|
17
|
Атразин
|
1912-24-9
|
NEG
|
—
|
—
|
10-10 – 10-4
|
—
|
4,64 × 10-4
|
Хетероциклено съединение
|
Хербицид
|
|
20
|
Спиронолактон
|
52-01-7
|
NEG
|
—
|
—
|
10-11 – 10-5
|
—
|
2,40 × 10-3
|
Лактон, стероид
|
Фармацевтични продукти
|
|
21
|
Кетоконазол
|
65277-42-1
|
NEG
|
—
|
—
|
10-11 – 10-5
|
—
|
9,41 × 10-5
|
Хетероциклено съединение
|
Фармацевтични продукти
|
|
22
|
Резерпин
|
50-55-5
|
NEG
|
—
|
—
|
10-11 – 10-5
|
—
|
1,64 × 10-3
|
Хетероциклено съединение, индол
|
Фармацевтичен продукт, ветеринарен агент
|
|
Съкращения: CASRN = Номер по Служба за химични индекси CAS; EC50 = половината от максимално ефективната концентрация на изпитваното вещество; NEG = отрицателен; POS = положителен; PC10 (и PC50) = концентрацията на изпитваното вещество, при което реакцията е 10 % (или 50 % за PC50) от реакцията, предизвикана от положителната контрола (E2, 1nM) във всяка плака.
|
Таблица 4
Списък на (10) вещества за изпитване на пригодност за изследване на антагонисти
|
|
Вещество (4)
|
CAS №
|
Изследване (55) ER STTA
|
Изследване (56) VM7Luc ER TA
|
ER STTA (55) кандидат, ефекти
|
ICCVAM (59) Консенсусна класификация
|
MeSH (60) Химичен клас
|
Клас на продукта (61)
|
|
Активност ER TA
|
IC50 (M)
|
Диапазон на конц. при изпитване (M)
|
Активност ER TA
|
IC50
(57) (M)
|
Най-висока конц. за определянето на обхвата (M) (58)
|
|
1
|
4-хидрокситамоксифен
|
68047-06-3
|
POS
|
3,97 × 10-9
|
10-12 – 10-7
|
POS
|
2,08 × 10-7
|
2,58 × 10-4
|
умерено POS
|
POS
|
Въглеводород (цикличен)
|
Фармацевтични продукти
|
|
2
|
Ралоксифен HCl
|
82640-04-8
|
POS
|
7,86 × 10-10
|
10-12 – 10-7
|
POS
|
1,19 × 10-9
|
1,96 × 10-4
|
умерено POS
|
POS
|
Въглеводород (цикличен)
|
Фармацевтични продукти
|
|
3
|
Тамоксифен
|
10540-29-1
|
POS
|
4,91 × 10-7
|
10-10 – 10-5
|
POS
|
8,17 × 10-7
|
2,69 × 10-4
|
POS
|
POS
|
Въглеводород (цикличен)
|
Фармацевтични продукти
|
|
4
|
17β-естрадиол
|
50-28-2
|
NEG
|
—
|
10-9 – 10-4
|
NEG
|
—
|
3,67 × 10-3
|
да бъде отрицателно (*4)
|
PN
|
Стероид
|
Фармацевтичен продукт, ветеринарен агент
|
|
5
|
Апигенин
|
520-36-5
|
NEG
|
—
|
10-9 – 10-4
|
NEG
|
—
|
3,70 × 10-4
|
NEG
|
NEG
|
Хетероциклено съединение
|
Оцветител, природен продукт, междинен фармацевтичен продукт
|
|
6
|
Ди-n-бутилфталат
|
84-74-2
|
NEG
|
—
|
10-8 – 10-3
|
NEG
|
—
|
3,59 × 10-3
|
NEG
|
NEG
|
Естер, Фталова киселина
|
Козметична съставка, индустриален химикал, пластификатор
|
|
7
|
Флавон
|
525-82-6
|
NEG
|
—
|
10-8 – 10-3
|
NEG
|
—
|
4,50 × 10-4
|
да бъде отрицателно (*4)
|
PN
|
Флавоноид, хетероциклено съединение
|
Природен продукт, фармацевтичен продукт
|
|
8
|
Генищайн
|
446-72-0
|
NEG
|
—
|
10-9 – 10-4
|
NEG
|
—
|
3,70 × 10-4
|
да бъде отрицателно (*4)
|
NEG
|
Флавоноид, хетероциклено съединение
|
Природен продукт, фармацевтичен продукт
|
|
9
|
p-n-нонилфенол
|
104-40-5
|
NEG
|
—
|
10-9 – 10-4
|
NEG
|
—
|
4,54 × 10-4
|
неизпитан
|
NEG
|
Фенол
|
Междинен химикал
|
|
10
|
Ресвератрол
|
501-36-0
|
NEG
|
—
|
10-8 – 10-3
|
NEG
|
—
|
4,38 × 10-4
|
да бъде отрицателно (*4)
|
NEG
|
Въглеводород (цикличен)
|
Природен продукт
|
|
Съкращения: CASRN = Номер по Служба за химични индекси CAS; M = моларна; IC50 = половината от максималната предизвикваща потискане концентрация на изпитваното вещество; NEG = отрицателен; PN = приема се за отрицателен; POS = положителен.
|
Критерии за приемливост на изпитвателната серия
|
16.
|
Приемането или отхвърлянето на изпитвателната серия се прави на базата на оценката на резултатите, получени за референтните еталони и използваните контроли за всеки експеримент. Стойностите за PC50 (EC50) или IC50 за референтните еталони следва да отговарят на критериите за приемливост, предвидени за избраното изследване (за STTA вж. допълнение 2, за VM7Luc ER TA вж. допълнение 3), като всички положителни/отрицателни контроли следва да бъдат правилно класифицирани за всеки приет експеримент. Способността за последователно изпълнение на изследването следва да бъде доказана чрез разработката и поддържането на база данни от предходни периоди относно референтните еталони и контроли (вж. точка 15). Стандартните отклонения (SD) или коефициентите на вариация (CV) за параметрите за изглаждане на кривата на референтните еталони от множество експерименти могат да бъдат използвани като мярка за вътрешнолабораторната възпроизводимост. Допълнително следва да бъдат изпълнени следните принципи относно критериите за приемливост:
|
—
|
Данните следва да бъдат достатъчни за количествената оценка на активирането на ER (за изследването с агонисти) или подтискането им (за изследването с антагонисти) (т.е. ефикасност и сила).
|
|
—
|
За да се осигури адекватна чувствителност, средната активност на репортерните гени за референтните концентрации на референтния естроген следва да бъде най-малко минималната специфицирана в изследванията по отношение на тази на контрола на носителя (разтворителя). За изследванията STTA и VM7Luc ER TA, тя е четири пъти тази на средната контрола на носителя на всяка плака.
|
|
—
|
Изпитваните концентрации следва да останат в рамките на обхвата на разтворимост на изпитваните химикали и да не демонстрират цитотоксичност.
|
|
Анализ на данните
|
17.
|
За класифициране на положителния и отрицателния отклик следва да се използва дефинираната процедура за интерпретиране на данни за всяко изследване.
|
|
18.
|
Изпълнението на критериите за приемливост (точка 16) показва, че изследването работи правилно, но не гарантира, че конкретна изпитвателна серия ще даде точни данни. Повторението на резултатите от първата серия е най-доброто указание, че се получават точни данни. Ако две серии дадат възпроизводими резултати (напр. резултатите от двете изпитвателни серии покажат, че изпитвания химикал е положителен), не е необходимо да се провежда трета серия.
|
|
19.
|
Ако две серии не дадат възпроизводими резултати (напр. изпитваният химикал е положителен при едната серия и отрицателен при другата) или ако се изисква по-висока степен на достоверност относно окончателния резултат от това изследване, следва да се проведат най-малко три независими серии. В този случай, класификацията се базира на двата сходни резултата от трите получени.
|
Общи критерии за тълкуване на данните
|
20.
|
Понастоящем не съществува универсално съгласуван метод за тълкуване на данните за ER TA. Въпреки това качествените (напр. положителен/отрицателен) и/или количествените (напр. EC50, PC50, IC50) оценки на ER-медиираната активност следва да бъдат базирани на емпирични данни и разумна научна преценка. Когато е възможно, положителните резултати следва да се характеризират както по големина на ефекта в сравнение с контролата на носителя (разтворителя) така и от референтния естроген и концентрацията, при която възниква ефектът (напр. EC50, PC50, RPCMax, IC50 и др.).
|
Протокол от изпитването
|
21.
|
Протоколът от изпитването следва да включва следната информация:
|
|
Изследване:
|
—
|
контрола/референтен еталон/изпитван химикал
|
|
—
|
източник, номер на партидата, крайна дата за употреба, ако има такава;
|
|
—
|
стабилност на самия изпитван химикал, ако е известна;
|
|
—
|
разтворимост и стабилност на изпитвания химикал в разтворител, ако са известни;
|
|
—
|
измерване на рН, осмолалитет и утайка в средата за отглеждане, към която е добавен изпитваният химикал, когато е уместно.
|
|
|
|
Вещество с една съставка:
|
—
|
външен вид, разтворимост във вода и допълнителни относими физични и химични свойства;
|
|
—
|
химична идентификация, като например наименование по IUPAC или CAS, CAS номер, код SMILES или InChI, структурна формула, чистота, химична идентичност на онечистванията, доколкото е целесъобразно и практически осъществимо, и т.н.
|
|
|
|
Вещество с повече съставки, UVCB и смеси:
|
—
|
определени, доколкото е възможно, с химичната си идентичност (вж. по-горе), количествения състав и относимите физични и химични свойства на съставките.
|
|
|
|
Разтворител/носител:
|
—
|
характеристики (естество, доставчик и партида);
|
|
—
|
обосновка за избора на разтворител/носител;
|
|
—
|
разтворимост и стабилност на изпитвания химикал в разтворител/носител, ако са известни;
|
|
|
|
Клетки:
|
—
|
тип и източник на клетките:
|
—
|
Експресира ли се ER ендогенно? Ако не, кой(кои) рецептор(и) са трансфектирани?
|
|
—
|
Използван(и) репортерен(-ни) модел(и) (включително биологичния вид на източника);
|
|
—
|
Метод на трансфектиране;
|
|
—
|
Метод за избор за поддържане на стабилно трансфектиране (когато е приложимо);
|
|
—
|
Методът на трансфектиране относим ли е за стабилните клетъчни линии?
|
|
|
—
|
брой на клетъчните преминавания (от топене);
|
|
—
|
номер на пасажа на клетките при размразяване;
|
|
—
|
методи за поддържане на клетъчните култури.
|
|
|
|
Условия на изпитването:
|
—
|
ограничения на разтворимостта;
|
|
—
|
описание на приложените методи за оценяване на жизнеспособността;
|
|
—
|
състав на средите, концентрация на CO2;
|
|
—
|
концентрации на изпитвания химикал;
|
|
—
|
обем на носителя и добавения изпитван химикал;
|
|
—
|
температура на инкубация и влажност;
|
|
—
|
времетраене на третирането;
|
|
—
|
клетъчна плътност в началото и по време на третирането;
|
|
—
|
референтни еталони за положителни и отрицателни вещества;
|
|
—
|
репортерни реагенти (наименование на продукта, доставчик и партида);
|
|
—
|
критерии за обявяване на изпитвателните серии за положителни, отрицателни или неясни.
|
|
|
|
Проверка за приемливост:
|
—
|
кратностите на индукциите за всяка плака от изследването и дали отговарят на минималните изисквания за изследването, на базата на контролите от предходни периоди;
|
|
—
|
реални стойности за критериите за приемливост, напр. log10EC50, log10PC50, logIC50 и стойности на наклона на кривата за паралелните положителни контроли/референтни еталони.
|
|
|
|
Резултати:
|
—
|
Необработени и нормализирани данни;
|
|
—
|
максимално ниво на кратност на индукцията;
|
|
—
|
данни за цитотоксичността;
|
|
—
|
ако съществува, най-ниската ефективна концентрация (LEC);
|
|
—
|
стойности на RPCMax, PCMax, PC50, IC50 и/или EC50, в зависимост от конкретния случай;
|
|
—
|
зависимостта концентрация—отклик, където е възможно;
|
|
—
|
статистически анализи, ако има, заедно с мярката за грешка и достоверност (напр. SEM, SD, CV или 95 % CI) и описание на начина за получаване на тези стойности.
|
|
|
ЛИТЕРАТУРА
|
(1)
|
OECD (2005). Guidance Document on the Validation and International Acceptance of New or Updated Test Methods for Hazard Assessment. Environment, Health and Safety Publications, Series on Testing and Assessment (No 34.), Organisation for Economic Cooperation and Development, Paris.
|
|
(2)
|
OECD (2015). Report of the Inter-Laboratory Validation for Stably Transfected Transactivation Assay to detect Estrogenic and Anti-estrogenic Activity. Environment, Health and Safety Publications, Series on Testing and Assessment (No 225), Organisation for Economic Cooperation and Development, Paris.
|
|
(3)
|
ICCVAM (2011). ICCVAM Test Method Evaluation Report on the LUMI-CELL® ER (BG1Luc ER TA) Test Method, an In Vitro Method for Identifying ER Agonists and Antagonists, National Institute of Environmental Health Sciences: Research Triangle Park, NC.
|
|
(4)
|
Pujol P. et al. (1998). Differential Expression of Estrogen Receptor-Alpha and -Beta Messenger RNAs as a Potential Marker of Ovarian Carcinogenesis, Cancer. Res., 58(23): p. 5367-73.
|
|
(5)
|
Rogers J.M. and Denison M.S. (2000). Recombinant Cell Bioassays for Endocrine Disruptors: Development of a Stably Transfected Human Ovarian Cell Line for the Detection of Estrogenic and Anti-Estrogenic Chemicals, In Vitro and Molecular Toxicology: Journal of Basic and Applied Research, 13(1): p. 67-82.
|
|
(6)
|
OECD (2012). Performance Standards For Stably Transfected Transactivation In Vitro Assay to Detect Estrogen Receptor Agonists (for TG 455). Environment, Health and Safety Publications, Series on Testing and Assessment (No 173.), Organisation for Economic Cooperation and Development, Paris.
|
|
(7)
|
OECD (2015). Performance Standards For Stably Transfected Transactivation In Vitro Assay to Detect Estrogen Receptor Antagonists. Environment, Health and Safety Publications, Series on Testing and Assessment (No 174.), Organisation for Economic Cooperation and Development, Paris.
|
|
(8)
|
OECD (2012). Guidance Document on Standardized Test Guidelines for Evaluating Chemicals for Endocrine Disruption. Environment, Health and Safety Publications, Series on Testing and Assessment (No 150.), Organisation for Economic Cooperation and Development, Paris.
|
|
(9)
|
Cavailles V. (2002). Estrogens and Receptors: an Evolving Concept. Climacteric, 5 Suppl 2: p. 20- 6.
|
|
(10)
|
Welboren W.J. et al. (2009). Genomic Actions of Estrogen Receptor Alpha: What are the Targets and how are they Regulated? Endocr. Relat. Cancer, 16(4): p. 1073-89.
|
|
(11)
|
Younes M. and Honma N. (2011). Estrogen Receptor Beta, Arch. Pathol. Lab. Med., 135(1): p. 63- 6.
|
|
(12)
|
Jefferson W.N., et al. (2002). Assessing Estrogenic Activity of Phytochemicals Using Transcriptional Activation and Immature Mouse Uterotrophic Responses, Journal of Chromatography B, 777(1-2): p. 179-189.
|
|
(13)
|
Sonneveld E. et al. (2006). Comparison of In Vitro and In Vivo Screening Models for Androgenic and Estrogenic Activities, Toxicol. Sci., 89(1): p. 173-187.
|
|
(14)
|
Takeyoshi M. et al. (2002). The Efficacy of Endocrine Disruptor Screening Tests in Detecting Anti- Estrogenic Effects Downstream of Receptor-Ligand Interactions, Toxicology Letters, 126(2): p. 91- 98.
|
|
(15)
|
Combes R.D. (2000). Endocrine Disruptors: a Critical Review of In Vitro and In Vivo Testing Strategies for Assessing their Toxic Hazard to Humans, ATLA Alternatives to Laboratory Animals,28(1): p. 81-118.
|
|
(16)
|
Escande A. et al. (2006). Evaluation of Ligand Selectivity Using Reporter Cell Lines Stably Expressing Estrogen Receptor Alpha or Beta, Biochem. Pharmacol,71(10): p. 1459-69.
|
|
(17)
|
Gray L.E. Jr. (1998). Tiered Screening and Testing Strategy for Xenoestrogens and Antiandrogens, Toxicol. Lett, 102-103, 677-680.
|
|
(18)
|
EDSTAC (1998). Endocrine Disruptor Screening and Testing Advisory Committee (EDSTAC) Final Report.
|
|
(19)
|
ICCVAM (2003). ICCVAM Evaluation of In Vitro Test Methods for Detecting Potential Endocrine Disruptors: Estrogen Receptor and Androgen Receptor Binding and Transcriptional Activation Assays.
|
|
(20)
|
Gustafsson J.Ö. (1999). Estrogen Receptor ß - A New Dimension in Estrogen Mechanism of Action, Journal of Endocrinology, 163(3): p. 379-383.
|
|
(21)
|
Ogawa S. et al. (1998). The Complete Primary Structure of Human Estrogen Receptor ß (hERß) and its Heterodimerization with ERαIn Vivo and In Vitro, Biochemical and Biophysical Research Communications, 243(1): p. 122-126.
|
|
(22)
|
Enmark E. et al. (1997). Human Estrogen Receptor ß-Gene Structure, Chromosomal Localization, and Expression Pattern, Journal of Clinical Endocrinology and Metabolism,82(12): p. 4258-4265.
|
|
(23)
|
Ball L.J. et al. (2009). Cell Type- and Estrogen Receptor-Subtype Specific Regulation of Selective Estrogen Receptor Modulator Regulatory Elements, Molecular and Cellular Endocrinology, 299(2): p. 204-211.
|
|
(24)
|
Barkhem T. et al. (1998). Differential Response of Estrogen Receptor Alpha and Estrogen Receptor Beta to Partial Estrogen Agonists/Antagonists, Mol. Pharmacol, 54(1): p. 105-12.
|
|
(25)
|
Deroo B.J. and Buensuceso A.V. (2010). Minireview: Estrogen Receptor-ß: Mechanistic Insights from Recent Studies, Molecular Endocrinology, 24(9): p. 1703-1714.
|
|
(26)
|
Harris D.M. et al. (2005). Phytoestrogens Induce Differential Estrogen Receptor Alpha- or Beta- Mediated Responses in Transfected Breast Cancer Cells, Experimental Biology and Medicine, 230(8): p. 558-568.
|
|
(27)
|
Anderson J.N. Clark J.H. and Peck E.J.Jr. (1972). The Relationship Between Nuclear Receptor- Estrogen Binding and Uterotrophic Responses, Biochemical and Biophysical Research Communications, 48(6): p. 1460-1468.
|
|
(28)
|
Toft D. (1972). The Interaction of Uterine Estrogen Receptors with DNA, Journal of Steroid Biochemistry, 3(3): p. 515-522.
|
|
(29)
|
Gorski J. et al. (1968), Hormone Receptors: Studies on the Interaction of Estrogen with the Uterus, Recent Progress in Hormone Research, 24: p. 45-80.
|
|
(30)
|
Jensen E.V. et al. (1967), Estrogen-Receptor Interactions in Target Tissues, Archives d’Anatomie Microscopique et de Morphologie Experimentale, 56(3):p. 547-569.
|
|
(31)
|
ICCVAM (2002). Background Review Document: Estrogen Receptor Transcriptional Activation (TA) Assay. Appendix D, Substances Tested in the ER TA Assay, NIH Publication Report (No 03-4505.).
|
|
(32)
|
Kanno J. et al. (2001). The OECD Program to Validate the Rat Uterotrophic Bioassay to Screen Compounds for In Vivo Estrogenic Responses: Phase 1, Environ. Health Persp., 109:785-94.
|
|
(33)
|
Kanno J. et al. (2003). The OECD Program to Validate the Rat Uterotrophic Bioassay: Phase Two Dose -Response Studies, Environ. Health Persp., 111:1530-1549.
|
|
(34)
|
Kanno J. et al. (2003), The OECD Program to Validate the Rat Uterotrophic Bioassay: Phase Two – Coded Single-Dose Studies, Environ. Health Persp., 111:1550-1558.
|
|
(35)
|
Geisinger et al. (1989) Characterization of a human ovarian carcinoma cell line with estrogen and progesterone receptors, Cancer 63, 280-288.
|
|
(36)
|
Baldwin et al. (1998) BG-1 ovarian cell line: an alternative model for examining estrogen-dependent growth in vitro, In Vitro Cell. Dev. Biol. – Animal, 34, 649-654.
|
|
(37)
|
Li, Y. et al. (2014) Research resource: STR DNA profile and gene expression comparisons of human BG-1 cells and a BG-1/MCF-7 clonal variant, Mol. Endo. 28, 2072-2081.
|
|
(38)
|
Rogers, J.M. and Denison, M.S. (2000) Recombinant cell bioassays for endocrine disruptors: development of a stably transfected human ovarian cell line for the detection of estrogenic and anti-estrogenic chemicals, In Vitro & Molec. Toxicol. 13, 67-82.
|
Допълнение 1
ОПРЕДЕЛЕНИЯ И СЪКРАЩЕНИЯ
Критерии за приемливост
: минималните стандарти за ефективност на експерименталните контроли и референтните еталони. Всички критерии за приемливост следва да бъдат изпълнени, за да може даден експеримент да се разглежда като валиден.
Точност (съответствие)
: степента на близост между резултатите от изследването и приетите референтни стойности. Това е мярка за ефективността на изследването и един от аспектите на неговата относимост. Терминът често се използва взаимозаменяемо с термина „съответствие“, за да се обозначи относителният дял на правилните резултати при дадено изследване (1).
Агонист
: Вещество, които предизвиква отклик, напр. транскрипция, когато се свързва със специфичен рецептор.
Антагонист
: Тип рецепторен лиганд или химикал, който не провокира биологичен отклик сам по себе си при свързване с даден рецептор, но блокира или смекчава отклиците, медиирани от агониста.
Анти-естрогенна активност
: способността на даден химикал да потиска действието на 17β-естрадиол, медииран чрез естрогенните рецептори.
Клетъчна морфология
: Формата и външният вид на клетките, отглеждани в един слой на единична ямка на плака с тъканна култура. Клетките, които умират, често пъти проявяват аномална клетъчна морфология.
CF
: Концептуалната рамка на ОИСР за изпитване и оценяване на вещества, нарушаващи функцията на ендокринната система.
Третиране с въглен/декстран
: Третиране на серума, използван в клетъчната култура. Третирането с въглен/декстран (често наричано “оголване”) премахва ендогенните хормони и хормон-свързващи протеини.
Химикал
: Вещество или смес.
Цитотоксичност
: Вредното въздействие върху клетъчната структура или функция, което в крайна сметка е в състояние да причини клетъчна смърт и се отразява чрез намаляване на броя на клетките, налични в ямката в края на периода на експозиция или намаляване на капацитета за измерване на клетъчната функция в сравнение с паралелната контрола на носителя.
CV
: Коефициент на вариация
DCC-FBS
: Фетален волски серум, обработен с въглен, покрит с декстран.
DMEM
: Модифицирана Йигъл среда на Дюлбеко
DMSO
: Диметилсулфоксид
E2
: 17β-естрадиол
EC50
: Половината от максимално ефективната концентрация на изпитвания химикал.
ED
: Ендокринни нарушения
hERα
: Човешки естрогенен рецептор алфа
hERß
: Човешки естрогенен рецептор бета
EFM
: Среда без естроген. Модифицирана от Дюлбеко среда на Йигъл (DMEM), допълнена с 4,5 % FBS, третиран с въглен/декстран, 1,9 % L-глутамин и 0,9 % Pen-Strep (пеницилин-стрептомицин).
ER
: Естрогенен рецептор
ERE
: Елемент на естрогенния отклик
Естрогенна активност
: Способността на даден химикал да наподобява 17β-естрадиол в способността му за свързване с естрогенни рецептори и активирането им. hERα-медиираната естрогенна активност може да се открие с помощта на този метод за изпитване.
ERTA
: Трансактивация на естрогенен рецептор
FBS
: Фетален волски серум
HeLa
: Човешка безсмъртна цервикална клетъчна линия
HeLa9903
: HeLa клетъчен субклон, в който стабилно се трансфектира hERαα и луцифераза репортерен ген
IC
50
: половината от максимално ефективната предизвикваща потискане концентрация на изпитван химикал.
ICCVAM
: междуведомствен координационен комитет за валидиране на алтернативни методи.
Междулабораторна възпроизводимост
: мярка за степента, в която различните квалифицирани лаборатории могат да получат качествено и количествено сходни резултати, като използват един и същ протокол и изпитват едни и същи вещества. Междулабораторната възпроизводимост се определя по време на предвалидационните и валидационните процедури и указва степента, в която дадено изследване може успешно да бъде прехвърлено между различни лаборатории, наричана също интерлабораторна възпроизводимост (1).
Вътрешнолабораторна възпроизводимост
: определяне на степента, в която квалифицирани служители в рамките на една и съща лаборатория могат да възпроизведат успешно резултатите, като използват конкретен протокол в различни моменти. Нарича се също „интралабораторна възпроизводимост“ (1).
LEC
: Най-ниската ефективна концентрация е най-ниската концентрация на изпитвания химикал, която предизвиква отклик (т.е. най-ниската концентрация на изпитвания химикал, при която кратността на индукцията е статистически различна от паралелната контрола на носителя).
Вариантен метод за изпитване (me-too test)
: Разговорен израз за изследване, който е структурно и функционално сходен с валидиран и приет референтен метод за изпитване. Използва се взаимозаменяемо със „сходен метод за изпитване“.
MT
: Металотионеин
MMTV
: Миши вирус на тумор на млечната жлеза
OHT
: 4-Хидрокситамоксифен
PBTG
: Насоки за изпитване, базирани на ефективност
PC (положителна контрола)
: силно активно вещество, за предпочитане 17ß-естрадиол, което е включено във всички изпитвания, за да гарантира правилното функциониране на изследването.
PC
10
: концентрацията на изпитван химикал, при която измерената активност в изследване с агонист е 10 % от максималната активност, индуцирана от ПК (E2 при 1nM за STTA изследването) във всяка плака.
PC
50
: концентрацията на изпитван химикал, при която измерената активност в изследване с агонист е 50 % от максималната активност, индуцирана от ПК (E2 при референтната концентрация, посочена в метода на изпитване) във всяка плака.
PC
Max
: концентрацията на изпитван химикал, включително стойността RPCMax
Стандарти за ефективност
: Стандарти, базирани на валидирано изследване, осигуряващи основата за оценка на сравнимостта на предложено изследване, което е по своя механизъм и функционално сходно. Включени са (1) съществени компоненти на изследването; (2) минимален списък на референтни химикали, подбрани измежду химикалите, използвани за демонстриране на приемливата ефективност на валидирания метод за изпитване; и (3) сравнимите нива за точност и надеждност въз основа на получените нива за валидирания метод на изпитване, които предложеното изследване следва да демонстрира при оценяването чрез използване на минималния списък на референтните химикали (1).
Вещества за изследване за пригодност
: Подмножество от референтни вещества, включени в Стандартите за ефективност, които може да се използват от лабораториите за доказване на техническа пригодност по отношение на стандартизиран метод за изпитване. Критериите за подбор на тези вещества обикновен включват изискването те да представляват обхвата от отговори, да се предлагат в търговската мрежа и да имат налични висококачествени референтни данни.
Пригодност
: Доказаната способност за правилно провеждане на изследване преди изпитване на неизвестни вещества.
Референтен естроген (положителна контрола, ПК)
: 17β-естрадиол (E2, CAS 50-28-2).
Референтен еталон
: референтно вещество, използвано за доказване на адекватността на изследване. 17β-естрадиолът е референтният еталон за изследванията STTA и VM7Luc ER TA.
Референтни методи на изпитване
: Изследванията, на които е базиран PBTG 455.
Относимост
: описание на взаимовръзката между изследването и ефекта, представляващ интерес, и дали тя е значима и полезна за определена цел. Това е степента, в която изследването правилно измерва или прогнозира биологичния ефект, представляващ интерес. Приложимостта включва разглеждането на точността (съответствието) на изследването (1).
Надеждност
: мярка за степента, в която дадено изследване може да се проведе възпроизводимо в една и съща лаборатория и в различни лаборатории с течение на времето, при използване на един и същ протокол. Определя се, като се изчислява вътрешно- и междулабораторната възпроизводимост.
RLU
: Относителни светлинни единици
РНК
: Рибонуклеинова киселина
RPC
Max
: максималното ниво на отклик, предизвикан от изпитван химикал, изразено като процентно съотношение на отклика, предизвикан от 1 nM E2 върху същата плака
RPMI
: Среда RPMI 1 640, допълнена с 0,9 % Pen-Strep и 8,0 % фетален волски серум (FBS)
Серия
: Индивидуален експеримент, който оценява химичното действие върху биологичния резултат от изследването. Всяка серия е пълен експеримент, извършен върху ямки с клетки за повторения върху плака, от общ сбор от клетки по едно и също време.
Независима серия
: Отделен, независим експеримент, който оценява химично действие върху биологичния резултат от изследването, използващ клетки от различен сбор, прясно разредени химикали, проведен в различни дни или в същия ден от различен персонал.
SD
: Стандартно отклонение.
Чувствителност
: Относителният дял от всички положителни/активни вещества, които са класифицирани правилно чрез изследването. Това е мярка за точността на изследването, която предоставя категорични резултати и е важен фактор при оценката на относимостта на изследване (1).
Специфичност
: Относителният дял от всички негативни/неактивни вещества, които са класифицирани правилно чрез изпитването. Това е мярка за точността на изследването, която предоставя категорични резултати и е важен фактор при оценката на относимостта на изследване (1).
Стабилна трансфекция
: Когато ДНК се трансфектира в култивирани клетки по такъв начин, че се интегрира устойчиво в клетъчния геном, което води до стабилна експресия на трансфектирани гени. Клонингите на стабилно трансфектирани клетки се избират по стабилни маркери (напр. устойчивост на G418).
STTA изследване
: Изследване за трансактивация чрез стабилна трансфекция, изследването за транскрипционна активация на ERα , използващо клетъчната линия HeLa 9 903.
Проучване
: Пълният обхват от експериментална работа, извършена за оценяване на едно специфично вещество, с използване на специфично изследване. Проучването обхваща всички стъпки, включващи изпитвания на разредено изпитвано вещество в изпитваната среда, предварителни серии за определяне на обхватите, всички необходими всеобхватни серии, анализи на данни, осигуряване на качество, оценки на цитотоксичността и др. Приключването на проучване позволява класифициране на действието на изпитвания химикал върху целта на токсичността (т.е. активно, неактивно или не неубедително), което се оценява от използваното изследване и от оценката на силата на действие във връзка с положителния референтен химикал.
Вещество
: Съгласно REACH (62), веществото се дефинира като химически елемент и неговите съединения в естествено състояние или получени чрез производствен процес, включително всяка добавка, необходима за запазване на стабилността му, както и всички примеси, извлечени от използвания процес, с изключение на всеки разтворител, който може да бъде отделен, без да се засяга стабилността на веществото или да се променя неговия състав. Много подобно определение е използвано в контекста на GHS на ООН (1).
TA (трансактивация)
: Инициирането на синтеза на иРНК в отговор на специфичен химичен сигнал, като например свързването на естроген с естрогенен рецептор
Изследване
: В рамките на контекста на настоящия метод за изпитване, изследване е една от методиките, приети за валидни поради това, че изпълняват очертаните критерии за ефективност. Компонентите на изследване включват например, специфичната клетъчна линия с асоциираните условия на растеж, специфична среда, в която се провежда изпитването, условията на постановката с плаки, подредба и разреждане на изпитваните химикали, заедно с някои други необходими мерки за контрол на качеството и свързаните стъпки за оценка на данните.
Изпитван химикал
: Всяко вещество или смес, изпитвано(а) с използване на настоящия метод за изпитване.
Транскрипция
: синтеза на иРНК
UVCB
: Химични вещества с неизвестен или променлив състав, сложни продукти от реакции и биологични материали.
Валидиран метод за изпитване
: изследване, за което са извършени изследването за валидиране за определяне на приложимостта (включително на точността) и надеждността му за конкретна цел. Важно е да се отбележи, че един валидиран метод на изпитване може да не притежава достатъчно ефективност по отношение на точността и надеждността си, за да се счита за приемлив за предлаганата цел (1).
Валидиране
: Процесът, чрез който надеждността и относимостта на конкретен подход, метод, изследване, процес или оценка се установяват за определена цел (1).
VC (контрол на носителя)
: Разтворителят, който се използва за разтваряне на изпитваните и контролните химикали се изпитва самостоятелно като носител, без разтворения химикал.
VM7
: Имортализирана клетка на аденокарцином, която експресира ендогенно естрогенен рецептор.
VM7Luc4E2
: Клетъчната линия VM7Luc4E2 е извлечена от VM7 имортализирани клетки на аденокарцином с човешки произход, които експресират ендогенно и двете форми на естрогенния рецептор (ERα и ERβ) и са устойчиво трансфектирани с плазмида pGudLuc7.ERE. Този плазмид съдържа четири копия на синтетичен олигонуклеотид, съдържащ елемента на естрогенния отклик, предшестващ вирусния промотор на миши тумор на млечната жлеза (MMTV), и гена на луцифераза на светулка.
Слаба положителна контрола
: Слабо активно вещество, избрано от списъка на референтните химикали, което е включено във всички изпитвания, за да гарантира правилното функциониране на изследването.
Допълнение 2
ИЗСЛЕДВАНЕ ЗА ТРАНСАКТИВАЦИЯ ЧРЕЗ СТАБИЛНО ТРАНСФЕКТИРАНЕ НА ЧОВЕШКИ ЕСТРОГЕНЕН РЕЦЕПТОР-Α, С ЦЕЛ ОТКРИВАНЕ НА АКТИВНОСТ НА ЕСТРОГЕННИ АГОНИСТИ И АНТАГОНИСТИ НА ХИМИКАЛИ, С ИЗПОЛЗВАНЕ НА КЛЕТЪЧНАТА ЛИНИЯ HERΑ-HELA-9903
ПЪРВОНАЧАЛНИ СЪОБРАЖЕНИЯ И ОГРАНИЧЕНИЯ (ВЖ. СЪЩО ОБЩО ВЪВЕДЕНИЕ)
|
1.
|
Настоящото изследване за трансактивация (TA) използва клетъчната линия hERα-HeLa-9 903 за откриване на активност на естрогенен агонист, медиирана от човешки естрогенен рецептор алфа (hERα). Изследването за валидиране на изследването за трансактивация чрез стабилно трансфектиране (STTA), проведено от Японския изследователски институт за оценка на химикали (CERI) с използване на клетъчната линия hERα-HeLa-9 903 за откриване на активност на агонисти и антагонисти, медиирана от човешки естрогенен рецептор алфа (hERα) доказа относимостта и надеждността на изследването за предвидената му цел (1).
|
|
2.
|
Това изследване е специално разработено за откриване hERα-медиирана TA чрез измерване на хемилуминесцентността като крайната точка. Въпреки това бяха докладвани луминесцентни сигнали, които не са медиирани от рецептори при концентрации на фитоестроген, по-високи от 1 μМ, и се дължат на свръхактивирането на луциферазния репортерен ген (2) (3). Тъй като кривата доза-отклик показва, че истинската активация на системата на ER възниква при по-ниски концентрации, експресията на луцифераза, получена при високи концентрации на фитоестрогени или подобни съединения, за които се подозира, че подобно на фитоестрогена предизвикват свръхактивация на луциферазния репортерен ген, трябва да се изследва внимателно в системи за изследване на ER TA чрез стабилно трансфектиране (допълнение 1).
|
|
3.
|
Разделите „ОБЩО ВЪВЕДЕНИЕ“ и „КОМПОНЕНТИ НА ИЗСЛЕДВАНЕТО ER TA“ следва да бъдат прочетени, преди да се използва настоящото изследване за регулаторни цели. Използваните определения и съкращения в рамките на настоящите TG са описани в допълнение 2.1.
|
ПРИНЦИП НА ИЗСЛЕДВАНЕТО (ВЖ. СЪЩО „ОБЩО ВЪВЕДЕНИЕ“)
|
4.
|
Изследването се използва за сигнализиране на свързването на естрогенния рецептор с лиганд. След свързването с лиганда, комплексът рецептор-лиганд се транслоцира в ядрото, където свързва специфични елементи на ДНК отклик и трансактивира луциферазния репортерен ген на светулка, което води до повишена клетъчна експресия на ензима луцифераза. Луциферинът е субстрат, който се трансформира от ензима луцифераза в биолуминесцентен продукт, който може да бъде количествено измерен с помощта на луминометър. Луциферазната активност може да бъде измерена бързо и евтино с множество тестови китове, налични в търговската мрежа.
|
|
5.
|
Системата за изпитване използва клетъчната линия hERα-HeLa-9 903, която е получена от човешки цервикален тумор, с два стабилно вмъкнати конструкта: i) конструктът за експресия на hERαα (кодиращ пълната дължина на човешкия рецептор), и ii) луциферазния репортерен конструкт от светулка, носител на пет тандемни повторения на елемент на естрогенен отклик (ERE) с вителогенин, управляван от миши металотионеин (MT) промоторен TATA елемент. Мишият MT TATA генен конструкт е с доказана най-добра ефективност и поради това се използва често. Вследствие на това, настоящата клетъчна линия hERα-HeLa-9 903 може да измери способността на изпитвания химикал да индуцира hERα-медиирана трансактивация на експресията на луциферазния ген.
|
|
6.
|
Когато се провежда изследване с агонисти на ER, тълкуването на данните се базира на това дали максималното ниво на отклик, предизвикано от изпитвания химикал, е равно или превишава отклика на агониста, равен на 10 % от предизвикания от максимално предизвикващата отклик (1 nM) концентрация на положителната контрола (ПК) 17β-естрадиол (E2) (т.е. стойността ПК10). Когато се провежда изследване с антагонисти на ER, тълкуването на данните се базира на това дали откликът показва най-малко 30 % намаление на активността спрямо отклика, предизвикан от контрола с добавка (25 pM E2) без цитотоксичност. Анализът и тълкуването на данните са обсъдени подробно в точки 34 - 48.
|
ПРОЦЕДУРА
Клетъчни линии
|
7.
|
За изследването следва да се използва стабилно трансфектираната клетъчна линия hERα-HeLa-9 903. Клетъчната линия може да се получи от клетъчната банка на Японската колекция от биоресурси за изследователски цели (JCRB) (63), след подписване на Договор за предаване на материал (MTA).
|
|
8.
|
Единствено клетки, характеризиращи се с липса на микоплазма следва да бъдат използвани за изпитванията. Избраният чувствителен метод за откриване на инфекции с микоплазма е RT-PCR (полимеразната верижна реакция в реално време) (4) (5) (6).
|
Стабилност на клетъчната линия
|
9.
|
За наблюдение на стабилността на клетъчната линия следва да се използват E2, 17α-естрадиол, 17α-метилтестостерон и кортикостерон като референтни еталони за изследването с агонисти, като поне веднъж при всяко изпълнение на изследването следва да се измерват стойностите за съставяне на пълна крива концентрация-отклик в рамките на обхвата на концентрацията на изпитването, посочен в таблица 1, а получените резултати следва да съответстват на резултатите, посочени в таблица 1.
|
|
10.
|
В случай на изследване с антагонисти, стойностите за съставяне на пълни графики за двата референтни еталона, тамоксифен и флутамид, следва да бъдат измервани едновременно с всяка серия. Трябва да се следи за правилната качествена класификация за двата химикала като положителен или отрицателен.
|
Клетъчна култура и условия на плаките за изследване
|
11.
|
Клетките следва да бъдат отглеждани в минималната есенциална среда на Eagle (EMEM) без фенолно червено, с добавка от 60 mg/l антибиотик канамицин и 10 % фетален волски серум с покритие от декстран и третиран въглен (DCC-FBS), в инкубатор с CO2 (5 % CO2) при температура 37±1°C. При достигане на 75 -90 % конфлуентност, клетките могат бъдат субкултивирани в съотношение 10 ml от 0,4 x 105 – 1 x 105 клетки/ml за 100 mm съд за клетъчни култури. Клетките следва да бъдат суспендирани с 10 % FBS-EMEM (който е същия като при EMEM с DCC-FBS) и след това разпределени в ямките на микроплака при плътност от 1 x 104 клетки/(100 μl x ямка). След това, клетките следва да бъдат предварително инкубирани в инкубатор с 5 % CO2 при 37°±1°C за 3 часа, преди експозиция на химикала. Пластмасовите съдове и пособия следва да нямат естрогенна активност.
|
|
12.
|
За да се поддържа цялостта на отклика, клетките следва да бъдат отглеждани за повече от един пасаж от замразения запас в кондиционираната среда и не следва да бъдат култивирани за повече от 40 пасажа. За клетъчната линия hERα-HeLa-9 903, това е по-малко от три месеца. Въпреки това, ефективността на клетките може да бъде намалена, ако бъдат отглеждани в неподходящи условия за култивиране.
|
|
13.
|
DCC-FBS може да бъде приготвен, както е описано в допълнение 2.2 или да бъде набавен от източници от търговската мрежа.
|
Критерии за приемливост
Референтни еталони за положителни и отрицателни вещества, за изследването с агонисти на ER
|
14.
|
Преди и по време на проучването следва да се провери способността за реагиране на изпитваната система като се използват подходящи концентрации на силен естроген: E2, слаб естроген (17α-естрадиол), много слаб агонист (17α-метилтестостерон), и отрицателно вещество (кортикостерон). Стойностите на приемливия обхват, получени от изследването за валидиране (1), са дадени в таблица 1. Тези 4 паралелни референтни еталона следва да бъдат включени във всеки експеримент и резултатите следва да попадат в рамките на дадените приемливи граници. В противен случай следва да бъде установена причината за неизпълнението на критериите за приемливост (напр. манипулиране на клетките, серума и антибиотиците с цел постигане на качество и концентрация), а изследването следва да бъде повторено. След като бъдат постигнати критериите за приемливост, за да се гарантирамаксимална изменчивост на стойностите за EC50, PC50 и PC10, от съществено значение е последователното използване на материалите за клетъчно култивиране. Четирите паралелни референтни еталона, които следва да бъдат включени във всеки експеримент (провеждан при същите условия, включително по отношение на материалите, нивото на пасаж на клетките, лабораторните техници), могат да осигурят чувствителността на изследването, защото стойностите за PC10 на трите положителни референтни еталона следва да попадат в рамките на приемливия обхват, както и тези за PC50 и EC50, когато могат да се изчислят такива (вж. таблица 1).
|
Таблица 1
Приемлив обхват на стойностите за четирите референтни еталона за изследването с агонисти на ER
|
Наименование
|
logPC50
|
logPC10
|
logEC50
|
Крива на Хил
|
Обхват на изпитването
|
|
17β-естрадиол (E2) CAS №: 50-28-2
|
-11,4~-10,1
|
<-11
|
-11,3~-10,1
|
0,7~1,5
|
10-14~10-8M
|
|
17α-естрадиол CAS №: 57-91-0
|
-9,6~-8,1
|
-10,7~-9,3
|
-9,6~-8,4
|
0,9~2,0
|
10-12~10-6M
|
|
Химикал CAS №: 50-22-6
|
—
|
—
|
—
|
—
|
10-10~10-4M
|
|
17α-метилтестостерон CAS №: 58-18-4
|
-6,0~-5,1
|
-8,0~-6,2
|
—
|
—
|
10-11~10-5M
|
Референтни еталони за положителни и отрицателни вещества за изследването с антагонисти на ER
|
15.
|
Преди и по време на проучването следва да се провери способността за реагиране на изпитваната система, като се използват подходящи концентрации на положително вещество (тамоксифен) и отрицателно вещество (флутамид). Стойностите на приемливия обхват, получени от изследването за валидиране (1), са дадени в таблица 2. Тези два паралелни референтни еталона следва да бъдат включени във всеки експеримент и резултатите следва да бъдат оценени като съответстващи, както е показано в критериите. В противен случай следва да бъде установена причината за неизпълнението на критериите (напр. манипулиране на клетките, серума и антибиотиците с цел постигане на качество и концентрация), а изследването следва да бъде повторено. В допълнение трябва да се изчислят стойностите за IC50 за положително вещество (тамоксифен) и резултатите следва да попадат в рамките на дадените приемливи граници. След като бъдат постигнати критериите за приемливост, за да се гарантира максимална изменчивост на стойностите за IC50 , от съществено значение е последователното използване на материалите за клетъчно култивиране. Двата паралелни референтни еталона, които следва да бъдат включени във всеки експеримент (провеждан при същите условия, включително по отношение на материалите, нивото на пасаж на клетките, лабораторните техници), могат да осигурят чувствителността на изследването (вж. таблица 2).
|
Таблица 2
Критерии и стойности на приемливия обхват на двата референтни еталона за изследването с антагонисти на ER
|
Наименование
|
Критерии
|
LogIC50
|
Обхват на изпитването
|
|
Тамоксифен CAS №: 10 540 -29-1
|
положителен: IC50 следва да се изчисли
|
-5,942-7,596
|
10-1010-5M
|
|
Флутамид CAS №: 13 311 -84-7
|
отрицателен: IC30 не следва да се изчислява
|
—
|
10-10 10-5M
|
Контроли на положителното вещество и носителя
|
16.
|
Положителната контрола (ПК) за изследването с агонисти на ER (1 nM E2) и за изследването с антагонисти на ER (10μМ TAM) следва да бъдат изпитвани най-малко три пъти във всяка плака. Носителят, който се използва за разтваряне на изпитвания химикал, следва да бъде изпитан като контрола на носителя (VC) най-малко три пъти във всяка плака. В допълнение към тази контрола на носителя (VC), ако ПК използва различен носител от изпитвания химикал, следва да се изпита друга VC най-малко три пъти в същата плака с ПК.
|
Критерии за качество за изследването с агонисти на ER
|
17.
|
Средната луциферазна активност на положителната контрола (1 nM E2) следва да бъде най-малко 4-кратно по-голяма от средната VC за всяка плака. Този критерий е установен на базата на надеждността на стойностите за крайните точки от изследването за валидиране (по данни от предходни периоди между четири и 30 пъти).
|
|
18.
|
По отношение на контрола на качеството на изследването, кратносттаа на индукцията, отговаряща на стойността за PC10 на паралелната ПК (1 nM E2), следва да бъде по-голяма от 1+2SD от стойността на кратността на индукцията (=1) на паралелната VC. С цел определяне на приоритети, стойността PC10 може да бъде полезна за опростяване на необходимия анализ на данните в сравнение със статистическия анализ. Въпреки че статистическият анализ предоставя информация за значимостта, този анализ не е количествен параметър по отношение на потенциала на базата на концентрацията и поради това е по-малко полезен за целите на определяне на приоритети.
|
Критерии за качество за изследването с антагонисти на ER
|
19.
|
Средната луциферазна активност на контрола с добавка (25 pM E2) следва да бъде най-малко 4-кратно по-голяма от средната VC за всяка плака. Този критерий е установен на базата на надеждността на стойностите за крайните точки от изследването за валидиране.
|
|
20.
|
По отношение на контрола на качеството на изследването, относителното активиране на транскрипцията (RTA) на 1 nM E2 следва да бъде повече от 100 %, RTA на 1μМ 4-хидрокситамоксифен(OHT) следва да бъде по-малко от 40,6 %, а RTA на 100 μМ дигитонин (Dig) следва да бъде по-малко от 0 %.
Доказване на пригодността
на лабораторията
(вж. точка 14 и таблици 3 и 4 в «КОМПОНЕНТИ НА ER TA ИЗСЛЕДВАНЕТО» от настоящия метод за изпитване).
|
Превозно средство
|
21.
|
В качеството си на паралелна контрола на носителя (VC) следва да бъде използван диметил сулфоксид (DMSO) или подходящ разтворител с концентрацията, използвана за различните положителни и отрицателни контроли и за изпитваните химикали. Изпитваните химикали следва да бъдат разтваряни в разтворител, който разтваря конкретния изпитван химикал и се смесва с клетъчната среда. Подходящи носители са вода, етанол (с чистота 95 % до 100 %) и DMSO. Ако се използва DMSO, нивото не следва да превишава 0,1 % (v/v). За всеки носител следва да се докаже, че максималният използван обем не е цитотоксичен и не оказва отрицателно влияние върху ефективността на изследването.
|
Приготвяне на изпитваните химикали
|
22.
|
По принцип изпитваните химикали следва да бъдат разтваряни в DMSO или друг подходящ разтворител и последователно разреждани със същия разтворител в обичайно използваното съотношение от 1:10 за приготвяне на разтвори за разреждане със среда.
|
Разтворимост и цитотоксичност: Съображение при определянето на обхвата.
|
23.
|
Следва да се проведе предварително изпитване за определяне на подходящия обхват на концентрацията на химикала, който ще бъде изпитван и за констатиране дали изпитваният химикал има вероятност да има проблеми с разтворимостта и цитотоксичността. Първоначално химикалите се изпитват до максимална концентрация от 1 μl/ml, 1 mg/ml или 1 mM, която от тези стойности е най-ниска. Въз основата на степента на цитотоксичност или на липсата на разтворимост, наблюдавани по време на предварителното изпитване, първата окончателна серия следва да изпитва химикала при логаритмични последователни разреждания, като се започне от максималната приемлива концентрация (напр. 1 mM, 100μM, 10μM и т.н.) като наличието на помътняване, преципитат или цитотоксичност следва да бъде отбелязвано. Концентрациите във втората и, ако е необходимо, в третата серия следва да бъдат коригирани, според случая с оглед по-добро характеризиране на кривата концентрация-отклик и недопускане на концентрации, за които е установено, че са неразтворими или предизвикват прекомерна цитотоксичност.
|
|
24.
|
При агонистите и антагонистите на ER, наличието на повишаващи се нива на цитотоксичност може значително да промени или да елиминира типичния сигмоидален отклик и следва да се вземе предвид при тълкуването на данните. Следва да се използват методи за изпитване на цитотоксичността, които могат да предоставят информация за 80 % клетъчна жизнеспособност, с помощта на подходящо изследване, базирано на лабораторния опит.
|
|
25.
|
Ако резултатите от изпитването за цитотоксичност покажат, че концентрацията на изпитвания химикал е намалила броя на клетките с 20 % или повече, тази концентрация следва да се разглежда като цитотоксична, а концентрациите, равни или над цитотоксичните концентрации, следва да бъдат изключвани от оценката.
|
Химическа експозиция и организация на плаката за изследването
|
26.
|
Процедурата за разрежданията на химикала (Стъпки 1 и 2) и експозицията на клетките (Стъпка 3) могат да бъдат провеждани както следва:
Стъпка 1: Всеки изпитван химикал следва да бъде разреден последователност в DMSO, или друг подходящ разтворител и добавен към ямките на микротитърна плака до постигане на окончателните серийни концентрации, определени от предварителното изпитване за определяне на обхвата (обикновено в следните серии, например 1 mM, 100 μM, 10 μM, 1μM, 100 nM, 10 nM, 1 nM, 100 pM и 10 pM (10-3-10-11 M)) за тройно изпитване.
Стъпка 2: Разреждане на химикала: Първо се разреждат 1,5 μl от изпитвания химикал в разтворителя до обем 500 μl от средата.
Стъпка 3: Експозиция на химикала на клетките: Добавят се 50 μl от разредения разтвор със средата (приготвени в Стъпка 2) към ямка за анализ, съдържаща 104 клетки/100 μl/ямка.
Препоръчителният окончателен обем на средата, необходима за всяка ямка е 150 μl. Изпитваните проби и референтните еталони могат да бъдат определени, както е показано в таблица 3 и таблица 4.
|
Таблица 3
Пример за определяне на концентрация на референтните еталони в плаката за анализ, като част от изследването с агонисти на ER
|
Ред
|
17α-метилтестостерон
|
Химикал
|
17α-естрадиол
|
E2
|
|
|
1
|
2
|
3
|
4
|
5
|
6
|
7
|
8
|
9
|
10
|
11
|
12
|
|
А
|
конц. 1 (10 μM)
|
→
|
→
|
100 μM
|
→
|
→
|
1 μM
|
→
|
→
|
10 nM
|
→
|
→
|
|
Б
|
конц. 2 (1 μM)
|
→
|
→
|
10 μM
|
→
|
→
|
100 nM
|
→
|
→
|
1 nM
|
→
|
→
|
|
В
|
конц. 3 (100 nM)
|
→
|
→
|
1 μM
|
→
|
→
|
10 nM
|
→
|
→
|
100 pM
|
→
|
→
|
|
Г
|
конц. 4 (10 nM)
|
→
|
→
|
100 nM
|
→
|
→
|
1 nM
|
→
|
→
|
10 pM
|
→
|
→
|
|
Д
|
конц. 5 (1 nM)
|
→
|
→
|
10 nM
|
→
|
→
|
100 pM
|
→
|
→
|
1 pM
|
→
|
→
|
|
Е
|
конц. 6 (100 pM)
|
→
|
→
|
1 nM
|
→
|
→
|
10 pM
|
→
|
→
|
0,1 pM
|
→
|
→
|
|
Ж
|
конц. 7 (10 pM)
|
→
|
→
|
100 pM
|
→
|
→
|
1 pM
|
→
|
→
|
0,01 pM
|
→
|
→
|
|
З
|
VC
|
→
|
→
|
→
|
→
|
→
|
ПК
|
→
|
→
|
→
|
→
|
→
|
|
VC: Контрола на носителя (0,1 % DMSO); ПК: положителна контрола (1 nM E2)
|
|
27.
|
Референтните еталони (E2, 17α-естрадиол, 17-ПК ямките, третирани с 1 nM E2, които могат да предизвикат максимална индукция на E2 и само ямките на VC, третирани с DMSO (или подходящ разтворител) следва да бъдат включени във всяка тестова плака за изследване (таблица 4). Когато клетки от различни източници (напр. различен номер на пасаж, различна партида и т.н.) се използват в рамките на един и същи експеримент, референтните еталони следва да бъдат изпитани за всеки клетъчен източник.
|
Таблица 4
Пример за определяне на концентрация на изпитваните химикали и химикалите за контрол в плаката за анализ, като част от изследването с антагонисти на ER
|
Ред
|
Изпитван химикал 1
|
Изпитван химикал 2
|
Изпитван химикал 3
|
Изпитван химикал 4
|
|
1
|
2
|
3
|
4
|
5
|
6
|
7
|
8
|
9
|
10
|
11
|
12
|
|
А
|
конц. 1 (10 μM)
|
→
|
→
|
1 mM
|
→
|
→
|
1 μM
|
→
|
→
|
10 nM
|
→
|
→
|
|
Б
|
конц. 2 (1 μM)
|
→
|
→
|
100 μM
|
→
|
→
|
100 nM
|
→
|
→
|
1 nM
|
→
|
→
|
|
В
|
конц. 3 (100 nM)
|
→
|
→
|
10 μM
|
→
|
→
|
10 nM
|
→
|
→
|
100 pM
|
→
|
→
|
|
Г
|
конц. 4 (10 nM)
|
→
|
→
|
1 μM
|
→
|
→
|
1 nM
|
→
|
→
|
10 pM
|
→
|
→
|
|
Д
|
конц. 5 (1 nM)
|
→
|
→
|
100 nM
|
→
|
→
|
100 pM
|
→
|
→
|
1 pM
|
→
|
→
|
|
Е
|
конц. 6 (100 pM)
|
→
|
→
|
10 nM
|
→
|
→
|
10 pM
|
→
|
→
|
0,1 pM
|
→
|
→
|
|
Ж
|
конц. 7 (10 pM)
|
→
|
→
|
1 nM
|
→
|
→
|
1 pM
|
→
|
→
|
0,01 pM
|
→
|
→
|
|
З
|
VC
|
→
|
→
|
→
|
→
|
→
|
ПК
|
→
|
→
|
→
|
→
|
→
|
|
VC: контрола на носителя (0,1 % DMSO); ПК: положителна контрола (1 nM E2)
|
Таблица 5
Пример за определяне на концентрация на референтните еталони в плаката за анализ, като част от изследването с антагонисти на ER
|
28.
|
За да се оцени антагонистичната активност на химикали, към ямките за анализ, разположени на редове от A до Ж, се добавя 25pM E2. Референтните еталони (тамоксифен и флутамид) следва да бъдат изпитвани във всяка серия. Във всяка плака за анализ от изпитването следва да бъдат включени ямки с ПК, третирани с 1 nM E2, който може да бъде използван като качествен контрол на клетъчната линия hERα-HeLa-9 903, ямки с VC, третирани с DMSO (или подходящ разтворител), ямки с 0,1 % DMSO, третирани с добавяне на DMSO към E2 , съответстващи на „Контрола с добавка“, ямки, третирани с окончателна концентрация от 1 μM OHT, и ямки, третирани с 100 μM Dig (таблица 5). Плаката за следващото изследване трябва да бъде със същото разположение на ямките без ямките за референтните еталони (таблица 6). Когато клетки от различни източници (напр. различен номер на пасаж, различна партида и т.н.) се използват в рамките на един и същи експеримент, референтните еталони следва да бъдат изпитани за всеки клетъчен източник.
|
Таблица 6
Пример за определяне на концентрация на изпитваните химикали и химикалите за контрол в плаката за анализ, като част от изследването с антагонисти на ER
|
29.
|
Следва да се потвърди отсъствието на гранични ефекти, ако е необходимо, а ако има подозрения за такива, разположението на ямките в плаката трябва да се промени, за да не се допуснат такива ефекти. Например, може да се използва плака с такова разпределение, което да изключва граничните ямки.
|
|
30.
|
След добавяне на химикалите, плаките за анализ следва да бъдат инкубирани в инкубатор с 5 % CO2 при 37±1oC за 20-24 часа за индуциране на продуктите на репортерния ген.
|
|
31.
|
По отношение на съединенията, които са силно летливи, трябва да се приложат специални съображения. В такива случаи, намиращите се в близост ямки с контроли могат да генерират фалшиви положителни резултати и това следва да се разглежда в светлината на очакваните и стойности на контролите от предходни контроли. В няколкото случая, в които летливостта може да бъде притеснителна, използването на „запечатващи филми за плаки“ може да помогне за ефективно изолиране на отделните ямки по време на изпитване, поради което се препоръчва в подобни случаи.
|
|
32.
|
С цел осигуряване на независимост на резултатите, следва да бъдат проведени повторни окончателни изпитвания за същия химикал в различни дни.
|
Изследване за луцифераза
|
33.
|
Наличният в търговската мрежа реагент за изследване на луцифераза [напр. Система за изследване на луцифераза Steady-Glo® (Promega, E2510 или еквивалентна)] или стандартна система за изследване на луцифераза (напр. Promega, E1500 или еквивалентна) могат да бъдат използвани за изследването при условие, че са изпълнени критериите за приемливост. Реагентите за изследването следва да бъдат избрани въз основа на чувствителността на използвания луминометър. При работа със стандартната система за изследване на луцифераза, преди добавяне на субстрата следва да се приложи лизиращ реагент за клетъчни култури (e.g. Promega, E1531 или еквивалентен). При прилагане на реагента луцифераза следва да се спазват инструкциите на производителя.
|
АНАЛИЗ НА ДАННИТЕ
Изследване с агонисти на ER
|
34.
|
Когато се провежда изследване с агонисти на ER, за да се получи относителната транскрипционна активност по отношение на ПК (1 nM E2), луминесцентните сигнали от същата плака могат да бъдат анализирани чрез следните стъпки (приемливи са и други еквивалентни математически процедури):
Стъпка 1. Изчисляване на средната стойност за VC.
Стъпка 2. Изваждане на средната стойност на VC от стойността на всяка ямка за нормализиране на данните.
Стъпка 3. Изчисляване на средната стойност за нормализирания ПК.
Стъпка 4. Разделяне на нормализираната стойност на всяка ямка в плаката на средната стойност на нормализираната ПК (ПК=100 %).
Окончателната стойност на всяка ямка е относителната транскрипционна активност за тази ямка, сравнена с отклика на ПК.
Стъпка 5. Изчисляване на средната стойност на относителната транскрипционна активност за всяка група по концентрация от изпитвания химикал. Съществуват две измерения на отклика: усреднената транскрипционна активност (отклик) и концентрацията, при която възниква откликът (вж. следващия раздел).
|
Съображения относно индукцията на EC50, PC50 и PC10
|
35.
|
За изчисляването на EC50 е необходима цялата крива концентрация-отклик, но това може не винаги да бъде постижимо или приложимо на практика, поради ограниченията на обхвата на изпитваната концентрация (например, поради проблеми, свързани с цитотоксичност или разтворимост). Все пак, тъй като EC50 и максималното ниво на индукция (съответстващо на максималната стойност на уравнението на Хил) са информативни параметри, те следва да бъдат отчитани, когато е възможно. За изчисляването на EC50 и максималното ниво на индукция следва да се използва подходящ софтуер за статистически изчисления (напр. статистическия софтуер Graphpad Prism). Ако логистичното уравнение на Хил е приложимо за данните за отклика в зависимост от концентрацията, EC50 следва да се изчислява съгласно следното уравнение (7):
Y=Най-ниско ниво + (Най-високо ниво-Най-ниско ниво) / (1+10 exp ((log EC50 -X) x крива на Хил) където:
X е логаритъмът от концентрацията; както и
Y е откликът, който започва в Най-ниското ниво и продължава до Най-високото ниво, следвайки сигмоидна крива. Най-ниското ниво е фиксирано на нула по логистичното уравнение на Хил.
|
|
36.
|
За всеки изпитван химикал трябва да се посочи следното:
RPCMax , което е максималното ниво на отклик, индуциран от изпитван химикал, изразено като процентно съотношение на отклика, индуциран от 1 nM E2 върху същата плака, както и PCMax (концентрацията, свързана с RPCMax); както и
За положителни химикали, концентрациите, които индуцират PC10 и, ако е необходимо, PC50.
|
|
37.
|
Стойността ПКx може да се изчисли чрез интерполиране между 2 точки по X-Y координатите, една непосредствено над и една непосредствено под дадена стойност на ПКx. Когато точките от данни, които лежат непосредствено над и под стойността на ПКx, са с координати съответно (a,b) и (c,d), тогава стойността на ПКx може да се изчисли с помощта на следното уравнение:
log[ПКx] = log[c]+(x-d)/(d-b)
|
|
38.
|
Описание на стойностите на ПК е дадено на фигура 1 по-долу.
Фигура 1
Пример за начин за определяне на стойности на ПК. ПК (1 nM E2) се включва във всяка плака за анализ

|
Изследване с антагонисти на ER
|
39.
|
Когато се провежда изследване с антагонисти на ER, за да се получи относителната транскрипционна активност (RTA) по отношение на контрола с добавка (25 pM E2), луминесцентните сигнали от същата плака могат да бъдат анализирани чрез следните стъпки (приемливи са и други еквивалентни математически процедури):
Стъпка 1. Изчисляване на средната стойност за VC.
Стъпка 2. Изваждане на средната стойност на VC от стойността на всяка ямка за нормализиране на данните. Стъпка 3. Изчисляване на средната стойност за нормализирания контрол с добавка.
Стъпка 4. Разделяне на нормализираната стойност на всяка ямка в плаката на средната стойност на нормализирания контрол с добавка (контрол с добавка=100 %).
Окончателната стойност на всяка ямка е относителната транскрипционна активност за тази ямка, сравнена с отклика на контрола с добавка.
Стъпка 5. Изчисляване на средната стойност на относителната транскрипционна активност за всяко третиране.
|
Съображения относно индукцията на IC30 и IC50
|
40.
|
За положителни химикали следва да бъдат посочени концентрациите, които индуцират IC30 и, ако е необходимо,IC50.
|
|
41.
|
Стойността ICx може да се изчисли чрез интерполиране между 2 точки по координатите X и Y, една непосредствено над и една непосредствено под дадена ICx стойност. Когато точките от данни, които лежат непосредствено над и под стойността ICx, са с координати съответно (a,b) и (c,d), тогава стойността ICx може да се изчисли с помощта на следното уравнение:
lin ICx = a-(b-(100-x)) (a-c) /(b-d)
Фигура 2
Пример за начин за определяне на IC стойности. Контролът с добавка (25 pM E2) се включва във всяка плака за анализ

RTA: относителна транскрипционна активност
|
|
42.
|
Резултатите следва да се базират на две (или три) независими серии. Ако две серии дават сравними и следователно възпроизводими резултати, не е необходимо да се извършва трета серия. За да са приемливи, резултатите следва:
|
—
|
да отговарят на критериите за приемливост (вж. точки 14-20 на критериите за приемливост),
|
|
Критерии за тълкуване на данните
Таблица 7
Критерии за вземане положително или отрицателно решение в изследването с агонисти на ER
|
За положително решение
|
Ако получената стойност RPCMax е равна или превишава 10 % от отклика на положителната контрола в най-малко две от две или две от три серии.
|
|
За отрицателно решение
|
Ако стойността RPCMax не достигне най-малко 10 % от отклика на положителната контрола в две от две или две от три серии.
|
Таблица 8
Критерии за вземане на положително или отрицателно решение в изследването с антагонисти на ER
|
За положително решение
|
Ако стойността IC30 е изчислена в най-малко две от две или две от три серии.
|
|
За отрицателно решение
|
Ако стойността IC30 не може да бъде изчислена в най-малко две от две или две от три серии.
|
|
43.
|
Критериите за тълкуване на данните са показани в таблици 7 и 8. Положителните резултати се характеризират както от големината на ефекта, така и от концентрацията, при която възниква този ефект. Изразяването на резултатите като концентрация, при която 50 % (PC50) или 10 % (PC10) от стойностите на ПК са достигнати за изследването с агонисти и 50 % (IC50) или 30 % (IC30) от стойностите на контролата с добавка са потиснати за изследването с антагонисти, постига и двете цели. Въпреки това, изпитваният химикал се определя като положителен, ако максималният отклик, предизвикан от изпитвания химикал (RPCMax), е равен или превишава 10 % от отклика на ПК в най-малко две от две или две от три серии, докато изпитваният химикал се разглежда като отрицателен, ако RPCMax не успее да достигне най-малко 10 % от отклика на положителната контрола в две от две или две от три серии.
|
|
44.
|
Изчисляването на PC10, PC50 и PCMax в изследването с агонисти на ER и IC30 и IC50 в изследването с антагонисти на ER може да се направи с използване на електронната таблица, предлагана заедно с Насоките за изпитване на публичния уеб сайт на ОИСР (64).
|
|
45.
|
Достатъчно би било да се получат стойностите за PC10 или PC50 и IC30 или IC50 най-малко два пъти. Все пак, ако получените базови стойности за данните в един същи обхват на концентрация показват изменчивост с неприемливо висок коефициент на вариация (CV; %) данните може да се разглеждат като ненадеждни и следва да се определи източникът на високия процент на изменчивост. CV на необработените данни от трикратното изпитване (т.е. данните за интензивността на луминесценцията) на точките с данни, които са използвани за изчисляването на PC10 , следва да бъде по-малък от 20 %.
|
|
46.
|
Изпълнението на критериите за приемливост показва, че системата за изследване функционира правилно, но не гарантира, че всяка конкретна серия ще даде точни данни. Дуплицирането на резултатите от първата серия е най-добрата гаранция, че се получават точни данни.
|
|
47.
|
В случай на изследване с агонисти на ER, където се изисква повече информация в допълнение към целите на скрининга и определянето на приоритети на настоящите TG за положителни изпитвани химикали, и по-точно за химикалите PC10-PC49, както и за онези, за които има подозрения, че предизвикват свръхстимулация на луцифераза, може да се потвърди, че наблюдаваната луциферазна активност е единствено ERα-специфичен отклик, използващ ERα антагонист (вж. допълнение 2.1).
|
ПРОТОКОЛ ОТ ИЗПИТВАНЕТО
|
48.
|
Вж. параграф 20 на „КОМПОНЕНТИ НА АНАЛИЗА ER TA“.
|
ЛИТЕРАТУРА
|
(1)
|
OECD (2015). Report of the Inter-Laboratory Validation for Stably Transfected Transactivation Assay to detect Estrogenic and Anti-estrogenic Activity. Environment, Health and Safety Publications, Series on Testing and Assessment (No 225), Organisation for Economic Cooperation and Development, Paris.
|
|
(2)
|
Escande A., et al. (2006). Evaluation of Ligand Selectivity Using Reporter Cell Lines Stably Expressing Estrogen Receptor Alpha or Beta, Biochem. Pharmacol., 71, 1 459-1 469.
|
|
(3)
|
Kuiper G.G., et al. (1998). Interaction of Estrogenic Chemicals and Phytoestrogens with Estrogen Receptor Beta, Endocrinol., 139, 4 252-4 263.
|
|
(4)
|
Spaepen M., et al. (1992). Detection of Bacterial and Mycoplasma Contamination in Cell Cultures by Polymerase Chain Reaction, FEMS Microbiol. Lett., 78(1), 89-94.
|
|
(5)
|
Kobayashi H., et al. (1995). Rapid Detection of Mycoplasma Contamination in Cell Cultures by Enzymatic Detection of Polymerase Chain Reaction (PCR) Products, J. Vet. Med. Sci., 57(4), 769- 71.
|
|
(6)
|
Dussurget O. and Roulland-Dussoix D. (1994). Rapid, Sensitive PCR-Based Detection of Mycoplasmas in Simulated Samples of Animal Sera, Appl. Environ. Microbiol., 60(3), 953-9.
|
|
(7)
|
De Lean A., Munson P.J. and Rodbard D. (1978). Simultaneous Analysis of Families of Sigmoidal Curves: Application to Bioassay, Radioligand Assay, and Physiological Dose-Response Curves, Am. J. Physiol., 235, E97-El02.
|
Допълнение 2,1
НЕВЕРНИ ПОЛОЖИТЕЛНИ РЕЗУЛТАТИ: ОЦЕНКА НА ЛУМИНЕСЦЕНТНИ СИГНАЛИ, НЕМЕДИИРАНИ ОТ РЕЦЕПТОРИ
|
1.
|
Неверните положителни резултати в изследването с агонисти на ER може да бъдат генерирани от немедиирана от ER активация на луциферазния ген или от директна активация на генния продукт, или друга, несвързана флуоресценция. За наличието на тези ефекти свидетелства непълна или необичайна крива доза-отклик. В случай на съмнение за подобни ефекти, трябва да се изследва ефектът на антагонистите на ER (напр. 4- хидрокситамоксифен (OHT) в нетоксична концентрация) върху отклика. Чистият антагонист ICI 1827 80 може да не бъде подходящ за тази цел, тъй като достатъчна концентрация на ICI 1827 80 може да намали стойността на VC, а това ще окаже влияние върху анализа на данните.
|
|
2.
|
За да се осигури валидност на този подход, трябва да се извърши изпитване в същата плака на:
|
—
|
Агонистичната активност на неизвестен химикал с / без 10 μM OHT
|
|
—
|
1 nM E2 (трикратно) като ПК за агонисти
|
|
—
|
1 nM E2 + OHT (трикратно)
|
|
Критерии за тълкуване на данните
Забележка: Всички ямки следва да бъдат третирани със една и съща концентрация на носителя.
|
—
|
Ако агонистичната активност на неизвестния химикал НЕ се повлияе от третирането с антагонист на ER, той се класифицира като “Отрицателен”.
|
|
—
|
Ако агонистичната активност на неизвестния химикал е напълно подтисната, следва да се приложат критериите за решение.
|
|
—
|
Ако агонистичната активност при най-ниската концентрация е равна на отклика на PC10 или го превишава, неизвестният химикал е потиснат в степен, равна на отклика на PC10 , или по-голяма. Изчислява се разликата в отговорите на нетретираните и третираните ямки с антагонист на ER, като тази разлика следва да се разглежда като отклик и следва да се използва за изчисляването на подходящите параметри, които позволяват вземането на решение за класификация.
|
Анализ на данните
Проверка на стандарта за ефективност.
Проверка на CV за ямки, третирани при едни и същи условия.
|
1.
|
Изчисляване на средната стойност на VC
|
|
2.
|
Изваждане на средната стойност за VC от стойността на всяка ямка, нетретирана с OHT
|
|
3.
|
Изчисляване на средната стойност на OHT
|
|
4.
|
Изваждане на средната стойност за VC от стойността на всяка ямка, третирана с OHT
|
|
5.
|
Изчисляване на средната стойност на ПК
|
|
6.
|
Изчисляване на относителната транскрипционна активност на всички други ямки спрямо ПК.
|
Допълнение 2.2
ПРИГОТВЯНЕ НА СЕРУМ, ТРЕТИРАН С ВЪГЛЕН С ПОКРИТИЕ ОТ ДЕКСТРАН (DCC)
|
1.
|
Третирането на серум с въглен с покритие от декстран (DCC) е общ метод за отстраняване на естрогенни съединения от серума, който се добавя към клетъчната среда, за да се изключи неверен отклик, свързан с наличието на остатъчни естрогени в серума. С тази процедура може да се третира 500 ml фетален волски серум (FBS).
|
Компоненти
|
2.
|
Ще бъдат необходими следните материали и оборудване:
|
Материали
|
Магнезиев хлорид хексахидрат (MgCl2·6H2O)
|
|
1 M HEPES буферен разтвор (pH 7.4)
|
|
Ултрапречистена вода от филтърна система
|
Оборудване
Стерилизиран в автоклав стъклен съд (размерът следва да се коригира според необходимостта) Лабораторна центрофуга с общо предназначение (която може да поддържа температура от 4 °C)
Процедура
|
3.
|
Следната процедура се коригира за работа с епруветки за центрофугиране от 50 ml:
[Ден 1] Приготвя се суспензия на въглен с декстраново покритие с 1 l ултрапречистена вода, съдържаща 1,5 mM MgCl2, 0,25 M захароза, 2,5 g въглен, 0,25 g декстран и 5 mM HEPES и се разбърква при 4 °C за една нощ.
[Ден 2] Разпределя се суспензията в епруветки за центрофугиране от 50 ml и се центрофугира със скорост от 10 000 об./мин. при 4 °C за 10 минути. Отстранява се супернатантът и се съхранява половината от въгленовия седимент при 4 °C за употреба през Ден 3. Суспендира се другата половина от въглена с FBS, който е бил плавно размразен, за да не се допусне преципитация и топлинно деактивиран при 56 °C за 30 минути, след което се прехвърля суспензията в стерилизирания в автоклав стъклен съд, напр. елренмайерова колба. Разбърква се леко суспензията при 4 °C цяла нощ.
[Ден 3] Суспензията с FBS се разпределя в епруветките и се центрофугира със скорост 10 000 об./мин. при 4 °C за 10 минути. Отстранява се FBS и се прехвърля в новия въгленов седимент, приготвен и съхранен през Ден 2. Суспендира се въгленовият седимент и се разбърква леко получената суспензия в стерилизиран в автоклав стъклен съд при 4 °C за една нощ.
[Ден 4] Разпределя се суспензията за центрофугиране със скорост 10 000 об./мин. при 4 °C за 10 минути и се стерилизира супернатантът чрез филтриране през 0,2 μm стерилен филтър. Този третиран с DCC FBS следва да бъде съхраняван при -20 °C може да се използва в период до една година.
|
Допълнение 3
VM7LUC ИЗСЛЕДВАНЕ ЗА ТРАНСАКТИВАЦИЯ НА ЕСТРОГЕНЕН РЕЦЕПТОР ЗА ОПРЕДЕЛЯНЕ НА АГОНИСТИ И АНТАГОНИСТИ НА ЕСТРОГЕННИЯ РЕЦЕПТОР
ПЪРВОНАЧАЛНИ СЪОБРАЖЕНИЯ И ОГРАНИЧЕНИЯ (ВЖ. СЪЩО „ОБЩО ВЪВЕДЕНИЕ“)
|
1.
|
Настоящото изследване използва клетъчната линия VM7Luc4E2 (65). То е валидирано от Националната програма по токсикология към Междуведомствения център за оценка на алтернативни токсикологични методи (NICEATM) и Междуведомствения координационен комитет по валидиране на алтернативни методи (ICCVAM) (1). Клетъчните линии VM7Luc преобладаващо експресират ендогенен ERα и минимално количество ендогенен ERβ (2) (3) (4).
|
|
2.
|
Настоящото изследване е приложимо за широк обхват от вещества, при условие че те могат да бъдат разтворени в диметилсулфоксид (DMSO; CASRN 67-68-5), не реагират с DMSO или със средата на клетъчната култура и не са цитотоксични в изпитваните концентрации. Ако употребата на DMSO е невъзможна, може да се използва друг носител като например етанол или вода (вж. точка 12). Доказаната ефективност на VM7Luc ER TA изследването за (ант)агонисти предполага, че данните, генерирани с това изследване, могат да дадат информация за ER-медиираните механизми на действие и биха могли да се разглеждат за целите на приоритизиране на веществата за по-нататъшни изпитвания.
|
|
3.
|
Това изследване е специално разработено за откриване на hERα и hERß-медиирана TA чрез измерване на хемилуминесцентността като крайна точка. Използването на хемилуминесценция в биологични изследвания е широко разпространено, защото луминесценцията има високо отношение сигнал-фон. Въпреки това, активността на луциферазата на светулка в изследвания, базирани на клетъчни култури, може да бъде смущавано от вещества, които потискат ензима луцифераза, причинявайки явно потискане и повишена луминесценция поради протеиновата стабилизация. Освен това, при някои основани на луциферазата изпитвания на ER с репортерни гени бяха докладвани немедиирани от рецептора луминисцентни сигнали, получени при по-висока от 1 μМ концентрация на фитоестроген, дължащи се на свръхактивиране на луциферазния репортерен ген (9) (11). Тъй като кривата доза-отклик показва, че истинската активация на ER системата възниква при по-ниски концентрации, експресията на луциферазата, получена при високи концентрации на фитоестрогени или подобни съединения, за които се подозира, че предизвикват подобно на фитоестрогена свръхактивиране на луциферазния репортерен ген, трябва да се изследва внимателно в системи за изследване на ER TA чрез стабилно трансфектиране.
|
|
4.
|
Разделите „ОБЩО ВЪВЕДЕНИЕ“ и „КОМПОНЕНТИ НА ИЗСЛЕДВАНЕТО ER TA“ следва да бъдат прочетени, преди да се използва настоящото изследване за регулаторни цели. Използваните определения и съкращения в рамките на настоящия метод на изпитване са описани в допълнение 1.
|
ПРИНЦИП НА ИЗСЛЕДВАНЕТО (ВЖ. СЪЩО „ОБЩО ВЪВЕДЕНИЕ“)
|
5.
|
Изследването се използва за доказване на свързване на ER с лиганда, последвано от транслокация на рецептор-лигандния комплекс в ядрото. След като попадне в клетъчното ядро, рецептор-лигандният комплекс се свързва със специфични елементи на ДНК отклика и трансактивира репортерния ген (luc), което води до производство на луцифераза и до последващото излъчване на светлина, което може да бъде определено количествено с помощта на луминометър. Луциферазната активност може да бъде измерена бързо и евтино с множество китове, налични в търговската мрежа. VM7Luc ER TA използва чувствителна към ER клетъчна линия от човешки аденокарцином на гърдата, VM7, която е била стабилно трансфектирана с модел на репортерен ген от светулка luc под контролана четири елемента на естрогенния отклик, разположени преди вирусния промотор на миши тумор на млечната жлеза (MMTV), за откриване на вещества с in vitro активност на агонисти или антагонисти на ER. Този промотор на MMTV показва незначителна кръстосана реактивност с други стероидни и нестероидни хормони (8). Критериите за тълкуване на данните са описани подробно в точка 41. Накратко, положителният отклик се определя от кривата концентрация-отклик, съдържаща най-малко три точки с неприпокриващи се ленти на грешката (средна стойност ± SD), както и промяна на амплитудата (нормализирани относителни светлинни единици [RLU]) от най-малко 20 % от максималната стойност за референтния еталон (17β-естрадиол [E2; CASRN 50-28-2] за изследването с агонисти, ралоксифен HCl [Ral; CASRN 84449-90-1]/E2 за изследването с антагонисти).
|
ПРОЦЕДУРА
Клетъчна линия
|
6.
|
За изследването следва да се използва стабилно трансфектираната клетъчна линия VM7Luc4E2. Понастоящем клетъчната линия е налична само при подписано технически лицензионно споразумение от Калифорнийския университет, Davis, Калифорния, САЩ (66) и от Xenobiotic Detection Systems Inc., Durham, Северна Каролина, САЩ (67).
|
Стабилност на клетъчната линия
|
7.
|
За да се поддържа стабилността и целостта на клетъчната линия, клетките следва да бъдат култивирани за повече от един пасагж от замразеното количество в средата за клетъчно култивиране (вж. точка 9). Клетките не следва да бъдат култивирани повече от 30 пасажа. За клетъчната линия VM7Luc4E2, 30 пасажа са приблизително три месеца.
|
Клетъчна култура и условия на плаките за изследване
|
8.
|
Процедурите, описани в Ръководството за добри практики при култивиране на клетки (5) (6), следва да бъдат спазвани, за да се осигури качеството на всички материали и методи с оглед поддържане на целостта, валидността и възпроизводимостта на всички провеждани процедури.
|
|
9.
|
Клетките VM7Luc4E2 се отглеждат в среда RPMI 1640 с добавени 0,9 % Pen-Strep и 8,0 % фетален волски серум (FBS) в специален инкубатор за тъканни култури при 37oC ± 1oC, 90 % ± 5 % влажност и 5,0 % ± 1 % CO2/въздух.
|
|
10.
|
При достигане на ~80 % сливане, клетките VM7Luc4E2 се субкултивират и кондиционират към среда без естроген в продължение на 48 часа, преди разпределянето им в 96-ямкови плаки за експозиция на изпитваните химикали и анализ на зависимата от естроген индукция на луциферазната активност. Средата без естроген (EFM) съдържа модифицирана от Дюлбеко среда на Игъл (DMEM) без фенолно червено, допълнена с 4,5 % FBS, третиран с въглен/декстран, 1,9 % L-глутамин и 0,9 % Pen-Strep (пеницилин-стрептомицин). Всички пластмасови съдове и пособия следва да нямат естрогенна активност [вж. подробния протокол (7)].
|
Критерии за приемливост
|
11.
|
Приемането или отхвърлянето на изпитване се прави на базата на оценката на резултатите, получени за референтните еталони и контролите от всеки експеримент, проведен върху 96-ямковата плака. Всеки референтен еталон се изпитвав множество концентрации, като има множество проби от всяка концентрация на референтно и контролно вещество. Резултатите се сравняват спрямо качествените контроли (КК) за тези параметри, които са получени от базите данни от изпитванията с агонисти и антагонисти от предходни периоди, генерирани от всяка лаборатория по време на доказването на пригодността. Базите данни от предходни периоди се актуализират непрекъснато със стойностите на референтните еталони и контролните вещества. Промените в оборудването или лабораторните условия може да наложат генериране на актуализирани бази данни от предходни периоди.
|
Изпитване с агонисти
Изпитване за определяне на обхват
|
•
|
Индукция: Индукцията в плаката следва да бъде измервана чрез разделяне на средната стойност на относителната светлинна единица (RLU) на референтен еталон с най-високо съдържание на E2 на средната RLU стойност на DMSO контрола. Обикновено се получава петкратна индукция, но за целите на приемането, индукцията следва да бъде четирикратна или по-голяма.
|
|
•
|
Резултати от контролата с DMSO: Стойностите на RLU за контролата на разтворителя следва да бъдат в рамките на 2,5 пъти стандартното отклонение на средната стойност на RLU от предходни периоди от контролата на разтворителя.
|
|
•
|
Експеримент, който не изпълни който и да било критерий за приемливост, следва да бъде бракуван и повторен.
|
Цялостно изпитване
То включва критерии за приемливост от изпитването за определяне на обхват на агонистите и следното:
|
•
|
Резултати за референтния еталон: Кривата концентрация-отклик на референтния еталон E2 следва да бъде сигмоидална по форма и да има най-малко три стойности в рамките на линейната си част.
|
|
•
|
Резултати от положителната контрола: Стойностите на RLU за контролата на метоксихлор следва да бъдат по-високи от средната стойност за DMSO плюс три пъти стандартното отклонение от средната стойност за DMSO.
|
|
•
|
Експеримент, който не изпълни всеки отделен критерий за приемливост, следва да бъде бракуван и повторен.
|
Изпитване с антагонисти
Изпитване за определяне на обхват
|
•
|
Редукция: Редукцията в плаката се измерва чрез разделяне на средната RLU стойност на референтен еталон с най-високо съдържание на Ral/E2 на средната RLU стойност на DMSO контрола. Обикновено се получава петкратна редукция, но за целите на приемането, редукцията следва да бъде по-голяма от или равна на три-кратна.
|
|
•
|
Резултати от контрола на E2: RLU стойностите за контрола на E2 следва да бъдат в рамките на 2,5 пъти стандартното отклонение на историческата средна RLU стойност на контрола на E2.
|
|
•
|
Резултати от контролата с DMSO: RLU стойностите за контрола на DMSO следва да бъдат в рамките на 2,5 пъти стандартното отклонение на историческата средна RLU стойност на контрола на разтворителя.
|
|
•
|
Експеримент, който не изпълни всеки отделен критерий за приемливост ще бъде бракуван и повторен.
|
Цялостно изпитване
То включва критерии за приемане от изпитването за определяне на обхват на антагонистите и следното:
|
•
|
Резултати за референтния еталон: Кривата концентрация-отклик на референтния еталон Ral/E2 следва да бъде сигмоидална по форма и да има най-малко три стойности в рамките на линейната част от нея.
|
|
•
|
Резултати от положителната контрола: RLU стойностите за контрола на тамоксифен/E2 следва да бъдат по-малки от средната стойност за контрола на E2 минус три пъти стандартното отклонение от средната стойност за контрола на E2.
|
|
•
|
Експеримент, който не изпълни всеки отделен критерий за приемливост, ще бъде бракуван и повторен.
|
Референтни еталони, положителни контроли и контроли на носителя
Контрол на носителя (изследвания с агонисти и антагонисти)
|
12.
|
Носителят, който е използван за разтваряне на изпитваните химикали, следва да бъде изпитан като контрола на носителя. Използваният носител по време на валидирането на изследването с VM7Luc ER TA беше 1 % (v/v) диметилсулфоксид (DMSO, CASRN 67-68-5) (вж. точка 24). Ако е използван носител, различен от DMSO, всички референтни еталони, контроли и изпитвани химикали следва да бъдат изпитани в същия носител, ако е приложимо.
|
Референтен еталон (за определяне на обхвата на агонисти)
|
13.
|
Референтният еталон е E2 (CASRN 50-28-2). За изпитвания за определяне на обхвата, референтният еталон се състои от серийно разреждане на четири концентрации на E2 (1,84 x 10-10, 4,59 x 10-11, 1,15 x 10-11 и 2,87 x 10-12 M), като всяка концентрация е изпитана в дублирани ямки.
|
Референтен еталон (цялостен за агонисти)
|
14.
|
E2 за цялостно изпитване се състои от 1:2 серийно разреждане, съставено от 11 концентрации (вариращи от 3,67 x 10-10 до 3,59 x 10-13 M) на E2 в дублирани ямки.
|
Референтен еталон (за определяне на обхвата на антагонисти)
|
15.
|
Референтният еталон е комбинация от Ral (CASRN 84449-90-1) и E2 (CASRN 50-28-2). Ral/E2 за изпитването за определяне на обхват се състои от серийно разреждане на три концентрации на Ral (3,06 x 10-9, 7,67 x 10-10 и 1,92 x 10-10M) плюс една фиксирана концентрация (9,18 × 10-11 M) на E2 в дублирани ямки.
|
Референтен еталон (цялостно изпитване с антагонисти)
|
16.
|
Ral/E2 за цялостно изпитване се състои от 1:2 серийно разреждане на Ral (вариращо от 2,45x 10-8 до 9,57 x 10-11M) плюс една фиксирана концентрация (9,18 × 10-11 M) на E2 съставена от девет концентрации на Ral/E2в дублирани ямки.
|
Слаба положителна контрола (агонисти)
|
17.
|
Слабата положителна контрола е 9,06 x 10-6 M p,p’-метоксихлор (метоксихлор; CASRN 72-43-5) в EFM.
|
Слаба положителна контрола (антагонисти)
|
18.
|
Слабата положителна контрола се състои от тамоксифен (CASRN 10540-29-1) 3,36 x 10-6 M с 9,18 × 10-11 M E2 в EFM.
|
Контрола на E2 (само за изследването с антагонисти)
|
19.
|
Контролата на E2 е 9,18 × 10-11 M E2 в EFM и се използва като базова стойност за отрицателната контрола.
|
Кратна индукция (агонисти)
|
20.
|
Индукцията на луциферазна активност на референтния еталон (E2) се измерва чрез разделяне на средната стойност на RLU на референтен еталон с най-високо съдържание на E2 на средната стойност на RLU на контролата с DMSO, като резултатът следва да бъде по-голям от четирикратен.
|
Кратна редукция (антагонисти)
|
21.
|
Средната луциферазна активност на референтния еталон (Ral/E2) се измерва чрез разделяне на средната стойност на RLU на референтен еталон с най-високо съдържание на Ral/E2 на средната стойност на RLU на контрола с DMSO, като резултатът следва да бъде по-голям от трикратен.
Доказване на пригодността на лабораторията (вж. точка 14 и таблици 3 и 4 в „КОМПОНЕНТИ НА ИЗСЛЕДВАНЕТО НА ER TA“ от настоящия метод за изпитване)
|
Носител
|
22.
|
Изпитваните химикали следва да бъдат разтваряни в разтворител, който разтваря изпитвания химикал и се смесва с клетъчната среда. Подходящи носители са вода, етанол (с чистота 95 % до 100 %) и DMSO. Ако се използва DMSO, нивото не следва да превишава 1 % (v/v). За всеки носител следва да се докаже, че максималния използва обем не е цитотоксичен и не оказва отрицателно влияние върху ефективността на изследването. Референтните еталони и контроли се разтварят в 100 % разтворител и след това се разреждат до подходящи концентрации в EFM.
|
Приготвяне на изпитваните химикали
|
23.
|
Изпитваните химикали се разтварят в 100 % DMSO (или подходящ разтворител) и след това се разреждат до подходящи концентрации в EFM. Всички изпитвани химикали следва да бъдат оставени да се аклиматизират към стайната температура, преди да бъдат разтваряни и разреждани. Разтворите на изпитваните химикали следва да бъдат прясно приготвени за всеки експеримент. Разтворите не следва да имат забележим преципитат или помътняване. Основните разтвори на референтните еталони и контролите могат да бъдат приготвени в по-големи количества; въпреки това, окончателните разреждания на референтния еталон, контролите и изпитваните химикали следва да бъдат прясно приготвени за всеки експеримент и използвани в рамките на 24 часа след приготвянето.
|
Разтворимост и цитотоксичност: Съображения при определянето на обхвати
|
24.
|
Изпитването за определяне на обхват се състои от седем точки - 1:10 серийни разреждания, изпълнени с дублиране. Първоначално изпитваните химикали се изпитват до максималната концентрация от 1 mg/ml (~1 mM) за изпитването на агонисти и 20 μg/ml (~10 μМ) за изпитването с антагонисти. Експериментите за определяне на обхвати се използват за определяне на следното:
|
|
—
|
Началните концентрации на изпитвания химикал, които ще бъдат използвани по време на цялостното изпитване
|
|
—
|
Началните разреждания на химикала (1:2 или 1:5), които ще бъдат използвани по време на цялостното изпитване
|
|
25.
|
Оценката на клетъчната жизнеспособност/цитотоксичност е включена в протоколите за изследване на агонисти и антагонисти (7) и е внедрена в изпитването за определяне на обхват и в цялостното изпитване. Методът за определяне на цитотоксичност, който е използван за оценяване на клетъчната жизнеспособност по време на валидирането на VM7Luc ER TA (1) е бил мащабиран метод за качествено визуално наблюдение; въпреки това може да се използва количествен метод за определяне на цитотоксичността (вж. протокол (7)). Данните от концентрациите на изпитвания химикал, които причиняват повече от 20 % редукция на жизнеспособността, не могат да бъдат използвани.
|
Експозиция на изпитвания химикал и организация на плаката за изследването
|
26.
|
Клетките се отброяват и поставят в EFM в 96-ямкови плаки за тъканни култури (2 x 105 клетки в ямка) и инкубират за 24 часа, за да се позволи на клетките да се прикрепят към плаката. EFM се отстранява и се подменя с изпитваните и референтните химикали в EFM и се инкубират за 19-24 часа. Относно веществата, които са силно летливи трябва да се приложат специални съображения, тъй като ямките с контроли в непосредствена близост могат да генерират неверни положителни резултати. В такива случаи, „запечатващите филми“ могат да помогнат за ефективно изолиране на отделните ямки по време на изпитване и по тази причина са препоръчителни.
|
Изпитвания за определяне на обхват
|
27.
|
Изпитването за определяне на обхват използва всички ямки на 96-ямковата плака за изпитване на до шест изпитвани химикала като седем точкови разреждания 1:10 изпълнени с дублиране (вж. фигури 1 и 2).
|
|
—
|
Изпитването за определяне на обхват на агонисти използва четири концентрации на E2 с дублиране, като референтния еталон и четири ямки за повторения за контролата с DMSO.
|
|
—
|
Изпитването за определяне на обхват на антагонисти използва три концентрации на Ral/E2 с 9,18 × 10-11 M E2 с дублиране, като референтния еталон, три ямки за повторения за E2 и за контролите с DMSO.
|
Фигура 1
Разположение на 96-ямковата плака за изпитването за определяне на обхват на агонисти
Съкращения: E2-1 до E2-4 = концентрации на референтния еталон E2 (от висока към ниска); TC1-1 до TC1-7 = концентрации (от висока към ниска) на изпитван химикал 1 (TC1); TC2-1 до TC2-7 = концентрации (от висока към ниска) на изпитван химикал 2 (TC2); TC3-1 до TC3-7 = концентрации (от висока към ниска) на изпитван химикал 3 (TC3); TC4-1 до TC4-7 = концентрации (от висока към ниска) на изпитван химикал 4 (TC4); TC5-1 до TC5-7 = концентрации (от висока към ниска) на изпитван химикал 5 (TC5); TC6-1 до TC6-7 = концентрации (от висока към ниска) на изпитван химикал 6 (TC6); VC = контрол на носителя (DMSO [1 % v/v EFM.]).
Фигура 2
Разположение на 96-ямковата плака за изпитването за определяне на обхват на антагонисти

Съкращения: E2 = E2 контрол; Ral-1 до Ral-3 = концентрации на референтния еталон Ралоксифен/E2 (от висока към ниска); TC1-1 до TC1-7 = концентрации (от висока към ниска) на изпитван химикал 1 (TC1); TC2-1 до TC2-7 = концентрации (от висока към ниска) на изпитван химикал 2 (TC2); TC3-1 до TC3-7 = концентрации (от висока към ниска) на изпитван химикал 3 (TC3); TC4-1 до TC4-7 = концентрации (от висока към ниска) на изпитван химикал 4 (TC4); TC5-1 до TC5-7 = концентрации (от висока към ниска) на изпитван химикал 5 (TC5); TC6-1 до TC6-7 = концентрации (от висока към ниска) на изпитван химикал 6 (TC6); VC = контрол на носителя (DMSO [1 % v/v EFM.]).
Забележка: Всички изпитвани химикали са изпитани в присъствието на 9,18 × 10-11 M E2.
|
28.
|
Препоръчителният окончателен обем на средата, необходима за всяка ямка,е 200 μl. Използват се само тестови плаки, в които клетките във всички ямки показват жизнеспособност от 80 % и повече.
|
|
29.
|
Определянето на началните концентрации за цялостното изпитване с
агонисти
е описано задълбочено в протокола с агонисти (7). Накратко, използват се следните критерии:
|
|
—
|
Ако на кривата на концентрацията на изпитвания химикал няма точки, които са по-големи от средната стойност плюс три пъти стандартното отклонение на контролата с DMSO, се провежда цялостно изпитване, като се използва 11-точково серийно разреждане 1:2, започващо от максималната разтворима концентрация.
|
|
—
|
Ако на кривата на концентрацията на изпитвания химикал има точки, които са по-големи от средната стойност плюс три пъти стандартното отклонение на контролата с DMSO, началната концентрация, която се използва за 11-точковата схема за разреждане при цялостното изпитване, следва да бъде един логаритъм по-висока от концентрацията, даваща най-високата коригирана стойност на RLU при изпитването за определяне на обхват. 11-точковата схема на разреждане е базирана на разреждания 1:2 или 1:5, в съответствие със следните критерии:
|
Следва да бъде използвано11-точково серийно разреждане 1:2, ако полученият обхват на концентрацията включва пълния диапазон от отклици на базата на кривата концентрация-отклик, генерирана в рамките на изпитването за определяне на обхват. В противен случай, се използва разреждане 1:5.
|
|
|
—
|
Ако даден изпитван химикал покаже двуфазна крива концентрация-отклик в рамките на изпитването за определяне на обхват, двете фази следва също да бъдат определени в цялостно изпитване.
|
|
30.
|
Определянето на началните концентрации за цялостното изпитване с
антагонисти
е описано задълбочено в протокола с антагонисти (7). Накратко, използват се следните критерии:
|
|
—
|
Ако на кривата на концентрацията на изпитвания химикал няма точки, които са по-малки от средната стойност минус три пъти стандартното отклонение на контролата на E2, се провежда цялостно контролно изпитване, като се използва 11-точково серийно разреждане 1:2, започващо от максималната разтворима концентрация.
|
|
—
|
Ако на кривата на концентрацията на изпитвания химикал има точки, които са по-малки от средната стойност минус три пъти стандартното отклонение на E2 контрола, началната концентрация, която се използва за 11-точковата схема за разреждане при цялостното изпитване следва да бъде една от следните:
|
—
|
Концентрацията, даваща най-ниската коригирана стойност на RLU в рамките на изпитването за определяне на обхват
|
|
—
|
Максималната концентрация на разтваряне (вж. протокола с антагонисти (7), фигура 14-2)
|
|
—
|
Най-ниската цитотоксична концентрация (Виж протокола с антагонисти (7), фигура 14-3 за свързан пример).
|
|
|
—
|
11-точковата схема на разреждане се базира на разреждания 1:2 или 1:5 серийно разреждане или разреждане в съответствие със следните критерии:
|
Следва да бъде използвано11-точково серийно разреждане 1:2, ако полученият обхват на концентрацията включва пълния диапазон от отклици на базата на кривата концентрация-отклик, генерирана в рамките на изпитването за определяне на обхват. В противен случай следва да се използва разреждане 1:5.
|
|
Цялостни изпитвания
|
31.
|
Цялостното изпитване се състои от 11-точкови серийни разреждания (серийни разреждания 1:2 или 1:5, базирани на началната концентрация за критериите на цялостното изпитване), като всяка концентрация се изпитва трикратно в 96-ямковата плака (вж. фигури 3 и 4).
|
|
—
|
Цялостното изпитване с агонисти използва 11 дублирани концентрации на E2 като референтен еталон. Във всяка плака са включени четири ямки за повторение за контролата с DMSO и четири ямки за повторение за контрола с метоксихлор (9,06 x 10-6 M).
|
|
—
|
Цялостното изпитване с антагонисти използва девет дублирани концентрации на Ral/E2 с 9,18 × 10-11 M E2 като референтен еталон, с четири ямки за повторение за контрола с E2 9,18 x 10-11 M, четири ямки за повторение за контроли с DMSO и четири ямки за повторение за тамоксифен 3,36 x 10-6M.
|
Фигура 3
Разположение на 96-ямковата плака за цялостното изпитване с агонисти
Съкращения: TC1-1 до TC1-11 = концентрации (от висока към ниска) на изпитван химикал 1; TC2-1 до TC2-11 = концентрации (от висока към ниска) на изпитван химикал 2; E2-1 до E2-11 = концентрации на референтния еталон E2 (от висока към ниска); Meth = p,p’ метоксихлор, слаба положителна контрола; VC = DMSO (1 % v/v) EFM контрол на носителя
Фигура 4
Разположение на 96-ямковата плака за цялостното изпитване на антагонисти
Съкращения: E2 = E2 контрол; Ral-1 до Ral-9 = концентрации на референтния еталон Ралоксифен/E2 (от висока към ниска); Tam = Тамоксифен/E2 слаба положителна контрола; TC1-1 до TC1-11 = концентрации (от висока към ниска) на изпитван химикал 1 (TC1); TC2-1 до TC2-11 = концентрации (от висока към ниска) на изпитван химикал 2 (TC2); VC = контрола на носителя (DMSO [1 % v/v EFM.]).
Забележка:Както бе отбелязано, всички ямки с референтния химикал и изпитваните вещества съдържат фиксирана концентрация на E2 (9,18 x 10-11M)
|
32.
|
С цел осигуряване на независимост на резултатите, следва да бъдат проведени повторни цялостни изпитвания за същия химикал в различни дни. Най-малко две цялостни изпитвания следва да бъдат проведени. Ако резултатите от изпитванията си противоречат (напр. едното изпитване е положително, а другото отрицателно) или ако едно от изпитванията е непригодно, следва да се проведе трето, допълнително изпитване.
|
Измерване на луминесценцията
|
33.
|
Луминесценцията се измерва в диапазона от 300 до 650 nm, с помощта на впръскващ луминометър и софтуер, който контролира обема за впръскване и интервала за измерване (7). Излъчването на светлина от всяка ямка се изразява като RLU на ямката.
|
АНАЛИЗ НА ДАННИТЕ
Определяне на EC50 /IC50
|
34.
|
Стойността на EC50 (половината от максимално ефективната концентрация на изпитвания химикал [агонисти]) и стойността на IC50 (половината от максималната потискаща концентрация на изпитвания химикал [антагонисти]) се определят от данните за отклика спрямо концентрацията. За изпитваните химикали, които са положителни в една или повече концентрации, концентрацията на изпитвания химикал, която причинява половината от максималния отклик (IC50 или EC50), се изчислява, като се използва анализ с функцията на Хил или друг подходящ алтернативен метод. Функцията на Хил е логистичен математически модел с четири параметъра, свързващ концентрацията на изпитвания химикал с отклика (обикновено се следва „S“-образна крива) като използва уравнението по-долу:
|

Където:
Y= отклик (т.е. RLU);
X= логаритъмът от концентрацията;
Най-ниско ниво (Bottom)= минималният отклик;
Най-високо ниво (Top)= максималният отклик;
lg EC50 (или lg IC50)= логаритъмът от X като отклик по средата между най-високото и най-ниското ниво;
Hillslope= наклонът на кривата.
Моделът изчислява най-подходящата стойност за параметрите „Най-високо ниво“, „Най-ниско ниво“, „Hillslope“, IC50 и EC50. За изчисляването на стойностите за EC50 и IC50 следва да се използва подходящ софтуер за статистически изчисления (напр. Graphpad PrismR).
Определяне на силно отклоняващи се стойности
|
35.
|
Добрата статистическа преценка може да бъде улеснена чрез включване (но не само) на Q-test (вж. протоколите с агонисти и антагонисти (7) за определяне на „неизползваемите“ ямки, които ще бъдат изключени от анализа на данните.
|
|
36.
|
За повторенията на референтния еталон E2 (двоен размер на пробата), всяка коригирана стойност на RLU за повторение при дадена концентрация на E2 се разглежда като силно отклоняваща се стойност, ако стойността ѝ е с повече от 20 % над или под коригираната стойност на RLU за тази концентрация в базата данни от предходни периоди.
|
Събиране и коригиране на данните от луминометъра за изпитването за определяне на обхват
|
37.
|
Необработените данни от луминометъра следва да бъдат прехвърлени в шаблон на електронна таблица, разработен специално за изследването. След това трябва да се определи дали има точки с данни със силно отклоняващи се стойности, които да бъдат премахнати. (Относно параметрите, които се определят в рамките на анализите, вж. критериите за приемане на изпитване.) Следва да се извършат следните изчисления:
|
Агонист
|
Стъпка 1
|
Изчислява се средната стойност за контролата на носителя (VC) DMSO.
|
|
Стъпка 2
|
Изважда се средната стойност на DMSO VC от стойността за всяка ямка за нормализиране на данните.
|
|
Стъпка 3
|
Изчислява се средната стойност за кратната индукция за референтния еталон (E2).
|
|
Стъпка 4
|
Изчислява се средната стойност на EC50 за изпитваните химикали.
|
Антагонист
|
Стъпка 1
|
Изчислява се средната стойност за DMSO VC.
|
|
Стъпка 2
|
Изважда се средната стойност на DMSO VC от стойността за всяка ямка за нормализиране на данните.
|
|
Стъпка 3
|
Изчисляване на средната стойност за кратната индукция за референтния еталон (Ral/E2).
|
|
Стъпка 4
|
Изчисляване на средната стойност за референтния еталон E2.
|
|
Стъпка 5
|
Изчисляване на средната стойност наIC50 за изпитваните химикали.
|
Събиране и коригиране на данните от луминометъра за цялостното изпитване
|
38.
|
Необработените данни от луминометъра следва да бъдат прехвърлени в шаблон на електронна таблица, разработен специално за изследването. След това трябва да се определи дали има точки с данни със силно отклоняващи се стойности, които да бъдат премахнати. (Относно параметрите, които се определят в рамките на анализите, вж. критериите за приемане на изпитване.) Следва да се извършат следните изчисления:
|
Агонист
|
Стъпка 1
|
Изчислява се средната стойност за DMSO VC.
|
|
Стъпка 2
|
Изважда се средната стойност на DMSO VC от стойността за всяка ямка за нормализиране на данните.
|
|
Стъпка 3
|
Изчислява се средната стойност за кратната индукция за референтния еталон (E2).
|
|
Стъпка 4
|
Изчисляване на средната стойност на EC50 за Е2 и изпитваните химикали.
|
|
Стъпка 5
|
Изчисляване на средната коригирана RLU стойност за метоксихлор.
|
Антагонист
|
Стъпка 1
|
Изчислява се средната стойност за DMSO VC.
|
|
Стъпка 2
|
Изважда се средната стойност на DMSO VC от стойността за всяка ямка за нормализиране на данните.
|
|
Стъпка 3
|
Изчислява се средната стойност за кратната индукция за референтния еталон (Ral/E2).
|
|
Стъпка 4
|
Изчислява се средната стойност на IC50 за Ral/E2 и изпитваните химикали.
|
|
Стъпка 5
|
Изчислява се средната коригирана стойност на RLU за тамоксифен.
|
|
Стъпка 6
|
Изчислява се средната стойност за референтния еталон E2.
|
Критерии за тълкуване на данните
|
39.
|
Изследването VM7Luc ER TA е предвидено да бъде част от подхода, основан на определяне на тежестта на доказателствата, който спомага за приоритизирането на веществата за изпитванията in vivo на нарушителите на функциите на ендокринната система. Част от тази процедура за приоритизиране е класифицирането на изпитвания химикал като положителен или отрицателен в активността му на агонист или антагонист на ER. Критериите за определяне на химикала като положителен или отрицателен, използвани в рамките на изследването за валидиране VM7Luc ER TA, са описани в таблица 1.
|
Таблица 1
Критерии за вземане на положително или отрицателно решение
|
|
|
АКТИВНОСТ НА АГОНИСТ
|
|
За положително решение
|
|
—
|
Кривата концентрация-отклик на всички изпитвани химикали, класифицирани като положителни за активност на агонист на ER, следва да се състои от основна линия, последвана от положителен наклон и завършваща с плато или с пик. В някои случаи могат да бъдат дефинирани само две от тези характеристики (основна линия–възходящ наклон или възходящ наклон–пик).
|
|
—
|
Линията, дефинираща положителния наклон, следва да съдържа най-малко три точки с неприпокриващи се ленти на грешка (средна стойност ± SD). Точките, образуващи основната линия се изключват, но линейният участък от кривата може да включва пика или първата точка от платото.
|
|
—
|
За положителна квалификация се изисква амплитуда на отклика (разликата между основната линия и пика) от най-малко 20 % от максималната стойност за референтния еталон E2 (т.е. 2000 RLU или повече, когато максималната стойност на отклика на референтните еталони [E2] е коригирана до 10000 RLU).
|
|
—
|
Ако е възможно, стойността EC50 следва да бъде изчислена за всеки положителен изпитван химикал.
|
|
|
За отрицателно решение
|
Средната коригирана стойност на RLU за дадена концентрация е равна на средната стойност на RLU за контролата с DMSO плюс три пъти стандартното отклонение на RLU за DMSO, или по-ниска.
|
|
Непригоден
|
Данни, които не могат да бъдат интерпретирани като валидни за демонстриране на наличието или отсъствието на активност, поради съществени качествени или количествени ограничения и се разглеждат като непригодни, и не могат да бъдат използвани за определяне на изпитвания химикал като положителен или отрицателен. Химикалите следва да бъдат изпитани повторно.
|
|
|
|
АКТИВНОСТ НА АНТАГОНИСТ
|
|
За положително решение
|
|
—
|
Данните за изпитвания химикал съставят крива на концентрация-отклик, състояща се от основна линия, която е последвана от крива с отрицателен (низходящ) наклон.
|
|
—
|
Линията, дефинираща отрицателния наклон, следва да съдържа най-малко три точки с неприпокриващи се ленти на грешка; Точките, образуващи основната линия, се изключват, но линейният участък от кривата може да включва първата точка от платото.
|
|
—
|
Трябва да има най-малко 20 % намаление в активността от максималната стойност на референтния еталон Ral/E2 (т.е. 8000 RLU или по-малко, когато максималната стойност на отклика на референтния еталон [Ral/E2] е коригирана до 10000 RLU).
|
|
—
|
Най-високите нетоксични концентрации на изпитвания химикал следва да бъдат равни на 1x10-5 M или по-ниски.
|
|
—
|
Ако е възможно, стойността IC50 следва да бъде изчислена за всеки положителен изпитван химикал.
|
|
|
За отрицателно решение
|
Всички точки с данни са над стойността на EC80 (80 % от отклика на E2 или 8000 RLU), при концентрации, по-ниски от 1,0 x 10-5 M.
|
|
Непригодни
|
Данните, които не могат да се интерпретират като валидни за доказване на наличието или отсъствието на активност, поради съществени качествени или количествени ограничения, се разглеждат като непригодни, и не могат да бъдат използвани за определяне на изпитвания химикал като положителен или отрицателен. Химикалът следва да бъде изпитан повторно.
|
|
40
|
Когато е възможно, положителните резултати се характеризират както от големината на ефекта, така и от концентрацията, при която възниква този ефект. Примери за положителни, отрицателни и непригодни данни са показани на фигури 5 и 6.
|
Фигура 5
Примери за агонисти: Положителни, отрицателни и непригодни данни

Пунктирната линия означава 20 % от отклика на E2, 2000 коригирани и нормализирани RLU.
Фигура 6
Примери за антагонисти: Положителни, отрицателни и непригодни данни

Пунктирната линия означава 80% от отклика на Ral/E2, 8000 коригирани и нормализирани RLU.
Плътната линия означава 1,00 x 10-5 M. За да се разглежда даден отклик като положителен, той следва да бъде под линията за 8000 RLU, и с концентрация, по-ниска от 1,00 x 10-5M.
Концентрациите, обозначени със звезда в графиката с мезо-хексестрол, означават степен на жизнеспособност от „2“ или по-висока.
Резултатите от изпитването на мезо-хексестрол се разглеждат като непригодни данни, защото единственият отклик, който е под 8000 RLU, възниква при 1,00 x 10-5M.
|
41.
|
Изчисляването на EC50 и IC50 може да бъде направено с помощта на функция на Хил с четири параметъра (за повече подробности, вж. протокола с агонисти и антагонисти (7)). Изпълнението на критериите за приемливост показва, че системата функционира правилно, но не гарантира, че всяка конкретна серия ще даде точни данни. Дуплицирането на резултатите от първата серия е най-добрата гаранция, че се получават точни данни (вж. точка 19 от “КОМПОНЕНТИ на ER TA изследването“).
|
ПРОТОКОЛ ОТ ИЗПИТВАНЕТО
|
42.
|
Вж. параграф 20 на „КОМПОНЕНТИ НА АНАЛИЗА ER TA“.
|
ЛИТЕРАТУРА
|
(1)
|
ICCVAM: 2011) ICCVAM Test Method Evaluation Report on the LUMI-CELL® ER (BG1Luc ER TA) Test Method: An In Vitro Method for Identifying ER Agonists and Antagonists, National Institute of Environmental Health Sciences: Research Triangle Park, NC.
|
|
(2)
|
Monje P., Boland R. (2001). Subcellular Distribution of Native Estrogen Receptor α and β Isoforms in Rabbit Uterus and Ovary, J. Cell Biochem., 82(3): 467-479.(3) Pujol P., et al. (1998). Differential
|
|
(3)
|
Expression of Estrogen Receptor-Alpha and -Beta Messenger RNAs as a Potential Marker of Ovarian Carcinogenesis, Cancer Res., 58(23): 5367-5373. Weihua Z., et al. (2000). Estrogen
|
|
(4)
|
Receptor (ER) β, a Modulator of ERα in the Uterus, Proceedings of the National Academy of Sciences of the United States of America 97(11): 936-5941.
|
|
(5)
|
Balls M., et al. (2006). The Importance of Good Cell Culture Practice (GCCP), ALTEX, 23(Suppl): p. 270-273.
|
|
(6)
|
Coecke S., et al. (2005). Guidance on Good Cell Culture Practice: a Report of the Second ECVAM Task Force on Good Cell Culture Practice, Alternatives to Laboratory Animals, 33: p. 261-287.
|
|
(7)
|
ICCVAM (2011). ICCVAM Test Method Evaluation Report, The LUMI-CELL® ER (BG1Luc ER TA) Test Method: An In Vitro Assay for Identifying Human Estrogen Receptor Agonist and Antagonist Activity of Chemicals, NIH Publication No 11-7850.
|
|
(8)
|
Rogers J.M., Denison M.S. (2000). Recombinant Cell Bioassays for Endocrine Disruptors: Development of a Stably Transfected Human Ovarian Cell Line for the Detection of Estrogenic and Anti-Estrogenic Chemicals, In Vitro Mol. Toxicol.,13(1):67-82.
|
|
(9)
|
Escande A., et al. (2006). Evaluation of Ligand Selectivity Using Reporter Cell Lines Stably Expressing Estrogen Receptor Alpha or Beta, Biochem. Pharmacol., 71(10):1459-69.
|
|
(10)
|
Thorne N., Inglese J., Auld D.S. (2010). Illuminating Insights into Firefly Luciferase and Other Bioluminescent Reporters Used in Chemical Biology, Chemistry and Biology,17(6):646-57.
|
|
(11)
|
Kuiper G.G, et al. (1998). Interaction of Estrogenic Chemicals and Phytoestrogens with Estrogen Receptor Beta, Endocrinology,139(10):4252-63.
|
|
(12)
|
Geisinger, et al. (1989). Characterization of a human ovarian carcinoma cell line with estrogen and progesterone receptors, Cancer 63, 280-288.
|
|
(13)
|
Baldwin, et al. (1998). BG-1 ovarian cell line: an alternative model for examining estrogen-dependent growth in vitro, In Vitro Cell. Dev. Biol. – Animal, 34, 649-654.
|
|
(14)
|
Li, Y., et al. (2014). Research resource: STR DNA profile and gene expression comparisons of human BG-1 cells and a BG-1/MCF-7 clonal variant, Mol. Endo. 28, 2072-2081.
|
|
(15)
|
Rogers, J.M. and Denison, M.S. (2000). Recombinant cell bioassays for endocrine disruptors:development of a stably transfected human ovarian cell line for the detection of estrogenicand anti-estrogenic chemicals, In Vitro & Molec. Toxicol. 13, 67-82.
|
Допълнение 4
ИЗСЛЕДВАНЕ ЗА ТРАНСАКТИВАЦИЯ ЧРЕЗ СТАБИЛНО ТРАНСФЕКТИРАНЕ НА ЧОВЕШКИ ЕСТРОГЕНЕН РЕЦЕПТОР-Α С ЦЕЛ ОТКРИВАНЕ НА АКТИВНОСТ НА ЕСТРОГЕННИ АГОНИСТИ И АНТАГОНИСТИ НА ХИМИКАЛИ С ИЗПОЛЗВАНЕ НА КЛЕТЪЧНАТА ЛИНИЯ ERΑ CALUX
ПЪРВОНАЧАЛНИ СЪОБРАЖЕНИЯ И ОГРАНИЧЕНИЯ (ВЖ. СЪЩО „ОБЩО ВЪВЕДЕНИЕ“)
|
1.
|
Настоящето изследване за трансактивация на ERα CALUX използва човешката клетъчната линия U2OS за откриване на активност на естрогенен агонист и антагонист, медиирана от човешки естрогенен рецептор алфа (hERα). Изследването за валидиране на биологичното изследване на стабилно трансфектиран ERα CALUX от BioDetection Systems BV (Амстердам, Нидерландия) доказа относимостта и надеждността на изследването за предвидената цел (1). Клетъчната линия ERα CALUX експресира само стабилно трансфектиран човешки ERα (2) (3).
|
|
2.
|
Настоящото изследване е специално разработено за откриване на hERα-медиирана трансактивация чрез измерване на биолуминесценцията като крайна точка. Използването на биолуминесценцията в биоизследванията е обичайно поради високото ѝ съотношение сигнал/шум (4).
|
|
3.
|
Докладвано е, че концентрации на фитоестроген, по-високи от 1 μM, предизвикват свръхактивиране на луциферазния репортерен ген, което води до луминесценция, която не е медиирана от рецептори (5) (6) (7). Ето защо по-високите концентрации на фитоестрогени или други подобни съединения, които могат да предизвикат свръхактивиране на експресията на луцифераза, трябва внимателно да се изследват при изследванията за трансактивация на стабилно трансфектирани ER (вж. допълнение 2).
|
|
4.
|
Разделите „ОБЩО ВЪВЕДЕНИЕ“ и „КОМПОНЕНТИ НА ИЗСЛЕДВАНЕТО ER TA“ следва да бъдат прочетени, преди да се използва настоящото изследване за регулаторни цели. Използваните определения и съкращения в рамките на настоящия метод за изпитване са описани в допълнение 1.
|
ПРИНЦИП НА ИЗСЛЕДВАНЕТО (ВЖ. СЪЩО „ОБЩО ВЪВЕДЕНИЕ“)
|
5.
|
Биоизследването се използва за оценяване на свързването на ER лиганда и последващата транслокация на рецептор-лигандния комплекс в ядрото. В клетъчното ядро, комплексът рецептор-лиганд се свързва със специфични елементи на ДНК отклик и трансактивира луциферазния репортерен ген на светулка, което води до повишена клетъчна експресия на ензима луцифераза. След добавяне на луциферазния субстрат луцеферин, последният се трансформира в биолуминесцентен продукт. Предизвиканата светлина може лесно да бъде открита и определена количествено с използване на луминометър.
|
|
6.
|
Изпитвателната система използва стабилно трансфектирани клетки ERα CALUX. Клетките ERα CALUX произлизат от клетъчната линия U2OS на човешки остеобластен остеосарком. Човешките клетки U2OS бяха стабилно трансфектирани с 3xHRE-TATA-Luc и pSG5-neo-hERα по метода на копреципитация с калциев фосфат. Клетъчната линия U2OS бе избрана като най-добър кандидат, който да служи като чувствителна към естроген (и други стероидни хоромони) репортерна клетъчна линия, въз основа на наблюденията, че клетъчната линия U2OS има незначителна или никаква активност като ендогенен рецептор. Отсъствието на ендогенни рецептори бе оценено единствено с използването на репортерни плазмиди за луцифераза, които не показаха активност, когато бяха добавени рецепторните лиганди. Освен това тази клетъчна линия поддържаше силни медиирани от хормони отклици, когато случайно бяха въвеждани сродни рецептори (2) (3) (8).
|
|
7.
|
Изпитването на химикали за естрогенна и анти-естрогенна активност с използване на клетъчната линия ERα CALUX включва предварителна скринингова серия и цялостна серия. По време на предварителната скринингова серия се определят разтворимостта, цитотоксичността и оптимизираните стойности на кривата концентрация-обхват на изпитваните химикали за цялостно изпитване. В сериите на цялостно изпитване оптимизираните стойности на кривата концентрация-обхват на изпитваните химикали се изпитват в рамките на биоизследванията с ERα CALUX, след което изпитваните химикали се класифицират според техния агонизъм или антагонизъм.
|
|
8.
|
Критериите за тълкуване на данните са описани подробно в точка 59. Накратко, даден изпитван химикал се разглежда като положителен за агонизъм в случай че най-малко две последователни концентрации на изпитвания химикал покажат отклик, който е равен или по-висок от 10 % от максималния отклик на референтния еталон 17β-естрадиол (PC10). Изпитван химикал се разглежда като положителен за антагонизъм в случай че най-малко две последователни концентрации на изпитвания химикал покажат отклик, който е равен или по-нисък от 80 % от максималния отклик на референтния еталон тамоксифен (PC80).
|
ПРОЦЕДУРА
Клетъчни линии
|
9.
|
За изследването следва да се използва стабилно трансфектираната клетъчна линия U2OS ERα CALUX. Клетъчната линия може да се набави от BioDetection Systems BV, Амстердам, Нидерландия чрез техническо лицензионно споразумение.
|
|
10.
|
Следва да се използват само клетъчни култури, които не съдържат микоплазма. Използваните клетъчни партиди следва да бъдат сертифицирани като отрицателни за замърсяване с микоплазма или преди употребата им да се извърши изпитване за микоплазма. RT-PCR (полимеразната верижна реакция в реално време) следва да се използва като чувствителен метод за откриване на инфекции с микоплазма (9).
|
Стабилност на клетъчната линия
|
11.
|
За да се поддържа стабилността и целостта на CALUX клетките, те следва да бъдат съхранявани в течен азот (-800C). След размразяване на клетките за стартиране на нова култура, клетките следва да бъдат субкултивирани най-малко два пъти, преди да бъдат използвани за оценяване на естрогенната агонистична и антагонистична активност на химикали. Клетките не следва да бъдат субкултивирани за повече от 30 пасажа.
|
|
12.
|
За да се следи стабилността на клетъчната линия във времето, следва да се провери способността за реагиране на системата за изпитване на агонистична и антагонистична активност, като се оцени EC50 или IC50 на референтния еталон. В допълнение следва да се наблюдава относителната индукция на пробата на положителната контрола (ПК) и на пробата на отрицателната контрола (NC). Резултатите следва да бъдат в съответствие с критериите за приемане за биоизследване с агонисти (таблица 3В) или антагонисти с ERα CALUX (таблица 4В). Референтните еталони, положителните и отрицателните контроли са дадени в таблица 1 и таблица 2 съответно за модела на изследване с антагонист и този на изследване с агонист.
|
Клетъчна култура и условия на плаките за изследване
|
13.
|
Клетките U2OS следва да бъдат култивирани в среда на растеж (среда DMEM/F12 (1:1) с фенолно червено като индикатор на pH, допълнена с фетален волски серум (7,5 %), аминокиселини, различни от незаменимите (1 %), 10 единици/ml пеницилин, стрептомицин и генетицин (G-418) като маркер за селекция). Клетките следва да бъдат поставени в инкубатор с CO2 (5 % CO2) при 370C и 100 % влажност. Когато се достигнат 85-95 % сливане, клетките следва или да бъдат субкултивирани, или да бъдат приготвени за посяване в 96-ямкови микротитърни плаки. В последния случай, клетките следва да бъдат повторно суспендирани при концентрация от 1x105 клетки/ml в среда за изследване без съдържание на естроген (среда DMEM/F12 (1:1) без фенолно червено, допълнена с фетален волски серум, третиран с въглен с покритие от декстран (5 % v/v), аминокиселини, различни от незаменимите (1 % v/v), 10 единици/ml пеницилин и стрептомицин), и разпределени в ямките на 96-ямковите микротитърни плаки (100 μl хомогенизирана клетъчна суспензия). Клетките следва да бъдат предварително инкубирани в инкубатор с CO2 (5 % CO2, 370 C, 100 % влажност) за 24 часа преди експозицията. Пластмасовите пособия не трябва да съдържат естроген.
|
Критерии за приемливост
|
14.
|
Агонистичната и антагонистичната активност на изпитвания(-те) химикал(и) се изпитва в рамките на изпитвателната серия. Изпитвателната серия се състои от максимум 6 микротитърни плаки. Всяка изпитвателна серия съдържа най-малко 1 пълна серия от разреждания на референтния еталон, проба на положителната контрола, проба на отрицателната контрола и контроли на разтворителя. Фигура 1 и 2 показват разпределението на плаките за изпитвателните серии с агонистична и антагонистична активност.
|
|
15.
|
Всяко разреждане на референтните еталони, изпитваните химикали, всички контроли на разтворители и положителните и отрицателните контроли следва да бъдат трикратно анализирани. Всеки от трикратните анализи следва да изпълнява изискванията, посочени в таблица 3A и таблица 4A.
|
|
16.
|
Пълна серия на разреждания на референтния еталон (17β-естрадиол за агонизъм; тамоксифен за антагонизъм) се измерва на първата плака във всяка изпитвана серия. За да могат да се сравняват резултатите от анализа на останалите 5 микротитърни плаки с първата микротитърна плака, съдържаща цялата крива концентрация-отклик на референтния еталон, всички плаки следва да съдържат 3 контролни проби: контрола на разтворителя, най-висока изпитана концентрация на референтния еталон и приблизителната EC50 (агонизъм), или IC50 (антагонизъм) концентрация на референтния еталон. Съотношението на усреднените стойности от контролните проби от първата плака и оставащите 5 плаки следва да изпълнява изискванията, дадени в таблица 3В (агонизъм) или таблица 4В (антагонизъм).
|
|
17.
|
За всяка от микротитърните плаки от изпитваната серия се изчислява z-коефициентът (10). Z-коефициентът се изчислява, като се използват отклиците на най-високата и най-ниската концентрация на референтния еталон. За валидна се смята микротитърна плака, която изпълнява изискванията, посочени в таблица 3В (агонизъм) или таблица 4В (антагонизъм).
|
|
18.
|
Референтният еталон следва да показва „S“-образна крива доза-отклик. Стойностите за EC50 или IC50, получени от отклика на серията разреждания на референтния еталон, следва да изпълняват изискванията, указани в таблица 3В (агонизъм) или таблица 4C (антагонизъм).
|
|
19.
|
Всяка изпитвана серия следва да съдържа проба за положителна контрола и проба за отрицателна контрола. Изчислената относителна индукция на двете проби за положителна и за отрицателна контрола следва да изпълнява изискванията, указани в таблица 3В (агонизъм) или таблица 4C (антагонизъм).
|
|
20.
|
По време на всички измервания следва да се измерва коефициентът на индукция на най-високата концентрация на референтния еталон, като се раздели средният отклик в относителни светлинни единици (RLU) на най-високата концентрация на референтния еталон 17β-естрадиол на средния отклик в RLU на контрола на референтния разтворител. Този коефициент на индукция следва да изпълнява минималните изисквания за кратна индукция, указани в таблица 3В (агонизъм) или таблица 4C (антагонизъм).
|
|
21.
|
Единствено микротитърните плаки, които изпълняват всички посочени по-горе критерии за приемане, се разглеждат като валидни и могат да се използват за оценяване на отклика на изпитваните химикали.
|
|
22.
|
Критериите за приемане са приложими за предварителните скринингови изпитвания и за цялостните изпитвания.
|
Таблица 1
Концентрации на референтния еталон, положителната контрола (ПК) и отрицателната контрола (NC) за биоизследването на CALUX с агонисти
|
|
Вещество
|
CAS №
|
Обхват на изпитване (M)
|
|
Референтен еталон
|
17β-естрадиол
|
50-28-2
|
1*10–13 –1*10–10
|
|
положителна контрола (ПК)
|
17α-метилтестостерон
|
58-18-4
|
3*10–6
|
|
отрицателна контрола (NC)
|
кортикостерон
|
50-22-6
|
1*10–8
|
Таблица 2
Концентрации на референтния еталон, положителната контрола (ПК) и отрицателната контрола (NC) за биоизследването на CALUX с антагонисти
|
|
Вещество
|
CAS №
|
Обхват на изпитване (M)
|
|
Референтен еталон
|
тамоксифен
|
10540-29-1
|
3*10–9 – 1*10–5
|
|
положителна контрола (ПК)
|
4-хидрокситамоксифен
|
68047-06-3
|
1*10–9
|
|
отрицателна контрола (NC)
|
ресвератрол
|
501-36-0
|
1*10–5
|
Таблица 3
Критерии за приемане за биоизследването на CALUX с агонисти на ERα
|
A - отделни проби в плака
|
Критерий
|
|
1
|
Максимално стандартно отклонение (%SD) на ямките за трикратно изпитване (за NC, ПК, всяко разреждане на изпитвания химикал и референтния еталон, с изключение на C0)
|
< 15 %
|
|
2
|
Максимално стандартно отклонение (%SD) на ямките за трикратно изпитване (за референтния еталон и контролите на разтворителя на изпитвания химикал (C0, SC))
|
< 30 %
|
|
3
|
Максимално изпускане на LDH, като мярка за цитотоксичност.
|
< 120 %
|
|
B - в рамките на единична микротитърна плака
|
|
|
4
|
Съотношение на контрола на разтворителя на референтния еталон (C0; плака 1) и контрола на разтворителя на изпитвания химикал (SC; плаки 2 до x)
|
0,5 до 2,0
|
|
5
|
Съотношение на прибл. EC50 и най-високите концентрации на референтния еталон в плака 1 и прибл. EC50 и най-високите концентрации на референтния еталон в плаки 2 до x (C4, C8)
|
0,70 до 1,30
|
|
6
|
Z-коефициент за всяка плака
|
>0,6
|
|
C - в рамките на единична серия на анализ (всички плаки в една серия)
|
|
|
7
|
Сигмоидална крива на референтния еталон
|
Да (17ß-естрадиол)
|
|
8
|
EC50 обхват на референтния еталон 17ß-естрадиол
|
4*10–12 – 4*10–11 M
|
|
9
|
Минимална кратна индукция на най-високата концентрация на 17ß-естрадиол, във връзка с контрола на разтворителя на референтния еталон.
|
5
|
|
10
|
Относителна индукция (%) ПК.
|
> 30 %
|
|
11
|
Относителна индукция (%) ПК
|
<10 %
|
Прибл.: приблизително; ПК: положителна контрола; NC: отрицателна контрола; SC: контрола на разтворителя на изпитвания химикал; C0: контрола на разтворителя на референтния еталон; SD: стандартно отклонение; LDH: лактат дехидрогеназа
Таблица 4
Критерии за приемане за биоизследването с CALUX с антагонисти на ERα
|
A - отделни проби в плака
|
Критерий
|
|
1
|
Максимално стандартно отклонение (%SD) на ямките за трикратно изпитване (за NC, ПК, всяко разреждане на изпитвания химикал и референтния еталон, контрол на разтворителя (C0))
|
< 15 %
|
|
2
|
Максимално стандартно отклонение (%SD) на ямките за трикратно изпитване (за контрол на носителя (VC) и най-висока концентрация на референтния еталон (C8))
|
< 30 %
|
|
3
|
Максимално изпускане на LDH, като мярка за цитотоксичност.
|
< 120 %
|
|
B - в рамките на единична микротитърна плака
|
|
|
4
|
Съотношение на контрола на разтворителя на референтния еталон (C0; плака 1) и контрола на разтворителя на изпитвания химикал (SC; плаки 2 до x)
|
0,70 до 1,30
|
|
5
|
Съотношение на прибл. IC50 на концентрациите на референтния еталон в плака 1 и прибл. IC50 на концентрациите на референтния еталон в плаки 2 до x (C4)
|
0,70 до 1,30
|
|
6
|
Съотношение на най-високите концентрации на референтния еталон в плака 1 и най-високите концентрации на референтния еталон в плаки 2 до x (C8)
|
0,50 до 2,0
|
|
7
|
Z-коефициент за всяка плака
|
>0,6
|
|
C - в рамките на единична серия на анализ (всички плаки в една серия)
|
|
|
8
|
Сигмоидална крива на референтния еталон
|
Да (Тамоксифен)
|
|
9
|
IC50 обхват на референтния еталон (Тамоксифен)
|
1*10–8 - 1*10–7 M
|
|
10
|
Минимална кратна индукция на контрола на разтворителя на референтния еталон във връзка с най-високата концентрация на Тамоксифен.
|
2,5
|
|
11
|
Относителна индукция (%) ПК.
|
<70 %
|
|
12
|
Относителна индукция (%) ПК
|
>85 %
|
Прибл.: приблизително; ПК: положителна контрола; NC: отрицателна контрола; VC: контрол на носителя (контрол на разтворителя без фиксираната концентрация на референтния еталон за агонисти); SC: контрола на разтворителя на изпитвания химикал; C0: контрола на разтворителя на референтния еталон; SD: стандартно отклонение; LDH: лактат дехидрогеназа
Контрол на разтворителя/носителя, референтни еталони, положителни контроли, отрицателни контроли
|
23.
|
Едни и същи контроли на разтворителя/носителя, референтни еталони, положителни контроли, отрицателни контроли следва да се използват както за предварителната скринингова серия, така и за сериите за цялостно изпитване. В допълнение, концентрацията на референтните еталони, положителните контроли и отрицателните контроли, следва да бъде една и съща.
|
Контрола на разтворителя
|
24.
|
Разтворителят, който е използван за разтваряне на изпитваните химикали, следва да бъде изпитан като контрола на разтворителя. Диметилсулфоксид (DMSO, 1 % (v/v); CASRN 67-68-5) се използва като носител по време на валидирането на биоизследването с CALUX за ERα. Ако се използва разтворител, различен от DMSO, всички референтни еталони, контроли и изпитвани химикали следва да бъдат изпитани в същия носител. Не трябва да се забравя, че контролата на разтворителя за изследванията с антагонисти съдържа фиксирана концентрация на референтния еталон за агонисти 17β-естрадиол (с концентрация приблизително EC50 ). За да се изпита разтворителят, използван за изследванията с агонисти, трябва да се приготви и изпита контрола на носителя.
|
Контрола на носителя (антагонизъм)
|
25.
|
За изпитване за антагонизъм, средата на изследването се допълва с фиксирана концентрация на референтния еталон за агонисти 17β-естрадиол (приблизително EC50 концентрация). За да се изпита разтворителят, използван за разтваряне на изпитваните химикали за антагонизъм, следва да се приготви среда на изследването без да се допълва с фиксирана концентрация на референтния еталон за агонисти 17β-естрадиол. Тази контролна проба е обозначена като контрола на носителя. Диметилсулфоксид (DMSO, 1 % (v/v); CASRN 67-68-5) се използва като носител по време на валидирането на биоизследването с CALUX за ERα. Ако се използва разтворител, различен от DMSO, всички референтни еталони, контроли и изпитвани химикали следва да бъдат изпитани в същия носител.
|
Референтни еталони
|
26.
|
Агонистичният референтен еталон е 17β-естрадиол (таблица 1). Референтните стандарти обхващат серия от разреждания на осем концентрации на 17β-естрадиол (1*10–13, 3*10–13, 1*10–12, 3*10–12, 6*10–12, 1*10–11, 3*10–11, 1*10–10 M).
|
|
27.
|
Антагонистичният референтен еталон е тамоксифен (таблица 2). Референтните стандарти обхващат серия от разреждания на осем концентрации на тамоксифен (3*10–9, 1*10–8, 3*10–8, 1*10–7, 3*10–7, 1*10–6, 3*10–6, 1*10–5 M). Всяка от концентрациите на антагонистичния референтен еталон се ко-инкубира с фиксирана концентрация на агонистичния референтен еталон 17β-естрадиол (3*10–12 M).
|
Положителна контрола
|
28.
|
Положителната контрола в агонистичните проучвания е 17α-метилтестостерон (таблица 1).
|
|
29.
|
Положителната контрола в антагонистичните проучвания е 4-хидрокситамоксифен (таблица 2). Положителната контрола за антагонизъм се коинкубира с фиксирана концентрация на агонистичния референтен еталон 17β-естрадиол (3*10–12 M).
|
Отрицателна контрола
|
30.
|
Отрицателната контрола в проучванията за агонизъм е кортикостерон (таблица 1).
|
|
31.
|
Отрицателната контрола в проучванията за антагонизъм ресвератрол (таблица 2). Отрицателната контрола за антагонизъм се коинкубира с фиксирана концентрация на агонистичния референтен еталон 17β-естрадиол (3*10–12 M).
|
Доказване на пригодността на лабораторията (вж. точка 14 и таблици 3 и 4 в «КОМПОНЕНТИ НА ER TA ИЗСЛЕДВАНЕТО» от настоящия метод за изпитване)
Носител
|
32.
|
Разтворителят, използван за разтваряне на изпитваните химикали следва да разтваря напълно изпитвания химикал и следва да може да се смесва с клетъчната среда. Подходящи разтворители са DMSO, вода и етанол (с чистота от 95 % до 100 %). В случай, че като разтворител се използва DMSO, максималната концентрация на DMSO по време на инкубация не трябва да превишава 1 % (v/v). Преди употреба, разтворителят следва да бъде изпитан за липса на цитотоксичност и влияние върху ефективността на изследванията.
|
Подготовка на референтните еталони, положителните контроли, отрицателните контроли и изпитваните химикали
|
33.
|
Референтните еталони, положителните контроли, отрицателните контроли и изпитваните химикали се разтварят в 100 % DMSO (или друг подходящ разтворител). След това трябва да се приготвят подходящи (серийни) разреждания в същия разтворител. Преди разтваряне всички изпитвани химикали следва да бъдат оставени да се аклиматизират към стайната температура. Прясно приготвените изходни разтвори на референтните еталони, положителните контроли, отрицателните контроли и изпитваните химикали не трябва да имат забележими признаци на преципитат или помътняване. Основните разтвори на референтните еталони и контролите могат да бъдат приготвени в по-големи количества. Изходните разтвори на изпитваните химикали следва да бъдат прясно приготвени преди всеки експеримент. Окончателните разреждания на референтния еталон, положителните контроли, отрицателните контроли и изпитваните химикали следва да бъдат прясно приготвени за всеки експеримент и използвани в рамките на 24 часа след приготвянето.
|
Разтворимост, цитотоксичност и определяне на обхват.
|
34.
|
По време на предварителната скринингова серия се определя разтворимостта на изпитваните химикали в избрания разтворител. Приготвя се основен разтвор с максимална концентрация от 0,1 M. В случай, че тази концентрация покаже проблеми с разтворимостта, следва да се приготвят изходни разтвори с по-ниска концентрация, докато изпитваните химикали се разтворят напълно. По време на предварителната скринингова серия се изпитват серийни разреждания от 1:10 на изпитвания химикал. Максималната концентрация на изследването за изпитване на агонисти или антагонисти е 1 mM. След предварителния скрининг се получава подходящ уточнен обхват на концентрацията за изпитваните химикали, който следва да се изпита по време на цялостните серии. Използваните разреждания за цялостното изпитване следва да бъдат 1x, 3x, 10x, 30x, 100x, 300x, 1000x и 3000x.
|
|
35.
|
Изпитването за цитотоксичност е включено в протокола на изследването с агонисти и антагонисти (11). Изпитването за цитотоксичност е част от серията за предварителен скрининг и от сериите за цялостно изпитване. Използваният метод за оценяване на цитотоксичността по време на валидирането на биоизследването на CALUX за ERα е изпитване на изпускане на лактат дехидрогеназа (LDH) в комбинация с качествена визуална инспекция на клетките (вж. допълнение 4.1) след излагане на изпитваните химикали. Въпреки това могат да се използва и други количествени методи за определяне на цитотоксичност (напр. колориметрично изследване базирано на тетразолиум (MTT) или биоизследване на CALUX за цитотоксичност). Като цяло, концентрации на изпитван химикал, които показват повече от 20 % намаляване на клетъчната жизнеспособност се разглеждат като цитотоксични и поради това не могат да се използват за оценка на данни. По отношение на изследването за изпускане на LDH, концентрацията на изпитвания химикал се смята за цитотоксична, когато процентът на изпускане на LDH е по-висок от 120 %.
|
Експозиция на изпитвания химикал и организация на плаката за изследването
|
36.
|
След трипсиниране на конфлуентните клетки в съда за култивиране, клетките се ре-суспендират в съотношение 1x105 клетки/ml в среда за изследване без съдържание на естроген. Сто μl от ре-суспендираните клетки се поставят във вътрешните ямки на 96-ямкова микротитътрна плака. Външните ямки се запълват с 200 μl фосфатно буфериран физиологичен разтвор (PBS) (вж. фигури 1 и 2). Поставените в плаката клетки се пред-инкубират за 24 часа в CO2 инкубатор (5 % CO2, 37°C, 100 % влажност).
|
|
37.
|
След предварителната инкубация, плаките се преглеждат за видими признаци на цитотоксичност (вж. допълнение 4.1), замърсяване и конфлуентност. Единствено плаките, които не показват цитотоксичност, замърсяване и са с минимум 85 % конфлуентност се използват за изпитване. Средата от вътрешните ямки се отстранява внимателно и се заменя с 200 μl среда за изследване без естроген, съдържаща подходящата серия на разреждане на референтните еталони, изпитваните химикали, положителните контроли, отрицателните контроли и контролите на разтворителя (таблица 5: проучвания на агонисти; Таблица 6: проучвания на антагонисти). Всички референтни еталони, изпитвани химикали, положителни контроли, отрицателни контроли и контроли на разтворителя се изпитват трикратно. На фигура1 се вижда разположението на плаката при изпитването с агонисти. На фигура 2 се вижда разположението на плаката при изпитването с антагонисти. Разположението на плаката за предварителното скринингово изпитване и цялостното изпитване е идентично. За изпитването с антагонисти, всички вътрешни ямки, с изключение на ямките за контрола на носителя (VC), съдържат и фиксирана концентрация на референтния еталон за агонисти 17β-естрадиол (3*10–12 M). Не бива да се забравя, че референтните еталони C8 и C4 следва да бъдат добавени към всяка TC плака.
|
|
38.
|
След експозиция на клетките на всички химикали, 96-ямковите микротитърни плаки следва да бъдат инкубирани за още 24 часа в инкубатор с CO2 (5 % CO2, 37°C, 100 % влажност).
|
Фигура 1
Разположение на ямките на 96-ямкови микротитърни плаки за предварителна проверка и оценяване на агонистичния ефект

C0 = разтворител на референтния еталон.
C(1-8) = серия разреждания (1-8, от ниска към висока концентрация) на референтния еталон.
ПК = положителна контрола.
NC = отрицателна контрола.
TCx-(1-8) = разреждания (1-8, от ниска към висока концентрация) на изпитвания химикал за серията за предварителна проверка и оценяване на агонистичния ефект на изпитван химикал x.
SC = контрол на разтворителя на изпитвания химикал (в оптималния вариант, същия разтворител като при C0, но може да е от друга партида).
Сиви клетки: = Външните ямки, запълнени с 200 μl PBS.
Фигура 2
Разположение на ямките на 96-ямкови микротитърни плаки за предварителен скрининг за антагонисти и оценяване на антагонистичния ефект

C0 = разтворител на референтния еталон.
C(1-8) = серия разреждания (1-8, от ниска към висока концентрация) на референтния еталон.
NC = отрицателна контрола.
ПК = положителна контрола.
TCx-(1-8) = разреждания (1-8, от ниска към висока концентрация) на изпитвания химикал за серията за предварителна проверка и оценяване на агонистичния ефект на изпитвания химикал x.
SC = контрол на разтворителя на изпитвания химикал (в оптималния вариант, същият разтворител като при C0, но може да е от друга партида).
VC = контрол на носителя (контрол на разтворителя без фиксираната концентрация на референтния еталон за агонисти 17β-естрадиол).
Сиви клетки: = Външните ямки, запълнени с 200 μl PBS.
Забележка: всички вътрешни ямки, с изключение на ямките за контрола на носителя (VC), съдържат и фиксирана концентрация на референтния еталон за агонисти 17β-естрадиол (3,0*10-12 M)
Измерване на луминесценцията
|
39.
|
Измерването на луминесценцията е описано подробно в протокола за изследване на агонисти и антагонисти (10). Средата от ямките следва да се отстрани и клетките да бъдат лизирани 24 часа след инкубация, за да се отворят клетъчните мембрани, което да позволи измерване на луциферазната активност.
|
|
40.
|
Процедурата за измерване на луминесценцията изисква луминометър, оборудван с 2 инжектора. Луциферазната реакция се активира с инжектирането на субстрата луциферин. Реакцията се спира с добавянето на 0,2 M NaOH. Спирането на реакцията предотвратява преноса на луминесценция от една ямка в друга.
|
|
41.
|
Светлината, излъчена от всяка ямка се изразява в относителни светлинни единици (RLU) на ямка.
|
Предварителна скринингова серия
|
42.
|
Резултатите от предскрининговия анализ се използват за определяне на оптимизираните стойности на кривата концентрация-обхват на изпитваните химикали за цялостното изпитване. Оценяването на резултатите от предскрининговия анализ и определянето на оптимизираните стойности на кривата концентрация-обхват на изпитваните химикали за цялостното изпитване са подробно описани в протокола за изследване на агонисти и антагонисти (10). Тук е представено кратко резюме на процедурите за определяне на обхвата на концентрацията на изпитваните химикали за изпитването на агонисти и антагонисти. Вж. таблици 5 и 6 за насоки относно постановката за серийно разреждане.
|
Избор на концентрации за оценка на агонистичните ефекти
|
43.
|
По време на предварителната скринингова серия, изпитваните химикали следва да бъдат изпитани, като се използва серията от разреждания, описани в таблица 5 (за агонизъм) и 6 (за антагонизъм). Всички концентрации следва да бъдат изпитани трикратно, съобразно разположението на ямките в плаката, както е указано на фигура 1 (агонизъм) или 2 (антагонизъм).
|
|
44.
|
Единствено резултатите от анализа, които изпълняват критериите за приемане (таблица 3) се разглеждат като валидни и могат да се използват за оценяване на отклика на изпитваните химикали. В случай че една или повече микротитърни плаки от една серия за анализ не изпълни критериите за приемане, съответните микротитърни плаки следва да бъдат анализирани повторно. В случай че първата плака, съдържаща пълната серия от разреждания на референтния еталон, не изпълни критериите за приемане, пълната серия за изпитване (6 плаки) трябва да бъде анализирана повторно.
|
|
45.
|
Първоначалните обхвати на концентрацията на изпитваните химикали следва да бъдат коригирани и серията на предварителния скрининг следва да бъде повторена, ако:
|
—
|
бъде забелязана цитотоксичност. Процедурата за предварителен скрининг следва да бъде повторена с по-ниски и нецитотоксични концентрации на изпитвания химикал.
|
|
—
|
предварителният скрининг на изпитвания химикал не покаже пълна крива доза-отклик, защото изпитаните концентрации генерират максимална индукция. Предварителната скринингова серия следва да бъде повторена, като се използват по-ниски концентрации на изпитвания химикал.
|
|
|
46.
|
Когато се наблюдава валиден отклик, свързан с дозата, следва да бъде избрана (най-ниската) концентрация, при която се наблюдава максимална индукция и при която не се проявява цитотоксичност. Най-високата концентрация на изпитвания химикал, която ще бъде изпитвана в рамките на сериите за цялостно изпитване, следва да бъде 3 пъти по-голяма от тази избрана концентрация.
|
|
47.
|
Следва да се подготви пълна серия на оптимизирано разреждане на изпитвания химикал, като се спазват стъпките за разреждане, описани в таблица 5, и се започне от най-високата концентрация, определена по-горе.
|
|
48.
|
Изпитван химикал, който не предизвиква никакъв агонистичен ефект, следва да бъде изпитан в сериите за цялостно изпитване, като се започне от най-високата, нецитотоксична концентрация, определена по време на предварителното скриниране.
|
Избор на концентрации за оценка на антагонистичните ефекти
|
49.
|
Единствено резултатите от анализа, които изпълняват критериите за приемане (таблица 4), се разглеждат като валидни и могат да се използват за оценяване на отклика на изпитваните химикали. В случай че една или повече микротитърни плаки от една серия за анализ не изпълни критериите за приемане, съответните микротитърни плаки следва да бъдат анализирани повторно. В случай че първата плака, съдържаща пълната серия от разреждания на референтния еталон, не изпълни критериите за приемане, пълната серия за изпитване (6 плаки) трябва да бъде анализирана повторно.
|
|
50.
|
Първоначалните обхвати на концентрацията на изпитваните химикали следва да бъдат коригирани и серията на предварителния скрининг следва да бъде повторена, ако:
|
—
|
бъде забелязана цитотоксичност. Процедурата за предварителен скрининг следва да бъде повторена с по-ниски и нецитотоксични концентрации на изпитвания химикал.
|
|
—
|
Предварителният скрининг на изпитвания химикал не покаже пълна крива доза-отклик, защото изпитаните концентрации генерират максимално потискане. Предварителният скрининг следва да бъде повторен, като се използват по-ниски концентрации на изпитвания химикал.
|
|
|
51.
|
Когато се открие валиден отклик, свързан с дозата, следва да бъде избрана (най-ниската) концентрация, при която се наблюдава максимално потискане и при която не се проявява цитотоксичност. Най-високата концентрация на изпитвания химикал, която ще бъде изпитвана в рамките на сериите за цялостно изпитване, следва да бъде 3 пъти по-голяма от тази избрана концентрация.
|
|
52.
|
Следва да се подготви пълна серия на оптимизирано разреждане на изпитвания химикал, като се спазват стъпките за разреждане, описани в таблица 6 и се започне от най-високата концентрация, определена по-горе.
|
|
53.
|
Изпитван химикал, който не предизвиква никакви антагонистични ефекти, следва да бъде изпитан в сериите за цялостно изпитване, като се започне от най-високата, нецитотоксична концентрация, изпитана по време на предварителното скриниране.
|
Серии на цялостно изпитване
|
54.
|
След избора на оптимизираните обхвати на концентрацията, изпитваните химикали следва да бъдат изпитани цялостно, като се използва серията от разреждания, описани в таблици 5 (агонизъм) и 6 (антагонизъм). Всички концентрации следва да бъдат изпитани трикратно, съобразно разположението на ямките в плаката, както е указано на фигура 1 (агонизъм) или 2 (антагонизъм).
|
|
55.
|
Единствено резултатите от анализа, които изпълняват критериите за приемане (таблица 3 и 4), се разглеждат като валидни и могат да се използват за оценяване на отклика на изпитваните химикали. В случай че една или повече микротитърни плаки от една серия за анализ не изпълни критериите за приемане, съответните микротитърни плаки следва да бъдат анализирани повторно. В случай че първата плака, съдържаща пълната серия от разреждания на референтния еталон, не изпълни критериите за приемане, пълната серия за изпитване (6 плаки) трябва да бъде анализирана повторно.
|
Таблица 5
Концентрации и разреждания на референтните еталони, контроли и изпитвани химикали, използвани за изпитване на агонисти
|
Референтен еталон 17β-естрадиол
|
TCx - предварителна скринингова серия
|
TCx - серия на цялостно изпитване
|
Контроли
|
|
конц. (M)
|
разреждане
|
разреждане
|
конц. (M)
|
|
C0
|
0
|
TCx-1
|
10 000 000 x
|
TCx-1
|
3000 x
|
ПК
|
3*10–6
|
|
C1
|
1*10–13
|
TCx-2
|
1 000 000 x
|
TCx-2
|
1000 x
|
NC
|
1*10–8
|
|
C2
|
3*10–13
|
TCx-3
|
100 000 x
|
TCx-3
|
300 x
|
C0
|
0
|
|
C3
|
1*10–12
|
TCx-4
|
10 000 x
|
TCx-4
|
100 x
|
SC
|
0
|
|
C4
|
3*10–12
|
TCx-5
|
1 000 x
|
TCx-5
|
30 x
|
|
|
|
C5
|
6*10–12
|
TCx-6
|
100 x
|
TCx-6
|
10 x
|
|
|
|
C6
|
1*10–11
|
TCx-7
|
10 x
|
TCx-7
|
3 x
|
|
|
|
C7
|
3*10–11
|
TCx-8
|
1 x
|
TCx-8
|
1 x
|
|
|
|
C8
|
1*10–10
|
|
|
|
|
|
|
|
TCx - изпитван химикал x
ПК - положителна контрола (17α-метилтестостерон)
NC - отрицателна контрола (кортикостерон)
C0 - контрол на разтворителя на референтния еталон
SC - контрол на разтворителя на изпитвания химикал
|
Таблица 6
Концентрации и разреждания на референтните еталони, контроли и изпитвани химикали, използвани за изпитване на антагонисти
|
Референтен еталон тамоксифен
|
TCx - предварителна скринингова серия
|
TCx - серия на цялостно изпитване
|
Контроли
|
|
конц. (M)
|
разреждане
|
разреждане
|
конц. (M)
|
|
C0
|
0
|
TCx-1
|
10 000 000 x
|
TCx-1
|
3000 x
|
ПК
|
1*10–9
|
|
C1
|
3*10–9
|
TCx-2
|
1 000 000 x
|
TCx-2
|
1000 x
|
NC
|
1*10–5
|
|
C2
|
1*10–8
|
TCx-3
|
100 000 x
|
TCx-3
|
300 x
|
C0
|
0
|
|
C3
|
3*10–8
|
TCx-4
|
10 000 x
|
TCx-4
|
100 x
|
SC
|
0
|
|
C4
|
1*10–7
|
TCx-5
|
1 000 x
|
TCx-5
|
30 x
|
|
|
|
C5
|
3*10-
|
TCx-6
|
100 x
|
TCx-6
|
10 x
|
Добавен агонист
|
|
C6
|
1*10–6
|
TCx-7
|
10 x
|
TCx-7
|
3 x
|
конц. (M)
|
|
C7
|
3*10–6
|
TCx-8
|
1 x
|
TCx-8
|
1 x
|
17β-естрадиол
|
3*10–12
|
|
C8
|
1*10–5
|
|
|
|
|
|
|
|
TCx - изпитван химикал x
ПК - положителна контрола (4-хидрокситамоксифен)
NC - отрицателна контрола (ресвератрол)
C0 - контрол на разтворителя на референтния еталон
SC - контрол на разтворителя на изпитвания химикал
VC - контрол на носителя (не съдържа фиксирана концентрация на референтния еталон за агонисти 17β-естрадиол) (3,0*10–12 M)
|
Събиране и анализ на данни
|
56.
|
След сериите за предварителен скрининг и цялостно изпитване следва да бъдат определени стойностите на EC10, EC50, PC10, PC50 и максималната индукция (TCxmax) на изпитвания химикал за изпитването на агонисти. За изпитването с антагонисти следва да се изчислят стойностите за IC20, IC50, PC80, PC50 и минималната индукция (TCxmin). Графично представяне на тези параметри е дадено на фигура 3 (агонизъм) и 4 (антагонизъм). Необходимите параметри се изчисляват на базата на относителната индукция на всеки изпитван химикал (спрямо максималната индукция на референтния еталон (=100 %)). За оценяването на данните следва да се използва нелинейна регресия (с променлив наклон, 4 параметъра), съгласно следното уравнение:
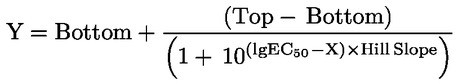
Където:
X = Логаритъм от дозата или концентрацията
Y = отклик (относителна индукция (%))
Top = Максимална индукция (%)
Bottom = Минимална индукция (%)
LogEC50 = Логаритъм на концентрацията, при която се наблюдава 50 % от максималния отклик
HillSlope = Коефициент на наклон или коефициент на Хил
|
|
57.
|
Необработените данни от луминометъра, изразени като относителни светлинни единици (RLU), следва да се прехвърлят в електронната таблица за анализ на данни, предназначена за предварителния скрининг и цялостните анализи. Необработените данни следва да отговарят на критериите за приемливост, както са посочени в Таблица 3А и 3Б (агонизъм) или 4А и 4Б (антагонизъм). В случай, че необработените данни отговарят на критериите за приемливост, се извършват следните изчисления за определяне на необходимите параметри:
|
Агонизъм
|
—
|
Изважда се средната стойност за RLU на референтния стандартен контролен разтвор от всички необработени данни от анализа на референтните еталони.
|
|
—
|
Изважда се средната стойност на RLU за контролния разтвор на изпитвания химикал от всички необработени данни от анализа на изпитваните химикали.
|
|
—
|
Изчислява се относителната индукция на всяка концентрация на референтния еталон. Определя се индукцията на най-високата концентрация на референтния еталон като 100 %.
|
|
—
|
Изчислява се относителната индукция на всяка концентрация на изпитвания химикал в сравнение с най-високата концентрация на референтния еталон, приета за 100 %.
|
|
—
|
Оценяват се резултатите от анализа след нелинейна регресия (променлив наклон, 4 параметъра).
|
|
—
|
Определят се EC50 и EC10 на референтния еталон.
|
|
—
|
Определят се EC50 и EC10 на изпитваните химикали.
|
|
—
|
Определя се максималната относителна индукция на изпитвания химикал (TCmax).
|
|
—
|
Определят се РC10 и РC50 на изпитваните химикали.
|
За някои изпитвани химикали цялата крива доза-отклик може да не бъде винаги налична, например поради проблеми с цитотоксичността или разтворимостта. Поради това ЕC50, ЕC10 и PC50 не могат да бъдат определени. В такъв случай могат да се определят само PC10 и TCmax.
Антагонизъм
|
—
|
Средната стойност на RLU на най-високата концентрация на референтния еталон се изважда от всички необработени данни от анализа на референтните еталони.
|
|
—
|
Средната стойност на RLU на най-високата концентрация на референтния еталон се изважда от всички необработени данни от анализа на изпитваните химикали.
|
|
—
|
Изчислява се относителната индукция на всяка концентрация на референтния еталон. Определя се индукцията на най-ниската концентрация на референтния еталон като 100 %.
|
|
—
|
Изчислява се относителната индукция на всяка концентрация на изпитвания химикал в сравнение с най-ниската концентрация на референтния еталон, приета за 100 %.
|
|
—
|
Оценяват се резултатите от анализа след нелинейна регресия (променлив наклон, 4 параметъра).
|
|
—
|
Определят се IC50 и IC20 на референтния еталон.
|
|
—
|
Определят се IC50 и IC20 на изпитваните химикали.
|
|
—
|
Определят се минималната относителна индукция на изпитвания химикал (TCmin).
|
|
—
|
Определят се РC80 и РC50 на изпитваните химикали.
|
Фигура 3
Общ преглед на параметрите, определени в изследването с агонист

EC10 = концентрация на вещество, при която се наблюдава 10 % от неговия максимален отклик.
EC50 = концентрация на вещество, при която се наблюдава 50 % от неговия максимален отклик.
PC10 = концентрация на изпитван химикал, при която неговият отклик е равен на ЕС10 на референтния еталон.
PC50 = концентрация на изпитван химикал, при която неговият отклик е равен на ЕС50 на референтния еталон.
TCxmax = максимална относителна индукция на изпитван химикал.
Фигура 4
Общ преглед на параметрите, определени в анализа на антагонист

IC20 = концентрация на веществото, при която се наблюдава 80 % от неговия максимален отклик (20 % инхибиране).
IC50 = концентрация на веществото, при която се наблюдава 50 % от неговия максимален отклик (50 % инхибиране).
PC80 = концентрация на изпитван химикал, при която неговият отклик е равен на IC20 на референтния стандарт.
PC50 = концентрация на изпитван химикал, при която неговият отклик е равен на IC50 на референтния стандарт.
TCxmin = минимална относителна индукция на изпитван химикал.
За някои изпитвани химикали цялата крива доза-отклик може да не бъде винаги налична, например поради проблеми с цитотоксичността или разтворимостта. Следователно IC50, IC20 и PC50 не могат да бъдат определени. В такъв случай могат да се определят само PC20 и TCmin.
|
58.
|
Резултатите следва да се базират на две (или три) независими серии. Ако две серии дават сравними и следователно възпроизводими резултати, не е необходимо да се извършва трета серия. За да са приемливи, резултатите следва:
|
—
|
Да отговарят на критериите за приемливост (вж. параграфи 14-22 на критериите за приемливост),
|
|
Критерии за тълкуване на данните
|
59.
|
За тълкуване на данните и вземане на решение дали изпитван химикал се счита за положителен или отрицателен следва да се използват следните критерии:
|
За всяка цялостна серия даден изпитван химикал се счита за положителен в случай, че:
|
1
|
TCmax е равна на или превишава 10 % от максималния отклик на референтния стандарт (REF10).
|
|
2
|
Поне 2 последователни концентрации на изпитвания химикал са равни на или превишават REF10.
|
|
|
За всяка цялостна серия даден изпитван химикал се счита за отрицателен в случай, че:
|
1
|
TCmax не превишава 10 % от максималния отклик на референтния стандарт (REF10).
|
|
2
|
По-малко от 2 последователни концентрации на изпитвания химикал са равни на или превишават REF10.
|
|
|
За всяка цялостна серия даден изпитван химикал се счита за положителен в случай, че:
|
1
|
TCmin е равна на или по-ниска от 80 % от максималния отклик на референтния стандарт (REF80 = 20 % инхибиране).
|
|
2
|
Поне 2 последователни концентрации на изпитвания химикал са равни на или по-ниски от REF80.
|
|
|
За всяка цялостна серия даден изпитван химикал се счита за отрицателен в случай, че:
|
1
|
TCmin превишава 80 % от максималния отклик на референтния стандарт (REF80 = 20 % инхибиране).
|
|
2
|
По-малко от 2 концентрации на изпитвания химикал са равни на или по-ниски от REF80.
|
|
|
|
60.
|
За да се характеризира силата на положителния отклик на даден изпитван химикал, следва да се съобщи величината на ефекта (агонизъм: TCmax; антагонизъм: TCmin) и концентрацията, при която ефектът възниква (агонизъм: EC10, EC50, PC10, PC50; антагонизъм: IC20, IC50, PC80, PC50).
|
ПРОТОКОЛ ОТ ИЗПИТВАНЕТО
|
61.
|
Вижте точка 20 на „КОМПОНЕНТИ НА АНАЛИЗА ER TA“
|
ЛИТЕРАТУРА
|
(1)
|
OECD (2016). Draft Validation report of the (anti-) ERα CALUX bioassay - transactivation bioassay for the detection of compounds with (anti)estrogenic potential. Environmental Health and Safety Publications, Series on Testing and Assessment (No 240). Organisation for Economic Cooperation and Development, Paris
|
|
(2)
|
Sonneveld E, Jansen HJ, Riteco JA, Brouwer A, van der Burg B. (2005). Development of androgen- and estrogen-responsive bioassays, members of a panel of human cell line-based highly selective steroid-responsive bioassays. Toxicol Sci. 83(1), 136-148.
|
|
(3)
|
Quaedackers ME, van den Brink CE, Wissink S, Schreurs RHMM, Gustafsson JA, van der Saag PT, and van der Burg B. (2001). 4-Hydroxytamoxifen trans-represses nuclear factor-kB Activity in human osteoblastic U2OS cells through estrogen receptor (ER)α and not through ERβ. Endocrinology 142(3), 1156-1166.
|
|
(4)
|
Thorne N, Inglese J and Auld DS. (2010). Illuminating Insights into Firefly Luciferase and Other Bioluminescent Reporters Used in Chemical Biology, Chemistry and Biology17(6):646-57.
|
|
(5)
|
Escande A, Pillon A, Servant N, Cravedi JP, Larrea F, Muhn P, Nicolas JC, Cavaillès V and Balaguer P. (2006). Evaluation of ligand selectivity using reporter cell lines stably expressing estrogen receptor alpha or beta. Biochem. Pharmacol., 71, 1459-1469.
|
|
(6)
|
Kuiper GG, Lemmen JG, Carlsson B, Corton JC, Safe SH, van der Saag PT, van der Burg B and Gustafsson JA. (1998). Interaction of estrogenic chemicals and phytoestrogens with estrogen receptor beta. Endocrinol., 139, 4252-4263.
|
|
(7)
|
Sotoca AM, Bovee TFH, Brand W, Velikova N, Boeren S, Murk AJ, Vervoort J, Rietjens IMCM. (2010). Superinduction of estrogen receptor mediated gene expression in luciferase based reporter gene assays is mediated by a post-transcriptional mechanism. J. Steroid. Biochem. Mol. Biol., 122, 204–211.
|
|
(8)
|
Sonneveld E, Riteco JAC, Jansen HJ, Pieterse B, Brouwer A, Schoonen WG, and van der Burg B. (2006). Comparison of in vitro and in vivo screening models for androgenic and estrogenic activities. Toxicol. Sci., 89(1), 173–187.
|
|
(9)
|
Kobayashi H, Yamamoto K, Eguchi M, Kubo M, Nakagami S, Wakisaka S, Kaizuka M and Ishii H. (1995). Rapid detection of mycoplasma contamination in cell cultures by enzymatic detection of polymerase chain reaction (PCR) products. J. Vet. Med. Sci., 57(4), 769-771.
|
|
(10)
|
Zhang J-H, Chung TDY, and Oldenburg KR. (1999). A simple statistical parameter for use in evaluation and validation of high throuphut screening assays. J. Biomol. Scr., 4, 67-73
|
|
(11)
|
Besselink H, Middelhof I, and Felzel, E. (2014). Transactivation assay for the detection of compounds with (anti)estrogenic potential using ERα CALUX cells. BioDetection Systems BV (BDS). Amsterdam, the Netherlands.
|
Appendix 4.1
ВИЗУАЛНА ИНСПЕКЦИЯ НА КЛЕТЪЧНАТА ЖИЗНЕСПОСОБНОСТ
Б.67 ИЗПИТВАНИЯ IN VITRO ЗА ГЕННИ МУТАЦИИ В КЛЕТКИ НА БОЗАЙНИЦИ С ИЗПОЛЗВАНЕ НА ГЕНА ТИМИДИН КИНАЗА
ВЪВЕДЕНИЕ
|
1.
|
Настоящият метод за изпитване (МИ) е еквивалентен на насоките за изпитване на ОИСР 490 (2016 г.). Методите за изпитване периодично се преразглеждат и ревизират в светлината на научния прогрес, регулаторните нужди и хуманното отношение към животните. Анализът на миши лимфом (MLA) и тестът ТК6, използващи локус на тимидин киназа (ТК), първоначално бяха включени в метода за изпитване Б.17. Впоследствие експертната работна група по MLA на Международните семинари относно изпитванията за генотоксичност (IWGT) разработи международно хармонизирани препоръки за критериите за приемливост на анализа и тълкуването на данните за MLA (1)(2)(3)(4)(5) и тези препоръки са включени в този нов метод на изпитване Б.67. Настоящият метод за изпитване е съставен за MLA и, тъй като използва и локус на ТК, теста ТК6. Докато MLA се използва широко за регулаторни цели, ТК6 се използва много по-рядко. Следва да се отбележи, че въпреки сходството между крайните точки, двете клетъчни линии не са взаимозаменяеми и някои регулаторни програми може основателно да изразят предпочитание към единия пред другия за дадена регулаторна употреба. Например валидирането на MLA демонстрира неговата уместност за откриване не само на генна мутация, но и на способността на даден изпитван химикал да предизвиква структурни хромозомни увреждания. Настоящият метод за изпитване е част от поредица от методи за изпитване от областта на генетичната токсикология. Разработен е документ на ОИСР, съдържащ ясна информация относно изпитванията в областта на генетичната токсикология, и преглед на последните промени, направени в насоките за изпитване на ОИСР за генетична токсичност (6).
|
|
2.
|
Целта на изпитванията in vitro за генни мутации в клетки на бозайници е откриването на генни мутации, предизвикани от химикали. Използваните в тези изпитвания клетъчни линии измерват правите мутации в репортерните гени, по-специално ендогенния ген на тимидин киназа (TK в човешки клетки и Tk в клетки на гризачи, наричани заедно в този метод на изпитване TK). Настоящият метод за изпитване е предназначен за употреба с две клетъчни линии: клетъчната линия L5178Y TK+/--3.7.2C на миши лимфом (обикновено наричана L5178Y) и клетъчната линия TK6 на човешки лимфобластоид (обикновено наричана TK6). Макар че двете клетъчни линии се различават по своя произход, клетъчен растеж, статус на p53, др., изпитванията за генни мутации TK могат да се провеждат по сходен начин и за двата вида клетки, както се описва в настоящия метод за изпитване.
|
|
3.
|
Автозомната и хетерозиготната природа на гена тимидин киназа позволява откриването на жизнени колонии, иито клетки имат дефицит на ензима тимидин киназа след мутация от TK+/-
до TK-/-
. Този дефицит може да е резултат от генетични събития, засягащи ТК гена, включващ както генни мутации (точкови мутации, мутации с изместване на рамката, малки делеции, др.), така и хромозомни събития (големи делеции, хромозомни пренареждания и митотична рекомбинация). Последните събития се изразяват като загуба на хетерозиготност, която е често срещана генетична промяна на тумор-супресорните гени в човешката туморогенеза. На теория загубата на цялата хромозома, носеща гена ТК, в резултат на увреждане на вретеното и/или митотична недизюнкция може да бъде открита при MLA. Действително, комбинацията от цитогенетичния и молекулярния анализ ясно показва, че някои MLA TK мутанти са резултат от недизюнкция. Въпреки това доказателствата показват, че изпитванията за мутация на TK гена не са надежден начин за откриване на анеугени, когато се прилагат стандартни критерии за цитотоксичност (каквито са описани в този метод на изпитване) и поради това използването на тези изпитвания за откриване на анеугени не е подходящо (7)(8)(9).
|
|
4.
|
В изпитванията за мутация на TK гена се генерират два отделни фенотипни класа на TK мутанти; нормално растящи мутанти, които растат със същата скорост като ТК хетерозиготните клетки, и бавно растящи мутанти, които растат с удължено време на удвояване. Нормално растящите и бавно растящите мутанти се разпознават като голяма колония и малка колония мутанти в MLA и като рано появяваща се колония и късно появяваща се колония мутанти в ТК6. Молекулярната и цитогенетичната природа на голямата и малката колония MLA мутанти е проучена подробно (8)(10)(11)(12)(13). Молекулярната и цитогенетичната природа на рано появяващите се и късно появяващите се ТК6 мутанти също са изследвани задълбочено (14)(15)(16)(17). Бавно растящите мутанти и на двата вида клетки претърпяват генетично увреждане, което включва предполагаем(и) ген(и), регулиращ(и) растежа близо до ТК локуса, което води до удължено време на удвояване и образуването на късно появяващи се или малки колонии (18). Индукцията на бавно растящи мутанти се свързва с химикали, които предизвикват големи структурни промени на хромозомно ниво. Клетките, чието увреждане не включва предполагаем(и) ген(и), регулиращ(и) растежа близо до ТК локуса, растат със скорост, подобна на родителските клетки, и се превръщат в нормално растящи мутанти. Индукцията предимно на нормално растящи мутанти се свързва с химикали, които действат преди всичко като точкови мутагени. Следователно е важно да се преброят както бавно растящите, така и нормално растящите мутанти, за да се възстановят всички мутанти и да се осигури някаква представа за вида(овете) на увреждането (мутагени спрямо кластогени), предизвикано от изпитвания химикал (10)(12)(18)(19).
|
|
5.
|
Методът на изпитване е организиран така, че да предоставя обща информация, която се отнася и за MLA, и за ТК6, както и специфични указания за отделните изпитвания.
|
|
6.
|
Използваните определения са дадени в допълнение 1.
|
ПЪРВОНАЧАЛНИ СЪОБРАЖЕНИЯ И ОГРАНИЧЕНИЯ
|
7.
|
In vitro изпитванията по принцип изискват екзогенен източник на метаболитно активиране. Екзогенната система на метаболитно активиране не се възпроизвежда изцяло в условия in vivo.
|
|
8.
|
Следва да се вземат мерки за избягване на условия, които биха довели до изкуствено възникнали положителни резултати (т.е. възможно взаимодействие със системата за изпитване), които не са причинени от прякото взаимодействие между изпитвания химикал и генетичния материал на клетката; такива условия включват промени в pH или осмолалитета, взаимодействие с компонентите на средата (20)(21) или прекомерно високи нива на цитотоксичност (22)(23)(24). Цитотоксичност, надвишаваща препоръчителните горни нива на цитотоксичност, както е определено в точка 28, се счита за прекомерна за MLA и TK6. Освен това следва да се отбележи, че изпитвани химикали, които са аналози на тимидин или се държат като аналози на тимидин, може да увеличат честотата на мутации чрез селективен растеж на спонтанните фонови мутанти по време на третирането на клетките и изискват допълнителни методи на изпитване с цел адекватна оценка (25).
|
|
9.
|
За произведени наноматериали може да е необходимо специфично адаптиране на настоящия метод за изпитване, но то не е описано в настоящия метод за изпитване.
|
|
10.
|
Преди използването на метода за изпитване по отношение на смес с цел генериране на данни за планирана регулаторна цел, следва да се разгледа въпросът дали той може да предостави адекватни резултати за тази цел и, ако е така, защо. Подобни съображения не са необходими, когато има регулаторно изискване за изпитване на сместа.
|
|
11.
|
Дефицитът на мутантни клетки в активността на ензима тимидин киназа поради мутация на TK
+/- до TK
-/- е резистентен на цитостатичните ефекти на аналога на пиримидин трифлуортимидин (TFT). Клетките с достатъчна ТК активност са чувствителни към TFT, което причинява инхибиране на клетъчния метаболизъм и спира по-нататъшното клетъчно делене. Следователно мутантните клетки са способни да пролиферират в присъствието на TFT и да образуват видими колонии, докато клетките, съдържащи ензима ТК, не са.
|
ПРИНЦИП НА ИЗПИТВАНЕТО
|
12.
|
Клетките в суспензия се експонират на изпитвания химикал както със, така и без екзогенен източник на метаболитно активиране (вж. точка 19), за подходящ период от време (вж. точка 33), след което се субкултивират, за да се определи цитотоксичността и да се даде възможност за фенотипна екпресия преди избора на мутант. Цитотоксичността се определя от относителния общ растеж (RTG – вж. точка 25) за MLA и от относителното оцеляване (RS – вж. точка 26) за TK6. Третираните култури се поддържат в среда на растеж в продължение на достатъчен период от време, характерен за всеки клетъчен вид (вж. точка 37), за да се осигури почти оптимална фенотипна експресия на предизвиканите мутации. След фенотипната експресия честотата на мутантите се определя чрез посяване на предварително известни количества клетки в среда, съдържаща селективния агент, за да се открият мутантни колонии, и в среда без селективен агент, за да се определи ефикасността на клониране (жизнеспособността). След подходящ инкубационен период колониите се преброяват. Честотата на мутантите се изчислява въз основа на броя на мутантните колонии, коригиран с ефикасността на клониране към момента на подбора на мутантите.
|
ОПИСАНИЕ НА МЕТОДА
Подготовка
Клетки
|
13.
|
За MLA: Тъй като MLA е разработен и характеризиран с използване на сублинията TK
+/- -3.7.2C на L5178Y клетки, тази специфична сублиния следва да се използва за MLA. Клетъчната линия L5178Y произлиза от метилхолантрен-индуциран тимусен лимфом от DBA-2 мишка (26). Clive и сътрудници третират L5178Y клетките (определениот Clive като TK+/+ -3) с етилметанов сулфонат и изолират TK-/- (определен като TK-/- -3.7) клон с помощта на бромодеоксиуридин като селективен агент. От TK-/- клона се изолират спонтанен TK+/- клон (определен като TK+/- -3.7.2.) и субклон (определен като TK+/--3.7.2C), които се характеризират за използване в MLA (27). Кариотипът на клетъчната линия е публикуван (28)(29)(30)(31). Модалният хромозомен брой е 40. Има една метацентрична хромозома (t12;13), която следва да се брои като една хромозома. Мишият ТК локус е разположен в дисталния край на хромозома 11. Клетъчната линия L5178Y TK
+/- -3.7.2C има мутации и в двата p53 алела и произвежда мутантен-p53 протеин (32) (33). p53 статусът на клетъчната линия TK+/--3.7.2C вероятно отговаря за способността на изпитването да открива мащабно увреждане (17).
|
|
14.
|
За ТК6: ТК6 е човешка лимфобластоидна клетъчна линия. Родителската клетъчна линия е трансформирана от вируса Epstein-Barr клетъчна линия, WI-L2, която първоначално произлиза от 5-годишен индивид от мъжки пол с наследствена сфероцитоза. Първият изолиран клон, HH4, е мутагенизиран с ICR191 и се генерира TK хетерозиготна клетъчна линия — TK6 (34). TK6 клетките са почти диплоидни и представителният кариотип е 47, XY, 13+, t(14; 20), t(3; 21) (35). Човешкият TK локус е разположен в дългото рамо на хромозома 17. TK6 е p53-компетентна клетъчна линия, защото притежава див тип p53 секвенция и в двата алела и експресира само див тип p53 протеин (36).
|
|
15.
|
И за MLA, и за ТК6, когато за пръв път се създава или попълва основен запас, се препоръчва лабораторията, провеждаща изпитването, да удостовери липсата на замърсяване с микоплазма, кариотипа на клетките или да оцвети клетките, които носят ТК локуса, и да провери времената за удвояване на популацията. Нормалната продължителност на клетъчния цикъл за клетките, използвана в лабораторията, провеждаща изпитването, следва да бъде установена и следва да съответства на публикуваните характеристики на клетките (16)(19)(37). Този основен запас следва да се съхранява при -150o C или по-ниска температура и да се използва за приготвяне на всички работни клетъчни запаси.
|
|
16.
|
Или преди създаването на голям брой криоконсервирани работни запаси, или непосредствено преди употреба в даден опит културата може да се наложи да бъде пречистена от предварително съществуващи мутантни клетки [освен ако контролната честота на мутации (MF) на разтвора вече не е в рамките на приемливия диапазон — вж. таблица 2 за MLA)]. Това се извършва с помощта на метотрексат (аминоптерин) за избор на клетки с дефицит на ТК и добавяне на тимидин, хипоксантин и глицин (L5178Y) или 2’-деоксицитидин (TK6) към културата, за да се гарантира оптимален растеж на ТК-компетентните клетки (19)(38)(39) и (40) за TK6). Общи съвети относно добрата практика за поддържане на клетъчни култури, както и специфични съвети за L5178Y и TK6 клетките, могат да бъдат намерени в (19)(31)(37)(39)(41). За лаборатории, които изискват основни клетъчни запаси за иницииране на MLA или TK6, или за получаване на нови основни клетъчни запаси, се предлага клетъчно хранилище с добре характеризирани клетки (37).
|
Среди и условия за отглеждане на културата
|
17.
|
И за двете изпитвания следва да се използват подходящи среда за отглеждане и инкубационни условия (съдове за отглеждане, влажна атмосфера с 5 % CO2 и температура на инкубиране от 37 °C) за поддържане на културите. Клетъчните култури следва винаги да се поддържат при условия, които осигуряват отглеждането им в логаритмична фаза. Особено важно е средите и условията за отглеждане на културите да се подберат така, че да осигуряват оптимален растеж на клетките по време на периода на експресия и клониране както за мутантни, така и за немутантни клетки. За MLA и ТК6 е важно също условията на отглеждане да гарантират оптимален растеж както на голяма/рано появяваща се колония, така и на малка/късно появяваща се колония на ТК мутанти. Още подробности за отглеждането, включително нуждата от правилно загряване на неактивиран конски серум, ако по време на селекцията се използва RPMI среда, могат да бъдат намерени в (19)(31)(38)(39)(40)(42).
|
Приготвяне на културите
|
18.
|
Размножават се клетки от изходни култури, посяват се в хранителна среда с такава гъстота, че суспензионните култури да продължават да растат експоненциално по време на периодите на третиране и експресия.
|
Метаболитно активиране
|
19.
|
При използването на L5178Y и TK6 клетки следва да се използват екзогенни метаболизиращи системи, тъй като те имат недостатъчен ендогенен капацитет за метаболизиране. Най-често използваната система, която се препоръчва по подразбиране, освен ако няма основание за друго, е постмитохондрична фракция (S9) с добавка на ко-фактор, приготвена от черен дроб на гризачи (обикновено плъхове), третирани с индуциращи ензими агенти като Aroclor 1254 (43) (44) (45) (46) или комбинация от фенобарбитал и β-нафтофлавон (46)(47)(48)(49)(50)(51).Последната комбинация не противоречи на Стокхолмската конвенция за устойчивите органични замърсители (52) и е доказано, че е също толкова ефективна, колкото Aroclor 1254, за индуцирането на оксидази със смесени функции (45)(46)(47)(48)(49). Фракцията S9 обикновено се използва в концентрации, вариращи от 1 до 2 %, но може да се увеличи до 10 % (v/v) в окончателната среда за изпитване. Изборът на типа и концентрацията на използваната екзогенна система за метаболитно активиране или метаболитен индуктор може да бъде повлиян от класа на изпитваните химикали.
|
Приготвяне на химикала за изпитване
|
20.
|
Твърдите изпитвани химикали следва да се подготвят в подходящи разтворители и, ако е необходимо, да се разредят преди третиране на клетките (вж. точка 21). Течните химикали за изпитване могат да се добавят директно в системата за изпитване и/или да се разредят преди третирането на системата за изпитване. Газообразните или летливите изпитвани химикали следва да бъдат изпитвани чрез подходящи изменения на стандартните протоколи, като например третиране в запечатани съдове за отглеждане (53)(54)(55). Приготвянето на изпитвания химикал следва да се извърши непосредствено преди третирането, освен ако данните за стабилността показват, че е допустимо съхранение.
|
УСЛОВИЯ НА ИЗПИТВАНЕ
Разтворители
|
21.
|
Разтворителят следва да бъде избран с цел оптимизиране на разтворимостта на изпитвания химикал, без да се оказва негативно влияние върху извършването на изпитването, като например промяна на клетъчния растеж, засягане на целостта на изпитвания химикал, реакция със съдовете за отглеждане, нарушаване на системата за метаболитно активиране. Препоръчва се винаги, когато е възможно, най-напред да се разглежда възможността за използване на вода като разтворител (или среда за отглеждане). Добре утвърдени разтворители са вода или диметилсулфоксид. Като цяло органичните разтворители не следва да надвишават 1 % (об./об.), а водните разтворители (физиологичен разтвор или вода) не трябва да превишават 10 % (об./об.) в окончателната среда за третиране. Ако се използват различни от добре утвърдените разтворители (напр. етанол или ацетон), използването им следва да бъде подкрепено от данни за съвместимостта им с изпитваните химикали и системата за изпитване, както и за това, че не са генетично токсични при използваната концентрация. При липса на подкрепящи данни е важно да се добавят нетретирани контроли (виж Допълнение 1, „Определения“), за да се докаже, че избраният разтворител не предизвиква някакви неблагоприятни или мутагенни ефекти.
|
ИЗМЕРВАНЕ НА ЦИТОТОКСИЧНОСТТА И ИЗБИРАНЕ НА КОНЦЕНТРАЦИИ НА ТРЕТИРАНЕ
|
22.
|
При определяне на най-високата концентрация на изпитвания химикал следва да се избягват концентрациите, които могат да доведат до изкуствено предизвикан положителен отклик, като например предизвикващите прекомерна цитотоксичност (вж. точка 28), утаяване (вж. точка 29) в средата за отглеждане или явни промени в pH или осмолалитета (вж. точка 8). Ако изпитваният химикал предизвиква явна промяна в pH на средата към момента на добавянето, pH може да бъде коригиран чрез буфериране на окончателната среда за третиране, за да се избегнат изкуствено предизвикани положителни резултати и да се поддържат подходящи условия за отглеждане.
|
|
23.
|
Изборът на концентрация се основава на цитотоксичността и на други съображения (вж. точки 27—30). Въпреки че оценката на цитотоксичността в първоначално изпитване може да бъде полезна за по-добро определяне на концентрациите, които ще се използват в главния опит, първоначалното изпитване не се изисква. Въпреки че се извършва първоначална оценка на цитотоксичността, измерването на цитотоксичността за всяка култура все още се изисква в основния експеримент. Ако се провежда опит за определяне на обхвата, той следва да обхваща широк диапазон на концентрации и може или да се прекрати в ден 1 след третиране, или да продължи с експресия на 2-я ден и до избор на мутант (ако изглежда, че използваните концентрации са подходящи).
|
|
24.
|
Цитотоксичността следва да се определя за всяка отделна изпитвана и контролна култура: методите за MLA (2) и TK6 (15) са определени от международно приета практика.
|
|
25.
|
И за двете версии на MLA с агар и микроямка: цитотоксичността следва да се оценява с помощта на относителен общ растеж (RTG), който първоначално е определен от Clive и Spector през 1975 г. (2). Това измерване включва относителния растеж на суспензията (RSG: изпитвана култура спрямо контрола на разтворител) по време на третиране на клетките, времето на експресия и относителната ефикасност на клониране (RCE: изпитвана култура спрямо контрола на разтворител) към момента на избор на мутанти (2). следва да се отбележи, че RSG включва всички загуби на клетки, възникващи в изпитваната култура по време на третиране (вж. Допълнение 2 за формулите).
|
|
26.
|
За ТК6: Цитотоксичността следва да се оценява с използване на относителна преживяемост (RS), т.е. ефикасността на клониране на клетки, които са посети непосредствено след третирането, с корекция за всички загуби на клетки по време на третирането, въз основа на броя на клетките, в сравнение с отрицателната контрола (на която е присвоена стойност 100 % за оцеляване) (вж. Допълнение 2 за формулата).
|
|
27.
|
следва да бъдат оценени най-малко четири концентрации на изпитване (без да се включват контролата на разтворителя и положителните контроли), които отговарят на критериите за приемливост (подходяща цитотоксичност, брой на клетки и т.н.). Използването на култури с едно повторение е препоръчително, но за всяка изпитвана концентрация може да се използват култури или без повторение, или с повторения. Резултатите, получени за култури с повторения при дадена концентрация, следва да се докладват отделно, но могат да бъдат обобщени за анализа на данните (55). За изпитваните химикали, показващи слаба цитотоксичност, или не показващи цитотоксичност, обичайно са подходящи приблизително 2 до 3-кратни концентрационни интервали. Там, където се среща цитотоксичност, избраните концентрации следва да обхващат диапазона на цитотоксичност от стойността, при която се получава цитотоксичност, както е описано в точка 28, до концентрации, при които е налице умерена, слаба или нулева цитотоксичност. Много изпитвани химикали показват стръмни криви на концентрация-отклик и за да се обхване целият диапазон на цитотоксичност, или да се проучи подробно връзката концентрация-отклик, може да е необходимо да се използват по-близко разположени концентрации и повече от четири концентрации, по-специално в ситуации, при които се изисква повтаряне на опита (вж. точка 70). Използването на повече от 4 концентрации може да бъде особено важно при използване на култури без повторение.
|
|
28.
|
Ако максималната концентрация се базира на цитотоксичност, най-високата концентрация следва да цели постигане на между 20 и 10 % RTG за MLA и между 20 и 10 % RS за ТК6 (точка 67).
|
|
29.
|
За малко разтворимите изпитвани химикали, които не са цитотоксични при концентрации, по-ниски от най-ниската неразтворима концентрация, най-високата анализирана концентрация трябва да предизвиква помътняване или утайка, видима с невъоръжено око или с помощта на инвертен микроскоп в края на третирането с изпитвания химикал. Дори ако цитотоксичността се наблюдава над най-ниската концентрация на неразтворимост, препоръчително е да се извърши изпитване само при една концентрация, която предизвиква помътняване или е с видима утайка, тъй като от утайката могат да се получат изкуствено предизвикани въздействия. Тъй като MLA и TK6 използват суспензионни култури, необходимо е особено внимание, за да се гарантира, че утайката не пречи на провеждането на изпитването. Определянето на разтворимостта в средата за отглеждане преди опита може също да е полезно.
|
|
30.
|
Ако не се наблюдава утайка или ограничаваща цитотоксичност, най-високата изпитвана концентрация следва да отговаря на 10 mM, 2 mg/ml или 2 μl/ml, която от стойностите е най-ниска (57)(58). Когато изпитваният химикал не е с определен състав, например вещество с неизвестен или променлив състав, сложни продукти от реакции или биологични материали [т.е. химични вещества с непознат или променлив състав (UVCB)] (5), екстракти от околната среда и други, може да е необходимо най-високата концентрация да е по-висока (напр. 5 mg/ml), при липсата на достатъчна цитотоксичност, за да се увеличи концентрацията на всеки от компонентите. Следва да се отбележи обаче, че тези изисквания могат да се различават по отношение на фармацевтични продукти за хуманна употреба (59).
|
Контроли
|
31.
|
За всяко опитно условие следва да се включат паралелни отрицателни контроли (вж. точка 21), състоящи се само от разтворител в средата за третиране, с които се работи по същия начин, както с третираните култури.
|
|
32.
|
Паралелни положителни контроли са необходими за доказване на способността на лабораторията да идентифицира мутагени при условията на използвания протокол на изпитването, ефективността на екзогенната система за метаболитно активиране (когато е приложимо) и за демонстриране на адекватно откриване както на малки/късно появяващи се, така и на големи/рано появяващи се ТК мутанти. Примери за положителни контроли са дадени в таблица 1 по-долу. Могат да бъдат използвани алтернативни вещества за положителни контроли, ако това е обосновано. Тъй като in vitro изпитванията на клетки от бозайници за генетична токсичност са достатъчно стандартизирани за краткотрайни третирания (3–4 часа), извършвани едновременно със и без метаболитно активиране с използване на същата продължителност на третиране, използването на положителни контроли може да се ограничи до мутаген, изискващ метаболитно активиране. В този случай откликът на тази положителна контрола без повторение ще докаже както активността на дадена система за метаболитно активиране, така и способността на изпитваната система за генериране на отклик. Ако се използва, дълготрайното третиране (т.е. 24 часа без S9) следва въпреки това да има собствена положителна контрола, тъй като продължителността на третиране ще се различава от тази на изпитването, което използва метаболитно активиране. Всяка положителна контрола следва да се използва при една или повече концентрации, които се очаква да породят възпроизводими и откриваеми увеличения спрямо фона, за да се докаже чувствителността на системата за изпитване, и откликът не следва да се компрометира от цитотоксичност, надхвърляща границите, определени в настоящия метод за изпитване (вж. точка 28).
|
Таблица 1
Препоръчителни референтни вещества за оценка на пригодността на лабораторията и за подбор на положителни контроли
|
Категория
|
Вещество
|
CAS №
|
|
1. Мутагени, активни без метаболитно активиране
|
|
Метилов метансулфонат
|
66-27-3
|
|
Митомицин С
|
50-07-7
|
|
4-нитрохинолин-N-оксид
|
56-57-5
|
|
2. Мутагени, изискващи метаболитно активиране
|
|
Бензо(a)пирен
|
50-32-8
|
|
Циклофосфамид (монохидрат)
|
50-18-0 (6055-19-2)
|
|
7,12-диметилбензантрацен
|
57-97-6
|
|
3-метилхолантрен
|
56-49-5
|
ПРОЦЕДУРА
Третиране с изпитвания химикал
|
33.
|
Пролифериращите клетки се третират с изпитвания химикал при наличието и в отсъствието на система за метаболитно активиране. Експозицията следва да е за подходящ период от време (обикновено 3—4 часа е достатъчно). Следва да се отбележи обаче, че тези изисквания могат да се различават по отношение на фармацевтични продукти за хуманна употреба (59). За MLA, в случаите, когато краткотрайното третиране води до отрицателни резултати и има информация, която предполага нуждата от по-дълго третиране [напр. нуклеозидни аналози, слабо разтворими химикали, (5)(59)], следва да се обмисли провеждане на изпитването с по-дълго третиране, т.е 24 часа без S9.
|
|
34.
|
Минималният брой клетки, използвани за всяка изпитвана (контролна и третирана) култура на всеки етап от изпитването, следва да се базира на честотата на спонтанните мутации. Общото указание е да се третират и прехвърлят достатъчно клетки във всяка опитна култура, така че да се поддържат поне 10, а в идеалния случай 100 спонтанни мутации, във всички фази на изпитването (третиране, фенотипна експресия и мутантен избор) (56).
|
|
35.
|
За MLA препоръчителната приемлива честота на спонтанни мутации е между 35-140 × 10-6 (за версия с агар) и 50-170 × 10-6 (за версия с микроямки) (вж. Таблица 2). За наличието на поне 10, а в идеалния случай 100 спонтанни мутации, оцеляващи след третиране, за всяка изпитвана култура, е необходимо да се третират поне 6 × 106 клетки. Третирането на такъв брой клетки и поддържането на достатъчно клетки по време на експресия и клониране за мутантен избор осигурява достатъчен брой спонтанни мутации (10 или повече) по време на всички фази на опита, дори за култури, третирани при концентрации, които водят до 90 % цитотоксичност (както е измерено от 10 % RTG) (19)(38)(39).
|
|
36.
|
За ТК6 честотата на спонтанните мутации като цяло е между 2 и 10 × 10-6. За наличието на поне 10 спонтанни мутации, оцеляващи след третиране, за всяка изпитвана култура, е необходимо да се третират поне 20 × 106 клетки. Третирането на такъв брой клетки осигурява достатъчен брой спонтанни мутации (10 или повече) дори за култури, третирани при концентрации, които причиняват 90 % цитотоксичност по време на третиране (10 % RS). Освен това достатъчен брой клетки следва да бъдат култивирани по време на периода на експресия и да бъдат посети за избор на мутант (60).
|
Време на фенотипна експресия и измерване на цитотоксичност и честотата на мутации
|
37.
|
В края на периода на третиране клетките се култивират за определено време, за да се позволи почти оптимална фенотипна експресия на новите индуцирани мутации; специфични за всяка клетъчна линия. За MLA периодът на фенотипна експресия е 2 дни. За ТК6 периодът на фенотипна експресия е 3—4 дни. Ако се използва 24-часово третиране, периодът на експресия започва след края на третирането.
|
|
38.
|
По време на периода на фенотипна експресия клетките се преброяват на ежедневна база. За MLA ежедневният брой клетки се използва за изчисляване на ежедневния растеж на суспензията (SG). След 2-дневния период на експресия клетките се суспендират в среда със и без селективно вещество за определяне съответно на броя на мутантите (плаки за селекция) и за ефикасност на клониране (плаки за жизнеспособност). За MLA има два в еднаква степен приемливи методи за клониране на мутантна селекция; единият използва мек агар, а другият използва течна среда в 96-ямкови плаки (19) (38) (39). Клонирането при ТК6 се провежда с използване на течна среда и 96-ямкови плаки (16).
|
|
39.
|
Трифлуоротимидин (TFT) е единственото препоръчително селективно вещество за TK мутанти (61).
|
|
40.
|
За MLA плаките с агар и плаките с микроямки се преброяват след 10—12 дни инкубация. За ТК6 колониите в плаките с микроямки се оценяват след 10—14 дни за рано появили се мутанти. За възстановяване на бавно растящите (късно появяващите се) ТК6 мутанти е необходимо повторно посяване на клетките в хранителна среда с TFT след преброяване на рано появилите се мутанти, след което плаките се инкубират допълнително още 7—10 дни (62). Вж. в точки 42 и 44 обсъждане, отнасящо се за преброяването на бавно и нормално растящи ТК мутанти.
|
|
41.
|
Съответните изчисления за двете изпитвания, включващи двата метода (агар и микроямки) за MLA са дадени в Допълнение 2. За методите с агар за MLA колониите се преброяват и броят на мутантните колонии се коригира с ефикасността на клониране за изчисляване на MF. За версията с микроямки на MLA и ТК6 ефикасността на клониране и за двете плаки — за селекция и ефикасност на клониране — се определя според разпределението на Поасон (63). MF се изчислява от тези две ефикасности на клониране.
|
Характеризиране на мутантна колония
|
42.
|
За MLA, ако изпитваният химикал е положителен (вж. точки 62-63), характеризиране на колонията по размер на колонията или растеж следва да се извърши за поне една от изпитваните култури (обикновено най-високата приемлива положителна концентрация) и за отрицателните и положителните контроли. Ако изпитваният химикал е отрицателен (вж. точка 64), характеризирането на мутантната колония следва да се извърши за отрицателните и положителните контроли. При метода с микроямки на MLA за малки колонии мутанти се определят тези, които обхващат по-малко от 25 % от диаметъра на ямката, а за големи тези, които обхващат повече от 25 % от диаметъра на ямката. При метода с агар се използва автоматичен брояч на колонии за преброяване на мутантните колонии и определяне на размера на колония. Подходите за определяне на размера на колония са описани подробно в литературата (19)(38)(40). Характеризиране на колонията на отрицателната и положителната контрола е необходимо, за да се демонстрира, че проучванията са проведени адекватно.
|
|
43.
|
Изпитваният химикал не може да се определи като отрицателен, ако мутанти и от голямата, и от малката колонии не се откриват адекватно в положителната контрола. Характеризирането на колония може да се използва за осигуряване на обща информация относно способността на изпитвания химикал да причинява точкови мутации и/или хромозомни събития (точка 4).
|
|
44.
|
TK6: Нормално растящите и бавно растящите мутанти се разграничават по разликата във времето на инкубация (вж. точка 40). За ТК6 обикновено се оценяват и рано, и късно появяващите се мутанти за всички култури, включително отрицателните и положителните контроли. Характеризиране на колонията на отрицателната и положителната контрола е необходимо, за да се демонстрира, че проучванията са проведени адекватно. Изпитваният химикал не може да се определи като отрицателен, ако и рано появяващите се, и късно появяващите се мутанти не се откриват адекватно в положителната контрола. Характеризирането на колония може да се използва за осигуряване на обща информация относно способността на изпитвания химикал да причинява точкови мутации и/или хромозомни събития (точка 4).
|
Пригодност на лабораторията
|
45.
|
За да се натрупа достатъчно опит по отношение на изпитването преди използването му за рутинно изпитване, лабораторията следва да е извършила поредица опити с референтни положителни вещества, действащи чрез различни механизми (най-малко по едно със и едно без метаболитно активиране, избрани от веществата, изброени в таблица 1) и различни отрицателни контроли (включително нетретирани култури и различни разтворители/носители). Положителният и отрицателният отклик при тези контроли следва да съответства на литературните източници. Това изискване не се прилага за лаборатории, които имат опит, т.е. които разполагат с налична база данни за предходни периоди, както е определено в точки 47-50. За MLA стойностите, получени за положителните и отрицателните контроли, следва да съответстват на препоръките на IWGT (вж. Таблица 2).
|
|
46.
|
Изборът на вещества за положителни контроли (вж. Таблица 1) следва да се изследва с краткотрайно и дълготрайно третиране (ако се използва дълготрайно третиране) при отсъствие на метаболитно активиране, и с краткотрайно третиране в присъствие на метаболитно активиране, за да се докаже пригодността за откриване на мутагенни химикали, да се определи ефективността на системата за метаболитно активиране и да се докаже целесъобразността на условията за растеж на клетките по време на третирането, фенотипната експресия и избора на мутанти, и на процедурите за оценяване. Следва да бъде избран обхват от концентрации на веществата от подбора така, че да се получават възпроизводими и свързани с концентрацията увеличения над фоновите стойности с цел да се демонстрират чувствителността и динамичният обхват на системата за изпитване.
|
Данни за контролите за предходни периоди
|
47.
|
Лабораторията следва да установи:
|
—
|
Размах и разпределение при положителните контроли от предходни периоди,
|
|
—
|
Размах и разпределение при отрицателните (без третиране и за разтворител) контроли от предходни периоди.
|
|
|
48.
|
При първото придобиване на данни за разпределението на отрицателните контроли за предходни периоди паралелните отрицателни контроли следва да съответстват на публикуваните данни за контролите. С добавянето на повече опитни данни към разпределението на контролите паралелните отрицателни контроли следва в идеалния случай да бъдат в рамките на 95 % от контролните граници на това разпределение (64)(65).
|
|
49.
|
Базата данни с отрицателни контроли от предходни периоди на лабораторията следва първоначално да се създаде с минимум 10 опита, но е за предпочитане тя да се състои най-малко от 20 опита, проведени при сравними опитни условия. Лабораториите следва да използват методи за контрол на качеството, като например контролни карти (напр. C-карти или карти за средна стойност (X-bar) (65)), за определяне на варирането на данните за положителните и отрицателните контроли и за доказване, че методологията е „под контрол“ в тяхната лаборатория (66). Допълнителни подробности и препоръки за създаването и използването на данни за предходни периоди може да се намерят в литературата (64).
|
|
50.
|
Данните за отрицателните контроли следва да се състоят от честоти на мутантите от култури без повторение или — за предпочитане — с повторения, както е описано в точка 27. Паралелните отрицателни контроли в идеалния случай следва да бъдат в рамките на граници за контрол 95 % от разпределението от базата данни на дадената лаборатория за отрицателните контроли за предходни периоди. Когато данните за отрицателни контроли попадат извън 95 %-та контролна граница, те могат да са приемливи за включване в разпределението на контролите за предходни периоди, при условие че тези данни не се отклоняват екстремално и има доказателства, че системата за изпитване е „под контрол“ (вж. точка 49), както и доказателства за липса на техническа или човешка грешка.
|
|
51.
|
Всички промени в опитния протокол следва да бъдат разглеждани от гледна точка на съгласуваността на данните със съществуващите бази данни на дадената лаборатория за контролите за предходни периоди. Всички значителни несъответствия следва да водят до създаването на нова база данни за контролите за предходни периоди.
|
ДАННИ И ПРОТОКОЛИРАНЕ
Представяне на резултатите
|
52.
|
Представянето на данни за MLA и TK6 следва да включва както за третираните, така и за контролните култури, необходимите данни за изчисляването на цитотоксичност (съответно RTG или RS) и честотата на мутации, както е описано по-долу.
|
|
53.
|
За MLA данните за отделна култура следва да включват RSG, RTG, ефикасността на клониране към момента на избор на мутант и броя на мутантните колонии (за версията с агар) или броя на празните ямки (за версията с микроямки). MF следва да се изрази като брой мутантни клетки на милион оцеляващи клетки. Ако откликът е положителен, следва да се предоставят MF на малка и голяма колония (и/или процентът на общата MF) за поне една концентрация на изпитвания химикал (обикновено най-високата положителна концентрация) и за отрицателните и положителните контроли. В случай на отрицателенотклик следва да се предоставят MF на малка и голяма колония за отрицателната контрола и положителната контрола.
|
|
54.
|
За ТК6 данните за отделна култура следва да включват RS, ефикасността на клониране към момента на избор на мутант и броя на празните ямки за рано появяващите се и късно появяващите се мутанти. MF следва да се изрази като брой мутантни клетки на брой брой оцеляващи клетки и следва да включва общата MF, както и MF (и/или процентът на общата MF) на рано и късно появяващите се мутанти.
|
Критерии за приемливост
|
55.
|
И за MLA, и за TK6 следва да са спазени следните критерии, преди да се определят общите резултати за специфичен изпитван химикал:
|
—
|
Изследвани са две опитни условия (т.е. краткотрайно третиране със и без метаболитно активиране – вж. точка 33), освен ако едно от тях не е довело до положителни резултати.
|
|
—
|
Следва да има достатъчен брой клетки и концентрации за анализ (вж. точки 27, 34-36).
|
|
—
|
Критериите за избор на най-високата концентрация са съвместими с тези, описани в точки 28-30.
|
|
Критерии за приемливост за отрицателни и положителни контроли
|
56.
|
Анализът на голямо количество MLA данни на експертната работна група по MLA на IWGT доведе до международен консенсус относно специфичните критерии за приемливост за MLA (1)(2)(3)(4)(5). Следователно този метод на изпитване предоставя специфични препоръки за определяне на приемливостта на отрицателните и положителните контроли и за оценка на резултатите за отделно вещество при MLA. За ТК6 има много по-малка база данни, която не е оценена от работна група.
|
|
57.
|
За MLA, всеки опит следва да се оценява според това дали нетретираната контрола/контролата на разтворителя отговаря на критериите за приемливост на работната група по MLA на IWGT ((4) и Таблица 2 по-долу) за: (1) MF (имайте предвид, че приемливите MF на IWGT са различни за версиите с агар и микроямки на MLA), (2) ефикасност на клониране (CE) към момента на избор на мутант и (3) растеж на суспензията (SG) за контролния разтворител (вж. Допълнение 2 за формулите).
Таблица 2
Критерии за приемливост за MLA
|
Параметър
|
Метод с мек агар
|
Метод с микроямки
|
|
Честота на мутациите
|
35 – 140 × 10-6
|
50 – 170 × 10-6
|
|
Ефикасност на клониране
|
65 – 120 %
|
65 – 120 %
|
|
Растеж на суспензията
|
8 – 32 пъти (3-4-часово третиране) 32 – 180 пъти (24-часово третиране, ако се провежда)
|
8 – 32 пъти (3-4-часово третиране) 32 – 180 пъти (24-часово третиране, ако се провежда)
|
|
|
58.
|
За MLA всяко изпитване следва да се оценява и за това дали положителната(ите) контрола(и) отговаря(т) на поне един от следните два критерия за приемливост, разработени от работната група на IWGT:
|
—
|
Положителната контрола следва да демонстрира абсолютно увеличение на общата MF, което означава увеличение над спонтанната фонова MF [индуцирана MF (IMF)] с поне 300 × 10-6. Поне 40 % от IMF следва да се отразява в MF на малката колония.
|
|
—
|
Положителната контрола има увеличение на MF в малката колония с поне 150 × 10-6 над това, наблюдавано в паралелната нетретирана контрола/контролата на разтворителя (IMF на малка колония 150 × 10-6).
|
|
|
59.
|
За ТК6 изпитването е приемливо, ако паралелната отрицателна контрола се смята за приемлива за добавяне към базата данни за предходни периоди на лабораторията за отрицателните контроли, както е описано в точки 48-49. Освен това паралелните положителни контроли (вж. точка 32) следва да предизвикват отклици, които са съвместими с генерираните в базата данни за предходни периоди за положителни контроли, и да генерират статистически значимо увеличение в сравнение с паралелната отрицателна контрола.
|
|
60.
|
И за двете изпитвания горната граница на цитотоксичност, наблюдавана в културата за положителна контрола, следва да е същата, като тази на опитните култури. Това означава, че RTG/RS следва да е не по-малко от 10 %. Достатъчно е да се използва една концентрация (или една от концентрациите на културите за положителна контрола, ако се използва повече от една концентрация), за да се демонстрира, че критериите за приемливост за положителната контрола са удовлетворени. Освен това MF на положителната контрола следва да е в рамките на приемливия диапазон, определен за лабораторията.
|
Оценка и тълкуване на резултатите
|
61.
|
За MLA експертната група по миши лимфом на IWGT е провела значителна работа по отношение на биологичната относимост и критериите за положителен отклик (4). Следователно този метод на изпитване предоставя специфични препоръки за тълкуването на резултатите MLA за изпитвания химикал (вж. точки 62-64). За ТК6 има много по-малка база данни, която не е оценена от работна група. Поради това препоръките за тълкуването на данните за ТК6 имат по-общ характер (вж. точки 65-66). И за двете изпитвания се прилагат допълнителни препоръки (вж. точки 67-71).
|
MLA
|
62.
|
Препоръчва се подход за определяне на положителните и отрицателните отговори, за да се гарантира, че увеличената MF е биологично относима. Вместо статистически анализ, какъвто обикновено се използва за други изпитвания, той разчита на използването на предварително определена предизвикана честота на мутации (т.е. увеличение на MF над паралелна контрола) и определя глобалния фактор за оценка (GEF), който се базира на анализа на разпределението на данните за MF за отрицателните контроли, получени от участващи лаборатории (4). За версията с агар на MLA GEF е 90 × 10-6, а за версията с микроямки на MLA GEF е 126 × 10-6.
|
|
63.
|
При условие че всички критерии за приемливост за изпълнени, даден изпитван химикал се счита за ясно положителен, ако, при всяко от изследваните опитни условия (вж. точка 33), увеличението на MF над паралелния фон превишава GEF и увеличението е свързано с концентрацията (напр. използване на трендов тест). След това изпитваният химикал се счита за способен да индуцира мутация в тази система за изпитване.
|
|
64.
|
При условие че всички критерии за приемливост за изпълнени, даден изпитван химикал се счита за ясно отрицателен, ако, при всички изследвани опитни условия (вж. точка 33), няма свързано с концентрацията увеличение или, ако има увеличение на MF, то не превишава GEF. След това изпитваният химикал се счита за неспособен да индуцира мутация в тази система за изпитване.
|
TK6
|
65.
|
При условие, че са изпълнени всички критерии за приемливост, изпитваният химикал се счита за ясно положителен, ако в някое от опитните условия (вж. точка 33):
|
—
|
при поне една от изпитваните концентрации се наблюдава статистически значимо увеличение в сравнение с паралелната отрицателна контрола
|
|
—
|
увеличението е свързано с концентрацията, когато се оценява с подходящ трендов тест (вж. точка 33)
|
|
—
|
някой от тези резултати е извън разпределението на данните за отрицателните контроли от предходни периоди (напр. на основата на разпределение на Поасон с граница за контрол 95 %). вж. точка 48).
|
Когато са изпълнени всички тези критерии се смята, че изпитваният химикал може да индуцира мутация в тази система за изпитване. Препоръки за най-подходящите статистически методи може да се намерят в литературата (66)(67).
|
|
66.
|
При условие, че са изпълнени всички критерии за приемливост, изпитваният химикал се счита за ясно отрицателен, ако във всички изследвани опитни условия (вж. точка 33):
|
—
|
в никоя от изпитваните концентрации не се наблюдава статистически значимо увеличение в сравнение с паралелната отрицателна контрола,
|
|
—
|
няма свързано с концентрацията увеличение, когато се оценява с подходящ трендов тест
|
|
—
|
всички резултати са в рамките на разпределението на данните за отрицателните контроли от предходни периоди (напр. на основата на разпределение на Поасон с граница за контрол 95 %); вж. точка 48).
|
След това изпитваният химикал се счита за неспособен да индуцира мутация в тази система за изпитване.
|
И за MLA, и за ТК6
|
67.
|
Ако максималната концентрация се базира на цитотоксичност, най-високата концентрация следва да цели постигане на между 20 и 10 % RTG/RS. Постигнат е консенсус, че е необходимо внимание при тълкуване на положителни резултати, които се откриват само между 20 и 10 % RTG/RS, и резултатът не следва да се счита за положителен, ако увеличението на MF възниква само при или под 10 % RTG/RS (ако се оценява) (2)(59).
|
|
68.
|
Има някои обстоятелства, при които допълнителна информация може да помогне за определянето дали даден изпитван химикал не е мутагенен, когато няма култура, показваща RTG стойност между 10-20 % RTG/RS. Тези ситуации са изложени както следва: (1) Няма доказателства за мутагенност (напр. няма отклик на доза, няма честоти на мутации над наблюдаваните при паралелната отрицателна контрола или фоновите диапазони за предходни периоди и т.н.) в поредица от точки с данни в обхвата от 100 % до 20 % RTG/RS и има поне една точка с данни между 20 и 25 % RTG/RS. (2) Няма доказателства за мутагенност (напр. няма отклик на доза, няма честоти на мутации над наблюдаваните при паралелната отрицателна контрола или фонови диапазони за предходни периоди, др.) в поредица от точки с данни от 100 % до 25 % RTG/RS и има отрицателна точка от данни малко под 10 % RTG/RS. И в двете ситуации може да се направи заключение, че изпитваният химикал е отрицателен.
|
|
69.
|
Няма изискване за потвърждаване на ясно положителен или ясно отрицателен отклик.
|
|
70.
|
В случаите, когато откликът не е нито ясно отрицателен, нито ясно положителен, както е описано по-горе, и/или с цел подпомагане на определянето на биологичната относимост на даден резултат, данните следва да се оценяват чрез експертна оценка и/или чрез допълнителни изследвания. Извършването на повторен опит с възможност за използване на променени опитни условия [напр. интервал на концентрация за увеличаване на вероятността за получаване на точки с данни в рамките на 10-20 % диапазон на RTG/RS, използване на други условия на метаболитно активиране (т.е. концентрация на S9 или произход на S9) и продължителност на третиране] може да се окаже полезно.
|
|
71.
|
В редки случаи дори след допълнителни изследвания наборът от данни не позволява стигането до заключение за положителни или отрицателни резултати. Поради това за отклика на изпитвания химикал следва да се заключи, че е неясен (тълкуван като еднакво вероятно да е положителен или отрицателен.
|
ПРОТОКОЛ ОТ ИЗПИТВАНЕТО
|
72.
|
Протоколът от изпитването следва да включва следната информация:
|
|
Изпитван химикал:
|
—
|
източник, номер на партидата, крайна дата за употреба, ако има такава;
|
|
—
|
стабилност на самия изпитван химикал, ако е известна;
|
|
—
|
разтворимост и стабилност на изпитвания химикал в разтворител, ако са известни;
|
|
—
|
измерване на рН, осмолалитет и утайка в средата за отглеждане, към която е добавен изпитваният химикал, когато е уместно.
|
|
|
Вещество с една съставка:
|
—
|
външен вид, разтворимост във вода и допълнителни относими физични и химични свойства;
|
|
—
|
химична идентификация, като например наименование по IUPAC или CAS, CAS номер, код SMILES или InChI, структурна формула, чистота, химична идентичност на онечистванията, доколкото е целесъобразно и практически осъществимо, и т.н.
|
|
|
|
Вещество с повече съставки, UVCB и смеси:
|
—
|
определени, доколкото е възможно, с химичната си идентичност (вж. по-горе), количествения състав и относимите физични и химични свойства на съставките.
|
|
|
|
|
Разтворител:
|
—
|
обосновка на избора на разтворител;
|
|
—
|
процентно съдържание на разтворителя в окончателната среда за отглеждане.
|
|
|
|
Клетки:
|
|
За първични култури в лабораторията:
|
—
|
вид и източник на клетките и история в изпитвателната лаборатория;
|
|
—
|
кариотипни характеристики и/или модален брой на хромозомите;
|
|
—
|
методи за поддържане на клетъчните култури;
|
|
—
|
отсъствие на микоплазма;
|
|
—
|
времена за удвояване на клетките.
|
|
|
|
|
Условия на изпитването:
|
—
|
обосновка за избора на концентрации и броя на клетъчните култури; включително напр. данни за цитотоксичност и ограничения на разтворимостта;
|
|
—
|
състав на средите, концентрация на CO2, влажност;
|
|
—
|
концентрация на изпитвания химикал, изразена като крайна концентрация в средата за отглеждане (напр. μg или mg/ml или mM от среда за отглеждане);
|
|
—
|
концентрация (и/или обем) на разтворителя и изпитвания химикал, добавени в средата за отглеждане;
|
|
—
|
температура на инкубиране;
|
|
—
|
времетраене на третирането;
|
|
—
|
гъстота на клетките по време на третиране;
|
|
—
|
тип и състав на системата за метаболитно активиране (източник на S9, метод на приготвяне на S9 микс, концентрация или обем на S9 микс и S9 в окончателната среда за отглеждане, контрол на качеството на S9);
|
|
—
|
вещества за положителни и отрицателни контроли, крайни концентрации за всяко условие на третиране;
|
|
—
|
продължителност на периода на експресия (в т.ч. брой на посетите клетки и субкултури и графици на подхранване, ако е уместно);
|
|
—
|
идентификационни данни на селективния агент и неговата концентрация;
|
|
—
|
за MLA следва да се посочи използваната версия (агар или микроямки)
|
|
—
|
критерии за приемливост на изпитванията;
|
|
—
|
използвани методи за преброяване на жизнеспособните и мутиралите клетки;
|
|
—
|
Методи, използвани за измерване на цитотоксичността;
|
|
—
|
всякаква допълнителна информация, относима към цитотоксичността и използвания метод;
|
|
—
|
продължителност на инкубационните периоди след посяване;
|
|
—
|
определение колонии с какъв размер и от какъв вид се вземат предвид (включително критерии за „малки“ и „големи“ колонии, както е подходящо);
|
|
—
|
критерии за считане на изследванията за положителни, отрицателни или неясни;
|
|
—
|
методи, използвани за определяне на pH, осмолалитет, ако са извършени и утайка, ако е подходящо.
|
|
|
|
Резултати:
|
—
|
брой на третираните клетки и брой на клетките, подложени на субкултура за всяка култура;
|
|
—
|
параметри на токсичност (RTG за MLA и RS за TK6);
|
|
—
|
признаци на утаяване и час на определяне;
|
|
—
|
броят клетки, посети в среда със селективен агент и в среда без селективен агент;
|
|
—
|
брой на колониите в среда без селективен агент и брой на резистентни колонии в среда със селективен агент, и свързаните честоти на мутации;
|
|
—
|
определяне на размера на колония за отрицателните и положителните контроли и, ако изпитваният химикал е положителен, поне една концентрация, и свързаните честоти на мутации;
|
|
—
|
зависимостта концентрация—отклик, където е възможно;
|
|
—
|
данни от паралелни отрицателни (разтворител) и положителни контроли (концентрации и разтворители);
|
|
—
|
данни за предходни периоди за отрицателни (разтворител) и положителни контроли (концентрации и разтворители) с диапазони, средни стойности и стандартни отклонения; брой изпитвания въз основа на които се базират контролите за предходни периоди;
|
|
—
|
статистически анализи (за единични култури и обобщени повторения, ако е подходящо) и p-стойности, ако има такива; и за MLA – оценка на GEF.
|
|
|
ЛИТЕРАТУРА
|
(1)
|
Moore, M.M., Honma, M. Clements, J. (Rapporteur), Awogi, T., Bolcsfoldi, G., Cole, J., Gollapudi, B., Harrington-Brock, K., Mitchell, A., Muster, W., Myhr, B., O'Donovan, M., Ouldelhkim, M-C., San, R., Shimada, H. and Stankowski, L.F. Jr. (2000). Mouse Lymphoma Thymidine Kinase Locus (TK) Gene Mutation Assay: International Workshop on Genotoxicity Test Procedures (IWGTP) Workgroup Report, Environ. Mol. Mutagen., 35 (3): 185-190.
|
|
(2)
|
Moore, M.M., Honma, M., Clements, J., Harrington-Brock, K., Awogi, T., Bolcsfoldi, G., Cifone, M., Collard, D., Fellows, M., Flanders, K., Gollapudi, B., Jenkinson, P., Kirby, P., Kirchner, S., Kraycer, J., McEnaney, S., Muster, W., Myhr, B., O’Donovan, M., Oliver, Ouldelhkim, M-C., Pant, K., Preston, R., Riach, C., San, R., Shimada, H. and Stankowski, L.F. Jr. (2002). Mouse Lymphoma Thymidine Kinase Locus Gene Mutation Assay: Follow-Up International Workshop on Genotoxicity Test Procedures, New Orleans, Louisiana, (April 2000), Environ. Mol. Mutagen., 40 (4): 292-299.
|
|
(3)
|
Moore, M.M., Honma, M., Clements, J., Bolcsfoldi, G., Cifone, M., Delongchamp, R., Fellows, M., Gollapudi, B., Jenkinson, P., Kirby, P., Kirchner, S., Muster, W., Myhr, B., O’Donovan, M., Ouldelhkim, M-C., Pant, K., Preston, R., Riach, C., San, R., Stankowski, L.F. Jr., Thakur, A., Wakuri, S. and Yoshimura, I. (2003). Mouse Lymphoma Thymidine Kinase Locus Gene Mutation Assay: International Workshop (Plymouth, UK) on Genotoxicity Test Procedures Workgroup Report, Mutation Res., 540: 127-140.
|
|
(4)
|
Moore, M.M., Honma, M., Clements, J., Bolcsfoldi, G., Burlinson, B., Cifone, M., Clarke, J., Delongchamp, R., Durward, R., Fellows, M., Gollapudi, B., Hou, S., Jenkinson, P., Lloyd, M., Majeska, J., Myhr, B., O’Donovan, M., Omori, T., Riach, C., San, R., Stankowski, L.F. Jr., Thakur, A.K., Van Goethem, F., Wakuri, S. and Yoshimura, I. (2006). Mouse Lymphoma Thymidine Kinase Gene Mutation Assay: Follow-Up Meeting of the International Workshop on Genotoxicity Tests – Aberdeen, Scotland, 2003 – Assay Acceptance Criteria, Positive Controls, and Data Evaluation, Environ. Mol. Mutagen., 47 (1): 1-5.
|
|
(5)
|
Moore, M.M., Honma, M., Clements, J., Bolcsfoldi, G., Burlinson, B., Cifone, M., Clarke, J., Clay, P., Doppalapudi, R., Fellows, M., Gollapudi, B., Hou, S., Jenkinson, P., Muster, W., Pant, K., Kidd, D.A., Lorge, E., Lloyd, M., Myhr, B., O’Donovan, M., Riach, C., Stankowski, L.F. Jr., Thakur A.K. and Van Goethem, F. (2007). Mouse Lymphoma Thymidine Kinase Mutation Assay: Meeting of the International Workshop on Genotoxicity Testing, San Francisco, 2005, Recommendations for 24-h Treatment, Mutation. Res., 627 (1): 36-40.
|
|
(6)
|
OECD (2016). Overview of the set of OECD Genetic Toxicology Test Guidelines and updates performed in 2014-2015. ENV Publications. Series on Testing and Assessment, No 234, OECD, Paris.
|
|
(7)
|
Fellows M.D., Luker, T., Cooper, A. and O'Donovan, M.R. (2012). Unusual Structure-Genotoxicity Relationship in Mouse Lymphoma Cells Observed with a Series of Kinase Inhibitors. Mutation, Res., 746 (1): 21-28.
|
|
(8)
|
Honma, M., Momose, M., Sakamoto, H., Sofuni, T. and Hayashi, M. (2001). Spindol Poisons Induce Allelic Loss in Mouse Lymphoma Cells Through Mitotic Non-Disjunction. Mutation Res., 493 (1-2): 101-114.
|
|
(9)
|
Wang, J., Sawyer, J.R., Chen, L., Chen, T., Honma, M., Mei, N. and Moore, M.M. (2009). The Mouse Lymphoma Assay Detects Recombination, Deletion, and Aneuploidy, Toxicol. Sci., 109 (1): 96-105.
|
|
(10)
|
Applegate, M.L., Moore, M.M., Broder, C.B., Burrell, A., and Hozier, J.C. (1990). Molecular Dissection of Mutations at the Heterozygous Thymidine Kinase Locus in Mouse Lymphoma Cells. Proc. National. Academy. Sci. USA, 87 (1): 51-55.
|
|
(11)
|
Hozier, J., Sawyer, J., Moore, M., Howard, B. and Clive, D. (1981). Cytogenetic Analysis of the L5178Y/TK+/- Leads to TK-/- Mouse Lymphoma Mutagenesis Assay System, Mutation Res., 84 (1): 169-181.
|
|
(12)
|
Hozier, J., Sawyer, J., Clive, D. and Moore, M.M. (1985). Chromosome 11 Aberrations in Small Colony L5178Y TK-/- Mutants Early in their Clonal History, Mutation Res., 147 (5): 237-242.
|
|
(13)
|
Moore, M.M., Clive, D., Hozier, J.C., Howard, B.E., Batson, A.G., Turner, N.T. and Sawyer, J. (1985). Analysis of Trifluorothymidine-Resistant (TFTr) Mutants of L5178Y/TK+/- Mouse Lymphoma Cells. Mutation Res., 151 (1): 161-174.
|
|
(14)
|
Liber H.L., Call K.M. and Little J.B. (1987). Molecular and Biochemical Analyses of Spontaneous and X-Ray-Induced Mutants in Human Lymphoblastoid Cells. Mutation Res., 178 (1): 143-153.
|
|
(15)
|
Li C.Y., Yandell D.W. and Little J.B. (1992). Molecular Mechanisms of Spontaneous and Induced Loss of Heterozygosity in Human Cells In Vitro. Somat. Cell Mol. Genet., 18 (1): 77-87.
|
|
(16)
|
Honma M., Hayashi M. and Sofuni T. (1997). Cytotoxic and Mutagenic Responses to X-Rays and Chemical Mutagens in Normal and P53-Mutated Human Lymphoblastoid Cells. Mutation. Res., 374 (1): 89-98.
|
|
(17)
|
Honma, M., Momose, M., Tanabe, H., Sakamoto, H., Yu, Y., Little, J.B., Sofuni, T. and Hayashi, M. (2000). Requirement of Wild-Type P53 Protein for Maintenance of Chromosomal Integrity. Mol. Carcinogen., 28 (4): 203-14.
|
|
(18)
|
Amundson S.A. and Liber H.L. (1992). A Comparison of Induced Mutation at Homologous Alleles of the TK Locus in Human Cells. II. Molecular Analysis of Mutants. Mutation Res., 267 (1): 89-95.
|
|
(19)
|
Schisler M.R., Moore M.M. and Gollapudi B.B. (2013). In Vitro Mouse Lymphoma (L5178Y TK+/- -3.7.2C) Forward Mutation Assay. In Protocols in Genotoxicity Assessment A. Dhawan and M. Bajpayee (Eds.), Springer Protocols, Humana Press: 27-50.
|
|
(20)
|
Long, L.H., Kirkland, D., Whitwell, J. and Halliwell, B. (2007). Different Cytotoxic and Clastogenic Effects of Epigallocatechin Gallate in Various Cell-Culture Media Due to Variable Rates of its Oxidation in the Culture Medium, Mutation Res., 634 (1-2): 177-183.
|
|
(21)
|
Nesslany, F., Simar-Meintieres, S., Watzinger, M., Talahari, I. and Marzin, D. (2008). Characterization of the Genotoxicity of Nitrilotriacetic Acid. Environ. Mol. Mutagen., 49 (6): 439-452.
|
|
(22)
|
Brusick D. (1986). Genotoxic Effects in Cultured Mammalian Cells Produced by Low pH Treatment Conditions and Increased Ion Concentrations. Environ. Mutagen., 8 (6): 879-886.
|
|
(23)
|
Morita, T., Nagaki, T., Fukuda, I. and Okumura, K. (1992). Clastogenicity of Low pH to Various Cultured Mammalian Cells. Mutation Res., 268 (2): 297-305.
|
|
(24)
|
Scott, D., Galloway, S.M., Marshall, R.R., Ishidate, M.Jr, Brusick, D., Ashby, J. and Myhr, B.C. (1991). Genotoxicity under Extreme Culture Conditions. A report from ICPEMC Task Group 9. Mutation Res., 257: 147-204.
|
|
(25)
|
Wang J., Heflich R.H. and Moore M.M. (2007). A Method to Distinguish Between the De Novo Induction of Thymidine Kinase Mutants and the Selection of Pre-Existing Thymidine Kinase Mutants in the Mouse Lymphoma Assay. Mutation Res., 626 (1-2): 185-190.
|
|
(26)
|
Fischer, G.A. (1958). Studies on the Culture of Leukemic Cells In Vitro. Ann. N.Y. Academy Sci., 76: 673-680.
|
|
(27)
|
Clive, D., Johnson, K.O., Spector, J.F.S., Batson, A.G. and Brown, M.M.M. (1979). Validation and Characterization of the L5178Y/TK+/– Mouse Lymphoma Mutagen Assay System. Mutation Res., 59(1): 61-108.
|
|
(28)
|
Sawyer, J., Moore, M.M., Clive, D. and Hozier, J. (1985). Cytogenetic Characterization of the L5178Y TK+/- 3.7.2C Mouse Lymphoma Cell Line, Mutation Res., 147 (5): 243-253.
|
|
(29)
|
Sawyer J.R., Moore M.M. and Hozier J.C. (1989). High-Resolution Cytogenetic Characterization of the L5178Y TK+/- Mouse Lymphoma Cell Line, Mutation Res., 214 (2): 181-193.
|
|
(30)
|
Sawyer, J.R., Binz, R.L., Wang, J. and Moore, M.M. (2006). Multicolor Spectral Karyotyping of the L5178Y TK+/--3.7.2C Mouse Lymphoma Cell Line, Environ. Mol. Mutagen., 47 (2): 127-131.
|
|
(31)
|
Fellows, M.D., McDermott, A., Clare, K.R., Doherty, A. and Aardema, M.J. (2014). The Spectral Karyotype of L5178Y TK+/- Mouse Lymphoma Cells Clone 3.7.2C and Factors Affecting Mutant Frequency at the Thymidine Kinase (TK) Locus in the Microtitre Mouse Lymphoma Assay, Environ. Mol. Mutagen., 55 (1): 35-42.
|
|
(32)
|
Storer, R.D., Jraynak, A.R., McKelvey, T.W., Elia, M.C., Goodrow, T.L. and DeLuca, J.G. (1997). The Mouse Lymphoma L5178Y TK+/- Cell Line is Heterozygous for a Codon 170 Mutation in the P53 Tumor Suppressor Gene. Mutation. Res., 373 (2): 157-165.
|
|
(33)
|
Clark L.S., Harrington-Brock, K., Wang, J., Sargent, L., Lowry, D., Reynolds, S.H. and Moore, M.M. (2004). Loss of P53 Heterozygosity is not Responsible for the Small Colony Thymidine Kinase Mutant Phenotype in L5178Y Mouse Lymphoma Cells. Mutagen., 19 (4): 263-268.
|
|
(34)
|
Skopek T.R., Liber, H.L., Penman, B.W. and Thilly, W.G. (1978). Isolation of a Human Lymphoblastoid Line Heterozygous at the Thymidine Kinase Locus: Possibility for a Rapid Human Cell Mutation Assay. Biochem. Biophys. Res. Commun., 84 (2): 411–416.
|
|
(35)
|
Honma M. (2005). Generation of Loss of Heterozygosity and its Dependency on P53 Status in Human Lymphoblastoid Cells. Environ. Mol. Mutagen., 45 (2-3): 162-176.
|
|
(36)
|
Xia, F., Wang, X., Wang, Y.H., Tsang, N.M., Yandell, D.W., Kelsey, K.T. and Liber, H.L. (1995). Altered P53 Status Correlates with Differences in Sensitivity to Radiation-Induced Mutation and Apoptosis in Two Closely Related Human Lymphoblast Lines. Cancer. Res., 55 (1): 12-15.
|
|
(37)
|
Lorge, E., M. Moore, J. Clements, M. O Donovan, M. Honma, A. Kohara, J. van Benthem, S. Galloway, M.J. Armstrong, V. Thybaud, B. Gollapudi, M. Aardema, J. Kim, A. Sutter, D.J. Kirkland (2015). Standardized Cell Sources and Recommendations for Good Cell Culture Practices in Genotoxicity Testing. (Manuscript in preparation).
|
|
(38)
|
Lloyd M. and Kidd D. (2012). The Mouse Lymphoma Assay. Springer Protocols: Methods in Molecular Biology 817, Genetic Toxicology Principles and Methods, ed. Parry and Parry, Humana Press. ISBN, 978-1-61779-420-9, 35-54.
|
|
(39)
|
Mei N., Guo X. and Moore M.M. (2014). Methods for Using the Mouse Lymphoma Assay to Screen for Chemical Mutagenicity and Photo-Mutagenicity. In: Optimization in Drug Discover: In Vitro Methods: Yan Z and Caldwell(Eds) , 2nd Edition, GW; Humana Press, Totowa, NJ.
|
|
(40)
|
Liber H.L. and Thilly W.G. (1982). Mutation Assay at the Thymidine Kinase Locus in Diploidhuman Lymphoblasts. Mutation Res., 94 (2): 467-485.
|
|
(41)
|
Coecke, S., Balls, M., Bowe, G., Davis, J., Gstraunthaler, G., Hartung, T., Hay, R., Merten, OW., Price, A., Schechtman, L., Stacey, G. and Stokes, W. (2005). Guidance on Good Cell Culture Practice. A Report of the Second ECVAM Task Force on Good Cell Culture Practice. ATLA, 33 (3): 261-287.
|
|
(42)
|
Moore M.M. and Howard B.E. (1982). Quantitation of Small Colony Trifluorothymidine-Resistant Mutants of L5178Y/TK+/- Mouse Lymphoma Cells in RPMI-1640 Medium, Mutation Res., 104 (4-5): 287-294.
|
|
(43)
|
Ames B.N., McCann J. and Yamasaki E. (1975). Methods for Detecting Carcinogens and Mutagens with the Salmonella/Mammalian Microsome Mutagenicity Test. Mutation Res., 31 (6): 347-364.
|
|
(44)
|
Maron D.M. and Ames B.N. (1983). Revised Methods for the Salmonella Mutagenicity Test. Mutation Res., 113 (3-4): 173-215.
|
|
(45)
|
Natarajan, A.T., Tates, A.D, Van Buul, P.P.W., Meijers, M. and De Vogel, N. (1976). Cytogenetic Effects of Mutagens/Carcinogens After Activation in a Microsomal System In Vitro, I. Induction of Chromosomal Aberrations and Sister Chromatid Exchanges by Diethylnitrosamine (DEN) and Dimethylnitrosamine (DMN) in CHO Cells in the Presence of Rat-Liver Microsomes. Mutation Res., 37 (1): 83-90.
|
|
(46)
|
Matsuoka A., Hayashi M. and Ishidate M. Jr. (1979). Chromosomal Aberration Tests on 29 Chemicals Combined with S9 Mix In Vitro. Mutation Res., 66 (3): 277-290.
|
|
(47)
|
Ong T.M., et al. (1980). Differential Effects of Cytochrome P450-Inducers on Promutagen Activation Capabilities and Enzymatic Activities of S-9 from Rat Liver, J. Environ. Pathol. Toxicol., 4 (1): 55-65
|
|
(48)
|
Elliott, B.M., Combes, R.D., Elcombe, C.R., Gatehouse, D.G., Gibson, G.G., Mackay, J.M. and Wolf, R.C. (1992). Report of UK Environmental Mutagen Society Working Party. Alternatives to Aroclor 1254-Induced S9 in In Vitro Genotoxicity Assays. Mutagen., 7 (3): 175-177.
|
|
(49)
|
Matsushima, T., Sawamura, M., Hara, K. and Sugimura, T. (1976). A Safe Substitute for Polychlorinated Biphenyls as an Inducer of Metabolic Activation Systems. In: In Vitro Metabolic Activation in Mutagenesis Testing. de Serres F.J., et al. (Eds, Elsevier, North-Holland, pp. 85-88.
|
|
(50)
|
Galloway S.M., et al. (1994). Report from Working Group on In Vitro Tests for Chromosomal Aberrations. Mutation Res., 312 (3): 241-261.
|
|
(51)
|
Johnson T.E., Umbenhauer D.R. and Galloway S.M. (1996). Human Liver S-9 Metabolic Activation: Proficiency in Cytogenetic Assays and Comparison with Phenobarbital/Beta-Naphthoflavone or Aroclor 1254 Induced Rat S-9, Environ. Mol. Mutagen., 28 (1): 51-59.
|
|
(52)
|
UNEP (2001). Stockholm Convention on Persistent Organic Pollutants, United Nations Environment Programme (UNEP).
|
|
(53)
|
Krahn D.F., Barsky F.C. and McCooey K.T. (1982). CHO/HGPRT Mutation Assay: Evaluation of Gases and Volatile Liquids. In: Genotoxic Effects of Airborne Agents Tice R.R., Costa D.L.and Schaich K.M. (Eds.). New York, Plenum, pp. 91-103.
|
|
(54)
|
Zamora, P.O., Benson, J.M., Li, A.P. and Brooks, A.L. (1983). Evaluation of an Exposure System Using Cells Grown on Collagen Gels for Detecting Highly Volatile Mutagens in the CHO/HGPRT Mutation Assay. Environ. Mutagen., 5 (6): 795-801.
|
|
(55)
|
Asakura M., Sasaki T., Sugiyama T., Arito H., Fukushima, S. and Matsushima, T. (2008). An Improved System for Exposure of Cultured Mammalian Cells to Gaseous Compounds in the Chromosomal Aberration Assay. Mutation Res., 652 (2): 122-130.
|
|
(56)
|
Arlett C.F., et al. (1989). Mammalian Cell Gene Mutation Assays Based upon Colony Formation. In: Statistical Evaluation of Mutagenicity Test Data, Kirkland, D.J. (Ed.), CambridgeUniversity Press, pp. 66-101.
|
|
(57)
|
Morita T., Honma M. and Morikawa K. (2012). Effect of Reducing the Top Concentration Used in the In Vitro Chromosomal Aberration Test in CHL Cells on the Evaluation of Industrial Chemical Genotoxicity. Mutation Res., 741 (1-2): 32-56.
|
|
(58)
|
Brookmire L., Chen J.J. and Levy D.D. (2013). Evaluation of the Highest Concentrations Used in the In Vitro Chromosome Aberrations Assay. Environ. Mol. Mutagen., 54 (1): 36-43.
|
|
(59)
|
USFDA (2012). International Conference on Harmonisation (ICH) Guidance S2 (R1) on Genotoxicity Testing and Data Interpretation for Pharmaceuticals Intended For Human Use. Available at: [https://www.federalregister.gov/a/2012-13774].
|
|
(60)
|
Honma M. and Hayashi M. (2011). Comparison of In Vitro Micronucleus and Gene Mutation Assay Results for P53-Competent Versus P53-Deficient Human Lymphoblastoid Cells. Environ. Mol. Mutagen., 52 (5): 373-384.
|
|
(61)
|
Moore-Brown, M.M., Clive, D., Howard, B.E., Batson, A.G. and Johnson, K.O. (1981). The Utilization of Trifluorothymidine (TFT) to Select for Thymidine Kinase-Deficient (TK-/-) Mutants from L5178Y/TK+/- Mouse Lymphoma Cells, Mutation Res., 85 (5): 363-378.
|
|
(62)
|
Liber H.L., Yandell D.W. and Little J.B. (1989). A Comparison of Mutation Induction at the TK and HRPT Loci in Human Lymphoblastoid Cells; Quantitative Differences are Due to an Additional Class of Mutations at the Autosomal TK locus. Mutation Res., 216 (1): 9-17.
|
|
(63)
|
Furth E.E., Thilly, W.G., Penman, B.W., Liber, H.L. and Rand, W.M. (1981). Quantitative Assay for Mutation in Diploid Human Lymphoblasts Using Microtiter Plates. Anal. Biochem., 110 (1): 1-8.
|
|
(64)
|
Hayashi, M, Dearfield, K., Kasper, P., Lovell, D., Martus, H. J. and Thybaud, V. (2011). Compilation and Use of Genetic Toxicity Historical Control Data, Mutation Res., 723 (2): 87-90.
|
|
(65)
|
Ryan T.P. (2000). Statistical Methods for Quality Improvement. John Wiley and Sons, New York 2nd Edition.
|
|
(66)
|
OECD (2014). Statistical analysis supporting the revision of the genotoxicity Test Guidelines. Environmental, Health and Safety Publications, Series on Testing and Assessment (No 199.), Organisation for Economic Cooperation and Development, Paris.
|
|
(67)
|
Fleiss J.L., Levin B. and Paik M.C. (2003). Statistical Methods for Rates and Proportions, Third Edition, New York: John Wiley & Sons.
|
Допълнение 1
ОПРЕДЕЛЕНИЯ
Анеуген
: всеки химикал или процес, който води до анеуплодия в клетките или организмите чрез взаимодействие с компонентите на митотичния и мейотичния цикъл на клетъчно делене.
Анеуплоидия
: всяко отклонение от обичайния диплоиден (или хаплоиден) брой на хромозомите с една или повече от една хромозома, но не и с цял набор (или набори) от хромозоми (полиплоидия).
Мутагени, предизвикващи заместване на двойка бази
: химикали, предизвикващи заместване на двойка бази в ДНК.
Химикал
: Вещество или смес.
Ефикасност на клониране
: Процентът на клетките, посети с ниска гъстота, които са в състояние да растат в колония, която може да бъде преброена.
Кластоген
: всеки химикал или процес, който причинява структурни аберации от хромозомен тип в популации от клетки или организми.
Цитотоксичност
: за изследванията, обхванати от настоящия метод за изпитване, цитотоксичността се определя като намаляване на относителния общ растеж (RTG) или относителното оцеляване (RS) съответно за MLA и TK6.
Мутация в поколението напред
: генна мутация от родителския тип в мутанта, от която възниква изменение или загуба на ензимна активност или на функцията на кодирания белтък.
Мутагени с изместване на рамката на разчитане
: химикали, които причиняват добавяне или делеция на една или много двойки бази в молекулата на ДНК.
Генотоксичен
: общ термин, обхващащ всички видове увреждане на ДНК или хромозоми, включително разкъсвания на ДНК, адукти, пренареждания, мутации, хромозомни аберации и анеуплоидия. Не всички типове генотоксични ефекти водят до мутации или трайно увреждане на хромозомите.
Митотична рекомбинация
: рекомбинация, по време на митозата, между хомоложни хроматиди, евентуално водеща до индуциране на разкъсвания на двойната спирала на ДНК или до загуба на хетерозиготност.
Мутагенен
: води до наследствена промяна на ДНК последователност(и) на базовите двойки в гени, или на структурата на хромозоми (хромозомни аберации).
Честота на мутации (MF)
: Броят на наблюдаваните мутантни клетки, разделен на броя на жизнеспособните клетки.
Време на фенотипно изразяване
: Времето след третирането, през което генетичното изменение се фиксира в генома, и всички съществуващи преди това генетични продукти се изчерпват до момента, в който фенотипният признак се променя.
Относителна преживяемост (RS)
: RS се използва като мярка за свързана с третирането цитотоксичност в ТК6. RS е ефикасността на клониране (CE) на клетки, които са посети непосредствено след третирането, с корекция за всяка загуба на клетки по време на третирането, в сравнение с ефикасността на клониране на отрицателната контрола.
Относителен растеж на суспензия (RSG)
: за MLA — относителният общ двудневен растеж на суспензията на изпитваната култура в сравнение с общия двудневен растеж на суспензията на отрицателната контрола/контролата на разтворителя (Clive and Spector, 1975). RSG следва да включва относителния растеж на изпитваната култура в сравнение с отрицателната/на разтворителя контрола по време на периода на третиране.
Относителен общ растеж (RTG)
: RTG се използва като мярка за свързана с третирането цитотоксичност при MLA. Той е мярка за относителен (спрямо контролния носител) растеж на изпитвани култури по време на фазите на третиране, двудневна експресия и избор на клониран мутант на изпитването. RSG на всяка изпитвана култура се умножава на относителната ефикасност на клониране на изпитваната култура към момента на избор на мутант и се изразява по отношение на ефикасността на клониране на отрицателната/на разтворителя контрола (Clive and Spector, 1975).
S9 фракции от черен дроб
: супернатант от хомогенат на черен дроб след центрофугиране при 9 000 g, т.е. необработен екстракт от черен дроб
S9 микс
: микс от S9 фракцията от черен дроб и кофактори, необходими за метаболитна ензимна активност.
Растеж на суспензия (SG)
: Увеличението в пъти на броя на клетките по време на фазите на третиране и експресия на MLA. SG се изчислява чрез умножаване на увеличението в пъти в ден 1 по увеличението в пъти в ден 2 за краткотрайно (3 или 4 часа) третиране. Ако се използва 24-часово третиране, SG е увеличението в пъти по време на 24-часовото третиране, умножено на увеличенията в пъти в дните 1 и 2 на експресия.
Контрола на разтворител
: общ термин за определяне на контролните култури, които са само на разтворителя, който се използва за разтваряне на изпитвания химикал.
Изпитван химикал
: Всяко вещество или смес, изпитвано(а) с използване на настоящия метод за изпитване.
Нетретирани контроли
: култури, които не се третират (т.е. без изпитвания химикал и без разтворител), но се обработват паралелно и по същия начин като културите, които се третират с изпитвания химикал.
Допълнение 2
ФОРМУЛИ
Цитотоксичност
И за двете версии на MLA (с агар и микроямка)
Цитотоксичността се определя като относителния общ растеж (RTG), който включва относителния растеж на суспензията (RSG) по време на 2-дневния период на експресия, и относителната ефикасност на клониране (RCE), получена към момента на избор на мутант. RTG, RSG и RCE се изразяват като проценти.
Изчисляване на RSG: Растеж на суспензията едно (SG1) е скоростта на растеж между ден 0 и ден 1 (клетъчна концентрация в ден 1 / клетъчна концентрация в ден 0), растеж на суспензията две (SG2) е скоростта на растеж между ден 1 и ден 2 (клетъчна концентрация в ден 2 / клетъчна концентрация в ден 1). RSG е общият SG (SG1 x SG2) за третираната култура в сравнение с нетретираната контрола/контролата на разтворителя. Т.е.: RSG = [SG1(изпитван) x SG2(изпитван)] / [SG1(контрола) x SG2(контрола)] SG1 следва да се изчисли от първоначалната клетъчна концентрация, използвана в началото на третирането на клетките. Това има значение за всяка диференциална цитотоксичност, която възниква в изпитваната(ите) култура(и) по време на третирането на клетките.
RCE е относителната ефикасност на клониране на изпитваната култура в сравнение с относителната ефикасност на клониране на нетретираната/на разтворителя контрола, получена към момента на избор на мутант.
Относителен общ растеж (RTG):
RTG=RSG x RCE
TK6
Относително оцеляване (RS):
Цитотоксичността се оценява чрез относително оцеляване, т.е. ефикасността на клониране (CE) на клетките, посети непосредствено след третирането, коригирана за всяка загуба на клетки по време на третирането, в сравнение с ефикасността на клониране при отрицателните контроли (на които е присвоена стойност 100 % за оцеляване). Коригирането за загуба на клетки по време на третиране може да се изчисли като:
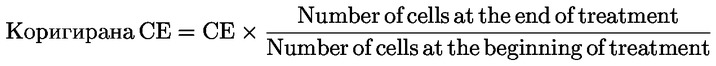
RS за култура, третирана с изпитван химикал, се изчислява като:

Честота на мутации и за MLA, и за TK6
Честотата на мутации (MF) е ефикасността на клониране на мутантни колонии в селективна среда (СЕМ), коригирана с ефикасността на клониране в неселективна среда към момента на избор на мутант (CEV). Т.е. MF=CEM/CEV. Изчисляването на тези две ефикасности на клониране се описва по-долу за методите на клониране с агар и микроямки.
Версия на MLA с агар: Във версията на MLA с мек агар броят на колониите върху плаката за избор на мутант (CM) и броят на колониите върху плаката за неселектирани или ефикасност на клониране (брой жизнеспособни) (CV) се получава чрез директно преброяване на клонингите. Когато се посяват 600 клетки в плаки за ефикасност на клониране (CE) за избор на мутант (CEM) и плаки за неселектирани или ефикасност на клониране (брой жизнеспособни) (CEV) и 3 x 106 клетки се използват за избор на мутант,
CEM = CM / (3 x 106) = (CM / 3) x 10-6
CEV = CV / 600
Версия на MLA и TK6 с микроямки:
Във версията на MLA с микроямки CM и CV се определят като продукта от общия брой микроямки (TW) и вероятния брой колонии на ямка (P) в плаките с микроямки.
CM = PM x TWM
CV = PV x TWV
От нулевата точка на разпределението на Поасон (Furth et al., 1981), P се получава чрез
P = - ln (EW / TW),
където EW е броят на празни ямки, а TW е общият брой ямки. Следователно:
CEM = CM / TM = (PM x TWM) / TM
CEV = CV / TV = (PV x TWV) / TV
За версията на MLA с микроямки честотите на малки и големи колонии мутанти се изчислява по същия начин, с използване на съответния брой празни ямки за малки и големи колонии.
За ТК6 честотите на малки и големи колонии мутанти се базират на рано появяващите се и късно появяващите се мутанти.
Б.68 IN VITRO МЕТОД ЗА ИЗПИТВАНЕ НА КРАТКОТРАЙНА ЕКСПОЗИЦИЯ ЗА ИДЕНТИФИЦИРАНЕ НА i) ХИМИКАЛИ, ПРИЧИНЯВАЩИ СЕРИОЗНО УВРЕЖДАНЕ НА ОЧИТЕ И ii) ХИМИКАЛИ, НЕИЗИСКВАЩИ КЛАСИФИЦИРАНЕ ЗА ДРАЗНЕЩО ДЕЙСТВИЕ ВЪРХУ ОЧИТЕ ИЛИ СЕРИОЗНО УВРЕЖДАНЕ НА ОЧИТЕ
ВЪВЕДЕНИЕ
|
1.
|
Настоящият метод за изпитване е еквивалентен на насоките за изпитване на ОИСР (ТG) 491 (2017). Методът за изпитване на краткотрайна експозиция (STE) е in vitro метод, който може да се използва при определени обстоятелства и със специфични ограничения за класифициране на опасността и етикетиране на химикали (вещества и смеси), които причиняват сериозно увреждане на очите, както и на тези, които не изискват класифициране за дразнещо действие върху очите или сериозно увреждане на очите, определено от Глобалната хармонизирана система на Организацията на обединените нации (ООН) за класифициране и етикетиране на химикали (GHS) (1) и Регламента на Европейския съюз (ЕС) 1272/2008 за класифициране, етикетиране и опаковане на вещества и смеси (CLP) (68).
|
|
2.
|
В продължение на много години потенциалната опасност за очите на химикали е оценявана предимно с използване на in vivo изпитване върху очи на зайци (TM B.5 (8), еквивалентен на ОИСР TG 405). Понастоящем е общоприето, че в обозримо бъдеще, нито едно алтернативно изпитване in vitro за дразнене на очите няма да е в състояние да замести напълно изпитването in vivo върху очи на зайци при прогнозирането в целия спектър на сериозно увреждане на очите/дразнене на очите, за различните класове химикали. При все това стратегическите комбинации от няколко алтернативни методи на изпитване в рамките на стратегия за (поетапно) изпитване може да са в състояние напълно да заменят изпитването върху очи на зайци (2). Подходът „отгоре надолу“ е създаден да се използва, когато въз основа на съществуваща информация за даден химикал се очаква висок потенциал за дразнещо действие или причиняване на сериозно увреждане на очите. От друга страна, подходът „отдолу нагоре“ е създаден да се използва, когато въз основа на съществуващата информация за даден химикал се очаква, че няма да причини дразнене на очите, което да изисква класифициране. Въпреки че методът за изпитване STE не се смята като заместващ напълно изпитването in vivo върху очи на зайци, той е подходящ за използване като част от стратегия за поетапно изпитване за регулаторно класифициране и етикетиране, като подхода „отгоре надолу“/„отдолу нагоре“, за идентифициране без последващо изпитване на (i) химикали, причиняващи сериозно увреждане на очите (категория 1 по GHS на ООН/CLP) и (ii) химикали (с изключение на силно летливи вещества и всички твърди химикали, различни от повърхностноактивни вещества), които не изискват класифициране за дразнене на очите или сериозно увреждане на очите (без категория по GHS на ООН/CLP) (1)(2). Независимо от това, даден химикал, за който не се прогнозира, че причинява сериозно увреждане на очите (категория 1 по GHS на ООН/CLP) или е без категория по GHS на ООН/CLP (не причинява сериозно увреждане на очите или дразнене на очите) с метода за изпитване STE, изисква допълнително изпитване за определяне на окончателно класифициране. Освен това е необходима консултация със съответните регулаторни органи, преди да се използва STE в подход „отдолу нагоре“ в схеми на класифициране, различни от тези на GHS на ООН/CLP. Изборът на най-подходящия метод за изпитване и използването на този метод за изпитване следва да се разглежда в контекста на насоките на ОИСР за интегрирани подходи за изпитване и оценка на сериозно увреждане на очите и дразнене на очите (14).
|
|
3.
|
Целта на настоящия метод за изпитване е да се опишат процедурите, използвани за оценка на потенциалната опасност за очите на даден изпитван химикал въз основа на неговата способност да предизвиква цитотоксичност при метода на изпитване с краткотрайна експозиция. Цитотоксичният ефект на химикали върху епителните клетки на роговицата е важен механизъм на действие (МОА), водещ до увреждане на роговичния епител и дразнене на очите. Клетъчната жизнеспособност в метода за изпитване STE се оценява чрез количествено измерване, след екстракция от клетки, на синя формазанова сол, която се произвежда от живите клетки чрез ензимно преобразуване на виталния оцветител МТТ (3-(4,5-диметилтиазол-2-ил)-2,5-дифенилтетразолиев бромид), известен също като тиазолил синьо тетразолиев бромид (3). Получената клетъчна жизнеспособност се сравнява с контролата на разтворителя (относителна жизнеспособност) и се използва за оценка на потенциалната опасност за очите на изпитвания химикал. Даден изпитван химикал се класифицира като категория 1 по GHS на ООН/CLP, когато и двете концентрации от 5 % и 0,05 % водят до клетъчна жизнеспособност по-малка или равна на (≤) 70 %. От друга страна, даден химикал се прогнозира, че е без категория по GHS на ООН/CLP, когато и двете концентрации от 5 % и 0,05 % водят до клетъчна жизнеспособност по-голяма от (>) 70 %.
|
|
4.
|
Терминът „изпитван химикал“ се използва в този метод за изпитване за обозначаване на това, какво се изпитва, и не е свързан с приложимостта на метода за изпитване STE за изпитване на вещества и/или смеси. Определенията са дадени в допълнение 1.
|
ПЪРВОНАЧАЛНИ СЪОБРАЖЕНИЯ И ОГРАНИЧЕНИЯ
|
5.
|
Този метод за изпитване се базира на протокола, разработен от Kao Corporation (4), който е предмет на две различни изпитвания за валидиране: едно от валидиращия комитет на Японското дружество за алтернативни методиза изпитване върху животни (JSAAE) (5) и друго от Японския център за валидиране на алтернативни методи (JaCVAM) (6). Извършена е партньорска проверка от NICEATM/ICCVAM въз основа на докладите от изпитването за валидиране и предходен преглед на документите на метода за изпитване (7).
|
|
6.
|
Когато се използва за идентифициране на химикали (вещества и смеси), предизвикващи сериозно увреждане на очите (категория 1 по GHS на ООН/CLP) (1), получените с метода на изпитване STE данни за 125 химикали (включващи и вещества, и смеси) са с обща точност от 83 % (104/125), процент неверни положителни резултати 1 % (1/86) и процент неверни отрицателни резултати 51 % (20/39) в сравнение с данните от метода за изпитване in vivo върху очи на зайци (7). Полученият процент неверни отрицателни резултати в настоящия контекст не е от критично значение, тъй като всички изпитвани химикали, които предизвикват клетъчна жизнеспособност ≤ 70 % при 5 % концентрация и > 70 % при 0,05 % концентрация, трябва да бъдат впоследствие изпитвани с други достатъчно валидирани in vitro методи за изпитване или, като последна възможност, с изпитването in vivo върху очи на зайци, в зависимост от регулаторните изисквания и в съответствие със стратегията за последователно изпитване и подходите за тежест на доказателства, които понастоящем се препоръчват (1) (8). Изпитвани са предимно вещества с една съставка, въпреки че има ограничени данни и за изпитването на смеси. Независимо от това методът за изпитване е технически приложим за изпитването на вещества с повече съставки и на смеси. Въпреки това, преди използването на настоящия метод за изпитване по отношение на смес с цел генериране на данни за планирана регулаторна цел, следва да се разгледа въпросът дали той може да даде адекватни резултати за тази цел и, ако е така, защо. Подобни съображения не са необходими, когато има регулаторно изискване за изпитване на сместа. Методът за изпитване STE не проявява други специфични недостатъци, когато се използва за идентифициране на изпитвани химикали от категория 1 по GHS на ООН/CLP. Изследователите биха могли да разгледат възможността за използване на този метод за изпитвани химикали, при които клетъчна жизнеспособност ≤ 70 % при 5 % и 0,05 % концентрация следва да се приеме като показателна за отклик, предизвикващ сериозно увреждане на очите, който следва да бъде класифициран като категория 1 по GHS на ООН/CLP без допълнително изпитване.
|
|
7.
|
Когато се използва за идентифициране на химикали (вещества и смеси), не изискващи класифициране за дразнене на очите и сериозно увреждане на очите (т.е. без категория по GHS на ООН/CLP), получените с метода за изпитване STE данни за 130 химикали (включващи и вещества, и смеси), са с обща точност от 85 % (110/130), процент неверни отрицателни резултати 12 % (9/73) и процент неверни положителни резултати 19 % (11/57) в сравнение с изпитването in vivo върху очи на зайци (7). Ако силно летливи вещества и твърди вещества, различни от повърхностноактивни вещества, се изключат от набора данни, общата точност се подобрява до 90 % (92/102), процентът неверни отрицателни резултати до 2 % (1/54) и процентът неверни положителни резултати до 19 % (9/48) (7). Вследствие на това потенциалните недостатъци на метода за изпитване на STE, когато се използва за идентифициране на изпитвани химикали, които не изискват класифициране за дразнене на очите и сериозно увреждане на очите (без категория по GHS на ООН/Регламент CLP), са с висок процент неверни отрицателни резултати за i) силно летливи вещества с налягане на пари над 6 kPa и ii) твърди химикали (вещества и смеси), различни от повърхностноактивни вещества, и смеси, съставени само от повърхностноактивни вещества. Тези химикали се изключват от областта на приложимост на метода за изпитване STE (7).
|
|
8.
|
Освен химикалите, споменати в точки 6 и 7, методът за изпитване STE генерира набор от данни, който съдържа и вътрешни данни за 40 смеси, които, при сравнение с изпитването in vivo на Draize за действие върху очите, демонстрират точност от 88 % (35/40), процент неверни положителни резултати 50 % (5/10) и процент неверни отрицателни резултати 0 % (0/30) за прогнозиране на смеси, които не изискват класифициране по системите за класификация GHS на ООН/CLP (9). Следователно методът за изпитване STE може да се прилага за идентифициране на смеси без категория по GHS на ООН/CLP при подход „отдолу нагоре“, с изключение на твърди смеси, различни от тези, които са съставени само от повърхностноактивни вещества, като разширение на неговото ограничение по отношение на твърди вещества. Освен това смесите, съдържащи вещества с налягане на пари по-високо от 6kPa, следва да се оценяват внимателно, за да се избегнат потенциални класифицирания на по-ниско ниво, и следва да се обосновават за всеки конкретен случай.
|
|
9.
|
Методът за изпитване STE не може да се използва за идентифициране на изпитвани химикали, които следва да бъдат класифицирани като категория 2 по GHS на ООН/CLP или категория 2А (дразнещи очите) по GHS на ООН/CLP, или 2В (леко дразнещи очите), поради значителния брой химикали от категория 1 по GHS на ООН/CLP, класифицирани на по-ниско ниво като категория 2, 2А или 2В или химикали без категория по GHS на ООН/CLP, класифицирани на по-високо ниво като 2, 2А или 2В (7). За тази цел може да се изисква допълнително изпитване с друг подходящ метод.
|
|
10.
|
Методът за изпитване STE е подходящ за изпитвани химикали, които се разтварят или суспендират равномерно в продължение на поне 5 минути във физиологичен разтвор, 5 % диметил сулфоксид (DMSO) във физиологичен разтвор или минерално масло. Методът за изпитване STE не е подходящ за изпитвани химикали, които са неразтворими или не могат да се суспендират равномерно в продължение на поне 5 минути във физиологичен разтвор, 5 % DMSO във физиологичен разтвор или минерално масло. Използването на минерално масло в метода за изпитване STE е възможно поради краткотрайната експозиция. Следователно методът за изпитване STE е подходящ за прогнозиране на потенциалната опасност за очите на разтворими във вода изпитвани химикали (напр. мастни алкохоли с дълга верига или кетони), при условие че те са смесими с поне един от трите предложени по-горе разтворители (4).
|
|
11.
|
Терминът „изпитван химикал“ се използва в този метод за изпитване за обозначаване на това, какво се изпитва (69), и не е свързан с приложимостта на метода за изпитване STE за изпитване на вещества и/или смеси.
|
ПРИНЦИП НА ИЗПИТВАНЕТО
|
12.
|
Методът за изпитване STE е базиращ се на цитотоксичност in vitro анализ, който се извършва върху сливащ се монослой на клетки от роговица на зайци на Statens Seruminstitut (SIRC), култивирани в 96-ямкова микроплака от поликарбонат (4). След петминутна експозиция на даден изпитван химикал цитотоксичността се измерва количествено като относителната жизнеспособност на SIRC клетките с използване на анализа МТТ (4). Намалена клетъчна жизнеспособност се използва за прогнозиране на потенциални нежелани ефекти, водещи до очно увреждане.
|
|
13.
|
Съобщава се, че 80 % от даден разтвор, капнат в око на зайци, се екскретира чрез конюктивалния сак в рамките на три до четири минути, докато повече от 80 % от даден разтвор, капнат в човешко око, се ескретира в рамките на една до две минути (10). Методът за изпитване STE опитва да апроксимира тези времена на експозиция и да използва цитотоксичността като крайна точка за оценка на степента на увреждане на SIRC клетките след петминутна експозиция на изпитвания химикал.
|
ДОКАЗВАНЕ НА КОМПЕТЕНТНОСТ
|
14.
|
Преди рутинната употреба на метода за изпитване STE, описан в настоящия метод за изпитване, лабораториите следва да демонстрират техническа компетентност чрез правилно класифициране на единадесетте вещества, препоръчани в Таблица 1. Тези вещества са избрани да представят целия диапазон на отговори за сериозно увреждане на очите или дразнене на очите въз основа на резултати от in vivo изпитвания върху очи на зайци (TG 405) и системата за класифициране GHS на ООН/CLP (1). Другите критерии за избор включват, че веществата следва да са достъпни на пазара, че следва да има налични висококачествени in vivo референтни данни и че висококачествените in vitro данни от метода за изпитване STE следва да са налични (3). При ситуации, в които дадено вписано вещество не е налично или когато е оправдано може да се използва друго вещество, за което има налични адекватни in vivo и in vitro референтни данни, при условие че се прилагат същите критерии, които са описани тук.
Таблица 1
Списък на опитните вещества
|
Вещество
|
CAS №
|
Химичен клас (70)
|
Агрегатно състояние
|
In Vivo GHS на ООН/CLP кат. (71)
|
Разтворител в изпитване STE
|
STE GHS по ООН/CLP кат.
|
|
Бензалкониев хлорид (10 %, воден разтвор)
|
8 001 -54-5
|
Ониево съединение
|
Течност
|
Категория 1
|
Физиологичен разтвор
|
Категория 1
|
|
Triton X-100 (100 %)
|
9 002 -93-1
|
Етер
|
Течност
|
Категория 1
|
Физиологичен разтвор
|
Категория 1
|
|
Кисело червено 92
|
18 472 -87-2
|
Хетероциклично съединение Бромно съединение Хлорирано съединение
|
Твърд
|
Категория 1
|
Физиологичен разтвор
|
Категория 1
|
|
Натриев хидроксид
|
1 310 -73-2
|
Основи; Неорганичен химикал
|
Твърд
|
Категория 1 (72)
|
Физиологичен разтвор
|
Категория 1
|
|
Бутиролактон
|
96-48-0
|
Лактон; Хетероциклично съединение
|
Течност
|
Категория 2A (Категория 2 по CLP)
|
Физиологичен разтвор
|
Не може да се направи прогноза
|
|
1-октанол
|
111-87-5
|
Алкохол
|
Течност
|
Категория 2A/B (73) (категория 2 по CLP)
|
Минерално масло
|
Не може да се направи прогноза
|
|
Циклопентанол
|
96-41-3
|
Алкохол; Въглеводород (цикличен)
|
Течност
|
Категория 2A/B (74) (категория 2 по CLP)
|
Физиологичен разтвор
|
Не може да се направи прогноза
|
|
2-етоксиетил ацетат
|
111-15-9
|
Алкохол; Етер
|
Течност
|
Без категория
|
Физиологичен разтвор
|
Без категория
|
|
Додекан
|
112-40-3
|
Въглеводород (ацикличен)
|
Течност
|
Без категория
|
Минерално масло
|
Без категория
|
|
Метилизобутилкетон
|
108-10-1
|
Кетон
|
Течност
|
Без категория
|
Минерално масло
|
Без категория
|
|
1,1-диметилгуанидин сулфат
|
598-65-2
|
Амидин; Сярно съединение
|
Твърд
|
Без категория
|
Физиологичен разтвор
|
Без категория
|
|
Съкращения: CAS RN = Номер в регистъра на Химическата реферативна служба
|
|
ПРОЦЕДУРА
Приготвяне на клетъчния монослой
|
15.
|
За провеждане на метода за изпитване STE следва да се използва клетъчната линия на роговица от зайци SIRC. Препоръчва се SIRC клетките да се получат от добре квалифицирана клетъчна банка, като CCL60 от Американската колекция на типови култури.
|
|
16.
|
SIRC клетките се култивират при 37 °C, 5 % CO2 и влажна атмосфера в съд за култивиране, съдържащ среда за отглеждане, която се състои от минимална основна среда на Eagle (MEM), допълнена с 10 % фетален говежди серум (FBS), 2 mM L-глутамин, 50–100 единици/ml пеницилин и 50–100 μg/ml стрептомицин. Клетките, които са се слели в съда за култивиране, следва да се разделят с използване на разтвор на трипсин-етилендиаминтетраоцетна киселина, със или без използване на прибор за остъргване на клетки. Клетките се размножават (напр. 2 до 3 преминавания) в съд за култивиране, преди да бъдат използвани за рутинно изпитване, и следва да преминат през не повече от 25 преминавания след размразяване.
|
|
17.
|
След това клетките, които са готови за изпитването STE, се приготвят с подходяща гъстота и се посяват в 96-ямкови плаки. Препоръчителната гъстота на посяване на клетките е 6,0 × 103 клетки на ямка, когато клетките се използват четири дни след посяване, или 3,0 × 103 клетки на ямка, когато клетките се използват пет дни след посяване, при обем на култура от 200 μl. Клетките, използвани за изпитването STE, които са посети в среда за отглеждане с подходящата гъстота, ще постигнат конфлуентност над 80 % към момента на изпитване, т.е. четири или пет дни след посяване.
|
Прилагане на изпитваните химикали и веществата за контроли
|
18.
|
Първият избор на разтворител за разтваряне или суспендиране на изпитвани химикали е физиологичен разтвор. Ако изпитваният химикал демонстрира слаба разтворимост или не може да се разтвори или суспендира равномерно в продължение на поне пет минути във физиологичен разтвор, се използва 5 % DMSO (CAS № 67-68-5) във физиологичен разтвор като втори избор на разтворител. За изпитвани химикали, които не могат да се разтворят или суспендират равномерно в продължение на поне пет минути или във физиологичен разтвор, или в 5 % DMSO във физиологичен разтвор, се използва минерално масло (CAS № 8042-47-5) като трети избор на разтворител.
|
|
19.
|
Изпитваните химикали се разтварят или суспендират равномерно в избрания разтворител при 5 % (w/w) концентрация и се разреждат допълнително чрез последователно 10-кратно разреждане до 0,5 % и 0,05 % концентрация. Всеки изпитван химикал трябва да се изпитва и при двете концентрации от 5 % и 0,05 %. Клетките, култивирани в 96-ямкова плака, се експонират на 200 μl/клетка при концентрация или 5 %, или 0,05 % на разтвора на изпитвания химикал (или суспензия) в продължение на пет минути при стайна температура. Изпитваните химикали (вещества с една съставка или вещества с повече съставки, или смеси) се разглеждат като вещества в чист вид и се разреждат или суспендират съгласно метода, независимо от тяхната чистота.
|
|
20.
|
Описаната в точка 16 среда за отглеждане се използва като контролна среда във всяка плака при всяко повторение. Освен това клетките трябва да се експонират и на проби на контрола на разтворител във всяка плака при всяко повторение. За изброените в точка 18 разтворители е потвърдено, че нямат нежелани ефекти върху жизнеспособността на SIRC клетките.
|
|
21.
|
В изпитването STE трябва да се използва 0,01 % натриев лаурил сулфат (SLS) във физиологичен разтвор като положителна контрола във всяка плака при всяко повторение. За да се изчисли клетъчната жизнеспособност на положителната контрола, всяка плака при всяка повторение трябва да включва и контрола на разтворител с физиологичен разтвор.
|
|
22.
|
Празна ямка е необходима, за да се определи компенсацията за оптична плътност, и трябва да се извърши в ямките, съдържащи само фосфатно буфериран физиологичен разтвор, без калций или магнезий (PBS-) или клетки.
|
|
23.
|
Всяка проба (изпитван химикал при 5 % и 0,05 %, контролна среда, контрола на разтворител и положителна контрола) следва да се изпитва три пъти при всяко повторение чрез експониране на клетките на 200 μl от съответния изпитван или контролен химикал в продължение на пет минути при стайна температура.
|
|
24.
|
Контролните сравнителни вещества са от полза за оценката на потенциала за дразнещо действие върху очите на неизвестни химикали от конкретен химичен или продуктов клас, или за оценка на съответния потенциал за дразнещо действие на вещества с дразнещо действие върху очите в рамките на конкретен диапазон от реакции на дразнене.
|
Измерване на клетъчната жизнеспособност
|
25.
|
След експозиция клетките се промиват два пъти с 200 μl РBS и се добавя 200 μl разтвор на MTT (0,5 mg MTT/ml от среда за отглеждане). След два часа време за реакция в инкубатор (37°C, 5 % CO2), разтворът на MTT се декантира, MTT формазан се екстрахира чрез добавяне на 200 μl 0,04 N солна киселина-изопропанол за 60 минути на тъмно при стайна температура и абсорбцията на разтвора MTT формазан се измерва при 570 nm с четец на плаки. Взаимодействие на изпитвани химикали с анализа MTT (чрез оцветители или вещества, които непосредствено намаляват MTT) възниква само ако значително количество от изпитван химикал остане в системата за изпитване при изплакване след експозиция, какъвто е случаят за 3D реконструирана човешка роговица или реконструирани човешки епидермални тъкани, но това не се отнася за 2D клетъчни култури, използвани за метода на изпитване STE.
|
Интерпретация на резултатите и модел за прогнозиране
|
26.
|
Стойностите на оптична плътност (OD), получени за всеки изпитван химикал, след това се използват за изчисляване на процента на клетъчна жизнеспособност спрямо контролата на разтворителя, който е определен на 100 %. Относителната клетъчна жизнеспособност се изразява като процент и се получава чрез разделяне на OD на изпитван химикал на OD на контролата на разтворителя след изваждане на OD на празна ямка от двете стойности.
По подобен начин, относителната клетъчна жизнеспособност на всяка контрола на разтворител се изразява като процент и се получава чрез разделяне на OD на всяка контрола на разтворител на OD на контролната среда след изваждане на OD на празна ямка от двете стойности.
|
|
27.
|
Следва да се извършат три независими повторения, всяко от които съдържа три репликирани ямки (т.е., n=9). Аритметичната средна стойност на трите ямки за всеки изпитван химикал и контрола на разтворител при всяко независимо повторение се използва за изчисляване на аритметичната средна стойност на относителната клетъчна жизнеспособност. Окончателната аритметична средна стойност на клетъчната жизнеспособност се изчислява от трите независими повторения.
|
|
28.
|
Граничните стойности на клетъчната жизнеспособност за идентифициране на изпитвани химикали, предизвикващи сериозно увреждане на очите (категория 1 по GHS на ООН/CLP), и изпитвани химикали, които не изискват класифициране за дразнене на очите или сериозно увреждане на очите (без категория по GHS на ООН/CLP) са дадени по-долу.
|
Таблица 2
Модел на прогнозиране за метода за изпитване STE
|
Клетъчна жизнеспособност
|
Класификация по GHS на ООН/CLP
|
Приложимост
|
|
При 5 %
|
При 0,05 %
|
|
> 70 %
|
> 70 %
|
Без категория
|
Вещества и смеси, с изключение на: i) силно летливи вещества с налягане на пари над 6 kPa (75) и ii) твърди химикали (вещества и смеси), различни от повърхностноактивни вещества, и смеси, съставени само от повърхностноактивни вещества
|
|
≤ 70 %
|
> 70 %
|
Не може да се направи прогноза
|
Не е приложимо
|
|
≤ 70 %
|
≤ 70 %
|
Категория 1
|
Вещества и смеси (76)
|
Критерии за приемливост
|
29.
|
Резултатите от изпитването се считат за приемливи, когато са изпълнени всички от следните критерии:
|
a)
|
Оптичната плътност на контролната среда (експонирана на средата на културата) следва да е 0,3 или по-висока след изваждане на оптичната плътност на празна ямка.
|
|
b)
|
Жизнеспособността на контролата на разтворителя следва да е 80 % или по-висока спрямо контролната среда. Ако при всяко повторение се използват множество контроли на разтворител, всяка от тези контроли следва да демонстрира клетъчна жизнеспособност по-голяма от 80 % за класифициране на изпитваните химикали, изпитвани с тези разтворители.
|
|
c)
|
Клетъчната жизнеспособност, получена с положителната контрола (0,01 % SLS) следва да е в рамките на две стандартни отклонения на средната стойност от предходни периоди. Горната и долната граници на приемливост за положителната контрола следва да се актуализират често, т.е. на всеки три месеца или всеки път, когато в лаборатории, в които рядко се провеждат изпитвания, е проведено приемливо изпитване (т.е. по-рядко от веднъж месечно). Когато дадена лаборатория не проведе достатъчен брой опити за установяване на статистически надеждно разпределение на положителна контрола, приемливо е да се използват горната и долната граници на приемливост, установени от разработчика на метода, т.е. между 21,1 % и 62,3 % според неговите лабораторни данни за предходни периоди, докато вътрешното разпределение се изгражда по време на първите рутинни изпитвания.
|
|
d)
|
Стандартното отклонение на окончателната клетъчна жизнеспособност, произтичащо от три независими повторения, следва да е по-малко от 15 % и за двете концентрации от 5 % и 0,05 % на изпитвания химикал.
Ако един или няколко от тези критерии не са изпълнени, резултатите следва да се отхвърлят и да се проведат други три независими повторения.
|
|
ДАННИ И ПРОТОКОЛИРАНЕ
Данни
|
30.
|
Данните за всяка отделна ямка (напр. стойности на клетъчна жизнеспособност) за всяко повторение, както и общата средна стойност, стандартното отклонение и класифицирането следва да се докладват.
|
Протокол от изпитването
|
31.
|
Протоколът от изпитването следва да включва следната информация:
|
|
Изпитван химикал и контролни вещества
|
—
|
Вещество с една съставка: химична идентификация, като наименование(я) по IUPAC или CAS, регистрационен(ни) номер(а) по CAS, SMILES или InChI код, структурна формула и/или други идентификатори;
|
|
—
|
Вещество с повече съставки, UVCB и смес: характеризиране, доколкото е възможно, чрез химическа идентичност (вж. по-горе), чистота, количествен състав и относимите физични и химични свойства (вж. по-горе) на съставките, доколкото са налични;
|
|
—
|
Агрегатно състояние, летливост, pH, LogP, молекулно тегло, химически клас и допълнителни относими физически и химични свойства, съответстващи на провеждането на проучването, доколкото са налични;
|
|
—
|
Чистота, химическа идентичност на онечистванията, както е целесъобразно и практически осъществимо и т.н.;
|
|
—
|
Третиране преди изпитване, ако е приложимо (напр. затопляне, смилане);
|
|
—
|
Условия на съхранение и стабилност, доколкото са налични.
|
|
|
|
Условия и процедури на метода за изпитване
|
—
|
Име и адрес на финансиращия, на изпитващата лаборатория и на ръководителя на изследването;
|
|
—
|
Описание на използвания метод за изпитване;
|
|
—
|
Използвана клетъчна линия, брой на преминавания и конфлуентност на клетките, използвани за изпитване;
|
|
—
|
Подробни данни за използваната процедура за изпитване;
|
|
—
|
Брой на извършените повтаряния и използвани повторения;
|
|
—
|
Използвани концентрации на изпитвания химикал (ако са различни от препоръчителните);
|
|
—
|
Обосновка за избора на разтворител за всеки изпитван химикал;
|
|
—
|
Продължителност на експозиция на изпитвания химикал (ако е различна от препоръчителната);
|
|
—
|
Описание на всякакви изменения на процедурата за изпитване;
|
|
—
|
Описание на използваните критерии за оценка и за решение;
|
|
—
|
Препратка към средна стойност на положителна контрола и стандартно отклонение (SD) за предходни периоди:
|
|
—
|
Доказване на компетентност на лабораторията за провеждане на метода за изпитване (напр. чрез изпитване на опитни вещества) или доказване на възпроизводима ефикасност на метода за изпитване във времето.
|
|
|
|
Резултати
|
—
|
Представяне в табличен вид, за всеки изпитван химикал и контролно вещество, и за всяка изпитвана концентрация, на отделните OD стойности за репликирана ямка, аритметичните средни OD стойности за всяко независимо повторение, % на клетъчна жизнеспособност за всяко независимо повторение и окончателният аритметичен среден % на клетъчна жизнеспособност и SD за трите повторения;
|
|
—
|
Резултатите за средата, разтворителя и положителната контрола доказват подходящи критерии за приемливост на проучването;
|
|
—
|
Описание на други наблюдавани ефекти;
|
|
—
|
Общата получена класификация по отношение на използвания модел за прогнозиране/критерии за вземане на решение.
|
|
|
ЛИТЕРАТУРА
|
(1)
|
United Nations UN (2013). Globally Harmonized System of Classification and Labelling of Chemicals (GHS). Fifth revised edition. New York & Geneva: United Nations Publications. ISBN: 978-92-1-117006-1. Available at: http://www.unece.org/trans/danger/publi/ghs/ghs_rev05/05files_e.html
|
|
(2)
|
Scott L, et al. (2010). A proposed Eye Irritation Testing Strategy to Reduce and Replace in vivo Studies Using Bottom-Up and Top-Down Approaches. Toxicol. In Vitro 24, 1-9.
|
|
(3)
|
Mosmann T. (1983). Rapid Colorimetric Assay for Cellular Growth and Survival: Application to 7 Proliferation and Cytotoxicity Assays. J. Immunol. Methods 65, 55-63.
|
|
(4)
|
Takahashi Y, et al. (2008). Development of the Short Time Exposure (STE) Test: an In Vitro Eye Irritation Test Using SIRC Cells. Toxicol. In Vitro 22,760-770.
|
|
(5)
|
Sakaguchi H, et al. (2011). Validation Study of the Short Time Exposure (STE) Test to Assess the Eye Irritation Potential of Chemicals. Toxicol. In Vitro 25,796-809.
|
|
(6)
|
Kojima H, et al. (2013). Second-Phase Validation of Short Time Exposure Tests for Assessment of Eye Irritation Potency of Chemicals. Toxicol. In Vitro 27, pp. 1855-1869.
|
|
(7)
|
ICCVAM (2013). Short Time Exposure (STE) Test Method Summary Review Document, NIH. Available at: [http://www.ntp.niehs.nih.gov/iccvam/docs/ocutox_docs/STE-SRD-NICEATM-508.pdf].
|
|
(8)
|
Глава Б.5 от настоящото Приложение, Остро дразнещо/корозивно действие върху очите.
|
|
(9)
|
Saito K, et al. (2015). Predictive Performance of the Short Time Exposure Test for Identifying Eye Irritation Potential of Chemical Mixtures.
|
|
(10)
|
Mikkelson TJ, Chrai SS and Robinson JR. (1973). Altered Bioavailability of Drugs in the Eye Due to Drug-Protein Interaction. J. Pharm. Sci. 1648-1653.
|
|
(11)
|
ECETOC (1998). Eye Irritation Reference Chemicals Data Bank. Technical Report (No 48. (2)), Brussels, Belgium.
|
|
(12)
|
Gautheron P, et al. (1992). Bovine Corneal Opacity and Permeability Test: an In Vitro Assay of Ocular Irritancy. Fundam Appl Toxicol. 18, 442–449.
|
|
(13)
|
OECD (2005). Guidance Document on the Validation and International Acceptance of New or Updated Test Methods for Hazard Assessment. Environmental, Health and Safety Publications, Series on Testing and Assessment (No 34). Organisation for Economic Cooperation and Development, Paris.
|
|
(14)
|
OECD (2017). Guidance Document on an Integrated Approaches on Testing and Assessment for Serious Eye Damage and Eye irritation. Environmental, Health and Safety Publications, Series on Testing and Assessment (No 263). Organisation for Economic Cooperation and Development, Paris.
|
Допълнение
ОПРЕДЕЛЕНИЯ
Точност
: степента на близост между резултатите от метода за изпитване и приетите референтни стойности. Това е мярка за ефективността на метода за изпитване и един от аспектите на неговата приложимост. Терминът често се използва взаимозаменяемо с термина „съответствие“, за да се обозначи относителният дял на правилните резултати при даден метод за изпитване (13).
Еталонно вещество
: Вещество, използвано като еталон за сравнение с изпитван химикал. Едно сравнително контролно вещество следва да притежава следните свойства; (i) постоянен и надежден източник (или източници); (ii) структурно и функционално сходство с класа на изпитваните химикали; (iii) известни физични/химични характеристики; (iv) потвърждаващи данни за известни въздействия и (v) известен потенциал в рамките на размаха на желания отклик.
Подход „отдолу нагоре“
: поетапен подход, използван за химикали, за които се предполага, че не изискват класифициране за дразнене сериозно увреждане на очите, който започва с определяне на химикали, за които не се изисква класифициране (отрицателни резултати) по отношение на други химикали (положителни резултати)
Химикал
: Вещество или смес.
Дразнене на очите
: Предизвикване на промени в очите вследствие на прилагане на изпитван химикал към предната повърхност на окото, които са напълно обратими в рамките на 21 дни след прилагането. Терминът е взаимозаменяем с „Обратими ефекти върху очите“ и с „Категория 2 по GHS на ООН/CLP“
Честота на фалшивонегативните резултати
: Делът на всички положителни химикали, невярно определени като отрицателни по даден метод за изпитване. Това е един от показателите за характеристиките на метода за изпитване.
Честота на фалшивопозитивните резултати
: Делът на всички отрицателни химикали, невярно определени като положителни по даден метод за изпитване. Това е един от показателите за характеристиките на метода за изпитване.
Опасност
: Вътрешно присъщо свойство на даден агент или ситуация, притежаващо потенциал да предизвика неблагоприятни ефекти когато един организъм, система или (суб-)популация бъдат експонирани на този агент.
Контролна среда
: Нетретирано повторение, съдържащо всички компоненти на дадена изпитвана система. Тази проба се обработва с пробите, третирани с изпитвания химикал, и с други контролни проби, за определяне дали разтворителят взаимодейства с изпитваната система.
Смес
: Смес или разтвор, съставен от две или повече вещества.
Вещество с една съставка
: Вещество, определено от неговия количествен състав, в който съдържанието на една основна съставка е най-малко 80 % (w/w).
MTT
: 3-(4,5-диметилтиазол-2-ил)-2,5-дифенилтетразолиев бромид; Тиазолил син тетразолиев бромид.
Вещество с повече съставки
: Вещество, определено от неговия количествен състав, в който повече от една основна съставка се съдържа в концентрация ≥ 10 % (w/w) и < 80 % (w/w). Веществото с повече съставки е резултат от производствен процес. Разликата между смес и вещество с повече съставки е, че дадена смес е получена чрез смесване на две или повече вещества без химична реакция. Веществото с повече съставки е резултат от химична реакция.
OD
: Оптична плътност.
Положителни контроли
: Повторение, съдържащо всички компоненти на дадена изпитвана система и третирано с вещество, за което е известно, че предизвиква положителен отклик. За да се гарантира, че може да се направи оценка на варирането на отклика в положителната контрола във времето, големината на положителния отклик не следва да е прекомерна.
Относимост
: описание на взаимовръзката между изпитването и ефекта, представляващ интерес, и дали тя е значима и полезна за определена цел. Това е степента, в която изпитването правилно измерва или прогнозира биологичния ефект, представляващ интерес. Относимостта включва разглеждането на точността (съответствието) на метода за изпитване (10).
Надеждност
: мярка за степента, в която даден метод за изпитване може да се приложи възпроизводимо в една и съща лаборатория и в различни лаборатории по различно време, при използване на един и същи протокол. Оценката за нея се прави, като се изчислява вътрешнолабораторната и междулабораторната възпроизводимост и вътрешнолабораторната повторяемост (13).
Чувствителност
: Относителният дял от всички положителни/активни химикали, които са класифицирани правилно чрез изпитването. Това е мярка за точността на метода за изпитване, който предоставя категорични резултати и е важен фактор при оценката на относимостта на метода за изпитване (10).
Сериозно увреждане на очите
: Предизвикване на тъканно увреждане на окото или сериозно физическо влошаване на зрението след прилагане на изпитван химикал към предната повърхност на окото, което не е напълно обратимо в рамките на 21 дни след прилагането. Терминът е взаимозаменяем с „Необратими ефекти върху очите“ и с „Категория 1 по GHS на ООН/CLP“.
Контрола на разтворител/носител
: Нетретирана проба, съдържаща всички компоненти на дадена изпитвана система, включително разтворителя или носителя, която се обработва с пробите, третирани с изпитвания химикал, и с други контролни проби, за определяне на базовия отклик при пробите, третирани с изпитвания химикал, разтворен в същия разтворител или носител. Когато се изпитва с паралелна контролна среда, тази проба показва също дали разтворителят или носителят взаимодействат с изпитваната система.
Специфичност
: Относителният дял от всички негативни/неактивни химикали, които са класифицирани правилно чрез изпитването. Това е мярка за точността на метода за изпитване, която предоставя категорични резултати и е важен фактор при оценката на метода за изпитване (13).
Вещество
: Химически елемент и неговите съединения в естествено състояние или получени чрез производствен процес, включително всяка добавка, необходима за запазване на стабилността му, както и всички примеси, извлечени от използвания процес, с изключение на всеки разтворител, който може да бъде отделен, без да се засяга стабилността на веществото или да се променя неговия състав.
Повърхностноактивно вещество
: също така наречено повърхностноактивен агент, представлява вещество, като например детергент, което е в състояние да намали повърхностното напрежение на дадена течност и по този начин да позволи образуването на пяна в течността или проникването и в твърди вещества; Известно е също като мокрител.
Изпитван химикал
: всяко вещество или смес, изпитвано(а) с използване на настоящия метод за изпитване.
Стратегия за поетапно изпитване
: стратегия за поетапно изпитване, при която цялата съществуваща информация за даден изпитван химикал се разглежда по конкретен ред, като на всеки етап се прилага процес, основан на тежестта на доказателствата, за определяне дали съществува достатъчно информация за вземане решение за класифициране с оглед на опасността, преди да се премине към следващия етап. Ако потенциалът за дразнещо действие на даден изпитван химикал може да се определи въз основа на съществуващата информация, не се налага допълнително изпитване. Ако потенциалът за дразнещо действие на даден изпитван химикал не може да се определи въз основа на съществуващата информация, се провежда процедура за поетапно последователно изпитване върху животни, докато стане възможно да се направи ясно класифициране.
Подход „отгоре надолу“
: поетапен подход, използван за химикал, за който се предполага, че предизвиква сериозно увреждане на очите, който започва с определяне на химикали, предизвикващи сериозно увреждане на очите (положителен резултат) по отношение на други химикали (отрицателен резултат).
Глобална хармонизирана система на Организацията на обединените нации за класифициране и етикетиране на химикали (GHS на ООН)
: Система, предлагаща класифициране на химикали (вещества и смеси) според стандартизирани видове и степени на физическа, здравна и екологична опасност и разглеждаща съответни съобщителни елементи, като например пиктограми, сигнални думи, съобщения за опасност, съобщения за предпазни мерки и информационни листовки за безопасност, така че те да съобщават информация за тяхното неблагоприятно въздействие с оглед защита на хората (включително работодатели, работещи, служители в транспорта, потребители и аварийни служители) и на околната среда (1).
Категория 1 по GHS на ООН/CLP
: Вж. „Сериозно увреждане на очите“.
Категория 2 по GHS на ООН/CLP
: Вж. „Дразнене на очите“.
„Без категория“ по GHS на ООН/CLP
: Химикали, които не отговарят на изискванията за класифициране като категория 1 или 2 (2А или 2В) по GHS на ООН/CLP.
UVCB
: вещества с неизвестен или променлив състав, сложни продукти от реакции или биологични материали.
Б.69 МЕТОД ЗА ИЗПИТВАНЕ НА РЕКОНСТРУИРАН ЧОВЕШКИ, ПРИЛИЧАЩ НА РОГОВИЦА ЕПИТЕЛ (RhCE) ЗА ИДЕНТИФИЦИРАНЕ НА ХИМИКАЛИ, НЕ ИЗИСКВАЩИ КЛАСИФИЦИРАНЕ И ЕТИКЕТИРАНЕ ЗА ДРАЗНЕНЕ НА ОЧИТЕ ИЛИ СЕРИОЗНО УВРЕЖДАНЕ НА ОЧИТЕ
ВЪВЕДЕНИЕ
|
1.
|
Настоящият метод за изпитване е еквивалентен на насоките за изпитване на ОИСР (ТG) 492 (2017). Сериозно увреждане на очите означава предизвикване на тъканно увреждане на очите или сериозно физическо увреждане на зрението след прилагане на изпитван химикал върху предната повърхност на очите, което не е напълно обратимо в рамките на 21 дни след прилагането, както е определено от Глобалната хармонизирана система за класификация и етикетиране на химикали (GHS) на Организацията на обединените нации (ООН) (1) и Регламент на Европейския съюз (ЕО) № 1272/2008 относно класифицирането, етикетирането и опаковането на вещества и смеси (CLP) (77). Също според GHS на ООН и CLP дразнене на очите означава предизвикване на промени в очите след прилагане на изпитван химикал към предната повърхност на очите, които са напълно обратими в рамките на 21 дни след прилагането. Изпитваните химикали, предизвикващи сериозно увреждане на очите, се класифицират като категория 1 по GHS на ООН и CLP, докато тези, които предизвикват дразнене на очите, се класифицират като категория 2 по GHS на ООН и CLP. Изпитвани химикали, некласифицирани за дразнене на очите или сериозно увреждане на очите, са онези, които не отговарят на изискванията за класифициране като категория 1 или 2 (2А или 2В) по GHS на ООН и CLP, т.е. те се определят като „без категория“ по GHS на ООН и CLP.
|
|
2.
|
Оценката на сериозно увреждане на очите/дразнене на очите обикновено включва използването на лабораторни животни (МИ B.5 (2)). Изборът на най-подходящия метод за изпитване и използването на този метод за изпитване следва да се разглежда в контекста на насоките на ОИСР за интегрирани подходи за изпитване и оценка на сериозно увреждане на очите и дразнене на очите (39).
|
|
3.
|
Настоящият метод за изпитване описва in vitro процедура, която позволява идентифицирането на химикали (вещества и смеси), които не изискват класифициране и етикетиране за дразнене на очите или сериозно увреждане на очите в съответствие с GHS на ООН и CLP. Той използва реконструиран човешки, приличащ на роговица епител (RhCE), който силно наподобява хистологичните, морфологичните, биохимичните и физиологичните свойства на човешкия роговичен епител. Четири други метода за изпитване in vitro са валидирани, считат се за научно потвърдени и са приети като МИ Б.47 (3), Б.48 (4), Б.61 (5) и Б.68 (6), за работа с крайните точки за човешкото здраве сериозно увреждане на очите/дразнене на очите.
|
|
4.
|
В този метод за изпитване са включени две валидирани, налични в търговската мрежа RhСE изпитвания. Изследвания за валидиране за оценка на дразнене на очите/сериозно увреждане на очите са проведени (7)(8)(9)(10)(11)(12)(13) с използване на изпитването за дразнене на очите (EIT) EpiOcular™ и теста за дразнене на очите (EIT) SkinEthic™ Human Corneal Epithelium (HCE). Всяко едно от тези изпитвания използва като система за изпитване предлагани в търговската мрежа тъканни модели на RhCE, които в текста по-нататък се наричат валидирани референтни методи — съответно VRM 1 и VRM2. В резултат на тези изследвания за валидиране и тяхната независима партньорска проверка (9)(12) се стигна до заключение, че EpiOcular™ EIT и SkinEthic™ HCE EIT са в състояние правилно да идентифицират химикали (вещества и смеси), не изискващи класифициране и етикетиране за дразнене за очите или сериозно увреждане на очите съгласно GHS на ООН, и тестовете се препоръчват като научно потвърдени за тази цел (13).
|
|
5.
|
Понастоящем е общоприето, че в обозримо бъдеще, нито един метод за изпитване in vitro няма да е в състояние да замести напълно изпитването in vivo на Draize (2)(14) при прогнозирането в целия спектър на сериозно увреждане на очите/дразнене на очите, за различните класове химикали. При все това стратегическите комбинации от няколко алтернативни методи за изпитване в рамките на стратегия за (поетапно) изпитване, като подходът „отгоре надолу“, може да са в състояние да заменят изпитването на очите на Draize (15). Подходът „отдолу нагоре“ (15) е създаден да се използва, когато въз основа на наличната информация се очаква химикалът да не причини достатъчно дразнене на очите, за да изисква класифициране, докато подходът „отгоре надолу“ (15) е създаден да се използва, когато въз основа на наличната информация се очаква даден химикал да причини сериозно увреждане на очите. EpiOcular™ EIT и SkinEthic™ HCE EIT се препоръчват за идентифициране на химикали, които не изискват класифициране за дразнене на очите или сериозно увреждане на очите според GHS на ООН/CLP (без категория) без допълнително изпитване, в рамките на стратегия за изпитване като подхода „отдолу нагоре/отдолу нагоре“, предложен от Scott et al., напр.като първоначална стъпка при подход „отдолу нагоре“ или като една от последните стъпки при подход „отгоре надолу“ (15). Въпреки това EpiOcular™ EIT и SkinEthic™ HCE EIT не са предназначени за разграничаване между категория 1 по GHS на ООН/CLP (сериозно увреждане на очите) и категория 2 по GHS на ООН/CLP (дразнене на очите). За такова разграничаване трябва да се работи в друг етап на стратегията за изпитване (15). Изпитван химикал, който се идентифицира като изискващ класификация за дразнене на очите/сериозно увреждане на очите с EpiOcular™ EIT или SkinEthic™ HCE EIT, следователно изисква допълнително изпитване (in vitro и/или in vivo), за да се стигне до окончателно заключение (без категория по GHS на ООН/CLP, категория 2 или категория 1), с използване напр. на МИ Б.47, Б.48, Б.61 или Б.68.
|
|
6.
|
Целта на настоящия метод за изпитване е да се опишат процедурите, използвани за оценка на потенциалната опасност за очите на даден изпитван химикал въз основа на неговата способност да предизвиква цитотоксичност при тъканен модел с RhCE, измерена чрез анализа МТТ (16) (вж. точка 21). Жизнеспособността на тъканта RhCE след експозиция на изпитван химикал се определя в сравнение с тъкани, третирани с вещество за отрицателна контрола (% на жизнеспособност), и след това се използва за прогнозиране на потенциалната опасност за очите на изпитвания химикал.
|
|
7.
|
Налични са стандарти за ефективност (17) за улесняване на валидирането на нови или модифицирани тестове in vitro на база RhCE, подобни на EpiOcular™ EIT и SkinEthic™ HCE EIT, в съответствие с принципите на Ръководство № 34 на ОИСР (18), които позволяват своевременна промяна на ОИСР TG 492 за тяхното включване. Взаимното приемане на данни (MAD) съгласно споразумението на ОИСР се гарантира само за изпитвания, които са валидирани според стандартите за ефективност, ако тези изпитвания са прегледани и включени в съответните насоки за изпитвания от ОИСР.
|
ОПРЕДЕЛЕНИЯ
|
8.
|
Определенията са дадени в допълнение 1.
|
ПЪРВОНАЧАЛНИ СЪОБРАЖЕНИЯ И ОГРАНИЧЕНИЯ
|
9.
|
Настоящият метод за изпитване се основава на търговски триизмерни тъканни модели на RhCE, които се произвеждат с използване или на първични човешки епидермални кератоцити (т.е. EpiOcular™ OCL-200), или на човешки безсмъртни роговични епителни клетки (т.е. SkinEthic™ HCE/S). Тъканните модели EpiOcular™ OCL-200 и SkinEthic™ HCE/S RhCE са подобни на in vivo триизмерната структура на роговичния епител и се произвеждат с използване на клетки от видове, представляващи интерес (19)(20). Освен това изпитванията измерват директно цитотоксичността, предизвикана от проникването на химикала през роговицата и увреждането на клетки и тъкани; след това цитотоксичният отклик определя общия резултат за in vivo сериозно увреждане на очите/дразнене на очите. Клетъчно увреждане може да възникне при различни механизми на действие (вж. точка 20), но цитотоксичността играе важна, ако не и основна, механистична роля за определяне на общия отклик на химикал за сериозно увреждане на очите/дразнене на очите, което се проявява in vivo предимно чрез непрозрачност на роговицата, ирит, зачервяване на конюктивата и/или конюктивален хемозис, независимо от физичните и химичните процеси на основно тъканно увреждане.
|
|
10.
|
В изследването за валидиране, лежащо в основата на този метод за изпитване, е изпитан широк спектър от химикали, обхващащи разнообразни химични видове, химични класове, молекулни тегла, LogP, химични структури и др. Базата данни за валидиране на EpiOcular™ EIT съдържа общо 113 химикали, обхващащи 95 различни органични функционални групи съгласно анализ на кутията с инструменти QSAR на ОИСР (8). Повечето от тези химикали представляват вещества с една съставка, но в проучването са включени и няколко вещества с повече съставки (включително 3 хомополимери, 5 съполимери и 10 квазиполимери). По отношение на агрегатното състояние и категориите по GHS на ООН/CLP 113-те изпитвани химикали се разпределят както следва: 13 течности от категория 1, 15 твърди вещества от категория 1, 6 течности от категория 2А, 10 твърди вещества от категория 2А, 7 течности от категория 2В, 7 твърди вещества от категория 2В, 27 течности без категория и 28 твърди вещества без категория (8). Базата данни за валидиране на SkinEthic™ HCE EIT съдържа общо 200 химикали, обхващащи 165 различни органични функционални групи (8)(10)(11). Повечето от тези химикали представляват вещества с една съставка, но в проучването са включени и няколко вещества с повече съставки (включително 10 полимера). По отношение на агрегатното състояние и категориите по GHS на ООН/CLP 200-те изпитвани химикали се разпределят както следва: 27 течности от категория 1, 24 твърди вещества от категория 1, 19 течности от категория 2А, 10 твърди вещества от категория 2А, 9 течности от категория 2В, 8 твърди вещества от категория 2В, 50 течности без категория и 53 твърди вещества без категория (10)(11).
|
|
11.
|
Този метод за изпитване е приложим за вещества и смеси, както и за твърди вещества, течности, полутвърди вещества и восъци. Течностите могат да бъдат или да не бъдат водни разтвори; твърдите вещества може да са разтворими или неразтворими във вода. Когато е възможно, твърдите вещества следва да бъдат смилани до фин прах преди прилагане; не се изисква друга предварителна обработка на пробата. Газове и аерозоли не са оценявани с изследване за валидиране. Макар да се предполага, че те могат да бъдат изпитвани с използване на технология с RhСE, настоящия метод за изпитване не разрешава изпитване на газове и аерозоли.
|
|
12.
|
Изпитваните химикали, поглъщащи светлина в същия диапазон като MTT формазан (по естествен път и след третиране) и изпитваните химикали, които могат пряко да редуцират виталния оцветител MTT (до MTT формазан), могат да попречат на измерванията на жизнеспособността на тъканта и изискват употребата на адаптирани контроли за корекции. Типът на адаптираните контроли, които може да са необходими, варира в зависимост от типа на интерференцията, предизвикана от изпитвания химикал и използваната процедура за количествено измерване на MTT формазан (вж. точки 36-42).
|
|
13.
|
Генерираните резултати в проучвания за предварително валидиране (21)(22) и пълно валидиране (8)(10)(11) показват, че и EpiOcular™ EIT, и SkinEthic™ HCE EIT могат да бъдат прехвърлени в лаборатории, за които се счита, че нямат опит в извършването на анализи, и са възпроизводими в рамките на лабораторията и между лабораториите. Въз основа на тези проучвания нивото на възпроизводимост по отношение на съгласуване на прогнозите, което може да се очаква при EpiOcular™ EIT от данни за 113 химикали, е от порядъка на 95 % в рамките на лабораторията и 93 % между лаборатории. Нивото на възпроизводимост по отношение на съгласуване на прогнозите, което може да се очаква при SkinEthic™ HCE EIT от данни за 120 химикали, е от порядъка на 92 % в рамките на лабораторията и 95 % между лаборатории.
|
|
14.
|
EpiOcular™ EIT може да се използва за идентифициране на химикали, които не изискват класифициране за дразнене на очите или сериозно увреждане на очите съгласно системата за класифициране GHS на ООН и CLP. Предвид данните, получени във изследването за валидиране (8), EpiOcular™ EIT има обща точност от 80 % (въз основа на 112 химикали), чувствителност 96 % (въз основа на 57 химикали), процент неверни отрицателни резултати 4 % (въз основа на 57 химикали), специфичност 63 % (въз основа на 55 химикали) и процент неверни положителни резултати 37 % (въз основа на 55 химикали) в сравнение с референтни данни от изпитване in vivo върху очи на зайци (МИ Б.5) (2)(14), класифицирани съгласно системата за класифициране GHS на ООН и CLP. Едно проучване, в което 97 течни агрохимични формули се изпитват с EpiOcular™ EIT, показва подобна ефективност на метода за изпитване за този вид смеси като получената във валидиращото проучване (23). 97-те формули се разпределят както следва: 21 от категория 1, 19 от категория 2A, 14 от категория 2B и 43 без категория, класифицирани съгласно системата за класифициране GHS на ООН въз основа на референтни данни от изпитване in vivo върху очи на зайци (МИ Б.5) (2)(14). Получена е обща точност от 82 % (въз основа на 97 формули), чувствителност 91 % (въз основа на 54 формули), процент неверни отрицателни резултати 9 % (въз основа на 54 формули), специфичност 72 % (въз основа на 43 формули) и процент неверни положителни резултати 28 % (въз основа на 43 формули) (23).
|
|
15.
|
SkinEthic™ HCE EIT може да се използва за идентифициране на химикали, които не изискват класифициране за дразнене на очите или сериозно увреждане на очите съгласно системата за класифициране GHS на ООН и CLP. Предвид данните, получени във изследването за валидиране (10)(11), SkinEthic™ HCE EIT има обща точност от 84 % (въз основа на 200 химикали), чувствителност 95 % (въз основа на 97 химикали), процент неверни отрицателни резултати 5 % (въз основа на 97 химикали), специфичност 72 % (въз основа на 103 химикали) и процент неверни положителни резултати 28 % (въз основа на 103 химикали) в сравнение с референтни данни от изпитване in vivo върху очи на зайци (МИ Б.5) (2)(14), класифицирани съгласно системата за класифициране GHS на ООН и CLP.
|
|
16.
|
Процентът на неверни отрицателни резултати, получен и при двете изпитвания с RhCE с вещества или смеси, попада в рамките на 12 % обща вероятност, че химикалите се идентифицират или като категория 2 по GHS на ООН и CLP, или като „без категория по GHS на ООН и CLP“ чрез изпитването in vivo на Draize, при повтарящи се изпитвания; това се дължи на присъщата за метода променливост в рамките на изпитването (24). Процентът на неверни положителни резултати, получени при двата метода на изпитване с RhCE с вещества или смеси, не е от решаващо значение в контекста на този метод за изпитване, тъй като всички изпитвани химикали, които предизвикват тъканна жизнеспособност равна или по-малка от установените гранични стойности (вж. точка 44) изискват по-подробно изпитванес други in vitro методи за изпитване, или като последен вариант — изпитване върху зайци, в зависимост от регулаторните изисквания, с използване на последователна стратегия за изпитване и подход за тежест на доказателства. Тези методи за изпитване могат да се използват за всички видове химикали, при които отрицателен резултат следва да се приеме като некласифициране на химикал за дразнене на очите и сериозно увреждане на очите (без категория по GHS на ООН и CLP). Необходима е консултация със съответните регулаторни органи, преди да се използват EpiOcular™ EIT и SkinEthic™ HCE EIT в схеми на класифициране, различни от тези на GHS на ООН/CLP.
|
|
17.
|
Ограничението на настоящия метод за изпитване е, че той не позволява разграничаване между дразнене на очите/обратими ефекти върху очите (категория 2) и сериозно увреждане на очите/необратими ефекти върху очите (категория 1), както са определени от GHS на ООН и CLP, нито между дразнещи очите (допълнителна категория 2А) и леко дразнещи очите (допълнителна категория 2В), както са определени от GHS на ООН (1). За тези цели се изисква допълнително изпитване с друг in vitro метод за изпитване.
|
|
18.
|
Терминът „изпитван химикал“ се използва в този метод за изпитване за обозначаване на това, какво се изпитва, (78) и не е свързан с приложимостта на метода за изпитване с RhCE за изпитване на вещества и/или смеси.
|
ПРИНЦИП НА ИЗПИТВАНЕТО
|
19.
|
Изпитваният химикал се прилага локално на минимум два триизмерни тъканни модели на RhCE и тъканната жизнеспособност се измерва след експозиция и инкубационния период след третиране. RhCE тъканите се реконструират от първични човешки епидермални кератоцити или човешки безсмъртни роговични епителни клетки, които са култивирани в продължение на няколко дни, за да образуват стратифициран, силно диференциран плоскоклетъчен епител, който е морфологично подобен на този, който се открива в човешката роговица. Тъканния модел на RhCE на EpiOcular™ RhCE се състои от поне 3 жизнеспособни слоя клетки и некератинизирана повърхност, която наподобява структурата на роговица, аналогична на откриваната in vivo. Тъканният модел на RhCE на SkinEthic™ HCE RhCE се състои от поне 4 жизнеспособни слоя клетки, включващи колонни базални клетки, преходни криловидни клетки и повърхностни сквамозни клетки, подобни на тези в нормалния човешки роговичен епител (20)(26).
|
|
20.
|
Предизвиканото по химичен начин сериозно увреждане на очите/дразнене на очите, което се проявява in vivo предимно чрез непрозрачност на роговицата, ирит, зачервяване на конюктивата и/или конюктивален хемозис, е резултат от поредица събития, започващи с проникване на химикала през роговицата и/или конюктивата и увреждане на клетките. Клетъчно увреждане може да възникне чрез няколко механизма на действие, включващи: лизис на клетъчната мембрана (напр. от повърхностноактивни вещества, органични разтворители); коагулация на макромолекули (по-специално протеини) (напр. от повърхностноактивни вещества, органични разтворители, основи и киселини); осапуняване на липиди (напр. от основи); и алкилиране или други ковалентни взаимодействия с макромолекули (напр. от избелващи вещества, пероксиди и алкилатори) (15)(27)(28). Въпреки това е доказано, че цитотоксичността играе важна, ако не и основна, механистична роля за определяне на общия отклик на химикал за сериозно увреждане на очите/дразнене на очите, независимо от физичните и химичните процеси на основно тъканно увреждане (29)(30). Освен това потенциалът на даден химикал за сериозно увреждане на очите/дразнене на очите принципно се определя от степента на първоначално увреждане (31), която корелира със степента на клетъчна смърт (29) и със степента на последващи отговори и евентуални резултати (32). Следователно слабите дразнители обикновено засягат само повърхностния роговичен епител, леките и умерените дразнители увреждат принципно епитела и повърхностната строма и силните дразнители увреждат епитела, дълбоката строма и понякога ендотелия на роговицата (30)(33). Измерването на жизнеспособността на тъканния модел на RhCE след локална експозиция на даден изпитван химикал за идентифициране на химикали, които не изискват класифициране за сериозно увреждане на очите/дразнене на очите (без категория по GHS на ООН и CLP), се основава на допускането, че всички химикали, предизвикващи сериозно увреждане на очите или дразнене на очите, предизвикват цитотоксичност в роговичния епител и/или конюктивата.
|
|
21.
|
Тъканната жизнеспособност на RhCE класически се измерва чрез ензимно превръщане на виталния оцветител MTT [3-(4,5-диметилтиазол-2-ил)-2,5- дифенилтетразолиев бромид; Тиазолил син тетразолиев бромид; CAS номер 298-93-1] от жизнеспособните клетки на тъканта в синя МТТ формазанова сол, която се измерва количествено след екстракция от тъканите (16). Химикалите, които не изискват класифициране и етикетиране съгласно GHS на ООН/CLP (без категория), се идентифицират като такива, които не намаляват тъканната жизнеспособност под определен праг (т.е. тъканна жизнеспособност > 60 % при EpiOcular™ EIT и SkinEthic™ HCE EITL (79) или > 50 % при SkinEthic™ HCE EITS (80)) (вж. точка 44).
|
ДОКАЗВАНЕ НА КОМПЕТЕНТНОСТ
|
22.
|
Преди рутинно използване на изпитвания с RhCE за регулаторни цели, лабораториите следва да демонстрират техническа компетентност чрез правилно прогнозиране на петнадесет химикали за изпитване за пригодност, изброени в Таблица 1. Тези химикали са избрани от химикалите, използвани във валидиращите проучвания на VRM (8)(10)(11). Изборът включва, до възможната степен, химикали, които: (i) обхващат различни агрегатни състояния; (ii) обхващат пълния спектър на in vivo отговори на сериозно увреждане на очите/дразнене на очите въз основа на висококачествени резултати, получени при референтно изпитване in vivo върху очи на зайци (МИ Б.5) (2)(14) и системата за класифициране GHS на ООН (т.е. категории 1, 2A, 2B или без категория) (1) и системата за класифициране CLP (т.е. категории 1, 2 или без категория); (iii) обхващат различни in vivo фактори за класифициране (24)(25); (iv) са представителни са за химичните класове, използвани във изследването за валидиране (8)(10)(11); (v) обхващат добро и обширно представяне на органични функционални групи (8)(10)(11); (iv) имат химически структури, които са добре определени (8)(10)(11); (vii) са оцветени и/или са преки редуктори на MTT; (viii) предизвикват възпроизводими резултати в методите за изпитване на RhCE по време на тяхното валидиране; (ix) са правилно предсказани от методите за изпитване на RhCE по време на техните изследвания за валидиране; (x) обхващат пълния спектър на in vitro отговори въз основа на висококачествени данни от методи за изпитване на RhCE (0 до 100 % жизнеспособност); (хi) се предлагат в търговската мрежа; и (хii) не са свързани с прекалено високи разходи за придобиване и/или обезвреждане. В ситуации, когато изброен химикал не е наличен или не може да се използва поради други обосновани причини, може да се използва друг химикал, който отговаря на гореописаните критерии, напр. от химикалите, използвани за валидиране на VRM. Тези отклонения обаче следва да са оправдани.
Таблица 1
Списък на химикалите за изпитване за пригодност
|
Химично наименование
|
CAS №
|
Органична функционална група (81)
|
Агрегатно състояние
|
VRM1 жизнеспособност (%) (82)
|
VRM2 жизнеспособност (%) (83)
|
VRM прогноза
|
Редуктор на MTT
|
Цветно взаимод.
|
|
In Vivo категория 1 (84)
|
|
|
Метилтиогликолат
|
2365-48-2
|
Естер на карбоксилна киселина; Тиоалкохол
|
L
|
10,9±6,4
|
5,5±7,4
|
Не може да се направи прогноза
|
Y (силен)
|
N
|
|
Хидроксиетил акрилат
|
818-61-1
|
Акрилат; Алкохол
|
L
|
7,5±4,7 (85)
|
1,6±1,0
|
Не може да се направи прогноза
|
N
|
N
|
|
2,5-диметил-2,5-хексанедиол
|
110-03-2
|
Алкохол
|
S
|
2,3±0,2
|
0,2±0,1
|
Не може да се направи прогноза
|
N
|
N
|
|
Натриев оксалат
|
62-76-0
|
Оксикарбоксилна киселина
|
S
|
29,0±1,2
|
5,3±4,1
|
Не може да се направи прогноза
|
N
|
N
|
|
In Vivo категория 2A (84)
|
|
|
2,4,11,13-тетраазатетрадекан-диимидимид, N,N″-бис(4-хлорфенил)-3,12-диимино-, ди-D-глюконат (20 %, воден разтвор) (86)
|
18472-51-0
|
Ароматен хетероцикличен халид; Арил халид; Дихидроксилова група; Гванидин
|
L
|
4,0±1,1
|
1,3±0,6
|
Не може да се направи прогноза
|
N
|
Y (слаб)
|
|
Натриев бензоат
|
532-32-1
|
Арил; Карбоксилна киселина
|
S
|
3,5±2,6
|
0,6±0,1
|
Не може да се направи прогноза
|
N
|
N
|
|
In Vivo категория 2B (84)
|
|
|
Диетил толуамид
|
134-62-3
|
Бензамид
|
L
|
15,6±6,3
|
2,8±0,9
|
Не може да се направи прогноза
|
N
|
N
|
|
2,2-диметил-3-метиленбицикло [2.2.1] хептан
|
79-92-5
|
Алкан, разклонен с третичен въглерод; Алкен; Бициклохептан; Карбоцикли с пръстен; Циклоалкан
|
S
|
4,7±1,5
|
15,8±1,1
|
Не може да се направи прогноза
|
N
|
N
|
|
In Vivo без категория (84)
|
|
|
1-етил-3-метилмидазолинов етилсулфат
|
342573-75-5
|
Алкокси; Амониева сол; Арил; Имидазол; Сулфати
|
L
|
79,9±6,4
|
79,4±6,2
|
Без категория
|
N
|
N
|
|
Дикаприлил етер
|
629-82-3
|
Алкокси; Етер
|
L
|
97,8±4,3
|
95,2±3,0
|
Без категория
|
N
|
N
|
|
Пиперонил бутоксид
|
51-03-6
|
Алкокси; Бензодиоксол; Бензил; Етер
|
L
|
104,2±4,2
|
96,5±3,5
|
Без категория
|
N
|
N
|
|
Полиетилен гликол (PEG-40) хидрогенирано рициново масло
|
61788-85-0
|
Ацилал; Алкохол; Алил; Етер
|
Вискозна
|
77,6±5,4
|
89,1±2,9
|
Без категория
|
N
|
N
|
|
1-(4-хлорфенил)-3-(3,4-дихлорфенил) урея
|
101-20-2
|
Ароматен хетероцикличен халид; Арил халид; Деривати на урея
|
S
|
106,7±5,3
|
101,9±6,6
|
Без категория
|
N
|
N
|
|
2,2′-метилен-бис-(6-(2H-бензотриазол-2-ил)-4-(1,1,3,3-тетраметилбутил)-фенол)
|
103597-45-1
|
Алкан, разклонен с четвъртичен въглерод; Слят, карбоцикличен, ароматен; Слети наситени хетероцикли; Прекурсори на хиноидни съединения; терт-бутил
|
S
|
102,7±13,4
|
97,7±5,6
|
Без категория
|
N
|
N
|
|
Калиев тетрафлуороборат
|
14075-53-7
|
Неорганична сол
|
S
|
88,6±3,3
|
92,9±5,1
|
Без категория
|
N
|
N
|
|
Съкращения:
CAS № = Номер по Служба за химични индекси CAS; UN GHS = Глобална хармонизирана система на Организацията на обединените нации за класифициране и етикетиране на химикали (GHS на ООН) (1); VRM1 = Валидиран референтен метод, EpiOcular™ EIT; VRM2 = Валидиран референтен метод, SkinEthic™ HCE EIT; Цветно взаимод. = цветно взаимодействие при измерване на стандартна абсорбция (оптична плътност (OD)) на MTT формазан.
|
|
|
23.
|
Като част от процедурата за изпитване на пригодността е препоръчително потребителите да проверят бариерните свойства на тъканите след получаване, както е посочено от производителя на тъканния модел RhСE (вж. точки 25, 27 и 30). Това е особено важно, ако тъканите са били превозвани на дълго разстояние или в продължение на дълги периоди от време. След като даден метод за изпитване е бил успешно създаден и пригодността за употребата му - установена и доказана, обикновено такава проверка няма да е необходима. Все пак, при редовно използване на даден метод за изпитване, е препоръчително да се продължи с оценяването на бариерните свойства на редовни интервали.
|
ПРОЦЕДУРА
|
24.
|
Обхванатите от този метод за изпитване тестове са валидните от научна гледка тестове EpiOcular™ EIT и SkinEthic™ HCE EIT (9)(12)(13), които по-нататък са наричани валидирани референтни методи (съответно VRM1 и VRM2). Стандартни оперативни процедури (СОП) за методите за изпитване на RhCE са налични и следва да се използват при внедряване и използване на тези методи за изпитване в лаборатория (34)(35). Следващите точки и Допълнение 2 описват основните компоненти и процедури на RhCE тестовете.
|
КОМПОНЕНТИ НА МЕТОДА ЗА ИЗПИТВАНЕ НА RНСE
Общи условия
|
25.
|
Следва да се използват подходящи клетки от човешки произход за реконструиране на наподобяваща роговица епителна триизмерна тъкан, която следва да е съставена от постепенно стратифицирани, но не вроговени клетки. Тъканният модел с RhCE се приготвя във вложки с порьозна синтетична мембрана, през която хранителни вещества могат да преминават в клетките. В реконструирания наподобяващ роговица епител трябва да присъстват множество слоеве от жизнени, некератинизирани епителни клетки. Тъканният модел на RhCE следва да има епителна повърхност, която влиза в директен контакт с въздух, така че да позволява непосредствена локална експозиция на изпитваните химикали по начин, който наподобява in vivo експозицията на роговичния епител. Тъканният модел на RhCE следва да образува функционална бариера, която е достатъчно здрава да устои на бързото проникване на цитотоксични вещества за сравнение, напр. Triton X-100 или натриев додецил сулфат (SDS). Бариерната функция следва да бъде доказана и може да бъде оценена чрез определяне или на времето на експозиция, необходимо за намаляване на тъканната жизнеспособност с 50 % (ET50) след приложение на вещество за сравнение със специфична, фиксирана концентрация (напр. 100 μl от 0,3 % (v/v) Triton X-100), или на концентрацията, при която дадено вещество за сравнение намалява жизнеспособността на тъканите с 50 % (IC50) след фиксирано време на експозиция (напр. 30 минути третиране с 50 μl SDS) (вж. точка 30). Задържащите свойства на модела на RhСE следва да предотвратяват преминаването на изпитван химикал около края на жизнеспособната тъкан, което би довело до незадоволително моделиране на експозицията на роговицата. Клетките от човешки произход, използвани за създаване на тъканния модел на RhCE, следва да не са замърсени с бактерии, вируси, микоплазма и гъбички. Стерилността на тъканния модел следва да се провери от доставчика за липса на замърсяване с гъбички и бактерии.
|
Функционални условия
Жизнеспособност
|
26.
|
Използваният анализ за количествено определяне на жизнеспособността на тъканта е анализът MTT (16). Жизнеспособните клетки на тъканния модел на RhСE редуцират виталния оцветител MTT в син формазанов преципитат на MTT, който след това се извлича от тъканта с помощта на изопропанол (или подобен разтворител). ИзвлечениятMTT формазан може да се определи количествено с помощта или на измерване на стандартната абсорбция (оптична плътност, (OD)), или чрез ВЕТХ/УВЕТХ спектрофотометрична процедура (36). OD само на разтворителя, използван за екстракцията, следва да бъде достатъчно ниска, т.е., OD < 0,1. Ползвателите на тъканния модел на RhСE следва да гарантират, че всяка използвана партида с тъканен модел на RhСE отговаря на определените критерии за отрицателната контрола. Границите на приемливост за OD стойностите на отрицателната контрола за VRM са дадени в Таблица 2. Ползвателят на ВЕТХ/УВЕТХ спектрофотометрия следва да използва интервалите за OD за отрицателната контрола, посочени в таблица 2, като критерий за приемливост за отрицателната контрола. В протокола за изпитването следва да се документира, че тъканите, третирани с веществото на отрицателната контрола, са стабилни в култура (показват сходни измервания за тъканна жизнеспособност) за периода на продължителност на експозицията. Подобна процедура следва да се спазва от производителя на тъканта като част от качествения контрол за освобождаване на партидата тъкани, но в този случай може да се прилагат критерии за приемливост, различни от посочените в Таблица 2. Разработчикът/доставчикът на тъканния модел на RhСE следва да определи интервал на приемливост (горна и долна граница) за стойностите на OD на отрицателната контрола (при условията за качествен контрол на метода за изпитване).
|
Таблица 2
Интервали на приемливост за стойности на OD за отрицателна контрола (за потребители на теста)
|
Тест
|
Долна граница на приемливост
|
Горна граница на приемливост
|
|
EpiOcular™ EIT (OCL-200) — VRM1 (за протоколите за течности и твърди вещества)
|
> 0,8 (87)
|
< 2,5
|
|
SkinEthic™ HCE EIT (HCE/S) — VRM2 (за протоколите за течности и твърди вещества)
|
> 1,0
|
≤ 2,5
|
Бариерна функция
|
27.
|
Тъканният модел на RhCE следва да е с достатъчна дебелина и здравина, за да устои на бързото проникване на цитотоксични вещества за сравнение, както е оценено напр. от ET50 (Triton X-100) или от IC50 (SDS) (Таблица 3). Бариерната функция на всяка партида от използвания тъканен модел на RhСE следва да бъде доказана от разработчика/доставчика на тъканния модел на RhСE при доставяне на тъканите на крайния потребител (вж. точка 30).
|
Морфология
|
28.
|
Хистологичното изследване на тъканния модел на RhCE следва да докаже наподобяваща човешка роговица епителна структура (включително поне 3 слоя от жизнеспособни епителни клетки и некератинизирана повърхност). За VRM подходяща морфология е определена от разработчика/доставчика, поради което няма нужда да се доказва отново от потребител на метода за изпитване за всяка използвана партида тъкани.
|
Възпроизводимост
|
29.
|
Резултатите от положителните и отрицателните контроли на метода за изпитване следва да докажат възпроизводимост във времето.
|
Контрол на качеството (КК)
|
30.
|
Тъканният модел на RhCE следва да се използва само ако разработчикът/доставчикът докаже, че всяка използвана партида тъканен модел на RhCЕотговаря на определените критерии за пускане в обръщение на продукция, сред които най-подходящи са тези за жизнеспособност (точка 26) и бариерна функция (вж. точка 27). Интервалът на приемливост (горна и долна граници) за бариерните функции, измерени от ET50 или IC50 (вж. точки 25 и 26) следва да бъдат определени от разработчика/доставчика на тъканния модел на RhCE. Интервалът на приемливост на ET50 и IC50, използван като критерий за КК за освобождаване на партида от разработчика/доставчика на тъканните модели на RhCE (използвани във VRM), е даден в Таблица 3. Разработчикът/доставчикът на тъканния модел на RhCE трябва да предостави на потребителите на метода за изпитване данни, доказващи съответствие с всички критерии за пускане в обръщение на продукция, така че те да са в състояние да включат тази информация в протокола за изпитването. Само резултати, получени с тъкани, които отговарят на всички тези критерии за пускане в обръщение на продукция, могат да бъдат приети като надеждна прогноза за химикали, не изискващи класифициране и етикетиране за дразнене на очите или сериозно увреждане на очите в съответствие с GHS на ООН/CLP.
|
Таблица 3
Критерии за КК за пускане в обръщение на партида
|
Тест
|
Долна граница на приемливост
|
Горна граница на приемливост
|
|
EpiOcular™ EIT (OCL-200) — VRM1 (100 μl от 0,3 % (v/v) Triton X-100)
|
ET50 = 12,2 min
|
ET50 = 37,5 min
|
|
SkinEthic™ HCE EIT (HCE/S) — VRM2 (30 минути третиране с 50 μl SDS)
|
IC50 = 1 mg/ml
|
IC50 = 3,2 mg/ml
|
Прилагане на изпитвания химикал и веществата за контроли
|
31.
|
За всеки изпитван химикал и за всяко вещество за контрола във всеки експеримент следва да се използват поне две повторения на тъкани. Използват се два различни протокола за третиране, един за течни изпитвани химикали и един за твърди изпитвани химикали (34)(35). И за двата метода и протокола повърхността на тъканния модел следва да се навлажни с фосфатно буфериран физиологичен разтвор на Дюлбеко без калций и магнезий (не съдържащ Ca2+/Mg2+ DPBS) преди прилагане на изпитваните химикали, за да се имитират влажните условия на човешкото око. Третирането на тъканите започва с експозиция на изпитвания(те) химикал(и) и веществата за контроли. И за двата протокола на третиране в двата VRM следва да се приложи достатъчно количество от изпитвания химикал или веществото за контрола, за да покрие равномерно епителната повърхност, като същевременно се избягва прилагане на неопределена доза (вж. точки 32 и 33) (Допълнение 2).
|
|
32.
|
Изпитвани химикали, които могат да се пипетират при 37 °C или по-ниски температури (с помощта на пипета с положително изместване, ако е необходимо), се третират като течности във VRM, в противен случай те следва да се третират като твърди вещества (вж. точка 33). Във VRM даден течен изпитван химикал се разпространява равномерно върху тъканната повърхност (т.е. приложение на минимум 60 μl/cm2) (вж. Допълнение 2, (33)(34)). Капилярните ефекти (ефектите на повърхностно напрежение), които може да възникнат поради малките обеми, приложени към вложката (върху тъканната повърхност), следва да се избягват до степента, до която е възможно, за да се гарантира правилното дозиране на тъканта. Тъканите, третирани с течни изпитвани химикали, се инкубират в продължение на 30 минути при стандартни условия на култивиране (37±2 °C, 5±1 % CO2, ≥ 95 % ОВ). В края на периода на експозиция течният изпитван химикал и веществата за контроли следва внимателно да се отстранят от тъканната повърхност чрез обилно изплакване с DPBS без Ca2+/Mg2+при стайна температура. След тази стъпка на изплакване следва потапяне в прясна среда при стайна температура след експозиция (за отстраняване на всяко количество изпитван химикал, абсорбирано в тъканта) в продължение на предварително определен период от време, който варира в зависимост от използвания VRM. Само за VMR1 инкубиране след експозиция в прясна среда при стандартни условия на култивиране се прилага преди извършването на МТТ анализа (вж. Допълнение 2, (34)(35)).
|
|
33.
|
Изпитвани химикали, които не могат да бъдат пипетирани при температури до 37 °C, се третират като твърди вещества във VRM. Количеството на приложения изпитван химикал следва да е достатъчно, за да покрие цялата повърхност на тъканта, т.е. следва да се приложи минимум 60 mg/cm2 (Допълнение 2). Когато е възможно, твърдите вещества следва да се изпитват под формата на фин прах. Тъкани, третирани с твърди изпитвани химикали, се инкубират в продължение на предварително определен период от време (в зависимост от използвания VRM) при стандартни условия на култивиране (вж. Допълнение 2, (34) (35)). В края на периода на експозиция твърдият изпитван химикал и веществата за контроли следва внимателно да се отстранят от тъканната повърхност чрез обилно изплакване с DPBS без Ca2+/Mg2+ при стайна температура. След тази стъпка на изплакване следва потапяне в прясна среда при стайна температура след експозиция (за отстраняване на всяко количество изпитван химикал, абсорбирано в тъканта) в продължение на предварително определен период от време, който варира в зависимост от използвания VRM, инкубирането след експозиция в прясна среда при стандартни условия на култивиране, преди извършването на МТТ анализа (вж. Допълнение 2, (34)(35)).
|
|
34.
|
Във всеки експеримент следва да се включват паралелни отрицателни и положителни контроли, за да се докаже, че жизнеспособността (определена с отрицателната контрола) и чувствителността (определена с положителната контрола) на тъканите са в рамките на интервалите на приемливост, определени въз основа на данни от предходни периоди. Паралелната отрицателна контрола осигурява също така изходното ниво (100 % тъканна жизнеспособност) за изчисляване на процента относителна жизнеспособност на тъканите, третирани с изпитвания химикал (изпитване за %жизнеспособност). Препоръчителното вещество за положителна контрола, което да се използва за VRM, е чист метил ацетат (CAS № 79-20-9, предлаган на пазара напр. от Sigma-Aldrich, кат. № 45997; течност). Препоръчителните вещества за отрицателна контрола, които да се използват за VRM1 и VRM2, са съответно свръхчиста H2O и DPBS без Ca2+/Mg2+. Това са веществата за контроли, използвани във валидиращите проучвания на VRM, за които съществуват най-много данни от предходни периоди. Използването на подходящи алтернативни вещества за положителна или отрицателна контрола следва да оправдано от научна гледка точка по подходящ начин. Отрицателните и положителните контроли следва да се изпитват с един и същ протокол(и) като този(тези), използвани за изпитваните химикали, включени в експеримента (т.е. за течности и/или твърди вещества). След това приложение следва да се извърши третирането, експозицията, изплакването, потапянето след експозиция и инкубирането след експозиция, където е приложимо, както е описано за експериментите с контроли, провеждани паралелно с тези на течни изпитвани химикали (вж. точка 32) или за експериментите с контроли, провеждани паралелно с тези на твърди изпитвани химикали (вж. точка 33), преди извършване на анализа МТТ (вж. точка 35) (34)(35). Един отделен набор от отрицателни и положителни контроли е достатъчен за всички изпитвани химикали с едно и също агрегатно състояние (течности или твърди вещества), включени в същия експеримент.
|
Измервания на тъканната жизнеспособност
|
35.
|
Анализът MTT е стандартизиран метод за количествено определяне (16), който следва да се използва за измерване на тъканната жизнеспособност при настоящия метод за изпитване. Той е съвместим с използване в триизмерни тъканни модели. Анализът МТТ се провежда веднага след периода на инкубиране след експозиция. Във VRM пробата на тъканен модел на RhCE се поставя в 0,3 ml разтвор на MTT при 1 mg/ml за 180±15 минути при стандартни условия на култивиране. Виталният оцветител МТТ се редуцира в син преципитат на МТТ формазан от жизнеспособните клетки на тъканния модел на RhCE. Утаеният син МТТ формазанов продукт след това се извлича от тъканта с помощта на подходящ обем изопропанол (или подобен разтворител) (34)(35). Тъканите, изпитвани с течни изпитвани химикали, следва да се извличат от горната и долната част на тъканите, докато тъканите, изпитвани с твърди изпитвани химикали и оцветени течности, следва да се извличат само от долната част на тъканта (за да се сведе до минимум всяко потенциално замърсяване на разтвора на изопропанол за извличане с някой изпитван химикал, който може да е останал в тъканта). Тъкани, третирани с течни изпитвани химикали, които не се измиват лесно, също може да се извличат само от долната част на тъканта. Паралелно изпитваните вещества за отрицателна и положителна контрола следва да се третират подобно на изпитвания химикал. Извлеченият MTT формазан може да се определи количествено или чрез измерване на стандартната абсорбция (OD) при 570 nm с използване на ширина на филтърната дължина на вълната максимум ±30 nm, или с помощта на процедура на ВЕТХ/УВЕТХ спектрофотометрия (вж. точка 42) (11)(36).
|
|
36.
|
Оптичните свойства на изпитвания химикал или неговото химично действие върху МТТ може да взаимодейства с измерването на МТТ формазан, което води до невярна оценка на тъканната жизнеспособност. Изпитваните химикали може да повлияят върху измерването на MTT формазан чрез пряка редукция на MTT в син MTT формазан и/или чрез цветова интерференция, ако изпитваният химикал абсорбира, по естествен път или в резултат на процедурите за третиране, в същия диапазон на OD, като MTT формазан (т.е. около 570 nm). Преди изпитване следва да се извършат предварителни проверки, за да се позволи идентифициране на потенциални преки редуктори на МТТ и/или химикали с цветова интерференция, и да се използват допълнителни контроли за откриване и коригиране на потенциалната интерференция на тези изпитвани химикали (вж. точки 37-41). Това е особено важно, когато специфичен изпитван химикал не бъде напълно отстранен от тъканния модел на RhCE чрез изплакване или когато той проникне в наподобяващия роговица епител и поради това присъства в тъканните модели на RhCE, когато се извършва МТТ анализ. За изпитвани химикали, които абсорбират светлина в същия диапазон, като MTT формазан (по естествен път или след третиране), които не са съвместими с измерването на стандартната абсорбция (OD) на MTT формазан поради прекалено силна интерференция, т.е. силна абсорбция при 570±30 nm, може да се използва процедура с ВЕТХ/УВЕТХ спектрофотометрия за измерване на МТТ формазан (вж. точки 41 и 42) (11)(36). Подробно описание на начина за откриване и коригиране на пряката редукция на MTT и интерференциите от оцветителите е налично в СОП за VRM (34)(35). Илюстративни диаграми, предоставящи указания за начина на идентифициране и работа с преки редуктори на MTT и/или химикали с цветова интерференция за VRM1 и VRM2 също са предоставени, съответно в Допълнение III и IV.
|
|
37.
|
За идентифициране на потенциална интерференция от изпитвани химикали, абсорбиращи светлина в същия диапазон, като МТТ формазан (по естествен път или след третиране), и вземане на решение за нуждата от допълнителни контроли изпитваният химикал се добавя към вода и/или изопропанол и се инкубира в продължение на подходящ период от време при стайна температура (вж. Допълнение 2, (34)(35)). Ако изпитваният химикал във вода и/или изопропанол абсорбира достатъчно светлина в диапазона 570±20 nm за VRM1 (вж. Допълнение 3) или ако при смесване на изпитвания химикал с вода за VRM2 се получава оцветен разтвор (вж. Допълнение 4), приема се, че изпитваният химикал влияе върху измерването на стандартната абсорбция (OD) на MTT формазан и следва да се използват допълнителни контроли с оцветители или, като алтернатива, следва да се използва процедура на ВЕТХ/УВЕТХ спектрофотометрия, в който случай тези контроли не са необходими (вж. точки 41 и 42 и Допълнения III и IV)(34)(35). Когато се извършва измерване на стандартната абсорбция (OD), всеки изпитван химикал, причиняващ интерференция, следва да се приложи върху поне две повторения на жизнеспособни тъкани, които се подлагат на цялата процедура за изпитване, но по време на етапа на инкубиране на MTT се инкубират със среда, вместо с разтвор на MTT, за да предизвикат неспецифичен цвят в живите тъкани (NSCliving) контрола (34)(35). NSCliving контролата трябва да се извърши паралелно с изпитването на оцветения изпитван химикал, а в случай на множество изпитвания трябва да се проведе независима NSCliving контрола за всяко извършено изпитване (във всяка серия), поради присъщата биологична изменчивост на живите тъкани. Реалната тъканна жизнеспособност се изчислява като: процента на тъканна жизнеспособност, получен от живи тъкани, изложени на изпитвания химикал, причиняващ интерференция, и инкубирани с разтвор на MTT (изпитване за процент жизнеспособност) минус процента неспецифичен цвят, получен от живи тъкани, изложени на изпитвания химикал, причиняващ интерференция, и инкубирани със среда без MTT, изпитвани паралелно с изпитването, което се коригира (%NSCliving), т.е. реалната тъканна жизнеспособност = [изпитване за % жизнеспособност] - [%NSCliving].
|
|
38.
|
За да се идентифицират преките редуктори на MTT, всеки изпитван химикал следва да бъде добавен към прясно приготвен разтвор на MTT. Към МТТ разтвора се добавя подходящо количество от изпитвания химикал и сместа се инкубира в продължение на около 3 часа при стандартни условия на култивиране (вж. Допълнения III и IV)(34)(35). Ако сместа с MTT, съдържаща изпитвания химика (или суспензия за неразтворими изпитвани химикали) стане синя/лилава, се приема, че изпитваният химикал пряко редуцира MTT и следва да се извърши допълнителна функционална проверка на нежизнеспособни тъканни модели на RhСE тъкани, независимо от използването на стандартното измерване на коефициента на поглъщане (OD) или ВЕТХ/УВЕТХ спектрофотометрична процедура. При тази допълнителна функционална проверка се използват умъртвени тъкани, които имат само остатъчна метаболитна активност, но поглъщат и задържат изпитвания химикал по начин, сходен с този на жизнеспособните тъкани. Умъртвените тъкани на VRM1 се приготвят чрез експозиция на ниска температура („умъртвени чрез замразяване“). Умъртвените тъкани на VRM2 се приготвят чрез удължено инкубиране (напр. поне 24±1 часа) във вода, последвано от съхранение при ниска температура („умъртвени с вода“). Всеки изпитван химикал, редуциращ MTT, се прилага върху поне две умъртвени тъканни повторения, които се подлагат на цялата процедура за изпитване, за да предизвикат неспецифична контрола на MTT редукция (NSMTT) (34)(35). Един NSMTT контрол е достатъчен за всеки изпитван химикал, независимо от броя на изпълнените независими изпитвания/серии. Реалната тъканна жизнеспособност се изчислява като: процента на тъканна жизнеспособност, получен от живи тъкани, изложени на въздействието на редуктор на MTT (изпитване за % жизнеспособност) минус процента неспецифична редукция на MTT, получена от умъртвени тъкани, изложени на същия редуктор на MTT, изчислена относително отрицателната контрола, извършена паралелно с изпитването (%NSMTT), т.е. реалната тъканна жизнеспособност = [изпитване за % жизнеспособност] - [%NSMTT].
|
|
39.
|
Изпитваните химикали, които се идентифицират като причиняващи цветова интерференция (вж. точка 37) и пряка редукция на МТТ (вж. точка 38), също изискват трети набор от контроли, когато се извършва измерване на стандартната абсорбция (OD), освен NSMTT и NSCliving контролите, описани в предходните точки. Обикновено такъв е случаят с тъмно оцветени изпитвани химикали, абсорбиращи светлина в диапазона 570±30 nm (напр., син, лилав, черен), защото техният присъщ цвят пречи на оценката на тяхната способност пряко да редуцират MTT, както е описано в точка 38. Това налага използването на NSMTT контроли по подразбиране, заедно с NSCliving контролите. Изпитваните химикали, за които се извършват NSMTT и NSCliving контроли, може да се абсорбират и задържат както от живи, така и от умъртвени тъкани. Поради това, в този случай, NSMTT контролата може не само да коригира потенциала за пряка редукция на MTT на изпитвания химикал, но и цветовата интерференция, произтичаща от абсорбцията и задържането на изпитвания химикал от умъртвени тъкани. Това би могло да доведе додвойна корекция за цветова интерференция, тъй като NSCliving контролата вече коригира цветовата интерференция, произтичаща от абсорбцията и задържането на изпитвания химикал от живи тъкани. За да се избегне възможна двойна корекция за цветова интерференция, трябва да се извърши трета контрола за неспецифичен цвят в умъртвени тъкани (NSCkilled) (вж. Допълнения III и IV)(34)(35). При този допълнителен контрол, изпитваният химикал се прилага върху най-малко две умъртвени тъканни повторения, които биват подложени на цялата процедура за изпитване, но по време на етапа за инкубация с MTT, се инкубират със среда, вместо с разтвор на MTT. Една NSCkilled контрола е достатъчна за всеки изпитван химикал, независимо от броя на извършените независими изпитвания/серии, но трябва да се изпълнява паралелно с NSMTT контролата и с една и съща партида тъкани. Реалната тъканна жизнеспособност се изчислява като: процента на тъканна жизнеспособност, получен от живи тъкани, изложени на изпитвания химикал (изпитване за % жизнеспособност) минус %NSMTT минус %NSCliving
плюс процента неспецифичен цвят, получен от умъртвени тъкани, изложени на изпитвания химикал, причиняващ интерференция, и инкубирани със среда без MTT, изчислена относително отрицателната контрола, извършена паралелно с изпитването, което се коригира (%NSCkilled), т.е. реалната тъканна жизнеспособност = [изпитване за % жизнеспособност] - [%NSMTT] - [%NSCliving] + [%NSCkilled].
|
|
40.
|
Важно е да се отбележи, че неспецифичната редукция на МТТ и неспецифичните цветови интерференции могат да повишат OD (когато се извършват измервания на стандартната абсорбция) на извлечената тъкан над линейния диапазон на спектрофотометъра и че неспецифичната редукция на МТТ може също така да увеличи пиковата площ на МТТ формазан (когато се извършват измервания чрез ВЕТХ/УВЕТХ спектрофотометрия) на извлечената тъкан над линейния диапазон на спектрофотометъра. Предвид това важно е всяка лаборатория да определи линейния диапазон на OD/пиковата площ на техния спектрофотометър, напр. с МТТ формазан (CAS № 57360-69-7), предлаган на пазара от напр. Sigma-Aldrich (кат. № M2003), преди да започне изпитването на изпитвани химикали за регулаторни цели.
|
|
41.
|
Измерването на стандартната абсорбция (OD) с помощта на спектрофотометър е подходящо за оценяване на преки редуктори на MTT и изпитвани химикали, причиняващи цветова интерференция, когато наблюдаваната интерференция при измерването с МТТ формазан не е прекалено силна (т.е. OD на тъканните модели, получени с изпитваните химикали без каквато и да е корекция за пряка редукция на MTT и/или цветова интерференция, са в рамките на линейния диапазон на спектрофотометъра). Независимо от това, резултатите за изпитвани химикали, предизвикващи %NSMTT и/или %NSCliving > 60 % (VRM1 и VRM2 за протокола за течности) или 50 % (VRM2 за протокола за твърди вещества) на отрицателната контрола, следва да се тълкуват предпазливо, тъй като това е установената гранична стойност, използвана във VRM за разграничаване на класифицирани от некласифицирани химикали (вж. точка 44). Стандартната абсорбция (OD) обаче не може да се измерва, когато интерференцията с измерването на MTT формазан е прекалено силна (т.е. водеща до неверни стойности на OD на изпитваните тъканни модели, които попадат извън линейния диапазон на спектрофотометъра). Оцветените изпитвани химикали или изпитвани химикали, които се оцветяват при контакт с вода или изопропанол, и които показват прекалено силна интерференция при измерване на стандартната абсорбция (OD) на MTT формазан, все пак могат да се оценяват с помощта на ВЕТХ/УВЕТХ спектрофотометрия (вж. Допълнения III и IV). Това е така, защото ВЕТХ/УВЕТХ системата позволява отделянето на MTT формазан от химикала преди неговото количествено определяне (36). Поради тази причина никога не се изискват NSCliving или NSCkilled контроли, когато се използва ВЕТХ/УВЕТХ спектрофотометрия, независимо от химикала, който се подлага на изпитване. NSMTT контроли никога не следва да се използват, ако има съмнение, че изпитваният химикал и пряк редуктор на MTT (спазвайки процедурата, описана в точка 38). NSMTT контроли следва да се използват и с изпитвани химикали с цвят (присъщ или появяващ се, когато са във вода), който пречи на оценката на тяхната способност пряко да редуцират МТТ, както е описано в точка 38. Когато се използва ВЕТХ/УВЕТХ спектрофотометрия за измерване на MTT формазан, процентът на жизнеспособност на тъканта се изчислява като процента на пиковата площ на MTT формазан, получен от живи тъкани, изложени на въздействието на изпитвания химикал спрямо пика на MTT формазан, получен при паралелната отрицателна контрола. За изпитвани химикали, които са в състояние пряко да редуцират МТТ, реалната тъканна жизнеспособност се изчислява като:изпитване за % жизнеспособност минус %NSMTT, както е описано в последното изречение на точка 38. И накрая следва да се отбележи, че преките редуктори на MTT или преките редуктори на МТТ, които освен това може да причиняват и цветова интерференция, които се задържат в тъканите след третиране и толкова силно редуцират MTT, че водят до стойности на OD (при използване на стандартно измерване на OD) или на пикови площи (при използване на ВЕТХ/УВЕТХ спектрофотометрия) на изпитваните тъканни модели, които попадат извън линейния диапазон на спектрофотометъра, не могат да се оценяват с методи за изпитване на RhC, въпреки че се очаква подобни случаи да възникват само в изключително редки ситуации.
|
|
42.
|
ВЕТХ/УВЕТХ спектрофотометрия може да се използва с всички видове изпитвани химикали (оцветени, неоцветени, редуктори на MTT и не-редуктори на MTT) за измерване на MTT формазан (11)(36). Поради разнообразието на системите за ВЕТХ/УВЕТХ спектрофотометрия е невъзможно всеки потребител да определи абсолютно еднакви условия на системата. Предвид това пригодността на системата за ВЕТХ/УВЕТХ спектрофотометрия следва да бъде доказана преди употребата й за количествено определяне на MTT формазан от тъканни модели чрез изпълнението на критериите за приемливост по отношение на набор от стандартни параметри, на базата на описаните в насоките на Администрацията по храни и лекарства на САЩ за индустрията по отношение на валидирането на био-аналитични методи (36)(38). Тези ключови параметри и техните критерии за приемливост са посочени в Допълнение 5. След като критериите за приемливост, дефинирани в Допълнение 5, са изпълнени, системата за ВЕТХ/УВЕТХ спектрофотометрия се счита за пригодна и готова за измерване на MTT формазан при експерименталните условия, описани в настоящия метод за изпитване.
|
Критерии за приемливост
|
43.
|
За всяка серия, използваща партиди на RhCE тъкан, които отговарят на качествения контрол (вж. точка 30), тъканите, третирани с вещество за отрицателна контрола, следва да показват OD, отразяваща състоянието на тъканите, които са преминали през етапите по транспортиране и приемане, както и през всички процедури по протокола, и не трябва да попадат извън границите, установени за предходни периоди, които са описани в Таблица 2 (вж. точка 26). По същия начин тъканите, третирани с веществото за положителна контрола, т.е. метил ацетат, следва да показват средна тъканна жизнеспособност < 50 % по отношение на отрицателната контрола във VRM1 според протоколите или за течности, или за твърди вещества, и ≤ 30 % (протокол за течности) или ≤ 20 % (протокол за твърди вещества) по отношение на отрицателната контрола във VRM2, отразявайки по този начин способността на тъканите да реагират на дразнещ изпитван химикал при условията на метода на изпитване (34)(35). Променливостта между тъканните повторения на изпитвания химикал и контролните вещества следва да попада в рамките на приетите лимити (т.е. разликата в жизнеспособността между две тъканни повторения следва да е по малка от 20 % или стандартното отклонение (СО) между три тъканни повторения не следва да превишава 18 %). Ако или отрицателната контрола, или положителната контрола, включени в дадена серия, попадат извън приетите граници, серията се счита за не отговаряща на изискванията и следва да се повтори. Ако променливостта между тъканните повторения на изпитван химикал е извън приетия интервал, изпитването трябва да са счита за не отговарящо на изискванията и следва да се повтори изпитването на изпитвания химикал.
|
Интерпретация на резултатите и модел за прогнозиране
|
44.
|
Стойностите на OD/пикови площи, получени с репликирани екстракти на тъкан за всеки изпитван химикал, следва да се използват за изчисляване на средния процент на тъканна жизнеспособност (средната стойност на тъканни повторения), нормализирана до отрицателната контрола, която е определена като 100 %. Процентната гранична стойност на тъканна жизнеспособност за идентифициране на изпитвани химикали, не изискващи класифициране за дразнене на очите или сериозно увреждане на очите („без категория“ по GHS на ООН и CLP), е дадена в Таблица 4. Резултатите следва да се тълкуват както следва:
|
—
|
Изпитваният химикал се идентифицира като не изискващ класифициране и етикетиране по GHS на ООН и CLP („без категория“), ако средният процент на тъканна жизнеспособност след експозиция и инкубиране след експозиция е по-голям от(>) установената процентна гранична стойност на тъканна жизнеспособност, както е посочена в Таблица 4. В този случай не се изисква допълнително изпитване с други методи за изпитване.
|
|
—
|
Ако средният процент на тъканна жизнеспособност след експозиция и инкубиране след експозиция е по-малък или равен на (≤) установената процентна гранична стойност на тъканна жизнеспособност, прогноза не може да се направи, както е показано в Таблица 4. В този случай се изисква допълнително изпитване с други методи на изпитване, тъй като методите за изпитване на RhCE демонстрират определен брой неверни положителни резултати (вж. точки 14-15) и не може да се вземе решение за отнасяне към категориите 1 и 2 на GHS на ООН и CLP (вж. точка 17).
|
Таблица 4
Прогнозни модели съгласно класификацията по GHS на ООН и CLP
|
VRM
|
Без категория
|
Не може да се направи прогноза
|
|
VRM 1 - EpiOcular™ EIT (и за двата протокола)
|
Средна тъканна жизнеспособност > 60 %
|
Средна тъканна жизнеспособност ≤ 60 %
|
|
VRM 2 - SkinEthic™ HCE EIT (за протокола за течности)
|
Средна тъканна жизнеспособност > 60 %
|
Средна тъканна жизнеспособност ≤ 60 %
|
|
VRM2 - SkinEthic™ HCE EIT (за протокола за твърди вещества)
|
Средна тъканна жизнеспособност > 50 %
|
Средна тъканна жизнеспособност ≤ 50 %
|
|
|
45.
|
Едно изпитване на поне две тъканни повторения следва да е достатъчно за даден изпитван химикал, когато полученият резултат е еднозначен. Въпреки това, при наличие на гранични резултати, като несъответстващи измервания на повторения и/или среден процент на тъканна жизнеспособност, равен на 60±5 % (VRM1 и VRM2 за протокола за течности) или на 50±5 % (VRM2 за протокола за твърди вещества), следва да се обмисли провеждане на второ изпитване, както и на трето, в случай на наличие на несъответстващи резултати между първите две изпитвания.
|
|
46.
|
Може да се обмислят различни процентни гранични стойности на тъканна жизнеспособност, разграничаващи класифицирани от некласифицирани изпитвани химикали, за специфични видове смеси, където е подходящо и оправдано, за да се увеличи общата ефективност на метода за изпитване за тези видове смеси (вж. точка 14). Химикалите за сравнение може да са полезни за оценяване на потенциала на неизвестен изпитван химикал или продуктов клас за сериозно увреждане на очите/дразнене на очите или за оценка на потенциала за относителна очна токсичност на класифициран химикал в рамките на специфичен диапазон на положителни отговори.
|
ДАННИ И ПРОТОКОЛИРАНЕ
Данни
|
47.
|
Данните от отделните тъканни повторения в серия (напр. стойности на OD/пикови площи на MTT формазан и изчислените данни за процент на тъканна жизнеспособност за изпитвания химикал и контролите, както и окончателната прогноза на метода за изпитване на RhCE) следва да се протоколират в табличен вид за всеки изпитван химикал, включително данните от повторени изпитвания, както е подходящо. Освен това средният процент на тъканна жизнеспособност и разликата на променливостта между две тъканни повторения (ако n=2 тъканни повторения) или СО (ако n≥ 3 тъканни повторения) за всеки отделен изпитван химикал и контрола следва да се протоколира. Всички наблюдавани интерференции на даден изпитван химикал с измерването на МТТ формазан чрез пряка редукция на МТТ и/или цветова интерференция следва да се протоколират за всеки изпитван химикал.
|
Протокол от изпитването
|
48.
|
Протоколът от изпитването следва да включва следната информация:
Изпитван химикал
|
|
Вещества с една съставка
|
—
|
Химична идентификация, като наименование(я) по IUPAC или CAS, регистрационен(ни) номер(а) по CAS, SMILES или InChI код, структурна формула и/или други идентификатори;
|
|
—
|
Агрегатно състояние, летливост, pH, LogP, молекулно тегло, химически клас и допълнителни относими физически и химични свойства, съответстващи на провеждането на проучването, доколкото са налични;
|
|
—
|
Чистота, химическа идентичност на примесите, както е целесъобразно и практически осъществимо, др.;
|
|
—
|
Третиране преди изпитване, ако е приложимо (напр. (напр. затопляне, смилане);
|
|
—
|
Условия на съхранение и стабилност, доколкото са налични.
|
|
|
|
Вещества с повече съставки, UVCB и смеси
|
—
|
характеризиране, доколкото е възможно, чрез химическата идентичност (вж. по-горе), чистотата, количествения състав и относимите физични и химични свойства на съставките (вж. по-горе), доколкото са налични;
|
|
—
|
Агрегатно състояние и допълнителни относими физически и химични свойства, съответстващи на провеждането на проучването, доколкото са налични;
|
|
—
|
Чистота, химическа идентичност на примесите, както е целесъобразно и практически осъществимо, др.;
|
|
—
|
Обработка преди изпитването, ако се прилага (напр. затопляне, смилане);
|
|
—
|
Условия на съхранение и стабилност, доколкото са налични.
|
|
Вещества за положителни и отрицателни контроли
|
—
|
Химична идентификация, като наименование(я) по IUPAC или CAS, регистрационен(ни) номер(а) по CAS, SMILES или InChI код, структурна формула и/или други идентификатори;
|
|
—
|
Агрегатно състояние, летливост, молекулно тегло, химически клас и допълнителни относими физически и химични свойства, съответстващи на провеждането на проучването, доколкото са налични;
|
|
—
|
Чистота, химическа идентичност на примесите, както е целесъобразно и практически осъществимо, др.;
|
|
—
|
Обработка преди изпитването, ако се прилага (напр. затопляне, смилане);
|
|
—
|
Условия на съхранение и стабилност, доколкото са налични;
|
|
—
|
Обосновка за използването на различна от ултрачиста H2O или DPBS без Ca 2+/Mg2+ отрицателна контрола, ако е приложимо;
|
|
—
|
Обосновка за използването на различна от чист метил ацетат положителна контрола, ако е приложимо;
|
|
—
|
Позоваване на резултати за положителни и отрицателни контроли от предходни периоди, демонстриращи подходящи критерии за приемливост на изпитването.
|
Информация за финансиращия и за изпитващата лаборатория
|
—
|
Име и адрес на финансиращия, на изпитващата лаборатория и на ръководителя на изследването.
|
|
—
|
Използвани тъканен модел на RhCE и протокол (предоставящи обосновка на избора, ако е приложимо)
|
Условия на метода за изпитване
|
—
|
Използван тъканен модел на RhCE, включително номер на партида;
|
|
—
|
Дължина на вълната и лента на пропускане (ако е приложимо), използвани за количествено определяне на MTT формазан, и линеен диапазон на измервателното устройство (напр. спектрофотометър);
|
|
—
|
Описание на метода, използван за количествено определяне на MTT формазан;
|
|
—
|
Описание на използваната система за ВЕТХ/УВЕТХ спектрофотометрия, ако е приложимо;
|
|
—
|
Пълна придружаваща информация за конкретния използван тъканен модел на RhСE, включително неговите характеристики. това следва да включва, но не се ограничава до:
|
i)
|
Качествен контрол на жизнеспособността (доставчик)
|
|
ii)
|
Жизнеспособност при условията на метода за изпитване (потребител);
|
|
iii)
|
Качествен контрол на бариерната функция;
|
|
iv)
|
Морфология, ако е приложимо;
|
|
v)
|
Възпроизводимост и способност за прогнозиране;
|
|
vi)
|
Други качествени контроли (КК) на тъканния модел на RhCE, ако е приложимо;
|
|
|
—
|
Препратка към данни за тъканния модел на RhCE от предходни периоди. това следва да включва, но не се ограничава до: Приемливост на данните за КК с позоваване на данни за партида от предходни периоди;
|
|
—
|
Декларация, че изпитващата лаборатория е демонстрирала пригодност за използването на метода за изпитване преди рутинна употреба чрез изпитване на химикали за изпитване за пригодност;
|
Критерии за приемливост на изпитвана серия и изпитване
|
—
|
Средни стойности и диапазони на приемливост на положителната и отрицателната контроли въз основа на данни от предходни периоди;
|
|
—
|
Допустимо вариране между тъканните повторения за положителни и отрицателни контроли;
|
|
—
|
Приемлива променливост между тъканните повторения за изпитвания химикал;
|
Процедура за изпитване
|
—
|
подробна информация за използваната процедура на изпитване;
|
|
—
|
Използвани дози на изпитвания химикал и контролните вещества;
|
|
—
|
Продължителност и температура на експозиция, периоди на потапяне след експозиция и инкубиране след експозиция (където е приложимо);
|
|
—
|
Описание на всякакви изменения на процедурата за изпитване;
|
|
—
|
Указание за използваните контроли за преки редуктори на MTT и/или оцветяващи изпитвани химикали, ако е приложимо;
|
|
—
|
Брой на използваните тъканни повторения за всеки изпитван химикал и контроли (положителна контрола, отрицателна контрола, NSMTT, NSCliving и NSCkilled, ако е приложимо);
|
Резултати
|
—
|
Представяне в табличен вид на данните за отделните изпитвани химикали и вещества за контроли за всяка серия (включително повторни експерименти, където е приложимо) и за всяко измерване на повторение, включително стойност на OD или пикова площ на MTT формазан, процент на тъканна жизнеспособност, среден процент на тъканна жизнеспособност, разлики между тъканни повторения или СО и окончателна прогноза;
|
|
—
|
Ако е приложимо, резултатите от използваните контроли за преки редуктори на MTT и/или оцветяващи изпитвани химикали, включително стойност на OD или пикова площ на MTT формазан, %NSMTT, %NSCliving, %NSCkilled, разлики между тъканни повторения или СО, окончателен верен процент на тъканна жизнеспособност и окончателна прогноза;
|
|
—
|
Резултатите, получени с изпитвания(ите) химикал(и) и веществата за контроли във връзка с определените критерии за приемливост на изпитваната серия и изпитването;
|
|
—
|
Описание на други наблюдавани ефекти, напр. оцветяване на тъканите от оцветен изпитван химикал;
|
Обсъждане на резултатите
Заключение
|
ЛИТЕРАТУРА
|
(1)
|
UN (2015). United Nations Globally Harmonized System of Classification and Labelling of Chemicals (GHS). ST/SG/AC.10/30/Rev.6, Sixth Revised Edition, New York and Geneva: United Nations. Available at: http://www.unece.org/fileadmin/DAM/trans/danger/publi/ghs/ghs_rev06/English/ST-SG-AC10-30-Rev6e.pdf.
|
|
(2)
|
Глава Б.5 от настоящото Приложение, Остро дразнещо/корозивно действие върху очите.
|
|
(3)
|
Глава Б.47 от настоящото приложение, Метод за изпитване на непрозрачност и пропускливост на говеждата роговица за идентификация на вещества с корозивно и дразнещо действие върху очите.
|
|
(4)
|
Глава Б.48 от настоящото приложение, Метод за изпитване с изолирани птичи очи за определяне на i) химикали, причиняващи сериозно увреждане на очите и ii) химикали, не изискващи класифициране за дразнещо действие върху очите или сериозно увреждане на очите.
|
|
(5)
|
Глава Б.61 от настоящото приложение, Метод за изпитване с пропускане на флуоресцеин за идентифициране на химикали с корозивно и силно дразнещо действие върху очите.
|
|
(6)
|
Глава Б.68 от настоящото приложение, Метод за изпитване In Vitro на краткотрайна експозиция за идентифициране на i) химикали, причиняващи сериозно увреждане на очите и ii) химикали, не изискващи класифициране за дразнещо действие върху очите или сериозно увреждане на очите.
|
|
(7)
|
Freeman, S.J., Alépée N., Barroso, J., Cole, T., Compagnoni, A., Rubingh, C., Eskes, C., Lammers, J., McNamee, P., Pfannenbecker, U., Zuang, V. (2010). Prospective Validation Study of Reconstructed Human Tissue Models for Eye Irritation Testing. ALTEX 27, Special Issue 2010,261-266.
|
|
(8)
|
EC EURL ECVAM (2014). The EURL ECVAM - Cosmetics Europe prospective validation study of Reconstructed human Cornea-like Epithelium (RhCE)-based test methods for identifying chemicals not requiring classification and labelling for serious eye damage/eye irritation: Validation Study Report. EUR 28125 EN; doi:10.2787/41680. (налично на адрес: http://publications.jrc.ec.europa.eu/repository/handle/JRC100280.
|
|
(9)
|
EURL ECVAM Science Advisory Committee (2014). ESAC Opinion on the EURL ECVAM Eye Irritation Validation Study (EIVS) on EpiOcular™ EIT and SkinEthic™ HCE and a related Cosmetics Europe study on HPLC/UPLC-spectrophotometry as an alternative endpoint detection system for MTT-formazan. ESAC Opinion No 2014-03 of 17 November 2014; EUR 28173 EN; doi: 10.2787/043697. (налично на адрес: http://publications.jrc.ec.europa.eu/repository/handle/JRC103702.
|
|
(10)
|
Alépée, N., Leblanc, V., Adriaens, E., Grandidier, M.H., Lelièvre, D, Meloni, M., Nardelli, L., Roper, C.S, Santirocco, E., Toner, F., Van Rompay, A., Vinall, J., Cotovio, J. (2016). Multi-laboratory validation of SkinEthic HCE test method for testing serious eye damage/eye irritation using liquid chemicals. Toxicol. In Vitro 31, 43-53.
|
|
(11)
|
Alépée, N., Adriaens, E., Grandidier, M.H., Meloni, M., Nardelli, L., Vinall, C.J., Toner, F., Roper, C.S, Van Rompay, A.R., Leblanc, V., Cotovio, J. (2016). Multi-laboratory evaluation of SkinEthic HCE test method for testing serious eye damage/eye irritation using solid chemicals and overall performance of the test method with regard to solid and liquid chemicals testing. Toxicol. In Vitro 34, 55-70.
|
|
(12)
|
EURL ECVAM Science Advisory Committee (2016). ESAC Opinion on the SkinEthic™ Human Corneal Epithelium (HCE) Eye Irritation Test (EIT). ESAC Opinion No 2016-02 of 24 June 2016; EUR 28175 EN; doi: 10.2787/390390. (налично на адрес: http://publications.jrc.ec.europa.eu/repository/handle/JRC103704.
|
|
(13)
|
EC EURL ECVAM (2016). Recommendation on the Use of the Reconstructed human Cornea-like Epithelium (RhCE) Test Methods for Identifying Chemicals not Requiring Classification and Labelling for Serious Eye Damage/Eye Irritation According to UN GHS. (Manuscript in Preparation).
|
|
(14)
|
Draize, J.H., Woodard, G., Calvery, H.O. (1944). Methods for the Study of Irritation and Toxicity of Substances Applied Topically to the Skin and Mucous Membranes. Journal of Pharmacol. and Exp. Therapeutics 82, 377-390.
|
|
(15)
|
Scott, L., Eskes, C., Hoffmann, S., Adriaens, E., Alépée, N., Bufo, M., Clothier, R., Facchini, D., Faller, C., Guest, R., Harbell, J., Hartung, T., Kamp, H., Le Varlet, B., Meloni, M., McNamee, P., Osborne, R., Pape, W., Pfannenbecker, U., Prinsen, M., Seaman, C., Spielman, H., Stokes, W., Trouba, K., Van den Berghe, C., Van Goethem, F., Vassallo, M., Vinardell, P., Zuang, V. (2010). A Proposed Eye Irritation Testing Strategy to Reduce and Replace In Vivo Studies Using Bottom-Up and Top-Down Approaches. Toxicol. In Vitro 24, 1-9.
|
|
(16)
|
Mosmann, T. (1983). Rapid Colorimetric Assay for Cellular Growth and Survival: Application to Proliferation and Cytotoxicity Assays. J. Immunol. Methods 65, 55-63.
|
|
(17)
|
OECD (2016). Series on Testing and Assessment No 216: Performance Standards for the Assessment of Proposed Similar or Modified In Vitro Reconstructed Human Cornea-Like Epithelium (RhCE) Test Methods for Identifying Chemicals not Requiring Classification and Labelling for Eye Irritation or Serious Eye Damage, Based on the Validated Reference Methods EpiOcular™ EIT and SkinEthic™ HCE EIT described in TG 492. Organisation for Economic Cooperation and Development, Paris.
|
|
(18)
|
OECD (2005). Series on Testing and Assessment No 34: Guidance Document on the Validation and International Acceptance of New or Updated Test Methods for Hazard Assessment. Organisation for Economic Cooperation and Development, Paris.
|
|
(19)
|
Kaluzhny, Y., Kandárová, H., Hayden, P., Kubilus, J., d’Argembeau-Thornton, L., Klausner, M. (2011). Development of the EpiOcular™ Eye Irritation Test for Hazard Identification and Labelling of Eye Irritating Chemicals in Response to the Requirements of the EU Cosmetics Directive and REACH Legislation. Altern. Lab. Anim. 39, 339-364.
|
|
(20)
|
Nguyen, D.H., Beuerman, R.W., De Wever, B., Rosdy, M. (2003). Three-dimensional construct of the human corneal epithelium for in vitro toxicology. In: Salem, H., Katz, S.A. (Eds), Alternative Toxicological Methods, CRC Press, pp. 147-159.
|
|
(21)
|
Pfannenbecker, U., Bessou-Touya, S., Faller, C., Harbell, J., Jacob, T., Raabe, H., Tailhardat, M., Alépée, N., De Smedt, A., De Wever, B., Jones, P., Kaluzhny, Y., Le Varlet, B., McNamee, P., Marrec-Fairley, M., Van Goethem, F. (2013). Cosmetics Europe multi-laboratory pre-validation of the EpiOcular™ reconstituted Human Tissue Test Method for the Prediction of Eye Irritation. Toxicol. In Vitro 27, 619-626.
|
|
(22)
|
Alépée, N., Bessou-Touya, S., Cotovio, J., de Smedt, A., de Wever, B., Faller, C., Jones, P., Le Varlet, B., Marrec-Fairley, M., Pfannenbecker, U., Tailhardat, M., van Goethem, F., McNamee, P. (2013). Cosmetics Europe Multi-Laboratory Pre-Validation of the SkinEthic™ Reconstituted Human Corneal Epithelium Test Method for the Prediction of Eye Irritation. Toxicol. In Vitro 27, 1476-1488.
|
|
(23)
|
Kolle, S.N., Moreno, M.C.R., Mayer, W., van Cott, A., van Ravenzwaay, B., Landsiedel, R. (2015). The EpiOcular™ Eye Irritation Test is the Method of Choice for In Vitro Eye Irritation Testing of Agrochemical Formulations: Correlation Analysis of EpiOcular™ Eye Irritation Test and BCOP Test Data to UN GHS, US EPA and Brazil ANIVSA Classifications. Altern. Lab. Anim. 43, 1-18.
|
|
(24)
|
Adriaens, E., Barroso, J., Eskes, C., Hoffmann, S., McNamee, P., Alépée, N., Bessou-Touya, S., De Smedt, A., De Wever, B., Pfannenbecker, U., Tailhardat, M., Zuang, V. (2014). Retrospective Analysis of the Draize Test for Serious Eye Damage/Eye Irritation: Importance of Understanding the In Vivo Endpoints Under UN GHS/EU CLP for the Development and Evaluation of In Vitro Test Methods. Arch. Toxicol. 88, 701-723.
|
|
(25)
|
Barroso, J., Pfannenbecker, U., Adriaens, E., Alépée, N., Cluzel, M., De Smedt, A., Hibatallah, J., Klaric, M., Mewes, K.R., Millet, M., Templier, M., McNamee, P. (2017). Cosmetics Europe compilation of historical serious eye damage/eye irritation in vivo data analysed by drivers of classification to support the selection of chemicals for development and evaluation of alternative methods/strategies: the Draize eye test Reference Database (DRD). Arch. Toxicol. 91, 521-547.
|
|
(26)
|
Meloni, M., De Servi, B., Marasco, D., Del Prete, S. (2011). Molecular mechanism of ocular surface damage: Application to an in vitro dry eye model on human corneal epithelium. Molecular Vision 17, 113-126.
|
|
(27)
|
Hackett, R.B., McDonald, T.O. (1991). Eye Irritation. In Advances in Modern Toxicology: Dermatoxicology Marzulli F.N.and Maibach H.I. (Eds.), 4th Edition, pp. 749–815. Washington, DC, USA: Hemisphere Publishing Corporation.
|
|
(28)
|
Fox, D.A., Boyes, W.K. (2008). Toxic Responses of the Ocular and Visual System. In Cassaret and Doull’s Toxicology: The Basic Science of Poisons Klaassen C.D.(Ed.), 7th Edition, pp. 665–697. Withby, ON, Canada: McGraw-Hill Ryerson.
|
|
(29)
|
Jester, J.V., Li, H.F., Petroll, W.M., Parker, R.D., Cavanagh, H.D., Carr, G.J., Smith, B., Maurer, J.K. (1998). Area and Depth of Surfactant Induced Corneal Injury Correlates with Cell Death. Invest. Ophthalmol. Vis. Sci. 39, 922–936.
|
|
(30)
|
Maurer, J.K., Parker, R.D., Jester, J.V. (2002). Extent of Corneal Injury as the Mechanistic Basis for Ocular Irritation: Key Findings and Recommendations for the Development of Alternative Assays. Reg. Tox. Pharmacol. 36, 106-117.
|
|
(31)
|
Jester, J.V., Li, L., Molai, A., Maurer, J.K. (2001). Extent of Corneal Injury as a Mechanistic Basis for Alternative Eye Irritation Tests. Toxicol. In Vitro 15, 115–130.
|
|
(32)
|
Jester, J.V., Petroll, W.M., Bean, J., Parker, R.D., Carr, G.J., Cavanagh, H.D., Maurer, J.K. (1998). Area and Depth of Surfactant-Induced Corneal Injury Predicts Extent of Subsequent Ocular Responses. Invest. Ophthalmol. Vis. Sci. 39, 2610–2625.
|
|
(33)
|
Jester, J.V. (2006). Extent of Corneal Injury as a Biomarker for Hazard Assessment and the Development of Alternative Models to the Draize Rabbit Eye Test. Cutan. Ocul. Toxicol. 25, 41–54.
|
|
(34)
|
EpiOcular™ EIT SOP, Version 8 (March 05, 2013). EpiOcular™ EIT for the Prediction of Acute Ocular Irritation of Chemicals. (налично на адрес: [https://ecvam-dbalm.jrc.ec.europa.eu/beta/index.cfm/methodsAndProtocols/index].
|
|
(35)
|
SkinEthic™ HCE EIT SOP, Version 1. (July 20, 2015). SkinEthic™ HCE Eye Irritation Test (EITL for Liquids, EITS for Solids) for the Prediction of Acute Ocular Irritation of Chemicals. (налично на адрес: https://ecvam-dbalm.jrc.ec.europa.eu/beta/index.cfm/methodsAndProtocols/index.
|
|
(36)
|
Alépée, N., Barroso, J., De Smedt, A., De Wever, B., Hibatallah, J., Klaric, M., Mewes, K.R., Millet, M., Pfannenbecker, U., Tailhardat, M., Templier, M., McNamee, P. (2015). Use of HPLC/UPLC-Spectrophotometry for Detection of Formazan in In Vitro Reconstructed Human Tissue (RhT)-Based Test Methods Employing the MTT-Reduction Assay to Expand their Applicability to Strongly Coloured Test Chemicals. Toxicol. In Vitro 29, 741-761.
|
|
(37)
|
Kaluzhny, Y., Kandárová, H., Handa, Y., DeLuca, J., Truong, T., Hunter, A., Kearney, P., d’Argembeau-Thornton, L., Klausner, M. (2015). EpiOcular™ Eye Irritation Test (EIT) for Hazard Identification and Labeling of Eye Irritating Chemicals: Protocol Optimization for Solid Materials and Extended Shipment Times. Altern. Lab Anim. 43, 101-127.
|
|
(38)
|
US FDA (2001). Guidance for Industry: Bioanalytical Method Validation. U.S. Department of Health and Human Services, Food and Drug Administration. May 2001. (налично на адрес: http://www.fda.gov/downloads/Drugs/Guidances/ucm070107.pdf.
|
|
(39)
|
OECD (2017). Guidance Document on an Integrated Approaches on Testing and Assessment for Serious Eye Damage and Eye irritation. Series on Testing and Assessment No 263. ENV Publications, Organisation for Economic Cooperation and Development, Paris.
|
Допълнение 1
ОПРЕДЕЛЕНИЯ
Точност
: степента на близост между резултатите от метода за изпитване и приетите референтни стойности. Това е мярка за ефективността на метода за изпитване и един от аспектите на неговата приложимост. Терминът често се използва взаимозаменяемо с термина „съответствие“, за да се обозначи относителният дял на правилните резултати при даден метод за изпитване (18).
Химикал за сравнение
: Химикал, използван като еталон за сравнение с изпитван химикал. Химикалът за сравнение следва да притежава следните свойства: (i) постоянен и надежден източник (или източници) за неговото идентифициране и характеризиране; (ii) структурно, функционално и/или химично сходство с класа на изпитвания(те) химикал(и); (iii) известни физични/химични характеристики; (iv) потвърждаващи данни за известни въздействия; и v) известна ефективност за постигане на желаната реакция.
Подход „отдолу нагоре“
: поетапен подход, използван за химикали, за които се предполага, че не изискват класифициране и етикетиране за дразнене сериозно увреждане на очите, който започва с определяне на химикали, за които не се изисква класифициране и етикетиране (отрицателни резултати) по отношение на други химикали (положителни резултати).
Химикал
: Вещество или смес.
Съответствие
: вж. „Точност“.
Роговица
: Прозрачната част на предната страна на очната ябълка, която покрива ириса и зеницата и пропуска светлината навътре.
CV
: Коефициент на вариация.
Откл.
: Отклонение.
EIT
: Изпитване за дразнене на очите.
EURL ECVAM
: Референтна лаборатория на Европейския съюз за алтернативи на опитите върху животни.
Дразнене на очите
: Предизвикване на промени в очите вследствие на прилагане на изпитван химикал към предната повърхност на окото, които са напълно обратими в рамките на 21 дни след прилагането. Терминът е взаимозаменяем с „Обратими ефекти върху очите“ и с „Категория 2 по GHS на ООН/CLP“.
ET50
: Необходимото време за експозиция за намаляване на тъканната жизнеспособност с 50 % при приложение на химикал за сравнение с определена фиксирана концентрация.
Честота на невярно определените отрицателни резултати
: Пропорцията на всички невярно определени отрицателни резултати по даден метод за изпитване относно вещества, предизвикващи положителен резултат. Това е един от показателите за характеристиките на метода за изпитване.
Честота на невярно определените позитивни резултати
: Пропорцията на всички невярно определени положителни резултати по даден метод за изпитване относно вещества, предизвикващи отрицателен резултат. Това е един от показателите за характеристиките на метода за изпитване.
Опасност
: Вътрешно присъщо свойство на даден агент или ситуация, притежаващо потенциал да предизвика неблагоприятни ефекти когато един организъм, система или (суб-)популация бъдат експонирани на този агент.
HCE
: SkinEthic™ човешки роговичен епител.
ВЕТХ
: Високоефективна течна хроматография.
IC50
: Концентрация, при която химикалът за сравнение намалява жизнеспособността на тъканите с 50 % след фиксирано време на експозиция (напр. 30 минути третиране със SDS).
Неопределена доза
: Количеството изпитван химикал, приложено върху тъканния модел на RhCE, превишаващо необходимото количество за пълно и равномерно покриване на епителната повърхност.
Необратими въздействия върху очите
: Вж. „Сериозно увреждане на очите“.
LLOQ
: Долна граница за количествено определяне.
LogP
: Логаритъм на коефициента на разпределение октанол-вода
Смес
: Смес или разтвор, съставен от две или повече вещества.
Вещество с една съставка
: Вещество, определено от неговия количествен състав, в който съдържанието на една основна съставка е най-малко 80 % (w/w).
Вещество с няколко съставки
: Вещество, определено от неговия количествен състав, в който повече от една основна съставка се съдържа в концентрация ≥ 10 % (w/w) и < 80 % (w/w). Веществото с повече съставки е резултат от производствен процес. Разликата между смес и вещество с повече съставки е, че дадена смес е получена чрез смесване на две или повече вещества без химична реакция. Веществото с повече съставки е резултат от химична реакция.
MTT
: 3-(4,5-диметилтиазол-2-ил)-2,5-дифенилтетразолиев бромид; Тиазолил син тетразолиев бромид.
Отрицателна контрола
: Проба, съдържаща всички компоненти на дадена изпитвана система и третирана с вещество, за което е известно, че не предизвиква положителен отклик в изпитваната система. Тази проба се обработва с пробите, третирани с изпитван химикал, и с други контролни проби, и се използва за определяне на 100 %-на тъканна жизнеспособност.
Некласифицирани
: химикали, които не са класифицирани за дразнене на очите (категория 2 по GHS на ООН/CLP, категория 2А или 2В по GHS на ООН/CLP) или сериозно увреждане на очите (категория 1 по GHS на ООН/CLP). Терминът е взаимозаменяем с „без категория по GHS на ООН/CLP“.
NSCkilled
: Неспецифичен цвят в умъртвени тъкани.
NSCliving
: Неспецифичен цвят в живи тъкани.
NSMTT
: Неспецифична редукция на MTT.
OD
: Оптична плътност.
Стандарти за ефективност
: стандарти въз основа на валидиран метод за изпитване, който се счита за валиден от научна гледна точка, осигуряващи основата за оценка на сравнимостта на предложен метод за изпитване, който е по своя механизъм и функционално сходен с валидирания метод. Включени са: i) съществени компоненти на метода за изпитване; ii) минимален списък на референтни химикали, подбрани между химикалите, използвани за демонстриране на приемливата ефективност на валидирания метод за изпитване; и iii) сходните нива за точност и надеждност въз основа на получените за валидирания метод за изпитване, които предложеният метод за изпитване следва да демонстрира при оценяването чрез използване на минималния списък на референтните химикали (18).
Положителни контроли
: Проба, съдържаща всички компоненти на дадена изпитвана система и третирана с вещество, за което е известно, че предизвиква положителен отклик в изпитваната система. Тази проба се обработва с пробите, третирани с изпитван химикал, и с други контролни проби. За да се гарантира, че може да се направи оценка на варирането на отклика в положителната контрола във времето, големината на положителния отклик не следва да е прекомерна.
Относимост
: описание на взаимовръзката между изпитването и ефекта, представляващ интерес, и дали тя е значима и полезна за определена цел. Това е степента, в която изпитването правилно измерва или прогнозира биологичния ефект, представляващ интерес. Относимостта включва разглеждането на точността (съответствието) на метода за изпитване (18).
Надеждност
: мярка за степента, в която даден метод за изпитване може да се приложи възпроизводимо в една и съща лаборатория и в различни лаборатории по различно време, при използване на един и същи протокол. Оценката за нея се прави, като се изчислява вътрешнолабораторната и междулабораторната възпроизводимост и вътрешнолабораторната повторяемост (18).
Заместващо изпитване
: изпитване, предназначено да замести изпитване, което се използва рутинно и е прието за определяне на опасността и/или оценка на риска, и за което е установено, че предоставя равностойна или по-добра защита на здравето на хората или животните или на околната среда, според приложимото, в сравнение с приетото изпитване за всички възможни изпитвани ситуации и химикали (18).
Възпроизводимост
: Съгласуваността между резултатите, получени чрез изследване на един и същ химикал, като се използва един и същ протокол за изпитване (вж. „Повторяемост“) (18).
Обратими въздействия върху очите
: Вж. „Дразнене на очите“.
RhCE
: Реконструиран, наподобяващ човешка роговица епител.
Серия
: Серията се състои от един или повече изпитвани химикали, подложени на паралелно изпитване с отрицателна контрола и с положителна контрола.
SD
: Стандартно отклонение.
Чувствителност
: делът на всички положителни/активни изпитвани химикали, които са класифицирани правилно чрез изпитването. Това е мярка за точността на метода за изпитване, който предоставя категорични резултати и е важен фактор при оценката на относимостта на метода за изпитване (18).
Сериозно увреждане на очите
: Предизвикване на тъканно увреждане на окото или сериозно физическо влошаване на зрението след прилагане на изпитвано вещество към предната повърхност на окото, което не е напълно обратимо в рамките на 21 дни след прилагането. Терминът е взаимозаменяем с „Необратими ефекти върху очите“ и с „Категория 1 по GHS на ООН и CLP“.
Стандартни оперативни процедури (СОП)
: Официални писмени процедури, които описват подробно как следва да се извършват рутинните и специфичните за изпитване лабораторни операции. Те се изискват от добрата лабораторна практика.
Специфичност
: делът на всички негативни/неактивни изпитвани химикали, които са класифицирани правилно чрез изпитването. Това е мярка за точността на метода за изпитване, която предоставя категорични резултати и е важен фактор при оценката на метода за изпитване (18).
Вещество
: Химически елемент и неговите съединения в естествено състояние или получени чрез производствен процес, включително всякакви добавки, необходими за запазване на стабилността му, както и всякакви онечиствания, произтичащи от използваната технология, но с изключение на разтворители, които могат да бъдат отделени, без да се засегне стабилността на веществото или да се промени съставът му.
Изпитване
: Единичен изпитван химикал, изпитван паралелно с поне две повторения на тъкан, както е определено в съответната СОП.
Тъканна жизнеспособност
: Параметър, измерващ общата активност на клетъчна популация в реконструирана тъкан като тяхната способност да редуцират виталния оцветител MTT, който, в зависимост от измерената крайна точка и използвания дизайн на изпитване, е свързан с общия брой и/или жизнеспособността на живите клетки.
Подход „отгоре надолу“
: Поетапен подход, използван за химикал, за който се предполага, че предизвиква сериозно увреждане на очите, който започва с определянето на химикали, предизвикващи сериозно увреждане на очите (положителен резултат) по отношение на други химикали (отрицателен резултат).
Изпитван химикал
: Всяко вещество или смес, изпитвано(а) с използване на настоящия метод за изпитване.
Стратегия за поетапно изпитване
: Стратегия за поетапно изпитване, която използва методи за изпитване по последователен начин. Цялата съществуваща информация за даден изпитван химикал се разглежда по конкретен ред, като на всеки етап се прилага процес, основан на тежестта на доказателствата, за определяне дали съществува достатъчно информация за вземане решение за класифициране с оглед на опасността, преди да се премине към следващия етап на стратегията. Ако потенциалът за опасност/действие на даден изпитван химикал може да се определи въз основа на съществуващата информация, не се налага допълнително изпитване (18).
ULOQ
: Горна граница за количествено определяне.
Глобална хармонизирана система на Организацията на обединените нации за класифициране и етикетиране на химикали (GHS на ООН)
: система, предлагаща класифициране на химикали (вещества и смеси) според стандартизирани видове и степени на физическа, здравна и екологична опасност и разглеждаща съответни съобщителни елементи, като например пиктограми, сигнални думи, предупреждения за опасност, препоръки за безопасност и информационни листове за безопасност, така че те да съобщават информация за тяхното неблагоприятно въздействие с оглед защита на хората (включително работодатели, работещи, служители в транспорта, потребители и аварийни служители) и на околната среда (1).
Категория 1 по GHS на ООН и CLP
: Вж. „Сериозно увреждане на очите“.
Категория 2 по GHS на ООН и CLP
: Вж. „Дразнене на очите“.
„Без категория“ по GHS на ООН и CLP
: Химикали, които не отговарят на изискванията за класифициране като категория 1 или 2 по GHS на ООН/ CLP (или категория 2А или 2В по GHS на ООН). Терминът е взаимозаменяем с „некласифицирани“.
УВЕТХ
: Ултра високоефективна течна хроматография.
UVCB
: вещества с неизвестен или променлив състав, сложни продукти от реакции или биологични материали.
Валиден метод за изпитване
: Метод за изпитване, за който се смята, че притежава достатъчна относимост и надеждност по отношение на конкретна цел и който се основава на научно обосновани принципи. Даден метод за изпитване никога не е валиден в абсолютен смисъл, а само по отношение на определена цел (18).
Валидиран метод за изпитване
: метод за изпитване, за който са извършени изследвания за валидиране за определяне на относимостта (включително на точността) и надеждността му за конкретна цел. Важно е да се отбележи, че един валидиран метод за изпитване може да не притежава достатъчно ефективност по отношение на точността и надеждността си, за да се счита за приложим за предлаганата цел (18).
VRM
: Валидиран референтен метод.
VRM1
: EpiOcular™ EIT се счита за валидирания референтен метод 1.
VRM2
: SkinEthic™ HCE EIT iсе счита за валидирания референтен метод 2.
Процес, основан на тежестта на доказателствата
: Процесът на отчитане на силните и слабите страни на различни видове информация за достигане до дадено заключение относно потенциалната опасност на даден химикал, и в подкрепа на това заключение.
Допълнение 2
ОСНОВНИ ИЗПИТВАНИ КОМПОНЕНТИ НА ТЕСТОВЕТЕ RHCE, ВАЛИДИРАНИ ЗА ИДЕНТИФИЦИРАНЕ НА ХИМИКАЛИ, НЕ ИЗИСКВАЩИ КЛАСИФИЦИРАНЕ И ЕТИКЕТИРАНЕ ЗА ДРАЗНЕНЕ НА ОЧИТЕ ИЛИ СЕРИОЗНО УВРЕЖДАНЕ НА ОЧИТЕ
|
Компоненти на теста
|
EpiOcular™ EIT
(VRM 1)
|
SkinEthic™ HCE EIT
(VRM 2)
|
|
Протоколи
|
Течности
(подлежащи на пипетиране при 37±1°C или по-ниски температури в продължение на 15 минути)
|
Твърди горива
(не подлежат на пипетиране)
|
Течности и вискозни вещества:
(подлежат на пипетиране)
|
Твърди горива
(не подлежат на пипетиране)
|
|
Повърхност на модела
|
0,6 cm2
|
0,6 cm2
|
0,5 cm2
|
0,5 cm2
|
|
Брой на тъканни повторения
|
Най-малко 2 сигнала
|
Най-малко 2 сигнала
|
Най-малко 2 сигнала
|
Най-малко 2 сигнала
|
|
Предварителна проверка за цветна интерференция
|
50 μl + 1 ml H2O в продължение на 60 min при 37±2°C, 5±1 % CO2, ≥95 % ОВ (неоцветени изпитвани химикали) или 50 μl + 2 ml изопропанол, смесен за 2-3 часа при RT (оцветени изпитвани химикали)
|
 ако OD на изпитвания химикал при 570±20 nm, след изваждане на OD за изопропанол или вода, е > 0,08 (която отговаря на приблизително 5 % от средната OD на отрицателната контрола), трябва да се извършат живи адаптирани контроли.
ако OD на изпитвания химикал при 570±20 nm, след изваждане на OD за изопропанол или вода, е > 0,08 (която отговаря на приблизително 5 % от средната OD на отрицателната контрола), трябва да се извършат живи адаптирани контроли.
|
|
50 mg + 1 ml H2O за 60 min при 37±2°C, 5±1 % CO2, ≥95 % ОВ
(неоцветен изпитван химикал) и/или
50 mg + 2 ml изопропанол, смесени за 2-3 часа при RT (оцветени и неоцветени изпитвани химикали)
|
 ако OD на изпитвания химикал при 570±20 nm, след изваждане на OD за изопропанол или вода, е > 0,08 (която отговаря на приблизително 5 % от средната OD на отрицателната контрола), трябва да се извършат живи адаптирани контроли.
ако OD на изпитвания химикал при 570±20 nm, след изваждане на OD за изопропанол или вода, е > 0,08 (която отговаря на приблизително 5 % от средната OD на отрицателната контрола), трябва да се извършат живи адаптирани контроли.
|
|
10 μl + 90 μl H2O, смесени за 30±2 min при стайна температура (RT, 18-28°C)
|
 ако изпитваният химикал се оцвети, трябва да се проведат живи адаптирани контроли
ако изпитваният химикал се оцвети, трябва да се проведат живи адаптирани контроли
|
|
10 mg + 90 μl H2O, смесени за 30±2 min при RT
|
 ако изпитваният химикал се оцвети, трябва да се проведат живи адаптирани контроли
ако изпитваният химикал се оцвети, трябва да се проведат живи адаптирани контроли
|
|
|
Предварителна проверка за пряка редукция на MTT
|
50 μl + 1 ml MTT 1 mg/ml разтвор за 180±15 min при 37±2°C, 5±1 % CO2, ≥95 % ОВ
|
 ако разтворът стане син/лилав, трябва да се проведат адаптирани контроли, умъртвени чрез замразяване
ако разтворът стане син/лилав, трябва да се проведат адаптирани контроли, умъртвени чрез замразяване
(50 μl стерилна дейонизирана вода в разтвор на MTT се използва като отрицателна контрола)
|
|
50 mg + 1 ml MTT 1 mg/ml разтвор за 180±15 min при 37±2°C, 5±1 % CO2, ≥95 % ОВ
|
 ако разтворът стане син/лилав, трябва да се проведат адаптирани контроли, умъртвени чрез замразяване
ако разтворът стане син/лилав, трябва да се проведат адаптирани контроли, умъртвени чрез замразяване
(50 μl стерилна дейонизирана вода в разтвор на MTT се използва като отрицателна контрола)
|
|
30 μl + 300 μl MTT 1 mg/ml разтвор за 180± 15 min при 37±2°C, 5±1 % CO2, ≥95 % ОВ
|
 ако разтворът стане син/лилав, трябва да се проведат адаптирани контроли, умъртвени с вода
ако разтворът стане син/лилав, трябва да се проведат адаптирани контроли, умъртвени с вода
(30 μl стерилна дейонизирана вода в разтвор на MTT се използва като отрицателна контрола)
|
|
30 mg + 300 μl MTT 1 mg/ml разтвор за 180± 15 min при 37±2°C, 5±1 % CO2, ≥95 % ОВ
|
 ако разтворът стане син/лилав, трябва да се проведат адаптирани контроли, умъртвени с вода
ако разтворът стане син/лилав, трябва да се проведат адаптирани контроли, умъртвени с вода
(30 μl стерилна дейонизирана вода в разтвор на MTT се използва като отрицателна контрола)
|
|
|
предварителна обработка
|
20 μl DPBS без Ca2+/Mg2+
за 30 ± 2 min при 37±2°C, 5±1 % CO2, ≥95 % ОВ, със защита от светлина.
|
20 μl DPBS без Ca2+/Mg2+
за 30±2 min при 37±2°C, 5±1 % CO2, ≥95 % ОВ, със защита от светлина.
|
—
|
—
|
|
Дози за третиране и приложение
|
50 μl (83,3 μl/cm2)
|
50 mg (83,3 mg/cm2) с използване на калибриран инструмент (напр. равна супена лъжица, калибрирана да носи 50 mg натриев хлорид).
|
10 μl DPBS без Ca2+/Mg2+ + 30 ± 2 μl (60 μl/cm2)
За вискозни вещества използвайте найлонова мрежа
|
30 μl DPBS без Ca2+/Mg2+ + 30 ± 2 mg (60 mg/cm2)
|
|
Експозиция време и температура
|
30 min (± 2 min)
в среда за отглеждане
при 37±2°C, 5±1 % CO2, ≥95 % ОВ
|
6 часа (± 0,25 ч.)
в среда за отглеждане
при 37±2°C, 5±1 % CO2, ≥95 % ОВ
|
30 min (± 2 min)
в среда за отглеждане
при 37±2°C, 5±1 % CO2, ≥95 % ОВ
|
4 часа (± 0,1 ч.)
в среда за отглеждане
при 37±2°C, 5±1 % CO2, ≥95 % ОВ
|
|
Изплакване на стайна температура
|
3 пъти в 100 ml DPBS без Ca2+/Mg2+
|
3 пъти в 100 ml DPBS без Ca2+/Mg2+
|
20 ml DPBS без Ca2+/Mg2+
|
25 ml DPBS без Ca2+/Mg2+
|
|
Потапяне след експозиция
|
12 min (± 2 min) при RT в среда за отглеждане
|
25 min (± 2 min) при RT в среда за отглеждане
|
30 min (± 2 min) при 37°C, 5 % CO2, 95 % ОВ, в среда за отглеждане
|
30 min (± 2 min) при RT в среда за отглеждане
|
|
Инкубиране след експозиция
|
120 min (± 15 min) в среда за отглеждане при 37±2°C, 5±1 % CO2, ≥95 % ОВ
|
18 ч. (± 0,25 ч.) в среда за отглеждане при 37±2°C, 5±1 % CO2, ≥95 % ОВ
|
Няма
|
18 ч. (± 0,5 ч.) в среда за отглеждане при 37±2°C, 5±1 % CO2, ≥95 % ОВ
|
|
Отрицателна контрола
|
50 μl H2O
Изпитвана паралелно
|
50 μl H2O
Изпитвана паралелно
|
30 ± 2μl DPBS без Ca2+/Mg2+
Изпитвана паралелно
|
30 ± 2μl DPBS без Ca2+/Mg2+
Изпитвана паралелно
|
|
Положителна контрола
|
50 μl метил ацетат
Изпитвана паралелно
|
50 μl метил ацетат
Изпитвана паралелно
|
30 ± 2μl метил ацетат
Изпитвана паралелно
|
30 ± 2μl метил ацетат
Изпитвана паралелно
|
|
Разтвор на MTT
|
300 μl 1 mg/ml
|
300 μl 1 mg/ml
|
300 μl 1 mg/ml
|
300 μl 1 mg/ml
|
|
Инкубация на MTT време и температура
|
180 min (± 15 min) при 37±2°C, 5±1 % CO2, ≥95 % ОВ
|
180 min (± 15 min) при 37±2°C, 5±1 % CO2, ≥95 % ОВ
|
180 min (± 15 min) при 37±2°C, 5±1 % CO2, ≥95 % ОВ
|
180 min (± 15 min) при 37±2°C, 5±1 % CO2, ≥95 % ОВ
|
|
Екстрахиращ разтворител
|
2 ml изопропанол
(екстракция от горната и долната част на вложката чрез пробиване на тъканта)
|
2 ml изопропанол
(екстракция от долната част на вложката чрез пробиване на тъканта)
|
1,5 ml изопропанол
(екстракция от горната и долната част на вставката)
|
1,5 ml изопропанол
(екстракция от долната част на вложката)
|
|
Време и температура на екстракция
|
2-3 ч. с разклащане (~120 rpm) при RT или през нощта при 4-10°C
|
2-3 ч. с разклащане (~120 rpm) при RT или през нощта при 4-10°C
|
4 ч. с разклащане (~120 rpm) при RT или поне през нощта без разклащане при 4-10°C
|
Поне 2 ч. с разклащане (~120 rpm) при RT
|
|
Показание за оптична плътност
|
570 nm (550 - 590 nm)
без еталонен филтър
|
570 nm (550-590 nm)
без еталонен филтър
|
570 nm (540 - 600 nm)
без еталонен филтър
|
570 nm (540 - 600 nm)
без еталонен филтър
|
|
Контрол на качеството на тъканта
|
Третиране с 100 μl 0,3 % (v/v) Triton X-100
12,2 min ≤ ET50 ≤ 37,5 min
|
Третиране с 100 μl 0,3 % (v/v) Triton X-100
12,2 min ≤ ET50 ≤ 37,5 min
|
30 min третиране със SDS (50 μl)
1,0 mg/ml ≤ IC50 ≤ 3,5 mg/ml
|
30 min третиране със SDS (50 μl)
1,0 mg/ml ≤ IC50 ≤ 3,2 mg/ml
|
|
Критерии за приемливост
|
|
1.
|
Средната OD на тъканните повторения, третирани с отрицателната контрола (NaCl), трябва да е > 0,8 и < 2,5
|
|
2.
|
Средната жизнеспособност на тъканните повторения, изложени за 30 минути на положителната контрола, изразена като % от отрицателната контрола, следва да е < 50 %
|
|
3.
|
Разликата в жизнеспособността между двете тъканни повторения следва да е по-малка от 20 %.
|
|
|
1.
|
Средната OD на тъканните повторения, третирани с отрицателната контрола (NaCl), трябва да е > 0,8 и < 2,5
|
|
2.
|
Средната жизнеспособност на тъканните повторения, изложени за 6 часа на положителната контрола, изразена като % от отрицателната контрола, следва да е < 50 %
|
|
3.
|
Разликата в жизнеспособността между двете тъканни повторения следва да е по-малка от 20 %.
|
|
|
1.
|
Средната OD на тъканните повторения, третирани с отрицателната контрола, следва да е > 1,0 и ≤ 2,5
|
|
2.
|
Средната жизнеспособност на тъканните повторения, изложени за 30 минути на положителната контрола, изразена като % от отрицателната контрола, следва да е ≤ 30 %
|
|
3.
|
Разликата в жизнеспособността между двете тъканни повторения следва да е по-малка от 20 %.
|
|
|
1.
|
Средната OD на тъканните повторения, третирани с отрицателната контрола, следва да е > 1,0 и ≤ 2,5
|
|
2.
|
Средната жизнеспособност на тъканните повторения, изложени за 4 часа на положителната контрола, изразена като % от отрицателната контрола, следва да е ≤ 20 %
|
|
3.
|
Разликата в жизнеспособността между двете тъканни повторения следва да е по-малка от 20 %.
|
|
Допълнение 3
ИЛЮСТРАТИВНИ ДИАГРАМИ, ПРЕДОСТАВЯЩИ УКАЗАНИЯ ЗА НАЧИНА НА ИДЕНТИФИЦИРАНЕ И РАБОТА С ПРЕКИ РЕДУКТОРИ НА МТТ И/ИЛИ ХИМИКАЛИ С ЦВЕТОВА ИНТЕРФЕРЕНЦИЯ ВЪЗ ОСНОВА НА СОП ЗА VRM1

Допълнение 4
ИЛЮСТРАТИВНИ ДИАГРАМИ, ПРЕДОСТАВЯЩИ УКАЗАНИЯ ЗА НАЧИНА НА ИДЕНТИФИЦИРАНЕ И РАБОТА С ПРЕКИ РЕДУКТОРИ НА МТТ И/ИЛИ ХИМИКАЛИ С ЦВЕТОВА ИНТЕРФЕРЕНЦИЯ ВЪЗ ОСНОВА НА СОП ЗА VRM2

Допълнение 5
КЛЮЧОВИ ПАРАМЕТРИ И КРИТЕРИИ ЗА ПРИЕМЛИВОСТ ЗА ОДОБРЯВАНЕ НА СИСТЕМА ЗА ВЕТХ/УВЕТХ СПЕКТРОФОТОМЕТРИЯ ЗА ИЗМЕРВАНЕ НА МТТ ФОРМАЗАН, ИЗВЛЕЧЕН ОТ ТЪКАННИ МОДЕЛИ НА RНСE
|
Параметър
|
Протокол, извлечен от Ръководството на FDA (36)(38)
|
Критерии за приемливост
|
|
Избирателност
|
Анализ на изопропанол, жива празна проба (изопропанолов екстракт от живи тъканни модели на RhСE, без каквото и да е третиране), мъртва празна проба (изопропанолов екстракт от умъртвени тъканни модели на RhСE, без каквото и да е третиране) и на оцветител (напр. метиленово синьо)
|
Площ наинтерференция ≤ 20 % от площтаLLOQ
(88)
|
|
Прецизност
|
Качествени контроли (т.е., MTT формазан с концентрация 1.6 μg/ml, 16 μg/ml и 160 μg/ml ) в изопропанол (n=5)
|
CV ≤ 15 % или ≤ 20 % за LLOQ
|
|
Точност
|
Качествени контроли в изопропанол (n=5)
|
%откл.≤ 15 % или ≤ 20 % за LLOQ
|
|
Матричен ефект
|
Качествени контроли в жива празна проба (n=5)
|
85 % ≤ %матричен ефект ≤ 115 %
|
|
Процент на пренасяне
|
Анализ на изопропанол съгласно ULOQ (89) стандарта
|
Площинтерференция ≤ 20 % от площтаLLOQ
|
|
Възпроизводимост (в рамките на деня)
|
3 независими калибрационни криви (базирани на 6 последователни 1/3 разреждания на MTT формазан в изопропанол, като се започне от ULOQ, т.е., 200 μg/ml);
Качествени контроли в изопропанол (n=5)
|
Калибрирационни криви: % откл.≤ 15 % или ≤ 20 % за LLOQ
Качествени контроли: % откл.≤ 15 % и CV ≤ 15 %
|
|
Възпроизводимост (между различни дни)
|
Ден 1 1 калибрационна крива и качествени контроли в изопропанол (n=3)
Ден 2 1 калибрационна крива и качествени контроли в изопропанол (n=3)
Ден 3 1 калибрационна крива и качествени контроли в изопропанол (n=3)
|
|
Краткосрочна стабилност на MTT формазан в тъканен екстракт на RhСE
|
Качествени контроли в жива празна проба (n=3), анализирана в деня на приготвяне и след 24 часа съхранение при стайна температура
|
%откл. ≤ 15 %
|
|
Дългосрочна стабилност на MTT формазан в тъканен екстракт на RhСE, ако се изисква
|
Качествени контроли в жива празна проба (n=3), анализирана в деня на приготвяне и след няколко дни съхранение при -20°C
|
%откл. ≤ 15 %
|
Б.70 ИЗПИТВАНИЯ IN VITRO НА ЧОВЕШКИ РЕКОМБИНАНТЕН ЕСТРОГЕНЕН РЕЦЕПТОР (hrER) ЗА ОТКРИВАНЕ НА ХИМИКАЛИ С АФИНИТЕТ НА СВЪРЗВАНЕ С ЕР
ОБЩО ВЪВЕДЕНИЕ
Насоки за изпитване на ОИСР, базирани на ефективност
|
1.
|
Настоящият метод за изпитване е еквивалентен на насоките за изпитване на ОИСР (ТG) 493 (2015). TG 493 представляват насоки за изпитване, базирани на ефективност (PBTG), които описват методологията за човешки рекомбинантни in vitro изследвания за откриване на вещества с афинитет за свързване на естрогенен рецептор (изпитвания за hrER свързване). Състоят се от две механистични и функционално сходни изследвания за идентифициране на свързващи естрогенен рецептор (т.е. ERα) вещества и следва да улеснят разработването на нови подобни или модифицирани изследвания в съответствие с принципите за валидиране, установени в Ръководството на ОИСР относно валидирането и международното приемане на нови или актуализирани методи за изпитване за оценка на опасности (1). Напълно валидираните референтни методи за изпитване (допълнение 2 и допълнение 3), които осигуряват основата за настоящитенасоки за изпитване, базирани на ефективност (PBTG), са:
|
—
|
Изследването In Vitro за свързване на естрогенен рецептор (ЕР) на Freyberger-Wilson (FW), използващо цялостен човешки рекомбинантен ERα (2), и
|
|
—
|
Изследването In Vitro за свързване на естрогенен рецептор на Изследователския институт за оценка на химикали (CERI), използващо човешки рекомбинантен лиганд-свързващ домейн на протеин (2).
|
Съществуват стандарти за ефективност (PS) (3), които улесняват разработката и валидирането на подобни методи за изпитване за същата крайна точка на опасност и позволяват своевременно изменение на PBTG 493, така че нови сходни изследвания да могат да бъдат добавени към актуализираните PBTG. Въпреки това сходни изследвания ще се добавят само след преглед и съгласуване от ОИСР, че отговарят на стандартите за ефективност. Включените в TG 493 изследвания могат да се използват, за да се изпълнят изискванията на държавите членки на ОИСР за резултати от изпитвания относно свързване на естрогенен рецептор, като същевременно се възползват от предимствата на взаимното признаване на данни на ОИСР.
|
Контекст и принципи на изследванията, включени в настоящия метод за изпитване
|
2.
|
През 1998 г. ОИСР инициира дейност с висок приоритет за преразглеждане на съществуващите и разработване на нови насоки за изпитване при провеждане на скрининг и изпитване на химикали, които имат потенциала да нарушават функциите на ендокринната система. Концептуалната рамка (КР) на ОИСР за изпитване и оценяване на химикали, които имат потенциала да нарушават функциите на ендокринната система бе преразгледана през 2012 г. Оригиналната и преразгледаната КР са включени в приложенията към Ръководството относно стандартизираните насоки за изпитване за оценка на химикали, нарушаващи функциите на ендокринната система (4). КР обхваща пет нива, като всяко ниво отговаря на различно ниво на биологична комплексност. Изследванията за свързване на ЕР, описани в настоящия метод на изпитване, са от ниво 2, което включва „in vitro изследвания, осигуряващи данни за избран(и) ендокринен(ни) механизъм(ми)/път(ища)“. Този метод на изпитване е за изследвания in vitro на свързването към рецептори, предназначени да идентифицират лиганди за човешкия естрогенен рецептор алфа (ЕРα).
|
|
3.
|
Уместността на изследването in vitro на свързването към ЕР по отношение на биологичните функции е доказана ясно. Изследванията на свързването към ЕР са предназначени да идентифицират химикали, които имат потенциала да нарушат пътя на естрогена, и са използвани широко през последните две десетилетия с цел да се характеризира разпределението на ЕР в тъканите, както и да се идентифицират агонисти/антагонисти на ЕР. Тези изследвания отразяват взаимодействието лиганд-рецептор, което е първоначалната стъпка на сигналния път на естрогена и е от съществено значение за репродуктивната функция при всички гръбначни животни.
|
|
4.
|
Взаимодействието на естрогените с ЕР може да окаже въздействие върху транскрипцията на естроген-контролираните гени и да предизвика негеномни ефекти, които могат да причинят индуциране или инхибиране на клетъчни процеси, включително тези, които са необходими за клетъчната пролиферация, нормалното развитие на зародиша и репродуктивната функция (5)(6)(7). Смущението на функцията на нормалните естрогенни системи може да причини евентуални неблагоприятни ефекти върху нормалното развитие (онтогенеза), репродуктивното здраве и целостта на репродуктивната система. Неподходящото сигнализиране на ЕР може да доведе до ефекти като повишен риск от хормонозависим рак, увредена плодовитост и изменения в растежа и развитието на зародиша (8).
|
|
5.
|
Изследванията in vitro на свързването са базирани на пряко взаимодействие на вещество със специфично място за свързване на рецептор и лиганд, което регулира генната транскрипция. Основният компонент на изследването на свързването към рекомбинантния човешки естрогенен рецептор алфа (hrERα) измерва способността на радиомаркиран лиганд ([3H]17β-естрадиол) да се свързва с ЕР в присъствието на нарастващи концентрации на изпитван химикал (т.е. конкурент). Изпитваните химикали, които имат висок афинитет към ЕР, се конкурират с радиомаркирания лиганд при по-ниска концентрация в сравнение с химикалите с по-нисък афинитет към рецептора. Това изследване се състои от два основни компонента: експеримент за свързване до насищане с цел характеризиране на параметрите на взаимодействието рецептор-лиганд и документиране на специфичността на ЕР, последван от експеримент за конкурентно свързване, който характеризира конкуренцията между изпитван химикал и радиомаркиран лиганд за свързване към ЕР.
|
|
6.
|
Валидационните изпитвания на CERI и изследванията за свързване на FW са доказали своята приложимост и надеждност спрямо предназначението си (2).
|
|
7.
|
Използваните определения и съкращения в рамките на настоящия метод за изпитване са описани в допълнение 1.
|
Обхват и ограничения, свързани с изследванията на свързването към рецептор
|
8.
|
Тези изследвания се предлагат за целите на скрининг изпитвания и определяне на приоритети, но могат да предоставят и информация за молекулно иницииращо събитие (МИС), която може да се използва при подход, при който се отчита значимостта на доказателствения материал. Те се занимават с химичното свързване към областта на ERα за свързване с лиганд в in vitro система. Поради това резултатите не бива да бъдат директно есктраполирани към комплексното сигнализиране и регулиране на здравата ендокринна система in vivo.
|
|
9.
|
Свързването на естествения лиганд 17β-естрадиол е първоначалната стъпка от поредица молекулни събития, която активира транскрипцията на прицелните гени и накрая кулминира с физиологична промяна (9). Поради тази причина свързването към областта на ERα за свързване на лиганд се разглежда като един от ключовите механизми за опосредстваното от ЕР нарушение на функциите на ендокринната система (ED), въпреки че има и други механизми, чрез които може да възникне ED, включително i) взаимодействия с места от ЕРα, различни от мястото за свързване с лиганд, ii) взаимодействия с други рецептори, относими спрямо естрогенното сигнализиране, ЕРβ и G-протеин свързан естрогенен рецептор, други рецептори и ензимни системи в рамките на ендокринната система, iii) синтез на хормони, iv) метаболитно активиране и/или деактивиране на хормони, v) разпределение на хормони в прицелните тъкани и vi) клиърънс на хормони от организма. Нито едно от изследванията съгласно този метод на изпитване не се занимава с тези режими на действие.
|
|
10.
|
Настоящият метод на изпитване се занимава със способността на вещества да се свързват с човешкия ERα и не разграничава между агонисти или антагонисти на ERα. Тези изследвания не се занимават и с по-нататъшни събития по веригата, като транскрипция на гени или физиологични промени. Като се има предвид, че по време на валидацията са използвани само единични вещества с една съставка, приложимостта върху изпитвани смеси не е разглеждана. Независимо от това, изследванията са теоретично приложими за изпитването на вещества с повече съставки и на смеси. Преди използването на метода за изпитване по отношение на смес с цел генериране на данни за планирана регулаторна цел, следва да се разгледа въпросът дали той може да даде адекватни резултати за тази цел и, ако е така, защо. Подобни съображения не са необходими, когато има регулаторно изискване за изпитване на сместа.
|
|
11.
|
Безклетъчните рецепторни системи нямат вътрешно присъща метаболитна способност и не са валидирани в съчетание с метаболитни ензимни системи. Въпреки това би могло да е възможно метаболитната дейност да се включи в проект на проучване, но за целта ще е необходимо допълнително валидиране.
|
|
12.
|
Химикали, които могат да денатурират протеина (т.е. протеина на рецептора), като повърхностно активни вещества или химикали, които могат да променят pH на буфера, използван в изследването, не могат да се изпитват или могат да се изпитват единствено при концентрации, при които не протичат такива взаимодействия. В противен случай концентрационният диапазон, който може да се изпита в изследванията за изпитван химикал, се ограничава от разтворимостта му в буфера, използван в изследването.
|
|
13.
|
За информационни цели таблица 1 предоставя резултатите от изпитване за 24-те вещества, които бяха изпитани в рамките на двете напълно валидирани изследвания, описани в настоящия метод на изпитване. От тези вещества, въз основа на публикуваните протоколи, 17 се класифицират като свързващи вещества към ЕР, а 6 са несвързващи вещества, включително при in vitro изследвания за транскрипционно активиране на ЕР и/или утеротрофичното изследване (9)(10)(11)(12)(13)(14)(15). Позовавайки се на данните, обобщени в таблица 1, беше наблюдавано почти 100 % съвпадение на резултатите от двете изследвания по отношение на класифицирането на всички вещества до 10-4M, като всяко вещество беше класифицирано правилно като свързващо или несвързващо вещество към ЕР. Допълнителна информация за тази група вещества, както и за допълнителните вещества, изпитвани в рамките на изследванията за свързване към ЕР по време на изследванията за валидиране, е предоставена в Стандартите за ефективност за изследването за свързване към hrER (3), допълнение 2 (таблици 1, 2 и 3).
Таблица 1
Класифициране на веществата като свързващи или несвързващи вещества към ЕР при изпитване в изследванията за свързване към hrER на FW и CERI в сравнение с очакван отклик
|
|
Наименование на веществото
|
CAS №
|
Очакван отклик
|
Изследване на FW
|
Изследване на CERI
|
Химичен клас в MESH
|
Продуктов клас
|
|
|
Концентрационен диапазон (M)
|
Класифициране
|
Концентрационен диапазон (M)
|
Класифициране
|
|
1
|
17β-Естрадиол
|
50-28-2
|
Свързващо вещество
|
1×10-11 – 1×10-6
|
Свързващо вещество
|
1×10-11 – 1×10-6
|
Свързващо вещество
|
Стероид
|
Фармацевтичен продукт, ветеринарен агент
|
|
2
|
Норетинодрел
|
68-23-5
|
Свързващо вещество
|
3×10-9 – 30×10-4
|
Свързващо вещество
|
3×10-9 – 30×10-4
|
Свързващо вещество
|
Стероид
|
Фармацевтичен продукт, ветеринарен агент
|
|
3
|
Норетиндрон
|
68-22-4
|
Свързващо вещество
|
3×10-9 – 30×10-4
|
Свързващо вещество
|
3×10-9 – 30×10-4
|
Свързващо вещество
|
Стероид
|
Фармацевтичен продукт, ветеринарен агент
|
|
4
|
Ди-n-бутил фталат
|
84-74-2
|
Несвързващо вещество (*5)
|
1×10-10 – 1×10-4
|
Несвързващо вещество (*6)
(1)
|
1×10-10 – 1×10-4
|
Несвързващо вещество (*6)
(1)
|
Въглеводород (цикличен), естер
|
Пластификатор, междинен химикал
|
|
5
|
Диетилстилбестрол
|
56-53-1
|
Свързващо вещество
|
1×10-10 – 1×10-3
|
Свързващо вещество
|
1×10-10 – 1×10-3
|
Свързващо вещество
|
Въглеводород (цикличен), фенол
|
Фармацевтичен продукт, ветеринарен агент
|
|
6
|
17α-етинилестрадиол
|
57-63-6
|
Свързващо вещество
|
1×10-10 – 1×10-3
|
Свързващо вещество
|
1×10-10 – 1×10-3
|
Свързващо вещество
|
Стероид
|
Фармацевтичен продукт, ветеринарен агент
|
|
7
|
Мезо-хексестрол
|
84-16-2
|
Свързващо вещество
|
1×10-10 – 1×10-3
|
Свързващо вещество
|
1×10-10 – 1×10-3
|
Свързващо вещество
|
Въглеводород (цикличен), фенол
|
Фармацевтичен продукт, ветеринарен агент
|
|
8
|
Генищайн
|
446-72-0
|
Свързващо вещество
|
1×10-10 – 1×10-3
|
Свързващо вещество
|
1×10-10 – 1×10-3
|
Свързващо вещество
|
Въглеводород (хетероцикличен), флавоноид
|
Природен продукт
|
|
9
|
Екуол
|
531-95-3
|
Свързващо вещество
|
1×10-10 – 1×10-3
|
Свързващо вещество
|
1×10-10 – 1×10-3
|
Свързващо вещество
|
Метаболит на фитоестроген
|
Природен продукт
|
|
10
|
Бутил парабен (n бутил-4-хидроксибензоат)
|
94-26-8
|
Свързващо вещество
|
1×10-10 – 1×10-3
|
Свързващо вещество
|
1×10-10 – 1×10-3
|
Свързващо вещество
|
Парабен
|
Консервант
|
|
11
|
Нонилфенол (смес)
|
84852-15-3
|
Свързващо вещество
|
1×10-10 – 1×10-3
|
Свързващо вещество
|
1×10-10 – 1×10-3
|
Свързващо вещество
|
Алкилфенол
|
Междинно съединение
|
|
12
|
o,p’-ДДТ
|
789-02-6
|
Свързващо вещество
|
1×10-10 – 1×10-3
|
Свързващо вещество
|
1×10-10 – 1×10-3
|
Свързващо вещество
|
Органохлор
|
Инсектицид
|
|
13
|
Химикал
|
50-22-6
|
Несвързващо вещество (*5)
|
1×10-10 – 1×10-4
|
Несвързващо вещество
|
1×10-10 – 1×10-4
|
Несвързващо вещество
|
Стероид
|
Природен продукт
|
|
14
|
Зеараленон
|
17924-92-4
|
Свързващо вещество
|
1×10-10 – 1×10-3
|
Свързващо вещество
|
1×10-10 – 1×10-3
|
Свързващо вещество
|
Въглеводород (хетероцикличен), лактон
|
Природен продукт
|
|
15
|
Тамоксифен
|
10540-29-1
|
Свързващо вещество
|
1×10-10 – 1×10-3
|
Свързващо вещество
|
1×10-10 – 1×10-3
|
Свързващо вещество
|
Въглеводород (цикличен)
|
Фармацевтичен продукт, ветеринарен агент
|
|
16
|
5α-дихидротестостерон
|
521-18-6
|
Свързващо вещество
|
1×10-10 – 1×10-3
|
Свързващо вещество
|
1×10-10 – 1×10-3
|
Свързващо вещество
|
Стероид, нефенолен
|
Природен продукт
|
|
17
|
Бисфенол A
|
80-05-7
|
Свързващо вещество
|
1×10-10 – 1×10-3
|
Свързващо вещество
|
1×10-10 – 1×10-3
|
Свързващо вещество
|
Фенол
|
Междинен химикал
|
|
18
|
4-n-хептилфенол
|
1987-50-4
|
Свързващо вещество
|
1×10-10 – 1×10-3
|
Нееднозначно вещество (5)
|
1×10-10 – 1×10-3
|
Свързващо вещество
|
Алкилфенол
|
Междинно вещество
|
|
19
|
Кепон (Хлордекон)
|
143-50-0
|
Свързващо вещество
|
1×10-10 – 1×10-3
|
Свързващо вещество
|
1×10-10 – 1×10-3
|
Свързващо вещество
|
Въглеводород (халогениран)
|
Пестицид
|
|
20
|
Бенз(а)антрацен
|
56-55-3
|
Несвързващо вещество
|
1×10-10 – 1×10-3
|
Несвързващо вещество (6)
|
1×10-10 – 1×10-3
|
Несвързващо вещество (6)
|
Ароматен въглеводород
|
Междинно вещество
|
|
21
|
Ентеролактон
|
78473-71-9
|
Свързващо вещество
|
1×10-10 – 1×10-3
|
Свързващо вещество
|
1×10-10 – 1×10-3
|
Свързващо вещество
|
Фитоестроген
|
Природен продукт
|
|
22
|
Прогестерон
|
57-83-0
|
Несвързващо вещество (*5)
|
1×10-10 – 1×10-4
|
Несвързващо вещество
|
1×10-10 – 1×10-4
|
Несвързващо вещество
|
Стероид
|
Природен продукт
|
|
23
|
Октилтриетоксисилан
|
2943-75-1
|
Несвързващо вещество
|
1×10-10 – 1×10-3
|
Несвързващо вещество
|
1×10-10 – 1×10-3
|
Несвързващо вещество
|
Силан
|
Модификатор на повърхността
|
|
24
|
Атразин
|
1912-24-9
|
Несвързващо вещество (*5)
|
1×10-10 – 1×10-4
|
Несвързващо вещество
|
1×10-10 – 1×10-4
|
Несвързващо вещество
|
Хетероциклично съединение
|
Хербицид
|
|
КОМПОНЕНТИ НА ИЗСЛЕДВАНЕТО ЗА СВЪРЗВАНЕ КЪМ hrЕР
Съществени компоненти на изследването
|
14.
|
Настоящият метод на изпитване се прилага за изследвания, при които се използва рецептор ЕР и достатъчно силен лиганд за рецептора, който може да се използва като маркер за изследването и да бъде изместен от нарастващи концентрации на изпитван химикал. Изследванията за свързване съдържат следните два основни компонента: 1) свързване до насищане и 2) конкурентно свързване. Изследването за свързване до насищане се използва за потвърждаване на специфичността и активността на рецепторните препарати, докато експериментът за конкурентно свързване се използва за оценяване на способността на изпитван химикал да се свързва с hrER.
|
Контроли
|
15.
|
Следва да се опише основанието за предлагания паралелен референтен естроген и контроли. Паралелните контроли (разтворител (носител), положителни (свързващо вещество към ЕР; силен и слаб афинитет), отрицателни (несвързващо вещество)), според случая, служат за индикация, че изследването се изпълнява съгласно условията за изпитване, и осигуряват база за сравнения между отделните експерименти; те обикновено са част от критериите за приемливост за даден експеримент (1). Пълните криви на концентрация за референтния естроген и контролите (т.е. слабо свързващо вещество и несвързващо вещество) следва да се използват в една плака по време на всяка серия. Всички останали плаки следва да съдържат: 1) висока (приблизително пълно изместване на радиомаркиран лиганд) и средна (приблизително IC50) концентрация на E2 и слабо свързващо вещество в по три повторения; 2) контрола на разтворителя и неспецифично свързване в по три повторения.
|
Стандартни процедури за контрол на качеството
|
16.
|
Стандартните процедури за контрол на качеството следва да се изпълняват, както е описано за всяко изследване, за да се гарантира, че активните рецептори, правилните концентрации на химикалите, границите на допустими отклонения остават стабилни при множество повторения и съхраняват способността си да предоставят очакваните отклик за свързване към ЕР във времето.
|
Доказване на пригодността на лабораторията
|
17.
|
Преди изпитване на неизвестни химикали с което и да било от изследванията в рамките на настоящия метод на изпитване, всяка лаборатория следва да докаже пригодността си за използване на изследването, като извърши изследвания за насищане, за да потвърди специфичността и активността на препарата на ЕР, и изследвания за конкурентно свързване с референтния естроген и контролите (слабо свързващо вещество и несвързващо вещество). Лабораторията следва да създаде историческа база данни с резултатите за референтния естроген и контролите, генерирани от 3—5 независими експеримента, проведени в различни дни. Тези експерименти ще бъдат основата за референтния естроген и контролите за предходни периоди за лабораторията и ще се използват като частична оценка на приемливостта на изследването за бъдещи серии.
|
|
18.
|
Способността за реагиране на изпитващата система ще бъде потвърдена и чрез изпитване на веществата за изпитване за пригодност, изброени в таблица 2. Списъкът на веществата за изпитване за пригодност е под-множество на референтните вещества, посочени в Стандартите за ефективност за изследванията за свързване към ЕР (3). Тези вещества могат да се намерят в търговската мрежа, представляват класове химикали, които обикновено се асоциират с активността при свързване към ЕР, проявяват подходящ диапазон на ефикасност, очакван за вещества с активност на свързване към ЕР (т.е. със силна до слаба активност за свързване към ЕР) и за вещества без такава активност (т.е. отрицателни контроли). За всяко вещество за изпитване за пригодност изпитваните концентрации следва да покриват диапазона, посочен в таблица 2. Следва да се извършат най-малко три експеримента за всяко вещество и резултатите следва да съответстват на очакваната химична активност. Всеки експеримент следва да се проведе независимо (т.е. с пресни разредени разтвори на рецептора, химикалите и реактивите), като за всяка концентрация се правят по три повторения. Пригодността се доказва чрез правилното класифициране (положително/отрицателно) на всяко опитно вещество. Изпитването за пригодност следва да бъде проведено от всеки техник, когато се запознава с изследванията.
Таблица 2
Списък на контролите и веществата за изпитване за пригодност за изследванията за конкурентно свързване към hrER
(90)
|
№
|
Наименование на веществото
|
CAS RN (91)
|
Очакван отклик (92)
(93)
|
Изпитван концентрационен диапазон (M)
|
Химичен клас в MeSH (94)
|
Продуктов клас (95)
|
|
Контроли (референтен естроген, слабо свързващо вещество, несвързващо вещество)
|
|
1
|
17-estradiol
|
50-28-2
|
Свързващо вещество
|
1×10-11 – 1×10-6
|
Стероид
|
Фармацевтичен продукт, ветеринарен агент
|
|
2
|
Норетинодрел (или) Норетиндрон
|
68-23-5 (или) 68-22-4
|
Свързващо вещество
|
3×10-9 – 30×10-6
|
Стероид
|
Фармацевтичен продукт, ветеринарен агент
|
|
3
|
Октилтриетоксисилан
|
2943-75-1
|
Несвързващо вещество
|
1×10-10 – 1×10-3
|
Силан
|
Модификатор на повърхността
|
|
Опитни вещества (95)
|
|
4
|
Диетилстилбестрол
|
56-53-1
|
Свързващо вещество
|
1×10-11 – 1×10-6
|
Въглеводород (цикличен), фенол
|
Фармацевтичен продукт, ветеринарен агент
|
|
5
|
17α-етинилестрадиол
|
57-63-6
|
Свързващо вещество
|
1×10-11 – 1×10-6
|
Стероид
|
Фармацевтичен продукт, ветеринарен агент
|
|
6
|
Мезо-хексестрол
|
84-16-2
|
Свързващо вещество
|
1×10-11 – 1×10-6
|
Въглеводород (цикличен), фенол
|
Фармацевтичен продукт, ветеринарен агент
|
|
7
|
Тамоксифен
|
10540-29-1
|
Свързващо вещество
|
1×10-11 – 1×10-6
|
Въглеводород (цикличен)
|
Фармацевтичен продукт, ветеринарен агент
|
|
8
|
Генищайн
|
446-72-0
|
Свързващо вещество
|
1×10-10 – 1×10-3
|
Хетероциклично съединение, флавоноид
|
Природен продукт
|
|
9
|
Бисфенол A
|
80-05-7
|
Свързващо вещество
|
1×10-10 – 1×10-3
|
Фенол
|
Междинен химикал
|
|
10
|
Зеаралонон
|
17924-92-4
|
Свързващо вещество
|
1×10-11 – 1×10-3
|
Хетероциклично съединение, лактон
|
Природен продукт
|
|
11
|
Бутил парабен
|
94-26-8
|
Свързващо вещество
|
1×10-11 – 1×10-3
|
Карбоксилна киселина, фенол
|
Консервант
|
|
12
|
Атразин
|
1912-24-9
|
Несвързващо вещество
|
1×10-11 – 1×10-6
|
Хетероциклично съединение
|
Хербицид
|
|
13
|
Ди-n-бутилфталат (ДБФ) (96)
|
84-74-2
|
Несвързващо вещество (97)
|
1×10-10 – 1×10-4
|
Въглеводород (цикличен), естер
|
Пластификатор, междинен химикал
|
|
14
|
Химикал
|
50-22-6
|
Несвързващо вещество
|
1×10-11 – 1×10-4
|
Стероид
|
Природен продукт
|
|
Изпитване на разтворимостта и установяване на концентрационния диапазон за изпитваните химикали
|
19.
|
Следва да се проведе предварително изпитване, за да се определи границата на разтворимост за всеки изпитван химикал и да се идентифицира подходящия концентрационен диапазон за използване при провеждането на изпитването. Границата на разтворимост на всеки изпитван химикал трябва първо да се определи в разтворителя и допълнително да се потвърди при условията на изследването. Крайната концентрация, изпитвана в изследването, не трябва да надвишава 1 mM. Изпитването за установяване на диапазона включва контрола на разтворител заедно с осем логаритмични последователни разреждания, започвайки с максималната приемлива концентрация (напр. 1 mM или по-ниска въз основа на границата на разтворимост), в присъствието на забелязана мътност или преципитация. Концентрациите във втория и третия експеримент следва да се коригират, както е подходящо, за да характеризират по-добре кривата отклик-концентрация.
|
Критерии за приемливост на изпитвателната серия
|
20.
|
Приемането или отхвърлянето на изпитвателна серия се прави на базата на оценката на резултатите, получени за референтния естроген и контролата, използвани за всеки експеримент. На първо място, за плака 1 пълните криви на концентрация за референтните контроли от всеки експеримент следва да съответстват на измерванията на ефективността с параметрите на налагане на кривата (напр. IC50 и наклон на Хил) въз основа на резултатите, докладвани за съответните протоколи за изследванията на CERI и на FW (допълнения 2 и 3), както и на данните за контроли от предходни периоди от лабораторията, провеждаща изпитването. Всички контроли (референтен естроген, слабо свързващо вещество и несвързващо вещество) следва да се класифицират правилно за всеки експеримент. На второ място, контролите на всички последващи плаки трябва да се оценят за последователност с плака 1. Следва да се използва достатъчен диапазон на концентрации на изпитвания химикал за ясно определяне на горната част на кривата на конкурентно свързване. Вариабилността между повторенията при всяка концентрация на изпитвания химикал, както и между трите независими серии следва да е разумна и обоснована от научна гледна точка. Способността за последователно изпълнение на изследването следва да бъде доказана чрез разработката и поддържането на база данни от предходни периоди относно референтния естроген и контроли. Стандартните отклонения (SD) или коефициентите на вариация (CV) за параметрите за налагане на кривите на средните стойности за референтния естроген и контролните слаби свързващи вещества от множество експерименти могат да бъдат използвани като мярка за вътрешнолабораторната възпроизводимост. Следва да се приложи професионална преценка, когато се преглеждат резултатите от контролите на плаката от всяка серия, както и за всеки изпитван химикал.
Допълнително следва да бъдат изпълнени следните принципи относно критериите за приемливост:
|
—
|
данните следва да са достатъчно, за да се направи количествена оценка на свързването към ЕР;
|
|
—
|
изпитваните концентрации следва да останат в рамките на диапазона на разтворимост на изпитвания химикал.
|
|
Анализ на данните
|
21.
|
Определената процедура за анализ на данните за свързването до насищане и конкурентно свързване следва да спазва основните принципи за характеризиране на взаимодействията рецептор-лиганд. Обикновено данните за свързването до насищане се анализират чрез модел на нелинейна регресия, който отчита общото и неспецифичното свързване. Може да се наложи корекция за изчерпване на лиганда (например Swillens, 1995 г. (19)), когато се определят Bmax и Kd. Данните от изследванията за конкурентно свързване обикновено се преобразуват (например процент на специфично свързване и концентрация на изпитвания химикал (log M)). Изчислените логаритмични стойности (IC50) за всеки изпитван химикал следва да се определят чрез подходящ софтуер за нелинейно налагане на крива с цел налагане на уравнение на Хил с четири параметъра. След първоначален анализ следва да се направи преглед на параметрите на налагане на кривата и да се направи визуален преглед доколко добре данните за свързването съвпадат с генерираната крива на конкурентно свързване. В някои случаи може да се наложи допълнителен анализ, за да се получи най-доброто налагане на кривата (напр. ограничаване на най-високата и/или най-ниската част на кривата, използване на правилото за 10 %, вж. допълнение 4 и справка 2 (раздел III.A.2).
|
|
22.
|
Изпълнението на критериите за приемливост (точка 20) показва, че системата на изследването работи правилно, но не гарантира, че конкретно изпитване ще даде точни данни. Повторението на правилните резултати от първото изпитване е най-доброто указание, че се получават точни данни.
|
Общи критерии за тълкуване на данните
|
23.
|
Понастоящем не съществува универсално съгласуван метод за тълкуване на данните за свързване към ЕР. Въпреки това и качествените (напр. свързващо/несвързващо вещество), и/или количествените (напр. log IC50, относителен афинитет за свързване (RBA) и т.н.) оценки на опосредстваната от hrER активност следва да бъдат базирани на емпирични данни и разумна научна преценка.
|
Протокол от изпитването
|
24.
|
Протоколът от изпитването следва да включва следната информация:
|
|
Контрола/референтно вещество/изпитван химикал
|
—
|
източник, номер на партидата, крайна дата за употреба, ако има такава
|
|
—
|
стабилност на самия изпитван химикал, ако е известна;
|
|
—
|
разтворимост и стабилност на изпитвания химикал в разтворител, ако са известни.
|
|
—
|
измерване на рН, осмолалитет и утайка в средата за отглеждане, към която е добавен изпитваният химикал, когато е уместно.
|
|
|
|
Вещество с една съставка:
|
—
|
външен вид, разтворимост във вода и допълнителни относими физични и химични свойства;
|
|
—
|
химична идентификация, като наименование по IUPAC или по CAS, CAS номер, SMILES или InChI код, структурна формула, чистота, химическа идентичност на онечистванията, както е целесъобразно и практически осъществимо, и т.н.
|
|
|
|
Вещество с повече съставки, UVCB и смеси:
|
—
|
определени, доколкото е възможно, с химичната си идентичност (вж. по-горе), количествения състав и относимите физични и химични свойства на съставките.
|
|
|
|
Разтворител/носител:
|
—
|
характеристики (естество, доставчик и партида);
|
|
—
|
обосновка за избора на разтворител/носител;
|
|
—
|
разтворимост и стабилност на изпитвания химикал в разтворител/носител, ако са известни.
|
|
|
|
Рецептори:
|
—
|
източник на рецепторите (доставчик, каталожен номер, партида, вид на рецептора, концентрация на активния рецептор, предоставена от доставчика, сертифициране от доставчика);
|
|
—
|
характеризиране на рецепторите (в това число резултатите от свързването до насищане): Kd, Bmax;
|
|
—
|
съхранение на рецепторите;
|
|
—
|
доставчик, каталожен номер, партида, специфична активност.
|
|
|
|
Условия на изпитването:
|
—
|
ограничения на разтворимостта при условията на изследването;
|
|
—
|
състав на свързващия буфер;
|
|
—
|
концентрация на рецептора;
|
|
—
|
концентрация на маркера (т.е. радиомаркирания лиганд);
|
|
—
|
концентрации на изпитвания химикал;
|
|
—
|
процент носител в окончателното изследване;
|
|
—
|
температура и време на инкубация;
|
|
—
|
метод на разделяне на свързано от свободно вещество;
|
|
—
|
положителни и отрицателни контроли/референтни вещества;
|
|
—
|
критерии за считане на изпитванията за положителни, отрицателни или нееднозначни.
|
|
|
|
Проверка за приемливост:
|
—
|
действителни стойности на IC50 и наклона на кривата за паралелни положителни контроли/референтни вещества.
|
|
|
|
Резултати:
|
—
|
сурови данни и данни за свързано вещество/свободно вещество;
|
|
—
|
проверка за потвърждение на денатуриране, ако е подходящо;
|
|
—
|
ако съществува, най-ниската ефективна концентрация (LEC);
|
|
—
|
стойностите на RBA и/или IC50 по целесъобразност;
|
|
—
|
зависимостта концентрация—отклик, където е възможно;
|
|
—
|
статистически анализи, ако има такива, заедно с мярката за грешка и достоверност (напр. SEM, SD, CV или 95 % доверителен интервал) и описание на начина за получаване на тези стойности.
|
|
|
|
Обсъждане на резултатите:
|
—
|
прилагане на правилото за 10 %.
|
|
Заключение
|
ЛИТЕРАТУРА
|
(1)
|
OECD (2005). Guidance Document on the Validation and International Acceptance of New or Updated Test Methods for Hazard Assessment. Environmental, Health and Safety Publications, Series on Testing and Assessment (No 34), Organisation for Economic Cooperation and Development, Paris.
|
|
(2)
|
OECD (2015). Integrated Summary Report: Validation of Two Binding Assays Using Human Recombinant Estrogen Receptor Alpha (hrERα), Health and Safety Publications, Series on Testing and Assessment (No 226), Organisation for Economic Cooperation and Development, Paris.
|
|
(3)
|
OECD (2015). Performance Standards for Binding Assays Using Human Recombinant Estrogen Receptor Alpha (hrERα), Health and Safety Publications, Series on Testing and Assessment (No 222), Organisation for Economic Cooperation and Development, Paris.
|
|
(4)
|
OECD (2012). Guidance Document on Standardized Test Guidelines for Evaluating Chemicals for Endocrine Disruption. Environmental, Health and Safety Publications, Series on Testing and Assessment (No 150), Organisation for Economic Cooperation and Development, Paris.
|
|
(5)
|
Cavailles V. (2002). Estrogens and Receptors: an Evolving Concept, Climacteric, 5 Suppl 2: p.20-6.
|
|
(6)
|
Welboren W.J., et al. (2009). Genomic Actions of Estrogen Receptor Alpha: What are the Targets and How are they Regulated? Endocr. Relat. Cancer., 16(4): p. 1073-89.
|
|
(7)
|
Younes M. and Honma N. (2011). Estrogen Receptor Beta, Arch. Pathol. Lab. Med., 135(1): p. 63-6.
|
|
(8)
|
Diamanti-Kandarakis et al. (2009). Endocrine-Disrupting Chemicals: an Endocrine Society Sci. Statement, Endo Rev 30(4):293-342.
|
|
(9)
|
ICCVAM (2002). Background Review Document. Current Status of Test Methods for Detecting Endocrine Disruptors: In Vitro Estrogen Receptor Binding Assays. (NIH Publication No 03-4504). National Institute of Environmental Health Sciences, Research Triangle Park, NC.
|
|
(10)
|
ICCVAM (2003). ICCVAM Evaluation of In Vitro Test Methods for Detecting Potential Endocrine Disruptors: Estrogen Receptor and Androgen Receptor Binding and Transcriptional Activation Assays.
|
|
(11)
|
ICCVAM (2006). ICCVAM Evaluation of In Vitro Test Methods for Detecting Potential Endocrine Disruptors: Estrogen Receptor and Androgen Receptor Binding and Transcriptional Activation Assays.
|
|
(12)
|
Akahori Y. et al. (2008). Relationship Between the Results of In Vitro Receptor Binding Assay to Human Estrogen Receptor Alpha and In Vivo Uterotrophic Assay: Comparative Study with 65 Selected Chemicals, Toxicol. In Vitro, 22(1): 225-231.
|
|
(13)
|
OECD (2007). Additional Data Supporting the Test Guideline on the Uterotrophic Bioassay in Rodents, Environment, Health and Safety Publications, Series on Testing and Assessment (No 67), Organisation for Economic Cooperation and Development, Paris.
|
|
(14)
|
Takeyoshi, M. (2006). Draft Report of Pre-validation and Inter-laboratory Validation For Stably Transfected Transcriptional Activation (TA) Assay to Detect Estrogenic Activity - The Human Estrogen Receptor Alpha Mediated Reporter Gene Assay Using hER-HeLa-9903 Cell Line, Chemicals Evaluation and Research Institute (CERI): Japan. p. 1-188.
|
|
(15)
|
Yamasaki, K; Noda, S; Imatanaka, N; Yakabe, Y. (2004). Comparative Study of the Uterotrophic Potency of 14 Chemicals in a Uterotrophic Assay and their Receptor-Binding Affinity, Toxicol. Letters, 146: 111-120.
|
|
(16)
|
Kummer V; Maskova, J; Zraly, Z; Neca, J; Simeckova, P; Vondracek, J; Machala, M. (2008). Estrogenic Activity of Environmental Polycyclic Aromatic Hydrocarbons in Uterus of Immature Wistar Rats. Toxicol. Letters, 180: 213-221.
|
|
(17)
|
Gozgit, JM; Nestor, KM; Fasco, MJ; Pentecost, BT; Arcaro, KF. (2004). Differential Action of Polycyclic Aromatic Hydrocarbons on Endogenous Estrogen-Responsive Genes and on a Transfected Estrogen-Responsive Reporter in MCF-7 Cells. Toxicol. and Applied Pharmacol., 196: 58-67.
|
|
(18)
|
Santodonato, J. (1997). Review of the Estrogenic and Antiestrogenic Activity of Polycyclic Aromatic Hydrocarbons: Relationship to Carcinogenicity. Chemosphere, 34: 835-848.
|
|
(19)
|
Swillens S (1995). Interpretation of Binding Curves Obtained with High Receptor Concentrations: Practical Aid for Computer Analysis, Mol Pharmacol 47(6):1197-1203.
|
Допълнение 1
ОПРЕДЕЛЕНИЯ И СЪКРАЩЕНИЯ
Правилото за 10 %: възможност за изключване от анализите на точки с данни, където средната стойност на повторенията за процента на специфично свързан [3H]17β-естрадиол е 10 % или повече над наблюдавания за средната стойност при по-ниска концентрация (вж. допълнение 4).
Критерии за приемливост: минималните стандарти за ефективност на експерименталните контроли и референтните еталони. Всички критерии за приемливост следва да бъдат изпълнени, за да може даден експеримент да се разглежда като валиден.
Точност (съответствие): степента на близост между резултатите от изследването и приетите референтни стойности. Това е мярка за ефективността на изследването и един от аспектите на неговата относимост. Терминът често се използва взаимозаменяемо с термина „съответствие“, за да се обозначи относителният дял на правилните резултати при дадено изследване (1).
CF: Концептуалната рамка на ОИСР за изпитване и оценяване на вещества, нарушаващи функцията на ендокринната система.
Химикал: Вещество или смес.
CV: Коефициент на вариация
E2: 17β-естрадиол
ED: Ендокринни нарушения
hERα: Човешки естрогенен рецептор алфа
ER: Естрогенен рецептор
Естрогенна активност: способността на химикал да имитира 17β-естрадиол в способността си да се свързва с естрогенни рецептори. С този метод на изпитване може да се открие свързване към hERα.
IC50: половината от максимално ефективната предизвикваща потискане концентрация на изпитван химикал.
ICCVAM: междуведомствен координационен комитет за валидиране на алтернативни методи.
Междулабораторна възпроизводимост: мярка за степента, в която различните квалифицирани лаборатории могат да получат качествено и количествено сходни резултати, като използват един и същ протокол и изпитват едни и същи вещества. Междулабораторната възпроизводимост се определя по време на предвалидационните и валидационните процедури и указва степента, в която дадено изследване може успешно да бъде прехвърлено между различни лаборатории, наричана също интерлабораторна възпроизводимост (1).
Вътрешнолабораторна възпроизводимост: определяне на степента, в която квалифицирани служители в рамките на една и съща лаборатория могат да възпроизведат успешно резултатите, като използват конкретен протокол в различни моменти. Нарича се също „интралабораторна възпроизводимост“ (1).
LEC: най-ниската ефективна концентрация е най-ниската концентрация на изпитвания химикал, която предизвиква отклик (т.е. най-ниската концентрация на изпитвания химикал, при която кратността на индукция е статистически различна от паралелната контрола на носителя).
Вариантен метод за изпитване (me-too test): Разговорен израз за изследване, който е структурно и функционално сходен с валидиран и приет референтен метод за изпитване. Използва се взаимозаменяемо със „сходен метод на изпитване“.
PBTG: Насоки за изпитване, базирани на ефективност
Стандарти за ефективност: стандарти въз основа на валидиран метод за изпитване, осигуряващи основата за оценка на сравнимостта на предложено изследване, което от гледна точка на своя механизъм и от функционална гледна точка е сходно с валидирания метод. Включени са (1) съществени компоненти на изследването; (2) минимален списък на референтни химикали, подбрани измежду химикалите, използвани за демонстриране на приемливата ефективност на валидирания метод за изпитване; и (3) сравнимите нива за точност и надеждност въз основа на получените нива за валидирания метод на изпитване, които предложеното изследване следва да демонстрира при оценяването чрез използване на минималния списък на референтните химикали (1).
Вещества за изследване за пригодност: подмножество от референтни вещества, включени в Стандартите за ефективност, които може да се използват от лабораториите за доказване на техническа пригодност по отношение на стандартизирано изследване. Критериите за подбор на тези вещества обикновен включват изискването те да представляват обхвата от отговори, да се предлагат в търговската мрежа и да имат налични висококачествени референтни данни.
Пригодност: Доказаната способност за правилно провеждане на изследване преди изпитване на неизвестни вещества.
Референтен естроген: 17ß-естрадиол (E2, CAS 50-28-2).
Референтни методи на изпитване: Изследванията, на които е базиран PBTG 493.
RBA: относителен афинитет за свързване. RBA на дадено вещество се изчислява като процент от логаритмичната стойност (IC50) за веществото спрямо логаритмичната стойност (IC50) за 17β-естрадиол.
Относимост: описание на взаимовръзката между изследването и ефекта, представляващ интерес, и дали тя е значима и полезна за определена цел. Това е степента, в която изследването правилно измерва или прогнозира биологичния ефект, представляващ интерес. Приложимостта включва разглеждането на точността (съответствието) на изследването (1).
Надеждност: мярка за степента, в която дадено изследване може да се проведе възпроизводимо в една и съща лаборатория и в различни лаборатории с течение на времето, при използване на един и същ протокол. Определя се, като се изчислява вътрешно- и междулабораторната възпроизводимост.
SD: Стандартно отклонение.
Изпитван химикал: Всяко вещество или смес, изпитвано(а) с използване на настоящия метод за изпитване.
Валидиран метод за изпитване: изследване, за което са извършени изследването за валидиране за определяне на приложимостта (включително на точността) и надеждността му за конкретна цел. Важно е да се отбележи, че един валидиран метод на изпитване може да не притежава достатъчно ефективност по отношение на точността и надеждността си, за да се счита за приемлив за предлаганата цел (1).
Валидиране: Процесът, чрез който надеждността и относимостта на конкретен подход, метод, изследване, процес или оценка се установяват за определена цел (1).
Допълнение 2
ИЗСЛЕДВАНИЯ IN VITRO НА FREYBERGER-WILSON ЗА СВЪРЗВАНЕ ДО НАСИЩАНЕ И КОНКУРЕНТНО СВЪРЗВАНЕ КЪМ ЕСТРОГЕНЕН РЕЦЕПТОР (ЕРΑ) С ИЗПОЛЗВАНЕ НА ЦЯЛОСТЕН РЕКОМБИНАНТЕН ЕРΑ
ПЪРВОНАЧАЛНИ СЪОБРАЖЕНИЯ И ОГРАНИЧЕНИЯ (ВЖ. СЪЩО „ОБЩО ВЪВЕДЕНИЕ“)
|
1.
|
В това изследване in vitro за свързване до насищане и конкурентно свързване към естрогенен рецептор (ЕРα) се използва цялостен човешки рецептор ЕРα(hrERα, който е произведен в и изолиран от заразени с baculovirus клетки на насекоми. Протоколът на CERI е преминал през международно многолабораторно изследване за валидиране (2), което е доказало неговата относимост и надеждност за целта на изследването.
|
|
2.
|
Това изследване представлява скринингова процедура за идентифициране на вещества, които могат да се свързват с hrERα. То се използва за определяне на способността на даден изпитван химикал да се конкурира със 17β-естрадиол за свързване с hrERα-LBD. Количествените резултати на изследването могат да включват IC50 (мярка за концентрацията на изпитван химикал, необходима за изместване на половината от [3H]-17β-естрадиол отhrERα) и относителните афинитети на свързване на изпитвани химикали за hrERα в сравнение със 17β-естрадиол. За целите на химичен скрининг приемливите качествени резултати на изследването могат да включват класификации на изпитвани химикали като свързващи hrERα, не свързващи или нееднозначни въз основа на критериите, описани за кривите на свързване.
|
|
3.
|
Изследването използва радиоактивен лиганд, който изисква лабораторията да притежава лиценз за радиоактивни материали. Всички процедури с радиоизотопи и опасни химикали следва да спазват разпоредбите и процедурите, както са описани от националното законодателство.
|
|
4.
|
Разделите „ОБЩО ВЪВЕДЕНИЕ“ и „КОМПОНЕНТИ НА ИЗСЛЕДВАНЕТО СВЪРЗВАНЕ НА ” hrER“ следва да бъдат прочетени, преди да се използва настоящото изследване за регулаторни цели. Използваните в настоящите TG определения и съкращения са описани в Допълнение 1.
|
ПРИНЦИП НА ИЗСЛЕДВАНЕТО (ВЖ. СЪЩО „ОБЩО ВЪВЕДЕНИЕ“)
|
5.
|
Изследването за свързване с hrERα измерва способността на радиомаркиран лиганд ([3H]17β-естрадиол) да се свързва с ЕР в присъствието на нарастващи концентрации на изпитван химикал (т.е. конкурент). Изпитваните химикали, които имат висок афинитет към ЕР, се конкурират с радиомаркирания лиганд при по-ниска концентрация в сравнение с химикалите с по-нисък афинитет към рецептора.
|
|
6.
|
Това изследване се състои от два основни компонента: експеримент за свързване до насищане с цел характеризиране на параметрите на взаимодействието рецептор – лиганд, последван от експеримент за конкурентно свързване, който характеризира конкуренцията между изпитван химикал и радиомаркиран лиганд за свързване към ЕР.
|
|
7.
|
Целта на експеримента за свързване до насищане е да характеризира специална партида рецептори за афинитет на свързване и брой при подготовка за експеримента на конкурентно свързване. Експериментът за свързване до насищане измерва, при равновесни условия, афинитета на фиксирана концентрация на естрогенния рецептор за неговия естествен лиганд (представен от дисоциационна константа, Kd) и концентрацията на активните места на рецептора (Bmax).
|
|
8.
|
Експериментът за свързване до насищане измерва афинитета на дадено вещество да се конкурира с [3H]17β-естрадиол за свързване с ЕР. Афинитетът се определя количествено от концентрацията на изпитван химикал, която, при равновесие, инхибира 50 % от специфичното свързване на [3H]17β-естрадиол (наричана „инхибиторна концентрация 50 %“ или IC50). Това може да се оцени и с използване на относителния афинитет на свързване (RBA, по отношение на IC50 на естрадиол, измерен отделно в същата серия). Експериментът за конкурентно свързване измерва свързването на [3H]17β-естрадиол при фиксирана концентрация в присъствието на широк диапазон (осем порядъка) на концентрации на изпитван химикал. След това данните се вписват, където е възможно, във форма на уравнение на Хил (Хил, 1910), което описва изместването на радиолиганда от конкурентно свързващо вещество на едно място. Степента на изместване на радиомаркирания естрадиол при равновесие се използва за характеризиране на изпитвания химикал като свързващ, несвързващ или предизвикващ нееднозначен отклик.
|
ПРОЦЕДУРА
Демонстриране на приемлива ефективност на hrERα протеин
|
9.
|
Преди рутинно провеждане на изследвания за насищане и конкурентно свързване, всяка нова партида hrERα следва да демонстрира правилно функциониране в лабораторията, в която ще се използва. За демонстриране на ефективност следва да се използва двустъпков процес. Тези стъпки са следните:
|
—
|
Проведете изследване за насищане и свързване на 3H]-17β-естрадиол, за да се демонстрира специфичност и насищане на hrERα. Анализът на нелинейна регресия на тези данни (напр. BioSoft; McPherson, 1985 г.; Motulsky, 1995) и последващата диаграма на Scatchard следва да документират афинитета на свързване с hrERα на [3H]-17β-естрадиол (Kd) и броя на рецепторите (Bmax) за определена партида на hrERα.
|
|
—
|
Провежда се изследване за конкурентно свързване с използване на контролните вещества (референтен естроген (17β-естрадиол)), слабо свързващо вещество (напр. норетинодрел или норетиндрон) и несвързващо вещество (октилтриетоксисилан, OTES). Всяка лаборатория следва да определи база данни от предходни периоди за документиране на последователността на IC50 и съответните стойности за референтния естроген и слабо свързващо вещество измежду експериментите и различните партиди на hrERα. Освен това параметрите на кривите на конкурентно свързване за контролните вещества следва да са в границите на 95 %-я доверителен интервал (вж. Таблица 1), които са разработени с използване на данни от лаборатории, които са участвали във изследване за валидиране на това изследване (2).
|
|
Таблица 1
Критерии за ефективност, разработени за референтния естроген и слабо свързващо вещество, изследване за свързване към hrER на FW
|
Вещество
|
Параметър
|
Средна стойност (7)
|
Стандартно отклонение (n)
|
95 % доверителни интервали (8)
|
|
Долна граница
|
Горна граница
|
|
17β-естрадиол
|
Най-високо (%)
|
100,44
|
10,84 (67)
|
97,8
|
103,1
|
|
Най-ниско (%)
|
0,29
|
1,25 (67)
|
-0,01
|
0,60
|
|
Наклон на Хил
|
-1,06
|
0,20 (67)
|
-1,11
|
-1,02
|
|
LogIC50 (M)
|
-8,92 (9)
|
0,18 (67)
|
-8,97
|
-8,88
|
|
Норетинодрел
|
Най-високо (%)
|
99,42
|
8,90 (68)
|
97,27
|
101,60
|
|
Най-ниско (%)
|
2,02
|
3,42 (68)
|
1,19
|
2,84
|
|
Наклон на Хил
|
-1,01
|
0,38 (68)
|
-1,10
|
-0,92
|
|
Log IC50 (M)
|
-6,39
|
0,27 (68)
|
-6,46
|
-6,33
|
|
Норетиндронв
(9)
|
Най-високо (%)
|
96,14
|
8,44 (27)
|
92,80
|
99,48
|
|
Най-ниско (%)
|
2,38
|
5,02 (27)
|
0,40
|
4,37
|
|
Наклон на Хил
|
-1,41
|
0,32 (27)
|
-1,53
|
-1,28
|
|
LogIC50(M)
|
-5,73
|
0,27 (27)
|
-5,84
|
-5,62
|
Доказване на пригодността на лабораторията
|
10.
|
Вж. точки 17 и 18 и таблица 2 в „КОМПОНЕНТИ НА ИЗСЛЕДВАНЕТО ЗА СВЪРЗВАНЕ КЪМ hrER“ от настоящия метод на изпитване. Всяко изследване (на насищане и конкурентно свързване) следва да се състои от три независими серии (т.е. с пресни разредени разтвори на рецептор, химикали и реактиви) в различни дни и всяка серия следва да съдържа три повторения.
|
Определяне на концентрацията на рецептора (hrERα)
|
11.
|
Концентрацията на активния рецептор леко варира според партидата и условията на съхранение. Поради тази причина концентрацията на активния рецептор следва да се определи така, както е получена от доставчика. Това ще доведе до подходящата концентрация на активния рецептор към момента на провеждане на серията.
|
|
12.
|
При условия, отговарящи на конкурентно свързване (т.е. 1 nM [3H]-естрадиол), номинални концентрации на рецептор от 0,25, 0,5, 0,75 и 1 nM следва да се инкубират в отсъствието (общо свързване) и присъствието (неспецифично свързване) на 1 μM немаркиран естрадиол. Специфичното свързване, изчислено като разликата между общото и неспецифичното свързване, се изчертава като зависимост спрямо номиналната концентрация на рецептора. Концентрацията на рецептор, която дава стойности на специфично свързване, съответстващи на 20 % от добавения радиомаркер, е свързана със съответната концентрация на рецептора и тази концентрация на рецептора следва да се използва в опитите за насищане и конкурентно свързване. Често на това условие отговаря окончателна концентрация на hrER от 0,5 nM.
|
|
13.
|
Ако критерият за 20 % не може да бъде удовлетворен, следва да се провери настройката на експеримента за възможни грешки. Невъзможността за постигане на критерия за 20 % може да сочи, че в рекомбинантната партида има много малко количество активен рецептор и тогава следва да се обмисли използването на друга партида рецептор.
|
Изследване на насищане
|
14.
|
Следва да се оценят три пъти осем увеличаващи се концентрации на [3H]17β-естрадиол при спазване на следните три условия (вж. Таблица 2):
|
—
|
В отсъствието на немаркиран 17β-естрадиол и присъствието на ЕР. Това е определянето на тоталното свързване чрез измерване на радиоактивността в ямките, в които има само [3H]17β-естрадиол.
|
|
—
|
В присъствието на 1 000 пъти по-голяма концентрация на немаркиран 17β-естрадиол спрямо маркиран 17β-естрадиол и присъствието на ЕР. Целта на това условие да се наситят местата на активно свързване с немаркиран 17β-естрадиол и чрез измерване на радиоактивността в тези ямки да се определи неспецифичното свързване. Всяко оставащо количество горещ естрадиол, който може да се свърже с рецептора, се счита за свързан в неспецифично място, тъй като студеният естрадиол следва да е с такава висока концентрация, при която той се свързва с всички налични специфични места на рецептора.
|
|
—
|
В отсъствието на немаркиран 17β-естрадиол и отсъствието на ЕР (определяне на обща радиоактивност)
|
|
Приготвяне на [3H]-17β-естрадиол, разтвори на немаркиран 17β-естрадиол и hrERα
|
15.
|
Разрежданията на [3H]-17β-естрадиол следва да се приготвят чрез добавяне на буфер, използван за изследването, към 12 nM изходен разтвор на [3H]-17β-естрадиол за получаване на концентрации, които първоначално варират от 0,12nM до 12 nM. Чрез добавяне на 40 μl от тези разтвори към съответните ямки за изследване на 96-ямкова микротитърна плака (при краен обем 160 μl) ще се получат крайните концентрации за изследването в диапазона от 0,03 до 3,0 nM. Приготвянето на буфера, използван в изследването, изходния разтвор на [3H]-17β-естрадиол и разрежданията, както и определянето на концентрациите са описани подробно в протокола за изследването на FW (2).
|
|
16.
|
Разредените разтвори на немаркиран 17β-естрадиол следва да се приготвят от 0.06 nM изходен разтвор на 17β-естрадиол чрез добавяне на буфер, използван в изследването, за да се постигнат осем увеличаващи се концентрации, които първоначално варират от 6 μM до 80 μM. Окончателните концентрации на изследването, вариращи от 80 до 96 nM, се получават чрез добавяне на 160 μl от тези разтвори към съответните ямки на 0-ямкова микротитърна плака (вж. Таблици 2 и 3). Крайната концентрация на немаркирания 17β-естрадиол в отделните ямки за изследване на неспецифичното свързване следва да е 1 000 пъти по-висока от концентрацията на маркирания [3H]-17β-естрадиол. Приготвянето на разредени разтвори на немаркиран 17β-естрадиол е описано подробно в протокола на CERI (2).
|
|
17.
|
Следва да се използва това количество на рецептор, което дава специфично свързвате от 20±5 % (вж. точки 12-13 на Допълнение 3). Разтворът на hrERα следва да се приготви непосредствено преди употреба.
|
|
18.
|
96-ямковите микротитърни плаки се приготвят както е показано в Таблица 2, с 3 повторения за всяка концентрация. Примери за определяне на концентрацията на плаката и обема на [3H]-17β-естрадиол, немаркиран 17β-естрадиол, буфера и рецептора са дадени в допълнение 2.2.
|
Таблица 2
Схема на микротитърна плака за изследване за свързване до насищане
|
|
1
|
2
|
3
|
4
|
5
|
6
|
7
|
8
|
9
|
10
|
11
|
12
|
|
|
А
|
0,03 nM [3H] E2 + ЕР
|
0,06 nM [3H] E2 + ЕР
|
0,08 nM [3H] E2 + ЕР
|
0,10 nM [3H] E2 + ЕР
|
Общо свързване (разтворител)
|
|
Б
|
0,30 nM [3H] E2 + ЕР
|
0,60 nM [3H] E2 + ЕР
|
1,0 nM [3H] E2 + ЕР
|
3,0 nM [3H] E2 + ЕР
|
|
В
|
|
|
|
|
|
|
Г
|
0,03 nM [3H] E2 + ЕР + 0,03 μM E2
|
0,06 nM [3H] E2 + ЕР + 0,06 μM E2
|
0,08 nM [3H] E2 + ЕР + 0,08 μM E2
|
0,10 nM [3H] E2 + ЕР+ 0,10 μM E2
|
Неспецифично свързване
|
|
Д
|
0,30 nM [3H] E2 + ЕР + 0,30 μM E2
|
0,60 nM [3H] E2 + ЕР + 0,60 μM E2
|
1,0 nM [3H] E2 + ЕР+ 1,0 μM E2
|
3,0 nM [3H] E2 + ЕР +3,0 μM E2
|
|
Е
|
|
|
|
|
|
|
Ж
|
|
|
|
|
|
|
З
|
|
|
|
|
|
[3H] E2: [3H]-17β-естрадиол
ER: естрогенен рецептор
E2: немаркиран 17β-естрадиол
|
|
19.
|
Микротитърните плаки за изследването следва да се инкубират при температура от 2° до 8°C в продължение на 16 до 20 часа и да се поставят в ротатор по време на инкубационния период.
|
Измерване на [3H]-17β-естрадиол, свързан с hrERα
|
20.
|
Свързаният с hrERα [3H]-17β-естрадиол следва да се отдели от свободния [3H]-17β-естрадиол чрез добавяне на 80 μl охладена суспензия на DCC във всяка ямка, разклащане на микротитърните плаки в продължение на 10 минути и центрофугиране в продължение на 10 минути при около 2 500 оборота в минута. За да се сведе до минимум разделянето на свързания с hrERα [3H]-17β-естрадиол по време на този процес, е изключително важно буферите и ямките за изследване да се държат при температура от 2 до 8°C и всяка стъпка да се провежда бързо. За ефективно и бързо обработване на плаките е необходима разклащаща машина за микротитърни плаки.
|
|
21.
|
50 μl от супернатанта, съдържащ свързан с hrERα [3H]-17β-естрадиол, след това следва да се отдели изключително внимателно, за да се избегне всяко замърсяване на ямките от контакт с DCC, и трябва да се използва за сцинтилационно броене.
|
|
22.
|
След това във всяка ямка (A1—B12 и D1—E12) следва да се добавят 200 μl от сцинтилационната течност, която е способна да преобразува кинетичната енергия на ядрените излъчвания в светлинна енергия. Ямки G1—H12 (идентифицирани като обща стойност на dpms) представляват последователни разреждания на [3H]-17β-естрадиол (40 μl), които следва да се доставят директно в сцинтилационната течност в ямките на измервателната плака, както е посочено в таблица 3, т.е. тези ямки съдържат само 200 μl сцинтилационна течност и подходящия разреден разтвор на [3H]-17β-естрадиол. Тези измервания демонстрират колко [3H]-17β-естрадиол в dpms е добавен към всеки набор от ямки за общото свързване и неспецифичното свързване.
|
Таблица 3
Схема на микротитърна плака за изследване за свързване до насищане, измерване на радиоактивността
|
|
1
|
2
|
3
|
4
|
5
|
6
|
7
|
8
|
9
|
10
|
11
|
12
|
|
|
А
|
0,03 nM [3H] E2 + ЕР
|
0,06 nM [3H] E2 + ЕР
|
0,08 nM [3H] E2 + ЕР
|
0,10 nM [3 H] E2 + ЕР
|
Общо свързване (разтворител)
|
|
Б
|
0,30 nM [3H] E2 + ЕР
|
0,60 nM [3H] E2 + ЕР
|
1,0 nM [3H] E2 + ЕР
|
3,0 nM [3H] E2 + ЕР
|
|
В
|
|
|
|
|
|
|
Г
|
0,03 nM [3H] E2 + ЕР + 0,03 μM E2
|
0,06 nM [3H] E2 + ЕР + 0,06 μM E2
|
0,08 nM [3H] E2 + ЕР + 0,08 μM E2
|
0,10 nM [3H] E2 + ЕР + 0,10 μM E2
|
Неспецифично свързване
|
|
Д
|
0,30 nM [3H] E2 + ЕР + 0,30 μM E2
|
0,60 nM [3H] E2 + ЕР + 0,60 μM E2
|
1,0 nM [3H] E2 + ЕР + 1,0 μM E2
|
3,0 nM [3H] E2 + ЕР + 3,0 μM E2
|
|
Е
|
|
|
|
|
|
|
Ж
|
0,03 nM [3H] E2(общо dpms)
|
0,06 nM [3H] E2
|
0,08 nM [3H] E2
|
0,10 nM [3H] E2
|
Общо dpms (*7)
|
|
З
|
0,30 nM [3H] E2
|
0,60 nM [3H] E2
|
1,0 nM [3H] E2
|
3,0 nM [3H] E2
|
|
[3H] E2: [3H]-17β-естрадиол
ER: естрогенен рецептор
E2: немаркиран 17β-естрадиол
dpms: разпадане в минута
|
|
23.
|
Измерването следва да започне с отстъп от най-малко 2 часа, а времето за преброяване следва да е 40 минути на ямка. За определянето на dpm/ямка с корекция на потискането следва да се използва сцинтилационен брояч за микротитърни плаки. Ако сцинтилационен брояч за микротитърни плаки не е на разположение, другата възможност е пробите да се измерват в конвенционален брояч. При тези условия може да се обмисли намаляване на времето за преброяване.
|
Изследване на конкурентно свързване
|
24.
|
Изследването за конкурентно свързване измерва свързването на една концентрация на [3H]-17β-естрадиол в присъствието на увеличаващи се концентрации на изпитван химикал. За всяка концентрация в рамките на една серия следва да се използват три паралелни повторения. Освен това следва да се проведат три непаралелни серии за всеки изпитван химикал. Изследването трябва да се проведе в една или повече 96-ямкови микротитърни плаки.
|
Контроли
|
25.
|
При провеждане на изследването във всеки експеримент следва да се включват паралелни разтворител и контроли (т.е. референтен естроген, слабо свързващо вещество и несвързващо вещество). Пълните криви на концентрация за референтния естроген и контролите (т.е. слабо свързващо вещество и несвързващо вещество) следва да се използват в една плака по време на всяка серия. Всички останали плаки следва да съдържат i) висока (максимално изместване) и средна (приблизително IC50) концентрация на E2 и слабо свързващо вещество в по три повторения; (ii) контрола на разтворителя и неспецифичното свързване, всяка в три повторения. Процедурите за приготвяне на използвания в изследването буфер, [3H]-17β-естрадиол, hrERα и разтворите на изпитван химикал са описани подробно в протокола на CERI (2).
|
Контрола на разтворител:
|
26.
|
Контролата на разтворителя показва, че разтворителят не взаимодейства с изпитваната система и измерва също така общото свързване (ТВ). Предпочитаният разтворител е DMSO. Като алтернатива, ако най-високата концентрация на изпитвания химикал не е разтворима в DMSO, може да се използва етанол. Концентрацията на етанол или DMSO, ако се използва, в ямките на крайното изследване е 1,5 % и не може да надвишава 2 %.
|
Контрола на буфер:
|
27.
|
Контролата на буфера (ВС) не трябва да съдържа нито разтворител, нито изпитван химикал, но трябва да включва всички други компоненти на изследването. Резултатите на контролата на буфера се сравняват с контролата на разтворителя, за да провери, че използваният разтворител не влияе върху системата на изследването.
|
Силно свързващо вещество (референтен естроген)
|
28.
|
17β-естрадиол (CAS № 50-28-2) е ендогенен лиганд и се свързва с висок афинитет с ЕР, подтип алфа. За всяко изследване на конкурентно свързване на hrERα следва да се приготви стандартна крива с използване на 17β-естрадиол, за да се позволи оценка на променливостта при провеждане на изследването във времето в рамките на една и съща лаборатория. Следва да се приготвят осем разтвора на немаркиран 17β-естрадиол в етанол, като концентрациите в ямките за изследване следва да варират в диапазона от 100 nM до 10 pM (-7[logM] до -11[logM]), разпределени, както следва: (-7[logM], -8[logM], -8,5[logM], -9[logM], - 9,5[logM], -10[logM], -11[logM]). Най-високата концентрация на немаркиран 17β-естрадиол (1 μM) следва да служи като индикатор за неспецифично свързване. Тази концентрация се разграничава чрез етикета „NSB“ в Таблица 4, въпреки че също е част от стандартната крива.
|
Слабо свързващо вещество
|
29.
|
Следва да се включи слабо свързващо вещество (норетинодрел (CAS № 68-23-5) или, като алтернатива, норетиндрон (CAS № 68-22-4)), за да докаже чувствителността на всеки експеримент и да се позволи оценка на променливостта при провеждане на изследването във времето. Следва да се приготвят осем разтвора на слабото свързващо вещество в етанол, като концентрациите в ямките за изследване варират от 3 nM до 30 μМ (-8,5[logM] до -4,5[logM]), разпределени, както следва: -4,5[logM], -5[logM], -5,5[logM], -6[logM], -6,5[logM], -7[logM],-7,5[logM], -8,5[logM].
|
Несвързващо вещество
|
30.
|
Като отрицателна контрола (несвързващо вещество) следва да се използва октитриетоксисилан (OTES, CAS № 2 943-75-1). Той гарантира, че изследването, както се провежда, открива изпитвани химикали, които не се свързват с hrERα. Следва да се приготвят осем разтвора на несвързващото вещество в етанол, като концентрациите в ямките за изследване следва да варират в диапазона от 0,1nM до 1 000 μМ (-10[logM] до -3[logM]) с логаритмична стъпка. Ди-n-бутил фталат (ДБФ) може да се използва като алтернативно контролно несвързващо вещество. Налице са данни, че максималната му разтворимост е -4[logM].
|
Концентрация на hrERα
|
31.
|
Следва да се използва това количество на рецептор, което дава специфично свързване от 20±5 % на 1 nM радиолиганд (вж. точки 12—13 на Допълнение 2). Разтворът на hrERα следва да се приготви непосредствено преди употреба.
|
[3H]-17β-естрадиол
|
32.
|
Окончателната концентрация на [3H]-17β-естрадиол в анализираните ямки следва да е 1,0 nM.
|
Изпитвани химикали
|
33.
|
На първо място е необходимо да се проведе тест за разтворимост, за да се определи границата на разтворимост за всеки изпитван химикал и да се идентифицира подходящия диапазон на концентрация за използване при изпълнение на протокола на изпитването. Границата на разтворимост на всеки изпитван химикал трябва първо да се определи в разтворителя и допълнително да се потвърди при условията на изследването. Крайната концентрация, изпитвана в изследването, не трябва да надвишава 1 mM. Изпитването за установяване на диапазона включва контрола на разтворител заедно с осем логаритмични последователни разреждания, започвайки с максималната приемлива концентрация (напр. 8 mM или по-ниска въз основа на границата на разтворимост), в присъствието на забелязана мътност или преципитация. Изпитваният химикал следва да се изпита, като се използват 8 криви на концентрации с логаритмична стъпка между тях, определени от предшестващото изпитване за установяване на диапазона. Концентрациите във втория и третия експеримент следва да се коригират, както е подходящо, за да характеризират по-добре кривата отклик-концентрация.
|
|
34.
|
Разрежданията на изпитвания химикал следва да се приготвят в подходящия разтворител (вж. точка 26 на Допълнение 2). Ако най-високата концентрация на изпитвания химикал не е разтворима в DMSO или етанол и добавянето на още разтворител би довело до това концентрацията на разтворителя в окончателната епруветка да е по-голяма от приемливата граница, най-високата концентрация може да бъде намалена до следващата по-ниска концентрация. В този случай може да се добави допълнителна концентрация в долната част на серията от концентрации. Другите концентрации в сериите следва да останат без промяна.
|
|
35.
|
Разтворите на изпитвания химикал следва да се наблюдават отблизо, когато се добавят в ямката, тъй като изпитваният химикал може да се утаи при добавянето му в ямката. Данните за всички ямки, съдържащи преципитат, следва да се изключат от налагането на кривата и да се отбележат причините за изключването на данните.
|
|
36.
|
Ако съществува предходна информация от други източници, която предоставя логаритъм (IC50) на даден изпитван химикал, може да е подходящо разрежданията да се разпределят геометрично по-близо около очаквания логаритъм (IC50) (т.е. 0,5 логаритмични единици). Окончателните резултати следва да демонстрират достатъчно разпределение на концентрациите от всяка страна на логаритъма (IC50), включително „горе“ и „долу“, така че кривата на свързването да може да бъде адекватно характеризирана.
|
Организация на изследваната плака
|
37.
|
Маркираните микротитърни плаки следва да се приготвят с използване на шестократни инкубации за контролата на разтворителя, най-високата концентрация на референтния естроген (Е2), която служи и като индикатор за неспецифично свързване (NSB), контролата на буфера, осемте концентрации на контролата за несвързващо вещество (октилтриетоксисилан), седемте по-ниски концентрации на референтния естроген (Е2), осемте концентрации на слабо свързващото вещество (норетинодрел или норетинодрон) и осемте концентрации на всеки изпитван химикал (ТС). Примерна схема на диаграмата на плаката за кривите на пълна концентрация за референтния естроген и контролите е дадена по-долу в Таблица 4. Допълнителни микротитърни плаки се използват за изпитвания химикал и следва да съдържат контроли на плаката, т.е. 1) висока (максимално изместване) и средна (приблизително IC50) концентрация на E2 и слабо свързващото вещество в три повторения; 2) контрола на разтворителя и неспецифично свързване в по шест повторения (Таблица 5). Пример за схема на микротитърна плака за работна таблица на конкурентно изследване е предоставен в Допълнение 2.3. Посочените в таблици 4 и 5 концентрации са крайните концентрации на изследването. Максималната концентрация за E2 следва да е 1×10-7 M, а за слабо свързващото вещество следва да се използва най-високата концентрация, използвана за слабото свързващо вещество в плака 1. Концентрацията IC50 следва да се определи от лабораторията въз основа на нейната база данни за контроли от предходни периоди. Очакването е, че тази стойност ще е сходна с наблюдаваната във изследванията за валидиране (вж. Таблица 1).
|
Таблица 4
Схема на микротитърна плака за изследване на конкурентно свързване
|
|
1
|
2
|
3
|
4
|
5
|
6
|
7
|
8
|
9
|
10
|
11
|
12
|
|
А
|
TB (само разтворител)
|
TB (само разтворител)
|
NSB
|
NSB
|
|
Б
|
E2 (1×10-7)
|
E2 (1×10-8)
|
E2 (1×10-8,5)
|
E2 (1×10-9)
|
|
В
|
E2 (1×10-9,5)
|
E2 (1×10-10)
|
E2 (1×10-11)
|
Празна (*8)
|
|
Г
|
NE (1×10-4,5)
|
NE (1×10-5)
|
NE (1×10-5,5)
|
NE (1×10-6)
|
|
Д
|
NE (1×10-6,5)
|
NE (1×10-7)
|
NE (1×10-7,5)
|
NE (1×10-8,5)
|
|
Е
|
OTES (1×10-3)
|
OTES (1×10-4)
|
OTES (1×10-5)
|
OTES (1×10-6)
|
|
Ж
|
OTES (1×10-7)
|
OTES (1×10-8)
|
OTES (1×10-9)
|
OTES (1×10-10)
|
|
З
|
Празна (за горещ) (*9)
|
Празна (за горещ) (*9)
|
Контрола на буфер
|
Контрола на буфер
|
|
В този пример слабото свързващо вещество е норетинодрел (NE)
|
Таблица 5
Схема на микротитърна плака за изследването на конкурентно свързване, пълни криви на концентрацията за изпитваните химикали и контролите на плаката
|
|
1
|
2
|
3
|
4
|
5
|
6
|
7
|
8
|
9
|
10
|
11
|
12
|
|
А
|
TB (само разтворител)
|
TB (само разтворител)
|
NSB
|
NSB
|
|
Б
|
TC1 (1×10-3)
|
TC1 (1×10-4)
|
TC1 (1×10-5)
|
TC1 (1×10-6)
|
|
В
|
TC1 (1×10-7)
|
TC1 (1×10-8)
|
TC1 (1×10-9)
|
TC1 (1×10-10)
|
|
Г
|
TC2 (1×10-3)
|
TC2 (1×10-4)
|
TC2 (1×10-5)
|
TC2 (1×10-6)
|
|
Д
|
TC2 (1×10-7)
|
TC2 (1×10-8)
|
TC2 (1×10-9)
|
TC2 (1×10-10)
|
|
Е
|
TC3 (1×10-3)
|
TC3 (1×10-4)
|
TC3 (1×10-5)
|
TC3 (1×10-6)
|
|
Ж
|
TC3 (1×10-7)
|
TC3 (1×10-8)
|
TC3 (1×10-9)
|
TC3 (1×10-10)
|
|
З
|
NE (IC50
|
NE (1×10-4,5)
|
E2 (IC50)
|
E2 (1×10-7)
|
|
В този пример слабото свързващо вещество е норетинодрел (NE)
|
Завършване на изследване на конкурентно свързване
|
38.
|
Както е показано в таблица 6, в ямките следва да се добавят 80 μl контрола на разтворителя, контрола на буфера, референтен естроген, слабо свързващо вещество, несвързващо вещество и изпитвани химикали, подготвени в буфер, предназначен за изследването. След това във всяка ямка следва да се добавят 40 μl от 4 nM разтвор на [3H]-17β-естрадиол. След леко завъртане в продължение на 10 до 15 минути при температура от 2° до 8°C, следва да се добавят 40 μl от разтвора на hrERα. Микротитърните плаки за изследването следва да се инкубират при температура от 2° до 8°C в продължение на 16 до 20 часа и да се поставят в ротатор по време на инкубационния период.
|
Таблица 6
Обем на компонентите на изследването на конкурентно свързване към hrER, микротитърни плаки
|
Обем (μl)
|
Съставка
|
|
80
|
Немаркиран 17β-естрадиол, норетинодрел, OTES, изпитвани химикали, разтворител или буфер
|
|
40
|
4 nM разтвор на [3H]-17β-естрадиол
|
|
40
|
разтвор на hrERα, концентрация според определеното
|
|
160
|
Общ обем във всяка ямка на изследването
|
|
39.
|
Количественото определяне на [3H]-17β-естрадиол, свързан с hrERα, след отделяне на [3H]-17β-естрадиол, свързан с hrERα, от свободен [3H]-17β-естрадиол чрез добавяне на 80 μl охладена суспензия на DCC във всяка ямка, следва да се извърши след това, както е описано в точки 20—23 за изследването на свързване до насищане.
|
|
40.
|
Ямки H1—6 (идентифицирани като празни (за горещо състояние) в таблица 4) представляват dpms на [3H]-маркиран естрадиол в 40 μl. 40 μl аликвотна част следва да се достави директно в сцинтилационната течност в ямки H1—H6.
|
Критерии за приемливост
Изследване на свързване до насищане
|
41.
|
Кривата на специфично свързване следва да достигне връх при използването на все по-големи концентрации на [3H]-17β-естрадиол, което показва насищане на hrERα с лиганд.
|
|
42.
|
Специфичното свързване при 1 nM от [3H]-17β-естрадиол следва да е в рамките на приемливия диапазон от 15 % до 25 % от средната измерена обща радиоактивност, добавена в сериите. Случайни леки отклонения извън този диапазон са допустими, но ако сериите последователно са извън този диапазон или дадена серия излиза значително извън този диапазон, концентрацията на протеин следва да се коригира и изследването за насищане да се повтори.
|
|
43.
|
Данните следва да произведат линейна диаграма на Scatchard.
|
|
44.
|
Неспецифичното свързване не трябва да е прекомерно. Стойността на неспецифичното свързване следва обикновено да е <35 % от общото свързване. Въпреки това съотношението може от време на време да надвишава тази граница, когато се измерва много ниска стойност на dpm за най-ниската изпитвана концентрация на радиомаркиран 17β-естрадиол.
|
Изследване на конкурентно свързване
|
45.
|
Увеличаващите се концентрации на немаркиран 17β-естрадиол следва да изместят [3H]-17β-естрадиол от рецептора по начин, съответстващ на конкурентно свързване в едно място.
|
|
46.
|
Стойността на IC50 за референтния естроген (т.е. 17β-естрадиол) следва да е приблизително равна на моларната концентрация на [3H]-17β-естрадиол плюс Kd, определена от изследването за свързване до насищане.
|
|
47.
|
Общото специфично свързване следва последователно да е в рамките на приемливия диапазон от 20 ± 5 %, когато средната измерена концентрация на общата радиоактивност, добавена във всяка ямка, е 1 nM в сериите. Случайни леки отклонения извън този диапазон са допустими, но ако сериите последователно са извън този диапазон или дадена серия излиза значително извън този диапазон, концентрацията на протеин следва да се коригира.
|
|
48.
|
Разтворителят не следва да променя чувствителността или възпроизводимостта на изследването. Резултатите на контролата на разтворителя (ямки на ТВ) се сравняват с контролата на буфера, за да се потвърди, че използваният разтворител не влияе върху системата на изследването. Резултатите на контролата за ТВ и на контролата на буфера следва да са сравними, ако разтворителят не оказва ефект върху изследването.
|
|
49.
|
Несвързващото вещество не следва да измести повече от 25 % от [3H]-17β-естрадиол от hrERα, когато се изпитва до 10-3 M (OTES) или 10-4 M (ДБФ).
|
|
50.
|
Критериите за ефективност са разработени за референтния естроген и две слаби свързващи вещества (напр. норетинодрел, норетиндрон) с използване на данни от изследването за валидиране на изследването за свързване към hrER на FW (приложение N към справка 2). 95 % доверителни интервали са предоставени за средната стойност (n) +/- SD за всички контролни серии сред лабораториите, участващи във изследването за валидиране. 95 % доверителни интервали са изчислени за параметрите на налагане на кривата (т.е. горна част, долна част, наклон на Хил, logIC50) за референтния естроген и слабо свързващи вещества и за log10RBA на слабо свързващите вещества по отношение на референтния естроген и са предоставени като критерии за ефективност за положителните контроли. В таблица 1 са дадени очакваните диапазони за параметрите на налагане на кривата, които могат да се използват като критерии за ефективност. На практика диапазонът на IC50 може леко да варира въз основа на Kd на рецепторния препарат и концентрацията на лиганда.
|
|
51.
|
Не са разработени критерии за ефективност за параметрите на налагане на кривата за изпитваните химикали поради широкия спектър на съществуващи потенциални изпитвани химикали и вариациите в потенциалните афинитети и резултати (напр. пълна крива, частична крива, без налагане на крива). Въпреки това следва да се приложи професионална преценка, когато се преглеждат резултатите от всяка серия за даден изпитван химикал. Следва да се използва достатъчен диапазон на концентрации на изпитвания химикал за ясно определяне на горната част (напр. 90—100 % свързване) на конкурентната крива. Варирането между повторенията при всяка концентрация на изпитван химикал, както и между трите непаралелни серии следва да е разумно и обосновано от научна гледна точка. Контролите от всяка серия за даден изпитван химикал следва да се доближават до измерванията на ефективността, съобщени за това изследване на FW, и да са последователни с данните за контроли от предходни периоди от всяка съответна лаборатория.
|
АНАЛИЗ НА ДАННИТЕ
Изследване на свързване до насищане
|
52.
|
Измерват се както общото, така и неспецифичното свързване. От тези стойности специфичното свързване на увеличаващи се концентрации на [3H]-17β-естрадиол при равновесни условия се изчислява чрез изваждане на неспецифичното от общото свързване. Графиката на специфичното свързване спрямо концентрацията на [3H]-17β-естрадиол следва да достигне върха за максималното специфично свързване, показателно за насищането на hrERα с [3H]-17β-естрадиол. Освен това анализът на данните следва да документира свързването на [3H]-17β- естрадиол с единично място на свързване с висок афинитет. Неспецифичното, общото и специфичното свързване следва да се показва на кривата за свързване до насищане. Допълнителният анализ на тези данни следва да използва анализ на нелинейна регресия (напр. BioSoft; McPherson, 1985 г.; Motulsky, 1995) с окончателно представяне на данните като диаграма на Scatchard.
|
|
53.
|
Анализът на данните следва да определи Bmax и Kd от самостоятелните данни за общо свързване с използване на предположението, че неспецифичното свързване е линейно, освен ако не е предоставена обосновка за използване на различен метод. Освен това следва да се използва надеждна регресия, когато се определя най-доброто съвпадение, освен ако не е предоставена обосновка. Избраният метод за надеждна регресия следва да бъде посочен. Корекция за изчерпване на лиганд (напр. използване на метода на Swillens 1995) следва винаги да се използва, когато се определя Bmax и Kd от данни за свързване до насищане.
|
Изследване на конкурентно свързване
|
54.
|
Кривата на конкурентно свързване се изчертава като специфично свързване на [3H]-17β- естрадиол спрямо концентрацията (log10 единици) на конкурента. Концентрацията на изпитвания химикал, която инхибира 50 % от максималното специфично свързване на [3H]-17β-естрадиол, е стойността на IC50.
|
|
55.
|
Оценките на логаритмичните(IC50) стойности за положителните контроли (напр. референтен естроген и слабо свързващо вещество) следва да се определят с използване на подходящ софтуер за нелинейно налагане на крива за вписване на уравнението на Хил с четири параметъра, (напр. BioSoft; McPherson, 1985 г.; Motulsky, 1995 г.). Горната част, долната част, наклонът и логаритмичната стойност (IC50) обикновено следва да се оставят неограничени при налагането на тези криви. Следва да се използва надеждна регресия, когато се определя най-доброто съвпадение, освен ако не е предоставена обосновка. Не следва да се използва корекция за изчерпване на лиганд. След първоначалния анализ всяка крива на свързване следва да се прегледа, за да се гарантира подходящо налагане върху модела. Относителният афинитет на свързване (RBA) за слабо свързващо вещество следва да се изчисли като процент на логаритъма (IC50) за слабото свързващо вещество по отношение на логаритъма (IC50) за 17β-естрадиол. Резултатите от положителните контроли и контролата на несвързващо вещество следва да се оценят с използване на измервания на ефективността на изследването, описани в точки 45-50 на настоящото Допълнение 2.
|
|
56.
|
Данните за всички изпитвани химикали следва да се анализират с използване на поетапен подход, за да се гарантира, че данните са анализирани по подходящ начин и че всяка крива на конкурентно свързване е правилно класифицирана. Препоръчва се всяка серия за даден изпитван химикал първо да премине през стандартизиран анализ на данни, който е идентичен на този, използван за референтния естроген и контролите на слабо свързващо вещество (вж. точка 55 на настоящото Допълнение 3). След приключването му следва да се извърши технически преглед на параметрите на налагане на кривата, както и визуален преглед доколко добре данните съвпадат с генерираната крива на конкурентно свързване за всяка серия. По време на този технически преглед наблюденията на зависимо от концентрацията намаляване на процента на специфично свързан [3H]-17β-естрадиол, слабата променливост измежду техническите повторения при всяка концентрация на химикала и съгласуваността на параметрите на налагане в трите серии са добър индикатор, че изследването и анализите на данни са проведени по подходящ начин.
|
Тълкуване на данните
|
57.
|
При условие че всички критерии за приемливост са изпълнени, даден изпитван химикал се счита за свързващ се с hrERα, ако кривата на свързване може да бъде наложена и най-ниската точка върху кривата на отклик в рамките на диапазона на данните е по-малка 50 % (Фигура 1).
|
|
58.
|
При условие че са изпълнени всички критерии за приемливост, даден изпитван химикал се счита за несвързващ се с hrERα, ако:
|
—
|
Кривата на свързване може да бъде наложена и най-ниската точка на наложената крива на отклик в рамките на диапазона на данните е над 75 % или
|
|
—
|
Кривата на свързване не може да бъде наложена и най-ниският неизгладен среден процент на свързване измежду групите на концентрация в данните е над 75 %.
|
|
|
59.
|
Изпитваните химикали се считат за нееднозначни, ако никое от горните условия не е изпълнено (напр. най-ниската точка на наложената крива на отклик е между 76 – 51 %).
|
Таблица 7
Критерии за присвояване на класификация въз основа на крива на конкурентно свързване за даден изпитван химикал
|
Класифициране
|
Критерии
|
|
Свързващо веществоa
|
Кривата на свързване може да бъде наложена.
Най-ниската точка върху кривата на отклик в рамките на диапазона на данните е по-малка от 50 %.
|
|
Несвързващо веществоб
|
Ако кривата на свързване може да бъде наложена,
най-ниската точка върху наложената крива на отклик в рамките на диапазона на данните е над 75 %.
Ако кривата на свързване не може да бъде наложена,
най-ниският неизгладен среден процент на свързване измежду групите на концентрация в данните е над 75 %.
|
|
Нееднозначно веществов
|
Всяка изпитвана серия, която не е нито свързващо, нито несвързващо вещество
(напр. най-ниската точка на наложената крива на отклик е между 76 – 51 %).
|
Фигура 1
Примери за класифициране на изпитван химикал с използване на крива на конкурентно свързване

|
60.
|
Многобройните проведени в лаборатория серии за изпитван химикал се комбинират с присвояване на цифрови стойности на всяка серия и се осредняват за сериите, както е показано в Таблица 8. Резултатите за комбинираните серии във всяка лаборатория се сравняват с очакваната класификация за всеки изпитван химикал.
|
Таблица 8
Метод за класифициране на изпитван химикал с използване на множество серии в дадена лаборатория
|
За присвояване на стойност на всяка серия:
|
|
|
Класифициране
|
Цифрова стойност
|
|
Свързващо вещество
|
2
|
|
Нееднозначно вещество
|
1
|
|
Несвързващо вещество
|
0
|
|
За класифициране на средна цифрова стойност за сериите:
|
|
|
Класифициране
|
Цифрова стойност
|
|
Свързващо вещество
|
Средна стойност ≥ 1,5
|
|
Нееднозначно вещество
|
0,5 ≤ средна стойност < 1,5
|
|
Несвързващо вещество
|
Средна стойност < 0,5
|
ПРОТОКОЛ ОТ ИЗПИТВАНЕТО
|
61.
|
Вж. точка 24 от „КОМПОНЕНТИ НА ИЗСЛЕДВАНЕТО ЗА СВЪРЗВАНЕ КЪМ hrER“ от настоящия метод на изпитване.
|
Допълнение 2.1
СПИСЪК НА ТЕРМИНИТЕ
[3H]E2: 17β-естрадиол, радиомаркиран с тритий.
DCC: въглен, покрит с декстран.
E2
: немаркиран 17β-естрадиол (инертен).
Буфер, използван в изследването: 10 mM тризаминометан, 10 mg говежди серумен албумин/ml, 2 mM дитиотреитол, 10 % глицерол, 0,2 mM левпептин, pH 7,5.
hrERα: Човешки рекомбинантен естрогенен рецептор алфа
Повторение: една от множество ямки, които съдържат едно и също съдържание с едни и същи концентрации и се изследват паралелно в рамките на единична серия. В настоящия протокол всяка концентрация на изпитван химикал се изпитва три пъти; т.е. има три повторения, които се изследват едновременно с всяка концентрация на изпитван химикал.
Серия: пълен набор от паралелно изпитвани ямки на микротитърни плаки за изследване, които осигуряват цялата информация, необходима за характеризиране на свързването на изпитван химикал към hrERα (а именно общ [3H]-17β-естрадиол, добавен в ямката на изследването, максимално свързване на [3H]-17β-естрадиол към hrERα, неспецифично свързване и общо свързване при различни концентрации на изпитвания химикал). Една серия може да се състои и само от една ямка за изследване (т.е. повторение) на концентрация, но тъй като настоящият протокол изисква трикратен анализ, една серия се състои от три ямки за изследване на концентрация. Освен това настоящият протокол изисква три независими (т.е. непаралелни) серии на химикал.
Допълнение 2,2
ТИПИЧНО ИЗСЛЕДВАНЕ ЗА НАСИЩАНЕ С [3H]-17Β-ЕСТРАДИОЛ С ТРИ ЯМКИ ЗА ПОВТОРЕНИЕ
|
Типично изследване за насищане с [3H]-17β-естрадиол с три ямки за повторение
|
|
Местоположение
|
Повторение
|
Код на тип ямка
|
Първоначална концентрация на горещ E2 (nM)
|
Обем на горещ E2 (μl)
|
Окончателна концентрация на горещ E2 (nM)
|
Първоначална концентрация на студен E2 (μМ)
|
Обем на студен E2 (μl)
|
Окончателна концентрация на студен E2 (μМ)
|
Обем на буфера (μl)
|
Обем на рецептора (μl)
|
Общ обем в ямките
|
|
A1
|
1
|
З
|
0,12
|
40
|
0,03
|
—
|
—
|
—
|
80
|
40
|
160
|
|
A2
|
2
|
З
|
0,12
|
40
|
0,03
|
—
|
—
|
—
|
80
|
40
|
160
|
|
A3
|
3
|
З
|
0,12
|
40
|
0,03
|
—
|
—
|
—
|
80
|
40
|
160
|
|
A4
|
1
|
З
|
0,24
|
40
|
0,06
|
—
|
—
|
—
|
80
|
40
|
160
|
|
A5
|
2
|
З
|
0,24
|
40
|
0,06
|
—
|
—
|
—
|
80
|
40
|
160
|
|
A6
|
3
|
З
|
0,24
|
40
|
0,06
|
—
|
—
|
—
|
80
|
40
|
160
|
|
A7
|
1
|
З
|
0,32
|
40
|
0,08
|
—
|
—
|
—
|
80
|
40
|
160
|
|
A8
|
2
|
З
|
0,32
|
40
|
0,08
|
—
|
—
|
—
|
80
|
40
|
160
|
|
A9
|
3
|
З
|
0,32
|
40
|
0,08
|
—
|
—
|
—
|
80
|
40
|
160
|
|
A10
|
1
|
З
|
0,40
|
40
|
0,10
|
—
|
—
|
—
|
80
|
40
|
160
|
|
A11
|
2
|
З
|
0,40
|
40
|
0,10
|
—
|
—
|
—
|
80
|
40
|
160
|
|
A12
|
3
|
З
|
0,40
|
40
|
0,10
|
—
|
—
|
—
|
80
|
40
|
160
|
|
B1
|
1
|
З
|
1,20
|
40
|
0,30
|
—
|
—
|
—
|
80
|
40
|
160
|
|
B2
|
2
|
З
|
1,20
|
40
|
0,30
|
—
|
—
|
—
|
80
|
40
|
160
|
|
B3
|
3
|
З
|
1,20
|
40
|
0,30
|
—
|
—
|
—
|
80
|
40
|
160
|
|
B4
|
1
|
З
|
2,40
|
40
|
0,60
|
—
|
—
|
—
|
80
|
40
|
160
|
|
B5
|
2
|
З
|
2,40
|
40
|
0,60
|
—
|
—
|
—
|
80
|
40
|
160
|
|
B6
|
3
|
З
|
2,40
|
40
|
0,60
|
—
|
—
|
—
|
80
|
40
|
160
|
|
B7
|
1
|
З
|
4,00
|
40
|
1,00
|
—
|
—
|
—
|
80
|
40
|
160
|
|
B8
|
2
|
З
|
4,00
|
40
|
1,00
|
—
|
—
|
—
|
80
|
40
|
160
|
|
B9
|
3
|
З
|
4,00
|
40
|
1,00
|
—
|
—
|
—
|
80
|
40
|
160
|
|
B10
|
1
|
З
|
12,00
|
40
|
3,00
|
—
|
—
|
—
|
80
|
40
|
160
|
|
B11
|
2
|
З
|
12,00
|
40
|
3,00
|
—
|
—
|
—
|
80
|
40
|
160
|
|
B12
|
3
|
З
|
12,00
|
40
|
3,00
|
—
|
—
|
—
|
80
|
40
|
160
|
|
D1
|
1
|
ГС
|
0,12
|
40
|
0,03
|
0,06
|
80
|
0,03
|
—
|
40
|
160
|
|
D2
|
2
|
ГС
|
0,12
|
40
|
0,03
|
0,06
|
80
|
0,03
|
—
|
40
|
160
|
|
D3
|
3
|
ГС
|
0,12
|
40
|
0,03
|
0,06
|
80
|
0,03
|
—
|
40
|
160
|
|
D4
|
1
|
ГС
|
0,24
|
40
|
0,06
|
0,12
|
80
|
0,06
|
—
|
40
|
160
|
|
D5
|
2
|
ГС
|
0,24
|
40
|
0,06
|
0,12
|
80
|
0,06
|
—
|
40
|
160
|
|
D6
|
3
|
ГС
|
0,24
|
40
|
0,06
|
0,12
|
80
|
0,06
|
—
|
40
|
160
|
|
D7
|
1
|
ГС
|
0,32
|
40
|
0,08
|
0,16
|
80
|
0,08
|
—
|
40
|
160
|
|
D8
|
2
|
ГС
|
0,32
|
40
|
0,08
|
0,16
|
80
|
0,08
|
—
|
40
|
160
|
|
D9
|
3
|
ГС
|
0,32
|
40
|
0,08
|
0,16
|
80
|
0,08
|
—
|
40
|
160
|
|
D10
|
1
|
ГС
|
0,40
|
40
|
0,10
|
0,2
|
80
|
0,1
|
—
|
40
|
160
|
|
D11
|
2
|
ГС
|
0,40
|
40
|
0,10
|
0,2
|
80
|
0,1
|
—
|
40
|
160
|
|
D12
|
3
|
ГС
|
0,40
|
40
|
0,10
|
0,2
|
80
|
0,1
|
—
|
40
|
160
|
|
E1
|
1
|
ГС
|
1,20
|
40
|
0,30
|
0,6
|
80
|
0,3
|
—
|
40
|
160
|
|
E2
|
2
|
ГС
|
1,20
|
40
|
0,30
|
0,6
|
80
|
0,3
|
—
|
40
|
160
|
|
E3
|
3
|
ГС
|
1,20
|
40
|
0,30
|
0,6
|
80
|
0,3
|
—
|
40
|
160
|
|
E4
|
1
|
ГС
|
2,40
|
40
|
0,60
|
1,2
|
80
|
0,6
|
—
|
40
|
160
|
|
E5
|
2
|
ГС
|
2,40
|
40
|
0,60
|
1,2
|
80
|
0,6
|
—
|
40
|
160
|
|
E6
|
3
|
ГС
|
2,40
|
40
|
0,60
|
1,2
|
80
|
0,6
|
—
|
40
|
160
|
|
E7
|
1
|
ГС
|
4,00
|
40
|
1,00
|
2
|
80
|
1
|
—
|
40
|
160
|
|
E8
|
2
|
ГС
|
4,00
|
40
|
1,00
|
2
|
80
|
1
|
—
|
40
|
160
|
|
E9
|
3
|
ГС
|
4,00
|
40
|
1,00
|
2
|
80
|
1
|
—
|
40
|
160
|
|
E10
|
1
|
ГС
|
12,00
|
40
|
3,00
|
6
|
80
|
3
|
—
|
40
|
160
|
|
E11
|
2
|
ГС
|
12,00
|
40
|
3,00
|
6
|
80
|
3
|
—
|
40
|
160
|
|
E12
|
3
|
ГС
|
12,00
|
40
|
3,00
|
6
|
80
|
3
|
—
|
40
|
160
|
|
G1
|
1
|
Горещ
|
0,12
|
40
|
0,03
|
—
|
—
|
—
|
—
|
—
|
40
|
|
G2
|
2
|
Горещ
|
0,12
|
40
|
0,03
|
—
|
—
|
—
|
—
|
—
|
40
|
|
G3
|
3
|
Горещ
|
0,12
|
40
|
0,03
|
—
|
—
|
—
|
—
|
—
|
40
|
|
G4
|
1
|
Горещ
|
0,24
|
40
|
0,06
|
—
|
—
|
—
|
—
|
—
|
40
|
|
G5
|
2
|
Горещ
|
0,24
|
40
|
0,06
|
—
|
—
|
—
|
—
|
—
|
40
|
|
G6
|
3
|
Горещ
|
0,24
|
40
|
0,06
|
—
|
—
|
—
|
—
|
—
|
40
|
|
G7
|
1
|
Горещ
|
0,32
|
40
|
0,08
|
—
|
—
|
—
|
—
|
—
|
40
|
|
G8
|
2
|
Горещ
|
0,32
|
40
|
0,08
|
—
|
—
|
—
|
—
|
—
|
40
|
|
G9
|
3
|
Горещ
|
0,32
|
40
|
0,08
|
—
|
—
|
—
|
—
|
—
|
40
|
|
G10
|
1
|
Горещ
|
0,40
|
40
|
0,10
|
—
|
—
|
—
|
—
|
—
|
40
|
|
G11
|
2
|
Горещ
|
0,40
|
40
|
0,10
|
—
|
—
|
—
|
—
|
—
|
40
|
|
G12
|
3
|
Горещ
|
0,40
|
40
|
0,10
|
—
|
—
|
—
|
—
|
—
|
40
|
|
H1
|
1
|
Горещ
|
1,20
|
40
|
0,30
|
—
|
—
|
—
|
—
|
—
|
40
|
|
H2
|
2
|
Горещ
|
1,20
|
40
|
0,30
|
—
|
—
|
—
|
—
|
—
|
40
|
|
H3
|
3
|
Горещ
|
1,20
|
40
|
0,30
|
—
|
—
|
—
|
—
|
—
|
40
|
|
H4
|
1
|
Горещ
|
2,40
|
40
|
0,60
|
—
|
—
|
—
|
—
|
—
|
40
|
|
H5
|
2
|
Горещ
|
2,40
|
40
|
0,60
|
—
|
—
|
—
|
—
|
—
|
40
|
|
H6
|
3
|
Горещ
|
2,40
|
40
|
0,60
|
—
|
—
|
—
|
—
|
—
|
40
|
|
H7
|
1
|
Горещ
|
4,00
|
40
|
1,00
|
—
|
—
|
—
|
—
|
—
|
40
|
|
H8
|
2
|
Горещ
|
4,00
|
40
|
1,00
|
—
|
—
|
—
|
—
|
—
|
40
|
|
H9
|
3
|
Горещ
|
4,00
|
40
|
1,00
|
—
|
—
|
—
|
—
|
—
|
40
|
|
H10
|
1
|
Горещ
|
12,00
|
40
|
3,00
|
—
|
—
|
—
|
—
|
—
|
40
|
|
H11
|
2
|
Горещ
|
12,00
|
40
|
3,00
|
—
|
—
|
—
|
—
|
—
|
40
|
|
H12
|
3
|
Горещ
|
12,00
|
40
|
3,00
|
—
|
—
|
—
|
—
|
—
|
40
|
|
Имайте предвид, че „горещите“ ямки са празни по време на инкубация. 40 μl се добавят само за сцинтилационно броене.
|
Допълнение 2.3
СХЕМА НА ЯМКА НА СРАВНИТЕЛНО ИЗСЛЕДВАНЕ НА СВЪРЗВАНЕ
|
Плоча
|
Местоположение
|
Повторение
|
Тип ямка
|
Код на ямка
|
Код на концентрация
|
Първоначална концентрация на конкурента (M)
|
Запас на hrER (μl)
|
Обем на буфера (μl)
|
Обем на маркера (Горещ E2) (μL)
|
Обем от плака за разреждане (μL)
|
Окончателен обем (μl)
|
Окончателна концентрация на конкурента (M)
|
|
S
|
A1
|
1
|
общо свързване
|
TB
|
TB1
|
—
|
40
|
|
40
|
80
|
160
|
—
|
|
S
|
A2
|
2
|
общо свързване
|
TB
|
TB2
|
—
|
40
|
|
40
|
80
|
160
|
—
|
|
S
|
A3
|
3
|
общо свързване
|
TB
|
TB3
|
—
|
40
|
|
40
|
80
|
160
|
—
|
|
S
|
A4
|
1
|
общо свързване
|
TB
|
TB4
|
—
|
40
|
|
40
|
80
|
160
|
—
|
|
S
|
A5
|
2
|
общо свързване
|
TB
|
TB5
|
—
|
40
|
|
40
|
80
|
160
|
—
|
|
S
|
A6
|
3
|
общо свързване
|
TB
|
TB6
|
—
|
40
|
|
40
|
80
|
160
|
—
|
|
S
|
A7
|
1
|
студен E2 (висок)
|
NSB
|
S0
|
2,00E-06
|
40
|
—
|
40
|
80
|
160
|
1,0E-06
|
|
S
|
A8
|
2
|
студен E2 (висок)
|
NSB
|
S0
|
2,00E-06
|
40
|
—
|
40
|
80
|
160
|
1,0E-06
|
|
S
|
A9
|
3
|
студен E2 (висок)
|
NSB
|
S0
|
2,00E-06
|
40
|
—
|
40
|
80
|
160
|
1,0E-06
|
|
S
|
A10
|
1
|
студен E2 (висок)
|
NSB
|
S0
|
2,00E-06
|
40
|
—
|
40
|
80
|
160
|
1,0E-06
|
|
S
|
A11
|
2
|
студен E2 (висок)
|
NSB
|
S0
|
2,00E-06
|
40
|
—
|
40
|
80
|
160
|
1,0E-06
|
|
S
|
A12
|
3
|
студен E2 (висок)
|
NSB
|
S0
|
2,00E-06
|
40
|
—
|
40
|
80
|
160
|
1,0E-06
|
|
S
|
B1
|
1
|
студен E2
|
S
|
S1
|
2,00E-07
|
40
|
—
|
40
|
80
|
160
|
1,0E-07
|
|
S
|
B2
|
2
|
студен E2
|
S
|
S1
|
2,00E-07
|
40
|
—
|
40
|
80
|
160
|
1,0E-07
|
|
S
|
B3
|
3
|
студен E2
|
S
|
S1
|
2,00E-07
|
40
|
—
|
40
|
80
|
160
|
1,0E-07
|
|
S
|
B4
|
1
|
студен E2
|
S
|
S2
|
2,00E-08
|
40
|
—
|
40
|
80
|
160
|
1,0E-08
|
|
S
|
B5
|
2
|
студен E2
|
S
|
S2
|
2,00E-08
|
40
|
—
|
40
|
80
|
160
|
1,0E-08
|
|
S
|
B6
|
3
|
студен E2
|
S
|
S2
|
2,00E-08
|
40
|
—
|
40
|
80
|
160
|
1,0E-08
|
|
S
|
B7
|
1
|
студен E2
|
S
|
S3
|
6,00E-09
|
40
|
—
|
40
|
80
|
160
|
3,0E-09
|
|
S
|
B8
|
2
|
студен E2
|
S
|
S3
|
6,00E-09
|
40
|
—
|
40
|
80
|
160
|
3,0E-09
|
|
S
|
B9
|
3
|
студен E2
|
S
|
S3
|
6,00E-09
|
40
|
—
|
40
|
80
|
160
|
3,0E-09
|
|
S
|
B10
|
1
|
студен E2
|
S
|
S4
|
2,00E-09
|
40
|
—
|
40
|
80
|
160
|
1,0E-09
|
|
S
|
B11
|
2
|
студен E2
|
S
|
S4
|
2,00E-09
|
40
|
—
|
40
|
80
|
160
|
1,0E-09
|
|
S
|
B12
|
3
|
студен E2
|
S
|
S4
|
2,00E-09
|
40
|
—
|
40
|
80
|
160
|
1,0E-09
|
|
S
|
C1
|
1
|
студен E2
|
S
|
S5
|
6,00E-10
|
40
|
—
|
40
|
80
|
160
|
3,0E-10
|
|
S
|
C2
|
2
|
студен E2
|
S
|
S5
|
6,00E-10
|
40
|
—
|
40
|
80
|
160
|
3,0E-10
|
|
S
|
C3
|
3
|
студен E2
|
S
|
S5
|
6,00E-10
|
40
|
—
|
40
|
80
|
160
|
3,0E-10
|
|
S
|
C4
|
1
|
студен E2
|
S
|
S6
|
2,00E-10
|
40
|
—
|
40
|
80
|
160
|
1,0E-10
|
|
S
|
C5
|
2
|
студен E2
|
S
|
S6
|
2,00E-10
|
40
|
—
|
40
|
80
|
160
|
1,0E-10
|
|
S
|
C6
|
3
|
студен E2
|
S
|
S6
|
2,00E-10
|
40
|
—
|
40
|
80
|
160
|
1,0E-10
|
|
S
|
C7
|
1
|
студен E2
|
S
|
S7
|
2,00E-11
|
40
|
—
|
40
|
80
|
160
|
1,0E-11
|
|
S
|
C8
|
2
|
студен E2
|
S
|
S7
|
2,00E-11
|
40
|
—
|
40
|
80
|
160
|
1,0E-11
|
|
S
|
C9
|
3
|
студен E2
|
S
|
S7
|
2,00E-11
|
40
|
—
|
40
|
80
|
160
|
1,0E-11
|
|
S
|
C10
|
1
|
празна
|
празна
|
B1
|
—
|
—
|
160
|
—
|
—
|
160
|
—
|
|
S
|
C11
|
2
|
празна
|
празна
|
B2
|
—
|
—
|
160
|
—
|
—
|
160
|
—
|
|
S
|
C12
|
3
|
празна
|
празна
|
B3
|
—
|
—
|
160
|
—
|
—
|
160
|
—
|
|
S
|
D1
|
1
|
норетинодрел
|
NE
|
WP1
|
6,00E-05
|
40
|
—
|
40
|
80
|
160
|
3,0E-05
|
|
S
|
D2
|
1
|
норетинодрел
|
NE
|
WP1
|
6,00E-05
|
40
|
—
|
40
|
80
|
160
|
3,0E-05
|
|
S
|
D3
|
1
|
норетинодрел
|
NE
|
WP1
|
6,00E-05
|
40
|
—
|
40
|
80
|
160
|
3,0E-05
|
|
S
|
D4
|
1
|
норетинодрел
|
NE
|
WP2
|
2,00E-05
|
40
|
—
|
40
|
80
|
160
|
1,0E-05
|
|
S
|
D5
|
1
|
норетинодрел
|
NE
|
WP2
|
2,00E-05
|
40
|
—
|
40
|
80
|
160
|
1,0E-05
|
|
S
|
D6
|
1
|
норетинодрел
|
NE
|
WP2
|
2,00E-05
|
40
|
—
|
40
|
80
|
160
|
1,0E-05
|
|
S
|
D7
|
1
|
норетинодрел
|
NE
|
WP3
|
6,00E-06
|
40
|
—
|
40
|
80
|
160
|
3,0E-06
|
|
S
|
D8
|
1
|
норетинодрел
|
NE
|
WP3
|
6,00E-06
|
40
|
—
|
40
|
80
|
160
|
3,0E-06
|
|
S
|
D9
|
1
|
норетинодрел
|
NE
|
WP3
|
6,00E-06
|
40
|
—
|
40
|
80
|
160
|
3,0E-06
|
|
S
|
D10
|
1
|
норетинодрел
|
NE
|
WP4
|
2,00E-06
|
40
|
—
|
40
|
80
|
160
|
1,0E-06
|
|
S
|
D11
|
1
|
норетинодрел
|
NE
|
WP4
|
2,00E-06
|
40
|
—
|
40
|
80
|
160
|
1,0E-06
|
|
S
|
D12
|
1
|
норетинодрел
|
NE
|
WP4
|
2,00E-06
|
40
|
—
|
40
|
80
|
160
|
1,0E-06
|
|
S
|
E1
|
1
|
норетинодрел
|
NE
|
WP
|
6,00E-07
|
40
|
|
40
|
80
|
160
|
3,0E-07
|
|
S
|
E2
|
2
|
норетинодрел
|
NE
|
WP
|
6,00E-07
|
40
|
|
40
|
80
|
160
|
3,0E-07
|
|
S
|
E3
|
3
|
норетинодрел
|
NE
|
WP
|
6,00E-07
|
40
|
|
40
|
80
|
160
|
3,0E-07
|
|
S
|
E4
|
1
|
норетинодрел
|
NE
|
WP
|
2,00E-07
|
40
|
|
40
|
80
|
160
|
1,0E-07
|
|
S
|
E5
|
2
|
норетинодрел
|
NE
|
WP
|
2,00E-07
|
40
|
|
40
|
80
|
160
|
1,0E-07
|
|
S
|
E6
|
3
|
норетинодрел
|
NE
|
WP
|
2,00E-07
|
40
|
|
40
|
80
|
160
|
1,0E-07
|
|
S
|
E7
|
1
|
норетинодрел
|
NE
|
WP
|
6,00E-08
|
40
|
—
|
40
|
80
|
160
|
3,0E-08
|
|
S
|
E8
|
2
|
норетинодрел
|
NE
|
WP
|
6,00E-08
|
40
|
—
|
40
|
80
|
160
|
3,0E-08
|
|
S
|
E9
|
3
|
норетинодрел
|
NE
|
WP
|
6,00E-08
|
40
|
—
|
40
|
80
|
160
|
3,0E-08
|
|
S
|
E10
|
1
|
норетинодрел
|
NE
|
WP
|
6,00E-09
|
40
|
—
|
40
|
80
|
160
|
3,0E-09
|
|
S
|
E11
|
2
|
норетинодрел
|
NE
|
WP
|
6,00E-09
|
40
|
—
|
40
|
80
|
160
|
3,0E-09
|
|
S
|
E12
|
3
|
норетинодрел
|
NE
|
WP
|
6,00E-09
|
40
|
—
|
40
|
80
|
160
|
3,0E-09
|
|
S
|
F1
|
1
|
OTES
|
N
|
OTES
|
2,00E-03
|
40
|
—
|
40
|
80
|
160
|
1,0E-03
|
|
S
|
F2
|
2
|
OTES
|
N
|
OTES
|
2,00E-03
|
40
|
—
|
40
|
80
|
160
|
1,0E-03
|
|
S
|
F3
|
3
|
OTES
|
N
|
OTES
|
2,00E-03
|
40
|
—
|
40
|
80
|
160
|
1,0E-03
|
|
S
|
F4
|
1
|
OTES
|
N
|
OTES
|
2,00E-04
|
40
|
—
|
40
|
80
|
160
|
1,0E-04
|
|
S
|
F5
|
2
|
OTES
|
N
|
OTES
|
2,00E-04
|
40
|
—
|
40
|
80
|
160
|
1,0E-04
|
|
S
|
F6
|
3
|
OTES
|
N
|
OTES
|
2,00E-04
|
40
|
—
|
40
|
80
|
160
|
1,0E-04
|
|
S
|
F7
|
1
|
OTES
|
N
|
OTES
|
2,00E-05
|
40
|
—
|
40
|
80
|
160
|
3,0E-05
|
|
S
|
F8
|
2
|
OTES
|
N
|
OTES
|
2,00E-05
|
40
|
—
|
40
|
80
|
160
|
3,0E-05
|
|
S
|
F9
|
3
|
OTES
|
N
|
OTES
|
2,00E-05
|
40
|
—
|
40
|
80
|
160
|
3,0E-05
|
|
S
|
F10
|
1
|
OTES
|
N
|
OTES
|
2,00E-06
|
40
|
—
|
40
|
80
|
160
|
1,0E-06
|
|
S
|
F11
|
2
|
OTES
|
N
|
OTES
|
2,00E-06
|
40
|
—
|
40
|
80
|
160
|
1,0E-06
|
|
S
|
F12
|
3
|
OTES
|
N
|
OTES
|
2,00E-06
|
40
|
—
|
40
|
80
|
160
|
1,0E-06
|
|
S
|
G1
|
1
|
OTES
|
N
|
OTES
|
2,00E-07
|
40
|
—
|
40
|
80
|
160
|
3,0E-07
|
|
S
|
G2
|
2
|
OTES
|
N
|
OTES
|
2,00E-07
|
40
|
—
|
40
|
80
|
160
|
3,0E-07
|
|
S
|
G3
|
3
|
OTES
|
N
|
OTES
|
2,00E-07
|
40
|
—
|
40
|
80
|
160
|
3,0E-07
|
|
S
|
G4
|
1
|
OTES
|
N
|
OTES
|
2,00E-08
|
40
|
—
|
40
|
80
|
160
|
1,0E-08
|
|
S
|
G5
|
2
|
OTES
|
N
|
OTES
|
2,00E-08
|
40
|
—
|
40
|
80
|
160
|
1,0E-08
|
|
S
|
G6
|
3
|
OTES
|
N
|
OTES
|
2,00E-08
|
40
|
—
|
40
|
80
|
160
|
1,0E-08
|
|
S
|
G7
|
1
|
OTES
|
N
|
OTES
|
2,00E-09
|
40
|
—
|
40
|
80
|
160
|
1,0E-09
|
|
S
|
G8
|
2
|
OTES
|
N
|
OTES
|
2,00E-09
|
40
|
—
|
40
|
80
|
160
|
1,0E-09
|
|
S
|
G9
|
3
|
OTES
|
N
|
OTES
|
2,00E-09
|
40
|
—
|
40
|
80
|
160
|
1,0E-09
|
|
S
|
G10
|
1
|
OTES
|
N
|
OTES
|
2,00E-10
|
40
|
—
|
40
|
—
|
160
|
1,0E-10
|
|
S
|
G11
|
2
|
OTES
|
N
|
OTES
|
2,00E-10
|
40
|
—
|
40
|
—
|
160
|
1,0E-10
|
|
S
|
G12
|
3
|
OTES
|
N
|
OTES
|
2,00E-10
|
40
|
—
|
40
|
—
|
160
|
1,0E-10
|
|
S
|
H1
|
1
|
горещ
|
З
|
З
|
—
|
—
|
—
|
40
|
—
|
40
|
—
|
|
S
|
H2
|
1
|
горещ
|
З
|
З
|
—
|
—
|
—
|
40
|
—
|
40
|
—
|
|
S
|
H3
|
1
|
горещ
|
З
|
З
|
—
|
—
|
—
|
40
|
—
|
40
|
—
|
|
S
|
H4
|
1
|
горещ
|
З
|
З
|
—
|
—
|
—
|
40
|
—
|
40
|
—
|
|
S
|
H5
|
1
|
горещ
|
З
|
З
|
—
|
—
|
—
|
40
|
—
|
40
|
—
|
|
S
|
H6
|
1
|
горещ
|
З
|
З
|
—
|
—
|
—
|
40
|
—
|
40
|
—
|
|
S
|
H7
|
1
|
контрола на буфер
|
BC
|
BC
|
—
|
40
|
80
|
40
|
—
|
160
|
—
|
|
S
|
H8
|
1
|
контрола на буфер
|
BC
|
BC
|
—
|
40
|
80
|
40
|
—
|
160
|
—
|
|
S
|
H9
|
1
|
контрола на буфер
|
BC
|
BC
|
—
|
40
|
80
|
40
|
—
|
160
|
—
|
|
S
|
H10
|
1
|
контрола на буфер
|
BC
|
BC
|
—
|
40
|
80
|
40
|
—
|
160
|
—
|
|
S
|
H11
|
1
|
контрола на буфер
|
BC
|
BC
|
—
|
40
|
80
|
40
|
—
|
160
|
—
|
|
S
|
H12
|
1
|
контрола на буфер
|
BC
|
BC
|
—
|
40
|
80
|
40
|
—
|
160
|
—
|
Имайте предвид, че „горещите“ ямки са празни по време на инкубация. 40 μl се добавят само за сцинтилационно броене.
|
Схема на ямка на сравнително изследване на свързване
|
|
Плоча
|
Местоположение
|
Повторение
|
Тип ямка
|
Код на ямка
|
Код на концентрация
|
Първоначална концентрация на конкурента (М)
|
Запас на hrER (μL)
|
Обем на буфера (μL)
|
Обем на маркера (Горещ E2) (μL)
|
Обем от плака за разреждане (μL)
|
Окончателен обем (μl)
|
Окончателна концентрация на конкурента (М)
|
|
P1
|
A1
|
1
|
общо свързване
|
TB
|
TBB1B1
|
—
|
40
|
—
|
40
|
80
|
160
|
—
|
|
P1
|
A2
|
2
|
общо свързване
|
TB
|
TB2
|
—
|
40
|
—
|
40
|
80
|
160
|
—
|
|
P1
|
A3
|
3
|
общо свързване
|
TB
|
TB3
|
—
|
40
|
—
|
40
|
80
|
160
|
—
|
|
P1
|
A4
|
1
|
общо свързване
|
TB
|
TB4
|
—
|
40
|
—
|
40
|
80
|
160
|
—
|
|
P1
|
A5
|
2
|
общо свързване
|
TB
|
TB5
|
—
|
40
|
—
|
40
|
80
|
160
|
—
|
|
P1
|
A6
|
3
|
общо свързване
|
TB
|
TB6
|
—
|
40
|
—
|
40
|
80
|
160
|
—
|
|
P1
|
A7
|
1
|
студен E2 (висок)
|
NSB
|
S0
|
2,00E-06
|
40
|
—
|
40
|
80
|
160
|
1,0E-06
|
|
P1
|
A8
|
2
|
студен E2 (висок)
|
NSB
|
S0
|
2,00E-06
|
40
|
—
|
40
|
80
|
160
|
1,0E-06
|
|
P1
|
A9
|
3
|
студен E2 (висок)
|
NSB
|
S0
|
2,00E-06
|
40
|
—
|
40
|
80
|
160
|
1,0E-06
|
|
P1
|
A10
|
1
|
студен E2 (висок)
|
NSB
|
S0
|
2,00E-06
|
40
|
—
|
40
|
80
|
160
|
1,0E-06
|
|
P1
|
A11
|
2
|
студен E2 (висок)
|
NSB
|
S0
|
2,00E-06
|
40
|
—
|
40
|
80
|
160
|
1,0E-06
|
|
P1
|
A12
|
3
|
студен E2 (висок)
|
NSB
|
S0
|
2,00E-06
|
40
|
—
|
40
|
80
|
160
|
1,0E-06
|
|
P1
|
B1
|
1
|
Изпитван химикал 1
|
TC1
|
1
|
2,00E-03
|
40
|
0
|
40
|
80
|
160
|
1,0E-03
|
|
P1
|
B2
|
2
|
Изпитван химикал 1
|
TC1
|
1
|
2,00E-03
|
40
|
0
|
40
|
80
|
160
|
1,0E-03
|
|
P1
|
B3
|
3
|
Изпитван химикал 1
|
TC1
|
1
|
2,00E-03
|
40
|
0
|
40
|
80
|
160
|
1,0E-03
|
|
P1
|
B4
|
1
|
Изпитван химикал 1
|
TC1
|
2
|
2,00E-04
|
40
|
0
|
40
|
80
|
160
|
1,0E-04
|
|
P1
|
B5
|
2
|
Изпитван химикал 1
|
TC1
|
2
|
2,00E-04
|
40
|
0
|
40
|
80
|
160
|
1,0E-04
|
|
P1
|
B6
|
3
|
Изпитван химикал 1
|
TC1
|
2
|
2,00E-04
|
40
|
0
|
40
|
80
|
160
|
1,0E-04
|
|
P1
|
B7
|
1
|
Изпитван химикал 1
|
TC1
|
3
|
2,00E-05
|
40
|
0
|
40
|
80
|
160
|
1,0E-05
|
|
P1
|
B8
|
2
|
Изпитван химикал 1
|
TC1
|
3
|
2,00E-05
|
40
|
0
|
40
|
80
|
160
|
1,0E-05
|
|
P1
|
B9
|
3
|
Изпитван химикал 1
|
TC1
|
3
|
2,00E-05
|
40
|
0
|
40
|
80
|
160
|
1,0E-05
|
|
P1
|
B10
|
1
|
Изпитван химикал 1
|
TC1
|
4
|
2,00E-06
|
40
|
0
|
40
|
80
|
160
|
1,0E-06
|
|
P1
|
B11
|
2
|
Изпитван химикал 1
|
TC1
|
4
|
2,00E-06
|
40
|
0
|
40
|
80
|
160
|
1,0E-06
|
|
P1
|
B12
|
3
|
Изпитван химикал 1
|
TC1
|
4
|
2,00E-06
|
40
|
0
|
40
|
80
|
160
|
1,0E-06
|
|
P1
|
C1
|
1
|
Изпитван химикал 1
|
TC1
|
5
|
2,00E-07
|
40
|
0
|
40
|
80
|
160
|
1,0E-07
|
|
P1
|
C2
|
2
|
Изпитван химикал 1
|
TC1
|
5
|
2,00E-07
|
40
|
0
|
40
|
80
|
160
|
1,0E-07
|
|
P1
|
C3
|
3
|
Изпитван химикал 1
|
TC1
|
5
|
2,00E-07
|
40
|
0
|
40
|
80
|
160
|
1,0E-07
|
|
P1
|
C4
|
1
|
Изпитван химикал 1
|
TC1
|
6
|
2,00E-08
|
40
|
0
|
40
|
80
|
160
|
1,0E-08
|
|
P1
|
C5
|
2
|
Изпитван химикал 1
|
TC1
|
6
|
2,00E-08
|
40
|
0
|
40
|
80
|
160
|
1,0E-08
|
|
P1
|
C6
|
3
|
Изпитван химикал 1
|
TC1
|
6
|
2,00E-08
|
40
|
0
|
40
|
80
|
160
|
1,0E-08
|
|
P1
|
C7
|
1
|
Изпитван химикал 1
|
TC1
|
7
|
2,00E-09
|
40
|
0
|
40
|
80
|
160
|
1,0E-09
|
|
P1
|
C8
|
2
|
Изпитван химикал 1
|
TC1
|
7
|
2,00E-09
|
40
|
0
|
40
|
80
|
160
|
1,0E-09
|
|
P1
|
C9
|
3
|
Изпитван химикал 1
|
TC1
|
7
|
2,00E-09
|
40
|
0
|
40
|
80
|
160
|
1,0E-09
|
|
P1
|
C10
|
1
|
Изпитван химикал 1
|
TC1
|
8
|
2,00E-10
|
40
|
0
|
40
|
80
|
160
|
1,0E-10
|
|
P1
|
C11
|
2
|
Изпитван химикал 1
|
TC1
|
8
|
2,00E-10
|
40
|
0
|
40
|
80
|
160
|
1,0E-10
|
|
P1
|
C12
|
3
|
Изпитван химикал 1
|
TC1
|
8
|
2,00E-10
|
40
|
0
|
40
|
80
|
160
|
1,0E-10
|
|
P1
|
D1
|
1
|
Изпитван химикал 2
|
TC2
|
1
|
2,00E-03
|
40
|
0
|
40
|
80
|
160
|
1,0E-03
|
|
P1
|
D2
|
2
|
Изпитван химикал 2
|
TC2
|
1
|
2,00E-03
|
40
|
0
|
40
|
80
|
160
|
1,0E-03
|
|
P1
|
D3
|
3
|
Изпитван химикал 2
|
TC2
|
1
|
2,00E-03
|
40
|
0
|
40
|
80
|
160
|
1,0E-03
|
|
P1
|
D4
|
1
|
Изпитван химикал 2
|
TC2
|
2
|
2,00E-04
|
40
|
0
|
40
|
80
|
160
|
1,0E-04
|
|
P1
|
D5
|
2
|
Изпитван химикал 2
|
TC2
|
2
|
2,00E-04
|
40
|
0
|
40
|
80
|
160
|
1,0E-04
|
|
P1
|
D6
|
3
|
Изпитван химикал 2
|
TC2
|
2
|
2,00E-04
|
40
|
0
|
40
|
80
|
160
|
1,0E-04
|
|
P1
|
D7
|
1
|
Изпитван химикал 2
|
TC2
|
3
|
2,00E-05
|
40
|
0
|
40
|
80
|
160
|
1,0E-05
|
|
P1
|
D8
|
2
|
Изпитван химикал 2
|
TC2
|
3
|
2,00E-05
|
40
|
0
|
40
|
80
|
160
|
1,0E-05
|
|
P1
|
D9
|
3
|
Изпитван химикал 2
|
TC2
|
3
|
2,00E-05
|
40
|
0
|
40
|
80
|
160
|
1,0E-05
|
|
P1
|
D10
|
1
|
Изпитван химикал 2
|
TC2
|
4
|
2,00E-06
|
40
|
0
|
40
|
80
|
160
|
1,0E-06
|
|
P1
|
D11
|
2
|
Изпитван химикал 2
|
TC2
|
4
|
2,00E-06
|
40
|
0
|
40
|
80
|
160
|
1,0E-06
|
|
P1
|
D12
|
3
|
Изпитван химикал 2
|
TC2
|
4
|
2,00E-06
|
40
|
0
|
40
|
80
|
160
|
1,0E-06
|
|
P1
|
E1
|
1
|
Изпитван химикал 2
|
TC2
|
5
|
2,00E-07
|
40
|
0
|
40
|
80
|
160
|
1,0E-07
|
|
P1
|
E2
|
2
|
Изпитван химикал 2
|
TC2
|
5
|
2,00E-07
|
40
|
0
|
40
|
80
|
160
|
1,0E-07
|
|
P1
|
E3
|
3
|
Изпитван химикал 2
|
TC2
|
5
|
2,00E-07
|
40
|
0
|
40
|
80
|
160
|
1,0E-07
|
|
P1
|
E4
|
1
|
Изпитван химикал 2
|
TC2
|
6
|
—
|
40
|
0
|
40
|
80
|
160
|
1,0E-08
|
|
P1
|
E5
|
2
|
Изпитван химикал 2
|
TC2
|
6
|
—
|
40
|
0
|
40
|
80
|
160
|
1,0E-08
|
|
P1
|
E6
|
3
|
Изпитван химикал 2
|
TC2
|
6
|
—
|
40
|
0
|
40
|
80
|
160
|
1,0E-08
|
|
P1
|
E7
|
1
|
Изпитван химикал 2
|
TC2
|
7
|
2,00E-06
|
40
|
0
|
40
|
80
|
160
|
1,0E-09
|
|
P1
|
E8
|
2
|
Изпитван химикал 2
|
TC2
|
7
|
2,00E-06
|
40
|
0
|
40
|
80
|
160
|
1,0E-09
|
|
P1
|
E9
|
3
|
Изпитван химикал 2
|
TC2
|
7
|
2,00E-06
|
40
|
0
|
40
|
80
|
160
|
1,0E-09
|
|
P1
|
E10
|
1
|
Изпитван химикал 2
|
TC2
|
8
|
2,00E-06
|
40
|
0
|
40
|
80
|
160
|
1,0E-10
|
|
P1
|
E11
|
2
|
Изпитван химикал 2
|
TC2
|
8
|
2,00E-06
|
40
|
0
|
40
|
80
|
160
|
1,0E-10
|
|
P1
|
E12
|
3
|
Изпитван химикал 2
|
TC2
|
8
|
2,00E-06
|
40
|
0
|
40
|
80
|
160
|
1,0E-10
|
|
P1
|
F1
|
1
|
Изпитван химикал 3
|
TC3
|
1
|
2,00E-03
|
40
|
0
|
40
|
80
|
160
|
1,0E-03
|
|
P1
|
F2
|
2
|
Изпитван химикал 3
|
TC3
|
1
|
2,00E-03
|
40
|
0
|
40
|
80
|
160
|
1,0E-03
|
|
P1
|
F3
|
3
|
Изпитван химикал 3
|
TC3
|
1
|
2,00E-03
|
40
|
0
|
40
|
80
|
160
|
1,0E-03
|
|
P1
|
F4
|
1
|
Изпитван химикал 3
|
TC3
|
2
|
2,00E-04
|
40
|
0
|
40
|
80
|
160
|
1,0E-04
|
|
P1
|
F5
|
2
|
Изпитван химикал 3
|
TC3
|
2
|
2,00E-04
|
40
|
0
|
40
|
80
|
160
|
1,0E-04
|
|
P1
|
F6
|
3
|
Изпитван химикал 3
|
TC3
|
2
|
2,00E-04
|
40
|
0
|
40
|
80
|
160
|
1,0E-04
|
|
P1
|
F7
|
1
|
Изпитван химикал 3
|
TC3
|
3
|
2,00E-05
|
40
|
0
|
40
|
80
|
160
|
1,0E-05
|
|
P1
|
F8
|
2
|
Изпитван химикал 3
|
TC3
|
3
|
2,00E-05
|
40
|
0
|
40
|
80
|
160
|
1,0E-05
|
|
P1
|
F9
|
3
|
Изпитван химикал 3
|
TC3
|
3
|
2,00E-05
|
40
|
0
|
40
|
80
|
160
|
1,0E-05
|
|
P1
|
F10
|
1
|
Изпитван химикал 3
|
TC3
|
4
|
2,00E-06
|
40
|
0
|
40
|
80
|
160
|
1,0E-06
|
|
P1
|
F11
|
2
|
Изпитван химикал 3
|
TC3
|
4
|
2,00E-06
|
40
|
0
|
40
|
80
|
160
|
1,0E-06
|
|
P1
|
F12
|
3
|
Изпитван химикал 3
|
TC3
|
4
|
2,00E-06
|
40
|
0
|
40
|
80
|
160
|
1,0E-06
|
|
P1
|
G1
|
1
|
Изпитван химикал 3
|
TC3
|
5
|
2,00E-07
|
40
|
0
|
40
|
80
|
160
|
1,0E-07
|
|
P1
|
G2
|
2
|
Изпитван химикал 3
|
TC3
|
5
|
2,00E-07
|
40
|
0
|
40
|
80
|
160
|
1,0E-07
|
|
P1
|
G3
|
3
|
Изпитван химикал 3
|
TC3
|
5
|
2,00E-07
|
40
|
0
|
40
|
80
|
160
|
1,0E-07
|
|
P1
|
G4
|
1
|
Изпитван химикал 3
|
TC3
|
6
|
2,00E-08
|
40
|
0
|
40
|
80
|
160
|
1,0E-08
|
|
P1
|
G5
|
2
|
Изпитван химикал 3
|
TC3
|
6
|
2,00E-08
|
40
|
0
|
40
|
80
|
160
|
1,0E-08
|
|
P1
|
G6
|
3
|
Изпитван химикал 3
|
TC3
|
6
|
2,00E-08
|
40
|
0
|
40
|
80
|
160
|
1,0E-08
|
|
P1
|
G7
|
1
|
Изпитван химикал 3
|
TC3
|
7
|
2,00E-09
|
40
|
0
|
40
|
80
|
160
|
1,0E-09
|
|
P1
|
G8
|
2
|
Изпитван химикал 3
|
TC3
|
7
|
2,00E-09
|
40
|
0
|
40
|
80
|
160
|
1,0E-09
|
|
P1
|
G9
|
3
|
Изпитван химикал 3
|
TC3
|
7
|
2,00E-09
|
40
|
0
|
40
|
80
|
160
|
1,0E-09
|
|
P1
|
G10
|
1
|
Изпитван химикал 3
|
TC3
|
8
|
2,00E-10
|
40
|
0
|
40
|
80
|
160
|
1,0E-10
|
|
P1
|
G11
|
2
|
Изпитван химикал 3
|
TC3
|
8
|
2,00E-10
|
40
|
0
|
40
|
80
|
160
|
1,0E-10
|
|
P1
|
G12
|
3
|
Изпитван химикал 3
|
TC3
|
8
|
2,00E-10
|
40
|
0
|
40
|
80
|
160
|
1,0E-10
|
|
P1
|
H1
|
1
|
норетинодрел
|
NE
|
|
IC50
|
40
|
0
|
40
|
80
|
160
|
|
|
P1
|
H2
|
2
|
норетинодрел
|
NE
|
|
IC50
|
40
|
0
|
40
|
80
|
160
|
|
|
P1
|
H3
|
3
|
норетинодрел
|
NE
|
|
IC50
|
40
|
0
|
40
|
80
|
160
|
|
|
P1
|
H4
|
1
|
норетинодрел
|
NE
|
|
1.00E-4.5
|
40
|
0
|
40
|
80
|
160
|
|
|
P1
|
H5
|
2
|
норетинодрел
|
NE
|
|
1.00E-4.5
|
40
|
0
|
40
|
80
|
160
|
|
|
P1
|
H6
|
3
|
норетинодрел
|
NE
|
|
1.00E-4.5
|
40
|
0
|
40
|
80
|
160
|
|
|
P1
|
H7
|
1
|
студен E2 S
|
|
|
IC50
|
40
|
0
|
40
|
80
|
160
|
|
|
P1
|
H8
|
2
|
студен E2 S
|
|
|
IC50
|
40
|
0
|
40
|
80
|
160
|
|
|
P1
|
H9
|
3
|
студен E2 S
|
|
|
IC50
|
40
|
0
|
40
|
80
|
160
|
|
|
P1
|
H10
|
1
|
студен E2 S
|
|
|
1,00 E-7
|
40
|
0
|
40
|
80
|
160
|
|
|
P1
|
H11
|
2
|
студен E2 S
|
|
|
1,00 E-7
|
40
|
0
|
40
|
80
|
160
|
|
|
P1
|
H12
|
3
|
студен E2 S
|
|
|
1,00 E-7
|
40
|
0
|
40
|
80
|
160
|
|
Допълнение 3
ИЗСЛЕДВАНЕ IN VITRO ЗА СВЪРЗВАНЕ НА ЕСТРОГЕНЕН РЕЦЕПТОР НА ИЗСЛЕДОВАТЕЛСКИЯ ИНСТИТУТ ЗА ОЦЕНКА НА ХИМИКАЛИ (CERI), ИЗПОЛЗВАЩО ЧОВЕШКИ РЕКОМБИНАНТЕН ERΑ ЛИГАНД-СВЪРЗВАЩ ДОМЕЙН НА ПРОТЕИН
ПЪРВОНАЧАЛНИ СЪОБРАЖЕНИЯ И ОГРАНИЧЕНИЯ (ВЖ. СЪЩО „ОБЩО ВЪВЕДЕНИЕ“)
|
1.
|
Това изследване in vitro на насищането на естрогенен рецептор (ERα) и на конкурентното свързване използва лиганд-свързващ домейн (LBD) на човешкия ERα (hrERα). Този модел на протеин е произведен от Изследователския институт за оценка на химикали (CERI), Япония, и съществува като глутатион-S-трансфераза (GST) фузионен протеин, който се експресира в E. coli. Протоколът на CERI е преминал през международно многолабораторно изследването за валидиране (2), което е доказало неговата относимост и надеждност за целта на изследването.
|
|
2.
|
Това изследване представлява скринингова процедура за идентифициране на вещества, които могат да се свързват с hrERα. То се използва за определяне на способността на даден изпитван химикал да се конкурира със 17β-естрадиол за свързване с hrERα-LBD. Количествените резултати на изследването могат да включват IC50 (мярка за концентрацията на изпитван химикал, необходима за изместване на половината от [3H]-17β-естрадиол отhrERα) и относителните афинитети на свързване на изпитвани химикали за hrERα в сравнение със 17β-естрадиол. За целите на химичен скрининг приемливите качествени резултати на изследването могат да включват класификации на изпитвани химикали като свързващи hrERα, не свързващи или нееднозначни въз основа на критериите, описани за кривите на свързване.
|
|
3.
|
Изследването използва радиоактивен лиганд, който изисква лабораторията да притежава лиценз за радиоактивни материали. Всички процедури с радиоизотопи и опасни химикали следва да спазват разпоредбите и процедурите, както са описани от националното законодателство.
|
|
4.
|
Разделите „ОБЩО ВЪВЕДЕНИЕ“ и „КОМПОНЕНТИ НА ИЗСЛЕДВАНЕТО СВЪРЗВАНЕ НА hrER“ следва да бъдат прочетени, преди да се използва настоящото изследване за регулаторни цели. Използваните в настоящите TG определения и съкращения са описани в Допълнение 1.
|
ПРИНЦИП НА ИЗСЛЕДВАНЕТО (ВЖ. СЪЩО „ОБЩО ВЪВЕДЕНИЕ“)
|
5.
|
Изследването за свързване с hrERα измерва способността на радиомаркиран лиганд ([3H]17β-естрадиол) да се свързва с ЕР в присъствието на нарастващи концентрации на изпитван химикал (т.е. конкурент). Изпитваните химикали, които имат висок афинитет към ЕР, се конкурират с радиомаркирания лиганд при по-ниска концентрация в сравнение с химикалите с по-нисък афинитет към рецептора.
|
|
6.
|
Това изследване се състои от два основни компонента: експеримент за свързване до насищане с цел характеризиране на параметрите на взаимодействието рецептор – лиганд, последван от експеримент за конкурентно свързване, който характеризира конкуренцията между изпитван химикал и радиомаркиран лиганд за свързване към ЕР.
|
|
7.
|
Целта на експеримента за свързване до насищане е да характеризира специална партида рецептори за афинитет на свързване и брой при подготовка за експеримента на конкурентно свързване. Експериментът за свързване до насищане измерва, при равновесни условия, афинитета на фиксирана концентрация на естрогенния рецептор за неговия естествен лиганд (представен от дисоциационна константа, Kd) и концентрацията на активните места на рецептора (Bmax).
|
|
8.
|
Експериментът за свързване до насищане измерва афинитета на дадено вещество да се конкурира с [3H]17β-естрадиол за свързване с ЕР. Афинитетът се определя количествено от концентрацията на изпитван химикал, която, при равновесие, инхибира 50 % от специфичното свързване на [3H]17β-естрадиол (наричана „инхибиторна концентрация 50 %“ или IC50). Това може да се оцени и с използване на относителния афинитет на свързване (RBA, по отношение на IC50 на естрадиол, измерен отделно в същата серия). Експериментът за конкурентно свързване измерва свързването на [3H]17β-естрадиол при фиксирана концентрация в присъствието на широк диапазон (осем порядъка) на концентрации на изпитван химикал. След това данните се вписват, където е възможно, във форма на уравнение на Хил (Хил, 1910), което описва изместването на радиолиганда от конкурентно свързващо вещество на едно място. Степента на изместване на радиомаркирания естрадиол при равновесие се използва за характеризиране на изпитвания химикал като свързващ, несвързващ или предизвикващ нееднозначен отклик.
|
ПРОЦЕДУРА
Демонстриране на приемлива ефективност на hrERα протеин
|
9.
|
Преди рутинно провеждане на изследвания за насищане и конкурентно свързване, всяка нова партида hrERα следва да демонстрира правилно функциониране в лабораторията, в която ще се използва. За демонстриране на ефективност следва да се използва двустъпков процес. Тези стъпки са следните:
|
—
|
Проведете изследване за насищане и свързване на 3H]-17β-естрадиол, за да се демонстрира специфичност и насищане на hrERα. Анализът на нелинейна регресия на тези данни (напр. BioSoft; McPherson, 1985 г.; Motulsky, 1995) и последващата диаграма на Scatchard следва да документират афинитета на свързване с hrERα на [3H]-17β-естрадиол (Kd) и броя на рецепторите (Bmax) за определена партида на hrERα.
|
|
—
|
Проведете изследване за конкурентно свързване с използване на контролните вещества (референтен естроген (17β-естрадиол), слабо свързващо вещество (напр. норетинодрел или норетиндрон) и несвързващо вещество (октилтриетоксисилан, OTES). Всяка лаборатория следва да определи база данни от предходни периоди за документиране на последователността на IC50 и съответните стойности за референтния естроген и слабо свързващо вещество измежду експериментите и различните партиди на hrERα. Освен това параметрите на кривите на конкурентно свързване за контролните вещества следва да са в границите на 95 %-я доверителен интервал (вж. Таблица 1), които са разработени с използване на данни от лаборатории, които са участвали във изследването за валидиране на това изследване (2).
|
Таблица 1
Критерии за ефективност, разработени за референтния естроген и слабо свързващо вещество, изследване за свързване на hrER на CERI
|
Вещество
|
Параметър
|
Средно (10)
|
Стандартно отклонение(n)
|
95 %-и доверителни интервали (11)
|
|
Долна граница
|
Горна граница
|
|
17β-естрадиол
|
Top
|
104,74
|
13,12 (70)
|
101,6
|
107,9
|
|
Bottom
|
0,85
|
2,41 (70)
|
0,28
|
1,43
|
|
HillSlope
|
–1,22
|
0,20 (70)
|
–1,27
|
–1,17
|
|
LogIC50
|
–8,93
|
0,23 (70)
|
–8,98
|
–8,87
|
|
Норетинодрел
|
Top
|
101,31
|
10,55 (68)
|
98,76
|
103,90
|
|
Bottom
|
2,39
|
5,01 (68)
|
1,18
|
3,60
|
|
HillSlope
|
–1,04
|
0,21 (68)
|
–1,09
|
–0,99
|
|
LogIC50
|
–6,19
|
0,40 (68)
|
–6,29
|
–6,10
|
|
Норетиндрон (12)
|
Top
|
92,27
|
7,79 (23)
|
88,90
|
95,63
|
|
Bottom
|
16,52
|
10,59 (23)
|
11,94
|
21,10
|
|
Наклон на Хил
|
–1,18
|
0,32 (23)
|
–1,31
|
–1,04
|
|
LogIC50
|
–6,01
|
0,54 (23)
|
–6,25
|
–5,78
|
|
Доказване на пригодността на лабораторията
|
10.
|
Вж. точки 17 и 18 и таблица 2 в „КОМПОНЕНТИ НА ИЗСЛЕДВАНЕТО ЗА СВЪРЗВАНЕ КЪМ hrER“ от настоящия метод на изпитване. Всяко изследване (на насищане и конкурентно свързване) следва да се състои от три независими серии (т.е. с пресни разредени разтвори на рецептор, химикали и реактиви) в различни дни и всяка серия следва да съдържа три повторения.
|
Определяне на концентрацията на рецептора (hrERα)
|
11.
|
Концентрацията на активния рецептор леко варира според партидата и условията на съхранение. Поради тази причина концентрацията на активния рецептор следва да се определи така, както е получена от доставчика. Това ще доведе до подходящата концентрация на активния рецептор към момента на провеждане на серията.
|
|
12.
|
При условия, отговарящи на конкурентно свързване (т.е. 0,5 nM [3H]-естрадиол), номинални концентрации на рецептор от 0,1, 0,2, 0,4 и 0,6 nM следва да се инкубират в отсъствието (общо свързване) и присъствието (неспецифично свързване) на 1 μM немаркиран естрадиол. Специфичното свързване, изчислено като разликата между общото и неспецифичното свързване, се изчертава като зависимост спрямо номиналната концентрация на рецептора. Концентрацията на рецептор, която дава стойности на специфично свързване, съответстващи на 40 % от добавения радиомаркер, е свързана със съответната концентрация на рецептора и тази концентрация на рецептора следва да се използва в опитите за насищане и конкурентно свързване. Често на това условие отговаря окончателна концентрация на hrER от 0,2 nM.
|
|
13.
|
Ако критерият за 40 % не може да бъде удовлетворен, следва да се провери настройката на експеримента за възможни грешки. Невъзможността за постигане на критерия за 40 % може да сочи, че в рекомбинантната партида има много малко количество активен рецептор и тогава следва да се обмисли използването на друга партида рецептор.
|
Изследване на насищане
|
14.
|
Следва да се оценят три пъти осем увеличаващи се концентрации на [3H]17β-естрадиол при спазване на следните три условия (вж. Таблица 2):
|
1.
|
В отсъствието на немаркиран 17β-естрадиол и присъствието на ЕР. Това е определянето на тоталното свързване чрез измерване на радиоактивността в ямките, в които има само [3H]17β-естрадиол.
|
|
2.
|
В присъствието на 2000 пъти по-голяма концентрация на немаркиран 17β-естрадиол спрямо маркиран 17β-естрадиол и присъствието на ЕР. Целта на това условие да се наситят местата на активно свързване с немаркиран 17β-естрадиол и чрез измерване на радиоактивността в тези ямки да се определи неспецифичното свързване. Всяко оставащо количество горещ естрадиол, който може да се свърже с рецептора, се счита за свързан в неспецифично място, тъй като студеният естрадиол следва да е с такава висока концентрация, при която той се свързва с всички налични специфични места на рецептора.
|
|
3.
|
В отсъствието на немаркиран 17β-естрадиол и отсъствието на ЕР (определяне на обща радиоактивност)
|
Приготвяне на [3H]-17β-естрадиол, разтвори на немаркиран 17β-естрадиол и hrERα
|
15.
|
40 nM разтвор на [3H]-17β-естрадиол следва да се приготви от 1 μM изходен разтвор на [3H]-17β-естрадиол в DMSO чрез добавяне на DMSO (за приготвяне на 200 nM) и буфер, използван за изследването, при стайна температура (за приготвяне на 40 nM). С използване на този 40 nM разтвор се приготвят сериите на разреден [3H]-17β-естрадиол, вариращи от 0,313 nM до 40 nM, с буфер, използван за изследването, при стайна температура (както е представено в ред 12 на Таблица 2). Окончателните концентрации на изследването, вариращи от 0,0313 до 4,0 nM, се получават чрез добавяне на 10 μl от тези разтвори към съответните ямки на 96-ямкова микротитърна плака (вж. Таблици 2 и 3). Приготвянето на буфера, използван в изследването, изчисляването на оригиналния изходен разтвор на [3H]-17β-естрадиол въз основа на неговата специфична активност, приготвянето на разредените разтвори и определянето на концентрациите са описани подробно в протокола на CERI (2).
|
|
16.
|
Разредените разтвори на немаркиран 17β-естрадиол следва да се приготвят от 1 nM изходен разтвор на 17β-естрадиол чрез добавяне на буфер, използван в изследването, за да се постигнат осем увеличаващи се концентрации, които първоначално варират от 0,625 μM до 80 μM. Окончателните концентрации на изследването, вариращи от 0,0625 до 8 μM, се получават чрез добавяне на 10 μl от тези разтвори към съответните ямки на 96-ямкова микротитърна плака, предназначена за измерване на неспецифично свързване (вж. Таблици 2 и 3). Приготвянето на разредени разтвори на немаркиран 17β-естрадиол е описано подробно в протокола на CERI (2).
|
|
17.
|
Следва да се използва концентрацията на рецептор, който дава 40±10 % специфично свързване (вж. точки 12-13). Разтворът на hrERα следва да се приготви с охладен с лед буфер, използван в изследването, непосредствено преди употреба, т.е. след като са приготвени всички ямки за общо свързване, неспецифично свързване и самостоятелен горещ лиганд.
|
|
18.
|
96-ямковите микротитърни плаки се приготвят както е показано в Таблица 2, с 3 повторения за всяка концентрация на [3H]-17β-естрадиол. Определянето на обема на [3H]-17β-естрадиол, немаркиран 17β-естрадиол, буфера и рецептора е дадено в Таблица 3.
Таблица 2
Схема на микротитърна плака за изследване за свързване до насищане
|
|
1 (*10)
|
2 (*10)
|
3 (*10)
|
4 (*10)
|
5 (*10)
|
6 (*10)
|
7 (*10)
|
8 (*10)
|
9 (*10)
|
10
|
11 (*11)
|
12 (*11)
|
|
За измерване на ТВ
|
За измерване на NSB
|
За определяне на самостоятелен горещ лиганд
|
|
Немаркирани разредени разтвори на Е2 за колони 4-6 на плаката
|
[3H]E2 разредени разтвори за колони 1-9 на плаката
|
|
А
|
0,0313 nM [3H] E2+ ЕР
|
0,0313 nM [3H] E2 + 0,0625 μМ E2+ ЕР
|
0,0313 nM
|
|
0,625 μМ
|
0,313 nM
|
|
Б
|
0,0625 nM [3H] E2+ ЕР
|
0,0625 nM [3H] E2+ 0,125 μМ E2+ ЕР
|
0,0625 nM
|
|
1,25 μМ
|
0,625 nM
|
|
В
|
0,125 nM [3H] E2+ ЕР
|
0,125 nM [3H] E2+ 0,25 μМ E2+ ЕР
|
0,125 nM
|
|
2,5 μМ
|
1,25 nM
|
|
Г
|
0,250 nM [3H] E2+ ЕР
|
0,250 nM [3H] E2+ 0,5 μМ E2+ ЕР
|
0,250 nM
|
|
5 μМ
|
2,5 nM
|
|
Д
|
0,50 nM [ H] E2+ ЕР
|
0,50 nM [3H] E2+ 1 μМ E2+ ЕР
|
0,50 nM
|
|
10 μМ
|
5 nM
|
|
Е
|
1,00 nM [3H] E2+ ЕР
|
1,00 nM [3H] E2+ 2 μМ E2+ ЕР
|
1,00 nM
|
|
20 μМ
|
10 nM
|
|
Ж
|
2,00 nM [3H] E2+ ЕР
|
2,00 nM [3H] E2+ 4 μМ E2+ ЕР
|
2,00 nM
|
|
40 μМ
|
20 nM
|
|
З
|
4,00 nM [3H] E2+ ЕР
|
4,00 nM [3H] E2+ 8 μМ E2+ ЕР
|
4,00 nM
|
|
80 μМ
|
40 nM
|
|
TB
|
:
|
общо свързване
|
|
NSB
|
:
|
неспецифично свързване
|
|
[3H] E2
|
:
|
[3H]17β-естрадиол
|
|
E2
|
:
|
немаркиран 17β-естрадиол
|
|
Таблица 3
Обеми на реактив за микротитърна плака за насищане
|
Номер на ред
|
1
|
2
|
3
|
4
|
5
|
6
|
7 (*12)
|
8 (*12)
|
9 (*12)
|
|
Подготвителни операции
|
Ямки за TB
|
Ямки за NSB
|
Самостоятелен горещ лиганд
|
|
Обем на компоненти за ямките за реакция отгоре и ред на добавяне
|
Буфер
|
60 μl
|
50 μl
|
90 μl
|
|
немаркиран E2 от ред 11 в Таблица 2
|
–
|
10 μl
|
–
|
|
[3H]E2 от ред 12 в Таблица 2
|
10 μl
|
10 μl
|
10 μl
|
|
hrERα
|
30 μl
|
30 μl
|
–
|
|
Общ обем на реакция
|
100 μl
|
100 μl
|
100 μl
|
|
Инкубация
|
РЕАКЦИЯ СЛЕД 2 ЧАСА ИНКУБАЦИЯ
|
Количествено определяне на радиоактивността веднага след приготвяне. Без инкубация
|
|
Третиране с 0,4 % DCC
|
Идентификационен номер 1.1.6.8. Не
|
Идентификационен номер 1.1.6.8. Не
|
№
|
|
Обем на 0,4 % DCC
|
100 μl
|
100 μl
|
–
|
|
Филтриране
|
Идентификационен номер 1.1.6.8. Не
|
Идентификационен номер 1.1.6.8. Не
|
№
|
|
ИЗМЕРВАНЕ НА DPMS
|
|
Количествено определяне на обема, добавян към сцинтилационен коктейл
|
100 μl (*13)
|
100 μl (*13)
|
50 μl
|
|
|
19.
|
Микротитърните плаки на изследването за определянето на общо свързване и неспецифично свързване следва да се инкубират при стайна температура (от 22°C до 28°C) в продължение на два часа.
|
|
Измерване на [3H]-17β-естрадиол, свързан с hrERα
|
20.
|
След двучасов период на инкубация, свързаният с hrERα [3H]-17β-естрадиол следва да се отдели от свободния [3H]-17β-естрадиол чрез добавяне на 100μl охладена с лед 0,4 % суспензия на DCC в ямките. След това плаките следва да се поставят върху лед за 10 минути и реакционната смес и суспензията на DCC следва да се филтрират чрез прехвърляне във филтър за микротитърна плака, за отстраняване на DCC. 100 μl от филтрата след това трябва да се добави към сцинтилационна течност в LSC флакони за определяне на разпадането в минута (dpms) на флакон чрез течно сцинтилационно броене.
|
|
21.
|
Като алтернатива, ако няма наличен филтър за микроплака, отстраняването на DCC може да се постигне чрез центрофугиране. 50 μl от супернатанта, съдържащ свързан с hrERα [3H]-17β-естрадиол, след това следва да се отдели изключително внимателно, за да се избегне всяко замърсяване на ямките от контакт с DCC, и трябва да се използва за сцинтилационно броене.
|
|
22.
|
Условието за самостоятелен горещ лиганд се използва за определяне на разпадането в минута (dpm) на [3H]-17β-естрадиол, добавен в анализираните ямки. Радиоактивността следва да се определи количествено веднага след приготвяне. Тези ямки не следва да се инкубират и не трябва да се третират със суспензия на DCC, но тяхното съдържание следва да се прехвърли директно в сцинтилационната течност. Тези измервания демонстрират колко [3H]-17β-естрадиол в dpms е добавен към всеки набор от ямки за общото свързване и неспецифичното свързване.
|
Изследване на конкурентно свързване
|
23.
|
Изследването за конкурентно свързване измерва свързването на една концентрация на [3H]-17β-естрадиол в присъствието на увеличаващи се концентрации на изпитван химикал. За всяка концентрация в рамките на една серия следва да се използват три паралелни повторения. Освен това следва да се проведат три непаралелни серии за всеки изпитван химикал. Изследването трябва да се проведе в една или повече 96-ямкови микротитърни плаки.
|
Контрол
|
24.
|
При провеждане на изследването във всеки експеримент следва да се включват паралелни разтворител и контроли (т.е. референтен естроген, слабо свързващо вещество и несвързващо вещество). Пълните криви на концентрация за референтния естроген и контролите (т.е. слабо свързващо вещество и несвързващо вещество) следва да се използват в една плака по време на всяка серия. Всички останали плаки следва да съдържат (i) висока (максимално изместване, т.е. приблизително пълно изместване на радиомаркиран лиганд) и средна (приблизително IC50) концентрация на E2 и слабо свързващо вещество в три повторения; (ii) контрола на разтворителя и неспецифичното свързване, всяка в три повторения. Процедурите за приготвяне на използвания в изследването буфер, [3H]-17β-естрадиол, hrERα и разтворите на изпитван химикал са описани подробно в протокола на CERI (2).
|
Контрола на разтворител:
|
25.
|
Контролата на разтворителя показва, че разтворителят не взаимодейства с изпитваната система и измерва също така общото свързване (ТВ). Предпочитаният разтворител е DMSO. Като алтернатива, ако най-високата концентрация на изпитвания химикал не е разтворима в DMSO, може да се използва етанол. Концентрацията на DMSO в окончателните анализирани ямки следва да е 2,05 % и може да бъде увеличена до 2,5 % в случай на липса на разтворимост на изпитвания химикал. Концентрации на DMSO над 2,5 % не следва да се използват поради интерференцията на по-високи концентрации на разтворителя с изследването. За изпитвани химикали, които не са разтворими в DMSO, но са разтворими в етанол, може да се използва максимум 2 % етанол в изследването без интерференция.
|
Контрола на буфер:
|
26.
|
Контролата на буфера (ВС) не трябва да съдържа нито разтворител, нито изпитван химикал, но трябва да включва всички други компоненти на изследването. Резултатите на контролата на буфера се сравняват с контролата на разтворителя, за да провери, че използваният разтворител не влияе върху системата на изследването.
|
Силно свързващо вещество (референтен естроген)
|
27.
|
17β-естрадиол (CAS № 50-28-2) е ендогенен лиганд и се свързва с висок афинитет с ЕР, подтип алфа. За всяко изследване на конкурентно свързване на hrERα следва да се приготви стандартна крива с използване на 17β-естрадиол, за да се позволи оценка на променливостта при провеждане на изследването във времето в рамките на една и съща лаборатория. Следва да се приготвят осем разтвора на немаркиран 17β-естрадиол в DMSO и използвания в изследването буфер, като окончателните концентрации в ямките, които се използват за окончателното разполагане на стандартната крива, са следните: 10–6, 10–7, 10–8, 10–8,5, 10–9, 10–9,5, 10–10, 10–11 M. Най–високата концентрация на немаркиран 17β–естрадиол (1 μM) следва да служи като индикатор за неспецифично свързване. Тази концентрация се разграничава чрез етикета „NSB“ в Таблица 4, въпреки че също е част от стандартната крива.
|
Слабо свързващо вещество
|
28.
|
Следва да се включи слабо свързващо вещество (норетинодрел (CAS № 68-23-5) или, като алтернатива, норетиндрон (CAS № 68-22-4)), за да докаже чувствителността на всеки експеримент и да се позволи оценка на променливостта при провеждане на изследването във времето. Следва да се приготвят осем разтвора на слабото свързващо вещество в DMSO и използвания в изследването буфер, като окончателните концентрации в ямките са следните: 10–4.5, 10–5,5, 10–6, 10–6,5, 10–7, 10–7,5, 10–8 и 10–9 M.
|
Несвързващо вещество
|
29.
|
Като отрицателна контрола (несвързващо вещество) следва да се използва октитриетоксисилан (OTES, CAS № 2943-75-1). Той гарантира, че изследването, както се провежда, открива изпитвани химикали, които не се свързват с hrERα. Следва да се приготвят осем разтвора на несвързващото вещество в DMSO и използвания в изследването буфер, като окончателните концентрации в ямките са следните: 10–3,10–4, 10–5, 10–6, 10–7, 10–8, 10–9, 10–10 M. Ди-ен-бутилфталат (DBP, CAS № 84-72-2) може да се използва като алтернативно несвързващо вещество, но да се изпитва само до 10–4M. Доказано е, че максималната разтворимост на DBP в изследването е 10–4M.
|
Концентрация на hrERα
|
30.
|
Следва да се използва това количество на рецептор, което дава специфично свързвате от 40±10 % (вж. точки 12-13 на Допълнение 3). Разтворът на hrERα следва да се приготви чрез разтваряне на функционалния hrERα в охладен с лед буфер, използван в изследването, непосредствено преди употреба.
|
[3H]-17β-естрадиол
|
31.
|
Окончателната концентрация на [3H]-17β-естрадиол в анализираните ямки следва да е 0,5 nM.
|
Изпитвани химикали
|
32.
|
На първо място е необходимо да се проведе тест за разтворимост, за да се определи границата на разтворимост за всеки изпитван химикал и да се идентифицира подходящия диапазон на концентрация за използване при изпълнение на протокола на изпитването. Границата на разтворимост на всеки изпитван химикал трябва първо да се определи в разтворителя и след това допълнително да се потвърди при условията на изследването. Окончателната концентрация, изпитвана в изследването, н следва да превишава 1mM. Изпитването за определяне на диапазон включва контрола на разтворител заедно с поне 8 логаритмични последователни разреждания, започвайки с максималната приемлива концентрация (напр. 1 mM или по-ниска, въз основа на границата на разтворимост), в присъствието на забелязана мътност или преципитация (вж. също точка 35 на Допълнение 3). След определяне на диапазона на концентрация за изпитването, даден изпитван химикал следва да се изпитва с използване на 8 логаритмични концентрации, разпределени по подходящ начин, както е определено в предходното изпитване за определяне на диапазон. Концентрациите, изпитвани във втория и третия експеримент, следва допълнително да се коригират, както е подходящо, за да характеризират по-добре кривата на отклик на концентрация, ако е необходимо.
|
|
33.
|
Разрежданията на изпитвания химикал следва да се приготвят в подходящия разтворител (вж. точка 25 на Допълнение 3). Ако най-високата концентрация на изпитвания химикал не е разтворима в DMSO или етанол и добавянето на още разтворител би довело до това концентрацията на разтворителя в окончателната епруветка да е по-голяма от приемливата граница, най-високата концентрация може да бъде намалена до следващата по-ниска концентрация. В този случай може да се добави допълнителна концентрация в долната част на серията от концентрации. Другите концентрации в сериите следва да останат без промяна.
|
|
34.
|
Разтворите на изпитвания химикал следва да се наблюдават отблизо, когато се добавят в ямката, тъй като изпитваният химикал може да се утаи при добавянето му в ямката. Данните за всички ямки, съдържащи преципитат, следва да се изключат от налагането на кривата и да се отбележат причините за изключването на данните.
|
|
35.
|
Ако съществува предходна информация от други източници, която предоставя логаритъм (IC50) на даден изпитван химикал, може да е подходящо разрежданията да се разпределят геометрично по-близо около очаквания логаритъм (IC50) (т.е. 0,5 логаритмични единици). Окончателните резултати следва да демонстрират достатъчно разпределение на концентрациите от всяка страна на логаритъма (IC50), включително „горе“ и „долу“, така че кривата на свързването да може да бъде адекватно характеризирана.
|
Организация на изследваната плака
|
36.
|
Маркираните микротитърни плаки следва да се приготвят с използване на шестократни инкубации за контролата на разтворителя, най-високата концентрация на референтния естроген (Е2), която служи и като индикатор за неспецифично свързване (NSB), контролата на буфера, трикратни инкубации за всяка от осемте концентрации на контролата за несвързващо вещество (октилтриетоксисилан), седемте по-ниски концентрации на референтния естроген (Е2), осемте концентрации на слабо свързващото вещество (норетинодрел или норетинодрон) и осемте концентрации на всеки изпитван химикал (ТС). Примерна схема на диаграмата на плаката за кривите на пълна концентрация за референтния естроген и контролите е дадена по-долу в Таблица 4. Допълнителни микротитърни плаки се използват за изпитвания химикал и следва да съдържат контроли на плаката (т.е. (i) висока (максимално изместване) и средна (приблизително IC50) концентрация на E2 и слабо свързващото вещество в три повторения; (ii) контрола на разтворителя (като общо свързване) и неспецифично свързване, всяка по шест пъти (Таблица 5). Пример за схема на микротитърна плака за работна таблица на конкурентно изследване е предоставен в Допълнение 3.3. Посочените в работната таблица концентрации, както е тези в Таблици 4 и 5, се отнасят за окончателните концентрации, използвани във всяка анализирана ямка. Максималната концентрация за E2 следва да е 1×10–7 M, а за слабо свързващото вещество следва да се използва най-високата концентрация, използвана за слабото свързващо вещество в плака 1. Концентрацията IC50 следва да се определи от лабораторията въз основа на нейната база данни за контроли от предходни периоди. Очакването е, че тази стойност ще е сходна с наблюдаваната във изследванията за валидиране (вж. Таблица 1).
|
Таблица 4
Схема на микротитърна плака за изследване на конкурентно свързване (98)
(99), пълни криви на концентрация за референтен естроген и контроли (плака 1)
|
|
1
|
2
|
3
|
4
|
5
|
6
|
7
|
8
|
9
|
10
|
11
|
12
|
|
Контрола на буфер и положителна контрола (Е2)
|
Слаба положителна контрола (Норетинодрел)
|
Отрицателна контрола (OTES)
|
TB и NSB
|
|
А
|
Празна (*14)
|
1×10–9 M
|
1×10–10 M
|
TB (контрола на разтворител) (2,05 % DMSO)
|
|
Б
|
E2 (1×10–11 M)
|
1×10–8 M
|
1×10–9 M
|
|
В
|
E2 (1×10–10 M)
|
1×10–7,5 M
|
1×10–8 M
|
NSB (10–6 M E2)
|
|
Г
|
E2 (1×10–9,5 M)
|
1×10–7 M
|
1×10–7 M
|
|
Д
|
E2 (1×10–9 M)
|
1×10–6,5 M
|
1×10–6 M
|
Контрола на буфер
|
|
Е
|
E2 (1×10–8,5 M)
|
1×10–6 M
|
1×10–5 M
|
|
Ж
|
E2 (1×10–8 M)
|
1×10–5,5 M
|
1×10–4 M
|
Празна (за горещ) (*15)
|
|
З
|
E2 (1×10–7 M)
|
1×10–4,5 M
|
1×10–3 M
|
Таблица 5
Схема на микротитърна плака за изследването на конкурентно свързване, допълнителни плаки за изпитвани химикали (ТС) и контроли на плака
|
|
1
|
2
|
3
|
4
|
5
|
6
|
7
|
8
|
9
|
10
|
11
|
12
|
|
|
Изпитван химикал-1 (TC-1)
|
Изпитван химикал-2 (TC-2)
|
Изпитван химикал-3 (TC-3)
|
Контрол
|
|
А
|
TC-1 (1×10–10 M)
|
TC-2 (1×10–10 M)
|
TC-3 (1×10–10 M)
|
E2 (1×10–7M)
|
|
Б
|
TC-1 (1×10–9 M)
|
TC-2 (1×10–9 M)
|
TC-3 (1×10–9 M)
|
E2 (IC50)
|
|
В
|
TC-1 (1×10–8 M)
|
TC-2 (1×10–8 M)
|
TC-3 (1×10–8 M)
|
NE (1×10
–4,5
M)
|
|
Г
|
TC-1 (1×10–7 M)
|
TC-2 (1×10–7 M)
|
TC-3 (1×10–7 M)
|
NE (IC50)
|
|
Д
|
TC-1 (1×10–6 M)
|
TC-2 (1×10–6 M)
|
TC-3 (1×10–6 M)
|
NSB (10–6 M E2)
|
|
Е
|
TC-1 (1×10–5 M)
|
TC-2 (1×10–5 M)
|
TC-3 (1×10–5 M)
|
|
Ж
|
TC-1 (1×10–4 M)
|
TC-2 (1×10–4 M)
|
TC-3 (1×10–4 M)
|
ТВ (Контрола на разтворител)
|
|
З
|
TC-1 (1×10–3 M)
|
TC-2 (1×10–3 M)
|
TC-3 (1×10–3 M)
|
|
В този пример слабото свързващо вещество е норетинодрел (NE)
|
Завършване на изследване на конкурентно свързване
|
37.
|
С изключение на ямките за общо свързване и празните ямки (за горещ), както е показано в Таблица 6, 50 μl от използвания в изследването буфер следва да се постави във всяка ямка и да се смеси с 10 μl от контролата на разтворителя, референтен естроген (E2), слабо свързващо вещество и изпитвани химикали, съответно 10 μl от разтвор на 5 nM [3H]-17β-естрадиол. След това във всяка плака се добавя 30μl охладен с лед разтвор на рецептор и внимателно се смесва. Разтворът на hrERα следва да е последния реактив за добавяне. Микротитърните плаки на изследването следва да се инкубират при стайна температура (от 22° до 28°C) в продължение на 2 часа.
Таблица 6
Обем на компонентите на изследването на конкурентно свързване към hrER, микротитърни плаки
|
Подготвителни операции
|
Различни от ямки за ТВ
|
Ямки за TB
|
Празна (за горещ)
|
|
Обем на компоненти за ямките за реакция отгоре и ред на добавяне
|
Използван в изследването буфер със стайна температура
|
50 μl
|
60 μl
|
90 μl
|
|
Немаркиран E2, слабо свързващо вещество, несвързващо вещество, разтворител и изпитвани химикали (*16)
|
10 μl
|
–
|
–
|
|
[3H]-17β-естрадиол за получаване на окончателна концентрация от 0,5 nM (т.е. 5 nM)
|
10 μl
|
10 μl
|
10 μl
|
|
Концентрация на hrERα, както е определена (вж. точки 12-13)
|
30 μl
|
30 μl
|
–
|
|
Общ обем във всяка ямка на изследването
|
100 μl
|
100 μl
|
100 μl
|
|
|
38.
|
Количественото определяне на [3H]-17β-естрадиол, свързан с hrERα, след отделяне на [3H]-17β-естрадиол, свързан с hrERα от свободен [3H]-17β-естрадиол чрез добавяне на 100 μl охладена с лед суспензия на DCC във всяка ямка, следва да се извърши след това както е описано в точки 21-23 на Допълнение 3 за изследването на свързване до насищане.
|
|
39.
|
Ямките G10-12 и H10-12 (идентифицирани като празни (за горещ) в Таблица 4) представят dpms на [3H]-маркиран естрадиол в 10 μl. 10 μl аликвотна част следва да се достави директно в сцинтилационната течност.
|
Критерии за приемливост
Изследване на свързване до насищане
|
40.
|
Кривата на специфично свързване следва да достигне върха като използваните увеличаващи се концентрации на [3H]-17β-естрадиол, което посочва насищане на hrERα с лиганд.
|
|
41.
|
Специфичното свързване при 0,5 nM от [3H]-17β-естрадиол следва да е в рамките на приемливия диапазон от 30 % до 50 % на средната измерена обща радиоактивност, добавена в сериите. Случайни леки отклонения извън този диапазон са допустими, но ако сериите последователно са извън този диапазон или дадена серия излиза значително извън този диапазон, концентрацията на протеин следва да се коригира и изследването за насищане да се повтори.
|
|
42.
|
Данните следва да произведат линейна диаграма на Scatchard.
|
|
43.
|
Неспецифичното свързване не трябва да е прекомерно. Стойността на неспецифичното свързване следва обикновено да е <35 % от общото свързване. Въпреки това съотношението може от време на време да надвишава тази граница, когато се измерва много нисък dpm за най-ниската концентрация на радиомаркиран изпитван 17β-естрадиол.
|
Изследване на конкурентно свързване
|
44.
|
Увеличаващите се концентрации на немаркиран 17β-естрадиол следва да изместят [3H]-17β-естрадиол от рецептора по начин, съответстващ на конкурентно свързване в едно място.
|
|
45.
|
Стойността на IC50 за референтния естроген (т.е. 17β-естрадиол) следва да е приблизително равна на моларната концентрация на [3H]-17β-естрадиол плюс Kd, определена от изследването за свързване до насищане.
|
|
46.
|
Общото специфично свързване следва да последователно да е в рамките на приемливия диапазон от 40 ± 10 %, когато средната измерена концентрация на общата радиоактивност, добавена във всяка ямка, е 0,5 nM в сериите. Случайни леки отклонения извън този диапазон са допустими, но ако сериите последователно са извън този диапазон или дадена серия излиза значително извън този диапазон, концентрацията на протеин следва да се коригира.
|
|
47.
|
Разтворителят не следва да променя чувствителността или възпроизводимостта на изследването. Резултатите на контролата на разтворителя (ямки на ТВ) се сравняват с контролата на буфера, за да се потвърди, че използваният разтворител не влияе върху системата на изследването. Резултатите на контролата за ТВ и на контролата на буфера следва да са сравними, ако разтворителят не оказва ефект върху изследването.
|
|
48.
|
Несвързващото вещество не следва да измести повече от 25 % от [3H]-17β-естрадиол от hrERα, когато се изпитва до 10–3 M (OTES) или 10–4 M (DBP).
|
|
49.
|
Критериите за ефективност са разработени за референтния естроген и две слаби свързващи вещества (напр. нороетинодрел, норетиндрон) с използване на данни от изследването за валидиране за изпитването за свързване на hrER на CERI (Приложение N на препратка 2). 95 % доверителни интервали са предоставени за средната стойност ± СО (n) на всички контроли измежду четири лаборатории, които са участвали във изследването за валидиране. 95 % доверителни интервали са изчислени за параметрите на налагане на кривата (т.е. горна част, долна част, наклон на кривата и Log IC50) за референтния естроген и слабо свързващи вещества и Log10RBA на слабо свързващи вещества по отношение на референтния естроген. В таблица 1 са дадени очакваните диапазони за параметрите на налагане на кривата, които могат да се използват като критерии за ефективност. На практика, диапазонът на IC50 може леко да варира въз основа на експериментално получената Kd на приготвянето на рецертора и концентрацията на лиганд, използвани за изследването.
|
|
50.
|
Не са разработени критерии за ефективност за параметрите на налагане на кривата за изпитваните химикали поради широкия спектър на съществуващи потенциални изпитвани химикали и вариациите в потенциалните афинитети и резултати (напр. пълна крива, частична крива, без налагане на крива). Въпреки това следва да се приложи професионална преценка, когато се преглеждат резултатите от всяка серия за даден изпитван химикал. Следва да се използва достатъчен диапазон на концентрации на изпитвания химикал за ясно определяне на горната част (напр. 90—100 % свързване) на конкурентната крива. Варирането между повторенията при всяка концентрация на изпитван химикал, както и между трите непаралелни серии следва да е разумно и обосновано от научна гледна точка. Контролите от всяка серия за даден изпитван химикал следва да се доближават до измерванията на ефективност, съобщени за това изследване на CERI, и да са последователни с данните за контроли от предходни периоди на всяка съответна лаборатория.
|
АНАЛИЗ НА ДАННИТЕ
Изследване на свързване до насищане
|
51.
|
Измерват се както общото, така и неспецифичното свързване. От тези стойности специфичното свързване на увеличаващи се концентрации на [3H]-17β-естрадиол при равновесни условия се изчислява чрез изваждане на неспецифичното от общото свързване. Графиката на специфичното свързване спрямо концентрацията на [3H]-17β-естрадиол следва да достигне върха за максималното специфично свързване, показателно за насищането на hrERα с [3H]-17β-естрадиол. Освен това анализът на данните следва да документира свързването на [3H]-17β- естрадиол с единично място на свързване с висок афинитет. Неспецифичното, общото и специфичното свързване следва да се показва на кривата за свързване до насищане. Допълнителният анализ на тези данни следва да използва анализ на нелинейна регресия (напр. BioSoft; McPherson, 1985 г.; Motulsky, 1995) с окончателно представяне на данните като диаграма на Scatchard.
|
|
52.
|
Анализът на данните следва да определи Bmax и Kd от самостоятелните данни за общо свързване с използване на предположението, че неспецифичното свързване е линейно, освен ако не е предоставена обосновка за използване на различен метод. Освен това следва да се използва надеждна регресия, когато се определя най-доброто съвпадение, освен ако не е предоставена обосновка. Избраният метод за надеждна регресия следва да бъде посочен. Корекция за изчерпване на лиганд (напр. използване на метода на Swillens 1995) следва винаги да се използва, когато се определя Bmax и Kd от данни за свързване до насищане.
|
Изследване на конкурентно свързване
|
53.
|
Кривата на конкурентно свързване се изчертава като специфично свързване на [3H]-17β- естрадиол спрямо концентрацията (log10 единици) на конкурента. Концентрацията на изпитвания химикал, която инхибира 50 % от максималното специфично свързване на [3H]-17β-естрадиол, е стойността на IC50.
|
|
54.
|
Оценките на логаритмичните(IC50) стойности за положителните контроли (напр. референтен естроген и слабо свързващо вещество) следва да се определят с използване на подходящ софтуер за нелинейно налагане на крива за вписване на уравнението на Хил с четири параметъра, (напр. BioSoft; McPherson, 1985 г.; Motulsky, 1995 г.). Горната част, долната част, наклонът и логаритмичната стойност (IC50) обикновено следва да се оставят неограничени при налагането на тези криви. Следва да се използва надеждна регресия, когато се определя най-доброто съвпадение, освен ако не е предоставена обосновка. Не следва да се използва корекция за изчерпване на лиганд. След първоначалния анализ всяка крива на свързване следва да се прегледа, за да се гарантира подходящо налагане върху модела. Относителният афинитет на свързване (RBA) за слабо свързващо вещество следва да се изчисли като процент на логаритъма (IC50) за слабото свързващо вещество по отношение на логаритъма (IC50) за 17β-естрадиол. Резултатите от положителните контроли и контролата на несвързващо вещество следва да се оценят с използване на измервания на ефективността на изследването, описани в точки 44-49 на настоящото Допълнение 3.
|
|
55.
|
Данните за всички изпитвани химикали следва да се анализират с използване на поетапен подход, за да се гарантира, че данните са анализирани по подходящ начин и че всяка крива на конкурентно свързване е правилно класифицирана. Препоръчва се всяка серия за даден изпитван химикал първо да премине през стандартизиран анализ на данни, който е идентичен на този, използван за референтния естроген и контролите на слабо свързващо вещество (вж. точка 54 на настоящото Допълнение 3). След приключването му следва да се извърши технически преглед на параметрите на налагане на кривата, както и визуален преглед доколко добре данните съвпадат с генерираната крива на конкурентно свързване за всяка серия. По време на този технически преглед наблюденията на зависимо от концентрацията намаляване на процента на специфично свързан [3H]-17β-естрадиол, слабата променливост измежду техническите повторения при всяка концентрация на изпитвания химикал и съгласуваността на параметрите на налагане в трите серии са добър индикатор, че изследването и анализите на данни са проведени по подходящ начин.
|
Тълкуване на данните
|
56.
|
При условие че всички критерии за приемливост са изпълнени, даден изпитван химикал се счита за свързващ се с hrERα, ако кривата на свързване може да бъде наложена и най-ниската точка върху кривата на отклик в рамките на диапазона на данните е по-малка 50 % (Фигура 1).
|
|
57.
|
При условие че са изпълнени всички критерии за приемливост, даден изпитван химикал се счита за несвързващ се с hrERα, ако:
|
—
|
Кривата на свързване може да бъде наложена и най-ниската точка на наложената крива на отклик в рамките на диапазона на данните е над 75 % или
|
|
—
|
Кривата на свързване не може да бъде наложена и най-ниският неизгладен среден процент на свързване измежду групите на концентрация в данните е над 75 %.
|
|
|
58.
|
Изпитваните химикали се считат за нееднозначни, ако никое от горните условия не е изпълнено (напр. най-ниската точка на наложената крива на отклик е между 76 – 51 %).
|
Таблица 7
Критерии за присвояване на класификация въз основа на крива на конкурентно свързване за даден изпитван химикал
|
Класифициране
|
Критерии
|
|
Свързващо веществоa
|
Кривата на свързване може да бъде наложена.
Най-ниската точка върху кривата на отклик в рамките на диапазона на данните е по-малка от 50 %.
|
|
Несвързващо веществоб
|
Ако кривата на свързване може да бъде наложена,
най-ниската точка върху наложената крива на отклик в рамките на диапазона на данните е над 75 %.
Ако кривата на свързване не може да бъде наложена,
най-ниският неизгладен среден процент на свързване измежду групите на концентрация в данните е над 75 %.
|
|
Нееднозначно веществов
|
Всяка изпитвана серия, която не е нито свързващо, нито несвързващо вещество
(напр. най-ниската точка на наложената крива на отклик е между 76 – 51 %).
|
Фигура 1
Примери за класифициране на изпитван химикал с използване на крива на конкурентно свързване
|
59.
|
Многобройните проведени в лаборатория серии за изпитван химикал се комбинират с присвояване на цифрови стойности на всяка серия и се осредняват за сериите, както е показано в Таблица 8. Резултатите за комбинираните серии във всяка лаборатория се сравняват с очакваната класификация за всеки изпитван химикал.
|
Таблица 8
Метод за класифициране на изпитван химикал с използване на множество серии в дадена лаборатория
|
За присвояване на стойност на всяка серия:
|
|
Класифициране
|
Цифрова стойност
|
|
Свързващо вещество
|
2
|
|
Нееднозначно вещество
|
1
|
|
Несвързващо вещество
|
0
|
|
За класифициране на средна цифрова стойност за сериите:
|
|
Класифициране
|
Цифрова стойност
|
|
Свързващо вещество
|
Средна стойност ≥ 1,5
|
|
Нееднозначно вещество
|
0,5 ≤ средна стойност < 1,5
|
|
Несвързващо вещество
|
Средна стойност < 0,5
|
ПРОТОКОЛ ОТ ИЗПИТВАНЕТО
|
60.
|
Вижте точка 24 от „КОМПОНЕНТИ НА ИЗСЛЕДВАНЕТО ЗА СВЪРЗВАНЕ НА hrER“ на настоящия метод за изпитване.
|
Допълнение 3,1
СПИСЪК НА ТЕРМИНИТЕ
[3H]E2
: 17β-естрадиол, радиомаркиран с тритий.
DCC
: въглен, покрит с декстран.
E2
: немаркиран 17β-естрадиол (инертен).
Буфер, използван в изследването
: 10 mM трис-HCl, pH 7,4, съдържащ 1 mM EDTA, 1mM EGTA, 1 mM NaVO3, 10 % глицерол, 0,2 mM левпептин, 1 mM дитиотреитол и 10 mg/ml говежди серумен албумин
hrERα
: човешки рекомбинантен естрогенен рецептор алфа (свързваща се с лиганд област).
Повторение
: една от множество ямки, които съдържат едно и също съдържание с едни и същи концентрации и се изследват паралелно в рамките на единична серия. В настоящия протокол всяка концентрация на изпитван химикал се изпитва три пъти; т.е. има три повторения, които се изследват едновременно с всяка концентрация на изпитван химикал.
Серия
: пълен набор от паралелно изпитвани ямки на микротитърни плаки за изследване, които осигуряват цялата информация, необходима за характеризиране на свързването на изпитван химикал към hrERα (а именно общ [3H]-17β-естрадиол, добавен в ямката на изследването, максимално свързване на [3H]-17β-естрадиол към hrERα, неспецифично свързване и общо свързване при различни концентрации на изпитвания химикал). Една серия може да се състои и само от една ямка за изследване (т.е. повторение) на концентрация, но тъй като настоящият протокол изисква трикратен анализ, една серия се състои от три ямки за изследване на концентрация. Освен това настоящият протокол изисква три независими (т.е. непаралелни) серии на химикал.
Допълнение 3.2
СХЕМА НА ЯМКА НА СРАВНИТЕЛНО ИЗСЛЕДВАНЕ НА СВЪРЗВАНЕ
|
Плоча
|
Местоположение
|
Повторение
|
Тип ямка
|
Код на ямка
|
Код на концентрация
|
Първоначална концентрация на конкурента (М)
|
Запас на hrER (μl)
|
Обем на буфера (μl)
|
Обем на маркера (Горещ E2) (μL)
|
Обем от плака за разреждане (μL)
|
Окончателен обем (μl)
|
Окончателна концентрация на конкурента (М)
|
|
S
|
A1
|
1
|
Празно
|
BK
|
BK1
|
—
|
—
|
—
|
—
|
—
|
—
|
—
|
|
S
|
A2
|
2
|
Празно
|
BK
|
BK2
|
—
|
—
|
—
|
—
|
—
|
—
|
—
|
|
S
|
A3
|
3
|
Празно
|
BK
|
BK3
|
—
|
—
|
—
|
—
|
—
|
—
|
—
|
|
S
|
B1
|
1
|
студен E2
|
S
|
S1
|
1.00E-10
|
30
|
50
|
10
|
10
|
100
|
1,0E-11
|
|
S
|
B2
|
2
|
студен E2
|
S
|
S1
|
1.00E-10
|
30
|
50
|
10
|
10
|
100
|
1,0E-11
|
|
S
|
B3
|
3
|
студен E2
|
S
|
S1
|
1.00E-10
|
30
|
50
|
10
|
10
|
100
|
1,0E-11
|
|
S
|
C1
|
1
|
студен E2
|
S
|
S2
|
1.00E-09
|
30
|
50
|
10
|
10
|
100
|
1,0E-10
|
|
S
|
C2
|
2
|
студен E2
|
S
|
S2
|
1.00E-09
|
30
|
50
|
10
|
10
|
100
|
1,0E-10
|
|
S
|
C3
|
3
|
студен E2
|
S
|
S2
|
1.00E-09
|
30
|
50
|
10
|
10
|
100
|
1,0E-10
|
|
S
|
D1
|
1
|
студен E2
|
S
|
S3
|
3.16E-09
|
30
|
50
|
10
|
10
|
100
|
3.2E-10
|
|
S
|
D2
|
2
|
студен E2
|
S
|
S3
|
3.16E-09
|
30
|
50
|
10
|
10
|
100
|
3.2E-10
|
|
S
|
D3
|
3
|
студен E2
|
S
|
S3
|
3.16E-09
|
30
|
50
|
10
|
10
|
100
|
3.2E-10
|
|
S
|
E1
|
1
|
студен E2
|
S
|
S4
|
1.00E-08
|
30
|
50
|
10
|
10
|
100
|
1,0E-09
|
|
S
|
E2
|
2
|
студен E2
|
S
|
S4
|
1.00E-08
|
30
|
50
|
10
|
10
|
100
|
1,0E-09
|
|
S
|
E3
|
3
|
студен E2
|
S
|
S4
|
1.00E-08
|
30
|
50
|
10
|
10
|
100
|
1,0E-09
|
|
S
|
F1
|
1
|
студен E2
|
S
|
S5
|
3.16E-08
|
30
|
50
|
10
|
10
|
100
|
3.2E-09
|
|
S
|
F2
|
2
|
студен E2
|
S
|
S5
|
3.16E-08
|
30
|
50
|
10
|
10
|
100
|
3.2E-09
|
|
S
|
F3
|
3
|
студен E2
|
S
|
S5
|
3.16E-08
|
30
|
50
|
10
|
10
|
100
|
3.2E-09
|
|
S
|
G1
|
1
|
студен E2
|
S
|
S6
|
1.00E-07
|
30
|
50
|
10
|
10
|
100
|
1,0E-08
|
|
S
|
G2
|
2
|
студен E2
|
S
|
S6
|
1.00E-07
|
30
|
50
|
10
|
10
|
100
|
1,0E-08
|
|
S
|
G3
|
3
|
студен E2
|
S
|
S6
|
1.00E-07
|
30
|
50
|
10
|
10
|
100
|
1,0E-08
|
|
S
|
H1
|
1
|
студен E2
|
S
|
S7
|
1.00E-06
|
30
|
50
|
10
|
10
|
100
|
1,0E-07
|
|
S
|
H2
|
2
|
студен E2
|
S
|
S7
|
1.00E-06
|
30
|
50
|
10
|
10
|
100
|
1,0E-07
|
|
S
|
H3
|
3
|
студен E2
|
S
|
S7
|
1.00E-06
|
30
|
50
|
10
|
10
|
100
|
1,0E-07
|
|
S
|
A4
|
1
|
норетинодрел
|
NE
|
WP1
|
1.00E-08
|
30
|
50
|
10
|
10
|
100
|
1,0E-09
|
|
S
|
A5
|
2
|
норетинодрел
|
NE
|
WP1
|
1.00E-08
|
30
|
50
|
10
|
10
|
100
|
1,0E-09
|
|
S
|
A6
|
3
|
норетинодрел
|
NE
|
WP1
|
1.00E-08
|
30
|
50
|
10
|
10
|
100
|
1,0E-09
|
|
S
|
B4
|
1
|
норетинодрел
|
NE
|
WP2
|
1.00E-07
|
30
|
50
|
10
|
10
|
100
|
1,0E-08
|
|
S
|
B5
|
2
|
норетинодрел
|
NE
|
WP2
|
1.00E-07
|
30
|
50
|
10
|
10
|
100
|
1,0E-08
|
|
S
|
B6
|
3
|
норетинодрел
|
NE
|
WP2
|
1.00E-07
|
30
|
50
|
10
|
10
|
100
|
1,0E-08
|
|
S
|
C4
|
1
|
норетинодрел
|
NE
|
WP3
|
3.16E-07
|
30
|
50
|
10
|
10
|
100
|
3.2E-08
|
|
S
|
C5
|
2
|
норетинодрел
|
NE
|
WP3
|
3.16E-07
|
30
|
50
|
10
|
10
|
100
|
3.2E-08
|
|
S
|
C6
|
3
|
норетинодрел
|
NE
|
WP3
|
3.16E-07
|
30
|
50
|
10
|
10
|
100
|
3.2E-08
|
|
S
|
D4
|
1
|
норетинодрел
|
NE
|
WP4
|
1.00E-06
|
30
|
50
|
10
|
10
|
100
|
1,0E-07
|
|
S
|
D5
|
2
|
норетинодрел
|
NE
|
WP4
|
1.00E-06
|
30
|
50
|
10
|
10
|
100
|
1,0E-07
|
|
S
|
D6
|
3
|
норетинодрел
|
NE
|
WP4
|
1.00E-06
|
30
|
50
|
10
|
10
|
100
|
1,0E-07
|
|
S
|
E4
|
1
|
норетинодрел
|
NE
|
WP5
|
3.16E-06
|
30
|
50
|
10
|
10
|
100
|
3.2E-07
|
|
S
|
E5
|
2
|
норетинодрел
|
NE
|
WP5
|
3.16E-06
|
30
|
50
|
10
|
10
|
100
|
3.2E-07
|
|
S
|
E6
|
3
|
норетинодрел
|
NE
|
WP5
|
3.16E-06
|
30
|
50
|
10
|
10
|
100
|
3.2E-07
|
|
S
|
F4
|
1
|
норетинодрел
|
NE
|
WP6
|
1.00E-05
|
30
|
50
|
10
|
10
|
100
|
1,0E-06
|
|
S
|
F5
|
2
|
норетинодрел
|
NE
|
WP6
|
1.00E-05
|
30
|
50
|
10
|
10
|
100
|
1,0E-06
|
|
S
|
F6
|
3
|
норетинодрел
|
NE
|
WP6
|
1.00E-05
|
30
|
50
|
10
|
10
|
100
|
1,0E-06
|
|
S
|
G4
|
1
|
норетинодрел
|
NE
|
WP7
|
3.16E-05
|
30
|
50
|
10
|
10
|
100
|
3.2E-06
|
|
S
|
G5
|
2
|
норетинодрел
|
NE
|
WP7
|
3.16E-05
|
30
|
50
|
10
|
10
|
100
|
3.2E-06
|
|
S
|
G6
|
3
|
норетинодрел
|
NE
|
WP7
|
3.16E-05
|
30
|
50
|
10
|
10
|
100
|
3.2E-06
|
|
S
|
H4
|
1
|
норетинодрел
|
NE
|
WP8
|
3.16E-04
|
30
|
50
|
10
|
10
|
100
|
3.2E-05
|
|
S
|
H5
|
2
|
норетинодрел
|
NE
|
WP8
|
3.16E-04
|
30
|
50
|
10
|
10
|
100
|
3.2E-05
|
|
S
|
H6
|
3
|
норетинодрел
|
NE
|
WP8
|
3.16E-04
|
30
|
50
|
10
|
10
|
100
|
3.2E-05
|
|
S
|
A7
|
1
|
OTES
|
N
|
OTES1
|
1.00E-09
|
30
|
50
|
10
|
10
|
100
|
1,0E-10
|
|
S
|
A8
|
2
|
OTES
|
N
|
OTES1
|
1.00E-09
|
30
|
50
|
10
|
10
|
100
|
1,0E-10
|
|
S
|
A9
|
3
|
OTES
|
N
|
OTES1
|
1.00E-09
|
30
|
50
|
10
|
10
|
100
|
1,0E-10
|
|
S
|
B7
|
1
|
OTES
|
N
|
OTES2
|
1.00E-08
|
30
|
50
|
10
|
10
|
100
|
1,0E-09
|
|
S
|
B8
|
2
|
OTES
|
N
|
OTES2
|
1.00E-08
|
30
|
50
|
10
|
10
|
100
|
1,0E-09
|
|
S
|
B9
|
3
|
OTES
|
N
|
OTES2
|
1.00E-08
|
30
|
50
|
10
|
10
|
100
|
1,0E-09
|
|
S
|
C7
|
1
|
OTES
|
N
|
OTES3
|
1.00E-07
|
30
|
50
|
10
|
10
|
100
|
1,0E-08
|
|
S
|
C8
|
2
|
OTES
|
N
|
OTES3
|
1.00E-07
|
30
|
50
|
10
|
10
|
100
|
1,0E-08
|
|
S
|
C9
|
3
|
OTES
|
N
|
OTES3
|
1.00E-07
|
30
|
50
|
10
|
10
|
100
|
1,0E-08
|
|
S
|
D7
|
1
|
OTES
|
N
|
OTES4
|
1.00E-06
|
30
|
50
|
10
|
10
|
100
|
1,0E-07
|
|
S
|
D8
|
2
|
OTES
|
N
|
OTES4
|
1.00E-06
|
30
|
50
|
10
|
10
|
100
|
1,0E-07
|
|
S
|
D9
|
3
|
OTES
|
N
|
OTES4
|
1.00E-06
|
30
|
50
|
10
|
10
|
100
|
1,0E-07
|
|
S
|
E7
|
1
|
OTES
|
N
|
OTES5
|
1.00E-05
|
30
|
50
|
10
|
10
|
100
|
1,0E-06
|
|
S
|
E8
|
2
|
OTES
|
N
|
OTES5
|
1.00E-05
|
30
|
50
|
10
|
10
|
100
|
1,0E-06
|
|
S
|
E9
|
3
|
OTES
|
N
|
OTES5
|
1.00E-05
|
30
|
50
|
10
|
10
|
100
|
1,0E-06
|
|
S
|
F7
|
1
|
OTES
|
N
|
OTES6
|
1.00E-04
|
30
|
50
|
10
|
10
|
100
|
1,0E-05
|
|
S
|
F8
|
2
|
OTES
|
N
|
OTES6
|
1.00E-04
|
30
|
50
|
10
|
10
|
100
|
1,0E-05
|
|
S
|
F9
|
3
|
OTES
|
N
|
OTES6
|
1.00E-04
|
30
|
50
|
10
|
10
|
100
|
1,0E-05
|
|
S
|
G7
|
1
|
OTES
|
N
|
OTES7
|
1.00E-03
|
30
|
50
|
10
|
10
|
100
|
1,0E-04
|
|
S
|
G8
|
2
|
OTES
|
N
|
OTES7
|
1.00E-03
|
30
|
50
|
10
|
10
|
100
|
1,0E-04
|
|
S
|
G9
|
3
|
OTES
|
N
|
OTES7
|
1.00E-03
|
30
|
50
|
10
|
10
|
100
|
1,0E-04
|
|
S
|
H7
|
1
|
OTES
|
N
|
OTES8DBP7
|
1.00E-02
|
30
|
50
|
10
|
10
|
100
|
1,0E-03
|
|
S
|
H8
|
2
|
OTES
|
N
|
OTES88
|
1.00E-02
|
30
|
50
|
10
|
10
|
100
|
1,0E-03
|
|
S
|
H9
|
3
|
OTES
|
N
|
OTES8
|
1.00E-02
|
30
|
50
|
10
|
10
|
100
|
1,0E-03
|
|
S
|
A10
|
1
|
общо свързване
|
TB
|
TB1
|
—
|
30
|
60
|
10
|
—
|
100
|
—
|
|
S
|
A11
|
2
|
общо свързване
|
TB
|
TB2
|
—
|
30
|
60
|
10
|
—
|
100
|
—
|
|
S
|
A12
|
3
|
общо свързване
|
TB
|
TB3
|
—
|
30
|
60
|
10
|
—
|
100
|
—
|
|
S
|
B10
|
4
|
общо свързване
|
TB
|
TB4
|
—
|
30
|
60
|
10
|
—
|
100
|
—
|
|
S
|
B11
|
5
|
общо свързване
|
TB
|
TB5
|
—
|
30
|
60
|
10
|
—
|
100
|
—
|
|
S
|
B12
|
6
|
общо свързване
|
TB
|
TB6
|
—
|
30
|
60
|
10
|
—
|
100
|
—
|
|
S
|
C10
|
1
|
студен E2 (висок)
|
NSB
|
S1
|
1.00E-05
|
30
|
50
|
10
|
10
|
100
|
1,0E-06
|
|
S
|
C11
|
2
|
студен E2 (висок)
|
NSB
|
S2
|
1.00E-05
|
30
|
50
|
10
|
10
|
100
|
1,0E-06
|
|
S
|
C12
|
3
|
студен E2 (висок)
|
NSB
|
S3
|
1.00E-05
|
30
|
50
|
10
|
10
|
100
|
1,0E-06
|
|
S
|
D10
|
4
|
студен E2 (висок)
|
NSB
|
S4
|
1.00E-05
|
30
|
50
|
10
|
10
|
100
|
1,0E-06
|
|
S
|
D11
|
5
|
студен E2 (висок)
|
NSB
|
S5
|
1.00E-05
|
30
|
50
|
10
|
10
|
100
|
1,0E-06
|
|
S
|
D12
|
6
|
студен E2 (висок)
|
NSB
|
S6
|
1.00E-05
|
30
|
50
|
10
|
10
|
100
|
1,0E-06
|
|
S
|
E10
|
1
|
Контрола на буфер
|
BC
|
BC1
|
—
|
—
|
100
|
—
|
—
|
100
|
—
|
|
S
|
E11
|
2
|
Контрола на буфер
|
BC
|
BC2
|
—
|
—
|
100
|
—
|
—
|
100
|
—
|
|
S
|
E12
|
3
|
Контрола на буфер
|
BC
|
BC3
|
—
|
—
|
100
|
—
|
—
|
100
|
—
|
|
S
|
F10
|
4
|
Контрола на буфер
|
BC
|
BC4
|
—
|
—
|
100
|
—
|
—
|
100
|
—
|
|
S
|
F11
|
5
|
Контрола на буфер
|
BC
|
BC5
|
—
|
—
|
100
|
—
|
—
|
100
|
—
|
|
S
|
F12
|
6
|
Контрола на буфер
|
BC
|
BC6
|
—
|
—
|
100
|
—
|
—
|
100
|
—
|
|
S
|
G10 (*17)
|
1
|
Празна (за горещ)
|
Горещ
|
H1
|
—
|
90
|
—
|
10
|
—
|
100
|
—
|
|
S
|
G11 (*17)
|
2
|
Празна (за горещ)
|
Горещ
|
H2
|
—
|
90
|
—
|
10
|
—
|
100
|
—
|
|
S
|
G12 (*17)
|
3
|
Празна (за горещ)
|
Горещ
|
H3
|
—
|
90
|
—
|
10
|
—
|
100
|
—
|
|
S
|
H10 (*17)
|
4
|
Празна (за горещ)
|
Горещ
|
H4
|
—
|
90
|
—
|
10
|
—
|
100
|
—
|
|
S
|
H11 (*17)
|
5
|
Празна (за горещ)
|
Горещ
|
H5
|
—
|
90
|
—
|
10
|
—
|
100
|
—
|
|
S
|
H12
|
6
|
Празна (за горещ)
|
Горещ
|
H6
|
—
|
90
|
—
|
10
|
—
|
100
|
—
|
|
P1
|
A1
|
1
|
Неизвестен 1
|
U1
|
1
|
1.00E-09
|
30
|
50
|
10
|
10
|
100
|
1,0E-10
|
|
P1
|
A2
|
2
|
Неизвестен 1
|
U1
|
1
|
1.00E-09
|
30
|
50
|
10
|
10
|
100
|
1,0E-10
|
|
P1
|
A3
|
3
|
Неизвестен 1
|
U1
|
1
|
1.00E-09
|
30
|
50
|
10
|
10
|
100
|
1,0E-10
|
|
P1
|
B1
|
1
|
Неизвестен 1
|
U1
|
2
|
1.00E-08
|
30
|
50
|
10
|
10
|
100
|
1,0E-09
|
|
P1
|
B2
|
2
|
Неизвестен 1
|
U1
|
2
|
1.00E-08
|
30
|
50
|
10
|
10
|
100
|
1,0E-09
|
|
P1
|
B3
|
3
|
Неизвестен 1
|
U1
|
2
|
1.00E-08
|
30
|
50
|
10
|
10
|
100
|
1,0E-09
|
|
P1
|
C1
|
1
|
Неизвестен 1
|
U1
|
3
|
1.00E-07
|
30
|
50
|
10
|
10
|
100
|
1,0E-08
|
|
P1
|
C2
|
2
|
Неизвестен 1
|
U1
|
3
|
1.00E-07
|
30
|
50
|
10
|
10
|
100
|
1,0E-08
|
|
P1
|
C3
|
3
|
Неизвестен 1
|
U1
|
3
|
1.00E-07
|
30
|
50
|
10
|
10
|
100
|
1,0E-08
|
|
P1
|
D1
|
1
|
Неизвестен 1
|
U1
|
4
|
1.00E-06
|
30
|
50
|
10
|
10
|
100
|
1,0E-07
|
|
P1
|
D2
|
2
|
Неизвестен 1
|
U1
|
4
|
1.00E-06
|
30
|
50
|
10
|
10
|
100
|
1,0E-07
|
|
P1
|
D3
|
3
|
Неизвестен 1
|
U1
|
4
|
1.00E-06
|
30
|
50
|
10
|
10
|
100
|
1,0E-07
|
|
P1
|
E1
|
1
|
Неизвестен 1
|
U1
|
5
|
1.00E-05
|
30
|
50
|
10
|
10
|
100
|
1,0E-06
|
|
P1
|
E2
|
2
|
Неизвестен 1
|
U1
|
5
|
1.00E-05
|
30
|
50
|
10
|
10
|
100
|
1,0E-06
|
|
P1
|
E3
|
3
|
Неизвестен 1
|
U1
|
5
|
1.00E-05
|
30
|
50
|
10
|
10
|
100
|
1,0E-06
|
|
P1
|
F1
|
1
|
Неизвестен 1
|
U1
|
6
|
1.00E-04
|
30
|
50
|
10
|
10
|
100
|
1,0E-05
|
|
P1
|
F2
|
2
|
Неизвестен 1
|
U1
|
6
|
1.00E-04
|
30
|
50
|
10
|
10
|
100
|
1,0E-05
|
|
P1
|
F3
|
3
|
Неизвестен 1
|
U1
|
6
|
1.00E-04
|
30
|
50
|
10
|
10
|
100
|
1,0E-05
|
|
P1
|
G1
|
1
|
Неизвестен 1
|
U1
|
7
|
1.00E-03
|
30
|
50
|
10
|
10
|
100
|
1,0E-04
|
|
P1
|
G2
|
2
|
Неизвестен 1
|
U1
|
7
|
1.00E-03
|
30
|
50
|
10
|
10
|
100
|
1,0E-04
|
|
P1
|
G3
|
3
|
Неизвестен 1
|
U1
|
7
|
1.00E-03
|
30
|
50
|
10
|
10
|
100
|
1,0E-04
|
|
P1
|
H1
|
1
|
Неизвестен 1
|
U1
|
8
|
1.00E-02
|
30
|
50
|
10
|
10
|
100
|
1,0E-03
|
|
P1
|
H2
|
2
|
Неизвестен 1
|
U1
|
8
|
1.00E-02
|
30
|
50
|
10
|
10
|
100
|
1,0E-03
|
|
P1
|
H3
|
3
|
Неизвестен 1
|
U1
|
8
|
1.00E-02
|
30
|
50
|
10
|
10
|
100
|
1,0E-03
|
|
P1
|
A4
|
1
|
Неизвестен 2
|
U2
|
1
|
1.00E-09
|
30
|
50
|
10
|
10
|
100
|
1,0E-10
|
|
P1
|
A5
|
2
|
Неизвестен 2
|
U2
|
1
|
1.00E-09
|
30
|
50
|
10
|
10
|
100
|
1,0E-10
|
|
P1
|
A6
|
3
|
Неизвестен 2
|
U2
|
1
|
1.00E-09
|
30
|
50
|
10
|
10
|
100
|
1,0E-10
|
|
P1
|
B4
|
1
|
Неизвестен 2
|
U2
|
2
|
1.00E-08
|
30
|
50
|
10
|
10
|
100
|
1,0E-09
|
|
P1
|
B5
|
2
|
Неизвестен 2
|
U2
|
2
|
1.00E-08
|
30
|
50
|
10
|
10
|
100
|
1,0E-09
|
|
P1
|
B6
|
3
|
Неизвестен 2
|
U2
|
2
|
1.00E-08
|
30
|
50
|
10
|
10
|
100
|
1,0E-09
|
|
P1
|
C4
|
1
|
Неизвестен 2
|
U2
|
3
|
1.00E-07
|
30
|
50
|
10
|
10
|
100
|
1,0E-08
|
|
P1
|
C5
|
2
|
Неизвестен 2
|
U2
|
3
|
1.00E-07
|
30
|
50
|
10
|
10
|
100
|
1,0E-08
|
|
P1
|
C6
|
3
|
Неизвестен 2
|
U2
|
3
|
1.00E-07
|
30
|
50
|
10
|
10
|
100
|
1,0E-08
|
|
P1
|
D4
|
1
|
Неизвестен 2
|
U2
|
4
|
1.00E-06
|
30
|
50
|
10
|
10
|
100
|
1,0E-07
|
|
P1
|
D5
|
2
|
Неизвестен 2
|
U2
|
4
|
1.00E-06
|
30
|
50
|
10
|
10
|
100
|
1,0E-07
|
|
P1
|
D6
|
3
|
Неизвестен 2
|
U2
|
4
|
1.00E-06
|
30
|
50
|
10
|
10
|
100
|
1,0E-07
|
|
P1
|
E4
|
1
|
Неизвестен 2
|
U2
|
5
|
1.00E-05
|
30
|
50
|
10
|
10
|
100
|
1,0E-06
|
|
P1
|
E5
|
2
|
Неизвестен 2
|
U2
|
5
|
1.00E-05
|
30
|
50
|
10
|
10
|
100
|
1,0E-06
|
|
P1
|
E6
|
3
|
Неизвестен 2
|
U2
|
5
|
1.00E-05
|
30
|
50
|
10
|
10
|
100
|
1,0E-06
|
|
P1
|
F4
|
1
|
Неизвестен 2
|
U2
|
6
|
1.00E-04
|
30
|
50
|
10
|
10
|
100
|
1,0E-05
|
|
P1
|
F5
|
2
|
Неизвестен 2
|
U2
|
6
|
1.00E-04
|
30
|
50
|
10
|
10
|
100
|
1,0E-05
|
|
P1
|
F6
|
3
|
Неизвестен 2
|
U2
|
6
|
1.00E-04
|
30
|
50
|
10
|
10
|
100
|
1,0E-05
|
|
P1
|
G4
|
1
|
Неизвестен 2
|
U2
|
7
|
1.00E-03
|
30
|
50
|
10
|
10
|
100
|
1,0E-04
|
|
P1
|
G5
|
2
|
Неизвестен 2
|
U2
|
7
|
1.00E-03
|
30
|
50
|
10
|
10
|
100
|
1,0E-04
|
|
P1
|
G6
|
3
|
Неизвестен 2
|
U2
|
7
|
1.00E-03
|
30
|
50
|
10
|
10
|
100
|
1,0E-04
|
|
P1
|
H4
|
1
|
Неизвестен 2
|
U2
|
8
|
1.00E-02
|
30
|
50
|
10
|
10
|
100
|
1,0E-03
|
|
P1
|
H5
|
2
|
Неизвестен 2
|
U2
|
8
|
1.00E-02
|
30
|
50
|
10
|
10
|
100
|
1,0E-03
|
|
P1
|
H6
|
3
|
Неизвестен 2
|
U2
|
8
|
1.00E-02
|
30
|
50
|
10
|
10
|
100
|
1,0E-03
|
|
P1
|
A7
|
1
|
Неизвестен 3
|
U3
|
1
|
1.00E-09
|
30
|
50
|
10
|
10
|
100
|
1,0E-10
|
|
P1
|
A8
|
2
|
Неизвестен 3
|
U3
|
1
|
1.00E-09
|
30
|
50
|
10
|
10
|
100
|
1,0E-10
|
|
P1
|
A9
|
3
|
Неизвестен 3
|
U3
|
1
|
1.00E-09
|
30
|
50
|
10
|
10
|
100
|
1,0E-10
|
|
P1
|
B7
|
1
|
Неизвестен 3
|
U3
|
2
|
1.00E-08
|
30
|
50
|
10
|
10
|
100
|
1,0E-09
|
|
P1
|
B8
|
2
|
Неизвестен 3
|
U3
|
2
|
1.00E-08
|
30
|
50
|
10
|
10
|
100
|
1,0E-09
|
|
P1
|
B9
|
3
|
Неизвестен 3
|
U3
|
2
|
1.00E-08
|
30
|
50
|
10
|
10
|
100
|
1,0E-09
|
|
P1
|
C7
|
1
|
Неизвестен 3
|
U3
|
3
|
1.00E-07
|
30
|
50
|
10
|
10
|
100
|
1,0E-08
|
|
P1
|
C8
|
2
|
Неизвестен 3
|
U3
|
3
|
1.00E-07
|
30
|
50
|
10
|
10
|
100
|
1,0E-08
|
|
P1
|
C9
|
3
|
Неизвестен 3
|
U3
|
3
|
1.00E-07
|
30
|
50
|
10
|
10
|
100
|
1,0E-08
|
|
P1
|
D7
|
1
|
Неизвестен 3
|
U3
|
4
|
1.00E-06
|
30
|
50
|
10
|
10
|
100
|
1,0E-07
|
|
P1
|
D8
|
2
|
Неизвестен 3
|
U3
|
4
|
1.00E-06
|
30
|
50
|
10
|
10
|
100
|
1,0E-07
|
|
P1
|
D9
|
3
|
Неизвестен 3
|
U3
|
4
|
1.00E-06
|
30
|
50
|
10
|
10
|
100
|
1,0E-07
|
|
P1
|
E7
|
1
|
Неизвестен 3
|
U3
|
5
|
1.00E-05
|
30
|
50
|
10
|
10
|
100
|
1,0E-06
|
|
P1
|
E8
|
2
|
Неизвестен 3
|
U3
|
5
|
1.00E-05
|
30
|
50
|
10
|
10
|
100
|
1,0E-06
|
|
P1
|
E9
|
3
|
Неизвестен 3
|
U3
|
5
|
1.00E-05
|
30
|
50
|
10
|
10
|
100
|
1,0E-06
|
|
P1
|
F7
|
1
|
Неизвестен 3
|
U3
|
6
|
1.00E-04
|
30
|
50
|
10
|
10
|
100
|
1,0E-05
|
|
P1
|
F8
|
2
|
Неизвестен 3
|
U3
|
6
|
1.00E-04
|
30
|
50
|
10
|
10
|
100
|
1,0E-05
|
|
P1
|
F9
|
3
|
Неизвестен 3
|
U3
|
6
|
1.00E-04
|
30
|
50
|
10
|
10
|
100
|
1,0E-05
|
|
P1
|
G7
|
1
|
Неизвестен 3
|
U3
|
7
|
1.00E-03
|
30
|
50
|
10
|
10
|
100
|
1,0E-04
|
|
P1
|
G8
|
2
|
Неизвестен 3
|
U3
|
7
|
1.00E-03
|
30
|
50
|
10
|
10
|
100
|
1,0E-04
|
|
P1
|
G9
|
3
|
Неизвестен 3
|
U3
|
7
|
1.00E-03
|
30
|
50
|
10
|
10
|
100
|
1,0E-04
|
|
P1
|
H7
|
1
|
Неизвестен 3
|
U3
|
8
|
1.00E-02
|
30
|
50
|
10
|
10
|
100
|
1,0E-03
|
|
P1
|
H8
|
2
|
Неизвестен 3
|
U3
|
8
|
1.00E-02
|
30
|
50
|
10
|
10
|
100
|
1,0E-03
|
|
P1
|
H9
|
3
|
Неизвестен 3
|
U3
|
8
|
1.00E-02
|
30
|
50
|
10
|
10
|
100
|
1,0E-03
|
|
P1
|
A10
|
1
|
Контрола E2 (макс.)
|
S
|
E2max1
|
1.00E-06
|
30
|
50
|
10
|
10
|
100
|
1.00E-07
|
|
P1
|
A11
|
2
|
Контрола E2 (макс.)
|
S
|
E2max2
|
1.00E-06
|
30
|
50
|
10
|
10
|
100
|
1.00E-07
|
|
P1
|
A12
|
3
|
Контрола E2 (макс.)
|
S
|
E2max3
|
1.00E-06
|
30
|
50
|
10
|
10
|
100
|
1.00E-07
|
|
P1
|
B10
|
1
|
Контрола E2 (IC50)
|
S
|
E2IC501
|
E2IC50x10
|
30
|
50
|
10
|
10
|
100
|
E2IC50
|
|
P1
|
B11
|
2
|
Контрола E2 (IC50)
|
S
|
E2IC502
|
E2IC50x10
|
30
|
50
|
10
|
10
|
100
|
E2IC50
|
|
P1
|
B12
|
3
|
Контрола E2 (IC50)
|
S
|
E2IC503
|
E2IC50x10
|
30
|
50
|
10
|
10
|
100
|
E2IC50
|
|
P1
|
C10
|
1
|
Контрола NE (макс.)
|
S
|
Nemax1
|
1.00E-3.5
|
30
|
50
|
10
|
10
|
100
|
1.00E-4.5
|
|
P1
|
C11
|
2
|
Контрола NE (макс.)
|
S
|
Nemax2
|
1.00E-3.5
|
30
|
50
|
10
|
10
|
100
|
1.00E-4.5
|
|
P1
|
C12
|
3
|
Контрола NE (макс.)
|
S
|
Nemax3
|
1.00E-3.5
|
30
|
50
|
10
|
10
|
100
|
1.00E-4.5
|
|
P1
|
D10
|
1
|
Контрола NE (IC50)
|
S
|
NEIC501
|
NEIC50 x10
|
30
|
50
|
10
|
10
|
100
|
NEIC50
|
|
P1
|
D11
|
2
|
Контрола NE (IC50)
|
S
|
NEIC502
|
NEIC50 x10
|
30
|
50
|
10
|
10
|
100
|
NEIC50
|
|
P1
|
D12
|
3
|
Контрола NE (IC50)
|
S
|
NEIC503
|
NEIC50 x10
|
30
|
50
|
10
|
10
|
100
|
NEIC50
|
|
P1
|
E10
|
1
|
студен E2 (висок)
|
NSB
|
S1
|
1.00E-05
|
30
|
50
|
10
|
10
|
100
|
1,0E-06
|
|
P1
|
E11
|
2
|
студен E2 (висок)
|
NSB
|
S2
|
1.00E-05
|
30
|
50
|
10
|
10
|
100
|
1,0E-06
|
|
P1
|
E12
|
3
|
студен E2 (висок)
|
NSB
|
S3
|
1.00E-05
|
30
|
50
|
10
|
10
|
100
|
1,0E-06
|
|
P1
|
F10
|
4
|
студен E2 (висок)
|
NSB
|
S4
|
1.00E-05
|
30
|
50
|
10
|
10
|
100
|
1,0E-06
|
|
P1
|
F11
|
5
|
студен E2 (висок)
|
NSB
|
S5
|
1.00E-05
|
30
|
50
|
10
|
10
|
100
|
1,0E-06
|
|
P1
|
F12
|
6
|
студен E2 (висок)
|
NSB
|
S6
|
1.00E-05
|
30
|
50
|
10
|
10
|
100
|
1,0E-06
|
|
P1
|
G10
|
1
|
общо свързване
|
TB
|
TB1
|
—
|
30
|
60
|
10
|
—
|
100
|
—
|
|
P1
|
G11
|
2
|
общо свързване
|
TB
|
TB2
|
—
|
30
|
60
|
10
|
—
|
100
|
—
|
|
P1
|
G12
|
3
|
общо свързване
|
TB
|
TB3
|
—
|
30
|
60
|
10
|
—
|
100
|
—
|
|
P1
|
H10
|
4
|
общо свързване
|
TB
|
TB4
|
—
|
30
|
60
|
10
|
—
|
100
|
—
|
|
P1
|
H11
|
5
|
общо свързване
|
TB
|
TB5
|
—
|
30
|
60
|
10
|
—
|
100
|
—
|
|
P1
|
H12
|
6
|
общо свързване
|
TB
|
TB6
|
—
|
30
|
60
|
10
|
—
|
100
|
—
|
Допълнение 4
СЪОБРАЖЕНИЯ ЗА АНАЛИЗА НА ДАННИ ОТ ИЗСЛЕДВАНЕТО ЗА КОНКУРЕНТНО СВЪРЗВАНЕ НА HRER
|
1.
|
Изследването за конкурентно свързване на hrERα измерва една концентрация на [3H]-17β-естрадиол в присъствието на увеличаващи се концентрации на изпитван химикал. Кривата на конкурентно свързване се изчертава като специфично свързване на [3H]-17β- естрадиол спрямо концентрацията (log10 единици) на конкурента. Концентрацията на изпитвания химикал, която инхибира 50 % от максималното специфично свързване на [3H]-17β-естрадиол, е IC50.
|
Анализ на данни за референтния естроген и слабо свързващо вещество (1)
|
2.
|
Данните от контролните серии се преобразуват (т.е. процент на специфично свързване на [3H]-17β-естрадиол и логаритмичната концентрация на контролния химикал) за допълнителен анализ. Оценките на логаритмичните(IC50) стойности за положителните контроли (напр. референтен естроген и слабо свързващо вещество) следва да се определят с използване на подходящ софтуер за нелинейно налагане на крива за вписване на уравнението на Хил с четири параметъра, (напр. BioSoft; GraphPad Prism) (2). Горната част, долната част, наклонът и логаритмичната стойност ( IC50) обикновено могат да се оставят неограничени при налагането на тези криви. Следва да се използва надеждна регресия, когато се определя най-доброто съвпадение, освен ако не е предоставена обосновка. Избраният метод за надеждна регресия следва да бъде посочен. Корекция за изчерпване на лиганд не е необходима за изследванията FW или CERI hrER, но може да се обмисли при нужда. След първоначалния анализ всяка крива на свързване следва да се прегледа, за да се гарантира подходящо налагане върху модела. Относителният афинитет на свързване (RBA) за слабо свързващо вещество може да се изчисли като процент на логаритъма (IC50) за слабото свързващо вещество по отношение на логаритъма (IC50) за 17β-естрадиол. Резултатите за положителните контроли и контролата на несвързващо вещество следва да се оценят с използване на измервания на ефективността на изследването и критериите за приемливост, както са описани в настоящия метод за изпитване (точка 20), Допълнение 2 (изследване на FW, точки 41-51) и Допълнение 3 (изследване на CERI, точки 41-51). Примери на 3 серии за референтния естроген и слабо свързващо вещество са показани на Фигура 1.
|
Фигура 1
Примери на криви на конкурентно свързване за референтния естроген и контролното слабо свързващо вещество

Анализ на данните за изпитвани химикали
|
3.
|
Данните за всички изпитвани химикали следва да се анализират с използване на поетапен подход, за да се гарантира, че данните са анализирани по подходящ начин и че всяка крива на конкурентно свързване е правилно класифицирана. Всяка серия за даден изпитван химикал следва първо да премине през стандартизиран анализ на данни, който е идентичен на този, използван за референтния естроген и контролите на слабо свързващо вещество. След приключването му следва да се извърши технически преглед на параметрите на налагане на кривата, както и визуален преглед доколко добре данните съвпадат с генерираната крива на конкурентно свързване за всяка серия. По време на този технически преглед наблюденията на зависимо от концентрацията намаляване на процента на специфично свързан [3H]-17β-естрадиол, слабата променливост измежду техническите повторения при всяка концентрация на химикала и съгласуваността на параметрите на налагане в трите серии са добър индикатор, че изследването и анализите на данни са проведени по подходящ начин. Необходимо е да се приложи професионална обосновка, когато се преглеждат резултатите от всяка серия за даден изпитван химикал и данните, използвани за класифициране на всеки изпитван химикал като свързващ или несвързващ, следва да са оправдани от научна гледна точка.
|
|
4.
|
От време на време е възможно да има примерни данни, които изискват допълнително внимание за подходящо анализиране и тълкуване на данните за свързване с hrER. Предишните проучвания демонстрираха случаи, при които анализът и тълкуването на данните за конкурентно свързване на рецептор могат да бъдат усложнени от промяна на процента на специфично свързване, когато се изпитват химикали при най-високи концентрации (Фигура 2). Това е добре известен проблем, който се среща при използване на протоколи за редица изследвания на конкурентно свързване на рецептор (3). В тези случаи зависимият от концентрацията отклик се наблюдава при по-ниски концентрации, но тъй като концентрацията на изпитвания химикал се доближава до границата на разтворимост, изместването на [3H]17β-естрадиол повече не намалява. В тези случаи данните за по-високи концентрации сочат, че е достигната биологичната граница на изследването. Например този феномен много пъти е свързван с химична неразтворимост и преципитация при високи концентрации или може да е също така отражение на превишаването на капацитета на покрития с декстран въглен да улови несвързания радиомаркиран лиганд по време на процедурата на отделяне при най-високите химични концентрации. Оставянето на тези точки с данни при налагане на данни за конкурентно свързване върху сигмоидна крива понякога може да доведе до неправилно класифициране на потенциала за свързване на ЕР за даден изпитван химикал (Фигура 2). За да се избегне това, протоколът на изследванията за свързване на hrER на FW и CERI hrER включва опция за изключване от анализите на точки с данни, където средната стойност на повторенията за процента на специфично свързан [3H]17β-естрадиол е 10 % или повече над средната наблюдавана стойност при по-ниска концентрация (т.е. това често се нарича правилото на 10 % процента). Това правило може да се използва само един път за дадена крива и трябва да има оставащи данни за поне 6 концентрации като тази, при която кривата може да бъде вярно класифицирана.
|
Фигура 2
Примери, криви на конкурентно свързване със и без използване на правилото на 10 %
|
5.
|
Подходящата употреба на правилото на 10 % за корекция на тези криви следва внимателно да се обмисли и да се запази за тези случаи, когато има силна индикация за свързващо се с hrER вещество. По време на провеждането на експерименти за изследването за валидиране на изследването за свързване на hrER на FW е наблюдавано, че правилото на 10% понякога води до нежелани и непредвидими последствия. Химикали, които не взаимодействат с рецептора (т.е. истински несвързващи се вещества), често показват вариабилност около 100 % свързване на радиолиганд, което е по-голямо от 10 % в диапазона на изпитваните концентрации. Ако се случи, че най-ниската стойност е при ниска концентрация, данните за всички по-високи концентрации вероятно могат да бъдат изтрити от анализа чрез използване на правилото на 10 %, макар че тези концентрации могат да са полезни за определянето, че химикалът не е свързващо се вещество. Фигура 3 показва примери, когато употребата на правилото на 10 % не е подходяща.
|
Фигура 3
Примери, данни за конкурентно свързване, когато употребата на правилото на 10 % не е подходящо


Препратки
|
(1)
|
OECD (2015). Integrated Summary Report: Validation of Two Binding Assays Using Human Recombinant Estrogen Receptor Alpha (hrERα), Health and Safety Publications, Series on Testing and Assessment (No 226), Organisation for Economic Cooperation and Development, Paris.
|
|
(2)
|
Motulsky H. and Christopoulos A. (2003). The law of mass action, In Fitting Models to Biological Data Using Linear and Non-linear Regression. GraphPad Software Inc., San Diego, CA, pp 187-191. Www.graphpad.com/manuals/Prism4/RegressionBook.pdf
|
|
(3)
|
Laws SC, Yavanhxay S, Cooper RL, Eldridge JC. (2006). Nature of the Binding Interaction for 50 Structurally Diverse Chemicals with Rat Estrogen Receptors. Toxicological Sci. 94(1):46-56.
|
Б.71 ИЗСЛЕДВАНИЯ IN VITRO ЗА КОЖНА СЕНСИБИЛИЗАЦИЯ, НАСОЧЕНИ КЪМ ОСНОВНОТО СЪБИТИЕ АКТИВИРАНЕ НА ДЕНДРИТНИ КЛЕТКИ ПО ПЪТЯ, ВОДЕЩ ДО НЕБЛАГОПРИЯТЕН ЕФЕКТ (AOP) ЗА КОЖНАТА СЕНСИБИЛИЗАЦИЯ
ОБЩО ВЪВЕДЕНИЕ
Метод на изпитване, основан на основното събитие активиране на дендритни клетки
|
1.
|
Кожният сенсибилизатор представлява вещество, което води до алергичен отговор след контакт с кожата, както е определено от Глобалната хармонизирана система на ООН за класифициране и етикетиране на химикали (GHS) (1) на Съвета за сигурност на ООН и от Регламент (ЕО) № 1272/2008 относно класифицирането, етикетирането и опаковането на вещества и смеси (CLP) (100). Налице е общо съгласие по отношение на основните биологични събития, които са в основата на кожната сенсибилизация. Настоящите познания за химичните и биологичните механизми, свързани с кожната сенсибилизация, са обобщени като път, водещ до неблагоприятен ефект в рамките на програмата на ОИСР за AOP (2), като се започне с молекулното иницииращо събитие, през междинните събития до неблагоприятния ефект, а именно алергичен контактен дерматит. В този случай молекулното иницииращо събитие (т.е. първото основно събитие) е ковалентното свързване на електрофилни вещества към нуклеофилни центрове в кожни белтъци. Второто основно събитие в този AOP се извършва в кератиноцитите и включва възпалителни реакции, както и промени в генната експресия, свързана със специфични сигнални пътища в клетките, като например пътищата, зависими от елемент за антиоксидантен отговор/отговор на електрофилно въздействие (ARE). Третото основно събитие е активирането на дендритни клетки (ДК), обичайно оценявано по експресията на специални повърхностни клетъчни маркери, хемокини и цитокини. Четвъртото основно събитие е активиране и пролиферация на Т-клетки, което се оценява непряко в изследването върху локални лимфни възли (LLNA) на мишки (3).
|
|
2.
|
Настоящият метод за изпитване (МИ) е еквивалентен на насоките за изпитване на ОИСР (ТG) 442E (2017). В него се описват изследванията in vitro, които са насочени към механизмите, описани в рамките на основното събитие, свързано с активирането на дендритни клетки на АОР за кожна сенсибилизация (2). МИ включва изпитвания, които трябва да се използват за подпомагане на разграничаването между кожни сенсибилизатори и непредизвикващи сенсибилизация вещества в съответствие с GHS на ООН и Регламента CLP.
|
|
Описаните в настоящия МИ изпитвания са:
|
—
|
Изпитване с активиране на човешка клетъчна линия (h-CLAT)
|
|
—
|
Изпитване с активиране на клетъчна линия U937 (U-SENS™)
|
|
—
|
Изследване за репортерен ген за интерлевкин-8 (изследване за IL-8 Luc)
|
|
|
|
3.
|
Изпитванията, включени в настоящия метод за изпитване, и съответните TG на ОИСР могат да се различават по отношение на процедурата, използвана за генериране на данните, и измерванията, но могат да се използват без разграничение в отговор на изискванията на държавите за резултати от изпитването на основното събитие, свързано с активирането на дендритните клетки на AOP за кожна сенсибилизация, като същевременно се извличат ползи от взаимното приемане на данни от ОИСР.
|
Контекст и принципи на изпитванията, включени в метода за изпитване, основан на основно събитие
|
4.
|
Оценката на кожната сенсибилизация обикновено е включвала използването на лабораторни животни. Класическите методи с използване на морски свинчета — изпитването на Magnusson—Kligman върху морски свинчета с максимизиране и изпитването на Buehler (метод за изпитване Б.6 (4)) — оценяват както индукционната фаза, така и фазата на проявяване на кожната сенсибилизация. Изпитванията с мишки, LLNA (МИ Б.42) (3) и двете му нерадиоактивни изменения, LLNA: DA (МИ Б.50 (5)) и LLNA: BrdU-ELISA (метод за изпитване Б.51 (6)), всички от които оценяват изключително индуктивния отговор, и също са възприети, тъй като спрямо изпитванията върху морски свинчета предоставят предимство по отношение на хуманното отношение към животните, заедно с обективно измерване на индукционната фаза на кожната сенсибилизация.
|
|
5.
|
Неотдавнашни механистично-базирани МИ in chemico и in vitro, насочени към първото основно събитие (МИ Б.59; Изследване за директна реакционна способност спрямо пептиди (7)) и второто основно събитие (МИ Б.60; Метод за изпитване ARE-Nrf2 луцифераза (8)) от AOP за кожна сенсибилизация, бяха приети, за да допринесат за оценката на потенциала на химикалите за опасност от кожна сенсибилизация.
|
|
6.
|
Изпитванията, описани в настоящия метод за изпитване, или определят количествено промяната в експресията на повърхностни клетъчни маркери, свързана с процеса на активиране на моноцитите и ДК след експозиция на сенсибилизатори (напр. CD54, CD86) или с промените в експресията на IL-8, цитокин, свързани с активирането на ДК. Докладвани са кожни сенсибилизатори, предизвикващи експресия на клетъчномембрани маркери, като CD40, CD54, CD80, CD83, и CD86, в допълнение към индуцирането на провъзпалителни цитокини, като например IL-1β и TNF-α, и няколко хемокини включително IL-8 (CXCL8) и CCL3 (9) (10) (11) (12), свързани с активиране на ДК (2).
|
|
7.
|
Въпреки това, тъй като активирането на ДК представлява само едно основно събитие от AOP за кожна сенсибилизация (2) (13), информацията, получена само от изпитванията за измерване на маркерите за активиране на ДК може да не е достатъчна, за да се направи заключение за наличието или липсата на потенциал за кожна сенсибилизация на химикалите. Поради това се предлага данните, получени с описаните в настоящия метод за изпитване изпитвания, да подкрепят разграничаването между кожни сенсибилизатори (т.е. категория 1 по GHS на ООН/Регламент CLP) и непредизвикващи сенсибилизация вещества, когато се използват в рамките на интегрирани подходи към изпитването и оценката (IATA), заедно с друга относима допълнителна информация, например изведена от изследвания in vitro, насочени към други основни събития от AOP за кожна сенсибилизация, както и от методи, различни от изпитвания, включително read-across от химически аналози (13). Примери за използването на данни, получени с тези изпитвания в рамките на определени подходи, т.е. подходи, които са стандартизирани както по отношение на набора от използвани източници на информация, така и по отношение на процедурата, прилагана към данните, с оглед извеждане на прогнози, са публикувани (13) и могат да бъдат използвани като полезни елементи в рамките на IATA.
|
|
8.
|
Изпитванията, описани в настоящия метод за изпитване, не могат да се използват самостоятелно, нито за подкатегоризация на кожните сенсибилизатори към подкатегории 1A и 1B, както е определено в GHS на ООН/Регламент CLP, за органите, които прилагат тези две незадължителни подкатегории, нито за прогнозиране на потенциала с оглед вземане на решения за оценка на безопасността. Независимо от това, в зависимост от регулаторната рамка, положителните резултати, получени по тези методи, могат да се използват самостоятелно за класифициране на даден химикал в категория 1 по GHS на ООН/Регламент CLP.
|
|
9.
|
Терминът „изпитван химикал“ се използва в настоящия метод за изпитване за обозначаване на това, което се изпитва (101), и не е свързан с приложимостта на изпитванията за изпитване на вещества с една съставка, вещества с повече съставки и/или смеси. Понастоящем има ограничена информация относно приложимостта на изпитванията за вещества, състоящи се от повече от една съставка/смеси (14) (15). Независимо от това изпитванията са технически приложими за изпитването на вещества с повече съставки и на смеси. Въпреки това, преди използването на настоящия метод за изпитване по отношение на смес с цел генериране на данни за планирана регулаторна цел, следва да се разгледа въпросът дали той може да даде адекватни резултати за тази цел и, ако е така, защо (102). Подобни съображения не са необходими, когато има регулаторно изискване за изпитване на сместа. Освен това, когато се извършва изпитване на вещества с повече съставки или на смеси, следва да се обърне внимание на възможно смущаване на наблюдаваните отклици от цитотоксични съставки.
|
ЛИТЕРАТУРА
|
(1)
|
United Nations UN (2015). Globally Harmonized System of Classification and Labelling of Chemicals (GHS). Sixth revised edition. New York & Geneva: United Nations Publications. ISBN: 978-92-1-117006-1. На разположение на адрес: https://www.unece.org/trans/danger/publi/ghs/ghs_rev06/06files_e.html.
|
|
(2)
|
OECD (2012). The Adverse Outcome Pathway for Skin Sensitisation Initiated by Covalent Binding to Proteins. Part 1: Scientific Evidence. Series on Testing and Assessment No. 168. На разположение на адрес: http://www.oecd.org/officialdocuments/publicdisplaydocumentpdf/?cote=ENV/JM/MONO(2012)10/PART1&docLanguage=En.
|
|
(3)
|
Глава Б.49 от настоящото приложение: Изследване на локалните лимфни възли
|
|
(4)
|
Глава Б.6 от настоящото приложение: Кожна сенсибилизация.
|
|
(5)
|
Глава Б.50 от настоящото приложение: Кожна сенсибилизация: изследване на локалните лимфни възли: DA.
|
|
(6)
|
Глава Б.51 от настоящото приложение: Кожна сенсибилизация: изследване на локалните лимфни възли: BrdU-ELISA.
|
|
(7)
|
Глава Б.59 от настоящото приложение: In chemico кожна сенсибилизация: Изследване за директна реакционна способност спрямо пептиди (DPRA).
|
|
(8)
|
Глава Б.60 от настоящото приложение: In vitro кожна сенсибилизация: Метод за изпитване ARE-Nrf2 луцифераза
|
|
(9)
|
Steinman RM. (1991). The dendritic cell system and its role in immunogenicity. Annu Rev Immunol 9:271-96.
|
|
(10)
|
Caux C, Vanbervliet B, Massacrier C, Azuma M, Okumura K, Lanier LL, and Banchereau J. (1994). B70/B7-2 is identical to CD86 and is the major functional ligand for CD28 expressed on human dendritic cells. J Exp Med 180:1841-7.
|
|
(11)
|
Aiba S, Terunuma A, Manome H, and Tagami H. (1997). Dendritic cells differently respond to haptens and irritants by their production of cytokines and expression of co-stimulatory molecules. Eur J Immunol 27:3031-8.
|
|
(12)
|
Aiba S, Manome H, Nakagawa S, Mollah ZU, Mizuashi M, Ohtani T, Yoshino Y, and Tagami. H. (2003). p38 mitogen-activated protein kinase and extracellular signal-regulated kinases play distinct roles in the activation of dendritic cells by two representative haptens, NiCl2 and DNCB. J Invest Dermatol 120:390-8.
|
|
(13)
|
OECD (2016). Series on Testing & Assessment No 256: Guidance Document On The Reporting Of Defined Approaches And Individual Information Sources To Be Used Within Integrated Approaches To Testing And Assessment (IATA) For Skin Sensitisation, Annex 1 and Annex 2. ENV/JM/HA(2016)29. Organisation for Economic Cooperation and Development, Paris. На разположение на адрес: https://community.oecd.org/community/iatass.
|
|
(14)
|
Ashikaga T, Sakaguchi H, Sono S, Kosaka N, Ishikawa M, Nukada Y, Miyazawa M, Ito Y, NishiyamaN, Itagaki H. (2010). A comparative evaluation of in vitro skin sensitisation tests: the human cell-line activation test (h-CLAT) versus the local lymph node assay (LLNA). Altern. Lab. Anim. 38, 275-284.
|
|
(15)
|
Piroird, C., Ovigne, J.M., Rousset, F., Martinozzi-Teissier, S., Gomes, C., Cotovio, J., Alépée, N. (2015). The Myeloid U937 Skin Sensitization Test (U-SENS) addresses the activation of dendritic cell event in the adverse outcome pathway for skin sensitization. Toxicol. In Vitro 29, 901-916.
|
Допълнение 1
IN VITRO КОЖНА СЕНСИБИЛИЗАЦИЯ: ИЗПИТВАНЕ С АКТИВИРАНЕ НА ЧОВЕШКА КЛЕТЪЧНА ЛИНИЯ (H-CLAT)
ПЪРВОНАЧАЛНИ СЪОБРАЖЕНИЯ И ОГРАНИЧЕНИЯ
|
1.
|
h-CLAT определя количествено промените в експресията на повърхностни клетъчни маркери, свързани с процеса на активиране на моноцити и дендритни клетки (ДК) (т.е. CD86 и CD54) в моноцитната клетъчна линия THP-1 от левкемия при човека след експозиция на сенсибилизиращи вещества (1) (2). Измерените нива на експресия на повърхностни клетъчни маркери CD86 и CD54 се използват след това за подпомагане на разграничаването между кожни сенсибилизатори и непредизвикващи сенсибилизация вещества.
|
|
2.
|
h-CLAT беше оценен в координирано от референтна лаборатория на Европейския съюз за алтернативи на изпитванията върху животни (EURL ECVAM) проучване за валидиране и последваща независима партньорска проверка от страна на Научния консултативен комитет на EURL ECVAM (ESAC). Като взе предвид всички налични доказателства и данни от регулаторните органи и заинтересованите страни, EURL ECVAM препоръча h-CLAT (3) за използване като част от IATA за подкрепа на разграничаването между сенсибилизатори и непредизвикващи сенсибилизация вещества за целите на класифицирането на опасността и етикетирането. Примери за използването на данни от h-CLAT в комбинация с друга информация са описани в литературата (4)(5)(6)(7)(8)(9)(10)(11).
|
|
3.
|
h-CLAT се оказа прехвърляем на лаборатории с опит в областта на техниките за клетъчна култура и анализ с поточна цитометрия. Нивото на възпроизводимостта в прогнозите, което може да се очаква от изпитването, е от порядъка на 80 % вътре в самата лаборатория и между лабораториите (3)(12).. Резултатите, получени в изследването за валидиране (13) и други публикувани изследвания (14) като цяло показват, че в сравнение с резултатите от LLNA точността при разграничаването на кожните сенсибилизатори (т.е. категория 1 по GHS на ООН/CLP) от непредизвикващи сенсибилизация вещества е 85 % (N = 142), с чувствителност от 93 % (94/101) и специфичност от 66 % (27/41) (въз основа на повторен анализ от EURL ECVAM (12), като се вземат предвид всички съществуващи данни, без да се разглеждат отрицателните резултати за химикали с Log Kow, по-голям от 3,5, както е описано в точка 4). Има по-голяма вероятност неверните отрицателни прогнози, свързани с h-CLAT, да се отнасят за химикали, които са с малък до умерен потенциал за кожна сенсибилизация (т.е. кожна сенсибилизация подкатегория 1B по GHS на ООН/Регламент CLP), отколкото при химикали, показващи голям потенциал за кожна сенсибилизация (т.е. подкатегория 1A по GHS на ООН/Регламент CLP) (4)(13)(15). Взета заедно, тази информация показва ползата от метода h-CLAT за подпомагане на определянето на опасността от кожна сенсибилизация. Независимо от това стойностите за точността, посочени тук за h-CLAT като самостоятелно изпитване, са само ориентировъчни, тъй като изпитването трябва да се разглежда в съчетание с други източници на информация в контекста на IATA и в съответствие с разпоредбите на точки 7 и 8 от Общото въведение. Освен това при оценяването на методи за сенсибилизация на кожата, които не използват животни, следва да се има предвид, че LLNA изпитването, както и други изпитвания върху животни, могат да не отразяват напълно положението при хората.
|
|
4.
|
Въз основа на наличните понастоящем данни бе показано, че методът h-CLAT е приложим по отношение на изпитвани химикали, които обхващат различни органични функционални групи, реакционни механизми, потенциал за сенсибилизация на кожата (както е определена в in vivo проучвания) и физични и химични свойства (3)(14)(15). Методът h-CLAT се прилага за изпитвани химикали, които са разтворими или образуват стабилна дисперсия (т.е. колоид или суспензия, в която изпитваният химикал не се утаява или отделя от разтворителя/носителя в различни фази) в подходящ разтворител/носител (вж. точка 14). Изпитваните химикали с Log Kow по-голямо от 3,5 обикновено дават неверни отрицателни резултати (14). Поради това отрицателните резултати, получени при изпитвани химикали с Log Kow, по-голямо от 3,5, следва да не се вземат под внимание. Все пак, положителните резултати, получени при изпитвани химикали с Log Kow, по-голямо от 3,5, могат да се използват, за да се подпомогне идентифицирането на изпитвания химикал като кожен сенсибилизатор. Освен това, поради ограничения метаболитен капацитет на използваната клетъчна линия (16) и поради условията на опита, про-хаптените (т.е. вещества, които изискват ензимно активиране, например чрез P450 ензими) и пре-хаптените (т.е. вещества, активирани чрез окисление), по-специално тези с малка скорост на окисление, могат също да дадат отрицателни резултати при h-CLAT (15). Флуоресцентните изпитвани химикали могат да бъдат оценени с h-CLAT (17), но независимо от това, силно флуоресцентните изпитвани химикали, които излъчват при същата дължина на вълната като флуоресцеиновия изотиоцианат (FITC) или като пропидиевия йодид (PI), ще предизвикат смущения при откриването с поточна цитометрия и следователно не могат да бъдат правилно оценени с помощта на FITC-конюгирани антитела или PI. В такъв случай могат да се използват други антитела, маркирани с флуорохром, или съответно други маркери за цитотоксичност, при условие че може да се покаже, че дават сходни резултати като маркираните с FITC антитела (вж. точка 24) или PI (вж. точка 18), напр. чрез изпитване на веществата за изпитване за пригодност от допълнение 1—2. С оглед на гореизложеното отрицателните резултати следва да се тълкуват в контекста на посочените ограничения и заедно с други източници на информация в рамките на IATA. В случаите, когато са налице доказателства за неприложимостта на h-CLAT метода по отношение на други специфични категории изпитвани химикали, той не следва да се използва за тези специфични категории.
|
|
5.
|
Както е описано по-горе, методът h-CLAT подкрепя разграничаването между кожни сенсибилизатори и непредизвикващи сенсибилизация вещества. Той обаче също така може потенциално да допринесе за оценка на потенциала за сенсибилизация (4)(5)(9), когато се използва в интегрирани подходи, като например IATA. Независимо от това се изисква по-нататъшна работа, за предпочитане въз основа на данни за човека, за определяне на това как резултатите от h-CLAT могат евентуално да послужат за информация за оценка на потенциала.
|
|
6.
|
Определенията са дадени в допълнение 1.1.
|
ПРИНЦИП НА ИЗПИТВАНЕТО
|
7.
|
Методът h-CLAT е in vitro изследване, което определя количествено промените в експресията на повърхностни клетъчни маркери (т.е. CD86 и CD54) в моноцитната клетъчна линия THP-1 от левкемия при човека, след 24 часова експозиция на изпитвания химикал. Тези повърхностни молекули са типични маркери за активиране на моноцитни THP-1 и могат да имитират активиране на ДК, което играе решаваща роля при праймирането на Т-клетките. Промените в експресията на повърхностните маркери се измерват чрез поточна цитометрия след клетъчно оцветяване с антитела, маркирани с флуорохром. Измерването на цитотоксичността се извършва също така едновременно, за да се оцени дали настъпва регулиране на експресията на повърхностните маркери в посока увеличаване при субцитотоксични концентрации. Относителният интензитет на флуоресценцията на повърхностните маркери в сравнение с контролата на разтворител/носител се изчислява и използва в модела за прогнозиране (вж. точка 26), за да се подкрепи разграничаването между сенсибилизатори и непредизвикващи сенсибилизация вещества
|
ДОКАЗВАНЕ НА ПРИГОДНОСТ
|
8.
|
Преди рутинното използване на изпитването, описано в настоящото допълнение към метод за изпитване Б.71, лабораториите следва да докажат техническата си пригодност, като използват 10-те вещества за изпитване на пригодност, изброени в допълнение 1.2. Освен това ползвателите на изпитвания следва да поддържат база данни с данни за предходни периоди, получени с проверките на реакционната способност (вж. точка 11) и с положителните контроли и контролите на разтворител/носител (вж. точки 20—22), и да използват тези данни, за да потвърдят, че възпроизводимостта на изпитването в тяхната лаборатория се запазва с течение на времето.
|
ПРОЦЕДУРА
|
9.
|
Това изпитване се основава на Протокол № 158 за h-CLAT от базата данни за методи, алтернативни на експериментите с животни (DB-ALM) (18), който представлява протоколът, използван за изследването за валидиране, координирано от EURL ECVAM. Препоръчва се този протокол да се използва при прилагането и използването на h-CLAT метода в лабораторни условия. Следва описание на основните компоненти и процедури за метода h-CLAT, който се състои от две стъпки: изследване за определяне на дозата и измерване на експресията на CD86/CD54.
|
Подготовка на клетките
|
10.
|
За извършване на метода h-CLAT следва да се използва моноцитната клетъчна линия THP-1 от левкемия при човека. Препоръчва се получаването на клетки (TIB-202 ™) от добре квалифицирана клетъчна банка, като например Американската банка за типови култури (American Type Culture Collection).
|
|
11.
|
Клетките THP-1 се култивират при 37oC, 5 % CO2 и овлажнена атмосфера в среда RPMI-1640, с добавка на 10 % фетален говежди серум (FBS), 0,05 mM 2-меркаптоетанол, 100 единици/ml пеницилин и 100 μg/ml стрептомицин. Употребата на пеницилин и на стрептомицин в хранителната среда може да бъде избегната. В такъв случай обаче ползвателите следва да се уверят, че липсата на антибиотици в средата за отглеждане не оказва въздействие върху резултатите, например чрез изпитване на веществата за пригодност, изброени в допълнение 1.2. Във всеки случай, за да се сведе до минимум рискът от замърсяване, трябва да се следват добрите практики при клетъчните култури, независимо от наличието или липсата на антибиотици в средата за отглеждане на клетки. THP-1 клетки се посяват редовно всеки 2—3 дни при плътност от 0,1 до 0,2 × 106 клетки/ml. Те трябва да се поддържат при плътности от 0,1 до 1,0 × 106 клетки/ml. Преди да бъдат използвани за изпитване, клетките следва да бъдат квалифицирани чрез извършване на проверка за реакционна способност. Проверката на реакционната способност на клетките следва да се извърши, като се използват положителните контроли, 2,4-динитрохлоробензен (DNCB) (CAS № 97-00-7, ≥ 99 % чистота) и никелов сулфат (NiSO4) (CAS № 10101-97-0, чистота ≥ 99 %) и отрицателната контрола, млечна киселина (LA) (CAS № 50-21-5, чистота ≥ 85 %), две седмици след размразяване. Както DNCB, така и NiSO4 следва да дадат положителен отклик при повърхностни клетъчни маркери както CD86, така и CD54, а LA следва да даде отрицателен отклик при повърхностни клетъчни маркери както CD86, така и CD54. За изследването се използват само клетките, които са преминали проверката за реакционна способност. Клетките могат да бъдат размножавани до два месеца след размразяването. Броят пасажи не трябва да надвишава 30. Проверката за реакционна способност следва да се извършва в съответствие с процедурите, описани в точки 20—24.
|
|
12.
|
За изпитване THP-1 клетките се посяват с плътност 0,1 × 106 клетки/ml или 0,2 × 106 клетки/ml, и се извършва предварително култивиране в колби за клетъчни култури съответно за 72 часа или 48 часа. Важно е плътността на клетките в колбата за за клетъчни култури непосредствено след периода на предварително култивиране да бъде възможно най-последователна във всеки експеримент (като се използва едно от двете условия на предварително култивиране, описани по-горе), тъй като плътността на клетките в колбата за култури непосредствено след предварителното култивиране би могла да засегне експресията на CD86/CD54, индуцирана от алергени (19). В деня на изпитването клетките, събрани от колбата за култури, се ресуспендират с прясна хранителна среда при 2 × 106 клетки/ml. След това клетките се разпределят в 24-ямкова плоскодънна плака (500 μl, 1 × 106 клетки/ямка) или 96-ямкова плоскодънна плака (80 μl, 1,6 × 105 клетки/ямка).
|
Изследване за определяне на дозата
|
13.
|
Извършва се изследване за определяне на дозата, за да се определи CV75, което е концентрацията на изпитвания химикал, която води до 75 % клетъчна жизнеспособност (CV) в сравнение с контролата на разтворител/носител. Стойността на CV75 се използва, за да се определи концентрацията на изпитваните химикали за измерване на експресията на CD86/CD54 (вж. точки 20—24).
|
Приготвяне на изпитваните химикали и веществата за контроли
|
14.
|
Изпитваните химикали и веществата за контроли се приготвят в деня на изпитването. По отношение на метода h-CLAT, изпитваните химикали се разтварят или трайно се диспергират (вж. също точка 4) във физиологичен разтвор или среда като първи вариант на разтворител/носител, или в диметилсулфоксид (DMSO, ≥ 99 % чистота) като втори вариант на разтворител/носител, ако изпитваният химикал не е разтворим или не образува стабилна дисперсия в предходните два разтворителя/носителя, до крайни концентрации от 100 mg/ml (във физиологичен разтвор или среда) или 500 mg/ml (в DMSO). Могат да се използват и други разтворители/носители, освен описаните по-горе, при предоставяне на достатъчна научна обосновка. Следва да се вземе предвид стабилността на изпитвания химикал в крайния разтворител/носител.
|
|
15.
|
Като се започне от изходните разтвори от 100 mg/ml (във физиологичен разтвор или среда) или 500 mg/ml (в DMSO) на изпитваните химикали, следва да се направят следните стъпки на разреждане:
|
—
|
За физиологичен разтвор или среда като разтворител/носител: Приготвят се осем изходни разтвора (осем концентрации) чрез серия от двукратни разреждания, като се използва съответният разтворител/носител. След това тези изходни разтвори се разреждат 50-кратно в среда за отглеждане (работни разтвори). Ако горната крайна концентрация в плаката от 1 000 μg/ml е нетоксична, максималната концентрация следва да се определи отново чрез провеждането на ново изпитване за цитотоксичност. Крайната концентрация в плаката не трябва да надвишава 5 000 μg/ml за изпитвани химикали, разтворени или стабилно диспергирани във физиологичен разтвор или среда.
|
|
—
|
За DMSO като разтворител/носител: Приготвят се осем изходни разтвора (осем концентрации) чрез серия от двукратни разреждания, като се използва съответният разтворител/носител. След това тези изходни разтвори се разреждат допълнително 250-кратно в среда за отглеждане (работни разтвори). Крайната концентрация в плаката не трябва да надвишава 1 000 μg/ml, дори ако тази концентрация не е токсична.
|
Работните разтвори в крайна сметка се използват за експозиция чрез добавяне на равен обем работен разтвор към обема на суспензията от THP-1 клетки в плаката (вж. също точка 17) за постигане на допълнително двукратно разреждане (обикновено, окончателният обхват на концентрациите в плаката е 7,81—1 000 μg/ml).
|
|
16.
|
Контролата на разтворител/носител, използвана в метода h-CLAT, е среда за отглеждане (за изпитвани химикали, които са разтворени или трайно диспергирани (вж. точка 4) или в среда, или във физиологичен разтвор), или DMSO (за изпитвани химикали, които са разтворени или трайно диспергирани в DMSO), изпитвана при единична крайна концентрация от 0,2 % в плаката. Тя се подлага на същото разреждане, както е описано за работните разтвори в точка 15.
|
Прилагане на изпитваните химикали и веществата за контроли
|
17.
|
Средата за отглеждане или работните разтвори, описани в точки 15 и 16, се смесват 1: 1 (v/v) с клетъчните суспензии, подготвени в 24-ямковата или 96-ямковата плоскодънна плака (вж. точка 12). След това третираните плаки се инкубират за 24 ± 0,5 часа при 37oC под 5 % CO2. Следва да се вземат мерки за избягване на изпаряването на летливи изпитвани химикали и на кръстосаното замърсяване с изпитвани химикали между ямките, като напр. плаките се запечатат преди инкубирането с изпитваните химикали (20).
|
Оцветяване с пропидиев йодид (PI)
|
18.
|
След 24 ± 0,5 часа експозиция клетките се прехвърлят в епруветки и се събират чрез центрофугиране. Супернатантите се изхвърлят и оставащите клетки се ресуспендират с 200 μl (в случай на 96-ямкова) или 600 μl (в случай на 24-ямкова плака) фосфатно буфериран физиологичен разтвор, съдържащ 0,1 % говежди серумен албумин (оцветяващ буфер). 200 μl от клетъчната суспензия се прехвърлят в 96-ямкова облодънна плака (в случай на 96-ямкова) или в микроепруветка (в случай на 24-ямкова) и се промиват два пъти с 200 μl (в случай на 96-ямкова) или 600 μl (в случай на 24-ямкова) оцветяващ буфер. Накрая, клетките се ресуспендират в оцветяващ буфер (напр. 400 μl) и се добавя PI разтвор (напр. 20 μl) (например, крайната концентрация на PI е 0,625 μg/ml). Други маркери за цитотоксичност, като 7-аминоактиномицин D (7-AAD), трипаново синьо или други, могат да бъдат използвани, ако може да се докаже, че алтернативните оцветители осигуряват сходни резултати като PI, например чрез изпитване на веществата за пригодност от допълнение 1.2.
|
Измерване на цитотоксичността чрез поточна цитометрия и оценка на стойността на CV75
|
19.
|
Фиксацията на PI се анализира с помощта на поточна цитометрия с канал за придобиване на данни FL-3. Придобиват се данни за общо 10 000 живи клетки (PI-отрицателни). Клетъчната жизнеспособност може да бъде изчислена с помощта на следната формула от програмата за анализ на цитометъра. Когато клетъчната жизнеспособност е малка, следва да се придобият данни за до 30 000 клетки, включително мъртви клетки. Друга възможност е данните да бъдат придобити в продължение на една минута след започването на анализа.

Стойността на CV75 (вж. точка 13), т.е. концентрация, показваща 75 % преживяемост на клетки (25 % цитотоксичност), се изчислява чрез log-линейна интерполация, като се използва следното уравнение:

където:
a е минималната стойност на клетъчната жизнеспособност над 75 %
c е максималната стойност на клетъчната жизнеспособност под 75 %
b и d са концентрациите, показващи съответно стойността на клетъчната жизнеспособност a и c

Могат да се използват други подходи за извеждане на CV75, при условие че се докаже, че това няма въздействие върху резултатите (напр. чрез изпитване на веществата за изпитване за пригодност).
|
Измерване на експресията на CD86/CD54
Приготвяне на изпитваните химикали и веществата за контроли
|
20.
|
Подходящият разтворител/носител (физиологичен разтвор, среда или DMSO; вж. точка 14) се използва за разтваряне или трайно диспергиране на изпитваните химикали. Изпитваните химикали се разреждат първо до концентрацията, съответстваща на 100-кратна (за физиологичен разтвор или среда) или 500-кратна (за DMSO) на 1,2 × CV75, определена в изследването за определяне на дозата (вж. точка 19). Ако не може да се определи CV75 (т.е. ако в изпитването за определяне на дозата не се наблюдава достатъчна цитотоксичност) , като начална концентрация следва да се използва концентрацията на най-голямата разтворимост или най-стабилна дисперсия на изпитвания химикал, приготвен с всеки разтворител/носител. Обърнете внимание, че крайната концентрация в плаката не следва да надвишава 5 000 μg/ml (в случай на физиологичен разтвор или среда) или 1 000 μg/ml (в случая на DMSO). След това се правят 1,2-кратни серийни разреждания със съответния разтворител/носител, за да се получат изходните разтвори (осем концентрации, обхващащи от 100 × 1,2 × CV75 до 100 × 0,335 × CV75 (за физиологичен разтвор или среда) или от 500×1,2 × CV75 до 500×0,335 × CV75 (за DMSO)), които да бъдат изпитани при метода h-CLAT (вж. DB-ALM протокол № 158 за пример на схема за дозиране). След това изходните разтвори се разреждат допълнително 50-кратно (за физиологичен разтвор или среда) или 250-кратно (за DMSO) в среда за отглеждане (работни разтвори). Тези работни разтвори в крайна сметка се използват за експозиция, с допълнително крайно двукратно разреждане в плаката. Ако резултатите не отговарят на критериите за приемливост, описани в точки 29 и 30, по отношение на клетъчната жизнеспособност, може да се повтори изследването за определяне на дозата, за да се определи по-точно CV75. Моля, имайте предвид, че само 24-ямкови плаки могат да бъдат използвани за измерване на експресия на CD86/CD54.
|
|
21.
|
Контролата на разтворител/носител се приготвя както е описано в точка 16. Положителната контрола, използвана в метода h-CLAT, е DNCB (вж. точка 11), за която изходните разтвори се приготвят в DMSO и се разреждат както е описано за изходните разтвори в точка 20. DNCB следва да се използва като положителната контрола за измерване на експресията на CD86/CD54 при крайна единична концентрация в плаката (обикновено 4,0 μg/ml). За получаване на концентрация на DNCB от 4,0 μg/ml в плаката се приготвя 2 mg/ml изходен разтвор на DNCB в DMSO и допълнително се разрежда 250-кратно със среда за отглеждане до 8 μg/ml работен разтвор. Като алтернатива, CV75 на DNCB, която се определя на всяко място за изпитване, също може да се използва като концентрация на положителна контрола. Могат да се използват други подходящи положителни контроли, ако има налични данни за предходни периоди, за извеждане на сравними критерии за приемливост на серията. За положителни контроли крайната единична концентрация в плаката не следва да надвишава 5 000 μg/ml (в случай на физиологичен разтвор или среда) или 1 000 μg/ml (в случая на DMSO). Критериите за приемливост на серията са същите като тези, които са описани за изпитвания химикал (вж. точка 29), с изключение на последния критерий за приемливост, тъй като положителната контрола се изпитва при една-единствена концентрация.
|
Прилагане на изпитваните химикали и веществата за контроли
|
22.
|
За всеки изпитван химикал и за вещество за контроли е необходим един експеримент, за да се получи прогноза. Всеки експеримент се състои от поне две независими серии за измерване на експресията на CD86/CD54 (вж. точки 26—28). Всяка независима серия се извършва в различен ден или в рамките на един и същи ден, при условие че за всяка серия: a) се приготвят независими пресни изходни разтвори и работни разтвори на изпитвания химикал и разтвори на антитела, и б) използват се независимо събрани клетки (т.е. клетките се събират от различни колби за отглеждане); въпреки това, клетките могат да са от един и същ пасаж. Изпитваните химикали и веществата за контроли, приготвени като работни разтвори (500 μl), се смесват с 500 μl от суспендираните клетки (1x106 клетки) при съотношение 1:1, и клетките се инкубират за 24 ± 0,5 часа, както е описано в точки 20 и 21. За всяка серия единично повторение за всяка концентрация на изпитвания химикал и вещество за контроли е достатъчно, защото прогноза се получава от най-малко две независими серии.
|
Оцветяване и анализ на клетките
|
23.
|
След 24 ± 0,5 часа експозиция клетките се прехвърлят от 24-ямкови плаки в епруветки, събират се чрез центрофугиране и след това се промиват два пъти с 1 ml оцветяващ буфер (ако е необходимо, могат да се предприемат допълнителни стъпки на промиване). След промиване клетките се блокират с 600 μl блокиращ разтвор (оцветяващ буфер, съдържащ 0,01 % (w/v) глобулин (Cohn фракция II, III, човешки; SIGMA, # G2388—10G или еквивалентен ) и се инкубират при температура 4oC в продължение на 15 минути. След блокирането клетките се разделят на три аликвотни части от по 180 μl в 96-ямкова облодънна плака или микроепруветка.
|
|
24.
|
След центрофугирането клетките се оцветяват с 50 μl маркирани с FITC анти-CD86, анти-CD54 или миши антитела от (изотип) IgG1 при 4oC в продължение на 30 минути. Антителата, описани в DB-ALM протокол № 158 за h-CLAT (18) следва да се използват чрез разреждане 3: 25 v/v (за CD86 (BD-PharMingen, # 555657; Clone: Fun-1)) или 3:50 v/v (за CD54 (DAKO, # F7143; Clone: 6.5B5) и IgG1 (DAKO, # X0927) с оцветяващ буфер. Тези коефициенти на разреждане на антитела са определени от разработилите изпитването като такива, които осигуряватнай-доброто съотношение сигнал/шум. Въз основа на опита на разработилите изпитването интензитетът на флуоресценцията на антителата обикновено е последователен между различните партиди. Въпреки това ползвателите могат да разгледат титруване на антитела в условията на собствената си лаборатория, за определяне най-добрите концентрации за използване. Могат да се използват и други маркирани с флуорхром анти-CD86 и/или анти-CD54 антитела, ако може да се докаже, че те осигуряват сходни резултати като FITC-конюгираните антитела, например чрез изпитване на веществата за пригодност от допълнение 1.2. Следва да се отбележи, че промяната на клона или доставчика на антителата, описани в DB-ALM протокол № 158 за h-CLAT (18), може да повлияе на резултатите. След промиване два или повече пъти с 150 μl оцветяващ буфер, клетките се ресуспендират в оцветяващ буфер (напр. 400 μl), и се добавя PI-разтворът (напр. 20 μl за получаване на крайна концентрация от 0,625 μg/ml) или друг разтвор на маркер за цитотоксичност (вж. точка 18). Нивата на експресията на CD86 и CD54 и клетъчната жизнеспособност се анализират с помощта на поточна цитометрия.
|
ДАННИ И ПРОТОКОЛИРАНЕ
Оценка на данните
|
25.
|
Експресията на CD86 и CD54 се анализира с помощта на поточна цитометрия с канал за придобиване на данни FL-1. Въз основа на средната геометрична стойност на интензитета на флуоресценция (MFI) по следното уравнение се изчислява относителният интензитет на флуоресценцията (RFI) на CD86 и CD54 за клетки от положителни контроли (ctrl) и третирани с химикал клетки:

Клетъчната жизнеспособност на клетките от контролата с изотип (ctrl) (които се оцветяват с миши антитела от (изотип) IgG1, се изчислява също в съответствие с формулата, описана в точка 19.
|
Модел на прогнозиране
|
26.
|
За измерване на експресията на CD86/CD54 всеки изпитван химикал се изпитва в най-малко две независими серии с цел да се изведе единична прогноза (ПОЛОЖИТЕЛНА или ОТРИЦАТЕЛНА). Прогнозата на h-CLAT се счита за ПОЛОЖИТЕЛНА, ако поне едно от следните условия е изпълнено през 2 от 2 или в най-малко 2 от 3 независими серии, в противен случай прогнозата на h-CLAT се счита за ОТРИЦАТЕЛНА (фигура 1):
|
—
|
RFI на CD86 е равен на 150 % или по-голям при всяка изпитвана концентрация (с клетъчна жизнеспособност ≥ 50 %);
|
|
—
|
RFI на CD54 е равен на 200 % или по-голям при всяка изпитвана концентрация (с клетъчна жизнеспособност ≥ 50 %);
|
|
|
27.
|
Въз основа на горното, ако и двете първи серии са едновременно положителни за CD86 и/или са едновременно положителни за CD54, прогнозата на h-CLAT се счита за ПОЛОЖИТЕЛНА и не е необходимо да се провежда трета серия. По подобен начин, ако първите две серии са отрицателни за двата маркера, прогнозата на h-CLAT се счита за ОТРИЦАТЕЛНА (при надлежно отчитане на предвиденото в точка 30), без да е необходимо да се провежда трета серия. Ако обаче първите две серии не са съгласувани за поне един от маркерите (CD54 или CD86), е необходима трета серия и окончателната прогноза ще се базира на мажоритарния резултат от трите отделни серии (т.е. 2 от 3). В това отношение следва да се отбележи, че ако се провеждат два независими серии и едната е положителна само за CD86 (наричана по-долу P1), а другата е положителна само за CD54 (наричана по-долу P2), е необходима трета серия. Ако тази трета серия е отрицателна и за двата маркера (наричана по-долу N), се счита, че прогнозата на h-CLAT е ОТРИЦАТЕЛНА. От друга страна, ако третата серия е положителна за който и да е маркер (P1 или P2), или и за двата маркера (наричана по-долу P12), прогнозата на h-CLAT се счита за ПОЛОЖИТЕЛНА.

Фигура 1: Модел на прогнозиране, използван при метода h-CLAT. Прогнозата по h-CLAT следва да се разглежда в рамките на IATA и в съответствие с разпоредбите на точки 7 и 8 от Общото въведение.
P1: серия с положителен само CD86; P2: серия с положителен само CD54; P12: серия с положителни както CD86, така и CD54; N: серия при която положителни не са нито CD86, нито CD54.
* Клетките показват относимите комбинации от резултатите от първите две серии, независимо от реда, в който могат да бъдат получени.
# Клетките показват относимите комбинации от резултатите от трите серии въз основа на резултатите, получени в първите две серии, показани в горната клетка, но не отразяват реда, в който могат да бъдат получени.
|
|
28.
|
За изпитваните химикали, прогнозирани като ПОЛОЖИТЕЛНИ по h-CLAT, по избор могат да бъдат определени две стойности на ефективни концентрации (EC) — EC150 за CD86 и EC200 за CD54, т.е. концентрацията, при която изпитваните химикали индуцират RFI от 150 или 200. Тези EC могат да допринесат за оценката на потенциала за сенсибилизация (9), когато се използват в интегрирани подходи, като например IATA (4) (5) (6) (7) (8). Те могат да бъдат изчислени по следните уравнения:
EC 150 (for CD86) = Bconc
+ [(150 - BRFI
)/ARFI - BRFI
) × (Aconc - Bconc
)]
EC 200 (for CD86) = Bconc
+ [(200 - BRFI
)/ARFI - BRFI
) × (Aconc - Bconc
)]
където:
Aconc е най-ниската концентрация в μg/ml с RFI > 150 (CD86) или 200 (CD54)
Bconc е най-високата концентрация в μg/ml с RFI < 150 (CD86) или 200 (CD54)
ARFI е RFI при най-ниската концентрация с RFI > 150 (CD86) или 200 (CD54)
BRFI е RFI при най-високата концентрация с RFI < 150 (CD86) или 200 (CD54)
За целите на по-точното извеждане на стойностите на EC150 и EC200 могат да се изискват три независими серии за измерване на експресията на CD86/CD54. Крайните стойности на EC150 и EC200 след това се определят като медианната стойност на EC, изчислена от трите независими серии. Когато само две от трите независими серии отговарят на критериите за положителност (вж. точки 26—27), се приема по-високата EC150 или EC200 от двете изчислени стойности.
|
Критерии за приемливост
|
29.
|
Когато се използва методът h-CLAT трябва да се спазват следните критерии за приемливост (22) (27).
|
—
|
Клетъчната жизнеспособност в средата и в контролите на разтворител/носител следва да е по-висока от 90 %.
|
|
—
|
В контролата на разтворител/носител RFI стойностите както на CD86, така и на CD54 не трябва да надвишават положителните критерии (CD86 RFI ≥ 150 % и CD54 RFI ≥ 200 %). RFI стойностите на контролата на разтворител/носител се изчисляват, като се използва формулата, описана в точка 25 („MFI на химикала“ следва да се замени с „MFI на разтворителя/носителя“, а „MFI на разтворителя/носителя“ следва да се замени с „MFI на контрола (на среда)“).
|
|
—
|
За контролите както на среда, така и на разтворител/носител, MFI отношението както на CD86, така и на CD54 към контролата с изотип, следва да бъде > 105 %.
|
|
—
|
При положителната контрола (DNCB) RFI стойностите както на CD86, така и на CD54, следва да отговарят на положителните критерии (CD86 RFI ≥ 150 и CD54 RFI ≥ 200) и клетъчната жизнеспособност следва да бъде повече от 50 %.
|
|
—
|
За изпитвания химикал клетъчната жизнеспособност следва да бъде повече от 50 % в най-малко четири изпитвани концентрации за всяка серия.
|
|
|
30.
|
Отрицателни резултати са допустими само за изпитвани химикали, при които клетъчната жизнеспособност е по-малка от 90 % при най-високата изпитвана концентрация (т.е. 1,2 × CV75 в съответствие със схемата за серийно разреждане, описана в точка 20). Ако клетъчната жизнеспособност при 1,2 × C75 е равна или по-висока от 90 %, отрицателният резултат следва да бъде отстранен. В такъв случай се препоръчва да се направи опит за прецизиране на избора на доза чрез повтаряне на определянето на CV75. Следва да се отбележи, че когато 5 000 μg/ml във физиологичен разтвор (или среда, или други разтворители/носители), 1 000 μg/ml в DMSO или най-високата разтворима концентрация се използват като максимална концентрация на изпитвания химикал, приемливо е наличието на отрицателен резултат, дори ако клетъчната жизнеспособност е над 90 %.
|
Протокол от изпитването
|
31.
|
Протоколът от изпитването следва да включва следната информация.
|
Изпитван химикал
|
Вещество с една съставка:
|
Химична идентификация, като наименование(я) по IUPAC или по CAS, CAS номер(а), SMILES или InChI код, структурна формула, и/или други идентификатори;
|
|
Външен вид, Log Kow, разтворимост във вода, разтворимост в DMSO, молекулно тегло и допълнителни относими физични и химични свойства, според наличността;
|
|
Чистота, химическа идентичност на онечистванията, както е целесъобразно и практически осъществимо и т.н.;
|
|
Обработка преди изпитването, ако се прилага (напр. затопляне, смилане);
|
|
Изпитвана концентрация (концентрации);
|
|
Условия на съхранение и стабилност, доколкото са налични;
|
|
Обосновка за избора на разтворител/носител за всеки изпитван химикал.
|
|
|
Вещество с повече съставки, UVCB и смес
|
характеризиране, доколкото е възможно, чрез химическата идентичност (вж. по-горе), чистотата, количествения състав и относимите физични и химични свойства на съставките (вж. по-горе), доколкото са налични.
|
|
Външен вид, разтворимост във вода, разтворимост в DMSO и допълнителни относими физични и химични свойства, според наличността;
|
|
Молекулно тегло или привидно молекулно тегло в случай на смеси/полимери с известен състав или друга информация, относима към провеждането на изследването;
|
|
Обработка преди изпитването, ако се прилага (напр. затопляне, смилане);
|
|
Изпитвана концентрация (концентрации);
|
|
Условия на съхранение и стабилност, доколкото са налични;
|
|
Обосновка за избора на разтворител/носител за всеки изпитван химикал.
|
|
|
|
Контроли
|
Положителна контрола
|
Химична идентификация, като наименование(я) по IUPAC или по CAS, CAS номер(а), SMILES или InChI код, структурна формула, и/или други идентификатори;
|
|
Външен вид, Log Kow, разтворимост във вода, разтворимост в DMSO, молекулно тегло и допълнителни относими физични и химични свойства, според наличността, където е приложимо;
|
|
Чистота, химическа идентичност на онечистванията, както е целесъобразно и практически осъществимо и т.н.;
|
|
Обработка преди изпитването, ако се прилага (напр. затопляне, смилане);
|
|
Изпитвана концентрация (концентрации);
|
|
Условия на съхранение и стабилност, доколкото са налични;
|
|
Препратка към резултатите за положителните контроли за предходни периоди, доказващи подходящи критерии за приемливост на серията, ако е приложимо.
|
|
|
|
Отрицателна контрола и контрола на разтворител/носител
|
Химична идентификация, като наименование(я) по IUPAC или по CAS, CAS номер(а), SMILES или InChI код, структурна формула, и/или други идентификатори;
|
|
Чистота, химическа идентичност на онечистванията, както е целесъобразно и практически осъществимо и т.н.;
|
|
Външен вид, молекулно тегло и допълнителни относими физични и химични свойства, в случай че са използвани разтворители/носители, различни от упоменатите в Насоките за изпитване и в зависимост от наличността;
|
|
Условия на съхранение и стабилност, доколкото са налични;
|
|
Обосновка за избора на разтворител/носител за всеки изпитван химикал.
|
|
|
Условия на изпитване
|
Име и адрес на финансиращия, на изпитващата лаборатория и на ръководителя на изследването;
|
|
Описание на използваното изпитване;
|
|
Използвана клетъчна линия, условия за нейното съхранение и източник (например мястото, от което са взети);
|
|
Използвана поточна цитометрия (например модел), включително инструментални настройки, глобулин, антитела и маркер за цитотоксичност;
|
|
Процедурата, използвана за доказване на пригодността на лабораторията за провеждане на изпитването чрез изпитване на вещества за изпитване за пригодност, и процедурата, използвана за доказване на възпроизводимостта на резултатите от изпитването с течение на времето, например данни за контролите за предходни периоди и/или данни от предходни проверки за реакционна способност.
|
|
|
Критерии за приемане на изпитването
|
Клетъчна жизнеспособност, MFI и RFI стойностти, получени с контролата на разтворител/носител в сравнение с допустимия размах;
|
|
Клетъчна жизнеспособност и RFI стойностти, получени с положителната контрола в сравнение с допустимия размах;
|
|
Клетъчна жизнеспособност при всички изпитвани концентрации на изпитвания химикал.
|
|
|
Процедура за изпитване
|
Брой на използваните серии;
|
|
Концентрации на изпитвания химикал, продължителност на прилагане и време на експозицията (при различие от препоръчаното)
|
|
Описание на използваните критерии за оценка и за решение;
|
|
Описание на всякакви изменения на процедурата за изпитване.
|
|
|
Резултати
|
Таблично представяне на данните, включително на CV75 (ако е приложимо), индивидуални геометрични MFI, RFI, стойности за клетъчната жизнеспособност, стойности EC150/EC200 (ако е приложимо), получени за изпитвания химикал и за положителната контрола за всяка серия, като се посочва рангът на изпитвания химикал в съответствие с модела на прогнозиране;
|
|
Описание на всякакви други относими наблюдения, ако е приложимо.
|
|
|
Обсъждане на резултатите
|
Обсъждане на резултатите, получени с метода h-CLAT;
|
|
Разглеждане на резултатите от изпитването в рамките на контекста на IATA, ако е налична друга относима информация.
|
|
|
ЛИТЕРАТУРА
|
(1)
|
Ashikaga T, Yoshida Y, Hirota M, Yoneyama K, Itagaki H, Sakaguchi H, Miyazawa M, Ito Y, Suzuki H, Toyoda H. (2006). Development of an in vitro skin sensitization test using human cell lines: The human Cell Line Activation Test (h-CLAT) I. Optimization of the h-CLAT protocol. Toxicol. In Vitro 20, 767–773.
|
|
(2)
|
Miyazawa M, Ito Y, Yoshida Y, Sakaguchi H, Suzuki H. (2007). Phenotypic alterations and cytokine production in THP-1 cells in response to allergens. Toxicol. In Vitro 21, 428-437.
|
|
(3)
|
EC EURL-ECVAM (2013). Recommendation on the human Cell Line Activation Test (h-CLAT) for skin sensitisation testing. Достъпно на: https://eurl-ecvam.jrc.ec.europa.eu/eurl-ecvam-recommendations
|
|
(4)
|
Takenouchi O, Fukui S, Okamoto K, Kurotani S, Imai N, Fujishiro M, Kyotani D, Kato Y, Kasahara T, Fujita M, Toyoda A, Sekiya D, Watanabe S, Seto H, Hirota M, Ashikaga T, Miyazawa M. (2015). Test battery with the human cell line activation test, direct peptide reactivity assay and DEREK based on a 139 chemical data set for predicting skin sensitizing potential and potency of chemicals. J Appl Toxicol. 35, 1318-1332.
|
|
(5)
|
Hirota M, Fukui S, Okamoto K, Kurotani S, Imai N, Fujishiro M, Kyotani D, Kato Y, Kasahara T, Fujita M, Toyoda A, Sekiya D, Watanabe S, Seto H, Takenouchi O, Ashikaga T, Miyazawa M. (2015). Evaluation of combinations of in vitro sensitization test descriptors for the artificial neural network-based risk assessment model of skin sensitization. J Appl Toxicol. 35, 1333-1347.
|
|
(6)
|
Bauch C, Kolle SN, Ramirez T, Fabian E, Mehling A, Teubner W, van Ravenzwaay B, Landsiedel R. (2012). Putting the parts together: combining in vitro methods to test for skin sensitizing potencials. Regul Toxicol Parmacol. 63, 489-504.
|
|
(7)
|
Van der Veen JW, Rorije E, Emter R, Natch A, van Loveren H, Ezendam J. (2014). Evaluating the performance of integrated approaches for hazard identification of skin sensitizing chemicals. Regul Toxicol Pharmacol. 69, 371-379.
|
|
(8)
|
Urbisch D, Mehling A, Guth K, Ramirez T, Honarvar N, Kolle S, Landsiedel R, Jaworska J, Kern PS, Gerberick F, Natsch A, Emter R, Ashikaga T, Miyazawa M, Sakaguchi H. (2015). Assessing skin sensitization hazard in mice and men using non-animal test methods. Regul Toxicol Parmacol. 71, 337-351.
|
|
(9)
|
Jaworska JS, Natsch A, Ryan C, Strickland J, Ashikaga T, Miyazawa M. (2015). Bayesian integrated testing strategy (ITS) for skin sensitization potency assessment: a decision support system for quantitative weight of evidence and adaptive testing strategy. Arch Toxicol. 89, 2355-2383.
|
|
(10)
|
Strickland J, Zang Q, Kleinstreuer N, Paris M, Lehmann DM, Choksi N, Matheson J, Jacobs A, Lowit A, Allen D, Casey W. (2016). Integrated decision strategies for skin sensitization hazard. J Appl Toxicol. DOI 10.1002/jat.3281.
|
|
(11)
|
Nukada Y, Ashikaga T, Miyazawa M, Hirota M, Sakaguchi H, Sasa H, Nishiyama N. (2012). Prediction of skin sensitization potency of chemicals by human Cell Line Activation Test (h-CLAT) and an attempt at classifying skin sensitization potency. Toxicol. In Vitro 26, 1150-60.
|
|
(12)
|
EC EURL ECVAM (2015). Re-analysis of the within and between laboratory reproducibility of the human Cell Line Activation Test (h-CLAT). Достъпно на: https://eurl-ecvam.jrc.ec.europa.eu/eurl-ecvam-recommendations/eurl-ecvam-recommendation-on-the-human-cell-line-activation-test-h-clat-for-skin-sensitisation-testing
|
|
(13)
|
EC EURL ECVAM (2012). human Cell Line Activation Test (h-CLAT) Validation Study Report Accessible at: https://eurl-ecvam.jrc.ec.europa.eu/eurl-ecvam-recommendations
|
|
(14)
|
Takenouchi O, Miyazawa M, Saito K, Ashikaga T, Sakaguchi H. (2013). Predictive performance of the human Cell Line Activation Test (h-CLAT) for lipophilic with high octanol-water partition coefficients. J. Toxicol. Sci. 38, 599-609.
|
|
(15)
|
Ashikaga T, Sakaguchi H, Sono S, Kosaka N, Ishikawa M, Nukada Y, Miyazawa M, Ito Y, NishiyamaN, Itagaki H. (2010). A comparative evaluation of in vitro skin sensitisation tests: the human cell-line activation test (h-CLAT) versus the local lymph node assay (LLNA). Altern. Lab. Anim. 38, 275-284.
|
|
(16)
|
Fabian E., Vogel D., Blatz V., Ramirez T., Kolle S., Eltze T., van Ravenzwaay B., Oesch F., Landsiedel R. (2013). Xenobiotic metabolizin enzyme activities in cells used for testing skin sensitization in vitro. Arch Toxicol 87, 1683-1969.
|
|
(17)
|
Okamoto K, Kato Y, Kosaka N, Mizuno M, Inaba H, Sono S, Ashikaga T, Nakamura T, Okamoto Y, Sakaguchi H, Kishi M, Kuwahara H, Ohno Y. (2010). The Japanese ring study of a human Cell Line Activation Test (h-CLAT) for predicting skin sensitization potential (6th report): A study for evaluating oxidative hair dye sensitization potential using h-CLAT. AATEX 15, 81-88.
|
|
(18)
|
DB-ALM (INVITTOX) (2014). Protocol 158: human Cell Line Activation Test (h-CLAT), 23pp. Достъпно на: http://ecvam-dbalm.jrc.ec.europa.eu/
|
|
(19)
|
Mizuno M, Yoshida M, Kodama T, Kosaka N, Okamato K, Sono S, Yamada T, Hasegawa S, Ashikaga T, Kuwahara H, Sakaguchi H, Sato J, Ota N, Okamoto Y, Ohno Y. (2008). Effects of pre-culture conditions on the human Cell Line Activation Test (h-CLAT) results; Results of the 4th Japanese inter-laboratory study. AATEX 13, 70-82.
|
|
(20)
|
Sono S, Mizuno M, Kosaka N, Okamoto K, Kato Y, Inaba H, , Nakamura T, Kishi M, Kuwahara H, Sakaguchi H, Okamoto Y, Ashikaga T, Ohno Y. (2010). The Japanese ring study of a human Cell Line Activation Test (h-CLAT) for predicting skin sensitization potential (7th report): Evaluation of volatile, poorly soluble fragrance materials. AATEX 15, 89-96.
|
|
(21)
|
OECD (2005). Guidance Document No 34 on The Validation and International Acceptance of New or Updated Test Methods for Hazard Assessment. OECD Series on Testing and Assessment. Organization for Economic Cooperation and Development, Paris, France, 2005, 96 pp.
|
|
(22)
|
OECD (2012). The Adverse Outcome Pathway for Skin Sensitisation Initiated by Covalent Binding to Proteins. Part 1: Scientific Evidence. Series on Testing and Assessment No 168. На разположение на адрес: http://www.oecd.org/officialdocuments/publicdisplaydocumentpdf/?cote=ENV/JM/MONO(2012)10/PART1&docLanguage=En
|
|
(23)
|
United Nations UN (2013). Globally Harmonized System of Classification and Labelling of Chemicals (GHS). Fifth revised edition. New York & Geneva: United Nations Publications. ISBN: 978-92-1-117006-1. На разположение на адрес: http://www.unece.org/trans/danger/publi/ghs/ghs_rev05/05files_e.html
|
|
(24)
|
ECETOC (2003). Contact sensitization: Classification according to potency. European Centre for Ecotoxicology & Toxicology of Chemicals (Technical Report No 87).
|
|
(25)
|
Ashikaga T, Sakaguchi H, Okamoto K, Mizuno M, Sato J, Yamada T, Yoshida M, Ota N, Hasegawa S, Kodama T, Okamoto Y, Kuwahara H, Kosaka N, Sono S, Ohno Y. (2008). Assessment of the human Cell Line Activation Test (h-CLAT) for Skin Sensitization; Results of the First Japanese Inter-laboratory Study. AATEX 13, 27-35.
|
Допълнение 1.1
ОПРЕДЕЛЕНИЯ
Точност
: степента на близост между резултатите от изпитването и приетите референтни стойности. Това е мярка за ефективността на изпитването и един от аспектите на неговата относимост. Терминът често се използва взаимозаменяемо с термина „съответствие“, за да се обозначи относителният дял на правилните резултати при дадено изпитване (21).
AOP (път, водещ до неблагоприятен ефект)
: последователност от събития от химичната структура на целеви химикал или група от сходни химикали през молекулното иницииращо събитие до представляващ интерес in vivo резултат (22).
Химикал
: Вещество или смес.
CV75
: Прогнозната концентрация, показваща клетъчна жизнеспособност 75 %.
EC150
: концентрациите, показващи стойности на RFI от 150 на експресията на CD86
EC200
: концентрациите, показващи стойности на RFI от 200 на експресията на CD54
Поточна цитометрия
: цитометрична техника, при която клетките, суспендирани в течност, се движат в поток една по една във времето през фокус на възбуждаща светлина, която се разсейва по начини, характерни за клетките и техните компоненти; клетките често се маркират с флуоресцентни маркери, така че светлината първо се абсорбира и след това се излъчва при изменени честоти.
Опасност
: Вътрешно присъщо свойство на даден агент или ситуация, притежаващо потенциал да предизвика неблагоприятни ефекти когато един организъм, система или (суб-)популация бъдат експонирани на този агент.
IATA (интегриран подход за изпитване и оценка)
: Структуриран подход, използван за идентифициране на опасност (потенциал), характеризиране на опасност (потенциал) и/или оценка на безопасността (потенциал и експозиция) на даден химикал или група от химикали, който стратегически интегрира и претегля всички данни, относими към предоставянето на информация за вземането на регулаторно решение по отношение на потенциалната опасност и/или риска, и/или необходимостта от допълнително целенасочено и следователно минимално изпитване.
Контрола на среда
: Нетретирано повторение, съдържащо всички компоненти на дадена изпитвана система. Тази проба се обработва с пробите, третирани с изпитвания химикал, и с други контролни проби, за определяне дали разтворителят/носителят взаимодейства с изпитваната система.
Смес
: смес или разтвор, съставени от две или повече вещества.
Вещество с една съставка
: Вещество, определено от неговия количествен състав, в който съдържанието на една основна съставка е най-малко 80 % (w/w).
Вещество с повече съставки
: Вещество, определено от неговия количествен състав, в който повече от една основна съставка се съдържа в концентрация ≥ 10 % (w/w) и < 80 % (w/w). Веществото с повече съставки е резултат от производствен процес. Разликата между смес и вещество с повече съставки е, че дадена смес е получена чрез смесване на две или повече вещества без химична реакция. Веществото с повече съставки е резултат от химична реакция.
Положителна контрола
: Повторение, съдържащо всички компоненти на дадена изпитвана система и третирано с вещество, за което е известно, че предизвиква положителен отклик. За да гарантира, че може да се направи оценка на варирането на отклика в положителната контрола във времето, големината на положителния отклик не следва да е прекомерна.
Пре-хаптени
: химикали, които стават сенсибилизатори чрез абиотично преобразуване
Про-хаптени
: химикали, които изискват ензимно активиране за реализирането на потенциал за кожна сенсибилизация
Относителен интензитет на флуоресценцията (RFI)
: Относителни стойности на средногеометричната стойност на интензитета на флуоресценция (MFI) в третираните с химикал клетки в сравнение с MFI в клетки, третирани с разтворител/носител.
Относимост
: описание на взаимовръзката между изпитването и ефекта, представляващ интерес, и дали тя е значима и полезна за определена цел. Това е степента, в която изпитването правилно измерва или прогнозира биологичния ефект, представляващ интерес. Относимостта включва разглеждане на точността (съответствието) на дадено изпитване (21).
Надеждност
: мярка за степента, в която дадено изпитване може да се извърши възпроизводимо в една и съща лаборатория и в различни лаборатории по различно време, като при извършването се използва един и същи протокол. Оценката за нея се прави, като се изчислява вътрешнолабораторната и междулабораторната възпроизводимост и вътрешнолабораторната повторяемост (21).
Серия
: Серията представлява един или повече изпитвани химикали, изпитвани едновременно с контрола на разтворител/носител и с положителна контрола.
Чувствителност
: Относителният дял от всички положителни/активни химикали, които са класифицирани правилно чрез изпитването. Това е мярка за точността на изпитване, което предоставя категорийни резултати и е важен фактор при оценката на относимостта на дадено изпитване.
Оцветяващ буфер
: Фосфатно буфериран физиологичен разтвор, съдържащ 0,1 % говежди серумен албумин.
Контрола на разтворител/носител
: Нетретирана проба, съдържаща всички компоненти на изпитваната система, с изключение на изпитвания химикал, но включваща разтворителя/носителя, който се използва. Използва се за определяне на базовия отклик за пробите, третирани с изпитвания химикал, разтворен или стабилно диспергиран в същия разтворител/носител. Когато се изпитва с паралелна контрола на среда, тази проба показва също дали разтворителят/носителят взаимодейства с изпитваната система.
Специфичност
: Относителният дял от всички отрицателни/неактивни химикали, които са класифицирани правилно чрез изпитването. Това е мярка за точността на изпитването, което предоставя категорийни резултати и е важен фактор при оценката на относимостта на дадено изпитване (21).
Вещество
: означава химичен елемент и неговите съединения в естествено състояние или получени чрез всеки производствен процес, включително всяка добавка, необходима за запазване на неговата стабилност и всеки примес, извлечен от използвания процес, с изключение на всеки разтворител, който може да бъде отделен, без да се засяга стабилността на веществото или да се променя неговият състав.
Изпитван химикал
: Всяко вещество или смес, изпитвано(а) с използване на настоящия метод.
Глобална хармонизирана система на Организацията на обединените нации за класифициране и етикетиране на химикали (GHS на ООН)
: система, предлагаща класифициране на химикали (вещества и смеси) според стандартизирани видове и степени на физическа, здравна и екологична опасност и разглеждаща съответни съобщителни елементи, като например пиктограми, сигнални думи, предупреждения за опасност, препоръки за безопасност и информационни листове за безопасност, така че те да съобщават информация за тяхното неблагоприятно въздействие с оглед защита на хората (включително работодатели, работещи, служители в транспорта, потребители и аварийни служители) и на околната среда (23).
UVCB
: вещества с неизвестен или променлив състав, сложни продукти от реакции или биологични материали.
Валидно изпитване
: Изпитване, за което се смята, че притежава достатъчна относимост и надеждност по отношение на конкретна цел и което се основава на научно обосновани принципи. Дадено изпитване никога не е валидно в абсолютен смисъл, а само по отношение на определена цел (21).
Допълнение 1.2
ВЕЩЕСТВА ЗА ИЗПИТВАНЕ ЗА ПРИГОДНОСТ
Преди рутинното използване на изпитването, описано в настоящото допълнение към метод за изпитване Б.71, лабораториите следва да докажат техническата си компетентност, като правилно получат очакваната прогноза по h-CLAT за 10-те вещества, препоръчани в Таблица 1, и чрез получаване на стойности за CV75, EC150 и EC200, които попадат в съответния референтен обхват за минимум 8 от 10-те вещества за изпитване за пригодност. Веществата за изпитване за пригодност са подбрани като представителни за размаха от отклици за опасностите от кожна сенсибилизация. Други критерии за подбор бяха веществата да са налични в търговската мрежа и да са налични висококачествени in vivo референтни данни, както и висококачествени in vitro данни, получени по метода h-CLAT. Също така са налични публикувани референтни данни за метода h-CLAT (3) (14).
Таблица 1
Препоръчвани вещества за доказване на техническа пригодност по отношение на метода h-CLAT
|
Вещества за изпитване за пригодност
|
CAS №
|
Агрегатно състояние
|
In vivo прогноза (103)
|
CV75 Позоваване Размах в μg/ml (104)
|
резултати от h-CLAT за CD86 (референтен за ЕС150 размах в μg/ml (104)
|
резултати от h-CLAT за CD54 (референтен за ЕС200 размах в μg/ml (104)
|
|
2,4-Динитрохлоробензен
|
97-00-7
|
Твърд
|
Сенсибилизатор (изключително силен)
|
2-12
|
Положително (0,5-10)
|
Положително (0,5-15)
|
|
4-Фенилендиамин
|
106-50-3
|
Твърд
|
Сенсибилизатор (силен)
|
5-95
|
Положително (<40)
|
Отрицателно (>1,5) (105)
|
|
Никелов сулфат
|
10101-97-0
|
Твърд
|
Сенсибилизатор (умерен)
|
30-500
|
Положително (<100)
|
Положително (10-100)
|
|
2-Меркаптобензотиазол
|
149-30-4
|
Твърд
|
Сенсибилизатор (умерен)
|
30-400
|
Отрицателно (>10) (105)
|
Положително (10-140)
|
|
R(+)-Лимонен
|
5989-27-5
|
Течност
|
Сенсибилизатор (слаб)
|
>20
|
Отрицателно (>5) (105)
|
Положително (<250)
|
|
Имидазолидинилуреа
|
39236-46-9
|
Твърд
|
Сенсибилизатор (слаб)
|
25-100
|
Положително (20-90)
|
Положително (20-75)
|
|
Изопропанол
|
67-63-0
|
Течност
|
Непредизвикващо сенсибилизация вещество
|
>5 000
|
Отрицателно (>5 000 )
|
Отрицателно (>5 000 )
|
|
Глицерол
|
56-81-5
|
Течност
|
Непредизвикващо сенсибилизация вещество
|
>5 000
|
Отрицателно (>5 000 )
|
Отрицателно (>5 000 )
|
|
Млечна киселина
|
50-21-5
|
Течност
|
Непредизвикващо сенсибилизация вещество
|
1500-5 000
|
Отрицателно (>5 000 )
|
Отрицателно (>5 000 )
|
|
4-Аминобензоена киселина
|
150-13-0
|
Твърд
|
Непредизвикващо сенсибилизация вещество
|
>1 000
|
Отрицателно (>1 000 )
|
Отрицателно (>1 000 )
|
|
Съкращения: CAS № = регистрационен номер по Службата за химични индекси.
|
Допълнение 2
IN VITRO КОЖНА СЕНСИБИЛИЗАЦИЯ: ИЗПИТВАНЕ С АКТИВИРАНЕ НА КЛЕТЪЧНА ЛИНИЯ U937 (U-SENS™)
ПЪРВОНАЧАЛНИ СЪОБРАЖЕНИЯ И ОГРАНИЧЕНИЯ
|
1.
|
U-SENS™ изпитването определя количествено промяната в експресията на повърхностен клетъчен маркер, свързана с процеса на активиране на моноцити и дендритни клетки (ДК) (т.е. CD86) в клетъчна линия U937 на хистиоцитен лимфом при човека след експозиция на сенсибилизиращи вещества (1). Измерените нива на експресия на повърхностния клетъчен маркер CD86 в клетъчна линия U937 се използват след това за подпомагане на разграничаването между кожни сенсибилизатори и непредизвикващи сенсибилизация вещества.
|
|
2.
|
U-SENS™ изпитването е било обект на оценка в изследване за валидиране (2), координирано от L’Oreal, и впоследствие на независима партньорска проверка от страна Научния консултативен комитет (ESAC) на референтната лаборатория на Европейския съюз за алтернативи на изпитванията върху животни (EURL ECVAM) (3). Като взе предвид всички налични доказателства и данни от регулаторните органи и заинтересованите страни, EURL ECVAM препоръча U-SENS™ (4) за използване като част от IATA за подкрепа на разграничаването между сенсибилизатори и непредизвикващи сенсибилизация вещества за целите на класифицирането на опасността и етикетирането. В своя документ с насоки относно протоколирането на структурирани подходи към интеграцията на данните и индивидуалните източници на информация, използвани в рамките на IATA за кожна сенсибилизация, понастоящем ОИСР обсъжда редица проучвания на конкретни случаи, описващи различни стратегии за изпитване и модели за прогнозиране. Един от различните определени подходи се основава на U-SENS изследването (5). Примери за използването на данни от U-SENS™ в комбинация с друга информация, включително данни за предходни периоди и налични валидни данни за човека (6), също са докладвани на друго място в литературата (4) (5) (7).
|
|
3.
|
Беше показано, че U-SENS™ изпитването може да се прехвърли на лаборатории с опит в областта на техниките за клетъчна култура и анализа с поточна цитометрия. Нивото на възпроизводимостта в прогнозите, което може да се очаква от изпитването, е от порядъка на 90 % и 84 % съответно вътре в самата лаборатория и между лабораториите (8). Резултатите, получени в изследването за валидиране (8) и други публикувани изследвания (1) като цяло показват, че в сравнение с резултатите от LLNA точността при разграничаването на кожните сенсибилизатори (т.е. категория 1 по GHS на ООН/CLP) от непредизвикващи сенсибилизация вещества е 86 % (N = 166), с чувствителност от 91 % (118/129) и специфичност от 65 % (24/37). В сравнение с резултатите при човека точността при разграничаването на кожни сенсибилизатори (т.е. категория 1 по GHS на ООН/CLP) от непредизвикващи сенсибилизация вещества е 77 % (N = 101), с чувствителност от 100 % (58/58) и специфичност от 47 % (20/43). Има по-голяма вероятност, в сравнение с LLNA, неверните отрицателни прогнози, свързани с U-SENS™, да се отнасят за химикали, които са с малък до умерен потенциал за кожна сенсибилизация (т.е. кожна сенсибилизация подкатегория 1B по GHS на ООН/CLP), отколкото за химикали, показващи голям потенциал за кожна сенсибилизация (т.е. подкатегория 1A по GHS на ООН/CLP) (1)(8)(9). Взета като цяло, тази информация показва ползата от U-SENS™ изпитването за подпомагане на определянето на опасностите от кожна сенсибилизация. Независимо от това стойностите за точността, посочени тук за U-SENS™ като самостоятелно изпитване, са само ориентировъчни, тъй като изпитването трябва да се разглежда в съчетание с други източници на информация в контекста на IATA и в съответствие с разпоредбите на точки 7 и 8 от Общото въведение. Освен това при оценяването на методи за сенсибилизация на кожата, които не използват животни, следва да се има предвид, че LLNA изпитването, както и други изпитвания върху животни, могат да не отразяват напълно положението при хората.
|
|
4.
|
Въз основа на наличните към момента данни е доказано, че U-SENS™ изпитването е приложимо за изпитвани химикали (включително за козметични съставки, напр. консерванти, повърхностноактивни вещества, активни вещества, багрила), които обхващат разнообразни органични функционални групи, физични и химични свойства, потенциали за кожна сенсибилизация (както са определени в in vivo изследвания) и спектъра от реакционни механизми, за които е известно, че са свързани с кожната сенсибилизация (т.е. акцептор на Михаел, образуване на Шифови бази, агент за ацилово нуклеофилно заместване, бимолекулно нуклеофилно заместване [SN2], или ароматно нуклеофилно заместване [SNAr]) (1) (8) (9) (10). U-SENS™ изпитването се прилага за изпитвани химикали, които са разтворими или образуват стабилна дисперсия (т.е. колоид или суспензия, в която изпитваният химикал не се утаява или отделя от разтворителя/носителя в различни фази) в подходящ разтворител/носител (вж. точка 13). Химикали в набора от данни, за които се съобщава, че са пре-хаптени (т.е. вещества, които се активират чрез окисляване) или про-хаптени (т.е. вещества, които изискват ензимно активиране, например посредством P450 ензими) са били прогнозирани правилно от U-SENS™ (1) (10). Веществата, разрушаващи мембрани, могат да доведат до неверни положителни резултати, дължащи се на неспецифично увеличаване на експресията наCD86, тъй като 3 от 7 неверни положителни резултата в сравнение с референтната класификация in vivo са повърхностноактивни вещества (1). Положителните резултати с повърхностноактивните вещества сами по себе си следва да се разглеждат предпазливо, докато отрицателните резултати с повърхностноактивни вещества могат все пак да се използват за подпомагане на идентифицирането на изпитвания химикал като непредизвикващо сенсибилизация вещество. Флуоресцентните изпитвани химикали могат да бъдат оценени с U-SENS™ (1), но независимо от това, силно флуоресцентните изпитвани химикали, които излъчват при същата дължина на вълната като флуоресцеиновия изотиоцианат (FITC) или като пропидиевия йодид (PI), ще предизвикат смущения при откриването с поточна цитометрия и следователно не могат да бъдат правилно оценени с помощта на FITC-конюгирани антитела (потенциално неверни отрицателни резултати) или PI (жизнеспособност, която не е измерима). В такъв случай могат да се използват други антитела, маркирани с флуорохром, или съответно други маркери за цитотоксичност, при условие че може да се покаже, че дават сходни резултати като маркираните с FITC антитела или PI (вж. точка 18), напр. чрез изпитване на веществата за изпитване за пригодност от допълнение 2.2. С оглед на гореизложеното положителните резултати от повърхностноактивните вещества и отрицателните резултати, получени при силно флуоресцентни изпитвани химикали следва да се тълкуват в контекста на посочените ограничения и заедно с други източници на информация в рамките на IATA. В случаите, когато са налице доказателства за неприложимостта на U-SENS™ изпитването по отношение на други специфични категории изпитвани химикали, той не следва да се използва за тези специфични категории.
|
|
5.
|
Както е описано по-горе, U-SENS™ изпитването подкрепя разграничаването между кожни сенсибилизатори и непредизвикващи сенсибилизация вещества. То обаче може да допринесе за оценка на потенциала за сенсибилизация, когато се използва в интегрирани подходи, като например IATA. Независимо от това се изисква по-нататъшна работа, за предпочитане въз основа на данни за човека, за определяне на това как резултатите от U-SENS™ могат евентуално да послужат за информация за оценка на потенциала.
|
|
6.
|
Определенията са дадени в допълнение 2.1.
|
ПРИНЦИП НА ИЗПИТВАНЕТО
|
7.
|
U-SENS™ изпитването е in vitro изследване, което определя количествено промените в експресията на повърхностния клетъчен маркер CD86 в клетъчна линия U937 на хистиоцитен лимфом при човека, след 45±3 часова експозиция на изпитвания химикал. Повърхностният маркер CD86 е типичен маркер за активиране на U937. За CD86 е известно, че е ко-стимулираща молекула, която може да имитира моноцитно активиране, което играе решаваща роля при праймирането на Т-клетките. Промените в експресията на повърхностния клетъчен маркер CD86 се измерват чрез поточна цитометрия след клетъчно оцветяване, обичайно с антитела, маркирани с флуоресцеинов изотиоцианат (FITC). Измерване на цитотоксичността (напр. чрез използване на PI) се извършва също така конкурентно, за да се оцени дали настъпва регулиране на експресията на повърхностния клетъчен маркер CD86 в посока увеличаване при субцитотоксични концентрации. Стимулационният индекс (SI) на повърхностния клетъчен маркер CD86 в сравнение с контролата на разтворител/носител се изчислява и използва в модела за прогнозиране (вж. точка 19), за да се подкрепи разграничаването между сенсибилизатори и непредизвикващи сенсибилизация вещества
|
ДОКАЗВАНЕ НА ПРИГОДНОСТ
|
8.
|
Преди рутинното използване на изпитването, описано в настоящото допълнение, за метод за изпитване Б.71, лабораториите следва да докажат техническата си пригодност, като използват 10-те вещества за изпитване на пригодност, изброени в допълнение 2.2, в съответствие с добрите практики за in vitro методите (11). Освен това ползвателите на изпитването следва да поддържат база данни с данни за преходни периоди, получени с проверките на реакционната способност (вж. точка 11) и с положителни контроли и контроли на разтворител/носител (вж. точки 15—16), и да използват тези данни, за да потвърдят, че възпроизводимостта на изпитването в тяхната лаборатория се запазва с течение на времето.
|
ПРОЦЕДУРА
|
9.
|
Това изпитване се основава на Протокол № 183 за U-SENS™ от базата данни за методи, алтернативни на експериментите с животни (DB-ALM) (12). Стандартните работни процедури (СРП) следва да се прилагат, когато се прилага и използва U-SENS™ изпитването в лабораторията. Може да се използва автоматизирана система за провеждане на U-SENS™, ако може да се докаже, че тя осигурява сходни резултати, например чрез изпитване на веществата за изпитване за пригодност от допълнение 2.2. По-долу е представено описание на основните компоненти и процедури за U-SENS™ изпитването.
|
Подготовка на клетките
|
10.
|
Клетъчната линия U937 на хистиоцитен лимфом при човека (13) следва да се използва за извършване на U-SENS™ изпитването. Клетките (клон CRL1593.2) следва да бъдат получени от добре квалифицирана клетъчна банка, като например Американската банка за типови култури (American Type Culture Collection).
|
|
11.
|
Клетките U937 се култивират при 37 °C, 5 % CO2 и овлажнена атмосфера в среда RPMI-1 640, с добавка на 10 % фетален телешки серум (FCS), 2 mM L-глутамин, 100 единици/ml пеницилин и 100 μg/ml стрептомицин (пълноценна среда). С U937 клетки се осъществяват рутинно пасажи на всеки 2—3 дни при плътност съответно от 1,5 или 3 × 105 клетки/ml. Клетъчната плътност не трябва да надвишава 2 × 106 клетки/ml, а клетъчната жизнеспособност, измерена чрез изключване с трипаново синьо, следва да бъде ≥ 90 % (да не се прилага при първия пасаж след размразяването). Преди да бъдат използвани за изпитване, всяка партида от клетки, FCS или антитела следва да бъде квалифицирана чрез извършване на проверка за реакционна способност. Проверката за реакционна способност на клетките следва да се извърши, като се използва положителната контрола, пикрилсулфонова киселина (2,4,6-тринитробензенсулфонова киселина: TNBS) (CAS № 2 508-19-2, ≥ 99 % чистота) и отрицателната контрола млечна киселина (LA) (CAS № 50-21-5, ≥ 85 % чистота), най-малко една седмица след размразяване. За проверката за реакционна способност следва да се изпитат шест крайни концентрации за всяка от 2-те контроли (TNBS: 1, 12,5, 25, 50, 75, 100μg/ml и LA: 1, 10, 20, 50, 100, 200μg/ml). TNBS, разтворим в пълноценна среда, следва да дава положителен и свързан с концентрацията отклик на CD86 (напр. когато положителна концентрация, SI на CD86 ≥ 150, е последвана от концентрация с увеличаващ се SI на CD86), а LA, разтворена в пълноценна среда, следва да дава отрицателен отклик на CD86 (вж. точка 21). За изследването следва да се използва само партидата клетки, преминала 2 пъти проверката за реакционна способност. Клетките могат да бъдат размножавани до седем седмици след размразяването. Броят пасажи не трябва да надвишава 21. Проверката за реакционна способност следва да се извършва в съответствие с процедурите, описани в точки 18—22.
|
|
12.
|
За изпитване U937 клетките се посяват с плътност 3 × 105 клетки/ml или 6 × 105 клетки/ml, и се извършва предварително култивиране в колби за клетъчни култури съответно 2 дни или 1 ден. Могат да се използват и условия на предварително култивиране, различни от описаните по-горе, ако е предоставена достатъчна научна обосновка и ако може да се докаже, че те осигуряват подобни резултати, например чрез изпитване на веществата за изпитване за пригодност от допълнение 2.2. В деня на изпитването клетките, събрани от колбата за култури, се ресуспендират с прясна хранителна среда при 5 × 105 клетки/ml. След това клетките се разпределят в 96-ямкова плоскодънна плака с 100 μl (крайна клетъчна плътност от 0,5 × 105 клетки/ямка).
|
Приготвяне на изпитваните химикали и веществата за контроли
|
13.
|
Оценката на разтворимостта се извършва преди изпитването. За тази цел изпитваните химикали се разтварят или стабилно се диспергират в концентрация 50 mg/ml в пълноценна среда като първи възможен разтворител, или диметилсулфоксид (DMSO, ≥ 99 % чистота), като втори вариант на разтворител/носител, ако изпитваният химикал не е разтворим в разтворител/носител пълноценна среда. За изпитването изпитваният химикал се разтваря в крайна концентрация от 0,4 mg/ml в пълноценна среда, ако химикалът е разтворим в този разтворител/носител. Ако химикалът е разтворим само в DMSO, химикалът се разтваря в концентрация от 50 mg/ml. Могат да се използват и други разтворители/носители, освен описаните по-горе, при предоставяне на достатъчна научна обосновка. Следва да се вземе предвид стабилността на изпитвания химикал в крайния разтворител/носител.
|
|
14.
|
Изпитваните химикали и веществата за контроли се приготвят в деня на изпитването. Тъй като не се провежда изследване за откриване на доза, за първата серия следва да се изпитат 6 крайни концентрации (1, 10, 20, 50, 100 и 200 μg/ml) в съответния разтворител/носител или в пълноценна среда, или в 0,4 % DMSO в среда. За последващите серии, като се започне от разтвори на изпитваните химикали от 0,4 mg/ml в пълноценна среда или 50 mg/ml в DMSO, се приготвят най-малко 4 работни разтвора (т.е. най-малко 4 концентрации), като се използва съответният разтворител/носител. Накрая, работните разтвори се използват за третиране чрез добавяне на равен обем суспензия от U937 клетки (вж. точка 11 по-горе) към обема на работния разтвор в плаката, за да се постигне допълнително 2-кратно разреждане (12). Концентрациите (най-малко 4 концентрации) за всяка следваща серия се избират въз основа на индивидуалните резултати от всички предишни серии (8). Използваемите крайни концентрации са 1, 2, 3, 4, 5, 7,5, 10, 12,5, 15, 20, 25, 30, 35, 40, 45, 50, 60, 70, 80, 90, 100, 120, 140, 160, 180 и 200 μg/ml. Максималната крайна концентрация е 200 μg/ml. В случай че се наблюдава положителна стойност на CD86 при 1 μg/ml, се прави оценка на 0,1 μg/ml, за да се открие концентрацията на изпитвания химикал, която не индуцира CD86 над положителния праг. За всяка серия се изчислява EC150 (концентрация, при която даден химикал достига положителния праг за CD86 от 150 %, вж. точка 19), ако се наблюдава положителнаконцентрация-отклик за CD86. Когато изпитваният химикал индуцира положителен отклик за CD86, който не е свързан с концентрацията, изчисляването на EC150 може да не е относимо, както е описано в протокол № 183 от DB-ALM за U-SENS™ (12). За всяка серия се изчислява CV70 (концентрация, при която даден химикал достига прага на цитотоксичност от 70 %, вж. точка 19), винаги когато е възможно (12). За да се изследва ефектът концентрация-отклик, от увеличение на CD86, всички концентрации от използваемите концентрации следва да се избират равномерно разположени между EC150 (или най-високата отрицателна концентрация на CD86, която не е цитотоксична), и CV70 (или най-високата разрешена концентрация, т.е. 200 μg/ml). Трябва да се изпитат минимум 4 концентрации на серия с най-малко 2 концентрации, които са общи с предишната серия или серии, за целите на сравняванто.
|
|
15.
|
Контролата на разтворител/носител, използвана в изпитването U-SENS™, е пълноценна среда (за изпитвани химикали, разтворени или стабилно диспергирани в пълноценна среда) (вж. точка 4) или 0,4 % DMSO в пълноценна среда (за изпитвани химикали, разтворени или стабилно диспергирани в DMSO).
|
|
16.
|
Положителната контрола, използвана в изпитването U-SENS™, е TNBS (вж. точка 11), приготвена в пълноценна среда. TNBS следва да се използва като положителната контрола за измерване на експресията на CD86 при крайна единична концентрация в плаката (50 μg/ml), при която се получава > 70 % клетъчна жизнеспособност. За получаване на концентрация на TNBS от 50 μg/ml в плаката, се приготвя 1 M (т.е. 293 mg/ml) изходен разтвор на TNBS в пълноценна среда и допълнително се разрежда 2 930-кратно с пълноценна среда до 100 μg/ml работен разтвор. Млечната киселина (LA, CAS № 50-21-5) следва да се използва като отрицателната контрола при 200 μg/ml, разтворена в пълноценна среда (от изходен разтвор от 0,4 mg/ml). Във всяка плака от всяка серия се приготвят три повторения на нетретирана контрола на пълноценна среда, контрола на разтворител/носител, отрицателни и положителни контроли (12). Могат да се използват други подходящи положителни контроли, ако има налични данни за предходни периоди, за извеждане на сравними критерии за приемливост на серията. Критериите за приемливост на серията при работа са същите като описаните за изпитвания химикал (вж. точка 12).
|
Прилагане на изпитваните химикали и веществата за контроли
|
17.
|
Контролата на разтворител/носител или работните разтвори, описани в точки 14-16, се смесват 1:1 (v/v) с клетъчните суспензии, подготвени в 24-ямковата или 96-ямковата плоскодънна плака (вж. точка 12). След това третираните плаки се инкубират за 45 ± 3 часа при 37 °C под 5 % CO2. Преди инкубирането плаките се запечатват с полупропусклива мембрана, за да се избегне изпаряване на летливи изпитвани химикали и кръстосано замърсяване между клетките, третирани с изпитваните химикали (12).
|
Оцветяване на клетките
|
18.
|
След 45 ± 3 часа експозиция клетките се прехвърлят във V-образна микротитърна плака и се събират чрез центрофугиране. Смущението на разтворимостта се определя като кристали или капки, наблюдавани под микроскоп, 45 ± 3 часа след третирането (преди клетъчното оцветяване). Супернатантите се изхвърлят и останалите клетки се промиват веднъж с 100 μl ледено студен фосфатно буфериран физиологичен разтвор (PBS), съдържащ 5 % фетален телешки серум (оцветяващ буфер). След центрофугирането клетките се ресуспендират със 100 μl оцветяващ буфер и се оцветяват с 5 μl (например 0,25 μg) маркирани с FITC анти-CD86 или миши антитела от (изотип) IgG1 при 4 °C за 30 минути със защита срещу светлина. Следва да се използват антителата, описани в протокол № 183 от DB-ALM за U-SENS™ (12) (за CD86: BD-PharMingen #5556 57 Clone: Fun-1 или Caltag/Invitrogen # MHCD8601 Clone: BU63; а за IgG1: BD-Pharmingen # 5557 48 или Caltag/Invitrogen # GM4992). Въз основа на опита на разработилите изпитването интензитетът на флуоресценцията на антителата обикновено е последователен между различните партиди. Други клонове или доставчик на антитела, които са преминали проверката за реакционна способност, могат да се използват за изследването (вж. точка 11). Въпреки това ползвателите могат да разгледат титруване на антитела в условията на собствената си лаборатория, за определяне на най-добрата концентрация за използване. Може да се използва и друга система за откриване, например маркирани с флуорхром анти-CD86 антитела, ако може дасе докаже, че те осигуряват сходни резултати като FITC-конюгираните антитела, например чрез изпитване на веществата за изпитване на пригодност от допълнение 2.2. След промиване с 100 μl оцветяващ буфер два пъти и веднъж с 100 μl ледено студен PBS, клетките се ресуспендират в ледено студен PBS (напр. 125 μl за проби, които се анализират ръчно епруветка по епруветка, или 50 μl при използване на плака с автоматично пробовземане) и се добавя PI разтвор (крайна концентрация от 3 μg/ml). Други маркери за цитотоксичност, като 7-аминоактиномицин D (7-AAD), или трипаново синьо, могат да бъдат използвани, ако може да се докаже, че алтернативните оцветители осигуряват сходни резултати като PI, например чрез изпитване на веществата за изпитване на пригодност от допълнение 2.2.
|
Анализ с поточна цитометрия
|
19.
|
Нивото на експресията на CD86 и клетъчната жизнеспособност се анализират с помощта на поточна цитометрия. Клетките се показват в точкова диаграма с големина (FSC) и гранулираност (SSC), изобразена в логаритмична скала, с цел ясно да се идентифицира популацията в първи аналитичен прозорец R1 и да се отстранят остатъците. Придобиват се данни за общо 10 000 клетки в аналитичен прозорец R1 за всяка ямка. Клетките от същия аналитичен прозорец R1 се визуализират в рамките на FL3 или FL4/SSC точкова диаграма. Жизнеспособните клетки се отграничават чрез поставяне на втори аналитичен прозорец R2, с който се избира популацията от клетки, отрицателни на пропидиев йодид (канал FL3 или FL4). Клетъчната жизнеспособност може да бъде изчислена с помощта на следната формула от програмата за анализ на цитометъра. Когато клетъчната жизнеспособност е малка, могат да се придобият данни за до 20 000 клетки, включително мъртви клетки. Друга възможност е данните да бъдат придобити в продължение на една минута след започването на анализа.

След това процентът на FL1-положителните клетки се измерва сред тези жизнеспособни клетки, включени в аналитичен прозорец R2 (в рамките на R1). Експресията на клетъчната повърхност на CD86 се анализира в FL1/SSC точова диаграма, с аналитичен прозорец към жизнеспособни клетки (R2).
За ямките с пълноценна среда/IgG1 прагът за анализа се определя близо до основната популация, така че контролите на пълноценна среда да са с IgG1 в рамките на целевата зона от 0,6 до 0,9 %.
Смущение в цвета се определя като отместване на точковата диаграма на маркирания с FITC IgG1 парцел (средногеометричен SI на IgG1 ≥ 150 %).
Стимулационният индекс (SI) на CD86 за клетки в контроли (нетретирана или в 0,4 % DMSO) и за третирани с химикал клетки се изчислява по следната формула:.

% на IgG1+ клетки в нетретирани контроли: отнася се за процента на FL1-положителните клетки, разпознаваеми от IgG1, определен с прага за анализа (приемлив диапазон ≥ 0,6 % и < 1,5 %, вж. точка 22) от жизнеспособните нетретирани клетки.
% на IgG1+/CD86+ клетки в контроли/третирани клетки: отнася се за процента на FL1-положителните клетки, разпознаваеми от IgG1/CD86, определени без придвижване на прага за анализа между жизнеспособните клетки в контроли/третираните клетки.
|
ДАННИ И ПРОТОКОЛИРАНЕ
Оценка на данните
|
20.
|
Следните параметри се изчисляват в рамките на U-SENS™ изпитването: стойност CV70, т.е. концентрация, при която се наблюдава 70 % преживяемост на U937 клетки (30 % цитотоксичност) и стойност EC150, т.е. концентрацията, при която изпитваните химикали индуцират стимулационен индекс на CD86 (SI) от 150 %.
CV70 се изчислява чрез лог-линейна интерполация, като се използва следната формула:
CV70 = C1 + [(V1 - 70) / (V1 – V2) * (C2 – C1)]
където:
V1 е минималната стойност на клетъчната жизнеспособност над 70 %
V2 е максималната стойност на клетъчната жизнеспособност под 70 %
C1 и C2 са концентрациите, показващи съответно стойностите на клетъчната жизнеспособност V1 и V2

Могат да се използват други подходи за извеждане на CV70, при условие че се докаже, че това няма въздействие върху резултатите (напр. чрез изпитване на веществата за изпитване за пригодност).
EC150 се изчислява чрез лог-линейна интерполация, като се използва следната формула:
EC150 = C1 + [(150 – S.I.1) / (S.I.2 – S.I.1) * (C2 – C1)]
където:
C1 е най-високата концентрация в μg/ml с CD86 SI < 150 % (SI 1).
C2 е най-ниската концентрация в μg/ml с CD86 SI ≥ 150 % (SI 2).

Стойностите на EC150 и CV70 се изчисляват
|
—
|
за всяка серия: отделните стойности EC150 и CV70 се използват като инструменти за изследване на ефекта концентрация-отклик от увеличението на CD86 (вж. точка 14),
|
|
—
|
въз основа на средните стойности на жизнеспособността се определя общият CV70 (12),
|
|
—
|
въз основа на средните стойности на SI на CD86 се определя общият ЕС150 за изпитвания химикал, прогнозиран като ПОЛОЖИТЕЛЕН с USENS™ (вж. точка 21) (12).
|
|
Модел на прогнозиране
|
21.
|
За измерване на експресията на CD86 всеки изпитван химикал се изпитва в най-малко четири концентрации и най-малко две независими серии (провеждани в различен ден) с цел да се изведе единична прогноза (ОТРИЦАТЕЛНА или ПОЛОЖИТЕЛНА).
|
—
|
Счита се, че индивидуалното заключение от U-SENS™ серия е отрицателно (наричано по-долу „N“), ако SI на CD86 е по-малко от 150 % при всички нецитотоксични концентрации (жизнеспособност на клетките ≥ 70 %) и ако не е наблюдавано смущение (цитотоксичност, разтворимост: вж. точка 18, или за цвят: вж. точка 19 независимо от нецитотоксичните концентрации, при които е открито смущението). Във всички други случаи: SI на CD86 по-висок или равен на 150 % и/или наблюдавани смущения, индивидуалното заключение от дадена U-SENS™ серия се счита за положително (наричано по-долу „P“).
|
|
—
|
Счита се, че дадена прогноза на USENS™ е ОТРИЦАТЕЛНА, ако поне две независими серии са отрицателни (N) (фигура 1). Ако и двете първи серии са отрицателни (N), прогнозата на U-SENS™ се счита за ОТРИЦАТЕЛНА и не е необходимо да се провежда трета серия.
|
|
—
|
Счита се, че дадена прогнозат на USENS™ е ПОЛОЖИТЕЛНА, ако поне две независими серии са положителни (P) (фигура 1). Ако и двете първи серии са положителни (P), прогнозата на U-SENS™ се счита за ПОЛОЖИТЕЛНА и не е необходимо да се провежда трета серия.
|
|
—
|
Тъй като не се провежда изследване за откриване на доза, съществува изключение, ако в първата серия SI на CD86 е по-висок или равен на 150 % само при най-високата концентрация, която не е цитотоксична. Тогава се счита, че серията НЕ ПОЗВОЛЯВА ЗАКЛЮЧЕНИЕ (NC) и следва допълнителни концентрации да се изпитат в допълнителни серии (между най-високата концентрация, която не е цитотоксична, и най-ниската концентрация на цитотоксичност — вж. точка 20). В случай че дадена серия е идентифицирана като NC, трябва да се проведат най-малко 2 допълнителни серии, както и четвърта серия в случай че серии 2 и 3 не се съгласуват (N и/или P независимо едно от друго) (фигура 1). Последващите серии ще се считат за положителни, дори ако само една концентрация, която не е цитотоксична, дава стойност на CD86, равна на 150 % или по-висока, тъй като определянето на концентрацията е било коригирано за конкретния изпитван химикал. Крайната прогноза ще се базира на предтавляващия мнозинство резултат от трите или четирите отделни серии (т.е. 2 от 3 или 2 от 4) (фигура 1).
|

Фигура 1: Модел на прогнозиране, използван в U-SENS™ изпитването. Прогнозата по U-SENS™ следва да се разглежда в рамките на IATA и в съответствие с предвиденото в точка 4 и в точки 7, 8 и 9 от Общото въведение.
N: Серия, в която не се наблюдава нито положителен CDR86, нито смущение;
P: Серия, в която се наблюдава положителен CDR86 и/или смущение(я);
NC: Не позволява заключение. Първа серия, която не позволява заключение при CD86 положителен само при най-високата нецитотоксична концентрация;
#: Индивидуално заключение „Не позволява заключение“ (NC), което се приписва само на първата серия, автоматично води до необходимост от трета серия с оглед достигане на мнозинство от Положителни (P) или Отрицателни (N) заключения в поне 2 от 3 независими серии.
$: #Клетките показват относимите комбинации от резултатите от трите серии въз основа на резултатите, получени в първите две серии, показани в горната клетка.
°: Клетките показват относимите комбинации от резултатите от четирите серии въз основа на резултатите, получени в първите три серии, показани в горната клетка.
|
Критерии за приемливост
|
22.
|
Когато се използва U-SENS™ изпитването трябва да се спазват следните критерии за приемливост (12).
|
—
|
В края на периода на експозиция от 45 ± 3 часа средната жизнеспособност на трите повторения с нетретирани U937 клетки трябва да бъде > 90 % и не трябва се наблюдава изместване при експресията на CD86. Базовата експресия на CD86 при нетретираните клетки U937 трябва да е в диапазона ≥ 2 % и ≤ 25 %.
|
|
—
|
Когато се използва като разтворител DMSO, валидността на контролата на носител DMSO се оценява чрез изчисляване на SI на DMSO в сравнение с нетретираните клетки, и оттам средната жизнеспособност на клетките от трите повторения трябва да бъде > 90 %. Контролата на носител DMSO е валидна, ако средната стойност на нейния SI на CD86 в трите повторения е по-малка от 250 % от средната стойност на SI на CD86 в трите повторения нетретирани U937 клетки.
|
|
—
|
Сериите се считат за валидни, ако най-малко две от три стойности на IgG1 за нетретирани клетки U937 са попаднали в диапазона ≥ 0,6 % и < 1,5 %.
|
|
—
|
Паралелната изпитвана отрицателна контрола (млечна киселина) се счита за валидна, ако най-малко две от трите повторения са били отрицателни (SI на CD86 < 150 %) и нецитотоксични (клетъчна жизнеспособност ≥ 70 %).
|
|
—
|
Положителната контрола (TNBS) се счита за валидна, ако най-малко две от трите повторения са били положителни (SI на CD86 ≥ 150 %) и нецитотоксични (клетъчна жизнеспособност ≥ 70 %).
|
|
Протокол от изпитването
|
23.
|
Протоколът от изпитването следва да включва следната информация.
|
Изпитван химикал
Вещество с една съставка:
|
Химична идентификация, като наименование(я) по IUPAC или по CAS, CAS номер(а), SMILES или InChI код, структурна формула, и/или други идентификатори;
|
|
Външен вид, пълна разтворимост в пълноценна среда, разтворимост в DMSO, молекулно тегло и допълнителни относими физични и химични свойства, според наличността;
|
|
Чистота, химическа идентичност на онечистванията, както е целесъобразно и практически осъществимо и т.н.;
|
|
Обработка преди изпитването, ако се прилага (напр. затопляне, смилане);
|
|
Изпитвана концентрация (концентрации);
|
|
Условия на съхранение и стабилност, доколкото са налични;
|
|
Обосновка за избора на разтворител/носител за всеки изпитван химикал.
|
Вещество с повече съставки, UVCB и смес:
|
характеризиране, доколкото е възможно, чрез химическата идентичност (вж. по-горе), чистотата, количествения състав и относимите физични и химични свойства на съставките (вж. по-горе), доколкото са налични.
|
|
Външен вид, пълна разтворимост в пълноценна среда, разтворимост в DMSO и допълнителни относими физични и химични свойства, според наличността;
|
|
Молекулно тегло или привидно молекулно тегло в случай на смеси/полимери с известен състав или друга информация, относима към провеждането на изследването;
|
|
Обработка преди изпитването, ако се прилага (напр. затопляне, смилане);
|
|
Изпитвана концентрация (концентрации);
|
|
Условия на съхранение и стабилност, доколкото са налични;
|
|
Обосновка за избора на разтворител/носител за всеки изпитван химикал.
|
|
|
Контроли
Положителна контрола
|
Химична идентификация, като наименование(я) по IUPAC или по CAS, CAS номер(а), SMILES или InChI код, структурна формула, и/или други идентификатори;
|
|
Външен вид, разтворимост в DMSO, молекулно тегло и допълнителни относими физични и химични свойства, според наличността, където е приложимо;
|
|
Чистота, химическа идентичност на онечистванията, както е целесъобразно и практически осъществимо и т.н.;
|
|
Обработка преди изпитването, ако се прилага (напр. затопляне, смилане);
|
|
Изпитвана концентрация (концентрации);
|
|
Условия на съхранение и стабилност, доколкото са налични;
|
|
Препратка към резултатите за положителните контроли за предходни периоди, доказващи подходящи критерии за приемливост на серията, ако е приложимо.
|
Отрицателна контрола и контрола на разтворител/носител
|
Химична идентификация, като наименование(я) по IUPAC или по CAS, CAS номер(а), SMILES или InChI код, структурна формула, и/или други идентификатори;
|
|
Чистота, химическа идентичност на онечистванията, както е целесъобразно и практически осъществимо и т.н.;
|
|
Външен вид, молекулно тегло и допълнителни относими физични и химични свойства, в случай че са използвани разтворители/носители, различни от упоменатите в Насоките за изпитване и в зависимост от наличността;
|
|
Условия на съхранение и стабилност, доколкото са налични;
|
|
Обосновка за избора на разтворител/носител за всеки изпитван химикал.
|
|
|
Условия на изпитване
|
Име и адрес на финансиращия, на изпитващата лаборатория и на ръководителя на изследването;
|
|
Описание на използваното изпитване;
|
|
Използвана клетъчна линия, условия за нейното съхранение и източник (например мястото, от което са взети);
|
|
Използвана поточна цитометрия (например модел), включително инструментални настройки, глобулин, антитела и маркер за цитотоксичност;
|
|
Процедурата, използвана за доказване на пригодността на лабораторията за провеждане на изпитването чрез изпитване на вещества за изпитване за пригодност, и процедурата, използвана за доказване на възпроизводимостта на резултатите от изпитването с течение на времето, например данни за контролите за предходни периоди и/или данни от предходни проверки за реакционна способност.
|
|
|
Критерии за приемливост на изпитването
|
Стойности на клетъчната жизнеспособност и SI на CD86, получени с контролата на разтворител/носител в сравнение с допустимия размах;
|
|
Стойности на клетъчната жизнеспособност и SI, получени с положителната контрола в сравнение с допустимия размах;
|
|
Клетъчна жизнеспособност при всички изпитвани концентрации на изпитвания химикал.
|
|
|
Процедура за изпитване
|
Брой на използваните серии;
|
|
Концентрации на изпитвания химикал, продължителност на прилагане и време на експозицията (при различие от препоръчаното)
|
|
Продължителност на експозицията;
|
|
Описание на използваните критерии за оценка и за решение;
|
|
Описание на всякакви изменения на процедурата за изпитване.
|
|
|
Резултати
|
Таблично представяне на данните, включително на стойности на CV70 (ако е приложимо), SI, клетъчна жизнеспособност, стойности на EC150 (ако е приложимо), получени за изпитвания химикал и за положителната контрола за всяка серия, като се посочва рангът на изпитвания химикал в съответствие с модела на прогнозиране;
|
|
Описание на всякакви други относими наблюдения, ако е приложимо.
|
|
|
Обсъждане на резултатите.
|
Обсъждане на резултатите, получени с U-SENS™ изпитването;
|
|
Разглеждане на резултатите от изпитването в рамките на контекста на IATA, ако е налична друга относима информация.
|
|
Заключения
|
ЛИТЕРАТУРА
|
(1)
|
Piroird, C., Ovigne, J.M., Rousset, F., Martinozzi-Teissier, S., Gomes, C., Cotovio, J., Alépée, N. (2015). The Myeloid U937 Skin Sensitization Test (U-SENS) addresses the activation of dendritic cell event in the adverse outcome pathway for skin sensitization. Toxicol. In Vitro 29, 901-916.
|
|
(2)
|
EURL ECVAM (2017). The U-SENS™ test method Validation Study Report. Достъпно на: http://ihcp.jrc.ec.europa.eu/our_labs/eurl-ecvam/eurl-ecvam-recommendations
|
|
(3)
|
EC EURL ECVAM (2016). ESAC Opinion No 2016-03 on the L'Oréal-coordinated study on the transferability and reliability of the U-SENS™ test method for skin sensitisation testing. EUR 28 178 EN; doi 10.2 787/8157 37. На разположение на адрес: [http://publications.jrc.ec.europa.eu/repository/handle/JRC103705].
|
|
(4)
|
EC EURL ECVAM (2017). EURL ECVAM Recommendation on the use of non-animal approaches for skin sensitisation testing. EUR 28 553 EN; doi 10.2 760/5889 55. На разположение на адрес: https://ec.europa.eu/jrc/en/publication/eur-scientific-and-technical-research-reports/eurl-ecvam-recommendation-use-non-animal-approaches-skin-sensitisation-testing.
|
|
(5)
|
Steiling, W. (2016). Safety Evaluation of Cosmetic Ingredients Regarding their Skin Sensitization Potential. doi:10.3390/cosmetics3020014.Cosmetics 3, 14.
|
|
(6)
|
OECD (2016). Guidance Document on The Reporting of Defined Approaches and Individual Information Sources to be Used Within Integrated Approaches to Testing and Assessment (IATA) For Skin Sensitisation, Series on Testing & Assessment No 256, ENV/JM/MONO(2016)29. Organisation for Economic Cooperation and Development, Paris. На разположение на адрес: [http://www.oecd.org/env/ehs/testing/series-testing-assessment-publications-number.htm.
|
|
(7)
|
Urbisch, D., Mehling, A., Guth, K., Ramirez, T., Honarvar, N., Kolle, S., Landsiedel, R., Jaworska, J., Kern, P.S., Gerberick, F., Natsch, A., Emter, R., Ashikaga, T., Miyazawa, M., Sakaguchi, H. (2015). Assessing skin sensitization hazard in mice and men using non-animal test methods. Regul. Toxicol. Pharmacol. 71, 337-351.
|
|
(8)
|
Alépée, N., Piroird, C., Aujoulat, M., Dreyfuss, S., Hoffmann, S., Hohenstein, A., Meloni, M., Nardelli, L., Gerbeix, C., Cotovio, J. (2015). Prospective multicentre study of the U-SENS test method for skin sensitization testing. Toxicol In Vitro 30, 373-382.
|
|
(9)
|
Reisinger, K., Hoffmann, S., Alépée, N., Ashikaga, T., Barroso, J., Elcombe, C., Gellatly, N., Galbiati, V., Gibbs, S., Groux, H., Hibatallah, J., Keller, D., Kern, P., Klaric, M., Kolle, S., Kuehnl, J., Lambrechts, N., Lindstedt, M., Millet, M., Martinozzi-Teissier, S., Natsch, A., Petersohn, D., Pike, I., Sakaguchi, H., Schepky, A., Tailhardat, M., Templier, M., van Vliet, E., Maxwell, G. (2014). Systematic evaluation of non-animal test methods for skin sensitisation safety assessment. Toxicol. In Vitro 29, 259-270.
|
|
(10)
|
Fabian, E., Vogel, D., Blatz, V., Ramirez, T., Kolle, S., Eltze, T., van Ravenzwaay, B., Oesch, F., Landsiedel, R. (2013). Xenobiotic metabolizin enzyme activities in cells used for testing skin sensitization in vitro. Arch. Toxicol. 87, 1 683-1 696.
|
|
(11)
|
OECD. (2018). Draft Guidance document: Good In Vitro Method Practices (GIVIMP) for the Development and Implementation of In Vitro Methods for Regulatory Use in Human Safety Assessment. Organisation for Economic Cooperation and Development, Paris. На разположение на адрес: http://www.oecd.org/env/ehs/testing/OECD Final Draft GIVIMP.pdf.
|
|
(12)
|
DB-ALM (2016). Protocol no 183: Myeloid U937 Skin Sensitization Test (U-SENS™), 33pp. Достъпно на: [http://ecvam-dbalm.jrc.ec.europa.eu/].
|
|
(13)
|
Sundström, C., Nilsson, K. (1976). Establishment and characterization of a human histiocytic lymphoma cell line (U-937). Int. J. Cancer 17, 565-577.
|
|
(14)
|
OECD (2005). Series on Testing and Assessment No. 34: Validation and International Acceptance of New or Updated Test Methods for Hazard Assessment. Organisation for Economic Cooperation and Development, Paris. На разположение на адрес: http://www.oecd.org/env/ehs/testing/series-testing-assessment-publications-number.htm.
|
|
(15)
|
United Nations UN (2015). Globally Harmonized System of Classification and Labelling of Chemicals (GHS). ST/SG/AC.10/30/Rev.6, Sixth Revised Edition, New York & Geneva: United Nations Publications. На разположение на адрес: http://www.unece.org/fileadmin/DAM/trans/danger/publi/ghs/ghs_rev06/English/ST-SG-AC10-30-Rev6e.pdf.
|
|
(16)
|
OECD (2012). Series on Testing and Assessment No 168: The Adverse Outcome Pathway for Skin Sensitisation Initiated by Covalent Binding to Proteins. Part 1: Scientific Evidence. Organisation for Economic Cooperation and Development, Paris. На разположение на адрес: http://www.oecd.org/env/ehs/testing/series-testing-assessment-publications-number.htm.
|
|
(17)
|
ECETOC (2003). Technical Report No 87: Contact sensitization: Classification according to potency. European Centre for Ecotoxicology & Toxicology of Chemicals, Brussels. На разположение на адрес: https://ftp.cdc.gov/pub/Documents/OEL/06.%20Dotson/References/ECETOC_2003-TR87.pdf.
|
Допълнение 2.1
ОПРЕДЕЛЕНИЯ
Точност
: степента на близост между резултатите от изпитването и приетите референтни стойности. Това е мярка за ефективността на изпитването и един от аспектите на неговата относимост. Терминът често се използва взаимозаменяемо с термина „съответствие“, за да се обозначи относителният дял на правилните резултати при дадено изпитване (14).
AOP (път, водещ до неблагоприятен ефект)
: последователност от събития от химичната структура на целеви химикал или група от сходни химикали през молекулното иницииращо събитие до представляващ интерес in vivo резултат (15).
Отклик на концентрацията при CD86
: Съществува зависимост от концентрацията (или отклик на концентрация), когато положителна концентрация (SI на CD86 ≥ 150) е последвана от концентрация с увеличаващ се SI на CD86.
Химикал
: Вещество или смес.
CV70
: Прогнозната концентрация, показваща клетъчна жизнеспособност 70 %.
Изместване
: Изместването се определя така i) коригираната стойност на CD86+ в % при повторение 3 за нетретираната контрола е по-малка от 50 % от средната на коригираните стойности на CD86+ в % при повторения 1 и 2 за нетретираната контрола; и ii) коригираната стойност на CD86+ в % при повторение 3 за отрицателната контрола е по-малка от 50 % от средната на коригираните стойности на CD86+ в % при повторения 1 и 2 за отрицателната контрола;
EC150
: прогнозните концентрации, показващи равен на 150 % SI на експресията на CD86.
Поточна цитометрия
: цитометрична техника, при която клетките, суспендирани в течност, се движат в поток една по една във времето през фокус на възбуждаща светлина, която се разсейва по начини, характерни за клетките и техните компоненти; клетките често се маркират с флуоресцентни маркери, така че светлината първо се абсорбира и след това се излъчва при изменени честоти.
Опасност
: Вътрешно присъщо свойство на даден агент или ситуация, притежаващо потенциал да предизвика неблагоприятни ефекти когато един организъм, система или (суб-)популация бъдат експонирани на този агент.
IATA (интегриран подход за изпитване и оценка)
: Структуриран подход, използван за идентифициране на опасност (потенциал), характеризиране на опасност (потенциал) и/или оценка на безопасността (потенциал и експозиция) на даден химикал или група от химикали, който стратегически интегрира и претегля всички данни, относими към предоставянето на информация за вземането на регулаторно решение по отношение на потенциалната опасност и/или риска, и/или необходимостта от допълнително целенасочено и следователно минимално изпитване.
Смес
: смес или разтвор, съставени от две или повече вещества.
Вещество с една съставка
: Вещество, определено от неговия количествен състав, в който съдържанието на една основна съставка е най-малко 80 % (w/w).
Вещество с повече съставки
: Вещество, определено от неговия количествен състав, в който повече от една основна съставка се съдържа в концентрация ≥ 10 % (w/w) и < 80 % (w/w). Веществото с повече съставки е резултат от производствен процес. Разликата между смес и вещество с повече съставки е, че дадена смес е получена чрез смесване на две или повече вещества без химична реакция. Веществото с повече съставки е резултат от химична реакция.
Положителна контрола
: Повторение, съдържащо всички компоненти на дадена изпитвана система и третирано с вещество, за което е известно, че предизвиква положителен отклик. За да гарантира, че може да се направи оценка на варирането на отклика в положителната контрола във времето, големината на положителния отклик не следва да е прекомерна.
Пре-хаптени
: химикали, които стават сенсибилизатори чрез абиотично преобразуване, напр. чрез окисление.
Про-хаптени
: химикали, които изискват ензимно активиране за реализирането на потенциал за кожна сенсибилизация.
Относимост
: описание на взаимовръзката между изпитването и ефекта, представляващ интерес, и дали тя е значима и полезна за определена цел. Това е степента, в която изпитването правилно измерва или прогнозира биологичния ефект, представляващ интерес. Относимостта включва разглеждане на точността (съответствието) на дадено изпитване (14).
Надеждност
: мярка за степента, в която дадено изпитване може да се извърши възпроизводимо в една и съща лаборатория и в различни лаборатории по различно време, като при извършването се използва един и същи протокол. Оценката за нея се прави, като се изчислява вътрешнолабораторната и междулабораторната възпроизводимост и вътрешнолабораторната повторяемост (14).
Серия
: Серията представлява един или повече изпитвани химикали, изпитвани едновременно с контрола на разтворител/носител и с положителна контрола.
Чувствителност
: Относителният дял от всички положителни/активни химикали, които са класифицирани правилно чрез изпитването. Това е мярка за точността на изпитването, което предоставя категорийни резултати, и е важен фактор при оценката на относимостта на дадено изпитване (14).
SI
: Стимулационен индекс. Относителни стойности на средногеометричната стойност на интензитета на флуоресценция в третираните с химикал клетки в сравнение с клетки, третирани с разтворител.
Контрола на разтворител/носител
: Нетретирана проба, съдържаща всички компоненти на изпитваната система, с изключение на изпитвания химикал, но включваща разтворителя/носителя, който се използва. Използва се за определяне на базовия отклик за пробите, третирани с изпитвания химикал, разтворен или стабилно диспергиран в същия разтворител/носител. Когато се изпитва с паралелна контрола на среда, тази проба показва също дали разтворителят/носителят взаимодейства с изпитваната система.
Специфичност
: Относителният дял от всички отрицателни/неактивни химикали, които са класифицирани правилно чрез изпитването. Това е мярка за точността на изпитването, което предоставя категорийни резултати и е важен фактор при оценката на относимостта на дадено изпитване (14).
Оцветяващ буфер
: Фосфатно буфериран физиологичен разтвор, съдържащ 5 % фетален телешки серум.
Вещество
: означава химичен елемент и неговите съединения в естествено състояние или получени чрез всеки производствен процес, включително всяка добавка, необходима за запазване на неговата стабилност и всеки примес, извлечен от използвания процес, с изключение на всеки разтворител, който може да бъде отделен, без да се засяга стабилността на веществото или да се променя неговият състав.
Изпитван химикал
: Всяко вещество или смес, изпитвано(а) с използване на настоящия метод.
Глобална хармонизирана система на Организацията на обединените нации за класифициране и етикетиране на химикали (GHS на ООН)
: система, предлагаща класифициране на химикали (вещества и смеси) според стандартизирани видове и степени на физическа, здравна и екологична опасност и разглеждаща съответни съобщителни елементи, като например пиктограми, сигнални думи, предупреждения за опасност, препоръки за безопасност и информационни листове за безопасност, така че те да съобщават информация за тяхното неблагоприятно въздействие с оглед защита на хората (включително работодатели, работещи, служители в транспорта, потребители и аварийни служители) и на околната среда (16).
UVCB
: вещества с неизвестен или променлив състав, сложни продукти от реакции или биологични материали.
Валидно изпитване
: Изпитване, за което се смята, че притежава достатъчна относимост и надеждност по отношение на конкретна цел и което се основава на научно обосновани принципи. Дадено изпитване никога не е валидно в абсолютен смисъл, а само по отношение на определена цел (14).
Допълнение 2.2
ВЕЩЕСТВА ЗА ИЗПИТВАНЕ ЗА ПРИГОДНОСТ
Преди рутинното използване на изпитването, описано в настоящото допълнение към метод за изпитване Б.71, лабораториите следва да докажат техническата си пригодност, като правилно получат очакваната прогноза по U-SENS™ за 10-те вещества, препоръчани в Таблица 1, и чрез получаване на стойности за CV70 и EC150, които попадат в съответния референтен обхват за минимум 8 от 10-те вещества за изпитване за пригодност. Веществата за изпитване за пригодност са подбрани като представителни за размаха от отклици за опасностите от кожна сенсибилизация. Други критерии за подбор бяха веществата да са налични в търговската мрежа и да са налични висококачествени in vivo референтни данни, както и висококачествени in vitro данни, получени по U-SENS™ изпитването. Също така са налични публикувани референтни данни за U-SENS™ изпитването (1) (8).
Таблица 1
Препоръчвани вещества за доказване на техническа пригодност по отношение на U-SENS™ изпитването
|
Вещества за изпитване за пригодност
|
CAS №
|
Агрегатно състояние
|
In vivo прогноза (106)
|
U-SENS™ Разтворител/Носител
|
U-SENS™ CV70 Позоваване Размах в μg/ml (107)
|
U-SENS™ референтен за ЕС150 размах в μg/ml (107)
|
|
4-Фенилендиамин
|
106-50-3
|
Твърд
|
Сенсибилизатор (силен)
|
Пълноценна среда (108)
|
<30
|
Положително (≤10)
|
|
Пикрилсулфонова киселина
|
2508-19-2
|
Течност
|
Сенсибилизатор (силен)
|
Пълноценна среда
|
>50
|
Положително (≤50)
|
|
Диетилов малеат
|
141-05-9
|
Течност
|
Сенсибилизатор (умерен)
|
DMSO
|
10-100
|
Положително (≤20)
|
|
Резорцинол
|
108-46-3
|
Твърд
|
Сенсибилизатор (умерен)
|
Пълноценна среда
|
>100
|
Положително (≤50)
|
|
Канелен алкохол
|
104-54-1
|
Твърд
|
Сенсибилизатор (слаб)
|
DMSO
|
>100
|
Положително (10-100)
|
|
4-Алиланизол
|
140-67-0
|
Течност
|
Сенсибилизатор (слаб)
|
DMSO
|
>100
|
Положително (<200)
|
|
Захарин
|
81-07-2
|
Твърд
|
Непредизвикващо сенсибилизация вещество
|
DMSO
|
>200
|
Отрицателно (>200)
|
|
Глицерол
|
56-81-5
|
Течност
|
Непредизвикващо сенсибилизация вещество
|
Пълноценна среда
|
>200
|
Отрицателно (>200)
|
|
Млечна киселина
|
50-21-5
|
Течност
|
Непредизвикващо сенсибилизация вещество
|
Пълноценна среда
|
>200
|
Отрицателно (>200)
|
|
Салицилова киселина
|
69-72-7
|
Твърд
|
Непредизвикващо сенсибилизация вещество
|
DMSO
|
>200
|
Отрицателно (>200)
|
|
Съкращения: CAS № = регистрационен номер по Службата за химични индекси.
|
Допълнение 3
IN VITRO КОЖНА СЕНСИБИЛИЗАЦИЯ: ИЗСЛЕДВАНЕ IL-8 LUC
ПЪРВОНАЧАЛНИ СЪОБРАЖЕНИЯ И ОГРАНИЧЕНИЯ
|
1.
|
За разлика от изследванията, при които се анализира експресията на маркерите на клетъчната повърхност, изследването IL-8 Luc измерва количествено измененията на експресията на IL-8 — цитокин, свързан с активирането на дендритни клетки (ДК). В получената от THP-1 IL-8 репортерна клетъчна линия (THP-G8, създадена от моноцитната клетъчна линия THP-1 от остра левкемия при човека) експресията на IL-8 се измерва след експозиция на сенсибилизатори (1). След това експресията на луциферазата се използва за подпомагане на разграничаването между кожни сенсибилизатори и непредизвикващи сенсибилизация вещества.
|
|
2.
|
IL-8 Luc изследването бе оценено чрез изследване за валидиране (2), проведено от Японския център за валидиране на алтернативни методи (JaCVAM), Министерството на икономиката, търговията и промишлеността (METI) и японското дружество за алтернативи на опитите с животни (JSAAE) и впоследствие бе подложено на независима партньорска проверка (3) под егидата на JaCVAM и на Министерството на здравеопазването, труда и благосъстоянието (MHLW) с подкрепата на Международната група за сътрудничество в областта на алтернативните методи за изпитване (ICATM ). Вземайки предвид всички налични доказателства и данни от регулаторните органи и заинтересованите страни, изследването IL-8 Luc се счита за полезно като част от IATA за подкрепа на разграничаването между сенсибилизатори и непредизвикващи сенсибилизация вещества за целите на класифицирането на опасността и етикетирането. Примери за използването на данни от изследването IL-8 Luc в комбинация с друга информация са описани в литературата (4) (5) (6).
|
|
3.
|
Бе показано, че изследването IL-8 Luc е прехвърляемо към лаборатории с опит в клетъчните култури и измерването на луциферазата. Вътрелаборната и междулабораторната възпроизводимост са били съответно 87,7 % и 87,5 % (2). Данните, получени в изследването за валидиране (2) и други публикувани изследвания (1) (6) показват, че в сравнение с LLNA изследването IL-8 Luc е заключило за 118 от 143 химикала като положителни или отрицателни, и за 25 химикала като не позволяващи да се даде заключение, и точността на изследването IL-8 Luc при разграничаването на кожните сенсибилизатори (категория 1 по GHS на ООН/Регламент CLP) от непредизвикващите сенсибилизация вещества (без категория по GHS на ООН/Регламент CLP) е 86 % (101/118), с чувствителност от 96 % (92/96) и специфичност от 41 % (9/22). С изключение на веществата извън областта на приложимост, описана по-долу (точка 5), изследването IL-8 Luc е заключило за 113 от 136 химикала като положителни или отрицателни, и за 23 химикала като не позволяващи да се даде заключение, и точността на изследването IL-8 Luc е 89 % (101/113), с чувствителност от 96 % (92/96) и специфичност от 53 % (9/17). С използване на данни за хора, цитирани в Urbisch et al. (7) изследването IL-8 Luc е заключило за 76 от 90 химикала като положителни или отрицателни, и за 14 химикала като не позволяващи да се даде заключение, и точността е 80 % (61/76), с чувствителност от 93 % (54/58) и специфичност от 39 % (7/18). С изключение на веществата извън областта на приложимост, изследването IL-8 Luc е заключило за 71 от 84 химикала като положителни или отрицателни, и за 13 химикала като не позволяващи да се даде заключение, и точността е 86 % (61/71), с чувствителност от 93 % (54/58) и специфичност от 54 % (7/13). Има по-голяма вероятност неверните отрицателни прогнози, свързани с изследването IL-8 Luc, да се отнасят за химикали, които са с малък/умерен потенциал за кожна сенсибилизация (кожна сенсибилизация подкатегория 1B по GHS на ООН/Регламент CLP), отколкото при химикали, показващи голям потенциал за кожна сенсибилизация (т.е. подкатегория 1A по GHS на ООН/Регламент CLP) (6). Взета заедно, информацията показва, че изследването IL-8 Luc оказва подкрепа за идентифициране на опасностите от кожна сенсибилизация. Точността на изследването IL-8 Luc като самостоятелно изпитване е само ориентировъчна, тъй като изпитването трябва да се разглежда в съчетание с други източници на информация в контекста на IATA и в съответствие с разпоредбите на точки 7 и 8 от Общото въведение. Освен това при оценяването на изпитвания за сенсибилизация на кожата, които не използват животни, следва да се има предвид, че LLNA и други изпитвания върху животни могат да не отразяват напълно положението при хората.
|
|
4.
|
Въз основа на наличните понастоящем данни бе показано, че изследването IL-8 Luc е приложимо по отношение на изпитвани химикали, които обхващат различни органични функционални групи, риакционни механизми, потенциал за сенсибилизация на кожата (както е определено в in vivo проучвания) и физични и химични свойства (2)(6).
|
|
5.
|
Въпреки че изследването IL-8 Luc използва като разтворител X-VIVOTM 15, с него правилно са оценени химикали с Log Kow >3,5 и тези с разтворимост във вода от около 100 μg/ml, изчислена с EPI SuiteTM, и неговата ефективност за откриване на сенсибилизатори със слаба разтворимост във вода е по-добра от тази на изследването IL-8 Luc с използване на диметилсулфоксид (DMSO) като разтворител (2). Въпреки това отрицателните резултати за изпитвани химикали, които не са разтворени при 20 mg/ml, могат да доведат до неверни отрицателни резултати, дължащи се на тяхната неспособност да се разтворят в X-VIVOTM 15. Поради това отрицателните резултати за тези химикали не следва да се вземат под внимание. В изследването за валидиране беше установено наличието на висок процент неверни отрицателни резултати за анхидриди. Освен това, поради ограничения метаболитен капацитет на клетъчната линия (8) и условията на опита про-хаптените (вещества, изискващи метаболитно активиране) и пре-хаптените (вещества, които се активират от окисление от въздуха) могат да дадат отрицателни резултати при изследването. Обаче независимо от това, че отрицателните резултати от съмнения за пре-/про-хаптени следва да се тълкуват предпазливо, изследването IL-8 Luc е оценило правилно 11 от 11 пре-хаптена, 6/6 про-хаптена, и 6/8 пре-/про-хаптена в набора от данни за изследването IL-8 Luc (2). Въз основа на неотдавнашния задълбочен преглед на три изпитвания, при които не се използват животни (DPRA, KeratinoSens ™ и h-CLAT), за установяване на пре- и про-хаптени (9), както и въз основа на факта, че клетките THP-G8, използвани в изпитването IL-8 Luc, са клетъчна линия, получена от THP-1, която се използва в h-CLAT, изпитването IL-8 Luc може също да допринесе за увеличаване на чувствителността на изпитванията за установяване на пре- и про-хаптени, в комбинация с други изпитвания. Повърхностноактивни вещества, изпитвани до момента, дават (неверни) положителни резултати, независимо от техния вид (напр. катионни, анионни или нейонни). Накрая, химикали, които смущават луциферазата, могат да доведат до объркване при нейната активност/измерване, като предизвикат привидно инхибиране или повишена луминесценция (10). Например за концентрации на фитоестрогени, по-високи от 1 μМ, има данни, че смущават сигналите за луминесценцията в други изпитвания с репортерни гени, основани на луциферазата, поради свръхактивиране на луциферазния репортерен ген. Вследствие на това експресията на луциферазата, получена при високи концентрации на фитоестрогени или съединения, за които се подозира, че предизвикват подобно на фитоестрогени активиране на луциферазния репортерен ген, трябва да бъде внимателно разгледана (11). Въз основа на горното повърхностноактивните вещества, анхидридите и химикалите, пречещи на луциферазата, са извън областта на приложимост на това изследване. В случаите, когато са налице доказателства за неприложимостта на изследването IL-8 Luc по отношение на други специфични категории изпитвани химикали, изпитването не следва да се използва за тези специфични категории.
|
|
6.
|
Както е описано по-горе, изследването IL-8 Luc подкрепя разграничаването между кожни сенсибилизатори и непредизвикващи сенсибилизация вещества. Необходима е допълнителна работа, за предпочитане въз основа на данни от хора, за да се определи дали резултатите от изследването IL-8 Luc могат да допринесат за оценката на потенциала, когато се разглеждат в комбинация с други източници на информация.
|
|
7.
|
Определенията са дадени в допълнение 3.1.
|
ПРИНЦИП НА ИЗПИТВАНЕТО
|
8.
|
При изследването IL-8 Luc се използва моноцитната клетъчна линия THP-1 от левкемия при човека, която е получена от Американската банка за типови култури (Manassas, VA, САЩ). При използването на тази клетъчна линия департаментът по дерматология на Tohoku University School of Medicine създаде получена от THP-1 IL-8 репортерна клетъчна линия, THP-G8, носеща гените на луциферази Stable Luciferase Orange (SLO) и Stable Luciferase Red (SLR), съответно под контрола на промоторите IL-8 и глицералдехид-3-фосфатдехидрогеназа ( GAPDH) (1). Това позволява количествено измерване на индукцията на луциферазния ген чрез откриване на луминесценция, получена от добре установени произвеждащи светлина луциферазни субстрати като показател за активността на IL-8 и GAPDH в клетките след експозиция на сенсибилизиращи химикали.
|
|
9.
|
Системата за изследване в два цвята включва луцифераза, излъчваща в оранжево (SLO; λmax = 580 nm) (12) за генната експресия на промотора IL-8, както и на излъчваща в червено луцифераза (SLR); λmax = 630 nm) (13) за генната експресия на промотора, използван като вътрешна контрола, GAPDH. Двете луциферзи излъчват различни цветове при реакция с d-луциферин от светулки и луминесценцията им се измерва едновременно в еднофазна реакция чрез разделяне на емисията от сместа за изследването, чрез използване на оптичен филтър (14) (допълнение 3.2).
|
|
10.
|
THP-G8 клетките се третират в продължение на 16 часа с изпитвания химикал, след което се измерват активността на луциферазата SLO (SLO-LA), която отразява активността на IL-8 промотора, и активността на луциферазата SLR (SLR-LA), която отразява активността на GAPDH промотора. За да станат съкращенията лесни за разбиране, SLO-LA и SLR-LA се обозначават съответно като IL8LA и GAPLA. В таблица 1 е дадено описание на термините, свързани с луциферазна активност в изследването IL-8 Luc. Измерените стойности се използват за изчисляване на нормализирана IL8LA (nIL8LA), която е отношението на IL8LA към GAPLA; индукцията на nIL8LA (Ind-IL8LA), която е отношението на средноаритметичните стойности на четирикратно измерените стойности на nIL8LA на THP-G8 клетките, третирани с изпитван химикал, към стойностите на nIL8LA на нетретирани клетки THP-G8; и инхибирането на GAPLA (Inh-GAPLA), което е отношението на средноаритметичните стойности на четирикратно измерените стойности на GAPLA на THP-G8 клетките, третирани с изпитван химикал, към стойностите на GAPLA на нетретирани клетки THP-G8, и се използва като индикатор за цитотоксичност.
Таблица 1
Описание на термините, свързани с луциферазната активност при изследването IL-8 Luc
|
Съкращения
|
Определение
|
|
GAPLA
|
луциферазна активност на SLR, отразяваща активността на GAPDH промотора
|
|
IL8LA
|
луциферазна активност на SLO, отразяваща активността на IL-8 промотора
|
|
nIL8LA
|
IL8LA / GAPLA
|
|
Ind-IL8LA
|
nIL8LA на THP-G8 клетки, третирани с химикали/nIL8LA на нетретирани клетки
|
|
Inh-GAPLA
|
GAPLA на THP-G8 клетки, третирани с химикали/GAPLA на нетретирани клетки
|
|
CV05
|
Най-ниската концентрация на химикала, при която Inh-GAPLA става < 0,05.
|
|
|
11.
|
На разположение са стандарти за ефективност (PS) (15), за да се улесни валидирането на модифицирани in vitro IL-8 изпитвания с луцифераза, подобни на изследването IL-8 Luc, и да се даде възможност за своевременно изменение на Насока за изпитване 442E на ОИСР с оглед на тяхното включване. Взаимното приемане на данни в ОИСР (MAD) ще бъде гарантирано само за изпитвания, валидирани съгласно PS, ако тези изпитвания са били проверени и включени в Насока за изпитване 442E от ОИСР (16).
|
ДОКАЗВАНЕ НА ПРИГОДНОСТ
|
12.
|
Преди рутинното използване на изпитването, описано в настоящото допълнение към метод за изпитване Б.71, лабораториите следва да докажат техническата си пригодност, като използват 10-те вещества за изпитване на пригодност, изброени в допълнение 3.3, в съответствие с добрите практики за in vitro методите (17). Освен това ползвателите на изпитването следва да поддържат база данни с данни за предходни периоди, получени с проверките на реакционната способност (вж. точка 15) и с положителни контроли и контроли на разтворител/носител (вж. точки 21—24), и да използват тези данни, за да потвърдят, че възпроизводимостта на изпитването в тяхната лаборатория се запазва с течение на времето.
|
ПРОЦЕДУРА
|
13.
|
Стандартната работна процедура (СРП) за изследването IL-8 Luc е налице и следва да бъде използвана при провеждане на изпитването (18). Лабораториите, които желаят да извършват изпитването, могат да получат рекомбинантната клетъчна линия THP-G8 от GPC Lab. Co. Ltd., Tottori, Япония, след подписване на Споразумение за трансферна материал (MTA) в съответствие с условията в образеца на ОИСР. В точките по-долу е представено описание на основните компоненти и процедури за изследването.
|
Подготовка на клетките
|
14.
|
Клетъчна линия THP-G8 от GPC Lab. Co. Ltd., Tottori, Япония, следва да се използва за провеждането на изследването IL-8 Luc (вж. точки 8 и 13). След получаване клетките се размножават (2—4 пасажа) и се съхраняват замразени като хомогенни изходни култури. Клетките от тези изходни култури могат да бъдат размножавани до максимум 12 пасажа или най-много 6 седмици. Средата, използвана за размножаване, е среда за отглеждане RPMI-1640, съдържаща 10 % фетален говежди серум (FBS), антибиотичен/антимикотичен разтвор (100U/ml пеницилин G, 100μg/ml стрептомицин и 0,25μg/ml амфотерицин B в 0,85 % физиологичен разтвор) (напр. GIBCO Cat#15240-062), 0,15μg/ml пуромицин (напр. CAS: 58-58-2) и 300 μg/ml G418 (напр. CAS:108321-42-2).
|
|
15.
|
Преди да бъдат използвани за изпитване, клетките следва да бъдат квалифицирани чрез извършване на проверка за реакционна способност. Тази проверка следва да бъде извършена 1—2 седмици или 2—4 пасажа след размразяването, като се използва положителната контрола, 4-нитробензилбромид (4-NBB) (CAS №: 100-11-8, чистота ≥ 99 %), и отрицателната контрола, млечна киселина (LA) (CAS:50-21-5, чистота ≥ 85 %). 4-NBB следва да даде положителен отклик на Ind-IL8LA (≥ 1,4), докато LA следва да даде отрицателен отклик на Ind-IL8LA (< 1,4). За изследването се използват само клетките, които преминават проверката за реакционна способност. Проверката следва да се извършва в съответствие с процедурите, описани в точки 22—24.
|
|
16.
|
За изпитване THP-G8 клетките се посяват с плътност 2 до 5 × 105 клетки/ml и се извършва предварително култивиране в колби за клетъчни култури съответно от 48 до 96 часа. В деня на провеждане на изпитването клетките, събрани от колбите за клетъчни култури, се промиват с RPMI-1640, съдържащ 10 % FBS, без никакви антибиотици, и след това се ресуспендират с RPMI-1640, съдържащ 10 % от FBS, без никакви антибиотици, при 1 × 106 клетки/ml. След това клетките се разпределят в 96-ямкова плоскодънна черна плака (напр. Costar Cat#3603) с 50μl (5 × 104 клетки/ямка).
|
Приготвяне на изпитвания химикал и веществата за контроли
|
17.
|
Изпитваният химикал и веществата за контроли се приготвят в деня на изпитването. По отношение на изследването IL-8 Luc изпитваните химикали се разтварят в X-VIVOTM 15, налична в търговската мрежа безсерумна среда (Lonza, 04-418Q) до крайната концентрация от 20 mg/ml. X-VIVOTM 15 се добавя към 20 mg изпитван химикал (независимо от разтворимостта на изпитвания химикал) в микроцентрофужна епруветка и се довежда до обем от 1 ml, след това се смесва енергично във вихров смесител и се разклаща на ротора при максимална скорост 8 rpm в продължение на 30 минути при температура на околната среда около 20oC. Освен това, ако твърдите химикали са все още неразтворими, епруветката минава през сонификация, докато химикалът се разтвори напълно или се диспергира стабилно. За изпитвани химикали, разтворими в X-VIVOTM 15, разтворът се разрежда с коефициент 5 с X-VIVOTM 15 и се използва като изходен разтвор в X-VIVOTM 15 на изпитвания химикал (4 mg/ml). За изпитвани химикали, които не са разтворими в X-VIVOTM 15, сместа се завърта отново за най-малко 30 min, след това се центрофугира при 15000 rpm (≈ 20 000g) за 5 min; полученият супернатант се използва като изходен разтвор в X-VIVOTM 15 на изпитвания химикал. Следва да се предостави научна обосновка за употребата на други разтворители, като например DMSO, вода или средата за отглеждане. Подробната процедура за разтваряне на химикали е описана в допълнение 3.5. Разтворите в X-VIVOTM 15, описани в точки 18-23, се смесват 1:1 (v/v) с клетъчните суспензии, подготвени в 96-ямкова плоскодънна черна плака (вж. точка 16).
|
|
18.
|
Първата изпитвателна серия има за цел да се определи цитотоксичната концентрация и да се изследва потенциалът за кожна сенсибилизация на химикалите. С помощта на X-VIVOTM 15 серийните разреждания на изходните разтвори на изпитваните химикали в X-VIVOTM 15 се приготвят с коефициент на разреждане от две (вж. допълнение 3.5), като се използва 96-ямков блок за изследване (напр. Costar Cat#EW-01729-03). След това 50 μl/ямка разреден разтвор се добавя към 50 μl от клетъчната суспензия в 96-ямкова плоскодънна черна плака. Така за изпитвани химикали, които са разтворими в X-VIVOTM 15, крайните концентрации на изпитваните химикали варират от 0,002 до 2 mg/ml (допълнение 3.5). За изпитвани химикали, които не са разтворими в X-VIVOTM 15 при 20 mg/ml, се определят само коефициенти на разреждане , които варират от 2 до 210, въпреки че действителните крайни концентрации на изпитваните химикали остават несигурни и зависят от концентрацията на наситените разтвори на изпитваните химикали в изходния разтвор X-VIVOTM 15.
|
|
19.
|
При последващите изпитвателни серии (т.е. второ, трето и четвърто повторения) изходният разтвор X-VIVOTM 15 се приготвя в концентрация 4 пъти по-висока от концентрацията на клетъчна жизнеспособност 05 (CV05; най-ниската концентрация, при която Inh-GAPLA става < 0,05) в първия опит. Ако при най-високата концентрация в първата серия Inh-GAPLA не намалява под 0,05, изходният разтвор X-VIVOTM 15 се приготвя при най-високата концентрация в първата серия. Концентрацията на CV05 се изчислява, като концентрацията на изходния разтвор в първата серия се раздели на коефициента на разреждане за CV05 (X) (коефициент на разреждане CV05 (X); коефициентът на разреждане, изискван за разреждане на изходния разтвор до CV05) (вж. допълнение 3.5). За изпитвани вещества, които не са разтворими в X VIVO при 20 mg/ml, CV05 се определя от концентрацията на изходния разтвор × 1/X. За серии от 2 до 4 се приготвя втори изходен разтвор като 4 × CV05 (допълнение 3.5).
|
|
20.
|
Серийните разреждания на вторите изходни разтвори на X-VIVOTM 15 се приготвят при коефициент на разреждане 1,5, като се използва 96-ямков блок за изследване. След това 50 μl/ямка разреден разтвор се добавя към 50 μl от клетъчната суспензия в ямките на 96-ямкова плоскодънна черна плака. Всяка концентрация на всеки изпитван химикал следва да бъде изпитана в 4 ямки. След това пробите се смесват с клатачна машина за плаки и се инкубират за 16 часа при 37oC и 5 % CO2, след което луциферазната активност се измерва, както е описано по-долу.
|
|
21.
|
Контролата на разтворител е сместа от 50 μl/ямка X-VIVOTM и 50 μl/ямка клетъчна суспензия в RPMI-1640 със съдържание на 10 % FBS.
|
|
22.
|
Препоръчителната положителна контрола е 4-NBB. 20 mg от 4-NBB се приготвят в микроцентрофужна епруветка от 1,5 ml, към която се добавя X-VIVOTM 15 до 1 ml. След това съдържанието на епруветката се смесва енергично във вихров смесител и се разклаща на ротор при максимална скорост 8 rpm в продължение на най-малко 30 минути. След центрофугиране при 20 000 g в продължение на 5 min, супернатантът се разрежда с коефициент 4 с X-VIVOTM 15 и 500 μl от разредения супернатант се прехвърлят към якмка в 96-ямков блок за изследване. Разреденият супернатант се разрежда допълнително с X-VIVOTM 15 с коефициенти 2 и 4 и 50 μl от разтвора се добавят към 50 μl суспензия от THP-G8 клетки в ямките на 96-ямкова плоскодънна черна плака (допълнение 3.6). Всяка концентрация на положителната контрола следва да бъде изпитана в 4 ямки. Плаката се разбърква на клатачна машина за плаки и се инкубира в CO2 инкубатор за 16 часа (37oC, 5 % CO2), след което луциферазна активност се измерва както е описано в точка 29.
|
|
23.
|
Препоръчителната положителна контрола е LA. 20 mg от LA се приготвят в микроцентрофужна епруветка от 1,5 ml, към която се добавя X-VIVOTM до 1 ml (20 mg/ml). Двадесет mg/ml разтвор на LA се разрежда с коефициент 5 с X-VIVOTM 15 (4 mg/ml); 500 μl от този 4 mg/ml разтвор на LA се прехвърлят към ямка от 96-ямков блок за изследване. Този разтвор се разрежда с коефициент 2 с X-VIVOTM 15 и след това се разрежда отново с коефициент 2, за да се получат разтвори от 2 mg/ml и 1 mg/ml. 50 μl от тези 3 разтвора и контрола на носител (X-VIVOTM 15) се добавят към 50 μl от суспензия от THP-G8 клетки в ямките на 96-ямкова плоскодънна черна плака. Всяка концентрация на отрицателната контрола се изпитва в 4 ямки. Плаката се разбърква на клатачна машина за плаки и се инкубира в CO2 инкубатор за 16 часа (37oC, 5 % CO2), след което луциферазната активност се измерва както е описано в точка 29.
|
|
24.
|
Могат да се използват други подходящи положителни или отрицателни контроли, ако има налични данни за предходни периоди, за извеждане на сравними критерии за приемливост на серията.
|
|
25.
|
Следва да се вземат мерки за избягване на изпаряването на летливи изпитвани химикали и на кръстосаното замърсяване с изпитвани химикали между ямките, като напр. плаките се запечатат преди инкубирането с изпитваните химикали.
|
|
26.
|
Изпитваните химикали и контролата на разтворител изискват от 2 до 4 серии, за да се изведе положителна или отрицателна прогноза (вж. таблица 2). Всяка серия се провежда в различен ден с пресен изходен разтвор в X-VIVOTM 15 на изпитваните химикали и независимо събрани клетки. Клетките могат да са от един и същ пасаж.
|
Измервания на луциферазната активност
|
27.
|
Луминесценцията се измерва с луминометър с 96-ямкова микроплака, оборудван с оптични филтри, напр. Phelios (ATTO, Токио, Япония), Tristan 941 (Berthold, Bad Wildbad, Германия) и серията ARVO (PerkinElmer, Waltham, MA, САЩ). Луминометърът трябва да се калибрира за всяко изпитване, за да се осигури възпроизводимост (19). За това калибриране на разположение са рекомбинантни луциферази, излъчващи в оранжево и в червено.
|
|
28.
|
100μl предварително затоплен реактив Tripluc® за изследване на луцифазаза (Tripluc) се прехвърлят към всяка ямка на плаката, съдържаща клетъчната суспензия, третирана със или без химикал. Плаката се разклаща в продължение на 10 минути при температура на околната среда от около 20oС. Плаката се поставя в луминометъра за измерване на луциферазната активност. Биолуминесценцията се измерва в продължение на 3 секунди, всеки път при отсъствие (F0) и наличие (F) на оптичния филтър. Трябва да се представи обосновка ако се използват алтернативни настройки, например в зависимост от използвания модел на луминометър.
|
|
29.
|
Параметрите за всяка концентрация се изчисляват въз основа на измерените стойности, напр. IL8LA, GAPLA, nIL8LA, Ind-IL8LA, Inh-GAPLA, средната ± SD на IL8LA, средната стойност ± SD на GAPLA, средната стойност ± SD на nIL8LA, средната ± SD of Ind-ILV8LA, средната ± SD of Inh-GAPLA, средната ± SD of Inh-GAPLA, и 95 %-ия доверителен интервал на Ind-IL8LA. Определенията на параметрите, използвани в настоящата точка, са дадени съответно в допълнения 3.1 и 3.4.
|
|
30.
|
Преди измерването, цветовото разграничаване при многоцветни изследвания на репортери обичайно се постига с помощта на детектори (луминометър и четец за плаки), оборудвани с оптични филтри, като дълговълнови или късовълнови пропускащи филтри с рязка граница между зоните, или лентово-пропускащи филтри. Коефициентите на пропускане на филтрите за всеки цвят на сигнала от биолуминесценция трябва да бъдат калибрирани преди изпитването, съгласно допълнение 3.2.
|
ДАННИ И ПРОТОКОЛИРАНЕ
Оценка на данните
|
31.
|
Критериите за положително/отрицателно решение изискват във всяка серия:
|
—
|
прогноза от IL-8 Luc изследване да бъде счетена за положителна ако даден изпитван химикал има Ind-IL8LA ≥ 1,4 и долната граница на доверителния 95 % интервал на Ind-IL8LA ≥ 1,0
|
|
—
|
прогноза от IL-8 Luc изследване да бъде счетена за отрицателна ако даден изпитван химикал има Ind-IL8LA < 1,4 и/или долната граница на доверителния 95 % интервал на Ind-IL8LA < 1,0
|
|
Модел на прогнозиране
|
32.
|
Изпитваните химикали, които дават два положителни резултата от 1-та , 2-та , 3-та или 4-та серия, се определят като положителни, а тези, които дават три отрицателни резултата от 1-та , 2-та , 3-та или 4-та серия, се определят като предполагаеми отрицателни (таблица 2). Сред предполагаемите отрицателни химикали, химикалите, които са разтворени при 20 mg/ml с X-VIVOTM 15, се оценяват като отрицателни, а химикалите, които не са разтворени при 20 mg/ml с X-VIVOTM 15, не следва да бъдат разглеждани (фигура 1).
Таблица 2
Критерии за определяне на положителни и предполагаеми отрицателни
|
1-а серия
|
2-а серия
|
3-а серия
|
4-а серия
|
Крайна прогноза
|
|
Положително
|
Положително
|
—
|
—
|
Положително
|
|
Отрицателно
|
Положително
|
—
|
Положително
|
|
Отрицателно
|
Положително
|
Положително
|
|
Отрицателно
|
Предполагаемо отрицателно
|
|
Отрицателно
|
Положително
|
Положително
|
—
|
Положително
|
|
Отрицателно
|
Положително
|
Положително
|
|
Отрицателно
|
Предполагаемо отрицателно
|
|
Отрицателно
|
Положително
|
Положително
|
Положително
|
|
Отрицателно
|
Предполагаемо отрицателно
|
|
Отрицателно
|
—
|
Предполагаемо отрицателно
|
|
Фигура 1
Модел за прогнозиране на крайна оценка
Критерии за приемливост
|
33.
|
Когато се използва IL-8 Luc изследване трябва да са спазени следните критерии за приемливост.
|
—
|
Ind-IL8LA следва да бъде повече от 5,0 поне в една концентрация на положителната контрола, 4-NBB, за всяка серия.
|
|
—
|
Ind-IL8LA следва да бъде по-малко от 1,4 във всяка концентрация на отрицателната контрола, млечна киселина, за всяка серия.
|
|
—
|
Данните от плаки, за които GAPLA от контролните ямки с клетки и Tripluc, но без химикали, е по-малко от 5 пъти стойността на ямката, която съдържа само среда за изпитване (50 μl/ямка с RPMI-1640, съдържаща 10 % FBS, и 50 μl/ямка с X-VIVOTM 15) следва да се отхвърлят.
|
|
—
|
Данните от плаки, за които стойността на Inh-GAPLA при всички концентрации на изпитваните или контролните химикали е по-малка от 0,05, следва да бъдат отхвърлени. В този случай първото изпитване трябва да се повтори, така че най-високата крайна концентрация от повторното изпитване да е най-ниската крайна концентрация от предходното изпитване.
|
|
Протокол от изпитването
|
34.
|
Протоколът от изпитването следва да включва следната информация:
|
Изпитвани химикали
Вещество с една съставка:
|
Химична идентификация, като наименование(я) по IUPAC или по CAS, CAS номер(а), SMILES или InChI код, структурна формула, и/или други идентификатори;
|
|
Външен вид, разтворимост във вода, молекулно тегло и допълнителни относими физични и химични свойства, според наличността;
|
|
Чистота, химическа идентичност на онечистванията, както е целесъобразно и практически осъществимо и т.н.;
|
|
Обработка преди изпитването, ако се прилага (напр. затопляне, смилане);
|
|
Разтворимост в X-VIVOTM 15. За химикали, които са неразтворими в X-VIVOTM 15., независимо дали след центрофугирането се наблюдава утаяване или флотация;
|
|
Изпитвана концентрация (концентрации);
|
|
Условия на съхранение и стабилност, доколкото са налични;
|
|
Обосновка за избора на разтворител/носител за всеки изпитван химикал, ако не е използван X-VIVOTM 15.
|
Вещество с повече съставки, UVCB и смес:
|
характеризиране, доколкото е възможно, чрез химическата идентичност (вж. по-горе), чистотата, количествения състав и относимите физични и химични свойства на съставките (вж. по-горе), доколкото са налични.
|
|
Външен вид, разтворимост във вода и допълнителни относими физични и химични свойства, според наличността;
|
|
Молекулно тегло или привидно молекулно тегло в случай на смеси/полимери с известен състав или друга информация, относима към провеждането на изследването;
|
|
Обработка преди изпитването, ако се прилага (напр. затопляне, смилане);
|
|
Разтворимост в X-VIVOTM 15. За химикали, които са неразтворими в X-VIVOTM 15., независимо дали след центрофугирането се наблюдава утаяване или флотация;
|
|
Изпитвана концентрация (концентрации);
|
|
Условия на съхранение и стабилност, доколкото са налични.
|
|
Обосновка за избора на разтворител/носител за всеки изпитван химикал, ако не е използван X-VIVOTM 15.
|
|
|
Контроли
Положителна контрола:
|
Химична идентификация, като наименование(я) по IUPAC или по CAS, CAS номер(а), SMILES или InChI код, структурна формула, и/или други идентификатори;
|
|
Външен вид, разтворимост във вода, молекулно тегло и допълнителни относими физични и химични свойства, според наличността, където е приложимо;
|
|
Чистота, химическа идентичност на онечистванията, както е целесъобразно и практически осъществимо и т.н.;
|
|
Обработка преди изпитването, ако се прилага (напр. затопляне, смилане);
|
|
Изпитвана концентрация (концентрации);
|
|
Условия на съхранение и стабилност, доколкото са налични;
|
|
Препратка към резултатите за положителните контроли за предходни периоди, доказващи подходящи критерии за приемливост, ако е приложимо.
|
Отрицателна контрола:
|
Химична идентификация, като наименование(я) по IUPAC или по CAS, CAS номер(а) и/или други идентификатори;
|
|
Чистота, химическа идентичност на онечистванията, както е целесъобразно и практически осъществимо и т.н.;
|
|
Външен вид, молекулно тегло и допълнителни относими физични и химични свойства, в случай че са използвани отрицателни контроли, различни от упоменатите в Насоките за изпитване и в зависимост от наличността;
|
|
Условия на съхранение и стабилност, доколкото са налични;
|
|
Обосновка за избора на разтворител за всеки изпитван химикал.
|
|
|
Условия на изпитване
|
Име и адрес на финансиращия, на изпитващата лаборатория и на ръководителя на изследването;
|
|
Описание на използваното изпитване;
|
|
Използвана клетъчна линия, условия за нейното съхранение и източник (например мястото, от което е взета);
|
|
Номер на партидата и произход на FBS, наименование на доставчика, номер на партидата на 96-ямковата плоскодънна черна плака и номер на партидата на реактива Tripluc;
|
|
Брой на пасажите и клетъчна плътност, използвани за изпитването;
|
|
Метод на преброяване на клетките, използван за посяване преди изпитването, и мерки, предприети за гарантиране на хомогенно разпределение на броя на клетките;
|
|
Използван луминометър (напр. модел), включително настройки, използван луциферазен субстрат, и доказване на съответни измервания на луминесценцията въз основа на контролното изпитване, описано в допълнение 3.2;
|
|
Процедурата, използвана за доказване на пригодността на лабораторията за извършване на изпитването (например чрез изпитване на вещества за изпитването за пригодност), или за доказване на възпроизводимостта на характеристиките на изпитването във времето.
|
|
|
Процедура за изпитване
|
Брой на извършените повторения и серии;
|
|
Концентрации на изпитвания химикал, процедура за прилагане и време на експозицията (ако са различни от препоръчаните)
|
|
Описание на използваните критерии за оценка и за решение;
|
|
Описание на използваните критерии за приемливост на изследването.
|
|
Описание на всякакви изменения на процедурата за изпитване.
|
|
|
Резултати
|
Измервания на IL8LA и GAPLA;
|
|
Изчисления за nIL8LA, Ind-IL8LA и Inh-GAPLA;
|
|
95 %-ият доверителен интервал на Ind-IL8LA;
|
|
Графика, показваща кривите доза-отклик за индукцията на луциферазна активност и за жизнеспособността;
|
|
Описание на всякакви други относими наблюдения, ако е приложимо.
|
|
|
Обсъждане на резултатите
|
Обсъждане на резултатите, получени с IL-8 Luc изследването;
|
|
Разглеждане на резултатите от изследването в контекста на IATA, ако е налична друга относима информация.
|
|
Заключение
|
ЛИТЕРАТУРА
|
(1)
|
Takahashi T, Kimura Y, Saito R, Nakajima Y, Ohmiya Y, Yamasaki K, and Aiba S. (2011). An in vitro test to screen skin sensitizers using a stable THP-1-derived IL-8 reporter cell line, THP-G8. Toxicol Sci 124:359-69.
|
|
(2)
|
OECD (2017). Validation report for the international validation study on the IL-8 Luc assay as a test evaluating the skin sensitizing potential of chemicals conducted by the IL-8 Luc Assay. Series on Testing and Assessment No 267, ENV/JM/MONO(2017)19. Organisation for Economic Cooperation and Development, Paris. На разположение на адрес: http://www.oecd.org/env/ehs/testing/series-testing-assessment-publications-number.htm.
|
|
(3)
|
OECD (2017). Report of the Peer Review Panel for the IL-8 Luciferase (IL-8 Luc) Assay for in vitro skin sensitisation. Series on Testing and Assessment No 258, ENV/JM/MONO(2017)20. Organisation for Economic Cooperation and Development, Paris. На разположение на адрес: http://www.oecd.org/env/ehs/testing/series-testing-assessment-publications-number.htm.
|
|
(4)
|
OECD (2016) Guidance Document On The Reporting Of Defined Approaches And Individual Information Sources To Be Used Within Integrated Approaches To Testing And Assessment (IATA) For Skin Sensitisation, Series on Testing & Assessment No 256, ENV/JM/MONO(2016)29. Organisation for Economic Cooperation and Development, Paris. На разположение на адрес: http://www.oecd.org/env/ehs/testing/series-testing-assessment-publications-number.htm.
|
|
(5)
|
van der Veen JW, Rorije E, Emter R, Natsch A, van Loveren H, and Ezendam J. (2014). Evaluating the performance of integrated approaches for hazard identification of skin sensitizing chemicals. Regul Toxicol Pharmacol 69:371-9.
|
|
(6)
|
Kimura Y, Fujimura C, Ito Y, Takahashi T, Nakajima Y, Ohmiya Y, and Aiba S. (2015). Optimization of the IL-8 Luc assay as an in vitro test for skin sensitization. Toxicol In Vitro 29:1816-30.
|
|
(7)
|
Urbisch D, Mehling A, Guth K, Ramirez T, Honarvar N, Kolle S, Landsiedel R, Jaworska J, Kern PS, Gerberick F, et al. (2015). Assessing skin sensitization hazard in mice and men using non-animal test methods. Regul Toxicol Pharmacol 71:337-51.
|
|
(8)
|
Ashikaga T, Sakaguchi H, Sono S, Kosaka N, Ishikawa M, Nukada Y, Miyazawa M, Ito Y, Nishiyama N, and Itagaki H. (2010). A comparative evaluation of in vitro skin sensitisation tests: the human cell-line activation test (h-CLAT) versus the local lymph node assay (LLNA). Alternatives to laboratory animals: ATLA 38:275-84.
|
|
(9)
|
Patlewicz G, Casati S, Basketter DA, Asturiol D, Roberts DW, Lepoittevin J-P, Worth A and Aschberger K (2016) Can currently available non-animal methods detect pre and pro haptens relevant for skin sensitisation? Regul Toxicol Pharmacol, 82:147-155.
|
|
(10)
|
Thorne N, Inglese J, and Auld DS. (2010). Illuminating insights into firefly luciferase and other bioluminescent reporters used in chemical biology. Chem Biol 17:646-57.
|
|
(11)
|
OECD (2016). Test No 455: Performance-Based Test Guideline for Stably Transfected Transactivation In Vitro Assays to Detect Estrogen Receptor Agonists and Antagonists, OECD Publishing, Paris. http://dx.doi.org/10.1787/9789264265295-en.
|
|
(12)
|
Viviani V, Uchida A, Suenaga N, Ryufuku M, and Ohmiya Y. (2001). Thr226 is a key residue for bioluminescence spectra determination in beetle luciferases. Biochem Biophys Res Commun 280:1286-91.
|
|
(13)
|
Viviani VR, Bechara EJ, and Ohmiya Y. (1999). Cloning, sequence analysis, and expression of active Phrixothrix railroad-worms luciferases: relationship between bioluminescence spectra and primary structures. Biochemistry 38:8271-9.
|
|
(14)
|
Nakajima Y, Kimura T, Sugata K, Enomoto T, Asakawa A, Kubota H, Ikeda M, and Ohmiya Y. (2005). Multicolor luciferase assay system: one-step monitoring of multiple gene expressions with a single substrate. Biotechniques 38:891-4.
|
|
(15)
|
OECD (2017). To be published - Performance Standards for the assessment of proposed similar or modified in vitro skin sensitisation IL-8 luc test methods. OECD Environment, Health and Safety Publications, Series on Testing and Assessment. OECD, Paris, France
|
|
(16)
|
OECD (2005). Guidance Document the Validation and International Acceptance of New or Updated Test Methods for Hazard Assessment. OECD Environment, Health and Safety publications, OECD Series on Testing and Assessment No 34. OECD, Paris, France.
|
|
(17)
|
OECD (2018). Draft Guidance document: Good In Vitro Method Practices (GIVIMP) for the Development and Implementation of In Vitro Methods for Regulatory Use in Human Safety Assessment. Organisation for Economic Cooperation and Development, Paris. На разположение на адрес: http://www.oecd.org/env/ehs/testing/OECD Final Draft GIVIMP.pdf.
|
|
(18)
|
JaCVAM (2016). IL-8 Luc assay protocol, Available at. http://www.jacvam.jp/en_effort/effort02.html.
|
|
(19)
|
Niwa K, Ichino Y, Kumata S, Nakajima Y, Hiraishi Y, Kato D, Viviani VR, and Ohmiya Y. (2010). Quantum yields and kinetics of the firefly bioluminescence reaction of beetle luciferases. Photochem Photobiol 86:1046-9.
|
|
(20)
|
OECD (2012). The Adverse Outcome Pathway for Skin Sensitisation Initiated by Covalent Binding to Proteins, Part 1: Scientific Evidence. OECD Environment, Health and Safety Publications, Series on Testing and Assessment No 168. OECD, Paris, France.
|
|
(21)
|
United Nations (2015). Globally Harmonized System of Classification and Labelling of Chemicals (GHS). Sixth revised edition. New York & Geneva: United Nations Publications. ISBN: 978-92-1-117006-1. На разположение на: http://www.unece.org/trans/danger/publi/ghs/ghs_rev05/05files_e.html.
|
Допълнение 3.1
ОПРЕДЕЛЕНИЯ
Точност
: степента на близост между резултатите от изпитването и приетите референтни стойности. Това е мярка за ефективността на изпитването и един от аспектите на неговата относимост. Терминът често се използва взаимозаменяемо с термина „съответствие“, за да се обозначи относителният дял на правилните резултати при дадено изпитване (16).
AOP (път, водещ до неблагоприятен ефект)
: последователност от събития от химичната структура на прицелен химикал или група от сходни химикали през молекулното иницииращо събитие до представляващ интерес in vivo резултат (20).
Химикал
: Вещество или смес.
CV05
: Клеъчна жизнеспособност 05, т.е. минимална концентрация, при която химикалите показват стойност на Inh-GAPLA по-малка от 0,05.
FInSLO-LA
: Съкращение, използвано в доклада за валидиране и в предишни публикации по отношение на IL-8 Luc изследването за позоваване на Ind-IL8LA. За определението вж. Ind-IL8LA.
GAPLA
: активност на Stable Luciferase Red (SLR) (λmax = 630 nm) , регулирана от GAPDH промотора и показваща клетъчната жизнеспособност и броя на жизнеспособните клетки.
Опасност
: Вътрешно присъщо свойство на даден агент или ситуация, притежаващо потенциал да предизвика неблагоприятни ефекти когато един организъм, система или (суб-)популация бъдат експонирани на този агент.
IATA (интегриран подход за изпитване и оценка)
: Структуриран подход, използван за идентифициране на опасност (потенциал), характеризиране на опасност (потенциал) и/или оценка на безопасността (потенциал и експозиция) на даден химикал или група от химикали, който стратегически интегрира и претегля всички данни, относими към предоставянето на информация за вземането на регулаторно решение по отношение на потенциалната опасност и/или риска, и/или необходимостта от допълнително целенасочено и следователно минимално изпитване.
II-SLR-LA
: Съкращение, използвано в доклада за валидиране и в предишни публикации по отношение на IL-8 Luc изследването за позоваване на Ind-GAPLA. За определението вж. Ind-GAPLA.
IL-8 (интерлевкин-8)
: Цитокин, получен от ендотелиални клетки, фибробласти, кератиноцити, макрофаги и моноцити, който причинява хемотаксис на неутрофили и Т-клетъчни лимфоцити.
IL8LA
: луциферазна активност на Stable Luciferase Orange (SLO) (λmax = 580 nm), която се регулира от IL-8 промотора.
Ind-IL8LA
: кратност на индукция на nIL8LA. Получава се чрез разделяне на nIL8LA на клетки THP-G8, третирани с химикали, на nIL8LA на нестимулирани THP-G8 клетки, и представлява индукцията на активността на IL-8 промотора от химикали.
Inh-GAPLA
: Инхибиране на GAPLA. Получава се чрез разделяне на GAPLA на клетки THP-G8, третирани с химикали, на GAPLA на нетретирани THP-G8 клетки, и представлява цитотоксичност на химикалите.
Минимален праг на индукция (MIT)
: най-ниската концентрация, при която даден химикал отговаря на положителните критерии
Смес
: смес или разтвор, съставени от две или повече вещества.
Вещество с една съставка
: Вещество, определено от неговия количествен състав, в който съдържанието на една основна съставка е най-малко 80 % (w/w).
Вещество с повече съставки
: Вещество, определено от неговия количествен състав, в който повече от една основна съставка се съдържа в концентрация ≥ 10 % (w/w) и < 80 % (w/w). Веществото с повече съставки е резултат от производствен процес. Разликата между смес и вещество с повече съставки е, че дадена смес е получена чрез смесване на две или повече вещества без химична реакция. Веществото с повече съставки е резултат от химична реакция.
nIL8LA
: луциферазната активност на SLO, отразяваща активността на IL-8 промотора (IL8LA), нормализирана с луциферазната активност на SLR луциферазата, отразяваща активността на GAPDH промотора (GAPLA). Представлява активността на IL-8 промотора след вземане предвид на клетъчната жизнеспособност или броя на клетките.
nSLO-LA
: Съкращение, използвано в доклада за валидиране и в предишни публикации по отношение на IL-8 Luc изследването за позоваване на nIL8LA. За определението вж. nIL8LA.
Положителна контрола
: Повторение, съдържащо всички компоненти на дадена изпитвана система и третирано с вещество, за което е известно, че предизвиква положителен отклик. За да гарантира, че може да се направи оценка на варирането на отклика в положителната контрола във времето, големината на положителния отклик не следва да е прекомерна.
Пре-хаптени
: химикали, които стават сенсибилизатори чрез абиотично преобразуване
Про-хаптени
: химикали, които изискват ензимно активиране за реализирането на потенциал за кожна сенсибилизация.
Относимост
: описание на взаимовръзката между изпитването и ефекта, представляващ интерес, и дали тя е значима и полезна за определена цел. Това е степента, в която изпитването правилно измерва или прогнозира биологичния ефект, представляващ интерес. Относимостта включва разглеждане на точността (съответствието) на дадено изпитване (16).
Надеждност
: мярка за степента, в която дадено изпитване може да се извърши възпроизводимо в една и съща лаборатория и в различни лаборатории по различно време, като при извършването се използва един и същи протокол. Оценката за нея се прави, като се изчислява вътрешнолабораторната и междулабораторната възпроизводимост и вътрешнолабораторната повторяемост (16).
Серия
: Серията представлява един или повече изпитвани химикали, изпитвани едновременно с контрола на разтворител/носител и с положителна контрола.
Чувствителност
: Относителният дял от всички положителни/активни химикали, които са класифицирани правилно чрез изпитването. Това е мярка за точността на изпитването, което предоставя категорийни резултати, и е важен фактор при оценката на относимостта на дадено изпитване (16).
SLO-LA
: Съкращение, използвано в доклада за валидиране и в предишни публикации по отношение на IL-8 Luc изследването за позоваване на IL8LA. За определението вж. IL8LA.
SLR-LA
: Съкращение, използвано в доклада за валидиране и в предишни публикации по отношение на IL-8 Luc изследването за позоваване на GAPLA. За определението вж. GAPLA.
Контрола на разтворител/носител
: Нетретирана проба, съдържаща всички компоненти на изпитваната система, с изключение на изпитвания химикал, но включваща разтворителя/носителя, който се използва. Използва се за определяне на базовия отклик за пробите, третирани с изпитвания химикал, разтворен или стабилно диспергиран в същия разтворител/носител. Когато се изпитва с паралелна контрола на среда, тази проба показва също дали разтворителят/носителят взаимодейства с изпитваната система.
Специфичност
: Относителният дял от всички отрицателни/неактивни химикали, които са класифицирани правилно чрез изпитването. Това е мярка за точността на изпитването, което предоставя категорийни резултати и е важен фактор при оценката на относимостта на дадено изпитване (16).
Вещество
: химичен елемент и неговите съединения в естествено състояние или получени чрез всеки производствен процес, включително всяка добавка, необходима за запазване на стабилността на продукта и всеки примес, извлечен от използвания процес, с изключение на всеки разтворител, който може да бъде отделен, без да се засяга стабилността на веществото или да се променя неговият състав.
Повърхностноактивно вещество
: също така наречено повърхностноактивен агент, представлява вещество, като например детергент, което е в състояние да намали повърхностното напрежение на дадена течност и по този начин да позволи образуването на пяна в течността или проникването ѝ в твърди вещества; Известно е също като мокрител. (TG437)
Изпитван химикал
: Всяко вещество или смес, изпитвано(а) с използване на настоящия метод.
THP-G8
: IL-8 репортерна клетъчна линия, използвана в IL-8 Luc изследване. Макрофагоподобната човешка клетъчна линия THP-1 е трансфектирана с гени на SLO и SLR луциферази, съответно под контрола на IL-8 и GAPDH промотори.
Глобална хармонизирана система на Организацията на обединените нации за класифициране и етикетиране на химикали (GHS на ООН)
: система, предлагаща класифициране на химикали (вещества и смеси) според стандартизирани видове и степени на физическа, здравна и екологична опасност и разглеждаща съответни съобщителни елементи, като например пиктограми, сигнални думи, предупреждения за опасност, препоръки за безопасност и информационни листове за безопасност, така че те да съобщават информация за тяхното неблагоприятно въздействие с оглед защита на хората (включително работодатели, работещи, служители в транспорта, потребители и аварийни служители) и на околната среда (21).
UVCB
: вещества с неизвестен или променлив състав, сложни продукти от реакции или биологични материали.
Валиден метод за изпитване
: Изпитване, за което се смята, че притежава достатъчна относимост и надеждност по отношение на конкретна цел и което се основава на научно обосновани принципи. Дадено изпитване никога не е валидно в абсолютен смисъл, а само по отношение на определена цел.
Допълнение 3.2
ПРИНЦИП НА ИЗМЕРВАНЕ НА ЛУЦИФЕРАЗНАТА АКТИВНОСТ И ОПРЕДЕЛЯНЕ НА КОЕФИЦИЕНТИТЕ НА ПРОПУСКАНЕ НА ОПТИЧНИТЕ ФИЛТРИ ЗА SLO И SLR
Мултирепортерната система за изследване Tripluc- може да се използва с луминометър от тип с микроплаки с многоцветна система за откриване, която може да бъде оборудвана с оптичен филтър (напр. Phelios AB-2350 (ATTO), ARVO (PerkinElmer), Tristar LB941 (Berthold)). Оптичният филтър, използван при измерването, е 600—620 nm дълговълнов или късовълнов филтър, или 600—700 nm лентово-пропускащ филтър.
Измерване на двуцветни луциферази с оптичен филтър.
Това е пример с използване на Phelios AB-2350 (ATTO). Този луминометър е оборудван с 600 nm дълговълнов филтър (R60 HOYA Co., 600 nm LP, Филтър 1) за разделяне на SLO (λmax = 580 nm) и SLR (λmax = 630 nm) луминесценцията.
За определяне коефициентите на пропускане на 600 nm LP, първо, с използването на пречистени ензими, SLO и SLR луциферази, се измерва i) интензитетът на биолуминесценцията на SLO и SLR без филтър (F0), ii) интензитетът на биолуминесценцията на SLO и SLR, която преминава през 600 nm LP (Филтър 1), и iii) изчисляват се коефициентите на пропускане на SNA и SLR на 600 nm LP за SLO и SLR, изброени по-долу.
|
Transmission coefficients
|
Abbreviation
|
Definition
|
|
SLO
|
Filter 1 Transmission coefficients
|
=κОR60
|
The filter’s transmission coefficient for the SLO
|
|
SLR
|
Filter 1 Transmission coefficients
|
κRR60
|
The filter’s transmission coefficient for the SLR
|
Когато интензитетът на SLO и SLR в изпитваната проба са определени съответно като O и R, i) светлинният интензитет без филтър (цялата оптична) F0 и ii) интензитетът на светлината, която се пропуска през 600 nm LP (Филтър 1) F1, са описани по-долу.
F0 = O+R
F1 = κОR60 x O + κRR60 x R
Тези формули могат да бъдат перифразирани по следния начин:

След това, като се използват изчислените коефициенти на пропускане (κОR60 и κRR60) и измерените F0 и F1, може да се изчисли O- и R-стойността, както следва:
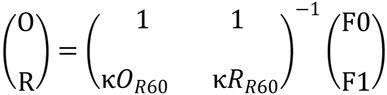
Материали и методи за определяне на коефициента на пропускане
|
(1)
|
Реактиви
Отделни пречистени луциферазни ензими:
|
Лиофилизиран пречистен SLO ензим
|
|
Лиофилизиран пречистен SLR ензим
|
|
(които за работата по валидирането са получени от GPC Lab. Co. Ltd., Tottori, Япония, с клетъчна линия THP-G8)
|
Реактив за изследването:
|
Реактив Tripluc® за изследване с луцифераза (например от TOYOBO Cat#MRA-301)
|
Среда за изследване с луцифераза (30 ml, съхранявана при 2—8 °C)
|
|
Реактив
|
Конц.
|
Крайна конц. в средата
|
Изисквано количество
|
|
RPMI-1640
|
—
|
—
|
27 ml
|
|
FBS
|
—
|
10 %
|
3 ml
|
|
(2)
|
Приготвяне на ензимен разтвор
В епруветка се разтваря лиофилизиран пречистен луциферазен ензим чрез добавяне на 200 μl 10 ~ 100 mM Tris/HCl или Hepes/HCl (pH 7,5 ~ 8,0), с добавка на 10 % (w/v) глицерол, ензимният разтвор се разделя на 10 μl-ови аликвотни части в 1,5 ml епруветки за еднократна употреба и те се съхраняват във фризер при -80 °C. Замразеният ензимен разтвор може да се използва за период до 6 месеца. Когато се използва, се добавя 1 ml от средата за изпитване с луцифераза (RPMI-1640 с 10 % FBS) към всяка епруветка, съдържаща ензимните разтвори (разреден ензимен разтвор), и се държат върху лед, за да се предотврати дезактивирането.
|
|
(3)
|
Измерване на биолуминесценцията
Реактивът Tripluc® за изследване с луцифазаза (Tripluc) се размразява и се държи при стайна температура или във водна баня, или при температурата на околния въздух. Луминометърът се включва 30 минути преди започване на измерването, за да се даде възможност за стабилизиране на фотоумножителя. 100 μl от разредения ензимен разтвор се прехвърлят в черна 96-ямкова плоскодънна плака (референтната SLO проба в # B1, # B2, # B3, референтната SLR проба в # D1, # D2, # D3). След това към всяка ямка на плаката, съдържаща разредения ензимен разтвор, с помощта на автоматична пипета се прехвърлят по100 μl предварително затоплен Tripluc. Плаката се разклаща в продължение на 10 минути при стайна температура (около 25 °C) с помощта на клатачна машина за плаки. Ако се появят мехурчета от разтворите в плаките, тези мехурчета се отстраняват. Плаката се поставя в луминометъра за измерване на луциферазна активност. Биолуминесценцията се измерва в продължение на 3 секунди, всеки път при отсъствие (F0) и наличие (F) на оптичния филтър.
Коефициентът на пропускане на оптичния филтър е изчислен, както следва:
Коефициент на пропускане (SLO (κОR60))= (#B1 от F1+ #B2 от F1+ #B3 от F1) / (#B1 от F0+ #B2 от F0+ #B3 от F0)
Коефициент на пропускане (SLR (κRR60))= (#D1 от F1+ #D2 от F1+ #D3 от F1) / (#D1 от F0+ #D2 от F0+ #D3 от F0)
Изчислените коефициенти на пропускане се използват за всички измервания, извършени с помощта на същия луминометър.
|
Контрол на качеството на оборудването
Следва да се използват процедурите, описани в протокола IL-8 Luc (18).
Допълнение 3.3
ВЕЩЕСТВА ЗА ИЗПИТВАНЕ ЗА ПРИГОДНОСТ
Преди рутинното използване на изпитването, описано в настоящото допълнение към метод за изпитване Б.71, лабораториите следва да докажат техническата си пригодност, като получат очакваната прогноза по IL-8 Luc изследването за 9-те вещества, препоръчани в Таблица 1, и чрез получаване на стойности, които попадат в съответния референтен обхват за минимум 8 от 9-те вещества за изпитване за пригодност (подбрани като представителни за размаха от отклици за опасностите от кожна сенсибилизация). Други критерии за подбор бяха веществата да са налични в търговската мрежа и да са налични висококачествени in vivo референтни данни, както и висококачествени in vitro данни, получени по IL-8 Luc изследването. Също така са налични публикувани референтни данни за IL-8 Luc изследването (6) (1).
Таблица 1
Препоръчвани вещества за доказване на техническа пригодност по отношение на IL-8 Luc изследването
|
Вещества за изпитване за пригодност
|
CAS №
|
Състояние
|
Разтворимост в X-VIVO15 при 20 mg/ml
|
In vivo прогноза (109)
|
IL-8 Luc прогноза (110)
|
Референен размах (μg/ml) (111)
|
|
|
CV05
(112)
|
IL-8 Luc MIT (113)
|
|
2,4-Динитрохлоробензен
|
97-00-7
|
Твърд
|
Неразтворим
|
Сенсибилизатор (изключително силен)
|
Положително
|
2,3-3,9
|
0,5-2,3
|
|
Формалдехид
|
50-00-0
|
Течност
|
Разтворим
|
Сенсибилизатор (силен)
|
Положително
|
9-30
|
4-9
|
|
2-Меркаптобензотиазол
|
149-30-4
|
Твърд
|
Неразтворим
|
Сенсибилизатор (умерен)
|
Положително
|
250-290
|
60-250
|
|
Етилендиамин
|
107-15-3
|
Течност
|
Разтворим
|
Сенсибилизатор (умерен)
|
Положително
|
500-700
|
0,1-0,4
|
|
Диметакрилат на етиленгликола
|
97-90-5
|
Течност
|
Неразтворим
|
Сенсибилизатор (слаб)
|
Положително
|
>2 000
|
0,04-0,1
|
|
4-Алиланизол (Естрагол)
|
140-67-0
|
Течност
|
Неразтворим
|
Сенсибилизатор (слаб)
|
Положително
|
>2 000
|
0,01-0,07
|
|
Стрептомицинсулфат
|
3810-74-0
|
Твърд
|
Разтворим
|
Непредизвикващо сенсибилизация вещество
|
Отрицателно
|
>2 000
|
>2 000
|
|
Глицерол
|
56-81-5
|
Течност
|
Разтворим
|
Непредизвикващо сенсибилизация вещество
|
Отрицателно
|
>2 000
|
>2 000
|
|
Изопропанол
|
67-63-0
|
Течност
|
Разтворим
|
Непредизвикващо сенсибилизация вещество
|
Отрицателно
|
>2 000
|
>2 000
|
|
Съкращения: CAS № = регистрационен номер по Службата за химични индекси.
|
Допълнение 3.4
ИНДЕКСИ И КРИТЕРИИ ЗА ОЦЕНКА
nIL8LA (nSLO-LA)
j-тото повторение (j = 1-4) на i-тата концентрация (i = 0-11) се измерва съответно за IL8LA (SLO-LA) и GAPLA (SLR-LA). Нормализираната IL8LA, посочена като nIL8LA (nSLO-LA), и се определя като:
nIL8LAij = IL8LAij/GAPLAij
Това е базовата единица на измерване в това изследване.
Ind-IL8LA (FInSLO-LA)
Кратното увеличение на усреднената nIL8LA (nSLO-LA) за повторението при i-тата концентрация, сравнена с него при концентрация 0, Ind-IL8LA, е първичната мярка на това изследване. Това съотношение се изчислява по следната формула:

Водещата лаборатория предложи стойност от 1,4 да съответства на положителен резултат за изпитвания химикал. Тази стойност се основава на проучването на данните на водещата лаборатория за предходни периоди. След това екипът за управление на данните използва тази стойност през всички фази на проучването за валидиране. Първичният резултат, Ind-IL8LA, е отношението на 2 средноаритметични стойности, както е показано в уравнението.
95 % доверителен интервал (95 % ДИ)
95 %-ят доверителен интервал (95 % ДИ) въз основа на съотношението може да се оцени, така че да се покаже точността на тази първична мярка за резултати. Долната граница на 95 % ДИ ≥ 1 показва, че nIL8LA при i-та концентрация е значимо по-голяма от тази при контролата на разтворител. Съществуват няколко начина за построяване на 95 % ДИ. В това проучване използвахме метода, познат като теорема на Fieller. Тази теорема за доверителен интервал от 95 % се получава по следната формула:

където:

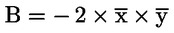



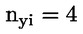


е 97,5 перцентил на централното t-разпределение с ν на степента на свобода, където

Inh-GAPLA (II-SLR-LA)
Inh-GAPLA е съотношение на усреднената GAPLA (SLR-LA) за повторението на i-тата концентрация в сравнение с това при контролата на разтворител, и се изписва с

Тъй като GAPLA е знаменателят на nIL8LA, една изключително малка стойност причинява голямо вариране в nIL8LA. Поради това стойностите на Ind-IL8LA с изключително малка стойност на Inh-GAPLA (под 0,05) могат да се считат за недобра прецизност.
Допълнение 3.5
СХЕМАТА НА МЕТОДИТЕ ЗА РАЗТВАРЯНЕ НА ХИМИКАЛИ ЗА IL-8 LUC ИЗСЛЕДВАНЕТО.
|
а)
|
За химикали, разтворени в X-VIVOTM 15 при 20 mg/ml

|
|
б)
|
За химикали, неразтворими в X-VIVOTM 15 при 20 mg/ml

|
Допълнение 3.6
СХЕМАТА НА МЕТОДА ЗА РАЗТВАРЯНЕ НА 4-NBB ЗА ПОЛОЖИТЕЛНАТА КОНТРОЛА ПРИ IL-8 LUC ИЗСЛЕДВАНЕТО.
“ |