|
03/ 45 |
BG |
Официален вестник на Европейския съюз |
3 |
32002D0657
|
L 221/8 |
ОФИЦИАЛЕН ВЕСТНИК НА ЕВРОПЕЙСКИЯ СЪЮЗ |
РЕШЕНИЕ НА КОМИСИЯТА
от 14 август 2002 година
за прилагане на Директива 96/23/EО на Съвета по отношение изпълнението на аналитични методи и тълкуването на резултати
(нотифицирано под номер C(2002) 3044)
(текст от значение за ЕИП)
(2002/657/EО)
КОМИСИЯТА НА ЕВРОПЕЙСКИТЕ ОБЩНОСТИ,
като взе предвид Договора за създаване на Европейската общност,
като взе предвид Директива 96/23/EО на Съвета от 29 април 1996 г. относно мерките за наблюдение на определени вещества и остатъци от тях в живи животни и животински продукти и за отмяна на Директиви 85/358/EИО и 86/469/EИО и Решения 89/187/EИО и 91/664/EИО (1), и по-специално член 15, параграф 1, втора алинея от нея,
като има предвид, че:
|
(1) |
Наличието на остатъци в продуктите от животински произход е въпрос от значение за общественото здраве. |
|
(2) |
Решение 98/179/EО на Комисията от 23 февруари 1998 г. за определяне на подробни правила за вземане на официални проби за мониторинга на определени вещества и остатъци от тях в живи животни и животински продукти (2) предвижда анализът на пробите да се извършва изключително само от одобрени от компетентните национални органи лаборатории за официален контрол на остатъците. |
|
(3) |
Качеството и сравнимостта на аналитичните резултати, получени от лаборатории, одобрени за официален контрол на остатъците, е необходимо да се гарантират. Това следва да се постигне, като се използват системи за гарантиране на качеството, и по-специално чрез прилагане на методи, утвърдени в съответствие с общи процедури и критерии за ефективност, и като се гарантира придържането към общи стандарти или към стандарти, които са общопризнати. |
|
(4) |
Директива 93/99/EИО на Съвета от 29 октомври 1993 г. относно допълнителни мерки, свързани с официалния контрол върху храните и Решение 98/179/EО (3) изискват лабораториите за официален контрол да бъдат акредитирани по ISO 17025 (1) считано от януари 2002 г. Съгласно Решение 98/179/EО от одобрените лаборатории се изисква да участват в международно призната външна оценка на качеството и схема за акредитация. Също така одобрените лаборатории следва да докажат компетентността си чрез редовно и успешно участие в подходящи схеми за изследване на уменията, признати или организирани от националните референтни лаборатории или от референтни лаборатории на Общността. |
|
(5) |
В съответствие с Директива 96/23/EО функционира мрежа от референтни лаборатории на Общността, национални референтни лаборатории и национални лаборатории за контрол, която цели повишаване на координацията. |
|
(6) |
В резултат на напредъка в аналитичната химия, настъпил след приемането на Директива 96/23/EО, концепцията за рутинните методи и референтните методи бе заменена от подход, основан на критерии, при който се създават критерии за ефективност и процедури за валидиране на скрининг метода и потвърдителния метод. |
|
(7) |
За да се гарантира хармонизираното прилагане на Директива 96/23/EО, е необходимо да се определят общи критерии за тълкуване на резултатите от изследванията на лабораториите за официален контрол. |
|
(8) |
За да се гарантира хармонизираното прилагане на Директива 96/23/EО, следва да се предвиди постепенно установяване на минимално изисквани граници на ефективност (МИГЕ) на аналитичен метод за вещества, за които няма допустима граница, и по-специално за тези вещества, чиято употреба не е разрешена или е специално забранена в Общността. |
|
(9) |
Решение 90/515/EИО на Комисията от 26 септември 1990 г. за определяне на референтни методи за откриване на остатъци от тежки метали и арсеник (4), Решение 93/256/EИО на Комисията от 14 май 1993 г. за определяне на методите, които следва да се използват за откриване на остатъци от вещества с хормонално или тиреостатично действие (5) и Решение 93/257/EИО на Комисията от 15 април 1993 г. за определяне на референтните методи и списъка на националните референтни лаборатории за откриване на остатъци (6), последно изменено с Решение 98/536/EО (7), бяха внимателно преразгледани, преди да бъдат обявени за неактуални по обхват и разпоредби предвид напредъка на научното и техническото знание, и съответно следва да бъдат отменени с настоящото решение. |
|
(10) |
За да се позволи методите за анализ на официални проби да бъдат адаптирани съобразно разпоредбите на настоящото решение, следва да се определи преходен период. |
|
(11) |
Мерките, предвидени в настоящото решение, са в съответствие със становището на Постоянния комитет по хранителната верига и здравето на животните, |
ПРИЕ НАСТОЯЩОТО РЕШЕНИЕ:
Член 1
Предмет и обхват
Настоящото решение определя правила за аналитичните методи, които следва да се използват при изследването на официални проби, взети в съответствие с член 15, параграф 1, второ изречение от Директива 96/23/EО, и уточнява общи критерии за тълкуването на аналитичните резултати от тези проби, получени в лабораториите за официален контрол.
Настоящото решение не се прилага за вещества, за които има определени подробни правила в други разпоредби на Общността.
Член 2
Дефиниции
За целите на настоящото решение се прилагат дефинициите в Директива 96/23/EО и в приложението към настоящото решение.
Член 3
Аналитични методи
Държавите-членки гарантират, че официалните проби, взети съгласно Директива 96/23/EО, се анализират чрез методи, които:
|
а) |
са документирани в инструкциите за теста, за предпочитане съгласно ISO 78-2 (6); |
|
б) |
отговарят на изискванията в част 2 от приложението към настоящото решение; |
|
в) |
са били валидирани съгласно процедурите, описани в част 3 от приложението; |
|
г) |
съответстват на минимално изискваните граници на ефективност (МИГЕ), които следва да се определят съгласно член 4. |
Член 4
Минимално изисквани граници на ефективност
Настоящото решение следва да бъде преразгледано с цел постепенно да се установят минимално изискваните граници на ефективност (МИГЕ) на аналитични методи, които да се приложат за вещества, за които не е определена допустима граница.
Член 5
Контрол на качеството
Държавите-членки гарантират качеството на резултатите от анализа на пробите, взети по реда на Директива 96/23/EО, в частност чрез наблюдение на тестовете и/или чрез резултати от калибриране съгласно глава 5.9 от ISO 17025 (1).
Член 6
Тълкуване на резултати
1. Резултатът от даден анализ се счита за резултат на несъответствие, ако критичната граница при потвърдителен метод за аналит се превиши.
2. Ако за дадено вещество е установена допустима граница, критичната граница е концентрацията, над която може да се реши със статистическа сигурност от 1 — α, че допустимата граница е била действително надхвърлена.
3. Ако за дадено вещество не е установена допустима граница, критичната граница е най-ниското ниво на концентрация, при което даден метод може да разграничи със статистическа сигурност от 1 — α наличието на конкретния аналит.
4. За веществата, изброени в група A от приложение I към Директива 96/23/EО, α-грешката е 1 % или по-малка. За всички други вещества α-грешката е 5 % или по-малка.
Член 7
Отмяна
Решения 90/515/EИО, 93/256/EИО и 93/257/EИО се отменят.
Член 8
Преходни разпоредби
Методите за анализ на официални проби от вещества, изброени в група A от приложение I към Директива 96/23/EО, които отговарят на критериите, установени в Решения 90/515/EИО, 93/256/ EИО и 93/257/EИО, могат да се използват в срок до две години след влизане в сила на настоящото решение. Методите, които в момента се прилагат за вещества, изброени в група Б от приложение I към Директива 96/23/EО, отговарят на разпоредбите на настоящото решение в срок най-късно до пет години след датата на прилагане на настоящото решение.
Член 9
Дата на прилагане
Настоящото решение се прилага от 1 септември 2002 г.
Член 10
Адресати
Адресати на настоящото решение са държавите-членки.
Съставено в Брюксел на 14 август 2002 година.
За Комисията
David BYRNE
Член на Комисията
(1) ОВ L 125, 23.5.1996 г., стр. 10.
(2) ОВ L 65, 5.3.1998 г., стр. 31.
(3) ОВ L 290, 24.11.1993 г., стр. 14.
(4) ОВ L 286, 18.10.1990 г., стр. 33.
(5) ОВ L 118, 14.5.1993 г., стр. 64.
(6) ОВ L 118, 14.5.1993 г., стр. 75.
(7) ОВ L 251, 11.9.1998 г., стр. 39.
ПРИЛОЖЕНИЕ
КРИТЕРИИ ЗА ЕФЕКТИВНОСТ, ДРУГИ ИЗИСКВАНИЯ И ПРОЦЕДУРИ ЗА АНАЛИТИЧНИ МЕТОДИ
1. ОПРЕДЕЛЕНИЯ
1.1. „Точност“ е най-голямата степен на близост на съответствие между резултата от теста и приетата референтна стойност (2). Определя се чрез определяне на верността и прецизността.
1.2. „Алфа (α) грешка“ е вероятността пробата от теста да съответства, дори и ако е получена чрез измерване, което не съответства (фалшиво решение за съответствие).
1.3. „Аналит“ е веществото, което следва да бъде открито, идентифицирано и/или определено количествено, както и производните, които се получават по време на неговия анализ.
1.4. „Бета (β) грешка“ е вероятността пробата от теста действително да не съответства, дори и ако е получена чрез измерване, което не съответства (фалшиво решение за съответствие).
1.5. „Отклонение“ е разликата между очаквания резултат от теста и приета референтна стойност (2).
1.6. „Стандарт за калибриране“ е средство за измерване, което представя количеството на съответното вещество по начин, който обвързва стойността му с референтна база.
1.7. „Сертифициран референтен материал (CRM)“ е материал, който има точно установено съдържание на аналит, предназначено за него.
1.8. „Кохроматография“ е процедура, при която екстрактът преди етапа(ите) на хроматография се разделя на две части. Първата част се хроматографира самостоятелно. Втората част се смесва със стандартния аналит, който следва да бъде измерен. След това тази смес също се хроматографира. Количеството добавен стандартен аналит следва да е близко до измереното количество на аналит в екстракта. Този метод има за цел да подобри идентифицирането на аналит, когато се използват хроматографски методи, особено когато не може да се използва подходящ вътрешен стандарт.
1.9. „Съвместно проучване“ е анализиране на една и съща проба с един и същ метод, за да се определят характеристиките на ефективност на метода. Проучването покрива случайна грешка при измерване и лабораторно отклонение.
1.10. „Метод за потвърждение“ означава методи, които предоставят цялостна или допълнителна информация, която да позволи веществото да бъде идентифицирано недвусмислено и ако е необходимо, да се определи количествено съобразно нивото от значение.
1.11. „Критична граница (CCα)“ е границата, към и над която може да се заключи с вероятност за грешка α, че дадена проба не съответства.
1.12. „Способност за откриване (CCβ)“ е най-малкото съдържание от вещество, което може да бъде открито, идентифицирано и/или определено количествено в проба с вероятност за грешка β. При вещества, за които няма установена допустима граница, способността за откриване е най-ниската концентрация, при която даден метод може да открие действително заразени проби със статистическа точност от 1 — β. При вещества с установена допустима граница способността за откриване е концентрацията, при която методът може да открие концентрации на допустимата граница със статистическа точност от 1 — β.
1.13. „Проба с обогатен материал“ е проба, обогатена с известно количество от аналита, който следва да бъде открит.
1.14. „Междулабораторно проучване (сравнение)“ е организиране, провеждане и оценяване на изпитване на една и съща проба от две или повече лаборатории съгласно предварително определени условия, за да се определи изпълнението на теста. Според своята цел проучването може да бъде съвместно проучване или проучване на способността.
1.15. „Вътрешен стандарт (ВС)“ е вещество, което не се съдържа в пробата, притежава физико-химични свойства, изключително близки до тези на аналита, който следва да бъде идентифициран, и се добавя към всяка проба и към всеки стандарт за калибриране.
1.16. „Лабораторна проба“ е проба, подготвена за изпращане до лаборатория и предназначена за инспекция или изпитване.
1.17. „Ниво от значение“ е концентрацията на вещество или аналит в дадена проба, която има значение за определяне на съответствието му със законодателството.
1.18. „Минимално изисквана граница на ефективност (МИГЕ)“ е минималното съдържание на аналит в проба, което следва най-малко да се открие и потвърди. Предназначението ѝ е да хармонизира ефективността на аналитичните методи при вещества, за които не е определена допустима граница.
1.19. „Характеристика на ефективността“ е функционалното качество, което може да се припише на даден аналитичен метод. Това може да бъде например специфичност, точност, вярност, прецизност, повторяемост, възпроизводимост, възстановяване, способност за откриване и износоустойчивост.
1.20. „Критерии за ефективност“ са изискванията за дадена характеристика на ефективност, съгласно които може да се прецени, че аналитичният метод е подходящ за целта и дава надеждни резултати.
1.21. „Допустима граница“ е максимално допустима граница на остатъчни вещества, максимално ниво или друга максимална допустимост на вещества, установени в други части от законодателството на Общността.
1.22. „Прецизност“ е близостта на съответствие между резултатите от независими изпитвания, получени при установени (предварително определени) условия. Мярката за прецизност обикновено се изразява като липса на прецизност и се изчислява като стандартно отклонение от резултатите от теста. По-малка прецизност се определя от по-голямо стандартно отклонение (2).
1.23. „Проучване на способността“ е анализиране на една и съща проба, която позволява на лабораториите да изберат собствените си методи, при условие че тези методи се използват в рутинни условия. Проучването следва да бъде извършено съгласно Ръководство ISO 43-1 (3) и 43-2 (4) и може да се използва за оценяване на възпроизводимостта на методите.
1.24. „Качествен метод“ е аналитичен метод, който идентифицира дадено вещество въз основа на неговите химични, биологични или физични качества.
1.25. „Количествен метод“ е аналитичен метод, който определя количеството или големината на частта от дадено вещество, така че да може да се изрази като цифрова стойност на подходящи единици.
1.26. „Контролен опит за определяне на реактива“ е завършена аналитична процедура, приложена без тестовата част или като се използва еквивалентно количество подходящ разтвор вместо тестовата част.
1.27. „Възстановяване“ е процент на истинската концентрация на вещество, възстановена по време на аналитична процедура. Определя се по време на валидирането, ако няма на разположение сертифициран референтен материал.
1.28. „Референтен материал“ е материал, едно или няколко качества от който са били потвърдени чрез валидиран метод, за да може да се използва за калибриране на апарат или за проверяване на метод за измерване.
1.29. „Повторяемост“ е прецизност при условия на повторяемост (2).
1.30. „Условия на повторяемост“ са условия, при които независимите резултати от теста са получени по един и същ метод върху идентични тестови единици в една и съща лаборатория от един и същ оператор, като е използвано едно и също оборудване (2).
1.31. „Възпроизводимост“ е прецизност при условия на възпроизводимост (2)(4).
1.32. „Условия на възпроизводимост“ са условия, при които резултатите от теста са получени по един и същи метод върху идентични тестови единици в различни лаборатории от различни оператори, като е използвано различно оборудване (2)(4).
1.33. „Износоустойчивост“ е податливостта на даден аналитичен метод към промени в експерименталните условия, които могат да се изразят като списък на материали за проби, аналити, условия за съхранение, условия на околната среда и/или условия за подготовка на пробата, при които методът може да се приложи, както е посочено или с определени минимални промени. По отношение на всички експериментални условия, които биха могли на практика да подлежат на вариране (например стабилност на реактиви, състав на пробата, pH, температура), всякакви различия, които биха могли да окажат въздействие върху аналитичния резултат, следва да бъдат посочени.
1.34. „Празно определяне на проба“ е цялостна аналитична процедура, приложена към част от тест, взета от проба, в която аналитът липсва.
1.35. „Скрининг метод“ означава метод, който се използват за откриване наличието на вещество или видове вещества на ниво от значение. Тези методи дават възможност за висок процент на анализ на пробите и се използват за отсяване на голям брой от пробите с потенциален резултат на несъответствие. Те са специално създадени, за да се избегнат фалшивите резултати на съответствие.
1.36. „Единично лабораторно проучване (вътрешно потвърждение)“ е аналитично проучване, при което се включва една лаборатория, използва се един метод, за да се анализират едни и същи или различни тестови материали при различни условия през умерено дълги интервали от време.
1.37. „Специфичност“ е способността на даден метод да различава аналита, който се измерва, от други вещества. Тази характеристика е предимно функция на описаната измервателна техника, но може да варира в зависимост от вида на сместа или свързващото вещество.
1.38. „Стандартна добавка“ е процедура, при която пробата за тест се разделя на две (или повече) части от проба. Едната част се анализира самостоятелно, а към другите части от проби преди анализа се добавят известни количества стандартен аналит. Количеството добавен стандартен аналит следва да е между два и пет пъти изчисленото количество аналит в пробата. Тази процедура е създадена, за да се определи съдържанието на аналит в дадена проба, като се има предвид възстановяването на аналитичната процедура.
1.39. „Стандартен аналит“ е аналит, чиито съдържание и чистота са известни и удостоверени и който при анализ се използва за референтен.
1.40. „Вещество“ е материя от конкретен или определен химичен състав и неговите метаболити.
1.41. „Част от проба“ е количеството материал, извлечено от пробата за тест, върху което се извършва тестът или наблюдението.
1.42. „Тестова проба“ е пробата, приготвена от лабораторната проба и от която се вземат части от пробата.
1.43. „Истинност“ е близостта на съгласуваност между средната стойност, получена от голяма поредица от резултати от тест, и приетата референтна стойност. Истинността обикновено се изразява като отклонение (2).
1.44. „Единици“ са единиците, които са описани в ISO 31 (20) и в Директива 71/354/EО (19).
1.45. „Валидиране“ означава потвърждаване чрез изследване и предоставяне на ефективно доказателство, че конкретните изисквания за специфично предназначена употреба са изпълнени (1).
1.46. „Вътрешнолабораторна възпроизводимост“ е прецизност, получена в една и съща лаборатория при предвидени (предварително одобрени) условия (отнасящи се например до метод, тестови материали, оператори, околна среда) през умерено дълги интервали от време.
2. КРИТЕРИИ ЗА ЕФЕКТИВНОСТ И ДРУГИ ИЗИСКВАНИЯ ЗА АНАЛИТИЧНИ МЕТОДИ
Аналитични методи или комбинации от методи, различни от описаните по-долу, могат да се използват само за скрининг или за потвърждаващи цели, ако се докаже, че отговарят на съответните изисквания, установени в настоящото решение.
2.1. ОБЩИ ИЗИСКВАНИЯ
2.1.1. Третиране на проби
Пробите се получават, третират и обработват, така че да има максимален шанс да се открие веществото. Процедурите за третиране на пробите предотвратяват възможността от случайна зараза или загуба на аналити.
2.1.2. Изпълнение на тестове
2.1.2.1. Възстановяване
По време на анализа на пробите възстановяването се определя във всяка партида проби, ако се използва фиксиран фактор за коригиране на възстановяването. Ако възстановяването е в рамките на границите, фиксираният фактор за коригиране може да се използва. В противен случай се използва факторът на възстановяване, получен за тази конкретна партида, освен ако не се приложи специфичният фактор на възстановяване на аналита в пробата, като в този случай за количественото определяне на аналит в дадена проба се използва процедурата за стандартна добавка (виж 3.5) или вътрешен стандарт.
2.1.2.2. Специфичност
При експериментални условия с метода може да прави разграничение между аналита и другите вещества. Необходимо е да се изчисли до каква степен е възможно това. Когато се използва описаната техника на измерване, е необходимо да се приложат стратегии за преодоляване на всяко смесване с вещества, което може да се предвиди, като например хомологии, аналогии, метаболични продукти от съответния остатък. От съществено значение е да се изследва смесването, което би могла да възникне от компоненти на свързващото вещество.
2.2. СКРИНИНГ МЕТОДИ
За целите на скрининга съгласно Директива 96/23/EО се използват само тези аналитични техники, за които може да се проследи по документален начин, че са валидирани и имат фалшива степен на съответствие от < 5 % (β-грешка) на нивото от значение. В случай на съмнение за резултат на несъответствие този резултат се потвърждава чрез потвърдителен метод.
2.3. МЕТОДИ ЗА ПОТВЪРЖДЕНИЕ ЗА ОРГАНИЧНИ ОСТАТЪЦИ И ЗАМЪРСИТЕЛИ
Потвърдителните методи за органични остатъци или замърсители предоставят информация за химичната структура на аналита. Следователно методи, основани само на хроматографски анализ, без използване на спектрометрично откриване, не са подходящи да се използват самостоятелно като потвърдителни методи. Независимо от това, ако на дадена техника ѝ липсва достатъчно специфичност, желаната специфичност се постига чрез аналитични процедури, състоящи се от подходящи комбинации от почистване, хроматографско отделяне (отделяния) и спектрометрично откриване.
Следните методи или комбинации от методи се считат за подходящи за идентифицирането на органични отпадъци или замърсители за посочените групи вещества:
Таблица 1
Подходящи методи за потвърждение за органични остатъци и замърсители
|
Техника за измерване |
Вещества приложение 1 96/23/EО |
Ограничения |
|
LC или GC с масово спектрометрично откриване |
групи A и Б |
Само ако следва постоянно или непостоянно (on-line или off-line) хроматографско отделяне Само ако са използвани техники за пълно сканиране или като се използват поне 3 (от група Б) или 4 (от група A) идентификационни точки за техники, които не регистрират всички масови спектри |
|
LC или GC с IR спектрометрично откриване |
групи A и B |
Необходимо е да се отговори на специфичните изисквания за абсорбция при IR-спектометрията |
|
LC — пълно сканиране DAD |
група Б |
Необходимо е да се отговори на специфичните изисквания за абсорбция при UV-спектрометрията |
|
LC — флуоресценция |
група Б |
Само за молекули, които проявяват естествена флуоресценция, и за молекули, които проявяват флуоресценция след трансформация или след дериватизация |
|
2-D TLC — пълно UV/VIS сканиране |
група Б |
Двуизмерната HPTLC-хроматография и кохроматографията са задължителни |
|
GC откриване с помощта на хващане на електрони |
група Б |
Само ако се използват две колони с различна полярност |
|
LC имунограма |
група Б |
Само ако се използват поне две различни хроматографски системи или втори независим метод за откриване |
|
LC-UV/VIS (единична дължина на вълна) |
Група Б |
Само ако се използват поне две различни хроматографски системи или втори независим метод за откриване |
2.3.1. Общи критерии и изисквания за ефективност
Потвърдителните методи предоставят информация за химичната структура на аналита. Ако повече от една съставка дава един и същ отговор, после методът не може да направи разлика между тези съставки. Методи, основани само на хроматографски анализ, без използване на спектрометрично откриване, не са подходящи да се използват самостоятелно като методи за потвърждение.
Ако се налага използването на подходящ вътрешен стандарт в метода, той се добавя към част от пробата в началото на процедурата на екстракция. В зависимост от възможността да се намерят, се използват или стабилни изотопни форми на аналита, които са изключително подходящи при масово спектрометрично откриване, или съединения, които са близки по структура до аналита.
Когато не може да се използва подходящ вътрешен стандарт, идентифицирането на аналита се потвърждава чрез кохроматография. В този случай се достига само един връх, като увеличената височина на върха (или областта) е еквивалентна на количеството добавен аналит. С газова хроматография (GC) или течна хроматография (LC) ширината на върха на половин максимална височина е в рамките на 90—110 % от обхвата на оригиналната широчина и периодите на задържане са идентични с разлика в рамките на 5 %. За методите на тънкослойната хроматография (TLC) само петното, за което се предполага, че се дължи на аналита, се подсилва; ново петно не се появява и визуалният изглед не се променя.
За предпочитане е референтният или обогатеният материал, съдържащ известни количества аналит, който е на или близо до допустимата граница или критичната граница (несъответстваща контролна проба), както и съответстващите контролни материали и празни проби за реактив да се подложат на цялата процедура, едновременно с анализирането на всяка партида проби за тест. Редът за инжектиране на екстрактите в аналитичния апарат е, както следва: празна проба за реактив, съответстваща контролна проба, проба(и), коя(и)то подлежи(ат) на потвърждение, съответстваща контролна проба отново и накрая — несъответстваща контролна проба. Всяко отклонение от тази последователност се обосновава.
2.3.2. Допълнителни критерии за ефективност и други изисквания за количествените методи за анализ
2.3.2.1. Вярност на количествените методи
В случай на повторни анализи на сертифициран референтен материал насочващият обхват на отклонението на експериментално определеното възстановяване, коригирано със средна масова фракция, от сертифицираната стойност е, както следва:
Таблица 2
Минимална вярност на количествените методи
|
Масова фракция |
Обхват |
|
≤ 1 μg/kg |
От - 50 % до + 20 % |
|
> 1 μg/kg до 10 μg/kg |
От -30 % дo + 10 % |
|
≥ 10 μg/kg |
От - 20 % до + 10 % |
Ако не са налични такива CRM, е прието верността на измерването да се оцени чрез възстановяване на добавки на познати количества аналит(и) към празна матрица. Данните, коригирани с възстановяване на средната стойност, са приемливи само когато попадат в обхватите, посочени в таблица 2.
2.3.2.2. Прецизност на количествените методи
Вътрешнолабораторният коефициент на вариация (CV) при повторен анализ на референтен или подсилен материал при условия на възпроизводимост не следва да надвишава нивото, изчислено с равенството на Хоруиц. Това равенство е:
![]()
където C е масовата фракция, изразена като валентност (степен) на 10 (например 1 mg/g = 10-3). В таблица 3 са представени примери.
Таблица 3
Примери за възпроизводимост на CV за количествени методи при обхват на аналитични масови фракции
|
Масова фракция |
Възпроизводимост CV(%) |
|
1 µg/kg |
|
|
10 µg/kg |
|
|
100 µg/kg |
23 |
|
1 000 µg/kg (1 mg/kg) |
16 |
За анализи, извършени при повторяеми условия, вътрешнолабораторният коефициент (CV) обикновено е между една втора и две трети от горепосочените стойности. За анализите, извършени при вътрешнолабораторни условия за възпроизводимост, вътрешнолабораторните CV не са по-големи от CV за възпроизводимост.
В случая на вещества с установена допустима граница методът постига вътрешнолабораторна възпроизводимост, която не е по-голяма от съответното CV за възпроизводимост при концентрация на 0,5 × допустимата граница.
2.3.3. Критерии за ефективност и други изисквания за масово спектрометрично откриване
Масовите спектрометрични методи са подходящи за разглеждане като потвърдителни методи само след постоянно или непостоянно хроматографско отделяне.
2.3.3.1. Хроматографско отделяне
За GC-MS процедурите газовото хроматографско отделяне се извършва, като се използват капилярни колони. За LC-MS процедурите хроматографското отделяне се извършва, като се използват подходящи LC колони. Във всеки случай минималното приемливо време на задържане за аналита, който се проверява, е два пъти времето за задържане, съответстващо на празния обем на колоната. Времето за задържане (или сравнителното време за задържане) на аналита в частта от проба съответства на калибрационния стандарт в рамките на специфичен времеви прозорец за задържане. Този прозорец за задържане е съизмерим с действащата мощност на хроматографската система. Съотношението на хроматографското време за задържане на аналита към това на вътрешния стандарт, т.е. съответното време за задържане на аналита съответства на това в калибрационния разтвор при отклонение от ± 0,5 % за GC и ± 2,5 % за LC.
2.3.3.2. Масово спектрометрично откриване
Масовото спектрометрично откриване се извършва, като се прилагат MS техники, като записване на цялостен спектър (пълно сканиране) или избран йонов мониторинг (ИЙМ), както и MS-MSn техники, като избран реактивен мониторинг (ИРМ) или други подходящи MS или MS-MSn техники в комбинация с подходящи йонизиращи модели. При масова спектрометрия с висока резолюция (МСВР) резолюцията като цяло е по-голяма от 10 000 за целия масов обхват от 10 %.
: Когато се извършва масово спектрометрично определяне чрез записване на спектър с пълно сканиране, наличието на всички измерени диагностични йони (молекулярните йони, характерните адукти на молекулярните йони, характерни фрагментни йони и изотопни йони) при сравнителен интензитет от повече от 10 % в референтния спектър на стандарта за калибриране е задължително.
: Когато се извършва масово спектрометрично определяне чрез фрагментография, за предпочитане е молекулярният йон да бъде един от избраните диагностични йони (молекулярния йон, характерни адукти на молекулярния йон, характерни фрагментни йони и всички техни изотопни йони). Избраните диагностични йони не следва да произхождат изцяло от същата част на молекулата. Съотношението сигнал към шум за всеки диагностичен йон е ≥ 3:1.
: Сравнителният интензитет на откритите йони, изразен като процент от интензитета на най-интензивния йон или като преход, съответства на тези от стандарта за калибриране или от разтвори на стандарт за калибриране, или от заострени мостри, при сравними концентрати, измерени при същите условия при следните отклонения:
Таблица 4
Максимално позволени отклонения за сравнителни йонови интензитети чрез използване на определен обхват масови спектрометрични техники
|
Сравнителен интензитет (% от базисния връх) |
EI-GC-MS (сравнителен) |
CI-GC-MS, GC-MSn LC-MS, LC-MSn (сравнителен) |
|
> 50 % |
±10 % |
±20 % |
|
> 20 % дo 50 % |
±15 % |
±25 % |
|
> 10 % дo 20 % |
±20 % |
±30 % |
|
≤ 10 % |
±50 % |
±50 % |
: сравнителният интензитет на диагностичните йони и/или прекурсори/продуктови йонни двойки се идентифицира, като се сравнят спектрите или като се интегрират сигналите на единните масови остатъци. Всеки път когато се прилага фонова корекция, тя се прилага еднакво през цялата партида (виж точка 2.3.1, четвърти параграф) и се посочва ясно.
: Когато се регистрира пълно сканиране на спектъра в единна масова спектрометрия, минимум четири йона са налични при сравнителен интензитет от ≥ 10 % от базовия връх. Молекулярният йон е включен, ако е наличен в референтния спектър със сравнителен интензитет от ≥ 10 %. Поне четири йона лежат в рамките на максимално позволените отклонения за сравнителни йонни интензитети (таблица 5). Може да се използва търсене в компютризирана библиотека. В този случай сравнението на масови спектрални данни в тестовите проби към тази на калибриращия разтвор следва да надхвърля критичния фактор на съвпадение. Този фактор следва да бъде определен по време на процеса на валидиране на всеки аналит въз основа на спектър, за който са изпълнени описаните по-долу критерии. Вариациите в спектъра, причинени от извадковата матрица, и детекторното изпълнение се проверяват.
: Когато се измерват масови фрагменти, като се използват други техники, различни от тези за пълно сканиране, за интерпретиране на данните се използва система на идентификационни точки. За потвърждаване на веществата в списъка в група А от приложение I към Директива 96/23/EО, се изискват минимум 4 идентификационни точки. За потвърждаване на веществата в списъка в група Б от приложение I към Директива 96/23/EО се изискват минимум 3 идентификационни точки. Таблицата по-долу показва номера на идентификационните точки, които може да получи всяка от основните масови спектрометрични техники. За да се направи класификация обаче на идентификационните точки, необходими за потвърждение, и сумата идентификационни точки, които да се изчислят, е необходимо:
|
а) |
да се измери минимум от поне едно йонно съотношение; |
|
б)всички съответни измерени йонни съотношения да отговарят на описаните по-горе критерии; ив) |
максимум три спектърни техники да могат да се комбинират с цел да се постигне минимален брой идентификационни точки. |
Таблица 5
Отношение между обхват от класове масов фрагмент и получени идентификационни точки
|
MS техника |
Идентификационни точки, получени за йон |
||||||||||
|
масова спектрометрия с ниска резолюция (LR) |
1,0 |
||||||||||
|
LR-MSn предкурсорен йон |
1,0 |
||||||||||
|
LR-MSn преходни продукти |
1,5 |
||||||||||
|
HRMS |
2,0 |
||||||||||
|
HR-MSn предкурсорен йон |
2,0 |
||||||||||
|
HR-MSn преходни продукти |
2,5 |
||||||||||
|
|||||||||||
Таблица 6
Примери на броя идентификационни точки, получени за определен обхват техники и комбинации от тях (n = цифра)
|
Техника(и) |
Брой йони |
Идентификационни точки |
|
GC-MS (EI или CI) |
N |
n |
|
GC-MS (EI и CI) |
2 (EI) + 2 (CI) |
4 |
|
GC-MS (EI или CI) 2 производни |
2 (производна A) + 2 (производна Б) |
4 |
|
LC-MS |
N |
n |
|
GC-MS-MS |
1 предкурсор и 2 дъщерни |
4 |
|
LC-MS-MS |
1 предкурсор и 2 дъщерни |
4 |
|
GC-MS-MS |
2 предкурсорни йона, всеки с 1 дъщеря |
5 |
|
LC-MS-MS |
2 предкурсорни йона, всеки с 1 дъщеря |
5 |
|
LC-MS-MS-MS |
1 предкурсор, 1 дъщеря и 2 от следващи поколения |
5,5 |
|
HRMS |
N |
2n |
|
GC-MS и LC-MS |
2 + 2 |
4 |
|
GC-MS и HRMS |
2 + 1 |
4 |
2.3.4. Критерии за ефективност и други изисквания за хроматография, обединени с инфрачервени открития
: подходящи върхове представляват максималните стойности на абсорбция в инфрачервения спектър на стандарта за калибриране, изпълняващи следните изисквания.
2.3.4.1. Инфрачервено откриване
: следва да е в обхвата на вълната 4 000—500 cm-1.
: не по-малко от или:
|
а) |
специфична моларна абсорбция от 40 по отношение на върховата базова линия; или |
|
б) |
относителна абсорбция от 12,5 % от абсорбцията на най-интензивния връх в обхват 4 000—500 cm-1, |
когато и двете са обект на измерване по отношение на нулева абсорбция и 5 % от абсорбирането на най-интензивния връх в обхват 4 000—500 cm-1, когато и двете се измерват по отношение на тяхната върхова базова линия.
Забележка: Въпреки че адекватните върхове съгласно буква a) може да се предпочитат от теоретична гледна точка, тези по буква б) са по-лесни за определяне на практика.
Определен е броят на върховете в инфрачервения спектър на аналита, чиито честоти отговарят на съответния връх в спектъра на стандарта за калибриране, с разлика от ± 1 cm-1.
2.3.4.2. Тълкуване на инфрачервени спектрални данни
Абсорбирането е налично във всички области от аналитния спектър, които отговарят на подходящ връх в референтния спектър от стандарта за калибриране. Минимум шест подходящи върха се изискват в инфрачервения спектър от стандарта за калибриране. Ако има по-малко от шест подходящи върха (7), съответният спектър не може да се използва като референтен спектър. „Резултатът“, т.е. процентът на подходящи върхове, установени в инфрачервения спектър на аналита, е поне 50. Когато няма точно съвпадение за подходящ връх, съответната област на аналитния спектър съответства на наличието на кореспондиращ връх. Процедурата е приложима само за върхове на абсорбиране в извадковия спектър с интензитет от поне три пъти върха до върховия шум.
2.3.5. Критерии за ефективност и други изисквания за определянето на аналит чрез използване на LC с други техники за откриване
2.3.5.1. Хроматографско отделяне
Необходимо е да се използва вътрешен стандарт, ако е наличен материал, подходящ за тази цел. За предпочитане е той да е от подобен стандарт с време на задържане, близко до това на аналита. Аналитът следва да се отлее при времето на задържане, което е типично за кореспондиращия стандарт за калибриране при същите експериментални условия. Минималното приемливо време на задържане за аналита следва да бъде два пъти времето на задържане, отговарящо на празния обем на колоната. Съотношението на времето за задържане на аналита към това на вътрешния стандарт, т.е. съответното време за задържане на аналита, е същото като това на стандарта за калибриране в подходящата матрица с разлика от ± 2,5 %.
2.3.5.2. Пълно сканиране UV/VIS откриване
Критериите за ефективност на LC методите следва да се изпълнят.
Максималните абсорбционни стойности в спектъра на аналита са със същата дължина на вълната като тази на стандарта за калибриране в рамките на обхват, определен от резолюцията на система за откриване. За диоден ред откриване обхватът е обикновено в рамките на ± 2 nm. Спектърът на аналита над 220 nm за тези части от двата спектъра със сравнителна абсорбция ≥ 10 % следва да не е видимо различен от спектъра на стандарта за калибриране. Този критерий е изпълнен, когато, на първо място, са налични същите максимални стойности и, на второ място, когато разликата между двата спектъра по никакъв начин не е по-голяма от 10 % от абсорбцията на стандарта за калибриране. Когато се използва компютърно подпомогнато търсене в база данни и откриване на съответствия, сравняването на спектралните данни в пробите за тест към тази на калибриращия разтвор следва да надхвърли критичния фактор на съвпадение. Този фактор се определя по време на процеса на валидиране за всеки аналит на базата на спектър, за който описаните по-горе критерии са изпълнени. Вариациите в спектъра, причинени от извадковата матрица и работата на детектора, се проверяват.
2.3.5.3. Критерии за ефективност на флуориметрично откриване
Критериите за ефективност на LC методите следва да се изпълнят.
Това се прилага при молекули, които проявяват естествена флуоресценция, и към молекули, които флуоресцират или след трансформация, или след дериватизация. Изборът на възбуждащите и емисионните дължини на вълните в комбинация с хроматографските условия се прави така, че да се минимизира появата на намесващи се компоненти в празните извадки от пробата.
Най-близкият върхов максимум в хроматограмата се отделя от обозначения аналитен връх с поне една пълна върхова ширина при 10 % от максималната височина на аналитния връх.
2.3.5.4. Критерии за ефективност за определяне на аналит с LC имунограма
Методът на LC имунограма не е подходящ за самостоятелно използване като потвърдителен метод.
Съответните критерии за LC методите следва да бъдат изпълнени.
Предварително определените параметри за контрол на качеството, например неспецифично свързване, относителното свързване на контролните проби и абсорбиращата стойност на бланката следва да са в рамките на границите, получени по време на валидирането на пробата.
Имунограмата следва да се състои от поне пет фракции.
Всяка фракция следва да е по-малко от половината ширина на върха.
Фракцията с максимално количество аналит следва да е една и съща за разглежданата проба, за несъответстващата контролна проба и за стандарта.
2.3.5.5. Определяне на аналит чрез използване на LC с UV/VIS откриване (единична дължина на вълната)
Методът на LC с UV/VIS откриване (единична дължина на вълната) не е подходящ за самостоятелно използване като потвърдителен метод.
Най-близкият върхов максимум в хроматограмата се отделя от определения аналитен връх с поне една пълна ширина на върха при 10 % от максималната височина на аналитния връх.
2.3.6. Критерии на ефективност и други изисквания за определянето на аналит с 2-D TLC, обединен до UV/VIS спектрометрично откриване с пълно сканиране
Двуизмерен HPTLC и кохроматография са задължителни.
RF стойностите на аналита съответстват на RF стойностите на стандартите в рамките на ± 5 %.
Визуалният външен вид на аналита не следва да се отличава от стандарта.
За петна с един и същ цвят центърът на най-близкото петно следва да е отделен от центъра на петното на аналита с поне половината от сумата на диаметрите на петната.
Спектърът на аналита не следва да е визуално различен от спектъра на стандарта съгласно описаното за всеобхватно UV/VIS откриване.
При използване на компютърно подпомогнато търсене в база данни и откриване на съответствия и комбиниране сравняването на спектралните данни в тестовите проби към тези на калибриращия разтвор следва да надхвърля фактора на критично съвпадение. Този фактор следва да се определи по време на валидиращия процес за всеки аналит на базата на спектър, за който са изпълнени критериите, описани по-горе. Вариации в спектъра, причинени от същата матрица, и изпълнението на детектора подлежат на проверка.
2.3.7. Критерии за ефективност и изисквания за определяне на аналит с GC в комбинация с детектор за улавяне на електрони (ECD)
Необходимо е да се използва вътрешен стандарт, ако е наличен материал, подходящ за тази цел. За предпочитане е това да е подобно вещество с време на задържане, близко до това на аналита. Аналитът следва да се отдели при време за задържане, което е типично за кореспондиращия стандарт за калибриране при същите условия. Минималното приемливо време за задържане за аналит следва да е два пъти времето за задържане, съответстващо на празния обем колона. Съотношението на времето за задържане на аналита към това на вътрешния стандарт, т.е. съответното време за задържане на аналита, следва да е същото като стандарта за калибриране в подходящата матрица, в рамките на обхват от ± 0,5 %. Най-близкият върхов максимум в хроматограмата следва да се отдели от определения аналитен връх с поне една цяла върхова ширина при 10 % от максималната височина на аналитния връх. За допълнителна информация може да се използва ко-хроматографията.
2.4. ПОТВЪРДИТЕЛНИ МЕТОДИ ЗА ЕЛЕМЕНТИ
Потвърждаващите анализи за химични елементи следва да се базират на концепцията за непротиворечива идентификация и точно и прецизно околичествяване посредством физико-химични качества, уникални за съответния химичен елемент (например характерна дължина на вълната на елемента от издадена или погълната радиация, атомна маса), на ниво от значение.
Следните методи или комбинации от методи се считат за подходящи за идентифициране на химичните елементи:
Таблица 7
Подходящи методи за потвърждение за химични елементи
|
Техника |
Измерен параметър |
|
Диференциална импулсна анодна волтаметрия с натрупване |
електрически сигнал |
|
Атомноабсорбционна спектрометрия |
|
|
Пламъкова |
дължина на вълната при абсорбция |
|
Генериране на хидриди |
дължина на вълната при абсорбция |
|
Студена пара |
дължина на вълната при абсорбция |
|
Електротермична атомизация (графитно покритие) |
дължина на вълната при абсорбция |
|
Атомноемисионна спектрометрия |
|
|
Индуктивно свързана плазма |
дължина на вълната при емисия |
|
Мас спектрометрия |
|
|
Индуктивно свързана плазма |
съотношение между маса и заряд |
2.4.1. Общи критерии за ефективност и други изисквания за методи за потвърждение
Референтен или обогатен материал, съдържащ известно количество аналит при или близо до максимално допустимата граница или до критичната граница (несъответстваща контролна проба), както и съответстваща контролна проба материали и реактивни бланки е препоръчително да преминат през цялата процедура едновременно с всяка анализирана партида тестови проби. Препоръчителният ред за инжектиране на екстрактите в аналитичния инструмент е: реактивна бланка, съответстваща контролна проба, проба, която следва да бъде потвърдена, и накрая несъответстваща контролна проба. Всяко отклонение от този ред се обосновава.
Като цяло повечето аналитични техники изискват цялостно преработване на органичната матрица, за да могат да се получат разтвори преди определянето на аналита. Това може да се постигне, като се използват микровълнови минерализационни процедури, които намаляват риска от загуба и/или зараза на аналита, който представлява интерес. Следва да се използват обеззаразени тефлонови съдове с добро качество. Ако други мокри или сухи методи на преработване се използват, е необходимо да се предостави документирано доказателство, което да изключва явления на потенциалната загуба или заразяване. Като алтернатива на преработването процедурите за отделяне (например извличане) могат при определени обстоятелства да бъдат избрани с цел да отделят аналита от компонентите на матрицата и/или да концентрират аналитите с цел да ги въведат в аналитичното оборудване.
По отношение на калибрирането, било то външно или базирано на стандартния допълващ метод, е необходимо да се вземат необходимите мерки да не се надвишава работният обхват, определен за анализа. В случая на външно калибриране е задължително да се подготвят стандарти за калибриране в разтвор, който съвпада възможно най-пълно със състава на разтвора на пробата. Необходимо е също да се приложи фонова корекция, ако се налага от специфични аналитични обстоятелства.
2.4.2. Допълнителни критерии за ефективност и други изисквания за количествени аналитични методи
2.4.2.1. Вярност на количествените методи
При повторен анализ на сертифициран референтен материал за елементи отклонението от експериментално определеното съдържание на средна стойност от сертифицираната стойност не следва да е извън границата ± 10 %. Когато не са налични такива CRM, е приемливо да се оцени верността на измерването чрез възстановяване на добавки от известни количества от елемента на неизвестните проби. Обръща се внимание на факта, че за разлика от аналита, добавеният елемент не е химически обвързан в реалната матрица и затова резултатите, получени посредством този подход, имат по-малко валидност от тези, постигнати чрез използване на CRM. Данните за възстановяване са приемливи само когато са в диапазона ± 10 % от целевата стойност.
2.4.2.2. Прецизност на количествените методи
В случай на повтарящ се анализ на проба, извършен при вътрешнолабораторни възпроизводствени условия, вътрешнолабораторният коефициент на вариация (CV) от средната стойност не следва да надвишава следните стойности:
Таблица 8
CV за количествени методи при обхват на масови фракции от елемента
|
Масови фракции |
CV (%) |
|
≥ 10 µg/kg до 100 µg/kg |
20 |
|
> 100 µg/kg до 1 000 µg/kg |
15 |
|
≥1 000 µg/kg |
10 |
2.4.3. Специфични изисквания за диференциална импулсна анодна волтаметрия с натрупване (DPASV)
Пълното унищожаване на органичната материя в проби преди определянето на DPASV е от най-голямо значение. Във волтамограмите няма да се забележат широки сигнали поради наличието на органични материали. Неорганични матрични съставки могат да повлияят върху височината на върховете в DPASV. Поради това е необходимо да се извърши околичествяване от метода на стандартните добавки. Образци на типични волтамограми на пробен разтвор се предоставят с метода.
2.4.4. Специфични изисквания за атомноабсорбционна спектрометрия (AAS)
Тази техника е предимно едноелементна и затова налага оптимизирането на експерименталните места в зависимост от елемента, който подлежи на околичествяване. При възможност резултатите следва да се проверят качествено и количествено, като се премине към алтернативни абсорбционни линии (в идеалния вариант следва да се изберат две различни линии). Стандартите на калибриране следва да се подготвят в матрица на разтвор, която да съвпада възможно най-пълно с тази на разтвора за измерване на пробата (например киселинна концентрация или модифицираща композиция). С цел да се минимизират празните стойности, всички реактиви следва да са с възможно най-голяма чистота. В зависимост от избрания модел за изпарение и/или атомизиране на пробата могат да се отличат различни видове AAS.
2.4.4.1. Специфични изисквания за пламъкова AAS
Мястото, на което се намира инструментът, следва да се оптимизира за всеки елемент. От особено значение е съставът на газа и потоците да се проверяват. Следва да се използва продължителен коректор на източника с цел да се избегнат намесите, причинени от фоново поглъщане. В случай на неизвестни матрици, следва да се направи проверка това дали е необходима корекция на фона.
2.4.4.2. Специфични изисквания за графитно покритие AAS
Заразата в лабораторията често засяга точността, когато се работи на ултранива в графитните покрития. Ето защо следва да се използват реактиви с висока чистота, дейонизирана вода и инертна пластмасова жица за проба и стандартна обработка. Мястото на инструментите за всеки елемент следва да се оптимизира. Особено предварителното третиране и атомизиращите условия (температура, време) и изменението на матрицата следва да се проверят.
Работата при изотермални атомизирани условия (например трансверсна 0-загрята графитна туба с интегрирана Лвов платформа (8) ще редуцира влиянието на матрицата, отнасяща се до атомизацията на аналита. В комбинация с модификация на матрицата и Зееман-фон корекция (9), ще се позволи околичествяването посредством калибрираща извивка, основана на измерване на воден стандартен разтвор.
2.4.5. Специфични изисквания за атомноабсорбционна спектрометрия при генериране на хидриди
Органични смеси, съдържащи елементи, като арсеник, бисмут, германий, олово, антимон, селен, калай и телур, могат да са много подходящи и изискват оксидиращо разпадане, за да се получат правилни резултати за общото съдържание на елемента. Ето защо обработването с микровълни или изпепеляването под високо налягане при силни оксидиращи условия е препоръчително. Най-голямо внимание следва да се обърне на цялостното и възпроизводимо преминаване на елементите в съответните им хидрати.
Формирането на арсениев хидрат в разтвор на хидрохлорна киселина с NaBH4 зависи от състоянието на оксидация на арсеника (As III: бързо формиране, As V: по-продължителен период за формиране). С цел да се избегне загуба на чувствителност за определянето на As V с инжекционна техника, причинена от времето за реакция в тази система, As V следва да се редуцира до As III след оксидиращото разпадане. Калиев йодид, аскорбинова киселина или цистеин са подходящи за тази цел. Бланки, калибриращи разтвори и пробни разтвори се третират по един и същи начин. Работата с партидна система позволява да се определят и двата арсениеви вида, без да се засегне точността. Поради забавеното формиране на As V-хидрат, калибрирането следва да се извърши посредством интегриране на върховата област. Мястото на инструмента следва да се оптимизира. Течният газ, който трансформира хидрата в атомизатора, е особено важен и следва да бъде проверен.
2.4.6. Специфични изисквания за атомноабсорбционна спектрометрия при студена пара
Студената пара се използва само при живак. Поради изпаряване и загуби при абсорбиране на елементен живак е необходимо специално внимание по време на целия анализ. Зараза от реактиви или околната среда следва старателно да се избягва.
Органични съединения, съдържащи живак, изискват оксидиращо разпадане с цел да се получат верни резултати за цялото съдържание на живак. За разпадането следва да се използват запечатани системи с микровълново обработване или изпепеляване под високо налягане. Необходимо е специално внимание за почистване на оборудването, което е имало допир до живака.
Работата с инжекционната техника е предимство. За по-ниските решаващи нива поглъщането на елементен живак върху златен/платинен абсорбатор, последвано от термично отделяне, е препоръчително. Контактът на абсорбатора или клетката с влага ще наруши измерването и следва да се избегне.
2.4.7. Специфични изисквания за индуктивно събрана плазма при спектрометрия с атомна емисия (ICP-AES)
Индуктивно събраната плазма при спектрометрия с атомна емисия (10) е многоелементен метод, който позволява едновременно измерване на различни елементи. За използването на ICP-AES, пробите следва първо да се преработят с цел да се разградят органичните матрици. Необходимо е да се използват запечатани системи с микровълново обработване или изпепеляване под високо налягане. За целенасочен ICP-AES анализ инструменталната калибрация и елементът или изборът на дължината на вълната имат важна роля. За инструменталната калибрация, в случая на линейни калибриращи закривявания, обикновено е необходимо да се измерят калибриращите разтвори на само два концентрата, защото ICP-AES калибрационните криви са като цяло линейни над четири до шест реда по скалата на концентрация. Калибрирането на ICP-AES системата следва да се извърши с многоелементен стандарт, който следва да се подготви в разтвор, който съдържа същата концентрация на киселина, както и разтворът за измерване. За линейната извивка следва да се проверят концентрациите на елемента.
Изборът на дължини на вълни за измерване на емисията аналити е подходящ за определяне концентрациите на елементите. Когато концентрацията на аналита падне извън работния обхват на емисионна линия, се налага да се използва различна емисионна линия. Първоначално следва да бъде избрана най-чувствителната емисионна линия (без намеса), след което се избира по-нечувствителна линия. Когато се работи при или близо до откриващия лимит, най-чувствителната линия за съответния аналит е обикновено най-добрият избор. Спектрални и фонови намеси причиняват основните затруднения в ICP-AES. Възможните намеси са например просто фоново изместване, склоново фоново изместване, директно спектрално припокриване и сложно фоново преместване. Всяка от тези намеси има собствени причинители и поправки. В зависимост от матриците следва да се приложат корекции на намесата и оптимизирането на опериращите параметри. Някои намеси могат да се избегнат с разреждане или с адаптиране на матриците. С всяка партида анализирани тестови мостри, референтен и подсилени материал, съдържащ известни количества от аналита, както и празен материал следва да се третират, както и тестовите мостри. За тестване за отклонение е необходимо да се провери стандартът, например след 10 проби. Всички реактиви и плазма газът следва да са от възможно най-висока чистота.
2.4.8. Специфични изисквания за индикативно събрана масова спекторметрия (ICP-MS)(11)
Определянето на остатъчни елементи от средна атомна маса, като хром, мед и никел, могат да са обект на силна намеса от други изобарни полиатомични йони. Това може да се заобиколи само ако е налична мощност на резолюция от поне 7 000—8 000. Затрудненията, свързани с MS техниките, включват инструментално отделяне, ефекти на матрицата и молекулярна йонна намеса (m/z < 80). Многобройна вътрешна стандартизация, покриваща същия масов обхват като елементите, които следва да се определят, е наложителна за коригиране на инструментално оттегляне и матрични ефекти.
Необходимо е цялостно разпадане на органично вещество преди измерването на ICP-MS. Както при AAS, след обработване в запечатани съдове изпаримите елементи, например йодин, следва да се превърнат в стабилно оксидно състояние. Най-сериозните резултати са от намеса от молекулярна йонна комбинация на аргон (плазма газ), водород, въглерод, азот и кислород (разтворни киселини, примеси на плазмен газ и атмосферни газове), както и извадковата матрица. Цялостно обработване, фоново измерване, подходящ избор на аналитични маси, понякога свързани с по-ниска обогатеност (по-бедна граница на откриване), и на разтворни киселини, например натриева киселина, са необходими с цел да се избегнат намесите.
За елементи, които предстои да бъдат определени, намесите следва да се изключат от подходящия избор на специфични аналитични маси, включително потвърждаване на изотопно съотношение. Инструментният отговор, като се разглеждат фано-факторите, следва да се провери за всяко измерване чрез използване на вътрешни стандарти.
3. ВАЛИДИРАНЕ
Валидирането следва да демонстрира, че аналитичният метод съответства на критериите, приложими за съответните характеристики за ефективност.
Различни контролни цели изискват различни категории методи. Следната таблица определя коя характеристика за ефективност да бъде потвърдена и за кой вид метод.
Таблица 9
Класификация на аналитични методи по характеристиките на ефективност, които следва да се определят
|
|
Граница на откриване CCβ |
Критична граница CCα |
Истинност/възстановяване |
Прецизност |
Избирателност/специфичност |
Приложимост/износоустойчивост/стабилност |
|
|
Качествени методи |
S |
+ |
– |
– |
– |
+ |
+ |
|
C |
+ |
+ |
– |
– |
+ |
+ |
|
|
Количествени методи |
S |
+ |
– |
– |
+ |
+ |
+ |
|
C |
+ |
+ |
+ |
+ |
+ |
+ |
|
|
S = скрининг методи; C = методи за потвърждение; + = определянето е задължително. |
|||||||
3.1. ПРОЦЕДУРИ ЗА ВАЛИДИРАНЕ
В главата са представени примери и/или справки за валидиращите процедури на аналитични методи. Могат да се използват други подходи, които демонстрират, че аналитичните методи съответстват на критериите за ефективност на характеристиките, при условие че постигат същото ниво и качество на информация.
Валидирането може да се извърши също и като се проведат вътрешнолабораторни проучвания, които са установени в Кодекс алиментариус, ISO или IUPAC (12), или съгласно алтернативните методи, като единични лабораторни проучвания или вътрешно валидиране (13)(14). Тази част се фокусира върху единични лабораторни проучвания (при вътрешно валидиране), като се използва модуларен подход. Този подход се състои от:
|
1. |
набор от общи характеристики на ефективност, независими от използвания валидационен модел, и |
|
2. |
по-специфични зависими от модела процедури, както са описани в таблица 10. |
Таблица 10
Параметри на изпълнение, зависими и независими от модела на валидиране
|
Валидиране |
||
|
Параметри на изпълнение, независими от модела |
Параметри на изпълнение, зависими от модела |
|
|
Общи характеристики за изпълнение (3.1.1) |
Конвенционален подход за валидиране (3.1.2) |
Вътрешен подход за валидиране (3.1.3) |
|
Специфичност |
Възстановяване |
Възстановяване |
|
Вярност |
Повторяемост |
Повторяемост |
|
Износоустойчивост: минимални промени |
Вътрешнолабораторна възпроизводимост |
Вътрешнолабораторна възпроизводимост |
|
Стабилност |
Възпроизводимост |
Възпроизводимост |
|
Критична граница (CCα) |
Критична граница (CCα) |
|
|
Възможност за откриване (CCβ) |
Възможност за откриване (CCβ) |
|
|
Калибриращи извивки |
Калибриращи извивки |
|
|
Износоустойчивост: основни промени |
Износоустойчивост |
|
3.1.1. Характеристики на изпълнение, независими от модела
Независимо от избрания подход за валидиране следните характеристики на изпълнение следва да бъдат определени. С цел да се минимизира големият обем работа, внимателно създаден и статистически стабилен подход може да се използва за комбиниране на експерименти, извършени с цел да се определят различните параметри.
3.1.1.1. Специфичност
За аналитичните методи е важна силата на дискриминация между аналита и близки до него вещества (изомери, метаболити, деградиращи продукти, ендогенни вещества, матрични съставки и т.н.). Необходими са два подхода за проверка за намеси.
Това налага веществата с потенциал за намеса да се изберат и да се анализират съответните празни мостри с цел да се установи наличието на възможни намеси и да се прецени ефектът на намеса:
|
— |
избира се набор от химически свързани съединения (метаболити, деривативи и т.н.) или други вещества, за които има вероятност да се съединят с вещества, които има вероятност да се съберат със съединението, което представлява интерес и което може да е налично в пробите, |
|
— |
анализират се подходящ брой представителни бланкови проби (n ≥ 20) и се проверява за намеси (сигнали, върхове, йонни следи) в района, който представлява интерес, където целевият аналит се очаква да се отдели, |
|
— |
в допълнение се подсилват представителни бланкови проби при съответната концентрация с вещества, които има вероятност да се намесят с идентификацията и/или околичествяването на аналита, |
|
— |
след анализа се преценява дали:
|
3.1.1.2. Вярност
В този параграф е описано определянето на верността (един компонент на акуратност). Верността може да се определи само посредством сертифициран референтен материал (CRM). CRM се използва при възможност и наличие. Процедурата е описана подробно в ISO 5725-4 (5). По-долу е представен пример:
|
— |
анализиране на шестте деления от CРМ в съответствие с тестовите инструкции за метода, |
|
— |
определяне концентрацията на наличния аналит във всяка проба от репликите, |
|
— |
изчисляване на средната стойност, стандартното отклонение и коефициента на вариация (%) за тези концентрации, |
|
— |
изчисляване на верността, като се раздели откритата средна стойност на концентрата на сертифицираната стойност (измерена като концентрат) и се умножи по 100, за да се изрази резултатът като процент. |
Вярност (%) = установена средна концентрация, коригирана при възстановяване × 100/сертифицирана стойност.
Ако не е наличен CRM, вместо вярност възстановяването може да се определи съгласно описанието в точка 4.1.2.1 по-долу.
3.1.1.3. Приложимост/износоустойчивост (незначителни промени)
Такива проучвания използват нарочното въвеждане на незначителни необходими вариации от лабораторията и наблюдението на последствията им.
Предварително проучващите изследвания следва да се извършат, като се изберат фактори от извадковата предварителна обработка, почистване и анализ, който може да повлияе на резултатите от измерването. Такива фактори могат да включват аналитика, източника и възрастта на реактивите, разтворители, стандарти и примерни проби, нивото на загряване, температурата, pH-стойността, както и много други фактори, които могат да възникнат в лабораторията. Тези фактори следва да бъдат модифицирани в поредност на скала, която да съвпада с отклоненията, които обикновено се срещат сред лабораториите:
|
— |
идентифициране на вероятни фактори, които могат да повлияят на резултатите, |
|
— |
леко видоизменяне на всеки фактор, |
|
— |
провеждане на тест за износоустойчивост, като се използва подходът на Юден (15)(16). (Други одобрени методи могат да се използват на този етап. Подходът на Юден обаче поддържа изискваното време и усилия до минимум.) Подходът на Юден е дробен факториален израз. Взаимодействията между различните фактори не може да се определи, |
|
— |
когато се установи, че даден фактор влияе значително върху резултатите от измерването, провеждане на допълнителни експерименти, за да се реши за границите на допустимост на този фактор, |
|
— |
фактори, които значително влияят върху резултатите, следва да се идентифицират ясно в протокола за метода. |
Основната идея не е да се проучват промените поетапно, а да се въведат няколко вариации едновременно. Като пример, нека А, Б, В, Г, Д, Е, Ж обозначават нормалните стойности за няколко различни фактора, които биха могли да повлияят на резултатите, ако нормалните им стойности малко се променят. Нека техните алтернативни стойности да се обозначат със съответните малки букви а, б, в, г, д, е и ж. Резултатът от това е 27, или 128 различни възможни комбинации.
Ако е възможно, следва да се избере подкомбинация от осем такива комбинации, които имат баланс между главни и малки букви (таблица 11). Следва да се направят осем определения, които ще използват комбинация от тези избрани фактори (A—Ж). Резултатите от определенията са показани в таблица 11 по-долу като Т-Щ.
Таблица 11
Експериментален модел за проучване на износоустойчивостта (минимални промени)
|
Фактор със стойност Е |
Комбинации от номера на определения |
|||||||
|
l |
2 |
3 |
4 |
5 |
6 |
7 |
8 |
|
|
A/a |
A |
A |
A |
A |
a |
a |
a |
a |
|
Б/б |
Б |
Б |
б |
б |
Б |
Б |
б |
б |
|
В/в |
В |
в |
В |
в |
В |
в |
В |
в |
|
Г/г |
Г |
Г |
г |
г |
г |
г |
Г |
Г |
|
Д/д |
Д |
д |
Д |
д |
д |
Д |
д |
Д |
|
Е/е |
Е |
е |
е |
Е |
Е |
е |
е |
Е |
|
Ж/ж |
Ж |
ж |
ж |
Ж |
ж |
Ж |
Ж |
ж |
|
Наблюдаван резултат R |
Т |
У |
Ф |
Х |
Ц |
Ч |
Ш |
Щ |
|
Изчисленията са посочени в примерните тествания за износоустойчивост в точка 3.3. |
||||||||
3.1.1.4. Стабилност
Наблюдавано е, че недостатъчната стабилност на аналита или съставките на матрицата в пробата по време на съхранението или анализа могат да доведат до значителни отклонения в резултата от анализа. Освен това стабилността на стандарта за калибриране в разтвора следва да бъде проверена. Обикновено аналитът е добре характеризиран при няколко условия за съхранение. Наблюдението на условията за съхранение ще представлява част от нормалната лабораторна акредитационна система. Когато това не е известно, по-долу са представени примери за това, как може да се определи стабилността.
Стабилност на аналита в разтвора:
|
— |
Подготвят се пресни разтвори на вещество от аналита(ите) и се разтваря, както е посочено в инструкциите за теста, за да се получат достатъчно аликвотни части (например 40) от всеки избран концентрат (около минимално изискваната граница на ефективност за вещества, за които не е създадена допустима граница или около допустимата граница за други вещества. Подготвят се и двата разтвора на аналита, използвани за засилване и приложени в окончателния анализ на разтвора и всеки друг вид разтвор, който може да представлява интерес (например стандарти от производни). |
|
— |
Измерва се съставът на аналита в прясно подготвения разтвор съгласно тестовите инструкции. |
|
— |
Поставят се подходящите обеми в съответните контейнери, поставят се етикети и се съхраняват съгласно схемата: Таблица 12 Схема за определяне стабилността на аналита в разтвор
|
|
— |
Времето за складиране може да се избере като една, две, три и четири седмици или повече, ако е необходимо, например до първите феномени на разпадане се наблюдават по време на идентификацията и околичествяването. Максималното време за складиране и оптималните условия за складиране се записват. |
|
— |
Изчисляването на концентрацията на аналита(ите) във всяка аликвотна част следва да се извършат, като се използва разтворът на прясно приготвения аналит към момента на анализа като 100 % .
Ci= концентрация при време point Cfresh= концентрация на пресен разтвор |
Стабилност на аналит(и) в матрица
|
— |
При всяка възможност е необходимо да се използват получени проби. Когато не е наличен такъв материал, следва да се използва матрица, засилена с аналит. |
|
— |
Когато е налице получен материал, концентрацията в материала следва да се определи, докато материалът е все още пресен. Други аликвотни части от материала могат да се вземат след една, две, четири и 20 седмици и след определяне на концентрациите. Тъканта следва да се съхранява при поне минус 20 °C или, ако е необходимо, и по-малко. |
|
— |
Ако няма наличен получен материал, се взема празен материал и се хомогенизира. Разделя се материалът на пет аликвотни части. Всяка аликвотна част се подсилва с аналита, който е за предпочитане да се подготви в малко количество воден разтвор. Анализира се незабавно една аликвотна част. Останалите аликвотни части се съхраняват при поне минус 20 °C или по-малко, ако е необходимо, и се анализират след една, две, четири и 20 седмици. |
3.1.1.5. Калибриращи криви
Когато се използват калибриращи криви за околичествяване:
|
— |
поне пет нива (включително и нулата) следва да се използват при създаването на кривата, |
|
— |
работният обхват на кривата следва да се опише, |
|
— |
математическата формула на кривата и доброто състояние на данните към кривата следва да се опишат, |
|
— |
следва да се опишат обхватите на кривата за параметрите на кривата. |
Когато е необходима серийна калибрация, базирана на стандартен разтвор, е наложително да се посочат нивата на приемливост за параметрите на калибриращата крива, която може да варира между сериите.
3.1.2. Конвенционални процедури за валидиране
Изчисляването на параметрите съгласно конвенционалните методи налага извършването на няколко индивидуални експеримента. Всяка характеристика на ефективността следва да се определи за всяка основна промяна (виж под приложимост/износоустойчивост по-горе). За многоаналитните методи могат едновременно да се анализират няколко аналита, доколкото съответните вероятни възможности се изключват предварително. По подобен начин могат да се определят няколко характеристики на изпълнение. И така, с цел да се минимизира количеството работа, е препоръчително да се комбинират експериментите възможно най-много (например повторяемост и вътрешнолабораторно възпроизвеждане със специфичност, анализ на празни проби за определяне критичната граница и тестване за специфичност).
3.1.2.1. Възстановяване
Ако няма налично CRM, възстановяването следва да се определи чрез експерименти, като се използва засилена празна матрица, например следната схема:
|
— |
избират се 18 аликвотни части от празен материал и четиридесет и шест аликвотни части всеки при 1, 1,5 и 2 пъти минимум изискван лимит за изпълнение или 0,5, 1 и 1,5 пъти допустимата граница, |
|
— |
анализират се пробите и се изчислява концентратът, наличен във всяка проба, |
|
— |
като се използва равенството по-долу, се изчислява възстановяването на всяка проба, |
|
— |
изчислява се средното възстановяване и CV от шестте резултата на всяко ниво, |
|
— |
% възстановяване = 100 × измерено ниво на съдържание/подсилване. |
Този конвенционален метод за определяне на възстановяването е вариант на стандартния добавъчен метод, описан в точка 3.5, когато:
|
— |
мострата се счита за празна вместо проба за анализиране, |
|
— |
се счита, че добивът (2) и възстановяването (3) са подобни за двете тестови части, |
|
— |
тестовите проби имат една и съща маса и тестовите части изваждат същите обеми, |
|
— |
количеството на калибриращия стандарт, който се добавя към втората (наострената) тестова част, се отбелязва xADD. (xADD = рА.VА), |
|
— |
x1 е измерената стойност за бланката и x2 измерената стойност за втората (наострената) тестова част, |
|
— |
тогава % възстановяване = 100 (x2 - x1)/xADD. |
Когато някое от посочените по-горе условия не е (или се предполага, че не е) постигнато, тогава пълната процедура за определяне на възстановяването посредством стандартния допълващ метод е описана в точка 3.5 и следва да се извърши.
3.1.2.2. Повторяемост
|
— |
Подготвя се набор от проби от идентични матрици, подсилени с аналита да дадат концентрат, равен на 1, 1,5 и 2 пъти минимално изискваната граница на ефективност или 0,5, 1 и 1,5 пъти допустимата граница. |
|
— |
При всяко ниво анализът следва да се извършва с поне шест реплики. |
|
— |
Анализират се пробите. |
|
— |
Изчислява се установеният концентрат във всяка проба. |
|
— |
Откриват се средната концентрация, стандартното отклонение и коефициентът на вариация (%) от подсилените проби. |
|
— |
Тези стъпки се повтарят при поне още два случая. |
|
— |
Изчислява се цялата средна стойност на концентратите и CV за подсилените проби. |
3.1.2.3. Вътрешнолабораторно възпроизвеждане
|
— |
Подготвя се набор от проби от специфицирани тестови материали (идентични или различни матрици), подсилени с аналита(ите) за получаване на концентрати, равни на 1, 1,5 и 2 пъти необходимата минимална граница на изпълнение или 0,5, 1 и 1,5 пъти допустимата граница. |
|
— |
На всяко ниво анализът следва да се извърши с поне шест деления. |
|
— |
Стъпките се повтарят при поне два други случая с различни оператори и различни условия на околната среда, например различни партиди реактиви, разтвори и т.н., различни температури на помещенията, различни инструменти и т.н., ако е възможно. |
|
— |
Анализират се пробите. |
|
— |
Изчислява се концентратът, установен във всяка проба. |
|
— |
Намират се средната концентрация, стандартното отклонение и коефициентът на вариация (%) от подсилените проби. |
3.1.2.4. Възпроизводимост
Когато следва да се потвърди възпроизводимост, лабораториите са длъжни да участват във взаимни проучвания по ISO 5725-2 (5).
3.1.2.5. Критична граница (CCα)
Критичната граница следва да се установи съгласно изискванията за идентификация или идентификация плюс околичествяване съгласно определението в „Критерии за изпълнение и други изисквания за аналитични методи“ (част 2).
При вещества, за които няма определена допустима граница, CCα може да се определи:
|
— |
или чрез процедура на калибриращата крива, или съгласно ISO 11843 (17) (тук посочено като критична стойност на разновидността в чисто състояние). В този случай следва да се използва празен материал, който се подсилва при и над минимално изискваната граница на ефективност в равно отдалечени стъпки. Пробите се анализират. След идентификацията се поставя сигнал срещу добавената концентрация. Кореспондиращата концентрация при пресечната точка „у“ плюс 2,33 пъти стандартното отклонение на вътрешнолабораторната възпроизводимост на пресечната точка се равнява на критичната граница. Това е приложимо само към количествени проби (α = 1 %), |
|
— |
или чрез анализиране на най-малко 20 празни материала на матрица, които да могат да изчислят сигналното към шумовото съотношение при времевия прозорец, при който се очаква аналитът. Три пъти съотношението сигнал към шум може да се използва като критична граница. Това е приложимо за количествени и качествени проби. |
В случая на вещества с установена допустима граница CCα може да се установи:
|
— |
или чрез процедурата по калибриращата крива по ISO 11843 (17) (тук уточнена като критична стойност на нетната разновидност). В този случай следва да се използва празен материал, който се засилва около допустимата граница при равни отстояния. Пробите се анализират. След идентификация сигналът се поставя срещу добавената концентрация. Кореспондиращата концентрация при позволеното ниво плюс 1,64 пъти стандартното отклонение на вътрешнолабораторната възпроизводимост се равнява на критичната граница (α = 5 %), |
|
— |
или чрез анализиране на най-малко 20 празни материала на матрица, подсилени с аналита(ите) при допустимата граница. Концентрацията при допустимата граница плюс 1,64 пъти съответстващото стандартно отклонение се равнява на критичната граница (α = 5 %). |
Виж също член 5 и точка 3.2.
3.1.2.6. Способност за откриване (CCβ)
Способността за откриване следва да се определи според изискванията за скрининг, идентификация или идентификация плюс околичествяване съгласно определението (виж част 2).
В случая на вещества, за които няма установена допустима граница, CCβ може да се определи като:
|
— |
процедурата за калибрираща крива съгласно ISO 11843 (17) (тук упомената като минимална стойност за установяване на променливата на чистото състояние). В този случай следва да се използва представителният празен материал, който се подсилва при и под минимално изискваната граница на ефективност при равни отстояния. Пробите се анализират. След идентификация се поставя сигнал срещу добавената концентрация. Кореспондиращата концентрация при нивото на решение плюс 1,64 пъти стандартното отклонение от вътрешнолабораторната възпроизводимост на средно измереното съдържание и при нивото на решение се равнява на възможността за откриване (β = 5 %), |
|
— |
анализ на поне 20 празни материала за матрица, подсилена с аналита(ите) при нивото на решение. Анализ на пробите и идентифициране на аналитите. Стойността на нивото на решение плюс 1,64 пъти стандартното отклонение от вътрешнолабораторната възпроизводимост от измереното съдържание се равнява на възможността за откриване (β = 5 %), |
|
— |
когато не са налични количествени резултати, възможността за откриване може да се определи, като се проучат подсилените материали при и над критичната граница. В този случай нивото на концентрация, където само ≤ 5 % остават резултати с погрешни оплаквания, се равнява на възможността за откриване на метода. Поради това следва да се направят поне 20 проучвания за поне едно ниво на концентрация с цел да се осигури надеждна база за това определяне. |
В случая на вещества, за които е била определена допустима граница, може да се създаде CCβ:
|
— |
или като се приложи процедурата за калибрираща крива съгласно ISO 11843 (17) (тук упомената като минимална откриваща стойност на променливата на чистото състояние). В този случай следва да се използва представителният празен материал, който е подсилен около допустимата граница на равни отстояния. Анализират се пробите и се идентифицира(т) аналитът(ите). Изчислява се стандартното отклонение от средно измереното съдържание при критичната граница. Кореспондиращата концентрация при стойността на критичната граница плюс 1,64 пъти стандартно отклонение от вътрешнолабораторната възпроизводимост се равнява на възможността за откриване (β = 5 %), |
|
— |
или като се анализират поне 20 празни материала за матрица, подсилени с аналита(ите) при нивото на решение. Стойността на нивото на решение плюс 1,64 пъти кореспондиращото стандартно отклонение се равнява на възможността за откриване (β = 5 %). |
Виж също раздел 3.2.
3.1.2.7. Износоустойчивост (главни промени)
Аналитичният метод следва да се тества при различни експериментални условия, които включват например различните видове, различните матрици или различните условия за проби. Въведените промени следва да са основни. Значимостта на тези промени може да се оцени например като се използва подходът на Юден (15)(16). Всяка характеристика на ефективност следва да се определи за всички основни промени, които са проявили значителен ефект при изпълнението на пробите.
3.1.3. Валидиране съгласно алтернативните модели
Когато се приложат алтернативни процедури за валидиране, подчертаващият модел и стратегията със съответните предпоставки, предположения и формули следва да се заложат във валидиращия протокол или най-малко на тяхно разположение следва да се предоставят референции. В следващата част е представен пример за алтернативен подход. Когато се прилага вътрешен модел на валидиране, характеристиките за изпълнение се определят по начин, който позволява валидиране за основни промени в рамките на същата процедура за валидиране. Това налага създаване на експериментален план за валидиране.
3.1.3.1. Експериментален план
Следва да се изготви експериментален план в зависимост от броя на различните видове и различните фактори за проучване. Оттук като първа стъпка на цялата процедура за валидиране следва да се разглеждат същите популации, които ще бъдат анализирани в лабораторията в бъдеще с цел да се изберат най-важните видове и тези фактори, които могат да повлияят върху резултатите от измерването. Впоследствие нивото на концентрация следва да се избере съгласно нивото от значение.
Пример:
|
— |
могат да се проучат едновременно няколко аналита с аналитичния метод, който се валидира, |
|
— |
идентифицирани са две вариации на ръководния фактор (A и Б). Ръководите фактори формират основата, на която се комбинират факторните нива. Тези ръководни фактори могат да включват фактори като видове или матрица. В този пример ръководният фактор е разнообразен на две нива, например два различни вида (видове A и Б) са разгледани. Като цяло е възможно да се разнообразят ръководните фактори на повече от няколко нива, които само намаляват броя на анализите, които се извършват, |
|
— |
избраните фактори следва да се разнообразят на две нива (посочени като + или -). |
Таблица 13
Примери на фактори, считани за важни за процедура за валидиране
|
Пол на животното |
(фактор 1) |
|
Порода |
(фактор 2) |
|
Транспортни условия |
(фактор 3) |
|
Условия за съхранение |
(фактор 4) |
|
Новост на пробата |
(фактор 5) |
|
Условия за угояване |
(фактор 6) |
|
Различни оператори с различен опит |
(фактор 7) |
Таблица 14
Вероятен експериментален план за посочения по-горе пример
|
Вид |
Фактор 1 |
Фактор 2 |
Фактор 3 |
Фактор 4 |
Фактор 5 |
Фактор 6 |
Фактор 7 |
№ на пробата |
|
A |
+ |
+ |
+ |
+ |
- |
+ |
- |
1 |
|
A |
+ |
+ |
- |
- |
+ |
- |
- |
2 |
|
A |
+ |
- |
+ |
- |
- |
- |
+ |
3 |
|
A |
+ |
- |
- |
+ |
+ |
+ |
+ |
4 |
|
A |
- |
+ |
+ |
- |
+ |
+ |
+ |
5 |
|
A |
- |
+ |
- |
+ |
- |
- |
+ |
6 |
|
A |
- |
- |
+ |
+ |
+ |
- |
- |
7 |
|
A |
- |
- |
- |
- |
- |
+ |
- |
8 |
|
Б |
+ |
+ |
+ |
+ |
+ |
- |
+ |
9 |
|
Б |
+ |
+ |
- |
- |
- |
+ |
+ |
10 |
|
Б |
+ |
- |
+ |
- |
+ |
+ |
- |
11 |
|
Б |
+ |
- |
- |
+ |
- |
- |
- |
12 |
|
Б |
- |
+ |
+ |
- |
- |
- |
- |
13 |
|
Б |
- |
+ |
- |
+ |
+ |
+ |
- |
14 |
|
Б |
- |
- |
+ |
+ |
- |
+ |
+ |
15 |
|
Б |
- |
- |
- |
- |
+ |
- |
+ |
16 |
Тъй като всяка проба (всяка комбинация на факторното ниво) следва да се наостри с четири различни концентрации около нивото на интерес и една празна проба следва да се анализира за всяко ниво, 5 × 16 = 80 анализа следва да се извършат за целия експеримент на валидиране.
За тези 80 резултата от измерването е възможно да се направи изчисление (13)(14).
Възстановяване:
|
— |
повторяемост за ниво на концентрация (sir), |
|
— |
вътрешнолабораторна възпроизводимост за ниво на концентрация (sir), |
|
— |
критична граница (CCα), |
|
— |
възможност за откриване (CCβ), |
|
— |
крива на мощност (β-ниво на грешка срещу концентрация (виж точка 3.1.3.2), |
|
— |
износоустойчивост на главните промени; износоустойчивостта на маловажните промени може да се определи съгласно параграф 3.1.1.3, |
|
— |
16 обвързани с пробата калибриращи криви, |
|
— |
една цялостна калибрираща крива, |
|
— |
интервал на предсказване на цялостната калибрираща крива, |
|
— |
матрично предизвикани отклонения (smat), |
|
— |
отклонения, предизвикани от действието (srun), |
|
— |
ефект от индивидуалните фактори при резултатите от измерването. |
Тези характеристики на изпълнението позволяват цялостната оценка на нивото на изпълнение на метода, тъй като се разглежда не само влиянието на индивидуалните фактори, а също и съответните комбинации от тях. С помощта на този експериментален дизайн е възможно да се реши дали един или друг от избраните фактори следва да бъде изключен от цялостната калибрираща крива, защото значително се отклонява от стандартните отклонения на другите фактори.
3.1.3.2. Крива на мощността
Кривата на мощността предоставя информация относно възможността за откриване на метода в рамките на избрания обхват на концентрация. Това се отнася до риска от β-грешка, когато се приложи разгледаният метод. Кривата на мощността позволява да се изчислят възможностите за откриване за съответните категории (скрининг, потвърждаване) или видове (качествени или количествени) на методи за определени β-грешки (например 5 %).
Фигура 1
Крива на мощността

3.1.3.3. Възпроизводимост
Определянето на възпроизводимостта на метода от концепцията за единните лабораторни проучвания (вътрешно потвърждаване) налага повтарящо се участие в професионалните проучвания съгласно ISO ръководство 43-1 (3) и 43-2 (4). На лабораториите се позволява да изберат собствени методи, при условие че тези методи се използват при рутинни условия. Стандартното отклонение от лабораторията може да се използва за оценка на възпроизводствения метод.
3.2. ГРАФИЧНО ПРЕДСТАВЯНЕ НА РАЗЛИЧНИТЕ АНАЛИТИЧНИ ОГРАНИЧЕНИЯ
Фигура 2
Вещества с неустановена допустима граница

Фигура 3
Вещества с установена допустима граница

3.3. ПРИМЕР ЗА ИЗЧИСЛЕНИЕ ЗА ТЕСТВАНЕ НА ИЗНОСОУСТОЙЧИВОСТТА НА НЕЗНАЧИТЕЛНИТЕ ПРОМЕНИ СЪГЛАСНО ПОДХОДА НА ЮДЕН (16)
Сравнения на средни стойности (A)
|
AA= Σ(Ai)/4 AB= Σ(Bi)/4 AC= Σ(Ci)/4 AD= Σ(Di)/4 AE= Σ(Ei)/4 AF= Σ(Ei)/4 AG= Σ(Gi)/4 Aa= Σ(ai)/4 Ab= Σ(bi)/4 Ac= Σ(ci)/4 Ad= Σ(di)/4 Ae= Σ(ei)/4 Af= Σ(fi)/4 Ag= Σ(gi)/4 |
Сравняват се средните стойности на главните букви (AA до AG) със средните на техните кореспондиращи малки букви(Aa до Ag). Ако даден фактор има ефект, разликата ще бъде значително по-голяма от разликите на другите фактори. Един износоустойчив метод не следва да бъде засегнат от промени, срещащи се почти със сигурност между лабораториите. Ако няма значителна разлика, най-реалистичното измерване на случайната грешка се дава от седемте разлика. |
|
Разлика (Di) |
Разлика на квадрат (Di 2) |
|
Da= A – a = Σ(Ai) – Σ(ai) Db= B – b = Σ(Bi) – Σ(bi) Dc= C – c = Σ(Ci) – Σ(ci) Dd= D – d = Σ(Di) – Σ(di) De= E – e = Σ(Ei) – Σ(ei) Df= F – f = Σ(Fi) – Σ(fi) Dg= G – g = Σ(Gi) – Σ(gi) |
Da 2= стойност a Db 2= стойност b Dc 2= стойност c Dd 2= стойност d De 2= стойност e Df 2= стойност f Dg 2= стойност g |
Стандартно отклонение от разликите Di (SDi):

Когато SDi е значително по-голямо от стандартното отклонение на метода, извършен при вътрешнолабораторни условия на възпроизводимост, както е указано по-горе, предварително може да се направи определено заключение, че всички фактори заедно имат ефект върху резултата, дори ако всеки отделен фактор не показва значително влияние и методът не е значително издръжлив срещу избраните модификации.
3.4. ПРИМЕРИ ЗА ИЗЧИСЛЕНИЕ ЗА ВЪТРЕШНОЛАБОРАТОРНИ ПРОЦЕДУРИ НА ВАЛИДИРАНЕ
Примери за изчисления за протокол за вътрешно валидиране съгласно описаното при валидиране съгласно алтернативните модели (3.1.3) (13) (14).
3.5. ПРИМЕРИ ЗА СТАНДАРТЕН ДОБАВЪЧЕН МЕТОД
Тестовата проба със съдържание T на аналита се разделя на две тестови части 1 и 2 със съответни маси m1 и m2. Тестова част 2 е заострена с обем VA от разтвор на концентрат ρA от аналита. Два екстракта от тестовите части на съответните обеми V1 и V2 се получават след екстракт и етапи на почистване на метода. Възстановяването на аналита се предполага, че е rc. И двата екстракта са проби с метод на измерване на чувствителността b и дават аналитичен отговор от x1 и x2, съответно.
Ако се приеме, че rc и b са същите за аналита в местната проба и в заострената проба, тогава съдържанието на T може да се изчисли като:
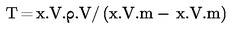
Методът ще позволи определянето на възстановяването rc. Тогава, в допълнение към пробата, описана по-горе, част от екстракта на частта от теста 1 (обем V3) се смесва с известно количество ρB.VB от аналита и се анализира. Аналитичният отговор е x3 и възстановяването е:
Също така е възможно да се изчисли чувствителността b, като:
Всички приложими условия и подробности са описани (18).
4. СПИСЪК НА СЪКРАЩЕНИЯТА
|
AAS |
атомноабсорбционна спектрометрия |
|
AES |
атомно-емисионна спектрометрия |
|
AOAC-I |
Асоциация на официалните аналитични химици — международна |
|
B |
обвързваща практика (имунопроби) |
|
CI |
химическа йонизация |
|
CRM |
сертифициран референтен материал |
|
CV |
коефициент на вариация |
|
2 D |
двуизмерен |
|
DAD |
откриване на диоден ред |
|
DPASV |
диференциална импулсна анодна оголваща волтаметрия |
|
ECD |
откриване на електронно улавяне |
|
EI |
йонизация на електронно влияние |
|
GC |
газова хроматография |
|
HPLC |
високо изпълняваща течна хроматография |
|
HPTLC |
високо изпълняващ тънък слой хроматография |
|
HRMS |
висока резолюция (мас спектрометрия) |
|
ICP-AES |
индуктивно свързана плазма - за атомно-емисионна спектрометрия |
|
ICP-MS |
индуктивно свързана плазма — мас спектрометрия |
|
IR |
инфрачервени |
|
ISO |
Международна организация по стандартизация |
|
LC |
течна хроматография |
|
LR(MS) |
ниска резолюция (мас спектрометрия) |
|
MИГЕ |
минимално изисквана граница на ефективност |
|
MS |
мас спектрометрия |
|
m/z |
съотношение маса/заряд |
|
RF |
сравнително мигриране към разтворимия фронт (TLC) |
|
RSDL |
сравнителни стандартни отклонения от лабораторията |
|
SIM |
избрано йонно наблюдение |
|
TLC |
тънък слой хроматография |
|
UV |
ултравиолетова светлина |
|
VIS |
видима светлина |
5. ИЗПОЛЗВАНА ЛИТЕРАТУРА
|
(1) |
ISO 17025: 1999 General requirement for the competence of calibration and testing laboratories. |
|
(2) |
ISO 3534-1: 1993 Statistical Methods for quality control — Vol. 1 vocabulary and symbols. |
|
(3) |
ISO Guide 43-1: 1997 Proficiency testing by interlaboratory comparisons — Part 1: Development and operation of proficiency testing schemes. |
|
(4) |
ISO Guide 43-2: 1997 Proficiency testing by interlaboratory comparisons — Part 2: Selection and use of proficiency testing schemes by laboratory accreditation bodies. |
|
(5) |
ISO 5725: 1994 Accuracy (trueness and precision) of measurement methods and results — Part 1: General principles and definitions; ISO 5725-2 Part 2: Basic method for the determination of repeatability and reproducibility of a standard measurement method; Part 4: Basic methods for the determination of the trueness of a standard measurement method. |
|
(6) |
ISO 78-2: 1999 Chemistry — Layouts for standards — Part 2: Methods of chemical analysis. |
|
(7) |
W.G de Ruig and J.M Weseman „A new approach to confirmation by infrared spectrometry“ J. Chemometrics 4 (1990) 61-77. |
|
(8) |
See e.g. May, T.W., Brumbaugh, W.G., 1982, Matrix modifier and L'vov platform for elimination of matrix interferences in the analysis of fish tissues for lead by graphite furnace atomic absorption spectrometry: Analytical Chemistry 54(7): 1032-1037 (90353). |
|
(9) |
Applications of Zeeman Graphite Furnace Atomic Absorption Spectrometry in the Chemical Laboratory and in Toxicology, C. Minoia, S. Caroli (Eds.), Pergamon Press (Oxford), 1992, pp. xxvi + 675. |
|
(10) |
Inductively Coupled Plasmas in Analytical Atomic Spectrometry, A. Montaser, D. W. Golighty (Eds.), VCH Publishers, Inc. (New York), 1992. |
|
(11) |
Plasma Source Mass Spectrometry Developments and Applications, G. Holland, S. D. Tanner (Eds.), The Royal Society of Chemistry, 1997, p. 329. |
|
(12) |
IUPAC (1995), Protocol for the design, conduct and interpretation of method-performance studies, Pure & Applied Chem, 67, 331. |
|
(13) |
Jülicher, B., Gowik, P. and Uhlig, S. (1998) Assessment of detection methods in trace analysis by means of a statistically based in-house validation concept. Analyst, 120, 173. |
|
(14) |
Gowik, P., Jülicher, B. and Uhlig, S. (1998) Multi-residue method for non-steroidal anti-inflammatory drugs in plasma using high performance liquid chromatography-photodiode-array detection. Method description and comprehensive in-house validation. J. Chromatogr., 716, 221. |
|
(15) |
OAC-I Peer Verified Methods, Policies and Procedures, 1993, AOAC International, 2200 Wilson Blvd., Suite 400, Arlington, Virginia 22201-3301, USA. |
|
(16) |
W.J. Youden; Steiner, E.H.; „Statistical Manual of the AOAC–Association of Official Analytical Chemists“, AOAC-I, Washington DC: 1975, p. 35 ff. |
|
(17) |
ISO 11843: 1997 Capability of detection — Part 1: Terms and definitions, Part 2: Methodology in the linear calibration case Part 2: Methodology in the linear calibration case. |
|
(18) |
R.W. Stephany & L.A. van Ginkel: „Yield or recovery: a world of difference“. Proceedings Eight Euro Food Chem, Vienna, Austria September 18-20 (1995) Federation of European Chemical Societies, Event 206. ISBN 3-900554-17X, page 2 to 9. |
|
(19) |
Директива 71/354/EИО от 18 октомври 1971 г. за хармонизиране на законодателството на държавите-членки, свързано с единици за измерване, ОВ L 243, 29.10.1971 г., стр. 29). |
|
(20) |
ISO 31-0: 1992 Quantities and units — Part 0: General principles |
(1) За масови фракции, по-ниски от 100 µg/kg, прилагането на равенството на Хоруиц дава неприемливо високи стойности. Поради това CV за концентрации, по-малки от 100 µg/kg, е възможно най-нисък.
(2) Резултат: тази част от масата аналит, съдържаща се в пробата, която присъства в окончателния екстракт.
(3) Възстановяване (тук): тази част от масата аналит, добавена към пробата, която присъства в екстракта. В останалата част от документа се приема, че резултатът и възстановяването са равни и затова се използва само терминът „възстановяване“.
