|
3.
|
Följande kapitel ska läggas till:
”B.49. MIKRONUKLEUSTEST AV DÄGGDJURSCELLER IN VITRO
INLEDNING
|
1.
|
Mikronukleustest in vitro (MNvit) är ett genotoxicitetstest för att upptäcka mikrokärnor i cytoplasmat i interfasceller. Mikrokärnor kan härröra från acentriska kromosomfragment (dvs. en centromer saknas) eller från hela kromosomer som inte kan migrera till polerna under det anafasa steget av celldelningen. Genom testet upptäcks aktiviteten hos klastogena och aneugena kemikalier (ämnen och blandningar) (1) (2) i celler som har genomgått celldelning under eller efter exponering för testämnet. Denna testmetod ger möjlighet att använda protokoll med och utan aktinpolymeriseringsinhibitorn cytokalasin B (cytoB). Tillsättning av cytoB före mitosen gör det möjligt att identifiera och selektivt analysera frekvensen mikrokärnor i celler som har slutfört en mitos eftersom sådana celler är binukleära (3) (4). Denna testmetod gör det också möjligt att använda protokoll utan blockering av cytokines, förutsatt att det finns belägg för att den analyserade cellpopulationen har genomgått mitos.
|
|
2.
|
Utöver MNvit-metoden för att identifiera kemikalier (ämnen och blandningar) som inducerar mikrokärnor kan man även använda blockering av cytokines, immunokemisk märkning av kinetochorer eller hybridisering med centromeriska/telomeriska prober (FISH-teknik, Fluorescens In Situ Hybridisation) för att få information om mekanismerna för kromosomskador och bildningen av mikrokärnor (5) (6) (7) (8) (9) (10) (11) (12) (13) (14) (15) (16). Märknings- och hybridiseringsteknik kan användas när det förekommer ökad bildning av mikrokärnor och man vill slå fast om ökningen är resultatet av klastogena och/eller aneugena händelser.
|
|
3.
|
Mikrokärnorna representerar skador som har förmedlats till dotterceller, medan kromosomavvikelser som detekteras i metafasceller inte alltid förmedlas. Eftersom mikrokärnor i interfasceller kan bedömas relativt objektivt, behöver laboratoriepersonalen endast bestämma huruvida cellerna har genomgått delning eller inte och hur många celler som innehåller mikrokärna. Därför kan detekteringen göras relativt snabbt och analysen kan automatiseras. Då kan man detektera tusentals i stället för hundratals celler per behandling, vilket ökar försökets tyngd. Eftersom mikrokärnor kan uppstå från isolerade kromosomer finns det också möjlighet att detektera aneuploidiframkallande ämnen som är svåra att undersöka genom konventionella kromosomavvikelsetest, t.ex. OECD:s testriktlinje 473 (kapitel B.10 i denna bilaga) (17). MNvit-metoden ger dock inte möjlighet att utan särskilda tekniker såsom FISH enligt beskrivningen i punkt 2 skilja mellan kemikalier som inducerar polyploidi från sådana kemikalier som inducerar klastogenicitet.
|
|
4.
|
MNvit-metoden är en in vitro-metod där man i regel använder odlade celler från människa eller gnagare. Metoden ger tillgång till en omfattande bas för undersökning av kromosomskadepotentialen in vitro eftersom både aneugener och klastogener kan upptäckas.
|
|
5.
|
MNvit-metoden är robust och effektiv för ett flertal celltyper, med eller utan tillsats av cytoB. Det finns omfattande data som stöd för att MNvit-metoden är giltig för ett flertal gnagarcellinjer (CHO, V79, CHL/IU och L5178Y) och lymfocyter från människa (18) (19) (20) (21) (22) (23) (24) (25) (26) (27) (28) (29) (30) (31). Dessa data kommer bland annat från internationella valideringsstudier samordnade av Société Française de Toxicologie Génétique (SFTG) (18) (19) (20) (21) (22) och rapporter från International Workshop on Genotoxicity Testing (4) (16). Tillgängliga data har också omvaliderats i en retrospektiv sammanvägd (weight-of-evidence) valideringsstudie vid Europiska centret för bestämning av alternativa metoder (ECVAM), och testmetoden har erkänts som vetenskapligt giltig av ECVAM:s vetenskapliga rådgivande kommitté (ESAC) (32) (33) (34). Man har beskrivit användningen av TK6 lymfoblastoidceller från människa (35), HepG2-celler (36) (37) och embryoceller från syrisk hamster (38), även om de inte har använts i valideringsstudier.
|
DEFINITIONER
|
6.
|
Definitioner av begreppen finns i tillägg 1.
|
INLEDANDE ÖVERVÄGANDEN
|
7.
|
För testning in vitro krävs i regel ett exogent system för metabolisk aktivering, om cellerna inte är metaboliskt kompetenta i fråga om de ämnen som testas. Det exogena metaboliska aktiveringssystemet efterliknar inte helt förhållandena in vivo. Man bör också se till att undvika sådana förhållanden som leder till artefaktiska positiva resultat som inte återspeglar den verkliga mutageniciteten och kan ha sin orsak i faktorer såsom markerade ändringar av pH eller osmolalitet eller hög cytotoxicitet (39) (40) (41). Om testkemikalien orsakar ändring av pH i mediet vid tillsättningstidpunkten bör pH justeras, företrädesvis genom buffring av stamlösningen så att alla volymer hålls lika stora vid alla testkoncentrationer och för alla kontroller.
|
|
8.
|
För analysen av induktion av mikrokärnor är det av central betydelse att mitos har inträffat i både de behandlade och obehandlade odlingarna. Det mest informativa stadiet för detektering av mikrokärnor är när cellerna har slutfört en mitos under eller efter behandling med testämnet.
|
PRINCIP FÖR TESTMETODEN
|
9.
|
Cellodlingar från människa eller däggdjur exponeras för testämnet både med och utan exogent metaboliskt aktiveringssystem, utom om de celler som används har tillräcklig metaboliserande kapacitet. Parallella kontroller med lösningsmedel/vehikel och positiva kontrollkemikalier ska ingå i alla test.
|
|
10.
|
Under eller efter exponering för testämnet odlas cellerna tillräckligt länge för att kromosomskador eller spindelfiberskador ska leda till att mikrokärnor bildas i interfascellerna. För induktion av aneuploidi bör testämnet i regel finnas närvarande under mitosen. Interfascellerna skördas och färgas in och närvaron av mikrokärnor analyseras. Idealiskt sett bör man för detekteringen av mikrokärnor endast använda celler som har slutfört mitosen under exponeringen för testämnet eller under perioden efter exponeringen, om en sådan används. I odlingar som har behandlats med en cytokinesinhibitor uppnås detta genom att endast analysera binukleära celler. Om blockering av cytokines inte används är det viktigt att påvisa att de analyserade cellerna sannolikt har genomgått celldelning under eller efter exponeringen för testämnet. Oavsett protokoll är det viktigt att påvisa att cellproliferation har skett både i kontrollodlingarna och de behandlade odlingarna, och omfattningen av testämnesinducerad cytotoxicitet eller cytostas ska bedömas i de odlingar (eller parallella odlingar) som är föremål för analys av mikrokärnor.
|
TESTBESKRIVNING
Förberedelser
|
11.
|
Man kan använda odlade primära perifera blodlymfocyter från människa (5) (19) (42) (43) och celler från ett antal gnagarcellinjer såsom CHO, V79, CHL/IU och L5178Y (18) (19) (20) (21) (22) (25) (26) (27) (28) (30). Användning av andra cellinjer och celltyper bör motiveras på grundval av deras påvisade prestanda med denna metod, enligt det som anges i avsnittet om kriterier för godkännande. Eftersom bakgrundsförekomsten av mikrokärnor påverkar försökets känslighet, rekommenderas att man använder celltyper med en låg stabil bakgrundsförekomst av mikrokärnbildning.
|
|
12.
|
De humana perifera blodlymfocyterna ska tas från unga (cirka 18–35 år) rökfria individer utan tidigare känd exponering för genotoxiska kemikalier eller strålning. Om celler från fler än en givare poolas för användning bör antalet givare anges. Förekomsten av mikrokärnor ökar med åldern och denna trend är mera markerad hos kvinnor än hos män (44), vilket bör beaktas när man väljer celler för poolning.
|
Medier och odlingsbetingelser
|
13.
|
Lämpliga odlingsmedier och inkubationsförhållanden (odlingskärl, CO2-halt, temperatur och luftfuktighet) bör väljas för odlingarna. Etablerade cellinjer och stammar bör rutinmässigt kontrolleras med avseende på det modala kromosomantalets stabilitet och frånvaro av kontaminerad mykoplasma, och får inte användas om kontamination har skett eller om det modala kromosomantalet har ändrats. Den normala cellcykeltiden vid de odlingsbetingelser som används i laboratoriet bör vara känd. Om blockering av cytokines används bör koncentrationen av cytokinesinhibitor vara optimerad för den berörda celltypen och bör även ha påvisats kunna ge en god skörd av binukleära celler för analys.
|
Beredning av odlingarna
|
14.
|
Etablerade cellinjer och stammar: celler dras upp från stamkulturer, inokuleras i ett odlingsmedium vars densitet är sådan att det inte sker sammanväxning i enkelskiktsodlingar och att suspensionsodlingar inte når för hög densitet före skörden, och inkuberas vid 37 °C.
|
|
15.
|
Lymfocyter: helblod behandlat med en antikoagulant (t.ex. heparin) eller separerade lymfocyter odlas i ett medium som innehåller en mitogen (t.ex. fytohemaglutinin) före exponering för testämnet och cytoB.
|
Metabolisk aktivering
|
16.
|
Exogena metaboliserande system bör användas när cellerna inte har tillräcklig endogen metabolisk kapacitet. Det mest använda systemet är en kofaktorstödd post-mitokondriell fraktion (S9) beredd ur lever från gnagare som behandlats med enzyminducerande ämnen såsom Aroclor 1254 (45) (46) eller en kombination av fenobarbiton och β-naftoflavon (46) (47) (48) (49). Den senare kombinationen bryter inte mot Stockholmkonventionen om långlivade organiska föroreningar (50) eller förordning (EG) nr 850/2004 om långlivade organiska föroreningar (66) och har visat sig vara lika effektiv som Aroclor 1254 när det gäller att inducera monooxygenaser (’mixed-function oxidases’) (46) (47) (48) (49). S9-fraktionen används typiskt i koncentrationer mellan 1–10 % (volym/volym) i det slutliga testmediet. Förhållandena i ett system för metabolisk aktivering kan bero på vilken klass av kemikalie som testas och i vissa fall kan det vara lämpligt att använda mer än en S9-koncentration.
|
|
17.
|
Genetiskt framställda cellinjer som uttrycker specifika aktiverande enzymer för människa eller gnagare kan eliminera behovet av ett exogent system för metabolisk aktivering och kan användas som testceller. I sådana fall bör valet av cellinjer underbyggas vetenskapligt, t.ex. genom monooxygenasernas relevans för testämnets metabolism (51) och deras respons på kända klastogener och aneugener (se avsnittet om kriterier för godkännande). Här bör konstateras att de ämnen som testas kanske inte metaboliseras av monooxygenaser, och i sådana fall indikerar negativa resultat inte att testämnet inte kan inducera mikrokärnor.
|
Beredning av testämnet
|
18.
|
Fasta kemikalier bör lösas upp i eller blandas med lämpliga lösningsmedel eller vehiklar och vid behov spädas ut innan cellerna behandlas. Flytande kemikalier kan tillsättas direkt till testsystemen och/eller spädas ut före behandlingen. Gaser eller flyktiga kemikalier bör testas genom lämpliga modifikationer av standardprotokollen, såsom behandling i slutna kärl (52) (53). Testämnesberedningarna bör vara färska, utom om stabilitetsdata visar att lagring kan godkännas.
|
Försöksbetingelser
Lösningsmedel/vehiklar
|
19.
|
Lösningsmedlet/vehikeln får inte reagera med testämnet, leda till att cellerna inte överlever eller att S9-aktiviteten inte kan upprätthållas vid den använda koncentrationen. Om andra än väletablerade lösningsmedel/vehiklar (t.ex. vatten, cellodlingsmedier, dimetylsulfoxid) används bör detta motiveras med data om deras förenlighet med testämnet och avsaknad av genetisk toxicitet. Det rekommenderas att man alltid när det är möjligt överväger vattenbaserade lösningsmedel/vehiklar som första alternativ.
|
Användning av cytoB som cytokinesinhibitor
|
20.
|
En av de viktigaste faktorerna när det gäller MNvit-metodens resultat är att se till att de celler som analyseras har slutfört mitosen under behandlingen eller under eventuell inkubationsperiod efter behandlingen. CytoB är det ämne som oftast används för blockering av cytokines eftersom det hindrar aktinbildning och således hindrar separationen av dotterceller efter mitosen så att binukleära celler kan bildas (5) (54) (55). Analysen av mikrokärnor kan då begränsas till celler som har genomgått mitos under eller efter behandlingen. Samtidigt kan man mäta testämnets effekt på cellproliferationskinetiken. CytoB ska användas som cytokinesinhibitor när humana lymfocyter används eftersom cellcykeltiderna varierar mellan odlingar och mellan givare och eftersom inte alla lymfocyter ger respons på fytohemaglutinin. Även andra metoder har använts vid testning av cellinjer för att bestämma om de celler som analyseras har delat sig; dessa metoder behandlas nedan (se punkt 26).
|
|
21.
|
Laboratoriet bör fastställa lämplig koncentration av cytoB för varje celltyp i syfte att nå optimal förekomst av binukleära celler i kontrollodlingarna för lösningsmedel/vehikel. Lämplig cytoB-koncentration är i regel 3–6 μg/ml.
|
Mätning av cellproliferation och cytotoxicitet och val av exponeringskoncentrationer
|
22.
|
Vid bestämning av den högsta testämneskoncentrationen ska man undvika koncentrationer som kan ge artefaktiska positiva resultat, t.ex. för kraftig cytotoxicitet, utfällning i odlingsmediet och markerade ändringar av pH eller osmolalitet (39) (40) (41).
|
|
23.
|
Cellproliferationen bör mätas för att se till att de behandlade cellerna har genomgått mitos under försöket och att behandlingarna görs på lämpliga nivåer av cytotoxicitet (se punkt 29). Om cytoB inte används ska cytotoxiciteten bestämmas med och utan metabolisk aktivering i celler som kräver metabolisk aktivering, med användning av den relativa ökningen av antalet celler eller relativ populationsfördubbling (se tillägg 2 för formler). När cytoB används kan cytotoxiciteten bestämmas med användning av replikeringsindex (se tillägg 2 för formel).
|
|
24.
|
Behandling av odlingarna med cytoB och mätning av de relativa förekomsterna av mononukleära, binukleära och multinukleära celler i odlingen är en exakt metod för kvantifiering av effekten på cellproliferationen och behandlingens cytotoxiska eller cytostatiska aktivitet (5) och säkerställer att endast celler som har delat sig under eller efter behandlingen analyseras.
|
|
25.
|
I försök där cytoB används kan cytostas/cytotoxicitet kvantifieras genom att bestämma proliferationsindex vid cytokinesinhibering (Cytokinesis-Block Proliferation Index, CBPI) (5) (26) (56) eller härledas från replikeringsindex (RI) från minst 500 celler per odling (se tillägg 2 för formler). När cytoB används för att bedöma cellproliferation bör CBPI eller RI bestämmas på grundval av minst 500 celler per odling. Dessa åtgärder och andra kan användas för att uppskatta cytotoxiciteten genom jämförelse av värden för behandlade odlingar och kontrollodlingar. Bedömning av andra markörer för cytotoxicitet (t.ex. sammanväxning, cellantal, apoptos, nekros, antalet metafaser) kan ge värdefull information.
|
|
26.
|
I studier utan cytoB är det nödvändigt att påvisa att de celler i odlingen som analyseras har genomgått delning under eller efter behandling med testämnet. Annars kan man få falsk negativ respons. Till de metoder som har använts för att se till att cellerna som analyseras har delat sig hör tillsättning och därpå följande detektering av bromodeoxiuridin (BrdU) för att identifiera celler som har delat sig (57), bildning av kloner när celler från permanenta cellinjer behandlas och analyseras in situ på ett objektglas (proliferationsindex (PI)) (25) (26) (27) (28) eller mätning av relativ populationsfördubbling (RPD) eller relativ ökning av celltal (RICC) eller andra prövade metoder (16) (56) (58) (59) (se tillägg 2 för formler). Bedömning av andra markörer för cytotoxicitet eller cytostas (t.ex. sammanväxning, celltal, apoptos, nekros, antal metafaser) kan ge värdefull information.
|
|
27.
|
Minst tre analyserbara testkoncentrationer bör utvärderas. För att uppnå detta kan det vara nödvändigt att genomföra försöket med ett större antal nära varandra liggande koncentrationer och analysera bildningen av mikrokärnor i de koncentrationer som ger en lämplig cytotoxicitetsskala. En alternativ strategi är att utföra ett preliminärt cytotoxicitetstest för att få ett snävare intervall för det definitiva testet.
|
|
28.
|
Den högsta koncentrationen ska vara avsedd att ge en cytotoxicitet på 55 ± 5 %. Högre nivåer kan inducera kromosomskada som en bieffekt av cytotoxicitet (60). När det uppstår cytotoxicitet ska de valda testkoncentrationerna täcka en skala som sträcker sig från den koncentration som ger 55 ± 5 % cytotoxicitet till den som ger liten eller ingen cytotoxicitet.
|
|
29.
|
Om ingen cytotoxicitet eller utfällning observeras ska de högsta testkoncentrationerna motsvara 0,01 M, 5 mg/ml eller 5 μl/ml, enligt det som är lägst. De koncentrationer som väljs för analys ska i regel vara separerade med högst 10. För testämnen som har en brant koncentration-responskurva kan det vara nödvändigt att använda närmare liggande testämneskoncentrationer så att odlingarna i intervallen för moderat och låg toxicitet också blir analyserade.
|
|
30.
|
Om lösligheten är en begränsande faktor ska den högsta koncentrationen, förutsatt att denna inte begränsas av cytotoxicitet, vara den lägsta koncentration vid vilken minimal utfällning är synlig i odlingarna, förutsatt att analysen inte störs. Utvärdering av utfällning bör göras med metoder som ljusmikroskopi med detektering av utfällning som består eller uppstår under odlingen (vid slutet av behandlingen).
|
Kontrollgrupper
|
31.
|
Parallella positiva och negativa kontroller (lösningsmedel eller vehikel), både med och utan metabolisk aktivering, bör inkluderas i varje försök.
|
|
32.
|
Positiva kontroller behövs för att påvisa cellernas och testprotokollets förmåga att identifiera klastogener och aneugener och att bekräfta S9-preparatets metaboliska förmåga. För den positiva kontrollen bör man använda kända inducerare av mikrokärnbildning vid koncentrationer som förväntas ge små men reproducerbara ökningar över bakgrundsnivån och påvisa testsystemets känslighet. Koncentrationerna för de positiva kontrollerna bör väljas på ett sådant sätt att effekterna är tydliga men inte omedelbart avslöjar de kodade objektglasens identitet för den som gör avläsningarna.
|
|
33.
|
En klastogen som kräver metabolisk aktivering (t.ex. cyklofosfamid, benso(a)pyren) bör användas för att påvisa den metaboliska förmågan och testsystemets förmåga att detektera klastogener. Om det är motiverat kan andra positiva kontroller användas. Eftersom vissa positiva kontroller som kräver metabolisk aktivering i vissa behandlingsförhållanden eller för vissa cellinjer kan vara aktiva utan exogen metabolisk aktivering, bör behovet av metabolisk aktivering och aktiviteten hos S9-preparaten testas i de valda cellinjerna och vid de valda koncentrationerna.
|
|
34.
|
För närvarande känner man inte till några aneugener som kräver metabolisk aktivering för sin genotoxiska aktivitet (16). Positiva kontroller som för närvarande är godkända för aneugen aktivitet är t.ex. kolchicin och vinblastin. Andra kemikalier kan användas om de endast eller primärt inducerar mikrokärnor genom aneugen aktivitet. För att det inte ska behövas två positiva kontroller (för klastogenicitet och aneugenicitet) utan metabolisk aktivering, kan aneugenicitetskontrollen tjäna som positiv kontroll utan S9, och klastogenicitetskontrollen kan användas för att testa lämpligheten för det system som används för metabolisk aktivering. De positiva kontrollerna för klastogenicitet och aneugenicitet bör användas för celler som inte kräver S9. Förslag på positiva kontroller finns i tillägg 3.
|
|
35.
|
Användning av positiva kontroller relaterade till kemisk klass kan också övervägas när lämpliga kemikalier finns att tillgå. Alla positiva kontroller bör vara lämpliga för celltypen och aktiveringsförhållandena.
|
|
36.
|
Lösningsmedels-/vehikelkontroller bör ingå för varje skördningstidpunkt. Dessutom bör man använda obehandlade negativa kontroller (utan lösningsmedel/vehikel), utom om det finns offentliggjorda data eller laboratoriet har historiska data som påvisar att det valda lösningsmedlet inte inducerar genotoxiska eller andra skadliga effekter vid de koncentrationer som används.
|
TESTFÖRFARANDE
Behandlingsschema
|
37.
|
För att maximera sannolikheten för att detektera en aneugen eller klastogen som agerar i ett visst skede av cellcykeln är det viktigt att tillräckligt antal celler behandlas med testämnet under cellcykelns alla skeden. Behandlingsschemat för cellinjer och primära cellodlingar kan därför avvika något från behandlingsschemat för lymfocyter som kräver mitogen stimulering för att inleda sin cellcykel; dessa behandlas i punkterna 41–43 (16).
|
|
38.
|
Teoretiska överväganden och offentliggjorda data (18) tyder på att de flesta aneugener och klastogener detekteras med en kortvarig behandlingsperiod på 3–6 timmar med och utan tillsats av S9, åtföljd av borttagning av testämnet och en tillväxtperiod på 1,5–2,0 cellcykler (6). Cellerna skördas vid en tidpunkt som motsvarar cirka 1,5–2,0 gånger den normala (dvs. obehandlade) cellcykelns längd efter att behandlingen inletts eller vid avslutad behandling (se tabell 1). Provtagnings- eller skördningstidpunkterna kan senareläggas om det är känt eller man misstänker att testämnet påverkar cellcykellängden (t.ex. vid testning av nukleosidanaloger).
|
|
39.
|
Eftersom S9-preparat är potentiellt cytotoxiska för odlade däggdjursceller används en förlängd exponeringsbehandling på 1,5–2,0 gånger normal cellcykel endast när S9 inte ingår. Vid förlängd behandling finns det alternativ där man har möjlighet att behandla cellerna med testkemikalie utan eller med cytoB. Dessa alternativ gäller för situationer där det kan finnas tvivel rörande eventuell växelverkan mellan testämnet och cytoB.
|
|
40.
|
Förslag till cellbehandlingsscheman finns i tabell 1. Dessa allmänna behandlingsscheman kan modifieras beroende på testämnets stabilitet eller reaktivitet eller de särskilda tillväxtegenskaper som gäller för de celler som används. Alla behandlingar bör inledas och avslutas medan cellerna är i exponentiell tillväxtfas. Dessa scheman presenteras i närmare detalj i punkterna 41–47 nedan.
Tabell 1
Tidpunkter för cellbehandling och skörd när MNvit-metoden används
|
Lymfocyter, primära celler och cellinjer behandlade med cytoB
|
+ S9
|
Behandla 3–6 timmar med tillsats av S9,
avlägsna S9 och behandlingsmedium,
tillsätt färskt medium och cytoB,
skörda efter 1,5–2,0 normala cellcykler.
|
|
– S9
Kort exponering
|
Behandla 3–6 timmar,
avlägsna behandlingsmedium,
tillsätt färskt medium och cytoB,
skörda efter 1,5–2,0 normala cellcykler.
|
|
– S9
Förlängd exponering
|
Alternativ A: Behandla under 1,5–2 normala cellcykler med tillsats av cytoB,
skörda i slutet av exponeringsperioden.
Alternativ B: Behandla under 1,5–2,0 normala cellcykler,
avlägsna testämnet,
tillsätt färskt medium och cytoB,
skörda efter 1,5–2,0 normala cellcykler.
|
|
Cellinjer som behandlas utan cytoB
(samma som de behandlingsscheman som anges ovan utom att cytoB inte tillsätts)
|
|
Lymfocyter, primära celler och cellinjer med cytoB
|
41.
|
För lymfocyter är det effektivast att starta exponeringen för testämnet 44–48 timmar efter stimulering med fytohemaglutinin, när cykelsynkroniseringen har fallit bort (5). I det första försöket behandlas cellerna 3–6 timmar med testämnet med och utan tillsats av S9. Behandlingsmediet avlägsnas och ersätts med färskt medium som innehåller cytoB och cellerna skördas efter 1,5–2,0 normala cellcykler.
|
|
42.
|
Om båda de initiala försöken med kort (3–6 timmar) behandling är negativa eller tvetydiga görs en efterföljande förlängd exponeringsbehandling utan S9. Det finns två likvärdigt godkända behandlingsalternativ att tillgå. Det kan dock vara lämpligare att använda alternativ A för stimulerade lymfocyter i fall där den exponentiella tillväxten kan minska vid 96 timmar efter stimulering. Likaså ska cellodlingarna inte ha nått sammanväxning vid den slutliga skördningstiden i alternativ B.
|
—
|
Alternativ A: Cellerna behandlas med testämnet under 1,5–2,0 normala cellcykler och skördas vid behandlingstidens slut.
|
|
—
|
Alternativ B: Cellerna behandlas med testämnet under 1,5–2,0 normala cellcykler. Behandlingsmediet avlägsnas och ersätts med färskt medium och cellerna skördas efter ytterligare 1,5–2,0 normala cellcykler.
|
|
|
43.
|
Primära celler och cellinjer ska behandlas på liknande sätt som lymfocyterna, utom att det inte är nödvändigt att stimulera dem med fytohemaglutinin under 44–48 timmar. Andra celler än lymfocyter ska exponeras så att cellerna vid försökets slut fortfarande är i logfas.
|
Cellinjer utan cytoB
|
44.
|
Cellerna behandlas 3–6 timmar med och utan tillsats av S9. Behandlingsmediet avlägsnas och ersätts med färskt medium och cellerna skördas efter 1,5–2,0 normala cellcykler.
|
|
45.
|
Om båda de initiala försöken med kort (3–6 timmar) behandling är negativa eller tvetydiga görs en efterföljande förlängd exponeringsbehandling (utan S9). Det finns två likvärdigt godkända behandlingsalternativ att tillgå.
|
—
|
Alternativ A: Cellerna behandlas med testämnet under 1,5–2,0 normala cellcykler och skördas vid behandlingstidens slut.
|
|
—
|
Alternativ B: Cellerna behandlas med testämnet under 1,5–2,0 normala cellcykler. Behandlingsmediet avlägsnas och ersätts med färskt medium och cellerna skördas efter ytterligare 1,5–2,0 normala cellcykler.
|
|
|
46.
|
I enkelskikt kan det finnas mitotiska celler (som identifieras genom att de är runda och lösgör sig från ytan) i slutet av behandlingen på 3–6 timmar. Eftersom dessa mitotiska celler enkelt lösgörs, kan de förloras när mediet som innehåller testämnet avlägsnas. Man bör vara uppmärksam på att samla in dem när odlingarna tvättas och återbörda dem till odlingarna, för att undvika förlust av celler som undergår mitos och är utsatta för risk för mikrokärnor vid tidpunkten för skörd.
|
Antal odlingar
|
47.
|
Dubbla odlingar bör användas för varje testämneskoncentration och för lösningsmedels-/vehikelodlingarna och de negativa kontrollerna. I fall där laboratoriets historiska data kan påvisa minimala variationer mellan dubbla odlingar kan det vara godtagbart att endast använda en odling. I så fall rekommenderas analys av ett högre antal koncentrationer.
|
Skörd av celler och beredning av objektglas
|
48.
|
Varje odling skördas och hanteras separat. Förberedningen av cellerna kan inbegripa hypoton behandling, men detta steg är inte nödvändigt om tillräcklig cellspridning erhålls på annat sätt. Olika tekniker kan användas för preparering av objektglasen förutsatt att man får cellpreparat av hög kvalitet för analysen. Cellernas cytoplasma bör behållas för att ge möjlighet att detektera mikrokärnor och (vid metoden med blockering av cytokines) tillförlitlig identifiering av binukleära celler.
|
|
49.
|
Objektglasen kan färgas med olika metoder, såsom Giemsa eller fluorescerande DNA-specifika färgämnen (59). Användningen av ett DNA-specifikt färgämne (t.ex. akridinorange (61) eller Hoechst 33258 plus pyronin-Y (62)) kan eliminera några av de artefakter som är associerade med färgämnen som inte är DNA-specifika. Anti-kinetochor-antikroppar, FISH-teknik med pancentromera DNA-prober eller primad in situ-märkning med pancentromerspecifik primer, tillsammans med lämplig DNA-motfärgning kan användas för att identifiera innehållet (kromosom/kromosomfragment) av mikrokärnor om man är intresserad av mekanismer för hur de bildas (15) (16). Andra metoder för differentiering mellan klastogener och aneugener kan användas om de har visat sig vara effektiva.
|
Analys
|
50.
|
Alla objektglas, inklusive glasen från lösningsmedels-/vehikelkontrollerna och de positiva och negativa kontrollerna, bör ges en oberoende kodning före analysen med mikroskop. Alternativt kan kodade prover analyseras med användning av ett validerat och automatiskt system för flödescytometri eller bildanalys.
|
|
51.
|
I cytoB-behandlade odlingar bör förekomsten av mikrokärnor analyseras i minst 2 000 binukleära celler per koncentration (minst 1 000 binukleära celler per odling, två odlingar per koncentration). Om enkla odlingar används bör minst 2 000 binukleära celler per koncentration analyseras ur odlingen. Om betydligt mindre antal än 1 000 binukleära celler per odling, eller 2 000 om en enkel odling används, finns att tillgå för analys för varje koncentration och en väsentlig ökning av mikrokärnor inte upptäcks, bör testet upprepas med användning av flera celler eller vid mindre toxiska koncentrationer, enligt det som är lämpligt. Man bör se upp med att inte analysera binukleära celler med oregelbunden form eller celler där de två kärnorna avviker betydligt i storlek, inte heller ska binukleära celler förväxlas med dåligt spridda multinukleära celler. Celler som innehåller fler än två huvudkärnor ska inte analyseras för mikrokärnor, eftersom bakgrundsförekomsten av mikrokärnor kan vara högre i dessa celler (63) (64). Analys av mononukleära celler kan godtas om testämnet påvisas störa cytoB-aktiviteten.
|
|
52.
|
För cellinjer som testas utan cytoB-behandling bör mikrokärnorna analyseras med minst 2 000 celler per koncentration (minst 1 000 celler per odling, två odlingar per koncentration). Om endast en odling per koncentration används bör minst 2 000 celler analyseras från odlingen.
|
|
53.
|
När cytoB används bör proliferationsindex vid cytokinesinhibering (CBPI) eller replikeringsindex (RI) bestämmas för bedömning av cellproliferationen (se tillägg 2) med användning av minst 500 celler per odling. När behandlingar görs utan tillsats av cytoB är det väsentligt att belägg ges för att de celler som analyseras har proliferat, enligt det som beskrivs i punkterna 24–27.
|
Kriterier för godkännande
|
54.
|
Ett laboratorium som vill använda MNvit-testning enligt beskrivningen i denna testmetod bör påvisa sin förmåga att på ett tillförlitligt och exakt sätt detektera kemikalier med känd aneugen och klastogen aktivitet, med och utan metabolisk aktivering, såväl som kända negativa kemikalier, med användning av referenskemikalierna enligt tillägg 3. För att påvisa sin förmåga att genomföra testmetoden korrekt bör laboratoriet tillhandahålla belägg för att de celler som analyseras för bildning av mikrokärnor har slutfört en kärndelning, om testningen genomförs utan användning av cytoB.
|
|
55.
|
De kemikalier som förtecknas i tillägg 3 rekommenderas som referenskemikalier. Ersättande eller ytterligare kemikalier kan ingå om deras aktivitet är känd och om de inducerar mikrokärnor enligt samma mekanismer och om de kan påvisas vara relevanta för de kemikalier som testas med MNvit-förfarandet. Motiveringen kan innehålla en valideringsstudie där man använt ett brett urval olika ämnen eller fokuserat på ett smalare spektrum baserat på testämnets kemiska klass eller mekanismen för den skada som undersöks.
|
|
56.
|
Lösningsmedels-/vehikelkontrollen och obehandlade odlingar bör ge reproducerbart låga och enhetliga förekomster av mikrokärnor (typiskt 5–25 mikrokärnor per 1 000 celler för de celltyper som anges i punkt 11). Andra celltyper kan ha annorlunda responsintervall, vilka bör bestämmas när celltyperna valideras för användning i ett MNvit-förfarande. Data från negativ kontroll, lösningsmedelskontroll och positiv kontroll bör användas för att etablera historiska kontrollintervall. Dessa värden bör användas för att bedöma den parallella negativa eller positiva kontrollens lämplighet för ett försök.
|
|
57.
|
Om det föreslås mindre ändringar av protokollet (t.ex. automatiska i stället för manuella analystekniker eller användning av en ny celltyp), bör ändringens effektivitet påvisas innan det modifierade protokollet kan anses vara godkänt för användning. När effektiviteten påvisas måste man också kunna visa att de centrala mekanismerna för kromosombrott och kromosomökning och -förlust kan detekteras och att lämpliga positiva och negativa resultat kan nås för klassen för det enskilda ämne eller för det breda urval av ämnen som ska testas.
|
DATA OCH RAPPORTERING
Behandling av resultaten
|
58.
|
Om blockering av cytokines används ska endast förekomsten av binukleära celler med mikrokärnor (oavsett antalet mikrokärnor per cell) användas för utvärderingen av inducering av mikrokärnor. Analys av antalet celler med en, två eller flera mikrokärnor kan ge användbar information men är inte obligatorisk.
|
|
59.
|
Parallella mått på cytotoxicitet och/eller cytostas för alla behandlade odlingar och kontrollodlingar med lösningsmedel/vehikel bör bestämmas (58). Proliferationsindex vid cytokinesinhibering (CBPI) eller replikeringsindex (RI) bör beräknas för alla behandlade odlingar och kontrollodlingar som mått på cellcykelfördröjning när blockering av cytokines används. Om cytoB inte tillsätts bör relativ populationsfördubbling (RPD) eller relativ ökning av celltal (RICC) eller proliferationsindex (PI) användas (se tillägg 2).
|
|
60.
|
Data bör anges per enskild odling. Dessutom bör alla data sammanfattas i tabellform.
|
|
61.
|
Att kemikalier inducerar mikrokärnor i MNvit-metoden kan bero på att de inducerar kromosombrott, kromosomförlust eller en kombination av dessa två. Ytterligare analys med användning av anti-kinetochor-antikroppar, centromerspecifika in situ-prober eller andra metoder kan göras för att avgöra huruvida mekanismen för inducering av mikrokärnor beror på klastogen och/eller aneugen aktivitet.
|
Utvärdering och tolkning av resultaten
|
62.
|
Det finns inget krav på verifiering genom ytterligare testning av ett tydligt positivt eller negativt svar. Tvetydiga resultat kan förtydligas genom analys av ytterligare 1 000 celler, som tas från alla odlingar för att bibehålla karaktären av blindförsök. Om detta inte klargör resultatet krävs ytterligare testning. Vid uppföljningstestning bör man överväga att modifiera undersökningsparametrarna så att man får en bredare eller snävare skala av förhållanden, enligt det som är lämpligt. Till de undersökningsparametrar som kanske måste ändras ingår koncentrationsintervall, tidpunkten för behandling och skörd av cellerna och/eller betingelserna för metabolisk aktivering.
|
|
63.
|
Det finns ett flertal kriterier för att fastställa ett positivt resultat, t.ex. en koncentrationsrelaterad ökning eller en statistiskt signifikant reproducerbar ökning av antalet celler som innehåller mikrokärnor. Man bör i första hand överväga resultatets biologiska relevans. Överväganden om huruvida de observerade värdena ligger inom eller utanför det historiska kontrollintervallet kan ge vägledning för utvärderingen av svarets biologiska betydelse. Lämpliga statistiska metoder kan vara till hjälp vid utvärdering av testresultaten (65). Resultaten från statistisk testning bör dock bedömas utifrån dos-responsförhållandet. Likaså bör reproducerbarhet och historiska data beaktas.
|
|
64.
|
Även om de flesta försök ger tydligt positiva eller negativa resultat, kan datauppsättningen i vissa fall inte ge möjlighet att göra en definitiv bedömning av testämnets aktivitet. Sådana tvetydiga eller tveksamma resultat kan uppstå oavsett hur många gånger försöket upprepas.
|
|
65.
|
Positiva resultat från MNvit-testning tyder på att testämnet inducerar kromosombrott eller kromosomförlust i odlade däggdjursceller. Negativa resultat indikerar att testämnet under de gällande försöksbetingelserna inte inducerar kromosombrott och/eller kromosomökning eller -förlust i de odlade däggdjurscellerna.
|
Testrapport
|
66.
|
Testrapporten ska innehålla minst följande uppgifter i den mån de är relevanta:
|
|
Testkemikalie
|
—
|
Identifieringsdata och CAS-nummer och EG-nummer.
|
|
—
|
Fysikalisk form och renhetsgrad.
|
|
—
|
Fysikalisk-kemiska egenskaper av betydelse för studien.
|
|
—
|
Testkemikaliens reaktionsbenägenhet med lösningsmedel/vehikel eller cellodlingsmedier.
|
|
|
|
Lösningsmedel/vehikel
|
—
|
Motivering för valet av lösningsmedel/vehikel.
|
|
—
|
Testämnets stabilitet och löslighet i lösningsmedlet/vehikeln.
|
|
|
|
Celler
|
—
|
Cellernas typ och källa.
|
|
—
|
Den använda celltypens lämplighet.
|
|
—
|
Frånvaro av mykoplasma, om detta är tillämpligt.
|
|
—
|
Uppgifter om cellcykelns längd, fördubblingstid eller proliferationsindex.
|
|
—
|
Om lymfocyter används, blodgivarnas kön, ålder och antal, om tillämpligt.
|
|
—
|
Om lymfocyter används, uppgift om huruvida helblod eller separata lymfocyter exponeras.
|
|
—
|
Antal passager, om detta är tillämpligt.
|
|
—
|
Metoder för underhållet av cellodlingar, om detta är tillämpligt.
|
|
—
|
Det modala antalet kromosomer.
|
|
—
|
Normal cellcykellängd (negativ kontroll).
|
|
|
|
Testbetingelser
|
—
|
Identitet för cytokinesinhiberande ämne (t.ex. cytoB), om sådant används, och ämnets koncentration samt cellexponeringens varaktighet
|
|
—
|
Motivering för val av koncentrationer och antalet odlingar, även data om cytotoxicitet och löslighetsbegränsningar i den mån dessa data finns att tillgå.
|
|
—
|
Mediesammansättning, CO2-koncentration, om tillämpligt.
|
|
—
|
Testämneskoncentrationer.
|
|
—
|
Koncentration (och/eller volym) för vehikel och tillsatt testämne.
|
|
—
|
Inkubationstemperatur och tid.
|
|
—
|
Behandlingens varaktighet.
|
|
—
|
Tidpunkten för skörd efter behandlingen.
|
|
—
|
Celldensitet vid inokulering, om tillämpligt.
|
|
—
|
Typ och sammansättning hos det system som används för metabolisk aktivering, även kriterier för godkännande.
|
|
—
|
Positiva kontrollkemikalier och negativa kontroller.
|
|
—
|
Metoder för preparering av objektglas och färgningsteknik.
|
|
—
|
Kriterier för identifiering av mikrokärnor.
|
|
—
|
Antalet analyserade celler.
|
|
—
|
Metoder för mätning av cytotoxicitet.
|
|
—
|
All tilläggsinformation som är relevant rörande cytotoxicitet.
|
|
—
|
Kriterier för att bedöma om testresultaten är positiva, negativa eller tvetydiga.
|
|
—
|
Metod(er) för statistisk analys.
|
|
—
|
Metoder, såsom användning av kinetochor-antikropp, för att fastställa om mikrokärnorna innehåller hela eller fragmenterade mikrokärnor, om tillämpligt.
|
|
|
|
Resultat
|
—
|
Mätningar av cytotoxicitet, t.ex. proliferationsindex (CBPI) eller replikeringsindex (RI) när cytokinesinhibering används, relativ ökning av celltal (RICC), relativ populationsfördubbling (RPD) eller proliferationsindex (PI) när cytokinesinhibering inte används, andra observationer om tillämpligt, t.ex. cellsammanväxning, apoptos, nekros, antalet metafaser, frekvensen binukleära celler.
|
|
—
|
Data om behandlingsmediets pH och osmolalitet, om uppmätt.
|
|
—
|
Definition av godkända celler för analys.
|
|
—
|
Fördelningen av mono-, bi- och multinukleära celler i fall där blockering av cytokines har använts.
|
|
—
|
Antalet celler med mikrokärnor angivet separat för varje behandlad odling och kontrollodling och, där det är lämpligt, angivelse av huruvida det gäller binukleära eller mononukleära celler.
|
|
—
|
Koncentration-responsförhållande, där det är möjligt.
|
|
—
|
Kemiska data om parallella negativa (lösningsmedel/vehikel) och positiva kontroller (koncentrationer och lösningsmedel).
|
|
—
|
Historiska data om negativa (lösningsmedel/vehikel) och positiva kontroller, med intervall, medelvärden och standardavvikelser samt tillförlitlighetsintervall (t.ex. 95 %).
|
|
—
|
Statistisk analys, p-värden om sådana finns.
|
|
|
HÄNVISNINGAR
|
(1)
|
Kirsch-Volders, M. (1997), Towards a validation of the micronucleus test. Mutation Res., 392, 1–4.
|
|
(2)
|
Parry, J.M. och Sors, A. (1993), The detection and assessment of the aneugenic potential of environmental chemicals: the European Community aneuploidy project, Mutation Res., 287, 3–15.
|
|
(3)
|
Fenech, M. och Morley, A.A. (1985), Solutions to the kinetic problem in the micronucleus assay, Cytobios., 43, 233–246.
|
|
(4)
|
Kirsch-Volders, M., Sofuni, T., Aardema, M., Albertini, S., Eastmond, D., Fenech, M., Ishidate, M. Jr, Lorge, E., Norppa, H., Surralles, J., von der Hude, W. och Wakata, A. (2000), Report from the In Vitro Micronucleus Assay Working Group, Environ. Mol. Mutagen., 35, 167–172.
|
|
(5)
|
Fenech, M. (2007), Cytokinesis-block micronucleus cytome assay, Nature Protocols, 2(5), 1084–1104.
|
|
(6)
|
Fenech, M. ochd Morley, A.A. (1986), Cytokinesis-block micronucleus method in human lymphocytes: effect of in-vivo ageing and low dose X-irradiation, Mutation Res., 161, 193–198.
|
|
(7)
|
Eastmond, D.A. och Tucker, J.D. (1989), Identification of aneuploidy-inducing agents using cytokinesis-blocked human lymphocytes and an antikinetochore antibody, Environ. Mol. Mutagen., 13, 34–43.
|
|
(8)
|
Eastmond, D.A. and Pinkel, D. (1990), Detection of aneuploidy and aneuploidy-inducing agents in human lymphocytes using fluorescence in-situ hybridisation with chromosome-specific DNA probes, Mutation Res., 234, 9–20.
|
|
(9)
|
Miller, B.M., Zitzelsberger, H.F., Weier, H.U. och Adler, I.D. (1991), Classification of micronuclei in murine erythrocytes: immunofluorescent staining using CREST antibodies compared to in situ hybridization with biotinylated gamma satellite DNA, Mutagenesis, 6, 297–302.
|
|
(10)
|
Farooqi, Z., Darroudi, F. och Natarajan, A.T. (1993), The use of fluorescence in-situ hybridisation for the detection of aneugens in cytokinesis-blocked mouse splenocytes, Mutagenesis, 8, 329–334.
|
|
(11)
|
Migliore, L., Bocciardi, R., Macri, C. och Lo Jacono, F. (1993), Cytogenetic damage induced in human lymphocytes by four vanadium compounds and micronucleus analysis by fluorescence in situ hybridization with a centromeric probe, Mutation Res., 319, 205–213.
|
|
(12)
|
Norppa, H., Renzi, L. och Lindholm, C. (1993), Detection of whole chromosomes in micronuclei of cytokinesis-blocked human lymphocytes by antikinetochore staining and in situ hybridization, Mutagenesis, 8, 519–525.
|
|
(13)
|
Eastmond, D.A, Rupa, D.S. och Hasegawa, L.S. (1994), Detection of hyperdiploidy and chromosome breakage in interphase human lymphocytes following exposure to the benzene metabolite hydroquinone using multicolor fluorescence in situ hybridization with DNA probes, Mutation Res., 322, 9–20.
|
|
(14)
|
Marshall, R.R., Murphy, M., Kirkland, D.J. och Bentley, K.S. (1996), Fluorescence in situ hybridisation (FISH) with chromosome-specific centromeric probes: a sensitive method to detect aneuploidy, Mutation Res., 372, 233–245.
|
|
(15)
|
Zijno, P., Leopardi, F., Marcon, R. och Crebelli, R. (1996), Analysis of chromosome segregation by means of fluorescence in situ hybridization: application to cytokinesis-blocked human lymphocytes, Mutation Res., 372, 211–219.
|
|
(16)
|
Kirsch-Volders, M., Sofuni, T., Aardema, M., Albertini, S., Eastmond, D., Fenech, M., Ishidate Jr., M., Lorge, E., Norppa, H., Surrallés, J., von der Hude, W. och Wakata, A. (2003), Report from the in vitro micronucleus assay working group. Mutation Res., 540, 153–163.
|
|
(17)
|
OECD (1997), In Vitro Mammalian Chromosome Aberration Test, Test Guideline No. 473, OECD Guidelines for Testing of Chemicals, OECD, Paris. Finns på: www.oecd.org/env/testguidelines
|
|
(18)
|
Lorge, E., Thybaud, V., Aardema, M.J., Oliver, J., Wakata, A., Lorenzon G. och Marzin, D. (2006), SFTG International collaborative Study on in vitro micronucleus test. I. General conditions and overall conclusions of the study, Mutation Res., 607, 13–36.
|
|
(19)
|
Clare, G., Lorenzon, G., Akhurst, L.C., Marzin, D., van Delft, J., Montero, R., Botta, A., Bertens, A., Cinelli, S., Thybaud, V. och Lorge, E. (2006), SFTG International collaborative study on the in vitro micronucleus test. II. Using human lymphocytes, Mutation Res., 607, 37–60.
|
|
(20)
|
Aardema, M.J., Snyder, R.D., Spicer, C., Divi, K., Morita, T., Mauthe, R.J., Gibson, D.P., Soelter, S., Curry, P.T., Thybaud, V., Lorenzon, G., Marzin, D. och Lorge, E. (2006), SFTG International collaborative study on the in vitro micronucleus test, III. Using CHO cells, Mutation Res., 607, 61–87.
|
|
(21)
|
Wakata, A., Matsuoka, A., Yamakage, K., Yoshida, J., Kubo, K., Kobayashi, K., Senjyu, N., Itoh, S., Miyajima, H., Hamada, S., Nishida, S., Araki, H., Yamamura, E., Matsui, A., Thybaud, V., Lorenzon, G., Marzin, D. och Lorge, E. (2006), SFTG International collaborative study on the in vitro micronucleus test, IV. Using CHO/IU cells, Mutation Res., 607, 88–124.
|
|
(22)
|
Oliver, J., Meunier, J.-R., Awogi, T., Elhajouji, A., Ouldelhkim, M.-C., Bichet, N., Thybaud, V., Lorenzon, G., Marzin, D. och Lorge, E. (2006), SFTG International collaborative study on the in vitro micronucleus test, V. Using L5178Y cells, Mutation Res., 607, 125–152.
|
|
(23)
|
Albertini, S., Miller, B., Chetelat, A.A. and Locher, F. (1997), Detailed data on in vitro MNT och in vitro CA: industrial experience, Mutation Res., 392, 187–208.
|
|
(24)
|
Miller, B., Albertini, S., Locher, F., Thybaud, V. och Lorge, E. (1997), Comparative evaluation of the in vitro micronucleus test and the in vitro chromosome aberration test: industrial experience, Mutation Res., 392, 45–59.
|
|
(25)
|
Miller, B., Potter-Locher, F., Seelbach, A., Stopper, H., Utesch, D. och Madle, S. (1998), Evaluation of the in vitro micronucleus test as an alternative to the in vitro chromosomal aberration assay: position of the GUM Working Group on the in vitro micronucleus test. Gesellschaft fur Umwelt-Mutations-forschung, Mutation Res., 410, 81–116.
|
|
(26)
|
Kalweit, S., Utesch, U., von der Hude, W. och Madle, S. (1999), Chemically induced micronucleus formation in V79 cells – comparison of three different test approaches, Mutation Res. 439, 183–190.
|
|
(27)
|
Kersten, B., Zhang, J., Brendler Schwaab, S.Y., Kasper, P. och Müller, L. (1999), The application of the micronucleus test in Chinese hamster V79 cells to detect drug-induced photogenotoxicity, Mutation Res. 445, 55–71.
|
|
(28)
|
von der Hude, W., Kalweit, S., Engelhardt, G., McKiernan, S., Kasper, P., Slacik-Erben, R., Miltenburger, H.G., Honarvar, N., Fahrig, R., Gorlitz, B., Albertini, S., Kirchner, S., Utesch, D., Potter-Locher, F., Stopper, H. och Madle, S. (2000), In vitro micronucleus assay with Chinese hamster V79 cells - results of a collaborative study with in situ exposure to 26 chemical substances, Mutation Res., 468, 137–163.
|
|
(29)
|
Garriott, M.L., Phelps, J.B. och Hoffman, W.P. (2002), A protocol for the in vitro micronucleus test, I. Contributions to the development of a protocol suitable for regulatory submissions from an examination of 16 chemicals with different mechanisms of action and different levels of activity, Mutation Res., 517, 123–134.
|
|
(30)
|
Matsushima, T., Hayashi, M., Matsuoka, A., Ishidate, M. Jr., Miura, K.F., Shimizu, H., Suzuki, Y., Morimoto, K., Ogura, H., Mure, K., Koshi, K. och Sofuni, T. (1999), Validation study of the in vitro micronucleus test in a Chinese hamster lung cell line (CHL/IU), Mutagenesis, 14, 569–580.
|
|
(31)
|
Elhajouji, A., och Lorge, E. (2006), Special Issue: SFTG International collaborative study on in vitro micronucleus test, Mutation Res., 607, 1–152.
|
|
(32)
|
ECVAM (2006), Statement by the European Centre for the Validation of Alternative Methods (ECVAM) Scientific Advisory Committee (ESAC) on the scientific validity of the in vitro micronucleus test as an alternative to the in vitro chromosome aberration assay for genotoxicity testing. ESAC 25th meeting, 16–17 November, 2006, Finns på: http://ecvam.jrc.it/index.htm
|
|
(33)
|
ESAC (2006), ECVAM Scientific Advisory Committee (ESAC) Peer Review, Retrospective Validation of the In Vitro Micronucleus Test, Summary and Conclusions of the Peer Review Panel, Finns på: http://ecvam.jrc.it/index.htm
|
|
(34)
|
Corvi, R., Albertini, S., Hartung, T., Hoffmann, S., Maurici, D., Pfuhler, S, van Benthem, J., Vanparys P. (2008), ECVAM Retrospective Validation of in vitro Micronucleus Test (MNT), Mutagenesis, 23, 271–283.
|
|
(35)
|
Zhang, L.S., Honma, M., Hayashi, M., Suzuki, T., Matsuoka, A. och Sofuni, T. (1995), A comparative study of TK6 human lymphoblastoid and L5178Y mouse lymphoma cell lines in the in vitro micronucleus test, Mutation Res., 347, 105–115.
|
|
(36)
|
Ehrlich, V., Darroudi, F., Uhl, M., Steinkellner, S., Zsivkovits, M. och Knasmeuller, S. (2002), Fumonisin B1 is genotoxic in human derived hepatoma (HepG2) cells, Mutagenesis, 17, 257–260.
|
|
(37)
|
Knasmüller, S., Mersch-Sundermann, V., Kevekordes, S., Darroudi, F., Huber, W.W., Hoelzl, C., Bichler, J. och Majer, B.J. (2004), Use of human-derived liver cell lines for the detection of environmental and dietary genotoxicants; current state of knowledge, Toxicol., 198, 315–328.
|
|
(38)
|
Gibson, D.P., Brauninger, R., Shaffi, H.S., Kerckaert, G.A., LeBoeuf, R.A., Isfort, R.J. och Aardema, M.J. (1997), Induction of micronuclei in Syrian hamster embryo cells: comparison to results in the SHE cell transformation assay for National Toxicology Program test chemicals, Mutation Res., 392, 61–70.
|
|
(39)
|
Scott, D., Galloway, S.M., Marshall, R.R., Ishidate, M. Jr., Brusick, D., Ashby, J. och Myhr, B.C. (1991), International Commission for Protection Against Environmental Mutagens and Carcinogens, Genotoxicity under extreme culture conditions. A report from ICPEMC Task Group 9, Mutation Res., 257, 147–205.
|
|
(40)
|
Morita, T., Nagaki, T., Fukuda, I. och Okumura, K. (1992), Clastogenicity of low pH to various cultured mammalian cells, Mutation Res., 268, 297–305.
|
|
(41)
|
Brusick, D. (1986), Genotoxic effects in cultured mammalian cells produced by low pH treatment conditions and increased ion concentrations, Environ. Mutagen., 8, 789–886.
|
|
(42)
|
Fenech, M. och Morley, A.A. (1985), Measurement of micronuclei in lymphocytes, Mutation Res., 147, 29–36.
|
|
(43)
|
Fenech, M. (1997), The advantages and disadvantages of cytokinesis-blood micronucleus method, Mutation Res., 392, 11–18.
|
|
(44)
|
Bonassi, S., Fenech, M., Lando, C., Lin, Y.P., Ceppi, M., Chang, W.P., Holland, N., Kirsch-Volders, M., Zeiger, E., Ban, S., Barale, R., Bigatti, M.P., Bolognesi, C., Jia, C., Di Giorgio, M., Ferguson, L.R., Fucic, A., Lima, O.G., Hrelia, P., Krishnaja, A.P., Lee, T.K., Migliore, L., Mikhalevich, L., Mirkova, E., Mosesso, P., Muller, W.U., Odagiri, Y., Scarffi, M.R., Szabova, E., Vorobtsova, I., Vral, A. och Zijno, A. (2001), HUman MicroNucleus Project: international database comparison for results with the cytokinesis-block micronucleus assay in human lymphocytes, I. Effect of laboratory protocol, scoring criteria and host factors on the frequency of micronuclei, Environ. Mol. Mutagen. 37, 31–45.
|
|
(45)
|
Maron, D.M. och Ames, B.N. (1983), Revised methods for the Salmonella mutagenicity test, Mutation Res., 113, 173–215.
|
|
(46)
|
Ong, T.-m., Mukhtar, M., Wolf, C.R. och Zeiger, E. (1980), Differential effects of cytochrome P450-inducers on promutagen activation capabilities and enzymatic activities of S-9 from rat liver, J. Environ. Pathol. Toxicol., 4, 55–65.
|
|
(47)
|
Elliott, B.M., Combes, R.D., Elcombe, C.R., Gatehouse, D.G., Gibson, G.G., Mackay, J.M. och Wolf, R.C. (1992), Alternatives to Aroclor 1254-induced S9 in in-vitro genotoxicity assays. Mutagenesis, 7, 175–177.
|
|
(48)
|
Matsushima, T., Sawamura, M., Hara, K. och Sugimura, T. (1976), A safe substitute for Polychlorinated Biphenyls as an Inducer of Metabolic Activation Systems, In: de Serres, F.J., Fouts, J. R., Bend, J.R. and Philpot, R.M. (eds), In Vitro Metabolic Activation in Mutagenesis Testing, Elsevier, North-Holland, pp. 85–88.
|
|
(49)
|
Johnson, T.E., Umbenhauer, D.R. och Galloway, S.M. (1996), Human liver S-9 metabolic activation: proficiency in cytogenetic assays and comparison with phenobarbital/beta-naphthoflavone or Aroclor 1254 induced rat S-9, Environ. Mol. Mutagen., 28, 51–59.
|
|
(50)
|
Unep (2001), Stockholm Convention on Persistent Organic Pollutants, United Nations Environment Programme (Unep). Finns på: http://www.pops.int/
|
|
(51)
|
Doherty, A.T., Ellard, S., Parry, E.M. och Parry, J.M. (1996), An investigation into the activation and deactivation of chlorinated hydrocarbons to genotoxins in metabolically competent human cells, Mutagenesis, 11, 247–274.
|
|
(52)
|
Krahn, D.F., Barsky, F.C. och McCooey, K.T. (1982), CHO/HGPRT Mutation Assay: Evaluation of Gases and Volatile Liquids, In: Tice, R.R., Costa, D.L. and Schaich, K.M. (eds), Genotoxic Effects of Airborne Agents. New York, Plenum, sidorna 91–103.
|
|
(53)
|
Zamora, P.O., Benson, J.M., Li, A.P. och Brooks, A.L. (1983), Evaluation of an exposure system using cells grown on collagen gels for detecting highly volatile mutagens in the CHO/HGPRT mutation assay, Environ. Mutagenesis 5, 795–801.
|
|
(54)
|
Fenech, M. (1993), The cytokinesis-block micronucleus technique: a detailed description of the method and its application to genotoxicity studies in human populations, Mutation Res., 285, 35–44.
|
|
(55)
|
Phelps, J.B., Garriott, M.L., och Hoffman, W.P. (2002), A protocol for the in vitro micronucleus test. II. Contributions to the validation of a protocol suitable for regulatory submissions from an examination of 10 chemicals with different mechanisms of action and different levels of activity, Mutation Res., 521, 103–112.
|
|
(56)
|
Kirsch-Volders, M., Sofuni, T., Aardema, M., Albertini, S., Eastmond, D., Fenech, M., Ishidate, M. Jr., Kirchner, S., Lorge, E., Morita, T., Norppa, H., Surralles, J., Vanhauwaert, A. och Wakata, A. (2004), Corrigendum to ’Report from the in vitro micronucleus assay working group’, Mutation Res., 564, 97–100.
|
|
(57)
|
Pincu, M., Bass, D. och Norman, A. (1984), An improved micronuclear assay in lymphocytes, Mutation Res., 139, 61–65.
|
|
(58)
|
Lorge, E., Hayashi, M., Albertini, S. och Kirkland, D. (2008), Comparison of different methods for an accurate assessment of cytotoxicity in the in vitro micronucleus test. I. Theoretical aspects, Mutation Res., 655, 1–3.
|
|
(59)
|
Surralles, J., Xamena, N., Creus, A., Catalan, J., Norppa, H. och Marcos, R. (1995), Induction of micronuclei by five pyrethroid insecticides in whole-blood and isolated human lymphocyte cultures, Mutation Res., 341, 169–184.
|
|
(60)
|
Galloway, S. (2000), Cytotoxicity and chromosome aberrations in vitro: Experience in industry and the case for an upper limit on toxicity in the aberration assay, Environ. Molec. Mutagenesis 35, 191–201.
|
|
(61)
|
Hayashi, M., Sofuni, T. och Ishidate, M. Jr. (1983), An Application of Acridine Orange Fluorescent Staining to the Micronucleus Test, Mutation Res., 120, 241–247.
|
|
(62)
|
MacGregor, J. T., Wehr, C. M. och Langlois, R. G. (1983), A Simple Fluorescent Staining Procedure for Micronuclei and RNA in Erythrocytes Using Hoechst 33258 and Pyronin Y, Mutation Res., 120, 269–275.
|
|
(63)
|
Hayashi, M., Sofuni, T. och Ishidate, M. Jr. (1983), An application of acridine orange fluorescent staining to the micronucleus test, Mutation Res., 120, 241–247.
|
|
(64)
|
Fenech, M., Chang, W.P., Kirsch-Volders, M., Holland, N., Bonassi, S. och Zeiger, E. (2003), HUMN project: detailed description of the scoring criteria for the cytokinesis-block micronucleus assay using isolated human lymphocyte cultures, Mutation Res., 534, 65–75.
|
|
(65)
|
Hoffman, W.P., Garriott, M.L. och Lee, C. (2003), In vitro micronucleus test, In: Encyclopedia of Biopharmaceutical Statistics, Second edition. S. Chow (ed.), Marcel Dekker, Inc. New York, NY, sidorna 463–467.
|
|
(66)
|
Europaparlamentets och rådets förordning (EG) nr 850/2004 av den 29 april 2004 om långlivade organiska föroreningar och om ändring av direktiv 79/117/EEG (EUT L 158, 30.4.2004, s. 7).
|
Tillägg 1
Definitioner
Aneugen: Ett ämne eller en process som genom växelverkan med komponenterna i den mitotiska och meiotiska celldelningscykeln leder till aneuploidi i celler eller organismer.
Aneuploidi: Alla avvikelser från det normala antalet diploida (eller haploida) kromosomer med en eller flera kromosomer, men inte med hela uppsättningar kromosomer (polyploidi).
Apoptos: Programmerad celldöd som karakteriseras av en serie steg som leder till sönderfall av celler till membranbundna partiklar som därefter elimineras genom fagocytos eller avstötning.
Cellproliferation: Ökning av antalet celler som resultat av mitotisk celldelning.
Centromer: DNA-regionen i en kromosom där båda kromatiderna är sammanhållna och på vilken båda kinetochorerna är fästa sida vid sida.
Klastogen: Alla ämnen eller processer som orsakar strukturella kromosomavvikelser i populationer av celler eller organismer.
Cytokines: Den celldelning som följer omedelbart efter mitosen vid vilken två dotterceller bildas med var sin enskild kärna.
Proliferationsindex vid cytokinesinhibering (CBPI): Förhållandet mellan antalet celler efter två delningar i den behandlade populationen och motsvarande antal i den obehandlade kontrollen (se tillägg 2 för formel).
Cytostas: Inhibering av celltillväxt (se tillägg 2 för formel).
Cytotoxicitet: Skadliga effekter på cellens struktur eller funktion som i slutändan leder till celldöd.
Genotoxisk: En allmän term som omfattar alla typer av DNA- eller kromosomskador, inklusive brott, adduktomfördelning, mutationer, kromosomavvikelser och aneuploidi. Inte alla typer av genotoxiska effekter leder till mutationer eller stabila kromosomskador.
Interfasceller: Celler som inte är i mitotisk fas.
Kinetochor: Ett protein som innehåller en struktur som fäster sig vid en kromosomcentromer till vilken spindelfibrer associerar under celldelning och ger upphov till en ordnad rörelse av dotterkromosomerna mot dottercellernas poler.
Mikrokärnor: Små extra kärnor som är separata från cellernas huvudkärna och som produceras under mitosens eller meiosens telofas från isolerade kromosomfragment eller hela kromosomer.
Mitos: Delning av cellkärnan, i regel bestående av profas, prometafas, metafas, anafas och telofas.
Mitotiskt index: Den andel celler som är i metafas dividerat med det totala antalet celler som kan observeras i en cellpopulation; en indikation på graden av cellproliferation hos den populationen.
Mutagent ämne: Ämne som producerar en ärftlig ändring av DNA-basparssekvensen (-sekvenserna) i gener eller i kromosomstrukturen (kromosomavvikelser).
Icke-disjunktion: När kromatidpar inte separeras och segregeras korrekt till dottercellerna under utveckling, vilket leder till dotterceller med abnormalt antal kromosomer.
Polyploidi: Numeriska kromosomavvikelser i celler eller organismer som inbegriper en eller flera hela kromosomuppsättningar, i motsats till en enskild kromosom eller kromosomer (aneuploidi).
Proliferationsindex (PI): Ett sätt att mäta cytotoxicitet när cytoB inte används (se tillägg 2 för formel).
Relativ ökning av celltal (RICC): Ett sätt att mäta cytotoxicitet när cytoB inte används (se tillägg 2 för formel).
Relativ populationsfördubbling (RPD): Ett sätt att mäta cytotoxicitet när cytoB inte används (se tillägg 2 för formel).
Replikeringsindex (RI): Andelen celldelningscykler som slutförts i en behandlad odling under exponeringsperioden och återhämtningen, i förhållande till den obehandlade kontrollen (se tillägg 2 för formel).
Testkemikalie (kallas också testämne): Alla ämnen eller blandningar som testas med denna testmetod.
Tillägg 2
Formler för bestämning av cytotoxicitet
|
1.
|
När cytoB används ska bedömningen av cytotoxicitet basera sig på proliferationsindex vid cytokinesinhibering (CBPI) eller replikeringsindex (RI) (16) (58). Proliferationsindex vid cytokinesinhibering indikerar det genomsnittliga antalet cellcykler per cell under exponeringsperioden för cytoB och kan användas för att beräkna cellproliferationen. Replikeringsindex indikerar det relativa antalet kärnor i behandlade odlingar i jämförelse med kontrollodlingar och kan användas för att beräkna procentandelen cytostas:
% cytostas = 100 – 100{(CBPIT – 1) ÷ (CBPIC – 1)}
och
|
T
|
=
|
odling behandlad med testkemikalie
|
|
C
|
=
|
vehikelkontrollodling
|
där
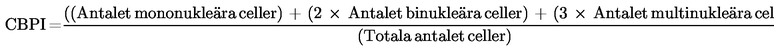
Således gäller att ett CBPI-värde på 1 (alla celler är mononukleära) motsvarar 100 % cytostas.
Cytostas = 100 – RI
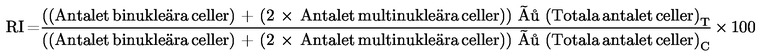
|
T
|
=
|
behandlade odlingar
|
|
C
|
=
|
kontrollodlingar
|
|
|
2.
|
Således betyder ett RI-värde på 53 % att, jämfört med antalet celler som har delat sig till binukleära och multinukleära celler i kontrollodlingen, endast 53 % av detta antal har delat sig i den behandlade odlingen, dvs. 47 % cytostas.
|
|
3.
|
När cytoB inte används, rekommenderas att cytotoxiciteten utvärderas på grundval av relativ ökning av celltal (RICC) eller relativ populationsfördubbling (RPD) (58), eftersom den andel av cellpopulationen som har delat sig beaktas i båda metoderna.
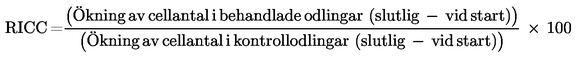
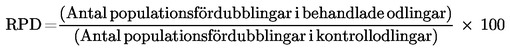
där
Populationsfördubbling = [log (Cellantal efter behandling ÷ Ursprungligt cellantal)] ÷ log 2
|
|
4.
|
Således indikerar ett RICC-värde eller ett RPD-värde på 53 % att det förekommer 47 % cytotoxicitet/cytostas.
|
|
5.
|
Genom att använda ett proliferationsindex (PI) kan cytotoxiciteten bestämmas genom att räkna antalet kloner som består av 1 cell (cl1), 2 celler (cl2), 3 till 4 celler (cl4) och 5 till 8 celler (cl8)
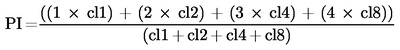
|
|
6.
|
PI har använts som en värdefull och tillförlitlig cytotoxicitetsparameter även för cellinjer som har odlats in situ utan tillsats av cytoB (25) (26) (27) (28).
|
Tillägg 3
Referenskemikalier som rekommenderas för bedömning av prestanda
(16)
|
Kategori
|
Kemikalie
|
CAS-nummer
|
EG-nummer
|
| 1. Klastogener utan metabolisk aktivering
|
|
|
Cytosinarabinosid
|
147-94-4
|
205-705-9
|
|
|
Mitomycin C
|
50-07-7
|
200-008-6
|
| 2. Klastogener som kräver metabolisk aktivering
|
|
|
Benso(a)pyren
|
50-32-8
|
200-028-5
|
|
|
Cyklofosfamid
|
50-18-0
|
200-015-4
|
| 3. Aneugener
|
|
|
Kolchicin
|
64-86-8
|
200-598-5
|
|
|
Vinblastin
|
143-67-9
|
205-606-0
|
| 4. Negativa ämnen
|
|
|
Di(2-etylhexyl)ftalat
|
117-81-7
|
204-211-0
|
|
|
Nalidixinsyra
|
389-08-2
|
206-864-7
|
|
|
Pyren
|
129-00-0
|
204-927-3
|
|
|
Natriumklorid
|
7647-14-5
|
231-598-3
|
B.50. HUDSENSIBILISERING: LLNA-METODEN (LOCAL LYMPH NODE ASSAY): DA
INLEDNING
|
1.
|
OECD:s riktlinjer för testning av kemikalier (OECD Guidelines for the Testing of Chemicals) och EU:s testmetoder ses regelbundet över med hänsyn till den vetenskapliga utvecklingen, nya lagstiftningsbehov och djurens välbefinnande. Den första testmetoden (B.42) för bestämning av hudsensibilisering hos möss (LLNA-metoden, OECD:s testriktlinje 429) har reviderats (1). Närmare uppgifter om valideringen av LLNA-metoden och en granskning av arbetet i samband med denna har offentliggjorts (2)(3)(4)(5)(6)(7)(8)(9). I LLNA-metoden används radioisotopiskt tymidin eller jod för att mäta lymfocytproliferation och metoden har därför begränsad användning när det förekommer problem med att anskaffa, använda eller bortskaffa radioaktivitet. LLNA: DA (som utvecklats av Daicel Chemical Industries, Ltd) är en icke-radioaktiv modifiering av LLNA enligt vilken mängden adenosintrifosfat (ATP) kvantifieras genom bioluminiscens som en indikator på lymfocytproliferation. LLNA: DA-testmetoden har validerats, granskats och rekommenderats av en internationell expertutvärderingspanel som konstaterade att den kan användas för att identifiera hudsensibiliserande och icke-hudsensibiliserande kemikalier, med vissa begränsningar (10)(11)(12)(13). Testmetoden är avsedd för bedömning av kemikaliers (ämnens och blandningars) potential att framkalla hudsensibilisering hos djur. Enligt kapitel B.6 i denna bilaga och i OECD:s testriktlinje 406 används test på marsvin, särskilt maximeringstest på marsvin och Buehler-test (14). LLNA (kapitel B.42 i denna bilaga, OECD:s testriktlinje 429) och de två icke-radioaktiva modifikationerna LLNA: DA (kapitel B.50 i denna bilaga, OECD:s testriktlinje 442 A) och LLNA: BrdU-ELISA (kapitel B.51 i denna bilaga, OECD:s testriktlinje 442 B) har fördelar jämfört med marsvinstestet enligt B.6 och OECD:s testriktlinje 406 (14) såtillvida att man kan minska antalet djur och använda dem på ett skonsammare sätt.
|
|
2.
|
I likhet med LLNA används LLNA: DA för att undersöka hudsensibiliseringens induktionsfas, vilket ger tillgång till kvantitativa data som är lämpliga för dos-responsbedömning. Vidare gäller att de här metodernas förmåga att detektera hudsensibiliserande kemikalier utan att det behövs radiomärkning för DNA eliminerar risken för yrkesmässig exponering för radioaktivitet och problem rörande bortskaffning av avfall. Detta kan i sin tur ge möjlighet till ökad användning av möss för att upptäcka hudsensibiliserande ämnen, vilket ytterligare kan minska användningen av marsvin för att testa kemikaliers hudsensibiliserande potential (dvs. B.6; OECD:s testriktlinje 406) (14).
|
DEFINITIONER
|
3.
|
Definitioner av begreppen finns i tillägg 1.
|
INLEDANDE ÖVERVÄGANDEN OCH BEGRÄNSNINGAR
|
4.
|
LLNA: DA är en modifierad LLNA-metod för identifiering av kemikalier med potential att framkalla hudsensibilisering, med specifika begränsningar. Detta betyder dock inte nödvändigtvis att LLNA: DA alltid ska användas i stället för LLNA eller test på marsvin (dvs. B.6; OECD:s testriktlinje 406) (14), utan att metoden är likvärdig och kan användas som ett alternativ i fall där positiva och negativa resultat i regel inte kräver någon ytterligare bekräftelse (10) (11). Testningslaboratoriet bör analysera all tillgänglig information om testämnet innan själva testningen genomförs. Sådan information inbegriper ämnets identitet och kemiska struktur, fysikalisk-kemiska egenskaper, resultaten från andra eventuella in vitro- eller in vivo-test avseende testämnets toxicitet samt toxikologiska data om strukturellt närbesläktade kemikalier. Denna information bör övervägas för att kunna fastställa om LLNA-metoden är lämplig för testämnet (mot bakgrund av att LLNA-metoden inte kan användas för vissa typer av kemikalier, se punkt 5) och som stöd för valet av dos.
|
|
5.
|
LLNA: DA är en in vivo-metod, och därför kan man inte undvika användningen av djur för bedömning av sensibilisering genom allergisk kontakt. Detta test erbjuder dock möjligheter till att minska antalet djur i jämförelse med test på marsvin (B.6; OECD:s testriktlinje 406) (14). Dessutom ger LLNA: DA möjlighet till en betydligt skonsammare behandling (mindre smärta och lidande) av de djur som används för testning av sensibilisering genom allergisk kontakt, eftersom man med LLNA: DA, till skillnad från B.6 och OECD:s testriktlinje 406 inte behöver framkalla provokationsinducerade dermala hypersensitivitetsreaktioner. Trots fördelarna med LLNA: DA i jämförelse med B.6 och OECD:s testriktlinje 406 (14), finns det vissa begränsningar som gör att man kanske måste använda B.6 eller OECD:s testriktlinje 406 (t.ex. testning av vissa metaller, falska positiva resultat med vissa hudirriterande ämnen (såsom vissa ytaktiva ämnen (6) (1 och kapitel B.42 i denna bilaga), testämnets löslighet)). Dessutom kan det för kemiska klasser eller ämnen som har funktionella grupper som påvisats agera som potentiella störfaktorer (16) vara nödvändigt att använda test på marsvin (dvs. B.6; OECD:s testriktlinje 406 (14)). Det rekommenderas att de begränsningar som anges för LLNA (1 och kapitel B.42 i denna bilaga) även ska gälla för LLNA: DA (10). Dessutom är LLNA: DA eventuellt inte lämplig för testning av ämnen som påverkar ATP-nivåerna (t.ex. ämnen som fungerar som ATP-inhibitorer) eller ämnen som stör exakt mätning av intracellulärt ATP (t.ex. förekomsten av ATP-degraderande enzymer eller förekomst av extracellulärt ATP i lymfnoden). Utöver sådana identifierade begränsningar bör LLNA: DA gå att använda för testning av alla ämnen, utom om ett ämne förknippas med egenskaper som kan påverka LLNA: DA-metodens exakthet. Dessutom bör överväganden göras rörande risken för positiva gränsresultat när man får värden för stimulationsindex (SI) mellan 1,8 och 2,5 (se punkterna 31–32). Detta grundar sig på valideringsdatabasen med 44 ämnen med användning av SI ≥ 1,8 (se punkt 6) för vilka LLNA: DA ger korrekt identifiering av alla 32 sensibiliserande ämnen enligt LLNA men felaktig identifiering av 3 av 12 icke-sensibiliserande ämnen enligt LLNA med SI-värden mellan 1,8 och 2,5 (dvs. på gränsen till positivt) (10). Eftersom samma datauppsättning användes för att fastställa SI-värdena och beräkna testmetodens förutsägande egenskaper kan dock de angivna resultaten vara en överuppskattning av de faktiska förutsägande egenskaperna.
|
PRINCIP FÖR TESTMETODEN
|
6.
|
LLNA: DA bygger på principen om att sensibiliserande ämnen inducerar en primär proliferation av lymfocyter i de lymfnoder som dränerar det område där testämnet appliceras. Denna proliferation står i proportion till den applicerade dosen och det allergiframkallande ämnets styrka, och är ett enkelt sätt att få en kvantitativ mätning av sensibiliseringen. Proliferationen mäts genom att jämföra medelproliferationen i varje testgrupp med medelproliferationen i den vehikelbehandlade kontrollgruppen. Förhållandet mellan medelproliferationen i respektive behandlingsgrupp och den parallella vehikelkontrollgruppen, kallat stimulationsindex (SI), bestäms och detta bör vara ≥ 1,8 för att det ska vara motiverat att klassificera testämnet som potentiellt hudsensibiliserande ämne. Det förfarande som beskrivs här grundar sig på mätning av ATP-innehållet (som korrelerar med antalet levande celler) genom bioluminiscens (17) i syfte att indikera ett ökat antal prolifererande celler i de aurikulära dränerande lymfnoderna (18) (19). I bioluminiscensmetoden används enzymet luciferas för att katalysera ombildningen till ljus från ATP och lucifering enligt följande reaktion:
ATP + luciferin + O
2
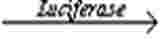 oxiluciferin + AMP + PPi
+ CO
2 + ljus
oxiluciferin + AMP + PPi
+ CO
2 + ljus
Det emitterade ljusets intensitet är linjärt relaterat till ATP-koncentrationen och uppmäts med en luminometer. Luciferin-luciferas-förfarandet är en känslig metod för ATP-kvantifiering som används i en mängd olika tillämpningar (20).
|
TESTBESKRIVNING
Val av djurart
|
7.
|
För detta test används mus. Valideringsstudier för LLNA: DA genomfördes uteslutande med stammen CBA/J som därför anses vara den stam som är att föredra (12) (13). I testet används unga vuxna mushonor som inte har fött ungar och som inte är dräktiga. När försöket inleds bör djuren vara 8–12 veckor gamla. Djurens viktvariation bör vara minimal och får inte överskrida 20 % av medelvikten. Övriga stammar eller hanar kan användas som alternativ förutsatt att det finns tillräckliga data som stöd för att det inte förekommer några betydande stam- eller könsberoende skillnader i LLNA: DA-respons.
|
Inhysnings- och utfodringsförhållanden
|
8.
|
Mössen ska inhysas i grupper (21), utom om det finns tillbörlig vetenskaplig motivering för individuell inhysning. Temperaturen i djurens utrymmen bör vara 22 ± 3 °C. Den relativa fuktigheten bör vara minst 30 % och ska helst inte överstiga 70 %, utom när rummet rengörs. Målvärdet bör dock vara 50–60 %. Belysningen bör vara artificiell med omväxlande 12 timmar ljus och 12 timmar mörker. För utfodringen kan konventionellt laboratoriefoder användas, och djuren bör ha obegränsad tillgång till dricksvatten.
|
Förberedelse av djuren
|
9.
|
Djuren väljs ut slumpmässigt och förses med märkning så att de kan identifieras individuellt (öronmärkning får dock inte användas). Därefter får djuren acklimatisera sig till laboratoriets förhållanden i sina burar i minst fem dagar före doseringsstart. Innan behandlingen inleds undersöks alla djur för att säkerställa att de inte har några synliga hudskador.
|
Förberedelse av doseringslösningar
|
10.
|
Kemikalier i fast form ska upplösas eller suspenderas i lösningsmedel/vehikel och vid behov spädas ut före appliceringen på mössens öron. Kemikalier i vätskeform kan appliceras i ren eller utspädd form. Olösliga kemikalier, av den typ som ofta används i medicintekniska produkter, ska före applicering på mössens öron extraheras kraftigt i lämpligt lösningsmedel för att få ut alla extraherbara beståndsdelar för testning. Testlösningarna ska beredas dagligen utom om stabilitetsdata visar att lagring kan accepteras.
|
Tillförlitlighetskontroll
|
11.
|
Positiva kontrollkemikalier används för att påvisa att testsystemet klarar av att ge respons med tillräcklig och reproducerbar känslighet för ett sensibiliserande testämne för vilket responsen har en välkarakteriserad styrka. Användningen av en parallell positiv kontroll rekommenderas eftersom den påvisar laboratoriets kompetens att genomföra testningar på ett framgångsrikt sätt och ger möjlighet att bedöma reproducerbarhet och jämförbarhet inom laboratoriet och mellan laboratorier. Vissa tillsynsmyndigheter kräver också att positiv kontroll används för varje försök, och därför rekommenderas samråd med relevanta myndigheter innan testning med LLNA: DA inleds. Likaså uppmuntras rutinanvändning av parallell positiv kontroll för att undvika tilläggstestning på djur i fall där myndigheterna inte godkänner periodiska positiva kontroller (se punkt 12). Den positiva kontrollen bör ge en positiv LLNA: DA-respons på den exponeringsnivå som förväntas ge en ökning av stimulationsindex (SI) som är större eller lika med 1,8 i förhållande till den negativa kontrollgruppen. Dosen för den positiva kontrollkemikalien ska väljas så att den inte orsakar särskilt kraftig hudirritation eller systemisk toxicitet och att induktionen är reproducerbar men inte för kraftig (t.ex. SI > 10 anses vara för högt värde). Rekommenderade ämnen för positiv kontroll är 25-procentig hexylkanelaldehyd (CAS-nummer 101-86-0) och 25-procentig eugenol (CAS-nummer 97-53-0) i aceton-olivoljeblandning i förhållandet 4:1 (volym/volym). I vissa fall kan även andra positiva kontrollkemikalier som uppfyller de ovan angivna kriterierna användas, förutsatt att tillbörlig motivering kan ges.
|
|
12.
|
Även om användning av en parallell positiv kontrollgrupp rekommenderas kan det finnas betingelser där det är tillräckligt med periodisk testning (dvs. minst var sjätte månad) av den positiva kontrollen i laboratorier som regelbundet genomför LLNA: DA (dvs. minst en gång i månaden) och som har en etablerad historisk databas över positiv kontroll som påvisar laboratoriets förmåga att få reproducerbara och exakta resultat från positiva kontroller. Tillräcklig kvalitet hos LLNA: DA kan framgångsrikt påvisas genom att enhetliga positiva resultat erhålls från den positiva kontrollen vid minst tio oberoende testningar som genomförs inom en skälig tidsperiod (kortare än ett år).
|
|
13.
|
Parallell positiv kontrollgrupp ska alltid användas när det sker ändringar rörande testningsförfarandet (t.ex. ny personal, nya material och/eller reagenser, annorlunda utrustning, byte av testdjurens ursprung), och sådana ändringar bör dokumenteras i laboratorierapporter. Man bör överväga om dessa ändringar påverkar den tidigare etablerade historiska databasens lämplighet, för att kunna avgöra om det behövs en ny historisk databas för dokumentering av den positiva kontrollens resultatenhetlighet.
|
|
14.
|
Man bör vara medveten om att beslut om att använda positiva kontroller med vissa intervaller i stället för parallellt har återverkningar på lämpligheten och godtagbarheten hos negativa undersökningsresultat som fås utan parallell positiv kontroll under den tid som löper mellan de periodiska undersökningarna av positiv kontroll. Om t.ex. falska negativa resultat fås vid ett periodiskt test av positiv kontroll, måste man kanske ifrågasätta resultat som erhållits mellan det sista godtagbara periodiska testet av positiv kontroll och ett icke-godtagbart periodiskt test med positiv kontroll. Följderna av dessa utfall bör övervägas noggrant när man avgör huruvida parallella positiva kontroller ska användas eller om de positiva kontrollerna enbart ska vara periodiska. Likaså bör man överväga användning av färre djur i den parallella positiva kontrollgruppen när detta är vetenskapligt motiverat och laboratoriet med stöd av laboratoriespecifika historiska data kan påvisa att färre möss kan användas (22).
|
|
15.
|
Även om det positiva kontrollämnet bör testas i en vehikel som veterligen framkallar en enhetlig respons (t.ex. aceton-olivoljeblandning i förhållandet 4:1 (volym/volym)), kan det förekomma vissa föreskriftsreglerade situationer där det även behövs testning i en icke-standardmässig vehikel (kliniskt och kemiskt relevant formulering) (23). Om det parallella positiva kontrollämnet testas i en annan vehikel än den som används för testämnet, bör en separat kontrollgrupp användas för den parallella positiva kontrollen.
|
|
16.
|
I fall där utvärdering görs av testämnen av en viss kemisk klass eller med en viss responsskala kan det också vara värdefullt med referensämnen för att påvisa att testmetoden fungerar tillfredsställande för bestämning av hudsensibiliseringspotentialen hos dessa typer av ämnen. Ett lämpligt referensämne ska ha följande egenskaper:
|
—
|
Strukturell och funktionell likhet med den ämnesklass som testas.
|
|
—
|
Kända fysikalisk-kemiska egenskaper.
|
|
—
|
Stödjande data från LLNA: DA.
|
|
—
|
Stödjande data från andra djurmodeller och/eller från människor.
|
|
TESTFÖRFARANDE
Antalet djur och dosnivåer
|
17.
|
Minst fyra djur per dosgrupp och minst tre koncentrationer av testämnet bör användas. Dessutom används en parallell negativ kontrollgrupp som endast behandlas med samma vehikel som används för testämnet och en positiv kontroll (parallell eller nyligen gjord, enligt laboratoriets principer för överväganden enligt punkterna 11–15). Man bör överväga flera upprepade doser av det positiva kontrollämnet, särskilt när den positiva kontrollen testas periodiskt. Med undantag för behandling med testämnet bör djuren i kontrollgrupperna hanteras på exakt samma sätt som djuren i dosgrupperna.
|
|
18.
|
Doserna och vehikeln bör väljas på grundval av rekommendationerna i hänvisningarna 0 och (24). Doserna väljs i regel enligt en lämplig koncentrationsserie såsom 100 %, 50 %, 25 %, 10 %, 5 %, 2,5 %, 1 %, 0,5 %, osv. Valet av koncentrationsserie ska ges tillbörlig vetenskaplig motivering. All existerande tillgänglig och relevant toxikologisk information (t.ex. akut toxicitet och hudirritation) samt strukturell och fysikalisk-kemisk information om testämnet (och/eller strukturellt besläktade ämnen) bör övervägas vid valet av de tre på varandra följande koncentrationerna så att den högsta koncentrationen ger maximal exponering samtidigt som systemisk toxicitet och särskilt kraftig lokal hudirritation undviks (24) (25). Om sådan information inte finns kan det vara nödvändigt att först genomföra ett preliminärt screeningtest (se punkterna 21–24).
|
|
19.
|
Vehikeln får inte störa eller snedvrida testresultaten och ska väljas med tanke på största möjliga löslighet för att få högsta möjliga koncentration samtidigt som man får en lösning/suspension som är lämplig för applicering av testämnet. Som vehikel rekommenderas aceton-olivoljeblandning (4:1 volym/volym), N,N-dimetylformamid, metyletylketon, propylenglykol och dimetylsulfoxid (6) men även andra vehiklar kan användas förutsatt att tillräcklig vetenskaplig motivering kan ges. I vissa situationer kan det vara nödvändigt att som extra kontroll använda ett kliniskt relevant lösningsmedel eller en kommersiellt tillgänglig blandning där testämnet ingår. Särskild vikt bör fästas vid att säkerställa att hydrofila ämnen inkorporeras i ett vehikelsystem som väter huden och som inte omedelbart rinner bort, genom att använda lämpliga solubiliserande ämnen (t.ex. 1-procentig Pluronic® L92). Det betyder att helt vattenbaserade vehiklar bör undvikas.
|
|
20.
|
Genom analys av lymfnoder från individuella möss kan man bestämma variationer djuren emellan och göra en statistisk jämförelse av skillnaden mellan mätningar för testämnet och mätningarna i kontrollgruppen (se punkt 33). Dessutom kan möjligheten att minska antalet möss i den positiva kontrollgruppen endast övervägas utifrån att data samlas in individuellt per djur (22). Vidare gäller att vissa tillsynsmyndigheter kräver att data insamlas per individuellt djur. Regelbunden insamling av individuella data per djur ger en fördel rörande djurens välbefinnande eftersom man kan undvika upprepad testning som annars skulle behövas om testämnesresultat som ursprungligen har samlats in på ett visst sätt (t.ex. genom poolade djurdata) måste granskas senare av tillsynsmyndigheter som har avvikande krav (såsom data per individuellt djur).
|
Preliminärt screeningtest
|
21.
|
Om det inte finns tillgång till information för att bestämma högsta testdosen (se punkt 18) bör ett preliminärt screeningtest genomföras för att definiera den lämpliga dosnivån för test i LLNA: DA. Syftet med det preliminära screeningtestet är att ge vägledning för valet av högsta dosnivå som ska användas i den huvudsakliga LLNA: DA-undersökningen i fall där det inte finns tillgång till information om vilken koncentration som inducerar systemisk toxicitet (se punkt 24) och/eller särskilt kraftig lokal hudirritation (se punkt 23). Den högsta dosnivå som testas ska vara 100 % testämne för vätskor eller högsta möjliga koncentration för fasta ämnen eller suspensioner.
|
|
22.
|
Det preliminära screeningtestet genomförs under identiska betingelser som vid den huvudsakliga LLNA: DA-undersökningen, utom att man inte utvärderar proliferationen i lymfnoderna och man kan använda färre djur i dosgruppen. Ett eller två djur per dosgrupp föreslås. Alla möss observeras dagligen för kliniska tecken på systemisk toxicitet eller lokal irritation på appliceringsstället. Djurens kroppsvikt registreras före testningen och efter avslutad testning (dag 8). På varje mus observeras båda öronen i fråga om hudrodnad och graderas enligt tabell 1 (25). Mätning av örontjocklek görs t.ex. med digital mikrometer eller Peacock Dial-instrument på dag 1 (före dosering), dag 3 (cirka 48 timmar efter den första dosen), dag 7 (24 timmar före avslut) och dag 8. Dessutom kan örontjockleken bestämmas på dag 8 genom vägning av en öronbit som stansats ut efter att djuren har avlivats skonsamt. Särskilt kraftig lokal hudirritation indikeras genom hudrodnadsgradering ≥ 3 och/eller ökning av örontjockleken med ≥ 25 % på någon av mätningsdagarna (26) (27). Den högsta dos som väljs för den huvudsakliga LLNA: DA-undersökningen blir nästa lägre dos i koncentrationsserien för den preliminära screeningen (se punkt 18) som inte inducerar systemisk toxicitet och/eller särskilt kraftig hudirritation.
Tabell 1
Grader av hudrodnad
|
Observation
|
Gradering
|
|
Ingen hudrodnad
|
0
|
|
Mycket svag rodnad (knappt skönjbar)
|
1
|
|
Väldefinierad hudrodnad
|
2
|
|
Moderat till svår hudrodnad
|
3
|
|
Svår hudrodnad (köttrodnad) till sårskorpebildning som utgör hinder för gradering av rodnaden
|
4
|
|
|
23.
|
Utöver en 25-procentig ökning av örontjocklek (26) (27) har en statistiskt signifikant ökning av örontjockleken hos behandlingsgruppens möss i jämförelse med kontrollgruppens möss också använts för att identifiera irriterande ämnen genom LLNA (28) (29) (30) (31) (32) (33) (34). Dock så gäller att även om statistiskt signifikanta ökningar kan inträffa när ökningen av örontjocklek är mindre än 25 %, har dessa inte specifikt förknippats med särskilt kraftig irritation (30) (31) (32) (33) (34).
|
|
24.
|
Följande kliniska observationer kan indikera systemisk toxicitet (35) när de används som del av en helhetsbedömning, och kan därför ge hänvisning om den högsta dosnivå som ska användas i det huvudsakliga LLNA: DA-försöket: ändringar av nervsystemets funktion (t.ex. upprest päls, ataxi, skälvningar och konvulsioner), ändringar av beteende (t.ex. aggressivitet, ändringar av putsningsbeteende, markerad ändring av aktivitetsnivå), ändringar av andningsmönster (dvs. ändringar av andningens frekvens och intensitet, såsom dyspné, gäspande och rassel) samt ändringar av födo- och vattenintag. Vid utvärderingen ska man också beakta tecken på letargi och/eller avsaknad av reaktioner, alla slags kliniska tecken på mer än lätt eller momentan smärta eller lidande, en minskning på mer än 5 % av kroppsvikten från dag 1 till dag 8 samt mortalitet. Döende djur och djur som visar tecken på svår smärta och lidande ska avlivas skonsamt (36).
|
Huvudundersökningens försöksschema
|
25.
|
För testet används följande försöksschema:
— Dag 1: Djuren märks individuellt och vikten samt eventuella kliniska observationer registreras. Applicera en 1-procentig vattenlösning av natriumlaurylsulfat (SLS) på baksidan av varje öra med en borste doppad i lösningen och täck örats hela baksida med fyra till fem strykningar. En timme efter SLS-behandlingen appliceras 25 μl lämpligt utspätt testämne, enbart vehikel eller positivt kontrollämne (parallell eller nyligen gjord positiv kontroll, i enlighet med laboratoriets principer rörande punkterna 11–15) på båda öronens baksida.
— Dagarna 2, 3 och 7: Upprepa förbehandlingen med 1-procentig SLS-vattenlösning och applicera testämnet på samma sätt som på dag 1.
— Dagarna 4, 5 och 6: Ingen behandling.
— Dag 8: Registrera vikten för varje djur och eventuella kliniska observationer. Avliva djuren skonsamt cirka 24–30 timmar efter appliceringsstarten dag 7. Skär bort de aurikulära dränerande lymfnoderna från varje musöra och behandla separat i fosfatbuffrad saltlösning (PBS) för varje djur. Närmare uppgifter och scheman för identifiering och dissekering av noder finns i hänvisning (22). För ytterligare kontroll av lokal hudrespons i huvudundersökningen kan tilläggsparametrar såsom gradering av örats hudrodnad eller mätningar av örats tjocklek (med tjockleksmätare eller genom vägning av utstansad bit vid obduceringen) införlivas i undersökningsprotokollet.
|
Beredning av cellsuspensioner
|
26.
|
För varje mus bereds en suspension med enskilda celler av de bilateralt bortskurna lymfnodscellerna (LNC) genom att placera lymfnoderna mellan två objektglas som trycks lätt samman för att krossa noderna. Kontrollera att vävnaden har spritt ut sig i ett tunt skikt och dra isär de två objektglasen. Bered en suspension av vävnaden på båda objektglasen genom att hålla dem ett i sänder i vinkel över en Petriskål och skölja med fosfatbuffrad saltlösning och skrapa samtidigt loss vävnaden med en cellskrapa. Observera att lymfnoderna i den negativa kontrollgruppens djur är små, och det är viktigt att iaktta stor försiktighet för att inte åstadkomma störande inverkan på SI-värdena. Totalt används 1 ml fosfatbuffrad saltlösning för sköljning av båda objektglasen. Gör en lätt homogenisering av LNC-suspensionen i Petriskålen med hjälp av cellskrapan. Dra upp en alikvot på 20 μl ur LNC-suspensionen med mikropipett och var försiktig med att inte ta upp membran som kan ses med blotta ögat, och blanda sedan med 1,98 ml fosfatbuffrad saltlösning till en provvolym på 2 ml. Bered sedan ett andra prov på 2 ml genom samma förfarande så att det finns två prov beredda per djur.
|
Bestämning av cellproliferation (mätning av lymfocyternas ATP-innehåll)
|
27.
|
Ökning av ATP-innehållet i lymfnoderna mäts genom luciferin-luciferas-metoden med användning av ett system för ATP-mätning som mäter bioluminiscensen som relativa ljusenheter (Relative Luminescence Units, RLU). Tiden mellan avlivning av ett djur fram till mätning av ATP-innehållet bör vara enhetlig, inom cirka 30 minuter, eftersom man anser att ATP-innehållet gradvis minskar med tiden efter avlivningen (12). Serien av förfaranden från bortskärning av de aurikulära lymfnoderna fram till ATP-mätning bör därför slutföras inom 20 minuter enligt det på förhand fastställda tidsschemat som är samma för varje djur. ATP-luminiscensen bör mätas för varje 2 ml-prov så att totalt två ATP-mätningar görs per djur. Genomsnittet för ATP-luminiscens bestäms och används därefter för beräkningarna (se punkt 30).
|
OBSERVATIONER
Kliniska observationer
|
28.
|
Varje mus bör observeras minst dagligen med tanke på eventuella kliniska tecken – lokal irritation vid appliceringsstället eller tecken på systemisk toxicitet. Alla observationer registreras systematiskt och individuellt per mus. Övervakningsplanerna bör inbegripa kriterier för omedelbar identifiering av möss som visar tecken på systemisk toxicitet, särskilt kraftig hudirritation eller hudkorrosion, så att de kan avlivas (36).
|
Kroppsvikt
|
29.
|
Såsom konstateras i punkt 25 ska de individuella djurens vikt registreras vid försökets början och strax före den schemalagda avlivningen.
|
BERÄKNING AV RESULTATEN
|
30.
|
Resultaten för varje behandlingsgrupp uttrycks som genomsnittet för stimulationsindex. Stimulationsindex erhålls genom att genomsnittligt värde för RLU/djur inom varje testämnesgrupp och den positiva kontrollgruppen divideras med genomsnittligt värde för RLU/djur för kontrollgruppen för lösningsmedel/vehikel. Genomsnittligt stimulationsindex för vehikelkontrollgrupperna är då 1.
|
|
31.
|
Vid bedömning av resultaten anses ett resultat vara positivt när SI ≥ 1,8 (10). Man kan dock även använda dos-responsförhållandets styrka samt den statistiska signifikansen och enhetligheten hos responsen i kontrollgruppen för lösningsmedel/vehikel och i den positiva kontrollen när man fastställer huruvida ett gränsresultat (dvs. SI-värde mellan 1,8 och 2,5) ska anses vara positivt 0 0 (37).
|
|
32.
|
För ett gränsresultat för positiv respons där SI-värdet ligger mellan 1,8 och 2,5 vill man kanske beakta tilläggsinformation såsom dos-responsförhållandet, tecken på systemisk toxicitet eller särskilt kraftig irritation och, där det är lämpligt, statistisk signifikans tillsammans med SI-värden för att bekräfta att resultaten är positiva (10). Likaså bör man beakta testämnets olika egenskaper, inbegripet eventuell strukturell släktskap med kända hudsensibiliserande ämnen, huruvida ämnet orsakar särskilt kraftig hudirritation hos mössen samt naturen hos det iakttagna förhållandet mellan dos och respons. Dessa och andra aspekter diskuteras närmare i andra källor (4).
|
|
33.
|
När data samlas in per enskild mus kan man göra en statistisk analys rörande förekomsten av dosrespons och graden för dos-responsförhållande. Alla statistiska analyser kan inbegripa en utvärdering av dos-responsförhållandet och en lämpligt anpassad jämförelse mellan testgrupper (t.ex. parvisa jämförelser mellan doseringsgrupp och parallell kontrollgrupp för lösningsmedel/vehikel). De statistiska analyserna kan inbegripa t.ex. linjär regression eller Williams test för utvärdering av dos-responstrender och Dunnetts test för parvisa jämförelser. Vid valet av en lämplig metod för den statistiska analysen bör man vara medveten om eventuella olikheter hos varianserna och andra relaterade problem som kan göra det nödvändigt att göra en datatransformation eller en icke-parametrisk statistisk analys. Det kan också vara nödvändigt att genomföra beräkningar av stimulationsindex och statistiska analyser med och utan vissa datapunkter (s.k. outliers – utanförliggande värden).
|
DATA OCH RAPPORTERING
Data
|
34.
|
Data sammanställs i tabellform med angivelse av RLU-värden per djur, gruppens medelvärde för RLU per djur, den associerade feltermen (t.ex. SD, SEM) och medelvärdet för stimulationsindex för varje dosgrupp jämfört med den parallella kontrollgruppen för lösningsmedel/vehikel.
|
Testrapport
|
35.
|
Testrapporten ska innehålla nedan angivna uppgifter.
|
|
Test- och kontrollkemikalier
|
—
|
Identifieringsdata (t.ex. CAS-nummer och EG-nummer, om tillgängligt, källa, renhet, kända orenheter, satsnummer).
|
|
—
|
Fysikalisk natur och fysikalisk-kemiska egenskaper (t.ex. flyktighet, stabilitet, löslighet).
|
|
—
|
För blandningar anges sammansättning och beståndsdelarnas relativa procentandelar.
|
|
|
|
Lösningsmedel/vehikel
|
—
|
Identifieringsdata (renhet, koncentration (om tillämpligt), volym som har använts).
|
|
—
|
Motivering för val av vehikel.
|
|
|
|
Testdjur
|
—
|
Mikrobiologiska data för djuren, om dessa data är kända.
|
|
—
|
Antal djur och deras ålder.
|
|
—
|
Ursprung, inhysningsförhållanden, foder och dylikt.
|
|
|
|
Testbetingelser
|
—
|
Ursprung, satsnummer och tillverkarens kvalitetsintyg/kvalitetskontrolldata för ATP-kit.
|
|
—
|
Detaljerade uppgifter om beredning och applicering av testämnet.
|
|
—
|
Motivering för valet av dos (även resultat från preliminär screening om sådan har genomförts).
|
|
—
|
Koncentrationerna för vehikel och testämne samt totala mängden testämne som applicerats.
|
|
—
|
Detaljerade uppgifter om foder- och vattenkvalitet (inbegripet diettyp/källa, vattenkälla).
|
|
—
|
Detaljerad beskrivning av behandlings- och provtagningsscheman.
|
|
—
|
Metoder för mätningar av toxicitet.
|
|
—
|
Kriterier för att fastställa om resultatet är positivt eller negativt.
|
|
—
|
Uppgifter rörande eventuella avvikelser från protokollet och redogörelse för hur avvikelsen påverkar undersökningens utformning och resultat.
|
|
|
|
Tillförlitlighetskontroll
|
—
|
Sammanfattning av resultaten från senaste tillförlitlighetskontroll, även information om testämne, koncentration och vehikel.
|
|
—
|
Data från parallella och/eller historiska positiva kontroller och parallella negativa kontroller (lösningsmedel/vehikel) för testningslaboratoriet.
|
|
—
|
Om försöket inte har omfattat parallell positiv kontroll, ange datum och laboratorierapport för senaste periodiska positiva kontroll och en rapport med uppgifter om laboratoriets historiska data för positiva kontroller som motivering för varför parallell positiv kontroll inte har använts.
|
|
|
|
Resultat
|
—
|
Mössens individuella vikt vid doseringsstart och vid schemalagd avlivning samt medelvärde och associerad felterm (t.ex. SD, SEM) för varje behandlingsgrupp.
|
|
—
|
Tidpunkten för första upptäckt av tecken på toxicitet, även eventuell hudirritation vid appliceringsstället, för varje försöksdjur.
|
|
—
|
Tidpunkten för avlivning och tidpunkten för ATP-mätning per djur.
|
|
—
|
En tabell med RLU-värden och SI-värden per djur för varje dosbehandlingsgrupp.
|
|
—
|
Medelvärde och associerad felterm (t.ex. SD, SEM) för RLU per mus för varje behandlingsgrupp och resultaten från analys av utanförliggande värden för varje behandlingsgrupp.
|
|
—
|
Beräknat stimulationsindex och ett lämpligt mått på variabilitet som beaktar variationen mellan djuren i testämnesgruppen och kontrollgruppen.
|
|
—
|
Förhållandet mellan dos och respons.
|
|
—
|
Statistisk analys, om tillämpligt.
|
|
|
|
Diskussion om resultaten
|
—
|
En kort kommentar om resultaten, dos-responsanalys och statistiska analyser (i tillämpliga fall), med en slutsats om huruvida testämnet ska anses vara hudsensibiliserande.
|
|
|
HÄNVISNINGAR
|
(1)
|
OECD (2010), Skin Sensitisation: Local Lymph Node Assay, Test Guideline No. 429, Guidelines for the Testing of Chemicals, OECD, Paris. Finns på: http://www.oecd.org/env/testguidelines
|
|
(2)
|
Chamberlain, M. och Basketter, D.A. (1996), The local lymph node assay: status of validation. Food Chem, Toxicol., 34, 999–1002.
|
|
(3)
|
Basketter, D.A., Gerberick, G.F., Kimber, I. och Loveless, S.E. (1996), The local lymph node assay: A viable alternative to currently accepted skin sensitisation tests. Food Chem, Toxicol., 34, 985–997.
|
|
(4)
|
Basketter, D.A., Gerberick, G.F. och Kimber, I. (1998), Strategies for identifying false positive responses in predictive sensitisation tests. Food Chem. Toxicol., 36, 327–333.
|
|
(5)
|
Van Och, F.M.M., Slob, W., De Jong, W.H., Vandebriel, R.J. och Van Loveren, H. (2000), A quantitative method for assessing the sensitising potency of low molecular weight chemicals using a local lymph node assay: employment of a regression method that includes determination of uncertainty margins. Toxicol., 146, 49–59.
|
|
(6)
|
ICCVAM (1999), The murine local lymph node Assay: A test method for assessing the allergic contact dermatitis potential of chemicals/compounds: The results of an independent peer review evaluation coordinated by the Interagency Coordinating Committee on the Validation of Alternative Methods (ICCVAM) and the National Toxicology Program Center for the Evaluation of Alternative Toxicological Methods (NICETAM). NIH Publication No: 99–4494. Research Triangle Park, N.C. Finns på: http://iccvam.niehs.nih.gov/docs/immunotox_docs/llna/llnarep.pdf
|
|
(7)
|
Dean, J.H., Twerdok, L.E., Tice, R.R., Sailstad, D.M., Hattan, D.G., Stokes, W.S. (2001), ICCVAM evaluation of the murine local lymph node assay: II. Conclusions and recommendations of an independent scientific peer review panel. Reg. Toxicol. Pharmacol. 34, 258–273.
|
|
(8)
|
Haneke, K.E., Tice, R.R., Carson, B.L., Margolin, B.H., Stokes, W.S. (2001), ICCVAM evaluation of the murine local lymph node assay: III. Data analyses completed by the national toxicology program interagency center for the evaluation of alternative toxicological methods. Reg. Toxicol. Pharmacol. 34, 274–286.
|
|
(9)
|
Sailstad, D.M., Hattan, D., Hill, R.N., Stokes, W.S. (2001), ICCVAM evaluation of the murine local lymph node assay: I. The ICCVAM review process. Reg. Toxicol. Pharmacol. 34, 249–257.
|
|
(10)
|
ICCVAM (2010), ICCVAM Test Method Evaluation Report. Nonradioactive local lymph node assay: modified by Daicel Chemical Industries, Ltd, based on ATP content test method protocol (LLNA: DA). NIH Publication No. 10–7551A/B. Research Triangle Park, NC: National Institute of Environmental Health Sciences. Finns på: http://iccvam.niehs.nih.gov/methods/immunotox/llna-DA/TMER.htm
|
|
(11)
|
ICCVAM (2009), Independent Scientific Peer Review Panel Report: Updated validation status of new versions and applications of the murine local lymph node assay: a test method for assessing the allergic contact dermatitis potential of chemicals and products. Research Triangle Park, NC: National Institute of Environmental Health Sciences. Finns på: http://iccvam.niehs.nih.gov/docs/immunotox_docs/LLNAPRPRept2009.pdf.
|
|
(12)
|
Idehara, K., Yamagishi, G., Yamashita, K. och Ito, M. (2008), Characterization and evaluation of a modified local lymph node assay using ATP content as a non-radio isotopic endpoint. J. Pharmacol. Toxicol. Meth., 58, 1–10.
|
|
(13)
|
Omori, T., Idehara, K., Kojima, H., Sozu, T., Arima, K., Goto, H., Hanada, T., Ikarashi, Y., Inoda, T., Kanazawa, Y., Kosaka, T., Maki, E., Morimoto, T., Shinoda, S., Shinoda, N., Takeyoshi, M., Tanaka, M., Uratani, M., Usami, M., Yamanaka, A., Yoneda, T., Yoshimura, I. och Yuasa, A. (2008), Interlaboratory validation of the modified murine local lymph node assay based on adenosine triphosphate measurement. J. Pharmacol. Toxicol. Meth., 58, 11–26.
|
|
(14)
|
OECD (1992), Skin Sensitisation, Test Guideline No. 406, Guidelines for Testing of Chemicals, OECD, Paris. Finns på: http://www.oecd.org/env/testguidelines
|
|
(15)
|
Kreiling, R., Hollnagel, H.M., Hareng, L., Eigler, L., Lee, M.S., Griem, P., Dreessen, B., Kleber, M., Albrecht, A., Garcia, C. och Wendel, A. (2008), Comparison of the skin sensitising potential of unsaturated compounds as assessed by the murine local lymph node assay (LLNA) and the guinea pig maximization test (GPMT). Food Chem. Toxicol., 46, 1896–1904.
|
|
(16)
|
Basketter, D., Ball, N., Cagen, S., Carrillo, J.C., Certa, H., Eigler, D., Garcia, C., Esch, H., Graham, C., Haux, C., Kreiling, R. och Mehling, A. (2009), Application of a weight of evidence approach to assessing discordant sensitisation datasets: implications for REACH. Reg. Toxicol. Pharmacol., 55, 90–96.
|
|
(17)
|
Crouch, S.P., Kozlowski, R., Slater, K.J. och Fletcher J. (1993), The use of ATP bioluminescence as a measure of cell proliferation and cytotoxicity. J. Immunol. Meth., 160, 81–88.
|
|
(18)
|
Ishizaka, A., Tono-oka, T. och Matsumoto, S. (1984), Evaluation of the proliferative response of lymphocytes by measurement of intracellular ATP. J. Immunol. Meth., 72, 127–132.
|
|
(19)
|
Dexter, S.J., Cámara, M., Davies, M. och Shakesheff, K.M. (2003), Development of a bioluminescent ATP assay to quantify mammalian and bacterial cell number from a mixed population. Biomat., 24, 27–34.
|
|
(20)
|
Lundin A. (2000), Use of firefly luciferase in ATP-related assays of biomass, enzymes, and metabolites. Meth. Enzymol., 305, 346–370.
|
|
(21)
|
ILAR (1996), Institute of Laboratory Animal Research (ILAR) Guide for the Care and Use of Laboratory Animals. 7th ed. Washington, DC: National Academies Press.
|
|
(22)
|
ICCVAM (2009), Recommended Performance Standards: Murine Local Lymph Node Assay, NIH Publication Number 09–7357, Research Triangle Park, NC: National Institute of Environmental Health Science. Finns på: http://iccvam.niehs.nih.gov/docs/immunotox_docs/llna-ps/LLNAPerfStds.pdf
|
|
(23)
|
McGarry, H.F. (2007), The murine local lymph node assay: regulatory and potency considerations under REACH. Toxicol., 238, 71–89.
|
|
(24)
|
Kimber, I., Dearman, R.J., Scholes E.W. och Basketter, D.A. (1994), The local lymph node assay: developments and applications. Toxicol., 93, 13–31.
|
|
(25)
|
OECD (2002), Acute Dermal Irritation/Corrosion, Test Guideline No. 404, Guidelines for Testing of Chemicals, OECD, Paris. Finns på: http://www.oecd.org/env/testguidelines
|
|
(26)
|
Reeder, M.K., Broomhead, Y.L., DiDonato, L. och DeGeorge, G.L. (2007), Use of an enhanced local lymph node assay to correctly classify irritants and false positive substances. Toxicologist, 96, 235.
|
|
(27)
|
ICCVAM (2009), Nonradioactive Murine Local Lymph Node Assay: Flow Cytometry Test Method Protocol (LLNA: BrdU-FC) Revised Draft Background Review Document. Research Triangle Park, NC: National Institute of Environmental Health Sciences. Finns på: http://iccvam.niehs.nih.gov/methods/immunotox/fcLLNA/BRDcomplete.pdf
|
|
(28)
|
Hayes, B.B., Gerber, P.C., Griffey, S.S. och Meade, B.J. (1998), Contact hypersensitivity to dicyclohexylcarbodiimide and diisopropylcarbodiimide in female B6C3F1 mice. Drug. Chem. Toxicol., 21, 195–206.
|
|
(29)
|
Homey, B., von Schilling, C., Blumel, J., Schuppe, H.C., Ruzicka, T., Ahr, H.J., Lehmann, P. och Vohr, V.W. (1998), An integrated model for the differentiation of chemical-induced allergic and irritant skin reactions. Toxicol. Appl. Pharmacol., 153, 83–94.
|
|
(30)
|
Woolhiser, M.R., Hayes, B.B. and Meade, B.J. (1998), A combined murine local lymph node and irritancy assay to predict sensitisation and irritancy potential of chemicals. Toxicol. Meth., 8, 245–256.
|
|
(31)
|
Hayes, B.B. och Meade, B.J. (1999), Contact sensitivity to selected acrylate compounds in B6C3F1 mice: relative potency, cross reactivity, and comparison of test methods. Drug Chem. Toxicol., 22, 491–506.
|
|
(32)
|
Ehling, G., Hecht, M., Heusener, A., Huesler, J., Gamer, A.O., van Loveren, H., Maurer, T., Riecke, K., Ullmann, L., Ulrich, P., Vandebriel, R. och Vohr, H.W. (2005), A European inter-laboratory validation of alternative endpoints of the murine local lymph node assay: first round. Toxicol., 212, 60–68.
|
|
(33)
|
Vohr, H.W. och Ahr, H.J. (2005), The local lymph node assay being too sensitive? Arch. Toxicol., 79, 721–728.
|
|
(34)
|
Patterson, R.M., Noga, E. och Germolec, D. (2007), Lack of evidence for contact sensitisation by Pfiesteria extract. Environ. Health Perspect., 115, 1023–1028.
|
|
(35)
|
ICCVAM (2009), Report on the ICCVAM-NICEATM/ECVAM/JaCVAM Scientific Workshop on Acute Chemical Safety Testing: Advancing In Vitro Approaches and Humane Endpoints for Systemic Toxicity Evaluations. Research Triangle Park, NC: National Institute of Environmental Health Sciences. Finns på: http://iccvam.niehs.nih.gov/methods/acutetox/Tox_workshop.htm
|
|
(36)
|
OECD (2000), Guidance Document on the Recognition, Assessment and Use of Clinical Signs as Humane Endpoints for Experimental Animals Used in Safety Evaluation, Environmental Health and Safety Monograph Series on Testing and Assessment No. 19, ENV/JM/MONO(2000)7, OECD, Paris. Finns på: http://www.oecd.org/env/testguidelines
|
|
(37)
|
Kimber, I., Hilton, J., Dearman, R.J., Gerberick, G.F., Ryan, C.A., Basketter, D.A., Lea, L., House, R.V., Ladies, G.S., Loveless, S.E. och Hastings, K.L. (1998), Assessment of the skin sensitisation potential of topical medicaments using the local lymph node assay: An interlaboratory exercise. J. Toxicol. Environ. Health, 53 563–79.
|
|
(38)
|
OECD (2005), Guidance Document on the Validation and International Acceptance of New or Updated Test Methods for Hazard Assessment, Environment, Health and Safety Monograph Series on Testing and Assessment No. 34, ENV/JM/MONO(2005)14, OECD, Paris. Finns på: http://www.oecd.org/env/testguidelines
|
Tillägg 1
DEFINITIONER
Exakthet: Mått på hur väl testmetodresultaten och de godtagna referensvärdena överensstämmer med varandra. Exakthet är ett mått på testmetodens prestanda och en relevansaspekt. Detta begrepp används ofta omväxlande med ’konkordans’ för att beskriva andelen korrekta resultat med en testmetod (38).
Referensämne: Sensibiliserande eller icke-sensibiliserande ämne som används som standard för jämförelse med ett testämne. Ett referensämne ska ha följande egenskaper: i) en eller flera enhetliga och tillförlitliga källor, ii) strukturell och funktionell likhet med den ämnesklass som testas, iii) kända fysikalisk-kemiska egenskaper, iv) stödjande data om kända effekter och v) känd styrka inom det önskade responsområdet.
Falsk negativt: Ett ämne som felaktigt har identifierats som negativt eller icke-aktivt genom en testmetod när ämnet i själva verket är positivt eller aktivt.
Falskt positivt: Ett ämne som felaktigt har identifierats som positivt eller aktivt genom en testmetod när ämnet i själva verket är negativt eller icke-aktivt.
Fara: Potentialen för skadliga verkningar på hälsa eller miljö. De skadliga verkningarna visar sig enbart om exponeringsnivån är tillräcklig.
Reproducerbarhet mellan laboratorier: Ett mått på i hur stor utsträckning olika kvalificerade laboratorier kan producera kvalitativt och kvantitativt liknande resultat, med användning av samma protokoll och samma testämnen. Reproducerbarheten mellan laboratorier fastställs under förvalideringen och valideringen och den indikerar i hur stor utsträckning ett test framgångsrikt kan överföras mellan laboratorier (38).
Reproducerbarhet inom laboratoriet: Ett mått på i vilken utsträckning kvalificerade personer inom samma laboratorium framgångsrikt kan upprepa resultat vid olika tidpunkter när de använder ett visst protokoll.
Utanförliggande värde: Ett utanförliggande värde (’outlier’) är en observation som markant skiljer sig från de övriga värdena i ett slumpmässigt urval från en population.
Kvalitetssäkring: En styrningsprocess där personer som är i oberoende ställning i förhållande till de personer som genomför testningen bedömer hur standarder, krav och registreringsförfaranden för laboratorietestning följs och hur exakt data förmedlas.
Tillförlitlighet: Ett mått på i vilken utsträckning en testmetod kan reproduceras inom och mellan laboratorier över tiden när metoden utförs med samma protokoll. Den bedöms genom att man beräknar reproducerbarheten inom och mellan laboratorier (38).
Hudsensibilisering: En immunologisk process som följer av att en mottaglig individ exponeras ytvägen för en inducerande kemisk allergen som provocerar en kutan immunrespons som kan leda till att det uppstår kontaktsensibilisering.
Stimulationsindex (SI): Ett värde som beräknas för att bedöma hudsensibiliseringspotentialen hos ett testämne och som är förhållandet mellan proliferationen i behandlingsgrupperna och proliferationen i den parallella vehikelkontrollgruppen.
Testämne (kallas också testkemikalie): Alla ämnen eller blandningar som testas med denna testmetod.
B.51. HUDSENSIBILISERING: LLNA-METODEN (LOCAL LYMPH NODE ASSAY): BrdU-ELISA
INLEDNING
|
1.
|
OECD:s riktlinjer för testning av kemikalier (OECD Guidelines for the Testing of Chemicals) och EU:s testmetoder ses regelbundet över med hänsyn till den vetenskapliga utvecklingen, nya lagstiftningsbehov och djurens välbefinnande. Den första testmetoden (B.42) för bestämning av hudsensibilisering hos möss (LLNA-metoden, OECD:s testriktlinje 429) har reviderats (1 och kapitel B.42 i denna bilaga). Närmare uppgifter om valideringen av LLNA och en översikt av arbetet i samband med denna har offentliggjorts (2) (3) (4) (5) (6) (7) (8) (9). I LLNA-metoden används radioisotopiskt tymidin eller jod för att mäta lymfocytproliferation och metoden har därför begränsad användning när det förekommer problem med att anskaffa, använda eller bortskaffa radioaktivitet. LLNA: BrdU-ELISA (Enzyme-Linked Immunosorbent Assay) är en icke-radioaktiv modifikation av LLNA-testmetoden och i den används icke-radiomärkt 5-bromo-2-deoxiuridin (BrdU) (CAS-nummer 59-14-3) i ett ELISA-baserat testsystem för mätning av lymfocytproliferation. LLNA: BrdU-ELISA har validerats, granskats och rekommenderats av en internationell oberoende expertutvärderingspanel som konstaterade att den kan användas för att identifiera hudsensibiliserande och icke-hudsensibiliserande kemikalier, med vissa begränsningar (10) (11) (12). Testmetoden är avsedd för bedömning av kemikaliers (ämnens och blandningars) potential att framkalla hudsensibilisering hos djur. Enligt kapitel B.6 i denna bilaga och i OECD:s testriktlinje 406 används test på marsvin, särskilt maximeringstest på marsvin och Buehler-test (13). LLNA (kapitel B.42 i denna bilaga; OECD:s testriktlinje 429) och de två icke-radioaktiva modifikationerna LLNA: BrdU-ELISA (kapitel B.51 i denna bilaga; OECD:s testriktlinje 442 B) och LLNA: DA (kapitel B.50 i denna bilaga; OECD:s testriktlinje 442 A) ger fördelar jämfört med marsvinstest enligt B.6 och OECD:s testriktlinje 406 (13) genom möjligheten att minska antalet djur och behandla dem på ett skonsammare sätt.
|
|
2.
|
I likhet med LLNA används LLNA: BrdU-ELISA för att undersöka hudsensibiliseringens induktionsfas, vilket ger tillgång till kvantitativa data som är lämpliga för dos-responsbedömning. Vidare gäller att de här metodernas förmåga att upptäcka hudsensibiliserande kemikalier utan att det behövs radiomärkning för DNA eliminerar risken för yrkesmässig exponering för radioaktivitet och problem rörande bortskaffning av avfall. Detta kan i sin tur ge möjlighet till ökad användning av möss för att upptäcka hudsensibiliserande ämnen, vilket ytterligare kan minska användningen av marsvin för att testa kemikaliers hudsensibiliserande potential (dvs. B.6; OECD:s testriktlinje 406) (13).
|
DEFINITIONER
|
3.
|
Definitioner av begreppen finns i tillägg 1.
|
INLEDANDE ÖVERVÄGANDEN OCH BEGRÄNSNINGAR
|
4.
|
LLNA: BrdU-ELISA är en modifierad LLNA-metod för identifiering av kemikalier med potential att framkalla hudsensibilisering, med specifika begränsningar. Detta betyder dock inte nödvändigtvis att LLNA: BrdU-ELISA alltid ska användas i stället för LLNA eller test på marsvin (dvs. B.6; OECD:s testriktlinje 406) (13), utan att metoden är likvärdig och kan användas som ett alternativ i fall där positiva och negativa resultat i regel inte kräver någon ytterligare bekräftelse (10) (11). Testningslaboratoriet bör analysera all tillgänglig information om testämnet innan själva testningen genomförs. Sådan information inbegriper ämnets identitet och kemiska struktur, fysikalisk-kemiska egenskaper, resultaten från andra eventuella in vitro- eller in vivo-test avseende toxicitet samt toxikologiska data om strukturellt närbesläktade kemikalier. Denna information bör övervägas för att kunna fastställa om LLNA: BrdU-ELISA-metoden är lämplig för testämnet (mot bakgrund av att metoden inte kan användas för vissa typer av kemikalier, se punkt 5) och som stöd för valet av dos.
|
|
5.
|
LLNA: BrdU-ELISA är en in vivo-metod, och därför kan man inte undvika användningen av djur för bedömning av sensibilisering genom allergisk kontakt. Metoden ger dock möjligheter att använda färre djur för detta ändamål i jämförelse med test på marsvin (B.6; OECD:s testriktlinje 406) (13). Dessutom ger LLNA: BrdU-ELISA möjlighet till en betydligt skonsammare behandling av djuren för testning av sensibilisering genom allergisk kontakt, eftersom man med LLNA: BrdU-ELISA, till skillnad från B.6 och OECD:s testriktlinje 406, inte behöver framkalla provokationsinducerade dermala hypersensitivitetsreaktioner. Dessutom behöver adjuvant inte användas, vilket är fallet vid maximeringstest på marsvin (kapitel B.6 i denna bilaga, 13). LLNA: BrdU-ELISA är därför en mindre plågsam metod för djuren. Trots fördelarna med LLNA: BrdU-ELISA i jämförelse med B.6 och OECD:s testriktlinje 406 (13) finns det vissa begränsningar som gör att man kanske måste använda B.6 eller OECD:s testriktlinje 406 (t.ex. testning av vissa metaller, falska positiva resultat med vissa hudirriterande ämnen (såsom vissa ytaktiva ämnen (6) (1 och kapitel B.42 i denna bilaga), testämnets löslighet)). Dessutom kan det för kemiska klasser eller ämnen som har funktionella grupper som påvisats agera som potentiella störfaktorer (15) vara nödvändigt att använda test på marsvin (dvs. B.6; OECD:s testriktlinje 406 (13)). Det rekommenderas att de begränsningar som anges för LLNA (1 och kapitel B.42 i denna bilaga) även ska gälla för LLNA: BrdU-ELISA (10). Utöver sådana identifierade begränsningar bör LLNA: BrdU-ELISA gå att använda för testning av alla kemikalier, utom om en kemikalie förknippas med egenskaper som kan påverka LLNA: BrdU-ELISA-metodens exakthet. Dessutom bör överväganden göras rörande möjligheten till positiva gränsresultat, när man erhåller värden för stimulationsindex mellan 1,6 och 1,9 (se punkterna 31–32). Detta grundar sig på valideringsdatabasen med 43 ämnen och användning av SI ≥ 1,6 (se punkt 6) för vilka LLNA: BrdU-ELISA ger korrekt identifiering av alla 32 sensibiliserande ämnen enligt LLNA men felaktig identifiering av 2 av 11 icke-sensibiliserande ämnen enligt LLNA med SI-värden mellan 1,6 och 1,9 (dvs. på gränsen till positivt) (10). Eftersom samma datauppsättning användes för att fastställa SI-värdena och beräkna testmetodens förutsägande egenskaper kan dock de angivna resultaten vara en överuppskattning av de faktiska förutsägande egenskaperna.
|
PRINCIP FÖR TESTMETODEN
|
6.
|
LLNA: BrdU-ELISA bygger på principen om att sensibiliserande ämnen inducerar en primär proliferation av lymfocyter i de lymfnoder som dränerar det område där testämnet appliceras. Denna proliferation står i proportion till den applicerade dosen och det allergiframkallande ämnets styrka, och är ett enkelt sätt att få en kvantitativ mätning av sensibiliseringen. Proliferationen mäts genom att jämföra medelproliferationen i varje testgrupp med medelproliferationen i den vehikelbehandlade kontrollgruppen. Förhållandet mellan medelproliferationen i respektive behandlingsgrupp och den parallella vehikelkontrollgruppen, kallat stimulationsindex (SI), bestäms och detta bör vara ≥ 1,6 för att det ska vara motiverat att klassificera testämnet som potentiellt hudsensibiliserande ämne. De förfaranden som beskrivs här grundar sig på mätning av BrdU-innehållet för att indikera ett ökat antal prolifererande celler i de aurikulära dränerande lymfnoderna. BrdU är ett analogt ämne med tymidin och inkorporeras på liknande sätt in i DNA i prolifererande celler. Inkorporeringen av BrdU mäts med ELISA med hjälp av en antikropp som är specifik för BrdU som också märks med peroxidas. När substratet tillsätts reagerar peroxidas med detta och det uppstår en färgad produkt som kvantifieras vid en specifik absorbans genom avläsning av en mikrotiterplatta.
|
TESTBESKRIVNING
Val av djurart
|
7.
|
För detta test används mus. Valideringsstudier för LLNA: BrdU-ELISA genomfördes uteslutande med stammen CBA/JN som därför anses vara den stam som är att föredra (10) (12). I testet används unga vuxna mushonor som inte har fött ungar och som inte är dräktiga. När försöket inleds bör djuren vara 8–12 veckor gamla. Djurens viktvariation bör vara minimal och får inte överskrida 20 % av medelvikten. Övriga stammar eller hanar kan användas som alternativ förutsatt att det finns tillräckliga data som stöd för att det inte förekommer några betydande stam- eller könsberoende skillnader i LLNA: BrdU-ELISA-respons.
|
Inhysnings- och utfodringsförhållanden
|
8.
|
Mössen ska inhysas i grupper (16), utom om det finns tillbörlig vetenskaplig motivering för individuell inhysning. Temperaturen i testdjurens utrymmen bör vara 22 ± 3 °C. Den relativa fuktigheten bör vara minst 30 % och ska helst inte överstiga 70 %, utom när rummet rengörs. Målvärdet bör dock vara 50–60 %. Belysningen bör vara artificiell med omväxlande 12 timmar ljus och 12 timmar mörker. För utfodringen kan konventionellt laboratoriefoder användas, och djuren bör ha obegränsad tillgång till dricksvatten.
|
Förberedelse av djuren
|
9.
|
Djuren väljs ut slumpmässigt och förses med märkning så att de kan identifieras individuellt (öronmärkning får dock inte användas). Därefter får djuren acklimatisera sig till laboratoriets förhållanden i sina burar i minst fem dagar före doseringsstart. Innan behandlingen inleds undersöks alla djur för att säkerställa att de inte har några synliga hudskador.
|
Förberedelse av doseringslösningar
|
10.
|
Kemikalier i fast form ska upplösas eller suspenderas i lösningsmedel/vehikel och vid behov spädas ut före appliceringen på mössens öron. Kemikalier i vätskeform kan appliceras i ren eller utspädd form. Olösliga kemikalier, av den typ som ofta används i medicintekniska produkter, ska extraheras kraftigt i lämpligt lösningsmedel så att alla extraherbara beståndsdelar för testning fås ut. Testlösningarna ska beredas dagligen utom om stabilitetsdata visar att lagring kan accepteras.
|
Tillförlitlighetskontroll
|
11.
|
Positiva kontrollkemikalier används för att påvisa att testsystemet har tillräcklig och reproducerbar känslighet för ett sensibiliserande testämne och en respons med välkarakteriserad styrka. Användningen av en parallell positiv kontroll rekommenderas eftersom den påvisar laboratoriets kompetens att genomföra testningar på ett framgångsrikt sätt och ger möjlighet till bedömning av reproducerbarhet och jämförbarhet inom laboratoriet och mellan laboratorier. Vissa tillsynsmyndigheter kräver också att positiv kontroll används för varje undersökning, och därför uppmuntras laboratorierna att samråda med relevanta myndigheter innan testning med LLNA: BrdU-ELISA inleds. Likaså uppmuntras rutinanvändning av parallell positiv kontroll för att undvika tilläggstestning på djur för att uppfylla krav som kan uppstå från användningen av periodisk positiv kontroll (se punkt 12). Den positiva kontrollen bör ge en positiv LLNA: BrdU-ELISA-respons på den exponeringsnivå som förväntas ge en ökning av stimulationsindex (SI) som är större eller lika med 1,6 i förhållande till negativa kontrollgruppen. Dosen av den positiva kontrollkemikalien ska väljas så att den inte orsakar särskilt kraftig hudirritation eller systemisk toxicitet och att induktionen är reproducerbar men inte för kraftig (t.ex. SI > 14 anses vara för högt värde). Rekommenderade ämnen för positiv kontroll är 25-procentig hexylkanelaldehyd (CAS-nummer 101-86-0) och 25-procentig eugenol (CAS-nummer 97-53-0) i aceton-olivoljeblandning i förhållandet 4:1 (volym/volym). I vissa fall kan även andra positiva kontrollkemikalier som uppfyller de ovan angivna kriterierna användas, förutsatt att tillbörlig motivering kan ges.
|
|
12.
|
Även om användning av en parallell positiv kontrollgrupp rekommenderas kan det finnas betingelser där det är tillräckligt med periodisk testning (dvs. minst var sjätte månad) av den positiva kontrollen i laboratorier som regelbundet genomför LLNA: BrdU-ELISA (dvs. minst en gång i månaden) och som har en etablerad historisk databas över positiv kontroll som påvisar laboratoriets förmåga att få reproducerbara och exakta resultat från positiva kontroller. Tillräcklig kvalitet hos LLNA: BrdU-ELISA kan framgångsrikt påvisas genom att enhetliga positiva resultat erhålls från den positiva kontrollen vid minst tio oberoende testningar som genomförs inom en skälig tidsperiod (dvs. kortare än ett år).
|
|
13.
|
Parallell positiv kontrollgrupp ska alltid användas när det sker ändringar rörande testningsförfarandet (t.ex. ny personal, nya material och/eller reagenser, annorlunda utrustning, byte av testdjurens ursprung), och sådana ändringar bör dokumenteras i laboratorierapporter. Man bör överväga om dessa ändringar påverkar den tidigare etablerade historiska databasens lämplighet, för att kunna avgöra om det behövs en ny historisk databas för dokumentering av den positiva kontrollens resultatenhetlighet.
|
|
14.
|
Man bör vara medveten om att beslut om att använda positiva kontroller med vissa intervaller i stället för parallellt har återverkningar på lämpligheten och godtagbarheten hos negativa undersökningsresultat som fås utan parallell positiv kontroll under den tid som löper mellan de periodiska undersökningarna med positiv kontroll. Om man t.ex. får falska negativa resultat vid ett periodiskt test av positiv kontroll, måste man kanske ifrågasätta resultat som erhållits mellan det sista godtagbara periodiska testet med positiv kontroll och ett icke-godtagbart periodiskt test med positiv kontroll. Följderna av dessa utfall bör övervägas noggrant när man avgör huruvida parallella positiva kontroller ska användas eller om de positiva kontrollerna enbart ska vara periodiska. Likaså bör man överväga användning av färre djur i den parallella positiva kontrollgruppen när detta är vetenskapligt motiverat och laboratoriet med stöd av laboratoriespecifika historiska data kan påvisa att färre möss kan användas (17).
|
|
15.
|
Även om det positiva kontrollämnet bör testas i en vehikel som veterligen framkallar en enhetlig respons (t.ex. aceton-olivoljeblandning i förhållandet 4:1 (volym/volym)), kan det förekomma vissa föreskriftsreglerade situationer där det även behövs testning i en icke-standardmässig vehikel (kliniskt och kemiskt relevant formulering) (18). Om det parallella positiva kontrollämnet testas i en annan vehikel än den som används för testämnet, bör en separat kontrollgrupp användas för den parallella positiva kontrollen.
|
|
16.
|
I fall där utvärdering görs av testämnen av en viss kemisk klass eller med en viss responsskala kan det också vara värdefullt med referensämnen för att påvisa att testmetoden fungerar tillfredsställande för bestämning av hudsensibiliseringspotentialen hos dessa typer av testämnen. Ett lämpligt referensämne ska ha följande egenskaper:
|
—
|
Strukturell och funktionell likhet med den ämnesklass som testas.
|
|
—
|
Kända fysikalisk-kemiska egenskaper.
|
|
—
|
Stödjande data från LLNA: BrdU-ELISA.
|
|
—
|
Stödjande data från andra djurmodeller och/eller från människor.
|
|
TESTFÖRFARANDE
Antalet djur och dosnivåer
|
17.
|
Minst fyra djur per dosgrupp och minst tre koncentrationer av testämnet bör användas. Dessutom används en parallell negativ kontrollgrupp som endast behandlas med samma vehikel som används för testämnet och en grupp för positiv kontroll (parallell eller nyligen gjord, enligt laboratoriets principer för överväganden enligt punkterna 11–15). Man bör överväga flera upprepade doser av det positiva kontrollämnet, särskilt när den positiva kontrollen testas periodiskt. Med undantag för behandling med testämnet bör djuren i kontrollgrupperna hanteras på exakt samma sätt som djuren i dosgrupperna.
|
|
18.
|
Doser och vehikel väljs på grundval av rekommendationerna i hänvisningarna 2 och 19. Doserna väljs i regel enligt en lämplig koncentrationsserie såsom 100 %, 50 %, 25 %, 10 %, 5 %, 2,5 %, 1 %, 0,5 %, osv. Valet av koncentrationsserie ska ges tillbörlig vetenskaplig motivering. All existerande tillgänglig och relevant toxikologisk information (t.ex. akut toxicitet och hudirritation) samt strukturell och fysikalisk-kemisk information om testämnet (och/eller strukturellt besläktade ämnen) bör övervägas vid valet av de tre på varandra följande koncentrationerna så att den högsta koncentrationen ger maximal exponering samtidigt som systemisk toxicitet och särskilt kraftig lokal hudirritation undviks (19) (20 och kapitel B.4 i denna bilaga). Om sådan information inte finns kan det vara nödvändigt att först genomföra ett preliminärt screeningtest (se punkterna 21–24).
|
|
19.
|
Vehikeln får inte störa eller snedvrida testresultaten och ska väljas med tanke på största möjliga löslighet för att få högsta möjliga koncentration samtidigt som man får en lösning/suspension som är lämplig för applicering av testämnet. Som vehikel rekommenderas aceton-olivoljeblandning (4:1 volym/volym), N,N-dimetylformamid, metyletylketon, propylenglykol och dimetylsulfoxid (6) men även andra vehiklar kan användas förutsatt att tillräcklig vetenskaplig motivering kan ges. I vissa situationer kan det vara nödvändigt att som extra kontroll använda ett kliniskt relevant lösningsmedel eller en kommersiellt tillgänglig blandning där testämnet ingår. Särskild vikt bör fästas vid att se till att hydrofila testämnen inkorporeras i ett vehikelsystem som väter huden och som inte omedelbart rinner bort, genom att använda lämpliga solubiliserande ämnen (t.ex. 1-procentig Pluronic® L92). Det betyder att helt vattenbaserade vehiklar bör undvikas.
|
|
20.
|
Genom analys av lymfnoder från individuella möss kan man bestämma variationer djuren emellan och göra en statistisk jämförelse av skillnaden mellan mätningar för testämnet och mätningarna i vehikelkontrollgruppen (se punkt 33). Dessutom kan möjligheten att minska antalet möss i den positiva kontrollgruppen endast övervägas utifrån att data samlas in individuellt per djur (17). Vidare gäller att vissa tillsynsmyndigheter kräver att data insamlas per individuellt djur. Regelbunden insamling av individuella data per djur ger en fördel rörande djurens välbefinnande eftersom upprepad testning undviks som annars skulle behövas om testämnesresultat som ursprungligen har samlats in på ett visst sätt (t.ex. genom poolade djurdata) måste granskas senare av tillsynsmyndigheter som har avvikande krav (såsom data per individuellt djur).
|
Preliminärt screeningtest
|
21.
|
Om det inte finns tillgång till information för att bestämma högsta testdosen (se punkt 18) bör ett preliminärt screeningtest genomföras för att definiera den lämpliga dosnivån för test i LLNA: BrdU-ELISA. Syftet med det preliminära screeningtestet är att få vägledning för valet av högsta dosnivå i huvudundersökningen, i fall där information saknas om vilken koncentration som inducerar systemisk toxicitet (se punkt 24) och/eller särskilt kraftig lokal hudirritation (se punkt 23). Den högsta dosnivå som testas ska vara 100 % testämne för vätskor eller högsta möjliga koncentration för fasta ämnen eller suspensioner.
|
|
22.
|
Det preliminära screeningtestet genomförs under identiska betingelser som vid den huvudsakliga LLNA: BrdU-ELISA-undersökningen, utom att man inte utvärderar proliferationen i lymfnoderna och man kan använda färre djur i dosgruppen. Ett eller två djur per dosgrupp föreslås. Alla möss observeras dagligen för kliniska tecken på systemisk toxicitet eller lokal irritation på appliceringsstället. Djurens kroppsvikt registreras före testningen och efter avslutad testning (dag 6). För varje mus observeras båda öronen för hudrodnad och graderas enligt tabell 1 (20 och kapitel B.4 i denna bilaga). Mätning av örontjocklek görs t.ex. med digital mikrometer eller Peacock Dial-instrument på dag 1 (före dosering), dag 3 (cirka 48 timmar efter den första dosen) och på dag 6. Dessutom kan örontjockleken bestämmas på dag 6 genom vägning av en öronbit som stansas ut efter skonsam avlivning. Särskilt kraftig lokal hudirritation indikeras genom hudrodnadsgradering ≥ 3 och/eller ökning av örontjockleken med ≥ 25 % på någon av mätningsdagarna (21) (22). Den högsta dos som väljs för huvudundersökningen blir nästa lägre dos i den preliminära screeningens koncentrationsserie (se punkt 18) som inte inducerar systemisk toxicitet och/eller särskilt kraftig hudirritation.
Tabell 1
Grader av hudrodnad
|
Observation
|
Gradering
|
|
Ingen hudrodnad
|
0
|
|
Mycket svag rodnad (knappt skönjbar)
|
1
|
|
Väldefinierad hudrodnad
|
2
|
|
Moderat till svår hudrodnad
|
3
|
|
Svår hudrodnad (köttrodnad) till sårskorpebildning som utgör hinder för gradering av rodnaden
|
4
|
|
|
23.
|
För identifiering av irriterande ämnen med LLNA har man utöver en 25-procentig ökning av örontjocklek (21) (22) också använt statistiskt signifikant ökning av örontjockleken hos behandlingsgruppens möss i jämförelse med kontrollgruppens möss (22) (23) (24) (25) (26) (27) (28). Dock så gäller att även om statistiskt signifikanta ökningar kan inträffa när ökningen av örontjocklek är mindre än 25 %, har dessa inte specifikt förknippats med särskilt kraftig irritation (25) (26) (27) (28) (29).
|
|
24.
|
Följande kliniska observationer kan indikera systemisk toxicitet (30) när de används som del av en helhetsbedömning och kan således ge indikation om den högsta dosnivå som ska användas i huvudundersökningen: ändringar av nervsystemets funktion (t.ex. upprest päls, ataxi, skälvningar och konvulsioner), ändringar av beteende (t.ex. aggressivitet, ändringar av putsningsbeteende, markerad ändring av aktivitetsnivå), ändringar av andningsmönster (dvs. ändringar av andningens frekvens och intensitet, såsom dyspné, gäspande och rassel) samt ändringar av födo- och vattenintag. Vid utvärderingen ska man också beakta tecken på letargi och/eller avsaknad av reaktioner, alla slags kliniska tecken på mer än lätt eller momentan smärta eller lidande, minskning av kroppsvikten med mer än 5 % från dag 1 till dag 6 samt mortalitet. Döende djur och djur som visar tecken på svår smärta och lidande ska avlivas skonsamt (31).
|
Huvudundersökningens försöksschema
|
25.
|
För testet används följande försöksschema:
— Dag 1: Djuren märks individuellt och vikten samt eventuella kliniska observationer registreras. Applicera 25 μl lämpligt utspätt testämne, ren vehikel eller positivt kontrollämne (parallell eller nyligen utförd positiv kontroll, i enlighet med laboratoriets principer när det gäller punkterna 11–15) på båda öronens baksida.
— Dagarna 2 och 3: Upprepa appliceringen enligt dag 1.
— Dag 4: Ingen behandling.
— Dag 5: Injicera 0,5 ml (5 mg/mus) BrdU-lösning (10 mg/ml) intraperitonealt.
— Dag 6: Registrera vikten för varje djur och eventuella kliniska observationer. Avliva djuren skonsamt cirka 24 timmar efter BrdU-injektionen. Skär bort de aurikulära dränerande lymfnoderna från varje musöra och behandla separat i fosfatbuffrad saltlösning (PBS) för varje djur. Närmare uppgifter och scheman för identifiering och dissekering av noder finns i hänvisning (17). För ytterligare kontroll av lokal hudrespons i huvudundersökningen kan tilläggsparametrar såsom gradering av örats hudrodnad eller mätningar av örats tjocklek (med tjockleksmätare eller genom vägning av utstansad bit vid obduceringen) införlivas i undersökningsprotokollet.
|
Beredning av cellsuspensioner
|
26.
|
För varje mus bereds en suspension med enskilda celler av lymfnodsceller (LNC) från de bilateralt bortskurna lymfnoderna genom försiktig mekanisk sönderdelning med hjälp av ett nät av rostfritt stål (200 mesh) eller annan godkänd teknik för beredning av en suspension med enskilda celler (t.ex. sönderdelning av lymfnoderna i en engångsmortel av plast åtföljt av passage genom ett nylonnät (70 mesh)). Förfarandet för beredning av LNC-suspensionen är kritiskt för detta test och därför bör varje laboratorium se till att färdigheterna övas in på förhand. Observera att lymfnoderna i den negativa kontrollgruppens djur är små, och det är viktigt att iaktta stor försiktighet för att undvika störande inverkan på SI-värdena. För varje fall ska målvolymen hos LNC-suspensionen justeras till en bestämd optimerad volym (cirka 15 ml). Den optimerade volymen baserar sig på att man för den negativa kontrollen ska få en genomsnittsabsorbans inom 0,1–0,2.
|
Bestämning av cellproliferation (mätning av BrdU-innehållet i lymfocyternas DNA)
|
27.
|
BrdU mäts i ELISA med hjälp av ett kommersiellt tillgängligt kit (t.ex. Roche Applied Science, Mannheim, Tyskland, katalognummer 11 647 229 001). Mätningen går i huvuddrag ut på att 100 μl LNC-suspension överförs till brunnarna i en flatbottnad mikrotiterplatta i tre uppsättningar. Efter fixering och denaturering av LNC tillsätts anti-BrdU-antikropp till varje brunn och tillåts reagera. Därefter avlägsnas anti-BrdU-antikroppen genom tvättning och substratlösning tillsätts så att kromogen kan bildas. Därefter mäts absorbansen vid 370 nm med en referensvåglängd på 492 nm. Under alla omständigheter gäller att testningsbetingelserna bör optimeras (se punkt 26).
|
OBSERVATIONER
Kliniska observationer
|
28.
|
Varje mus bör observeras minst dagligen med tanke på eventuella kliniska tecken – lokal irritation vid appliceringsstället eller tecken på systemisk toxicitet. Alla observationer registreras systematiskt och individuellt per mus. Övervakningsplanerna bör inbegripa kriterier för omedelbar identifiering av tecken på systemisk toxicitet, särskilt kraftig hudirritation eller hudkorrosion så att berörda djur kan avlivas (31).
|
Kroppsvikt
|
29.
|
Enligt det som anges i punkt 25 ska de individuella djurens vikt registreras vid försökets början och strax före den schemalagda avlivningen.
|
BERÄKNING AV RESULTATEN
|
30.
|
Resultaten för varje behandlingsgrupp uttrycks som genomsnittet för stimulationsindex. Stimulationsindex erhålls genom att genomsnittligt värde för BrdU-märkningsindex/djur inom varje testämnesgrupp och den positiva kontrollgruppen divideras med genomsnittligt värde för BrdU-märkningsindex för kontrollgruppen för lösningsmedel/vehikel. Det genomsnittliga stimulationsindexet för vehikelkontrollgrupperna har då värdet 1.
BrdU-märkningsindex definieras enligt följande:
BrdU-märkningsindex = (ABSem – ABS blankem) – (ABSref – ABS blankref)
där em = emissionsvåglängd och ref = referensvåglängd.
|
|
31.
|
Vid bedömning av resultaten anses ett resultat vara positivt när SI ≥ 1,6 (10). Man kan dock även använda styrkan på dos-responsförhållandets samt den statistiska signifikansen och enhetligheten hos responsen i kontrollgruppen för lösningsmedel/vehikel och i den positiva kontrollen när man fastställer huruvida ett gränsresultat (dvs. SI-värde mellan 1,6 och 1,9) ska anses vara positivt (3) (6) (32).
|
|
32.
|
För ett gränsresultat för positiv respons där SI-värdet ligger mellan 1,6 och 1,9 vill man kanske beakta tilläggsinformation såsom dos-responsförhållandet, tecken på systemisk toxicitet eller särskilt kraftig irritation och, där det är lämpligt, statistisk signifikans tillsammans med SI-värden för att bekräfta att resultaten är positiva (10). Likaså bör man beakta testämnets olika egenskaper, inbegripet eventuell strukturell släktskap med kända hudsensibiliserande ämnen, huruvida ämnet orsakar särskilt kraftig hudirritation hos mössen samt naturen hos det iakttagna förhållandet mellan dos och respons. Dessa och andra aspekter diskuteras närmare i andra källor (4).
|
|
33.
|
När data samlas in per enskild mus kan man göra en statistisk analys rörande förekomsten av dosrespons och graden för dos-responsförhållande. Alla statistiska analyser kan inbegripa en utvärdering av dos-responsförhållandet och en lämpligt anpassad jämförelse mellan testgrupper (t.ex. parvisa jämförelser mellan doseringsgrupp och parallell kontrollgrupp för lösningsmedel/vehikel). De statistiska analyserna kan inbegripa t.ex. linjär regression eller Williams test för utvärdering av dos-responstrender och Dunnetts test för parvisa jämförelser. Vid valet av en lämplig metod för den statistiska analysen bör man vara medveten om eventuella olikheter hos varianserna och andra relaterade problem som kan göra det nödvändigt att göra en datatransformation eller en icke-parametrisk statistisk analys. Det kan också vara nödvändigt att genomföra beräkningar av stimulationsindex och statistiska analyser med och utan vissa datapunkter (s.k. outliers – utanförliggande värden).
|
DATA OCH RAPPORTERING
Data
|
34.
|
Data sammanställs i tabellform med angivelse av värdena för BrdU-märkningsindex per djur, gruppens medelvärde för BrdU-märkningsindex per djur, den associerade feltermen (t.ex. SD, SEM) och medelvärdet för stimulationsindex för varje dosgrupp jämfört med den parallella kontrollgruppen för lösningsmedel/vehikel.
|
Testrapport
|
35.
|
Testrapporten ska innehålla nedan angivna uppgifter.
|
|
Test- och kontrollkemikalier:
|
—
|
Identifieringsdata (t.ex. CAS-nummer och EG-nummer, om tillgängligt, källa, renhet, kända orenheter, satsnummer).
|
|
—
|
Fysikalisk natur och fysikalisk-kemiska egenskaper (t.ex. flyktighet, stabilitet, löslighet).
|
|
—
|
För blandningar anges sammansättning och beståndsdelarnas relativa procentandelar.
|
|
|
|
Lösningsmedel/vehikel:
|
—
|
Identifieringsdata (renhet, koncentration (om tillämpligt), volym som har använts).
|
|
—
|
Motivering för val av vehikel.
|
|
|
|
Testdjur:
|
—
|
Mikrobiologiska data för djuren, om dessa data är kända.
|
|
—
|
Antal djur och deras ålder.
|
|
—
|
Ursprung, inhysningsförhållanden, foder och dylikt.
|
|
|
|
Testbetingelser:
|
—
|
ELISA-satsens ursprung, satsnummer och kvalitetsförsäkran/kvalitetskontrolldata (antikroppskänslighet samt detektionsspecificitet och -gräns).
|
|
—
|
Detaljerade uppgifter om beredning och applicering av testämnet.
|
|
—
|
Motivering för valet av dos (även resultat från preliminär screening om sådan har genomförts).
|
|
—
|
Koncentrationerna för vehikel och testämne samt totala mängden testämne som applicerats.
|
|
—
|
Detaljerade uppgifter om foder- och vattenkvalitet (inbegripet diettyp/källa, vattenkälla).
|
|
—
|
Detaljerad beskrivning av behandlings- och provtagningsscheman.
|
|
—
|
Metoder för mätningar av toxicitet.
|
|
—
|
Kriterier för att fastställa om resultatet är positivt eller negativt.
|
|
—
|
Uppgifter rörande eventuella avvikelser från protokollet och redogörelse för hur avvikelsen påverkar undersökningens utformning och resultat.
|
|
|
|
Tillförlitlighetskontroll:
|
—
|
Sammanfattning av resultaten från senaste tillförlitlighetskontroll, även information om testämne, koncentration och vehikel.
|
|
—
|
Data från parallella och/eller historiska positiva kontroller och parallella negativa kontroller (lösningsmedel/vehikel) för testningslaboratoriet.
|
|
—
|
Om försöket inte har omfattat parallell positiv kontroll, ange datum och laboratorierapport för senaste periodiska positiva kontroll och en rapport med uppgifter om laboratoriets historiska data för positiva kontroller som motivering för varför parallell positiv kontroll inte har använts.
|
|
|
|
Resultat:
|
—
|
Mössens individuella vikt vid doseringsstart och vid schemalagd skonsam avlivning samt medelvärde och associerad felterm (t.ex. SD, SEM) för varje behandlingsgrupp.
|
|
—
|
Tidpunkten för första upptäckt av tecken på toxicitet, även eventuell hudirritation vid appliceringsstället, för varje försöksdjur.
|
|
—
|
En tabell med BrdU-märkningsindex per mus och SI-värden per behandlingsgrupp.
|
|
—
|
Medelvärde och associerad felterm (t.ex. SD, SEM) för BrdU-märkningsindex per mus för varje behandlingsgrupp och resultaten från analys av utanförliggande värden för varje behandlingsgrupp.
|
|
—
|
Beräknat stimulationsindex och ett lämpligt mått på variabilitet som beaktar variationen mellan djuren i testämnesgruppen och kontrollgruppen.
|
|
—
|
Förhållandet mellan dos och respons.
|
|
—
|
Statistisk analys, om tillämpligt.
|
|
|
|
Diskussion om resultaten:
|
—
|
En kort kommentar om resultaten, dos-responsanalys och statistiska analyser (i tillämpliga fall), med en slutsats om huruvida testämnet ska anses vara hudsensibiliserande.
|
|
|
HÄNVISNINGAR
|
(1)
|
OECD (2010), Skin Sensitisation: Local Lymph Node Assay, Test Guideline No. 429, Guidelines for the Testing of Chemicals, OECD, Paris. Finns på: http://www.oecd.org/env/testguidelines
|
|
(2)
|
Chamberlain, M. och Basketter, D.A. (1996), The local lymph node assay: status of validation. Food Chem. Toxicol., 34, 999–1002.
|
|
(3)
|
Basketter, D.A., Gerberick, G.F., Kimber, I. och Loveless, S.E. (1996), The local lymph node assay: A viable alternative to currently accepted skin sensitisation tests. Food Chem. Toxicol., 34, 985–997.
|
|
(4)
|
Basketter, D.A., Gerberick, G.F. och Kimber, I. (1998), Strategies for identifying false positive responses in predictive sensitisation tests. Food Chem. Toxicol., 36, 327–33.
|
|
(5)
|
Van Och, F.M.M., Slob, W., De Jong, W.H., Vandebriel, R.J. och Van Loveren, H. (2000), A quantitative method for assessing the sensitising potency of low molecular weight chemicals using a local lymph node assay: employment of a regression method that includes determination of uncertainty margins. Toxicol., 146, 49–59.
|
|
(6)
|
ICCVAM (1999), The murine local lymph node Assay: A test method for assessing the allergic contact dermatitis potential of chemicals/compounds: The results of an independent peer review evaluation coordinated by the Interagency Coordinating Committee on the Validation of Alternative Methods (ICCVAM) and the National Toxicology Program Center for the Evaluation of Alternative Toxicological Methods (NICETAM). NIH Publication No: 99–4494. Research Triangle Park, N.C. Finns på: http://iccvam.niehs.nih.gov/docs/immunotox_docs/llna/llnarep.pdf
|
|
(7)
|
Dean, J.H., Twerdok, L.E., Tice, R.R., Sailstad, D.M., Hattan, D.G., Stokes, W.S. (2001), ICCVAM evaluation of the murine local lymph node assay: II. Conclusions and recommendations of an independent scientific peer review panel. Reg. Toxicol. Pharmacol., 34(3), 258–273.
|
|
(8)
|
Haneke, K.E., Tice, R.R., Carson, B.L., Margolin, B.H., Stokes, W.S. (2001), ICCVAM evaluation of the murine local lymph node assay: III. Data analyses completed by the national toxicology program interagency center for the evaluation of alternative toxicological methods. Reg. Toxicol. Pharmacol., 34(3), 274–286.
|
|
(9)
|
Sailstad, D.M., Hattan, D., Hill, R.N., Stokes, W.S. (2001), ICCVAM evaluation of the murine local lymph node assay: I. The ICCVAM review process. Reg. Toxicol. Pharmacol., 34(3), 249–257.
|
|
(10)
|
ICCVAM (2010), ICCVAM Test Method Evaluation Report. Nonradioactive local lymph node assay: BrdU-ELISA Test Method Protocol (LLNA: BrdU-ELISA). NIH Publication No. 10–7552A/B. Research Triangle Park, NC: National Institute of Environmental Health Sciences. Finns på: http://iccvam.niehs.nih.gov/methods/immunotox/llna-ELISA/TMER.htm
|
|
(11)
|
ICCVAM (2009), Independent Scientific Peer Review Panel Report: Updated validation status of new versions and applications of the murine local lymph node assay: a test method for assessing the allergic contact dermatitis potential of chemicals and products. Research Triangle Park, NC: National Institute of Environmental Health Sciences. Finns på: http://iccvam.niehs.nih.gov/docs/immunotox_docs/LLNAPRPRept2009.pdf
|
|
(12)
|
Takeyoshi, M., Iida, K., Shiraishi, K. och Hoshuyama, S. (2005), Novel approach for classifying chemicals according to skin sensitising potency by non-radioisotopic modification of the local lymph node assay. J. Appl. Toxicol., 25, 129–134.
|
|
(13)
|
OECD (1992), Skin Sensitisation, Test Guideline No. 406, Guidelines for Testing of Chemicals, OECD, Paris. Finns på: http://www.oecd.org/env/testguidelines
|
|
(14)
|
Kreiling, R., Hollnagel, H.M., Hareng, L., Eigler, L., Lee, M.S., Griem, P., Dreessen, B., Kleber, M., Albrecht, A., Garcia, C. och Wendel, A. (2008), Comparison of the skin sensitising potential of unsaturated compounds as assessed by the murine local lymph node assay (LLNA) and the guinea pig maximization test (GPMT). Food Chem. Toxicol., 46, 1896–1904.
|
|
(15)
|
Basketter, D., Ball, N., Cagen, S., Carrilo, J.C., Certa, H., Eigler, D., Garcia, C., Esch, H., Graham, C., Haux, C., Kreiling, R. och Mehling, A. (2009), Application of a weight of evidence approach to assessing discordant sensitisation datasets: implications for REACH. Reg. Toxicol. Pharmacol., 55, 90–96.
|
|
(16)
|
ILAR (1996), Institute of Laboratory Animal Research (ILAR) Guide for the Care and Use of Laboratory Animals. 7th ed. Washington, DC: National Academies Press.
|
|
(17)
|
ICCVAM (2009), Recommended Performance Standards: Murine Local Lymph Node Assay. NIH Publication Number 09–7357. Research Triangle Park, NC: National Institute of Environmental Health Sciences. Finns på: http://iccvam.niehs.nih.gov/docs/immunotox_docs/llna-ps/LLNAPerfStds.pdf
|
|
(18)
|
McGarry, H.F. (2007), The murine local lymph node assay: regulatory and potency considerations under REACH. Toxicol., 238, 71–89.
|
|
(19)
|
Kimber, I., Dearman, R.J., Scholes E.W. och Basketter, D.A. (1994), The local lymph node assay: developments and applications. Toxicol., 93, 13–31.
|
|
(20)
|
OECD (2002), Acute Dermal Irritation/Corrosion, Test Guideline No. 404, Guidelines for Testing of Chemicals, OECD, Paris. Finns på: http://www.oecd.org/env/testguidelines
|
|
(21)
|
Reeder, M.K., Broomhead, Y.L., DiDonato, L. och DeGeorge, G.L. (2007), Use of an enhanced local lymph node assay to correctly classify irritants and false positive substances. Toxicologist, 96, 235.
|
|
(22)
|
ICCVAM (2009), Nonradioactive Murine Local Lymph Node Assay: Flow Cytometry Test Method Protocol (LLNA: BrdU-FC) Revised Draft Background Review Document. Research Triangle Park, NC: National Institute of Environmental Health Sciences. Finns på: http://iccvam.niehs.nih.gov/methods/immunotox/fcLLNA/BRDcomplete.pdf.
|
|
(23)
|
Hayes, B.B., Gerber, P.C., Griffey, S.S. och Meade, B.J. (1998), Contact hypersensitivity to dicyclohexylcarbodiimide and diisopropylcarbodiimide in female B6C3F1 mice. Drug Chem. Toxicol., 21, 195–206.
|
|
(24)
|
Homey, B., von Schilling, C., Blumel, J., Schuppe, H.C., Ruzicka, T., Ahr, H.J., Lehmann, P. och Vohr, V.W. (1998), An integrated model for the differentiation of chemical-induced allergic and irritant skin reactions. Toxicol. Appl. Pharmacol., 153, 83–94.
|
|
(25)
|
Woolhiser, M.R., Hayes, B.B. och Meade, B.J. (1998), A combined murine local lymph node and irritancy assay to predict sensitisation and irritancy potential of chemicals. Toxicol. Meth., 8, 245–256.
|
|
(26)
|
Hayes, B.B. och Meade, B.J. (1999), Contact sensitivity to selected acrylate compounds in B6C3F1 mice: relative potency, cross reactivity, and comparison of test methods. Drug. Chem. Toxicol., 22, 491–506.
|
|
(27)
|
Ehling, G., Hecht, M., Heusener, A., Huesler, J., Gamer, A.O., van Loveren, H., Maurer, T., Riecke, K., Ullmann, L., Ulrich, P., Vandebriel, R. och Vohr, H.W. (2005), A European inter- laboratory validation of alternative endpoints of the murine local lymph node assay: first round. Toxicol., 212, 60–68.
|
|
(28)
|
Vohr, H.W. och Ahr, H.J. (2005), The local lymph node assay being too sensitive? Arch. Toxicol., 79, 721–728.
|
|
(29)
|
Patterson, R.M., Noga, E. och Germolec D. (2007), Lack of evidence for contact sensitisation by Pfiesteria extract. Environ. Health Perspect., 115, 1023–1028.
|
|
(30)
|
ICCVAM (2009), Report on the ICCVAM-NICEATM/ECVAM/JaCVAM Scientific Workshop on Acute Chemical Safety Testing: Advancing In Vitro Approaches and Humane Endpoints for Systemic Toxicity Evaluations. Research Triangle Park, NC: National Institute of Environmental Health Sciences. Finns på: http://iccvam.niehs.nih.gov/methods/acutetox/Tox_workshop.htm.
|
|
(31)
|
OECD (2000), Guidance Document on the Recognition, Assessment and Use of Clinical Signs as Humane Endpoints for Experimental Animals Used in Safety Evaluation, Environmental Health and Safety Monograph Series on Testing and Assessment No. 19, ENV/JM/MONO(2000)7, OECD, Paris. Finns på: http://www.oecd.org/env/testguidelines
|
|
(32)
|
Kimber, I., Hilton, J., Dearman, R.J., Gerberick, G.F., Ryan, C.A., Basketter, D.A., Lea, L., House, R.V., Ladies, G.S., Loveless, S.E. och Hastings, K.L. (1998), Assessment of the skin sensitisation potential of topical medicaments using the local lymph node assay: An interlaboratory exercise. J. Toxicol. Environ.l Health, 53, 563–79.
|
|
(33)
|
OECD (2005), Guidance Document on the Validation and International Acceptance of New or Updated Test Methods for Hazard Assessment, Environment, Health and Safety Monograph Series on Testing and Assessment No. 34, ENV/JM/MONO(2005)14, OECD, Paris. Finns på: http://www.oecd.org/env/testguidelines
|
Tillägg 1
DEFINITIONER
Exakthet: Ett mått på hur väl testmetodresultaten och de godtagna referensvärdena överensstämmer med varandra. Exakthet är ett mått på testmetodens prestanda och en relevansaspekt. Detta begrepp används ofta omväxlande med ’konkordans’ för att beskriva andelen korrekta resultat med en testmetod (33).
Referensämne: Ett sensibiliserande eller icke-sensibiliserande ämne som används som standard för jämförelse med ett testämne. Ett referensämne ska ha följande egenskaper: i) en eller flera enhetliga och tillförlitliga källor, ii) strukturell och funktionell likhet med den ämnesklass som testas, iii) kända fysikalisk-kemiska egenskaper, iv) stödjande data om kända effekter och v) känd styrka inom det önskade responsområdet.
Falsk negativt: Ett testämne som felaktigt har identifierats som negativt eller icke-aktivt genom en testmetod när ämne i själva verket är positivt eller aktivt (33).
Falskt positivt: Ett testämne som felaktigt har identifierats som positivt eller aktivt genom en testmetod när ämne i själva verket är negativt eller icke-aktivt (33).
Fara: Potentialen för skada på hälsa eller miljö. De skadliga verkningarna visar sig enbart om exponeringsnivån är tillräcklig.
Reproducerbarhet mellan laboratorier: Ett mått på i hur stor utsträckning olika kvalificerade laboratorier kan producera kvalitativt och kvantitativt liknande resultat, med användning av samma protokoll och samma testämne. Reproducerbarheten mellan laboratorier fastställs under förvalideringen och valideringen och den indikerar i hur stor utsträckning ett test framgångsrikt kan överföras mellan laboratorier (33).
Reproducerbarhet inom laboratoriet: Ett mått på i vilken utsträckning kvalificerade personer inom samma laboratorium framgångsrikt kan upprepa resultat vid olika tidpunkter när de använder ett visst protokoll (33).
Utanförliggande värde: Ett utanförliggande värde (’outlier’) är en observation som markant skiljer sig från de övriga värdena i ett slumpmässigt urval från en population.
Kvalitetssäkring: En styrningsprocess där personer som är i oberoende ställning i förhållande till de personer som genomför testningen bedömer hur standarder, krav och registreringsförfaranden för laboratorietestning följs och hur exakt data förmedlas.
Tillförlitlighet: Ett mått på i vilken utsträckning en testmetod kan reproduceras inom och mellan laboratorier över tiden när metoden utförs med samma protokoll. Den bedöms genom att man beräknar reproducerbarheten inom och mellan laboratorier (33).
Hudsensibilisering: En immunologisk process som följer av att en mottaglig individ exponeras ytvägen för en inducerande kemisk allergen som provocerar en kutan immunrespons som kan leda till att det uppstår kontaktsensibilisering.
Stimulationsindex (SI): Ett värde som beräknas för att bedöma hudsensibiliseringspotentialen hos ett testämne och som är förhållandet mellan proliferationen i behandlingsgrupperna och proliferationen i den parallella vehikelkontrollgruppen.
Testämne (kallas också testkemikalie): Alla ämnen eller blandningar som testas med denna testmetod.
” |