

EUR-Lex Access to European Union law
This document is an excerpt from the EUR-Lex website
Document 31985L0490
Fourth Commission Directive 85/490/EEC of 11 October 1985 on the approximation of the laws of the Member States relating to methods of analysis necessary for checking the composition of cosmetic products
Четвърта Директива на Комисията от 11 октомври 1985 година за сближаване на законодателствата на държавите-членки относно методите за анализ, необходими за контрола върху състава на козметичните продукти
Четвърта Директива на Комисията от 11 октомври 1985 година за сближаване на законодателствата на държавите-членки относно методите за анализ, необходими за контрола върху състава на козметичните продукти
OJ L 295, 7.11.1985, p. 30–45
(DA, DE, EL, EN, FR, IT, NL)
Spanish special edition: Chapter 15 Volume 006 P. 115 - 130
Portuguese special edition: Chapter 15 Volume 006 P. 115 - 130
Special edition in Finnish: Chapter 13 Volume 014 P. 192 - 207
Special edition in Swedish: Chapter 13 Volume 014 P. 192 - 207
Special edition in Czech: Chapter 13 Volume 008 P. 46 - 61
Special edition in Estonian: Chapter 13 Volume 008 P. 46 - 61
Special edition in Latvian: Chapter 13 Volume 008 P. 46 - 61
Special edition in Lithuanian: Chapter 13 Volume 008 P. 46 - 61
Special edition in Hungarian Chapter 13 Volume 008 P. 46 - 61
Special edition in Maltese: Chapter 13 Volume 008 P. 46 - 61
Special edition in Polish: Chapter 13 Volume 008 P. 46 - 61
Special edition in Slovak: Chapter 13 Volume 008 P. 46 - 61
Special edition in Slovene: Chapter 13 Volume 008 P. 46 - 61
Special edition in Bulgarian: Chapter 13 Volume 007 P. 43 - 58
Special edition in Romanian: Chapter 13 Volume 007 P. 43 - 58
Special edition in Croatian: Chapter 13 Volume 009 P. 113 - 128
 In force
In force
|
13/ 07 |
BG |
Официален вестник на Европейския съюз |
43 |
31985L0490
|
L 295/30 |
ОФИЦИАЛЕН ВЕСТНИК НА ЕВРОПЕЙСКИЯ СЪЮЗ |
ЧЕТВЪРТА ДИРЕКТИВА НА КОМИСИЯТА
от 11 октомври 1985 година
за сближаване на законодателствата на държавите-членки относно методите за анализ, необходими за контрола върху състава на козметичните продукти
(85/490/ЕИО)
КОМИСИЯТА НА ЕВРОПЕЙСКИТЕ ОБЩНОСТИ,
като взе предвид Договора за създаване на Европейската икономическа общност,
като взе предвид Директива 76/768/ЕИО на Съвета от 27 юли 1976 г. за сближаване на законодателствата на държавите-членки относно козметичните продукти (1), последно изменена с Директива 85/391/ЕИО (2), и по-специално член 8, параграф 1 от нея,
като има предвид, че Директива 76/768/ЕИО предвижда официални проверки на козметичните продукти с цел да се установи спазването на условията, предвидени в общностните разпоредби относно състава на козметичните продукти;
като има предвид, че следва да се установят възможно най-бързо всички необходими за целите на анализите методи; като има предвид, че вече бяха предприети три стъпки към реализирането на тази цел с определянето на някои методи в Директиви 80/1335/ЕИО (3), 82/434/ЕИО (4) и 83/514/ЕИО (5) на Комисията, а четвъртата стъпка се състои в определянето на методи за идентификация и дозировка на глицерол 1-(4-аминобензоат), дозировка на хлорбутанол, идентификация и дозировка на хинин, идентификация и дозировка на неорганични сулфити и бисулфити, идентификация и дозировка на хлорати на алкалните метали и идентификация и дозировка на натриев йодат;
като има предвид, че мерките, предвидените в настоящата директива, са в съответствие със становището на Комитета за привеждане в съответствие с техническия прогрес на Директива 76/768/ЕИО,
ПРИЕ НАСТОЯЩАТА ДИРЕКТИВА:
Член 1
Държавите-членки предприемат всички необходими мерки, за да гарантират, че в хода на официалните проверки на козметичните продукти:
|
— |
идентификацията и дозировката на глицерол 1-(4-аминобензоат), |
|
— |
дозировката на хлорбутанол, |
|
— |
идентификацията и количественото определяне на хинина, |
|
— |
идентификацията и дозировката на неорганичните сулфити и бисулфити, |
|
— |
идентификацията и дозировката на хлорати на алкалните метали, и |
|
— |
идентификацията и дозировката на натриев йодат |
ще се извършват съгласно описаните в приложението методи.
Член 2
Държавите-членки въвеждат в сила законовите, подзаконовите и административните разпоредби, необходими, за да се съобразят с настоящата директива, най-късно до 31 декември 1986 г.
Държавите-членки незабавно уведомяват Комисията за това.
Член 3
Адресати на настоящата директива са държавите-членки.
Съставено в Брюксел на 11 октомври 1985 година.
За Комисията
Stanley CLINTON-DAVIS
Член на Комисията
(1) ОВ L 262, 27.9.1976 г., стр. 169.
(2) ОВ L 224, 22.8.1985 г., стр. 40.
(3) ОВ L 383, 31.12.1980 г., стр. 27.
(4) ОВ L 185, 30.6.1982 г., стр. 1.
(5) ОВ L 291, 24.10.1983 г., стр. 9.
ПРИЛОЖЕНИЕ
ИДЕНТИФИКАЦИЯ И ДОЗИРОВКА НА ГЛИЦЕРОЛ 1-(4-АМИНОБЕНЗОАТ)
А. ИДЕНТИФИКАЦИЯ
1. ЦЕЛ И ПРИЛОЖНО ПОЛЕ
Този метод е предназначен за откриване на алфа-моноглицерил 4-аминобензоат (глицерол 1-(4-аминобензоат). Той позволява да се идентифицира и етил 4-аминобензоат (бензокаин МОН), който може да се съдържа като примес.
2. ПРИНЦИП
Идентификацията се извършва чрез тънкослойна хроматография върху силикагел в присъствие на флуоресциращ индикатор и откриване на свободната първична аминогрупа чрез образуване на диазо-багрило върху плаката.
3. РЕАКТИВИ
Всички реактиви трябва да бъдат с аналитична чистота.
3.1. Смес от разтворители: циклохексан/изопропанол/стабилизиран дихлорметан 48/64/9 (v/v/v).
3.2. Проявяващ разтворител: разтвор на петролев етер (40-60)/бензен/ацетон/амониев хидроокис (минимум 25 % амоняк): 35/35/35/1 (v/v/v/v).
|
Проявяващ разтвор: |
|
3.4. Стандартни разтвори:
алфа-моноглицерил 4-аминобензоат: 0,05 г в 100 мл от смесения разтворител 3.1;
етил 4-аминобензоат: 0,05 г в 100 мл от смесения разтворител 3.1.
3.5. Плаки с покритие от силикагел 60 F254 с дебелина 0,25 мм и формат 200 мм х 200 мм.
4. АПАРАТУРА
4.1. Стандартно оборудване за тънкослойна хроматография.
4.2. Ултразвуков вибратор.
4.3. Микропорест филтър FH 0,5 мкм или еквивалентен.
5. ПРОЦЕДУРА
5.1. Приготвяне на пробата
В 10-милилитров пикнометър със запушалка се претеглят 1,5 г от предназначения за анализ продукт. Долива се до марката с разтворителя 3.1. Поставя се запушалката и колбата се оставя в продължение на един час на стайна температура в ултразвуковия вибратор (4.2). Разтворът се филтрира през микропорестия филтър (4.3) и филтратът се използва за целите на хроматографския анализ.
5.2. Тънкослойна хроматография
Върху плаката (3.5) се нанасят 10 мкл от разтвора на пробата (5.1) и от всеки от стандартните разтвори (3.4).
Хроматограмата се развива до височина 150 мм в предварително наситената с разтворителя 3.2 камера. Плаката се оставя да изсъхне на стайна температура.
5.3. Проявяване
5.3.1. Плаката се изследва на ултравиолетова светлина при 254 нм.
5.3.2. Напълно изсъхналата плака се напръсква с разтвора 3.3 а).
Плаката се оставя да изсъхне на стайна температура в продължение на 1 минута, след което незабавно се напръсква с разтвора 3.3 б).
Плаката се изсушава в сушилня при 60 °С. Появяват се оранжево оцветени петна. Алфа-моноглицерил 4-аминобензоат: RF 0,07; етил 4-аминобензоат: RF 0,55.
Б. ДОЗИРОВКА
1. ЦЕЛ И ПРИЛОЖНО ПОЛЕ
Този метод определя дозировката на глицерол 1-(4-аминобензоат) алфа-моноглицерил 4-аминобензоат. Той определя и съдържанието на етил 4-аминобензоат. С метода не може да се определят повече от 5 % (m/m) от алфа-моноглицерил 4-аминобензоат и 1 % (m/m) от етил 4-аминобензоат.
2. ОПРЕДЕЛЯНЕ
Съдържанието на алфа-моноглицерил 4-аминобензоат и на етил 4-аминобензоат, измерено по този метод, се изразява в масови проценти (m/m) от продукта.
3. ПРИНЦИП
Предназначеният за анализ продукт се суспендира в метанол и след подходяща обработка на пробата същата се анализира чрез високоефективна течна хроматография (ВЕТХ).
4. РЕАКТИВИ
Всички реактиви трябва да бъдат с аналитична чистота и трябва да бъдат подходящи за ВЕТХ.
4.1. Метанол.
4.2. Калиев хидрогенортофосфат (KH2PO4).
4.3. Цинков ди(ацетат) (Zn(CH3COO)2.2H2О).
4.4. Оцетна киселина (d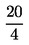 = 1,05).
= 1,05).
4.5. Тетракалиев хексацианоферат (K4(Fe(CN)6).3H2O).
4.6. Етил 4-хидроксибензоат.
4.7. Алфа-моноглицерил 4-аминобензоат.
4.8. Етил 4-аминобензоат.
4.9. Фосфатен буферен разтвор (0,02 М): 2,72 г калиев хидрогенортофосфат (4.2) се разтварят в един литър вода.
4.10. Елуиращ разтвор: фосфатен буферен разтвор (4.9)/метанол (4.1) 61/39 (v/v).
Съставът на подвижната фаза може да се променя, за да се постигне разделителна способност R ≥ 1,5.

където:
|
R1 и R2 |
= |
времена на задържане на пиковете, в минути, |
|
W1 и W2 |
= |
ширини на пиковете при полувисочината, в милиметри, |
|
d′ |
= |
скорост на въртене на хартията, в милиметри за минута. |
4.11. Изходен разтвор на алфа-моноглицерил 4-аминобензоат: претеглят се прецизно около 40 мг алфа-моноглицерил 4-аминобензоат и се поставят в 100-милилитров пикнометър. Разтваря се в 40 мл метанол (4.1). Долива се до марката с буферен разтвор (4.9) и се хомогенизира.
4.12. Изходен разтвор на етил 4-аминобензоат: претеглят се прецизно около 40 мг етил 4-аминобензоат и се поставят в 100-милилитров пикнометър. Разтваря в 40 мл метанол (4.1). Долива се до марката с буферния разтвор (4.9) и се хомогенизира.
4.13. Вътрешен стандартен разтвор: претеглят се прецизно около 50 мг етил 4-хидроксибензоат (4.6), прехвърлят се в 100-милилитрова стандартна колба, разтварят се в 40 мл метанол (4.1), извършва се доливане до марката с буферния разтвор (4.9) и се хомогенизира.
Стандартен разтвор: приготвят се четири стандартни разтвора чрез разтваряне в 100 мл елюиращ агент (4.10) в съответствие със следната таблица:
|
Стандартен разтвор |
Алфа-моноглицерил 4-аминобензоат |
Етил 4-аминобензоат |
Етил 4-хидроксибензоат |
|||
|
(мкг/мл) (1) |
мл (4.11) |
(мкг/мл) (1) |
мл (4.12) |
(мкг/мл) (1) |
мл (4.13) |
|
|
I |
8 |
2 |
8 |
2 |
50 |
10 |
|
II |
16 |
4 |
12 |
3 |
50 |
10 |
|
III |
24 |
6 |
16 |
4 |
50 |
10 |
|
IV |
40 |
10 |
20 |
5 |
50 |
10 |
4.15. Разтвор Carrez I: 26,5 г тетракалиев ферохексацианид (4.5) се разтварят във вода и се извършва доливане до 250 мл.
4.16. Разтвор Carrez II: 54,9 г цинков ди(ацетат) (4.3) и 7,5 мл оцетна киселина (4.4) се разтварят във вода и се извършва доливане до 250 мл.
4.17. Merck Lichrosorb RP-18 или еквивалентен със среден размер на частиците 5 мкм.
5. АПАРАТУРА
5.1. Обичайното лабораторно оборудване.
5.2. Високоефективен течен хроматограф с UV детектор с променлива дължина на вълната и термостатирана камера, настроена за 45 °С.
5.3. Колона от неръждаема стомана с дължина 250 мм; вътрешен диаметър 4,6 мм; пълнеж: Lichrosorb RP-18 (4.17).
5.4. Ултразвукова баня.
6. ПРОЦЕДУРА
6.1. Приготвяне на пробата
6.1.1. В 100-милилитрова бехерова чаша се претегля прецизно около 1 г от пробата и се добавят 10 мл метанол (4.1).
6.1.2. Бехеровата чашата се поставя в ултразвуковата баня (5.4) за 20 минути за получаване на суспензия. Така получената суспензия се прехвърля количествено в 100-милилитрова стандартна колба с не повече от 75 мл от елюиращия агент (4.10).
Добавят се последователно 1 мл от разтвора Carrez I (4.15) и 1 мл от разтвора Carrez II (4.16), като след всяко добавяне се извършва хомогенизиране. Долива се до марката с елюиращия агент (4.10), извършва се ново хомогенизиране и филтриране през нагънатата филтърна хартия.
6.1.3. С помощта на пипета 3,0 мл от получения в 6.1.2 филтрат и 5,0 мл от вътрешния стандартен разтвор (4.13) се прехвърлят в 50-милилитрова стандартна колба. Извършва се доливане до марката с елюиращ агент (4.10) и хомогенизиране. Така полученият разтвор се използва за целите на хроматографския анализ, описан в 6.2.
6.2. Условия на хроматографията
6.2.1. Потокът на подвижната фаза (4.10) се настройва на 1,2 мл/мин, а температурата на колоната — на 45TС.
6.2.2. Детекторът (5.2) се настройва на 274 нм.
6.2.3. С помощта на микроспринцовка в хроматографа се впръскват минимум два пъти 20 мкл от разтвора (6.1.3) и се измерват площите на пиковете.
6.3. Калибровъчна крива
6.3.1. Впръскват се по 20 мкл от всеки от стандартните разтвори (4.14) и се измерва площта на пика.
6.3.2. За всяка концентрация се изчислява съотношението между площите на пиковете на алфа-моноглицерил 4-аминобензоат и площите на пиковете на вътрешния стандарт. Това съотношение се нанася върху абсцисата, а върху ординатата се нанася съотношението между съответстващите маси.
6.3.3. Същата процедура се повтаря по отношение на етил 4-хидроксибензоата.
7. ИЗЧИСЛЯВАНЕ
7.1. От получената в 6.3 калибровъчна крива се отчитат масовите съотношения (RP1, RP2), съответстващи на съотношенията между площите на пиковете, изчислени в 6.2.3, където
RP1= масата на алфа-моноглицерил 4-аминобензоат/масата на етил 4-хидроксибензоат,
RP2= масата на етил 4-аминобензоат/масата на етил 4-хидроксибензоат
7.2. От получените по този начин масови съотношения с помощта на следните формули се изчисляват съдържанията на алфа-моноглицерил 4-аминобензоат и етил 4-аминобензоат в масови проценти (% m/m):
|
|
RP % (m/m) алфа-моноглицерил 4-аминобензоат |

|
|
RP % (m/m) етил 4-аминобензоат |

|
|
където: q = претегленото в 4.12 количество етил 4-хидроксибензоат (вътрешния стандарт), в милиграми, |
|
|
p = количеството от пробата, в грамове, претеглено в подточка 6.1.1. |
8. ПОВТАРЯЕМОСТ (2)
8.1. За съдържание на алфа-моноглицерил 4-аминобензоат от 5 % (m/m) разликата между резултатите от две успоредни определяния, извършени върху една и съща проба, не трябва да надхвърля 0,25 %.
8.2. За съдържание на етил 4-аминобензоат от 1 % (m/m) разликата между резултатите от две успоредни определяния, извършени върху една и съща проба, не трябва да надхвърля 0,10 %.
9. ЗАБЕЛЕЖКИ
9.1. Преди провеждането на анализа се проверява дали пробата не съдържа вещества, които могат да застъпят пика на вътрешния стандарт (етил 4-аминобензоат) върху хроматограмата.
9.2. За целите на проверката за липсата на интерференции определянето се повтаря след промяна на относителната част на метанола в подвижната фаза с 10 %.
ДОЗИРОВКА НА ХЛОРБУТАНОЛ
1. ЦЕЛ И ПРИЛОЖНО ПОЛЕ
Настоящият метод е подходящ за определянето на дозировката на хлорбутанол (МОН) до максимална концентрация 0,5 % (m/m) във всички козметични продукти, с изключение на аерозолните.
2. ОПРЕДЕЛЯНЕ
Съдържанието на хлорбутанол, измерено по този метод, се изразява в масови проценти (% m/m) от продукта.
3. ПРИНЦИП
След подходяща обработка на предназначения за анализ продукт се извършва определяне чрез газова хроматография, като в качеството на вътрешен стандарт се използва 2,2,2-трихлоретанол.
4. РЕАКТИВИ
Всички реактиви трябва да бъдат с аналитична чистота.
4.1. Хлорбутанол (1,1,1-трихлор-2-метилпропанол-2).
4.2. 2,2,2-трихлоретанол.
4.3. Абсолютен етанол.
4.4. Стандартен разтвор на хлорбутанол: 0,025 г в 100 мл етанол (4.3) (m/v).
4.5. Стандартен разтвор на 2,2,2-трихлоретанол: 4 мг в 100 мл етанол (4.3) (m/v).
5. АПАРАТУРА
5.1. Стандартно лабораторно оборудване.
5.2. Газов хроматограф с електронен детектор, Ni 63.
6. ПРОЦЕДУРА
6.1. Приготвяне на пробата
Претеглят се прецизно между 0,1 и 0,3 г (p g) от пробата. Това количество се поставя в 100-милилитрова мерителна колба. Количеството се разтваря в етанол (4.3), добавя се 1 мл от вътрешния стандартен разтвор (4.5) и се долива до марката с етанол (4.3).
6.2. Условия на газхроматографския анализ
6.2.1. Работните условия трябва да осигурят разделителна способност R ≥ 1,5.

където:
|
R1 и R2 |
= |
времена на задържане на пиковете, в минути, |
|
W1 и W2 |
= |
ширини на пиковете при полувисочината, в милиметри, |
|
d′ |
= |
скорост на въртене на хартията, в милиметри за минута. |
Като пример, необходимата разделителна способност се осигурява при следните работни условия:
|
Колона |
I |
II |
|
Материал |
Стъкло |
Неръждаема стомана |
|
Дължина |
1,80 м |
3 м |
|
Диаметър |
3 мм |
3 мм |
|
Неподвижна фаза |
10 % Carbowax 20 M TPA върху Gaschrom Q 80-100 меша |
5 % OV 17 върху Chromosorb WAW DMCS 80-100 меша |
|
Кондициониране |
от 2 до 3 дни при 190 °С |
|
|
Температура: |
|
|
|
— инжектор |
200 °С |
150 °С |
|
— колона |
150 °С |
100 °С |
|
— детектор |
200 °С |
150 °С |
|
Газ-носител |
Азот |
Аргон/метан (95/5 v/v) |
|
Дебит |
35 мл/мин |
35 мл/мин |
6.3. Стандартна крива
В пет 100-милилитрови мерителни колби се поставят по 1 мл от стандартния разтвор (4.5) и съответно 0,20; 0,30; 0,40; 0,50 и 0,60 мл от разтвора 4.4, извършва се доливане до марките с етанол (4.3) и хомогенизиране. В хроматографа се впръсква по 1 мкл от всеки от тези разтвори при спазване на работните условия, описани в подточка 6.2.2, и се построява калибровъчна крива чрез нанасяне върху абсцисата на съотношението между масата на хлорбутанола и масата на 2,2,2-трихлоретанола, и върху ординатата – съотношението между съответстващите площи на пиковете.
6.4. Впръсква се 1 мкл от получения в 6.1 разтвор и се процедира в съответствие с описаните в подточка 6.2.2 условия.
7. ИЗЧИСЛЯВАНЕ
7.1. От стандартната крива (6.3) се изчислява количеството „а“, изразено като мкг хлорбутанол, в разтвора (6.1).
7.2. Съдържанието на хлорбутанол в пробата се изчислява по следната формула:
хлорбутанол =
8. ПОВТАРЯЕМОСТ (2)
За съдържание на хлорбутанол 0,5 % (m/m) разликата между резултатите от две успоредни дозировки, извършени върху една и съща проба, не трябва да надхвърля 0,01 %.
Бележка
Ако резултатът е равен на или по-голям от максимално допустимата концентрация, е необходимо да се провери дали няма интерференции.
ИДЕНТИФИКАЦИЯ И ДОЗИРОВКА НА ХИНИН
А. ИДЕНТИФИКАЦИЯ
1. ЦЕЛ И ПРИЛОЖНО ПОЛЕ
Този метод е предназначен за откриване на наличието на хинин в шампоаните и лосионите за коса.
2. ПРИНЦИП
Идентификацията се извършва по метода на тънкослойната хроматография върху силикагел. Наличието на хинин се разпознава по синята флуоресценция на хинина в киселинни условия при 360 нм.
За потвърждаване на резултата флуоресценцията може да бъде елиминирана с бромни пари, като амонячните пари ще предизвикат появата на жълтеникава флуоресценция.
3. РЕАКТИВИ
Всички реактиви трябва да бъдат с аналитична чистота.
3.1. Плаки с покритие от силикагел, без флуоресцентни индикатори, с дебелина 0,25 мм и формат 200 мм × 200 мм.
3.2. Проявяващ разтворител: толуол/диетилов етер/дихлорметан/диетиламин/ 20/20/20/8 (v/v/v/v).
3.3. Метанол.
3.4. Сярна киселина (96 %; d = 1,84).
= 1,84).
3.5. Диетилов етер.
3.6. Проявяващ агент: към 95 мл диетилов етер (3.5) в охладен съд се добавят внимателно 5 мл сярна киселина (3.4).
3.7. Бром.
3.8. Разтвор на амониев хидроокис (28 %; d = 0,90).
= 0,90).
3.9. Хинин, безводен.
3.10. Стандартен разтвор: в стандартна колба се претеглят прецизно около 100,0 мг безводен хинин (3.9) и се разтварят в 100 мл метанол (3.3).
4. АПАРАТУРА
4.1. Стандартно оборудване за тънкослойна хроматография.
4.2. Ултразвукова баня.
4.3. Микропорест филтър, FH 0,5 fm или еквивалентен с подходящо филтриращо приспособление.
5. ПРОЦЕДУРА
5.1. Приготвяне на пробата
В 100-милилитрова стандартна колба се претегля прецизно такова количество от пробата, което съдържа приблизително 100 мг хинин, разтваря се и колбата се долива до марката с метанол (3.3).
Колбата се затваря и се оставя в продължение на един час на стайна температура в ултразвуковия вибратор (4.2). Филтрира се (4.3) и филтратът се използва за целите на хроматографския анализ.
5.2. Тънкослойна хроматография
Върху плаката с покритие от силикагел (3.1) се нанасят 1,0 мкл от стандартния разтвор (3.10) и 1,0 мкл от разтвора на пробата (5.1). Хроматограмата се развива на разстояние 150 мм с помощта на разтворителя 3.2 в наситена преди това с разтворител (3.2) камера.
5.3. Проявяване
5.3.1. Плаката се суши на стайна температура.
5.3.2. Напръсква се с реактива (3.6).
5.3.3. Плаката се оставя да съхне в продължение на един час на стайна температура.
5.3.4. Плаката се изследва на светлината на ултравиолетова лампа, настроена за дължина на вълната 360 нм. Хининът се откроява като интензивно флуоресциращо синьо петно.
Като пример в долната таблица са представени RF стойностите на основните хининоподобни алкалоиди при проявяване с разтворителя 3.2.
|
Алкалоид |
Rf |
|
Хинин |
0,20 |
|
Хинидин |
0,29 |
|
Цинхонин |
0,33 |
|
Цинхонидин |
0,27 |
|
Хидрохинидин |
0,17 |
5.3.5. За допълнителна идентификация на наличието на хинин плаката се въвежда в контакт с бромни пари (3.7) в продължение на приблизително един час. Флуоресценцията изчезва. Когато същата плака се подложи на въздействието на амонячни пари (3.8), петната се появяват отново в кафяво оцветяване, а когато плаката се изследва отново на ултравиолетовата светлина при 360 нм, може да се появи жълтеникава флуоресценция.
Идентификационен праг: 0,1 мкг хинин.
Б. ДОЗИРОВКА
1. ЦЕЛ И ПРИЛОЖНО ПОЛЕ
Този метод описва дозировката на хинина. Той може да се използва за определяне на максимално допустимата концентрация от 0,5 % (m/m) в шампоаните и 0,2 % в лосионите за коса.
2. ОПРЕДЕЛЯНЕ
Съдържанието на хинин, определено по този метод, се изразява в масови проценти (% m/m) от продукта.
3. ПРИНЦИП
След подходяща обработка на предназначения за анализ продукт, определянето се извършва по метода на високоефективната течна хроматография (ВЕТХ).
4. РЕАКТИВИ
Всички реактиви трябва да бъдат с аналитична чистота и подходящи за ВЕТХ.
4.1. Ацетонитрил.
4.2. Калиев хидрогенортофосфат (KH2PO4).
4.3. Ортофосфорна киселина (85 %; d = 1,7).
= 1,7).
4.4. Тетраметиламониев бромид.
4.5. Хинин, безводен.
4.6. Метанол.
4.7. Разтвор на ортофосфорна киселина (0,1 М): в пикнометър се претеглят 11,53 г ортофосфорна киселина (4.3) и се разтварят в 1 000 мл дестилирана вода.
4.8. Разтвор на калиев хидрогенортофосфат (0,1 М): в пикнометър се претеглят 13,6 г калиев хидрогенортофосфат (4.2) и се разтварят в 1 000 мл дестилирана вода.
4.9. Разтвор на тетраметиламониев бромид: 15,40 г тетраметиламониев бромид (4.4) се разтварят в 1 000 мл дестилирана вода в мерителна колба.
4.10. Елюиращ агент: ортофосфорна киселина (4.7)/калиев хидрогенортофосфат (4.8)/тетраметиламониев бромид (4.9)/вода за ВЕТХ/ацетонитрил (4.1) 10/50/100/340/90 (v/v/v/v/v).
Съставът на тази подвижна фаза може да се промени, за да се постигне разделителна способност R ≥ 1,5.

където:
|
R1 и R2 |
= |
времена на задържане на пиковете, в минути, |
|
W1 и W2 |
= |
ширини на пиковете при полувисочината, в милиметри, |
|
d′ |
= |
скорост на въртене на хартията, в милиметри за минута. |
4.11. Силициев диоксид, обработен с октадецилсилан, 10 мкм.
4.12. Стандартни разтвори: в поредица от 100-милилитрови стандартни колби се претеглят прецизно приблизително 5,0; 10,0; 15,0 и 20,0 мг от безводния хинин (4.5). Извършва се доливане до марките с метанол (4.6) и съдържанието на колбите се разклаща до разтварянето на хинина. Всяка от пробите се филтрира върху 0,5 мкм филтър.
5. АПАРАТУРА
5.1. Стандартно лабораторно оборудване.
5.2. Ултразвукова баня.
5.3. Високоефективен течен хроматограф с детектор с променлива дължина на вълната.
5.4. Колона от неръждаема стомана с дължина 250 мм, вътрешен диаметър 4,6 мм, пълнеж: силициев диоксид (4.11).
5.5. Микропорест филтър FH 0,5 мкм или еквивалентен, с подходящо филтриращо приспособление.
6. ПРОЦЕДУРА
6.1. Приготвяне на пробата
В 100-милилитров пикнометър се претегля прецизно такова количество от пробата, което е достатъчно, за да съдържа 10,0 мг безводен хинин. Добавят се 20 мл метанол (4.6) и колбата се поставя в ултразвуковата баня (5.2) за 20 минути. Долива се до марката с метанол (4.6). Разтворът се разбърква, след което от него се филтрира една аликвотна част (5.5).
6.2. Хроматографски анализ
Дебит на подвижната фаза: 1,0 мл/мин.
Дължина на вълната на детектора (5.3): 332 нм.
Впръскван обем: 10 мл от филтрирания разтвор (6.1).
Измерване: площ на пика.
6.3. Калибровъчна крива
Най-малко три пъти се впръскват 10,0 мкл от всеки от контролните разтвори (4.12), измерва се площта на пиковете и се изчислява средната площ за всяка от концентрациите.
Получава се калибровъчната крива и се проверява дали същата има праволинеен ход.
7. ИЗЧИСЛЯВАНЕ
7.1. От калибровъчната крива (6.3) се изчислява количеството от безводния хинин, изразено в мкг, съдържащо се във впръскания обем (6.2).
7.2. Концентрацията на безводния хинин в пробата, в масови проценти (% m/m), се получава от следната формула:
% (m/m) безводен хинин:
където:
В– количеството, в микрограма, от безводния хинин, определено в десетте микролитра от филтрирания разтвор (6.1).
А– масата на пробата в грамове (6.1).
8. ПОВТАРЯЕМОСТ (2)
За съдържание на безводен хинин от 0,5 % (m/m) разликата между резултатите от две успоредни дозировки, извършени върху една и съща проба, не трябва да надхвърля 0,02 %.
За съдържание на безводен хинин от 0,2 % (m/m) разликата между резултатите от две успоредни дозировки, извършени върху една и съща проба, не трябва да надхвърля 0,01 %.
ИДЕНТИФИКАЦИЯ И ДОЗИРОВКА НА НЕОРГАНИЧНИ СУЛФИТИ И БИСУЛФИТИ
ЦЕЛ И ПРИЛОЖНО ПОЛЕ
Методът описва идентификацията и дозировката на неорганични сулфити и хидрогенсулфити в козметичните продукти. Той е приложим само за продуктите, които съдържат водна или алкохолна фаза и за концентрации до 0,2 % серен диоксид.
А. ИДЕНТИФИКАЦИЯ
1. ПРИНЦИП
Пробата се подгрява в солна киселина и отделеният серен диоксид се идентифицира или по неговия мирис, или по ефекта му върху индикаторна хартия.
2. РЕАКТИВИ
Всички реактиви трябва да бъдат с аналитична чистота.
2.1. Солна киселина (4 М).
2.2. Индикаторна хартия с калиев йодат и скорбяла или друга индикаторна хартия.
3. АПАРАТУРА
3.1. Стандартно лабораторно оборудване.
3.2. Колба (25 мл), снабдена с къс обратен хладник.
4. ПРОЦЕДУРА
4.1. В колбата (3.2) се поставят около 2,5 г от пробата, заедно с 10 мл солна киселина (2.1).
4.2. Разтворът се разбърква и подгрява до кипване.
4.3. Извършва се проверка за отделяне на серен диоксид или по мириса, или чрез индикаторна хартия (2.2).
Б. ДОЗИРОВКА
1. ОПРЕДЕЛЯНЕ
Съдържанието на сулфити или на хидрогенсулфити в пробата, определено в съответствие с този метод, се изразява в масови проценти серен диоксид.
2. ПРИНЦИП
След подкисляване на пробата отделеният серен диоксид се подлага на дестилация с разтвор на водороден прекис. Образувалата се сярна киселина се титрува със стандартен разтвор на натриева основа.
3. РЕАКТИВИ
Всички реактиви трябва да бъдат с аналитична чистота.
3.1. Водороден прекис 0,2 % (m/v). Приготвя се ежедневно.
3.2. Ортофосфорна киселина (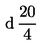 = 1,75).
= 1,75).
3.3. Метанол.
3.4. Стандартен разтвор на натриева основа (0,01 М).
3.5. Азот.
3.6. Индикатор: смес 1:1 (v/v) на метил руж (0,03 % m/v в етанол) и метиленово синьо (0,05 % m/v в етанол). Разтворът се филтрира.
4. АПАРАТУРА
4.1. Стандартно лабораторно оборудване.
4.2. Дестилационен апарат (виж фигурата).
5. ПРОЦЕДУРА
5.1. В дестилационната колба А (виж фигурата) се претеглят прецизно около 2,5 г от пробата.
5.2. Добавят се 60 мл вода и 50 мл метанол (3.3) и се смесват.
5.3. В дестилационния приемник D (виж фигурата) се поставят 10 мл водороден прекис (3.1), 60 мл вода и няколко капки от индикатора (3.6). Добавят се няколко капки натриева основа (3.4) до оцветяването на индикатора в зелено.
5.4. Стъпка 5.3 се повтаря за промивна бутилка Е (виж фигурата).
5.5. Апаратурата се свързва и потокът от азот (3.5) се настройва на около 60 мехурчета за минута.
5.6. През фунията в дестилационната колба А се изливат 15 мл ортофосфорна киселина (3.2).
5.7. Извършва се бързо подгряване до кипене, след което колбата се оставя да извира равномерно в продължение на 30 минути.
5.8. Отделя се дестилационният приемник D. Тръбата се промива и се извършва титруване с разтвор на натриева основа (3.4) до промяна на индикатора в зелено (3.6).
6. ИЗЧИСЛЯВАНЕ
Съдържанието на сулфити или хидрогенсулфити в пробата, в масови проценти, се изчислява по следната формула:
% (m/m) серен диоксид =
където:
M= моларната концентрация на разтвора на натриевата основа (3.4),
V= обемът на натриевата основа (3.4), необходим за титруването (5.8), в милилитри,
m= масата на пробата (5.1), в грамове.
7. ПОВТАРЯЕМОСТ (2)
За съдържание на серен диоксид 0,2 % m/m разликата между резултатите от две успоредни дозировки, извършени върху една и съща проба, не трябва да бъде по-голяма от 0,006 %.
Апарат на Танер за дестилация на серен диоксид
Всички размери са дадени в милиметри

ИДЕНТИФИКАЦИЯ И ДОЗИРОВКА НА ХЛОРАТИ НА АЛКАЛНИ МЕТАЛИ
ЦЕЛ И ПРИЛОЖНО ПОЛЕ
Този метод описва идентификацията и дозировката на хлорати в пастите за зъби и други козметични продукти.
А. ИДЕНТИФИКАЦИЯ
1. ПРИНЦИП
Хлоратите се разделят от другите халогенати чрез тънкослойна хроматография и се идентифицират чрез окисление на калиевия йодид до йод.
2. РЕАКТИВИ
Всички реактиви трябва да бъдат с аналитична чистота.
2.1. Стандартни разтвори: водни разтвори на калиев хлорат, бромат и йодад (0,2 % m/v), прясно приготвени.
2.2. Проявяващ разтворител: амонячен разтвор (28 % m/v) ацетон/бутанол (60/130/30 v/v/v).
2.3. Калиев йодид, воден разтвор (5 % m/v).
2.4. Скорбялов разтвор (от 1 до 5 % m/v).
2.5. Солна киселина (1 М).
2.6. Готови плаки за тънкослойна хроматография от целулоза (0,25 мм).
3. АПАРАТУРА
Стандартно оборудване за тънкослойна хроматография.
4. ПРОЦЕДУРА
4.1. С вода се извлича около 1 г от пробата, филтрира се и се разрежда до около 25 мл.
4.2. Върху плаката (2.6) се нанасят около 2 мкл от разтвора (4.1) заедно с аликвотни части 2 мкл от всеки от трите стандартни разтвора (2.1).
4.3. Плаката се поставя във вана и чрез възходяща хроматография с помощта на разтворителя 2.2 се развиват около три четвърти от дължината на плаката (2.6).
4.4. Плаката се изважда от ваната и се изчаква изпаряването на разтворителя. (Забележка: Това може да продължи до два часа).
4.5. Плаката се напръсква с калиев йодид (2.3) и се оставя да съхне в продължение на около пет минути.
4.6. Плаката се напръсква със скорбялов разтвор (2.4) и се оставя да съхне в продължение на около пет минути.
4.7. Плаката се напръсква със солна киселина (2.5).
5. ОЦЕНКА
Ако има наличие на хлорати, след около половин час ще се появи синьо петно (може и кафяво) при RF стойност приблизително 0,7—0,8.
|
Халати |
RF |
|
Йодат |
от 0 до 0,2 |
|
Бромат |
от 0,5 до 0,6 |
|
Хлорат |
от 0,7 до 0,8 |
Трябва да се отбележи, че броматите и йодатите предизвикват незабавна реакция. Да се внимава да не се получи объркване между петната от бромати и хлорати.
Б. ДОЗИРОВКА
1. ОПРЕДЕЛЯНЕ
Съдържанието на хлорати в пробата, определено чрез настоящия метод, се изразява в масови проценти хлорати.
2. ПРИНЦИП
Хлоратът се редуцира с цинков прах в кисела среда. Образуваният хлорид се измерва чрез потенциометрично титруване с разтвор на сребърен нитрат. Подобна дозировка преди редукцията позволява да се определи евентуалното съдържание на халогениди.
3. РЕАКТИВИ
Всички реактиви трябва да бъдат с аналитична чистота.
3.1. Оцетна киселина, 80 % (m/m).
3.2. Цинков прах.
3.3. Стандартен разтвор на сребърен нитрат (0,1 М).
4. АПАРАТУРА
4.1. Стандартно лабораторно оборудване.
4.2. Потенциометър, снабден със сребърен индикаторен електрод.
5. ПРОЦЕДУРА
5.1. Приготвяне на пробата
В центрофужна епруветка се претегля прецизно количество „m“, равно на около 2 г. Добавят се около 15 мл оцетна киселина (3.1) и се извършва щателно хомогенизиране. Изчаква се 30 минути и се центрофугира при 2 000 оборота в минута в продължение на 15 минути. Повърхностният течен слой се прехвърля в 50-милилитров пикнометър. Центрофугирането се повтаря два пъти при добавяне към остатъка на 15 мл оцетна киселина (3.1). Разтворът, в който има съдържание на хлорат, се събира в същия пикнометър. Долива се до марката с оцетна киселина (3.1).
5.2. Редуциране на хлората
Вземат се 20 мл от разтвора 5.1 и към тях се добавят 0,6 г цинков прах (3.2). Разтворът се довежда до кипене в колба, снабдена с обратен хладник. След кипене в продължение на 30 минути разтворът се охлажда и филтрира. Колбата се изплаква с вода. Извършва се филтриране и филтратът се събира с промивните води.
5.3. Определяне на хлорида
20 мл от разтвора 5.2 се титруват със сребърен нитрат (3.3) с помощта на потенциометъра (4.2). По същия начин със сребърен нитрат (3.3) се титруват 20 мл от разтвора 5.1.
Забележка: Ако продуктът съдържа бромни или йодни производни, които могат да отделят бромиди или йодиди след редукцията, върху кривата на титруването ще има няколко инфлексни точки. В този случай обемът на титрувания разтвор (3.3), съответстващ на хлорида, представлява разликата между последната и предпоследната инфлексна точка.
6. ИЗЧИСЛЯВАНЕ
Съдържанието на хлорат в пробата (% m/m) се изчислява по следната формула:
Хлорат =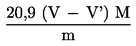
където:
|
V |
= |
обема на разтвора на сребърния нитрат (3.3), използван за титруване на разтвор 5.2, в милилитри, |
|
V′ |
= |
обема на разтвора на сребърния нитрат (3.3), използван за титруване на 20 милилитра от разтвор 5.1, в милилитри, |
|
М |
= |
моларността на стандартния разтвор на сребърния нитрат (3.3), |
|
m |
= |
масата на пробата, в грамове. |
7. ПОВТАРЯЕМОСТ (2)
За съдържание на хлорат от 3 до 5 % m/m разликата между резултатите от две успоредни дозировки, извършени върху една и съща проба, не трябва да бъде по-голяма от 0,07 % m/m.
ИДЕНТИФИКАЦИЯ И ДОЗИРОВКА НА НАТРИЕВ ЙОДАТ
ЦЕЛ И ПРИЛОЖНО ПОЛЕ
Методът се отнася за идентификацията и дозировката на натриев йодат в козметичните продукти.
А. ИДЕНТИФИКАЦИЯ
1. ПРИНЦИП
Натриевият йодат се разделя от останалите халогенати по метода на тънкослойната хроматография и се разпознава по окислението на йодида, при което се образува йод.
2. РЕАКТИВИ
Всички реактиви трябва да бъдат с аналитична чистота.
2.1. Стандартни разтвори. Водни разтвори на калиев хлорат, бромат и йодад (0,01 % m/v), прясно приготвени.
2.2. Проявяващ разтворител.
Амонячен разтвор (28 % m/v)/ацетон/бутанол (60/130/30 v/v/v).
2.3. Воден разтвор на калиев йодид (5 % m/v).
2.4. Разтвор на скорбяла (от 1 до 5 % m/v).
2.5. Солна киселина (1 М).
3. АПАРАТУРА
3.1. Готови целулозни плаки за тънкослойна хроматография (0,25 мм).
3.2. Стандартно оборудване за тънкослойна хроматография.
4. ПРОЦЕДУРА
4.1. С вода се извлича около 1 г от пробата, разтворът се филтрира и разрежда до около 10 мл.
4.2. Върху стартовата линия на плаката (3.1) се нанасят около 2 мкл от този разтвор заедно с аликвотни части 2 мкл от всеки от трите контролни разтвора (2.1).
4.3. Плаката се поставя във вана и чрез възходяща хроматография се развиват около три четвърти от дължината на плаката с помощта на разтворителя 2.2.
4.4. Плаката се изважда от ваната и се изчаква изпаряването на разтворителя на стайна температура. (Забележка: Това може да продължи до два часа).
4.5. Плаката се напръсква с калиев йодид (2.3) и се оставя да съхне в продължение на около пет минути.
4.6. Плаката се напръсква с разтвор на скорбяла (2.4) и се оставя да съхне в продължение на около пет минути.
4.7. Накрая плаката се напръсква със солна киселина (2.5).
5. ОЦЕНКА
При наличие на йодат незабавно се появява синьо петно (цветът може да бъде кафяв или да стане кафяв при престояване) с RF стойност приблизително от 0 до 0,2.
Трябва да се отбележи, че броматите предизвикват незабавни реакции при RF стойности приблизително от 0,5 до 0,6, а хлоратите – след около 30 минути при RF стойности съответно от 0,7 до 0,8.
Б. ДОЗИРОВКА
1. ОПРЕДЕЛЯНЕ
Съдържанието на натриев йодат, определено чрез настоящия метод, се изразява в масови проценти натриев йодат.
2. ПРИНЦИП
Натриевият йодат се разтваря във вода и се определя количествено по метода на високоефективната течна хроматография с помощта последователно свързани колони с обърнати фази С18 и анионообменна колона.
3. РЕАКТИВИ
Всички реактиви трябва да бъдат с аналитична чистота и подчертано подходящи за високоефективна течна хроматография (ВЕТХ).
3.1. Солна киселина (4 М).
3.2. Натриев сулфит, воден разтвор, 5 % m/v.
3.3. Изходен разтвор на натриев йодат.
Приготвя се изходен разтвор, съдържащ 50 мг натриев йодат на всеки 100 мл вода.
3.4. Калиев хидрогенортофосфат.
3.5. Динатриев ортокисел фосфат 2H2О.
3.6. Подвижна фаза за високоефективна течна хроматография: 3,88 г калиев дихидрогенортофосфат (3.4) и 1,19 г динатриев хидрогенортофосфат 2H2О (3.5) се разтварят в 1 литър вода.
рH на така получения разтвор е 6,2.
3.7. Универсална индикаторна хартия, pH 1—11.
4. АПАРАТУРА
4.1. Стандартно лабораторно оборудване.
4.2. Кръгла филтърна хартия с диаметър 110 мм, Schleicher and Sсhull № 575, или еквивалентна.
4.3. Високоефективен течен хроматограф с детектор с променлива дължина на вълната.
4.4. Колони с дължина 120 мм и с вътрешен диаметър 4,6 мм; два броя, свързани последователно; първа колона – Nucleosil® 5 C18 или еквивалентна; втора колона – Vydac™ — 301 — SB или еквивалентна.
5. ПРОЦЕДУРА
5.1. Приготвяне на пробата
5.1.1. Течни проби (шампоани)
В 10-милилитрова снабдена със стъклена запушалка градуирана епруветка или пикнометър се претегля прецизно порция от пробата за анализ с приблизителна маса 1,0 г.
Извършва се доливане до марката с вода и хомогенизиране.
При необходимост, разтворът се филтрира.
Йодатът във филтрата се определя по метода на ВЕТХ така, както е описано в раздел 5.2.
5.1.2. Твърди проби (сапун)
Внимателно се разделя част от пробата и в 100-милилитров снабден със стъклена запушалка мерителен цилиндър се претегля прецизно порция за анализ с приблизителна маса 1,0 г. Долива се до 50 мл с вода и енергично се разклаща в продължение на една минута.
Сместа се центрофугира и се филтрира през филтърна хартия (4.1) или се оставя да престои в продължение на най-малко една нощ.
Гелообразният разтвор се разклаща енергично и се филтрира през филтърна хартия (4.1).
Йодатът се дозира във филтрата по метода на ВЕТХ, съгласно подточка 5.2.
5.2. Хроматография
Дебит: 1 мл/мин.
Дължина на вълната на детектора (4.2): 210 нм.
Впръскван обем: 10 мкл.
Измерване: площ на пика.
5.3. Калибриране
С помощта на пипета в 50-милилитрови мерителни колби се поставят съответно 1,0; 2,0; 5,0; 10,0 и 20,0 мл от изходния разтвор на натриевия йодат (3.3). Долива се до марката и се разбърква.
Така получените разтвори съдържат съответно 0,01; 0,02; 0,05; 0,10 и 0,20 мг натриев йодат на един милилитър.
В течния хроматограф (4.2) се впръскват 10 мкл от всеки стандартен йодатен разтвор и се получава хроматограмата. Определя се площта на пика на йодата и се построява стандартна крива от площите на пиковете спрямо концентрациите на натриевия йодат.
6. ИЗЧИСЛЯВАНЕ
Съдържанието на натриев йодат, в масови проценти (% m/m), се изчислява по следната формула:
% (m/m) натриев йодат =
където
m= масата на пробата (5.1), в грамове,
V= общият обем на разтвора на пробата, получен според описаното в 5.1, в милилитри,
с= концентрацията, определена от калибровъчната крива (5.3), в милиграма натриев йодат за милилитър.
7. ПОВТАРЯЕМОСТ (2)
За съдържание на натриев йодат от 0,1 % (m/m) разликата между резултатите от две успоредни дозировки, извършени върху една и съща проба, не трябва да бъде по-голяма от 0,002 %.
8. ПОТВЪРЖДАВАНЕ НА РЕЗУЛТАТА
8.1. Принцип
В подкисления разтвор на даден козметичен продукт йодатът (IO3-) се редуцира чрез сулфит до йодид (I-) и така полученият разтвор се анализира по метода на ВЕТХ. Ако даден пик, чието време на задържане съответства на времето на задържане на йодата, изчезне след обработка със сулфита, първоначалният пик може да се припише с голяма доза вероятност на йодата.
8.2. Процедура
С помощта на пипета в конична колба се поставят 5 мл от разтвора на пробата, получен съгласно точка 5.1.
Стойността на рH на разтвора се довежда до 3 или по-малка стойност със солна киселина (3.1); универсална индикаторна хартия (3.7).
Добавят се три капки от разтвора на натриевия сулфит (3.2) и се разбърква.
В течния хроматограф (4.2) се впръскват 10 мкл от разтвора.
Така получената хроматограма се сравнява с хроматограмата, получена по отношение на същата проба съгласно указанията в точка 5.
(1) Тези стойности са дадени за сведение и съответстват на точните маси от 4.11, 4.12 и 4.13.
Забележка: Тези разтвори могат да се приготвят по друг начин.
(2) Съгласно стандарт ISO 5725.