ПРИЛОЖЕНИЕ
Приложенията към Регламент (ЕИО) № 2568/91 се изменят, както следва:
|
1. |
Резюмето се изменя както следва:
|
|
2. |
Допълнение I се заменя със следния текст: „ПРИЛОЖЕНИЕ I ХАРАКТЕРИСТИКИ НA ВИДОВЕТE МАСЛИНОВО МАСЛО Забележки:
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
3. |
Допълнение 1 се изменя, както следва:
|
|
4. |
Заглавието на приложение II се заменя със следното: |
|
5. |
Приложение IV се заменя със следния текст: „ПРИЛОЖЕНИЕ IV ОПРЕДЕЛЯНЕ НА СЪДЪРЖАНИЕТО НА ПАРАФИНИ ЧРЕЗ КАПИЛЯРНА ГАЗОВА ХРОМАТОГРАФИЯ 1. ПРЕДМЕТ Този метод описва способ за определяне на съдържанието на парафини в маслиново масло. Парафините се анализират съобразно броя въглеродни атоми. Методът може да се използва по-специално за диференциране на маслиновото масло, получено чрез пресоване, от това, което е получено чрез екстракция (маслиново масло от остатъчен материал). 2. ПРИНЦИП Мастното вещество, към което е добавен подходящ вътрешен стандарт, се фракционира чрез хроматография върху колонка от хидратиран силикагел; снема се фракцията, която първа се елуира в опитните условия (при полярност, по-ниска от тази на триглицеридите), след което се анализира директно чрез капилярна газова хроматография. 3. ОБОРУДВАНЕ 3.1. Ерленмайерова колба от 25 ml. 3.2. Стъклена колонка за газова хроматография с вътрешен диаметър 15 mm, височина 30 до 40 cm, оборудвана с кранче. 3.3. Апарат за газова хроматография, приспособен за функциониране с капилярна колонка, оборудван със система за директно въвеждане в колонката, състояща се от: 3.3.1. Термостатна камера за колонките, оборудвана с температурен програматор. 3.3.2. Инжектор за студено пускане — за директно вкарване в колонката. 3.3.3. Пламъчно-йонизационен детектор и усилвателен преобразувател. 3.3.4. Записващ интегратор, пригоден за работа с усилвателен преобразувател (3.3.3), с време на отговор под 1 секунда и променлива скорост на подаване на хартията. (Възможно е да се използват и информационни системи, които позволяват компютризирано събиране на данните от газовата хроматография.) 3.3.5. Капилярна колонка от стъкло или кварц, с дължина от 8 до 12 m, вътрешен диаметър от 0,25 до 0,32 mm, покрита отвътре със стационарна фаза, с еднородна дебелина между 0,10 и 0,30 μm. (Стационарни фази от тип SE-52 или SE-54, годни за употреба, се намират в търговската мрежа.) 3.4. Микроспринцовка с вместимост 10 μl, за директно подаване в колонката, снабдена със закалена игла. 3.5. Електровибратор. 3.6. Ротационен изпарител. 3.7. Муфелна пещ. 3.8. Аналитични везни, гарантиращи точност на измерване до ± 0,1 mg. 3.9. Обикновена лабораторна стъклария. 4. РЕАКТИВИ 4.1. Силикагел с гранулометричен състав между 60 и 200 μm. Силикагелът трябва да престои в пещ при 500 °C в продължение на минимум 4 часа. След охлаждане добавете към него вода, равна на 2 % от снетото количество силикагел. Разбъркайте добре, за да хомогенизирате сместа. Оставете я на тъмно в продължение на минимум 12 часа преди употреба. 4.2. n-хексан за хроматография. 4.3. Етилов етер за хроматография. 4.4. n-хептан за хроматография. 4.5. Стандартен разтвор на лауринов арахидат — 0,1 % (m/v) в хексан (вътрешен стандарт). (Възможно е да се използва и цетилов палмитат или миристилов стеарат.) 4.5.1. Судан 1 (1-фенил-азо-2-нафтол). 4.6. Газ носител: чист водород или хелий, за газова хроматография. 4.7. Спомагателни газове:
5. РАБОТЕН РЕЖИМ 5.1. Подготовка на хроматографската колонка Потопете 15 g силикагел (4.1) в n-хексан (4.2) и го поставете в колонката (3.2). Компенсирайте бързата седиментация с помощта на електрическа бъркалка (3.5), за да направите хроматографския пласт по-хомогенен. Перколирайте 30 ml n-хексан, за да отстраните евентуалните примеси. Претеглете с помощта на везните (3.8) точно 500 mg от пробата в ерленмайеровата колба с вместимост 25 ml (3.1), добавете нужното количество вътрешен стандарт (4.5), в зависимост от предполагаемото съдържание на парафини. Например, когато се касае за маслиново масло, добавете 0,1 mg лауринов арахидат, а когато става въпрос за маслиново масло от остатъчен материал — 0,25 до 0,5 mg. Прехвърлете така приготвената проба в хроматографската колонка с помощта на две дози n-хексан, всяка по 2 ml (4.2). Оставете разтворителя да изтече, така че да достигне на 1 mm от горното ниво на абсорбента, след което перколирайте допълнително 70 ml n-хексан, за да отстраните естествено наличните n-алкани. После започнете хроматографското елуиране, събирайки 180 ml от сместа n-хексан/етилов етер, в съотношение 99:1, като поддържате дебит от около 15 капки на 10 секунди. Елуирането на пробата трябва да се извърши при стайна температура 22 ± 4 °C. Бележки:
Изсушете така получената фракция в ротационен изпарител (3.6) до практически пълното отстраняване на разтворителя. Последните 2 ml от разтворителя се отстраняват с помощта на слаб азотен поток; след това добавете 2-4 ml n-хептан. 5.2. Анализ чрез газова хроматография 5.2.1. Предварителни операции Поставете колонката в газовия хроматограф (3.3), свързвайки входния извод към системата on-column, а изходния — към детектора. Извършете основните проверки на апаратурата за газова хроматография (здравина на газовите контури, ефективност на детектора и на записващата система и т. н.). Ако колонката се използва за първи път, се препоръчва да пристъпите към кондиционирането ѝ. Прокарайте през колонката малко газ, след което включете апаратурата за газова хроматография. Нагрейте постепенно до достигане, след около 4 часа, на температура 350 °C. Поддържайте тази температура в продължение на минимум 2 часа, след което пристъпете към настройката на апаратурата към условията на работа (настройка на газовия поток, запалване на пламъка, включване към електронното записващо устройство (3.3.4), настройка на температурата на камерата за колонката, на детектора и т. н.) и настройте сигнала на чувствителност поне 2 пъти по-висока от предвидената за провеждане на анализа. Ходът на основната линия трябва да бъде линеен, без пикове от каквото и да е естество, и не трябва да показва отклонения. Отрицателното праволинейно отклонение е показател за неизправни връзки на колонката; положителното отклонение е показател за недостатъчно кондициониране на колонката. 5.2.2. Избор на работните условия Общо взето, работните условия, които следва да се спазват, са следните:
Тези условия могат да варират в зависимост от характеристиките на колонката и на газовия хроматограф по такъв начин, че да се постигне анализиране на всички парафини, задоволителна разделителна способност на пиковете (виж фигурата); времето на задържане на вътрешния стандарт C32 следва да бъде 18 ± 3 минути. Най-изразеният пик на парафините следва да се намира на минимум 60 % височина от основата на скалата. Параметрите на интегриране на пиковете следва да се определят по начин, който позволява правилно изчисление на лицата на пиковете, взети под внимание. Бележка. Като се има предвид високата крайна температура, се допуска положително отклонение, което не бива да надвишава 10 % височина от основата на скалата. 5.3. Провеждане на анализа С помощта на микроспринцовка с вместимост 10 μl вземете 1 μl от разтвора; издърпайте буталото на спринцовката, така че иглата да остане празна. Вкарайте иглата в инжекционния прибор и след 1—2 секунди бързо инжектирайте разтвора; след около 5 секунди бавно извадете иглата. Проведете запис на анализа до пълното елуиране на парафините. Основната линия следва винаги да отговаря на необходимите условия. 5.4. Идентифициране на пиковете Идентифицирането на различните пикове следва да се извършва въз основа на времената на задържане и в сравнение със смеси от парафини с познати времена на задържане, анализирани при същите условия. Фигурата представя хроматограма на парафините в необработено маслиново масло. 5.5. Количествено изчисление С помощта на интегратора пристъпете към изчислението на лицата на пиковете на вътрешния стандарт и на алифатните естери от C40 до C46. Изчислете съдържанието на парафин във всеки един от естерите, в mg/kg мастно вещество, по формулата:
където:
6. ИЗРАЗЯВАНЕ НА РЕЗУЛТАТИТЕ Отбележете сумата на съдържанията на отделните парафини от C40 до C46, в mg/kg мастно вещество (ppm). Забележка. Компонентите, които трябва да се определят, се отнасят до пиковете с четно въглеродно число между естерите C40 и C46, съгласно примера на хроматограмата на парафините в маслиновото масло, посочена на фигурата по-долу. Ако естерът C46 се появи двойно, се препоръчва за идентифицирането му да се анализира фракцията на парафините в маслиново масло от остатъчен материал, където пикът C46 се идентифицира лесно, тъй като е най-изразеният. Резултатите се дават с точност до една десета. Фигура Хроматограма на парафините в маслиново масло (9)
Допълнение Определяне на линейната скорост на газа Инжектирайте от 1 до 3 μl метан (или пропан) в газовия хроматограф след настройката му към нормални условия на работа. Засечете с хронометър времето, необходимо на газа да премине през колонката, от момента на инжектирането му до момента, в който пикът се очертае (tM). Линейната скорост, в cm/s, се изчислява по формулата L/tM, където L е дължината на колонката в сантиметри, а tM е засеченото време в секунди. |


|
6. |
Приложение VII се заменя със следния текст: „ПРИЛОЖЕНИЕ VII ОПРЕДЕЛЯНЕ НА ПРОЦЕНТНОТО СЪДЪРЖАНИЕ НА 2-ГЛИЦЕРИЛ МОНОПАЛМИТАТ 1. ПРЕДМЕТ И СФЕРА НА ПРИЛОЖЕНИЕ Този метод описва аналитичната процедура за определяне на процентното съдържание на палмитинова киселина в позиция 2 на триглицеридите посредством анализа на 2-глицерил монопалмитат. Методът е приложим за течни растителни масла при стайна температура (20 °C). 2. ПРИНЦИП След подготовката ѝ маслената проба се подлага на въздействието на панкреатична липаза: частична хидролиза, специфична за позиции 1 и 3 на молекулата на триглицерида, води до появата на моноглицериди в позиция 2. Процентното съдържание на 2-глицерил монопалмитат в моноглицеридната фракция се определя, след силилация, посредством капилярна газова хроматография. 3. АПАРАТУРА И ЛАБОРАТОРНО ОБОРУДВАНЕ 3.1. Ерленмайерова колба от 25 ml. 3.2. Бехерови чаши от 100, 250 и 300 ml. 3.3. Стъклена колонка за хроматография, с вътрешен диаметър 21—23 mm, дължина 400 mm, оборудвана с диск с поресто дъно и кранче. 3.4. Градуирани епруветки от 10, 50, 100 и 200 ml. 3.5. Колби с вместимост 100 и 250 ml. 3.6. Ротационен изпарител. 3.7. Центрофужни епруветки с конично дъно, с вместимост 10 ml, с шлифована запушалка. 3.8. Центрофуга за епруветки от 10 и 100 ml. 3.9. Термостат, който позволява поддържане на постоянна температура от 40 °C ± 0,5 °C. 3.10. Калибровани пипети от 1 и 2 ml. 3.11. Хиподермична спринцовка от 1 ml. 3.12. Микроспринцовка от 100 μl. 3.13. Фуния от 1 000 ml. 3.14. Газов хроматограф за капилярни колонки, оборудван със система за студено инжектиране „on column“, за директно вкарване на пробата в колонката и с пещ, която е в състояние да поддържа желаната температура с точност до 1 °C. 3.15. Инжектор за студено пускане „on column“ за директно вкарване на пробата в колонката. 3.16. Пламъчно-йонизационен детектор и електрометър. 3.17. Записващ интегратор, адаптиран към електрометъра, с време на отговор под 1 секунда и променлива скорост на подаване на хартията. 3.18. Капилярна колонка от стъкло или кварц, с дължина 8—12 метра, с вътрешен диаметър от 0,25 до 0,32 mm, с покритие от метилполисилоксан или фенил метилполисилоксан 5 %, с дебелина 0,10—0,30 μm, която може да се използва при 370 °C. 3.19. Микроспринцовка с вместимост 10 μl, снабдена със закалена игла, с минимална дължина 7,5 cm, за директно инжектиране върху колонката. 4. РЕАКТИВИ 4.1. Силикагел с гранулометричен състав между 0,063 и 0,200 mm (70/280 mesh), приготвен по следния начин: поставете силикагела в порцеланова капсула, изсушете в пещ при 160 °C в продължение на 4 часа, след което оставете да се охлади на стайна температура в ексикатор. Добавете обем вода, равен на 5 % от теглото на силикагела, като процедирате по следния начин: в ерленмайерова колба от 500 ml претеглете 152 g силикагел и добавете 8 g дестилирана вода, запушете с тапа и разклатете внимателно до равномерно разпределяне на водата. Оставете да престои минимум 12 часа преди употреба. 4.2. n-хексан (за хроматография). 4.3. Изопропанол. 4.4. Воден разтвор на изопропанол, в обемно съотношение 1/1. 4.5. Панкреатична липаза. Използваната липаза трябва да има активност между 2,0 и 10 липазни единици/mg. (В търговската мрежа се предлагат панкреатични липази с активност между 2 и 10 единици на милиграм ензим.) 4.6. Буферен разтвор: 1 M воден разтвор на трисхидрокси-метиламинометан, доведен до pH 8 (проверява се с потенциометър), чрез добавяне на концентрирана солна киселина (1/1 v/v). 4.7. Натриев холат с ензимно качество — воден разтвор 0,1 % (разтворът трябва да се използва до 15 дни след приготвянето му). 4.8. Калциев хлорид, воден разтвор 22 %. 4.9. Диетилов етер за хроматография. 4.10. Проявяващ разтворител: смес n-хексан/диетилов етер (87/13) (v/v). 4.11. Натриев хидроокис, разтвор 12 тегловни процента. 4.12. Разтвор в етилов алкохол на 1 % фенолфталеин. 4.13. Газ носител: водород или хелий, за газова хроматография. 4.14. Спомагателни газове: водород, минимум 99 %, свободен от влага и органични примеси, и въздух, за газова хроматография, със същата чистота. 4.15. Реактив за силанизация: смес пиридин(хексаметилдисилазан) триметилхлоросилан 9/3/1 (v/v/v). (В търговската мрежа се предлагат готови за употреба разтвори. Могат да се използват други реактиви за силилация като бис-триметилсилил трифлуорацетамид + 1 % триметилхлоросилан, разреден със същия обем безводен пиридин.) 4.16. Сравнителни проби: чисти моноглицериди или смеси от моноглицериди с известен състав на процентното съдържание, подобен на този на пробата. 5. ПРОЦЕДУРА 5.1. Подготовка на пробата 5.1.1. Маслото със свободна киселинност под 3 % не се нуждае от неутрализация преди хроматографията върху колонката със силикагел. Маслото със свободна киселинност над 3 % трябва да се подложи на неутрализация, както е описано в точка 5.1.1.1. 5.1.1.1. Във фунията с вместимост 1 000 ml (3.13) налейте 50 g масло и 200 ml n-хексан. Добавете 100 ml изопропанол и такова количество от разтвора на натриевия хидроокис 12 % (4.11), което съответства на свободната киселинност на маслото плюс добавка от 5 %. Разклатете енергично в продължение на една минута. Добавете 100 ml дестилирана вода, разбъркайте отново и оставете в покой. След декантирането отстранете долния слой от сапуни. Отстранете евентуалните междинни слоеве (клей и неразтворими вещества). Промийте хексановия разтвор от неутрализираната киселина с последователни дози от по 50—60 ml от разтвора изопропанол/вода 1/1 (v/v) (4.4) до изчезване на розовото оцветяване на фенолфталеина. Отстранете по-голямата част от хексана чрез дестилация под вакуум (използвайте например ротационен изпарител) и прехвърлете маслото в колба от 100 ml (3.5). Изсушете маслото под вакуум до пълното отстраняване на разтворителя. В края на тази операция киселинността на маслото трябва да бъде под 0,5 %. 5.1.2. Поставете 1,0 g масло, приготвено по горепосочения начин, в ерленмайерова колба от 25 ml (3.1) и разтворете в 10 ml проявяваща смес (4.10). Оставете разтвора да престои в продължение на минимум 15 минути преди хроматографията върху колонка със силикагел. Ако разтворът е мътен, го центрофугирайте, за да гарантирате оптимални условия за хроматография. (Могат да се използват готови за употреба патрони силикагел SPE от 500 mg.) 5.1.3. Подготовка на хроматографската колонка Сипете в колонката (3.3) около 30 ml от проявяващия разтвор (4.10), поставете парче памук в долната част на колонката с помощта на стъклена пръчица; натиснете, за да отстраните въздуха. В бехерова чаша пригответе суспензия от 25 g силикагел (4.1) в около 80 ml проявяващ разтвор и я сипете в колонката с помощта на фуния. Проверете дали цялото количество силикагел е постъпило в колонката; промийте с проявяващия разтвор (4.10), отворете кранчето и оставете нивото на течността да стигне до около 2 mm над горното ниво на силикагела. 5.1.4. Хроматография върху колонка В ерленмайерова колба от 25 ml (3.1) претеглете точно 1,0 g от пробата, приготвена по описания в точка 5.1 начин. Разтворете пробата в 10 ml проявяващ разтвор (4.10). Сипете разтвора, приготвен по указанията в точка 5.1.3, в хроматографската колонка. Избягвайте движението на повърхността на колонката. Отворете кранчето и оставете разтвора на пробата да изтече, докато достигне нивото на силикагела. Проявете със 150 ml проявяващ разтвор. Регулирайте дебита на 2 ml/min (така че 150 ml да изтекат в колонката за около 60—70 минути). Съберете елуата в предварително претеглена колба с вместимост 250 ml. Изпарете разтворителя под вакуум и отстранете последните следи от него под азотен поток. Претеглете колбата и изчислете събрания екстракт. (В случай че използвате готови за употреба SPE патрони от силикагел, процедирайте по следния начин: поставете 1 ml разтвор (5.1.2) в предварително приготвените с 3 ml n-хексан патрони.) След перколация на разтвора проявете с 4 ml n-хексан/диетилов етер 9/1 (v/v). Съберете елуата в епруветка от 10 ml и го подложете на изпарение под азотен поток до изсъхване. Подложете сухия остатък на въздействие на панкреатична липаза (5.2). От основно значение е да проверите съдържанието на мастни киселини преди и след преминаването върху SPE патрона). 5.2. Хидролиза с панкреатична липаза 5.2.1. В центрофужна епруветка претеглете 0,1 g масло, приготвено съгласно точка 5.1. Добавете 2 ml буферен разтвор (4.6), 0,5 ml разтвор на натриев холат (4.7) и 0,2 ml разтвор на калциев хлорид, като разклащате добре след всяко добавяне. Затворете епруветката с шлифованата запушалка и я поставете в термостата при 40 ± 0,5 °C. 5.2.2. Добавете 20 mg липаза, разклатете старателно (като внимавате да не намокрите запушалката) и поставете епруветката в термостата за точно 2 минути, след което я извадете, разклатете енергично в продължение точно на 1 минута и оставете да се охлади. 5.2.3. Добавете 1 ml диетилов етер, запушете с тапата и разклатете енергично, след което центрофугирайте и прехвърлете етеровия разтвор в чиста и суха епруветка с помощта на микроспринцовка. 5.3. Подготовка на силанизираните деривати и на газовата хроматография 5.3.1. С помощта на микроспринцовка поставете 100 μl разтвор (5.2.3) в епруветка с конично дъно от 10 ml. 5.3.2. Отстранете разтворителя под слаб азотен поток, добавете 200 μl реактив за силанизация (4.15), запушете епруветката и оставете да престои 20 минути. 5.3.3. След 20 минути добавете от 1 до 5 ml n-хексан (в зависимост от хроматографските условия): полученият разтвор е готов за газова хроматография. 5.4. Газова хроматография Условията на работа са следните:
5.4.1. Идентифициране на пиковете Идентифицирането на индивидуалните моноглицериди се извършва в съответствие с получените времена на задържане и в сравнение със стандартните смеси моноглицериди, анализирани при същите условия. 5.4.2. Количествена преценка Лицето на всеки пик се изчислява с помощта на електронен интегратор. 6. ИЗРАЗЯВАНЕ НА РЕЗУЛТАТИТЕ Процентното съдържание на глицерил монопалмитат се изчислява от съотношението между лицето на съответния пик и сбора от лицата на пиковете на всички моноглицериди (виж фигура 2) по формулата: Глицерил монопалмитат (%): където:
Резултатът се дава с точност до една десета. 7. ПРОТОКОЛ ОТ АНАЛИЗА Протоколът от анализа следва да посочва:
Фигура 1 Хроматограма на продуктите от реакцията на силанизация, получени чрез действието на липазата върху рафинирано маслиново масло, към което е добавено 20 % естерифицирано масло (100 %)
Фигура 2 Хроматограма на: A) неестерифицирано маслиново масло, след липаза; след силанизация; в тези условия (капилярна колонка 8—12 m) парафиновата фракция се елуира едновременно с диглицеридната фракция или малко след това. След липаза съдържанието на триглицериди не би следвало да надвишава 15 %.
Хроматограма на: Б) естерифицирано масло след липаза; след силанизация; в тези условия (капилярна колонка 8—12 m), парафиновата фракция се елуира едновременно с диглицеридната фракция или малко след това. След липаза съдържанието на триглицериди не би следвало да надвишава 15 %.
8. БЕЛЕЖКИ Бележка 1. ПРИГОТВЯНЕ НА ЛИПАЗАТА В търговската мрежа се предлагат липази със задоволителна липазна активност. Възможно е и да бъдат приготвени лабораторно по следния начин: Охладете до 0 °C 5 kg пресен свински панкреас. Отстранете околните твърда мазнина и съединителна тъкан и раздробете добре в ножова мелница до получаване на гладка кремообразна смес. Разбъркайте в продължение на 4 до 6 часа с 2,5 литра безводен ацетон, след което центрофугирайте. Екстрахирайте утайката още три пъти със същото количество безводен ацетон, след това два пъти със смес от равни части ацетон и диетилов етер и два пъти с диетилов етер. Изсушете остатъка под вакуум за 48 часа до получаването на устойчив прах, който трябва да се съхранява в хладилник и на сухо. Бележка 2. ПРОВЕРЯВАНЕ НА АКТИВНОСТТА НА ЛИПАЗАТА Пригответе емулсия от маслиново масло, както следва: Разбъркайте в смесител в продължение на 10 минути смес от 165 ml от разтвор на гуми арабика при 100 g/l, 15 g натрошен лед и 20 ml предварително неутрализирано маслиново масло. Сипете последователно в бехерова чаша с вместимост 50 ml 10 ml от тази емулсия, а след това 0,3 ml разтвор на натриев холат при 0,2 g/ml и 20 ml дестилирана вода. Поставете бехеровата чаша в термостат, настроен на 37 °C; потопете електродите на pH метъра и спиралната бъркалка. Прибавете, капка по капка, с помощта на бюрета, разтвор на натриев хидроокис (0,1 N), докато pH достигне 8,3. Добавете един обем водна суспензия на липазен прах (0,1 g/ml липаза). Веднага щом pH метърът покаже pH 8,3, включете хронометъра и добавете разтвора на натриевия хидроокис, капка по капка, така че да се поддържа pH от 8,3. Отчитайте всяка минута обема на консумирания разтвор. Нанесете данните в координатна система, чиято абсциса съответства на времето, а ординатата ѝ — на милилитрите алкален разтвор (0,1 N), нужни за поддържането на постоянно pH. Трябва да се получи линейна графика. Активността на липазата, измерена в липазни единици/mg, се изчислява по следната формула:
където:
Единица липаза се дефинира като количеството ензим, което отделя 10 микроеквивалента киселина в минута.“ |
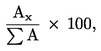




|
7. |
Точка 6.2 от приложение X A се заменя със следния текст:
|
(1) Сбор от изомерите, които могат (или не могат) да се отделят чрез капилярна колона.
(2) Или когато средната стойност на дефектите е по-малка или равна на 2,5, а средната стойност на плодовия аромат е равна на 0.
(3) Масло със съдържание на парафини между 300 mg/kg и 350 mg/kg се определя като масло за осветление, ако общото съдържание на алифатни алкохоли е по-малко или равно на 350 mg/kg или ако процентното съдържание на еритродиол и уваол е по-малко или равно на 3,5 %.
(4) Масло със съдържание на парафини между 300 mg/kg и 350 mg/kg се определя като сурово маслиново масло от остатъчен материал, ако общото съдържание на алифатни алкохоли е над 350 mg/kg и ако процентното съдържание на еритродиол и уваол е над 3,5 %.
(5) Съдържание на други мастни киселини (%): палмитинова: 7,5—20,0; палмитолеинова: 0,3—3,5; хептадеканова: ≤ 0,3; хептадецинова: ≤ 0,3; стеаринова: 0,5—5,0; олеинова: 55,0—83,0; линолова: 3,5—21,0.
(6) Сбор от : делта-5,23-сигмастадиенол+хлеростерол+бета-ситостерол+ситостанол+делта-5-авенастерол+делта-5,24-стигмастадиенол.
(7) Масло със съдържание на парафини между 300 mg/kg и 350 mg/kg се счита за маслиново масло за осветление, ако общото съдържание на алифатни алкохоли е по-малко или равно на 350 mg/kg или ако процентното съдържание на еритродиол и уваол е по-малко или равно на 3,5.
(8) Масло със съдържание на парафини между 300 mg/kg и 350 mg/kg се счита за сурово маслиново масло от остатъчен материал, ако общото съдържание на алифатни алкохоли е над 350 mg/kg и ако процентното съдържание на еритродиол и уваол е над 3,5.
(9) След елуирането на стероловите естери хроматографският ход не бива да съдържа изразени пикове (триглицериди).