
|
03/ 34 |
BG |
Официален вестник на Европейския съюз |
7 |
32000L0045
|
L 174/32 |
ОФИЦИАЛЕН ВЕСТНИК НА ЕВРОПЕЙСКИЯ СЪЮЗ |
ДИРЕКТИВА 2000/45/ЕО НА КОМИСИЯТА
от 6 юли 2000 година
относно установяване на общностни методи за анализ с оглед определяне на витамин А, витамин Е и триптофан в храните за животни
(текст от значение за ЕИП)
КОМИСИЯТА НА ЕВРОПЕЙСКИТЕ ОБЩНОСТИ,
като взе предвид Договора за създаването на Европейската общност,
като взе предвид Директива 70/373/ЕИО на Съвета от 20 юли 1970 г. относно въвеждането на общностни методи за вземане на проби и анализ за целите на официалния контрол на храните за животни (1), изменена последно с Акта за присъединяване на Австрия, Финландия и Швеция (2), и по-специално член 2 от нея,
като има предвид, че:
|
(1) |
Директива 70/373/ЕИО предвижда, че официалният контрол храните за животни, който има за цел да установи дали се спазват условията, изисквани по смисъла на законовите, подзаконовите и административните разпоредби, относно качеството и състава на храните за животни, трябва да се извършва съгласно общностните методи за вземане на проби и анализ. |
|
(2) |
Директива 70/524/ЕИО на Съвета от 23 ноември 1970 г. относно добавките при храненето на животни (3), изменена последно с Регламент (ЕО) № 2439/1999 на Комисията (4) предвижда, че съдържанието на витамин А и витамин Е трябва да бъде посочено на етикета, когато тези вещества се прибавят в премиксите и във фуражите. |
|
(3) |
Директива 79/373/ЕИО на Съвета от 2 април 1979 г. относно търговията с комбинирани фуражи (5), последно изменена с Директива 2000/16/ЕО (6), както и Директива 93/74/ЕИО на Съвета от 13 септември 1993 г. относно фуражите със специфични хранителни цели (7), последно изменена с Директива 96/25/ЕО (8), постановяват, че аминокиселините трябва да се вписват при етикетирането на храните за животни. |
|
(4) |
Трябва да се разработят общностни методи за анализ на тези вещества. |
|
(5) |
Предвидените в настоящата Директива мерки са съобразени със становището на Постоянния комитет по храните за животни, |
ПРИЕ НАСТОЯЩАТА ДИРЕКТИВА:
Член 1
Държавите-членки гарантират, че при анализите, извършени с цел официален контрол на съдържанието в храните за животни и в предварителните смеси на витамин А, витамин Е и триптофан, се спазва описаният в приложението метод.
Член 2
Преди 31 август 2000 г. държавите-членки въвеждат в сила законовите, подзаконовите и административните разпоредби, необходими за да се съобразят с настоящата директива. Те незабавно информират Комисията за това.
Държавите-членки прилагат тези разпоредби, считано от 1 септември 2000 г.
Когато държавите-членки приемат тези мерки, в тях се съдържа позоваване на настоящата директива или то се извършва при официалното им публикуване. Условията и редът на позоваване се определят от държавите-членки.
Член 3
Настоящата директива влиза в сила на двадесетия ден след публикуването ѝ в Официален вестник на Европейските общности.
Член 4
Адресати на настоящата директива са държавите-членки.
Съставено в Брюксел на 6 юли 2000 година.
За Комисията
David BYRNE
Член на Комисията
(1) ОВ L 170, 3.8.1970 г., стр. 2.
(2) ОВ L 241, 29.8.1994 г., стр. 1.
(3) ОВ L 270, 14.12.1970 г., стр. 1.
(4) ОВ L 297, 18.11.1999 г., стр. 8.
(5) ОВ L 86, 6.4.1979 г., стр. 30.
(6) ОВ L 105, 3.5.2000 г., стр. 36.
(7) ОВ L 237, 22.9.1993 г., стр. 23.
(8) ОВ L 125, 23.5.1996 г., стр. 35.
ПРИЛОЖЕНИЕ
ЧАСТ А
ДОЗИРАНЕ НА ВИТАМИН А
1. Цел и обхват
Методът дава възможност да се определи съдържанието на витамин А (ретинол) в храните за животни и предварителните смеси. Витамин А включва ретинол-all-trans и неговите цис-изомери(cis), които се определят по този метод. Съдържанието на витамин А се изразява в международни единици (UI) на кг. Една международна единица (UI) отговаря на активността на 0,300 µg витамин А алкохол-all-trans или на 0,344 µg витамин А ацетат all-trans, или на 0,550 µg витамин А палмитат all-trans.
Прагът на значимост е 2 000 UI витамин А/кг.
2. Принцип
Пробата се хидролизира с разтвор от етанолов калиев хидроксид и витамин А се извлича в петролен етер. Разтворителят се елиминира чрез изпаряване; утайката се разтваря в метанол (метилов спирт) и, ако е необходимо, се разрежда до изискваната концентрация. Съдържанието на витамин А се определя чрез течна високорезултатна хроматография в инверсна (обратна) фаза (CLHP-PI) с помощта на УВ или флуорометричен датчик. Параметрите на хроматографията се подбират така че да се получи отделяне между витамин А алкохол- all-trans и неговите цис-изомери.
3. Реактиви
3.1. Етанол (етилов спирт), σ = 96 %
3.2. Петролен етер, интервал на температура на кипене 40 °С—60 °С
3.3. Метанол (метилов спирт)
3.4. Разтвор на калиев хидроксид, β = 50 г/100 мл
3.5. Разтвор на натриев аскорбат, β = 10 г/100 мл (виж забележката в точка 7.7.)
Натриев сулфид, Na2S · x H2O (x = 7-9)
3.6.1. Разтвор на натриев сулфид, с = 0,5 мол/л в глицерин, β = 120 г/л (за х = 9) (виж. забележката в точка 7.8)
3.7. Разтвор на фенолфталеин, β = 2 г/100 мл в етанол (етилов спирт) (3.1)
3.8. Пропанол-2
3.9. Мобилна фаза за CLHP: смес от метанол (метилов спирт) (3.3) и вода, например: 980 + 20 (v + v). Точните пропорции се определят от характеристиките на използваната колона.
3.10. Азот, свободен от кислород.
Витамин А ацетат all-trans, с изключителна чистота, гарантирана активност, например 2,80 ха 10 на 6 UI/г
3.11.1. Матерен разтвор на витамин А ацетат all-trans ce претегля приблизително до 0,1 мг 50 мг витамин А ацетат (3.11) в колба, градуирана на 100 мл. Разтваря ce в пропанол-2 (3.8) и ce допълва до отметката със същия разтворител. Номиналната концентрация на този разтвор е 1 400 UI витамин А на мл. Точното съдържание се определя по точка 5.6.3.1.
Витамин А all-trans, с изключителна чистота, гарантирана активност, например 1,80 × 10 на 6 UI/г.
3.12.1. Матерен разтвор на витамин А ацпалмитат all-trans ce претегля приблизително до 0,1 мг, 80 мг витамин А палмитат (3.12) в колба, градуирана на 100 мл. Разтваряте в пропанол-2 (3.8) и допълвате до отметката със същия разтворител. Номиналната концентрация на този разтвор е 1 400 UI витамин А на мл. Точното съдържание се определя по точка. 5.6.3.2.
3.13. ВНТ(ди-терт/tert/-бутил-2,6-метил-4-фенол) (виж забележката в точка. 7.5)
4. Апаратура
4.1. Ротативен вакуумен изпарител
Непрозрачни стъклени лабораторни съдове
4.2.1. Сферични или конусовидни колби, 500 мл, с шлифовано стъклено гърло
4.2.2. Градуирани колби с шлифована стъклена запушалка, тясно гърло, 10, 25, 100 и 500 мл
4.2.3. Конусовидни декантационни колби, 1 000 мл, с шлифована стъклена запушалка
4.2.4. Крушообразни колби, 250 мл, с шлифовано стъклено гърло
4.3. Охладител (хладилник) Allihn, полезна дължина 300 мм, с шлифовано стъклено съединение за захранваща тръба с газ
4.4. Гофрирана филтрова хартия за сепарация на фазите, диаметър 185 мм (например: Schleicher & Schuell 597 HY 1/2)
Комплект CLHP с инжекторна система
4.5.1. Колона за течна хроматография, 250 мм × 4 мм, С18, частици 5 или 10 µm, или еквивалент (коефициент на параметри: само един пик за всички ретинолови изомери при условия CLHP)
4.5.2. УВ- или флуорометричен датчик с променлива дължина на вълната
4.6. Спектрофотометър с 10-милиметрови кварцови (фото)елементи
4.7. Вана за водна баня с магнитен миксер
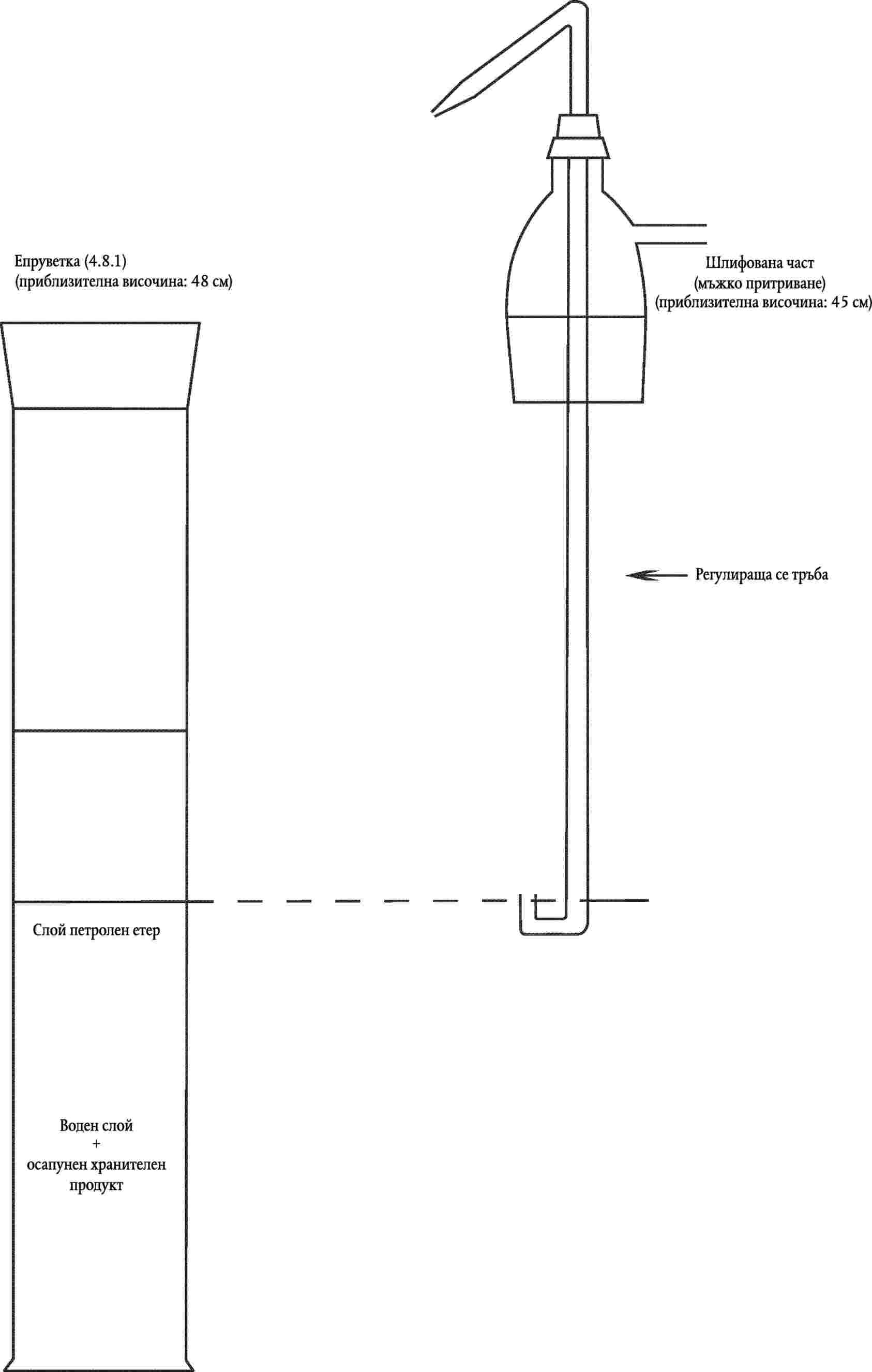
Екстрактор (виж фиг.1), състоящ се от:
4.8.1. Епруветка с обем 1 л, с шлифовани стъклени гърло и запушалка
4.8.2. Шлифована част (мъжко притриване), снабдена със странична тръбичка и регулираща се тръба по средата. Долната част на тръбата е с форма на U и с дюза в противоположния край, така че горният течен слой в епруветката да може да се прехвърли в декантационната колба
5. Процедура
Забележка: Витамин А е чувствителен на светлина (УВ) и податлив на окисляване. Всички манипулации трябва да се извършват на тъмно (в непрозрачно стъкло или покрито с алуминиево фолио) и в отсъствие на кислород (елиминира се с азот). По време на екстракцията въздухът над течността трябва да се замени с азот (да се избягва прекалено голямото налягане, като от време на време запушалката трябва да се отваря).
5.1. Обработка на пробата
Пробата се смила, така че да може да премине през сито с отвори 1 мм, като се избягва нагряване. Смилането трябва да стане непосредствено преди претеглянето и осапуняването, в противен случай съществува риск от загуби на витамин А.
5.2. Осапуняване
Според тегловото съдържание на витамин А се претегля с точност до 0,01 г, от 2 до 25 г от пробата в сферична или конусовидна колба от 500 мл (4.2.1.). Прибавя ce последователно при постоянно размесване 130 мл етанол (етилов спирт) (3.1), около 100 мг ВНТ (3.13), 2 мл разтвор на натриев аскорбат (3.5) и 2 мл разтвор на натриев сулфид (3.6). Монтира ce охладителя (хладилника) (4.3) на колбата и ce потапя във вана за водна баня с магнитен миксер (4.7). Нагрявате до кипене и оставяте да се отвърне за 5 мин. Прибавя ce след това 25 мл разтвор на калиев хидроксид (3.4) през охладителя (4.3) и ce оставя отново да се отвърне за 25 мин., като ce бърка при слаб приток на азот. Изплаква ce охладителя с около 20 мл вода и ce оставя съдържанието на колбата да изстине до околната температура.
5.3. Екстракция
Прехвърля ce чрез декантация количеството осапуняващ разтвор, като ce изплаква с целия обем от 250 мл вода, в декантационната колба от 1 000 мл (4.2.3) или в екстрактора (4.8). Изплаква ce последователно колбата за осапуняване с 25 мл етилов спирт (3.1) и 100 мл петролен етер (3.2) и ce прехвърля течността за изплакване в декантационната колба или в екстрактора. Пропорцията на водата и на етиловия спирт при комбинираните разтвори трябва да е около 2:1. Разклаща ce силно в продължение на 2 мин. и ce оставя в покой за 2 мин.
5.3.1. Екстракция с помощта на декантационна колба (4.2.3)
Когато слоевете се отделят (виж забележката в точка 7.3), прехвърля ce слоя петролен етер в друга декантационна колба (4.2.3). Повтаря ce два пъти операцията с 100 мл петролен етер (3.2), а след това два пъти с 50 мл петролен етер (3.2).
Промива ce два пъти и ce комбинира екстракти в декантационната колба като ce разклаща леко (за да се избегне образуването на емулсия) с части по 100 мл вода и отново ce разклаща с нови части по 100 мл вода, докато последната стане безцветна след добавянето на разтвор на фенолфталеин (3.7) (обикновено са достатъчни четири промивания). Филтрира ce промития екстракт със сух гофриран филтър за разделяне на фазите (4.4), за да се отстрани остатъчната вода, и ce прехвърля в градуирана 500-милилитрова колба (4.2.2). Изплаква ce декантационната колба и филтъра с 50 мл петролен етер (3.2), долива ce до делението с петролен етер (3.2) и ce разбърква добре.
5.3.2. Екстракция с помощта на екстрактор (4.8)
Когато слоевете се разделят (виж забележката в точка 7.3), ce поставя отново запушалката на епруветката (4.8.1) в шлифованата част (мъжко притриване) (4.8.2) и ce поставя долния край във форма на U на регулиращата се тръба, така че тя да се окаже точно над нивото на граничната повърхност. През страничната тръбичка ce подава налягане чрез азота, прехвърля ce горния слой петролен етер в декантационна колба от 1 000 мл (4.2.3). Прибавя ce 100 мл петролен етер (3.2) в стъкления цилиндър, затваря ce запушалката и ce разклаща силно. Оставят се слоевете да се разделят и ce прехвърля горния слой в декантационната колба, както е посочено по-горе. Повтаря ce процедурата на екстракция с нови 100 мл петролен етер (3.2), след това още два пъти с 50 мл петролен етер (3.2) и ce прибавя слоевете петролен етер в декантационната колба.
Промиват ce комбинираните екстракти от петролен етер съгласно процедурата, описана в т. 5.3.1, и ce действа както е посочено в тази точка.
5.4. Подготовка на пробния разтвор за CLHP
Накапва ce с пипета аликвотна част от разтвора на петролен етер (от 5.3.1 или 5.3.2) в крушообразна колба от 250 мл (4.2.4). Оставя ce разтворителят да се изпари почти изцяло в ротативния изпарител (4.1) при понижено налягане, при температура на ваната не по-висока от 40 °С. Възстановява ce атмосферното налягане, като ce пусне азот (3.10), и ce изважда колбата от изпарителя. Отстранявате останалото количество разтворител в поток азот (3.10) и незабавно ce разтваря утайката в дадения обем (10—100 мл) метанол (метилов спирт) (3.3) (концентрацията на витамин А трябва да е в порядъка от 5 UI/мл до 30 UI/мл).
Дозиране чрез CLHP
Витамин А се отделя на колона С18 в инверсна (обратна) фаза (4.5.1) и концентрацията се измерва с помощта на УВ-датчик (325 nm) или флуорометричен датчик (възбуждане: 325 nm; емисия: 475 nm) (4.5.2)
Впръсква се аликвотна част (например 20 µl) от получения метанолов разтвор (виж точка 5.4) и елюирате с мобилната фаза (3.9). Изчислява се средната височина на пика (повърхност) на няколко инжектирания със същия пробен разтвор и средните височини на пиковете (провърхност) на няколко инжектирания с еталонните разтвори (5.6.2).
Условия CLHP
За ориентиране се предлагат следните условия; и други условия могат да се прилагат, стига да дават еквивалентни резултати:
|
Колона за течна хроматография (4.5.1): |
250ммх4 мм, С18, частици от 5 или 10 µm, или еквивалент |
|
Мобилна фаза (3.9): |
смес от метилов спирт(3.3) и вода, например 980 + 20(v + v) |
|
Дебит: |
1—2 мл/мин. |
|
Датчик (4.5.2): |
УВ-детектор (325 nm) или флуо рометричен датчик (възбужда-не: 325 nm/емисия: 475 nm) |
5.6. Изготвяне на сравнителен разтвор
5.6.1. Изготвяне на работните сравнителни (стандартни) разтвори
Капва ce с пипета 20 мл от матерния разтвор на витамин А ацетат (3.11.1) или 20 мл от матерния разтвор на витамин А палмитат (3.12.1) в колба с плоско или конусообразно дъно с обем 500 мл (4.2.1) и хидролизирате както е описано в точка 5.2, но без да ce прибавя ВНТ. Пристъпва ce след това към екстракция с петролен етер (3.2) (виж точка 5.3) и се допълва до 500 мл с петролен етер (3.2). Оставя ce 100 мл от този екстракт да се изпари почти напълно на ротативния изпарител (виж. 5.4), отстранява ce останалото количество разтворител в поток от азот (3.10) и отново ce разтваря утайката в 10,0 мл метанол (метилов спирт) (3.3). Номиналната концентрация на този разтвор е 560 UI витамин А на мл. Точното съдържание трябва да се определи съгласно точка 5.6.3.3. Работният сравнителен разтвор се изготвя за всяка отделна манипулация.
Капва ce 2,0 мл от работния сравнителен разтвор в градуирана колба от 20 мл, допълва ce до делението с метанол (3.3) и ce размесва. Номиналната концентрация на този разреден работен сравнителен разтвор е 56 UI витамин А на мл.
5.6.2. Изготвяне на сравнителните разтвори и начертаване на градуираната крива
Прехвърля ce 1,0, 2,0, 5,0 и 10, 0 мл от разредения работен сравнителен разтвор в няколко градуирани на 20 мл колби, допълва ce до съответното деление с метанол (3.3) и ce смесва. Номиналните концентрации на тези разтвори са 2,8, 5,6, 14,0 и 28,0 UI витамин А на мл.
Впръсква ce няколко пъти 20 мл от всеки сравнителен разтвор и ce определя средните височини на пиковете (повърхности). Според средните височини на пиковете (повърхности), начертава ce градуирана крива, като се вземат предвид резултатите от УВ-контрола (5.6.3.3).
5.6.3. Калибриране чрез УВ на сравнителните разтвори
5.6.3.1. Матерен разтвор на витамин А ацетат
Капва сe пипета 2,0 мл от материния разтвор на витамин А ацетат (3.11.1) в градуирана колба от 50 мл (4.2.2) и ce допълва до съответното деление с пропанол-2 (3.8). Номиналната концентрация на този разтвор е 56 UI витамин А на мл. Капва ce с пипета 3,0 мл от този разреден разтвор на витамин А ацетат в градуирана колба от 25 мл и допълвате до съответното деление с пропанол-2 (3.8). Номиналната концентрация на този разтвор е 6,72 UI витамин А на мл. Измерва ce УВ-спектъра на разтвора по отношение на пропанол-2 (3.8) със спектрометъра (4.6) между 300 nm и 400 nm. Максималното затихване трябва да е между 325 nm и 327 nm.
Изчисление на съдържанието ва витамин А:
5.6.3.2. Матерен разтвор на витамин А палмитат
Капва ce с пипета 2,0 мл от матерния разтвор на витамин А палмитат (3.12.1) в градуирана колба от 50 мл (4.2.2) и ce допълва до съответното деление с пропанол-2 (3.8). Номиналната концентрация на този разтвор е 56 UI витамин А на мл. Капва ce с пипета 3,0 мл от този разреден разтвор на витамин А палмитат в градуирана колба от 25 мл и ce допълва до съответното деление с пропанол-2 (3.8). Номиналната концентрация на разтвора е 6,72 UI витамин А на мл. Измерва ce УВ-спектъра на разтвора по отношение на пропанол-2 (3.8) със спектрометъра (4.6) между 300 nm и 400 nm. Максималното затихване трябва да е между 325 nm и 327 nm.
Изчисление на съдържанието на витамин А:

5.6.3.3. Работен сравнителен разтвор на витамин А
Капва ce с пипета 3,0 мл неразреден работен сравнителен разтвор на витамин А, изготвен по точка 5.6.1, в градуирана колба от 50 мл (4.2.2) и ce допълва до съответното деление с пропанол-2 (3.8). Капва ce с пипета 5,0 мл от този разтвор в градуирана колба от 25 мл и ce допълва до съответното деление с пропанол-2 (3.8). Измерва ce УВ-спектъра на разтвора по отношение на пропанол-2 (3.8) със спектрометъра (4.6) между 300 nm и 400 nm. Максималното затихване трябва да е между 325 nm и 327 nm.
Изчисление на съдържанието на витамин А:

6. Изчисление на резултатите
Като изхождате от средната височина (повърхност) на пиковете на витамин А на пробния разтвор, ce определя концентрацията на този разтвор в UI/мл по показатели на градуираната крива (5.6.2).
Съдържанието w на витамин А в пробата, изразено в IU/кг, е дадено от следната формула:

в която:
|
β |
= |
съдържанието на витамин А в пробния разтвор (5.4) в IU/мл |
|
V1 |
= |
обема на пробния разтвор (5.4) в мл |
|
V2 |
= |
обема на взетата аликвотна част по т. 5.4 в мл |
|
m |
= |
масата на опитния материал в г |
7. Забележки
7.1. При пробите с ниско съдържание на витамин А би било полезно да се обединят екстрактите в петролен етер, получени от две осапунявания (претеглено количество: 25 г), в един пробен разтвор за дозиране чрез CLHP.
7.2. Взетата за анализ проба не трябва да съдържа повече от 2 г мастни вещества.
7.3. Ако няма разделяне на фазите,ce прибавя около 10 мл етанол (етилов спирт) (3.1), за да елиминирате емулсията.
7.4. С рибено масло и други мастни вещества времето на осапуняване трябва да достигне 45—60 мин.
7.5. ВНТ може да се замени с хидрохинон (n-диоксибензол).
7.6. Като се използва колоната в директна фаза, ретиноловите изомери могат да се разделят.
7.7. Разтворът на натриев аскорбат може да се замени с около 150 мг аскорбинова киселина.
7.8. Разтворът на натриев сулфид може да се замени с около 50 мг EDTA.
8. Повторяемост
Разликата в резултатите от двете паралелни дозирания на една и съща проба не трябва да надхвърля 15 % по отношение на върховия резултат.
9 Резултати от вътрешнолабораторно проучване (1)
|
|
Предварител-на смес |
Минерален концентрат |
Протеинов хранителен продукт |
Храна за малки прасета |
|
|
L |
13 |
12 |
13 |
12 |
13 |
|
n |
48 |
45 |
47 |
46 |
49 |
|
средно [UI/ug] |
17,02 × 106 |
1,21 × 106 |
537 100 |
151 800 |
18 070 |
|
sr [UI/ug] |
0,51 × 106 |
0,039 × 106 |
22 080 |
12 280 |
682 |
|
r [UI/ug] |
1,43 × 106 |
0,109 × 106 |
61 824 |
34 384 |
1 910 |
|
CVr [%] |
3,0 |
3,5 |
4,1 |
8,1 |
3,8 |
|
SR [UI/ug] |
1,36 × 106 |
0,069 × 106 |
46 300 |
23 060 |
3 614 |
|
R [UI/ug] |
3,81 × 106 |
0,193 × 106 |
129 568 |
64 568 |
10 119 |
|
CVR [%] |
8,0 |
6,2 |
8,6 |
15 |
20 |
Фигура 1: Екстрактор (4.8)

ЧАСТ Б
ДОЗИРАНЕ НА ВИТАМИН Е
1. Цел и обхват
Методът дава възможност да се определи съдържанието на витамин Е в храните за животни и предварителните смеси. Съдържанието на витамин Е се изразява в мг DL-α-токоферолов ацетат на кг. 1 мг DL-α-токоферолов ацетат отговаря на 0,91 мг DL-α-токоферол (витамин Е).
Прагът на значимост е 2 мг витамин Е/кг.
2. Принцип
Пробата се хидролизира с разтвор от етанолов калиев хидроксид и витамин Е се извлича в петролен етер. Разтворителят се елиминира чрез изпаряване; утайката се разтваря в метанол (метилов спирт) и, ако е необходимо, се разрежда до изискваната концентрация. Съдържанието на витамин Е се определя чрез течна високорезултатна хроматография в инверсна (обратна) фаза (CLHP-PI) с помощта на УВ- или флуорометричен датчик.
3. Реактиви
3.1. Етанол (етилов спирт), σ = 96 %
3.2. Петролен етер, интервал на температура на кипене 40 °С—60 °С
3.3. Метанол (метилов спирт)
3.4. Разтвор на калиев хидроксид, β = 50 г/100 мл
3.5. Разтвор на натриев аскорбат, β = 10 г/100 мл (виж. забележката в тока 7.7.)
Натриев сулфид, Na2 S xH2O (х = 7-9)
3.6.1. Разтвор на натриев сулфид, с = 0,5 мол/л в глицерин, β = 120 г/л (за х = 9 (виж забележката в точка 7.8)
3.7. Разтвор на фенолфталеин, β = 2 г/100 мл в етанол (етилов спирт) (3.1)
3.8. Мобилна фаза за CLHP: смес от метанол (метилов спирт) (3.3) и вода, например: 980 + 20 (v + v). Точните пропорции се определят от характеристиките на използваната колона.
3.9. Азот, свободен от кислород.
DL-α-токоферолов ацетат, извънредно чист, с гарантирана активност
3.10.1. Матерен разтвор на DL-α-токоферолов ацетат ce претегля приблизително до 0,1 мг, 100 мг DL-α-токоферолов ацетат (3.10) в колба, градуирана на 100 мл. Разтваря ce в етанол (3.1) и ce допълвавa до делението със същия разтворител. 1 мл от този разтвор съдържа 1 мг DL-α-токоферолов ацетат (за УВ-контрол: виж точка 5.6.1.3; за стабилизация: виж. забележката в тока 7.4)
DL-α-токоферол, с изключителна чистота, с гарантирана активност
3.11.1. Претегля ce приблизително до 0,1 мг 100 мг DL-α-токоферолов ацетат (3.10) в колба, градуирана на 100 мл. Разтваря ce в етанол (3.1) и ce допълва до делението със същия разтворител. 1 мл от този разтвор съдържа 1 мг DL-α-токоферол (за УВ-контрол: виж точка 5.6.2.3; за стабилизация: виж забележката в точка 7.4)
3.12. ВНТ(ди-терт/tert/-бутил-2,6-метил-4-фенол) (виж забележката в точка 7.5)
4. Апаратура
4.1. Ротативен вакуумен изпарител
Непрозрачни стъклени лабораторни съдове
4.2.1. Сферични или конусовидни колби, 500 мл, с шлифовано стъклено гърло
4.2.2. Градуирани колби с шлифована стъклена запушалка, тясно гърло, 10, 25, 100 и 500 мл
4.2.3. Конусовидни декантационна колби, 1 000 мл, с шлифована стъклена запушалка
4.2.4. Крушообразни колби, 250 мл, с шлифовано стъклено гърло
4.3. Охладител (хладилник) Allihn, полезна дължина 300 мм, с шлифовано стъклено съединение за захранваща тръба с газ
4.4. Гофрирана филтрова хартия за сепарация на фазите, диаметър 185 мм (например: Schleicher & Schuell 597 HY 1/2)
Комплект CLHP с инжекторна система
4.5.1. Колона за течна хроматография, 250 мм х 4 мм, С18, частици 5 или 10 µm, или еквивалент
4.5.2. УВ- или флуорометричен датчик с променлива дължина на вълната
4.6. Спектрофотометър с 10-милиметрови кварцови (фото)елементи
4.7. Вана за водна баня с магнитен миксер
Екстрактор (виж фиг.1), състоящ се от:
4.8.1. Епруветка с обем 1 л, с шлифовани стъклени гърло и запушалка
4.8.2. Шлифована част (мъжко притриване), снабдена със странична тръбичка и регулираща се тръба по средата. Долната част на тръбата е с форма на U и с дюза в противоположния край, така че горният течен слой в епруветката да може да се прехвърли в декантационната колба
5. Процедура
Забележка: Витамин Е е чувствителен на светлина (УВ) и податлив на окисляване. Всички манипулации трябва да се извършват на тъмно (в непрозрачно стъкло или покрито с алуминиево фолио) и в отсъствие на кислород (елиминира се с азот). По време на екстракцията въздухът над течността трябва да се замени с азот (да се избягва прекалено голямото налягане, като от време на време запушалката трябва да се отваря).
5.1. Обработка на пробата
Пробата се смила, така че да може да премине през сито с отвори 1 мм, като се избягва нагряване. Смилането трябва да стане непосредствено преди претеглянето и осапуняването, в противен случай съществува риск от загуби на витамин Е.
5.2. Осапуняване
Според тегловото съдържание на витамин Е, ce претегля с точност до 0,01 г, от 2 до 25 г от пробата в сферична или конусовидна колба от 500 мл (4.2.1.). Прибавя ce последователно при постоянно размесване 130 мл етанол (етилов спирт) (3.1), около 100 мг ВНТ (3.12), 2 мл разтвор на натриев аскорбат (3.5) и 2 мл разтвор на натриев сулфид (3.6). Монтира ce охладителя (4.3) на колбата и ce потапя във вана за водна баня с магнитен миксер (4.7). Нагрява ce до кипене и ce оставя да се отвърне за 5 мин. Прибавя ce след това 25 мл разтвор на калиев хидроксид (3.4) през охладителя (4.3) и ce оставя отново да се отвърне за 25 мин., като бъркате при слаб приток на азот. Изплаква ce охладителя с около 20 мл вода и ce оставя съдържанието на колбата да изстине до околната температура.
5.3. Екстракция
Прехвърля ce чрез декантация осапуняващия разтвор,като ce изплаква с целия обем от 250 мл вода в декантационната колба от 1 000 мл (4.2.3) или в екстрактора (4.8). Изплаква ce последователно колбата за осапуняване с 25 мл етилов спирт (3.1) и 100 мл петролен етер (3.2) и ce прехвърля течността за изплакване в декантационната колба или в екстрактора. Пропорцията на водата и на етиловия спирт при комбинираните разтвори трябва да е около 2:1. Раклащва ce силно в продължение на 2 мин. и ce оставя в покой за 2 мин.
5.3.1. Екстракция с декантационна колба (4.2.3)
Когато слоевете се отделят (виж забележката в точка 7.3), ce прехвърля слоя петролен етер в друга декантационна колба (4.2.3). Повтаря ce два пъти операцията с 100 мл петролен етер (3.2), а след това два пъти с 50 мл петролен етер (3.2).
Промива ce два пъти комбинираните екстракти в декантационната колба като разклащате леко (за да се избегне образуването на емулсия) с части по 100 мл вода и отново ce разклащва с нови части по 100 мл вода, докато последната стане безцветна след добавянето на разтвор на фенолфталеин (3.7) (обикновено са достатъчни четири промивания). Филтрира ce промития екстракт със сух гофриран филтър за отделяне на фазите (4.4), за да се отстрани остатъчната вода, и ce прехвърля в градуирана 500-милилитрова колба (4.2.2). Изплаква ce декантационната колба и филтъра с 50 мл петролен етер (3.2), доливате до делението с петролен етер (3.2) и ce разбърква добре.
5.3.2. Екстракция с помощта на екстрактор (4.8)
Когато слоевете се отделят (виж забележката в точка 7.3), ce поставя отново запушалката на епруветката (4.8.1) в шлифованата част (мъжко притриване) (4.8.2) и ce поставя долния край във форма на U на регулиращата се тръба, така че тя да се окаже точно над нивото на граничната повърхност. През страничната тръбичка ce подава налягане чрез азота, прехвърля ce горния слой петролен етер в декантационна колба от 1 000 мл (4.2.3). Прибавя ce 100 мл петролен етер (3.2) в стъкления цилиндър, затваря ce запушалката и ce разклаща силно. Оставят ce слоевете да се отделят и ce прехвърля горния слой в декантационната колба, както е посочено по-горе. Повтаря ce процедурата на екстракция с нови 100 мл петролен етер (3.2), след това още два пъти с 50 мл петролен етер (3.2) и ce прибавят слоевете петролен етер в декантационната колба.
Промива ce комбинираните екстракти от петролен етер съгласно процедурата, описана в точка 5.3.1 и ce действа както е посочено в тази точка.
5.4. Подготовка на пробния разтвор за CLHP
Накапва ce с пипета аликвотна част от разтвора на петролен етер (от 5.3.1 или 5.3.2) в крушообразна колба от 250 мл (4.2.4). Оставя ce разтворителят да се изпари почти изцяло в ротативния изпарител (4.1) при понижено налягане, при температура на ваната не по-висока от 40 °С. Възстановява ce атмосферното налягане, като ce пусне азот (3.9) и ce изважда колбата от изпарителя. Отстранява ce останалото количество разтворител в поток азот (3.9) и незабавно разтваря ce утайката в дадения обем (10—100 мл) метанол (метилов спирт) (3.3) (концентрацията на DL-α-токоферол трябва да е в порядъка от 5 µg/мл до 30 µg (мл).
Дозиране чрез CLHP
Витамин Е се отделя на колона С18 в инверсна (обратна) фаза (4.5.1) и концентрацията се измерва с помощта на флуорометричен датчик (възбуждане: 295 nm; емисия: 330 nm) (4.5.2)
Впръсква ce аликвотна част (например 20 µl) от получения метанолов разтвор (виж точка 5.4) и ce елюира с мобилната фаза (3.8). Изчислява ce средните височини на пика (повърхности) на няколко впръсквания със същия пробен разтвор и средните височини на пиковете (провърхност) на няколко впръсквания с еталонните разтвори (5.6.2).
Условия CLHP
За ориентиране се предлагат следните условия; и други условия могат да се прилагат, стига да дават еквивалентни резултати:
|
Колона за течна хроматография (4.5.1): |
250мм х 4 мм, С18, частици от 5 или 10 µm, или еквивалент |
|
Мобилна фаза (3. 8): |
смес от метилов спирт(3.3) и вода, например 980 + 20(v + v) |
|
Дебит: |
1-2 мл/мин. |
|
Датчик (4.5.2): |
флуорометричен датчик (възбуждане: 295 nm/емисия: 330 nm) или УВ-датчик (292 nm) |
5.6. Изготвяне на сравнитеилен (стандартен) разтвор (DL-α-токоферолон ацетат или DL-α-токоферол)
5.6.1. Изготвяне на сравнителен разтвор на DL-α-токоферол
5.6.1.1. Изготвяне на работен сравнителен разтвор
Капва ce с пипета 25 мл от матерния разтвор на DL-α-токоферолов ацетат (3.10.1) в колба с плоско или конусообразно дъно с обем 500 мл (4.2.1) и хидролизира ce както е описано в точка 5.2. Пристъпва ce след това към екстракция с петролен етер (3.2) (виж точка 5.3) и ce допълва до 500 мл с петролен етер (3.2). Оставя ce 25 мл от този екстракт да се изпари почти напълно на ротативния изпарител (виж 5.4), отстранява ce останалото количество разтворител в поток от азот (3.9) и отново ce разтваря утайката в 25,0 мл метанол (метилов спирт) (3.3). Номиналната концентрация на този разтвор е 45,5 µg на DL-α-токоферол на ml, равно на 50 мg. DL- a- токофенол ацетат на ml. Работният сравнителен разтвор се изготвя за всяка отделна манипулация.
5.6.1.2. Изготвяне на сравнителните разтвори и начертаване на градуираната крива
Прехвърля ce 1,0, 2,0, 4,0 и 10, 0 мл от работния сравнителен разтвор в няколко градуирани на 20 мл колби, допълва ce до съответното деление с метанол (3.3) и ce смесва. Номиналните концентрации на тези разтвори са 2,5, 5,0, 10.0 и 25,0 µg/мл DL-α-токоферолон ацетат, или 2,28, 4,55, 9,10 µg DL-α-токоферол.
Впръсква ce няколко пъти 20 млот всеки сравнителен разтвор и ce определят средните височини на пиковете (повърхности). Според средните височини на пиковете (повърхности) ce начертава градуирана крива.
5.6.1.3. Ув-калибриране на матерния разтвор на DL-α-токоферол (3.10.1)
Разтваря ce 5,0 мл матерен разтвор на DL-α-токоферол (3.10.1), допълва se до 25,0 мл в етанол и ce измерва УВ-спектъра на този разтвор по отношение на етанола (3.1) в спектрофотометър (4.6) между 250 nm и 320 nm.
Максималната абсорбция трябва да е 284 nm.
![]()
При това разреждане трябва да се получи стойност на затихване от 0,84 до 0,88.
5.6.2. Сравнителен разтвор на DL-α-токоферол
5.6.2.1. Изготвяне на работния сравнителен разтвор на DL-α-токоферол
Капва ce с пипета 2,0 мл от сравнителния разтвор на DL-α-токоферол (3.11.1) в градуирана колба от 50 мл, разтваря ce в метанол (3.3) и ce допълва до съответното деление с метанол. Номиналната концентрация на този разтвор е 44,0 µg DL-α-токоферол на мл, еквивалент на 44,0 µg DL-α-токоферолон ацетат на мл. Работният сравнителен разтвор се изготвя за всяка отделна манипулация.
5.6.2.2. Изготвяне на сравнителните разтвори и начертаване на градуираната крива
Прехвърля ce 1,0, 2,0, 4,0 и 10, 0 мл от работния сравнителен разтвор в няколко градуирани на 20 мл колби, допълва ce до съответното деление с метанол (3.3) и ce смесва. Номиналните концентрации на тези разтвори са 2,0, 8,0 и 20,0 µg/мл DL-α-токоферол, или 2,20, 4,40, 8,79 µg DL-α-токоферолов ацетат.
Впръсква се няколко лъти 20 мл от всеки сравнителен разтвор и се определат средните височини на пиковетe (повърхности). Споpед среднитe височинк нa пиковетe (повърхноcти) се начертава градуирана крива.
5.6.2.3. УВ-калибриране на матерния разтвор на DL-α-токоферол (3.11.1)
Разтваря ce 2,0 мл матерен разтвор на DL-α-токоферол (3.10.1), допълва ce до 25,0 мл в етанол и ce измерва УВ-спектъра на този разтвор по отношение на етанола (3.1) в спектрофотометър (4.6) между 250 nm и 320 nm. Максималната абсорбция трябва да е 292 nm:

При това разреждане трябва да се получи стойност на затихване от 0,6
6. Изчисление на резултатите
Като ce изхожда от средната височина (повърхност) на пиковете на витамин Е на пробния разтвор,ce определя концентрацията на този разтвор в µg/мл (изразена в DL-α-токоферолов ацетат) по показатели на градуираната крива (5.6.1.2 или 5.6.2.2.).
Съдържанието w на витамин Е в пробата, изразено в мг/кг е дадено от следната формула:

в която:
|
β |
= |
съдържанието на витамин Е в пробния разтвор (5.4) в µg/мл |
|
V1 |
= |
обема на пробния разтвор (5.4) в мл |
|
V2 |
= |
обема на взетата аликвотна част по точка 5.4 в мл |
|
m |
= |
масата на опитния материал в г |
7. Забележки
7.1. При пробите с ниско съдържание на витамин Е е добре да се обединят екстрактите в петролен етер, получени от две осапунявания (претеглено количество: 25 г), в един пробен разтвор за дозиране чрез CLHP.
7.2. Взетата за анализ проба не трябва да съдържа повече от 2 г мастни вещества.
7.3. Ако няма разделяне на фазите, ce прибавя около 10 мл етанол (етилов спирт) (3.1), за да ce елиминира емулсията.
7.4. След спектрофотометричното измерване на разтвора от DL-α-токоферолов ацетат или на DL-α-токоферола съгласно точка 5.6.1.3 или 5.6.2.3 ce прибавя около 10 мг ВНТ (3.12) към разтвора (3.10.1 или 3.10.2) и ce оставя този разтвор в охладителя (хладилника)(максимален срок на годност: четири седмици).
7.5. ВНТ може да се замени с хидрохинон (n-диоксибензол).
7.6. Колоната може да се използва в директна фаза, за да се разделят токоферолите α-, β-, χ- и δ.
7.7. Разтворът на натриев аскорбат може да се замени с около 150 мг аскорбинова киселина.
7.8. Разтворът на натриев сулфид може да се замени с около 50 мг EDTA.
8. Повторяемост
Разликата в резултатите от двете паралелни дозирания на една и съща проба не трябва да надхвърля 15 % по отношение на върховия резултат.
9. Резултати от вътрешнолабораторно проучване (1)
|
|
Предварител-на смес |
Минерален концентрат |
Протеинов хранителен продукт |
Храна за малки прасета |
|
|
L |
12 |
12 |
12 |
12 |
12 |
|
n |
48 |
48 |
48 |
48 |
48 |
|
средно [мг/кг] |
17 380 |
1 187 |
926 |
315 |
61,3 |
|
sr [мг/кг] |
384 |
45,3 |
25,2 |
13,0 |
2,3 |
|
r [мг/кг] |
1 075 |
126,8 |
70,6 |
36,4 |
6,4 |
|
CVr [%] |
2,2 |
3,8 |
2,7 |
4,1 |
3,8 |
|
SR [мг/гg] |
830 |
65,0 |
55,5 |
18,9 |
7,8 |
|
R [мг/кг] |
2 324 |
182,0 |
155,4 |
52,9 |
21,8 |
|
CVR [%] |
4,8 |
5,5 |
6,0 |
6,0 |
12,7 |
Фигура 1: Екстрактор (4.8)
ЧАСТ В
ДОЗИРАНЕ НА ТРИПТОФАНА
1. Цел и обхват
Методът дава възможност да се определи общия и свободен триптофан в храните за животни. Методът не прави разлика между форми D и L.
2. Принцип
За да се определи общия триптофан, пробата се хидролизира в алкална среда с разтвор на наситен бариев хидроксид и се загрява до 110 °С за двадесет часа. След хидролизата се прибавя вътрешен еталон (еталонно вещество, примесено към прахообразен образец - бел. прев.)
За да се определи свободния триптофан, пробата се екстрахира в кисела среда при наличие на вътрешен еталон.
Триптофанът и вътрешният еталон в хидролизата или в екстракта се определят от CLHP с флуорометричен датчик.
3. Реактиви
3.1. Използва ce двойно дестилирана вода или вода с равностойно качество (проводимост < 10 µS/см)
3.2. Еталонно вещество: триптофан (чистота/съдържание ≥ 99 %), изсушен във вакуум при наличие на дифосфорен петоокис (дифосфор пентоксид)
3.3. Вътрешен еталон в точното му значение: α-метил-триптофан (чистота/съдържание ≥ 99 %), изсушен във вакуум при наличие на дифосфорен петоокис (дифосфор пентоксид)
3.4. Октахидратиран бариев хидроксид (да се внимава Ва(ОН)2. 8Н2О да не се излага прекалено дълго на въздух, за да се избегне образуването на ВаСО3, който може да попречи на дозирането) (вж. забележката в точка 9.3)
3.5. Натриев хидроксид (хидроокис)
3.6. Ортофосфорна киселина, w = 85 %
3.7. Солна киселина, ρ20 = 1,19 г/мл
3.8. Метанол, качество CLHP
3.9. Петролен етер, температура на кипене: 40—60 °С
3.10. Разтвор на натриев хидроксид, с = 1 мол/л:
Разтваря ce 40,0 г NaOH (3.5) във вода и ce долива с вода (3.1) до 1 литър
3.11. Солна киселина, с = 6 мол/л:
Взима ce 492 мл НСl (3.7) и ce долива с вода до 1 литър
3.12. Солна киселина, с = 1 мол/л:
Взима ce 82 мл НСl (3.7) и доливате с вода до 1 литър
3.13. Солна киселина, с = 1 мол/л:
Взима ce 8,2 мл НСl (3.7) и ce долива с вода до 1 литър
3.14. Ортофосфорна киселина, с = 0,5 мол/л:
Взима ce 34 мл ортофосфорна киселина (3.6) и ce долива с вода (3.1) до 1 литър
3.15. Матерен разтвор на триптофан (3.2), с = 2,50 µmol/ml
В градуирана колба от 500 мл ce разтваря 0,2553 г триптофан (3.2) в солна киселина (3.13) и ce допълва до съответното деление със солна киселина (3.13). Съхранява ce при - 18 oС в продължение на четири седмици най-много.
3.16. Концентриран вътрешен еталонен (сравнителен) разтвор, с = 2,50 µmol/ml
В градуирана колба от 500 мл разтваря ce 0,2728 г α-метил-триптофан (3.3) в солна киселина (3.13) и ce допълва до съответното деление със солна киселина (3.13). Съхранява ce при - 18 oС в продължение на четири седмици най-много.
3.17. Сравнителен разтвор за калибриране на триптофан и вътрешен еталон
Взима ce 2,00 мл матерен разтвор на триптофан (3.15) и 2,00 мл матерен вътрешен сравнителен разтвор (на α-метил-триптофан) (3.16). Разрежда ce с вода (3.1) и метанол (3.8) приблизително до същия обем и приблизително до същата концентрация на метанол (10—3 %) като готовия хидролизат.
Този разтвор се изготвя при всяка манипулация.
Да се осигури защита срещу пряка светлина по време на изготвянето на разтвора.
3.18. Оцетна киселина
3.19. Трихлор-1,1,1-метил-2-пропанол-2
3.20. Етаноламин (коламин) > 98 %
3.21. 1 г разтвор на трихлор-1,1,1-метил-2-пропанол-2 (3.19) в 100 мл метанол (3.8)
3.22. Мобилна фаза за CLHP: 3,00 г оцетна киселина (3.18) + 900 мл вода (3.1) + 50 мл разтвор (3.21) на трихлор-1,1,1-метил-2-пропанол-2 (3.19) в метанол (3.8) (1 г/100 мл). рН трябва да достигне 5,00 с етаноламин (3.20). Долива ce до 1 000 мл с вода (3.1).
4. Апаратура
4.1. Инсталация за CLHP със спектрофлуорометрично детектиране
4.2. Колона за течна хроматография, 125 мм х 4 мм, С18, частици от 3 µm ири еквивалент
4.3. рН-метър
4.4. Пропиленова колба, обем 125мл с широко гърло и винтова запушалка
4.5. Мембранен филтър, 0,45 µm
4.6. Автоклав, 110 (±2) °С, 1,4 (± 0,1) бара
4.7. Механичен или магнитен миксер
4.8. Миксер Vortex
5. Процедура
5.1. Изготвяне на пробите
Смелва ce пробата, за да премине през сито с отвори 0,5 мм. Пробите с голяма влажност трябва да се изсушат на въздух при температура 50 °С максимум или чрез лиофилизация преди смилането. Преди смилането пробите с голямо съдържание на мастни вещества трябва да се екстрахират с петролен етер (3.9).
5.2. Определяне на свободния триптофан (екстракт)
Претегля ce близо 1 мг подходящо количество (1—5 г) от изготвената проба (5.1) в конусовидна колба. Прибавя ce 100,0 мл солна киселина, с = 0,1 мол/л (3.13) и 5,00 мл вътрешен сравнителен матерен разтвор (3.16). Разбърква ce или ce размесва в продължение на 60 мин. с механичния или магнитен миксер (4.7). Оставя ce седимента да се избистри чрез утаяване и ce прибавя с пипета 10,0 мл от повърхностния слой в бехерова чаша. Прибавя ce 5 мл ортофосфорна киселина, с = 0,5 мол/л (3.14). Достига ce рН 3 с натриев хидроксид, с = 1,0 мол/л (3.10). Прибавя ce достатъчно количество метанол (3.8), за да постигнете концентрация между 10 и 30 % метанол в крайния обем. Прехвърля ce в градуирана колба с подходящ обем и ce разрежда с достатъчен обем вода, необходим за хроматографията почти еднакъв обем с този на контролния разтвор за еталониране (3.17).
Филтрира ce няколко милилитра от разтвора през мембранен филтър 0,45 µm, преди да пристъпите към впръскването в колона CLHP. Преминава ce към етапа на хроматографията според точка 5.4.
Небходима е защита на сравнителния разтвор и на екстрактите срещу пряка слънчева светлина. Ако няма възможност да се анализират в същия ден, екстрактите трябва да са съхраняват при температура 5 °С за максимум три дни.
5.3. Дозиране на общия триптофан (хидролизат)
Претегля ce приблизително 0,2 мг (между 0,1 и 1 г) от подготвената проба (5.1) в пропиленова колба (4.4). Претеглената част трябва да съдържа около 10 мг азот. Пребавя ce 8,4 г октахидратиран бариев хидроксид (3.4) и 10 мл вода. Размесва ce с миксер Vortex (4.8) или с магнитен миксер (бъркалка) (4.7). Оставя ce магнита с тефлоново покритие в сместа. Измива ce стените на съда с 4 мл вода. Поставя ce винтовата запушалка и ce затваря колбата без да ce затяга. Прехвърля ce в автоклав (4.6) с вряща вода и пара за 30 до 60 мин. Затваря ce автоклава и ce включва при температура 110 (±2) °С в продължение на двадесет часа.
Преди да ce отворя автоклава, ce намалява температурата малко под 100 °С. За да се избегне кристализация на Ва(ОН)2. 8Н 2О, ce прибавя към загрятата смес 30 мл вода със стайна температура. Леко ce разклаща или ceразбърква. Прибавя ce 2,00 мл от вътрешния матерен сравнителен разтвор (α-метил-триптофан) (3.16). Охлаждат cе съдовете във вана с ледена вода в продължение на 15 мин.
Тогава ce прибавя 5 мл ортофосфорва киселина, с = 0,5 мол/л (3.14). Държa ce съда в охлаждаща вана и ce неутрализира солната киселина, с = 6 мола/л (3.11) след това, като ce разклаща, след коетто ce регулира рН на 3,0 с HCl, с = 1 мол/л (3.12). Прибавя ce достатъчно количество метанол, за да ce достигнет концентрация между 10 и 30 % метанол в крайния обем. Прехвърля ce в градуирана колба с подходящ капацитет и ce разрежда с обема, необходим за хроматографията (например 100 мл). Добавянето на метанол не трябва да предизвика утаяване.
Филтрира ce няколко милилитра от разтвора през мембранен филтър 0,45 µm (4.5) преди да ce впръска (инжектира ce) в колона CLHP. Пристъпва ce към етапа на хроматографията съгласно точка 5.4.
Небходима е защита на сравнителния разтвор и на екстрактите срещу пряка слънчева светлина. Ако няма възможност да се анализират в същия ден, екстрактите трябва да са съхраняват при температура 5 °С за максимум три дни.
5.4. HPLC дозиране
За ориентиране се предлагат следните условия за елюиране; и други условия могат да се прилагат, стига за дадат еквивалентни резултати (виж също така забележките в точки 9.1 и 9.2.
|
Колона за хроматография (4.2) |
125 мм х 4 мм, С18, частици 3 µm или еквивалент |
|
Температура на колоната |
Стайна температура (на околната среда) |
|
Мобилна фаза (3.22) |
3,00 г оцетна киселина (3.18) + 900 мл вода (3.1)+50 мл разтвор(3.21) на три-хлор-1,1,1-метил-2-пропанол-2(3.19) (1 г/100 мл). Регулирайте рН на 5,00 с етаноламин (3.20). Долейте до 1 000 мл с вода (3.1) |
|
Дебит |
1 мл/мин. |
|
Общо време на емулсия |
около 34 мин. |
|
Дължина на вълната на детектиране |
възбуждане: 280 nm; емисия: 356 nm |
|
Инжекционен обем |
20 µl |
6. Изчисляване на резутатите
![]()
|
A |
= |
повърхнина на пика на вътрешния еталон, еталонен разтвор (за еталониране) (3.17) |
|
B |
= |
повърхнина на пика на триптофана, ексракт (5.2) или хидролизат (5.3) |
|
С |
= |
обем в мл (2 мл) на матерния разтвор на триптофан (3.15), прибавен към разтвора за еталониране (3.17) |
|
D |
= |
концентрация в µмол/мл (2,50) на матерния разтвор на триптофан (3.15), прибавен към разтвора за еталониране (3.17) |
|
E |
= |
обем в мл на вътрешния матерен разтвор (3.16), прибавен към екстракта (5.2) (= 5,00 мл) или към хидролизата (5.3) (= 200 мл) |
|
F |
= |
повърхнина на пика на вътрешния еталон, екстракт (5.2) или хидролизат (5.3) |
|
G |
= |
повърхнина на пика на триптофана, разтвор за еталониране (3.17) |
|
H |
= |
обем в мл (200 мл) от вътрешния матерен еталонен разтвор (3.16), прибавен към разтвора за еталониране |
|
W |
= |
тегло на пробата в г (коригирано, за да се получи началното тегло, ако продуктът е изсушен и/или обезмаслен) |
|
MW |
= |
молекулно тегло на триптофана ( 204,23) |
7. Повторяемост
Разликата в резултатите от двете паралелни дозирания на една и съща проба не трябва да надхвърля 10 % по отношение на най-високия резултат.
8. Резултати от вътрешнолабораторно изпитание
Проведено е междулаборaтоpно изпитание в рамките на Общноcтта (четвъртo cрaвнение между лабopатoрите); анализирани cа тpи проби (обpазци) от 12 лаборатории, за дa се легализира метод за хидролиза. Нa всякo излитание се направени пет анализа. Резулатите са дадени в следващата таблица:
|
|
Проба 1 Храна за свине |
Проба 2 Храна за свине, с до-бавка на L-триптофан |
Проба 3 Концентри- рана храна за свине |
|
L |
12 |
12 |
12 |
|
n |
50 |
55 |
50 |
|
средно [г/кг] |
2,42 |
3,40 |
4,22 |
|
sr [г/кг] |
0,05 |
0,05 |
0,08 |
|
r [г/кг] |
0,14 |
0,14 |
0,22 |
|
CVr [%] |
1,9 |
1,6 |
1,9 |
|
SR [г/гg] |
0,15 |
0,20 |
0,09 |
|
R [г/кг] |
0,42 |
0,56 |
0,25 |
|
CVR [%] |
6,3 |
6,0 |
2,2 |
Проведено е друго междулабораторно изпитание в рамките на Общността (трето сравнение м/у лабораториите); анализирани са две проби (образци) от 13 лаборатории, за да се легализира екстракционния метод за свободния триптофан. На всяко изпитание се направени пет анализа. Резултатите са дадени в следващата таблица:
|
|
Проба 4 Смес от пшеница и соя |
Проба 5 Смес от пшеница и соя (= проба 4) с добавка на триптофан (0,457 г/кг |
|
L |
12 |
12 |
|
n |
55 |
60 |
|
средно [г/кг] |
0,391 |
0,931 |
|
sr [г/кг] |
0,005 |
0,012 |
|
r [г/кг] |
0,014 |
0,034 |
|
CVr [%] |
1,34 |
1,34 |
|
SR [г/гg] |
0,018 |
0,048 |
|
R [г/кг] |
0,050 |
0,134 |
|
CVR [%] |
4,71 |
5,11 |
Проведено е още едно междулабораторно изпитание в рамките на Общността; анализирани са четири проби (образци) от седем лаборатории, за да се легализира Метода чрез хидролиза. На всяко изпитание сa направени пет анализа. Резултатите са дадени в следващата таблица:
|
|
Проба 1 Смесена храна за свине (CRM 117) |
Проба 2 Рибено брашно с ниско съдържание на мастни вещества (CRM 118) |
Проба 3 Соево брашно (CRM 119) |
Проба 4 Обезмаслено мляко на прах (CRM 120) |
|
L |
7 |
7 |
7 |
7 |
|
n |
25 |
30 |
30 |
30 |
|
средно [г/кг] |
2,064 |
8,801 |
6,882 |
5,236 |
|
sr [г/кг] |
0,021 |
0,101 |
0,089 |
0,040 |
|
r [г/кг] |
0,059 |
0,283 |
0,249 |
0,112 |
|
CVr [%] |
1,04 |
1,15 |
1,30 |
0,76 |
|
SR [г/гg] |
0,031 |
0,413 |
0,283 |
0,221 |
|
R [г/кг] |
0,087 |
1,156 |
0,792 |
0,619 |
|
CVR [%] |
1,48 |
4,69 |
4,11 |
4,22 |
9. Забележки
9.1. Следните специални условия за хроматография могат да доведат до по-добро разделяне на триптофана и α-метил-триптофана.
Изократично елуиране, последвано от почистване на колоната по градиент:
|
Колона за течна хроматография: |
125мм х 4мм, С18, частици 5 µm, или еквивалент |
||
|
Температура на колоната: |
32 °С |
||
|
Мобилна фаза: |
А: смес 95/5 КН2РО4 с 0,01 мола/л) метанол (V + V) |
||
|
Б: метанол |
|||
|
Програма на градиента: |
0 мин. |
100 %А |
0 % Б |
|
15 мин. |
100 % А |
0 % Б |
|
|
17 мин. |
60 % А |
40 % Б |
|
|
19 мин. |
60 % А |
40 % Б |
|
|
21 мин. |
100 % Ь |
0 % Б |
|
|
33 мин. |
100 % А |
0 % Б |
|
|
Дебит: |
1,2 мл/мин. |
||
|
Общо време на елуиране: |
около 33 мин. |
||
9.2. Хроматографията варира според типа CLHP и според напълването на колоната. Избраната система трябва да доведе до връщане на базисния ред между пиковете на триптофана и на вътрешния еталон. Освен това е важно продуктите на разлагането определено да са разделени от триптофана и вътрешния еталон. Трябва да се пристъпи към изпитание без вътрешен еталон, така че да провери, че няма примеси на базисния ред на нивото на вътрешния еталон. От значение също така е времето за елуиране да е достатъчно продължително, за да има възможност за елуиране на всички продукти на разлагане, в противен случай късните пикове на елуиране могат да интерферират със следващите операции по хроматографията.
В поредицата от операции хроматорафската система трябва да даде линейна реакция. Тази реакция трябва да се измери при постоянна концентрация (т.е. нормалната концентрация) на вътрешния еталон и при различни концентрации на триптофана. Важно е височината на пиковете на триптофана и на вътрешния еталон да се намира в линейния диапазон на системата CLHP/флуоресценция. Ако пикът или пиковете на триптофана и/или на вътрешния еталон са прекалено ниски или прекалено високи, анализът трябва да се повтори с проба с друг размер и/или изменен краен обем.
9.3. Бариев хидроксид (хидроокис)
С течение на времето бариевият хидроксид все по-трудно се разтваря. Това води до мътен разтвор при определяне по CLHP, което пък ще означава слаби резултати за триптофана.
(1) Проучване, проведено от работната група по храните за животни на Verband Deutscher Landwirtschaftlicher Untersuchungs- und Forschungsanstalten (VDLUFA).