|
13/ 16
|
BG
|
Официален вестник на Европейския съюз
|
126
|
31995L0032
|
L 178/20
|
ОФИЦИАЛЕН ВЕСТНИК НА ЕВРОПЕЙСКИЯ СЪЮЗ
|
|
ШЕСТА ДИРЕКТИВА 95/32/ЕО НА КОМИСИЯТА
от 7 юли 1995 година
относно методите за анализ, необходими за проверка на състава на козметичните продукти
(текст от значение за ЕИП)
КОМИСИЯТА НА ЕВРОПЕЙСКИТЕ ОБЩНОСТИ,
като взе предвид Договора за създаване на Европейската общност,
като взе предвид Директива 76/768/ЕИО на Съвета от 27 юли 1976 г. относно сближаването на законодателствата на държавите-членки, свързана с козметични продукти (1), последно изменена с Директива 94/32/ЕО на Комисията (2), и по-специално член 8, параграф 1 от нея,
като има предвид, че Директива 76/768/ЕИО регламентира условията за официално изследване на козметичните продукти с цел да се гарантира, че разпоредбите на Комисията по отношение състава на козметичните продукти са изпълнени;
като има предвид, че всички необходими методи за анализ трябва да бъдат въведени в най-кратък срок; като има предвид, че такива методи са вече приети с Директива 80/1335/ЕИО на Комисията (3), изменена с Директива 87/143/ЕИО (4), 82/434/ЕИО (5), изменена с Директиви 90/207/ЕИО (6), 83/514/ЕИО (7), 85/490/ЕИО (8) и 93/73/ЕИО (9);
като има предвид, че идентификацията и определянето на бензоена киселина, 4-хидроксибензоена киселина, сорбинова киселина, салицилова киселина и пропионова киселина, както и идентификацията и определянето на хидрохинон, хидрохинон монометилов етер, хидрохинон моноетилов етер и хидрохинон монобензилов етер представляват една шеста стъпка в тази насока;
като има предвид, че предвидените в тази директива мерки са в съответствие със становището на Комитета по адаптиране на Директива 76/768/ЕИО към техническия прогрес,
ПРИЕ НАСТОЯЩАТА ДИРЕКТИВА:
Член 1
Държавите-членки предприемат всички необходими действия, с които да гарантират, че по време на официалното изследване на козметичните продукти:
|
—
|
идентификацията и определянето на бензоена киселина, 4-хидроксибензоена киселина, сорбинова киселина, салицилова киселина и пропионова киселина,
|
|
—
|
идентификацията и определянето на хидрохинон, хидрохинон монометилов етер, хидрохинон моноетилов етер и хидрохинон монобензилов етер,
|
се провеждат в съответствие с описаните в приложението методи.
Член 2
1. Държавите-членки въвеждат в сила всички необходими законови, подзаконови и административни разпоредби, за да се съобразят с настоящата директива, не по-късно от 30 септември 1996 г. Те уведомяват незабавно Комисията за това.
Когато държавите-членки приемат тези разпоредби, в тях се съдържа позоваване на тази директива или то се прави по време на официалното им публикуване. Начинът на това позоваване се определя от държавите-членки.
2. Държавите-членки съобщават на Комисията разпоредбите от националните си законодателства, които те приемат в областта, обхваната от настоящата директива.
Член 3
Настоящата директива влиза в сила на двадесетия ден след публикуването ѝ в Официален вестник на Европейските общности.
Член 4
Адресати на настоящата директива са държавите-членки.
Съставено в Брюксел на 7 юли 1995 година.
За Комисията
Emma BONINO
Член на Комисията
(1) ОВ L 262, 27.9.1976 г., стр. 169.
(2) ОВ L 181, 15.7.1994 г., стр. 31.
(3) ОВ L 383, 31.12.1980 г., стр. 27.
(4) ОВ L 57, 27.2.1987 г., стр. 56.
(5) ОВ L 185, 30.6.1982 г., стр. 1.
(6) ОВ L 108, 28.4.1990 г., стр. 92.
(7) ОВ L 291, 24.10.1983 г., стр. 9.
(8) ОВ L 295, 7.11.1985 г., стр. 30.
(9) ОВ L 231, 14.9.1993 г., стр. 34.
ПРИЛОЖЕНИЕ
I. ИДЕНТИФИКАЦИЯ И ОПРЕДЕЛЯНЕ НА БЕНЗОЕНА КИСЕЛИНА, 4-ХИДРОКСИБЕНЗОЕНА КИСЕЛИНА, СОРБИНОВА КИСЕЛИНА, САЛИЦИЛОВА КИСЕЛИНА И ПРОПИОНОВА КИСЕЛИНА В КОЗМЕТИЧНИ ПРОДУКТИ
1. Обхват и приложно поле
Методът се прилага за идентификация и определяне на бензоена киселина, 4-хидроксибензоена киселина, сорбинова киселина, салицилова киселина и пропионова киселина в козметични продукти. Отделни процедури описват идентификацията на тези консерванти; определянето на пропионова киселина, 4-хидроксибензоена киселина, салицилова киселина, сорбинова киселина и бензоена киселина.
2. Дефиниция
Количествата на бензоена киселина, 4-хидроксибензоена киселина, сорбинова киселина, салицилова киселина и пропионова киселина, определени по този метод, се изразяват като процент от масата на свободните киселини.
А. ИДЕНТИФИКАЦИЯ
1. Принцип
След киселинно-основна екстракция на консервантите екстрактът се анализира чрез тънкослойна хроматография (ТСХ), използвайки непосредствена дериватизация. В зависимост от резултатите идентификацията се потвърждава чрез високоефективна течна хроматография (ВЕТХ) или за пропионовата киселина - чрез газова хроматография.
2. Реактиви
2.1. Общо изискване
Всички реактиви трябва да са с квалификация „чист за анализ“ (ч.з.а.), водата - дестилирана или с еквивалентна чистота.
2.2. Ацетон
2.3. Диетилов етер
2.4. Ацетонитрил
2.5. Толуен
2.6. n-хексан
2.7. Парафин, течен
2.8. Солна киселина - 4 М
2.9. Калиев хидроксид - 4 М
2.10. Калциев хлорид, CaCl2.2H2O
2.11. Литиев карбонат, Li2CO3
2.12. 2-бром-2′-ацетонафтон
2.13. 4-хидроксибензоена киселина
2.14. Салицилова киселина
2.15. Бензоена киселина
2.16. Сорбинова киселина
2.17. Пропионова киселина
2.18. Сравнителни разтвори
Приготвят се 0,1 % (m/V) разтвори (100 mg/100 ml) от всеки от петте консерванта (2.13 до 2.17) в диетилов етер.
2.19. Реактив за дериватизация
0,5 % (m/V) разтвор на 2-бром-2′-ацетонафтон (2.12) в ацетонитрил (2.4) (50 mg/10 ml). Този разтвор се приготвя всяка седмица и се съхранява в хладилник.
2.20. Разтвор на катализатор
0,3 % (m/V) разтвор на литиев карбонат (2.11) във вода (300 mg/100 ml). Този разтвор се приготвя непосредствено преди употреба.
2.21. Подвижна фаза
Толуен (2.5)/ацетон (2.2) = 20:0,5 (V/V).
2.22. Течен парафин (2.7)/n-хексан (2.6) = 1:2 (V/V).
3. Апаратура
Стандартно лабораторно оборудване
3.1. Водна баня, която може да поддържа температура 60 °С
3.2. Хроматографска вана
3.3. Източник на УВ светлина, 254 и 366 nm
3.4. Плаки за тънкослойна хроматография Kieselgel 60 - без флуоресцентен индикатор, 20 × 20 cm, дебелина на слоя 0,25 mm, с концентрираща зона 2,5 × 20 cm (Merck 11 845 или еквивалентни)
3.5. Микроспринцовка - 10 µl
3.6. Микроспринцовка - 25 µl
3.7. Сушилня, която може да поддържа температура 105 °С
3.8. Стъклени епруветки 50 ml със запушалки на винт
3.9. Филтърна хартия - диаметър 90 mm, Schleicher & Schull, Weissband № 5892 или еквивалентна
3.10. Универсална рН-индикаторна хартия - рН 1-11
3.11. Стъклени шишенца за проби 5 ml
3.12. Ротационен изпарител (Rotavapor или еквивалентен)
3.13. Нагревателна плоча
4. Процедура
4.1. Приготвяне на пробата
В стъклена епруветка 50 ml (3.8) се претегля около 1 g от пробата. Прибавят се 4 капки 4 M солна киселина (2.8) и 40 ml ацетон (2.2). За силно алкални продукти като тоалетни сапуни е необходимо да се прибавят 20 капки 4 М солна киселина (2.8). Проверява се стойността на рН дали е около 2 с индикаторна хартия (3.10). Епруветката се запушва и се разклаща енергично в продължение на 1 min.
Ако е необходимо да се улесни екстрахирането на консервантите в ацетоновата фаза, сместа се нагрява леко до около 60 °С за стопяване на течната фаза.
Разтворът се охлажда до стайна температура и се филтрува през филтърна хартия (3.9) в конична колба.
20 ml от филтрата се прехвърлят в конична колба 200 ml, прибавят се 20 ml вода и се разбърква. рН на сместа се довежда до около 10 с 4 М калиева основа (2.9), като за измерване на рН се използва индикаторна хартия (3.10).
Прибавя се 1 g калциев хлорид (2.10) и се разклаща енергично. Филтрува се през филтърна хартия (3.9) в делителна фуния от 250 ml, съдържаща 75 ml диетилов етер (2.3), и се разклаща енергично в продължение на 1 min. Оставя се да се разделят слоевете и водният слой се излива в конична колба 250 ml. Етерният слой се отстранява. Като се използва индикаторна хартия (3.10), рН на водния слой се довежда до около 2 с помощта на 4 М солна киселина (2.8). Прибавят се 10 ml диетилов етер (2.3), колбата се запушва и се разбърква енергично в продължение на 1 min. Оставя се да се разделят слоевете и етерният слой се прехвърля в ротационен изпарител (3.12). Водният слой се изхвърля.
Етерният слой се изпарява почти до сухо и остатъкът се разтваря в 1 ml диетилов етер (2.3). Разтворът се прехвърля в шишенце за проби (3.11).
4.2. Тънкослойна хроматография
За всички стандартни разтвори и от проби, които ще се хроматографират, се нанасят със спринцовка (3.5) по около 3 µl от разтвора на литиев карбонат (2.20) на еднакви разстояния върху стартовата линия на концентриращата зона на плаката за ТСХ (3.4) и тя се изсушава в поток от студен въздух.
Плаката се поставя върху нагревателната плоча (3.13), загрята до 40 °С, за да останат петната колкото е възможно по-малки. С микроспринцовка (3.5) точно върху петната от литиев карбонат се нанасят по 10 µl от стандартните разтвори (2.18) и от разтвора на пробата (4.1) върху стартовата линия на плаката.
Накрая се нанасят по около 15 µl от реактива за дериватизация (2.19) (2-бром-2′-ацетонафтон) отново точно върху петната, където са нанесени стандартните разтвори/разтворът на пробата, както и разтворът на литиев карбонат.
Плаката се нагрява в сушилня (3.7) при температура 80 °С в продължение на 45 min. След охлаждане плаката се поставя в хроматографската вана (3.2), която предварително е доведена до равновесие за 15 min (без да се прилага облицоване с филтърна хартия) с подвижната фаза 2.21 (толуен/ацетон). Плаката престоява, докато фронтът на разтворителя достигне разстояние 15 cm от стартовата линия (това може да отнеме приблизително 80 min).
Плаката се изсушава в поток от студен въздух и получените петна се изследват под УВ светлина (3.3). За засилване на флуоресценцията на бледите петна плаката може да се потопи в смес от течен парафин и n-хексан (2.22).
5. Идентификация/отчитане на резултатите
Изчислява се Rf-стойността на всяко петно.
Сравнява се Rf-стойността и поведението при УВ-облъчване на петната от пробата и петната от стандартните разтвори.
Прави се предварително заключение относно присъствието и идентичността на наличните консерванти. Провежда се ВЕТХ, описана в раздел Б, или ако се докаже наличието на пропионова киселина - газова хроматография (ГХ), описана в раздел В. Сравняват се получените времена на задържане с тези на стандартните разтвори.
Обобщават се резултатите от ТСХ, ВЕТХ или ГХ и на тази база се определя окончателната идентификация на присъстващите в пробата консерванти.
Б. ОПРЕДЕЛЯНЕ НА БЕНЗОЕНА КИСЕЛИНА, 4-ХИДРОКСИБЕНЗОЕНА КИСЕЛИНА, СОРБИНОВА КИСЕЛИНА И САЛИЦИЛОВА КИСЕЛИНА
1. Принцип
След подкисляване пробата се екстрахира със смес от етанол и вода. След филтруване консервантите се определят посредством високоефективна течна хроматография (ВЕТХ).
2. Реактиви
2.1. Всички реактиви трябва да са с квалификация „чист за анализ“ (ч.з.а.), а където е необходимо - с чистота „за ВЕТХ“. Използваната вода трябва да бъде дестилирана или с еквивалентна чистота.
2.2. Етанол, абсолютен
2.3. 4-хидроксибензоена киселина
2.4. Салицилова киселина
2.5. Бензоена киселина
2.6. Сорбинова киселина
2.7. Натриев ацетат (CH3COONa.3H2O)
2.8. Оцетна киселина, (α)4
20= 1,05 g/ml
2.9. Ацетонитрил
2.10. Сярна киселина - 2 М
2.11. Калиев хидроксид, течен - 0,2 М
2.12. 2-метоксибензоена киселина
2.13. Смес етанол-вода
Смесват се девет обема етанол (2.2) и един обем вода (2.1)
2.14. Разтвор за вътрешен стандарт
Приготвя се разтвор, който съдържа около 1 g 2-метоксибензоена киселина (2.12) в 500 ml смес етанол/вода (2.13).
Подвижна фаза за ВЕТХ
2.15.1. Ацетатен буфер: към 1 l вода се прибавят 6,35 g натриев ацетат (2.7) и 20,0 ml оцетна киселина (2.8) и се разбърква.
2.15.2. Подвижната фаза се приготвя чрез смесване на 9 обема ацетатен буфер (2.15.1) и един обем ацетонитрил (2.9).
2.16. Основен разтвор на консервантите
Претеглят се с точност 0,001 g около 0,05 g 4-хидроксибензоена киселина (2.3), 0,2 g салицилова киселина (2.4), 0,2 g бензоена киселина (2.5) и 0,05 g сорбинова киселина (2.6) в мерителна колба от 50 ml и се долива до марката със смес етанол/вода (2.13). Разтворът се съхранява в хладилник и е стабилен в продължение на една седмица.
2.17. Стандартни разтвори на консервантите
В серия от мерителни колби от 20 ml се поставят съответно 8,00, 4,00, 2,00, 1,00 и 0,50 ml от основния разтвор (2.16). Във всяка колба се прибавят по 10,00 ml от разтвора за вътрешен стандарт (2.14) и по 0,5 ml 2 М сярна киселина (2.10). Допълва се до марката със смес етанол/вода (2.13). Тези разтвори се приготвят непосредствено преди употреба.
3. Апаратура
Стандартно лабораторно оборудване и:
3.1. Водна баня, нагласена на 60 °С.
3.2. Високоефективен течен хроматограф с УВ детектор с променлива дължина на вълната и 10 µl дозираща капиляра.
3.3. Аналитична колона
Неръждаема стомана, дължина: 12,5 до 25 cm, вътрешен диаметър: 4,6 mm, пълнеж: Nucleosil 5C18 или еквивалентен.
3.4. Филтърна хартия - диаметър 90 mm, Schleicher & Schull, Wiessband № 5892 или еквивалентна.
3.5. Стъклени епруветки от 50 ml със запушалки на винт
3.6. Стъклени шишенца за проби от 5 ml
3.7. Парченца карборунд за кипене, размер 2 до 4 mm или еквивалентни
4. Процедура
Приготвяне на пробата
4.1.1. Приготвяне на проба без добавяне на вътрешен стандарт
В стъклена епруветка (3.5) 50 ml се претегля 1 g от пробата с точност до 0,001 g. С пипета в епруветката се прибавя 1,00 ml 2 М сярна киселина (2.10) и 40 ml смес етанол/вода (2.13). Прибавя се около 1 g карборунд (3.7), епруветката се затваря и се разклаща енергично поне 1 минута, докато се получи хомогенна суспензия. За улесняване екстракцията на консервантите в етанолната фаза епруветката се поставя във водна баня (3.1), нагласена на 60 °С, точно за 5 минути.
Епруветката незабавно се охлажда със студена вода и екстрактът престоява 1 час при температура 5 °С, след което се филтрува през филтърна хартия (3.4).
Около 2 ml от екстракта се прехвърлят в шишенце за проби (3.6). Екстрактът се съхранява при 5 °С и се анализира чрез ВЕТХ в рамките на 24 часа от приготвянето.
4.1.2. Приготвяне на проба с добавяне на вътрешен стандарт
В стъклена епруветка от 50 ml (3.5) се претегля 1 g от пробата с точност до 0,001 g. С пипета в епруветката се прибавя 1,00 ml 2 М сярна киселина (2.10) и 30 ml смес етанол/вода (2.13). Прибавя се около 1 g карборунд (3.7) и 10,00 ml разтвор за вътрешен стандарт (2.14), епруветката се затваря и се разклаща енергично поне 1 минута, докато се получи хомогенна суспензия. За улесняване екстракцията на консервантите в етанолната фаза епруветката се поставя във водна баня при 60 °С точно за 5 минути.
Епруветката незабавно се охлажда със студена вода и екстрактът престоява 1 час при температура 5 °С,
след което се филтрува през филтърна хартия (3.4). Около 2 ml от екстракта се прехвърлят в шишенце за проби (3.6). Екстрактът се съхранява при 5 °С и се анализира чрез ВЕТХ в рамките на 24 часа от приготвянето.
Високоефективна течна хроматография
Подвижна фаза - ацетонитрил/ацетатен буфер (2.15).
Скоростта на подвижната фаза през колоната се нагласява на 2,0 ± 0,5 ml/min. Детекторът се настройва на 240 nm.
4.2.1. Стандартна крива
В течния хроматограф (3.2) се дозират по 10 µl от всеки от стандартните разтвори на консервантите (2.17). За всеки разтвор се определя отношението между височината на пиковете на изследваните консерванти към височината на пика на вътрешния стандарт, получени от хроматограмите. Построява се графика за всеки консервант, като се нанасят отношенията на височините на пика спрямо съответните концентрации на всеки стандартен разтвор.
Проверява се дали получените зависимости за всеки стандартен разтвор са линейни.
4.2.2. Определяне
В течния хроматограф (3.2) се дозират 10 µl от екстракта на пробата (4.1.1) и се записва хроматограмата. Дозират се 10 µl от стандартния разтвор на консерванти (2.17) и се записва хроматограмата. Сравняват се получените хроматограми. Ако в хроматограмата на екстракта на пробата (4.1.1) няма пик с приблизително същото време на задържане както това на 2-метоксибензоената киселина (препоръчвания вътрешен стандарт), в течния хроматограф се дозират 10 µl от екстракта на пробата с добавен вътрешен стандарт (4.1.2) и се записва хроматограмата.
Ако на хроматограмата на екстракта от пробата (4.1.1) се наблюдава пречещ пик, който има време на задържане като на 2-метоксибензоената киселина, трябва да се избере друг подходящ вътрешен стандарт. (Ако един от изследваните консерванти отсъства от хроматограмата, този консервант може да бъде използван като вътрешен стандарт).
Получените хроматограми на стандартния разтвор и на разтвора на пробата трябва да отговарят на следните изисквания:
|
—
|
Разделянето на пиковете в най-лошо разделената двойка трябва да бъде не по-малко от 0,90 (за дефиниция на коефициента на разделяне виж фиг.1).
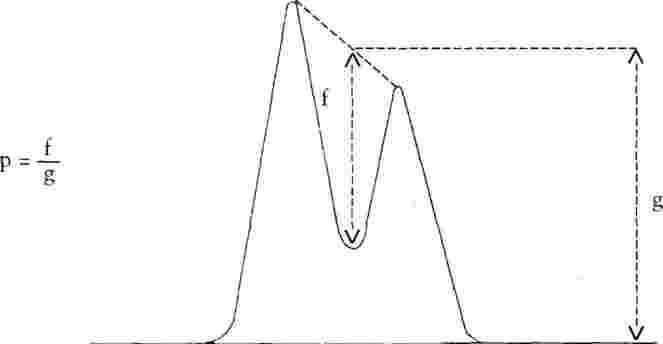
Ако не е постигнато необходимото разделяне, трябва да се използва по-ефективна колона или друга по състав подвижна фаза, чийто подбор продължава до изпълнение на изискванията.
|
|
—
|
Коефициентът на асиметрия (As) на всички получени пикове трябва да бъде в интервала от 0,9 до 1,5 (за дефиниция на коефициента на асиметрия виж фиг. 2). Препоръчва се хроматограмата за определяне коефициента на асиметрия да се записва при скорост най-малко 2 cm/min.
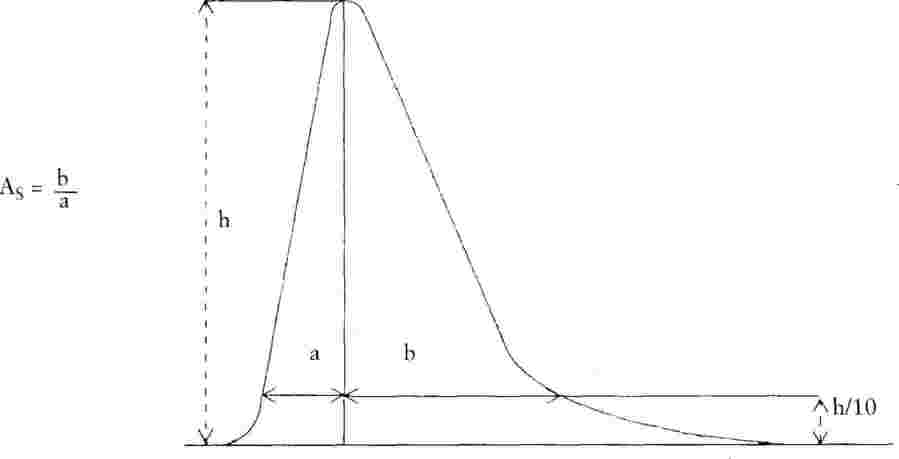
|
|
—
|
Базовата линия трябва да бъде стабилна.
|
5. Изчисления
За изчисляване на концентрацията на киселините-консерванти в разтвора на пробата се използват отношенията на височините на пиковете на изследваните консерванти към височината на пика на вътрешния стандарт (2-метоксибензоена киселина) от стандартната крива. Съдържанието на бензоена киселина, 4-хидроксибензоена киселина, сорбинова киселина или салицилова киселина се изчислява в проценти от масата (xi) по формулата:
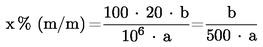
където:
a= масата на изпитваната проба (4.1.2) в g,
b= концентрацията на консерванта в екстракта на пробата (4.1.2), получена от стандартната крива, в mg/ml.
6. Повторяемост (1)
За съдържание на 4-хидроксибензоена киселина от 0,40 % разликата между резултатите от две успоредни определяния на една и съща проба не трябва да надхвърля по абсолютна стойност 0,035 %.
За съдържание на бензоена киселина от 0,50 % разликата между резултатите от две успоредни определяния на една и съща проба не трябва да надхвърля по абсолютна стойност 0,050 %.
За съдържание на салицилова киселина от 0,50 % разликата между резултатите от две успоредни определяния на една и съща проба не трябва да надхвърля по абсолютна стойност 0,045 %.
За съдържание на сорбинова киселина от 0,60 % разликата между резултатите от две успоредни определяния на една и съща проба не трябва да надхвърля по абсолютна стойност 0,035 %.
7. Забележки
7.1. Резултатите от проведения тест за допустими отклонения от методиката показват, че количеството сярна киселина, която се добавя при екстракцията на киселините от пробата, е критично и границите за количеството на обработваната проба трябва да се поддържат в посочения интервал.
7.2. По желание може да се използва подходяща предколона.
В. ОПРЕДЕЛЯНЕ НА ПРОПИОНОВА КИСЕЛИНА
1. Обхват и приложно поле
Този метод е подходящ за определяне на пропионова киселина с максимална концентрация 2 % (m/m) в козметични продукти.
2. Дефиниция
Концентрацията на пропионова киселина, измерена по този метод, се изразява като процент (m/m) от масата на продукта.
3. Принцип
След екстракция на пропионовата киселина от продукта определянето се извършва чрез газова хроматография с използване на 2-метилпропионова киселина като вътрешен стандарт.
4. Реактиви
Всички реактиви трябва да са с квалификация „чисти за анализ“ (ч.з.а.). Използваната вода е дестилирана или с еквивалентна чистота.
4.1. Етанол - 96 % (V/V)
4.2. Пропионова киселина
4.3. 2-метилпропионова киселина
4.4. Ортофосфорна киселина - 10 % (m/V)
4.5. Стандартен разтвор на пропионова киселина
В мерителна колба 50 ml се претегля около 1,00 g (p грама) с точност 0,001 g пропионова киселина и се долива до марката с етанол (4.1).
4.6. Разтвор за вътрешен стандарт
В мерителна колба 50 ml се претегля около 1,00 g (е грама) с точност 0,001 g 2-метилпропионова киселина и се долива до марката с етанол (4.1).
5. Апаратура
5.1. Стандартно лабораторно оборудване и:
5.2. Газов хроматограф с пламъчно-йонизационен детектор
5.3. Стъклена епруветка (2 × 15 cm) със запушалка на винт
5.4. Водна баня, нагласена на 60 °С
5.5. Стъклена спринцовка 10 ml с мембранен филтър (диаметър на порите 0,45 µm)
6. Процедура
Приготвяне на пробата
6.1.1. Приготвяне на проба без вътрешен стандарт
В стъклена епруветка (5.3) се претегля около 1 g с точност 0,001 g от пробата. Прибавят се 0,5 ml фосфорна киселина (4.4) и 9,5 ml етанол (4.1).
Епруветката се затваря и се разклаща енергично. Ако е необходимо, епруветката се поставя на водна баня, нагрята до 60 °С (5.4), за 5 min за пълно разтваряне на липидната фаза. Охлажда се бързо на течаща вода. Част от разтвора се филтрува през мембранен филтър (5.5). Филтратът се хроматографира същия ден.
6.1.2. Приготвяне на проба с вътрешен стандарт
В стъклена епруветка (5.3) се претегля 1 g (а грама) с точност 0,001 g от пробата. Прибавят се 0,5 ml фосфорна киселина (4.4), 0,50 ml от разтвора за вътрешен стандарт (4.6) и 9,0 ml етанол (4.1).
Епруветката се затваря и се разклаща енергично. Ако е необходимо, епруветката се поставя на водна баня, нагрята до 60 °С (5.4), за 5 min за разтваряне на липидната фаза. Охлажда се бързо на течаща вода. Част от разтвора се филтрува през мембранен филтър (5.5). Филтратът се хроматографира същия ден.
Условия за газова хроматография
Препоръчват се следните работни условия:
Колона
|
|
|
Тип
|
неръждаема стомана
|
|
Дължина
|
2 m
|
|
Диаметър
|
1/8
|
|
Пълнеж
|
10 % SPTM 1000 (или еквивалентна) + 1 % H3PO4 върху Хромосорб, WAW 100 до 120 mesh
|
|
Температура
|
|
|
Инжектор
|
200 °С
|
|
Колона
|
120 °С
|
|
Детектор
|
200 ° С
|
|
Газ носител
|
азот
|
|
Скорост на потока
|
25 ml/min
|
|
Хроматография
6.3.1. Стандартна крива
В серия мерителни колби от 20 ml се поставят съответно 0,25, 0,50, 1,00, 2,00 и 4,00 ml разтвор на пропионова киселина (4.5). Във всяка колба се добавя с пипета 1,00 ml от разтвора за вътрешен стандарт (4.6), долива се до марката с етанол (4.1) и се разбърква. Приготвените по този начин разтвори съдържат е mg/ml 2-метилпропионова киселина като вътрешен стандарт (т.е. 1 mg/ml, ако е = 1 000) и р/4, р/2, р, 2р и 4р mg/ml пропионова киселина (т.е. 0,25, 0,50, 1,00, 2,00 и 4,00 mg/ml, ако р = 1 000).
Инжектират се по 1 µl от всеки от тези разтвори и се построява стандартна крива, като на абсцисата се нанасят отношенията на масите на пропионовата киселина към 2-метилпропионовата киселина, а по ординатата - отношенията на съответните площи на пиковете им.
За всеки разтвор се правят по три инжектирания и се изчислява средната площ на пиковете.
6.3.2. Определяне
Инжектира се 1 µl от филтрата на пробата 6.1.1. Получената хроматограма се сравнява с тези на стандартните разтвори (6.3.1). Ако пикът има приблизително същото време на задържане както 2-метилпропионовата киселина, се сменя вътрешният стандарт. Ако не се наблюдава интерференция, се инжектира 1 µl от филтрата на пробата (6.1.2) и се измерват площите на пика на пропионовата киселина и на пика на вътрешния стандарт.
Правят се три инжектирания за всеки разтвор и се изчислява средната площ на пиковете.
7. Изчисления
7.1. От стандартната крива, получена в 6.3.1, се отчита отношението на масите (К), съответстващо на отношението на площите на пиковете, получени в 6.3.2.
7.2. От така полученото отношение на масите се изчислява съдържанието на пропионова киселина (X) в проценти от масата, като се използва формулата:

където:
К= съотношение, изчислено в 7.1,
е= масата на вътрешния стандарт, получена в 4.6, в g,
а= масата на пробата, претеглена в 6.1.2, в g.
Резултатът се закръглява до втория знак.
8. Повторяемост (2)
За съдържание на пропионова киселина от 2 % (m/m) разликата между резултатите от две успоредни определяния на една и съща проба не трябва да надвишава 0,12 %.
II. ИДЕНТИФИКАЦИЯ И ОПРЕДЕЛЯНЕ НА ХИДРОХИНОН, ХИДРОХИНОН МОНОМЕТИЛОВ ЕТЕР, ХИДРОХИНОН МОНОЕТИЛОВ ЕТЕР И ХИДРОХИНОН МОНОБЕНЗИЛОВ ЕТЕР В КОЗМЕТИЧНИ ПРОДУКТИ
А. ИДЕНТИФИКАЦИЯ
1. Обхват и приложно поле
Методът се прилага за откриване и идентификация на хидрохинон, хидрохинон монометилов етер, хидрохинон моноетилов етер и хидрохинон монобензилов етер (монобензон) в козметични продукти за избелване на кожата.
2. Принцип
Хидрохинонът и неговите етери се идентифицират чрез тънкослойна хроматография (ТСХ).
3. Реактиви
Всички реактиви трябва да са с квалификация „чист за анализ“ (ч.з.а.).
3.1. Етанол - 96 % (V/V)
3.2. Хлороформ
3.3. Диетилов етер
3.4. Подвижна фаза:
Хлороформ/диетилов етер = 66:33 (V/V)
3.5. Амоняк, 25 % (m/m) (d4
20 = 0,91 g/ml)
3.6. Аскорбинова киселина
3.7. Хидрохинон
3.8. Хидрохинон монометилов етер
3.9. Хидрохинон моноетилов етер
3.10. Хидрохинон монобензилов етер (монобензон)
Стандартни (референтни) разтвори
Стандартните разтвори трябва да бъдат приготвени непосредствено преди употреба и са стабилни един ден.
3.11.1. В градуирана епруветка от 10 ml се претеглят 0,05 g хидрохинон (3.7). Прибавят се 0,250 g аскорбинова киселина (3.6) и 5 ml етанол (3.1). Добавя се амоняк (3.5), докато рН достигне 10, и се долива до обем 10 ml с етанол (3.1).
3.11.2. В градуирана епруветка от 10 ml се претеглят 0,05 g хидрохинон монометилов етер (3.8). Прибавят се 0,250 g аскорбинова киселина (3.6) и 5 ml етанол (3.1). Добавя се амоняк (3.5), докато рН достигне 10, и се долива до обем 10 ml с етанол (3.1).
3.11.3. В градуирана епруветка от 10 ml се претеглят 0,05 g хидрохинон моноетилов етер (3.9). Прибавят се 0,250 g аскорбинова киселина (3.6) и 5 ml етанол (3.1). Добавя се амоняк (3.5), докато рН достигне 10, и се долива до обем 10 ml с етанол (3.1).
3.11.4. В градуирана епруветка от 10 ml се претеглят 0,05 g хидрохинон монобензилов етер (3.10). Прибавят се 0,250 g аскорбинова киселина (3.6) и 5 ml етанол (3.1). Добавя се се амоняк (3.5), докато рН достигне 10, и се долива до обем 10 ml с етанол (3.1).
3.12. Сребърен нитрат
3.13. 12-молибденфосфорна киселина
3.14. Калиев ферицианид хексахидрат
3.15. Ферихлорид хексахидрат
Реактиви за проявяване на петната
Към 5 % (m/V) воден разтвор на сребърен нитрат (3.12) се прибавя амоняк (3.5), докато образувалата се утайка се разтвори.
Внимание:
Разтворът става експлозивно нестабилен при престояване и след употреба трябва да се изхвърли.
3.16.2. 10 % (m/V) разтвор на 12-молибденфосфорна киселина (3.13) в етанол (3.1).
3.16.3. Приготвя се 1 % (m/V) воден разтвор на калиев ферицианид (3.14) и 2 % (m/V) воден разтвор на ферихлорид (3.15). Преди употреба се смесват равни части от двата разтвора.
4. Апаратура
Стандартно лабораторно оборудване и:
4.1. Стандартно оборудване за ТСХ
4.2. Готови плаки за ТСХ - силикагел GHR/UV254; 20 × 20 cm (Machery, Nagel или еквивалентни); дебелина на слоя: 0,25 mm
4.3. Ултразвукова вана
4.4. Центрофуга
4.5. Ултравиолетова лампа - 254 nm
5. Процедура
5.1. Приготвяне на пробата
В градуирана епруветка от 10 ml се претеглят 3,0 g от пробата. Прибавят се 0,250 g аскорбинова киселина (3.6) и 5 ml етанол (3.1). Добавя се амоняк (3.5), докато рН достигне 10, и се долива до обем 10 ml с етанол (3.1). Епруветката се затваря със запушалка и се хомогенизира в ултразвукова вана за 10 min. Филтрува се през филтърна хартия или се центрофугира при 3 000 rpm.
Тънкослойна хроматография
5.2.1. Хроматографската вана се насища с подвижна фаза (3.4).
5.2.2. Върху плаката се нанасят по 2 µl от стандартните разтвори (3.11) и 2 µl от разтвора на пробата (5.1). Хроматограмата се проявява на тъмно при стайна температура, докато фронтът на разтворителя достигне разстояние 15 cm от стартовата линия.
5.2.3. Плаката се изважда и се изсушава при стайна температура.
Проявяване на петната
5.3.1. Плаката се изследва под УВ светлина при 254 nm и се отбелязва разположението на петната.
5.3.2. Плаката се напръсква с един от следните реактиви:
|
—
|
сребърен нитрат (3.16.1),
|
|
—
|
12-молибденфосфорна киселина (3.16.2); нагрява се до 120 °С,
|
|
—
|
разтвор на калиев ферицианид и разтвор на ферихлорид (3.16.3).
|
6. Отчитане на резултатите
Изчислява се Rf-стойността на всяко петно.
Сравняват се петната, получени от разтвора на пробата, с петната на стандартните разтвори по отношение на: техните Rf-стойности, цвета на петната при УВ облъчване и цвета на петната след оцветяване с реактивите за напръскване.
Прилага се ВЕТХ, описана в раздел (Б), и се сравняват времената на задържане на пика (пиковете) на пробата с тези на стандартните разтвори.
Обобщават се резултатите от ТСХ и ВЕТХ, за да се потвърди присъствието на хидрохинон и/или неговите етери.
7. Забележки
При описаните условия се наблюдават следните Rf-стойности:
|
хидрохинон
|
0,32
|
|
хидрохинон монометилов етер
|
0,53
|
|
хидрохинон моноетилов етер
|
0,55
|
|
хидрохинон монобензилов етер
|
0,58
|
Б. ОПРЕДЕЛЯНЕ
1. Обхват и приложно поле
Методът се прилага за определяне на хидрохинон, хидрохинон монометилов етер, хидрохинон моноетилов етер и хидрохинон монобензилов етер в козметични продукти за избелване на кожата.
2. Принцип
Пробата се екстрахира със смес вода/метанол при слабо нагряване до стопяване на всички липидни материали. Всички търсени вещества се определят посредством течна хроматография с обърнати фази и УВ детектор.
3. Реактиви
3.1. Всички реактиви трябва да са с квалификация „чисти за анализ“ (ч..з.а.), а водата – дестилирана или поне с еквивалентна чистота.
3.2. Метанол
3.3. Хидрохинон
3.4. Хидрохинон монометилов етер
3.5. Хидрохинон моноетилов етер
3.6. Хидрохинон монобензилов етер (монобензон)
3.7. Тетрахидрофуран, „ВЕТХ“ чистота
3.8. Смес вода/метанол = 1:1 (V/V). Смесват се един обем вода с един обем метанол (3.2).
3.9. Подвижна фаза - смес: тетрахидрофуран/вода = 45:55 (V/V). Смесват се 45 обема тетрахидрофуран (3.7) и 55 обема вода.
3.10. Стандартен (референтен) разтвор.
В мерителна колба 50 ml се претеглят с точност 0,06 g хидрохинон (3.3), 0,08 g хидрохинон монометилов етер (3.4), 0,10 g хидрохинон моноетилов етер (3.5) и 0,12 g хидрохинон монобензилов етер (3.6). Разтварят се и се долива до марката с метанол (3.2). Стандартният разтвор се приготвя чрез разреждане на 10,00 ml от този разтвор до 50,00 ml със смес вода/метанол (3.8). Тези разтвори се приготвят непосредствено преди употреба.
4. Апаратура
Стандартно лабораторно оборудване и:
4.1. Водна баня, поддържаща температура 60 °С
4.2. Високоефективен течен хроматограф с УВ детектор с променяща се дължина на вълната и дозатор с 10 µl капиляра
4.3. Аналитична колона
Хроматографска колона от неръждаема стомана, дължина 250 mm, вътрешен диаметър 4,6 mm, пълнеж Zorbax фенил (химически свързан фенетилсилан върху Zorbax SIL, допълнително дезактивиран (end-capped) с триметилхлорсилан), размер на частиците 6 µm или еквивалентен. Да не се използва предколона, различна от „phenyl guard“ или еквивалентна.
4.4. Филтърна хартия, диаметър 90 mm, Schleicher & Schull, Weissband, № 5892 или еквивалентна.
5. Процедура
5.1. Приготвяне на пробата
В мерителна колба 50 ml се претегля 1 g от пробата (а грама) с точност до 0,001 g. Пробата се диспергира в 25 ml смес вода/метанол (3.8). Колбата се запушва и се разклаща енергично до получаване на хомогенна суспензия. Разклаща се в продължение поне на 1 min. Колбата се поставя във водна баня (4.1) при 60 °С за ускоряване на екстракцията, след което се охлажда и долива до марката със смес вода/метанол (3.8). Екстрактът се филтрува с филтърна хартия (4.4). ВЕТХ определянето трябва да се извърши в рамките на 24 часа от приготвянето на екстракта.
Високоефективна течна хроматография
5.2.1. Скоростта на подвижната фаза (3.9) се нагласява на 1,0 ml/min, а дължината на вълната на детектора се настройва на 295 nm.
5.2.2. Дозират се 10 µl от разтвора на пробата, получен съгласно описанието в 5.1, и се записва хроматограмата. Измерва се площта на пиковете. Извършва се калибриране, както е описано в 5.2.3. Сравняват се хроматограмите, получени за пробата и за стандартния разтвор. Използват се площите на пиковете и коефициентите на детектора (RF), получени съгласно 5.2.3, за изчисляване на концентрациите на анализираните вещества в разтвора на пробата.
5.2.3. Стандартна крива
Инжектират се 10 µl от стандартния разтвор (3.10) и се записва хроматограмата. Инжектирането продължава, докато се получи постоянна площ на пика.
Определя се коефициентът на детектора RFi:

където:
|
pi
|
=
|
площта на пика на хидрохинона, хидрохинон монометиловия етер, хидрохинон моноетиловия етер или хидрохинон монобензиловия етер,
|
|
c i
|
=
|
концентрацията на стандартния разтвор (3.10) на хидрохинона, хидрохинон монометиловия етер, хидрохинон моноетиловия етер или хидрохинон монобензиловия етер в g/50 ml.
|
Получените хроматограми на стандартния разтвор и на разтвора на пробата трябва да отговарят на следните изисквания:
|
—
|
разделянето на пиковете в най-лошо разделената двойка трябва да бъде не по-малко от 0,90 (за дефиниция на коефициента на разделяне виж фиг. 1).

Ако не е постигнато необходимото разделяне, трябва да се използва по-ефективна колона или друга по състав подвижна фаза до получаване на добро разделяне.
|
|
—
|
Коефициентът на асиметрия (AS) на всички получени пикове трябва да бъде в интервала от 0,9 до 1,5 (за дефиниция на коефициента на асиметрия виж фиг. 2). Препоръчва се хроматограмата за определяне коефициента на асиметрия да се записва при скорост най-малко 2 cm/min.
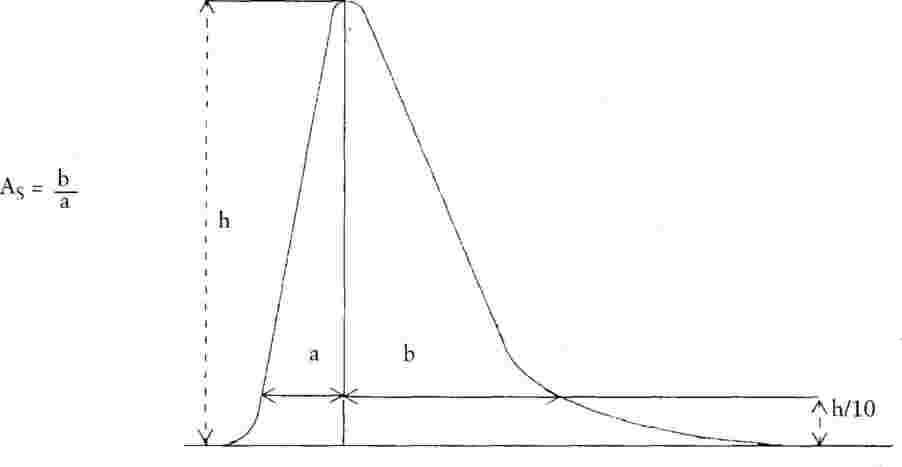
|
|
—
|
Базовата линия трябва да е стабилна.
|
6. Изчисления
За изчисляване на концентрацията (концентрациите) на анализираното вещество (вещества) в пробата се използват площите на пиковете на анализираните вещества. Концентрацията на анализираното вещество в пробата се изчислява като процент от масата (xi) по формулата:
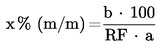
където:
|
а
|
=
|
масата на пробата в g;
|
|
bi
|
=
|
площта на пика на анализираното вещество в пробата.
|
7. Повторяемост (2)
7.1. За съдържание на хидрохинон от 2,0 % разликата между резултатите от две успоредни определяния на една и съща проба не трябва да надвишава по абсолютна стойност 0,13 %.
7.2. За съдържание на хидрохинон монометилов етер от 1,0 % разликата между резултатите от две успоредни определяния на една и съща проба не трябва да надвишава по абсолютна стойност 0,1 %.
7.3. За съдържание на хидрохинон моноетилов етер от 1,0 % разликата между резултатите от две успоредни определяния на една и съща проба не трябва да надвишава по абсолютна стойност 0,11 %.
7.4. За съдържание на хидрохинон монобензилов етер от 1,0 % разликата между резултатите от две успоредни определяния на една и съща проба не трябва да надвишава по абсолютна стойност 0,11 %.
8. Възпроизводимост (2)
8.1. За съдържание на хидрохинон от 2,0 % разликата между резултатите от две успоредни определяния на една и съща проба при различни условия (различни лаборатории, различни аналитици, различни апарати и/или различно време) не трябва да надвишава по абсолютна стойност 0,37 %.
8.2. За съдържание на хидрохинон монометилов етер от 1,0 % разликата между резултатите от две успоредни определяния на една и съща проба при различни условия (различни лаборатории, различни аналитици, различни апарати и/или различно време) не трябва да надвишава по абсолютна стойност 0,21 %.
8.3. За съдържание на хидрохинон моноетилов етер от 1,0 % разликата между резултатите от две успоредни определяния на една и съща проба при различни условия (различни лаборатории, различни аналитици, различни апарати и/или различно време) не трябва да надвишава по абсолютна стойност 0,19 %.
8.4. За съдържание на хидрохинон монобензилов етер от 1,0 % разликата между резултатите от две успоредни определяния на една и съща проба при различни условия (различни лаборатории, различни аналитици, различни апарати и/или различно време) не трябва да надвишава по абсолютна стойност 0,11 %.
9. Забележки
9.1. Когато се установи съдържание на хидрохинон, значително по-голямо от 2 %, и се изисква по-прецизна оценка на съдържанието му, екстрактът от пробата (5.1) трябва да се разреди до концентрацията, която би се получила от проба, съдържаща 2 % хидрохинон, след което определянето се повтаря.
(При някои апарати абсорбцията може да бъде извън интервала на линейност на детектора при високи концентрации на хидрохинон.)
9.2. Интерференции
Описаният метод дава възможност за определяне на хидрохинон и неговите етери при единично изократично хроматографиране. Използването на „фенил“ колона осигурява достатъчно време на задържане на хидрохинона, което не може да се гарантира при използването на С18 колона с описаната подвижна фаза.
Освен това методът е податлив на интерференции от редица парабени. В тези случаи определянето се повтаря, като се използват различни системи подвижна/неподвижна фаза. Подходящи методи могат да бъдат намерени в позовавания 1 и 2, а именно:
Колона: Zorbax ODS, 4,6 mm × 25 cm, или еквивалентна:
|
|
за хидрохинон – метанол/вода = 5:95 (V/V)
|
|
|
за хидрохинон монометилов етер – метанол/вода = 30:70 (V/V)
|
|
|
за хидрохинон монобензилов етер – метанол/вода = 80:20 (V/V) (3).
|
Колона: Spherisorb S5-ODS, или еквивалентна:
|
|
подвижна фаза – вода/метанол = 90:10 (V/V)
|
Тези условия са подходящи за хидрохинона (4).
(1) Виж ISO 5725.
(2) ISO 5725.
(3) M. Herpol-Borremans et M.-O. Masse, Identification et dosage de l'hydroquinone et de ses éthers méthylique et benzylique dans les produits cosmétiques pour blanchir la peau. Int. j. Cosmet. Sci. 8-203-214 (1986).
(4) J. Firth and I. Rix, Determination of hydroquinone in skin toning creams, Analyst (1986), 111, p. 129.