ANEXO
O anexo do Regulamento (CE) n.o 440/2008 é alterado do seguinte modo:
São inseridos os seguintes capítulos B.53, B.54, B.55, B.56, B.57 e B.58:
«B.53. ESTUDO DE NEUROTOXICIDADE DURANTE O DESENVOLVIMENTO
INTRODUÇÃO
1. Este método é equivalente ao Test Guideline TG 426 (2007) da OCDE. Em junho de 1995, um grupo de trabalho da OCDE que se ocupa dos efeitos tóxicos durante a reprodução e o desenvolvimento debateu em Copenhaga a necessidade de atualizar os Test Guidelines relativos à toxicidade durante a reprodução e o desenvolvimento existentes à data, bem como de elaborar novas orientações para parâmetros ainda não abrangidos (1). Esse grupo de trabalho recomendou que se elaborasse um Test Guideline relativo à neurotoxicidade durante o desenvolvimento, com base nas orientações da Agência de Proteção do Ambiente (EPA) dos E.U.A., as quais foram entretanto revistas (2). Em junho de 1996, realizou-se em Copenhaga uma segunda reunião de consultas com o objetivo de fornecer ao Secretariado orientações sobre as linhas gerais de um novo Test Guideline relativo à neurotoxicidade durante o desenvolvimento, designadamente acerca dos principais elementos do mesmo, como a escolha da espécie animal, o período de administração, o período de ensaio, os parâmetros a avaliar e os critérios de avaliação dos resultados. Em 1998, foram publicadas orientações da referida agência para a avaliação do risco de neurotoxicidade (3). Em outubro de 2000, realizaram-se paralelamente uma reunião de consulta de peritos da OCDE e jornadas do Instituto de Ciências do Risco do International Life Sciences Institute (ILSI). Em 2005, realizou-se em Tóquio uma reunião de consulta de peritos. Estas reuniões visaram debater as questões científicas e técnicas relativas ao Test Guideline em causa, tendo as recomendações delas emanadas (4)(5)(6)(7) sido tidas em conta na elaboração do presente método. Os documentos de orientações da OCDE n.o 43 (ensaio e avaliação de efeitos tóxicos na reprodução) (8) e n.o 20 (ensaios de neurotoxicidade) (9) contêm informações adicionais sobre a execução do presente método, a interpretação dos resultados do mesmo e a terminologia nele utilizada.
CONSIDERAÇÕES INICIAIS
2. É sabido que diversos produtos químicos têm efeitos neurotóxicos durante o desenvolvimento, na espécie humana e noutras espécies (10)(11)(12)(13). A fim de se determinarem e avaliarem as características tóxicas de um produto químico, pode ser necessário determinar a neurotoxicidade potencial do mesmo durante o desenvolvimento. Os estudos de neurotoxicidade durante o desenvolvimento visam gerar dados, nomeadamente de caracterização da resposta à dosagem, relativos aos efeitos funcionais e morfológicos potenciais no sistema nervoso em desenvolvimento da progenitura, após exposição intrauterina ou nos primeiros estádios de vida após o nascimento.
3. Os estudos de neurotoxicidade durante o desenvolvimento podem realizar-se separadamente, ser integrados em estudos de toxicidade durante a reprodução e/ou de neurotoxicidade em indivíduos adultos — por exemplo, os métodos B.34 (14), B.35 (15), B.43 (16) — ou ser complementares de estudos de toxicidade durante o desenvolvimento pré-natal (por exemplo, o método B.31 — (17). Se o estudo de neurotoxicidade durante o desenvolvimento for incorporado noutro estudo ou o complementar. é imperativo manter a integridade de ambos os tipos de estudos. Os ensaios devem respeitar a legislação ou as orientações estatais ou institucionais aplicáveis no que respeita à utilização de animais de laboratório em investigação — por exemplo (18).
4. Antes de efetuarem o estudo, os laboratórios devem ponderar todas as informações disponíveis sobre o produto químico em causa, nomeadamente a identidade e a estrutura do mesmo, as propriedades físico-químicas do produto químico, os resultados de outros ensaios de toxicidade in vitro ou in vivo deste, dados toxicológicos disponíveis sobre produtos químicos estruturalmente afins e ainda a utilização ou utilizações previstas do produto químico em questão. Estas informações ajudarão a escolher uma dose inicial adequada e são necessárias para comprovar a pertinência do ensaio para a proteção da saúde humana.
PRINCÍPIO DO MÉTODO
5. Administra-se o produto químico em estudo a animais durante os períodos de gestação e de lactação. Efetuam-se ensaios nas progenitoras para avaliar os efeitos nas fêmeas grávidas e em lactação, podendo igualmente obter-se informações comparativas entre aquelas e a sua progenitura. Procede-se à avaliação de neurotoxicidade em progenitura selecionada aleatoriamente das ninhadas. A avaliação consiste em exames destinados a detetar anomalias neurológicas macroscópicas e comportamentais, nomeadamente ao nível do desenvolvimento físico, da ontogenia comportamental, da atividade motora, das funções motoras e sensoriais, da aprendizagem e da memória, bem como na determinação da evolução ponderal do cérebro e na avaliação de neuropatologias durante o desenvolvimento pós-natal e na idade adulta.
6. Se a aplicação deste método constituir um estudo separado, podem submeter-se animais disponíveis em cada grupo a protocolos neurocomportamentais, neuropatológicos, neuroquímicos ou eletrofisiológicos específicos suscetíveis de complementar os dados fornecidos pelos exames recomendados no presente método (16)(19)(20)(21). Esses protocolos podem revelar-se particularmente úteis se observações empíricas, efeitos previstos ou o mecanismo ou modo de ação apontarem para um determinado tipo de neurotoxicidade. Os protocolos em questão podem ser aplicados às progenitoras ou à progenitura. Podem igualmente executar-se protocolos ex vivo ou in vitro complementares, desde que estes não alterem a integridade dos protocolos in vivo.
PREPARATIVOS PARA O ENSAIO
Escolha da espécie animal
7. A espécie preferida é o rato, mas podem utilizar-se outras espécies adequadas. Importa, porém, salientar que os dias de gestação e de desenvolvimento pós-natal especificados neste método correspondem às estirpes de ratos mais utilizadas, devendo escolher-se durações comparáveis se forem utilizadas estirpes inabituais de ratos ou espécies diferentes. Se forem utilizadas outras espécies, será necessário justificá-lo com base em dados toxicológicos, farmacocinéticos e/ou outros. A justificação da opção tomada deve apoiar-se em avaliações neurocomportamentais e neuropatológicas pós-natais já disponíveis, específicas da espécie em causa. Se um ensaio anterior tiver gerado resultados preocupantes, deve ser ponderada a utilização da espécie ou estirpe que os gerou. Dado que as diversas estirpes de ratos têm desempenhos diferentes, importa comprovar a adequação da fecundidade e da reatividade da estirpe selecionada. Se forem utilizadas outras espécies, é necessário documentar a fiabilidade e sensibilidade da deteção da neurotoxicidade durante o desenvolvimento das espécies em causa.
Condições de alojamento e de alimentação
8. A temperatura do biotério deve ser de 22 °C ± 3 °C. A humidade relativa deve estar compreendida entre 50 % e 60 %, embora sejam aceitáveis valores compreendidos entre 30 %, no mínimo, e um valor máximo que, preferencialmente, não deve exceder 70 %, salvo durante os períodos de limpeza do biotério. A iluminação deve ser artificial, com uma sequência de 12 horas de luz seguidas de 12 horas de escuridão. Também se pode inverter o ciclo de iluminação antes do acasalamento e durante todo o estudo, a fim de se proceder à avaliação dos parâmetros funcionais e comportamentais durante o período de obscuridade (sob luz vermelha), isto é, durante o período de atividade normal dos animais (22). As alterações do ciclo iluminação-obscuridade devem ter um período de aclimatação adequado, que permita aos animais adaptarem-se ao novo ciclo. Na alimentação, podem utilizar-se dietas convencionais de laboratório e deve ser fornecida água de beber sem restrições. Deve indicar-se no relatório o tipo de alimentação e de água e analisar-se a presença de contaminantes em ambos.
9. Os animais podem ser alojados individualmente ou em pequenos grupos do mesmo sexo. O acasalamento deve ocorrer em gaiolas adequadas para o efeito. Uma vez comprovado o acasalamento ou, o mais tardar, no 15.o dia de gestação, as fêmeas cobertas devem ser alojadas separadamente em gaiolas de maternidade. As gaiolas devem ser dispostas de forma a minimizar possíveis efeitos derivados do seu posicionamento. As fêmeas cobertas que se aproximem do termo da gestação devem dispor de materiais de nidificação definidos apropriados. É sabido que manipulações inadequadas ou agitação durante a gravidez podem ter efeitos indesejados, nomeadamente abortos espontâneos ou alterações do desenvolvimento fetal ou pós-natal. A fim de evitar mortalidade fetal devida a fatores não relacionados com a exposição, os animais devem ser cuidadosamente manipulados durante a gestação e deve evitar-se qualquer agitação provocada por fatores exteriores, como ruído excessivo
Preparação dos animais
10. Os animais utilizados devem ser saudáveis, ter sido aclimatados às condições laboratoriais e não devem ter participado em experiências anteriores, salvo se o estudo estiver integrado noutro (ver o ponto 3). É necessário caracterizar a espécie, a estirpe, a proveniência, o sexo, o peso e a idade dos animais utilizados no estudo. Deve atribuir-se a cada animal um número de identificação único, com o qual o animal seja marcado. Os animais de todos os grupos estudados devem, tanto quanto possível, ter peso e idade uniformes e pertencer à gama normal da espécie e estirpe em estudo. As fêmeas utilizadas para todos os níveis de dosagem devem ser jovens adultas nulíparas. Não devem acasalar-se entre si animais da mesma ninhada, sendo necessário tomar precauções para o evitar. Considera-se dia 0 de gravidez aquele em que se observa um rolhão vaginal e/ou esperma. No caso de se comprarem fêmeas grávidas a um fornecedor, deve proporcionar-se-lhes um período de aclimatação adequado (2 a 3 dias, por exemplo). As fêmeas cobertas devem ser distribuídas aleatória e, o mais possível, uniformemente pelos grupos de controlo e pelos grupos expostos (recomenda-se, por exemplo, a aplicação de um protocolo de distribuição aleatória estratificada, nomeadamente baseado no peso corporal, para uniformizar a distribuição por todos os grupos). Devem repartir-se as fêmeas cobertas pelo mesmo macho uniformemente por todos os grupos.
PROCEDIMENTO DE ENSAIO
Número e sexo dos animais
11. Cada grupo exposto e cada grupo de controlo deve compreender um número de fêmeas grávidas a expor ao produto químico em estudo suficiente para que a progenitura por elas gerada seja em número adequado para a avaliação de toxicidade. Para cada nível de dosagem recomendam-se 20 ninhadas. Admite-se o recurso a modelos de grupos escalonados ou de administração em replicado, desde que se atinja o número total previsto de ninhadas por grupo e se utilizem modelos estatísticos adequados para tratar os replicados.
12. O mais tardar ao quarto dia após o nascimento, deve ajustar-se o tamanho de cada ninhada eliminando aleatoriamente as crias em excesso, de modo que todas as ninhadas tenham o mesmo número de animais (23). As ninhadas não devem ter mais crias do que a média da estirpe de roedor utilizada (8-12). Tanto quanto possível, as ninhadas devem ter o mesmo número de machos e de fêmeas. Não é admissível uma eliminação seletiva de crias, por exemplo, baseada no peso corporal. Após a normalização das ninhadas por eliminação de crias e antes de passar ao ensaio dos parâmetros funcionais, é necessário identificar individualmente as crias destinadas aos ensaios a decorrer antes ou depois do desmame, recorrendo a um método não-agressivo de identificação que lhes seja aplicável (ver, por exemplo, a referência 24).
Afetação dos animais aos ensaios funcionais e comportamentais, às determinações do peso cerebral e às avaliações neuropatológicas
13. O presente método admite várias abordagens no que respeita à afetação dos animais expostos in utero e por via do aleitamento aos ensaios funcionais e comportamentais, à determinação da maturidade sexual, à pesagem do cérebro e às avaliações neuropatológicas (25). Desde que a integridade dos ensaios iniciais exigidos não fique comprometida, é possível acrescentar os outros ensaios — da função neurocomportamental (por exemplo, do comportamento social), neuroquímicos ou neuropatológicos — que em cada caso se justifiquem.
14. Selecionam-se em cada grupo de dosagem as crias a destinar às avaliações de parâmetros ao quarto dia após o nascimento ou posteriormente. Tanto quanto possível, a seleção das crias deve ser de molde a que participe em cada ensaio o mesmo número de machos e de fêmeas de cada ninhada integrada em cada grupo de dosagem. No caso dos ensaios da atividade motora, deve utilizar-se o mesmo par macho e fêmea em todas as idades anteriores ao desmame (ver o ponto 35). Em todos os outros ensaios, podem afetar-se a cada ensaio comportamental o mesmo par de animais ou pares diferentes. Para evitar a confusão dos efeitos da idade com treino adquirido nas medições já efetuadas, pode ser necessário utilizar crias diferentes nos ensaios de comparação da função cognitiva entre animais recentemente desmamados e animais adultos (26)(27). Na ocasião do desmame (ao 21.o dia após o nascimento), as crias não selecionadas para os ensaios podem ser eutanasiadas. As alterações de afetações de crias devem ser mencionadas no relatório. A unidade de medida estatística deve ser a ninhada (ou a progenitora) e não a cria.
15. Há diferentes maneiras de afetar crias aos exames anteriores ou posteriores ao desmame, aos testes cognitivos, aos exames patológicos, etc. (ver um modelo geral na figura 1 e exemplos no apêndice 1). É o seguinte o número mínimo de animais recomendado em cada grupo de dosagem para os exames anteriores ou posteriores ao desmame:
|
Exames clínicos e peso corporal |
Todos os animais |
|
Exames clínicos aprofundados |
20 de cada sexo (1 de cada sexo por ninhada) |
|
Pesagem cerebral (após fixação) 11 a 22 dias após o nascimento |
10 de cada sexo (1 de cada ninhada) |
|
Pesagem cerebral (sem fixação) aproximadamente 70 dias após o nascimento |
10 de cada sexo (1 de cada ninhada) |
|
Neuropatologia (fixação por perfusão ou imersão) 11 a 22 dias após o nascimento |
10 de cada sexo (1 de cada ninhada) |
|
Neuropatologia (fixação por perfusão) aproximadamente 70 dias após o nascimento |
10 de cada sexo (1 de cada ninhada) |
|
Maturidade sexual |
20 de cada sexo (1 de cada sexo por ninhada) |
|
Outros indicadores do desenvolvimento (facultativo) |
Todos os animais |
|
Ontogenia comportamental |
20 de cada sexo (1 de cada sexo por ninhada) |
|
Atividade motora |
20 de cada sexo (1 de cada sexo por ninhada) |
|
Funções motoras e sensoriais |
20 de cada sexo (1 de cada sexo por ninhada) |
|
Aprendizagem e memória |
10 de cada sexo (1) (1 de cada ninhada) |
Dosagem
16. Devem utilizar-se pelo menos três níveis de dosagem e um grupo de controlo em paralelo. Os níveis de dosagem devem ser espaçados de forma a produzir uma gradação de efeitos tóxicos. Salvo se as doses estiverem limitadas pela natureza físico-química ou pelas propriedades biológicas do produto químico, deve selecionar-se para dose mais elevada um nível que induza alguma toxicidade nas progenitoras (por exemplo, sinais clínicos, diminuição do ganho de peso corporal — mas não mais de 10 % — e/ou indícios de toxicidade limitada pela dose num órgão-alvo). Com algumas exceções, a dose mais elevada não deve exceder 1 000 mg/kg de peso corporal/dia. Por exemplo, a exposição humana previsível pode aconselhar uma dose mais elevada. Em alternativa, podem efetuar-se estudos-piloto ou estudos exploratórios preliminares para determinar a dosagem mais elevada que deve ser utilizada para produzir um determinado nível mínimo de toxicidade nas progenitoras. Se tiver sido demonstrado, num estudo normal de toxicidade durante o desenvolvimento ou num estudo-piloto, que o produto químico em estudo tem efeitos tóxicos durante o desenvolvimento, a dose mais elevada deve ser a dose máxima que não induz na progenitura níveis excessivos de toxicidade, nem malformações ou mortalidade intrauterina ou neonatal, passíveis de impedir uma avaliação significativa da neurotoxicidade. A dose mais baixa não deve gerar nenhum indício de toxicidade, incluindo neurotoxicidade, nas progenitoras nem durante o desenvolvimento. Devem selecionar-se uma sequência decrescente de doses, que permita correlacionar as respostas observadas com as doses administradas e evidenciar um nível sem observação de efeitos adversos (NOAEL), ou doses próximas do limite de deteção, que permitam determinar uma dose de referência. O intervalo ótimo entre doses consecutivas é frequentemente definido por um fator de 2 a 4. A inclusão de um quarto grupo de dosagem é muitas vezes preferível ao uso de intervalos muito grandes (por exemplo, fatores superiores a 10) entre as dosagens.
17. Na escolha das doses devem ter-se em conta os dados disponíveis sobre a toxicidade do produto químico em estudo ou de matérias afins, bem como informações adicionais sobre o metabolismo e a toxicocinética dos mesmos. Estas informações também poderão servir para demonstrar a adequação do programa de dosagem. Deve ponderar-se a possibilidade de administração direta às crias, com base nas informações relativas à exposição e nos dados farmacocinéticos disponíveis (28)(29). Antes de efetuar estudos por administração direta das doses, é necessário pesar cuidadosamente os prós e os contras (30).
18. O grupo de controlo em paralelo deve ser um grupo ficticiamente exposto ao produto químico em estudo ou, se for usado um veículo para administrar o produto químico, um grupo de controlo do veículo. Normalmente, deve administrar-se a todos os animais o mesmo volume do produto químico em estudo ou do veículo, proporcionalmente ao peso corporal. Se for usado um veículo ou outro aditivo para facilitar a administração das doses, devem ter-se em conta as seguintes características do veículo ou aditivo: efeitos na absorção, na distribuição, no metabolismo ou na retenção do produto químico em estudo, efeitos nas propriedades químicas do produto químico em estudo suscetíveis de alterarem as características tóxicas do mesmo e efeitos no consumo de alimentos, na ingestão de água ou no estado nutricional dos animais. O veículo não deve ter efeitos que possam interferir na interpretação do estudo, ter toxicidade neurocomportamental ou ter efeitos na reprodução ou no desenvolvimento. Ao utilizarem-se veículos novos, o grupo de controlo do veículo deve ser complementado por um grupo ficticiamente exposto ao produto químico em estudo. Os animais do(s) grupo(s) de controlo devem ser tratados de forma idêntica aos animais dos grupos expostos.
Administração das doses
19. O produto químico em estudo e o veículo devem ser administrados pela via mais representativa da exposição humana potencial e com base nos dados metabólicos e de distribuição relativos aos animais utilizados no estudo. Prefere-se, em geral, a via oral (por exemplo sonda esofágica, incorporação na dieta ou incorporação na água de beber), mas podem utilizar-se outras vias (por exemplo via dérmica ou inalação), justificáveis por determinadas características ou pelas vias conhecidas ou previsíveis de exposição humana — para mais orientações, ver o documento de orientações n.o 43 (8). É necessário justificar a via de administração escolhida. O produto químico em estudo deve ser administrado todos os dias aproximadamente à mesma hora.
20. Normalmente, a dose administrada a cada animal é calculada com base na determinação mais recente do peso corporal do animal. O ajustamento das doses durante o último terço da gravidez deve, porém, ser feito com precaução. Se for observada toxicidade excessiva em progenitoras expostas, os animais em questão devem ser eutanasiados.
21. No mínimo, o produto químico em estudo e o veículo devem ser administrados diariamente às fêmeas cobertas, desde a implantação (sexto dia de gravidez) até ao final da lactação (21 dias após o nascimento), a fim de expor as crias ao mesmo durante o desenvolvimento neurológico pré-natal e pós-natal. A idade à qual se inicia a administração das doses e a duração e a frequência desta podem ser ajustadas se os dados disponíveis evidenciarem que outro modelo experimental corresponde melhor à exposição humana. No caso de outras espécies, as durações da administração das doses devem ser ajustadas de modo a garantir a exposição ao produto químico em todos os períodos precoces do desenvolvimento do cérebro (nos períodos equivalentes ao crescimento pré-natal e pós-natal precoce do cérebro humano). A administração das doses pode começar no início da gravidez (dia zero de gravidez), embora deva ponderar-se a possibilidade de o produto químico em estudo provocar mortalidade antes mesmo da implantação. O início da administração das doses ao sexto dia de gravidez evitará esse risco, mas não haverá então exposição ao produto químico em estudo nos estádios de desenvolvimento compreendidos entre o dia zero e o sexto dia de gravidez. Se o laboratório comprar fêmeas cobertas em data determinada, não será possível iniciar a administração do produto químico no dia zero de gravidez, pelo que o sexto dia de gravidez se afigura uma boa opção. O laboratório que efetua os ensaios deve estabelecer o programa de administração das doses em função das informações pertinentes de que disponha sobre os efeitos do produto químico em estudo, da experiência adquirida e de considerações logísticas. Nessa ótica, o programa definido pode prever que a administração das doses se prolongue para além do desmame. O produto químico em estudo não deve ser administrado no dia do parto aos animais que ainda não tenham parido toda a progenitura. Em geral, presume-se que as crias são expostas através do leite materno. Todavia, deve ponderar-se a administração direta de doses às crias, se faltarem indícios de que a progenitura está continuamente exposta. Esses indícios podem provir, por exemplo, de dados farmacocinéticos, da toxicidade eventualmente detetada na progenitura ou de alterações de biomarcadores (28).
EXAMES
Exame das progenitoras
22. Pelo menos uma vez por dia, deve verificar-se cuidadosamente o estado de saúde de todas as progenitoras, incluindo os casos de morbidez ou de mortalidade.
23. Durante os períodos de exposição e de observação, devem efetuar-se com regularidade exames clínicos mais aprofundados (pelo menos duas vezes durante o período de administração durante a gestação e duas vezes durante o período de administração durante a lactação) a, pelo menos, dez progenitoras por nível de dose. Os animais devem ser examinados fora da gaiola onde se encontram alojados, por pessoal formado que não esteja a par da exposição a que os animais foram sujeitos. Os técnicos em causa devem aplicar protocolos normalizados de minimização dos erros de apreciação induzidos pela agitação dos animais e de maximização da reprodutibilidade de técnico para técnico. Tanto quanto possível, é recomendável que os exames realizados num determinado estudo o sejam pelo mesmo técnico.
24. Os sinais observados devem ser mencionados no relatório. Sempre que possível, também deve ser mencionada no relatório a intensidade desses sinais. Entre os exames a realizar, contam-se a observação de alterações da pele, da pelagem, dos olhos e das mucosas, bem como da ocorrência de secreções ou de reações neurovegetativas (por exemplo, lacrimação, horripilação, alterações da dimensão pupilar, respiração anormal e/ou respiração pela boca e sinais inabituais ao nível da micção ou da defecação).
25. Devem igualmente ser indicadas no relatório quaisquer respostas inabituais relativas à posição corporal, ao nível de atividade (por exemplo maior ou menor exploração da zona-padrão) e à coordenação dos movimentos. Também devem ser mencionadas no relatório as alterações na marcha (por exemplo, marcha bamboleante ou ataxia), na postura (por exemplo, dorso arqueado) e na reatividade à manipulação, à colocação ou a outros estímulos ambientais, bem como a ocorrência de movimentos clónicos ou tónicos, de convulsões ou tremores, de comportamentos estereotipados (por exemplo, atos de higiene demasiado repetitivos, movimentos de cabeça pouco usuais ou movimentação repetitiva em círculo) ou estranhos (por exemplo, lamber excessivamente ou morder, automutilação, movimentação para trás, vocalização) ou de atos de agressão.
26. Devem indicar-se no relatório os sinais de toxicidade observados, o grau e a duração destes e o dia e a hora em que apareceram.
27. Os animais devem ser pesados, quando da administração da dose, pelo menos semanalmente durante o estudo, no dia do parto ou próximo deste e 21 dias após o nascimento (dia do desmame). Nos estudos que recorram a sonda gástrica, as progenitoras devem ser pesadas, pelo menos, duas vezes por semana. As doses devem ser ajustadas quanto seja necessário quando de cada pesagem. O consumo de alimentos durante a gestação e a lactação deve determinar-se pelo menos semanalmente. Se a exposição se fizer através da água de beber, o consumo de água deve ser determinado pelo menos semanalmente.
Exame da progenitura
28. Deve examinar-se cuidadosamente toda a progenitura pelo menos uma vez por dia para detetar eventuais sinais de toxicidade, incluindo casos de morbidez ou de mortalidade.
29. Durante os períodos de exposição e de observação, devem efetuar-se exames clínicos mais aprofundados da progenitura. A progenitura (pelo menos uma cria de cada sexo por ninhada) deve ser examinada por pessoal formado que não esteja a par da exposição a que os animais foram sujeitos. Os técnicos em causa devem aplicar protocolos normalizados de minimização dos erros de apreciação e de maximização da reprodutibilidade de técnico para técnico. Tanto quanto possível, é recomendável que os exames sejam realizados pelo mesmo técnico. No mínimo, devem examinar-se os parâmetros referidos nos pontos 24 e 25 adequados ao estádio de desenvolvimento em observação.
30. Devem indicar-se no relatório os sinais de toxicidade observados na progenitura, o grau e a duração destes e o dia e a hora em que apareceram.
Indicadores físicos e de desenvolvimento
31. As alterações dos indicadores de desenvolvimento anteriores ao desmame (abertura do pavilhão auditivo, abertura dos olhos, nascimento dos incisivos) estão estreitamente correlacionadas com o peso corporal (30)(31). Muitas vezes, o peso corporal é, portanto, o melhor indicador do desenvolvimento físico. Só se recomenda a medição de outros indicadores do desenvolvimento quando haja indícios anteriores de que os parâmetros em causa proporcionarão informações adicionais. Indica-se no quadro 1 quando esses parâmetros devem ser avaliados. Consoante os efeitos previstos e os resultados das primeiras medições, pode ser aconselhável aumentar o número de ocasiões de avaliação ou efetuar as medições noutros estádios do desenvolvimento.
32. Ao avaliar o desenvolvimento físico, é preferível utilizar a idade pós-coital em vez da idade pós-natal (33). Se as crias forem examinadas no dia do desmame, recomenda-se que o exame seja efetuado antes daquele, para evitar confusões com o efeito de agitação associado ao desmame. Além disso, os exames a realizar após o desmame não devem ser realizados nos dois dias imediatos.
Quadro 1
Ocasiões da avaliação dos indicadores físicos e de desenvolvimento e parâmetros funcionais e comportamentais (2)
|
Faixas etárias Parâmetros |
Pré-desmame (3) |
Adolescência (3) |
Adultos jovens (3) |
|
Indicadores físicos e de desenvolvimento |
|||
|
Peso corporal e exames clínicos |
Semanalmente (4) |
pelo menos de duas em duas semanas |
pelo menos de duas em duas semanas |
|
Peso cerebral |
22 dias após o nascimento (5) |
no termo |
|
|
Neuropatologia |
22 dias após o nascimento (5) |
no termo |
|
|
Maturidade sexual |
— |
oportunamente |
— |
|
Outros indicadores de desenvolvimento (6) |
oportunamente |
— |
— |
|
Parâmetros funcionais e comportamentais |
|||
|
Ontogenia comportamental |
pelo menos duas medições |
|
|
|
Atividade motora (incluindo a habituação) |
1-3 vezes (7) |
— |
uma vez |
|
Funções motoras e sensoriais |
— |
uma vez |
uma vez |
|
Aprendizagem e memória |
— |
uma vez |
uma vez |
33. Depois de contadas as crias vivas, determina-se o sexo das mesmas, por exemplo por exame visual ou medição da distância anogenital (34)(35), e pesa-se cada cria de cada ninhada à nascença ou pouco depois, pelo menos uma vez por semana durante a lactação e, em seguida, pelo menos de duas em duas semanas. Quando da avaliação da maturidade sexual, deve determinar-se a idade e o peso corporal de, pelo menos, uma fêmea com permeabilidade vaginal (36) e um macho com descolamento do prepúcio (37) de cada ninhada.
Ontogenia comportamental
34. Deve medir-se a ontogenia dos comportamentos escolhidos em, pelo menos, uma cria de cada sexo por ninhada durante o período etário adequado. As crias utilizadas para avaliar os comportamentos em causa devem ser sempre as mesmas em todos os dias de avaliação. Os dias em que se procede a medições devem ser distribuídos uniformemente ao longo do período de avaliação, a fim de determinar se a ontogenia de cada comportamento avaliado é normal ou se há nela alterações decorrentes da exposição (38). Seguem-se alguns exemplos de comportamentos cuja ontogenia pode ser avaliada: reflexo postural labiríntico, geotaxia negativa e atividade motora (38)(39)(40).
Atividade motora
35. Deve monitorizar-se a atividade motora (41)(42)(43)(44)(45) antes do desmame e na idade adulta. No tocante ao dia do desmame, ver o ponto 32. A duração de cada sessão de avaliação deve ser suficiente para revelar a habituação dos animais não-expostos, de controlo, durante a sessão. Recomenda-se vivamente o recurso à atividade motora para avaliar a ontogenia comportamental. No caso de as observações terem essa finalidade, os animais utilizados nas sessões anteriores ao desmame devem ser sempre os mesmos. A frequência das sessões deve ser suficiente para avaliar a ontogenia da habituação em cada sessão (44). Para isso, até ao desmame, inclusive, podem ser necessárias três ou mais sessões de avaliação (por exemplo, 13, 17 e 21 dias após o nascimento). Devem examinar-se os mesmos animais, ou animais da mesma ninhada, também na idade adulta, perto do termo do estudo (por exemplo, 60 a 70 dias após o nascimento). Se necessário, podem efetuar-se sessões de avaliação noutros dias. Deve monitorizar-se a atividade motora recorrendo a um aparelho de registo automático com capacidade para detetar aumentos e decréscimos de atividade (a atividade de base mensurável com o dispositivo não deve ser demasiado reduzida — ao ponto de inviabilizar a deteção de decréscimos de atividade — nem demasiado elevada — ao ponto de inviabilizar a deteção de acréscimos de atividade). Deve aplicar-se a cada dispositivo um protocolo normalizado que, tanto quanto possível, assegure a reprodutibilidade entre dispositivos e entre sessões de avaliação. Os grupos expostos devem ser distribuídos pelos vários dispositivos da maneira mais equilibrada possível. Cada animal deve ser avaliado individualmente. Os grupos expostos devem ser distribuídos ao longo de cada sessão, para evitar confusões com os ritmos de atividade circadianos. As variações das condições experimentais devem ser reduzidas ao mínimo e não devem ter nenhuma relação sistemática com a exposição dos animais. Entre as variáveis que podem afetar muitas medições do comportamento, incluindo as da atividade motora, contam-se o nível sonoro, a dimensão e a forma da gaiola de ensaio, a temperatura, a humidade relativa, as condições de luminosidade, os cheiros, a utilização no ensaio da gaiola de alojamento ou de outra gaiola e distrações ambientais.
Funções motoras e sensoriais
36. Devem examinar-se aprofundadamente as funções motoras e sensoriais pelo menos uma vez no período da adolescência e uma vez na idade adulta jovem (por exemplo, 60 a 70 dias após o nascimento). No tocante ao dia do desmame, ver o ponto 32. O número de experiências deve ser suficiente para que se disponha de uma quantidade adequada de amostras dos modos sensoriais (por exemplo, somatossensorial e vestibular) e das funções motoras (por exemplo força e coordenação). Alguns exemplos de testes das funções motoras e sensoriais: resposta ao estiramento (46), reflexo postural labiríntico (47)(48), habituação ao sobressalto auditivo (40)(49)(50)(51)(52)(53)(54) e potenciais evocados (55).
Testes de aprendizagem e memória
37. Deve efetuar-se um teste de memória e aprendizagem associativa depois do desmame (por exemplo 25 ± 2 dias após o nascimento) e a adultos jovens (60 ou mais dias após o nascimento). No tocante ao dia do desmame, ver o ponto 32. Podem utilizar-se o mesmo teste ou testes distintos nestes dois estádios de desenvolvimento. Existe uma certa flexibilidade na escolha do teste ou testes de aprendizagem e de memória a realizar nos ratos recentemente desmamados e nos ratos adultos, mas o(s) teste(s) escolhido(s) deve(m) satisfazer dois critérios. O primeiro é que a avaliação da aprendizagem deve ser efetuada em termos das alterações ocorridas ao longo de diversas experiências, ou sessões, repetidas de aprendizagem ou, caso o teste compreenda uma única experiência, relativamente a uma condição que controle os efeitos não associativos do treino. O segundo é que o teste ou testes devem compreender uma medição da memória (de curto ou de longo prazo) além da aprendizagem inicial (aquisição), mas esta medição da memória só deve ser incluída no relatório se for acompanhada de uma medição da aquisição obtida no mesmo teste. Se o teste ou testes de aprendizagem e memória revelar(em) efeitos do produto químico em estudo, importa ponderar a realização de outros testes, que permitam excluir qualquer interpretação fundada em alterações das capacidades sensoriais, de motivação e/ou motoras. Além destes dois critérios, recomenda-se que a escolha do teste de aprendizagem e memória se baseie na sensibilidade comprovada do mesmo à classe de produtos químicos em estudo, se tal informação estiver disponível nas fontes bibliográficas. Na falta dessa informação, seguem-se alguns exemplos de testes que podem satisfazer os critérios referidos: evitação passiva (43)(56)(57), adaptação espacial retardada no rato adulto (58) e no rato não desmamado (59), condicionamento olfativo (43)(60), labirinto aquático de Morris (61)(62)(63), labirinto de Biel ou de Cincinnati (64)(65), labirinto de braços radiais (66), labirinto em T (43) e aquisição e conservação de comportamentos programados (26)(67)(68). As referências bibliográficas descrevem outros testes para ratos recentemente desmamados (26)(27) e ratos adultos (19)(20).
Autópsia
38. As progenitoras podem ser eutanasiadas depois do desmame da progenitura.
39. Procede-se à avaliação neuropatológica da progenitura utilizando tecidos de animais eutanasiados 22 dias após o nascimento ou anteriormente, entre 11 e 22 dias após o nascimento, bem como no termo do estudo. No caso da progenitura eutanasiada até 22 dias após o nascimento, inclusive, examinam-se tecidos cerebrais. No caso dos animais eutanasiados no termo do estudo, examinam-se tecidos do sistema nervoso central e tecidos do sistema nervoso periférico. Os animais eutanasiados até 22 dias após o nascimento, inclusive, podem ser fixados por imersão ou perfusão. Os animais eutanasiados no termo do estudo devem ser fixados por perfusão. Todos os aspetos da preparação das amostras de tecidos, desde a perfusão dos animais à dissecação das amostras, ao tratamento dos tecidos e à coloração das lâminas devem inserir-se num modelo experimental equilibrado, em que cada lote contenha amostras representativas de cada grupo de dosagem. O documento de orientações n.o 20 (9) contém mais orientações sobre neuropatologias; ver igualmente a referência 103.
Tratamento das amostras de tecidos
40. Devem registar-se todas as anomalias macroscópicas detetadas na autópsia. As amostras de tecidos colhidas devem representar as principais regiões do sistema nervoso. As amostras de tecidos devem ser conservadas num fixador adequado e ser tratadas segundo protocolos histológicos normalizados já publicados (69)(70)(71)(103). A incorporação em parafina é aceitável para tecidos do sistema nervoso central e do sistema nervoso periférico. Se for necessária maior resolução (por exemplo no caso dos nervos periféricos, quando se suspeite de neuropatias periféricas, ou para a análise morfométrica de nervos periféricos), pode ser melhor utilizar ósmio na pós-fixação, juntamente com a incorporação numa resina epoxídica. Os tecidos cerebrais colhidos para análises morfométricas devem ser incorporados num meio adequado ao mesmo tempo para todos os níveis de dosagem, a fim de evitar os erros de contração por vezes decorrentes da conservação prolongada em fixadores (6).
Exame neuropatológico
41. Os objetivos deste exame qualitativo são os seguintes:
|
i) |
Identificar as regiões do sistema nervoso que evidenciam alterações neuropatológicas; |
|
ii) |
Identificar os tipos de alterações neuropatológicas resultantes da exposição ao produto químico em estudo; |
|
iii) |
Determinar a gravidade das alterações neuropatológicas. |
Um patologista convenientemente formado deve examinar ao microscópio cortes histológicos representativos das amostras de tecidos, a fim de detetar alterações neuropatológicas. Deve ser atribuído a cada uma dessas alterações um grau subjetivo de gravidade. A coloração com hematoxilina e eosina pode ser suficiente para o exame dos cortes cerebrais de animais eutanasiados até 22 dias após o nascimento, inclusive. No entanto, para examinar cortes de tecidos dos sistemas nervosos central e periférico de animais eutanasiados no termo do estudo, recomenda-se a coloração da mielina (por exemplo, com azul rápido de luxol/violeta de cresilo) e uma coloração com prata (por exemplo a coloração de Bielschowsky ou de Bodians). Cabe ao patologista avaliar, com base na sua experiência profissional e no tipo das alterações observadas, se é conveniente utilizar outros tipos de coloração para identificar e caracterizar determinados tipos de alterações — por exemplo proteína ácida fibrilar glial (GFAP) ou histoquímica da lecitina para examinar alterações gliais e microgliais (72), fluoro-jade para deteção de necroses (73)(74) ou colorações com prata específicas da degenerescência neural (75).
42. Deve efetuar-se uma avaliação morfométrica (quantitativa), pois esses dados podem contribuir para detetar efeitos da exposição e são úteis na interpretação das diferenças no peso cerebral ou morfológicas devidas à exposição (76)(77). Devem colher-se amostras dos tecidos nervosos e os tecidos devem ser preparados para esta avaliação. As avaliações morfométricas podem compreender, por exemplo, medições lineares ou da superfície de determinadas regiões do cérebro (78). Ambos os tipos de medições devem ser praticados em cortes homólogos cuidadosamente selecionados com base em localizadores microscópicos fiáveis (6). O recurso à estereologia pode permitir identificar efeitos da exposição em parâmetros como o volume ou o número de células de determinadas regiões neuroanatómicas (79)(80)(81)(82)(83)(84).
43. Devem examinar-se os cérebros para localizar eventuais indícios de alterações neuropatológicas relacionadas com a exposição, colhendo amostras adequadas das principais regiões cerebrais — por exemplo, bolbos olfativos, córtex cerebral, hipocampo, núcleos da base, tálamo, hipotálamo, mesencéfalo (teto, tegumento e pedúnculos cerebrais), protuberância anelar, bolbo raquidiano, cerebelo) — para assegurar um exame aprofundado. É importante que os cortes sejam efetuados no mesmo plano em todos os animais. Devem constituir-se amostras representativas de cortes da medula espinal e do sistema nervoso periférico dos animais adultos eutanasiados no termo do estudo. As zonas examinadas devem compreender os olhos, com o nervo ótico e a retina, a medula espinal, ao nível das dilatações cervical e lombar, fibras das raízes dorsal e ventral, o nervo ciático proximal, o nervo tibial proximal (ao nível do joelho) e as ramificações do nervo tibial ao nível dos músculos da barriga da perna. Devem ser examinados cortes transversais e longitudinais da medula espinal e dos nervos periféricos.
44. A avaliação neuropatológica deve compreender um exame com vista à deteção de indícios de alterações do desenvolvimento do sistema nervoso (6)(85)(86)(87)(88)(89), além das alterações ao nível celular (por exemplo vacuolização neuronal, degeneração ou necrose) e dos tecidos (por exemplo gliose, infiltração leucocitária ou formação de quistos). Para isto, é importante distinguir os efeitos relacionados com a exposição, por um lado, das ocorrências reconhecidamente normais do desenvolvimento no estádio correspondente à idade do animal no momento da eutanásia, por outro (90). Seguem-se alguns exemplos de alterações significativas indicadoras de que o desenvolvimento foi prejudicado:
|
— |
alterações das dimensões ou da forma dos bolbos olfativos, dos hemisférios cerebrais ou do cerebelo; |
|
— |
alterações das dimensões relativas das diversas regiões do cérebro, designadamente aumento ou diminuição das dimensões de regiões em consequência da perda ou da persistência de populações normalmente transitórias de células ou de projeções axónicas (por exemplo camada germinal externa do cerebelo, corpo caloso); |
|
— |
alterações de proliferação, migração ou diferenciação reveladas por zonas de apoptose ou necrose excessivas, agregados ou populações dispersas de neurões etópicos, mal orientados ou mal formados ou alterações das dimensões relativas das diversas camadas das estruturas corticais; |
|
— |
alterações dos padrões de mielinização, nomeadamente redução dimensional global ou alterações da coloração das estruturas mielinizadas; |
|
— |
indícios de hidrocefalia, nomeadamente dilatação ventricular, estenose do aqueduto cerebral e emagrecimento dos hemisférios cerebrais. |
Análise da relação entre as alterações neuropatológicas e a dosagem
45. Recomenda-se o seguinte protocolo sequencial para os exames neuropatológicos qualitativos e quantitativos: Começa-se por comparar cortes do grupo exposto à dose mais elevada com cortes do grupo de controlo. Se não se detetarem indícios de alterações neuropatológicas nos animais do grupo exposto à dose mais elevada, não são necessários mais exames. Caso se detetem alterações neuropatológicas nesse grupo, examinam-se animais dos grupos expostos às doses intermédia e mais baixa. Se o estudo do grupo exposto à dose mais elevada for interrompido devido à morte dos animais ou a efeitos tóxicos não relacionados com a exposição ao produto químico em estudo, devem examinar-se os grupos expostos à dose mais elevada e à dose intermédia para verificar se existem alterações neuropatológicas. Se forem detetados indícios de neurotoxicidade nos grupos expostos às doses mais baixas, esses grupos devem ser objeto de exames neuropatológicos. Se, nos exames qualitativos ou quantitativos, for detetada alguma alteração neuropatológica relacionada com a exposição ao produto químico em estudo, deve determinar-se, com base na avaliação de todos os animais de todos os grupos de dosagem, a relação entre a dose e a incidência, frequência e gravidade das lesões ou das alterações morfométricas. Esta avaliação deve incidir em todas as regiões do cérebro que evidenciem alguma alteração neuropatológica. Para cada tipo de lesão, devem descrever-se as características em que se baseiam os vários graus de gravidade, indicando os critérios utilizados para os diferenciar. Devem ser registados a frequência de cada tipo de lesão e o respetivo grau de gravidade e deve ser efetuada uma análise estatística de avaliação da natureza da resposta à dosagem. Recomenda-se o uso de lâminas codificadas (91).
DADOS E RELATÓRIOS
Dados
46. Os dados devem ser individualizados e também resumidos em quadros, indicando, para cada grupo estudado, os tipos de alterações e o número de progenitoras, progenitura, por sexo, e ninhadas que apresentam cada tipo de alteração. Caso se tenha procedido à exposição pós-natal direta da progenitura, devem indicar-se a via, a duração e o período de exposição.
Avaliação e interpretação dos resultados
47. O objetivo dos estudos de neurotoxicidade durante o desenvolvimento é fornecer informações sobre os efeitos da exposição repetida ao produto químico durante o desenvolvimento intrauterino e pós-natal. Uma vez que o estudo incide tanto na toxicidade geral como na neurotoxicidade durante o desenvolvimento, os resultados obtidos permitem estabelecer uma distinção entre efeitos neurotóxicos durante o desenvolvimento não associados a toxicidade materna em geral e efeitos neurotóxicos durante o desenvolvimento apenas induzidos a níveis também tóxicos para as progenitoras. Em virtude da complexidade das interdependências entre o modelo do estudo, a análise estatística e o significado biológico dos dados, a interpretação correta de dados de neurotoxicidade durante o desenvolvimento exige a apreciação de um especialista na matéria (107)(109). A interpretação dos resultados dos ensaios deve basear-se na ponderação da suficiência da prova (20)(92)(93)(94). Devem discutir-se os tipos de efeitos comportamentais ou morfológicos eventualmente observados, bem como os indícios de uma relação entre a dose e a resposta. Esta caracterização deve ter em conta os dados de todos os estudos disponíveis pertinentes para a avaliação da neurotoxicidade durante o desenvolvimento, nomeadamente estudos epidemiológicos no ser humano ou relatórios de casos paradigmáticos, bem como estudos experimentais em animais (por exemplo, dados toxicocinéticos, informações de relações estrutura-atividade e dados de outros estudos de toxicidade). Deve igualmente ser contemplada a relação entre as doses do produto químico em estudo e a presença, ausência, incidência e intensidade dos eventuais efeitos neurotóxicos em cada sexo (20)(95).
48. A avaliação dos dados deve compreender uma discussão do significado biológico e do significado estatístico. A análise estatística deve ser entendida como um instrumento mais orientador do que determinante na interpretação dos dados. A falta de significado estatístico não deve ser o único fundamento para concluir pela ausência de efeitos ligados à exposição, tal como a existência de significado estatístico não deve constituir a única justificação para concluir pela ocorrência de efeitos ligados à exposição. Para evitar resultados falsos negativos e as dificuldades inerentes à demonstração de resultados negativos, devem incluir-se na discussão dados históricos de controlo e dados de controlo positivos, sobretudo se não ocorrerem efeitos relacionados com a exposição (102)(106). A probabilidade da obtenção de resultados falsos positivos deve ser discutida no âmbito da avaliação estatística geral dos resultados (96). A avaliação deve abranger a relação eventualmente existente entre as alterações neuropatológicas e comportamentais observadas.
49. Os resultados devem ser todos analisados com base em modelos estatísticos adaptados ao modelo experimental (108). A opção por uma análise paramétrica ou não-paramétrica deve fundamentar-se na ponderação de fatores como a natureza dos dados (transformados ou não) e a distribuição dos mesmos, assim como na robustez relativa da análise estatística escolhida. O objetivo e o modelo do estudo devem orientar a escolha de uma análises estatística que minimize os erros do tipo I (falsos positivos) e do tipo II (falsos negativos) (96)(97)(104)(105). Nos estudos do desenvolvimento em espécies multíparas nos quais se estudem várias crias por ninhada, o modelo estatístico deve incluir a ninhada, para evitar uma inflação de erros do tipo I (98)(99)(100)(101). A unidade de medida estatística deve ser a ninhada e não a cria e as experiências devem ser concebidas de modo que crias da mesma ninhada não sejam consideradas observações independentes. Os parâmetros que sejam medidos várias vezes no mesmo sujeito devem ser analisados por recurso a modelos estatísticos que tenham em conta o facto de essas medições não serem independentes.
Relatório dos ensaios
50. Elementos a constar do relatório dos ensaios:
Produto químico em estudo:
|
— |
natureza física e propriedades físico-químicas pertinentes; |
|
— |
dados de identificação, incluindo a proveniência; |
|
— |
grau de pureza da preparação e impurezas conhecidas e/ou previsíveis. |
Veículo (se for o caso):
|
— |
justificação da escolha do veículo, se não for água nem soro fisiológico. |
Animais estudados:
|
— |
espécie e estirpe utilizadas e, caso não sejam utilizados ratos, justificação da utilização de outra espécie; |
|
— |
fornecedor dos animais; |
|
— |
número, idade no início do estudo e sexo dos animais; |
|
— |
proveniência, condições de alojamento, dieta, água de beber, etc.; |
|
— |
peso de cada animal no início do ensaio. |
Condições de realização dos ensaios:
|
— |
fundamentação da escolha das doses; |
|
— |
fundamentação da via e do período de administração; |
|
— |
caracterização das doses administradas, incluindo elementos sobre o veículo, o volume e a forma física do administrado; |
|
— |
elementos relativos à formulação do produto químico em estudo/à incorporação do mesmo na dieta dos animais; concentração atingida, estabilidade e homogeneidade da preparação; |
|
— |
método utilizado para identificar individualmente as progenitoras e a progenitura; |
|
— |
descrição pormenorizada do(s) protocolo(s) de aleatorização utilizados para integrar as progenitoras nos grupos expostos, selecionar as crias a eliminar das ninhadas e integrar as crias restantes nos grupos estudados; |
|
— |
elementos relativos à administração do produto químico em estudo; |
|
— |
se aplicável, equivalência entre a concentração do produto químico em estudo na dieta/na água de beber ou inalado, expressa em ppm, e a dose real, expressa em mg/kg de peso corporal/dia; |
|
— |
condições ambientais; |
|
— |
elementos relativos à qualidade dos alimentos e da água (por exemplo, da torneira ou destilada); |
|
— |
datas de início e termo do estudo. |
Protocolos de exame e protocolos experimentais:
|
— |
descrição pormenorizada dos protocolos utilizados para normalizar exames e protocolos; definições utilizadas na pontuação das observações; |
|
— |
lista dos protocolos experimentais utilizados e justificação da escolha dos mesmos; |
|
— |
elementos relativos aos protocolos comportamentais/funcionais, patológicos, neuroquímicos e eletrofisiológicos utilizados, incluindo informações e demais elementos sobre os aparelhos automáticos; |
|
— |
protocolos de calibração e de verificação da equivalência dos aparelhos utilizados; protocolos utilizados para garantir o equilíbrio dos grupos expostos na execução experimental; |
|
— |
breve justificação das eventuais decisões fundamentadas na experiência profissional. |
Resultados (individualizados e resumidos, incluindo a média e a variância, quando se justifique):
|
— |
número de animais no início do estudo e número de animais no final do estudo; |
|
— |
número de animais e de ninhadas utilizados em cada método de ensaio; |
|
— |
número de identificação de cada animal e da ninhada de que provém; |
|
— |
número de animais e peso médio por sexo à nascença, de cada ninhada; |
|
— |
peso corporal e alterações de peso corporal, incluindo o peso corporal das progenitoras e da progenitura no termo dos ensaios; |
|
— |
dados do consumo de alimentos e, se pertinente, do consumo de água (por exemplo se o produto químico em estudo for administrado pela água de beber); |
|
— |
dados das respostas tóxicas por sexo e por nível de dosagem, incluindo sinais de toxicidade ou mortalidade (e, se for o caso, o momento e a causa da morte); |
|
— |
natureza, gravidade, duração, dia do aparecimento, hora do dia e evolução ulterior das observações dos exames clínicos aprofundados; |
|
— |
pontuação de cada indicador de desenvolvimento (peso, maturidade sexual e ontogenia comportamental) em cada ocasião de observação; |
|
— |
descrição pormenorizada das observações comportamentais, funcionais, neuropatológicas, neuroquímicas e eletrofisiológicas efetuadas, por sexo, incluindo os acréscimos e as reduções em relação ao(s) grupo(s) de controlo; |
|
— |
resultados das autópsias; |
|
— |
pesos cerebrais; |
|
— |
diagnósticos decorrentes dos sinais e lesões neurológicos, incluindo doenças ou estados naturais; |
|
— |
imagens de observações representativas; |
|
— |
imagens pouco aumentadas que permitiram aferir da homologia dos cortes utilizados para a morfometria; |
|
— |
dados de absorção e metabólicos, incluindo dados complementares de estudos toxicocinéticos realizados separadamente, se disponíveis; |
|
— |
tratamento estatístico dos resultados (incluindo os modelos estatísticos utilizados para analisar os dados) e resultados, sejam estes significativos ou não; |
|
— |
lista do pessoal participante no estudo, incluindo a respetiva formação profissional. |
Discussão dos resultados:
|
— |
informações relativas à relação entre a dose e a resposta, por sexo e por grupo; |
|
— |
relação entre quaisquer outros efeitos tóxicos e as conclusões acerca do potencial neurotóxico do produto químico em estudo, por sexo e por grupo; |
|
— |
influência de eventuais informações toxicocinéticas nas conclusões; |
|
— |
similitude de efeitos com os de qualquer neurotóxico conhecido; |
|
— |
dados corroborantes da fiabilidade e sensibilidade do método de ensaio (dados históricos de controlo e dados de controlo positivos); |
|
— |
eventuais relações entre efeitos neuropatológicos e funcionais; |
|
— |
nível sem observação de efeitos adversos (NOAEL) ou doses de referência para as progenitoras e a progenitura, por sexo e por grupo. |
Conclusões:
|
— |
discussão da interpretação geral dos dados com base nos resultados, incluindo o NOAEL e uma conclusão sobre a neurotoxicidade, ou não, durante o desenvolvimento, do produto químico em estudo. |
REFERÊNCIAS
|
1. |
OCDE (1995). Draft Report of the OECD Ad Hoc Working Group on Reproduction and Developmental Toxicity. Copenhaga, Dinamarca, 13 e 14 de junho de 1995. |
|
2. |
US EPA (1998). U.S. Environmental Protection Agency Health Effects Test Guidelines. OPPTS 870.6300. Developmental Neurotoxicity Study. US EPA 712-C-98-239. Acessível em: http://www.epa.gov/opptsfrs/OPPTS_Harmonized/870_Health_Effects_Test_Guidelines/Series/ |
|
3. |
US EPA (1998). Guidelines for Neurotoxicity Risk Assessment. US EPA 630/R-95/001F. Acessível em: http://cfpub.epa.gov/ncea/cfm/recordisplay.cfm?PrintVersion=True&deid=12479 |
|
4. |
Cory-Slechta, D.A., Crofton, K.M., Foran, J.A., Ross, J.F., Sheets, L.P., Weiss, B., Mileson, B. (2001). Methods to identify and characterize developmental neurotoxicity for human health risk assessment: I. Behavioral effects. Environ. Health Perspect. 109:79-91. |
|
5. |
Dorman, D.C., Allen, S.L., Byczkowski, J.Z., Claudio, L., Fisher, J.E. Jr., Fisher, J.W., Harry, G.J., Li, A.A., Makris, S.L., Padilla, S., Sultatos, L.G., Mileson, B.E. (2001). Methods to identify and characterize developmental neurotoxicity for human health risk assessment: III. Pharmacokinetic and pharmacodynamic considerations. Environ. Health Perspect. 109:101-111. |
|
6. |
Garman, R.H., Fix, A.S., Jortner, B.S., Jensen, K.F., Hardisty, J.F., Claudio, L., Ferenc, S. (2001). Methods to identify and characterize developmental neurotoxicity for human health risk assessment: II. Neuropathology. Environ. Health Perspect. 109:93-100. |
|
7. |
OCDE (2003). Report of the OECD Expert Consultation Meeting on Developmental Neurotoxicity Testing. Washington D.C., EUA, 23 a 25 de outubro de 2000. |
|
8. |
OCDE (2008). OECD Environment, Health and Safety Publications Series on Testing and Assessment No. 43. Guidance Document on Mammalian Reproductive Toxicity Testing and Assessment. Environment Directorate, OCDE, Paris, julho de 2008. Acessível em: http://search.oecd.org/officialdocuments/displaydocumentpdf/?cote=env/jm/mono(2008)16&doclanguage=en |
|
9. |
OCDE (2003). OECD Environment, Health and Safety Publications Series on Testing and Assessment No. 20. Guidance Document for Neurotoxicity Testing. Environment Directorate, OCDE, Paris, setembro de 2003. Acessível em: http://www.oecd.org/document/22/0,2340,en_2649_34377_1916054_1_1_1_1,00.html |
|
10. |
Kimmel, C.A., Rees, D.C., Francis, E.Z. (1990). Qualitative and quantitative comparability of human and animal developmental neurotoxicity. Neurotoxicol. Teratol. 12:173-292. |
|
11. |
Spencer, P.S., Schaumburg, H.H., Ludolph, A.C. (2000). Experimental and Clinical Neurotoxicology, 2.a edição, ISBN 0195084772. Oxford University Press, Nova Iorque. |
|
12. |
Mendola, P., Selevan, S.G., Gutter, S., Rice, D. (2002). Environmental factors associated with a spectrum of neurodevelopmental deficits. Ment. Retard. Dev. Disabil. Res. Rev. 8:188-197. |
|
13. |
Slikker, W.B., Chang, L.W. (1998). Handbook of Developmental Neurotoxicology, 1.a edição, ISBN 0126488606. Academic Press, Nova Iorque. |
|
14. |
Capítulo B.34 deste anexo: Teste de toxicidade sobre a reprodução em uma geração. |
|
15. |
Capítulo B.35 deste anexo: Estudo de toxicidade sobre a reprodução em duas gerações. |
|
16. |
Capítulo B.43 deste anexo: Estudo de neurotoxicidade em roedores. |
|
17. |
Capítulo B.31 deste anexo: Estudo de toxicidade sobre o desenvolvimento pré-natal. |
|
18. |
Diretiva 2010/63/UE do Parlamento Europeu e do Conselho, de 22 de setembro de 2010, relativa à proteção dos animais utilizados para fins científicos (JO L 276 de 20.10.2010, p. 33). |
|
19. |
OMS (1986). Principles and Methods for the Assessment of Neurotoxicity Associated with Exposure to Chemicals (Environmental Health Criteria 60). Albany, Nova Iorque: World Health Organization Publications Center, EUA. Acessível em: http://www.inchem.org/documents/ehc/ehc/ehc060.htm |
|
20. |
OMS (2001). Neurotoxicity Risk Assessment for Human Health:Principles and Approaches (Environmental Health Criteria 223). World Health Organization Publications, Genebra. Acessível em: http://www.intox.org/databank/documents/supplem/supp/ehc223.htm |
|
21. |
Chang, L.W., Slikker, W. (1995). Neurotoxicology:Approaches and Methods, 1.a edição, ISBN 012168055X. Academic Press, Nova Iorque. |
|
22. |
De Cabo, C., Viveros, M.P. (1997). Effects of neonatal naltrexone on neurological and somatic development in rats of both genders. Neurotoxicol. Teratol. 19:499-509. |
|
23. |
Agnish, N.D., Keller, K.A. (1997). The rationale for culling of rodent litters. Fundam. Appl. Toxicol. 38:2-6. |
|
24. |
Avery, D.L., Spyker, J.M. (1977). Foot tattoo of neonatal mice. Lab. Animal Sci. 27:110-112. |
|
25. |
Wier, P.J., Guerriero, F.J., Walker, R.F. (1989). Implementation of a primary screen for developmental neurotoxicity. Fundam. Appl. Toxicol. 13:118-136. |
|
26. |
Spear, N.E., Campbell, B.A. (1979). Ontogeny of Learning and Memory. ISBN 0470268492. Erlbaum Associates, New Jersey. |
|
27. |
Krasnegor, N.A., Blass, E.M., Hofer, M.A., Smotherman, W. (1987). Perinatal Development:A Psychobiological Perspective. Academic Press, Orlando. |
|
28. |
Zoetis, T., Walls, I. (2003). Principles and Practices for Direct Dosing of Pre-Weaning Mammals in Toxicity Testing and Research. ILSI Press, Washington, DC. |
|
29. |
Moser, V., Walls, I., Zoetis, T. (2005). Direct dosing of preweaning rodents in toxicity testing and research: Deliberations of an ILSI RSI expert working group. Int. J. Toxicol. 24:87-94. |
|
30. |
Conolly, R.B., Beck, B.D., Goodman, J.I. (1999). Stimulating research to improve the scientific basis of risk assessment. Toxicol. Sci. 49:1-4. |
|
31. |
ICH (1993). ICH Harmonised Tripartite Guideline: Detection of Toxicity to Reproduction for Medical Products (S5A). International Conference on Harmonisation of Technical Requirements for Registration of Phamaceuticals for Human Use. |
|
32. |
Lochry, E.A. (1987). Concurrent use of behavioral/functional testing in existing reproductive and developmental toxicity screens: Practical considerations. J. Am. Coll. Toxicol. 6:433-439. |
|
33. |
Tachibana, T., Narita, H., Ogawa, T., Tanimura, T. (1998). Using postnatal age to determine test dates leads to misinterpretation when treatments alter gestation length, results from a collaborative behavioral teratology study in Japan. Neurotoxicol. Teratol. 20:449-457. |
|
34. |
Gallavan, R.H. Jr., Holson, J.F., Stump, D.G., Knapp, J.F., Reynolds, V.L. (1999). Interpreting the toxicologic significance of alterations in anogenital distance: potential for confounding effects of progeny body weights. Reprod. Toxicol. 13:383-390. |
|
35. |
Gray, L.E. Jr., Ostby, J., Furr, J., Price, M., Veeramachaneni, D.N., Parks, L. (2000). Perinatal exposure to the phthalates DEHP, BBP, and DINP, but not DEP, DMP, or DOTP, alters sexual differentiation of the male rat. Toxicol. Sci. 58:350-365. |
|
36. |
Adams, J., Buelke-Sam, J., Kimmel, C.A., Nelson, C.J., Reiter, L.W., Sobotka, T.J., Tilson, H.A., Nelson, B.K. (1985). Collaborative behavioral teratology study: Protocol design and testing procedure. Neurobehav. Toxicol. Teratol. 7:579-586. |
|
37. |
Korenbrot, C.C., Huhtaniemi, I.T., Weiner, R.W. (1977). Preputial separation as an external sign of pubertal development in the male rat. Biol. Reprod. 17:298-303. |
|
38. |
Spear, L.P. (1990). Neurobehavioral assessment during the early postnatal period. Neurotoxicol. Teratol. 12:489-95. |
|
39. |
Altman, J., Sudarshan, K. (1975). Postnatal development of locomotion in the laboratory rat. Anim. Behav. 23:896-920. |
|
40. |
Adams, J. (1986). Methods in Behavioral Teratology. Em: Handbook of Behavioral Teratology. Riley, E.P., Vorhees, C.V. (editores) Plenum Press, Nova Iorque, p. 67-100. |
|
41. |
Reiter, L.W., MacPhail, R.C. (1979). Motor activity: A survey of methods with potential use in toxicity testing. Neurobehav. Toxicol. 1:53-66. |
|
42. |
Robbins, T.W. (1977). A critique of the methods available for the measurement of spontaneous motor activity, Handbook of Psychopharmacology, volume 7. Iverson, L.L., Iverson, D.S., Snyder, S.H. (editores). Plenum Press, Nova Iorque, p. 37-82. |
|
43. |
Crofton, K.M., Peele, D.B., Stanton, M.E. (1993). Developmental neurotoxicity following neonatal exposure to 3,3'-iminodipropionitrile in the rat. Neurotoxicol. Teratol. 15:117-129. |
|
44. |
Ruppert, P.H., Dean, K.F., Reiter, L.W. (1985). Development of locomotor activity of rat pups in figure-eight mazes. Dev. Psychobiol. 18:247-260. |
|
45. |
Crofton, K.M., Howard, J.L., Moser, V.C., Gill, M.W., Reiter, L.W., Tilson, H.A., MacPhail, R.C. (1991). Interlaboratory comparison of motor activity experiments: Implications for neurotoxicological assessments. Neurotoxicol. Teratol. 13:599-609. |
|
46. |
Ross, J.F., Handley, D.E., Fix, A.S., Lawhorn, G.T., Carr, G.J. (1997). Quantification of the hind-limb extensor thrust response in rats. Neurotoxicol. Teratol. 19:1997.405-411. |
|
47. |
Handley, D.E., Ross, J.F., Carr, G.J. (1998). A force plate system for measuring low-magnitude reaction forces in small laboratory animals. Physiol. Behav. 64:661-669. |
|
48. |
Edwards, P.M., Parker, V.H. (1977). A simple, sensitive, and objective method for early assessment of acrylamide neuropathy in rats. Toxicol. Appl. Pharmacol. 40:589-591. |
|
49. |
Davis, M. (1984). The mammalian startle response. Em: Neural Mechanisms of Startle Behavior. Eaton, R.C. (editores). Plenum Press, Nova Iorque, p. 287-351. |
|
50. |
Koch, M. (1999). The neurobiology of startle. Prog. Neurobiol. 59:107-128. |
|
51. |
Crofton, K.M. (1992). Reflex modification and the assessment of sensory dysfunction. Em: Target Organ Toxicology Series:Neurotoxicology. Tilson, H., Mitchell, C. (editores). Raven Press, Nova Iorque, p. 181-211. |
|
52. |
Crofton, K.M., Sheets, L.P. (1989). Evaluation of sensory system function using reflex modification of the startle response. J. Am. Coll. Toxicol. 8:199-211. |
|
53. |
Crofton, K.M, Lassiter, T.L, Rebert, C.S. (1994). Solvent-induced ototoxicity in rats: An atypical selective mid-frequency hearing deficit. Hear. Res. 80:25-30. |
|
54. |
Ison, J.R. (1984). Reflex modification as an objective test for sensory processing following toxicant exposure. Neurobehav. Toxicol. Teratol. 6:437–445. |
|
55. |
Mattsson, J.L., Boyes, W.K., Ross, J.F. (1992). Incorporating evoked potentials into neurotoxicity test schemes. Em: Target Organ Toxicology Series: Neurotoxicity. Tilson, H., Mitchell, C., (editores). Raven Press, Nova Iorque. p. 125-145. |
|
56. |
Peele, D.B., Allison, S.D., Crofton, K.M. (1990). Learning and memory deficits in rats following exposure to 3,3'-iminopropionitrile. Toxicol. Appl. Pharmacol. 105:321-332. |
|
57. |
Bammer, G. (1982). Pharmacological investigations of neurotransmitter involvement in passive avoidance responding: A review and some new results. Neurosci. Behav. Rev. 6:247-296. |
|
58. |
Bushnell, P.J. (1988). Effects of delay, intertrial interval, delay behavior and trimethyltin on spatial delayed response in rats. Neurotoxicol. Teratol. 10:237-244. |
|
59. |
Green, R.J., Stanton, M.E. (1989). Differential ontogeny of working memory and reference memory in the rat. Behav. Neurosci. 103:98-105. |
|
60. |
Kucharski, D., Spear, N.E. (1984). Conditioning of aversion to an odor paired with peripheral shock in the developing rat. Develop. Psychobiol. 17:465-479. |
|
61. |
Morris, R. (1984). Developments of a water-maze procedure for studying spatial learning in the rat. J. Neurosci. Methods 11:47-60. |
|
62. |
Brandeis, R., Brandys, Y., Yehuda, S. (1989). The use of the Morris water maze in the study of memory and learning. Int. J. Neurosci. 48:29-69. |
|
63. |
D'Hooge, R., De Deyn, P.P. (2001). Applications of the Morris water maze in the study of learning and memory. Brain Res. Rev. 36:60-90. |
|
64. |
Vorhees, C.V. (1987). Maze learning in rats: A comparison of performance in two water mazes in progeny prenatally exposed to different doses of phenytoin. Neurotoxicol. Teratol. 9:235-241. |
|
65. |
Vorhees, C.V. (1997). Methods for detecting long-term CNS dysfunction after prenatal exposure to neurotoxins. Drug Chem. Toxicol. 20:387-399. |
|
66. |
Akaike, M., Tanaka, K., Goto, M., Sakaguchi, T. (1988). Impaired Biel and Radial arm maze learning in rats with methyl-nitrosurea induced microcephaly. Neurotoxicol. Teratol. 10:327-332. |
|
67. |
Cory-Slechta, D.A., Weiss, B., Cox, C. (1983). Delayed behavioral toxicity of lead with increasing exposure concentration. Toxicol. Appl. Pharmacol. 71:342-352. |
|
68. |
Campbell, B.A., Haroutunian, V. (1981). Effects of age on long-term memory: Retention of fixed interval responding. J. Gerontol. 36:338-341. |
|
69. |
Fix, A.S, Garman, R.H. (2000). Practical aspects of neuropathology: A technical guide for working with the nervous system. Toxicol. Pathol. 28:122-131. |
|
70. |
Prophet, E.B., Mills, B., Arrington, J.B., Sobin, L.H. (1994). Laboratory Methods in Histotechnology. American Registry of Pathology, Washington, DC, p. 84-107. |
|
71. |
Bancroft, J.D., Gamble, M. (2002). Theory and Practice of Histological Techniques, 5.a edição. Churchill Livingstone, Londres. |
|
72. |
Fix, A.S., Ross, J.F., Stitzel, S.R., Switzer, R.C. (1996). Integrated evaluation of central nervous system lesions: stains for neurons, astrocytes, and microglia reveal the spatial and temporal features of MK-801-induced neuronal necrosis in the rat cerebral cortex. Toxicol. Pathol. 24:291-304. |
|
73. |
Schmued, L.C., Hopkins, K.J. (2000). Fluoro-Jade B: A high affinity tracer for the localization of neuronal degeneration. Brain Res. 874:123-130. |
|
74. |
Krinke, G.J., Classen, W., Vidotto, N., Suter, E., Wurmlin, C.H. (2001). Detecting necrotic neurons with fluoro-jade stain. Exp. Toxic. Pathol. 53:365-372. |
|
75. |
De Olmos, I.S., Beltramino, C.A., de Olmos de Lorenzo, S. (1994). Use of an amino-cupric-silver technique for the detection of early and semiacute neuronal degeneration caused by neurotoxicants, hypoxia and physical trauma. Neurotoxicol. Teratol. 16, 545-561. |
|
76. |
De Groot, D.M.G., Bos-Kuijpers, M.H.M., Kaufmann, W.S.H., Lammers, J.H.C.M., O'Callaghan, J.P., Pakkenberg, B., Pelgrim, M.T.M., Waalkens-Berendsen, I.D.H., Waanders, M.M., Gundersen, H.J. (2005a). Regulatory developmental neurotoxicity testing: A model study focusing on conventional neuropathology endpoints and other perspectives. Environ. Toxicol. Pharmacol. 19:745-755. |
|
77. |
De Groot, D.M.G., Hartgring, S., van de Horst, L., Moerkens, M., Otto, M., Bos-Kuijpers, M.H.M., Kaufmann, W.S.H., Lammers, J.H.C.M., O'Callaghan, J.P., Waalkens-Berendsen, I.D.H., Pakkenberg, B., Gundersen, H.J. (2005b). 2D and 3D assessment of neuropathology in rat brain after prenatal exposure to methylazoxymethanol, a model for developmental neurotoxicity. Reprod. Toxicol. 20:417-432. |
|
78. |
Rodier, P.M., Gramann, W.J. (1979). Morphologic effects of interference with cell proliferation in the early fetal period. Neurobehav. Toxicol. 1:129-135. |
|
79. |
Howard, C.V., Reed, M.G. (1998). Unbiased Stereology:Three-Dimensional Measurement in Microscopy. Springer-Verlag, Nova Iorque. |
|
80. |
Hyman, B.T., Gomez-Isla, T., Irizarry, M.C. (1998). Stereology: A practical primer for neuropathology. J. Neuropathol. Exp. Neurol. 57:305-310. |
|
81. |
Korbo, L., Andersen, B.B., Ladefoged, O., Møller, A. (1993). Total numbers of various cell types in rat cerebellar cortex estimated using an unbiased stereological method. Brain Res. 609:262-268. |
|
82. |
Schmitz, C. (1997). Towards more readily comprehensible procedures in disector stereology. J. Neurocytol. 26:707-710. |
|
83. |
West, M.J. (1999). Stereological methods for estimating the total number of neurons and synapses: Issues of precision and bias. Trends Neurosci. 22:51-61. |
|
84. |
Schmitz, C., Hof, P.R. (2005). Design-based stereology in neuroscience. Neuroscience 130:813-831. |
|
85. |
Gavin, C.E., Kates, B., Gerken, L.A., Rodier, P.M. (1994). Patterns of growth deficiency in rats exposed in utero to undernutrition, ethanol, or the neuroteratogen methylazoxymethanol (MAM). Teratology 49:113-121. |
|
86. |
Ohno, M., Aotani, H., Shimada, M. (1995). Glial responses to hypoxic/ischemic encephalopathy in neonatal rat cerebrum. Develop. Brain Res. 84:294-298. |
|
87. |
Jensen K.F., Catalano S.M. (1998). Brain morphogenesis and developmental neurotoxicology. Em: Handbook of Developmental Neurotoxicology. Slikker, Jr. W., Chang, L.W. (editores). Academic Press, Nova Iorque, p. 3-41. |
|
88. |
Ikonomidou, C., Bosch, F., Miksa, M., Bittigau, P., Vöckler, J., Dikranian, K., Tenkova, T.I., Stefovska, V., Turski, L., Olney, J.W. (1999). Blockade of NMDA receptors and apoptotic neurodegeneration in the developing brain. Science 283:70-74. |
|
89. |
Ikonomidou, C., Bittigau, P., Ishimaru, M.J., Wozniak, D.F., Koch, C., Genz, K., Price, M.T., Sefovska, V., Hörster, F., Tenkova, T., Dikranian, K., Olney, J.W. (2000). Ethanol-induced apoptotic degeneration and fetal alcohol syndrome. Science 287:1056-1060. |
|
90. |
Friede, R. L. (1989). Developmental Neuropathology. Segunda edição. Springer-Verlag, Berlim. |
|
91. |
House, D.E., Berman, E., Seeley, J.C., Simmons, J.E. (1992). Comparison of open and blind histopathologic evaluation of hepatic lesions. Toxicol. Let. 63:127-133. |
|
92. |
Tilson, H.A., MacPhail, R.C., Crofton, K.M. (1996). Setting exposure standards: a decision process. Environ. Health Perspect., 104:401-405. |
|
93. |
US EPA (2005). Guidelines for Carcinogen Risk Assessment. US EPA NCEA-F-0644A. |
|
94. |
US EPA (1996). Guidelines for Reproductive Toxicity Risk Assessment. Federal Register 61(212):56274-56322. |
|
95. |
Danish Environmental Protection Agency (1995). Neurotoxicology. Review of Definitions, Methodology, and Criteria. Miljøprojekt n.o 282. Ladefoged, O., Lam, H.R., Østergaard, G., Nielsen, E., Arlien-Søborg, P. |
|
96. |
Muller, K.E., Barton, C.N., Benignus, V.A. (1984). Recommendations for appropriate statistical practice in toxicologic experiments. Neurotoxicology 5:113-126. |
|
97. |
Gad, S.C. (1989). Principles of screening in toxicology with special emphasis on applications to Neurotoxicology. J. Am. Coll. Toxicol. 8:21-27. |
|
98. |
Abby, H., Howard, E. (1973). Statistical procedures in developmental studies on a species with multiple offspring. Dev. Psychobiol. 6:329-335. |
|
99. |
Haseman, J.K., Hogan, M.D. (1975). Selection of the experimental unit in teratology studies. Teratology 12:165-172. |
|
100. |
Holson, R.R., Pearce, B. (1992). Principles and pitfalls in the analysis of prenatal treatment effects in multiparous species. Neurotoxicol. Teratol. 14:221-228. |
|
101. |
Nelson, C.J., Felton, R.P., Kimmel, C.A., Buelke-Sam, J., Adams, J. (1985). Collaborative Behavioral Teratology Study: Statistical approach. Neurobehav. Toxicol. Teratol. 7:587-90. |
|
102. |
Crofton, K.M., Makris, S.L., Sette, W.F., Mendez, E., Raffaele, K.C. (2004). A qualitative retrospective analysis of positive control data in developmental neurotoxicity studies. Neurotoxicol. Teratol. 26:345-352. |
|
103. |
Bolon, B., Garman, R., Jensen, K., Krinke, G., Stuart, B., e um grupo de trabalho ad hoc do Scientific and Regulatory Policy Committee da STP. (2006). A 'best practices' approach to neuropathological assessment in developmental neurotoxicity testing — for today. Toxicol. Pathol. 34:296-313. |
|
104. |
Tamura, R.N., Buelke-Sam, J. (1992). The use of repeated measures analysis in developmental toxicology studies. Neurotoxicol. Teratol. 14(3):205-210. |
|
105. |
Tukey, J.W., Ciminera, J.L., Heyse, J.F. (1985). Testing the statistical certainty of a response to increasing doses of a drug. Biometrics 41:295-301. |
|
106. |
Crofton, K.M., Foss, J.A., Haas, U., Jensen, K., Levin, E.D., Parker, S.P. (2008). Undertaking positive control studies as part of developmental neurotoxicity testing: report from the ILSI Research Foundation/Risk Science Institute expert working group on neurodevelopmental endpoints. Neurotoxicology and Teratology 30(4):266-287. |
|
107. |
Raffaele, K.C., Fisher, E., Hancock, S., Hazelden, K., Sobrian, S.K. (2008). Determining normal variability in a developmental neurotoxicity test: report from the ILSI Research Foundation/Risk Science Institute expert working group on neurodevelopmental endpoints. Neurotoxicology and Teratology 30(4):288-325. |
|
108. |
Holson, R.R., Freshwater, L., Maurissen, J.P.J., Moser, V.C., Phang, W. (2008). Statistical issues and techniques appropriate for developmental neurotoxicity testing: a report from the ILSI Research Foundation/Risk Science Institute expert working group on neurodevelopmental endpoints. Neurotoxicology and Teratology 30(4):326-348. |
|
109. |
Tyl, R.W., Crofton, K.M., Moretto, A., Moser, V.C., Sheets, L.P., Sobotka, T.J. (2008). Identification and interpretation of developmental neurotoxicity effects: a report from the ILSI Research Foundation/Risk Science Institute expert working group on neurodevelopmental endpoints. Neurotoxicology and Teratology 30(4):349-381. |
Figura 1
Diagrama de resumo dos ensaios funcionais e comportamentais, da avaliação neuropatológica e da pesagem cerebral. Baseia-se na descrição constante dos pontos 13 a 15. O apêndice 1 contém exemplos da repartição dos animais

Apêndice 1
|
1. |
Descrevem-se a seguir e resumem-se em quadros alguns exemplos de repartição dos animais. Estes exemplos visam ilustrar que os animais estudados podem ser integrados de diversas maneiras nos vários modelos de ensaio. |
Exemplo 1
|
2. |
Utilizam-se 20 crias de cada sexo por nível de dose (um macho e uma fêmea de cada ninhada) nos testes da ontogenia comportamental anteriores ao desmame. Destes animais, eutanasiam-se 10 crias de cada sexo por nível de dose (um macho ou uma fêmea de cada ninhada) 22 dias após o nascimento. Remove-se o cérebro dos animais eutanasiados, após o que se pesa cada cérebro e se prepara o mesmo para a avaliação histopatológica. Registam-se ainda os dados ponderais dos cérebros não fixados dos restantes 10 machos e 10 fêmeas por nível de dose. |
|
3. |
Utilizam-se outros 20 animais de cada sexo por nível de dose (um macho e uma fêmea de cada ninhada) nos testes funcionais/comportamentais posteriores ao desmame (exames clínicos aprofundados, atividade motora, sobressalto auditivo e ensaio da função cognitiva na adolescência) e na determinação da idade da maturidade sexual. Destes, no termo do estudo (aproximadamente 70 dias após o nascimento), anestesiam-se 10 dos animais de cada sexo expostos a cada nível de dose (um macho ou uma fêmea de cada ninhada) e fixam-se por perfusão. Após nova fixação in situ, remove-se o cérebro de cada um destes animais e prepara-se o mesmo para a avaliação neuropatológica. |
|
4. |
Utilizam-se outras 20 crias de cada sexo por nível de dose (um macho e uma fêmea de cada ninhada) nos testes da função cognitiva em adultos jovens (por exemplo, 60 a 70 dias após o nascimento). Destes, no termo do estudo, eutanasiam-se 10 dos animais de cada sexo expostos a cada nível de dose (um macho ou uma fêmea de cada ninhada), removem-se os seus cérebros e pesam-se estes últimos. |
|
5. |
Os restantes 20 animais de cada sexo por grupo ficam de reserva para outros testes eventualmente a realizar. Quadro 1
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Exemplo 2
|
6. |
Utilizam-se 20 crias de cada sexo por nível de dose (um macho e uma fêmea de cada ninhada) nos testes de ontogenia comportamental anteriores ao desmame. Destes animais, eutanasiam-se 10 crias de cada sexo por nível de dose (um macho ou uma fêmea de cada ninhada) 11 dias após o nascimento. Remove-se o cérebro dos animais eutanasiados, após o que se pesa cada cérebro e se prepara o mesmo para a avaliação histopatológica. |
|
7. |
Utilizam-se outros 20 animais de cada sexo por nível de dose (um macho e uma fêmea de cada ninhada) nos exames posteriores ao desmame (exames clínicos aprofundados, atividade motora, determinação da idade da maturidade sexual e funções motoras e sensoriais). Destes, no termo do estudo (aproximadamente 70 dias após o nascimento), anestesiam-se 10 dos animais de cada sexo expostos a cada nível de dose (um macho ou uma fêmea de cada ninhada) e fixam-se por perfusão. Após nova fixação in situ, remove-se o cérebro de cada um destes animais e prepara-se o mesmo para a avaliação neuropatológica. |
|
8. |
Utilizam-se 10 crias de cada sexo por nível de dose (um macho ou uma fêmea de cada ninhada) nos testes da função cognitiva na adolescência e em adultos jovens. Utilizam-se animais diferentes nos testes da função cognitiva efetuados 23 dias após o nascimento e a adultos jovens. No termo do estudo, eutanasiam-se 10 animais de cada sexo em cada grupo de animais adultos ensaiados, removem-se os seus cérebros e pesam-se estes últimos. |
|
9. |
Os restantes 20 animais de cada sexo por grupo, não selecionados para os testes, são eutanasiados e eliminados quando do desmame. Quadro 2
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Exemplo 3
|
10. |
Utilizam-se 20 crias de cada sexo por nível de dose (um macho e uma fêmea de cada ninhada) na determinação dos pesos cerebrais e na avaliação neuropatológica realizadas 11 dias após o nascimento. Destes animais, eutanasiam-se 10 crias de cada sexo por nível de dose (um macho ou uma fêmea de cada ninhada) 11 dias após o nascimento. Remove-se-lhes o cérebro, após o que se pesa cada cérebro e se prepara o mesmo para a avaliação histopatológica. Registam-se ainda os dados ponderais dos cérebros não fixados dos restantes 10 machos e 10 fêmeas por nível de dose. |
|
11. |
Utilizam-se outros 20 animais de cada sexo por nível de dose (um macho e uma fêmea de cada ninhada) nos testes de ontogenia comportamental (atividade motora), nas avaliações após o desmame (atividade motora e determinação da idade de maturidade sexual) e no ensaio da função cognitiva na adolescência. |
|
12. |
Utilizam-se outros 20 animais de cada sexo por nível de dose (um macho e uma fêmea de cada ninhada) nos testes das funções motoras e sensoriais (sobressalto auditivo) e para os exames clínicos aprofundados. Destes, no termo do estudo (aproximadamente 70 dias após o nascimento), anestesiam-se 10 dos animais de cada sexo expostos a cada nível de dose (um macho ou uma fêmea de cada ninhada) e fixam-se por perfusão. Após nova fixação in situ, remove-se o cérebro de cada um destes animais, pesa-se e prepara-se o mesmo para a avaliação neuropatológica. |
|
13. |
Utilizam-se outras 20 crias de cada sexo por nível de dose (um macho e uma fêmea de cada ninhada) nos testes da função cognitiva em adultos jovens. Destes, no termo do estudo, eutanasiam-se 10 animais de cada sexo por grupo (um macho ou uma fêmea de cada ninhada), removem-se os seus cérebros e pesam-se estes últimos. Quadro 3
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Apêndice 2
Definições
Produto químico: substância ou mistura.
Produto químico em estudo: qualquer substância ou mistura à qual seja aplicado este método de ensaio.
B.54. BIOENSAIO UTEROTRÓFICO EM ROEDORES: ENSAIO DE DESPISTAGEM A CURTO PRAZO DE PROPRIEDADES ESTROGÉNICAS
INTRODUÇÃO
1. Este método é equivalente ao Test Guideline TG 440 (2007) da OCDE. Em 1998, a OCDE deu início a uma ação prioritária com vista à revisão das orientações existentes e à elaboração de novas orientações para despistagem e ensaio de potenciais desreguladores do sistema endócrino (1). Um dos elementos dessa ação foi a elaboração de um Test Guideline para o bioensaio uterotrófico em roedores. Este bioensaio foi, em seguida, objeto de um vasto programa de validação, que incluiu a compilação de um documento-base pormenorizado (2)(3) e a realização de amplos estudos intra e interlaboratoriais destinados a examinar a pertinência e a reprodutibilidade do bioensaio com um estrogéneo de referência muito ativo, agonistas fracos de recetores de estrogénios, um antagonista forte de recetores de estrogénios e um produto químico de referência negativo (4)(5)(6)(7)(8)(9). O presente método B.54 constitui o corolário da experiência adquirida durante o programa de validação do ensaio, bem como dos resultados nele obtidos relativamente aos agonistas de estrogénios.
2. O bioensaio uterotrófico constitui um ensaio de despistagem a curto prazo que remonta aos anos 30 (27)(28) e foi pela primeira vez normalizado para efeitos de despistagem, por uma comissão de peritos, em 1962 (32)(35). O ensaio baseia-se no aumento do peso uterino, designado por “resposta uterotrófica” (ver a referência 29). Avalia-se a capacidade de um determinado produto químico de induzir uma atividade biológica análoga à dos agonistas ou antagonistas dos estrogéneos naturais (por exemplo, o 17ß-estradiol), mas o ensaio é muito menos utilizado na deteção de antagonistas do que de agonistas. O útero responde aos estrogéneos de duas maneiras. A resposta inicial é um aumento do peso, devido à absorção de água. Segue-se um aumento de peso causado pelo crescimento dos tecidos (30). A reação do útero na fêmea de rato é comparável à que se verifica na ratinha.
3. Este bioensaio serve de ensaio de despistagem in vivo, enquadrando-se a sua aplicação no contexto do Quadro conceptual da OCDE para ensaio e avaliação de produtos químicos desreguladores do sistema endócrino (apêndice 2). Nesse quadro conceptual, o bioensaio uterotrófico inscreve-se no nível 3, enquanto ensaio in vivo que fornece dados acerca de um único mecanismo endócrino, a atividade estrogénica.
4. O bioensaio uterotrófico destina-se a ser incluído numa série de ensaios in vitro e in vivo que visam identificar produtos químicos suscetíveis de interagir com o sistema endócrino, tendo em vista a avaliação dos riscos para a saúde humana e para o ambiente. No programa de validação realizado pela OCDE, utilizaram-se agonistas fortes e fracos de estrogénios para avaliar a eficácia do ensaio na identificação de produtos químicos estrogénicos (4)(5)(6)(7)(8). Foi assim possível comprovar a sensibilidade do protocolo experimental aos agonistas de estrogénios, bem como a boa reprodutibilidade intra e interlaboratorial do mesmo.
5. No que respeita a produtos químicos “negativos”, apenas foi incluído no programa de validação um produto químico de referência “negativo” já identificado como tal num ensaio uterotrófico e em ensaios in vitro de recetores e de ligação a recetores. No entanto, avaliaram-se outros dados experimentais, não relacionados com o programa de validação da OCDE, que corroboraram a especificidade do bioensaio uterotrófico para a despistagem de agonistas de estrogénios (16).
CONSIDERAÇÕES INICIAIS E LIMITAÇÕES
6. Os agonistas e antagonistas de estrogénios agem como ligandos dos recetores dos estrogénios a e b e podem ativar ou inibir, respetivamente, a atividade de transcrição dos recetores. Daí podem advir consequências negativas para a saúde, nomeadamente efeitos com incidências na reprodução e no desenvolvimento. É, pois, necessário poder determinar e avaliar rapidamente a capacidade agonista ou antagonista de estrogénios revelada pelos produtos químicos. Embora constituam informações úteis, a afinidade de um ligando por um recetor de estrogénios e a ativação da transcrição de genes relatores in vitro são apenas dois dos fatores determinantes de eventuais perigos. Outros fatores determinantes possíveis são a ativação ou desativação metabólicas após a entrada no organismo, a distribuição do produto químico pelos tecidos-alvo e a eliminação do produto químico do organismo, as quais, pelo menos em parte, dependem da via de administração e do produto químico em estudo. Daqui decorre a necessidade de despistar a eventual atividade dos produtos químicos in vivo em condições adequadas, a não ser que as características ADME (absorção, distribuição, metabolismo e eliminação) do produto químico já forneçam informações suficientes. Os tecidos uterinos reagem com um crescimento rápido e vigoroso à estimulação com estrogénios, designadamente em roedores de laboratório, cujo ciclo estral dura aproximadamente 4 dias. Os roedores, nomeadamente o rato, também são amplamente utilizados nos estudos de toxicidade para caracterização de perigos. O útero dos roedores é, portanto, um órgão-alvo adequado para a despistagem in vivo de agonistas e antagonistas de estrogénios.
7. O presente método baseia-se nos protocolos utilizados no estudo de validação realizado pela OCDE que se revelaram fiáveis e reprodutíveis nos estudos intra e interlaboratoriais efetuados (5)(7). Estão disponíveis dois métodos: um método no qual se utilizam fêmeas adultas ovariectomizadas e um método no qual se utilizam fêmeas imaturas não-ovariectomizadas. O programa de validação realizado pela OCDE mostrou que a sensibilidade e a reprodutibilidade dos dois métodos são comparáveis. Todavia, o método que utiliza fêmeas imaturas, cujo eixo hipotálamo-hipófise-gónadas está intacto, é um pouco menos específico, embora o seu campo de investigação seja mais amplo do que o do método que utiliza fêmeas ovariectomizadas, pois o método com fêmeas imaturas é sensível aos produtos químicos que interagem com o referido eixo e não unicamente aos produtos químicos que interagem com os recetores de estrogénios. No rato, o eixo hipotálamo-hipófise-gónadas está funcional aproximadamente aos 15 dias de idade. Antes desta idade, não é possível acelerar a puberdade com tratamentos como o da GnRH. Com a aproximação da puberdade, antes da abertura da vagina, as fêmeas têm vários ciclos silenciosos, sem abertura vaginal nem ovulação, mas que se manifestam por flutuações hormonais. Se um produto químico estimular direta ou indiretamente o eixo hipotálamo-hipófise-gónadas, a puberdade e a ovulação são antecipadas e dá-se uma aceleração da abertura da vagina. Os produtos químicos que agem sobre o referido eixo produzem este efeito, mas algumas dietas, com níveis de energia metabolizável mais elevados do que o normal, embora não sejam estrogénicas, estimulam o crescimento e aceleram a abertura vaginal. Esses produtos químicos não induzem respostas uterotróficas em animais adultos ovariectomizados, cujo eixo hipotálamo-hipófise-gónadas não funciona.
8. Numa perspetiva de bem-estar animal, deve privilegiar-se o método que utiliza animais imaturos, para evitar o pré-tratamento cirúrgico dos mesmos e a eventual impossibilidade de utilizar os animais que evidenciem sinais de entrada no ciclo estral (ver o ponto 30).
9. A resposta uterotrófica não é inteiramente de origem estrogénica, pois determinados produtos químicos, que não são agonistas nem antagonistas de estrogénios, podem igualmente provocá-la. Por exemplo, doses relativamente elevadas de progesterona, testosterona ou diversas progestinas sintéticas também podem induzir uma estimulação (30). Pode efetuar-se uma análise histológica das respostas obtidas para pesquisar sinais de queratinização ou de cornificação da vagina (30). Independentemente da possível origem da resposta, um resultado positivo num bioensaio uterotrófico deve normalmente dar lugar a uma pesquisa mais aprofundada. O poder estrogénico pode ser confirmado por ensaios in vitro, como os ensaios de ligação aos recetores dose estrogénios e os ensaios de ativação da transcrição, ou por outros ensaios in vivo, como o ensaio de puberdade em fêmeas.
10. Atendendo a que o bioensaio uterotrófico constitui um ensaio de despistagem in vivo, o método de validação adotado favoreceu o bem-estar animal e optou por uma estratégia de ensaio por etapas. Para o efeito, o esforço concentrou-se na validação rigorosa da reprodutibilidade e da sensibilidade da despistagem da atividade estrogénica — o principal problema associado a muitos produtos químicos –, mas pouco trabalho foi consagrado à componente antiestrogénica do ensaio. Dado que o número de produtos químicos claramente antiestrogénicos (com atividade não dissimulada por atividade estrogénica) é muito reduzido, só foi ensaiado um produto químico antiestrogénico (com forte atividade). O presente método é, pois, dedicado ao protocolo estrogénico, enquanto o protocolo que descreve o modo antagonista do ensaio consta de um documento de orientações (37). A reprodutibilidade e a sensibilidade do ensaio para produtos químicos com atividade puramente antiestrogénica serão definidas com maior clareza ulteriormente, depois da aplicação regular do correspondente protocolo durante algum tempo e de se terem identificado mais produtos químicos com este modo de ação.
11. Pressupõe-se que todos os procedimentos com animais respeitarão as normas locais de manipulação de animais. As descrições, neste protocolo, dos cuidados a ter com os animais e do tratamento a dar aos animais são normas mínimas, prevalecendo, se for mais estrita, a regulamentação local, como a decorrente da Diretiva 2010/63/UE do Parlamento Europeu e do Conselho, de 22 de setembro de 2010, relativa à proteção dos animais utilizados para fins científicos (38). Para mais orientações sobre o tratamento ético de animais, ver o documento da OCDE com a referência 25.
12. Tal como em qualquer ensaio com animais vivos, é essencial que, antes de se iniciar este ensaio, se tenha a certeza de que os dados pretendidos são realmente necessários. Seguem-se dois exemplos de situações nas quais podem sê-lo:
|
— |
potencial de exposição elevado (nível 1 do quadro conceptual descrito no apêndice 2) ou indícios de atividade estrogénica (nível 2) que justifiquem a necessidade de investigar o ocorrência de tais efeitos in vivo; |
|
— |
efeitos reveladores de atividade estrogénica em ensaios in vivo dos níveis 4 ou 5, que justifiquem a necessidade de investigar se os efeitos em causa se relacionam com um mecanismo estrogénico não elucidável em ensaios in vitro. |
13. No apêndice 1 definem-se alguns conceitos utilizados neste método.
PRINCÍPIO DO MÉTODO
14. A sensibilidade do bioensaio uterotrófico assenta num dispositivo experimental constituído por animais cujo eixo hipotálamo-hipófise-ovários não funciona, daí resultando baixos níveis endógenos de estrogéneos em circulação. Esta situação proporciona pesos uterinos baixos à partida e maximiza a amplitude possível da resposta aos estrogénios administrados. Os roedores fêmeas preenchem estas condições nos seguintes estados de sensibilidade aos estrogénios:
|
i) |
Fêmeas imaturas após o desmame, mas antes da puberdade; |
|
ii) |
Fêmeas adultas jovens ovariectomizadas, transcorrido um período adequado para a regressão dos tecidos uterinos. |
15. Administra-se diariamente o produto químico em estudo por sonda esofágica ou injeção subcutânea. Administram-se doses crescentes constantes do produto químico em estudo a, pelo menos, dois grupos de animais (ver orientações no ponto 35), expondo cada grupo a um nível de dose. O período de administração é de três dias consecutivos, no caso do método com fêmeas imaturas, e de, pelo menos, três dias consecutivos, no caso do método com fêmeas adultas ovariectomizadas. Autopsiam-se os animais cerca de 24 horas após lhes ser administrada a última dose. Para o despiste de agonistas de estrogénios, compara-se o peso uterino médio dos grupos de animais expostos com o do grupo de animais aos quais foi administrado apenas o veículo, para determinar se ocorreu algum aumento com significado estatístico. Um aumento com significado estatístico do peso uterino médio de um grupo de animais expostos ao produto químico em estudo é revelador de uma resposta positiva neste bioensaio.
DESCRIÇÃO DO MÉTODO
Escolha da espécie animal
16. Podem utilizar-se as estirpes habituais de roedores de laboratório, por exemplo as estirpes de ratos Sprague-Dawley e Wistar a que se recorreu na validação. Não devem utilizar-se estirpes cuja resposta uterina seja comprovadamente menor ou no caso de se suspeitar disso. O laboratório deve demonstrar a sensibilidade da estirpe utilizada conforme se explica nos pontos 26 e 27.
17. Desde os anos 30 que se têm utilizado fêmeas de rato e ratinhas nos bioensaios uterotróficos. Os estudos de validação realizados pela OCDE foram-no apenas em ratos, com base no pressuposto da equivalência entre essas espécies e, portanto, na boa probabilidade de que a utilização de apenas uma delas seria suficiente para a validação a nível mundial, permitindo economizar recursos e preservar animais. O rato é a espécie escolhida na maior parte dos estudos relativos à reprodução e ao desenvolvimento. Atendendo a que se dispõe de uma vasta base de dados históricos relativos ao ratinho e, portanto, para alargar o campo de aplicação do bioensaio uterotrófico nos roedores à utilização dessa espécie, efetuou-se um estudo de validação complementar limitado em ratinhas (16). Mantendo o propósito inicial de economizar recursos e preservar animais, adotou-se uma abordagem comparativa, no âmbito da qual se utilizou um número reduzido de produtos químicos, participou um pequeno número de laboratórios e as amostras não foram codificadas. Esse estudo de validação comparativo mostrou que existe uma boa correspondência, qualitativa e quantitativa, entre os dados obtidos nos bioensaios uterotróficos realizados em ratinhas adultas jovens ovariectomizadas e os dados obtidos em bioensaios análogos com fêmeas de rato. Quando os resultados do bioensaio uterotrófico se inserirem numa etapa preliminar de um estudo a longo prazo, torna-se possível utilizar animais da mesma estirpe e proveniência em ambos os estudos. A abordagem comparativa circunscreveu-se a ratinhas ovariectomizadas e o respetivo relatório não contém um conjunto de dados suficientemente sólido para validar o modelo que utiliza animais imaturos, razão pela qual a aplicação desse modelo a ratinhas não é abrangida pelo presente método.
18. Conclui-se que, em alguns casos, podem ser utilizadas ratinhas em vez de fêmeas de rato. Nessa eventualidade, haverá que justificar a utilização dessa outra espécie, com base em critérios toxicológicos, farmacocinéticos e/ou outros. Poderá ser necessário alterar o protocolo se forem utilizadas ratinhas. Por exemplo, o consumo alimentar do ratinho, relativamente ao peso corporal, é superior ao do rato, pelo que o teor de fitoestrogénios dos alimentos dos ratinhos deve ser menor do que o teor de fitoestrogénios dos alimentos dos ratos (9)(20)(22).
Condições de alojamento e de alimentação
19. Todos os procedimentos devem respeitar as normas locais de manipulação de animais de laboratório. As descrições, neste protocolo, dos cuidados a ter com os animais e do tratamento a dar aos animais são normas mínimas, prevalecendo, se for mais estrita, a regulamentação local, como a decorrente da Diretiva 2010/63/UE do Parlamento Europeu e do Conselho, de 22 de setembro de 2010, relativa à proteção dos animais utilizados para fins científicos (38). A temperatura do biotério deve ser de 22 °C (± 3 °C). A humidade relativa não deve ser inferior a 30 % e, de preferência, não deve exceder 70 %, exceto durante a limpeza do biotério. Idealmente, deve procurar manter-se a humidade relativa entre 50 % e 60 %. A iluminação deve ser artificial, com uma sequência de 12 horas de luz seguidas de 12 horas de escuridão.
20. Os animais devem receber ad libitum uma dieta de laboratório e água de beber. Os animais adultos jovens podem ser alojados individualmente ou em pequenos grupos de não mais de três por gaiola. Por serem juvenis, recomenda-se que os animais imaturos sejam alojados em gaiolas coletivas.
21. Sabe-se que teores elevados de fitoestrogénios nas dietas de laboratório aumentam o peso uterino em roedores num grau passível de interferir com os bioensaios uterotróficos (13)(14)(15). Teores elevados de fitoestrogénios nas dietas de laboratório e níveis elevados de energia metabolizável nas mesmas também podem induzir puberdade precoce, se forem utilizados animais imaturos. A presença de fitoestrogénios resulta, sobretudo, da incorporação de produtos de soja e de luzerna nas dietas de laboratório, tendo-se verificado que as concentrações de fitoestrogénios variam de lote para lote das dietas de laboratório (23). O peso corporal é uma variável importante, dado que a quantidade de alimentos ingerida está relacionada com este parâmetro. Por conseguinte, a dose de fitoestrogénio proveniente do consumo da mesma dieta pode variar de espécie para espécie e dependem da faixa etária (9). Proporcionalmente ao peso corporal, o consumo de alimentos de fêmeas de rato imaturas pode ser aproximadamente duplo do consumo de alimentos de fêmeas adultas jovens ovariectomizadas. Ainda proporcionalmente ao peso corporal, o consumo de alimentos de ratinhas adultas jovens pode ser aproximadamente quádruplo do consumo de alimentos de fêmeas adultas jovens de rato ovariectomizadas.
22. Os resultados dos bioensaios uterotróficos (9)(17)(18)(19) mostram, porém, que são aceitáveis pequenas quantidades de fitoestrogénios na dieta, as quais não reduzem a sensibilidade do ensaio. A título indicativo, no caso das fêmeas imaturas de rato das estirpes Sprague-Dawley e Wistar, o teor de fitoestrogénios da dieta de laboratório não deve exceder 350 μg de equivalente de genisteína por grama de dieta (6)(9). Uma dieta com tais características deve ser igualmente adequada para o estudo de fêmeas adultas jovens de rato ovariectomizadas, uma vez que, proporcionalmente ao peso corporal, o consumo de alimentos dos adultos jovens é inferior ao dos animais imaturos. Se forem utilizadas ratinhas adultas ovariectomizadas ou fêmeas de rato mais sensíveis aos fitoestrogénios, importará reduzir proporcionalmente os níveis de fitoestrogénios da dieta (20). Por outro lado, diferenças entre dietas ao nível da energia metabolizável disponível podem gerar desfasamentos no início da puberdade (21)(22).
23. Antes de iniciar o estudo, é necessário escolher cuidadosamente uma dieta cujo nível de fitoestrogénios não seja elevado (ver orientações nas referências 6 e 9) e cujo nível de energia metabolizável não seja excessivo, dado que, caso contrário, os resultados podem ser falseados (15)(17)(19)(22)(36). A confirmação de que o dispositivo de ensaio utilizado pelo laboratório satisfaz os critérios de desempenho como se refere nos pontos 26 e 27 constitui uma verificação importante destes dois fatores. A título cautelar, em aplicação das boas práticas de laboratório, deve colher-se, para eventual análise do teor de fitoestrogénios, uma amostra representativa de cada lote de dieta fornecido durante o estudo — por exemplo, no caso de o peso uterino dos animais do grupo de controlo exceder os valores históricos de controlo ou se a resposta ao estrogénio de referência (17α-etinilestradiol) for inadequada. As partes alíquotas constituídas devem ser integradas no estudo e analisadas ou ser congeladas a –20 °C ou conservadas de um modo que permita evitar a decomposição da amostra antes da análise.
24. Algumas camas podem conter produtos químicos estrogénicos ou antiestrogénicos naturais (por exemplo, é sabido que as maçarocas de milho afetam os ciclos das fêmeas de rato e são antiestrogénicas). As camas utilizadas devem conter o mínimo de fitoestrogénios possível.
Preparação dos animais
25. Distribuem-se aleatoriamente animais de laboratório sem indícios de qualquer doença ou anomalia física pelos grupos de controlo e pelos grupos expostos. As gaiolas devem ser dispostas de forma a minimizar possíveis efeitos derivados do seu posicionamento. Os animais devem ser identificados individualmente. Durante o período de aclimatação, os animais imaturos devem permanecer em gaiolas acompanhados das respetivas mães ou de outras fêmeas aleitantes, até serem desmamados. O período de aclimatação antes do início do estudo deve ser de cerca de 5 dias, no caso dos animais adultos jovens e dos animais imaturos acompanhados das respetivas mães ou de outras fêmeas aleitantes. Se os animais imaturos a utilizar já tiverem sido desmamados e não estiverem acompanhados das respetivas mães ou de outras fêmeas aleitantes, pode ser necessário reduzir o período de aclimatação, dado que a administração das doses deve iniciar-se imediatamente após o desmame (ver o ponto 29).
PROCEDIMENTO DE ENSAIO
Verificação da competência técnica do laboratório
26. A competência dos laboratórios pode ser verificada de duas maneiras:
|
— |
Através de uma verificação periódica, por referência a um estudo de base inicial de controlo positivo (ver o ponto 27). Pelo menos de seis em seis meses e sempre que ocorram alterações suscetíveis de influenciar os resultados do ensaio (por exemplo, nova formulação da dieta, mudança do pessoal que faz as dissecações, mudança da estirpe ou do fornecedor dos animais, etc.), deve verificar-se a resposta do dispositivo de ensaio (o modelo animal) a uma dose adequada (baseada no estudo de base inicial de controlo positivo referido no ponto 27) de um estrogénio de referência: o 17a-etinilestradiol (n.o CAS 57-63-6). |
|
— |
Através da utilização de grupos de controlo paralelos, incluindo em cada ensaio um grupo ao qual é administrada uma dose adequada do estrogénio de referência. |
Se o dispositivo não responder como previsto, devem examinar-se as condições experimentais, adaptando-as em conformidade. Recomenda-se, em ambos os casos, que a dose do estrogénio de referência corresponda a DE70-80 (DE: dose eficaz).
27. Estudo de base inicial de controlo positivo — Antes de um determinado laboratório efetuar um estudo no qual aplique o presente método pela primeira vez, é necessário demonstrar a competência técnica do laboratório determinando a resposta do modelo animal a um mínimo de quatro doses de um estrogénio de referência: 17a-etinilestradiol (n.o CAS 57-63-6). Compara-se o efeito no peso uterino com dados históricos atestados (ver a referência 5). Se os resultados deste estudo de base inicial não forem os previstos, devem examinar-se as condições experimentais, adaptando-as em conformidade.
Número e estado dos animais
28. O número mínimo de animais de cada grupo exposto ou de controlo é de seis, tanto no caso do protocolo com fêmeas imaturas como do protocolo com fêmeas adultas ovariectomizadas.
Idade dos animais imaturos
29. É necessário indicar o dia de nascimento dos animais utilizados na variante do bioensaio uterotrófico que recorre a animais imaturos. A administração das doses deve iniciar-se suficientemente cedo, para garantir que, no final da administração do produto químico em estudo, ainda não tenha ocorrido o aumento fisiológico dos estrogénios endógenos associado à puberdade. Por outro lado, há indícios de que os animais muito jovens podem ser menos sensíveis. Para definir a idade ótima, cada laboratório deve atender aos seus dados históricos sobre a maturidade sexual dos animais.
Como regra geral, a administração das doses às fêmeas de rato pode iniciar-se imediatamente após um desmame precoce, 18 dias após o nascimento (para este efeito, o dia do nascimento é o dia 0). A administração de doses a fêmeas de rato deve terminar, de preferência, 21 dias após o nascimento, mas sempre antes do 25.o dia após o nascimento. Com efeito, depois disso, o eixo hipotálamo-hipófise-ovários torna-se funcional e os níveis de estrogénios endógenos podem começar a subir, aumentado concomitantemente os pesos uterinos médios de base e aumentando também os desvios-padrão dos grupos (2)(3)(10)(11)(12).
Ovariectomia
30. A ovariectomia das ratinhas e das fêmeas de rato dos grupos expostos e de controlo deve ocorrer entre a sexta e a oitava semanas de idade. No caso das fêmeas de rato, o período mínimo entre a ovariectomia e o primeiro dia de administração das doses é de 14 dias, a fim de permitir que o útero readquira um peso mínimo de base estável. No caso das ratinhas, o período mínimo entre a ovariectomia e o primeiro dia de administração das doses é de 7 dias. Uma vez que bastam pequenas quantidades de tecido dos ovários para gerar níveis significativos de estrogénios em circulação (3), os animais devem ser testados antes de serem utilizados, mediante a observação de células epiteliais de esfregaços vaginais obtidos em, pelo menos, cinco dias consecutivos (por exemplo, no caso das fêmeas de rato, do décimo ao décimo quarto dia após a ovariectomia). Os animais que apresentarem algum indício de entrada no ciclo estral não devem ser utilizados. Posteriormente, quando da autópsia, deve verificar-se se está presente algum tecido dos ovários nos cotos cicatriciais da ovariectomia. Se assim for, os dados relativos ao animal em causa não devem ser utilizados nos cálculos (3).
31. A ovariectomia é efetuada com o animal apoiado no ventre, depois de convenientemente anestesiado. Efetua-se uma incisão na zona dorsal lateral da parede abdominal, com cerca de 1 cm de extensão, no ponto intermédio entre o limite costal inferior e a crista ilíaca, afastada lateralmente alguns milímetros da orla lateral do músculo lombar. Extraem-se os ovários da cavidade abdominal, depositam-se num campo assético e efetua-se o corte na junção entre o oviduto e o corpo uterino. Depois de confirmar que não se verifica nenhuma hemorragia importante, sutura-se a parede abdominal e fecha-se a abertura na pele com agrafos ou com uma sutura adequada. A figura 1 ilustra esquematicamente os pontos de laqueação. Deve efetuar-se uma analgesia pós-operatória adequada, recomendada por um veterinário com experiência em roedores.
Peso corporal
32. No método da ovariectomização de fêmeas adultas, não existe uma correlação entre o peso corporal e o peso uterino, porque este é afetado por hormonas como os estrogénios, mas não pelos fatores de crescimento que regulam o tamanho do corpo. No modelo que utiliza fêmeas imaturas, em contrapartida, durante o período de maturação o peso corporal está relacionado com o peso uterino (34). Por conseguinte, no início do estudo por aplicação do modelo que recorre a fêmeas imaturas, as diferenças de peso entre os animais utilizados devem ser mínimas, não se desviando mais de 20 % do peso médio. Para isso, o criador deve normalizar o tamanho das ninhadas, a fim de garantir que a progenitura das diversas progenitoras é alimentada sensivelmente do mesmo modo. Integram-se os animais nos grupos expostos e de controlo procedendo a uma distribuição aleatória de pesos, de maneira que o peso corporal médio de cada grupo não difira estatisticamente do peso corporal médio de qualquer outro grupo. Deve evitar-se integrar animais da mesma ninhada no mesmo grupo exposto, tanto quanto possível sem aumentar o número de ninhadas utilizadas no estudo.
Dosagem
33. Normalmente, dois grupos de dosagem e um grupo de controlo são suficientes para determinar se um produto químico pode ter atividade estrogénica in vivo. Por razões de bem-estar animal, privilegia-se, portanto, este modelo. No caso de se pretender traçar uma curva da relação entre a dose administrada e a resposta obtida ou efetuar uma extrapolação para doses mais baixas, são necessários, pelo menos, três grupos de dosagem. No caso de se pretender obter informações que vão além de uma identificação de atividade estrogénica (como, por exemplo, uma estimativa do poder estrogénico), deve ponderar-se um regime de dosagem diferente. Salvo no que respeita à exposição ao produto químico em estudo, os animais do grupo de controlo devem ser tratados do mesmo modo que os animais dos grupos ensaiados. Se for utilizado um veículo para administrar o produto químico em estudo, os animais do grupo de controlo devem receber a mesma quantidade de veículo utilizada nos grupos expostos (ou a maior quantidade de veículo utilizada nos grupos ensaiados, se aquela variar de grupo para grupo).
34. O objetivo, no caso do bioensaio uterotrófico, é selecionar doses que garantam a sobrevivência dos animais e não lhes provoquem efeitos tóxicos nem sofrimento significativos após três dias consecutivos de administração do produto químico, até à dose máxima diária de 1 000 mg/kg. Na escolha das doses devem ter-se em conta os dados eventualmente disponíveis sobre a toxicidade e a (toxico)cinética do produto químico em estudo ou de matérias afins. Para se estabelecer o nível de dose mais elevado, deve atender-se, em primeiro lugar, à DL50 e/ou aos dados sobre toxicidade aguda de que se disponha, a fim de evitar a morte dos animais e de os poupar a grande sofrimento ou aflição (24)(25)(26). A dose mais elevada deve ser a dose máxima tolerada (DMT), podendo também ser aceite um estudo realizado a um nível de dosagem que tenha induzido uma resposta uterotrófica positiva. A título exploratório, aceitam-se geralmente intervalos largos (por exemplo, meia unidade logarítmica, correspondente a um fator de progressão das doses de 3,2, ou mesmo uma unidade logarítmica) entre as doses administradas. Se não se dispuser de dados adequados para o efeito, pode efetuar-se um estudo exploratório, para facilitar a determinação das doses a utilizar.
35. Em alternativa, se for possível estimar a atividade estrogénica de um agonista com base em dados in vitro (ou in silico), esses dados podem ser tidos em conta na escolha das doses. Por exemplo, obtém-se uma estimativa da quantidade do produto químico em estudo que gerará respostas uterotróficas equivalentes às do agonista de referência (etinilestradiol) determinando a correspondente atividade in vitro relativamente à do etinilestradiol. A dose mais elevada a ensaiar resultará da multiplicação dessa dose equivalente por um fator adequado (10 ou 100, por exemplo).
Estudo exploratório
36. Se necessário, pode efetuar-se um estudo exploratório preliminar com poucos animais. Para o efeito, pode utilizar-se o documento de orientações n.o 19 da OCDE (25), que define os sinais clínicos da toxicidade ou do sofrimento provocados em animais. Se for possível fazê-lo nesse estudo exploratório após três dias de administração das doses, procede-se à excisão e pesagem dos úteros cerca de 24 horas após a administração da última dose. Estes dados podem ser depois utilizados para afinar o estudo principal (escolha de uma dose máxima e de uma dose inferior aceitáveis e recomendação do número de grupos de dosagem).
Administração das doses
37. Administra-se o produto químico em estudo por sonda esofágica ou por injeção subcutânea. Ao escolher a via de administração, é necessário atender aos aspetos do bem-estar animal e a aspetos toxicológicos como a relação com a via de exposição humana ao produto químico (por exemplo, sonda esofágica para exposição por ingestão e injeção subcutânea para exposição por inalação ou por adsorção cutânea), tendo ainda em conta as propriedades físico-químicas do produto químico em estudo e, em especial, as informações toxicológicas e os dados metabólicos e cinéticos disponíveis (por exemplo, a necessidade de evitar metabolismos de primeira passagem ou melhor eficiência de uma determinada via de administração).
38. Sempre que possível, deve começar por ponderar-se a possibilidade de se utilizarem soluções/suspensões aquosas. Porém, dado que os ligandos de estrogénios ou os precursores metabólicos dos mesmos são geralmente hidrófobos, normalmente utiliza-se uma solução ou suspensão oleosa (por exemplo, em óleo de milho, de amendoim ou de gergelim ou em azeite). No entanto, uma vez que estes óleos têm valores calóricos e teores de gorduras diferentes, o veículo pode influenciar a absorção total de energia metabolizável e, em consequência disso, pode alterar as medições dos parâmetros determinados, designadamente o peso uterino, em especial no protocolo que utiliza fêmeas imaturas (33). Antes da realização do estudo é, portanto, necessário ensaiar o veículo previsto relativamente a grupos de controlo sem veículo. Pode dissolver-se o produto químico em estudo numa quantidade mínima de etanol a 95 % ou de outro solvente adequado, diluindo-se depois até à concentração final no veículo a utilizar nos ensaios. É necessário conhecer as características de toxicidade do solvente e verificá-las num grupo de controlo apenas exposto ao solvente. Se o produto químico em estudo for considerado estável, pode favorecer-se a sua dissolução aquecendo ligeiramente e agitando vigorosamente. Deve determinar-se a estabilidade do produto químico em estudo no veículo utilizado. Se o produto químico em estudo se mantiver estável durante todo o estudo, podem preparar-se uma alíquota inicial do mesmo e, diariamente, as diluições às doses necessárias.
39. A cronologia da administração das doses depende do modelo experimental (ver o ponto 29 para o protocolo com fêmeas imaturas e o ponto 30 para o protocolo com fêmeas adultas ovariectomizadas). No caso das fêmeas de rato imaturas, administra-se o produto químico em estudo três dias consecutivos. No caso das fêmeas de rato ovariectomizadas, também se recomendam exposições de três dias, mas são aceitáveis exposições mais longas, que podem melhorar a deteção de produtos químicos fracamente ativos. No caso das ratinhas ovariectomizadas, três dias de exposição são normalmente suficientes, sem que haja vantagens significativas no prolongamento da mesma até sete dias quando se trate de agonistas fortes de estrogéneos. Todavia, no caso dos estrogéneos fracos, tal não ficou demonstrado no estudo de validação (16), pelo que a administração das doses a ratinhas adultas ovariectomizadas deve prolongar-se por sete dias consecutivos.As doses devem ser administradas à mesma hora do dia e ser adaptadas de modo a manter um nível de dose constante em relação ao peso corporal de cada animal (por exemplo miligramas diários do produto químico em estudo por quilograma de peso corporal. Deve ainda minimizar-se a variabilidade do volume administrado proporcionalmente ao peso corporal, ajustando a concentração das soluções utilizadas de modo que, proporcionalmente ao peso corporal, o volume administrado seja o mesmo para todos os níveis de dose e todas as vias de administração.
40. A administração forçada aos animais através de sonda esofágica deve efetuar-se numa dose diária única, por meio de um tubo estomacal ou de uma cânula de intubação adequada. O volume máximo de líquido que pode ser administrado de cada vez depende do tamanho do animal. Devem respeitar-se as normas locais de manipulação de animais, mas o volume administrado não deve exceder 5 ml por quilograma de peso corporal, salvo no caso das soluções aquosas, em que se permitem 10 ml por quilograma de peso corporal.
41. Se o produto químico em estudo for administrado por injeção subcutânea, deve ser injetada nos animais uma dose diária única. As doses devem ser administradas na região escapular dorsal ou na região lombar, por meio de uma seringa do tipo utilizado no teste da tuberculina (calibre 23 ou 25), dotada de uma agulha esterilizada. É facultativo rapar a pelagem dos animais no local da injeção. Devem registar-se as perdas ou escorrimentos nesse local, bem como todas as administrações incompletas de doses. O volume injetado diariamente a um rato não deve exceder 5 ml por quilograma de peso corporal, divididos por dois locais de injeção, salvo no caso das soluções aquosas, em que se permitem 10 ml por quilograma de peso corporal.
Exames
Exames gerais e clínicos
42. Deve efetuar-se um exame clínico geral pelo menos uma vez por dia; se forem detetados sinais de toxicidade, o exame deve ser mais frequente. De preferência, os exames devem ser efetuados à mesma hora ou às mesmas horas do dia, tendo em atenção o período previsto de efeitos mais acentuados após a administração do produto químico. Devem examinar-se todos os animais de modo a detetar os casos de mortalidade e morbilidade e sinais clínicos gerais como mudanças de comportamento, alterações da pele, da pelagem, dos olhos ou das mucosas e a ocorrência de secreções ou excreções e de reações neurovegetativas (por exemplo lacrimação, horripilação, alterações da dimensão pupilar, respiração anormal, etc.).
Peso corporal e consumo de alimentos
43. Deve pesar-se diariamente cada animal com a aproximação de 0,1 g, começando imediatamente antes do início da exposição, isto é, quando da repartição dos animais pelos grupos. A quantidade de alimentos consumida em cada gaiola durante o período de exposição pode também ser determinada por gaiola, a título facultativo, pesando os distribuidores de alimentos. Os resultados de consumo de alimentos devem ser expressos em gramas diários por animal.
Dissecação e pesagem dos úteros
44. Os animais devem ser eutanasiados 24 h após a última exposição. Idealmente, a ordem das autópsias deve ser aleatória no universo dos grupos, evitando seguir uma ordem crescente ou decrescente das doses, método que poderia afetar subtilmente os resultados. Constitui objetivo deste bioensaio determinar o peso do útero fresco e do útero enxuto. O peso fresco inclui o útero e o fluido luminal; o peso enxuto determina-se depois de pesado e removido o fluido luminal do útero.
45. Antes da dissecação, examina-se a vagina da cada animal imaturo, para determinar em que estado de abertura se encontra. A dissecação começa pela abertura da parede abdominal, com início na sínfise púbica. Em seguida, separam-se os cornos uterinos e os ovários (se presentes) da parede abdominal dorsal. Separam-se também a bexiga urinária e os ureteres das faces laterais e ventral do útero e da vagina. Libertam-se as aderências fibrosas entre o reto e a vagina, até ser possível identificar a junção do orifício vaginal e da pele perineal. Separam-se o útero e a vagina do resto do corpo efetuando uma incisão na parede vaginal imediatamente acima da junção com a pele perineal, conforme se ilustra na figura 2. Retira-se o útero do corpo cortando cuidadosamente a ligação do mesentério uterino ao longo da face dorsal lateral de cada corno uterino. Uma vez extraído o útero do corpo, a manipulação destes tecidos deve ser suficientemente rápida para evitar que os mesmos sequem. A perda de peso por secagem adquire maior importância no caso de tecidos pequenos, como o útero (23). Se presentes, retiram-se os ovários, por corte ao nível do oviduto, procedendo de modo a evitar perdas de fluido luminal pelos cornos uterinos. Se o animal tiver sido ovariectomizado, deve verificar-se se os cotos cicatriciais contêm vestígios de tecido dos ovários. Devem eliminar-se os restos de gordura e de tecido conjuntivo. Separa-se a vagina do útero imediatamente abaixo do colo do útero, de modo que este fique solidário com o corpo uterino, como se ilustra na figura 2.
46. Deposita-se cada útero num recipiente tarado e com uma marcação individualizada (por exemplo uma placa de Petri ou uma cápsula de pesagem de plástico), continuando a tomar as precauções necessárias para impedir a secagem antes da pesagem (por exemplo, pode pôr-se em cada recipiente um papel de filtro ligeiramente embebido em soro fisiológico). Pesam-se o útero e o fluido luminal com a aproximação de 0,1 mg (peso húmido do útero).
47. Procede-se em seguida à eliminação do fluido luminal de cada útero. Furam-se ambos os cornos uterinos ou cortam-se os mesmos longitudinalmente. Coloca-se cada útero sobre papel de filtro (por exemplo, Whatman n.o 3) ligeiramente humedecido e comprime-se cuidadosamente com um segundo papel de filtro, também ligeiramente humedecido, para remover completamente o fluido luminal. Pesa-se o útero sem o fluido luminal com a aproximação de 0,1 mg (peso enxuto do útero).
48. Pode utilizar-se o peso do útero no termo do ensaio para verificar se a idade adequada das fêmeas de rato imaturas não foi excedida, embora os dados históricos da estirpe de ratos utilizada pelo laboratório sejam determinantes quanto a este aspeto (para a interpretação dos resultados, ver o ponto 56).
Estudos facultativos
49. Após pesagem, pode colocar-se cada útero em formol a 10 % tamponado a pH neutro, para posterior exame histopatológico após coloração com hematoxilina e eosina. Pode examinar-se também a vagina (ver o ponto 9). Podem efetuar-se ainda medições morfométricas do epitélio do endométrio, para comparações quantitativas.
DADOS E RELATÓRIOS
Dados
50. Dados do estudo a fornecer:
|
— |
número de animais no início do ensaio; |
|
— |
número e identidade dos animais que morreram ou foram eutanasiados durante o ensaio e data e hora de cada morte ou eutanásia; |
|
— |
número e identidade dos animais com sinais de toxicidade e descrição dos sinais de toxicidade observados, nomeadamente o momento do seu aparecimento e a duração e intensidade dos efeitos tóxicos detetados; |
|
— |
número e identidade dos animais com lesões e descrição dos tipos de lesões. |
51. Registam-se os dados correspondentes a cada animal no que respeita a peso corporal, peso uterino húmido e peso uterino enxuto. Deve recorrer-se a análises estatísticas unilaterais para determinar se a administração do produto químico em estudo provocou um aumento com significado estatístico (p < 0,05) do peso do útero, provocado por um agonista. Devem efetuar-se análises estatísticas que permitam determinar se o peso húmido e o peso enxuto do útero sofreram alterações em consequência da exposição. Por exemplo, pode efetuar-se uma análise de covariância dos dados (ANCOVA), sendo o peso corporal na autópsia a covariável. Antes da análise dos dados, pode efetuar-se uma transformação logarítmica de estabilização da variância dos dados uterinos. O teste de Dunnett e Hsu pode servir para estabelecer comparações par a par entre cada grupo exposto e os grupos de controlo do veículo e para calcular os intervalos de confiança. Pode recorrer-se a traçados de resíduos estudantizados para detetar eventuais valores aberrantes e avaliar a homogeneidade das variâncias. Aplicaram-se estes métodos no programa de validação realizado pela OCDE, com recurso ao processo dos modelos lineares gerais (PROC GLM) do sistema de análise estatística (SAS Institute, Cary, NC, EUA), versão 8 (6)(7).
52. Informações a constar do relatório final:
Instalações nas quais se realizaram os ensaios:
|
— |
pessoal responsável e responsabilidades de cada um no estudo; |
|
— |
dados do estudo de base inicial de controlo positivo e dados periódicos de controlo positivo (ver os pontos 26 e 27). |
Produto químico em estudo:
|
— |
caracterização dos produtos químicos em estudo; |
|
— |
natureza física e propriedades físico-químicas pertinentes; |
|
— |
método e frequência de preparação de diluições; |
|
— |
dados obtidos sobre a estabilidade; |
|
— |
análises das soluções administradas. |
Veículo:
|
— |
caracterização do veículo utilizado no estudo (natureza, fornecedor e lote); |
|
— |
justificação da escolha do veículo (se não for água). |
Animais estudados:
|
— |
espécie e estirpe utilizadas e justificação das opções tomadas; |
|
— |
fornecedor e instalações específicas do fornecedor; |
|
— |
idades na entrega e datas de nascimento; |
|
— |
no caso dos animais imaturos, se são fornecidos com as mães ou outras fêmeas aleitantes e data do desmame; |
|
— |
elementos relativos ao processo de aclimatação dos animais; |
|
— |
número de animais por gaiola; |
|
— |
método e elementos relativos à identificação de cada animal e de cada grupo. |
Condições de realização dos ensaios:
|
— |
elementos relativos ao processo de aleatorização (método utilizado); |
|
— |
fundamentação da escolha das doses; |
|
— |
elementos relativos à formulação, concentrações obtidas, estabilidade e homogeneidade do produto químico em estudo; |
|
— |
elementos relativos à administração do produto químico em estudo e fundamentação da via de exposição escolhida; |
|
— |
dieta (nome, tipo, fornecedor, composição e, se conhecidos, níveis de fitoestrogénios); |
|
— |
água fornecida (por exemplo, água da torneira ou água filtrada) e modo de fornecimento (tubagem a partir de um reservatório grande, garrafas, etc.); |
|
— |
camas (nome, tipo, fornecedor e composição); |
|
— |
registos das condições de engaiolamento, do período de iluminação, da temperatura e humidade ambientes, da limpeza do biotério; |
|
— |
descrição pormenorizada das autópsias e da pesagem dos úteros; |
|
— |
descrição dos métodos estatísticos utilizados. |
Resultados
Por animal:
|
— |
todos os pesos corporais diários (desde a integração num grupo até à autópsia do animal), com a aproximação de 0,1 g; |
|
— |
idade do animal (em dias, considerando dia 0 o do nascimento) no início da administração do produto químico em estudo; |
|
— |
data e hora da administração de cada dose; |
|
— |
volume e dose calculados administrados e registos de eventuais perdas durante ou depois da administração das doses; |
|
— |
estado diário do animal, incluindo os exames efetuados e os sintomas detetados; |
|
— |
causa presumida de morte (se o animal for encontrado moribundo ou morto durante o estudo); |
|
— |
data e hora da eutanásia e período decorrido desde a administração da última dose; |
|
— |
peso uterino húmido (com a aproximação de 0,1 mg) e eventuais registos de perdas de fluido luminal durante a dissecação e a preparação para a pesagem; |
|
— |
peso uterino enxuto (com a aproximação de 0,1 mg). |
Por grupo de animais:
|
— |
pesos corporais médios diários (com a aproximação de 0,1 mg) e respetivos desvios-padrão (desde a integração nos grupos até às autópsias); |
|
— |
pesos uterinos húmidos médios e pesos uterinos enxutos médios (com a aproximação de 0,1 mg) e respetivos desvios-padrão; |
|
— |
se for determinado, consumo diário de alimentos (calculado em gramas de alimentos consumidos por animal); |
|
— |
resultados das análises estatísticas comparativas dos pesos uterinos húmido e enxuto dos grupos expostos com as determinações correspondentes nos grupos de controlo do veículo; |
|
— |
resultados das análises estatísticas comparativas do peso corporal total e do ganho de peso corporal dos grupos expostos com as determinações correspondentes nos grupos de controlo do veículo. |
53. Resumo das principais orientações deste método de ensaio
|
|
Rato |
Ratinho |
|
Animais |
||
|
Estirpe |
Estirpe habituais de roedores de laboratório |
|
|
Número de animais |
Pelo menos seis animais por grupo de dosagem |
|
|
Número de grupos |
Pelo menos dois grupos expostos (ver orientações no ponto 33) e um grupo de controlo negativo Ver orientações relativas aos grupos de controlo positivo nos pontos 26 e 27 |
|
|
Condições de alojamento e de alimentação |
||
|
Temperatura do biotério |
22 °C ± 3 °C |
|
|
Humidade relativa |
50-60 %; não inferior a 30 % nem superior a 70 % |
|
|
Sequência de luz e de escuridão |
12 horas de luz seguidas de 12 horas de escuridão |
|
|
Dieta e água de beber |
Ad libitum |
|
|
Alojamento |
Individual ou em grupos de não mais de três animais (recomenda-se que os animais imaturos sejam alojados em grupo) |
|
|
Dieta e camas |
Baixo nível de fitoestrogénios recomendado na dieta e nas camas |
|
|
Protocolo |
||
|
Método |
Método (preferido) que recorre a fêmeas imaturas não ovariectomizadas Método que recorre a fêmeas adultas ovariectomizadas |
Método que recorre a fêmeas adultas ovariectomizadas |
|
Idade da exposição dos animais imaturos |
Não antes de 18 dias após o nascimento. Administração das doses a completar antes de transcorridos 25 dias após o nascimento. |
Não se aplica à presente versão do método. |
|
Idade da ovariectomia |
Entre 6 e 8 semanas |
|
|
Idade da exposição dos animais ovariectomizados |
Pelo menos 14 dias entre a ovariectomia e o primeiro dia de administração das doses |
Pelo menos 7 dias entre a ovariectomia e o primeiro dia de administração das doses |
|
Peso corporal |
A variação deste parâmetro deve ser mínima, não devendo exceder 20 % do peso médio |
|
|
Administração das doses |
||
|
Via de administração |
Sonda esofágica ou injeção subcutânea |
|
|
Frequência da administração |
Uma dose diária |
|
|
Volume administrado por sonda esofágica ou por injeção |
≤ 5 ml/kg de peso corporal (ou até 10 ml/kg de peso corporal no caso das soluções aquosas) (injetados em dois locais, no caso da via subcutânea) |
|
|
Duração da administração |
3 dias consecutivos no caso do modelo que recorre a fêmeas imaturas Pelo menos 3 dias consecutivos no caso do modelo que recorre a fêmeas adultas ovariectomizadas |
7 dias consecutivos no caso do modelo que recorre a fêmeas adultas ovariectomizadas |
|
Autópsia |
Cerca de 24 horas após a última dose |
|
|
Resultados |
||
|
Resposta positiva |
Aumento estatisticamente significativo do peso uterino médio (húmido e/ou enxuto) |
|
|
Estrogénio de referência |
17α-etinilestradiol |
|
ORIENTAÇÕES PARA INTERPRETAÇÃO E ACEITAÇÃO DOS RESULTADOS
54. Em geral, deve considerar-se que um estudo de atividade estrogénica deu resultado positivo se, pelo menos no grupo exposto à dose mais elevada, ocorrer um aumento do peso uterino estatisticamente significativo (p< 0,05), comparativamente ao grupo de controlo do solvente. Corrobora ainda um resultado positivo a demonstração de uma relação biologicamente plausível entre a dose e a intensidade da resposta, tendo em atenção que a conjugação dos efeitos estrogénicos e antiestrogénicos do produto químico em estudo pode afetar a forma da curva de resposta à dosagem.
55. A dose máxima tolerada não deve ser excedida, a fim de possibilitar uma interpretação com significado dos dados. Nessa perspetiva, devem avaliar-se cuidadosamente as reduções de peso corporal, os sinais clínicos e outros dados obtidos.
56. Um aspeto importante a ponderar para a aceitação dos dados de um bioensaio uterotrófico é a questão dos pesos uterinos dos animais do grupo de controlo do veículo. Se forem detetados pesos elevados no grupo de controlo, podem ficar comprometidas a sensibilidade do bioensaio e a capacidade de deteção de agonistas muito fracos de estrogénios. As referências bibliográficas e os dados obtidos na validação do bioensaio uterotrófico indicam a possibilidade de ocorrência espontânea de pesos médios elevados nos grupos de controlo, em especial quando se trata de animais imaturos (2)(3)(6)(9). Uma vez que o peso uterino das fêmeas de rato imaturas depende de muitas variáveis, como a estirpe e o peso corporal, não é possível estabelecer um limite superior preciso para esse parâmetro. A título indicativo, se o peso do útero enxuto das fêmeas de rato imaturas de controlo estiver compreendido entre 40 e 45 mg, os resultados devem ser considerados suspeitos; se esse peso exceder 45 mg, o ensaio poderá ter de ser repetido. Todavia, este aspeto tem de ser analisado caso a caso (3)(6)(8). No ensaio de fêmeas de rato adultas, uma ovariectomia incompleta deixará restos de tecidos dos ovários, que podem produzir estrogénios endógenos e atrasar a regressão do peso uterino.
57. A obtenção de pesos uterinos enxutos no grupo de controlo do veículo inferiores a 0,09 % do peso corporal, para fêmeas de rato imaturas, e inferiores a 0,04 % do peso corporal, para fêmeas adultas jovens ovariectomizadas, parece gerar resultados aceitáveis (ver o quadro 31 da referência 2). Se os pesos uterinos dos animais do grupo de controlo excederem estes valores, devem examinar-se vários fatores, nomeadamente a idade dos animais, a qualidade da ovariectomia, a ocorrência de fitoestrogénios na dieta, etc., e deve encarar-se com precaução um resultado negativo no ensaio (nenhuma indicação de atividade estrogénica).
58. Os laboratórios devem conservar dados históricos dos grupos de controlo do veículo. Devem conservar igualmente dados históricos das respostas a estrogénios de referência positivos, como o 17a-etinilestradiol. Os laboratórios podem igualmente ensaiar a resposta a agonistas reconhecidamente fracos de estrogénios. Para garantir que os métodos do laboratório em causa têm sensibilidade suficiente, podem comparar-se todos estes dados com os dados disponíveis (2)(3)(4)(5)(6)(7)(8).
59. No estudo de validação realizado pela OCDE, os pesos uterinos enxutos evidenciaram menos variabilidade do que os pesos uterinos húmidos (6)(7). Todavia, considera-se que uma resposta significativa em qualquer destes parâmetros indica que o produto químico em estudo tem atividade estrogénica.
60. A resposta uterotrófica não é inteiramente de origem estrogénica. Porém, um resultado positivo num bioensaio uterotrófico deve, geralmente, ser interpretado como prova de atividade estrogénica in vivo e, normalmente, deve dar lugar a uma pesquisa mais aprofundada (ver o ponto 9 e o Quadro conceptual da OCDE para ensaio e avaliação de produtos químicos desreguladores do sistema endócrino — apêndice 2).
Figura 1
Desenho esquemático ilustrativo da ablação dos ovários

Começa-se por efetuar uma incisão na zona dorsal lateral da parede abdominal, no ponto intermédio entre o limite costal inferior e a crista ilíaca, afastada lateralmente alguns milímetros da orla lateral do músculo lombar. Localizam-se os ovários na cavidade abdominal. Retiram-se os ovários da cavidade abdominal para um campo assético, laqueia-se o canal entre cada ovário e o útero, para conter a hemorragia, e separam-se os ovários efetuando uma incisão acima do ponto de laqueação situado na junção de cada oviduto com o corno uterino. Depois de confirmar que não se verifica nenhuma hemorragia importante, sutura-se a parede abdominal e fecha-se a abertura na pele, por exemplo com agrafos ou por sutura. Os animais devem poder restabelecer-se e o peso do útero deve poder regredir durante, pelo menos, 14 dias, antes da utilização.
Figura 2
Remoção e preparação dos tecidos uterinos para a pesagem

Começa-se por abrir a parede abdominal na sínfise púbica. Em seguida, separam-se ambos os ovários (se estiverem presentes) e os cornos uterinos da parede abdominal dorsal. Separam-se também a bexiga urinária e os ureteres das faces laterais e ventral do útero e da vagina. Libertam-se as aderências fibrosas entre o reto e a vagina, até ser possível identificar a junção do orifício vaginal e da pele perineal. Separam-se o útero e a vagina do resto do corpo efetuando uma incisão na parede vaginal imediatamente acima da junção com a pele perineal, conforme se ilustra na figura. Retira-se o útero do corpo cortando cuidadosamente a ligação do mesentério uterino ao longo da face dorsal lateral de cada corno uterino. Uma vez extraído o útero do corpo, eliminam-se os restos de gordura e de tecido conjuntivo. Se presentes, retiram-se os ovários por corte ao nível do oviduto, procedendo de modo a evitar perdas de fluido luminal pelos cornos uterinos. Se o animal tiver sido ovariectomizado, deve verificar-se se os cotos cicatriciais contêm vestígios de tecido dos ovários. Separa-se a vagina do útero imediatamente abaixo do colo do útero, de modo que este fique solidário com o corpo uterino, como se ilustra na figura. Pode então pesar-se o útero.
Apêndice 1
DEFINIÇÕES
Atividade antiestrogénica : capacidade do produto químico de suprimir a ação do 17ß-estradiol num mamífero.
Produto químico : substância ou mistura.
Data de nascimento : dia 0 após o nascimento.
Dosagem : termo geral que abrange a dose, a frequência desta e a duração da administração da mesma.
Dose : quantidade de produto químico em estudo administrada. No bioensaio uterotrófico, exprime-se em peso diário do produto químico em estudo por unidade de peso corporal do animal ensaiado (por exemplo mg/kg de peso corporal/dia).
Dose máxima tolerável (DMT) : quantidade máxima do produto químico que, quando introduzida no organismo, não mata os animais em estudo (indicada por DL0) (IUPAC, 1993).
Atividade estrogénica : capacidade do produto químico de agir como o 17ß-estradiol num mamífero.
X dias após o nascimento : número X de dias de vida após o dia de nascimento.
Sensibilidade : proporção dos produtos químicos positivos/ativos que são corretamente classificados pelo ensaio. Constitui uma medida da exatidão dos métodos de ensaio que geram resultados categóricos e é um aspeto importante a ter em conta na avaliação da pertinência de um método de ensaio.
Especificidade : proporção dos produtos químicos negativos/inativos que são corretamente classificados pelo ensaio. Constitui uma medida da exatidão dos métodos de ensaio que geram resultados categóricos e é um aspeto importante a ter em conta na avaliação da pertinência de um método de ensaio.
Produto químico em estudo : qualquer substância ou mistura à qual seja aplicado este método de ensaio.
Uterotrófico : termo usado para designar uma influência positiva no crescimento dos tecidos uterinos.
Validação : processo científico concebido para caracterizar as limitações e as condições operatórias dos métodos de ensaio e para demonstrar a fiabilidade e a pertinência dos mesmos para determinado fim.
Apêndice 2

NOTAS EXPLICATIVAS DO QUADRO CONCEPTUAL
|
Nota 1: |
É possível entrar no quadro e dele sair em qualquer nível, consoante a natureza das informações necessárias para a avaliação dos perigos e dos riscos. |
|
Nota 2: |
No nível 5, os métodos ecotoxicológicos devem integrar parâmetros indicativos dos mecanismos dos efeitos indesejados e dos mecanismos das consequências negativas potenciais nas populações. |
|
Nota 3: |
Se um modelo multimodal abranger vários dos ensaios de parâmetro único, esse modelo substituirá estes últimos. |
|
Nota 4: |
A avaliação de cada produto químico deve basear-se numa análise caso a caso que tenha em conta todas as informações disponíveis, tendo presente a função dos níveis do quadro. |
|
Nota 5: |
Esta versão do quadro não deve ser considerada exaustiva. Nos níveis 3, 4 e 5, integra ensaios já disponíveis ou em validação. Estes últimos são incluídos a título provisório. Logo que estejam afinados e validados, serão formalmente integrados no quadro. |
|
Nota 6: |
Os ensaios integrados no nível 5 não devem considerar-se unicamente de caráter definitivo. Os ensaios em questão contribuem para a avaliação geral dos perigos e dos riscos. |
REFERÊNCIAS
|
1. |
OCDE (1998). Report of the First Meeting of the OECD Endocrine Disrupter Testing and Assessment (EDTA) Task Force, 10 e 11 de março de 1998. ENV/MC/CHEM/RA(98)5. |
|
2. |
OCDE (2003). Detailed Background Review of the Uterotrophic Bioassay: Summary of the Available Literature in Support of the Project of the OECD Task Force on Endocrine Disrupters Testing and Assessment (EDTA) to Standardise and Validate the Uterotrophic Bioassay. OECD Environment, Health and Safety Publications Series on Testing and Assessment No. 38. ENV/JM/MONO(2003)1. |
|
3. |
Owens J.W., Ashby J. (2002). Critical Review and Evaluation of the Uterotrophic Bioassay for the Identification of Possible Estrogen Agonists and Antagonists: In Support of the Validation of the OECD Uterotrophic Protocols for the Laboratory Rodent. Crit. Rev. Toxicol. 32:445-520. |
|
4. |
OCDE (2006). OECD Report of the Initial Work Towards the Validation of the Rodent Uterotrophic Assay — Phase 1. OECD Environmental Health and Safety Publication Series on Testing and Assessment No. 65. ENV/JM/MONO(2006)33. |
|
5. |
Kanno, J., Onyon L., Haseman J., Fenner-Crisp P., Ashby J., Owens W. (2001). The OECD program to validate the rat uterotrophic bioassay to screen compounds for in vivo estrogenic responses: Phase 1. Environ Health Perspect. 109:785-94. |
|
6. |
OCDE (2006). OECD Report of the Validation of the Rodent Uterotrophic Bioassay: Phase 2 — Testing of Potent and Weak Oestrogen Agonists by Multiple Laboratories. OECD Environmental Health and Safety Publication Series on Testing and Assessment No. 66. ENV/JM/MONO(2006)34. |
|
7. |
Kanno J., Onyon L., Peddada S., Ashby J., Jacob E., Owens W. (2003). The OECD program to validate the rat uterotrophic bioassay: Phase Two — Dose Response Studies. Environ. Health Persp. 111:1530-1549. |
|
8. |
Kanno J., Onyon L., Peddada S., Ashby J., Jacob E., Owens W. (2003). The OECD program to validate the rat uterotrophic bioassay: Phase Two — Coded Single Dose Studies. Environ. Health Persp. 111:1550-1558. |
|
9. |
Owens W., Ashby J., Odum J., Onyon L. (2003). The OECD program to validate the rat uterotrophic bioassay: Phase Two — Dietary phytoestrogen analyses. Environ. Health Persp. 111:1559-1567. |
|
10. |
Ogasawara Y., Okamoto S., Kitamura Y., Matsumoto K. (1983). Proliferative pattern of uterine cells from birth to adulthood in intact, neonatally castrated, and/or adrenalectomized mice assayed by incorporation of [I125]iododeoxyuridine. Endocrinology 113:582-587. |
|
11. |
Branham W.S., Sheehan D.M., Zehr D.R., Ridlon E., Nelson C.J. (1985). The postnatal ontogeny of rat uterine glands and age-related effects of 17b-estradiol. Endocrinology 117:2229-2237. |
|
12. |
Schlumpf M., Berger L., Cotton B., Conscience-Egli M., Durrer S., Fleischmann I., Haller V., Maerkel K., Lichtensteiger W. (2001). Estrogen active UV screens. SÖFW-J. 127:10-15. |
|
13. |
Zarrow M.X., Lazo-Wasem E.A., Shoger R.L. (1953). Estrogenic activity in a commercial animal ration. Science 118:650-651. |
|
14. |
Drane H.M., Patterson D.S.P., Roberts B.A., Saba N. (1975). The chance discovery of oestrogenic activity in laboratory rat cake. Fd. Cosmet. Toxicol. 13:425-427. |
|
15. |
Boettger-Tong H., Murphy L., Chiappetta C., Kirkland J.L., Goodwin B., Adlercreutz H., Stancel G.M., Makela S. (1998). A case of a laboratory animal feed with high estrogenic activity and its impact on in vivo responses to exogenously administered estrogens. Environ. Health Perspec. 106:369-373. |
|
16. |
OCDE (2007). Additional data supporting the Test Guideline on the Uterotrophic Bioassay in rodents. OECD Environmental Health and Safety Publication Series on Testing and Assessment No. 67. |
|
17. |
Degen G.H., Janning P., Diel P., Bolt H.M. (2002). Estrogenic isoflavones in rodent diets. Toxicol. Lett. 128:145-157. |
|
18. |
Wade M.G., Lee A., McMahon A., Cooke G., Curran I. (2003). The influence of dietary isoflavone on the uterotrophic response in juvenile rats. Food Chem. Toxicol. 41:1517-1525. |
|
19. |
Yamasaki K., Sawaki M., Noda S., Wada T., Hara T., Takatsuki M. (2002). Immature uterotrophic assay of estrogenic compounds in rats given different phytoestrogen content diets and the ovarian changes in the immature rat uterotrophic of estrogenic compounds with ICI 182,780 or antide. Arch. Toxicol. 76:613-620. |
|
20. |
Thigpen J.E., Haseman J.K., Saunders H.E., Setchell K.D.R., Grant M.F., Forsythe D. (2003). Dietary phytoestrogens accelerate the time of vaginal opening in immature CD-1 mice. Comp. Med. 53:477-485. |
|
21. |
Ashby J., Tinwell H., Odum J., Kimber I., Brooks A.N., Pate I., Boyle C.C. (2000). Diet and the aetiology of temporal advances in human and rodent sexual development. J. Appl. Toxicol. 20:343-347. |
|
22. |
Thigpen J.E., Lockear J., Haseman J., Saunders H.E., Caviness G., Grant M.F., Forsythe D.B. (2002). Dietary factors affecting uterine weights of immature CD-1 mice used in uterotrophic bioassays. Cancer Detect. Prev. 26:381-393. |
|
23. |
Thigpen J.E., Li L.-A., Richter C.B., Lebetkin E.H., Jameson C.W. (1987). The mouse bioassay for the detection of estrogenic activity in rodent diets: I. A standardized method for conducting the mouse bioassay. Lab. Anim. Sci. 37:596-601. |
|
24. |
OCDE (2008). Acute oral toxicity — up-and-down procedure. OECD Guideline for the Testing of Chemicals No 425. Paris. |
|
25. |
OCDE (2000). Guidance document on the recognition, assessment and use of clinical signs as humane endpoints for experimental animals used in safety evaluation. Environmental Health and Safety Monograph Series on Testing and Assessment No 19. ENV/JM/MONO(2000)7. |
|
26. |
OCDE (2001). Guidance document on acute oral toxicity. Environmental Health and Safety Monograph Series on Testing and Assessment No 24. ENV/JM/MONO(2001)4. |
|
27. |
Bulbring, E., Burn, J.H. (1935). The estimation of oestrin and of male hormone in oily solution. J. Physiol. 85:320-333. |
|
28. |
Dorfman, R.I., Gallagher, T.F. Koch, F.C. (1936). The nature of the estrogenic substance in human male urine and bull testis. Endocrinology 19:33-41. |
|
29. |
Reel, J.R., Lamb IV, J.C., Neal, B.H. (1996). Survey and assessment of mammalian estrogen biological assays for hazard characterization. Fundam. Appl. Toxicol. 34:288-305. |
|
30. |
Jones, R.C., Edgren, R.A. (1973). The effects of various steroid on the vaginal histology in the rat. Fertil. Steril. 24:284-291. |
|
31. |
OCDE (1982). Organization for Economic Co-operation and Development — Principles of Good Laboratory Practice. ISBN 92-64-12367-9. Paris. |
|
32. |
Dorfman R.I. (1962). Methods in Hormone Research, volume II, parte IV: Standard Methods Adopted by Official Organization. Academic Press, Nova Iorque. |
|
33. |
Thigpen J. E. et al., (2004). Selecting the appropriate rodent diet for endocrine disruptor research and testing studies. ILAR J. 45(4):401-416. |
|
34. |
Gray L.E., Ostby J. (1998). Effects of pesticides and toxic substances on behavioral and morphological reproductive development: endocrine versus non-endocrine mechanism. Toxicol. Ind. Health 14(1-2):159-184. |
|
35. |
Booth A.N., Bickoff E.M., Kohler G.O. (1960). Estrogen-like activity in vegetable oils and mill by-products. Science 131:1807-1808. |
|
36. |
Kato H., Iwata T., Katsu Y., Watanabe H., Ohta Y., Iguchi T. (2004). Evaluation of estrogenic activity in diets for experimental animals using in vitro assay. J. Agric. Food Chem. 52, 1410-1414. |
|
37. |
OCDE (2007). Guidance Document on the Uterotrophic Bioassay Procedure to Test for Antioestrogenicity. Series on Testing and Assessment, No. 71. |
|
38. |
Diretiva 2010/63/UE do Parlamento Europeu e do Conselho, de 22 de setembro de 2010, relativa à proteção dos animais utilizados para fins científicos (JO L 276 de 20.10.2010, p. 33). |
B.55. BIOENSAIO DE HERSHBERGER NO RATO: ENSAIO DE DESPISTAGEM A CURTO PRAZO DE PROPRIEDADES (ANTI)ANDROGÉNICAS
INTRODUÇÃO
1. Este método é equivalente ao Test Guideline TG 441 (2009) da OCDE. Em 1998, a OCDE deu início a uma ação prioritária com vista à revisão das orientações existentes e à elaboração de novas orientações para despistagem e ensaio de potenciais desreguladores do sistema endócrino (1). Um dos elementos dessa ação foi a elaboração de um Test Guideline para o bioensaio de Hershberger no rato. Após várias décadas de utilização pela indústria farmacêutica, este ensaio foi pela primeira vez normalizado em 1962, por uma comissão de peritos oficiais, como meio de despistagem de produtos químicos androgénicos (2). Entre 2001 e 2007, o bioensaio de Hershberger no rato foi objeto de um vasto programa de validação, que incluiu a elaboração de um documento-base de recensão (23), a compilação de um artigo pormenorizado sobre o método (3), a elaboração de um guia de dissecação (21) e a realização de amplos estudos intra e interlaboratoriais para verificar a fiabilidade e reprodutibilidade deste bioensaio. Os estudos de validação realizaram-se com um androgénio de referência muito ativo (o propionato de testosterona), dois androgénios sintéticos muito ativos (o acetato de trembolona e a metiltestosterona), um produto farmacêutico antiandrogénico muito ativo (a flutamida), um inibidor muito ativo da síntese da di-hidrotestosterona, o androgénio natural (a finasterida), vários pesticidas com fraca atividade antiandrogénica (linurão, vinclozolina, procimidona, p,p'-DDE), um inibidor muito ativo da 5α-redutase (a finasterida) e dois produtos químicos reconhecidamente inativos (dinitrofenol e nonilfenol) (4)(5)(6)(7)(8). O presente método de ensaio resulta da longa experiência histórica na aplicação do bioensaio de Hershberger, da experiência adquirida no programa de validação do mesmo e da apreciação dos resultados obtidos nesse programa.
2. O bioensaio de Hershberger constitui um ensaio de despistagem in vivo a curto prazo em tecidos acessórios do aparelho reprodutor masculino. O ensaio remonta aos anos 30 e foi modificado nos anos 40 pela inclusão dos músculos do referido aparelho que respondem aos androgénios (2)(9-15). Nos anos 60, avaliaram-se mais de 700 androgénios potenciais, recorrendo a uma versão normalizada do protocolo (2)(14). Ainda nos anos 60, o método foi considerado um ensaio normalizado para androgénios e antiandrogénios (2)(15). O bioensaio atual baseia-se nas variações ponderais de cinco tipos de tecido dependentes dos androgénios em ratos peripúberes castrados. Avalia-se a capacidade de um produto químico de induzir uma atividade biológica análoga às dos agonistas ou antagonistas de androgénios ou dos inibidores da 5α-redutase. Os cinco tipos de tecido dependentes dos androgénios utilizados neste método são a próstata ventral, as vesículas seminais (incluindo os fluidos e as glândulas coagulantes), o complexo formado pelos músculos elevatórios do ânus e bulbocavernosos, o par de glândulas de Cowper e a glande peniana. No rato peripúbere castrado, estes cinco tipos de tecido reagem aos androgénios aumentando de peso absoluto. Quando se estimulam os mesmos tecidos a aumentarem de peso por meio da administração de um androgénio de referência muito ativo, todos eles reagem aos antiandrogénios diminuindo de peso absoluto. O modelo primário do bioensaio de Hershberger, validado nas fases 1, 2 e 3 do programa de validação do método, é o macho peripúbere castrado cirurgicamente.
3. O bioensaio de Hershberger serve de ensaio de despistagem mecanístico in vivo de agonistas de androgénios, antagonistas de androgénios e inibidores da 5a-redutase, enquadrando-se a sua aplicação no contexto do Quadro conceptual da OCDE para ensaio e avaliação de produtos químicos desreguladores do sistema endócrino (apêndice 2). Nesse quadro conceptual, o bioensaio de Hershberger inscreve-se no nível 3, enquanto ensaio in vivo que fornece dados acerca de um único mecanismo endócrino, a (anti)androgenicidade. O bioensaio de Hershberger destina-se a ser incluído numa série de ensaios in vitro e in vivo que visam identificar produtos químicos suscetíveis de interagir com o sistema endócrino, tendo em vista a avaliação dos riscos para a saúde humana e para o ambiente.
4. Devido aos problemas de bem-estar animal colocados pela castração e a fim de evitar essa etapa, estudou-se como modelo alternativo para o ensaio de Hershberger o macho completo (não castrado) recentemente desmamado estimulado. O método com animais estimulados a seguir ao desmame foi validado (24). Porém, nos estudos de validação, a versão do ensaio de Hershberger que recorre a animais recentemente desmamados não se revelou adequada para detetar capazmente os efeitos ponderais das doses de antiandrogénios fracos ensaiadas em órgãos dependentes dos androgénios, não tendo, portanto, sido incluída no presente método. Todavia, considerando que a sua utilização pode ser vantajosa, não apenas no plano do bem-estar animal, mas também como fonte de informação sobre outros modos de ação, a referida versão do método de Hershberger consta do documento de orientações n.o 115 da OCDE (25).
CONSIDERAÇÕES INICIAIS E LIMITAÇÕES
5. Os agonistas e os antagonistas de androgénios agem como ligandos do recetor de androgénios e podem ativar ou inibir, respetivamente, a transcrição de genes controlada pelo recetor. Por outro lado, alguns produtos químicos (inibidores da 5a-redutase) inibem a conversão da testosterona no androgénio natural mais ativo di-hidrotestosterona em alguns tecidos-alvo dos androgénios. Desses produtos químicos podem advir consequências negativas para a saúde, nomeadamente efeitos com incidências na reprodução e no desenvolvimento. É, pois, necessário, do ponto de vista da regulamentação, poder determinar e avaliar rapidamente a capacidade agonista ou antagonista de androgénios, ou inibidora da 5a-redutase, revelada pelos produtos químicos. Embora constituam informações úteis, a afinidade de um ligando por um recetor de androgénios, medida pela ligação ao recetor, e a ativação da transcrição de genes relatores in vitro não são os únicos fatores determinantes de eventuais perigos. Constituem outros fatores determinantes a ativação ou desativação metabólicas após a entrada no organismo, a distribuição do produto químico pelos tecidos-alvo e a eliminação do produto químico do organismo. Daqui decorre a necessidade de despistar a eventual atividade dos produtos químicos in vivo em condições experimentais e de exposição adequadas. As avaliações in vivo são menos críticas se forem conhecidas as características ADME (absorção, distribuição, metabolismo e eliminação) do produto químico. Os tecidos dependentes dos androgénios reagem com um crescimento rápido e vigoroso à estimulação com androgénios, designadamente em ratos peripúberes castrados. Os roedores, nomeadamente o rato, também são amplamente utilizados nos estudos de toxicidade para caracterização de perigos. A versão do ensaio em ratos peripúberes castrados e nos cinco tecidos-alvo indicados adequa-se, pois, à despistagem in vivo de agonistas e antagonistas de androgénios e de inibidores da 5a-redutase.
6. O presente método baseia-se nos protocolos utilizados no estudo de validação realizado pela OCDE que se revelaram fiáveis e reprodutíveis nos estudos intra e interlaboratoriais efetuados (4)(5)(6)(7)(8). O presente método contempla um protocolo para androgénios e um protocolo para antiandrogénios
7. Embora a dose de propionato de testosterona utilizada para detetar antiandrogénios no programa de validação do bioensaio de Hershberger realizado pela OCDE tenha sido diferente de laboratório para laboratório (0,2 e 0,4 mg diários por quilograma, por injeção subcutânea), a capacidade de deteção de atividade antiandrogénica fraca ou forte pouco diferiu nestas duas variantes do protocolo. Todavia, é evidente que a dose de propionato de testosterona não deve ser tão elevada que bloqueie os efeitos dos antagonistas fracos de recetores de androgénios nem tão baixa que os tecidos androgénicos pouco cresçam, mesmo sem coadministração de um antiandrogénio.
8. A reação de crescimento de cada tecido dependente dos androgénios não é inteiramente de origem androgénica, pois há outros produtos químicos, além dos agonistas de androgénios, que podem alterar o peso de determinados tecidos. Porém, uma reação concomitante de crescimento de vários tecidos corrobora a hipótese de um mecanismo mais específico dos androgénios. Por exemplo, doses elevadas de estrogénios muito ativos podem aumentar o peso das vesículas seminais, mas os outros tecidos dependentes dos androgénios, utilizados no ensaio, não reagirão da mesma maneira. Os produtos químicos antiandrogénicos podem agir como antagonistas dos recetores de androgénios ou como inibidores da 5a-redutase. Os efeitos dos inibidores da 5a-redutase são variáveis, porque a conversão na di-hidrotestosterona, mais ativa, depende do tecido. Os antiandrogénios que inibem a 5α — redutase, como a finasterida, têm efeitos mais pronunciados na próstata ventral do que noutros tecidos, comparativamente a antagonistas muito ativos de recetores de androgénios, como a flutamida. Esta diferença na resposta dos tecidos pode ser utilizada para distinguir modos de ação mediados por recetores de androgénios de modos de ação mediados pela 5α — redutase. Além disso, os recetores de androgénios estão relacionados, em termos evolutivos, com os recetores de outras hormonas esteroides e outras hormonas há que, quando administradas em doses suprafisiológicas elevadas, podem ligar-se a eles e desempenhar um papel antagonista dos efeitos do propionato de testosterona como ativador do crescimento (13). É ainda plausível que o reforço do metabolismo dos esteroides e a consequente diminuição da testosterona sérica reduzam o crescimento dos tecidos dependente dos androgénios. Por conseguinte, um resultado positivo no bioensaio de Hershberger deve, normalmente, ser avaliado com base na ponderação da suficiência de prova também em ensaios in vitro, como ensaios de ligação aos recetores de androgénios e aos recetores de estrogénios e os correspondentes ensaios de ativação da transcrição, ou noutros ensaios in vivo, nos quais se examinem tecidos-alvo similares dos androgénios, como o ensaio no macho púbere, o ensaio de 15 dias no macho adulto completo ou estudos com repetição da dose a 28 dias ou a 90 dias.
9. A experiência adquirida revela que os androgénios xenobióticos são mais raros do que os antiandrogénios xenobióticos. Consequentemente, é de prever que o bioensaio de Hershberger seja utilizado com maior frequência na despistagem de antiandrogénios. Todavia, pode recomendar-se a aplicação do protocolo de ensaio relativo aos androgénios a esteroides e a produtos químicos análogos dos esteroides, bem como a produtos químicos para os quais a aplicação de métodos dos níveis 1 ou 2 do quadro conceptual (apêndice 2) tenha dado indicações de possíveis efeitos androgénicos. Analogamente, os ensaios do nível 5 podem permitir observar efeitos indesejáveis associados a perfis (anti)androgénicos, que aconselhem uma avaliação destinada a verificar se o produto químico em causa age por via endócrina.
10. Pressupõe-se que todos os procedimentos com animais respeitarão as normas locais de manipulação de animais. As descrições, nestes protocolos, dos cuidados a ter com os animais e do tratamento a dar aos animais são normas mínimas, prevalecendo, se for mais estrita, a regulamentação local, como a decorrente da Diretiva 2010/63/UE do Parlamento Europeu e do Conselho, de 22 de setembro de 2010, relativa à proteção dos animais utilizados para fins científicos (26). Para mais orientações sobre o tratamento ético de animais, ver o documento da OCDE com a referência 17.
11. Tal como em qualquer bioensaio no qual se utilizem animais, deve ponderar-se cuidadosamente a necessidade de efetuar o presente estudo. São fundamentalmente duas as razões em que pode fundamentar-se a decisão de realizar o estudo:
|
— |
potencial de exposição elevado (nível 1 do quadro conceptual) ou indícios de atividade (anti)androgénica em ensaios in vitro (nível 2) que justifiquem a necessidade de investigar a ocorrência de tais efeitos in vivo; |
|
— |
efeitos de natureza (anti)androgénica em ensaios in vivo dos níveis 4 ou 5, que justifiquem a necessidade de investigar o modo de ação, por exemplo a fim de determinar se os efeitos em causa se devem a um mecanismo (anti)androgénico. |
12. No apêndice 1 definem-se alguns conceitos utilizados neste método.
PRINCÍPIO DO MÉTODO
13. A sensibilidade do bioensaio de Hershberger radica na utilização de machos cuja produção de androgénios endógenos é mínima. Utilizam-se, para isso, machos castrados, prevendo um período adequado após a castração para que os tecidos visados readquiram um peso de base mínimo uniforme. Nestas circunstâncias, ao efetuar a despistagem da eventual atividade androgénica, os níveis de androgénios endógenos em circulação são baixos, o eixo hipotálamo-hipófise-gónadas é incapaz de os repor por mecanismos de retroação, a capacidade de resposta dos tecidos é máxima e a variabilidade do peso inicial dos tecidos é mínima. Ao efetuar a despistagem da eventual atividade antiandrogénica, consegue-se que o ganho de peso dos tecidos seja maior se estes forem estimulados com um androgénio de referência. Em consequência disto, no bioensaio de Hershberger só são necessários seis animais por grupo de dosagem, ao passo que noutros ensaios, que recorram a machos púberes ou adultos completos, se recomendam 15 machos por grupo de dosagem.
14. A castração dos ratos peripúberes deve realizar-se de um modo adequado, mediante a aplicação de técnicas de anestesia e de assepsia aprovadas. Para eliminar o desconforto pós-operatório, devem administrar-se analgésicos nos primeiros dias após a cirurgia. Ao eliminar os mecanismos de retroação endócrinos compensatórios presentes no animal completo, que podem atenuar os efeitos dos androgénios e antiandrogénios administrados, e concomitantemente também a grande variabilidade dos níveis séricos de testosterona que se verificam de indivíduo para indivíduo, a castração melhora a precisão do ensaio na deteção de androgénios e antiandrogénios fracos. A castração reduz, portanto, o número dos animais necessários à despistagem das atividades endócrinas em apreço.
15. Na despistagem de uma possível atividade androgénica, administra-se o produto químico em estudo diariamente, por sonda esofágica ou injeção subcutânea, durante dez dias consecutivos, a, pelo menos, dois grupos de animais, sendo cada grupo exposto a um nível de dose diferente. Eutanasiam-se os animais cerca de 24 horas após lhes ser administrada a última dose. Um aumento com significado estatístico do peso de dois ou mais órgãos-alvo no caso dos grupos expostos ao produto químico em estudo, comparativamente ao grupo de controlo do veículo, constitui uma indicação de atividade androgénica potencial do produto químico em causa (ver o ponto 60). Androgénios como a trembolona, que não sofrem os efeitos da 5α — redutase, afetam mais o complexo formado pelos músculos elevatórios do ânus e bulbocavernosos e a glande peniana do que o propionato de testosterona, mas todos os tecidos devem aumentar de peso.
16. No rastreio de uma possível atividade antiandrogénica, administra-se o produto químico em estudo diariamente, por sonda esofágica ou injeção subcutânea, durante dez dias consecutivos, juntamente com doses diárias de propionato de testosterona (0,2 ou 0,4 mg diários por quilograma), por injeção subcutânea. Determinou-se no programa de validação que podem utilizar-se 0,2 ou 0,4 mg diários de propionato de testosterona por quilograma, dado que ambas as quantidades permitem detetar os antiandrogénios, devendo escolher-se uma destas doses para o ensaio. Administram-se doses crescentes constantes do produto químico em estudo a, pelo menos, três grupos de animais, sendo cada grupo exposto a um nível de dose diferente. Eutanasiam-se os animais cerca de 24 horas após lhes ser administrada a última dose. Um decréscimo com significado estatístico do peso de dois ou mais órgãos-alvo no caso dos grupos expostos ao produto químico em estudo e ao propionato de testosterona, comparativamente ao grupo de controlo ao qual apenas é administrado propionato de testosterona, constitui uma indicação de atividade antiandrogénica potencial do produto químico em causa (ver o ponto 61).
DESCRIÇÃO DO MÉTODO
Escolha da espécie e da estirpe
17. Desde os anos 30 que se tem utilizado o rato nos bioensaios de Hershberger. Embora, em termos biológicos, seja plausível que as respostas do rato e do ratinho sejam semelhantes, optou-se pelo rato para os ensaios de Hershberger para tirar partido de 70 anos de experiência com o modelo do rato. Além disso, dado que os resultados do bioensaio de Hershberger podem constituir os preliminares de um estudo plurigeracional a longo prazo, torna-se possível utilizar animais da mesma espécie, estirpe e proveniência em ambos os estudos.
18. Este protocolo permite que os laboratórios escolham a estirpe de rato que utilizam no ensaio, em geral a que cada laboratório utiliza normalmente. Podem utilizar-se as estirpes habituais de ratos de laboratório, mas não devem utilizar-se estirpes que atinjam a maturidade substancialmente mais tarde do que aos 42 dias de idade, dado que a castração de animais com 42 dias poderia inviabilizar a pesagem da glande peniana, que só pode ser efetuada depois da descolagem do prepúcio do resto de pénis. Por conseguinte, salvo raras exceções, não devem utilizar-se estirpes derivadas do rato Fisher 344, cujo desenvolvimento sexual tem uma cronologia diferente da de outras estirpes mais frequentemente utilizadas, como a Sprague-Dawley ou a Wistar (16). Se for utilizada uma estirpe derivada do rato Fisher 344, o laboratório deve castrar os ratos com uma idade ligeiramente maior e terá de demonstrar a sensibilidade da estirpe por que tiver optado. Compete a cada laboratório justificar com clareza a escolha da estirpe de rato. Se o ensaio de despistagem constituir uma etapa preliminar de um estudo por via oral com repetição da dose, de um estudo dos efeitos na reprodução ou no desenvolvimento ou de um estudo a longo prazo, devem utilizar-se animais da mesma estirpe e proveniência em todos os casos.
Condições de alojamento e de alimentação
19. Todos os procedimentos devem respeitar as normas locais de manipulação de animais de laboratório. As descrições, nestes protocolos, dos cuidados a ter com os animais e do tratamento a dar aos animais são normas mínimas, prevalecendo, se for mais estrita, a regulamentação local, como a decorrente da Diretiva 2010/63/UE do Parlamento Europeu e do Conselho, de 22 de setembro de 2010, relativa à proteção dos animais utilizados para fins científicos (26). A temperatura do biotério deve ser de 22 °C (± 3 °C). A humidade relativa não deve ser inferior a 30 % e, de preferência, não deve exceder 70 %, exceto durante a limpeza do biotério. Idealmente, deve procurar manter-se a humidade relativa entre 50 % e 60 %. A iluminação deve ser artificial, com uma sequência de 12 horas de luz seguidas de 12 horas de escuridão.
20. Por serem juvenis e porque os ratos são animais sociais, recomenda-se o alojamento em gaiolas coletivas e não individualmente. O alojamento de dois ou três animais por gaiola evita a concentração e as tensões que lhe estão associadas, as quais podem interferir no controlo hormonal do desenvolvimento dos tecidos acessórios do aparelho sexual. As gaiolas devem ser cuidadosamente limpas para remover eventuais contaminantes e ser dispostas de forma a minimizar possíveis efeitos derivados do seu posicionamento. Devem ter tamanho suficiente para os animais que nelas se alojam (superfície de aproximadamente 2 000 cm2).
21. Deve identificar-se individualmente cada animal recorrendo a um método não-agressivo (por exemplo por marcação ou etiqueta auricular). O método de identificação deve ser mencionado no relatório.
22. Os animais devem receber ad libitum uma dieta de laboratório e água de beber. O laboratório deve utilizar no bioensaio de Hershberger a dieta de laboratório a que normalmente recorre no seu trabalho experimental com produtos químicos. Nos estudos de validação deste bioensaio, não se detetaram efeitos nem variabilidades atribuíveis à dieta. A dieta utilizada deve ser referida no relatório. Deve conservar-se uma amostra dessa dieta para a realização das análises que possam vir a ser necessárias.
Requisitos aplicáveis ao peso dos órgãos dependentes dos androgénios
23. No estudo de validação, não houve indícios de que uma diminuição do peso corporal tivesse incidências no aumento ou na diminuição do ganho de peso dos tecidos-alvo (os tecidos que está previsto pesar neste estudo).
24. O peso dos órgãos dependentes dos androgénios das várias estirpes de rato utilizadas com êxito no programa de validação revelou-se maior nas estirpes mais corpulentas do que nas estirpes mais leves. Por conseguinte, não se incluem nos requisitos do bioensaio de Hershberger pesos absolutos previsíveis dos órgãos dos animais de controlo positivo e negativo.
25. Uma vez que o coeficiente de variação de um tecido é inversamente proporcional ao poder estatístico, os requisitos do bioensaio de Hershberger baseiam-se em coeficientes de variação máximos admissíveis para cada tecido (quadro 1). Os coeficientes de variação indicados no quadro resultaram dos estudos de validação realizados pela OCDE. No caso de resultados negativos, o laboratório deve examinar os coeficientes de variação do grupo de controlo e do grupo exposto à dose mais elevada, a fim de determinar se os requisitos de coeficiente de variação máximo foram excedidos.
26. Deve repetir-se o estudo nos seguintes casos: 1) três ou mais dos dez coeficientes de variação possíveis no grupo de controlo e no grupo exposto à dose mais elevada excedem os máximos indicados nos quadros 1 para os estudos de agonistas e de antagonistas e 2) pelo menos dois tecidos-alvo são marginalmente não-significativos, isto é, apresentam valores de r compreendidos entre 0,05 e 0,10.
Quadro 1
Coeficientes de variação máximos admissíveis, determinados nos estudos de validação realizados pela OCDE, para os tecidos-alvo acessórios do aparelho sexual, aplicáveis ao modelo com ratos castrados (12)
|
Tecido |
Efeitos antiandrogénicos |
Efeitos androgénicos |
|
Vesículas seminais |
40 % |
40 % |
|
Próstata ventral |
40 % |
45 % |
|
Complexo formado pelos músculos elevatórios do ânus e bulbocavernosos |
20 % |
30 % |
|
Glândulas de Cowper |
35 % |
55 % |
|
Glande peniana |
17 % |
22 % |
PROCEDIMENTO DE ENSAIO
Respeito da regulamentação e verificações no laboratório
27. Ao contrário do que se passa com o ensaio uterotrófico (capítulo B.54 deste anexo), no caso do bioensaio de Hershberger é desnecessário demonstrar a competência do laboratório antes do início do estudo, porque este ensaio compreende, em paralelo, o ensaio de um controlo positivo (propionato de testosterona e flutamida) e de um controlo negativo.
Número e estado dos animais
28. O número mínimo de animais de cada grupo exposto ou de controlo é de seis, tanto no caso do protocolo para androgénios como do protocolo para antiandrogénios.
Castração
29. Após a receção dos animais, deve existir um período inicial de aclimatação de vários dias, para verificar se os mesmos se encontram em boas condições de saúde e de vitalidade. Dado que, nos animais castrados com menos de 42 dias de idade (ou antes de transcorridos 42 dias após o nascimento), o descolamento do prepúcio pode não ter ocorrido, a castração deve realizar-se 42 dias após o nascimento ou em data posterior. Os animais são castrados sob anestesia. Efetua-se uma incisão no escroto, removem-se os testículos e os epidídimos e laqueiam-se os vasos sanguíneos e os canais seminíferos. Depois de confirmar que não há sangramento, fecha-se o escroto por sutura ou com agrafos. Para eliminar o desconforto pós-operatório, devem administrar-se analgésicos aos animais nos primeiros dias após a cirurgia. Se os animais forem adquiridos a um fornecedor já castrados, este deve garantir a idade e o estádio de maturidade sexual dos mesmos.
Aclimatação após castração
30. Para possibilitar a regressão do peso dos tecidos-alvo, os animais devem prosseguir a aclimatação às condições do laboratório durante um período mínimo de sete dias após a castração. Devem ser examinados diariamente e os que apresentem sinais de doenças ou de anomalias físicas devem ser retirados. A exposição às doses em estudo pode, portanto, iniciar-se 49 dias após o nascimento, mas não depois dos 60 dias de idade. A idade dos animais no dia da autópsia não deve ser superior a 70 dias. Esta flexibilidade permite que os laboratórios programem com eficiência o trabalho experimental.
Aleatorização do peso corporal e dos grupos
31. As diferenças de peso corporal de rato para rato são uma fonte de variabilidade do peso dos tecidos em cada grupo e entre os grupos de animais. O aumento da variabilidade do peso dos tecidos faz aumentar o coeficiente de variação e diminui o poder estatístico do ensaio (por vezes designado “sensibilidade do ensaio”). Importa, portanto, limitar as variações de peso corporal, tanto no plano experimental como no plano estatístico.
32. No plano experimental, é necessário reduzir as diferenças de peso corporal em cada grupo e entre os grupos em estudo. Em primeiro lugar, os animais anormalmente pequenos ou anormalmente grandes não devem participar no estudo. No início do estudo, as diferenças de peso entre animais não devem exceder ± 20 % do peso médio (por exemplo, 175 g ± 35 g no caso dos ratos peripúberes castrados). Em segundo lugar, integram-se os animais nos grupos expostos e de controlo procedendo a uma distribuição aleatória de pesos, de maneira que o peso corporal médio de cada grupo não difira estatisticamente do peso corporal médio de qualquer outro grupo. Deve mencionar-se no relatório o protocolo de aleatorização por blocos utilizado.
33. Dado que a toxicidade pode diminuir o peso corporal dos animais dos grupos expostos comparativamente ao dos animais dos grupos de controlo, pode utilizar-se como covariável estatística o peso corporal no primeiro dia de administração do produto químico em estudo — e não o peso corporal no dia da autópsia.
Dosagem
34. Normalmente, dois grupos de dosagem do produto químico em estudo, juntamente com um grupo de controlo positivo e um grupo de controlo do veículo (negativo) (ver o ponto 43), são suficientes para determinar se o produto químico pode ter atividade androgénica in vivo. Por razões de bem-estar animal, privilegia-se, portanto, este modelo. No caso de se pretender traçar uma curva da relação entre a dose administrada e a resposta obtida ou efetuar uma extrapolação para doses mais baixas, são necessários, pelo menos, três grupos de dosagem. No caso de se pretender obter informações que vão além de uma identificação de atividade androgénica (como, por exemplo, uma estimativa do poder androgénico), deve ponderar-se um regime de dosagem diferente. Na despistagem de antiandrogénios, administra-se o produto químico em estudo juntamente com um agonista de androgénios de referência. Neste caso, devem utilizar-se, pelo menos, três grupos, expostos a doses diferentes do produto químico em estudo, juntamente com um grupo de controlo positivo e um grupo de controlo negativo (ver o ponto 44). Salvo no que respeita à exposição ao produto químico em estudo, os animais dos grupos de controlo devem ser tratados do mesmo modo que os animais dos grupos ensaiados. Se for utilizado um veículo para administrar o produto químico em estudo, os animais do grupo de controlo devem receber o volume máximo de veículo utilizado nos grupos ensaiados.
35. Na escolha das doses devem ter-se em conta os dados eventualmente disponíveis sobre a toxicidade e a (toxico)cinética do produto químico em estudo ou de matérias afins. Para se estabelecer o nível de dose mais elevado, deve atender-se, em primeiro lugar, à DL50 e/ou aos dados sobre toxicidade aguda de que se disponha, a fim de evitar a morte dos animais e de os poupar a grande sofrimento ou aflição (17)(18)(19)(20), e, em segundo lugar, às informações disponíveis sobre as doses utilizadas em estudos de toxicidade subcrónica ou crónica. Em geral, a dose mais elevada não deve provocar uma redução do peso corporal final dos animais superior a 10 % do peso dos animais do grupo de controlo. A dose mais elevada deve ser 1) a dose máxima à qual os animais sobrevivem e que não lhes causa efeitos tóxicos nem sofrimento significativos após dez dias consecutivos de administração, sem exceder a dose máxima diária de 1 000 mg/kg (ver o ponto 36), ou 2) uma dose inferior indutora de efeitos (anti)androgénicos. A título exploratório, aceitam-se intervalos largos (por exemplo, meia unidade logarítmica, correspondente a um fator de progressão das doses de 3,2, ou mesmo uma unidade logarítmica) entre as doses administradas. Se não se dispuser de dados adequados para o efeito, pode efetuar-se um estudo exploratório (ver o ponto 37), para facilitar a determinação das doses a utilizar.
Dose-limite
36. Se o ensaio da dose-limite de 1 000 mg diários por quilograma de peso corporal e de uma dose inferior a esta, aplicando os protocolos descritos para este estudo, não gerar uma diferença estatisticamente significativa entre os pesos correspondentes dos órgãos reprodutores, pode considerar-se desnecessário ensaiar outras doses. A dose-limite aplica-se a todos os casos, exceto se os dados relativos à exposição humana aconselharem o ensaio de doses superiores.
Estudo exploratório
37. Se necessário, pode efetuar-se um estudo exploratório preliminar com poucos animais, para escolher os grupos de dosagem apropriados [recorrendo aos métodos de ensaio da toxicidade aguda — capítulos B.1.bis e B.1.tris deste anexo (27), Test Guideline 425 da OCDE (19)]. O objetivo, no caso do bioensaio de Hershberger, é selecionar doses que garantam a sobrevivência dos animais e não lhes provoquem efeitos tóxicos nem sofrimento significativos após dez dias consecutivos de administração do produto químico, até à dose-limite diária de 1 000 mg/kg como referido nos pontos 35 e 36. Para o efeito, pode utilizar-se um documento de orientações da OCDE (17) que define os sinais clínicos da toxicidade ou do sofrimento provocados em animais. Se for possível fazê-lo nesse estudo exploratório, após dez dias de administração das doses procede-se à excisão e pesagem dos tecidos-alvo cerca de 24 horas após a última dose. Estes dados podem ser depois utilizados para escolher as doses a utilizar no estudo principal.
Produtos químicos de referência e veículo
38. O agonista de androgénios de referência é o propionato de testosterona (n.o CAS 57-82-5). A dose deste agonista de referência pode ser 0,2 mg ou 0,4 mg diários por quilograma de peso corporal. O antagonista de androgénios de referência é a flutamida (n.o CAS 1311-84-7). A dose deste antagonista de referência, coadministrada com a dose do propionato de testosterona de referência, deve ser de 3 mg diários por quilograma de peso corporal.
39. Sempre que possível, deve começar por ponderar-se a possibilidade de se utilizarem soluções/suspensões aquosas. Porém, dado que os ligandos de androgénios ou os precursores metabólicos dos mesmos são geralmente hidrófobos, normalmente utiliza-se uma solução ou suspensão oleosa (por exemplo, em óleo de milho, de amendoim ou de gergelim ou em azeite). Pode dissolver-se o produto químico em estudo numa quantidade mínima de etanol a 95 % ou de outro solvente adequado, diluindo-se depois até à concentração final no veículo a utilizar nos ensaios. É necessário conhecer as características de toxicidade do solvente e verificá-las num grupo de controlo apenas exposto ao solvente. Se o produto químico em estudo for considerado estável, pode favorecer-se a sua dissolução aquecendo ligeiramente e agitando vigorosamente. Deve determinar-se a estabilidade do produto químico em estudo no veículo utilizado. Se o produto químico em estudo se mantiver estável durante todo o estudo, podem preparar-se uma alíquota inicial do mesmo e, diariamente, as diluições às doses necessárias, tomando as precauções necessárias para evitar a contaminação ou a deterioração das amostras.
Administração das doses
40. Administra-se o propionato de testosterona por injeção subcutânea e a flutamida por sonda esofágica.
41. Administra-se o produto químico em estudo por sonda esofágica ou por injeção subcutânea. Ao escolher a via de administração, é necessário atender aos aspetos do bem-estar animal e ter em conta as propriedades físico-químicas do produto químico em estudo. Além disso, caso se obtenham resultados positivos por injeção, deve atender-se a aspetos toxicológicos como a relação com a via de exposição humana ao produto químico (por exemplo, sonda esofágica para exposição por ingestão e injeção subcutânea para exposição por inalação ou por adsorção cutânea), bem como às informações toxicológicas e aos dados metabólicos e cinéticos disponíveis (por exemplo, a necessidade de evitar metabolismos de primeira passagem ou melhor eficiência de uma determinada via de administração), antes de efetuar ensaios aprofundados a longo prazo.
42. Devem administrar-se as doses aos animais da mesma maneira e com a mesma cronologia durante dez dias consecutivos, com intervalos de aproximadamente 24 horas. A dosagem deve ajustar-se diariamente com base nas determinações quotidianas concomitantes do peso corporal. Deve registar-se o volume e o momento da administração de cada dose em cada dia de exposição A dose máxima indicada no ponto 35 não deve ser excedida, a fim de possibilitar uma interpretação com significado dos dados. Nessa perspetiva, devem avaliar-se cuidadosamente as reduções de peso corporal, os sinais clínicos e outros dados obtidos. A administração forçada aos animais através de sonda esofágica deve efetuar-se por meio de um tubo estomacal ou de uma cânula de intubação adequada. O volume máximo de líquido que pode ser administrado de cada vez depende do tamanho do animal. Devem respeitar-se as normas locais de manipulação de animais, mas o volume administrado não deve exceder 5 ml por quilograma de peso corporal, salvo no caso das soluções aquosas, em que se permitem 10 ml por quilograma de peso corporal. No caso das injeções subcutâneas, as doses devem ser administradas na região escapular dorsal ou na região lombar, por meio de uma seringa do tipo utilizado no teste da tuberculina (calibre 23 ou 25), dotada de uma agulha esterilizada. É facultativo rapar a pelagem dos animais no local da injeção. Devem registar-se as perdas ou escorrimentos nesse local, bem como todas as administrações incompletas de doses. O volume injetado diariamente a um rato não deve exceder 0,5 ml por quilograma de peso corporal.
Protocolo específico para agonistas de androgénios
43. No ensaio de agonistas de androgénios, o veículo deve ser utilizado como controlo negativo e o grupo exposto ao propionato de testosterona como controlo positivo. Verifica-se se o produto químico em estudo tem atividade biológica de agonista de androgénios administrando as doses escolhidas do mesmo a grupos de animais durante dez dias consecutivos. Compara-se o peso de cada um dos cinco tecidos acessórios do aparelho sexual dos animais dos grupos expostos ao produto químico em estudo com o dos animais do grupo de controlo do veículo, a fim de determinar se houve algum aumento de peso com significado estatístico.
Protocolo específico para antagonistas de androgénios e inibidores da 5α-redutase
44. No ensaio de antagonistas de androgénios e de inibidores da 5α-redutase, o grupo exposto ao propionato de testosterona constitui o controlo negativo e o grupo coadministrado com doses de referência de propionato de testosterona e flutamida constitui o controlo positivo. Verifica-se a existência de atividade biológica de antagonista de androgénios e de inibidor da 5α-redutase administrando o produto químico em estudo durante dez dias consecutivos, juntamente com uma dose de referência de propionato de testosterona. Compara-se o peso de cada um dos cinco tecidos acessórios do aparelho sexual dos animais dos grupos expostos ao produto químico em estudo e ao propionato de testosterona com o dos animais do grupo exposto unicamente ao propionato de testosterona de referência, a fim de determinar se houve alguma diminuição de peso com significado estatístico.
EXAMES
Exames clínicos
45. Deve efetuar-se um exame clínico geral pelo menos uma vez por dia; se forem detetados sinais de toxicidade, o exame deve ser mais frequente. De preferência, os exames devem ser efetuados à mesma hora ou às mesmas horas do dia, tendo em atenção o período previsto de efeitos mais acentuados após a administração do produto químico. Devem examinar-se todos os animais de modo a detetar os casos de mortalidade e morbilidade e sinais clínicos gerais como mudanças de comportamento, alterações da pele, da pelagem, dos olhos ou das mucosas e a ocorrência de secreções ou excreções e de reações neurovegetativas (por exemplo lacrimação, horripilação, alterações da dimensão pupilar, respiração anormal, etc.).
46. Os animais que morram devem ser retirados e eliminados sem análise de dados. Devem indicar-se no relatório do estudo a mortalidade de animais antes da autópsia e as razões aparentes de cada morte. Os animais moribundos devem ser eutanasiados. Os animais eutanasiados por estarem moribundos devem ser indicados no relatório do estudo, juntamente com as razões aparentes da morbilidade.
Peso corporal e consumo de alimentos
47. Deve pesar-se diariamente cada animal com a aproximação de 0,1 g, começando imediatamente antes do início da exposição, isto é, quando da repartição dos animais pelos grupos. A quantidade de alimentos consumida em cada gaiola durante o período de exposição pode também ser determinada por gaiola, a título facultativo, pesando os distribuidores de alimentos. Os resultados de consumo de alimentos devem ser expressos em gramas diários por animal.
Dissecação e pesagem dos tecidos e órgãos
48. Aproximadamente 24 horas após a última administração do produto químico em estudo, eutanasiam-se e sangram-se os ratos pelo procedimento normal do laboratório, passando-se em seguida à autópsia. O método de eutanásia deve ser referido no relatório do laboratório.
49. Idealmente, a ordem das autópsias deve ser aleatória no universo dos grupos, evitando seguir uma ordem crescente ou decrescente das doses, método que poderia afetar os resultados. As constatações da autópsia, por exemplo modificações patológicas e lesões visíveis, devem ser anotadas e posteriormente referidas no relatório.
50. Devem pesar-se os cinco tecidos dependentes dos androgénios (próstata ventral, vesículas seminais, incluindo as glândulas coagulantes, complexo formado pelos músculos elevatórios do ânus e bulbocavernosos, glândulas de Cowper e glande peniana). Para isso, excisam-se os tecidos em causa, retiram-se cuidadosamente os outros tecidos e a gordura aderentes e determinam-se os respetivos pesos em fresco (tecidos não fixados). Cada tecido deve ser manipulado com cuidados especiais para evitar perdas de fluidos e a secagem do mesmo, que, ao reduzirem os pesos registados, podem introduzir erros e variabilidade significativos. Alguns tecidos podem ter dimensões muito reduzidas ou ser difíceis de dissecar, ambas as circunstâncias fonte de variabilidade. É, portanto, importante que os dissecadores dos tecidos acessórios do aparelho sexual estejam familiarizados com procedimentos normalizados de dissecação dos mesmos. A OCDE publicou um manual de procedimentos normalizados de dissecação (21). Uma formação cuidadosa baseada nesse manual minimizará esta fonte potencial de variabilidade no estudo. Idealmente, o prossector de um dado tecido deve ser sempre o mesmo, para eliminar as diferenças de pessoa para pessoa no tratamento dos tecidos. Se isso não for possível, o programa das autópsias deve ser organizado de modo que cada prossector fique incumbido de dissecar um determinado tecido de todos os grupos expostos e não de modo que um prossector fique incumbido de dissecar todos os tecidos do(s) grupo(s) de controlo, ficando outro incumbido dos grupos expostos. Deve pesar-se cada tecido acessório do aparelho sexual com a aproximação de 0,1 mg, sem o desumedecer previamente, registando os pesos obtidos para cada animal.
51. Alguns tecidos podem ter dimensões muito reduzidas ou ser difíceis de dissecar, ambas as circunstâncias fonte de variabilidade. Estudos anteriores revelaram que o intervalo dos coeficientes de variação obtidos parece depender da competência do laboratório. Em alguns casos, obtiveram-se grandes diferenças nos pesos absolutos dos tecidos, nomeadamente da próstata ventral e das glândulas de Cowper, determinados no mesmo laboratório.
52. A pesagem do fígado, dos rins e das glândulas suprarrenais é facultativa. Novamente, devem retirar-se todas as aderências de tecidos fasciais ou de gordura. O fígado deve ser pesado e o seu peso registado com a aproximação de 0,1 g; os rins e as glândulas suprarrenais, com a aproximação de 0,1 mg. O fígado, os rins e as glândulas suprarrenais não são influenciadas pelos androgénios, mas também fornecem indicadores úteis de toxicidade sistémica.
53. A determinação dos teores séricos de hormona luteinizante (LH), de hormona foliculoestimulante (FSH) e de testosterona (T) é facultativa. Os teores séricos de testosterona são úteis para determinar se o produto químico em estudo induz o metabolismo hepático da testosterona, baixando os níveis séricos desta. Sem estes dados relativos à testosterona, esse efeito poderia parecer advir de um mecanismo antiandrogénico. Os teores de hormona luteinizante dão informações acerca da capacidade de um antiandrogénio, não apenas de reduzir o peso dos órgãos, mas também de afetar a função do eixo hipotálamo-hipófise, o que, segundo estudos a longo prazo, pode induzir tumores nos testículos. A hormona foliculoestimulante é importante na espermatogénese. A determinação dos teores séricos de T4 e T3 são igualmente análises facultativas capazes de fornecer informações complementares úteis acerca da capacidade de desregulação da homeostase das hormonas tiroideias. No caso de se pretender efetuar análises hormonais, anestesiam-se os ratos antes da autópsia e colhem-se amostras de sangue por punção cardíaca. O método de anestesia deve ser escolhido com cuidado, para que não afete as análises hormonais. Devem indicar-se no relatório o método de preparação do soro, a proveniência dos kits para os ensaios radioimunológicos ou para outras técnicas de medição, os protocolos analíticos e os resultados obtidos. Os teores de hormona luteinizante e de testosterona devem ser indicados em nanogramas por mililitro de soro.
54. Descreve-se a seguir o modo de proceder à dissecação dos tecidos, baseado no guia pormenorizado da dissecação, com fotografias, que foi publicado como suplemento no âmbito do programa de validação (21). A página Web da Food and Drug Administration da Coreia contém igualmente uma hiperligação para uma filmagem de uma dissecação (22).
|
— |
Com o ventre do animal voltado para cima, verificar se o prepúcio está descolado da glande peniana. Se assim for, recolher o prepúcio e cortar a glande, pesando-a com a aproximação de 0,1 mg e registando o peso determinado. |
|
— |
Efetuar um corte na pele e na parede abdominais, de modo a expor as vísceras. Se estiver prevista a pesagem dos órgãos facultativos, retirar o fígado, pesando-o com a aproximação de 0,1 g; em seguida, retirar o estômago e os intestinos e depois os rins e as glândulas suprarrenais, pesando-as aos pares com a aproximação de 0,1 mg. Esta dissecação expõe a bexiga e constitui o início da dissecação dos tecidos-alvo acessórios do aparelho sexual. |
|
— |
Para dissecar a próstata ventral, separar a bexiga da camada muscular ventral cortando o tecido conjuntivo ao longo da linha mediana. Deslocar a bexiga no sentido anterior, para o lado das vesículas seminais, de modo a mostrar os lóbulos esquerdo e direito da próstata ventral (cobertos por uma camada de gordura). Raspar cuidadosamente a gordura dos lóbulos esquerdo e direito da próstata ventral. Afastar cuidadosamente da uretra o lóbulo direito, dissecando-o daquela. Segurando o lóbulo direito da próstata ventral, afastar cuidadosamente da uretra o lóbulo esquerdo, dissecando-o igualmente daquela. Pesar com a aproximação de 0,1 mg e registar o peso. |
|
— |
Para dissecar as vesículas seminais juntamente com as glândulas coagulantes, deslocar a bexiga no sentido caudal, expondo os canais deferentes e os lóbulos esquerdo e direito do conjunto formado pelas vesículas seminais e pelas glândulas coagulantes. Para evitar fugas de fluidos, aplicar uma pinça homeostática na base deste conjunto, na confluência com a uretra. Dissecar cuidadosamente as vesículas seminais e as glândulas coagulantes; mantendo as pinças aplicadas, remover a gordura e os tecidos aderentes; colocar os tecidos-alvo numa cápsula de pesagem previamente tarada, retirar as pinças e pesar com a aproximação de 0,1 mg, registando o peso obtido. |
|
— |
Para dissecar o complexo formado pelos músculos elevatórios do ânus e bulbocavernosos, é necessário expor esses músculos e a base do pénis. Os músculos elevatórios do ânus rodeiam o cólon, ao passo que a parte anterior dos mesmos e os músculos bulbocavernosos estão ligados aos bolbos penianos. Remove-se a pele e os tecidos aderentes da região perianal que se estende da base do pénis à extremidade anterior do ânus. Dissecam-se gradualmente os músculos bulbocavernosos dos bolbos penianos e dos outros tecidos. Corta-se o cólon em dois, podendo assim dissecar-se e remover-se todo o complexo formado pelos músculos elevatórios do ânus e bulbocavernosos. Removem-se a gordura e os tecidos aderentes a esses músculos e pesam-se estes últimos com a aproximação de 0,1 mg, registando o peso obtido. |
|
— |
Depois de removido o complexo formado pelos músculos elevatórios do ânus e bulbocavernosos, ficam visíveis as glândulas de Cowper ou bulbouretrais, de forma arredondada, situadas na base dos bolbos penianos, em posição ligeiramente dorsal. É necessário proceder cuidadosamente à dissecação, para evitar danificar a cápsula, cuja superfície é fina, e assim impedir a fuga de fluidos. Pesar o conjunto das duas glândulas de Cowper com aproximação de 0,1 mg e registar o peso obtido. |
|
— |
Se houver alguma perda de fluidos de alguma glândula durante a autópsia e a dissecação é necessário referi-lo no relatório. |
55. Se a avaliação de um produto químico exigir a autópsia de mais animais do que é razoável programar para um só dia, o início do estudo pode ser estendido por dois dias consecutivos, o mesmo sucedendo às autópsias e às operações conexas destas. No caso de se proceder deste modo, deve utilizar-se por dia metade dos animais de cada grupo exposto.
56. Depois da autópsia, as carcaças devem ser eliminadas de modo adequado.
RELATÓRIOS
Dados
57. Os dados devem ser individualizados (isto é, devem ser indicados por animal o peso corporal, o peso de cada tecido acessório do aparelho sexual, as medições facultativas e as outras respostas e observações) e por grupo de animais (com indicação das médias e dos desvios-padrão de todas as medições efetuadas). Devem ser resumidos em quadros e deles devem constar o número de animais no início do ensaio, o número de animais que morreram ou apresentaram sinais de toxicidade durante o ensaio e uma descrição dos sinais de toxicidade observados, nomeadamente o momento do seu aparecimento e a sua duração e intensidade.
58. Informações a constar do relatório final:
Instalações nas quais se realizaram os ensaios
|
— |
nome e localização das instalações; |
|
— |
diretor do estudo e outro pessoal e responsabilidades de cada um no estudo; |
|
— |
datas de início e termo do estudo, isto é, primeiro dia de administração do produto químico e último dia de autópsia. |
Produto químico em estudo
|
— |
proveniência, número de lote, identidade, grau de pureza, endereço completo do fornecedor e caracterização do produto ou produtos químicos em estudo; |
|
— |
natureza física e propriedades físico-químicas pertinentes; |
|
— |
condições de armazenagem; método e frequência de preparação de diluições; |
|
— |
dados obtidos sobre a estabilidade; |
|
— |
análises das soluções ou suspensões administradas. |
Veículo
|
— |
caracterização do veículo utilizado no estudo (identidade, fornecedor e número do lote); |
|
— |
justificação da escolha do veículo (se não for água). |
Animais estudados e condições em que são mantidos
|
— |
espécie e estirpe utilizadas e justificação das opções tomadas; |
|
— |
proveniência ou fornecedor dos animais, incluindo o endereço completo; |
|
— |
número e idade dos animais fornecidos; |
|
— |
condições de alojamento (temperatura, iluminação, etc.); |
|
— |
dieta (nome, tipo, fornecedor, número de lote, composição e, se conhecidos, níveis de fitoestrogénios); |
|
— |
camas (nome, tipo, fornecedor e composição); |
|
— |
condições de engaiolamento e número de animais por gaiola. |
Condições de realização dos ensaios
|
— |
idade dos animais no momento da castração e duração da aclimatação após a castração; |
|
— |
peso de cada animal no início do estudo (com a aproximação de 0,1 g); |
|
— |
processo de aleatorização, registo da integração dos animais nos grupos do veículo, do produto químico de referência e do produto químico em estudo e registo da distribuição dos animais pelas gaiolas; |
|
— |
média e desvio-padrão dos pesos corporais correspondentes a cada grupo, para cada dia de pesagem ao longo do estudo; |
|
— |
fundamentação da escolha das doses; |
|
— |
via de administração do produto químico em estudo e fundamentação da via de exposição escolhida; |
|
— |
ensaios de antiandrogenicidade: dose e volume utilizados de propionato de testosterona; |
|
— |
doses e volumes utilizados para a exposição ao produto químico em estudo; |
|
— |
cronologia da administração das doses; |
|
— |
procedimentos de autópsia, incluindo os meios de sangramento e as anestesias; |
|
— |
se forem efetuadas análises do soro, elementos relativos aos métodos utilizados; por exemplo, caso seja utilizado um método radioimunométrico (RIA), indicar a proveniência dos kits, a data de validade destes, o método de contagem da cintilação e o método de normalização. |
Resultados
|
— |
Exame diário da cada animal durante o período de administração das doses, incluindo, nomeadamente: |
|
— |
peso corporal (com a aproximação de 0,1 g), |
|
— |
eventuais sinais clínicos, |
|
— |
medições ou anotações relativas ao consumo de alimentos; |
|
— |
Autópsia de cada animal, incluindo, nomeadamente: |
|
— |
data da autópsia, |
|
— |
grupo de exposição no qual o animal estava integrado, |
|
— |
identificação do animal, |
|
— |
prossector, |
|
— |
hora da realização da autópsia e da dissecação, |
|
— |
idade do animal, |
|
— |
peso corporal final no momento da autópsia (anotar algum aumento ou diminuição com significado estatístico), |
|
— |
ordem de sangramento e dissecação dos animais na autópsia, |
|
— |
peso de cada um dos cinco tecidos-alvo dependentes dos androgénios: |
|
— |
próstata ventral (com a aproximação de 0,1 mg), |
|
— |
vesículas seminais e glândulas coagulantes, incluindo os fluidos (o par, com a aproximação de 0,1 mg), |
|
— |
complexo dos músculos elevatórios do ânus e bulbocavernosos (com a aproximação de 0,1 mg), |
|
— |
glândulas de Cowper (peso fresco — o par, com a aproximação de 0,1 mg), |
|
— |
glande peniana (peso fresco, com a aproximação de 0,1 mg), |
|
— |
peso de cada tecido facultativo, se determinado: |
|
— |
fígado (com a aproximação de 0,1 g), |
|
— |
rins (o par, com a aproximação de 0,1 mg), |
|
— |
glândulas suprarrenais (o par, com a aproximação de 0,1 mg), |
|
— |
comentários e observações gerais; |
|
— |
Análises das hormonas séricas, se efetuadas:
|
|
— |
comentários e observações gerais. |
Resumo dos dados
Os dados devem ser resumidos em quadros que indiquem a dimensão da amostra para cada grupo, o valor médio e o desvio-padrão ou o erro-padrão da média. Os quadros devem incluir os pesos corporais na autópsia, as variações do peso corporal desde o início da exposição até à autópsia, os pesos dos tecidos-alvo acessórios do aparelho sexual e os eventuais pesos de órgãos facultativos.
Discussão dos resultados
Análise dos resultados
59. Os pesos corporais e dos órgãos apurados na autópsia devem ser objeto de uma análise estatística destinada a determinar características como a homogeneidade da variância, se necessário mediante uma transformação adequada dos dados. Devem comparar-se os grupos expostos com um grupo de controlo, recorrendo a técnicas como a ANOVA (análise da variância), seguida de comparações par a par (por exemplo, o teste unilateral de Dunnett) e da aplicação de um critério de diferença estatística, por exemplo p ≤ 0,05. Devem identificar-se os grupos com significado estatístico. Devem evitar-se pesos de órgãos “relativos”, dada a invalidade dos pressupostos estatísticos em que se fundamenta esta manipulação de dados.
60. O grupo de controlo a utilizar no estudo do agonismo de androgénios deve ser um grupo exposto unicamente ao veículo. As características do modo de ação de um determinado produto químico podem gerar respostas relativas diferentes de tecido para tecido. A trembolona, por exemplo, que não sofre os efeitos da 5α-redutase, afeta mais o complexo formado pelos músculos elevatórios do ânus e bulbocavernosos e a glande peniana do que o propionato de testosterona. Um aumento com significado estatístico (p ≤ 0,05) do peso de quaisquer dois ou mais dos cinco tecidos-alvo dependentes dos androgénios (próstata ventral, complexo formado pelos músculos elevatórios do ânus e bulbocavernosos, glande peniana, glândulas de Cowper e vesículas seminais, com as glândulas coagulantes) deve ser considerado um resultado positivo de agonista de androgénios (nesse caso, todos os tecidos-alvo devem apresentar algum grau de aumento do crescimento). Pode realizar-se uma avaliação combinada das respostas dos tecidos dos órgãos acessórios do aparelho sexual efetuando uma análise multivariada dos dados. Este método pode servir para afinar as conclusões, sobretudo nos casos em que apenas um tipo de tecido dê uma resposta com significado estatístico.
61. O grupo de controlo a utilizar no estudo do antagonismo de androgénios deve ser um grupo exposto ao androgénio de referência (unicamente propionato de testosterona). As características do modo de ação de um determinado produto químico podem gerar respostas relativas diferentes de tecido para tecido. Comparativamente a antagonistas muito ativos de recetores de androgénios, como a flutamida, os inibidores da 5α-redutase, como a finasterida, afetam mais a próstata ventral do que os outros tecidos. Uma redução com significado estatístico (p ≤ 0,05) do peso de quaisquer dois ou mais dos cinco tecidos-alvo dependentes dos androgénios (próstata ventral, complexo formado pelos músculos elevatórios do ânus e bulbocavernosos, glande peniana, glândulas de Cowper e vesículas seminais, com as glândulas coagulantes), comparativamente à exposição unicamente ao propionato de testosterona, deve ser considerada um resultado positivo de antagonista de androgénios (nesse caso, todos os tecidos-alvo devem apresentar algum grau de diminuição do crescimento). Pode realizar-se uma avaliação combinada das respostas dos tecidos dos órgãos acessórios do aparelho sexual efetuando uma análise multivariada dos dados. Este método pode servir para afinar as conclusões, sobretudo nos casos em que apenas um tipo de tecido dê uma resposta com significado estatístico.
62. Os dados devem ser resumidos em quadros que indiquem o valor médio, o erro-padrão da média (o desvio-padrão também é aceitável) e a dimensão da amostra para cada grupo. Devem ser igualmente facultados quadros com os dados individualizados. Devem analisar-se os valores individualizados, as médias, os erros-padrão (ou desvios-padrão) e os coeficientes de variação dos dados relativos aos grupos de controlo, para determinar se satisfazem critérios aceitáveis de coerência com os valores históricos correspondentes. Se os coeficientes de variação excederem os valores indicados no quadro 1 (ver os pontos 25 e 26) para o peso de cada órgão, deve verificar-se se há erros no registo ou na transcrição dos dados ou se o laboratório ainda não domina a dissecação correta dos tecidos dependentes dos androgénios, sendo necessária mais prática ou mais formação. Em geral, os coeficientes de variação (quociente do desvio-padrão pelo peso médio do órgão) são reprodutíveis de laboratório para laboratório e de estudo para estudo. Os dados apresentados devem ser, pelo menos, os seguintes: pesos da próstata ventral, das vesículas seminais, do complexo formado pelos músculos elevatórios do ânus e bulbocavernosos, das glândulas de Cowper, da glande peniana e do fígado, peso corporal e variação deste entre o início da exposição e a autópsia. Os dados também podem ser apresentados depois de um ajustamento de covariância em função do peso corporal, mas tal não dispensa a apresentação dos dados não-ajustados. Além disso, se em algum dos grupos não tiver ocorrido o descolamento do prepúcio, deve registar-se a incidência desse descolamento e comparar-se estatisticamente esta última com a do grupo de controlo, utilizando um teste exato de Fisher.
63. Ao verificar a concordância entre os dados inseridos no computador e os constantes das folhas de dados originais, deve estar-se especialmente atento a valores de pesos de órgãos que não sejam biologicamente plausíveis ou se afastem mais de três desvios-padrão da média pertinente do grupo exposto em causa, podendo ser necessário rejeitá-los por serem provavelmente erros de registo.
64. A comparação dos resultados do estudo com os valores de coeficiente de variação determinados pela OCDE (quadro 1) constitui frequentemente uma etapa importante na interpretação da validade dos resultados do estudo. Os laboratórios devem conservar dados históricos dos grupos de controlo do veículo. Devem conservar igualmente dados históricos das respostas a produtos químicos de referência positivos, como o propionato de testosterona e a flutamida. Os laboratórios podem determinar periodicamente a resposta a agonistas e antagonistas reconhecidamente fracos de androgénios, devendo, nesse caso, conservar também tais dados. Estes dados podem ser comparados com dados disponíveis da OCDE, para garantir que os métodos aplicados pelo laboratório têm precisão e poder estatísticos suficientes.
Apêndice 1
DEFINIÇÕES
Androgénico : termo usado para designar uma influência positiva no crescimento de tecidos dependentes dos androgénios.
Antiandrogénico : produto químico capaz de suprimir a ação do propionato de testosterona num mamífero.
Produto químico : substância ou mistura.
Data de nascimento : dia 0 após o nascimento.
Dose : quantidade de produto químico em estudo administrada. No bioensaio de Hershberger, exprime-se em peso diário do produto químico em estudo por unidade de peso corporal do animal ensaiado (por exemplo, mg/kg de peso corporal/dia).
Dosagem : termo geral que abrange a dose, a frequência desta e a duração da administração da mesma.
Moribundo : termo usado para designar um animal que está quase a morrer.
X dias após o nascimento : número X de dias de vida após o dia de nascimento.
Sensibilidade : capacidade de um método de ensaio de identificar corretamente produtos químicos que possuam a propriedade em estudo.
Especificidade : capacidade de um método de ensaio de identificar corretamente produtos químicos que não possuam a propriedade em estudo.
Produto químico em estudo : qualquer substância ou mistura à qual seja aplicado este método de ensaio.
Validação : processo científico concebido para caracterizar as limitações e as condições operatórias dos métodos de ensaio e para demonstrar a fiabilidade e a pertinência dos mesmos para determinado fim.
Apêndice 2

NOTAS EXPLICATIVAS DO QUADRO CONCEPTUAL
|
Nota 1: |
É possível entrar no quadro e dele sair em qualquer nível, consoante a natureza das informações necessárias para a avaliação dos perigos e dos riscos. |
|
Nota 2: |
No nível 5, os métodos ecotoxicológicos devem integrar parâmetros indicativos dos mecanismos dos efeitos indesejáveis e dos mecanismos das consequências negativas potenciais nas populações. |
|
Nota 3: |
Se um modelo multimodal abranger vários dos ensaios de parâmetro único, esse modelo substituirá estes últimos. |
|
Nota 4: |
A avaliação de cada produto químico deve basear-se numa análise caso a caso que tenha em conta todas as informações disponíveis, tendo presente a função dos níveis do quadro. |
|
Nota 5: |
Esta versão do quadro não deve ser considerada exaustiva. Nos níveis 3, 4 e 5, integra ensaios já disponíveis ou em validação. Estes últimos são incluídos a título provisório. Logo que estejam afinados e validados, serão formalmente integrados no quadro. |
|
Nota 6: |
Os ensaios integrados no nível 5 não devem considerar-se unicamente de caráter definitivo. Os ensaios em questão contribuem para a avaliação geral dos perigos e dos riscos. |
REFERÊNCIAS
|
1. |
OCDE (1998). Report of the First Meeting of the OECD Endocrine Disrupter Testing and Assessment (EDTA) Task Force. 10 e 11 de março de 1998. ENV/MC/CHEM/RA(98)5. |
|
2. |
Dorfman R.I. (1962). Standard methods adopted by official organization. Academic Press, NY. |
|
3. |
Gray L.E. Jr., Furr J., Ostby J.S. (2005). Hershberger assay to investigate the effects of endocrine disrupting compounds with androgenic and antiandrogenic activity in castrate-immature male rats. Em: Current Protocols in Toxicology 16.9.1-16.9.15. J Wiley and Sons Inc. |
|
4. |
OCDE (2006). Final OECD report of the initial work towards the validation of the rat Hershberger assay. Phase 1. Androgenic response to testosterone propionate and anti-androgenic effects of flutamide. Environmental Health and Safety, Monograph Series on Testing and Assessment N.o 62. ENV/JM/MONO(2006)30. |
|
5. |
OCDE (2008). Report of the OECD Validation of the Rat Hershberger Bioassay: Phase 2: Testing of Androgen Agonists, Androgen Antagonists and a 5a-Reductase Inhibitor in Dose Response Studies by Multiple Laboratories. Environmental Health and Safety, Monograph Series on Testing and Assessment N.o 86. ENV/JM/MONO(2008)3. |
|
6. |
OCDE (2007). Report of the Validation of the Rat Hershberger Assay: Phase 3: Coded Testing of Androgen Agonists, Androgen Antagonists and Negative Reference Chemicals by Multiple Laboratories. Surgical Castrate Model Protocol. Environmental Health and Safety, Monograph Series on Testing and Assessment N.o 73. ENV/JM/MONO(2007)20. |
|
7. |
Owens, W., Zeiger E., Walker M., Ashby J., Onyon L., Gray, Jr., L.E. (2006). The OECD programme to validate the rat Hershberger bioassay to screen compounds for in vivo androgen and antiandrogen responses. Phase 1: Use of a potent agonist and a potent antagonist to test the standardized protocol. Env. Health Persp. 114:1265-1269. |
|
8. |
Owens W., Gray L.E., Zeiger E., Walker M., Yamasaki K., Ashby J., Jacob E. (2007). The OECD program to validate the rat Hershberger bioassay to screen compounds for in vivo androgen and antiandrogen responses: phase 2 dose-response studies. Environ Health Perspect. 115(5):671-8. |
|
9. |
Korenchevsky V. (1932). The assay of testicular hormone preparations. Biochem J. 26:413-422. |
|
10. |
Korenchevsky V., Dennison M., Schalit R. (1932). The response of castrated male rats to the injection of the testicular hormone. Biochem J. 26:1306-1314. |
|
11. |
Eisenberg E., Gordan G.S. (1950). The levator ani muscle of the rat as an index of myotrophic activity of steroidal hormones. J. Pharmacol. Exp. Therap. 99:38-44. |
|
12. |
Eisenberg E., Gordan G.S., Elliott H.W. (1949). Testosterone and tissue respiration of the castrate male rat with a possible test for mytrophic activity. Endocrinology 45:113-119. |
|
13. |
Hershberger L., Shipley E., Meyer R. (1953). Myotrophic activity of 19-nortestosterone and other steroids determined by modified levator ani muscle method. Proc. Soc. Exp. Biol. Med. 83:175-180. |
|
14. |
Hilgar A.G., Vollmer E.P. (1964). Endocrine bioassay data: Androgenic and myogenic. United States Public Health Service, Washington DC. |
|
15. |
Dorfman R.I. (1969). Androgens and anabolic agents. Em: Methods in Hormone Research, volume IIA. (Dorfman R.I., editores). New York:Academic Press, 151-220. |
|
16. |
Massaro E.J. (2002). Handbook of Neurotoxicology, volume I. New York:Humana Press, p. 38. |
|
17. |
OCDE (2000). Guidance document on the recognition, assessment and use of clinical signs as humane endpoints for experimental animals used in safety evaluation. Environmental Health and Safety Monograph Series on Testing and Assessment No 19. ENV/JM/MONO(2000)7. |
|
18. |
OCDE (1982). Organization for Economic Co-operation and Development — Principles of Good Laboratory Practice. ISBN 92-64-12367-9. Paris. |
|
19. |
OCDE (2008). Acute oral toxicity — up-and-down procedure. OECD Guideline for the testing of chemicals No 425. |
|
20. |
OCDE (2001). Guidance document on acute oral toxicity. Environmental Health and Safety Monograph Series on Testing and Assessment No 24. ENV/JM/MONO(2001)4. |
|
21. |
Supplemental materials for Owens et al. (2006). The OECD programme to validate the rat Hershberger bioassay to screen compounds for in vivo androgen and antiandrogen responses. Phase 1: Use of a potent agonist and a potent antagonist to test the standardized protocol. Env. Health Persp. 114:1265-1269. Ver a secção II: The dissection guidance provided to the laboratories: http://www.ehponline.org/docs/2006/8751/suppl.pdf |
|
22. |
Korea Food and Drug Administration. Visual reference guide on Hershberger assay procedure, including a dissection video: http://rndmoa.kfda.go.kr/endocrine/reference/education_fr.html |
|
23. |
OCDE (2008). Background Review Document on the Rodent Hershberger Bioassay. Environmental Health and Safety Monograph Series on Testing and Assessment No 90. ENV/JM/MONO(2008)17. |
|
24. |
OCDE (2008). Draft Validation report of the Intact, Stimulated, Weanling Male Rat Version of the Hershberger Bioassay. |
|
25. |
OCDE (2009). Guidance Document on the Weanling Hershberger Bioassay in rats: A shortterm screening assay for (anti)androgenic properties. Series on Testing and Assessment, Number 115. |
|
26. |
Diretiva 2010/63/UE do Parlamento Europeu e do Conselho, de 22 de setembro de 2010, relativa à proteção dos animais utilizados para fins científicos (JO L 276 de 20.10.2010, p. 33). |
|
27. |
Os seguintes capítulos do presente anexo:
|
B.56 ESTUDO ALARGADO DE TOXICIDADE DURANTE A REPRODUÇÃO NUMA GERAÇÃO
INTRODUÇÃO
1. Este método de ensaio é equivalente ao Test Guideline TG 443 (2012) da OCDE. Baseia-se na proposta do Comité Técnico para a Avaliação da Segurança dos Produtos Químicos Agrícolas (ACSA) do International Life Science Institute (ILSI) — Health and Environmental Sciences Institute (HESI), para um estudo alargado da reprodução numa geração, no estádio do ciclo de vida F1, como o publicado por Cooper et al., 2006 (1). O protocolo seguido no estudo foi melhorado e esclarecido de modo a proporcionar flexibilidade e a salientar a importância de se partir dos conhecimentos existentes, utilizando observações em vida para orientar e adaptar os ensaios. Este método de ensaio fornece uma descrição detalhada da realização prática de um estudo alargado de toxicidade durante a reprodução numa geração. O método de ensaio descreve três coortes de animais da geração F1:
Coorte 1: avalia os parâmetros(endpoints) de reprodução/desenvolvimento; esta coorte pode ser alargada para incluir uma geração F2.
Coorte 2: avalia o potencial impacto da exposição a produtos químicos no desenvolvimento do sistema nervoso.
Coorte 3: avalia o potencial impacto da exposição a produtos químicos no desenvolvimento do sistema imunitário.
2. As decisões sobre a avaliação ou não da segunda geração e sobre a eventual omissão das coortes para estudo dos efeitos da neurotoxicidade e/ou da imunotoxicidade durante o desenvolvimento devem refletir o conhecimento existente quanto aos produtos químicos a avaliar, bem como as necessidades das diferentes autoridades reguladoras. O objetivo do método de ensaio é apresentar detalhes sobre a forma como o estudo poderá ser realizado e como cada coorte deverá ser avaliada.
3. O procedimento para decidir sobre o desencadeamento interno da produção de uma 2.a geração encontra-se descrito no documento de orientações n.o 117 da OCDE (39), para as autoridades reguladoras que utilizem desencadeadores internos.
OBJETIVOS E CONSIDERAÇÕES INICIAIS
4. O principal objetivo do estudo alargado de toxicidade durante a reprodução numa geração é avaliar estádios específicos do ciclo de vida não cobertos por outros tipos de estudos de toxicidade e testar os efeitos que podem ocorrer em resultado da exposição pré- e pós-natal a produtos químicos. No que se refere aos parâmetros da reprodução, a expetativa é que, numa primeira fase e quando disponível, a informação proveniente de estudos de dosagem repetida [incluindo estudos de despistagem de toxicidade durante a reprodução — p. ex.: TG 422 da OCDE (32)] ou de ensaios de despistagem de desreguladores endócrinos a curto prazo [p. ex.: ensaio uterotrófico — método de ensaio B.54 (36), e ensaio de Hershberger — método de ensaio B.55 (37)] seja utilizada para detetar efeitos nos órgãos reprodutores de machos e fêmeas. Isso pode incluir a espermatogénese (histopatologia testicular) nos machos e os ciclos éstricos, as contagens de folículos/maturação dos ovócitos e integridade dos ovários (histopatologia) para as fêmeas. O estudo alargado de toxicidade durante a reprodução numa geração funcionará portanto como um ensaio de parâmetros da reprodução que requerem a interação de machos com fêmeas, fêmeas com conceptos e fêmeas com a descendência e com a geração F1 até depois da maturidade sexual [consultar o documento de orientações n.o 151 da OCDE (40), que apoia o presente método de ensaio].
5. O método de ensaio é concebido para permitir uma avaliação dos efeitos pré- e pós-natais de produtos químicos no desenvolvimento, bem como uma avaliação rigorosa da toxicidade sistémica em fêmeas grávidas e em lactação, assim como na descendência jovem e adulta. A análise detalhada dos principais parâmetros de desenvolvimento, como a viabilidade da descendência, a saúde neonatal, o estado de desenvolvimento à nascença e o desenvolvimento físico e funcional até à idade adulta, deverá permitir identificar os órgãos a analisar na descendência. Além disso, o estudo fornecerá e/ou confirmará informações sobre os efeitos de um determinado produto químico sobre a integridade e o funcionamento do sistema reprodutor dos machos e fêmeas adultos. São analisados, especificamente mas não exclusivamente, os seguintes parâmetros: função das gónadas, ciclo éstrico, maturação dos espermatozóides no epidídimo, comportamento de acasalamento, conceção, gravidez, parto e lactação. Além disso, a informação obtida nas avaliações da neurotoxicidade no desenvolvimento e imunotoxicidade no desenvolvimento permitirá caracterizar os potenciais efeitos nesses sistemas. Os dados derivados destes ensaios deverão permitir a determinação dos níveis sem efeitos adversos observáveis (NOAEL), dos níveis mínimos com efeitos adversos observáveis (LOAEL) e/ou das doses de referência para os vários parâmetros e/ou ser utilizados para caracterizar efeitos detetados em anteriores estudos de dose repetida e/ou funcionar como orientação para ensaios subsequentes.
6. Na figura 1 é apresentado um esquema do protocolo. O produto químico em estudo é administrado continuamente em doses graduadas a vários grupos de machos e fêmeas na maturidade sexual. Esta geração parental (P) recebe doses do produto durante um período definido pré-acasalamento (selecionado com base na informação disponível sobre o produto químico em estudo, mas com um mínimo de duas semanas) e durante um período de acasalamento de duas semanas. Os machos P continuam a receber tratamento pelo menos até ao desmame da descendência F1. Devem ser tratados durante um mínimo de 10 semanas. Podem ser tratados durante mais tempo, se necessário para esclarecer certos efeitos sobre a reprodução. O tratamento das fêmeas P é continuado durante a gravidez e a amamentação, terminando após o desmame das respetivas ninhadas (ou seja, 8 a 10 semanas de tratamento). A descendência F1 continua a ser tratada com o produto químico em estudo desde o desmame até à idade adulta. Se se pretender avaliar uma segunda geração [consultar o documento de orientações 117 da OCDE (39)], a descendência F1 será mantida em tratamento até ao desmame da geração F2, ou até à conclusão do estudo.
7. As observações clínicas e os exames patológicos são realizados em todos os animais para deteção de sinais de toxicidade, com especial ênfase sobre a integridade e o funcionamento dos sistemas reprodutores masculino e feminino e sobre a saúde, crescimento, desenvolvimento e funções da descendência. No momento do desmame, a descendência selecionada é atribuída a subgrupos específicos (coortes 1-3; ver os pontos 33 e 34 e a figura 1) para a continuação dos estudos, incluindo a maturação sexual, as funções e integridade dos órgãos reprodutores, os parãmetros neurológicos e comportamentais ou as funções imunitárias.
8. Na realização do estudo, deverão ser seguidos os princípios de orientação e as considerações descritas no documento de orientações 19 da OCDE quanto ao reconhecimento, avaliação e utilização dos sinais clínicos como parâmetros de tratamento humano para experiências com animais utilizados em avaliações de segurança (34).
9. Quando estiver disponível um número suficiente de estudos para verificar o impacto deste novo protocolo, o método de ensaio será reanalisado e, se necessário, revisto à luz da experiência adquirida.
Figura 1
Esquema do “Estudo alargado de toxicidade durante a reprodução numa geração”
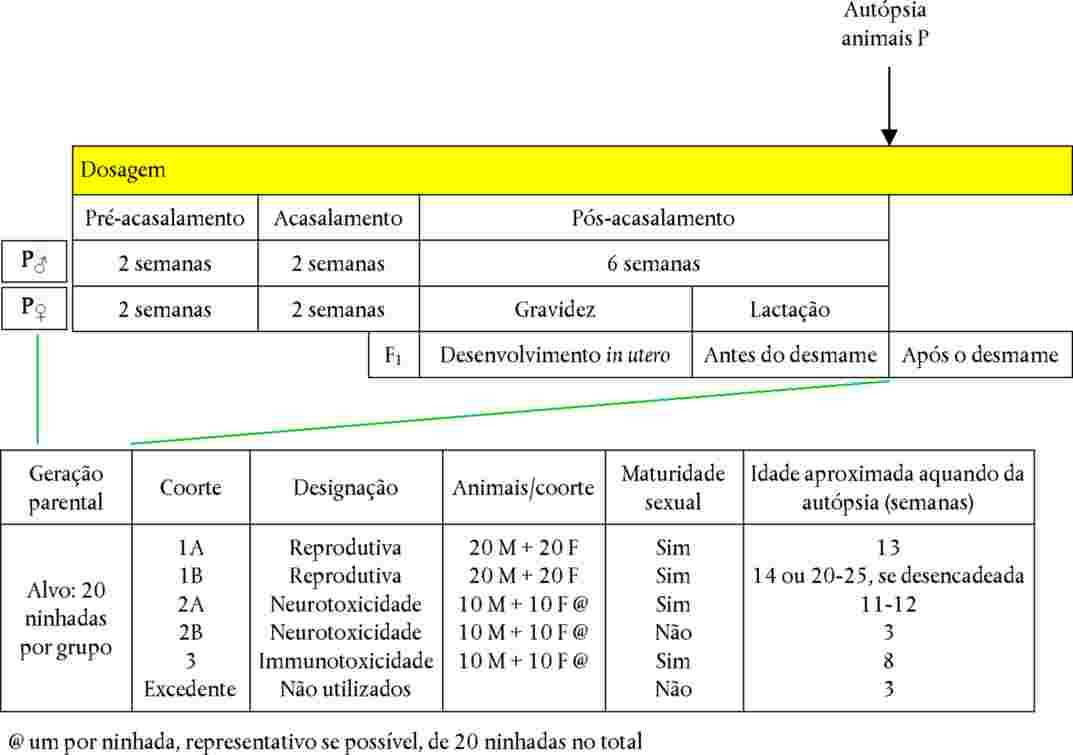
DESCRIÇÃO DO MÉTODO/PREPARAÇÃO PARA O ENSAIO
Animais
Seleção da estirpe e espécie animal
10. A escolha da espécie para o ensaio de toxicidade durante a reprodução deve ser cuidadosamente considerada à luz de todas as informações disponíveis. Contudo, tendo em conta a grande quantidade de dados contextuais e a comparabilidade com ensaios de toxicidade geral, o rato é normalmente a espécie preferida, pelo que os critérios e recomendações fornecidos neste método de ensaio se referem a esta espécie. Se for utilizada outra espécie, deve ser fornecida uma justificação e será necessário proceder às modificações adequadas ao protocolo. As estirpes com níveis baixos de fecundidade ou com elevada incidência bem conhecida de defeitos de desenvolvimento espontâneos não devem ser utilizadas.
Idade, peso corporal e critérios de inclusão
11. Devem ser utilizados animais progenitores saudáveis, que não tenham sido sujeitos a procedimentos experimentais anteriores. Os estudos deverão incluir machos e fêmeas, sendo que as fêmeas devem ser nulíparas e não devem estar grávidas. Os animais P devem ser animais maturos, de peso semelhante (considerando o respetivo sexo) no início da administração do produto químico, com uma idade semelhante (aproximadamente 90 dias) no momento do acasalamento e representativos da espécie e estirpe em estudo. Os animais devem ser aclimatizados durante pelo menos 5 dias após a chegada. Os animais são aleatoriamente distribuídos pelos grupos de controlo e tratamento, de tal forma que resulte em valores médios de peso corporal comparáveis entre os grupos (ou seja, ± 20 % da média).
Condições de alojamento e alimentação
12. A temperatura na sala onde se encontram os animais do estudo deve ser de 22 °C (± 3 °C). A humidade relativa deve estar entre 30-70 %, com uma gama ideal de 50-60 %. A iluminação artificial deve seguir um regime de 12 horas com luz e 12 horas sem luz. Podem ser usadas dietas convencionais de laboratório, com acesso ilimitado a água potável. Deve ser dada especial atenção ao teor de fitoestrogénios na dieta, uma vez que um elevado nível de fitoestrogénios na dieta poderá afetar alguns dos parâmetros de reprodução. Recomendam-se dietas normalizadas de fórmula aberta com utilização reduzida de produtos químicos estrogénicos (2) (30). A escolha da dieta pode ser influenciada pela necessidade de garantir uma mistura adequada do produto químico em estudo, quando administrado por essa via. Devem verificar-se o teor, a homogeneidade e a estabilidade do produto químico em estudo na dieta. Os alimentos e a água potável devem ser analisados regulamente para detetar eventuais contaminantes. Devem ser mantidas amostras de cada lote da dieta utilizada durante o estudo, em condições adequadas (p. ex.: congeladas a –20 °C), até à finalização do relatório, para o caso de os resultados exigirem análises mais aprofundadas dos ingredientes da dieta.
13. Os animais devem ser colocados em gaiolas em pequenos grupos do mesmo sexo e grupo de tratamento. Podem ser alojados individualmente para evitar potenciais ferimentos (p. ex.: machos após o período de acasalamento). O acasalamento deve ocorrer em gaiolas adequadas. Constatada a copulação, as fêmeas que se presuma estarem grávidas devem ser alojadas em separado, em gaiolas de maternidade ou parto, onde lhes devem ser fornecidos materiais adequados e definidos para nidificação. As ninhadas devem ser alojadas com as respetivas progenitoras até ao desmame. Os animais F1 devem ser alojados em pequenos grupos do mesmo sexo e grupo de tratamento, desde o desmame até à conclusão do estudo. Caso se justifique do ponto de vista científico, os animais podem ser alojados individualmente. O nível de fitoestrogénios presente no material para nidificação deve ser mínimo.
Número e identificação dos animais
14. Normalmente, cada grupo de ensaio e de controlo deve conter um número de pares de animais em fase de acasalamento suficiente para gerar, pelo menos, 20 fêmeas grávidas por grupo de dosagem. O objetivo é produzir gravidezes suficientes para assegurar uma avaliação significativa do potencial que o produto químico tem para afetar a fertilidade, a gravidez e o comportamento materno da geração P e o crescimento e desenvolvimento da descendência F1, desde a conceção até à maturidade. O facto de não se obter o número desejado de fêmeas grávidas não invalida necessariamente o estudo, decisão que deverá ser tomada caso a caso, tendo em consideração uma potencial relação causal com o produto químico em estudo.
15. Cada animal P recebe um número de identificação único, antes do início da administração da dose prevsta do produto. Se existirem dados de histórico laboratorial que sugiram que uma percentagem significativa de fêmeas poderá não apresentar ciclos éstricos regulares (4 ou 5 dias), aconselha-se a realização de uma avaliação dos ciclos éstricos antes do início do tratamento. Em alternativa, a dimensão do grupo pode ser aumentada de modo a assegurar que pelo menos 20 fêmeas em cada grupo apresentem ciclos éstricos regulares (4 ou 5 dias) no início do tratamento. Todos os idivíduos da geração F1 são identificados com um número único quando os neonatos são examinados pela primeira vez no dia 0 ou 1 após o nascimento (PND). Devem ser mantidos registos com indicação das ninhadas e da sua origem para todos os animais F1, e F2, quando aplicável, durante todo o estudo.
Produto químico em estudo
Informações disponíveis sobre o produto químico em estudo
16. A análise das informações existentes é importante para as decisões sobre a via de administração, a escolha do veículo, a seleção da espécie animal, a seleção das dosagens e as potenciais modificações do esquema de dosagem. Assim, ao planear o “Estudo alargado de toxicidade durante a reprodução numa geração”, devem ser tidas em consideração todas as informações disponíveis relevantes sobre o produto químico em estudo, ou seja, propriedades físico-químicas, toxicocinéticas (incluindo o metabolismo específico da espécie) e toxicodinâmicas, relações estrutura/atividade (SAR), processos metabólicos in vitro, resultados de estudos de toxicidade anteriores e informações relevantes sobre produtos com uma estrutura análoga. As informações preliminares sobre a absorção, distribuição, metabolismo e eliminação (ADME) e sobre a bioacumulação podem ser derivadas a partir da estrutura química, de dados físico-químicos, da importância das ligações às proteínas plasmáticas ou de estudos toxicocinéticos (TK), ao passo que os resultados de estudos de toxicidade poderão fornecer informações adicionais, por exemplo, sobre os NOAEL, o metabolismo ou a indução desse mesmo metabolismo.
Consideração de dados de toxicocinética
17. Embora não sejam necessários, os dados de toxicocinética de estudos anteriormente realizados para a determinação de intervalos de dosagem ou outros são extremamente úteis para o planeamento da conceção do estudo, escolha dos níveis de dosagem e interpretação de resultados. Têm especial utilidade os dados que: 1) confirmam a exposição dos fetos em desenvolvimento e das crias ao produto químico em estudo (ou aos metabolitos relevantes); 2) fornecem uma estimativa da dosimetria interna; e 3) avaliam a potencial saturação dependente da dosagem dos processos cinéticos. Os dados toxicocinéticos adicionais, tais como perfis de metabolitos, dados de concentração ao longo do tempo, etc., devem também ser considerados, se disponíveis. Podem ainda ser recolhidos dados toxicocinéticos suplementares durante o estudo principal, desde que tal não interfira com a recolha e interpretação dos parâmetros do estudo principal.
Como orientação geral, o seguinte conjunto de dados toxicocinéticos seria útil no planeamento do “Estudo alargado de toxicidade durante a reprodução numa geração”:
|
— |
Fase final da gravidez (p. ex.: 20.o dia de gestação) — sangue materno e sangue fetal; |
|
— |
Fase intermédia de amamentação (10.o dia após o nascimento) — sangue materno, sangue da cria e/ou leite; |
|
— |
Fase inicial do pós-desmame (p. ex.: 28.o dia após o nascimento) — amostras de sangue em fase de desmame. |
Deve haver flexibilidade na determinação das substâncias específicas a analisar (p. ex.: produto químico principal e/ou metabolitos) e do regime de amostragem. Por exemplo, o número e o momento de colheita das amostras num determinado dia de amostragem dependerão da via de exposição e do conhecimento prévio das propriedades toxicocinéticas em animais não grávidos. Em estudos sobre a dieta, será suficiente uma amostra sempre à mesma hora em cada dia, ao passo que a administração por gavagem poderá exigir amostragens adicionais de modo a obter uma melhor estimativa do intervalo de dosagem interna. Contudo, não é necessário produzir um gráfico completo da dosagem ao longo do tempo para todos os dias em que são recolhidas amostras. Se necessário, o sangue pode ser agregado por sexo dentro de cada ninhada para as análises fetais e neonatais.
Via de administração
18. A seleção da via deve ter em consideração a(s) vias(s) mais relevante(s) para a exposição humana. Embora o protocolo seja concebido para administração do produto químico em estudo através da dieta, pode ser modificado para administração por outras vias (água potável, gavagem, inalação, derme), dependendo das características do produto químico e das informações que se pretendem obter.
Escolha do veículo
19. Quando necessário, o produto químico em estudo pode ser dissolvido ou suspenso num veículo adequado. Recomenda-se que, quando possível, seja considerada primeiro a utilização de uma solução/suspensão aquosa, antes da possibilidade de uma solução/suspensão oleosa (p. ex., óleo de milho). No que se refere a outros veículos que não a água, devem ser conhecidas as características tóxicas do veículo. Deve ser evitada a utilização de veículos com potencial toxicidade intrínseca (p. ex., acetona, DMSO). A estabilidade do produto químico em estudo no veículo deve ser determinada. Se for utilizado um veículo ou outro aditivo para facilitar a dosagem, devem consideradar-se as seguintes características: efeitos na absorção, distribuição, metabolismo ou retenção do produto químico em estudo; efeitos sobre as propriedades químicas do produto químico em estudo que possam alterar as respetivas características de toxicidade; e efeitos sobre o consumo de alimentos ou água ou o estado nutricional dos animais.
Seleção da dose
20. Normalmente, o estudo deve incluir, pelo menos, três níveis de dosagem e um controlo simultâneo. Ao selecionar os níveis de dosagem adequados, o investigador deve considerar todas as informações disponíveis, incluindo as informações de dosagem de estudos anteriores, dados toxicocinéticos em animais grávidos ou não grávidos, importância da transferência na amamentação e estimativas da exposição humana. Se estiverem disponíveis dados de toxicocinética que indiquem uma saturação dos processos toxicocinéticos dependente da dosagem, deve ter-se o cuidado de evitar dosagens elevadas que resultem claramente em saturação, desde que, como é evidente, se preveja que a exposição dos seres humanos seja muito inferior ao ponto de saturação. Nesses casos, a dosagem mais elevada deverá ser igual ou ligeiramente superior ao ponto de inflexão na transição para um comportamento toxicocinético não-linear.
21. Na ausência de dados toxicocinéticos relevantes, as dosagens devem basear-se nos efeitos tóxicos, salvo quando existam limitações devidas à natureza físico-química do produto químico em estudo. Se os níveis de dosagem se basearem na toxicidade, a dose mais elevada deve ser escolhida com o objetivo de induzir alguma toxicidade sistémica, mas não a morte ou grande sofrimento dos animais.
22. Deve ser selecionada uma sequência descendente de níveis de dosagem para demonstrar qualquer efeito relacionado com a dose e para estabelecer os NOAEL ou dosagens próximas do limite de deteção que permitam deduzir uma dose de referência para o(s) parâmetro(s) mais sensíveis. Para evitar um grande espaçamento de doses entre os NOAEL e os LOAEL, o ideal será normalmente utilizar intervalos com uma ordem de grandeza do dobro ou do quádruplo. Em geral, será preferível acrescentar um quarto grupo de ensaio do que utilizar intervalos demasiado grandes (p. ex.: com um fator de multiplicação superior a 10) entre as doses.
23. Exceto no que se refere ao tratamento com o produto químico em estudo, os animais no grupo de controlo são tratados de forma idêntica aos indivíduos no grupo de ensaio. O grupo de controlo não deverá receber tratamento, deverá receber um tratamento similado ou constituirá um grupo de controlo do veículo em caso de utilização de um veículo para administração do produto químico em estudo. Se for utilizado um veículo, o grupo de controlo deve receber o volume mais elevado desse veículo utilizado no estudo.
Ensaio de limite
24. Caso não haja evidência de toxicidade com uma dose de pelo menos 1 000 mg/kg de peso corporal/dia em estudos de dose repetida, ou se não for esperada toxicidade com base nos dados de produtos químicos estrutural e/ou metabolicamente relacionados, indicando similaridade nas propriedades metabólicas in vivo/in vitro, poderá não ser necessário realizar um estudo com várias dosagens. Nesses casos, o “Estudo alargado de toxicidade durante a reprodução numa geração” pode ser realizado utilizando um grupo de controlo e uma dose única pelo menos igual a 1 000 mg/kg de peso corporal/dia. Contudo, caso sejam detetadas evidências de toxicidade durante a reprodução ou no desenvolvimento com este limite de dosagem, serão necessários mais estudos com dosagens inferiores para identificação de um NOAEL. Estas considerações sobre os ensaios de limite aplicam-se apenas quando a exposição humana não indicar a necessidade de ensaiar uma dosagem superior.
PROCEDIMENTOS
Exposição da descendência
25. A exposição na dieta é o método de administração preferencial. Se forem realizados estudos com gavagem, importa notar que normalmente as crias apenas receberão o produto químico em estudo de forma indireta, através do leite, até ao início da respetiva administração direta, a partir do desmame. Em estudos com administração na dieta ou na água potável, as crias receberão adicionalmente o produto químico em estudo de forma direta quando começarem a alimentar-se por si próprias, durante a última semana do período de amamentação. Devem ser consideradas modificações à conceção do estudo nos casos em que a excreção do produto químico em estudo no leite for fraca e em que haja falta de evidência para uma exposição contínua da descendência. Nestes casos, a dosagem direta às crias durante o período de amamentação deve ser considerada com base nas informações de toxicocinética disponíveis, toxicidade na descendência ou alterações em biomarcadores (3) (4). Os benefícios e as desvantagens d evem ser cuidadosamente considerados antes da realização de estudos de dosagem direta em crias em aleitamento (5).
Esquema de dosagem e administração das doses
26. Poderão existir informações disponíveis sobre ciclos éstricos, histopatologia do aparelho reprodutor masculino e feminino e análise de esperma nos testículos/epidídimo de estudos anteriores de toxicidade de dose repetida com duração adequada. A duração do tratamento pré-acasalamento no “Estudo alargado de toxicidade durante a reprodução numa geração” visa, portanto, detetar os efeitos de alterações funcionais que possam interferir com o comportamento de acasalamento e fertilização. O tratamento pré-acasalamento deve ser suficientemente longo para atingir condições de exposição em estado de equilíbrio nos machos e fêmeas da geração P. Um tratamento pré-acasalamento de 2 semanas para ambos os sexos é considerado adequado na maioria dos casos. Para as fêmeas, esse período abrange 3-4 ciclos éstricos completos e deve ser suficiente para detetar quaisquer efeitos adversos nesses ciclos. Para os machos, equivale ao tempo necessário para o trânsito dos espermatozóides em maturação pelo epidídimo e deverá permitir detetar efeitos pós-testiculares no esperma (durante as fases finais da espermiação e maturação dos espermatozóides no epidídimo) no acasalamento. No momento terminal, para o qual estejam programados os parâmetros de análise do esperma e de histopatologia dos testículos e epidídimo, os machos P e F1 terão estado expostos durante pelo menos um ciclo espermatogénico completo [(6) (7) (8) (9) e documento de orientações 151 da OCDE (40)].
27. Os cenários de exposição pré-acasalamento para os machos podem ser adaptados se estudos anteriores tiverem identificado claramente toxicidade testicular (espermatogénese negativamente afetada) ou efeitos na integridade e função do esperma. Da mesma forma, para as fêmeas, os efeitos conhecidos do produto químico em estudo sobre o ciclo éstrico e, por conseguinte, sobre a recetividade sexual, poderão justificar diferentes cenários de exposição pré-acasalamento. Em casos especiais, poderá ser aceitável que o tratamento das fêmeas P seja iniciado apenas após a obtenção de um esfregaço com presença de esperma [ver documento de orientações 151 da OCDE (40)].
28. Quando o período de dosagem pré-acasalamento estiver estabelecido, os animais devem ser tratados continuamente com o produto químico em estudo 7 dias/semana até à autópsia. Todos os animais devem receber a dosagem pelo mesmo método. A dosagem deve prosseguir durante o período de acasalamento de 2 semanas e, no caso das fêmeas P, ao longo da gestação e da amamentação até ao dia do término após o desmame. Os machos devem ser tratados da mesma forma até ao término, no momento em que os animais F1 são desmamados. Para a autópsia, deve ser dada prioridade às fêmeas, que devem ser autopsiadas no próprio dia ou próximo do dia da lactação. A autópsia dos machos pode ser distribuída por vários dias, em função das instalações laboratoriais. Salvo nos casos em que já tenha sido iniciada durante o período de amamentação, a dosagem direta dos machos e fêmeas F1 selecionados deve ser iniciada aquando do desmame e continuar até à autópsia programada, dependendo da distribuição pelas coortes.
29. No caso de produtos químicos administrados através da dieta ou da água potável, é importante assegurar que as quantidades do produto químico em estudo envolvidas não interferem com a nutrição normal ou com a composição da água. Quando o produto químico em estudo é administrado na dieta, pode aplicar-se uma concentração constante (ppm) ou um nível constante de dosagem em termos do peso corporal do animal; a opção escolhida deve ser especificada.
30. Quando o produto químico em estudo é administrado por gavagem, o volume de líquido administrado de uma só vez não deverá normalmente exceder 1 ml/100 g de peso corporal (0,4 ml/100 g de peso corporal é o máximo no caso de utilização de óleos, por exemplo óleo de milho). Exceto no caso de produtos químicos irritantes ou corrosivos, que normalmente revelam efeitos exacerbados em concentrações superiores, a variabilidade no volume de ensaio deve ser minimizada ajustando a concentração de modo a garantir um volume constante em todos os níveis de dosagem. O tratamento deve ser fornecido aproximadamente à mesma hora em cada dia. Normalmente, a dose para cada animal deve basear-se na determinação mais recente do peso corporal de cada indivíduo e decverá ser ajustada pelo menos uma vez por semana nos machos adultos e nas fêmeas adultas não grávidas, e a cada dois dias para as fêmeas grávidas e os animais F1, quando a administração for efetuada antes do desmame e durante as 2 semanas após o desmame. Se os dados de toxicocinética indicarem uma baixa transferência placentária do produto químico em estudo, poderá ser necessário ajustar a dose de gavagem durante a última semana da gravidez, de modo a evitar a administração de uma dose excessivamente tóxica à progenitora. As fêmeas não devem ser tratadas por gavagem ou por qualquer outra via de tratamento em que o animal tenha de ser manuseado, no dia do parto; a omissão da administração do produto químico em estudo nesse dia é preferível à perturbação do processo de parto.
Acasalamento
31. Cada fêmea P deve ser colocada com um único macho não aparentado e escolhido aleatoriamente do mesmo grupo de dosagem (emparelhamento 1:1) até que seja observada evidência de copulação ou que tenham passado 2 semanas. Se não houver machos suficientes, por exemplo devido a morte de machos antes do emparelhamento, o(s) macho(s) que já tenha(m) acasalado pode(m) ser emparelhado(s) (1:1) com outra(s) fêmea(s), de modo a que todas as fêmeas sejam emparelhadas. O dia 0 da gravidez é definido como o dia em que é confirmada a evidência de acasalamento (deteção de rolhão vaginal ou esperma). Os animais devem ser separados logo que possível após observação da evidência de copulação. Se não tiver ocorrido acasalamento após 2 semanas, os animais devem ser separados sem que haja mais oportunidade de acasalamento. Os pares acasalados devem ser claramente identificados nos dados.
Dimensão da ninhada
32. No 4.o dia após o nascimento, a dimensão de cada ninhada pode ser ajustada eliminando as crias em excesso por seleção aleatória, de modo a obter o valor mais próximo possível de cinco machos e cinco fêmeas por ninhada. A eliminação seletiva de crias, por exemplo com base no peso corporal, não será adequada. Sempre que o número de crias macho ou fêmea não permitir a existência de cinco de cada sexo por ninhada, poderá proceder-se a um ajuste parcial (p. ex.: seis machos e quatro fêmeas).
Seleção de crias para estudos pós-desmame (ver Figura 1)
33. Aquando do desmame (cerca do 21.o dia após o nascimento), são selecionadas para exames posteriores crias de todas as ninhadas disponíveis, até 20 por grupo de controlo e dose, que serão mantidas até à maturação sexual (a não ser que sejam necessários ensaios mais precoces). As crias são selecionadas aleatoriamente, com a exceção de que não devem ser incluídos animais de tamanho claramente inferior ao normal (animais com peso corporal mais de dois desvios padrão abaixo do peso corporal médio das crias da respetiva ninhada), na medida em que não serão provavelmente representativos do grupo de tratamento.
No 21.o dia após o nascimento, as crias F1 selecionadas são atribuídas aleatoriamente a uma das três coortes de animais, da seguinte forma:
Coorte 1 (1A e 1B)= Ensaio de toxicidade durante a reprodução/no desenvolvimento
Coorte 2 (2A e 2B)= Ensaio de neurotoxicidade no desenvolvimento
Coorte 3= Ensaio de imunotoxicidade no desenvolvimento
Coorte 1A: Um macho e uma fêmea/ninhada/grupo (20/sexo/grupo): seleção prioritária para avaliação primária dos efeitos sobre os sistemas reprodutivos e da toxicidade geral.
Coorte 1B: Um macho e uma fêmea/ninhada/grupo (20/sexo/grupo): seleção prioritária para avaliação e seguimento do comportamento reprodutivo através do acasalamento de animais F1, quando avaliados [consultar o documento de orientações 117 da OCDE (39)] e para obtenção de dados adicionais de histopatologia em casos de suspeita de tóxicos para o sistema reprodutivo ou endócrino, ou quando os resultados da coorte 1A não forem inequívocos.
Coorte 2A: Total de 20 crias por grupo (10 machos e 10 fêmeas por grupo; um macho ou uma fêmea por ninhada) destinadas a ensaios neurocomportamentais, seguidos de avaliação neuro-histopatológica quando chegados à fase adulta.
Coorte 2B: Total de 20 crias por grupo (10 machos e 10 fêmeas por grupo; um macho ou uma fêmea por ninhada) destinadas a avaliação neuro-histopatológica no desmame (21.o ou 22.o dia após o nascimento). Se o número de animais for insuficiente, deve dar-se preferência à colocação dos animais na Coorte 2A.
Coorte 3: Total de 20 crias por grupo (10 machos e 10 fêmeas por grupo; um por ninhada, quando possível). Poderá ser necessário utilizar mais crias do grupo de controlo como animais de controlo positivo no ensaio de resposta de anticorpos dependentes de células T (TDAR) no 56.o ± 3 dia após o nascimento.
34. Caso não exista um número insuficiente de crias numa ninhada para atribuição a todas as coortes, a coorte 1 tem precedência, já que poderá ser conservada para produzir uma geração F2. Podem ser atribuídas mais crias a qualquer uma das coortes em caso de preocupações específicas; por exemplo, se houver suspeita de que um produto químico é para o sistema nervoso, imunitário ou reprodutivo. Estas crias podem ser utilizadas para exames em diferentes momentos ou para a avaliação de parâmetros suplementares. As crias não afetadas a uma dterminada coorte serão analisadas na ótica da bioquímica clínica (parágrafo 55) e sujeitas a uma autópsia macroscópica (parágrafo 68).
Segundo acasalamento dos animais P
35. Normalmente, não se recomenda um segundo acasalamento dos animais P, já que isso poderá implicar a perda de informações importantes sobre o número de locais de implantação (e, portanto, de dados sobre as perdas pós-implantação e perinatais, indicadoras de um possível potencial teratogénico) na primeira ninhada. A necessidade de verificar ou elucidar um determinado efeito em fêmeas expostas terá uma melhor resposta alargando o estudo de modo a incluir um acasalamento da geração F1. Contudo, um segundo acasalamento dos machos P com fêmeas não tratadas será sempre uma opção para esclarecer os resultados que não sejam inequívocos ou para uma caracterização mais aprofundada dos efeitos sobre a fertilidade observados no primeiro acasalamento.
OBSERVAÇÕES EM VIDA
Observações clínicas
36. Para os animais P e os animais F1 selecionados, é realizada uma observação clínica geral uma vez por dia. Em caso de administração por gavagem, as observações clínicas deverão ter lugar antes e depois da administração (para possíveis sinais de toxicidade associada ao pico de concentração plasmática). Registam-se as alterações comportamentais pertinentes, os sinais de parto difícil ou prolongado e todos os sinais de toxicidade. Duas vezes por dia (durante o fim de semana, uma vez por dia), todos os animais devrão ser observados em termos de toxicidade grave, morbilidade e mortalidade.
37. Além disso, deverá realizar-se semanalmente um exame mais detalhado de todos os animais P e F1 (após o desmame), que para maior conveniência poderá realizar-se numa ocasião em que o animal seja pesado, o que minimiza o stress do manuseamento. As observações deverão ser cuidadosamente realizadas e registadas utilizando sistemas de classificação definidos pelo laboratório de ensaio. Devem ser envidados esforços para garantir um mínimo de variação nas condições de ensaio. Os sinais a ter em conta devem incluir, sem caráter limitativo, alterações na pele, pêlo, olhos, membranas mucosas, ocorrência de secreções e excreções e as atividades autónomas (p. ex., lacrimação, piloereção, tamanho das pupilas, padrão respiratório anómalo). Devem também ser registadas as alterações na marcha, postura, resposta ao manuseamento, bem como a presença de movimentos clónicos ou tónicos, estereotipia (p. ex., excesso de limpeza, movimentos repetitivos em círculo) ou comportamentos estranhos (p. ex., automutilação, marcha para a retaguarda).
Peso corporal e consumo de alimentos/água
38. Os animais P são pesados no primeiro dia da dosagem e, depois disso, pelo menos uma vez por semana. Adicionalmente, as fêmeas P são pesadas durante a amamentação nos mesmos dias da pesagem das crias das respetivas ninhadas (ver parágrafo 44). Todos os animais F1 são pesados individualmente no desmame (21.o dia após o nascimento) e, depois disso, pelo menos uma vez por semana. O peso corporal deverá também ser registado no dia em que atingem a puberdade (conclusão da separação prepucial ou abertura da vagina). Todos os animais são pesados aquando do sacrifício.
39. Durante o estudo, o consumo de alimentos e água (caso a administração do produto químico em estudo seja feita na água potável) é registado pelo menos uma vez por semana, nos mesmos dias das pesagens dos animais (exceto durante a coabitação). O consumo de alimentos de cada gaiola de animais F1 é registado semanalmente, a partir da colocação numa determinada coorte.
Ciclos éstricos
40. Poderão já estar disponíveis informações preliminares sobre os efeitos relacionados com o produto químico em estudo no ciclo éstrico, obtidas em anteriores estudos de toxicidade de dose repetida, que podem ser utilizadas na conceção de um protocolo de ensaio específico para o “Estudo alargado de toxicidade durante a reprodução numa geração” do produto químico em estudo. Normalmente, a avaliação do ciclo éstrico (por citologia vaginal) começará no início do período de tratamento e continuará até confirmação do acasalamento ou até ao final do período de acasalamento de 2 semanas. Se as fêmeas tiverem sido sujeitas a despistagem para determinar se o seu ciclo éstrico é normal antes do tratamento, será útil continuar a obter esfregaços após o início do tratamento, mas se existirem preocupações quanto a efeitos não específicos no início do tratamento (nomeadamente uma redução inicial marcada no consumo de alimentos), pode permitir-se que os animais se adaptem ao tratamento durante um período máximo de duas semanas antes de iníciar o período de recolha de esfregaços nas 2 semanas anteriores ao emparelhamento. Se o período de tratamento da fêmea for alargado desta forma (ou seja, com um tratamento pré-acasalamento de 4 semanas), deve considerar-se a aquisição de animais mais jovens e o alargamento do período de tratamento dos machos antes do emparelhamento. Ao obter células vaginais/cervicais, deve ter-se o cuidado de evitar perturbações na mucosa e, por consequência, a indução de pseudogravidezes (10) (11).
41. Os esfregaços vaginais devem ser examinados diariamente para todas as fêmeas F1 da coorte 1A, desde o início da abertura da vagina até que se registe o primeiro esfregaço cornificado, de modo a determinar o período entre os dois eventos. Os ciclos éstricos de todas as fêmeas F1 na coorte 1A devem também ser acompanhados durante um período de duas semanas, iniciado por volta do 75.o dia após o nascimento. Além disso, caso seja necessário o acasalamento da geração F1, a citologia vaginal na coorte 1B será seguida desde o momento do emparelhamento até que seja detetada evidência de acasalamento.
Acasalamento e gravidez
42. Além dos parâmetros padrão (p. ex., peso corporal, consumo de alimentos, observações clínicas, incluindo verificações da mortalidade/morbilidade), são registadas as datas de emparelhamento, a data de inseminação e a data de parto, e calculados o intervalo pré-coital (do emparelhamento até à inseminação) e a duração da gravidez (da inseminação até ao parto). As fêmeas P devem ser examinadas cuidadosamente para sinais de distócia na altura prevista para o parto. Quaisquer anomalias no comportamento de nidificação ou no aleitamento devem ser registadas.
43. O dia do parto é o dia 0 de amamentação (LD 0) para a progenitora e o dia 0 após o nascimento (PND 0) para a descendência. Em alternativa, todas as comparações poderão também basear-se no tempo pós-coital, para evitar que os dados do desenvolvimento pós-natal sejam afetados por diferenças na duração da gravidez; contudo, deve também ser registado o período decorrido desde o parto. Isso será especialmente importante quando o produto químico em estudo tem influência sobre a duração da gravidez.
Parâmetros de descendência
44. Cada ninhada deve ser examinada o mais cedo possível após o nascimento (dia 0 ou 1 após o nascimento) de modo a estabelecer o número e o sexo das crias, de nados-mortos, de nados-vivos e a presença de anomalias graves (anomalias visíveis externamente, incluindo fenda palatina, hemorragias subcutâneas, textura ou cor anómala da pele, presença de cordão umbilical, ausência de leite no estômago, presença de secreções secas). Além disso, o primeiro exame clínico dos neonatos deve incluir uma avaliação qualitativa da temperatura corporal, do estado de atividade e da reação ao manuseamento. As crias que sejam encontradas mortas no dia 0 após o nascimento ou mais tarde devem ser examinadas quanto a possíveis deficiências e às causas de morte. As crias vivas devem ser contadas e pesadas individualmente no dia 0 ou 1 após o nascimento e, depois disso, regularmente, pelo menos no 4.o, 7.o, 14.o e 21.o dia após o nascimento. Os exames clínicos aplicáveis à idade dos animais devem ser repetidos quando a descendência for pesada ou com maior frequência caso tenham sido feitas constatações específicas à nascença. Os sinais a ter em conta incluem, sem caráter limitativo: anomalias externas, alterações na pele, pelo, olhos, membranas mucosas, ocorrência de secreções e excreções e atividade autónoma. Também devem ser registadas as alterações na marcha, postura, resposta ao manuseamento, bem como a presença de movimentos clónicos ou tónicos, estereotipia ou comportamento estranho.
45. A distância anogenital (AGD) de cada cria deve ser medida pelo menos uma vez entre o dia 0 após o nascimento e o dia 4 após o nascimento. O peso corporal da cria deve ser recolhido no dia em que a AGD for medida e as AGD devem ser normalizadas de acordo com uma medida do tamanho da cria, preferencialmente a raiz cúbica do peso corporal (12). A presença de mamilos/auréolas nas crias macho deve ser verificada no 12.o ou 13.o dia após o nascimento.
46. Todos os animais F1 selecionados são avaliados diariamente quanto à separação balanoprepucial ou abertura da vagina nos machos e fêmeas, respetivamente, com início antes do dia previsto para o surgimento desses sinais, de modo a detetar se a maturação sexual ocorre prematuramente. Devem ter-se em atenção quaisquer anomalias dos órgãos genitais, tais como filamentos vaginais persistentes, hipospadia ou epispádia do pénis. A maturidade sexual dos animais F1 é comparada com o desenvolvimento físico através da determinação da idade e do peso corporal aquando da separação balanoprepucial ou da abertura da vagina nos machos e fêmeas, respetivamente (13).
Avaliação da potencial neurotoxicidade no desenvolvimento (coortes 2A e 2B)
47. Os animais da coorte 2A (10 machos e 10 fêmeas) e da coorte 2B (10 machos e 10 fêmeas) de cada grupo de tratamento (para cada coorte: 1 macho ou 1 fêmea por ninhada; todas as ninhadas representadas por pelo menos 1 cria; seleção aleatória) devem ser utilizados para avaliações de neurotoxicidade. Os animais da coorte 2A devem ser sujeitos a avaliações da resposta auditiva, de um conjunto de observações funcionais, da atividade motora (ver parágrafos 48-50) e da neuropatologia (ver parágrafos 74-75). Devem ser envidados esforços para garantir que as variações em todas as condições de ensaio sejam mínimas e não estejam sistematicamente relacionadas com o tratamento. Entre as variáveis que podem afetar o comportamento encontram-se o nível sonoro (p. ex., ruído intermitente), a temperatura, a humidade, a iluminação, os odores, a hora do dia e os fatores ambientais. Os resultados dos ensaios de neurotoxicidade devem ser interpretados em relação a valores de referência histórica de controlo adequados. Os animais da coorte 2B devem ser utilizados para avaliação neuropatológica no 21.o ou 22.o dia após o nascimento (ver parágrafos 74-75).
48. Deve ser realizado um ensaio de resposta auditiva no 24.o dia após o nascimento (± 1 dia) utilizando animais da coorte 2A. O dia do ensaio deve ser distribuído de forma equilibrada no interior dos grupos tratados e de controlo. Cada sessão é constituída por 50 ensaios. Na realização do ensaio de resposta auditiva, deve ser determinada a amplitude média de resposta em cada bloco de 10 ensaios (5 blocos de 10 ensaios), com condições de ensaio otimizadas para produzir habituação entre sessões. Estes procedimentos devem estar em conformidade com o método de ensaio B.53 (35).
49. Num momento adequeado, entre o 63.o e o 75.o dia após o nascimento, os animais da coorte 2A são sujeitos a um conjunto de observações funcionais e a um ensaio automatizado da atividade motora. Estes procedimentos devem ser conformes com os métodos de ensaio B.43 (33) e B.53 (35). O conjunto de observações funcionais inclui uma descrição aprofundada do aspeto, comportamento e integridade funcional do indivíduo. Esses elementos são avaliados através de observações na gaiola onde o animal é conservado, após remoção para uma área padrão de observação (campo aberto) em que o animal se move livremente e através de ensaios com manipulação. Devem ser examinados primeiro os animais menos interativos, acabando nos mais interativos. No apêndice 1 é apresentada uma lista das medições. Todos os animais devem ser observados cuidadosamente, por observadores formados que desconheçam o seu estado de tratamento e utilizando procedimentos normalizados, para minimizar a variabilidade devida ao observador. Quando possível, aconselha-se que seja sempre o mesmo observador a avaliar os animais num determinado ensaio. Se isso não for possível, é necessário demonstrar a fiabilidade entre observadores. Para cada parâmetro do conjunto de ensaios comportamentais, devem ser utilizadas escalas e critérios de classificação explícitos e definidos em termos operacionais. Se possível, devem ser desenvolvidas medidas quantitativas objetivas para os parâmetros observáveis que envolvem uma classificação subjetiva. No que se refere à atividade motora, cada animal é sujeito ao ensaio individualmente. A sessão de ensaio deve ser suficientemente longa para revelar a habituação entre sessões para efeitos de controlo. A atividade motora deve ser monitorizada por um aparelho automático de registo de atividade, capaz de detetar tanto aumentos como diminuições na atividade (ou seja, a atividade basal conforme medida pelo dispositivo não deve ser tão baixa a ponto de impedir a deteção de diminuições, nem tão elevada a ponto de impedir a deteção de aumentos na atividade). Cada dispositivo deve ser testado através de procedimentos padrão para assegurar, tanto quanto possível, a fiabilidade do funcionamento entre dispositivos e nos vários dias. Sempre que possível, os grupos de tratamento devem ser distribuídos de forma equilibrada pelos diferentes dispositivos. Os grupos de tratamento devem ser ensaiados de acordo com uma distribuição equilibrada ao longo do dia, de modo a evitar influências devidas aos ritmos circadianos de atividade.
50. Se as informações existentes indicarem a necessidade de outros ensaios funcionais (p. ex., sensoriais, sociais ou cognitivos), estes devem ser integrados sem comprometer a integridade das outras avaliações realizadas no estudo. Se este ensaio for realizado nos mesmos animais utilizados para os ensaios de resposta auditiva padrão, do conjunto de observações funcionais e da atividade motora, os diferentes ensaios devem ser programados de modo a minimizar o risco de comprometer a sua integridade. Estes procedimentos suplementares poderão ser particularmente úteis quando a observação empírica, os efeitos previstos ou os dados mecanísticos/modo de ação indicarem um tipo específico de neurotoxicidade.
Avaliação de potencial imunotoxicidade no desenvolvimento (coorte 3)
51. No 56.o dia após o nascimento (± 3 dias), 10 machos e 10 fêmeas dos animais da coorte 3 de cada grupo de tratamento (1 macho ou 1 fêmea por ninhada; todas as ninhadas representadas por, pelo menos, 1 cria; seleção aleatória) devem ser usados num ensaio da resposta dos anticorpos dependente das células T, ou seja, da primeira resposta dos anticorpos IgM a um antigénio dependente de células T, como o ensaio com glóbulos vermelhos de ovelha (SRBC) ou o ensaio com hemocianina de lapa californiana (KLH), em conformidade com os atuais procedimentos de ensaio da imunotoxicidade (14) (15). A resposta pode ser avaliada através da contagem de células formadoras de placa (PFC) no baço ou da determinação do título de anticorpos IgM específicos para SRBC ou KLH no soro ELISA, no pico da resposta. O pico da resposta ocorre normalmente 4 (resposta PFC) ou 5 (ELISA) dias após a imunização intravenosa. Se a primeira resposta dos anticorpos for analisada através da contagem de células formadoras de placa, poderão avaliar-se subgrupos de animais em dias separados, desde que: a imunização e sacrifício do subgrupo sejam planeados de modo a que as PFC sejam contadas no pico da resposta; esses subgrupos contenham um número igual de descendentes do género masculino e feminino de todos os grupos de dose, incluindo os grupos de controlo; e esses subgrupos sejam avaliados aproximadamente com a mesma idade após o nascimento.A exposição ao produto químico em estudo continuará até ao dia anterior à colheita dos baços para o ensaio PFC ou de soro para o ensaio ELISA.
Avaliação de acompanhamento de potencial toxicidade durante a reprodução (coorte 1B)
52. Os animais da coorte 1B podem ser mantidos em tratamento para além do 90.o dia após o nascimento e, se necessário, procriados para obter uma geração F2. Os machos e as fêmeas do mesmo grupo de admnistração devem coabitar (evitando o acasalamento entre irmãos) durante duas semanas, com início no 90.o dia após o nascimento ou após esse dia, mas não ultrapassando o 120.o dia após o nascimento. Os procedimentos devem ser os mesmos que para os animais P. No entanto, com base na suficiência de prova, poderá ser suficiente terminar as ninhadas no 4.o dia após o nascimento em vez de continuar a acompanhá-las até ao desmame ou para além disso.
OBSERVAÇÕES FINAIS
Bioquímica clínica/Hematologia
53. Os efeitos sistémicos devem ser monitorizados nos animais P. No término do estudo, são colhidas amostras de sangue em jejum de um local definido de dez machos e fêmeas P de cada grupo de dosagem, selecionados aleatoriamente. Essas amostras são armazenadas em condições adequadas e sujeitas a hematologia parcial ou exaustiva, bioquímica clínica, ensaio de T4 e TSH ou outros exames sugeridos pelo perfil de efeitos conhecido do produto químico em estudo [ver documento de orientações 151 da OCDE (40)]. Devem ser examinados os seguintes parâmetros hematológicos: hematócrito, concentração de hemoglobina, contagem de eritrócitos, contagem total e diferencial de leucócitos, contagem de plaquetas e tempo/potencial de coagulação sanguínea. Os exames ao plasma ou soro devem incluir: glicose, colesterol total, ureia, creatinina, proteína total, albumina e pelo menos duas enzimas indicativas dos efeitos hepatocelulares (tais como alanina aminotransferase, aspartato aminotransferase, fosfatase alcalina, gamaglutamiltranspeptidase e sorbitol desidrogenase). Em algumas circunstâncias, a medição de enzimas adicionais e ácidos biliares pode providenciar informações úteis. Para além disso, deve proceder-se à colheita e armazenamento de sangue de todos os animais para possíveis análises posteriores, de forma a ajudar a clarificar efeitos ambíguos ou a gerar dados internos de exposição. Se não se pretender um segundo acasalamento dos animais P, as amostras de sangue devem ser obtidas imediatamente antes do sacrifício programado ou durante esse procedimento. Se os animais envolvidos no estudo forem conservados, as amostras de sangue devem ser colhidas alguns dias antes de os animais acasalarem pela segunda vez. A não ser que os dados existentes dos estudos com dose repetida indiquem que o parâmetro não é afetado pelo produto químico em estudo, a análise à urina deve ser realizada antes do término e os seguintes parâmetros avaliados: aspeto, volume, osmolalidade ou gravidade específica, pH, proteína, glicose, sangue e células sanguíneas, vestígios celulares. Também se pode proceder à colheita de urina para monitorizar a excreção do produto químico em estudo e/ou do(s) seu(s) metabolito(s).
54. Os efeitos sistémicos também devem ser monitorizados nos animais F1. No término do estudo, são colhidas amostras de sangue em jejum de um local definido de dez machos e fêmeas da coorte 1A de cada grupo de dosagem, selecionados aleatoriamente. As amostras são armazenadas em condições adequadas e sujeitas a bioquímica clínica padrão, incluindo a avaliação de níveis séricos para hormonas da tiróide (T4 e TSH), hematologia (contagem total e diferencial de leucócitos e eritrócitos) e avaliações da análise à urina.
55. As crias excedentes no 4.o dia após o nascimento devem ser sujeitas a uma autópsia macroscópica e deve considerar-se a medição das concentrações séricas das hormonas tiroideias (T4). Se necessário, pode proceder-se à colheita de sangue neonatal (4.o dia após o nascimento) por ninhadas para análises bioquímicas/da tiróide. Também é colhido sangue para análises T4 e TSH de animais desmamados sujeitos a autópsia macroscópica no 22.o dia após o nascimento (crias F1 não selecionadas para coortes).
Parâmetros do esperma
56. Os parâmetros do esperma devem ser medidos em todos os machos da geração P, a não ser que existam dados que demonstrem que os parâmetros do esperma não são afetados num estudo de 90 dias. O exame dos parâmetros do esperma deve ser realizado em todos os machos da coorte 1A.
57. No término do estudo, é registado o peso dos testículos e do epidídimo de todos os machos P e F1 (coorte 1A). Pelo menos um testículo e um epidídimo são reservados para exame histopatológico. O outro epidídimo é utilizado para a contagem das reservas de esperma na cauda do epidídimo (16) (17). Além disso, é colhido esperma da cauda do epidídimo (ou ducto deferente) utilizando métodos que minimizem os danos na avaliação da mobilidade e morfologia dos espermatozóides (18).
58. A mobilidade dos espermatozóides pode ser avaliada imediatamente após o sacrifício ou registada para análise posterior. A percentagem de espermatozóides com mobilidade progressiva pode ser determinada de forma subjetiva ou objetiva, através da análise de movimento assistida por computador (19) (20) (21) (22) (23) (24). Para a avaliação da morfologia dos espermatozóides, deve examinar-se uma amostra de esperma do epidídimo (ou ducto deferente) com preparações fixas ou húmidas (25) e, pelo menos, 200 espermatozóides por amostra classificados como normais (a cabeça e o flagelo/cauda têm uma aparência normal) ou anormais. São exemplo de anomalias morfológicas dos espermatozóides a fusão, cabeças isoladas e cabeças e/ou caudas disformes (26). Cabeças de espermatozóides disformes ou grandes podem indicar defeitos na espermiação.
59. Se as amostras de esperma forem congeladas, os esfregaços fixos e as imagens para a análise de mobilidade dos espermatozóides registadas na altura da autópsia (27), as análises subsequentes podem limitar-se aos machos dos grupos de controlo e das dosagens mais elevadas. No entanto, se forem observados efeitos associados ao tratamento, os grupos de dose mais baixa também devem ser avaliados.
Autópsia macroscópica
60. Na altura do término do estudo ou em caso de morte prematura, todos os animais P e F1 são sujeitos a autópsia e examinados macroscopicamente no que se refere a anomalias estruturais ou alterações patológicas. Deve prestar-se especial atenção aos órgãos do sistema reprodutivo. As crias eutanasiadas por se encontrarem moribundas e as crias mortas devem ser registadas e, se não estiverem maceradas, examinadas quanto a potenciais defeitos e/ou causa de morte, e preservadas.
61. Nas fêmeas P e F1 adultas, é examinado um esfregaço vaginal no dia da autópsia para determinar o estádio do ciclo éstrico e permitir a correlação com a histopatologia dos órgãos reprodutivos. Os úteros de todas as fêmeas P (e fêmeas F1, se aplicável) são examinados relativamente à presença e número de locais de implantação, sem comprometer a avaliação histopatológica.
Pesagem dos órgãos e preservação de tecido — animais adultos P e F1
62. Na altura do término do estudo, os pesos corporais e os pesos húmidos dos órgãos abaixo listados de todos os animais P e todos os adultos F1 das coortes relevantes (como descrito em baixo), são determinados logo que possível após a dissecação, para evitar que sequem. Posteriormente, estes órgãos devem ser preservados em condições adequadas. Salvo indicação em contrário, os órgãos pares podem ser pesados individualmente ou em conjunto, de forma consistente com a prática comum do laboratório em questão.
|
— |
Útero (com ovidutos e colo do útero), ovários; |
|
— |
Testículos, epidídimos (total e cauda para as amostras utilizadas para contagens de espermatozóides); |
|
— |
Próstata (partes dorsolateral e ventral em conjunto). Impõe-se grande prudência durante o corte do complexo da próstata, de forma a evitar a perfuração das vesículas seminais repletas de fluido. Se se verificar um efeito associado ao tratamento no peso total da próstata, os segmentos dorsolateral e ventral devem ser cuidadosamente dissecados após a fixação e pesados separadamente. |
|
— |
Vesículas seminais com glândulas de coagulação e respetivos fluidos (como uma unidade); |
|
— |
Cérebro, fígado, rins, coração, baço, timo, hipófise, tiróide (pós-fixação), glândulas suprarrenais e órgãos e tecidos-alvo conhecidos. |
63. Para além dos órgãos acima listados, deve preservar-se amostras dos nervos periféricos, músculo, medula espinal, olho e nervo ótico, trato gastrointestinal, bexiga, pulmão, traqueia (com a tiróide e paratiróide), medula óssea, ducto deferente (machos), glândula mamária (machos e fêmeas) e vagina, em condições adequadas.
64. Todos os órgãos dos animais da coorte 1A devem ser pesados e preservados para histopatologia.
65. Para a investigação de efeitos imunotóxicos induzidos antes e depois do parto, 10 machos e 10 fêmeas dos animais da coorte 1A de cada grupo de tratamento (1 macho ou 1 fêmea por ninhada; todas as ninhadas representadas por pelo menos 1 cria; seleção aleatória) serão sujeitos ao seguinte procedimento no término do estudo:
|
— |
pesagem dos nós linfáticos associados e distantes da via de exposição (para além da pesagem das glândulas suprarrenais, do timo e do baço, já realizada em todos os animais da coorte 1A); |
|
— |
análise da subpopulação de linfócitos esplénicos (linfócitos T CD4+ e CD8+, linfócitos B e células exterminadoras naturais), utilizando uma metade do baço; a outra metade do baço é preservada para avaliação histopatológica. |
A análise de subpopulações de linfócitos esplénicos em animais não imunizados (coorte 1A) determinará se a exposição está relacionada com uma mudança na distribuição estável imunológica de linfócitos auxiliares (CD4+) ou citotóxicos (CD8+) provenientes do timo ou células exterminadoras naturais (NK) (respostas rápidas às células neoplásicas e agentes patogénicos).
66. Os animais da coorte 1B devem ter os seguintes órgãos pesados e os tecidos correspondentes processados para a fase de bloco:
|
— |
Vagina (não pesada) |
|
— |
Útero com colo do útero |
|
— |
Ovários |
|
— |
Testículos (pelo menos um) |
|
— |
Epidídimos |
|
— |
Vesículas seminais e glândulas de coagulação |
|
— |
Próstata |
|
— |
Hipófise |
|
— |
Órgãos-alvo identificados |
Deve proceder-se à histopatologia na coorte 1B se os resultados da coorte 1A não forem inequívocos, ou em casos de suspeita de tóxicos para o sistema reprodutivo ou endócrino.
67. Coortes 2A e 2B: Ensaio de neurotoxicidade no desenvolvimento (21.o ou 22.o dia pós-natal e descendência adulta). O estudo dos animais da coorte 2A termina após o ensaio comportamental, com pesagem de cérebro registada e neuro-histopatologia completa para efeitos de avaliação de neurotoxicidade. O estudo dos animais da coorte 2B termina no 21.o ou 22.o dia após o nascimento, com a pesagem de cérebro registada e o exame microscópico do mesmo para efeitos de avaliação de neurotoxicidade. A fixação com perfusão é necessária para os animais da coorte 2A e opcional para os animais da coorte 2B, como descrito no método de ensaio B.53 (35).
Pesagem de órgãos e preservação de tecido — animais F1 desmamados
68. O estudo das crias não selecionadas para coortes, incluindo as de tamanho inferior ao normal, termina após o desmame, no 22.o dia após o nascimento, a não ser que os resultados apontem para a necessidade de investigação subsequente em vida. As crias cujo estudo já tenha terminado são sujeitas a autópsia macroscópica, incluindo uma avaliação dos órgãos reprodutores, consoante descrito nos parágrafos 62 e 63. O cérebro, o baço e o timo de até 10 crias por sexo e por grupo, do maior número possível de ninhadas, devem ser pesados e preservados em condições adequadas. Para além disso, os tecidos mamários destas crias do sexo masculino e feminino podem ser preservados para posterior análise microscópica (13) [ver documento de orientações 151 da OCDE (40)]. As anomalias graves e os tecidos-alvo devem ser preservados para possível exame histológico.
Histopatologia — animais P
69. É realizada uma histopatologia completa dos órgãos listados nos parágrafos 62 e 63 para todos os animais P de controlo e sujeitos a uma dose elevada. Os órgãos que apresentem alterações associadas ao tratamento devem também ser examinados em todos os animais nos grupos de dosagem inferior para ajudar a determinar um NOAEL. Para além disso, os órgãos reprodutores de todos os animais com suspeita de fertilidade reduzida, por exemplo aqueles que não conseguiram acasalar, conceber, reproduzir-se ou dar à luz descendência saudável, ou cujo ciclo éstrico ou quantidade, mobilidade ou morfologia dos espermatozóides tenham sido afetados, bem como todas as lesões graves, devem ser sujeitos a avaliação histopatológica.
Histopatologia — animais F1
Animais da coorte 1
70. É realizada uma histopatologia completa dos órgãos listados nos parágrafos 62 e 63 para todos os animais adultos de controlo e sujeitos a uma dose elevada da coorte 1A. Todas as ninhadas devem estar representadas por pelo menos 1 cria por sexo. Os órgãos e tecidos que apresentem alterações associadas ao tratamento e todas as lesões graves devem também ser examinados em todos os animais dos grupos de dosagem inferior para ajudar a determinar um NOAEL. Para a avaliação de efeitos induzidos antes e depois do parto nos órgãos linfóides, deve ser avaliada a histopatologia dos nós linfáticos e medula óssea colhidos de 10 machos e 10 fêmeas da coorte 1a, juntamente com a avaliação histopatológica do timo, baço e glândulas suprarrenais já realizada em todos os animais 1A.
71. Os tecidos reprodutivos e endócrinos de todos os animais da coorte 1B, processados para a fase de bloco, consoante descrito no parágrafo 66, devem ser alvo de exame histopatológico em caso de suspeita de tóxicos para o sistema reprodutor ou endócrino. A coorte 1B deve também ser sujeita a exame histológico se os resultados da coorte 1A não forem inequívocos.
72. Os ovários de fêmeas adultas devem conter folículos primordiais e em crescimento, assim como corpos lúteos; assim, um exame histopatológico deve ter como objetivo uma avaliação quantitativa dos folículos primordiais e de baixo crescimento, assim como dos corpos lúteos, em fêmeas F1; o número de animais, a seleção da secção do ovário e o tamanho da amostra da secção devem ser estatisticamente adequados para o procedimento de avaliação utilizado. A enumeração folicular deve ser realizada em primeiro lugar em animais de controlo e sujeitos a dosagem elevada e, se se verificar um efeito adverso nos últimos, devem ser examinadas as dosagens inferiores. O exame deve incluir a enumeração da quantidade de folículos primordiais, que podem ser combinados com folículos de baixo crescimento, para comparação entre os ovários dos animais tratados e dos animais de controlo [ver documento de orientações 151 da OCDE (40)]. A avaliação dos corpos lúteos deve ser realizada em paralelo com o ensaio ao ciclo éstrico, de modo a que o estádio do ciclo possa ser tido em consideração na avaliação. O oviduto, o útero e a vagina são examinados quanto ao desenvolvimento típico do órgão.
73. São realizados exames de histopatologia testicular detalhados nos machos F1 de modo a identificar efeitos associados ao tratamento na diferenciação e desenvolvimento dos testículos e na espermatogénese (38). Quando possível, devem ser examinadas secções da rede testicular. A cabeça, corpo e cauda do epidídimo e o ducto deferente são examinados quanto ao desenvolvimento típico do órgão, assim como quanto aos parâmetros requeridos para os machos P.
Animais da coorte 2
74. É realizada uma neuro-histopatologia de todos os animais de controlo e sujeitos a uma dose elevada da coorte 2A, por sexo, a seguir ao término do ensaio neurocomportamental (depois do 75.o mas antes do 90.o dia após o nascimento). É realizada a histopatologia do cérebro de todos os animais de controlo e sujeitos a uma dose elevada da coorte 2B, por sexo, no 21.o ou 22.o dia após o nascimento. Os órgãos ou tecidos que apresentem alterações associadas ao tratamento devem também ser examinados nos animais dos grupos de dosagem inferior, para ajudar a determinar um NOAEL. Nos animais das coortes 2A e 2B, são examinadas várias secções do cérebro para permitir o exame dos bolbos olfativos, córtex cerebral, hipocampo, gânglios basais, tálamo, hipotálamo, centro do cérebro (forame cego, calote e pedúnculos cerebrais), tronco cerebral e cerebelo. Apenas na coorte 2A, são examinados os olhos (retina e nervo ótico) e amostras dos nervos periféricos, músculo e medula espinal. Todos os procedimentos neuro-histológicos devem estar em conformidade com o método de ensaio B.53 (35).
75. As avaliações morfométricas (quantitativas) devem ser realizadas em áreas representativas do cérebro (secções homólogas selecionadas cuidadosamente com base em marcos microscópicos) e podem incluir medições lineares ou espaciais de regiões específicas do cérebro. Pelo menos três secções consecutivas devem ser retiradas em cada marco (nível), de modo a selecionar a secção mais homóloga e representativa do cérebro específico a avaliar. O neuropatologista deve verificar com cuidado se as secções preparadas para a medição são equivalentes às outras do conjunto de amostras e, assim, indicadas para inclusão, uma vez que as medições lineares, em particular, podem variar ao longo de uma distância relativamente curta (28). As secções não homólogas não devem ser utilizadas. O objetivo é colher amostras de todos os animais reservados para este propósito (10/sexo/nível de dose); porém, pode também ser adequada uma quantidade menor. No entanto, amostras de menos de 6 animais/sexo/nível de dose não são geralmente consideradas suficientes para efeitos deste método de ensaio. Pode recorrer-se à estereologia para identificar efeitos associados ao tratamento em parâmetros como o volume ou o número de células em regiões neuroanatómicas específicas. Todos os aspetos da preparação de amostras de tecido, desde a fixação do tecido, dissecação das amostras de tecido, processamento do tecido até à coloração de lâminas, devem seguir uma conceção equilibrada para que cada lote contenha amostras representativas de cada grupo de dosagem. Quando se utilizarem análises morfométricas ou estereológicas, o tecido do cérebro deve ser envolvido em meios adequados em todos os níveis de dose em simultâneo, de modo a evitar artefactos de contração associados a um armazenamento prolongado no fixador.
RELATÓRIOS
Dados
76. Os dados são registados individualmente e resumidos sob a forma de tabela. Se for caso disso, para cada grupo de ensaio e cada geração, deve registar-se o seguinte: número de animais no início do ensaio, número de animais encontrados mortos durante o ensaio ou eutanasiados, hora de todos os óbitos, naturais ou por eutanásia, número de animais férteis, número de fêmeas grávidas, número de fêmeas a dar à luz uma ninhada e número de animais que apresenta sinais de toxicidade. Também deve ser registada uma descrição da toxicidade, incluindo a hora de início, duração e gravidade.
77. Os resultados numéricos devem ser avaliados através de um método estatístico adequado e aceite. Os métodos estatísticos devem ser selecionados no âmbito da definição do estudo e mencionar, de modo adequado, os dados fora do normal (p. ex.: dados de contagem), não considerados (p. ex.: tempo de observação limitado), não independentes (p. ex.: efeitos da ninhada e medições repetidas) e as variâncias desiguais. Os modelos lineares generalizados mistos e modelos de dose-resposta cobrem uma vasta classe de ferramentas analíticas que podem ser adequadas para os dados gerados no âmbito deste método de ensaio. O relatório deve incluir informações suficientes sobre os métodos de análise e o programa informático utilizado, para que o revisor/estatístico independente possa avaliar/reavaliar a análise.
Avaliação de resultados
78. As conclusões devem ser avaliadas em termos de efeitos observados, incluindo as autópsias e as constatações microscópicas. A avaliação inclui a relação, ou a falta dela, entre a dose e a presença, incidência e gravidade das anomalias, incluindo lesões graves. Os órgãos-alvo, fertilidade, anomalias clínicas, desempenho reprodutivo e da ninhada, alterações do peso corporal, mortalidade e quaisquer outros efeitos tóxicos e no desenvolvimento também devem ser avaliados. Deve prestar-se especial atenção às alterações específicas do sexo. As propriedades físico-químicas do produto químico em estudo e, se disponíveis, os dados de toxicocinética, incluindo transferência placentária e excreção no leite, devem ser tidos em consideração durante a avaliação dos resultados do ensaio.
Relatório de ensaio
79. O relatório de ensaio deve incluir as seguintes informações obtidas no presente estudo a partir dos animais P, F1 e F2 (quando relevante):
Produto químico em estudo:
|
— |
Todas as informações disponíveis e relevantes sobre o produto químico em estudo e sobre as suas propriedades toxicocinéticas e toxicodinâmicas; |
|
— |
Dados de identificação do produto; |
|
— |
Pureza; |
Veículo (se adequado):
|
— |
Justificação da escolha do veículo, se outro que não água; |
Animais de ensaio:
|
— |
Espécie/estirpe utilizada; |
|
— |
Número, idade e sexo dos animais; |
|
— |
Origem, condições de alojamento, dieta, materiais de nidificação, etc.; |
|
— |
Peso individual dos animais no início do ensaio; |
|
— |
Dados do esfregaço vaginal para as fêmeas P antes do início do tratamento (se forem recolhidos dados nessa altura); |
|
— |
Registos de emparelhamento da geração P, indicando o macho e a fêmea de um acasalamento e o grau de sucesso do mesmo; |
|
— |
Registos da ninhada de origem para os adultos da geração F1; |
Condições de ensaio:
|
— |
Justificação para a escolha do nível de dosagem; |
|
— |
Detalhes sobre a formulação do produto químico em estudo/preparação da dieta, concentrações alcançadas; |
|
— |
Estabilidade e homogeneidade da preparação no veículo ou agente de transporte (p. ex.: dieta, água potável), no sangue e/ou no leite, nas condições de utilização e armazenamento entre as utilizações; |
|
— |
Detalhes da administração do produto químico em estudo; |
|
— |
Conversão da concentração do produto químico em estudo (ppm) na dieta/água potável para a dose alcançada (mg/kg de peso corporal/dia), se aplicável; |
|
— |
Detalhes sobre a qualidade dos alimentos e da água (incluindo composição da dieta, se disponível); |
|
— |
Descrição detalhada sobre os procedimentos de randomização na seleção das crias para abate e na atribuição das crias aos grupos de ensaio; |
|
— |
Condições ambientais; |
|
— |
Lista do pessoal que participa no estudo, incluindo a respetiva formação profissional; |
Resultados (resumo e dados individuais por sexo e dose):
|
— |
Consumo de alimentos, consumo de água, se disponível, eficiência alimentar (aumento do peso corporal por grama de alimentos consumidos, exceto para o período de coabitação e durante a amamentação) e consumo do produto químico em estudo (administração na dieta/água potável) para os animais P e F1; |
|
— |
Dados de absorção (se disponíveis); |
|
— |
Dados sobre o peso corporal dos animais P; |
|
— |
Dados sobre o peso corporal dos animais F1 selecionados, após o desmame; |
|
— |
Hora do óbito durante o estudo ou indicação de que os animais sobreviveram até ao término do estudo; |
|
— |
Natureza, gravidade e duração das observações clínicas (reversíveis ou não); |
|
— |
Hematologia, análises à urina e dados químicos clínicos, incluindo TSH e T4; |
|
— |
Análise fenotípica das células do baço (células T, B, NK); |
|
— |
Celularidade da medula óssea; |
|
— |
Dados de resposta tóxica; |
|
— |
Número de fêmeas P e F1 com ciclo éstrico normal ou anormal e duração do ciclo; |
|
— |
Tempo até ao acasalamento (intervalo pré-coito, número de dias entre o emparelhamento e o acasalamento); |
|
— |
Efeitos tóxicos ou outros na reprodução, incluindo números e percentagens de animais que alcançaram o acasalamento, gravidez, parto e amamentação, de machos que induziram gravidez, de fêmeas com sinais de distócia/parto prolongado ou difícil; |
|
— |
Duração da gravidez e, se disponível, do parto; |
|
— |
Número de implantações, tamanho da ninhada e percentagem de crias do sexo masculino; |
|
— |
Número e percentagem de perda pós-implantação, nados-vivos e nados-mortos; |
|
— |
Dados sobre o peso da ninhada e das crias (machos, fêmeas e combinados), número de animais de tamanho inferior ao normal, se constatado; |
|
— |
Número de crias com anomalias manifestamente visíveis; |
|
— |
Efeitos tóxicos ou outros na descendência, crescimento pós-natal, viabilidade, etc.; |
|
— |
Dados sobre marcos físicos em crias e outros dados sobre o desenvolvimento pós-natal; |
|
— |
Dados sobre a maturação sexual dos animais F1; |
|
— |
Dados sobre observações funcionais em crias e adultos, quando aplicável; |
|
— |
Peso corporal no momento do sacrifício e peso relativo e absoluto dos órgãos dos animais P e animais adultos F1; |
|
— |
Dados obtidos por autópsia; |
|
— |
Descrição detalhada de todos os achados histopatológicos; |
|
— |
Número total de espermatozóides na cauda do epidídimo, percentagem de espermatozóides com mobilidade progressiva; percentagem de espermatozóides com morfologia normal e percentagem de espermatozóides com cada uma das anomalias identificadas nos machos P e F1; |
|
— |
Números e estádios de maturação dos folículos contidos nos ovários das fêmeas P e F1, quando aplicável; |
|
— |
Enumeração de corpos lúteos nos ovários das fêmeas F1; |
|
— |
Tratamento estatístico dos resultados, quando adequado; |
Parâmetros da coorte 2:
|
— |
Descrição detalhada dos métodos utilizados para uniformizar observações e procedimentos, assim como definições operacionais para as observações de classificação; |
|
— |
Lista de todos os procedimentos de ensaio utilizados e justificação da respetiva utilização; |
|
— |
Detalhes de procedimentos comportamentais/funcionais, neuropatológicos e morfométricos utilizados, incluindo informações e detalhes sobre dispositivos automáticos; |
|
— |
Procedimentos para calibração e garantia de equivalência de dispositivos e equilibragem dos grupos de tratamento em procedimentos de ensaio; |
|
— |
Breve justificação que explique quaisquer decisões que envolvam julgamento profissional; |
|
— |
Descrição detalhada de todos os achados comportamentais/funcionais, neuropatológicos e morfométricos por sexo e grupo de dosagem, incluindo aumentos e diminuições em relação aos controlos; |
|
— |
Peso do cérebro; |
|
— |
Quaisquer diagnósticos decorrentes de sinais e lesões neurológicas, incluindo doenças ou condições naturais; |
|
— |
Imagens de constatações exemplares; |
|
— |
Imagens de baixa resolução para comprovar a homologia das secções utilizadas para morfometria; |
|
— |
Tratamento estatístico dos resultados, incluindo modelos estatísticos utilizados para analisar os dados e os resultados, independentemente de terem sido significativos ou não; |
|
— |
Relação de quaisquer outros efeitos tóxicos para uma conclusão sobre o potencial neurotóxico do produto químico em estudo, por sexo e grupo de dosagem; |
|
— |
Impacto de quaisquer informações toxicocinéticas nas conclusões; |
|
— |
Dados que suportam a fiabilidade e sensibilidade do método de ensaio (ou seja, dados positivos e históricos de controlo); |
|
— |
Relações, se existentes, entre os efeitos neuropatológicos e funcionais; |
|
— |
NOAEL ou dose de referência para crias e descendentes, por sexo e grupo de dosagem; |
|
— |
Discussão da interpretação global dos dados com base nos resultados, incluindo uma conclusão sobre se o produto químico provocou, ou não, neurotoxicidade no desenvolvimento e o NOAEL; |
Parâmetros da coorte 3:
|
— |
Títulos de anticorpos IgM séricos (sensibilização a SRBC ou KLH), ou unidades PFC IgM esplénicas (sensibilização a SRBC); |
|
— |
O desempenho do método TDAR deve ser confirmado como parte do processo de otimização nos laboratórios que realizam o ensaio pela primeira vez e periodicamente (p. ex.: anualmente) por todos os laboratórios; |
|
— |
Discussão da interpretação global dos dados com base nos resultados, incluindo uma conclusão sobre se o produto químico provocou, ou não, imunotoxicidade no desenvolvimento e NOAEL; |
Discussão dos resultados
Conclusões, incluindo valores de NOAEL nos progenitores e na descendência
Todas as informações que não tenham sido obtidas durante o estudo, mas que sejam úteis para a interpretação dos resultados (p. ex.: semelhanças com os efeitos de quaisquer neurotóxicos conhecidos) devem também ser disponibilizadas.
Interpretação dos Resultados
80. Um “Estudo alargado de toxicidade durante a reprodução numa geração” proporcionará informações sobre os efeitos da exposição repetida a produtos químicos durante todas as fases do ciclo reprodutivo, conforme necessário. Em particular, o estudo providencia informações sobre o sistema reprodutivo e sobre os parâmetros de desenvolvimento, crescimento, sobrevivência e funcionais da descendência até ao 90.o dia após o nascimento.
81. A interpretação dos resultados do estudo deve ter em consideração todas as informações disponíveis sobre o produto químico, incluindo propriedades físico-químicas, toxicocinéticas e toxicodinâmicas, informações relevantes disponíveis sobre análogos estruturais e resultados de estudos de toxicidade anteriores com o produto químico em estudo (p. ex.: toxicidade aguda, toxicidade após aplicação repetida, estudos mecanísticos e estudos que avaliam se existem diferenças qualitativas e quantitativas substanciais entre espécies no que respeita às propriedades metabólicas in vivo/in vitro). Os resultados da autópsia macroscópica e da pesagem dos órgãos devem ser avaliados no contexto das observações de outros estudos com dose repetida, quando tal for viável. As reduções no crescimento das crias podem ser consideradas relativamente a uma influência do produto químico em estudo na composição do leite (29).
Coorte 2 (Neurotoxicidade no desenvolvimento)
82. Os efeitos neurocomportamentais e neuropatológicos podem ser interpretados no contexto de todos os achados, recorrendo a uma abordagem de suficiência de prova com parecer de peritos. Devem ser discutidos os padrões das constatações comportamentais ou morfológicas, se presentes, assim como a evidência de uma relação dose-resposta. A avaliação de neurotoxicidade no desenvolvimento, incluindo estudos epidemiológicos em humanos ou relatórios de caso e estudos experimentais em animais (p. ex.: dados toxicocinéticos, informação sobre estrutura/atividade e dados de outros estudos de toxicidade), deve ser incluída nesta caracterização. A avaliação dos dados deve incluir uma discussão da significância biológica e estatística. A avaliação deve incluir a relação, se existente, entre alterações neuropatológicas e comportamentais. Para orientações quanto à interpretação dos resultados de neurotoxicidade no desenvolvimento, consultar o método de ensaio B.53 (35) e Tyl et al., 2008 (31).
Coorte 3 (Imunotoxicidade no desenvolvimento)
83. A supressão ou o reforço da função imunitária, conforme estipulado pelas TDAR (respostas de anticorpos dependentes de células T) deve avaliar-se no contexto de todas as observações constatadas. A significância dos resultados de TDAR deve ser sustentada por outros efeitos em indicadores relacionados com a imunologia (p. ex.: celularidade da medula óssea, peso e histopatologia dos tecidos linfoides, distribuição do subgrupo de linfócitos). Os efeitos estabelecidos por TDAR podem ser menos significativos no caso de outras toxicidades constatadas em concentrações de menor exposição.
84. O documento de orientações 43 da OCDE deve ser consultado como fonte de ajuda na interpretação dos resultados sobre a reprodução e da neurotoxicidade (26).
REFERÊNCIAS
|
1. |
Cooper, R.L., J.C. Lamb, S.M. Barlow, K. Bentley, A.M. Brady, N. Doerr, D.L. Eisenbrandt, P.A. Fenner-Crisp, R.N. Hines, L.F.H. Irvine, C.A. Kimmel, H. Koeter, A.A. Li, S.L. Makris, L.P. Sheets, G.J.A. Speijers and K.E. Whitby (2006), “A Tiered Approach to Life Stages Testing for Agricultural Chemical Safety Assessment”, Critical Reviews in Toxicology, 36, 69-98. |
|
2. |
Thigpen, J.E., K.D.R. Setchell, K.B. Ahlmark, J. Locklear, T. Spahr, G.F. Leviness, M.F. Goelz, J.K. Haseman, R.R. Newbold, and D.B. Forsythe (1999), “Phytoestrogen Content of Purified Open and Closed Formula Laboratory Animal Diets”, Lab. Anim. Sci., 49, 530- 536. |
|
3. |
Zoetis, T. and I. Walls (2003), Principles and Practices for Direct Dosing of Pre-Weaning Mammals in Toxicity Testing and Research, ILSI Press, Washington, DC. |
|
4. |
Moser, V.C., I. Walls and T. Zoetis (2005), “Direct Dosing of Preweaning Rodents in Toxicity Testing and Research: Deliberations of an ILSI RSI Expert Working Group”, International Journal of Toxicology, 24, 87-94. |
|
5. |
Conolly, R.B., B.D. Beck, and J.I. Goodman (1999), “Stimulating Research to Improve the Scientific Basis of Risk Assessment”, Toxicological Sciences, 49, 1-4. |
|
6. |
Ulbrich, B. and A.K. Palmer (1995), “Detection of Effects on Male Reproduction — a Literature Survey”, Journal of the American College of Toxicologists, 14, 293-327. |
|
7. |
Mangelsdorf, I., J. Buschmann and B. Orthen (2003), “Some Aspects Relating to the Evaluation of the Effects of Chemicals on Male Fertility”, Regulatory Toxicology and Pharmacology, 37, 356-369. |
|
8. |
Sakai, T., M. Takahashi, K. Mitsumori, K. Yasuhara, K. Kawashima, H. Mayahara and Y. Ohno (2000). “Collaborative work to evaluate toxicity on male reproductive organs by repeated dose studies in rats — overview of the studies”, Journal of Toxicological Sciences, 25, 1-21. |
|
9. |
Creasy, D.M. (2003), “Evaluation of Testicular Toxicology: A Synopsis and Discussion of the Recommendations Proposed by the Society of Toxicologic Pathology”, Birth Defects Research, Part B, 68, 408-415. |
|
10. |
Goldman, J.M., A.S. Murr, A.R. Buckalew, J.M. Ferrell and R.L. Cooper (2007), “The Rodent Estrous Cycle: Characterization of Vaginal Cytology and its Utility in Toxicological Studies”, Birth Defects Research, Part B, 80 (2), 84-97. |
|
11. |
Sadleir, R.M.F.S. (1979), “Cycles and Seasons”, in C.R. Auston and R.V. Short (eds.), Reproduction in Mammals: I. Germ Cells and Fertilization, Cambridge, New York. |
|
12. |
Gallavan, R.H. Jr, J.F. Holson, D.G. Stump, J.F. Knapp and V.L. Reynolds (1999), “Interpreting the Toxicologic Significance of Alterations in Anogenital Distance: Potential for Confounding Effects of Progeny Body Weights”, Reproductive Toxicology, 13: 383-390. |
|
13. |
Korenbrot, C.C., I.T. Huhtaniemi and R.I. Weiner (1977), “Preputial Separation as an External Sign of Pubertal Development in the Male Rat”, Biological Reproduction, 17, 298-303. |
|
14. |
Ladics, G.S. (2007), “Use of SRBC Antibody Responses for Immunotoxicity Testing”, Methods, 41, 9-19. |
|
15. |
Gore, E.R., J. Gower, E. Kurali, J.L. Sui, J. Bynum, D. Ennulat and D.J. Herzyk (2004), “Primary Antibody Response to Keyhole Limpet Hemocyanin in Rat as a Model for Immunotoxicity Evaluation”, Toxicology, 197, 23-35. |
|
16. |
Gray, L.E., J. Ostby, J. Ferrell, G. Rehnberg, R. Linder, R. Cooper, J. Goldman, V. Slott and J. Laskey (1989), “A Dose-Response Analysis of Methoxychlor-Induced Alterations of Reproductive Development and Function in the Rat”, Fundamental and Applied Toxicology, 12, 92-108. |
|
17. |
Robb, G.W., R.P. Amann and G.J. Killian (1978), “Daily Sperm Production and Epididymal Sperm Reserves of Pubertal and Adult Rats”, Journal of Reproduction and Fertility,54, 103-107. |
|
18. |
Klinefelter, G.R., L.E. Jr Gray and J.D. Suarez (1991), “The Method of Sperm Collection Significantly Influences Sperm Motion Parameters Following Ethane Dimethanesulfonate Administration in the Rat”. Reproductive Toxicology,5, 39-44. |
|
19. |
Seed, J., R.E. Chapin, E.D. Clegg., L.A. Dostal, R.H. Foote, M.E. Hurtt, G.R. Klinefelter, S.L. Makris, S.D. Perreault, S. Schrader, D. Seyler, R. Sprando, K.A. Treinen, D.N. Veeramachaneni and L.D. Wise (1996), “Methods for Assessing Sperm Motility, Morphology, and Counts in the Rat, Rabbit, and Dog: a Consensus Report”, Reproductive Toxicology, 10, 237- 244. |
|
20. |
Chapin, R.E., R.S. Filler, D. Gulati, J.J. Heindel, D.F. Katz, C.A. Mebus, F. Obasaju, S.D. Perreault, S.R. Russell and S. Schrader (1992), “Methods for Assessing Rat Sperm Motility”, Reproductive Toxicology, 6, 267-273. |
|
21. |
Klinefelter, G.R., N.L. Roberts and J.D. Suarez (1992), “Direct Effects of Ethane Dimethanesulphonate on Epididymal Function in Adult Rats: an In Vitro Demonstration”, Journal of Andrology, 13, 409-421. |
|
22. |
Slott, V.L., J.D. Suarez and S.D. Perreault (1991), “Rat Sperm Motility Analysis: Methodologic Considerations”, Reproductive Toxicology, 5, 449-458. |
|
23. |
Slott, V.L., and S.D. Perreault (1993), “Computer-Assisted Sperm Analysis of Rodent Epididymal Sperm Motility Using the Hamilton-Thorn Motility Analyzer”, Methods in Toxicology, Part A, Academic, Orlando, Florida. p. 319-333. |
|
24. |
Toth, G.P., J.A. Stober, E.J. Read, H. Zenick and M.K. Smith (1989), “The Automated Analysis of Rat Sperm Motility Following Subchronic Epichlorhydrin Administration: Methodologic and Statistical Considerations”, Journal of Andrology, 10, 401-415. |
|
25. |
Linder, R.E., L.F. Strader, V.L. Slott and J.D. Suarez (1992), “Endpoints of Spermatoxicity in the Rat After Short Duration Exposures to Fourteen Reproductive Toxicants”, Reproductive Toxicology, 6, 491-505. |
|
26. |
OCDe (2008), Guidance Document on Mammalian Reproductive Toxicity Testing and Assessment, Series on Testing and Assessment, No. 43, ENV/JM/MONO(2008)16, OECD, Paris. |
|
27. |
Working, P.K., M. Hurtt (1987), “Computerized Videomicrographic Analysis of Rat Sperm Motility”, Journal of Andrology, 8, 330-337. |
|
28. |
Bolin, B., R. Garman, K. Jensen, G. Krinke, B. Stuart, and an ad Hoc Working Group of the STP Scientific and Regulatory Policy Committee (2006), “A 'Best Practices' Approach to Neuropathologic Assessment in Developmental Neurotoxicity Testing — for Today”, Toxicological Pathology, 34, 296-313. |
|
29. |
Stütz, N., B. Bongiovanni, M. Rassetto, A. Ferri, A.M. Evangelista de Duffard, and R. Duffard (2006), “Detection of 2,4-dichlorophenoxyacetic Acid in Rat Milk of Dams Exposed During Lactation and Milk Analysis of their Major Components”, Food Chemicals Toxicology, 44, 8-16. |
|
30. |
Thigpen, JE, K.D.R. Setchell, J.K. Haseman, H.E. Saunders, G.F. Caviness, G.E. Kissling, M.G. Grant and D.B. Forsythe (2007), “Variations in Phytoestrogen Content between Different Mill Dates of the Same Diet Produces Significant Differences in the Time of Vaginal Opening in CD-1 Mice and F344 Rats but not in CD Sprague Dawley Rats”, Environmental health perspectives, 115(12), 1717-1726. |
|
31. |
Tyl, R.W., K. Crofton, A. Moretto, V. Moser, L.P. Sheets and T.J. Sobotka (2008), “Identification and Interpretation of Developmental Neurotoxicity Effects: a Report from the ILSI Research Foundation/Risk Science Institute Expert Working Group on Neurodevelopmental Endpoints”, Neurotoxicology and Teratology, 30: 349-381. |
|
32. |
OECD (1996), Combined Repeated Dose Toxicity Study with the Reproduction/Developmental Toxicity Screening Test, OECD Guideline for Testing of Chemicals, No. 422, OECD, Paris. |
|
33. |
Capítulo B.43 do presente anexo, “Ensaio de neurotoxicidade em roedores”. |
|
34. |
OCDE (2000), Guidance Document on the recognition, assessment, and use of clinical signs as humane endpoints for experimental animals used in safety evaluations, Series on Testing and Assessment, No. 19, ENV/JM/MONO(2000)7, OECD, Paris. |
|
35. |
Capítulo B.53 do presente anexo, “Estudo de neurotoxicidade durante o desenvolvimento”. |
|
36. |
Capítulo B.54 do presente anexo, “Bioensaio uterotrófico em roedores: Ensaio de despistagem a curto prazo de propriedades estrogénicas”. |
|
37. |
Capítulo B.55 do presente anexo, “Bioensaio de Hershberger no rato: Ensaio de despistagem a curto prazo de propriedades (anti)androgénicas”. |
|
38. |
OCDE (2009), Guidance Document for Histologic Evalution of Endocrine and Reproductive Test in Rodents, Series on Testing and Assessment, No. 106, OECD, Paris. |
|
39. |
OCDE (2011), Guidance Document on the Current Implementation of Internal Triggers in the Extended One Generation Reproductive Toxicity Study in the United States and Canada, Series on Testing and Assessment, No. 117, ENV/JM/MONO(2011)21, OECD, Paris. |
|
40. |
OCDE (2013), Guidance Document supporting TG 443: Extended One Generation Reproductive Toxicity Study, Series on Testing and Assessment, No. 151, OECD, Paris. |
Apêndice 1
Medidas e observações incluídas na bateria de observação funcional (coorte 2a)
|
Gaiola e Campo Aberto |
Manipulativas |
Fisiológicas |
|
Postura |
Facilidade de remoção |
Temperatura |
|
Tonicoclónico involuntário |
Facilidade de manipulação |
Peso corporal |
|
Fechamento das pálpebras |
Tónus muscular |
Reação pupilar |
|
Piloereção |
Reação à aproximação |
Tamanho das pupilas |
|
Salivação |
Reação ao toque |
|
|
Lacrimação |
Resposta auditiva |
|
|
Vocalizações |
Reação ao beliscão na cauda |
|
|
Levantar nas patas traseiras |
Reação de endireitamento |
|
|
Anormalidades da marcha |
Abdução das patas ao aterrar |
|
|
Excitação |
Força de preensão dos membros dianteiros |
|
|
Estereotipia |
Força de preensão dos membros traseiros |
|
|
Comportamento estranho |
|
|
|
Manchas |
|
|
|
Anormalidades respiratórias |
|
|
Apêndice 2
DEFINIÇÕES
Produto químico : uma substância ou uma mistura.
Produto químico em estudo : qualquer substância ou mistura estudada utilizando este método de ensaio.
B.57. ENSAIO DE ESTEROIDOGÉNESE H295R
INTRODUÇÃO
1. Este método é equivalente ao Test Guideline TG 456 (2011) da OCDE. A OCDE iniciou em 1998 uma atividade altamente prioritária com o objetivo de rever as diretrizes em vigor para a pesquisa e o ensaio de produtos químicos com possíveis efeitos de perturbação endócrina e desenvolver novas diretrizes. O Conceptual Framework for Testing and Assessment of Endocrine Disrupting Chemicals da OCDE, (2002) compreende cinco níveis, cada um dos quis corresponde a um nível diferente de complexidade biológica (1). O ensaio de esteroidogénese H295R in vitro descrito no presente método de ensaio utiliza uma linhagem celular do adenocarcinoma humano (células NCI-H295R) e constitui um ensaio in vitro de nível 2, proporcionando dados mecanísticos a utilizar para fins de monitorização e priorização. O desenvolvimento e a normalização do ensaio para a pesquisa dos efeitos dos produtos químicos na esteroidogénese, designadamente na produção de 17β-estradiol (E2) e de testosterona (T), resultaram de um processo em várias fases. O ensaio H295R foi otimizado e validado (2) (3) (4) (5).
2. O objetivo do ensaio de esteroidogénese H295R consiste em detetar produtos químicos que afetem a produção de E2 e T. O ensaio H295R tem por objetivo identificar substâncias xenobióticas que tenham por alvo(s) os componentes endógenos que constituem o mecanismo bioquímico intracelular que, numa sequência de reações, conduz à produção de E2 e/ou T a partir do colesterol. O ensaio H295R não tem por objetivo identificar produtos químicos que afetem a esteroidogénese através de efeitos no sistema hipotálamo-hipófise-gónadas (HPG). O objetivo consiste apenas em obter uma resposta POSITIVA/NEGATIVA quanto ao potencial de um produto químico de induzir ou inibir a produção de T e E2; podem, contudo, obter-se resultados quantitativos em certos casos (ver pontos 53 e 54). Os resultados do ensaio são expressos em alterações relativas da produção de hormonas relativamente aos ensaios de controlo com solvente (SC). O objetivo não consiste em obter informações específicas sobre os mecanismos implicados na interação do produto químico em estudo com o sistema endócrino. Foram realizadas investigações com a linhagem celular tendo em vista identificar efeitos nas enzimas específicos e nas hormonas intermediárias, como a progesterona (2).
3. As definições e abreviaturas utilizadas no presente método de ensaio são descritas no apêndice. O apêndice I-III do documento da OCDE intitulado Multi-Laboratory Validation of the H295R Steroidogenesis Assay to Identify Modulators of Testosterone and Estradiol Production (4) inclui um protocolo pormenorizado com instruções para a preparação das soluções, a cultura das células e a execução das várias fases do ensaio.
CONSIDERAÇÕES INICIAIS E LIMITAÇÕES
4. Na biossíntese das hormonas sexuais esteroides participam cinco enzimas diferentes, que catalisam seis reações específicas. A conversão enzimática do colesterol em pregnenolona, promovida pela enzima CYP11A de clivagem da cadeia lateral do colesterol do citocromo P450, constitui o passo inicial numa série de reações bioquímicas que conduzem à síntese de produtos finais esteroides. Consoante a ordem das duas reações seguintes, o mecanismo da esteroidogénese divide-se em duas vias, a via dos Δ5-hidroxiesteroides e a via dos Δ4-cetoesteroides, que convergem na produção de androstenodiona (figura 1).
5. A androstenodiona é convertida em testosterona (T) por ação da desidrogenase dos 17β-hidroxiesteroides (17β-HSD). A testosterona é simultaneamente um intermediário e um produto final (hormona). Nos machos, pode ser convertida em di-hidrotestosterona (DHT) por ação da 5α-redutase presente na membrana celular, no invólucro nuclear e no retículo endoplásmico dos tecidos-alvo da ação androgénica, como a próstata e as vesículas seminais. A DHT é um androgénio significativamente mais potente que a testosterona, sendo também considerada um produto final hormonal. O ensaio H295R não determina a DHT (ver ponto 10).
6. A aromatase (CYP19) é a enzima que, no processo da esteroidogénese, promove a conversão das substâncias androgénicas em estrogénicas, nomeadamente a testosterona (T) em 17β-estradiol (E2) e a androstenodiona em estrona. A E2 e a T são consideradas produtos finais hormonais no processo da esteroidogénese.
7. A especificidade da atividade de liase da CYP17 relativamente aos substratos intermediários difere em função das espécies. No homem, a enzima favorece os substratos da via dos Δ5-hidroxiesteroides (pregnenolona), enquanto, no rato, são favorecidos os substratos da via dos Δ4-cetoesteroides (progesterona) (19). Estas diferenças na atividade de liase da CYP17 permitem explicar algumas diferenças específicas das espécies na resposta aos produtos químicos que alteram a esteroidogénese in vivo (6). Observou-se que as células H295 refletem mais estreitamente a expressão das enzimas adrenais e o mecanismo de produção de esteroides no homem adulto (20), embora, no respeitante à síntese de androgénios, produzam enzimas para as vias dos Δ5-hidroxiesteroides e dos Δ4-cetoesteroides (7) (11) (13) (15).
Figura 1
Mecanismo da esteroidogénese nas células H295R
Nota:
As enzimas são indicadas em itálico, as hormonas em negrito e as setas indicam a direção do processo. O fundo cinzento indica as vias e os produtos corticosteróides. As hormonas sexuais e respetivas vias estão envoltas por uma linha circular. CYP = citocromo P450; HSD = desidrogenase dos hidroxiesteroides; DHEA = desidroepiandrosterona
8. A linhagem celular do adenocarcinoma humano H295R constitui um modelo útil para a investigação in vitro dos efeitos na síntese de hormonas esteroides (2) (7) (8) (9) (10). A linhagem celular H295R exprime genes que codificam todas as enzimas-chave da esteroidogénese atrás referidas (11) (15) (figura 1). Trata-se de uma propriedade única, dado que a expressão in vivo desses genes é específica dos tecidos e das fases de desenvolvimento, sendo que, em geral, um só tecido ou uma única fase de desenvolvimento não exprime todos os genes implicados na esteroidogénese (2). As células H295R têm características fisiológicas de células adrenais fetais humanas não diferenciadas por zonas (11). As células constituem um sistema in vitro específico, na medida em que têm a capacidade de produzir todas as hormonas esteroides do córtex adrenal e das gónadas dos adultos, permitindo observar, em simultâneo, efeitos na síntese de corticosteróides e de hormonas sexuais esteroides como os androgénios e os estrogénios, embora o ensaio tenha sido validado apenas para a deteção de T e E2. As alterações registadas pelo sistema de ensaio, expressas em alterações na produção de T e E2, podem decorrer duma vasta gama de interações diversas dos produtos químicos em estudo com as funções esteroidogénicas expressas pelas células H295R. Essas alterações abrangem a modulação da expressão, a síntese ou a ação das enzimas implicadas na produção, transformação ou eliminação das hormonas esteroides (12) (13) (14). A inibição da produção hormonal pode ser devida à ligação competitiva direta a uma enzima implicada no processo, ao impacto em cofatores como o NADPH (fosfato de dinucleótido nicotinamida-adenina) e o cAMP (monofosfato de adenosina cíclica), e/ou a um aumento do metabolismo dos esteroides ou à supressão da expressão dos genes que codificam determinadas enzimas no mecanismo da esteroidogénese. Enquanto a inibição pode resultar de processos diretos ou indiretos implicados na produção das hormonas, a indução é, em geral, de natureza indireta — por exemplo, afetando cofatores como o NADPH e o cAMP (caso da forskolina), reduzindo o metabolismo dos esteroides (13) e/ou regulando positivamente a expressão dos genes esteroidogénicos.
9. O ensaio H295R apresenta várias vantagens:
|
— |
Permite a deteção de aumentos e reduções na produção de T e E2; |
|
— |
Permite a avaliação direta do possível impacto de um produto químico na viabilidade das células ou na citotoxicidade. Esta característica é importante, na medida em que permite a distinção entre os efeitos devidos à citotoxicidade e os efeitos devidos à interação direta dos produtos químicos com o mecanismo da esteroidogénese, o que não é possível por recurso a sistemas de explantes de tecidos constituídos por vários tipos de células com sensibilidades e funcionalidades diversas; |
|
— |
Não exige a utilização de animais; |
|
— |
A linhagem celular H295R encontra-se disponível no comércio. |
10. As principais limitações do ensaio são as seguintes:
|
— |
A sua capacidade metabólica é desconhecida, sendo, provavelmente, bastante limitada, pelo que o ensaio não permite identificar a ação de produtos químicos que necessitem de ativação metabólica. |
|
— |
Uma vez que é obtida a partir de tecido adrenal, a linhagem H295R contém as enzimas que promovem a produção dos glucocorticoides e dos mineralocorticoides, bem como das hormonas sexuais; os efeitos na produção de glucocorticoides e mineralocorticoides podem condicionar os teores de T e E2 observados no ensaio. |
|
— |
Não determina a DHT, pelo que, em princípio, não permite detetar produtos químicos que inibem a 5α-redutase, caso em que pode recorrer-se ao ensaio de Hershberger (16). |
|
— |
Não permite detetar produtos químicos que interfiram com a esteroidogénese ao nível do eixo hipotálamo-pituitária-gónadas (HPG), caso que só pode ser estudado com animais intactos. |
PRINCÍPIO DO ENSAIO
11. O objetivo do ensaio consiste na deteção de produtos químicos que afetem a produção de testosterona e de E2. A testosterona é também um intermediário na produção de E2. O ensaio permite detetar, em especial, produtos químicos que inibam ou induzam as enzimas que promovem a esteroidogénese.
12. Em geral, o ensaio é realizado em condições normalizadas de cultura celular, em placas de cultura com 24 alvéolos. Em alternativa, podem utilizar-se placas de outras dimensões; contudo, a inoculação e as condições experimentais devem ser ajustadas em conformidade, de forma a manter o respeito dos critérios de desempenho.
13. Após um período de aclimação de 24 horas em placas multialvéolos, as células são expostas durante 48 horas a sete concentrações do produto químico em estudo, pelo menos em triplicado. Adicionam-se concentrações fixas de um solvente e de um inibidor ou promotor conhecido da produção de hormonas, na qualidade de controlos negativos e positivos. No final do período de exposição, é removido o meio de cada alvéolo. A viabilidade das células em cada alvéolo é analisada imediatamente após a remoção do meio. As concentrações de hormonas no meio podem ser medidas por diversos métodos, nomeadamente por recurso a conjuntos de determinação de hormonas disponíveis no comércio e/ou técnicas instrumentais como a cromatografia em fase líquida acoplada com espetrometria de massa (LC-MS). Os dados são expressos em fatores de alteração relativamente ao controlo com solvente e à menor concentração com efeito observável (LOEC — Lowest Observed Effect Concentration). Se o ensaio for negativo, a concentração mais elevada objeto de ensaio é referida como concentração sem efeitos observáveis (NOEC — No Observed Effect Concentration). As conclusões quanto à capacidade do produto químico de afetar a esteroidogénese devem basear-se em, pelo menos, dois ensaios independentes. O primeiro ensaio pode consistir num rastreio de gamas de concentração, procedendo-se ao ajustamento das concentrações nos dois ensaios posteriores, se pertinente, caso se registem problemas de solubilidade ou citotoxicidade ou se a atividade do produto químico se situar no limite da gama de concentrações de ensaio.
PROCEDIMENTO DE CULTURA CELULAR
Linhagem celular
14. As células NCI-H295R são comercializadas pela American Type Culture Collections (ATCC), mediante a assinatura de um contrato (Material Transfer Agreement — MTA) (14).
Introdução
15. Devido às alterações da capacidade de produção de E2 das células com a idade ou o número de passagens (2), as células devem ser cultivadas com base num protocolo específico antes de serem utilizadas, devendo registar-se o número de passagens desde o descongelamento das células, bem como o número da passagem na qual as mesmas são congeladas e armazenadas em azoto líquido. O primeiro número indica o número real da passagem; o segundo descreve o número da passagem na qual as células foram congeladas e armazenadas. Por exemplo, células que tenham sido congeladas após a passagem n.o 5, posteriormente descongeladas e divididas três vezes (quatro passagens, sendo a primeira constituída pelas células recém-descongeladas), sendo subsequentemente sujeitas a nova cultura, serão classificadas como células de passagem 4,5. O apêndice 1 do relatório de validação (4) apresenta um exemplo do sistema de numeração.
16. O meio de partida é utilizado como base para os meios suplementado e de congelamento. O meio suplementado é um componente necessário à cultura celular. O meio de congelamento destina-se especificamente a permitir o congelamento sem impacto nas células para armazenagem a longo prazo. Antes da utilização, deve analisar-se oNu-serum (ou um soro semelhante com propriedades análogas, relativamente ao qual se tenha demonstrado que produz dados conformes com os requisitos de desempenho e controlo da qualidade do ensaio), componente do meio suplementado, para pesquisar concentrações de fundo de T e E2. A preparação destas soluções é descrita no apêndice II do relatório de validação (4).
17. Após o início da cultura de células H295R a partir de um lote ATCC original, as células devem ser sujeitas a cinco passagens (ou seja, quatro divisões). As células da passagem n.o 5 são então congeladas em azoto líquido, para armazenagem. Antes do congelamento, procede-se ao ensaio de uma amostra das células da passagem n.o 4 numa placa de controlo da qualidade (ver pontos 36 e 37), para verificar se a produção basal de hormonas e a resposta aos produtos químicos de controlo positivo cumprem os critérios de controlo da qualidade definidos no quadro 5.
18. Após a cultura, as células H295R têm de ser congeladas e armazenadas em azoto líquido, de forma a garantir que são sempre da passagem ou idade adequada à cultura e à utilização. O número máximo de passagens após a inoculação na cultura de um lote de células novo (15) ou congelado (16) aceitável para utilização no ensaio H295R não deve exceder 10. Por exemplo, o número aceitável de passagens em culturas de células a partir de um lote congelado na passagem n.o 5 estará compreendido entre 4,5 e 10,5. No caso das células colhidas de lotes congelados, deve seguir-se o procedimento descrito no ponto 19. Estas células devem ser cultivadas por, pelo menos, quatro passagens adicionais (passagem 4,5) antes de serem utilizadas no ensaio.
Preparação das células a partir do material congelado
19. Utiliza-se o procedimento de inoculação das células a partir do material congelado sempre que um novo lote de células é removido do azoto líquido para fins de cultura e ensaio. O procedimento pormenorizado encontra-se descrito no apêndice III do relatório de validação (4). As células são removidas do azoto líquido, descongeladas rapidamente e colocadas num meio suplementado, num tubo de centrifugadora, sendo centrifugadas à temperatura ambiente, ressuspendidas em meio suplementado e transferidas para um recipiente de cultura. O meio de cultura deve ser substituído no dia seguinte. As células H295R são cultivadas numa incubadora a 37 °C, em ar atmosférico com 5 % CO2, sendo o meio renovado duas a três vezes por semana. Quando as células exibirem uma confluência de aproximadamente 85-90 %, devem ser divididas. A divisão é necessária para assegurar o bom estado e o crescimento das células e conservá-las até à realização dos bioensaios. As células são lavadas três vezes com uma solução-tampão salina de fosfatos (PBS, sem Ca2+ nem Mg2+.) e retiradas do recipiente de cultura através da adição de uma enzima de desprendimento adequada, por exemplo tripsina, dissolvida em PBS (sem Ca2+ nem Mg2+). Imediatamente após a remoção das células do recipiente de cultura, deve suspender-se a ação da enzima através da adição de um meio suplementado na proporção de três vezes o volume utilizado no tratamento enzimático. As células são colocadas num tubo de centrifugadora e centrifugadas à temperatura ambiente; o sobrenadante é removido e o aglomerado de células ressuspendido no meio suplementado. Coloca-se no novo recipiente de cultura a quantidade adequada da solução com as células. A quantidade desta solução deve ser ajustada de forma que as células mostrem confluência num período de 5-7 dias. O rácio de subcultura recomendado é de 1:3 a 1:4. A placa deve ser rotulada cuidadosamente. As células ficam, assim, prontas para utilização no ensaio, devendo as células em excesso ser congeladas em azoto líquido do modo descrito no ponto 20.
Congelamento das células H295R (preparação para armazenamento em azoto líquido)
20. Para preparar as células H295R para congelamento, deve seguir-se o procedimento atrás descrito para a divisão das mesmas, até à etapa de ressuspensão do aglomerado de células presentes no fundo do tubo de centrifugadora, que é efetuada em meio de congelamento. A solução é transferida para um balão criogénico devidamente rotulado e congelada a –80°C durante 24 horas, período após o qual o balão criogénico é transferido para o recipiente com azoto líquido, para armazenagem. O apêndice III do relatório de validação (4) contém os pormenores do procedimento a seguir.
Preparação das placas e pré-incubação das células para ensaio
21. O número necessário de placas de 24 alvéolos, preparadas do modo descrito no ponto 19, depende do número de produtos químicos a ensaiar, bem como da confluência das células nos recipientes de cultura. Em geral, um recipiente de cultura (75 cm2) com 80-90 % de células confluentes fornece um número de células suficiente para 1 a 1,5 placas de 24 alvéolos, à densidade-alvo de 200 000 a 300 000 células por ml de meio, resultando numa confluência de, aproximadamente, 50-60 % nos alvéolos em 24 horas (figura 2). Esta é, normalmente, a densidade celular ótima para a produção de hormonas no ensaio. A densidades mais elevadas, são alterados os perfis de produção de T e de E2. Antes de realizar o ensaio pela primeira vez, recomenda-se que sejam ensaiadas várias densidades de inoculação compreendidas entre 200 000 e 300 000 células por ml, devendo selecionar-se para os ensaios posteriores a densidade que determine uma confluência de 50-60 % nos alvéolos em 24 horas.
Figura 2
Fotomicrografia de células H295R à densidade de inoculação de 50 %, numa placa de cultura de 24 alvéolos, às 24 horas, no bordo (A) e no centro (B) de um alvéolo

22. O meio é removido com uma pipeta do recipiente de cultura, sendo as células lavadas três vezes com PBS estéril (sem Ca2+ nem Mg2+). Para desprender as células do recipiente de cultura, adiciona-se uma solução de enzima em PBS. Após um período adequado ao desprendimento das células, suspende-se a ação enzimática através da adição de meio suplementado à razão de três vezes o volume utilizado no tratamento enzimático. Colocam-se as células num tubo de centrifugadora e centrifuga-se à temperatura ambiente; seguidamente, remove-se o sobrenadante e ressuspende-se o aglomerado de células em meio suplementado. Calcula-se a densidade celular, utilizando, p.ex. um hemocitómetro ou um contador celular. A solução com as células deve ser diluída para a densidade pretendida na placa e homogeneizada de forma a garantir uma densidade celular homogénea. As células são colocadas na placa com 1 ml de solução por alvéolo, devendo rotular-se as placas e os alvéolos. As placas inoculadas são incubadas durante 24 horas a 37 °C, em ar atmosférico com 5 % de CO2, de forma a permitir a aderência das células aos alvéolos.
REQUISITOS DE CONTROLO DA QUALIDADE
23. Durante a dosagem, é indispensável que sejam colocados nos alvéolos volumes exatos das soluções e amostras, dado que esses volumes determinam as concentrações utilizadas no cálculo dos resultados.
24. Antes do início da cultura celular e de qualquer ensaio posterior, cada laboratório deve demonstrar a sensibilidade do seu sistema de determinação das hormonas (pontos 29-31).
25. Caso se utilizem métodos de determinação de hormonas baseados em anticorpos, deve averiguar-se, antes de iniciar o ensaio, o potencial de interferência dos produtos químicos a ensaiar com o sistema de medida utilizado para quantificar a T e a E2, como referido no ponto 32.
26. O DMSO é o solvente recomendado para o ensaio. Caso se utilize um solvente alternativo, devem determinar-se os seguintes parâmetros:
|
— |
Solubilidade do produto químico em estudo, da forskolina e do procloraz no solvente; e |
|
— |
Citotoxicidade em função da concentração do solvente. |
Recomenda-se que a concentração máxima permissível do solvente não exceda uma diluição de 10 vezes a concentração menos citotóxica do solvente.
27. Antes de realizar o ensaio pela primeira vez, o laboratório deve realizar um ensaio de qualificação que demonstre a sua capacidade de adotar e manter as condições de cultura celular e condições experimentais adequadas ao ensaio do produto químico em conformidade com os pontos 33-35.
28. Deve realizar-se um ensaio de controlo, como descrito nos pontos 36 e 37, antes da utilização de um novo lote de células, com vista a avaliar o desempenho das mesmas.
Desempenho do sistema de determinação das hormonas
Sensibilidade, exatidão e precisão do método; reatividade cruzada com a matriz da amostra
29. Para a análise da T e da E2 produzidas pelas células H295R, cada laboratório pode utilizar um sistema de determinação de hormonas da sua escolha, desde que o mesmo satisfaça os critérios de desempenho, nomeadamente os limites de quantificação (LOQ). Estes são, geralmente, de 100 pg/ml no caso da testosterona e de 10 pg/ml no caso da E2, valores que se baseiam nos níveis de hormonas basais observados nos estudos de validação. Podem, contudo, justificar-se níveis mais elevados ou mais baixos, em função dos níveis basais de hormonas obtidos no laboratório em causa. Antes de iniciar a verificação do controlo da qualidade e os ensaios, o laboratório deve demonstrar que o método a utilizar na determinação das hormonas permite medir as respetivas concentrações no meio suplementado com exatidão e precisão suficientes para o cumprimento dos critérios de controlo de qualidade especificados nos quadros 1 e 5, analisando o meio suplementado enriquecido com hormona (controlo interno). O meio suplementado é enriquecido com, pelo menos, três concentrações de cada hormona (p.ex. 100, 500 e 2 500 pg/ml de T; 10, 50 e 250 pg/ml de E2; em alternativa, podem utilizar-se as concentrações mais baixas possíveis em função dos limites de deteção do sistema de determinação escolhido para as concentrações enriquecidas mais baixas para a T e a E2) e, seguidamente, analisado. As concentrações de hormonas determinadas nas amostras não extraídas não devem divergir em mais de 30 % das concentrações nominais e a variação entre determinações replicadas da mesma amostra não deve exceder 25 % (o quadro 8 contém critérios adicionais de controlo da qualidade). Caso estes critérios sejam satisfeitos, considera-se que o método de determinação das hormonas é suficientemente exato e preciso, não se prevendo a ocorrência de reações cruzadas com os componentes do meio (matriz da amostra) que possam influir de forma significativa nos resultados. Nesse caso, não é necessário proceder à extração das amostras antes da determinação das hormonas.
30. Se não forem satisfeitos os critérios de controlo da qualidade constantes dos quadros 1 e 8, pode estar a ocorrer um efeito matricial significativo, devendo realizar-se um ensaio com meio enriquecido extraído. O apêndice II do relatório de validação (4) apresenta um exemplo do procedimento de extração. A determinação das concentrações de hormonas nas amostras extraídas deve efetuar-se em triplicado (17). Se, após a extração, for possível demonstrar que os componentes do meio não interferem com o método de deteção das hormonas definido pelos critérios de controlo da qualidade, todos os ensaios subsequentes devem ser realizados com amostras extraídas. Se os critérios de controlo da qualidade não forem cumpridos após a extração, o sistema de determinação das hormonas utilizado não é adequado para os fins do ensaio de esteroidogénese H295R, devendo recorrer-se a um método alternativo.
Curva-padrão
31. As concentrações de hormonas nos controlos com solvente devem situar-se no troço linear da curva-padrão, devendo, preferencialmente, ser próximas do valor correspondente ao ponto médio do referido troço, para que se possa determinar a indução ou a inibição da síntese das hormonas. As diluições do meio (ou dos extratos) a determinar devem ser selecionadas em conformidade. A relação linear deve ser definida por um método estatístico adequado.
Ensaio de determinação da interferência de produtos químicos
32. Se, para a determinação das hormonas, forem utilizados ensaios baseados em anticorpos, como ensaios de imunoabsorção enzimática (ELISA) e ensaios radioimunológicos (RIA), deve averiguar-se a possível interferência de cada um dos produtos químicos em estudo com o sistema de determinação de hormonas a utilizar, antes do início do ensaio do produto em causa [apêndice III do relatório de validação (4)], dado que alguns produtos químicos podem interferir com os processos de ensaio (17). Caso a análise demonstre uma interferência traduzida num aumento da produção de T e/ou E2 ≥ 20 % relativamente à produção basal destas hormonas, deve efetuar-se um ensaio de interferência dos produtos químicos na determinação das hormonas (do tipo descrito no apêndice III do relatório de validação (4), secção 5.0) com todas as diluições do produto químico em estudo, para identificar a dose-limiar à qual ocorre uma interferência significativa (≥ 20 %). Se a interferência for inferior a 30 %, os resultados podem ser corrigidos em conformidade. Se exceder 30 %, os dados não são válidos, devendo as concentrações em causa ser eliminadas do estudo. Se ocorrer uma interferência significativa da substância em estudo com o sistema de determinação das hormonas a mais de uma concentração não citotóxica, deve utilizar-se outro sistema de determinação. Para evitar interferências de produtos químicos contaminantes, recomenda-se que as hormonas sejam extraídas do meio por recurso a solventes adequados; o relatório de validação descreve métodos possíveis para esse efeito (4).
Quadro 1
Critérios de desempenho para os sistemas de determinação de hormonas
|
Parâmetro |
Critério |
|
Sensibilidade do método de determinação |
Limite de quantificação (LOQ) T: 100 pg/ml; E2: 10 pg/ml (18) |
|
Eficiência de extração das hormonas (apenas se esta for necessária) |
As taxas médias de recuperação (com base em determinações em triplicado) para as quantidades de hormonas adicionadas não devem exibir um desvio superior a 30 % da quantidade adicionada. |
|
Interferência química (apenas para sistemas baseados em anticorpos) |
Não deve registar-se uma reatividade cruzada substancial (≥ 30 % da produção basal da hormona em causa) com nenhuma das hormonas produzidas pelas células (19) (20) |
Ensaio de proficiência laboratorial
33. Antes de ensaiar produtos químicos desconhecidos, um laboratório deve demonstrar, por recurso ao ensaio de proficiência laboratorial, a sua capacidade de adotar e manter as condições de cultura celular e condições experimentais adequadas à boa realização do ensaio. Dado que o desempenho de um ensaio está diretamente ligado ao pessoal do laboratório que o realiza, o procedimento deve ser parcialmente repetido caso ocorram mudanças no pessoal.
34. O ensaio de proficiência deve ser realizado nas condições descritas nos pontos 38 a 40, expondo células a sete concentrações crescentes de indutores e inibidores fortes, moderados e ligeiros, bem como a um produto químico negativo (ver quadro 2). Especificamente, os produtos químicos a ensaiar incluem o indutor forte forskolina (n.o CAS 66575-29-9); o inibidor forte procloraz (n.o CAS 67747-09-5); o indutor moderado atrazina (n.o CAS 1912-24-9); o inibidor moderado aminoglutetimida (n.o CAS 125-84-8); o indutor fraco (produção de E2) e o inibidor fraco (produção de T) bisfenol A (n.o CAS 80-05-7), bem como o produto químico negativo gonadotrofina coriónica humana (HCG) (n.o CAS 9002-61-3), como se ilustra no quadro 2. Utilizam-se placas distintas para cada produto químico, de acordo com o modelo que se ilustra no quadro 6. Cada ensaio diário dos produtos químicos para o esnaio de proficiência deve incluir uma placa de controlo da qualidade (quadro 4, pontos 36-37).
Quadro 2
Produtos químicos para o ensaio de proficiência e concentrações de exposição
|
Produto químico |
Concentrações de ensaio [μM] |
|
Procloraz |
0 (21), 0,01, 0,03, 0,1, 0,3, 1, 3, 10 |
|
Forskolina |
0 (21), 0,03, 0,1, 0,3, 1, 3, 10, 30 |
|
Atrazina |
0 (21), 0,03, 0,1, 1, 3, 10, 30, 100 |
|
Aminoglutetimida |
0 (21), 0,03, 0,1, 1, 3, 10, 30, 100 |
|
Bisfenol A |
0 (21), 0,03, 0,1, 1, 3, 10, 30, 100 |
|
HCG |
0 (21), 0,03, 0,1, 1, 3, 10, 30, 100 |
No ensaio de proficiência laboratorial, a exposição das células H295R aos produtos químicos é efetuada em placas de 24 alvéolos. A dosagem é expressa em μM no respeitante a todas as doses de produtos químicos em estudo. As doses devem ser administradas em DMSO a 0,1 % (v/v) por alvéolo. Todas as concentrações de ensaio devem ser ensaiadas em triplicado (quadro 6). Utilizam-se placas distintas para cada produto químico. Cada série de ensaios diária deve incluir uma placa de controlo da qualidade.
35. As análises da viabilidade celular e das hormonas devem ser efetuadas do modo descrito nos pontos 42 a 46. O valor-limiar (menor concentração com efeito observável, LOEC) e a decisão de classificação devem ser comunicados e comparados com os valores constantes do quadro 3. Os dados são considerados aceitáveis se forem conformes com o LOEC e a decisão de classificação constantes do quadro 3.
Quadro 3
Valores-limiar (LOEC) e decisões de classificação respeitantes aos produtos químicos para o ensaio de proficiência
|
|
N.o CAS |
LOEC [μM] |
Decisão de classificação |
||
|
T |
E2 |
T |
E2 |
||
|
Procloraz |
67747-09-5 |
≤ 0,1 |
≤ 1,0 |
+ (22) (Inibição) |
+ (Inibição) |
|
Forskolina |
66575-29-9 |
≤ 10 |
≤ 0,1 |
+ (Indução) |
+ (Indução) |
|
Atrazina |
1912-24-9 |
≤ 100 |
≤ 10 |
+ (Indução) |
+ (Indução) |
|
Aminoglutetimida |
125-84-8 |
≤ 100 |
≤ 100 |
+ (Inibição) |
+ (Inibição) |
|
Bisfenol A |
80-05-7 |
≤ 10 |
≤ 10 |
+ (Inibição) |
+ (Indução) |
|
HCG |
9002-61-3 |
n/a |
n/a |
Negativo |
Negativo |
Placa de controlo da qualidade
36. A placa de controlo da qualidade é utilizada para verificar o desempenho das células H295R em condições de cultura normalizadas, bem como para criar uma base de dados históricos de concentrações das hormonas nos solventes de controlo, nos controlos positivos e negativos e nos outros ensaios de controlo da qualidade que forem sendo efetuados.
|
— |
O desempenho das células H295R deve ser avaliado por recurso a uma placa de controlo da qualidade para cada novo lote ATCC, ou após a utilização, pela primeira vez, de um lote de células previamente congeladas, exceto se tiver sido realizado um ensaio de proficiência laboratorial (pontos 32-34) com esse lote de células. |
|
— |
A placa de controlo da qualidade proporciona uma avaliação completa das condições de ensaio de produtos químicos (p.ex. viabilidade celular, controlos com solvente, controlos negativos e positivos, variabilidade intra e interensaios), devendo constituir parte integrante de cada ensaio. |
37. O ensaio de controlo da qualidade é realizado numa placa de 24 alvéolos, seguindo os mesmos procedimentos em matéria de incubação, dosagem, viabilidade celular/citotoxicidade, extração e análise das hormonas descritos nos pontos 38 a 46 para o ensaio dos produtos químicos. A placa de controlo da qualidade contém brancos, controlos com solvente e duas concentrações de um indutor (forskolina, 1, 10 μM) e de um inibidor (procloraz, 0,1, 1 μM) conhecido da síntese de E2 e T. Utiliza-se também, em alguns alvéolos, MeOH (controlo positivo do ensaio de viabilidade celular/citotoxicidade). O quadro 4 apresenta uma descrição pormenorizada da configuração da placa. Os critérios que esta deve satisfazer são enumerados no quadro 5. Tanto no controlo com solvente como nos alvéolos em branco deve observar-se a produção basal mínima de hormonas (T e E2).
Quadro 4
Configuração da placa de controlo da qualidade para o ensaio do desempenho de células H295R não expostas e de células expostas a inibidores (PRO = procloraz) e promotores (FOR = forskolina) conhecidos da produção de E2 e T. Após o termo da exposição e a remoção do meio, adiciona-se uma solução de metanol a 70 % a todos os alvéolos com MeOH, que servirá de controlo positivo da citotoxicidade [ver o ensaio de citotoxicidade no apêndice III do relatório de validação (4)]
|
|
1 |
2 |
3 |
4 |
5 |
6 |
|
A |
Branco (23) |
Branco (23) |
Branco (23) |
Branco (23) (+ MeOH) (24) |
Branco (23) (+ MeOH) (24) |
Branco (23) (+ MeOH) (24) |
|
B |
DMSO (25) 1 μl |
DMSO (25) 1 μl |
DMSO (25) 1 μl |
DMSO (25) 1 μl (+ MeOH) (24) |
DMSO (25) 1 μl (+ MeOH) (24) |
DMSO (25) 1 μl (+ MeOH) (24) |
|
C |
FOR 1 μM |
FOR 1 μM |
FOR 1 μM |
PRO 0,1 μM |
PRO 0,1 μM |
PRO 0,1 μM |
|
D |
FOR 10 μM |
FOR 10 μM |
FOR 10 μM |
PRO 1 μM |
PRO 1 μM |
PRO 1 μM |
Quadro 5
Critérios de desempenho para a placa de controlo da qualidade
|
|
T |
E2 |
|
Produção basal de hormonas no controlo com solvente (CS) |
≥ 5 × LOQ |
≥ 2,5 × LOQ |
|
Indução (10 μM forskolina) |
≥ 1,5 × CS |
≥ 7,5 × CS |
|
Inibição (1μM procloraz) |
≤ 0,5 × CS |
≤ 0,5 × CS |
PROCEDIMENTO DE ExposIÇÃO AOS PRODUTOS QUÍMICOS
38. As células pré-incubadas são tiradas da incubadora (ponto 21) e observadas ao microscópio, para assegurar que se encontram em boas condições (aderência, morfologia), antes da dosagem.
39. As células são colocadas num espaço confinado biosseguro, sendo o meio suplementado removido e substituído por um novo meio suplementado (1 ml/alvéolo). O DMSO é o solvente preferido neste método de ensaio. Contudo, se existirem motivos para utilizar outros solventes, deve apresentar-se a respetiva fundamentação científica. As células são expostas ao produto químico em estudo através da adição de 1 μl da solução-mãe adequada em DMSO [ver apêndice II do relatório de validação (4)] por mililitro de meio suplementado (volume do alvéolo). A concentração final de DMSO nos alvéolos será, assim, de 0,1 %. Para assegurar a homogeneização, é geralmente preferível que a solução-mãe adequada do produto químico em estudo em DMSO seja misturada com meio suplementado de forma a produzir a concentração final pretendida para cada dose, sendo a mistura resultante adicionada em cada alvéolo imediatamente após a remoção do meio anterior. Se se utilizar esta opção, a concentração de DMSO (0,1 %) deverá ser a mesma em todos os alvéolos. Os alvéolos que contenham as duas concentrações mais elevadas são inspecionados visualmente, por recurso a um estereomicroscópio, para averiguar a eventual formação de precipitados ou turbidez, que constituem indicações de uma solubilização incompleta do produto químico em estudo. Caso se observem essas condições (turbidez, formação de precipitados), os alvéolos que contêm as concentrações inferiores (e assim sucessivamente) são também examinados; as concentrações que não tenham conduzido a uma dissolução completa são excluídas da avaliação e análise complementar. Recoloca-se a placa na incubadora a 37 °C, com ar atmosférico com 5 % de CO2, durante 48 horas. O quadro 6 ilustra a configuração da placa com a substância em estudo. Os lotes 1-7 mostram a distribuição na placa das doses crescentes de produto químico em estudo.
Quadro 6
Esquema de dosagem para a exposição de células H295R a produtos químicos em estudo, numa placa de 24 alvéolos
|
|
1 |
2 |
3 |
4 |
5 |
6 |
|
A |
DMSO |
DMSO |
DMSO |
Lote 4 |
Lote 4 |
Lote 4 |
|
B |
Lote 1 |
Lote 1 |
Lote 1 |
Lote 5 |
Lote 5 |
Lote 5 |
|
C |
Lote 2 |
Lote 2 |
Lote 2 |
Lote 6 |
Lote 6 |
Lote 6 |
|
D |
Lote 3 |
Lote 3 |
Lote 3 |
Lote 7 |
Lote 7 |
Lote 7 |
40. Após 48 horas, as placas são removidas da incubadora, sendo cada alvéolo sujeito à inspeção microscópica com vista a determinar o estado das células (aderência, morfologia, grau de confluência) e quaisquer sinais de citotoxicidade. O meio presente em cada alvéolo é dividido em duas porções iguais, de aproximadamente 490 μl, e transferido para dois recipientes devidamente rotulados (uma das alíquotas constitui uma amostra de reserva para cada alvéolo). Para evitar a secagem das células, o meio é removido por filas ou colunas e substituído com o meio para o ensaio de viabilidade celular/citotoxicidade. Se a viabilidade celular ou citotoxicidade não for determinada de imediato, adiciona-se a cada alvéolo 200 μl de PBS com Ca2+ e Mg2+. Os meios são congelados a – 80 °C até se proceder à análise das concentrações de hormonas (ver pontos 44-46). Embora, num meio mantido a – 80 °C, a T e a E2 se mantenham, em geral, estáveis por, pelo menos, três meses, cada laboratório deve documentar a estabilidade das hormonas durante a armazenagem.
41. A viabilidade celular/citotoxicidade em cada placa é determinada imediatamente após a remoção do meio.
Determinação da viabilidade celular
42. Para determinar o impacto potencial do produto químico em estudo, pode utilizar-se um ensaio de viabilidade celular/citotoxicidade da escolha do laboratório. O ensaio deve ter capacidade para proporcionar uma medição correta da percentagem de células viáveis presentes num alvéolo; em alternativa, deve demonstrar-se a sua comparabilidade direta (expressa numa função linear) com o ensaio Live/Dead® [ver o apêndice III do relatório de validação (4)]. Um ensaio alternativo que se verificou funcionar bem é o ensaio com MTT [brometo de 3-(4,5-dimetiltiazol-2-il)-2,5-difeniltetrazólio] (18). A avaliação da viabilidade celular por recurso aos referidos métodos constitui uma medida relativa que não apresenta necessariamente uma relação linear com o número absoluto de células num alvéolo. O analista deve, pois, efetuar, em paralelo, um exame visual subjetivo de cada alvéolo, devendo também, se necessário, obter e arquivar imagens digitais dos controlos com solvente (SC — Solvent Controls) e das duas concentrações não citotóxicas mais elevadas, para permitir a avaliação posterior da densidade celular real. Se a inspeção visual mostrar, ou o ensaio de viabilidade celular/citotoxicidade demonstrar, um aumento aparente do número de células, é necessário verificá-lo. Se esse aumento for comprovado, o facto deve constar do relatório do ensaio. A viabilidade celular é expressa em relação à resposta média nos SC (100 % de células viáveis), e é calculada por um método adequado ao ensaio de viabilidade celular/citotoxicidade em causa. No caso do ensaio com MTT, pode utilizar-se a seguinte fórmula:
% células viáveis = (resposta no alvéolo – resposta média nos alvéolos tratados com MeOH [= 100 % células mortas]) – (resposta média nos alvéolos SC – resposta média nos alvéolos tratados com MeOH [= 100 % células mortas)
43. Os alvéolos cuja viabilidade celular seja inferior a 80 % relativamente à viabilidade média nos SC (= 100 %) não devem ser incluídos na análise final dos dados. A inibição da esteroidogénese que se observa em cerca de 20 % dos casos de citotoxicidade deve ser cuidadosamente avaliada, de forma a garantir que a citotoxicidade não constitui a causa da inibição.
Análise das hormonas
44. Para a análise da T e da E2, cada laboratório pode utilizar um sistema de determinação de hormonas da sua escolha. Podem utilizar-se alíquotas de reserva do meio de cada grupo de tratamento para preparar diluições destinadas a obter uma concentração no troço linear da curva-padrão. Como referido no ponto 29, cada laboratório deve demonstrar a conformidade do sistema de determinação de hormonas que utilizar (p.ex. ELISA, RIA, LC-MS, LC-MS/MS) com os critérios de controlo da qualidade, analisando, para o efeito, meio suplementado enriquecido com um controlo interno de hormona, antes de efetuar os ensaios de controlo da qualidade ou de analisar os produtos químicos. Para garantir que as componentes do sistema de ensaio não interferem com a determinação das hormonas, estas podem ter de ser extraídas do meio antes da análise (as condições nas quais a extração é ou não necessária são descritas no ponto 30). Recomenda-se que a extração seja efetuada de acordo com os procedimentos descritos no apêndice III do relatório de validação (4).
45. Se for utilizado um conjunto de ensaio comercial para medir a produção de hormonas, a análise deve ser efetuada do modo especificado nos manuais de instruções fornecidos pelo fabricante. A maioria dos fabricantes tem um procedimento único para a realização das análises hormonais. As diluições das amostras têm de ser ajustadas para que as concentrações de hormonas previstas nos controlos com solvente se situem na zona média do troço linear da curva-padrão do ensaio individual [apêndice III do relatório de validação (4)]. Os valores que se situem fora do referido troço linear devem ser rejeitados.
46. As concentrações finais das hormonas são calculadas do seguinte modo:
Exemplo:
|
Quantidade extraída: |
450 μl de meio |
|
Reconstituição em: |
250 μl de tampão |
|
Diluição no ensaio: |
1:10 (para situar a amostra no troço linear da curva-padrão) |
|
Concentração de hormonas obtida no ensaio: |
150 pg/ml (já ajustada à concentração por ml de amostra utilizada no ensaio) |
|
Recuperação: |
89 % |
|
Concentração final de hormonas = |
(Concentração de hormonas (por ml) ÷ recuperação) (fator de diluição) |
|
Concentração final de hormonas = |
(150 pg/ml) ÷ (0,89) × (250 μl/450 μl) × 10 = 936,3 pg/ml |
Seleção das concentrações de ensaio
47. Devem efetuar-se, no mínimo, duas séries independentes de ensaios. Exceto se existirem informações, nomeadamente sobre os limites de solubilidade ou a citotoxicidade, que permitam selecionar as concentrações de ensaio, recomenda-se que as concentrações do ensaio inicial sejam espaçadas por fatores de log10, sendo 10–3 M a concentração máxima. Se o produto químico for solúvel e não citotóxico a qualquer das concentrações ensaiadas e a primeira série de ensaios produzir resultados negativos para todas as concentrações, há que proceder a uma confirmação com uma nova série de ensaios, utilizando as mesmas condições que na primeira série (quadro 7). Se os resultados da primeira série de ensaios forem equívocos (ou seja, o fator de alteração em relação ao SC for estatisticamente significativa apenas a uma concentração) ou positivos (ou seja, o fator de alteração entre duas ou mais concentrações adjacentes for estatisticamente significativo), o ensaio deve ser repetido da forma indicada no quadro 7, refinando as concentrações de ensaio selecionadas. As concentrações de ensaio na segunda e terceira séries (se for caso disso) devem ser ajustadas com base nos resultados das concentrações da série de ensaios inicial que produziram um efeito, utilizando intervalos de concentração semilogarítmicos (p. ex., se, na série original de 0,001, 0,01, 0,1, 1, 10, 100, 1 000 μM se observaram induções a 1 e 10 μM, as concentrações ensaiadas na segunda série devem ser de 0,1, 0,3, 1, 3, 10, 30 e 100 μM), exceto se tiverem de ser utilizadas concentrações mais baixas para atingir uma LOEC. Neste caso, devem utilizar-se na segunda série de ensaios, pelo menos, cinco concentrações inferiores à concentração mais baixa ensaiada na primeira série, de acordo com uma escala semilogarítmica. Se os resultados da segunda série de ensaios não confirmarem os da primeira (isto é, se não ocorrer significância estatística à concentração com resultados positivos no ensaio anterior ± 1 incremento de concentração), é necessário efetuar uma terceira série de ensaios utilizando as condições originais. Os resultados equívocos da primeira série consideram-se negativos se o efeito observado não puder ser confirmado em nenhuma das séries de ensaios subsequentes. Os resultados equívocos são considerados respostas positivas (efeito) se for possível confirmar a resposta em, pelo menos, uma série de ensaios complementar com um incremento de concentração de ± 1 (ver o ponto 55 do procedimento para a interpretação dos dados).
Quadro 7
Matriz de decisões para possíveis cenários de resultados
|
Série 1 |
Série 2 |
Série 3 |
Decisão |
|||
|
Cenário |
Decisão |
Cenário |
Decisão |
Cenário |
Positivo |
Negativo |
|
Negativo |
Confirmar (26) |
Negativo |
Parar |
|
|
X |
|
Negativo |
Confirmar (26) |
Positivo |
Refinar (27) |
Negativo |
|
X |
|
Equívoco (28) |
Refinar (27) |
Negativo |
Confirmar (26) |
Negativo |
|
X |
|
Equívoco (28) |
Refinar (27) |
Negativo |
Confirmar (26) |
Positivo |
X |
|
|
Equívoco (28) |
Refinar (27) |
Positivo |
|
|
X |
|
|
Positivo |
Refinar (27) |
Negativo |
Confirmar (26) |
Positivo |
X |
|
|
Negativo |
Confirmar (26) |
Positivo |
Refinar (27) |
Positivo |
X |
|
|
Positivo |
Refinar (27) |
Positivo |
Parar |
|
X |
|
Controlo de qualidade da placa de ensaio
48. Além dos critérios aplicáveis à placa de controlo da qualidade, apresentam-se no quadro 8 outros critérios de qualidade que devem ser cumpridos, respeitantes, designadamente, à variação aceitável entre alvéolos replicados, ensaios replicados, linearidade e sensibilidade dos sistemas de determinação de hormonas, variabilidade entre as determinações replicadas de hormonas na mesma amostra e percentagem de recuperação das amostras enriquecidas em hormonas após a extração do meio (se pertinente — ver o ponto 30 para os requisitos em matéria de extração). Para serem tidos em conta na avaliação posterior, os dados devem situar-se nas gamas aceitáveis definidas para cada parâmetro. Se isso não suceder, deve assinalar-se que os critérios de controlo da qualidade não foram cumpridos no caso da amostra em questão, devendo a mesma ser reanalisada ou eliminada do conjunto de dados.
Quadro 8
Gamas e/ou variação (%) aceitáveis para os parâmetros da placa do ensaio H295R.
(LOQ: Limite de quantificação do sistema de determinação de hormonas. CV: Coeficiente de variação; SC: Controlo com solvente; DPM: Desintegrações por minuto)
|
|
Comparação |
T |
E2 |
|
Produção basal de hormonas nos SC |
Fator multiplicador em relação à LOQ |
≥ 5 vezes |
≥ 2,5 vezes |
|
Ensaios de exposição — CV para os SC intraplacas (alvéolos replicados) |
Concentrações absolutas |
≤ 30 % |
≤ 30 % |
|
Ensaios de exposição — CV para os SC interplacas (ensaios replicados) |
Fator multiplicador |
≤ 30 % |
≤ 30 % |
|
Sistema de determinação das hormonas –Sensibilidade |
Fator de redução detetável em relação ao SC |
≥ 5 vezes |
≥ 2,5 vezes |
|
Sistema de determinação das hormonas — determinações replicadas de CV para os SC (29) |
Concentrações absolutas |
≤ 25 % |
≤ 25 % |
|
Extração do meio — Recuperação do padrão interno 3H (se pertinente) |
DPM |
≥ 65 % Nominal |
|
ANÁLISE E APRESENTAÇÃO DOS RESULTADOS
Análise dos dados
49. Para avaliar o aumento/decréscimo relativos da produção de hormona induzidos pelos produtos químicos, os resultados devem ser normalizados em relação ao valor médio de SC de cada placa de ensaio, sendo os resultados expressos em alterações relativamente ao SC em cada placa. Todos os dados devem ser expressos em média ± 1 desvio-padrão (SD).
50. A análise dos dados deve apenas abranger dados relativos às hormonas dos alvéolos nos quais se observou uma citotoxicidade inferior a 20 %. As alterações relativas são calculadas do seguinte modo:
Alteração relativa = (Concentração de hormonas em cada alvéolo) ÷ (Concentração média de hormonas em todos os alvéolos de controlo com solvente).
51. Se a inspeção visual do alvéolo ou o ensaio de viabilidade/citotoxicidade descrito no ponto 42 indiciarem um aumento do número de células, este aumento aparente deve ser verificado. Caso se confirme, o facto deve ser referido no relatório de ensaio.
52. Antes de proceder às análises estatísticas, importa avaliar os pressupostos de normalidade e homogeneidade das variâncias. A normalidade deve ser avaliada por recurso a gráficos de probabilidade-padrão ou por outro método estatístico adequado (p.ex. ensaio de Shapiro-Wilk). Se os dados (fatores de alteração) não apresentarem uma distribuição normal, deve procurar-se transformá-los para que se aproximem da mesma. Se os dados apresentarem uma distribuição normal ou próxima desta, devem analisar-se as diferenças entre os grupos sujeitos a concentrações do produto químico e os SC, por recurso a um ensaio paramétrico (p. ex., ensaio de Dunnett), tomando a concentração como variável independente e a resposta (fator de alteração) como variável dependente. Se os dados não apresentarem uma distribuição normal, deve utilizar-se um ensaio não paramétrico adequado (p. ex., ensaio de Kruskal-Wallis ou ensaio de Steel). Consideram-se as diferenças significativas se p ≤ 0,05. As avaliações estatísticas são efetuadas com base nos valores médios dos alvéolos que representam dados replicados independentes. Devido ao grande intervalo entre as doses na primeira série de ensaios (escala de log10), prevê-se que, em muitos casos, não seja possível definir claramente relações concentração-resposta se as duas doses mais elevadas se situarem no troço linear da curva sigmóide. Assim, no caso dos dados da primeira série de ensaios ou de qualquer outra série de dados em que tal ocorra (p.ex., se não puder ser estimada a eficácia máxima), aplicam-se estatísticas de variável fixa do tipo I, da forma atrás descrita.
53. Se, pelo menos, dois pontos se situarem no troço linear da curva e puderem ser calculadas as eficácias máximas — como se prevê no caso de alguns ensaios da 2a série efetuados utilizando intervalos semilogarítmicos entre as concentrações de exposição — a devem utilizar-se métodos probit, logit ou outros modelos adequados de regressão linear (p. ex., EC50 e EC20) para o cálculo das concentrações efetivas.
54. Os resultados devem ser apresentados em forma gráfica (gráfico de barras representando a média ± 1 desvio-padrão) e tabular (LOEC/NOEC, tipo de efeito e intensidade da resposta máxima na componente dose-resposta dos dados) (a figura 3 apresenta um exemplo). A avaliação dos dados apenas se considera válida se for baseada em, pelo menos, duas séries de ensaios independentes. Um ensaio, ou série de ensaios, é considerado independente se tiver sido efetuado numa data diferente, utilizando uma nova série de soluções e de controlos. A gama de concentrações utilizada nas séries de ensaios 2 e 3 (se necessário) pode ser adaptada com base nos resultados da série 1, de forma a definir melhor a gama de respostas que abrange a LOEC (ver ponto 47).
Figura 3
Exemplo de apresentação e avaliação de dados obtidos com a execução do ensaio H295R, em forma gráfica e tabular
[Os asteriscos indicam diferenças estatisticamente significativas em relação ao controlo com solvente (p< 0,05). LOEC: Menor concentração com efeito observável; Alteração máx: Intensidade máxima da resposta observada a qualquer concentração relativamente à resposta média do SC (= 1)]

|
Produto químico |
LOEC |
Alteração máx. |
|
Forskolina |
0,01 |
0,15 vezes |
|
Letrozole |
0,001 |
29 vezes |
Procedimento de interpretação dos dados
55. Um produto químico é considerado positivo se o fator multiplicativo de indução for estatisticamente diferente (p ≤ 0,05) entre duas concentrações adjacentes do controlo com solvente em, pelo menos, duas séries de ensaios independentes (quadro 7). Um produto químico é considerado negativo na sequência de duas séries de ensaios independentes negativas, ou de três séries em que duas se revelaram negativas e uma equívoca ou positiva. Se os dados produzidos por três ensaios independentes não cumprirem os critérios de decisão enumerados no quadro 7, os resultados experimentais não são interpretáveis. Os resultados obtidos com concentrações superiores aos limites de solubilidade ou concentrações citotóxicas não devem ser incluídos na interpretação dos resultados.
Relatório de ensaio
56. O relatório de ensaio deve incluir as seguintes informações:
Estabelecimento de ensaio
|
— |
Nome e endereço do estabelecimento; |
|
— |
Diretor do estudo e outras pessoas envolvidas, com especificação das suas responsabilidades; |
|
— |
Datas de início e termo do estudo; |
Produto químico em estudo, reagentes e substâncias de controlo
|
— |
Identidade (nome/n.o CAS se pertinente), fonte, número de lote ou série, pureza, fornecedor e caracterização do produto químico em estudo, dos reagentes e das substâncias de controlo; |
|
— |
Natureza física e propriedades fisicoquímicas relevantes do produto químico em estudo; |
|
— |
Condições de armazenagem; método e frequência de preparação do produto químico em estudo, dos reagentes e das substâncias de controlo; |
|
— |
Estabilidade do produto químico em estudo; |
Células
|
— |
Fonte e tipo de células; |
|
— |
Número (identificador) de passagens celulares das células utilizadas no ensaio; |
|
— |
Descrição dos procedimentos de conservação das culturas celulares; |
Requisitos prévios à realização do ensaio (se pertinente)
|
— |
Descrição e resultados do ensaio de interferência produto químico-hormona; |
|
— |
Descrição e resultados das determinações de eficiência de extração das hormonas; |
|
— |
Curvas-padrão e de calibração correspondentes a todos os ensaios analíticos a realizar; |
|
— |
Limites de deteção dos métodos analíticos escolhidos; |
Condições de ensaio
|
— |
Composição do meio; |
|
— |
Concentração do produto químico em estudo; |
|
— |
Densidade celular (concentrações celulares estimadas ou determinadas a 24 e 48 horas) |
|
— |
Solubilidade do produto químico em estudo (limite de solubilidade, se determinado); |
|
— |
Tempo e condições de incubação; |
Resultados do ensaio
|
— |
Dados em bruto para cada alvéolo (substâncias de controlo e produtos químicos em estudo) -- cada determinação replicada na forma dos dados originais fornecidos pelo instrumento utilizado para determinar a produção de hormonas (p.ex. OD, unidades de fluorescência, DPM, etc.); |
|
— |
Validação da normalidade ou explicação da transformação dos dados; |
|
— |
Respostas médias ± 1 desvio-padrão para cada alvéolo determinado; |
|
— |
Dados de citotoxicidade (concentrações de ensaio que causaram citotoxicidade); |
|
— |
Confirmação do cumprimento dos requisitos de controlo da qualidade; |
|
— |
Alteração relativamente ao controlo com solvente, com correção para ter em conta a citotoxicidade; |
|
— |
Gráfico de barras ilustrativo das alterações relativas (fatores multiplicativos) para cada concentração, da SD e da significância estatística, como referido nos pontos 49-54; |
Interpretação dos dados
|
— |
Aplicação do procedimento de interpretação dos dados aos resultados e discussão das constatações; |
Discussão
|
— |
Existem algumas indicações, decorrentes do estudo, quanto à possibilidade de os dados relativos à T/E2 terem sido influenciados por efeitos indiretos nas vias dos glucocorticoides e mineralocorticoides? |
Conclusões
REFERÊNCIAS
|
1. |
OCDE (2002), OECD Conceptual Framework for the Testing and Assessment of Endocrine Disrupting Chemicals (apêndice 2 do capítulo B.54 do presente anexo) |
|
2. |
Hecker, M., Newsted, J.L., Murphy, M.B., Higley, E.B., Jones, P.D., Wu, R. and Giesy, J.P. (2006), Human adrenocarcinoma (H295R) cells for rapid in vitro determination of effects on steroidogenesis: Hormone production, Toxicol. Appl. Pharmacol., 217, 114-124. |
|
3. |
Hecker, M., Hollert, H., Cooper, R., Vinggaard, A.-M., Akahori, Y., Murphy, M., Nellemann, C., Higley, E., Newsted, J., Wu, R., Lam, P., Laskey, J., Buckalew, A., Grund, S., Nakai, M., Timm, G., and Giesy, J. P. (2007), The OECD validation program of the H295R steroidgenesis assay for the identification of in vitro inhibitors or inducers of testosterone and estradiol production, Phase 2: inter laboratory pre-validation studies. Env. Sci. Pollut. Res., 14, 23 — 30. |
|
4. |
OECD (2010), Multi-Laboratory Validation of the H295R Steroidogenesis Assay to Identify Modulators of Testosterone and Estradiol Production, OECD Series of Testing and Assessment No. 132, ENV/JM/MONO(2010)31, Paris. Disponível em [http://www.oecd.org/document/30/0,3746,en_2649_34377_1916638_1_1_1_1,00.html] |
|
5. |
OECD (2010), Peer Review Report of the H295R Cell-Based Assay for Steroidogenesis, OECD Series of Testing and Assessment No. 133, ENV/JM/MONO(2010)32, Paris. Disponível em: [http://www.oecd.org/document/30/0,3746,en_2649_34377_1916638_1_1_1_1,00.html] |
|
6. |
Battelle (2005), Detailed Review Paper on Steroidogenesis. Disponível em: [http://www.epa.gov/endo/pubs/edmvs/steroidogenesis_drp_final_3_29_05.pdf] |
|
7. |
Hilscherova, K., Jones, P. D., Gracia, T., Newsted, J. L., Zhang, X., Sanderson, J. T., Yu, R. M. K., Wu, R. S. S. and Giesy, J. P. (2004), Assessment of the Effects of Chemicals on the Expression of Ten Steroidogenic Genes in the H295R Cell Line Using Real-Time PCR, Toxicol. Sci., 81, 78-89. |
|
8. |
Sanderson, J. T., Boerma, J., Lansbergen, G. and Van den Berg, M. (2002), Induction and inhibition of aromatase (CYP19) activity by various classes of pesticides in H295R human adrenocortical carcinoma cells, Toxicol. Appl. Pharmacol., 182, 44-54. |
|
9. |
Breen, M.S., Breen, M., Terasaki, N., Yamazaki, M. and Conolly, R.B. (2010), Computational model of steroidogenesis in human H295R cells to predict biochemical response to endocrine-active chemicals: Model development for metyrapone, Environ. Health Perspect., 118: 265-272. |
|
10. |
Higley, E.B., Newsted, J.L., Zhang, X., Giesy, J.P. and Hecker, M. (2010), Assessment of chemical effects on aromatase activity using the H295R cell line, Environ. Sci. Poll. Res., 17:1137-1148. |
|
11. |
Gazdar, A. F., Oie, H. K., Shackleton, C. H., Chen, T. R., Triche, T. J., Myers, C. E., Chrousos, G. P., Brennan, M. F., Stein, C. A. and La Rocca, R. V. (1990), Establishment and characterization of a human adrenocortical carcinoma cell line that expresses Multiple pathways of steroid biosynthesis, Cancer Res., 50, 5488-5496. |
|
12. |
He, Y.H., Wiseman, S.B., Zhang, X.W., Hecker, M., Jones, P.D., El-Din, M.G., Martin, J.W. and Giesy, J.P. (2010), Ozonation attenuates the steroidogenic disruptive effects of sediment free oil sands process water in the H295R cell line, Chemosphere, 80:578-584. |
|
13. |
Zhang, X.W., Yu, R.M.K., Jones, P.D., Lam, G.K.W., Newsted, J.L., Gracia, T., Hecker, M., Hilscherova, K., Sanderson, J.T., Wu, R.S.S. and Giesy, J.P. (2005), Quantitative RT-PCR methods for evaluating toxicant-induced effects on steroidogenesis using the H295R cell line, Environ. Sci. Technol., 39:2777-2785. |
|
14. |
Higley, E.B., Newsted, J.L., Zhang, X., Giesy, J.P. and Hecker, M. (2010), Differential assessment of chemical effects on aromatase activity, and E2 and T production using the H295R cell line, Environ. Sci. Pol. Res., 17:1137-1148. |
|
15. |
Rainey, W. E., Bird, I. M., Sawetawan, C., Hanley, N. A., Mccarthy, J. L., Mcgee, E. A., Wester, R. and Mason, J. I. (1993), Regulation of human adrenal carcinoma cell (NCI-H295) production of C19 steroids, J. Clin. Endocrinol. Metab., 77, 731-737. |
|
16. |
Chapter B.55 of this Annex: Hershberger Bioassay in Rats: A short-term Screening Assay for (Anti)Androgenic Properties. |
|
17. |
Shapiro, R., and Page, L.B. (1976), Interference by 2,3-dimercapto-1-propanol (BAL) in angiotensin I radioimmunoassay, J. Lab. Clin. Med., 2, 222-231. |
|
18. |
Mosmann, T. (1983), Rapid colorimetric assay for growth and survival: application to proliferation and cytotoxicity assays, J. Immunol. Methods., 65, 55-63. |
|
19. |
Brock, B.J., Waterman, M.R. (1999). Biochemical differences between rat and human cytochrome P450c17 support the different steroidogenic needs of these two species, Biochemistry. 38:1598-1606. |
|
20. |
Oskarsson, A., Ulleras, E., Plant, K., Hinson, J. Goldfarb, P.S., (2006), Steroidogenic gene expression in H295R cells and the human adrenal gland: adrenotoxic effects of lindane in vitro, J. Appl. Toxicol., 26:484-492. |
Apêndice
DEFINIÇÕES
|
|
Confluência refere-se à cobertura ou proliferação das células sobre ou no meio de cultura. |
|
|
Produto químico designa uma substância ou mistura. |
|
|
CV designa o coeficiente de variação, definido como a razão entre o desvio-padrão de uma distribuição e a sua média aritmética. |
|
|
CYP designa as mono-oxigenases do citocromo P450, enzimas produzidas por uma família de genes, que catalisam uma vasta gama de reações bioquímicas, nomeadamente a síntese e o metabolismo das hormonas esteroides. |
|
|
DPM significa desintegrações por minuto. Consiste no número de átomos numa determinada quantidade de matéria radioativa que sofre decaimento num minuto. |
|
|
E2 designa o 17β-estradiol, estrogénio mais importante nos mamíferos. |
|
|
As células H295R são células de adenocarcinoma humano, que têm características fisiológicas de células adrenais do feto humano sem diferenciação por zonas que exprimem todas as enzimas implicadas na esteroidogénese. Podem ser obtidas junto do ATCC. |
|
|
O meio de congelamento é utilizado para congelar as células e armazenar as células congeladas. É constituído por meio de partida ao qual se adiciona NuSerum BD e dimetilssulfóxido. |
|
|
Troço linear é o troço da curva-padrão do sistema de determinação de hormonas em que os resultados são proporcionais à concentração de substância a analisar presente na amostra. |
|
|
LOQ designa o “limite de quantificação”, que consiste na quantidade mais baixa de um produto químico distinguível da ausência desse produto (ensaio em branco), com um dado intervalo de confiança. Para os fins do presente método, e salvo especificação em contrário, o LOQ é, em geral, definido pelo fabricante do sistema de ensaio. |
|
|
LOEC designa a menor concentração com efeito observável, concentração mais baixa à qual a resposta no ensaio é estatisticamente diferente da do controlo com solvente. |
|
|
NOEC designa a concentração sem efeitos observáveis, concentração mais elevada utilizada no ensaio sem que se observe uma resposta positiva. |
|
|
Passagem designa o número de vezes que as células são divididas após o início de uma cultura a partir de material congelado. À passagem inicial a partir do material congelado é atribuído o número um (1). As células resultantes da primeira divisão correspondem à passagem n.o 2, etc. |
|
|
PBS designa a solução-tampão salina de fosfatos de Dulbecco. |
|
|
Controlo de qualidade (CQ) designa as determinações necessárias para assegurar a validade dos dados. |
|
|
Placa de controlo de qualidade é uma placa de 24 alvéolos com duas concentrações dos controlos positivo e negativo, destinada a monitorizar o desempenho de um novo lote de células ou a fornecer os controlos positivos para o ensaio dos produtos químicos. |
|
|
Série é um ensaio independente caracterizado por um novo conjunto de soluções e controlos. |
|
|
Meio de partida é a base para a preparação dos restantes reagentes. Consiste numa mistura na proporção de 1:1 de meio de Eagle modificado de Dulbecco e mistura de nutrientes F-12 de Ham (DMEM/F12) em tampão HEPES 15 mM, sem adição de vermelho de fenol nem bicarbonato de sódio. Este último é adicionado como tampão — ver o apêndice II do relatório de validação (4). |
|
|
O meio suplementado é constituído por meio de partida ao qual é adicionado Nu-Serum BD e ITS+ premium mix — ver o apêndice II do relatório de validação (4). |
|
|
Esteroidogénese é a via metabólica através da qual o colesterol é transformado nas várias hormonas esteroides. Alguns intermediários no processo, como a progesterona e a testosterona, são hormonas importantes por si só, mas constituem também precursores de outras hormonas, a jusante no processo metabólico. |
|
|
T designaa testosterona, um dos dois androgénios mais importantes nos mamíferos. |
|
|
Produto químico em estudo designa qualquer substância ou mistura ensaiada por recurso ao presente método de ensaio. |
|
|
Placa de ensaio é a placa na qual as células H295R são expostas aos produtos químicos em estudo. As placas de ensaio contêm o solvente de controlo e sete concentrações do produto químico em estudo, em triplicado. |
|
|
Tripsina 1X é a uma solução diluída da enzima tripsina, protease do suco pancreático, utilizada para remover as células de uma placa de cultura — ver o apêndice III do relatório de validação (4). |
B.58. ENSAIOS DE MUTAÇÕES GENÉTICAS DAS CÉLULAS GERMINATIVAS E SOMÁTICAS DE ROEDORES TRANSGÉNICOS
INTRODUÇÃO
1. Este método de ensaio é equivalente ao ao Test Guideline TG 488 (2013) da OCDE. Existem métodos de ensaio da UE para uma ampla variedade de ensaios de mutações in vitro, capazes de detetar mutações ao nível dos cromossomas e/ou dos genes. Existem métodos de ensaio para parâmetros in vivo (ou seja, aberrações cromossomáticas e síntese não programada de ADN); no entanto, estes métodos não medem as mutações dos genes. Os ensaios de mutações em Roedores Transgénicos (TGR) preenchem a necessidade de ensaios in vivo práticos e amplamente disponíveis para mutações dos genes.
2. Os ensaios de mutações em TGR foram analisados de forma aprofundada (24) (33). Usam ratos e ratinhos transgénicos que contêm várias cópias de vetores de transporte de plasmídeos ou fagos integrados nos cromossomas. Os transgenes contêm genes repórter para a deteção de vários tipos de mutações induzidas in vivo por produtos químicos.
3. As mutações que surjam num roedor são classificadas através da recuperação do transgene e da análise do fenótipo do gene repórter num hospedeiro bacteriano com o gene repórter ausente. Os ensaios de mutações genéticas em TGR medem as mutações induzidas em genes neutros recuperados de praticamente qualquer tecido do roedor. Portanto, estes ensaios contornam muitas das limitações atualmente associadas ao estudo das mutações genéticas in vivo em genes endógenos (p. ex.: número limitado de tecidos adequados à análise, seleção negativa/positiva contra mutações).
4. A experiência adquirida sugere que os transgenes respondem aos agentes mutagénicos de modo semelhante aos genes endógenos, especialmente no que respeita à deteção de substituições de pares de bases, de mutações por deslocamento e de pequenas supressões e inserções(24).
5. Os International Workshops on Genotoxicity Testing (IWGT) subscreveram a inclusão dos ensaios de mutações genéticas em TGR para a deteção in vivo de mutações genéticas e recomendaram um protocolo para a sua aplicação (15) (29). O presente método de ensaio baseia-se nessas recomendações. A referência (16) inclui análises adicionais que fundamentam a utilização deste protocolo.
6. Espera-se que, no futuro, venha a ser possível combinar um ensaio de mutações genéticas em TGR com um estudo de toxicidade de dose repetida (Capítulo B.7 do presente Anexo). No entanto, são necessários dados para garantir que a sensibilidade dos ensaios de mutações genéticas em TGR não seja afetada pelo período de tempo inferior usado nos estudos de toxicologia de dose repetida: um dia entre o final do período de administração e o momento da amostragem, em comparação com os 3 dias usados nos ensaios de mutações genéticas em TGR. São também necessários mais dados para verificar se o resultado dos ensaios de dose repetida não é negativamente afetado pela utilização de uma estirpe de roedores transgénicos em vez das estirpes de roedores tradicionais. Quando esses dados estiverem disponíveis, o presente método de ensaio será atualizado.
7. Os termos-chave encontram-se definidos no Apêndice.
CONSIDERAÇÕES INICIAIS
8. Os ensaios de mutações genéticas em TGR relativamente aos quais existem dados suficientes para apoiar o seu uso neste método de ensaio são: bacteriófago lacZ do ratinho (Muta™Mouse); plasmídeo lacZ do ratinho; delta gpt (gpt e Spi–) do ratinho e do rato; lacI do ratinho e do rato (Big Blue®), quando conduzidos em condições padrão. Além disso, o ensaio de seleção positiva cII poderá ser usado para avaliar mutações nos modelosBig Blue® e Muta™Mouse. Normalmente, a mutagénese é avaliada nos modelos TGR em termos de frequência das mutações; se necessário, no entanto, a análise molecular das mutações pode fornecer informação adicional (ver parágrafo 24).
9. Estes ensaios in vivo de mutações genéticas em roedores são especialmente relevantes para a avaliação de riscos mutagénicos, uma vez que as respostas do ensaio dependem do metabolismo in vivo, da farmacocinética, dos processos de reparação do ADN e da síntese de ADN translesão, embora estes fatores possam variar conforme as espécies, tecidos e tipos de lesão do ADN. Um ensaio in vivo de mutações genéticas poderá ser útil para uma investigação mais aprofundada de um efeito mutagénico detetado num sistema in vitro e em complemento dos resultados de ensaios que usem outros parâmetros in vivo (24). Para além de ter relação causal com a indução do cancro, a mutação genética é um parâmetro relevante para a previsão de doenças não-oncológicas com base em mutações nalguns tecidos somáticos (12) (13), bem como de doenças transmitidas através da linha germinativa.
10. Se houver evidência de que o produto químico em estudo, ou um metabolito relevante, não atingirá nenhum dos tecidos de interesse, não deverá ser realizado um ensaio de mutações genéticas em TGR.
PRINCÍPIO DO ENSAIO
11. Nos ensaios descritos no parágrafo 8, o gene-alvo é de origem bacteriana ou bacteriofágica e a forma como é recuperado do ADN genómico do roedor é através da incorporação do transgene num vetor de transporte λ bacteriófágico ou plasmídeo. Este procedimento envolve a extração do ADN genómico do tecido de interesse do roedor, o processamento in vitro desse ADN genómico (ou seja, empacotamento de vetores λ, ou ligação e eletroporação de plasmídeos para recuperar o vetor de transporte) e a deteção subsequente de mutações em hospedeiros bacterianos, em condições adequadas. Os ensaios empregam transgenes neutros, facilmente obtíveis da maioria dos tecidos.
12. As experiências básicas de ensaio de mutações genéticas em TGR envolvem o tratamento do animal com um produto químico durante um determinado período. Os produtos químicos podem ser administrados por qualquer via adequada, incluindo a implantação (p. ex.: ensaio de dispositivos médicos). O período total durante o qual um animal receberá doses do produto é designado por período de administração. A administração é geralmente seguida de um período, antes do sacrifício, durante o qual o produto químico não é administrado e as lesões do ADN não reparadas se fixam em mutações estáveis. Na literatura, este período tem sido designado por tempo de manifestação, tempo de fixação ou tempo de expressão; o final deste período é o momento de amostragem (15) (29). Depois de o animal ser sacrificado, o ADN genómico é isolado do(s) tecido(s) de interesse e purificado.
13. Os dados respeitantes a um único tecido por animal e a vários empacotamentos/ligações são geralmente agregados, e a frequência de mutação é geralmente avaliada usando um total de entre 105 e 107 unidades formadoras de placas ou formadoras de colónias. Quando se usam métodos de seleção positiva, as unidades formadoras de placas totais são determinadas com um conjunto separado de placas não seletivas.
14. Os métodos de seleção positiva foram desenvolvidos para facilitar a deteção de mutações tanto no gene gpt [gpt delta em ratinhos e ratos, fenótipo gpt – (20) (22) (28)] como no gene lacZ [Muta™Mouse ou plasmídeo lacZ no ratinho (3) (10) (11) (30)]; por seu lado, as mutações no gene lacI em animais Big Blue® são detetadas através de um método não seletivo que identifica mutações através da geração de placas coloridas (azuis). A metodologia de seleção positiva é também usada para detetar mutações pontuais que surjam no gene cII do vetor de transporte λ bacteriofágico [Big Blue® no ratinho ou rato e Muta™Mouse (17)] e mutações de supressão nos genes λ red e gam [seleção Spi – no delta gpt no ratinho e no rato (21) (22) (28)]. A frequência de mutação é calculada dividindo o número de placas/plasmídeos que apresentam mutações no transgene pelo número total de placas/plasmídeos recuperados da mesma amostra de ADN. Nos estudos de mutações genéticas em TGR, a frequência de mutação é o parâmetro comunicado. Além disso, pode determinar-se uma frequência de mutação como a proporção de células que apresentam mutações independentes; este cálculo exige correção para expansão clonal através da sequenciação dos mutantes recuperados(24).
15. As mutações classificadas nos ensaios de mutações pontuais lacI, lacZ, cII e gpt consistem principalmente em mutações por substituição de um par de bases, mutações por deslocamento e pequenas inserções/supressões. A proporção relativa destes tipos de mutações entre as mutações espontâneas é semelhante à verificada no gene Hprt endógeno. As grandes supressões só são detetadas nos ensaios de seleção Spi – e ensaios com plasmídeos lacZ (24). As mutações de interesse são mutações in vivo que surgem no rato ou ratinho. As mutações in vitro e ex vivo, que podem surgir durante a recuperação, replicação ou reparação dos fagos/plasmídeos, são relativamente raras e, nalguns sistemas, podem ser especificamente identificadas ou excluídas pelo sistema de hospedeiro bacteriano/seleção positiva.
DESCRIÇÃO DO MÉTODO
Preparações
Seleção da espécie animal
16. Atualmente, estão disponíveis vários modelos de deteção de mutações génicas em ratinhos transgénicos, mais amplamente usados do que os modelos com ratos transgénicos. Se o rato for claramente um modelo mais adequado do que o ratinho (p. ex.: na investigação do mecanismo de carcinogénese de um tumor apenas observado em ratos, para correlacionar com um estudo de toxicidade em ratos ou se o metabolismo do rato for reconhecidamente mais representativo do metabolismo humano), deve considerar-se a utilização de modelos com ratos transgénicos.
Condições de alojamento e alimentação
17. A temperatura na sala onde são conservados os animais para experiências deve ser, idealmente, de 22 °C (± 3 °C). Embora a humidade relativa deva ser pelo menos de 30 % e, de preferência, não deva exceder os 70 %, exceto durante a limpeza da sala, o objetivo deve ser a manutenção de uma humidade relativa entre os 50 % e os 60 %. A iluminação deve ser artificial, com um ciclo diário de 12 horas de luz seguidas de 12 horas de escuridão. Para a alimentação, podem ser usadas dietas convencionais de laboratório, com acesso ilimitado a água potável. A escolha da dieta pode ser influenciada pela necessidade de garantir uma mistura adequada de um produto químico em estudo, quando administrado por esta via. Os animais devem ser mantidos em pequenos grupos (não mais do que cinco) do mesmo sexo, se não for previsivel um comportamento agressivo. Os animais podem ser alojados individualmente se tal se justificar do ponto de vista científico.
Preparação dos animais
18. Os animais jovens e saudáveis, adultos e sexualmente maturos (8 a 12 semanas de idade no início do tratamento) são aleatoriamente afetados aos grupos de controlo e de tratamento. Os animais são identificados por um código único. Devem ser aclimatizados às condições do laboratório durante pelo menos cinco dias. As gaiolas devem estar dispostas de forma a minimizar os possíveis efeitos devidos à localização. No início do estudo, a diferença de peso entre os animais deve ser mínima e não deve exceder ± 20 % do peso médio de cada sexo.
Preparação das doses
19. Os produtos químicos em estudo na forma sólida devem ser dissolvidos ou suspensos em solventes ou veículos adequados, ou misturados na dieta ou na água potável antes da administração aos animais. Os produtos químicos em estudo na forma líquida podem ser administrados diretamente ou diluídos antes da administração. Para exposições por inalação, os produtos químicos em estudo podem ser administrados sob a forma de gás, vapor ou aerossol sólido/líquido, dependendo das respetivas propriedades físico-químicas. Devem ser utilizadas preparações frescas do produto químico em estudo, a menos que os dados de estabilidade demonstrem que o mesmo pode ser armazenado.
Condições de Ensaio
Solvente/veículo
20. O solvente/veículo não deve produzir efeitos tóxicos nos volumes utilizados e não devem existir indícios de reação química com o produto químico em estudo. Se forem usados solventes/veículos pouco conhecidos, a sua utilização deverá ser fundamentada por dados de referência que indiquem a sua compatibilidade. Recomenda-se que, sempre que possível, se considere em primeiro lugar a utilização de um solvente/veículo aquoso.
Controlos Positivos
21. Normalmente, devem ser usados animais de controlo positivo em simultâneo. No entanto, para laboratórios que tenham demonstrado competência (ver parágrafo 23) e que usem rotineiramente estes ensaios, o ADN de animais anteriormente utilizados como controlo positivo pode ser incluído em cada estudo para confirmar o sucesso do método. Esse ADN de experiências anteriores deve ser obtido a partir da mesma espécie e dos mesmos tecidos de interesse e armazenado adequadamente (ver parágrafo 36). Quando são usados controlos positivos em simultâneo, não é necessário administrá-los pela mesma via usada para o produto químico em estudo; no entanto, os controlos positivos devem induzir comprovadamente mutações em um ou mais tecidos de interesse para o produto químico em estudo. As doses dos produtos químicos do controlo positivo devem ser selecionadas de modo a produzir efeitos fracos ou moderados que permitam avaliar criticamente o funcionamento e a sensibilidade do ensaio. O Quadro 1 inclui exemplos de produtos químicos de controlo positivo e de alguns dos respetivos tecidos-alvo.
Quadro 1
Exemplos de produtos químicos de controlo positivo e de alguns dos respetivos tecidos-alvo
|
Produto Químico de Controlo Positivo e N.o CAS |
Nome e N.o EINECS |
Características |
Tecido-alvo de Mutação |
|
|
Rato |
Ratinho |
|||
|
N-Etilo-N-nitrosureia [N.o CAS 759-73-9] |
N-Etilo-N-nitrosureia [212-072-2] |
Agente mutagénico de ação direta |
Fígado, pulmão |
Medula óssea, cólon, epitélio do cólon, intestino, fígado, pulmão, baço, rim, células granulosas do ovário, células germinativas masculinas |
|
Carbamato de etilo (uretano) [N.o CAS 51-79-6] |
Uretano [200-123-1] |
Agente mutagénico, exige metabolismo mas produz apenas efeitos fracos |
|
Medula óssea, pré-estômago, intestino delgado, fígado, pulmão, baço |
|
2,4-Diaminotolueno [N.o CAS 95-80-7] |
4-Metil-m-fenilenodiamina [202-453-1] |
Agente mutagénico, exige metabolismo, também positivo no ensaio Spi– |
Fígado |
Fígado |
|
Benzo[a]pireno [N.o CAS 50-32-8] |
Benzo[def]criseno [200-028-5] |
Agente mutagénico, exige metabolismo |
Fígado, omento |
Medula óssea, mama, cólon, pré-estômago, estômago glandular, coração, fígado, pulmão, células germinativas masculinas |
Controlos negativos
22. Cada amostragem deve incluir controlos negativos, tratados apenas com solvente ou veículo e em todos os outros aspetos tratados da mesma forma que os grupos de tratamento. Na ausência de dados de controlo históricos ou publicados que demonstrem que o solvente/veículo escolhido não induz nenhum efeito prejudicial ou mutagénico, cada amostragem deverá também incluir controlos sem tratamento, de modo a estabelecer a aceitabilidade do controlo do veículo.
Verificação da proficiência do laboratório
23. A competência nestes ensaios deve ser estabelecida através da demonstração da capacidade de reproduzir os resultados esperados a partir de dados publicados (24) para: 1) frequências de mutação com produtos químicos de controlo positivo (incluindo respostas fracas) como os listados no quadro 1, não-mutagénicos e controlos do veículo; e 2) recuperação de transgenes do ADN genómico (p. ex.: eficiência de empacotamento).
Sequenciação de mutações
24. Para aplicações normativas, não é exigida a sequenciação do ADN dos mutantes, especialmente quando for obtido um resultado positivo ou negativo claro. No entanto, os dados de sequenciação podem ser úteis quando se observa uma elevada variação interindividual. Nestes casos, a sequenciação pode ser usada para excluir a possibilidade de jackpots ou eventos de clonagem, ao identificar a proporção de mutantes únicos de um tecido em particular. A sequenciação de cerca de 10 mutantes por tecido e por animal deverá ser suficiente para determinar simplesmente se a clonagem contribuiu para a frequência de mutação; poderá ser necessário sequenciar até 25 mutantes para corrigir matematicamente a frequência de mutação para os efeitos da clonagem clonagem. A sequenciação dos mutantes pode também ser considerada quando se constatarem pequenos aumentos (ou seja, pouco acima dos valores do controlo não tratado) nas frequências de mutação. As diferenças no espectro de mutantes entre as colónias mutantes de animais tratados e não tratados podem apoiar as suspeitas de um efeito mutagénico (29). Além disso, os espectros de mutações poderão ser úteis para desenvolver hipóteses mecanísticas. Quando for necessário incluir a sequenciação no protocolo do estudo, deve dedicar-se particular atenção à conceção do mesmo, em particular no que se refere ao número de mutantes sequenciados por amostra, de modo a assegurar uma cobertura suficiente de acordo com o modelo estatístico usado (ver parágrafo 43).
PROCEDIMENTO
Número e Sexo dos Animais
25. O número de animais por grupo deve ser predeterminado de modo a ser suficiente para proporcionar a validade estatística necessária para detetar pelo menos uma duplicação na frequência de mutação. Os grupos terão pelo menos cinco animais; no entanto, se a validade estatística não for suficiente, o número de animais deve ser aumentado conforme necessário. Normalmente, devem ser usados machos. Pode haver casos em que se justifique testar apenas fêmeas, por exemplo no ensaio de medicamentos específicos para mulheres ou na investigação do metabolismo específico do sexo feminino. Se houver diferenças significativas entre os sexos em termos de toxicidade ou de metabolismo, será necessário utilizar machos e fêmeas.
Período de Administração
26. Com base em observações que demonstram a acumulação de mutações com cada tratamento, é necessário aplicar um regime de dose repetida, com tratamentos diários por um período de 28 dias. Este período é geralmente considerado aceitável, tanto para produzir uma acumulação suficiente de mutações por agentes mutagénicos fracos como para oferecer um tempo de exposição adequado para deteção de mutações em órgãos de proliferação lenta. Para algumas avaliações, podem ser adequados regimes de tratamento alternativos, cujos esquemas de doseamento alternativos devem ser cientificamente justificados no protocolo. Os tratamentos não devem ser mais curtos do que o tempo necessário para completar a indução de todas as enzimas metabólicas relevantes e os tratamentos mais curtos poderão exigir a utilização de vários pontos de amostragem adequados para órgãos com diferentes taxas de proliferação. De qualquer modo, toda a informação disponível (p. ex.: sobre toxicidade geral ou sobre o metabolismo e a farmacocinética) deve ser usada na justificação de um protocolo, especialmente quando este se desviar das recomendações padrão supracitadas. Embora possam aumentar a sensibilidade, os tratamentos mais longos do que 8 semanas devem ser claramente explicados e justificados, uma vez que tempos de tratamento prolongados podem produzir um aumento aparente na frequência de mutação através da expansão clonal (29).
Níveis de Dosagem
27. Os níveis de dosagem devem ser baseados nos resultados de um estudo de determinação de intervalos de dosagem que meça a toxicidade geral e que tenha sido conduzido pela mesma via de exposição, ou nos resultados de estudos de toxicidade subaguda pré-existentes. Podem usar-se animais não transgénicos da mesma estirpe de roedores para a determinação dos intervalos de dosagem. No ensaio principal, e a fim de obter informação sobre a resposta à dosagem, um estudo completo deve incluir um grupo de controlo negativo (ver parágrafo 22) e um mínimo de três níveis de dose, adequadamente espaçados, exceto quando for utilizada uma dose limite (ver parágrafo 28). A dose superior deve ser a Dose Máxima Tolerada (DMT). A DMT é definida como a dose que produz sinais de toxicidade tais que níveis de dose superiores, baseados no mesmo regime de doseamento, seriam letais. Os produtos químicos com atividades biológicas específicas em doses baixas não tóxicas (como hormonas e agentes mitogénicos) e produtos químicos que apresentem saturação de propriedades toxicocinéticas podem ser exceções aos critérios de definição da dosagem e devem ser avaliados caso a caso. Os níveis de dose usados devem abarcar um intervalo desde a toxicidade máxima até uma toxicidade reduzida ou ausente.
Ensaio de Limite
28. Se as experiências de determinação de intervalos de dosagem, ou dados existentes de estirpes relacionadas de roedores, indicarem que um regime de tratamento com, pelo menos, a dose limite (ver abaixo) não produz efeitos tóxicos observáveis,e se não for de esperar genotoxicidade com base em dados relativos a produtos químicos estruturalmente relacionados, poderá não ser considerado necessário um estudo completo com três níveis de dose. Para um período de administração de 28 dias (ou seja, 28 tratamentos diários), a dose limite é de 1 000 mg/kg de peso corporal/dia. Para períodos de administração de 14 dias ou menos, a dose limite é de 2 000 mg/kg/de peso corporal/dia (os esquemas de doseamento que se afastem dos 28 tratamentos diários devem ser cientificamente justificados no protocolo; ver parágrafo 26).
Administração das Doses
29. O produto químico em estudo é geralmente administrado por gavagem, usando uma sonda gástrica ou uma cânula de intubação adequada. De modo geral, a via antecipada de exposição humana deve ser considerada na conceção de um ensaio. Portanto, outras vias de exposição (tais como água potável, via subcutânea, intravenosa, tópica, por inalação, intratraqueal, na dieta ou por implantação) podem ser aceitáveis, quando puderem ser justificadas. A injeção intraperitonial não é recomendada, uma vez que não é uma via fisiologicamente relevante de exposição humana. O volume máximo de líquido que pode ser administrado por gavagem ou injeção de uma só vez depende do tamanho do animal de ensaio. O volume não deve exceder os 2 ml/100 g de peso corporal. A utilização de volumes superiores deverá ser justificada. Exceto no caso de produtos químicos irritantes ou corrosivos, que normalmente revelam efeitos exacerbados em concentrações superiores, a variabilidade no volume de ensaio deve ser minimizada ajustando a concentração de modo a garantir um volume constante em todos os níveis de dose.
Momento de Amostragem
Células Somáticas
30. O momento da amostragem é uma variável crítica, porque é determinado pelo tempo necessário para que as mutações se fixem. Este período é específico para cada tecido e parece estar relacionado com o tempo de substituição da população de células, sendo que a medula óssea e o intestino são tecidos de resposta rápida e o fígado apresenta uma resposta muito mais lenta. Um compromisso aceitável para a medição de frequências de mutação tanto em tecidos de proliferação rápida como lenta é 28 tratamentos diários consecutivos (conforme indicado no parágrafo 26), com amostragem três dias depois do tratamento final, embora a frequência de mutação máxima possa não se manifestar em tecidos de proliferação lenta nestas condições. Se os tecidos de proliferação lenta assumirem particular importância, poderá ser mais adequada uma amostragem mais tardia, 28 dias após o período de administração de 28 dias (16) (29). Nesses casos, a amostragem mais tardia substituirá a amostragem aos 3 dias e exigirá justificação científica.
Células Germinativas
31. Os ensaios em TGR são adequados para o estudo da indução de mutações genéticas em células germinativas masculinas (7) (8) (27), em que o tempo e a cinética da espermatogénese tenham sido bem definidos (27). O número reduzido de óvulos disponíveis para análise, mesmo depois de superovulação, e o facto de não haver síntese de ADN no ovócito impedem a determinação de mutações em células germinativas femininas usando ensaios transgénicos (31).
32. Os momentos de amostragem para as células germinativas masculinas devem ser selecionados de modo a recolher amostras de todos os tipos de células expostas durante o desenvolvimento das células germinativas e a que o estádio-alvo da amostragem tenha recebido uma exposição suficiente. O tempo necessário para a progressão das células germinativas em desenvolvimento desde a fase das células estaminais espermatogoniais até à fase dos espermatozóides maduros que chegam ao ducto deferente/cauda do epidídimo é de~49 dias no ratinho (36) e de ~70 dias no rato (34) (35). Após uma exposição de 28 dias com um período subsequente de amostragem de três dias, o esperma acumulado, colhido no ducto deferente/cauda do epidídimo (7) (8), representa uma população de células expostas durante aproximadamente a última metade da espermatogénese, que inclui o período mitótico e pós-mitótico, mas não o período espermatogonial ou as células estaminais. Para obter amostras adequadas das células presentes no ducto deferente/cauda do epidídimo que estavam no estádio de células estaminais espermatogoniais durante o período de exposição, é necessário um momento de amostragem adicional, no mínimo às 7 semanas (ratinhos) ou 10 semanas (rato) após o final do tratamento.
33. As células extraídas dos túbulos seminíferos após um regime de 28 + 3 dias contêm uma população mista enriquecida para todos os estádios das células germinativas em desenvolvimento (7) (8). Colher amostras destas células para deteção de mutações genéticas não fornece uma avaliação tão precisa dos estádios nos quais as mutações das células germinais foram induzidas, como a que pode ser obtida através da colheita de amostras de espermatozóides do ducto deferente/cauda do epidídimo (uma vez que as células germinativas colhidas nos túbulos são de diferentes tipos e haverá algumas células somáticas a contaminar esta população celular). No entanto, a colheita de amostras de células dos túbulos seminíferos, para além da recolha de espermatozóides do ducto deferente/cauda do epidídimo após um regime de amostragem de apenas 28 + 3 dias, permitirá alguma cobertura das células expostas ao longo da maior parte das fases de desenvolvimento das células germinativas e poderá ser útil para detetar alguns agentes mutagénicos de células germinativas.
Observações
34. Devem ser feitas observações clínicas gerais pelo menos uma vez por dia, de preferência à(s) mesma(s) hora(s) todos os dias e tendo em consideração o período de pico de efeitos antecipados após a administração da dose. A condição de saúde dos animais deve ser registada. Pelo menos duas vezes por dia, todos os animais devem ser observados quanto a sinais de morbilidade e mortalidade. Todos os animais devem ser pesados pelo menos uma vez por semana e no momento do sacrifício. Pelo menos uma vez por semana, devem ser realizadas medições do consumo de alimentos. Se o produto químico em estudo for administrado através de água potável, o consumo de água deve ser medido em cada mudança de água e pelo menos uma vez por semana. Os animais que exibam indicadores não letais de toxicidade excessiva devem ser eutanasiados antes da conclusão do período de ensaio (23).
Colheita de tecidos
35. A fundamentação lógica para a colheita de tecidos deve ser definida de forma clara. Uma vez que é possível estudar a indução de mutações em praticamente qualquer tecido, a seleção dos tecidos a colher deve basear-se no motivo para a realização do estudo e quaisquer dados de mutagenicidade, carcinogenicidade ou toxicidade existentes para o produto químico que esteja a ser investigado. Nos fatores importantes a considerar devem incluir-se a via de administração [com base na(s) via(s) de exposição humana provável(is)], a distribuição prevista do tecido e o possível mecanismo de ação. Na ausência de qualquer informação de contexto, deverão ser colhidos vários tecidos somáticos que poderão apresentar interesse, respresentativos dos tecidos de proliferação rápida, de proliferação lenta e dos locais de contacto. Além disso, os espermatozóides do ducto deferente/cauda do epidídimo e as células germinativas em desenvolvimento provenientes dos túbulos seminíferos (como descrito nos parágrafos 32 e 33) devem ser colhidos e armazenados para a eventualidade de que seja necessária uma análise futura da mutagenicidade das células germinativas. Deve ser obtido o peso dos órgãos e, para os órgãos de maiores dimensões, a mesma área do órgão deverá ser recolhida em todos os animais.
Armazenamento de tecidos e ADN
36. Os tecidos (ou homogeneizados de tecido) devem ser armazenados a uma temperatura de – 70 °C ou inferior e ser utilizados para isolamento do ADN no prazo de 5 anos. O ADN isolado, armazenado refrigerado a 4 °C em tampão adequado, deverá idealmente ser utilizado para análise das mutações no prazo de 1 ano.
Seleção de tecidos para análise das mutações
37. A escolha de tecidos deve basear-se em considerações como: 1) a via de administração ou o local de primeiro contacto (p. ex.: estômago glandular, se a administração for oral, pulmão, se a administração for por inalação, ou pele, se for utilizada uma aplicação tópica); e 2) os parâmetros farmacocinéticos observados em estudos de toxicidade geral que indiquem disposição, retenção ou acumulação nos tecidos ou órgãos-alvo para a toxicidade. Se forem realizados estudos na sequência de estudos de carcinogenicidade, devem ser considerados os tecidos-alvo dessa carcinogenicidade. A escolha dos tecidos para análise deve maximizar a deteção dos produtos químicos que sejam mutagénicos de ação direta in vitro, rapidamente metabolizados, altamente reativos ou mal absorvidos, ou aqueles para os quais o tecido-alvo é determinado pela via de administração (6).
38. Na ausência de informação contextual e tendo em consideração o local de contacto decorrente da via de administração, o fígado e pelo menos um tecido de divisão rápida (p. ex.: estômago glandular, medula óssea) devem ser avaliados em termos de mutagenicidade. Na maioria dos casos, os requisitos acima podem ser preenchidos a partir de análises de dois tecidos cuidadosamente selecionados, mas nalguns casos será necessário analisar três ou mais tecidos diferentes. Se houver motivos de preocupação específica com os efeitos sobre as células germinativas, incluindo respostas positivas em células somáticas, os tecidos de células germinativas devem ser avaliados para mutações.
Métodos de Medição
39. Existem métodos laboratoriais padrão ou que se encontram publicados para a deteção de mutações nos modelos transgénicos recomendados: bacteriófago e plasmídeo lambda lacZ (30); lacI no ratinho (2) (18); delta gpt no ratinho (22); delta gpt no rato (28); cII (17). As modificações devem ser justificadas e devidamente documentadas. Os dados de vários pacotes podem ser agregados e utilizados para se obter um número adequado de placas ou colónias. No entanto, a necessidade de um grande número de reações de empacotamento para se obter um número adequado de placas pode ser uma indicação de qualidade insuficiente do ADN. Nesses casos, os dados devem ser considerados com precaução, pois podem não ser fiáveis. O número total ideal de placas ou colónias por amostra de ADN é regido pela probabilidade estatística de deteção de números suficientes de mutações numa determinada frequência de mutação espontânea. Geralmente é necessário um mínimo de 125 000 a 300 000 placas se a frequência de mutação espontânea estiver na ordem de 3 × 10–5 (15). Para o ensaio Big Blue® lacI, é importante demonstrar que é possível detetar toda a gama de fenótipos mutantes na côr, através da inclusão de controlos de côr adequados em cada conjunto de placas. Os tecidos e as amostras (ou órgãos) resultantes devem ser processados e analisados utilizando uma conceção em bloco, na qual as amostras provenientes do grupo de controlo veículo/solvente, do grupo de controlo positivo (se for utilizado) ou do ADN de controlo positivo (quando apropriado) e de cada grupo de tratamento sejam processados em conjunto.
DADOS E RELATÓRIOS
Tratamento dos resultados
40. Os dados relativos a cada animal devem ser apresentados num quadro. A unidade experimental é o animal. O relatório deve incluir o número total de unidades formadoras de placas (pfu) ou unidades formadoras de colónias (cfu), o número de mutações e a frequência de mutação para cada tecido de cada animal. Se houver várias reações empacotamento/resgate, deve ser comunicado o número de reações por amostra de ADN. Embora devam ser conservados os dados respeitantes a cada reação individual, só é necessário comunicar os totais de pfu ou cfu. Devem ser comunicados os dados relativos à toxicidade e aos sinais clínicos, em conformidade com o parágrafo 34. Todos os resultados de sequenciamento devem ser apresentados para cada mutação analisada e devem ser apresentados os cálculos da frequência de mutação resultantes para cada animal e tecido.
Avaliação e interpretação estatística dos resultados
41. Existem vários critérios para determinar um resultado positivo, tais como um aumento na frequência de mutação relacionado com a dose ou um claro aumento na frequência de mutação de um único grupo de dosagem comparativamente ao grupo de controlo de solvente/veículo. Devem ser analisados pelo menos três grupos tratados, de modo a proporcionar dados suficientes para uma análise da resposta à dosagem. Embora a relevância biológica dos resultados deva ser a principal consideração, podem ser utilizados métodos estatísticos adequados auxiliares da avaliação dos resultados dos ensaios (4) (14) (15) (25) (26). Os testes estatísticos utilizados devem ter em consideração o animal como unidade experimental.
42. Um produto químico em estudo para o qual os resultados não cumpram os critérios acima em qualquer tecido é considerado não mutagénico neste ensaio. Para a relevância biológica de um resultado negativo, a exposição do tecido deve ser confirmada.
43. Para as análises de sequenciamento de ADN, está disponível uma série de abordagens estatísticas para auxiliar na interpretação dos resultados (1) (5) (9) (19).
44. A consideração sobre se os valores observados estão dentro ou fora da faixa de controlo histórico pode fornecer orientações para a avaliação da significância biológica da resposta (32).
Relatório de ensaio
45. O relatório de ensaio deve incluir a seguinte informação:
Produto químico em estudo:
|
— |
dados de identificação e n.o CAS, se for conhecido; |
|
— |
fonte, número do lote, se disponível; |
|
— |
natureza física e pureza; |
|
— |
propriedades físico-químicas relevantes para a realização do estudo; |
|
— |
estabilidade do produto químico em estudo, se conhecida; |
Solvente/veículo:
|
— |
justificação da escolha do veículo; |
|
— |
solubilidade e estabilidade do produto químico em estudo no solvente/veículo, se conhecidas; |
|
— |
preparação de fórmulas alimentares, na água potável ou inaláveis; |
|
— |
determinações analíticas sobre essas fórmulas (p. ex.: estabilidade, homogeneidade, concentrações nominais); |
Animais de ensaio:
|
— |
espécie/estirpe utilizada e justificação da escolha; |
|
— |
número, idade e sexo dos animais; |
|
— |
proveniência, condições de alojamento, dieta administrada, etc.; |
|
— |
peso de cada animal no início do ensaio, incluindo o intervalo de pesos corporais, a média e o desvio-padrão para cada grupo; |
Condições de ensaio:
|
— |
dados de controlo positivos e negativos (veículo/solvente); |
|
— |
dados do estudo de determinação de intervalos de dosagem; |
|
— |
motivo da escolha da gama de dosagem; |
|
— |
detalhes da preparação do produto químico em estudo; |
|
— |
detalhes relativos à administração do produto químico em estudo; |
|
— |
justificação da via de administração escolhida; |
|
— |
métodos de medição da toxicidade para os animais, incluindo, sempre que disponíveis, análises histopatológicas ou hematológicas e a frequência com que foram realizadas observações dos animais e dos respetivos pesos corporais; |
|
— |
métodos de verificação de que o produto químico em estudo atingiu o tecido-alvo, ou a circulação geral, se os resultados forem negativos; |
|
— |
dose real (mg/kg de peso corporal/dia), calculada a partir da concentração do produto químico em estudo nos alimentos/água potável (ppm) e do consumo, se aplicável; |
|
— |
pormenores relativos à qualidade dos alimentos e da água; |
|
— |
descrição detalhada do tratamento e calendários de amostragem e justificação das escolhas feitas; |
|
— |
método de eutanásia; |
|
— |
procedimentos para o isolamento e preservação de tecidos; |
|
— |
métodos para o isolamento de ADN genómico de roedor, resgate do transgene a partir de ADN genómico e transferência do ADN transgénico para um hospedeiro bacteriano; |
|
— |
números de origem e lote de todas as células, kits e reagentes (quando aplicável); |
|
— |
métodos para a enumeração das mutações; |
|
— |
métodos de análise molecular das mutações e utilização na correção para a clonalidade e/ou para o cálculo das frequências de mutação, se aplicável; |
Resultados:
|
— |
condição do animal antes e durante o período de ensaio, incluindo sinais de toxicidade; |
|
— |
pesos corporal e dos órgãos no momento do sacrifício; |
|
— |
para cada tecido/animal, número de mutações, número de placas ou colónias avaliadas e frequência das mutações; |
|
— |
para cada grupo de tecidos/animais, número de reações de empacotamento por amostra de ADN, número total de mutações, frequência média de mutação, desvio padrão; |
|
— |
relação dose-resposta, quando possível; |
|
— |
para cada tecido/animal, número de mutações independentes e frequência de mutação média, sempre que tenha sido realizada uma análise molecular das mutações; |
|
— |
dados históricos e simultâneos sobre o controlo negativo, com intervalos, médias e desvios-padrão; |
|
— |
dados de controlo positivo simultâneos (ou controlo positivo de ADN não simultâneo); |
|
— |
determinações analíticas, se disponíveis (p. ex.: concentrações de ADN utilizadas nos empacotamentos, dados de sequenciação do ADN); |
|
— |
análise estatística e métodos aplicados; |
Discussão dos resultados
Conclusão
REFERÊNCIAS
|
1. |
Adams, W.T. and T.R. Skopek (1987), “Statistical Test for the Comparison of Samples from Mutational Spectra”, J. Mol. Biol., 194: 391-396. |
|
2. |
Bielas, J.H. (2002), “A more Efficient Big Blue® Protocol Improves Transgene Rescue and Accuracy in an Adduct and Mutation Measurement”, Mutation Res., 518: 107–112. |
|
3. |
Boerrigter, M.E., M.E. Dollé, H.-J. Martus, J.A. Gossen and J. Vijg (1995), “Plasmid-based Transgenic Mouse Model for Studying in vivo Mutations”, Nature, 377(6550): 657–659 |
|
4. |
Carr, G.J. and N.J. Gorelick (1995), “Statistical Design and Analysis of Mutation Studies in Transgenic Mice”, Environ. Mol. Mutagen, 25(3): 246–255. |
|
5. |
Carr, G.J. and N.J. Gorelick (1996), “Mutational Spectra in Transgenic Animal Research: Data Analysis and Study Design Based upon the Mutant or Mutation Frequency”, Environ. Mol. Mutagen, 28: 405–413. |
|
6. |
Dean, S.W., T.M. Brooks, B. Burlinson, J. Mirsalis, B. Myhr, L. Recio and V. Thybaud (1999), “Transgenic Mouse Mutation Assay Systems can Play an important Role in Regulatory Mutagenicity Testing in vivo for the Detection of Site-of-contact Mutagens”, Mutagenesis, 14(1): 141–151. |
|
7. |
Douglas, G.R., J. Jiao, J.D. Gingerich, J.A. Gossen and L.M. Soper(1995), “Temporal and Molecular Characteristics of Mutations Induced by Ethylnitrosourea in Germ Cells Isolated from Seminiferous Tubules and in Spermatozoa of lacZ Transgenic Mice”, Proc. Natl. Acad. Sci. USA, 92: 7485-7489. |
|
8. |
Douglas, G.R., J.D. Gingerich, L.M. Soper and J. Jiao (1997), “Toward an Understanding of the Use of Transgenic Mice for the Detection of Gene Mutations in Germ Cells”, Mutation Res., 388(2-3): 197-212. |
|
9. |
Dunson, D.B. and K.R. Tindall (2000), “Bayesian Analysis of Mutational Spectra”, Genetics, 156: 1411–1418. |
|
10. |
Gossen, J.A., W.J. de Leeuw, C.H. Tan, E.C. Zwarthoff, F. Berends, P.H. Lohman, D.L. Knook and J. Vijg(1989), “Efficient Rescue of Integrated Shuttle Vectors from Transgenic Mice: a Model for Studying Mutations in vivo”, Proc. Natl. Acad. Sci. USA, 86(20): 7971–7975. |
|
11. |
Gossen, J.A. and J. Vijg (1993), “A Selective System for lacZ-Phage using a Galactose-sensitive E. coli Host”, Biotechniques, 14(3): 326, 330. |
|
12. |
Erikson, R.P. (2003), “Somatic Gene Mutation and Human Disease other than Cancer”, Mutation Res., 543: 125-136. |
|
13. |
Erikson, R.P. (2010), “Somatic Gene Mutation and Human Disease other than Cancer: an Update”, Mutation Res., 705: 96-106. |
|
14. |
Fung, K.Y., G.R. Douglas and D. Krewski (1998), “Statistical Analysis of lacZ Mutant Frequency Data from Muta™Mouse Mutagenicity Assays”, Mutagenesis, 13(3): 249–255. |
|
15. |
Heddle, J.A., S. Dean, T. Nohmi, M. Boerrigter, D. Casciano, G.R. Douglas, B.W. Glickman, N.J. Gorelick, J.C. Mirsalis, H.-J Martus, T.R. Skopek, V. Thybaud, K.R.Tindall and N. Yajima (2000), “In vivo Transgenic Mutation Assays”, Environ. Mol. Mutagen., 35: 253-259. |
|
16. |
Heddle, J.A., H.-J. Martus and G.R. Douglas (2003), “Treatment and Sampling Protocols for Transgenic Mutation Assays”, Environ. Mol. Mutagen., 41: 1-6. |
|
17. |
Jakubczak, J.L., G. Merlino, J.E. French, W.J. Muller, B. Paul, S. Adhya and S. Garges (1996), “Analysis of Genetic Instability during Mammary Tumor Progression using a novel Selection-based Assay for in vivo Mutations in a Bacteriophage λ Transgene Target”, Proc. Natl. Acad. Sci. USA, 93(17): 9073–9078. |
|
18. |
Kohler, S.W., G.S. Provost, P.L. Kretz, A. Fieck, J.A. Sorge and J.M. Short (1990), “The Use of Transgenic Mice for Short-term, in vivo Mutagenicity Testing”, Genet. Anal. Tech. Appl., 7(8): 212–218. |
|
19. |
Lewis P.D., B. Manshian, M.N. Routledge, G.B. Scott and P.A. Burns (2008), “Comparison of Induced and Cancer-associated Mutational Spectra using Multivariate Data Analysis”, Carcinogenesis, 29(4): 772-778. |
|
20. |
Nohmi, T., M. Katoh, H. Suzuki, M. Matsui, M. Yamada, M. Watanabe, M. Suzuki, N. Horiya, O. Ueda, T. Shibuya, H. Ikeda and T. Sofuni (1996), “A new Transgenic Mouse Mutagenesis Test System using Spi — and 6-thioguanine Selections”, Environ. Mol. Mutagen., 28(4): 465–470. |
|
21. |
Nohmi, T., M. Suzuki, K. Masumura, M. Yamada, K. Matsui, O. Ueda, H. Suzuki, M. Katoh, H. Ikeda and T. Sofuni (1999), “Spi – Selection: an Efficient Method to Detect γ-ray-induced Deletions in Transgenic Mice”, Environ. Mol. Mutagen., 34(1): 9–15. |
|
22. |
Nohmi, T., T. Suzuki and K.I. Masumura (2000), “Recent Advances in the Protocols of Transgenic Mouse Mutation Assays”, Mutation Res., 455(1–2): 191–215. |
|
23. |
OCDE (2000), Guidance Document on the Recognition, Assessment and Use of Clinical Signs as Humane Endpoints for Experimental Animals Used in Safety Evaluation, Series on Testing and Assessment, N.o 19, ENV/JM/MONO(2000)7, OECD, Paris. |
|
24. |
OCDE (2009), Detailed Review Paper on Transgenic Rodent Mutation Assays, Series on Testing and Assessment, N.o 103, ENV/JM/MONO(2009)7, OECD, Paris. |
|
25. |
Piegorsch, W.W., B.H. Margolin, M.D. Shelby, A. Johnson, J.E. French, R.W. Tennant and K.R. Tindall (1995), “Study Design and Sample Sizes for a lacI Transgenic Mouse Mutation Assay”, Environ. Mol. Mutagen., 25(3): 231–245. |
|
26. |
Piegorsch, W.W., A.C. Lockhart, G.J. Carr, B.H. Margolin, T. Brooks, … G.R. Douglas, U.M. Liegibel, T. Suzuki, V. Thybaud, J.H. van Delft and N.J. Gorelick (1997), “Sources of Variability in Data from a Positive Selection lacZ Transgenic Mouse Mutation Assay: an Interlaboratory Study”, Mutation. Res., 388(2–3): 249–289. |
|
27. |
Singer, T.M., I.B. Lambert, A. Williams, G.R. Douglas and C.L. Yauk (2006), “Detection of Induced Male Germline Mutation: Correlations and Comparisons between Traditional Germline Mutation Assays, Transgenic Rodent Assays and Expanded Simple Tandem Repeat Instability Assays”, Mutation. Res., 598: 164-193. |
|
28. |
Toyoda-Hokaiwado, N., T. Inoue, K. Masumura, H. Hayashi, Y. Kawamura, Y. Kurata, M. Takamune, M. Yamada, H. Sanada, T. Umemura, A. Nishikawa and T. Nohmi (2010), “Integration of in vivo Genotoxicity and Short-term Carcinogenicity Assays using F344 gpt delta Transgenic Rats: in vivo Mutagenicity of 2,4-diaminotoluene and 2,6-diaminotoluene Structural Isomers”, Toxicol. Sci., 114(1): 71-78. |
|
29. |
Thybaud, V., S. Dean, T. Nohmi, J. de Boer, G.R. Douglas, B.W. Glickman, N.J. Gorelick, J.A. Heddle, R.H. Heflich, I. Lambert, H.-J. Martus, J.C. Mirsalis, T. Suzuki and N. Yajima (2003), “In vivo Transgenic Mutation Assays”, Mutation Res., 540: 141-151. |
|
30. |
Vijg, J. and G.R. Douglas (1996), “Bacteriophage λ and Plasmid lacZ Transgenic Mice for studying Mutations in vivo” in: G. Pfeifer (ed.), Technologies for Detection of DNA Damage and Mutations, Part II, Plenum Press, New York, NY, USA, p. 391–410. |
|
31. |
Yauk, C.L., J.D. Gingerich, L. Soper, A. MacMahon, W.G. Foster and G.R. Douglas (2005), “A lacZ Transgenic Mouse Assay for the Detection of Mutations in Follicular Granulosa Cells”, Mutation Res., 578(1-2): 117-123. |
|
32. |
Hayashi, M., K. Dearfield, P. Kasper, D. Lovell, H.-J. Martus, V. Thybaud (2011), “Compilation and Use of Genetic Toxicity Historical Control Data”, Mutation Res., doi:10.1016/j.mrgentox.2010.09.007. |
|
33. |
OCDE (2011), Retrospective Performance Assessment of OECD Test Guideline on Transgenic Rodent Somatic and Germ Cell Gene Mutation Assays, Series on Testing and Assessment, N.o 145, ENV/JM/MONO(2011)20, OECD, Paris. |
|
34. |
Clermont, Y. (1972), “Kinetics of spermatogenesis in mammals seminiferous epithelium cycle and spermatogonial renewal”. Physiol. Rev. 52: 198-236. |
|
35. |
Robaire, B., Hinton, B.T., and Oregbin-Crist, M.-C. (2006), “The Epididymis”, in Neil, J.D., Pfaff, D.W., Chalis, J.R.G., de Kretser, D.M., Richards, J.S., and P. M, Wassarman (eds.), Physiology of Reproduction, Elsevier, the Netherlands, p. 1071-1148. |
|
36. |
Russell, L.B. (2004), “Effects of male germ-cell stage on the frequency, nature, and spectrum of induced specific-locus mutations in the mouse”, Genetica, 122: 25–36. |
Apêndice
DEFINIÇÕES
Período de administração : o período total durante o qual um animal é exposto a uma dose do produto químico.
Substituição de um par de bases : um tipo de mutação que provoca a substituição de uma única base de nucleotídeos do ADN por outra base de nucleotídeos do ADN.
Cápside : o invólucro proteico que envolve uma partícula viral.
Produto químico : uma substância ou uma mistura.
Expansão clonal : a produção de muitas células a partir de uma única célula (mutante).
Unidades formadoras de colónias (cfu) : uma medida do número de bactérias viáveis.
Concatâmero : uma biomolécula contínua e longa constituída por várias cópias idênticas ligadas em série.
Local cos : um segmento de 12 nucleótidos de ADN de cadeia simples que existe em ambas as extremidades do genoma de cadeia dupla do bacteriófago lambda.
Supressão : uma mutação na qual um ou mais nucleótidos (sequenciais) desaparecem do genoma.
Eletroporação : a aplicação de impulsos elétricos para aumentar a permeabilidade das membranas celulares.
Gene endógeno : um gene nativo do genoma.
Variação extra-binomial : maior variabilidade das estimativas repetidas de uma proporção da população do que seria de esperar se essa população tivesse uma distribuição binomial.
Mutação por deslocamento : uma mutação genética causada por inserções ou supressões de um número de nucleótidos que não é divisível uniformemente por três dentro de uma sequência de ADN que codifica para uma proteína/péptido.
Inserção : o acréscimo de um ou mais pares de bases de nucleótidos numa sequência de ADN.
Jackpot : um grande número de mutações que surgiram através da expansão clonal de uma única mutação.
Grandes supressões : supressões no ADN auperiores a vários milhares de bases (detetadas de forma eficaz pelos ensaios de seleção Spi – e do plasmídeo lacZ).
“Colagem” : a ligação covalente de duas extremidades de moléculas de ADN utilizando ligases.
Mitogénio : uma substância química que estimula uma célula a iniciar a divisão celular, desencadeando a mitose (ou seja, a divisão celular).
Gene neutro : um gene que não é afetado pelas pressões seletivas positivas ou negativas.
Empacotamento : a síntese de partículas de fagos infeciosos a partir de uma preparação de proteínas da cápside e da cauda do fago e um concatâmero de moléculas de ADN de fágicas. Geralmente utilizado para empacotar o ADN clonado num vetor lambda (separado por locais cos) em partículas lambda infeciosas.
Eficiência de empacotamento : a eficiência com a qual os bacteriófagos empacotados são recuperados em bactérias hospedeiras.
Unidade formadora de placas (pfu) : uma medida de números de bacteriófagos viáveis.
Mutação pontual : um termo geral para uma mutação que afeta apenas uma pequena sequência de ADN, incluindo pequenas inserções, supressões e substituições de pares de bases.
Seleção positiva : um método que permite que sobrevivam apenas os indivíduos nos quais se verificaram mutações.
Gene repórter : um gene cujo resultado mutante pode ser facilmente detetado.
Período de amostragem : o final do período, antes do sacrifício, durante o qual a substância química não é administrada e as lesões do ADN não processadas são fixadas em mutações estáveis.
Vetor de transporte : um vetor construído de modo a poder propagar-se em duas espécies hospedeiras diferentes; assim, o ADN inserido num vetor de transporte pode ser testado ou manipulado em dois tipos de células diferentes ou dois organismos diferentes.
Produto químico em estudo : qualquer substância ou mistura estudada utilizando este método de ensaio.
Transgénico : de, relativo a, ou organismo cujo genoma foi modificado através da transferência de um gene ou genes de outras espécies.
(1) Consoante a sensibilidade dos testes da função cognitiva, pode ponderar-se utilizar no estudo um número maior de animais, por exemplo, um macho e uma fêmea de cada ninhada (ver a afetação dos animais no apêndice 1). O documento de orientações n.o 43 da OCDE (8) fornece mais indicações sobre a dimensão das amostras.
(2) Este quadro apresenta o número mínimo de vezes que as medições devem ser realizadas. Consoante os efeitos previstos e os resultados das primeiras medições, pode ser aconselhável aumentar o número de ocasiões de avaliação (por exemplo de animais mais velhos) ou efetuar as medições noutros estádios de desenvolvimento.
(3) Recomenda-se que as crias não sejam examinadas nos dois dias após o desmame (ver o ponto 32). As idades de avaliação do estádio da adolescência são as seguintes: aprendizagem e memória = 25 ± 2 dias após o nascimento; funções motoras e sensoriais = 25 ± 2 dias após o nascimento. A idade recomendada para avaliar adultos jovens é de 60 a 70 dias após o nascimento.
(4) Nos períodos de aumento rápido do peso corporal, este deve ser determinado, pelo menos, duas vezes por semana nos casos em que as doses sejam administradas diretamente às crias, para que possam ajustar-se as doses.
(5) Se necessário, o peso cerebral pode ser determinado e as manifestações neuropatológicas podem ser avaliadas mais cedo (por exemplo 11 dias após o nascimento — ver o ponto 39).
(6) Se for caso disso, indicar-se-ão os outros indicadores de desenvolvimento utilizados, além do peso corporal (por exemplo, a abertura dos olhos — ver o ponto 31).
(7) Ver o ponto 35.
(8) Neste exemplo, as ninhadas são reduzidas a 4 machos e 4 fêmeas, por eliminação dos restantes animais; as crias do sexo masculino são numeradas de 1 a 4; as do sexo feminino, de 5 a 8.
(9) Neste exemplo, as ninhadas são reduzidas a 4 machos e 4 fêmeas, por eliminação dos restantes animais; as crias do sexo masculino são numeradas de 1 a 4; as do sexo feminino, de 5 a 8.
(10) As crias utilizadas nos testes cognitivos efetuados 23 dias após o nascimento não são as mesmas utilizadas na realização desses testes em adultos jovens (por exemplo, ninhadas pares num caso e ímpares no outro, do total de 20).
(11) Neste exemplo, as ninhadas são reduzidas a 4 machos e 4 fêmeas, por eliminação dos restantes animais; as crias do sexo masculino são numeradas de 1 a 4; as do sexo feminino, de 5 a 8.
(12) Determinou-se o limite máximo de coeficiente de variação correspondente a cada tecido a partir de um gráfico dos valores dos coeficientes de variação — organizados sequencialmente do menor ao maior — correspondentes a todas as médias de todas as experiências realizadas a um determinado modelo (agonista ou antagonista) durante os estudos de validação. Considerou-se que o coeficiente de variação máximo correspondia ao ponto — dito “de viragem” — a partir do qual os incrementos entre os coeficientes de variação mais elevados imediatos da série passavam a ser claramente maiores do que os existentes entre os coeficientes de variação imediatamente anteriores na série. Importa referir que, embora esta análise tenha permitido identificar pontos de viragem relativamente fiáveis para o modelo “antagonista” do ensaio, as curvas dos coeficientes de variação dos ensaios de agonistas revelaram incrementos mais uniformes, pelo que os coeficientes de variação máximos identificados por este método são algo arbitrários neste último caso.
(13) Os estudos mostram que a glândula mamária, especialmente durante o seu desenvolvimento precoce, constitui um parâmetrosensível para a ação do estrogénio. Recomenda-se que os parâmetros que envolvam glândulas mamárias de crias de ambos os sexos sejam incluídos neste método de ensaio, quando validados.
(14) ATCC CRL-2128; ATCC, Manassas, VA, USA, [http://www.lgcstandards-atcc.org/].
(15) A expressão “novo lote” refere-se a um novo lote de células recebidas do ATCC.
(16) A expressão “lote congelado” referes-se a células anteriormente cultivadas e, de seguida, congeladas num laboratório diverso do ATCC.
Nota: Se a extração for necessária, devem efetuar-se três medições em replicado para cada extrato. Cada amostra deve ser extraída apenas uma vez.
Nota: Os limites de determinação do método derivam dos valores de produção basal de hormonas constantes do quadro 5 e baseiam-se no desempenho. Os limites podem ser superiores se a produção basal de hormonas for superior.
(19) A percentagens superiores, alguns anticorpos da T e da E2 podem reagir de forma cruzada com a androstenodiona e a estrona, respetivamente. Nessas condições, não é possível determinar com exatidão os efeitos na 17β-HSD. Apesar disso, os dados podem proporcionar informações úteis sobre os efeitos na produção de estrogénios ou androgénios em geral. Nesses casos, os dados devem ser expressos em androgénios/estrogénios produzidos, em vez de E2 e T.
(20) Designadamente: colesterol, pregnenolona, progesterona, 11-desoxicorticosterona, corticosterona, aldosterona, 17α-pregnenolona,17α-progesterona, desoxicortisol, cortisol, DHEA, androstenodiona, estrona.
(21) Controlo com solvente (DMSO) (0), 1 μl DMSO/alvéolo.
(22) +, positivo
n/a: não aplicável, dado que não deverão ocorrer alterações após a exposição a concentrações não citotóxicas do controlo negativo.
(23) As células nos alvéolos correspondentes ao branco recebem apenas meio (sem solvente).
(24) O metanol (MeOH) é adicionado após o termo da exposição, sendo o meio removido dos alvéolos.
(25) Controlo com solvente DMSO (1 μl/alvéolo).
(26) Confirmar o ensaio anterior utilizando as mesmas condições experimentais.
(27) Reefetuar o ensaio com intervalos de concentração semilogarítmicos (em torno da concentração que produziu resultados significativamente diferentes no ensaio anterior).
(28) Estatisticamente, a uma dada concentração, o fator multiplicador é significativamente diferente do SC
(29) Refere-se a determinações replicadas da mesma amostra.