
|
13/ 06 |
BG |
Официален вестник на Европейския съюз |
136 |
31983L0514
|
L 291/9 |
ОФИЦИАЛЕН ВЕСТНИК НА ЕВРОПЕЙСКИЯ СЪЮЗ |
ТРЕТА ДИРЕКТИВА НА КОМИСИЯТА
от 27 септември 1983 година
за сближаване на законодателствата на държавите-членки относно методите за анализ, необходими за контрола върху състава на козметичните продукти
(83/514/ЕИО)
КОМИСИЯТА НА ЕВРОПЕЙСКИТЕ ОБЩНОСТИ,
като взе предвид Договора за създаване на Европейската икономическа общност,
като взе предвид Директива 76/768/ЕИО на Съвета от 27 юли 1976 г. за сближаване на законодателствата на държавите-членки относно козметичните продукти (1), последно изменена с Директива 83/341/ЕИО (2), и по-специално член 8, параграф 1 от нея,
като има предвид, че Директива 76/768/ЕИО предвижда официални проверки на козметичните продукти, с цел да се установи спазването на условията, предвидени в общностните разпоредби относно състава на козметичните продукти;
като има предвид, че следва да се установят възможно най-бързо всички необходими методи за анализ; като има предвид, че две стъпки към реализирането на тази цел вече бяха предприети с определянето на някои методи в Директиви 80/1335/ЕИО (3) и 82/434/ЕИО (4) на Комисията, а третата стъпка се състои в определянето на методи за определяне на дихлорметан и 1,1,1-трихлоретан, за идентификация и определяне на хинолин-8-ол и бис(8-хидроксихинолин) сулфат, за определяне на амоняк, за идентификация и определяне на нитрометан, за идентификация и определяне на меркаптооцетната киселина в състава на продуктите за къдрене на коса, за изправяне на косъма и депилатоарите, за идентификация и определяне на хексахлорофен, за определяне на тозилхлорамид натрий, за определяне на общия флуор в пастите за зъби, за идентификация и определяне на органоживачни съединения, за определяне на алкални и алкалоземни сулфиди;
като има предвид, че мерките, предвидени в настоящата директива, са в съответствие със становището на Комитета за привеждане в съответствие с техническия прогрес на Директива 76/768/ЕИО,
ПРИЕ НАСТОЯЩАТА ДИРЕКТИВА:
Член 1
Държавите-членки приемат всички необходими мерки, за да гарантират, че по време на официалните проверки на козметичните продукти:
|
— |
определянето на дихлорметана и на 1,1,1-трихлоретана, |
|
— |
идентификацията и определянето на хинолин-8-ола и на бис(8-хидроксихинолин) сулфата, |
|
— |
определянето на амоняка, |
|
— |
идентификацията и определянето на нитрометана, |
|
— |
идентификацията и определянето на меркаптооцетната киселина в състава на продуктите за къдрене на коса, за изправяне на косъма и депилатоарите, |
|
— |
идентификацията и определянето на хексахлорофена (МНН), |
|
— |
определянето на тозилхлорамид натрия, |
|
— |
определянето на общия флуор в пастите за зъби, |
|
— |
идентификацията и определянето на органоживачните съединения, |
|
— |
определянето на алкалните и на алкалоземните сулфиди, |
да се извършват съгласно методите, описани в приложението.
Член 2
Държавите-членки въвеждат в сила законовите, подзаконовите и административните разпоредби, необходими за да се съобразят с настоящата директива, най-късно до 31 декември 1984 г.
Те незабавно уведомяват Комисията за това.
Член 3
Адресати на настоящата директива са държавите-членки.
Съставено в Брюксел на 27 септември 1983 година.
За Комисията
Frans ANDRIESSEN
Член на Комисията
(1) ОВ L 262, 27.9.1976 г., стр. 169.
(2) ОВ L 188, 13.7.1983 г., стр. 15.
(3) ОВ L 383, 31.12.1980 г., стр. 27.
(4) ОВ L 185, 30.6.1982 г., стр. 1.
ПРИЛОЖЕНИЕ
ОПРЕДЕЛЯНЕ НА ДИХЛОРМЕТАН И НА 1,1,1-ТРИХЛОРЕТАН
1. ЦЕЛ И ПРИЛОЖНО ПОЛЕ
Този метод описва определянето на дихлорметан (метилен хлорид) и на 1,1,1-трихлоретан (метил хлороформ) във всички козметични продукти, които могат да съдържат тези разтворители.
2. ОПРЕДЕЛЕНИЕ
Съдържанието на дихлорметан и на 1,1,1-трихлоретан в пробата, определено съгласно този метод, се изразява в проценти спрямо масата.
3. ПРИНЦИП
Методът използва газхроматографски анализ с трихлорметан (хлороформ) като вътрешен стандарт.
4. РЕАКТИВИ
Всички реактиви трябва да са с аналитична чистота.
4.1. Хлороформ (CHCl3).
4.2. Тетрахлорметан (CCl4).
4.3. Дихлорметан (CH2Cl2).
4.4. 1,1,1-трихлоретан (CH3CCl3).
4.5. Ацетон.
4.6. Азот.
5. АПАРАТУРА
5.1. Стандартно лабораторно оборудване.
5.2. Газов хроматограф, снабден с детектор за топлопроводимост.
5.3. Преливен флакон, 50 до 100 ml (виж метода за вземане на проби в 5.3) (1).
5.4. Спринцовка с газ под налягане, 25 до 50 μl (виж метода за вземане на проби в 5.4.2.2) (1).
6. НАЧИН НА РАБОТА
6.1. Проба, която не е под налягане: пробата се претегля точно в конична колба със запушалка. Като вътрешен стандарт се вкарва точно претеглено количество хлороформ (4.1), еквивалентно на съдържащото се в пробата предполагаемо количество дихлорметан и 1,1,1-трихлоретан. Разбърква се добре.
Проба под налягане: използва се методът за вземане на проби, описан в раздела „вземане на проби“, при следните допълнителни уточнения:
6.2.1. След прехвърляне на пробата в преливния флакон (5.3) като вътрешен стандарт в него се вкарва допълнителен обем хлороформ (4.1), еквивалентен на съдържащото се в пробата предполагаемо количество дихлорметан и/или 1,1,1-трихлоретан. Разбърква се добре. Мъртвият обем на вентила се промива с 0,5 ml тетрахлорметан (4.2). След изсушаване по разликата се определя точно добавената маса на вътрешния стандарт.
6.2.2. След напълване на спринцовката с проба дюзата на спринцовката се продухва с азот (4.6) така, че да няма остатъци преди впръскване в хроматографа.
6.2.3. След всяко вземане на проба накрайникът на вентила или на преливния флакон трябва няколкократно да се промие с ацетон (4.5) (със спринцовка за подкожни инжекции) и след това напълно се подсушава с азот (4.6).
6.2.4. За всеки анализ се правят измервания върху два различни преливни флакони, като се провеждат по пет измервания на флакон.
7. ХРОМАТОГРАФСКИ УСЛОВИЯ
7.1. Предколона
Тръба: неръждаема стомана.
Дължина: 300 mm.
Диаметър: 3 или 6 mm.
Пълнеж: същият материал като използвания за пълнежа на аналитичната колона.
7.2. Колона
Неподвижната фаза е изработена от Hallcomid M 18 или Chromosorb. Колоната трябва да осигурява разделителна способност R, равна на или по-голяма от 1,5:
където:
|
r1 и r2 |
= |
времена на задържане (в минути), |
|
W1 и W2 |
= |
широчини на пиковете при полувисочината (в милиметри), |
|
d′ |
= |
скорост на въртене на хартията (в милиметра за минута). |
Като пример, следните работни условия дават търсените резултати:
|
Колона |
I |
II |
|
Материал: |
Тръба от неръждаема стомана |
Тръба от неръждаема стомана |
|
Дължина: |
350 cm |
400 cm |
|
Диаметър: |
3 mm |
6 mm |
|
Носител: |
||
|
сhromosorb: |
WAW |
WAW-DMCS-HP |
|
ситов анализ: |
100 до 120 mesh |
60 до 80 mesh |
|
Неподвижна фаза: |
Hallcomid M 18, 10 % |
Hallcomid M 18, 20 % |
Температурните условия могат да варират в зависимост от типа на апарата. В примерите те са, както следва:
|
Колона |
I |
II |
|
Температури: |
||
|
колона: |
65 °С |
75 °С |
|
инжектор: |
150 °С |
125 °С |
|
детектор: |
150 °С |
200 °С |
|
Газ носител: |
||
|
дебит на хелия: |
45 ml/min |
60 ml/min |
|
входно налягане: |
2,5 bar |
2,0 bar |
|
Впръскване: |
15 μl |
15 μl |
8. СМЕС ЗА ОПРЕДЕЛЯНЕ НА КОЕФИЦИЕНТИТЕ НА ЧУВСТВИТЕЛНОСТ
В конична колба със запушалка се приготвя следната точно претеглена смес:
|
|
Дихлорметан (4.3), 30 % (m/m). |
|
|
1,1,1-трихлоретан (4.4), 35 % (m/m). |
|
|
Хлороформ (4.1), 35 % (m/m). |
9. ИЗЧИСЛЕНИЯ
9.1. Изчисляване на коефициента на чувствителност за вещество „р“ по отношение на вещество „а“, избрано като вътрешен стандарт
Нека първото вещество е „р“, където:
|
kp |
= |
неговия коефициент на чувствителност, |
|
mp |
= |
неговата маса в сместа, |
|
Ap |
= |
площта на неговия пик. |
Нека второто вещество е „а“, където:
|
ka |
= |
неговия коефициент на чувствителност (приравнен към единица), |
|
Ma |
= |
неговата маса в сместа, |
|
Aa |
= |
площта на неговия пик, |
откъдето:
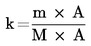
Като пример, бяха получени следните коефициенти на чувствителност (за хлороформа: к = 1):
|
Дихлорметан: |
к1 = 0,78 ± 0,03 |
|
1,1,1-трихлоретан: |
к2 = 1,00 ± 0,03 |
9.2. Изчисляване на съдържащите се % (m/m) на дихлорметан и на 1,1,1-трихлоретан в подлежащата на изследване проба
Нека:
|
ma |
= |
масата (в грама) на въведения хлороформ, |
|
Ms |
= |
масата (в грама) на изследваната проба, |
|
Aa |
= |
площта на пика на хлороформа, |
|
A1 |
= |
площта на пика на дихлорметана, |
|
A2 |
= |
площта на пика на 1,1,1-трихлоретана, |
откъдето:


10. ПОВТОРЯЕМОСТ (2)
За съдържание на дихлорметан и/или 1,1,1-трихлоретан от 25 % (m/m) разликата между резултатите от две успоредни изпитвания, извършени върху една и съща проба, не трябва да надхвърля абсолютната стойност от 2,5 % (m/m).
ИДЕНТИФИКАЦИЯ И ОПРЕДЕЛЯНЕ НА ХИНОЛИН-8-ОЛ И БИС(8-ХИДРОКСИХИНОЛИН) СУЛФАТ
1. ЦЕЛ И ПРИЛОЖНО ПОЛЕ
Този метод описва идентификацията и определянето на хинолин-8-ол на неговия сулфат.
2. ОПРЕДЕЛЕНИЕ
Съдържанието на хинолин-8-ол и на бис(8-хидроксихинолин) сулфат в пробата, определено съгласно този метод, се изразява като проценти спрямо масата на хинолин-8-ол.
3. ПРИНЦИП
3.1. Идентификация
Идентификацията се извършва по метода на тънкослойната хроматография.
3.2. Определяне
Определянето се извършва чрез спектрофотометричен анализ при 410 nm на комплекса, получен в резултат на взаимодействието с Fehling разтвор.
4. РЕАКТИВИ
Всички реактиви трябва да са с аналитична чистота.
4.1. Хинолин-8-ол.
4.2. Бензол. Предвид токсичността на продукта, при работа с бензола трябва да се действа изключително предпазливо.
4.3. Хлороформ.
4.4. Разтвор на натриев хидроксид, 50 % (m/m).
4.5. Меден сулфат пентахидрат.
4.6. Калиево-натриев тартарат.
4.7. Солна киселина 1 М.
4.8. Сярна киселина 0,5 М.
4.9. Разтвор на натриева основа 1 М.
4.10. Етанол.
4.11. Бутан-1-ол.
4.12. Ледена оцетна киселина.
4.13. Солна киселина 0,1 N.
4.14. „Целит 545“ или еквивалентен.
4.15. Стандартни разтвори
4.15.1. В 100 милилитров пикнометър се претеглят 100 mg хинолин-8-ол (4.1). Разтваря се в малко сярна киселина (4.8). Долива се до марката със сярна киселина (4.8).
4.15.2. В 100 милилитров пикнометър се претеглят 100 mg хинолин-8-ол. Разтваря в етанол (4.10). Долива се до марката с етанол (4.10) и се разбърква.
4.16. Fehling разтвор
Разтвор А
В 100 милилитров пикнометър се претеглят 7 g меден сулфат пентахидрат (4.5). Разтваря се в малко вода. Долива се до марката с вода и се разбърква.
Разтвор Б
В 100 милилитров пикнометър се претеглят 35 g калиево-натриев тартарат (4.6). Разтваря се в 50 ml вода. Добавят се 20 ml натриев хидроксид (4.4). Долива се до марката с вода и се разбърква. Непосредствено преди употреба, в 100 милилитров пикнометър с пипета се накапват 10 ml от разтвор А и 10 ml от разтвор Б. Долива се до марката и се разбърква.
4.17. Подвижни фази за тънкослойна хроматография
|
I |
: |
Бутан-1-ол (4.11)/оцетна киселина (4.12)/вода (80:20:20; v/v/v). |
|
II |
: |
Хлороформ (4.13)/оцетна киселина (4.12) (95:5; v/v). |
4.18. 2,6-дихлоро-4-(хлоримино)циклохекса-2,5-диенон, 1 % (m/v) разтвор в етанол (4.10).
4.19. Натриев карбонат, 1 % (m/v) разтвор във вода.
4.20. Етанол (4.10), 30 % (v/v) разтвор във вода.
4.21. Динатриев дихидроген етилендиаминтетраацетат, 5 % (m/v) разтвор във вода.
4.22. Буферен разтвор, pH 7
В еднолитров пикнометър се претегля 27 g безводен калиев дихидрогенортофосфат и 70 g дикалиев хидрогенортофосфат трихидрат. Долива се до марката с вода.
4.23. Готови плаки за тънкослойна хроматография
Готови плаки за тънкослойна хроматография с дебелина 0,25 mm (например Merck Kieselgel 60 или еквивалентни). Преди употреба плаките се напръскват с 10 ml реактив (4.21) и се сушат при 80 °С.
5. АПАРАТУРА
5.1. 100 милилитрова облодънна колба с гърло от шлифовано стъкло.
5.2. Стандартни колби.
5.3. Градуирани пипети, 10 и 5 ml.
5.4. Фол-пипети, 20, 15, 10 и 5 ml.
5.5. Делителни фунии, 100, 50 и 25 ml.
5.6. Нагъната филтърна хартия, диаметър 90 mm.
5.7. Ротационен изпарител.
5.8. Обратен хладник с шлиф.
5.9. Спектрофотометър.
5.10. Кювети с дебелина на слоя 10 mm.
5.11. Бъркалка с нагряване.
5.12. Стъклена хроматографска колона: дължина 160 mm и диаметър 8 mm, долна част със стеснение, запълнено с тампон от стъклена вата, и горна част с адаптер за прилагане на налягане.
6. НАЧИН НА РАБОТА
6.1. Идентификация
6.1.1. Течни проби
6.1.1.1. Довежда се рН на част от пробата за изпитване до 7, след което от нея се нанасят 5 и 10 μl върху стартовата линия на предварително третираната плака за тънкослойна хроматография (4.23).
6.1.1.2. Накапват се 10 и 30 μl от стандартния разтвор (4.15.2) на още две точки на стартовата линия, след което плаката се поставя в една от двете подвижни фази (4.17).
6.1.1.3. Когато фронтът на разтворителя се придвижи на 15 cm, плаката се суши при 110 °С (в продължение на 15 min). На УВ светлина (366 nm) петната на хинолин-8-ол флуоресцират в жълто.
6.1.1.4. Плаката се напръсква с разтвор на натриев карбонат (4.19). Изсушава се и се напръсква с разтвор на 2,6-дихлоро-4-(хлоримино)циклохекса-2,5-диенон (4.18). Хинолин-8-олът се проявява като синьо петно.
6.1.2. Твърди проби или кремове
6.1.2.1. 1 g от пробата се диспергира в 5 ml буферен разтвор (4.22). След това, се прехвърля с 10 ml хлороформ (4.3) в делителна фуния и се разклаща. След отделянето на слоя хлороформ водният слой се екстрахира още два пъти с 10 ml хлороформ (4.3). Събраните заедно и филтрирани екстракти от хлороформа се изпаряват до почти сухо състояние в 100-милилитрова облодънна колба (5.1) във въртящия изпарител (5.7). Остатъкът се разтваря в 2 ml хлороформ (4.3) и от така получения разтвор върху плака за тънкокслойна хроматография със силикагел се нанасят 10 и 30 μl (4.23) в съответствие с описания в 6.1.1.1 метод.
6.1.2.2. Върху плаката се нанасят 10 и 30 μl от стандартния разтвор (4.15.2) и анализът продължава по начина, описан в точки 6.1.1.2 до 6.1.1.4.
6.2. Определяне
6.2.1. Течни проби
6.2.1.1. В 100-милилитрова облодънна колба се претеглят 5 g от пробата. Добавя се 1 ml разтвор на сярна киселина (4.8) и сместа се изпарява почти до сухо състояние при понижено налягане и температура от 50 °С.
6.2.1.2. Този остатък се разтваря в 20 ml топла вода. Разтворът се прехвърля в 100 милилитрова стандартна колба. Промива се три пъти с по 20 ml вода. Долива се до 100 ml с вода и се разбърква.
6.2.1.3. С пипета се вземат 5 ml от разтвора и се поставят в делителна фуния от 50 ml (5.5). Прибавят се 10 ml Fehling разтвор (4.16). Медният комплекс на хинолин-8-ола [оксин мед (ISO)] се екстрахира три пъти с по 8 ml хлороформ (4.3).
6.2.1.4. Филтрираните хлороформени фази се събират в 25-милилитрова стандартна колба (5.2). Долива се до марката с хлороформ (4.3) и се разклаща. Измерва се оптическата плътност на жълтия разтвор при 410 nm по отношение на хлороформа.
6.2.2. Твърди проби или кремове
6.2.2.1. В 100 милилитрова облодънна колба (5.1) се претеглят 0,500 g от пробата. Добавят се 30 ml бензол (4.2) и 20 ml солна киселина (4.7). Съдържанието на колбата се кипва в обратен хладник в продължение на 30 минути с разбъркване.
6.2.2.2. Съдържанието на колбата се прехвърля в 100-милилитровата делителна фуния (5.5). Промива се с 5 ml 1N разтвор на солна киселина (4.7). Водната фаза се прехвърля в облодънна колба (5.1), а бензоловата фаза се промива с 5 ml 1N солна киселина (4.7).
6.2.2.3. При емулсиите, които не са подходящи за продължаването на анализа, 0,500 g от пробата се смесват с 2 g Целит 545 (4.14), при което се получава свободно сипещ се прах. Сместа се прехвърля на малки порции в стъклена хроматографска колона (5.12).
След всяко добавяне пълнежът на колоната се уплътнява. Веднага след прехвърлянето на цялата смес в колоната, тя се елуира със солна киселина (4.13) така че 10 ml елуат да се получат за около 10 минути (при необходимост може да се елуира в атмосфера от азот под слабо налягане). По време на елуирането трябва да е сигурно, че над пълнежа на колоната постоянно има известно количество солна киселина. Първите 10 ml от елуата по-нататък се обработват по начина, описан в 6.2.2.4.
6.2.2.4. Събраните водни фази (6.2.2.2) или елуатът (6.2.2.3) се изпаряват почти до сухо на ротационен вакуум изпарител при понижено налягане.
6.2.2.5. Остатъкът се разтваря в 6 ml от разтвора на натриевата основа (4.9). Добавят се 20 ml Фелингов разтвор (4.16) и съдържанието на колбата се прехвърля в 50 милилитрова делителна фуния (5.5). Колбата се промива с 8 ml хлороформ (4.3). Разклаща се и фазата на хлороформа се филтрира в стандартна 50-милилитрова колба (5.2).
6.2.2.6. Екстракцията се извършва три пъти с по 8 ml хлороформ (4.3). Фазите на хлороформа се филтрират и събират в 50-милилитрова колба. Долива се до марката с хлороформ (4.3) и се разклаща. Измерва се оптическата плътност на жълтия разтвор при 410 nm по отношение на хлороформа (4.3).
7. СТАНДАРТНА КРИВА
В четири 100-милилитрови облодънни колби (5.1), всяка от които съдържа 3 ml 30 % воден разтвор на етанол (4.20), с пипета се прехвърлят 5, 10, 15 и 20 ml порции от стандартния разтвор(4.15.1), които съответстват на 5, 10, 15 и 20 mg хинолин-8-ол. Анализът продължава в съответствие с описанието в 6.2.1.
8. ИЗЧИСЛЕНИЯ
8.1. Течни проби

където:
|
a |
= |
милиграми хинолин-8-ол върху стандартната крива (7), |
|
m |
= |
масата (в милиграми) на изпитваната проба (6.2.1.1.). |
8.2. Твърди проби или кремове

където:
|
a |
= |
милиграми хинолин-8-ол върху стандартната крива (7), |
|
m |
= |
масата (в милиграми) на изпитваната проба (6.2.1.1). |
9. ПОВТОРЯЕМОСТ (2)
За съдържание на хинолин-8-ол около 0,3 % разликата между резултатите от две успоредни изпитвания, проведени с една и съща проба, не трябва да надхвърля по абсолютна стойност 0,02 %.
ОПРЕДЕЛЯНЕ НА АМОНЯК
1. ЦЕЛ И ПРИЛОЖНО ПОЛЕ
Този метод описва определянето на свободен амоняк в козметичните продукти.
2. ОПРЕДЕЛЕНИЕ
Съдържанието на амоняк в пробата, определено в съответствие с настоящия метод, се изразява в проценти спрямо масата амоняк.
3. ПРИНЦИП
Разтвор на бариев хлорид се добавя към частта от пробата за изпитване на козметичния продукт във водно-метанолова среда. Утайката, която може да се образува се филтрира или центрофугира. С тази процедура, по време на парната дестилация, се избягва загубата на амоняк от някои амониеви соли като карбонат и бикарбонати и кисели карбонати и солите на мастните киселини с изключение на амониевия ацетат.
Амонякът от филтрата или повърхностната фаза се подлага на парна дестилация и се определя чрез потенциометрично или друго титруване.
4. РЕАКТИВИ
Всички реактиви трябва да са с аналитична чистота.
4.1. Метанол.
4.2. Бариев хлорид дихидрат, 25 % (m/v) разтвор.
4.3. Ортоборна киселина, 4 % (m/v) разтвор.
4.4. Сярна киселина, 0,25 М стандартен разтвор.
4.5. Антиразпенваща течност
4.6. Натриева основа, 0,5 М стандартен разтвор
4.7. Индикатор, ако такъв е необходим: 5 ml 0,1 % (m/v) разтвор на метилрот в етанол се смесват с 2 ml 0,1 % (m/v) воден разтвор на метиленово синьо.
5. АПАРАТУРА
5.1. Стандартно лабораторно оборудване.
5.2. Центрофуга със затворени епруветки за 100 ml.
5.3. Апарат за парна дестилация.
5.4. Потенциометър.
5.5. Измерващ стъклен електрод и сравнителен каломелов електрод.
6. НАЧИН НА РАБОТА
6.1. В 100 милилитров пикнометър се претегля такава маса (m) от пробата, която съответства на максимум 150 mg амоняк.
6.2. Добавят се 10 ml вода, 10 ml метанол (4.1) и 10 ml разтвор на бариев хлорид (4.2). Долива се до 100 ml с метанол (4.1).
6.3. Разбърква се и се оставя една нощ в хладилника (5 °С).
6.4. След това все още студеният разтвор се филтрира или центрофугира в затворени епруветки в продължение на 10 минути, така че да се получи бистър филтрат или повърхностен слой.
6.5. В апарата за парната дестилация (5.3) с пипета се прехвърлят 40 ml от така избистрения разтвор, последвано евентуално от 0,5 ml антиразпенваща течност(4.5).
6.6. Дестилира се и 200 ml от дестилата се събират в 250 милилитрова бехерова чаша, съдържаща 10 ml стандартна сярна киселина (4.4) и 0,1 ml индикатор (4.7).
6.7. Излишъкът от киселината се титрува обратно със стандартен разтвор на натриева основа (4.6).
NB: За потенциометричното определяне, 200 ml от дестилата се събират в 250 милилитрова бехерова чаша, съдържаща 25 ml от разтвора на ортоборната киселина (4.3), и се титрува със стандартна сярна киселина (4.4) като се записва кривата на неутрализация.
7. ИЗЧИСЛЕНИЯ
7.1. Изчисления в случай на обратно титруване
Нека:
|
V1 |
= |
обемът (в милилитри) на разтвора на употребената натриева основа (4.6), |
|
M1 |
= |
неговата действителна моларност (4.6), |
|
M2 |
= |
коефициентът на действителна моларност на разтвора на сярната киселина (4.4), |
|
m |
= |
масата (в милиграми) на взетата за целите на анализа част (6.1). |
оттук:
![]()
7.2. Изчисляване в случай на директното потенциометрично титруване
Където:
|
V2 |
= |
обемът (в милилитри) на разтвора на употребената сярна киселина (4.4), |
|
M2 |
= |
неговата действителна моларност (4.4), |
|
m |
= |
масата (в милиграми) на взетата за изпитване част (6.1). |
откъдето:

8. ПОВТОРЯЕМОСТ (2)
За съдържание на амоняк от коло 6 % разликата между резултатите от две успоредни изпитвания, проведени с една и съща проба, не трябва да надхвърля по абсолютна стойност от 0,6 %.
ИДЕНТИФИКАЦИЯ И ОПРЕДЕЛЯНЕ НА НИТРОМЕТАН
1. ЦЕЛ И ПРИЛОЖНО ПОЛЕ
Този метод се прилага за идентификация и определяне по-ниска или равна на 0,3 % нитрометан в козметични продукти в аерозолни опаковки.
2. ОПРЕДЕЛЕНИЕ
Съдържанието на нитрометан в пробата, определено по този метод, се изразява като проценти спрямо масата нитрометан от общото съдържание на аерозолната опаковка.
3. ПРИНЦИП
Нитрометанът се идентифицира чрез цветна реакция. Нитрометанът се определя с газова хроматография след добавяне на вътрешен стандарт.
4. ИДЕНТИФИКАЦИЯ
4.1. Реактиви
Всички реактиви трябва да са с аналитична чистота.
4.1.1. Натриев хидроксид, 0,5 М разтвор.
4.1.2. Реактив на Folin.
0,1 g натриев 3,4-дихидро-3,4-диоксонафталин-1-сулфонат се разтварят във вода и разтворът се разрежда до 100 ml.
4.2. Начин на работа
Към 1 ml от пробата се добавят 10 ml от разтвора от 4.1.1 и 1 ml от разтвора от 4.1.2. Оцветяването в лилаво е доказателство за наличие на нитрометан.
5. ОПРЕДЕЛЯНЕ
5.1. Реактиви
Всички реактиви трябва да са с аналитична чистота.
5.1.1. Хлороформ (вътрешен стандарт 1).
5.1.2. 2,4-диметилхептан (вътрешен стандарт 2).
5.1.3. Етанол, 95 %.
5.1.4. Нитрометан.
5.1.5. Референтен разтвор на хлороформ
В предварително тариран 25 милилитров пикнометър се поставят около 650 mg хлороформ (5.1.1). Колбата и съдържанието се претеглят отново с точност. Долива се до 25 ml с 95 % етанол (5.1.3). Претегля се и се изчислява съдържанието на хлороформ в този разтвор в %.
5.1.6. Референтен разтвор на 2,4-диметилхептан
Приготвя се по същия начин както еталонния разтвор на хлороформа, но в 25 милилитров пикнометър се претеглят 270 mg 2,4-диметилхептан (5.1.2).
5.2. Апаратура
5.2.1. Газов хроматограф с пламъчно-йонизационен детектор.
5.2.2. Апаратура за вземане на проби от аерозоли (преливен флакон, микроспринцовки, съединители и т.н.) както е описана в глава II от приложението към Директива 80/1335/ЕИО на Комисията от 22 декември 1980 г. (1).
5.2.3. Стандартно лабораторно оборудване.
5.3. Начин на работа
5.3.1. Подготвяне на пробата
В 100 милилитров предварително тариран преливен флакон, почистена и вакуумирана в съответствие с процедурата, описана в точка 5.4 от глава II от приложението към горепосочената директива се поставят около 5 ml от единия от разтворите на вътрешен стандарт (5.1.5 или 5.1.6). Използва се стъклена спринцовка от 10 или 20 ml, без игла, приспособена към преливния флакон по метода, описан в параграф 5, раздел II от горепосочената директива на Комисията. Претегля се отново, за да се определи вкараното количество от пробата. Като се използва същата техника, в този флакон се вкарват около 50 g от съдържанието на пробата от аерозолната опаковка. Отново се претегля, за да се определи количеството на прехвърлената проба. Разбърква се добре.
С посочената микроспринцовка (5.2.2) се впръскват около 10 μl . Правят се пет впръсквания.
5.3.2. Приготвяне на стандарта
В 50 милилитров пикнометър се претеглят с точност около 500 mg нитрометан (5.1.4) с 500 mg хлороформ (5.1.1) или 210 mg 2,4-диметилхептан (5.1.2). Долива се с 95 % етанол (5.1.3). Разбърква се добре. 5 ml от този разтвор се поставят в 20-милилитров пикнометър. Долива се с 95 % етанол (5.1.3).
С посочената микроспринцовка (5.2.2) се впръскват около 10 μl . Правят се пет впръсквания.
5.3.3. Газхроматографски условия
5.3.3.1. Колона
Състои се от две части, първата с пълнеж от дидецилфталат върху Gas Chrom Q, а втората – Ucon 50 HB 280X върху Gas Chrom Q. Приготвената комбинирана колона трябва да осигурява разделителна способност R, равна или по-голяма от 1,5, където:
![]()
нека:
|
r1 и r2 |
= |
времената на задържане (в минути), |
|
W1 и W2 |
= |
ширините на пиковете при полувисочината (в милиметри), |
|
d′ |
= |
скорост на въртене на хартията (в милиметри за минута). |
Като примери, търсената разделителна способност се осигурява при следните две части:
Колона А:
|
|
Материал: неръждаема стомана. |
|
|
Дължина: 1,5 м. |
|
|
Диаметър: 3 mm. |
|
|
Пълнеж: 20 % дидецилфталат върху Gas Chrom Q (100 до 120 mesh). |
Колона Б:
|
|
Материал: неръждаема стомана. |
|
|
Дължина: 1,5 м. |
|
|
Диаметър: 3 mm. |
|
|
Пълнеж: 20 % Ucon 50 HB 280X върху Gas Chrom Q (100 до 120 mesh). |
5.3.3.2. Детектор
Подходяща настройка за чувствителността на електроизмервателя на пламъчно -йонизационния детектор е 8 × 10–10 А.
5.3.3.3. Температурни условия
Подходящи са следните условия:
|
|
Инжектор: 150 °С. |
|
|
Детектор: 150 °С. |
|
|
Колона: между 50 и 80 °С в зависимост от отделните колони и апарата. |
5.3.3.4. Газ
Газ-носител: азот.
|
|
Налягане: 2,1 bar. |
|
|
Дебит: 40 ml/min. |
|
|
Захранване на детектора: както е посочено от производителя на детектора. |
6. ИЗЧИСЛЕНИЯ
6.1. Корекционен коефициент на нитрометан, изчислен спрямо използвания вътрешен стандарт
Ако „n“ е нитрометанът:
където:
|
kn |
= |
неговият коефициент на чувствителност, |
|
m′n |
= |
неговата маса (в грама) в сместа, |
|
S′n |
= |
площта на неговия пик. |
Ако „с“ е вътрешният стандарт хлороформ или 2,4-диметилхептан:
където:
|
m′c |
= |
неговата маса (в грама) в сместа, |
|
S′c |
= |
площта на неговия пик, |
тогава:
![]()
(kn e функция на апарата).
6.2. Концентрация на нитрометана в пробата
Ако „n“ е нитрометанът:
където:
|
kn |
= |
неговия коефициент на чувствителност, |
|
Sn |
= |
площта на неговия пик. |
Ако „с“ е вътрешният стандарт хлороформ или 2,4-диметилхептан:
където:
|
mc |
= |
неговата маса (в грама) в сместа, |
|
Sc |
= |
площта на неговия пик, |
|
M |
= |
масата (в грама) на прехвърления аерозол, |
оттук процентното съдържание (m/m) на нитрометана в пробата е:

7. ПОВТОРЯЕМОСТ (2)
За съдържание на нитрометан от около 0,3 % (m/m) разликата между резултатите от две успоредни изпитвания, проведени с една и съща проба, не трябва да надхвърля по абсолютна стойност от 0,03 % (m/m).
ИДЕНТИФИКАЦИЯ И ОПРЕДЕЛЯНЕ НА МЕРКАПТООЦЕТНАТА КИСЕЛИНА В ПРОДУКТИТЕ ЗА КЪДРЕНЕ И ИЗПРАВЯНЕ НА КОСАТА И В ДЕПИЛАТОАРИ
1. ЦЕЛ И ПРИЛОЖНО ПОЛЕ
Този метод описва идентификацията и определянето на меркаптооцетната киселина в продуктите за къдрене и изправяне на коса и в депилатоарите, които могат да съдържат и други редуциращи агенти.
2. ОПРЕДЕЛЕНИЕ
Съдържанието на меркаптооцетна киселина в пробата, определено в съответствие с настоящия метод, се изразява в проценти спрямо масата меркаптооцетна киселина.
3. ПРИНЦИП
Идентификацията на меркаптооцетната киселина се извършва чрез капков анализ и тънкослойна хроматография, а определянето се извършва йодометрично или чрез газова хроматография.
4. ИДЕНТИФИКАЦИЯ
4.1. Идентификация чрез капков анализ
4.1.1. Реактиви
Всички реактиви трябва да са с аналитична чистота.
4.1.1.1. Индикаторна хартия с оловен ди(ацетат).
4.1.1.2. Разтвор на солна киселина (една обемна единица концентрирана солна киселина плюс една обемна единица вода).
4.1.2. Начин на работа
4.1.2.1. Идентификация на меркаптооцетна киселина чрез цветна реакция с оловен ди (ацетат)
Една капка от пробата за анализ се нанася върху хартия с оловен ди (ацетат) (4.1.1.1). Ако се получи интензивно жълто оцветяване, е възможно наличие на меркаптооцетна киселина.
Чувствителност: 0,5 %.
4.1.2.2. Характеризиране на неорганични сулфиди чрез образуването на сероводород при подкисляване
В епруветка се поставят няколко милиграма от пробата за анализ. Добавят се 2 ml дестилирана вода и 1 ml солна киселина (4.1.1.2). Образува се сероводород, който се разпознава по специфичния му мирис, а върху хартия с оловен ди(ацетат) (4.1.1.1) се отлага черна утайка от оловен сулфид.
Чувствителност: 50 ppm.
4.1.2.3. Характеризиране на сулфити чрез образуването на серен диоксид при подкисляване
Процедира се както е описано в 4.1.2.2. Довежда се до кипене. Серният диоксид се разпознава по неговия мирис и по редукционните му свойства, например, по отношение на перманганатните йони.
4.2. Идентификация с тънкослойна хроматография
4.2.1. Реактиви
Всички реактиви, освен ако не е посочено друго, трябва да са с аналитична чистота.
4.2.1.1. Меркаптооцетна киселина (тиогликолова киселина), минимална чистота 98 %, определена йодометрично.
4.2.1.2. 2,2′-дитиоди(оцетна) киселина, минимална чистота 99 %, определена йодометрично.
4.2.1.3. 2-меркаптопропионова киселина (тиомлечна киселина), минимална чистота 95 %, определена йодометрично.
4.2.1.4. 3-меркаптопропионова киселина, минимална чистота 98 %, определена йодометрично.
4.2.1.5. 3-меркаптопропан-1,2-диол (1-тиоглицерин), минимална чистота 98 %, определена йодометрично.
4.2.1.6. Готови плаки за тънкослойна хроматография със силикагел с дебелина 0,25 mm.
4.2.1.7. Готови плаки за тънкослойна хроматография с алуминиев оксид, Merck F254 E или еквивалентни.
4.2.1.8. Солна киселина, концентрирана,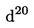 = 1,19 g/ml.
= 1,19 g/ml.
4.2.1.9. Етилацетат.
4.2.1.10. Хлороформ.
4.2.1.11. Диизопропилов етер.
4.2.1.12. Тетрахлорметан.
4.2.1.13. Ледена оцетна киселина.
4.2.1.14. Калиев йодид, 1 % (m/v) разтвор във вода.
4.2.1.15. Платинов тетрахлорид, 0,1 % (m/v) разтвор във вода.
4.2.1.16. Подвижни фази.
4.2.1.16.1. Етилацетат (4.2.1.9), хлороформ (4.2.1.10), диизопропилов етер (4.2.1.11), оцетна киселина (4.2.1.13) (20:20:10:10, обемно).
4.2.1.16.2. Хлороформ (4.2.1.10), оцетна киселина (4.2.1.13) (90:20, обемно).
4.2.1.17. Реактиви за откриване
4.2.1.17.1. Непосредствено преди употреба се смесват равни обеми от разтвор (4.2.1.14) и разтвор (4.2.1.15).
4.2.1.17.2. Разтвор на бром, 5 % (m/v):
5 g бром се разтварят в 100 ml тетрахлорметан (4.2.1.12).
4.2.1.17.3. Разтвор на флуоресцеин, 0,1 % (m/v):
100 ml флуоресцеин се разтварят в 100 ml етанол.
4.2.1.17.4. Хексаамониев хептамолибдат, 10 % (m/v) разтвор във вода.
4.2.1.18. Стандартни разтвори.
4.2.1.18.1. Меркаптооцетна киселина (4.2.1.1), 0,4 % (m/v) разтвор във вода.
4.2.1.18.2. 2,2′-дитиоди(оцетна киселина) (4.2.1.2), 0,4 % (m/v) разтвор във вода.
4.2.1.18.3. 2-меркаптопропионова киселина (4.2.1.3), 0,4 % (m/v) разтвор във вода.
4.2.1.18.4. 3-меркаптопропионова киселина (4.2.1.4), 0,4 % (m/v) разтвор във вода.
4.2.1.18.5. 3-меркаптопропан-1,2-диол (4.2.1.5), 0,4 % (m/v) разтвор във вода.
4.2.2. Апаратура
Стандартна апаратура за тънкослойна хроматография.
4.2.3. Начин на работа
4.2.3.1. Обработка на пробите
Подкислява се до pH 1 с няколко капки солна киселина (4.2.1.8) и, ако е необходимо, се филтрира.
В някои случаи може да е целесъобразно пробата да се разреди. За тази цел, преди разреждането същата се подкислява със солна киселина.
4.2.3.2. Хроматографиране
Върху плаката се нанася 1 μl от разтвора на пробата (4.2.3.1) и по 1 μl от всеки от петте стандартни разтвора (4.2.1.18). Внимателно се изсушава на слаба азотна струя и плаката се развива в подвижната фаза (4.2.1.16.1 или 4.2.1.16.2). Плаката се изсушава по възможно най-бързия начин, за да се избегне окислението на тиолите.
4.2.3.3. Проявяване на петната
Плаката се напръсква с един от трите реактива (4.2.1.17.1, 4.2.1.17.3 или 4.2.1.17.4). Ако плаката се напръска с реактив (4.2.1.17.3), след това се обработва с бромни пари (например в камера, която съдържа малка чаша с реактив (4.2.1.17.2), докато петната станат видими. Откриването с помощта на реактива за напръскване (4.2.1.17.4) ще бъде задоволително, само ако времето на сушене на тънкия слой не е повече от 30 минути.
4.2.3.4. Разчитане
Сравняват се Rf-стойностите и цветът на стандартните разтвори с тези на пробата. Средните Rf-стойности, представени по-долу за груба ориентация, имат единствено сравнителна стойност. Те зависят от:
|
— |
степента на активираност на тънкия слой по време на хроматографирането, |
|
— |
температурата на хроматографската камера. |
Таблица на Rf -стойностите, получени върху слоя от силикагел
|
|
Елюиращи разтворители |
|
|
4.2.1.16.1 |
4.2.1.16.2 |
|
|
Меркаптооцетна киселина |
0,25 |
0,80 |
|
2-меркаптопропионова киселина |
0,40 |
0,95 |
|
2,2′-дитиоди(оцетна) киселина |
0,00 |
0,35 |
|
3-меркаптопропионова киселина |
0,45 |
0,95 |
|
3-меркаптопропан-1,2-диол |
0,45 |
0,35 |
5. ОПРЕДЕЛЯНЕ (3)
Определянето трябва да започва задължително с йодометричната процедура.
5.1. Йодометрично определяне
5.1.1. Принцип
Определянето се извършва чрез окисление на „-SH“ групата с йод в кисела среда в съответствие с уравнението:
2 HOOC-CH2SH + I2 → (HOOC-CH2-S)2 + 2I–+2H+
5.1.2. Реактиви
Йод, 0,05 М стандартен разтвор.
5.1.3. Апаратура
Стандартно лабораторно оборудване.
5.1.4. Начин на работа
В 150-милилитрова конична колба със запушалка, съдържаща 50 ml дестилирана вода, се претегля точно 0,5 до 1 g от пробата. Добавят се 5 ml солна киселина (4.1.1.2) (pH на разтвора — около 0) и се титрува с разтвор на йод (5.1.2) до появата на жълто оцветяване. Ако е необходимо, се използва индикатор (например разтвор на скорбяла или тетрахлорметан).
5.1.5. Изчисления
Съдържанието на меркаптооцетна киселина се изчислява по формулата:
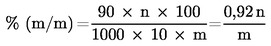
където:
|
m |
= |
масата (в грамове) на частта от пробата за анализ, |
|
n |
= |
използвания обем от разтвор на йод (5.1.2). |
5.1.6. Забележка
Ако резултатът, изчислен като меркаптооцетна киселина, е с 0,1 % или повече под разрешената максимална концентрация, извършването на допълнителни определяния не е необходимо. Ако резултатът е равен или по-голям от допустимите максимални концентрации и идентификацията е показала наличие на няколко редуциращи агента, е необходимо да се извърши газ хроматографско определяне.
5.2. Газова хроматография
5.2.1. Принцип
Меркаптооцетната киселина се отделя от ексципиента чрез утаяване с разтвор на кадмиев ди (ацетат). След метилиране с диазометан, приготвен или in situ или предварително в разтвор на диетилов етер, метиловото производно на меркаптооцетната киселина се дозира чрез газово-течна хроматография, като в качеството на вътрешен стандарт се използва метил октаноат.
5.2.2. Реактиви
Всички реактиви трябва да са с аналитична чистота.
5.2.2.1. Меркаптооцетна киселина, 98 %.
5.2.2.2. Солна киселина, = 1,19 g/ml.
= 1,19 g/ml.
5.2.2.3. Метанол.
5.2.2.4. Кадмиев ди(ацетат) дихидрат, 10 % (m/v) разтвор във вода.
5.2.2.5. Метилоктаноат, 2 % (m/v) разтвор в метанол.
5.2.2.6. Ацетатен буферен разтвор (pH 5):
Натриев ацетат трихидрат, 77 g.
Ледена оцетна киселина, 27,5 g.
Деминерализирана вода за получаване на краен обем от един литър.
5.2.2.7. Солна киселина, 3 М разтвор в метанол (5.2.2.3), прясно приготвена.
5.2.2.8. 1-метил-3-нитро-1-нитрозогуанидин.
5.2.2.9. Натриев хидроокис, 5 М разтвор.
5.2.2.10. Йод, 0,05 М стандартен разтвор.
5.2.2.11. Диетилов етер.
5.2.2.12. Разтвор на диазометан, приготвен от N-метил-N-нитрозотолуол-4-сулфонамид (Fieser, Реактиви за органичния синтез (Wiley), 1967)
Полученият разтвор съдържа около 1,5 g диазометан в 100 ml диетилов етер. Поради токсичността и изключителната нестабилност на газообразния диазометан всички експерименти трябва да се извършват в лабораторна камина с мощно засмукване, като се избягва употребата на апаратура от шлифовано стъкло (за целта има специални комплекти).
5.2.3. Апаратура
5.2.3.1. Стандартно лабораторно оборудване.
5.2.3.2. Апаратура за приготвяне на диазометан за метилиране in situ (виж Fales, Н.М., Jaouni, Т.М. and Babashak, J.F., Analyt. Chem 1973, 45, 2302).
5.2.3.3. Апаратура за предварително приготвяне на диазометан (Fieser).
5.2.4. Приготвяне на пробата
В 50-милилитрова центрофужна епруветка се претегля точно достатъчно количество от пробата с оглед на отчитането на предполагаемо количество от 50 до 70 mg меркаптооцетна киселина. Подкислява се с няколко капки солна киселина (5.2.2.2) за получаване на pH около 3.
Добавят се 5 ml деминерализирана вода и 10 ml от ацетатен буферен разтвор (5.2.2.6).
С помощта на индикаторна хартия се удостоверява, че стойността на pH е около 5. След това се добавят 5 ml разтвор на кадмиев ди(ацетат) (5.2.2.4).
Изчаква се 10 минути, след което се центрофугира в продължение на най-малко 15 минути при 4 000 g. Горният течен слой, който може да съдържа неразтворима мазнина (в случая на кремообразните продукти), се отстранява. Тези мазнини не трябва да се бъркат с тиолите, които се отлагат като компактна маса на дъното на епруветката. Проверява се да няма утаяване при прибавяне на няколко капки разтвор на кадмиевия ди(ацетат) (5.2.2.4) към повърхностния слой.
Ако предходните идентификации не са показали наличие на други редуциращи агенти освен тиолите, чрез йодометрично определяне се проверява дали съдържащите се в повърхностния слой тиоли не надхвърлят 6 до 8 % от първоначалното количество.
В съдържащата утайката центрофужна епруветка се въвеждат 10 ml метанол (5.2.2.3) и утайката се диспергира фино чрез разбъркване с пръчка. Отново се центрофугира най-малко 15 минути при 4 000 g. Повърхностният слой се отлива и се проверява за отсъствие на тиоли.
Утайката се промива втори път по същия начин.
Пак в същата центрофужна епруветка се добавят:
|
— |
2 ml разтвор на метил октаноат (5.2.2.5), |
|
— |
5 ml солна киселина в метанол (5.2.2.7). |
Тиолите се разтварят напълно (може да остане малко неразтворимо вещество от ексцепиента). Този разтвор се обозначава с „S“.
С аликвотна част от този разтвор се извършва йодометрична проверка за съдържанието на тиоли, което трябва да е най-малко 90 % от полученото в 5.1.
5.2.5. Метилиране
Метилирането се провежда или чрез процедура in situ (5.2.5.1), или с предварително приготвен разтвор на диазометан (5.2.5.2).
5.2.5.1. Метилиране in situ
В апарата за метилиране (5.2.3.2), съдържащ 1 ml етер (5.2.2.11), се въвеждат 50 μl разтвор „S“ и се метилират по метода (5.2.3.2) с около 300 mg 1-метил-3-нитро-1-нитрозогуанидин (5.2.2.8). След 15 минути (разтворът на етера трябва да е жълт, което е доказателство за наличие на излишък от диазометан) разтворът на пробата се прехвърля в 2-милилитрова бутилка с херметична запушалка. Оставя се за една нощ в хладилната камера. Метилират се две проби едновременно.
5.2.5.2. Метилиране с предварително приготвен разтвор на диазометан (5.2.2.12)
В 5-милилитрова колба със запушалка се поставя 1 ml от разтвора на диазометана (5.2.2.12), след което се добавят 50 μl от разтвор „S“. Оставя се за една нощ в хладилната камера.
5.2.6. Приготвяне на стандарта
Приготвя се стандартен разтвор на меркаптооцетна киселина (5.2.2.1) с позната концентрация, съдържащ около 60 mg чиста меркаптооцетна киселина (5.2.2.1) в 2 ml.
Това е разтвор „Е“.
Утаяването, проверката и метилирането се извършват съгласно описаното в точка 5.2.4 и 5.2.5.
5.2.7. Газхроматографски условия
5.2.7.1. Колона
Тип: неръждаема стомана.
Дължина: 2 m
Диаметър: 3 mm
5.2.7.2. Пълнеж
20 % дидецил фталат/хромосорб, WAW 80 до 100 mesh.
5.2.7.3. Детектор
Пламъчно-йонизационен. Подходящата зададена чувствителност за електроизмервателя на пламъчно-йонизационния детектор е 8 × 10–10 А.
5.2.7.4. Подавани газове
Газ-носител: азот.
|
|
Налягане: 2,2 bar. |
|
|
Дебит: 35 ml/мин. |
Спомагателен газ: водород.
|
|
Налягане: 1,8 bar. |
|
|
Дебит: 15 ml/мин. |
Захранване на детектора: съгласно указанията на производителя на апарата.
5.2.7.5. Температури
Инжектор: 200 °С.
Детектор: 200 °С.
Колона: 90 °С.
5.2.7.6. Скорост на хартията на самописеца
5 mm/min.
5.2.7.7. Количество за впръскване
3 μl. Правят се пет впръсквания.
5.2.7.8. Хроматографските условия са дадени с ориентировъчна цел. Те позволяват да се получи разделителна способност „R“≥ 1,5, където:

където:
|
r1 и r2 |
= |
времена на задържане (в минути), |
|
W1 и W2 |
= |
ширини на пиковете при половината от височината (в милиметри), |
|
d′ |
= |
скорост на хартията (в милиметри за минута). |
Препоръчва се хроматографирането да завърши с регулиране на температурата от 90 до 150 °С със скорост 10 °С за минута, за да се премахне възможността за влияние на лабилните вещества при следващите измервания.
5.2.8. Изчисления
5.2.8.1. Коефициент на пропорционалност за меркаптооцетната киселина
Изчислява се по отношение на метилоктаноата на базата на стандартна смес.
Ако „t“ е меркаптооцетната киселина:
където:
|
kt |
= |
нейния коефициент на чувствителност, |
|
m′t |
= |
нейната маса (в милиграми) в сместа, |
|
S′t |
= |
площта на нейния пик. |
Ако „с“ е метилоктаноатът:
където:
|
m′c |
= |
неговата маса (в милиграми) в сместа, |
|
S′c |
= |
площта на неговия пик, |
тогава:
![]()
Този коефициент варира според използваната апаратура.
5.2.8.2. Концентрация на меркаптооцетната киселина в пробата
Ако „t“ е меркаптооцетната киселина:
където:
|
kt |
= |
нейния коефициент на чувствителност, |
|
St |
= |
площта на нейния пик. |
Ако „с“ е метилоктаноатът:
където:
|
mc |
= |
неговата маса (в милиграми) в сместа, |
|
Sc |
= |
площта на неговия пик, |
|
М |
= |
масата (в милиграми) на първоначалната част от пробата за изследване, |
тогава % (m/m), на меркаптооцетната киселинае в пробата, е:

6. ПОВТОРЯЕМОСТ (2)
За съдържание на меркаптооцетна киселина 8 % (m/m) разликата между резултатите от две успоредни изпитвания, проведени с една и съща проба, не трябва да надхвърля по абсолютна стойност от 0,8 % (m/m).
ИДЕНТИФИКАЦИЯ И ОПРЕДЕЛЯНЕ НА ХЕКСАХЛОРОФЕН
А. ИДЕНТИФИКАЦИЯ
1. ЦЕЛ И ПРИЛОЖНО ПОЛЕ
Този метод е подходящ за всички козметични продукти.
2. ПРИНЦИП
Съдържащият се в пробата хексахлорофен се екстрахира с етилацетат и се идентифицира чрез тънкослойна хроматография.
3. РЕАКТИВИ
Всички реактиви трябва да са с аналитична чистота.
3.1. Сярна киселина, 4 М разтвор.
3.2. Целит AW.
3.3. Етилацетат.
3.4. Подвижна фаза: бензен, съдържащ 1 % (v/v) ледена оцетна киселина.
3.5. Реактив за проявяване на петната I:
Разтвор на родамин В: 100 mg родамин В се разтварят в смес от 150 ml диетилов етер, 70 ml абсолютен етанол и 16 ml вода.
3.6. Реактив за проявяване на петната II:
Разтвор на 2,6-дибром-4-(хлоримино)циклохекса-2,5-диенон: 400 mg 2,6-дибром-4-(хлоримино)циклохекса-2,5-диенон се разтварят в 100 ml метанол (да се приготвя ежедневно).
Разтвор на натриев карбонат: 10 g натриев карбонат се разтварят в 100 ml деминерализирана вода.
3.7. Стандартен разтвор:
Хексахлорофен, 0,05 % (m/v) разтвор в етилацетат.
4. АПАРАТУРА
4.1. Плаки от кизелгел 254 ТСХ, 200 × 200 mm (или еквивалентни).
4.2. Стандартно оборудване за тънкослойна хроматография.
4.3. Термостатирана при 26 °С баня за поместване на хроматографската камера.
5. ПРИГОТВЯНЕ НА ПРОБАТА ЗА ИЗПИТВАНЕ
5.1. Смесват се добре 1 g от хомогенизираната проба с 1 g Целит AW (3.2) и 1 ml сярна киселина (3.1).
5.2. Суши се два часа при 100 °С.
5.3. Изсушеният остатък се охлажда и се стрива на фин прах.
5.4. Екстрахира се двукратно с 10 ml етилацетат (3.3), след всяка екстракция се центрофугира и слоевете етилацетат се обединяват.
5.5. Изпарява се при 60 °С.
5.6. Остатъкът се разтваря в 2 ml етилацетат (3.3).
6. НАЧИН НА РАБОТА
6.1. Върху плаката за тънкослойната хроматография (4.1) се нанасят 2 μl от разтвора на пробата за изпитване (5.6) и 2 μl от стандартния разтвор (3.7).
6.2. Камерата (4.3) се насища с подвижната фаза (3.4).
6.3. Плаката се поставя в камерата и се развива до 150 mm.
6.4. Плаката се изважда и изсушава в снабдена с вентилатор сушилня при температура около 105 °С.
6.5. Проявяване на петната
Петната от хексахлорофен върху плаката за тънкослойната хроматография се визуализират, както е посочено в точка 6.5.1 или 6.5.2.
6.5.1. Плаката се напръсква равномерно с проявяващ реактив I (3.5). След 30 минути плаката се изследва на ултравиолетова светлина при 254 nm.
6.5.2. Плаката се напръсква равномерно с разтвора на визуализиращия агент II (3.6) в 2,6-дибром-4-(хлоримино)циклохекса-2,5-диенон. След това плаката се напръсква с разтвора на натриевия карбонат (3.6). Плаката се изследва на дневна светлина след 10-минутно сушене при стайна температура.
7. ТЪЛКУВАНЕ
7.1. Проявяващ реактив I (3.5):
Хексахлорофенът се откроява като синкаво петно върху жълто-оранжевия флуоресциращ фон и има Rf-стойност приблизително 0,5.
7.2. Проявяващ реактив II (3.6):
Хексахлорофенът се откроява като небесносиньо до тюркоазено оцветено петно върху белия фон и има Rf-стойност приблизително 0,5.
Б. ОПРЕДЕЛЯНЕ
1. ЦЕЛ И ПРИЛОЖНО ПОЛЕ
Този метод се прилага по отношение на всички козметични продукти.
2. ОПРЕДЕЛЕНИЕ
Съдържанието на хексахлорофен в пробата, определено по този метод, се изразява в проценти спрямо масата хексахлорофен.
3. ПРИНЦИП
Хексахлорофенът се определя след превръщане в метилово производно, чрез газова хроматография с електрон-улавящ детектор.
4. РЕАКТИВИ
Всички реактиви трябва да са с аналитична чистота.
4.1. Етилацетат.
4.2. N-метил-N-нитрозо-p-толуолсулфонамид (диазалд).
4.3. Диетилов етер.
4.4. Метанол.
4.5. 2-(2-етоксиетокси)етанол(карбитол).
4.6. Мравчена киселина.
4.7. Калиев хидроокис, 50 % (m/m) воден разтвор (приготвя се всеки ден).
4.8. Хексан за спектроскопия.
4.9. Бромхлорофен (стандарт № 1).
4.10. 4,4′,6,6′-тетрахлор-2,2′-тиодифенол (стандарт № 2).
4.11. 2,4,4′-трихлор-2-хидрокси-дифенилов етер (стандарт № 3).
4.12. Ацетон.
4.13. Сярна киселина, 4 М.
4.14. Целит AW.
4.15. Мравчена киселина/етилацетат, 10 % (v/v) разтвор.
4.16. Хексахлорофен.
5. АПАРАТУРА
5.1. Стандартно лабораторно оборудване.
5.2. Миниапарат за приготвяне на диазометан (Analyt. Chem., 1973, 45, 2302-2).
5.3. Газов хроматограф, снабден с електрон-улавящ детектор с източник 63 Ni.
6. НАЧИН НА РАБОТА
6.1. Приготвяне на стандартни разтвори
Стандартът се подбира така, че да не взаимодейства с никое вещество, съдържащо се в ексцепиента на подложения на анализ продукт. Обикновено най-подходящ е стандарт № 1 (4.9).
6.1.1. В 100-милилитрова мерителна колба се претеглят точно около 50 mg от стандарт № 1, 2 или 3 (4.9, 4.10 или 4.11) и 50 mg хексахлорофен (4.16). Долива се до пълния обем с етилацетат (4.1) (разтвор А). 10 ml от разтвор А се разреждат до 100 ml с етилацетат (4.1) (разтвор Б).
6.1.2. В 100-милилитрова мерителна колба се претеглят точно около 50 mg от стандарт № 1, 2 или 3 (4.9, 4.10 или 4.11). Долива се до пълния обем с етилацетат (4.1) (разтвор В).
6.2. Приготвяне на пробата (4)
Претегля се точно 1 g от хомогенизираната проба и това количество се смесва щателно с 1 ml сярна киселина (4.13), 15 ml ацетон (4.12) и 8 g Целит AW (4.14). Сместа се суши до въздушно сухо състояние на парна баня в продължение на 30 минути, след което се суши час и половина в снабдена с вентилатор сушилня. Остатъкът се охлажда, раздробява се на ситно и се прехвърля в стъклена колона. Елуира се с етилацетат (4.1) и се събират 100 ml. Добавят се 2 ml от вътрешния стандарт (разтвор В) (6.1.2).
6.3. Метилиране на пробата
Всички реактиви и апаратурата се охлаждат от 0 до 4 °С в продължение на два часа. Във външното отделение на апарата за получаване на диазометана се вкарват 1,2 ml от разтвора, получен в 6.2, и 0,1 ml метанол (4.4). В централния резервоар се въвеждат около 200 mg диазалд (4.2), добавя се 1 ml карбитол (4.5) и 1 ml диетилов етер (4.3) и се разтваря. Апаратът се свързва, потапя се наполовина в баня при 0 °С и със спринцовка в централния резервоар се впръсква около 1 ml охладен разтвор на калиев хидроокис (4.7). Проверява се дали появилият се в резултат на образуването на диазометана жълт цвят е траен. Ако жълтото оцветяване не е трайно, метилирането се повтаря с нови 200 mg диазалд (4.2) (5).
След 15 минути апаратът се изважда от банята, след което се оставя затворен 12 часа при стайна температура. Апаратът се отваря, излишъкът от диазометан се въвежда в реакция чрез добавяне на няколко капки 10 % (v/v) разтвор на мравчена киселина в етилацетат (4.15) и органичният разтвор се прехвърля в 25 милилитров пикнометър. Долива се до пълния обем с хексан (4.8).
В хроматографа се впръскват 1,5 μl от този разтвор.
6.4. Метилиране на стандарта
Всички реактиви и апаратурата се охлаждат от 0 до 4 °С в продължение на два часа. Във външното отделение на апарата за диазометана се въвеждат:
|
|
0,2 ml разтвор Б (6.1.1), |
|
|
1 ml етилацетат (4.1), |
|
|
0,1 ml метанол (4.4). |
Метилирането продължава в съответствие с описанието от 6.3. В хроматографа се впръскват 1,5 μl от получения разтвор.
7. ГАЗОВА ХРОМАТОГРАФИЯ
Колоната трябва да осигурява разделителна способност „R“, най-малко равна или по-голяма от 1,5, където:
![]()
където:
|
r1 и r2 |
= |
времената на задържане (в минути), |
|
W1 и W2 |
= |
широчините на пиковете при полувисочината (в милиметри), |
|
d′ |
= |
скоростта на хартията (в милиметри за минута). |
Като подходящи са установени следните условия на газова хроматография:
Колона: неръждаема стомана.
Дължина: 1,7 m.
Диаметър: 3 mm.
Носител:
|
|
chromosorb: WAW |
|
|
ситов анализ: 80 до 100 mesh. |
Неподвижна фаза: 10 % OV 17.
Температури:
|
|
колона: 280 °С. |
|
|
инжектор: 280 °С. |
|
|
детектор: 280 °С. |
Газ-носител: азот, несъдържащ кислород.
Налягане: 2,3 bar.
Дебит: 30 ml/мин.
8. ИЗЧИСЛЕНИЯ
8.1. Коефициент на пропорционалност на хексахлорофена
Коефициентът се изчислява спрямо избрания стандарт по отношение на стандартната смес.
Където:
|
h |
= |
хексахлорофен, |
|
kh |
= |
неговия коефициент на пропорционалност, |
|
m′h |
= |
неговата маса (в грама) в сместа, |
|
A′h |
= |
площта на неговия пик, |
|
s |
= |
избрания стандарт, |
|
m′s |
= |
неговата маса (в грама) в сместа, |
|
A′s |
= |
площта на неговия пик, |
тогава:
![]()
8.2. Количеството на хексахлорофен в пробата
Където:
|
h |
= |
хексахлорофен, |
|
kh |
= |
неговия коефициент на пропорционалност, |
|
Ah |
= |
площта на неговия пик, |
|
s |
= |
избрания стандарт, |
|
ms |
= |
неговата маса (в грама) в сместа, |
|
As |
= |
площта на неговия пик, |
|
М |
= |
масата (в грама) на взетата проба, |
тогава % (m/m) на хексахлорофена в пробата е:

9. ПОВТОРЯЕМОСТ (2)
За съдържание на хексахлорофен 0,1 % (m/m) разликата между резултатите от две успоредни изпитвания, проведени с една и съща проба, не трябва да надхвърля по абсолютна стойност 0,005 % (m/m).
ОПРЕДЕЛЯНЕ НА НАТРИЕВ ТОЗИЛХЛОРАМИД (ХЛОРАМИН-Т)
1. ЦЕЛ И ПРИЛОЖНО ПОЛЕ
Този метод се отнася за определянето на натриевия тозилхлорамид (хлорамин-Т) в козметичните продукти чрез тънкослойна хроматография.
2. ОПРЕДЕЛЕНИЕ
Съдържанието на хлорамин-Т в пробата, определено в съответствие с настоящия метод, се изразява в проценти спрямо масата (m/m).
3. ПРИНЦИП
Хлорамин-Т се хидролизира напълно до 4-толуолсулфонамид чрез кипене със солна киселина.
Количеството на образувалия се 4-толуолсулфонамид се определя фотоденсиометрично чрез тънкослойна хроматография.
4. РЕАКТИВИ
Всички реактиви трябва да са с аналитична чистота.
4.1. Натриев тозилхлорамид (хлорамин-Т).
4.2. Стандартен разтвор на 4-толуолсулфонамид: 50 mg 4-толуолсулфонамид в 100 ml етанол (4.5).
4.3. Солна киселина, 37 % (m/m), = 1,18 g/ml.
= 1,18 g/ml.
4.4. Диетилов етер.
4.5. Етанол, 96 % (v/v).
4.6. Подвижна фаза
4.6.1. 1-бутанол етанол (4.5) вода (40: 4: 9; v/v/v), или
4.6.2. Хлороформ ацетон (6: 4; v/v).
4.7. Готови ТСХ плаки, силикагел 60, без флуоресциращ индикатор.
4.8. Калиев перманганат.
4.9. Солна киселина, 15 % (m/m).
4.10. Реактив за разпръскване: 2-толуидин, 1 % (m/v) разтвор в етанол (4.5).
5. АПАРАТУРА
5.1. Стандартна лабораторна апаратура.
5.2. Стандартно оборудване за тънкослойна хроматография.
5.3. Фотоденситометър.
6. НАЧИН НА РАБОТА
6.1. Хидролиза
В 50-милилитрова облодънна колба се претегля точно около 1 g от пробата (m). Добавят се 5 ml вода и 5 ml солна киселина (4.3) и се кипва на обратен хладник в продължение на един час. Топлата суспензия незабавно се прехвърля с вода в 50-милилитров пикнометър. Оставя се да се охлади и се долива до марката с вода. Центрофугира се най-малко 5 минути на 3 000 оборота в минута и повърхностният слой се филтрира.
6.2. Екстракция
6.2.1. Вземат се 30 ml от филтрата и се подлагат на трикратна екстракция с 15 ml диетилов етер (4.4). При необходимост етерните фази се изсушават и се събират в 50-милилитров пикнометър. Долива се до марката с диетилов етер (4.4).
6.2.2. Вземат се 25 ml от сухите етерни екстракти и се изпаряват до сухо в поток от азот. Остатъкът се разтваря отново с 1 ml етанол (4.5).
6.3. Тънкослойна хроматография
6.3.1. Върху плаката за тънкослойна хроматография (4.7) се нанасят 20 μl от етаноловия остатък (6.2).
В същото време и по същия начин се нанасят съответно 8, 12, 16 и 20 μl от стандартния разтвор на 4-толуолсулфонамида (4.2).
6.3.2. След това се развива на височина от около 150 mm с развиващия разтворител (4.6.1 или 4.6.2).
6.3.3. След пълното изпаряване на развиващия разтворител плаката се поставя за 2—3 минути в атмосфера от хлорни пари, която се получава чрез изливане на около 100 ml солна киселина (4.9) върху около 2 g калиев перманганат (4.8) в затворен съд. Излишъкът от хлор се отстранява чрез загряване на плаката до 100 °С за 5 минути. След това плаката се напръсква с реактив (4.10).
6.4. Измерване
След около 1 час се измерват лилавите петна с помощта на фотоденситометър при 525 nm.
6.5. Построяване на калибровъчната крива
Нанасят се стойностите на максималната височина на получените пикове, установени за четирите 4-толуолсулфонамидни петна, спрямо съответните количества 4-толуолсулфонамид (например, 4, 6, 8, 10 μg 4-толуолсулфонамиди на петно).
7. ЗАБЕЛЕЖКА
Точността на метода може да се провери чрез работа с 0,1 или 0,2 % (m/v) разтвор на хлорамин-Т (4.1), обработен по същия начин както пробата (6).
8. ИЗЧИСЛЕНИЯ
Съдържанието на хлорамин-Т в пробата, изразено в проценти спрямо масата, се изчислява както следва:
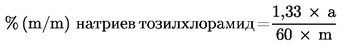
където:
|
1,33 |
= |
коефициента на превръщане за 4-толуолсулфонамид на хлорамин-Т, |
|
а |
= |
количеството (в μg) 4-толуолсулфонамид в пробата според отчетеното върху калибровъчните криви, |
|
m |
= |
масата (в грамове) на взетата проба. |
9. ПОВТОРЯЕМОСТ (2)
За съдържание на хлорамин-Т около 0,2 % (m/m) разликата между резултатите две успоредни изпитвания, проведени с една и съща проба, не трябва да надхвърля по абсолютна стойност 0,03 % (m/m).
ОПРЕДЕЛЯНЕ НА ОБЩИЯ ФЛУОР В ПАСТИТЕ ЗА ЗЪБИ
1. ЦЕЛ И ПРИЛОЖНО ПОЛЕ
Този метод е предназначен за определяне на общия флуор в пастите за зъби. Методът е подходящ за концентрации, ненадхвърлящи 0,25 %.
2. ОПРЕДЕЛЕНИЕ
Съдържанието на флуор в пробата, определено в съответствие с настоящия метод, се изразява в проценти.
3. ПРИНЦИП
Определянето се извършва чрез газова хроматография. Флуорът от флуор-съдържащите съединения се превръща в кисела среда с хлортриетилсилан (TECS) в триетилфлуорсилан (ТЕFS) и едновременно с това се екстрахира с ксилол, съдържащ като вътрешен стандарт циклохексан.
4. РЕАКТИВИ
Всички реактиви трябва да са с аналитична чистота.
4.1. Натриев флуорид, изсушен при 120 °С до постоянна маса.
4.2. Вода, двойно дестилирана или с еквивалентно качество.
4.3. Солна киселина, = 1,19 g/ml.
= 1,19 g/ml.
4.4. Циклохексан (СН).
4.5. Ксилен без пикове в хроматограмата непосредствено преди пика на разтворителя, когато последният се хроматографира при същите условия както пробата (6.1). При необходимост се пречиства чрез дестилация (5.8).
4.6. Хлортриетилсилан (TECS Merck или еквивалентен).
4.7. Стандартни разтвори на флуор
4.7.1. Изходен разтвор, 0,250 mg F-ml. Претеглят се прецизно 138,1 mg натриев флуорид (4.1) и се разтварят във вода (4.2). Разтворът се прехвърля количествено в 250-милилитров пикнометър (5.5). Разрежда се до марката с вода (4.2) и се разбърква.
4.7.2. Разреден изходен разтвор, 0,050 mg F-ml. С пипета 20 ml от изходния разтвор (4.7.1) се прехвърлят в 100-милилитров пикнометър (5.5). Разрежда се до марката с вода и се разбърква.
4.8. Разтвор на вътрешен стандарт
Смесват се 1 ml циклохексан (4.4) и 5 ml ксилол (4.5).
4.9. Хлоротриетилсилан — разтвор на вътрешен стандарт
В 10-милилитров пикнометър с пипета (5.7) се прехвърлят 0,6 ml от TECS (4.6) и 0,12 ml от разтвора на вътрешен стандарт (4.8). Разрежда се с ксилол (4.5) до марката и се разбърква. Разтворът се приготвя ежедневно.
4.10. Перхлорна киселина, 70 % (m/v).
4.11. Перхлорна киселина, 20 % (m/v) във вода (4.2).
5. АПАРАТУРА
5.1. Стандартно лабораторно оборудване.
5.2. Газов хроматограф, снабден с пламъчно-йонизационен детектор.
5.3. Хомогенизатор Vortex или еквивалентен апарат.
5.4. Клатачна машина Buhler, тип SMB1 или еквивалентен.
5.5. Пикнометри, 100 и 250 ml, от полипропилен.
5.6. Центрофужни епруветки (стъклени); 20 ml, с винтови запушалки с тефлоново покритие, тип Sovirel 611-56 или еквивалентни. Епруветките и резбовите капачки се почистват в продължение на няколко часа в перхлорна киселина (4.11), изплакват се пет пъти с вода (4.2) и се сушат при 100 °С.
5.7. Регулируеми пипети, които могат да осигуряват обеми от 50 до 200 μl, с пластмасови накрайници за еднократна употреба.
5.8. Апарат за дестилация, снабден с колона Schneider с три сфери или еквивалентна колона Vigreux.
6. НАЧИН НА РАБОТА
6.1. Анализ на пробата
6.1.1. Избира се неотваряна туба с паста за зъби, разрязва се и цялото съдържание се прехвърля в пластмасов съд, разбърква се добре и сместа се съхранява в условия, които изключват възможността от влошаване на нейните качества.
6.1.2. В центрофужна епруветка (5.6) се претеглят точно 150 mg (m) от пробата, добавят се 5 ml вода (4.2) и се хомогенизира (5.3).
6.1.3. Добавя се 1 ml ксилол (4.5).
6.1.4. На капки се добавят 5 ml солна киселина (4.3) и се хомогенизира (5.3).
6.1.5. В центрофужната епруветка (5.6) с пипета се добавят 0,5 ml разтвор на вътрешен стандарт на хлортриетилсилан (4.9).
6.1.6. Епруветката се затваря с винтова капачка (5.6) и се хомогенизира в продължение на 45 минути на вибратор (5.4), настроен за 150 разклащания за минута.
6.1.7. Центрофугира се в продължение на 10 минути при такава скорост, че да се получи ясно разделяне на фазите, капачката на епруветката се отвинтва, органичният слой се отстранява и върху колоната на газовияхроматограф (5.2) се впръскват 3 μl от органичната фаза.
Забележка: За елуирането на всички компоненти са необходими около 20 минути.
6.1.8. Впръскването се повтаря, изчислява се средното съотношение между площите на пиковете (АТЕFS/АCH) и от калибровъчната крива (6.3) се отчита съответното количество флуор (в милиграми (m1)).
6.1.9. Общото съдържание на флуор в пробата (в проценти спрямо масата флуор) се изчислява съгласно посоченото в точка 7.
6.2. Хроматографски условия
6.2.1. Колона: неръждаема стомана.
Дължина: 1,8.
Диаметър: 3 mm.
Носител: Gaschrom Q 80 до 100 mesh.
Неподвижна фаза: силиконово масло DC 200 или еквивалентно, 20 %. Колоната се кондиционира в нощта преди анализа при 100 °С (дебит на газ-носителя: 25 ml азот за минута).Тази операция се повтаря всяка нощ. След всяко четвърто или пето впръскване колоната се рекондиционира чрез нагряване при 100 °С в продължение на 30 минути.
Температури:
|
|
Колона: 70 °С, |
|
|
Инжектор: 150 °С, |
|
|
Детектор: 250 °С. |
Дебит на газ-носителя: 35 ml азот за минута.
6.3. Калибровъчна крива
6.3.1. В поредица от шест центрофужни епруветки (5.6) с пипета се капват съответно 0, 1, 2, 3, 4 и 5 ml от разредения стандартен разтвор на флуор (4.7.2). Обемът във всяка епруветка се довежда до 5 ml с вода (4.2).
6.3.2. Процедира се в съответствие с описаното в точки от 6.1.3 до 6.1.6 включително.
6.3.3. Върху колоната на газовия хроматографа (5.2) се впръскват 3 μl от органичната фаза.
6.3.4. Впръскването се повтаря и се изчислява средното съотношение между площите на пиковете (АТЕFS/АCH).
6.3.5. Построява се калибровъчна крива, корелираща масата на флуора (в милиграми) в стандартните разтвори (6.3.1) със съотношението на площите на пиковете (АТЕFS/АCH), измерено в съответствие с 6.3.4. Точките от графиката се свързват с най-добре съответстващата права линия, изчислена чрез регресионен анализ.
7. ИЗЧИСЛЕНИЕ
Общото съдържание на флуор в пробата (като проценти спрямо масата флуор), (% (m/m)) се дава от:

където:
|
m |
= |
частта от пробата за изследване (в милиграми) (6.1.2), |
|
m1 |
= |
количеството F (в милиграми), отчетено от калибровъчната крива (6.1.8). |
8. ПОВТОРЯЕМОСТ (2)
За съдържанието на флуор около 0,15 % (m/m) разликата между резултатите от две успоредни изпитвания, проведени с една и съща проба, не трябва да надхвърля по абсолютна стойност 0,012 % (m/m).
ИДЕНТИФИКАЦИЯ И ОПРЕДЕЛЯНЕ НА ОРГАНОЖИВАЧНИ СЪЕДИНЕНИЯ
ЦЕЛ И ПРИЛОЖНО ПОЛЕ
Този метод може да се използва за идентификация и определяне на органоживачните производни, употребявани като консерванти в състава на козметичните продукти за очи. Методът е приложим към тиомерсал (МНН) (натриев 2-(етилмеркуритио)бензоат) и фенилмеркури и неговите соли.
А. ИДЕНТИФИКАЦИЯ
1. ПРИНЦИП
Органоживачните съединения образуват комплекс с 1,5-дифенил-3-тиокарбазон. След екстракция на дитизоната с тетрахлорметан се провежда тънкослойна хроматография върху силикагел. Петната на дитизонатите са оранжево оцветени.
2. РЕАКТИВИ
Всички реактиви трябва да са с аналитична чистота.
2.1. Сярна киселина, 25 % (v/v).
2.2. 1,5-дифенил-3-тиокарбазон (дитизон): 0,8 mg в 100 ml тетрахлорметан (2.4).
2.3. Азот.
2.4. Тетрахлорметан.
2.5. Подвижна фаза: хексан/ацетон, 90:10 (v/v).
2.6. Стандартен разтвор, 0,001 % във вода, на:
натриев 2-(етилмеркуритио)бензоат,
етилмеркурихлорид или метилмеркурихлорид,
фенилмеркуринитрат или фенилмеркуриацетат,
меркури дихлорид или меркури ди(ацетат).
2.7. Готови ТСХ плаки със силикагел (например Merck 5721 или еквивалентни).
2.8. Натриев хлорид.
3. АПАРАТУРА
3.1. Стандартно лабораторо оборудване.
3.2. Стандартно оборудване за тънкослойна хроматография.
3.3. Филтър за разделяне на фазите.
4. НАЧИН НА РАБОТА
4.1. Екстракция
4.1.1. 1 g от пробата се разрежда чрез титруване с 20 ml дестилирана вода в центрофужна епруветка. Постига се максимално диспергиране и се загрява до 60 °С на водна баня. Добавят се 4 g натриев хлорид (2.8). Разклаща се. Оставя се да се охлади.
4.1.2. Центрофугира се най-малко 20 минути при 4 500 оборота за минута с цел отделяне на по-голямата част от твърдата фаза от течността. Филтрира се в делителна фуния и се добавят 0,25 ml разтвор на сярна киселина (2.1).
4.1.3. Екстрахира се няколко пъти с 2 или 3 ml разтвор на дитизон (2.2), докато последната органична фаза остане зелена.
4.1.4. Последователно се филтрира всяка органична фаза през филтъра за разделяне на фазите (3.3).
4.1.5. Изпарява се до сухо в поток от азот (2.3).
4.1.6. Разтваря се с 0,5 ml тетрахлорметан (2.4). Този разтвор се нанася незабавно така, както е посочено в 4.2.1.
4.2. Разделяне и идентификация
4.2.1. Върху плаката с покритие от силикагел (2.7) се нанасят незабавно 50 μl от получения в 4.1.6 разтвор на тетрахлорметан. Едновременно с това се обработват 10 ml от стандартния разтвор (2.6), както е посочено в 4.1 и върху същата плака се нанасят 50 μl от получения в 4.1.6 разтвор.
4.2.2. Плаката се поставя в разтворителя (2.5), като на последния се позволява да се издигне на 150 mm. Органоживачните съединения се открояват като оцветени петна, чийто цвят е стабилен, ако веднага след изпаряване на разтворителя плаката се покрие със стъклена плоча.
Като пример, се получават следните Rf-стойности:
|
|
Rf |
Цвят |
|
Тиомерсал |
0,33 |
Оранжев |
|
Етилмеркурихлорид |
0,29 |
Оранжев |
|
Метилмеркурихлорид |
0,29 |
Оранжев |
|
Фенилмеркурихлорид |
0,21 |
Оранжев |
|
Живачни (II) соли |
0,10 |
Оранжев |
|
Меркури ди(ацетат) |
0,10 |
Оранжев |
|
1,5-дифенил-3-тиокарбазон |
0,09 |
Розов |
Б. ОПРЕДЕЛЯНЕ
1. ОПРЕДЕЛЕНИЕ
Съдържанието на органоживачни съединения, определено по този метод, се изразява като проценти спрямо масата (m/m) живак в пробата.
2. ПРИНЦИП
Методът се състои в измерване на количеството на общия наличен живак. Поради това на първо място е необходимо да се установи, че в пробата няма съдържание на живак в неорганично състояние и да се идентифицира органоживачното производно, което се съдържа в пробата. След минерализиране освободеният живак се измерва чрез безпламъкова атомна абсорбция.
3. РЕАКТИВИ
Всички реактиви трябва да са с аналитична чистота.
3.1. Концентрирана азотна киселина, = 1,41 g/ml.
= 1,41 g/ml.
3.2. Концентрирана сярна киселина,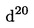 = 1,84 g/ml.
= 1,84 g/ml.
3.3. Двойно дестилирана вода.
3.4. Калиев перманганат, 7 % (m/v) разтвор.
3.5. Хидроксиламониев хлорид, 1,5 % (m/v) разтвор.
3.6. Дикалиев пероксодисулфат, 5 % (m/v) разтвор.
3.7. Калаен дихлорид, 10 % (m/v) разтвор.
3.8. Концентрирана солна киселина,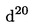 = 1,18 g/сm3 (ml)
= 1,18 g/сm3 (ml)
3.9. Импрегнирана с паладиев дихлорид стъклена вата, 1 % (m/m).
4. АПАРАТУРА
4.1. Стандартно лабораторно оборудване.
4.2. Апарат за безпламъково атомно-абсорбционно определяне на живак (метод с охладени пари), включително необходимата стъклария. Оптичната траектория на клетката трябва да бъде най-малко 100 mm.
5. НАЧИН НА РАБОТА
Трябва да се вземат всички обичайни предпазни мерки, отнасящи се до анализите на следи от живак.
5.1. Процедура
5.1.1. Претеглят се точно150 mg от пробата (m). Добавят се 10 ml азотна киселина (3.1) и разтворът се оставя да се извари в продължение на три часа в херметично затворена колба на водна баня при 55 °С, като се разклаща през равномерни интервали от време. В същото време се извършва анализ на празна проба с реактивите.
5.1.2. След охлаждане се добавят 10 ml сярна киселина (3.2) и разтворът се връща на водната баня при 55 °С, където престоява в продължение на 30 минути.
5.1.3. Колбата се поставя в ледена баня и внимателно се добавят 20 ml вода (3.3).
5.1.4. Добавят се 2 ml аликвотни части от 7 % разтвор на калиев перманганат (3.4), докато разтворът се оцвети. Връща се отново на водна баня при 55 °С за още 15 минути.
5.1.5. Добавят се 4 ml от разтвора на дикалиевия пероксодисулфат (3.6). Затоплянето на водната баня продължава при 55 °С за 30 минути.
5.1.6. Оставя се да се охлади и съдържанието на колбата се прехвърля в 100-милилитрова стандартна колба. Колбата се промива с 5 ml хидроксиламониев хлорид (3.5), след което четирикратно се изплаква с 10 ml вода (3.3). Разтворът трябва да бъде напълно обезцветен. Долива се до марката с вода (3.3).
5.2. Определяне
5.2.1. 10 ml от предназначения за анализ разтвор (5.1.6) се поставят в стъкления съд за определяне на живака по метода с охладените пари (4.2). Разрежда се със 100 ml вода (3.3) и след това с 5 ml сярна киселина (3.2) и 5 ml разтвор на калаен дихлорид (3.7). След всяко добавяне се хомогенизира. Изчакват се 30 секунди за редуциране на всички живачни йони до метално състояние и данните се отчитат (n).
5.2.2. Между съда за редуциране на живака и клетката за потока на прибора (4.2) се поставя известно количество импрегнирана с паладиев дихлорид стъклена вата (3.9). Повтаря се процедурата от 5.2.1 и данните се записват. Ако показанието не е нула, минерализацията е била непълна и анализът трябва да се повтори.
6. ИЗЧИСЛЕНИЯ
Където:
|
m |
= |
масата (в милиграми) на изследваната проба, |
|
n |
= |
количеството живак (в µg), отчетено от прибора. |
Количеството на живака, изразено като живак в проценти, се изчислява в съответствие със следната формула:
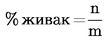
7. ЗАБЕЛЕЖКИ
7.1. За да се подобри минерализацията, може да е необходимо да се започне с разреждане на пробата.
7.2. Ако има подозрения за протичаща абсорбция на живака от субстрата, трябва да се извърши определяне по метода на стандартните добавяния.
8. ПОВТОРЯЕМОСТ (2)
За концентрации на живака от 0,007 % разликата между резултатите от две успоредни изпитвания, извършени върху пробата, не трябва да надхвърля абсолютната стойност от 0,00035 %.
ОПРЕДЕЛЯНЕ НА АЛКАЛНИ И АЛКАЛОЗЕМНИ СУЛФИДИ
1. ЦЕЛ И ПРИЛОЖНО ПОЛЕ
Този метод описва определянето на сулфидите в състава на козметичните продукти. Наличието на тиоли или други редуциращи агенти (включително сулфити) не пречи.
2. ОПРЕДЕЛЕНИЕ
Концентрацията на сулфиди, определена по този метод, се изразява като съдържание на сяра в проценти.
3. ПРИНЦИП
След подкисляване на средата, сероводородът се увлича с поток от азот, след което се фиксира под формата на кадмиев сулфид. Последният се филтрира и промива, след което неговото съдържание се определя по йодометричен път.
4. РЕАКТИВИ
Всички реактиви трябва да са с аналитична чистота.
4.1. Концентрирана солна киселина,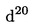 = 1,19 g/ml.
= 1,19 g/ml.
4.2. Натриев тиосулфат, 0,1 М стандартен разтвор.
4.3. Йод, 0,05 М стандартен разтвор.
4.4. Динатриев сулфид.
4.5. Кадмиев ди(ацетат).
4.6. Концентриран амоняк, = 0,90 g/ml.
= 0,90 g/ml.
4.7. Амонячен разтвор на кадмиев ди(ацетат): 10 g кадмиев ди(ацетат) (4.5) се рзтварят в около 50 ml вода. Добавя се амоняк (4.6) докато утайката се разтвори отново (т.е. приблизително 20 ml). Долива се с вода до 100 ml.
4.8. Азот.
4.9. Разтвор на амоняк М.
5. АПАРАТУРА
5.1. Стандартно лабораторно оборудване.
5.2. 100 милилитрова облодънна колба с три стандартни гърла от шлифовано стъкло.
5.3. Две 150-милилитрови конични колби с гърла от шлифовано стъкло, снабдени с приспособление, включващо потапяща тръба, и странична тръба за извеждане на засмукания газ.
5.4. Една фуния с дълго стеснение.
6. НАЧИН НА РАБОТА
6.1. Засмукване на сулфидите
6.1.1. Взема се опаковка, която не е била отваряна преди това. В облодънната колба (5.2) се претегля прецизно такава маса (m) (изразена в грамове) от продукта, която съответства на не повече от 30 mg сулфидни йони. Добавят се 60 ml вода и две капки пеногасител.
6.1.2. Във всяка от двете конични колби (5.3) се прехвърлят по 50 ml от разтвора (4.7).
6.1.3. Облодънната колба (5.2) се съединява с фунията, потапящата тръба и изходящата тръба. Изходящата тръба се съединява с конични колби (5.3), свързани последователно с помощта на тръби от PVC.
NB: Апаратът за засмукване на газа трябва да бъде подложен на следното изпитване за херметичност: симулирайки условията на изпитването, предназначеният за анализ продукт се подменя с 10 ml сулфиден разтвор (приготвен от 4.4), съдържащ „Х“ mg сулфид (определен йодометрично). Нека „Y“ бъде броят милиграми от сулфида, засечени в края на тази операция. Разликата между количеството „Х“ и количеството „Y“ не трябва да бъде по-голяма от 3 %.
6.1.4. Пропуска се азот (4.8) в продължение на 15 минути със скорост две мехурчета за секунда за изкарване на въздуха, съдържащ се в облодънната колба (5.2).
6.1.5. Облодънната колба се подгрява до 85 ± 5 °С.
6.1.6. Подаването на азота (4.8) се преустановява и капка по капка се добавят 40 ml солна киселина (4.1).
6.1.7. Подаването на азот (4.8) се възобновява към момента на прехвърлянето на почти цялото количество на киселината, като се остави минимално течностно уплътнение, което да предотврати изтичането на сероводорода.
6.1.8. Подгряването се прекратява след 30 минути. Колбата (5.2) се оставя да се охлади и пропускането на азота (4.8) продължава поне още един час и половина.
6.2. Титруване
6.2.1. Кадмиевият сулфид се филтрира с помощта на фунията с дългото стеснение (5.4).
6.2.2. Коничните колби (5.3) се промиват с амонячен разтвор (4.9) и съдържанието се излива върху филтъра. След това се промиват с дестилирана вода и тази вода се използва за промиване на утайката върху филтъра преципитат.
6.2.3. Промиването на утайката завършва със 100 ml вода.
6.2.4. Хартиеният филтър се поставя в първата конична колба, в която се е намирала утайката. Добавят се 25 ml (n1) разтвор на йод (4.3), приблизително 20 ml солна киселина (4.1) и 50 ml дестилирана вода.
6.2.5. Определя се излишъкът на йод с помощта на разтвора на натриевия тиосулфат (n2) (4.2).
7. ИЗЧИСЛЕНИЯ
Съдържанието на сулфида в пробата, изразено като сяра в проценти, се изчислява по следната формула:

където:
|
n1 |
= |
броя милилитри от използвания стандартен разтвор на йод (4.3), |
|
x1 |
= |
моларността на този разтвор, |
|
n2 |
= |
броя милилитри от използвания стандартен разтвор на натриев тиосулфат (4.2), |
|
x2 |
= |
моларността на този разтвор, |
|
m |
= |
масата (в грама) на изследваната проба. |
8. ПОВТОРЯЕМОСТ (2)
За съдържание на сулфид около 2 % (m/m) разликата между резултатите от две успоредни изпитвания, извършени върху една и съща проба, не трябва да надхвърля абсолютната стойност от 0,2 % (m/m).
(1) ОВ L 383, 31.12.1980 г., стр. 27.
(2) Съгласно стандарт ISO 5725.
NB: Определянето на меркаптооцетната киселина трябва да се извърши върху неупотребяван продукт от току-що отворени контейнери с оглед предотвратяването на окислението.
(4) Поради голямото разнообразие на видовете продукти, в които може да се съдържа хексахлорофен, от значение е първо да се провери извличането на хексахлорофена от пробата по този метод, преди да се запишат резултатите. Ако извлечените количества са малки, със съгласието на заинтересованите страни могат да се направят промени като смяна на разтворителя (бензол вместо етилацетат и т.н.).
(5) Устойчивостта на това жълто оцветяване доказва наличието на излишък от диазометан, необходим за осигуряването на пълното метилиране на пробата.