BILAGA
I Bilaga V till Förordning (EG) nr 152/2009 Ska del B ”BESTÄMNING AV HALTER AV DIOXINER (PCDD/PCDF) OCH PCB” ersättas med följande:
”B. BESTÄMNING AV HALTER AV DIOXINER (PCDD/PCDF) OCH PCB
KAPITEL I
Provtagningsmetoder och tolkning av analysresultat
1. Syfte och tillämpningsområde
Provtagning för offentlig kontroll av halter av polyklorerade dibenso-p-dioxiner (PCDD) och polyklorerade dibensofuraner (PCDF) (nedan kallade PCDD/PCDF), dioxinlika polyklorerade bifenyler (nedan kallade dioxinlika PCB) (1) och icke-dioxinlika PCB i foder ska göras i enlighet med bilaga I. De kvantitativa kraven i fråga om kontroll av ämnen eller produkter som är jämnt fördelade i fodret enligt punkt 5.1 i bilaga I ska uppfyllas. De erhållna samlingsproven ska betraktas som representativa för de partier eller delpartier från vilka de tas. De halter som bestäms i laboratorieproven ska ligga till grund för bedömningen av om proven är överensstämmande med gränsvärdena enligt direktiv 2002/32/EG.
I denna del B ska definitionerna i bilaga I till kommissionens beslut 2002/657/EG (2) gälla.
Dessutom gäller följande definitioner:
|
|
screeningmetoder: metoder för att påvisa prov med halter av PCDD/PCDF och dioxinlika PCB som överskrider gränsvärdena eller åtgärdsgränserna. De ska ge en kostnadseffektiv och hög analyskapacitet och därigenom öka möjligheterna att upptäcka nya fall som medför hög exponering och hälsorisker för konsumenterna. Screeningmetoder ska baseras på bioanalytiska metoder eller GC-MS-metoder. Provresultat som överskrider brytpunkten vid kontroll av överensstämmelsen med gränsvärdet ska kontrolleras genom en fullständig ny analys av det ursprungliga provet med en konfirmeringsmetod. |
|
|
konfirmeringsmetoder: metoder för att få fram fullständig eller kompletterande information som möjliggör en entydig identifiering och kvantifiering av PCDD/PCDF och dioxinlika PCB vid gränsvärdet, eller vid behov vid åtgärdsgränsen. Sådana metoder bygger på gaskromatografi i kombination med högupplösande masspektrometri (GC-HRMS) eller gaskromatografi i kombination med tandem-masspektrometri (GC-MS/MS). |
2. Partiets eller delpartiets överensstämmelse med gränsvärdet
2.1 Icke-dioxinlika PCB
Partiet är överensstämmande med gränsvärdet om analysresultatet inte överskrider gränsvärdet för icke-dioxinlika PCB enligt direktiv 2002/32/EG, med beaktande av mätosäkerheten.
Partiet är icke-överensstämmande med gränsvärdet om analysresultatet avseende den övre koncentrationen (3), bekräftat genom dubbelprov (4), överskrider gränsvärdet enligt direktiv 2002/32/EG, med beaktande av mätosäkerheten. Medelvärdet av två bestämningar, där mätosäkerheten beaktas, används för kontroll av överensstämmelsen.
Mätosäkerheten ska beaktas enligt någon av följande metoder:
|
— |
Genom att fastställa den utvidgade mätosäkerheten med användning av en täckningsfaktor på 2, vilket ger en konfidensgrad på cirka 95 %. Ett parti eller delparti är icke-överensstämmande om det uppmätta värdet minus U överstiger gränsvärdet. |
|
— |
Genom att fastställa beslutsgränsen (CCα) enligt punkt 3.1.2.5 i bilaga I till beslut 2002/657/EG. Ett parti eller delparti är icke-överensstämmande om det uppmätta värdet är lika med eller högre än CCα. |
Styckena 1, 2 och 3 ska tillämpas på de analysresultat som erhålls vid provtagning för offentlig kontroll. När det gäller analys för överklagande eller referensändamål ska nationella regler gälla.
2.2 Dioxiner (PCDD/PCDF) och dioxinlika PCB
Partiet är överensstämmande med gränsvärdena i följande fall:
|
— |
En enda analys med en screeningmetod för vilken andelen prov som felaktigt bedömts vara överensstämmande med gränsvärdet är lägre än 5 % ger ett resultat som visar att halten inte överskrider respektive gränsvärde för PCDD/PCDF och summan av PCDD/PCDF och dioxinlika PCB enligt direktiv 2002/32/EG. |
|
— |
En enda analys med en konfirmeringsmetod ger ett resultat som inte överskrider respektive gränsvärde för PCDD/PCDF och summan av PCDD/PCDF och dioxinlika PCB enligt direktiv 2002/32/EG, med beaktande av mätosäkerheten. |
För screeninganalyser ska en brytpunkt fastställas för bedömningen av om provet/n är överensstämmande med det gränsvärde som anges för PCDD/PCDF respektive för summan av PCDD/PCDF och dioxinlika PCB.
Partiet är icke-överensstämmande med gränsvärdet om analysresultatet avseende den övre koncentrationen (5), fastställt med en konfirmeringsmetod och bekräftat genom dubbelprov, överskrider gränsvärdet enligt direktiv 2002/32/EG, med beaktande av mätosäkerheten (6). Medelvärdet av två bestämningar, där mätosäkerheten beaktas, används för kontroll av överensstämmelsen.
Mätosäkerheten ska beaktas enligt någon av följande metoder:
|
— |
Genom att fastställa den utvidgade mätosäkerheten med användning av en täckningsfaktor på 2, vilket ger en konfidensgrad på cirka 95 %. Ett parti eller delparti är icke-överensstämmande om det uppmätta värdet minus U överstiger gränsvärdet. Om det har gjorts en separat bestämning av PCDD/PCDF och av dioxinlika PCB ska summan av den skattade utvidgade mätosäkerheten för de enskilda analysresultaten användas när man anger summan av PCDD/PCDF och dioxinlika PCB. |
|
— |
Genom att fastställa beslutsgränsen (CCα) enligt punkt 3.1.2.5 i bilaga I till beslut 2002/657/EG. Ett parti eller delparti är icke-överensstämmande om det uppmätta värdet är lika med eller högre än CCα. |
Styckena 1–4 ska tillämpas på de analysresultat som erhålls vid provtagning för offentlig kontroll. När det gäller analys för överklagande eller referensändamål ska nationella regler gälla.
3. Resultat som överskrider de åtgärdsgränser som anges i bilaga II till direktiv 2002/32/EG
Åtgärdsgränserna fungerar som ett redskap för provurval i de fall där det är nödvändigt att identifiera en föroreningskälla och att vidta åtgärder för att minska eller eliminera den. Screeningmetoderna ska fastställa lämpliga brytpunkter för urvalet av dessa prov. Om det behövs betydande insatser för att identifiera en föroreningskälla och minska eller eliminera föroreningen kan det vara lämpligt att bekräfta om åtgärdsgränsen överskridits genom att analysera dubbelprov med en konfirmeringsmetod, med beaktande av mätosäkerheten (7).
KAPITEL II
Provberedning och krav på analysmetoder för offentlig kontroll av halter av dioxiner (PCDD/PCDF) och dioxinlika PCB i foder
1. Tillämpningsområde
Kraven i detta kapitel ska tillämpas när foder analyseras för offentlig kontroll av halterna av 2,3,7,8-substituerade polyklorerade dibenso-p-dioxiner och polyklorerade dibensofuraner (nedan kallade PCDD/PCDF) samt av dioxinlika polyklorerade bifenyler (nedan kallade dioxinlika PCB) och för andra lagstadgade ändamål.
Övervakningen av förekomsten av PCDD/PCDF och dioxinlika PCB i foder kan utföras med två olika typer av analysmetoder:
|
a) |
Screeningmetoder Syftet med screeningmetoder är att påvisa prov med halter av PCDD/PCDF och dioxinlika PCB som överskrider gränsvärdena eller åtgärdsgränserna. Screeningmetoder bör ge en kostnadseffektiv och hög analyskapacitet och därigenom öka möjligheterna att upptäcka nya fall som medför hög exponering och hälsorisker för konsumenterna. När dessa metoder används bör man sträva efter att undvika resultat som felaktigt bedömts vara överensstämmande. De kan bland annat bestå av bioanalytiska metoder och GC-MS-metoder. Screeningmetoder jämför analysresultatet med en brytpunkt och ger ett ja/nej-svar för bedömning av om gränsvärdet eller åtgärdsgränsen eventuellt har överskridits. Koncentrationen av PCDD/PCDF och summan av PCDD/PCDF och dioxinlika PCB i prov som misstänks vara icke-överensstämmande med gränsvärdet måste bestämmas/bekräftas med en konfirmeringsmetod. Screeningmetoderna kan dessutom ge en indikation på halterna av de PCDD/PCDF och dioxinlika PCB som förekommer i provet. Om bioanalytiska screeningmetoder används ska resultatet uttryckas som bioanalytiska ekvivalenter (BEQ), medan om fysikalisk-kemiska GC-MS-metoder används ska resultatet uttryckas som toxicitetsekvivalenter (TEQ). Screeningmetodernas resultat i form av numeriska värden är lämpliga för att demonstrera att proven är överensstämmande eller misstänkt icke-överensstämmande eller att åtgärdsgränsen överskridits, och ger en indikation på koncentrationsintervallet om proven följs upp med konfirmeringsmetoder. Däremot är de inte lämpliga för utvärdering av bakgrundshalter, uppskattning av intag, uppföljning av tidstrender avseende halter eller omprövning av åtgärdsgränser och gränsvärden. |
|
b) |
Konfirmeringsmetoder Konfirmeringsmetoder möjliggör en entydig identifiering och kvantifiering av de PCDD/PCDF och dioxinlika PCB som förekommer i ett prov och ger fullständig information på kongennivå. Dessa metoder möjliggör därför kontroll av gränsvärden och åtgärdsgränser, inklusive konfirmering av de resultat som fåtts med screeningmetoder. Dessutom kan resultaten användas för andra ändamål, t.ex. bestämning av låga bakgrundshalter vid foderövervakning, uppföljning av tidstrender, exponeringsbedömning samt för uppbyggnad av en databas om eventuell omprövning av åtgärdsgränser och gränsvärden. De är också viktiga för att fastställa kongenmönster så att källan till en eventuell förorening kan identifieras. Sådana metoder bygger på GC-HRMS. För att bekräfta överensstämmelse eller icke-överensstämmelse med gränsvärdet kan även GC-MS/MS användas. |
2. Bakgrund
För att beräkna den totala koncentrationen av dioxinlika föreningar i ett prov, den så kallade toxicitetsekvivalenten (TEQ), ska koncentrationen av varje enskilt ämne i ett givet prov multipliceras med respektive ämnes toxicitetsekvivalensfaktor (TEF) (se fotnot (1)* i kapitel I) och därefter adderas.
I denna del B i bilaga V avses med den accepterade särskilda kvantifieringsgränsen för en enskild kongen den lägsta analythalten som med rimlig statistisk säkerhet kan mätas och som uppfyller de identifieringskriterier som beskrivs i internationellt erkända standarder, t.ex. standarden EN 16215:2012 (Djurfoder – Bestämning av dioxiner och dioxinlika PCB:er med GC/HRMS och av indikator-PCB:er med GC/HRMS) och/eller i EPA-metoderna 1613 och 1668, med senare revideringar.
Kvantifieringsgränsen för en enskild kongen kan identifieras
|
a) |
som den koncentration av en analyt i ett provextrakt som ger en instrumentrespons för de två skilda joner som ska övervakas, med ett S/N-förhållande (signal-brusförhållande) på 3:1 för den mindre känsliga rådatasignalen, eller |
|
b) |
om beräkningen av signal-brusförhållandet på grund av tekniska skäl inte ger tillförlitliga resultat, som den lägsta koncentrationen på en kalibreringskurva som ger en godtagbar (≤ 30 %) och konsekvent (uppmätt åtminstone i början och slutet av en provserie) avvikelse till den genomsnittliga relativa responsfaktorn som beräknats för alla punkter på kalibreringskurvan för varje provserie. Kvantifieringsgränsen beräknas utifrån den lägsta koncentrationen med beaktande av utbytet för interna standarder och mängden prov. |
Bioanalytiska screeningmetoder ger inte resultat på kongennivå utan endast en indikation (8) på TEQ-värdet, uttryckt som bioanalytiska ekvivalenter (BEQ), på grund av att det i ett provextrakt även kan finnas föreningar som ger en respons i analysen men som inte uppfyller alla krav i TEQ-principen.
Screeningmetoder och konfirmeringsmetoder får endast användas för kontroll av en viss matris om metoderna har tillräckligt hög känslighet för att på ett tillförlitligt sätt påvisa halter vid åtgärdsgränsen eller gränsvärdet.
3. Krav på kvalitetssäkring
3.1 Åtgärder ska vidtas för att undvika korskontaminering under alla moment vid provtagning och analys.
3.2 Proven ska förvaras och transporteras i behållare av glas, aluminium, polypropen eller polyeten som är lämpade för förvaring och inte påverkar halterna av PCDD/PCDF och dioxinlika PCB i proven. Spår av pappersdamm ska avlägsnas från provbehållaren.
3.3 Proven ska förvaras och transporteras på ett sådant sätt att foderprovet bibehålls i oförändrat tillstånd.
3.4 Varje laboratorieprov ska vid behov finmalas och blandas omsorgsfullt enligt en metod som garanterar fullständig homogenisering (t.ex. så att de passerar en 1 mm sikt). Om vattenhalten är för hög ska proven torkas innan de mals.
3.5 Reagenser, glasvaror och utrustning ska kontrolleras för om de eventuellt kan påverka de TEQ- eller BEQ-baserade resultaten.
3.6 En analys av ett blankprov ska göras genom att man utför hela analysen men utelämnar provet.
3.7 För bioanalytiska metoder ska alla glasvaror och lösningsmedel som används i analysen vara garanterat fria från ämnen som kan störa påvisandet av målföreningar i mätområdet. Glasvaror ska sköljas med lösningsmedel eller upphettas till de temperaturer som krävs för att avlägsna spår av PCDD/PCDF, dioxinlika föreningar och störande föreningar från dess yta.
3.8 Det prov som extraheras ska vara tillräckligt stort för att uppfylla kraven vad gäller ett tillräckligt lågt mätområde som inkluderar gränsvärden eller åtgärdsgränser.
3.9 De särskilda provberedningsförfaranden som används för de givna produkterna ska följa internationellt accepterade riktlinjer.
4. Krav på laboratorier
4.1 I enlighet med förordning (EG) nr 882/2004 ska laboratorierna ackrediteras av ett godkänt ackrediteringsorgan som verkar enligt ISO Guide 58 för att säkerställa att de tillämpar ett system för kvalitetssäkring av analysverksamheten. Laboratorierna ska ackrediteras enligt standarden EN ISO/IEC 17025.
4.2 Laboratoriekompetensen ska bevisas genom fortlöpande medverkan i kollaborativa studier för bestämning av halten av PCDD/PCDF och dioxinlika PCB i relevanta fodermatriser och koncentrationsintervall.
4.3 Laboratorier som använder screeningmetoder vid rutinkontroll av prov ska ha ett nära samarbete med laboratorier som använder konfirmeringsmetoden, både för kvalitetskontroll och för att bekräfta analysresultatet för misstänkta prov.
5. Grundläggande krav på analysmetoder för dioxiner (PCDD/PCDF) och dioxinlika PCB
5.1 Lågt mätområde och låga kvantifieringsgränser
Eftersom vissa typer av PCDD/PCDF är extremt toxiska ska detektionsgränsen för dessa föreningar vara i storleksordningen femtogram (10–15 g). För de flesta PCB-kongener räcker det att kvantifieringsgränsen är i storleksordningen nanogram (10–9 g). Vid mätning av de mer toxiska dioxinlika PCB-kongenerna (särskilt kongener som inte är substituerade på orto-position, s.k. non-orto kongener) ska dock de lägsta halterna i mätområdet vara i storleksordningen pikogram (10–12 g). För alla andra PCB-kongener räcker det att kvantifieringsgränsen är i storleksordningen nanogram (10–9 g).
5.2 Hög selektivitet (specificitet)
5.2.1 PCDD/PCDF och dioxinlika PCB måste kunna skiljas från en stor mängd andra föreningar som extraheras samtidigt och ingår i koncentrationer flerfaldigt högre än hos de givna analyterna och som kan störa analysen. GC-MS-metoder måste kunna särskilja mellan olika kongener, exempelvis mellan de toxiska kongenerna (t.ex. de sjutton 2,3,7,8-substituerade PCDD/PCDF och de tolv dioxinlika PCB) och andra kongener.
5.2.2 Bioanalytiska metoder ska kunna påvisa målföreningar och ange dem som summan av PCDD/PCDF och/eller dioxinlika PCB. Upprening av prov ska syfta till att avlägsna föreningar som orsakar resultat som felaktigt bedömts vara icke-överensstämmande eller föreningar som kan minska responsen, vilket orsakar resultat som felaktigt bedömts vara överensstämmande.
5.3 Hög noggrannhet (riktighet och precision, skenbart utbyte för bioassay)
5.3.1 Bestämning med GC-MS-metoder ska ge en giltig uppskattning av den faktiska koncentrationen i ett prov. En hög noggrannhet krävs för att resultatet av en provanalys inte ska avvisas på grund av att det fastställda TEQ-värdet har för dålig tillförlitlighet. Noggrannheten uttrycks som riktighet (skillnaden mellan det medelvärde som har uppmätts för en analyt i ett certifierat material och dess certifierade värde, uttryckt som ett procenttal av detta värde) och precision (RSDR, dvs. den relativa standardavvikelsen beräknad utifrån de resultat som fåtts under reproducerbara förhållanden).
5.3.2 För bioanalytiska metoder ska det skenbara utbytet för bioassay bestämmas. Det skenbara utbytet för bioassay motsvaras av det BEQ-värde som beräknats från kalibreringskurvan för TCDD eller PCB 126, korrigerats för blank och därefter delats med det TEQ-värde som fastställts med konfirmeringsmetoden. Syftet är att korrigera för faktorer som till exempel förlusten av PCDD/PCDF och dioxinlika föreningar under extraktions- och uppreningsstegen, föreningar som extraherats ut samtidigt och ökar eller minskar responsen (agonistiska och antagonistiska effekter), kvaliteten på kurvpassningen eller skillnader mellan TEF-värden och REP-värden (relativ potensfaktor). Det skenbara utbytet för bioassay beräknas utifrån lämpliga referensprov med representativa kongenmönster kring den givna nivån.
5.4 Validering i intervallet för gränsvärdet och allmänna åtgärder för kvalitetskontroll
5.4.1 Laboratorierna ska påvisa en metods prestanda i intervallet för gränsvärdet, t.ex. 0,5 gång, 1 gång och 2 gånger gränsvärdet med en godtagbar variationskoefficient för upprepade analyser, under valideringsförfarandet och rutinanalyser.
5.4.2 Laboratorierna ska regelbundet analysera blankprov och spikade prov eller kontrollprov (helst certifierat referensmaterial om sådant finns) som en åtgärd för intern kvalitetskontroll. Kontrollkort för analyser av blankprov och spikade prov eller kontrollprov ska framställas och kontrolleras för att säkerställa att den analytiska prestandan uppfyller kraven.
5.5 Kvantifieringsgräns
5.5.1 För en bioanalytisk screeningmetod är det inte ett absolut krav att kvantifieringsgränsen fastställs, men metoden ska bevisligen kunna särskilja mellan blankprovet och brytpunkten. När ett BEQ-värde anges ska en rapporteringsnivå fastställas för att hantera prov som ger en respons under denna nivå. Rapporteringsnivån ska bevisligen vara skild från metodens blankprov med åtminstone en faktor på tre och ge respons vid halter under mätområdet. Den ska därför beräknas utifrån prov med halter av målföreningarna kring de miniminivåer som krävs och inte utifrån ett S/N-förhållande (signal-brusförhållande) eller ett blankprov.
5.5.2 Kvantifieringsgränsen för en konfirmeringsmetod ska motsvara ungefär en femtedel av gränsvärdet.
5.6 Analyskriterier
För att resultaten från konfirmeringsmetoder eller screeningmetoder ska vara tillförlitliga ska följande kriterier vara uppfyllda i intervallet för gränsvärdet eller åtgärdsgränsen för TEQ-värdet respektive BEQ-värdet, vilka kan bestämmas antingen som det totala TEQ-värdet (dvs. summan av PCDD/PCDF och dioxinlika PCB) eller separat för PCDD/PCDF och för dioxinlika PCB:
|
|
Screening med bioanalytiska eller fysikalisk-kemiska metoder |
Konfirmeringsmetoder |
|
Andelen prov som felaktigt bedömts vara överensstämmande (9) |
< 5 % |
|
|
Riktighet |
|
–20 % till +20 % |
|
Repeterbarhet (RSDr) |
< 20 % |
|
|
Reproducerbarhet inom laboratoriet (RSDR) |
< 25 % |
< 15 % |
5.7 Särskilda krav på screeningmetoder
5.7.1 Både GC-MS och bioanalytiska metoder får användas för screening. För GC-MS-metoder ska kraven i punkt 6 uppfyllas. För cellbaserade bioanalytiska metoder anges särskilda krav i punkt 7.
5.7.2 Laboratorier som använder screeningmetoder vid rutinkontroll av prov ska ha ett nära samarbete med laboratorier som använder konfirmeringsmetoden.
5.7.3 Screeningmetodens prestanda ska kontrolleras vid rutinanalyser genom analytisk kvalitetskontroll och löpande metodvalidering. Det ska finnas ett fortlöpande program för att kontrollera överensstämmande resultat.
5.7.4 Kontroll av en eventuell hämning av cellens respons och cytotoxicitet
Vid rutinmässig screening ska 20 % av provextrakten analyseras med och utan tillsats av 2,3,7,8-TCDD i en halt som motsvarar gränsvärdet eller åtgärdsgränsen för att kontrollera om responsen eventuellt hämmas av störande ämnen i provextraktet. Den uppmätta koncentrationen i det spikade provet ska jämföras med summan av koncentrationen i det ej spikade provet och den tillsatta koncentrationen. Om den uppmätta koncentrationen är mer än 25 % lägre än den beräknade koncentrationen (summan) tyder detta på att signalen eventuellt hämmas och det berörda provet ska genomgå konfirmeringsanalys med GC-HRMS. Resultaten ska följas upp med kontrollkort.
5.7.5 Kvalitetskontroll av överensstämmande prov
Ungefär 2–10 % av de överensstämmande proven ska bekräftas med GC-HRMS. Antalet prov beror på provmatrisen och laboratoriets erfarenhetsnivå.
5.7.6 Kvalitetskontrolluppgifter som grund för bestämning av andelen prov som felaktigt bedömts vara överensstämmande
Efter screening av prov vars halter ligger under eller över gränsvärdena eller åtgärdsgränserna ska man bestämma andelen prov som felaktigt bedömts vara överensstämmande med kraven. Den faktiska andelen prov som felaktigt bedömts vara överensstämmande ska vara lägre än 5 %. När man vid kvalitetskontrollen av överensstämmande prov har tillgång till minst 20 bekräftade resultat per matris eller matrisgrupp ska dessa uppgifter användas som grund för att dra slutsatser om andelen prov som felaktigt bedömts vara överensstämmande. Resultat från prov som analyserats i ringtest eller vid föroreningsincidenter och som täcker koncentrationsintervall upp till exempelvis två gånger gränsvärdet får också tas med i de 20 resultat som minst ska ingå i utvärderingen. Proven ska täcka de vanligaste kongenmönstren och komma från olika källor.
Även om det främsta syftet med screeningmetoder är att påvisa prov som överskrider åtgärdsgränsen, är kriteriet för att fastställa andelen prov som felaktigt bedömts vara överensstämmande gränsvärdet, med beaktande av mätosäkerheten för konfirmeringsmetoden.
5.7.7 Screeningresultat som eventuellt är icke-överensstämmande ska alltid kontrolleras genom en fullständig ny analys av det ursprungliga provet med en konfirmeringsmetod. Dessa provresultat kan också användas för att utvärdera andelen prov som felaktigt bedömts vara icke-överensstämmande. För screeningmetoder motsvaras andelen prov som felaktigt bedömts vara icke-överensstämmande av den andel prov som med hjälp av konfirmeringsanalyser konstaterats vara överensstämmande och som vid tidigare screening hade konstaterats vara eventuellt icke-överensstämmande. Utvärderingen av hur väl screeningmetoden fungerar ska göras på grundval av en jämförelse mellan antalet prov som felaktigt bedömts vara icke-överensstämmande och det totala antalet prov som kontrollerats. Detta värde ska vara så lågt att det lönar sig att använda en screeningmetod.
5.7.8 Bioanalytiska metoder ska, åtminstone under valideringsbetingelser, ge en godtagbar indikation på TEQ-värdet, beräknat och uttryckt som BEQ.
Även för bioanalytiska metoder som används under repeterbarhetsbetingelser bör normalt sett den interna repeterbarheten (RSDr) vara mindre än reproducerbarheten (RSDR).
6. Särskilda krav på GC-MS-metoder för screening eller konfirmering
6.1 Godtagbara skillnader mellan WHO-TEQ-värdena avseende den övre och den lägre koncentrationen
Det får inte skilja mer än 20 % mellan den övre och den lägre koncentrationen vid konfirmering av att gränsvärdet, eller vid behov åtgärdsgränsen, överskridits.
6.2 Kontroll av utbytet
6.2.1 För validering av analysmetoden ska interna standarder i form av 13C-märkta 2,3,7,8-klorsubstituerade PCDD/PCDF och 13C-märkta dioxinlika PCB tillsättas alldeles i början av analysen, t.ex. före extraktionen. Minst en kongen för varje tetra- till oktaklorerad homolog grupp av PCDD/PCDF och minst en kongen för varje homolog grupp av dioxinlika PCB ska tillsättas (alternativt tillsätts minst en kongen för varje masspektrometrisk SIR-funktion [selected ion recording] som används för att övervaka PCDD/PCDF och dioxinlika PCB). För konfirmeringsmetoder ska alla de sjutton 13C-märkta 2,3,7,8-substituerade interna PCDD/PCDF-standarderna och alla de tolv 13C-märkta interna dioxinlika PCB-standarderna användas.
6.2.2 Med hjälp av lämpliga kalibreringslösningar ska de relativa responsfaktorerna också bestämmas för de kongener till vilka ingen 13C-märkt analog tillsätts.
6.2.3 För foder av vegetabiliskt eller animaliskt ursprung som innehåller mindre än 10 % fett ska interna standarder tillsättas innan extraktionen görs. För foder av animaliskt ursprung som innehåller mer än 10 % fett ska interna standarder tillsättas antingen före eller efter fettextraktionen. Extraktionens effektivitet ska valideras på lämpligt sätt, med hänsyn till när de interna standarderna tillsätts och till om resultaten anges per gram produkt eller per gram fett.
6.2.4 Innan en GC-MS-analys utförs ska en eller två utbytesstandarder (surrogat) tillsättas.
6.2.5 Det krävs en kontroll av utbytet. För konfirmeringsmetoder ska utbytet av de enskilda interna standarderna ligga i intervallet 60–120 %. Lägre eller högre utbytesgrad för enskilda kongener – särskilt för vissa hepta- och oktaklorerade dibenso-p-dioxiner och dibensofuraner – ska godtas under förutsättning att deras bidrag till det totala TEQ-värdet inte överstiger 10 % (baserat på summan av PCDD/PCDF och dioxinlika PCB). För GC-MS-screeningmetoder ska utbytet ligga i intervallet 30–140 %.
6.3 Avlägsnande av störande ämnen
|
— |
PCDD/PCDF ska separeras från störande klorföreningar, såsom icke-dioxinlika PCB och klorerade difenyletrar, med hjälp av lämpliga kromatografiska tekniker (lämpligtvis med en florisil-, aluminiumoxid- och/eller kolkolonn). |
|
— |
Gaskromatografisk separation av isomerer ska ha mindre än 25 % mellan två toppar för 1,2,3,4,7,8-HxCDF och 1,2,3,6,7,8-HxCDF. |
6.4 Kalibrering med standardkurva
Kalibreringskurvan ska omfatta det relevanta intervallet för gränsvärdet eller åtgärdsgränsen.
6.5 Särskilda kriterier för konfirmeringsmetoder
|
— |
För GC-HRMS:
|
|
— |
För GC-MS/MS:
|
7. Särskilda krav på bioanalytiska metoder
Bioanalytiska metoder bygger på biologiska principer såsom cellbaserade metoder, receptorbaserade metoder eller immunologiska metoder. I denna punkt (punkt 7) fastställs krav på bioanalytiska metoder i allmänhet.
Med en screeningmetod klassificeras i princip ett prov antingen som överensstämmande eller som misstänkt icke-överensstämmande genom en jämförelse mellan det beräknade BEQ-värdet och brytpunkten (se punkt 7.3). Prov under brytpunkten är överensstämmande, medan prov som är lika med eller över brytpunkten är misstänkt icke-överensstämmande och kräver analys med en konfirmeringsmetod. I praktiken kan ett BEQ-värde som motsvarar 2/3 av gränsvärdet användas som brytpunkt, förutsatt att det kan säkerställas att andelen prov som felaktigt bedömts vara överensstämmande är lägre än 5 % och att andelen prov som felaktigt bedömts vara icke-överensstämmande är godtagbar. Om man använder separata gränsvärden för PCDD/PCDF och för summan av PCDD/PCDF och dioxinlika PCB krävs lämpliga brytpunkter för bioassay av PCDD/PCDF för att utan fraktionering kunna kontrollera provens överensstämmelse. För kontroll av prov som överskrider åtgärdsgränserna skulle en lämplig procentandel av respektive åtgärdsgräns lämpa sig som brytpunkt.
När det gäller vissa bioanalytiska metoder får ett vägledande värde uttryckt i BEQ ges för prov i mätområdet som överstiger rapporteringsgränsen (se punkt 7.1.1 och 7.1.6).
7.1 Utvärdering av försöksrespons
7.1.1 Allmänna krav
|
— |
När man beräknar koncentrationer utifrån en kalibreringskurva för TCDD kommer de lägsta och högsta koncentrationerna i kurvan att ha en hög variation (hög variationskoefficient). Mätområdet är det område där variationskoefficienten är mindre än 15 %. Den lägsta koncentrationen i mätområdet (rapporteringsgränsen) ska ligga över metodens blankvärde med åtminstone en faktor på tre. Den högsta koncentrationen i mätområdet motsvaras vanligtvis av EC70-värdet (70 % av den maximalt effektiva koncentrationen), men den är lägre om variationskoefficienten överstiger 15 % i detta område. Mätområdet ska fastställas under valideringen. Brytpunkterna (se punkt 7.3) ska med god marginal ligga inom mätområdet. |
|
— |
Standardlösningar och provextrakt ska åtminstone analyseras som dubbelprov. När dubbelprov analyseras ska en standardlösning eller ett kontrollextrakt som provas i 4–6 brunnar (fördelade över plattan) ge en respons eller en koncentration (endast möjligt i mätområdet) som har en variationskoefficient mindre än 15 %. |
7.1.2 Kalibrering
7.1.2.1 Kalibrering med standardkurva
|
— |
Provhalter ska skattas genom att försöksresponsen jämförs med en kalibreringskurva för TCDD (eller PCB 126 eller en standardblandning med PCDD/PCDF och dioxinlika PCB) utifrån vilken BEQ-värdet kan beräknas i extraktet och därefter i provet. |
|
— |
Kalibreringskurvor ska bestå av 8–12 koncentrationer (åtminstone som dubbelprov) och ha tillräckligt med koncentrationer i den nedre delen av kurvan (mätområdet). Särskild uppmärksamhet ska ägnas kvaliteten på kurvpassningen i mätområdet. När man ska bedöma anpassningsgraden vid icke-linjär regression är R2-värdet i sig inte viktigt. Man uppnår en bättre kurvpassning genom att minimera skillnaden mellan beräknade och observerade koncentrationer inom kurvans mätområde, t.ex. genom att minimera residualkvadratsumman. |
|
— |
Den skattade halten i provextraktet korrigeras därefter för BEQ-värdet hos ett blankprov för matrisen eller lösningsmedlet (för att beakta föroreningar från lösningsmedel och andra kemikalier som använts) och för det skenbara utbytet (som beräknats från BEQ-värdet för lämpliga referensprov med representativa kongenmönster kring gränsvärdet eller åtgärdsgränsen). När man korrigerar för ett utbyte ska det skenbara utbytet ligga inom det intervall som krävs (se punkt 7.1.4). Referensprov som används för att korrigera utbytet ska uppfylla de krav som anges i punkt 7.2. |
7.1.2.2 Kalibrering med referensprov
Man kan också använda en kalibreringskurva som genererats från minst fyra referensprov (se punkt 7.2.4: ett blankprov för matrisen och tre referensprov på 0,5 gång, 1 gång och 2 gånger gränsvärdet eller åtgärdsgränsen), varigenom man inte behöver korrigera för blank och utbyte. I detta fall får den försöksrespons som motsvarar 2/3 av gränsvärdet (se punkt 7.3) beräknas direkt från dessa prov och användas som brytpunkt. För kontroll av prov som överskrider åtgärdsgränserna skulle en lämplig procentandel av dessa åtgärdsgränser lämpa sig som brytpunkt.
7.1.3 Separat bestämning av PCDD/PCDF och dioxinlika PCB
Extrakt får delas upp i fraktioner som innehåller dels PCDD/PCDF, dels dioxinlika PCB, vilket gör att man kan ange separata TEQ-värden för PCDD/PCDF och för dioxinlika PCB (uttryckta som BEQ). Företrädesvis ska man använda en kalibreringskurva för PCB 126-standard för att utvärdera resultaten för den fraktion som innehåller dioxinlika PCB.
7.1.4 Skenbart utbyte för bioassay
Det skenbara utbytet för bioassay ska beräknas utifrån lämpliga referensprov med representativa kongenmönster kring gränsvärdet eller åtgärdsgränsen och uttryckas som en procentandel av BEQ-värdet i jämförelse med TEQ-värdet. Beroende på vilken typ av analysmetod och vilka TEF-värden (10) som används kan skillnaderna mellan TEF- och REP-värdena för dioxinlika PCB ge upphov till låga skenbara utbyten för dioxinlika PCB i jämförelse med för PCDD/PCDF. Om en separat bestämning av PCDD/PCDF och dioxinlika PCB utförs ska därför de skenbara utbytena för bioassay ligga i intervallet 20–60 % för dioxinlika PCB och i intervallet 50–130 % för PCDD/PCDF (intervallen gäller för kalibreringskurvan för TCDD). Bidraget från dioxinlika PCB till summan av PCDD/PCDF och dioxinlika PCB kan variera mellan olika matriser och prov. Dessa variationer återspeglas i de skenbara utbytena för bioassay, vilka ska ligga i intervallet 30–130 % för summan av PCDD/PCDF och dioxinlika PCB. Om TEF-värdena för PCDD/PCDF och dioxinlika PCB i EU-lagstiftningen skulle ändras väsentligt, måste dessa intervall granskas.
7.1.5 Kontroll av utbytet efter upprening
Förlusten av föreningar under uppreningen ska kontrolleras under valideringen. Ett blankprov som spikats med en blandning av olika kongener ska genomgå upprening (minst n = 3) och därefter ska utbyte och variabilitet kontrolleras med en konfirmeringsmetod. Utbytet ska ligga i intervallet 60–120 %, särskilt för kongener som i olika blandningar bidrar med mer än 10 % till TEQ-värdet.
7.1.6 Rapporteringsgräns
Vid rapportering av BEQ-värden ska en rapporteringsgräns bestämmas utifrån relevanta matrisprov som omfattar typiska kongenmönster. Däremot ska inte kalibreringskurvan för standarder användas eftersom precisionen är låg för den nedre delen av kurvan. Även effekter från extraktion och upprening ska beaktas. Rapporteringsgränsen ska ligga över metodens blankvärde med åtminstone en faktor på tre.
7.2 Användning av referensprov
7.2.1 Referensproven ska representera provmatriser, kongenmönster och koncentrationsintervall för PCDD/PCDF och dioxinlika PCB kring gränsvärdet eller åtgärdsgränsen.
7.2.2 Ett blankprov för matrisen, eller när detta inte är möjligt ett blankprov för metoden, och ett referensprov vid gränsvärdet eller åtgärdsgränsen ska ingå i varje försöksserie. Dessa prov ska extraheras och analyseras samtidigt under identiska betingelser. Referensprovet ska ha en klart högre respons än blankprovet eftersom detta säkerställer metodens lämplighet. Dessa prov kan användas för att korrigera för blank och utbyte.
7.2.3 De referensprov som valts ut för att korrigera för utbyte ska vara representativa för alla proven, vilket innebär att kongenmönstrena inte ska resultera i att halterna underskattas.
7.2.4 För kontroll av gränsvärdet eller åtgärdsgränsen får ytterligare referensprov på exempelvis 0,5 gång och 2 gånger gränsvärdet eller åtgärdsgränsen tas med i syfte att påvisa analysens tillförlitlighet inom det givna intervallet. Dessa prov får kombineras för att beräkna BEQ-värdet i prov (se punkt 7.1.2.2).
7.3 Bestämning av brytpunkter
Förhållandet mellan bioanalytiska resultat angivna som BEQ och resultat från konfirmeringsmetoden angivna som TEQ ska fastställas, till exempel genom matrisanpassade kalibreringsexperiment som omfattar referensprov spikade med 0 gång, 0,5 gång, 1 gång och 2 gånger gränsvärdet och 6 prov per halt (totalt n = 24). Korrektionsfaktorer (blank och utbyte) får beräknas från detta förhållande, men de ska kontrolleras i enlighet med punkt 7.2.2.
Brytpunkter ska fastställas för bedömning av om prov överensstämmer med gränsvärdena, eller vid behov för kontroll av åtgärdsgränserna, med respektive gränsvärden eller åtgärdsgränser som fastställts för antingen PCDD/F och dioxinlika PCB var för sig, eller för summan av PCDD/F och dioxinlika PCB. De representeras av den nedre gränsen för fördelningen av de bioanalytiska resultaten (korrigerade för blank och utbyte) som motsvarar beslutsgränsen för konfirmeringsmetoden med en konfidensgrad på 95 %, vilket innebär att andelen prov som felaktigt bedömts vara överensstämmande är mindre än 5 % och RSDR är mindre än 25 %. Beslutsgränsen för konfirmeringsmetoden motsvaras av gränsvärdet, med beaktande av mätosäkerheten.
Brytpunkten (uttryckt som BEQ) kan beräknas enligt ett av de sätt som anges i punkt 7.3.1, 7.3.2 och 7.3.3 (se figur 1).
7.3.1 Med användning av det nedre konfidensbandet i det 95-procentiga prediktionsintervallet för beslutsgränsen för konfirmeringsmetoden och med formeln
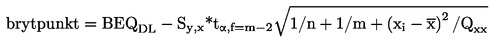
där
|
BEQDL |
BEQ-värdet som motsvarar beslutsgränsen för konfirmeringsmetoden, vilket är gränsvärdet inklusive mätosäkerhet |
|
sy,x |
residualstandardavvikelse |
|
t α,f = m-2 |
Students t-faktor (α = 5 %, f = frihetsgrader, ensidiga) |
|
m |
totalt antal kalibreringspunkter (index j) |
|
n |
antal prov per koncentration (repetitioner) |
|
xi |
provkoncentration för kalibreringspunkt i analyserad med en konfirmeringsmetod (uttryckt som TEQ) |
|
|
medelvärdet av koncentrationerna för alla kalibreringsprov (uttryckt som TEQ) |
|
|
parametern kvadratsumma, i = index för kalibreringspunkt i. |
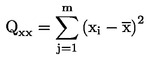
7.3.2 Beräknat utifrån de bioanalytiska resultaten (korrigerade för blank och utbyte) från flera analyser av prov (n ≥ 6) som förorenats med en halt som motsvarar beslutsgränsen för konfirmeringsmetoden, vilken är den nedre gränsen för fördelningen vid motsvarande medelvärde för BEQ, och med formeln
brytpunkt = BEQDL – 1,64 × SDR
där
|
SDR |
standardavvikelse för resultat från bioassay vid BEQDL, mätt under reproducerbara betingelser inom laboratoriet. |
7.3.3 Beräknat som medelvärdet av de bioanalytiska resultaten (uttryckta som BEQ, korrigerade för blank och utbyte) från flera analyser av prov (n ≥ 6) som förorenats med en halt som motsvarar 2/3 av gränsvärdet eller åtgärdsgränsen. Detta grundas på att denna halt kommer att ligga omkring den brytpunkt som bestämts enligt punkt 7.3.1 eller 7.3.2.
Figur 1

Figur 1: Tillvägagångssätt för beräkning av brytpunkter som bygger på en 95-procentig konfidensgrad vilket innebär att andelen prov som felaktigt bedömts vara överensstämmande är mindre än 5 % och RSDR är mindre än 25 %.
|
1. |
Utifrån det nedre konfidensbandet i det 95-procentiga prediktionsintervallet för beslutsgränsen för konfirmeringsmetoden. |
|
2. |
Utifrån flera analyser av prov (n ≥ 6) som förorenats med en halt som motsvarar beslutsgränsen för konfirmeringsmetoden, vilket är den nedre gränsen för fördelningen (en klockformad kurva i figuren) vid motsvarande medelvärde för BEQ. |
7.3.4 Begränsningar för brytpunkter
De BEQ-baserade brytpunkter som beräknats utifrån den RSDR som fastställts under valideringen med hjälp av ett begränsat antal prov med olika matriser eller kongenmönster kan vara högre än gränsvärden eller åtgärdsgränser som baserats på TEQ-värden, eftersom en bättre precision kan uppnås vid validering än vid rutinanalyser då ett okänt spektrum av möjliga kongenmönster måste kontrolleras. I sådana fall ska brytpunkterna beräknas utifrån en RSDR som motsvarar 25 % eller helst 2/3 av gränsvärdet eller åtgärdsgränsen.
7.4 Prestanda
7.4.1 Eftersom inga interna standarder kan användas i bioanalytiska metoder ska försök som kontrollerar repeterbarheten hos dessa metoder utföras i syfte att bedöma standardavvikelsen inom en försöksserie och mellan försöksserier. Repeterbarheten ska vara under 20 % och reproducerbarheten inom laboratoriet under 25 %. Detta ska grundas på de beräknade halterna uttryckta som BEQ efter korrigering för blank och utbyte.
7.4.2 Under valideringsförfarandet ska försöket visa att metoden kan skilja mellan ett blankprov och ett prov med en halt som motsvarar brytpunkten, så att prov som ligger över den relevanta brytpunkten kan identifieras (se punkt 7.1.2).
7.4.3 Målföreningar, möjliga interferenser och högsta godtagbara värden för blankprov ska definieras.
7.4.4 Vid analys av ett provextrakt (n = 3) ska standardavvikelsen inte överstiga 15 % för en respons eller en koncentration som beräknats utifrån responsen (endast möjligt i mätområdet).
7.4.5 De okorrigerade resultaten från referensprov uttryckta som BEQ (blank och vid gränsvärdet eller åtgärdsgränsen) ska användas vid utvärderingen av den bioanalytiska metodens prestanda under en konstant tidsperiod.
7.4.6 Kontrollkort för metodens blankprov och för varje typ av referensprov ska framställas och kontrolleras för att säkerställa att den analytiska prestandan uppfyller kraven, särskilt för metodens blankprov när det gäller kravet på minsta skillnad till de lägsta koncentrationerna i mätområdet och för referensprov när det gäller reproducerbarheten inom laboratoriet. Metodens blankprov ska kontrolleras så att man när blankprovet dras av från provresultatet inte får resultat som felaktigt bedöms vara överensstämmande.
7.4.7 Resultaten från analyserna med konfirmeringsmetoderna av misstänkta prov och av 2–10 % av de överensstämmande proven (minst 20 prov per matris) ska samlas in och användas för att utvärdera screeningmetodens prestanda och förhållandet mellan BEQ- och TEQ-värdena. Dessa data kan användas för omprövning av de brytpunkter som tillämpas på rutinprov för validerade matriser.
7.4.8 Att en metod har en bra prestanda kan också styrkas genom deltagande i ringtest. Om ett laboratorium kan styrka ett framgångsrikt deltagande får resultat från prov som analyserats i ringtest och som täcker ett koncentrationsintervall upp till exempelvis två gånger gränsvärdet tas med i utvärderingen av andelen prov som felaktigt bedömts vara överensstämmande. Proven ska täcka de vanligaste kongenmönstren och komma från olika källor.
7.4.9 Vid incidenter får brytpunkter omprövas för att återspegla den specifika matrisen och kongenmönstret för denna enda incident.
8. Rapportering av resultat
8.1 Konfirmeringsmetoder
8.1.1 Om den använda analysmetoden medger det ska analysresultaten innehålla halterna av de enskilda PCDD/PCDF- och dioxinlika PCB-kongenerna angivna som lägre koncentrationer, övre koncentrationer och mellanvärden. Rapporteringen av resultat ska på så sätt ge maximalt med information och därigenom göra det möjligt att tolka resultaten utifrån särskilda krav.
8.1.2 Rapporten ska ange den metod som använts vid extraktionen av PCDD/PCDF och dioxinlika PCB.
8.1.3 Utbytet för de enskilda interna standarderna ska redovisas om det ligger utanför det intervall som anges i punkt 6.2.5 eller när gränsvärdet överskrids (i så fall ska utbytet för ett av de två dubbelproven anges). I övriga fall ska det redovisas på begäran.
8.1.4 Eftersom mätosäkerheten ska beaktas vid bedömning av om ett prov är överensstämmande ska denna parameter anges. Analysresultatet ska rapporteras som x +/– U, där x är analysresultatet och U den utvidgade mätosäkerheten beräknad med en täckningsfaktor på 2, vilket ger en konfidensgrad på cirka 95 %. Om det har gjorts en separat bestämning av PCDD/PCDF och av dioxinlika PCB ska summan av den skattade utvidgade mätosäkerheten för de enskilda analysresultaten användas när man anger summan av PCDD/PCDF och dioxinlika PCB.
8.1.5 Om mätosäkerheten har beaktats genom att beslutsgränsen (CCα) har fastställts (se punkt 2.2 i del B kapitel I) ska denna parameter anges.
8.1.6 Resultaten ska anges i samma enheter och med minst samma antal signifikanta siffror som används för att ange gränsvärden i direktiv 2002/32/EG.
8.2 Bioanalytiska screeningmetoder
8.2.1 Resultaten från screeningen ska anges som överensstämmande eller som misstänkt icke-överensstämmande.
8.2.2 Dessutom får ett resultat för PCDD/PCDF och/eller dioxinlika PCB, uttryckt som BEQ och inte som TEQ, anges.
8.2.3 Prov vars respons ligger under rapporteringsgränsen ska benämnas ’under rapporteringsgränsen’.
8.2.4 För varje typ av provmatris ska rapporten innehålla uppgifter om det gränsvärde eller den åtgärdsgräns som utvärderingen baseras på.
8.2.5 Rapporten ska innehålla uppgifter om typ av försök som har gjorts, den grundläggande principen för försöket och typ av kalibrering.
8.2.6 Rapporten ska ange den metod som använts vid extraktionen av PCDD/PCDF och dioxinlika PCB.
8.2.7 För prov som misstänks vara icke-överensstämmande ska rapporten innehålla information om de åtgärder som ska vidtas. Koncentrationen av PCDD/PCDF och summan av PCDD/PCDF och dioxinlika PCB i proven med förhöjda halter ska bestämmas/bekräftas med en konfirmeringsmetod.
KAPITEL III
Provberedning och krav på analysmetoder för offentlig kontroll av halten av icke-dioxinlika PCB (PCB 28, 52, 101, 138, 153, 180)
1. Tillämpningsområde
Kraven i detta kapitel ska tillämpas när foder analyseras för offentlig kontroll av halterna av icke-dioxinlika polyklorerade bifenyler (nedan kallade icke-dioxinlika PCB) och för andra lagstadgade ändamål.
2. Detektionsmetoder som ska användas
Gaskromatografi i kombination med elektroninfångningsdetektion (GC-ECD), GC-LRMS, GC-MS/MS, GC-HRMS eller likvärdiga metoder.
3. Identifiering och konfirmering av givna analyter
3.1 Relativ retentionstid i förhållande till interna standarder eller referensstandarder (godtagbar avvikelse +/–0,25 %).
3.2 Gaskromatografisk separation av alla sex indikator-PCB (PCB 28, PCB 52, PCB 101, PCB 138, PCB 153 och PCB 180) från störande ämnen, särskilt andra PCB som elueras samtidigt, i synnerhet om provhalterna ligger i nivå med lagstadgade gränsvärden och det ska konfirmeras att proverna är icke-överensstämmande.
[Kongener som ofta elueras samtidigt är t.ex. PCB 28/31, PCB 52/69 och PCB 138/163/164. När det gäller GC-MS ska även eventuella interferenser från fragment av mer högklorerade kongener beaktas.]
3.3 Krav på GC-MS-metoder
Övervakning av åtminstone
|
a) |
två specifika joner för HRMS, |
|
b) |
två specifika joner med m/z > 200 eller tre specifika joner med m/z > 100 för LRMS, |
|
c) |
en moderjon och två dotterjoner för MS/MS. |
Högsta tillåtna toleranser för kvoten för förekomsten av utvalda massfragment:
Relativ avvikelse mellan kvoten för förekomsten av utvalda massfragment från den teoretiska förekomsten eller kalibreringsstandarden för måljon (den vanligast förekommande jonen som övervakats) och konfirmeringsjon(er).
|
Konfirmeringsjonens (-jonernas) relativa intensitet jämfört med måljonens |
GC-EI-MS (relativ avvikelse) |
GC-CI-MS, GC-MSn (relativ avvikelse) |
|
> 50 % |
± 10 % |
± 20 % |
|
> 20 % till 50 % |
± 15 % |
± 25 % |
|
> 10 % till 20 % |
± 20 % |
± 30 % |
|
≤ 10 % |
± 50 % (11) |
± 50 % (11) |
3.4 Krav på GC-ECD-metoder
Resultat som överskrider gränsvärdet ska konfirmeras med hjälp av två GC-kolonner som har stationära faser med olika polaritet.
4. Bevis för metodens prestanda
Metodens prestanda ska valideras i intervallet för gränsvärdet (0,5–2 gånger gränsvärdet) med en godtagbar variationskoefficient för upprepade analyser (se krav på intermediärt precisionsmått i punkt 9).
5. Kvantifieringsgräns
Blankprovets värden ska vara högst 30 % av den föroreningshalt som motsvarar gränsvärdet (12).
6. Kvalitetskontroll
Laboratorierna ska regelbundet kontrollera blankprov, analysera spikade prov och prov för kvalitetskontroll samt medverka i kollaborativa studier med relevanta matriser.
7. Kontroll av utbytet
7.1 Lämpliga interna standarder med fysikalisk-kemiska egenskaper som är jämförbara med de givna analyternas ska användas.
7.2 Tillsats av intern standard:
Ska tillsättas produkter före extraktion och upprening.
7.3 Krav på metoder som använder alla sex isotopmärkta indikator-PCB-kongener:
|
a) |
Resultaten ska korrigeras för de interna standardernas utbyten. |
|
b) |
Utbytet för isotopmärkta interna standarder ska ligga i intervallet 50–120 %. |
|
c) |
Ett lägre eller högre utbyte för enskilda kongener vars bidrag till summan av de sex indikator-PCB är mindre än 10 % är godtagbart. |
7.4 Krav på metoder som inte använder alla sex isotopmärkta interna standarder eller använder andra interna standarder:
|
a) |
Utbytet för interna standarder ska kontrolleras för varje prov. |
|
b) |
Utbytet för interna standarder ska ligga i intervallet 60–120 %. |
|
c) |
Resultaten ska korrigeras för utbytet för interna standarder. |
7.5 Utbytet för omärkta kongener ska kontrolleras genom spikade prov eller prov för kvalitetskontroll vilka har koncentrationer i intervallet för gränsvärdet. Godtagbara utbyten för dessa kongener ska ligga i intervallet 70–120 %.
8. Krav på laboratorier
I enlighet med förordning (EG) nr 882/2004 ska laboratorierna ackrediteras av ett godkänt ackrediteringsorgan som verkar enligt ISO Guide 58 för att säkerställa att de tillämpar ett system för kvalitetssäkring av analysverksamheten. Laboratorierna ska ackrediteras enligt standarden EN ISO/IEC 17025.
9. Prestanda: kriterier för summan av de sex indikator-PCB vid gränsvärdet
|
Riktighet |
–30 till +30 % |
|
Intermediärt precisionsmått (RSD %) |
≤ 20 % |
|
Skillnaden mellan beräkningen av övre och lägre koncentration |
≤ 20 % |
10. Rapportering av resultat
10.1 Om den använda analysmetoden medger det ska analysresultaten innehålla halterna av de enskilda PCB-kongenerna angivna som lägre koncentrationer, övre koncentrationer och mellanvärden. Rapporteringen av resultat ska på så sätt ge maximalt med information och därigenom göra det möjligt att tolka resultaten utifrån särskilda krav.
10.2 Rapporten ska ange den metod som använts vid extraktionen av PCB och lipider.
10.3 Utbytet av de enskilda interna standarderna ska redovisas om det ligger utanför det intervall som anges i punkt 7 eller när gränsvärdet överskrids. I övriga fall ska det redovisas på begäran.
10.4 Eftersom mätosäkerheten ska beaktas vid bedömning av om ett prov är överensstämmande ska också denna parameter anges. Analysresultatet ska rapporteras som x +/– U, där x är analysresultatet och U den utvidgade mätosäkerheten beräknad med en täckningsfaktor på 2, vilket ger en konfidensgrad på cirka 95 %.
10.5 Om mätosäkerheten har beaktats genom att beslutsgränsen (CCα) har fastställts (se punkt 2.1 i kapitel I) ska denna parameter anges.
10.6 Resultaten ska anges i samma enheter och med minst samma antal signifikanta siffror som används för att ange gränsvärden i direktiv 2002/32/EG.
(9) Med avseende på gränsvärdena.
(11) Eftersom det finns ett tillräckligt antal massfragment med relativ intensitet större än 10 % bör man inte använda konfirmeringsjon(er) med en relativ intensitet mindre än 10 % jämfört med måljonens.