ISSN 1725-2601
doi:10.3000/17252601.L_2009.220.por
Jornal Oficial
da União Europeia
L 220

Edição em língua portuguesa
Legislação
52.o ano
24 de Agosto de 2009
|
ISSN 1725-2601 doi:10.3000/17252601.L_2009.220.por |
||
|
Jornal Oficial da União Europeia |
L 220 |
|
|
|
||
|
Edição em língua portuguesa |
Legislação |
52.o ano |
|
Índice |
|
I Actos aprovados ao abrigo dos Tratados CE/Euratom cuja publicação é obrigatória |
Página |
|
|
|
REGULAMENTOS |
|
|
|
* |
Regulamento (CE) n.o 761/2009 da Comissão, de 23 de Julho de 2009, que altera, tendo em vista a adaptação ao progresso técnico, o Regulamento (CE) n.o 440/2008 que estabelece métodos de ensaio nos termos do Regulamento (CE) n.o 1907/2006 do Parlamento Europeu e do Conselho relativo ao registo, avaliação, autorização e restrição de substâncias químicas (REACH) ( 1 ) |
|
|
|
|
|
(1) Texto relevante para efeitos do EEE |
|
PT |
Os actos cujos títulos são impressos em tipo fino são actos de gestão corrente adoptados no âmbito da política agrícola e que têm, em geral, um período de validade limitado. Os actos cujos títulos são impressos em tipo negro e precedidos de um asterisco são todos os restantes. |
I Actos aprovados ao abrigo dos Tratados CE/Euratom cuja publicação é obrigatória
REGULAMENTOS
|
24.8.2009 |
PT |
Jornal Oficial da União Europeia |
L 220/1 |
REGULAMENTO (CE) N.o 761/2009 DA COMISSÃO
de 23 de Julho de 2009
que altera, tendo em vista a adaptação ao progresso técnico, o Regulamento (CE) n.o 440/2008 que estabelece métodos de ensaio nos termos do Regulamento (CE) n.o 1907/2006 do Parlamento Europeu e do Conselho relativo ao registo, avaliação, autorização e restrição de substâncias químicas (REACH)
(Texto relevante para efeitos do EEE)
A COMISSÃO DAS COMUNIDADES EUROPEIAS,
Tendo em conta o Tratado que institui a Comunidade Europeia,
Tendo em conta o Regulamento (CE) n.o 1907/2006 do Parlamento Europeu e do Conselho, de 18 de Dezembro de 2006, relativo ao registo, avaliação, autorização e restrição dos produtos químicos (REACH), que cria a Agência Europeia dos Produtos Químicos, que altera a Directiva 1999/45/CE e revoga o Regulamento (CEE) n.o 793/93 do Conselho e o Regulamento (CE) n.o 1488/94 da Comissão, bem como a Directiva 76/769/CEE do Conselho e as Directivas 91/155/CEE, 93/67/CEE, 93/105/CE e 2000/21/CE da Comissão (1), nomeadamente o n.o 3 do artigo 13.o,
Considerando o seguinte:
|
(1) |
O Regulamento (CE) n.o 440/2008 da Comissão (2) estabelece os métodos de ensaio a aplicar para os fins do Regulamento (CE) n.o 1907/2006 com vista à determinação das propriedades físico-químicas, da toxicidade e da ecotoxicidade das substâncias. |
|
(2) |
É necessário actualizar o Regulamento (CE) n.o 440/2008 modificando determinados métodos de ensaio e aditando vários métodos novos adoptados pela OCDE. Foram consultadas as partes interessadas sobre a presente proposta. Estas alterações adaptam os métodos em questão ao progresso científico e técnico. |
|
(3) |
Há que rever as disposições relativas à pressão de vapor de modo a incluir o novo método de efusão. |
|
(4) |
É necessário aditar um novo método de medição do diâmetro geométrico médio, ponderado em função do comprimento, das fibras. |
|
(5) |
Para reduzir o número de animais utilizados para fins experimentais, em conformidade com a Directiva 86/609/CEE do Conselho, de 24 de Novembro de 1986, relativa à aproximação das disposições legislativas, regulamentares e administrativas dos Estados-Membros respeitantes à protecção dos animais utilizados para fins experimentais e outros fins científicos (3), é conveniente actualizar o Regulamento (CE) n.o 440/2008 aditando, com carácter prioritário, um novo método de ensaio in vitro de irritação cutânea. Embora esse método in vitro ainda esteja a ser discutido no âmbito da OCDE, neste caso excepcional é conveniente aditar o método B.46 ao presente regulamento. Este método deve ser actualizado o mais rapidamente possível, logo que se chegue a um acordo no âmbito da OCDE ou caso surjam novas informações que justifiquem a revisão do método. |
|
(6) |
É necessário rever as disposições relativas ao teste de inibição para algas, de modo a incluir outras espécies e a satisfazer os requisitos de avaliação dos perigos e de classificação dos produtos químicos. |
|
(7) |
É necessário aditar dois novos métodos, um para medir a mineralização aeróbica das águas de superfície através de um ensaio de simulação de biodegradação e o outro para determinar a toxicidade para o género Lemna através de um ensaio de inibição de crescimento. |
|
(8) |
O Regulamento (CE) n.o 440/2008 deve, portanto, ser alterado em conformidade. |
|
(9) |
As medidas previstas no presente regulamento estão em conformidade com o parecer do comité instituído pelo artigo 133.o do Regulamento (CE) n.o 1907/2006, |
ADOPTOU O PRESENTE REGULAMENTO:
Artigo 1.o
O anexo do Regulamento (CE) n.o 440/2008 é alterado do seguinte modo:
|
1. |
A parte A é alterada do seguinte modo:
|
|
2. |
A parte B é alterada do seguinte modo: É aditado o capítulo B.46 constante do anexo III do presente regulamento. |
|
3. |
A parte C é alterada do seguinte modo:
|
Artigo 2.o
O presente regulamento entra em vigor no terceiro dia seguinte ao da sua publicação no Jornal Oficial da União Europeia.
O presente regulamento é obrigatório em todos os seus elementos e directamente aplicável em todos os Estados-Membros.
Feito em Bruxelas, em 23 de Julho de 2009.
Pela Comissão
Stavros DIMAS
Membro da Comissão
(1) JO L 396 de 30.12.2006, p. 1.
(2) JO L 142 de 31.5.2008, p. 1.
(3) JO L 358 de 18.12.1986, p. 1.
ANEXO I
|
A.4. |
PRESSÃO DE VAPOR |
1. MÉTODO
O presente método é equivalente ao método OCDE TG 104 (2004).
1.1. INTRODUÇÃO
A presente versão revista do Método A.4 (1) inclui um método adicional, o método de efusão: termogravimetria isotérmica, concebido para produtos químicos com pressões de vapor muito baixas (até 10–10 Pa). Tendo em conta a necessidade de procedimentos, em especial para a determinação da pressão de vapor de substâncias com uma pressão de vapor muito baixa, as gamas de aplicação de outros procedimentos deste método foram reavaliadas.
Em equilíbrio termodinâmico, a pressão de vapor de uma substância pura é exclusivamente função da temperatura. Os princípios fundamentais são descritos noutro local (2) (3).
Não existe nenhum procedimento único de medição que seja aplicável a toda a gama de pressões de vapor, desde menos de 10–10 Pa até 105 Pa. O presente método inclui oito métodos diferentes de medição da pressão de vapor, que podem ser aplicados em função da gama de pressões em causa. A aplicação e a gama de medição dos diferentes métodos são comparadas no quadro 1. Estes métodos só podem ser aplicados para compostos que não sejam degradados nas condições de ensaio. Nos casos em que os métodos experimentais não sejam aplicáveis por motivos técnicos, a pressão de vapor pode também ser estimada, e o anexo apresenta um método recomendado para essa estimativa.
1.2. DEFINIÇÕES E UNIDADES
A pressão de vapor de uma substância é a pressão de saturação exercida sobre uma substância sólida ou líquida.
Deve ser utilizada a unidade SI de pressão, o Pascal (Pa). Apresentam-se a seguir outras unidades que têm sido utilizadas, com os respectivos factores de conversão:
|
1 Torr |
= |
1 mm Hg |
= |
1,333 × 102 Pa |
|
1 atmosfera |
= |
1,013 × 105 Pa |
|
|
|
1 bar |
= |
105 Pa |
|
|
A unidade SI de temperatura é o Kelvin (K). A conversão de graus centígrados em graus Kelvin é dada pela fórmula:
T = t +273,15
onde T é a temperatura Kelvin ou temperatura termodinâmica e t é a temperatura em graus centígrados.
Quadro 1
|
Método de medição |
Substâncias |
Repetibilidade estimada |
Reproductividade estimada |
Gama recomendada: |
|
|
Sólidas |
Líquidas |
||||
|
Método dinâmico |
Fusão a baixa temperatura |
Sim |
Até 25 % 1 a 5 % |
Até 25 % 1 a 5 % |
103 Pa a 2 × 103 Pa 2 × 103 Pa a 105 Pa |
|
Método estático |
Sim |
Sim |
5 a 10 % |
5 a 10 % |
10 Pa a 105 Pa 10–2 Pa a 105 Pa (1) |
|
Método do isoteniscópio |
Sim |
Sim |
5 a 10 % |
5 a 10 % |
102 Pa a 105 Pa |
|
Método de efusão: equilíbrio de pressão de vapor |
Sim |
Sim |
5 a 20 % |
Até 50 % |
10–3 Pa a 1 Pa |
|
Método de efusão: célula de Knudsen |
Sim |
Sim |
10 a 30 % |
— |
10–10 Pa a 1 Pa |
|
Método de efusão: termogravimetria isotérmica |
Sim |
Sim |
5 a 30 % |
Até 50 % |
10–10 Pa a 1 Pa |
|
Método da saturação de gases |
Sim |
Sim |
10 a 30 % |
Até 50 % |
10–10 Pa a 103 Pa |
|
Método de rotação |
Sim |
Sim |
10 a 20 % |
— |
10–4 Pa a 0,5 Pa |
1.3. PRINCÍPIO DO ENSAIO
Em geral, a pressão de vapor é determinada a diferentes temperaturas. Numa gama limitada de temperaturas, o logaritmo da pressão de vapor de uma substância pura é função linear do inverso da temperatura termodinâmica, de acordo com a equação de Clapeyron-Clausius simplificada:

onde:
|
p |
= |
pressão de vapor, em Pascal |
|
ΔHv |
= |
calor de vaporização, em J mol–1 |
|
R |
= |
constante universal dos gases perfeitos, 8,314 J mol–1 K–1 |
|
T |
= |
temperatura, em K |
1.4. SUBSTÂNCIAS DE REFERÊNCIA
Não é necessário utilizar substâncias de referência. Essas substâncias servem fundamentalmente para verificar periodicamente o desempenho de um método, permitindo ainda a comparação entre os resultados de diferentes métodos.
1.5. DESCRIÇÃO DO MÉTODO
1.5.1. Método dinâmico (método de Cottrell)
1.5.1.1. Princípio
A pressão de vapor é determinada medindo a temperatura de ebulição da substância a diferentes pressões pré-estabelecidas, mais ou menos entre 103 a 105 Pascal. Este método é igualmente recomendado para a determinação da temperatura de ebulição. Para esse efeito, pode ser utilizado até 600 K. As temperaturas de ebulição dos líquidos são aproximadamente 0,1 °C mais elevadas a uma profundidade de 3 a 4 cm do que à superfície, devido à pressão hidrostática da coluna de líquido. No caso do método de Cottrell (4), o termómetro é colocado no vapor, acima da superfície do líquido, e o líquido em ebulição é bombeado em contínuo sobre a ponta do termómetro. Uma fina camada do líquido, que se encontra em equilíbrio com o vapor à pressão atmosférica, forma-se em torno da ponta do termómetro. Deste modo, o termómetro lê o verdadeiro ponto de ebulição, sem erros devidos ao sobreaquecimento ou à pressão hidrostática. A bomba originalmente utilizada por Cottrell é apresentada na figura 1. O tubo A contém o líquido em ebulição. O fio de platina B, selado no fundo do tubo, facilita uma ebulição uniforme. O tubo lateral C conduz a um condensador, enquanto que a manga D evita que o condensado frio atinja o termómetro E. Quando o líquido contido em A entra em ebulição, as bolhas e o líquido recolhidos pelo funil são transportados pelos dois braços da bomba F até à ponta do termómetro.
|
Figura 1
|
Figura 2
|
Bomba de Cottrell (4)
|
A: |
Termopar |
|
B: |
Volume tampão de vácuo |
|
C: |
Manómetro |
|
D: |
Vácuo |
|
E: |
Ponto de medição |
|
F: |
Resistência, cerca de 150 W |
1.5.1.2. Equipamento
A figura 2 mostra um aparelho de alta precisão que utiliza o princípio de Cottrell. O aparelho consiste num tubo com uma secção inferior para a ebulição, um condensador na parte intermédia e uma saída com flange na parte superior. A bomba de Cottrell é colocada na secção de ebulição, obtida através de uma resistência eléctrica. A temperatura é medida por um termopar coberto ou por um termómetro de resistência, inseridos através da flange, na parte superior. A saída é ligada a um sistema de regulação da pressão, composto por uma bomba de vácuo, um volume tampão, um manostato de admissão de azoto para a regulação da pressão e um manómetro.
1.5.1.3. Procedimento
A substância é colocada na secção de ebulição. É possível que surjam problemas com sólidos não pulverizados, mas esta questão pode por vezes ser resolvida aquecendo a água da manga de arrefecimento. O aparelho é selado a nível da flange e a substância é desgaseificada. Este processo não é aplicável às substâncias que provocam a formação de espuma.
Estabiliza-se a pressão ao nível mais baixo a que se pretende efectuar a leitura e o sistema de aquecimento é posto a funcionar. Ao mesmo tempo, o sensor de temperatura é ligado a um registador.
O equilíbrio é atingido quando se regista uma temperatura de ebulição constante a pressão constante. É necessário ter o cuidado de evitar que o aparelho seja sacudido durante a ebulição. Além disso, deve garantir-se a condensação total no sistema de arrefecimento. Para a determinação da pressão de vapor de sólidos com baixo ponto de fusão, é necessário tomar precauções para evitar o bloqueio do condensador.
Depois de registar este ponto de equilíbrio, passa-se para uma pressão mais elevada. Repete-se este processo até se atingir a pressão de 105 Pa (aproximadamente 5 a 10 pontos de medição na totalidade). Para fins de verificação, os pontos de equilíbrio deverão ser repetidos para valores decrescentes de pressão.
1.5.2. Método estático
1.5.2.1. Princípio
No método estático (5), a pressão de vapor em equilíbrio termodinâmico é determinada a uma dada temperatura. Este método é aplicável para substâncias e para líquidos e sólidos compostos numa gama de pressões entre os 10–1 e os 105 Pa, podendo, com os devidos cuidados, ser também utilizado na gama de 1 a 10 Pa.
1.5.2.2. Equipamento
O equipamento consiste num banho a temperatura constante (precisão de ±0,2 K), num recipiente para a amostra, ligado a uma linha de vácuo, num manómetro e num sistema de regulação da pressão. A câmara que contém a amostra (figura 3a) é ligada à linha de vácuo através de uma válvula e de um manómetro diferencial (tubo em U cheio com um fluido manométrico apropriado), que servirá de indicador do zero. O manómetro diferencial poderá ser cheio com mercúrio, silicones ou ftalatos, dependendo da gama de pressões e do comportamento químico da substância em estudo. No entanto, a utilização de mercúrio deve, sempre que possível, ser evitada por razões ambientais. A substância em estudo não deve dissolver-se de forma perceptível nem reagir com o fluido contido no tubo em U. Em vez de um tubo em U, pode utilizar-se um manómetro com indicador da pressão (figura 3b). No caso do manómetro, o mercúrio pode ser utilizado para pressões entre a pressão normal e os 102 Pa, enquanto que os fluidos de silicone e os ftalatos são apropriados para pressões abaixo dos 102 Pa, até 10 Pa. Existem outros manómetros que podem ser utilizados abaixo dos 102 Pa e os manómetros de membrana aquecível podem mesmo ser utilizados para valores inferiores a 10–1 Pa. A temperatura é medida na parede externa do recipiente que contém a amostra ou no interior do mesmo.
1.5.2.3. Procedimento
Utilizando o aparelho descrito na figura 3a, enche-se o tubo em U com o líquido escolhido, que deve ser desgaseificado a temperatura elevada antes de se proceder às leituras. A substância em estudo é colocada no aparelho e desgaseificada a baixa temperatura. Caso a amostra seja um composto, a temperatura deverá ser suficientemente baixa para evitar qualquer alteração da composição do material. O equilíbrio pode ser estabelecido mais rapidamente por agitação. A amostra pode ser arrefecida com azoto líquido ou com gelo seco, mas deve ter-se o cuidado de evitar a condensação do ar ou do fluido da bomba. Com a válvula de cima do recipiente da amostra aberta, aplica-se sucção durante vários minutos, para remoção do ar. Se necessário, a operação de desgasificação deve ser repetida várias vezes.
|
Figura 3a
|
Figura 3b
|
Quando a amostra é aquecida com a válvula fechada, a pressão de vapor aumenta, o que altera o equilíbrio do fluido no tubo em U. Para compensar este fenómeno, permite-se a entrada de azoto ou de ar no aparelho até que o indicador de pressão diferencial marque novamente zero. A pressão necessária para que tal ocorra pode ser lida no manómetro ou através de um instrumento de maior precisão. Essa pressão corresponde à pressão de vapor da substância para essa temperatura de medição. Utilizando o aparelho descrito na Figura 3b, a pressão de vapor pode ser lida directamente.
A pressão de vapor é determinada para pequenos intervalos adequados de temperatura (aproximadamente 5 a 10 pontos de medição na totalidade), até à temperatura máxima pretendida.
Para fins de verificação, as leituras a baixas temperaturas devem ser repetidas. Caso os valores obtidos na repetição das leituras não coincidam com a curva obtida para os valores crescentes de temperatura, isso poderá dever-se às seguintes razões:
|
i) |
a amostra ainda contém ar (p.ex.: materiais de elevada viscosidade) ou substâncias com um ponto de fusão mais baixo, que são libertadas durante o aquecimento; |
|
ii) |
a substância sofreu uma reacção química no intervalo de temperatura estudado (p. ex.: decomposição, polimerização). |
1.5.3. Método do isoteniscópio
1.5.3.1. Princípio
O isoteniscópio (6) baseia-se no princípio do método estático. O método implica a colocação da amostra numa ampola que é mantida a temperatura constante e que está ligada a um manómetro e a uma bomba de vácuo. As impurezas mais voláteis do que a substância em estudo são eliminadas por desgasificação a baixa pressão. A pressão de vapor da amostra às temperaturas seleccionadas será equilibrada contra uma pressão conhecida de um gás inerte. O isoteniscópio foi desenvolvido para medição da pressão de vapor de determinados hidrocarbonetos líquidos, mas também é aplicável para o estudo de sólidos. Normalmente, este método não é adequado para sistemas com múltiplos componentes. Os resultados estarão sujeitos a ligeiros erros, caso as amostras contenham impurezas não voláteis. A gama recomendada é de 102 a 105 Pa.
1.5.3.2. Equipamento
A figura 4 mostra um exemplo típico de dispositivo de medição. A descrição completa pode ser consultada no ATSM D 2879-86 (6).
1.5.3.3. Procedimento
No caso dos líquidos, a própria substância serve como fluido para o manómetro diferencial. Coloca-se no isoteniscópio uma quantidade de líquido suficiente para encher a ampola e o segmento mais curto do manómetro. O isoteniscópio é ligado a um sistema de vácuo e purgado, sendo depois enchido com azoto. Repete-se duas vezes a evacuação e a purga do sistema, para garantir a remoção do oxigénio residual. Coloca-se o isoteniscópio cheio em posição horizontal, de modo a que a amostra se espalhe numa camada fina sobre a ampola da amostra e o manómetro. Reduz-se a pressão do sistema para 133 Pa e aquece-se suavemente a amostra até entrar em ligeira ebulição (remoção dos gases dissolvidos). O isoteniscópio é depois colocado de forma a que a amostra regresse à ampola e encha a secção mais curta do manómetro. A pressão é mantida a 133 Pa. A parte saliente da ampola é aquecida em chama fraca até que o vapor libertado pela amostra se expanda suficientemente para deslocar parte da amostra da parte superior da ampola para o interior do manómetro, criando um espaço cheio de vapor da amostra e livre de azoto. Coloca-se depois o isoteniscópio num banho a temperatura constante, ajustando a pressão do azoto até ser idêntica à da amostra. Em situação de equilíbrio, a pressão de azoto é igual à pressão de vapor da substância.
Figura 4
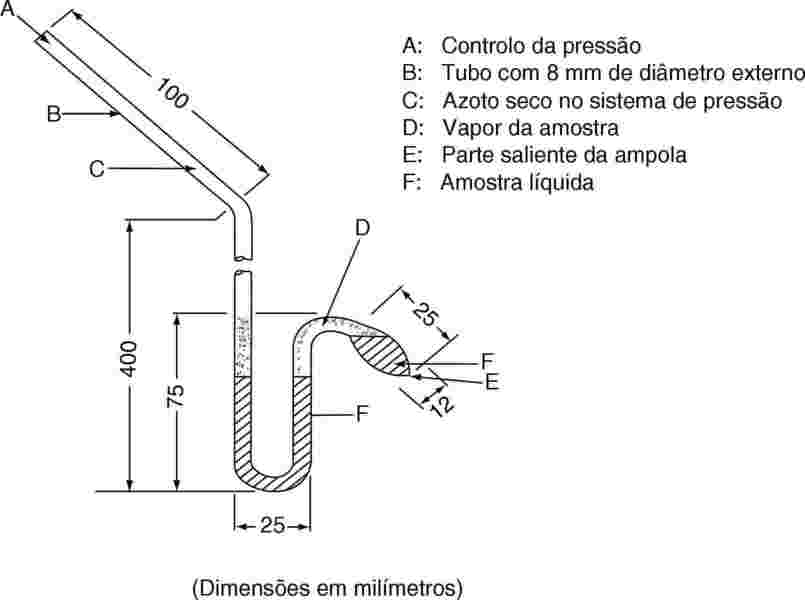
No caso dos sólidos, em função das gamas de pressão e de temperatura, poderão ser utilizados no manómetro líquidos como os fluidos de silicone ou os ftalatos. O líquido do manómetro, depois de desgaseificado, é utilizado para encher uma protuberância existente no segmento mais longo do isoteniscópio. Depois, coloca-se na ampola o sólido em estudo e desgaseifica-se a alta temperatura. O isoteniscópio é então inclinado de modo a que o líquido do manómetro possa correr para o tubo em U.
1.5.4. Método de efusão: equilíbrio de pressão de vapor (7)
1.5.4.1. Princípio
Uma amostra da substância em estudo é aquecida num pequeno forno e colocada numa campânula onde o ar foi esgotado. O forno é coberto com uma tampa, com pequenos orifícios de diâmetro conhecido. O vapor libertado pela substância e que se escapa por um dos orifícios é dirigido para o prato de uma balança de precisão, que também se deverá encontrar no interior da mesma campânula. Em certas montagens, o prato da balança pode estar rodeado por uma caixa de refrigeração que conduza o calor dissipado para o exterior por condução térmica, sendo arrefecido por radiação de modo a condensar o vapor que se escapa da substância. O momento gerado pelo jacto de vapor actua como uma força sobre o prato da balança. A pressão de vapor pode ser deduzida de duas formas: directamente a partir da força exercida sobre o prato da balança ou através da velocidade de evaporação, utilizando a equação de Hertz-Knudsen (2):

onde:
|
G |
= |
velocidade de evaporação (kg s–1 m–2) |
|
M |
= |
massa molar (g mol–1) |
|
T |
= |
temperatura (K) |
|
R |
= |
constante universal dos gases perfeitos (J mol–1 K–1) |
|
P |
= |
pressão de vapor (Pa) |
A gama recomendada é de 10–3 a 1 Pa.
1.5.4.2. Equipamento
O princípio geral do aparelho é ilustrado na figura 5.
Figura 5
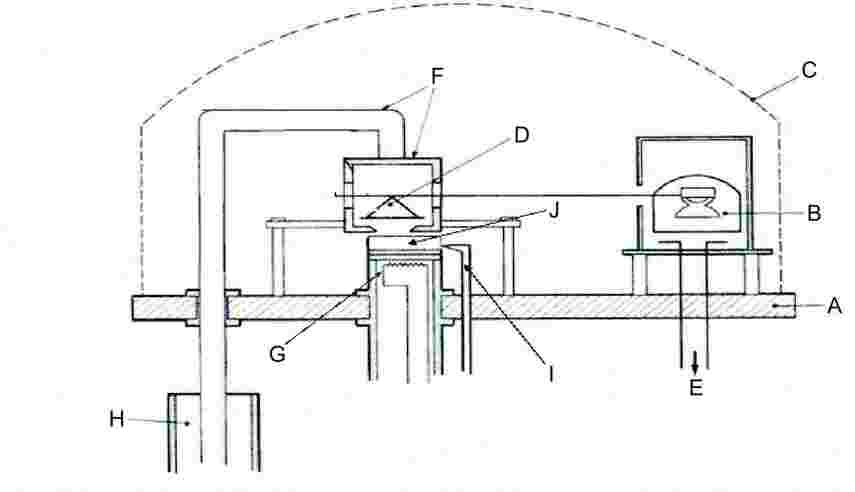
|
A: |
Base plate |
F: |
Refrigeration box and cooling bar |
|
B: |
Moving coil instrument |
G: |
Evaporator furnace |
|
C: |
Bell jar |
H: |
Dewar flask with liquid nitrogen |
|
D: |
Balance with scale pan |
I: |
Measurement of temperature of sample |
|
E: |
Vacuum measuring device |
J: |
Test Substance |
1.5.5. Método de efusão: célula de Knudsen
1.5.5.1. Princípio
O método baseia-se na estimativa da massa da substância em estudo que flui para o exterior a partir de uma célula de Knudsen em cada unidade de tempo (8) sob a forma de vapor, através de um microorifício, em condições de ultravácuo. A massa de vapor efluente pode ser calculada através da determinação da massa perdida pela célula ou condensando o vapor a baixa temperatura e determinando a quantidade de substância que se volatilizou, por cromatografia. A pressão de vapor é calculada através da relação de Heinz-Knudsen (ver o ponto 1.5.4.1), com aplicação de factores de correcção que dependem dos parâmetros do aparelho (9). A gama recomendada é de 10–10 to 1 Pa (10)(11)(12)(13)(14).
1.5.5.2. Equipamento
O princípio geral do aparelho é ilustrado na figura 6.
Figura 6

|
1: |
Connection to vacuum |
7: |
Threaded lid |
|
2: |
Wells from platinum resistance thermometer or temperature measurement and control |
8: |
Butterfly nuts |
|
3: |
Lid for vacuum tank |
9: |
Bolts |
|
4: |
O-ring |
10: |
Stainless steel effusion cells |
|
5: |
Aluminum vacuum tank |
11: |
Heater cartridge |
|
6: |
Device for installing and removing the effusion cells |
|
|
1.5.6. Método de efusão: termogravimetria isotérmica
1.5.6.1. Princípio
O método baseia-se na determinação das velocidades de evaporação aceleradas das substâncias em estudo a alta temperatura e à pressão ambiente, utilizando a termogravimetria (10)(15)(16)(17)(18)(19)(20). As velocidades de evaporação vT são calculadas expondo o composto seleccionado a um fluxo atmosférico lento de gás inerte e procedendo ao seguimento da diminuição do seu peso a temperaturas isotérmicas pré-determinadas T, em Kelvin, ao longo de um período adequado. As pressões de vapor pT são calculadas a partir dos valores de vT, aplicando a relação linear entre o logaritmo da pressão de vapor e o logaritmo da velocidade de evaporação. Se necessário, pode proceder-se a uma extrapolação para as temperaturas de 20 e de 25 °C, por análise da regressão de log pT vs. 1/T. Este método é adequado para substâncias a pressões de vapor tão baixas quanto 10–10 Pa (10–12 mbar) e com um grau de pureza tão próximo quanto possível de 100 %, de modo a evitar a incorrecta interpretação da diminuição de peso constatada.
1.5.6.2. Equipamento
O princípio geral do aparelho experimental é apresentado na figura 7.
Figura 7

A placa de suporte com a amostra, pendurada de uma microbalança no interior de uma câmara a temperatura controlada, é sujeita a um fluxo de azoto seco, que transporta consigo as moléculas vaporizadas da substância em estudo. Depois de sair da câmara, o fluxo de gás é purificado numa unidade de adsorção.
1.5.6.3. Procedimento
A substância em estudo é aplicada sobre a superfície de uma placa de vidro rugoso, numa camada homogénea. No caso dos sólidos, a placa é molhada de forma uniforme numa solução da substância, utilizando um solvente adequado, e seca em atmosfera inerte. Para a medição, a placa revestida é pendurada no analisador termogravimétrico e a subsequente diminuição do seu peso é medida em contínuo, em função do tempo.
A velocidade de evaporação vT a uma determinada temperatura é calculada a partir da perda de peso Δm da placa com a amostra através da seguinte equação:

onde F é a área do revestimento da substância em estudo, que normalmente corresponde à área da própria placa, e t é o tempo ao longo do qual se verificou a perda de peso Δm.
A pressão de vapor pT é calculada em função da velocidade de evaporação vT pela seguinte equação:
Log pT = C + D log vT
onde C e D são constantes específicas do aparelho experimental utilizado, dependentes do diâmetro da câmara de medição e da intensidade do fluxo de gás. Essas constantes têm de ser determinadas uma vez para cada aparelho experimental, através da medição e da regressão de log pT vs. log vT para uma série de compostos com pressão de vapor conhecida (11)(21)(22).
A relação entre a pressão de vapor pT e a temperatura T, em Kelvin, é dada pela seguinte equação:
Log pT = A + B 1/T
onde A e B são as constantes obtidas pela regressão de log pT vs. 1/T. Através desta equação, a pressão de vapor pode ser calculada para qualquer outra temperatura, por extrapolação.
1.5.7. Método da saturação de gases (23)
1.5.7.1. Princípio
Um gás inerte é passado, à temperatura ambiente e com um caudal conhecido e suficientemente lento para garantir a saturação, através ou sobre uma amostra da substância em estudo. É particularmente importante garantir a saturação da fase gasosa. A substância assim transportada é capturada, normalmente utilizando um adsorvente, e a sua quantidade é determinada. Em alternativa à captura e posterior análise do vapor, podem utilizar-se diversas técnicas analíticas em linha, como por exemplo a cromatografia gasosa, para determinar quantitativamente a quantidade de material transportado. A pressão de vapor é calculada partindo do pressuposto de que as leis dos gases ideais são aplicáveis e de que a pressão total de uma mistura de gases é igual à soma das pressões dos gases componentes da mistura. A pressão parcial da substância em estudo, ou seja, a pressão de vapor, é calculada a partir do volume total conhecido dos gases e do peso do material transportado.
O procedimento de saturação dos gases é aplicável a substâncias químicas líquidas ou sólidas. Pode ser utilizado para pressões de vapor superiores a 10–10 Pa (10)(11)(12)(13)(14). O método é mais fiável para as pressões de vapor inferiores a 103 Pa. Acima dos 103 Pa, as pressões de vapor são geralmente sobrestimadas, provavelmente devido à formação de aerossóis. Na medida em que as medições da pressão de vapor são efectuadas à temperatura ambiente, não é necessário extrapolar dados obtidos a temperaturas mais elevadas, sendo evitado esse passo que pode frequentemente acarretar erros importantes.
1.5.7.2. Equipamento
O procedimento exige a utilização de uma câmara a temperatura constante. O esquema da figura 8 mostra uma câmara que contém três placas para amostras sólidas e três câmaras para amostras líquidas, o que permite a análise em triplicado de amostras sólidas ou líquidas. A temperatura é controlada com uma precisão mínima de ±0,5 °C.
Figura 8

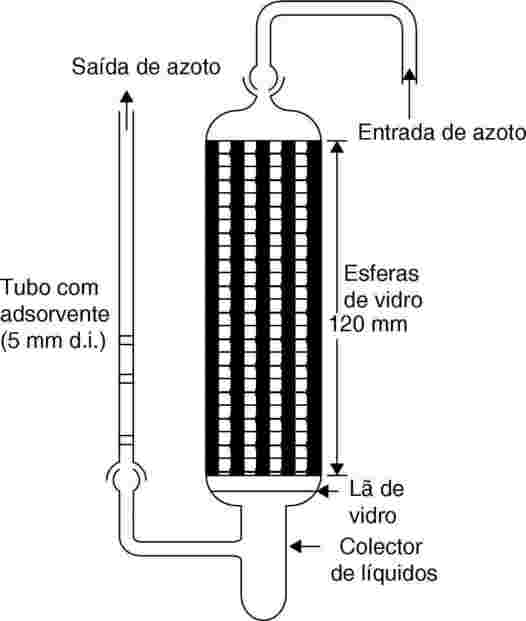
Normalmente, o gás inerte utilizado como transportador é o azoto, mas por vezes pode ser necessário utilizar outro gás (24). O gás transportador tem de ser seco. O fluxo de gás é dividido em 6 partes, controladas por válvulas de agulha (orifício de aproximadamente 0,79 mm), sendo transportado para a câmara por tubagens de cobre com 3,8 mm de diâmetro interno. Depois de alcançado o equilíbrio térmico, o gás flui pela amostra e pelo colector adsorvente, após o que sai da câmara.
As amostras sólidas são introduzidas em tubos de vidro com diâmetro interno de 5 mm, com rolhas de lã de vidro nas duas extremidades (ver a figura 9). A figura 10 mostra um sistema para amostras líquidas com colector adsorvente. Para a medição da pressão de vapor de líquidos, o método mais reprodutível passa pelo revestimento de esferas de vidro ou de um adsorvente inerte, por exemplo sílica, com o líquido em estudo e pelo enchimento do recipiente com essas esferas. Em alternativa, o gás transportador pode ser passado por uma frita grosseira e borbulhado através de uma coluna da substância líquida em estudo.
|
Figura 9
|
Figura 10
|
O sistema adsorvente inclui duas secções, uma das quais funciona como sistema de apoio (backup). Nos casos em que a pressão de vapor seja muito baixa, o adsorvente só irá reter quantidades ínfimas da substância, pelo que a adsorção sobre a lã de vidro e sobre a tubagem de vidro entre a amostra e o adsorvente poderá representar um problema sério.
A utilização de colectores arrefecidos com CO2 sólido é outra forma eficaz de recolha do material vaporizado. Esse tipo de colectores não provoca retornos de pressão na coluna de saturação, facilitando assim a remoção e quantificação do material recolhido.
1.5.7.3. Procedimento
O caudal do gás transportador à saída é medido à temperatura ambiente. O caudal deve ser verificado frequentemente durante a realização da experiência, de forma a garantir que se obtém um valor preciso para o volume total de gás transportador. O ideal é proceder à medição em contínuo, utilizando um fluxómetro mássico. A saturação da fase gasosa pode exigir um tempo de contacto considerável, o que implica a utilização de fluxos bastante baixos (25).
No final da experiência, as duas secções adsorventes (principal e de apoio) são analisadas separadamente. O componente adsorvido em cada secção é retirado por adição de um solvente. As soluções resultantes são sujeitas a análise quantitativa, de forma a determinar o peso total dessorvido em cada secção. A escolha do método analítico (tal como a escolha do adsorvente e do solvente utilizado para a dessorção) será ditada pela natureza do material em estudo. A eficiência da dessorção é determinada injectando uma quantidade conhecida de amostra no adsorvente e procedendo depois à sua dessorção e à análise da quantidade recuperada. É importante que a eficiência da dessorção seja verificada para uma concentração idêntica ou semelhante à da amostra, em condições de ensaio.
Para garantir a saturação do gás transportador com a substância em estudo, são utilizados três caudais diferentes de gás. Se a pressão de vapor calculada não se mostrar dependente do caudal utilizado, presume-se que o gás está saturado.
A pressão de vapor é calculada pela seguinte equação:
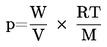
onde:
|
p |
= |
pressão de vapor (Pa) |
|
W |
= |
massa da substância em estudo evaporada (g) |
|
V |
= |
volume de gás saturado (m3) |
|
R |
= |
constante universal dos gases perfeitos 8,314 (J mol–1 K–1) |
|
T |
= |
temperatura (K) |
|
M |
= |
massa molar da substância em estudo (g mol–1) |
Os volumes medidos devem ser corrigidos para tomar em conta as diferenças de pressão e de temperatura entre o fluxómetro e o saturador.
1.5.8. Método de rotação
1.5.8.1. Princípio
Este método utiliza uma sonda de viscosidade rotativa, na qual o elemento de medição é uma pequena esfera de aço que é suspensa num campo magnético e forçada a rodar por campos rotativos (26)(27)(28). A velocidade de rotação é medida por bobinas de captação. Quando a esfera atinge uma determinada velocidade de rotação, normalmente cerca de 400 rotações por segundo, interrompe-se o fornecimento de energia e dá-se uma desaceleração devida ao atrito com o gás. A diminuição da velocidade de rotação em função do tempo é medida. A pressão de vapor é deduzida a partir da desaceleração da esfera de aço, que depende da pressão. A gama recomendada é de 10–4 a 0,5 Pa.
1.5.8.2. Equipamento
A figura 11 representa esquematicamente o aparelho experimental. O sensor de medição é colocado numa câmara a temperatura constante, regulada com uma precisão mínima de 0,1 °C. A célula com a amostra é colocada noutra câmara, com a temperatura igualmente regulada com uma precisão mínima de 0,1 °C. As restantes partes do aparelho experimental são mantidas a uma temperatura superior, para evitar a condensação. O aparelho é ligado a um sistema de alto vácuo.
Figura 11

2. DADOS E RELATÓRIOS
2.1. DADOS
Qualquer que seja o método escolhido, a pressão do vapor deve ser determinada pelo menos a duas temperaturas. É preferível proceder à determinação para três ou mais valores de temperatura entre 0 e 50 °C, para verificar a linearidade da curva de pressão de vapor. No caso do método de efusão (célula de Knudsen e termogravimetria isotérmica) e do método de saturação dos gases, recomenda-se a utilização da gama de temperaturas de 120 a 150 °C, em vez da gama de 0 a 50 °C.
2.2. RELATÓRIO DO ENSAIO
O relatório do ensaio deve incluir as seguintes informações:
|
— |
método utilizado, |
|
— |
especificação exacta da substância (identificação e impurezas) e passos prévios de purificação, se for caso disso, |
|
— |
são necessários pelo menos dois — e de preferência três ou mais — valores de pressão de vapor e de temperatura na gama de 0 a 50 °C (ou de 120 a 150 °C), |
|
— |
pelo menos uma das temperaturas deverá ser igual ou inferior a 25 °C, se tal for tecnicamente possível em função do método de ensaio escolhido, |
|
— |
todos os dados originais, |
|
— |
a curva de log p em função de 1/T, |
|
— |
uma estimativa da pressão de vapor a 20 ou a 25 °C. |
Caso se observe um fenómeno de transição (mudança de estado, decomposição), deverá ser acrescentada a seguinte informação:
|
— |
natureza da alteração, |
|
— |
temperatura à qual ocorre a alteração, à pressão atmosférica, |
|
— |
pressão de vapor a 10 e a 20 °C abaixo da temperatura de transição, bem como a 10 C e a 20 °C acima dessa temperatura (excepto no caso de passagem do estado sólido ao estado gasoso). |
Devem ser fornecidas todas as informações e observações de interesse para a interpretação dos resultados, nomeadamente no que se refere às impurezas e ao estado físico da substância.
3. BIBLIOGRAFIA
|
(1) |
Jornal Oficial das Comunidades Europeias L 383 A, 26-47 (1992). |
|
(2) |
Ambrose, D. (1975). Experimental Thermodynamics, Vol. II, Le Neindre, B., e Vodar, B., Eds., Butterworths, Londres. |
|
(3) |
Weissberger R., ed. (1959). Technique of organic Chemistry, Physical Methods of Organic Chemistry, 3rd ed., Interscience Publ., Nova Iorque, 1959, vol. I, parte I, capítulo IX. |
|
(4) |
Glasstone, S. (1946). Textbook of Physical Chemistry, 2nd ed., Van Nostrand Company, Nova Iorque. |
|
(5) |
NF T 20-048 AFNOR (Setembro de 1985). Chemical products for industrial use — Determination of vapour pressure of solids and liquids within range from 10–1 to 105 Pa — Static method. |
|
(6) |
ASTM D 2879-86, Standard test method for vapour pressure — temperature relationship and initial decomposition temperature of liquids by isoteniscope. |
|
(7) |
NF T 20-047 AFNOR (Setembro de 1985). Chemical products for industrial use — Determination of vapour pressure of solids and liquids within range from 10–3 to 1 Pa — Vapour pressure balance method. |
|
(8) |
Knudsen, M. (1909). Ann. Phys. Lpz., 29, 1979; (1911), 34, 593. |
|
(9) |
Ambrose, D., Lawrenson, I.J., Sprake, C.H.S. (1975). J. Chem. Thermodynamics 7, 1173. |
|
(10) |
Schmuckler, M.E., Barefoot, A.C., Kleier, D.A., Cobranchi, D.P. (2000), Vapor pressures of sulfonylurea herbicides; Pest Management Science 56, 521-532. |
|
(11) |
Tomlin, C.D.S. (ed.), The Pesticide Manual, Twelfth Edition (2000). |
|
(12) |
Friedrich, K., Stammbach, K., Gas chromatographic determination of small vapour pressures determination of the vapour pressures of some triazine herbicides. J. Chromatog. 16 (1964), 22-28. |
|
(13) |
Grayson, B.T., Fosbraey, L.A., Pesticide Science 16 (1982), 269-278. |
|
(14) |
Rordorf, B.F., Prediction of vapor pressures, boiling points and enthalpies of fusion for twenty-nine halogenated dibenzo-p-dioxins, Thermochimia Acta 112 Issue 1 (1987), 117-122. |
|
(15) |
Gückel, W., Synnatschke, G., Ritttig, R., A Method for Determining the Volatility of Active Ingredients Used in Plant Protection; Pesticide Science 4 (1973) 137-147. |
|
(16) |
Gückel, W., Synnatschke, G., Ritttig, R., A Method for Determining the Volatility of Active Ingredients Used in Plant Protection II. Application to Formulated Products; Pesticide Science 5 (1974) 393-400. |
|
(17) |
Gückel, W., Kaestel, R., Lewerenz, J., Synnatschke, G., A Method for Determining the Volatility of Active Ingredients Used in Plant Protection. Part III: The Temperature Relationship between Vapour Pressure and Evaporation Rate; Pesticide Science 13 (1982) 161-168. |
|
(18) |
Gückel, W., Kaestel, R., Kroehl, T., Parg, A., Methods for Determining the Vapour Pressure of Active Ingredients Used in Crop Protection. Part IV: An Improved Thermogravimetric Determination Based on Evaporation Rate; Pesticide Science 45 (1995) 27-31. |
|
(19) |
Kroehl, T., Kaestel, R., Koenig, W., Ziegler, H., Koehle, H., Parg, A., Methods for Determining the Vapour Pressure of Active Ingredients Used in Crop Protection. Part V: Thermogravimetry Combined with Solid Phase MicroExtraction (SPME); Pesticide Science, 53 (1998) 300-310. |
|
(20) |
Tesconi, M., Yalkowsky, S.H., A Novel Thermogravimetric Method for Estimating the Saturated Vapor Pressure of Low-Volatility Compounds; Journal of Pharmaceutical Science 87(12) (1998) 1512-20. |
|
(21) |
Lide, D.R. (ed.), CRC Handbook of Chemistry and Physics, 81th ed.(2000), Vapour Pressure in the Range -25 °C to 150 °C. |
|
(22) |
Meister, R.T. (ed.), Farm Chemicals Handbook, Vol. 88 (2002). |
|
(23) |
40 CFR, 796. (1993). pp 148-153, Office of the Federal Register, Washington DC. |
|
(24) |
Rordorf B.F. (1985). Thermochimica Acta 85, 435. |
|
(25) |
Westcott et al. (1981). Environ. Sci. Technol. 15, 1375. |
|
(26) |
Messer G., Röhl, P., Grosse G., and Jitschin W. (1987). J. Vac. Sci. Technol. (A), 5(4), 2440. |
|
(27) |
Comsa G., Fremerey J.K., and Lindenau, B. (1980). J. Vac. Sci. Technol. 17(2), 642. |
|
(28) |
Fremerey, J.K. (1985). J. Vac. Sci. Technol. (A), 3(3), 1715. |
(1) Utilizando um manómetro de capacitância.
Apêndice
Método de estimação
INTRODUÇÃO
Um valor estimado da pressão de vapor pode ser utilizado para:
|
— |
decidir qual o método experimental mais apropriado, |
|
— |
apresentar uma estimativa ou um valor limite para os casos em que os métodos experimentais não podem ser aplicados por razões técnicas. |
MÉTODO DE ESTIMAÇÃO
A pressão de vapor de líquidos e de sólidos pode ser estimada utilizando a correlação de Watson modificada (a). O único dado experimental necessário é o ponto de ebulição normal. O método é aplicável na gama de pressões entre 105 Pa e 10–5 Pa.
Para mais informações sobre o método, consultar o «Handbook of Chemical Property Estimation Methods» (b). Ver igualmente a Environmental Monograph n.o 67 (c) da OCDE.
PROCEDIMENTO DE CÁLCULO
A pressão de vapor é calculada do seguinte modo:

onde:
|
T |
= |
temperatura de interesse |
|
Tb |
= |
ponto de ebulição normal |
|
PVP |
= |
pressão de vapor à temperatura T |
|
ΔHVb |
= |
calor de vaporização |
|
ΔZb |
= |
factor de compressibilidade (estimado em 0,97) |
|
m |
= |
factor empírico, dependente do estado físico à temperatura de interesse |
Para além do que,

onde KF é um factor empírico que permite tomar em consideração a polaridade da substância. Os factores KF estão listados, para diferentes tipos de compostos, na referência bibliográfica (b).
É bastante frequente que existam dados disponíveis que apresentam os pontos de fusão a pressões reduzidas. Nesses casos, a pressão de vapor é calculada do seguinte modo:

onde T1 é o ponto de ebulição à pressão reduzida P1.
RELATÓRIO
Quando for utilizado o método de estimativa, o respectivo relatório incluirá uma documentação exaustiva do método de cálculo utilizado.
BIBLIOGRAFIA
|
(a) |
Watson, K.M. (1943). Ind. Eng. Chem, 35, 398. |
|
(b) |
Lyman, W.J., Reehl, W.F., Rosenblatt, D.H. (1982). Handbook of Chemical Property Estimation Methods, McGraw-Hill. |
|
(c) |
OCDE Environmental Monograph N.o 67. Application of Structure-Activity Relationships to the Estimation of Properties Important in Exposure Assessment (1993). |
ANEXO II
|
A.22. |
DIÂMETRO MÉDIO GEOMÉTRICO PONDERADO EM FUNÇÃO DO COMPRIMENTO DE FIBRAS |
1. MÉTODO
1.1. INTRODUÇÃO
O presente método descreve um procedimento para medição do diâmetro médio geométrico ponderado em função do comprimento (LWGMD) de lotes de fibras minerais artificiais (FMA). Dado que o LWGMD do material terá uma probabilidade de 95 % de se encontrar no interior dos níveis de confiança a 95 % (LWGMD ± dois desvios-padrão) da amostra, o valor apresentado (resultado do teste) será o valor inferior do intervalo de confiança a 95 % da amostra (ou seja, LWGMD — 2 desvios-padrão). O método baseia-se numa actualização (Junho de 1994) de um projecto de procedimento industrial do HSE (Departamento de Saúde e Segurança Britânico) adoptado numa reunião entre o ECFIA e o HSE em Chester, em 26/9/93, e desenvolvido para e por um segundo ensaio interlaboratorial (1, 2). Este método de medição pode ser utilizado para caracterizar o diâmetro das fibras em lotes de substâncias ou produtos que contenham FMA, nomeadamente fibras cerâmicas refractárias (FCR), fibras vítricas artificiais (FVA), fibras cristalinas e fibras policristalinas.
A ponderação em função do comprimento é uma forma de compensar o efeito de distribuição dos diâmetros causada pela quebra das fibras longas durante a amostragem ou manuseamento dos materiais. Na medição da distribuição por tamanhos do diâmetro das FMA utilizam-se métodos estatísticos geométricos (média geométrica), visto que essa distribuição se aproxima geralmente da distribuição logarítmica normal.
A medição do comprimento em complemento da medição do diâmetro é tediosa e demorada, mas se se medirem apenas as fibras que tocam uma linha infinitamente fina em cada campo de visão do miscroscópio electrónico, a probabilidade de que uma determinada fibra seja seleccionada é proporcional ao seu comprimento. Na medida em que esse facto resolve a questão do comprimento e da sua ponderação nos cálculos, a única medição necessária é a do diâmetro, e o LWGMD-2SD pode ser calculado como se descreve.
1.2. DEFINIÇÕES
Partícula: Um objecto em que a relação do comprimento com a espessura é menor do que 3:1.
Fibra: Um objecto em que a relação do comprimento com a espessura (razão de forma) é igual ou superior a 3:1.
1.3. ÂMBITO E LIMITAÇÕES
O método foi concebido para a análise da distribuição dos diâmetros nos casos em que o diâmetro médio varia entre 0,5 μm e 6 μm. Os diâmetros superiores podem ser medidos utilizando a microscopia electrónica com menor ampliação, mas o método terá limitações cada vez maiores quanto mais finas sejam as fibras, pelo que se recomenda, quando o diâmetro médio for inferior a 0,5 μm, a utilização de um microscópio electrónico de transmissão (MET) para a medição do diâmetro das fibras.
1.4. PRINCÍPIO DO MÉTODO DE ENSAIO
Retira-se um determinado número de amostras representativas das mantas de fibra ou dos lotes de fibras soltas. Reduz-se o comprimento das fibras através de um procedimento de esmagamento e dilui-se em água uma subamostra representativa. Extraem-se aliquotas que são filtradas em filtro de policarbonato com uma dimensão de poro de 0,2 μm e preparadas para serem examinadas utilizando técnicas de microscopia electrónica de varrimento (MEV). O diâmetro das fibras é medido com uma ampliação de 10 000 x ou superior (1) utilizando o método da intersecção com uma linha para obtenção de uma estimativa do diâmetro médio não afectada de erro sistemático. O limite inferior do intervalo de confiança a 95 % (com base num teste unicaudal) é calculado para obter uma estimativa do valor inferior da média geométrica do diâmetro das fibras presentes no material.
1.5. DESCRIÇÃO DO MÉTODO DE ENSAIO
1.5.1. Segurança/precauções
A exposição física às fibras em suspensão no ar deve ser reduzida ao mínimo, pelo que o manuseamento das fibras secas deve ser feito em câmara de fluxo laminar (hotte) ou caixa de luvas. A exposição física deve ser objecto de seguimento periódico que permita avaliar a eficácia dos métodos de controlo. Durante o manuseamento de FMA, devem ser utilizadas luvas descartáveis, para reduzir a irritação da pele e para evitar a contaminação cruzada.
1.5.2. Aparelhos / equipmento
|
— |
Prensa (capaz de produzir 10 MPa) e cadinhos, |
|
— |
Filtros capilares de policarbonato com diâmetro de poro de 0,2 μm (25 mm de diâmetro), |
|
— |
Filtro de membranas em éster de celulose com diâmetro de poro de 5 μm, para suporte durante a filtração, |
|
— |
Aparelho de filtração em vidro (ou sistema de filtração descartável) que aceite filtros de 25 mm de diâmetro (p.ex.: sistema de microanálise em vidro da Millipore, tipo XX10 025 00), |
|
— |
Água recém-destilada filtrada a 0,2 μm para remoção dos microorganismos, |
|
— |
Aparelho de revestimento a quente com eléctrodo em ouro ou ouro/paládio, |
|
— |
Microscópio electrónico de varrimento com capacidade de resolução a 10 nm e com ampliação de 10 000 x, |
|
— |
Diversos: espátulas, lâmina de bisturi tipo 24, tesouras, tubos de MEV, cola ou fita adesiva de carbono, prata coloidal, |
|
— |
Sonda de ultra-sons ou banho ultra-sónico de bancada, |
|
— |
Amostrador ou sonda para recolha de amostras cilíndricas das mantas de FMA. |
1.5.3. Procedimento de ensaio
1.5.3.1. Amostragem
No caso das mantas e placas, um amostrador ou sonda de 25 mm de diâmetro é utilizado para a recolha de amostras em toda a secção. Essas amostras devem ser igualmente espaçadas por toda a largura nas peças pequenas ou retiradas aleatoriamente de diversas áreas quando as peças sejam longas. O mesmo equipamento pode ser utilizado para retirar amostras aleatórias no caso de fibras soltas. Quando possível, devem ser retiradas seis amostras, por forma a reflectir as variações espaciais do material.
Esmaga-se as seis amostras num cadinho de 50 mm de diâmetro a 10 MPa. O material é re-homogeneizado com uma espátula e prensado uma segunda vez a 10 Mpa, podendo depois ser removido do cadinho e armazenado num frasco de vidro selado.
1.5.3.2. Preparação das amostras
Se necessário, qualquer ligante orgânico poderá ser removido colocando a fibra numa estufa a 450 °C durante cerca de uma hora.
Formar um cone e dividir em quatro para obter subamostras (este passo deve ser realizado em câmara de fluxo laminar).
Utilizando uma espátula, adicionar uma pequena quantidade (< 0,5 g) de amostra a 100 ml de água acabada de destilar e filtrada através de um filtro de membrana de 0,2 μm (poderão ser utilizadas fontes alternativas de água ultrapura, desde que se demonstre que a sua qualidade é satisfatória). Dispersar totalmente utilizando uma sonda de ultra-sons com uma potência de 100 W regulada de forma a produzir cavitação. (Se esse tipo de sonda não estiver disponível, utilizar o seguinte método: agitar e inverter a amostra repetidamente durante 30 segundos; passar por ultra-sons num banho ultra-sónico de bancada durante cinco minutos; voltar a agitar e inverter repetidamente durante mais 30 segundos).
Imediatamente após a dispersão da fibra, remover um determinado número de aliquotas (p.ex.: três aliquotas de 3, 6 e 10 ml) utilizando uma pipeta de boca larga (com uma capacidade de 2 a 5 ml).
Filtrar todas as aliquotas em vácuo através de um filtro de policarbonato com poro de 0,2 μm, suportado por um filtro de suporte MEC com poro de 5 µm, utilizando um funil de filtração em vidro de 25 mm com um reservatório cilíndrico. Colocam-se primeiro cerca de 5 ml da água destilada filtrada no funil e a aliquota é depois lentamente pipetada para a água com a ponta da pipeta abaixo do menisco. A pipeta e o reservatório devem ser cuidadosamente lavadas depois de pipetar, dado que as fibras mais finas têm tendência para se concentrarem perto da superfície.
Remover cuidadosamente o filtro e separá-lo do filtro de suporte antes de o colocar num recipiente para secagem.
Cortar uma secção de um quarto ou metade do filtrado com uma lâmina de bisturi de tipo 24, fazendo movimentos laterais durante o corte. Colar cuidadosamente a secção cortada num suporte de SEM utilizando fita ou cola de carbono. Aplicar prata coloidal em pelo menos três sítios para melhorar o contacto eléctrico entre a borda do filtro e o suporte. Quando a cola/prata coloidal estiverem secos, revestir a superfície do depósito a quente com aproximadamente 50 nm de ouro ou de ouro/paládio.
1.5.3.3. Calibração e operação do MEV
1.5.3.3.1. Calibração
A calibração do MEV deve ser verificada pelo menos uma vez por semana (idealmente uma vez por dia) utilizando uma grelha de calibração normalizada. A calibração deve ser verificada utilizando uma amostra-padrão, e se o valor medido (MEV) não se encontrar num intervalo de ±2 % do respectivo valor, o MEV deve ser novamente calibrado e o procedimento de verificação deve ser repetido.
O MEV deve ter uma resolução que permita um diâmetro mínimo de campo de pelo menos 0,2 µm, utilizando uma matriz de amostra real, com uma ampliação de 2 000 x.
1.5.3.3.2. Operação
O MEV deve ser operado com uma ampliação de 10 000 x (2) em condições que permitam uma boa resolução, com imagem aceitável, a baixas velocidades de varrimento, por exemplo de 5 segundos por imagem. Embora os parâmetros operacionais de diferentes MEV possam variar, em geral obtém-se a melhor visibilidade e resolução, em materiais de peso atómico relativamente baixo, utilizando voltagens de aceleração de 5-10 keV e trabalhando com um pequeno diâmetro de feixe (spot size) e com uma distância de trabalho curta. Dado que se vai utilizar a linha mediana como referência, deve utilizar-se uma inclinação de 0o, para evitar a necessidade de voltar a focar ou, no caso de MEV com um módulo eucêntrico, utilizar a distância de trabalho eucêntrica. Pode trabalhar-se com menor ampliação desde que o material não contenha nenhuma fibra de pequeno diâmetro e que o diâmetro das fibras seja grande (> 5 μm).
1.5.3.4. Medição do diâmetro
1.5.3.4.1. Exame com baixa ampliação para avaliação da amostra
A amostra deve ser inicialmente examinada com baixa ampliação para verificar se não existem aglomerados de fibras de grandes dimensões e para avaliar a densidade de fibras na amostra. Caso haja muita aglomeração, recomenda-se a preparação de uma nova amostra.
Para fins de exactidão estatística, é necessário proceder à medição de um número mínimo de fibras, pelo que poderá parecer desejável dispor de maior densidade de fibras, na medida em que o exame de campos vazios leva tempo e não contribui em nada para a análise. No entanto, se o filtro estiver sobrecarregado, torna-se difícil medir o diâmetro de todas as fibras e as fibras de menor diâmetro podem ser escondidas por outras de maior diâmetro.
Uma densidade de fibras superior a 150 fibras por milímetro linear poderá resultar numa sobreestimação do LWGMD. Por outro lado, a baixa concentração de fibras aumentará o tempo necessário para a análise, pelo que frequentemente é mais útil preparar uma nova amostra com uma densidade de fibras próxima da ideal do que persistir nas contagens de filtros em que a concentração de fibras é baixa. A densidade ideal das fibras deverá resultar, em média, em uma ou duas fibras que podem ser medidas por cada campo de visão, com uma ampliação de 5 000 x. A densidade ideal depende, contudo, do diâmetro das fibras, pelo que é necessário que o operador utilize a sua experiência e discernimento para decidir se a densidade de fibras está ou não próxima do ideal.
1.5.3.4.2. Ponderação do diâmetro das fibras em função do comprimento
Só devem ser contadas as fibras que toquem (ou interceptem) uma linha (infinitamente) fina traçada no ecrã do MEV. Para tal, deve ser traçada no centro do ecrã uma linha horizontal (ou vertical).
Em alternativa, pode colocar-se um ponto no centro do ecrã e proceder depois a um exame linear contínuo ao longo do filtro numa determinada direcção. O diâmetro de todas as fibras com uma razão de forma superior a 3:1 que toquem ou interceptem esse ponto deverá ser medido e registado.
1.5.3.4.3. Medição das fibras
Recomenda-se que sejam medidas pelo menos 300 fibras. Cada fibra deve ser medida uma única vez no ponto de intersecção com o ponto ou linha traçados no ecrã (ou perto do ponto de intersecção se a parede exterior da fibra estiver escondida). Caso se encontrem fibras de secção irregular, deve ser feita uma medição que represente o diâmetro médio da fibra. Os pontos de medição na parede exterior da fibra devem ser cuidadosamente definidos antes da medição da distância que separa os dois pontos opostos. A medição pode ser efectuada em linha, no ecrã, ou a posteriori, em imagens armazenadas ou fotografias. Recomenda-se a utilização de sistemas semi-automáticos de medição em imagens que transferem os dados directamente para uma folha de cálculo, na medida em que permitem poupar tempo, eliminar os erros de transcrição e automatizar o processo de cálculo.
As extremidades das fibras mais longas devem ser observadas a baixa ampliação para verificar se não estão a encaracolar de volta para o campo de visão, por forma a garantir que só sejam medidas uma única vez.
2. DADOS
2.1. TRATAMENTO DOS RESULTADOS
Geralmente, os diâmetros das fibras não apresentam uma distribuição normal. No entanto, é possível obter uma distribuição que se aproxima da normal através de uma transformação logarítmica.
Calcular a média aritmética (lnD médio) e o desvio-padrão (SDlnD) dos valores do logaritmo neperiano (lnD) dos n diâmetros de fibra (D).
|
|
(1) |
|
|
(2) |
O desvio-padrão é dividido pela raíz quadrada do número de medições (n) para obter o erro-padrão (SElnD).
|
|
(3) |
Substrair o dobro do erro-padrão da média e calcular o exponencial desse valor (média menos dois erros-padrão) para obter a média geométrica menos dois erros-padrão geométricos.
|
|
(4) |
3. APRESENTAÇÃO DOS RESULTADOS
RELATÓRIO DE ENSAIO
O relatório de ensaio deve incluir pelo menos a seguinte informação:
|
— |
Valor de LWGMD-2SE. |
|
— |
Todos os desvios, em particular os que possam ter efeitos sobre a precisão ou a fiabilidade dos resultados, com a devida justificação. |
4. BILBLIOGRAFIA
|
1. |
B. Tylee SOP MF 240. Health and Safety Executive. February 1999. |
|
2. |
G. Burdett and G. Revell. Development of a standard method to measure the length-weigthed geometric mean fibre diameter: Results of the Second inter-laboratory exchange. IR/L/MF/94/07. Project R42.75 HPD. Health and Safety Executive. Research and Laboratory Services Division. 1994. |
(1) Esta é a ampliação indicada para as fibras de 3 µm; no caso das fibras de 6 µm poderá ser mais indicado utilizar uma ampliação de 5 000 x.
(2) Para as fibras de 3 μm, ver a nota de pé de página anterior.
ANEXO III
|
B.46. |
IRRITAÇÃO CUTÂNEA IN VITRO: ENSAIO EM MODELOS DE EPIDERME HUMANA RECONSTRUÍDA |
1. MÉTODO
1.1. INTRODUÇÃO
Entende-se por irritação cutânea a produção de danos reversíveis na pele por aplicação de uma substância em estudo durante um máximo de quatro horas [definição que consta do sistema harmonizado a nível mundial da ONU para a classificação e rotulagem de produtos químicos (GHS)] (1). O presente método de ensaio descreve uma técnica in vitro que, consoante as informações necessárias, pode servir para determinar o poder de irritação cutânea de substâncias, enquanto ensaio alternativo independente no âmbito de uma estratégia de ensaio pautada pela suficiência de prova (2).
A determinação da irritação cutânea tem normalmente feito uso de animais de laboratório (ver o método B.4) (3). No contexto da preocupação com o bem-estar animal, o método B.4 permite determinar a corrosão/irritação cutânea por aplicação de uma estratégia de ensaio sequencial com base em métodos in vitro e ex vivo validados, assim se evitando a dor e o sofrimento de animais. Há três métodos ou directrizes de ensaio in vitro validados, B.40, B.40.A e TG 435 (4, 5, 6), com utilidade para a parte de corrosividade da estratégia de ensaio sequencial do método B.4.
O presente método baseia-se em modelos de epiderme humana reconstruída, cuja concepção geral (utilização de queratinócitos de epiderme humana como fonte celular, tecido representativo e citoarquitectura) permite reproduzir com muita fidelidade as propriedades bioquímicas e fisiológicas das camadas superiores da pele humana, isto é, da epiderme. A metodologia descrita neste método de ensaio permite identificar substâncias irritantes perigosas correspondentes à categoria 2 do sistema GHS da ONU. O método inclui ainda uma série de normas de desempenho para a avaliação de métodos de ensaio similares e modificados baseados em modelos de epiderme humana reconstruída (7).
Foram efectuados estudos de pré-validação, optimização e validação de dois métodos de ensaio in vitro (8, 9, 10, 11, 12, 13, 14, 15, 16, 17) que utilizam modelos de epiderme humana reconstruída, disponíveis no mercado sob as designações EpiSkin™ e EpiDerm™. Estas referências basearam-se na frase R38. A referência 25 aborda determinados aspectos de recálculo para efeitos do sistema GHS. Os métodos com desempenho equivalente ao do método EpiSkin™ (método de referência validado 1) são recomendados como métodos de ensaio independentes, alternativos ao ensaio in vivo no coelho, para a classificação de substâncias irritantes na categoria 2 do sistema GHS. Os métodos com desempenho equivalente ao do método EpiDerm™ (método de referência validado 2) só são recomendados para a classificação de substâncias irritantes na categoria 2 do sistema GHS como métodos de rastreio ou integrados numa estratégia de ensaio sequencial, num contexto de suficiência de prova. Antes de se poder utilizar um ensaio in vitro proposto, baseado num modelo de epiderme humana reconstruída, para avaliar a irritação cutânea numa perspectiva normativa, há que determinar a fiabilidade, a relevância (exactidão) e as limitações do método na utilização proposta, para garantir que o mesmo é comparável ao método de referência validado 1, em conformidade com as normas de desempenho estabelecidas no apêndice ao presente método.
Com base nos requisitos do presente método, foram validados dois outros métodos de ensaio in vitro baseados num modelo de epiderme humana reconstruída, que dão resultados similares aos do método de referência validado 1 (18). Trata-se do método EpiDerm™ modificado (método de referência 2 modificado) e do método SkinEthic RHE™ [método similar (em inglês «me-too» 1)].
1.2. DEFINIÇÕES
Para efeitos do presente método, entende-se por:
Exactidão: o grau de acordo entre os resultados do método de ensaio e os valores de referência aceites. Constitui uma medida do desempenho do método e um dos aspectos da relevância. Este termo e o termo «concordância» são muitas vezes utilizados indistintamente para indicar a proporção de resultados correctos de um método de ensaio.
Substância de controlo de lote: uma substância de aferição à qual o tecido reage com viabilidade celular intermédia.
Viabilidade celular: um parâmetro que mede a actividade total de uma população celular (por exemplo, em termos de capacidade das desidrogenases mitocondriais celulares para reduzirem o pigmento vital MTT (brometo de [3-(4,5-dimetiltiazol-2-il)-2,5-difeniltetrazólio, azul de tiazolilo)); consoante o indicador determinado e o tipo de ensaio realizado, é possível correlacionar a viabilidade celular com o número total de células vivas e/ou a vitalidade dessas células.
TE50 : o tempo de exposição necessário para reduzir a viabilidade celular em 50 %, após aplicação de uma dada concentração fixa de uma substância marcadora; ver também CI50.
Taxa de falsos negativos: a proporção das substâncias positivas que um método de ensaio considera erradamente negativas. É um dos indicadores de desempenho dos métodos de ensaio.
Taxa de falsos positivos: a proporção das substâncias negativas (sem actividade) que um método de ensaio considera erradamente positivas. É um dos indicadores de desempenho dos métodos de ensaio.
Dose infinita: uma quantidade da substância em estudo aplicada à pele que excede a quantidade necessária para recobrir completa e uniformemente a superfície cutânea.
GHS (Sistema Mundial Harmonizado de Classificação e Rotulagem de Produtos Químicos): um sistema que propõe a classificação das substâncias e misturas em função de tipos e níveis normalizados de perigos físicos, sanitários e ambientais e trata ainda dos elementos de comunicação correspondentes, como pictogramas, palavras-sinal, advertências de perigo, recomendações de prudência e fichas de dados de segurança, de modo a transmitir informações sobre os efeitos indesejáveis das substâncias e misturas, tendo em vista à protecção das pessoas (empregadores, trabalhadores, transportadores, consumidores, pessoal dos serviços de emergência, etc.) e do ambiente (1); foi incorporado na legislação comunitária pelo Regulamento (CE) n.o 1272/2008.
CI50 : a concentração à qual uma substância marcadora reduz a viabilidade dos tecidos em 50 %, após um tempo de exposição fixo; ver também TE50.
Normas de desempenho: normas, associadas a um método de referência validado, com base nas quais pode ser avaliada a comparabilidade de um método de ensaio proposto que seja mecanística e funcionalmente similar. Integram essas normas: I) os componentes essenciais do método de ensaio; II) uma lista mínima de substâncias de referência, seleccionadas entre as substâncias utilizadas para demonstrar a aceitabilidade do desempenho do método de referência validado; e III) os níveis de exactidão e de fiabilidade, comparáveis aos obtidos com o método de referência validado, que o método de ensaio proposto deve evidenciar ao ser avaliado utilizando a lista mínima de substâncias de referência.
Fiabilidade: a medida em que, utilizando o mesmo protocolo, um método de ensaio pode ser continuadamente reproduzido no mesmo laboratório e em laboratórios diferentes. A fiabilidade é avaliada com base nos valores calculados das reprodutibilidades intralaboratorial e interlaboratorial.
Sensibilidade: a proporção das substâncias positivas/activas que são correctamente classificadas pelo ensaio. Constitui uma medida da exactidão de um método de ensaio cujos resultados sejam estabelecidos em função de categorias e é um aspecto importante a ter em conta na avaliação da relevância de um método de ensaio.
Especificidade: a proporção das substâncias negativas/sem actividade que são correctamente classificadas pelo ensaio. Constitui uma medida da exactidão de um método de ensaio cujos resultados sejam apresentados em função de categorias e é um aspecto importante a ter em conta na avaliação da relevância de um método de ensaio.
Irritação cutânea: a produção de danos reversíveis na pele por aplicação de uma substância em estudo durante um máximo de quatro horas. A irritação cutânea é uma reacção local, não imunogénica, que surge pouco depois do estímulo (24). A sua característica principal é a reversibilidade do processo, no qual se produzem reacções inflamatórias e surge a maior parte dos sinais clínicos característicos das irritações associadas a processos inflamatórios (eritema, edema, prurido e dor).
1.3. ÂMBITO E LIMITAÇÕES
Os ensaios no quadro do presente método, baseados num modelo de epiderme humana reconstruída, têm como limitação o facto de só permitirem atribuir a classificação de substância irritante da pele correspondente à categoria 2 do sistema GHS da ONU. Como não permitem classificar substâncias na categoria 3 facultativa do sistema GHS, todas as substâncias não classificadas na categoria 2 ficarão por classificar (nenhuma categoria). É de prever que o presente método tenha de ser revisto em função das necessidades normativas, da futura inclusão de novos indicadores, dos aperfeiçoamentos que venham a ser efectuados ou de novos ensaios similares que surjam.
O presente método permite identificar produtos irritantes perigosos constituídos por uma única substância (19), mas não fornece informações adequadas sobre a corrosão da pele. O método não pode ser aplicado a gases e aerossóis; as misturas ainda não foram objecto de um estudo de validação.
1.4. PRINCÍPIO DO MÉTODO DE ENSAIO
A substância em estudo é aplicada localmente num modelo tridimensional de epiderme humana reconstruída, constituído por queratinócitos de epiderme humana normais, cultivados de modo a formarem um modelo de epiderme humana com várias camadas e altamente diferenciado. O modelo é constituído por uma camada basal, uma camada espinhosa e uma camada granulosa organizadas e por um stratum corneum em várias camadas, com uma estrutura lipídica lamelar intercelular semelhante ao observado in vivo.
O princípio dos ensaios em modelos de epiderme humana reconstruída baseia-se na hipótese de que as substâncias irritantes conseguem penetrar o stratum corneum por difusão, revelando-se citotóxicas para as células das camadas subjacentes. A viabilidade celular é medida pela conversão, pela desidrogenase, do corante vital MTT [brometo de 3-(4,5-dimetiltiazol-2-il)-2,5-difeniltetrazólio, azul de tiazolilo, n.o EINECS 206-069-5, n.o CAS 298-93-1] num sal azul de formazano, que é determinado quantitativamente depois de extraído dos tecidos (20). As substâncias irritantes são identificadas com base na sua capacidade de redução da viabilidade celular para valores inferiores a limites definidos (≤ 50 % no caso das substâncias irritantes classificadas na categoria 2 do sistema GHS da ONU). As substâncias a que estejam associadas viabilidades celulares acima do limite definido não são classificadas (ou seja, nenhuma categoria é atribuída para viabilidades celulares superiores a 50 %).
Os sistemas baseados em modelos de epiderme humana reconstruída podem sem aplicados a sólidos, líquidos, semi-sólidos e ceras. Os líquidos podem ser aquosos ou não e os sólidos podem ser solúveis ou insolúveis em água. Sempre que possível, os sólidos devem ser ensaiados sob a forma de pó fino. Dado que, na validação dos sistemas de ensaio baseados em modelos de epiderme humana reconstruída, foram utilizadas 58 substâncias cuidadosamente seleccionadas, representativas de uma vasta gama de classes químicas, é de prever que os métodos sejam genericamente aplicáveis ao universo de classes químicas (16). A validação inclui 13 substâncias irritantes da categoria 2 do sistema GHS. Não foram incluídos ácidos, bases, sais e outras substâncias inorgânicas não corrosivos; algumas classes de substâncias orgânicas reconhecidamente irritantes, como os hidroperóxidos, os fenóis e os tensioactivos, também não foram incluídas na validação ou foram-no apenas numa escala limitada.
1.5. DEMONSTRAÇÃO DE COMPETÊNCIA
Antes de um método validado conforme ao presente método de ensaio passar a ser utilizado por rotina, os laboratórios podem optar por comprovar a sua competência técnica, utilizando para o efeito as dez substâncias recomendadas no quadro 1. No âmbito do presente método, a categoria 3 facultativa do sistema GHS da ONU é considerada «nenhuma categoria». Para que possam utilizar-se, em ensaios normativos, novos métodos de ensaio similares (me-too) que tenham sido desenvolvidos com base no presente método e que sejam estrutural e funcionalmente similares aos métodos de referência validados ou para que possa fazer-se uso de modificações de métodos validados, há que demonstrar previamente, na óptica das normas de desempenho constantes do apêndice ao presente método, que a fiabilidade e a exactidão do novo método de ensaio são comparáveis às dos métodos validados.
Quadro 1
Substâncias de referência, constantes do apêndice, destinadas à comprovação de competência
|
Substância |
Número CAS |
Pontuação in vivo |
Estado físico |
Categoria GHS |
|
Ácido naftalenoacético |
86-87-3 |
0 |
S |
Nenhuma |
|
Isopropanol |
67-63-0 |
0,3 |
L |
Nenhuma |
|
Estearato de metilo |
112-61-8 |
1 |
S |
Nenhuma |
|
Butirato de heptilo |
5870-93-9 |
1,7 |
L |
Categoria 3 facultativa |
|
Salicilato de hexilo |
6259-76-3 |
2 |
L |
Categoria 3 facultativa |
|
Ciclamenaldeído |
103-95-7 |
2,3 |
L |
Categoria 2 |
|
1-Bromo-hexano |
111-25-1 |
2,7 |
L |
Categoria 2 |
|
Metacrilato de butilo |
97-88-1 |
3 |
L |
Categoria 2 |
|
1-Metil-3-fenil-1-piperazina |
5271-27-2 |
3,3 |
S |
Categoria 2 |
|
Heptanal |
111-71-7 |
4 |
L |
Categoria 2 |
1.6. DESCRIÇÃO DO MÉTODO
Descreve-se a seguir os componentes e procedimentos de um ensaio baseado num modelo de epiderme humana reconstruída para a avaliação da irritação cutânea. O modelo de epiderme humana pode ser construído, preparado ou obtido comercialmente (EpiSkin™, EpiDerm™ e SkinEthic RHE™, por exemplo). Os protocolos normalizados do método de ensaio correspondentes aos modelos EpiSkin™, EpiDerm™ e SkinEthic RHE™ podem ser obtidos no endereço [http://ecvam.jrc.ec.europa.eu] (21, 22, 23). Os ensaios devem ser efectuados em observância do seguinte:
1.6.1. Componentes dos modelos de epiderme humana reconstruída
1.6.1.1. Condições gerais do modelo
Na construção do epitélio devem ser utilizados queratinócitos humanos normais. Devem estar presentes várias camadas de células epiteliais viáveis (camada basal, camada espinhosa, camada granulosa), sob um stratum corneum funcional. Este deve ter várias camadas e possuir o perfil lipídico essencial para estabelecer uma barreira funcional suficientemente robusta para resistir a uma penetração rápida de substâncias marcadoras citotóxicas, por exemplo dodecilsulfato de sódio (SDS) ou Triton X-100. A função de barreira pode ser avaliada determinando a concentração à qual a substância marcadora reduz a viabilidade dos tecidos em 50 % (CI50), após um tempo de exposição fixo, ou determinando o tempo de exposição necessário para reduzir a viabilidade celular em 50 % (TE50), após aplicação de uma dada concentração fixa da substância marcadora. As propriedades de contenção do modelo devem impedir a passagem de matéria para o tecido viável por contorno do stratum corneum, que redundaria numa modelação deficiente da exposição cutânea. O modelo cutâneo não deve estar contaminado por bactérias, vírus, micoplasmas ou fungos.
1.6.1.2. Condições funcionais do modelo
1.6.1.2.1. Viabilidade
Para quantificar a viabilidade recorre-se, de preferência, ao método do MTT (20). A densidade óptica do corante extraído (por solubilização) do tecido tratado com a substância de controlo negativa deve ser pelo menos vinte vezes maior do que a densidade óptica do solvente de extracção por si só. O tecido tratado com a substância de controlo negativa deve, comprovadamente, manter-se estável em cultura (deve permitir obter medições de viabilidade semelhantes) durante todo o período de exposição do ensaio.
1.6.1.2.2. Função de barreira
O stratum corneum e a sua composição lipídica devem ser suficientes para resistir a uma penetração rápida de substâncias marcadoras citotóxicas (por exemplo, SDS ou Triton X-100), estimada com base na CI50 ou no TE50.
1.6.1.2.3. Morfologia
Um exame histológico da pele/epiderme reconstruída, a efectuar por pessoal devidamente habilitado, deve revelar uma estrutura de pele/epiderme humana (incluindo um stratum corneum com várias camadas).
1.6.1.2.4. Reprodutibilidade
Os resultados obtidos por aplicação do método a um dado modelo devem mostrar-se continuadamente reprodutíveis, utilizando de preferência, para o efeito, uma substância de controlo de lote (substância de aferição) adequada (ver o apêndice).
1.6.1.2.5. Controlo de qualidade do modelo
Cada lote de modelo de epiderme utilizado deve satisfazer determinados critérios de liberação de produção, dos quais os mais importantes são o de viabilidade (ponto 1.6.1.2.1) e o de função de barreira (ponto 1.6.1.2.2). O fornecedor do modelo cutâneo (ou o investigador, quando utilize modelo próprio) deve definir um intervalo de aceitabilidade (limites inferior e superior) para a CI50 ou o TE50. Ao receberem tecidos, os laboratórios devem verificar as propriedades de barreira dos mesmos. Apenas resultados obtidos com tecidos que possuam as características exigidas podem ser utilizados com fiabilidade na previsão de efeitos de irritação. A título de exemplo, apresentam-se a seguir os intervalos de aceitabilidade dos métodos de referência validados.
Quadro 2
Exemplos de critérios de controlo de qualidade para a liberação de lotes
|
|
Limite inferior de aceitabilidade |
Valor médio do intervalo de aceitabilidade |
Limite superior de aceitabilidade |
|
Método de referência validado 1 (18 horas de tratamento com SDS) |
CI50 = 1,0 mg/ml |
CI50 = 2,32 mg/ml |
CI50 = 3,0 mg/ml |
|
Método de referência validado 2 (Triton X-100 a 1 %) |
TE50 = 4,8 h |
TE50 = 6,7 h |
TE50 = 8,7 h |
1.6.1.3. Aplicação das substâncias em estudo e de controlo
Deve utilizar-se um número suficiente de replicados de tecido para cada tratamento e para as substâncias de controlo (pelo menos três replicados em cada série de ensaios). Tanto no caso das substâncias líquidas como das substâncias sólidas deve aplicar-se uma quantidade de substância em estudo suficiente para cobrir uniformemente a superfície de pele, evitando uma dose infinita (ver o ponto 1.2, «Definições»), ou seja, no mínimo 25 μl/cm2 ou 25 mg/cm2. No caso das substâncias sólidas, a superfície da epiderme deve ser humidificada, com água desionizada ou destilada, antes da aplicação, para garantir um bom contacto com a pele. Sempre que possível, os sólidos devem ser ensaiados sob a forma de pó fino. Após o período de exposição, a substância em estudo deve ser cuidadosamente removida da superfície da pele, por lavagem com uma solução-tampão aquosa ou com uma solução a 0,9 % de NaCl. Consoante o modelo de epiderme humana reconstruída que for utilizado, o período de exposição pode variar entre 15 minutos e 60 minutos e a temperatura de incubação entre 20 °C e 37 °C. Para mais pormenores, ver os procedimentos operacionais normalizados dos três métodos (21, 22, 23).
Deve utilizar-se em cada estudo substâncias de controlo negativas e positivas, para demonstrar que a viabilidade (controlo negativo), a função de barreira e a sensibilidade resultante (controlo positivo) dos tecidos se situam no intervalo histórico de aceitabilidade definido. Sugere-se a utilização de solução aquosa a 5 % de SDS como substância de controlo positiva. Como substância de controlo negativa, sugere-se a utilização de água ou de solução-tampão de fosfato (PBS).
1.6.1.4. Medição da viabilidade celular
O elemento mais importante do procedimento de ensaio é que as medições de viabilidade não sejam efectuadas imediatamente após a exposição às substâncias em estudo, mas sim após um período suficientemente longo de incubação dos tecidos lavados em meio fresco, depois do tratamento. Esse período permite tanto a recuperação de efeitos irritantes fracos como a manifestação de efeitos citotóxicos claros. Na fase de optimização do ensaio (9, 10, 11, 12, 13), verificou-se que a duração óptima do período de incubação depois do tratamento é de 42 horas, assim se tendo procedido na validação dos métodos de ensaio de referência.
Para medir a viabilidade celular, deve utilizar-se o método quantitativo validado de conversão do MTT, que é compatível com uma arquitectura de tecidos a três dimensões. Coloca-se a amostra de pele numa solução de MTT de concentração apropriada (por exemplo, 0,3-1 mg/ml), durante 3 horas. O precipitado azul de formazano é depois extraído do tecido com um solvente (por exemplo, isopropanol ou isopropanol acidificado), medindo-se a concentração de formazano por determinação da densidade óptica ao comprimento de onda de 570 nm (gama de comprimentos de onda: máximo ±30 nm).
As propriedades ópticas da substância em estudo ou a reacção química desta com o MTT podem interferir no ensaio, gerando estimativas erróneas de viabilidade (caso a substância em estudo evite ou reverta a coloração ou produza coloração). Isto pode suceder se a substância em estudo não for completamente removida da pele por lavagem ou se penetrar na epiderme. Se a substância em estudo agir directamente no MTT, for naturalmente corada ou adquirir coloração durante o tratamento dos tecidos, devem ser utilizadas substâncias de controlo suplementares, que permitam detectar interferências da substância em estudo na técnica de medição da viabilidade e corrigir esse efeito. Para uma descrição pormenorizada do modo como efectuar os ensaios de redução directa do MTT, consultar os protocolos dos métodos de referência validados (21, 22, 23). A coloração inespecífica devida a essas interferências não deve exceder 30 % do correspondente à substância de controlo negativa (para se efectuarem correcções). Se a coloração inespecífica exceder 30 %, a substância em estudo é considerada incompatível com o ensaio.
1.6.1.5. Critérios de aceitabilidade dos ensaios
Em cada ensaio que utilize lotes válidos (ver o ponto 1.6.1.2.5), a densidade óptica dos tecidos tratados com a substância de controlo negativa deve reflectir a qualidade de tecidos sujeitos a todas as etapas de expedição e recepção e a todo o processo do protocolo de irritação. A densidade óptica das substâncias de controlo não deve ser inferior aos limites inferiores históricos estabelecidos. Analogamente, os tecidos tratados com a substância de controlo positiva — ou seja, com solução aquosa a 5 % de SDS — devem manifestar a sensibilidade própria dos tecidos e a capacidade de reacção destes à substância irritante nas condições de cada ensaio (por exemplo, viabilidade ≤ 40 % no caso do método de referência validado 1 e viabilidade ≤ 20 % no caso do método de referência validado 2). Devem ainda ser definidas estimativas conexas adequadas da variabilidade dos resultados dos replicados de tecidos (por exemplo, se se recorrer a desvios-padrão, não devem exceder 18 %).
2. DADOS
2.1. DADOS
Para cada tratamento, devem ser apresentados num quadro os dados (por exemplo, valores de densidade óptica e percentagens de viabilidade celular calculadas para cada substância em estudo, bem como a classificação correspondente) correspondentes a cada replicado de amostra em estudo, incluindo os dados relativos às repetições de ensaios eventualmente efectuadas. Deve igualmente ser indicada para cada ensaio a média ± desvio-padrão. Devem referir-se ainda, em relação a cada substância estudada, as interacções observadas com o MTT e as substâncias estudadas coradas.
2.2. INTERPRETAÇÃO DOS RESULTADOS
Os valores de densidade óptica obtidos para cada amostra em estudo podem ser utilizados para calcular uma percentagem de viabilidade em relação à amostra de controlo negativa, que é fixada em 100 %. A percentagem de viabilidade celular que estabelece a diferenciação entre as substâncias em estudo irritantes e não classificadas e o(s) método(s) estatístico(s) utilizado(s) para avaliar os resultados e identificar as substâncias irritantes devem ser claramente definidos e documentados, devendo ainda demonstrar-se que são adequados. Indicam-se a seguir os valores limite de diferenciação para a previsão de efeitos irritantes, associados aos métodos de referência validados.
Considera-se a substância em estudo irritante da pele da categoria 2 do sistema GHS da ONU:
|
i) |
se a viabilidade dos tecidos depois da exposição e da incubação subsequente ao tratamento for inferior ou igual (≤) a 50 %; |
Considera-se que a substância em estudo não se insere em nenhuma categoria:
|
ii) |
se a viabilidade dos tecidos depois da exposição e da incubação subsequente ao tratamento for superior (>) a 50 %. |
3. RELATÓRIOS
3.1. RELATÓRIO DO ENSAIO
O relatório do ensaio deve incluir as seguintes informações:
Substâncias em estudo e de controlo:
|
— |
Denominação ou denominações químicas, como as denominações IUPAC ou CAS, e o n.o CAS, se forem conhecidos, |
|
— |
Grau de pureza e composição da substância (em percentagem ponderal), |
|
— |
Propriedades físico-químicas (por exemplo, estado físico, estabilidade e volatilidade, pH, hidrossolubilidade, se forem conhecidos) relevantes para o estudo, |
|
— |
Pré-tratamento das substâncias em estudo/de controlo, se for o caso (aquecimento ou moagem, por exemplo), |
|
— |
Condições de armazenagem. |
Justificação do modelo cutâneo e do protocolo utilizados.
Condições de ensaio:
|
— |
Sistema celular utilizado, |
|
— |
Informações sobre a calibração do equipamento de medição e a gama de comprimentos de onda utilizados para medir a viabilidade celular (por exemplo, espectrofotómetro), |
|
— |
Informações completas que substanciem o modelo cutâneo específico utilizado, incluindo sobre o desempenho do mesmo, por exemplo:
|
|
— |
Pormenores sobre o procedimento de ensaio seguido, |
|
— |
Doses de ensaio utilizadas, duração da exposição e do período de incubação depois do tratamento, |
|
— |
Descrição de eventuais modificações do procedimento de ensaio, |
|
— |
Referência a dados históricos sobre o modelo, por exemplo:
|
|
— |
Descrição dos critérios de avaliação utilizados, incluindo a justificação do(s) ponto(s) de diferenciação seleccionado(s) para o modelo preditivo. |
Resultados:
|
— |
Quadro dos resultados correspondentes a cada amostra em estudo, |
|
— |
Descrição de outros efeitos eventualmente observados. |
Discussão dos resultados.
Conclusões.
4. REFERÊNCIAS
|
1. |
Nações Unidas (ONU) (2007). Globally Harmonized System of Classification and Labelling of Chemicals (GHS), Second revised edition, UN New York and Geneva, 2007. Acessível em: http://www.unece.org/trans/danger/publi/ghs/ghs_rev02/02files_e.html. |
|
2. |
REACH: Guidance on Information Requirements and Chemical Safety Assessment. Acessível em: http://guidance.echa.europa.eu/docs/guidance_document/information_requirements_en.htm?time=1232447649. |
|
3. |
Método de ensaio B.4. TOXICIDADE AGUDA: IRRITAÇÃO/CORROSÃO DÉRMICA. |
|
4. |
Método de ensaio B.40. CORROSÃO DA PELE IN VITRO: ENSAIO DA RESISTÊNCIA ELÉCTRICA TRANSCUTÂNEA (RET). |
|
5. |
Método de ensaio B.40.A. CORROSÃO DA PELE IN VITRO: ENSAIO EM MODELOS DE PELE HUMANA. |
|
6. |
OCDE (2006). Test Guideline 435. OECD Guideline for the Testing of Chemicals. In Vitro Membrane Barrier Test Method. Adoptado em 19 de Julho de 2006. Acessível em: http://www.oecd.org/document/22/0,2340,en_2649_34377_1916054_1_1_1_1,00.html. |
|
7. |
ECVAM (2009). Performance Standards for applying human skin models to in vitro skin irritation. Acessível através de «Download Study Documents» em: http://ecvam.jrc.ec.europa.eu. |
|
8. |
Fentem, J. H., Briggs, D., Chesné, C., Elliot, G. R., Harbell, J. W., Heylings, J. R., Portes, P., Roguet, R., van de Sandt, J. J. M., Botham, P. (2001). A prevalidation study on in vitro tests for acute skin irritation. Results and evaluation by the Management Team. Toxicology in Vitro 15, 57-93. |
|
9. |
Portes, P., Grandidier, M. H., Cohen, C., Roguet, R. (2002). Refinement of the EPISKIN protocol for the assessment of acute skin irritation of chemicals: follow-up to the ECVAM prevalidation study. Toxicology in Vitro 16, 765-770. |
|
10. |
Kandárová, H., Liebsch, M., Genschow, E., Gerner, I., Traue, D., Slawik, B., Spielmann, H. (2004). Optimisation of the EpiDerm test protocol for the upcoming ECVAM validation study on in vitro skin irritation tests. ALTEX 21, 107-114. |
|
11. |
Kandárová, H., Liebsch, M., Gerner, I., Schmidt, E., Genschow, E., Traue, D., Spielmann H. (2005). The EpiDerm Test Protocol fort the Upcoming ECVAM Validation Study on In Vitro Skin Irritation Tests — An Assessment of the Performance of the Optimised Test. ATLA 33, 351-367. |
|
12. |
Cotovió, J., Grandidier, M.-H., Portes, P., Roguet, R., G. Rubinsteen (2005). The In Vitro Acute Skin Irritation of Chemicals: Optimisation of the EPISKIN Prediction Model within the Framework of the ECVAM Validation Process. ATLA 33, 329-249. |
|
13. |
Zuang, V., Balls, M., Botham, P. A., Coquette, A., Corsini, E., Curren, R. D., Elliot, G. R., Fentem, J. H., Heylings, J. R., Liebsch, M., Medina, J., Roguet, R., van De Sandt, J. J. M., Wiemann, C., Worth, A. (2002). Follow-up to the ECVAM prevalidation study on in vitro tests for acute skin irritation. ECVAM Skin Irritation Task Force Report 2. ATLA 30,109-129. |
|
14. |
Spielmann, H., Hoffmann, S., Liebsch, M., Botham, P., Fentem, J., Eskes, C., Roguet, R., Cotovió, J., Cole, T., Worth, A., Heylings, J., Jones, P., Robles, C., Kandárová, H., Gamer, A., Remmele, M., Curren, R., Raabe, H., Cockshott, A., Gerner, I., Zuang, V. (2007). The ECVAM International Validation Study on In Vitro Tests for Acute Skin Irritation: Report on the Validity of the EPISKIN and EpiDerm Assays and on the Skin Integrity Function Test. ATLA 35, 559-601. |
|
15. |
Hoffmann, S. (2006). ECVAM Skin Irritation Validation Study Phase II: Analysis of the Primary Endpoint MTT and the Secondary Endpoint IL1-α. 135 páginas mais anexos. Acessível através de «Download Study Documents» em: http://ecvam.jrc.ec.europa.eu. |
|
16. |
Eskes, C., Cole, T., Hoffmann, S., Worth, A., Cockshott, A., Gerner, I., Zuang V. (2007). ECVAM International Validation Study on In Vitro Tests for Acute Skin Irritation: Selection of Test Chemicals. ATLA 35, 603-619. |
|
17. |
J. Cotovió, M.-H. Grandidier, D. Lelièvre, R. Roguet, E. Tinois-Tessonneaud, J. Leclaire (2007). In vitro acute skin irritancy of chemicals using the validated EPISKIN model in a tiered strategy — Results and performances with 184 cosmetic ingredients, AATEX, Special Issue-proceedings from WC6. Vol. 14, 351-358. |
|
18. |
Informação do ESAC sobre o método EpiDerm melhorado e o método similar SkinEthic (5 de Novembro de 2008). |
|
19. |
CE (2006). Regulamento (CE) n.o 1907/2006 do Parlamento Europeu e do Conselho, de 18 de Dezembro de 2006, relativo ao registo, avaliação, autorização e restrição dos produtos químicos (REACH), que cria a Agência Europeia dos Produtos Químicos, que altera a Directiva 1999/45/CE e revoga o Regulamento (CEE) n.o 793/93 do Conselho e o Regulamento (CE) n.o 1488/94 da Comissão, bem como a Directiva 76/769/CEE do Conselho e as Directivas 91/155/CEE, 93/67/CEE, 93/105/CE e 2000/21/CE da Comissão. Jornal Oficial da União Europeia L 396 de 30.12.2006, p. 1. OP, Luxemburgo. |
|
20. |
Mosman, T. (1983). Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. Journal of Immunological Methods 65, 55-63. |
|
21. |
EpiSkin™ SOP, Version 1.6 (Janeiro de 2005). Validation of the EpiSkin Skin Irritation Test — 42 Hours Assay For The Prediction of Acute Skin Irritation of Chemicals. Acessível através de «Download Study Documents» em: http://ecvam.jrc.ec.europa.eu. |
|
22. |
EpiDerm™ SOP, Version 5.0 (Outubro de 2004). Draft Standard Operating Procedure. In Vitro Skin Irritation Test: Human Skin Model. Model: EpiDerm™-200. Acessível através de «Download Study Documents» em: http://ecvam.jrc.ec.europa.eu. |
|
23. |
SkinEthic RHE™ SOP. Ficará acessível através de «Download Study Documents» em: http://ecvam.jrc.ec.europa.eu. |
|
24. |
Harvell, J. D., Lammintausta, K., Maibach H. I. (1995). Irritant contact dermatitis em: Guin J. D., Practical Contact Dermatitis, Mc Graw-Hill, New York, pp. 7-18. |
|
25. |
Griesinger C., Barroso J., Zuang V.: ECVAM background document on the recent adaptations of the ECVAM performance standards for in vitro skin irritation testing in the context of the drafting process of an EU test method and an OECD draft test guideline. Ispra, 13 de Novembro de 2008. |
Apêndice
Avaliação das características de desempenho de modelos de epiderme humana reconstruída propostos para ensaios de irritação cutânea in vitro
INTRODUÇÃO
Os procedimentos cuja utilização no âmbito do presente método de ensaio seja proposta devem ser avaliados, com vista à determinação da sua fiabilidade e exactidão, utilizando substâncias representativas de toda a gama da escala de irritação de Draize. Ao serem avaliados utilizando as 20 substâncias de referência recomendadas (quadro 2), os procedimentos propostos devem exibir valores de fiabilidade e exactidão comparáveis aos do método de referência validado 1 (quadro 3) (1). Indicam-se nos pontos II e III os valores normais de exactidão e de fiabilidade a obter. Para que a fiabilidade e o desempenho (sensibilidade, especificidade, taxa de falsos negativos, taxa de falsos positivos e exactidão) do método de ensaio proposto possam ser comparados com os do método de referência validado 1, incluem-se substâncias não classificadas e substâncias classificadas (categoria 2 do sistema GHS da ONU) representativas de classes químicas relevantes. Antes de se utilizar o método para o ensaio de novas substâncias, deve determinar-se a sua fiabilidade e a capacidade do mesmo para identificar correctamente substâncias irritantes da categoria 2 do sistema GHS.
NORMAS DE DESEMPENHO
As normas de desempenho compreendem os seguintes elementos: I) Componentes essenciais do método de ensaio; II) Substâncias de referência; III) Valores definidos de exactidão e de fiabilidade (2). Estas normas baseiam-se nas normas de desempenho estabelecidas depois de concluído o estudo de validação relativo à irritação cutânea efectuado pelo ECVAM (3).
I) Componentes essenciais do método de ensaio
Condições gerais do modelo
Na construção do epitélio devem ser utilizados queratinócitos humanos normais. Devem estar presentes várias camadas de células epiteliais viáveis (camada basal, camada espinhosa, camada granulosa), sob um stratum corneum funcional. Este deve ter várias camadas e possuir o perfil lipídico essencial para estabelecer uma barreira funcional suficientemente robusta para resistir a uma penetração rápida de substâncias marcadoras citotóxicas, por exemplo SDS ou Triton X-100. A função de barreira pode ser avaliada determinando a concentração à qual a substância marcadora reduz a viabilidade dos tecidos em 50 % (CI50), após um tempo de exposição fixo, ou determinando o tempo de exposição necessário para reduzir a viabilidade celular em 50 % (TE50), após aplicação de uma dada concentração fixa da substância marcadora. As propriedades de contenção do modelo devem impedir a passagem de matéria para o tecido viável por contorno do stratum corneum, que redundaria numa modelação deficiente da exposição cutânea. O modelo cutâneo não deve estar contaminado por bactérias, vírus, micoplasmas ou fungos.
Condições funcionais do modelo
Viabilidade
Para quantificar a viabilidade recorre-se, de preferência, ao método do MTT (4). A densidade óptica do corante extraído (por solubilização) do tecido tratado com a substância de controlo negativa deve ser pelo menos vinte vezes maior do que a densidade óptica do solvente de extracção por si só. O tecido tratado com a substância de controlo negativa deve, comprovadamente, manter-se estável em cultura (deve permitir obter medições de viabilidade semelhantes) durante todo o período de exposição do ensaio.
Função de barreira
O stratum corneum e a sua composição lipídica devem ser suficientes para resistir a uma penetração rápida de substâncias marcadoras citotóxicas (por exemplo, SDS ou Triton X-100), estimada com base na CI50 ou no TE50.
Morfologia
Um exame histológico da pele/epiderme reconstruída, a efectuar por pessoal devidamente habilitado, deve revelar uma estrutura de pele/epiderme humana (incluindo um stratum corneum com várias camadas).
Reprodutibilidade
Os resultados obtidos por aplicação do método a um dado modelo devem mostrar-se continuadamente reprodutíveis, utilizando de preferência, para o efeito, uma substância de controlo de lote (substância de aferição) adequada (ver as definições no ponto 1.2).
Controlo de qualidade do modelo
Cada lote de modelo de epiderme utilizado deve satisfazer determinados critérios de liberação da produção, dos quais os mais importantes são o de «viabilidade» e o de «função de barreira». O fornecedor do modelo cutâneo (ou o investigador, quando utilize modelo próprio) deve definir um intervalo de aceitabilidade (limites inferior e superior) para a CI50 ou o TE50. Ao receberem tecidos, os laboratórios devem verificar as propriedades de barreira dos mesmos. Apenas resultados obtidos com tecidos que possuam as características exigidas podem ser utilizados com fiabilidade na previsão de efeitos de irritação. A título de exemplo, apresentam-se a seguir os intervalos de aceitabilidade dos métodos de referência validados.
Quadro 1
Exemplos de critérios de controlo de qualidade para a liberação de lotes
|
|
Limite inferior de aceitabilidade |
Valor médio do intervalo de aceitabilidade |
Limite superior de aceitabilidade |
|
Método de referência validado 1 (18 horas de tratamento com SDS) |
CI50 = 1,0 mg/ml |
CI50 = 2,32 mg/ml |
CI50 = 3,0 mg/ml |
|
Método de referência validado 2 (Triton X-100 a 1 %) |
TE50 = 4,8 h |
TE50 = 6,7 h |
TE50 = 8,7 h |
II) Substâncias de referência
Utilizam-se substâncias de referência para determinar se a fiabilidade e a exactidão de um novo método de ensaio in vitro proposto, baseado num modelo de epiderme humana reconstruída, que comprovadamente seja suficientemente similar, em termos estruturais e funcionais, aos métodos de referência validados ou que constitua uma modificação menor de um método de referência validado, indiciam um desempenho comparável ao do método de referência validado 1 (1). As 20 substâncias de referência indicadas no quadro 2 são representativas de várias classes químicas com interesse, estando algumas classificadas na categoria 2 do sistema GHS da ONU. As substâncias constantes da lista compreendem 10 da categoria 2 do sistema GHS, 3 da categoria 3 facultativa do mesmo sistema e 7 não classificadas em nenhuma categoria. No âmbito do presente método, a categoria 3 facultativa é considerada «nenhuma categoria». Estas substâncias de referência constituem o número mínimo que deve ser utilizado para avaliar a exactidão e a fiabilidade de métodos que sejam propostos para ensaios de irritação cutânea baseados em modelos de epiderme humana reconstruída. Se alguma das substâncias indicadas não estiver disponível, podem utilizar-se outras, para as quais se disponha de dados de referência in vivo adequados. Para avaliar melhor a exactidão de um método de ensaio proposto, podem acrescentar-se outras substâncias à lista mínima de substâncias de referência, que sejam representativas de outras classes químicas e para as quais se disponha de dados de referência in vivo adequados.
Quadro 2
Substâncias de referência para determinação da exactidão e da fiabilidade de modelos de epiderme humana reconstruída destinados a ensaios de irritação cutânea
|
Substância (1) |
Número CAS |
Número Einecs |
Estado físico |
Pontuação in vivo |
Categoria GHS in vitro |
Categoria GHS in vivo |
|
1-Bromo-4-clorobutano |
6940-78-9 |
230-089-3 |
L |
0 |
Categoria 2 |
Nenhuma |
|
Ftalato de dietilo |
84-66-2 |
201-550-6 |
L |
0 |
Nenhuma |
Nenhuma |
|
Ácido naftalenoacético |
86-87-3 |
201-705-8 |
S |
0 |
Nenhuma |
Nenhuma |
|
Fenoxiacetato de alilo |
7493-74-5 |
231-335-2 |
L |
0,3 |
Nenhuma |
Nenhuma |
|
Isopropanol |
67-63-0 |
200-661-7 |
L |
0,3 |
Nenhuma |
Nenhuma |
|
4-(Metiltio)benzaldeído |
3446-89-7 |
222-365-7 |
L |
1 |
Categoria 2 |
Nenhuma |
|
Estearato de metilo |
112-61-8 |
203-990-4 |
S |
1 |
Nenhuma |
Nenhuma |
|
Butirato de heptilo |
5870-93-9 |
227-526-5 |
L |
1,7 |
Nenhuma |
Categoria 3 facultativa |
|
Salicilato de hexilo |
6259-76-3 |
228-408-6 |
L |
2 |
Nenhuma |
Categoria 3 facultativa |
|
Fosfato de triisobutilo |
126-71-6 |
204-798-3 |
L |
2 |
Categoria 2 |
Categoria 3 facultativa |
|
1-Decanol |
112-30-1 |
203-956-9 |
L |
2,3 |
Categoria 2 |
Categoria 2 |
|
Ciclamenaldeído |
103-95-7 |
203-161-7 |
L |
2,3 |
Categoria 2 |
Categoria 2 |
|
1-Bromo-hexano |
111-25-1 |
203-850-2 |
L |
2,7 |
Categoria 2 |
Categoria 2 |
|
Cloridrato de 2-clorometil-3,5-dimetil-4-metoxipiridina |
86604-75-3 |
434-680-9 |
S |
2,7 |
Categoria 2 |
Categoria 2 |
|
alfa-Terpineol |
98-55-5 |
202-680-6 |
L |
2,7 |
Categoria 2 |
Categoria 2 |
|
Dissulfureto de di-n-propilo |
629-19-6 |
211-079-8 |
L |
3 |
Nenhuma |
Categoria 2 |
|
Metacrilato de butilo |
97-88-1 |
202-615-1 |
L |
3 |
Categoria 2 |
Categoria 2 |
|
5-(1,1-Dimetiletil)-2-metilbenzenotiol |
7340-90-1 |
438-520-9 |
L |
3,3 |
Categoria 2 |
Categoria 2 |
|
1-Metil-3-fenil-1-piperazina |
5271-27-2 |
431-180-2 |
S |
3,3 |
Categoria 2 |
Categoria 2 |
|
Heptanal |
111-71-7 |
203-898-4 |
L |
4 |
Categoria 2 |
Categoria 2 |
As substâncias indicadas no quadro 2 constituem uma distribuição representativa das 58 utilizadas no estudo internacional de validação relativo à irritação cutânea efectuado pelo ECVAM (1). Essas substâncias foram seleccionadas com base nos seguintes critérios:
|
— |
disponibilidade comercial, |
|
— |
representatividade no que respeita a toda a gama da escala de irritação de Draize (de não irritante a fortemente irritante), |
|
— |
estrutura química bem definida, |
|
— |
representatividade comparativamente à reprodutibilidade e à capacidade preditiva do método validado, determinadas no estudo de validação efectuado pelo ECVAM, |
|
— |
representatividade ao nível da funcionalidade química utilizada no processo de validação, |
|
— |
exclusão de perfis extremamente tóxicos (por exemplo, carcinogenicidade ou toxicidade para o aparelho reprodutor) e de custos proibitivos de eliminação. |
III) Valores definidos de exactidão e de fiabilidade
O desempenho (sensibilidade, especificidade, taxa de falsos negativos, taxa de falsos positivos e exactidão) do método de ensaio proposto deve ser comparável ao do método de referência validado 1 (quadro 3), ou seja, a sensibilidade deve ser igual ou superior a 80 %, a especificidade deve ser igual ou superior a 70 % e a exactidão deve ser igual ou superior a 75 %. O cálculo do desempenho deve ser efectuado com base em todas as classificações obtidas para as 20 substâncias nos vários laboratórios participantes. Para se estabelecer a classificação da cada substância em cada laboratório deve utilizar-se o valor médio de viabilidade correspondente às várias séries de ensaios efectuadas (no mínimo três séries de ensaios válidas).
Quadro 3
Desempenho do método de referência validado 1 (2)
|
Método de ensaio |
Número de substâncias |
Sensibilidade |
Especificidade |
Taxa de falsos negativos |
Taxa de falsos positivos |
Exactidão |
|
Método de referência validado 1 (3) |
58 |
87,2 % (4) |
71,1 % (5) |
12,8 % |
29,9 % |
74,7 % |
|
Método de referência validado 1 (3) |
20 |
90 % |
73,3 % |
10 % |
26,7 % |
81,7 % |
A fiabilidade do método de ensaio proposto deve ser comparável à dos métodos de referência validados.
Reprodutibilidade intralaboratorial
Ao determinar-se a variabilidade intralaboratorial num laboratório, a concordância, nesse laboratório, das classificações (categoria 2/nenhuma categoria) obtidas em séries de ensaios diferentes e independentes realizadas às 20 substâncias de referência deve ser igual ou superior a 90 %.
Reprodutibilidade interlaboratorial
Se o método de ensaio proposto se destinar a ser utilizado num único laboratório, não é essencial determinar a reprodutibilidade interlaboratorial. No caso dos métodos a transferir para outros laboratórios, a concordância entre, de preferência, pelo menos três laboratórios das classificações (categoria 2/nenhuma categoria) obtidas em séries de ensaios diferentes e independentes realizadas às 20 substâncias de referência deve ser igual ou superior a 80 %.
REFERÊNCIAS
|
1. |
Spielmann, H., Hoffmann, S., Liebsch, M., Botham, P., Fentem, J., Eskes, C., Roguet, R., Cotovió, J., Cole, T., Worth, A., Heylings, J., Jones, P., Robles, C., Kandárová, H., Gamer, A., Remmele, M., Curren, R., Raabe, H., Cockshott, A., Gerner, I., Zuang, V. (2007). The ECVAM International Validation Study on In Vitro Tests for Acute Skin Irritation: Report on the Validity of the EPISKIN and EpiDerm Assays and on the Skin Integrity Function Test. ATLA 35, 559-601. |
|
2. |
OCDE (2005). Guidance Document N.o 34 on the validation and international acceptance of new or updated test methods for hazard assessment. OCDE, Paris. |
|
3. |
ECVAM (2007). Performance Standards for applying human skin models to in vitro skin irritation. Acessível através de «Download Study Documents» em: http://ecvam.jrc.ec.europa.eu. Consultado em 27.10.2008. |
|
4. |
Mosman, T. (1983). Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. Journal of Immunological Methods 65, 55-63. |
|
5. |
Eskes, C., Cole, T., Hoffmann, S., Worth, A., Cockshott, A., Gerner, I., Zuang. V (2007). ECVAM International Validation Study on In Vitro Tests for Acute Skin Irritation: Selection of Test Chemicals. ATLA 35, 603-619. |
(1) As 20 substâncias de referência constituem uma selecção representativa das 58 que foram utilizadas para validar o método de referência 1 (EpiSkin™). A referência (5) contém uma lista completa das substâncias estudadas e indica os critérios que foram utilizados para as seleccionar.
(2) Indica-se neste quadro o desempenho do método de referência validado 1 no que respeita à capacidade do método para identificar correctamente as substâncias irritantes (categoria 2 do sistema GHS da ONU) e as substâncias não classificadas (sem nenhuma categoria, incluindo a categoria 3 facultativa), de entre as 58 ou 20 (quadro 2) substâncias de referência.
(3) EpiSkin™.
(4) Com base em 13 substâncias irritantes da categoria 2 do sistema GHS.
(5) Com base em 45 substâncias irritantes da categoria 3, ou substâncias químicas sem nenhuma categoria de classificação, no âmbito do sistema GHS.
ANEXO IV
|
C.3. |
CIANOBACTÉRIAS E ALGAS DE ÁGUA DOCE, ENSAIO DE INIBIÇÃO DE CRESCIMENTO |
1. MÉTODO
Este método é equivalente ao OECD TG 201 (Orientação de ensaio 201 da OCDE, 2006) (1).
1.1. INTRODUÇÃO
Os métodos de ensaio são revistos e actualizados periodicamente à luz dos progressos científicos. O método de ensaio C.3 precisava de ser revisto a fim de incluir mais espécies e de garantir o cumprimento das exigências em matéria de avaliação de riscos e de classificação dos produtos químicos. A revisão foi concluída com base na extensa experiência prática, nos progressos científicos no domínio dos estudos de toxicidade com algas e na extensa utilização no domínio regulamentar desde a adopção do método original.
1.2. DEFINIÇÔES
Para efeitos do presente método de ensaio, foram utilizadas as seguintes definições e abreviaturas:
Biomassa: peso seco de matéria viva presente numa população, expresso por unidade de volume; ou seja, mg de algas/litro de solução de ensaio. Normalmente, a «biomassa» é definida como uma massa, mas no presente ensaio o termo é utilizado em relação à massa por unidade de volume. Ainda no presente ensaio, alguns valores alternativos de biomassa, como as contagens de células, a fluorescência, etc., são normalmente medidos, sendo o termo «biomassa» utilizado também para essas medições alternativas.
Coeficiente de variação: é uma medida adimensional da variabilidade de um parâmetro, definida como a relação entre o desvio-padrão e a média. Esta medida pode igualmente ser expressa em termos de percentagem. O coeficiente de variação médio da taxa de crescimento específica média em culturas de controlo replicadas deve ser calculado do seguinte modo:
|
1. |
Calcular o CV, em percentagem, da taxa média de crescimento específico de cada replicado a partir das taxas de crescimento diárias/em cada secção do ensaio. |
|
2. |
Calcular o valor médio de todos os valores calculados de acordo com o ponto 1, a fim de obter o coeficiente de variação médio das taxas de crescimento específico diárias/em cada secção do ensaio dos replicados das culturas de controlo. |
ECx : é a concentração da substância em estudo que, dissolvida no meio de ensaio, resulta numa redução de x % (p. ex.: 50 %) do crescimento do organismo de ensaio, após um determinado período de exposição (que deverá ser explicitamente mencionado nos casos em que se afaste da duração total ou normal do ensaio). A fim de indicar de forma inequívoca se o valor de EC se refere à taxa de crescimento ou ao rendimento, serão utilizados, respectivamente, os símbolos «ErC» e «EyC».
Meio de crescimento: é o meio de cultura sintético completo em que as algas de ensaio são cultivadas quando expostas à substância em estudo. A substância em estudo será normalmente dissolvida no meio de ensaio.
Taxa de crescimento (taxa média de crescimento específico): é o aumento logarítmico da biomassa durante o período de exposição.
Menor concentração com efeito observável (LOEC): é a concentração mais baixa à qual se observa que a substância em estudo tem um efeito estatisticamente significativo (para p < 0,05) de redução do crescimento, quando comparada com o controlo, para um determinado período de exposição. No entanto, todas as concentrações de ensaio superiores à LOEC devem ter um efeito prejudicial igual ou superior ao verificado com a LOEC. Quando estas duas condições não estiverem satisfeitas, deverá ser fornecida uma explicação pormenorizada sobre a forma como se determinou a LOEC (e, consequentemente, a NOEC).
Concentração sem efeitos observáveis (NOEC): é a concentração de ensaio imediatamente inferior à LOEC.
Variável de resposta: é uma variável de estimação da toxicidade, derivada de qualquer dos parâmetros medidos que descrevem a biomassa nos diferentes métodos de cálculo. No caso do presente método, as taxas de crescimento e os rendimentos são variáveis de resposta derivadas da medição directa da biomassa ou de qualquer dos métodos alternativos mencionados.
Taxa de crescimento específico: é uma variável de resposta definida como o quociente da diferença entre os logaritmos naturais de um parâmetro de observação (no presente método, a biomassa) e o respectivo período.
Rendimento: é o valor de uma variável de medição no final do período de exposição menos o valor da variável de medição no início do período de exposição, que exprime o aumento da biomassa durante o ensaio.
1.3. APLICABILIDADE DO ENSAIO
O presente método de ensaio será de aplicação mais fácil para substâncias hidrossolúveis que, nas condições de ensaio, permaneçam em solução na água. Para o ensaio de substâncias voláteis, fortemente adsorventes, coradas, com baixa hidrossolubilidade ou que possam afectar a disponibilidade de nutrientes ou de minerais no meio de ensaio, poderá ser necessário proceder a algumas alterações do procedimento descrito (p. ex.: utilização de sistemas fechados, condicionamento dos recipientes de ensaio). Para mais orientações sobre algumas alterações adequadas, consultar as referências bibliográficas (2), (3) e (4).
1.4. PRINCÍPIO DO ENSAIO
O objectivo do presente ensaio é determinar os efeitos de uma substância sobre o crescimento de cianobactérias e/ou algas de água doce. Os organismos de ensaio em fase de crescimento exponencial são expostos à substância em estudo em culturas em meio líquido, normalmente durante 72 horas. Apesar da duração relativamente curta do ensaio, podem avaliar-se os efeitos ao longo de diversas gerações.
A resposta do sistema consiste na redução do crescimento numa série de culturas de algas (unidades de ensaio) expostas a diferentes concentrações da substância em estudo. Essa resposta é avaliada em função da concentração de exposição, por comparação com o crescimento médio de culturas de controlo replicadas e não expostas à substância em estudo. A fim de obter uma caracterização total da resposta do sistema aos efeitos tóxicos (optimização da sensibilidade), permite-se o crescimento exponencial não limitado das culturas, com uma quantidade suficiente de nutrientes e em condições de iluminação constante, durante um período suficiente para que se possa medir a redução da taxa de crescimento específica.
O crescimento, bem como a respectiva inibição, é quantificado através da medição da biomassa das algas ao longo do tempo. A biomassa das algas é definida como o peso seco por unidade de volume, ou seja, ml de algas/litro de solução de ensaio. A determinação do peso seco é, contudo, difícil, pelo que se utilizam parâmetros alternativos. Entre esses, o mais utilizado é a contagem de células. Outros parâmetros alternativos incluem o volume de células, a fluorescência, a densidade óptica, etc. Deve ser conhecido um factor de conversão entre o parâmetro alternativo medido e a biomassa.
O ponto final do ensaio ocorre quando se dá inibição do crescimento, expresso como o aumento logarítmico da biomassa (taxa média de crescimento específico) durante o período de exposição. A partir das taxas médias de crescimento específico registadas numa série de soluções de ensaio, a concentração que causa uma inibição da taxa de crescimento de x % (p. ex.: 50 %) é determinada e expressa como ErCx (nesse caso, ErC50).
Para efeitos da aplicação do presente método no âmbito do quadro regulamentar da UE, o cálculo dos resultados deverá basear-se numa taxa média de crescimento específico, pelos motivos abaixo expostos, no ponto 2.2. Uma variável de resposta adicional utilizada no presente método de ensaio é o rendimento, que pode ser necessário para dar cumprimento a exigências regulamentares específicas de alguns países. Essa variável é definida como a biomassa no final do período de exposição menos a biomassa no início do mesmo período. A partir do rendimento registado numa série de soluções de ensaio, a concentração que causa uma inibição do rendimento de x % (p. ex.: 50 %) é determinada e expressa como EyCx (nesse caso, EyC50).
Por outro lado, a menor concentração com efeito observável (LOEC) e a concentração sem efeitos observáveis (NOEC) podem ser determinadas estatisticamente.
1.5. INFORMAÇÃO SOBRE A SUBSTÂNCIA EM ESTUDO
A informação sobre a substância em estudo que poderá ser útil para estabelecer as condições do ensaio inclui a sua fórmula estrutural, pureza, estabilidade à luz, estabilidade nas condições de ensaio, propriedades de absorção da luz, pKa e os resultados dos estudos de transformação, incluindo a biodegradabilidade na água.
A hidrossolubilidade, o coeficiente de partição octanol/água (Pow) e a pressão de vapor da substância em estudo deverão ser conhecidos, devendo estar disponível um método validado para a quantificação da substância nas soluções de ensaio com uma eficiência de recuperação e um limite de detecção conhecidos.
1.6. SUBSTÂNCIA DE REFERÊNCIA
Para verificação do procedimento de ensaio, pode(m) ser ensaiada(s) (uma) substância(s) de referência, como por exemplo o 3,5-diclorofenol, utilizado na prova internacional do anel (4). O dicromato de potássio pode também ser utilizado como substância de referência para as algas verdes. Será desejável proceder ao ensaio de uma substância de referência pelo menos duas vezes por ano.
1.7. VALIDADE DO ENSAIO
Um ensaio é considerado válido quando satisfaz os seguintes critérios de desempenho:
|
— |
A biomassa das culturas de controlo deve ter aumentado exponencialmente num factor de pelo menos 16 durante as 72 horas do período de ensaio. Esse valor corresponde a uma taxa de crescimento específica de 0,92 dia–1. A taxa de crescimento das espécies mais frequentemente utilizadas é geralmente muito mais elevada (ver o apêndice 1). Este critério não poderá ser cumprido quando se utilizarem espécies de crescimento mais lento do que as espécies que constam do apêndice 1. Nesses casos, o período de ensaio deverá ser alargado para permitir um factor de crescimento mínimo de 16 nas culturas de controlo, devendo esse crescimento ser exponencial durante todo o período de ensaio. O período de ensaio poderá ser reduzido até ao mínimo de 48 horas, de modo a manter um crescimento exponencial não limitado durante todo o período de ensaio, desde que o factor mínimo de multiplicação por 16 seja atingido, |
|
— |
O coeficiente médio de variação das taxas de crescimento específico em cada secção do ensaio (dias 0-1, 1-2 e 2-3, no caso dos ensaios de 72 horas) nas culturas de controlo (ver no ponto 1.2 a definição de «coeficiente de variação») não deve exceder os 35 %. No que respeita ao cálculo das taxas de crescimento específico em cada secção do ensaio, ver o segundo parágrafo do ponto 2.2.1. Este critério é aplicável ao valor médio dos coeficientes de variação calculados para os replicados das culturas de controlo, |
|
— |
O coeficiente de variação das taxas médias de crescimento específico dos replicados das culturas de ensaio, durante todo o período de ensaio, não deve exceder 7 % nos ensaios com Pseudokirchneriella subcapitata e com Desmodesmus subspicatus. Em relação a outras espécies menos utilizadas, esse valor não deve exceder os 10 %. |
1.8. DESCRIÇÃO DO MÉTODO
1.8.1. Equipamento
Os recipientes de ensaio e outros equipamentos que entrem em contacto com as soluções de ensaio devem ser totalmente construídos em vidro ou noutro material quimicamente inerte. Todo o material deve ser cuidadosamente lavado, de forma a garantir que nenhum contaminante orgânico ou inorgânico possa interferir com o crescimento das algas ou com a composição das soluções de ensaio.
Os recipientes de ensaio serão normalmente frascos de vidro dimensionados de forma a permitir a recolha dos volumes de cultura necessários para as medições ao longo do ensaio e a garantir uma superfície de contacto suficiente para a permuta de CO2 com a atmosfera (ver o segundo parágrafo do ponto 1.8.9). De notar que o volume de líquido deve ser suficiente para as necessárias determinações analíticas (ver o quinto parágrafo do ponto 1.8.11).
Adicionalmente, será necessário o seguinte equipamento:
|
— |
Aparelho de cultura: recomenda-se a utilização de um armário ou câmara onde a temperatura de incubação escolhida possa ser mantida com uma precisão de ±2 °C, |
|
— |
Instrumentos de medição da luz: é importante notar que o método escolhido para a medição da intensidade luminosa e, em especial, o tipo de receptor (colector) terá influência sobre o valor medido. As medições deverão ser feitas, de preferência, utilizando um receptor esférico (4 π, que responda à luz directa e à luz reflectida a partir de todos os ângulos acima e abaixo do plano de medição) ou um receptor 2 π (que responda à luz proveniente de todos os ângulos acima do plano de medição), |
|
— |
Aparelho para determinação da biomassa das algas: A contagem de células, que é o parâmetro alternativo mais frequentemente utilizado para a biomassa das algas, poderá ser feita utilizando um contador electrónico de partículas, um microscópio com câmara de contagem ou um citómetro de fluxo. Outros métodos alternativos possíveis são a medição com um citómetro de massa, com um fluorímetro, com um espectrofotómetro ou com um colorímetro. Será conveniente calcular um factor de conversão que relacione a contagem de células com o peso seco. A fim de conseguir medições úteis para as baixas concentrações de biomassa, quando se utilize um espectrofotómetro poderá ser necessário utilizar células com um percurso óptico mínimo de 4 cm. |
1.8.2. Organismos de ensaio
Podem ser utilizadas diversas espécies de cianobactérias e de microalgas de água livre. As estirpes que constam da lista do apêndice 1 mostraram-se apropriadas para utilização no procedimento especificado no presente método de ensaio.
Se forem utilizadas outras espécies, deverá ser comunicada a respectiva estirpe e/ou origem. É necessário confirmar que o crescimento exponencial das algas seleccionadas para o ensaio pode ser mantido ao longo de todo o período de ensaio, nas condições vigentes.
1.8.3. Meio de crescimento
São recomendados dois meios de crescimento alternativo, o meio OCDE e o meio AAP. As composições desses meios são apresentadas no apêndice 2. De notar que o valor inicial de pH e a capacidade tampão (regulação do aumento do pH) dos dois meios são diferentes. Logo, os resultados dos ensaios poderão ser diferentes em função do meio utilizado, em especial quando estiverem em estudo substâncias ionizantes.
Poderá ser necessário modificar o meio de crescimento para determinados efeitos, como por exemplo quando se pretender proceder ao ensaio de metais ou de agentes quelantes ou quando se pretender proceder a ensaios a diferentes valores de pH. A utilização de um meio modificado deve ser descrita em pormenor e justificada (3)(4).
1.8.4. Concentração inicial de biomassa
A biomassa inicial das culturas de ensaio deve ser a mesma em todas as culturas e suficientemente baixa para permitir um crescimento exponencial ao longo de todo o período de incubação, sem risco de esgotamento dos nutrientes. A biomassa inicial não deve ser superior a 0,5 mg/l em peso seco. São recomendadas as seguintes concentrações iniciais de células:
|
Pseudokirchneriella subcapitata: |
5 x 103-104 |
células/ml |
|
Desmodesmus subspicatus |
2-5 x 103 |
células/ml |
|
Navicula pelliculosa |
104 |
células/ml |
|
Anabaena flos-aquae |
104 |
células/ml |
|
Synechococcus leopoliensis |
5 x 104-105 |
células/ml |
1.8.5. Concentrações da substância em estudo
A gama de concentrações na qual é provável a ocorrência de efeitos pode ser determinada com base nos resultados de ensaios de determinação da gama de concentrações a utilizar. Para o ensaio final e definitivo, devem ser seleccionadas pelo menos cinco concentrações, organizadas numa série geométrica com um factor que não exceda os 3,2. Para as substâncias em estudo que mostrem uma curva de resposta à concentração invariável, poderá justificar-se um factor mais elevado. As séries de concentrações deverão, de preferência, abranger uma gama que cause uma inibição de 5-75 % da taxa de crescimento das algas.
1.8.6. Replicados e controlos
O ensaio deve ser conduzido com três replicados de cada concentração em estudo. Caso não seja necessário determinar o NOEC, o ensaio poderá ser alterado de modo a aumentar o número de concentrações estudadas, reduzindo o número de replicados por concentração. O controlo deve ter, no mínimo, três replicados e o ideal será utilizar para o controlo o dobro do número de replicados utilizados para cada concentração estudada.
Poderá ser preparado um conjunto separado de soluções de ensaio para as determinações analíticas das concentrações da substância em estudo (ver o quarto e o sexto parágrafos do ponto 1.8.11).
Quando for utilizado um solvente para solubilização da substância em estudo, o ensaio deverá incluir controlos adicionais que contenham esse solvente à mesma concentração usada nas culturas de ensaio.
1.8.7. Preparação da cultura de inóculo
Para adaptar as algas às condições de ensaio e garantir que se encontram na fase de crescimento exponencial quando são utilizadas para inocular as soluções em estudo, prepara-se uma cultura de inóculo, em meio de ensaio, 2-4 dias antes do início do mesmo. A biomassa de algas deve ser ajustada de modo a permitir que o inóculo se mantenha em crescimento exponencial até ao início do ensaio. A cultura de inóculo deve ser incubada nas mesmas condições que as culturas de ensaio. O aumento da biomassa da cultura de inóculo será medido, para garantir que o seu crescimento se encontra dentro dos padrões normais para a estirpe de ensaio nas condições de cultura. Um exemplo do procedimento a utilizar para a cultura de algas é apresentado no apêndice 3. Para evitar uma situação de divisões celulares sincronizadas durante o ensaio, poderá ser necessário executar um segundo passo de propagação da cultura de inóculo.
1.8.8. Preparação das soluções em estudo
Todas as soluções em estudo devem conter a mesma concentração de meio de crescimento e a mesma biomassa inicial de algas de ensaio. As soluções de ensaio às concentrações escolhidas são normalmente preparadas misturando uma solução de reserva da substância em estudo com meio de crescimento e com a cultura de inóculo. As soluções de reserva são normalmente preparadas por dissolução da substância no meio de ensaio.
Nos casos em que se pretenda adicionar ao meio de ensaio substâncias pouco hidrossolúveis, poderão ser utilizados solventes como a acetona, o álcool t-butílico ou a dimetilformamida (2)(3). A concentração de solvente, que deve ser acrescentado a todas as culturas (incluindo as culturas de controlo) do ensaio à mesma concentração, não deve ultrapassar os 100 µl/l.
1.8.9. Incubação
Tapar os frascos de ensaio com rolhas permeáveis ao ar. Os frascos são agitados e colocados no aparelho de incubação. Durante o ensaio, é necessário manter as algas em suspensão e facilitar as transferências de CO2. Para tal, deve utilizar-se uma agitação ou um movimento constante. As culturas devem ser mantidas a uma temperatura entre os 21 e os 24 °C, controlada com uma precisão de ±2 °C. Quando forem utilizadas espécies diferentes das que constam da lista do apêndice 1, por exemplo tropicais, poderão ser necessário utilizar temperaturas mais elevadas, desde que sejam cumpridos os critérios de validade. Recomenda-se que os frascos sejam colocados na incubadora de forma aleatória e que a respectiva posição seja mudada diariamente.
O pH do meio de controlo não deve aumentar mais do que 1,5 unidades durante o ensaio. Para os metais e compostos que ionizam parcialmente a um pH próximo do pH do ensaio, poderá ser necessário limitar a variação do pH, a fim de obter resultados reprodutíveis e bem definidos. Uma variação inferior a 0,5 unidades de pH é tecnicamente realizável e pode ser conseguida se se garantir uma taxa de transferência mássica de CO2 da atmosfera para a solução de ensaio, por exemplo aumentando a agitação. Outra hipótese passa pela redução das necessidades de CO2, reduzindo a biomassa inicial ou a duração do ensaio.
A superfície em que as culturas são incubadas deve receber uma iluminação fluorescente contínua e uniforme, do tipo «branco frio» ou «luz do dia». As diferentes estirpes de algas e cianobactérias têm diferentes necessidades em termos de luz. A intensidade da luz deve, portanto, ser adaptada ao organismo de ensaio utilizado. Para as espécies recomendadas de algas verdes, a intensidade da luz ao nível da solução de ensaio será seleccionada na gama dos 60-120 µE·m–2·s–1, medidos na gama de comprimentos de onda necessária para a fotossíntese, entre 400-700 nm, utilizando um receptor adequado. Algumas espécies, em especial a Anabaena flos-aquae, crescem bem com menor intensidade de luz, podendo mesmo ser prejudicadas por uma luz demasiado intensa. Para essas espécies, deve ser utilizada intensidade luminosa média de 40-60 µE·m–2·s–1 (para os instrumentos de medição da luz calibrados em lux, uma gama equivalente a 4 440-8 880 lux para a luz branca corresponde aproximadamente à intensidade luminosa recomendada de 60-120 µE·m–2·s–1). A intensidade luminosa não deve variar mais de ±15 % em relação à intensidade média sobre a zona de incubação.
1.8.10. Duração do ensaio
A duração normal dos ensaios é de 72 horas. No entanto, poderão ser efectuados ensaios de maior ou menor duração, desde que estejam cumpridos todos os critérios de validade referidos no ponto 1.7.
1.8.11. Medições e determinações analíticas
A biomassa de algas em cada frasco será determinada pelo menos uma vez por dia, ao longo do período de ensaio. Se as medições forem efectuadas em pequenos volumes retirados da solução de ensaio por meio de uma pipeta, esses volumes não devem ser repostos.
A medição da biomassa é efectuada por contagem manual de células num microscópio ou num contador electrónico de partículas (contagem de células e/ou biovolume). Podem ser utilizadas outras técnicas alternativas, como a citometria de fluxo, a fluorescência clorofílica in vitro ou in vivo (6)(7) ou a densidade óptica, desde que se possa demonstrar uma boa correlação com a biomassa ao longo de toda a gama de concentrações de biomassa prevista durante o ensaio.
O pH das soluções será medido no início e no final do ensaio.
Desde que exista um procedimento analítico para a determinação da substância em estudo na gama de concentrações utilizada, as soluções de ensaio deverão ser analisadas para verificação da concentração inicial e da manutenção da concentração de exposição ao longo do ensaio.
A análise da concentração da substância em estudo no início e no final do ensaio, para as concentrações de ensaio mais elevadas e mais baixas e para uma concentração próxima do EC50 previsto, poderá ser suficiente quando não for esperada uma variação das concentrações de exposição superior a 20 % em relação ao respectivo valor nominal ao longo do ensaio. Nos casos em que seja improvável que as concentrações se mantenham no intervalo de 80-120 % da concentração nominal, recomenda-se a análise de todas as concentrações de ensaio no início e no final do mesmo. Para as substâncias em estudo voláteis, instáveis ou fortemente adsorventes, recomenda-se a realização de amostragens suplementares para análise a intervalos de 24 horas durante o período de exposição, de modo a caracterizar melhor a diminuição da concentração da substância em estudo. Para essas substâncias, será necessário utilizar um número maior de replicados. Em qualquer dos casos, a determinação das concentrações da substância em estudo só é necessária num dos frascos replicados para cada concentração de ensaio (ou no conteúdo misturado de todos os frascos replicados).
O meio de ensaio preparado especificamente para a análise das concentrações de exposição durante o ensaio deverá ser tratado da mesma forma que o utilizado no ensaio, ou seja, deve ser inoculado com algas e incubado em condições idênticas. Se for necessário analisar a concentração da substância em estudo dissolvida, as algas poderão ter de ser separadas do meio de crescimento. Essa separação deverá, de preferência, ser feita por centrifugação a baixa velocidade, suficiente para garantir a deposição das algas.
Se existirem provas de que a concentração da substância de ensaio não variou mais de 20 %, durante o ensaio, em relação ao valor da concentração nominal ou da concentração inicial medida, a análise dos resultados poderá basear-se nos valores nominais ou iniciais medidos. Se a variação em relação à concentração nominal ou à concentração inicial medida for superior a ±20 %, a análise dos resultados deverá basear-se na média geométrica da concentração durante a exposição ou em modelos que descrevam a diminuição da concentração da substância em estudo (3)(8).
O ensaio de inibição de crescimento de algas representa um sistema de ensaio mais dinâmico do que a maior parte dos outros tipos de ensaio de toxicidade a curto prazo em meio aquático. Logo, as concentrações reais de exposição poderão ser difíceis de determinar, especialmente para as substâncias mais adsorventes e ensaiadas a concentrações mais baixas. Nesses casos, o desaparecimento da substância da solução, por adsorção à cada vez maior biomassa de algas, não significa que a substância tenha desaparecido do sistema de ensaio. Quando os resultados do ensaio forem analisados, deve verificar-se se a diminuição da concentração da substância em estudo ao longo do mesmo é ou não acompanhada de uma diminuição da inibição do crescimento. Se for esse o caso, poderá ser considerada a possibilidade de utilização de um modelo que descreva a diminuição da concentração da substância em estudo (8). Senão, o melhor poderá ser basear a análise dos resultados nas concentrações iniciais (nominais ou medidas).
1.8.12. Outras observações
O inóculo deverá ser observado ao microscópio para verificar se a cultura apresenta um aspecto normal e saudável, tal como as culturas de ensaio, no final do mesmo, para verificar se se observa algum aspecto anormal das algas que possa ser causado pela exposição à substância em estudo.
1.8.13. Ensaio limite
Em determinadas circunstâncias, ou seja, quando um ensaio preliminar indicar que a substância em estudo não apresenta efeitos tóxicos em concentrações até aos 100 mg·l–1 ou até ao limite de solubilidade da substância no meio de ensaio (conforme a que seja menor), poderá proceder-se a um ensaio limite com comparação das respostas de um grupo de controlo e de um grupo exposto à substância em estudo (a uma concentração de 100 mg·l–1 ou que corresponda ao limite de solubilidade da substância no meio de ensaio). É fortemente recomendado que se confirme a ausência de toxicidade através da análise da concentração de exposição. Todas as condições de ensaio e critérios de validade anteriormente descritos são aplicáveis aos ensaios limite, com excepção da necessidade de utilizar, no mínimo, seis replicados dos frascos expostos à substância. As variáveis de resposta dos grupos de controlo e de exposição podem ser analisadas através de um ensaio estatístico de comparação das médias, como por exemplo o teste t-Student. Caso as variâncias dos dois grupos sejam diferentes, deverá ser realizado um teste t-Student ajustado para diferenças das variâncias.
1.8.14. Modificação para substâncias fortemente coradas
Deve ser utilizada uma irradiação (intensidade luminosa) na parte superior da gama prevista pelo presente método de ensaio: 120 µE·m–2·s–1 ou mais.
O percurso óptico deve ser encurtado, reduzindo o volume das soluções de ensaio (cerca de 5-25 ml).
Deve proceder-se a uma agitação suficiente, por exemplo sacudindo ligeiramente, de modo a obter uma frequência elevada de exposição das algas a altos níveis de irradiação na superfície da cultura.
2. DADOS
2.1. APRESENTAÇÃO DAS CURVAS DE CRESCIMENTO
A biomassa dos frascos de ensaio pode ser expressa nas unidades do parâmetro alternativo que tenha sido utilizado para as medições (p. ex.: número de células, fluorescência).
Criar tabelas com a concentração estimada da biomassa das culturas de ensaio e dos controlos, em função das concentrações do material em estudo e do momento da amostragem, arredondado às horas, de modo a produzir gráficos das curvas de crescimento. Numa primeira fase poderá ser útil utilizar tanto a escala geométrica como a logarítmica, mas a segunda é obrigatória e resulta geralmente numa melhor representação das variações do padrão de crescimento durante o período de ensaio. De notar que o crescimento exponencial, quando apresentado em escala logarítmica, resulta numa recta cuja inclinação (declive) indica a taxa de crescimento específico.
Utilizando os gráficos, verificar se as culturas de controlo cresceram exponencialmente à taxa prevista ao longo de todo o ensaio. São factores críticos a análise de todos os pontos, do aspecto global dos gráficos e a verificação dos dados brutos e dos procedimentos aplicados, a fim de detectar eventuais erros. Verificar, em particular, qualquer dos pontos que pareça ser afectado por erros sistemáticos. Se se identificarem e/ou forem altamente prováveis erros de procedimento, o ponto em causa será identificado como um ponto aberrante e não deverá ser incluído na análise estatística subsequente (uma concentração de algas igual a zero num dos dois ou três replicados pode indicar que o frasco não foi correctamente inoculado, ou que não estava suficientemente limpo). Os motivos para a rejeição de um ponto considerado como aberrante devem ser claramente indicados no respectivo relatório de ensaio. Os motivos aceites são exclusivamente os (raros) erros de procedimento e não a simples falta de precisão. Os procedimentos estatísticos para a identificação dos pontos aberrantes apresentam uma utilidade limitada para este tipo de problemas, não substituindo nunca a opinião abalizada de um perito. Os pontos aberrantes (assinalados como tal) deverão, de preferência, ser conservados entre os dados apresentados posteriormente em qualquer gráfico ou tabela.
2.2. VARIÁVEIS DE RESPOSTA
O objectivo do ensaio é determinar os efeitos da substância em estudo sobre o crescimento das algas. O método de ensaio descreve duas variáveis de resposta, já que os Estados-Membros têm diferentes preferências e necessidades regulamentares. Para que os resultados dos ensaios possam ser aceitáveis em todos os Estados-Membros, os efeitos terão de ser avaliados utilizando ambas as variáveis de resposta, a) e b), a seguir descritas.
|
a) |
Taxa média de crescimento específico: esta variável é calculada com base no aumento logarítmico diário da biomassa durante o período de ensaio. |
|
b) |
Rendimento: a variável de resposta é a biomassa no final do ensaio menos a biomassa no início do mesmo. |
Para efeitos da aplicação do presente método no âmbito do quadro regulamentar da UE, o cálculo dos resultados deverá basear-se numa taxa média de crescimento específico, pelos motivos abaixo expostos. Cabe aqui notar que os valores de toxicidade calculados através destas duas variáveis de resposta não são comparáveis e que essa diferença tem de ser reconhecida para efeitos da utilização dos resultados do ensaio. Os valores de ECx baseados na taxa média de crescimento específico (ErCx) serão geralmente mais elevados do que os resultados baseados no rendimento (EyCx), se forem seguidas as condições de ensaio apresentadas no presente método de ensaio, devido à base matemática das respectivas abordagens. Esse facto não deve ser interpretado como uma diferença de sensibilidade entre as duas variáveis de resposta, mas simplesmente como uma diferença matemática entre os valores. O conceito de taxa média de crescimento específico baseia-se no padrão geral de crescimento exponencial das algas em culturas não sujeitas a limitações, sendo a toxicidade estimada com base nos efeitos sobre a taxa de crescimento, e não depende do valor absoluto da taxa de crescimento específico dos controlos, do declive da curva de concentração-resposta ou da duração do ensaio. Em contraste, os resultados baseados no rendimento enquanto variável de resposta são dependentes de todas essas variáveis. O valor de EyCx é dependente da taxa de crescimento específico da espécie de algas utilizada em cada ensaio e da taxa máxima de crescimento específico, que pode ser diferente para as diferentes espécies ou mesmo para as diferentes estirpes de algas. Esta variável de resposta não deve ser utilizada para comparar a sensibilidade das diferentes espécies ou mesmo das diferentes estirpes de algas aos produtos tóxicos. Embora seja preferível, do ponto de vista científico, utilizar a taxa média de crescimento específico para a estimação da toxicidade, as estimativas da toxicidade com base no rendimento foram também incluídas no presente método de ensaio para satisfazer as actuais exigências regulamentares de alguns países.
2.2.1. Taxa média de crescimento
A taxa média de crescimento específico para um determinado período é calculada como o aumento logarítmico da biomassa em cada um dos frascos de controlo e de ensaio, a partir da seguinte equação:
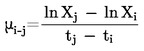 (dia–1)
(dia–1)
onde:
|
µ i-j : |
é a taxa média de crescimento específico entre o momento i e o momento j |
|
X i : |
é a biomassa no momento i |
|
X j : |
é a biomassa no momento j |
Para cada um dos grupos de exposição e de controlo, calcular um valor médio da taxa de crescimento, associado às respectivas estimativas da variância.
Calcular a taxa média de crescimento específico ao longo de todo o ensaio (normalmente nos dias 0-3), utilizando o valor nominal da biomassa inoculada como valor inicial, de preferência à utilização de um valor medido nesse momento, já que dessa forma se consegue geralmente uma precisão mais elevada. Se o equipamento utilizado para a medição da biomassa permitir uma determinação suficientemente precisa dos níveis reduzidos de biomassa presentes após a inoculação (p. ex.: citómetro de fluxo), poderá utilizar-se o valor medido de concentração inicial da biomassa. Verificar igualmente as taxas de crescimento em cada secção do ensaio, calculadas como as taxas de crescimento específico em cada dia do ensaio (dias 0-1, 1-2 e 2-3), verificando se a taxa de crescimento dos controlos se mantém constante (ver os critérios de validade, ponto 1.7). Uma taxa de crescimento específico significativamente mais baixa no primeiro dia do que a taxa média de crescimento específico da totalidade do ensaio pode indicar uma fase de latência. Embora a fase de latência possa ser minimizada ou mesmo praticamente eliminada nas culturas de controlo através de uma propagação adequada da cultura de arranque, essa fase de latência nas culturas expostas à substância em estudo pode indicar uma recuperação após a fase inicial de stress por toxicidade ou uma menor exposição à substância em estudo devido ao seu desaparecimento (incluindo a eventual adsorção à biomassa das algas) após a exposição inicial. Logo, a taxa de crescimento em cada secção do ensaio pode ser avaliada para verificar os efeitos da substância em estudo durante o período de exposição. A existência de diferenças substanciais entre as taxas de crescimento de cada secção do ensaio e a taxa média de crescimento indica um desvio em relação à situação de crescimento exponencial constante, exigindo portanto uma análise mais pormenorizada das curvas de crescimento.
Calcular a percentagem de inibição da taxa de crescimento para cada um dos replicados expostos, utilizando a seguinte equação:

onde:
|
%Ir: |
percentagem de inibição da taxa média de crescimento específico |
|
µC: |
valor médio das taxas médias de crescimento específico (µ) no grupo de controlo |
|
µT: |
taxa média de crescimento específico dos replicados do grupo exposto |
Quando forem utilizados solventes na preparação das soluções de ensaio, devem utilizar-se para o cálculo da percentagem de inibição os controlos com adição de solvente, em vez dos controlos apenas do meio de cultura.
2.2.2. Rendimento
O rendimento é calculado como a biomassa no final do ensaio menos a biomassa inicial, em cada um dos frascos de controlo e de exposição. Para cada uma das concentrações de ensaio e de controlo, calcular um valor médio de rendimento, associado às respectivas estimativas da variância. A percentagem de inibição do rendimento (% Iy) pode ser calculada, para cada replicado exposto, do seguinte modo:

onde:
|
% Iy: |
percentagem de inibição do rendimento |
|
YC: |
valor médio de rendimento no grupo de controlo |
|
YT: |
valor de rendimento para os replicados expostos |
2.3. GRÁFICO DA CURVA DE RESPOSTA À CONCENTRAÇÃO
Desenhar o gráfico da percentagem de inibição em função do logaritmo da concentração da substância em estudo e analisar o gráfico cuidadosamente, não entrando em consideração com os pontos que tenham sido identificados como aberrantes na primeira fase. Ajustar uma curva aproximada aos pontos experimentais, à vista ou por interpolação informática, de modo a obter uma primeira impressão da relação de resposta à concentração, após o que se deverá proceder à definição de uma curva mais exacta, de preferência utilizando métodos estatísticos computorizados. Dependendo da utilização que se pretenda dar aos dados, a qualidade (precisão) e a quantidade de dados, bem como a disponibilidade de ferramentas de análise dos mesmos, poderá decidir-se (por vezes, justificadamente) nesta fase não continuar a análise dos dados e limitar as leituras à determinação dos valores-chave EC50 e EC10 (e/ou EC20) a partir da curva ajustada à vista (ver também o ponto seguinte, em relação aos efeitos de estimulação). Entre as razões válidas para a não utilização de um método estatístico podem referir-se:
|
— |
Os dados não permitem obter, a partir de um método computorizado, resultados mais fiáveis do que os que se conseguem obter por opinião abalizada — nessas situações, alguns programas informáticos poderão mesmo ser incapazes de apresentar qualquer solução fiável (iterações divergentes, etc.), |
|
— |
Os casos em que a respostas é a estimulação do crescimento não são bem descritos pelos programas informáticos disponíveis (ver abaixo). |
2.4. PROCEDIMENTOS ESTATÍSTICOS
O objectivo é descrever de forma quantitativa, por análise de regressão, a relação concentração-resposta. Pode utilizar-se uma regressão linear ponderada, antecedida de uma transformação de linearização dos dados de resposta — por exemplo para unidades probit, logit ou de Weibull (9), mas a técnica preferida é a aplicação de procedimentos de regressão não linear, que permitem lidar melhor com as inevitáveis irregularidades dos dados e com os desvios em relação a uma boa distribuição. Nas zonas próximas da inibição nula ou total, essas irregularidades podem mesmo ser magnificadas pela transformação, o que irá interferir com a análise (9). Cabe aqui notar que os métodos padrão de análise que utilizam os transformados probit, logit ou Weibull se destinam à análise de dados quantais (p. ex.: mortalidade e sobrevivência), pelo que terão de ser alterados para lidar com dados de crescimento ou de biomassa. Alguns métodos para a determinação dos valores de ECx a partir de dados contínuos podem ser consultados em (10), (11) e (12). A utilização da análise de regressão não linear é apresentada com mais pormenor no apêndice 4.
Para cada uma das variáveis de resposta em estudo, utilizar a relação concentração-resposta para estimar os valores de ECx. Sempre que possível, devem ser determinados os limites do intervalo de confiança a 95 %. Para cada estimativa, a adequação do ajustamento da curva estimada pelo modelo de regressão aos dados de resposta deve ser avaliada, graficamente ou por métodos estatísticos. A análise de regressão deve ser efectuada utilizando os valores de cada replicado e não o valor médio para cada grupo exposto. No entanto, se o ajustamento de um gráfico não linear se revelar difícil ou não for possível devido à grande dispersão dos dados, o problema poderá ser contornado pela aplicação da regressão aos valores médios de cada grupo, como forma prática de reduzir a influência dos pontos que sejam provavelmente aberrantes. A utilização desse método deve ser referida no relatório de ensaio como um desvio em relação ao procedimento normal, por não ter sido possível ajustar uma curva aos valores individuais de todos os replicados com bons resultados.
As estimativas do EC50 e os respectivos intervalos de confiança podem também ser obtidos utilizando uma interpolação linear com bootstrapping (13), quando os modelos/métodos de regressão disponíveis não forem aplicáveis aos dados existentes.
Para a estimação do LOEC e, portanto, do NOEC, bem como dos efeitos da substância em estudo sobre a taxa de crescimento, será necessário analisar as médias dos frascos expostos utilizando técnicas de análise da variância (ANOVA). A média para cada concentração deve então ser comparada com a média observada no controlo, utilizando um método apropriado de comparação múltipla ou de análise das tendências. Os testes de Dunnett ou de Williams (14)(15)(16)(17)(18) poderão ser úteis para esse efeito. É necessário verificar se está garantida a presunção de homogeneidade da variância das análises ANOVA. Essa verificação poderá ser realizada graficamente ou através de um teste formal (18). Podem ser utilizados os testes de Levene ou de Bartlett. O problema do não cumprimento da presunção de homogeneidade das variâncias pode por vezes ser corrigido através da transformação logarítmica dos dados. Caso a heterogeneidade das variâncias seja extrema e não possa ser corrigida por transformação, deverá ser considerada a possibilidade de utilização de métodos de análise das tendências como por exemplo o método degressivo de Jonkheere. Para mais orientações sobre a determinação do NOEC, consultar a referência (12).
Os mais recentes desenvolvimentos científicos conduziram à recomendação de que fosse abandonado o conceito de NOEC, que deverá ser substituído pela estimação de valores de ECx por regressão. No caso do presente ensaio com algas, não foi definido nenhum valor ideal para o x. A gama entre 10-20 % parece ser apropriada (dependendo da variável de resposta escolhida), devendo ser comunicados, de preferência, tanto os valores de EC10 como de EC20.
2.5. ESTIMULAÇÃO DO CRESCIMENTO
Por vezes, pode observa-se uma estimulação do crescimento (inibição negativa) a baixas concentrações. Esse fenómeno pode resultar tanto de hormese («estimulação por tóxicos») como da adição de factores estimulantes do crescimento associados ao material em estudo que é adicionado ao meio de reserva. Cabe aqui notar que a adição de nutrientes inorgânicos não deverá, em princípio, ter qualquer efeito directo sobre o ensaio, na medida em que o meio de ensaio deve, de qualquer modo, garantir uma situação de excedente de nutrientes ao longo de todo o ensaio. A estimulação a baixas concentrações pode geralmente ser ignorada para efeitos do cálculo de EC50, a não nos casos em que é extrema. Nesses casos de estimulação extrema, ou quando o valor de x em ECx é muito baixo, poderá ser necessário utilizar procedimentos especiais. A eliminação pura e simples das respostas de estimulação para efeitos da análise dos dados deve ser evitada sempre que possível e, nos casos em que os programas de ajustamento de curvas aos dados não consigam lidar com as consequências da estimulação a baixas concentrações, pode recorrer-se à interpolação linear com bootstrapping. Se a estimulação seja extrema, pode considerar-se a possibilidade de utilizar um modelo de hormese (19).
2.6. INIBIÇÃO NÃO TÓXICA DO CRESCIMENTO
Os materiais em estudo que absorvam a luz podem ocasionar uma diminuição da taxa de crescimento por efeito da redução da quantidade de luz disponível. Esse efeitos de tipo físico devem ser calculados de forma independente dos efeitos tóxicos, se necessário alterando as condições do ensaio, e apresentados separadamente. Para mais orientação sobre esta questão, ver as referências (2) e (3).
3. RELATÓRIOS
3.1. RELATÓRIO DE ENSAIO
O relatório do ensaio deve incluir a seguinte informação:
Substância em estudo:
|
— |
natureza física e propriedades fisico-químicas relevantes, incluindo o limite de hidrossolubilidade, |
|
— |
dados relativos à identificação química, incluindo a pureza. |
Espécie de ensaio:
|
— |
estirpe, fornecedor ou fonte do organismo e condições de cultura utilizadas. |
Condições de ensaio:
|
— |
data de início do ensaio e respectiva duração, |
|
— |
descrição do planeamento do ensaio: recipientes de ensaio, volume das culturas, densidade da biomassa no início do ensaio, |
|
— |
composição do meio, |
|
— |
concentrações de ensaio e replicados (p. ex.: número de replicados, número de concentrações ensaiadas e progressão geométrica utilizada), |
|
— |
descrição da preparação das soluções de ensaio, incluindo a eventual utilização de solventes, etc., |
|
— |
equipamento de incubação, |
|
— |
intensidade e qualidade da luz (fonte, homogeneidade), |
|
— |
temperatura, |
|
— |
concentrações ensaiadas: concentrações nominais do ensaio e resultados das análises para determinação da concentração da substância em estudo nos frascos de ensaio. Devem ser comunicados a eficácia de recuperação e o limite de quantificação do método na matriz de ensaio, |
|
— |
todos os desvios em relação ao presente método de ensaio, |
|
— |
método de determinação da biomassa e provas da correlação entre o parâmetro medido e o peso seco. |
Resultados:
|
— |
valores do pH no início e no final do ensaio, para todos os frascos expostos, |
|
— |
biomassa em cada frasco e em cada ponto de medição, bem como o método utilizado para a sua medição, |
|
— |
curvas de crescimento (gráficos da biomassa em função do tempo), |
|
— |
variáveis de resposta calculadas para cada replicado exposto, com os respectivos valores médios, e coeficiente de variação dos replicados, |
|
— |
representação gráfica da relação concentração/efeito, |
|
— |
estimativas da toxicidade para as variáveis de resposta, p. ex.: EC50, EC10, EC20, e os intervalos de confiança associados. Quando forem calculados, valores do LOEC e do NOEC e métodos estatísticos utilizados para a respectiva determinação, |
|
— |
caso tenha sido utilizada uma análise ANOVA, dimensão do efeito detectado (p. ex.:, diferenças menos significativas), |
|
— |
ocorrência de estimulação do crescimento que tenha sido verificada em qualquer dos grupos expostos, |
|
— |
qualquer outro efeito observado, como por exemplo a alteração da morfologia das algas, |
|
— |
discussão dos resultados, incluindo qualquer influência sobre os resultado do ensaio que seja decorrente das alterações efectuadas em relação ao presente método de ensaio. |
4. BIBLIOGRAFIA
|
(1) |
OECD TG 201 (2006) Freshwater Alga and Cyanobacteria, Growth Inhibition Test |
|
(2) |
ISO 1998: Water quality — Guidance for algal growth inhibition tests with poorly soluble materials, volatile compounds, metals and waster water. ISO/DIS 14442 |
|
(3) |
OCDE 2000: Guidance Document on Aquatic Toxicity Testing of Difficult Substances and mixtures. Environmental Health and Safety Publications. Series on Testing and Assessment, n.o 23 |
|
(4) |
ISO 1998: Water quality — Sampling — Part 16: General Guidance for Biotesting. ISO 5667-16 |
|
(5) |
ISO 1993: Water quality — Algal growth inhibition test. ISO 8692 |
|
(6) |
Mayer, P., Cuhel, R. and Nyholm, N. (1997). A simple in vitro fluorescence method for biomass measurements in algal growth inhibition tests. Water Research 31: 2525-2531 |
|
(7) |
Slovacey, R.E. and Hanna, P.J. In vivo fluorescence determinations of phytoplancton chlorophyll, Limnology & Oceanography 22,5 (1977), pp.919-925 |
|
(8) |
Simpson, S.L., Roland, M.G.E., Stauber, J.L. and Batley, G.E. (2003) Effect of declining toxicant concentrations on algal bioassay endpoints. Environ. Toxicol. Chem 22, 2073-2079 |
|
(9) |
Christensen, E.R., Nyholm, N. (1984): Ecotoxicological Assays with Algae: Weibull Dose-Response Curves. Env. Sci. Technol. 19, 713-718 |
|
(10) |
Nyholm, N. Sørensen, P.S., Kusk, K.O. and Christensen, E.R. (1992): Statistical treatment of data from microbial toxicity tests. Environ. Toxicol. Chem. 11, 157-167 |
|
(11) |
Bruce, R.D., and Versteeg, D.J. (1992). A statistical procedure for modelling continuous toxicity data. Env. Toxicol. Chem. 11:1485-1494 |
|
(12) |
OCDE. (2004). Guidance Document on Statistical Analysis of Ecotoxicity Data |
|
(13) |
Norberg-King T.J. (1988) An interpolation estimate for chronic toxicity: The ICp approach. National Effluent Toxicity Assessment Center Technical Report 05-88. USEPA, Duluth, MN |
|
(14) |
Dunnett, C.W. (1955) A multiple comparisons procedure for comparing several treatments with a control. J. Amer. Statist. Assoc. 50: 1096-1121 |
|
(15) |
Dunnett, C.W. (1964) New tables for multiple comparisons with a control. Biometrics 20: 482-491 |
|
(16) |
Williams, D.A. (1971) A test for differences between treatment means when several dose levels are compared with a zero dose control. Biometrics 27: 103-117 |
|
(17) |
Williams, D.A. (1972) The comparison of several dose levels with a zero dose control. Biometrics 28: 510-531 |
|
(18) |
Draper, N.R. and Smith, H. (1981). Applied Regression Analysis, second edition. Wiley, New York |
|
(19) |
Brain P. and Cousens R. (1989). An equation to describe dose-responses where there is stimulation of growth at low doses. Weed Research, 29, 93-96 |
Apêndice 1
Estirpes que se revelaram adequadas para o ensaio
Algas verdes
|
— |
Pseudokirchneriella subcapitata (anteriormente designada por Selenastrum capricornutum), ATCC 22662, CCAP 278/4, 61.81 SAG |
|
— |
Desmodesmus subspicatus (anteriormente designada por Scenedesmus subspicatus) 86.81 SAG |
Diatomáceas
|
— |
Navicula pelliculosa, UTEX 664 |
Cianobactérias
|
— |
Anabaena flos-aquae, UTEX 1444, ATCC 29413, CCAP 1403/13A |
|
— |
Synechococcus leopoliensis, UTEX 625, CCAP 1405/1 |
Origem das estirpes
As estirpes recomendadas estão disponíveis sob a forma de culturas puras nas seguintes colecções (por ordem alfabética):
|
|
|
|
|
|
|
|
|
|
|
|
Aspecto e características das espécies recomendadas
|
|
P. subcapitata |
D. subspicatus |
N. pelliculosa |
A. flos-aquae |
S. leopoliensis |
|
Aspecto |
Células isoladas, curvadas e torcidas |
Células ovais, na sua maioria isoladas |
Bastonetes |
Cadeias de células ovais |
Bastonetes |
|
Dimensões (C × L), em µm |
8-14 × 2-3 |
7-15 × 3-12 |
7,1 × 3,7 |
4,5 × 3 |
6 × 1 |
|
Volume celular (µm3/célula) |
40-60 (1) |
60-80 (1) |
40-50 (1) |
30-40 (1) |
2,5 (2) |
|
Peso seco das células (mg/célula) |
2-3 × 10–8 |
3-4 × 10–8 |
3-4 × 10–8 |
1-2 × 10–8 |
2-3 × 10–9 |
|
Taxa de crescimento (3) (dia–1) |
1,5-1,7 |
1,2-1,5 |
1,4 |
1,1-1,4 |
2,0-2,4 |
Recomendações específicas quanto à cultura e ao manuseamento das espécies de ensaio recomendadas
Pseudokirchneriella subcapitata e Desmodesmus subspicatus
Estas algas verdes são geralmente fáceis de conservar em diferentes meios de cultura. Toda a informação sobre os meios mais adequados está disponível junto das colecções de culturas. As células apresentam-se normalmente isoladas e a sua densidade pode ser facilmente medida utilizando um contador electrónico de partículas ou um microscópio.
Anabaena flos-aquae
Diferentes meios de cultura podem ser utilizados para manter uma cultura de arranque. É particularmente importante evitar que a cultura em meio líquido vá para além da fase de crescimento exponencial, já que a recuperação se torna difícil para lá desse ponto.
A Anabaena flos-aquae desenvolve agregados de cadeias de células. A dimensão desses agregados pode variar em função das condições de cultura. Para se poder proceder à contagem de células com um contador electrónico de partículas ou com um microscópio, com vista à determinação da biomassa, poderá ser necessário quebrar esses agregados.
As amostras podem ter de ser divididas em subamostras e tratadas com ultra-sons para quebrar as cadeias e reduzir a variabilidade das contagens. O tratamento com ultra-sons para quebrar as cadeias pode destruir as células se for demasiado prolongado. A intensidade e a duração do tratamento com ultra-sons devem ser idênticos para todas as amostras tratadas.
Contar as células num número suficiente de campos do hemocitómetro (pelo menos 400 células) para compensar a variabilidade. A fiabilidade das determinações através da densidade microscópica será assim maior.
O volume total de Anabaena pode ser determinado com um contador electrónico de partículas, depois de quebrar as cadeias de células através de um tratamento cuidadoso com ultra-sons. A energia dos ultra-sons deve ser ajustada de forma a evitar provocar danos nas células.
Utilizar um agitador magnético de rotação ou outro método semelhante que permita garantir que a suspensão de algas utilizada para inocular os frascos de ensaio esteja bem misturada e seja homogénea.
Os frascos de ensaio devem ser colocados num agitador orbital ou lateral, a cerca de 150 rotações por minuto. Em alternativa, pode utilizar-se uma agitação intermitente para reduzir a tendência para a criação de agregados da Anabaena. Se houver agregação, deve ter-se o cuidado de retirar amostras representativas para as medições de biomassa. Poderá ser necessário agitar vigorosamente os frascos imediatamente antes da mostra, de modo a desfazer os agregados de algas.
Synechococcus leopoliensis
Diferentes meios de cultura podem ser utilizados para manter uma cultura de arranque. Toda a informação sobre os meios mais adequados está disponível junto das colecções de culturas.
A Synechococcus leopoliensis apresenta células isoladas, em forma de bastonete. As células são muito pequenas, o que complica a utilização das contagens ao microscópio para a medição da biomassa. Poderá utilizar-se um contador electrónico de partículas equipado para a contagem de partículas com uma dimensão mínima aproximada de 1 µm. Outra possibilidade é medições fluorimétricas in vitro.
Navicula pelliculosa
Podem ser utilizados diferentes meios de cultura para manter uma cultura de arranque. Toda a informação sobre os meios mais adequados está disponível junto das colecções de culturas. De notar que o meio terá de conter silicatos.
A Navicula pelliculosa pode, em certas condições de cultura, formar agregados. Uma vez que produzem lípidos, as células têm por vezes tendência a acumularem-se na película superficial. Nessas circunstâncias, terão de se adoptar medidas especiais, aquando da recolha das subamostras para determinação da biomassa, de modo a obter amostras representativas. Poderá ser necessária uma agitação vigorosa, por exemplo com um agitador magnético de rotação.
(1) Medido com um contador electrónico de partículas.
(2) Calculado a partir das dimensões medidas.
(3) Taxa de crescimento mais frequentemente observada em meio OCDE, com incubação a 21 °C com uma intensidade luminosa de aproximadamente 70 µE·m–2·s–1.
Apêndice 2
Meio de cultura
Pode utilizar-se um dos dois meios de cultura seguintes:
Meio OCDE: meio original do método OECD TG 201, norma ISO 8692;
Meio US EPA AAP, norma ASTM.
Para a preparação destes meios, deve utilizar-se reagentes e produtos químicos de qualidade analítica e água desionizada.
Composição do meio AAP (US. EPA) e do meio OECD TG 201
|
Composto |
EPA |
OCDE |
||
|
|
mg/l |
mM |
mg/l |
mM |
|
NaHCO3 |
15,0 |
0,179 |
50,0 |
0,595 |
|
NaNO3 |
25,5 |
0,300 |
|
|
|
NH4Cl |
|
|
15,0 |
0,280 |
|
MgCl2·6(H2O) |
12,16 |
0,0598 |
12,0 |
0,0590 |
|
CaCl2·2(H2O) |
4,41 |
0,0300 |
18,0 |
0,122 |
|
MgSO4·7(H2O) |
14,6 |
0,0592 |
15,0 |
0,0609 |
|
K2HPO4 |
1,044 |
0,00599 |
|
|
|
KH2PO4 |
|
|
1,60 |
0,00919 |
|
FeCl3·6(H2O) |
0,160 |
0,000591 |
0,0640 |
0,000237 |
|
Na2EDTA·2(H2O) |
0,300 |
0,000806 |
0,100 |
0,000269 (1) |
|
H3BO3 |
0,186 |
0,00300 |
0,185 |
0,00299 |
|
MnCl2·4(H2O) |
0,415 |
0,00201 |
0,415 |
0,00210 |
|
ZnCl2 |
0,00327 |
0,000024 |
0,00300 |
0,0000220 |
|
CoCl2·6(H2O) |
0,00143 |
0,000006 |
0,00150 |
0,00000630 |
|
Na2MoO4·2(H2O) |
0,00726 |
0,000030 |
0,00700 |
0,0000289 |
|
CuCl2.2(H2O) |
0,000012 |
0,00000007 |
0,00001 |
0,00000006 |
|
pH |
7,5 |
8,1 |
||
Nos ensaios com a diatomácea Navicula pelliculosa, ambos os meios devem ser complementados com Na2SiO3·9H20, na quantidade necessária para obter uma concentração final de 1,4 mg Si/l.
O pH do meio é função do equilíbrio entre o sistema carbonato do meio e a pressão parcial de CO2 no ar atmosférico. A relação aproximada entre o pH a 25 °C e a concentração molar de bicarbonato é dada por:
pHeq = 11,30 + log [HCO3]
Com 15 mg/l de NaHCO3, pHeq = 7,5 (meio U.S. EPA) e com 50 mg/l NaHCO3, pHeq = 8,1 (meio OCDE).
Composição elementar dos meios de ensaio
|
Elemento |
EPA |
OCDE |
|
|
mg/l |
mg/l |
|
C |
2,144 |
7,148 |
|
N |
4,202 |
3,927 |
|
P |
0,186 |
0,285 |
|
K |
0,469 |
0,459 |
|
Na |
11,044 |
13,704 |
|
Ca |
1,202 |
4,905 |
|
Mg |
2,909 |
2,913 |
|
Fe |
0,033 |
0,017 |
|
Mn |
0,115 |
0,115 |
Preparação do meio OCDE
|
Nutriente |
Concentração na solução de reserva |
|
Solução de reserva 1: macronutrientes |
|
|
NH4Cl MgCl2·6H2O CaCl2·2H2O MgSO4·7H2O KH2PO4 |
1,5 g·l–1 1,2 g·l–1 1,8 g·l–1 1,5 g·l–1 0,16 g·l–1 |
|
Solução de reserva 2: ferro |
|
|
FeCl3·6H2O Na2EDTA·2H2O |
64 mg·l-–1 100 mg·l-–1 |
|
Solução de reserva 3: oligoelementos |
|
|
H3BO3 MnCl2·4H2O ZnCl2 CoCl2·6H2O CuCl2·2H2O Na2MoO4·2H2O |
185 mg·l–1 415 mg·l–1 3 mg·l–1 1,5 mg·l–1 0,01 mg·l–1 7 mg·l–1 |
|
Solução de reserva 4: bicarbonato |
|
|
NaHCO3 |
50 g·l–1 |
|
Na2SiO3·9H20 |
|
Esterilizar as soluções de reserva por filtração através de membrana (diâmetro de poro de 0,2 µm) ou em autoclave (120 °C, 15 min). Armazenar as soluções ao abrigo da luz, a 4 °C.
Não esterilizar as soluções 2 e 4 em autoclave, mas sim por filtração através de membrana.
Preparar o meio de cultura adicionando um volume apropriado das soluções de reserva 1-4 à água.
Adicionar a 500 ml de água esterilizada:
|
— |
10 ml da solução de reserva 1 |
|
— |
1 ml da solução de reserva 2 |
|
— |
1 ml da solução de reserva 3 |
|
— |
1 ml da solução de reserva 4 |
Completar o volume até 1 000 ml, com água esterilizada.
Permitir que decorra um tempo suficiente para que o meio atinja o equilíbrio com o CO2 atmosférico, se necessário borbulhando com ar estéril filtrado durante algumas horas.
Preparação do meio AAP
|
A1.1. |
Adicionar 1 ml de cada uma das soluções de reserva descritas em A1.2.1-A1.2.7 a cerca de 900 ml de água desionizada ou destilada, corrigindo depois o volume até 1 l. |
|
A1.2. |
As soluções de reserva de macronutrientes são preparadas dissolvendo os seguintes elementos em 500 ml de água desionizada ou destilada. Os reagentes A1.2.1, A1.2.2, A1.2.3 e A1.2.4 podem ser combinados numa única solução de reserva. |
|
A1.2.1. |
NaNO3 – 12,750 g. |
|
A1.2.2. |
MgCl2 ·6H2O – 6,082 g. |
|
A1.2.3. |
CaCl2 ·2H2O – 2,205 g. |
|
A1.2.4. |
Solução de reserva Micronutrientes (ver o ponto A1.3). |
|
A1.2.5. |
MgSO4·7H2O – 7,350 g. |
|
A1.2.6. |
K2HPO4 – 0,522 g. |
|
A1.2.7. |
NaHCO3 – 7,500 g. |
|
A1.2.8. |
Na2SiO3·9H2O — Ver o ponto A1.1. Nota A1.1 — Utilizar exclusivamente quando a espécie de ensaio for uma diatomácea. O composto pode ser acrescentado directamente (202,4 mg) ou utilizando uma solução de reserva que permita obter uma concentração final de Si de 20 mg/l no meio |
|
A1.3. |
As soluções de reserva de micronutrientes são preparadas dissolvendo os seguintes elementos em 500 ml de água desionizada ou destilada. |
|
A1.3.1. |
H3BO3 – 92,760 mg. |
|
A1.3.2. |
MnCl2·4H2O – 207,690 mg. |
|
A1.3.3. |
ZnCl2 – 1,635 mg. |
|
A1.3.4. |
FeCl3·6H2O – 79,880 mg. |
|
A1.3.5. |
CoCl2·6H2O – 0,714 mg. |
|
A1.3.6. |
Na2MoO4·2H2O – 3,630 mg. |
|
A1.3.7. |
CuCl2·2H2O – 0,006 mg. |
|
A1.3.8. |
Na2EDTA·2H2O – 150,000 mg. [(Etilenodinitrilo)tetraacetato de dissódio]. |
|
A1.3.9. |
Na2SeO4·5H2O – 0,005 mg — Ver o ponto A1.2. Nota A1.2 — Utilizar exclusivamente no meio para as culturas de arranque de espécies de diatomáceas. |
|
A1.4. |
Ajustar o pH a 7,5 ± 0,1 com HCl ou NaOH 0,1 N ou 1,0 N. |
|
A1.5. |
Filtrar os meios para um recipiente esterilizado através de um filtro de membrana com um diâmetro de poro de 0,22 μm, se se pretender utilizar um contador electrónico de partículas, ou de 0,45 μm, se não for esse o caso. |
|
A1.6. |
Armazenar o meio ao abrigo da luz, a 4 °C, até ao momento da utilização. |
(1) A relação molar do EDTA para o ferro é ligeiramente superior a 1, o que permite evitar a precipitação do ferro e, simultaneamente, minimizar a quelação dos iões de metais pesados.
Apêndice 3
Exemplo de um procedimento para a cultura de algas
Observações gerais
O objectivo da cultura de acordo com o procedimento a seguir descrito é a preparação de culturas de algas para a realização de ensaios de toxicidade.
Devem ser utilizados métodos que permitam garantir que as culturas de algas não sejam infectadas por bactérias. Mesmo que se pretendam utilizar culturas puras, devem preparar-se e ser conservadas culturas de cada uma das diferentes espécies de algas.
Todas as operações devem ser realizadas em condições de esterilidade, de modo a evitar a contaminação por bactérias ou por outras algas.
Equipamento e material
Ver o ponto «Método de Ensaio»: Equipamento.
Procedimentos para a obtenção de culturas de algas
Preparação das soluções nutrientes (meios):
Todos os sais nutrientes do meio são preparados como soluções de reserva concentradas e armazenados ao abrigo da luz e ao frio. As soluções são esterilizadas por filtração ou em autoclave.
Os meios são preparados misturando a quantidade correcta de solução de reserva com água destilada esterilizada, tomando o cuidado de evitar infecções. Para os meios sólidos, é acrescentado àgar-àgar à concentração de 0,8 %.
Culturas de arranque:
As culturas de arranque são culturas de algas com pequeno volume que são regularmente transferidas para meio fresco, de modo a que possam ser utilizadas como inóculo para os ensaios. Se as culturas não forem regularmente utilizadas, devem ser conservadas por inoculação em tubos com meio de ágar-ágar inclinado. As culturas serão transferidas para meio fresco pelo menos uma vez de dois em dois meses.
As culturas de arranque são feitas em frascos cónicos (com um volume de cerca de 100 ml), com o meio apropriado. Quando as algas forem incubadas a 20 °C com iluminação contínua, será necessário transferi-las a cada semana.
Durante a transferência, uma determinada quantidade da cultura «antiga» é transferida com uma pipeta esterilizada para um frasco com meio fresco, de modo a obter, para as espécies de crescimento rápido, uma concentração inicial cerca de 100 vezes inferior à que existia na cultura original.
A taxa de crescimento de uma espécie pode ser determinada a partir da sua curva de crescimento. A partir do momento em que seja conhecida, é possível estimar a densidade a que a cultura deve ser transferida para o meio fresco. A transferência deve ser feita antes de a cultura atingir a fase de morte celular.
Pré-cultura:
O objectivo da pré-cultura é obter uma quantidade de algas adequada para a inoculação das culturas de ensaio. A pré-cultura será incubada em condições de ensaio e utilizada enquanto se encontra na fase exponencial, normalmente após um período de incubação de 2 a 4 dias. Quando as culturas de algas apresentarem células deformadas ou anormais, devem ser descartadas.
Apêndice 4
Análise dos dados por regressão não linear
Considerações de carácter geral
A resposta dos ensaios com algas e de outros ensaios de crescimento microbiano — o aumento da biomassa é, por natureza, uma variável contínua ou métrica — terá a forma de uma taxa quando se utilizar a taxa de crescimento ou de um integral em função do tempo se se escolher utilizar a quantidade de biomassa. Ambas as variáveis serão referenciadas em função da resposta média dos replicados de controlo, não expostos, que apresentem uma resposta mais elevada às condições vigentes — sendo que a temperatura e a luz são os principais factores determinantes nos ensaios com algas. O sistema é distribuído ou homogéneo, e a biomassa pode ser vista como um valor contínuo, sem tomar em consideração as células individuais. A distribuição das variâncias do tipo de resposta está, nesses sistemas, exclusivamente relacionada com os factores experimentais (tipicamente descritos pela curva log-normal ou pela distribuição normal dos erros). Esta situação contrasta com as respostas típicas dos bioensaios com dados quantais, nos quais a tolerância (tipicamente distribuída de forma binomial) de cada organismo é frequentemente assumida como a componente dominante da variância. As respostas dos controlos são, nesse caso, nulas ou correspondentes à linha de base.
Numa situação sem complicações, a resposta normalizada ou relativa, r, diminui de forma monotónica entre 1 (inibição nula) e 0 (inibição a 100 %). De notar que todas as respostas têm um erro associado, pelo que se poderão constatar inibições negativas apenas por efeito dos erros aleatórios.
Análise de regressão
Modelos
O objectivo da análise de regressão é a descrição quantitativa da curva de concentração-resposta, sob a forma de uma função matemática de regressão Y = f (C) ou, mais frequentemente, F (Z), onde Z = log C. A utilização da função inversa C = f–1 (Y) permite o cálculo dos valores de ECx, incluindo EC50, EC10 e EC20, bem como dos respectivos limites de confiança a 95 %. Diversas funções matemáticas simples podem ser utilizadas para uma boa descrição da relação concentração-resposta obtida em ensaios de inibição de crescimento com algas. Essas funções incluem, por exemplo, a equação logística, a equação assimétrica de Weibull e a função de distribuição log normal, todas curvas sigmóides que se aproximam da assímptota 1 quando C → 0 e da assímptota 0 quando C → infinito.
A utilização de modelos com uma função de limite contínuo (p. ex.: o modelo Kooijman «para a inibição do crescimento da população», Kooijman et al. 1996) foi recentemente proposta como alternativa aos modelos assimptóticos. Esse modelo assume que não há qualquer efeito às concentrações que se encontram abaixo de um determinado limite, EC0+, estimado por extrapolação, a partir da relação concentração-resposta, do ponto de intercepção do eixo das concentrações, aplicando uma função contínua simples que não é diferenciada no ponto inicial.
De notar que a análise em causa poderá ser uma simples minimização da soma dos mínimos quadrados (assumindo uma variância constante) ou dos quadrados ponderados, quando for necessário compensar para uma variância heterogénea.
Procedimento
O procedimento pode ser esquematizado do seguinte modo: seleccionar uma equação Y = f (C) apropriada e ajustá-la aos dados por regressão não linear. Usar, de preferência, as medições de cada frasco, em vez do valor médio dos replicados, de modo a extrair tanta informação quanto possível dos dados disponíveis. Por outro lado, se a variância for elevada, a experiência prática sugere que os valores médios dos replicados podem resultar numa estimativa matematicamente mais robusta e menos influenciada por erros sistemáticos que possam afectar dados do que acontece quando se utilizam os valores medidos em cada frasco.
Desenhar a curva ajustada e marcar os pontos medidos, verificando se há um bom ajustamento. A análise dos mínimos quadrados pode ser um instrumento particularmente útil para esse fim. Se a função escolhida para o ajustamento da curva de concentração-resposta não descrever a totalidade da curva ou de alguma das suas partes essenciais, como por exemplo a resposta a baixas concentrações, seleccionar outra opção de ajustamento da curva — p. ex.: uma curva não simétrica, como a equação de Weibull, em vez de uma curva simétrica. A inibição negativa pode, por exemplo no caso da função de distribuição log-normal, constituir um problema que também poderá tornar necessária a utilização de uma função de regressão alternativa. Não se recomenda a atribuição de um valor zero ou de um valor positivo baixo a esses valores negativos, já que esse procedimento iria distorcer a distribuição dos erros. Poderá ser indicado proceder a diferentes ajustamentos para cada parte da curva, por exemplo na parte da baixa inibição, para estimativa dos valores de ECx baixo. Calcular, a partir da equação ajustada (por «estimativa inversa», C = f–1 (Y)) as estimativas de alguns pontos ECx característicos, comunicando, no mínimo, o valor de EC50 e o valor estimado de um ou de dois EC x baixo. A experiência com ensaios práticos demonstra que a precisão dos ensaios com algas permite normalmente uma estimativa razoavelmente correcta do nível de 10 % de inibição, se se dispuser de um número suficiente de pontos medidos — a não ser nos casos em que haja estimulação a baixas concentrações, que poderá ser factor de confusão. A precisão das estimativas de EC20 é muitas vezes consideravelmente maior do que para o EC10, porque o ponto EC20 se encontra normalmente na parte central, quase linear, da curva de concentração-resposta. Por vezes, o valor de EC10 pode ser difícil de estimar, devido à estimulação do crescimento a baixas concentrações. Assim, embora o EC10 possa normalmente ser obtido com uma precisão suficiente, recomenda-se que seja sempre comunicado também o EC20.
Factores de ponderação
Normalmente, a variância experimental não é constante e, na maior parte dos casos, inclui uma componente proporcional, pelo que há vantagem em realizar, por rotina, uma regressão ponderada. Geralmente, assume-se que os factores de ponderação utilizados nesse tipo de análise são inversamente proporcionais da variância:
Wi = 1/Var(ri)
Diversos programas de regressão permitem a opção de uma análise de regressão ponderada com factores de conversão que são apresentados sob a forma de listas. O mais conveniente será normalizar os factores de ponderação, isto é, multiplicá-los por n/Σ wi (em que n é o número de pontos experimentais) de modo a que a respectiva soma seja igual a 1.
Respostas de normalização
A normalização em função da resposta média dos controlos suscita problemas de princípio e tem como resultado uma estrutura de variâncias que é bastante complicada. Ao dividir as respostas pelo valor médio da resposta dos controlos, para obtenção da percentagem de inibição, é introduzido um erro adicional, causado pelo erro que afecta a média dos controlos. A não ser nos casos em que esse erro é negligenciável, os factores de ponderação da regressão e dos limites de confiança terão de ser corrigidos em relação à co-variância com o controlo (17). Cabe aqui notar que é importante obter estimativas de alta precisão do valor médio da resposta dos controlos, de modo a minimizar a variância global para a resposta relativa. Essa variância pode ser descrita do seguinte modo:
(o i em índice faz referência ao nível de concentração i, enquanto que o 0 em índice se refere aos controlos)
Yi = Resposta relativa = ri/r0 = 1 — I = f (Ci)
com uma variância de:
Var (Yi) = Var (ri/r0) ≅ (∂Yi / ∂ ri)2·Var(ri) + (∂ Yi/ ∂ r0)2·Var (r0)
e, tendo em conta que:
(∂ Yi/ ∂ ri) = 1/r0 e (∂ Yi / ∂ r0) = ri/r0 2
com uma distribuição normal e replicados mi e m0:
Var(ri) = σ2/mi
a variância total da resposta relativa, Yi, passa portanto a ser:
Var(Yi) = σ2/(r0 2 mi) + ri 2·σ2/r0 4 m0
O erro da média dos controlos é inversamente proporcional à raiz quadrada do número de replicados do controlo utilizados para o cálculo dessa média, podendo por vezes justificar-se a inclusão de dados históricos para reduzir fortemente o erro. Outro procedimento alternativo será não normalizar os dados e fazer o ajustamento das respostas absolutas, incluindo os dados de resposta dos controlos, mas introduzindo como parâmetro adicional a ajustar por regressão não linear o valor de resposta dos controlos. No caso das equações de regressão habitualmente utilizadas, com 2 parâmetros, este método exige o ajustamento de 3 parâmetros, pelo que necessita de mais pontos do que a regressão não linear de dados que tenham sido normalizados utilizando uma resposta do controlo pré-definida.
Intervalos de confiança inversos
O cálculo de intervalos de confiança da regressão não linear por estimativa inversa é bastante complexo e não é normalmente uma opção disponível nos pacotes estatísticos informáticos mais comuns. É possível obter uma aproximação dos limites de confiança utilizando programas normais de regressão não linear com reparametrização (Bruce and Versteeg, 1992), o que implica voltar a escrever a equação matemática com os pontos que se pretende estimar, ou seja, EC10 e EC50, como os parâmetros a determinar. (Partindo da função I = f (α, β, concentração), utilizar as relações de definição f (α, β, EC10) = 0,1 e f (α, β, EC50) = 0,5 para substituir f (α, β, concentração) por uma função equivalente g (EC10, EC50, concentração).
Um cálculo mais directo (Andersen et al, 1998) poderá ser efectuado mantendo a equação original e utilizando uma expansão de Taylor em redor dos valores médios de ri e de r0.
Recentemente, os métodos de «boot strapping» têm vindo a ganhar popularidade. Esses métodos utilizam os dados medidos e uma reamostragem frequente, determinada por um gerador de números aleatórios, para estimar uma distribuição empírica da variância.
Bibliografia
Kooijman, S.A.L.M.; Hanstveit, A.O.; Nyholm, N. (1996): No-effect concentrations in algal growth inhibition tests. Water Research, 30, 1625-1632
Bruce, R.D. and Versteeg, D.J.(1992) A Statistical Procedure for Modelling Continuous Ecotoxicity Data. Env. Toxicol. Chem.11, 1485-1494
Andersen, J.S., Holst, H., Spliid, H., Andersen, H., Baun, A. & Nyholm, N. (1998): Continuous ecotoxicological data evaluated relative to a control response. Journal of Agricultural, Biological and Environmental Statistics, 3, 405-420
ANEXO V
|
C.25. |
MINERALIZAÇÃO AERÓBICA EM ÁGUAS DE SUPERFÍCIE — ENSAIO DE SIMULAÇÃO DE BIODEGRADAÇÃO |
1. MÉTODO
Este método é equivalente ao OCDE TG 309 (2004) (1).
1.1. INTRODUÇÃO
O objectivo do presente ensaio é medir a evolução da biodegradação de uma substância em estudo, a baixa concentração, em águas naturais aeróbicas, bem como quantificar as observações efectuadas sob a forma de expressões com taxas cinéticas. O presente ensaio de simulação é realizado em regime descontínuo (batch), em grupos de frascos colocados num agitador de laboratório, para determinação das taxas de degradação aeróbica de substâncias orgânicas em amostras de águas de superfície naturais (água doce, água salobra ou água do mar). O ensaio baseia-se na norma ISO/DIS 14592-1 (2), incluindo ainda elementos dos métodos de ensaio C.23 e C.24 (3)(4). A título facultativo, nos ensaios de longa duração, poderá ser utilizado o regime semicontínuo, em vez do regime descontínuo, para evitar a deterioração do organismo de ensaio. O principal objectivo do ensaio de simulação é determinar a mineralização da substância de ensaio nas águas superficiais, sendo essa mineralização utilizada como base para a expressão da cinética de degradação. O ensaio poderá ainda, a título facultativo e como objectivo secundário, ser utilizado para obter informação sobre a degradação primária e sobre a formação dos principais metabolitos. A identificação dos metabolitos e, quando possível, a quantificação das respectivas concentrações, são particularmente importantes para as substâncias que mineralizam de forma muito lenta (p.ex.: com uma meia-vida para o 14C residual total superior a 60 dias). Devido às limitações analíticas, a identificação e quantificação dos principais metabolitos exigirá normalmente a utilização de concentrações elevadas da substância em estudo (p.ex.: > 100 μg/l).
No contexto do presente ensaio, uma baixa concentração significa uma concentração (p.ex.: de menos de 1 μg/l a 100 μg/l) suficientemente baixa para garantir que a cinética de biodegradação obtida pelo ensaio reflecte o que seria de esperar no meio natural. Por comparação com a massa total de substratos de carbono biodegradáveis disponíveis na água natural utilizada para o ensaio, a substância em estudo, presente a baixa concentração, servirá como substrato secundário, pelo que será de esperar uma cinética de biodegradação de primeira ordem (cinética de «não crescimento») com a substância em estudo a ser degradada por «co-metabolismo». Um cinética de primeira ordem implica que a taxa de degradação (mg/l/dia) é proporcional à concentração de substrato, que diminui ao longo do tempo. Numa verdadeira cinética de primeira ordem, a taxa específica constante de degradação, k, é independente do tempo e da concentração, ou seja, k não varia de forma apreciável durante o decurso de uma experiência e não se altera com as concentrações adicionadas entre as diferentes fases da mesma. Por definição, a taxa específica constante de degradação é igual à alteração relativa da concentração por unidade de tempo: k = (1/C) · (dC/dt). Embora seja de esperar, nas condições descritas, uma cinética de primeira ordem, poderão existir circunstâncias em que outro tipo de cinética seja mais apropriado. Podem observar-se desvios em relação a uma cinética de primeira ordem, por exemplo, no caso em que certos fenómenos de transferência de massa como a taxa de difusão, e não a taxa da reacção biológica, são o factor limitante da biotransformação No entanto, os dados quase sempre podem ser descritos por uma cinética de pseudo primeira ordem, com uma taxa constante dependente da concentração.
A informação sobre a biodegradabilidade da substância em estudo a concentrações mais elevadas (p.ex.: a partir de ensaios-padrão de identificação), bem como sobre a degradabilidade abiótica, os metabolitos e as propriedades físico-químicas relevantes, deve estar disponível antes da realização do ensaio, de modo a permitir a planificação da experiência e a interpretação dos resultados. O estudo de substâncias marcadas com 14C e a determinação da distribuição do 14C pelas fases no final do ensaio permitem a determinação da biodegradabilidade global. Quando se utilizam substâncias não marcadas, a biodegradabilidade global só pode ser estimada se se proceder ao ensaio de concentrações mais elevadas e se todos os metabolitos forem conhecidos.
1.2. DEFINIÇÕES
Biodegradação primária: alteração estrutural (transformação) de uma substância química por microorganismos, resultando na perda da identidade química.
Biodegradação funcional: alteração estrutural (transformação) de uma substância química por microorganismos, resultando na perda de uma determinada propriedade.
Biodegradação aeróbica completa: desnaturação de uma substância química por microorganismos, na presença de oxigénio, de que resulta a formação de dióxido de carbono, água e sais minerais dos restantes elementos presente (mineralização) e a produção de biomassa e de produtos orgânicos de biossíntese microbiana.
Mineralização: desnaturação de uma substância química ou de matéria orgânica por microorganismos, na presença de oxigénio, de que resulta a formação de dióxido de carbono, água e sais minerais dos restantes elementos presentes.
Fase de latência: período que decorre desde o início do ensaio até à adaptação do microorganismo responsável pela degradação e ao aparecimento de um grau detectável de biodegradação (p.ex.: 10 % da biodegradação teórica máxima, ou menos, dependendo da precisão da técnica de medição) de uma substância química ou de matéria orgânica.
Nível máximo de biodegradação: nível de biodegradação de uma substância química ou matéria orgânica num ensaio, registado em percentagem, acima do qual não ocorre mais biodegradação durante o ensaio.
Substrato primário: conjunto de fontes naturais de carbono e de energia que permitem o crescimento e a sobrevivência da biomassa microbiana.
Substrato secundário: componente do substrato presente numa concentração tão baixa que a sua degradação apenas fornece aos microorganismos presentes quantidades insignificantes de carbono e de energia, quando comparadas com o carbono e energia fornecidos pela degradação dos componentes principais do substrato (substratos primários).
Taxa constante de degradação: taxa cinética constante de primeira ordem ou de pseudo- primeira ordem, k (d–1), que indica a taxa dos processos de degradação. Numa experiência em modo descontínuo, k é estimado a partir da parte inicial da curva de degradação, após o final da fase de latência.
Meia-vida, t1/2 (d): termo utilizado para caracterizar a taxa de uma reacção de primeira ordem. É o intervalo de tempo que corresponde a uma divisão da concentração por 2. A meia-vida e a taxa constante de degradação são relacionados pela equação t1/2 = ln 2/k.
Tempo de meia degradação, DT50 (d): termo utilizado para quantificar o resultado dos ensaios de biodegradação. É o período, incluindo a fase de latência, necessário para se atingir um valor de 50 % de degradação.
Limite de detecção (LOD) e limite de quantificação (LOQ): o limite de detecção (LOD) é a concentração de uma substância abaixo da qual a identidade da mesma não pode ser distinguida da linha de base do aparelho analítico utilizado. O limite de quantificação (LOQ) é a concentração de uma substância abaixo da qual a concentração não pode ser determinada com uma precisão aceitável.
Carbono Orgânico Dissolvido (COD): é a parte do carbono orgânico presente numa amostra de água que não pode ser removida por separação de fases, por exemplo por centrifugação a 40 000 ms–2 durante 15 minutos ou por filtração através de membrana com um diâmetro de poro de 0,2 μm-0,45 μm.
Actividade total 14C orgânico (TOA): a actividade total do 14C associado ao carbono orgânico.
Actividade 14C orgânico dissolvido (DOA): a actividade total do 14C associado ao carbono orgânico dissolvido.
Actividade 14C orgânico particulado (POA): a actividade total do 14C associado ao carbono orgânico particulado.
1.3. APLICABILIDADE DO ENSAIO
O ensaio de simulação é aplicável a substâncias orgânicas não voláteis ou pouco voláteis, ensaiadas a baixas concentrações. Utilizando frascos abertos para a atmosfera (p.ex.: tapados com rolha de lã de vidro), as substâncias com uma constante de Henry inferior a cerca de 1 Pa·m3/mol (aprox. 10–5 atm·m3/mol) podem ser consideradas, em termos práticos, como não voláteis. Utilizando frascos fechados, com algum espaço livre na parte superior, é possível ensaiar substâncias ligeiramente voláteis (com uma constante de Henry < 100 Pa·m3/mol ou < 10–3 atm·m3/mol) sem perdas a partir do sistema de ensaio. A perda de substâncias marcadas com 14C pode ocorrer, se não se tomarem as devidas precauções, durante a purga do CO2. Nessas situações, poderá ser necessário capturar o CO2 num absorvente alcalino interno ou utilizar um sistema externo de absorção do CO2 (determinação directa do 14CO2; ver o anexo 3). Para a determinação da cinética de biodegradação, a concentração da substância em estudo deve ser inferior à sua hidrossolubilidade máxima. Cabe aqui notar, contudo, que os valores de hidrossolubilidade existentes na literatura podem ser consideravelmente mais elevados do que a solubilidade da substância em estudo em águas naturais. A título facultativo, a solubilidade de substâncias em estudo particularmente difíceis de dissolver em água poderá ser determinada utilizando as águas naturais em estudo.
O método pode ser utilizado para simular a biodegradação em águas de superfície livres de partículas grosseiras (ensaio pelágico) ou em águas de superfície turvas, como as que se podem encontrar, por exemplo, junto ao interface água/sedimento (ensaio com sedimento em suspensão).
1.4. PRINCÍPIO DO ENSAIO
O ensaio é realizado em modo descontínuo, incubando a substância em estudo apenas com água de superfície (ensaio pelágico) ou com água de superfície com sólidos/sedimentos em suspensão numa concentração entre 0,01 e 1 g/l de peso seco (ensaio com sedimento em suspensão), para simular uma massa de água com sólidos em suspensão ou com sedimentos re-suspendidos. Uma concentração de sólidos/sedimentos na parte inferior do intervalo referido é típica na maior parte das águas superficiais. Os frascos de ensaio são incubados em ambiente escuro, à temperatura ambiente, em condições aeróbicas e com agitação. Para a determinação da cinética de degradação, devem ser utilizadas pelo menos duas concentrações da substância em estudo, diferentes entre si num factor de 5 a 10 e que devem corresponder à gama de concentrações presentes no ambiente natural. A concentração máxima da substância em estudo não deve ultrapassar os 100 µg/l, mas será preferível utilizar concentrações máximas abaixo dos 10 µg/l, de modo a garantir a biodegradação de acordo com uma cinética de primeira ordem. A concentração mais baixa não deverá exceder os 10 μg/l, mas será preferível utilizar concentrações na gama dos 1-2 μg/l ou mesmo inferiores a 1 μg/l. Normalmente, é possível proceder a uma análise adequada de concentrações dessa ordem de grandeza utilizando substâncias marcadas com 14C disponíveis no comércio. Devido a limitações analíticas, a medição da concentração da substância em estudo com a precisão exigida é muitas vezes impossível, quando a concentração utilizada é ≤ 100 µg/l (ver o segundo parágrafo do ponto 1.7.2). Uma concentração mais elevada da substância em estudo (> 100 µg/l e, por vezes, > 1 mg/l) pode ser utilizada para a identificação e quantificação dos principais metabolitos ou para os casos em que não existe um método analítico específico com um limite de detecção suficientemente baixo. Quando se ensaiarem concentrações elevadas da substância em estudo, poderá não ser possível utilizar os resultados para estimar a constante de degradação de primeira ordem ou a meia-vida, já que a degradação não se fará provavelmente de acordo com uma cinética de primeira ordem.
A degradação será acompanhada a intervalos apropriados, por medição do 14C residual ou da concentração residual da substância em estudo, quando se utilizar um método específico de análise química. A marcação com 14C das partes mais estáveis da molécula em estudo garante a determinação da mineralização total, enquanto que a marcação com 14C das partes menos estáveis da mesma molécula, tal como a utilização de um método de análise específico, só permite avaliar a biodegradação primária. No entanto, a parte mais estável não inclui necessariamente a fracção funcionalmente relevante da molécula (que pode ser relacionada com uma determinada propriedade, como a toxicidade, a bioacumulação, etc). Se for esse o caso, poderá ser melhor utilizar no estudo uma substância marcada com 14C na sua parte funcional, de modo a poder acompanhar a eliminação da propriedade que lhe esteja associada.
1.5. INFORMAÇÃO SOBRE A SUBSTÂNCIA EM ESTUDO
No presente ensaio, a substância em estudo utilizada tanto pode ser marcada como não marcada. A marcação, para a qual se recomenda a técnica do 14C, deve ser normalmente aplicada na(s) parte(s) mais estável(is) da molécula (ver também o ponto 1.4). No caso das substâncias com mais do que um anel aromático, deverão ser marcados, de preferência, um ou mais átomos de carbono em cada anel. Por outro lado, deverão igualmente ser marcados, de preferência com 14C, um ou mais átomos de carbono em ambos os lados das ligações mais facilmente degradáveis. A pureza química e/ou radioquímica da substância em estudo deve ser > 95 %. No caso das substâncias marcadas com radioisótopos, será preferível uma actividade específica da ordem dos μCi/mg (1,85 MBq) ou mais, de modo a facilitar as medições do 14C nos ensaios com baixas concentrações iniciais. Deve ser fornecida a seguinte informação em relação à substância em estudo:
|
— |
hidrossolubilidade [método de ensaio A.6], |
|
— |
solubilidade em solvente(s) orgânico(s) (substâncias aplicadas com um solvente ou de baixa hidrossolubilidade), |
|
— |
constante de dissociação (pKa), nos casos em que a substância seja propensa a protonização ou desprotonização [OCED TG 112] (5), |
|
— |
pressão de vapor [método de ensaio A.4] e/ou constante de Henry, |
|
— |
estabilidade química na água e em ambiente escuro (hidrólise) [método C.7]. |
Quando se proceder ao ensaio de substâncias com baixa hidrossolubilidade em água do mar, poderá também ser útil conhecer a constante de salinização (ou «constante de Setschenow»), Ks, que é definida pela seguinte equação: log (S/S’) = Ks Cm, onde S e S’ representam a solubilidade da substância em água doce e salgada, respectivamente, e Cm é a concentração molar de sal.
Se o ensaio for conduzido como um «ensaio com sedimento em suspensão», deverá igualmente ser fornecida a seguinte informação:
|
— |
coeficiente de partição n-octanol/água [método de ensaio A.8], |
|
— |
coeficiente de adsorção [método C.18]. |
Outra informação que poderá ser útil inclui:
|
— |
a concentração no ambiente natural, quando for conhecida ou puder ser estimada, |
|
— |
a toxicidade da substância em estudo para o microorganismo [método C.11], |
|
— |
a biodegradabilidade imediata e/ou inerente [métodos C.4 A-F, C.12, C.9, OCDE TG 302] (5), |
|
— |
a biodegradabilidade aeróbica ou anaeróbica em estudos de transformação no solo ou em sedimentos/água [métodos C.23 e C.24]. |
1.6. SUBSTÂNCIA DE REFERÊNCIA
Uma substância, que normalmente deverá ser facilmente degradada em condições aeróbicas (p.ex.: anilina ou benzoato de sódio), deverá ser utilizada como substância de referência. O tempo previsto para a degradação da anilina ou do benzoato de sódio é normalmente inferior a 2 semanas. A substância de referência é utilizada para garantir que a actividade microbiana da água em estudo se encontra dentro de certos limites; ou seja, que a água contém uma população microbiana activa.
1.7. CRITÉRIOS DE QUALIDADE
1.7.1. Recuperação
Imediatamente após a adição da substância em estudo, as concentrações iniciais de ensaio devem ser verificadas por medição da actividade do 14C ou por análise química, no caso das substâncias não marcadas, em amostras pelo menos duplicadas. Essa verificação permitirá obter informação sobre a aplicabilidade e reproductividade do método analítico e ainda sobre a homogeneidade da distribuição da substância em estudo. Normalmente, o valor a utilizar posteriormente na análise dos dados será o valor inicial medido da actividade do 14C ou da concentração da substância em estudo, e não a concentração nominal, o que permite eliminar os eventuais erros devido às perdas por adsorção e a erros de doseamento. No caso de uma substância marcada com 14C, o nível de recuperação no final da experiência é dado pelo balanço de massas (ver o último parágrafo do ponto 1.8.9.4). Em termos ideais, o balanço de massas das substâncias marcadas com 14C deve situar-se entre 90 % e 110 %, enquanto que a precisão analítica deverá resultar numa recuperação inicial de entre 70 % e 110 % para as substâncias em estudo não marcadas. Estes intervalos devem ser interpretados como um objectivo, não devendo ser utilizados como critério de aceitação do ensaio. A título facultativo, poderá ser determinada a precisão analítica para concentrações inferiores da substância em estudo e para os principais metabolitos.
1.7.2. Reproductividade e sensibilidade do método analítico
A reproductividade do método analítico (incluindo a eficiência da extracção inicial) em termos da quantificação da substância em estudo e, se aplicável, dos respectivos metabolitos, deverá ser verificada através de cinco análises replicadas de extractos da água de superfície.
O limite de detecção (LOD) do método analítico para a substância em estudo e para os seus metabolitos deverá ser, quando possível, pelo menos 1 % da quantidade inicial aplicada no sistema de ensaio. O limite de quantificação (LOQ) deverá ser igual ou menor do que 10 % da concentração aplicada. A análise química de muitas substância orgânicas e dos respectivos metabolitos exige frequentemente que a substância em estudo seja aplicada numa concentração relativamente elevada, ou seja, > 100 μg/l.
1.8. DESCRIÇÃO DO MÉTODO DE ENSAIO
1.8.1. Equipamento
O ensaio pode ser conduzido em frascos cónicos ou cilíndricos de capacidade apropriada (p.ex.: 0,5 l ou 1 l) tapados com rolhas de silicone ou de borracha, ou ainda em frascos de soro com tampas impermeáveis ao CO2 (p.ex.: rolha de borracha butílica). Outra possibilidade será efectuar o ensaio utilizando diversos frascos para cada concentração e recolhendo, para cada amostra, um frasco inteiro, pelo menos em duplicado (ver o último parágrafo do ponto 1.8.9.1). No caso das substâncias não voláteis e não marcadas com um radioisótopo, não será necessário utilizar rolhas ou tampas impermeáveis aos gases, bastando utilizar rolhas de algodão que evitem a contaminação aérea (ver o segundo parágrafo do ponto 1.8.9.1). As substâncias ligeiramente voláteis devem ser ensaiadas num sistema de tipo biométrico, com ligeira agitação das águas de superfície em estudo. A fim de garantir a ausência de contaminação bacteriana, os frascos poderão, a título facultativo, ser esterilizados por aquecimento ou em autoclave, antes do início do ensaio. Será utilizado o seguinte equipamento de laboratório:
|
— |
mesa com agitação ou agitadores magnéticos, para agitação contínua dos frascos de ensaio, |
|
— |
centrifugadora, |
|
— |
medidor de pH, |
|
— |
turbidímetro para medições nefelométricas da turbidez, |
|
— |
forno ou micro-ondas, para as determinações do peso seco, |
|
— |
aparelho de filtração por membrana, |
|
— |
autoclave ou forno para a esterilização por calor do material de vidro, |
|
— |
instalações para o manuseamento de substâncias marcadas com 14C, |
|
— |
equipamento para quantificar a actividade do 14C em amostras das soluções com o CO2 precipitado e, se necessário, de amostras de sedimento, |
|
— |
equipamento analítico para o doseamento da substância em estudo (e da substância de referência) através de análise química específica (p.ex.: cromatografia de gás, HPLC). |
1.8.2. Soluções de reserva da substância em estudo
Utilizando água desionizada, preparam-se soluções de reserva da substância em estudo e da substância de referência (ver o primeiro parágrafo do ponto 1.8.7). A água desionizada deve estar isenta de qualquer substância que possa ser tóxica para os microorganismos e não deverá ter um teor de carbono orgânico dissolvido (COD) superior a 1 mg/l (6).
1.8.3. Recolha e transporte da água de superfície
O local de amostragem para a recolha da água de superfície deve ser seleccionado, independentemente da situação, de acordo com o objectivo do ensaio. Para a escolha dos locais de amostragem, deverão ser tomados em conta os eventuais efeitos da agricultura, da indústria ou das actividades domésticas. Se um determinado ambiente aquático tiver comprovadamente sido contaminado com a substância em estudo ou com outras substâncias estruturalmente análogas durante os quatro anos anteriores, não deverá ser utilizado para a recolha de amostras de água, a não ser que o investigador esteja expressamente interessado na investigação das taxas de degradação em locais previamente expostos à substância. O pH e a temperatura da água devem ser medidos no local de colheita. Devem igualmente ser registadas a profundidade a que foi feita a amostra e a aparência (p.ex.: cor e turbidez) da mesma (ver o ponto 3). A concentração de oxigénio e/ou o potencial redox da água e da camada superficial do sedimento devem ser medidos de modo a demonstrar a existência de condições aeróbicas, a não ser que essas condições sejam óbvias a julgar pela aparência e pela experiência já adquirida no local. A água de superfície deve ser transportada num recipiente cuidadosamente lavado. Durante o transporte, a temperatura da amostra não deve ultrapassar significativamente a temperatura que irá ser utilizada no ensaio. Quando a duração do transporte ultrapassar as 2 a 3 horas, recomenda-se o arrefecimento a 4o C. A amostra de água não deve ser congelada.
1.8.4. Armazenamento e preparação da água de superfície
O ensaio deve, de preferência, ser iniciado no prazo de um dia a partir da colheita da amostra. O armazenamento da água, quando necessário, deve ser reduzido ao mínimo possível e não deve, em qualquer caso, ultrapassar um máximo de 4 semanas. A amostra de água deve ser conservada a 4o C, com arejamento, até ser utilizada. Antes disso, devem remover-se as partículas mais grosseiras, por exemplo por filtração através de um filtro de nylon com um poro de cerca de 100 μm, de papel de filtro grosso ou ainda por sedimentação.
1.8.5. Preparação da água corrigida com o sedimento (facultativo)
Para os ensaios com sedimento em suspensão, o sedimento superficial é acrescentado a frascos contendo água natural (filtrada para remoção das partículas mais grosseiras, como se descreve no ponto 1.8.4), de modo a obter uma suspensão; a concentração de sólidos em suspensão deve ser de 0,01 a 1 g/l. O sedimento superficial deve ser proveniente do mesmo local de onde foi recolhida a amostra de água. Dependendo do tipo de ambiente aquático em causa, o sedimento superficial poderá ser caracterizado por um teor elevado de carbono orgânico (2,5-7,6 %) e por uma textura fina ou por um baixo teor de carbono orgânico (0,5-2,5 %) e por uma textura mais grosseira (3). O sedimento superficial pode ser preparado do seguinte modo: extrair diversas amostras em profundidade do sedimento, utilizando um tubo de plástico transparente, separando as camadas superiores aeróbicas (desde a superfície até uma profundidade máxima de 5 cm) imediatamente após a amostragem e juntando-as numa só amostra. Essa amostra do sedimento deve ser transportada num recipiente com muito espaço livre, de modo a que o sedimento se mantenha em condições aeróbicas (arrefecer a 4o C se o transporte durar mais do que 2-3 horas). A amostra de sedimento deverá ser suspensa na água de ensaio numa razão de 1:10 e mantida a 4o C, com arejamento, até ao momento da utilização. O armazenamento do sedimento, quando necessário, deve ser reduzido ao mínimo possível e não deve, em qualquer caso, ultrapassar um máximo de 4 semanas.
1.8.6. Procedimento semicontínuo (facultativo)
Nos casos em que a fase de latência, até que seja possível medir uma degradação significativa da substância em estudo, seja longa poderá ser necessária uma incubação prolongada (diversos meses). Se essa característica já for conhecida de anteriores ensaios da substância, o ensaio poderá ser iniciado utilizando um procedimento semicontínuo, que permite a renovação periódica de parte da água ou da suspensão em estudo (ver o anexo 2). Se não for esse o caso, o ensaio normal, em modo descontínuo, poderá ser transformado num ensaio semicontínuo se não se observar nenhuma degradação da substância em estudo durante aproximadamente 60 dias de ensaio em descontínuo (ver o segundo parágrafo do ponto 1.8.8.3).
1.8.7. Adição da substância em estudo (ou de referência)
Para as substâncias altamente hidrossolúveis (> 1 mg/l) e pouco voláteis (constante de Henry < 1 Pa·m3/mol ou < 10–5 atm·m3/mol), poderá preparar-se uma solução de reserva em água ionizada (ver o ponto 1.8.2); essa solução deverá ser adicionada aos frascos de ensaio no volume necessário para obter a concentração pretendida. O volume de qualquer solução de reserva adicionada deverá ser limitado ao mínimo que seja prático (sempre que possível, < 10 % do volume final de líquido). Outro procedimento passa pela dissolução da substância em estudo num volume maior de água de ensaio, que poderá ser utilizada em alternativa à utilização de solventes orgânicos.
Quando não for possível evitá-lo, as soluções de reserva de substâncias não voláteis e de fraca hidrossolubilidade podem ser preparadas com um solvente orgânico volátil, mas a quantidade adicionada desse solvente não deve exceder 1 % v/v e não deve ter qualquer efeito adverso sobre a actividade microbiana. O solvente não deverá afectar a estabilidade da substância em estudo em meio aquático. O solvente deverá ser escorrido até que só reste uma quantidade extremamente pequena, de maneira a não aumentar significativamente o COD da água ou da suspensão em estudo, factor que deverá ser verificado por análise específica da substância em causa ou, se possível, por análise do COD (6). Deve tomar-se o cuidado de limitar a quantidade de solvente transferido ao mínimo estritamente necessário, bem como de garantir que a quantidade pretendida da substância em estudo possa ser dissolvida no volume final de água previsto para o ensaio. Poderão ser utilizadas outras técnicas para a introdução da substância em estudo nos frascos de ensaio, como se descreve em (7) e (8). Quando se utilizar um solvente orgânico para a aplicação da substância em estudo, frascos de controlo do solvente com a água de ensaio (sem mais nenhuma substância adicionada) e com a água de ensaio e a substância de referência deverão ser tratados da mesma forma que os frascos de ensaio propriamente ditos, aos quais foi acrescentada a substância em estudo, dissolvida num solvente. O objectivo dos controlos do solvente é analisar a possibilidade de ocorrência de efeitos adversos causados pelo solvente sobre a população microbiana e que tenham efeitos sobre a degradação da substância de referência.
1.8.8. Condições de ensaio
1.8.8.1. Temperatura de ensaio
A incubação deve ser feita em local escuro (de preferência) ou com luz difusa e a temperatura controlada (± 2 °C), que poderá ser a temperatura de campo ou uma temperatura normalizada de 20-25o C. A temperatura de campo pode ser entendida como a temperatura real da amostra no momento da colheita ou como a temperatura média no local da recolha.
1.8.8.2. Agitação
Os frascos devem ser agitados ou mexidos em contínuo, para que as partículas e microorganismos se mantenham em suspensão. A agitação servirá igualmente para facilitar a transferência de oxigénio a partir do espaço livre acima do líquido, de modo a que as condições aeróbicas sejam mantidas de forma adequada. Colocar os frascos numa mesa de agitação (aproximadamente 100 rpm) ou utilizar agitação magnética. A agitação deve ser contínua, mas tão suave quanto possível, desde que permita manter uma suspensão homogénea.
1.8.8.3. Duração do ensaio
A duração do ensaio não deverá normalmente ultrapassar os 60 dias, a não ser que se utilize um regime de ensaio semicontínuo, com renovação periódica da suspensão de ensaio (ver o ponto 1.8.6 e o anexo 2). O período dos ensaios em descontínuo poderá, no entanto, ser alargado até um máximo de 90 dias, se a degradação da substância em estudo se tiver iniciado durante os primeiros 60 dias. A degradação será acompanhada, a intervalos apropriados, pela determinação da actividade residual do 14C ou da evolução do 14CO2 (ver o ponto 1.8.9.4) e/ou por análise química (ver o ponto 1.8.9.5). O período de incubação deve ser suficientemente longo para permitir a avaliação do processo de degradação. A degradação deverá, de preferência, ultrapassar os 50 %; no caso das substâncias de degradação lenta, a percentagem de degradação deve ser suficiente (normalmente superior a 20 %) para permitir a estimação de uma taxa constante de degradação cinética.
O pH e a concentração de oxigénio do sistema de ensaio devem ser medidos periodicamente, a não ser quando a experiência anterior de ensaios semelhantes com amostras de água e de sedimentos recolhidos no mesmo local faça com que isso não seja necessário. Em certas condições, o metabolismo dos substratos primários presentes a concentrações muito elevadas na água ou nos sedimentos poderá eventualmente resultar numa produção de CO2 e numa depleção do oxigénio suficientes para alterar de forma significativa as condições experimentais durante o ensaio.
1.8.9. Procedimento
1.8.9.1. Preparação dos frascos para o ensaio pelágico
Transferir um volume adequado de água de ensaio para os frascos, enchendo até cerca de um terço do volume total, pelo menos com 100 ml. Se se utilizarem diversos frascos (de forma a permitir a recolha de um frasco inteiro em cada amostra), o volume de água de ensaio mais indicado será também de cerca de 100 ml, já que a utilização de volumes menores poderá ter influência sobre a duração da fase de latência. A substância em estudo é adicionada a partir de uma solução de reserva, como se descreve nos pontos 1.8.2 e 1.8.7. Para a determinação da cinética de degradação e para o cálculo da taxa constante de degradação cinética, devem ser utilizadas pelo menos duas concentrações diferentes da substância em estudo, com um factor de concentração entre 5 e 10. Ambas as concentrações devem ser inferiores a 100 µg/l e, de preferência, estar compreendidas no intervalo entre <1 e 10 µg/l.
Fechar os frascos com rolhas ou tampas impermeáveis ao ar e ao CO2. No caso dos ensaios de substâncias químicas não marcadas com 14C e não voláteis, podem ser utilizadas simples tampas de algodão que evitem a contaminação (ver o ponto 1.8.1), desde que se saiba que os principais metabolitos também não são voláteis e que se utilize um método indirecto para a determinação do CO2 (ver o anexo 3).
Incubar os frascos à temperatura escolhida (ver o ponto 1.8.8.1). Retirar amostras para a análise química ou para a medição do 14C no início do ensaio (ou seja, antes de começar a biodegradação, ver o ponto 1.7.1) e depois a intervalos adequados durante o ensaio. A amostragem pode ser efectuada retirando subamostras (p.ex.: aliquotas de 5 ml) de cada replicado ou recolhendo um frasco inteiro em cada momento de amostra. A mineralização da substância em estudo pode ser determinada de forma directa ou indirecta (ver o anexo 3). Normalmente, uma estimativa fiável da constante de degradação exige, no mínimo, 5 pontos de amostragem durante a fase de degradação (ou seja, após o final da fase de latência), a não ser quando se possa provar que três pontos são suficientes para as substâncias de degradação rápida. Quanto às substâncias que não são rapidamente degradadas, a obtenção de mais pontos durante a fase de degradação não deve constituir nenhuma dificuldade, pelo que se deverão utilizar mais pontos de amostragem para a estimativa de k.. Não é possível indicar nenhum calendário fixo de amostragem, dada a variação das taxas de biodegradação; no entanto a recomendação, nos casos em que a degradação seja lenta, é que se efectuem amostras uma vez por semana. Se a substância em estudo se degradar rapidamente, deve ser feita uma amostra por dia durante os primeiros três dias e, depois disso, uma amostra a cada dois ou três dias. Em determinadas circunstâncias, como por exemplo substâncias de hidrólise rápida, poderá ser necessário proceder a uma amostragem de hora a hora. Recomenda-se a realização de um estudo prévio ao ensaio, de modo a determinar o intervalo de amostragem mais adequado. Se as amostras tiverem de ser conservadas para posterior realização de análises específicas, será aconselhável retirar um número maior de amostras e depois, no final do estudo, seleccionar as que serão analisadas de trás para a frente, ou seja, analisando primeiro as últimas amostras (ver o segundo parágrafo do ponto 1.8.9.5 para mais orientações sobre a estabilidade das amostras durante o armazenamento).
1.8.9.2. Número de frascos e de amostras
Preparar um número suficiente de frascos de ensaio para incluir:
|
— |
frascos de ensaio; frascos pelo menos em duplicado (e, de preferência, um mínimo de três) para cada concentração da substância em estudo ou diversos frascos por concentração, quando se recolherem frascos inteiros em cada momento de amostragem (representados pelo símbolo FT), |
|
— |
frascos de ensaio para o cálculo do balanço de massas; frascos pelo menos em duplicado para cada concentração em estudo (representados pelo símbolo FM), |
|
— |
controlos «brancos», sem adição da substância em estudo; pelo menos um frasco de ensaio «branco», contendo apenas água de ensaio (representado pelo símbolo FB), |
|
— |
controlo de referência; frascos em duplicado, com a substância de referência (p.ex.: anilina ou benzoato de sódio, a 10 µg/l) (representados pelo símbolo FC). O objectivo do controlo de referência é confirmar a existência de uma actividade microbiana mínima. Quando for conveniente, poderá ser utilizada uma substância de referência marcada com um isótopo, mesmo nos casos em que a degradação da substância em estudo seja acompanhada por análise química, |
|
— |
controlo estéril; um ou dois frascos com água de ensaio esterilizada, para analisar a possível degradação abiótica ou outro tipo de eliminação não-biológica da substância em estudo (representados pelo símbolo FS); A actividade biológica pode ser interrompida por tratamento em autoclave (121o C; 20 min) da água de ensaio, pela adição de um produto tóxico (p.ex.: azoteto de sódio (NaN3) a 10-20 g/l, cloreto de mercúrio (HgCl2) a 100 mg/l ou formalina a 100 mg/l) ou por irradiação com raios gama. Se se utilizar HgCl2, deve ser eliminado como resíduo tóxico. No caso de águas com grande quantidade de sedimento, nem sempre é fácil conseguir obter condições de esterilidade; nesses casos, recomenda-se a repetição do tratamento em autoclave (p.ex.: três vezes). Deve ter-se em atenção que as características de adsorção do sedimento poderão ser alteradas pelo tratamento em autoclave, |
|
— |
controlos do solvente, contendo água de ensaio e água de ensaio com a substância de referência; frascos em duplicado, tratados com a mesma quantidade de solvente e sujeitos ao mesmo procedimento que os frascos a que foi adicionada a substância em estudo. O objectivo é verificar os eventuais efeitos adversos do solvente, através da determinação da degradação da substância de referência. |
Para a definição do método de ensaio, o investigador deverá tomar em consideração a importância relativa do aumento do número de replicados contra o aumento do número de pontos de amostragem. O número exacto de frascos que serão necessários dependerá do método utilizado para a medição da degradação (ver o terceiro parágrafo do ponto 1.8.9.1, o ponto 1.8.9.4 e o apêndice 3).
Em cada momento de amostragem, devem ser retiradas de cada frasco de ensaio duas subamostras (p.ex.: aliquotas de 5 ml). Se se utilizar um regime com amostragem de frascos inteiros em cada momento de amostragem, devem ser sacrificados dois frascos para cada amostra (ver o primeiro parágrafo do ponto 1.8.9.1).
1.8.9.3. Preparação dos frascos para o ensaio com sedimento em suspensão (facultativo)
Acrescentar os volumes necessários de água de ensaio e de sedimento, se for o caso, aos frascos de ensaio (ver o ponto 1.8.5). A preparação dos frascos para o ensaio com sedimento em suspensão é idêntica à dos frascos para o ensaio pelágico (ver os pontos 1.8.9.1 e 1.8.9.2). Utilizar, de preferência, frascos de soro ou de forma semelhante. Colocar os frascos num agitador, em posição horizontal. Como é óbvio, os frascos abertos, utilizados para as substâncias não voláteis e não marcadas com 14C devem ser colocados em posição vertical; nesse caso, recomenda-se a utilização de agitação magnética, com barras de agitação revestidas a vidro. Se necessário, arejar os frascos para manter condições aeróbicas adequadas.
1.8.9.4. Determinações radioquímicas
O 14CO2 resultante da degradação é medido de forma directa e indirecta (ver o apêndice 3). O 14CO2 é determinado indirectamente pela diferença entre a actividade 14C inicial na água ou suspensão de ensaio e a actividade total residual no momento da amostra, medida após acidificação da amostra a pH 2-3 e purga do CO2. Dessa forma, o carbono inorgânico é removido, pelo que a actividade residual medida provém do material orgânico. A determinação indirecta do 14CO2 não deve ser utilizada quando os principais metabolitos da degradação da substância de ensaio sejam voláteis (ver o apêndice 3). Se possível, a evolução do 14CO2 deverá ser medida de forma directa (ver o apêndice 3), em cada momento de amostragem, em pelo menos um frasco de ensaio; esse procedimento permite, por um lado, obter os balanços mássicos e, por outro, verificar a evolução do processo de degradação, mas está limitado aos ensaios conduzidos com frascos fechados.
Se o 14CO2 resultante da degradação for medido directamente durante o ensaio, devem ser previstos, no início do mesmo, um número suficiente de frascos para o efeito. A determinação directa do 14CO2 é recomendada nos casos em que os principais metabolitos resultantes da transformação da substância em estudo sejam voláteis. Em cada ponto de medição, os frascos adicionais serão acidificados até pH de 2-3 e o 14CO2 será recolhido num adsorvente interno ou externo (ver o apêndice 3).
A título facultativo, as concentrações da substância em estudo marcada com 14C e dos principais metabolitos poderão ser determinadas por radiocromatografia (p.ex.: cromatografia em camada fina, RAD-TLC) ou por HPLC com detector de radionuclídeos.
Ainda a título facultativo, poderá determinar-se a distribuição da radioactividade remanescente pelas diferentes fases (ver o apêndice 1) e a concentração residual da substância em estudo e dos metabolitos.
No final do ensaio, o balanço mássico deverá ser determinado através de medição directa do 14CO2, utilizando frascos de ensaio diferentes, dos quais não tenham sido retiradas amostras durante o ensaio (ver o apêndice 3).
1.8.9.5. Análise química específica
Se existir um método analítico específico com uma sensibilidade suficiente, a biodegradação primária poderá ser avaliada medindo a concentração residual total da substância de ensaio, em vez de utilizar técnicas de radiomarcação. Se for utilizada uma substância marcada com um radioisótopo (para medição da mineralização total), poderão ser efectuadas em paralelo análises químicas específicas, a fim de obter informação adicional que possa ser útil e de verificar o procedimento utilizado. As análises químicas específicas poderão igualmente ser utilizadas para a medição dos metabolitos formados durante a degradação da substância em estudo, sendo recomendadas para as substâncias que mineralizam com uma meia-vida superior a 60 dias. A concentração da substância em estudo e dos respectivos metabolitos deve ser determinada e comunicada para cada momento de amostragem (sob a forma de uma concentração e, quando aplicável, de uma percentagem). Em geral, os metabolitos detectados com uma concentração ≥ 10 % da concentração aplicada em qualquer momento do ensaio devem ser identificados, a não ser que a sua presença possa ser razoavelmente justificada por qualquer outro mecanismo que não a degradação da substância em estudo. Deverá considerar-se a hipótese de identificação de todos os metabolitos cuja concentração aumente continuamente ao longo do ensaio, mesmo que a sua concentração nunca ultrapasse o limite acima indicado, já que essa característica poderá indicar que se trata de produtos persistentes. A análise dos metabolitos nos controlos estéreis deve ser ponderada nos casos em que se considera possível a ocorrência de transformação biótica rápida (hidrólise) da substância em estudo. A necessidade de quantificação e de identificação dos metabolitos deve ser ponderada numa base casuística, devendo, quando for realizada, ser justificada no relatório de ensaio. As técnicas de extracção com solventes orgânicos devem ser aplicadas de acordo com as instruções apresentadas nos respectivos procedimentos analíticos.
As amostras devem ser conservadas hermeticamente fechadas entre 2 e 4 °C, se a análise for realizada num prazo de 24 horas (preferível). Para um armazenamento mais prolongado, as amostras devem ser congeladas abaixo dos –18 °C ou conservadas com adição de químicos. A acidificação não é um método recomendado para a conservação das amostras, na medida em que as amostras acidificadas poderão ser instáveis. Se as amostras não forem analisadas num prazo de 24 horas e forem armazenadas por um período prolongado, deverá proceder-se a um estudo de estabilidade durante o armazenamento, a fim de demonstrar a estabilidade das substâncias de interesse em condições de armazenamento a –18 °C ou com conservantes químicos. Se o método analítico implicar a extracção com solventes ou em fase sólida (SPE), essa extracção deverá ser efectuada imediatamente após a amostragem ou depois de conservar a amostra refrigerada por um prazo máximo de 24 horas.
Dependendo da sensibilidade do método analítico, poderá ser necessário recolher volumes superiores aos que são indicados no ponto 1.8.1. O ensaio pode ser realizado de forma simples com volumes de ensaio de 1 l, em frascos com uma capacidade de 2-3 l, o que torna possível a recolha de amostras de aproximadamente 100 ml.
2. RESULTADOS E APRESENTAÇÃO DE RELATÓRIOS
2.1. TRATAMENTO DOS RESULTADOS
2.1.1. Apresentação dos resultados
Arredondar os momentos de amostragem às horas (a não ser nos casos em que a substância em estudo se degrade substancialmente numa questão de minutos ou de horas), mas não aos dias. Marcar num gráfico as estimativas da actividade residual da substância em estudo (para as substâncias marcadas com 14C) ou da concentração residual (para as substâncias não marcadas) em função do tempo, tanto em papel milimétrico como em papel semilogarítmico (ver as figuras 1a, 1b). Se tiver ocorrido degradação, comparar os resultados dos frascos FT com os dos frascos FS. Se as médias dos resultados dos frascos com a substância de ensaio (FT) e dos frascos estéreis (FS) apresentarem um desvio inferior a 10 %, pode assumir-se que a degradação observada se deve principalmente a factores abióticos. Se a degradação for menor nos frascos FS, os respectivos valores poderão ser utilizados para corrigir (por subtracção) os valores relativos aos frascos FT, de modo a poder estimar a dimensão da biodegradação. Quando se efectuarem análises facultativas dos principais metabolitos, os gráficos da sua formação e destruição devem ser apresentados, juntamente com um gráfico da evolução da concentração da substância em estudo.
Estimar a duração da fase de latência, tL, a partir da curva de degradação (gráfico semilogarítmico), extrapolando a sua parte linear até ao ponto de degradação zero ou, em alternativa, determinando o tempo necessário para que ocorra uma degradação de cerca de 10 % (ver as figuras 1a e 1b). A partir do gráfico semilogarítmico, estimar a constante de primeira ordem k e o respectivo erro-padrão, por regressão do ln (valor residual da actividade 14C ou da concentração da substância) em função do tempo. Em relação às medições do 14C, em particular, utilizar apenas os dados da parte inicial linear da curva, após o final da fase de latência, sendo preferível que se seleccionem poucos pontos que sejam significativos do que um número maior de pontos afectados de maior incerteza. Os factores de incerteza incluem, nesse caso, os erros inerentes à medição directa da actividade residual do 14C, que é recomendada (ver abaixo). Por vezes, poderá ser relevante calcular duas constantes diferentes, quando a degradação apresentar duas fases bem distintas. Para tal, devem ser definidas as duas fases da curva de degradação. Os cálculos da taxa constante k e da meia-vida t½ = ln2/k devem ser efectuados para cada um dos frascos replicados, quando forem retiradas subamostras de um mesmo frasco, ou utilizando valores médios, quando se recolherem frascos inteiros em cada momento de amostragem (ver o último parágrafo do ponto 1.8.9.2). Quando for utilizado o primeiro desses procedimentos, deverão apresentar-se a taxa constante e a meia-vida para cada um dos frascos replicados e ainda o seu valor médio, com o respectivo desvio padrão. Se se tiverem usado concentrações elevadas da substância em estudo, a curva de degradação poderá desviar-se consideravelmente de uma linha recta (gráfico semilogarítmico), caso em que não será válida uma cinética de primeira ordem. Logo, a definição a meia-vida deixará de fazer sentido. É possível, no entanto, que se possa aplicar uma cinética de pseudo primeira ordem numa gama limitada de concentrações, o que poderá permitir a definição do meio-tempo de degradação DT50 (tempo necessário para uma degradação de 50 %). Contudo, deve tomar-se em consideração que o decurso da degradação para lá da gama de concentrações seleccionada não pode ser previsto utilizando esse DT50, que apenas descreve um conjunto específico de resultados. Existem diversos instrumentos analíticos que podem facilitar os cálculos estatísticos e o ajustamento das curvas, sendo recomendada a utilização de programas informáticos desse tipo.
Se forem realizadas análises químicas específicas, calcular as taxas constantes e as meia-vida da degradação primária, como se descreve acima, para o caso da mineralização total. Se a degradação primária constituir o processo limitante, poderá ser possível, em certos casos, utilizar pontos de toda a curva de degradação. Isto acontece porque a medição, ao contrário do que acontece com as medições da actividade do 14C, é directa.
Se forem utilizadas substâncias marcadas com 14C, deve ser apresentado, pelo menos em relação ao momento final do ensaio, um balanço de massas expresso em percentagem da concentração inicial aplicada.
2.1.2. Actividade residual
Quando a parte de uma substância orgânica que está marcada com 14C é biodegradada, a maior parte do 14C é convertido em 14CO2, enquanto que outra parte é utilizada para a produção de biomassa e/ou para a síntese de metabolitos extracelulares. Logo, a degradação completa «final» de uma substância não resulta numa conversão de 100 % do respectivo carbono em 14CO2. O 14C integrado em metabolitos da biosíntese é posteriormente libertado, de forma lenta, como 14CO2, num processo de «mineralização secundária». Por esse motivo, a actividade residual do 14C orgânico (medida após purga do CO2) ou do 14CO2 produzido em função do tempo apresenta uma parte final com um declive muito baixo, após a conclusão da degradação. Esse factor vem complicar a interpretação cinética dos dados e, portanto, só a parte inicial da curva (a partir do final da fase de latência e até à degradação de aproximadamente 50 % da substância presente) deverá normalmente ser utilizada para a estimação de uma taxa constante de degradação. Se a substância em estudo for degradada, a actividade total do 14C orgânico residual será sempre superior à actividade do 14C associada à substância em estudo que permanece intacta. Se a substância em estudo for degradada segundo uma cinética de primeira ordem e se uma fracção constante, α, for mineralizada em CO2, o declive inicial da curva de desaparecimento do 14C (14C orgânico total em função do tempo) será α vezes superior ao declive da correspondente curva de concentração da substância em estudo (ou, em termos mais precisos, da parte da substância em estudo marcada com 14C). Se se utilizarem as medições não corrigidas da actividade total de 14C orgânico, a taxa constante de degradação calculada será, portanto, subavaliada. A literatura especializada descreve alguns procedimentos para a estimação das concentrações da substância em estudo a partir da actividade radioquímica medida, com base em diversos pressupostos de simplificação (2)(9)(10)(11). Esses procedimentos serão mais facilmente aplicáveis às substâncias de degradação rápida.
2.2. INTERPRETAÇÃO DOS RESULTADOS
Se se constatar que k é independente da concentração adicionada (ou seja, se o k calculado for aproximadamente o mesmo para todas as concentrações da substância em estudo), poderá assumir-se que a taxa constante de primeira ordem é representativa das condições de ensaio utilizadas, ou seja, da substância em estudo, da amostra de água e da temperatura de ensaio. Só a experiência permitirá decidir até que ponto os resultados podem ser generalizados ou extrapolados para outros sistemas. Se se utilizar uma concentração elevada da substância em estudo e se a degradação não ocorrer, portanto, segundo uma cinética de primeira ordem, os dados não poderão ser utilizados para a estimação directa de uma taxa constante de primeira ordem ou da correspondente meia-vida. No entanto, os dados recolhidos a partir de um ensaio com elevada concentração da substância em estudo poderão ainda ser utilizáveis para a estimação do grau de mineralização total e/ou dos limites de detecção e de quantificação dos metabolitos.
Se as taxas de outros processos, diferentes da biodegradação, que causam a perda da substância (p.ex.: hidrólise ou volatilização) forem conhecidas, poderão ser subtraídas da taxa líquida de desaparecimento da substância observada ao longo do ensaio, de modo a obter uma estimativa aproximada da taxa de biodegradação. Os dados relativos à hidrólise podem se obtidos, por exemplo, a partir dos frascos de controlo estéreis ou de ensaios paralelos com uma concentração mais elevada da substância em estudo.
A determinação, directa e indirecta, do 14CO2 (ponto 1.8.9.4 e anexo 3) só pode ser utilizada para medir o grau de mineralização da substância em estudo em CO2. A radiocromatografia (RAD-TLC) ou o HPLC podem ser utilizados para analisar as concentrações da substância de ensaio marcada com 14C e a formação dos principais metabolitos (ver o terceiro parágrafo do ponto 1.8.9.4). A fim de permitir a estimação directa da meia-vida, é necessário que não esteja presente nenhum dos principais metabolitos (definidos como os metabolitos cuja concentração é ≥ 10 % da quantidade da substância em estudo utilizada. Se estiverem presentes metabolitos principais, de acordo com essa definição, será necessário proceder a uma análise pormenorizada dos dados. Essa análise poderá incluir a repetição de ensaios e/ou a identificação dos metabolitos (ver o primeiro parágrafo do ponto 1.8.9.5), a não ser quando o destino dos produtos de transformação possa ser razoavelmente avaliado com base na experiência adquirida (p.ex.: informação sobre a sequência de reacções da degradação). Na medida em que a proporção do carbono da substância em estudo que é transformada em CO2 varia (dependendo, em grande medida, da concentração da substância e de outros substratos disponíveis, das condições de ensaio e da população microbiana), este ensaio não permite uma estimação simples da biodegradação final, como acontece com o ensaio de redução gradual do COD, mas o seu resultado é semelhante ao obtido no caso dos ensaios respirométricos. O grau de mineralização será, portanto, menor ou igual do que o nível mínimo da degradação final. Para conseguir obter uma imagem mais completa da biodegradação final (mineralização e incorporação sob a forma de biomassa), deverá ser efectuada, no final do ensaio, uma análise da distribuição do 14C pelas diferentes fases (ver o apêndice 1). O 14C presente na fase particulada será composto pelo 14C incorporado na biomassa bacteriana e pelo 14C adsorvido sobre outras partículas orgânicas.
2.3. VALIDADE DO ENSAIO
Se a substância de referência não for degradada durante o período em que isso seria de esperar (no caso da anilina e do benzoato de sódio, normalmente menos de duas semanas), a validade do ensaio será duvidosa e deverá ser verificada de forma mais aprofundada; em alternativa, o ensaio deverá ser repetido com uma nova amostra de água. Durante um ensaio de anel ISO do presente método, com a participação de sete laboratórios de toda a Europa, as constantes da taxa de degradação para a anilina variaram entre 0,3 e 1,7 dia–1, com uma média de 0,8 d–1 a 20 oC e um desvio-padrão de ±0,4 d–1 (t½ = 0,9 dias). A fase de latência tipicamente observada é de 1 a 7 dias. As águas analisadas tinham, de acordo com os relatórios apresentados, uma biomassa bacteriana correspondente a 103 -104 unidades formadoras de colónias (CFU) por ml. As taxas de degradação medidas nas águas ricas em nutrientes da Europa Central foram superiores às das águas nórdicas, oligotróficas, o que poderá dever-se a uma situação trófica diferente ou a uma exposição anterior a substâncias químicas.
A recuperação total (balanço de massas) no final da experiência deverá situar-se entre os 90-110 % para as substâncias marcadas com um radioisótopo, enquanto que a recuperação inicial no início da experiência se deverá situar entre os 70-110 % para as substâncias não marcadas. No entanto, estes intervalos devem ser interpretados como um objectivo, não devendo ser utilizados como critério de aceitação do ensaio.
3. RELATÓRIO DE ENSAIO
O relatório deve indicar claramente o tipo de ensaio, ou seja, se se trata de um ensaio pelágico ou com sedimento em suspensão, para além da seguinte informação:
Substância em estudo e substância(s) de referência:
|
— |
nomes comuns, nomes químicos (nomes IUPAC e/ou CAS recomendados), números CAS, fórmulas estruturais (indicando a posição do 14C, quando se utilizarem substâncias marcadas) e propriedades físico-químicas relevantes da substância em estudo e da substância de referência (ver os pontos 1.5 e 1.6), |
|
— |
nomes químicos, números CAS, fórmulas estruturais (indicando a posição do 14C, quando se utilizarem substâncias marcadas) e propriedades físico-químicas relevantes das substâncias utilizadas como padrão para a identificação e quantificação dos metabolitos, |
|
— |
pureza (impurezas) da substância em estudo e das substâncias de referência, |
|
— |
pureza radioquímica da substância marcada e respectiva actividade específica (se aplicável). |
Águas de superfície:
Deve ser fornecida, no mínimo, a seguinte informação em relação à amostra de água:
|
— |
localização e descrição do local de amostragem, incluindo, se possível, os antecedentes conhecidos em termos de poluição, |
|
— |
data e hora da recolha da amostra, |
|
— |
nutrientes (N total, amónio, nitritos, nitratos, P total, ortofosfatos dissolvidos), |
|
— |
profundidade da colheita, |
|
— |
aspecto da amostra (p.ex.: cor e turbidez), |
|
— |
COD e COT, |
|
— |
CBO, |
|
— |
temperatura e pH no local e momento da amostragem, |
|
— |
oxigénio ou potencial redox (obrigatório apenas quando as condições aeróbicas não sejam óbvias), |
|
— |
salinidade e condutividade (no caso da água do mar e da água salobra), |
|
— |
sólidos em suspensão (caso a amostra se apresente turva), |
|
— |
eventualmente, outra informação relevante acerca do local de amostragem no momento da mesma (p.ex.: dados actuais ou passados sobre o caudal dos rios ou correntes marinhas, descargas importantes que se encontram próximas e o respectivo tipo, condições meteorológicas que precederam a recolha da amostra), |
e, facultativamente:
|
— |
biomassa microbiana (p.ex.: contagem directa com laranja de acridina ou número de unidades formadoras de colónias), |
|
— |
carbono inorgânico, |
|
— |
concentração de clorofila-a, como estimativa específica da biomassa de algas. |
Quando se proceder a um ensaio com sedimentos em suspensão, deverá também ser fornecida a seguinte informação sobre os sedimentos:
|
— |
profundidade da colheita de sedimento, |
|
— |
aparência do sedimento (p.ex.: colorido, lamacento, lodoso ou arenoso), |
|
— |
textura (p.ex.: % de areão, de areia fina, de lodo e de argilas), |
|
— |
peso seco dos sólidos em suspensão, em g/l, concentração do COT ou perda de peso após combustão, como medição do conteúdo em matéria orgânica, |
|
— |
pH, |
|
— |
oxigénio ou potencial redox (obrigatório apenas quando as condições aeróbicas não sejam óbvias). |
Condições de ensaio:
|
— |
período decorrido entre a recolha das amostras e a respectiva utilização no ensaio, condições de armazenamento e de pré-tratamento das amostras, data de realização dos estudos, |
|
— |
quantidade da substância em estudo aplicada, concentrações de ensaio e substância de referência, |
|
— |
método de aplicação da substância em estudo, incluindo a eventual utilização de solventes, |
|
— |
volume de água de superfície e de sedimentos (se aplicável) utilizados, e volume recolhido em cada amostra para fins de análise, |
|
— |
descrição do sistema de ensaio utilizado. |
Se as culturas não forem mantidas em ambiente escuro, informação sobre as condições de «luz difusa»:
|
— |
informação sobre o(s) método(s) utilizado(s) para a preparação dos controlos estéreis (p.ex.: temperatura, duração e número de tratamentos em autoclave), |
|
— |
temperatura de incubação, |
|
— |
informação sobre as técnicas analíticas e sobre o(s) método(s) utilizado(s) para as medições da actividade radioquímica, para a verificação dos balanços de massa e para a medição da distribuição pelas fases (se for efectuada), |
|
— |
número de replicados. |
Resultados:
|
— |
percentagens de recuperação (ver o ponto 1.7.1), |
|
— |
reproductividade e sensibilidade dos métodos analíticos utilizados, incluindo os respectivos limites de detecção (LOD) e de quantificação (LOQ) (ver o ponto 1.7.2), |
|
— |
todos os dados medidos (incluindo o momento das amostragens), tabela com os valores calculados e curvas de degradação; para cada concentração estudada e para cada frasco replicado, apresentar o coeficiente de correlação linear correspondente ao declive do gráfico logarítmico, a duração estimada da fase de latência e (se possível) uma taxa constante de primeira ordem ou de pseudo primeira ordem, bem como a correspondente meia-vida (ou período de meia-vida, t50), |
|
— |
comunicar os valores relevantes, p.ex.: duração da fase de latência, taxa constante e meia-vida de degradação (ou t50), sob a forma de uma média dos resultados observados nos diferentes replicados, |
|
— |
classificar o sistema como adaptado ou não-adaptado, a julgar pelo aspecto da curva de degradação e pela possível influência da concentração de ensaio, |
|
— |
resultados da verificação final dos balanços de massa e das medições da distribuição pelas diferentes fases (se efectuadas), |
|
— |
fracção do 14C mineralizada e, caso sejam utilizadas análises específicas, nível final da degradação primária, |
|
— |
identificação, concentração molar e percentagem da substância aplicada e dos principais metabolitos (ver o primeiro parágrafo do ponto 1.8.9.5), quando aplicável, |
|
— |
proposta de esquema da sequência de reacções, quando aplicável, |
|
— |
discussão dos resultados. |
4. BIBLIOGRAFIA
|
1. |
OECD TG 309 (2004) Aerobic Mineralisation in surface water — Simulation Biodegradation Test. |
|
2. |
ISO/DIS 14592-1 (1999) Water quality — Evaluation of the aerobic biodegradability of organic compounds at low concentrations — Part 1: Shake flask batch test with surface water or surface water/sediment suspensions. |
|
3. |
Testing Method C.23. Aerobic and anaerobic transformation in soil. |
|
4. |
Testing Method C.24. Aerobic and anaerobic transformation in aquatic sediments. |
|
5. |
OECD (1993). Guidelines for the Testing of Chemicals. OECD, Paris. |
|
6. |
ISO 8245 (1999). Water quality — Guidelines on the determination of total organic carbon (TOC) and dissolved organic carbon (DOC). |
|
7. |
ISO 10634 (1995). Water quality — Guidance for the preparation and treatment of poorly water-soluble organic compounds for the subsequent evaluation of their biodegradability in an aqueous medium. |
|
8. |
OECD draft (2000). Guidance Document on aquatic toxicity testing of difficult substances and mixtures. Environmental Health and Safety Publications. Series on Testing and Assessment, N.o 22. |
|
9. |
Simkins, S. and Alexander, M. (1984). Models for mineralization kinetics with the variables of substrate concentration and population density. Appl. Environ. Microbiol.47, 394-401. |
|
10. |
Ingerslev, F. and N. Nyholm. (2000). Shake-flask test for determination of biodegradation rates of 14C-labeled chemicals at low concentrations in surface water systems. Ecotoxicol. Environ. Saf. 45, 274-283. |
|
11. |
ISO/CD 14592-1 (1999). Ring test report: Water Quality — Evaluation of the aerobic biodegradability of organic compounds at low concentrations part 1 — report of 1998/1999 ring-test. Shake flask batch test with surface water or surface water/sediment suspensions. |
Apêndice 1
Distribuição do 14C pelas diferentes fases
Para efeitos da verificação do procedimento, as medições rotineiras da actividade residual do 14C orgânico total (TOA) devem ser complementadas por medições dos balanços de massa, que indicam a determinação directa do 14CO2 produzido após captura num adsorvente (ver o anexo 3). A formação de 14CO2 representa, por si só, uma prova directa de biodegradação, por oposição à degradação abiótica ou a outros mecanismos de redução da concentração da substância, como a volatilização ou a adsorção. Alguma informação útil para a caracterização do comportamento em termos de biodegradabilidade pode ser obtida medindo a distribuição do TOA entre a fase de solução (actividade do 14C orgânico dissolvido, DOA) e a fase particulada (actividade do 14C orgânico particulado, POA), após separação das partículas por filtração através de membrana ou por centrifugação. A POA consiste na substância adsorvida sobre a biomassa microbiana e sobre outras partículas, para além do carbono marcado proveniente da substância em estudo e que tenha sido utilizado para a síntese de novo material celular, sendo assim incorporado na fracção da biomassa particulada. A formação de material orgânico dissolvido com 14C pode ser estimada pela DOA no final da biodegradação (parte em que a curva da degradação em função do tempo passa a ser paralela ao eixo dos ×).
Estimar a distribuição do 14C pelas fases em amostras seleccionadas, por filtração das amostras através de um filtro de membrana com diâmetro de poro de 0,22 μm ou de 0,45 μm, num material que não seja significativamente adsorvente em relação à substância em estudo (os filtros de policarbonato poderão servir). Caso a adsorção da substância em estudo no filtro seja demasiado importante para que possa ser ignorada (a verificar antes de iniciar a experiência) poderá utilizar-se, em vez da filtração, um procedimento de centrifugação a alta velocidade (2 000 g, 10 min).
Proceder à filtração ou à centrifugação de amostras não filtradas, como se descreve no anexo 3. Dissolver os filtros de membrana num fluído de cintilação adequado e proceder à contagem do modo habitual, utilizando apenas o método do rácio externo padrão para corrigir a atenuação, ou utilizar um oxidante das amostras. Se se tiver procedido a centrifugação, ressuspender o pellet formado pela fracção particulada em 1-2 ml de água destilada e transferir para uma célula de cintilómetro. Lavar duas vezes com 1 ml de água destilada e transferir a água da lavagem para a célula. Se necessário, a suspensão poderá ser colocada num gel para a contagem por cintilação em meio líquido.
Apêndice 2
Procedimento semicontínuo
Para conseguir chegar a uma degradação suficiente das substâncias mais recalcitrantes, poderá ser necessário incubar as culturas durante períodos que poderão chegar a vários meses. A duração do ensaio não deverá normalmente ultrapassar 60 dias, a não ser quando as características da amostra original da água possam ser mantidas através da renovação da suspensão de ensaio. O período de ensaio poderá, no entanto, ser alargado até um máximo de 90 dias, sem renovação da suspensão de ensaio, se a degradação da substância em estudo se tiver iniciado durante os primeiros 60 dias.
Durante a incubação por longos períodos, a diversidade da comunidade microbiana poderá reduzir-se devido a diferentes mecanismos de perda e também ao esgotamento de nutrientes essenciais e substratos primários de carbono na água de ensaio. Assim, recomenda-se a utilização de um ensaio em semicontínuo, a fim de determinar de forma apropriada a taxa de degradação das substâncias em que esse processo é mais lento. O ensaio deve ser iniciado utilizando um procedimento semicontínuo quando a experiência anterior mostrar que será necessário um período de incubação de três meses para conseguir a degradação de 20 % da substância presente. Se não for esse o caso, o ensaio normal, em modo descontínuo, poderá ser transformado num ensaio semicontínuo se não se observar nenhuma degradação da substância em estudo durante aproximadamente 60 dias de ensaio em descontínuo. Pode interromper-se o procedimento semicontínuo e continuar o ensaio em descontínuo a partir do momento em que se tenha registado uma degradação substancial (p.ex.: > 20 %).
No ensaio semicontínuo, cerca de um terço do volume da suspensão de ensaio será substituída, a cada quinze dias, por água acabada de recolher à qual foi adicionada a substância em estudo à concentração inicial pretendida. Se o ensaio for realizado com sedimento em suspensão, o sedimento será igualmente adicionado à água de substituição, com a mesma concentração inicial do ensaio (entre 0,01 e 1 g/l). Para efeitos da realização de um ensaio com sólidos sedimentares em suspensão, é importante manter o sistema em suspensão permanente, mesmo durante a mudança de água, e um tempo de residência seja idêntico para os sólidos e para a água, já que de outra forma a pretendida similaridade com um sistema aquoso homogéneo sem fases definidas não estará garantida. Por essas razões, quando se utilizar o regime semicontínuo será preferível utilizar uma concentração inicial de sólidos em suspensão na parte inferior do intervalo de concentrações sugerido.
A substância em estudo deve ser adicionada com uma concentração que faça com que a concentração inicial não seja ultrapassada aquando da renovação parcial da suspensão de ensaio, o que permitirá evitar os fenómenos de adaptação que ocorrem frequentemente a altas concentrações da substância em estudo. Na medida em que o procedimento implica tanto uma reinoculação como a compensação dos nutrientes e substratos primários entretanto esgotados, a diversidade microbiana original é reposta, pelo que o ensaio poderá, teoricamente, prolongar-se até ao infinito. Quando se utiliza o procedimento semicontínuo, é importante notar que a concentração residual da substância em estudo deve ser corrigida em função das quantidades removidas e adicionadas em cada renovação. A concentração total e a concentração em solução da substância de ensaio podem ser tratadas como equivalentes no caso dos compostos com baixa adsorção. A adsorção é insignificante (< 5 %), nas condições de ensaio (0,1-1 g sólidos/l) para as substâncias em que log Kow < 3 (válido para compostos lipofílicos neutros). Essa característica é ilustrada pelo seguinte exemplo: 0,1 g/l de sólidos correspondem, mais ou menos, a 10 mg de carbono por litro (fracção carbono, fC = 0,01). Assumindo que:
Log Kow (da substância de ensaio) = 3
Koc = 0,42 × Kow
Coeficiente de partição, Kd = fC × Koc
logo, a fracção dissolvida da concentração total (C-água (Cw)/C-total (Ct) será:
Cw/Ct = 1/(1 + Kd × SS) = 1(1 + Koc × fC × SS) = 1/(1 + 0,42 × 103 × 0,01 × 0,1 × 10–3) = 0,999
Apêndice 3
Determinação do 14CO2
Determinação indirecta do 14CO2
Para as medições de rotina, o método indirecto é geralmente o mais rápido e preciso, quando a substância de ensaio e os respectivos metabolitos não forem voláteis. Transferir simplesmente amostras não filtradas, por exemplo com um volume de 5 ml, para células de cintiloscópio. A actividade das amostras deverá localizar-se inicialmente entre os 5 000 dpm-10 000 dpm (80-170 Bq), sendo a actividade mínima inicial, normalmente, da ordem dos 1 000 dpm. O CO2 deve ser purgado após acidificação das amostras a pH 2-3 com uma ou duas gotas de HCl ou H3PO4 concentrado. A purga do CO2 pode ser feita por borbulhamento com ar durante cerca de ½-1 hora. Em alternativa, agitar vigorosamente as células (p.ex.: em agitador orbital) durante 1-2 horas ou, com agitação menos vigorosa, de um dia para o outro. A eficiência da purga de CO2 deve ser verificada (prolongando o período de arejamento ou de agitação). Adicionar depois um líquido de cintilação adequado para a contagem de amostras aquosas, homogeneizar a amostra num agitador orbital e determinar a radioactividade por contagem de cintilação do líquido, subtraindo a actividade de base encontrada nos brancos utilizados no ensaio (FB). A não ser nos casos em que a água de ensaio seja muito corada ou contenha uma elevada concentração de partículas, as amostras devem normalmente apresentar uma atenuação uniforme, pelo que será suficiente proceder a correcções da atenuação utilizando um padrão externo. Se a água de ensaio se apresentar muito corada, poderá ser necessário proceder a uma correcção adicional da atenuação utilizando um padrão interno. Se a concentração de partículas for elevada poderá ser difícil obter uma solução ou gel homogéneo, ou poderão ocorrer variações de atenuação demasiado grandes de amostra para amostra. Nesse caso, poderá ser utilizado o método de contagem descrito abaixo para as lamas. Se o ensaio for realizado com sedimento em suspensão, a medição do 14CO2 poderá ser efectuada indirectamente retirando uma amostra homogénea de 10 ml da água/suspensão de ensaio e separando as respectivas fases por centrifugação a uma velocidade apropriada (p.ex.: 40 000 m/s2 durante 15 min). A fase aquosa deverá então ser tratada como se descreve acima. Para a determinação da actividade de 14C da fase particulada (POA), ressuspender o sedimento num pequeno volume de água destilada, transferir para as células de cintiloscópio e adicionar líquido de cintilação de modo a formar um gel (existem líquidos de cintilação especiais para esse efeito). Dependendo da natureza das partículas (nomeadamente do respectivo teor de matéria orgânica), poderá ser viável proceder à digestão da amostra, de um dia para o outro, numa solução que dissolva os tecidos e depois à respectiva homogeneização em agitador orbital, antes de adicionar o líquido de cintilação. Em alternativa, a POA pode ser determinada por combustão em presença de oxigénio excedentário, utilizando um oxidador de amostras. Para a contagem, devem ser sempre incluídos padrões internos, podendo ser necessário proceder a correcções da atenuação por adição de um padrão interno a cada uma das amostras.
Determinação directa do 14CO2
Se o 14CO2 produzido for medido directamente, será necessário prever, no início do ensaio, um número suficiente de frascos, que serão recolhidos em cada momento de amostragem e acidificados a pH 2-3, sendo o 14CO2 recolhido num adsorvente interno (colocado em cada frasco de ensaio no início do mesmo) ou externo. Como agente adsorvente, podem utilizar-se produtos alcalinos (p.ex.: solução 1 N ou pellets de NaOH), etanolamina ou um adsorvente baseado na etanolamina que se encontre disponível no mercado. Quando as medições do 14CO2 sejam feitas de forma directa, os frascos de ensaio devem ser hermeticamente fechados, por exemplo com rolhas de borracha butírica.
Figura 1a
Exemplo de um gráfico aritmético dos dados (actividade residual em função do tempo)

Figura 1b
Exemplo de um gráfico semilogarítmico dos dados (ln da actividade residual em função do tempo)

ANEXO VI
|
C.26. |
ENSAIO DE INIBIÇÃO DE CRESCIMENTO COM LEMNA SPP. |
1. MÉTODO
Este método é equivalente ao OECD TG 221 (2006). As autoridades da UE estão de acordo em afirmar que o ensaio com Lemna constitui uma alternativa adequada para o ensaio com algas de substâncias fortemente coradas (2)(3).
1.1. INTRODUÇÃO
O presente método de ensaio foi concebido para avaliar a toxicidade de determinadas substâncias para as plantas aquáticas do Género Lemna (lentilha-de-água). O método é baseado em orientações existentes (4)(5)(6)(7)(8)(9), mas inclui modificações desses métodos para tomar em conta a investigação mais recente e as consultas realizadas em relação a uma série de questões. O método proposto foi validado por um ensaio de anel internacional (10).
O presente método de ensaio descreve ensaios de toxicidade que utilizam as espécies Lemna gibba e Lemna minor, que foram extensamente estudadas e são objecto das normas acima referidas. A taxonomia das Lemna spp. é difícil, sendo complicada pela existência de diversos fenótipos. Embora a resposta das Lemna aos agentes tóxicos possa ser afectada por variabilidade genética, a informação actualmente existente sobre essa fonte de variabilidade não é suficiente para permitir que se recomende um clone específico para a utilização no presente método de ensaio. Cabe aqui notar que embora o ensaio não seja realizado com culturas puras, são tomadas medidas, em diversas fases, para que a contaminação por outros organismos seja reduzida ao mínimo.
São descritos os pormenores do ensaio com renovação (semiestático e em contínuo) e sem renovação (estático) da solução de ensaio. Dependendo dos objectivos do ensaio e das exigências regulamentares, é recomendado que se analise a possibilidade de aplicação dos métodos semiestático e em contínuo, nomeadamente para as substâncias que desaparecem rapidamente da solução por efeito de volatilização, de fotodegradação, de precipitação ou de biodegradação. Para mais orientações, consultar a referência bibliográfica (11).
1.2. DEFINIÇÕES
Para efeitos do presente método de ensaio, foram utilizadas as seguintes definições e abreviaturas:
Biomassa: é o peso seco da matéria viva presente numa população. No presente ensaio são utilizadas algumas formas alternativas de medição da biomassa, como o número ou a superfície das frondes, sendo o termo «biomassa» utilizado também para essas medições alternativas.
Clorose: é o amarelecimento do tecido das frondes.
Clone: é um organismo ou célula obtido a partir de um único indivíduo por reprodução assexuada. Os indivíduos de um mesmo clone são, portanto, geneticamente idênticos.
Colónia: é um agregado de frondes (geralmente 2 a 4) progenitoras e descendentes, ligadas entre si. As colónias são por vezes designadas por «planta».
ECx : é a concentração da substância em estudo que, dissolvida no meio de ensaio, resulta numa redução de x % (p. ex.: 50 %) do crescimento da Lemna, após um determinado período de exposição (que deverá ser explicitamente mencionado nos casos em que se afaste da duração total ou normal do ensaio). A fim de indicar de forma inequívoca se o valor de EC se refere à taxa de crescimento ou ao rendimento, o símbolo «ErC» é utilizado no primeiro caso e o símbolo «EyC» no segundo, sendo seguidos da designação da variável de medição utilizada, p. ex.: ErC (número de frondes).
Ensaio em contínuo: é um ensaio em que a solução em estudo é continuamente substituída.
Fronde: é uma estrutura individual/única, em «forma de folha», de uma planta de lentilha-de-água. Trata-se da mais pequena unidade, ou seja, indivíduo, capaz de reprodução.
Giba: é uma fronde que apresenta um aspecto irregular ou inchado.
Crescimento: é o aumento da variável de medição, que pode ser o número de frondes, o peso seco, o peso húmido ou a superfície das frondes, ao longo do período de ensaio.
Taxa de crescimento (taxa média de crescimento específico): é o aumento logarítmico da biomassa durante o período de exposição.
Menor concentração com efeito observável (LOEC): é a concentração mais baixa à qual se observa que a substância em estudo tem um efeito estatisticamente significativo (p < 0,05) de redução do crescimento, quando comparada com o controlo, para um determinado período de exposição. No entanto, todas as concentrações de ensaio superiores à LOEC devem ter um efeito prejudicial igual ou superior ao verificado com a LOEC. Quando estas duas condições não puderem ser satisfeitas, deverá ser fornecida uma explicação pormenorizada sobre a forma como se determinou a LOEC (e, consequentemente, a NOEC).
Variáveis de medição: é qualquer tipo de variável que seja medida para permitir a determinação do ponto final do ensaio, utilizando uma ou mais do que uma variáveis de resposta. No presente método, o número de frondes, a superfície das frondes, o peso fresco e o peso seco são variáveis de resposta.
Monocultura: é uma cultura com uma única espécie de planta.
Necrose: é o tecido da fronde morto (ou seja, branco ou empapado em água).
Concentração sem efeitos observáveis (NOEC): é a concentração de ensaio imediatamente inferior à LOEC.
Fenótipo: são as características observáveis de um organismo, determinadas pela interacção dos seus genes com o ambiente em que vive.
Variáveis de resposta: são variáveis de estimação da toxicidade, derivadas de qualquer dos parâmetros medidos que descrevem a biomassa nos diferentes métodos de cálculo. No presente método, as taxas de crescimento e o rendimento são variáveis de resposta derivadas de variáveis de medição como o número de frondes, a superfície das frondes, o peso fresco ou o peso seco.
Ensaio semiestático (com renovação do meio): é um ensaio em que a solução de ensaio é periodicamente substituída, em determinados momentos do ensaio.
Ensaio estático: é um método de ensaio durante o qual não há renovação da solução de ensaio.
Ponto final do ensaio: descreve, em termos gerais, o factor que será alterado pela substância química em estudo, por comparação com o controlo, cuja identificação é o objectivo do ensaio. No presente método, o ponto final é a inibição do crescimento, que pode ser expressa por diferentes variáveis de resposta, baseadas em uma ou mais variáveis de medição.
Meio de ensaio: é o meio sintético completo de crescimento em que as plantas de ensaio são cultivadas quando expostas à substância em estudo. A substância em estudo será normalmente dissolvida no meio de ensaio.
Rendimento: é o valor de uma variável de medição que exprime a biomassa no final do período de exposição menos o valor dessa variável de medição no início do período de exposição.
1.3. PRINCÍPIO DO ENSAIO
Utilizando monoculturas de plantas do género Lemna em crescimento exponencial, são feitas culturas com diferentes concentrações da substância em estudo, durante sete dias. O objectivo do ensaio é quantificar os efeitos da substância em estudo sobre o crescimento vegetativo durante esse período, com base na avaliação de determinadas variáveis de medição. O número de frondes é a variável de medição primária. Pelo menos mais uma variável de medição (superfície total das frondes, peso seco ou peso fresco) será também medida, já que certas substâncias podem afectar outras variáveis de medição de forma muito mais severa do que o número de frondes. Para quantificar os efeitos relacionados com a substância em estudo, o crescimento das culturas de ensaio é comparado com o dos controlos, sendo determinada a concentração que causa uma inibição do crescimento de x % (p. ex.: 50 %), expressa como ECx (p. ex.: EC50).
O ponto final do ensaio é a inibição do crescimento, expresso como o aumento logarítmico da variável de medição (taxa média de crescimento específico) durante o período de exposição. A partir das taxas médias de crescimento específico registadas numa série de soluções de ensaio, a concentração que causa uma inibição da taxa de crescimento de x % (p. ex.: 50 %) é determinada e expressa como ErCx (nesse caso, ErC50)
Outra variável de resposta utilizada no presente método de ensaio é o rendimento, que pode ser necessário para dar cumprimento a exigências regulamentares específicas de alguns países. O rendimento é definido como o valor da variável de medição no final do período de exposição menos o valor da variável de medição no início do período de exposição A partir dos rendimentos registados numa série de soluções de ensaio, a concentração que causa uma inibição do rendimento de x % (p. ex.: 50 %) é determinada e expressa como EyCx (nesse caso, EyC50).
Por outro lado, a menor concentração com efeito observável (LOEC) e a concentração sem efeitos observáveis (NOEC) podem ser determinadas estatisticamente.
1.4. INFORMAÇÃO SOBRE A SUBSTÂNCIA EM ESTUDO
Deve existir um método analítico que apresente uma sensibilidade adequada para a quantificação da substância no meio de ensaio.
A informação sobre a substância em estudo que poderá ser útil para estabelecer as condições do ensaio inclui a sua fórmula estrutural, pureza, hidrossolubilidade, estabilidade à luz e em água, pKa, Kow, pressão de vapor e biodegradabilidade. A hidrossolubilidade e a pressão de vapor podem ser utilizadas para calcular a constante da Lei de Henry, o que permitirá verificar a probabilidade de perdas significativas da substância em estudo durante o período de ensaio. Dessa forma, poderá avaliar-se se é necessário adoptar medidas específicas para o controlo dessas perdas. Nos casos em que a informação sobre a solubilidade e a estabilidade da substância de ensaio for incerta, recomenda-se que as mesmas sejam avaliadas nas condições de ensaio, ou seja, com o mesmo meio de crescimento, temperatura e regime de iluminação que irá ser utilizado no ensaio.
O controlo do pH do meio de ensaio é particularmente importante, nomeadamente no caso dos ensaios de metais ou de substâncias hidroliticamente instáveis, pelo que se recomenda, nesses casos, a adição de um tampão ao meio de crescimento (ver o primeiro parágrafo do ponto 1.7.4. Para mais orientações sobre o ensaio de substâncias cujas propriedades físico-químicas dificultam o ensaio, consultar a referência bibliográfica (11).
1.5. SUBSTÂNCIA DE REFERÊNCIA
Para verificação do procedimento de ensaio, pode(m) ser ensaiada(s) (uma) substância(s) de referência, como por exemplo o 3,5-diclorofenol, utilizado no ensaio de anel internacional (10). Será desejável proceder ao ensaio de uma substância de referência pelo menos duas vezes por ano ou, quando os ensaios forem realizados com menos frequência, em paralelo com a determinação da toxicidade das substâncias em estudo.
1.6. VALIDADE DO ENSAIO
Para que o ensaio seja válido, o tempo de duplicação do número de frondes no controlo deve ser inferior a 2,5 dias (60 horas), o que corresponde aproximadamente a uma multiplicação por sete em sete dias e a uma taxa média de crescimento específico de 0,275 d–1. Utilizando os meios e as condições de ensaio descritas no presente método, esse critério pode ser cumprido num regime de ensaio estático (8). Parte-se igualmente do princípio de que esse critério poderá ser cumprido nos ensaios semiestáticos ou contínuos. O cálculo do tempo de duplicação é apresentado no ponto 2.1.
1.7. DESCRIÇÃO DO MÉTODO
1.7.1. Equipamento
Todos os equipamentos que entrem em contacto com as soluções de ensaio devem ser totalmente construídos em vidro ou noutro material quimicamente inerte O material de vidro utilizado para as culturas e ensaios deve ser limpo de todos os contaminantes químicos que possam lixiviar para o meio de ensaio e esterilizado. Os frascos de ensaio devem ser suficientemente largos para que as frondes das diferentes colónias dos frascos de controlo possam crescer sem se sobreporem até ao final do ensaio. Não importa se as raízes tocarem o fundo dos frascos de ensaio, mas será aconselhável utilizar frascos com uma profundidade mínima de 20 mm e com um volume mínimo de 100 ml. A escolha do melhor tipo de frascos não é crítica, desde que esses requisitos sejam cumpridos. O ensaio já foi realizado com sucesso em frascos de vidro, caixas de cristalização ou caixas de Petri de dimensões apropriadas. Os frascos de ensaio devem ser cobertos de modo a minimizar a evaporação e a contaminação acidental, sem pôr em causa as necessárias trocas gasosas. Os frascos e, em especial, as tampas mais indicadas não provocarão zonas de sombra ou alterações das características do espectro luminoso.
As culturas e os frascos de ensaio não devem ser conservados juntos. Para tal, o melhor será utilizar câmaras de crescimento, incubadoras ou mesmo salas separadas. A iluminação e a temperatura devem ser controláveis e mantidas a um nível constante (ver o ponto 1.7.8).
1.7.2. Organismo de ensaio
O presente ensaio pode ser realizado com Lemna gibba ou com Lemna minor. O apêndice 1 apresenta uma breve descrição das espécies de lentilha-de-água que já foram utilizadas para ensaios de toxicidade. O material vegetal pode ser obtido junto de uma colecção de culturas, de outro laboratório ou no campo. Quando forem recolhidas no campo, as plantas devem ser mantidas em cultura no mesmo meio que irá ser utilizado para o ensaio durante pelo menos oito semanas antes do mesmo. Os locais de colheita no campo onde se faz a recolha de culturas de arranque devem estar isentos de fontes evidentes de poluição. Caso sejam obtidas de outro laboratório ou de uma colecção de culturas, as plantas devem ser mantidas da mesma forma durante pelo menos três semanas. A fonte do material vegetal e da espécie ou clone (se conhecido) utilizado para o ensaio deve ser sempre indicada.
Devem ser utilizadas monoculturas visivelmente isentas de contaminação por outros organismos, como algas ou protozoários. As plantas saudáveis de L. minor são formadas por colónias com duas a cinco frondes, enquanto que as colónias saudáveis de L. gibba podem apresentar até sete frondes.
A qualidade e a uniformidade das plantas utilizadas para o ensaio terão uma influência significativa sobre o resultado do mesmo, pelo que as plantas devem ser cuidadosamente seleccionadas. Devem ser utilizadas plantas jovens, de crescimento rápido e sem lesões ou descoloração (clorose) visíveis. Uma cultura de boa qualidade é caracterizada por uma elevada incidência de colónias com pelo menos duas frondes. Um número elevado de frondes isoladas é indicativo de stress ambiental, nomeadamente por limitação de nutrientes, pelo que o material vegetal das culturas com essas características não deverá ser utilizado para os ensaios.
1.7.3. Cultura
Para reduzir a frequência das transferências para conservação da cultura (p. ex.: durante os períodos em que não estejam previstos ensaios com Lemna) as culturas poderão ser conservadas em ambiente escuro e a baixa temperatura (4-10 °C). O apêndice 2 apresenta mais informação sobre as técnicas de cultura. O surgimento de sinais evidentes de contaminação por algas ou por outros organismos exigirá a esterilização superficial de uma subamostra de frondes de Lemna, seguida de transferência para meio fresco (ver o apêndice 2). Nesse caso, a parte restante da cultura contaminada deverá ser descartada.
Pelo menos sete dias antes do ensaio, um número suficiente de colónias será transferido em condições assépticas para meio fresco esterilizado e cultivado durante 7-10 dias nas condições de ensaio.
1.7.4. Meio de ensaio
Existem diversos meios recomendados para as culturas de Lemna minor e Lemna gibba, como se descreve a seguir. A inclusão de um tampão de pH no meio de ensaio [MOPS (ácido 4-morfolinepropanosulfónico, N.o CAS: 1132-61-2; N.o EINECS: 214-478-5) no meio para L. minor ou NaHCO3 no meio para L. gibba] deve ser cuidadosamente ponderada nos casos em que possa haver interacção com a substância em estudo que influencie a expressão da sua toxicidade. O meio de Steinberg (12) também poderá ser utilizado, desde que estejam cumpridos os critérios de validade do ensaio.
Para as culturas e ensaios com L. minor, recomenda-se a utilização do meio Swedish Standard (SIS) para Lemna, modificado. A composição desse meio é dada no apêndice 3.
O meio de crescimento 20X — AAP, descrito no apêndice 3, é recomendado para as culturas e ensaios com L. gibba.
O meio de Steinberg, tal como descrito no apêndice 3, também é indicado para a L. minor, mas pode ser utilizado para a L. gibba, desde que estejam cumpridos os critérios de validade do ensaio.
1.7.5. Soluções de ensaio
As soluções de ensaio são geralmente preparadas através da diluição de soluções de reserva. As soluções de reserva da substância em estudo são normalmente preparadas por dissolução da substância em meio de crescimento.
A concentração mais elevada da substância em estudo no ensaio não deverá normalmente ser superior ao limite de solubilidade da substância em água, nas condições de ensaio. No entanto, cabe aqui notar que as Lemna spp. flutuam à superfície, podendo ser expostas a substâncias recolhidas na interface água-ar (p. ex.: substâncias de baixa hidrossolubilidade, hidrófobas ou tensioactivas). Nessas circunstâncias, terá lugar uma exposição a materiais diferentes dos que se encontram em solução e as concentrações de ensaio podem, dependendo das características da substância em estudo, ultrapassar o limite de hidrossolubilidade. Para as substância com baixa hidrossolubilidade, poderá ser necessário preparar uma solução de reserva concentrada ou dispersar a substância por meio de um dispersante ou solvente orgânico, de modo a facilitar a adição de quantidades exactas da substância em estudo ao meio de ensaio e a respectiva dispersão e dissolução. Devem ser desenvolvidos todos os esforços para evitar utilizar materiais desse tipo. A utilização de dispersantes ou de solventes auxiliares não deverá acarretar qualquer fitotoxicidade. Assim, por exemplo, os solventes mais comuns que não causam fitotoxicidade em concentrações até aos 100 ml·l–1 incluem a acetona e a dimetilformamida. Se for utilizado um dispersante ou um solvente, a sua concentração final, tão baixa quanto possível (≤ 100 ml·l–1), deve ser comunicada e todos os frascos de exposição e de controlo devem ser expostos à mesma concentração. Para mais orientações sobre a utilização de dispersantes, consultar a referência bibliográfica (11).
1.7.6. Grupos de ensaio e de controlo
O conhecimento prévio da toxicidade da substância em estudo para a Lemna, por exemplo determinada com base nos resultados de ensaios de determinação da gama de concentrações, permitirá uma mais fácil escolha das concentrações a utilizar no ensaio. No ensaio de toxicidade definitivo, deverão normalmente ser estudadas cinco concentrações de ensaio, em progressão geométrica. De preferência, o factor de separação entre as concentrações de ensaio não deve ultrapassar os 3,2, mas poderá ser utilizado um valor mais elevado quando a curva de concentração-resposta for pouco pronunciada. Quando se utilizarem menos de cinco concentrações, deve ser apresentada uma justificação. Devem ser utilizados pelo menos três replicados de cada concentração de ensaio.
Para efeitos da definição da gama de concentrações (tanto nos ensaios de determinação da gama quanto nos ensaios definitivos de toxicidade) deve tomar-se em consideração os seguintes elementos:
|
— |
Para a determinação do ECx, as concentrações de ensaio devem abranger o valor de ECx, de modo a garantir um nível de confiança apropriado. Assim, para a determinação do EC50, por exemplo, a concentração de ensaio mais elevada deve ser superior ao valor de EC50. Se o valor de EC50 se encontrar fora da gama de concentrações ensaiadas, os intervalos de confiança associados serão maiores, o que poderá impossibilitar o ajustamento de um modelo estatístico, |
|
— |
Se o objectivo for a determinação do LOEC/NOEC, a concentração mais baixa ensaiada deve ser suficientemente baixa para que o seu crescimento não seja significativamente inferior ao dos controlos. Por outro lado, a concentração mais elevada a ensaiar deve ser suficientemente alta para que o seu crescimento seja significativamente inferior ao do controlo. Se não for esse o caso, o ensaio terá de ser repetido com uma gama de concentrações diferente (a não ser quando a concentração máxima já se encontre perto do limite de solubilidade ou da concentração máxima pretendida, 100 mg·l–1). |
Todos os ensaios devem incluir controlos compostos pelo mesmo meio nutriente, número de frondes e de colónias e sujeitos às mesmas condições ambientais e procedimentos que os frascos de ensaio, mas sem adição da substância em estudo. Se for utilizado um dispersante ou solvente auxiliar, o ensaio deverá incluir um controlo suplementar, com a mesma concentração de solvente/dispersante que os frascos com a substância em estudo. O número de frascos com os replicados dos controlos (e de frascos com solvente, se aplicável) deve ser pelo menos igual e idealmente o dobro do número de frascos utilizados para cada concentração de ensaio.
Caso não seja necessário determinar o NOEC, o ensaio poderá ser alterado de modo a aumentar o número de concentrações estudadas, reduzindo o número de replicados por concentração. O número de replicados dos controlos deve, no entanto, ser de pelo menos três.
1.7.7. Exposição
As colónias, com 2 a 4 frondes visíveis, são transferidas da cultura de inóculo e distribuídas de forma aleatória pelos frascos de ensaio, em condições assépticas. Cada frasco de ensaio deve ser inoculado com um total de 9 a 12 frondes. O número de frondes e de colónias deve ser o mesmo em todos os frascos de ensaio. A experiência já obtida com o presente método e os dados dos ensaios de anel indicam que a utilização de três replicados de cada concentração em estudo, com 9 a 12 frondes em cada replicado, são suficientes para detectar diferenças de crescimento da ordem dos 4 a 7 %, para a inibição calculada a partir da taxa de crescimento (e dos 10 a 15 % a partir do rendimento), entre as diferentes concentrações (10).
A colocação aleatória dos frascos de ensaio na incubadora é necessária para reduzir ao mínimo a influência das diferenças espaciais em termos de intensidade da luz ou de temperatura. É igualmente necessário mudar a posição dos frascos, por blocos ou de forma aleatória, após as amostragens (ou mais frequentemente).
Se um ensaio preliminar de estabilidade mostrar que a concentração da substância em estudo não pode ser mantida (ou seja, se a concentração medida diminuir mais de 80 % em relação à concentração inicial medida) durante o período de ensaio (7 dias), recomenda-se a utilização de um regime de ensaio semiestático. Nesse caso, as colónias devem ser expostas a soluções frescas de ensaio e de controlo em pelo menos duas ocasiões durante o ensaio (p. ex.: nos dias 3 e 5). A frequência da exposição ao meio fresco dependerá da estabilidade da substância em estudo; para manter concentrações quase constantes de substâncias altamente instáveis ou voláteis, poderá ser necessária uma frequência maior. Em certos casos, poderá ser necessário utilizar um procedimento em contínuo (11)(13).
A possibilidade de exposição através de aplicação foliar (aspersão) não está prevista no presente método de ensaio, para esse método ver a referência bibliográfica (14).
1.7.8. Condições de incubação
Deve ser utilizada uma iluminação fluorescente constante com luz branca, quente ou fria, de modo a proporcionar às culturas uma intensidade luminosa seleccionada na gama dos 85-135 μE·m–2·s–1, medida no comprimento de onda da fotossíntese (400-700 nm) em pontos que se encontrem à mesma distância da fonte luminosa que as frondes de Lemna (intensidade equivalente a 6 500-10 000 lux). As diferenças em relação à intensidade luminosa escolhida não devem ultrapassar, em toda a zona de ensaio, ± 15 %. O método de detecção e medição da luz e, em particular, o tipo de sensor utilizado, afectarão o valor medido. Os sensores esféricos (que respondem à luz proveniente de todos os ângulos acima e abaixo do plano de medição) e os sensores «de co-seno» (que respondem à luz proveniente de todos os ângulos acima do plano de medição) são preferíveis aos sensores unidireccionais e darão leituras mais elevadas para as fontes múltiplas de luz do tipo aqui descrito.
A temperatura dos frascos de ensaio deve ser mantida a 24 ± 2 °C. O pH do meio de controlo não deve aumentar mais de 1,5 unidades durante o ensaio. No entanto, um desvio superior a 1,5 unidades de pH não invalidará o ensaio quando se possa demonstrar que os critérios de validade são cumpridos. Em certos caos, contudo, como por exemplo nos ensaios de substâncias instáveis ou de metais, poderá ser necessário adoptar cuidados adicionais em relação à variação de pH. Para mais informações, consultar a referência bibliográfica (11).
1.7.9. Duração
O ensaio terminará 7 dias depois de as plantas terem sido transferidas para os frascos de ensaio.
1.7.10. Medições e determinações analíticas
No início do ensaio, o número de frondes em cada frasco de ensaio será registado, com o cuidado de contar todas as frondes evidentes e distintamente visíveis. O número de frondes que apresentam uma aparência normal e anormal deve ser determinado no início do ensaio e pelo menos mais uma vez a cada 3 dias durante o período de exposição (ou seja, pelo menos duas vezes ao longo dos 7 dias do ensaio) e no final do mesmo. Qualquer alteração observada no desenvolvimento das plantas, por exemplo no que respeita à dimensão e aparência das frondes, a indicações de necrose, clorose ou da formação de gibas, à desagregação ou perda de flutuabilidade das colónias ou à dimensão e aspecto das raízes, deverá ser registada. As características mais significativas do meio de ensaio (p. ex.: presença de materiais em suspensão, crescimento de algas nos frascos de ensaio) devem também ser registadas.
Para além da determinação do número de frondes durante o ensaio, devem também ser avaliados os efeitos da substância em estudo sobre uma (ou mais) das seguintes variáveis de medição:
|
i) |
superfície total das frondes, |
|
ii) |
peso seco, |
|
iii) |
peso fresco. |
A superfície total das frondes, enquanto variável, tem a vantagem de poder ser determinada para cada frasco de ensaio e de controlo no início, durante e no final do ensaio. O peso seco ou fresco deverá ser determinado no início do ensaio a partir de uma amostra da cultura de inóculo, representativa do inóculo utilizado para lançar o ensaio, e no final do mesmo, com o material retirado das plantas de cada frasco de ensaio e de controlo. Se a superfície das frondes não for medida, o seguimento do peso seco será preferível ao do peso fresco.
A superfície total das frondes, o peso seco e o peso fresco podem ser determinados do seguinte modo:
|
i) |
Superfície total das frondes: a superfície total das frondes de todas as colónias pode ser determinada por análise visual. Uma silhueta do frasco e das plantas de ensaio pode ser filmada com uma câmara de vídeo (p. ex.: colocando o frasco numa mesa de luz) e digitalizada. A partir de uma calibração utilizando formas planas com uma superfície conhecida, a superfície total das frondes em cada frasco de ensaio pode ser determinada. Deve ter-se o cuidado de excluir a interferência causada pela borda do frasco de ensaio. Uma abordagem alternativa, mais trabalhosa, será tirar uma fotografia dos frascos e plantas de ensaio, recortando depois a silhueta resultante das colónias e determinando a respectiva superfície utilizando um integrador ou papel milimétrico. Outras técnicas (p. ex.: a relação entre o peso da superfície de papel equivalente à silhueta das colónias e o peso de uma superfície de papel unitária) poderão igualmente revelar-se apropriadas; |
|
ii) |
Peso seco: todas as colónias de cada frasco de ensaio são recolhidas e lavadas com água destilada ou desionizada. Retira-se a água em excesso e seca-se a 60 °C até obter um peso constante. Os fragmentos de raízes que se encontrem presentes também devem ser incluídos. O peso seco deverá ser expresso com uma precisão mínima de 0,1 mg; |
|
iii) |
Peso fresco: as colónias são transferidas para tubos de plástico (ou de outro material inerte) de peso conhecido e com pequenos orifícios (1 mm) no fundo. Os tubos são depois centrifugados a 3 000 rpm durante 10 minutos, à temperatura ambiente. Os tubos com as colónias, agora secas, voltam a ser pesados e o peso fresco é calculado por subtracção do peso dos tubos vazios. |
1.7.10.1. Frequência das medições e determinações analíticas
Se se utilizar um método de ensaio estático, o pH de cada frasco de ensaio deve ser medido no início e no final do ensaio. No caso dos ensaios semiestáticos, deve ser medido o pH da solução de ensaio «fresca», antes da sua utilização, bem como o pH das correspondentes soluções «usadas».
A intensidade luminosa deve ser medida, na câmara de crescimento, incubadora ou sala, em pontos que estejam à mesma distância da fonte de luz que as frondes de Lemna. A medição deverá ser efectuada pelo menos uma vez durante o ensaio. Deve ser registada diariamente a temperatura do meio contido num frasco especialmente destinado para esse efeito e mantido nas mesmas condições na câmara de crescimento, incubadora ou sala utilizada para o ensaio.
Durante o ensaio, as concentrações da substância em estudo deverão ser determinadas a intervalos regulares. Nos ensaios estáticos, a exigência mínima é que as concentrações sejam determinadas no início e no final do ensaio.
No caso dos ensaios semiestáticos em que não seja de esperar que a concentração da substância em estudo se mantenha no intervalo de ± 20 % da concentração nominal, será necessário analisar todas as soluções de ensaio no momento em que sejam preparadas e também imediatamente antes da respectiva utilização (ver o terceiro parágrafo do ponto 1.7.7). No entanto, para os ensaios em que a concentração inicial medida da substância em estudo não se encontra no intervalo de ± 20 % do valor nominal, mas em que se possam fornecer provas suficientes de que as concentrações iniciais são reprodutíveis e estáveis (isto é, se mantêm dentro da gama dos 80-120 % das concentrações iniciais), as determinações químicas poderão ser limitadas à maior e à menor concentração usadas no ensaio. Em qualquer dos casos, a determinação das concentrações da substância em estudo antes da sua utilização só é necessária num dos frascos replicados para cada concentração de ensaio (ou no conteúdo misturado de todos os frascos replicados).
Nos ensaios em contínuo, poderá ser utilizado um regime de amostragem semelhante ao descrito para os ensaios semiestáticos, com análises no início, no meio e no final do ensaio, sem que seja necessário, nesse caso, medir as concentrações nas soluções «usadas». Nesse tipo de ensaios, a taxa de diluição da mistura do diluente ou da solução de reserva com a substância em estudo deverá ser verificada diariamente.
Se existirem provas de que a concentração da substância de ensaio não variou mais de ± 20 %, durante o ensaio, em relação ao valor da concentração nominal ou da concentração inicial medida, a análise dos resultados poderá basear-se nos valores nominais ou iniciais medidos. Se a variação em relação à concentração nominal ou à concentração inicial medida for superior a ± 20 %, a análise dos resultados deverá basear-se na média geométrica da concentração durante a exposição ou em modelos que descrevam a diminuição da concentração da substância em estudo (11).
1.7.11. Ensaio limite
Em determinadas circunstâncias, ou seja, quando um ensaio preliminar indicar que a substância em estudo não apresenta efeitos tóxicos em concentrações até aos 100 mgl–1 ou até ao limite de solubilidade da substância no meio de ensaio (conforme a que seja menor), poderá proceder-se a um ensaio limite com comparação das respostas de um grupo de controlo e de um grupo exposto à substância em estudo (a uma concentração de 100 mgl–1 ou que corresponda ao limite de solubilidade da substância no meio de ensaio). É fortemente recomendado que a ausência de toxicidade seja confirmada por análise da concentração de exposição. Todas as condições de ensaio e critérios de validade anteriormente descritos são aplicáveis aos ensaios limite, com excepção da necessidade de utilizar o dobro do número de replicados dos frascos expostos à substância. O crescimento dos grupos de controlo e de exposição pode ser analisado através de um ensaio estatístico de comparação das médias, como por exemplo o teste t-Student.
2. RESULTADOS E RELATÓRIO
2.1. TEMPO DE DUPLICAÇÃO
Para determinar o tempo de duplicação (Td) do número de frondes e verificar se o ensaio cumpre o critério de validade correspondente (ponto 1.6), a seguinte fórmula deverá ser aplicada aos dados obtidos a partir dos frascos de controlo:
Td = ln 2/μ
onde μ é a taxa média de crescimento específico, determinada de acordo com o primeiro e segundo parágrafos do ponto 2.2.1.
2.2. VARIÁVEIS DE RESPOSTA
O objectivo do ensaio é determinar os efeitos da substância em estudo sobre o crescimento vegetativo da Lemna. O método de ensaio descreve duas variáveis de resposta, já que os Estados-Membros têm diferentes preferências e necessidades regulamentares. Para que os resultados dos ensaios possam ser aceitáveis em todos os Estados-Membros, os efeitos terão de ser avaliados utilizando ambas as variáveis de resposta, a) e b), a seguir descritas:
|
a) |
Taxa média de crescimento específico: esta variável é calculada com base no aumento logarítmico do número de frondes e, adicionalmente, na evolução logarítmica de outro parâmetro de medição (superfície total das frondes, peso seco ou peso fresco) em função do tempo (por dia), tanto nos frascos de controlo como nos grupos tratados com a substância em estudo. É por vezes designada por taxa de crescimento relativa (15); |
|
b) |
Rendimento: esta variável de resposta é calculada com base no aumento do número de frondes e, adicionalmente, na evolução de outro parâmetro de medição (superfície total das frondes, peso seco ou peso fresco), tanto nos frascos de controlo como nos grupos tratados com a substância em estudo, até ao final do ensaio. |
Cabe aqui notar que os valores de toxicidade calculados através destas duas variáveis de resposta não são comparáveis e que essa diferença tem de ser reconhecida para efeitos da utilização dos resultados do ensaio. Os valores de ECx baseados na taxa média de crescimento específico (ErCx) serão geralmente mais elevados do que os resultados baseados no rendimento (EyCx), se forem seguidas as condições de ensaio apresentadas no presente método de ensaio, devido à base matemática das respectivas abordagens. Esse facto não deve ser interpretado como uma diferença de sensibilidade entre as duas variáveis de resposta, mas simplesmente como uma diferença matemática entre os valores. O conceito de taxa média de crescimento específico baseia-se no padrão geral de crescimento exponencial da lentilha-de-água em culturas não sujeitas a limitações, sendo a toxicidade estimada com base nos efeitos sobre a taxa de crescimento, e não depende do valor absoluto da taxa de crescimento específico dos controlos, do declive da curva de concentração-resposta ou da duração do ensaio. Em contraste, os resultados baseados no rendimento enquanto variável de resposta são dependentes de todas essas variáveis. O valor de EyCx é dependente da taxa de crescimento específico da espécie de lentilha-de-água utilizada em cada ensaio e da taxa máxima de crescimento específico, que pode ser diferente para as diferentes espécies ou mesmo para os diferentes clones. Esta variável de resposta não deve ser utilizada para comparar a sensibilidade das diferentes espécies ou mesmo dos diferentes clones de lentilha-de-água aos produtos tóxicos. Embora seja preferível, do ponto de vista científico, utilizar a taxa média de crescimento específico para a estimação da toxicidade, as estimativas da toxicidade com base no rendimento foram também incluídas no presente método de ensaio para satisfazer as actuais exigências regulamentares de alguns países.
As estimativas da toxicidade deverão ser baseadas no número de frondes e numa variável de medição adicional (superfície total das frondes, peso seco ou peso fresco), já que certas substâncias poderão afectar outras variáveis de medição de forma muito mais severa do que o número de frondes. Esse efeito passará despercebido se o cálculo se basear exclusivamente no número de frondes.
O número de frondes, bem como o valor de qualquer outra variável de resposta medida, como a superfície total das frondes, o peso seco ou o peso fresco, será registado numa tabela, juntamente com as concentrações da substância em estudo em cada momento de medição. A subsequente análise dos dados, por exemplo para determinação do LOEC, do NOEC ou dos ECx, deverá basear-se nos valores obtidos para cada replicado e não em médias calculadas para cada grupo exposto à substância em estudo.
2.2.1. Taxa média de crescimento específico
A taxa média de crescimento específico durante um determinado período é calculada como o aumento logarítmico das variáveis de crescimento — número de frondes e outra variável de medição (superfície total das frondes, peso seco ou peso fresco) — aplicando a fórmula a seguir apresentada aos valores obtidos para cada replicado dos controlos e dos frascos expostos à substância em estudo.

onde:
|
— |
μi-j: |
taxa média de crescimento específico entre o momento i e o momento j |
|
— |
Ni: |
variável de medição no frasco de ensaio ou de controlo no momento i |
|
— |
Nj: |
variável de medição no frasco de ensaio ou de controlo no momento j |
|
— |
t: |
período decorrido entre i e j |
Para cada um dos grupos de exposição e de controlo, calcular um valor médio da taxa de crescimento, associado às respectivas estimativas da variância.
A taxa média de crescimento específico deve ser calculada para a totalidade do período de ensaio (na fórmula acima, «i» é o momento inicial e «j» o momento final do ensaio). Para cada uma das concentrações de ensaio e de controlo, calcular o valor médio da taxa média de crescimento específico, associado às respectivas estimativas da variância. Por outro lado, a taxa de crescimento em cada secção do ensaio deve ser avaliada para verificar os efeitos da substância em estudo durante o período de exposição (p. ex.: através da análise das curvas de crescimento após transformação logarítmica). A existência de diferenças substanciais entre as taxas de crescimento de cada secção do ensaio e a taxa média de crescimento indica um desvio em relação à situação de crescimento exponencial constante, exigindo portanto uma análise mais pormenorizada das curvas de crescimento. Nesses casos, a abordagem mais cautelosa passa pela comparação entre as taxas de crescimento específico das culturas expostas durante o período de máxima inibição e as taxas observadas nos controlos durante o mesmo período.
A percentagem de inibição da taxa de crescimento (Ir) poderá então ser calculada para cada concentração de ensaio (grupos expostos), de acordo com a seguinte fórmula:

onde:
|
— |
% Ir: |
percentagem de inibição da taxa média de crescimento específico |
|
— |
μC: |
valor médio de μ nos controlos |
|
— |
μT: |
valor médio de μ no grupo exposto |
2.2.2. Rendimento
Os efeitos sobre o rendimento são determinados com base em duas variáveis de medição, o número de frondes e outra variável (superfície total das frondes, peso seco ou peso fresco), para cada frasco de ensaio, no início e no final do mesmo. No que respeita ao peso seco e ao peso fresco, a biomassa de partida é determinada com base numa amostra das frondes retirada da cultura utilizada para inocular os frascos de ensaio (ver o segundo parágrafo do ponto 1.7.3). Para cada uma das concentrações de ensaio e para os controlos, calcular um valor médio de rendimento, associado às respectivas estimativas da variância. A percentagem média de inibição do rendimento (% Iy) pode ser calculada, para cada grupo exposto, do seguinte modo:
onde:
|
— |
% Iy: |
percentagem de redução do rendimento |
|
— |
bC: |
biomassa final menos biomassa inicial do grupo de controlo |
|
— |
bT: |
biomassa final menos biomassa inicial do grupo exposto |
2.2.3. Representação gráfica da curva de concentração-resposta
Devem ser representadas as curvas de concentração-resposta, que relacionam a percentagem média de inibição da variável de resposta (Ir, ou Iy, calculadas como se indica no último parágrafo do ponto 2.2.1 ou no ponto 2.2.2) com o logaritmo da concentração da substância em estudo.
2.2.4. Estimação do ECx
As estimativas do ECx (p. ex.: EC50) devem ser baseadas tanto na taxa média de crescimento específico (ErCx) como no rendimento (EyCx), devendo cada uma dessas medidas ser, por seu lado, baseada no número de frondes e numa variável de medição adicional (superfície total das frondes, peso seco ou peso fresco), já que certas substâncias afectam de forma diferente o número de frondes e outras variáveis de medição. Os parâmetros de toxicidade pretendidos são, portanto, quatro valores de ECx para cada um dos níveis de inibição x calculados: ErCx (número de frondes); ErCx (superfície total das frondes, peso seco ou peso fresco); EyCx (número de frondes); e EyCx (superfície total das frondes, peso seco ou peso fresco).
2.3. PROCEDIMENTOS ESTATÍSTICOS
O objectivo é descrever de forma quantitativa, por análise de regressão, a relação concentração-resposta. Pode utilizar-se uma regressão linear ponderada, antecedida de uma transformação de linearização dos dados de resposta — por exemplo para unidades probit, logit ou de Weibull (16) –, mas a técnica preferida é a aplicação de procedimentos de regressão não linear, que permitem lidar melhor com as inevitáveis irregularidades dos dados e com os desvios em relação a uma boa distribuição. Nas zonas próximas da inibição nula ou total, essas irregularidades podem mesmo ser aumentadas pela transformação, o que irá interferir com a análise (16). Cabe aqui notar que os métodos padrão de análise que utilizam os transformados probit, logit ou de Weibull se destinam à análise de dados quantais (p. ex.: mortalidade ou sobrevivência), pelo que terão de ser alterados para lidar com os dados relativos à taxa de crescimento ou ao rendimento. Alguns métodos para a determinação dos valores de ECx a partir de dados contínuos podem ser consultados em (17), (18) e (19).
Para cada uma das variáveis de resposta em análise, utilizar a relação concentração-resposta para estimar os valores pontuais de ECx. Sempre que possível, deverão ser determinados os limites de confiança a 95 % para cada estimativa. A adequação do ajustamento da curva estimada pelo modelo de regressão aos dados de resposta deve ser avaliada, graficamente ou por métodos estatísticos. A análise de regressão deve ser efectuada utilizando os valores de cada replicado e não o valor médio para cada grupo exposto.
As estimativas do EC50 e os respectivos intervalos de confiança podem também ser obtidos utilizando uma interpolação linear com bootstrapping (20), quando os métodos/modelos de regressão disponíveis não forem aplicáveis aos dados existentes.
Para a estimação do LOEC e, portanto, do NOEC, será necessário comparar as médias dos frascos expostos utilizando técnicas de análise da variância (ANOVA). A média para cada concentração deve então ser comparada com a média observada nos controlos, utilizando um método apropriado de comparação múltipla ou de análise das tendências. Os testes de Dunnett ou de Williams (21)(22)(23)(24) poderão ser úteis para esse efeito. É necessário verificar se o pressuposto de homogeneidade da variância das análises ANOVA é cumprido. Essa verificação poderá ser realizada graficamente ou através de um teste formal (25). Para tal, podem utilizar-se os testes de Levene ou de Bartlett. O problema do não cumprimento do pressuposto de homogeneidade das variâncias pode por vezes ser corrigido através da transformação logarítmica dos dados. Caso a heterogeneidade das variâncias seja extrema e não possa ser corrigida por transformação, deverá ser considerada a possibilidade de utilização de métodos de análise das tendências como, por exemplo, o método degressivo de Jonkheere. Para mais orientações sobre a determinação do NOEC, consultar a referência (19).
Os mais recentes desenvolvimentos científicos conduziram à recomendação de que fosse abandonado o conceito de NOEC, que deverá ser substituído pela estimação dos valores de ECx por regressão. No caso do presente ensaio com Lemna, não foi definido nenhum valor ideal para o x. No entanto, a gama entre 10-20 % parece ser apropriada (dependendo da variável de resposta escolhida), devendo ser comunicados, de preferência, tanto os valores de EC10 como de EC20.
3. RELATÓRIOS
3.1. RELATÓRIO DO ENSAIO
O relatório do ensaio deve incluir a seguinte informação:
Substância de ensaio:
|
— |
natureza física e propriedades físico-químicas, incluindo o limite de hidrossolubilidade, |
|
— |
dados de identificação química (p. ex.: Número CAS), incluindo a respectiva pureza. |
Espécie de ensaio:
|
— |
nome científico, clone (se for conhecido) e fonte. |
Condições de ensaio:
|
— |
procedimento de ensaio utilizado (estático, semiestático ou contínuo), |
|
— |
data de início do ensaio e respectiva duração, |
|
— |
meio de ensaio, |
|
— |
descrição do método experimental: frascos e tampas utilizados no ensaio, volumes das soluções, número de colónias e de frondes em cada frasco de ensaio, no início do mesmo, |
|
— |
concentrações de ensaio (nominais ou medidas, conforme apropriado) e número de replicados por concentração, |
|
— |
métodos de preparação das soluções de reserva e de ensaio, incluindo a eventual utilização de solventes ou dispersantes, |
|
— |
temperatura durante o ensaio, |
|
— |
fonte luminosa, intensidade e homogeneidade da luz, |
|
— |
valores de pH dos meios de controlo e de ensaio, |
|
— |
concentrações da substância em estudo e respectivo método de análise, juntamente com os dados apropriados para a avaliação da qualidade do método (estudos de validação, desvios-padrão ou limites de confiança das análises), |
|
— |
métodos utilizados para a determinação do número de frondes ou de outras variáveis de medição, como o peso seco, o peso fresco ou a superfície das frondes, |
|
— |
todos os desvios em relação ao presente método de ensaio. |
Resultados:
|
— |
dados brutos: número de frondes e outras variáveis de medição em cada frasco de ensaio e de controlo, para cada momento de amostragem, e momento de realização da análise, |
|
— |
médias e desvios-padrão para cada variável de medição, |
|
— |
curvas de crescimento para cada concentração (recomenda-se a transformação logarítmica da variável de medição, ver o segundo parágrafo do ponto 2.2.1, |
|
— |
tempo de duplicação/taxa de crescimento dos controlos, com base no número de frondes, |
|
— |
variáveis de resposta calculadas para cada replicado exposto, com os respectivos valores médios, e coeficiente de variação dos replicados, |
|
— |
representação gráfica da relação concentração/efeito, |
|
— |
estimativas dos pontos finais de toxicidade para as variáveis de resposta, p. ex.: EC50, EC10, EC20, e os intervalos de confiança associados. Quando forem calculados, valores do LOEC e/ou do NOEC e métodos estatísticos utilizados para a respectiva determinação, |
|
— |
caso tenha sido utilizada uma análise ANOVA, limite de detecção do efeito (p. ex.: diferenças menos significativas), |
|
— |
ocorrência de estimulação do crescimento que tenha sido verificada em qualquer dos grupos expostos, |
|
— |
sinais visuais de fitotoxicidade, bem como quaisquer observações em relação às soluções de ensaio, |
|
— |
discussão dos resultados, incluindo qualquer influência sobre os resultado do ensaio que seja decorrente das alterações efectuadas em relação ao presente método de ensaio. |
4. BIBLIOGRAFIA
|
(1) |
OECD TG 221 (2006) Lemna spp. Growth Inhibition Test |
|
(2) |
Recurso a estudos com Lemna para substâncias coradas é descrito na secção 13.5.3 do EU Manual of Decisions, de Julho de 2006, disponível no endereço: http://ecb.jrc.ec.europa.eu/new-chemicals |
|
(3) |
Guidance on information requirements and chemical safety assessment — Chapter R.7b: Endpoint specific guidance; Table 7.8.3 Summary of difficult substance testing issues, disponível no endereço: http://guidance.echa.europa.eu/docs/guidance_document/information_requirements_en.htm?time=1234958685#A |
|
(4) |
ASTM International. (2003). Standard Guide for Conducting Static Toxicity Test With Lemna gibba G3. E 1415-91 (Reapproved 1998). pp. 733-742. In, Annual Book of ASTM Standards, Vol. 11.05 Biological Effects and Environmental Fate; Biotechnology; Pesticides, ASTM, West Conshohocken, PA |
|
(5) |
USEPA — United States Environmental Protection Agency. (1996). OPPTS 850.4400 Aquatic Plant Toxicity Test Using Lemna spp., «Public draft». EPA 712-C-96-156. 8 pp |
|
(6) |
AFNOR — Association Française de Normalisation. (1996). XP T 90-337: Détermination de l’inhibition de la croissance de Lemna minor. 10 pp |
|
(7) |
SSI — Swedish Standards Institute. (1995). Water quality — Determination of growth inhibition (7-d) Lemna minor, duckweed. SS 02 82 13. 15 pp. (em sueco) |
|
(8) |
Environment Canada (1999). Biological Test Method: Test for Measuring the Inhibition of Growth Using the Freshwater Macrophyte, Lemna minor. EPS 1/RM/37-120 pp |
|
(9) |
Environment Canada (1993). Proposed Guidelines for Registration of Chemical Pesticides: Non-Target Plant Testing and Evaluation. Canadian Wildlife Service, Technical Report Series N.o 145 |
|
(10) |
Sims I., Whitehouse P., and Lacey R. (1999). The OECD Lemna Growth Inhibition Test. Development and Ring-testing of draft OECD Test Guideline. R&D Technical Report EMA 003. WRc plc — Environment Agency |
|
(11) |
OCDE (2000). Guidance Document on Aquatic Toxicity Testing of Difficult Substances and mixtures. OCDE Environmental Health and Safety Publications, Series on Testing and Assessment N.o 23 |
|
(12) |
ISO DIS 20079. Water Quality — Determination of the Toxic Effect of Water Constituents and Waste Water to Duckweed (Lemna minor) — Duckweed Growth Inhibition Test |
|
(13) |
Walbridge C. T. (1977). A flow-through testing procedure with duckweed (Lemna minor L.). Environmental Research Laboratory — Duluth, Minnesota 55804. US EPA Report No. EPA-600/3-77 108. Setembro de 1977 |
|
(14) |
Lockhart W. L., Billeck B. N. and Baron C. L. (1989). Bioassays with a floating plant (Lemna minor) for effects of sprayed and dissolved glyphosate. Hydrobiologia, 118/119, 353-359 |
|
(15) |
Huebert, D.B. and Shay J.M. (1993). Considerations in the assessment of toxicity using duckweeds. Environmental Toxicology and Chemistry, 12, 481-483 |
|
(16) |
Christensen, E.R., Nyholm, N. (1984): Ecotoxicological Assays with Algae: Weibull Dose-Response Curves. Env. Sci. Technol. 19, 713-718 |
|
(17) |
Nyholm, N. Sørensen, P.S., Kusk, K.O. and Christensen, E.R. (1992): Statistical treatment of data from microbial toxicity tests. Environ. Toxicol. Chem. 11, 157-167 |
|
(18) |
Bruce R.D. and Versteeg D.J. (1992). A statistical procedure for modelling continuous toxicity data. Environmental Toxicology and Chemistry, 11, 1485-1494 |
|
(19) |
OCDE (2004). Guidance Document on Statistical Analysis of Ecotoxicity Data |
|
(20) |
Norberg-King T.J. (1988). An interpolation estimate for chronic toxicity: The ICp approach. National Effluent Toxicity Assessment Center Technical Report 05-88. USEPA, Duluth, MN |
|
(21) |
Dunnett, C.W. (1955). A multiple comparisons procedure for comparing several treatments with a control. J. Amer. Statist. Assoc., 50, 1096-1121 |
|
(22) |
Dunnett, C.W. (1964). New tables for multiple comparisons with a control. Biometrics, 20, 482-491 |
|
(23) |
Williams, D.A. (1971). A test for differences between treatment means when several dose levels are compared with a zero dose control. Biometrics, 27: 103-117 |
|
(24) |
Williams, D.A. (1972). The comparison of several dose levels with a zero dose control. Biometrics, 28: 510-531 |
|
(25) |
Brain P. and Cousens R. (1989). An equation to describe dose-responses where there is stimulation of growth at low doses. Weed Research, 29, 93-96 |
Apêndice 1
Descrição de Lemna spp.
A planta aquática que tem o nome comum de lentilha-de-água, Lemna spp., pertence à Família Lemnaceae, composta por quatro Géneros com espécies distribuídas por todo o mundo. As suas diferentes formas e taxonomia já se encontram descritas de forma exaustiva (1)(2). A Lemna gibba e a Lemna minor são espécies representativas das zonas temperadas, muito utilizadas em ensaios de toxicidade. Ambas as espécies apresentam um caule discóide (fronde), flutuante ou submerso, e uma raiz muito fina que parte do centro da face inferior de cada fronde. As Lemna spp. raramente produzem flores, sendo a reprodução garantida pela produção vegetativa de novas frondes (3). Em comparação com as plantas mais velhas, as plantas jovens tendem a ser mais claras, com raízes mais curtas e são constituídas por duas ou três frondes de tamanhos diferentes. O pequeno tamanho, a estrutura simples, a reprodução assexuada e o tempo de duplicação curto das Lemna fazem com que estas plantas sejam muito utilizadas em ensaios de laboratório (4)(5).
Devido a variações de sensibilidade, provavelmente interespecíficas, só são válidas as comparações de sensibilidade para uma mesma espécie.
Exemplos de espécies de Lemna que já foram utilizadas em ensaios laboratoriais: referências das espécies
Lemna aequinoctialis: Eklund, B. (1996). The use of the red alga Ceramium strictum and the duckweed Lemna aequinoctialis in aquatic ecotoxicological bioassays. Licentiate in Philosophy Thesis 1996:2. Dep. of Systems Ecology, Stockholm University.
Lemna major: Clark, N. A. (1925). The rate of reproduction of Lemna major as a function of intensity and duration of light. J. phys. Chem., 29: 935-941.
Lemna minor: United States Environmental Protection Agency (USEPA). (1996). OPPTS 850.4400 Aquatic Plant Toxicity Test Using Lemna spp., «Public draft». EPA 712-C-96-156. 8 pp.
Association Française de Normalisation (AFNOR). (1996). XP T 90-337: Détermination de l’inhibition de la croissance de Lemna minor. 10 pp.
Swedish Standards Institute (SIS). (1995). Water quality — Determination of growth inhibition (7-d) Lemna minor, duckweed. SS 02 82 13. 15 pp. (em sueco).
Lemna gibba: ASTM International. (2003). Standard Guide for Conducting Static Toxicity Test With Lemna gibba G3. E 1415-91 (Reapproved 1998). pp. 733-742.
United States Environmental Protection Agency (USEPA). (1996). OPPTS 850.4400 Aquatic Plant Toxicity Test Using Lemna spp., «Public draft». EPA 712-C-96-156. 8 pp.
Lemna paucicostata: Nasu, Y., Kugimoto, M. (1981). Lemna (duckweed) as an indicator of water pollution. I. The sensitivity of Lemna paucicostata to heavy metals. Arch. Environ. Contam. Toxicol., 10:1959-1969.
Lemna perpusilla: Clark, J. R. et al. (1981). Accumulation and depuration of metals by duckweed (Lemna perpusilla). Ecotoxicol. Environ. Saf., 5:87-96.
Lemna trisulca: Huebert, D. B., Shay, J. M. (1993). Considerations in the assessment of toxicity using duckweeds. Environ. Toxicol. and Chem., 12:481-483.
Lemna valdiviana: Hutchinson, T.C., Czyrska, H. (1975). Heavy metal toxicity and synergism to floating aquatic weeds. Verh.-Int. Ver. Limnol., 19:2102-2111.
Fontes para obtenção de plantas de Lemna
|
University of Toronto Culture Collection of Algae and Cyanobacteria |
|
Department of Botany, University of Toronto |
|
Toronto, Ontario, CANADÁ, M5S 3 B2 |
|
Tel: +1-416-978-3641 |
|
Fax:+1-416-978-5878 |
|
e-mail: jacreman@botany.utoronto.ca |
|
http://www.botany.utoronto.ca/utcc |
|
North Carolina State University |
|
Forestry Dept |
|
Duckweed Culture Collection |
|
Campus Box 8002 |
|
Raleigh, NC 27695-8002 |
|
EUA |
|
phone 001 (919) 515-7572 |
|
astomp@unity.ncsu.edu |
|
Institute of Applied Environmental Research (ITM) Stockholm University |
|
SE-106 91 STOCKHOLM |
|
SUÉCIA |
|
Tel: +46 8 674 7240 |
|
Fax +46 8 674 7636 |
|
Federal Environmental Agency (UBA) |
|
FG III 3.4 |
|
Schichauweg 58 |
|
12307 Berlin |
|
ALEMANHA |
|
e-mail: lemna@uba.de |
|
http://www.umweltbundesamt.de/contact.htm |
Bibliografia
|
(1) |
Hillman, W.S. (1961). The Lemnaceae or duckweeds: A review of the descriptive and experimental literature. The Botanical Review, 27:221-287 |
|
(2) |
Landolt, E. (1986). Biosystematic investigations in the family of duckweed (Lemnaceae). Vol. 2. Geobotanischen Inst. ETH, Stiftung Rubel, Zürich, Switzerland |
|
(3) |
Björndahl, G. (1982). Growth performance, nutrient uptake and human utilization of duckweeds (Lemnaceae family). ISBN 82-991150-0-0. The Agricultural Research Council of Norway, University of Oslo |
|
(4) |
Wang, W. (1986). Toxicity tests of aquatic pollutants by using common duckweed. Environmental Pollution, Ser B, 11:1-14 |
|
(5) |
Wang, W. (1990). Literature review on duckweed toxicity testing. Environmental Research, 52:7-22 |
Apêndice 2
Conservação das culturas de reserva
As culturas de reserva podem ser conservadas a baixa temperatura (4-10 °C) por longos períodos, sem que seja necessário proceder à sua transferência periódica. O meio de crescimento da Lemna pode ser o mesmo que é utilizado para os ensaios, mas as culturas de reserva também podem ser conservadas noutros meios nutrientes enriquecidos.
Periodicamente, um certo número de plantas jovens, verdes claras, são removidas e novamente cultivadas, através de uma técnica asséptica, em frascos com meio fresco. Nas condições de baixa temperatura aqui sugeridas, a transferência das culturas pode ser realizada a intervalos que poderão ir até aos três meses.
Devem utilizar-se frascos de cultura quimicamente limpos (lavados com ácido) e estéreis e técnicas de manuseamento em condições de assepsia. Caso se verifique uma contaminação da cultura de reserve, por exemplo por outras algas ou por um fungo, será necessário adoptar medidas para eliminar os organismos contaminantes. No caso das algas e da maior parte dos restantes organismos contaminantes, isso será possível por esterilização superficial. Retira-se uma amostra da planta contaminada e cortam-se as raízes. O material é depois vigorosamente agitado em água limpa, sendo depois mergulhado numa solução a 0,5 % (v/v) de hipoclorito de sódio durante um período de 30 segundos a 5 minutos. O material vegetal é depois lavado com água esterilizada e transferido para uma série de frascos de cultura com meio de crescimento fresco. Muitas frondes acabarão por morrer como resultado deste tratamento, principalmente quando os períodos de exposição utilizados forem mais longos, mas algumas delas sobreviverão e ficarão normalmente isentas de qualquer contaminação. As frondes sobreviventes podem ser utilizadas para reinocular novas culturas.
Apêndice 3
Meios
Existem diversos meios de crescimento recomendados para L. minor e L. gibba. Para a L. minor, o meio recomendado é o meio Swedish Standard (SIS) modificado, enquanto que para L. gibba o meio recomendado é o meio 20X AAP. A composição de ambos os meios é apresentada a seguir. Para a preparação destes meios, devem utilizar-se reagentes e produtos químicos de qualidade analítica e água desionizada.
Meio de crescimento Swedish Standard (SIS) para Lemna
|
— |
As soluções de reserva I-V são esterilizadas em autoclave (120 °C, 15 minutos) ou por filtração através de membrana (aproximadamente 0,2 μm de diâmetro de poro), |
|
— |
As soluções VI e (opcionalmente) VII são esterilizadas exclusivamente por filtração através de membrana; estas soluções não devem ser esterilizadas em autoclave, |
|
— |
As soluções de reserva esterilizadas devem ser armazenadas em local fresco e ao abrigo da luz. As soluções I-V devem ser descartadas ao fim de seis meses, enquanto que a solução VI (e, opcionalmente, também a VII) têm um período de conservação de um mês,
|
||||||||||||||||||||||||||||||||||||||||||||||||||
|
— |
Para preparar um litro de meio SIS, adicionar os seguintes elementos a 900 ml de água desionizada:
Nota: A solução de reserva VII (tampão MOPS) poderá ser necessária para o estudo de determinadas substâncias (ver o último parágrafo do ponto 1.4). |
|
— |
Ajustar o pH a 6,5 ± 0,2 com HCl ou NaOH 0,1 N ou 1,0 N, completando o volume até 1 l com água desionizada. |
Meio de crescimento 20X–AAP
As soluções de reserva são preparadas com água esterilizada ou desionizada.
As soluções de reserva esterilizadas devem ser armazenadas em local fresco e ao abrigo da luz. Nessas condições, terão um tempo de conservação mínimo de 6-8 semanas.
Para o meio 20X–AAP, prepara-se cinco soluções de reserva de nutrientes (A1, A2, A3, B e C), utilizando produtos químicos de qualidade analítica. Para produzir o meio de crescimento, adicionar 20 ml de cada solução de reserva de nutrientes a cerca de 850 ml de água desionizada. Ajustar o pH a 7,5 ± 0,1 com HCl ou NaOH 0,1 N ou 1,0 N, completando o volume até 1 l com água desionizada. Filtrar o meio assim obtido por uma membrana com diâmetro de poro de (aproximadamente) 0,2 μm para um recipiente esterilizado.
O meio de crescimento para os ensaios deve ser preparado com 1-2 dias de antecedência, de modo a permitir a estabilização do pH. O pH do meio de crescimento deve ser verificado antes da sua utilização e, se necessário, reajustado com HCl ou NaOH 0,1 N ou 1,0 N, como se descreve acima.
|
N.o da solução de reserva |
Substância |
Concentração na solução de reserva (g·l–1) (1) |
Concentração no meio preparado (g·l–1) (1) |
Meio preparado |
|
|
Elemento |
Concentração (mg·l–1) (1) |
||||
|
A1 |
NaNO3 MgCl2·6H2O CaCl2.2H2O |
26 12 4,4 |
510 240 90 |
Na; N Mg Ca |
190; 84 58,08 24,04 |
|
A2 |
MgSO4.7H2O |
15 |
290 |
S |
38,22 |
|
A3 |
K2HPO4·3H2O |
1,4 |
30 |
K; P |
9,4;3,7 |
|
B |
H3BO3 MnCl2.4H2O FeCl3.6H2O Na2EDTA·2H2O ZnCl2 CoCl2·6H2O Na2MoO4.2H2O CuCl2·2H2O |
0,19 0,42 0,16 0,30 3,3 mg·l–1 1,4 mg·l–1 7,3 mg·l–1 0,012 mg·l–1 |
3,7 8,3 3,2 6,0 66 μg·l–1 29 μg·l–1 145 μg·l–1 0,24 μg·l–1 |
B Mn Fe — Zn Co Mo Cu |
0,65 2,3 0,66 — 31 μg·l–1 7,1 μg·l–1 58 μg·l–1 0,080 μg·l–1 |
|
C |
NaHCO3 |
15 |
300 |
Na; C |
220; 43 |
Nota: A concentração final teoricamente ideal de bicarbonato (que evita qualquer variação significativa do pH) é de 15 mg/l e não de 300 mg/l. No entanto, a utilização habitual do meio 20X–AAP, nomeadamente no ensaio de anel para o presente método, baseia-se numa concentração de 300 mg/l [I. Sims, P. Whitehouse e R. Lacey. (1999)]. The OECD Lemna Growth Inhibition Test. Development and Ring-testing of draft OECD Test Guideline. R&D Technical Report EMA 003. WRc plc — Environment Agency.
Meio de STEINBERG (derivado do método ISO 20079)
Concentrações e soluções de reserva
|
— |
O meio de Steinberg modificado é utilizado no método ISSO 20079 apenas para a Lemna minor (já que o método só utiliza essa espécie) mas o ensaio mostrou bons resultados também com Lemna gibba. |
|
— |
Para a preparação do meio, deve utilizar-se reagentes e produtos químicos de qualidade analítica e água desionizada. |
|
— |
Preparar o meio nutriente a partir das soluções de reserva ou do meio 10 vezes mais concentrado, que é a máxima concentração que se pode obter sem que ocorra precipitação. |
Quadro 1
Meio de Steinberg com pH estabilizado (modificado segundo Altenburger)
|
Substância |
Meio nutriente |
||
|
Macroelementos |
peso molecular |
mg/l |
mmol/l |
|
KNO3 |
101,12 |
350,00 |
3,46 |
|
Ca(NO3)2.4H2O |
236,15 |
295,00 |
1,25 |
|
KH2PO4 |
136,09 |
90,00 |
0,66 |
|
K2HPO4 |
174,18 |
12,60 |
0,072 |
|
MgSO4.7H2O |
246,37 |
100,00 |
0,41 |
|
Microelementos |
peso molecular |
µg/l |
µmol/l |
|
H3BO3 |
61,83 |
120,00 |
1,94 |
|
ZnSO4.7H2O |
287,43 |
180,00 |
0,63 |
|
Na2MoO4.2H2O |
241,92 |
44,00 |
0,18 |
|
MnCl2.4H2O |
197,84 |
180,00 |
0,91 |
|
FeCl3.6H2O |
270,21 |
760,00 |
2,81 |
|
EDTA disódico bi-hidratado |
372,24 |
1 500,00 |
4,03 |
Quadro 2
Soluções de reserva (Macroelementos)
|
g/l |
||
|
Solução de reserva 1: |
|||
|
KNO3 |
17,50 |
||
|
KH2PO4 |
4,5 |
||
|
K2HPO4 |
0,63 |
||
|
Solução de reserva 2: |
|||
|
MgSO4.7H2O |
5,00 |
||
|
Solução de reserva 3: |
|||
|
Ca(NO3)2.4H2O |
14,75 |
||
Quadro 3
Soluções de reserva (Microelementos)
|
mg/l |
||
|
Solução de reserva 4: |
|||
|
H3BO3 |
120,0 |
||
|
Solução de reserva 5: |
|||
|
ZnSO4.7H2O |
180,0 |
||
|
Solução de reserva 6: |
|||
|
Na2MoO4.2H2O |
44,0 |
||
|
Solução de reserva 7: |
|||
|
MnCl2.4H2O |
180,0 |
||
|
Solução de reserva 8: |
|||
|
FeCl3.6H2O |
760,00 |
||
|
EDTA dissódico bi-hidratado |
1 500,00 |
||
|
— |
As soluções de reserva 2 e 3 e, em separado, 4 a 7, podem ser misturadas (tomando em conta as concentrações pretendidas). |
|
— |
Para aumentar o período de conservação, tratar as soluções em autoclave a 121 °C durante 20 min ou, em alternativa, esterilizar por filtração através de membrana (0,2 µm). Para a solução de reserva 8, é fortemente recomendada a esterilização por filtração (0,2 µm). |
Preparação da concentração final do meio de STEINBERG (modificado)
|
— |
Juntar 20 ml das soluções de reserva 1, 2 e 3 (ver o quadro 2) a cerca de 900 ml de água desionizada, para evitar a precipitação. |
|
— |
Juntar 1,0 ml das soluções de reserva 4, 5, 6, 7 e 8 (ver o quadro 3). |
|
— |
O pH deverá ser de 5,5 ± 0,2 (ajustar por adição de um volume tão pequeno quanto possível de solução de NaOH ou de HCl). |
|
— |
Completar com água até 1 000 ml. |
|
— |
Se as soluções de reserva tiverem sido esterilizadas e se se utilizar uma água apropriada, não é necessário proceder a mais nenhuma esterilização. Se o meio final for esterilizado, a solução de reserva 8 deve ser acrescentada depois do tratamento em autoclave (a 121 °C durante 20 min). |
Preparação do meio de STEINBERG (modificado) 10 vezes concentrado, para armazenamento temporário
|
— |
Juntar 20 ml das soluções de reserva 1, 2 e 3 (ver o quadro 2) a cerca de 30 ml de água para evitar a precipitação. |
|
— |
Juntar 1,0 ml das soluções de reserva 4, 5, 6, 7 e 8 (ver o quadro 3). Completar com água até 100 ml. |
|
— |
Se as soluções de reserva tiverem sido esterilizadas e se utilizar uma água apropriada, não é necessário proceder a mais nenhuma esterilização. Se o meio final for esterilizado, a solução de reserva 8 deve ser acrescentada depois do tratamento em autoclave (a 121 °C durante 20 min). |
|
— |
O pH do meio (concentração final) deverá ser de 5,5 ± 0,2. |
(1) Excepto nos casos assinalados