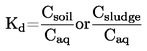ISSN 1725-5139
Dziennik Urzędowy
Unii Europejskiej
L 142

Wydanie polskie
Legislacja
Tom 51
31 maja 2008
|
ISSN 1725-5139 |
||
|
Dziennik Urzędowy Unii Europejskiej |
L 142 |
|
|
|
||
|
Wydanie polskie |
Legislacja |
Tom 51 |
|
Spis treści |
|
I Akty przyjęte na mocy Traktatów WE/Euratom, których publikacja jest obowiązkowa |
Strona |
|
|
|
ROZPORZĄDZENIA |
|
|
|
* |
Rozporządzenie Komisji (WE) nr 440/2008 z dnia 30 maja 2008 r. ustalające metody badań zgodnie z rozporządzeniem (WE) nr 1907/2006 Parlamentu Europejskiego i Rady w sprawie rejestracji, oceny, udzielania zezwoleń i stosowanych ograniczeń w zakresie chemikaliów (REACH) ( 1 ) |
|
|
|
|
|
(1) Tekst mający znaczenie dla EOG |
|
PL |
Akty, których tytuły wydrukowano zwykłą czcionką, odnoszą się do bieżącego zarządzania sprawami rolnictwa i generalnie zachowują ważność przez określony czas. Tytuły wszystkich innych aktów poprzedza gwiazdka, a drukuje się je czcionką pogrubioną. |
I Akty przyjęte na mocy Traktatów WE/Euratom, których publikacja jest obowiązkowa
ROZPORZĄDZENIA
|
31.5.2008 |
PL |
Dziennik Urzędowy Unii Europejskiej |
L 142/1 |
ROZPORZĄDZENIE KOMISJI (WE) NR 440/2008
z dnia 30 maja 2008 r.
ustalające metody badań zgodnie z rozporządzeniem (WE) nr 1907/2006 Parlamentu Europejskiego i Rady w sprawie rejestracji, oceny, udzielania zezwoleń i stosowanych ograniczeń w zakresie chemikaliów (REACH)
(Tekst mający znaczenie dla EOG)
KOMISJA WSPÓLNOT EUROPEJSKICH,
uwzględniając Traktat ustanawiający Wspólnotę Europejską,
uwzględniając rozporządzenie (WE) nr 1907/2006 Parlamentu Europejskiego i Rady z dnia 18 grudnia 2006 r. w sprawie rejestracji, oceny, udzielania zezwoleń i stosowanych ograniczeń w zakresie chemikaliów (REACH), utworzenia Europejskiej Agencji Chemikaliów, zmieniające dyrektywę 1999/45/WE oraz uchylające rozporządzenie Rady (EWG) nr 793/93 i rozporządzenie Komisji (WE) nr 1488/94, jak również dyrektywę Rady 76/769/EWG i dyrektywy Komisji 91/155/EWG, 93/67/EWG, 93/105/WE i 2000/21/WE (1), w szczególności jego art. 13 ust. 3,
a także mając na uwadze, co następuje:
|
(1) |
Zgodnie z rozporządzeniem (WE) nr 1907/2006, jeżeli dla wygenerowania informacji o swoistych właściwościach substancji wymagane są badania, przeprowadza się je przy użyciu metod badań określonych na poziomie Wspólnoty. |
|
(2) |
Dyrektywa Rady 67/548/EWG z dnia 27 czerwca 1967 r. w sprawie zbliżenia przepisów ustawodawczych, wykonawczych i administracyjnych odnoszących się do klasyfikacji, pakowania i etykietowania substancji niebezpiecznych (2) ustanawia w załączniku V metody określania właściwości fizyko-chemicznych, toksyczności oraz ekotoksyczności substancji i preparatów. Załącznik V do dyrektywy 67/548/EWG został skreślony dyrektywą 2006/121/WE Parlamentu Europejskiego i Rady ze skutkiem od dnia 1 czerwca 2008 r. |
|
(3) |
Należy włączyć do niniejszego rozporządzenia metody badań zawarte w załączniku V do dyrektywy 67/548/EWG. |
|
(4) |
Niniejsze rozporządzenie nie wyklucza stosowania innych metod badań, pod warunkiem że ich stosowanie jest zgodne z art. 13. ust. 3 rozporządzenia 1907/2006. |
|
(5) |
Przy opracowywaniu metod badań należy w pełni uwzględnić zasady zastępowania badań na zwierzętach innymi metodami, ograniczania skali badań na zwierzętach i doskonalenia metod tego rodzaju badań, zwłaszcza jeśli dostępne są odpowiednie zatwierdzone metody zastępowania, ograniczania i doskonalenia badań na zwierzętach. |
|
(6) |
Przepisy niniejszego rozporządzenia są zgodne z opinią Komitetu powołanego na mocy art. 133 rozporządzenia (WE) nr 1907/2006, |
PRZYJMUJE NINIEJSZE ROZPORZĄDZENIE:
Artykuł 1
Metody badań stosowanych do celów rozporządzenia 1907/2006/WE określa się w załączniku do niniejszego rozporządzenia.
Artykuł 2
W stosownych przypadkach Komisja dokona przeglądu metod badań określonych w niniejszym rozporządzeniu pod kątem możliwości zastępowania, ograniczania i doskonalenia badań na zwierzętach kręgowych.
Artykuł 3
Wszelkie odesłania do załącznika V do dyrektywy 67/548/EWG są traktowane jako odesłania do niniejszego rozporządzenia.
Artykuł 4
Niniejsze rozporządzenie wchodzi w życie następnego dnia po jego opublikowaniu w Dzienniku Urzędowym Unii Europejskiej.
Niniejsze rozporządzenie stosuje się od dnia 1 czerwca 2008 r.
Sporządzono w Brukseli dnia 30 maja 2008 r.
W imieniu Komisji
Stavros DIMAS
Członek Komisji
(1) Dz.U. L 396 z 30.12.2006, s. 1; sprostowanie w Dz.U. L 136 z 29.5.2007, s. 3.
(2) Dz.U. 196 z 16.8.1967, s. 1. Dyrektywa ostatnio zmieniona dyrektywą 2006/121/WE Parlamentu Europejskiego i Rady (Dz.U. L 396 z 30.12.2006, s. 850; sprostowanie w Dz.U. L 136 z 29.5.2007, s. 281).
ZAŁĄCZNIK
CZĘŚĆ A: METODY USTALANIA WŁAŚCIWOŚCI FIZYKO-CHEMICZNYCH
SPIS TREŚCI
|
A.1. |
TEMPERATURA TOPNIENIA/KRZEPNIĘCIA |
|
A.2. |
TEMPERATURA WRZENIA |
|
A.3. |
GĘSTOŚĆ WZGLĘDNA |
|
A.4. |
PRĘŻNOŚĆ PARY |
|
A.5. |
NAPIĘCIE POWIERZCHNIOWE |
|
A.6. |
ROZPUSZCZALNOŚĆ W WODZIE |
|
A.8. |
WSPÓŁCZYNNIK PODZIAŁU |
|
A.9. |
TEMPERATURA ZAPŁONU |
|
A.10. |
ZAPALNOŚĆ (CIAŁA STAŁE) |
|
A.11. |
ZAPALNOŚĆ (GAZY) |
|
A.12. |
ZAPALNOŚĆ (W KONTAKCIE Z WODĄ) |
|
A.13. |
WŁAŚCIWOŚCI PIROFORYCZNE CIAŁ STAŁYCH I CIECZY |
|
A.14. |
WŁAŚCIWOŚCI WYBUCHOWE |
|
A.15. |
TEMPERATURA SAMOZAPŁONU (CIECZE I GAZY) |
|
A.16. |
WZGLĘDNA TEMPERATURA SAMOZAPŁONU DLA CIAŁ STAŁYCH |
|
A.17. |
WŁAŚCIWOŚCI UTLENIAJĄCE (CIAŁA STAŁE) |
|
A.18. |
ŚREDNIA LICZBOWA MASA CZĄSTECZKOWA I ROZKŁAD MASY CZĄSTECZKOWEJ POLIMERÓW |
|
A.19. |
ZAWARTOŚĆ POLIMERÓW O MAŁEJ MASIE CZĄSTECZKOWEJ |
|
A.20. |
ZACHOWANIE POLIMERÓW W WODZIE – ROZPUSZCZANIE/EKSTRAKCJA |
|
A.21. |
WŁAŚCIWOŚCI UTLENIAJĄCE (CIECZE) |
A.1. TEMPERATURA TOPNIENIA/KRZEPNIĘCIA
1. METODA
Większość opisanych metod oparto na wytycznych OECD dotyczących badań (1). Podstawowe zasady podano w pozycjach bibliograficznych (2) i (3).
1.1. WPROWADZENIE
Opisane metody i urządzenia powinny być stosowane do ustalania temperatury topnienia substancji, bez ograniczeń związanych z ich stopniem czystości.
Wybór metody zależy od charakteru badanej substancji. W konsekwencji, czynnikiem ograniczającym będzie to, czy substancja łatwo, z trudnością czy też wcale nie poddaje się sproszkowaniu.
W przypadku niektórych substancji bardziej właściwe jest oznaczanie temperatury zamarzania lub krzepnięcia, przy czym w wytycznych uwzględniono także normy dla tych oznaczeń.
Jeżeli z powodu szczególnych właściwości substancji żaden z powyższych parametrów nie może być zmierzony w sposób konwencjonalny, może być stosowne określenie temperatury płynięcia.
1.2. DEFINICJE I JEDNOSTKI
Temperaturę topnienia definiuje się jako temperaturę, w której przejście fazowe ze stanu stałego do stanu ciekłego zachodzi pod ciśnieniem atmosferycznym i niniejsza temperatura idealnie odpowiada temperaturze krzepnięcia.
Ponieważ przechodzenie z jednego stanu skupienia do drugiego w przypadku wielu substancji zachodzi w szerokim zakresie temperatury, często opisuje się go jako zakres temperatury topnienia.
Jednostki konwersji (ze stopni K na oC)
t = T – 273,15
|
t |
: |
temperatura Celsjusza, stopień Celsjusza (oC) |
|
T |
: |
temperatura termodynamiczna, stopień Kelwina (K) |
1.3. SUBSTANCJE ODNIESIENIA
Nie ma konieczności stosowania substancji odniesienia za każdym razem, gdy przeprowadzane są badania nowej substancji. Powinny one służyć głównie sprawdzeniu od czasu do czasu wydajności metody i pozwolić na porównanie wyników z innymi metodami.
Niektóre substancje kalibracyjne wymieniono w pozycji bibliograficznej (4).
1.4. ZASADA METODY BADANIA
Temperatura (zakres temperatur) przejścia fazowego ze stanu stałego do stanu ciekłego lub ze stanu ciekłego w stan stały jest ustalona. W praktyce podczas podgrzewania/schładzania próbki substancji badanej pod ciśnieniem atmosferycznym ustalane są temperatury etapu początkowego topnienia/krzepnięcia i końcowego etapu topnienia/krzepnięcia. Opisano pięć rodzajów metod, mianowicie metodę kapilarną, metody ze stolikami grzewczymi, ustalanie temperatur krzepnięcia, metody analiz termicznych, i ustalanie temperatury płynięcia (według rozwiniętych opracowań dla olejów naftowych).
W niektórych przypadkach wygodniejsze jest zmierzenie temperatury krzepnięcia w miejsce temperatury topnienia.
1.4.1. Metoda kapilarna
1.4.1.1. Przyrządy do oznaczania temperatury topnienia z łaźnią cieczową
Niewielką ilość drobno zmielonej substancji umieszcza się w rurce kapilarnej i ciasno upakowuje. Rurkę podgrzewa się łącznie z termometrem, regulując wzrost temperatury w ten sposób, aby wynosił mniej niż 1 K/min w trakcie topnienia. Oznacza się początkową i końcową temperaturę topnienia.
1.4.1.2. Przyrządy do oznaczania temperatury topnienia z blokiem metalu
Zgodnie z opisem w ppkt 1.4.1.1, z tym że rurka kapilarna i termometr są umieszczone w podgrzewanym bloku metalu, przy czym można je obserwować przez znajdujące się w nim otwory.
1.4.1.3. Wykrywanie przy pomocy komórki fotoelektrycznej
Próbka w rurce kapilarnej jest automatycznie podgrzewana w cylindrze metalowym. Wiązka światła jest automatycznie kierowana przez substancję, poprzez dziurę w cylindrze, do precyzyjnie skalibrowanej komórki fotoelektrycznej. Właściwości optyczne większości substancji zmieniają się z nieprzezroczystych na przezroczyste podczas topnienia. Natężenie światła docierającego do komórki fotoelektrycznej zwiększa się, w wyniku czego wysyłany jest sygnał zatrzymania do cyfrowego wskaźnika odczytującego temperaturę z platynowego termometru oporowego umieszczonego w komorze grzejnej. Metoda ta nie ma zastosowania do niektórych intensywnie zabarwionych substancji.
1.4.2. Płyty grzewcze
1.4.2.1. Płyta grzewcza Koflera
W metodzie płyty grzewczej Koflera wykorzystuje się dwa kawałki metalu o różnym przewodnictwie cieplnym, podgrzewane elektrycznie, przy czym stolik jest zaprojektowany w taki sposób, aby gradient temperatury był prawie liniowy wzdłuż jego długości. Temperatura rozgrzanej płyty może dochodzić od 283 do 573 K, przy czym odczytywana jest ona przy pomocy specjalnego urządzenia do odczytywania temperatury, składającego się z suwaka ze wskaźnikiem i klapki przeznaczonej dla odpowiedniego stolika. W celu ustalenia temperatury topnienia, substancję umieszcza się cienką warstwą bezpośrednio na powierzchni gorącej płyty. W ciągu kilku sekund pokazuje się ostra linia oddzielająca ciecz od fazy stałej. Temperaturę na wysokości linii dzielącej odczytuje się przez ustawienie wskaźnika w taki sposób, aby spoczywał na tej linii.
1.4.2.2. Mikroskop do badania topnienia
Szereg mikroskopowych płyt grzewczych wykorzystuje się do ustalania temperatury przy użyciu bardzo małych ilości materiału. W większości płyt grzewczych temperaturę mierzy się przy pomocy czułego termoogniwa, jednak czasem wykorzystywane są także termometry rtęciowe. Typowy przyrząd mikroskopowy do oznaczania temperatury topnienia metodą podgrzewania płyty jest wyposażony w komorę grzewczą, która zawiera płytkę metalową, na której umieszczana jest próbka na szkiełku. W środku płytki metalowej znajduje się otwór, przez którą wchodzi światło z lusterka oświetlającego mikroskopu. W trakcie użycia, komora jest zamknięta przez szklaną płytę w celu odcięcia powietrza od obszaru próbki.
Podgrzewanie próbki jest regulowane za pomocą reostatu. Dla bardzo precyzyjnych pomiarów dla substancji optycznie anizotropowych, stosuje się światło spolaryzowane.
1.4.2.3. Metoda menisku
Metodę tę wykorzystuje się głównie w odniesieniu do poliamidów.
W sposób wizualny oznacza się temperaturę, przy której zachodzi przemieszczenie menisku oleju silikonowego, zawartego między płytą grzewczą i szklaną przykrywką opartą na próbce badanego poliamidu.
1.4.3. Metoda oznaczania temperatury krzepnięcia
Próbkę umieszcza się w specjalnej probówce i umieszcza w przyrządzie dla oznaczania temperatury krzepnięcia. Próbka jest delikatnie i ciągle mieszana w trakcie chłodzenia, a temperatura jest mierzona w odpowiednich przedziałach. Gdy tylko temperatura pozostaje na stałym poziomie podczas kilku kolejnych odczytów (skorygowanych o błąd termometru), zapisuje się ją jako temperaturę krzepnięcia.
Należy zapobiegać szybkiemu schładzaniu przez utrzymywanie równowagi między fazą stałą i fazą ciekłą.
1.4.4. Analiza termiczna
1.4.4.1. Różnicowa analiza termiczna (DTA)
Niniejsza technika rejestruje różnicę w temperaturach między daną substancją i materiałem odniesienia w funkcji temperatury, w trakcie poddawania danej substancji i materiału odniesienia temu samemu programowi kontrolowania temperatury. W momencie gdy próbka ulega przemianie pociągającej zmianę entalpii, zmiana ta jest wskazywana przez odchylenie endotermiczne (topnienie) lub egzotermiczne (krzepnięcie) od linii bazowej zapisu temperatury.
1.4.4.2. Różnicowa kalorymetria skaningowa (DSC)
Niniejsza technika rejestruje różnicę w energii pobranej przez daną substancję i materiał odniesienia, w funkcji temperatury, w trakcie poddawania danej substancji i materiału odniesienia temu samemu programowi kontrolowania temperatury. Niniejsza energia jest energią konieczną do osiągnięcia zerowej różnicy temperatury między daną substancją a materiałem odniesienia. W momencie, gdy próbka ulega przemianie pociągającej zmianę entalpii, zmiana ta jest wskazywana przez odchylenie endotermiczne (topnienie) lub egzotermiczne (krzepnięcie) od linii bazowej zapisu przepływu ciepła.
1.4.5. Temperatura płynięcia
Niniejsza metoda została opracowana dla olejów naftowych i jest właściwa do zastosowania dla substancji olejowych o niskich temperaturach topnienia.
Po ogrzaniu wstępnym próbka jest chłodzona z właściwą szybkością i badane są w odstępach co 3 K charakterystyki przepływu. Najniższa temperatura, przy której jest obserwowany ruch substancji, jest odczytywana jako temperatura płynięcia.
1.5. KRYTERIA JAKOŚCI
Stosowalność i dokładność różnych metod wykorzystywanych do oznaczania temperatury topnienia/zakres temperatur topnienia przedstawiono w poniższej tabeli:
TABELA: STOSOWALNOŚĆ METOD
A. Metody kapilarne
|
Metoda pomiaru |
Substancje łatwo proszkowalne |
Substancje trudno proszkowalne |
Zakres temperatury |
Założona dokładność (1) |
Istniejąca norma |
|
Przyrządy do pomiaru temperatury topnienia z łaźnią cieczową |
tak |
Tylko niektóre |
273 do 573 K |
±0,3 K |
JIS K 0064 |
|
Przyrządy do pomiaru temperatury topnienia z blokiem metalu |
tak |
Tylko niektóre |
293 do > 573 K |
±0,5 K |
ISO 1218 (E) |
|
Wykrywanie przy pomocy komórki fotoelektrycznej |
tak |
Poszczególne z zastosowaniem urządzeń |
253 do 573 K |
±0,5 K |
|
B. Metody temperaturowe i schładzania
|
Metoda pomiaru |
Substancje łatwo proszkowalne |
Substancje trudno proszkowalne |
Zakres temperatury |
Założona dokładność (2) |
Istniejąca norma |
|
Płyta grzewcza Koflera |
tak |
nie |
283 do > 573 K |
± 1 K |
ANSI/ASTM D 3451-76 |
|
Mikroskop do badania topnienia |
tak |
Tylko niektóre |
273 do > 573 K |
±0,5 K |
DIN 53736 |
|
Metoda menisku |
nie |
Specyficzna dla poliamidów |
293 do > 573 K |
±0,5 K |
ISO 1218 (E) |
|
Metody badania temperatur krzepnięcia |
tak |
tak |
223 do 573 K |
±0,5 K |
e.g. BS 4695 |
C. Analizy termiczne
|
Metoda pomiaru |
Substancje łatwo proszkowalne |
Substancje trudno proszkowalne |
Zakres temperatury |
Założona dokładnoś (3) |
Istniejąca norma |
|
Różnicowa analiza termiczna |
tak |
tak |
173 do 1 273 K |
do 600 K ±0,5 K do 1 273 K ±2,0 K |
ASTM E 537-76 |
|
Rożnicowa kalorymetria skaningowa |
tak |
tak |
173 do 1 273 K |
do 600 K ±0,5 K do 1 273 K ±2,0 K |
ASTM E 537-76 |
D. Temperatura płynięcia
|
Metoda pomiaru |
Substancje latwo proszkowalne |
Substancje trudno proszkowalne |
Zakres temperatury |
Założona dokładnoś (4) |
Istniejąca norma |
|
Temperatura płynięcia |
Dla olejów naftowych i substancji oleistych |
Dla olejów naftowych i substancji oleistych |
223 do 323 K |
±0,3 K |
ASTM D 97-66 |
1.6. OPIS METOD
Procedury przeprowadzenia oznaczeń według prawie wszystkich metod opisano w normach międzynarodowych i krajowych (zob. dodatek 1).
1.6.1. Metody z wykorzystaniem rurki kapilarnej
Drobno sproszkowane substancje poddawane powolnemu wzrostowi temperatury, zwykle wykazują etapy topnienia przedstawione na rys. 1.
Rysunek 1

W trakcie ustalania temperatury tapnienia odnotowuje się temperatury na początku topnienia i na etapie końcowym.
1.6.1.1. Przyrząd do badania temperatury topnienia z łaźnią cieczową
Na rysunku 2 przedstawiono rodzaj znormalizowanego przyrządu do badania temperatury topnienia wykonanego ze szkła (JTS K 0064); wszystkie wymiary podano w milimetrach.
Rysunek 2
Łaźnia cieczowa:
Należy wybrać odpowiednią ciecz. Wybór cieczy zależy od temperatury topnienia, która ma być oznaczona, na przykład ciekła parafina dla temperatur topnienia nie wyższych od 473 K, olej silikonowy dla temperatur topnienia nie wyższych niż 573 K.
Dla temperatur topnienia powyżej 523 K można wykorzystać mieszaninę składającą się z trzech części kwasu siarkowego i dwóch części siarczanu potasu (wagowo). Należy podjąć stosowne środki ostrożności przy stosowaniu mieszaniny takiej jak niniejsza.
Termometr:
Należy używać wyłącznie termometrów spełniających wymagania następujących norm lub norm równoważnych:
ASTM E 1-71, DIN 12770, JIS K 8001.
Procedura:
Suchą substancję należy drobno sproszkować w moździerzu i wprowadzić do rurki kapilarnej zatopionej na końcu, tak aby poziom napełnienia wynosił około 3 mm po ciasnym upakowaniu. Aby uzyskać jednolicie upakowaną próbkę, należy spuścić rurkę kapilarną z wysokości około 700 mm przez rurkę szklaną, pionowo na szkiełko zegarkowe.
Napełnioną rurkę kapilarną umieszcza się w łaźni tak, by środkowa część rtęciowej bańki termometru stykała się z rurką kapilarną w części, w której umieszczona jest próbka. Zwykle rurkę kapilarną wprowadza się do przyrządu o temperaturze o około 10 K niższej od temperatury topnienia.
Ciecz łaźni jest podgrzewana w ten sposób, aby temperatura rosła o około 3 K/min. Ciecz należy mieszać. Przy około 10 K poniżej oczekiwanej temperatury topnienia szybkość przyrostu temperatury ustawia się na maksimum 1 K/min.
Obliczenie:
Temperaturę topnienia oblicza się na podstawie następującego wzoru:
T = TD + 0,00016 (TD – TE) n
gdzie:
|
T |
= |
skorygowana temperatura topnienia w stopniach K, |
|
TD |
= |
odczyt temperatury z termometru D w stopniach K, |
|
TE |
= |
odczyt temperatury z termometru E w stopniach K, |
|
n = |
= |
liczba kresek podziałki słupka rtęci na termometrze D przy rurce wyjściowej. |
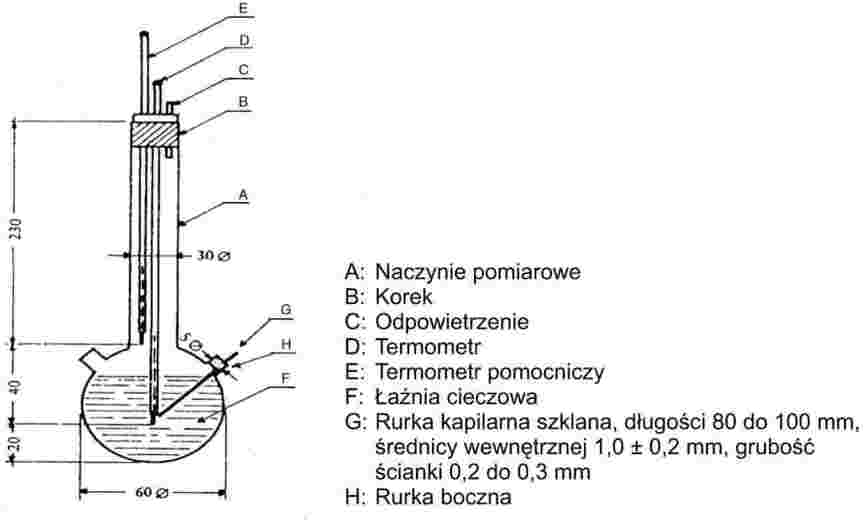
1.6.1.2. Przyrządy do pomiaru temperatury topnienia z metalowym blokiem
Przyrząd:
Składa się z:
|
— |
cylindrycznego bloku metalowego, którego górna część jest wydrążona i tworzy komorę (zob. rysunek 3), |
|
— |
zatyczki metalowej z dwiema lub większą liczbą dziur, która pozwala na montowanie probówek na bloku metalowym, |
|
— |
systemu grzejnego do podgrzewania bloku metalowego, wyposażonego na przykład w grzałki oporowe elektryczne zamknięty w bloku, |
|
— |
reostatu do regulacji zasilania, jeżeli stosowane jest podgrzewanie elektryczne, |
|
— |
czterech okienek ze szkła żaroodpornego na poprzecznych ścianach komory, zorientowanych pod kątem prostym względem siebie. Przed każdym z okienek zamontowany jest okular do obserwacji rurki kapilarnej. Pozostałe trzy okienka są wykorzystywane do oświetlania wnętrza zamknięcia przy pomocy lamp, |
|
— |
rurki kapilarnej ze szkła żaroodpornego, zamkniętej na jednym końcu (zob. ppkt 1.6.1.1). |
Zob. normy wspomniane w ppkt 1.6.1.1. Stosuje się także termoelektryczne przyrządy pomiarowe o porównywalnej dokładności.
Rysunek 3
1.6.1.3. Wykrywanie przy pomocy komórki fotoelektrycznej
Przyrząd i procedura:
Przyrząd składa się z komory metalowej z automatycznym systemem grzewczym. Trzy rurki kapilarne są napełniane zgodnie z ppkt 1.6.1.1 i umieszczane w piecu.
Możliwych jest kilka liniowych ustawień narastania temperatury w celu skalibrowania przyrządu, przy czym właściwy wzrost temperatury jest regulowany elektrycznie na z góry określoną stałą i szybkość liniową. Rejestratory pokazują rzeczywistą temperaturę pieca i temperaturę substancji w rurkach kapilarnych.
1.6.2. Płyty grzewcze
1.6.2.1. Płyta grzewcza Koflera
Zob. dodatek.
1.6.2.2. Mikroskop do badania topnienia
Zob. dodatek.
1.6.2.3. Metoda menisku (poliamidy)
Zob. dodatek.
Szybkość grzania w obrębie temperatury topnienia powinna być mniejsza niż 1 K/min.
1.6.3. Metody oznaczania temperatury krzepnięcia
Zob. dodatek.
1.6.4. Analiza termiczna
1.6.4.1. Różnicowa analiza termiczna
Zob. dodatek.
1.6.4.2. Różnicowa kalorymetria skaningowa
Zob. dodatek.
1.6.5. Oznaczanie temperatury płynięcia
Zob. dodatek.
2. DANE
W niektórych przypadkach konieczna jest korekcja termometru.
3. SPORZĄDZANIE SPRAWOZDANIA
Sprawozdanie z badań zawiera w miarę możliwości następujące informacje:
|
— |
zastosowana metoda, |
|
— |
precyzyjna specyfikacja substancji (tożsamość i zanieczyszczenia) i wstępny etap oczyszczenia, jeśli wykonano, |
|
— |
ocena dokładności. |
Jako temperaturę topnienia przedstawia się średnią z co najmniej dwóch pomiarów znajdujących się w granicach oszacowanej dokładności pomiarowej (zob. tabele).
Jeżeli różnica między temperaturą na początku i na końcowym etapie topnienia znajduje się w granicach dokładności metody, za temperaturę topnienia przyjmuje się temperaturę na końcowym etapie topnienia; w innym przypadku podaje się obie temperatury.
Jeżeli substancja przed osiągnięciem temperatury topnienia rozkłada się lub sublimuje, podawana jest temperatura, w której obserwowane są te efekty.
Należy podać wszystkie informacje i uwagi istotne dla interpretacji wyników, w szczególności w odniesieniu do zanieczyszczeń i stanu skupienia substancji.
4. LITERATURA
|
(1) |
OECD, Paris, 1981, Test Guideline 102, Decision of the Council C(81) 30 final. |
|
(2) |
IUPAC, B. Le Neindre, B. Vodar, eds. Experimental thermodynamics, Butterworths, London 1975, vol. II, 803–834. |
|
(3) |
R. Weissberger éd.: Technique of organic Chemistry, Physical Methods of Organic Chemistry, 3rd ed., Interscience Publ., New York, 1959, vol. I, Part I, Chapter VII. |
|
(4) |
IUPAC, Physicochemical measurements: Catalogue of reference materials from national laboratories, Pure and applied chemistry, 1976, vol. 48, 505–515. |
Dodatek
W celu uzyskania dodatkowych szczegółów technicznych można zapoznać się na przykład z następującymi normami.
1. Metody kapilarne
1.1. Urządzenia do pomiaru temperatury topnienia z łaźnią cieczową
|
ASTM E 324-69 |
Standard test method for relative initial and final melting points and the melting range of organic chemicals |
|
BS 4634 |
Method for the determination of melting point and/or melting range |
|
DIN 53181 |
Bestimmung des Schmelzintervalles von Harzen nach Kapilarverfarehn |
|
JIS K 00-64 |
Testing methods for melting point of chemical products. |
1.2. Urządzenia do pomiaru temperatury topnienia z blokiem metalowym
|
DIN 53736 |
Visuelle Bestimmung der Schmelztemperatur von teilkristallinen Kunststoffen |
|
ISO 1218 (E) |
Plastics- polyamides -determination of ‘melting point’ |
2. Płyty grzewcze
2.1. Płyta grzewcza Koflera
|
ANSI/ASTM D 3451-76 |
Standard recommended practices for testing polymeric powder coatings |
2.2. Mikroskop do badania topnienia
|
DIN 53736 |
Visuelle Bestimmung der Schmelztemperatur von teilkristallinen Kunststoffen |
2.3. Metoda menisku (dla poliamidów)
|
ISO 1218 (E) |
Plastics – polyamides – determination of ‘melting point’ |
|
ANSI/ASTM D 2133-66 |
Standard specification for acetal resin injection moulding and extrusion materials |
|
NF T 51-050 |
Resines de polyamides. Determination du ‘point de fusion’ méthode du menisque |
3. Metody ustalania temperatury krzepnięcia
|
BS 4633 |
Method for the determination of crystallizing point |
|
BS 4695 |
Method for Determination of Melting Point of petroleum wax (Cooling Curve) |
|
DIN 51421 |
Bestimmung des Gefrierpunktes von Flugkraftstoffen, Ottokraftstoffen und Motorenbenzolen |
|
ISO 2207 |
Cires de petrole: determination de la température de figeage |
|
DIN 53175 |
Bestimmung des Erstarrungspunktes von Fettsiiuren |
|
NF T 60-114 |
Point de fusion des paraffines |
|
NF T 20-051 |
Méthode de détermination du point de cristallisation (point de congélation) |
|
ISO 1392 |
Method for the determination of the freezing point |
4. Analiza termiczna
4.1. Różnicowa analiza termiczna
|
ASTM E 537-76 |
Standard method for assessing the thermal stability of chemicals by methods of differential thermal analysis |
|
ASTM E 473-85 |
Standard definitions of terms relating to thermal analysis |
|
ASTM E 472-86 |
Standard practice for reporting thermoanalytical data |
|
DIN 51005 |
Thermische Analyse, Begriffe |
4.2. Różnicowa kalorymetria skaningowa
|
ASTM E 537-76 |
Standard method for assessing the thermal stability of chemicals by methods of differential thermal analysis |
|
ASTM E 473-85 |
Standard definitions of terms relating to thermal analysis |
|
ASTM E 472-86 |
Standard practice for reporting thermoanalytical data |
|
DIN 51005 |
Thermische Analyse, Begriffe |
5. Oznaczanie temperatury płynięcia
|
NBN 52014 |
Echantillonnage et analyse des produits du petrole: Point de trouble et point d'ecoulement limite – Monsterneming en ontleding van aardolieproducten: Troebelingspunt en vloeipunt |
|
ASTM D 97-66 |
Standard test method for pour point of petroleum oils |
|
ISO 3016 |
Petroleum oils – Determination of pour point |
A.2. TEMPERATURA WRZENIA
1. METODA
Większość opisanych metod oparto na wytycznych OECD dotyczących badań (1). Podstawowe zasady podano w (2) i (3).
1.1. WPROWADZENIE
Urządzenia i metody tutaj opisane stosuje się do cieczy i nisko topliwych substancji, pod warunkiem że nie ulegają one reakcji chemicznej poniżej ich temperatury wrzenia (jak na przykład: samoutlenieniu, wtórnemu przekształceniu, rozkładowi i tak dalej). Metody te mogą być stosowane do czystych i zanieczyszczonych ciekłych substancji.
Kładzie się nacisk na metody wykorzystujące wykrywanie za pomocą komórki fotoelektrycznej i analizę termiczną, ponieważ metody te pozwalają na oznaczenie zarówno temperatury topnienia jak i temperatury wrzenia. Ponadto, pomiary mogą być wykonywane w sposób automatyczny.
„Metoda dynamiczna” ma tę zaletę, że można ją także wykorzystywać do oznaczenia ciśnienia pary i nie jest konieczna korekcja temperatury wrzenia do ciśnienia normalnego (101,325 kPa), gdyż można regulować ciśnienie normalne podczas pomiaru.
Uwagi:
Wpływ zanieczyszczeń na oznaczanie temperatury wrzenia w dużym stopniu zależy od charakteru zanieczyszczenia. Jeżeli w próbce znajdują się lotne zanieczyszczenia mogące wpływać na wyniki, badaną substancję należy oczyścić.
1.2. DEFINICJE I JEDNOSTKI
Temperatura wrzenia normalna jest zdefiniowana jako temperatura, przy której prężność pary cieczy wynosi 101,325 kPa.
Jeżeli temperatura wrzenia nie jest mierzona w normalnym ciśnieniu atmosferycznym, zależność prężności pary od temperatury opisuje się równaniem Clausiusa – Clapeyrona:

gdzie:
|
P |
= |
prężność pary substancji w paskalach, |
|
ΔHV |
= |
jej ciepło parowania w J mol-1, |
|
R |
= |
uniwersalna molowa stała gazowa = 8,314 J mol-1 K-1, |
|
T |
= |
temperatura termodynamiczna w stopniach K. |
Temperatura wrzenia jest ustalona w odniesieniu do ciśnienia otoczenia w czasie trwania pomiaru.
Konwersje
Ciśnienie (jednostki: kPa)
|
100 kPa |
= |
l bar = 0,1 MPa (jednostka „bar” jest wciąż dozwolona, ale nie jest zalecana) |
|
133 Pa |
= |
1 mm Hg = 1 tor (jednostki „mm H” i „Torr” nie są dozwolone) |
|
1 atm |
= |
standardowa atmosfera = 101 325 Pa (jednostka „atm” jest niedozwolona) |
Temperatura (jednostki: K)
t = T - 273,15
|
t |
: |
temperatura Celsjusza, stopień Celsjusza (oC) |
|
T |
: |
temperatura termodynamiczna, Kelwin (K) |
1.3. SUBSTANCJE ODNIESIENIA
Nie ma konieczności stosowania substancji odniesienia za każdym razem, gdy przeprowadzane są badania nowej substancji. Powinny one służyć głównie sprawdzeniu od czasu do czasu wydajności metody i pozwolić na porównanie wyników z innymi metodami.
Niektóre substancje kalibracyjne wymieniono w opisach metod wymienionych w dodatku.
1.4. ZASADA METODY BADANIA
Pięć metod ustalania temperatury wrzenia (zakresu wrzenia) jest oparte na pomiarze temperatury wrzenia, dwie inne oparte są o analizę termiczną.
1.4.1. Wyznaczanie za pomocą ebuliometru
Ebuliometry zostały pierwotnie opracowane w celu oznaczania masy cząsteczkowej poprzez podwyższanie temperatury wrzenia, jednak nadają się także do dokładnych pomiarów tej temperatury. Bardzo prosty przyrząd opisano w ASTM D 1120-72 (zob. dodatek). Ciecz jest podgrzewana w tym przyrządzie w warunkach równowagi, pod ciśnieniem atmosferycznym, do wrzenia.
1.4.2. Metoda dynamiczna
Metoda ta obejmuje pomiar temperatury ponownej kondensacji pary przy pomocy właściwego termometru w wykroplinach podczas wrzenia. Podczas stosowania tej metody ciśnienie może być zmienne.
1.4.3. Metoda destylacji dla wyznaczania temperatury wrzenia
Metoda ta obejmuje destylację cieczy i pomiar temperatury ponownej kondensacji pary oraz oznaczenie ilości destylatu.
1.4.4. Metoda według Siwolobowa
Próbkę podgrzewa się w probówce, która jest zanurzona w cieczy w gorącej łaźni. W probówce zanurzona jest z kolei zatopiona na końcu rurka kapilarna, zawierająca pęcherzyk powietrza w dolnej części.
1.4.5. Wykrywanie przy pomocy komórki fotoelektrycznej
Zgodnie z zasadą według Siwolobowa wykonuje się automatyczny pomiar fotoelektryczny z wykorzystaniem unoszących się pęcherzyków.
1.4.6. Różnicowa analiza termiczna
Niniejsza technika rejestruje różnicę w temperaturach między substancją i materiałem odniesienia jako funkcję temperatury, gdy substancja oraz materiał odniesienia podlegają temu samemu programowi kontrolowania temperatury. W momencie gdy próbka podlega przejściu pociągającym za sobą zmianę entalpii, zmiana ta jest wskazywana przez endotermiczne odchylenie (wrzenie) od linii bazowej zapisu temperatury.
1.4.7. Różnicowa kalorymetria skaningowa
Niniejsza technika rejestruje różnicę w energii pobranej przez substancję oraz materiał odniesienia, jako funkcję temperatury, gdy substancja oraz materiał odniesienia podlegają temu samemu programowi kontrolowania temperatury. Niniejsza energia jest energią konieczną do ustalenia zerowej różnicy temperatury między substancją i materiałem odniesienia. W momencie gdy próbka podlega przejściu pociągającym za sobą zmianę entalpii, zmiana ta jest wskazywana przez endotermiczne odchylenie (wrzenie) od linii bazowej zapisu przepływu ciepła.
1.5. KRYTERIA JAKOŚCI
Zastosowanie i dokładność różnych metod wykorzystywanych do ustalania temperatury wrzenia/zakres temperatury wrzenia przedstawiono w tabeli 1.
Tabela 1
Porównanie metod
|
Metoda pomiaru |
Założona dokładność |
Istniejąca norma |
|
Ebuliometr |
ASTM D 1120-72 (5) |
|
|
Metody dynamiczne |
±0,5 K (do 600 K) (6) |
|
|
Proces destylacji (zakres wrzenia) |
±0,5 K (do 600 K) |
ISO/R 918, DIN 53171, BS 4591/71 |
|
Według Siwolobowa |
± 2 K (do 600 K) (6) |
|
|
Wykrywanie za pomocą komórki fotoelektrycznej |
±0,3 K (do 373 K) (6) |
|
|
Różnicowa kalorymetria termiczna |
±0,5 K (do 600 K) ±2,0 K (do 1 273 K) |
ASTM E 537-76 |
|
Różnicowa kalorymetria skaningowa |
±0,5 K (do 600 K) ±2,0 K (do 1 273 K) |
ASTM E 537-76 |
1.6. OPIS METOD
Procedury dotyczące niektórych metod badania opisano w normach międzynarodowych i krajowych (zob. dodatek).
1.6.1. Ebuliometr
Zob. dodatek
1.6.2. Metoda dynamiczna
Zob. metoda badania A.4 dla oznaczania prężności pary.
Odnotowuje się temperaturę wrzenia stwierdzoną pod przyłożonym ciśnieniem 101,325 kPa.
1.6.3. Proces destylacji (zakres wrzenia)
Zob. dodatek.
1.6.4. Metoda według Siwolobowa
Próbka jest podgrzewana w przyrządzie do oznaczania temperatury topnienia w probówce o średnicy około 5 mm (rysunek 1).
Rysunek 1 pokazuje rodzaj znormalizowanego przyrządu do badania temperatury topnienia i wrzenia (JIS K 0064) (wykonanego ze szkła, wszystkie wymiary w milimetrach).
Rysunek 1

Rurkę kapilarną (kapilara do badania wrzenia), zatopioną na wysokości około 1 cm powyżej dolnego końca, umieszcza się w probówce. Badaną substancję wprowadza się w ten sposób, aby zatopiony odcinek kapilary znalazł się poniżej powierzchni cieczy. Probówkę zawierającą kapilarę albo przymocowuje się do termometru przy pomocy taśmy gumowej, albo umocowuje z boku na wsporniku (zob. rysunek 2).
|
Rysunek 2 Zasada według Siwolobowa |
Rysunek 3 Zmodyfikowana zasada |
|
|
|
Ciecz do łaźni należy wybrać w zależności od temperatury wrzenia. Przy temperaturach do 573 K stosuje się olej silikonowy. Ciekła parafina jest stosowana tylko do 473 K. Początkowo grzanie łaźni cieczowej ustawia się na wzrost temperatury 3 K/min. Ciecz łaźni musi być mieszana. W temperaturze około 10 K poniżej spodziewanej temperatury wrzenia szybkość wzrostu temperatury ustawia się na maksimum 1 K/min. Przy zbliżaniu się do temperatury wrzenia z wrzącej zawartości kapilary zaczynają unosić się pęcherzyki.
Temperatura wrzenia to temperatura, w której przy chwilowym schłodzeniu łańcuszek pęcherzyków przestaje powstawać i ciecz w kapilarze nagle zaczyna się unosić. Odpowiadający temu odczyt temperatury to temperatura wrzenia substancji.
W zmodyfikowanej zasadzie pomiaru (rysunek 3) temperatura wrzenia jest oznaczana w kapilarze od ustalania temperatury topnienia. Jest ona wyciągnięta do przewężenia o długości około 2 cm (a) i zassana jest w niej mała ilość próbki. Otwarty koniec cienkiej kapilary jest zamknięty poprzez obtopienie w taki sposób, że na jej końcu znajduje się mały pęcherzyk powietrza. Podczas ogrzewania w aparacie do ustalania temperatury topnienia (b), pęcherzyk powietrza rozszerza się. Temperaturze wrzenia odpowiada taka temperatura, przy której zatyczka z substancji badanej osiąga poziom powierzchni cieczy łaźni (c).
1.6.5. Wykrywanie za pomocą komórki fotoelektrycznej
Próbka jest ogrzewana w rurce kapilarnej umieszczonej wewnątrz podgrzewanego bloku metalowego.
Wiązka światła przechodzi poprzez odpowiednie otwory w bloku, poprzez badaną substancję na precyzyjnie skalibrowaną komórkę fotoelektryczną.
W czasie wzrostu temperatury próbki pojedyncze pęcherzyki powietrza pojawiają się w kapilarze. Gdy osiągnięta jest temperatura wrzenia, ilość pęcherzyków gwałtownie wzrasta. Powoduje to zmianę intensywności światła rejestrowanego przez komórkę fotoelektryczną i powoduje wysłanie sygnału zatrzymania do wskaźnika odczytującego temperaturę z platynowego termometru rezystancyjnego umieszczonego w bloku.
Niniejsza metoda jest szczególnie użyteczna, gdyż pozwala na oznaczania poniżej temperatury pokojowej do 253,15 K (-20 oC) bez żadnych zmian w przyrządzie. Przyrząd umieszcza się jedynie w łaźni chłodzącej.
1.6.6. Analiza termiczna
1.6.6.1. Różnicowa analiza termiczna
Zob. dodatek.
1.6.6.2. Różnicowa kalorymetria skaningowa
Zob. dodatek.
2. DANE
Przy niewielkich odchyleniach od ciśnienia normalnego (maksimum ± 5 kPa) temperaturę wrzenia normalizuje się do Tn na podstawie następującego wzoru równania Sidneya-Younga:
Tn = T + (fT × Δp)
gdzie:
|
Δp |
= |
(101,325 – p) [uwaga na znak], |
|
P |
= |
pomiar ciśnienia w kPa, |
|
fT |
= |
szybkość zmian temperatury wrzenia z ciśnieniem w K/Kpa, |
|
T |
= |
zmierzona temperatura wrzenia w K, |
|
Tn |
= |
temperatura wrzenia skorygowana do ciśnienia normalnego w K. |
Współczynniki korekcji temperatury, fT, i wzory na ich przybliżone obliczanie zawarto we wspomnianych wyżej normach międzynarodowych i krajowych dla wielu substancji.
Na przykład w metodzie DIN 53171 wymieniono następujące, przybliżone poprawki dla rozpuszczalników zawartych w farbach:
Tabela 2
Temperatura – współczynniki korekcji fT
|
Temperatura T (K) |
Współczynnik korekcji fT (K/kPa) |
|
323,15 |
0,26 |
|
348,15 |
0,28 |
|
373,15 |
0,31 |
|
398,15 |
0,33 |
|
423,15 |
0,35 |
|
448,15 |
0,37 |
|
473,15 |
0,39 |
|
498,15 |
0,41 |
|
523,15 |
0,44 |
|
548,15 |
0,45 |
|
573,15 |
0,47 |
3. SPORZĄDZANIE SPRAWOZDANIA
Sprawozdanie z badań zawiera w miarę możliwości następujące informacje:
|
— |
zastosowana metoda, |
|
— |
precyzyjna specyfikacja substancji (tożsamość i zanieczyszczenia) i wstępny etap oczyszczenia, jeśli wykonano, |
|
— |
ocena dokładności. |
Średnią co najmniej dwóch pomiarów znajdujących się w zakresie oszacowanej dokładności (zob. tabela 1) przedstawia się jako temperaturę wrzenia.
Należy podać zmierzone temperatury wrzenia i ich średnią, a także ciśnienie (ciśnienia), w którym (których) były wykonywane pomiary, w kPa. Najkorzystniej jest, gdy ciśnienie jest zbliżone do normalnego ciśnienia atmosferycznego.
Należy podać wszystkie informacje i uwagi istotne dla interpretacji wyników, w szczególności w odniesieniu do zanieczyszczeń i stanu skupienia substancji.
4. LITERATURA
|
(1) |
OECD, Paris, 1981, Test Guideline 103, Decision of the Council C (81) 30 final. |
|
(2) |
IUPAC, B. Le Neindre, B. Vodar, editions. Experimental thermodynamics, Butterworths, London 1975, vol. II. |
|
(3) |
R. Weissberger edition: Technique of organic chemistry, Physical methods of organic chemistry, Third Edition, Interscience Publications, New York, 1959, vol. I, part I, chapter VIII. |
Dodatek
W celu uzyskania dodatkowych szczegółów technicznych można zapoznać się na przykład z następującymi normami.
1. Ebuliometr
|
1.1. |
Urządzenia do pomiaru temperatury topnienia z łaźnią cieczową |
|
ASTM D 1120-72 |
Standard test method for boiling point of engine anti-freezes |
2. Proces destylacji (zakres wrzenia)
|
ISO/R 918 |
Test Method for Distillation (Distillation Yield and Distillation Range) |
|
BS 4349/68 |
Method for determination of distillation of petroleum products |
|
BS 4591/71 |
Method for the determination of distillation characteristics |
|
DIN 53171 |
Losungsmittel fur Anstrichstoffe, Bestimmung des Siedeverlaufes |
|
NF T 20-608 |
Distillation: determination du rendement et de l’intervalle de distillation |
3. Różnicowa analiza termiczna i różnicowa kalorymetria skaningowa
|
ASTM E 537-76 |
Standard method for assessing the thermal stability of chemicals by methods of differential thermal analysis |
|
ASTM E 473-85 |
Standard definitions of terms relating to thermal analysis |
|
ASTM E 472-86 |
Standard practice for reporting thermoanalytical data |
|
DIN 51005 |
Thermische Analyse, Begriffe |
A.3. GĘSTOŚĆ WZGLĘDNA
1. METODA
Większość opisanych metod oparto na wytycznych OECD dotyczących badań (1). Podstawowe zasady podano w pozycjach bibliograficznych (2) i (3).
1.1. WPROWADZENIE
Opisane metody oznaczania gęstości względnej stosują się do substancji stałych i płynnych, bez ograniczeń związanych z ich stopniem czystości. Różne metody możliwe do zastosowania wymieniono w tabeli 1.
1.2. DEFINICJE I JEDNOSTKI
Gęstość względna ciał stałych lub cieczy jest stosunkiem między masą objętości badanej substancji, ustaloną w 20 oC, oraz masą tej samej objętości wody, ustaloną w 4 oC. Gęstość względna jest bezwymiarowa.
Gęstość, ρ, substancji jest ilorazem masy, m, i jej objętości, v.
Gęstość, ρ, w układzie jednostek SI jest dana w kg/m3.
1.3. SUBSTANCJE ODNIESIENIA (1), (3)
Nie ma konieczności stosowania substancji odniesienia za każdym razem, gdy przeprowadzane są badania nowej substancji. Powinny one służyć głównie sprawdzeniu od czasu do czasu wydajności metody i pozwolić na porównanie wyników z innymi metodami.
1.4. ZASADA METOD
Stosowane są cztery klasy metod.
1.4.1. Metody wyporu hydrostatycznego
1.4.1.1. Areometr (dla substancji ciekłych)
Wystarczająco dokładne i szybkie oznaczenia gęstości można uzyskać przy użyciu areometrów pływakowych, które pozwalają na obliczenie gęstości cieczy na podstawie głębokości zanurzenia, przez odczyt na skali z podziałką.
1.4.1.2. Waga hydrostatyczna (dla substancji płynnych i stałych)
Różnicę między wagą badanej próbki zmierzoną w powietrzu i w odpowiedniej cieczy (na przykład w wodzie) wykorzystuje się do oznaczenia jej gęstości.
W przypadku ciał stałych zmierzona gęstość jest charakterystyczna jedynie dla określonej, zastosowanej próbki. W przypadku oznaczania gęstości cieczy, ich ilość o znanej objętości, v, waży się najpierw w powietrzu, a potem w cieczy.
1.4.1.3. Metoda zanurzeniowa (dla substancji ciekłych) (4)
Metoda ta polega na oznaczaniu gęstości cieczy na podstawie różnicy między wynikami zważenia cieczy przed i po zanurzeniu w nim ciała o znanej objętości.
1.4.2. Metody piknometryczne
W przypadku ciał stałych lub cieczy można się posłużyć piknometrami o różnych kształtach i o znanych wymiarach. Gęstość oblicza się na podstawie różnicy masy między napełnionym i pustym piknometrem oraz jego znanej objętości.
1.4.3. Piknometr wykonujący pomiar porównawczy w powietrzu (dla ciał stałych)
Gęstość ciała stałego w dowolnej formie można zmierzyć w temperaturze pokojowej przy użyciu piknometru wykonującego pomiary porównawcze w gazie. Objętość substancji mierzy się w powietrzu lub w gazie obojętnym w cylindrze o zmiennej, wykalibrowanej objętości. Dla obliczenia gęstości wykonuje się jeden pomiar masy po zakończeniu pomiaru objętości.
1.4.4. Densytometr oscylacyjny (5, 6, 7)
Gęstość cieczy można zmierzyć przy pomocy densytometru oscylacyjnego. Mechaniczny oscylator skonstruowany w postaci U-rurki drga w częstotliwości rezonansowej zależnej od jego masy. Wprowadzenie badanej próbki zmienia częstotliwość rezonansową oscylatora. Przyrząd należy skalibrować przy użyciu dwóch substancji płynnych o znanej gęstości. Najkorzystniej, aby substancje te wybrać tak, aby ich gęstości były bliskie zakresowi, który ma być mierzony.
1.5. KRYTERIA JAKOŚCI
Zastosowania poszczególnych metod wykorzystywanych do oznaczania gęstości względnej wymieniono w tabeli.
1.6. OPIS METOD
W dodatku załączono normy podane jako przykłady, z którymi należy się zapoznać w celu uzyskania informacji o dodatkowych szczegółach technicznych.
Badania należy przeprowadzać w temperaturze 20 oC, przy czym konieczne jest wykonanie co najmniej dwóch pomiarów.
2. DANE
Zob. normy.
3. SPORZĄDZANIE SPRAWOZDANIA
Sprawozdanie z badań zawiera w miarę możliwości następujące informacje:
|
— |
zastosowana metoda, |
|
— |
precyzyjna specyfikacja substancji (tożsamość i zanieczyszczenia) i wstępny etap oczyszczania, jeśli wykonano. |
Gęstość względną,  , należy podawać w sposób określony w ppkt 1.2, łącznie ze stanem skupienia mierzonej substancji.
, należy podawać w sposób określony w ppkt 1.2, łącznie ze stanem skupienia mierzonej substancji.
Należy podać wszystkie informacje i uwagi istotne dla interpretacji wyników, w szczególności w odniesieniu do zanieczyszczeń i stanu skupienia substancji.
Tabela
Zastosowanie metod
|
Metoda pomiaru |
Gęstość |
Maksymalna możliwa lepkość dynamiczna |
Istniejące normy |
|||
|
ciało stałe |
ciecz |
|||||
|
|
tak |
5 Pa s |
ISO 387, ISO 649-2, NF T 20-050 |
||
|
|
|
|
|
||
|
tak |
|
|
ISO 1183 (A) |
||
|
|
tak |
5 Pa s |
ISO 901 i 758 |
||
|
|
tak |
20 Pa s |
DIN 53217 |
||
|
|
|
|
ISO 3507 |
||
|
tak |
|
|
ISO 1183(B), NF T 20-053 |
||
|
|
tak |
500 Pa s |
ISO 758 |
||
|
tak |
|
|
DIN 55990 Teil 3, DIN 53243 |
||
|
|
tak |
5 Pa s |
|
||
4. LITERATURA
|
(1) |
OECD, Paris, 1981, Test Guideline 109, Decision of the Council C(81) 30 final. |
|
(2) |
R. Weissberger ed., Technique of Organic Chemistry, Physical Methods of Organic Chemistry, 3rd ed., chapter IV, Interscience Publ. , New York, 1959, vol. I, part 1. |
|
(3) |
IUPAC, Recommended reference materials for realization of physico-chemical properties, Pure and applied chemistry, 1976, vol. 48, 508. |
|
(4) |
Wagenbreth, H., Die Tauchkugel zur Bestimmung der Dichte von Flüssigkeiten, Technisches Messen tm, 1979, vol. 11, 427–430. |
|
(5) |
Leopold, H., Die digitale Messung von Flüssigkeiten, Elektronik, 1970, vol. 19, 297-302. |
|
(6) |
Baumgarten, D., Füllmengenkontrolle bei vorgepackten Erzeugnissen – Verfahren zur Dichtebestimmung bei flüssigen Produkten und ihre praktische Anwendung, Die Pharmazeutische Industrie, 1975, vol. 37, 717–726. |
|
(7) |
Riemann, J., Der Einsatz der digital en Dichtemessung im Brauereilaboratorium, Brauwissenschaft, 1976, vol. 9, 253–255. |
Dodatek
Dla uzyskania dodatkowych szczegółów technicznych można zapoznać się na przykład z następującymi normami.
1. Metody wyporu hydrostatycznego
1.1. Aerometr
|
DIN 12790, ISO 387 |
Hydrometer; general instructions |
|
DIN 12791 |
Part I: Density hydrometers; construction, adjustment and use Part II: Density hydrometers; standardized sizes, designation Part III: Use and test |
|
ISO 649-2 |
Laboratory glassware: Density hydrometers for general purpose |
|
NF T 20-050 |
Chemical products for industrial use – Determination of density of liquids – Areometric method |
|
DIN 12793 |
Laboratory glassware: range find hydrometers |
1.2. Waga hydrostatyczna
Dla substancji stałych
|
ISO 1183 |
Method A: Methods for determining the density and relative density of plastics excluding cellular plastics |
|
NF T 20-049 |
Chemical products for industrial use – Determination of the density of solids other than powders and cellular products – Hydrostatic balance method |
|
ASTM-D-792 |
Specific gravity and density of plastics by displacement |
|
DIN 53479 |
Testing of plastics and elastomers; determination of density |
Dla substancji ciekłych
|
ISO 901 |
ISO 758 |
|
DIN 51757 |
Testing of mineral oils and related materials; determination of density |
|
ASTM D 941-55, ASTM D 1296-67 i ASTM D 1481-62 |
|
|
ASTM D 1298 |
Density, specific gravity or API gravity of crude petroleum and liquid petroleum products by hydrometer method |
|
BS 4714 |
Density, specific gravity or API gravity of crude petroleum and liquid petroleum products by hydrometer method |
1.3. Metoda zanurzeniowa
|
DIN 53217 |
Testing of paints, varnishes and similar coating materials; determination of density; immersed body method |
2. Metody piknometryczne
2.1. Dla substancji ciekłych
|
ISO 3507 |
Pycnometers |
|
ISO 758 |
Liquid chemical products; determination of density at 20 oC |
|
DIN 12797 |
Gay-Lussac pycnometer (for non-volatile liquids which are not too viscous) |
|
DIN 12798 |
Lipkin pycnometer (for liquids with a kinematic viscosity of less than 100,10-6 m2 s-1 at 15 oC) |
|
DIN 12800 |
Sprengel pycnometer (for liquids as DIN 12798) |
|
DIN 12801 |
Reischauer pycnometer (for liquids with a kinematic viscosity of less than 100,10-6 m2 s-1 at 20 oC, applicable in particular also to hydrocarbons and aqueous solutions as well as to liquids with higher vapour pressure, approximately 1 bar at 90 oC) |
|
DIN 12806 |
Hubbard pycnometer (for viscous liquids of all types which do not have too high a vapour pressure, in particular also for paints, varnishes and bitumen) |
|
DIN 12807 |
Bingham pycnometer (for liquids, as in DIN 12801) |
|
DIN 12808 |
Jaulmes pycnometer (in particular for ethanol – water mixture) |
|
DIN 12809 |
Pycnometer with ground-in thermometer and capillary side tube (for liquids which are not too viscous) |
|
DIN 53217 |
Testing of paints, varnishes and similar products; determination of density by pycnometer |
|
DIN 51757 |
Point 7: Testing of mineral oils and related materials; determination of density |
|
ASTM D 297 |
Section 15: Rubber products – chemical analysis |
|
ASTM D 2111 |
Method C: Halogenated organic compounds |
|
BS 4699 |
Method for determination of specific gravity and density of petroleum products (graduated bicapillary pycnometer method) |
|
BS 5903 |
Method for determination of relative density and density of petroleum products by the capillary- stoppered pycnometer method |
|
NF T 20-053 |
Chemical products for industrial use – Determination of density of solids in powder and liquids – Pyknometric method |
2.2. Dla substancji stałych
|
ISO 1183 |
Method B: Methods for determining the density and relative density of plastics excluding cellular plastics |
|
NF T 20-053 |
Chemical products for industrial use – Determination of density of solids in powder and liquids – Pyknometric method |
|
DIN 19683 |
Determination of the density of soils |
3. Piknometr wykonujący pomiar porównawczy w powietrzu
|
DIN 55990 |
Part 3: Prüfung von Anstrichstoffen und ähnlichen Beschichrungsstoffen; Pulverlack; Bestimmung der Dichte |
|
DIN 53243 |
Anstrichstoffe; Chlorhaltige Polymere; Prüfung |
A.4. PRĘŻNOŚĆ PARY
1. METODA
Większość opisanych metod oparto na wytycznych OECD dotyczących badań (1). Podstawowe zasady podano w pozycjach bibliograficznych (2) i (3).
1.1. WPROWADZENIE
Do przeprowadzenia tego oznaczenia przydatne jest posiadanie wstępnych informacji na temat budowy, temperatury topnienia i temperatury wrzenia substancji.
Nie istnieje procedura pojedynczego pomiaru dająca się zastosować do całego zakresu prężności par. Dlatego zalecane jest kilka metod stosowanych do pomiaru prężności par w zakresie od < 10-4 do 105 Pa.
Zanieczyszczenia zwykle wpływają na prężność pary do pewnego stopnia, zależącego głównie od rodzaju zanieczyszczenia.
Jeżeli w próbce znajdują się zanieczyszczenia lotne, które mogą wpływać na wynik, substancję należy oczyścić. Może być także właściwe powołanie się na prężność pary materiału czystości technicznej.
Kilka opisanych tu metod wykorzystuje przyrząd składający się z metalowych części, co należy rozważyć przy badaniu substancji powodujących korozję.
1.2. DEFINICJE I JEDNOSTKI
Prężność pary substancji określa się jako ciśnienie nasycenia ponad substancją w stanie stałym lub płynnym. W stanie równowagi termodynamicznej prężność pary czystej substancji jest funkcją jedynie temperatury.
Jednostką ciśnienia układu SI, którą należy stosować, jest paskal (Pa).
Jednostki, które stosowano w przeszłości, łącznie z ich współczynnikami konwersji:
|
1 tor (≡ 1 mm Hg) |
= 1,333 ×102 Pa |
|
1 atmosfera |
= 1,013 × 105 Pa |
|
1 bar |
= 105 Pa |
Jednostką temperatury w układzie SI jest kelwin (K).
Uniwersalna molowa stała gazowa R wynosi 8,314 Jmol-1 K-1
Temperaturowa zależność prężności pary jest opisana równaniem Clausiusa-Clapeyrona:
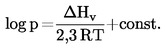
gdzie:
|
p |
= |
prężność pary substancji w Pa, |
|
ΔHv |
= |
jej ciepło parowania w Jmol-l, |
|
R |
= |
uniwersalna molowa stała gazowa w Jmol-l K-1, |
|
T |
= |
temperatura termodynamiczna w K. |
1.3. SUBSTANCJE ODNIESIENIA
Nie ma konieczności stosowania substancji odniesienia za każdym razem, gdy przeprowadzane są badania nowej substancji. Powinny one służyć głównie sprawdzeniu od czasu do czasu wydajności metody i pozwolić na porównanie wyników z innymi metodami.
1.4. ZASADA METOD BADAŃ
Do oznaczania prężności pary proponuje się siedem metod, które można stosować przy różnych zakresach prężności pary. W przypadku każdej metody prężność pary określa się w różnych temperaturach. W ograniczonym zakresie temperatur logarytm prężności pary czystej substancji jest liniową funkcją odwrotności temperatury.
1.4.1. Metoda dynamiczna
Przy użyciu metody dynamicznej dokonuje się pomiaru temperatury wrzenia, która odpowiada określonemu ciśnieniu.
Zalecany zakres:
Od 103 do 105 Pa.
Niniejsza metoda jest także zalecana do oznaczania normalnej temperatury wrzenia i jest użyteczna w tym celu do 600 K.
1.4.2. Metoda statyczna
W procesie statycznym w punkcie równowagi termodynamicznej oznacza się prężność pary ustaloną w zamkniętym układzie, w określonej temperaturze. Metoda ta jest właściwa dla jednoskładnikowych i wieloskładnikowych ciał stałych i cieczy.
Zalecany zakres:
Od 103 do 105 Pa.
Stosując należytą uwagę, niniejszą metodę można zastosować także w zakresie od 1 do 10 Pa.
1.4.3. Izoteniskop
Ta znormalizowana metoda jest także metodą statyczną, jednak na ogół nie jest właściwa dla układów wieloskładnikowych. Dodatkowe informacje są dostępne w metodzie ASTM D-2879-86.
Zalecany zakres:
Od 100 do 105 Pa.
1.4.4. Metoda efuzji: waga do oceny prężności pary
Ustala się ilość substancji opuszczającej komórkę w jednostce czasu przez szczelinę o znanym rozmiarze, w warunkach próżni, w ten sposób, że powrót substancji do komórki jest nieznaczny (np. przez pomiar pulsu generowanego na czułej wadze przez strumień pary lub przez pomiar utraty masy).
Zalecany zakres:
Od 10-3 do 1 Pa.
1.4.5. Metoda efuzji: pomiar poprzez utratę masy lub przez pułapkowanie oparów
Metoda oparta jest na oznaczeniu masy badanej substancji ulatniającej się na jednostkę czasu z komórki Knudsena (4) w postaci oparów przez mikrootwór w warunkach wysokiej próżni. Masę oparów, które uległy efuzji, uzyskuje się albo przez oznaczenie utraty masy komórki, albo przez kondensację oparów w niskiej temperaturze i oznaczenie ilości ulotnionej substancji za pomocą analizy chromatograficznej. Prężność pary jest obliczana za pomocą zależności Hertza-Knudsena.
Zalecany zakres:
Od 10-3 do 1 Pa.
1.4.6. Metoda nasycenia gazem
Strumień obojętnego gazu nośnikowego jest przepuszczany nad substancją w taki sposób, że powoduje to nasycenie jego parą. Ilość przeniesionego materiału przez znaną ilość gazu nośnikowego mierzy się albo przez zbieranie go w odpowiedniej pułapce, albo impulsową techniką analityczną. Jest to następnie stosowane do obliczenia prężności pary w danej temperaturze.
Zalecany zakres:
Od 10-4 do 1 Pa.
Stosując należytą uwagę, niniejszą metodę można zastosować także w zakresie od 1 do 10 Pa.
1.4.7. Metoda wirującego rotora
W mierniku wirującego rotora, rzeczywistym elementem pomiarowym jest mała stalowa kula zawieszona w polu magnetycznym, obracająca się z dużą szybkością. Ciśnienie gazu jest wyprowadzane z zależnego od ciśnienia, spadku szybkości stalowej kuli.
Zalecany zakres:
Od 10-4 do 0,5 Pa.
1.5. KRYTERIA JAKOŚCI
Porównano poszczególne metody oznaczania prężności pary pod względem ich zastosowania, powtarzalności, odtwarzalności, zakresu pomiaru, istnienia norm. Porównanie to przedstawiono w tabeli poniżej.
Tabela
Kryteria jakości
|
Metoda pomiarowa |
Substancje |
Szacowana powtarzalność (7) |
Szacowana odtwarzalność (7) |
Zalecany zakres |
Istniejące normy |
|||
|
stałe |
ciekłe |
|||||||
|
nisko topliwe |
tak |
do 25 % |
do 25 % |
103 Pa do 2 × 103 Pa |
— |
||
|
|
|
|
1 do 5 % |
1 do 5 % |
2 × 103 Pa do 105 Pa |
— |
||
|
tak |
tak |
5 do 10 % |
5 do 10 % |
10 Pa do 105 Pa (8) |
NFT 20-048 (5) |
||
|
tak |
tak |
5 do 10 % |
5 do 10 % |
102 Pa do 105 Pa |
ASTM-D 2879-86 |
||
|
tak |
tak |
5 do 20 % |
do 50 % |
10-3 Pa do 1 Pa |
NFT 20-047 (6) |
||
|
tak |
tak |
10 do 30 % |
— |
10-3 Pa do 1 Pa |
— |
||
|
tak |
tak |
10 do 30 % |
do 50 % |
10-4 Pa do 1 Pa (8) |
— |
||
|
tak |
tak |
10 do 20 % |
— |
10-4 Pa do 0,5 Pa |
— |
||
1.6. OPIS METOD
1.6.1. Pomiar dynamiczny
1.6.1.1. Przyrząd
Zwykle przyrząd pomiarowy składa się z naczynia do gotowania z dołączoną chłodnicą wykonaną ze szkła lub metalu (rysunek 1), urządzenia do pomiaru temperatury i urządzenia do regulowania i pomiaru ciśnienia. Typowy przyrząd pomiarowy pokazany na rysunku jest wykonany z żaroodpornego szkła i składa się z pięciu części:
Duża, częściowo dwuścienna rura składa się ze złącza szlifowanego z płaszczem, chłodnicy, naczynia chłodzącego i otworu wlotowego.
Do części rury, w której odbywa się wrzenie, dołączony jest szklany cylinder z „pompką” Cottrella, który ma nierówną powierzchnię tłuczonego szkła w celu uniknięcia burzliwości w procesie wrzenia.
Temperatura jest mierzona odpowiednim czujnikiem temperatury (na przykład termometrem oporowym, szczelnym termoogniwem) umieszczonym w aparacie w punkcie pomiaru (nr 5, rysunek 1) za pomocą odpowiedniego wlotu (na przykład złączem szlifowym).
Wykonane są niezbędne połączenia do regulacji ciśnienia i oprzyrządowania pomiarowego.
Bańka, która działa jako objętość buforowa, jest połączona z przyrządem pomiarowym poprzez rurkę kapilarną.
Naczynie przeznaczone do wrzenia jest ogrzewane za pomocą elementu grzejnego (na przykład grzałką) włożonego do szklanego przyrządu od dołu. Wymagany prąd grzania jest ustawiany i regulowany poprzez termoogniwo.
Konieczna próżnia w zakresie między 102 Pa i około 105 Pa jest wytwarzana za pomocą pompy próżniowej.
Stosuje się odpowiedni zawór do liczników powietrza lub azotu dla regulacji ciśnienia (zakres pomiarowy około 102 do 105 Pa) oraz wentylację.
Ciśnienie jest mierzone za pomocą manometru.
1.6.1.2. Procedura pomiarowa
Prężność pary jest mierzona przez ustalenie temperatury wrzenia próbki przy różnych określonych ciśnieniach między prawie 103 a 105 Pa. Ustalona temperatura pod stałym ciśnieniem wskazuje, że uzyskano temperaturę wrzenia. Metoda ta nie jest użyteczna do pomiaru ciśnienia pary substancji, które się pienią.
Substancja jest umieszczona w czystym, suchym naczyniu do próbek. Można się natknąć na problemy z niesproszkowanymi ciałami stałymi, lecz czasem można je rozwiązać przez podgrzanie płaszcza chłodzącego. Po napełnieniu naczynia przyrząd jest uszczelniany na kołnierzu i odgazowuje się substancję. Następnie ustawia się najniższe pożądane ciśnienie i włącza układ grzejny. W tym samym czasie czujnik temperatury jest podłączany do rejestratora.
Stan równowagi jest osiągany, gdy rejestruje się stałą temperaturę wrzenia przy stałym ciśnieniu. Należy zwrócić szczególną uwagę, by zapobiec wrzeniu burzliwemu podczas wrzenia. Dodatkowo musi zachodzić całkowita kondensacja w chłodnicy. Przy oznaczaniu prężności pary niskotopliwych ciał stałych należy zwrócić uwagę na uniknięcie zablokowania skraplacza.
Po zapisaniu parametrów punktu równowagi ustawia się wyższe ciśnienie. Proces kontynuuje się w ten sposób do osiągnięcia ciśnienia 105 Pa (ogółem od około 5 do 10 punktów pomiarowych). W ramach sprawdzenia można powtórzyć oznaczanie punktów równowagi w trakcie obniżania ciśnienia.
1.6.2. Pomiar statyczny
1.6.2.1. Przyrząd
Przyrząd zawiera naczynie na próbkę, system grzejny i chłodzący do regulacji temperatury próbki i pomiaru temperatury. Przyrząd zawiera również przyrządy do ustawiania i pomiaru ciśnienia. Rysunki 2a i 2b obrazują zastosowane podstawowe zasady.
Komora próbki (rysunek 2a) jest podłączona z jednej strony odpowiednim zaworem dla wysokiej próżni. U-rurka zawierająca odpowiednią ciecz manometryczną jest dołączona z drugiej strony. Jeden z końców rozgałęzienia U-rurki odcina pompę próżniową, cylinder azotu lub zawór wentylacji oraz manometr.
Zamiast U-rurki można zastosować manometr ze wskaźnikiem ciśnienia (rysunek 2b).
W celu regulacji temperatury próbki, umieszcza się naczynie próbki wraz z zaworem i U-rurką lub manometrem w łaźni, w której utrzymywana jest stała temperatura z dokładnością ±0,2 K. Pomiary temperatury są prowadzone na zewnętrznej ściance naczynia zawierającego próbkę lub w samym naczyniu.
Pompa próżniowa z pułapką chłodzącą opary jest używana do odpowietrzania przyrządu.
W metodzie 2a prężność pary substancji jest mierzona pośrednio, stosując wskaźnik zera. Wynika to z wzięcia pod uwagę faktu, że gęstość płynu w U-rurce zmienia się przy znacznej zmianie temperatury.
W zależności od zakresu ciśnień oraz chemicznego zachowania się badanej substancji, następujące płyny są odpowiednie do użycia jako wskaźniki zera dla U-rurki: płyny silikonowe, ftalany. Badana substancja nie może w znaczący sposób rozpuszczać się lub reagować z płynem U-rurki.
W zakresie normalnego ciśnienia do 102 Pa, do manometru stosuje się rtęć, podczas gdy płyny silikonowe i ftalany są odpowiednie do użycia poniżej 102. Pa w dół do 10 Pa. Manometry pojemnościowe o podgrzewanej membranie mogą być używane nawet do poniżej 10-1 Pa. Istnieją również czujniki ciśnienia, które mogą być stosowane poniżej 102 Pa.
1.6.2.2. Procedura pomiarowa
Przed pomiarami, wszystkie podzespoły przyrządu pokazanego na rysunku 2 muszą być gruntownie oczyszczone i wysuszone.
W metodzie 2a należy napełnić U-rurkę wybranym płynem, który musi być odgazowany w podwyższonej temperaturze przed rozpoczęciem odczytów.
Badaną substancję umieszcza się w przyrządzie, który jest następnie zamykany, i zmniejsza się wystarczająco temperaturę w celu odgazowania. Temperatura musi być wystarczająco niska dla zapewnienia usunięcia powietrza, ale w przypadku wielokrotnego systemu składników, nie może powodować zmiany składu materiału. Równowaga, jeżeli zachodzi potrzeba, może być uzyskana znacznie szybciej przy pomocy mieszania.
Próbkę można przechłodzić za pomocą ciekłego azotu (uważać, by zapobiec kondensacji powietrza lub płynu pompy) lub mieszaniny etanolu i suchego lodu. Przy pomiarach niskotemperaturowych używać łaźni z regulowaną temperaturą podłączonej do ultrakriomatu.
Otwarcie zaworu nad naczyniem z próbką powoduje włączenie na kilka minut ssania w celu usunięcia powietrza. Następnie zamyka się zawór i zmniejsza temperaturę próbki do najniższego żądanego poziomu. O ile zachodzi taka konieczność, proces odgazowania musi być powtórzony kilka razy.
Gdy próbka jest podgrzewana, prężność pary wzrasta. Zmienia to równowagę cieczy w U-rurce. Celem kompensacji tego zjawiska azot jest podawany do przyrządu poprzez zawór do momentu, gdy wskaźnik ciśnienia płynu ponownie osiągnie zero. Ciśnienie wymagane do tego jest odczytywane precyzyjnym manometrem w temperaturze pokojowej. Niniejsze ciśnienie odpowiada prężności pary substancji w określonej temperaturze pomiaru.
Metoda 2b jest podobna, lecz prężność pary jest odczytywana bezpośrednio.
Zależność temperaturowa prężności pary jest oznaczana w odpowiednio małych przedziałach (około 5 do 10 punktów pomiarowych całego zakresu) do pożądanego maksimum. Sprawdzeniem jest powtórzenie odczytów niskotemperaturowych.
Jeżeli wartości z powtórzonych odczytów nie pokrywają się z krzywą uzyskaną dla wzrostu temperatury, może być to spowodowane jedną z następujących przyczyn:
|
1. |
Próbka wciąż zawiera powietrze (na przykład materiały o wysokiej lepkości) lub nisko wrzące substancje, które jest/są uwalniane w trakcie ogrzewania i mogą być usunięte przez odpompowanie po przechłodzeniu. |
|
2. |
Temperatura chłodzenia nie jest wystarczająco niska. W tym przypadku jako środek chłodzący jest używany ciekły azot. O ile zachodzi przyczyna 1 lub 2, pomiary muszą być powtórzone. |
|
3. |
Substancja ulega reakcji chemicznej w badanym zakresie temperatur (na przykład rozkładowi, polimeryzacji). |
1.6.3. Izoteniskop
Pełny opis tej metody można znaleźć w pozycji bibliograficznej (7). Zasadę działania urządzenia pomiarowego pokazano na rysunku 3. Podobnie jak w przypadku metody statycznej opisanej w ppkt 1.6.2, izoteniskop nadaje się do badań ciał stałych lub cieczy.
W przypadku cieczy sama substancja pełni rolę cieczy, którym napełniony jest pomocniczy manometr. Ilość cieczy, wystarczająca do napełnienia kolby i krótkiego odgałęzienia sekcji manometru, jest umieszczana w izoteniskopie. Izoteniskop podłącza się do systemu próżniowego i odpompowuje, a następnie napełnia się go azotem. Odpompowanie i czyszczenie systemu jest powtarzane dwukrotnie w celu usunięcia resztkowego tlenu. Napełniony izoteniskop umieszcza się w pozycji poziomej, tak by próbka rozpłynęła się cienką warstwą w naczyniu próbki i sekcji manometru (część U). Ciśnienie systemu zmniejsza się do 133 Pa i podgrzewa delikatnie próbkę do rozpoczęcia wrzenia (usunięcie rozpuszczonych związanych gazów). Następnie umieszcza się izoteniskop tak, że próbka powraca do naczynia i krótszego rozgałęzienia manometru, tak by były całkowicie wypełnione cieczą. Ciśnienie utrzymuje się takie jak dla odgazowania; wyciągnięta końcówka naczynia próbki jest podgrzewana małym płomieniem do czasu, gdy uwalniane opary próbki rozszerzą się wystarczająco do wyparcia części próbki z górnej części kolby i ramienia manometru do sekcji manometru izoteniskopu, tworząc wypełnioną parami, wolną od azotu przestrzeń.
Izoteniskop jest następnie umieszczany w łaźni o stałej temperaturze, a ciśnienie azotu ustawia się do momentu wyrównania go z ciśnieniem próbki. Równowaga ciśnień jest wskazywana przez sekcję manometryczną izoteniskopu. W punkcie równowagi, prężność pary azotu jest równa prężności pary substancji.
W przypadku ciał stałych stosowane ciecze manometryczne w zależności od zakresu i temperatury podano w ppkt 1.6.2.1. Odgazowaną cieczą manometryczną napełnia się wybrzuszenie na dłuższym ramieniu izoteniskopu. Następnie ciało stałe, które ma być badane, jest umieszczane w naczyniu i jest odgazowywane w podwyższonej temperaturze. Po tym izoteniskop jest nachylany tak, by ciecz manometryczna mogła przepłynąć do U-rurki. Pomiar prężności pary w funkcji temperatury jest wykonywany zgodnie z ppkt 1.6.2.
1.6.4. Metoda efuzji: waga do oceny prężności pary
1.6.4.1. Przyrząd
Różne wersje przyrządów opisano w literaturze (1). Przyrząd opisany tutaj obejmuje przedstawienie ogólnej zasady (rysunek 4). Rysunek 4 pokazuje główne części przyrządu, zawierającego naczynie do wysokiej próżni ze stali nierdzewnej lub szkła, osprzęt do wytwarzania i pomiaru próżni i wbudowane podzespoły do pomiaru prężności pary w równowadze. W przyrządzie znajdują się wbudowane następujące podzespoły:
|
— |
wyparka z kołnierzem i obrotowym wlotem. Wyparka jest cylindrycznym naczyniem, wykonanym na przykład z miedzi lub ze stopu chemicznie odpornego o dobrym termicznym przewodnictwie. Można również użyć szklane naczynie z miedzianą ścianką. Wyparka posiada średnicę około 3 do 5 cm i jest wysoka na 2 do 5 cm. Znajdują się w niej od jednego do trzech otworów o różnych wymiarach dla strumienia pary. Wyparka jest podgrzewana albo płytą grzejną od spodu, albo spiralą grzejną dookoła powierzchni zewnętrznej. Aby zapobiec rozpraszaniu ciepła do płyty dennej, grzejnik jest podłączony do płyty dennej poprzez metal o niskim przewodnictwie cieplnym (stal niklowosrebrowa lub chromoniklowa), na przykład przez rurkę niklowosrebrową połączoną z obrotowym wlotem, jeśli używa się wyparki o kilku otworach. Takie rozmieszczenie umożliwia wprowadzenie prętów miedzianych, co pozwala na chłodzenie zewnętrzne za pomocą łaźni chłodzącej, |
|
— |
jeżeli wieko miedziane wyparki posiada trzy otwory o różnych średnicach, rozmieszczonych co 90o, można objąć różne zakresy prężności pary z całkowitego zakresu pomiarowego (otwory o średnicy między około 0,30 i 4,50 mm). Duże otwory są stosowane do niskiej prężności pary i odwrotnie. Przez obracanie wyparki można ustawić pożądany otwór lub pozycję pośrednią w strumieniu cząsteczek (otwór wyparki – osłona – szala wagi) uwalnianych lub odchylanych przez otwór wyparki na szalę wagi. W celu pomiaru temperatury substancji umieszcza się termoogniwo lub termometr oporowy w stosownym punkcie, |
|
— |
powyżej osłony znajduje się przedłużenie szalki wagi dla mikrowagi o wysokiej czułości (zob. poniżej). Szala wagi posiada średnicę około 30 mm. Odpowiednim materiałem jest aluminium pokryte złotem, |
|
— |
szala wagi jest otoczona przez cylindryczną, wykonaną z brązu lub miedzi skrzynkę chłodniczą. W zależności od typu wagi posiada ona otwory dla belki wagi i osłonięty otwór dla strumienia cząsteczek i powinna gwarantować całkowitą kondensację par na szalce wagi. Rozpraszanie ciepła na zewnątrz jest zapewnione na przykład przez pręt miedziany połączony ze skrzynką chłodniczą. Pręt jest przeprowadzony przez płytę denną i termicznie od niej odizolowany, na przykład za pomocą rury ze stali chromoniklowej. Pręt jest zanurzony w naczyniu Dewara zawierającym ciekły azot pod płytą denną, bądź ciekły azot cyrkuluje przez pręt. Skrzynka chłodnicza jest utrzymywana w temperaturze około - 120 oC. Szala wagi jest chłodzona wyłącznie przez promieniowanie, co jest zadawalające dla zakresu ciśnień stosowanych do badań (chłodzenie około jednej godziny przed rozpoczęciem pomiarów), |
|
— |
waga jest umieszczana powyżej skrzynki chłodniczej. Odpowiednimi wagami są na przykład wysokoczułe, dwuramienne elektroniczne mikrowagi (8) lub wysokoczuły przyrząd z ruchomą cewką (zob. wytyczne OECD dotyczące badań 104, wydanie z 12.5.81), |
|
— |
płyta denna zawiera również elektryczne połączenia dla termoogniw (lub termometrów oporowych) oraz wężownicę grzejną, |
|
— |
próżnia jest wytwarzana w naczyniu za pomocą pompy podciśnieniowej lub pompy wysokiej próżni (wymagana jest próżnia około 1 do 2 × 10-3 Pa, uzyskiwana po 2 godzinach pompowania). Ciśnienie reguluje się stosownym manometrem jonizacyjnym. |
1.6.4.2. Procedura pomiarowa
Naczynie napełnia się badaną substancją i zamyka pokrywę. Osłona i skrzynka chłodnicza są przesunięte w poprzek wyparki. Przyrząd zamyka się i włącza pompy próżniowe. Ciśnienie końcowe przed rozpoczęciem pomiarów powinno być około 10-4 Pa. Chłodzenie skrzynki chłodniczej rozpoczyna się przy 10-2 Pa.
Po uzyskaniu żądanej próżni rozpoczyna się serie kalibracji od najniższej wymaganej temperatury. Ustawia się odpowiedni otwór w pokrywie, strumień par przechodzi przez osłonę bezpośrednio powyżej otworu i uderza w ochłodzoną szalę wagi. Szala wagi musi być wystarczająco duża dla zapewnienia, że całkowity strumień przechodzący przez osłonę uderzy w nią. Pęd strumienia par działa jako siła na szalę wagi i cząsteczki kondensują się na jej ochłodzonej powierzchni.
Pęd i równoczesna kondensacja wytwarzają sygnał na rejestratorze. Ocena sygnałów dostarcza dwóch rodzajów informacji:
|
1. |
W przyrządzie opisanym tutaj prężność pary jest oznaczana bezpośrednio z pędu na szali wagi (nie jest konieczna do tego znajomość masy cząsteczkowej (2)). Należy wziąć pod uwagę czynniki geometryczne, takie jak otwór wyparki i kąt strumienia cząsteczek przy ocenie odczytów. |
|
2. |
W tym samym czasie można zmierzyć masę kondensatu i obliczyć z niej szybkość parowania. Prężność pary można także obliczyć z szybkości parowania i masę cząsteczkową, stosując równanie Hertza (2). |

gdzie:
|
G |
= |
szybkość parowania (kg s-l m-2), |
|
M |
= |
masa molowa (g mol-l), |
|
T |
= |
temperatura (K), |
|
R |
= |
uniwersalna molowa stała gazowa (Jmol-l K-l), |
|
p |
= |
prężność pary (Pa). |
Po uzyskaniu niezbędnej próżni rozpoczyna się wykonywanie serii pomiarów przy najniższej pożądanej temperaturze pomiarowej.
Dla dalszych pomiarów zwiększa się temperaturę małymi przedziałami do momentu uzyskania maksymalnej pożądanej wartości temperatury. Próbka następnie jest ponownie schładzana i można dokonać zapisu drugiej krzywej ciśnienia pary. Jeżeli w drugiej serii nie uda się potwierdzić wyników pierwszej, możliwe jest, że substancja może ulegać rozkładowi w zakresie temperatur, w których wykonuje się pomiar.
1.6.5. Metoda efuzji – przez utratę masy
1.6.5.1. Przyrząd
Przyrząd efuzyjny składa się z następujących podstawowych części:
|
— |
zbiornik, który może być termostatowany i odpompowywany, w którym umieszczone są komórki efuzyjne, |
|
— |
pompa wysokiej próżni (np. pompa dyfuzyjna lub turbomolekularna) z próżniomierzem, |
|
— |
pułapka wykorzystująca skroplony azot lub suchy lód. |
Elektrycznie podgrzewany aluminiowy zbiornik próżniowy z 4 komórkami efuzyjnymi ze stali nierdzewnej przedstawiono przykładowo na rysunku 5. Folia ze stali nierdzewnej grubości około 0,3 mm posiada otwór efuzyjny o średnicy od 0,2 do 1,0 mm i jest połączona z komórką efuzyjną gwintowaną pokrywą.
1.6.5.2. Procedura pomiarowa
Substancjami, badaną i odniesienia, napełniono każdą komórkę efuzyjną, metalowa membrana z otworem jest zabezpieczona gwintowaną pokrywą, a każda komórka jest ważona z dokładnością 0,1 mg. Komórkę umieszcza się w termostatowanym przyrządzie, który jest następnie odpompowywany do jednej dziesiątej oczekiwanego ciśnienia. W określonych odstępach czasowych z zakresu od 5 do 30 godzin wpuszcza się do przyrządu powietrze i określa utratę masy komórki efuzyjnej przez ponowne ważenie.
W celu zapewnienia, że wyniki nie są zakłócone przez zanieczyszczenia lotne, komórka jest ponownie ważona w określonych odstępach czasowych, celem sprawdzenia, czy szybkość parowania jest stała w co najmniej dwóch takich odstępach czasowych.
Prężność pary p w komórce efuzyjnej jest dana przez:

gdzie :
|
p |
= |
prężność pary (Pa), |
|
m |
= |
masa substancji opuszczającej komórkę w ciągu czasu t (kg), |
|
t |
= |
czas (s), |
|
A |
= |
powierzchnia otworu (m2), |
|
K |
= |
współczynnik korekcji, |
|
R |
= |
uniwersalna stała gazowa (Jmol-l K-1), |
|
T |
= |
temperatura (K), |
|
M |
= |
masa cząsteczkowa (kg mol-1). |
Współczynnik korekcji K zależy od stosunku długości do promienia otworu cylindrycznego:
|
stosunek |
0,1 |
0,2 |
0,6 |
1,0 |
2,0 |
|
K |
0,952 |
0,909 |
0,771 |
0,672 |
0,514 |
Powyższe równanie może być zapisane:

gdzie:  jest stałą komórki efuzyjnej.
jest stałą komórki efuzyjnej.
Niniejsza stała komórki efuzyjnej może być oznaczona przy pomocy substancji odniesienia (2,9), używając następującego równania:

gdzie:
|
p(r) |
= |
prężność pary substancji odniesienia (Pa), |
|
M(r) |
= |
masa cząsteczkowa substancji odniesienia (kg mol-l). |
1.6.6. Metoda nasycenia gazem
1.6.6.1. Przyrząd
Typowy przyrząd stosowany do wykonywania tego badania składa się z szeregu elementów podanych na rysunku 6a i opisanych poniżej (1).
Gaz obojętny:
Gaz nośnikowy nie może wchodzić w reakcje chemiczne z substancją badaną. Na ogół do tego celu wystarcza azot, jednak czasem mogą być wymagane inne gazy (10). Zastosowany gaz musi być suchy (zob. rysunek 6a, pozycja 4: czujnik wilgotności względnej).
Kontrola przepływu:
Wymagany jest odpowiedni system kontroli przepływu gazu, aby zapewnić stały i odpowiednio dobrany przepływ przez kolumnę saturatora.
Pułapki do zbierania pary:
Zależą one od charakterystyki określonej próbki oraz wybranej metody analizy. Pary powinny być zbierane w sposób ilościowy, w postaci umożliwiającej późniejszą analizę. Do niektórych badanych substancji nadają się pułapki zawierające ciecze, takie jak heksan lub glikol etylenowy. W przypadku innych mogą być stosowane odpowiednie absorbenty w stałym stanie skupienia.
Jako alternatywę dla pułapkowania par i dalszych analiz można zastosować techniki analityczne transportowe, takie jak chromatografia, do oznaczania ilościowego ilości materiału przeniesionego przez znaną ilość gazu nośnikowego. Następnie mierzona jest utrata masy badanej próbki.
Wymiennik ciepła:
W celu wykonywania pomiarów w różnych temperaturach może być konieczne włączenie do zestawu wymiennika ciepła.
Kolumna saturatora:
Badana substancja jest osadzana z roztworu na odpowiedni obojętny nośnik. Powleczonym nośnikiem jest wypełniana kolumna saturatora, której wymiary i szybkość przepływu powinny być takie, aby zapewnić pełne nasycenie gazu nośnikowego. Kolumna saturatora musi być termostatowana. Przy pomiarach powyżej temperatury pokojowej obszar między kolumną saturatora i pułapkami musi być podgrzewany, aby zapobiec kondensacji badanej substancji.
W celu zmniejszenia masy przenoszonej wskutek dyfuzji, należy umieścić kapilarę za kolumną saturatora (rysunek 6b).
1.6.6.2. Procedura pomiarowa
Przygotowanie kolumny saturatora:
Do odpowiedniej ilości nośnika dodaje się roztwór substancji badanej w wysoce lotnym rozpuszczalniku. Należy dodać wystarczającą ilość substancji badanej do utrzymania nasycenia przez cały czas trwania badania. Rozpuszczalnik ulega całkowitemu odparowaniu w powietrzu lub w obrotowym przyrządzie wyparnym, a starannie zmieszany materiał jest dodawany do kolumny saturatora. Po termostatycznym ustaleniu temperatury próbki przez przyrząd przepuszcza się suchy azot.
Pomiar:
Pułapki lub impulsowy wskaźnik są przyłączone do linii wycieku kolumny i zapisywanego czasu. Sprawdza się szybkość przepływu na początku i w regularnych odstępach czasu w trakcie eksperymentu, przy użyciu licznika pęcherzyków (lub w sposób ciągły przy użyciu przepływomierza masowego).
Należy wykonywać pomiary ciśnienia u wylotu do saturatora. Można to wykonać albo:
|
a) |
przez włączenie miernika ciśnienia pomiędzy saturator i pułapki (może być to niezadowalające, ponieważ powoduje to wzrost martwej przestrzeni i powierzchni adsorbującej); lub |
|
b) |
przez ustalenie spadków ciśnienia w określonym układzie pułapek jako funkcji przepływu w oddzielnym eksperymencie (metoda ta może nie sprawdzać się zbyt dobrze w przypadku pułapek płynnych). |
Czas wymagany do zebrania ilości substancji badanej niezbędnej dla różnych metod analitycznych zostaje ustalony we wstępnych seriach badań lub szacunkowo. Alternatywą dla zbierania substancji do dalszej analizy jest ilościowa technika analityczna impulsowa (np. chromatografia). Przed obliczeniem prężności pary w określonej temperaturze należy przeprowadzić wstępne serie badań w celu ustalenia maksymalnego przepływu, który doprowadzi do pełnego nasycenia gazu nośnikowego parami substancji. Jest to gwarantowane, jeżeli gaz nośnikowy przepuści się przez saturator wystarczająco powoli, tak aby obniżenie szybkości nie powodowało już zwiększenia obliczonej prężności pary.
Określona metoda analityczna zostaje ustalona w zależności od charakteru badanej substancji (np. chromatografia gazowa lub grawimetria).
Oznacza się ilość substancji transportowaną przez znaną objętość gazu nośnikowego.
1.6.6.3. Obliczenie prężności pary
Prężność pary oblicza się na podstawie gęstości pary, W/V, przy użyciu następującego wzoru:

gdzie:
|
P |
= |
prężność pary (Pa), |
|
W |
= |
masa odparowanej substancji badanej (g), |
|
V |
= |
objętość nasyconego gazu (m3), |
|
R |
= |
uniwersalna molowa stała gazowa (Jmol-l K-1), |
|
T |
= |
temperatura (K), |
|
M |
= |
masa molowa badanej substancji (g mol-l). |
Zmierzone objętości należy skorygować względem różnic ciśnienia i temperatury między przepływomierzem i termostatowanym saturatorem. Jeżeli przepływomierz jest zlokalizowany w kierunku z prądem od pułapki pary, mogą być niezbędne poprawki w celu uwzględnienia wszelkich odparowanych składników pułapki (1).
1.6.7. Metoda wirującego rotora (8, 11, 13)
1.6.7.1. Przyrząd
Technika wirującego rotora jest wykonywana poprzez użycie miernika lepkości z wirującym rotorem, jak pokazano na rysunku 8. Schematyczny szkic doświadczalnego ustawienia pokazano na rysunku 7.
Przyrząd pomiarowy typowo składa się z głowicy pomiarowej wirującego rotora, umieszczonej w termostatowanej obudowie (regulowanej z dokładnością w zakresie 0,1 oC), pojemnika próbki umieszczonego w termostatowanej obudowie (regulacja z dokładnością w zakresie 0,01 oC) oraz wszystkich innych części do ustawienia, które utrzymywane są w wyższej temperaturze, by zapobiec kondensacji. Pompa wysokiej próżni jest włączona do systemu za pomocą zaworów wysokiej próżni.
Głowica pomiarowa wirującego rotora składa się ze stalowej kuli (o średnicy od 4 do 5 mm) w rurze. Kula jest zawieszona i stabilizowana w polu magnetycznym, głównie za pomocą kombinacji stałych pól magnetycznych i cewek sterujących.
Kula jest wprowadzana w wirowanie polami obrotowymi wytwarzanymi przez cewki. Cewki czujnikowe, mierzące zawsze obecne poprzeczne niskie namagnesowanie kuli, pozwalają na mierzenie szybkości wirowania.
1.6.7.2. Procedura pomiarowa
Gdy kula osiąga daną szybkość obrotową v(o) (zwykle około 400 obrotów na sekundę), dalsze pobudzanie jest wstrzymywane i zachodzi zmniejszanie szybkości z powodu tarcia gazu.
Spadek szybkości obrotowej jest mierzony w funkcji czasu. Ponieważ tarcie spowodowane zawieszeniem magnetycznym jest mało istotne w porównaniu z tarciem gazu, ciśnienie gazu p jest dane przez:
![]()
gdzie:
|
|
= |
średnia szybkość cząsteczek gazu, |
|
r |
= |
promień kuli, |
|
ρ |
= |
gęstość kuli, |
|
σ |
= |
współczynnik przeniesienia stycznego pędu (ε = 1 dla idealnie sferycznej powierzchni kuli), |
|
t |
= |
czas, |
|
v(t) |
= |
szybkość obrotowa po czasie t, |
|
v(o) |
= |
początkowa szybkość obrotowa, |
zatem równanie można zapisać także:

gdzie tn, tn-1 są czasami wymaganymi dla danej liczby N obrotów. Te przedziały czasowe tn i tn-1 następują jeden po drugim oraz tn > tn-1
Średnia szybkość cząsteczki gazu  jest dana przez:
jest dana przez:

gdzie:
|
T |
= |
temperatura, |
|
R |
= |
uniwersalna molowa stała gazowa, |
|
M |
= |
masa molowa. |
2. DANE
Ciśnienie pary obliczone zgodnie z dowolną z poprzednio opisanych metod należy określić dla co najmniej dwóch temperatur. Najkorzystniejsze jest wykonanie trzech lub większej liczby pomiarów w amplitudzie od 0 do 50 oC w celu sprawdzenia liniowości krzywej zmian prężności pary.
3. SPORZĄDZANIE SPRAWOZDANIA
Sprawozdanie z badań zawiera w miarę możliwości następujące informacje:
|
— |
zastosowana metoda, |
|
— |
precyzyjna specyfikacja substancji (tożsamość i zanieczyszczenia) i wstępny etap oczyszczenia, jeśli wykonano, |
|
— |
co najmniej dwie wartości prężność pary i temperatury, najlepiej z zakresu od 0 do 50 oC, |
|
— |
wszystkie dane pierwotne, |
|
— |
krzywa zależności log p od 1/T, |
|
— |
oszacowanie prężności pary w 20 lub 25 oC. |
Jeżeli obserwowane jest przejście (zmiana stanu skupienia, rozkład), należy odnotować następujące informacje:
|
— |
charakter zmiany, |
|
— |
temperatura, w której dochodzi do zmiany pod ciśnieniem atmosferycznym, |
|
— |
prężność pary w temperaturze o 10 i 20 oC niższej od temperatury przejścia oraz w temperaturze wyższej o 10 i 20 oC od tej temperatury (chyba że przejście następuje ze stanu stałego do gazowego). |
Należy podać wszystkie informacje i uwagi istotne dla interpretacji wyników, w szczególności w odniesieniu do zanieczyszczeń i stanu skupienia substancji.
4. LITERATURA
|
(1) |
OECD, Paris, 1981, Test Guideline 104, Decision of the Council C(81) 30 final. |
|
(2) |
Ambrose, D. in B. Le Neindre, B. Vodar, (Eds.): Experimental Thermodynamics, Butterworths, London, 1975, vol. II. |
|
(3) |
R. Weissberger ed.: Technique of Organic Chemistry, Physical Methods of Organic Chemistry, 3rd ed. Chapter IX, Interscience Publ., New York, 1959, vol. I, part I. |
|
(4) |
Knudsen, M. Ann. Phys. Lpz., 1909, vol. 29, 1979; 1911, vol. 34, 593. |
|
(5) |
NF T 20-048 AFNOR (Sept. 85). Chemical products for industrial use -Determination of vapour pressure of solids and liquids within range from 10-1 to 105 Pa – Static method. |
|
(6) |
NF T 20-047 AFNOR (Sept. 85). Chemical products for industrial use -Determination of vapour pressure of solids and liquids within range from 10-3 to 1 Pa – Vapour pressure balance method. |
|
(7) |
ASTM D 2879-86, Standard test method for vapour pressure- temperature relationship and initial decomposition temperature of liquids by isoteniscope. |
|
(8) |
G. Messer, P. Rohl, G. Grosse and W. Jitschin. J. Vac. Sci. Technol. (A), 1987, vol. 5 (4), 2440. |
|
(9) |
Ambrose, D.; Lawrenson, I.J.; Sprake, C.H.S. J. Chem. Thermodynamics 1975, vol. 7, 1173. |
|
(10) |
B.F. Rordorf. Thermochimica Acta, 1985, vol. 85, 435. |
|
(11) |
G. Comsa, J.K. Fremerey and B. Lindenau. J. Vac. Sci. Technol., 1980, vol. 17 (2), 642. |
|
(12) |
G. Reich. J. Vac. Sci. Technol., 1982, vol. 20 (4), 1148. |
|
(13) |
J.K. Fremerey. J. Vac. Sci. Technol. (A), 1985, vol. 3 (3), 1715. |
Dodatek 1
Metoda szacowania
WPROWADZENIE
Obliczone wartości prężności pary stosuje się:
|
— |
w celu zdecydowania, która z metod doświadczalnych jest odpowiednia, |
|
— |
w celu dostarczenia oszacowania lub granicznej wartości w przypadkach, gdy metoda doświadczalna nie może być zastosowana z powodu przyczyn technicznych (włączając te przypadki, gdzie prężność pary jest bardzo niska), |
|
— |
do pomocy w identyfikacji tych przypadków, gdzie pominięcie pomiarów doświadczalnych jest uzasadnione ponieważ prężność pary wynosi prawdopodobnie < 10-5 Pa w temperaturze otoczenia. |
METODA SZACOWANIA
Prężność pary cieczy i ciał stałych można oszacować poprzez użycie zmodyfikowanej korelacji Watsona (a). Jedyną tylko wymaganą daną doświadczalną jest temperatura wrzenia w warunkach normalnych. Metoda ma zastosowanie w zakresie ciśnień od 105 do 10-5 Pa.
Szczegółowe informacje na temat metody podano w „Handbook of Chemical Property Estimation Methods” (b).
PROCEDURA OBLICZENIA
Zgodnie z pozycją (b) prężność pary jest obliczana, jak następuje:

gdzie:
|
T |
= temperatura będąca przedmiotem zainteresowania, |
|
Tb |
= temperatura wrzenia w warunkach normalnych, |
|
Pvp |
= prężność pary w temperaturze T, |
|
ΔHvb |
= ciepło parowania, |
|
ΔZb |
= współczynnik ściśliwości (oszacowany na 0,97), |
|
m |
= empiryczny współczynnik zależny od stanu fizycznego w temperaturze będącej przedmiotem zainteresowania. |
Dalej

gdzie KF jest współczynnikiem empirycznym biorącym pod uwagę polarność substancji. Dla kilku rodzajów związków współczynniki KF są zamieszczone w (b).
Całkiem często są dostępne dane, w których podano temperaturę wrzenia pod zredukowanym ciśnieniem. W takim przypadku, zgodnie z (b), prężność pary oblicza się w następujący sposób:
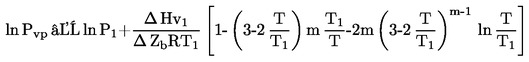
gdzie T1 jest temperaturą wrzenia pod zredukowanym ciśnieniem P1.
SPRAWOZDANIE
Jeżeli zastosowano metodę szacowania, sprawozdanie powinno zawierać obszerną dokumentację obliczeń.
LITERATURA
|
(a) |
K.M. Watson, Ind. Eng. Chem; 1943, vol. 35, 398. |
|
(b) |
W.J. Lyman, W.F. Reehl, D.H. Rosenblatt. Handbook of Chemical Property Estimation Methods, Mc Graw-Hill, 1982. |
Dodatek 2
Rysunek 1
Przyrząd do wyznaczania krzywej prężności pary zgodnie z metodą dynamiczną

Rysunek 2a
Przyrząd dla wyznaczania krzywej prężności pary zgodnie z metodą statyczną (z użyciemmanometru w postaci U-rurki)

Rysunek 2b
Przyrząd dla wyznaczania krzywej prężności pary zgodnie z metodą statyczną (z użyciemwskaźnika ciśnienia)

Rysunek 3
Izoteniskop (zob. pozycja bibliograficzna 7)
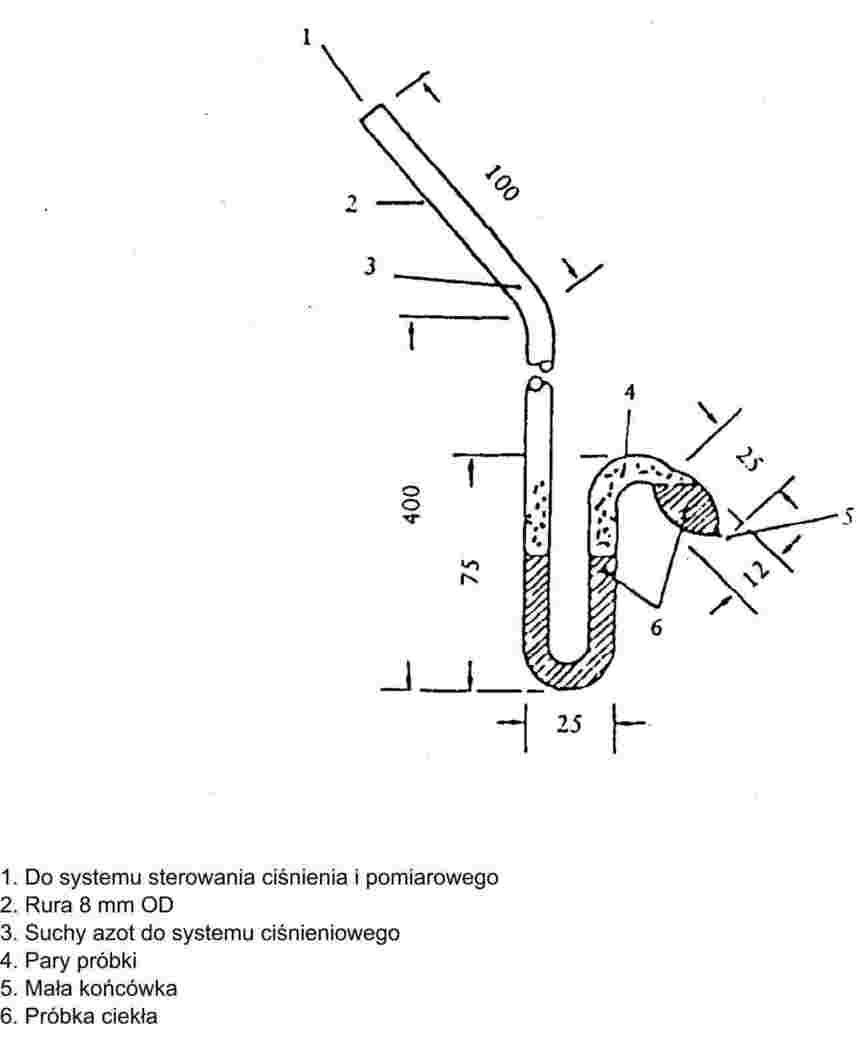
Rysunek 4
Przyrząd dla wyznaczania krzywej prężności pary zgodnie z metodą wagową

Rysunek 5
Przykład przyrządu do odparowania w niskim ciśnieniu metodą efuzji, z komórką efuzyjną o objętości 8 cm3

Rysunek 6a
Przykład systemu przepływu do oznaczania prężności pary metodą saturacji gazem

Rysunek 6b
Przykład systemu do oznaczania prężności pary metodą saturacji gazem, z kapilarą umieszczoną za komorą saturacji
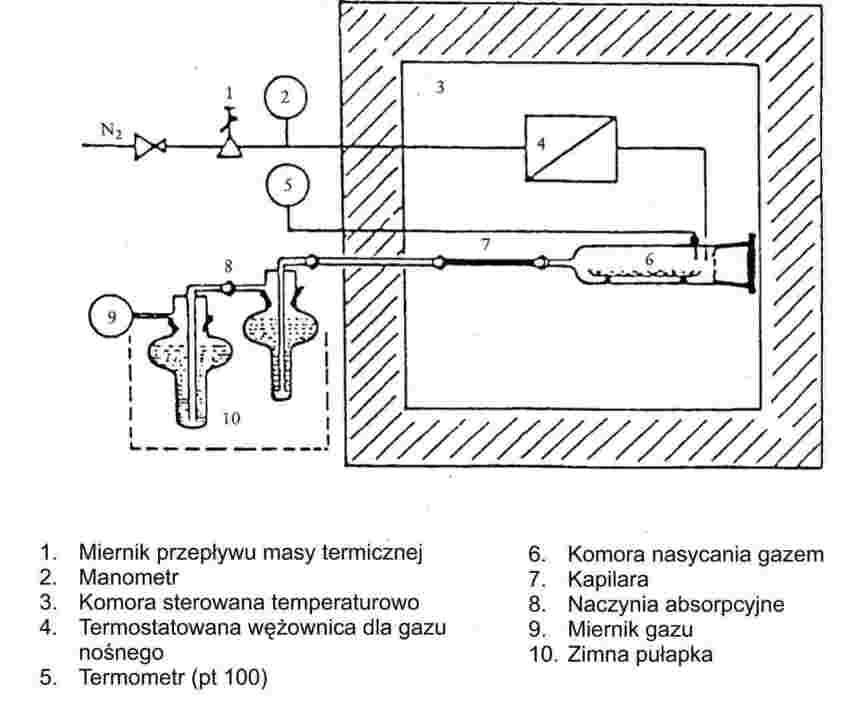
Rysunek 7
Przykład doświadczalnego zestawu dla metody wirującego rotora
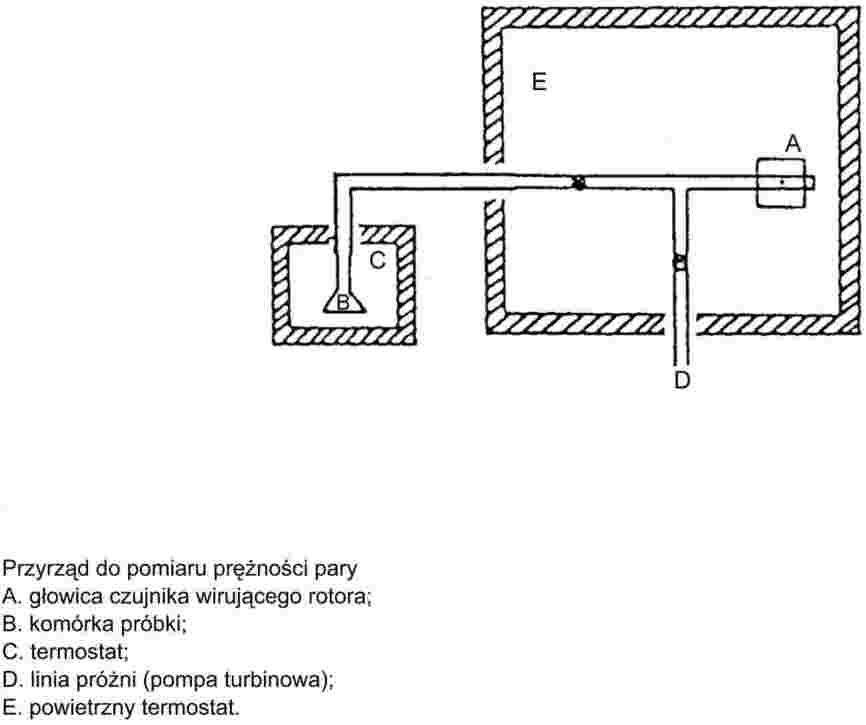
Rysunek 8
Przykład głowicy pomiarowej wirującego rotora

A.5. NAPIĘCIE POWIERZCHNIOWE
1. METODA
Większość opisanych metod oparto na wytycznych OECD dotyczących badań (1). Podstawowe zasady podano w pozycji bibliograficznej (2).
1.1. WPROWADZENIE
Opisywane metody są stosowane do pomiarów napięcia powierzchniowego roztworów wodnych.
Przed przeprowadzeniem tych badań dobrze jest mieć wstępne informacje na temat rozpuszczalności w wodzie, budowy, właściwości hydrolitycznych i stężenia krytycznego dla tworzenia się micelli substancji.
Poniższe metody można stosować w odniesieniu do większości substancji chemicznych, bez jakichkolwiek ograniczeń związanych z ich stopniem czystości.
Pomiar napięcia powierzchniowego metodą tensjometru pierścieniowego jest zastrzeżony wyłącznie dla roztworów wodnych o lepkości dynamicznej mniejszej niż około 200 mPa s.
1.2. DEFINICJE IJEDNOSTKI
Jako napięcie powierzchniowe określa się entalpię wolnej powierzchni na jednostkę pola powierzchni.
Napięcie powierzchniowe jest podawane jako:
N/rn (układ SI) lub
mN/m (układ SI)
1 N/m = 103 dyn/cm
1 mN/m = 1 dyna/cm w nieważnym systemie CGS
1.3. SUBSTANCJE ODNIESIENIA
Nie ma konieczności stosowania substancji odniesienia za każdym razem, gdy przeprowadzane są badania nowej substancji. Powinny one służyć głównie sprawdzeniu od czasu do czasu wydajności metody i pozwolić na porównanie wyników z innymi metodami.
Substancje odniesienia, które obejmują szeroki zakres napięć powierzchniowych, podano w pozycjach bibliograficznych (1) i (3).
1.4. ZASADA METOD
Metody te opierają się na pomiarach maksymalnej siły, jaką trzeba wywierać pionowo na mieszadło lub pierścień w kontakcie z powierzchnią badanej cieczy umieszczonej naczyniu miarowym, aby oddzielić ją od tej powierzchni, bądź też na płytkę, której krawędź wchodzi w kontakt z powierzchnią, aby pociągnąć do góry utworzoną błonkę.
Substancje o rozpuszczalności w wodzie przynajmniej w stężeniu 1 mg/l badane są w roztworach wodnych w pojedynczych stężeniach.
1.5. KRYTERIA JAKOŚCI
Metody te zapewniają większą precyzję niż wymagana na ogół do ocen przy przeglądach środowiskowych.
1.6. OPIS METOD
Roztwór substancji przygotowuje się w wodzie destylowanej. Stężenie tego roztworu powinno wynosić 90 % rozpuszczalności nasycenia w wodzie, jeżeli to stężenie przekracza 1 g/l, do badania stosuje się stężenie 1 g/l. Nie podlegają badaniu substancje, których rozpuszczalność w wodzie jest mniejsza niż 1 mg/l.
1.6.1. Metoda płytkowa
Zob. ISO 304 i NF T 73-060 (Surface active agents – determination of surface tension by drawing up liquid films).
1.6.2. Metoda zawieszki
Zob. ISO 304 and NF T 73-060 (Surface active agents – determination of surface tension by drawing up liquid films).
1.6.3. Metoda pierścieniowa
Zob. ISO 304 and NF T 73-060 (Surface active agents – determination of surface tension by drawing up liquid films).
1.6.4. Zharmonizowana metoda pierścieniowa OECD
1.6.4.1. Przyrząd
Do tego pomiaru można wykorzystać dostępne w handlu tensjometry. Składają się one z następujących elementów:
|
— |
ruchomy stół próbki, |
|
— |
system pomiaru siły, |
|
— |
ciało pomiarowe (pierścień), |
|
— |
naczynie pomiarowe. |
1.6.4.1.1.
Ruchomy stół z próbką jest wykorzystywany jako wspornik dla naczynia pomiarowego z kontrolowaną temperaturą, w którym znajduje się badana ciecz. Razem z układem do pomiaru siły stół ten jest montowany na stojaku.
1.6.4.1.2.
Układ do pomiaru siły (zob. rysunek) mieści się ponad stołem z próbką. Błąd pomiaru siły nie może przekraczać ± 10-6 N, co odpowiada granicy błędu wynoszącej ±0,1 mg w przypadku pomiaru masy. W większości przypadków skalę pomiarową dostępnych na rynku tensjometrów kalibruje się w mN/m, tak aby napięcie powierzchniowe można było odczytywać bezpośrednio w mN/m, z dokładnością do 0,1 mN/m.
1.6.4.1.3.
Pierścień na ogół jest wykonany z drutu platynowo-irydowego o grubości około 0,4 mm i średnim obwodzie 60 mm. Pierścień drutu zwiesza się horyzontalnie z metalowego sworznia i drucianego wspornika, tak aby stworzyć połączenie z układem do pomiaru siły (zob. rysunek).
Rysunek
Ciało pomiarowe
(Wszystkie wymiary w milimetrach)

1.6.4.1.4.
Naczynie pomiarowe z badanym roztworem musi być naczyniem szklanym z kontrolowaną temperaturą. Musi być tak zaprojektowane, aby w trakcie pomiaru temperatura roztworu badanego i fazy gazowej nad jego powierzchnią pozostały stałe, a próbka nie mogła wyparować. Dopuszczalne są cylindryczne, szklane naczynia o średnicy wewnętrznej nie mniejszej niż 45 mm.
1.6.4.2. Przygotowanie przyrządu
1.6.4.2.1.
Szklane naczynia muszą być starannie oczyszczone. W razie potrzeby należy je przepłukać gorącym kwasem chromowo-siarkowym, a następnie kwasem fosforowym o konsystencji syropu (od 83 do 98 % wagowo H3PO4), potem starannie przepłukać pod wodą z kranu, a wreszcie umyć dwukrotnie destylowaną wodą do uzyskania reakcji obojętnej, a na końcu wysuszyć lub przepłukać częścią płynnej próbki mierzonej.
Pierścień należy najpierw starannie opłukać w wodzie w celu usunięcia wszelkich substancji rozpuszczalnych w tej cieczy, następnie na krótki czas zanurzyć w kwasie chromowo-siarkowym, później umyć dwukrotnie destylowaną wodą do uzyskania reakcji obojętnej, a na końcu krótko podgrzać nad płomieniem metanolowym.
Uwaga:
Zanieczyszczenia substancjami, które nie ulegają rozpuszczeniu ani zniszczeniu wskutek oddziaływania kwasu chromo-siarkowego lub fosforowego, takimi jak silikony, należy usunąć przy pomocy odpowiedniego rozpuszczalnika organicznego.
1.6.4.2.2.
Weryfikacja przyrządu polega na sprawdzeniu punktu zerowego i takim jego wyregulowaniu, aby wskazania instrumentu pozwalały na niezawodne oznaczenia w mN/m.
Montaż:
Przyrząd musi być wypoziomowany, na przykład przy pomocy poziomnicy alkoholowej, u podstawy tensjometru, przez wyregulowanie śrub poziomujących u podstawy.
Ustawienie punktu zero:
Po zamontowaniu pierścienia na przyrządzie, przed jego zanurzeniem w cieczy, wskazania tensjometru należy wyregulować do zera i sprawdzić, czy pierścień jest ustawiony równolegle do powierzchni cieczy. W tym celu powierzchnię cieczy można wykorzystać jako lustro.
Kalibracje:
Kalibrację przy pomocy rzeczywistego badania można dokonać przy pomocy jednej z dwóch procedur:
|
a) |
używając masę: procedura wykorzystująca koniki wagi o znanej masie od 0,1 do 1,0 g umieszczane na pierścieniu. Współczynnik kalibracyjny Φa, przez który należy pomnożyć wszystkie wskazania instrumentu, należy ustalić według następującego wzoru (1):
gdzie:
|
|
b) |
używając wodę: procedura stosująca czystą wodę, której napięcie powierzchniowe przy na przykład 23 oC jest równe 72,3 mN/m. Niniejsza procedura jest wykonywana szybciej niż kalibracja wagowa, lecz występuje przy niej zawsze niebezpieczeństwo, że napięcie powierzchniowe wody jest zafałszowane śladowymi zanieczyszczeniami związków powierzchniowo czynnych. Współczynnik kalibracyjny Φb, przez który należy pomnożyć wszystkie wskazania instrumentu, należy ustalić według następującego wzoru (2):
gdzie:
|
1.6.4.3. Przygotowanie próbek
Należy sporządzić roztwory wodne badanych substancji, używając odpowiednich stężeń w wodzie. Nie mogą one zawierać substancji w stanie nierozpuszczonym.
Roztwór należy utrzymywać w stałej temperaturze (±0,5 oC). Ponieważ napięcie powierzchniowe roztworu w naczyniu pomiarowym zmienia się w czasie, należy wykonać kilka pomiarów w różnych momentach i nakreślić krzywą pokazującą napięcie powierzchniowe w funkcji czasu. Brak jakichkolwiek dalszych zmian oznacza, że osiągnięto stan równowagi.
Zanieczyszczenie pyłami i gazami innych substancji przeszkadza w pomiarze. Dlatego badanie musi być prowadzone pod przykrywą ochronną.
1.6.5. Warunki badania
Pomiar należy przeprowadzić w temperaturze około 20 oC, która musi być kontrolowana w ten sposób, aby utrzymywała się w zakresie ±0,5 oC.
1.6.6. Wykonanie badania
Roztwory do pomiaru należy wprowadzić do starannie wyczyszczonego naczynia pomiarowego, dokładając wszelkich starań, aby uniknąć wytwarzania piany, a następnie naczynie umieszcza się na stoliku przyrządu do badań. Blat stolika z naczyniem pomiarowym należy unieść do momentu zanurzenia pierścienia poniżej powierzchni roztworu badanego. Następnie blat stolika opuszcza się stopniowo i równo (z szybkością około 0,5 cm/min), tak aby oddzielić pierścień od powierzchni, do momentu osiągnięcia maksymalnej siły. Warstwa cieczy dołączona do pierścienia nie może się od niego oddzielić. Po wykonaniu pomiarów pierścień należy ponownie zanurzyć poniżej powierzchni i powtórzyć pomiary, aż zostanie osiągnięta stała wartość napięcia powierzchniowego. W przypadku każdego oznaczenia należy zapisać czas odprzeniesienia roztworu do naczynia pomiarowego. Odczyty należy wykonywać przy maksymalnej sile potrzebnej do oderwania pierścienia od powierzchni cieczy.
2. DANE
W celu obliczenia napięcia powierzchniowego wartość odczytaną w mN/m na przyrządzie należy na początku pomnożyć przez współczynnik kalibracyjny Φa lub Φb (zależnie od zastosowanej procedury kalibracyjnej). Da to wartość jedynie przybliżoną, dlatego wymaga korekcji.
Harkins i Jordan (4) empirycznie ustalili współczynniki korekcji dla wartości napięcia powierzchniowego zmierzonych metodą pierścienia. Zależą one od wymiarów pierścienia, gęstości cieczy i napięcia powierzchniowego tej ostatniej.
Ponieważ ustalanie współczynnika korekcji dla każdego kolejnego pomiaru na podstawie tabel Harkinsa i Jordana jest pracochłonne, do obliczenia napięcia powierzchniowego roztworów wodnych można wykorzystać procedurę uproszczoną wykonywania odczytów skorygowanych wartości napięcia powierzchniowego bezpośrednio z tabeli. (W przypadku gdy odczytane wartości znajdują się między tymi, które podano w tabeli, należy zastosować interpolację).
Tabela
Korekcja zmierzonego napięcia powierzchniowego
Tylko dla roztworów wodnych, ρ ≅ 1 g/cm3
|
r |
= 9,55 mm (średni promień pierścienia) |
|
r |
= 0,185 mm (promień drutu pierścienia) |
|
Wartość doświadczalna (mN/m) |
Wartość skorygowana (mN/m) |
|
|
Kalibracja wagowa (zob. 1.6.4.2.2 lit. a)) |
Kalibracja wodą (zob. 1.6.4.2.2 lit.b)) |
|
|
20 |
16,9 |
18,1 |
|
22 |
18,7 |
20,1 |
|
24 |
20,6 |
22,1 |
|
26 |
22,4 |
24,1 |
|
28 |
24,3 |
26,1 |
|
30 |
26,2 |
28,1 | |
|
32 |
28,1 |
30,1 |
|
34 |
29,9 |
32,1 |
|
36 |
31,8 |
34,1 |
|
38 |
33,7 |
36,1 |
|
40 |
35,6 |
38,2 |
|
42 |
37,6 |
40,3 |
|
44 |
39,5 |
42,3 |
|
46 |
41,4 |
44,4 |
|
48 |
43,4 |
46,5 |
|
50 |
45,3 |
48,6 |
|
52 |
47,3 |
50,7 |
|
54 |
49,3 |
52,8 |
|
56 |
51,2 |
54,9 |
|
58 |
53,2 |
57,0 |
|
60 |
55,2 |
59,1 |
|
62 |
57,2 |
61,3 |
|
64 |
59,2 |
63,4 |
|
66 |
61,2 |
65,5 |
|
68 |
63,2 |
67,7 |
|
70 |
65,2 |
69,9 |
|
72 |
67,2 |
72,0 |
|
74 |
69,2 |
— |
|
76 |
71,2 |
— |
|
78 |
73,2 |
— |
Niniejsza tabela została zestawiona na podstawie poprawki Harkinsa-Jordana. Jest ona zbliżona do tej w normie (DIN 53914) dla wody i roztworów wodnych (gęstość ρ = 1 g/cm3) i dla dostępnych w handlu pierścieni o wymiarach R = 9,55 mm (średni promień pierścienia) i r = 0,185 mm (promień drutu pierścienia). W tabeli podano skorygowane wartości pomiarów napięcia powierzchniowego wykonanych po kalibracji odważnikowej lub przy pomocy wody.
Alternatywnie, bez poprzednio opisanej kalibracji, napięcie powierzchniowe można obliczyć na podstawie następującego wzoru:

gdzie:
|
F |
= |
siła zmierzona dynamometrem przy zerwaniu błonki, |
|
R |
= |
promień pierścienia, |
|
f |
= |
współczynnik korekcji (1). |
3. SPORZĄDZENIE SPRAWOZDANIA
3.1. SPRAWOZDANIE Z BADANIA
Sprawozdanie z badania zawiera w miarę możliwości następujące informacje:
|
— |
zastosowana metoda, |
|
— |
rodzaj zastosowanej wody lub zastosowanego roztworu, |
|
— |
precyzyjna specyfikacja substancji (tożsamość i zanieczyszczenia), |
|
— |
wyniki pomiaru: napięcie powierzchniowe (odczyt), z podaniem zarówno poszczególnych odczytów, jak i ich średniej arytmetycznej oraz skorygowanej średniej (z wzięciem pod uwagę współczynnika przyrządu i tabeli korekcji), |
|
— |
stężenie roztworu, |
|
— |
temperatura badania, |
|
— |
wiek zastosowanego roztworu; w szczególności czas od przygotowania do zmierzenia właściwości roztworu, |
|
— |
opis zależności napięcia powierzchniowego od czasu po przeniesieniu roztworu do naczynia pomiarowego, |
|
— |
należy podać wszystkie informacje i uwagi istotne dla interpretacji wyników, w szczególności w odniesieniu do zanieczyszczeń i stanu skupienia substancji. |
3.2. INTERPRETACJA WYNIKÓW
Zważywszy że destylowana woda posiada napięcie powierzchniowe 72,75 mN/m w 20 oC, substancje wykazujące napięcie powierzchniowe niższe niż 60 mN/m zgodnie z warunkami niniejszej metody należy uznać za materiały będące powierzchniowo czynnymi.
4. LITERATURA
|
(1) |
OECD, Paris, 1981, Test Guideline 115, Decision of the Council C(81) 30 final. |
|
(2) |
R. Weissberger ed.: Technique of Organic Chemistry, Physical Methods of Organic Chemistry, 3rd ed., Interscience Publ., New York, 1959, vol. I, part I, chapter XIV. |
|
(3) |
Pure Appl. Chem., 1976, vol. 48, 511. |
|
(4) |
Harkins, W.D., Jordan, H.F., J. Amer. Chem. Soc., 1930, vol. 52, 1751. |
A.6. ROZPUSZCZALNOŚĆ W WODZIE
1. METODA
Opisane metody są oparte na wytycznych OECD dotyczących badań (1).
1.1. WPROWADZENIE
Do przeprowadzenia tego badania przydatne jest posiadanie wstępnych informacji na temat wzoru strukturalnego, ciśnienia pary, stałej dysocjacji i hydrolizy (jako funkcji pH) substancji.
Nie ma pojedynczej metody, która objęłaby cały zakres rozpuszczalności w wodzie.
Dwie metody badań opisane poniżej obejmują cały zakres rozpuszczalności, lecz nie stosuje sie ich do substancji lotnych:
|
— |
jedna z nich dotyczy zasadniczo czystych substancji o niskiej rozpuszczalności (< 10-2 g/l) które są stabilne w wodzie; określa się ją jako „metodę wymywania kolumnowego”, |
|
— |
druga dotyczy zasadniczo czystych substancji o wyższej rozpuszczalności (> 10-2 g/l), które są stabilne w wodzie; określa się ją jako „metodę kolby”. |
Na rozpuszczalność w wodzie badanej substancji w sposób istotny może wpłynąć obecność zanieczyszczeń.
1.2. DEFINICJA I JEDNOSTKI
Rozpuszczalność w wodzie substancji podaje się jako stężenie substancji w wodzie przy nasyceniu masowym w określonej temperaturze. Wartość tę określa się w jednostkach masy na objętość roztworu. Jednostką w układzie SI jest kg/m3 (można także stosować gramy na litr).
1.3. SUBSTANCJE ODNIESIENIA
Nie ma konieczności stosowania substancji odniesienia za każdym razem, gdy przeprowadzane są badania nowej substancji. Powinny one służyć głównie sprawdzeniu od czasu do czasu wydajności metody i pozwolić na porównanie wyników z innymi metodami.
1.4. ZASADA METODYBADANIA
W prostym badaniu wstępnym należy ustalić przybliżoną ilość próbki i czas niezbędny do uzyskania stężenia nasycenia masowego.
1.4.1. Metoda wymywania kolumnowego
Niniejsza metoda jest oparta na wymywaniu wodą substancji badanej z mikrokolumny wypełnionej obojętnym materiałem nośnika, takim jak pręty szklane lub piasek, pokrytych nadmiarem badanej substancji. Rozpuszczalność w wodzie ustala się, gdy stężenie masowe eluatu jest stałe. Przejawia się to wystąpieniem plateau stężenia w funkcji czasu.
1.4.2. Metoda kolby
W ramach zastosowania tej metody substancja (ciała stałe muszą zostać sproszkowane) jest rozpuszczana w wodzie w temperaturze nieco wyższej od temperatury badania. Po uzyskaniu nasycenia mieszaninę schładza się i utrzymuje w temperaturze badania, mieszając ją, jeżeli jest to potrzebne do uzyskania stanu równowagi. Alternatywnie pomiar może być wykonywany bezpośrednio w badanej temperaturze, jeżeli jest zapewniony przez właściwe pobieranie próbek tak by uzyskać równowagę nasycenia. Następnie oznacza się stężenie masowe substancji w roztworze wodnym, który nie może zawierać żadnych nierozpuszczonych cząstek, przy pomocy odpowiedniej metody analitycznej.
1.5. KRYTERIA JAKOŚCI
1.5.1. Powtarzalność
Dla metody wymywania kolumny < 30 % jest osiągalne; dla metody kolby < 15 % powinno być obserwowane.
1.5.2. Czułość
Zależy ona od metody analizy, lecz oznaczenie stężenia masowego do 10-6 gram na litr może być uzyskane.
1.6. OPIS METODY
1.6.1. Warunki badania
Najkorzystniej jest przeprowadzić badanie w temperaturze 20 ±0,5 oC. Jeżeli podejrzewa się istnienie zależności rozpuszczalności od temperatury (> 3 % na oC), należy także zastosować dwie inne wartości temperatury, o co najmniej 10 oC wyższe i niższe od początkowo wybranej temperatury. W takim przypadku należy kontrolować temperaturę w granicach ±0,1 oC. Wybrana temperatura powinna być utrzymywana na stałym poziomie we wszystkich istotnych częściach sprzętu.
1.6.2. Badanie wstępne
Do około 0,1 g próbki (substancje stałe należy sproszkować) w zamkniętym korkiem szklanym cylindrze miarowym o pojemności 10 ml dodaje się wzrastające ilości wody destylowanej w temperaturze pokojowej, wykonując kolejne etapy pokazane w poniższej tabeli:
|
0,1 g rozpuszczone w „x” ml wody |
0,1 |
0,5 |
1 |
2 |
10 |
100 |
> 100 |
|
Przybliżona rozpuszczalność (gramy na litr) |
> 1 000 |
1 000 do 200 |
200 do 100 |
100 do 50 |
50 do 10 |
10 do 1 |
< 1 |
Po każdym dodaniu wskazanej ilości wody mieszaninę wstrząsa się energicznie przez 10 minut i kontroluje wzrokowo pod kątem jakichkolwiek nierozpuszczalnych części próbki. Jeżeli po dodaniu 10 ml wody próbka lub jej część pozostają nierozpuszczone, doświadczenie należy powtórzyć w 100 ml cylindrze miarowym z większą objętością wody. Przy niskiej rozpuszczalności czas wymagany do rozpuszczenia substancji musi być znacząco dłuższy (co najmniej 24 godziny). Przybliżoną rozpuszczalność podano w tabeli pod taką objętością dodanej wody, w jakiej dochodzi do pełnego rozpuszczenia próbki. Jeżeli substancja jest wciąż nierozpuszczalna, należy zastosować dłuższy czas niż 24 godziny (maksymalnie 96 godzin) lub należy podjąć dalsze rozcieńczanie celem ustalenia, czy może być użyta metoda ustalania rozpuszczalności przez wymywanie kolumnowe czy metoda kolby.
1.6.3. Metoda wymywania kolumnowego
1.6.3.1. Materiał nośnikowy, rozpuszczalnik i eluent
Materiał nośnikowy stosowany w metodzie wymywania kolumnowego powinien być obojętny. Materiałami, które można zastosować, są szklane pręty i krzemionka. Należy zastosować odpowiedni, lotny rozpuszczalnik o jakości odczynnika analitycznego do naniesienia substancji badanej na materiał nośnikowy. Jako eluent należy użyć podwójnie destylowaną w szklanym lub kwarcowym przyrządzie wodę.
Uwaga:
Nie można stosować wody pochodzącej bezpośrednio z wymieniacza jonowego.
1.6.3.2. Załadowywanie materiału nośnikowego
Odważa się około 600 mg materiału nośnikowego i przenosi do kolby okrągłodennej o pojemności 50 ml.
Odpowiednią, odważoną ilość substancji badanej rozpuszcza się w wybranym rozpuszczalniku. Właściwą ilość niniejszego roztworu dodaje się do materiału nośnikowego. Rozpuszczalnik musi zostać całkowicie odparowany, np. w obrotowym przyrządzie wyparnym; w innym razie nie zostanie uzyskane nasycenie wodą nośnika ze względu na zjawiska podziału na powierzchni materiału nośnikowego.
Załadowanie materiału nośnikowego może stwarzać problemy (błędne wyniki), jeżeli substancja badana ulega osadzeniu jako olej lub w innej fazie krystalicznej. Problem ten należy przebadać doświadczalnie i wykonać szczegółowe sprawozdanie.
Załadowany materiał nośnikowy pozostawia się do nasączenia wodą na około dwie godziny w około 5 ml wody, a następnie dodawany jest on do mikrokolumny. Alternatywnie do mikrokolumny można wsypać suchy, załadowany materiał nośnikowy, a następnie wypełnić mikrokolumnę wodą i całość pozostawić do uzyskania stanu równowagi na około dwie godziny.
Procedura badania
Wymywanie substancji z materiału nośnikowego można uzyskać jedną z dwóch metod:
|
— |
przy pomocy pompy recyrkulacyjnej (zob. rysunek 1), |
|
— |
przy pomocy naczynia poziomującego (zob. rysunek 4). |
1.6.3.3. Metoda wymywania kolumnowego z pompą recyrkulacyjn
Przyrząd
Schematyczne rozmieszczenie typowego systemu przedstawiono na rysunku 1. Odpowiednia mikrokolumna jest pokazana na rysunku 2, dopuszczalny jest również każdy rozmiar zapewniający spełnienie kryteriów powtarzalności i czułości. Kolumna musi zapewnić przestrzeń słupa cieczy równą co najmniej pięciu objętościom wypełnienia wodą i powinna być zdolna do utrzymania minimum pięciu próbek. Alternatywnie jej wymiar może być zmniejszony, jeżeli użyty zostanie gotowy rozpuszczalnik zamieniający początkowe pięć objętości wypełnienia usuniętych wraz z zanieczyszczeniami.
Kolumnę należy podłączyć do pompy recyrkulacyjnej zdolnej do kontrolowanego wydatku około 25 ml/godz. Pompę podłącza się przy pomocy połączeń z politetrafluoroetylenu (PTFE) i/lub szklanych. Kolumna i pompa po złożeniu powinny mieć wyposażenie umożliwiające pobieranie próbek odcieku i powinny wyrównywać przestrzeń czołową pod ciśnieniem atmosferycznym. Materiał wypełniający kolumnę musi być podtrzymany małą (5 mm) zatyczką z waty szklanej, która służy także do odfiltrowywania cząstek. Pompa recyrkulacyjna może być na przykład pompą perystaltyczną (należy uważać, aby nie doszło do zanieczyszczenia i/lub adsorpcji na materiale rury) lub membranową.
Procedura pomiarowa
Uruchamia się przepływ przez kolumnę. Zalecane jest stosowanie szybkości przepływu około 25 ml/godz. (odpowiada to 10 wypełnieniom złoża na godzinę dla opisanej tu kolumny). Objętość pierwszych pięciu wypełnień (minimum) odrzuca się w celu usunięcia zanieczyszczeń rozpuszczalnych w wodzie. Następnie uruchamia się pompę recyrkulacyjną, aż do uzyskania równowagi, którą definiuje się jako pięć kolejnych próbek, których stężenie nie różni się o więcej niż ± 30 % w sposób losowy. Próbki te powinny zostać od siebie oddzielone w odstępach czasowych odpowiadających przejściu eluentu w ilości co najmniej 10 wypełnień złoża.
1.6.3.4. Metoda wymywania kolumnowego z naczyniem poziomującym
Przyrząd (zob. rysunki 4 i 3)
Naczynie poziomujące: Podłączenie do naczynia poziomującego wykonuje się przy użyciu szlifowanego złącza szklanego podłączonego przez rurkę z PTFE. Zaleca się stosowanie szybkości przepływu około 25 ml/godz. Kolejne wymywane frakcje należy zebrać i wykonać analizę wybraną metodą.
Procedura pomiarowa
Do ustalenia rozpuszczalności w wodzie wykorzystuje się frakcje ze środkowego zakresu odcieku, gdzie stężenia są stałe (± 30 %) w co najmniej pięciu kolejnych frakcjach.
W obu przypadkach (stosując pompę recyrkulacyjną lub naczynie poziomujące), drugą serię wykonuje się przy połowie szybkości przepływu pierwszej. Jeżeli wyniki obu serii są zgodne, badanie jest zadowalające; jeżeli istnieje większa pozorna rozpuszczalność przy niższej prędkości przepływu, wtedy znowu należy stosować zmniejszenie tej prędkości o połowę, aż dwie kolejne serie dadzą taką samą rozpuszczalność.
W obu przypadkach (przy użyciu pompy recyrkulacyjnej lub naczynia poziomującego) należy skontrolować frakcje pod kątem obecności substancji koloidalnej przez sprawdzenie, czy nie występuje efekt Tyndalla (rozproszenie światła). Obecność takich cząstek czyni wyniki nieważnymi i badanie należy powtórzyć, poprawiając działanie filtrujące kolumny.
Należy odnotowywać pH każdej próbki. Drugą serię oznaczeń należy przeprowadzić w tej samej temperaturze.
1.6.4. Metoda kolby
1.6.4.1. Przyrząd
Do zastosowania metody kolby potrzebne są następujące materiały:
|
— |
zwykłe, laboratoryjne wyroby szklane i instrumenty, |
|
— |
urządzenie służące do mieszania roztworów w kontrolowanej, stałej temperaturze, |
|
— |
wirówka (najlepiej termostatowana), o ile jest potrzebna w przypadku emulsji, oraz |
|
— |
urządzenia do oznaczeń analitycznych. |
1.6.4.2. Procedura pomiarowa
Na podstawie badania wstępnego szacuje się ilość materiału niezbędną do nasycenia pożądanej objętości wody. Wymagana ilość wody będzie zależeć od metody analitycznej i zakresu rozpuszczalności. Do każdego z trzech naczyń wyposażonych w korki szklane (np. probówek wirówkowych, kolb) odważa się po około pięciokrotności wyżej ustalonej ilości materiału. Wybraną objętość wody dodaje się do każdego naczynia, po czym naczynia należy szczelnie zakorkować. Zamknięte naczynia wytrząsa się następnie w temperaturze 30 oC. (Należy użyć wytrząsarki lub mieszarki, która może działać w stałej temperaturze, np. magnetyczne mieszanie w łaźni wodnej z temperaturą kontrolowaną termostatycznie). Po jednym dniu jedno z naczyń jest usuwane i pozostawione do ustalenia się nowej równowagi przez 24 godziny w temperaturze badania, przy czym od czasu do czasu należy je wstrząsnąć. Zawartość naczynia poddaje się następnie wirowaniu w temperaturze badania i oznacza stężenie klarownej fazy wodnej przy pomocy odpowiedniej metody analitycznej. Pozostałe dwie kolby traktuje się podobnie po początkowym ustaleniu równowagi w temperaturze 30 oC przez, odpowiednio, dwa i trzy dni. Jeżeli wyniki oznaczenia stężenia w co najmniej ostatnich dwóch naczyniach cechują się wymaganą odtwarzalnością, badanie należy uznać za zadowalające. Całe badanie należy powtórzyć, stosując dłuższe okresy ustalania równowagi, jeżeli wyniki dla naczyń 1, 2 i 3 wykazują tendencję do wzrastających wartości.
Procedura pomiarowa może być również wykonana bez wstępnej inkubacji w 30 oC. W celu oceny szybkości ustalania się równowagi nasycenia pobierane są próbki do chwili, gdy czas mieszania nie wywiera już wpływu na stężenie badanego roztworu.
Należy odnotowywać pH każdej próbki.
1.6.5. Analiza
Dla tych oznaczeń najkorzystniejsza jest metoda analityczna specyficzna dla substancji, gdyż małe ilości rozpuszczalnych zanieczyszczeń mogą spowodować duże błędy wyników pomiarów rozpuszczalności. Przykładami takich metod są: chromatografia gazowa lub cieczowa, metody miareczkowania, metody fotometryczne, metody woltametryczne.
2. DANE
2.1. METODA WYMYWANIA KOLUMNOWEGO
Dla każdej serii należy obliczyć średnią wartość dla co najmniej pięciu kolejnych próbek pobranych z plateau nasycenia, podobnie jak odchylenie standardowe. Wyniki należy podać w jednostkach masy na objętość roztworu.
Średnie obliczone w dwóch badaniach przy różnych przepływach są porównywane i powinny mieć powtarzalność mniejszą niż 30 %.
2.2. METODA KOLBY
Należy podać poszczególne wyniki dla każdej z trzech kolb i powinny być one uznane za stałe(powtarzalność mniejsza niż 15 %), należy je uśrednić i podać w jednostkach masy na objętość roztworu. Może to wymagać przeliczenia jednostek masy na jednostki objętości, za pomocą gęstości, gdy rozpuszczalność jest bardzo duża (> 100 gramów na litr).
3. SPORZĄDZANIE SPRAWOZDANIA
3.1. METODA WYMYWANIA KOLUMNOWEGO
Sprawozdanie z badań zawiera w miarę możliwości następujące informacje:
|
— |
wyniki badania wstępnego, |
|
— |
precyzyjna specyfikacja substancji (tożsamość i zanieczyszczenia), |
|
— |
poszczególne stężenia, prędkości przepływu i pH każdej próbki, |
|
— |
średnie i odchylenia standardowe dla co najmniej pięciu próbek z plateau nasycenia dla każdej serii, |
|
— |
średnia z dwóch kolejnych, możliwych do przyjęcia serii, |
|
— |
temperatura wody w czasie procesu nasycania, |
|
— |
zastosowana metoda analityczna, |
|
— |
charakter zastosowanego materiału nośnikowego, |
|
— |
załadowanie materiału nośnikowego, |
|
— |
zastosowany rozpuszczalnik, |
|
— |
dowody niestabilności chemicznej substancji w trakcie badania oraz zastosowana metoda, |
|
— |
wszystkie informacje stosowne dla interpretacji wyników, szczególnie w odniesieniu do zanieczyszczeń i stanu fizycznego substancji. |
3.2. METODA KOLBY
Sprawozdanie z badań zawiera w miarę możliwości następujące informacje:
|
— |
wyniki badania wstępnego, |
|
— |
precyzyjna specyfikacja substancji (tożsamość i zanieczyszczenia), |
|
— |
poszczególne oznaczenia analityczne i ich średnia, w przypadku gdy więcej niż jedna wartość była oznaczana dla każdej kolby, |
|
— |
pH każdej próbki, |
|
— |
średnia wartości dla poszczególnych kolb, które były zgodne, |
|
— |
temperatura badania, |
|
— |
zastosowana metoda analityczna, |
|
— |
dowody niestabilności chemicznej substancji w trakcie badania oraz zastosowana metoda, |
|
— |
wszystkie informacje stosowne dla interpretacji wyników, szczególnie w odniesieniu do zanieczyszczeń i stanu fizycznego substancji. |
4. LITERATURA
|
(1) |
OECD, Paris, 1981, Test Guideline 105, Decision of the Council C(81) 30 final. |
|
(2) |
NF T 20-045 (AFNOR) (Sept. 85). Chemical products for industrial use -Determination of water solubility of solids and liquids with low solubility -Column elution method. |
|
(3) |
NF T 20-046 (AFNOR) (Sept. 85). Chemical products for industrial use -Determination of water solubility of solids and liquids with high solubility -Flask method. |
Dodatek
Rysunek 1
Metoda wymywania kolumnowego z pompą recyrkulacyjną

Rysunek 2
Typowa mikrokolumna
(wszystkie wymiary w milimetrach)

Rysunek 3
Typowa mikrokolumna
(wszystkie wymiary w milimetrach)

Rysunek 4
Metoda wymywania kolumnowego z naczyniem poziomującym

A.8. WSPÓŁCZYNNIK PODZIAŁU
1. METODA
Opisana metoda „wstrząsania kolby” jest oparta na wytycznych OECD dotyczących badań (1).
1.1. WPROWADZENIE
Dla wykonania niniejszego badania korzystne jest posiadanie informacji wstępnych dotyczących wzoru strukturalnego, stałej dysocjacji, rozpuszczalności w wodzie, hydrolizy, rozpuszczalności w n-oktanolu i napięcia powierzchniowego substancji.
Pomiary na substancjach ulegających jonizacji powinny być wykonane tylko na ich niezjonizowanej postaci (wolny kwas lub wolna zasada) uzyskanej przez użycie właściwego buforu o pH co najmniej jedną jednostkę mniejszą (wolny kwas) lub większą (wolna zasada) od pK.
Niniejsza metoda badania zawiera dwie oddzielne procedury: metoda wstrząsania kolby i chromatografii cieczowej wysokiej wydajności (HPLC). Pierwszą stosuje się gdy wartość log Pow (zob. definicje poniżej) wypada w zakresie od - 2 do 4, drugą w zakresie od 0 do 6. Przed przeprowadzeniem procedury doświadczalnej, należy wpierw wstępnie uzyskać oszacowanie współczynnika podziału.
Metodę wstrząsania kolby stosuje się jedynie do zasadniczo czystych substancji rozpuszczalnych w wodzie i n-oktanolu. Nie stosuje się jej do materiałów powierzchniowo czynnych (dla takich należy podać wartość obliczoną lub oszacowaną opartą na rozpuszczalności danej substancji w n-oktanolu i w wodzie).
Metody HPLC nie stosuje się dla silnych kwasów i zasad, związków kompleksowych metali, materiałów powierzchniowo czynnych lub substancji, które reagują z rozpuszczalnikiem wymywającym. Dla takich materiałów należy podać wartość obliczoną lub oszacowaną opartą na rozpuszczalności danej substancji w n-oktanolu i w wodzie.
Metoda HPLC jest mniej czuła na obecność zanieczyszczeń w badanym związku niż metoda wstrząsania kolby. Pomimo tego, zanieczyszczenia w niektórych przypadkach mogą czynić interpretację wyników trudną, ponieważ wyznaczenie piku jest niepewne. Dla mieszanin dających nieczytelne pasmo, należy ustalić górną i dolną granicę log P.
1.2. DEFINICJA I JEDNOSTKI
Współczynnik podziału (P) definiuje się jako stosunek stężeń równowagi (ci) substancji rozpuszczonej w układzie dwufazowym, składającym się z dwóch zasadniczo niemieszających się ze sobą rozpuszczalników. W przypadku n-oktanolu i wody:

Dlatego współczynnik podziału (P) jest ilorazem dwóch stężeń, przy czym na ogół podaje się go w postaci jego logarytmu o podstawie 10 (log P).
1.3. SUBSTANCJE ODNIESIENIA
Metoda wstrząsania kolby
Nie ma konieczności stosowania substancji odniesienia za każdym razem, gdy przeprowadzane są badania nowej substancji. Powinny one służyć głównie sprawdzeniu od czasu do czasu wydajności metody i pozwolić na porównanie wyników z innymi metodami.
Metoda HPLC
W celu skorelowania danych zmierzonych metodą HPLC danego związku z jego wartością P należy ustalić krzywą kalibracji log P w zależności od danych chromatograficznych, używając co najmniej 6 punktów odniesienia. Jest zadaniem użytkownika wybór właściwych substancji odniesienia. W każdym przypadku, gdy będzie możliwe, co najmniej jedna substancja odniesienia powinna posiadać Pow powyżej tego dla substancji badanej, a druga powinna posiadać Pow poniżej substancji badanej. Dla wartości log P mniejszej niż 4 kalibracja musi być oparta o dane uzyskane metodą wstrząsania kolby. Dla wartości log P większej od 4 kalibracja powinna być oparta na ważnych wartościach literaturowych, jeżeli są one zgodne z wartościami wyliczonymi. Celem uzyskania lepszej dokładności, zalecane jest wybór takich substancji odniesienia, które są strukturalnie zbliżone do badanej substancji.
Rozszerzone wykazu wartości log Pow dla wielu grup związków chemicznych są dostępne w pozycjach bibliograficznych (2) i (3). Jeżeli dane współczynnika podziału strukturalnie zbliżonych związków nie są dostępne, należy zastosować bardziej ogólną kalibrację, ustaloną w oparciu o inne związki odniesienia.
Wykaz zalecanych substancji odniesienia i ich wartości Pow podano w dodatku 2.
1.4. ZASADA METODY
1.4.1. Metoda wstrząsania kolby
W celu ustalenia współczynnika podziału należy uzyskać równowagę między wszystkimi wchodzącymi w interakcje składnikami układu, przy czym należy ustalić stężenie substancji rozpuszczonych w dwóch fazach. Studia w zakresie literatury na ten temat wskazują, że możliwe jest użycie kilku różnych technik dla rozwiązania niniejszego problemu, to jest poprzez zmieszanie dwóch faz oraz kolejno ich rozdzielenie w celu ustalenia stężenia równowagi badanej substancji.
1.4.2. Metoda HPLC
HPLC jest wykonywana na kolumnach analitycznych wypełnionych handlowo dostępną fazą stałą zawierającą długo łańcuchowe węglowodory (np. C8, C18) chemicznie związane na krzemionce. Związki chemiczne wstrzyknięte do takiej kolumny przesuwają się w niej z różnymi szybkościami z powodu różnych stopni ich podziału między fazą ruchomą oraz węglowodorową fazą stacjonarną. Mieszaniny związków chemicznych są wymywane w kolejności ich hydrofobowości, pierwsze wymywane są związki chemiczne rozpuszczalne w wodzie, a na końcu związki chemiczne rozpuszczalne w olejach, proporcjonalnie do ich współczynnika podziału między węglowodory oraz wodę. Umożliwia to uzyskanie zależności między czasem retencji na takiej kolumnie (odwrócona faza) i ustalenie współczynnika podziału n-oktanol/woda. Współczynnik podziału jest wyprowadzany ze współczynnika objętości k, podanego wyrażeniem:

w której tr = czas retencji badanej substancji, to = średni czas przejścia cząsteczek rozpuszczalnika przez kolumnę (czas martwy).
Nie są wymagane analizy ilościowe, a konieczne jest tylko oznaczenie czasu wymywania.
1.5. KRYTERIA JAKOŚCI
1.5.1. Powtarzalnoś
Metoda wstrząsania kolby
W celu zapewnienia precyzji współczynnika podziału należy przeprowadzić dwukrotne oznaczenia w trzech różnych warunkach badania, przy czym można zmieniać ilość oznaczanej substancji oraz stosunek objętości rozpuszczalników. Ustalone wartości współczynnika podziału wyrażone jako ich wspólne logarytmy powinny zawierać się w przedziale ±0,3 jednostek logarytmicznych.
Metoda HPLC
W celu zwiększenia poziomu ufności pomiaru, należy wykonać podwójne oznaczenia. Wartości log P wyprowadzone z poszczególnych pomiarów powinny mieścić się w zakresie ±0,1 jednostek logarytmicznych.
1.5.2. Czułość
Metoda wstrząsania kolby
Zakres pomiarowy metody ustala się na podstawie granicy wykrywalności procedury analitycznej. Powinno to pozwalać na ocenę wartości log Pow w zakresie od - 2 do 4 (wyjątkowo, gdy zezwalają warunki, niniejszy zakres może być rozszerzony do log Pow do 5), gdy stężenie rozpuszczenia w jednej z faz jest nie większe niż 0,01 mola na litr.
Metoda HPLC
Metoda HPLC umożliwia ocenę współczynnika podziału w zakresie log Pow od 0 do 6.
Zwykle współczynnik podziału danego związku może być oszacowany z dokładnością w obrębie zakresu ± 1 jednostki logarytmicznej wartości uzyskanej z metody wstrząsania kolby. Typowe korelacje można znaleźć w pozycjach bibliograficznych (4), (5), (6), (7) i (8). Wyższą dokładność można zwykle uzyskać, gdy krzywe korelacji są oparte na strukturalnie zbliżonych związkach odniesienia (9).
1.5.3. Specyficznoś
Metoda wstrząsania kolby
Prawo podziału Nernsta obowiązuje jedynie w warunkach stałej temperatury, stałego ciśnienia i stałego pH, w przypadku rozcieńczonych roztworów. W sposób ścisły dotyczy czystej substancji zdyspergowanej między dwoma czystymi rozpuszczalnikami. Istnienie kilku różnych substancji rozpuszczonych w jednej lub dwóch fazach może wpłynąć na uzyskane wyniki.
Zjawiska dysocjacji lub asocjacji rozpuszczonych cząsteczek prowadzą do odchyleń od prawa podziału Nernsta. Na takie odchylenia wskazuje fakt, że współczynnik podziału staje się zależny od stężenia roztworu.
Ze względu na szereg różnych badanych stanów równowagi, ta metoda badania nie powinna być stosowana w odniesieniu do związków ulegających jonizacji bez zastosowania korekcji. Należy rozważyć użycie roztworów buforowych w miejsce wody dla takich związków; pH buforu powinno różne przynajmniej o jedną jednostkę pH od pKa substancji i być w zgodności z pH badanego środowiska.
1.6. OPIS METODY
1.6.1. Wstępna ocena współczynnika podziału
Bardziej wskazana jest ocena współczynnika podziału za pomocą metody obliczeniowej (zob. dodatek 1) lub, jeśli stosowne, ze stosunków rozpuszczalności badanej substancji w czystych rozpuszczalnikach (10).
1.6.2. Metoda wstrząsania kolby
1.6.2.1. Przygotowanie
N-oktanol: oznaczenie współczynnika podziału należy wykonać przy użyciu odczynnika jakości analitycznej o wysokiej czystości.
Woda: należy użyć wody destylowanej lub dwa razy destylowanej z przyrządu szklanego lub kwarcowego. Jeżeli jest uzasadnione, dla związków ulegających jonizacji, należy użyć roztworów buforowych w miejsce wody.
Uwaga:
Nie można stosować wody pochodzącej bezpośrednio z wymieniacza jonowego.
1.6.2.1.1.
Przed ustaleniem współczynnika podziału wykonuje się wzajemne nasycenie faz układu rozpuszczalników przez wytrząsanie w temperaturze prowadzenia eksperymentu. Aby to uzyskać, wygodnie jest zastosować wytrząsanie dwóch dużych butli magazynowych z n-oktanolem lub wodą wysokiej czystości do analiz z wystarczającą ilością drugiego rozpuszczalnika, przez 24 godziny, na mechanicznej wytrząsarce, a następnie odstawienie ich na okres tak długi, aby doszło do rozdzielenia faz i uzyskania stanu nasycenia.
1.6.2.1.2.
Cała objętość układu dwufazowego powinna prawie wypełniać naczynie do badania. Pomoże to w niedopuszczeniu do utraty materiału z powodu jego ulatniania się. Stosunek objętościowy i ilości substancji, które należy zastosować, ustala się na podstawie następujących informacji:
|
— |
wstępna ocena współczynnika podziału (zob. powyżej), |
|
— |
minimalna ilość substancji badanej wymagana do procedury analitycznej, oraz |
|
— |
ograniczenie maksymalnego stężenia w każdej z faz do 0,01 mola na litr. |
Przeprowadza się trzy badania. W pierwszym używa się wyliczonej wielkości stosunku n-oktanolu do wody; w drugiej ten stosunek dzieli się przez dwa; i w trzecim stosunek ten mnoży się przez dwa (np. 1:1, 1:2, 2:1).
1.6.2.1.3.
Roztwór podstawowy przygotowuje się w n-oktanolu wstępnie nasyconym wodą. Stężenie takiego roztworu podstawowego musi być precyzyjnie oznaczone, przed użyciem go w oznaczaniu współczynnika podziału. Roztwór ten powinien być przechowywany w warunkach zapewniających jego stabilność.
1.6.2.2. Warunki badania
Powinna być utrzymywana stała temperatura badania (± 1 oC) i mieścić się w zakresie od 20 do 25 oC.
1.6.2.3. Procedura pomiarowa
1.6.2.3.1.
Dla każdego z warunków badania należy przygotować po dwa naczynia do badań zawierające wymagane, dokładnie odmierzone ilości obu rozpuszczalników, łącznie z niezbędną ilością roztworu podstawowego.
Fazy n-oktanolu powinny być mierzone objętościowo. Naczynia do badania należy albo ustawić na odpowiedniej wytrząsarce, albo wytrząsać w ręce. Przy zastosowaniu wirowania probówki zalecaną metodą jest obrócenie szybko probówki o około 180o względem jej poprzecznej osi, tak by nie doprowadzić do jakiegokolwiek wzrostu ilości powietrza w obu fazach. Doświadczenie pokazało, że 50 takich obrotów jest zwykle wystarczające dla ustalenia równowagi podziału. Dla pewności zaleca się 100 obrotów w ciągu pięciu minut.
1.6.2.3.2.
Jeżeli jest konieczne w celu rozdzielenia faz, należy zastosować wirowanie mieszaniny. Do tego celu należy użyć wirówki laboratoryjnej utrzymywanej w temperaturze pokojowej bądź też, w razie wykorzystywania wirówki o niekontrolowanej temperaturze, należy zapewnić uzyskanie ponownego stanu równowagi probówek do wirówki w temperaturze badania na co najmniej jedną godzinę przed analizą.
1.6.2.4. Analiza
W celu oznaczenia współczynnika podziału konieczne jest ustalenie stężenia substancji badanej w obu fazach. Można tego dokonać przez pobranie podwielokrotności każdej z obu faz z każdej z probówek dla każdych warunków badania i poddanie tych podwielokrotności analizie ich przy użyciu wybranej procedury. Należy obliczyć całkowitą ilość substancji obecnej w obu fazach i porównać ją z ilością pierwotnie wprowadzonej substancji.
Należy pobrać próbki fazy wodnej przy użyciu procedury zmniejszającej do minimum ryzyko pobrania śladów n-oktanolu: do pobrania próbki fazy wodnej można użyć szklanej strzykawki z wyjmowaną igłą. Strzykawka powinna zostać na początku częściowo wypełniona powietrzem. Należy delikatnie wypchnąć powietrze, wprowadzając igłę przez warstwę n-oktanolu. Pobiera się odpowiednią objętość fazy wodnej do strzykawki. Następnie strzykawkę szybko wyjmuje się z roztworu i odłącza igłę. Zawartość strzykawki można wykorzystać jako próbkę fazy wodnej. Najkorzystniej jest ustalić stężenie w dwóch rozdzielonych fazach przy użyciu metody specyficznej dla substancji. Przykładami właściwych metod analitycznych są:
|
— |
metody fotometryczne, |
|
— |
chromatografia gazowa, |
|
— |
wysokosprawna chromatografia cieczowa. |
1.6.3. Metoda HPLC
1.6.3.1. Przygotowanie
Przyrząd
Wymagany jest chromatograf cieczowy, wyposażony w bezimpulsową pompę i odpowiednie urządzenie do wykrywania. Zalecane jest użycie zaworu wtryskowego z obiegiem wtrysku. Obecność grup polarnych w fazie stacjonarnej może poważnie zakłócić działanie kolumny HPLC. Zatem fazy stacjonarne powinny posiadać minimalną zawartość grup polarnych (11). Można stosować dostępne w handlu mikrocząsteczkowe wypełnienia dla fazy odwróconej lub gotowe wypełnione kolumny. Kolumna ochronna powinna być umieszczona między systemem wtrysku a kolumną analityczną.
Faza ruchoma
Do przygotowania rozpuszczalnika wymywającego, odgazowywanego przed użyciem, używa się metanolu oraz wody o czystości HPLC. Wykorzystuje się elucję izokratyczną. Należy zastosować stosunki metanol/woda o minimalnej zawartości wody 25 %. Mieszanina metanol-woda o typowym stosunku 3:1 (obj.) jest zadawalająca do wymywania związków o log P 6 w ciągu jednej godziny, przy szybkości przepływu 1 ml/mm. Dla związków o wysokim log P może być konieczne skrócenie czasu elucji (oraz związków odniesienia) poprzez zmniejszenie polarności fazy ruchomej lub długości kolumny.
Substancje o bardzo niskiej rozpuszczalności w n-oktanolu wykazują tendencję do dawania nienormalnie niskich wartości log Pow przy metodzie HPLC; piki takich związków czasem towarzyszą czołu rozpuszczalnika. Zjawisko to jest prawdopodobnie związane z faktem, że proces podziału jest za powolny do uzyskania równowagi w czasie branym pod uwagę przy zwykłym rozdzielaniu metodą HPLC. Zmniejszenie szybkości przepływu i/lub obniżenie stosunku metanol/woda może być zatem w tym przypadku skuteczne do osiągnięcia rzeczywistej wartości.
Związki badane i odniesienia muszą być rozpuszczalne w fazie ruchomej w stężeniach wystarczających do umożliwienia ich wykrycia. Tylko w wyjątkowych przypadkach można zastosować dodatki do mieszaniny metanol/woda, ponieważ dodatki zmieniają właściwości kolumny. Dla chromatogramów z dodatkami obowiązkowe jest użycie oddzielnej kolumny tego samego typu. Jeśli układ metanol-woda nie jest odpowiedni, można użyć innych mieszanin organicznych rozpuszczalników, na przykład etanol-woda lub acetonitryl-woda.
Wartość pH eluentu jest krytyczna dla związków ulegających jonizacji. Powinno być ono w obrębie zakresu roboczego pH kolumny, które zwykle wynosi między 2 i 8. Zalecane jest buforowanie. Należy zachować ostrożność, aby nie dopuścić do wytrącania się soli i zniszczenia kolumny, co zachodzi dla niektórych mieszanin faza organiczna/bufor. Pomiary HPLC faz stacjonarnych opartych na krzemionce powyżej pH 8 nie są polecane, gdyż użycie zasadowej fazy ruchomej powoduje gwałtowne pogorszenie się wydajności kolumny.
Substancje rozpuszczone
Związki odniesienia powinny być o najwyższej dostępnej czystości. Związki używane do celów badania lub kalibracji są rozpuszczone, o ile to możliwe, w fazie ruchomej.
Warunki badania
Temperatura w trakcie wykonywania pomiarów nie może zmieniać się więcej niż ± 2 K.
1.6.3.2. Pomiar
Obliczenie czasu martwego to
Czas martwy to ustala się albo przez zastosowanie szeregów homologicznych (np. n-alkilometyloketonów), albo nieustalonych związków organicznych (np. tiomocznik lub formamid). Dla obliczenia czasu martwego to z użyciem szeregów homologicznych, zestaw co najmniej siedmiu reprezentantów szeregu jest wtryskiwany i oznacza się poszczególne czasy retencji. Wstępne czasy retencji tr(nc + 1) są wykreślane w funkcji tr(n c), oraz punkt przecięcia a oraz nachylenie b równania regresji:
tr (nc + 1) = a + b tr (n c)
są ustalane (nc = ilość atomów węgla). Czas martwy to jest zatem dany przez:
to = a/(1 – b)
Krzywa kalibracji
Następnym krokiem jest sporządzenie wykresu korelacji wartości log k w zależności od log P dla właściwych związków odniesienia. W praktyce zestaw między 5 a 10 standartowych związków odniesienia, których log P jest wokół oczekiwanego zakresu jest kolejno wtryskiwany i oznaczane są czasy retencji, najkorzystniej przez integrator rejestrujący podłączony do systemu detekcji. Odpowiadające logarytmy masowych stosunków podziałów, log k, są wyliczane i wykreślane w funkcji log P ustalonego metodą wstrząsania kolby. Kalibracja jest prowadzona w regularnych odstępach czasu co najmniej raz dziennie, tak aby możliwe zmiany wydajności kolumny mogły być akceptowane.
Oznaczanie masowego stosunku podziału badanej substancji
Badana substancja jest wtryskiwana do tak małej ilości fazy ruchomej jak to możliwe. Oznacza się czas retencji (podwójnie), pozwalający na obliczenie masowego stosunku podziału k. Z wykresu korelacji związków odniesienia, można interpolować współczynnik podziału badanej substancji. Dla bardzo niskich oraz dla bardzo wysokich współczynników podziału konieczna jest ekstrapolacja. W tych przypadkach należy zwrócić szczególną uwagę na granice ufności linii regresji.
2. DANE
Metoda wstrząsania kolby
Niezawodność ustalonych wartości P można sprawdzić przez porównanie średnich wyników dwukrotnych oznaczeń ze średnią wszystkich wyników.
3. SPORZĄDZANIE SPRAWOZDANIA
Sprawozdanie z badań zawiera w miarę możliwości następujące informacje:
|
— |
precyzyjna specyfikacja substancji (tożsamość i zanieczyszczenia), |
|
— |
tam gdzie metody nie nadają się do zastosowania (np. dla materiałów powierzchniowo czynnych), należy przedstawić obliczoną lub oszacowaną wartość w oparciu o poszczególne rozpuszczalności w n-oktanolu i wodzie, |
|
— |
należy podać wszystkie informacje i uwagi istotne dla interpretacji wyników, w szczególności w odniesieniu do zanieczyszczeń i stanu skupienia substancji. |
Dla metody wstrząsania kolby:
|
— |
wynik wstępnej oceny, o ile wykonano, |
|
— |
temperatura oznaczania, |
|
— |
dane na temat procedur analitycznych zastosowanych do oznaczenia stężeń, |
|
— |
czas i szybkość wirowania, o ile zastosowano, |
|
— |
zmierzone stężenia w obu fazach dla każdego oznaczenia (co oznacza, że całkowita liczba 12 stężeń znajdzie się w sprawozdaniu), |
|
— |
waga substancji badanej, objętość każdej fazy zastosowanej w każdym naczyniu oraz całkowita obliczona objętość substancji badanej obecnej w każdej z faz po uzyskaniu stanu równowagi, |
|
— |
należy podać obliczone wartości współczynnika podziału (P) oraz średniej dla każdego zestawu warunków badania, podobnie jak średnie z wszystkich oznaczeń. W przypadku gdy istnieją dane przemawiające za zależnością współczynnika podziału od stężenia, należy to odnotować w sprawozdaniu, |
|
— |
należy podać odchylenia standardowe poszczególnych wartości P ponad ich średnią, |
|
— |
średnie wartości P uzyskane na podstawie wszystkich oznaczeń należy także podać jako ich logarytm (o podstawie 10), |
|
— |
teoretyczne obliczenie Pow, gdy wartość ta została oznaczona lub gdy zmierzona wartość jest > 104, |
|
— |
pH używanej wody oraz fazy wodnej w czasie trwania eksperymentu, |
|
— |
jeżeli używane są bufory, należy podąć uzasadnienie dla ich użycia w miejsce wody, skład, stężenia i pH buforów, pH fazy wodnej przed i po eksperymencie. |
Dla metody HPLC:
|
— |
wynik wstępnej oceny, o ile wykonano, |
|
— |
substancje badane i odniesienia oraz ich czystość, |
|
— |
zakres temperatury oznaczeń, |
|
— |
pH, przy którym wykonano oznaczenia, |
|
— |
szczegóły kolumn analitycznej i ochronnej, fazy ruchomej i środków detekcji, |
|
— |
dane retencji oraz literaturowe wartości log P dla związków odniesienia użytych w kalibracji, |
|
— |
szczegóły dopasowania linii regresji (log k w zależności od log P), |
|
— |
dane średniej retencji i interpolowana wartość log P badanego związku, |
|
— |
opis wyposażenia i warunki pracy, |
|
— |
profile wymywania, |
|
— |
ilość badań oraz substancje odniesienia substancji wprowadzonych do kolumny, |
|
— |
czas martwy i sposób jego pomiaru. |
4. LITERATURA
|
(1) |
OECD, Paris, 1981, Test Guideline 107, Decision of the Council C(81) 30 final. |
|
(2) |
C. Hansch and A.J. Leo, Substituent Constants for Correlation Analysis in Chemistry and Biology, John Wiley, New York 1979. |
|
(3) |
Log P and Parameter Database, A tool for the quantitative prediction of bioactivity (C. Hansch, chairman, A.J. Leo, dir.) – Available from Pomona College Medical Chemistry Project 1982, Pomona College, Claremont, California 91711. |
|
(4) |
L. Renberg, G. Sundström and K. Sundh-Nygärd, Chemosphere, 1980, vol. 80, 683. |
|
(5) |
H. Ellgehausen, C. D’Hondt and R. Fuerer, Pestic. Sci., 1981, vol. 12, 219. |
|
(6) |
B. McDuffie, Chemosphere, 1981, vol. 10, 73. |
|
(7) |
W.E. Hammers et al., J. Chromatogr., 1982, vol. 247, 1. |
|
(8) |
J.E. Haky and A.M. Young, J. Liq. Chromat., 1984, vol. 7, 675. |
|
(9) |
S. Fujisawa and E. Masuhara, J. Biomed. Mat. Res., 1981, vol. 15, 787. |
|
(10) |
O. Jubermann, Verteilen und Extrahieren, in Methoden der Organischen Chemie (Houben Weyl), Allgemeine Laboratoriumpraxis (edited by E.Muller), Georg Thieme Verlag, Stuttgart, 1958, Band I/1, 223–339. |
|
(11) |
R.F. Rekker and H.M. de Kort, Euro. J. Med. Chem., 1979, vol. 14, 479. |
|
(12) |
A. Leo, C. Hansch and D. Elkins, Partition coefficients and their uses. Chem. Rev., 1971, vol. 71, 525. |
|
(13) |
R.F. Rekker, The Hydrophobic Fragmental Constant, Elsevier, Amsterdam, 1977. |
|
(14) |
NF T 20-043 AFNOR (1985). Chemical prqducts for industrial use -Determination of partition coefficient – Flask shaking method. |
|
(15) |
C.V. Eadsforth and P. Moser, Chemosphere, 1983, vol. 12, 1459. |
|
(16) |
A. Leo, C. Hansch and D. Elkins, Chem. Rev., 1971, vol. 71, 525. |
|
(17) |
C. Hansch, A. Leo, S.H. Unger, K.H. Kim, D. Nikaitani and E.J. Lien, J. Med. Chem., 1973, vol. 16, 1207. |
|
(18) |
W.B. Neely, D.R. Branson and G.E. Blau, Environ. Sci. Technol., 1974, vol. 8, 1113. |
|
(19) |
D.S. Brown and E.W. Flagg, J. Environ. Qual., 1981, vol. 10, 382. |
|
(20) |
J.K. Seydel and K.J. Schaper, Chemische Struktur und biologische Aktivität von Wirkstoffen, Verlag Chemie, Weinheim, New York 1979. |
|
(21) |
R. Franke, Theoretical Drug Design Methods, Elsevier, Amsterdam 1984, |
|
(22) |
Y.C. Martin, Quantitative Drug Design, Marcel Dekker, New York, Base11978. |
|
(23) |
N.S. Nirrlees, S.J. Noulton, C.T. Murphy, P.J. Taylor; J. Med. Chem., 1976, vol. 19, 615. |
Dodatek 1
Metody obliczenia/szacowania
WPROWADZENIE
Ogólne wprowadzenie do metod obliczeniowych, dane i przykłady są przedstawione w pozycji „Handbook of Chemical Property Estimation Methods” (a).
Stosuje się obliczoną wartość Pow:
|
— |
w celu zadecydowania która z metod doświadczalnych jest właściwa (zakres metody wstrząsania kolby: log Pow: od - 2 do 4, zakres metody HPLC: log Pow: od 0 do 6), |
|
— |
do wyboru właściwych warunków badania (np. substancji odniesienia dla procedur HPLC, stosunek objętości n-oktanolu do wody dla metody wytrząsania kolby), |
|
— |
jako wewnętrzne laboratoryjne sprawdzenie możliwych błędów doświadczalnych, |
|
— |
dla uzyskania oszacowanej Pow w przypadkach, gdy z przyczyn technicznych metody doświadczalne nie mogą być stosowane. |
METODA SZACOWANIA
Wstępne oszacowanie współczynnika podziału
Wartość współczynnika podziału może być oszacowana, stosując rozpuszczalności badanej substancji w czystych rozpuszczalnikach:
Do tego:

METODY OBLICZEŃ
Zasada metod obliczeniowych
Wszystkie metody obliczeniowe są oparte o formalną fragmentację cząsteczki na odpowiednie podstruktury, dla których pewne przyrosty log Pow są znane. Log Pow całej cząsteczki jest następnie obliczany jako suma wartości, odpowiadając jej fragmentom plus suma składników korekcji dla oddziaływań wewnątrzcząsteczkowych.
Wykaz stałych fragmentów i składników korekcji są dostępne w pozycjach bibliograficznych (b), (c), (d) i (e). Niektóre są regularnie aktualizowane (b).
Kryteria jakości
Ogólnie, wiarygodność metod obliczeniowych obniża się ze wzrostem złożoności badanych związków. W przypadku prostych cząsteczek o niskiej masie cząsteczkowej i jednej lub dwu grupach funkcyjnych, można oczekiwać odchylenia od 0,1 do 0,3 jednostek log Pow między wynikami różnych metod fragmentaryzacji i wartościami pomierzonymi. W przypadku bardziej złożonych cząsteczek margines błędu może być większy. Zależy to od rzetelności i dostępności stałych fragmentu, jak również zdolności rozpoznania oddziaływań wewnątrzcząsteczkowych (np. wiązania wodorowe) i poprawności użycia składników korekcji (oprócz problemów z oprogramowaniem komputerowym CLOGP-3) (b). W przypadku związków zjonizowanych ważne jest właściwe rozważenie ładunku lub stopnia jonizacji.
Procedury obliczeń
π-Metoda Hanscha
Oryginalna stała podstawienia hydrofobowego, wprowadzona przez Fujita i współpracowników (f) jest zdefiniowana jako:
πx = log Pow (PhX) – log Pow (PhH)
gdzie Pow (PhX) jest współczynnikiem podziału pochodnej aromatycznej, a Pow (PhH) związku macierzystego
(np. πCl = log Pow (C6H5Cl) – log Pow (C6H6) = 2,84 – 2,13 = 0,71).
Zgodnie z tą definicją π-metoda stosowana jest głównie dla podstawienia aromatycznego. N-wartości dla większej liczby podstawników zostało stabelaryzowane w pozycjach bibliograficznych (b), (c) i (d). Są one stosowane dla obliczeń log Pow cząsteczek aromatycznych lub podstruktur.
Metoda Rekkera
Zgodnie z Rekkerem (g) wartość log Pow jest obliczana, jak następuje:
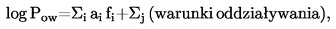
gdzie fi reprezentuje różne stałe fragmentów cząsteczki, natomiast ai – częstotliwość ich występowania w badanej cząsteczce. Składniki korekcji mogą być wyrażone jako całkowita wielokrotność pojedynczej stałej Cm (tak zwana „stała magiczna”). Stałe fragmentu fi, i Cm zostały ustalone z wykazu 1 054 doświadczalnych wartości Pow (825 związków) poprzez wielokrotną analizę regresyjną (c), (h). Ustalenie składników oddziaływań przeprowadzono zgodnie z zestawem zasad opisanych w pozycjach bibliograficznych (e), (h) i (i).
Metoda Hansch-Leo
Zgodnie z Hansch i Leo (c) wartość log Pow jest obliczana z:
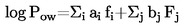
gdzie fi przedstawia różne stałe fragmentu cząsteczkowego, Fj składniki korekcji oraz ai, bi odpowiadające częstotliwości występowania. Wyprowadzony z doświadczalnych wartości Pow, wykaz atomowych i fragmentów grup wartości oraz wykaz składników korekcji Fj (tak zwanych „współczynników”) zostały ustalone metodą prób i błędów. Składniki korekcji zostały uporządkowane w kilka różnych klas (a), (c). Wzięcie pod uwagę wszystkich zasad i składników korekcji jest względnie skomplikowane i zużywające czas. Opracowano pakiet oprogramowania (b).
Metoda połączona
Obliczenie log Pow złożonych cząsteczek może być znacząco poprawione, jeżeli cząsteczka zostanie rozcięta w większe podstruktury, dla których godne zaufania wartości log Pow są dostępne, albo z tabeli z pozycji bibliograficznych (b), (c), albo z własnych pomiarów. Takie fragmenty (np. heterocykle, antrachinon, azobenzen) mogą być zatem połączone z wartościami π Hanscha lub ze stałymi fragmentów Rekkera lub Leo.
Uwagi:
|
(i) |
metody obliczeniowe mogą być stosowane tylko do częściowo lub całkowicie zjonizowanych związków, gdzie możliwe jest branie pod uwagę koniecznych współczynników korekcji; |
|
(ii) |
jeżeli można założyć wewnątrzcząsteczkowe wiązania wodorowe, odpowiadające składniki korekcji (od około +0,6 do +1,0 log Pow, jednostek) muszą być dodane (a). Wskazania obecności takich wiązań można uzyskać z przestrzennych modeli lub danych spektroskopowych cząsteczki; |
|
(iii) |
jeżeli jest możliwe występowanie kilku postaci tautomerycznych, podstawą do obliczeń powinna być najbardziej prawdopodobna postać; |
|
(iv) |
należy starannie prowadzić weryfikacje wykazu stałych dla fragmentów. |
Sprawozdanie
Jeżeli stosuje się metody obliczenia/oceny, raport z badań powinien, o ile to możliwe, zawierać następujące informacje:
|
— |
opis substancji (mieszanina, zanieczyszczenia itp.), |
|
— |
wskazanie wszystkich możliwych wewnątrzcząsteczkowych wiązań wodorowych, dysocjacji, ładunkui wszystkich innych niezwykłych oddziaływań (np. tautomeria), |
|
— |
opis metody obliczenia, |
|
— |
identyfikacja lub zasób bazy danych, |
|
— |
cechy wyróżniające wyboru fragmentów, |
|
— |
pełna dokumentacja obliczenia. |
LITERATURA
|
(a) |
W.J. Lyman, W.F. Reehl and D.H. Rosenblatt (ed.), Handbook of Chemical Property Estimation Methods, McGraw-Hill, New York, 1983. |
|
(b) |
Pomona College, Medicinal Chemistry Project, Claremont, California 91711, USA, Log P Database and Med. Chem. Software (Program CLOGP-3). |
|
(c) |
C. Hansch, A.J. Leo, Substituent Constants for Correlation Analysis in Chemistry and Biology, John Wiley, New York, 1979. |
|
(d) |
A. Leo, C. Hansch, D. Elkins, Chem. Rev., 1971, vol. 71, 525. |
|
(e) |
R.F. Rekker, H.M. de Kort, Eur. J. Med. Chem. -Chill. Ther. 1979, vol. 14, 479. |
|
(f) |
T. Fujita, J. Iwasa and C. Hansch, J. Amer. Chem. Soc., 1964, vol. 86, 5175. |
|
(g) |
R.F. Rekker, The Hydrophobic Fragmental Constant, Pharmacochemistry Library, Elsevier, New York, 1977, vol. 1. |
|
(h) |
C.V. Eadsforth, P. Moser, Chemosphere, 1983, vol. 12, 1459. |
|
(i) |
R.A. Scherrer, ACS, American Chemical Society, Washington D.C., 1984, Symposium Series 255, p. 225. |
Dodatek 2
Zalecane substancje odniesienia dla metody HPLC
|
Nr |
substancja odniesienia |
log Pow |
pKa |
|
1 |
2-butanon |
0,3 |
|
|
2 |
4-acetylopirydyna |
0,5 |
|
|
3 |
anilina |
0,9 |
|
|
4 |
acetanilid |
1,0 |
|
|
5 |
alkohol benzylowy |
1,1 |
|
|
6 |
p-metoksyfenol |
1,3 |
pKa = 10,26 |
|
7 |
kwas fenoksyoctowy |
1,4 |
pKa = 3,12 |
|
8 |
fenol |
1,5 |
pKa = 9,92 |
|
9 |
2,4-dinitrofenol |
1,5 |
pKa = 3,96 |
|
10 |
benzonitryl |
1,6 |
|
|
11 |
fenyloacetonitryl |
1,6 |
|
|
12 |
alkohol 4-metylobenzylowy |
1,6 |
|
|
13 |
acetofenon |
1,7 |
|
|
14 |
2-nitrofenol |
1,8 |
pKa = 7,17 |
|
15 |
kwas 3-nitrobenzoesowy |
1,8 |
pKa = 3,47 |
|
16 |
4-chloroanilina |
1,8 |
pKa = 4,15 |
|
17 |
nitrobenzen |
1,9 |
|
|
18 |
alkohol cynamonowy |
1,9 |
|
|
19 |
kwas benzoesowy |
1,9 |
pKa = 4,19 |
|
20 |
p-krezol |
1,9 |
pKa = 10,17 |
|
21 |
kwas cynamonowy |
2,1 |
pKa = 3,89 cis 4,44 trans |
|
22 |
anizol |
2,1 |
|
|
23 |
benzoesan metylu |
2,1 |
|
|
24 |
benzen |
2,1 |
|
|
25 |
kwas 3-metylobenzoesowy |
2,4 |
pKa = 4,27 |
|
26 |
4-chlorofenol |
2,4 |
pKa = 9,1 |
|
27 |
trichloroetylen |
2,4 |
|
|
28 |
atrazyna |
2,6 |
|
|
29 |
benzoesan etylu |
2,6 |
|
|
30 |
2,6-dichlorobenzonitryl |
2,6 |
|
|
31 |
kwas 3-chlorobenzoesowy |
2,7 |
pKa = 3,82 |
|
32 |
toluen |
2,7 |
|
|
33 |
1-naftol |
2,7 |
pKa = 9,34 |
|
34 |
2,3-dichloroanilina |
2,8 |
|
|
35 |
chlorobenzen |
2,8 |
|
|
36 |
allilofenyloeter |
2,9 |
|
|
37 |
bromobenzen |
3,0 |
|
|
38 |
etylobenzen |
3,2 |
|
|
39 |
benzofenon |
3,2 |
|
|
40 |
4-fenylofenol |
3,2 |
pKa = 9,54 |
|
41 |
tymol |
3,3 |
|
|
42 |
1,4-dichlorobenzen |
3,4 |
|
|
43 |
difenyloamina |
3,4 |
pKa = 0,79 |
|
44 |
naftalen |
3,6 |
|
|
45 |
benzoesan fenylu |
3,6 |
|
|
46 |
izopropylobenzen |
3,7 |
|
|
47 |
2,4,6-trichlorofenol |
3,7 |
pKa = 6 |
|
48 |
bifenyl |
4,0 |
|
|
49 |
benzoesan benzylu |
4,0 |
|
|
50 |
2,4-dinitro-6-sec-butylofenol |
4,1 |
|
|
51 |
1,2,4-trichlorobenzen |
4,2 |
|
|
52 |
kwas dodekanowy |
4,2 |
|
|
53 |
difenyloeter |
4,2 |
|
|
54 |
n-butylobenzen |
4,5 |
|
|
55 |
fenantren |
4,5 |
|
|
56 |
fluoranten |
4,7 |
|
|
57 |
dibenzyl |
4,8 |
|
|
58 |
2,6-difenylopirydyna |
4,9 |
|
|
59 |
trifenyloamina |
5,7 |
|
|
60 |
DDT |
6,2 |
|
|
Inne substancje odniesienia o niskim log Pow |
|||
|
1 |
kwas nikotynowy |
-0,07 |
|
A.9. TEMPERATURAZAPŁONU
1. METODA
1.1. WPROWADZENIE
Jest użyteczne posiadanie wstępnych informacji na temat zapalności substancji przed przeprowadzeniem niniejszego badania. Procedura badania jest odpowiednia dla ciekłych substancji, które mogą być zapalone źródłem zapłonu. Metody badania zamieszczone w niniejszym tekście są godne zaufania tylko dla zakresów zapłonu, które są wyszczególnione w pojedynczych metodach.
Przy wyborze stosowanej metody należy rozważyć możliwość chemicznych reakcji substancją a uchwytem próbki.
1.2. DEFINICJE I JEDNOSTKI
Temperatura zapłonu jest najniższą temperaturą, skorygowaną do ciśnienia 101,325 kPa, w której ciecz wydziela pary, w warunkach określonych w metodzie badania, w takiej ilości, że wytwarzana jest mieszanina palna para/powietrze w naczyniu badawczym.
Jednostki: oC
t = T – 273,15
(t w oC i T w oK)
1.3. SUBSTANCJE ODNIESIENIA
Nie ma konieczności stosowania substancji odniesienia za każdym razem, gdy przeprowadzane są badania nowej substancji. Powinny one służyć głównie sprawdzeniu od czasu do czasu wydajności metody i pozwolić na porównanie wyników z innymi metodami.
1.4. ZASADA METODY
Substancja jest umieszczana w naczyniu badawczym i podgrzewana lub chłodzona to temperatury badania zgodnej z procedurą opisaną w indywidualnej metodzie badania. Próby zapłonu są przeprowadzane w celu stwierdzenia, czy próbka zapłonie lub nie w temperaturze badania.
1.5. KRYTERIA JAKOŚCI
1.5.1. Powtarzalność
Powtarzalność zmienia się zgodnie z zakresem temperatury zapłonu i zastosowanej metody badania; maksymalnie 2 oC.
1.5.2. Czułość
Czułość zależy od zastosowanej metody badania.
1.5.3. Specyficzność
Specyficzność niektórych metod badania jest ograniczona do niektórych amplitud temperatury zapłonu i zależy od cech samej substancji (np. wysoka lepkość).
1.6. OPIS METODY
1.6.1. Przygotowania
Próbka badanej substancji jest umieszczana w przyrządzie badawczym zgodnie z 1.6.3.1 i/lub 1.6.3.2.
Dla bezpieczeństwa zalecane jest, by w metodzie używać próbki o małej liczności. Około 2 cm3 stosuje się dla substancji wysokoenergetycznych lub toksycznych.
1.6.2. Warunki badania
Przyrząd powinien, tak dalece jak jest to zgodne z bezpieczeństwem, być umieszczony w pozycji wolnej od przeciągów powietrza.
1.6.3. Przeprowadzenie badania
1.6.3.1. Metoda równowagi
Zob. ISO 1516, ISO 3680, ISO 1523, ISO 3679.
1.6.3.2. Metoda nierównowagi
Przyrząd Abela:
Zob. BS 2000 część 170, NF M07-011, NF T66-009.
Przyrząd Abel-Pensky:
Zob. EN 57, DIN 51755 część 1 (dla temperatur od 5 do 65 oC), DIN 51755 część 2 (dla temperatur poniżej 5 oC), NF M07-036.
Przyrząd Tag:
Zob. ASTM D 56.
Przyrząd Pensky-Martens:
Zob. ISO 2719, EN 11, DIN 51758, ASTM D 93, BS 2000-34, NF M07-019.
Uwagi:
Jeżeli temperatura zapłonu, oznaczona metoda nierównowagi, według 1.6.3.2, wynosi 0 ± 2 oC, 21 ± 2 oC lub 55 ± 2 oC, należy je potwierdzić metodą równowagi, stosując ten sam przyrząd.
Tylko te metody, które podają temperaturę jako temperaturę zapłonu, stosuje się do zgłoszenia.
Dla oznaczenia temperatury zapłonu lepkich cieczy (farby, gumy oraz podobne) zawierających rozpuszczalniki można używać tylko przyrządów i metod badań stosownych dla oznaczania temperatury zapłonu cieczy lepkich.
Zob. ISO 3679, ISO 3680, ISO 1523, DIN 53213 część 1.
|
2. |
DAN |
3. SPORZĄDZANIE SPRAWOZDANIA
Sprawozdanie z badań zawiera w miarę możliwości następujące informacje:
|
— |
precyzyjna specyfikacja substancji (tożsamość i zanieczyszczenia), |
|
— |
stwierdzenie zastosowania danej metody, jak również o wszystkich możliwych odchyleniach, |
|
— |
wyniki i wszystkie dodatkowe uwagi dotyczące interpretacji wyników. |
4. LITERATURA
Brak.
A.10. ZAPALNOŚĆ (CIAŁA STAŁE)
1. METODA
1.1. WPROWADZENIE
Dla przeprowadzenia badania przydatne jest posiadanie wstępnych informacji na temat ewentualnych właściwości wybuchowych substancji.
Niniejsze badanie należy stosować jedynie do substancji sproszkowanych, ziarnistych i pastopodobnych.
Aby nie uwzględniać wszystkich substancji, które można zapalić, a tylko te, które palą się szybko lub te, których zachowanie się przy spalaniu jest z jakichkolwiek względów szczególnie niebezpieczne, jedynie substancje, których szybkość spalania przekracza pewne wartości graniczne, uważa się za wysoce łatwopalne.
Jest szczególnie niebezpieczne, jeżeli inkandescencja rozchodzi się poprzez proszek metalu, z powodu trudności z ugaszeniem ognia. Proszki metali należy uznać za wysoce łatwopalne, jeżeli podtrzymują one rozszerzanie inkandescencji w masie w czasie określonego czasu.
1.2. DEFINICJA I JEDNOSTKI
Czas spalania jest wyrażony w sekundach.
1.3. SUBSTANCJE ODNIESIENIA
Nie wyszczególniono.
1.4. ZASADA METODY
Substancja jest formowana w nieprzerwaną wstęgę lub proszkową smugę o długości około 250 mm i przeprowadza się wstępne badanie klasyfikacyjne do określenia, czy zachodzi zapłon przez płomień gazu, rozprzestrzenianie przez przypalanie płomieniem lub tlenie się. Jeżeli zachodzi rozprzestrzenianie ponad 200 mm w sformowanym materiale w obrębie określonego czasu, przeprowadzany jest program pełnego badania w celu określenia szybkości spalania.
1.5. KRYTERIA JAKOŚCI
Nie ustalono.
1.6. OPIS METODY
1.6.1. Wstępne badanie klasyfikacyjne
Substancja jest formowana w nieprzerwaną wstęgę lub proszkową smugę o długości około 250 mm, 20 mm szerokości i 10 mm wysokości na niepalnej, nieporowatej płycie bazowej o niskim przewodnictwie ciepła. Gorącym płomieniem palnika gazowego (minimalna średnica 5mm), działa się na jeden z końców smugi proszku do momentu zapalenia się proszku lub przez maksimum 2 minuty (5 minut dla proszków metali lub stopów metali). Należy zanotować, czy spalanie rozchodzi wzdłuż 200 mm smugi w obrębie czasu badania 4 minut (lub 40 minut dla proszków metali). Jeżeli substancja nie zapala się i nie podtrzymuje spalania albo przez spalanie z płomieniem lub z tleniem się wzdłuż 200 mm smugi proszku w obrębie czasu 4 minut (lub 40 minut) czasu trwania badania, wtedy substancji nie uważa się za wysoce łatwopalną i dalsze badania nie są wymagane. Jeżeli substancja podtrzymuje spalenie się smugi proszku długości 200 mm w czasie mniejszym od 4 minut, lub mniejszym od 40 minut dla proszków metali, należy przeprowadzić procedurę niżej opisaną (ppkt 1.6.2. i następne).
1.6.2. Badanie szybkości spalania
1.6.2.1. Przygotowanie
Sproszkowanymi lub ziarnistymi substancjami swobodnie napełnia się formę długą na 250 mm, o trójkątnym przekroju wewnętrznej wysokości 10 mm i szerokości 20 mm. Z obu stron formy w kierunku wzdłużnym montuje się dwie metalowe płyty z ograniczeniami poprzecznymi, które wystają 2 mm poza górną krawędź obszaru przekroju trójkątnego (rysunek). Formę zrzuca się następnie trzykrotnie z wysokości 2 cm na twardą powierzchnię. W razie potrzeby napełnia się wtedy formę ponownie. Poprzeczne ograniczenia są następnie zdejmowane i zeskrobuje się nadmiar substancji. Płytę bazową, niepalną, nieporowatą i o niskim przewodnictwie cieplnym umieszcza się na górze formy, przyrząd odwraca się i zdejmuje formę.
Substancjami pastopodobnymi powleka się w postaci liny o długości 250 mm i przekroju około 1 cm2, niepalną, nieporowatą i o niskim przewodnictwie cieplnym płytę bazową.
1.6.2.2. Warunki badania
W przypadku substancji wrażliwych na wilgoć badanie należy przeprowadzić jak najszybciej po ich wyjęciu z pojemnika.
1.6.2.3. Przeprowadzenie badania
Ustawić stos w kierunku prostopadłym do ciągu powietrza w szafie wyciągowej.
Szybkość przepływu powietrza powinna być wystarczająca do zapobieżenia przedostaniu się dymu do laboratorium i nie powinna być zmieniana w trakcie badania. Wokół przyrządu należy ustawić ekran chroniący przed ciągiem powietrza.
Stosuje się gorący płomień z palnika gazowego (minimalna średnica 5 mm) do zapalenia z jednego końca stosu. Gdy stos spali się na długości 80 mm, na następnych 100 mm mierzy się szybkość spalania.
Badanie przeprowadza się sześć razy, używając za każdym razem czystej, chłodnej płyty, niezależnie od wcześniej obserwowanego pozytywnego wyniku.
2. DANE
Istotne z punktu widzenia oceny są czas spalania z wstępnego badania klasyfikacyjnego (1.6.1) i najkrótszy z sześciu zbadanych (1.6.2.3) czas spalania.
3. SPORZĄDZANIE SPRAWOZDANIA
3.1. SPRAWOZDANIE Z BADANIA
Sprawozdanie z badań zawiera w miarę możliwości następujące informacje:
|
— |
precyzyjna specyfikacja substancji (tożsamość i zanieczyszczenia), |
|
— |
opis substancji badanej i jej stanu fizycznego, w tym zawartości wilgoci, |
|
— |
wyniki z wstępnego badania klasyfikacyjnego i z badania szybkości spalania jeśli przeprowadzono, |
|
— |
wszelkie dodatkowe uwagi istotne dla interpretacji wyników. |
3.2. INTERPRETACJA WYNIKÓW
Sproszkowane, ziarniste lub pastopodobne substancje są uważane za wysoce łatwopalne, jeżeli czas spalania w jakimkolwiek badaniu przeprowadzonym zgodnie z procedurą opisaną w 1.6.2 jest mniejszy niż 45 sekund. Proszki metali lub stopów metali, uważa się za wysoce łatwopalne, jeżeli zapalają się i płomień lub strefa reakcji rozszerza się w całej próbce w czasie 10 minut lub mniejszym.
4. LITERATURA
NF T 20-042 (SEPT 85). Chemical products for industrial use. Determination of the flammability of solids.
Dodatek
Rysunek
Forma i akcesoria do przygotowania stosu
(wszystkie wymiary w milimetrach)
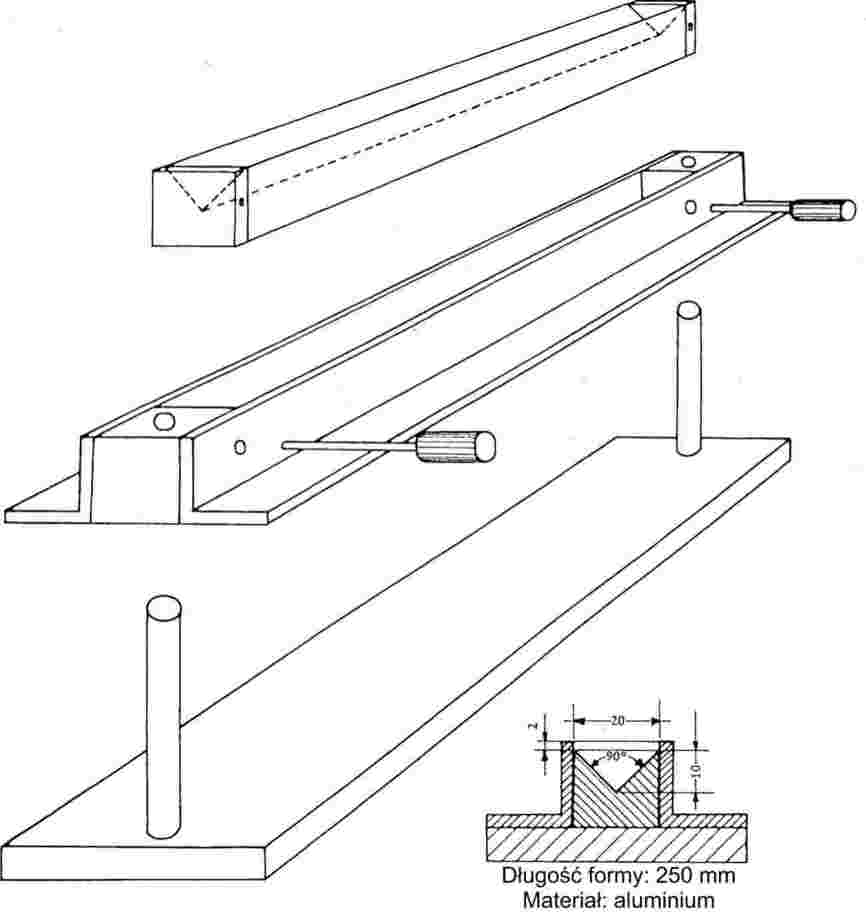
A.11. ZAPALNOŚĆ (GAZY)
1. METODA
1.1. WPROWADZENIE
Niniejsza metoda pozwala na oznaczenie, czy gazy zmieszane z powietrzem w temperaturze pokojowej (około 20 oC) i pod ciśnieniem atmosferycznym są palne, a jeśli tak, to w jakim zakresie stężeń. Mieszaniny badanego gazu z powietrzem we wzrastającym stężeniu poddaje się działaniu iskry elektrycznej i obserwuje się, czy dochodzi do zapłonu.
1.2. DEFINICJA I JEDNOSTKI
Zakres palności to zakres stężenia między dolną i górną granicą wybuchowości. Dolna i górna granica wybuchowości to granice stężenia palnego gazu zmieszanego z powietrzem, przy których nie dochodzi do propagacji płomienia.
1.3. SUBSTANCJE ODNIESIENIA
Nie wyszczególniono.
1.4. ZASADA METODY
Stężenie gazu w powietrzu zwiększa się w kolejnych etapach, przy czym na każdym etapie mieszaninę tę poddaje się działaniu iskry elektrycznej.
1.5. KRYTERIA JAKOŚCI
Nie ustalono.
1.6. OPIS METODY
1.6.1. Przyrząd
Naczynie do badania to prosty cylinder szklany o minimalnej średnicy wewnętrznej wynoszącej 50 mm i minimalnej wysokości 300 mm. Elektrody zapłonowe są umieszczone w odległości 3 do 5 mm od siebie, 60 mm ponad dnem cylindra. Cylinder jest wyposażony w otwór dekompresyjny. Przyrząd musi być osłonięty w taki sposób, aby ograniczyć wszelkie szkody związane z ewentualnym wybuchem.
Iskra o stałej indukcji o czasie trwania 0,5 s, generowana z wysokonapięciowego transformatora o napięciu wyjściowym od 10 do 15 kV (maksymalna moc zasilająca 300 W), stosowana jest jako źródło zapłonu. Przykład odpowiedniego przyrządu opisano w pozycji bibliograficznej (2).
1.6.2. Warunki badania
Badanie należy wykonać w temperaturze pokojowej (około 20 oC).
1.6.3. Przeprowadzenie badania
Przy użyciu pomp dozujących do szklanego cylindra wprowadza się znane stężenia gazu w powietrzu. Przez mieszaninę przepuszcza się iskrę i obserwuje się, czy od źródła zapłonu oddzieli się płomień i czy będzie on ulegać niezależnej propagacji. Stężenie gazu zmienia się etapowo o 1 % obj., aż dojdzie do zapłonu w sposób opisany powyżej.
Jeżeli budowa chemiczna gazu wskazuje, że może on być niepalny, a skład mieszaniny stechiometrycznej z powietrzem może być obliczony, należy zbadać tylko mieszaniny w zakresie 10 % poniżej składu stechiometrycznego oraz 10 % powyżej tego składu, w krokach co 1 %.
2. DANE
Jedyną istotną informacją dla ustalenia omawianej właściwości jest informacja na temat propagacji płomienia.
3. SPORZĄDZANIE SPRAWOZDANIA
Sprawozdanie z badań zawiera w miarę możliwości następujące informacje:
|
— |
precyzyjna specyfikacja substancji (tożsamość i zanieczyszczenia), |
|
— |
opis zastosowanego przyrządu, z podaniem wymiarów, |
|
— |
temperatura, w której wykonano badanie, |
|
— |
stężenia badane i uzyskane wyniki, |
|
— |
wynik badania: gaz niepalny lub gaz wysoce łatwopalny, |
|
— |
jeżeli ustalono, że gaz jest niepalny, należy wtedy potwierdzić zakres stężenia, w którym badano go w odstępach co 1 %, |
|
— |
należy przedstawić wszelkie informacje i uwagi istotne dla interpretacji wyników. |
4. LITERATURA
|
(1) |
NF T 20-041 (SEPT 85). Chemical products for industrial use. Determination of the flammability of gases. |
|
(2) |
W.Berthold, D.Conrad, T.Grewer, H.Grosse-Wortmann, T.Redeker und H.Schacke. „Entwicklung einer Standard-Apparatur zur Messung von Explosionsgrenzen”. Chem.-Ing.-Tech. 1984, vol. 56, 2, 126-127. |
A.12. ZAPALNOŚĆ (W KONTAKCIE Z WODĄ)
1. METODA
1.1. WPROWADZENIE
Niniejszą metodę badania można wykorzystywać do ustalenia, czy reakcja substancji z wodą prowadzi do tworzenia się niebezpiecznych ilości gazu lub gazów, które mogą być wysoce łatwopalne.
Metodę badania można stosować w odniesieniu zarówno do ciał stałych, jak i cieczy. Metoda ta nie ma zastosowania do substancji, które ulegają spontanicznemu zapłonowi w kontakcie z powietrzem.
1.2. DEFINICJE IJEDNOSTKI
Wysoce łatwopalny: Substancje i preparaty, które, w kontakcie z wodą lub wilgotnym powietrzem, uwalniają wysoce łatwopalne gazy w niebezpiecznych ilościach z minimalną szybkością 1 litr/kg na godzinę.
1.3. ZASADA METODY
Substancję bada się w wieloetapowej sekwencji opisanej poniżej; gdy dojdzie do zapłonu na dowolnym z etapów, kontynuowanie badania nie jest potrzebne. Jeżeli wiadomo, że substancja nie reaguje gwałtownie z wodą, należy postępować zgodnie z etapem 4 (1.3.4).
1.3.1. Etap 1
Substancję badaną umieszcza się w rynience z wodą destylowaną w temperaturze 20 oC i zapisuje się, czy uwolniony gaz zapali się, czy nie.
1.3.2. Etap 2
Substancję badaną umieszcza się na bibule filtracyjnej pływającej na powierzchni naczynia zawierającego wodę destylowaną w temperaturze 20 oC, po czym zapisuje się, czy uwolniony gaz zapali się, czy nie. Rolą bibuły filtracyjnej jest jedynie utrzymywanie substancji w jednym miejscu, tak aby zwiększyć szanse na uzyskanie zapłonu.
1.3.3. Etap 3
Z substancji badanej tworzy się stos o wysokości około 2 cm i średnicy 3 cm. Do stosu dodaje się kilka kropli wody i zapisuje się, czy dochodzi do zapalenia się uwolnionego gazu, czy nie.
1.3.4. Etap 4
Substancję badaną miesza się z wodą destylowaną w temperaturze 20 oC, po czym mierzy się szybkość uwalniania gazu w czasie siedmiu godzin w odstępach jednogodzinnych. Jeżeli oznaczana szybkość uwalniania wykazuje niesystematyczne wahania lub wzrasta po siedmiu godzinach, czas pomiaru należy wydłużyć do maksimum pięciu dni. Badanie można przerwać, gdy mierzona szybkość w dowolnym momencie przekroczy 1 litr/kg na godzinę.
1.4. SUBSTANCJE ODNIESIENIA
Nie wyszczególniono.
1.5. KRYTERIA JAKOŚCI
Nie ustalono.
1.6. OPIS METOD
1.6.1. Etap 1
1.6.1.1. Warunki badania
Badanie wykonywane jest w temperaturze pokojowej (około 20 oC).
1.6.1.2. Wykonanie badania
Niewielką ilość (o średnicy około 2 mm) substancji badanej należy umieścić w rynience zawierającej wodę destylowaną. Należy zapisać, czy (i) uwalnia się jakikolwiek gaz oraz czy (ii) dochodzi do zapalenia się gazu. W przypadku gdy dochodzi do zapalenia się gazu, nie są potrzebne dalsze badania substancji, gdyż substancję taką uważa się za niebezpieczną.
1.6.2. Etap 2
1.6.2.1. Przyrząd
Na powierzchni wody destylowanej w dowolnym, właściwym naczyniu, np. w parownicy o średnicy 100 mm, kładzie się płasko bibułę filtracyjną.
1.6.2.2. Warunki badania
Badanie wykonywane jest w temperaturze pokojowej (około 20 oC).
1.6.2.3. Przeprowadzenie badania
Niewielką ilość substancji badanej (o średnicy około 2 mm) umieszcza się na środku bibuły filtracyjnej. Należy zapisać, czy (i) uwalnia się jakikolwiek gaz oraz czy (ii) dochodzi do zapalenia się gazu. W przypadku gdy dochodzi do zapalenia się gazu, nie są potrzebne dalsze badania substancji, gdyż substancję taką uważa się za niebezpieczną.
1.6.3. Etap 3
1.6.3.1. Warunki badania
Badanie wykonywane jest w temperaturze pokojowe (około 20 oC).
1.6.3.2. Przeprowadzenie badania
Z substancji badanej należy zrobić stos o wysokości około 2 cm i średnicy 3 cm, z wrębem u góry. Do wgłębienia dodaje się kilka kropli wody i zapisuje się, czy: (i) uwalnia się jakikolwiek gaz oraz czy (ii) dochodzi do zapalenia się gazu. W przypadku gdy dochodzi do zapalenia się gazu, nie są potrzebne dalsze badania substancji, gdyż substancję taką uważa się za niebezpieczną.
1.6.4. Etap 4
1.6.4.1. Przyrząd
Przyrząd jest ustawiony jak pokazano na rysunku.
1.6.4.2. Warunki badania
Sprawdzić pojemnik substancji badanej na jakikolwiek proszek < 500 μm (wielkość ziarna). Jeżeli proszek tworzy więcej niż 1 % wagowo całości lub jeżeli próbka jest krucha, wtedy całą substancję należy zmielić na proszek przed badaniem dla umożliwienia zmniejszenia wielkości ziarna w czasie przechowywania i posługiwania się nią; w innych przypadkach bada się substancję taką, jak otrzymano. Badanie należy wykonać w temperaturze pokojowej (około 20 oC) i pod ciśnieniem atmosferycznym.
1.6.4.3. Przeprowadzenie badania
Od 10 do 20 ml wody umieszcza się we wkraplaczu przyrządu, a 10 g substancji umieszcza się w kolbie stożkowej. Objętość uwalniającego się gazu można zmierzyć dowolną, właściwą metodą. Otwiera się kurek wkraplacza w celu wpuszczenia wody do kolby stożkowej i uruchamia się stoper. Wydzielanie gazu mierzone jest co godzinę w trakcie okresu siedmiu godzin. Jeżeli w ciągu tego okresu czasu, wydzielanie gazu jest nierówne, lub pod koniec tego okresu, szybkość wydzielania gazu wzrasta, wtedy pomiary należy kontynuować do pięciu dni. Jeżeli w dowolnym momencie pomiaru szybkość wydzielania gazu przekracza 1 litr/kg na godzinę, należy przerwać badanie. Badanie należy przeprowadzić trzykrotnie.
Jeżeli nie jest znana chemiczna tożsamość gazu, należy go poddać analizie. Jeżeli gaz zawiera wysoce łatwopalne składniki i nie wiadomo, czy mieszanina jest wysoce łatwopalna, należy przygotować mieszaninę o takim samym składzie i poddać badaniu przy użyciu metody A.11.
2. DANE
Substancja jest uważana za niebezpieczną, jeżeli:
|
— |
samozapłon ma miejsce na każdy etapie procedury badania, lub |
|
— |
występuje wydzielanie gazu palnego o szybkości większej niż 1 litr/kg substancji na godzinę. |
3. SPORZĄDZANIE SPRAWOZDANIA
Sprawozdanie z badań zawiera w miarę możliwości następujące informacje:
|
— |
precyzyjna specyfikacja substancji (tożsamość i zanieczyszczenia), |
|
— |
szczegóły każdego wstępnego przygotowania badanej substancji, |
|
— |
wyniki badań (etapy 1, 2, 3 i 4), |
|
— |
tożsamość chemiczna wydzielanego gazu, |
|
— |
szybkość wydzielania gazu, jeżeli przeprowadzono etap 4 (1.6.4), |
|
— |
wszystkie dodatkowe uwagi stosowne do interpretacji wyników. |
4. LITERATURA
|
(1) |
Recommendations on the transport of dangerous goods, test and criteria, 1990, United Nations, New York. |
|
(2) |
NF T 20-040 (SEPT 85). Chemical products for industrial use. Determination of the flammability of gases formed by the hydrolysis of solid and liquid products. |
Dodatek
Rysunek
Przyrząd
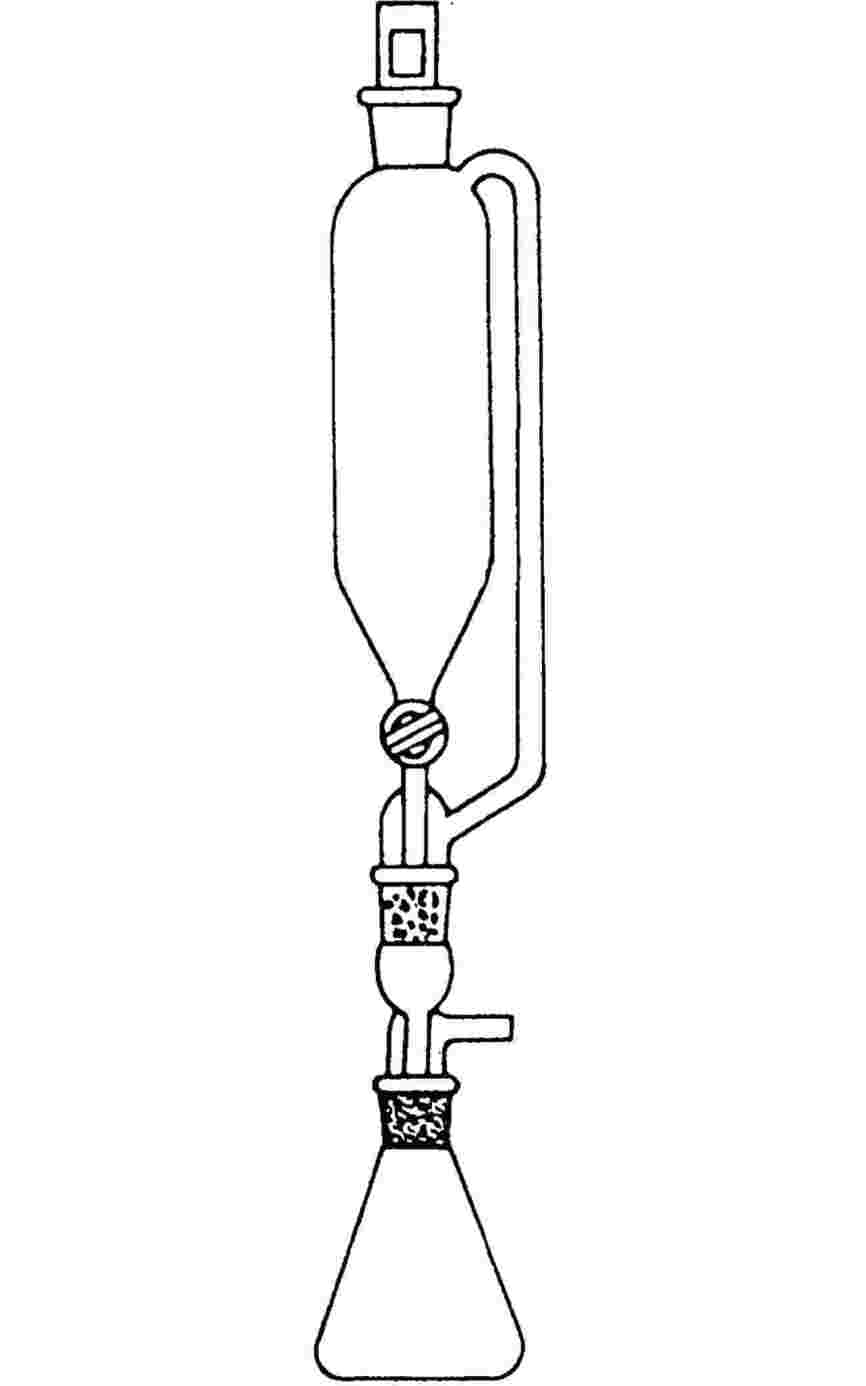
A.13. WŁAŚCIWOŚCI PIROFORYCZNE CIAŁ STAŁYCH I CIECZY
1. METODA
1.1. WPROWADZENIE
Procedura badań ma zastosowanie do stałych lub ciekłych substancji, które w małych ilościach samorzutnie zapalają się w krótkim czasie po wejściu z powietrzem w temperaturze pokojowej (około 20 oC).
Substancje, które wymagają wystawienia na powietrze na okres godzin lub dni w temperaturze pokojowej lub w podwyższonej temperaturze, zanim zajdzie zapłon nie są objęte niniejszą metodą badania.
1.2. DEFINICJE I JEDNOSTKI
Substancje uważa się za posiadające właściwości piroforyczne, jeżeli zapalają się lub ulegają zwęglaniu zgodnie z warunkami opisanymi w 1.6.
Samozapalność cieczy może wymagać również zbadania za pomocą metody A.15 Temperatura samozapłonu (ciecze i gazy).
1.3. SUBSTANCJE ODNIESIENIA
Nie wyszczególniono.
1.4. ZASADA METODY
Substancja, czy w postaci ciała stałego czy cieczy, jest dodawana do obojętnego nośnika i poddawana kontaktowi z powietrzem w temperaturze otoczenia na okres pięciu minut. Jeżeli substancje nie zapalają się, absorbuje się je na bibule filtracyjnej i wystawia na powietrze w temperaturze otoczenia (około 20 oC) na czas pięciu minut. Jeżeli ciało stałe lub ciecz zapala się, lub ciecz zapala się lub zwęgla bibułę filtracyjną, wtedy substancję uważa się za piroforyczną.
1.5. KRYTERIA JAKOŚCI
Powtarzalność: ze względów istotnych dla bezpieczeństwa pojedynczy pozytywny wynik jest wystarczający do uznania substancji za piroforyczną.
1.6. OPIS METODY BADANIA
1.6.1. Przyrząd
Tygiel porcelanowy o średnicy około 10 cm napełnia się ziemią okrzemkową do wysokości około 5 mm w temperaturze pokojowej.
Uwaga:
Ziemię okrzemkową lub dowolną inną porównywalną, obojętną substancję, która jest powszechnie dostępna, bierze się jako reprezentacyjną dla gleby, na którą badana substancja może się uwolnić w razie wypadku.
Do badania cieczy, które nie zapalają się w kontakcie z powietrzem, gdy poddaje się je kontaktowi z obojętnym nośnikiem, wymagana jest sucha bibuła filtracyjna.
1.6.2. Przeprowadzenie badania
a) Sproszkowane ciała stałe
Od 1 do 2 cm3 badanej substancji w proszku sypie się z wysokości około 1 m na niepalną powierzchnię i obserwuje się, czy substancja ta ulegnie zapaleniu podczas spadania lub w ciągu pięciu minut od opadnięcia.
Badanie przeprowadza się sześć razy, niezależnie od wystąpienia zapalenia się.
b) Ciecze
Około 5 cm3 badanej cieczy wlewa się do przygotowanego tygla porcelanowego i obserwuje się, czy substancja zapali się w ciągu pięciu minut.
Jeżeli w sześciu badaniach nie nastąpi zapłon, należy przeprowadzić następujące badania:
0,5 ml badanej próbki za pomocą strzykawki przenosi się na wgniecioną bibułę filtracyjną i obserwuje się, czy nastąpi zapalenie się lub zwęglenie bibuły filtracyjnej w czasie pięciu minut po naniesieniu cieczy. Niezależnie od tego, czy zachodzi zapalenie się lub zwęglenie, badanie przeprowadza się trzy razy.
2. DANE
2.1. OPRACOWANIE WYNIKÓW
Wykonywania badań można zaprzestać, gdy tylko w jednym z nich uzyska się dodatni wynik.
2.2. OCENA
Jeżeli substancja zapala się w czasie pięciu minut od dodania obojętnego nośnika i wystawienia na powietrze, lub ciekła substancja zwęgla lub zapala bibułę filtracyjną w czasie pięciu minut po naniesieniu i wystawieniu na powietrze, jest uważana za piroforyczną.
3. SPORZĄDZANIE SPRAWOZDANIA
Sprawozdanie z badań zawiera w miarę możliwości następujące informacje:
|
— |
precyzyjna specyfikacja substancji (tożsamość i zanieczyszczenia), |
|
— |
wyniki badań, |
|
— |
wszystkie dodatkowe uwagi dotyczące interpretacji wyników. |
4. LITERATURA
|
(1) |
NF T 20-039 (SEPT 85). Chemical products for industrial use. Determination of the spontaneous flammability of solids and liquids. |
|
(2) |
Recommendations on the Transport of Dangerous Goods, Test and criteria, 1990, United Nations, New York. |
A.14. WŁAŚCIWOŚCI WYBUCHOWE
1. METODA
1.1. WPROWADZENIE
Metoda dostarcza schematu badania do określenia, czy ciało stałe lub substancja pastopodobna przedstawia niebezpieczeństwo wybuchu, gdy poddaje się ją działaniu płomienia (wrażliwość cieplna) lub uderzeniu lub tarciu (wrażliwość na bodźce mechaniczne), i czy ciekła substancja przedstawia niebezpieczeństwo wybuchu, gdy podda się ją działaniu płomienia lub uderzeniu.
Metoda składa się z trzech części:
|
a) |
badanie wrażliwości cieplnej (1); |
|
b) |
badanie wrażliwości mechanicznej na uderzenia (1); |
|
c) |
badanie wrażliwości mechanicznej na tarcie (1). |
Metoda pozwala uzyskać dane pozwalające na ocenę prawdopodobieństwa zainicjowania wybuchu przy pomocy niektórych powszechnie występujących bodźców. Metoda nie jest przeznaczona do stwierdzenia, czy substancja jest zdolna do wybuchu w każdych warunkach.
Metoda jest właściwa do określenia, czy substancja może przedstawiać niebezpieczeństwo wybuchu (wrażliwość cieplna i mechaniczna) w szczególnych warunkach określonych w dyrektywie. Jest oparta o pewną ilość typów przyrządów, które są szeroko międzynarodowo stosowane (1) i które zwykle dają w pełni znaczące wyniki. Uznane jest, że metoda nie jest ostateczna. Można stosować alternatywne przyrządy do tych celów, zapewniając że są znane międzynarodowo i wyniki mogą być stosownie skorelowane z tymi z wymaganych przyrządów.
Nie ma potrzeby przeprowadzania badań, gdy dostępne są informacje termodynamiczne (np. ciepło tworzenia, ciepło rozkładu) i/lub nieobecne są pewne reaktywne grupy (2) we wzorze strukturalnym, ustanawiające poza wszelkimi wątpliwościami, że substancja jest niezdolna do gwałtownego rozkładu z wydzieleniem gazów lub uwolnieniem ciepła (to jest, materiał nie przedstawia żadnego ryzyka wybuchu). Badanie wrażliwości mechanicznej w odniesieniu do tarcia nie jest wymagane dla cieczy.
1.2. DEFINICJE I JEDNOSTKI
Materiały wybuchowe:
Substancje, które wybuchają pod działaniem płomienia lub które są wrażliwe na uderzenie lub tarcie w wymaganych przyrządach (lub bardziej wrażliwe mechanicznie niż 1,3-dinitrobenzen w alternatywnym przyrządzie).
1.3. SUBSTANCJE ODNIESIENIA
1,3-dinitrobenzen, techniczny, krystaliczny, przesiany przez sito 0,5 mm, dla metod tarcia i uderzenia.
Perhydro-1,3,5-trinitro-1,3,5-triazyna (RDX, heksogen, cyklonit-CAS 121-82-4), rekrystalizowany z wodnego cykloheksanonu, przesiewany na mokro przez sito 250 mikrometrów (μm) i zatrzymywany na sicie 150 μm oraz suszony w 103 ± 2 oC (przez 4 godziny) do drugiej serii badań na tarcie i uderzenia.
1.4. ZASADA METODY
Badania wstępne są konieczne dla ustalenia bezpiecznych warunków przeprowadzenia trzech badań wrażliwości.
1.4.1. Badania bezpiecznego postępowania z substancją (3)
Ze względów bezpieczeństwa, przed przeprowadzeniem głównych badań, bardzo małe próbki (około 10 mg) substancji są poddawane ogrzaniu bez zamknięcia w płomieniu gazu, wstrząsowi w dogodnym przyrządzie i tarciu przez użycie młotka i kowadła lub jakiejkolwiek maszyny ciernej. Celem jest stwierdzenie, czy substancja jest tak wrażliwa i wybuchowa, że przepisane badania wrażliwości, szczególnie wrażliwość cieplna, powinny być prowadzone ze specjalnymi środkami bezpieczeństwa, by zapobiec zranieniu operatora.
1.4.2. Wrażliwość cieplna
Metoda obejmuje podgrzewanie substancji w stalowej rurze, zamkniętej kryzami z otworami o różnej średnicy, dla określenia, czy substancja jest podatna na wybuch w warunkach intensywnego ogrzewania pod określonym przykryciem.
1.4.3. Wrażliwość mechaniczna (uderzenie)
Metoda obejmuje poddawanie substancji uderzeniu spowodowanemu przez zrzucenie określonej masy z określonej wysokości.
1.4.4. Wrażliwość mechaniczna (tarcie)
Metoda obejmuje poddawanie ciała stałego lub substancji pastopodobnych tarciu między znormalizowanymi powierzchniami w określonych warunkach obciążenia i ruchu względnego.
1.5. KRYTERIA JAKOŚCI
Nie ustalono.
1.6. OPIS METODY
1.6.1. Wrażliwość cieplna (działanie płomienia)
1.6.1.1. Przyrząd
Przyrząd składa się z nienadającej się do ponownego zastosowania rury stalowej z zamknięciami wielokrotnego użytku (rysunek 1), zainstalowanej w urządzeniu do podgrzewania i zabezpieczającym. Każda rura jest wykonana z arkusza stali głęboko tłoczonej (zob. dodatek) i posiada wewnętrzną średnicę 24 mm, długość 75 mm i grubość ścianek 0,5 mm. Rury są kołnierzowane na otwartych końcach dla umożliwienia ich zamknięcia przez zestaw płyty kryzowej. Składa się on z odpornej na ciśnienie płyty kryzy, z centralnie położonym otworem, przykręconej mocno do rury za pomocą dwuczęściowych złącz śrubowych (nakrętka i gwintowany pierścień). Nakrętka i gwintowany pierścień są wykonane ze stali chromomanganowej (zob. dodatek), beziskrowej do temperatury 800 oC. Kryzy są grubości 6 mm, wykonane ze stali żaroodpornej (zob. dodatek), i są dostępne w zakresie różnych średnic otworów.
1.6.1.2. Warunki badania
Zwykle substancje są badane tak jak je otrzymano, chociaż w niektórych przypadkach, na przykład gdy są sprasowane, odlewane lub w inny sposób skondensowane, może być konieczne badanie substancji po skruszeniu. Dla ciał stałych masa materiału użytego w każdym badaniu jest określana w dwuetapowej procedurze przebiegu próbnego. Wytarowana rura jest napełniana 9 cm3 substancji i substancja jest ubijana siłą 80 N przyłożoną w poprzek całkowitego przekroju rury. Z powodów bezpieczeństwa lub w przypadkach, gdzie postać fizyczna próbki zmienia się przez ściśnięcie, można użyć inną procedurę napełniania; przykładowo, gdy substancja jest bardzo wrażliwa na tarcie, wtedy ubijanie nie jest odpowiednie. Jeżeli materiał jest ściśliwy, wtedy dodaje się go więcej i ubija do momentu wypełnienia rury do 55 mm od góry. Całkowita masa użyta do wypełnienia rury do poziomu 55 mm jest określona i dodaje się następnie dwie dodatkowe porcje, każda ubijana z siłą 80 N. Następnie materiał jest albo dodawany z ubijaniem, albo wyjmowany, w zależności od wymagań, celem pozostawienia rury wypełnionej do poziomu 15 mm od góry. Wykonuje się drugi przebieg próbny, rozpoczynając z ubitą ilością trzeciej masy całkowitej znalezionej w pierwszym etapie przebiegu próbnego. Do tych przyrostów dodaje się dwa więcej z ubijaniem o sile 80 N i poziomem substancji w rurze ustawionym na 15 mm od góry, poprzez dodawanie lub ujmowanie materiału zgodnie z wymaganiem. Ilość ciała stałego ustalona w drugim etapie przebiegu próbnego jest stosowana do każdej próby. Napełnianie przeprowadzono w trzech równych ilościach, każdej ściśniętej do 9 cm3 z konieczną siłą (można to uprościć przez zastosowanie pierścieni dystansowych).
Ciecze i żele są ładowane do rury na wysokość 60 mm, zwracając szczególną uwagę na żele, by zapobiec tworzeniu się pustych przestrzeni. Gwintowany pierścień jest wsuwany w rurę od dołu, umieszcza się stosowną płytę kryzową i dokręca nakrętkę po nasmarowaniu małą ilością smaru na bazie disiarczku molibdenu. Jest podstawową
zasadą sprawdzenie, czy nie uwięziono substancji między kołnierzem i płytą, lub w gwincie.
Podgrzewanie jest prowadzone za pomocą propanu pobieranego z butli przemysłowej, wyposażonej w zawór regulacyjny ciśnienia (60 do 70 mbar), poprzez miernik i równomiernie rozprowadzany (co wskazuje wizualna obserwacja płomieni z palników) poprzez dysze do czterech palników. Palniki są umieszczone dookoła komory badawczej, jak pokazano na rysunku 1. Cztery palniki posiadają łączne zużycie około 3,2 litra propanu na minutę. Można zastosować alternatywne gazy palne i palniki, ale szybkość ogrzewania musi być jak wyspecyfikowano na rysunku 3. Dla wszystkich przyrządów szybkość ogrzewania musi być okresowo sprawdzana używając rury wypełnione ftalanem dibutylu, jak wskazano na rysunku 3.
1.6.1.3. Przeprowadzenie badania
Każde badanie jest przeprowadzane do momentu albo rozerwania rury, albo ogrzania w czasie pięciu minut. Badanie, wynikiem którego jest fragmentacja rury na trzy lub więcej części, które w niektórych przypadkach mogą być połączone jedna do drugiej wąskimi tasiemkami metalu jak pokazano na rysunku 2, ocenia się jako dające wybuch. Badanie wynikiem którego jest mniej fragmentów lub wcale, jest uważane jako niedające wybuchu.
Wpierw wykonuje się serie trzech badań z płytą kryzową o średnicy 6,0 mm, jeśli nie uzyska się wybuchów, przeprowadza się drugą serię trzech badań z płyta kryzową o średnicy 2,0 mm. Jeżeli wybuch zachodzi w jednej z dwóch serii badań, niewymagane są następne badania.
1.6.1.4. Ocena
Wynik badania jest uważany za pozytywny, jeżeli wybuch zachodzi w jednej z dwóch powyższych serii badań.
1.6.2. Wrażliwość mechaniczna (uderzenie)
1.6.2.1. Przyrząd (rysunek 4)
Podstawowe części przyrządu spadowego młota są wykonane z bloku ze staliwa z podstawą, kowadła, kolumny, prowadnic, bijaków, mechanizmu zwalniającego i uchwytu próbki. Kowadło stalowe 100 mm (średnica) × 70 mm (wysokość) jest przykręcone do góry bloku stalowego 230 mm (długość) × 250 mm (szerokość) × 200 mm (wysokość) z podstawą staliwną 450 mm (długość) × 450 mm (szerokość) × 60 mm (wysokość). Kolumna wykonana z bezszwowej ciągnionej rury stalowej, jest zabezpieczona w uchwycie przykręconym do tylnej ściany bloku stalowego. Cztery śruby mocują przyrząd do stałego betonowego bloku 60 × 60 × 60 cm w taki sposób, że szyny prowadnicy są całkowicie pionowe i bijak opada swobodnie. Stosownymi do użycia są bijaki 5 oraz 10 kg, wykonane z stali uspokojonej. Głowica uderzeniowa każdego bijaka jest z utwardzonej stali HRC 60 do 63, i posiada minimalną średnicę 25 mm.
Próbka podlegająca badaniu jest zawarta w przyrządzie uderzeniowym składającym się z dwóch współosiowych cylindrów ze stali uspokojonej, jeden powyżej drugiego, w wydrążonym stalowym pierścieniu prowadzącym. Cylindry ze stali uspokojonej powinny posiadać średnicę 10 (-0,003, -0,005) mm i wysokość 10 mm oraz posiadać polerowane powierzchnie, zaokrąglone krawędzie (promień krzywizny 0,5 mm) i twardość HRC 58 do 65. Drążony cylinder musi posiadać zewnętrzną średnicę 16 mm, polerowany otwór wiertniczy 10 (+0,005, +0,010) mm i wysokość 13 mm. Przyrząd uderzeniowy jest wyposażony w pośrednie kowadło (średnica 26 mm i 26 mm wysokość) wykonane ze stali i centrowane pierścieniem z otworami, pozwalającymi na ucieczkę dymów.
1.6.2.2. Warunki badania
Objętość próbki powinna wynosić 40 mm3 lub objętość właściwą dla dowolnego alternatywnego przyrządu. Stałe substancje powinny być badane w stanie suchym i przygotowywane, jak następuje:
|
a) |
sproszkowane substancje należy przesiać (wielkość oczek sita: 0,5 mm); do badania wykorzystuje się całą ilość substancji, która przeszła przez sito; |
|
b) |
sprasowane, odlewane lub w inny sposób skondensowane substancje są kruszone w małe kawałki i przesiewane; przesiane frakcje średnicy od 0,5 do 1 mm są używane do badania i muszą być reprezentatywne dla substancji oryginalnej. |
Substancje zwykle dostarczane jako pasty, powinny być badane w stanie suchym, tam gdzie to możliwe, lub w każdym razie, po usunięciu maksymalnie możliwej ilości rozpuszczalnika. Ciekłe substancje są badane ze szczeliną 1mm między dolnym i górnym stalowym cylindrem.
1.6.2.3. Przeprowadzenie badań
Przeprowadza się serie sześciu badań, spuszczając masę 10 kg z wysokości 0,40 m (40 J). Jeżeli w trakcie sześciu badań przy 40 J uzyska się wybuch, należy wykonać serię dalszych sześciu badań, spuszczając masę 5 kg z 0,15 m (7,5 J). W innym przyrządzie próbka jest porównywana z wybraną substancją odniesienia, stosując ustaloną procedurę (np. technikę w górę-w dół itd.).
1.6.2.4. Ocena
Wynik badania uważa się za pozytywny, jeżeli zachodzi wybuch (zapalenie się gwałtownym płomieniem jest równoważne wybuchowi i przedstawiane w sprawozdaniu) przynajmniej raz we wszystkich badaniach z określonym przyrządem uderzeniowym lub próbka jest bardziej wrażliwa niż 1,3-dinitrobenzen lub RDX w alternatywnym badaniu na uderzenie.
1.6.3. Wrażliwość mechaniczna (tarcie)
1.6.3.1. Przyrząd (rysunek 5)
Przyrząd do tarcia składa się z płyty bazowej staliwnej, na której zamocowany jest przyrząd tarcia. Składa się on z umocowanych porcelanowych prętów i ruchomej porcelanowej płyty. Płyta porcelanowa jest umieszczona w wózku poruszającym się w dwóch prowadnicach. Wózek jest dołączony do silnika elektrycznego poprzez korbowód, krzywkę mimośrodową i odpowiednią przekładnię, tak że porcelanowa płyta przesuwa się tylko raz, w tył i naprzód poniżej porcelanowego pręta w odległości 10 mm. Pręt porcelanowy może być obciążony przykładowo 120 lub 360 N.
Płaska porcelanowa płyta jest wykonana z porcelany technicznej (chropowatość 9 do 32 μm) i posiada wymiary 25 mm (długość) × 25 mm (szerokość) × 5 mm (wysokość). Cylindryczny pręt porcelanowy jest również wykonany z białej technicznej porcelany i ma długość 15 mm, średnicę 10 mm oraz schropowaconą końcową powierzchnię sferyczną o promieniu krzywizny 10 mm.
1.6.3.2. Warunki badania
Objętość próbki powinna wynosić 10 mm3 lub objętość odpowiednią dla każdego przyrządu alternatywnego.
Stałe substancje powinny być badane w stanie suchym i przygotowywane jak następuje:
|
a) |
sproszkowane substancje należy przesiać (wielkość oczek sita: 0,5 mm); do badania wykorzystuje się całą ilość substancji, która przeszła przez sito; |
|
b) |
sprasowane, odlewane lub w inny sposób skondensowane substancje są kruszone w małe kawałki i przesiewane; do badań używane są przesiane frakcje średnicy mniejszej od 0,5 mm. |
Substancje zwykle dostarczane jako pasty powinny być badane w stanie suchym, tam gdzie to możliwe. Jeżeli substancja nie może być przygotowana w stanie suchym, pasta (po usunięciu maksymalnie możliwej ilości rozpuszczalnika) jest badana jako warstwa 0,5 mm grubości, 2 mm szerokości, 10 mm długości, przygotowana przy użyciu szablonu.
1.6.3.3. Przeprowadzenie badań
Pręt porcelanowy jest położony na badaną próbkę i obciążany. Podczas przeprowadzania badania ślady gąbki na płytce porcelanowej muszą być położone poprzecznie w stosunku do kierunku ruchu. Należy uważać, aby pręt spoczywał na próbce, aby wystarczająco dużo materiału badanego znajdowało się pod prętem i aby płytka poruszała się prawidłowo pod prętem. Dla substancji w postaci past do nanoszenia substancji na płytę stosuje się szablon o grubości 0,5 mm ze szczeliną 2 × 10 mm. Porcelanowa płyta przesuwa się o 10 mm wprzód i do tyłu pod porcelanowym prętem w czasie 0,44 sekundy. Każda część powierzchni płyty i pręta używana jest tylko raz; dwa końce każdego pręta służą dla dwóch prób, a dwie powierzchnie płyty służą każda do trzech prób.
Serię sześciu badań przeprowadza się z obciążeniem 360 N. Jeżeli uzyskuje się pozytywne zdarzenie w czasie trwania tych sześciu badań, następną serię sześciu badań należy wykonać z obciążeniem 120 N. W innych przyrządach, próbka jest porównywana z wybraną substancją odniesienia, stosując ustaloną procedurę (np. technikę „w górę-w dół” itp.).
1.6.3.4. Ocena
Wynik badania jest uważany za pozytywny, jeżeli zaszedł wybuch (zapalenie się gwałtownym płomieniem jest równoważne wybuchowi i przedstawiane w sprawozdaniu) przynajmniej raz w jakimkolwiek z badań przy użyciu wyspecyfikowanego przyrządu do tarcia lub odpowiada kryteriom równoważności w alternatywnym badaniu przez tarcie.
2. DANE
W zasadzie, w sensie dyrektywy, substancja jest uważana za przedstawiając niebezpieczeństwo wybuchu, jeżeli uzyskano pozytywny wynik w badaniach wrażliwości na ciepło, uderzenie i tarcie.
3. SPORZĄDZANIE SPRAWOZDANIA
3.1. SPRAWOZDANIE Z BADANIA
Sprawozdanie z badań zawiera w miarę możliwości następujące informacje:
|
— |
tożsamość, skład, czystość, zawartość wilgoci itd., badanej substancji, |
|
— |
postać fizyczna próbki i czy lub nie była kruszona, rozdrabniana i/lub przesiewana, |
|
— |
obserwacje z czasu trwania badań wrażliwości cieplnej (np. masa próbki, ilość fragmentów itp.), |
|
— |
obserwacje z czasu trwania badań wrażliwości mechanicznej (np. tworzenie znacznych ilości dymu lub całkowity rozkład bez detonacji, płomienie, iskry, detonacja, gwałtowne zapalenie itp.), |
|
— |
wyniki każdego rodzaju badania, |
|
— |
w przypadku gdy wykorzystano alternatywny przyrząd, należy podać uzasadnienie naukowe oraz dowody na korelację między wynikami uzyskanymi przy pomocy podanego przyrządu i przyrządu równoważnego, |
|
— |
wszelkie użyteczne uwagi, takie jak odniesienie do badań produktów podobnych, które mogą być istotne dla prawidłowej interpretacji wyników. |
|
— |
wszelkie dodatkowe uwagi istotne dla interpretacji wyników. |
3.2. INTERPRETACJA I OCENA WYNIKÓW
W sprawozdaniu z badań należy podać wszelkie wyniki uznane za fałszywe, stanowiące anomalię lub niereprezentatywne. W przypadku gdyby którykolwiek z wyników powinien zostać odrzucony, należy podać wszelkie alternatywne lub dodatkowe badania. Pomimo wytłumaczenia anomalnych wyników muszą być zaakceptowane ich wartość nominalne i zastosowane do zgodnej z nimi klasyfikacji substancji.
4. LITERATURA
|
(1) |
Recommendations on the Transport of Dangerous Goods: Tests and criteria, 1990, United Nations, New York. |
|
(2) |
Bretherick, L., Handbook of Reactive Chemical Hazards, 4th edition, Butterworths, London, ISBN 0-750-60103-5, 1990. |
|
(3) |
Koenen, H., Ide, K.H. and Swart, K.H., Explosivstoffe, 1961, vol. 3, 6-13 and 30-42. |
|
(4) |
NF T 20-038 (September 85). Chemical products for industrial use –Determination of explosion risk. |
Dodatek
Przykłady specyfikacji materiałowych do badania wrażliwości cieplnej (zob. DIN 1623)
|
(1) |
Tube: Material specification No 1.0336.505 g |
|
(2) |
Orifice plate: Material specification No 1.4873 |
|
(3) |
Threaded collar and nut: Material specification No 1.3817 |
Rysunek 1
Przyrząd badawczy do wrażliwości cieplnej
(wszystkie wymiary w mm)

Rysunek 2
Badanie wrażliwości cieplnej
(przykłady fragmentacji)
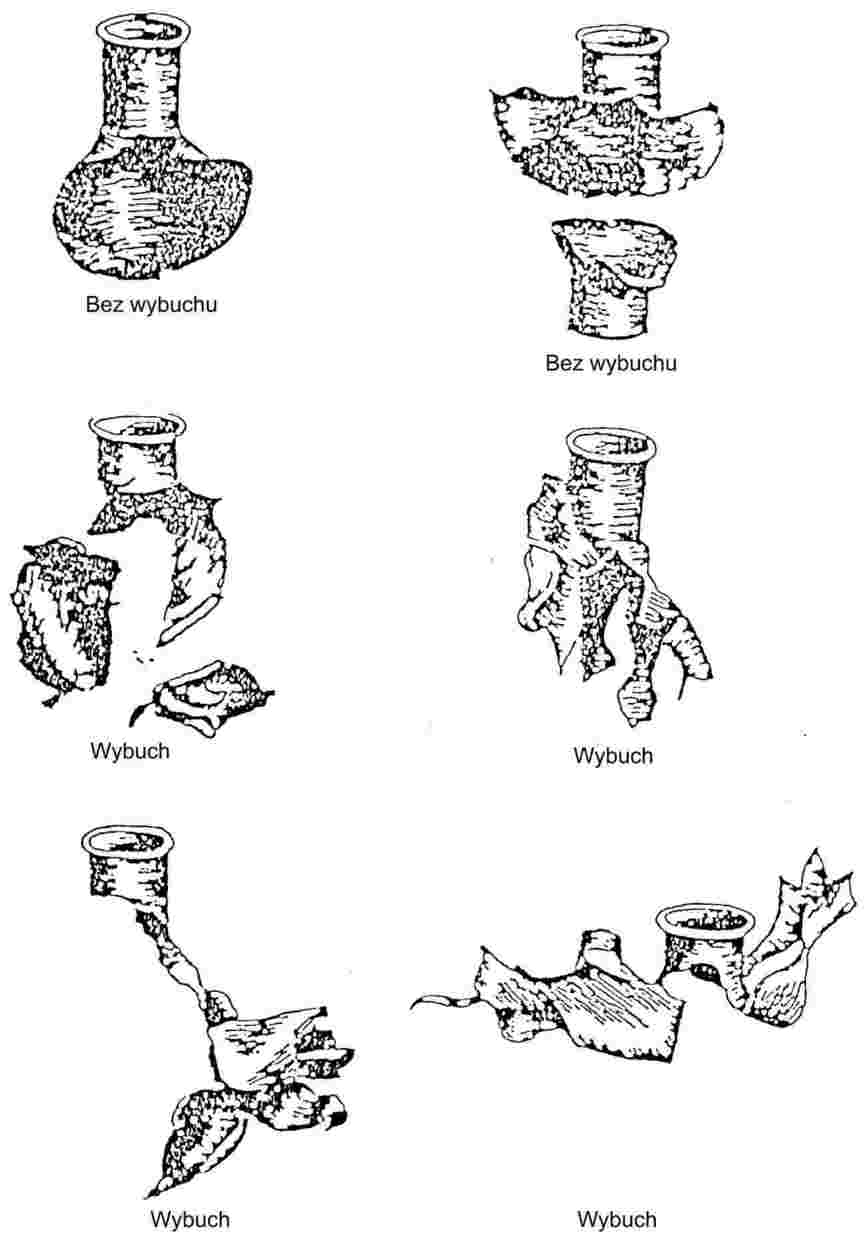
Rysunek 3
Kalibracja szybkości podgrzewania dla badania wrażliwości cieplnej

Krzywa temperatura/czas uzyskana przy podgrzewaniu ftalanu dibutylu (27 cm3) w zamkniętej rurze (1,5 mm płyta kryzowa) przy użyciu szybkości przeprywu propanu 3,2 litra/min. Temperaturę pomierzono termoogniwem chromel/alumel w osłonie ze stali nierdzewnej średnicy l mm, umieszczonego centrycznie 43 mm poniżej obrzeża rury. Szybkość grzania między 135 oC i 285 oC powinna wynosić między 185 i 215 K/min.
Rysunek 4
Przyrząd do badania wrażliwości na uderzenie
(wszystkie wymiary w mm)

Rysunek 4
Ciąg dalszy
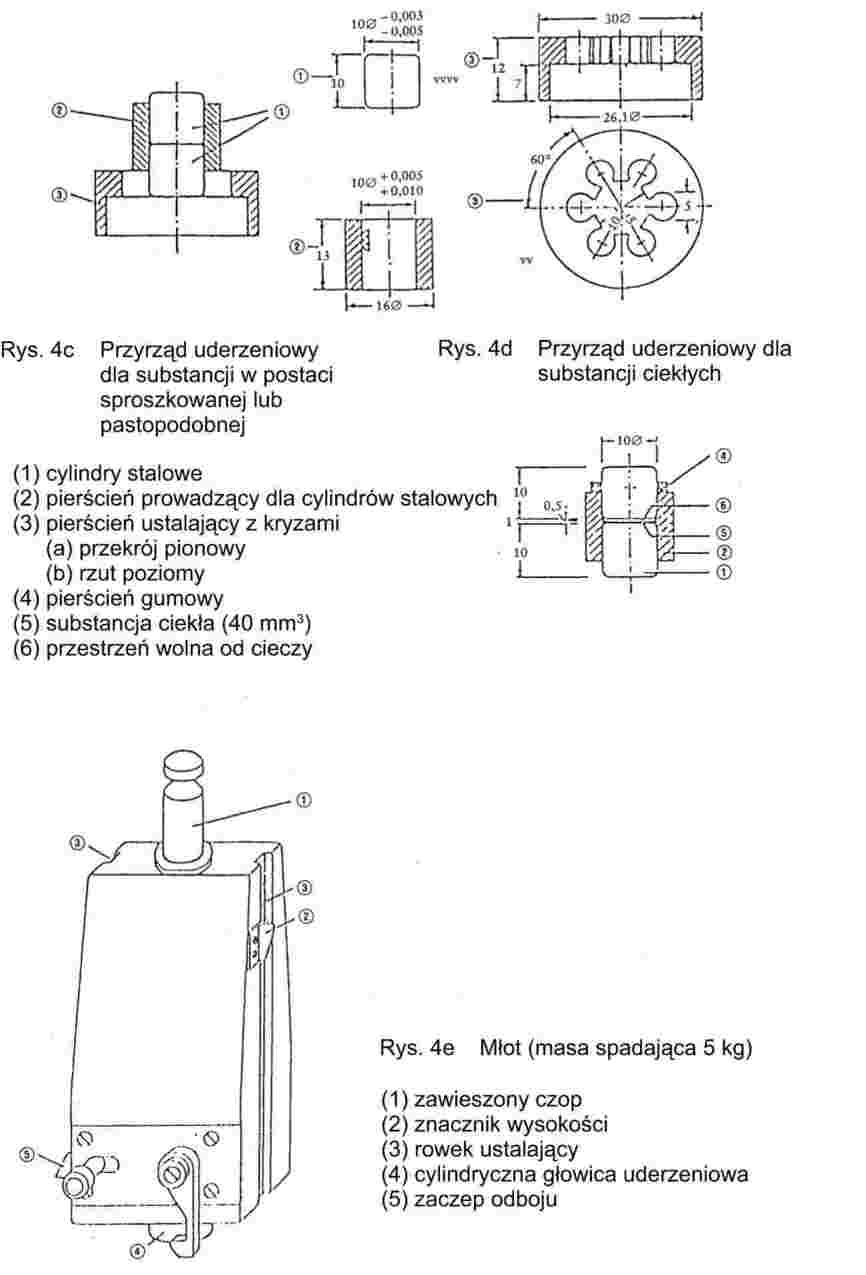
Rysunek 5
Przyrząd do badania wrażliwości na tarcie

A.15. TEMPERATURA SAMOZAPŁONU (CIECZE I GAZY)
1. METODA
1.1. WPROWADZENIE
Badaniu temu należy poddać substancje wybuchowe i substancje ulegające samoistnemu zapaleniu w kontakcie z powietrzem w temperaturze otoczenia. Procedura badania nadaje się do stosowania dla gazów, cieczy i par, które w obecności powietrza mogą zapalić się od gorącej powierzchni.
Temperatura samozapłonu może ulec znacznemu zmniejszeniu w obecności zanieczyszczeń katalitycznych, przez powierzchnię materiału lub większą objętość naczynia do badania.
1.2. DEFINICJE I JEDNOSTKI
Stopień samozapalności jest wyrażony w warunkach temperatury samozapłonu. Temperatura ta to najniższa temperatura, w której badana substancja zapali się po zmieszaniu jej z powietrzem w warunkach podanych w opisie metody badania.
1.3. SUBSTANCJE ODNIESIENIA
Substancje odniesienia zacytowano w normach (zob. 1.6.3). Powinny one służyć głównie do sprawdzenia od czasu do czasu zdolności metody i umożliwiać porównanie z wynikami uzyskanymi innymi metodami.
1.4. ZASADA METODY
Metoda wyznacza minimalną temperaturę powierzchni wewnętrznej obudowy, która powoduje zapalenie się gazu, pary lub cieczy wtryśniętej do obudowy.
1.5. KRYTERIA JAKOŚCI
Powtarzalność zmienia się w zależności zakresu temperatur samozapłonu i zastosowanej metody badania.
Czułość i specyficzność zależą od zastosowanej metody badania.
1.6. OPIS METODY
1.6.1. Przyrząd
Przyrząd jest opisany w metodzie podanej w 1.6.3.
1.6.2. Warunki badania
Badanie próbki substancji badanej przeprowadza się zgodnie z metodą określoną w 1.6.3.
1.6.3. Przeprowadzenie badania
Zob. IEC 79-4, DIN 51794, ASTM-E 659-78, BS 4056, NF T 20-037.
2. DANE
Zapisać temperaturę badania, ciśnienie atmosferyczne, ilość wykorzystanej próbki, odstęp czasowy do uzyskania zapłonu.
3. SPORZĄDZANIE SPRAWOZDANIA
Sprawozdanie z badań zawiera w miarę możliwości następujące informacje:
|
— |
precyzyjna specyfikacja substancji (tożsamość i zanieczyszczenia), |
|
— |
ilość użytej próbki, ciśnienie atmosferyczne, |
|
— |
zastosowany przyrząd, |
|
— |
wyniki pomiarów (temperatury badania, wyniki dotyczące zapłonu, odpowiednie opóźnienia), |
|
— |
wszelkie dodatkowe uwagi istotne dla interpretacji wyników. |
4. LITERATURA
Brak.
A.16. WZGLĘDNA TEMPERATURA SAMOZAPŁONU DLA CIAŁSTAŁYCH
1. METODA
1.1. WPROWADZENIE
Badaniu temu nie należy poddawać substancji wybuchowych i substancji ulegających samoistnemu zapaleniu w kontakcie z powietrzem w temperaturze otoczenia.
Celem niniejszego badania jest przedstawienie wstępnych informacji na temat samozapłonu substancji stałych w podwyższonych temperaturach.
W przypadku gdy temperatura powstała albo w reakcji substancji z tlenem lub podczas rozkładu egzotermicznego nie zostaje wystarczająco szybko utracona do otoczenia, dochodzi do samopodgrzania, co z kolei prowadzi do samozapłonu. Do samozapłonu dochodzi więc wtedy, gdy szybkość wytwarzania ciepła przekracza szybkość jego utraty.
Procedura badania jest przydatna jako wstępne badanie klasyfikacyjne substancji stałych. Ze względu na złożony charakter zapłonu i spalania się ciał stałych temperatura samozapłonu ustalona przy użyciu tej metody badania powinna być wykorzystywana wyłącznie do celów porównawczych.
1.2. DEFINICJE I JEDNOSTKI
Temperatura samozapłonu uzyskana przy użyciu tej metody to minimalna temperatura otoczenia wyrażona w oC, w której pewna objętość substancji będzie ulegać zapaleniu w określonych warunkach.
1.3. SUBSTANCJE ODNIESIENIA
Brak.
1.4. ZASADA METODY
Pewna ilość substancji do badań jest umieszczana w piecu w temperaturze pokojowej; krzywa temperatura/czas wiążąca warunki w środku próbki jest rejestrowana do momentu, gdy temperatura pieca wzrośnie do 400 oC, lub do temperatury topnienia, jeśli jest niższa, z szybkością 0,5 oC/min. Do celu niniejszego badania, temperatura pieca w której temperatura próbki osiągnie 400 oC przez samopodgrzanie jest zwana temperaturą samozapłonu.
1.5. KRYTERIA JAKOŚCI
Brak.
1.6. OPIS METODY
1.6.1. Przyrząd
1.6.1.1. Piec
Piec laboratoryjny (o objętości około 2 litrów) z programatorem temperatury wyposażony w obieg naturalnego powietrza i zawór przeciwwybuchowy. W celu zapobieżenia ewentualnemu ryzyku wybuchu wszystkie gazy pochodzące z rozkładu nie mogą wejść w kontakt z elementami ogrzewania elektrycznego.
1.6.1.2. Kostka z siatki drucianej
Kawałek drucianej siatki stalowej o otworach oczek 0,045 mm należy wyciąć zgodnie z szablonem przedstawionym na rysunku 1. Siatkę składa się i zabezpiecza drutem u góry otwartego kubka.
1.6.1.3. Termoogniwa
Odpowiednie termoogniwa.
1.6.1.4. Rejestrator
Dowolny rejestrator dwukanałowy, wykalibrowany od 0 do 600 oC lub o odpowiednim napięciu.
1.6.2. Warunki badania
Substancje są badane w stanie otrzymanym.
1.6.3. Przeprowadzenie badania
Kostkę napełnia się badaną substancją i łagodnie uklepuje, dodając substancję, aż kostka będzie całkowicie wypełniona. Następnie próbkę zawiesza się pośrodku pieca w temperaturze pokojowej. Jedno termoogniwo umieszcza się pośrodku kostki, a drugie między kostką i ścianą pieca w celu zapisywania temperatury w piecu.
Prowadzi się ciągły zapis temperatury pieca i próbki podczas podwyższania temperatury pieca do 400 oC lub do temperatury topnienia substancji stałej, jeżeli ta ostatnia jest niższa, z szybkością 0,5 oC/min.
Gdy substancja się zapali, termoogniwo w próbce pokaże bardzo ostry wzrost temperatury powyżej temperatury w piecu.
2. DANE
Temperatura pieca przy której temperatura próbki osiąga 400 oC przez samopodgrzanie jest stosowana do obliczeń (zob. rysunek 2).
3. SPORZĄDZANIE SPRAWOZDANIA
Sprawozdanie z badań zawiera w miarę możliwości następujące informacje:
|
— |
opis badanej substancji, |
|
— |
wyniki pomiarów, włącznie z krzywą zależności temperatura/czas, |
|
— |
wszelkie dodatkowe uwagi istotne dla interpretacji wyników. |
4. BIBLIOGRAFIA
NF T 20-036 (September 85). Chemical products for industrial use. Determination of the relative temperature of the spontaneous flammability of solids.
Rysunek 1
Szablon kostki do badań 20 mm

Rysunek 2
Typowa krzywa zależności temperatura/czas
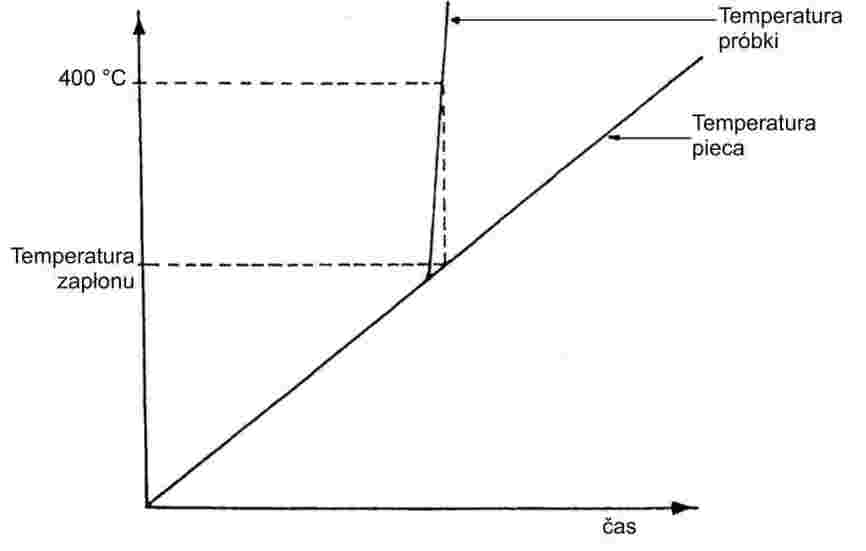
A.17. WŁAŚCIWOŚCI UTLENIAJĄCE (CIAŁA STAŁE)
1. METODA
1.1. WPROWADZENIE
Do przeprowadzenia badania przydatne jest posiadanie wstępnych informacji na temat ewentualnych właściwości wybuchowych substancji.
Niniejsze badanie nie jest przeznaczone dla cieczy, gazów, substancji wysoce łatwopalnych i wybuchowych lub nadtlenków organicznych.
Niniejsze badanie nie musi być prowadzone, gdy analiza wzoru strukturalnego chemicznego ustala ponad wszelkie wątpliwości, że substancja jest niezdolna do egzotermicznego reagowania z materiałem palnym.
W celu upewnienia się, czy badanie nie powinno być przeprowadzone z zastosowaniem szczególnych środków ostrożności, należy wykonać badanie wstępne.
1.2. DEFINICJAI JEDNOSTKI
Czas spalania: czas reakcji, w sekundach, jaki zabiera strefie reakcyjnej przejście wzdłuż stosu, zgodnie z procedurą określoną w 1.6.
Szybkość spalania: wyrażana w milimetrach na sekundę.
Maksymalna szybkość spalania: największe wartości szybkości spalania uzyskane w przypadku mieszanin zawierających od 10 do 90 % wagowych utleniacza.
1.3. SUBSTANCJE ODNIESIENIA
Azotan baru (czystości analitycznej) jest używany jako substancja odniesienia w próbie właściwej i w badaniu wstępnym.
Mieszaniną odniesienia jest ta mieszanina azotanu baru ze sproszkowaną celulozą, przygotowana zgodnie z ppkt 1.6, która charakteryzuje się maksymalną szybkością spalania (zazwyczaj mieszanina z 60 % azotanem baru wagowo).
1.4. ZASADA METODY
Dla zachowania bezpieczeństwa przeprowadza się badanie wstępne. Nie wymaga się prowadzenia dalszych badań, gdy wstępne badanie wyraźnie wskazuje, że badana substancja posiada właściwości utleniające. Jeżeli to nie jest ten przypadek, substancja musi być przedmiotem pełnego badania.
W pełnym badaniu substancję badaną i określoną substancję palną należy mieszać z różną szybkością. Z każdej mieszaniny formuje się następnie stos, który jest zapalany z jednej strony. Maksymalną ustaloną szybkość spalania porównuje się z maksymalną szybkością spalania mieszaniny odniesienia.
1.5. KRYTERIA JAKOŚCI
W razie potrzeby można zastosować dowolną metodę mielenia i mieszania, o ile różnica maksymalnej szybkości spalania w sześciu oddzielnych badaniach będzie odbiegać od wartości średniej arytmetycznej o nie więcej niż 10 %.
1.6. OPIS METODY
1.6.1. Przygotowanie
1.6.1.1. Substancja badana
Zmniejszyć wielkość ziarna badanej próbki do wielkości ziarna < 0,125 mm, stosując następującą procedurę: przesiać badaną substancję, rozdrobnić pozostałą frakcję, powtarzać procedurę do momentu, gdy cała porcja do badań przejdzie przez sito.
Można zastosować dowolną metodę mielenia lub przesiewania spełniającą kryteria jakościowe.
Przed przygotowaniem mieszaniny substancję suszy się w temperaturze 105 oC, do uzyskania stałej masy. W przypadku rozkładu badanej substancji w temperaturze poniżej 105 oC substancję tę należy wysuszyć w odpowiedniej, niższej temperaturze.
1.6.1.2. Substancja palna
Jako substancję palną wykorzystuje się sproszkowaną celulozę. Celuloza powinna być rodzaju stosowanego w chromatografii cienkowarstwowej lub chromatografii kolumnowej. Odpowiednim sprawdzonym typem jest celuloza z więcej niż 80 % włókien o długości między 0,020 i 0,075 mm. Proszek celulozy jest przesiewany przez sito o wymiarze oczka 0,125 mm. Poprzez całe badanie stosuje się tę samą partię celulozy.
Przed przygotowaniem mieszaniny sproszkowana celuloza jest suszona w 105 oC do chwili uzyskania stałej masy.
Jeżeli w badaniu wstępnym stosowana jest mączka drzewna, należy przygotować mączkę drzewną z miękkiego drewna, poprzez zebranie porcji, która przechodzi oczko siatki 1,6 mm, energicznie wymieszać, a następnie suszyć w 105 oC przez cztery w warstwie nie grubszej niż 25 mm. Ochłodzić i przechowywać w hermetycznym pojemniku, w pełni wypełnionym jak to praktycznie wymagane, najlepiej w obrębie czasu 24 godzin od wysuszenia.
1.6.1.3. Źródło zapłonu
Jako źródła zapłonu należy użyć gorącego płomienia gazowego palnika (średnicy minimum 5 mm). Jeżeli stosuje się inne źródło zapłonu (np. przy badaniu w atmosferze obojętnej), należy przedstawić w sprawozdaniu opis i uzasadnienie.
1.6.2. Przeprowadzenie badania
Uwaga:
Mieszaniny utleniaczy z celulozą lub mączką drzewną muszą być traktowane jako potencjalnie wybuchowe i należy obchodzić się z nimi z należytą ostrożnością.
1.6.2.1. Badanie wstępne
Wysuszona substancja jest wymieszana z wysuszoną celulozą lub mączką drzewną w proporcjach 2 części substancji badanej na 1 część celulozy lub mączki drzewnej wagowo, po czym z mieszaniny kształtuje się mały, stożkowaty stos o wymiarach 3,5 cm (średnica podstawy) × 2,5 cm (wysokość) przez napełnienie, bez ubijania, stożkowatej formy (np. szklanego lejka laboratoryjnego z zatkaną szyjką).
Stos umieszcza się na chłodnej, niepalnej, nieporowatej płycie bazowej o niskim przewodnictwie cieplnym. Badanie należy przeprowadzić w szafie wyciągowej, tak jak opisano w 1.6.2.2.
Źródło zapłonu wchodzi w kontakt ze stożkiem. Należy zaobserwować i zapisać intensywność i czas trwania ostatecznej reakcji.
Substancja jest uważana za utleniającą, jeżeli reakcja zachodzi gwałtownie.
W każdym przypadku, gdy wynik można poddać w wątpliwość, konieczne jest przeprowadzenie pełnej serii badań opisanych powyżej.
1.6.2.2. Badanie impulsowe
Przygotować kolejne porcje mieszaniny utleniacza/celulozy zawierające od 10 do 90 % wagowych części utleniacza w odstępach co 10 %. Dla granicznych przypadków należy użyć mieszanin pośrednich utleniacz/celuloza, dla uzyskania maksymalnej szybkości spalania bardziej precyzyjnie.
Należy uformować stos przy pomocy formy. Forma jest wykonana z metalu, ma długość 250 mm i trójkątny przekrój, przy czym jej wewnętrzna wysokość wynosi 10 mm, a wewnętrzna szerokość – 20 mm. Po obu stronach formy w kierunku wzdłużnym montuje się dwie blaszki jako ograniczenia poprzeczne wystające na 2 mm poza górną krawędź trójkątnego przekroju (rysunek). Niniejszy układ jest luźno napełniany lekkim nadmiarem mieszaniny. Po jednokrotnym upuszczeniu formy z wysokości 2 cm na twardą powierzchnię pozostały nadmiar substancji należy zeskrobać przy pomocy skośnie ustawionego arkusza. Następnie ograniczenia poprzeczne należy usunąć i wygładzić pozostały proszek przy pomocy wałka. Następnie na górze formy umieszcza się niepalną, nieporowatą o niskim przewodnictwie cieplnym, płytę bazową, odwraca przyrząd i zdejmuje formę.
Ustawić stos w kierunku prostopadłym do ciągu powietrza w szafie wyciągowej.
Szybkość przepływu powietrza powinna być wystarczająca do zapobieżenia przedostaniu się dymu do laboratorium i nie powinna być zmieniana w trakcie badania. Wokół przyrządu należy ustawić ekran chroniący przed ciągiem powietrza.
Ze względu na higroskopijność celulozy i badanych substancji, badanie należy przeprowadzić jak najszybciej.
Zapalić jeden z końców stosu przez dotknięcie płomieniem.
Zmierzyć czas reakcji na odległości 200 mm po tym, jak strefa reakcji rozprzestrzeni się z początkowej odległości 30 mm.
Badanie przeprowadza się z substancją odniesienia i przynajmniej raz dla każdego z zakresów mieszanin badanej substancji z celulozą.
Jeżeli maksymalna szybkość spalania okazuje się być znacząco większa niż ta z mieszaniny odniesienia, badanie należy wstrzymać; poza tym badanie musi być powtarzane pięć razy dla każdej z trzech mieszanin dających największą szybkość spalania.
Jeżeli wynik podejrzewany jest o nieprawdziwie pozytywny, należy powtórzyć badanie, stosując obojętną substancję o podobnej wielkości ziarna, taką jak diatomit w miejsce celulozy. Alternatywnie mieszanina badana substancja/celuloza o największej szybkości spalania powinna być ponownie zbadana w obojętnej atmosferze (< 2 % wagowych zawartości tlenu).
2. DANE
Ze względów bezpieczeństwa maksymalna szybkość spalania – nie jej wartość średnia – musi być uważana za charakterystyczną właściwością utleniającą substancji badanej.
Istotne znaczenie dla oceny ma najwyższa wartość szybkości spalania w serii sześciu badań danej mieszaniny.
Sporządzić wykres najwyższej wartości szybkości spalania dla każdej mieszaniny w zależności od stężenia utleniacza. Z wykresu należy wziąć maksymalną szybkość spalania.
Sześć zmierzonych wartości szybkości spalania w obrębie serii uzyskanej z mieszaniną z maksymalną szybkością spalania nie może odbiegać od wartości średniej arytmetycznej o więcej niż 10 %; w innym przypadku należy poprawić metody mielenia i mieszania.
Należy porównać maksymalną uzyskaną szybkość spalania z maksymalną szybkością spalania mieszaniny odniesienia (zob. 1.3).
Jeżeli badania przeprowadzono w obojętnej atmosferze, maksymalną szybkość reakcji porównuje się z tą z mieszaniny odniesienia w atmosferze obojętnej.
3. SPRAWOZDANIE
3.1. SPRAWOZDANIE Z BADANIA
Sprawozdanie z badań zawiera w miarę możliwości następujące informacje:
|
— |
tożsamość, skład, czystość, zawartość wilgoci i tym podobne dane badanej substancji, |
|
— |
wszelkie procesy przetwarzania, którym była poddawana próbka badana (np. mielenie, suszenie itp.), |
|
— |
źródło zapłonu używane w badaniach, |
|
— |
wyniki pomiarów, |
|
— |
sposób zachodzenia reakcji (np. błyskawiczne spalanie na powierzchni, spalanie w całej masie, wszelkie informacje dotyczące produktów spalania itp.), |
|
— |
wszelkie dodatkowe uwagi istotne dla interpretacji wyników, włącznie z opisem intensywności reakcji (płomienie, iskry, dymy, powolne tlenie się itp.) oraz przybliżony czas trwania uzyskany we wstępnym badaniu ze względów bezpieczeństwa/klasyfikacyjnym w przypadku zarówno substancji badanej, jak i substancji odniesienia, |
|
— |
wyniki badań substancji obojętnej, jeśli stosowano, |
|
— |
wyniki z badań w obojętnej atmosferze, jeśli stosowano. |
3.2. INTERPRETACJA WYNIKU
Substancja jest uważana za substancję utleniającą, gdy:
|
a) |
we wstępnym badaniu zachodzi gwałtowna reakcja; |
|
b) |
w pełnym badaniu maksymalna szybkość spalania badanych mieszanin jest wyższa lub równa maksymalnej szybkości spalania mieszaniny odniesienia z celulozy i azotanu baru. |
W celu zapobieżenia wynikom nieprawdziwie pozytywnym wyniki uzyskane przy badaniu substancji zmieszanej z materiałem obojętnym i/lub przy badaniu w atmosferze obojętnej muszą być także rozważone przy interpretacji wyników.
4. LITERATURA
NF T 20-035 (September 85). Chemical products for industrial use. Determination of the oxidizing properties of solids.
Dodatek
Rysunek
Forma i akcesoria do przygotowania stosu
(wszystkie wymiary w milimetrach)

A.18. ŚREDNIA LICZBOWA MASA CZĄSTECZKOWA I ROZKŁAD MASY CZĄSTECZKOWEJ POLIMERÓW
1. METODA
Niniejsza metoda chromatografii żelowo-permeacyjnej (GPC) jest powtórzeniem OECD TG 118 (1996). Podstawowe zasady i dalsze informacje techniczne są podane w pozycji bibliograficznej (1).
1.1. WPROWADZENIE
Skoro właściwości polimerów są tak zróżnicowane, nie istnieje możliwość opisania jednej metody ustalającej dokładnie warunki rozdzielania i oceny, która obejmuje wszystkie ewentualności i specyficzne przypadki występujące przy rozdzielaniu polimerów. W szczególności złożone systemy polimerów nie ulegają często chromatografii żelowo-permeacyjnej (GPC). Jeśli GPC nie jest praktykowana, masa cząsteczkowa może być oznaczona za pomocą innych metod (zob. załącznik). W takich przypadkach powinny zostać podane pełne dane szczegółowe i uzasadnienia dla wykorzystanych metod.
Opisana metoda oparta jest na normie DIN 55672 (1). Szczegółowe informacje o tym, w jaki sposób mają być przeprowadzane doświadczenia oraz w jaki sposób oceniać dane, można znaleźć w powyższej normie DIN. W przypadku gdy niezbędne są modyfikacje warunków doświadczalnych, zmiany te muszą być uzasadnione. Inne normy mogą być używane jedynie, gdy istnieją do nich pełne odniesienia. Opisana metoda wykorzystuje próbki polistyrenu o znanej polidyspersyjności do kalibracji i może istnieć konieczność zmodyfikowania jej, aby była odpowiednia dla niektórych polimerów, np. rozpuszczalnych w wodzie oraz polimerów o długich rozgałęzionych łańcuchach.
1.2. DEFINICJE I JEDNOSTKI
Średnia liczbowo masa cząsteczkowa Mn i średnia wagowo masa cząsteczkowa Mw są oznaczane, wykorzystując następujące równania:
|
|
|
gdzie:
Hi jest poziomem sygnału detektora z linii podstawowej dla objętości retencji Vi,
Mi jest masą cząsteczkową polimerowej frakcji przy objętości retencji Vi, oraz
n jest liczbą punktów pomiarowych.
Rozpiętość rozkładu masy cząsteczkowej, która jest miarą stopnia dyspersji systemu, jest podana na podstawie stosunku Mw/Mn.
1.3. SUBSTANCJE ODNIESIENIA
GPC jest metodą względną i w związku z tym musi zostać podjęta kalibracja. Zwykle do tego celu jest wykorzystywany polistyren w niewielkim stopniu rozproszony, o liniowej budowie, posiadający średnie masy cząsteczkowe Mn i Mw oraz znany rozkład masy cząsteczkowej. Krzywa kalibracji może być wykorzystywana w oznaczaniu masy cząsteczkowej nieznanej próbki wyłącznie, jeżeli warunki rozdzielenia próbki oraz wzorce zostały wybrane w identyczny sposób.
Określony związek między masą cząsteczkową oraz objętością elucji jest ważny wyłącznie w specyficznych warunkach, w których przeprowadzane jest konkretne doświadczenie. Warunki obejmują przede wszystkim temperaturę, rozpuszczalnik (lub mieszaninę rozpuszczalników), warunki chromatografii oraz kolumnę rozdzielającą lub systemy kolumn.
Masy cząsteczkowe próbki oznaczone w ten sposób są odpowiednimi wartościami oraz są opisane jako „ciężar równoważnikowy polistyrenu”. Oznacza to, że w zależności od strukturalnych i chemicznych różnic między próbką a wzorcami masy cząsteczkowe mogą odchylać się od wartości absolutnych/całkowitych w większym lub mniejszym stopniu. Jeżeli wykorzystywane są inne wzorce, np. glikolu polietylenowego, tlenku polietylenu, metaakrylanu polimetylu, kwasu poliakrylowego, należy podać odpowiednie przyczyny.
1.4. ZASADA METODY BADAWCZEJ
Zarówno rozkład masy cząsteczkowej próbki jak i średnie masy cząsteczkowe (Mn, M w) mogą być oznaczane, wykorzystując GPC. GPC jest szczególnym rodzajem chromatografii cieczowej, w której próbka jest rozdzielana według hydrodynamicznych objętości pojedynczych składników (2).
Rozdzielenie następuje, kiedy próbka przechodzi przez kolumnę, która jest wypełniona porowatym materiałem – zazwyczaj organicznym żelem. Małe cząsteczki mogą wnikać w pory, podczas gdy duże cząsteczki są wykluczane. Droga dużych cząsteczek jest z tego względu krótsza i to one są poddane procesowi elucji w pierwszej kolejności. Średnich rozmiarów cząsteczki wnikają w niektóre pory i są później poddawane elucji. Najmniejsze cząsteczki o średnim hydrodynamicznym promieniu, mniejszym niż pory żelu, mogą wnikać we wszystkie pory. Poddawane są elucji na końcu.
W sytuacji idealnej rozdzielanie jest kontrolowane wyłącznie przez wielkość czasteczki gatunku, ale w praktyce trudno jest uniknąć co najmniej niewielkich zakłócających efektów absorbcyjnych. Nierówne wypełnienia kolumny oraz objętości martwe mogą pogorszyć sytuację (2).
Detekcji dokonuje się przez np. współczynnik załamania światła lub pochłaniania promieni UV i daje w wyniku nieskomplikowaną krzywą rozkładu. Jednakże w celu przypisania rzeczywistych wartości masy cząsteczkowej do krzywej niezbędne jest poddanie kolumny kalibracji przez przepuszczanie polimerów o znanej masie cząsteczkowej oraz, w idealnym przypadku, o podobnej strukturze, np. różnych wzorców polistyrenowych. Krzywa Gaussa, powstająca zazwyczaj w ten sposób, czasami wypaczona jest małymi „ogonami” skierowanymi w stronę małych mas cząsteczkowych, oś pionowa wskazuje jakość w masie różnych wyeluowanych odmian polistyrenu, a oś pozioma wskazuje logarytm z masy cząsteczkowej.
1.5. KRYTERIA JAKOŚCIOWE
Powtarzalność (względne odchylenie standardowe: RSD) objętości eluowania powinna być lepsza niż 0,3 %. Wymagana powtarzalność analizy musi być zapewniona przez korektę dokonywaną poprzez wzorce wewnętrzne, jeżeli chromatogram jest uzyskiwany w zależności od czasu i nie odpowiada wyżej wspomnianym kryteriom (1). Polidyspersyjności są zależne od mas cząsteczkowych wzorców. W przypadku wzorców polistyrenowych typowe wartości to:
|
Mp < 2 000 |
Mw/Mn < 1,20 |
|
2 000 ≤ Mp ≤ 106 |
Mw/Mn < 1,05 |
|
Mp > 106 |
Mw/Mn < 1,20 |
(Mp jest masą cząsteczkową wzorca w maksimum piku)
1.6. OPIS METODY BADAWCZEJ
1.6.1. Przygotowanie roztworów wzorcowych polistyrenu
Wzorce polistyrenu są rozpuszczane przez ostrożne mieszanie w wybranym eluencie. Zalecenia wytwórcy muszą być uwzględnione w trakcie przygotowania roztworu.
Stężenia wybranych wzorców są zależne od wielu czynników, np. od objętości wstrzykiwanej substancji, lepkości roztworu oraz czułości detektora analitycznego. Maksymalna objętość injekcji musi być dostosowana do długości kolumny, w celu uniknięcia przeładowania. Typowe objętości injekcji dla analitycznego rozdzielenia przy użyciu GPC z kolumną o wymiarach 30 cm × 7,8 mm wynoszą na ogół 40–100 μl. Wyższe objętości są możliwe, ale nie powinny przekraczać 250 μl. Optymalny stosunek między objętością injekcji a stężeniem musi być określony przed rzeczywistą kalibracją kolumny.
1.6.2. Przygotowywanie roztworu próbki
Zasadniczo te same wymagania stosuje się do przygotowywania roztworów próbek. Próbka jest rozpuszczana w odpowiednim rozpuszczalniku, np. tetrahydrofuranie (THF), przez lekkie wstrząsanie. W żadnym okolicznościach nie powinno się rozpuszczać próbki w wannie ultradźwiękowej. W razie konieczności próbka roztworu jest oczyszczana poprzez membranowy filtr posiadający pory o wymiarach między 0,2 a 2 μ m.
Obecność nierozpuszczalnych cząstek musi być zapisana w sprawozdaniu końcowym, ponieważ może to być odpowiednie dla próbek o dużej masie cząsteczkowej. Powinna zostać wykorzystana odpowiednia metoda w celu określenia udziału procentowego w masie rozpuszczonych cząstek. Roztwór powinien zostać wykorzystany w ciągu 24 godzin.
1.6.3. Aparatura:
|
— |
zbiornik na rozpuszczalnik, |
|
— |
odgazowywacz (w danym przypadku), |
|
— |
pompa, |
|
— |
nawilżacz pulsowy (w danym przypadku), |
|
— |
system wtryskowy, |
|
— |
kolumny chromatograficzne, |
|
— |
detektor, |
|
— |
przepływomierz (w danym przypadku), |
|
— |
urządzenie rejestrujące dane, |
|
— |
naczynie na odpady. |
Należy zapewnić, że system GPC jest obojętny w odniesieniu do stosowanych rozpuszczalników (np. przez wykorzystanie stalowych kapilar w przypadku rozpuszczalnika THF).
1.6.4. System wtryskowy i system dostarczający rozpuszczalnik
Określona objętość roztworu próbki jest umieszczona w kolumnie albo wykorzystując automatyczny próbnik, albo ręcznie w ściśle określonej strefie. Zbyt szybkie wycofywanie lub zniżanie tłoka nurnikowego, jeśli jest wykonywane ręcznie, może spowodować zmiany w obserwowanym rozkładzie masy cząsteczkowej. System dostarczający rozpuszczalnik, na ile jest to możliwe, powinien być wolny od pulsacji, co można osiągnąć przez wbudowanie do niego tłumika pulsacji. Szybkość przepływu wynosi 1 ml/min.
1.6.5. Kolumna
W zależności od próbki właściwości polimeru oznacza się albo za pomocą prostej kolumny, albo kilku kolumn połączonych w sekwencję. Pewna ilość materiałów porowatych kolumn o określonych właściwościach dostępna jest w ogólnej sprzedaży (np. rozmiar porów, granica ekskluzji). Wybór żelu rozdzielającego lub długość kolumny jest uzależniona zarówno od właściwości próbki (objętości hydrodynamiczne, rozkład masy cząsteczkowej), jak i od specyficznych warunków rozdzielania, takich jak rozpuszczalnik, temperatura i prędkość przepływu (1) (2) (3).
1.6.6. Półki teoretyczne
Kolumna lub połączenie kolumn wykorzystywanych do rozdzielania musi być scharakteryzowana przez ilość półek teoretycznych. Obejmuje to, w przypadku THF jako rozpuszczalnikia eluencyjnego, umieszczenie roztworu etylobenzenu lub innych odpowiednich niepolarnych substancji rozpuszczonych w kolumnie o znanej długości. Liczba półek teoretycznych jest uzyskiwana na podstawie następującego równania:
|
|
lub |
|
gdzie:
|
N |
= |
jest liczbą półek teoretycznych, |
|
Ve |
= |
jest objętością eluecyjną w maksimum piku, |
|
W |
= |
jest podstawową szerokością piku, |
|
W1/2 |
= |
jest szerokością piku w połowie wysokości. |
1.6.7. Wydajność rozdzielania
Oprócz liczby półek teoretycznych, która jest określającą ilościowo szerokością pasma, ważną rolę odgrywa także wydajność rozdzielenia, będąca określana przez nachylenie krzywej kalibracyjnej. Wydajność rozdzielania kolumny jest uzyskiwana z następującego stosunku:

gdzie:
|
Ve, Mx |
= |
jest objętością elucyjną dla polistyrenu o masie cząsteczkowej Mx, |
|
Ve,(10.Mx) |
= |
jest objętością elucyjną dla polistyrenu o 10-krotnie większej masie cząsteczkowej Mx. |
Rozkład systemu jest zwykle określany w następujący sposób:

Gdzie:
|
Ve1, Ve2 |
= |
są objętościami eluencyjnymi dwóch wzorców polistyrenowych w maksimum piku, |
|
W1, W2 |
= |
są szerokościami piku linii podstawowej, |
|
M1, M2 |
= |
są masami cząsteczkowymi w maksimum piku (powinny różnić się współczynnikiem o 10). |
Wartość R dla systemu kolumn powinna być większa niż 1, 7 (4).
1.6.8. Rozpuszczalniki
Wszystkie rozpuszczalniki muszą posiadać wysoką czystość (dla THF używana jest czystość 99,5 %). Zbiornik na rozpuszczalnik (o ile jest to konieczne – w atmosferze gazu obojętnego) musi być odpowiednio duży dla kalibracji kolumny i analiz wielu próbek. Rozpuszczalnik musi być odgazowany zanim zostanie przeniesiony do kolumny za pomocą pompy.
1.6.9. Kontrola temperatury
Temperatura krytycznych komponentów wewnętrznych (pętla wtryskowa, kolumny, detektor i przewody) powinna być stała i zgodna z wyborem rozpuszczalnika.
1.6.10. Detektor
Zadaniem detektora jest rejestracja ilościowa stężenia próbki eluowanej z kolumny. W celu uniknięcia niepotrzebnego poszerzenia pików, objętość kuwety komórki detektora musi być tak mała, jak to tylko jest możliwe. Nie powinna być większa niż 10 μl, z wyjątkiem detektorów mierzących światło rozproszone oraz lepkość. W celu detekcji zazwyczaj używana jest refraktometria różnicowa. Jednakże jeżeli wymagają tego właściwości próbki lub właściwości eluacyjne rozpuszczalnika, mogą być używane inne rodzaje detektora, np. UV/VIS, IR, detektory lepkości itd.
2. DANE I SPORZĄDZANIE SPRAWOZDAŃ
2.1. DANE
Powinno nastąpić odniesienie do normy DIN (1) w celu uzyskania szczegółowych kryteriów oceny, jak również do wymogów odnoszących się do zbierania i przetwarzania danych.
Dla każdej próbki muszą zostać przeprowadzone dwa niezależne doświadczenia. Próbki muszą być analizowane pojedynczo.
Dla każdego pomiaru muszą być zapewnione Mn, Mw, Mw/Mn, Mp.. Niezbędne jest wyraźne wskazanie, że zmierzone wartości są wartościami względnymi, równoważnymi masom cząsteczkowym użytych wzorców.
Po określeniu objętości retencji lub czasów retencji (możliwie poprawionych, wykorzystując wzorzec wewnętrzny) wartości logarytmu Mp (Mp będącego maksymalną wartością piku wzorca kalibracji) są wykreślane w oparciu o jedną z tych wielkości. Niezbędne są co najmniej dwa punkty kalibracji na dekadę masy cząsteczkowej oraz wymagane jest co najmniej pięć punktów pomiarowych dla całkowitej krzywej, która powinna obejmować oszacowaną masę cząsteczkową próbki. Niska wartość punktu końcowego masy cząsteczkowej krzywej wzorcowania jest oznaczana za pomocą n-heksylobenzenu lub innego odpowiedniego niepolarnego roztworu. Średnia liczbowo i średnia wagowo masa cząsteczkowa są zwykle określane za pomocą przetwarzania danych elektronicznych, w oparciu o wzory z sekcji 1.2. W przypadku gdy używana jest ręczna digitalizacji, można powołać się na ASTMD 3536-91 (3).
Krzywa rozkładu musi być dostarczona w formie tabeli lub ilustracji (różnicowa częstotliwość lub procent sumy w oparciu o logarytm M). Na wizerunku graficznym jedna dekada masy cząsteczkowej powinna mieć normalnie około 4 cm szerokości, a maksymalna wartość szczytowa około 8 cm wysokości. W przypadku krzywych całkowego rozkładu różnica w rzędnych między 0 a 100 % powinna wynosić około 10 cm.
2.2. SPRAWOZDANIE Z BADANIA
Sprawozdanie z badania musi zawierać następujące informacje:
2.2.1. Substancja badana:
|
— |
dostępne informacje dotyczące substancji badanej (tożsamość, dodatki, zanieczyszczenia), |
|
— |
opis przetwarzania próbki, uwagi, problemy. |
2.2.2. Oprzyrządowanie:
|
— |
zbiornik z eluentem, obojętny gaz, odgazowywanie eluenta, skład eluenta, zanieczyszczenia, |
|
— |
pompa, pulsowy tłumik, system wtryskowy, |
|
— |
kolumny rozdzielające (wytwórca, wszystkie informacje dotyczące właściwościach kolumn, takie jak rozmiar poru, rodzaj rozdzielającego materiału itd., liczba, długość oraz kolejność użytych kolumn), |
|
— |
liczba teoretycznych półek kolumny (lub połączonych kolumn), wydajność rozdzielania, (rozdzielczość tego systemu), |
|
— |
informacja dotycząca symetrii pików, |
|
— |
temperatura kolumny, rodzaj sterowania temperaturą, |
|
— |
detektor (zasada pomiarów, rodzaj, objętość kuwety), |
|
— |
przepływomierz w przypadkach, kiedy jest używany (wytwórca, zasada pomiarów), |
|
— |
system umożliwiający rejestrację i przetwarzanie danych (osprzęt i oprogramowanie). |
2.2.3. Kalibracja systemu:
|
— |
szczegółowy opis metody używanej w celu skonstruowania krzywej kalibracji, |
|
— |
informacja dotycząca kryteriów jakościowych dla tej metody (np. współczynnik korelacji, błąd sumy kwadratów itd.), |
|
— |
informacje dotyczące ekstrapolacji, założenia i przybliżenia, poczynione w trakcie procedury doświadczalnej oraz szacowania i przetwarzania danych, |
|
— |
wszystkie pomiary przeprowadzone w celu konstrukcji krzywej kalibracji powinny być udokumentowane w tabelach, które zawierają następujące informacje dla każdego z punktów kalibracji:
|
2.2.4. Ocena:
|
— |
ocena na podstawie czasu: wszystkie metody mające na celu zapewnienie wymaganej odtwarzalności (metoda poprawiania, wzorce wewnętrzne itd.) |
|
— |
informacja dotycząca tego, czy ocena została przeprowadzona na podstawie objętości eluencyjnej lub czasu retencji, |
|
— |
informacja dotycząca ograniczeń oceny, jeżeli pik nie został poddany analizie w całości, |
|
— |
opis metod wygładzających, jeżeli są wykorzystywane, |
|
— |
procedury przygotowawcze oraz wstępnego przetworzenia próbki, |
|
— |
obecność nierozpuszczalnych cząstek, jeżeli istnieją, |
|
— |
objętość wtrysku (μl) oraz stężenie wtrysku (mg/ml), |
|
— |
uwagi wskazujące skutki, które prowadzą do odchyleń od idealnego profilu GPC, |
|
— |
szczegółowy opis wszystkich zmian w procedurach badawczych, |
|
— |
szczegóły dotyczące zakresów błędów, |
|
— |
wszystkie inne informacje oraz uwagi istotne w odniesieniu do interpretacji wyników. |
3. BIBLIOGRAFIA
|
(1) |
DIN 55672 (1995). Gelpermeationschromatographie (GPC) mit Tertrahydrofuran (THF) als Elutionsmittel, Teil 1. |
|
(2) |
Yau, W. W., Kirkland, J. J., and Bly, D. D. eds, (1979). Modern Size Exclusion Liquid Chromatography, J. Wiley and Sons. |
|
(3) |
ASTM D 3536-91, (1991). Standard Test Method for Molecular Weight Averages and Molecular Weight Distribution by Liquid Exclusion Chromatography (Gel Permeation Chromatography-GPC). American Society for Testing and Materials, Philadelphia, Pennsylvania. |
|
(4) |
ASTM D 5296-92, (1992). Standard Test Method for Molecular Weight Averages and MolecularWeight Distribution of Polystyrene by High Performance Size-Exclusion Chromatography. American Society for Testing and Materials, Philadelphia, Pennsylvania. |
Dodatek
Przykłady innych metod określania średniej liczbowo masy cząsteczkowej (Mn) w odniesieniu do polimerów
Chromatografia żelowo-permeacyjna (GPC) jest preferowaną metodą określania Mn, w szczególności kiedy dostępny jest zestaw wzorców, których struktura jest porównywalna ze strukturą polimerów. Jednakże, w przypadku gdy istnieją trudności praktyczne w wykorzystywaniu GPC lub już istnieje przypuszczenie, że substancja nie będzie podlegała kryterium regulującym Mn (a które wymaga potwierdzenia), dostępne są metody alternatywne, takie jak:
1. Wykorzystywanie własności koligatywnych
|
1.1. |
Ebuliometria/kriometria: obejmuje pomiar podwyższenia temperatury wrzenia (ebuliometria) lub spadku temperatury zamarzania (kriometria) rozpuszczalnika, kiedy dodany zostaje polimer. Metoda oparta jest na fakcie, że skutek rozpuszczonego polimeru na temperaturę wrzenia/zamarzania płynu zależy od masy cząsteczkowej polimeru (1) (2). Zastosowanie: Mn < 20 000. |
|
1.2. |
Zmniejszanie ciśnienia pary: obejmuje pomiar ciśnienia pary wybranego płynu odniesienia przed i po dodaniu znanych ilości polimeru (1) (2). Zastosowanie: Mn < 20 000 (teoretycznie; jednakże w praktyce wartości są ograniczone). |
|
1.3. |
Osmometria membranowa: opiera się na zasadzie osmozy, tzn. naturalnej skłonności cząsteczek rozpuszczalnika do przenikania przez półprzepuszczalną membranę z rozcieńczonego do stężonego roztworu aż do osiągnięcia równowagi. W trakcie badania rozcieńczony roztwór posiada zerowe stężenie polimeru, natomiast stężony roztwór zawiera polimer. Skutkiem przepuszczenia rozpuszczalnika przez membranę jest zróżnicowanie ciśnienia, które zależne jest od stężenia i masy cząsteczkowej polimeru (1) (3)(4). Zastosowanie: Mn w zakresie 20 000–200 000. |
|
1.4. |
Osmometria fazy parowania: obejmuje porównanie wskaźnika parowania czystego rozpuszczalnika aerozolu do co najmniej trzech aerozoli zawierających polimer o różnych stężeniach (1) (5) (6). Zastosowanie: Mn < 20 000. |
2. Analiza grupy końcowej
W celu wykorzystania tej metody potrzeba wiedzy zarówno o całkowitej strukturze polimeru, jak i o charakterze łańcucha kończącego grupy końcowe (która musi być możliwa do odróżnienia od głównego szkieletu, np. przez NMR lub miareczkowanie/różniczkowanie). Oznaczenie stężenia cząsteczkowego grup końcowych obecnych w polimerze może prowadzić do wyprowadzenia wartości masy cząsteczkowej (7) (8) (9).
Zastosowanie, Mn do 50 000 (ze zmniejszającą się wiarygodnością).
3. Bibliografia
|
(1) |
Billmeyer, F.W. Jr., Textbook of Polymer Science, 3rd Edn., John Wiley, New York, 1984. |
|
(2) |
Glover, C.A., Absolute Colligative Property Methods. Chapter 4. In: Polymer Molecular Weights, Part I P.E. Slade, Jr. ed., Marcel Dekker, New York, 1975. |
|
(3) |
ASTM D 3750-79, Standard Practice for Determination of Number-Average Molecular Weight of Polymers by Membrane Osmometry. American Society for Testing and Materials, Philadelphia, Pennsylvania, 1979. |
|
(4) |
Coll, H., Membrane Osmometry. In: Determination of Molecular Weight, A.R. Cooper ed., J. Wiley and Sons, 1989, pp. 25–52. |
|
(5) |
ASTM 3592-77, (1977). Standard Recommended Practice for Determination of Molecular Weight by Vapour Pressure, American Society for Testing and Materials, Philadelphia, Pennsylvania. |
|
(6) |
Morris, C.E.M., (1989). Vapour Pressure osmometry. In: Determinationn of Molecular Weight, A.R. Cooper ed., John Wiley and Sons. |
|
(7) |
Schröder, E., Müller, G., and Arndt, K-F., (1989). Polymer Characterisation, Carl Hanser Verlag, Munich. |
|
(8) |
Garmon, R.G., (1975). End-Group Determinations, Chapter 3 In: Polymer Molecular Weights, Part I, P.E. Slade, Jr. ed. Marcel Dekker, New York. |
|
(9) |
Amiya, S., et al. (1990). Pure and Applied Chemistry, 62, 2139-2146. |
A.19. ZAWARTOŚĆ POLIMERÓW O MAŁEJ MASIE CZĄSTECZKOWEJ
1. METODA
Niniejsza metoda chromatografii żelowo-permeacyjnej jest powtórzeniem OECD TG 119 (1996). Podstawowe zasady i dalsze informacje techniczne są podane w odniesieniach.
1.1. WPROWADZENIE
Ze względu na to, że właściwości polimerów są tak zróżnicowane, nie istnieje możliwość opisania pojedynczej metody ustalającej dokładnie warunki rozdzielenia i oceny, która obejmuje wszystkie ewentualności i właściwości pojawiające się w trakcie rozdzielania polimerów. W szczególności złożone systemy polimerów często nie ulegają często chromatografii żelowo-permeacyjnej (GPC). Jeśli GPC nie jest stosowane, masa cząsteczkowa może być ustalona za pomocą innych metod (zob. załącznik). W takich przypadkach powinny zostać podane wszystkie szczegóły i uzasadnienia dla wykorzystanych metod.
Opisana metoda oparta jest na normie DIN 55672 (1). Szczegółowe informacje dotyczące tego, w jaki sposób przeprowadzać doświadczenia oraz w jaki sposób oceniać dane, można znaleźć w niniejszej normie DIN. W przypadku gdy niezbędne są zmiany warunków doświadczeń, wspomniane zmiany muszą być uzasadnione. Inne normy mogą być wykorzystywane tylko w przypadku, gdy istnieją do nich pełne odniesienia. Opisana metoda wykorzystuje próbki polistyrenu o znanej polidyspersyjności w odniesieniu do kalibracji oraz może wystąpić konieczność jej zostać zmodyfikowania, tak aby była odpowiednia dla niektórych polimerów, np. polimerów rozpuszczalnych w wodzie i polimerów o długich rozgałęzieniach łańcucha.
1.2. DEFINICJE I JEDNOSTKI
Mała masa cząsteczkowa jest arbitralnie określana jako masa cząsteczkowa poniżej 1 000 daltonów.
Średnia liczbowo masa cząsteczkowa Mn i średnia wagowo masa cząsteczkowa Mw jest określana przy użyciu następujących równań: :
|
|
|
gdzie:
|
HI |
= |
jest poziomem detektora sygnału z linii podstawowej dla objętości retencyjnej Vi, |
|
Mi |
= |
jest masą cząsteczkową polimerowej frakcji przy objętości retencyjnej Vi, i n jest liczbą punktów pomiarowych. |
Szerokość rozkładu masy cząsteczkowej, który jest miarą dyspersyjności systemu, jest podana na podstawie stosunku Mw/Mn.
1.3. SUBSTANCJE ODNIESIENIA
Ze względu na to, że GPC jest metodą względną, musi zostać podjęta kalibracja. Zwykle do tego celu używane są wzorce polistyrenowe mało rozproszone, o linearnej budowie, posiadające średnie masy cząsteczkowe Mn i Mw oraz znany rozkład masy cząsteczkowej. Krzywa kalibracji może być wykorzystywana wyłącznie w celu określania masy cząsteczkowej nieznanej próbki, jeżeli warunki rozdzielenia próbki oraz wzorca zostały dobrane w identyczny sposób.
Ustalony związek między masą cząsteczkową oraz objętością elucji jest ważny wyłącznie w specyficznych warunkach danego doświadczenia. Warunki obejmują przede wszystkim temperaturę, rozpuszczalnik (lub mieszaninę rozpuszczalników), warunki chromatografii oraz kolumnę rozdzielającą lub systemy kolumn.
Masy cząsteczkowe próbki ustalone w ten sposób są względnymi wartościami oraz są opisane jako równoważnik mas cząsteczkowych polistyrenowych. Oznacza to, że w zależności od strukturalnych i chemicznych różnic między próbką a wzorcami, masy cząsteczkowe mogą odchylać się od wartości absolutnych w większym lub mniejszym stopniu. Jeśli wykorzystywane są inne wzorce, np. glikolu polietylenu, tlenku polietylenu, metaakrylanu polimetylu czy kwasu poliakrylowego, powinny zostać podane odpowiednie przyczyny.
1.4. ZASADA METODY BADAWCZEJ
Zarówno rozkład masy cząsteczkowej próbki, jak i średnie masy cząsteczkowe (Mn, Mw) mogą być ustalane, wykorzystując GPC. GPC jest szczególnym rodzajem chromatografii cieczowej, w której próbka jest oddzielana według hydrodynamicznych wielkości pojedynczych składników (2).
Rozdzielenie następuje wówczas, gdy próbka przechodzi przez kolumnę, która jest wypełniona porowatym materiałem – jest to zazwyczaj organiczny żel. Małe cząsteczki mogą wnikać w pory, podczas gdy duże cząsteczki są wykluczane. Ścieżka dużych cząsteczek jest niniejszym krótsza i to one są najpierw poddane procesowi eluacji. Średnie cząsteczki wnikają w niektóre pory i są później poddawane eluacji. Najmniejsze cząsteczki o średnim hydrodynamicznym promieniu, mniejszym niż pory żelu, mogą wnikać we wszystkie pory. Są poddawane eluacji na końcu.
W sytuacji idealnej oddzielanie jest kontrolowane całkowicie rozmiarem odmian cząsteczkowych, ale w praktyce trudno jest uniknąć co najmniej niewielkich efektów absorbcyjnych. Nierówne wypełnienie kolumny oraz objętość martwa mogą tylko pogorszyć sytuację (2).
Na detekcję ma wpływ np. współczynnik załamania lub pochłanianie promieni UV oraz wydajności nieskomplikowanej krzywej rozkładu. Jednakże w celu przypisania rzeczywistych wartości masy cząsteczkowej do krzywej niezbędne jest poddanie kolumny kalibracji przez przepuszczanie polimerów o znanej masie cząsteczkowej oraz, w idealnym przypadku, o podobnej strukturze, np. różnych wzorców polistyrenowych. Krzywa Gaussa, powstająca zazwyczaj w ten sposób, czasami wypaczona jest małymi „ogonami” skierowanymi w stronę małych mas cząsteczkowych, oś pionowa wskazuje jakość w masie różnych wyeluowanych odmian polistyrenu, a oś pozioma wskazuje logarytm z masy cząsteczkowej.
Zawartość niskich mas cząsteczkowych jest uzyskiwana na podstawie krzywej. Obliczenia mogą być tylko wtedy dokładne, gdy odmiany o małych masach cząsteczkowych odpowiadają polimerowi jako całości.
1.5. KRYTERIA JAKOŚCIOWE
Powtarzalność (względne odchylenie standardowe: RSD) objętości eluowania powinna być lepsza niż 0,3 %. Wymagana powtarzalność analizy musi być zapewniona przez korektę dokonywaną poprzez wewnętrzne wzorce, jeżeli chromatogram jest oceniany w oparciu o czas i nie odpowiada wyżej wspomnianym kryteriom (1). Polidyspersyjności są zależne od mas cząsteczkowych wzorców. W przypadku wzorców polistyrenowych typowe wartości to:
|
Mp < 2 000 |
Mw/Mn < 1,20 |
|
2 000 ≤ Mp ≤ 106 |
Mw/Mn < 1,05 |
|
Mp > 106 |
Mw/Mn < 1,20 |
(Mp jest masą cząsteczkową wzorca w maksimum piku)
1.6. OPIS METODY BADAWCZEJ
1.6.1. Przygotowanie wzorcowych roztworów polistyrenu
Wzorce polistyrenu są rozpuszczane przez ostrożne mieszanie w wybranym eluencie. Zalecenia wytwórcy muszą zostać uwzględnione w trakcie przygotowywania roztworów.
Stężenia wybranych wzorców są zależne od wielu czynników, np. od objętości injekcji, lepkości roztworu oraz czułości detektora analitycznego. Maksymalna objętość injekcji musi być dostosowana do długości kolumny, w celu uniknięcia przeładowania. Typowe objętości injekcji dla analitycznych rozdzieleń, wykorzystując GPC z kolumną o wymiarach 30 cm × 7,8 mm, wynoszą zazwyczaj 40–100 μl. Wyższe objętości są możliwe, ale nie powinny przekraczać 250 μl. Optymalny stosunek między objętością injekcji oraz stężeniem musi być określony przed rzeczywistą kalibracją kolumny.
1.6.2. Przygotowywanie roztworu próbki
Zasadniczo te same wymagania stosuje się do przygotowywania roztworów próbek. Próbka jest rozpuszczana w odpowiednim rozpuszczalniku, np. tetrahydrofuranie (THF), przez lekkie wstrząsanie. W żadnych okolicznościach nie powinno się rozpuszczać próbki w wannie ultradźwiękowej. W razie konieczności, próbka roztworu jest oczyszczana przez membranowy filtr posiadający pory o wymiarach między 0,2 i 2 μ m.
Obecność nierozpuszczalnych cząstek musi być uwzględniona w sprawozdaniu końcowym, jako że może być to ważne w odniesieniu do próbek o wysokiej masie cząsteczkowej. Powinna zostać wykorzystana odpowiednia metoda w celu określenia procentu wagowego nierozpuszczonych cząstek. Roztwór musi zostać wykorzystany w ciągu 24 godzin.
1.6.3. Korekta w odniesieniu do zawartości zanieczyszczeń oraz dodatków
Zazwyczaj niezbędna jest korekta udziału zawartości odmian M < 1 000 w odniesieniu do udziału niepolimerowych szczególnych obecnych składników (np. zanieczyszczenia i/lub dodatki), chyba że zmierzona zawartość wynosi już < 1 %. Osiągane jest to przez bezpośrednią analizę roztworu polimerowego lub eluentu metodą GPC.
W przypadkach gdy eluent po przejściu przez kolumnę jest za bardzo rozcieńczony, aby można go było poddać dalszej analizie, musi zostać zagęszczony. Może okazać się niezbędne odparowanie eluentu do sucha oraz ponowne jego rozpuszczenie. Stężenie eluentu musi zostać uzyskane w warunkach, które zapewniają, że nie pojawią się żadne zmiany w eluencie. Obróbka eluentu po fazie GPC jest zależna od wykorzystanej metody analitycznej wykorzystywanej do oznaczania ilościowego.
1.6.4. Aparatura
Aparatura GPC składa się z następujących części składowych:
|
— |
zbiornik na rozpuszczalnik, |
|
— |
odgazowywacz (w miarę potrzeb), |
|
— |
pompa, |
|
— |
nawilżacz pulsowy (w miarę potrzeb), |
|
— |
system wtryskowy, |
|
— |
kolumny chromatograficzne, |
|
— |
detektor, |
|
— |
przepływomierz (w miarę potrzeb), |
|
— |
urządzenia rejestrujące dane, |
|
— |
naczynie na odpady. |
Musi zostać zapewnione, że system GPC będzie jest obojętny w odniesieniu do stosowanych rozpuszczalników (np. przez wykorzystanie stalowych kapilar w przypadku rozpuszczalnika THF).
1.6.5. System wtryskowy i system dostarczający rozpuszczalnik
Określona objętość roztworu próbki jest umieszczona w kolumnie albo przy użyciu automatycznego próbnika, albo ręcznie do ściśle określonej strefy. Zbyt szybkie wstrzykiwanie, jeśli jest wykonywane ręcznie, może spowodować zmiany w obserwowalnym rozkładzie masy cząsteczkowej. System dostarczający rozpuszczalnik, na ile jest to możliwe, powinien być wolny od pulsacji, co można osiągnąć poprzez wbudowanie do niego tłumika pulsacji. Prawidłowa prędkość przepływu wynosi 1 ml/min.
1.6.6. Kolumna
W zależności od próbki właściwości polimeru określa się albo za pomocą prostej kolumny, albo kilku kolumn połączonych w sekwencję. Pewna ilość materiałów porowatych kolumn o określonych właściwościach dostępna jest w ogólnej sprzedaży (np. rozmiar porów, granica ekskluzji). Wybór żelu rozdzielającego lub długość kolumny jest uzależniona zarówno od właściwości próbki (objętości hydrodynamiczne, rozkład masy cząsteczkowej), jak i od specyficznych warunków rozdzielania, takich jak rozpuszczalnik, temperatura i prędkość przepływu (1) (2) (3).
1.6.7. Półki teoretyczne
Kolumna lub połączenie kolumn wykorzystywanych do rozdzielania musi być scherakteryzowana przez ilość półek teoretycznych. Obejmuje to, w przypadku THF jako rozpuszczalnikia eluencyjnego, umieszczenie roztworu etylobenzenu lub innych odpowiednich niepolarnych substancji rozpuszczonych w kolumnie o znanej długości. Liczba półek teoretycznych jest uzyskiwana na podstawie następującego równania:
|
|
lub |
|
gdzie:
|
N |
= |
jest liczbą półek teoretycznych, |
|
Ve |
= |
jest objętością eluencyjną w maksimum piku, |
|
W |
= |
jest wyjściową szerokością piku, |
|
W1/2 |
= |
jest szerokością piku w połowie wysokości. |
1.6.8. Wydajność rozdzielania
Oprócz liczby półek teoretycznych, która jest określającą ilościowo szerokością pasma, ważną rolę odgrywa także wydajność rozdzielenia, będąca określana przez nachylenie krzywej kalibracyjnej. Wydajność rozdzielania kolumny jest uzyskiwana z następującego stosunku:

gdzie:
|
Ve, Mx |
= |
jest objętością eluencyjną dla polistyrenu o masie cząsteczkowej Mx, |
|
Ve, (10.Mx) |
= |
jest objętością eluencyjną dla polistyrenu o 10-krotnie większej masie cząsteczkowej. |
Rozdzielczość systemu jest zwykle określana w następujący sposób:

gdzie:
|
Ve1, Ve2 |
= |
są objętościami eluencyjnymi dwóch wzorców polistyrenowych w maksimum piku; |
|
W1, W2 |
= |
są największymi szerokościami piku w lini podstawowej; |
|
M1, M2 |
= |
są masami cząsteczkowymi przy maksimum piku (powinny różnić się współczynnikiem o 10). |
Wartość R w odniesieni do systemu kolumn powinna być wyższa niż 1,7 (4).
1.6.9. Rozpuszczalniki
Wszystkie rozpuszczalniki muszą posiadać wysoką czystość (dla THF używana jest czystość 99,5 %). Zbiornik na rozpuszczalnik (o ile jest to konieczne – w atmosferze gazu obojętnego) musi być odpowiednio duży dla kalibracji kolumny i analiz wielu próbek. Rozpuszczalnik musi być odgazowany zanim zostanie przeniesiony do kolumny za pomocą pompy.
1.6.10. Kontrola temperatury
Temperatura krytycznych komponentów wewnętrznych (pętla wtryskowa, kolumny, detektor i przewody) powinna być stała i zgodna z wyborem rozpuszczalnika.
1.6.11. Detektor
Zadaniem detektora jest rejestracja ilościowa stężenia próbki eluowanej z kolumny. W celu uniknięcia niepotrzebnego poszerzenia pików objętość kuwety komórki detektora musi być tak mała, jak to tylko jest możliwe. Nie powinna być większa niż 10 μl, z wyjątkiem detektorów mierzących światło rozproszone oraz lepkość. W celu detekcji zazwyczaj używana jest refraktometria różnicowa. Jednakże jeżeli wymagają tego właściwości próbki lub właściwości eluacyjne rozpuszczalnika, mogą być używane inne rodzaje detektora, np. UV/VIS, IR, detektory lepkości itd.
2. DANE I SPORZĄDZANIE SPRAWOZDAŃ
2.1. DANE
Powinno nastąpić odniesienie do normy DIN (1) w celu uzyskania szczegółowych kryteriów oceny, jak również do wymogów odnoszących się do zbierania i przetwarzania danych.
Dla każdej próbki muszą zostać przeprowadzone dwa niezależne doświadczenia. Próbki muszą być analizowane pojedynczo. We wszystkich przypadkach zasadnicze jest ustalenie danych również z prób ślepych, poddawanych przetworzeniu w takich samych warunkach co próbka.
Niezbędne jest wyraźne wskazanie, że zmierzone wartości są wartościami względnymi, równoważnymi masom cząsteczkowym użytych wzorców.
Po określeniu objętości retencji lub czasów retencji (możliwie poprawionych wykorzystując wzorzec wewnętrzny) wartości logarytmu Mp (Mp będącego maksymalną wartością piku wzorca kalibracji) są wykreślane w oparciu o jedną z tych wielkości. Niezbędne są co najmniej dwa punkty kalibracji na dekadę masy cząsteczkowej oraz wymagane jest co najmniej pięć punktów pomiarowych dla całkowitej krzywej, która powinna obejmować oszacowaną masę cząsteczkową próbki. Niska wartość punktu końcowego masy cząsteczkowej krzywej wzorcowania jest określana za pomocą n-heksylobenzenu lub innego odpowiedniego niepolarnego roztworu. Część krzywej odpowiadająca masom cząsteczkowym poniżej 1 000 jest ustalana i korygowana w razie konieczności w odniesieniu do zanieczyszczeń oraz dodatków. Krzywe eluacji są zazwyczaj oceniane za pomocą elektronicznego przetwarzania danych W przypadku gdy używana jest ręczna digitalizacja, można powołać się na ASTMD 3536-91 (3).
Jeśli jakikolwiek nierozpuszczalny polimer jest zachowany w kolumnie, jego masa cząsteczkowa może być wyższa niż masa cząsteczkowa frakcji rozpuszczalnej, a jeśli nie jest brana pod uwagę może skutkować nadmiernym oszacowaniem zawartości niskiej masy cząsteczkowej. Wskazówki w odniesieniu do korygowania zawartości niskiej masy cząsteczkowej w odniesieniu do nierozpuszczalnego polimeru są przewidziane w załączniku.
Krzywa rozkładu musi być przewidziana w formie tabeli lub ilustracji (różnicowa częstotliwość lub procent sumy w oparciu o logarytm M). Na wizerunku graficznym jedna dekada masy cząsteczkowej powinna mieć normalnie około 4 cm szerokości, a maksymalna wartość szczytowa około 8 cm wysokości. W przypadku krzywych całkowego rozkładu różnica w rzędnych między 0 a 100 % powinna wynosić około 10 cm.
2.2. SPRAWOZDANIE Z BADANIA
Sprawozdanie z badania musi zawierać następujące informacje:
2.2.1. Substancja badana:
|
— |
dostępne informacje dotyczące badanej substancji (tożsamość, dodatki, zanieczyszczenia), |
|
— |
opis przetwarzania próbki, uwagi, problemy. |
2.2.2. Oprzyrządowanie:
|
— |
zbiornik z eluentem, obojętny gaz, odgazowywanie eluentu, skład eluentu, zanieczyszczenia, |
|
— |
pompa, pulsowy tłumik, system wtryskowy, |
|
— |
kolumny rozdzielające (wytwórca, wszystkie informacje dotyczące właściwości kolumn, takie jak rozmiar poru, rodzaj rozdzielającego materiału itd., liczba, długość oraz kolejność użytych kolumn), |
|
— |
liczba półek teoretycznych kolumny (lub połączonych), wydajność rozdzielania, (rozdzielczość systemu), |
|
— |
informacja dotycząca symetrii pików, |
|
— |
temperatura kolumny, rodzaj sterowania temperaturą, |
|
— |
detektor (zasada pomiarów, rodzaj, objętość kuwetowa), |
|
— |
przepływomierz w przypadkach, gdy jest używany (wytwórca, zasada pomiarów), |
|
— |
system umożliwiający rejestrację i przetwarzanie danych (osprzęt i oprogramowanie). |
2.2.3. Kalibracja systemu
|
— |
szczegółowy opis metody używanej do przygotowania krzywej kalibracji, |
|
— |
informacja dotycząca kryteriów jakościowych dla niniejszej metody (np. współczynnik korelacji, błąd sumy kwadratów itd.), |
|
— |
informacja dotycząca ekstrapolacji, założenia i przybliżenia poczynione w trakcie procedury doświadczalnej oraz oszacowanie i przetwarzanie danych, |
|
— |
wszystkie pomiary przeprowadzone w celu przygotowania krzywej kalibracji powinny być udokumentowane w tabelach, które zawierają następujące informacje dla każdego punktu kalibracji:
|
2.2.4. Informacja dotycząca zawartości polimerów o niskiej masie cząsteczkowej:
|
— |
opis metod stosowanych w analizach i sposobie, w jaki przeprowadzone zostały doświadczenia, |
|
— |
informacja dotycząca zawartości procentowej frakcji o małej masie cząsteczkowej w odniesieniu do masy całkowitej próbki, |
|
— |
informacja dotycząca zanieczyszczeń, dodatków i innych niepolimerowych składnikach w procentach w odniesieniu do masy całkowitej próbki. |
2.2.5. Ocena:
|
— |
ocena na podstawie czasu: wszystkie metody mające na celu zapewnienie wymaganej odtwarzalności (metoda poprawiania, wzorce wewnętrzne itd.), |
|
— |
informacja dotycząca tego, czy ocena została przeprowadzona na podstawie objętości eluencyjnej lub czasu retencji, |
|
— |
informacja dotycząca ograniczeń oceny, jeżeli pik nie został poddany analizie w całości, |
|
— |
opis metod wygładzających, jeżeli są wykorzystywane, |
|
— |
procedury przygotowawcze oraz wstępnego przetworzenia próbki, |
|
— |
obecność nierozpuszczalnych cząstek, jeżeli istnieją, |
|
— |
objętość wtrysku (μl) oraz stężenie wtrysku (mg/ml), |
|
— |
uwagi wskazujące skutki, które prowadzą do odchyleń od idealnego profilu GPC, |
|
— |
szczegółowy opis wszystkich zmian w procedurach badawczych, |
|
— |
szczegóły dotyczące zakresów błędów, |
|
— |
wszystkie inne informacje oraz uwagi istotne w odniesieniu do interpretacji wyników. |
3. BIBLIOGRAFIA
|
(1) |
DIN 55672 (1995) Gelpermeationschromatographie (GPC) mit Tetrahydrofuran (THF) als Elutionsmittel, Teil 1. |
|
(2) |
Yau, W.W., Kirkland, J.J., and Bly, D.D. eds. (1979). Modern Size Exclusion Liquid Chromatography, J. Wiley and Sons. |
|
(3) |
ASTM D 3536-91, (1991). Standard Test method for Molecular Weight Averages and Molecular Weight Distribution by Liquid Exclusion Chromatography (Gel Permeation Chromatography-GPC). American Society for Testing and Materials, Philadelphia, Pennsylvania. |
|
(4) |
ASTM D 5296-92, (1992). Standard Test method for Molecular Weight Averages and Molecular Weight Distribution of Polystyrene by High Performance Size-Exclusion Chromatography. American Society for Testing and Materials, Philadelphia, Pennsylvania. |
Załącznik
Wytyczne dotyczące korygowania zawartości związków o niskiej masie cząsteczkowej w odniesieniu do obecności nierozpuszczalnych polimerów
Jeśli w próbce obecny jest nierozpuszczalny polimer, skutkiem tego jest utrata masy podczas analizy GPC. Nierozpuszczalny polimer pozostaje nieodwracalnie w kolumnie lub na filtrze próbkowym, podczas gdy rozpuszczalna część próbki przechodzi przez kolumnę. W przypadku gdy inkrement współczynnika załamania światła (dn/dc) polimeru może zostać oszacowany lub zmierzony, można oszacować utratę masy próbki na kolumnie. W takim przypadku przeprowadza się korektę, wykorzystując zewnętrzną kalibrację wzorcowymi materiałami o znanym stężeniu oraz dn/dc w celu kalibracji reakcji refraktometru. W poniższym przykładzie wykorzystywany jest wzorzec metaakrylanu polimetylu (pMMA).
W zewnętrznej kalibracji, w celu analizy akrylowych polimerów, wzorzec pMMA, o znanym stężeniu w tetrahydrofuranie, jest analizowany przez GPC, a otrzymane dane są wykorzystywane w celu znalezienia stałej refraktometru zgodnie ze wzorem:
K = R/(C × V × dn/dc)
gdzie:
|
K |
= |
stała refraktometru (w mikrowoltosekundach/ml), |
|
R |
= |
wskaźnik wzorca pMMA (w mikrowoltosekundach), |
|
C |
= |
stężenie wzorca pMMA (w mg/ml), |
|
V |
= |
objętość dozowana (w ml), oraz |
|
dn/dc |
= |
inkrement współczynnika załamania światła dla pMMA w tetrahydrofuranie (w ml/mg) |
Następujące dane są typowe dla wzorca pMMA:
|
R |
= |
2 937 891 |
|
C |
= |
1,07 mg/ml |
|
V |
= |
0,1 ml |
|
dn/dc |
= |
9 × 10-5 ml/mg |
Uzyskana wartość K, 3,05 × 1011, jest następnie wykorzystywana w celu obliczania teoretycznej reakcji detektora, jeżeli 100 % wtryśniętego polimeru eluowało przez detektor.
A.20. ZACHOWANIE POLIMERÓW W WODZIE – ROZPUSZCZANIE/EKSTRAKCJA
1. METODA
Opisana metoda jest powtórzeniem wersji poprawionej OECD WT 120 (1997). Dalsze informacje techniczne podane są w pozycji bibliograficznej (1).
1.1. WPROWADZENIE
W odniesieniu do niektórych polimerów, takich jak np. polimerów emulsyjnych, zanim można zastosować metodę opisaną poniżej, konieczne mogą być wstępne prace przygotowawcze. Metoda ta nie ma zastosowania do polimerów płynnych oraz do polimerów, które reagują z woda w warunkach badania.
Jeżeli metoda nie jest praktyczna lub nie jest możliwe jej zastosowanie, zachowanie rozpuszczanie/ekstrakcja może zostać zbadane za pomocą innych metod. W takich przypadkach należy podać pełne szczegóły oraz uzasadnienie dla zastosowania metody.
1.2. SUBSTANCJE ODNIESIENIA
Brak.
1.3. ZASADA METODY BADAWCZEJ
Zachowanie polimerów rozpuszczanie/ekstrakcja w środowisku wodnym ustalane jest przy wykorzystaniu metody kolbowej (zob. A.6. Rozpuszczalność w wodzie, metoda kolbowa) ze zmianami opisanymi poniżej.
1.4. KRYTERIA JAKOŚCIOWE
Brak.
1.5. OPIS METODY BADAWCZEJ
1.5.1. Sprzęt
W odniesieniu do tej metody wymagany jest następujący sprzęt:
|
— |
przyrząd zgniatający, np. młyn do wytwarzania cząstek o znanym rozmiarze, |
|
— |
aparatura do wstrząsania z możliwością kontroli temperatury, |
|
— |
system filtrowania membranowego, |
|
— |
właściwy sprzęt analityczny, |
|
— |
znormalizowane sita. |
1.5.2. Przygotowanie próbki
Próbka reprezentatywna musi zostać najpierw zmniejszona do wielkości cząsteczek między 0,125 i 0,25 mm, wykorzystując właściwe sita. W odniesieniu do stabilności próbki lub w odniesieniu do procesu mielenia wymagane może być chłodzenie. Materiały o właściwościach podobnych do gumy mogą być kruszone w temperaturze ciekłego azotu (1).
Jeżeli wymagana frakcja wielkości cząsteczki jest nieosiągalna, powinno zostać podjęte działanie w celu zmniejszenia wielkości cząsteczek do najmniejszej możliwej wielkości oraz złożone sprawozdanie o wyniku. W sprawozdaniu niezbędne jest wskazanie sposobu, w jaki skruszona próbka była składowana przed badaniem.
1.5.3. Procedura
Trzy próbki o masie 10 g są odważane do trzech naczyń wyposażonych w szklane korki i dodaje się po 1 000 ml wody do każdego z naczyń. Jeżeli zastosowanie ilości 10 g polimeru okazuje się niewykonalne, powinna zostać wykorzystana największa najbliższa tej wartości ilość, jaką można zastosować, a objętość wody powinna zostać odpowiednio dostosowana.
Naczynia są szczelnie zamykane korkiem, a następnie wstrząsane w temperaturze 20 oC. Powinno zostać wykorzystane urządzenie wstrząsające lub mieszające, działające w stałej temperaturze. Po okresie 24 godzin zawartość każdego naczynia jest odwirowywana lub filtrowana, a stężenie polimeru w czystej fazie wodnej ustalane jest za pomocą odpowiedniej metody. Jeżeli odpowiednie metody analityczne w odniesieniu do fazy wodnej nie są dostępne, całkowitą rozpuszczalność/podatność na ekstrakcję można oszacować z suchej masy pozostałości filtracyjnej lub z odwirowanego strątu.
Zazwyczaj niezbędne jest ilościowe zróżnicowanie między zanieczyszczeniami i dodatkami, z jednej strony, oraz odmianami o małej masie cząsteczkowej, z drugiej strony. W przypadku oznaczania grawimetrycznego ważne jest także przeprowadzenie próby ślepej bez wykorzystania substancji badanej, w celu uwzględnienia pozostałości powstających w wyniku metody doświadczalnej.
Zachowanie polimerów rozpuszczanie/ekstrakcja w wodzie w temperaturze 37 oC przy pH 2 i pH 9 może zostać ustalone w ten sam sposób, jak opisano w odniesieniu do przeprowadzania doświadczenia w temperaturze 20 oC. Wartości pH można osiągnąć przez dodanie albo odpowiednich roztworów buforowych, albo odpowiednich kwasów lub zasad, takich jak kwas solny, kwas octowy, wodorotlenek sodu lub potasu o czystości analitycznej lub NH3.
W zależności od wykorzystanej metody analizy, należy przeprowadzić jedno lub dwa badania. Jeżeli dostępne są wystarczająco szczegółowe metody w odniesieniu do bezpośredniej analizy fazy wodnej dla komponentu polimerowego, wystarczy przeprowadzić jedno badanie, jak opisano powyżej. Jednakże jeżeli takie metody nie są dostępne, a określenie zachowania rozpuszczania/ekstrakcji polimeru jest ograniczone do analizy pośredniej przez ustalanie tylko całkowitej zawartości węgla organicznego (TOC) ekstraktu wodnego, powinno zostać przeprowadzone dodatkowe badanie. Takie dodatkowe badanie należy także przeprowadzić trzykrotnie, stosując dziesięciokrotnie mniejsze próbki polimeru oraz takie same ilości wody jak ilości wykorzystane w pierwszym badaniu.
1.5.4. Analiza
1.5.4.1. Badanie przeprowadzane z próbką jednego rozmiaru
Mogą być dostępne metody analizy bezpośredniej komponentów polimerowych w fazie wodnej. Alternatywnie można rozważyć pośrednią analizę rozpuszczonych/ekstrahowanych komponentów polimerowych przez określenie całkowitej zawartości części rozpuszczalnych i korektę w odniesieniu do komponentów, które nie są właściwe dla polimerów.
Możliwa jest analiza fazy wodnej dla ogółu odmian polimerowych:
albo przez wystarczającą czułą metodę, np.:
|
— |
TOC, wykorzystującą ekstrahowanie na ciepło nadsiarczanu lub dwuchromianu, w celu otrzymania CO2, a następnie wykonanie oznaczenia za pomocą podczerwieni IR lub analizy chemicznej, |
|
— |
spektometrie absorpcji atomowej (AAS) lub jej równoważnik emisji indukcyjnie wzbudzonej plazmy (ICP) dla krzemu lub polimerów zawierających metale, |
|
— |
pochłanianie UV lub spektrofluorometrie dla polimerów arylu, |
|
— |
LC-MS dla próbek o małej masie cząsteczkowej, |
lub przez odparowywanie w warunkach próżniowych do sucha ekstraktu wodnego i analizę spektroskopową (IR, UV itd.) lub analizę AAS/ICP pozostałości.
Jeżeli niemożliwa jest analiza samej fazy wodnej, roztwór wodny należy ekstrahować za pomocą organicznego rozpuszczalnika niemieszającego się z woda, np. chlorowanego węglowodoru. Rozpuszczalnik jest następnie odparowywany, a pozostałość analizowana pod kątem podanej zawartości polimeru w sposób określony powyżej. Wszelkie komponenty w tej pozostałości, które są identyfikowane jako zanieczyszczenia lub dodatki, mają zostać odjęte w celu określenia stopnia rozpuszczenia/ekstrakcji samego polimeru.
Jeżeli obecne są względnie duże ilości takich substancji, może wystąpić konieczność poddania pozostałości np. analizie HPLC lub GC, w celu odróżnienia zanieczyszczeń od obecnego monomeru i frakcji pochodnych monomeru, tak aby można było ustalić ich rzeczywistą zawartość.
W niektórych przypadkach wystarczające może być zwykłe odparowanie organicznego rozpuszczalnika do sucha i zważenie suchej pozostałości.
1.5.4.2. Badanie przeprowadzane z dwoma próbkami o różnych rozmiarach cząsteczek
Wszystkie ekstrakty wodne analizuje w odniesieniu do TOC.
Oznaczanie grawimetryczne przeprowadzane jest na nierozpuszczonej/niewyekstrahowanej części próbki. Jeżeli po odwirowaniu lub filtrowaniu zawartości każdego z naczyń pozostałości polimerów pozostają przytwierdzone do ścianek naczynia, naczynie powinno zostać przepłukane filtratem do momentu oczyszczenia naczynia ze wszystkich widocznych pozostałości. Następnie filtrat ponownie jest ponownie odwirowywany oraz filtrowany. Pozostałości na filtrze lub w rurze wirnikowej są suszone w temperaturze 40 oC w próżni oraz ważone. Suszenie kontynuuje się do momentu osiągnięcia stałej wagi.
2. DANE
2.1. BADANIE PRZEPROWADZANE Z PRÓBKĄ JEDNEGO ROZMIARU CZĄSTEK
Powinny zostać podane poszczególne wyniki dla każdej z trzech kolb oraz wartości średnie, wyrażone jednostkach masy na objętość roztworu (zwykle mg/l) lub masy na masę próbki polimeru (zwykle mg/g). Dodatkowo powinna zostać również podana utrata wagi próbki (obliczona jako masa substancji rozpuszczonej podzielona przez masę pierwszej próbki). Błędy względne powinny zostać odliczone (RSD). Dla całej substancji (polimer + niezbędne substancje dodane itd.) oraz dla samego polimeru (tj. po odjęciu substancji dodanych) należy podąć oddzielne dane liczbowe.
2.2. BADANIE PRZEPROWADZANE Z DWOMA PRÓBKAMI O RÓŻNYCH ROZMIARACH CZĄSTECZKI
Powinny zostać podane poszczególne wartości TOC ekstraktów wodnych dwóch trzykrotnie przeprowadzanych doświadczeń oraz średnich wartość dla każdego doświadczenia, wyrażone w jednostkach masy na objętość roztworu (zwykle mg/l) oraz w jednostkach masy na wagę pierwszej próbki (zwykle mgC/g).
Jeżeli nie istnieje różnica między wynikami w wysokich oraz niskich współczynnikach proporcji próbka/woda, może to wskazywać na to, że wszystkie możliwe do wyekstrahowania komponenty zostały faktycznie wyekstrahowane. W takim przypadku analiza bezpośrednia zazwyczaj nie byłaby konieczna. Powinny zostać podane masy poszczególnych pozostałości, wyrażone jako udział początkowych mas próbek.
Dla każdego eksperymentu powinny zostać obliczone wartości średnie. Różnice pomiędzy wartością 100 i otrzymanymi procentami wyrażają procenty materiału rozpuszczonego i ekstrahowanego w pierwotnej próbce.
3. SPORZĄDZANIE SPRAWOZDAŃ
3.1. SPRAWOZDANIE Z BADANIA
Sprawozdanie z badania musi zawierać następujące informacje:
3.1.1. Substancja badana:
|
— |
dostępne informacje dotyczące substancji badanej (tożsamość, substancje dodane, zanieczyszczenia, zawartość odmian o malej masie cząsteczkowej). |
3.1.2. Warunki przeprowadzania doświadczenia
|
— |
opis wykorzystanych procedur i warunków przeprowadzania doświadczenia, |
|
— |
opis metod analizy i wykrywania. |
3.1.3. Wyniki:
|
— |
wyniki rozpuszczalności/podatności na ekstrakcję w mg/l; wartości poszczególne i średnie dla badań ekstrakcji w poszczególnych roztworach rozbite na zawartość polimerów oraz zanieczyszczeń, dodatki itd., |
|
— |
wyniki rozpuszczalności/podatności na ekstrakcję w mg/g polimeru, |
|
— |
wartości TOC ekstraktów wodnych, masa substancji rozpuszczonej oraz udziały procentowe jeżeli zostały zmierzone, |
|
— |
pH każdej próbki, |
|
— |
informacje dotyczące wartości ślepej próby, |
|
— |
w miarę potrzeb odniesienia do niestabilności chemicznej substancji badanej, zarówno podczas procesu badania, jak i podczas procesu analitycznego, |
|
— |
wszystkie informacje które są ważne w odniesieniu do interpretacji wyników. |
4. BIBLIOGRAFIA
|
(1) |
DIN 53733 (1976) Zerkleinerung von Kunststofferzeugnissen fur Prufzwecke. |
A.21. WŁAŚCIWOŚCI UTLENIAJĄCE (CIECZE)
1. METODA
1.1. WPROWADZENIE
Ta metoda badawcza jest przeznaczona do pomiaru potencjału substancji ciekłej do zwiększenia szybkości spalania oraz intensywności spalania materiałów palnych lub do tworzenia mieszanin z materiałami palnymi, które spontanicznie zapalają się, kiedy są ze sobą dokładnie wymieszane. Jest oparta na badaniu ONZ dla cieczy utleniających (1) i jest z nim równoważna, jakkolwiek metoda A.21 jest przede wszystkim zaprojektowana, aby spełniać wymagania rozporządzenia 1907/2006, gdzie wymagane jest porównanie tylko z jedną substancją odniesienia. Konieczne może być badanie i porównanie z dodatkowymi substancjami odniesienia, gdyby wyniki badań miały być wykorzystywane do innych celów (9).
To badanie nie musi być przeprowadzone, jeżeli badanie wzoru strukturalnego wyjaśnia ponad wszelką zasadną wątpliwość, że ta substancja nie jest zdolna do reakcji egzotermicznej z materiałem palnym.
Przed przeprowadzeniem tego badania przydatne jest posiadanie wstępnych informacji na temat jakichkolwiek potencjalnych właściwości wybuchowych substancji.
Badanie to nie ma zastosowania do ciał stałych, gazów, substancji wybuchowych lub łatwopalnych bądź nadtlenków organicznych.
Można nie przeprowadzać tego badania, jeżeli są już dostępne dla substancji badanej wyniki próby ONZ dla cieczy utleniających (1).
1.2. DEFINICJE IJEDNOSTKI
Średni czas wzrostu ciśnienia jest wartością średnią mierzonych czasów, w jakich badana mieszanina spowoduje wzrost ciśnienia z 690 kPa do 2 070 kPa powyżej ciśnienia atmosferycznego.
1.3. SUBSTANCJAODNIESIENIA
Jako substancja odniesienia wymagany jest 65 % (m/m) roztwór wodny kwasu azotowego (czysty do analizy) (10).
Ewentualnie, jeżeli osoba badająca przewiduje, że wyniki tego badania będą mogły ostatecznie zostać użyte do innych celów, może być również właściwe badanie dodatkowych substancji odniesienia (11).
1.4. ZASADY METODY BADAWCZEJ
Badana ciecz jest mieszana w stosunku wagowym 1 do 1 z celulozą włóknistą i wprowadzana do komory ciśnieniowej. Jeżeli podczas mieszania lub napełniania następuje spontaniczny zapłon, nie są konieczne dalsze badania.
Jeżeli nie następuje spontaniczny zapłon, przeprowadza się pełne badanie. Mieszanina jest ogrzewana w komorze ciśnieniowej i mierzony jest średni czas potrzebny do podniesienia ciśnienia od 690 kPa do 2 070 kPa powyżej ciśnienia atmosferycznego. Otrzymana wartość jest porównywana ze średnim czasem wzrostu ciśnienia dla mieszaniny substancji odniesienia i celulozy w stosunku 1:1.
1.5. KRYTERIA JAKOŚCIOWE
W serii pięciu prób przeprowadzonych na pojedynczej substancji żaden wynik nie powinien odbiegać od średniej arytmetycznej o więcej niż 30 %. Wyniki różniące się od średniej o więcej niż 30 % powinny zostać odrzucone, procedura mieszania i napełniania powinna zostać poprawiona, a badanie powtórzone.
1.6. OPIS METODY
1.6.1. Przygotowanie
1.6.1.1. Substancja palna
Jako materiał palny jest używana sucha, włóknista celuloza o długości włókna od 50 do 250 μm i średniej średnicy równej 25 μm (12). Jest suszona do stałej masy, w warstwie o grubości nieprzekraczającej 25 mm w temperaturze 105 oC przez 4 godziny i trzymana w suszarce, ze środkiem suszącym, aż do ochłodzenia i do momentu, gdy będzie potrzebna do użycia. Zawartość wody w suchej celulozie powinna być mniejsza niż 0,5 % suchej masy (13). W razie potrzeby należy przedłużyć czas suszenia aż do osiągnięcia tego stanu (14). W całym badaniu musi być używana ta sama partia celulozy.
1.6.1.2. Aparatura
1.6.1.2.1.
Potrzebna jest komora ciśnieniowa. Komora ta składa się z cylindrycznego, stalowego zbiornika ciśnieniowego o długości 89 mm i średnicy zewnętrznej 60 mm (zob. rysunek 1). Po przeciwległych stronach cylindra są ścięte dwie płaszczyzny (zmniejszające przekrój poprzeczny komory do 50 mm), aby ułatwić trzymanie podczas montażu zapłonnika i wkładki odpowietrzającej. Komora, która ma otwór o średnicy 20 mm, jest od wewnątrz na obu końcach rozwiercona do głębokości 19 mm oraz nagwintowana, tak aby można było wkręcić w nią rurę o średnicy 1" według norm British Standard Pipe (BSP) lub odpowiednik według układu metrycznego. Urządzenie do pomiaru ciśnienia, w postaci ramienia bocznego, jest przykręcone do zakrzywionej powierzchni zewnętrznej komory ciśnieniowej w odległości 35 mm od jednego końca, pod kątem 90° do ściętej płaszczyzny. Gniazdo dla niego jest nawiercone na głębokość 12 mm i nagwintowane, tak aby można było wkręcić w nie gwint o średnicy 1/2" według BSP (lub odpowiednik według układu metrycznego), znajdujący się na końcu ramienia bocznego. W razie potrzeby stosuje się obojętną uszczelkę w celu zapewnienia gazoszczelności. Ramię boczne wystaje na 55 mm poza korpus komory ciśnieniowej i ma wywiercony otwór o średnicy 6 mm. Koniec ramienia bocznego jest rozwiercony i nagwintowany, tak aby można było osadzić manometr przeponowy. Może być stosowane dowolne urządzenie do pomiaru ciśnienia, pod warunkiem że będzie odporne na działanie gorących gazów lub produktów rozkładu i będzie zdolne reagować na zmiany wzrostu ciśnienia w zakresie 690–2 070 kPa w czasie nie dłuższym niż 5 ms.
Koniec komory ciśnieniowej najbardziej oddalony od ramienia bocznego jest zamknięty zapłonnikiem, który jest wyposażony w dwie elektrody, jedną izolowaną, a drugą uziemioną do jego korpusu. Drugi koniec komory ciśnieniowej jest zamknięty membraną bezpieczeństwa (ciśnienie uderzeniowe wynosi około 2 200 kPa) utrzymywaną w swojej pozycji przy pomocy wkładki ustalającej, która ma otwór o średnicy 20 mm. W razie potrzeby, w celu zapewnienia dopasowania gazoszczelnego, przy zapłonniku stosowana jest uszczelka obojętna. Stojak (rysunek 2) utrzymuje zestaw podczas użytkowania na odpowiedniej wysokości. Składa się on zwykle z płytki podstawy, wykonanej z miękkiej stali, o wymiarach 235 mm × 184 mm × 6 mm i z profilu kwadratowego o długości 185 mm i wymiarach 70 mm × 70 mm × 4 mm.
Z każdego z przeciwległych boków na jednym końcu na długości profilu kwadratowego wycięto fragment, tak że powstała konstrukcja mająca dwie nóżki o płaskich ściankach, na której jest nienaruszony przekrój skrzynkowy o długości 86 mm. Końce tych płaskich ścianek są przycięte pod kątem 60° do poziomu i przyspawane do płytki podstawy. W jednym boku górnego końca głównego profilu wycięto szczelinę o szerokości 22 mm i głębokości 46 mm, żeby podczas wkładania zestawu komory ciśnieniowej, najpierw koniec z zapłonnikiem, do uchwytu z przekroju skrzynkowego ramię boczne mieściło się w szczelinie. Kawałek stali o szerokości 30 mm i grubości 6 mm jest przyspawany do dolnej wewnętrznej ścianki przekroju skrzynkowego i działa jako rozpórka. Dwie śruby skrzydełkowe o średnicy 7 mm, przykręcone do przeciwległej ścianki, służą do trzymania komory ciśnieniowej mocno na miejscu. Dwa paski o szerokości 12 mm ze stali o grubości 6 mm, przyspawane do części bocznych, przylegając do podstawy przekroju skrzynkowego, podpierają komorę ciśnieniową od dołu.
1.6.1.2.2.
System zapłonu składa się drutu Ni/Cr o długości 25 cm o średnicy 0,6 mm i oporze 3,85 om/m. Drut jest zwinięty, przy pomocy pręta o średnicy 5 mm, w kształt cewki i podłączony do elektrod zapłonnika. Cewka powinna mieć jedną z konfiguracji przedstawionych na rysunku 3. Odstęp pomiędzy dnem komory a spodem cewki zapłonowej powinien wynosić 20 mm. Jeżeli elektrody nie są nastawne, końce przewodu zapłonowego pomiędzy cewką a dnem komory powinny być izolowane osłoną ceramiczną. Przewód jest ogrzewany prądem stałym o natężeniu przynajmniej 10 A.
1.6.2. Prowadzenie badania (15)
Aparatura, złożona w całości z przetwornikiem ciśnieniowym i systemem ogrzewania, ale bez założonej membrany bezpieczeństwa, jest trzymana końcem z zapłonnikiem na dół. 2,5 g cieczy przeznaczonej do badania jest mieszane z 2,5 g wysuszonej celulozy w zlewce szklanej przy pomocy szklanej bagietki (16). Dla bezpieczeństwa mieszanie powinno być prowadzone za osłoną bezpieczeństwa umieszczoną między operatorem a mieszaniną. Jeżeli mieszanina zapali się podczas mieszania lub napełniania, nie będą konieczne żadne dalsze badania. Mieszanina jest podawana do komory ciśnieniowej małymi porcjami z lekkim ubijaniem. Należy przy tym zapewnić, żeby była dobrze upakowana wokół cewki zapłonowej i żeby się z nią dobrze stykała. Ważne jest, żeby nie poruszyć cewki podczas procesu upakowywania, ponieważ może to prowadzić do błędnych wyników (17). Membrana bezpieczeństwa jest umieszczana na swoim miejscu, a nakrętka zabezpieczająca jest mocno przykręcana. Naładowana komora jest przenoszona membraną bezpieczeństwa do góry do stojaka, który powinien być umieszczony w odpowiednim, opancerzonym dygestorium lub komorze zapłonowej. Do zewnętrznych końcówek zapłonnika podłącza się zasilanie i podaje prąd o natężeniu 10 A. Czas od rozpoczęcia mieszania do włączenia zasilania nie powinien przekraczać 10 min.
Sygnał generowany przez przetwornik ciśnieniowy jest zapisywany przez odpowiedni system, który pozwala zarówno na ocenę, jak i tworzenie ciągłego zapisu profilu zmiany ciśnienia w czasie (np. przejściowy rejestrator podłączony do rejestratora graficznego). Mieszanina jest ogrzewana aż do przerwania membrany bezpieczeństwa lub do upływu 60 s. Jeżeli membrana bezpieczeństwa nie zostanie zerwana, należy pozwolić mieszaninie ostygnąć przed ostrożnym demontażem aparatury, podejmując środki bezpieczeństwa na wypadek wzrostu ciśnienia. Z substancją badaną i z substancją(-ami) odniesienia wykonuje się pięć prób. Zapisywany jest czas potrzebny na zwiększenie ciśnienia od 690 kPa do 2 070 kPa powyżej ciśnienia atmosferycznego. Obliczany jest średni czas wzrostu ciśnienia.
W niektórych przypadkach substancje mogą wytwarzać wzrost ciśnienia (zbyt duży lub zbyt mały) wskutek reakcji chemicznych niecharakteryzujących właściwości utleniających substancji. W tych przypadkach do wyjaśnienia przebiegu reakcji może być konieczne powtórzenie badania z substancją obojętną np. diatomitem (ziemia okrzemkowa) zamiast celulozy.
2. DANE
Czasy wzrostu ciśnienia zarówno dla substancji badanej, jak i substancji odniesienia. Czasy wzrostu ciśnienia dla badań z substancją obojętną, jeżeli zostały przeprowadzone.
2.1. OPRACOWANIE WYNIKÓW
Oblicza się średnie czasy wzrostu ciśnienia zarówno dla substancji badanej, jak i dla substancji odniesienia.
Oblicza się średni czas wzrostu ciśnienia dla badań z substancją obojętną (jeżeli przeprowadzono).
Przykładowe wyniki zostały przedstawione w tabeli 1.
Tabela 1
Przykładowe wyniki (18)
|
Substancja (19) |
Średnia wartość wzrostu ciśnienia dla mieszaniny 1:1 z celulozą (ms) |
|
Dwuchromian amonu; nasycony roztwór wodny |
20 800 |
|
Azotan amonu, nasycony roztwór wodny |
6 700 |
|
Azotan żelaza, nasycony roztwór wodny |
4 133 |
|
Nadchloran litu, nasycony roztwór wodny |
1 686 |
|
Nadchloran magnezu, nasycony roztwór wodny |
777 |
|
Azotan niklu, nasycony roztwór wodny |
6 250 |
|
Kwas azotowy, 65 % |
4 767 (20) |
|
Kwas nadchlorowy, 50 % |
121 (20) |
|
Kwas nadchlorowy, 55 % |
59 |
|
Azotan potasu, 30 % roztwór wodny |
26 690 |
|
Azotan srebra, nasycony roztwór wodny |
|
|
Chloran sodu, 40 % roztwór wodny |
2 555 (20) |
|
Azotan sodu, 45 % roztwór wodny |
4 133 |
|
Substancja obojętna |
|
|
Woda: celuloza |
3. SPRAWOZDANIE
3.1. SPRAWOZDANIE Z BADAŃ
Sprawozdanie z badań powinno zawierać następujące informacje:
|
— |
identyfikację, skład, stopień czystości itp. badanej substancji, |
|
— |
stężenie badanej substancji, |
|
— |
procedurę suszenia celulozy, |
|
— |
zawartość wody w użytej celulozie, |
|
— |
wyniki pomiarów, |
|
— |
ewentualne wyniki badań z zastosowaniem substancji obojętnej, |
|
— |
obliczone średnie czasy wzrostu ciśnienia, |
|
— |
jakiekolwiek odchylenia od tej metody i ich przyczyny, |
|
— |
wszystkie dodatkowe informacje lub uwagi istotne dla interpretacji wyników. |
3.2. INTERPRETACJA WYNIKÓW (22)
Wyniki są oceniane na podstawie:
|
a) |
czy mieszanina substancji badanej i celulozy zapala się spontanicznie; oraz |
|
b) |
porównania średniego czasu potrzebnego do wzrostu ciśnienia od 690 kPa do 2 070 kPa z tą samą wartością dla substancji odniesienia. |
Ciecz jest uznawana za utleniacz, jeżeli:
|
a) |
mieszanina w stosunku wagowym 1:1 badanej substancji i celulozy zapala się spontanicznie; lub |
|
b) |
mieszanina w stosunku wagowym 1:1 substancji badanej i celulozy wykazuje średni czas wzrostu ciśnienia niższy lub równy średniemu czasowi wzrostu ciśnienia mieszaniny 1:1, wagowo, dla 65 % (m/m) roztworu wodnego kwasu azotowego i celulozy. |
Aby uniknąć fałszywych wyników pozytywnych, w razie potrzeby przy interpretacji wyników należy również uwzględnić wyniki otrzymane w badaniu substancji z materiałem obojętnym.
4. BIBILIOGRAFIA
|
(1) |
Recommendations on the Transport of Dangerous Goods, Manual of Tests and Criteria. 3rd revised edition. Publikacja ONZ Nr: ST/SG/AC.10/11/Rev. 3, 1999, s. 342. Badanie O.2: Badanie na utlenianie cieczy. |
Rysunek 1
Komora ciśnieniowa

Rysunek 2
Stojak
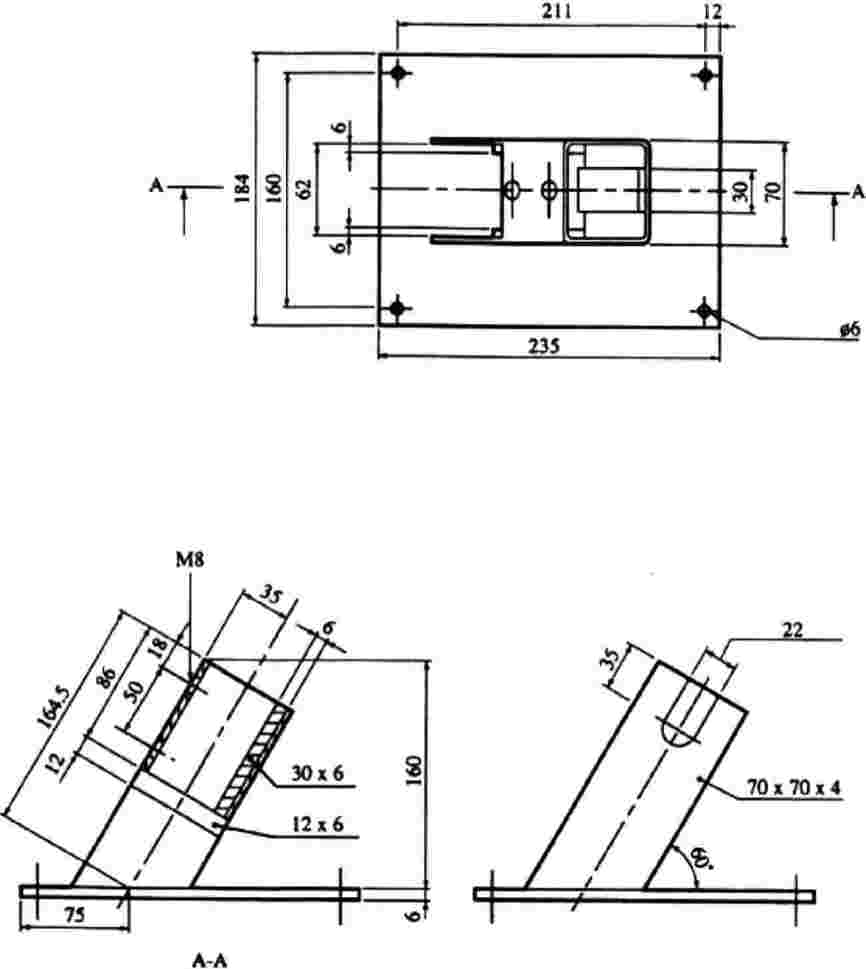
Rysunek 3
Układ zapłonu

Uwaga: Może zostać użyta którakolwiek z tych konfiguracji.
(1) W zależności od typu przyrządu i stopnia czystości substancji.
(2) W zależności od typu przyrządu i stopnia czystości substancji.
(3) W zależności od typu przyrządu stopnia czystości substancji.
(4) W zależności od typu przyrządu i stopnia czystości substancji.
(5) Niniejsza dokładność obowiązuje tylko dla prostego urządzenia, jak na przykład opisano w ASTM Dl 120-72; można ją poprawić bardziej złożonymi urządzeniami ebuliometru.
(6) Ważna jedynie dla czystych substancji. Użycie w innych okolicznościach powinno być uzasadnione.
(7) W zależności od stopnia czystości.
(8) Metody te mogą także być użyte w zakresie od 1 do 10 Pa pod warunkiem podjęcia środków ostrożności.
(9) Jak na przykład w ramach przepisów transportowych ONZ.
(10) Kwas powinien być miareczkowany przed badaniem, aby potwierdzić jego stężenie.
(11) Np. 50 % (m/m) kwasu chlorowego (VII) i 40 % (m/m) chlorku sodowego stosuje się w pozycji bibliograficznej (1).
(12) Np. proszek celulozowy do kolumn chromatograficznych Whatmana CF 11, nr katalogowy 4021 050.
(13) Potwierdzone przez np. miareczkowanie metodą Karla-Fishera.
(14) Alternatywnie, tę zawartość wody można również osiągnąć przez np. ogrzewanie w 105 o C w próżni przez 24 godz.
(15) Mieszaniny utleniaczy z celulozą muszą być traktowane jako potencjalnie wybuchowe i należy z nimi postępować z należytą ostrożnością.
(16) Można to osiągnąć w praktyce, przygotowując mieszaninę w stosunku 1:1 cieczy przeznaczonej do badania i celulozy z ilości większej, niż jest potrzebna, do próby i przenosząc 5 ±0,1 g do komory ciśnieniowej. Mieszanina powinna być świeżo przygotowywana do każdej próby.
(17) W szczególności należy unikać zetknięcia sąsiednich zwojów cewki.
(18) Zob. poz. lit. (1) dla klasyfikacji według programu transportu ONZ.
(19) Roztwór nasycony powinien zostać przygotowany w 20 oC.
(20) Wartość średnia dla międzylaboratoryjnych prób porównawczych.
(21) Nie osiągnięto maksymalnego ciśnienia 2 070 kPa.
(22) Zob. pozycja bibliograficzna (1) w celu interpretacji wyników według programu transportu ONZ.
CZĘŚĆ B: METODY OZNACZANIA TOKSYCZNOŚCI I INNYCH SKUTKÓW ZDROWOTNYCH
SPIS TREŚCI
WPROWADZENIE OGÓLNE
|
B.1 bis. |
TOKSYCZNOŚĆ OSTRA POKARMOWA – PROCEDURA Z WYKORZYSTANIEM STAŁEJ DAWKI |
|
B.1 ter. |
TOKSYCZNOŚĆ OSTRA POKARMOWA – METODA KLAS OSTREJ TOKSYCZNOŚCI |
|
B.2. |
TOKSYCZNOŚĆ OSTRA (INHALACYJNA) |
|
B.3. |
TOKSYCZNOŚĆ OSTRA (DERMALNA) |
|
B.4. |
OSTRA TOKSYCZNOŚĆ: PODRAŻNIENIA/KOROZJA SKÓRY |
|
B.5. |
TOKSYCZNOŚĆ OSTRA: PODRAŻNIENIE/KOROZJA OCZU |
|
B.6. |
SENSYBILIZACJA SKÓRY |
|
B.7. |
TOKSYCZNOŚĆ POWTARZANEJ (28 DNI) DAWKI (DOUSTNA) |
|
B.8. |
TOKSYCZNOŚĆ POWTARZANEJ (28 DNI) DAWKI (INHALACYJNA) |
|
B.9. |
TOKSYCZNOŚĆ POWTARZANEJ (28 DNI) DAWKI (DERMALNA) |
|
B.10. |
MUTAGENNOŚĆ – BADANIE ABERRACJI CHROMOSOMOWEJ SSAKÓW IN VITRO |
|
B.11. |
MUTAGENNOŚĆ – BADANIE ABERRACJI CHROMOSOMOWEJ SZPIKU KOSTNEGO IN VIVO |
|
B.12. |
MUTAGENNOŚĆ – BADANIE MIKROJĄDROWE ERYTROCYTÓW IN VIVO U SSAKÓW |
|
B.13/14. |
MUTAGENNOŚĆ: BADANIE MUTACJI POWROTNYCH W KOMÓRKACH BAKTERYJNYCH |
|
B.15 |
BADANIE MUTAGENNOŚCI I BADANIE PRZESIEWOWE NA RAKOTWÓRCZOŚĆ MUTACJA GENU –SACCHAROMYCES CEREVISIAE |
|
B.16. |
BADANIE REKOMBINACJI GENETYCZNEJ – SACCHAROMYCES CEREVISIAE |
|
B.17. |
MUTAGENNOŚĆ – BADANIE MUTACJI GENETYCZNEJ IN VITRO U SSAKÓW |
|
B.18. |
USZKODZENIE I NAPRAWA DNA – NIEREGULARNA SYNTEZA DNA – KOMÓRKI SSAKÓW IN VITRO |
|
B.19. |
TEST WYMIANY CHROMATYD SIOSTRZANYCH IN VITRO |
|
B.20. |
BADANIA SPRZĘŻONYCH Z PŁCIĄ RECESYWNYCH CECH LETALNYCH U DROSOPHILA MELANOGASTER |
|
B.21. |
BADANIE PRZEMIANY KOMÓRKOWEJ SSAKÓW IN VITRO |
|
B.22. |
BADANIE DOMINUJĄCEGO GENU LETALNEGO GRYZONIA |
|
B.23. |
BADANIE ABERRACJI CHROMOSOMOWEJ SPERMATOGONIÓW U SSAKÓW |
|
B.24. |
TEST PLAMKOWY U MYSZY |
|
B.25. |
TRANSLOKACJA DZIEDZICZNOŚCI U MYSZY |
|
B.26. |
BADANIE TOKSYCZNOŚCI PODPRZEWLEKŁEJ DROGĄ POKARMOWĄ – STUDIUM TOKSYCZNOŚCI NA GRYZONIACH METODĄ POWTARZANEJ 90-DNIOWEJ DAWKI DROGĄ POKARMOWĄ |
|
B.27. |
BADANIE TOKSYCZNOŚCI PODPRZEWLEKŁEJ DROGĄ POKARMOWĄ – BADANIA TOKSYCZNOŚCI NA NIEGRYZONIACH METODĄ POWTARZANEJ 90-DNIOWEJ DAWKI DROGĄ POKARMOWĄ |
|
B.28. |
BADANIE PODCHRONICZNEJ TOKSYCZNOŚCI SKÓRY – 90-DNIOWE BADANIA POWTARZANEGO DAWKOWANIA NA SKÓRZE Z WYKORZYSTANIEM GATUNKÓW GRYZONI |
|
B.29. |
BADANIE PODCHRONICZNEJ TOKSYCZNOŚCI INHALACYJNEJ 90-DNIOWE – BADANIA INHALACYJNE Z WYKORZYSTANIEM GATUNKÓW GRYZONI |
|
B.30. |
BADANIE TOKSYCZNOŚCI CHRONICZNEJ |
|
B.31. |
BADANIE PRZEDURODZENIOWEJ TOKSYCZNOŚCI ROZWOJOWEJ |
|
B.32. |
BADANIE RAKOTWÓRCZOŚCI |
|
B.33. |
POŁĄCZONE BADANIE TOKSYCZNOŚCI CHRONICZNEJ/RAKOTWÓRCZOŚCI |
|
B.34. |
BADANIE TOKSYCZNOŚCI REPRODUKCJI JEDNEGO POKOLENIA |
|
B.35. |
DWUPOKOLENIOWE BADANIE TOKSYCZNOŚCI REPRODUKCYJNEJ |
|
B.36. |
TOKSYKOKINETYKA |
|
B.37. |
OPÓŹNIONA NEUROTOKSYCZNOŚĆ SUBSTANCJI FOSFOROORGANICZNYCH PO DUŻEJ EKSPOZYCJI |
|
B.38. |
OPÓŹNIONA NEUROTOKSYCZNOŚĆ SUBSTANCJI FOSFOROORGANICZNYCH – 28-DNIOWE BADANIE WIELOKROTNEJ DAWKI |
|
B.39. |
TEST POREPERACYJNEJ SYNTEZY DNA Z KOMÓRKAMI WĄTROBY SSAKÓW IN VIVO |
|
B.40. |
BADANIE NISZCZENIA SKÓRY METODĄ IN VITRO: TEST PRZEZSKÓRNEGO OPORU ELEKTRYCZNEGO (TER) |
|
B.40 bis. |
BADANIE ZNISZCZENIA SKÓRY METODĄ IN VITRO: BADANIE MODELU SKÓRY LUDZKIEJ |
|
B.41. |
BADANIE FOTOTOKSYCZNOŚCI 3T3 NRU IN VITRO |
|
B.42. |
UCZULENIE SKÓRY: TEST LOKALNYCH WĘZŁÓW CHŁONNYCH |
|
B.43. |
BADANIE NEUROTOKSYCZNOŚCI U GRYZONI |
|
B.44. |
ABSORPCJA PRZEZ SKÓRĘ: METODA IN VIVO |
|
B.45. |
ABSORPCJA PRZEZ SKÓRĘ: METODA IN VITRO |
WPROWADZENIE OGÓLNE
A. CHARAKTERYSTYKA SUBSTANCJI TESTOWANEJ
Skład substancji testowanej, w tym główne zanieczyszczenia oraz jej istotne właściwości fizyko-chemiczne włącznie z trwałością, powinny być znane przed rozpoczęciem każdego badania toksyczności.
Właściwości fizyko-chemiczne substancji testowanej dostarczają ważnych informacji przy wyborze drogi aplikowania, projektowaniu każdego poszczególnego badania oraz posługiwaniu się i przechowywaniu substancji testowanej.
Rozpoczęcie badania powinno poprzedzać zastosowanie metody analitycznej w celu jakościowego i ilościowego oznaczenia substancji testowanej (wraz z głównymi zanieczyszczeniami, jeżeli możliwe) w ośrodku dawkującym i materiale biologicznym.
Wszystkie informacje dotyczące identyfikacji, właściwości fizyko-chemicznych, czystości i zachowania substancji testowanej powinny być zawarte w sprawozdaniu z testu.
B. UTRZYMANIE ZWIERZĄT
Rygorystyczna kontrola warunków otoczenia i właściwe techniki utrzymania zwierząt są niezbędne przy testowaniu toksyczności.
(i) Warunki, w których przebywają zwierzęta
Warunki otoczenia w pomieszczeniach dla zwierząt doświadczalnych i zagrodach powinny być odpowiednie dla badanego gatunku. Odpowiednimi warunkami dla szczurów, myszy i świnek morskich są: temperatura pokojowa wynosząca 22 oC ± 3 oC i wilgotność względna 30–70 %; dla królików temperatura powinna wynosić 20 oC ± 3 oC przy wilgotności względnej 30–70 %.
Niektóre techniki eksperymentalne są szczególnie wrażliwe na wpływ temperatury i w takich przypadkach szczegóły odpowiednich warunków są zawarte w opisie metody badania. We wszystkich badaniach działania toksycznego temperatura i wilgotność powinny być monitorowane, rejestrowane i zawarte w końcowym sprawozdaniu z badania.
Oświetlenie powinno być sztuczne, w cyklu 12 godzin jasno, 12 godzin ciemno. Szczegóły form oświetlenia powinny być rejestrowane i zawarte w końcowym sprawozdaniu z badania.
Przy braku innych wskazań w danej metodzie zwierzęta mogą być umieszczane pojedynczo, lub umieszczane w klatkach w małych grupach o tej samej płci; w przypadku umieszczania grupowego, nie więcej niż pięć zwierząt powinno być umieszczanych w jednej klatce.
W sprawozdaniach z eksperymentów na zwierzętach, ważne jest wskazanie stosowanego typu umieszczania zwierząt w klatkach i liczby zwierząt umieszczanych w każdej klatce zarówno podczas ekspozycji na związek chemiczny, jak i w późniejszym okresie obserwacji.
(ii) Warunki żywienia
Diety powinny w pełni spełniać zapotrzebowanie pokarmowe gatunku poddanego testowi. W przypadkach gdy substancje testowane są podawane zwierzętom w ich pożywieniu, wartość pokarmowa może być zredukowana przez interakcję tej substancji ze składnikiem pokarmowym. Możliwość takiej reakcji powinna być rozważona przy interpretacji wyników testów. Konwencjonalne diety laboratoryjne mogą być stosowane przy nieograniczonym dostarczaniu wody pitnej. Na wybór diety może mieć wpływ potrzeba zapewnienia odpowiedniej domieszki substancji testowanej przy podawaniu powyższą metodą.
Pokarmowe substancje zanieczyszczające, o których wiadomo, że wpływają na toksyczność, nie powinny być obecne w stężeniach przeszkadzających.
C. TESTOWANIE ALTERNATYWNE
Unia Europejska jest zaangażowana w promowanie rozwijania i zatwierdzania technik alternatywnych, które mogą dostarczyć ten sam poziom informacji jak obecne testy na zwierzętach, ale które stosują mniej zwierząt, powodują mniejsze cierpienie lub całkowicie unikają wykorzystania zwierząt.
Takie metody, w przypadku gdy staną się dostępne, muszą być rozważane, gdziekolwiek jest to możliwe, przy charakteryzowaniu zagrożenia i późniejszym klasyfikowaniu i etykietowaniu wewnętrznych zagrożeń.
D. OCENA I INTERPRETACJA
Podczas oceny i interpretacji testów, muszą być rozważone ograniczenia zakresu, w którym wyniki badań na zwierzętach i in vitro mogą być odniesione bezpośrednio do człowieka i dlatego tam, gdzie dostępne są dowody niekorzystnych skutków dla ludzi, mogą one być użyte do potwierdzenia wyników testowania.
D. BIBLIOGRAFIA
Większość tych metod jest opracowana w ramach programu OECD – wskazówki dotyczące testowania, i powinny być one realizowane zgodnie z zasadami dobrej praktyki laboratoryjnej, aby zapewnić możliwie najszerszą „wzajemną akceptację danych”.
Dodatkowe informacje mogą być odnalezione w bibliografii wskazówek OECD i odpowiedniej literaturze publikowanej w innych źródłach.
B.1 bis. TOKSYCZNOŚĆ OSTRA POKARMOWA – PROCEDURA Z WYKORZYSTANIEM STAŁEJ DAWKI
1. METODA
Niniejsza metoda jest kopią OECD TG 420 (2001)
1.1. WPROWADZENIE
Tradycyjne metody oceny ostrej toksyczności wykorzystują śmierć zwierząt jako punkt końcowy. W 1984 r. Brytyjskie Towarzystwo Toksykologiczne zaproponowało nowe podejście do określania ostrej toksyczności, oparte na podawaniu serii dawek o stałym stężeniu (1). To podejście umożliwia uniknięcie śmierci zwierząt jako punktu końcowego, wykorzystując jako alternatywę obserwacje wyraźnych objawów zatrucia na danym etapie podawania serii dawek o stałym stężeniu. W następstwie badań weryfikacyjnych in vivo, przeprowadzonych przez Zjednoczone Królestwo (2) oraz na szczeblu międzynarodowym (3), procedura ta została przyjęta jako metoda badawcza w 1992 r. Jednocześnie, wykorzystując w szeregu badań modele matematyczne, poddano ocenie właściwości statystyczne Procedury stałych dawek (4)(5)(6). Zarówno badania in vivo, jak i badania z wykorzystaniem modeli wykazały, że procedura ta jest powtarzalna, wymaga mniejszej liczby zwierząt i powoduje mniej cierpień niż metody tradycyjne oraz że możliwa jest dzięki niej ocena substancji w podobny sposób, jak to ma miejsce z wykorzystaniem innych metod.
Wytyczne dotyczące wyboru najbardziej odpowiedniej metody badawczej dla indywidualnych przypadków zostały zamieszczone w Wytycznych dotyczących badania ostrej toksyczności drogą pokarmową (7). Wytyczne te zawierają także dodatkowe informacje na temat przeprowadzania i interpretacji metody badawczej B.1 bis.
Zasadniczo w badaniu głównym z wykorzystaniem niniejszej metody stosuje się jedynie umiarkowane toksycznie dawki oraz unika się dawek mogących spowodować śmierć. Nie jest również konieczne podawanie dawek mogących powodować silny ból lub stres wynikający z działania żrącego lub podrażniającego. Zwierzęta w stanie agonalnym lub w oczywisty sposób cierpiące albo chore i przeżywające strach powinny zostać uśmiercone w humanitarny sposób i uwzględniane w interpretacji wyników badań jako zwierzęta padłe w trakcie badania. Kryteria podejmowania decyzji co do uśmiercenia zwierząt w stanie agonalnym lub cierpiących ból oraz wytyczne dotyczące rozpoznania przewidywanego lub zbliżającego się zgonu są zawarte w oddzielnych Wytycznych (8).
Metoda umożliwia przedstawienie informacji na temat niebezpiecznych właściwości danej substancji i pozwala na ocenę i klasyfikację zgodną z Globalnym Zharmonizowanym Systemem (GHS) klasyfikującym związki chemiczne pod względem ich ostrej toksyczności (9).
Laboratorium badawcze powinno uwzględnić wszystkie dostępne informacje na temat badanej substancji przed rozpoczęciem badań. Informacje te powinny obejmować nazwę i budowę chemiczną substancji; właściwości fizyko-chemiczne; wyniki innych badań toksyczności in vitro lub in vivo; dane dotyczące toksyczności substancji zbliżonych pod względem budowy; przewidywane zastosowanie (zastosowania) tej substancji. Informacje te są niezbędne celem spełnienia wszelkich wymogów dotyczących ochrony zdrowia ludzkiego, i pomocne w wyborze odpowiedniej dawki wyjściowej.
1.2. DEFINICJE
Toksyczność ostra pokarmowa: oznacza niepożądane skutki pojawiające się w następstwie podania doustnego pojedynczej dawki substancji lub wielu dawek w okresie 24 godzin.
Opóźniona śmierć: oznacza, że zwierzę nie umiera, ani nie wydaje się być w agonii przez 48 godzin, lecz pada później w 14-dniowym okresie obserwacyjnym.
Dawka: oznacza ilość podawanej substancji badanej. Dawka jest wyrażana jako masa próbki na jednostkę masy zwierzęcia (np. mg/kg).
Wyraźne działanie toksyczne: jest ogólnym terminem opisującym wyraźne objawy zatrucia występujące w następstwie podania substancji badanej (zob. (3) celem uzyskania przykładów), pozwalające oczekiwać, że u większości zwierząt następna najwyższa stała dawka spowoduje bądź silny ból, przewlekłe oznaki strachu, stan agonalny (kryteria są przedstawione w Wytycznych dotyczących humanitarnego punktu końcowego (8)), bądź zgon u większości badanych zwierząt.
GHS: Globalny zharmonizowany system klasyfikacji substancji i mieszanin chemicznych. Połączone działania OECD (w dziedzinie zdrowia ludzkiego i ochrony środowiska), Komitetu Ekspertów ONZ w sprawie transportu towarów niebezpiecznych (w zakresie właściwości fizyko-chemicznych) i Międzynarodowej Organizacji Pracy (ILO) (w zakresie wymiany informacji na temat niebezpieczeństw), koordynowane w ramach Międzyorganizacyjnego programu na rzecz właściwego zarządzania związkami chemicznymi (IOMC).
Zagrażająca śmierć: występuje w przypadku gdy przewiduje się wystąpienie stanu agonalnego lub śmierci przed następnym zaplanowanym czasem obserwacji. Objawy wskazujące na ten stan mogą obejmować u gryzonia konwulsje, ułożenie na boku, pozycja leżąca oraz drgawki (zob. Wytyczne dotyczące humanitarnego punktu końcowego (9) celem uzyskania szczegółowych informacji).
LD 50 (średnia dawka śmiertelna): jest statystycznie wyprowadzoną pojedynczą dawką substancji, która powoduje zgon 50 % zwierząt przy podaniu drogą pokarmową. Wartość LD50 jest wyrażona w wadze substancji badanej na jednostkę masy badanego zwierzęcia (mg/kg).
Dawka graniczna: oznacza górną dawkę graniczną w badaniach (2 000 lub 5 000 mg/kg).
Stan agonalny: stan śmierci lub niezdolności do przeżycia nawet w przypadku zastosowania leczenia (zob. Wytyczne dotyczące humanitarnego punktu końcowego (8) w celu uzyskania danych szczegółowych).
Przewidywalna śmierć: obecność objawów klinicznych wskazujących na nadchodzący zgon przed planowanym zakończeniem eksperymentu, na przykład: niezdolność do dotarcia do wody lub pożywienia (zob. Wytyczne dotyczące humanitarnego punktu końcowego (8) w celu uzyskania danych szczegółowych).
1.3. ZASADA METODY BADAWCZEJ
Grupom zwierząt tej samej płci podaje się, zgodnie z procedurą stopniową, stałe dawki na poziomie 5, 50, 300 i 2 000 mg/kg (w wyjątkowych wypadkach można rozważyć podanie dodatkowej stałej dawki wynoszącej 5 000 mg/kg, zob. sekcja 1.6.2). Poziom pierwszej stałej dawki jest określany na podstawie badania poglądowego na poziomie, który spowoduje wystąpienie określonych objawów zatrucia, lecz nie spowoduje ciężkich zatruć lub śmierci. Objawy kliniczne oraz objawy kojarzone z bólem, cierpieniem i zbliżającą się śmiercią zostały opisane w oddzielnych Wytycznych OECD (8). Następnej grupie zwierząt może zostać podana wyższa bądź niższa stała dawka, w zależności od obecności lub braku objawów zatrucia lub śmiertelności. Taka procedura jest powtarzana aż do osiągnięcia dawki powodującej wyraźne zatrucie lub powodującej nie więcej niż jeden przypadek śmiertelny, lub w przypadku braku skutków w następstwie podania najwyższej dawki, bądź w przypadku zgonu przy najniższej dawce.
1.4. OPIS METODY BADAWCZEJ
1.4.1. Wybór gatunków zwierząt
Preferowanym gatunkiem gryzonia jest szczur, choć dopuszcza się wykorzystywanie innych gatunków gryzoni. Na ogół w doświadczeniach wykorzystuję się samice (7). Wynika to z faktu, znajdującego swoje potwierdzenie w literaturze dotyczącej konwencjonalnych badań LD50, że różnice między płciami są znikome, lecz tam gdzie występują, samice są z reguły nieznacznie bardziej wrażliwe (10). Jednakże jeżeli wiedza na temat właściwości toksykologicznych i toksykokinetycznych związków o pokrewnej budowie wskazuje, że samce mogą być bardziej wrażliwe, w takim przypadku należy wybrać do badania tę płeć. Jeżeli badanie jest przeprowadzane na samcach, należy podać uzasadnienie.
Zaleca się wykorzystanie zdrowych, młodych, dorosłych zwierząt pochodzących ze szczepów na ogół wykorzystywanych w laboratorium. Samice powinny być bezdzietne i nieciężarne. Każde zwierzę na początku cyklu laboratoryjnego powinno być w wieku między 8 a 12 tygodniem, a jego masa nie powinna przekraczać lub być mniejsza o ± 20 % średniej masy wszystkich wykorzystywanych uprzednio zwierząt.
1.4.2. Warunki przebywania i warunki żywieniowe
Temperatura w badawczych pomieszczeniach hodowlanych powinna wynosić 22o C (±3 oC). Chociaż wilgotność względna powinna wynosić co najmniej 30 % i pożądane jest, żeby nie przekraczała 70 % poza czasem czyszczenia pomieszczenia, celem powinno być utrzymywanie jej na poziomie 50–60 %. Oświetlenie powinno być sztuczne, w cyklu 12 godzin jasno, 12 godzin ciemno. Do żywienia można stosować konwencjonalne pasze laboratoryjne z nieograniczonym dostępem do wody pitnej. Zwierzęta mogą być grupowane w klatkach w sposób nieograniczający łatwej obserwacji pojedynczych sztuk.
1.4.3. Przygotowanie zwierząt
Zwierzęta są wybierane losowo, oznaczane w sposób umożliwiający identyfikację poszczególnych osobników, i przetrzymywane w swoich klatkach przynajmniej przez 5 dni przed podaniem pierwszej dawki, co umożliwia aklimatyzację do warunków laboratoryjnych.
1.4.4. Przygotowanie dawek
Na ogół badana substancja powinna być podawana w dawkach o stałej objętości przy zmianie stężenia dawkowanego preparatu. Jednakże w przypadku badania końcowego produktu będącego cieczą lub mieszaniny, dla celów oceny niebezpiecznego działania tej substancji, bardziej stosowne może okazać się wykorzystanie substancji nierozcieńczonej. tj. substancji o stałym stężeniu. Wymóg ten może być ponadto określony w niektórych przepisach wykonawczych. W żadnym razie niedopuszczalne jest przekroczenie maksymalnej objętości przewidzianej do podania. Maksymalna objętość cieczy podanej jednorazowo zależy od wielkości badanego zwierzęcia. Dla gryzoni objętość ta nie powinna przekraczać 1 ml/100 g masy ciała: jednakże w przypadku roztworów wodnych dopuszczalne jest przyjęcie dawki 2 ml/100 g masy ciała. Jeżeli chodzi o przygotowywanie dozowanego preparatu, należy użyć, o ile to możliwe, roztworów/zawiesin/emulsji wodnych, następnie rozważyć przygotowanie roztworów/zawiesin/emulsji w oleju (np. oleju kukurydzianym), a dopiero w następnej kolejności roztworów innych nośników. W przypadkach stosowania nośnika różnego od wody muszą być znane jego właściwości toksykologiczne. Dawki muszą być przygotowywane na krótko przed podaniem, chyba że stabilność przygotowanego preparatu w okresie w jakim ma on być stosowany jest znana i możliwa do przyjęcia.
1.5. PROCEDURA
1.5.1. Podawanie dawki
Substancja badana jest podawana w formie pojedynczej dawki przez zgłębnik przy użyciu sondy przełykowej lub odpowiedniej kaniuli intubacyjnej. W nietypowych przypadkach, gdy niemożliwe jest podanie pojedynczej dawki, jednorazowa dawka może zostać rozbita na mniejsze porcje i podawana w odstępach czasowych w okresie nieprzekraczającym 24 godzin.
Zwierzęta powinny być wyposzczone przed podaniem dawki (np. szczurom należy wstrzymać podawanie jedzenia przez noc, lecz nie wstrzymywać wody, myszom należy wstrzymać jedzenie przez 3–4 godziny, lecz nie wstrzymywać wody). Następnie, po wyposzczeniu, zwierzęta należy zważyć oraz podać im substancję badaną. W przypadku szczurów, po podaniu substancji badanej można wstrzymać podawanie jedzenia przez 3–4 godziny, a w przypadku myszy przez 1–2 godziny. Jeżeli dawka jest podawana porcjami przez określony czas, może być konieczne podanie pokarmu i wody w zależności od długości okresu podawania dawki.
1.5.2. Badania poglądowe
Celem badań poglądowych jest wybranie odpowiedniej dawki początkowej dla badań zasadniczych. Substancja badana jest podawana poszczególnym zwierzętom w sposób sekwencyjny, zgodnie ze schematem postępowania zawartym w załączniku 1. Badania poglądowe są przerywane, gdy możliwe jest podjęcie decyzji co do ilości dawki wyjściowej (lub następuje śmierć w wyniku podania najmniejszej stałej dawki).
Dawka początkowa wyznaczona do badania poglądowego danej substancji jest wybierana w oparciu o dane eksperymentalne z badań in vivo i in vitro tej samej substancji lub substancji pokrewnych pod względem budowy, spośród stałych dawek określonych na poziomie 5, 50, 300, i 2 000 mg/kg, jako dawka, która powinna wywołać wyraźną toksyczność. W przypadku braku danych na temat takich substancji należy ustalić dawkę wyjściową na poziomie 300 mg/kg.
Dopuszczalna jest nie krótsza niż 24-godzinna przerwa w podawaniu dawki jednemu zwierzęciu. Wszystkie zwierzęta należy objąć obserwacją przez przynajmniej 14 dni.
W wyjątkowych okolicznościach i wyłącznie jeżeli wymagają tego określone potrzeby regulacyjne, można rozważyć podanie dawki dodatkowej stałej na górnym poziomie 5 000 mg/kg (zob. załącznik 3). Z uwagi na dobrostan zwierząt badania na zwierzętach w zakresie kategorii 5 GSH (2 000 i 5 000 mg/kg) są odradzane i powinny być rozważane wyłącznie w przypadku znacznego prawdopodobieństwa, że wyniki takich badań przyniosą bezpośrednie korzyści dla ochrony zdrowia ludzkiego, zwierząt lub środowiska naturalnego.
W przypadku gdy zwierzę, któremu w badaniach poglądowych została podana najniższa stała dawka (5 mg/kg), ginie, normalną procedurą jest przerwanie badań i przypisanie substancji do kategorii 1 GSH (jak to przedstawiono w załączniku 1). Jednakże w przypadku gdy wymagane jest potwierdzenie klasyfikacji wstępnej, możliwe jest przeprowadzenie procedury dodatkowej, zgodnie z poniższym opisem. Drugie zwierzę otrzymuje dawkę 5 mg/kg. Jeżeli również ono pada, przypisanie do kategorii 1 GHS zostaje potwierdzone, a badanie zostaje natychmiastowo przerwane. Jeżeli drugie zwierzę przeżyje, dawkę 5 mg/kg podaje się maksymalnie trzem kolejnym zwierzętom. Z uwagi na wysokie ryzyko śmiertelności dawki powinny być podawane sekwencyjnie, co zapewni optymalną ochronę zwierząt. Przerwy w podawaniu dawek każdemu zwierzęciu powinny pozwalać na uzyskanie pewności, że poprzednie zwierzę przeżyje. W momencie wystąpienia drugiego przypadku śmiertelnego sekwencyjne podawanie dawek zostaje natychmiast przerwane, a podawanie dawek kolejnym zwierzętom wstrzymane. Ponieważ wystąpienie drugiego przypadku śmiertelnego (niezależnie od liczby zwierząt poddanych badaniu do momentu ich przerwania) mieści się w wyniku A (2 lub więcej przypadków śmiertelnych), stosowana jest zasada klasyfikacji określona w załączniku 2, zgodnie z którą stężenie stałej dawki wynosi 5 mg/kg. Dodatkowo, do momentu przyjęcia nowego GHS, obowiązujące są wytyczne zawarte w załączniku 4, dotyczące klasyfikacji substancji w ramach systemu WE.
1.5.3. Badania zasadnicze
1.5.3.1. Liczba zwierząt i poziomy dawek
Działania, jakie mają być podjęte w następstwie przeprowadzenia badań przy pomocy dawek wyjściowych, są przedstawione na schemacie postępowania zawartym w załączniku 2. Wymagany jest jeden z trzech schematów postępowania; bądź przerwanie testów i przypisanie do jednej z klas klasyfikacji niebezpieczeństwa, zbadanie zwierzęcia przy pomocy wyższej stałej dawki, bądź jego zbadanie przy pomocy niższej stałej dawki. Jednakże w celu ochrony zwierząt poziom, który spowodował śmierć zwierzęcia w trakcie badań poglądowych, nie będzie powtórnie wykorzystany w badaniach zasadniczych (zob. załącznik 2). Doświadczenie wykazuje, że najbardziej prawdopodobny wynik na poziomie wyjściowym stałej dawki umożliwi klasyfikację substancji, skutkiem czego nie będą konieczne dalsze badania.
Normalnie wykorzystuje się ogólną liczbę pięciu zwierząt jednej płci dla każdego badanego poziomu dawki. Liczba ta obejmuje jedno zwierzę zbadane w trakcie badań poglądowych przy pomocy wybranej dawki oraz cztery inne zwierzęta (z wyjątkiem sytuacji, w których poziom dawki wykorzystanej w badaniach zasadniczych nie był wcześniej zastosowany w badaniach poglądowych).
Odstępy między podawaniem dawek na każdym poziomie określa się na podstawie czasu wystąpienia, długości trwania i dokuczliwości objawów zatrucia. Podanie zwierzęciu następnej dawki powinno zostać opóźnione do momentu uzyskania pewności co do możliwości przeżycia zwierząt, którym podano ostatnią dawkę. Zalecana jest przerwa rzędu 3 lub 4 dni między podaniem kolejnych dawek, o ile wystąpi taka potrzeba, w celu umożliwienia obserwacji opóźnionego zatrucia. Odstęp ten może zostać przedłużony np. w przypadku niejednoznacznej reakcji.
W przypadku stosowania górnej dawki granicznej na poziomie 5 000 mg/kg należy stosować procedurę przedstawioną w załączniku 3 (zob. także sekcja 1.6.2).
1.5.3.2. Badanie graniczne
Badanie graniczne jest stosowane w sytuacji gdy badający posiada informacje sugerujące, że badany materiał może być nietoksyczny np. posiada toksyczność na poziomie przekraczającym nieznacznie dawki graniczne określone przepisami wykonawczymi. Informacje na temat toksyczności można uzyskać na podstawie wiedzy na temat podobnych zbadanych związków lub podobnych badanych mieszanin lub produktów, z uwzględnieniem tożsamości i zawartości procentowej składników, o których wiadomo, że mają znaczenie z punktu widzenia toksyczności. W sytuacji niewystarczającej ilości lub braku informacji na temat toksyczności materiału badanego lub w przypadku gdy oczekuje się, że badany materiał może być toksyczny, należy przeprowadzić badanie zasadnicze.
W przypadku zastosowania normalnej procedury badanie graniczne dla potrzeb niniejszych wytycznych polega na podaniu dawki wyjściowej badania poglądowego, a następnie podaniu substancji czterem pozostałym zwierzętom.
1.6. OBSERWACJE
Zwierzęta są poddawane obserwacji osobniczej przynajmniej raz w ciągu pierwszych 30 minut po podaniu dawki, okresowo przez pierwsze 24 godziny, ze szczególnym uwzględnieniem pierwszych 4 godzin, a następnie codziennie, ogólnie przez 14 dni, z wyjątkiem sytuacji, w których muszą one zostać wyeliminowane z badań i uśmiercone w sposób humanitarny z uwagi na ich dobrostan, lub gdy padły. Jednakże czas trwania obserwacji nie powinien być ustalany według ścisłych reguł. Powinien zależeć od działania toksycznego, czasu wystąpienia skutków i długości okresu powrotu do stanu normalnego, i może skutkiem tego być wydłużony, w przypadku gdy zostanie to uznane za stosowne. Czas, w jakim pojawiają się i zanikają objawy zatrucia, jest istotny, szczególnie w przypadku występowania opóźnionych objawów zatrucia (11). Wszystkie spostrzeżenia są systematycznie zapisywane w ewidencji prowadzonej osobno dla każdego osobnika.
Dodatkowe obserwacje są wymagane, jeżeli zwierzęta wciąż wykazują objawy zatrucia. Obserwacje powinny dotyczyć zmian na skórze i futrze, oczach i błonach śluzowych, a także w układach oddechowym, krążenia, autonomicznym i centralnym układzie nerwowym oraz zmian w funkcjonowaniu somatomotorycznym i schematach zachowań. Szczególną uwagę należy poświęcić obserwacjom drżenia, konwulsji, ślinienia się, biegunki, letargu, snu i śpiączki. Należy przestrzegać zasad i kryteriów streszczonych w Wytycznych dotyczących humanitarnego punktu końcowego (8). Zwierzęta, u których stwierdzono stan agonalny, oraz zwierzęta wykazujące objawy silnego bólu, powinny zostać uśmiercone w sposób humanitarny. W przypadku uśmiercenia zwierząt z przyczyn humanitarnych lub ich zgonu, czas zgonu powinien zostać zapisany najdokładniej jak to możliwe.
1.6.1. Masa ciała
Masa każdego zwierzęcia powinna zostać określona na krótko przed podaniem substancji badanej i przynajmniej w odstępie tygodnia od jej podania. Zmiany masy powinny zostać obliczone i zapisane. Po zakończeniu badań zwierzęta, które przeżyły, są ważone i uśmiercane w sposób humanitarny.
1.6.2. Patologia
Wszystkie badane zwierzęta (włączając zwierzęta, które padły podczas badań lub zostały wyeliminowane z badań ze względu na ich dobrostan) powinny zostać poddane całkowitej sekcji zwłok. Wszystkie całkowite zmiany patologiczne powinny zostać zapisane oddzielnie dla każdego zwierzęcia. Badanie mikroskopowe organów wykazujących wyraźne zmiany patologiczne u zwierząt, które przeżyły 24 lub więcej godzin po podaniu pierwszej dawki, mogą również zostać rozważone, ponieważ mogą one dostarczyć użytecznych informacji.
2. DANE
Należy podać dane dla każdego zwierzęcia. Dodatkowo, wszystkie dane powinny zostać streszczone w tabeli, pokazującej dla każdej badanej grupy liczbę wykorzystanych sztuk, liczbę zwierząt wykazujących objawy zatrucia, liczbę zwierząt padłych w trakcie badań lub uśmierconych z przyczyn humanitarnych, czas zgonu poszczególnych zwierząt, opis oraz przebieg w czasie skutków zatrucia oraz powrotu do stanu normalnego, a także wyniki sekcji zwłok.
3. SPRAWOZDANIE
3.1. SPRAWOZDANIE Z BADAŃ
Sprawozdanie z badań musi zawierać następujące informacje, jeśli są one stosowne:
|
|
Substancja badana:
|
|
|
Nośnik (jeśli był użyty):
|
|
|
Zwierzęta badane:
|
|
|
Warunki badań:
|
|
|
Wyniki:
|
|
|
Omówienie i interpretacja wyników. |
|
|
Wnioski. |
4. BIBLIOGRAFIA
|
(1) |
British Toxicology Society Working Party on Toxicity (1984). Special report: a new approach to the classification of substances and preparation on the basis of their acute toxicity. Human Toxicol., 3, 85-92. |
|
(2) |
Van den Heuvel, M.J., Dayan, A.D. and Shillaker, R.O. (1987). Evaluation of the BTS approach to the testing of substances and preparation for their acute toxicity. Human Toxicol., 6, 279–291. |
|
(3) |
Van den Heuvel, M.J., Clark, D.G., Fielder, R.J., Koundakjian, P.P., Oliver, G.J.A., Pelling, D., Tomlinson, N.J. and Walker, A.P. (1990). The international validation of a fixed-dose procedure as an alternative to the classical LD50 test. Fd. Chem. Toxicol. 28, 469–482. |
|
(4) |
Whitehead, A. and Curnow, R.N. (1992). Statistical evaluation of the fixed-dose procedure. Fd. Chem.Toxicol., 30, 313–324. |
|
(5) |
Stallard, N. and Whitehead, A. (1995). Reducing numbers in the fixed-dose procedure. Human Exptl.Toxicol. 14, 315–323. Human Exptl. Toxicol. |
|
(6) |
Stallard, N., Whitehead, A. and Ridgeway, P. (2002). Statistical evaluation of the revised fixed doseprocedure. Hum. Exp. Toxicol., 21, 183–196. |
|
(7) |
OECD (2001). Guidance Document on Acute Oral Toxicity Testing. Environmental Health and Safety Monograph Series on Testing and Assessment N. 24. Paris. |
|
(8) |
OECD (2000). Guidance Document on the Recognition, Assessment and Use of Clinical Signs as Humane Endpoints for Experimental Animals Used in Safety Evaluation. Environmental Health and Safety Monograph Series on Testing and Assesment N. 19. |
|
(9) |
OECD (1998). Harmonised Integrated Hazard Classification for Human Health and Environmental Effects of Chemical Substances as endorsed by the 28th Joint Meeting of the Chemicals Committee and the Working Party on Chemicals in November 1998, Part 2, p.11 [http://webnet1.oecd.org/oecd/pages/home/displaygeneral/0,3380,EN-documents-521-14-no-24-no-0,FF.html]. |
|
(10) |
Lipnick, R.L., Cotruvo, J.A., Hill, R.N., Bruce, R.D., Stitzel, K.A., Walker, A.P., Chu, I., Goddard, M., Segal, L., Springer, J.A. and Myers, R.C. (1995). Comparison of the Up-and-Down, Conventional LD50, and Fixed-Dose Acute Toxicity Procedures. Fd. Chem. Toxicol. 33, 223–231. |
|
(11) |
Chan P.K and A.W. Hayes (1994) Chapter 16 Acute Toxicity and Eye Irritation. In: Principles and Methods of Toxicology. 3 rd Edition. A.W. Hayes, Editor. Raven Press, Ltd. New York, USA. |
ZAŁĄCZNIK 1
SCHEMAT DZIAŁAŃ DLA BADAŃ POGLĄDOWYCH


ZAŁĄCZNIK 2
SCHEMAT DZIAŁAŃ DLA BADAŃ ZASADNICZYCH
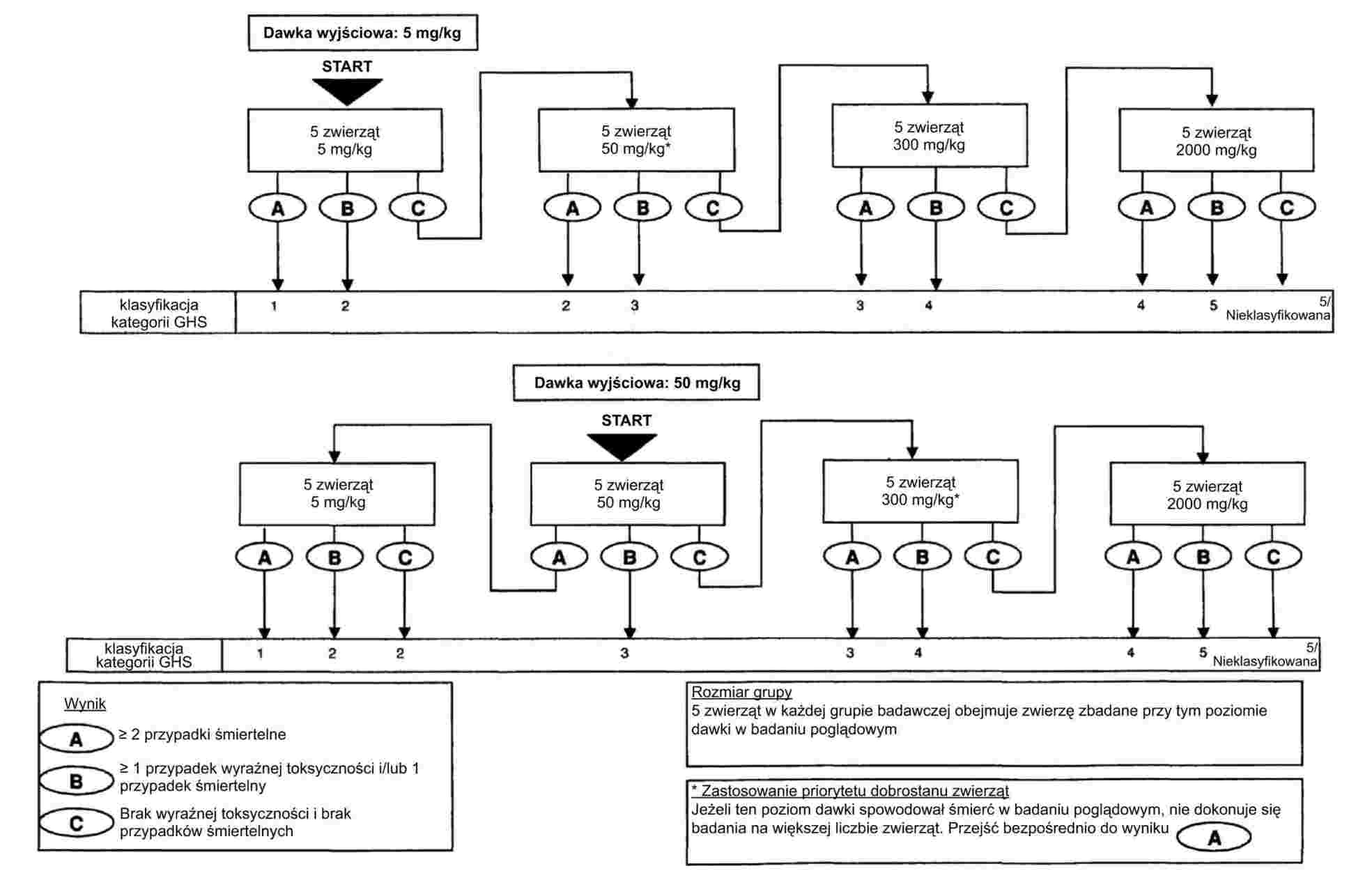
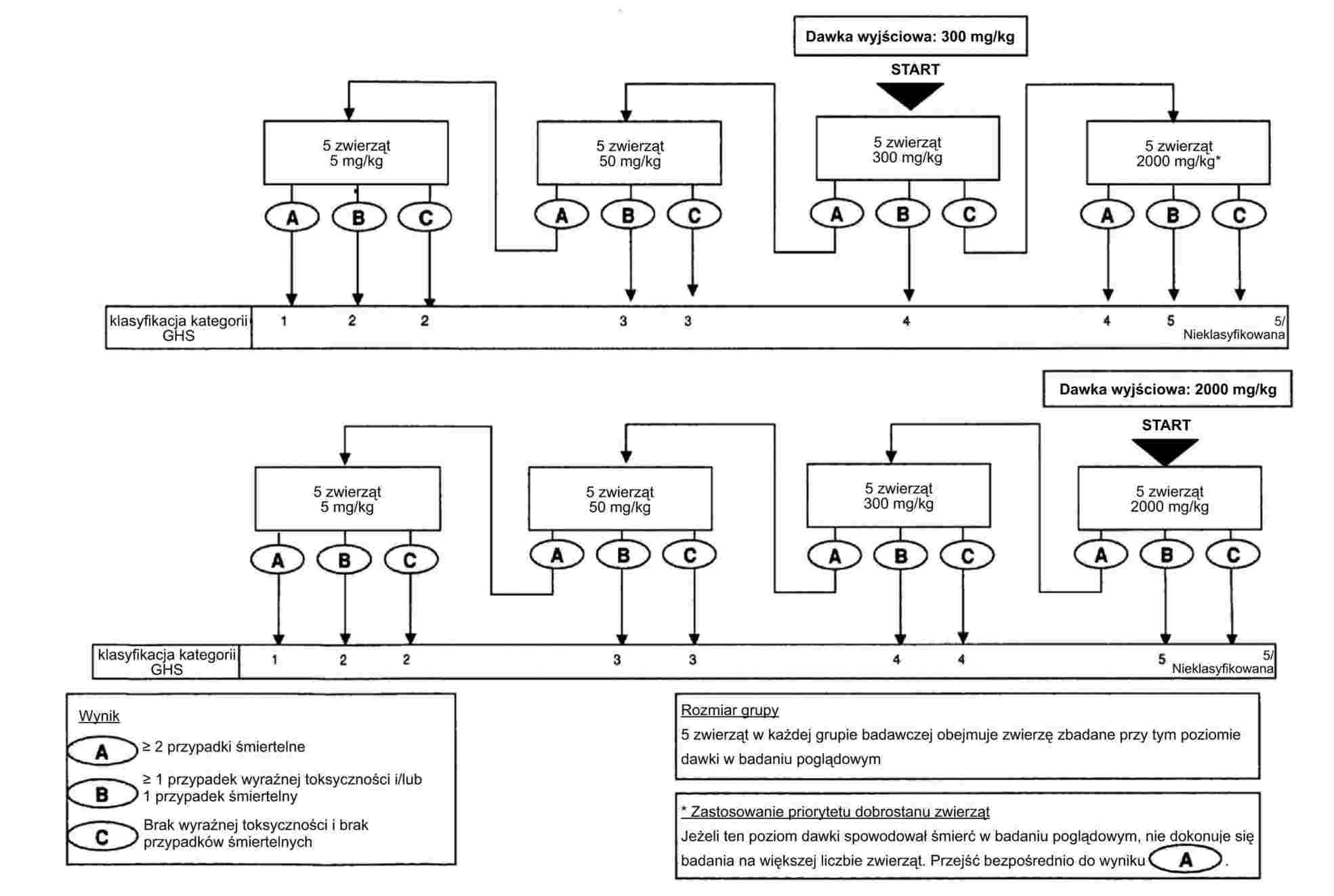
ZAŁĄCZNIK 3
KRYTERIA KLASYFIKACJI SUBSTANCJI BADANYCH O OCZEKIWANYCH WARTOŚCIACH LD 50 PRZEKRACZAJĄCYCH 2 000 MG/KG, Z POMINIĘCIEM BADAŃ
Kryteria kategorii niebezpieczeństwa 5 mają w zamierzeniu umożliwiać identyfikację substancji badanych, które charakteryzują się niskim niebezpieczeństwem ostrej toksyczności, lecz które w określonych okolicznościach mogą okazać się niebezpieczne dla populacji podatnych na zatrucie. Przewiduje się, że takie substancje charakteryzują się pokarmowym lub skórnym LD50 mieszczącym się w zakresie 2 000–5 000 mg/kg lub odpowiadają analogicznym dawkom w przypadku innych szlaków kontaktu z substancją. Substancja może zostać zaklasyfikowana do kategorii niebezpieczeństwa przy następujących cechach: 2 000 mg/kg < LD50 < 5 000 mg/kg (kategoria 5 według GHS) w następujących przypadkach:
|
a) |
jeżeli została przypisana do tej kategorii w wyniku zastosowania jednego ze schematów przedstawionych w załączniku 2, w oparciu o poziom śmiertelności; |
|
b) |
jeżeli dostępne są wiarygodne dowody wskazujące, że LD50 powinno znaleźć się w obrębie kategorii 5; bądź jeżeli inne badania na zwierzętach lub ostre skutki u ludzi sugerują niebezpieczeństwo dla zdrowia ludzkiego spowodowane ostrym działaniem; |
|
c) |
jeżeli okazuje się, w wyniku przeprowadzenia ekstrapolacji, szacunków lub pomiaru danych, że przypisanie do wyższej klasy zagrożenia nie jest uzasadnione oraz:
|
BADANIA PRZY DAWKACH PRZEKRACZAJĄCYCH 2 000 MG/KG
W wyjątkowych okolicznościach i wyłącznie jeżeli wymagają tego określone potrzeby regulacyjne, można rozważyć podanie dawki dodatkowej stałej na górnym poziomie 5 000 mg/kg. Z uwagi na dobrostan zwierząt badania przy 5 000 mg/kg są odradzane i powinny być brane pod uwagę wyłącznie w przypadku znacznego prawdopodobieństwa, że wyniki takich badań przyniosą bezpośrednie korzyści dla ochrony zdrowia ludzkiego, zwierząt lub środowiska naturalnego (9).
Badanie poglądowe
Zakres przepisów niniejszej decyzji dotyczących procedury sekwencyjnej określonej w załączniku 1 zostaje rozszerzony o poziom dawki wynoszący 5 000 mg/kg. W związku z tym, jeżeli w przypadku zastosowania w badaniu poglądowym dawki początkowej wynoszącej 5 000 mg/kg wystąpi wynik A (zgon), należy zbadać następne zwierzę z wykorzystaniem dawki 2 000 mg/kg; pojawienie się wyników B i C (wyraźne zatrucie lub brak zatrucia) pozwoli na wybranie dawki 5 000 mg/kg jako dawki wyjściowej ma być: jako dawki wyjściowej badania głównego. Podobnie, jeżeli zostanie użyta inna dawka niż 5 000 mg/kg, badanie będzie zmierzało do zastosowania dawki 5 000 mg/kg w przypadku wystąpienia wyników B lub C przy 2 000 mg/kg; pojawienie się następnie wyniku A przy dawce 5 000 mg/kg wyznaczy dawkę wyjściową w badaniu zasadniczym przy 2 000 mg/kg, natomiast pojawienie się wyników B lub C wyznaczy dawkę wyjściową w badaniu głównym na 5 000 mg/kg.
Badanie główne
Zakres przepisów niniejszej decyzji dotyczących procedury sekwencyjnej określonej w załączniku 2 zostaje rozszerzony o poziom dawki wynoszący 5 000 mg/kg. W związku z tym, w przypadku gdy dawka wyjściowa badania zasadniczego wynosi 5 000 mg/kg, wynik A (≥ 2 przypadki śmiertelne) będzie wymagał zbadania drugiej grupy przy 2 000 mg/kg; wynik B (wyraźna toksyczność i/lub ≤ 1) lub wynik C (brak toksyczności) spowoduje niesklasyfikowanie substancji w ramach GHS. Podobnie, w przypadku zastosowania innej dawki początkowej niż 5 000 mg/kg, badanie będzie zmierzało do 5 000 mg/kg przy wyniku C przy 2 000 mg/kg; pojawienie się następnie wyniku A przy 5 000 mg/kg spowoduje przypisanie substancji do kategorii 5 GHS, natomiast pojawienie się wyników B lub C spowoduje niesklasyfikowanie substancji.
ZAŁĄCZNIK 4
METODA BADAWCZA B.1 bis
Wytyczne dotyczące klasyfikacji zgodnie z systemem WE obowiązującej w okresie przejściowym do momentu pełnego wdrożenia Globalnego Zharmonizowanego Systemu Klasyfikacji (GHS) (wytyczne pochodzą z pozycji bibliograficznej (8))


B.1 ter. TOKSYCZNOŚĆ OSTRA POKARMOWA – METODA KLAS OSTREJ TOKSYCZNOŚCI
1. METODA
Niniejsza metoda badawcza jest kopią OECD TG 423 (2001).
1.1. WPROWADZENIE
Metoda klas ostrej toksyczności (1) przyjęta w tym badaniu jest procedurą stopniową, w której dla każdego stopnia wykorzystywane są trzy osobniki zwierząt jednej płci. Zależnie od śmiertelności oraz/lub stanu agonalnego zwierząt do stwierdzenia ostrej toksyczności badanej substancji konieczne mogą być średnio 2–4 etapy. Niniejsza procedura jest powtarzalna, w jej toku wykorzystuje się niewiele zwierząt i daje ona możliwość klasyfikacji substancji w sposób podobny do innych metod badania ostrej toksyczności. Metoda klas ostrej toksyczności opiera się na ocenach biometrycznych (2)(3)(4)(5) przeprowadzonych z wykorzystaniem stałych dawek, oddzielonych w odpowiedni sposób umożliwiający ocenę substancji dla celów klasyfikacji i oceny niebezpieczeństwa. Metoda ta została przyjęta w 1996 r. i była poddawana intensywnej weryfikacji in vivo w oparciu o dane o LD50 uzyskane z literatury, zarówno na szczeblu narodowym (6), jak i międzynarodowym (7).
Wytyczne dotyczące wyboru najbardziej odpowiedniej metody badawczej dla danego przypadku zostały zamieszczone w Wytycznych dotyczących badania toksyczności ostrej pokarmowej (8). Wytyczne te zawierają ponadto dodatkowe informacje na temat przeprowadzania i interpretacji Metody badawczej B.1ter.
Nie jest również konieczne podawanie dawek mogących powodować silny ból lub strach wynikający z działania żrącego lub podrażniającego. Zwierzęta w stanie agonalnym lub w oczywisty sposób cierpiące albo chore i doświadczające strachu powinny zostać uśmiercone w humanitarny sposób i uwzględniane w interpretacji wyników badań jako zwierzęta padłe w trakcie badania. Kryteria podejmowania decyzji co do uśmiercenia zwierząt w stanie agonalnym lub cierpiących ból oraz wytyczne dotyczące rozpoznawania przewidywalnego lub zbliżającego się zgonu, są zawarte w oddzielnych Wytycznych (9).
Metoda opiera się na wcześniej określonych dawkach, a wyniki uzyskane w wyniku jej zastosowania pozwalają na ocenę i klasyfikację zgodną z Globalnym Zharmonizowanym Systemem klasyfikacji związków chemicznych pod względem ich ostrej toksyczności (10).
W zasadzie niniejsza metoda nie pozwala na dokładne obliczenie LD50, lecz pozwala na określenie zakresu ekspozycji, tam gdzie oczekiwana jest śmiertelność, ponieważ śmierć określonego odsetka zwierząt nadal stanowi jeden z głównych punktów końcowych badania. Metoda umożliwia określenie wartości LD50 wyłącznie w przypadku, gdy przynajmniej dwie dawki spowodowały śmiertelność wyższą niż 0 %, lecz mniejszą niż 100 %. Wykorzystanie selekcji wcześniej określonych dawek, niezależnie od substancji badanej, której klasyfikacja jest wyraźnie powiązana z liczbą zwierząt obserwowanych na poszczególnych etapach, poprawia możliwości laboratoriom w zakresie konsekwencji i powtarzalności sprawozdań laboratoryjnych.
Laboratorium badawcze powinno uwzględniać wszystkie dostępne informacje na temat badanej substancji przed rozpoczęciem badań. Informacje te powinny obejmować tożsamość i budowę chemiczną substancji; właściwości fizyko-chemiczne; wyniki innych badań toksyczności in vitro lub in vivo substancji; dane dotyczące toksyczności substancji pokrewnych pod względem budowy; przewidywane zastosowanie (zastosowania) tej substancji. Informacje te są niezbędne celem spełnienia wszelkich wymogów dotyczących ochrony zdrowia ludzkiego i umożliwienia wyboru najbardziej odpowiedniej dawki wyjściowej.
1.2. DEFINICJE
Toksyczność ostra pokarmowa: oznacza niepożądane skutki pojawiające się w następstwie podania doustnego pojedynczej dawki substancji lub wielu dawek w okresie 24 godzin.
Opóźniona śmierć: oznacza, że zwierzę nie umiera, ani nie wydaje się być w agonii przez 48 godzin, lecz pada później w 14-dniowym okresie obserwacyjnym.
Dawka: oznacza ilość podanej substancji badanej. Dawka jest wyrażana w wadze próbki na jednostkę masy zwierzęcia (np. mg/kg).
GHS: Globalny Zharmonizowany System Klasyfikacji Substancji i Mieszanin Chemicznych. Połączone działania OECD (w dziedzinie zdrowia ludzkiego i ochrony środowiska), Komitetu Ekspertów ONZ w sprawie transportu towarów niebezpiecznych (w zakresie właściwości fizyko-chemicznych) i Międzynarodowej Organizacji Pracy (ILO) (w zakresie wymiany informacji na temat niebezpieczeństw), koordynowane w ramach Międzyorganizacyjnego programu na rzecz właściwego zarządzania związkami chemicznymi (IOMC).
Zagrażająca śmierć: występuje w przypadku, gdy przewiduje się wystąpienie stanu agonalnego lub śmierć przed następnym zaplanowanym punktem czasowym obserwacji. Objawy wskazujące na ten stan mogą obejmować u gryzonia konwulsje, ułożenie na boku, pozycję leżącą oraz drgawki (zob. Wytyczne dotyczące humanitarnego punktu końcowego (9) celem uzyskania szczegółowych informacji).
LD 50 (średnia dawka śmiertelna): jest statystycznie wyprowadzoną pojedynczą dawką substancji, która powoduje zgon 50 procent zwierząt, przy podaniu drogą pokarmową. Wartość LD50 jest wyrażona w wadze substancji badanej na jednostkę masy badanego zwierzęcia (mg/kg).
Dawka graniczna: oznacza górną dawkę graniczną w badaniach (2 000 lub 5 000 mg/kg).
Stan agonalny: stan śmierci lub niezdolności do przeżycia nawet w przypadku zastosowania leczenia (zob. Wytyczne metodyczne dotyczące humanitarnego punktu końcowego (9) w celu uzyskania danych szczegółowych).
Przewidywana śmierć: obecność objawów klinicznych wskazujących na nadchodzący zgon w przewidzianym czasie w przyszłości przed planowanym zakończeniem eksperymentu, na przykład: niezdolność do dotarcia do wody lub pożywienia (zob. Wytyczne metodyczne dotyczące humanitarnego punktu końcowego (9) w celu uzyskania danych szczegółowych).
1.3. ZASADA METODY BADANIA
Zasadą badania jest to, że w oparciu o procedurę etapową z użyciem minimalnej liczby zwierząt na każdym etapie uzyskiwane są wystarczające informacje na temat ostrej toksyczności substancji badanej w celu jej zaklasyfikowania. Substancja jest podawana doustnie grupie zwierząt laboratoryjnych w jednej z określonych dawek. Substancja jest badana przy użyciu procedury etapowej, na każdym etapie wykorzystywane są trzy zwierzęta jednej płci (na ogół samice). Brak lub obecność śmiertelności związanej z podaniem odczynnika zwierzętom determinuje następny etap, tj.:
|
— |
dalsze badania są zbyteczne, |
|
— |
dawka zostaje podana trzem następnym zwierzętom, |
|
— |
dawka na następnym niższym bądź wyższym poziomie zostaje podana trzem następnym zwierzętom. |
Szczegóły procedury są opisane w załączniku 1. Metoda pozwala na ocenę w odniesieniu do przypisania substancji badanej do jednej z szeregu klas toksyczności określonych poprzez ustalone wartości odcinania D50.
1.4. OPIS METODY BADAWCZEJ
1.4.1. Wybór gatunków zwierząt
Preferowanym gatunkiem gryzonia jest szczur, choć dopuszcza się wykorzystywanie innych gatunków gryzoni. Na ogół w doświadczeniach wykorzystuję się samice (9). Wynika to z faktu, znajdującego swoje potwierdzenie w literaturze na temat konwencjonalnych badań LD50, że różnice między płciami są znikome, lecz tam gdzie występują, samice są z reguły nieznacznie bardziej wrażliwe (11). Jednakże jeżeli wiedza na temat właściwości toksykologicznych i toksokinetycznych związków o podobnej budowie wskazuje na to, że samce mogą być bardziej wrażliwe, w takim przypadku należy wybrać do badania tę płeć. Jeżeli badanie jest przeprowadzane na samcach, należy podać uzasadnienie.
Zaleca się wykorzystanie zdrowych, młodych, dorosłych zwierząt pochodzących ze szczepów na ogół wykorzystywanych w laboratorium. Samice powinny być bezdzietne i nieciężarne. Każde zwierzę na początku cyklu laboratoryjnego powinno być w wieku między 8 a 12 tygodniem, a jego masa nie powinna przekraczać lub być mniejsza o ± 20 % średniej masy wszystkich wykorzystywanych uprzednio zwierząt.
1.4.2. Warunki przebywania i warunki żywieniowe
Temperatura w badawczych pomieszczeniach hodowlanych powinna wynosić 20 oC (± 3 oC). Chociaż wilgotność względna powinna wynosić co najmniej 30 % i pożądane jest, żeby nie przekraczała 70 % poza czasem czyszczenia pomieszczenia, celem powinno być utrzymywanie jej na poziomie 50–60 %. Oświetlenie powinno być sztuczne, w cyklu 12 godzin jasno, 12 godzin ciemno. Do żywienia można stosować konwencjonalne pasze laboratoryjne z nieograniczonym dostępem do wody pitnej. Zwierzęta mogą być grupowane w klatkach w sposób nieograniczający łatwej obserwacji pojedynczych sztuk.
1.4.3. Przygotowanie zwierząt
Zwierzęta są wybierane losowo, oznaczane w sposób umożliwiający identyfikację pojedynczych osobników i przetrzymywane w swoich klatkach przynajmniej przez 5 dni przed podaniem pierwszej dawki, co umożliwia aklimatyzację do warunków laboratoryjnych.
1.4.4. Przygotowanie dawek
Na ogół badana substancja powinna być podawana w dawkach o stałej objętości przy zmianie stężenia dawkowanego preparatu. Jednakże w przypadku badania końcowego produktu będącego cieczą lub mieszaniną dla celów oceny ryzyka tej substancji bardziej stosowne może okazać się wykorzystanie w badaniach substancji nierozcieńczonej. tj. substancji o stałym stężeniu. Wymóg ten może być ponadto określony w niektórych przepisach wykonawczych. W żadnym razie niedopuszczalne jest przekroczenie maksymalnej objętości przewidzianej do podania. Maksymalna objętość cieczy podanej jednorazowo zależy od wielkości badanego zwierzęcia. Dla gryzoni objętość ta nie powinna przekraczać 1 ml/100g masy ciała: jednakże w przypadku roztworów wodnych dopuszczalne jest przyjęcie dawki 2 ml/100 g masy ciała. Jeżeli chodzi o przygotowywanie dozowanego preparatu należy użyć, o ile to możliwe, roztworów/zawiesin/emulsji wodnych, następnie rozważyć przygotowanie roztworów/zawiesin/emulsji w oleju (np. oleju kukurydzianym), a dopiero w następnej kolejności roztworów innych nośników. W przypadkach stosowania nośnika różnego od wody muszą być znane jego właściwości toksykologiczne. Dawki muszą być przygotowywane na krótko przed podaniem, chyba że stabilność przygotowanego preparatu w okresie w jakim ma on być stosowany jest znana i możliwa do przyjęcia.
1.5. PROCEDURA
1.5.1. Podawanie dawki
Substancja badana jest podawana w formie pojedynczej dawki przez zgłębnik przy użyciu sondy przełykowej lub odpowiedniej kaniuli intubacyjnej. W nietypowych przypadkach, gdy niemożliwe jest podanie pojedynczej dawki, jednorazowa dawka może zostać rozbita na mniejsze porcje i podawana w odstępach czasowych w okresie nieprzekraczającym 24 godzin.
Zwierzęta powinny być wyposzczone przed podaniem dawki (np. szczurom należy wstrzymać podawanie jedzenia przez noc, lecz nie wstrzymywać podawania wody, myszom należy wstrzymać podawanie jedzenia przez 3–4 godziny, lecz nie wstrzymywać wody). Następnie, po wyposzczeniu, zwierzęta należy zważyć oraz podać im substancję badaną. W przypadku szczurów, po podaniu substancji badanej można wstrzymać podawanie jedzenia przez 3–4 godziny, a w przypadku myszy przez 1–2 godziny. Jeżeli dawka jest podawana porcjami przez określony czas, może być konieczne podanie pokarmu i wody w zależności od długości okresu podawania dawki.
1.5.2. Liczba zwierząt i poziomy dawki
Na każdym etapie wykorzystywane są trzy zwierzęta. Poziom dawki, która ma być wykorzystana jako dawka wyjściowa, jest wybierana z jednego z czterech stałych poziomów 5, 50, 300 i 2 000 mg/kg masy ciała. Poziom wyjściowy dawki powinien być taki, żeby powodował śmiertelność u niektórych zwierząt, którym ją zaaplikowano. Schemat postępowania zamieszczony w załączniku 1 opisuje procedurę, którą należy podjąć dla każdej dawki wyjściowej. Dodatkowo, do momentu przyjęcia nowego GHS, obowiązujące są wytyczne zawarte w załączniku 4, dotyczące klasyfikacji substancji w ramach systemu WE.
Jeżeli dostępne informacje sugerują brak prawdopodobieństwa śmiertelności przy najwyższym poziomie dawki (2 000 mg/kg masy ciała), powinno zostać przeprowadzone badanie graniczne. W przypadku braku informacji na temat danej substancji, która ma być badana, z uwagi na dobrostan zwierząt zalecane jest zastosowanie dawki wyjściowej 300 mg/kg masy ciała.
Odstępy między podawaniem dawek na każdym poziomie określa się na podstawie czasu wystąpienia, długości trwania i dokuczliwości objawów zatrucia. Podanie zwierzęciu następnej dawki powinno zostać opóźnione do momentu uzyskania pewności co do możliwości przeżycia zwierząt, którym podano ostatnią dawkę.
W wyjątkowych okolicznościach i wyłącznie jeżeli wymagają tego określone potrzeby regulacyjne, można rozważyć podanie dawki dodatkowej ustalonej na górnym poziomie 5 000 mg/kg (zob. załącznik 3). Z uwagi na dobrostan zwierząt badania na zwierzętach w zakresie kategorii 5 GSH (2 000 i 5 000 mg/kg) są odradzane i powinny być rozważane wyłącznie w przypadku znacznego prawdopodobieństwa, że wyniki takich badań przyniosą bezpośrednie korzyści dla ochrony zdrowia ludzkiego, zwierząt lub środowiska naturalnego.
1.5.3. Badanie graniczne
Badanie graniczne jest stosowane w sytuacji gdy badający posiada informacje sugerujące, że badany materiał może być nietoksyczny np. posiada toksyczność na poziomie przekraczającym nieznacznie dawki graniczne określone przepisami wykonawczymi. Informacje na temat toksyczności mogą zostać uzyskane na podstawie wiedzy na temat podobnych zbadanych związków lub podobnych zbadanych mieszanin lub produktów, z uwzględnieniem tożsamości i zawartości procentowej składników, o których wiadomo, że mają znaczenia z punktu widzenia toksyczności. W sytuacji niewystarczającej ilości lub braku informacji na temat toksyczności materiału testowego lub w przypadku gdy, zgodnie z oczekiwaniami, badany materiał może być toksyczny, należy przeprowadzić badanie zasadnicze.
Badanie graniczne dla jednej dawki na poziomie 2 000 mg/kg masy ciała może zostać przeprowadzone na sześciu zwierzętach (trzy na każdym etapie). W wyjątkowych okolicznościach dopuszczalne jest przeprowadzenie badania granicznego przy jednej dawce na poziomie 5 000 mg/kg na trzech zwierzętach (zob. załącznik 2). W przypadku wystąpienia śmiertelności związanej z substancją badaną konieczne może okazać się przeprowadzenie badania na następnym niższym poziomie.
1.6. OBSERWACJE
Zwierzęta są poddawane obserwacji osobniczej przynajmniej raz w ciągu pierwszych 30 min. po podaniu dawki, okresowo przez pierwsze 24 godziny, ze szczególnym uwzględnieniem pierwszych 4 godzin, a następnie codziennie ogólnie przez 14 dni, z wyjątkiem sytuacji w których muszą one zostać wyeliminowane z badań i humanitarnie zabite ze względu na ich dobro, lub zostały znalezione martwe. Jednakże czas trwania obserwacji nie powinien być ustalany według ścisłych reguł. Powinien on być zależny od działania toksycznego, czasu wystąpienia skutków i długości okresu powrotu do stanu normalnego, i może skutkiem tego być przedłużany, w przypadku gdy zostanie to uznane za stosowne. Czas w jakim pojawiają się i zanikają objawy zatrucia jest istotny, szczególnie w przypadku występowania opóźnionych objawów zatrucia (11). Wszystkie obserwacje są systematycznie zapisywane w ewidencji prowadzonej osobno dla każdego zwierzęcia.
Dodatkowe obserwacje będą wymagane, jeżeli zwierzęta wciąż wykazują objawy zatrucia. Obserwacje powinny dotyczyć zmian na skórze i futrze, oczach i śluzówce a także w układach oddechowym, krążenia, autonomicznym i centralnym układzie nerwowym oraz zmian w funkcjonowaniu somatomotorycznym i schematach zachowania. Szczególną uwagę należy poświęcić obserwacjom drżenia, drgawek, ślinienia się, biegunki, letargu, snu i śpiączki. Należy przestrzegać zasad i kryteriów streszczonych w Wytycznych dotyczących humanitarnego punktu końcowego. Zwierzęta u których stwierdzono stan agonalny oraz zwierzęta wykazujące objawy silnego bólu powinny zostać uśmiercone w sposób humanitarny. W przypadku uśmiercenia zwierząt z przyczyn humanitarnych lub stwierdzenia u nich zgonu, czas zgonu powinien zostać zapisany najdokładniej, jak to jest możliwe.
1.6.1. Masa ciała
Masa każdego zwierzęcia powinna zostać określona na krótko przed podaniem substancji badanej i przynajmniej w odstępie tygodnia od jej podania. Zmiany masy powinny zostać obliczone i zapisane. Po zakończeniu badań zwierzęta, które przeżyły, są ważone i humanitarnie uśmiercane.
1.6.2. Patologia
Wszystkie badane zwierzęta (włączając zwierzęta, które padły podczas badań lub zostały wyeliminowane z badań ze względu na ich dobro) powinny zostać poddane całkowitej sekcji zwłok. Wszystkie całkowite zmiany patologiczne powinny zostać zapisane oddzielnie dla każdego zwierzęcia. Badanie mikroskopowe organów wykazujących ewidentne zmiany patologiczne u zwierząt, które przeżyły 24 lub więcej godzin po podaniu pierwszej dawki, mogą również zostać wzięte pod uwagę, ponieważ mogą one dostarczyć użytecznych informacji.
2. DANE
Należy podać dane dla każdego zwierzęcia. Dodatkowo, wszystkie dane powinny zostać streszczone w tabeli pokazującej dla każdej badanej grupy liczbę wykorzystanych sztuk, liczbę zwierząt wykazujących objawy zatrucia, liczbę zwierząt, u których stwierdzono zgon w trakcie badań lub uśmierconych z przyczyn humanitarnych, czas zgonu poszczególnych zwierząt, opis oraz przebieg w czasie objawów zatrucia oraz powrotu do stanu normalnego, a także wyniki sekcji.
3. SPRAWOZDANIE
3.1. Sprawozdanie z badań
Sprawozdanie z badań musi zawierać następujące informacje, o ile są one stosowne:
|
|
Substancja badana:
|
|
|
Nośnik (jeśli był użyty):
|
|
|
Zwierzęta badane:
|
|
|
Warunki badań:
|
|
|
Wyniki:
|
|
|
Omówienie i interpretacja wyników. |
|
|
Wnioski. |
4. BIBLIOGRAFIA
|
(1) |
Roll R., Höfer-Bosse Th. And Kayser D. (1986). New Perspectives in Acute Toxicity Testing of Chemicals. Toxicol. Lett., Suppl. 31, 86. |
|
(2) |
Roll R., Riebschläger M., Mischke U. and Kayser D. (1989). Neue Wege zur Bestimmung der akuten Toxizität von Chemikalien. Bundesgesundheitsblatt 32, 336–341. |
|
(3) |
Diener W., Sichha L., Mischke U., Kayser D. and Schlede E. (1994). The Biometric Evaluation of the Acute- Toxic-Class Metoda (Oral). Arch. Toxicol. 68, 559–610. |
|
(4) |
Diener W., Mischke U., Kayser D. and Schlede E. (1995). The Biometric Evaluation of the OECD Modified Version of the Acute-Toxic-Class Method (Oral). Arch. Toxicol. 69, 729–734. |
|
(5) |
Diener W., and Schlede E. (1999) Acute Toxicity Class Metodas: Alterations to LD/LC50 Tests. ALTEX16, 129–134. |
|
(6) |
Schlede E., Mischke U., Roll R. and Kayser D. (1992). A National Validation Study of the Acute-Toxic- Class Method. An Alternative to the LD50 Test. Arch. Toxicol. 66, 455–470. |
|
(7) |
Schlede E., Mischke U., Diener W. and Kayser D. (1994). The International Validation Study of the Acute- Toxic-Class Method (Oral). Arch. Toxicol. 69, 659–670. |
|
(8) |
OECD (2001) Guidance Document on Acute Oral Toxicity Testing. Environmental Health and Safety Monograph Series on Testing and Grade N. 24. Paris. |
|
(9) |
OECD (2000) Guidance Document on the Recognition, Grade and Use of Clinical Signs as Humane Endpoints for Experimental Animals Used in Safety Evaluation. Environmental Health and Safety Monograph Series on Testing and Assessment N 19. |
|
(10) |
OECD (1998) Harmonized Integrated Hazard Classification System For Human Health And Environmental Effects Of Chemical Substances as endorsed by the 28th Joint Meeting of the Chemicals Committee and the Working Party on Chemicals in November 1998, Part 2, p. 11 [http://webnet1.oecd.org/oecd/stronas/home/displaygeneral/0,3380,EN-documents-521-14-no-24-no-0,FF.html] |
|
(11) |
Lipnick R L, Cotruvo, J A, Hill R N, Bruce R D, Stitzel K A, Walker A P, Chu I; Goddard M, Segal L, Springer J A and Myers R C (1995) Comparison of the Up-and Down, Conventional LD50, and Fixed Dose Acute Toxicity Procedures. Fd. Chem. Toxicol 33, 223–231. |
|
(12) |
Chan P.K. and A.W. Hayes. (1994). Chap. 16. Acute Toxicity and Eye Irritancy. Principles and Methods of Toxicology. Third Edition. A.W. Hayes, Editor. Raven Press, Ltd., New York, USA. |
ZAŁĄCZNIK 1
PROCEDURA REALIZOWANA W PRZYPADKU DAWEK WYJŚCIOWYCH
UWAGI OGÓLNE
Dla każdej dawki wyjściowej, odpowiednie schematy badań zawarte w niniejszym załączniku przedstawiają procedurę, jaką należy zastosować.
|
— |
ZAŁĄCZNIK 1a: Dawka początkowa wynosi 5 mg/kg. |
|
— |
ZAŁĄCZNIK 1b: Dawka początkowa wynosi 50 mg/kg. |
|
— |
ZAŁĄCZNIK 1c: Dawka początkowa wynosi 300 mg/kg. |
|
— |
ZAŁĄCZNIK 1d: Dawka początkowa wynosi 2 000 mg/kg. |
W zależności od liczby zwierząt uśmierconych w sposób humanitarny lub martwych zwierząt procedura badań przebiega zgodnie z kierunkiem wyznaczanym przez strzałki.
ZAŁĄCZNIK 1A
PROCEDURA BADANIA PRZY DAWCE WYJŚCIOWEJ 5 MG/KG MASY CIAŁA

ZAŁĄCZNIK 1B
PROCEDURA BADANIA PRZY DAWCE WYJŚCIOWEJ 50 MG/KG MASY CIAŁA

ZAŁĄCZNIK 1C
PROCEDURA BADANIA PRZY DAWCE WYJŚCIOWEJ 300 MG/KG MASY CIAŁA
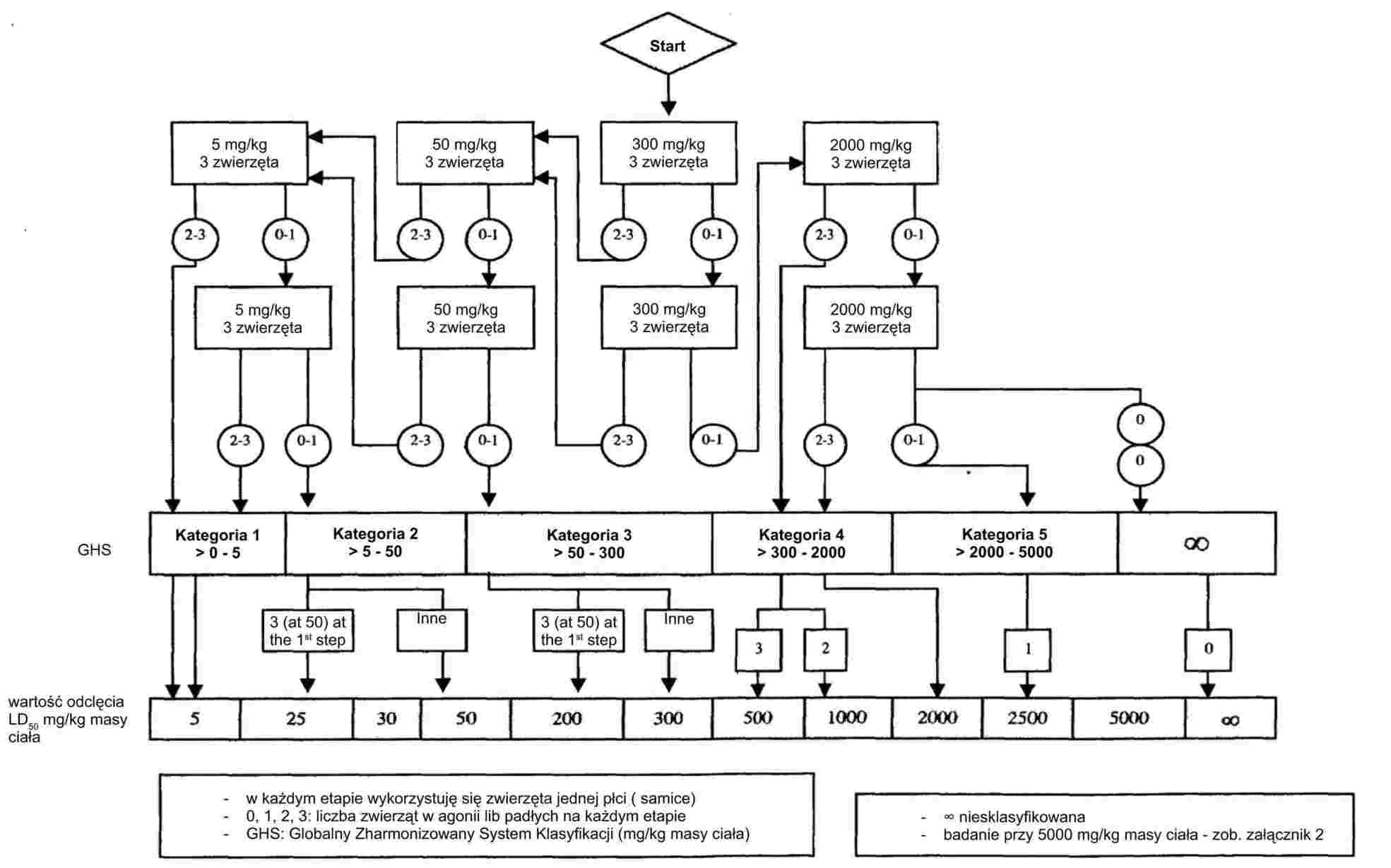
ZAŁĄCZNIK 1D
PROCEDURA BADANIA PRZY DAWCE WYJŚCIOWEJ 2 000 MG/KG MASY CIAŁA
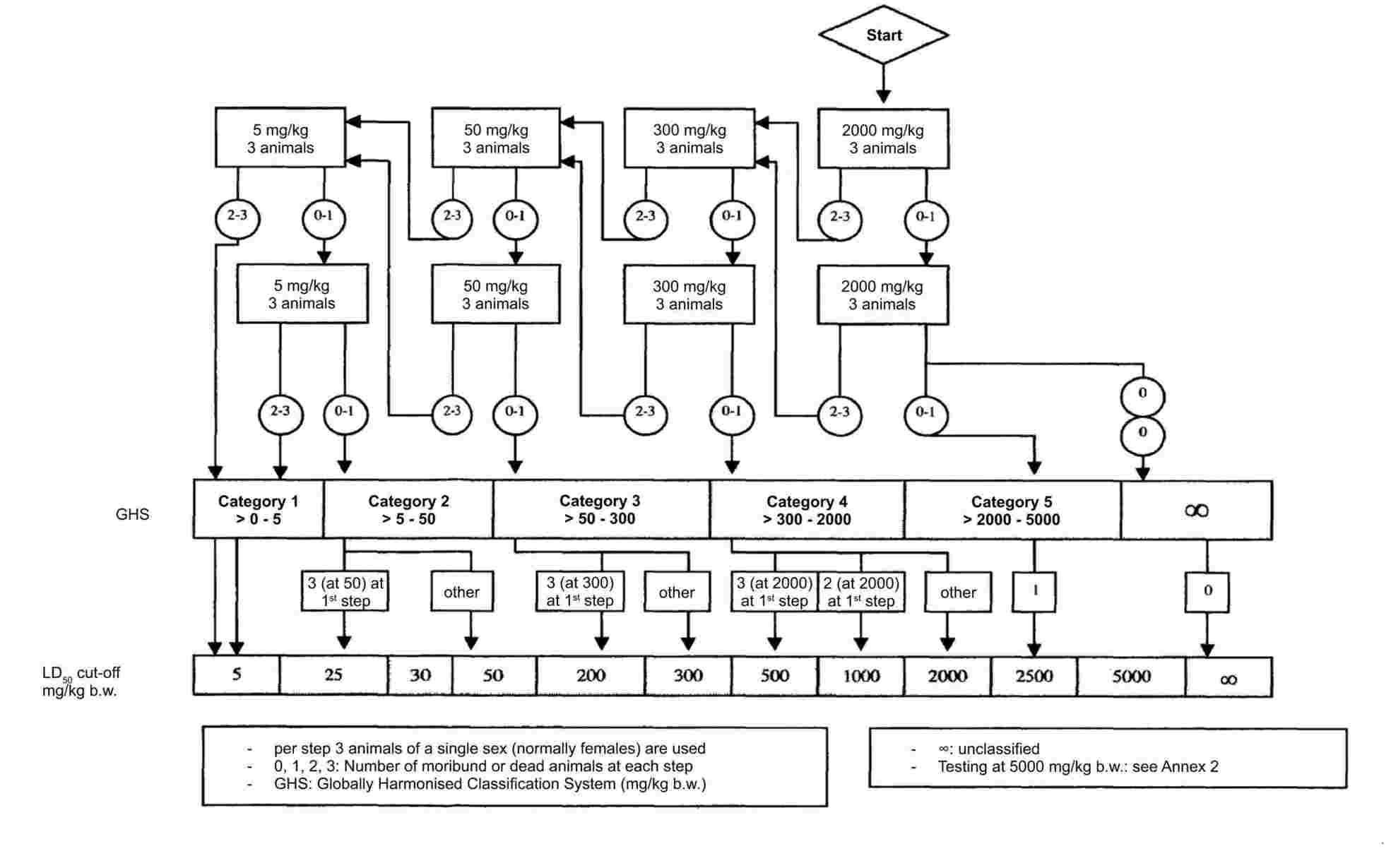
ZAŁĄCZNIK 2
KRYTERIA KLASYFIKACJI SUBSTANCJI BADANYCH O OCZEKIWANYCH WARTOŚCIACH LD50 PRZEKRACZAJĄCYCH 2 000 mg/kg, Z POMINIĘCIEM BADAŃ
Kryteria kategorii niebezpieczeństwa 5 mają w zamierzeniu umożliwiać identyfikację substancji badanych, które charakteryzują się niskim niebezpieczeństwem ostrej toksyczności, lecz które, w określonych okolicznościach mogą okazać się niebezpieczne dla populacji podatnych na zatrucie. Przewiduje się, że takie substancje charakteryzują się pokarmowym lub skórnym LD50 mieszczącym się w zakresie 2 000–5 000 mg/kg lub odpowiadają analogicznym dawkom w przypadku innych szlaków kontaktu z substancją. Substancja może zostać zaklasyfikowana do kategorii niebezpieczeństwa przy następujących cechach: 2 000 mg/kg < LD50 < 5 000 mg/kg (kategoria 5 według GHS) w następujących przypadkach:
|
a) |
jeżeli została przypisana do tej kategorii w wyniku zastosowania jednego ze schematów przedstawionych w załącznikach 1a–1d, w oparciu o poziom śmiertelności; |
|
b) |
jeżeli dostępne są wiarygodne dowody wskazujące, że LD50 powinno znaleźć się w obrębie kategorii 5; bądź jeżeli inne badania na zwierzętach lub ostre skutki u ludzi sugerują niebezpieczeństwo dla zdrowia ludzkiego spowodowane ostrym działaniem; |
|
c) |
jeżeli okazuje się, w wyniku przeprowadzenia ekstrapolacji, szacunków lub pomiaru danych, że przypisanie do wyższej klasy niebezpieczeństwa nie jest uzasadnione oraz:
|
BADANIA PRZY DAWKACH PRZEKRACZAJĄCYCH 2 000 MG/KG
Uznając potrzebę dbałości o dobrostan zwierząt, badanie zwierząt w ramach kategorii 5 (5 000 mg/kg) jest odradzane i powinno być rozważane, wyłącznie jeżeli występuje wysokie prawdopodobieństwo, że wynik takich badań będzie miał bezpośredni wpływ na bezpieczeństwo i zdrowie ludzi i zwierząt (10). Nie należy prowadzić badań przy zastosowaniu wyższych poziomów dawek.
W przypadku konieczności przeprowadzenia badania przy zastosowaniu dawki 5 000 mg/kg wymagane jest przeprowadzenie jednego etapu (tj. przy wykorzystaniu trzech zwierząt). Jeżeli pierwsze zwierzę padnie, następnemu podaje się 2 000 mg/kg, zgodnie ze schematem działań zamieszczonym w załączniku 1. Jeżeli pierwsze zwierzę przeżyje, dawka podawana jest dwóm następnym. Jeżeli pada wyłącznie jedno zwierzę, przyjmuje się, że wartość LD50 przekracza 5 000 mg/kg. Jeżeli padają oba zwierzęta, podawanie kontynuuje się na poziomie 2 000 mg/kg.
ZAŁĄCZNIK 3
METODA BADAWCZA B.1 ter Wytyczne dotyczące klasyfikacji zgodnie z systemem WE obowiązującej w okresie przejściowym do momentu pełnego wdrożenia Globalnego Zharmonizowanego Systemu Klasyfikacji (GHS) (wytyczne pochodzą z pozycji bibliograficznej (8))


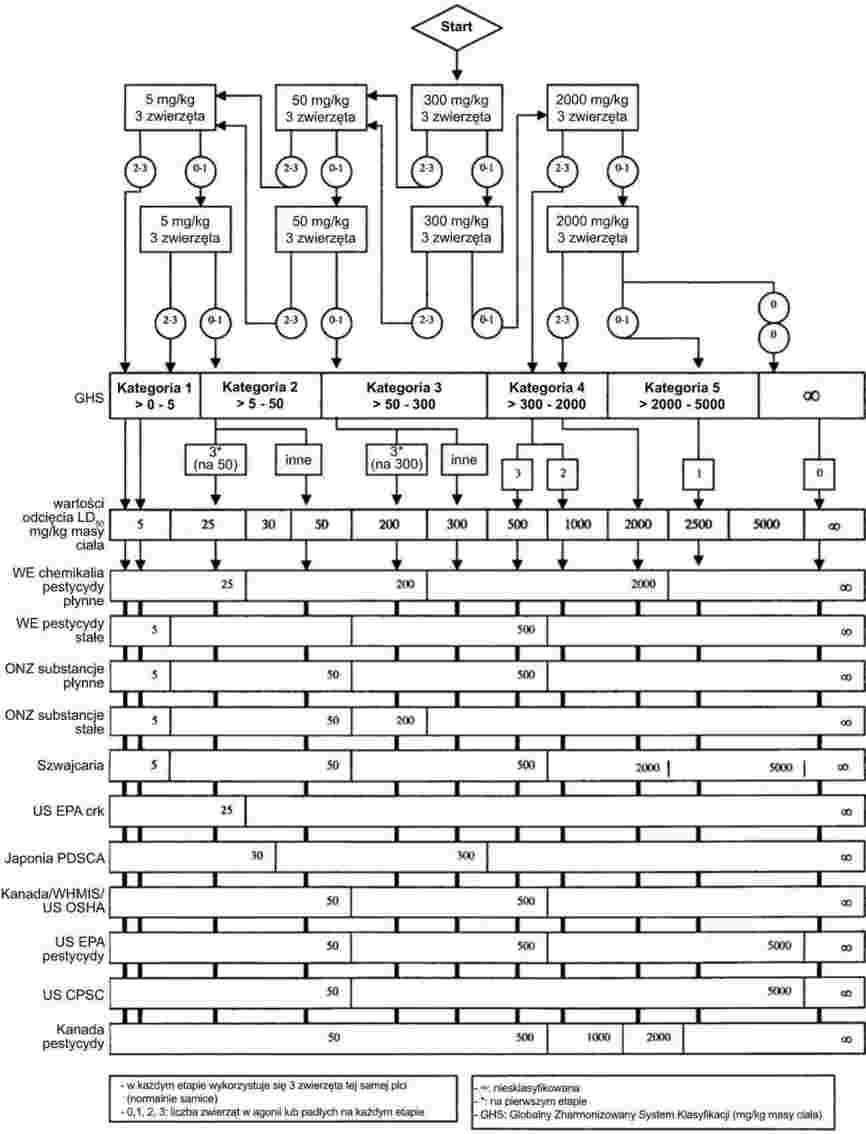

B.2. TOKSYCZNOŚĆ OSTRA (INHALACYJNA)
1. METODA
1.1. WPROWADZENIE
Jest użyteczne posiadanie wstępnych informacji na temat rozkładu wielkości ziarna, prężności pary, temperatury topnienia, temperatury wrzenia, temperatury zapłonu w wybuchowości (o ile dotyczy) substancji.
Zob. także Wprowadzenie ogólne część B lit. A).
1.2. DEFINICJE
Zob. Wprowadzenie ogólne część B lit. B).
1.3. SUBSTANCJE ODNIESIENIA
Brak.
1.4. ZASADA METODY BADANIA
Kilka grup zwierząt doświadczalnych poddaje się ekspozycji przez określony okres czasu na substancję badaną w stopniowanych stężeniach, po jednym stężeniu na grupę. Następnie przeprowadza się obserwację skutków podania substancji i zgonów. Zwierzęta, które padną w trakcie badania, i te, które przeżyją po jego zakończeniu, poddaje się sekcji zwłok.
Zwierzęta wykazujące ciężkie i trwałe oznaki stanu zagrożenia i bólu mogą wymagać humanitarnego uśmiercenia. Dozowanie badanych substancji w sposób znany z powodowania znaczącego bólu i stanu zagrożenia z powodu właściwości żrących lub poważnie drażniących nie może być przeprowadzane.
1.5. KRYTERIA JAKOŚCI
Brak.
1.6. OPIS METODY BADANIA
1.6.1. Przygotowania
Zwierzęta muszą być trzymane w doświadczalnych warunkach pobytu i karmienia przez co najmniej pięć dni przed przeprowadzeniem badania. Przed przeprowadzeniem badania zdrowe, młode, dorosłe zwierzęta są wybierane losowo i przydzielane do wymaganej liczby grup otrzymujących substancję. Nie muszą być poddawane symulowanej ekspozycji, chyba że jest to wskazane z powodu wykorzystania określonego przyrządu zapewniającego ekspozycję.
Badane substancje w postaci ciała stałego wymagają mikronizacji w celu uzyskania właściwego wymiaru cząstki.
W miarę potrzeby do badanej substancji można dodać odpowiednie podłoże, aby można było zapewnić właściwe stężenie tej substancji w powietrzu – w takim przypadku należy zastosować grupę kontrolną, która będzie otrzymywać samo podłoże. W razie wykorzystania podłoża lub innych substancji pomocniczych w celu ułatwienia dawkowania, musi być wiadomo, że nie wykazująone działania toksycznego. O ile będzie to właściwe, można wykorzystać dane historyczne.
1.6.2. Warunki badania
1.6.2.1. Zwierzęta doświadczalne
O ile nie ma przeciwwskazań, preferowanym gatunkiem jest szczur. Należy wykorzystywać powszechnie stosowane szczepy laboratoryjne. Dla każdej płci, na początku badania, zakres zmian wagi używanych zwierząt nie może przekraczać ± 20 % właściwej wartości średniej.
1.6.2.2. Liczba i płeć
Wykorzystuje się po co najmniej 10 gryzoni (pięć samic i pięciu samców) w przypadku każdego poziomu dawkowania. Samice powinny być nieródkami i nie mogą być w ciąży.
Uwaga: W badaniach ostrej toksyczności u zwierząt wyższego rzędu niż gryzonie, należy rozważyć użycie mniejszej ilości. Dawki muszą być starannie wybrane oraz należy dołożyć wszelkich starań by nie przekraczać dawek średniej toksyczności. Należy unikać podawania w takich badaniach dawek śmiertelnych substancji badanej.
1.6.2.3. Stężenia ekspozycyjne
Powinno się zapewnić odpowiednią liczbę wartości tych stężeń – co najmniej trzy – przy czym należy zachować odpowiednie odstępy między nimi, tak aby uzyskać grupy badane różniące się istotnie efektami toksycznymi i stopniem śmiertelności. Dane powinny wystarczyć do nakreślenia krzywej zależności między stężeniem a śmiertelnością oraz w miarę możliwości powinny zapewnić możliwe do przyjęcia obliczenie LC50.
1.6.2.4. Badanie graniczne
W przypadku gdy ekspozycja pięciu samców i pięciu samic badanych zwierząt na 20 mg na litr gazu lub 5 mg na litr aerozolu lub cząstek substancji przez cztery godziny (lub, jeżeli jest to niemożliwe ze względu na fizyczne lub chemiczne, w tym wybuchowe, właściwości substancji badanej, na maksymalne możliwe do uzyskania stężenie) nie spowoduje związanych z danym związkiem zgonów w ciągu 14 dni, dalsze badania można uznać za zbędne.
1.6.2.5. Czas ekspozycji
Okres czasu ekspozycji powinien być cztery godziny.
1.6.2.6. Wyposażenie
Zwierzęta należy poddać badaniu przy użyciu przyrządów do inhalacji zapewniających przepływ dynamiczny powietrza co najmniej 12 wymian na godzinę, aby zapewnić odpowiednią zawartość tlenu i równomierną dystrybucję powietrza ekspozycji. W przypadku wykorzystania komory należy ją tak zaprojektować, aby zmniejszyć do minimum stłoczenie zwierząt badanych i zwiększyć do maksimum ich ekspozycję drogą inhalacyjną na substancję badaną. Z reguły, aby zapewnić stabilność powietrza w komorze, całkowita „objętość” zwierząt badanych nie powinna przekraczać 5 % objętości komory do badań. Można zastosować ekspozycję doustno-nosową, ekspozycję jedynie głowy lub ekspozycję całego ciała w pojedynczych komorach, przy czym te pierwsze dwa sposoby pomogą w zminimalizowaniu przechodzenia substancji badanej do organizmu innymi drogami.
1.6.2.7. Okres obserwacji
Okres obserwacji powinien wynosić co najmniej 14 dni. Jednakże nie powinien on być sztywno ustalony. Powinien zależeć od reakcji toksycznych, szybkości ich pojawiania się oraz okresu powrotu do zdrowia po ich wystąpieniu; w związku z tym można go wydłużyć, jeżeli uzna się to za konieczne. Ważny jest czas, po jakim pojawiają się i znikają objawy toksyczności oraz czas zgonu, w szczególności gdy istnieje tendencja do opóźnionych zgonów.
1.6.3. Procedura
Tuż przed ekspozycją zwierzęta waży się, a następnie poddaje ekspozycji na stężenie badane w wyznaczonej aparaturze przez minimalny okres czterech godzin, po zapewnieniu równowagi stężenia substancji w komorze. Czas do uzyskania równowagi powinien być krótki. Temperatura przeprowadzania badania powinna być utrzymywana na poziomie 22 oC ± 3 oC. Idealnie wilgotność względną należy utrzymywać w przedziale między 30 i 70 %, jednak w niektórych przypadkach (np. badania niektórych aerozoli) może to nie być możliwe ze względów praktycznych. Utrzymanie lekkiego podciśnienia wewnątrz komory (ok. 5 mm słupa wody) zapobiega wyciekowi substancji badanej do otaczającego obszaru. W trakcie ekspozycji należy zaprzestać podawania zwierzętom karmy i wody. Należy użyć układów generacji i monitorowania powietrza stosowanego do badań. Układ powinien zapewnić jak najszybsze uzyskanie stabilnych warunków ekspozycji. Komora musi być zaprojektowana i działać w taki sposób, by utrzymywać jednorodny rozkład badanego powietrza w jej obrębie.
Należy prowadzić pomiary lub monitorować:
|
(a) |
szybkość przepływu powietrza (w sposób ciągły), |
|
(b) |
rzeczywiste stężenie substancji badanej mierzone w obszarze wdychania co najmniej trzy razy w czasie trwania ekspozycji (niektóre atmosfery, na przykład aerozole o wysokich stężeniach mogą wymagać częstszej kontroli). W czasie trwania okresu ekspozycji, stężenie nie powinno zmieniać się więcej niż ± 15 % od wartości średniej. W przypadku niektórych aerozoli ten poziom kontroli możne być niemożliwy do uzyskania, przy czym dopuszczalny jest wtedy szerszy zakres. Dla aerozoli należy przeprowadzić analizę wielkości cząsteczki (co najmniej raz na badaną grupę), |
|
(c) |
temperaturę i wilgotność, o ile możliwe, w sposób ciągły. |
W trakcie ekspozycji i po jej zakończeniu należy prowadzić systematyczne obserwacje, zapisując ich wyniki; należy prowadzić oddzielne zapisy dla każdego zwierzęcia. Obserwacja powinna być częsta w ciągu pierwszego dnia. Staranne badanie kliniczne należy przeprowadzać co najmniej raz każdego dnia roboczego, inne obserwacje należy przeprowadzać codziennie, podejmując odpowiednie działania mające na celu zmniejszenie do minimum straty zwierząt podczas badania, np. sekcję zwłok lub zamrażanie tych zwierząt, które zostaną znalezione martwe, oraz izolację lub usypianie zwierząt słabych lub śmiertelnie chorych.
Obserwacje powinny obejmować zmiany skóry i sierści, oczu i błon śluzowych oraz zmiany układu oddechowego, krążenia, autonomicznego i ośrodkowego układu nerwowego, jak też wzorców aktywności somatomotorycznej i wzorca zachowania. Szczególną uwagę należy zwrócić na obserwacje drżeń, drgawek, zwiększonego wydzielania śliny, biegunki, stanów zwiększonej senności, snu i śpiączki. Należy jak najprecyzyjniej odnotować termin zgonu. Masy ciała poszczególnych zwierząt należy ustalać co tydzień po ekspozycji oraz w momencie zgonu.
Zwierzęta, które zdechną w trakcie badania, i te, które przeżyją po jego zakończeniu, poddaje się sekcji zwłok, ze szczególnym zwróceniem uwagi na wszelkie zmiany w obrębie górnych i dolnych dróg oddechowych. Należy odnotować wszelkie makroskopowe zmiany patologiczne. O ile jest to wskazane, należy pobrać wycinki tkanek do badania histopatologicznego.
2. DANE
Dane należy przedstawić w skrócie w formie tabelarycznej, z podaniem – dla każdej badanej grupy – liczby zwierząt na początku badania, terminów zgonu poszczególnych zwierząt, liczby zwierząt wykazujących inne objawy toksyczności, opisu efektów toksycznych i wyniku sekcji zwłok. Należy obliczać zmiany masy ciała i zapisywać je, gdy przeżycie przekroczy jedną dobę. Zwierzęta, które zostały humanitarnie uśmiercone z powodów związanych ze stanami zagrożenia i bólem, są zapisywane jako zgony związane z substancją. Należy ustalić LC50 przy użyciu uznanej metody. Ocena danych powinna objąć istnienie ewentualnej współzależności między ekspozycją zwierząt na substancję badaną i częstością i ciężkością wszelkich zaburzeń, włącznie z zaburzeniami zachowania i klinicznymi, zmianami makroskopowymi, zmianami masy ciała, umieralnością i wszelkimi innymi efektami toksycznymi.
3. SPORZĄDZANIE SPRAWOZDANIA
3.1. SPRAWOZDANIE Z BADANIA
Sprawozdanie z badań zawiera w miarę możliwości następujące informacje:
|
— |
gatunek, szczep, źródło, warunki środowiska, dieta itp., |
|
— |
warunki badania: opis przyrządu do ekspozycji, włączając konstrukcję, typ, wymiary, źródło powietrza, układ wytwarzania aerozoli, metody klimatyzacji powietrza i metoda przetrzymywania zwierząt w komorze badawczej, gdy jest używana. Należy opisać wyposażenie do pomiaru temperatury, wilgotności, stężenia aerozolu i rozkładu wielkości ziaren. |
Dane ekspozycji
Powinny być przedstawione w formie tabeli, z podaniem wartości średniej i miary zmienności (np. odchylenia standardowego) i powinny, jeśli możliwe, zawierać:
|
a) |
szybkości przepływu powietrza przez urządzenia do inhalacji; |
|
b) |
temperaturę i wilgotność powietrza; |
|
c) |
nominalne stężenia (całkowita ilość badanej substancji dostarczona do wyposażenia inhalacyjnego, podzielona przez objętość powietrza); |
|
d) |
rodzaj podłoża, o ile jest wykorzystane; |
|
e) |
rzeczywiste stężenia w badawczej strefie wdychania; |
|
f) |
średnicę aerodynamiczną średniej masy (MMAD) i geometryczne standardowe odchylenie (GSD); |
|
g) |
czas równoważenia; |
|
h) |
okres ekspozycji;
|
3.2. OCENA I INTERPRETACJA
Zob. Wprowadzenie ogólne część B lit. D).
4. LITERATURA
Zob. Wprowadzenie ogólne część B lit. E).
B.3. TOKSYCZNOŚĆ OSTRA (DERMALNA)
1. METODA
1.1. WPROWADZENIE
Zob. Wprowadzenie ogólne część B lit. A).
1.2. DEFINICJA
Zob. Wprowadzenie ogólne część B lit. B).
1.3. SUBSTANCJE ODNIESIENIA
Brak.
1.4. ZASADA METODY BADANIA
Substancję badaną nakłada się na skórę w stopniowanych dawkach kilku grupom zwierząt doświadczalnych, po jednej dawce na grupę. Następnie obserwuje się skutki podania substancji i zgony. Zwierzęta, które zdechną w trakcie badania, i te, które przeżyją po jego zakończeniu, poddaje się sekcji zwłok.
Zwierzęta wykazujące ciężkie i trwałe oznaki stanu zagrożenia i bólu mogą wymagać humanitarnego uśmiercenia. Dozowanie badanych substancji na drodze znanej z powodowania znaczącego bólu i stany zagrożenia z powodu właściwości żrących lub drażniących nie może być przeprowadzane.
1.5. KRYTERIA JAKOŚCI
Brak.
1.6. OPIS METODY BADANIA
1.6.1. Przygotowania
Zwierzęta muszą być trzymane w doświadczalnych warunkach pobytu i karmienia przez co najmniej pięć dni przed przeprowadzeniem badania. Przed przeprowadzeniem badania zdrowe, młode, dorosłe zwierzęta są wybierane losowo i przydzielane do poszczególnych grup otrzymujących substancję. Na około 24 godziny przed przeprowadzeniem badania należy usunąć sierść z grzbietowej powierzchni tułowia zwierząt przez przycięcie lub ogolenie. Podczas przycinania lub golenia sierści należy uważać, aby nie uszkodzić skóry, gdyż mogłoby to zmienić jej przepuszczalność. Nie mniej niż 10 % powierzchni ciała powinno być wolne do zastosowania substancji badanej. W przypadku badania ciał stałych, które powinny zostać sproszkowane, o ile jest to właściwe, substancja badana powinna zostać wystarczająco zwilżona wodą lub w razie konieczności odpowiednim podłożem zapewniającym dobry kontakt ze skórą. W przypadku wykorzystania podłoża należy uwzględnić jego wpływ na penetrację substancji badanej przez skórę. Płynne substancje badane stosuje się na ogół w stanie nierozcieńczonym.
1.6.2. Warunki badania
1.6.2.1. Zwierzęta doświadczalne
Można wykorzystać dorosłe szczury lub króliki. Można również wykorzystać inne gatunki, jednak wymaga to uzasadnienia. Należy wykorzystywać powszechnie stosowane szczepy laboratoryjne. Dla każdej płci na początku badania zakres zmienności wagowej użytych zwierząt nie powinien przekraczać ± 20 % właściwej wartości średniej.
1.6.2.2. Liczba i płeć
Co najmniej pięć zwierząt jest używanych dla każdej wielkości dawki. Powinny być one wszystkie jednakowej płci. Jeżeli stosuje się samice, powinny być one nieródkami i nie mogą być w ciąży. Jeżeli dostępna jest informacja wskazująca, że dana płeć jest znacząco bardziej czuła, należy użyć do dawkowania zwierząt tej płci.
Uwaga: W badaniach ostrej toksyczności u zwierząt wyższego rzędu niż gryzonie należy rozważyć użycie mniejszej ilości. Dawki muszą być starannie wybrane oraz należy dołożyć wszelkich starań by nie przekraczać dawek średniej toksyczności. Należy unikać podawania w takich badaniach dawek śmiertelnych substancji badanej.
1.6.2.3. Poziomy dawkowania
Powinno się zapewnić odpowiednią liczbę, co najmniej trzy, przy czym należy zachować odpowiednie odstępy między nimi, tak aby uzyskać grupy badane różniące się istotnie efektami toksycznymi i umieralnością. Przy podejmowaniu decyzji co do wielkości dawek należy uwzględniać wszelkie działania drażniące i żrące. Dane powinny być wystarczające do opracowania krzywej zależności odpowiedzi od dawki oraz, o ile to będzie możliwe, do ustalenia w możliwy do przyjęcia sposób wartości LD50.
1.6.2.4. Badanie graniczne
Przeprowadza się badanie graniczne przy jednej wielkości dawki, przynajmniej 2 000 mg/kg masy ciała, w grupie składającej się z pięciu samców i pięciu samic, stosując procedurę powyżej opisaną. Jeżeli zachodzi związek śmiertelności z badaną substancją, należy rozważyć wykonanie pełnego badania.
1.6.2.5. Okres obserwacji
Okres obserwacji powinien wynosić co najmniej 14 dni. Jednakże nie powinien on być sztywno ustalony. Powinien zależeć od reakcji toksycznych, szybkości ich pojawiania się oraz okresu powrotu do zdrowia po ich wystąpieniu; w związku z tym można go wydłużyć, jeżeli uzna się to za konieczne. Ważny jest czas, po jakim pojawiają się i znikają objawy toksyczności, oraz czas zgonu, zwłaszcza gdy istnieje tendencja do opóźnionych zgonów.
1.6.3. Procedura
Zwierzęta powinny być trzymane w oddzielnych klatkach. Substancja badana powinna zostać nałożona w sposób równomierny na obszar równy około 10 % całkowitej powierzchni ciała. W przypadku substancji wysoce toksycznych można je nanieść na mniejszą powierzchnię, jednak należy dążyć do tego, aby pokryć jak największy obszar jak najcieńszą i jak najbardziej równomierną warstwą badanego związku.
Substancje badane powinny być utrzymywane w kontakcie ze skórą dzięki zastosowaniu porowatego opatrunku z gazy i niedrażniącej taśmy w trakcie ekspozycji zapewnianej przez 24 godziny. Miejsce badania należy dodatkowo przykryć w taki sposób, aby utrzymać opatrunek z gazy i substancję badaną i zapewnić, aby zwierzęta nie mogły zjeść tej ostatniej. W celu zapobieżenia spożyciu substancji badanej można uwiązać zwierzę, jednak pełna immobilizacja nie jest zalecaną metodą.
Pod koniec okresu ekspozycji należy usunąć resztki badanej substancji, w miarę możliwości stosując wodę lub jakieś inne, właściwe metody oczyszczania skóry.
Obserwacje należy odnotowywać w sposób systematyczny, w miarę ich zbierania. Należy prowadzić oddzielne zapisy dla każdego zwierzęcia. Obserwacja powinna być częsta w ciągu pierwszego dnia. Staranne badanie kliniczne należy przeprowadzać co najmniej raz każdego dnia roboczego. Inne obserwacje należy przeprowadzać codziennie, podejmując odpowiednie działania mające na celu zmniejszenie do minimum utraty zwierząt podczas badania, np. sekcję zwłok lub zamrażanie tych zwierząt, które zostaną znalezione martwe, oraz izolację lub usypianie zwierząt słabych lub śmiertelnie chorych.
Obserwacje przy klatce zwierzęcia powinny obejmować zmiany sierści, traktowanej substancją skóry, oczu i błon śluzowych oraz zmiany układu oddechowego, krążenia, autonomicznego i ośrodkowego układu nerwowego, jak też wzorców aktywności somatomotorycznej i wzorca zachowania. Szczególną uwagę należy zwrócić na obserwacje drżeń, drgawek, zwiększonego wydzielania śliny, biegunki, stanów zwiększonej senności, snu i śpiączki. Należy jak najprecyzyjniej odnotować termin zgonu. Zwierzęta, które zdechną w trakcie badania, i te, które przeżyją po jego zakończeniu, poddaje się sekcji zwłok. Należy odnotować wszelkie makroskopowe zmiany patologiczne. O ile jest to wskazane, należy pobrać wycinki tkanek do badania histopatologicznego.
Ocena toksyczności dla innej płci
Po zakończeniu badań jednej płci, przeprowadza się dawkowanie na przynajmniej jednej grupie pięciu zwierząt płci przeciwnej, w celu ustalenia czy zwierzęta tej płci nie są znacząco bardziej wrażliwe na substancję badaną. Użycie mniejszej ilości zwierząt musi być uzasadnione w indywidualnych okolicznościach. Jeżeli dostępna jest stosowna informacja pokazująca, że zwierzęta badanej płci są znacznie bardziej wrażliwe, drugą płeć można pominąć.
2. DANE
Dane należy przedstawić w skrócie w formie tabelarycznej, z podaniem – dla każdej badanej grupy – liczby zwierząt na początku badania, terminów zgonu poszczególnych zwierząt, liczby zwierząt wykazujących inne objawy toksyczności, opisu efektów toksycznych i wyniku sekcji zwłok. Należy określić masę ciała poszczególnych zwierząt i zapisać ją tuż przed podaniem badanej substancji, a później co tydzień i w momencie zgonu. Należy obliczać zmiany masy ciała i zapisywać je, gdy zwierzę przeżyje więcej niż jedną dobę. Zwierzęta humanitarnie zabite z powodu bólu i zagrożenia związanego z daną substancją są zapisywane jako uśmiercone w związku z substancją. Można ustalić LD50 przy użyciu uznanej metody.
Ocena danych powinna objąć istnienie ewentualnej współzależności pomiędzy ekspozycją zwierząt na substancję badaną a częstością i ciężkością wszelkich zaburzeń, włącznie z zaburzeniami zachowania i klinicznymi, zmianami makroskopowymi, zmianami masy ciała, umieralnością i wszelkimi innymi efektami toksycznymi.
3. SPORZĄDZANIE SPRAWOZDANIA
3.1. SPRAWOZDANIE Z BADANIA
Sprawozdanie z badań zawiera w miarę możliwości następujące informacje:
|
— |
gatunek, szczep, źródło, warunki środowiska, dieta itp., |
|
— |
warunki badania (włączając metodę oczyszczania skóry i typ opatrunku: okluzyjny lub nieokluzyjny), |
|
— |
wielkości dawek (wraz z podłożem, o ile jest wykorzystane, i stężeniami), |
|
— |
płeć zwierząt poddanych dawkowaniu, |
|
— |
tabelaryczne przedstawienie danych reakcji toksycznej w rozbiciu na płcie i wielkość dawki (to jest liczba padłych zwierząt lub zabitych w czasie trwania badania, liczba zwierząt wykazujących oznaki działania toksycznego, liczba zwierząt poddanych ekspozycji), |
|
— |
czas zgonu po dawkowaniu, przyczyny i kryteria przyjęte przy humanitarnym zabijaniu zwierząt, |
|
— |
wszystkie obserwacje, |
|
— |
wartość LD50 dla płci poddanej pełnym badaniom ustalonym na 14 dni (wraz z określoną metodą oznaczania), |
|
— |
95 % przedział ufności dla LD50 (o ile można go przedstawić), |
|
— |
krzywa zależności umieralności od dawki i jej nachylenie (o ile metoda oznaczania pozwala na ustalenie tych danych), |
|
— |
wyniki sekcji zwłok, |
|
— |
wszelkie wyniki badań histopatologicznych, |
|
— |
wyniki wszystkich badań płci przeciwnej, |
|
— |
omówienie wyników (należy zwrócić szczególną uwagę, że humanitarne zabicie zwierząt w czasie trwania badania wpływa na obliczoną wartość LD50), |
|
— |
interpretacja wyników. |
3.2. OCENA I INTERPRETACJA
Zob. Wprowadzenie ogólne część B lit. D).
4. LITERATURA
Zob. Wprowadzenie ogólne część B lit. E).
B.4. OSTRA TOKSYCZNOŚĆ: PODRAŻNIENIA/KOROZJA SKÓRY
1. METODA
Niniejsza metoda jest kopią OECD TG 404 (2002).
1.1. WPROWADZENIE
W przygotowywaniu niniejszej zaktualizowanej metody specjalną uwagę poświęcono możliwym ulepszeniom w zakresie zagadnień dotyczących dobrostanu zwierząt, a także ocenie wszystkich dostępnych informacji na temat substancji badanej, w celu uniknięcia niepotrzebnych badań na zwierzętach laboratoryjnych. Niniejsza metoda zawiera zalecenie, zgodnie z którym przed przystąpieniem do opisywanego badania in vivo na korozję/podrażnienia skórne wywołane przez daną substancję, zostanie przeprowadzona analiza wagi dowodów uzyskanych na podstawie istniejących danych. Jeżeli dostępne dane są niewystarczające, mogą zostać opracowane przy pomocy badań sekwencyjnych (1). Strategia badań zawiera wykaz zweryfikowanych i zaakceptowanych badań in vitro i stanowi załącznik do niniejszej metody. Dodatkowo, tam gdzie to stosowne, zamiast jednoczesnej aplikacji zwierzęciu trzech łat testowych, zalecane jest stosowanie aplikacji sekwencyjnej, zamiast symultanicznej, we wstępnych badaniach in vivo.
Z uwagi na dobrze pojęty interes nauki i dobrostan zwierząt badania in vivo nie powinny być podejmowane, jeżeli wszystkie dostępne dane odnoszące się do potencjalnego działania korozyjnego/podrażniającego skórę w wyniku działania substancji nie zostały uzyskane z analizy wagi dowodów. Takie dane będą zawierały dowody z wykonanych badań na ludziach oraz/lub zwierzętach laboratoryjnych, dowody korozji/podrażnień wywołanych przez jedną lub wiele substancji pokrewnych pod względem budowy lub mieszanin takich substancji, dane wskazujące na silną kwasowość lub alkaliczność substancji (2)(3) oraz wyniki zweryfikowanych i zaakceptowanych badań in vivo lub ex vivo (4)(5)(5a). Takie analizy powinny ograniczyć potrzebę badań in vivo pod kątem korozji/podrażnień skórnych wywołane przez substancje, w odniesieniu do których już istnieje wystarczający materiał dowodowy w odniesieniu do dwóch odnośnych punktów docelowych, zgromadzony w wyniku prowadzenia innych badań.
Preferowaną strategię badań sekwencyjnych, obejmującą przeprowadzenie zweryfikowanych i zaakceptowanych badań in vivo i ex vivo działania korozyjnego/podrażniającego, zawiera załącznik do niniejszej metody. Strategia ta została opracowana i jednomyślnie zalecona przez uczestników warsztatów OECD (6), a następnie zatwierdzona jako zalecana strategia badań w ramach Globalnego Zharmonizowanego Systemu Klasyfikacji Związków Chemicznych (GHS) (7). Zaleca się realizację tej strategii badań przed podjęciem badań in vivo. Dla nowych substancji strategia ta jest zalecanym stopniowym podejściem badawczym służącym opracowywaniu danych wartościowych z naukowego punktu widzenia dotyczących korozyjnego/podrażniającego działania substancji na skórę. W odniesieniu do istniejących substancji, na temat których brak wystarczających danych co do ich działania korozyjnego/podrażniającego, strategia ta powinna zostać wykorzystana w celu uzupełnienia brakujących danych. Użycie innych strategii badawczych bądź procedur lub decyzja o nieprzyjęciu stopniowego podejścia badawczego wymaga uzasadnienia.
Jeżeli określenie korozyjności lub podrażnienia nie może zostać przeprowadzone przy użyciu analizy wagi dowodów, która jest zbieżna ze strategią badań sekwencyjnych, należy rozważyć badanie in vivo (zob. załącznik).
1.2. DEFINICJE
Podrażnienie skórne: jest wynikiem odwracalnych uszkodzeń skóry wynikających z zastosowania substancji badanej przez okres czterech godzin.
Korozja skórna: jest wynikiem nieodwracalnego uszkodzenia skóry; mianowicie widocznej martwicy przebiegającej przez naskórek w głąb skóry, spowodowanej zaaplikowaniem substancji badanej na okres nieprzekraczający czterech godzin. Rodzaje korozji obejmują wrzody, krwawienie, strupy oraz – po zakończeniu 14-dniowej obserwacji – odbarwienia wynikłe ze zbielenia skóry, obszary jednolitej alopecji, a także blizny. Celem oceny niejasnych zmian można przeprowadzić badanie histopatologiczne.
1.3. ZASADA METODY BADAWCZEJ
Substancja, która ma być zbadana, jest nakładana na skórę zwierzęcia doświadczalnego w pojedynczej dawce, część skóry niepoddana zabiegowi służy jako część kontrolna. Stopień korozji/podrażnienia jest oceniany przy pomocy punktacji w określonych odstępach i zapisywany, a następnie dodatkowo opisywany w celu całkowitego oszacowania skutków działania. Czas trwania badania powinien być wystarczający do oceny odwracalności lub nieodwracalności zaobserwowanych skutków.
Zwierzęta wykazujące ciągłe objawy silnego strachu i/lub bólu na jakimkolwiek etapie badań powinny zostać uśmiercone w sposób humanitarny, a substancja oceniona stosownie do zaobserwowanych skutków. Kryteria podejmowania decyzji co do uśmiercenia zwierząt w stanie agonalnym lub cierpiących ból zostały określone w załączniku (8).
1.4. OPIS METODY BADAWCZEJ
1.4.1. Przygotowanie do badań in vivo
1.4.1.1. Wybór gatunku zwierząt
Biały królik jest preferowanym gatunkiem zwierząt laboratoryjnych. Wykorzystywane są zdrowe młode dorosłe króliki. W przypadku wykorzystania innych gatunków należy podać uzasadnienie.
1.4.1.2. Przygotowanie zwierząt
Na około 24 godziny przed badaniem należy usunąć futro z tułowia zwierzęcia poprzez gładkie wygolenie obszaru skóry na grzbiecie. Należy unikać otarć naskórka. Należy wykorzystywać wyłącznie zwierzęta ze zdrową nienaruszoną skórą.
Niektóre szczepy królików posiadają kępy futra, których gęstość jest większa w określonych porach roku. Takie obszary gęstego futra nie powinny być używane jako miejsca badań.
1.4.1.3. Warunki przebywania i warunki żywieniowe
Temperatura w badawczych pomieszczeniach hodowlanych powinna wynosić 20 oC (± 3o) dla królików. Chociaż wilgotność względna powinna wynosić co najmniej 30 % i pożądane jest, żeby nie przekraczała 70 % poza czasem czyszczenia pomieszczenia, celem powinno być utrzymywanie jej na poziomie 50–60 %. Oświetlenie powinno być sztuczne, w cyklu 12 godzin jasno, 12 godzin ciemno. Do żywienia można stosować konwencjonalne pasze laboratoryjne z nieograniczonym dostępem do wody pitnej.
1.4.2. Procedura badań
1.4.2.1. Podanie substancji badanej
Substancja badana powinna zostać zaaplikowana na małym obszarze skóry (około 6 cm2) i przykryta łatą gazy przytrzymywaną na miejscu przy pomocy niepodrażniającego plastra. W przypadku gdy zaaplikowanie substancji bezpośrednio nie jest niemożliwe (np. w przypadku cieczy lub niektórych past), substancja badana powinna zostać najpierw nałożona na gazę, a następnie na skórę. Gaza powinna luźno przylegać do skóry i powinna być przyczepiona do skóry na czas trwania ekspozycji przy pomocy odpowiedniego opatrunku półokluzyjnego. Jeżeli substancja badana została podana na łacie, powinna ona być przyczepiona do skóry w sposób zapewniający dobry kontakt i równomierne rozprowadzenie substancji na skórze. Należy zapobiec dostępowi zwierzęcia do gazy oraz spożyciu lub inhalacji substancji badanej przez zwierzę.
Ciekłe substancje badane są na ogół stosowane w stanie nierozcieńczonym. W przypadku badania substancji stałych (których postać można zmienić na postać proszkową, o ile zachodzi tak konieczność) substancja badana powinna zostać rozcieńczona przy użyciu minimalnej ilości wody (lub, o ile to konieczne, innego odpowiedniego nośnika), wystarczającej do zapewnienia dobrego kontaktu ze skórą. Jeżeli stosowany jest inny nośnik niż woda, potencjalny wpływ na podrażnienia skóry przez tą substancję powinien być minimalny lub żaden.
O ile to możliwe, na zakończenie okresu ekspozycji, który na ogół trwa cztery godziny, pozostałości substancji badanej powinny zostać usunięte przy użyciu wody lub innego odpowiedniego rozcieńczalnika, w sposób pozwalający na uniknięcie zmian w zaistniałej reakcji oraz nienaruszenie naskórka.
1.4.2.2. Poziom dawki
Aplikowana jest dawka 0,5 ml cieczy lub 0,5 g ciała stałego lub pasty na badany obszar skóry.
1.4.2.3. Badanie wstępne (Badanie in vivo podrażnienia/korozji skóry przy użyciu jednego zwierzęcia)
Stanowczo zaleca się, aby badania in vivo były przeprowadzane wstępnie przy użyciu jednego zwierzęcia, zwłaszcza w przypadku podejrzeń, że substancja jest potencjalnie korozyjna. Jest to zgodne z sekwencyjną strategią badawczą (zob. załącznik 1).
Jeżeli oceniono, że substancja może mieć właściwości korozyjne na podstawie analizy wagi dowodów, nie są wymagane dalsze badania na zwierzętach. W przypadku większości substancji, w odniesieniu do których istnieje podejrzenie korozyjności, nie wymagane są zazwyczaj dodatkowe badania in vivo. Jednakże w przypadkach gdzie istnieje potrzeba dostarczenia dodatkowych danych z uwagi na niewystarczające dowody, mogą zostać przeprowadzone ograniczone badania na zwierzętach z zastosowaniem następującej procedury: na skórę zwierzęcia nakłada się maksymalnie do trzech łat testowych. Pierwsza łata jest usuwana po trzech minutach. Jeżeli nie zaobserwowano żadnych poważnych reakcji skórnych, nakładana jest druga łata i usuwana po godzinie. Jeżeli obserwacje na tym etapie wskazują na to, że ekspozycja może być humanitarnie przeprowadzana, należy wydłużyć jej czas do czterech godzin, trzecia łata jest nakładana i zdejmowana po czterech godzinach, a reakcja punktowana.
Jeżeli działanie korozyjne jest stwierdzone po jednej z trzech sekwencyjnych ekspozycji, badanie jest natychmiast przerywane. W przypadku niestwierdzenia działania korozyjnego po usunięciu ostatniej łaty, zwierzę jest poddawane obserwacji w okresie 14 dni, o ile wcześniej nie rozwinie się korozja.
W przypadku gdy nie przewiduje się, że substancja badana może powodować korozję, lecz może wywoływać podrażnienia, powinna ona zostać zaaplikowana jednemu zwierzęciu na czas czterech godzin.
1.4.2.4. Badanie potwierdzające (Badanie in vivo podrażnienia skórnego przy wykorzystaniu dodatkowych zwierząt)
W przypadku niestwierdzenia działań korozyjnych w badaniach wstępnych działanie podrażniające lub jego brak należy potwierdzić przy wykorzystaniu maksymalnie dwóch dodatkowych zwierząt. Każde powinno zostać zbadane przy pomocy jednej łaty w czterogodzinnym okresie ekspozycji. W przypadku stwierdzenia działania podrażniającego w badaniu wstępnym, badanie potwierdzające może zostać przeprowadzone w sposób sekwencyjny lub poprzez ekspozycję dwóch dodatkowych zwierząt jednocześnie. W wyjątkowym przypadku nieprzeprowadzania badania wstępnego, można zbadać dwa lub trzy zwierzęta, z których każde zostanie poddane działaniu jednej łaty, usuniętej następnie po czterech godzinach. Jeżeli w przypadku wykorzystywania dwóch zwierząt, obydwa wykazują te same reakcje, nie wymaga się przeprowadzania dalszych badań. W innym wypadku zbadane zostaje również trzecie zwierzę. Niejednoznaczna reakcja może wymagać oceny z wykorzystaniem dodatkowych zwierząt.
1.4.2.5. Okres obserwacji
Długość trwania obserwacji powinien być wystarczający do pełnej oceny odwracalności zaobserwowanych efektów. Jednakże eksperyment powinien zostać przerwany, skoro tylko zwierzę zacznie wykazywać trwałe objawy silnego bólu lub strachu. W celu określenia odwracalności skutków zwierzę powinno być obserwowane maksymalnie przez okres 14 dni od usunięcia łat. W przypadku zaobserwowania odwracalności przed upływem 14 dni, eksperyment powinien zostać przerwany.
1.4.2.6. Obserwacja kliniczna i ocena punktowa reakcji skóry
Zwierzęta powinny zostać zbadane pod kątem wystąpienia objawów rumienia i obrzęku oraz reakcji odnotowanych po 60 minutach, a następnie po 24, 48 oraz 72 godzinach od usunięcia łaty. W badaniu wstępnym przeprowadzanym na jednym zwierzęciu miejsce zaaplikowania substancji badanej jest badane również natychmiast po usunięciu łaty. Reakcje skórne są oceniane przy pomocy wartości punktowych i zapisywane zgodnie z punktacją podaną w tabeli poniżej. W przypadku uszkodzenia naskórka, którego nie można zidentyfikować jako podrażnienia ani korozji po 72 godzinach, w celu określenia odwracalności efektów konieczne może okazać się prowadzenie obserwacji do 14 dnia. Oprócz spostrzeżeń na temat podrażnień należy w pełni opisać i zarejestrować wszelkie miejscowe skutki toksyczne, takie jak odtłuszczenie skóry oraz wszelkie ogólnoustrojowe niekorzystne skutki (np. skutki klinicznych objawów zatrucia i utrata masy ciała). Celem wyjaśnienia niejednoznacznych reakcji można posłużyć się badaniem histopatologicznym.
Ocena punktowa reakcji skóry jest z konieczności subiektywna. Celem propagowania harmonizacji oceny punktowej reakcji skóry oraz zapewnienia pomocy laboratoriom badawczym oraz osobom zaangażowanym w prowadzenie oraz interpretację obserwacji, personel prowadzący obserwacje powinien być odpowiednio przeszkolony w zakresie stosowanego systemu oceny (zob. tabela poniżej). Pomocne mogą okazać się ilustrowane instrukcje dotyczące klasyfikacji podrażnień i innych zmian skóry (9).
2. DANE
2.1. PREZENTACJA WYNIKÓW
Streszczenie wyników badań powinno zostać przedstawione w formie tabeli w sprawozdaniu końcowym z badań, która powinna zawierać wszystkie pozycje wymienione w sekcji 3.1.
2.2. OCENA WYNIKÓW
Wartości punktowe podrażnienia skóry winny być ocenianie stosownie do natury oraz natężenia zmian, oraz w świetle ich odwracalności lub jej braku. Poszczególne wyniki nie stanowią normy bezwzględnej właściwości podrażniających danego materiału, ponieważ ocenie poddawane są również pozostałe skutki ekspozycji na badany materiał. Alternatywnie, poszczególne wyniki powinny być traktowane jako wartości odniesienia, które muszą być oceniane w świetle wszystkich pozostałych spostrzeżeń z badań.
W ocenie reakcji podrażnienia pod uwagę powinna być brana odwracalność zmian skóry. Jeżeli reakcje takie jak alopecja (na ograniczonej powierzchni), hiperkeratoza, hiperplazja i łuszczenie są nadal obecne wraz z upływem 14-dniowego okresu obserwacji, substancja badana powinna zostać uznana za drażniącą.
3. SPRAWOZDANIE
3.1. SPRAWOZDANIE Z BADAŃ
Sprawozdanie z badań musi zawierać następujące informacje:
|
|
Uzasadnienie badań in vivo: analizę wagi dowodów, włączając wyniki realizacji strategii badań sekwencyjnych:
|
|
|
Substancja badana:
|
|
|
Nośnik:
|
|
|
Badane zwierzęta:
|
|
|
Warunki badań:
|
|
|
Wyniki:
|
|
|
Omówienie wyników |
4. BIBLIOGRAFIA
|
(1) |
Barratt, M.D., Castell, J.V., Chamberlain, M., Combes, R.D., Dearden, J.C., Fentem, J.H., Gerner, I.,Giuliani, A., Gray, T.J.B., Livingston, D.J., Provan, W.M., Rutten, F.A.J.J.L., Verhaar, H.J.M., Zbinden, P.(1995) The Integrated Use of Alternative Approaches for Predicting Toxic Hazard. ECVAM Workshop Report 8. ATLA 23,410–429. |
|
(2) |
Young, J.R., How, M.J., Walker, A.P., Worth W.M.H. (1988) Classification as Corrosive or Irritant to Skin of Preparations Containing Acidic or Alkaline Substance Without Testing on Animals. Toxicol. InVitro, 2, 19–26. |
|
(3) |
Worth, A.P., Fentem, J.H., Balls, M., Botham, P.A., Curren, R.D., Earl, L.K., Esdaile, D.J., Liebsch, M. (1998) Evaluation of the proposed OECD Testing Strategy for skin corrosion. ATLA 26, 709–720. |
|
(4) |
ECETOC (1990) Monograph No. 15, „Skin Irritation”, European Chemical Industry, Ecology and Toxicology Centre, Brussels. |
|
(5) |
Fentem, J.H., Archer, G.E.B., Balls, M., Botham, P.A., Curren, R.D., Earl, L.K., Esdaile, D.J., Holzhutter, H.G. and Liebsch, M. (1998) The ECVAM international validation study on in vitro tests for skin corrosivity. 2. Results and evaluation by the Management Team. Toxicology in Vitro 12, pp. 483–524. |
|
(5a) |
Testing Method B.40 Skin Corrosion. |
|
(6) |
OECD (1996) OECD Test Guidelines Programme: Final Report of the OECD Workshop on Harmonization of Validation and Acceptance Criteria for Alternative Toxicological Test Methods. Held in Solna, Sweden, 22–24 January 1996 (http://www1.oecd.org/ehs/test/background.htm). |
|
(7) |
OECD (1998) Harmonized Integrated Hazard Classification System for Human Health and Environmental Effects of Chemical Substances, as endorsed by the 28th Joint Meeting of the Chemicals Committee and the Working Party on Chemicals, November 1998 (http://www1.oecd.org/ehs/Class/HCL6.htm). |
|
(8) |
OECD (2000). Guidance Document on the Recognition, Grade and Use of Clinical Signs as Humane Endpoints for Experimental Animals Used in Safety Evaluation. OECD Environmental Health and Safety Publications. Series on Testing and Grading No. 19 (http://www1.oecd.org/ehs/test/monos.htm). |
|
(9) |
EPA (1990). Atlas of Dermal Lesions, (20T-2004). United States Environmental Protection Agency, Office of Pesticides and Toxic Substances, Washington, DC, August 1990. [Dostępne na wniosek w sekretariacie OECD]. |
Tabela I
PUNKTOWA OCENA REAKCJI SKÓRY
Tworzenie rumienia i obrzęku
|
Brak rumienia … |
0 |
|
Bardzo lekki rumień (prawie niewidoczny)… |
1 |
|
Wyraźny rumień… |
2 |
|
Rumień umiarkowany do mocnego… |
3 |
|
Mocny rumień (czerwień przypominająca barwę mięsa czerwonego) do form obrzęku wykluczających klasyfikację zmiany jako rumienia… |
4 |
Maksymalna możliwa wartość punktowa: 4
Tworzenie się obrzęku
|
Brak obrzęku… |
0 |
|
Bardzo słaby obrzęk (prawie niewidoczny)… |
1 |
|
Lekki obrzęk (dobrze wyodrębniony)… |
2 |
|
Umiarkowany obrzęk (wzniesienie na około 1 mm)… |
3 |
|
Mocny obrzęk (wniesienie ponad 1 mm, sięgający poza obszar eksponowania) … |
4 |
Maksymalna możliwa wartość punktowa: 4
Celem wyjaśnienia reakcji niejednoznacznych można przeprowadzić badanie histopatologiczne.
ZAŁĄCZNIK
Strategia badań sekwencyjnych podrażnienia i korozji skóry
ROZWAŻANIA OGÓLNE
Zarówno w dobrze pojętym interesie nauki, jak i dobrostanu zwierząt, istotne jest uniknięcie niepotrzebnego wykorzystywania zwierząt oraz minimalizacja badań, które mogą powodować silne reakcje u zwierząt. Wszystkie informacje na temat danej substancji ważne z punktu widzenia jej potencjalnego działania podrażniającego/korozyjnego na skórę powinny zostać poddane ocenie przed podjęciem badań in vivo. Wystarczające dowody, pozwalające na sklasyfikowanie substancji badanej jako potencjalnie korozyjnej lub żrącej, mogą być dostępne na dzień dzisiejszy, skutkiem czego zbyteczne będzie przeprowadzanie badań na zwierzętach laboratoryjnych. Dlatego wykorzystanie analizy wagi dowodów pochodzących z istniejących danych oraz strategii badań sekwencyjnych zminimalizuje potrzebę badań in vivo, zwłaszcza jeżeli substancja może powodować silne reakcje.
Zalecane jest wykorzystanie analizy wagi dowodów celem oceny istniejących informacji dotyczących podrażnienia i korozji skóry przez substancje, a także celem stwierdzenia, czy należy przeprowadzić dodatkowe badania, inne niż badania skóry in vivo, celem scharakteryzowania takiego potencjalnego działania substancji. Jeżeli niezbędne są dalsze badania, zaleca się realizowanie strategii badań sekwencyjnych celem uzyskania odpowiednich danych doświadczalnych. W odniesieniu do substancji, które nie posiadają historii badań, strategia badań sekwencyjnych powinna zostać zastosowana w celu uzyskania kompletu danych potrzebnych do oceny potencjalnego działania korozyjnego/podrażniającego danej substancji na skórę. Strategia badań opisana w niniejszym załączniku została opracowana w trakcie warsztatów OECD (1), a następnie potwierdzona i rozszerzona w ramach Zharmonizowanego Zintegrowanego Systemu klasyfikacji substancji chemicznych zagrażających zdrowiu ludzkiemu i środowisku, zatwierdzonym w wyniku prac w ramach 28. Wspólnego spotkania Komitetu ds. Związków Chemicznych i Grupy Roboczej ds. Związków Chemicznych w listopadzie 1998 roku (2).
Pomimo że strategia badań sekwencyjnych nie jest integralną częścią metody badawczej B.4, utożsamia ona zalecane podejście do określania cech korozji/podrażnienia skóry. Wspomniane podejście reprezentuje zarówno najlepszą praktykę, jak i wzorzec etyczny dla badań in vivo podrażnień/korozji skóry. Omawiana metoda badawcza stanowi wytyczne dotyczące prowadzenia badań in vivo i streszcza czynniki, jakim należy poświęcić uwagę przed rozpoczęciem takich badań. Strategia określa podejście do oceny istniejących danych na temat właściwości podrażniających/korozyjnych substancji badanej na skórę oraz podejście warstwowe do generowania odpowiednich danych dotyczących substancji wymagających dodatkowych badań lub takich, w odniesieniu do których nie przeprowadzono jak dotąd badań. W ramach strategii zalecane jest ponadto przeprowadzenie zweryfikowanych i zaakceptowanych badań in vivo lub ex vivo korozji/podrażnienia skóry w określonych okolicznościach.
OPIS STRATEGII OCENY I BADAŃ
Przed podjęciem badań w ramach strategii badań sekwencyjnych (rysunek) należy poddać ocenie wszystkie dostępne informacje w celu określenia potrzeby prowadzenia badań in vivo na skórze. Chociaż istotne informacje mogą zostać uzyskane na podstawie oceny pojedynczych parametrów (np. wartości skrajnych pH), należy wziąć pod uwagę ogół istniejących informacji. Należy poddać ocenie wszystkie dane dotyczące skutków działania danej substancji lub substancji analogicznych, w podejmowaniu decyzji dotyczącej wagi dowodów, oraz należy przedstawić racjonalne uzasadnienie powziętej decyzji. Główny nacisk należy położyć na istniejące dane na temat działania substancji na ludzi oraz zwierzęta, a w następnej kolejności na wyniki badań in vivo lub ex vivo. O ile to możliwe, należy uniknąć badań in vivo substancji powodujących korozję. Czynniki uwzględniane w omawianej strategii badań obejmują:
Ocenę istniejących danych na temat działania substancji na ludzi i zwierzęta (etap 1). W pierwszej kolejności należy wziąć pod uwagę istniejące dane dotyczące działania na ludzi, np. badania kliniczne lub dane na temat stanowisk pracy, a także studia konkretnych przypadków oraz/lub dane pochodzące z badań przeprowadzonych na zwierzętach, np. pojedyncze lub wielokrotne badania ekspozycji na substancje toksyczne, ponieważ dostarczają informacji bezpośrednio związanych z oddziaływaniem na skórę. Substancje o znanym działaniu podrażniającym lub korozyjnym, a także substancje wyraźnie niewywierające skutków korozyjnych lub podrażniających nie muszą być badane in vivo.
Analiza zależności aktywności i struktury (SAR) (etap 2). Należy uwzględnić wyniki badań substancji pokrewnych pod względem budowy, o ile są one dostępne. W przypadku dostępności wystarczających danych na temat oddziałania na ludzi i/lub zwierzęta substancji pokrewnych pod względem budowy lub mieszanin takich substancji, wskazujących na ich potencjalne działanie korozyjne/podrażniające na skórę, można domniemywać, że substancja badana poddawana ocenie spowoduje taką samą reakcję. W takich przypadkach substancja ta nie musi być poddawana badaniu. Negatywne wyniki z badań na temat substancji pokrewnych pod względem budowy lub mieszanin takich substancji nie są wystarczającym dowodem na brak działań podrażniających lub korozyjnych danej substancji w świetle strategii badań sekwencyjnych. Zweryfikowane i zaakceptowane podejście SAR powinno zostać wykorzystane w celu zidentyfikowania potencjalnego oddziałania korozyjnego i podrażniającego na skórę.
Właściwości fizyko-chemiczne i reaktywność chemiczna (etap 3). Substancje charakteryzujące się skrajnym pH ≥ 2,0 i ≤ 11,5 mogą mieć silne działanie miejscowe. Jeżeli takie skrajne pH jest podstawą do identyfikacji substancji jako korozyjnej dla skóry, możliwe jest wykorzystanie jej rezerwy kwasowej/alkalicznej (tj. zdolności buforowej) (3)(4). Jeżeli zdolność buforowa wskazuje na to, że substancja może nie być korozyjna dla skóry, należy przeprowadzić dalsze badania w celu potwierdzenia tej cechy, najlepiej przy użyciu zweryfikowanych i zaakceptowanych badań in vivo lub ex vivo (zob. etapy 5 i 6).
Toksyczność skórna (etap 4). Jeżeli dana substancja chemiczna okazała się być bardzo toksyczna na skórę, badanie in vivo podrażnień/korozji skórnej może okazać się niemożliwe do przeprowadzenia, ponieważ ilość substancji badanej normalnie podawanej mogłaby przekroczyć bardzo toksyczną dawkę, co w konsekwencji powodowałoby śmierć lub silne cierpienie zwierząt. Dodatkowo, jeżeli badania toksyczności skórnej na białych królikach już zostały przeprowadzone do poziomu dawki granicznej 2 000 mg/kg masy ciała lub wyższego, i nie zaobserwowano skutków korozyjnych lub podrażniających, dodatkowe badania podrażnienia/korozji skóry mogą okazać się niepotrzebne. Należy uwzględnić wiele czynników w trakcie oceny ostrej toksyczności skórnej w przeprowadzonych uprzednio badaniach. Na przykład przedstawione informacje na temat zmian skóry mogą być niekompletne. Badania i obserwacje mogły być przeprowadzone na innych niż króliki gatunkach, które mogą wykazywać znaczące różnice w zakresie wrażliwości reakcji. Także postać substancji badanej zaaplikowanej zwierzętom mogła być nieodpowiednia do oceny działania korozyjnego/podrażniającego na skórę (np. poziom rozcieńczenia substancji do badań toksyczności skórnej (5)). Jednakże w przypadku właściwie zaprojektowanych i przeprowadzonych badań toksyczności u królików wyniki negatywne mogą stanowić wystarczające dowody na to, że dana substancja nie jest korozyjna lub podrażniająca.
Wyniki badań in vitro lub ex vivo (etap 5 i 6). Substancje, które wykazują właściwości korozyjne lub silnie podrażniające w zweryfikowanych i zaakceptowanych badaniach in vitro lub ex vivo (6)(7), przewidziane do oceny tych specyficznych skutków, nie muszą być badane na zwierzętach. Można przyjąć, że substancje te dadzą podobne wyniki in vivo.
Badania in vivo na królikach (etap 7 i 8). W przypadku podjęcia, na podstawie analizy wagi dowodów, decyzji dotyczącej przeprowadzenia badań in vivo, badanie takie powinno rozpocząć się badaniem wstępnym na jednym zwierzęciu. Jeżeli wyniki tego badania wskazują, że substancja ma działanie korozyjne na skórę, dalsze badania powinny zostać wstrzymane. W przypadku niestwierdzenia działania korozyjnego w badaniu wstępnym reakcja podrażniająca lub negatywna powinna zostać potwierdzona przy użyciu maksymalnie dwóch dodatkowych zwierząt poddanych ekspozycji przez czas czterech godzin. W przypadku wystąpienia podrażnienia we wstępnym badaniu należy przeprowadzić sekwencyjne badanie potwierdzające lub jednoczesną ekspozycję obydwu zwierząt.
BIBLIOGRAFIA
|
(1) |
OECD (1996). Test Guidelines Programme: Final Report on the OECD Workshop on Harmonization of Validation and Acceptance Criteria for Alternative Toxicological Test method. Held on Solna, Sweden, 22–24 January 1996 (http://www 1 .oecd.org/ehs/test/background.htm). |
|
(2) |
OECD (1998). Harmonized Integrated Hazard Classification System for Human Health and Environmental Effects of Chemical Substances, as endorsed by the 28th Joint Meeting of the Chemicals Committee and the Working Party on Chemicals, November 1998 (http://www1.oecd.org/ehs/Class/HCL6.htm). |
|
(3) |
Worth, A.P., Fentem J.H., Balls M., Botham P.A., Curren R.D., Earl L.K., Esdail D.J., Liebsch M. (1998). An Evaluation of the Proposed OECD Testing Strategy for Skin Corrosion. ATLA 26, 709–720. |
|
(4) |
Young, J.R., How, M.J., Walker, A.P., Worth, W.M.H. (1988). Classification as Corrosive or Irritant to Skin of Preparations Containing Acidic or Alkaline Substances, Without Testing on Animals. Toxic In Vitro, 2 (1) pp 19–26. |
|
(5) |
Patil, S.M., Patrick, E., Maibach, H.I. (1996) Animal, Human, and In Vitro Test Method for Predicting Skin Irritation, in: Francis N. Marzulli and Howard I. Maibach (editors): Dermatotoxicology. Fifth Edition ISBN 1- 56032-356-6, Chapter 31,411–436. |
|
(6) |
Testing Method B.40. |
|
(7) |
Fentem, J.H., Archer, G.E.B., Balls, M., Botham, P.A., Curren, R.D., Earl, L.K., Edsail, D.J., Holzhutter, H.G. and Liebsch, M. (1998) The ECVAM international validation study on in vitro tests for skin corrosivity. 2. Results and evaluation by the Management Team. Toxicology in Vitro 12, pp. 483–524. |
RYSUNEK STRATEGIA BADAŃ I OCENY PODRAŻNIEŃ/KOROZJI SKÓRY

B.5. TOKSYCZNOŚĆ OSTRA: PODRAŻNIENIE/KOROZJA OCZU
1. METODA
Niniejsza metoda jest kopią OECD TG 405 (2002).
1.1. WPROWADZENIE
W przygotowywaniu niniejszej zaktualizowanej metody specjalną uwagę poświęcono możliwym ulepszeniom w zakresie zagadnień dotyczących dobrostanu zwierząt, a także ocenie wszystkich dostępnych informacji na temat substancji badanej, w celu uniknięcia niepotrzebnych badań na zwierzętach laboratoryjnych. Niniejsza metoda zawiera zalecenie, zgodnie z którym przed przystąpieniem do opisywanego badania in vivo na korozję/podrażnienia oka wywołane przez daną substancję zostanie przeprowadzona analiza wagi dowodów uzyskanych na podstawie istniejących danych (1). Jeżeli dostępne dane są niewystarczające, mogą zostać opracowane przy pomocy badań sekwencyjnych (2)(3). Strategia badań zawiera wykaz zweryfikowanych i zaakceptowanych badań in vitro i stanowi załącznik do niniejszej metody. Dodatkowo zalecane jest przeprowadzenie badań in vivo podrażnień/korozji skóry w celu oszacowania korozji oka przed przystąpieniem do badań in vivo oka.
Z uwagi na dobrze pojęty interes nauki i dobrostan zwierząt badania in vivo nie powinny być podejmowane, jeżeli wszystkie dostępne dane odnoszące się do potencjalnego działania korozyjnego/podrażniającego skórę w wyniku działania substancji nie zostały uzyskane z analizy wagi dowodów. Takie dane będą zawierały dowody z wykonanych badań na ludziach oraz/lub zwierzętach laboratoryjnych, dowody korozji/podrażnień wywołanych przez jedną lub wiele substancji pokrewnych pod względem budowy lub mieszanin takich substancji, dane wskazujące na silną kwasowość lub alkaliczność substancji (4)(5) oraz wyniki zweryfikowanych i zaakceptowanych badań in vivo lub ex vivo (6)(6a). Takie badania mogą być przeprowadzone przed lub w następstwie analizy wagi dowodów.
W przypadku pewnych substancji takie analizy mogą wskazywać na potrzebę badań in vivo potencjalnego korozyjnego/drażniącego działania danej substancji na oko. W każdym takim przypadku przed podjęciem badań in vivo oka zostaną przeprowadzone najpierw badania in vivo skutków działania substancji na skórę, zgodnie z metodą badawczą B.4 (7). Zastosowanie analizy wagi dowodów i strategii badań sekwencyjnych powinno ograniczyć potrzebę prowadzenia badań in vivo pod kątem korozji/podrażnienia oka przez substancje, w odniesieniu do których już istnieje wystarczający materiał dowodowy, zgromadzony w wyniku prowadzenia innych badań. Jeżeli nie jest możliwe określenie potencjalnego działania korozyjnego lub drażniącego na oka przy zastosowaniu strategii badań sekwencyjnych, nawet po przeprowadzeniu badań in vivo pod kątem korozji i podrażnienia skóry, można przeprowadzić badania in vivo pod kątem korozji/podrażnienia oka.
Załącznik do niniejszej metody zawiera preferowaną strategię badań sekwencyjnych, obejmującą przeprowadzenie zweryfikowanych i zaakceptowanych badań in vivo i ex vivo działania korozyjnego/podrażniającego. Strategia ta została opracowana i jednomyślnie zalecona przez uczestników warsztatów OECD (8), a następnie zatwierdzona jako zalecana strategia badań w ramach Globalnego Zharmonizowanego Systemu klasyfikacji związków chemicznych (GHS) (9). Zaleca się realizację tej strategii badań przed podjęciem badań in vivo. Dla nowych substancji strategia ta jest zalecanym stopniowym podejściem badawczym służącym opracowywaniu wartościowych z naukowego punktu widzenia danych dotyczących korozyjnego/podrażniającego działania substancji. W odniesieniu do istniejących substancji, na temat których brak wystarczających danych co do ich działania korozyjnego/podrażniającego, strategia ta powinna zostać wykorzystana w celu uzupełnienia brakujących danych. Użycie innych strategii badawczych bądź procedur lub decyzja o nieprzyjęciu stopniowego podejścia badawczego wymaga uzasadnienia.
1.2. DEFINICJE
Podrażnienie oka: zmiany w oku spowodowane zaaplikowaniem substancji badanej na wierzchnią warstwę oka, które w pełni odwracalne w ciągu 21 dni po zaaplikowaniu.
Korozja oka: uszkodzenie tkanki oka lub poważne fizyczne uszkodzenie wzroku, spowodowane zaaplikowaniem substancji badanej na wierzchnią warstwę oka, które nie jest w pełni odwracalne w ciągu 21 dni po zaaplikowaniu.
1.3. ZASADA METODY BADAWCZEJ
Substancja, która ma być zbadana, jest nakładana na jedno oko zwierzęcia doświadczalnego, drugie oko jest wykorzystywane do badania kontrolnego. Stopień korozji/podrażnienia jest oceniany poprzez ocenę punktową zmian spojówki, rogówki oraz tęczówki w określonych odstępach. Inne zmiany w oku i niekorzystne skutki ogólnoustrojowe są również opisywane w celu uzyskania kompletnej oceny skutków. Długość trwania badania powinien być odpowiedni do oszacowania odwracalności lub nieodwracalności obserwowanych skutków.
Zwierzęta wykazujące ciągłe objawy silnego strachu i/lub bólu na jakimkolwiek etapie badania powinny zostać uśmiercone w sposób humanitarny, a substancja odpowiednio oceniona. Kryteria dotyczące podjęcia decyzji o uśmierceniu w sposób humanitarny zwierząt w stanie agonalnym i silnie cierpiących zawiera załącznik (10).
1.4. OPIS METODY BADAWCZEJ
1.4.1. Przygotowanie do badań in vivo
1.4.1.1. Wybór gatunków zwierząt
Biały królik jest preferowanym gatunkiem zwierząt laboratoryjnych. Wykorzystywane są zdrowe młode dorosłe króliki. W przypadku wykorzystania innych gatunków należy podać uzasadnienie.
1.4.1.2. Przygotowanie zwierząt
Obydwoje oczu zwierzęcia doświadczalnego wstępnie wybranego do badania powinny być zbadane na 24 godziny przed przystąpieniem do badań. Zwierzęta z objawami podrażnienia oczu, wad wzroku lub uszkodzeń rogówki nie powinny być wykorzystywane w badaniu.
1.4.1.3. Warunki przetrzymywani i karmienia
Zwierzęta powinny być przetrzymywane oddzielnie. Temperatura w badawczych pomieszczeniach hodowlanych powinna wynosić 20 oC (± 3 oC) dla królików. Chociaż wilgotność względna powinna wynosić co najmniej 30 % i pożądane jest, żeby nie przekraczała 70 % poza czasem czyszczenia pomieszczenia, celem powinno być utrzymywanie jej na poziomie 50–60 %. Oświetlenie powinno być sztuczne, w cyklu 12 godzin jasno, 12 godzin ciemno. Do żywienia można stosować konwencjonalne pasze laboratoryjne z nieograniczonym dostępem do wody pitnej.
1.4.2. Procedura badań
1.4.2.1. Podawanie substancji
Substancja badana powinna zostać umieszczona na spojówce jednego oka każdego ze zwierząt po delikatnym odsunięciu powieki z gałki ocznej. Powieki są następnie delikatnie zamykane na około jedną sekundę w celu zapobieżenia utraty materiału badanego. Drugie, nienaruszone oko jest przedmiotem badania kontrolnego.
1.4.2.2. Nawilżanie
Oczy zwierząt badanych nie powinny być przepłukiwane przez przynajmniej 24 godziny po wkropleniu substancji badanej, z wyjątkiem badania ciał stałych (zob. sekcja 1.4.2.3.2) i przypadków natychmiastowego działania korozyjnego lub podrażniającego. O ile to stosowne, po 24 godzinach można przepłukać oczy.
Włączanie dodatkowej satelickiej grupy zwierząt w celu zbadania skutków przepłukiwania nie jest zalecane, chyba że ma to uzasadnienie naukowe. W przypadku potrzeby grupy satelickiej należy użyć dwóch królików. Warunki przepłukiwania powinny być dokładnie dokumentowane np. czas przepłukiwania, skład i temperatura roztworu, długość trwania, objętość i szybkość aplikowania.
1.4.2.3. Poziom dawki
1.4.2.3.1. Badanie cieczy
W badaniu cieczy stosuje się dawkę o objętości 0,1 ml. Nie powinno się stosować cieczy pod ciśnieniem do aplikowania substancji bezpośrednio na oko. Struga cieczy powinna zostać rozprężona i zebrana w zbiorniku przed aplikowaniem jej 0,1 ml na oko.
1.4.2.3.2. Badanie substancji stałych
W badaniu ciał stałych, past i cząstek stałych stosowana ilość powinna mieć objętość 0,1 ml lub wagę nie większą niż 100 mg. Badany materiał powinien zostać sproszkowany. Objętość ciała stałego powinna być mierzona po delikatnym zbiciu np. poprzez ostukanie pojemnika pomiarowego. Jeżeli substancja badana w formie ciała stałego nie została usunięta w wyniku działania mechanizmów fizjologicznych w pierwszym punkcie czasowym obserwacji po upłynięciu godziny, oko może zostać przepłukane roztworem soli fizjologicznej lub wodą destylowaną.
1.4.2.3.3. Badanie aerozoli
Zaleca się, aby wszystkie strugi cieczy pod ciśnieniem i aerozole zostały rozprężone i zebrane przed osadzeniem na oku. Wyjątek stanowią substancje w aerozolach znajdujące się w pojemnikach pod ciśnieniem, które nie mogą zostać zebrane z powodu ich parowania. W takich przypadkach należy przytrzymać oko w pozycji otwartej i zaaplikować substancję badaną do oka jednym podmuchem, trwającym około sekundy, z odległości 10 cm prostopadle do oka. Ta odległość może zależeć w znacznym stopniu od ciśnienia w rozpylaczu i jego zawartości. Należy uważać, aby nie doszło do uszkodzenia oka z powodu ciśnienia w rozpylaczu. W odpowiednich przypadkach, może być niezbędna ocena możliwości potencjalnego „mechanicznego” uszkodzenia oka spowodowanego siłą rozpylacza.
Oszacowanie dawki aerozolu może być wykonane poprzez następującą symulację badania: substancja jest napylana na papierek wagowy przez otwór o rozmiarze oka królika znajdujący się bezpośrednio przed papierkiem. Poziom wzrostu wagi papierka wykorzystuje się celem określenia przybliżonej ilości jaka byłaby zaaplikowana do oka. Dla substancji lotnych, dawka może zostać oszacowana poprzez zważenie pojemnika odbiorczego przed i po badanego materiału.
1.4.2.4. Badanie wstępne (Badanie in vivo podrażnienia/korozji oka z wykorzystaniem jednego zwierzęcia)
Zgodnie ze strategią badań sekwencyjnych (zob. załącznik 1) stanowczo zaleca się, aby badania in vivo były przeprowadzane wstępnie przy użyciu jednego zwierzęcia
Jeżeli wyniki takich badań wskazują, że substancja ma właściwości korozyjne lub powoduje silne podrażnienia oka, procedura przewiduje zaprzestanie dalszych badań podrażnień oczu.
1.4.2.5. Znieczulenie miejscowe
Znieczulenie miejscowe może zostać zastosowane na podstawie analizy wyników uzyskanych w indywidualnych przypadkach. Jeżeli analiza wagi dowodów wskazuje na to, że substancja może potencjalnie powodować ból, lub badania wstępne pozwalają sądzić, że pojawią się bolesne reakcje, można zastosować miejscowe znieczulenie przed zaaplikowaniem substancji badanej. Rodzaj, stężenie i dawka znieczulenia miejscowego powinny zostać uważnie wybrane, tak aby zapewnić, że różnice w reakcji na substancję badaną nie są wynikiem zastosowania znieczulenia. Oko kontrolne powinno również zostać znieczulone.
1.4.2.6. Badanie potwierdzające (Badanie in vivo podrażnienia oka na dodatkowych zwierzętach)
W przypadku niestwierdzenia działań korozyjnych w badaniu wstępnym działanie podrażniające lub jego brak należy potwierdzić przy wykorzystaniu maksymalnie dwóch dodatkowych zwierząt. Jeżeli obserwuje się silne działanie podrażniające w badaniu wstępnym, zaleca się, aby badanie potwierdzające zostało przeprowadzone w sposób sekwencyjny na jednym zwierzęciu, a niejednocześnie na dwóch zwierzętach. Jeżeli działanie na drugim zwierzęciu okaże się korozyjne lub silnie podrażniające, badanie nie będzie kontynuowane. Dodatkowe zwierzę może być potrzebne w celu potwierdzenia słabego lub średniego działania podrażniającego.
1.4.2.7. Okres obserwacji
Długość trwania obserwacji powinien być wystarczający do oceny pełnej odwracalności obserwowanych efektów. Jednakże eksperyment powinien zostać przerwany w momencie, gdy zwierzę wykazuje objawy silnego bólu lub strachu (9). W celu określenia odwracalności efektów w normalnych okolicznościach zwierzę powinno być obserwowane przez 21 dni po zaaplikowaniu substancji badanej. Jeżeli odwracalność jest obserwowana przed upływem 21 dni, eksperyment powinien zostać przerwany w tym momencie.
1.4.2.7.1. Obserwacje kliniczne i ocena reakcji oka
Oczy powinny zostać zbadane po 1, 24, 48 i 72 godzinach od podania substancji badanej. Zwierzęta powinny być pod obserwacją nie dłużej niż to konieczne do uzyskania wszystkich informacji. Zwierzęta wykazujące objawy ciągłego silnego bólu lub strachu powinny zostać bezzwłocznie uśmiercone w sposób humanitarny, a substancja odpowiednio oceniona. Uśmiercone w sposób humanitarny powinny zostać zwierzęta z następującymi zmianami oka zaszłymi w następstwie zaaplikowania substancji: perforacja lub owrzodzenie rogówki, włączając garbiak; krwawienie z tylnej komory oka; zmętnienie rogówki równe 4 utrzymujące się 48 godzin; brak odbicia światła (reakcja rogówki o wartości punktowej 2) utrzymujący się 72 godziny; owrzodzenie błony spojówki; martwica spojówki lub błony mrużnej; lub oddzielająca się tkanka martwicza. Wynika to z faktu, że tego typu zmiany są na ogół nieodwracalne.
Zwierzęta, u których nie wystąpiły zmiany oczne, mogą zostać uśmiercone nie wcześniej niż 3 dni od zaaplikowania substancji. Zwierzęta z łagodnymi lub umiarkowanymi zmianami powinny przebywać pod obserwacją do czasu zaniknięcia zmian lub przez 21 dni, po których badanie zostaje przerwane. Obserwacje powinny być prowadzone 7., 14. i 21. dnia w celu określenia stanu zmian oraz ich odwracalności lub nieodwracalności.
Wartości punktowe reakcji oka (spojówki, rogówki i tęczówki) powinny być zapisywane dla każdego badania (tabela I). Należy również podać informacje na temat innych zmian oka (np. przekrwienie, plamy) lub innych niekorzystnych skutków ogólnoustrojowych.
Badanie reakcji może zostać wykonane przy zastosowaniu lupy stereoskopowej, ręcznej lampy szczelinowej, biomikroskopu lub innego odpowiedniego urządzenia. Po zapisaniu obserwacji w 24 godzinie oko może zostać poddane dalszym badaniom przy pomocy fluoresceiny.
Ocena odpowiedzi ocznej jest z konieczności subiektywna. W celu zharmonizowania oceny reakcji ocznej i w celu wsparcia laboratoriów badawczych i osób biorących udział w interpretacjach i obserwacjach należy przeprowadzić odpowiednie szkolenia w zakresie stosowanego systemu.
2. DANE
2.2. OCENA WYNIKÓW
Wartości punktowe podrażnienia skóry winny być ocenianie stosownie do natury oraz natężenia zmian, oraz w świetle ich odwracalności lub jej braku. Poszczególne wyniki nie stanowią normy bezwzględnej właściwości podrażniających danego materiału, ponieważ ocenie poddawane są również pozostałe skutki ekspozycji na badany materiał. Alternatywnie, poszczególne wyniki powinny być traktowane jako wartości odniesienia, które muszą być oceniane w świetle wszystkich pozostałych spostrzeżeń z badań.
3. SPRAWOZDANIE
3.1. SPRAWOZDANIE Z BADAŃ
Sprawozdanie z badań musi zawierać następujące informacje:
|
|
Uzasadnienie badań in vivo: analizę wagi dowodów, włączając wyniki realizacji strategii badań sekwencyjnych:
|
|
|
Substancja badana:
|
|
|
Nośnik:
|
|
|
Badane zwierzęta:
|
|
|
Warunki badań:
|
|
|
Omówienie wyników. |
3.2. INTERPRETACJA WYNIKÓW
Transponowanie wyników badań podrażnienia oka u zwierząt laboratoryjnych na ludzi ma sens tylko w ograniczonym stopniu. W wielu przypadkach białe króliki są bardziej wrażliwe niż ludzie na podrażnienia lub korozję oka.
W interpretacji danych należy zadbać o pominięcie podrażnienia oka będącego wynikiem infekcji wtórnej.
4. BIBLIOGRAFIA
|
(1) |
Barratt, M.D., Castell, J.V., Chamberlain, M., Combes, R.D., Dearden, J.C., Fentem, J.H., Gerner, I., Giuliani, A., Gray, T.J.B., Livingston, D.J., Provan, W.M., Rutten, F.A.J.J.L., Verhaar, H.J.M., Zbinden, P. (1995) The Integrated Use of Alternative Approaches for Predicting Toxic Hazard. ECVAM Workshop Report 8. ATLA 23,410–429. |
|
(2) |
de Silva, O., Cottin, M., Dami, N., Roguet, R., Catroux, P., Toufic, A., Sicard, C., Dossou, K.G., Gerner, I., Schlede, E., Spielmann, H., Gupta, K.C., Hill, R.N. (1997) Evaluation of Eye Irritation Potential: Statistical Analysis and Tier Testing Strategies. Food Chem. Toxicol 35,159–164. |
|
(3) |
Worth A.P. and Fentem J.H. (1999) A general approach for evaluating stepwise testing strategies ATLA 27, 161–177. |
|
(4) |
Young, J.R., How, M.J., Walker, A.P., Worth W.M.H. (1988) Classification as Corrosive or Irritant to Skin of Preparations Containing Acidic or Alkaline Substance Without Testing on Animals. Toxicol. In Vitro, 2, 19–26. |
|
(5) |
Neun, D.J. (1993) Effects of Alkalinity on the Eye Irritation Potential of Solutions Prepared at a Single pH. J. Toxicol. Cut. Ocular Toxicol. 12,227–231. |
|
(6) |
Fentem, J.H., Archer, G.E.B., Balls, M., Botham, P.A., Curren, R.D., Earl, L.K., Edsaile, D.J., Holzhutter, H.G. and Liebsch, M. (1998) The ECVAM international validation study on in vitro tests for skin corrosivity. 2. Results and evaluation by the Management Team. Toxicology in Vitro 12, pp. 483–524. |
|
(6a) |
Testing Method B.40 Skin Corrosion. |
|
(7) |
Testing Method B.4. Acute toxicity: dermal irritation/corrosion. |
|
(8) |
OECD (1996) OECD Test Guidelines Programme: Final Report of the OECD Workshop on Harmonization of Validation and Acceptance Criteria for Alternative Toxicological Test Methods. Held in Solna, Sweden, 22–24 January 1996 (http://www.oecd.org/ehs/test/background.htm). |
|
(9) |
OECD (1998) Harmonized Integrated Hazard Classification System for Human Health and Environmental Effects of Chemical Substances, as endorsed by the 28th Joint Meeting of the Chemicals Committee and the Working Party on Chemicals, November 1998 (http://www.oecd.org/ehs/Class/HCL6.htm). |
|
(10) |
Guidance Document on the Recognition, Grade and Use of Clinical Signs as Humane Endpoints for Experimental Animals Used in Safety Evaluation. OECD Environmental Health and Safety Publications. Series on Testing and Grading No. 19 (http://www.oecd.org/ehs/test/monos.htm). |
Tabela I
OCENA PUNKTOWA ZMIAN OKA
Rogówka
Zmętnienie: stopień gęstości (odczyt powinien dotyczyć obszaru o największej gęstości) (1)
|
Brak owrzodzenia lub zmętnienia |
0 |
|
Rozrzucone lub rozmyte obszary zmętnienia (inne od lekkiego zmatowienia normalnego połysku); |
1 |
|
wyraźnie widoczne szczegóły tęczówki Jasno określone obszary półprzezroczyste; lekko zaciemnione detale tęczówki |
2 |
|
Obszary o kolorze perłowym; brak widocznych szczegółów tęczówki; rozmiar źrenicy trudny do określenia |
3 |
|
Zmatowienie rogówka; tęczówka niewidoczna z powodu zmętnienia |
4 |
Maksymalna możliwa wartość punktowa: 4
UWAGI
Tęczówka
|
W normie |
0 |
|
Głęboko pomarszczona, zastój krwi, opuchlizna, przekrwienie okołorogówkowe; lub; tęczówka reaguje na światło (przyjmuje się, że powolna reakcja stanowi skutek) |
1 |
|
Krwawienie, silne zniszczenia, lub brak reakcji na światło |
2 |
Maksymalna możliwa wartość punktowa: 2
Spojówka
Zaczerwienienie (dotyczy spojówki powieki i gałki ocznej; wyłączając rogówkę i tęczówkę)
|
w normie |
0 |
|
Niektóre naczynia krwionośne przekrwione |
1 |
|
Rozmycie, kolor karmazynowy; pojedyncze naczynia trudno widoczne |
2 |
|
Rozmycie, kolor mięsa czerwonego |
3 |
Maksymalna możliwa wartość punktowa: 3
Obrzęk spojówki
Opuchlizna (dotyczy powiek i/lub błony mrużnej)
|
W normie |
0 |
|
Niewielka opuchlizna powyżej normy |
1 |
|
Widoczna opuchlizna z częściowym wynicowaniem powieki |
2 |
|
Opuchlizna, z powiekami zamkniętymi w połowie |
3 |
|
Opuchlizna, z powiekami zamkniętymi więcej niż w połowie |
4 |
Maksymalna możliwa wartość punktowa: 4
ZAŁĄCZNIK
Strategia badania sekwencyjnego podrażnień i korozji oka
ROZWAŻANIA OGÓLNE
Zarówno w dobrze pojętym interesie nauki, jak i z uwagi na dobrostan zwierząt, istotne jest uniknięcie niepotrzebnego wykorzystywania zwierząt oraz minimalizacja badań mogących powodować silne reakcje u zwierząt. Wszystkie informacje na temat danej substancji ważne z punktu widzenia jej potencjalnego działania podrażniającego/korozyjnego na oko, powinny zostać poddane ocenie przed podjęciem badań in vivo. Wystarczające dowody, pozwalające na sklasyfikowanie substancji badanej jako potencjalnie korozyjnej lub żrącej, mogą być dostępne już teraz, skutkiem czego zbyteczne będzie przeprowadzanie badań na zwierzętach laboratoryjnych. Dlatego wykorzystanie analizy wagi dowodów pochodzących z istniejących danych oraz strategii badań sekwencyjnych zminimalizuje potrzebę badań in vivo, zwłaszcza jeżeli substancja może powodować silne reakcje.
Zalecane jest wykorzystanie analizy wagi dowodów celem oceny istniejących informacji dotyczących podrażnienia i korozji skóry przez substancje, a także celem stwierdzenia, czy należy przeprowadzić dodatkowe badania, inne niż badania in vivo, celem scharakteryzowania takiego potencjalnego działania substancji. Jeżeli niezbędne są dalsze badania, zaleca się realizowanie strategii badań sekwencyjnych celem uzyskania odpowiednich danych doświadczalnych. W odniesieniu do substancji, które nie posiadają historii badań, strategia badań sekwencyjnych powinna zostać zastosowana w celu uzyskania kompletu danych potrzebnych do oceny potencjalnego działania korozyjnego/podrażniającego danej substancji na oczy. Strategia badań opisana w niniejszym załączniku została opracowana w trakcie warsztatów OECD (1), a następnie potwierdzona i rozszerzona w ramach Zharmonizowanego Zintegrowanego Systemu klasyfikacji substancji chemicznych zagrażających zdrowiu ludzkiemu i środowisku, zatwierdzonym w wyniku prac w ramach 28. Wspólnego spotkania Komitetu ds. Związków Chemicznych i Grupy Roboczej ds. Związków Chemicznych w listopadzie 1998 r. (2).
Pomimo że strategia badań sekwencyjnych nie jest integralną częścią metody badawczej B.5, utożsamia ona zalecane podejście do określania cech korozji/podrażnienia oczu. Wspomniane podejście reprezentuje zarówno najlepszą praktykę, jak i wzorzec etyczny dla badań in vivo podrażnień/korozji. Omawiana metoda badawcza stanowi wytyczne dotyczące prowadzenia badań in vivo i streszcza czynniki, jakim należy poświęcić uwagę przed rozpoczęciem takich badań. Strategia określa podejście do oceny istniejących danych na temat właściwości podrażniających/korozyjnych substancji badanej na oczy oraz podejście warstwowe do generowania odpowiednich danych dotyczących substancji wymagających dodatkowych badań, lub takich, w odniesieniu do których nie przeprowadzono jak dotąd badań. W ramach strategii zalecane jest ponadto przeprowadzenie zweryfikowanych i zaakceptowanych badań in vivo lub ex vivo, a dopiero w następnej kolejności zastosowanie metody badawczej badania korozji/podrażniania skóry B.4 w specyficznych okolicznościach (3)(4).
OPIS KROKOWEJ STRATEGII BADAŃ
Przed podjęciem badań w ramach strategii badań sekwencyjnych (rysunek), należy poddać ocenie wszystkie dostępne informacje w celu określenia potrzeby prowadzenia badań in vivo na oku. Chociaż istotne informacje mogą zostać uzyskane na podstawie oceny pojedynczych parametrów (np. wartości skrajnych pH), należy wziąć pod uwagę ogół istniejących informacji. Należy poddać ocenie wszystkie dane dotyczące skutków działania danej substancji, lub substancji analogicznych, w podejmowaniu decyzji dotyczącej wagi dowodów, oraz należy przedstawić racjonalne uzasadnienie powziętej decyzji. Główny nacisk należy położyć na istniejące dane na temat działania substancji na ludzi oraz zwierzęta, a w następnej kolejności na wyniki badań in vivo lub ex vivo. O ile to możliwe, należy uniknąć badań in vivo substancji powodujących korozję. Czynniki uwzględniane w omawianej strategii badań obejmują:
Oszacowanie wyników dotyczących ludzi i zwierząt (etap 1). W pierwszej kolejności należy wziąć pod uwagę istniejące dane dotyczące działania na ludzi, np. badania kliniczne lub dane na temat stanowisk pracy a także studia konkretnych przypadków oraz/lub dane pochodzące z badań przeprowadzonych na zwierzętach, np. pojedyncze lub wielokrotne badania ekspozycji na substancje toksyczne, ponieważ dostarczają informacji bezpośrednio związanych z oddziaływaniem na oko. Następnie należy poddać ocenie dostępne dane uzyskane w wyniku badań na ludziach/zwierzętach dotyczących działania drażniącego/korozji skóry. Substancje o znanym działaniu korozyjnym lub powodujących dotkliwe podrażnienia oczu nie powinny być aplikowane do oczu zwierząt, podobnie substancje powodujące działanie korozyjne i/lub podrażniające dla skóry. Takie substancje również powinny zostać uznane za korozyjne i/lub podrażniające oczy. Substancje, wobec których istnieją wystarczające dowody, uzyskane w poprzednich badaniach okulistycznych, świadczące o braku działania korozyjnego lub podrażniającego, również nie powinny być poddawane badaniu in vivo na oku.
Analiza zależności między strukturą a aktywnością (SAR) (etap 2). Należy uwzględnić wyniki badań substancji pokrewnych pod względem budowy, o ile są one dostępne. W przypadku dostępności wystarczających danych na temat oddziałania na ludzi i/lub zwierzęta substancji pokrewnych pod względem budowy lub mieszanin takich substancji, wskazujących na ich potencjalne działanie korozyjne/podrażniające na oko, można domniemywać, że substancja badana poddawana ocenie spowoduje taką samą reakcję. W takich przypadkach substancja ta nie musi być poddawana badaniu. Negatywne wyniki z badań na temat substancji pokrewnych pod względem budowy lub mieszanin takich substancji nie są wystarczającym dowodem na brak działań podrażniających lub korozyjnych danej substancji w świetle strategii badań sekwencyjnych. Zweryfikowane i zaakceptowane podejście SAR powinno zostać wykorzystane w celu zidentyfikowania potencjalnego oddziałania korozyjnego i podrażniającego na skórę i oko.
Właściwości fizyko-chemiczne i reaktywność chemiczna (etap 3). Substancje charakteryzujące się skrajnym pH ≥ 2,0 i ≤ 11,5 mogą mieć silne działanie miejscowe. Jeżeli takie skrajne pH jest podstawą do identyfikacji substancji jako korozyjnej dla oka, możliwe jest wykorzystanie jej rezerwy kwasowej/alkalicznej (tj. zdolności buforowej) (5)(6). Jeżeli zdolność buforowa wskazuje na to, że substancja może nie być korozyjna dla oka, należy przeprowadzić dalsze badania w celu potwierdzenia tej cechy, najlepiej przy użyciu zweryfikowanych i zaakceptowanych badań in vivo lub ex vivo (zob. krok 5 i 6).
Uwzględnienie innych istniejących informacji (etap 4). Wszystkie dostępne informacje na temat ogólnoustrojowej toksyczności skórnej powinny zostać poddane ocenie na tym etapie. Ponadto należy uwzględnić ostrą toksyczność skórną powodowaną przez substancję badaną. Jeżeli wykazano, że substancja badana jest bardzo toksyczna w kontakcie ze skórą, badanie na oku może okazać się zbyteczne. Pomimo że zależność między ostrą toksycznością skórną a działaniem podrażniającym/korozyjnym na oko nie jest zawsze oczywista, można założyć, że jeśli reagent jest bardzo toksyczny w kontakcie ze skórą, będzie wykazywał wysoką toksyczność po zaaplikowaniu na oko. Dane na ten temat mogą również zostać rozpatrzone między realizacją etapów 2 i 3.
Wyniki z badań in vitro lub ex vivo (etapy 5 i 6). Substancje, które wykazują właściwości korozyjne lub silnie podrażniające w zweryfikowanych i zaakceptowanych badaniach in vitro lub ex vivo (7)(8), które były zweryfikowane i zaakceptowane na potrzeby oceny działania korozyjnego/podrażniającego na skórę lub oko, nie muszą być badane na zwierzętach. Można założyć, że takie substancje powodują podobnie silne reakcje in vivo. Jeżeli zweryfikowane i zaakceptowane badania in vivo/ex vivo nie są dostępne, należy pominąć etap 5 i 6, a następnie przejść bezpośrednio do etapu 7.
Ocena badań in vivo podrażnień lub korozji skórnej w wyniku działania substancji (etap 7). Jeżeli nie istnieją wystarczające dowody do przeprowadzenia jednoznacznej analizy wagi dowodów na temat potencjalnego działania korozyjnego/podrażniającego na oko, w oparciu o na wynikach badań przedstawionych powyżej, w pierwszej kolejności należy ocenić potencjalne działanie podrażniające/korozyjne na skórę, z wykorzystaniem metody badawczej B.4 (4) i towarzyszącego jej załącznika (9). Jeżeli w wyniku badań okaże się, że substancja powoduje korozję lub silne podrażnienia skóry, należy uznać ją za korozyjną lub podrażniającą dla oka, chyba że istnieją inne informacje prowadzące do przeciwnych wniosków. W świetle tych informacji badanie in vivo może okazać się zbyteczne. Jeżeli substancja jest niekorozyjna lub nie powoduje silnego podrażnienia skóry, badania in vivo oka powinny zostać przeprowadzone.
Badania in vivo na królikach (etap 8 i 9): Okulistyczne badania in vivo powinny rozpoczynać się od badań wstępnych z wykorzystaniem jednego zwierzęcia. Jeżeli wyniki tych badań świadczą o silnym działaniu podrażniającym lub korozyjnym dla oka, dalsze badania powinny zostać wstrzymane. Jeżeli to badanie nie wskazuje na żadne korozyjne lub silnie podrażniające skutki, należy przeprowadzić badanie potwierdzające na dwóch dodatkowych zwierzętach.
BIBLIOGRAFIA
|
(1) |
OECD (1996) OECD Test Guidelines Programme: Final Report of the OECD Workshop on Harmonization of Validation and Acceptance Criteria for Alternative Toxicological Testing Methods. Held in Solna, Sweden, 22–24 January 1996 (http://www1.oecd.org/ehs/test/background.htm). |
|
(2) |
OECD (1998) Harmonized Integrated Hazard Classification System for Human Health and Environmental Effects of Chemical Substances, as endorsed by the 28th Joint Meeting of the Chemicals Committee and the Working Party on Chemicals, November 1998 (http://www1.oecd.org/ehs/Class/HCL6.htm). |
|
(3) |
Worth, A.P. and Fentem J.H. (1999). A General Approach for Evaluating Stepwise Testing Strategies. ATLA 27, 161–177. |
|
(4) |
Testing method B.4. Acute Toxicity: dermal irritation/corrosion. |
|
(5) |
Young, J.R., How, M.J., Walker, A.P., Worth W.M.H. (1988) Classification as Corrosive or Irritant to Skin of Preparations Containing Acidic or Alkaline Substance Without Testing on Animals. Toxicol. In Vitro, 2, 19–26. |
|
(6) |
Neun, D.J. (1993) Effects of Alkalinity on the Eye Irritation Potential of Solutions Prepared at a Single pH. J. Toxicol. Cut. Ocular Toxicol. 12, 227–231. |
|
(7) |
Fentem, J.H., Archer, G.E.B., Balls, M., Botham, P.A., Curren, R.D., Earl, L.K., Edsail, D.J., Holzhutter, H.G. and Liebsch, M. (1998) The ECVAM international validation study on in vitro tests for skin corrosivity. 2. Results and evaluation by the Management Team. Toxicology in Vitro 12, pp. 483.524. |
|
(8) |
Testing Method B.40 Skin Corrosion. |
|
(9) |
Annex to Testing Method B.4: A Sequential Testing Strategy for Skin Irritation and Corrosion. |
(2) Rysunek
Strategia badań i oceny podrażnień/korozji oka
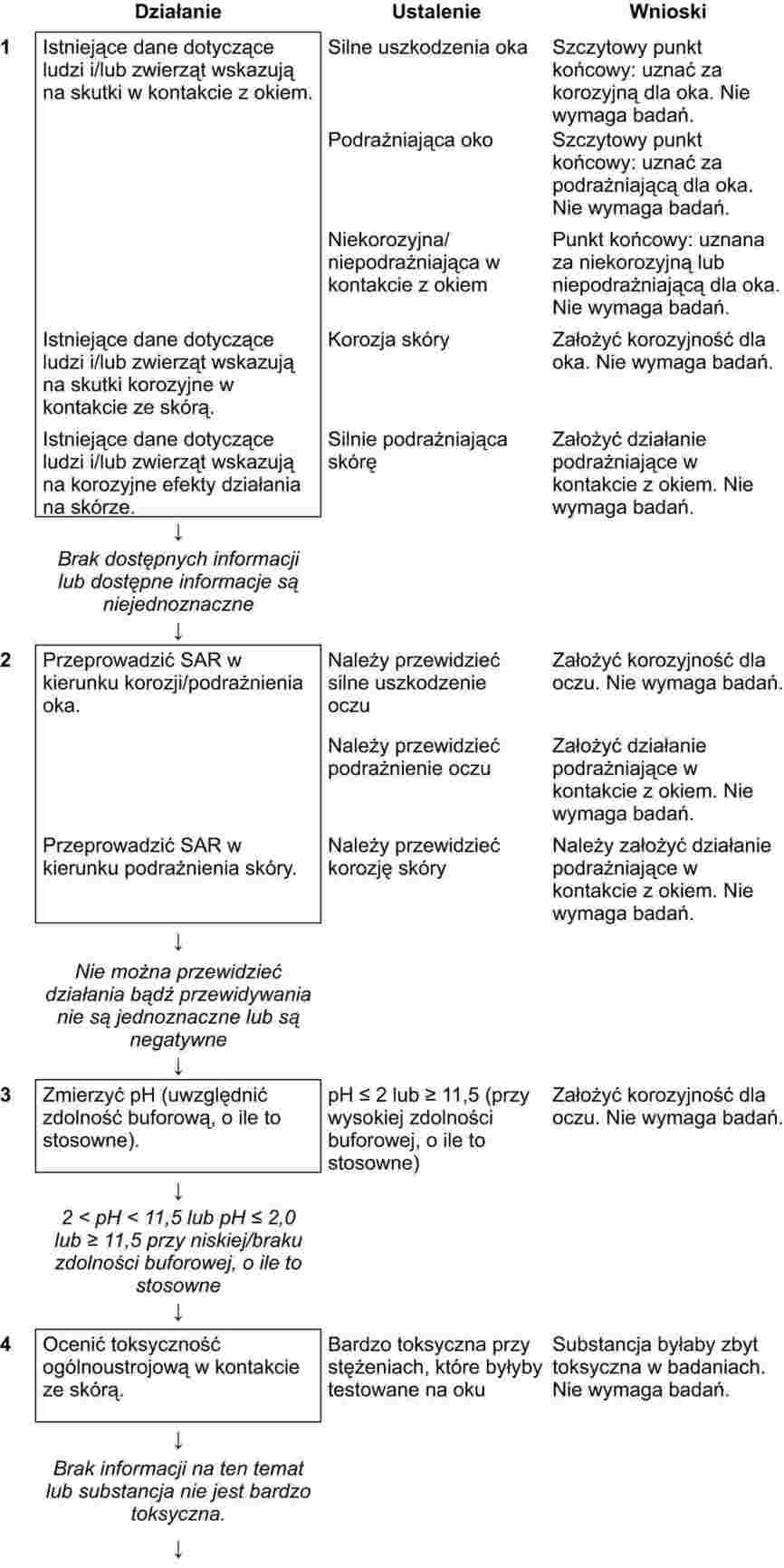

B.6. SENSYBILIZACJA SKÓRY
1. METODA
1.1. WPROWADZENIE
Uwagi:
Czułość i zdolność testów do wykrywania potencjalnych sensybilizatorów ludzkiej skóry uważa się za ważne w systemie klasyfikacji toksyczności odnoszącej się do zdrowia publicznego.
Nie ma jednej metody badania, która właściwie zidentyfikuje wszystkie substancje z potencjałem do sensybilizowania ludzkiej skóry i która odnosi się do wszystkich substancji.
Czynniki, takie jak fizyczne charakterystyki substancji, w tym jej zdolność do wnikania w skórę, muszą być wzięte pod uwagę przy wyborze testu.
Są wykorzystywane dwa typy testów stosujących świnki morskie: testy ze środkiem wspomagającym, w których stan alergiczny jest potęgowany przez rozpuszczenie lub utworzenie zawiesiny substancji testowanej w kompletnym środku wspomagającym Freunda (FCA), i testy niewspomagane.
Testy ze środkiem wspomagającym mogą być bardziej precyzyjne w przewidywaniu prawdopodobnego działania substancji prowadzącego do sensybilizacji skóry u ludzi niż metody nie stosujące kompletnego środka wspomagającego Freunda i są zatem metodami preferowanymi.
Test maksymalizacyjny świnki morskiej (GPMT) jest szeroko stosowanym testem ze środkiem wspomagającym. Pomimo iż kilka innych metod może być wykorzystanych w celu wykrycia potencjału substancji do wywołania reakcji sensybilizacji skóry, GPMT uważa się za preferowaną technikę wspomagającą.
W przypadku wielu kategorii związków chemicznych testy niewspomagane (preferowany jest test Buehlera) uważa się za mniej czułe.
W niektórych przypadkach mogą istnieć słuszne powody wyboru testu Buehlera, wymagającego aplikowania miejscowego zamiast iniekcji śródskórnej, wykorzystywanej w teście maksymalizacyjnym świnki morskiej. W przypadku stosowania testu Buehlera powinno być podane naukowe uzasadnienie.
Test maksymalizacyjny świnki morskiej (GPMT) i test Buehlera są opisane w niniejszej metodzie. Inne metody mogą być stosowane, pod warunkiem że są potwierdzone i podane jest naukowe uzasadnienie.
Jeżeli uzyskano wynik pozytywny w ogólnie przyjętym badaniu sortującym, substancja testowana może być nazwana potencjalnym sensybilizatorem i przeprowadzanie testu świnki morskiej może nie być konieczne. Jednakże jeżeli uzyskano wynik negatywny w takim teście, test świnki morskiej musi zostać przeprowadzony, z zastosowaniem procedury opisanej w tej metodzie badania.
Zob. także Wprowadzenie ogólne w części B.
1.2. DEFINICJE
Sensybilizacja skóry: (alergiczne kontaktowe zapalenie skóry) jest immunologiczną reakcją skórną na substancję. U ludzi reakcje mogą charakteryzować się swędzeniem, rumieniem, obrzękiem, grudkami, pęcherzykami, wzniesieniami naskórka wypełnionymi przezroczystą, wodną cieczą lub połączeniem tych objawów. W przypadku innych gatunków reakcje mogą się różnić i mogą być zaobserwowane tylko rumień i obrzęk.
Ekspozycja indukcyjna: eksperymentalna ekspozycja podmiotu na substancję testowaną z zamiarem wywołania stanu nadwrażliwego.
Okres indukcji: okres co najmniej jednego tygodnia następujący po ekspozycji indukcyjnej, podczas którego stan nadwrażliwy może się rozwinąć.
Ekspozycja prowokacyjna: eksperymentalna ekspozycja podmiotu poddanego wcześniej działaniu substancji testowanej, następująca po okresie indukcji w celu ustalenia, czy podmiot reaguje w sposób nadwrażliwy.
1.3. SUBSTANCJE ODNIESIENIA
Czułość i niezawodność stosowanej techniki eksperymentalnej powinna być oceniana co sześć miesięcy przez zastosowanie substancji, o których wiadomo, że mają właściwości od słabej do miarkowanej sensybilizacji skóry.
We właściwie przeprowadzonym teście powinno się spodziewać reakcji co najmniej 30 % w teście ze środkiem wspomagającym i co najmniej 15 % w teście niewspomaganym dla słabych/umiarkowanych sensybilizatorów.
Preferowane są następujące substancje:
|
Numery CAS |
Numery EINECS |
Nazwy EINECS |
Nazwy zwyczajowe |
|
101-86-0 |
202-983-3 |
aldehyd α-heksylocynamonowy |
aldehyd α-heksylocynamonowy |
|
149-30-4 |
205-736-8 |
benzotiazolo-2-tiol (2-merkaptobenzotiazol) |
kaptaks |
|
94-07-7 |
202-303-5 |
anestezyna |
nordcaine |
Mogą istnieć sytuacje, w których, biorąc pod uwagę zadowalające uzasadnienie, mogą być stosowane inne substancje kontrolne spełniające powyższe kryteria.
1.4. ZASADA METODY BADANIA
Zwierzęta badane są początkowo poddane działaniu substancji testowanej przez śródskórne iniekcje i/lub aplikowanie naskórkowe (ekspozycja indukcyjna). W następstwie 10- do 14-dniowego okresu odpoczynku (okres indukcji), podczas którego może się rozwinąć reakcja odpornościowa, zwierzęta są poddane działaniu dawki prowokacyjnej. Rozmiar i stopień reakcji skórnej na ekspozycję prowokacyjną u zwierząt badanych jest porównywany ze wykazywanym przez zwierzęta kontrolne, które są poddawane pozorowanej terapii podczas indukcji i ulegają ekspozycji prowokacyjnej.
1.5. OPIS METOD BADANIA
Jeżeli uważa się za konieczne usunięcie substancji testowanej, powinno ono być dokonane przy użyciu wody lub odpowiedniego rozpuszczalnika, bez zmiany istniejącej reakcji lub integralności naskórka.
1.5.1. Test maksymalizacyjny świnki morskiej (GPMT)
1.5.1.1.
Zdrowe, młode, dorosłe, albinotyczne osobniki świnki morskiej są aklimatyzowane do warunków laboratoryjnych przez co najmniej 5 dni przed rozpoczęciem testu. Przed testem zwierzęta są losowo przydzielane do grup. Sierść jest usuwana przez strzyżenie, golenie lub możliwie przez chemiczną depilację, zależnie od stosowanej metody badania. Przedmiotem troski powinno być unikanie otarcia skóry. Zwierzęta są ważone przed rozpoczęciem i na końcu testu.
1.5.1.2.
1.5.1.2.1. Zwierzęta badane
Wykorzystywane są powszechnie używane laboratoryjne rasy albinotyczne świnek morskich.
1.5.1.2.2. Liczba i płeć
Samce i/lub samice zwierząt mogą być wykorzystane. Jeżeli wykorzystane są samice, powinny one być nieciężarne i takie, które nie rodziły.
W grupie badanej jest wykorzystane co najmniej 10 zwierząt i co najmniej 5 zwierząt w grupie kontrolnej. Jeżeli użyto mniej niż 20 badanych i 10 kontrolnych świnek morskich i nie jest możliwy wniosek, że testowana substancja jest sensybilizatorem, zdecydowanie zalecane jest badanie dodatkowych zwierząt, aby otrzymać w sumie co najmniej 20 badanych i 10 kontrolnych zwierząt.
1.5.1.2.3. Poziomy dawki
Stężenie substancji testowanej stosowane przy każdej ekspozycji indukcyjnej powinno być systematycznie dobrze tolerowane i powinno być najwyższe, powodujące od słabego do umiarkowanego podrażnienia skóry. Stężenie stosowane przy ekspozycji prowokacyjnej powinno być najwyższą dawką niedrażniącą. Jeżeli konieczne, właściwe stężenia mogą być określone na podstawie badania pilotażowego wykorzystującego dwa lub trzy zwierzęta. W tym celu powinno być rozważone wykorzystanie zwierząt poddanych działaniu FCA.
1.5.1.3.
1.5.1.3.1. Indukcja
Dzień 0 – grupa badana
Trzy pary śródskórnych iniekcji o objętości 0,1 ml są podawane w okolicę barku, która jest oczyszczona z sierści tak, aby iniekcje każdej pary leżały po obu stronach linii środkowej.
Iniekcja 1: mieszanina 1:1 (objętościowo) FCA/woda lub fizjologiczny roztwór soli.
Iniekcja 2: substancja testowana w odpowiednim nośniku w wybranym stężeniu.
Iniekcja 3: substancja testowana przygotowana w wybranym stężeniu w mieszaninie 1:1 (objętościowo) FCA/woda lub fizjologicznym roztworze soli.
W iniekcji 3 substancje rozpuszczalne w wodzie są rozpuszczane w fazie wodnej przed zmieszaniem z FCA. Przygotowuje się zawiesinę w FCA substancji rozpuszczalnych w tłuszczach i nierozpuszczalnych przed połączeniem z fazą wodną. Końcowe stężenie substancji testowanej musi być równe stężeniu użytemu w iniekcji 2.
Iniekcje 1 i 2 są podawane blisko siebie i najbliżej głowy, podczas gdy 3 jest podawana w kierunku części ogonowej powierzchni badanej.
Dzień 0 – grupa kontrolna
Trzy pary śródskórnych iniekcji o objętości 0,1 ml są podawane w te same miejsca, co u zwierząt badanych.
Iniekcja 1: mieszanina 1:1 (objętościowo) FCA/woda lub fizjologiczny roztwór soli.
Iniekcja 2: nierozcieńczony nośnik.
Iniekcja 3: Preparat nośnika o stężeniu wagowym 50 % w mieszaninie 1:1 (objętościowo) FCA/woda lub fizjologicznym roztworze soli.
Dzień 5–7 – grupy badana i kontrolna
Na około 24 godziny przed zastosowaniem miejscowej indukcji, jeżeli substancja nie jest środkiem drażniącym skórę, badana powierzchnia, po dokładnym strzyżeniu i/lub goleniu, jest poddawana działaniu 0,5 ml laurylosiarczanu (VI) sodu w wazelinie o stężeniu 10 %, w celu wywołania miejscowego podrażnienia.
Dzień 6–8 – grupa badana
Powierzchnia badana jest ponownie oczyszczana z sierści. Bibuła filtracyjna (2 × 4 cm) jest dokładnie pokrywana substancją testowaną w odpowiednim nośniku oraz nakładana na powierzchnię badaną i utrzymywana w kontakcie za pomocą szczelnego opatrunku przez 48 godzin. Wybór nośnika powinien być uzasadniony. Substancje stałe są drobno rozcierane i łączone z odpowiednim nośnikiem. Ciecze mogą być stosowane bez rozcieńczania, jeżeli właściwe.
Dzień 6–8 – grupa kontrolna
Powierzchnia badana jest ponownie oczyszczana z sierści. Nakładany jest tylko nośnik, w podobny sposób na powierzchnię badaną i utrzymywany w kontakcie za pomocą szczelnego opatrunku przez 48 godzin.
1.5.1.3.2. Prowokacja
Dzień 20–22 – grupa badana i kontrolna
Boki badanych i kontrolnych zwierząt są oczyszczane z sierści. Łata lub komora wypełniona substancją testowaną jest nakładana na jeden bok zwierząt oraz, w odpowiednim przypadku, łata lub komora wypełniona tylko nośnikiem może być nałożona na drugi bok. Łaty są utrzymywane w kontakcie za pomocą szczelnego opatrunku przez 24 godziny.
1.5.1.3.3. Obserwacja i klasyfikacja: grupy: badana i kontrolna:
|
— |
około 21 godzin po usunięciu łaty powierzchnia prowokacji jest czyszczona i dokładnie strzyżona i/lub golona i, jeżeli to konieczne, depilowana, |
|
— |
około 3 godzin później (w przybliżeniu 48 godzin od początku stosowania prowokacji) reakcja skóry jest obserwowana i odnotowywana zgodnie z klasami wskazanymi w dodatku, |
|
— |
około 24 godziny po tej obserwacji przeprowadza się drugą obserwację (72 godziny) i ponownie odnotowuje. |
Ślepy odczyt wyników badań zwierząt badanych i kontrolnych jest wskazany.
Jeżeli konieczne jest wyjaśnienie wyników otrzymanych w pierwszej prowokacji, powinna być rozważona druga prowokacja (tj. powtórna prowokacja), gdzie właściwe z nową grupą kontrolną, w przybliżeniu jeden tydzień po pierwszej. Powtórna prowokacja może być również przeprowadzona na pierwotnej grupie kontrolnej.
Wszystkie reakcje skórne i jakiekolwiek niezwykłe wyniki badań, w tym reakcje ogólnoustrojowe, wynikające z procedur indukcji i prowokacji powinny być obserwowane i odnotowywane zgodnie ze skalą klasyfikacyjną Magnusson/Kligman (zob. dodatek). W celu wyjaśnienia wątpliwych reakcji mogą być przeprowadzone inne procedury, np. badanie histopatologiczne, pomiar grubości fałdy skórnej.
1.5.2. Test Buehlera
1.5.2.1.
Zdrowe, młode, dorosłe, albinotyczne osobniki świnki morskiej są aklimatyzowane do warunków laboratoryjnych przez co najmniej 5 dni przed rozpoczęciem testu. Przed testem zwierzęta są losowo przydzielane do grup. Sierść jest usuwana przez strzyżenie, golenie lub możliwie przez chemiczną depilację, zależnie od stosowanej metody badania. Należy uważać, aby uniknąć otarcia skóry. Zwierzęta są ważone przed rozpoczęciem i na końcu testu.
1.5.2.2.
1.5.2.2.1. Zwierzęta badane
Wykorzystywane są powszechnie używane laboratoryjne rasy albinotyczne świnek morskich.
1.5.2.2.2. Liczba i płeć
Mogą być wykorzystane samce i/lub samice zwierząt. Jeżeli wykorzystane są samice, powinny one być nieciężarne i takie, które nie rodziły.
W grupie badanej jest wykorzystanych co najmniej 20 zwierząt i co najmniej 10 zwierząt w grupie kontrolnej.
1.5.2.2.3. Poziomy dawki
Stężenie substancji testowanej stosowane przy każdej ekspozycji indukcyjnej powinno być możliwie najwyższe, by spowodować słabe, ale nie nadmierne podrażnienie. Stężenie stosowane przy ekspozycji prowokacyjnej powinno być najwyższą dawką niedrażniącą. Jeżeli konieczne, właściwe stężenia mogą być wyznaczone na podstawie badania pilotażowego wykorzystującego dwa lub trzy zwierzęta.
W przypadku materiałów testowanych rozpuszczalnych w wodzie, właściwe jest stosowanie jako nośnika wody lub rozcieńczonego niedrażniącego roztworu środka powierzchniowo czynnego. W przypadku innych materiałów testowanych 80 % roztwór etanol/woda jest preferowany dla indukcji i aceton dla prowokacji.
1.5.2.3.
1.5.2.3.1. Indukcja
Dzień 0 – grupa badana
Jeden bok jest oczyszczany z sierści (dokładne strzyżenie). System łat powinien być całkowicie wypełniony substancją testowaną w odpowiednim nośniku (wybór nośnika powinien być uzasadniony; ciekłe substancje testowane mogą być stosowane bez rozcieńczania, jeżeli właściwe).
System łat jest nakładany na powierzchnię testowaną i utrzymywany w kontakcie ze skórą za pomocą szczelnej łaty lub komory i odpowiedniego opatrunku przez 6 godzin.
System łat musi być szczelny. Odpowiedni jest bawełniany tampon, który może być okrągły lub kwadratowy, o przybliżonej powierzchni 4–6 cm2. W celu zapewnienia szczelności preferowane jest unieruchomienie badanego osobnika przy użyciu odpowiedniego elementu unieruchamiającego. Jeżeli zastosowane jest owijanie, mogą być potrzebne dodatkowe ekspozycje.
Dzień 0 – grupa kontrolna
Jeden bok jest oczyszczany z sierści (dokładne strzyżenie). Nakładany jest tylko nośnik, w sposób podobny do stosowanego w doniesieniu do grupy badanej. System łat jest utrzymywany w kontakcie ze skórą za pomocą szczelnej łaty lub komory i odpowiedniego opatrunku przez 6 godzin. Jeżeli można wykazać, że nie jest konieczna symulowana grupa kontrolna, można się posłużyć naturalną grupą kontrolną.
Dzień 6–8 i 13–15 – grupa badana i kontrolna
Przeprowadza się takie samo aplikowanie jak w dniu 0, na tę samą powierzchnię badaną (oczyszczenie z sierści w razie konieczności) tego samego boku, w dniu 6–8 i ponownie w dniu 13–15.
1.5.2.3.2. Prowokacja
Dzień 27–29 – grupa badana i kontrolna
Bok zwierząt badanych i kontrolnych, który nie był wykorzystywany do badania, jest oczyszczany z sierści (dokładne strzyżenie). Na zadnią część nieużywanego boku zwierząt badanych i kontrolnych nakładana jest łata lub komora, zawierająca odpowiednią ilość substancji testowanej w maksymalnym niedrażniącym stężeniu.
Jeżeli ma to znaczenie, szczelna łata lub komora tylko z nośnikiem jest również nakładana na przednią część nieużywanego boku zarówno badanych, jak i kontrolnych zwierząt. Łaty i komory są utrzymywane w kontakcie ze skórą za pomocą odpowiedniego opatrunku przez 6 godzin.
1.5.2.3.3. Obserwacja i klasyfikacja:
|
— |
około 21 godzin po usunięciu łaty powierzchnia prowokowana jest oczyszczana z sierści, |
|
— |
około 3 godziny później (w przybliżeniu 30 godzin po przyłożeniu łaty prowokującej) reakcje skórne są obserwowane i odnotowywane zgodnie z klasami wskazanymi w dodatku, |
|
— |
około 24 godziny później po 30-godzinnej obserwacji (w przybliżeniu 54 godziny po przyłożeniu łaty prowokującej) reakcje skórne są ponownie obserwowane i odnotowywane. |
Ślepy odczyt zwierząt badanych i kontrolnych jest wskazany.
Jeżeli konieczne jest wyjaśnienie wyników otrzymanych z pierwszej prowokacji, powinna być rozważona druga prowokacja (tj. powtórna prowokacja), gdzie właściwe, z nową grupą kontrolną, w przybliżeniu jeden tydzień po pierwszej. Powtórna prowokacja może być również przeprowadzona na pierwotnej grupie kontrolnej.
Wszystkie reakcje skórne i jakiekolwiek nietypowe wyniki badań, w tym reakcje ogólnoustrojowe, wynikające z procedur indukcji i prowokacji powinny być zaobserwowane i odnotowywane zgodnie ze skalą klasyfikacyjną Magnusson/Kligman (zob. dodatek). W celu wyjaśnienia wątpliwych reakcji mogą być przeprowadzone inne procedury, np. badanie histopatologiczne, pomiar grubości fałdy skórnej.
2. DANE (GPMT i test buehlera)
Dane powinny być podsumowane w formie tabeli, przedstawiając reakcje skórne każdego zwierzęcia podczas każdej obserwacji.
3. SPORZĄDZANIE SPRAWOZDANIA (GPMT I TEST BUEHLERA)
Jeżeli jest wykonana próba sortująca przed testem świnki morskiej, muszą być podane opis i odniesienie do testu (np. miejscowa próba węzła chłonnego (LLNA), test obrzęku ucha myszy (MEST)), w tym szczegóły procedury, wraz z wynikami testu, oraz wynikami zastosowania substancji odniesienia.
Sprawozdanie z testu (GMPT i test Buehlera)
Sprawozdanie z testu obejmuje, jeżeli możliwe, następujące informacje:
|
|
Zwierzęta badane:
|
|
|
Warunki testu:
|
|
|
Wyniki:
|
|
|
Rozpatrzenie wyników. |
|
|
Wnioski. |
4. BIBLIOGRAFIA
Niniejsza metoda jest analogiczna do OECD TG 423.
Dodatek
TABELA
Skala klasyfikacji Magnusson/Kligman do oceny szkodliwych reakcji na łatę prowokacyjną
0= brak widocznych zmian
1= odosobniony lub niejednolity rumień
2= umiarkowany i zlewający się rumień
3= silny rumień i obrzęk
B.7. TOKSYCZNOŚĆ POWTARZANEJ (28 DNI) DAWKI (DOUSTNA)
1. METODA
1.1. WPROWADZENIE
Zob. Wprowadzenie ogólne w części B.
1.2. DEFINICJE
Zob. Wprowadzenie ogólne w części B.
1.3. ZASADA METODY BADANIA
Substancja testowana jest podawana kilku grupom zwierząt doświadczalnych doustnie, codziennie, w stopniowanych dawkach, jeden poziom dawki na grupę przez okres 28 dni. Podczas okresu dawkowania każdego dnia zwierzęta są dokładnie obserwowane w celu wykrycia oznak toksyczności. Zwierzęta, które zdychają lub są zabijane podczas testu, poddaje się sekcji zwłok, a na zakończenie testu zwierzęta, które przeżyły, są zabijane i poddawane sekcji zwłok.
Niniejsza metoda kładzie większy nacisk na objawy neurologiczne jako określony punkt końcowy, i podkreślana jest konieczność uważnych obserwacji klinicznych zwierząt, w celu otrzymania możliwie najwięcej informacji. Metoda ta powinna identyfikować związki chemiczne o potencjale neurotoksycznym, co może dawać podstawy do dalszego dogłębnego badania tego aspektu. Dodatkowo, metoda może wykazywać skutki immunologiczne i toksyczność organów rozrodczych.
1.4. OPIS METODY BADANIA
1.4.1. Przygotowania
Zdrowe, młode, dorosłe zwierzęta są losowo przydzielane do grupy kontrolnej i badanej. Klatki powinny być ustawione w taki sposób, żeby zminimalizować możliwy wpływ położenia klatki. Zwierzęta są identyfikowane jednoznacznie i przetrzymywane w klatkach przez co najmniej pięć dni przed rozpoczęciem badania, żeby umożliwić aklimatyzację do warunków laboratoryjnych.
Substancja testowana jest podawana bezpośrednio do żołądka, w pożywieniu lub w wodzie pitnej. Metoda aplikowania zależy od celu badania i właściwości fizycznych/chemicznych substancji.
W przypadku gdy to konieczne, substancja testowana jest rozpuszczona lub ma postać zawiesiny w odpowiednim nośniku. Zaleca się, aby jeżeli możliwe, najpierw było rozważone użycie wodnego roztworu/zawiesiny, następnie roztworu/emulsji w oleju (np. olej kukurydziany) i potem możliwy roztwór w innym nośnikach. W przypadku nośników innych niż woda musi być znana charakterystyka toksyczna nośnika. Powinna być określona trwałość testowanej substancji w nośniku.
1.4.2. Warunki badania
1.4.2.1. Zwierzęta badane
Preferowanym gatunkiem gryzonia jest szczur, chociaż inne gatunki gryzoni mogą być wykorzystane. Powinny być wykorzystywane powszechnie używane laboratoryjne rasy młodych, zdrowych, dorosłych zwierząt. Samice powinny być nieciężarne i takie, które nie rodziły. Dawkowanie powinno się rozpocząć jak najszybciej po odsadzeniu od matki, a w każdym przypadku przed ukończeniem dziewięciu tygodni.
Na początku badania wahania wagi zwierząt powinny być minimalne i nie przekraczać ± 20 % średniej wagi dla każdej płci.
W przypadku gdy badanie wielokrotnej dawki doustnej jest przeprowadzane jako badanie wstępne do badania długoterminowego, preferowane do obu badań są zwierzęta tej samej rasy i z tego samego źródła.
1.4.2.2. Liczba i płeć
Co najmniej 10 zwierząt (pięć samic i pięć samców) powinno być użytych dla każdego poziomu dawki. Jeżeli planowane jest uśmiercanie w międzyczasie, liczba powinna być powiększona o liczbę zwierząt, które według planu mają być zabite przed ukończeniem badania.
Dodatkowo grupa satelicka 10 zwierząt (pięć zwierząt każdej płci) może być poddana działaniu wysokiego poziomu dawki przez 28 dni i obserwowana, przez 14 dni po dawkowaniu, pod kątem odwracalności, trwałości, i opóźnionego wystąpienia objawów zatrucia. Jest także stosowana grupa satelicka 10 zwierząt kontrolnych (pięć zwierząt każdej płci).
1.4.2.3. Poziomy dawki
Zasadniczo powinny być wykorzystane co najmniej trzy grupy badane i grupa kontrolna. Z wyjątkiem zastosowania substancji testowanej, ze zwierzętami grupy kontrolnej powinno się obchodzić w sposób identyczny, jak z podmiotami grupy badanej. Jeżeli przy podawaniu substancji testowanej jest stosowany nośnik, grupa kontrolna powinna otrzymać największą użytą objętość nośnika.
Jeżeli z oceny innych danych należałoby oczekiwać braku objawów przy dawce 1 000 mg/kg masy ciała/dzień, może być przeprowadzony test graniczny. Jeżeli nie są dostępne odpowiednie dane, może być wykonane badanie wyników zakresowych, pomagające określić dawki, które mają być użyte.
Poziomy dawki powinny być wybierane, biorąc pod uwagę jakiekolwiek istniejące dane o toksyczności i dane toksykokinetczne osiągalne dla substancji testowanej lub materiałów pokrewnych. Powinien zostać wybrany najwyższy poziom dawki, w celu wywołania objawów zatrucia, ale nie śmierci lub poważnego cierpienia. Od tego czasu malejąca sekwencja poziomów dawki powinna być dobierana w taki sposób, aby wykazać wszystkie reakcje powiązane z dawkowaniem i brak obserwowanych objawów niekorzystnych przy najmniejszym poziomie dawki (NOAEL). Dwie lub cztery ograniczone przerwy są często optymalne dla ustalenia malejących poziomów dawki i dodanie czwartej grupy badanej jest często bardziej pożądane niż stosowanie bardzo dużych przerw (np. współczynnik większy od 10) między dawkowaniem.
W przypadku substancji podawanych w pożywieniu lub wodzie pitnej ważne jest zapewnienie, aby wymagane ilości substancji testowanej nie kolidowały z prawidłową równowagą odżywiania lub wody. W przypadku gdy substancja testowana jest podawana w pożywieniu, może być stosowane stałe stężenie żywieniowe (ppm) albo stały poziom dawki odniesionej do masy ciała zwierząt; użyta opcja musi być wyszczególniona. W przypadku substancji podawanej bezpośrednio do żołądka dawka powinna być podawana każdego dnia o podobnym czasie i w razie konieczności modyfikowana, żeby utrzymać stały poziom dawki w odniesieniu do masy ciała zwierzęcia.
W przypadku gdy badanie wielokrotnej dawki jest przeprowadzane jako badanie wstępne do badania długoterminowego, w obu badaniach powinna być stosowana podobna dieta.
1.4.2.4. Test graniczny
Jeżeli test przy jednym poziomie dawki równym co najmniej 1 000 mg/kg masy ciała/dzień lub, w przypadku podawania w pożywieniu lub wodzie pitnej, równoważny procent w pożywieniu lub wodzie pitnej (na podstawie ustalonej masy ciała), stosując procedury opisane dla tego badania, nie wywołuje widocznych objawów zatrucia i jeżeli nie należałoby spodziewać się toksyczności na podstawie danych, dotyczących substancji strukturalnie pokrewnych, wtedy pełne badanie przy użyciu trzech poziomów dawki może nie być konieczne. Niniejszy test graniczny ma zastosowanie, z wyłączeniem tych przypadków, w których ekspozycja ludzi wskazuje na konieczność użycia wyższego poziomu dawki.
1.4.2.5. Okres obserwacji
Okres obserwacji powinien wynosić 28 dni. Zwierzęta w grupie satelickiej wyznaczone do obserwacji uzupełniających powinny być przetrzymane przez co najmniej 14 dalszych dni bez terapii, w celu wykrycia opóźnionych objawów zatrucia, ich trwałości lub powrotu do zdrowia.
1.4.3. Procedura
Substancja testowana jest dawkowana zwierzętom codziennie przez siedem dni każdego tygodnia w okresie 28 dni; zastosowanie pięciodniowego tygodniowego reżimu dawkowania musi być uzasadnione. W przypadku gdy substancja testowana jest podawana bezpośrednio do żołądka, powinno to być zrobione w jednej dawce przy użyciu rurki lub odpowiedniej kaniuli dotchawicznej. Maksymalna objętość cieczy, która może być podana jednorazowo, zależy od rozmiarów badanego zwierzęcia. Objętość nie powinna przekraczać 1 ml/100 g masy ciała, oprócz roztworów wodnych, w przypadku których można użyć 2 ml/100 g masy ciała. Poza substancjami drażniącymi i korozyjnymi, które normalnie ujawniają zaostrzone objawy przy wyższych stężeniach, zmienność objętości w badaniu powinna być zminimalizowana przez dostosowanie stężenia, aby zapewnić stałą objętość przy wszystkich poziomach dawki.
1.4.3.1. Ogólne obserwacje
Ogólne obserwacje kliniczne powinny być przeprowadzane co najmniej raz dziennie, najlepiej każdego dnia w tym samym czasie, biorąc pod uwagę szczytowy okres przewidywanych objawów po dawkowaniu. Stan zdrowia zwierząt powinien być odnotowywany. Co najmniej dwa razy dziennie wszystkie zwierzęta są obserwowane pod kątem zachorowalności i śmiertelności. Zwierzęta konające oraz zwierzęta przejawiające ostre cierpienie i ból powinny być usuwane, gdy zostanie to zauważone, humanitarnie zabite i poddane sekcji zwłok.
Jeden raz przed pierwszą ekspozycją (aby uwzględnić porównanie w obrębie podmiotów), i od tego czasu co najmniej raz w tygodniu, powinny być przeprowadzane szczegółowe obserwacje kliniczne wszystkich zwierząt. Obserwacje te powinny być przeprowadzane na zewnątrz klatki macierzystej na znormalizowanej powierzchni i najlepiej za każdym razem w tym samym czasie. Powinny one być starannie odnotowywane, najlepiej przy użyciu systemów zapisywania, wyraźnie zdefiniowanych przez laboratorium testujące. Powinny być podjęte wysiłki, aby zagwarantować, że zmiany warunków badania są minimalne i że obserwacje są przeprowadzane najlepiej przez obserwatorów nieświadomych terapii. Zanotowane oznaki powinny obejmować, ale nie powinny być do nich ograniczone, zmiany w skórze, futrze, oczach, błonach śluzowych, wystąpienie wydzielin i wydalin aktywności wegetatywnej (np. łzawienie, podniesienie sierści, rozmiar źrenicy, niezwykła forma oddychania). Powinny być także odnotowywane zmiany w sposobie chodzenia, postawie, i reakcji na dotyk, jak również obecność ruchów drgawkowych lub kurczowych, stereotypów (np. nadmierne czyszczenie się, powtarzające się krążenie w kółko) oraz dziwne zachowanie (np. samookaleczanie, chodzenie do tyłu).
W czwartym tygodniu ekspozycji powinna być przeprowadzona ocena reaktywności sensorycznej na różnego typu bodźce (np. bodźce słuchowe, wzrokowe i proprioreceptoryczne), ocena siły uchwytu i aktywności motorycznej. Dalsze szczegóły procedur, które mogłyby być zastosowane, są podane w literaturze (zob. Wprowadzenie ogólne w część B).
Obserwacje funkcjonalne przeprowadzane w czwartym tygodniu ekspozycji mogą być pominięte, gdy badanie prowadzone jest jako badanie wstępne do późniejszego badania (90-dniowego) podchronicznego. W takim przypadku obserwacje funkcjonalne powinny być zawarte w tym uzupełniającym badaniu. Z drugiej strony, dostępność danych dotyczących obserwacji funkcjonalnych z badania wielokrotnej dawki może poprawić możliwość wyboru poziomów dawki do celów późniejszego badania podchronicznego.
Wyjątkowo obserwacje funkcjonalne mogą być pominięte w przypadku grup, które w przeciwnym przypadku ujawniają oznaki zatrucia w stopniu, który mógłby znacząco kolidować z wykonaniem testu funkcjonalnego.
1.4.3.2. Masa ciała i spożycie żywności/wody
Wszystkie zwierzęta powinny być ważone co najmniej raz w tygodniu. Pomiary spożycia żywności i wody powinny być wykonywane co najmniej raz na tydzień. Jeżeli substancja testowana podawana jest z wodą pitną, spożycie wody powinno być również mierzone co najmniej raz na tydzień.
1.4.3.3. Hematologia
Na końcu okresu badania powinny być przeprowadzone następujące analizy hematologiczne: hematokryt, stężenie hemoglobiny, liczba erytrocytów, całkowita i różnicowa liczba leukocytów, liczba płytek krwi i pomiar krzepliwości krwi czas/potencjał.
Próbki krwi powinny być pobierane z wyznaczonego miejsca tuż przed lub jako część procedury zabijania zwierząt i przechowywane w odpowiednich warunkach.
1.4.3.4. Biochemia kliniczna
Oznaczenia biochemii klinicznej w celu zbadania znaczących skutków toksycznych w tkankach, a ściśle – skutków w nerkach i wątrobie, powinny być wykonane na otrzymanych próbkach krwi wszystkich zwierząt tuż przed lub jako część procedury zabijania zwierząt (z wyjątkiem tych, które znaleziono konające i/lub zabite w międzyczasie). Zalecane jest wstrzymanie podawania pożywienia dla zwierząt przez noc poprzedzającą pobranie krwi (2). Badania osocza i surowicy muszą obejmować sód, potas, glukozę, całkowity cholesterol, mocznik, kreatyninę, całkowite białko i albuminę, co najmniej dwa enzymy wskazujące na skutki komórkowe w wątrobie (takie jak aminotransferaza alaninowa, aminotransferaza asparaginowa, alkaliczna fosfataza, transpeptydaza gamma-glutamylowa i dehydrogenaza sorbitowa). Pomiary dodatkowych enzymów (wątrobowego lub innego pochodzenia) i kwasów żółciowych mogą dostarczyć przydatnych informacji w danych okolicznościach.
Fakultatywnie, stosując czasowe objętościowe zbieranie moczu podczas ostatniego tygodnia badań, mogłyby być wykonane następujące oznaczenia analizy moczu: wygląd, objętość, osmolarność i ciężar właściwy, pH, białko, glukoza i krew/komórki krwi.
Dodatkowo powinno być rozważone badanie markerów surowicy dotyczących ogólnego uszkodzenia tkanki. Inne oznaczenia, które powinny być przeprowadzone, jeżeli znane właściwości substancji testowanej mogą lub podejrzewa się, że mogą wpływać na pokrewne profile metaboliczne, obejmują wapń, fosforan, triglicerydy po poście, hormony specyficzne, metahemoglobinę i cholinoesterazę. Powyższe musi zostać rozpoznane w odniesieniu do substancji w niektórych klasach lub na zasadzie analizy przypadku.
Ogólnie, istnieje konieczność elastycznego podejścia, zależnie od gatunku i obserwowanych i/lub spodziewanych skutków danej substancji.
Jeżeli dane poziomu tolerancji z przeszłości są niedostateczne, powinno się rozważyć oznaczenie zmiennych hematologicznych i biochemii klinicznej zanim rozpocznie się dawkowanie.
1.4.3.5. Sekcja zwłok
Wszystkie zwierzęta uczestniczące w badaniu muszą być poddane pełnej, szczegółowej sekcji zwłok, która obejmuje staranne badanie zewnętrznych powierzchni ciała, wszystkich otworów i jam czaszkowych, piersiowych i brzusznych oraz ich zawartości. Wątroba, nerki, nadnercza, jądra, gruczoły opuszkowo-cewkowe, grasica, śledziona, mózg i serce wszystkich zwierząt powinny być właściwie oczyszczone z jakiejkolwiek przylegającej tkanki i zważone mokre, jak najszybciej po przeprowadzeniu sekcji, aby uniknąć wysuszenia.
Następujące tkanki powinny być zakonserwowane w utrwalającym ośrodku najbardziej odpowiednim zarówno dla typu tkanki, jak i zamierzonego późniejszego badania histopatologicznego: wszystkie duże zmiany patologiczne, mózg (reprezentatywne rejony w tym mózg, móżdżek i pons), rdzeń kręgowy, żołądek, jelito cienkie i grube (w tym kępki Peyera), wątroba, nerki, nadnercza, śledziona, serce, grasica, tarczyca, tchawica i płuca (zakonserwowane przez napełnienie środkiem utrwalającym i następnie zanurzenie), gruczoły płciowe, pomocnicze narządy płciowe (np. macica, prostata), pęcherz moczowy, węzły chłonne (najlepiej jeden węzeł chłonny obejmujący drogę podawania i inny węzeł odległy od drogi podawania, aby uwzględnić skutki ogólnoustrojowe), nerw obwodowy (kulszowy lub piszczelowy), najlepiej w bliskiej odległości od mięśnia, i część szpiku kostnego (lub, ewentualnie, świeżo pobrana mała objętość szpiku kostnego). Wyniki badań klinicznych i innych mogą wskazywać na konieczność zbadania dodatkowych tkanek. Powinny również zostać zakonserwowane inne narządy, na które może być skierowane działanie substancji testowanej, opierając się na jej znanych właściwościach.
1.4.3.6. Badanie histopatologiczne
Pełne badanie histopatologiczne powinno być przeprowadzone na zakonserwowanych narządach i tkankach wszystkich zwierząt z grupy kontrolnej i grupy wysokiej dawki. Badania te powinny być rozszerzone na zwierzęta z grup wszystkich innych dawek, jeżeli w grupie wysokiej dawki obserwowane są zmiany związane z działaniami badawczymi.
Wszystkie duże zmiany patologiczne muszą być zbadane.
W przypadku gdy stosowana jest grupa satelicka, badania histopatologiczne powinny być przeprowadzone na tkankach i narządach rozpoznanych jako wykazujące skutki w grupach badanych.
2. DANE
Powinny zostać dostarczone dane indywidualne zwierząt. Dodatkowo wszystkie dane powinny być podsumowane w formie tabeli, przedstawiając dla każdej grupy badanej liczbę zwierząt na początku testu, liczbę zwierząt, które znaleziono martwe podczas testu lub zabito z powodów humanitarnych i czas każdej śmierci lub humanitarnego zabicia, liczbę zwierząt wykazujących oznaki toksyczności, opis obserwowanych oznak zatrucia, w tym czas rozpoczęcia, okres trwania, ostrość wszystkich objawów zatrucia, liczbę zwierząt wykazujących zmiany patologiczne, typ tych zmian i procent zwierząt przejawiających każdy typ zmian patologicznych.
W przypadkach gdy jest to możliwe, wyniki liczbowe powinny być ocenione za pomocą odpowiedniej i ogólnie akceptowanej metody statystycznej. Metody statystyczne powinny być wybrane podczas planowania badania.
3. SPORZĄDZANIE SPRAWOZDANIA
SPRAWOZDANIE Z TESTU
Sprawozdanie z testu obejmuje, jeżeli możliwe, następujące informacje:
|
|
Zwierzęta badane:
|
|
|
Warunki testu:
|
|
|
Wyniki:
|
|
|
Rozpatrzenie wyników. |
|
|
Wnioski. |
4. BIBLIOGRAFIA
Niniejsza metoda jest analogiczna do OECD TG 407.
B.8. TOKSYCZNOŚĆ POWTARZANEJ (28 DNI) DAWKI (INHALACYJNA)
1. METODA
1.1. WPROWADZENIE
Przydatne jest posiadanie wstępnych informacji na temat rozkładu wielkości cząstek, prężności pary, temperatury topnienia, temperatury wrzenia, temperatury zapłonu i wybuchowości (o ile stosowalne) substancji badanej.
Patrz także: Wprowadzenie ogólne część B lit. A).
1.2. DEFINICJA
Zob. Wprowadzenie ogólne część B lit. B).
1.3. SUBSTANCJE ODNIESIENIA
Brak.
1.4. ZASADA METODY BADANIA
Kilka grup zwierząt doświadczalnych poddaje się ekspozycji przez określony okres czasu na substancję badaną w stopniowanych stężeniach, po jednym stężeniu na grupę, przez 28 dni. W przypadku gdy zastosuje się podłoże w celu zapewnienia właściwego stężenia substancji badanej w powietrzu, należy zastosować grupę kontrolną otrzymującą wyłącznie takie podłoże. W okresie podawania prowadzi się obserwację zwierząt w celu wykrycia wszelkich objawów toksyczności. Zwierzęta, które padną w trakcie badania, i te, które przeżyją po jego zakończeniu, poddaje się sekcji zwłok.
1.5. KRYTERIA JAKOŚCI
Brak.
1.6. OPIS METODY BADANIA
1.6.1. Przygotowania
Zwierzęta muszą być trzymane w doświadczalnych warunkach pobytu i karmienia przez co najmniej pięć dni przed przeprowadzeniem badania. Przed przeprowadzeniem badania zdrowe, młode, dorosłe zwierzęta są wybierane losowo i przydzielane do wymaganej liczby grup. W razie potrzeby do badanej substancji można dodać odpowiednie podłoże, aby można było zapewnić właściwe stężenie tej substancji w powietrzu. W razie zastosowania podłoża lub innych substancji pomocniczych w celu ułatwienia dawkowania musi być wiadomo, że nie wykazują one działania toksycznego. O ile będzie to właściwe, można wykorzystać dane historyczne.
1.6.2. Warunki badania
1.6.2.1. Zwierzęta doświadczalne
O ile nie ma przeciwwskazań, preferowanym gatunkiem jest szczur. Należy wykorzystać powszechnie stosowane szczepy laboratoryjne młodych, zdrowych zwierząt.
Na początku badania zmienność masy ciała zastosowanych zwierząt nie powinna przekraczać ± 20 % odpowiedniej wartości średniej.
1.6.2.2. Liczba i płeć
Wykorzystuje się po co najmniej 10 zwierząt (pięć samic i pięciu samców) w każdej grupie badanej. Samice powinny być nieródkami i nie mogą być w ciąży. Jeżeli planuje się uśmiercanie zwierząt w międzyczasie, ogólna ich liczba powinna być zwiększona o liczbę zwierząt zaplanowanych do uśmiercenia przed zakończeniem badania. Ponadto satelicka grupa 10 zwierząt (po pięcioro zwierząt każdej płci) może otrzymywać większą dawkę przez 28 dni i można prowadzić w niej obserwację pod kątem odwracalności, utrzymywania się i opóźnionego pojawiania się efektów toksycznych przez 14 dni od zastosowania substancji. Jest także stosowana grupa satelicka 10 zwierząt kontrolnych (pięć zwierząt każdej płci).
1.6.2.3. Stężenie ekspozycyjne
Konieczne jest zapewnienie co najmniej trzech stężeń, wraz z kontrolą i kontrolą otrzymującą podłoże (w stężeniu odpowiadającym stężeniu podłożu przy najwyższym poziomie dawkowania substancji), o ile to ostatnie jest stosowane. Z wyjątkiem podawania substancji badanej, zwierzęta w grupie kontrolnej powinny być traktowane w taki sam sposób jak zwierzęta w grupach badanych. Najwyższe stężenie powinno być tak dobrane, aby wywierało działanie toksyczne, jednak nie prowadziło do zgonów lub prowadziło do jedynie niewielu zgonów. Najniższe stężenie nie powinno dawać żadnych objawów toksyczności. W przypadku gdy istnieje możliwa do wykorzystania, oszacowana wielkość ekspozycji u ludzi, najniższe stężenie powinno ją przewyższać. W idealnej sytuacji pośrednie stężenie powinno prowadzić do uzyskania minimalnych dostrzegalnych efektów toksycznych. W przypadku gdy zostanie wykorzystane więcej niż jedno stężenie pośrednie, wielkości stężeń powinny być tak rozstawione, aby uzyskać stopniowanie działań toksycznych. W grupach otrzymujących niskie i pośrednie stężenia oraz w grupach kontrolnych częstość występowania zgonów powinna być niska, aby umożliwić znaczącą ocenę wyników.
1.6.2.4. Czas ekspozycji
Czas codziennej ekspozycji powinien wynosić sześć godzin, jednak mogą być potrzebne inne okresy, aby spełnić szczególne wymagania.
1.6.2.5. Wyposażenie
Zwierzęta należy poddać badaniu przy użyciu przyrządów do inhalacji zapewniających przepływ dynamiczny powietrza co najmniej 12 wymian na godzinę, aby zapewnić odpowiednią zawartość tlenu i równomierną dystrybucję powietrza ekspozycji. W przypadku zastosowania komory należy ją tak zaprojektować, aby zmniejszyć do minimum stłoczenie zwierząt badanych i zwiększyć do maksimum ich ekspozycję drogą inhalacyjną na substancję badaną. Z reguły, aby zapewnić stabilność powietrza w komorze, całkowita „objętość” zwierząt badanych nie powinna przekraczać 5 % objętości komory do badań. Można zastosować ekspozycję ustno-nosową, ekspozycję jedynie głowy lub ekspozycję całego ciała w pojedynczych komorach, przy czym te pierwsze dwa sposoby pomogą w zminimalizowaniu przechodzenia substancji badanej do organizmu innymi drogami.
1.6.2.6. Okres obserwacji
Zwierzęta doświadczalne należy obserwować codziennie pod kątem występowania objawów toksyczności w całym okresie otrzymywania substancji i zdrowienia. Należy zapisać czas zgonu zwierzęcia i czas pojawienia się i ustąpienia objawów toksyczności.
1.6.3. Procedura
Zwierzęta poddaje się działaniu substancji badanej codziennie, przez pięć do siedmiu dni w tygodniu, przez okres 28 dni. Zwierzęta we wszelkich grupach satelickich, u których planuje się obserwację po zakończeniu eksperymentu, należy przetrzymać przez następnych 14 dni bez podawania im substancji, aby wykryć ustępowanie lub utrzymywanie się efektów toksycznych. Temperatura przeprowadzania badania powinna być utrzymywana na poziomie 22 ± 3 oC.
Wilgotność względną należy utrzymywać najlepiej w przedziale pomiędzy 30 a 70 %, jednak w niektórych przypadkach (np. badania aerozoli) może to nie być możliwe ze względów praktycznych. Utrzymywanie lekkiego podciśnienia w komorze (≤ 5 mm słupa wody) zapobiega wyciekowi substancji badanej do otaczającego obszaru. W trakcie ekspozycji należy zaprzestać podawania zwierzętom karmy i wody.
Należy stosować dynamiczny układ inhalacyjny z odpowiednim analitycznym systemem regulacji stężenia. Zalecane jest przeprowadzenie badania próbnego w celu ustalenia właściwych stężeń ekspozycji. Szybkość przepływu powietrza należy tak wyregulować, aby warunki w całej komorze były jednorodne. Układ powinien zapewnić jak najszybsze uzyskanie stabilnych warunków ekspozycji.
Należy prowadzić pomiary lub monitorować następujące:
|
a) |
szybkość przepływu powietrza (w sposób ciągły); |
|
b) |
rzeczywiste stężenie substancji badanej mierzone w strefie oddechowej. W okresie ekspozycji stężenie nie powinno się zmieniać o więcej niż ± 15 % wartości średniej. W przypadku niektórych aerozoli ten poziom kontroli możne być niemożliwy do uzyskania, przy czym dopuszczalny jest wtedy szerszy zakres. W trakcie całego badania stężenia w kolejnych dniach powinny być utrzymywane na poziomie na tyle niezmiennym, na ile to będzie praktycznie możliwe. Dla aerozoli należy wykonać cotygodniowo co najmniej jedną analizę wielkości cząsteczek dla badanej grupy; |
|
c) |
temperaturę oraz wilgotność, o ile możliwe w sposób ciągły. |
W trakcie ekspozycji i po jej zakończeniu należy prowadzić systematyczne obserwacje, zapisując ich wyniki; należy prowadzić oddzielne zapisy dla każdego zwierzęcia. Wszystkie zwierzęta należy obserwować codziennie i odnotowywać występujące u nich objawy toksyczności, włącznie z czasem ich pojawienia się, stopniem ich ciężkości i czasem trwania. Obserwacje powinny obejmować zmiany skóry i sierści, oczu i błon śluzowych oraz zmiany układu oddechowego, krążenia, autonomicznego i ośrodkowego układu nerwowego, jak też wzorców aktywności somatomotorycznej i wzorca zachowania. Co tydzień należy ważyć zwierzęta. Zaleca się także przeprowadzanie cotygodniowych pomiarów wielkości spożycia karmy. Regularna obserwacja zwierząt jest niezbędna do tego, aby w możliwym zakresie nie stracić zwierząt badanych z takich przyczyn, jak kanibalizm, autoliza tkanek lub przemieszczenie. Pod koniec okresu badania wszystkie osobniki, które przeżyły w grupach innych niż satelicka, poddaje się sekcji zwłok. Zwierzęta konające oraz zwierzęta przejawiające ostre cierpienie i ból powinny być usuwane, gdy zostanie to zauważone, humanitarnie zabite i poddane sekcji zwłok.
Następujące badania należy wykonać pod koniec okresu badania u wszystkich zwierząt, włącznie z osobnikami w grupie kontrolnej:
|
(i) |
badania hematologiczne, w tym co najmniej hematokryt, stężenie hemoglobiny, liczba erytrocytów, liczba leukocytów z rozmazem i oznaczenie parametrów krzepnięcia; |
|
(ii) |
laboratoryjne badania biochemiczne, włączając co najmniej jeden parametr funkcyjny wątroby i nerek: aktywność aminotransferazy alaninowej (uprzednio nazywanej transferazą glutaminianowo-pirogronową), aktywność aminotransferazy asparaginianowej (uprzednio nazywanej transferazą glutaminianowo-szczawiooctową) w surowicy, stężenie azotu mocznikowego, albumin, kreatyniny we krwi, całkowite stężenie bilirubiny i całkowite stężenie białka w surowicy. |
Do innych oznaczeń, które mogą okazać się niezbędne do odpowiedniej oceny toksykologicznej, należą oznaczenia stężenia wapnia, fosforu, chlorków, sodu, potasu, glukozy na czczo, lipidogram, stężenia hormonów i oznaczenia równowagi kwasowo-zasadowej, stężenia methemoglobiny oraz aktywności cholinesterazy.
W razie potrzeby można zastosować dodatkowe oznaczenia biochemiczne, aby rozszerzyć badania przyczyn zaobserwowanych działań toksycznych.
1.6.3.1. Pełne badanie patomorfologiczne
Wszystkie zwierzęta objęte badaniem powinny zostać poddane pełnemu makroskopowemu badaniu patomorfologicznemu. Co najmniej wątroba, nerki, nadnercza, płuca i organy badane należy zważyć w stanie wilgotnym tak szybko jak to możliwe po wycięciu, aby zapobiec wysuszeniu. Narządy i tkanki (wątrobę, nerki, śledzionę, nadnercza i serce, jak też wszelkie narządy wykazujące makroskopowe zmiany patologiczne lub o nieprawidłowych wymiarach) należy zakonserwować w odpowiednim środku w celu przeprowadzenia ewentualnych, przyszłych badań histopatologicznych. Płuca należy usunąć w stanie nienaruszonym, zważyć i zakonserwować w odpowiedniej substancji utrwalającej dla zapewnienia utrzymania ich struktury.
1.6.3.2. Badania histopatologiczne
W grupie otrzymującej najwyższe stężenie i w grupie kontrolnej (grupach kontrolnych) należy wykonać badanie histologiczne zakonserwowanych narządów i tkanek. We wszystkich grupach otrzymujących niższe dawki należy przeprowadzić badanie tych narządów i tkanek, w których stwierdzono zmiany przypisywane badanej substancji przy zastosowaniu najwyższych dawek. Należy również przeprowadzić badanie histologiczne zwierząt we wszelkich grupach satelickich, ze szczególnym naciskiem na te narządy i tkanki, w których stwierdzono skutki podania substancji w innych badanych grupach.
2. DANE
Dane należy przedstawić w skrócie w formie tabelarycznej, z podaniem – dla każdej badanej grupy – liczby zwierząt na początku badania oraz liczby zwierząt z każdym rodzajem zmiany.
Wszystkie zaobserwowane wyniki należy ocenić przy pomocy właściwej metody statystycznej. Można zastosować dowolną, uznaną metodę statystyczną.
3. SPORZĄDZANIE SPRAWOZDANIA
3.1. SPRAWOZDANIE Z BADANIA
Sprawozdanie z badań zawiera w miarę możliwości następujące informacje:
|
— |
gatunek, szczep, źródło, warunki środowiska, dieta itp., |
|
— |
warunki badania: Opis urządzenia użytego do zapewnienia ekspozycji włącznie z jego konstrukcją, rodzajem, wymiarami, źródłem powietrza, systemem generowania cząstek i aerozoli, metodą klimatyzacji i metodą trzymania zwierząt w komorze do badań w razie jej wykorzystania. Należy opisać urządzenia zastosowane do pomiaru temperatury, wilgotności i, gdzie sytuacja tego wymaga, stabilności stężenia i rozkładu wielkości cząstek w aerozolu. Dane ekspozycji: Powinny być przedstawione w formie tabeli, z podaniem wartości średniej i miary zmienności (np. odchylenia standardowego) i powinny, o ile to możliwe zawierać:
|
|
— |
dane reakcji toksycznej w rozbiciu na płcie i stężenia, |
|
— |
czas zgonu w trakcie badania lub fakt, że zwierzęta przeżyły do jego zakończenia, |
|
— |
opis działań toksycznych lub innych; poziom niewywierający działania, |
|
— |
czas zaobserwowania każdego nieprawidłowego objawu i późniejszy przebieg, |
|
— |
dane na temat karmy i masy ciała, |
|
— |
zastosowane badania hematologiczne i ich wyniki, |
|
— |
zastosowane laboratoryjne badania biochemiczne i ich wyniki, |
|
— |
wyniki sekcji zwłok, |
|
— |
szczegółowy opis wszystkich wyników histopatologicznych, |
|
— |
o ile to możliwe, statystyczna obróbka wyników, |
|
— |
omówienie wyników, |
|
— |
interpretacja wyników. |
3.2. OCENA I INTERPRETACJA
Zob. Wprowadzenie ogólne część B lit. D).
4. LITERATURA
Zeb. Wprowadzenie ogólne część B lit. E).
B.9. TOKSYCZNOŚĆ POWTARZANEJ (28 DNI) DAWKI (DERMALNA)
1. METODA
1.1. WPROWADZENIE
Zob. Wprowadzenie ogólne część B lit. A).
1.2. DEFINICJE
Zob. Wprowadzenie ogólne część B lit. B).
1.3. SUBSTANCJE ODNIESIENIA
Brak.
1.4. ZASADA METODY BADANIA
Substancję badaną podaje się na skórę w stopniowanych dawkach kilku grupom zwierząt doświadczalnych, po jednej dawce na grupę, przez okres 28 dni. W okresie podawania prowadzi się codzienną obserwację zwierząt w celu wykrycia objawów toksyczności. Zwierzęta, które zdechną w trakcie badania, i te, które przeżyją po jego zakończeniu, poddaje się sekcji zwłok.
1.5. KRYTERIA JAKOŚCI
Brak.
1.6. OPIS METODY BADANIA
1.6.1. Przygotowania
Zwierzęta muszą być trzymane w doświadczalnych warunkach pobytu i karmienia przez co najmniej pięć dni przed przeprowadzeniem badania. Przed przeprowadzeniem badania zdrowe, młode, dorosłe zwierzęta są wybierane losowo i przydzielane do poszczególnych grup poddanych działaniu substancji i kontrolnych. Na krótko przed rozpoczęciem eksperymentu należy usunąć sierść z grzbietowej powierzchni tułowia zwierząt przez przycięcie lub ogolenie. Można zastosować ogolenie, jednak należy je przeprowadzić na około 24 godziny przed rozpoczęciem badania. Na ogół konieczne jest powtarzanie przycinania lub golenia sierści w odstępach około jednego tygodnia. Podczas przycinania lub golenia sierści należy uważać, aby nie uszkodzić skóry. Nie mniej niż 10 % powierzchni ciała powinno być wolne w celu zastosowania substancji badanej. Przy podejmowaniu decyzji o wielkości powierzchni ciała, jaką należy oczyścić, oraz o rozmiarach pokrycia tej powierzchni substancją należy uwzględnić masę ciała zwierzęcia. Przy badaniu ciała stałego, które można sproszkować gdy to stosowne, substancja badana powinna być wystarczająco nawilżona wodą lub, w miarę potrzeby, stosownym podłożem dla zapewnienia dobrego kontaktu ze skórą. Płynne substancje badane stosuje się na ogół w stanie nierozcieńczonym. Substancję stosuje się codziennie przez pięć do siedmiu dni w tygodniu.
1.6.2. Warunki badania
1.6.2.1. Zwierzęta doświadczalne
Można wykorzystać dorosłe szczury, króliki lub świnki morskie. Można stosować inne gatunki, lecz ich użycie wymaga uzasadnienia.
Na początku badania zmienność masy ciała zastosowanych zwierząt nie powinna przekraczać ± 20 % odpowiedniej wartości średniej.
1.6.2.2. Liczba i płeć
Wykorzystuje się co najmniej 10 zwierząt (pięć samic i pięciu samców) ze zdrową, nienaruszoną skórą w przypadku każdego poziomu dawkowania. Samice powinny być nieródkami i nie mogą być w ciąży. Jeżeli planuje się uśmiercanie zwierząt w międzyczasie, ogólna ich liczba powinna być zwiększona o liczbę zwierząt zaplanowanych do uśmiercenia przed zakończeniem badania. Dodatkowo, grupa satelicka 10 zwierząt (pięć zwierząt każdej płci) może być poddana działaniu wysokiego poziomu dawki przez 28 dni i obserwowana, przez 14 dni po dawkowaniu, pod kątem odwracalności, trwałości i opóźnionego wystąpienia objawów zatrucia. Jest także stosowana grupa satelicka 10 zwierząt kontrolnych (pięć zwierząt każdej płci).
1.6.2.3. Wielkości dawek
Należy zastosować co najmniej trzy wielkości dawek z grupą kontrolną lub grupą kontrolną otrzymującą podłoże, o ile to ostatnie jest stosowane. Czas ekspozycji powinien wynosić co najmniej sześć godzin na dobę. Substancję badaną należy podawać o podobnej porze każdego dnia, przy czym wielkości dawek należy tak dostosowywać w odpowiednich odstępach czasu (co tydzień lub co dwa tygodnie), aby utrzymać stałą wielkość dawki w przeliczeniu na masę ciała zwierzęcia. Z wyjątkiem zastosowania substancji badanej, ze zwierzętami grupy kontrolnej powinno się obchodzić w sposób identyczny jak z podmiotami grupy badanej. W przypadku stosowania podłoża w celu ułatwienia dawkowania, u zwierząt kontrolnych otrzymujących podłoże należy stosować samo to podłoże w taki sam sposób, jak w grupach otrzymujących substancję badaną, w takiej samej dawce, jak stosowana w grupie otrzymującej najwyższą dawkę. Najwyższa dawka powinna być tak dobrana, aby wywierała działanie toksyczne, jednak nie prowadziła do zgonów lub prowadziła do jedynie niewielu zgonów. Najniższa dawka nie powinna dawać żadnych objawów toksyczności. W przypadku gdy istnieje możliwa do wykorzystania, oszacowana wielkość ekspozycji u ludzi, najniższy poziom dawkowania powinien ją przewyższać. W idealnej sytuacji pośredni poziom dawkowania powinien prowadzić do uzyskania minimalnych dostrzegalnych efektów toksycznych. W przypadku gdy zostanie wykorzystana więcej niż jedna dawka pośrednia, wielkości dawek powinny być tak rozstawione, aby uzyskać stopniowanie działań toksycznych. W grupach otrzymujących niskie i pośrednie dawki oraz w grupach kontrolnych częstość występowania zgonów powinna być niska, aby umożliwić znaczącą ocenę wyników.
W przypadku gdy zastosowanie substancji badanej wiąże się z ciężkim działaniem drażniącym na skórę, stężenia należy zmniejszyć, przy czym może to spowodować zmniejszenie pojawiania się lub brak innych skutków toksycznych przy najwyższym poziomie dawkowania. Ponadto w przypadku znacznego uszkodzenia skóry może być konieczne przerwanie badania i podjęcie nowego eksperymentu z zastosowaniem niższych stężeń.
1.6.2.4. Czas ekspozycji
W przypadku gdy w badaniu wstępnym z zastosowaniem dawki 1 000 mg/kg lub wyższej, odpowiadającej potencjalnej ekspozycji u ludzi, o ile taki poziom jest znany, nie zostaną stwierdzone żadne efekty toksyczne, można uznać, że dalsze badania nie są potrzebne.
1.6.2.5. Okres obserwacji
Zwierzęta doświadczalne należy obserwować codziennie pod kątem występowania objawów toksyczności. Należy zapisać czas zgonu zwierzęcia i czas pojawienia się i ustąpienia objawów toksyczności.
1.6.3. Procedura
Zwierzęta powinny być trzymane w oddzielnych klatkach. W idealnej sytuacji podaje się im substancję badaną przez siedem dni w tygodniu przez okres 28 dni. Zwierzęta w grupach satelickich, w których planuje się późniejszą obserwację, powinny zostać przetrzymane przez następnych 14 dni bez podawania im substancji w celu stwierdzenia ustępowania lub utrzymywania się działań toksycznych. Czas ekspozycji powinien wynosić co najmniej sześć godzin na dobę.
Substancja badana powinna zostać nałożona w sposób równomierny na obszar równy około 10 % całkowitej powierzchni ciała. W przypadku substancji wysoce toksycznych można je nanieść na mniejszą powierzchnię, jednak należy dążyć do tego, aby pokryć jak największy obszar jak najcieńszą i jak najbardziej równomierną warstwą badanego związku.
W trakcie ekspozycji substancja badana jest utrzymywana w kontakcie ze skórą dzięki zastosowaniu porowatego opatrunku z gazy i niedrażniącej taśmy. Miejsce badania należy dodatkowo przykryć w taki sposób, aby utrzymać opatrunek z gazy i substancję badaną i zapewnić, aby zwierzęta nie mogły zjeść tej ostatniej. W celu zapobieżenia spożyciu substancji badanej można uwiązać zwierzę, jednak pełna immobilizacja nie jest zalecaną metodą. Jako alternatywę można zastosować „kołnierzowy przyrząd zabezpieczający”.
Pod koniec okresu ekspozycji należy usunąć resztki badanej substancji, w miarę możliwości stosując wodę lub jakieś inne, właściwe metody oczyszczania skóry.
Wszystkie zwierzęta należy obserwować codziennie i odnotowywać występujące u nich objawy toksyczności, włącznie z czasem ich pojawienia się, stopniem ich ciężkości i czasem trwania. Obserwacje przy klatce zwierzęcia powinny obejmować zmiany skóry i sierści, oczu i błon śluzowych oraz zmiany układu oddechowego, krążenia, autonomicznego i ośrodkowego układu nerwowego, jak też wzorców aktywności somatomotorycznej i wzorca zachowania. Co tydzień należy ważyć zwierzęta. Zaleca się także przeprowadzanie cotygodniowych pomiarów wielkości spożycia karmy. Regularna obserwacja zwierząt jest niezbędna do tego, aby w możliwym zakresie nie stracić zwierząt badanych z takich przyczyn, jak kanibalizm, autoliza tkanek lub przemieszczenie. Pod koniec okresu badania wszystkie osobniki, które przeżyły w grupach innych niż satelicka, poddaje się sekcji zwłok. Zwierzęta konające oraz zwierzęta przejawiające ostre cierpienie i ból powinny być usuwane, gdy zostanie to zauważone, humanitarnie zabite i poddane sekcji zwłok.
Następujące badania należy wykonać pod koniec okresu badania u wszystkich zwierząt, włącznie z osobnikami w grupie kontrolnej:
|
1) |
badania hematologiczne, w tym co najmniej hematokryt, stężenie hemoglobiny, liczba erytrocytów, liczba leukocytów z rozmazem i oznaczenie parametrów krzepnięcia; |
|
2) |
laboratoryjne badania biochemiczne krwi zawierające co najmniej jeden parametr funkcji wątroby i nerek: aktywność aminotransferazy alaninowej (uprzednio nazywanej transferazą glutaminianowo-piro- gronową) w surowicy, aktywność aminotransferazy asparaginianowej (uprzednio nazywanej transferazą glutaminianowo-szczawiooctową) w surowicy, stężenie azotu mocznikowego, albumin, kreatyniny we krwi, całkowite stężenie bilirubiny i całkowite stężenie białka w surowicy. |
Do innych oznaczeń, które mogą okazać się niezbędne do odpowiedniej oceny toksykologicznej, należą oznaczenia stężenia wapnia, fosforu, chlorków, sodu, potasu, glukozy na czczo, lipidogram, stężenia hormonów i oznaczenia równowagi kwasowo-zasadowej, stężenia methemoglobiny oraz aktywności cholinesterazy.
W razie potrzeby można zastosować dodatkowe oznaczenia biochemiczne, aby rozszerzyć badania zaobserwowanych działań toksycznych.
1.6.4. Pełne badanie patomorfologiczne
Wszystkie zwierzęta objęte badaniem powinny zostać poddane pełnemu makroskopowemu badaniu patomorfologicznemu. Należy zważyć wątrobę, nerki, nadnercza i jądra w stanie wilgotnym, najszybciej, jak to będzie możliwe po rozcięciu zwłok, aby uniknąć wysuszenia. Narządy i tkanki, tj. prawidłową i poddawaną ekspozycji na substancję skórę, wątrobę, nerki i narządy docelowe (czyli narządy wykazujące makroskopowe zmiany patologiczne lub o nieprawidłowych wymiarach) należy zakonserwować w odpowiednim środku w celu przeprowadzenia ewentualnych, przyszłych badań histopatologicznych.
1.6.5. Badania histopatologiczne
W grupie otrzymującej najwyższą dawkę i w grupie kontrolnej należy wykonać badanie histologiczne zakonserwowanych narządów i tkanek. We wszystkich grupach otrzymujących niższe dawki należy przeprowadzić badanie tych narządów i tkanek, w których stwierdzono zmiany przypisywane badanej substancji przy zastosowaniu najwyższych dawek. Należy również przeprowadzić badanie histologiczne zwierząt w grupie satelickiej, ze szczególnym naciskiem na te narządy i tkanki, w których stwierdzono skutki podania substancji w innych badanych grupach.
2. DANE
Dane należy przedstawić w skrócie w formie tabelarycznej, z podaniem – dla każdej badanej grupy – liczby zwierząt na początku badania oraz liczby zwierząt z każdym rodzajem zmiany patologicznej.
Wszystkie zaobserwowane zmiany należy ocenić przy pomocy właściwej metody statystycznej. Można zastosować dowolną, uznaną metodę statystyczną.
3. SPORZĄDZANIE SPRAWOZDANIA
3.1. SPRAWOZDANIE Z BADANIA
Sprawozdanie z badań zawiera w miarę możliwości następujące informacje:
|
— |
dane zwierząt (gatunek, szczep, źródło, warunki środowiska, dieta itp.), |
|
— |
warunki badania (włączając typ opatrunku: okluzyjny lub nieokluzyjny), |
|
— |
wielkości dawek (włączając podłoże, o ile jest wykorzystane) i stężenia, |
|
— |
poziom niewywierający działania, o ile to możliwe, |
|
— |
dane reakcji toksycznej w rozbiciu na płci i dawki, |
|
— |
czas zgonu w trakcie badania lub fakt, że zwierzęta przeżyły do jego zakończenia, |
|
— |
działania toksyczne lub inne, |
|
— |
czas zaobserwowania każdego nieprawidłowego objawu i późniejszy przebieg zmian, |
|
— |
dane na temat karmy i masy ciała, |
|
— |
zastosowane badania hematologiczne i ich wyniki, |
|
— |
zastosowane laboratoryjne badania biochemiczne i ich wyniki, |
|
— |
wyniki sekcji zwłok, |
|
— |
szczegółowy opis wszystkich wyników histopatologicznych, |
|
— |
gdzie to możliwe, statystyczna obróbka wyników, |
|
— |
omówienie wyników, |
|
— |
interpretacja wyników. |
3.2. OCENA I INTERPRETACJA
Zob. Wprowadzenie ogólne część B lit. D).
4. LITERATURA
Zob. Wprowadzenie ogólne część B lit. E).
B.10. MUTAGENNOŚĆ – BADANIE ABERRACJI CHROMOSOMOWEJ SSAKÓW IN VITRO
1. METODA
Metoda ta jest powtórzeniem OECD TG 473, metody badania aberracji chromosomowej ssaków in vitro (1997).
1.1. WPROWADZENIE
Celem badania chromosomowej aberracji u ssaków jest zidentyfikowanie czynników, które powodują strukturalne chromosomowe aberracje w hodowanych komórkach ssaków (1)(2)(3). Strukturalne aberracje mogą występować w dwóch typach, chromosomowym lub chromatydowym. W przypadku mutagenów chemicznych większość wywołanych aberracji jest typu chromatydowego, ale mogą się pojawiać aberracje typu chromosomowego. Wzrost poliploidalności może wskazywać, że substancja chemiczna ma potencjał do wywoływania aberracji liczbowej. Jednakże metoda ta nie jest wykorzystywana do pomiarów aberracji liczbowych i nie jest rutynowo wykorzystywana do tego celu. Mutacje chromosomowe oraz powiązane z nimi zdarzenia są przyczyną wielu chorób genetycznych u ludzi oraz istnieje dowód na to, że mutacje chromosomowe oraz związane z nimi zdarzenia powodujące zmiany w onkogenach oraz genach hamowania nowotworów komórek somatycznych są odpowiedzialne za wywoływanie raka u ludzi oraz zwierząt doświadczalnych.
Aberracja chromosomowa in vitro może wykorzystywać ustanowione linie komórkowe, łańcuchy komórkowe lub pierwotne hodowle komórek. Wykorzystane komórki są wybierane na podstawie możliwości wzrostu hodowli, stabilności kariotypu, liczby chromosomów, zróżnicowania chromosomów oraz spontanicznych częstotliwości aberracji.
Badania przeprowadzane in vitro wymagają wykorzystania źródła egzogennego aktywacji metabolicznej. Ten system aktywacji metabolicznej nie może imitować w całości warunków in vivo w odniesieniu do ssaków. Powinna zostać zachowana ostrożność w celu uniknięcia warunków, które mogłyby prowadzić do pozytywnych wyników, które nie odzwierciedlają wewnętrznej mutagenności oraz mogą wynikać ze zmian w pH, stężeniu osmolalnym lub wysokich poziomów cytotoksyczności (4)(5).
Badanie to jest wykorzystywane w celu sprawdzenia możliwych czynników mutagennych oraz rakotwórczych dla ssaków. Wiele związków, które mają charakter dodatni w tym badaniu, są czynnikami rakotwórczymi dla ssaków, jednakże nie istnieje idealne powiązanie między tym badaniem a rakotwórczością. Powiązanie zależne jest od klasy chemicznej oraz jest coraz więcej dowodów na to, że istnieją czynniki rakotwórcze, które nie są wykrywane w drodze tego badania, ponieważ wydaje się, iż działają one poprzez mechanizmy inne niż bezpośrednie niszczenie DNA.
Zob. również Ogólne wprowadzenie do części B.
1.2. DEFINICJE
Aberracja typu chromatydowego: strukturalne uszkodzenie chromosomu, wyrażone jako pękanie pojedynczych chromatydów lub pękanie oraz ponowne łączenie między chromatydami.
Aberracja typu chromosomowego: strukturalne uszkodzenie chromosomu, wyrażone jako pękanie pojedynczych chromatydów lub pękanie oraz ponowne łączenie obu chromatydów w identycznej lokalizacji.
Endoreduplikacja: proces, w którym po okresie replikacji DNA jądro nie ulega mitozie, ale rozpoczyna nowy okres S. Wynikiem są chromosomy o 4, 8, 16, … chromatydach.
Szczelina: achromatyczne uszkodzenie mniejsze niż szerokość chromatydu, powodujące minimalne wypaczenie chromatydu.
Indeks mitotyczny: stosunek komórek w metafazie podzielony przez całkowitą liczba komórek zaobserwowany w populacji komórek; wskazanie stopnia proliferacji tej populacji.
Aberracja liczbowa: zmiana w ilości chromosomów w odniesieniu do normalnej ilości charakterystycznej dla wykorzystywanych komórek.
Poliploidalność: wielokrotność ilości chromosomów haploidalnych (n) inna niż liczba diploidalna (np. 3n, 4n itd.).
Aberracja strukturalna: zmiana w strukturze chromosomu, wykrywalna przez badanie mikroskopowe etapu metafazy podziału komórek, zaobserwowana jako usunięcia oraz fragmenty, zmiany (zachodzące – wewnątrz oraz między chromosomami).
1.3. ZASADA METODY BADAWCZEJ
Hodowle komórek są poddane działaniu substancji badanej zarówno z aktywacją, jak i bez aktywacji metabolicznej. Substancje są poddawane działaniu metafazy zatrzymującej (np. Colcemid® lub kolchicyna) substancje we wcześniej określonych odstępach czasu po poddaniu hodowli komórek działaniu substancji badanej, komórki pobrane, zabarwione oraz komórki metafazy są analizowane mikroskopowo na obecność aberracji chromosomowych.
1.4. OPIS METODY BADAWCZEJ
1.4.1. Preparaty
1.4.1.1. Komórki
Wykorzystywane mogą być rozmaite linie komórkowe, łańcuchy lub pierwotne hodowle komórek łącznie z komórkami ludzkimi (np. fibroblasty chomika chińskiego, limfocyty krwi obwodowej ludzi lub ssaków).
1.4.1.2. Warunki dotyczące podłoża hodowli i hodowli
W utrzymywaniu hodowli powinny być stosowane odpowiednie warunki podłoża hodowli oraz inkubacji (naczynia do hodowli, stężenie CO2, temperatura oraz wilgotność). Ustanowione linie komórkowe oraz szczepy powinny być rutynowo sprawdzane pod kątem stabilności zmiennej ilości chromosomów oraz nieobecności zanieczyszczenia mykoplazmą oraz nie powinny być wykorzystywane, jeżeli są zanieczyszczone. Normalny czas cyklów komórkowych oraz wykorzystywane warunki dotyczące hodowli powinny być znane.
1.4.1.3. Preparaty hodowli
Ustanowione linie komórkowe oraz łańcuchy: komórki są rozmnażane z hodowli podstawowej, siane na podłożu hodowli w takiej gęstości, że hodowle nie osiągną zrastania się przed czasem pobierania, oraz są inkubowane w temperaturze 37 oC.
Limfocyty: krew pełna jest poddawana działaniu antykoagulantu (np. heparyny) lub oddzielone limfocyty uzyskane ze zdrowych przedmiotów są dodawane do podłoża hodowli zawierającego mitogen (np. phytohaemagglutinin) oraz inkubowane w temperaturze 37 oC.
1.4.1.4. Aktywacja metaboliczna
Komórki powinny zostać poddane działaniu substancji badanej zarówno w obecności, jak i w nieobecności odpowiedniego systemu aktywacji metabolicznej. Najpowszechniej wykorzystywanym systemem jest frakcja przesączu znad mitochondriów uzupełniona o kofaktor, przygotowana z wątroby gryzoni poddanej działaniu substancji, czynnikami pobudzającymi enzymy, takimi jak Aroklor 1254 (6)(7)(8)(9) lub mieszanina fanobarbitonu oraz β-naftoflawonu (10)(11)(12).
Frakcja przesączu znad mitochondriów jest zazwyczaj wykorzystywana w stężeniach w zakresie 1–10 % objętościowo w końcowym nośniku badania. Warunki systemu aktywacji metabolicznej mogą być uzależnione od klasy badanej substancji chemicznej. W niektórych przypadkach właściwe może być wykorzystanie więcej niż jednego stężenia frakcji przesączu znad mitochondriów.
Pewna liczba ulepszeń, w tym konstrukcja genetycznie modyfikowanych linii komórkowych wyrażających szczególne enzymy aktywujące, mogą zapewnić możliwość aktywacji endogenicznej. Wybór wykorzystanych linii komórkowych powinien być uzasadniony naukowo (np. przez znaczenie izoenzymu cyto-chromowego P450 dla metabolizmu substancji).
1.4.1.5. Substancja badana/przygotowanie
Stała substancja badana powinna zostać rozpuszczona lub zawieszona w odpowiednich rozpuszczalnikach lub nośnikach oraz rozcieńczona, jeżeli jest to odpowiednie, przed poddaniem jej działaniu komórek. Płynne substancje badane mogą być dodawane bezpośrednio do systemu badawczego i/lub rozcieńczane przed poddaniem ich działaniu komórek. Powinny zostać wykorzystane świeżo przygotowywane substancje, chyba że dane dotyczące stabilności wskazują, iż dopuszczalne jest składowanie substancji.
1.4.2. Warunki badania
1.4.2.1. Rozpuszczalnik/nośnik
Rozpuszczalnik/nośnik nie powinien mieć możliwości reagowania chemicznego z substancją badaną oraz powinien być zgodny z przeżywaniem komórek oraz aktywnością S9. Jeśli inne niż dobrze znane rozpuszczalniki/nośniki są wykorzystywane, ich włączenie do badania powinno być wsparte danymi wskazującymi ich zgodność. Zaleca się, o ile to możliwe, aby w pierwszej kolejności rozważone zostało wykorzystanie wodnych rozpuszczalników/nośników. Badając substancje nietrwałe w wodzie, wykorzystane rozpuszczalniki organiczne powinny być wolne od wody. Woda może zostać usunięta przez zastosowanie sita molekularnego.
1.4.2.2. Stężenia poddania działaniu substancji
Wśród kryteriów, które mają zostać uwzględnione przy określaniu najwyższego stężenia, znajduje się cytotoksyczność, rozpuszczalność w systemie oraz zmiany pH lub osmolalność.
Cytotoksyczność powinna zostać określona z aktywacją lub bez aktywacji metabolicznej w ramach głównego doświadczenia, przy wykorzystaniu odpowiedniego wskazania dotyczącego integralności komórki oraz wzrostu, takiego, jak stopień zrastania się, liczba komórek zdolnych do życia lub indeksy mitotyczne. Może to być użyteczne do oznaczenia cytotoksyczności oraz rozpuszczalności w doświadczeniu wstępnym.
Powinny zostać wykorzystane co najmniej trzy nadające się do analizy stężenia. W przypadku gdy pojawia się cytotoksyczność, stężenia te powinny obejmować zakres od najwyższej do małej toksyczności lub jej braku; będzie to zazwyczaj oznaczać, że stężenia powinny zostać rozdzielone przez nie więcej niż jeden czynnik między 2 a √10. W czasie pobierania wyhodowanych komórek najwyższe stężenia powinny wskazywać znaczące zmniejszenie w stopniu zrastania się, liczbie komórek lub indeksie mitotycznym (wszystkie wyższe niż 50 %). Indeks mitotyczny jest jedynie bezpośrednią miarą skutków cytotoksycznych/cytostatycznych oraz jest zależny od czasu po poddaniu działaniu substancji. Jednakże indeks mitotyczny jest dopuszczalny w odniesieniu do hodowli zawiesinowych, w których inne pomiary toksyczności mogą być niewygodne i niepraktyczne. Informacje w sprawie kinetyki komórek, takie jak średnie czasy tworzenia się (AGT), mogą być wykorzystywane jako informacje dodatkowe. Jednakże AGT jest ogólną średnią, która nie zawsze ujawnia istnienie opóźnionych podpopulacji, a nawet delikatne wzrosty średnich czasów tworzenia się mogą być związane z bardzo zasadniczym opóźnieniem w czasie optymalnych wydajności aberracji.
W odniesieniu do substancji względnie niecytotoksycznych maksymalne stężenie badawcze powinno wykosić 5 μl/ml, 5 mg/ml lub 0,01 M, w zależności od tego, które z nich jest najniższe.
W odniesieniu do substancji względnie nierozpuszczalnych, które nie są toksyczne w stężeniach niższych niż stężenia nierozpuszczalne, najwyższa wykorzystana dawka powinna być stężeniem powyżej limitu rozpuszczalności w końcowym podłożu hodowli na koniec okresu poddawania działaniu substancji. W niektórych przypadkach (np. kiedy toksyczność pojawia się wyłącznie przy wyższym niż najniższe stężeniu nierozpuszczalnym) słuszne jest przeprowadzenie badania na jednym większym stężeniu z widocznym strącaniem się. Użyteczna może być ocena rozpuszczalności na początku oraz na końcu poddawania działaniu substancji, ze względu na to, że rozpuszczalność może zmieniać się w trakcie trwania poddania działaniu substancji w systemie badawczym spowodowana obecnością komórek, surowicy S9 itp. Nierozpuszczalność może zostać wykryta okiem nieuzbrojonym. Stężanie nie powinno zakłócać oceny.
1.4.2.3. Kontrole pozytywne i negatywne
Równoległe kontrole pozytywne i negatywne (rozpuszczalnik lub nośnik), zarówno z aktywacją metaboliczną, jak i bez niej, powinny zostać objęte każdym doświadczeniem. Kiedy wykorzystywana jest aktywacja metaboliczna, pozytywna kontrolna substancja chemiczna jest substancją, która wymaga aktywacji w celu wywołania reakcji mutagennej.
Pozytywne kontrole powinny wykorzystywać znany elastogen przy poziomie poddania działaniu substancji, od którego oczekuje się wytworzenia odtwarzalnego oraz wykrywalnego wzrostu poza tło, które wskazuje czułość systemu.
Stężenia pozytywnych kontroli powinny zostać wybrane tak, aby były jasne, ale by badającemu nie ujawniały bezpośrednio tożsamości zakodowanych szkiełek laboratoryjnych. Przykłady pozytywnych substancji kontrolnych obejmują:
|
Warunki aktywacji metabolicznej |
Substancja |
Nr CAS |
Nr Einecs |
|
Nieobecność egzogenicznej aktywacji metabolicznej |
Metanosulfonat metylu |
66-27-3 |
200-625-0 |
|
Metanosulfonat etylu |
62-50-0 |
200-536-7 |
|
|
Nitrozomocznik etylu |
759-73-9 |
212-072-2 |
|
|
Mitomycyna C |
50-07-7 |
200-008-6 |
|
|
N-tlenek-4-nitrokwinolinu |
56-57-5 |
200-281-1 |
|
|
Obecność egzogenicznej aktywacji metabolicznej |
Benzo[a]piren |
50-32-8 |
200-028-5 |
|
Cyklofosfamid Cyklofosfamid monohydrat |
50-18-0 6055-19-2 |
200-015-4 |
Mogą być wykorzystane inne właściwe substancje kontroli pozytywnych. Wykorzystanie substancji chemicznej z klasy powiązanej z kontrolą pozytywną powinno zostać rozważone, jeżeli jest dostępne.
Kontrole negatywne, składające się z rozpuszczalników lub samego nośnika w podłożu poddania działaniu substancji, oraz poddanie działaniu substancji w ten sam sposób co hodowle poddane działaniu substancji, powinny zostać włączone w ramy każdego okresu pobierania hodowli. Dodatkowo wykorzystywane powinny być kontrole, w ramach których nie poddaje się działaniu substancji, chyba że istnieją dane historyczne dotyczące kontroli wskazujące na to, że inne skutki szkodliwe lub mutagenne są wywołane wybranym rozpuszczalnikiem.
1.4.3. Procedura
1.4.3.1. Poddanie działaniu substancji badanej
Komórki rozmnażające się wegetatywnie są poddawane działaniu, wykorzystując substancję badaną w obecności oraz nieobecności systemu aktywacji metabolicznej. Przetwarzanie limfocytów powinno następować w około 48 godzin po stymulacji mitogenicznej.
|
1.4.3.2. |
Hodowle reprodukcyjne powinny normalnie być wykorzystywane przy każdym stężeniu oraz są one silnie zalecane w odniesieniu do negatywnych/rozpuszczalnikowych hodowli kontrolnych. W przypadku gdy na podstawie danych historycznych mogą zostać przedstawione minimalne zmiany między hodowlami reprodukcyjnymi (13)(14), dopuszczalne może być, aby pojedyncze hodowle miały być wykorzystywane przy każdym badaniu. Substancje gazowe lub lotne powinny być badane odpowiednia metodą, taką jak badanie w szczelnie zamkniętych naczyniach do hodowli (15)(16). |
1.4.3.3. Czas pobierania hodowli
W pierwszym doświadczeniu komórka powinna być poddana działaniu substancji badanej, zarówno z aktywacja metaboliczną, jak i bez niej, przez 3–6 godzin oraz próbki powinny zostać pobrane w czasie równoważnym długości około 1,5 cyklu komórkowego po rozpoczęciu poddawania działaniu substancji (12). Jeżeli ten protokół daje negatywne wyniki, zarówno z aktywacją, jak i bez niej, powinno zostać przeprowadzone dodatkowe doświadczenie, z ciągłym poddawaniem działaniu substancji badanej do momentu pobrania próbek w czasie równoważnym długości 1,5 cyklu komórkowego. Niektóre substancje chemiczne mogą być wykryte z mniejszym trudem przez czasy poddawania działaniu substancji badanej/pobierania próbek dłuższe niż długości 1,5 cyklu. Wyniki negatywne z metaboliczną aktywacją muszą zostać ustalone na zasadzie jednostkowych przypadków. W tych przypadkach, gdy potwierdzenie negatywnych wyników nie jest uznawane za niezbędne, powinno zostać zapewnione uzasadnienie.
1.4.3.4. Preparaty chromosomów
Hodowle komórek są poddawane działaniu Colcemidu® lub kolchicyny zazwyczaj przez 1–3 godzin przed pobraniem hodowli. Każda hodowla komórek jest pobierana oraz poddawana działaniu substancji oddzielnie w celu przygotowania chromosomów. Przygotowanie chromosomów obejmuje hipotoniczne przetwarzanie komórek, wiązanie oraz barwienie.
1.4.3.5. Analiza
Wszystkie szkiełka mikroskopowe, włącznie z tymi dotyczącymi pozytywnych oraz negatywnych kontroli, powinny zostać odpowiednio zakodowane przed analizą mikroskopową. Ze względu na to, że procedury wiązania, utrwalania barwnika często skutkują przerwaniem stosunku komórek metafazy do straty chromosomów, oceniane komórki powinny z tego względu zawierać liczbę centromerów równą zmiennej liczbie ± 2 dla wszystkich rodzajów komórek. Powinno zostać ocenione przynajmniej 200 dobrze rozciągniętych metafaz na stężenie oraz kontrole, równo podzielonych między wtórniki, jeżeli ma to zastosowanie. Liczba ta może zostać zredukowana, jeśli zaobserwowana jest wysoka liczba aberracji.
Chociaż celem badania jest wykrycie strukturalnych aberracji chromosomowych, ważne jest odnotowanie poliploidalności oraz endoreduplikacji, jeśli zaobserwowane są te zdarzenia.
2. DANE
2.1. PRZETWARZANIE WYNIKÓW
Jednostką doświadczalną jest komórka, a z tego względu udział procentowy komórek ze strukturalną aberracją chromosomową powinien zostać poddany ocenie. Różne typy strukturalnej aberracji chromosomowej powinny zostać wymienione wraz z ich ilościami oraz częstotliwościami w odniesieniu do hodowli doświadczalnych oraz kontrolnych. Szczeliny są oddzielnie odnotowywane oraz zgłaszane, ale ogólnie nie są włączane do częstotliwości aberracji ogółem.
Równoległe pomiary cytotoksyczności w odniesieniu do wszystkich przetwarzanych oraz negatywnych hodowli kontrolnych w ramach głównych doświadczeń aberracji powinny również zostać odnotowane.
Powinny zostać dostarczone dane dotyczące poszczególnych hodowli. Dodatkowo wszelkie dane powinny zostać podsumowane w formie tabelarycznej.
Nie istnieje wymóg jasnej reakcji pozytywnej w odniesieniu do weryfikacji. Niejednoznaczne wyniki powinny zostać wyjaśnione w drodze dalszych badań, w szczególności z wykorzystaniem modyfikacji warunków doświadczalnych. Potrzeba potwierdzenia wyników negatywnych została omówiona w ppkt 1.4.3.3. Modyfikacje parametrów badawczych w celu poszerzenia zakresu ocenionych warunków powinna zostać rozważona w dalszych doświadczeniach. Parametry badawcze, które mogą być zmodyfikowane, obejmują zakres stężenia oraz warunki aktywacji metabolicznej.
2.2. OCENA ORAZ INTERPRETACJA WYNIKÓW
Istnieją liczne kryteria oznaczania wyników pozytywnych, takie jak związany ze stężeniem wzrost odtwarzalności w pewnej ilości komórek z aberracjami chromosomowymi. Biologiczne znaczenie wyników powinno zostać wzięte pod uwagę w pierwszej kolejności. Metody statystyczne mogą zostać wykorzystane jako metody pomocnicze w przeprowadzaniu oceny wyników (3)(13). Znaczenie statystyczne nie powinno być jedynym czynnikiem określającym w odniesieniu do reakcji pozytywnej.
Wzrost ilości komórek poliploidalnych może wskazywać, że substancja badana ma potencjał do wstrzymywania procesów mitotycznych oraz pobudzania liczbowych aberracji chromosomowych. Wzrost liczby komórek z endoreduplikowanymi chromosomami może wskazywać, że substancje badane mają potencjał wstrzymywania postępowania cyklu komórkowego (17)(18).
Substancja badana, w odniesieniu do której wyniki nie spełniają powyższych kryteriów, uważana jest za niemutagenną w tym systemie.
Chociaż większość doświadczeń będzie dawało w jasny sposób pozytywne lub negatywne wyniki, w rzadkich przypadkach zestaw danych będzie uniemożliwiał dokonanie ostatecznej oceny dotyczącej aktywności substancji badanej. Wyniki mogą pozostać niejednoznaczne lub sporne, niezależnie od tego, ile razy doświadczenia były powtarzane.
Wyniki pozytywne z badania chromosomowej aberracji in vitro wskazują, że substancja badana wywołuje aberracje. Wyniki negatywne wskazują, że w warunkach badawczych substancja badana nie wywołuje aberracji chromosomowej hodowanych komórek somatycznych ssaków.
3. SPRAWOZDAWCZOŚĆ
SPRAWOZDANIE Z BADANIA
Sprawozdanie z badania musi zawierać następujące informacje:
|
|
Rozpuszczalnik/nośnik:
|
|
|
Komórki:
|
|
|
Warunki badania:
|
|
|
Wyniki:
|
|
|
Omówienie wyników. |
|
|
Wnioski. |
4. ODNIESIENIA
|
(1) |
Evans, H. J. (1976), Cytological Methods for Detecting Chemical Mutagens, w: Chemical mutagens, Principles and Methods for their Detection, Vol. 4, Hollaender, A. (ed) Plenum Press, New York and London, pp. 1–29. |
|
(2) |
Ishidate, M. Jr. and Sofumi, T. (19 8 5), The In vitro Chromosomal Aberration Test Using Chinese Hamster Lung (CHL) Fibroblast Cells in Culture, in: Progress in Mutation Research, Vol. 5, Ashby, J. et al., (eds) Elsevier Science Publishers, Amsterdam-New York-Oxford, pp. 427–432. |
|
(3) |
Galloway, S. M., Armstrong, M. J., Reuben, C, Colman, S., Brown, B., Cannon, C, Bloom, A. D., Naka-mura, F., Ahmed, M., Duk, S., Rimpo, J., Margolin, G. H., Resnick, M. A., Anderson, G. and Zeiger E. (1978), Chromosome aberration and sister chromatic exchanges in Chinese hamster ovary cellS: Evaluation of 108 chemicals, Environs, Molec. Mutagen 10 (suppl.10), pp. 1–175. |
|
(4) |
Scott, D., Galloway, S. M., Marshall, R. R., Ishidate, M. Jr., Brusick, D., Ashby, J. and Myhr, B. C. (1991), Genotoxicity under Extreme Culture Conditions. A report from ICPEMC Task Group 9, Mutation Res., 257, pp. 147–204. |
|
(5) |
Morita, T., Nagaki, T., Fukuda, I. and Okumura, K., (1992), Clastogenicity of low pH to Various Cultured Mammalian Cells, Mutation Res., 268, pp. 297–305. |
|
(6) |
Ames, B. N., McCann, J. and Yamasaki, E. (1975), Methods for Detecting Carcinogens and Mutagens with the Salmonella/Mammalian Microsome Mutagenicity Test, Mutation Res., 31, pp. 347–364. |
|
(7) |
Maron, D. M. and Ames, B. N. (1983), Revised Methods for the Salmonella Mutagenicity Test, Mutation Res., 113, pp. 173–215. |
|
(1) (8) |
Natarajan, A. T., Tates, A. D., van Buul, P. P. W., Meijers, M. and de Vogel, N. (1976), Cytogenetic Effects of Mutagen/Carcinogens after Activation in a Mircrosomal System In vitro, I. Induction of Chromosome Aberrations and Sister Chromatid Exchange by Diethylnitrosamine (DEN) and Dime-thylnitrosamine (DMN) in CHO Cells in the Presence of Rat-Liver Microsomes, Mutation Res., 37, pp. 83–90. |
|
(9) |
Matsuoka, A., Hayashi, M. and Ishidate, M. Jr. (1979), Chromosomal Aberration Tests on 29 Chemicals Combined with S9 Mix In vitro, Mutation Res., 66, pp. 277–290. |
|
(10) |
Elliot, B. M., Combes, R. D., Elcombe, C. R., Gatehouse, D. G., Gibson, G. G., Mackay, J. M. and Wolf, R. C. (1992), Report of UK Environmental Mutagen Society Working Party. Alternative to Aroclor 1254-induced S9 in In vitro Genotoxicity Assays, Mutagenesis, 7, pp. 175–177. |
|
(11) |
Matsushima, T., Sawamura, M., Hara, K. and Sugimura, T. (1976), A. Safe Substitute for Polychlori-nated Biphenyls as an Inducer of Metabolic Activation Systems, in: de Serres, F. J. Fouts, J. R. Bend, J. R. and Philpot, R. M. (eds), In vitro Metabolic Activation in Mutagenesis Testing, Elsevier, North-Holland, pp. 85–88. |
|
(12) |
Galloway, S. M., Aardema, M. J., Ishidate, M. Jr., Ivett, J. L., Kirkland, D. J. Morita, T., Mosesso, P., Sofu-mi, T. (1994), Report from Working Group on in In vitro Tests for Chromosomal Aberrations, Mutation Res., 312, pp. 241–261. |
|
(13) |
Richardson, C, Williams, D. A., Allen, J. A., Amphlett, G., Chanter, D. O. and Phillips, B. (1989), Analysis of Data from In vitro Cytogenetic Assays, w: Statistical Evaluation of Mutagenicity Test Data, Kirkland, D. J., (cd) Cambridge University Press, Cambridge, pp. 141–154. |
|
(14) |
Soper, K. A. and Galloway, S. M. (1994), Replicate Flasks are not Necessary for In vitro Chromosome Aberration Assays in CHO Cells, Mutation Res., 312, pp. 139–149. |
|
(15) |
Krahn, D. F., Barsky, F. C. and McCooey, K. T. (1982), CHO/HGPRT Mutation Assay: Evaluation of Gases and Volatile Liquids, in: Tice, R. R., Costa, D. L., Schaich, K. M. (eds), Genotoxic Effects of Airborne Agents, New York, Plenum, pp. 91–103. |
|
(16) |
Zamora, P. O., Benson, J. M., Li, A. P. and Brooks, A. L. (1983), Evaluation of an Exposure System Using Cells Grown on Collagen Gels for Detecting Highly Volatile Mutagens in the CHO/HGPRT Mutation Assay, Environmental Mutagenesis, 5, pp. 795–801. |
|
(17) |
Locke-Huhle, C. (1983), Endoreduplication in Chinese hamster cells during alpha-radiation induced G2 arrest, Mutation Res., 119, pp. 403–413. |
|
(18) |
Huang, Y., Change, C. and Trosko, J. E. (1983), Aphidicolin-induced endoreduplication in Chinese hamster cells, Cancer Res., 43, pp. 1362–1364. |
B.11. MUTAGENNOŚĆ – BADANIE ABERRACJI CHROMOSOMOWEJ SZPIKU KOSTNEGO IN VIVO
1. METODA
Metoda ta jest powtórzeniem OECD TG 475, metody badania aberracji chromosomowej szpiku kostnego (1997).
1.1. WPROWADZENIE
Badanie aberracji chromosomowej ssaków in vivo jest wykorzystywane w celu wykrycia strukturalnych aberracji chromosomowych komórek szpiku kostnego zwierząt, zwykle gryzoni, wywołanych przez substancje badane (1)(2)(3)(4). Strukturalne aberracje mogą występować w dwóch typach, chromosomowym lub chromatydowym. Wzrost poliploidalności może wskazywać, że substancja chemiczna ma potencjał do wywoływania aberracji liczbowej. W przypadku mutagenów chemicznych większość wywołanych aberracji jest typu chromatydowego, ale mogą pojawiać się również aberracje typu chromosomowego. Mutacje chromosomowe oraz powiązane z nimi zdarzenia są przyczyną wielu ludzkich chorób genetycznych oraz istnieje dowód na to, że mutacje chromosomowe oraz związane z nimi zdarzenia powodujące zmiany w onkogenach oraz genach hamowania nowotworów komórek somatycznych są odpowiedzialne za wywoływanie raka u ludzi oraz zwierząt doświadczalnych.
Rutynowo w badaniach tych wykorzystuje się gryzonie. Docelową komórką w tym badaniu jest szpik kostny, ze względu na to, że jest on wysoce unaczynioną tkanką oraz zawiera populacje komórek o szybkich cyklach, które mogą być bez trudu wyizolowane oraz poddane działaniu substancji. Inne gatunki oraz tkanki docelowe nie są przedmiotem wykorzystania tej metody.
To badanie chromosomowej aberracji ma szczególne znaczenie w odniesieniu do oceny zagrożenia mutagennego, w związku z czym pozwala ono na wykorzystanie stężenia czynników metabolizmu in vivo, farmakokinetyków oraz procesów naprawy DNA, chociaż mogą one różnić się od siebie w zależności od gatunku oraz tkanek. Badanie in vivo jest również użyteczne w odniesieniu do dalszych badań wyników mutagennych wykrytych w badaniu in vitro.
Jeżeli istnieje dowód na to, że substancja badana lub metabolit reakcyjny nie dotrze do tkanki docelowej, niewłaściwe jest wykorzystanie tej metody.
Zob. również Ogólne wprowadzenie do części B.
1.2. DEFINICJE
Aberracja typu chromatydowego: strukturalne uszkodzenie chromosomu, wyrażone jako pękanie pojedynczych chromatydów lub pękanie oraz ponowne łączenie między chromatydami.
Aberracja typu chromosomowego: strukturalne uszkodzenie chromosomu, wyrażone jako pękanie pojedynczych chromatydów lub pękanie oraz ponowne łączenie obu chromatydów w identycznej lokalizacji.
Endoreduplikacja: proces, w którym po okresie replikacji DNA jądro nie ulega mitozie, ale rozpoczyna nowy okres S. Wynikiem są chromosomy o 4, 8, 16, ... chromatydach.
Szczelina: achromatyczne uszkodzenie mniejsze niż szerokość chromatydu, powodująca minimalne wypaczeniu chromatydu(-ów).
Aberracja liczbowa: zmiana w ilości chromosomów w odniesieniu do normalnej ilości charakterystycznej dla wykorzystywanych komórek.
Poliploidalność: wielokrotność ilości chromosomów haploidalnych (n) inna niż liczba diploidalna (np. 3n, 4n itd.).
Aberracja strukturalna: zmiana w strukturze chromosomu, wykrywalna przez badanie mikroskopowe etapu metafazy podziału komórek, zaobserwowana jako usunięcia oraz fragmenty, zmiany (zachodzące – wewnątrz oraz między chromosomami).
1.3. ZASADA METODY BADAWCZEJ
Zwierzęta są poddane działaniu substancji badanej odpowiednią drogą oraz są uśmiercane w odpowiednim czasie po badaniu. Przed uśmierceniem zwierzęta są poddane działaniu metafazy zatrzymującej substancje (np. Colcemid® lub kolchicyna). Następnie dokonuje się przygotowania chromosomu ze szpiku kostnego oraz zabarwienia, a komórki metafazy są analizowane w odniesieniu do aberracji chromosomowej.
1.4. OPIS METODY BADAWCZEJ
1.4.1. Preparaty
1.4.1.1. Wybór gatunku zwierząt
Szczury, myszy i chomiki chińskie są powszechnie wykorzystywane, chociaż może zostać wykorzystany każdy odpowiedni gatunek ssaków. Powszechnie wykorzystywane laboratoryjne szczepy młodych, zdrowych, dorosłych zwierząt powinny zostać wykorzystane w badaniu. W czasie przeprowadzania badania różnice w masie zwierząt powinny być minimalne oraz nie powinny przekraczać ± 20 % średniej masy każdej płci.
1.4.1.2. Warunki pobytu i karmienia
Ogólne warunki określone w ogólnym wprowadzeniu do części B mają zastosowanie, chociaż celem w odniesieniu do wilgotności powinien być poziom wilgotności 50–60 %.
1.4.1.3. Preparaty zwierzęce
Zdrowe młode dorosłe osobniki zwierząt są losowo przypisane do grup kontrolnych oraz grup poddanych działaniu substancji. Klatki powinny zostać przygotowane w sposób minimalizujący możliwe skutki negatywne wynikające z umieszczenia w klatce. Zwierzęta są identyfikowane oraz aklimatyzowane do warunków laboratoryjnych przynajmniej przez okres pięciu dni.
1.4.1.4. Przygotowanie dawek
Substancje badane stałe powinny zostać rozpuszczone lub zawieszone w odpowiednich rozpuszczalnikach lub nośnikach oraz rozcieńczone, jeżeli stosowne, przed dozowaniem zwierzętom. Substancje badane płynne mogą być dawkowane bezpośrednio lub mogą zostać rozpuszczone przed dawkowaniem. Powinny zostać wykorzystane świeżo przygotowane substancje, chyba że dane dotyczące stabilności wskazują, iż dopuszczalne jest składowanie substancji.
1.4.2. Warunki badania
1.4.2.1. Rozpuszczalnik/nośnik
Rozpuszczalnik/nośnik nie powinien wywoływać skutków toksycznych przy wykorzystywanych dawkach oraz nie powinien mieć możliwości reagowania chemicznego z substancją badaną. Jeśli inne niż dobrze znane rozpuszczalniki/nośniki są wykorzystywane, ich włączenie do doświadczenia powinno być wsparte danymi wskazującymi ich zgodność. Zaleca się, o ile to możliwe, aby w pierwszej kolejności rozważone zostało wykorzystanie wodnych rozpuszczalników/nośników.
1.4.2.2. Kontrole
Równoległe kontrole pozytywne i negatywne (rozpuszczalnik/nośnik) powinny zostać przeprowadzone w odniesieniu do każdej płci w ramach każdego badania. Z wyjątkiem poddawania działaniu substancji badanej zwierzęta w grupie kontrolnej powinny być przedmiotem identycznych działań w porównaniu ze zwierzętami w grupie poddanej działaniu substancji.
Kontrole pozytywne powinny wywoływać strukturalne aberracje in vivo przy poziomie poddania działaniu substancji, w odniesieniu do którego oczekuje się wykrywalnego wzrostu powyżej tło. Dawki kontroli pozytywnych powinny zostać dobrane tak, aby wyniki były jasne, ale by badającemu nie ujawniały bezpośrednio tożsamości sporządzonych szkiełek mikroskopowych. Dopuszczalne jest, aby pozytywne kontrole podawane były inną drogą niż substancje badane oraz aby ich próbki pobierane były tylko jeden raz. Wykorzystanie substancji chemicznych kontroli pozytywnych powiązanych z klasą chemiczną może być brane pod uwagę, jeżeli są one dostępne. Przykłady substancji pozytywnych kontroli obejmują:
|
Substancja |
Nr CAS |
Nr Einecs |
|
Metanosulfonat etylu |
62-50-0 |
200-536-7 |
|
Nitrozomocznik etylu |
759-73-9 |
212-072-2 |
|
Mitomycyna C |
50-07-7 |
200-008-6 |
|
Cyklofosfamid Cyklofosfamid monohydrat |
50-18-0 6055-19-2 |
200-015-4 |
|
Trietylenomelamina |
51-18-3 |
200-083-5 |
Negatywne kontrole z poddaniem działaniu samego rozpuszczalnika lub nośnika, a w innych przypadkach poddanie działaniu w ten sam sposób jak grupy poddane działaniu substancji, powinny zostać objęte każdorazowym pobieraniem próbek, chyba że zmienność oraz częstotliwości komórek z aberracjami chromosomowymi są dostępne w historycznych danych dotyczących kontroli. Jeżeli stosowane jest jednokrotne pobieranie próbek w odniesieniu do negatywnych kontroli, najbardziej odpowiednim czasem jest pierwsze pobranie próbek. Dodatkowo kontrole bez poddania działaniu substancji powinny być również wykorzystywane, chyba że istnieją historyczne lub opublikowane dane dotyczące kontroli wykazujące, że wybrany rozpuszczalnik/nośnik nie wywołuje żadnych skutków szkodliwych lub mutagennych.
1.5. PROCEDURA
1.5.1. Liczba oraz płeć zwierząt
Każda z poddanych działaniu substancji oraz kontrolnych grup obejmuje przynajmniej pięć nadających się do poddania analizie zwierząt każdej płci. Jeżeli w czasie badania istnieją dostępne dane z badań naukowych dotyczących tego samego gatunku oraz z wykorzystaniem tej samej drogi poddania działania substancji, które wykazują, że nie istnieją zasadnicze różnice w toksyczności między płciami, wówczas badanie na jednej płci będzie wystarczające. W przypadku gdy poddanie człowieka działaniu substancji chemicznej może być zależne od płci, jak na przykład w odniesieniu do niektórych czynników środków farmaceutycznych, badanie powinno zostać przeprowadzone na zwierzętach odpowiedniej płci.
1.5.2. Harmonogram poddawania działaniu substancji
Substancje badane są podawane przede wszystkim jako pojedynczy zabieg. Substancje badane mogą być również podawane jako dawka rozbita na części, np. dwa zabiegi, tego samego dnia w odstępie nie dłuższym niż kilku godzin, w celu ułatwienia podawania jako duża objętość substancji. Inne reżimy dawki powinny zostać naukowo uzasadnione.
Próbki powinny być pobierane dwukrotnie w ciągu jednego dnia po poddaniu działaniu substancji. W odniesieniu do gryzoni pierwszy odstęp czasu między pobieraniem trwa 1,5 długości normalnego cyklu komórkowego (ten ostatni trwa przeciętnie 12–18 godzin) po poddaniu działaniu substancji. Ze względu na to, że czas wymagany na pobór oraz przemianę materii substancji badanej, jak również jej skutki na kinetykę cyklu komórkowego może wpływać na optymalny czas wykrywania aberracji chromosomowej, zalecane jest późniejsze pobieranie próbek w 24 godziny po pobraniu pierwszej próbki. Jeżeli wykorzystywane są reżimy dawki większe niż jeden dzień, zastosowany powinien zostać jeden okres pobierania próbek w czasie1,5 długości trwania normalnego cyklu komórkowego po przeprowadzeniu końcowego poddania działaniu substancji.
Przed uśmierceniem zwierzętom wstrzykuje się wewnątrzotrzewnowo odpowiednią dawkę czynnika zatrzymującego metafazę (np. Colcemid® lub kolchicyna). W następnej kolejności pobiera się próbki od zwierząt w odpowiednich odstępach czasu. W odniesieniu do myszy odstęp czasu między pobieraniem próbek wynosi 3–5 godzin; w odniesieniu do chomików ten odstęp czasu wynosi 4–5 godzin. Komórki są pobierane ze szpiku kostnego oraz analizowane pod kątem występowania aberracji chromosomowych.
1.5.3. Poziomy dawek
Jeżeli zakres badań jest przeprowadzanych ze względu na to, że nie istnieją dostępne odpowiednie dane, powinny być one prowadzone w tym samym laboratorium, wykorzystując ten sam reżim gatunku, szczepu, płci oraz poddawania działaniu substancji, który ma zostać zastosowany do głównego badania (5). Jeżeli istnieje toksyczność, trzy poziomy dawek są stosowane w odniesieniu do pierwszego okresu pobierania próbek. Trzy poziomy dawek powinny objąć zakres od maksimum do niskiej toksyczności lub do jej braku. W trakcie późniejszego okresu pobierania próbek muszą zostać wykorzystane jedynie najwyższe dawki. Najwyższa dawka jest określona jako dawka wywołująca oznaki toksyczności, a wyższe poziomy dawek oparte na tym samym reżimie dawkowania mogłyby być śmiertelne. Substancje o szczególnej aktywności biologicznej przy niskich nietoksycznych dawkach (takie jak hormony i mitogeny) mogą stanowić wyjątek w odniesieniu do kryteriów ustalania dawki i powinny być oceniane na zasadzie jednostkowych przypadków. Najwyższa dawka może być również określona jako dawka, która wywołuje pewne wskazania toksyczności w szpiku kostnym (np. wyższa niż 50 % redukcja indeksu mitotycznego).
1.5.4. Badanie ograniczone
Jeżeli badanie w oparciu o jeden poziom dawek o przynajmniej 2 000 mg/kg masy ciała, wykorzystujące pojedyncze poddanie działaniu substancji, lub jak dwa zabiegi poddania działaniu substancji tego samego dnia, nie daje dostrzegalnych wyników toksycznych, oraz jeżeli nie podejrzewa się genotoksyczności w oparciu o dane dotyczące substancji powiązanych strukturalnie, wówczas przeprowadzenie pełnego badania może nie być konieczne. W odniesieniu do badań o dłuższym czasie trwania ograniczenie dawki wynosi 2 000 mg/kg/masa ciała/dzień w odniesieniu do poddawania działaniu substancji trwającego do 14 dni oraz 1 000 mg/kg/masa ciała/dzień w odniesieniu do poddawania działaniu substancji trwającego dłużej niż 14 dni. Spodziewa się, iż poddanie człowieka działaniu substancji może wykazać potrzebę wykorzystania większej dawki w badaniu ograniczonym.
1.5.5. Dawkowanie
Substancja badana jest zazwyczaj podawana przez zgłębnik żołądkowy lub odpowiednią rurkę intubacyjną, lub przez wstrzyknięcie wewnątrzotrzewnowe. Inne drogi poddania działaniu substancji mogą być dopuszczalne w przypadku, gdy ich wykorzystanie jest uzasadnione. Maksymalna objętość płynu, jaka może być podana przez zgłębnik lub wstrzyknięta jednorazowo, zależy od wielkości badanego zwierzęcia. Objętość nie powinna przekraczać 2 ml/100 g masy ciała. Wykorzystanie objętości wyższych niż te musi być uzasadnione. Z wyjątkiem podrażniających lub żrących substancji, które zazwyczaj ujawniają zwiększone skutki o dużym stężeniu, zmienności w objętości substancji badanej powinny zostać zminimalizowane przez dostosowanie stężenia w celu zapewnienia stałej objętości przy wszystkich poziomach dawek.
1.5.6. Preparaty chromosomów
Zaraz po uśmierceniu zwierzęcia, uzyskuje się szpik kostny, poddany działaniu roztworu hipotonicznego oraz ustabilizowany. Komórki są następnie rozmieszczane na szkiełku mikroskopu i barwione.
1.5.7. Analiza
Indeks mitotyczny powinien zostać oznaczony jako miara cytotoksyczności w przynajmniej 1 000 komórek na zwierzę w odniesieniu do wszystkich poddanych działaniu substancji zwierząt (łącznie z pozytywnymi kontrolami) lub kontrolowanych zwierząt, które nie zostały poddane działaniu substancji.
Przynajmniej 100 komórek powinno zostać poddane analizie w odniesieniu do każdego zwierzęcia. Liczba ta może zostać zmniejszona, jeśli zaobserwowano dużą liczbę aberracji. Wszystkie szkiełka mikroskopowe, w tym dotyczące negatywnych i pozytywnych kontroli, powinny być niezależnie zakodowane przed analizą mikroskopową. Ponieważ procedury przygotowania szkiełka mikroskopowego często skutkują przerwaniem proporcji między metafazą ze stratą chromosomów, oceniane komórki powinny z tego względu zawierać pewną liczbę centromerów równą 2n ± 2.
2. DANE
2.1. PRZETWARZANIE WYNIKÓW
Dane dotyczące poszczególnych zwierząt powinny zostać przedstawione w formie tabelarycznej. Jednostką doświadczalną jest zwierzę. W odniesieniu do każdego zwierzęcia ocenione powinny zostać: liczba ocenionych komórek, liczba aberracji przypadających na komórkę oraz udział procentowy komórek ze strukturalnymi aberracjami chromosomowymi. Różne typy strukturalnej aberracji chromosomowej powinny być wymienione wraz z ich ilościami oraz częstotliwościami w odniesieniu do grupy poddanej działaniu substancji oraz kontrolnej. Szczeliny są oddzielnie odnotowywane oraz zgłaszane, ale ogólnie nie są włączane do częstotliwości aberracji ogółem. Jeżeli nie ma dowodu na różnicę między płciami, dane dotyczące obu płci mogą zostać połączone dla przeprowadzenia analizy statystycznej.
2.2. OCENA ORAZ INTERPRETACJA WYNIKÓW
Istnieją liczne kryteria oznaczania wyników pozytywnych, takie jak związany z dawką wzrost odpowiedniej ilości komórek z aberracjami chromosomowymi lub wyraźny wzrost w ilości komórek z aberracjami w grupie pojedynczej dawki przy jednym okresie pobierania próbek. Biologiczne znaczenie wyników powinno zostać wzięte pod uwagę w pierwszej kolejności. Metody statystyczne mogą zostać wykorzystane jako metody pomocnicze w przeprowadzaniu oceny wyników (6). Znaczenie statystyczne nie powinno być jedynym czynnikiem określającym w odniesieniu do reakcji pozytywnej. Wyniki niejednoznaczne powinny zostać wyjaśnione przez dalsze badanie, w szczególności z wykorzystaniem modyfikacji warunków doświadczalnych.
Wzrost liczby komórek poliploidalnych może wskazywać, że substancja badana ma potencjał wywoływania liczbowych aberracji chromosomowych. Wzrost endoreduplikacji chromosomami może wskazywać, że substancje badane mają potencjał wstrzymywania postępowania cyklu komórkowego (7)(8).
Substancja badana, w odniesieniu do której wyniki nie spełniają powyższych kryteriów, uważana jest za niemutagenną w tym badaniu.
Chociaż większość doświadczeń będzie dawała w jasny sposób pozytywne lub negatywne wyniki, w rzadkich przypadkach zestaw danych wyklucza dokonanie ostatecznej oceny dotyczącej aktywności badanej substancji. Wyniki mogą pozostać niejednoznaczne lub sporne niezależnie od liczby powtórzonych doświadczeń.
Wyniki pozytywne z badania aberracji chromosomowej wskazują, że substancja badana wywołuje chromosomową aberrację w szpiku kostnym badanych gatunków. Wyniki negatywne wskazują, że w warunkach badawczych substancja badana nie pobudza aberracji chromosomowej w szpiku kostnym badanych zwierząt.
Prawdopodobieństwo, że substancja badana lub jej metabolity docierają do ogólnego obiegu lub w szczególności do tkanki docelowej (np. toksyczność systemowa), powinno zostać wzięte pod uwagę.
3. SPRAWOZDAWCZOŚĆ
3.1. SPRAWOZDANIE Z BADANIA
Sprawozdanie z badania musi zawierać następujące informacje:
|
|
Rozpuszczalnik/nośnik:
|
|
|
Badane zwierzęta:
|
|
|
Warunki badania:
|
|
|
Wyniki:
|
|
|
Omówienie wyników. |
|
|
Wnioski. |
4. ODNIESIENIA
|
(1) |
Adler, I. D. (1984), Cytogenetic Tests in Mammals, w: Mutagenicity Testing: a Practical Approach, S. Venitt and J. M. Parry (eds), IRL Press, Oxford, Washington D. C, pp. 275–306. |
|
(2) |
Preston, R. J., Dean, B. J., Galloway, S., Holden, H., McFee, A. F. and Shelby, M. (1987), Mammalian In vivo Cytogenetic AssayS: Analysis of Chromosome Aberrations in Bone Marrow Cells, Mutation Res., 189, pp. 157–165. |
|
(1) (3) |
Richold, M., Chandley, A., Ashby, J., Gatehouse, D. G., Bootman, J. and Henderson, L. (1990), In vivo Cytogenetic Assays, w: D. J. Kirkland (ed.), Basic Mutagenicity Tests, UKEMS Recommended Procedures. UKEMS Sub-Committee on Guidelines for Mutagenicity Testing. Report, Part I revised, Cambridge University Press, Cambridge, New York, Port Chester, Melbourne, Sydney, pp. 115–141. |
|
(4) |
Tice, R. R., Hayashi, M., MacGregaro, J. T., Anderson, D., Blakey, D. H., Holden, H. E., Kirsch-Volders, M., Oleson Jr., F. B., Pacchierotti, F., Preston, R. J., Romagna, F., Shimada, H., Sutou, S. and Vannier, B. (1994), Report from the Working Group on the in vivo Mammalian Bone Marrow Chromosomal Aberration Test, Mutation Res., 312, pp. 305–312. |
|
(5) |
Fielder, R. J., Allen, J. A., Boobis, A. R., Botham, P. A., Doe, J., Esdaile, D. J., Gatehouse, D. G., Hodson-Walker, G., Morton, D. B., Kirkland, D. J. and Richold, M. (1992), Report of British Toxicology Society/UK Environmental Mutagen Society Working Group: Dose setting in In vivo Mutagenicity Assays, Mutagenesis, 7, pp. 313–319. |
|
(6) |
Lovell, D. P., Anderson, D., Albanese, R., Amphlett, G. E., Clare, G., Ferguson, R., Richold, M., Papworth, D. G. and Savage, J. R. K. (19 8 9), Statistical Analysis of In vivo Cytogenetic Assays, w: UKEMS Sub-Committee on Guidelines for Mutagenicity Testing, Report Part III. Statistical Evaluation of Mutagenicity Test Data, D. J. Kirkland (ed.) Cambridge University Press, Cambridge, pp. 184–232. |
|
(7) |
Locke-Huhle, C. (1983), Endoreduplication in Chinese hamster cells during alpha-radiation-induced G2 arrest, Mutation Res., 119, pp. 403–413. |
|
(8) |
Huang, Y., Change, C. and Trosko, J. E. (1983), Aphidicolin-induced endoreduplication in Chinese hamster cells, Cancer Res., 43, pp. 1362–1364. |
B.12. MUTAGENNOŚĆ – BADANIE MIKROJĄDROWE ERYTRYOCYTOW IN VIVO U SSAKÓW
1. METODA
Metoda ta jest powtórzeniem OECD TG 474, metody badania mikrojądrowego erytrocytów in vivo u ssaków (1997).
1.1. WPROWADZENIE
Badanie mikrojądrowe erytrocytów u ssaków in vivo jest wykorzystywane w celu wykrycia uszkodzeń wywołanych przez chromosomy lub aparat mitotyczny erytroblastów przez analizę erytrocytu pobranego jako próbka szpiku kostnego lub komórek krwi obwodowej zwierząt, zazwyczaj gryzoni.
Celem badania mikrojądrowego jest identyfikacja substancji powodujących cytogenetyczne uszkodzenia, które skutkują formowaniem się mikrojądra zawierającego okrywające fragmenty chromosomów lub całe chromosomy.
Kiedy w erytroblaście szpiku kostnego rozwija się w erytrocyt polichromatyczny, główne jądro jest wyrzucane; każde mikrojądro, które zostało uformowane, może pozostać poza cytoplazmą poddaną w inny sposób tworzeniu się zarodników. Wizualizacja mikrojądra jest ułatwiona w tych komórkach, ponieważ brak im głównego jądra. Wzrost częstotliwości mikrojądrowych polichromicznych erytrocytów w zwierzętach poddanych działaniu substancji jest wskazaniem wywołanego uszkodzenia chromosomu.
Szpik kostny gryzoni jest rutynowo wykorzystywany w tych badaniach ze względu na to, że erytrocyty polichromiczne produkowane są w tej tkance. Pomiar mikrojądrowych niedojrzałych (polichromicznych) erytrocytów w krwi obwodowej jest na równi dopuszczalny w odniesieniu do jakiegokolwiek gatunku, co do którego wykazana została niezdolność śledziony do usunięcia mikrojądrowego erytrocytu lub który wskazał odpowiednią czułość w celu wykrywania czynników, które powodują strukturalne lub liczbowe aberracje chromosomowe. Mikrojądro może być wyróżnione w oparciu o różne kryteria. Obejmują one identyfikacje obecności lub nieobecności kinetochoru lub centromeru DNA w mikrojądrze. Częstotliwość mikrojądrowych niedojrzałych (polichromicznych) erytrocytów jest głównym punktem końcowym. Liczba dojrzałych erytrocytów w krwi obwodowej, które zawierają mikrojądro, pośród danej ilości dojrzałych erytrocytów może być również wykorzystana jako punkt końcowy oznaczania, jeśli zwierzęta są poddawane działaniu substancji przez cztery tygodnie lub dłużej.
To badanie mikrojądrowe erytrocytów u ssaków in vivo ma szczególne znaczenie dla oceniania zagrożenia, w związku z czym pozwala ono na stężenie czynników metabolizmu farmakokinetyków oraz procesów naprawy DNA chociaż mogą one różnić się wzajemnie w zależności od gatunku oraz tkanek, oraz genetycznych punktów końcowych. Badanie in vivo jest również użyteczne w odniesieniu do dalszych badań skutków mutagennych wykrytych w systemie in vitro.
Jeżeli istnieje dowód na to, że substancja badana lub metabolit reakcyjny nie dotrze do tkanki docelowej, niewłaściwe jest wykorzystanie tego badania.
Zob. również Ogólne wprowadzenie do części B.
1.2. DEFINICJE
Centromer (Kinetochor): region(-y) chromosomu, w którym(-ch) mikrotubule wrzeciona podziałowego są asocjowane w trakcie podziału komórki, dopuszczając uporządkowane przemieszczanie się chromosomów potomnych do biegunów komórek potomnych.
Mikrojądra: małe jądra, oddzielone oraz występujące dodatkowo w stosunku do jądra głównego komórek, wytwarzane w trakcie telofazy mitozy (mejozy) przez okrywające fragmenty chromosomów lub całe chromosomy.
Erytrocyt normochromiczny: dojrzały erytrocyt, któremu brakuje rybosomów oraz może być odróżniony od niedojrzałego, polichromatycznych erytrocytów przez szczepy wybiórczo w odniesieniu dla rybosomów.
Erytrocyt polichromiczny: niedojrzały erytrocyt, w pośrednim stadium rozwoju, który nadal zawiera rybosomy i z tego względu może zostać odróżniony od dojrzałych, normochromicznych erytrocytów, dzięki plamom selektywnym dla rybosomów.
1.3. ZASADA METODY BADAWCZEJ
Zwierzęta są poddane działaniu substancji badanej odpowiednią drogą. Jeżeli wykorzystywany jest szpik kostny, zwierzęta są uśmiercane w odpowiednim czasie po poddaniu działaniu substancji, pobiera się szpik kostny, sporządzane są preparaty oraz dokonuje się barwienia (1)(2)(3)(4)(5)(6)(7). Kiedy wykorzystywana jest krew obwodowa, krew pobierana jest w odpowiednim czasie po poddaniu działaniu substancji oraz sporządzane są i barwione preparaty wymazowe (4)(8)(9)(10). W odniesieniu do badań z krwią obwodową powinno upłynąć możliwie jak najmniej czasu między ostatnim poddaniem działaniu substancji a pobraniem komórek. Preparaty są analizowane pod kątem obecności mikrojądra.
1.4. OPIS METODY BADAWCZEJ
1.4.1. Preparaty
1.4.1.1. Wybór gatunku zwierząt
Szczury lub myszy są zalecane, jeśli wykorzystywany jest szpik kostny, chociaż może zostać wykorzystany każdy odpowiedni gatunek ssaków. Jeśli wykorzystywana jest krew obwodowa, zalecane są myszy. Jednakże może zostać wykorzystany każdy odpowiedni gatunek ssaków pod warunkiem, że jest gatunkiem, w którym śledziona nie usuwa mikrojądrowych erytrocytów lub gatunkiem, który wykazał czułość odpowiednią do wykrycia czynników powodujących strukturalne lub liczbowe aberracje chromosomowe. Powszechnie wykorzystywane laboratoryjne szczepy młodych zdrowych dorosłych zwierząt powinny zostać wykorzystane w badaniu. W czasie przeprowadzania badania różnice w masie zwierząt powinny być minimalne i nie powinny przekraczać ± 20 % średniej masy każdej płci.
1.4.1.2. Warunki przetrzymywania i karmienia
Ogólne warunki określone w ogólnym wprowadzeniu do części B są stosowane, chociaż celem w odniesieniu do wilgotności powinien być poziom 50–60 %.
1.4.1.3. Przygotowanie zwierząt
Zdrowe młode dorosłe osobniki zwierząt są losowo przypisane do grup kontrolnych oraz grup poddanych działaniu substancji. Zwierzęta są identyfikowane w niepowtarzalny sposób. Zwierzęta są aklimatyzowane do warunków laboratoryjnych przynajmniej przez pięć dni. Klatki powinny zostać przygotowane w sposób minimalizujący możliwe negatywne skutki wynikające z umieszczenia w klatce.
1.4.1.4. Przygotowanie dawek
Substancje badane stałe powinny zostać rozpuszczone lub zawieszone w odpowiednich rozpuszczalnikach lub nośnikach oraz rozcieńczone, jeżeli to stosowne, przed dawkowaniem zwierzętom. Substancje badane płynne mogą być dawkowane bezpośrednio lub mogą zostać rozpuszczone przed dawkowaniem. Powinny zostać wykorzystane świeżo przygotowywane substancje, chyba że dane dotyczące stabilności wskazują, iż dopuszczalne jest składowanie substancji.
1.4.2. Warunki badania
1.4.2.1. Rozpuszczalnik/nośnik
Rozpuszczalnik/nośnik nie powinien wywoływać skutków toksycznych przy wykorzystywanych poziomach dawek oraz nie powinien mieć możliwości reagowania chemicznego z substancją badaną. Jeśli inne niż dobrze znane rozpuszczalniki/nośniki są wykorzystywane, ich włączenie do doświadczenia powinno być wsparte danymi wskazującymi ich zgodność. Zaleca się, o ile to możliwe, aby w pierwszej kolejności rozważone zostało wykorzystanie wodnych rozpuszczalników/nośników.
1.4.2.2. Kontrole
Równoległe kontrole pozytywne i negatywne (rozpuszczalnik/nośnik) powinny zostać przeprowadzone w odniesieniu do każdej płci w ramach każdego badania. Z wyjątkiem poddawania działaniu substancji badanej zwierzęta w grupie kontrolnej powinny być przedmiotem identycznych działań w porównaniu ze zwierzętami w grupie poddanej działaniu substancji.
Kontrole pozytywne powinny wytwarzać mikrojądro przy poziomie poddania działaniu substancji, w odniesieniu do którego oczekuje się wykrywalnego wzrostu względem tła. Dawki kontroli pozytywnych powinny zostać wybrane tak, aby wyniki były jasne, ale nie ujawniały bezpośrednio tożsamości zakodowanych szkiełek mikroskopowych badającemu. Dopuszczalne jest, aby pozytywne kontrole podawane były inną drogą niż substancje badane oraz aby ich próbki pobierane były tylko jeden raz. Dodatkowo wykorzystanie substancji chemicznych pozytywnych powiązanych z klasą chemiczną kontroli może być brane pod uwagę, jeżeli są one dostępne. Przykłady pozytywnych substancji kontrolnych obejmują:
|
Substancja |
Nr CAS |
Nr Einecs |
|
Etyl metanosulfonowy |
62-50-0 |
200-536-7 |
|
N-etylo-N-nitrozomocznik |
759-73-9 |
212-072-2 |
|
Mitomycyna C |
50-07-7 |
200-008-6 |
|
Cyklofosfamid Cyclofosfamid monohydrat |
50-18-0 6055-19-2 |
200-015-4 |
|
Trietyleneomelamina |
51-18-3 |
200-083-5 |
Negatywne kontrole z poddaniem działaniu samego rozpuszczalnika lub nośnika, a w innych przypadkach poddane działaniu w ten sam sposób jak grupy poddane działaniu substancji, powinny zostać objęte każdorazowym pobieraniem próbek, chyba że dopuszczalna zmienność oraz częstotliwości komórek z aberracjami chromosomowymi są dostępne z historycznych danych dotyczących kontroli. Jeżeli stosowane jest jednokrotne pobieranie próbek w odniesieniu do negatywnych kontroli, najbardziej odpowiednim czasem jest pierwszy okres pobrania próbek. Dodatkowo kontrole bez poddania działaniu substancji powinny być również wykorzystywane, chyba że istnieją historyczne lub opublikowane dane dotyczące kontroli wykazujące, że wybrany rozpuszczalnik/nośnik nie wywołuje żadnych skutków szkodliwych lub mutagennych.
Jeżeli wykorzystywana jest krew obwodowa, dopuszczalna może być również próbka sprzed poddania działaniu substancji jako równoległa kontroli negatywnej, ale tylko w krótkich badaniach krwi obwodowej (np. 1–3 zabiegów poddania działaniu substancji), jeśli wynikające z badania dane mieszczą się w spodziewanym zakresie w odniesieniu do kontroli historycznych.
1.5. PROCEDURA
1.5.1. Liczba oraz płeć zwierząt
Każda z poddanych działaniu substancji oraz kontrolnych grup obejmuje przynajmniej pięć nadających się do poddania analizie zwierząt z każdej płci (11). Jeżeli w czasie badania istnieją dostępne dane z badań naukowych dotyczących tego samego gatunku oraz z wykorzystaniem tej samej drogi poddania działania substancji, które wykazują, że nie istnieją zasadnicze różnice w toksyczności między płciami, wówczas badanie na jednej płci będzie wystarczające. Gdy poddanie człowieka działaniu substancji chemicznej może być zależne od płci, jak na przykład w odniesieniu do niektórych środków farmaceutycznych, badanie powinno zostać przeprowadzone na zwierzętach odpowiedniej płci.
1.5.2. Harmonogram poddawania działaniu substancji
Nie można zalecić żadnego harmonogramu poddawania działaniu substancji (np. jeden, dwa albo więcej zabiegów poddawania działaniu substancji w 24-godzinnych odstępach czasu). Próbki z rozszerzonych reżimów dawkowania są dopuszczalne, o ile wykazane zostały pozytywne wyniki w odniesieniu do tego badania, w odniesieniu do badania negatywnego, o ile wykazana została toksyczność lub została wykorzystana dawka ograniczona oraz kontynuowano dawkowanie do momentu okresu pobierania próbek. Substancja badana może być również podawana jako dawka podzielona, np. dwa zabiegi poddania działaniu substancji tego samego dnia, w odstępie nie więcej niż kilku godzin, w celu ułatwienia podawania dużej objętości substancji.
Badanie można przeprowadzić na dwa sposoby:
|
a) |
zwierzęta są poddawane działaniu substancji badanej jeden raz. Próbki szpiku kostnego są pobierane przynajmniej dwukrotnie, rozpoczynając nie wcześniej niż w 24 godziny po poddaniu działaniu substancji badanej, ale nie przedłużając poza okres 48 godzin po poddaniu działaniu substancji badanej z odpowiednimi odstępami czasu między pobieraniem próbek. Stosowanie próbek wcześniej niż 24 godziny po leczeniu powinno być uzasadnione. Próbki krwi obwodowej są pobierane przynajmniej dwukrotnie, rozpoczynając nie wcześniej niż w 36 godzin po poddaniu działaniu substancji badanej z odpowiednimi odstępami czasu po pobraniu pierwszej próbki, ale nieprzedłużającymi się powyżej 72 godzin. Jeżeli rozpoznano pozytywną reakcję przy jednym okresie pobierania próbek, nie jest wymagane dodatkowe pobieranie próbek; |
|
b) |
jeżeli wykorzystywane są dwa lub więcej zabiegów poddawania działaniu substancji badanej dziennie (np. dwa lub więcej zabiegów poddawania działaniu substancji przy 24-godzinnych odstępach czasu), próbki powinny zostać zebrane w okresie między 18 a 24 godziną po końcowym poddaniu działaniu substancji w odniesieniu do szpiku kostnego oraz między 36 a 48 godziną po poddaniu działaniu substancji w odniesieniu do krwi obwodowej (12). |
Można stosować dodatkowo inne okresy pobierania próbek, jeżeli są one odpowiednie.
1.5.3. Poziomy dawek
Jeżeli studium określające zakres badania jest przeprowadzane ze względu na to, że nie istnieją dostępne odpowiednie dane, powinno być ono prowadzane w tym samym laboratorium, wykorzystując ten sam reżim gatunku, szczepu, płci oraz poddawania działaniu substancji, który ma zostać zastosowany do głównego badania (13). Jeżeli istnieje toksyczność, trzy poziomy dawek są wykorzystywane w odniesieniu do pierwszego okresu pobierania próbek. Te poziomy dawek powinny objąć zakres od najwyższej do niskiej toksyczności lub do jej braku. W trakcie późniejszego okresu pobierania próbek muszą zostać wykorzystane jedynie najwyższe dawki. Najwyższa dawka jest określona jako dawka wywołująca oznaki toksyczności, a wyższe poziomy dawek oparte na tych samych reżimach dotyczących dawkowania mogłyby być zabójcze. Substancje o szczególnej aktywności biologicznej przy niskich nietoksycznych dawkach (takie jak hormony i mitogeny) mogą stanowić wyjątek w odniesieniu do kryteriów ustalania dawki i powinny być oceniane na zasadzie jednostkowych przypadków. Najwyższa dawka może być również określona jako dawka, która wywołuje pewne wskazania toksyczności w szpiku kostnym (np. redukcja udziału niedojrzałych erytrocytów pośród erytrocytów ogółem w szpiku kostnym lub krwi obwodowej).
1.5.4. Badanie ograniczone
Jeżeli badanie w oparciu o jeden poziom dawek w odniesieniu do zwierząt o przynajmniej 2 000 mg/kg masy ciała, wykorzystujące pojedyncze poddanie działaniu substancji lub dwa zabiegi poddania działaniu substancji tego samego dnia, nie daje dostrzegalnych skutków toksycznych, oraz jeżeli nie podejrzewa się genotoksyczności w oparciu o dane dotyczące substancji powiązanych strukturalnie, wówczas przeprowadzenie pełnego badania może nie być konieczne. W odniesieniu do badań o dłuższym czasie trwania, ograniczenie dawki wynosi 2 000 mg/kg/masa ciała/dzień w odniesieniu do poddania działaniu substancji trwającego do 14 dni oraz 1 000 mg/kg/masa ciała/dzień w odniesieniu do poddania działaniu substancji trwającego dłużej niż 14 dni. Spodziewane poddanie człowieka działaniu substancji może wykazać potrzebę wykorzystania większej dawki w badaniu ograniczonym.
1.5.5. Dawkowanie
Substancja badana jest zazwyczaj podawana przez zgłębnik żołądkowy lub odpowiednią rurkę intubacyjną lub wstrzyknięcie wewnątrzotrzewnowe. Inne drogi poddania działaniu substancji mogą być dopuszczalne, w przypadku gdy ich wykorzystanie może być uzasadnione. Maksymalna objętość płynu, jaka może być podana przez zgłębnik lub wstrzyknięta jednorazowo, zależy od wielkości badanego zwierzęcia. Objętość nie powinna przekraczać 2 ml/100 g masy ciała. Wykorzystanie objętości wyższych musi być uzasadnione. Z wyjątkiem podrażniających lub żrących substancji, które zazwyczaj ujawniają zwiększone skutki o wyższym stężeniu, zmienności w objętości substancji badanej powinny zostać zminimalizowane przez dostosowanie stężenia w celu zapewnienia stałej objętości przy wszystkich poziomach dawek.
1.5.6. Przygotowanie szpiku kostnego/krwi
Komórki szpiku kostnego są zazwyczaj uzyskiwane z kości udowych lub piszczeli bezpośrednio po uśmierceniu. Powszechnie komórki są usuwane z kości udowych lub piszczeli, preparowane oraz barwione, wykorzystując ustalone metody. Krew obwodowa jest uzyskiwana z tętnicy szyjnej lub innego odpowiedniego naczynia krwionośnego. Komórki krwi są natychmiast barwione (8)(9)(10) lub sporządzane są preparaty wymazowe, a następnie są barwione. Wykorzystanie szczególnego barwnika DNA (np. oranż akrydynowy (14) lub Hoechst 33258 plus pyronina-Y (15)) może wyeliminować niektóre spośród artefaktów związanych z barwnikiem nienadającym się do szczególnego szczepu DNA. Ta korzyść nie wyklucza zastosowania zwykłych barwników (np. Giemsa). Dodatkowe systemy (np. kolumny celulozowe w celu usunięcia komórek jądrowych (16)) mogą być również wykorzystane pod warunkiem, że wykazano, iż systemy te odpowiednio działają w odniesieniu do przygotowywania mikrojądra w laboratorium.
1.5.7. Analiza
Udział niedojrzałych erytrocytów pośród ogółu (dojrzałych i niedojrzałych) erytrocytów jest oznaczana podliczając ogół przynajmniej 200 erytrocytów w odniesieniu do szpiku kostnego oraz 1 000 erytrocytów w odniesieniu do krwi obwodowej (17). Wszystkie szkiełka mikroskopowe, łącznie z tymi dotyczącymi pozytywnych i negatywnych kontroli, powinny być niezależnie kodowane przed przeprowadzeniem analizy mikroskopowej. Przynajmniej 2 000 niedojrzałych erytrocytów przypadających na zwierzę jest oceniane w odniesieniu do zapadalności niedojrzałych erytrocytów. Analizując szkiełka mikroskopowe, udział niedojrzałych erytrocytów nie powinien być mniejszy niż 20 % wartości kontrolnej. Jeżeli zwierzęta są poddawane działaniu substancji nieprzerwanie przez cztery tygodnie lub dłużej, przynajmniej 2 000 dojrzałych erytrocytów na zwierzę może zostać ocenione w odniesieniu do częstości występowania mikrojądra. Systemy automatycznej analizy (analiza obrazowa oraz przepływowa analiza cytometryczna zawiesin komórkowych) są dopuszczalne alternatywnie w odniesieniu do to oceny manualnej, jeżeli są odpowiednio uzasadnione oraz jeżeli stwierdzono ich ważność.
2. DANE
2.1. PRZETWARZANIE WYNIKÓW
Dane dotyczące poszczególnych zwierząt powinny zostać przedstawione w formie tabelarycznej. Liczba ocenionych niedojrzałych erytrocytów, liczba mikrojądrowych niedojrzałych erytrocytów oraz liczba niedojrzałych erytrocytów pośród erytrocytów ogółem powinna zostać wymieniona oddzielnie w odniesieniu do każdego zwierzęcia poddanego analizie. Jeżeli zwierzęta są poddawane działaniu substancji nieprzerwanie przez cztery tygodnie lub dłużej, dane dotyczące dojrzałych erytrocytów powinny zostać podane, jeżeli zostały one zebrane. Udział niedojrzałych erytrocytów pośród erytrocytów ogółem oraz, jeżeli uznaje się to za mające zastosowanie, udział procentowy mikrojądrowych erytrocytów podawany jest dla każdego zwierzęcia. Jeżeli nie ma dowodu na różnicę między płciami, dane dotyczące obu płci mogą zostać połączone celem przeprowadzenia analizy statystycznej.
2.2. OCENA ORAZ INTERPRETACJA WYNIKÓW
Istnieją liczne kryteria oznaczania wyników pozytywnych, takie jak związany z dawką wzrost w odpowiedniej ilości komórek z aberracjami chromosomowymi lub wyraźny wzrost w ilości komórek z aberracjami w grupie pojedynczej dawki przy jednym okresie pobierania próbek. Biologiczne znaczenie wyników powinno zostać wzięte pod uwagę w pierwszej kolejności. Metody statystyczne mogą zostać wykorzystane jako metody pomocnicze w przeprowadzaniu oceny wyników (18)(19). Znaczenie statystyczne nie powinno być jedynym czynnikiem określającym w odniesieniu do reakcji pozytywnej. Wyniki niejednoznaczne powinny zostać wyjaśnione przez dalsze badanie, w szczególności z wykorzystaniem modyfikacji warunków doświadczalnych.
Substancja badana, w odniesieniu do której wyniki nie spełniają powyższych kryteriów, uważana jest za niemutagenną w tym badaniu.
Chociaż większość doświadczeń będzie dawało w jasny sposób pozytywne lub negatywne wyniki, w rzadkich przypadkach zestaw danych będzie wykluczał dokonanie ostatecznej oceny dotyczącej aktywności substancji badanej. Wyniki mogą pozostać niejednoznaczne lub sporne niezależnie od ilości powtórzeń doświadczenia.
Wyniki pozytywne z badania mikrojądrowego wskazują, że substancja badana wywołuje powstanie mikrojądra, które jest wynikiem uszkodzenia chromosomowego albo uszkodzenia w erytroblaście badanego gatunku. Wyniki negatywne wskazują, że w warunkach badawczych substancja badana nie produkuje mikrojądra w niedojrzałym erytrocycie badanego gatunku.
Prawdopodobieństwo, że substancja badana lub jej metabolity docierają do ogólnego obiegu lub w szczególności tkanki docelowej (np. toksyczność systemowa), powinno zostać rozpatrzone.
3. SPRAWOZDAWCZOŚĆ
SPRAWOZDANIE Z BADANIA
Sprawozdanie z badania musi zawierać następujące informacje:
|
|
Rozpuszczalnik/nośnik:
|
|
|
Badane zwierzęta:
|
|
|
Warunki badania:
|
|
|
Wyniki:
|
|
|
Omówienie wyników. |
|
|
Wnioski. |
4. ODNIESIENIA
|
(1) |
Heddle, J. A. (1973), A Rapid In vivo Test for Chromosomal Damage, Mutation Res., 18, pp. 187–190. |
|
(2) |
Schmid, W. (1975), The Micronucleus Test, Mutation Res., 31, pp. 9–15. |
|
(3) |
Heddle, J. A., Salamone, M. F., Hite, M., Kirkhart, B., Mavournin, K, MacGregor, J. G. and Newell, G. W. (198 3), The Induction of Micronuclei as a Measure of Genotoxicity, Mutation Res. 123, pp. 61–118. |
|
(4) |
Mavournin, K H., Blakey, D. H., Cimino, M. C, Salamone, M. F. and Heddle, J. A. (1990), The In vivo Micronucleus Assay in Mammalian Bone Marrow and Peripheral Blood. A report of the U. S. Environmental Protection Agency Gene-Tox Program, Mutation Res., 239, pp. 29–80. |
|
(5) |
MacGregor, J. T., Schlegel, R., Choy, W. N. and Wehr, C. M. (1983), Micronuclei in Circulating Erythrocytes: A Rapid Screen for Chromosomal Damage During Routine Toxicity Testing in Mice, w: Developments in Science and Practice of Toxicology, ed. A. W. Hayes, R. C. Schnell and T. S. Miya. Elsevier, Amsterdam, pp. 555–558. |
|
(6) |
MacGregor, J. T., Heddle, J. A. Hite, M., Margolin, G. H., Ramel, C, Salamone, M. F., Tice, R. R., and Wild, D. (19 8 7), Guidelines for the Conduct of Micronucleus Assays in Mammalian Bone Marrow Erythrocytes, Mutation Res., 189, pp. 103–112. |
|
(7) |
MacGregor, J. T., Wehr, C. M., Henika, P. R. and Shelby, M. E. (1990), The in vivo Erythrocyte Micronucleus Test: Measurement at Steady State Increases Assay Efficiency and Permits Integration with Toxicity Studies, Fund. Appl. Toxicol. 14, pp. 513–522. |
|
(8) |
Hayashi, M., Morita, T., Kodama, Y., Sofumi, T. and Ishidate, M. Jr. (1990), The Micronucleus Assay with Mouse Peripheral Blood Reticulocytes Using Acridine Orange-Coated Slides, Mutation Res., 245, pp. 245–249. |
|
(9) |
The Collaborative Study Group for the Micronucleus Test (1992). Micronucleus Test with Mouse Peripheral Blood Erythrocytes by Acridime Orange Supravital Staining: The Summary Report of the 5th Collaborative Study by CSGMT/JEMMS, MMS, Mutation Res., 278, pp. 83–98. |
|
(10) |
The Collaborative Study Group for the Micronucleus Test (CSGMT/JEMMS, MMS: The Mammalian Mutagenesis Study Group of the Environmental Mutagen Society of Japan)(1995), Protocol recommended for the short-term mouse peripheral blood micronucleus test, Mutagenesis, 10, pp. 53–159. |
|
(11) |
Hayashi, M., Tice, R. R., MacGregor, J. T., Anderson, D., Blackey, D. H., Kirsch-Volders, M., Oleson, Jr. F. B., Pacchicrotti, F., Romagna, F., Shimada, H. Sutou, S. and Vannier, B. (19 94), in vivo Rodent Erythrocyte Micronucleus Assay, Mutation Res., 312, pp. 293–304. |
|
(12) |
Higashikuni, N. and Sutou, S. (1995), An optimal, generalised sampling time of 30 ± 6 h after double dosing in the mouse peripheral micronucleus test, Mutagenesis, 10, pp. 313–319. |
|
(13) |
Fielder, R. J., Allen, J. A., Boobis, A. R., Botham, P. A., Doe, J., Esdaile, D. J., Gatehouse, D. G., Hodson-Walker, G., Morton, D. B., Kirkland, D. J. and Rochold, M. (1992), Report of British Toxicology Society/UK Environmental Mutagen Society Working Group: Dose Setting in In vivo Mutagenicity Assays, Mutagenesis, 7, pp. 313–319. |
|
(14) |
Hayashi, M., Sofumi, T. and Ishidate, M. Jr. (1983), An Application of Acridine Orange Fluorescent Staining to the Micronucleus Test, Mutation Res., 120, pp. 241–247. |
|
(15) |
MacGregor, J. T., Wehr, C. M. and Langlois, R. G. (1983), A Simple Fluorescent Staining Procedure for Micronuclei and RNA in Erythrocytes Using Hoechst 33258 and Pyromin Y, Mutation Res., 120, pp. 269–275. |
|
(16) |
Romagna, F. and Staniforth, C. D. (1989), The automated bone marrow micronucleus test, Mutation Res., 213, pp. 91–104. |
|
(17) |
Gollapudi, B. and McFadden, L. G. (1995), Sample size for the estimation of polychromatic to normochromatic erythrocyte ratio in the bone marrow micronucleus test, Mutation Res., 347, pp. 97–99. |
|
(18) |
Richold, M., Ashby, J., Bootman, J., Chandley, A., Gatehouse, D. G. and Henderson, L. (1990), In vivo Cytogenetics Assay, w: D. J. Kirkland (ed.), Basic Mutagenicity tests, UKEMS Recommended Procedures, UKEMS Sub-Committee on Guidelines for Mutagenicity Testing. Report, Part I, revised, Cambridge University Press, Cambridge, New York, Port Chester, Melbourne, Sydney, pp. 115–141. |
|
(19) |
Lovell, D. P., Anderson, D., Albanese, R., Amphlett, G. E., Clare, G., Ferguson R., Richold, M., Papworth, D. G. and Savage, J. R. K (1989), Staticical Analysis of In vivo Cytogenetic Assays, w: D. J. Kirkland (ed.), Statistical Evaluation of Mutagenicity Test Data. UKEMS Sub-Committee on Guidelines for Mutagenicity Testing. Report, Part III, Cambridge University Press, Cambridge, New York, Port Chester, Melbourne, Sydney, pp. 184–232. |
B.13/14. MUTAGENNOŚĆ – BADANIE MUTACJI POWROTNYCH W KOMORKACH BAKTERYJNYCH
1. METODA
Metoda ta jest powtórzeniem OECD TG 471, badania mutacji powrotnych w komórkach bakteryjnych (1997).
1.1. WPROWADZENIE
Badania mutacji powrotnych w komórkach bakteryjnych wykorzystuje aminokwasy wymagające szczepów Salmonella typhimurium oraz Escherichia coli w celu wykrycia mutacji punktowych, które obejmują zastępowanie (substytucja), dodanie lub usunięcie jednego lub większej liczby par zasad DNA (1)(2)(3). Zasadą tego badania mutacji powrotnych w komórkach bakteryjnych jest to, że wykrywa ono mutacje, które powodują powrót mutacji obecnych w badanych szczepach oraz przywracają funkcjonalną zdolność bakterii do syntezowania podstawowego aminokwasu. Odpowiednie bakterie są wykrywane przez ich zdolność do wzrostu w razie nieobecności aminokwasu wymaganego przez macierzysty badany szczep.
Mutacje punktowe są przyczyną wielu chorób genetycznych u ludzi oraz istnieje podstawowy dowód na to, że mutacje punktowe w onkogenach i genach hamujących nowotwory komórek somatycznych są odpowiedzialne za nowotwory u ludzi oraz zwierząt badanych. Badanie mutacji powrotnych w komórkach bakteryjnych jest szybkie, tanie oraz względnie proste do przeprowadzenia. Wiele szczepów badanych posiada wiele cech, które czynią je bardziej czułymi na wykrywanie mutacji łącznie z reakcyjnymi sekwencjami DNA w lokalizacjach powrotnych, wzmożoną przenikalnością do dużych cząsteczek oraz eliminacją systemów naprawy DNA lub wzmocnieniem procesu naprawy DNA skłonnego do błędów. Specyficzność badanych szczepów może zapewnić pewne użyteczne informacje w sprawie typów mutacji, które są wywołane czynnikami genotoksycznymi. Bardzo duża baza danych wyników w odniesieniu do szerokiej różnorodności struktur jest dostępna w odniesieniu do badania mutacji powrotnych w komórkach bakteryjnych oraz dobrze ustalone metodologie zostały opracowane w odniesieniu do badanych substancji chemicznych o różnych właściwościach fizyczno-chemicznych łącznie ze związkami lotnymi.
Zob. również Ogólne wprowadzenie do części B.
1.2. DEFINICJE
Badanie mutacji powrotnych w komórkach bakteryjnych w Salmonella typhimurium albo Escherichia coli wykrywa mutacje w szczepie wymagającym aminokwasów (histydyny lub tryptofanu, odpowiednio) w celu wytworzenia szczepu niezależnie od zewnętrznych dostaw aminokwasów.
Pary zasad podstawiania mutagenów są czynnikami, które powodują zasadowe zmiany w DNA. W badaniu powrotnym ta zmiana może wystąpić w miejscu oryginalnej mutacji lub w drugim miejscu w genomie bakteryjnym.
Mutageny powodujące zmiany fazy odczytu są czynnikami, które powodują dodanie lub usunięcie jednej lub większej liczby par zasad w DNA, zmieniając w ten sposób odczyt RNA.
1.3. WSTĘPNE ROZWAŻANIA
Badanie mutacji powrotnych w komórkach bakteryjnych wykorzystuje komórki prokariotyczne, które różnią się od komórek ssaków takimi czynnikami jak pobór, przemiana materii, struktura chromosomowa procesy naprawy DNA. Badania przeprowadzane in vitro ogólnie wymagają wykorzystania egzogenicznego źródła aktywacji metabolicznej. Systemy aktywacji metabolicznej in vitro nie mogą naśladować całkowicie warunków in vivo. Badanie z tego względu nie dostarcza bezpośrednich informacji w sprawie potencjału mutagenności oraz rakotwórczości substancji u ssaków.
Badanie mutacji powrotnych w komórkach bakteryjnych jest powszechnie wykorzystywane jako wstępny przegląd aktywności genotoksycznej oraz, w szczególności w odniesieniu aktywacji wywołującej mutacje punktowe. Rozległa baza danych wykazała, że wiele substancji chemicznych, które są pozytywne w tym badaniu, ujawnia również aktywność mutagenną w innych badaniach. Istnieją przykłady czynników mutagennych, które nie są wykryte przez to badanie, powody tych niedoborów mogą zostać przypisane szczególnemu charakterowi wykrytego punktu końcowego, różnicom między aktywacją metaboliczną lub różnicom w biodostępności. Z drugiej strony, czynniki, które wzmacniają wrażliwość badania mutacji powrotnych w komórkach bakteryjnych, mogą prowadzić do zbyt wysokiego oszacowania aktywność mutagennej.
Badanie mutacji powrotnych w komórkach bakteryjnych może być nieodpowiednie w odniesieniu do oceny niektórych klas substancji chemicznych, na przykład wysoce bakteriobójczych związków (np. niektóre antybiotyki) oraz tych, które uznaje się za (które znane są) zakłócające szczególnie system replikacji komórek ssaków (np. niektóre czynniki hamujące topoizomerazę oraz niektóre nuklozydowe substancje analogiczne). W takich przypadkach badanie mutacji u ssaków może być bardziej odpowiednie.
Chociaż wiele związków, które są pozytywne w tym badaniu, jest czynnikami rakotwórczymi dla ssaków, korelacja nie jest zupełna. Zależy to od klasy substancji chemicznej oraz istnieją czynniki rakotwórcze, które nie są wykrywane przez substancje badaną, ponieważ działają one przez inne niegenotoksyczne mechanizmy lub mechanizmy nieobecne w komórkach bakteryjnych.
1.4. ZASADA METODY BADAWCZEJ
Zawiesiny komórek bakteryjnych są poddane działaniu substancji badanej, w obecności oraz w nieobecności egzogenicznego systemu aktywacji metabolicznej. W metodzie włączania płytki te zawiesiny są zmieszane z nałożonym agarem oraz nakładane na minimalne podłoże. W odniesieniu do obu technik, po dwóch lub trzech dniach inkubacji, powrotne hodowle są podliczane oraz porównywane z ilością spontanicznie powracających hodowli na rozpuszczalnikowych płytkach kontrolnych.
Zostało opisanych wiele procedur w odniesieniu do przeprowadzania badania mutacji powrotnych w komórkach bakteryjnych. Wśród tych powszechnie wykorzystywanych znajdują się metoda włączająca płytkę (1)(2)(3)(4), metoda uprzedniej inkubacji (2)(3)(5)(6)(7)(8), metoda fluktuacji (9)(10) oraz metoda zawiesinowa (11). Zostały opisane modyfikacje w odniesieniu do badania gazów lub oparów (12).
Procedury opisane w tej metodzie odnoszą się przede wszystkim do metod włączenia płytki oraz uprzedniej inkubacji. Żadna z nich nie jest dopuszczalna w odniesieniu do przeprowadzania doświadczeń zarówno z aktywacją metaboliczną, jak i bez niej. Niektóre substancje mogą zostać wykryte w bardziej skuteczny sposób, wykorzystując metodę uprzedniej inkubacji. Substancje te należą do klas chemicznych, które obejmują nitrozaminy o krótkich łańcuchach, metale dwuwartościowe, aldehydy, barwniki azowe oraz związki diazowe oraz alkaloidy pirolizydynowe, związki allilowe oraz związki azotowe (3). Rozpoznano także, że niektóre klasy mutagenów nie są zawsze wykrywane, wykorzystując procedury standardowe, takie jak metoda włączająca płytki lub metoda uprzedniej inkubacji. Powinny być one uznawane za „szczególne przypadki” i silnie zalecane jest, iż powinny zostać wykorzystane alternatywne procedury. Następujące „szczególne przypadki” mogą zostać zidentyfikowane (wraz z przykładami procedur, które mogłyby zostać wykorzystane w odniesieniu do ich wykrycia) barwniki azowe oraz związki diazowe (3)(5)(6)(13), gazy oraz lotne substancje chemiczne (12)(14)(15)(16) oraz glikozydy (17)(18). Odchylenie od standardowej procedury musi zostać naukowo uzasadnione.
1.5. OPIS METODY BADAWCZEJ
1.5.1. Preparaty
1.5.1.1. Bakterie
Świeże kultury bakterii powinny być hodowane do późnej fazy wykładniczej lub wczesnej fazy stacjonarnej wzrostu (w przybliżeniu 109 komórek na ml). Kultury bakteryjne w późnej fazie stacjonarnej nie powinny być wykorzystywane. Zasadnicze jest, aby kultury bakteryjne wykorzystywane w badaniu zawierały wysokie miano bakterii zdolnych do życia. Miano może być wykazane zarówno z historycznych danych dotyczących kontroli krzywych wzrostu, w każdym oznaczaniu przez określenie ilości zdolnych do życia komórek przez doświadczenie platerowania.
Zalecaną temperaturą inkubacji jest 37 oC.
Powinno zostać wykorzystane przynajmniej pięć szczepów bakterii. Powinno to obejmować cztery szczepy S. typhimurium (TA 1535; TA 1537 lub TA97a lub TA97; TA98; oraz TA100), które zostały pokazane jako mające być rzetelne oraz odtwarzalnie reagujące między laboratoriami. Te cztery szczepy S. typhimurium posiadają GC pary bazowe w miejscu podstawowego powrotu oraz wiadome jest, że nie mogą one wykrywać utleniających mutagenów, czynników sieciujących oraz hydrazyny. Takie substancje mogą być wykrywane przez szczepy E. coli WP2 lub S. typhimurium TA1 02 (19), które posiadają pary zasad AT w miejscu podstawowego powrotu. Z tego względu zalecaną kombinacją szczepów jest:
|
— |
S. typhimurium TA1 535, oraz |
|
— |
S. typhimurium TA1 537 lub TA97, lub TA97a, oraz |
|
— |
S. typhimurium TA98, oraz |
|
— |
S. typhimurium TA100, oraz |
|
— |
E. coli WP2 uvrA lub E. coli WP2 uvrA (pKM101), lub S. typhimurium TA102. |
W celu wykrycia mutagenów sieciujących preferowane może być objęcie TA102 lub dodanie wprawnych w naprawianie DNA szczepów E. coli (np. E. coli WP2 lub E. coli WP2 (pKM101)).
Powinny być wykorzystywane ustalone procedury przygotowania hodowli podstawowych, weryfikacja markerem oraz składowanie. Wymogi w odniesieniu do wzrostu, dotyczące aminokwasu, powinny być wykazane dla każdego zamrożonego preparatu hodowli podstawowych (histydyna dla szczepów S. typhimurium oraz tryptofan dla szczepów E. coli). Inne właściwości fenotypowe powinny zostać sprawdzone w podobny sposób, mianowicie: obecność lub nieobecność plazmidów czynnika R, gdzie stosowne (np. odporność na ampicylinę w szczepach TA98, TA100 oraz TA97a lub TA97, WP2 uvrA oraz WP2 uvrA (pKM101), oraz odporność na ampicylinę + tetracyklinę w szczepie TA 102); (np. rfa mutacja w S. typhimurium przez wrażliwość na fiolet krystaliczny oraz uvrA mutacja w E. coli lub uvrB mutacja w S. typhimurium przez wrażliwość na światło ultrafioletowe) (2)(3). Szczepy powinny również wytwarzać spontaniczne kolonie powrotne w ramach zakresów częstotliwości oczekiwanych na podstawie historycznych danych laboratoryjnych oraz przeważnie w zakresie odnotowanym w literaturze.
1.5.1.2. Podłoże
Wykorzystywany jest odpowiedni minimalny agar (np. zawierające Vogel-Bonner minimalne podłoże E oraz glukoza) oraz powlekający agar zawierający histydynę oraz biotynę lub tryptofan w celu umożliwienia podziału kilku komórek (1)(2)(9).
1.5.1.3. Aktywacja metaboliczna
Bakterie powinny być poddane działaniu substancji badanej zarówno w obecności, jak i w nieobecności odpowiedniego systemu aktywacji metabolicznej. Najbardziej powszechnie wykorzystywanym systemem jest uzupełniana współczynnikiem frakcja pomitochondrialna (S9) przygotowana z wątroby gryzoni poddanych działaniu czynników pobudzających enzymy takimi jak Aroklor 1254 (1)(2) lub kombinacją feno-barbitonu oraz B nafoflavonu (18)(20)(21). Frakcja pomitochondrialna jest zazwyczaj wykorzystywana w stężeniach w zakresie od 5 do 30 % objętościowo w mieszaninie S9. Wybór oraz warunki systemu metabolicznej aktywacji mogą być uzależnione od klasy badanej substancji chemicznej. W niektórych przypadkach właściwe może być wykorzystanie więcej niż jednego stężenia frakcji pomitochondrialnej. W odniesieniu do barwników azowych oraz związków diazowych, przy wykorzystaniu systemu aktywacji metabolicznej, może być bardziej odpowiednie (6)(13).
1.5.1.4. Substancja testowa/preparat
Stała substancja badana powinna zostać rozpuszczona lub zawieszona w odpowiednich rozpuszczalnikach lub nośnikach oraz rozcieńczona, jeżeli jest to odpowiednie przed poddaniem bakterii działaniu substancji. Płynne substancje badane mogą być dodawane bezpośrednio do systemu badawczego i/lub rozcieńczane przed poddaniem działaniu substancji. Powinny zostać wykorzystane świeże preparaty, chyba że dane dotyczące stabilności wykazują dopuszczalność składowania.
Rozpuszczalnik/nośnik nie powinien mieć możliwości reagowania chemicznego z substancją badaną oraz powinien być zgodny z przeżywaniem bakterii oraz aktywnością S9. Jeśli inne niż dobrze znane rozpuszczalniki/nośniki są wykorzystywane, ich włączenie do doświadczenia powinno być wsparte danymi wskazującymi ich zgodność. Zaleca się, o ile to możliwe, aby w pierwszej kolejności rozważone zostało wykorzystanie wodnych rozpuszczalników/nośników. Badając substancje niestabilne w wodzie, wykorzystane rozpuszczalniki organiczne powinny być wolne od wody.
1.5.2. Warunki badawcze
1.5.2.1. Szczepy badane (zob. ppkt 1.5.1.1)
1.5.2.2. Stężenie poddania działaniu substancji
Wśród kryteriów, które mają być uwzględnione, oznaczając najwyższą ilość substancji badanej, które ma zostać wykorzystana, jest cytotoksyczność oraz rozpuszczalność w końcowej mieszaninie, której działaniu poddawane są szczepy.
Użyteczne może być oznaczenie toksyczności oraz nierozpuszczalności w doświadczeniu wstępnym. Cytotoksyczność może zostać wykryta przez redukcję ilości powrotnych hodowli, oczyszczenie lub zmniejszenie podłoża lub stopień przeżywania poddanych działaniu substancji hodowli. Cytotoksyczność substancji może być zmieniana w obecności systemów aktywacji metabolicznej. Nierozpuszczalność powinna zostać oceniona jako wytrącanie w końcowej mieszaninie w rzeczywistych warunkach badania oraz oczywistych dla oka nieuzbrojonego.
Zalecane najwyższe stężenie badawcze w odniesieniu do rozpuszczalnych niecytotoksycznych substancji stanowi 5 mg/płytkę lub 5 μl/płytkę. W odniesieniu do niecytotoksycznych substancji, które nie są rozpuszczalne przy 5 mg/płytkę lub 5 μl/płytkę, jedno lub więcej badanych stężeń powinno być nierozpuszczalne w końcowej mieszaninie cytotoksycznej. Strącanie nie powinno zakłócać oceny.
Przynajmniej pięć różnych nadających się do analizowania stężeń substancji badanej powinno zostać wykorzystane przy w przybliżeniu połowie logarytmu (np. √1 0) odstępów czasu między punktami badawczymi w odniesieniu do wstępnego doświadczenia. Krótsze odstępy czasu mogą być odpowiednie, jeśli reakcja stężenia jest badana. Badanie powyżej stężenia 5 mg/płytkę lub 5 μl/płytkę może być brane pod uwagę, oceniając substancje zawierające podstawowe ilości potencjalnie mutagennych zanieczyszczeń.
1.5.2.3. Negatywne i pozytywne kontrole
Równoległe, specyficzne co do szczepu pozytywne i negatywne (rozpuszczalnik lub nośnik) kontrole, zarówno z aktywacją metaboliczną, jak i bez niej, powinny być włączone w ramy każdego oznaczania. Powinien zostać przeprowadzony wybór uwzględniania pozytywnych kontroli, które wykazują skuteczne działanie każdego oznaczania.
W odniesieniu do oznaczeń wykorzystujących system metabolicznej aktywacji, substancja(-e) wzorcowa(-e) kontroli pozytywnych powinna(-y) zostać wybrana(-e) na podstawie typu wykorzystanego szczepu bakterii.
Następujące substancje stanowią przykłady odpowiednich substancji pozytywnych kontroli w odniesieniu do oznaczania z metaboliczna aktywacją:
|
Substancja |
Nr CAS |
Nr Einecs |
|
9,10-dimetyloantracen |
781-43-1 |
212-308-4 |
|
7,12-dimetylobenzo[a]antracen |
57-97-6 |
200-359-5 |
|
Benzo[a]piren |
50-32-8 |
200-028-5 |
|
2-aminoantracen |
613-13-8 |
210-330-9 |
|
Cyklofosfamid |
50-18-0 |
200-015-4 |
|
Cyklofosfamid monohydrat |
6055-19-2 |
|
Następująca substancja jest odpowiednią kontrolą pozytywną w odniesieniu do metody redukcyjnej aktywacji metabolicznej:
|
Substancja |
Nr CAS |
Nr Einecs |
|
Congo Red |
573-58-0 |
209-358-4 |
2-aminoantracen nie powinien być wykorzystywany jako wyłączny wskaźnik skuteczności mieszaniny S9. Jeśli wykorzystywany jest 2-aminoantracen, każda partia S9 powinna być również charakteryzowana mutagenem, który wymaga metabolicznej aktywacji przez enzymy mikrosomowe, np. benzo[a]piren, dimetylobenzoantracen.
Następujące substancje są przykładami specyficznych co do szczepu kontroli pozytywnych dla badań przeprowadzanych bez egzogenicznego systemu aktywacji metabolicznej:
|
Substancje |
Nr CAS |
Einecs No |
Szczep |
|
Azydek sodu |
26628-22-8 |
247-852-1 |
TA 1535 i TA 100 |
|
2-nitrofluoren |
607-57-8 |
210-138-5 |
TA 98 |
|
9-aminoakrydyna |
90-45-9 |
201-995-6 |
TA 1537, TA 97 i TA 97a |
|
ICR 191 |
17070-45-0 |
241-129-4 |
TA 1537, TA 97 i TA 97a |
|
Hydronadtlenek kumenu |
80-15-9 |
201-254-7 |
TA 102 |
|
Mitomycyna C |
50-07-7 |
200-008-6 |
WP2 uvrA i TA 102 |
|
N-etylo-N-nitro-N-nitrozoguanidyna |
70-25-7 |
200-730-1 |
WP2, WP2uvrA i WP2uvrA (pKM101) |
|
1-tlenek 4-nitrokwinolinu |
56-57-5 |
200-281-1 |
WP2, WP2uvrA i WP2uvrA (pKM101) |
|
Furylofuramid (AF2) |
|
3688-53-7 |
Szczepy zawierające plazmidy |
Mogą być wykorzystane inne odpowiednie substancje kontroli pozytywnych. Wykorzystanie substancji powiązanych klasą z substancjami kontroli pozytywnych powinno zostać uwzględnione, jeżeli są one dostępne.
Negatywne kontrole, składające się z samego rozpuszczalnika lub nośnika bez substancji badanej, oraz w innych przypadkach poddane działaniu w ten sam sposób jak grupy poddane działaniu substancji powinny zostać objęte kontrolą. Dodatkowo kontrole bez poddania działaniu substancji powinny być również wykorzystywane, chyba że istnieją historyczne dane dotyczące kontroli wykazujące, że brak szkodliwych lub mutagennych skutków wywoływanych wybranym rozpuszczalnikiem.
1.5.3. Procedura
W odniesieniu do metody włączającej płytkę (1)(2)(3)(4), bez aktywacji metabolicznej, zazwyczaj 0,05 ml lub 0,1 ml stężenia badawczego, 0,1 ml świeżej hodowli bakteryjnej (zawierającej w przybliżeniu 108 zdolnych do życia komórek) oraz 0,5 ml sterylnego bufora jest mieszane z 2,0 ml pokrywającego agaru. W odniesieniu do oznaczania z aktywacją metaboliczną zazwyczaj 0,5 ml mieszaniny aktywacji metabolicznej zawierającej odpowiednie ilości pomitochondrialnej frakcji (w zakresie 5–30 % objętościowo w mieszaninie aktywacji metabolicznej) są mieszane z pokrywającym agarem (2,0 ml), łącznie z roztworem substancji badanej. Zawartości każdej z rurek są mieszane oraz wylewane na powierzchnię minimalnej płytki agarowej. Agar pokrywający krzepnie przed inkubacją.
W odniesieniu do metody uprzedniej inkubacji (2)(3)(5)(6) substancja badana/roztwór badany jest uprzednio inkubowany ze szczepem badanym (zawierającym w przybliżeniu 108 zdolnych do życia komórek) oraz sterylnym roztworem buforowym lub systemem metabolicznej aktywacji (0,5 ml) zazwyczaj przez 20 minut lub dłużej w temperaturze 30–37 oC przed mieszaniem z pokrywającym agarem oraz polewając na powierzchnię minimalnej płytki agarowej. Zazwyczaj 0,05 lub 0,1 ml substancji badanej/roztworu badanego, 0,1 ml i bakterii oraz 0,5 ml mieszaniny S9 lub sterylnego roztworu buforowego są mieszane z 2,0 ml pokrywającego agaru. Rurki powinny zostać napowietrzone w trakcie uprzedniej inkubacji, wykorzystując wstrząsarkę.
W odniesieniu do odpowiedniego szacowania zmienności trzykrotne pokrywanie płytkami powinno zostać wykorzystane przy każdej dawce badanej. Wykorzystanie podwójnego pokrywania płytkami jest dopuszczalne, jeżeli jest to naukowo uzasadnione. Okazjonalna strata płytki niekoniecznie czyni oznaczanie nieważnym.
Gazowe lub lotne substancje powinny być badane odpowiednimi metodami, takimi jak np. w szczelnie zamkniętych naczyniach (12)(14)(15)(16).
1.5.4. Inkubacja
Wszystkie płytki w danym oznaczeniu powinny być inkubowane w temperaturze 37 oC przez 48–72 godzin. Po okresie inkubacji podliczana jest liczba powrotnych kolonii.
2. DANE
2.1. PRZETWARZANIE WYNIKÓW
Dane powinny zostać przedstawione jako liczba kolonii przypadająca na płytkę. Liczba powrotnych kolonii zarówno w płytkach negatywnych (kontrola rozpuszczalnikowa oraz kontrola bez poddania działaniu substancji, jeżeli zostały wykorzystane), jak i pozytywnych kontroli powinny zostać również podane. Podlicza się poszczególne płytki, średnia liczba powrotnych kolonii przypadająca na płytkę powinna zostać przedstawiona w odniesieniu do substancji badanej oraz pozytywnej i negatywnej (bez poddania działaniu substancji badanej i/lub rozpuszczalnikowej) kontroli.
Nie istnieje wymóg jasnej reakcji pozytywnej w odniesieniu do weryfikacji. Niejednoznaczne wyniki powinny zostać wyjaśnione w drodze dalszych badań, w szczególności z wykorzystaniem modyfikacji warunków doświadczalnych. Wyniki negatywne muszą zostać potwierdzone na zasadzie jednostkowych przypadków. Modyfikacja parametrów badawczych w celu poszerzenia zakresu ocenionych warunków powinna zostać rozważona w dalszych doświadczeniach. Parametry badawcze, które mogą być zmodyfikowane, obejmują zakres stężenia, metodę poddania działaniu substancji badanej (włączenie płytek lub uprzednia inkubacja) oraz warunki aktywacji metabolicznej.
2.2. OCENA ORAZ INTERPRETACJA WYNIKÓW
Istnieją liczne kryteria oznaczania wyników pozytywnych, takie jak związany z dawką wzrost ilości powrotnych kolonii powyżej zakres badanych i/lub odtwarzalności przy jednym lub większej ilości stężeń, przypadających na płytkę w przynajmniej jednym szczepie z lub bez systemu aktywacji metabolicznej. Biologiczne znaczenie wyników powinno zostać wzięte pod uwagę w pierwszej kolejności. Metody statystyczne mogą zostać wykorzystane jako metody pomocnicze w przeprowadzaniu oceny wyników (24). Jednakże znaczenie statystyczne nie powinno być jedynym czynnikiem określającym w odniesieniu do reakcji pozytywnej.
Substancja badana, w odniesieniu do której wyniki nie spełniają powyższych kryteriów, uważana jest za niemutagenną w tym badaniu.
Chociaż większość doświadczeń będzie dawało w jasny sposób pozytywne lub negatywne wyniki, w rzadkich przypadkach zestaw danych będzie wykluczał dokonanie ostatecznej oceny dotyczącej aktywności badanej substancji. Wyniki mogą pozostać niejednoznaczne lub sporne niezależnie od liczby powtórzonych doświadczeń.
Wyniki pozytywne z badania mutacji powrotnych w komórkach bakteryjnych wskazują, że substancja badana wywołuje mutacje punktowe przez zastąpienie lub mutacje fazy odczytu w genomie albo Salmonella typhimurium i/lub Escherichia coli. Wyniki negatywne wskazują, że w warunkach badawczych substancja badana nie jest mutagenna w badanym gatunku.
3. SPRAWOZDAWCZOŚĆ
SPRAWOZDANIE Z BADANIA
Sprawozdanie z badania musi zawierać następujące informacje:
|
|
Rozpuszczalnik/nośnik:
|
|
|
Szczepy:
|
|
|
Warunki badania:
|
|
|
Wyniki:
|
|
|
Omówienie wyników. |
|
|
Wnioski. |
4. ODNIESIENIA
|
(1) |
Ames, B. N., McCann, J. and Yamasaki E. (1975), Methods of Detecting Carcinogens and Mutagens with the Salmonella/Mammalian-Microsome Mutagenicity Test, Mutation Res., 31, pp. 347–364. |
|
(2) |
Maron, D. M. and Ames, B. N. (1983), Revised Methods for the Salmonella Mutagenicity Test, Mutation Res., 113, pp. 173–215. |
|
(3) |
Gatehouse, D., Haworth, S., Cebula, T., Gocke, E., Kier, L., Matsushima, T., Melcion, C., Nohmi, T., Venitt, S. and Zeiger, E. (1994), Recommendations for the Performance of Bacterial Mutation Assays, Mutation Res., 312, pp. 217–233. |
|
(4) |
Kier, L. D., Brusick D.J., Auletta, A. E., Von Halle, E. S., Brown, M. M., Simmon, V. F., Dunkel, V., McCann, J., Mortelmans, K., Prival, M., Rao, T. K. and Ray V. (1986), The Salmonella typhimurium/Mammalian Microsomal Assay: A Report of the U. S. Environmental Protection Agency Gene-Tox Program, Mutation Res., 168, pp. 69–240. |
|
(5) |
Yahagi, T., Degawa, M., Seino, Y. Y., Matsushima, T., Nagao, M., Sugimura, T. and Hashimoto, Y. (1975), Mutagenicity of Carcinogen Azo Dyes and their Derivatives, Cancer Letters 1, pp. 91–96. |
|
(6) |
Matsushima, M., Sugimura, T., Nagao, M., Yahagi, T., Shirai, A. and Sawamura, M. (1980), Factors Modulating Mutagenicity Microbial Tests, w: Short-term Test Systems for Detecting Carcinogens, ed. Norpoth K. H. and Garner, R. C., Springer, Berlin-Heidelberg-New York, pp. 273–285. |
|
(7) |
Gatehouse, D. G., Rowland, I. R., Wilcox, P., Callender, R. D. and Foster, R. (1980), Bacterial Muta tion Assays, w: Basic Mutagenicity TestS: UKEMS Part 1 Revised, ed. D.J. Kirkland, Cambridge University Press, pp. 13–61. |
|
(8) |
Aeschacher, H. U., Wolleb, U. and Porchet, L. (1987), Liquid Preincubation Mutagenicity Test for Foods, J. Food Safety, 8, pp. 167–177. |
|
(9) |
Green, M. H. L., Muriel, W. J. and Bridges, B. A. (1976), Use of a simplified fluctuation test to detect low levels of mutagens, Mutation Res., 38, pp. 33–42. |
|
(10) |
Hubbard, S. A., Green, M. H. L., Gatehouse, D. and Bridges, J. W. (1984), The Fluctuation Test in Bacteria, w: Handbook of Mutagenicity Test Procedures, 2nd Edition, ed. Kilbey, B.J., Legator, M., Nichols, W. and Ramel, C., Elsevier, Amsterdam-New York-Oxford, pp. 141–161. |
|
(11) |
Thompson, E. D. and Melampy, P. J. (1981), An Examination of the Quantitative Suspension Assay for Mutagenesis with Strains of Salmonella typhimurium, Environmental Mutagenesis, 3, pp. 453–465. |
|
(12) |
Araki, A., Noguchi, T., Kato, F. and Matsushima, T. (1994), Improved Method for Mutagenicity Testing of Gaseous Compounds by Using a Gas Sampling Bag, Mutation Res., 307, pp. 335–344. |
|
(13) |
Prival, M.J., Bell, S.J., Mitchell, V. D., Reipert, M. D. and Vaughan, V. L. (1984), Mutagenicity of Benzidine and Benzidine-Congener Dyes and Selected Monoazo Dyes in a Modified Salmonella Assay, Mutation Res., 136, pp. 33–47. |
|
(14) |
Zeiger, E., Anderson, B. E., Haworth, S., Lawlor, T. and Mortelmans, K. (1992), Salmonella Mutagenicity Tests. V. Results from the Testing of 311 Chemicals, Environ. Mol. Mutagen., 19, pp. 2–141. |
|
(15) |
Simmon, V., Kauhanen, K. and Tardiff, R. G. (1977), Mutagenic Activity of Chemicals Identified in Drinking Water, in Progress in Genetic Toxicology, D. Scott, B. Bridges and F., Sobels (eds.) Elsevier, Amsterdam, pp. 249–258. |
|
(16) |
Hughes, T.J., Simmons, D. M., Monteith, I. G. and Claxton, L. D. (1987), Vaporisation Technique to Measure Mutagenic Activity of Volatile Organic Chemicals in the Ames/Salmonella Assay, Environmental Mutagenesis, 9, pp. 421–441. |
|
(17) |
Matsushima, T., Matsumoto, A., Shirai, M., Sawamura, M., and Sugimura, T. (19 79), Mutagenicity of the Naturally Occurring Carcinogen Cycasin and Synthetic Methylazoxy Methane Conjugates in Salmonella typhimurium, Cancer Res., 39, pp. 3780–3782. |
|
(18) |
Tamura, G., Gold, C., Ferro-Luzzi, A. and Ames, B. N. (1980), Fecalase: A Model for Activation of Dietary Glycosides to Mutagens by Intestinal Flora, Proc. Natl. Acad. Sci. USA, 77, pp. 4961–4965. |
|
(19) |
Wilcox, P., Naidoo, A., Wedd, D. J. and Gatehouse, D. G. (1990), Comparison of Salmonella typhimu rium TA 102 with Escherichia coli WP2 Tester strains, Mutagenesis, 5, pp. 285–291. |
|
(20) |
Matsushima, T., Sawamura, M., Hara, K. and Sugimura, T. (1976), A Safe Substitute for Polychlorinated Biphenyls as an Inducer or Metabolic Activation Systems, w: In vitro Metabolic Activation in Mutagenesis Testing, eds. F. J. de Serres et al. Elsevier, North Holland, pp. 85–88. |
|
(21) |
Elliot, B. M., Combes, R. D., Elcombe, C. R., Gatehouse, D. G., Gibson, G. G., Mackay, J. M. and Wolf, R. C. (1992), Alternatives to Aroclor 1254-inducedS9 ininvitro Genotoxicity Assays, Mutagenesis, 7, pp. 175–177. |
|
(22) |
Maron, D., Katzenellenbogen, J. and Ames, B. N. (1981), Compatibility of Organic Solvents with the Salmonella/Microsome Test, Mutation Res., 88, pp. 343–350. |
|
(23) |
Claxton, L. D., Allen, J., Auletta, A., Mortelmans, K., Nestmann, E. and Zeiger, E. (1987), Guide for the Salmonella typhimurium/Mammalian Microsome Tests for Bacterial Mutagenicity, Mutation Res., 189, pp. 83–91. |
|
(24) |
Mahon, G. A. T., Green, M. H. L., Middleton, B., Mitchel, I., Robinson, W. D. and Tweats, D. J. (1989), Analysis of Data from Microbial Colony Assays, w: UKEMS Sub-Committee on Guidelines for Mutagenicity Testing, Part II. Statistical Evaluation of Mutagenicity Test Data, ed. Kirkland, D.J., Cambridge University Press, pp. 28–65. |
B.15. BADANIE MUTAGENNOŚCI I BADANIE PRZESIEWOWE NA RAKOTWÓRCZOŚĆ MUTACJA GENU — SACCHAROMYCES CEREVISIAE
1. METODA
1.1. WSTĘP
Zob. Wprowadzenie ogólne część B.
1.2. DEFINICJE
Zob. Wprowadzenie ogólne część B.
1.3. SUBSTANCJE ODNIESIENIA
Brak.
1.4. ZASADA METODY BADAWCZEJ
Odmiana haploidalnych i diploidalnych szczepów bakterii drożdży Saccharomyces cerevisiae może zostać użyta w celu dokonania pomiaru produkcji mutacji genu wywołanych czynnikami chemicznymi z udziałem oraz bez aktywacji metabolitycznej.
Używane były wczesne systemy mutacji w szczepach haploidalnych, takie jak pomiar mutacji od czerwonych mutantów wymagających do wzrostu adeniny (ade-1, ade-2) do białych mutantów z podwójną mutacją adeninową oraz selektywne systemy, takie jak indukcja oporności na kanawainę i cykloheksimid.
Najszerzej uznawany system mutacji wstecznych (rewersji) wymaga użycia haploidalnego szczepu XV 185–14C, posiadający nonsensowną mutację ochre ade 2–1, arg 4–17, lys 1–1 i trp 5–48, który może rewertować głównie przez mutacje zastępcze indukujące mutacje specyficzne co do miejsca lub mutacje supresora ochre. XV 185-14C posiada także marker his 1–7, niezmysłową mutację, która może rewertować głównie przez mutacje zewnątrzgenowe, i marker hom 3–10 rewertowany przez mutageny powodujące zmianę fazy odczytu.
Wśród diploidalnych szczepów S. cerevisiae jedynym szeroko używanym szczepem jest D7, który jest homozygotyczny względem ilv 1–92.
1.5. KRYTERIA JAKOŚCIOWE
Brak.
1.6. OPIS METODY BADAWCZEJ
Przygotowania
Roztwory testowanych substancji chemicznych i próbki kontrolne powinny być przygotowywane tuż przed testem, przy użyciu odpowiednich nośników. W przypadku związków organicznych, które nie są rozpuszczalne w wodzie, należy używać nie więcej niż 2-procentowych (objętościowo) roztworów rozpuszczalników organicznych, takich jak etanol, aceton, czy dimetylosulfotlenek (DMSO). Końcowe stężenie nośnika nie powinno znacząco wpływać na żywotność komórek i charakterystykę wzrostu.
Komórki należy poddać oddziaływaniu testowych substancji chemicznych zarówno w przypadku obecności, jak i braku obecności właściwego egzogenicznego systemu aktywacji metabolicznej.
Najpowszechniej używanym systemem jest współczynnik uzupełniony postmitochondrialną frakcją z wątrób gryzoni wcześniej przebadanych enzymem wywołującym czynniki. Wykorzystanie innych gatunków, tkanek, postmitochondrialnych frakcji lub procedur również może być właściwe dla aktywacji matabolicznej.
Warunki badania
Haploidalny szczep XV185–14C i diploidalny szczep D7 są najczęściej używanymi szczepami w badaniach mutacji genu. Inne szczepy również mogą być właściwe.
Wykorzystuje się właściwe pożywki do hodowli szczepu celem ustalenia liczby komórek, które zachowały żywotność i komórek zmutowanych.
Próbki kontrolne pozytywne, niepoddane działaniu substancji i z dodatkiem rozpuszczalnika, muszą być przygotowane jednocześnie. Odpowiednie substancje do kontroli pozytywnej powinny być użyte na zakończenie każdej mutacji.
Należy użyć co najmniej pięć odpowiednio rozmieszczonych stężeń substancji testowej. W odniesieniu do substancji toksycznych, najwyższa wielkość testowego stężenia nie powinna obniżać liczby komórek, które zachowały żywotność poniżej poziomu 5–10 %. Odpowiednio, należy zbadać substancje rozpuszczalne w wodzie do granicy ich rozpuszczalności, wykorzystując właściwe procedury. W odniesieniu do nietoksycznych substancji łatwo rozpuszczalnych w wodzie, najwyższe stężenie należy ustalić dla każdego przypadku oddzielnie.
Płytki inkubują się cztery do siedmiu dni w temperaturze 28–30 oC bez dostępu światła.
Należy użyć subkultury z normalnym zakresem częstotliwości mutacji samorzutnej.
Należy użyć co najmniej trzech szalek replikacji na dane stężenie dla oznaczenia prototrofów produkowanych przez mutację genu i żywotności komórki. W przypadku badań z wykorzystaniem znaczników takich jak horn 3–10 o niskim wskaźniku mutacji liczba szalek musi zostać powiększona w celu dostarczenia istotnych danych statystycznych.
Procedura
Badanie szczepów S. cerevisiae zazwyczaj przeprowadzane jest według procedury badania w środowisku płynnym włączając albo komórki stacjonarne albo wzrostowe. Badania wstępne należy przeprowadzić na komórkach wzrostowych: 1–5 × 107 komórek/ml jest poddanych ekspozycji na testową substancję chemiczną przez 18 godzin przy temperaturze 28–37 oC ze wstrząsaniem; podczas czynności badawczych, w odpowiednich przypadkach, dodawana jest odpowiednia ilość substancji systemu aktywacji metabolicznej. Na zakończenie badań komórki poddawane są odwirowaniu, oczyszczaniu i umieszczeniu na właściwej pożywce do hodowli szczepu. Po okresie inkubacji płytki poddawane są analizie w celu ustalenia ilości komórek, które zachowały żywotność i komórek wywołujących mutację genu. Jeżeli pierwsze badanie daje negatywny wynik, następne doświadczenie powinno być przeprowadzane z użyciem komórek znajdujących się w fazie stacjonarnej. Jeżeli pierwsze badanie daje wynik pozytywny, to potwierdzane jest następnym, niezależnym badaniem.
2. DANE
Dane należy przedstawić w formie tabeli, wskazując liczbę zliczonych kolonii, liczbę mutantów, liczbę komórek żywych i mutantów. Wszystkie wyniki należy potwierdzić w odpowiednim niezależnym badaniu. Dane należy ocenić za pomocą właściwych metod statystycznych.
3. SPRAWOZDANIE
3.1. SPRAWOZDANIE Z BADAŃ
Sprawozdanie z badań powinno, jeżeli możliwe, zawierać następujące informacje:
|
— |
użyty szczep, |
|
— |
warunki testu: komórki w fazie stacjonarnej bądź w fazie wzrostu, skład pożywki, temperatura i czas inkubacji, system aktywacji metabolicznej, |
|
— |
warunki badania: wielkość stężenia substancji podczas ekspozycji na jej działanie, procedura i czas trwania badania, temperatura zastosowana do badania, pozytywne i negatywne kontrole, |
|
— |
liczba zliczonych kolonii, liczba mutantów, liczba komórek żywych i mutantów, stosunek dawki do reakcji w odpowiednich przypadkach, dane oceny statystycznej, |
|
— |
analiza wyników, |
|
— |
interpretacja wyników. |
3.2. ANALIZA I INTERPRETACJA
Zob. Wprowadzenie ogólne część B.
4. ODNIESIENIA
Zob. Wprowadzenie ogólne część B.
B.16. BADANIE REKOMBINACJI GENETYCZNEJ – SACCHAROMYCES CEREVISIAE
1. METODA
1.1. WSTĘP
Zob. Wprowadzenie ogólne część B.
1.2. DEFINICJE
Zob. Wprowadzenie ogólne część B.
1.3. SUBSTANCJE ODNIESIENIA
Brak.
1.4. ZASADA METODY BADAWCZEJ
Mitotyczna rekombinacja u Saccharomyces cerevisiae może być wykryta między genami (lub ogólnie między genem a jego centromerem) i wewnątrz genów. Ten pierwszy przypadek posiada nazwę „mitotyczne crossing-over” i generuje symetryczny produkt, podczas gdy drugi przypadek najczęściej generuje niesymetryczne produkty i posiada nazwę „konwersja genów”. Zajście crossing-over jest widoczne dzięki wytwarzaniu się kolonii homozygot recesywnych, lub sektorów wytworzonych w szczepach heterozygotycznych, podczas gdy konwersja genów uwidacznia się przez wytworzenie prototroficznych rewertantów w auksotroficznych, heteroallelicznych szczepach posiadających dwa różne nieprawidłowe allele tego samego genu. Do detekcji mitotycznej konwersji genów najczęściej używa się szczepów D4 (heteroalleliczny pod względem ade 2 i trp 5), D7 (heteroalleliczny pod względem trp 5), BZ34 (heteroalleliczny pod względem arg 4) i JD1 (heteroalleliczny pod względem his 4 i trp 5). Mitotyczne crossing-over, którego skutkiem jest powstanie czerwonych i różowych sektorów komórek homozygotycznych, może być wykrywane przy użyciu szczepów D5 lub D7 (które również wskazują na mitotyczną konwersję genów i wsteczną mutację przy ilv 1–92). Obydwa szczepy są heteroalleliczne pod względem odpowiadających sobie alleli ade 2.
1.5. KRYTERIA JAKOŚCIOWE
Brak.
1.6. OPIS METODY BADAWCZEJ
Przygotowania
Roztwory testowych substancji chemicznych i próbki kontrolne powinny być przygotowywane tuż przed badaniem, przy użyciu odpowiednich nośników. W przypadku związków organicznych, które nie są rozpuszczalne w wodzie, należy używać nie więcej niż 2 % (objętościowo) roztworów rozpuszczalników organicznych, takich jak etanol, aceton, czy dimetylosulfotlenek (DMSO). Końcowe stężenie nośnika nie powinno w sposób znaczący wpływać na żywotność komórek i charakterystykę wzrostu.
Komórki należy poddać oddziaływaniu testowych substancji chemicznych zarówno w przypadku obecności, jak i braku obecności właściwego egzogenicznego systemu aktywacji metabolicznej. Najpowszechniej używanym systemem jest współczynnik uzupełniony postmitochondrialną frakcją z wątrób gryzoni wcześniej przebadanych enzymem wywołującym czynniki. Wykorzystanie innych gatunków, tkanek, postmitochondrialnych frakcji lub procedur również może być właściwe dla aktywacji metabolicznej.
Warunki badania
Najczęściej wykorzystywanymi szczepami są szczepy diploidalne D4, D5, D7 i JD1. Inne szczepy również mogą być właściwe.
Wykorzystuje się właściwe pożywki do hodowli szczepu celem ustalenia liczby komórek pozostałych przy życiu i częstotliwości genetycznych rekombinacji.
Próbki kontrolne pozytywne, niepoddane działaniu substancji i z dodatkiem rozpuszczalnika, muszą być przygotowane jednocześnie. Odpowiednie substancje do kontroli pozytywnej powinny być użyte na zakończenie każdej mutacji.
Należy użyć co najmniej pięć odpowiednio rozmieszczonych stężeń substancji testowej. Należy uwzględnić między innymi takie czynniki, jak cytotoksyczność i rozpuszczalność. Najniższe stężenie nie może wpływać na żywotność komórki. W odniesieniu do substancji toksycznych najwyższa wielkość badanego stężenia nie powinna obniżać liczby komórek, które zachowały żywotność poniżej poziomu 5–10 %. Odpowiednio, należy zbadać substancje rozpuszczalne w wodzie do granicy ich rozpuszczalności, wykorzystując właściwe procedury. W odniesieniu do nietoksycznych substancji łatwo rozpuszczalnych w wodzie najwyższe stężenie należy ustalić dla każdego przypadku oddzielnie.
Komórki mogą zostać poddane oddziaływaniu substancji testowej albo w fazie stacjonarnej, albo podczas wzrastania przez okres do 18 godzin. Jednakże, w przypadku długotrwałych badań, kultury należy poddać badaniu mikroskopowemu w celu wykrycia formacji zarodnika, którego obecność unieważnia badanie.
Płytki są inkubowane w ciemności od czterech do siedmiu dni w temperaturze 28–30 oC. Płytki użyte do oznaczenia czerwonych i różowych sektorów homozygot wytworzonych przez mitotyczny crossing-over powinny być przechowywane w lodówce (w temperaturze około 4 oC) przez dalsze 1–2 dni przed oznaczeniem wyników, aby umożliwić rozwój odpowiednich zabarwionych kolonii.
Należy użyć subkultur z normalnym zakresem częstotliwości samorzutnej rekombinacji genetycznej.
Powinny być użyte co najmniej trzy szalki na określone stężenie, w celu oznaczenia prototrofów wytworzonych za pomocą mitotycznej konwersji genów i w celu oznaczenia żywotności komórek. W przypadku oznaczania recesywnych homozygot wytworzonych przez mitotyczny crossing-over liczba płytek powinna być zwiększona, aby dostarczyć odpowiednią liczbę kolonii.
Procedura
Badanie szczepów S. cerevisiae zazwyczaj przeprowadzane jest według procedury badania w środowisku płynnym, włączając komórki stacjonarne albo wzrostowe. Początkowe badania należy przeprowadzić na komórkach wzrostowych: 1–5 × 107 komórek/ml jest poddanych ekspozycji na testową substancję chemiczną przez 18 godzin przy temperaturze 28–37 oC ze wstrząsaniem; w okresie trwania badań, w odpowiednich przypadkach, dodawana jest odpowiednia ilość systemu aktywacji metabolicznej.
Na zakończenie badań komórki poddawane są odwirowaniu, oczyszczaniu i umieszczane na właściwej pożywce do hodowli szczepu. Po okresie inkubacji płytki poddawane są analizie w celu ustalenia liczby komórek, które zachowały żywotność i komórek wywołujących mutację genu.
Jeżeli pierwsze badanie daje wynik negatywny, następne doświadczenie powinno być przeprowadzane z użyciem komórek znajdujących się w fazie stacjonarnej. Jeżeli pierwsze badanie daje wynik pozytywny, to potwierdzane jest następnym, niezależnym badaniem.
2. DANE
Dane powinny być przedstawione w formie tabeli, która wskazuje liczbę zliczonych kolonii, ilość rekombinantów, liczbę komórek żywych i częstotliwość wystąpienia rekombinacji. Wyniki powinny być potwierdzone niezależnym badaniem. Dane powinny być ocenione przy użyciu odpowiednich metod statystycznych.
Dane należy ocenić za pomocą właściwych metod statystycznych.
3. SPRAWOZDANIE
3.1. SPRAWOZDANIE Z BADAŃ
Sprawozdanie z badań powinno, jeżeli możliwe, zawierać następujące informacje:
|
— |
użyty szczep, |
|
— |
warunki badań: komórki fazy stacjonarnej lub wzrostowej, skład pożywki, temperatura i okres trwania inkubacji, system aktywacji metabolicznej, |
|
— |
warunki badania: wielkość stężenia substancji, procedura i czas trwania badania, temperatura zastosowana do badania, pozytywne i negatywne grupy kontrolne, |
|
— |
liczba zliczonych kolonii, liczba rekombinantów, liczba komórek żywych i częstotliwość wystąpienia rekombinacji, w odpowiednich przypadkach stosunek dawki do reakcji, dane analizy statystycznej, |
|
— |
analiza wyników, |
|
— |
interpretacja wyników. |
3.2. ANALIZA I INTERPRETACJA
Zob. Wprowadzenie ogólne część B.
4. ODNIESIENIA
Zob. Wprowadzenie ogólne część B.
B.17. MUTAGENNOŚĆ – BADANIE MUTACJI GENETYCZNEJ IN VITRO U SSAKÓW
1. METODA
Metoda ta jest powtórzeniem OECD TG 476, badania mutacji genetycznej in vitro u ssaków (1997).
1.1. WPROWADZENIE
Badanie mutacji genetycznej in vivo u ssaków może być wykorzystane w celu wykrycia mutacji genu wywołanej przez substancje chemiczne. Odpowiednie linie komórkowe, włącznie z L5178Y mysimi komórkami chłoniaków, linie komórkowe chomika chińskiego CHO, CHO-AS52 oraz V79, oraz ludzkie komórki TK6 limfoblastoidu (1). W tych liniach komórkowych najbardziej powszechnie wykorzystywane są pomiary genetycznych punktów końcowych przy kinazie tymidynowej (TK) oraz transferazie fosforybozylowej hypoksantyny-guaniny (HPRT) oraz transgenie transferazy fosforybozylowej ksantyny-guaniny (XPRT). Badania mutacji TK, HPRT oraz XPRT wykrywają różne widma zdarzeń genetycznych. Autosomalne umiejscowienie TK oraz XPRT może pozwolić na wykrycie zdarzeń genetycznych (np. dużych delecji) nie wykrywanych przy HPRT locus na X chromosomach (2)(3)(4)(5)(6).
W badaniu mutacji genetycznej in vitro u ssaków, mogą być wykorzystane hodowle ustanowionych linii komórkowych lub szczepy komórkowe. Wykorzystywane komórki wybierane są na podstawie zdolności do wzrostu w hodowli oraz stabilności spontanicznych częstotliwości mutacji.
Badania przeprowadzane in vitro wymagają ogólnie wykorzystania egzogenicznego źródła aktywacji metabolicznej. Ten system aktywacji metabolicznej nie może naśladować w całości warunków in vivo u ssaków. Powinna zostać zachowana ostrożność w celu uniknięcia warunków, które prowadziłyby do wyników nie-odzwierciedlających wrodzonej mutagenności. Pozytywne wyniki, które nie odzwierciedlają wrodzonej mutagenności, mogą wynikać ze zmian w pH, osmolalności lub wysokiego poziomu cytotoksyczności (7).
Badanie to jest wykorzystywane w celu wykrycia obecności możliwych mutagenów oraz czynników rakotwórczych dla ssaków. Wiele związków, które są pozytywne w tym badaniu są czynnikami rakotwórczymi dla ssaków, jednakże brak idealnego powiązania między tym badaniem oraz rakotwórczością. Powiązanie to jest zależne od klasy substancji chemicznej oraz istnieje wzrastający dowód na to, że istnieją czynniki rakotwórcze, które nie są wykrywane przez to badanie, ponieważ wydaje się, iż działają one przez inne, niegenotoksyczne mechanizmy lub mechanizmy, których brak w komórkach bakteryjnych (6).
Zob. także Ogólne wprowadzenie do części B
1.2. DEFINICJE
Mutacja powodująca zmianę bądź utratę aktywności białka: mutacja genu z rodzaju macierzystego mutanta, który powoduje zmianę lub stratę aktywności enzymatycznej funkcji zakodowanego białka.
Mutageny powodujące substytucję: substancje, które powodują zastąpienie jednego lub wielu par zasad DNA.
Mutageny powodujące mutacje zmiany fazy odczytu: Substancje, które powodują dodanie lub skreślenie pojedynczej lub wielu par nukleotydów w cząstce DNA.
Czas ekspresji fenotypowej: okres, w trakcie którego produkty genu niezmienionego są oddzielane od świeżo zmutowanej komórki.
Częstotliwość mutacji: liczba zaobserwowanych zmutowanych komórek podzielona przez liczbę komórek zdolnych do życia.
Względny wzrost całkowity: wzrost liczby komórek w czasie w porównaniu z populacją kontrolną komórek; obliczony jako stosunek względnego wzrostu czasu skuteczności kloningu negatywnych kontroli względem kontroli negatywnych.
Względny wzrost zawieszania: wzrost liczby komórek powyżej okresu ekspresji względem kontroli negatywnych.
Zdolność do życia: skuteczność kloningu komórek poddanych działaniu substancji w czasie umieszczania w warunkach selektywnych po okresie ekspresji.
Przeżywanie: skuteczność kloningu komórek poddanych działaniu substancji, kiedy są umieszczane na koniec okresu poddawania działaniu substancji; przeżywanie zazwyczaj wyrażane jest w odniesieniu do przeżywania populacji komórek kontrolnych.
1.3. ZASADA METODY BADAWCZEJ
Brakujące komórki w kinazie tymidynowej (TK) z powodu mutacji TK+/- → TK-/- są odporne na skutki cytotoksyczne substancji analogicznej pirymidynie-trifluorotymidynie (TFT). Zdrowe komórki kinazy tymidynowej są wrażliwe na TFT, która powoduje wstrzymanie komórkowej przemiany materii oraz zatrzymuje dalsze dzielenie się komórek. W ten sposób zmutowane komórki są w stanie rozmnażać się wege-tatywnie w obecności TFT, podczas gdy normalne komórki, które zawierają kinazę tymidynową, nie są do tego zdolne. Podobnie komórki, którym brakuje HPRT lub XPRT, są wybierane w oparciu o odporność na 6-tioguaninę (TG) lub 8-azaguaninę (AG). Właściwości substancji badanej powinny być ostrożnie wzięte pod uwagę, jeżeli zasadowy element analogiczny lub związek związany z selektywnym czynnikiem jest badany w ramach jakiegokolwiek badania mutacji genów u ssaków. Na przykład jakakolwiek podejrzewana toksyczność selektywna ze strony substancji badanej w odniesieniu do komórek zmutowanych i niezmutowanych powinna zostać zbadana. W ten sposób przeprowadzenie wyboru system/czynnik musi być potwierdzone, badając substancje chemiczne strukturalnie zawiązane z czynnikami selektywnymi (8).
Komórki w zawiesinie lub w hodowli jednocząsteczkowej są poddawane działaniu substancji badanej, zarówno z aktywacją, jak i bez aktywacji metabolicznej, przez odpowiedni okres oraz hodowane wtórnie w celu oznaczenia cytotoksyczności oraz w celu dopuszczenia do ekspresji fenotypowej przed wybraniem mutanta (9)(10)(11)(12)(13). Cytotoksyczność jest zazwyczaj oznaczana przez pomiar odpowiedniej skuteczności kloningu (przeżywania) lub odpowiedni wzrost hodowli ogółem po okresie poddania działaniu substancji badanej. Hodowle, które zostały poddane działaniu substancji badanej, są utrzymywane w podłożu wzrostu przez wystarczający okres czasu, charakterystyczny dla każdego wybranego locus oraz rodzaju komórki, w celu dopuszczenia do prawie optymalnej ekspresji fenotypowej wywołanych mutacji. Częstotliwość mutacji jest oznaczana przez osadzanie znanej ilości komórek w podłożu zawierającym czynnik selektywny w celu wykrycia zmutowanych komórek oraz w podłożu bez czynnika selektywnego w celu oznaczenia skuteczności kloningu (zdolność do życia). Po odpowiednim okresie inkubacji, hodowle są podliczane. Częstotliwość mutacji jest uzyskiwana z ilości hodowli mutantów w selektywnym podłożu oraz ilości hodowli w podłożu nieselektywnym.
1.4. OPIS METODY BADAWCZEJ
1.4.1. Preparaty
1.4.1.1. Komórki
Różnorodność rodzajów komórek jest dostępna do wykorzystania w tym badaniu, łącznie z subklonami L5171Y, CHO, CHO-AS52, V79 lub komórkami TK6. Rodzaje komórek wykorzystane w tym badaniu powinny posiadać wykazaną czułość na mutanty chemiczne, wysoką skuteczność kloningu oraz stabilną częstotliwość spontanicznej mutacji. Komórki powinny być sprawdzone w odniesieniu do zanieczyszczenia mykoplazmy oraz w przypadku zanieczyszczenia nie powinny być wykorzystywane.
Badanie powinno być tak opracowane, aby miało wcześniej oznaczoną czułość. Liczba wykorzystanych komórek, hodowli oraz stężeń substancji badanej powinna odzwierciedlać te określone parametry (14). Minimalna liczba zdolnych do życia komórek, które przeżywają poddanie działaniu substancji oraz wykorzystywanych na każdym etapie w badaniu, powinna być oparta na częstotliwości mutacji spontanicznej. Ogólną wskazówką jest wykorzystanie liczby komórek, która jest przynajmniej 10-krotnością odwrotności częstotliwości mutacji spontanicznej. Jednakże zaleca się wykorzystanie przynajmniej 106 komórek. Odpowiednie dane historyczne dotyczące wykorzystanego systemu komórkowego powinny być dostępne w celu wskazania spójnego sposobu przeprowadzenia badania.
1.4.1.2. Warunki dotyczące podłoża oraz hodowli
Odpowiednie podłoże hodowli oraz warunki inkubacji (naczynia do hodowli, temperatura, stężenie CO2 oraz wilgotność) powinny być wykorzystane. Podłoże powinno zostać wybrane według systemu selektywnego oraz wykorzystanego rodzaju komórek w badaniu. Jest w szczególności ważne, aby zostały wybrane warunki hodowli, które zapewniają optymalny wzrost komórek w trakcie okresu ekspresji oraz zdolności do formowania kolonii zarówno komórek zmutowanych, jak i niezmutowanych.
1.4.1.3. Przygotowanie hodowli
Komórki są rozmnażane z hodowli podstawowych, osadzonych w podłożu hodowli oraz inkubowanych w temperaturze 37 oC. Przed wykorzystaniem w tym badaniu, może istnieć potrzeba oczyszczenia hodowli z istniejących już wcześniej komórek zmutowanych.
1.4.1.4. Aktywacja metaboliczna
Komórki powinny zostać poddane działaniu substancji badanej zarówno w obecności, jak i w nieobecności odpowiedniego systemu aktywacji metabolicznej. Najbardziej powszechnie wykorzystywanym systemem jest uzupełniana współczynnikiem frakcja pomitochondrialna (S9) przygotowana z wątroby gryzoni, otrzymujących czynniki pobudzające enzymy takie jak Aroklor 1254 (15)(16)(17)(18) lub kombinacja fenobarbitonu oraz β nafoflavonu (19)(20).
Frakcja pomitochondrialna jest zazwyczaj wykorzystywana w stężeniach w zakresie 1–10 % objętościowo w końcowym podłożu badawczym. Wybór oraz warunki systemu aktywacji metabolicznej mogą być uzależnione od klasy badanej substancji chemicznej. W niektórych przypadkach właściwe może być wykorzystanie więcej niż jednego stężenia frakcji pomitochondrialnej.
Ilość rozwijania się komórek, w tym budowa genetycznie skonstruowanych linii komórkowych stanowiących ekspresję szczególnych enzymów aktywujących, może przewidywać potencjał aktywacji endogenicznej. Wybór wykorzystywanych linii komórkowych powinien być naukowo uzasadniony (np. przez relewancję izoenzymu cytochromu P450 dla przemiany materii substancji badanej).
1.4.1.5. Preparat substancji badanej
Stałe substancje badane powinny zostać rozpuszczone lub zawieszone w odpowiednich rozpuszczalnikach lub nośnikach oraz rozcieńczone, jeżeli jest to odpowiednie, zanim komórki zostaną poddane działaniu substancji badanej. Płynne substancje badane mogą być dodawane bezpośrednio do systemu badawczego i/lub rozcieńczane przed poddaniem działaniu substancji badanej. Powinny zostać wykorzystane świeże preparaty, chyba że dane dotyczące stabilności wykazują dopuszczalność ich składowania.
1.4.2. Warunki badania
1.4.2.1. Rozpuszczalnik/nośnik
Rozpuszczalnik/nośnik nie powinien mieć możliwości reagowania chemicznego z substancją badaną oraz powinien być zgodny z przeżywaniem komórek oraz aktywnością S9. Jeśli inne niż dobrze znane rozpuszczalniki/nośniki są wykorzystywane, ich włączenie do doświadczenia powinno być wsparte danymi wskazującymi ich zgodność. Zaleca się, o ile to możliwe, aby w pierwszej kolejności rozważone zostało wykorzystanie wodnych rozpuszczalników/nośników. Badając substancje niestabilne w wodzie, wykorzystane organiczne rozpuszczalniki powinny być wolne od wody. Woda może zostać usunięta przez dodanie sita cząsteczkowego.
1.4.2.2. Stężenia poddania działaniu substancji
Wśród kryteriów, które mają zostać uwzględnione, oznaczając najwyższe stężenia, znajduje się cytotoksyczność, rozpuszczalność w systemie badawczym, oraz zmiany pH lub osmolalności.
Cytotoksyczność powinna być oznaczana z aktywacją metaboliczną oraz bez aktywacji metabolicznej w głównym doświadczeniu, wykorzystując odpowiednie wskazanie integralności komórki oraz wzrostu, takie jak odpowiedni kloning (przeżywanie) lub odpowiedni wzrost całkowity. Użyteczne może być oznaczenie cytotoksyczności oraz rozpuszczalności w doświadczeniu wstępnym.
Powinny zostać wykorzystane przynajmniej cztery nadające się do analizy stężenia. W przypadku gdy istnieje cytotoksyczność, stężenia te powinny obejmować zakres od maksymalnej do minimalnej toksyczności lub jej braku, będzie to zazwyczaj oznaczać, że poziomy stężenia powinny zostać oddzielone przez nie więcej niż jeden czynnik między 2 a √10. Jeżeli maksymalne stężenie jest oparte na cytotoksyczności, wynikiem tego powinien być wzrost w przybliżeniu 10–20 % (ale nie mniejszy niż 10 %) względnego przeżywania (względna skuteczność kloningu) lub względny wzrost. W odniesieniu do względnie nietoksycznych substancji maksymalne stężenie badania powinno wynosić 5 mg/ml, 5 μl/ml, lub 1,01 M, w zależności od tego, które ze stężeń jest najniższe.
Względnie nierozpuszczalne substancje powinny być badane do granic ich rozpuszczalności lub poza granice rozpuszczalności w warunkach hodowli. Dowody na nierozpuszczalność powinny być oznaczane w końcowym podłożu podania substancji, działaniu której komórki są poddawane. Użyteczna może być ocena rozpuszczalności na początku oraz na końcu podawania substancji, ze względu na to, że rozpuszczalność może się zmieniać w czasie trwania poddania działaniu substancji w systemie badawczym w wyniku obecności komórek, S9, surowicy itd. Nierozpuszczalność może być wykryta przez wykorzystanie oka nieuzbrojonego. Strącanie się nie powinno zakłócać oceny.
1.4.2.3. Kontrole
Równoległe pozytywne i negatywne (rozpuszczalnik lub nośnik) kontrole, zarówno z aktywacją metaboliczną, jak i bez aktywacji metabolicznej powinny zostać objęte każdym doświadczeniem. Jeżeli wykorzystywana jest aktywacja metaboliczna, pozytywna kontrolna substancja chemiczna powinna być substancją, która wymaga aktywacji w celu wywołania reakcji mutagennej.
Przykłady pozytywnych substancji kontrolnych obejmują:
|
Warunki aktywacji metabolicznej |
Locus |
Substancja |
Nr CAS |
Nr Einecs |
|
Brak egzogenicznej aktywacji metabolicznej |
HPRT |
Etyl metanosulfonowy |
62-50-0 |
200-536-7 |
|
Etyl nitrozomocznikowy |
759-73-9 |
212-072-2 |
||
|
TK (małe i duże hodowle) |
Metyl metanosulfonowy |
66-27-3 |
200-625-0 |
|
|
XPRT |
Etyl metanosulfonowy |
62-50-0 |
200-536-7 |
|
|
Etyl nitrozomocznikowy |
759-73-9 |
212-072-2 |
||
|
Obecność egzogenicznej aktywacji metabolicznej |
HPRT |
3-Metylocholantren |
56-49-5 |
200-276-4 |
|
N-Nitrosodimethylamine |
62-75-9 |
200-549-8 |
||
|
7,12-antracen dimetylobenzenu |
57-97-6 |
200-359-5 |
||
|
TK (małe i duże hodowle) |
Cyklofosfamid |
50-18-0 |
200-015-4 |
|
|
Cyklofosfamidomonohydrat |
6055-19-2 |
|
||
|
Benzo[a]piren |
50-32-8 |
200-028-5 |
||
|
3-Metylocholantren |
56-49-5 |
200-276-5 |
||
|
XPRT |
N-Nitrozodometyloamina (dla wysokich poziomów S-9) |
62-75-9 |
200-549-8 |
|
|
Benzo[a]piren |
50-32-8 |
200-028-5 |
Mogą zostać wykorzystane inne odpowiednie wzorcowe pozytywne substancje kontrolne, np. jeśli laboratorium posiada dane historyczne w oparciu o 5-bromo 2'-deoksyurydynę (nr CAS 59-14-3, nr Einecs 200-415-9), ta substancja wzorcowa mogłaby zostać również wykorzystana. Powinno zostać uwzględnione wykorzystanie pozytywnych substancji kontrolnych związanych z klasą substancji chemicznych, jeżeli są dostępne.
Powinny zostać objęte badaniem negatywne kontrole, składające się z samego rozpuszczalnika lub nośnika w podłożu, do którego dodawana jest substancja, oraz w odniesieniu do których stosuje się identyczne działania jak w odniesieniu do grupy poddanej działaniu substancji badanej. Dodatkowo kontrole bez poddania działaniu substancji powinny być również wykorzystane, chyba że istnieją historyczne dane dotyczące kontroli wykazujące, że wybrany rozpuszczalnik nie wywołuje żadnych szkodliwych lub mutagennych skutków.
1.4.3. Procedura
1.4.3.1. Poddawanie działaniu substancji badanej
Rozmnażające się komórki powinny zostać poddane działaniu substancji badanej zarówno z aktywacją metaboliczną, jak i bez aktywacji metabolicznej. Poddanie działaniu substancji powinno trwać odpowiedni okres czasu (zazwyczaj skuteczny jest okres 3–6 godzin). Czas poddania działaniu substancji badanej może zostać przedłużony na okres jednego lub większej liczby cykli komórkowych.
Albo komórki poddawane pojedynczo działaniu substancji, albo poddawane podwojonemu działaniu substancji badanej mogą być wykorzystane przy każdym badanym stężeniu. Jeżeli wykorzystywane są pojedyncze hodowle, ilość stężeń powinna zostać zwiększona w celu zapewnienia odpowiedniej ilości hodowli do analizy (np. przynajmniej osiem stężeń nadających się do analizy). Powinny zostać wykorzystane podwojone negatywne (rozpuszczalnik) hodowle kontrolne.
Gazowe lub lotne substancje powinny być badane odpowiednimi metodami, takimi jak w szczelnie zamkniętych naczyniach do hodowli (21)(22).
1.4.3.2. Pomiar przeżywania, zdolności do życia oraz częstotliwości mutacji
Na koniec okresu poddawania działaniu substancji komórki są wymywane oraz hodowane w celu oznaczenia przeżywania oraz w celu umożliwienia ekspresji genotypowej mutanta. Pomiar cytotoksyczności przez oznaczanie względnej skuteczności kloningu (przeżywanie) lub względny wzrost całkowity zazwyczaj rozpoczyna się po okresie poddawania działaniu substancji.
Każdy locus posiada określony wymóg w celu dopuszczania do prawie optymalnej ekspresji fenotypowej nowo powstałych mutantów (HPRT oraz XPRT wymaga przynajmniej 6–8 dni, a TK przynajmniej 2 dni). Komórki są hodowane w podłożu z czynnikami selektywnymi lub bez czynników selektywnych w odniesieniu do oznaczania ilości mutantów oraz wydajności klonowania, odpowiednio. Pomiary zdolności do życia (wykorzystywane do obliczania częstotliwości mutacji) rozpoczynane są na koniec okresu ekspresji przez umieszczanie w nieselektywnym podłożu.
Jeżeli substancja badana jest pozytywna w badaniu L5178Y TK+/-, powinno zostać przeprowadzone sortowanie według rozmiarów na przynajmniej jednej z badanych kolonii (najwyższe pozytywne stężenie) oraz na pozytywnych i negatywnych kontrolach. Jeżeli substancja badana jest negatywna w badaniu L5178Y TK+/-, powinno zostać przeprowadzone sortowanie według rozmiarów na negatywnych i pozytywnych kontrolach. W badaniach wykorzystujących TK6TK+/- może być również przeprowadzone sortowanie według rozmiarów kolonii.
2. DANE
2.1. PRZETWARZANIE WYNIKÓW
Dane powinny obejmować oznaczenie cytotoksyczności oraz zdolności do życia, liczbę koloni oraz częstotliwości mutacji w odniesieniu do hodowli, które zostały poddane działaniu substancji oraz hodowli kontrolnych. W przypadku pozytywnej reakcji w badaniu L5178Y TK+/-, kolonie są oceniane wykorzystując kryteria małych oraz dużych kolonii na przynajmniej jednym stężeniu substancji badanej (najwyższe pozytywne stężenie) oraz na pozytywnych i negatywnych kontrolach. Charakter cytogenetyczny oraz charakter budowy cząsteczkowej mutantów, zarówno małych, jak i dużych kolonii, został szczegółowo zbadany (23)(24). W badaniu TK+/- kolonie są oceniane, wykorzystując kryteria normalnego wzrostu (duże) oraz wolnego wzrostu (małe) koloni (25). Zmutowane komórki, które najbardziej dotkliwie odczuły uszkodzenie genetyczne, przedłużyły podwajanie okresów, a zatem tworzą małe kolonie. Uszkodzenie to zazwyczaj waha się w zakresie od strat całego genu do kariotypów widocznych aberracji chromosomowych. Wprowadzenie mutantów małych kolonii zostało powiązane z substancjami chemicznymi, które wywołują całkowite aberracje chromosomowe (26). Mniej poważnie dotknięte zmutowane komórki wzrastają w zakresie podobnym do komórek macierzystych oraz tworzą duże kolonie.
Powinno zostać podane przeżywanie (względna skuteczność kloningu) lub względny wzrost całkowity. Częstotliwość mutacji powinna być wyrażona jako liczba komórek mutantów przypadających na liczbę komórek przeżywających.
Powinny zostać dostarczone dane dotyczące poszczególnych hodowli. Dodatkowo wszelkie dane powinny zostać podsumowane w formie tabelarycznej.
Nie istnieje wymóg sprawdzania jasnej reakcji pozytywnej. Niejednoznaczne wyniki powinny zostać wyjaśnione przede wszystkim wykorzystując modyfikacje warunków doświadczalnych. Wyniki negatywne muszą zostać potwierdzone na zasadzie jednostkowych przypadków. W przypadkach gdy potwierdzenie negatywnych wyników nie jest uznawane za niezbędne, powinno zostać zapewnione uzasadnienie. Modyfikacje parametrów badawczych w celu rozszerzenia zakresu ocenianych warunków powinno być rozważone w dalszych doświadczeniach w odniesieniu do niejednoznacznych lub negatywnych wyników. Parametry badawcze, które mogą zostać zmodyfikowane, obejmują zakres stężeń oraz warunki aktywacji metabolicznej.
2.2. OCENA I INTERPRETACJA WYNIKÓW
Istnieją liczne kryteria oznaczania wyników pozytywnych, takie jak związany z dawką wzrost lub wzrost odtwarzalności częstotliwości mutacji. Biologiczne znaczenie wyników powinno zostać wzięte pod uwagę w pierwszej kolejności. Metody statystyczne mogą zostać wykorzystane jako metody pomocnicze w przeprowadzaniu oceny wyników (24). Znaczenie statystyczne nie powinno być jedynym czynnikiem określającym w odniesieniu do reakcji pozytywnej.
Substancja badana, w odniesieniu do której wyniki nie spełniają powyższych kryteriów, uważana jest za niemutagenną w tym systemie.
Chociaż większość doświadczeń będzie dawała jasne pozytywne lub negatywne wyniki, w rzadkich przypadkach zestaw danych będzie wykluczał dokonanie ostatecznej oceny dotyczącej aktywności substancji badanej. Wyniki mogą pozostać niejednoznaczne lub sporne niezależnie od liczby powtórzeń doświadczenia.
Wyniki pozytywne z badania mutacji genetycznej u ssaków wskazują, że substancja badana wywołuje mutacje genowe w hodowanych komórkach. Największe znaczenie ma reakcja pozytywna stężenia. Wyniki negatywne wskazują, że w warunkach badawczych, substancja badana nie wywołuje mutacji genetycznych w wykorzystanych hodowanych komórkach ssaków.
3. SPRAWOZDAWCZOŚĆ
SPRAWOZDANIE Z BADANIA
Sprawozdanie z badania musi obejmować następujące informacje:
|
|
Rozpuszczalnik/nośnik:
|
|
|
Komórki:
|
|
|
Warunki badania:
|
|
|
Wyniki:
|
4. ODNIESIENIA
|
(1) |
Moore, M. M., DeMarini, D. M., DeSerres, F. J. and Tindal, K. R. (eds.) (1987), Banbury Report 28; Mammalian Cell Mutagenesis, Cold Spring Harbor Laboratory, New York. |
|
(2) |
Chu, E. H. Y. and Malling H. V. (1968), Mammalian Cell Genetics. II. Chemical Induction of Specific Locus Mutations in Chinese Hamster Cells In vitro, Proc. Natl. Acad. Sci. USA, 61, pp. 1306–1312. |
|
(3) |
Liber, H. L. and Thilly, W. G. (1982), Mutation Assay at the Thymidine Kinase Locus in Diploid Human Lymphoblasts, Mutation Res. 94, pp. 467–485. |
|
(4) |
Moore, M. M., Harington-Brock, K., Doerr, C. L. and Dearfield, K. L. (1989), Differential Mutant Quantitation at the Mouse Lymphoma TK and CHO HGPRT Loci, Mutagenesis, 4, pp. 394–403. |
|
(5) |
Aaron, C. S., and Stankowski, Jr. L. F., (1989), Comparison of the AS52/XPRT and the CHO/HPRT AssayS: Evaluation of Six Drug Candidates, Mutation Res. 223, pp. 121–128. |
|
(6) |
Aaron, C. S., Bolcsfoldi, G., Glatt, H. R., Moore, M., Nishi, Y., Stankowski, Jr. L. F., Theiss, J. and Thompson, E. (1994), Mammalian Cell Gene Mutation Assays Working Group Report. Report of the International Workshop on Standardisation of Genotoxicity Test Procedures. Mutation Res. 312, pp. 235–239. |
|
(7) |
Scott, D., Galloway, S. M., Marshall, R. R., Ishidate, M., Brusick, D., Ashby, J. and Myhr, B. C. (1991), Genotoxicity Under Extreme Culture Conditions. A report from ICPEMC Task Group 9, Mutation Res. 257, pp. 147–204. |
|
(8) |
Clive, D., McCuen, R., Spector, J. F. S., Piper, C. and Mavournin, K. H. (1983), Specific Gene Muta tions in L5178Y Cells in Culture. A Report of the U. S. Environmental Protection Agency Gene-Tox Program, Mutation Res., 115, pp. 225–251. |
|
(9) |
Li, A. P., Gupta, R. S., Heflich, R. H. and Wasson, J. S. (1988), A Review and Analysis of the Chinese Hamster Ovary/Hypoxanthine Guanine Phosphoribosyl Transferase System to Determine the Mutagenicity of Chemical AgentS: A Report of Phase III of the U. S. Environmental Protections Agency Gene-Tox Program, Mutation Res., 196, pp. 17–36. |
|
(10) |
Li, A. P., Carver, J. H., Choy, W. N, Hsie, A. W., Gupta, R. S., Loveday, K. S., ÓNeill, J. P., Riddle, J. C, Stankowski, L. F. Jr. and Yang, L. L. (1987), A Guide for the Performance of the Chinese Hamster Ovary Cell/Hypoxanthine-Guanine Phosphoribosyl Transferase Gene Mutation Assay, Mutation Res., 189, pp. 135–141. |
|
(11) |
Liber, H. L., Yandell, D. W. and Little, J. B. (1989), A Comparison of Mutation Induction at the TK and HPRT Loci in Human Lymphoblastoid CellS: Quantitative Differences are Due to an Additional Class of Mutations at the Autosomal TK Locus, Mutation Res., 216, pp. 9–17. |
|
(12) |
Stankowski, L. F. Jr., Tindal,, K. R., and Hsie, A. W. (1986), Quantitative and Molecular Analyses of Ethyl Methanosulphonate – and ICR 191 -Induced Molecular Analyses of Ethyl Methanosulphonate – and ICR 191- Induced Mutation in AS52 Cells, Mutation Res., 160, pp. 133–147. |
|
(13) |
Turner, N. T., Batson, A. G. and Clive, D. (19 84), Procedure for the L5178Y/TK+/- – TK+/- Mouse Lymphoma Cell Mutagenicity Assay, w: Kilbey, B. J. et al (eds.) Handbook of Mutagenicity Test Procedures, Elsevier Science Publishers, New York, pp. 239–268. |
|
(14) |
Arlett, C. F., Smith, D. M., Clarke, G. M., Green, M. H. L., Cole, J., McGregor, D. B. and Asquith S. C. (1989), Mammalian Cell Gene Mutation Assays Based upon Colony Formation, w: Statistical Evaluation of Mutagenicity Test Data, Kirkland, D. J., ed., Cambirdge University Press, pp. 66–101. |
|
(15) |
Abbondandolo, A., Bonatti, S., Corti, G., Fiorio, R., Loprieno, N. and Mazzaccoro, A. (1977), Induction of 6-Thioguanine-Resistant Mutants in V79 Chinese Hamster Cells by Mouse-Liver Microsome-Activated Dimethylnitrosamine, Mutation Res. 46, pp. 365–373. |
|
(16) |
Ames, B. N., McCann, J. and Yamasaki, E. (1975), Methods for Detecting Carcinogens and Mutagens with the Salmonella/Mammalian-Microsome Mutagenicity Test, Mutation Res. 31, pp. 347–364. |
|
(17) |
Clive, D., Johnson, K. O., Spector, J. F. S., Batson A. G. and Brown M. M. M. (1979), Validation and Characterisation of the L5178Y/TK – Mouse Lymphoma Mutagen Assay System, Mutat. Res. 59, pp. 61–108. |
|
(18) |
Maron, D. M. and Ames, B. N. (1983), Revised Methods for the Salmonella Mutagenicity Test, Mutation Res. 113, pp. 173–215. |
|
(19) |
Elliott, B. M., Combes, R. D., Elcombe, C. R., Gatehouse, D. G., Gibson, G. G., Mackay, J. M. and Wolf, R. C. (1992), Alternatives to Aroclor 1254-Induced S9 in: In vitro Genotoxicity Assays, Mutagenesis 7, pp. 175–177. |
|
(20) |
Matsushima, T., Sawamura, M., Hara, K. and Sugimura, T. (1976), A Safe Substitute for Polycholrinated Biphenyls as an Inducer of Metabolic Activation Systems, in: In vitro Metabolic Activation in Mutagenesis Testing, de Serres, F. J., Fouts, J. R., Bend, J. R. and Philpot, R. M. (eds), Elsevier, North-Holland, pp. 85–88. |
|
(21) |
Krahn, D. F., Barsky, F. C. and McCooey, K. T. (1982), CHO/HGPRT Mutation Assay: Evaluation of Gases and Volatile Liquids, in: Ticc, R. R., Costa, D. L., Schaich, K. M. (eds), Genotoxic Effects of Airborne Agents, New York, Plenum, pp. 91–103. |
|
(22) |
Zamora, P. O., Benson, J. M., Li, A. P. and Brooks, A. L. (1983), Evaluation of an Exposure System Using Cells Grown on Collagen Gels for Detecting Highly Volatile Mutagens in the CHO/HGPRT Mutation Assay, Environmental Mutagenesis, 5, pp. 795–801. |
|
(23) |
Applegate, M. L., Moore, M. M., Broder, C. B., Burrell, A. and Hozier, J. C. (1990), Molecular Dissection of Mutations at the Heterozygous Thymidine Kinase Locus in Mouse Lymphoma Cells, Proc. Natl. Acad. Sci. USA, 87, pp. 51–55. |
|
(24) |
Moore, M. M., Clive, D. Hozier, J. C, Howard, B. E., Batson, A. G., Turner, N. T. and Sawyer, J. (1985), Analysis of Trifluoronthymidine, Resistant (TFT+) Mutants of L5178Y/TK+/- – Mouse Lymphoma Cells, Mutation Res. 151, pp. 161–174. |
|
(25) |
Yandell, D. W., Dryja, T. P. and Little, J. B. (1990), Molecular Genetic Analysis of Recessive Mutations at a Heterozygous Autosomal Locus in Human Cells, Mutation Res. 229, pp. 89–102. |
|
(26) |
Moore, M. M. and Doerr, C. L. (1990), Comparison of Chromosome Aberration Frequency and Small Colony TK-Deficient Mutant Frequency in L5178Y/TK+/- – 3.7.2C Mouse Lymphoma Cells, Mutagenesis, 5, pp. 609–614. |
B.18. USZKODZENIE I NAPRAWA DNA – NIEREGULARNA SYNTEZA DNA – KOMÓRKI SSAKÓW IN VITRO
1. METODA
1.1. WSTĘP
Zob. Wprowadzenie ogólne część B.
1.2. DEFINICJE
Zob. Wprowadzenie ogólne część B.
1.3. SUBSTANCJE ODNIESIENIA
Brak.
1.4. ZASADA METODY BADAWCZEJ
Test niezaplanowanej syntezy DNA (UDS) mierzy syntezę DNA po wycięciu i usunięciu odcinka DNA zawierającego obszar uszkodzony przez czynniki chemiczne lub fizyczne. Ten test jest oparty na inkorporacji tymidyny znakowanej trytem (3H-TdR) do DNA komórek ssaków, które nie znajdują się w fazie S cyklu komórkowego. Przyjęcie 3H-TdR może być oznaczone metodą autoradiografii lub zliczania w ciekłym scyntylatorze (LSC) DNA badanych komórek. Komórki ssaków w kulturach, o ile nie są to pierwotne hepatocyty szczura, są poddawane działaniu czynników testowych z obecnością lub bez obecności egzogennych systemów aktywacji metabolicznej. UDS może być także wykonywany w systemach in vivo.
1.5. KRYTERIA JAKOŚCIOWE
Brak.
1.6. OPIS METODY BADAWCZEJ
Przygotowania
Substancje testowe i próbki kontrolne bądź próbki odniesienia powinny być przygotowywane w pożywce wzrostowej, bądź rozpuszczone lub zawieszone w odpowiednich nośnikach, a następnie rozcieńczone w pożywce wzrostowej i użyte do oznaczenia. Końcowe stężenie nośnika nie powinno wpływać na żywotność komórki.
Kultury hepatocytów szczura, ludzkich limfocytów lub ustalonych linii komórkowych (np. ludzkich diploidalnych fibroblastów) mogą być użyte do oznaczeń.
Komórki należy poddać działaniu substancji testowej zarówno w obecności, jak i przy braku obecności właściwego systemu aktywacji metabolicznej.
Warunki badania
Konieczne jest wykorzystanie w każdym punkcie badania co najmniej dwóch kultur komórkowych do autoradiografii i sześciu kultur (lub mniej w przypadku naukowego uzasadnienia) dla określenia UDS za pomocą LSC.
Do każdego badania należy włączyć równoległe pozytywne i negatywne grupy kontrolne (niezbadane i/lub którym podawano nośnik) przy obecności, jak i przy braku aktywacji metabolicznej.
Przykładami związków, które mogą być dodawane do próbek kontrolnych pozytywnych, mogą być 7, 12-dimetylobenzatracen (7,12-DMBA) lub 2-acetylaminofluoren (2-AAF). W przypadku ustalonych linii komórkowych 4-nitrochinolino-N-tlenek (4-NQO) jest przykładem związku mogącego stanowić pozytywną kontrolę zarówno dla autoradiografii, jak i oznaczeń LSC wykonywanych bez aktywacji metabolicznej komórek; N-dimetylonitrozamina jest przykładem związku dodawanego do próbek kontrolnych, kiedy używane są systemy aktywacji metabolizmu.
Należy użyć wielu stężeń substancji testowej ponad zakres odpowiedni w celu określenia reakcji. Najwyższe stężenia powinny wywołać pewne objawy cytotoksyczności. Odpowiednio, substancje nierozpuszczalne w wodzie należy badać do granicy ich rozpuszczalności. Dla substancji nietoksycznych łatwo rozpuszczalnych w wodzie wyższe stężenie substancji testowej należy rozpatrywać dla każdego przypadku oddzielnie.
W celu utrzymania kultury należy zastosować właściwą pożywkę, stężenie CO2, temperaturę oraz wilgotność. Ustalone linie komórkowe należy okresowo sprawdzać w celu wykrycia skażenia mikoplazmą.
System aktywacji metabolicznej nie jest używany w przypadku pierwotnych kultur hepatocytów. Ustalone linie komórkowe i limfocyty poddane ekspozycji na substancję testową zarówno przy obecności, jak i przy braku odpowiedniego systemu aktywacji metabolizmu.
Procedura
Ustalone linie komórkowe są tworzone z kultur zapasowych (np. przez trypsynizację bądź wytrząsanie), pasażowanych w odpowiedniej gęstości do naczyń i inkubowanych w temperaturze 37 oC.
Krótkoterminowe kultury hepatocytów szczura zakładane są przez umożliwienie hepatocytom świeżo rozdzielonym na właściwej pożywce, zagnieżdżenie się na powierzchni wzrostowej.
Kultury ludzkich limfocytów zakładane są za pomocą właściwych technik.
Pierwotne hypocyty szczura
Świeżo izolowane hepatocyty szczura są przez odpowiedni czas poddawane działaniu substancji testowej w pożywce zawierającej 3H-TdR. Pod koniec tego okresu pożywka powinna zostać odsączona, a komórki przepłukane, przygotowane i wysuszone. Szkiełka powinny być zanurzone w emulsji autoradiograficznej (ewentualnie można użyć filmu), naświetlone, wywołane, wybarwione i zliczone.
Ustalone linie komórkowe i limfocytowe
Techniki autoradiograficzne: Kultury komórkowe są poddane ekspozycji na substancję testową przez odpowiedni okres czasu po zastosowaniu 3H-TdR. Czas trwania ekspozycji na działanie substancji zależy od charakteru substancji, aktywności systemów metabolizujących oraz rodzaju komórek. W celu wykrycia szczytu UDS należy dodać 3H-TdR równocześnie z substancją testową albo w przeciągu kilku minut po ekspozycji na substancje testową. Wybór między tymi dwiema procedurami będzie uwarunkowany ewentualnymi interakcjami między substancją testową a 3H-TdR. Aby odróżnić między UDS i semikonserwatywną replikacją DNA, to ostatnie powinno być zahamowane, na przykład przez użycie pożywki bez zawartości argininy, niewielkiej zawartości surowicy lub przez dodatek hydroksymocznika w pożywce.
Badanie UDS przy pomocy LSC: Przed zastosowaniem substancji testowej powinno zostać zablokowane wejście komórek w fazę S w taki sposób, jak opisano powyżej; komórki powinny być poddawane działaniu substancji testowych tak, jak opisano dla autoradiografii. Po zakończeniu okres inkubacji DNA należy wydobyć z komórek oraz należy ustalić ogólną zawartość DNA oraz stopień zawartości 3H-TdR.
Należy zauważyć, że – w przypadku użycia ludzkich limfocytów w powyższych technikach – wymagane jest wyeliminowanie półkonserwatywnej replikacji DNA w niestymulowanych kulturach.
Analiza
Przy oznaczaniu UDS w komórkach danej kultury jądra znajdujące się w fazie S nie są zliczane. Należy zliczyć, co najmniej 50 komórek na skupienie. Szkiełka powinny być opisywane przed zliczaniem. Kilka wyraźnie oddzielonych losowych pól powinno być policzonych na każdym szkiełku. Ilość 3H-TdR wbudowanego w cytoplazmę powinna być oznaczona przez liczenie trzech terenów o kształcie jądra w cytoplazmie każdej ze zliczanych komórek.
Oznaczając LSC UDS, należy zastosować odpowiednią liczbę kultur do każdej wielkości stężenia.
Wszystkie wyniki należy potwierdzić niezależnie przeprowadzonym badaniem.
2. DANE
Dane należy przedstawić w formie tabeli.
2.1. USTALENIA AUTORADIOGRAFICZNE
Należy oddzielnie odnotować stopień zawartości 3H-TdR w cytoplazmie oraz liczbę ziaren wykrytą w jądrze komórki.
Średnia, mediana i tryb mogą być użyte, aby opisać dystrybucję i stopień wbudowania 3H-TdR do cytoplazmy i liczbę ziaren na jądro.
2.2. OZNACZENIE LSC
Dla oznaczenia LSC należy zgłaszać zawartość 3H-TdR jako dpm/ug DNA. Można zastosować średnią dpm/ug DNA z odchyleniem standardowym w celu opisania dystrybucji zawartości.
Dane należy ocenić za pomocą właściwych metod statystycznych.
3. SPRAWOZDANIE
3.1. SPRAWOZDANIE Z BADAŃ
Sprawozdanie z badań powinno, jeżeli możliwe, zawierać następujące informacje:
|
— |
zastosowane komórki, gęstość oraz liczba pasaży trakcie badania, liczba kultur komórek, |
|
— |
metody użyte w celu utrzymania kultury komórek, włączając pożywkę, temperaturę i stężenie CO2, |
|
— |
substancja badana, nośnik, stężenia i racjonalna podstawa wyboru wielkości zastosowanych stężeń w próbie, |
|
— |
szczegóły systemów aktywacji metabolicznej, |
|
— |
zasady badania, |
|
— |
pozytywne i negatywne grupy kontrolne, |
|
— |
zastosowane techniki autoradiograficzne, |
|
— |
procedury użyte w celu zablokowania wejścia komórek do fazy S, |
|
— |
procedury zastosowane w celu wydobycia DNA oraz ustalenia ogólnej zawartości DNA w oznaczaniu LSC, |
|
— |
stosunek dawki do reakcji, jeżeli to możliwe, |
|
— |
analiza statystyczna, |
|
— |
analiza wyników, |
|
— |
interpretacja wyników. |
3.2. ANALIZA I INTERPRETACJA
Zob. Wprowadzenie ogólne część B.
4. ODNIESIENIA
Zob. Wprowadzenie ogólne część B.
B.19. TEST WYMIANY CHROMATYD SIOSTRZANYCH IN VITRO
1. METODA
1.1. WSTĘP
Zob. Wprowadzenie ogólne część B.
1.2. DEFINICJE
Zob. Wprowadzenie ogólne część B.
1.3. SUBSTANCJE ODNIESIENIA
Brak.
1.4. ZASADA METODY BADAWCZEJ
Test wymiany chromatyd siostrzanych (SCE) jest krótkoterminowym badaniem mającym na celu wykrywanie wzajemnych wymian DNA między dwoma chromatydami siostrzanymi powielającego się chromosomu. SCE stanowi wymianę produktów replikacji DNA w pozornie homologicznych miejscach. Proces wymiany przypuszczalnie obejmuje rozerwanie i ponowne połączenie DNA, chociaż niewiele poznano na temat jego molekularnej podstawy. Wykrywanie SCE wymaga niektórych sposobów odmiennego etykietowania chromatyd siostrzanych, co można osiągnąć przez włączenie bromodeoksyurydyna (BrdU) do chromosomowego DNA dla dwóch cykli komórkowych.
Komórki ssaków in vitro poddane są ekspozycji na substancje testowe z użyciem lub bez użycia egzogennego systemu aktywacji metabolicznej, jeżeli jest to odpowiednia metoda i hodowane przez okres dwóch cykli replikacyjnych w pożywce zawierającej BrdU. Po dodaniu inhibitora hamującego tworzenie się wrzeciona kariokinetycznego (np. kolchicyny), umożliwiającego nagromadzenie się komórek w stadium mitozy podobnym do metafazy (metafaza c), komórki są zbierane i przygotowywane są preparaty chromosomów.
1.5. KRYTERIA JAKOŚCIOWE
Brak.
1.6. OPIS METODY BADAWCZEJ
Przygotowania:
|
— |
w teście mogą zostać wykorzystane pierwotne kultury, (ludzkie limfocyty) lub ustalone linie komórkowe (np. komórki jajnika chomika chińskiego). Linie komórkowe należy sprawdzić na skażenie mikoplazmą, |
|
— |
należy zastosować właściwą pożywkę hodowlaną i warunki inkubacji (np. temperatura, zbiorniki hodowlane, stężenie CO2 i wilgotność), |
|
— |
substancje testowe można przygotować na pożywce hodowlanej lub rozpuścić lub zawiesić w odpowiednich nośnikach przed poddaniem badaniu właściwych komórek. Stężenie końcowe nośnika w systemie hodowli nie powinno znacznie wpłynąć na żywotność komórki ani na tempo wzrostu oraz należy monitorować wpływ na częstotliwość SCE przez kontrolę somatyczną, |
|
— |
komórki należy poddać działaniu substancji testowej zarówno w obecności, jak i braku obecności egzogenicznego systemu aktywacji metabolicznej ssaków. Alternatywnie, w przypadku użycia rodzaju komórek z wrodzonym systemem aktywności metabolicznej, tempo oraz charakter aktywności powinny być właściwe w odniesieniu z do badanej klasy chemicznej. |
Warunki badania
Należy użyć co najmniej podwójne kultury dla każdego punktu badania.
Pozytywne grupy kontrolne, używając zarówno bezpośrednio działającego związku, jak i związku wymagającego metabolicznej aktywacji, należy włączyć do każdego badania; należy uwzględnić również grupę kontrolną, której podawany jest nośnik.
Poniższe substancje są przykładami substancji, które mogą być użyte jako pozytywne grupy kontrolne:
|
— |
bezpośrednio działające związki:
|
|
— |
pośrednio działające związki:
|
W odpowiednich przypadkach można włączyć dodatkową pozytywną grupę kontrolną tej samej klasy chemicznej co substancja chemiczna używana w badaniu.
Należy użyć co najmniej trzy odpowiednio rozmieszczone substancje testowe. Najwyższe stężenie powinno wywołać znaczne objawy toksyczności, ale umożliwiać musi odpowiednią replikację komórki. Odpowiednio, substancje nierozpuszczalne w wodzie należy badać do granicy ich rozpuszczalności, wykorzystując właściwe procedury. Dla substancji nietoksycznych łatwo rozpuszczalnych w wodzie wyższe stężenie substancji testowej należy rozpatrywać dla każdego przypadku oddzielnie.
Procedura
Ustalone linie komórkowe są tworzone z kultur zapasowych (np. przez trypsynizację lub wytrząsanie), pasażowanych w odpowiedniej gęstości do naczyń i inkubowanych w temperaturze 37 oC. Dla kultur monowarstwowych ilość komórek na naczynie powinna być regulowana w ten sposób, że kultury stanowią niewiele więcej niż 50 % płynu w czasie zbierania komórek. Ewentualnie komórki mogą być użyte w kulturze zawieszonej. Kultury ludzkich limfocytów są zakładane z heparynizowanej krwi z użyciem odpowiednich technik i inkubowane w temperaturze 37 oC.
Komórki w fazie gwałtownego wzrostu poddawane są ekspozycji na substancję testową przez odpowiedni okres czasu; w większości wypadków wystarczy jedna lub dwie godziny w celu uzyskania pożądanych skutków, ale w niektórych przypadkach czas badania może być przedłużony do dwóch kompletnych cyklów komórkowych. Komórki nieposiadające odpowiedniego wrodzonego systemu aktywności metabolicznej należy poddać działaniu testowej substancji chemicznej w obecności lub braku obecności odpowiedniego systemu aktywności metabolicznej. Po zakończeniu okresu ekspozycji komórki są oczyszczane z substancji testowej i hodowane przez okres dwóch cykli replikacyjnych w obecności BrdU. Alternatywnie, komórki można poddać procedurze równoczesnej ekspozycji na substancję testową oraz BrdU przez całkowity okres hodowli dwóch cyklów komórkowych.
Kultury ludzkich limfocytów poddane są badaniu w chwili, gdy znajdują się w stadium półsynchronicznym.
Komórki analizowane są podczas ich drugiego podziału, po poddaniu działaniu substancji, co zapewnia, iż najbardziej wrażliwe fazy podziału komórki zostały poddane działaniu testowej substancji chemicznej. Wszystkie kultury, do których dodano BrdU, należy utrzymywać w środowisku braku światła lub świetle przyćmionym pochodzącym z żarówek, do chwili zbioru komórek w celu zminimalizowania fotolizy DNA zawierającego BrdU.
Kultury komórkowe poddawane są działaniu inhibitora (np. kolchiny) na godzinę do czterech godzin przed zbiorem. Każda kultura jest zbierana i przetwarzana oddzielnie w celu przygotowania chromosomów.
Przygotowanie chromosomu dokonywane jest za pomocą standardowych technik cytogenetycznych. Barwienie preparatów w celu wykazania SCE można przeprowadzić za pomocą kilku technik (np. metoda fluorescencyjna plus Giemsa).
Liczba komórek przeznaczonych do analizy powinna być oparta o częstotliwość samorzutnej kontroli SCE. Zazwyczaj co najmniej 25 rozprzestrzeniających się metafaz na kulturę jest analizowanych pod względem SCE. Preparty są oznaczane przed rozpoczęciem analiz. W odniesieniu do ludzkich limfocytów jedynie metafazy zawierające 46 centromerów są poddawane analizie. W ustalonych liniach komórkowych jedynie metafazy zawierające ± 2 centromery liczby modalnej są poddawane analizie. Powinno zostać stwierdzone, czy centromeryczny przełącznik znacznika jest oznaczony jako SCE. Wszystkie wyniki należy potwierdzić niezależnie przeprowadzonym badaniem.
2. DANE
Dane należy przedstawić w formie tabeli. Liczba SCE dla każdej metafazy oraz liczba SCE na dany chromosom dla każdej metafazy powinna zostać oddzielnie wyszczególniona dla wszystkich kultur badanych i kontrolnych.
Dane należy oszacować za pomocą odpowiednich metod statystycznych.
3. SPRAWOZDANIE
3.1. SPRAWOZDANIE Z BADAŃ
Sprawozdanie z badań powinno, jeżeli możliwe, zawierać następujące informacje:
|
— |
użyte komórki, metody utrzymywania kultur komórek, |
|
— |
warunki badań: skład pożywki, stężenie CO2, stężenie substancji testowej, użyty nośnik, temperatura inkubacji, czas poddania działaniu substancji, użyty inhibitor, jego stężenie oraz czas trwania badania z jego użyciem, rodzaj użytego systemu metabolicznej aktywacji ssaków, pozytywne i negatywne grupy kontrolne, |
|
— |
liczba kultur komórkowych w danym punkcie badania, |
|
— |
szczegóły dotyczące techniki zastosowanej w celu przygotowania preparatu, |
|
— |
liczba metafaz poddanych analizie (dane przedstawione oddzielnie dla każdej kultury), |
|
— |
średnia liczba SCE w danej komórce i w danym chromosomie (dane przedstawione oddzielnie dla każdej kultury), |
|
— |
kryteria obliczania SCE, |
|
— |
racjonalna podstawa wyboru dawki, |
|
— |
stosunek dawki do reakcji, w odpowiednich przypadkach, |
|
— |
statystyczna analiza, |
|
— |
analiza wyników, |
|
— |
interpretacja wyników. |
3.2. ANALIZA I INTERPRETACJA
Zob. Wprowadzenie ogólne część B.
4. ODNIESIENIA
Zob. Wprowadzenie ogólne część B.
B.20. BADANIA SPRZĘŻONYCH Z PŁCIĄ RECESYWNYCH CECH LETALNYCH U DROSOPHILA MELANOGASTER
1. METODA
1.1. WSTĘP
Zob. Wprowadzenie ogólne część B.
1.2. DEFINICJE
Zob. Wprowadzenie ogólne część B.
1.3. SUBSTANCJE ODNIESIENIA
Brak.
1.4. ZASADA METODY BADAWCZEJ
Badanie sprzężonych z płcią recesywnych cech letalnych (SLRL) u Drosopbila melanogaster wykrywa istnienie mutacji, zarówno punktowych, jak i niewielkich delecji, w linii zarodkowej owada. Niniejsze badanie jest poprzedzone testem mutacji zdolnym zbadać około 800 miejsc na chromosomie X; stanowi to około 80 % wszystkich miejsc chromosomu X. Chromosom X stanowi około jednej piątej całego genomu haploidalnego.
Mutacje w chromosomie X u Drosophila melanogaster fenotypowo ujawniają się u samców przenoszących gen nurtujący. Kiedy mutacja jest letalna u homozygot, jej obecność można wnioskować na podstawie nieobecności jednej klasy męskiego potomstwa, podczas gdy heterozygotyczne samice normalnie wydają dwa rodzaje męskiego potomstwa. Badanie SLRL wykorzystuje te aspekty za pomocą specjalnie oznaczonych i ułożonych chromosomów.
1.5. KRYTERIA JAKOŚCIOWE
Brak.
1.6. OPIS METODY BADAWCZEJ
Przygotowania
Samce pochodzące z dobrze zdefiniowanych zasobów dzikiego typu i samice z zasobu Muller-5 mogą być używane. Inne odpowiednio oznaczone samice z wielokrotną inwersją chromosomu X także mogą być używane.
Substancje testowe powinny być rozpuszczone w wodzie. Substancje nierozpuszczalne w wodzie mogą być rozpuszczone lub zawieszone w odpowiednich nośnikach (np. mieszaninie etanolu i Tween-60 lub 80) i rozcieńczone w wodzie lub roztworze soli przed podaniem. Dimetylosulfoksyd nie powinien być używany jako nośnik.
Test powinien być zaprojektowany z ustalonymi z góry czułością i siłą. Częstotliwość samorzutnej mutacji obserwowana w odpowiedniej grupie kontrolnej w dużym stopniu wpłynie na liczbę badanych chromosomów, które należy poddać analizie.
Poddanie działaniu substancji może odbywać się drogą pokarmową, przez wstrzyknięcie lub przez ekspozycję na gazy lub opary. Substancja testowa może być podawana w roztworze cukru. W przypadku gdy zaistnieje taka konieczność, substancje mogą zostać rozpuszczone w 0,7 % roztworze NaCl oraz wstrzyknięte do klatki piersiowej lub brzucha.
Należy rozważyć użycie negatywnych (użycie nośnika) i pozytywnych grup kontrolnych. Jednakże w przypadku dostępności do odpowiednich laboratoryjnych danych bazowych nie jest wymagane wprowadzenie równoczesnych grup kontrolnych.
Należy zastosować trzy poziomy dawek. W celu dokonania wstępnej analizy można zastosować jeden poziom dawki substancji testowej, której wielkość będzie oscylować na minimalnym tolerowanym poziomie stężenia lub poziomie stężenia wywołującym pewne objawów i toksyczności. Odnośnie do substancji nietoksycznych należy użyć maksymalną stosowaną wielkość stężenia substancji testowej.
Dzikie samce (trzy lub pięciodniowe) poddawane są działaniu substancji testowej oraz indywidualnie używane do krycia dziewiczych samic pochodzących z zasobu Muller-5 lub innego właściwie oznaczonego zasobu (z wielokrotnymi odwróconymi chromosomami X). Samice zastępowane są nowymi dziewiczymi samicami co dwa lub trzy dni, w celu objęcia całego cyklu komórek zarodkowych. Potomstwo tych samic jest analizowane pod względem wystąpienia skutków śmiertelności odpowiadających wpływowi na dojrzałe plemniki, spermatydy średniego i późnego stadium, spermatydy wczesnego stadium, spermatocyty i spermatogonia w czasie poddania działaniu.
Heterozygotyczne samice F1 pochodzące z powyższych krzyżówek kryte są indywidualnie (tj. jedna samica na fiolkę) ich braćmi. W pokoleniu F2 każda kultura liczona jest pod względem braku dzikich samców. Jeżeli okaże się, iż kultura powstała od samic F1 przenoszących cechę śmiertelności w rodzicielskim chromosomie X (tj. nie zaobserwowano żadnych samców z badanym chromosomem), córki takich samic z tym samym genotypem należy zbadać w celu ustalenia, iż cecha śmiertelności jest powtarzana w następnym pokoleniu.
2. DANE
Dane należy przedstawić w formie tabeli w celu wykazania liczby zbadanych chromosomów X, liczby niepłodnych samców oraz liczby chromosomów z cechą letalną przy każdej wielkości stężenia oraz w każdym okresie krycia dla każdego samca poddanego działaniu substancji testowej. Należy odnotować liczbę klastrów różnego rozmiaru na danego samca. Niniejsze wyniki powinny zostać potwierdzone oddzielnym badaniem.
Odpowiednie metody statystyczne powinny być użyte w ocenie cech letalnych sprzężonych z płcią. Skupianie się recesywnych cech letalnych pochodzących od jednego samca powinno być wzięte pod uwagę i przeanalizowane przy pomocy odpowiedniej metody statystycznej.
3. SPRAWOZDANIE
3.1. SPRAWOZDANIE Z BADAŃ
Sprawozdanie z badań powinno, jeżeli możliwe, zawierać następujące informacje:
|
— |
zasoby: zastosowane zasoby lub szczepy Drosophila, wiek owadów, liczba samców poddanych badaniu, liczba bezpłodnych samców, liczba ustalonych kultur F2, liczba kultur F2 bez potomstwa, liczba chromosomów przenoszących cechę letalną, wykrytą w każdej fazie komórki zarodkowej, |
|
— |
kryteria dla określenia wielkości badanych grup, |
|
— |
warunki badania: szczegółowy opis badania i zasada pobierania próbek, poziomy ekspozycji, dane dotyczące toksyczności, negatywne (rozpuszczalnik) i pozytywne grupy kontrolne, odpowiednio do potrzeb, |
|
— |
kryteria służące ocenie mutacji letalnych, |
|
— |
zależność między objawami a poziomem ekspozycji, jeżeli to możliwe, |
|
— |
dane dotyczące analizy, |
|
— |
analiza wyników, |
|
— |
interpretacja wyników. |
3.2. ANALIZA I INTERPRETACJA
Zob. Wprowadzenie ogólne część B.
4. ODNIESIENIA
Zob. Wprowadzenie ogólne część B.
B.21. BADANIE PRZEMIANY KOMÓRKOWEJ SSAKÓW IN VITRO
1. METODA
1.1. WSTĘP
Zob. Wprowadzenie ogólne część B.
1.2. DEFINICJE
Zob. Wprowadzenie ogólne część B.
1.3. SUBSTANCJE ODNIESIENIA
Brak.
1.4. ZASADA METODY BADAWCZEJ
Systemy hodowli komórkowej ssaków mogą być zastosowane w celu wykrycia zmian fenotypowych in vitro wywołanych substancjami chemicznymi związanymi ze złośliwymi przemianami in vivo. Powszechnie używane komórki, takie jak C3H10TV2, 3T3, SHE, szczura Fischera i testowe polegają na ocenie zmian w morfologii komórki, formowania skupisk czy zależności od zakotwiczenia w półstałym agarze. Istnieją systemy rzadziej stosowane, które pozwalają wykryć inne zmiany fizjologiczne lub morfologiczne w komórkach w następstwie ekspozycji na substancję rakotwórczą. Żadne testy końcowe badania in vitro nie mają ustalonego bezpośredniego powiązania z nowotworem. Niektóre systemy badawcze są w stanie wykryć aktywatory guzów. Cytototoksyczność można ustalić przez określenie wpływu badanego materiału na zdolności formowania kolonii (wydajność klonowania) lub wskaźniki wzrostu kultury. Pomiar cytotoksyczności ma na celu ustalenie, że ekspozycja na działanie substancji testowej jest istotna pod względem toksykologicznym, ale nie może być wykorzystywana w celu obliczenia częstotliwości przemian we wszystkich próbach, ponieważ niektóre mogą obejmować długotrwałą inkubację i/lub pasażowanie.
1.5. KRYTERIA JAKOŚCIOWE
Brak.
1.6. OPIS METODY BADAWCZEJ
Przygotowania
W zależności od stosowanego badania przemian dostępne są różne linie komórkowe lub komórki podstawowe. Badacz musi zapewnić, iż komórki wykorzystywane w przeprowadzanym badaniu wykazują właściwe zmiany fenotypowe w następstwie poddania działaniu znanym czynnikom rakotwórczym oraz że badanie, przeprowadzane w laboratorium badacza, jest potwierdzone i posiada udokumentowaną wiarygodność.
Należy zastosować pożywkę oraz warunki doświadczalne właściwe dla prób przemiany komórkowej.
Substancja testowa może zostać przygotowana na pożywce hodowlanej lub rozpuszczona lub zawieszona we właściwych nośnikach przed rozpoczęciem badania komórek. Stężenie końcowe nośnika w systemie hodowli nie powinno wpływać na żywotność komórki, tempo wzrostu ani na częstotliwość przemian.
Komórki należy poddać działaniu substancji testowej zarówno w obecności jak i przy braku obecności właściwego systemu aktywacji metabolicznej. Alternatywnie, w przypadku użycia rodzaju komórek z wrodzoną aktywacją metaboliczną, wskaźnik charakteru aktywności powinien być znany, aby był właściwy dla badanej klasy chemicznej.
Warunki badania
Pozytywne grupy kontrolne, używając zarówno bezpośrednio działającego związku, jak i związku wymagającego metabolicznej aktywacji, należy włączyć do każdego badania; należy uwzględnić również negatywną grupę kontrolną, której podawany jest nośnik.
Poniższe substancje są przykładami substancji, które mogą być użyte jako pozytywne grupy kontrolne:
|
— |
związki działające bezpośrednio:
|
|
— |
związki wymagające aktywacji metabolicznej:
|
W odpowiednich przypadkach można włączyć dodatkową pozytywną grupę kontrolną tej samej klasy chemicznej co związek używany w badaniu.
Należy użyć kilka stężeń substancji testowej. Niniejsze stężenia powinny wywołać objawy toksyczności z nimi związane, przy najwyższym stężeniu powodować niski poziom komórek pozostałych przy życiu oraz poziom żywotności komórek przy najniższym stężeniu, oscylujący w przybliżeniu na tym samym poziomie, co przy stężeniu użytym w negatywnej grupie kontrolnej. Odpowiednio, substancje nierozpuszczalne w wodzie należy badać do granicy ich rozpuszczalności, wykorzystując właściwe procedury. Dla substancji nietoksycznych łatwo rozpuszczalnych w wodzie wyższe stężenie substancji testowej należy rozpatrywać dla każdego przypadku oddzielnie.
Procedura
Komórki należy poddać działaniu substancji przez określony okres czasu, w zależności od stosowanego systemu badań, może to dotyczyć ponownego dawkowania wraz ze zmianą pożywki do hodowli (i w miarę potrzeby, świeżą mieszaninę aktywacji metabolicznej) w przypadku długotrwałej ekspozycji na działanie substancji testowej. Komórki nieposiadające odpowiedniej wrodzonej aktywności metabolicznej należy poddać ekspozycji na substancję testową w obecności i bez obecności właściwego systemu aktywności metabolicznej. Pod koniec okresu ekspozycji komórki są przepłukiwane w celu usunięcia substancji testowej i hodowane w warunkach odpowiednich dla monitorowania momentu pojawienia się zmienionego fenotypu i zasięgu transformacji. Wszelkie wyniki potwierdzane są niezależnym badaniem.
2. DANE
Dane powinny być opracowane w formie tabeli i mogą wymagać uwzględnienia różnorodności form zgodnie z metodą oznaczenia, która została użyta, np. zliczanie płytek, płytki pozytywne bądź ilość stransformowanych komórek. Jeżeli jest to odpowiednia metoda, ilość komórek, które przeżyły, powinna być wyrażona jako procent poziomu kontroli i częstotliwości występowania ekspresji wyrażonej jako stosunek ilości komórek stransformowanych do komórek, które przeżyły ekspozycję. Dane należy przeanalizować za pomocą właściwych metod statystycznych.
3. SPRAWOZDANIE
3.1. SPRAWOZDANIE Z BADAŃ
Sprawozdanie z badań powinno, jeżeli możliwe, zawierać następujące informacje:
|
— |
rodzaj użytej komórki, liczba kultur komórkowych, metody utrzymania kultur komórkowych, |
|
— |
warunki badania: stężenie substancji, użyty nośnik, czas inkubacji, czas trwania oraz częstotliwość badania, gęstość komórkowa podczas badania, rodzaj użytego egzogenicznego systemu aktywacji metabolicznej, pozytywne i negatywne grupy kontrolne, specyfikacja kontrolowanego fenotypu, zastosowany system wyboru (odpowiednio do potrzeb), racjonalna podstawa wyboru dawkowania, |
|
— |
metody stosowane do liczenia żywych i zmienionych komórek, |
|
— |
statystyczna analiza, |
|
— |
analiza wyników, |
|
— |
interpretacja wyników. |
3.2. ANALIZA I INTERPRETACJA
Zob. Wprowadzenie ogólne część B.
4. ODNIESIENIA
Zob. Wprowadzenie ogólne część B.
B.22. BADANIE DOMINUJĄCEGO GENU LETALNEGO GRYZONIA
1. METODA
1.1. WSTĘP
Zob. Wprowadzenie ogólne część B.
1.2. DEFINICJE
Zob. Wprowadzenie ogólne część B.
1.3. SUBSTANCJE ODNIESIENIA
Brak.
1.4. ZASADA METODY BADAWCZEJ
Dominujące cechy letalne powodują śmierć embrionu lub płodu. Indukcja dominujących cech letalnych przez ekspozycję na substancję chemiczną wskazuje na fakt, iż substancja zaatakowała tkankę zarodkową badanego gatunku. Ogólnie przyjmuje się, iż dominujące cechy letalne spowodowane są uszkodzeniami chromosomowymi (nieprawidłowości strukturalne i liczbowe). Śmierć zarodkowa, w przypadku badania samic, może również wynikać ze skutków toksyczności.
Ogólnie, samce poddawane są działaniu badanego związku oraz wykorzystywane do krycia niebadanych dziewiczych samic. Różne fazy komórki zarodkowej można poddać oddzielnym badaniom przez zastosowanie sekwencyjnego krycia w odpowiednich odstępach czasu. Wzrost liczby martwych implantów na samicę w grupie badanej ponad liczbę martwych implantów na samicę w grupie kontrolnej odzwierciedla straty poimplantacyjne. Straty przedimplantacyjne można ocenić w oparciu o liczbę ciałek żółtych lub przez porównanie całkowitej ilości implantów danej samicy w grupie badanej i grupach kontrolnych. Całkowity efekt dominujących cech letalnych jest sumą przedimplantacyjnych i poimplantacyjnych strat. Obliczenie całkowitego skutku dominujących cech letalnych jest oparte na porównaniu żywych implantów danej samicy w grupie badanej do żywych implantów danej samicy w grupie kontrolnej. Zmniejszanie się ilości implantów w pewnych odstępach czasu może być skutkiem uśmiercania komórek (tj. spermatocytów i/lub spermatogoniów).
1.5. KRYTERIA JAKOŚCIOWE
Brak.
1.6. OPIS METODY BADAWCZEJ
Przygotowania
Jeżeli to możliwe, substancje testowe należy rozpuścić lub zawiesić w izotonicznym roztworze soli. Substancje chemiczne nierozpuszczalne w wodzie mogą być rozpuszczone lub zawieszone we właściwych nośnikach. Użyty nośnik nie powinien ani kolidować z testową substancją chemiczną, ani powodować jakichkolwiek objawów toksyczności. Należy zastosować świeże preparaty testowej substancji chemicznej.
Warunki badania
Testowy związek chemiczny ogólnie należy podać tylko raz. W oparciu o informacje badań toksykologicznych można zastosować powtórzony harmonogram badań. Zwyczajowymi drogami podawania substancji testowej jest intubacja drogą pokarmową lub przez iniekcję dootrzewnową. Inne drogi podawania substancji również mogą być właściwe.
Gatunkami zalecanymi do wykorzystania w badaniu są szczury i myszy. Zdrowe, w pełni dojrzałe zwierzęta są losowo przydzielane do grupy badanej i grup kontrolnych.
Należy wykorzystać odpowiednią liczbę samców, uwzględniając samoczynne zmiany cech biologicznych. Wybrana ilość powinna być oparta na wcześniej oznaczonej czułości detekcji i sile znaczenia. Na przykład w typowym badaniu liczba samców w każdej grupie dawkowania powinna być wystarczająca, aby zapewnić 30–50 ciężarnych samic w danym okresie krycia.
Ogólnie, do każdego badania należy włączyć równoległe negatywne i pozytywne grupy kontrolne (dawkowanie z użyciem nośnika). W przypadku dostępności wyników pozytywnej grupy kontrolnej z badań ostatnio przeprowadzanych w tym samym laboratorium niniejsze wyniki można zastosować zamiast równoległej pozytywnej grupy kontrolnej. Należy użyć pozytywnych substancji kontrolnych przy odpowiednio niskiej dawce (np. MMS, dootrzewnowo, przy 10 mg/kilogram) w celu wykazania czułości badania.
Zwyczajowo należy zastosować trzy poziomy dawek. Najwyższa wielkość dawki powinna powodować objawów i toksyczności lub obniżoną płodność u badanych zwierząt. W niektórych przypadkach jednorazowe dawkowanie może okazać się wystarczające.
Substancje nietoksyczne należy testować przy 5 g/kilogram przy jednorazowym podaniu dawki lub przy 1 g/kilogram/dzień przy powtarzalnym podawaniu dawki.
Procedura
Dostępne jest kilka harmonogramów badania. Najszerzej stosowane jest jednorazowe podawanie substancji testowej. Można stosować inne odpowiednie harmonogramy badań.
Poszczególne samce wykorzystywane są do krycia jednej lub dwóch niebadanych, dziewiczych samic sekwencyjnie w odpowiednich odstępach czasu po poddaniu działaniu substancji. Samice należy pozostawić w obecności samca co najmniej przez czas trwania jednego cyklu rujowego lub do czasu wystąpienia krycia ustalonego przez obecność spermy w pochwie lub obecność czopu nasienia w pochwie.
Liczba kryć następujących po zakończeniu badania jest regulowana harmonogramem badania i należy zapewnić, iż zostaną pobrane próbki wszystkich faz komórki zarodkowej po zakończeniu badania.
Samice uśmiercane są w drugiej połowie okresu ciąży, a zawartość macicy poddawana jest badaniu w celu ustalenia liczby żywych oraz martwych implantów. Można przeprowadzić badanie jajników w celu ustalenia liczby ciałek żółtych.
2. DANE
Dane należy przedstawić w formie tabeli, wskazując liczbę samców, liczbę ciężarnych samic, oraz liczbę nieciężarnych samic. Wyniki krycia, włączając identyfikację każdego samca i samicy, należy odnotować indywidualnie. W odniesieniu do każdej samicy należy odnotować tydzień, w którym nastąpiło krycie, wielkość dawki otrzymanej przez samca oraz ilość martwych i żywych implantów.
Obliczenie całkowitego skutku dominujących cech letalnych jest oparte na porównaniu żywych implantów przypadających na samicę w grupie badanej do żywych implantów przypadających na samicę w grupie kontrolnej. Stosunek martwych implantów do żywych implantów w grupie badanej w porównaniu do tego samego stosunku w grupie kontrolnej jest analizowany w celu wykazania strat poimplantacyjnych.
Jeżeli dane odnotowane sąjako zgony wczesne i późne, należy ten fakt jasno sprecyzować w tabeli. Jeżeli dokonywana jest ocena strat przedimplantacyjnych, taki fakt należy odnotować. Straty przedimplantacyjne można obliczyć jako różnicę między liczbą ciałek żółtych oraz liczbą implantów lub jako obniżenie średniej liczby implantów na daną macicę w porównaniu z kryciami kontrolnymi.
Dane szacowane są z wykorzystaniem odpowiednich metod statystycznych.
3. SPRAWOZDANIE
3.1. SPRAWOZDANIE Z BADAŃ
Sprawozdanie z badań powinno, jeżeli możliwe, zawierać następujące informacje:
|
— |
gatunki, szczep, wiek i waga badanych zwierząt, liczba zwierząt każdej płci w grupie badanej i grupach kontrolnych, |
|
— |
substancja testowa, nośnik, badane poziomy dawek oraz racjonalna podstawa wyboru dawki, negatywne i pozytywne grupy kontrolne, dane dotyczące toksyczności, |
|
— |
droga oraz harmonogram badań, |
|
— |
harmonogram krycia, |
|
— |
metody stosowane w celu ustalenia zaistnienia krycia, |
|
— |
czas uśmiercenia zwierząt, |
|
— |
kryteria, według których oznaczane są dominujące cechy letalne, |
|
— |
związek wysokości dawkowania z reakcją, w odpowiednich przypadkach, |
|
— |
analiza statystyczna, |
|
— |
analiza wyników, |
|
— |
interpretacja wyników. |
3.2. ANALIZA I INTERPRETACJA
Zob. Wprowadzenie ogólne część B.
4. ODNIESIENIA
Zob. Wprowadzenie ogólne część B.
B.23. BADANIE ABERRACJI CHROMOSOMOWEJ SPERMATOGONIOW U SSAKÓW
1. METODA
Metoda ta jest powtórzeniem OECD TG 483, badania aberracji chromosomowej spermatogoniów u ssaków (1997).
1.1. WPROWADZENIE
Celem badania aberracji chromosomowej spermatogoniów u ssaków in vivo jest zidentyfikowanie substancji, które powodują strukturalne aberracje chromosomowe w spermatogoniach ssaków (1)(2)(3)(4)(5). Strukturalne aberracje mogą występować w dwóch typach, chromosomowym lub chromatydowym. W przypadku mutagenów chemicznych większość wywołanych aberracji jest typu chromatydowego, ale mogą pojawiać się aberracje typu chromosomowego. Metoda ta jest przeznaczona do pomiaru aberracji liczbowych oraz nie jest rutynowo wykorzystywana do tego celu. Mutacje chromosomowe oraz powiązane z nimi zdarzenia są przyczyną wielu ludzkich chorób genetycznych.
Badanie to dokonuje pomiaru zdarzeń chromosomowych w komórkach zarodków oraz z tego względu podejrzewa się, iż przewiduje wprowadzenie dziedzicznych mutacji w komórkach zarodków.
Zazwyczaj w tym badaniu wykorzystywane są gryzonie. Niniejsze badanie cytogenetyczne in vivo wykrywa aberracje chromosomowe podczas mitotycznego podziału spermatogoniów. Inne komórki docelowe nie podlegają tej metodzie.
W celu wykrycia aberracji typu chromatydowego w spermatogoniach pierwszy podział komórki mitotycznej następujący po poddaniu działaniu substancji powinien zostać zbadany, zanim te zmiany patologiczne są zgubione w toku dalszych podziałów komórkowych. Dalsze informacje ze spermatogoniów macierzystych mogą być uzyskane w drodze mejotycznej analizy w odniesieniu do aberracji typu chromosomowego w metafazie diakinezy, jeżeli komórki poddane działaniu substancji stają się spermatocytami.
Niniejsze badanie in vivo przeznaczone jest do zbadania, czy mutageny komórek somatycznych są również czynne w komórkach zarodników. Dodatkowo badanie spermatogoniów jest właściwe do celów oceny zagrożenia mutagenności, w związku z czym pozwala na uwzględnienie czynników metabolizmu in vivo, farmakokinetyków oraz procesów naprawy DNA.
Pewna ilość generacji spermatogoniów jest obecna w badaniu z widmem czułości na przetwarzanie chemiczne. Zatem wykryte aberracje przedstawiają zagregowaną reakcję poddanych działaniu substancji populacji spermatogoniów z większą ilością zróżnicowanych spermatogoniów przeważających. W zależności od ich pozycji w jądrze różne generacje spermatogoniów mogą być narażone na ogólny obieg, ze względu na barierę komórek Sertoli oraz barierę krew-jądro.
Jeżeli istnieją dowody na to, że substancja lub odpowiedni metabolit oddziałujący nie dotrze do komórki docelowej, właściwe jest wykorzystanie tego badania.
Zob. także Ogólne wprowadzenie do części B.
1.2. DEFINICJE
Aberracja typu chromatydowego: strukturalne uszkodzenie chromosomu, wyrażone jako pękanie pojedynczych chromatydów lub pękanie oraz ponowne łączenie między chromatydami.
Aberracja typu chromosomowego: strukturalne uszkodzenie chromosomu, wyrażone jako pękanie pojedynczych chromatydów lub pękanie oraz ponowne łączenie obu chromatydów w identycznej lokalizacji.
Szczelina: achromatyczne uszkodzenie mniejsze niż szerokość chromatydu, powodujące minimalne wypaczeniu chromatydu.
Aberracja liczbowa: zmiana w liczbie chromosomów w odniesieniu do normalnej liczby charakterystycznej dla wykorzystywanych komórek.
Poliploidalność: wielokrotność liczby chromosomów haploidalnych (n) inna niż liczba diploidalna (np. 3n, 4n itd.).
Aberracja strukturalna: zmiana w strukturze chromosomu, wykrywalna przez badanie mikroskopowe etapu metafazy podziału komórek, zaobserwowana jako usunięcia oraz fragmenty, zmiany (zachodzące – wewnątrz oraz między chromosomami).
1.3. ZASADA METODY BADAWCZEJ
Zwierzęta są poddane działaniu substancji badanej odpowiednią drogą poddania działaniu substancji oraz są uśmiercane w odpowiednim czasie po poddaniu ich działaniu substancji badanej. Przed uśmierceniem zwierzęta są poddawane działaniu substancji zatrzymującej metafazę (np. kolchicyna lub Colcemid®). Preparaty chromosomowe są sporządzane z komórek zarodników oraz barwione, a komórki metafazy są analizowane pod kątem aberracji chromosomowych.
1.4. OPIS METODY BADAWCZEJ
1.4.1. Preparaty
1.4.1.1. Wybór gatunków zwierząt
Powszechnie wykorzystywane są samce chomika chińskiego oraz myszy. Jednakże mogą być wykorzystane w badaniu samce innych odpowiednich gatunków saków. Powszechnie wykorzystywane szczepy laboratoryjne młodych, zdrowych, dojrzałych zwierząt powinny zostać wykorzystane. W trakcie przeprowadzania badania różnice w masie ciała zwierząt powinny być minimalne oraz nie powinny przekraczać ± 20 % średniej masy.
1.4.1.2. Warunki przetrzymywania i karmienia
Ogólne warunki określone w Ogólnym wprowadzeniu do części B są stosowane, chociaż celem w odniesieniu do wilgotności powinien być poziom 50–60 %.
1.4.1.3. Preparaty zwierzęce
Zdrowe, młode, dojrzałe samce są losowo przypisywane do grup kontrolnych oraz grup poddawanych działaniu substancji badanej. Klatki powinny być urządzone w taki sposób, aby możliwe skutki negatywne wynikające z umieszczenia w klatce były zminimalizowane. Zwierzęta są identyfikowane w niepowtarzalny sposób. Zwierzęta są aklimatyzowane do warunków laboratoryjnych przez przynajmniej pięć dni przed rozpoczęciem badania.
1.4.1.4. Przygotowanie dawek
Substancje badane stałe powinny zostać rozpuszczone lub zawieszone w odpowiednich rozpuszczalnikach lub nośnikach oraz rozcieńczone, jeżeli stosowne, przed dawkowaniem zwierzętom. Substancje badane płynne mogą być dawkowane bezpośrednio lub mogą zostać rozpuszczone przed dawkowaniem. Powinny zostać wykorzystane świeżo przygotowywane substancje, chyba że dane dotyczące stabilności wskazują iż dopuszczalne jest składowanie substancji.
1.4.2. Warunki badawcze
1.4.2.1 Rozpuszczalnik/nośnik
Rozpuszczalnik/nośnik nie powinien wywoływać skutków toksycznych przy wykorzystywanych dawkach oraz nie powinien mieć możliwości reagowania chemicznego z substancją badaną. Jeśli wykorzystywane są inne niż dobrze znane rozpuszczalniki/nośniki, ich włączenie do doświadczenia powinno być wsparte danymi wskazującymi ich zgodność. Zaleca się, o ile to możliwe, aby w pierwszej kolejności rozważone zostało wykorzystanie wodnych rozpuszczalników/nośników.
1.4.2.2. Kontrole
Równoległe pozytywne i negatywne (rozpuszczalnik/nośnik) kontrole powinny zostać objęte każdym badaniem. Z wyjątkiem poddawania działaniu substancji badanej, zwierzęta w grupie kontrolnej powinny być przedmiotem identycznych działań w porównaniu ze zwierzętami w grupie poddanej działaniu substancji.
Kontrole pozytywne powinny wywoływać strukturalne aberracje w spermatogoniach, jeżeli są podawane na poziomie poddania działaniu substancji, w odniesieniu do którego oczekuje się wykrywalnego wzrostu powyżej tło.
Dawki kontroli pozytywnych powinny zostać dobrane tak, aby wyniki były jasne, ale nie ujawniały bezpośrednio tożsamości zakodowanych szkiełek mikroskopowych badającemu. Dopuszczalne jest, aby pozytywne kontrole podawane były inną drogą niż substancje badane oraz aby ich próbki pobierane były tylko jeden raz. Wykorzystanie substancji chemicznych kontroli pozytywnych powiązanych z klasą chemiczną może być brane pod uwagę, jeżeli są one dostępne. Przykłady substancji pozytywnych kontroli obejmują:
|
Substancja |
Nr CAS |
Nr Einecs |
|
Cyklofosfamid Cyklophosfamid monohydrat |
50-18-0 6055-19-2 |
200-015-4 |
|
Cykloheksyloamina |
108-91-8 |
203-629-0 |
|
Mitomycyna C |
50-07-7 |
200-008-6 |
|
Krylamid monomeryczny |
79-06-1 |
201-173-7 |
|
Trietylenomelamina |
51-18-3 |
200-083-5 |
Negatywne kontrole, poddane działaniu rozpuszczalnika lub nośnika oraz w innym przypadku poddane działaniu w ten sam sposób co grupy poddane działaniu substancji, powinny zostać objęte badaniem w odniesieniu do każdego pobierania próbek, chyba że dopuszczalna jest wewnątrz zwierzęca zdolność do życia oraz częstotliwości komórek z aberracjami chromosomowymi są wykazane przez dane historyczne. Dodatkowo, powinny zostać również wykorzystane kontrole bez poddawania działaniu substancji, chyba że istnieją historyczne lub opublikowane dane wykazujące, że przez wybrany rozpuszczalnik/nośnik nie są wywoływane żadne szkodliwe lub mutagenne skutki.
1.5. PROCEDURA
1.5.1. Liczba zwierząt
Każda grupa poddana działaniu substancji oraz grupa kontrolna musi obejmować przynajmniej pięć nadających się do analizy samców.
1.5.2. Harmonogram poddania działaniu substancji
Substancje badane są zazwyczaj podawane raz lub dwa razy (np. jako pojedyncza dawka lub jako dwa zabiegi poddawania działaniu substancji). Substancje badane mogą być również podawane jako dawka podzielona, np. dwa zabiegi poddania działaniu substancji tego samego dnia w odstępie czasu nie większym niż kilka godzin, w celu ułatwienia podawania dużych objętości substancji. Inne reżimy dawki powinny być naukowo uzasadnione.
W grupie najwyższej dawki wykorzystywane są dwa okresy pobierania próbek po poddaniu działaniu substancji badanej. Ze względu na to, że na kinetykę komórkową może mieć wpływ substancja badana, wykorzystywany jest jeden wczesny oraz jeden późny okres pobierania próbek w około 24–48 godzin po poddaniu działania substancji badanej. W odniesieniu do dawek innych niż dawka najwyższa powinien zostać uwzględniony 24-godzinny okres pobierania próbek lub 1,5 długości trwania cyklu komórkowego, chyba że w odniesieniu do innych okresów pobierania próbek uważa się, iż są one odpowiednie dla wykrycia skutków (6).
Dodatkowo mogą zostać wykorzystane inne okresy pobierania próbek. Na przykład w przypadku substancji chemicznych, które mogą wywoływać aberracje opóźnione lub mogą wywierać skutki niezależne od S, odpowiednie mogą być wcześniejsze okresy pobierania próbek (1).
Odpowiedniość powtarzanych harmonogramów poddawania działaniu substancji musi zostać zidentyfikowana na zasadzie jednostkowych przypadków. Po powtarzanym zabiegu – poddania działaniu substancji – zwierzę powinno następnie zostać uśmiercone w 24 godziny (1,5 długości cyklu komórkowego) po ostatnim zabiegu poddania działaniu substancji. Dodatkowe okresy pobierania próbek mogą zostać wykorzystane w przypadku, gdy jest to stosowne.
Przed uśmierceniem zwierzętom wstrzykuje się wewnątrzotrzewnowo odpowiednią dawkę substancji zatrzymującej metafazę (np. Colcemid® lub kolchicynę). Następnie pobiera się próbkę ze zwierząt w odpowiednich odstępach czasu. W odniesieniu do myszy ten odstęp czasu wynosi 3–5 godzin, w odniesieniu do chomików chińskich ten odstęp czasu wynosi w przybliżeniu 4–5 godzin.
1.5.3. Poziomy dawek
Jeżeli przeprowadzane jest badanie ustalające dawkę, ponieważ nie istnieją dostępne odpowiednie dane, powinno zostać ono przeprowadzone w tym samym laboratorium, wykorzystując ten sam reżim gatunku, szczepu oraz poddania działaniu substancji, który ma zostać wykorzystany w badaniu głównym (7). Jeżeli istnieje toksyczność, wykorzystywane są trzy poziomy dawek w odniesieniu do pierwszego okresu pobierania próbek. Te poziomy dawek powinny obejmować zakres od maksymalnej do małej toksyczności lub do jej braku. W czasie późniejszego okresu pobierania próbek muszą zostać wykorzystane jedynie najwyższe dawki. Najwyższa dawka określana jest jako dawka wywołująca oznaki toksyczności, tak że wyższe dawki oparte na tym samym reżimie dawkowania mogłyby wywołać śmiertelność.
Substancje o szczególnej aktywności biologicznej przy niskotoksycznych dawkach (takie jak hormony oraz mitogeny) mogą stanowić wyjątki w odniesieniu do kryteriów ustalania dawek oraz powinny być oceniane na zasadzie jednostkowych przypadków. Najwyższa dawka może być również określona jako dawka wytwarzająca niektóre wskazania toksyczności w spermatogoniach (np. zmniejszenie wskaźnika mitotycznego podziału spermatogoniów do pierwszej oraz drugiej fazy mejotycznej; redukcja ta nie powinna przekroczyć 50 %).
1.5.4. Badanie ograniczone
Jeżeli badanie w oparciu o jeden poziom dawek w odniesieniu do zwierząt o przynajmniej 2 000 mg/kg masy ciała, wykorzystujące pojedyncze poddanie działaniu substancji lub dwa zabiegi poddania działaniu substancji tego samego dnia, nie daje dostrzegalnych skutków toksycznych oraz jeżeli nie podejrzewa się genotoksyczności w oparciu o dane dotyczące substancji powiązanych strukturalnie, wówczas przeprowadzenie pełnego badania może nie być konieczne. Spodziewane poddanie człowieka działaniu substancji może wykazać potrzebę wykorzystania większej dawki w badaniu ograniczonym.
1.5.5. Dawkowanie
Badana substancja jest zazwyczaj podawana przez zgłębnik żołądkowy lub odpowiednią rurkę intubacyjną, lub przez wstrzyknięcie wewnątrzotrzewnowe. Mogą być dopuszczalne inne drogi poddania działaniu substancji, w przypadku gdy mogą być uzasadnione. Maksymalna objętość płynu, jaka może być podana przez zgłębnik lub wstrzyknięcie jednorazowo zależy od wielkości badanego zwierzęcia. Objętość nie powinna przekraczać 2 ml/100 kg masy ciała. Wykorzystanie pojemności wyższych niż te musi być uzasadnione. Z wyjątkiem substancji podrażniających lub żrących, które będą w normalnych warunkach ujawniać pogorszone skutki z większymi stężeniami, zmienność w badanej objętości powinna zostać zminimalizowana przez dostosowanie stężenia w celu zapewnienia stałej objętości przy wszystkich poziomach dawek.
1.5.6. Przygotowanie chromosomów
Bezpośrednio po uśmierceniu zawiesiny komórkowe uzyskiwane są z jednego lub obu badań, poddanego działaniu roztworu hipotonicznego oraz stabilizowane. Następnie komórki są rozprowadzane na szkiełkach mikroskopowych oraz barwione.
1.5.7. Analiza
W odniesieniu do każdego zwierzęcia powinno zostać poddane analizie przynajmniej 100 dobrze rozprowadzonych metafaz (np. minimum 500 metafaz na grupę). Liczba ta może zostać zmniejszona, jeżeli zaobserwowane są duże liczby aberracji. Wszystkie szkiełka mikroskopowe, włącznie z tymi z pozytywnych i negatywnych kontroli, powinny być niezależnie kodowane przed przeprowadzeniem analizy mikroskopowej. Ze względu na to, że skutkiem procedur stabilizacji często jest zerwanie proporcji udziału metafaz ze stratą chromosomów, komórki oceniane powinny zawierać liczbę centromerów równą 2n ± 2.
2. DANE
2.1. PRZETWARZANIE WYNIKÓW
Dane dotyczące poszczególnych zwierząt powinny zostać przedstawione w formie tabelarycznej. Jednostką doświadczalną jest zwierzę. W odniesieniu do każdego zwierzęcia liczba komórek z aberracjami chromosomowymi oraz liczba aberracji przypadających na komórkę powinny zostać ocenione. Różne typy strukturalnej aberracji chromosomowej powinny być wymienione wraz z ich ilościami oraz częstotliwościami w odniesieniu do grupy poddanej działaniu substancji oraz grupy kontrolnej. Szczeliny są oddzielnie odnotowywane oraz zgłaszane, ale ogólnie nie są włączane do częstotliwości aberracji ogółem.
Jeżeli obserwuje się zarówno mitozę, jak i mejozę, powinien zostać oznaczony wskaźnik podziału mitotycznego spermatogoniów w odniesieniu do pierwszej i drugiej metafazy majotycznej, jako miara cytotoksyczności dla wszystkich poddanych działaniu substancji oraz zwierząt poddanych kontroli negatywnej w całkowitej próbce 100 dzielących się komórek na zwierzę, w celu ustalenia możliwego skutku cytotoksycznego. Jeżeli zaobserwowana jest jedynie mitoza, wskaźnik mitozy powinien zostać oznaczony w przynajmniej 1 000 komórek przypadających na zwierzę.
2.2. OCENA ORAZ INTERPRETACJA WYNIKÓW
Istnieją liczne kryteria oznaczania wyników pozytywnych, takie jak związany z dawką wzrost względnej ilości komórek z aberracjami chromosomowymi lub wyraźny wzrost w ilości komórek z aberracjami w pojedynczej dawce przy jednym pobieraniu próbek. Biologiczne znaczenie wyników powinno zostać wzięte pod uwagę w pierwszej kolejności. Metody statystyczne mogą zostać wykorzystane jako metody pomocnicze w przeprowadzaniu oceny wyników (8). Jednakże znaczenie statystyczne nie powinno być jedynym czynnikiem określającym w odniesieniu do reakcji pozytywnej. Wyniki niejednoznaczne powinny zostać wyjaśnione przez dalsze badanie, w szczególności wykorzystując modyfikację warunków doświadczalnych.
Substancja badana, w odniesieniu do której wyniki nie spełniają powyższych kryteriów, uważana jest za niemutagenną w tym badaniu.
Chociaż większość doświadczeń będzie dawała w jasny sposób pozytywne lub negatywne wyniki, w rzadkich przypadkach zestaw danych będzie wykluczał dokonanie ostatecznej oceny dotyczącej aktywności badanej substancji. Wyniki mogą pozostać niejednoznaczne lub sporne niezależnie od liczby powtórzonych doświadczeń.
Wyniki pozytywne z badania aberracji chromosomowych spermatogoniów in vivo wskazują, że substancja badana wywołuje aberracje chromosomowe w komórkach zarodków badanych gatunków. Wyniki negatywne wskazują, że w warunkach badawczych substancja badana nie jest mutagenna w badanym gatunku.
Prawdopodobieństwo, że substancja badana lub jej metabolity docierają do tkanki docelowej, powinno zostać omówione.
3. SPRAWOZDAWCZOŚĆ
SPRAWOZDANIE Z BADANIA
Sprawozdanie z badania musi zawierać następujące informacje:
|
|
Rozpuszczalnik/nośnik:
|
|
|
Badane zwierzęta:
|
|
|
Warunki badania:
|
|
|
Wyniki:
|
|
|
Omówienie wyników. |
|
|
Wnioski. |
4. ODNIESIENIA
|
(1) |
Adler, I. D., (1986), Clastogenic Potential in Mouse Spermatogonia of Chemical Mutagens Related to their Cell-Cycle Specifications, w: Genetic Toxicology of Environmental Chemicals, Part B: Genetic Effects and Applied Mutagenesis, Ramel, C, Lambert, B. and Magnusson, J. (eds) Liss, New York, pp. 477–484. |
|
(2) |
Adler, I. D., (1984), Cytogenetic tests in Mammals, w: Mutagenicity Testing: a Practical Approach, (ed.) S. Venitt and J. M. Parry, IRL Press, Oxford, Washington DC, pp. 275–306. |
|
(3) |
Evans, E. P., Breckon, G. and Ford, C. E. (1964), An Air-drying Method for Meiotic Preparations from Mammalian Testes, Cytogenetics and Cell Genetics, 3, pp. 289–294. |
|
(1) (4) |
Richold, M., Ashby, J., Chandley, A., Gatehouse, D. G. and Henderson, L. (1990), In vivo Cytogenetic Assays, in: D. J. Kirkland (ed.), Basic Mutagenicity Tests, UKEMS Recommended Procedures. UKEMS Subcommittee on Guidelines for Mutagenicity Testing. Report. Part I revised, Cambridge University Press, Cambridge, New York, Port Chester, Melbourne, Sydney, pp. 115–141. |
|
(5) |
Yamamoto, K. and Kikuchi, Y. (1978), A New Method for Preparation of Mammalian Spermatogonial Chromosomes, Mutation Res., 52, pp. 207–209. |
|
(6) |
Adler, I. D., Shelby M. D., Bootman, J., Favor, J., Generoso, W., Pacchierotti, F., Shibuya, T. and Tanaka N. (1994), International Workshop on Standardisation of Genotoxicity Test Procedures. Summary Report of the Working Group on Mammalian Germ Cell Tests, Mutation Res., 312, pp. 313–318. |
|
(7) |
Fielder, R. J., Allen, J. A., Boobis, A. R., Botham, P. A., Doe, J., Esdaile, D. J., Gatehouse, D. G., Hodson-Walker, G., Morton, D. B., Kirkland, D. J. and Richold, M. (1992), Report of British Toxicology Society/UK Environmental Mutagen Society Working Group: Dose setting in In vivo Mutagenicity Assays, Mutagenesis, 7, pp. 313–319. |
|
(8) |
Lovell, D. P., Anderson, D., Albanese, R., Amphlett, G. E., Clarc, G., Ferguson, R., Richold, M., Pap-worth, D. G. and Savage J. R. K. (1989), Statistical Analysis of In vivo Cytogenetic Assays, in: D. J. Kirkland (ed.), Statistical Evaluation of Mutagenicity Test Data. UKEMS Subcommittee on Guidelines for Mutagenicity Testing, report, Part III. Cambridge University Press, Cambridge, New York, Port Chester, Melbourne, Sydney, pp. 184–232. |
B.24. TEST PLAMKOWY U MYSZY
1. METODA
1.1. WSTĘP
Zob. Wprowadzenie ogólne część E.
1.2. DEFINICJE
Zob. Wprowadzenie ogólne część E.
1.3. SUBSTANCJE ODNIESIENIA
Brak.
1.4. ZASADA METODY BADAWCZEJ
Niniejszy test jest testem in vivo u myszy, u których rozwijające się zarodki eksponowane są na działanie substancji chemicznych. Komórkami docelowymi u rozwijających się zarodków są melanoblasty, a docelowe geny są genami, które kontrolują pigmentację sierści. Rozwijające się zarodki są heterozygotyczne dla wielu takich genów odpowiedzialnych za kolor sierści. Mutacja wewnątrz lub strata (przez różnorodne procesy) allelu dominującego takiego genu w melanoblaście prowadzi do ekspresji fenotypu recesywnego w komórkach potomnych, ujawniając się w postaci plamki o zmienionym kolorze na futrze myszy. Liczona jest liczba potomstwa z takimi plamkami, mutacjami, a ich częstotliwość porównywana jest z potomstwem wywodzącym się od zarodków poddanych jedynie działaniu rozpuszczalnika. Test plamkowy u myszy wykrywa domniemane somatyczne mutacje w komórkach płodów.
1.5. KRYTERIA JAKOŚCIOWE
Brak.
1.6. OPIS METODY BADAWCZEJ
Przygotowania
Jeżeli to możliwe, substancje testowe rozpuszczane są lub zawieszane w izotonicznym roztworze soli. Substancje chemiczne nierozpuszczalne w wodzie są rozpuszczane lub zawieszane we właściwych nośnikach. Zastosowany nośnik nie powinien ani kolidować z testową substancją chemiczną, ani powodować jakichkolwiek objawów toksyczności. Należy zastosować świeże preparaty testowej substancji chemicznej.
Myszy szczepu T (nie-agouti, a/a; szynszyla, różowe oczy, cchp/cchp; brązowe, b/b; jaśniejsze, o krótkich uszach, d se/d se; łaciate, s/s) są krzyżowane zarówno ze szczepem HT (jasne, nie-agouti, brachypodia, pa a bp/pa a bp; grafitowe kędzierzawe ln fz/ln fz; perłowe pe/pe), jak i ze szczepem C57 BL (nie-agouti, a/a). Inne właściwie krzyżówki, takie jak między NMR1 (nie-agouti, a/a; albinos, c/c) a DBA (nie-agouti, a/a; brązowe, b/b; jaśniejsze d/d) mogą być stosowane pod warunkiem, że ich potomstwo będzie typu nie-agouti.
Odpowiednia liczna ciężarnych samic poddana jest badaniu w celu uzyskania właściwej liczby żywego potomstwa w odniesieniu do każdego poziomu użytej dawki. Właściwy rozmiar próbki jest regulowany liczbą plamek zaobserwowanych u badanych myszy oraz skalą danych kontrolnych. Negatywny wynik akceptowany jest jedynie wtedy, gdy w wynikach uwzględniono co najmniej 300 osobników potomstwa pochodzących od samic poddanych działaniu substancji testowej o najwyższej wielkości dawki.
Powinny zostać udostępnione dane z równoległej grupy kontrolnej w odniesieniu do myszy poddanych jedynie działaniu nośnika (negatywne grupy kontrolne). Dane bazowe grup kontrolnych, pochodzące z tego samego laboratorium, mogą zostać włączone w celu podniesienia czułości testu, pod warunkiem iż są one jednorodne. Należy udostępnić dane z pozytywnej grupy kontrolnej ostatnio uzyskane w tym samym laboratorium w związku z poddaniem działaniu substancji chemicznej, o której wiadomo, iż wywołuje objawy mutageniczności, w przypadku braku wykrycia mutageniczności związanej z działaniem testowej substancji chemicznej.
Zwyczajowe drogi podawania substancji to intubacja drogą pokarmową lub przez iniekcję dootrzewnową stosowane u ciężarnych samic. W odpowiednich przypadkach stosuje się podawanie substancji przez drogi inhalacyjne lub inne drogi podawania substancji testowej.
Stosuje się co najmniej dwa poziomy dawek, włączając jeden skutkujący objawami toksyczności lub obniżoną wielkością miotu. W przypadku nietoksycznych substancji chemicznych należy zastosować ekspozycję na maksymalny stosowany poziom dawki.
Procedura
Poddanie działaniu dawki jednorazowej zazwyczaj stosowane jest w 8, 9 lub 10 dniu ciąży, licząc dzień 1 jako dzień, w którym po raz pierwszy zaobserwowano czop nasienia w pochwie. Dni te odpowiadają 7,25, 8,25 i 9,25 dniom po poczęciu. Po tym okresie można zastosować kolejne podanie dawki.
Potomstwo jest oznaczane oraz liczone pod względem plamek między trzecim i czwartym tygodniem po urodzeniu. Wyróżnia się trzy klasy plamek:
|
a) |
białe plamki do 5 mm linii środkowo-brzusznej, które przypuszczalnie powodowane są uśmiercaniem komórek (WMVS); |
|
b) |
żółte, podobne do agouti, cętka występujące na sutku, genitaliach, gardle, w okolicach pach i pachwin oraz w okolicach środka czoła, które przypuszczalnie są wynikiem braku różnicowania (MDS); oraz |
|
c) |
zabarwione oraz białe plamki losowo rozmieszczone na sierści, które przypuszczalnie wywodzą się z mutacji somatycznych (RS). |
Wszystkie trzy klasy są odnotowane jako wyniki, ale jedynie ostatnia, RS, posiada przydatność genetyczną. Problemy z rozróżnieniem między MDS a RS można rozwiązać przez fluorescencyjną mikroskopię próbek sierści.
Należy odnotować oczywiste rażące nieprawidłowości morfologiczne występujące u potomstwa.
2. DANE
Dane przedstawiane są jako ogólna liczba liczonego potomstwa oraz liczba osobników, u których zaobserwowano jedną lub więcej plamek mutacji somatycznej. Dane z grupy badanej oraz negatywnej są porównywane za pomocą odpowiednich metod. Dane przedstawiane są również na tle poszczególnych miotów.
3. SPRAWOZDAWCZOŚĆ
3.1. SPRAWOZDANIE Z BADAŃ
Sprawozdanie z badań powinno, jeżeli możliwe, zawierać następujące informacje:
|
— |
szczepy zastosowane w krzyżówkach, |
|
— |
liczba ciężarnych samic w grupie badanej i grupach kontrolnych, |
|
— |
średnia wielkość miotu w grupie badanej i grupach kontrolnych w chwili urodzenia oraz po zakończeniu ssania, |
|
— |
wielkość dawki (dawek) testowej substancji chemicznej, |
|
— |
zastosowany rozpuszczalnik, |
|
— |
dzień ciąży, w którym podano substancję testową, |
|
— |
droga podawania substancji testowej, |
|
— |
ogólna liczba badanego potomstwa oraz liczba potomstwa z WMVS, MDS i RS w grupie badanej i grupach kontrolnych, |
|
— |
rażące nieprawidłowości morfologiczne, |
|
— |
związek między poziomem dawki a reakcją osobników z RS, jeżeli to możliwe, |
|
— |
analiza statystyczna, |
|
— |
analiza wyników, |
|
— |
interpretacja wyników. |
3.2. ANALIZA I INTERPRETACJA
Zob. Wprowadzenie ogólne część B.
4. ODNIESIENIA
Zob. Wprowadzenie ogólne część B.
B.25. TRANSLOKACJA DZIEDZICZNOŚCI U MYSZY
1. METODA
1.1. WSTĘP
Zob. Wprowadzenie ogólne część B.
1.2. DEFINICJE
Zob. Wprowadzenie ogólne część B.
1.3. SUBSTANCJE ODNIESIENIA
Brak.
1.4. ZASADA METODY BADAWCZEJ
Badanie translokacji dziedziczności u myszy wykrywa strukturalne i ilościowe zmiany chromosomowe w komórkach zarodkowych ssaków, zregenerowane w pierwszym pokoleniu potomstwa. Typy wykrytych zmian chromosomowych to obustronne translokacje oraz, jeżeli włącza się potomstwo płci żeńskiej, utrata chromosomu X. Nośniki translokacji i samice o genotypie XO wykazują zmniejszoną płodność, która jest używana do wyboru potomstwa F1 do celów analizy cytogenetycznej. Całkowita bezpłodność jest spowodowana przez pewne typy translokacji (chromosom X-autosom i typu c-t). Translokacje są obserwowane w komórkach mejotycznych w diakinezie metafazy I u osobników męskich, również F1, samców lub męskiego potomstwa samic F1. Samice o genotypie XO są identyfikowane cytogenetycznie dzięki obecności tylko 39 chromosomów w komórkach szpiku kostnego przechodzących mitozę.
1.5. KRYTERIA JAKOŚCIOWE
Brak.
1.6. OPIS METODY BADAWCZEJ
Przygotowania
Substancje testowe rozpuszczane są w izotonicznym roztworze soli. Substancje chemiczne nierozpuszczalne w wodzie są rozpuszczane lub zawieszane we właściwych nośnikach. Należy zastosować świeże roztwory testowego związku chemicznego. Zastosowany nośnik nie powinien kolidować z testową substancją chemiczną ani powodować jakichkolwiek objawów toksyczności.
Zwyczajowe drogi podawania substancji to intubacja drogą pokarmową lub iniekcja dootrzewnowa. Inne drogi podawania substancji testowej mogą być również właściwe.
W celu ułatwienia hodowli i cytologicznej weryfikacji niniejsze badania przeprowadzane są na myszach. Nie jest wymagany szczególny szczep myszy. Jednakże średni rozmiar miotu powinien być większy niż osiem osobników i powinien być stosunkowo stały.
Należy wykorzystać zdrowe, dojrzałe płciowo zwierzęta.
Wymagana liczba zwierząt zależy od częstotliwości samorzutnej translokacji oraz minimalnego stopnia wywoływania objawów, w celu uzyskania wyniku pozytywnego.
Badanie zazwyczaj przeprowadzane jest przez analizę potomstwa samców F1. Należy poddać badaniu co najmniej 500 samców potomstwa F1 w danej grupie dawkowania. W przypadku włączenia samic potomstwa F1 wymagane jest 300 samców i 300 samic.
Powinny zostać udostępnione odpowiednie dane uzyskane z równoległych i bazowych grup kontrolnych. W przypadku dostępności wyników pozytywnej grupy kontrolnej, uzyskanych w z badań ostatnio przeprowadzanych w tym samym laboratorium, wyniki te można zastosować zamiast równoległej pozytywnej grupy kontrolnej.
Badany jest jeden poziom dawki, zazwyczaj najwyższy poziom dawki związany jest z produkcją minimalnych objawów toksyczności, ale nie ma wpływu na zachowania związane z rozrodczością lub przeżyciem osobników. W celu ustalenia zależności między poziomem dawki a reakcją wymagane jest zastosowanie dwóch dodatkowych poziomów dawek. W przypadku nietoksycznych substancji chemicznych należy zastosować ekspozycję na najmniejszy stosowany poziom dawki.
Procedura
Wymagane są dwa harmonogramy podawania substancji. Najszerzej stosowane jest jednorazowe podanie substancji testowej. Stosować można również podawanie substancji testowej siedem dni w tygodniu przez okres 35 dni. Liczba kryć po zakończeniu podawania substancji regulowana jest harmonogramem badań w celu zapewnienia uzyskania próbek z wszystkich faz komórki zarodkowej. Po zakończeniu okresu krycia samice należy umieścić w pojedynczych klatkach. W przypadku porodu samic należy odnotować datę, wielkość miotu oraz płeć potomstwa. Całe potomstwo zawierające osobniki samców jest odzwyczajane od ssania, a potomstwo zawierające samice jest odrzucane, chyba że jest włączone do badania.
Stosowana jest jedna z dwóch możliwych metod:
|
— |
badanie płodności potomstwa F1 i późniejsza weryfikacja ewentualnych nośników translokacji przez przeprowadzenie analiz cytogenicznych, |
|
— |
analizy cytogenetyczne całego męskiego potomstwa F1 bez wcześniejszej selekcji przez badanie płodności. |
|
a) |
Badanie płodności Zmniejszona płodność osobnika F1 może być stwierdzona na podstawie obserwacji rozmiarów potomstwa i/lub analizy zawartości macic samic. Należy określić kryteria dla ustalania normalnej i obniżonej płodności wykorzystanego szczepu myszy. Obserwacje wielkości miotu: samce F1 przeznaczone do badania umieszczane są w klatkach pojedynczo z samicami zarówno z tego samego badania, jak i z koloni. Klatki kontroluje się codziennie, zaczynając od 18 dnia po kryciu. Wielkość miotu oraz płeć potomstwa F2 odnotowane są po urodzeniu, a następnie mioty są odrzucane. W przypadku badania potomstwa żeńskiego F1 potomstwo F2 małych miotów jest zachowywane w celu przeprowadzenia dalszych badań. Żeńskie nośniki translokacji są weryfikowane za pomocą analiz translokacji u ich męskiego potomstwa. Samice XO są rozpoznawane przez zmianę wskaźnika płci u ich potomstwa wynoszącego od 1:1 do 1:2 osobników samczych do samic. W procedurze sekwencyjnej normalne zwierzęta F1 są eliminowane z dalszych badań, jeżeli pierwszy miot F2 osiągnął lub przekroczył wcześniej ustaloną wartość; w przeciwnym razie obserwacjom poddawany jest drugi lub trzeci miot F2. Zwierzęta F1, których nie można zaklasyfikować jako normalne po zakończeniu obserwacji trzech miotów F2, zostają poddane dalszemu badaniu przez analizę zawartości macicznych albo bezpośrednio poddawane analizom cytogenetycznym. Analiza zawartości macicznej: Redukcja rozmiaru miotu z nośnikami translokacji spowodowana jest śmiercią zarodkową, tak że wysoka liczba martwych implantów jest przejawem obecności translokacji u badanych zwierząt. Samce F1, mające być poddane badaniu, wykorzystywane są do krycia dwóch lub trzech samic każdy. Poczęcie ustalane jest przez codzienną poranną kontrolę czopu nasienia w pochwie. Samice uśmiercane są 14–16 dni później a żywe lub martwe implanty w ich macicach są odnotowywane. |
|
b) |
Analiza cytogeniczna Preparaty z jąder przygotowywane są techniką suszenia powietrzem. Nośniki translokacji rozpoznawane są przez obecność wielowartościowych konfiguracji w diakinezie metafazy I w podstawowych spermatocytach. Obserwacja co najmniej dwóch komórek z wielowartościowymi związkami stanowi wymagane dowody na potwierdzenie faktu, iż zwierzę posiada nośnik translokacji. Wszystkie samce F1 należy zbadać cytogenetycznie, jeżeli nie została przeprowadzona żadna selekcja podczas rozmnażania. Należy mikroskopowo oznaczyć minimum 25 komórek diakinezy metafazy I danego samca. Analiza metafaz mitotycznych w spermatogoniach bądź szpiku kostnym jest wymagana u samców F1 z małymi jądrami i defektem mejozy przed diakinezą lub z samic F1 z podejrzeniem genotypu XO. Obecność niezwykle długiego i/lub krótkiego chromosomu w każdej z 10 komórek jest dowodem na szczególną męską translokację prowadzącą do niepłodności (typ c-t). Niektóre translokacje chromosom X-autosom, które powodują męską niepłodność mogą być zidentyfikowane tylko na podstawie analizy pasmowej chromosomów mitotycznych. Obecność 39 chromosomów we wszystkich 10 mitozach jest dowodem na istnienie XO u samicy. |
2. DANE
Dane należy przedstawić w formie tabeli.
W każdym okresie krycia należy odnotować średni rozmiar miotu oraz stosunek płci w potomstwie pochodzącym z rodzicielskiej pary przy urodzeniu i w okresie odzwyczajania od ssania.
Dla oszacowania płodności zwierząt F1 przedstawiane są średnie wielkości potomstwa pochodzącego z normalnego krycia oraz ilość żyjących i nieżywych implantów z każdego krycia osobników F1, gdzie odnotowano występowanie nośników translokacji. W celu dokonania analizy zawartości macicznej należy odnotować średnią liczbę żywych i martwych implantów potomstwa pochodzącego z normalnego krycia oraz poszczególne liczby żyjących i martwych implantów dla każdego potomstwa gdzie odnotowano występowanie nośników translokacji.
Aby dokonać analizy cytogenetycznej diakinezy metafazy I, ilość typów wielowartościowych konfiguracji oraz całkowita liczba parzeń i czasu trwania krycia są odnotowywane.
W odniesieniu do bezpłodnych osobników F1 należy odnotować ogólną liczbę kryć oraz czas trwania okresu krycia. Należy podać wagę jąder oraz szczegóły wyników analizy cytogenetycznej.
W odniesieniu do samic XO odnotowana jest średnia wielkość miotu, stosunek płci potomstwa F2 oraz wyniki analizy cytogenetycznej.
Jeżeli możliwe, potomstwo F1 z nośnikami translokacji poddawane jest wcześniejszej selekcji przez badania płodności, tabele muszą zawierać informację, ile z nich zostało potwierdzonych jako translokacje heterozygotyczne.
Należy odnotować dane z badań negatywnych i pozytywnych grup kontrolnych.
3. SPRAWOZDAWCZOŚĆ
3.1. SPRAWOZDANIE Z BADAŃ
Sprawozdanie z badań powinno, jeżeli możliwe, zawierać następujące informacje:
|
— |
szczep myszy, wiek zwierząt, waga badanych zwierząt, |
|
— |
liczba rodzicielskich zwierząt każdej płci w grupie badanej i grupach kontrolnych, |
|
— |
warunki badania, szczegółowy opis badania, wielkości dawkowania, rozpuszczalniki, harmonogram krycia, |
|
— |
liczba i płeć potomstwa danej samicy, liczba i płeć potomstwa hodowanego do analizy translokacji, |
|
— |
czas i kryteria analizy translokacyjnej, |
|
— |
liczba oraz szczegółowy opis nośników translokacji, włączając dane dotyczące rozmnażania i dane dotyczące zawartości macicznej, w odpowiednich przypadkach, |
|
— |
procedury cytogenetyczne i szczegóły analizy mikroskopowej, najlepiej wraz ze zdjęciami, |
|
— |
statystyczna analiza, |
|
— |
analiza wyników, |
|
— |
interpretacja wyników. |
3.2. ANALIZA I INTERPRETACJA
Zob. Wprowadzenie ogólne część B.
4. ODNIESIENIA
Zob. Wprowadzenie ogólne część B.
B.26. BADANIE TOKSYCZNOŚCI PODPRZEWLEKŁEJ DROGĄ POKARMOWĄ – STUDIUM TOKSYCZNOŚCI NA GRYZONIACH METODĄ POWTARZANEJ 90-DNIOWEJ DAWKI DROGĄ POKARMOWĄ
1. METODA
Niniejsza metoda badania toksyczności podprzewlekłej drogą pokarmową jest kopią OECD TG 408 (1998).
1.1. WPROWADZENIE
Przy oszacowaniu i ocenie cech toksyczności substancji chemicznej oznaczenie podprzewlekłej toksyczności drogą pokarmową, stosując powtarzane dawki, jest prowadzone po początkowej informacji o toksyczności, uzyskanej z 28-dniowych badań toksyczności ostrej lub powtarzanej. 90-dniowe badanie dostarcza informacji o możliwym niebezpieczeństwie dla zdrowia, które prawdopodobnie powstanie z powtarzanego narażenia w ciągu przedłużonego okresu czasu, obejmującego moment odstawienia od piersi, poprzez rozwój do dojrzałości. Badanie dostarczy informacji na temat głównych toksycznych działań, wskaże zaatakowane organy i możliwości akumulacji, dostarczy oceny poziomu ekspozycji na badania niepowodującego negatywnych skutków, którą stosuje się przy wyborze poziomów dawki dla badań przewlekłych i ustalania kryteriów bezpieczeństwa dla narażenia ludzi.
Metoda kładzie dodatkowy nacisk na aspekt neurologiczny i wskazuje oddziaływania na układ immunologiczny i rozrodczy. Potrzeba starannej klinicznej obserwacji zwierząt, aby uzyskać tak wiele informacji, jak to możliwe, jest również stresująca. Niniejsze badanie powinno umożliwić określenie substancji chemicznych, które mogą potencjalnie powodować działania neurotoksyczne, immunologiczne lub wpływać na organy rozrodcze oraz uzasadniające dalsze dogłębne ich zbadanie.
Zob. także Wprowadzenie ogólne część B.
1.2. DEFINICJE
Dawka: jest ilością podawanej substancji badanej. Dawka jest wyrażana wagowo (g, mg) lub jako waga substancji badanej na jednostkę wagi badanego zwierzęcia (np. mg/kg), lub jako stężenie stałe żywieniowe (ppm).
Dawkowanie: jest ogólnym pojęciem obejmującym dawkę, jej częstotliwość i czas podawania dawki.
NOAEL: jest skrótem od poziomu, przy którym nie obserwuje się niekorzystnych skutków, oraz jest najwyższym poziomem dawki, gdzie nie obserwuje się niekorzystnych efektów zależnych od podawanej substancji.
1.3. ZASADA METODY BADANIA
Badana substancja jest codziennie podawana doustnie w stopniowanych dawkach kilku grupom zwierząt doświadczalnych, jeden poziom dawki na grupę przez okres 90 dni. W czasie okresu podawania zwierzęta są ściśle obserwowane w zakresie oznak zatrucia. Zwierzęta, które zmarły lub zostały zabite w czasie trwania badania, są poddawane sekcji zwłok, po zakończeniu badania zwierzęta, które przeżyły, są również zabijane i poddawane sekcji zwłok.
1.4. OPIS METODY BADANIA
1.4.1. Przygotowanie zwierząt
Muszą zostać użyte zdrowe zwierzęta, zaaklimatyzowane w warunkach laboratoryjnych przez przynajmniej pięć dni i niebędące przedmiotem wcześniejszych procedur eksperymentalnych. Zwierzęta badane muszą charakteryzować się gatunkiem, szczepem, źródłem, płcią, wagą i/lub wiekiem. Zwierzęta muszą być losowo wyznaczane do doświadczeń kontrolnych i grup traktowanych. Klatki muszą być rozmieszczone w taki sposób, aby możliwy wpływ wynikający z ich przemieszczenia był minimalny. Każde zwierzę musi być oznakowane niepowtarzalnym numerem identyfikacyjnym.
1.4.2. Przygotowanie dawek
Substancja badana jest podawana przez zgłębnik lub w postaci pokarmu lub wody pitnej. Metoda podawania drogą pokarmową jest zależna od celu badania oraz właściwości fizycznych/chemicznych badanego materiału.
Tam gdzie jest to konieczne, badaną substancje rozpuszcza się lub wykonuje zawiesinę w odpowiednim nośniku. Zalecane jest, aby tam gdzie to jest możliwe, rozważyć wpierw użycie roztworu lub zawiesiny wodnej, a następnie rozważyć wykonanie roztworu/zawiesiny w oleju (np. w oleju kukurydzianym) i dopiero następnie w innych nośnikach. Dla nośników różnych od wody muszą być znane cechy toksyczności. Powinna być określona stabilność badanej substancji w warunkach podawania dawek.
1.4.3. Warunki badania
1.4.3.1. Zwierzęta eksperymentalne
Zalecanym gatunkiem jest szczur, ale także inne gatunki gryzoni, np. mysz. Powinny być wykorzystane zwykłe stosowane laboratoryjne szczepy zdrowych dorosłych zwierząt. Samice muszą być zeroroczne, nieciężarne. Dozowanie musi rozpocząć się tak szybko, jak to możliwe po odłączeniu od matki, a w każdym przypadku przed osiągnięciem wieku dziewięciu tygodni. Przy rozpoczęciu badań, zmiany wagi wykorzystanych zwierząt muszą być minimalne i nie przekraczać ± 20 % średniej wagi w każdej płci. W przypadku gdy badania są prowadzone jako wstępne do badań toksyczności przewlekłej długoterminowej, do obu badań należy wykorzystać zwierzęta tego samego szczepu i źródła.
1.4.3.2. Ilość i płeć
Co najmniej 20 zwierząt (10 samic i 10 samców) należy wykorzystać w odniesieniu do każdego poziomu dawki. Jeśli planuje się zabijanie pośrednie, liczba musi być zwiększona o przewidzianą ilość zwierząt do zabicia przed zakończeniem badań. W oparciu o wcześniejszą wiedzę o substancjach chemicznych i bliskich odpowiednikach należy rozważyć włączenie dodatkowej satelickiej grupy 10 zwierząt (po 5 każdej płci) do grupy kontrolnej i grupy najwyżej dawkowanej, do celów obserwacji, po okresie traktowania, odwracalności lub trwałości jakichkolwiek skutków toksycznych. Czas trwania tego okresu po zakończeniu traktowania powinien być ustalony odpowiednio w odniesieniu do obserwowanych skutków.
1.4.3.3. Poziomy dawek
Stosuje się co najmniej trzy poziomy dawek i równoczesne doświadczenie kontrolne, poza przypadkiem, gdy prowadzone jest badanie graniczne (zob. 1.4.3.4). Poziomy dawek mogą być oparte na wynikach powtarzanej dawki lub badaniach ustalania zakres oraz powinny uwzględniać wszelkie istniejące dane toksykologiczne i toksykokinetyczne, dostępne w odniesieniu do badanej substancji lub związanych z nią materiałów. Poza ograniczeniami natury fizykochemicznej i działaniami biologicznymi substancji badanej, poziom najwyższej dawki musi być wybrany w celu wzbudzenia objawów toksycznych, ale nie spowodowania śmierci czy też poważnych cierpień. Opadająca sekwencja poziomu dawki musi być wybrana w celu pokazania reakcji związanej z dawkowaniem i poziomem, przy którym nie obserwuje się negatywnych skutków (NOAEL) przy najniższym poziomie dawki. Dwa do czterech zachodzące przedziały są często optymalne do ustalenia opadających poziomów dawki i dodanie czwartej grupy badanej jest często polecane dla zastosowania bardzo dużych odstępów (np. więcej niż współczynnik 6–10) między dawkowaniami.
Grupa kontrolna musi być grupą nietraktowaną lub grupą kontrolną nośnika, jeśli nośnik jest używany do podawania substancji badanej. Zwierzęta w grupie kontrolnej, poza nietraktowaniem jej substancją badaną, są obsługiwane w identyczny sposób jak w zwierzęta w grupach badanych. Jeśli stosowany jest nośnik, grupa kontrolna musi otrzymywać nośnik w maksymalnie stosowanej objętości. Gdy substancja badana jest podawana w diecie i powoduje zmniejszenie zapotrzebowania żywieniowego, wtedy grupa kontrolna z pary żywieniowej może być pomocna w rozróżnieniu między zmniejszeniem spowodowanym smacznością lub zmianami toksykologicznymi w modelu badania.
Następujące cechy nośnika i innych dodatków muszą być rozważone jako właściwe: wpływ wchłaniania, rozkładu, metabolizmu, retencji badanej substancji, wpływ właściwości chemicznych substancji badanej, mogące zmienić jej cechy toksyczności oraz wpływy zapotrzebowania na żywność i wodę lub żywieniowy status zwierząt.
1.4.3.4. Badanie graniczne
Jeśli badanie przy jednym poziomie dawki, równoważnym co najmniej 1 000 mg/kg masy ciała/dzień, stosując procedurę opisaną w odniesieniu do niniejszego badania, powoduje, że nie obserwuje się negatywnych skutków oraz jeśli nie oczekuje się toksyczności na podstawie danych substancji strukturalnie związanych, wtedy pełne badania z zastosowaniem trzech poziomów nie wydają się konieczne. Badanie graniczne ma zastosowanie, z wyjątkiem sytuacji, gdy narażenie ludzi wskazuje potrzebę zastosowania wyższego poziomu dawki.
1.5. PROCEDURA
1.5.1. Podawanie dawek
Zwierzętom dozuje się substancję badaną przez siedem dni każdego tygodnia w okresie 90 dni. Każdy inny sposób dozowania, na przykład pięć dni w tygodniu, wymaga uzasadnienia. Gdy substancja badana jest podawana przez zgłębnik, musi być to wykonane w pojedynczej dawce, przy użyciu sondy przełykowej lub odpowiedniej kaniuli intubacyjnej. Maksymalna objętość cieczy, która może być podana w jednym momencie, zależy od rozmiarów zwierzęcia badanego. Objętość nie może przekraczać 1 ml/100 g masy ciała, poza przypadkiem wodnych roztworów, gdzie można zastosować 2 ml/100 g masy ciała. Z wyjątkiem substancji drażniących i żrących, które zwykle ujawniają silniejsze działanie przy wyższym stężeniu, zmienności w objętości badanej muszą być zminimalizowane przez ustawienie stężenia tak, aby zapewnić stałą objętość we wszystkich poziomach dozowania.
W odniesieniu do substancji podawanych w pokarmie lub wodzie pitnej istotne jest zapewnienie, aby ilości danej substancji badanej nie kolidowały z normalnym odżywianiem lub równowagą wodną. Gdy substancja badana jest podawana w pokarmie – w stałym stężeniu w pokarmie (ppm) albo w stałym poziomie dawki – może być zastosowana w odniesieniu do masy ciała zwierzęcia; wykorzystana alternatywa musi być określona. W odniesieniu do substancji podawanej przez zgłębnik dawka musi być podana w podobnej porze każdego dnia i regulowana tak, by utrzymać stały poziom dawki w odniesieniu do masy ciała zwierzęcia. W przypadku gdy badanie 90-dniowe jest stosowane jako wstępne do badań długoterminowych toksyczności przewlekłej, w obu badaniach powinien być użyty podobny pokarm.
1.5.2. Obserwacje
Okres obserwacji powinien wynosić przynajmniej 90 dni. Zwierzęta w grupie satelickiej, zaplanowanej do dalszej obserwacji, muszą być trzymane przez stosowny okres bez traktowania w celu wykrycia wytrzymałości na lub powrotu do normy od działań toksycznych.
Ogólne kliniczne obserwacje powinny być dokonywane przynajmniej raz na dzień, najlepiej o tej (tych) samej(-ych) porze (porach) każdego dnia, uwzględniając szczytowy okres przewidywanego działania po dozowaniu. Stan kliniczny zwierząt powinien być rejestrowany. Co najmniej dwa razy dziennie, zwykle na początku i pod koniec każdego dnia, wszystkie zwierzęta są kontrolowane w zakresie oznak zachorowań i śmiertelności.
Przynajmniej raz przed pierwszą ekspozycją (uwzględnić w granicach porównania) i raz tygodniowo później należy dokonać szczegółowej obserwacji klinicznej na wszystkich zwierzętach. Obserwacje te powinny być dokonywane poza klatką domową, najlepiej w warunkach standardowych za każdym razem w podobnych porach. Powinny one być starannie zapisane, najlepiej przez zastosowanie systemów punktacji, wyraźnie określonych przez laboratorium badawcze. Należy dołożyć starań w celu zapewnienia, aby zmiany w warunkach obserwacji były minimalne. Oznaki zauważone powinny obejmować zmiany na skórze, futrze, oczach, błonach śluzowych, występowania wydzielin i czynności autonomiczne (np. łzawienie, piloerekcja, rozmiar źrenic, zmieniony rytm oddychania), lecz nie powinny być ograniczone tylko do nich. Zmiany w chodzie, postawie i reakcja na obchodzenie się, jak również obecność zachowań klonicznych i tonicznych, stereotypów (np. nadmierne zajmują się sobą, kołowacizna) lub dziwaczne zachowanie (np. samookaleczanie, chodzenie wstecz) także muszą być zarejestrowane (1).
Badanie oftalmologiczne przy użyciu oftalmoskopu lub równoważnego odpowiedniego przyrządu musi być wykonane przed podawaniem dawek badanej substancji i przy ukończeniu badań, najlepiej na wszystkich zwierzętach, ale przynajmniej w grupach wysokiej dawki i kontrolnej. Jeśli wykryto zmiany w oczach, należy przebadać wszystkie zwierzęta.
Odnośnie do końca okresu ekspozycji, a w żadnym przypadku nie wcześniej niż w 11 tygodniu, należy przeprowadzić badania reaktywności sensorycznej na bodźce różnych typów (1) (np. na bodźce słuchowe, optyczne i proprioreceptoryczne) (2)(3)(4), ocenę siły chwytu (5) i ocenę aktywności motorycznej (6). Dalsze szczegóły procedur do przeprowadzenia są podane w odpowiednich odniesieniach. Jednakże można także zastosować procedury alternatywne do tych, do których dokonano odniesień.
Obserwacje funkcjonalne prowadzone odnośnie do końca badań mogą być pominięte, jeśli dane dotyczące obserwacji funkcjonalnych są dostępne z innych badań i codzienne kliniczne obserwacje nie ujawniają żadnych deficytów funkcjonalnych.
Wyjątkowo, obserwacje funkcjonalne mogą być także pominięte w odniesieniu do grup, które w inny sposób przejawiają oznaki zatrucia do tego stopnia, że mogłyby znacznie zakłócić wykonanie badania funkcjonalnego.
1.5.2.1. Waga ciała i spożycie pokarmu/wody
Wszystkie zwierzęta powinny być ważone przynajmniej raz na tydzień. Pomiary spożycia pokarmu powinny być dokonywane przynajmniej co tydzień. Jeśli substancja badana jest podawana w wodzie pitnej, spożycie wody musi być także mierzone przynajmniej cotygodniowo. Spożycie wody może być także rozważone w odniesieniu do badań pokarmowych lub zgłębnika, w czasie których pragnienie może się zmienić.
1.5.2.2. Biochemia kliniczna i hematologia
Próbki krwi powinny być pobrane z nazwanego siedliska i przechowywane, jeśli ma to zastosowanie, we właściwych warunkach. Na końcu okresu badania próbki są zbierane tuż przed procedurą zabijania zwierząt lub jako jej część.
Następujące badania hematologiczne powinny być wykonane pod koniec okresu badania, a także gdy wszystkie pośrednie próbki krwi muszą zostać zebrane: hematokryt, stężenie hemoglobiny, liczba erytrocytów, całkowita i różnicowa liczba leukocytów, liczba płytek krwi i pomiar czasu/potencjał krzepnięcia krwi.
Oznaczenia biochemii klinicznej do badań głównych skutków toksycznych w tkankach, w szczególności działań na nerki i wątrobę, powinny być wykonane na próbkach krwi, uzyskanych od każdego zwierzęcia tuż przed lub jako część procedury zabijania zwierząt (oddzielnie od tych, które okazały się konające i/lub zabite w międzyczasie). W sposób podobny do badań hematologicznych może być wykonane pobieranie próbek w trakcie badań dla przeprowadzenia klinicznych badań biochemicznych. Zaleca się przetrzymanie zwierząt przez noc, przed pobraniem próbek krwi (3). Oznaczenia w plazmie lub surowicy muszą zawierać sód, potas, glukozę, całkowity cholesterol, mocznik, azot mocznikowy we krwi, kreatyninę, całkowite proteiny i albuminy i więcej niż dwa enzymy charakterystyczne efektów wątrobokomórkowych (takich jak alanina, aminotransferaza, asparaginian aminotransferazy, fosfataza alkaliczna, gamma glutamylotranspeptydaza i dehydrogenaza sorbitolowa). Można także włączyć pomiary dodatkowych enzymów (wątrobowego lub innego pochodzenia) i kwasów żółciowych, które pod pewnymi warunkami mogą dostarczyć użytecznych informacji.
Fakultatywnie można przeprowadzić następujące oznaczenia analityczne w moczu, w ciągu ostatniego tygodnia badań, stosując zebraną czasowo objętość moczu: przejrzystość, objętość, osmolality lub ciężar właściwy, pH, białka, glukoza i komórki krwi.
Dodatkowo należy rozważyć zastosowanie do badań surowicy znaczników ogólnego uszkodzenia tkanek. Inne oznaczenia, które powinny być dokonane, jeśli znane właściwości substancji badanej mogą lub są podejrzane o wpływ na przebieg metabolizmu, to wapń, fosfor, trwałe trójglicerydy, specyficzne hormony, metahemoglobina i cholinoesteraza. Mogą być one potrzebne do określenia niektórych związków chemicznych w niektórych klasach lub na zasadzie jednostkowych przypadków.
Ogólnie, potrzebne jest elastyczne podejście, w zależności od gatunku oraz obserwowanych i/lub spodziewanych skutków pochodzących od danej substancji.
Jeśli historyczne dane bazowe są nieodpowiednie należy rozważyć czy hematologiczne i kliniczne biochemiczne zmiany powinny być określone przed rozpoczęciem dawkowania; zasadniczo nie jest zalecane, aby te dane były gromadzone przed traktowaniem (7).
1.5.2.3. Całościowa sekcja zwłok
Wszystkie zwierzęta w badaniu podlegają pełnej, szczegółowej całościowej sekcji zwłok, która obejmuje staranne badanie zewnętrznej powierzchni organu, wszystkie otwory i czaszkowe, piersiowe i brzuszne jamy i ich zawartości. Wątroba, nerka, nadnercza, jądra, najądrza, macica, jajniki, grasica, śledziona, mózg i serce wszystkich zwierząt (oddzielnie od tych, które okazały się konające i/lub zabite w międzyczasie) powinny być okrojone z wszelkich przylegających tkanek, odpowiednio, i należy tak szybko jak to możliwe pomierzyć ich mokrą wagę po sekcji w celu zapobieżenia wyschnięciu.
Następujące tkanki powinny być zakonserwowane w najwłaściwym środku utrwalającym dla obu typów tkanek i zamierzonych kolejnych badań histopatologicznych: wszystkie całkowite uszkodzenia, mózg (reprezentatywne obszary, włączając płaszcz mózgu, móżdżek i szpik/mostek), rdzeń kręgowy (na trzech poziomach: karkowym, środkowo-piersiowym i lędźwiowym), przysadka, tarczyca, przytarczyczka, grasica, przełyk, gruczoły ślinowe, żołądek, małe i duże jelita (włączając plamy Peyera), wątroba, trzustka, nerki, nadnercza, śledziona, serce, tchawica i płuca (zabezpieczone przez inflację utrwaloną a następnie zanurzoną), aorta, gonady, macica, dodatkowe organy płciowe, żeńskie gruczoły sutkowe, prostata, pęcherz moczowy, pęcherzyk żółciowy (mysz), węzły limfatyczne (najlepiej jeden węzeł pokrywający drogę podawania i drugi odległy od drogi podawania aby objąć działania na układ), nerw peryferyjny (kulszowy lub piszczelowy) zalecany w dużej bliskości od mięśnia, fragment kości szpikowej (i/lub świeża kość z zassanym szpikiem), skóra i oczy (jeśli zaobserwowano zmiany podczas badań oftalmologicznych). Kliniczne i inne wyniki badań mogą zasugerować konieczność zbadania dodatkowych tkanek. Także jakiekolwiek organy uznane za mogące być celem w oparciu o znane właściwości badanej substancji powinny być zakonserwowane.
1.5.2.4. Histopatologia
Pełna histopatologia powinna być przeprowadzona na zakonserwowanych ciałach i tkankach wszystkich zwierząt z grupy kontrolnej i wysokodawkowanej. Badania te powinny być rozciągnięte na wszystkie inne dawkowane grupy, jeśli w wysokodawkowanej grupie obserwuje się zmiany związane z działaniem środka.
Należy przebadać wszystkie zmiany patologiczne.
Jeśli używa się grupy satelickiej, histopatologia powinna być przeprowadzona na tkankach i ciałach określonych jako wykazujące działanie, w grupach traktowanych.
2. DANE I SPRAWOZDAWCZOŚĆ
2.1. DANE
Należy przedstawić dane osobnika. Dodatkowo wszystkie dane powinny być zestawione w postaci tabelarycznej, przedstawiając w odniesieniu do każdej badanej grupy liczbę zwierząt na początku badania, liczbę zwierząt, które poniosły śmierć podczas trwania badania lub które zabito z przyczyn humanitarnych wraz z czasem, w którym nastąpiła śmierć lub humanitarne zabicie, liczbę wykazującą oznaki zatrucia, opis obserwowanych oznak zatrucia, włączając czas zaistnienia, czas trwania, stopień ciężkości każdego działania toksycznego, liczbę zwierząt wykazującą zmiany patologiczne, typ zmian patologicznych i procent zwierząt obrazujący każdy typ zmiany patologicznej.
Gdy ma to zastosowanie, wyniki liczbowe powinny być szacowane właściwymi i ogólnie przyjętymi metodami statystycznymi. Metody statystyczne i dane poddawane analizie powinny być wybrane w czasie przygotowania projektu badań.
2.2. SPRAWOZDANIE Z BADANIA
Sprawozdanie dotyczące badań musi zawierać następujące informacje:
2.2.1. Substancja użyta w badaniu:
|
— |
charakter fizyczny, czystość i właściwości fizyko-chemiczne, |
|
— |
dane identyfikacyjne, |
|
— |
nośnik (jeśli jest to stosowne): uzasadnienie wyboru nośnika, jeśli jest różny od wody. |
2.2.2. Gatunki użyte w badaniu:
|
— |
użyte gatunki i szczep, |
|
— |
ilość, wiek i płeć zwierząt, |
|
— |
źródło, warunki zamieszkiwania, pokarm itp., |
|
— |
waga osobnicza zwierząt przy rozpoczęciu badania. |
2.2.3. Warunki badania:
|
— |
racjonalne uzasadnienie wybranego poziomu dawkowania, |
|
— |
szczegółowe informacje dotyczące składu badanej substancji/przygotowanie pokarmu, uzyskane stężenie, stabilność i jednorodność przygotowania, |
|
— |
szczegółowe informacje dotyczące podawania substancji badanej, |
|
— |
dawka rzeczywista (mg/kg waga ciała/dzień), wskaźnik konwersji pokarm/woda pitna stężenia badanej substancji (ppm) do dawki rzeczywistej, jeśli ma to zastosowanie, |
|
— |
szczegółowe informacje dotyczące jakości żywności i wody. |
2.2.4. Wyniki:
|
— |
waga ciała oraz zmiany wagi ciała, |
|
— |
spożycie żywności i wody, jeśli ma to zastosowanie, |
|
— |
dane reakcji na zatrucie według płci i poziomu dawki, włączając oznaki zatrucia, |
|
— |
charakter, surowość i czas trwania obserwacji klinicznych (stan odwracalny czy nie), |
|
— |
wyniki badania oftalmologicznego, |
|
— |
ocena czynności sensorycznych, siły zacisku i czynności motorycznych (gdy jest to dostępne), |
|
— |
badania hematologiczne z odpowiednimi wartościami poziomów tolerancji, |
|
— |
biochemiczne badania kliniczne z odpowiednimi wartościami poziomów tolerancji, |
|
— |
końcowa waga ciała, waga organów i proporcje wagi organ/ciało, |
|
— |
ustalenia sekcji zwłok, |
|
— |
szczegółowy opis wszystkich histopatologicznych wyników badań, |
|
— |
dane dotyczące absorpcji, jeśli jest to dostępne, |
|
— |
statystyczne opracowywanie wyników, tam gdzie jest to stosowne. |
Omówienie wyników.
Wnioski.
3. BIBLIOGRAFIA
|
(1) |
IPCS (1986). Principles and Methods for the Assessment of Neurotoxicity Associated with Exposure to Chemicals. Environmental Health Criteria Document No 60. |
|
(2) |
Tupper, D. E., Wallace, R.B. (1980). Utility of the Neurologic Examination in Rats. Acta Neurobiol. Exp., 40, pp. 999–1003. |
|
(3) |
Gad, S. C. (1982). A Neuromuscular Screen for Use in Industrial Toxicology. J. Toxicol. Environ. Health, 9, pp. 691–704. |
|
(4) |
Moser, V. C, Mc Daniel, K. M., Phillips, P. M. (1991). Rat Strain and Stock Comparisons Using a Functional Observational Battery: Baseline Values and Effects of Amitraz. Toxicol. Appl. Pharmacol., 108, pp. 267–283. |
|
(5) |
Meyer O. A., Tilson H. A., Byrd W. C, Riley M. T. (1979). A Method for the Routine Assessment of Fore- and Hind- limb grip Strength of Rats and Mice. Neurobehav. Toxivol., 1, pp. 233–236. |
|
(6) |
Crofton K. M., Howard J. L., Moser V. C, Gill M. W., Reiter L. W., Tilson H. A., MacPhail R. C. (1991). Interlaboratory Comparison of Motor Activity Experiments: Implication for Neurotoxicological Assessments. Neurotoxicol. Teratol., 13, pp. 599–609. |
|
(7) |
Weingand K., Brown G., Hall R. et al. (1996). „Harmonisation of Animal Clinic Pathology Testing in Toxicity and Safety Studies”, Fundam. & Appl. Toxicol., 29, pp. 198–201. |
B.27. BADANIE TOKSYCZNOŚCI PODPRZEWLEKŁEJ DROGĄ POKARMOWĄ – BADANIA TOKSYCZNOŚCI NA NIEGRYZONIACH METODĄ POWTARZANEJ 90-DNIOWEJ DAWKI DROGĄ POKARMOWĄ
1. METODA
Niniejsza metoda badania toksyczności podprzewlekłej drogą pokarmową jest kopią OECD TG 409 (1998).
1.1. WPROWADZENIE
Przy oszacowaniu i ocenie cech toksyczności substancji chemicznej oznaczenie podprzewlekłej toksyczności drogą pokarmową, stosując powtarzane dawki, może być prowadzone po początkowej informacji o toksyczności, uzyskanej z 28-dniowych badań toksyczności ostrej lub powtarzanej. 90-dniowe badanie dostarcza informacji o możliwym niebezpieczeństwie dla zdrowia, które prawdopodobnie powstanie z powtarzanego narażenia przez okres szybkiego rozwoju do wczesnej dorosłości. Badanie dostarczy informacji na temat głównych toksycznych działań, wskaże zaatakowane organy i możliwości akumulacji, dostarczy oceny poziomu ekspozycji nie powodującego negatywnych skutków, którą stosuje się przy wyborze poziomów dawki dla badań przewlekłych i ustalania kryteriów bezpieczeństwa dla narażenia ludzi.
Metoda badania umożliwia określenie u gatunków należących do niegryzoni negatywnych skutków ekspozycji na działanie chemiczne oraz powinna być wykorzystywana tylko:
|
— |
w przypadku gdy skutki zaobserwowane w innych badaniach wymagają wyjaśnienia/charakterystyki dla drugiego gatunku niegryzoni, lub |
|
— |
w przypadku gdy badania toksykokinetyczne wskazują, że wykorzystanie właściwego gatunku niegryzoni jest najbardziej trafnym wyborem ze zwierząt laboratoryjnych, lub |
|
— |
w przypadku gdy inne właściwe przyczyny usprawiedliwiają wykorzystanie gatunku niegryzoni. |
Zob. także Ogólne wprowadzenie część B.
1.2. DEFINICJE
Dawka: jest ilością podawanej substancji badanej. Dawka jest wyrażana wagowo (g, mg) lub jako waga substancji badanej na jednostkę wagi badanego zwierzęcia (np. mg/kg), lub jako stężenie stałe żywieniowe (ppm).
Dawkowanie: jest ogólnym pojęciem obejmującym dawkę, jej częstotliwość i czas podawania dawki.
NOAEL: jest skrótem od poziomu, przy którym nie obserwuje się niekorzystnych skutków, oraz jest najwyższym poziomem dawki, gdzie nie obserwuje się niekorzystnych efektów zależnych od podawanej substancji.
1.3. ZASADA METODY BADANIA
Substancja badana jest codziennie podawana doustnie w stopniowanych dawkach, kilku grupom zwierząt doświadczalnych, jeden poziom dawki na grupę przez okres 90 dni. W czasie okresu podawania zwierzęta są ściśle obserwowane na oznaki zatrucia. Zwierzęta, które zmarly lub zostały zabite w czasie trwania badania, są poddawane sekcji zwłok po zakończeniu badania; zwierzęta, które przeżyły są również zabijane i poddawane sekcji zwłok.
1.4. OPIS METODY BADANIA
1.4.1. Dobór gatunków zwierząt
Zwykle wykorzystywanym gatunkiem niegryzoni jest pies, który powinien być określonej rasy; często wykorzystywany jest pies gończy. Inne gatunki, np. małe świnie, mogą być również wykorzystane. Naczelne nie są zalecane, a ich wykorzystanie powinno być uzasadnione. Powinny być wykorzystane młode, zdrowe zwierzęta, a w przypadku psa wskazane jest rozpoczęcie dawkowania w wieku 4–6 miesięcy i nie późniejszym niż dziewięć miesięcy. W przypadku gdy badanie jest prowadzone jako wstępne do badań toksyczności przewlekłej długoterminowej, w obu badaniach należy wykorzystać ten sam gatunek/rasę.
1.4.2. Przygotowanie zwierząt
Powinny być wykorzystane młode zdrowe zwierzęta, zaaklimatyzowane w warunkach laboratoryjnych i nie będące przedmiotem wcześniejszych procedur eksperymentalnych. Czas trwania aklimatyzacji zależy od wybranych gatunków badanych i ich źródła pochodzenia. Zalecane jest przynajmniej pięć dni w odniesieniu do psów albo celowo hodowanej trzody od rezydenta kolonii i co najmniej dwa tygodnie w odniesieniu do zwierząt z zewnętrznych źródeł. Zwierzęta badane powinny charakteryzować się gatunkiem, szczepem, źródłem, płcią, wagą i/lub wiekiem. Zwierzęta powinny być losowo wyznaczane do doświadczeń kontrolnych i grup traktowanych. Klatki powinny być rozmieszczone w taki sposób, aby możliwy wpływ wynikający z ich umieszczania był zminimalizowany. Każde zwierzę musi być oznakowane niepowtarzalnym numerem identyfikacyjnym.
1.4.3. Przygotowanie dawek
Substancja badana jest podawana w postaci pokarmu lub wody pitnej, przez zgłębnik lub w kapsułkach. Metoda podawania drogą pokarmową jest zależna od celu badania oraz właściwości fizyko-chemicznych badanego materiału.
Tam gdzie jest to konieczne, badaną substancje rozpuszcza się lub wykonuje zawiesinę w odpowiednim nośniku. Zalecane jest, aby tam gdzie to jest możliwe, rozważyć wpierw użycie roztworu lub zawiesiny wodnej, a następnie rozważyć wykonanie roztworu/zawiesiny w oleju (np. w oleju kukurydzianym) i dopiero następnie w innych nośnikach. Dla nośników różnych od wody muszą być znane cechy toksyczności. Powinna być określona stabilność badanej substancji w warunkach podawania dawek.
1.5. PROCEDURA
1.5.1. Ilość i płeć zwierząt
Co najmniej osiem zwierząt (cztery samice i cztery samce) należy wykorzystać w odniesieniu do każdego poziomu dawki. Jeśli planuje się zabijanie w trakcie, liczba powinna być zwiększona o przewidzianą ilość zwierząt do zabicia przed zakończeniem badań. Liczba zwierząt na końcu badania musi być odpowiednia do celów wnikliwego oszacowania toksycznych skutków. W oparciu o uprzednią znajomość substancji lub bliskiego odpowiednika należy rozważyć włączenie dodatkowej, satelickiej grupy ośmiu zwierząt (po cztery każdej płci) do grupy kontrolnej i grupy najwyżej dawkowanej do celów obserwacji po okresie czasu traktowania, odwracalności lub trwałości jakichkolwiek skutków toksycznych. Czas trwania tego okresu po traktowaniu powinien być ustalony odpowiednio w odniesieniu do obserwowanych skutków.
1.5.2. Poziomy dawek
Stosuje się co najmniej trzy poziomy dawek i równoczesne doświadczenie kontrolne, poza przypadkiem gdy prowadzone jest badanie graniczne (zob. 1.5.3). Poziomy dawek mogą być oparte na wynikach powtarzanej dawki lub badaniach ustalania zakresu oraz powinny uwzględniać wszelkie istniejące dane toksykologiczne i toksykokinetyczne, dostępne w odniesieniu do badanej substancji lub związanych z nią materiałów. Poza ograniczeniami natury fizykochemicznej i działaniami biologicznymi substancji badanej, poziom najwyższej dawki musi być wybrany w celu wzbudzenia objawów toksycznych, ale nie spowodowania śmierci czy też poważnych cierpień. Opadająca sekwencja poziomu dawki musi być wybrana w celu pokazania reakcji związanej z dawkowaniem i poziomem, przy którym nie obserwuje się negatywnych skutków (NOAEL) przy najniższym poziomie dawki. Dwa do czterech zachodzące przedzialy są często optymalne do ustalenia opadających poziomów dawki i dodanie czwartej grupy badanej jest często polecane dla zastosowania bardzo dużych odstępów (np. więcej niż współczynnik 6–10) między dawkowaniami.
Grupa kontrolna musi być grupą nietraktowaną lub grupą kontrolną nośnika, jeśli nośnik jest używany do podawania substancji badanej. Zwierzęta w grupie kontrolnej, poza nietraktowaniem jej substancją badaną, są obsługiwane w identyczny sposób jak w zwierzęta w grupach badanych. Jeśli stosowany jest nośnik, grupa kontrolna musi otrzymywać nośnik w maksymalnie stosowanej objętości. Gdy substancja badana jest podawana w diecie i powoduje zmniejszenie zapotrzebowania żywieniowego, wtedy grupa kontrolna z pary żywieniowej może być pomocna w rozróżnieniu między zmniejszeniem spowodowanym odczuwaniem smaku lub zmianami toksykologicznymi w modelu badania.
Następujące cechy nośnika i innych dodatków muszą być rozważone jako właściwe: wpływ wchłaniania, rozkładu, metabolizmu, retencji badanej substancji, wpływ właściwości chemicznych substancji badanej, mogące zmienić jej cechy toksyczności, oraz wpływy zapotrzebowania na żywność i wodę lub żywieniowy status zwierząt.
1.5.3. Badanie graniczne
Jeśli badanie przy jednym poziomie dawki, równoważnym co najmniej 1 000 mg/kg masy ciała/dzień, stosując procedurę opisaną w odniesieniu do niniejszego badania, powoduje, że nie obserwuje się negatywnych skutków oraz jeśli nie oczekuje się toksyczności na podstawie danych substancji strukturalnie związanych, wtedy pełne badania z zastosowaniem trzech poziomów nie wydają się konieczne. Badanie graniczne ma zastosowanie z wyjątkiem sytuacji, gdy narażenie ludzi wskazuje potrzebę zastosowania wyższego poziomu dawki.
1.5.4. Podawanie dawek
Zwierzętom dozuje się substancję badaną przez siedem dni każdego tygodnia w okresie 90 dni. Każdy inny sposób dozowania, na przykład pięć dni w tygodniu, wymaga uzasadnienia. Gdy substancja badana jest podawana przez zgłębnik, musi być to wykonane w pojedynczej dawce, przy użyciu sondy przelykowej lub odpowiedniej kaniuli intubacyjnej. Maksymalna objętość cieczy, która może być podana w jednym momencie, zależy od rozmiarów zwierzęcia badanego. Zwykle objętość powinna być utrzymywana na możliwe najmniejszym poziomie. Z wyjątkiem substancji drażniących i żrących, które zwykle ujawniają silniejsze działanie przy wyższym stężeniu, zmienności w objętości badanej muszą być zminimalizowane przez ustawienie stężenia tak, aby zapewnić stałą objętość we wszystkich poziomach dozowania.
W odniesieniu do substancji podawanych w pokarmie lub wodzie pitnej istotne jest zapewnienie, aby ilości danej substancji badanej nie kolidowały z normalnym odżywianiem lub równowagą wodną. Gdy substancja badana jest podawana w pokarmie – w stałym stężeniu w pokarmie (ppm) albo w stałym poziomie dawki – może być zastosowana w odniesieniu do masy ciała zwierzęcia; wykorzystana alternatywa musi być określona. W odniesieniu do substancji podawanej przez zgłębnik, dawka musi być podana w podobnej porze każdego dnia i regulowana tak by utrzymać stały poziom dawki w odniesieniu do masy ciała zwierzęcia. W przypadku gdy badanie 90-dniowe jest stosowane jako wstępne do badań długoterminowych toksyczności przewlekłej, w obu badaniach powinien być użyty podobny pokarm.
1.5.5. Obserwacje
Okres obserwacji powinien wynosić przynajmniej 90 dni. Zwierzęta w grupie satelickiej, zaplanowanej do dalszej obserwacji, muszą być trzymane przez stosowny okres bez traktowania w celu wykrycia wytrzymałości na lub powrotu do normy od działań toksycznych.
Ogólne kliniczne obserwacje powinny być dokonywane przynajmniej raz na dzień, najlepiej o tej (tych) samej(-ych) porze (porach) każdego dnia, uwzględniając szczytowy okres przewidywanego działania po dozowaniu. Stan kliniczny zwierząt powinien być rejestrowany. Co najmniej dwa razy dziennie, zwykle na początku i pod koniec każdego dnia, wszystkie zwierzęta są kontrolowane w zakresie oznak zachorowań i śmiertelności.
Przynajmniej raz przed pierwszą ekspozycją (uwzględnić w granicach porównania) i raz tygodniowo później należy dokonać szczegółowej obserwacji klinicznej na wszystkich zwierzętach. Obserwacje te powinny być dokonywane poza klatką domową, najlepiej w warunkach standardowych za każdym razem w podobnych porach. Powinny one być starannie zapisane, najlepiej przez zastosowanie systemów punktacji, wyraźnie określonych przez laboratorium badawcze. Należy dołożyć starań w celu zapewnienia, aby zmiany w warunkach obserwacji były minimalne. Oznaki zauważone powinny obejmować zmiany na skórze, futrze, oczach, błonach śluzowych, występowania wydzielin i czynności autonomiczne (np. łzawienie, piloerekcja, rozmiar źrenic, zmieniony rytm oddychania), lecz nie powinny być ograniczone tylko do nich. Zmiany w chodzie, postawie i reakcja na obchodzenie się, jak również obecność zachowań klonicznych i tonicznych, stereotypów (np. nadmierne zajmowanie się sobą, kolowacizna) lub dziwaczne zachowanie (np. samookaleczanie, chodzenie wstecz) także muszą być zarejestrowane.
Badanie oftalmologiczne przy użyciu oftalmoskopu lub równoważnego odpowiedniego przyrządu musi być wykonane przed podawaniem dawek badanej substancji i przy ukończeniu badań, najlepiej na wszystkich zwierzętach, ale przynajmniej w grupach wysokiej dawki i kontrolnej. Jeśli wykryto zmiany w oczach, należy przebadać wszystkie zwierzęta.
1.5.5.1. Waga ciała i i spożycie pokarmu/wody
Wszystkie zwierzęta powinny być ważone przynajmniej raz na tydzień. Pomiary spożycia pokarmu powinny być dokonywane przynajmniej co tydzień. Jeśli substancja badana jest podawana w wodzie pitnej, spożycie wody musi być także mierzone przynajmniej cotygodniowo. Spożycie wody może być także rozważone w odniesieniu do badań pokarmowych lub zgłębnika, w czasie których pragnienie może się zmienić.
1.5.5.2. Biochemia kliniczna i hematologia
Próbki krwi powinny być pobrane z nazwanego siedliska i przechowywane, jeśli ma to zastosowanie, we właściwych warunkach. Na końcu okresu badania próbki są zbierane tuż przed procedurą zabijania zwierząt lub jako jej część.
Hematologia, włączając hematokryt, stężenie hemoglobiny, liczba erytrocytów, całkowita i różnicowa liczba leukocytów, liczba płytek krwi i pomiar potencjału krzepliwości takiego jak czas krzepliwości, czas protrombinowy i tromboplastynowy, powinna być zbadana na początku badania, następnie albo w odstępach miesięcznych albo w środku okresu badania oraz ostatecznie pod koniec okresu badania.
Oznaczenia biochemii klinicznej do badań głównych skutków toksycznych w tkankach, w szczególności działań na nerki i wątrobę, powinny być wykonane na próbkach krwi, uzyskanych od każdego zwierzęcia na początku, następnie albo odstępach miesięcznych albo w środku okresu badania oraz ostatecznie pod koniec okresu badania. Dziedzinami badań, które muszą być rozważone, są równowaga elektrolitów, metabolizm węglowodanów oraz funkcje wątroby i nerek. Na wybór specyficznych badań wpływają obserwacje sposobu działania substancji badanej. Zwierzęta powinny być przetrzymane przez okres odpowiedni do gatunku przed pobieraniem próbek krwi. Zalecane oznaczenia obejmują wapń, fosfor, chlorki, sód, potas, glukozę, aminotransferazę alaninową, aminotransferaze asparaginową, dekarboksylazę ornityny, gamma glutamylową transpeptydazę, azot mocznikowy, albuminy, kreatyninę krwi, całkowitą bilirubinę i całkowitą surowicę białkową.
Oznaczenia analityczne w moczu powinny być wykonane przynajmniej na początku, następnie w środku i pod koniec badania, stosując zebraną czasowo objętość moczu. Oznaczenia moczu obejmują przejrzystość, objętość, osmolality lub ciężar właściwy, pH, białka, glukoza i komórki krwi. Inne parametry dodatkowo mogą być oznaczane w miarę potrzeb rozszerzenia badania zaobserwowanego(-ych) skutku(-ów).
Ponadto należy rozważyć zastosowanie do badań surowicy znaczników ogólnego uszkodzenia tkanek. Inne oznaczenia, które mogą być niezbędne do odpowiedniej oceny toksykologicznej, obejmują lipidy, hormony, równowagę kwasowo-zasadową, metahemoglobinę, inhibitory cholinoesterazy. Dodatkowe badania kliniczne mogą być wykorzystywane, gdy niezbędne jest rozszerzenie badania zaobserwowanych skutków. Mogą być one potrzebne do określenia niektórych związków chemicznych w niektórych klasach lub na zasadzie jednostkowych przypadków.
Ogólnie, potrzebne jest elastyczne podejście, w zależności od gatunku oraz obserwowanych i/lub spodziewanych skutków pochodzących od danej substancji.
1.5.5.3. Całościowa sekcja zwłok
Wszystkie zwierzęta w badaniu podlegają pełnej, szczegółowej brutto sekcji zwłok, która obejmuje staranne badanie zewnętrznej powierzchni organu, wszystkie otwory i czaszkowe, piersiowe i brzuszne jamy i ich zawartości. Wątroba, nerka, nadnercza, jądra, najądrza, macica, jajniki, grasica, śledziona, mózg i serce wszystkich zwierząt (oddzielnie od tych, które okazały się konające i/lub zabite w międzyczasie) powinny być okrojone z wszelkich przylegających tkanek, odpowiednio, i należy tak szybko jak to możliwe pomierzyć ich mokrą wagę po sekcji w celu zapobieżenia wyschnięciu.
Następujące tkanki powinny być zakonserwowane w najwłaściwszym środku utrwalającym dla obu typów tkanek i zamierzonych kolejnych badań histopatologicznych: wszystkie całkowite uszkodzenia, mózg (reprezentatywne obszary, włączając płaszcz mózgowy, móżdżek i szpik/mostek), rdzeń kręgowy (na trzech poziomach: karkowym, środkowo-piersiowym i lędźwiowym), przysadka, tarczyca, przytarczyczka, grasica, przełyk, gruczoły ślinowe, żołądek, małe i duże jelita (włączając plamy Peyera), wątroba, trzustka, nerki, nadnercza, śledziona, serce, tchawica i płuca (zabezpieczone przez inflację utrwaloną a następnie zanurzoną), aorta, gonady, macica, dodatkowe organy płciowe, żeńskie gruczoły sutkowe, prostata, pęcherz moczowy, pęcherzyk żółciowy (mysz), węzły limfatyczne (najlepiej jeden węzeł pokrywający drogę podawania i drugi odległy od drogi podawania, aby objąć działania na układ), nerw peryferyjny (kulszowy lub piszczelowy) zalecany w dużej bliskości od mięśnia, fragment kości szpikowej (i/lub świeża kość z zassanym szpikiem), skóra i oczy (jeśli zaobserwowano zmiany podczas badań oftalmologicznych). Kliniczne i inne wyniki badań mogą zasugerować konieczność zbadania dodatkowych tkanek. Także jakiekolwiek organy uznane za mogące być celem w oparciu o znane właściwości badanej substancji powinny być zakonserwowane.
1.5.5.4. Histopatologia
Pełna histopatologia powinna być przeprowadzona na zakonserwowanych ciałach i tkankach wszystkich zwierząt z grupy kontrolnej i wysokodawkowanej. Badania te powinny być rozciągnięte na wszystkie inne dawkowane grupy, jeśli w wysokodawkowanej grupie obserwuje się zmiany związane z działaniem środka.
Należy przebadać wszystkie zmiany patologiczne.
Jeśli używa się grupy satelickiej, histopatologia powinna być przeprowadzona na tkankach i ciałach, określonych jako wykazujące działanie, w grupach traktowanych.
2. DANE I SPRAWOZDAWCZOŚĆ
2.1. DANE
Należy przedstawić dane osobnika. Dodatkowo, wszystkie dane powinny być zestawione w postaci tabelarycznej, przedstawiając w odniesieniu do każdej badanej grupy liczbę zwierząt na początku badania, liczbę zwierząt, które poniosły śmierć podczas trwania badania lub które zabito z przyczyn humanitarnych wraz z czasem, w którym nastąpiła śmierć lub humanitarne zabicie, liczbę wykazującą oznaki zatrucia, opis obserwowanych oznak zatrucia, włączając czas zaistnienia, czas trwania, stopień ciężkości każdego działania toksycznego, liczbę zwierząt wykazującą zmiany patologiczne, typ zmian patologicznych i procent zwierząt obrazujący każdy typ zmiany patologicznej.
Gdy ma to zastosowanie, wyniki liczbowe powinny być szacowane właściwymi i ogólnie przyjętymi metodami statystycznymi. Metody statystyczne i dane poddawane analizie, powinny być wybrane w czasie przygotowania projektu badań.
2.2. SPRAWOZDANIE DOTYCZĄCE BADAŃ
Sprawozdanie dotyczące badań musi zawierać następujące informacje:
2.2.1. Substancja użyta w badaniu:
|
— |
charakter fizyczny, czystość i właściwości fizyko-chemiczne, |
|
— |
dane identyfikacyjne, |
|
— |
nośnik (jeśli jest to stosowne): uzasadnienie wyboru nośnika, jeśli jest różny od wody. |
2.2.2. Gatunki użyte w badaniu:
|
— |
użyte gatunki i szczep, |
|
— |
ilość, wiek i płeć zwierząt, |
|
— |
źródło, warunki zamieszkiwania, pokarm itp., |
|
— |
waga osobnicza zwierząt przy rozpoczęciu badania. |
2.2.3. Warunki badania:
|
— |
racjonalne uzasadnienie wybranego poziomu dawkowania, |
|
— |
szczegółowe informacje dotyczące składu badanej substancji/przygotowanie pokarmu, uzyskane stężenie, stabilność i jednorodność przygotowania, |
|
— |
szczegółowe informacje dotyczące podawania substancji badanej, |
|
— |
dawka rzeczywista (mg/kg waga ciała/dzień), wskaźnik konwersji pokarm/woda pitna stężenia badanej substancji (ppm) do dawki rzeczywistej, jeśli ma to zastosowanie, |
|
— |
szczegółowe informacje dotyczące jakości żywności i wody. |
2.2.4. Wyniki:
|
— |
waga ciała oraz zmiany wagi ciała, |
|
— |
spożycie żywności i wody, jeśli ma to zastosowanie, |
|
— |
dane reakcji na zatrucie według płci i poziomu dawki, włączając oznaki zatrucia, |
|
— |
charakter, surowość i czas trwania obserwacji klinicznych (stan odwracalny czy nie), |
|
— |
wyniki badania oftalmologicznego, |
|
— |
ocena czynności sensorycznych, siły zacisku i czynności motorycznych (gdy jest to dostępne), |
|
— |
badania hematologiczne z odpowiednimi wartościami poziomów tolerancji, |
|
— |
biochemiczne badania kliniczne z odpowiednimi wartościami poziomów tolerancji, |
|
— |
końcowa waga ciała, waga organów i proporcje wagi organ/ciało, |
|
— |
ustalenia sekcji zwłok, |
|
— |
szczegółowy opis wszystkich histopatologicznych wyników badań, |
|
— |
dane dotyczące absorpcji, jeśli jest to dostępne, |
|
— |
statystyczne opracowanie wyników, tam gdzie jest to stosowne. |
Omówienie wyników.
Wnioski.
B.28. BADANIE PODCHRONICZNEJ TOKSYCZNOŚCI SKÓRY – 90-DNIOWE BADANIA POWTARZANEGO DAWKOWANIA NA SKÓRZE Z WYKORZYSTANIEM GATUNKÓW GRYZONI
1. METODA
1.1. WSTĘP
Zob. Wprowadzenie ogólne część B.
1.2. DEFINICJE
Zob. Wprowadzenie ogólne część B.
1.3. SUBSTANCJE ODNIESIENIA
Brak
1.4. ZASADA METODY BADAWCZEJ
Substancja testowa jest nanoszona codziennie na skórę, w stopniowanych dawkach, kilku grupom zwierząt badanych, jedna dawka na grupę przez okres 90 dni. W okresie podawania dawki zwierzęta obserwowane są codziennie celem wykrycia objawów toksyczności. Zwierzęta, które zdechną w czasie trwania badania, poddawane są sekcji zwłok, a na zakończenie badania zwierzęta pozostałe przy życiu są zabijane i również poddawane sekcji zwłok.
1.5. KRYTERIA JAKOŚCIOWE
Brak.
1.6. OPIS METODY BADAWCZEJ
Przygotowania
Zwierzęta podlegają warunkom przebywania i żywieniowym ustalonym do celów badania przez co najmniej pięć dni przed rozpoczęciem badania. Przed rozpoczęciem badania młode zdrowe zwierzęta są losowo przydzielane do grup badanych i kontrolnych. Tuż przed rozpoczęciem badania futro zostaje ostrzyżone w części grzbietowej tułowia badanych zwierząt. Można zastosować golenie, ale powinno być przeprowadzone około 24 godziny przed rozpoczęciem badania. Ponowne strzyżenie lub golenie jest zazwyczaj potrzebne w przybliżeniu co tydzień. Podczas strzyżenia lub golenia futra należy uważać, aby nie otrzeć skóry. Nie mniej niż 10 % powierzchni skóry powinno zostać oczyszczone w celu podania substancji testowej. Należy uwzględnić wagę zwierząt w chwili podjęcia decyzji o powierzchni oczyszczanej skóry oraz o powierzchni jej pokrycia. Badając substancje stałe, które mogą być ścierane w odpowiednich przypadkach, substancja testowa powinna być odpowiednio nawilżana wodą lub, w miarę potrzeby, za pomocą odpowiedniego nośnika w celu zapewnienia dobrego kontaktu ze skórą. Płynne substancje testowe na ogół są aplikowane nierozcieńczone. Substancja aplikowana jest zasadniczo przez pięć do siedmiu dni w tygodniu.
Warunki badania
Wykorzystywać można dorosłe szczury, króliki lub świnki morskie. W chwili rozpoczęcia badania rozpiętość różnicy wagowej powinna wynosić ± 20 % wagi średniej. W przypadku przeprowadzania podchronicznych badań na skórze jako badań wstępnych do badań długoterminowych ten sam gatunek i szczep powinny być wykorzystane w obu badaniach.
Co najmniej 20 zwierząt (10 samic i 10 samców) ze zdrową skórą powinno być wykorzystanych na każdym z poziomów dawek. Samice powinny być nieciężarne i takie, które nie rodziły. Jeżeli planowane są zgony zwierząt, ich liczba powinna zostać powiększona o liczbę zwierząt planowanych do uśmiercenia przed zakończeniem badań. Ponadto grupa satelitarna w ilości 20 zwierząt (10 zwierząt każdej płci) może zostać poddana wysokiej dawce przez okres 90 dni i obserwowana pod względem odwracalności, odporności, lub opóźnionego wystąpienia toksycznych skutków przez 28 dni po przeprowadzeniu czynności badawczych.
Powinny zostać wykorzystane co najmniej trzy poziomy dawek łącznie z dawką kontrolną lub kontrolą nośnika, jeżeli jest stosowany. Okres ekspozycji na substancję powinien wynosić co najmniej sześć godzin dziennie. Substancję należy stosować o podobnych porach każdego dnia, a ilość stosowanej substancji dostosowana jest do odpowiednich odstępów czasu (tygodniowych lub dwutygodniowych), w celu utrzymania stałej dawki w odniesieniu do wagi ciała zwierząt. Oprócz stosowania substancji testowej, zwierzęta grupy kontrolnej powinny być traktowane w identyczny sposób co obiekty grupy badanej. W przypadku użycia nośnika w celu usprawnienia dawkowania grupa kontrolna przyjmuje dawki w ten sam sposób co grupy badane oraz otrzymuje tę samą ilość nośnika co ilość podawana grupie otrzymującej najwyższą dawkę. Najwyższa dawka powinna wywołać objawy toksyczności, ale nie powinna powodować lub powodować niewiele przypadków śmiertelności. Najniższa dawka nie powinna powodować żadnych objawów toksyczności. W przypadku rozważenia ekspozycji człowieka najniższa dawka powinna ją przekraczać. Najlepiej było by, gdyby średnia dawka wywoływała minimalne, dające się zaobserwować skutki toksyczności. W przypadku użycia więcej niż jednej średniej dawki należy rozszerzyć stosowane poziomy dawek w celu wywołania stopniowania skutków toksyczności. W grupach otrzymujących niskie oraz średnie dawki, oraz w grupach kontrolnych jakiekolwiek przypadki śmiertelności powinny być nieliczne w celu umożliwienia dokonania miarodajnej oceny wyników badań.
W przypadku gdy aplikowanie substancji testowej powoduje poważne podrażnienia skóry, stężenie powinno zostać obniżone, co może doprowadzić do zmniejszenia lub braku innych objawów toksyczności w grupie najwyższego dawkowania. Jeżeli skóra została poważnie uszkodzona, konieczne może okazać się zakończenie bieżących badań i podjęcie nowych badań z niższymi stężeniami.
Test graniczny
Jeżeli wstępne badanie przy dawce w wysokości 1 000 mg/kilogram lub przy wyższej dawce związanej z ewentualną ekspozycją człowieka wywołuje żadnych objawów toksyczności, dalsze badanie może być uznane za niepotrzebne.
Okres obserwacji
Zwierzęta badane powinny być codziennie obserwowane w celu wykrycia objawów toksyczności. Czas zgonu oraz czas pojawienia się i zaniku objawów toksyczności należy odnotować.
Procedura
Zwierzęta powinny zostać umieszczone w osobnych klatkach. Zwierzęta należy poddać działaniu substancji testowej najlepiej siedem dni w tygodniu, przez okres 90 dni.
Zwierzęta z grup satelitarnych przeznaczone do obserwacji uzupełniających powinny być trzymane przez dodatkowy okres 28 dni bez podawania jakichkolwiek substancji w celu stwierdzenia regeneracji lub odporności na skutki toksyczności. Czas ekspozycji powinien wynosić sześć godzin dziennie.
Substancja testowa powinna być aplikowana w sposób jednolity na powierzchni, która stanowi około 10 % ogólnej powierzchni ciała. W odniesieniu do substancji wysokotoksycznych pokryty obszar powierzchni ciała może być mniejszy ale pozostała powierzchnia powinna być pokryta tak cienką i jednolitą warstwą jak to możliwe.
W czasie ekspozycji substancja testowa wchodzi w kontakt ze skórą za pomocą porowatego opatrunku z gazy i nie drażniącej taśmy. Następnie badane miejsce powinno być w odpowiedni sposób okryte w celu utrzymania opatrunku z gazy oraz substancji testowej i zapewnienia, że zwierzę nie będzie w stanie spożyć substancji testowej. W celu zapobieżenia spożycia substancji testowej mogą zostać zastosowane środki zabezpieczające ale całkowite unieruchomienie nie jest zalecaną metodą.
Na zakończenie okresu ekspozycji, pozostała substancja testowa powinna zostać usunięta, w przypadku gdy to możliwe, za pomocą wody lub innej właściwej metody oczyszczania skóry.
Wszystkie zwierzęta powinny być obserwowane codziennie a objawy toksyczności powinny być odnotowane włączając porę ich rozpoczęcia, stopień oraz czas trwania. Obserwacje zwierząt w klatkach powinny obejmować zmiany zachodzące na ich skórze i futrze, zmiany oczu i błony śluzowej a także zmiany układu oddechowego, krążenia oraz autonomicznego i centralnego układu nerwowego, czynności somatomotorycznych oraz schematów zachowań. Powinny być dokonywane pomiary spożycia żywności oraz pomiary wagi ciała zwierząt raz w tygodniu. Regularne obserwacje zwierząt są konieczne w celu zapewnienia, że nie dochodzi do strat na zwierzętach w wyniku takich przypadków jak kanibalizm, autoliza tkanek lub nieprawidłowe umieszczenie. Na końcu okresu badania wszystkie zwierzęta pozostałe przy życiu z niesatelitarnej grupy badanej zostają zabite i poddane sekcji zwłok. Zwierzęta konające powinny być usuwane i poddawane sekcji zwłok po stwierdzeniu takiego faktu.
Wszystkie zwierzęta, włączając zwierzęta z grup kontrolnych, zwyczajowo poddawane są następującym badaniom:
|
a) |
Badanie oftalmologiczne z wykorzystaniem oftalmoskopu lub podobnego sprzętu, powinno zostać przeprowadzone przed zastosowaniem substancji testowej oraz zakończeniem badań, najlepiej na wszystkich zwierzętach, a co najmniej na zwierzętach z grupy najwyższej dawki i grupy kontrolnej. W przypadku wykrycia zmian w oczach należy przebadać wszystkie zwierzęta. |
|
b) |
Hematologia, włączając hematokryt, badanie stężenia hemoglobiny, ilości krwinek czerwonych, ogólnej ilości krwinek białych i obraz białokrwinkowy, pomiar wskaźników krzepliwości krwi takich jak czas krzepnięcia, czas protrombinowy, czas tromboplastynowy, czy ilość płytek krwi powinny zostać zbadane na końcu okresu badania. |
|
c) |
Powinno zostać przeprowadzone oznaczenie klinicznej biochemii krwi na końcu okresu badania. Obszarami zainteresowania uważanymi za właściwe dla wszystkich badań są: równowaga elektrolityczna, metabolizm węglowodanów, funkcje wątroby i nerek. Wybór szczególnych badań będzie uzależniony od obserwacji formy i działania substancji. Sugerowane ustalenia to: pomiar wapnia, fosforu, chlorku, sodu, potasu, stężenie glukozy na czczo (z okresem głodzenia stosownym do gatunku zwierzęcia), transaminaza pirogronianowo-glutaminowa surowicy (4), transaminaza szczawiowooctowoglutamylowa surowicy (5), dekarboksylazy ornityny, transpeptydaza gammaglutamylowa, mocznika azotu, albuminy, kreatyny krwi całkowitej bilirubiny oraz całkowitej surowicy białka. Inne ustalenia, które mogą być niezbędne do właściwej analizy toksykologicznej obejmują badania lipidów, hormonów, równowagi kwasowo-zasadowej, czynności metahemoglobiny i cholinoesteraza. Dodatkowa biochemia kliniczna może zostać zastosowana w miarę potrzeby w celu rozszerzenia badań odnośnie obserwowanych skutków. |
|
d) |
Analiza moczu nie jest wymagana jako badanie rutynowe, ale jedynie w przypadku spodziewanej lub obserwowanej toksyczności. |
Jeżeli dane bazowe są niewłaściwe, należy zwrócić uwagę na określenie parametrów hematologicznych i klinicznej biochemii przed rozpoczęciem dawkowania.
Wszystkie zwierzęta powinny być poddane całkowitej sekcji zwłok, która obejmuje badanie zewnętrznej powierzchni ciała, wszelkich otworów, jamy czaszkowej, klatki piersiowej i jamy brzusznej oraz ich zawartości. Wątroba, nerki, nadnercza oraz jądra powinny być zważone niezwłocznie po przeprowadzeniu sekcji w celu uniknięcia wyschnięcia. Poniższe organy i tkanki powinny być zachowane w odpowiednim pojemniku w celu ewentualnego przyszłego badania histopatologicznego: wszelkie zmiany patologiczne, mózg – włączając części rdzenia/most, móżdżek i kora mózgowa, przysadka mózgowa, tarczyca/przytarczyca, tkanki grasicy, (tchawica), płuca, serce, aorta, gruczoły ślinowe, wątroba, śledziona, nerki, nadnercza, trzustka, gruczoły płciowe, macica, dodatkowe organy genitaliów, woreczek żółciowy (jeżeli obecny), przełyk, żołądek, dwunastnica, jelito czcze, talerz, jelito ślepe, okrężnica, odbytnica, pęcherz moczowy, przedstawiciel pachowych węzłów chłonnych, (żeńskie gruczoły malene), (umięśnienie uda), nerwy obwodowe, (oczy), (mostek wraz ze szpikiem kostnym), (kość udowa, włączając powierzchnie stawowe), i (rdzeń kręgowy w trzech poziomach – szyjny, śródpiersiowy i lędźwiowy) i (oczodołowe gruczoły łzo we). Tkanki wymienione w nawiasach będą wymagały przebadania w przypadku powstania objawów toksyczności lub zaangażowania danych organów.
|
a) |
Pełna histopatologia powinna zostać przeprowadzona na normalnej i badanej skórze oraz na organach i tkankach zwierząt w grupie kontrolnej i grupie najwyższego dawkowania. |
|
b) |
Wszelkie zmiany patologiczne powinny zostać zbadane. |
|
c) |
Organy docelowe działania substancji w grupach innego dawkowania powinny zostać zbadane. |
|
d) |
W przypadku gdy wykorzystywane są szczury, płuca zwierząt w grupach niskiego i średniego dawkowania powinny zostać poddane badaniu histopatologicznemu w celu stwierdzenia dowodów infekcji, ponieważ badanie to dostarcza dogodnej analizy stanu zdrowia zwierząt. Przeprowadzanie dalszych badań histopatologicznych nie musi być wymagane jako rutynowe w odniesieniu do zwierząt w tych grupach ale zawsze musi być przeprowadzane na organach, które wykazują objawów zmian patologicznych w grupie najwyższego dawkowania. |
|
e) |
W przypadku wykorzystywania grupy satelitarnej histopatologia powinna zostać przeprowadzona na tkankach i organach zidentyfikowanych jako te wykazujące skutki w grupach poddanych badaniu. |
2. DANE
Dane powinny zostać podsumowane w formie tabeli, wykazującej liczbę zwierząt każdej badanej grupy na początku badań, liczbę zwierząt wykazujących zmiany patologiczne, rodzaj zmian oraz procent zwierząt wykazujących każdy z rodzajów zmian. Wyniki powinny zostać oszacowane właściwą metodą statystyczną. Można zastosować każdą uznaną metodę statystyczną.
3. SPRAWOZDANIE
3.1. SPRAWOZDANIE Z BADAŃ
Sprawozdanie z badań zawiera, w miarę możliwości, następujące informacje:
|
— |
gatunek lub szczep, źródło, warunki środowiska, pożywienie, |
|
— |
warunki badania, |
|
— |
dawkowanie (włączając nośnik, jeżeli został użyty) oraz jego stężenie, |
|
— |
dane dotyczące reakcji na toksyczność ze względu na płeć i dawkę, |
|
— |
brak skutków, |
|
— |
czas zgonu podczas badań lub informacja o tym, czy zwierzęta przetrwały do czasu ich zakończenia, |
|
— |
opis skutków toksyczności lub innych, |
|
— |
okres obserwacji każdego nietypowego objawu i późniejszego postępowania, |
|
— |
brak skutków, |
|
— |
dane dotyczące wagi ciała i pożywienia, |
|
— |
wyniki badań oftalmologicznych, |
|
— |
zastosowane badania hematologiczne oraz wszystkie wyniki, |
|
— |
zastosowane kliniczne badania biochemiczne oraz wszystkie wyniki (włączając wyniki wszelkich analiz moczu), |
|
— |
wyniki badań sekcji zwłok, |
|
— |
szczegółowy opis wszystkich wyników badań histopatologicznych, |
|
— |
statystyczne ujęcie wyników, jeżeli to możliwe, |
|
— |
analiza wyników, |
|
— |
interpretacja wyników. |
3.2 ANALIZA I INTERPRETACJA
Zob. Wprowadzenie ogólne część B.
4. ODNIESIENIA
Zob. Wprowadzenie ogólne część B.
B.29. BADANIE PODCHRONICZNEJ TOKSYCZNOŚCI INHALACYJNEJ – 90-DNIOWE BADANIA INHALACYJNE Z WYKORZYSTANIEM GATUNKÓW GRYZONI
1. METODA
1.1. WSTĘP
Zob. Wprowadzenie ogólne część B.
1.2. DEFINICJE
Zob. Wprowadzenie ogólne część B.
1.3. SUBSTANCJE ODNIESIENIA
Brak.
1.4. ZASADA METODY BADAWCZEJ
Kilka grup zwierząt badanych poddane jest codziennemu oddziaływaniu substancji testowej w stopniowanych stężeniach przez wyznaczony okres czasu, jeden rodzaj stężenia jest używany w danej grupie przez okres 90 dni. W przypadku wykorzystania nośnika w celu wytworzenia właściwego stężenia substancji testowej w powietrzu, należy wykorzystać grupę kontrolną do oceny nośnika. W okresie podawania dawki, zwierzęta obserwowane są codziennie celem wykrycia objawów toksyczności. Zwierzęta które padną w czasie trwania badania poddawane są sekcji zwłok, a przy zakończeniu badania, zwierzęta pozostałe przy życiu również należy zabić i poddać sekcji zwłok.
1.5. KRYTERIA JAKOŚCIOWE
Brak.
6. OPIS METODY BADAWCZEJ
1.6.1. Przygotowania
Zwierzęta podlegają warunkom przebywania i żywieniowym ustalonym do celów badania przez co najmniej 5 dni przed rozpoczęciem badania. Przed rozpoczęciem badania młode zdrowe zwierzęta są losowo przydzielane do grup badanych i kontrolnych. W miarę potrzeby do substancji testowej może zostać dodany odpowiedni nośnik celem wspomożenia wytworzenia właściwego stężenia substancji w powietrzu. W przypadku użycia jakiegokolwiek nośnika lub innych dodatków w celu usprawnienia dawkowania należy upewnić się, iż nie powodują żadnych skutków toksyczności. Dane bazowe mogą być wykorzystane stosownie do potrzeb.
1.6.2. Warunki badania
Jeżeli nie ma przeciwwskazań, preferowanym gatunkiem jest szczur. Należy zastosować powszechnie wykorzystywane laboratoryjne szczepy młodych, zdrowych zwierząt. Na początku badania różnica w wadze wykorzystywanych zwierząt nie powinna przekraczać ± 20 % odpowiedniej średniej wartości. W przypadku przeprowadzania testów podchroniczności doustnej jako wstęp do badań długoterminowych ten sam gatunek i szczep powinny być wykorzystane w obu badaniach.
Co najmniej 20 zwierząt (10 samic i 10 samców) powinno być poddanych ekspozycji każdej wielkości stężenia substancji. Samice powinny być nieciężarne i takie, które nie rodziły. Jeżeli planowane są zgony zwierząt, ich liczba powinna zostać powiększona o liczbę zwierząt planowanych do uśmiercenia przed zakończeniem badań. Ponadto grupa satelitarna w ilości 20 zwierząt (10 zwierząt każdej płci) może zostać poddana działaniu wysokiego stężenia przez okres 90 dni i obserwowana pod względem odwracalności, odporności lub opóźnionego pojawienia się toksycznych skutków, przez 28 dni po zakończeniu czynności badawczych.
Wymagane są co najmniej trzy rodzaje stężenia, dla grupy kontrolnej i w grupy kontrolnej do oceny nośnika (odpowiadające stężeniu nośnika przy najwyższej wielkości dawki), w przypadku stosowania nośnika. Z wyjątkiem zastosowania substancji testowej, zwierzęta z grupy kontrolnej powinny być traktowane w identyczny sposób co podmioty z grupy badanej. Najwyższe stężenie powinno wywołać skutki toksyczności, ale żadnych lub kilka przypadków śmiertelności. W przypadku rozważenia ekspozycji człowieka, najniższa dawka powinna ją przekraczać. Najlepiej byłoby, gdyby średnie stężenie wywoływało minimalne dające się zaobserwować skutki toksyczności. W przypadku użycia więcej niż jednego średniego stężenia stężenia powinny zostać rozszerzone w celu wywołania stopniowania skutków toksyczności. W grupach poddanych niskiemu i średniemu stężeniu oraz w grupach kontrolnych, jakiekolwiek przypadki śmiertelności powinny być nieliczne, w celu umożliwienia dokonania miarodajnej oceny wyników badań.
Dzienny czas trwania ekspozycji powinien wynosić sześć godzin po zrównoważeniu stężeń w komorach. Inne długości czasu trwania ekspozycji mogą zostać zastosowane w celu spełnienia szczególnych wymagań.
Zwierzęta powinny być badane z użyciem sprzętu inhalacyjnego zaprojektowanego w celu podtrzymania dynamicznego przepływu powietrza przy co najmniej 12 wymianach powietrza na godzinę w celu zapewnienia odpowiedniej zawartości tlenu oraz równomiernie dystrybuowanego powietrza z zawartością substancji. W przypadku stosowania komory, jej projekt powinien zminimalizować tłoczenie się badanych zwierząt i zminimalizować, przez inhalację, ich narażenie na substancję testową. Ogólnie rzecz biorąc, w celu zapewnienia stabilności komory powietrznej, ogólna liczba (objętość) badanych zwierząt nie powinna przekraczać 5 % objętości komory badawczej. Wykorzystywane mogą być komory ustno-nosowe, obejmujące cała głowę lub całe ciało; pierwsze dwie komory minimalizują pobieranie substancji innymi drogami.
Badane zwierzęta powinny być obserwowane codziennie w celu wykrycia objawów toksyczności podczas całego okresu badania oraz okresu regeneracji. Należy odnotować czas zgonu i czasu pojawienia się i zaniku objawów toksyczności.
1.6.3. Procedura
Zwierzęta poddane są działaniu substancji przez cały dzień, pięć do siedmiu dni w tygodniu, przez okres 90 dni. Zwierzęta jakiejkolwiek grupy satelitarnej przeznaczone do obserwacji uzupełniających powinny być trzymane przez dalsze 28 dni bez podawania dawek w celu stwierdzenia odwracalności lub odporności na skutki toksyczności. Wysokość temperatury, w jakiej przeprowadzane jest badanie należy utrzymywać na poziomie 22 ± 3 oC. Należy utrzymywać względną wilgotność między 30 % i 70 %, ale w niektórych przypadkach (np. badania aerozoli) stosowanie powyższego nie jest wymagane. Należy wstrzymać wydawanie pożywienia i wody w czasie ekspozycji na działanie substancji.
Konieczne jest wykorzystanie dynamicznego systemu inhalacji z odpowiednim analitycznym systemem kontroli stężenia. W celu ustalenia odpowiedniego stężenia zaleca się przeprowadzenie testu próbnego. Należy dostosować przepływ powietrza w celu zapewnienia jednolitych warunków w komorze. System powinien zapewnić jak najszybsze osiągnięcie stabilnych warunków działania czynnika.
Pomiary lub kontrole powinny obejmować:
|
a) |
tempo przepływu powietrza (stale); |
|
b) |
rzeczywiste stężenie substancji testowej mierzone w strefie oddychania. W czasie oddziaływania substancji, stężenie nie powinno ulegać wahaniom wyższym niż ± 15 % średniej wartości. Jednakże w przypadku pyłów i aerozoli niniejsza wielkość kontrolna może nie zostać osiągnięta i akceptuje się stosowanie szerszego zakresu. W czasie całego czasu trwania badań, dzienne stężenia powinny być utrzymywane na możliwie stałym poziomie. Rozwijając system wytwarzania, należy przeprowadzić analizę granulometryczną w celu ustalenia stabilności stężenia aerozoli. W czasie działania substancji należy przeprowadzać analizy tak często, jak będzie to konieczne celem ustalenia spójności dystrybucji granulometrycznej; |
|
c) |
temperatura i wilgotność; |
|
d) |
w czasie trwania działania substancji, należy systematycznie odnotować uwagi; należy prowadzić indywidualne rejestry dla każdego zwierzęcia. Wszystkie zwierzęta powinny być codziennie obserwowane a objawy toksyczności odnotowane włączając czas ich rozpoczęcia, stopień oraz czas trwania. Obserwacje zwierząt w klatkach powinny obejmować: zmiany zachodzące na ich skórze i futrze, zmiany oczu i błonie śluzowej a także zmiany układu oddechowego, krążenia oraz autonomicznego i centralnego układu nerwowego, czynności somatomotorycznych oraz schematów zachowań. Powinny być dokonywane pomiary spożycia żywności oraz pomiary wagi ciała zwierząt raz w tygodniu. Regularne obserwacje zwierząt są konieczne w celu zapewnienia, że nie występują straty w zwierzętach w wyniku takich przypadków, jak kanibalizm, autoliza tkanek lub nieprawidłowego umieszczenia. Na końcu okresu ekspozycji wszystkie zwierzęta pozostałe przy życiu zostają poddane sekcji zwłok. Zwierzęta konające powinny być usuwane i poddawane sekcji zwłok po stwierdzeniu takiego faktu. |
Wszystkie zwierzęta, włączając zwierzęta z grup kontrolnych, zwyczajowo poddawane są następującym badaniom:
|
a) |
badanie oftalmologiczne z wykorzystaniem oftalmoskopu lub podobnego sprzętu, powinny zostać wykonane przed poddaniem działaniu substancji testowej oraz zakończeniem badań, najlepiej na wszystkich zwierzętach, a co najmniej na zwierzętach z grupy najwyższej dawki i grupy kontrolnej. W przypadku wykrycia zmian w oczach należy przebadać wszystkie zwierzęta; |
|
b) |
hematologia, włączając hematokryt, stężenie hemoglobiny, badanie ilości krwinek czerwonych, całkowitej ilości krwinek białych i obraz białokrwinkowy, pomiar wskaźników krzepliwości krwi takich jak czas krzepnięcia, czas protrombinowy, czas tromboplastynowy czy ilość płytek krwi powinny zostać zbadane na końcu okresu badania; |
|
c) |
powinno zostać przeprowadzone określenie klinicznej biochemii krwi na końcu okresu badania. Obszarami uważanymi za właściwe dla wszystkich badań są: równowaga elektrolityczna, metabolizm węglowodanów, funkcje wątroby i nerek. Wybór szczególnych badań będzie uzależniony od obserwacji formy i działania substancji. Sugerowane ustalenia to: pomiar wapnia, fosforu, chlorku, sodu, potasu, stężenie glukozy na czczo (z okresem głodzenia stosownym do gatunku zwierzęcia), transaminaza pirogronianowo-glutaminowa surowicy (4), transaminaza szczawiowooctowoglutamylowa surowicy (5), dekarboksylazy ornityny, transpeptydaza gammaglutamylowa, mocznika azotu, albuminy, kreatyny krwi całkowitej bilirubiny oraz całkowitej surowicy białka. Inne ustalenia, które mogą być niezbędne do właściwej analizy toksykologicznej obejmują analizy lipidów, hormonów, równowagi kwasowo-zasadowej, czynności metahemoglobiny i cholinoesteraza. Dodatkowa biochemia kliniczna może zostać zastosowana w miarę potrzeby w celu rozszerzenia badań w zakresie obserwowanych skutków; |
|
d) |
analiza moczu nie jest wymagana jako badanie rutynowe, ale jedynie w przypadku spodziewanej lub zaobserwowanej toksyczności. |
Jeżeli dane bazowe są niewystarczające, należy rozważyć określenie parametrów hematologicznych i biochemii klinicznej przed rozpoczęciem dawkowania.
Wszystkie zwierzęta powinny być poddane sekcji zwłok, która obejmuje badanie zewnętrznej powierzchni ciała, wszelkich otworów, jamy czaszkowej, klatki piersiowej i jamy brzusznej oraz ich zawartości. Wątroba, nerki, nadnercza oraz jądra powinny być zważone niezwłocznie po przeprowadzeniu sekcji w celu uniknięcia wyschnięcia. Poniższe organy i tkanki powinny być zachowane w odpowiednim pojemniku w celu ewentualnego przyszłego badania histopatologicznego: wszystkie zmiany patologiczne, płuca, które należy usunąć w stanie nienaruszonym, należy zważyć i poddać działaniu odpowiedniego utrwalacza celem zapewnienia utrzymania struktury płuc (przepłukiwanie odpowiednim utrwalaczem uważane jest za skuteczną procedurę), tkanki nosogardzieli, mózg – włączając części rdzenia/most, móżdżek i kora mózgowa, przysadka mózgowa, tarczyca/przytarczyca, tkanki grasicy, tchawica, płuca, serce, aorta, gruczoły ślinowe, wątroba, śledziona, nerki, nadnercza, trzustka, gruczoły płciowe, macica, (dodatkowe organy genitaliów), (skóra) woreczek żółciowy (jeżeli obecny), przełyk, żołądek, dwunastnica, jelito czcze, talerz, jelito ślepe, okrężnica, odbytnica, pęcherz moczowy, przedstawiciel pachowych węzłów chłonnych, (żeńskie gruczoły malene), (umięśnienie uda), nerwy obwodowe, (oczy), mostek wraz ze szpikiem kostnym, (kość udowa, włączając powierzchnie stawowe) i (rdzeń kręgowy w trzech poziomach – szyjny, śródpiersiowy i lędźwiowy). Tkanki wymienione w nawiasach będą wymagały przebadania w przypadku powstania objawów toksyczności lub stanu organów, na które skierowane jest działanie substancji.
|
a) |
Pełna histopatologia powinna zostać przeprowadzona na drogach oddechowych oraz na organach i tkankach zwierząt w grupie kontrolnej i grupie najwyższego dawkowania. |
|
b) |
Wszelkie zmiany patologiczne powinny zostać zbadane. |
|
c) |
Organy, na które skierowane jest działanie substancji, w grupach innego dawkowania powinny zostać zbadane. |
|
d) |
Płuca zwierząt w grupach niskiego i średniego dawkowania powinny zostać poddane badaniu histopatologicznemu, ponieważ niniejsze badania dostarcza dogodnej oceny stanu zdrowia zwierząt. Przeprowadzanie dalszych badań histopatologicznych nie musi być wymagane jako rutynowe w odniesieniu do zwierząt w tych grupach, ale zawsze musi być przeprowadzane na organach, które wykazują dowody zmian patologicznych w grupie najwyższego dawkowania. |
|
e) |
W przypadku wykorzystywania grupy satelitarnej, histopatologia powinna zostać przeprowadzona na tkankach i organach zidentyfikowanych jako wykazujące zmiany patologiczne w grupach poddanych badaniu. |
2. DANE
Dane powinny zostać podsumowane w formie tabeli, wskazującej liczbę zwierząt w każdej badanej grupie na początku badań, liczbę zwierząt wykazujących zmiany patologiczne, rodzaje amin oraz odsetek zwierząt wykazujących każdy z rodzajów zmian. Wyniki powinny zostać oszacowane właściwą metodą statystyczną. Można zastosować każdą uznaną metodę statystyczną.
3. SPRAWOZDAWCZOŚĆ
3.1. SPRAWOZDANIE Z BADAŃ
Sprawozdanie z badań zawiera, w miarę możliwości, następujące informacje:
|
— |
gatunek lub szczep, źródło, |
|
— |
warunki środowiska, pożywienie, |
|
— |
warunki badania, Opis urządzeń badawczych: włączając projekt, typ, wymiary, źródło powietrza, system wytwarzania cząstek stałych zawieszonych w gazie i aerozoli, metoda klimatyzowania powietrza, oczyszczanie powietrza z komory i metoda umieszczania zwierząt w komorze badawczej w razie jej stosowania. Należy opisać sprzęt do pomiaru temperatury, wilgotności, w miarę potrzeb, stabilność stężenia aerozoli lub wielkości cząsteczki. Dane dotyczące badania: powinny zostać umieszczone w tabeli i przedstawione jako wartość średnia wraz z pomiarem zmiennej (np. odchylenie standardowe) i powinny zawierać:
|
|
— |
dane dotyczące reakcji na toksyczność ze względu na płeć i stężenie, |
|
— |
brak skutków, |
|
— |
czas zgonu podczas badań lub informacja o tym czy zwierzęta przetrwały do czasu ich zakończenia, |
|
— |
opis skutków toksyczności lub innych, |
|
— |
okres obserwacji każdego nietypowego objawu i późniejszego postępowania, |
|
— |
dane dotyczące wagi ciała i pożywienia, |
|
— |
wyniki badań oftalmologicznych, |
|
— |
zastosowane badania hematologiczne oraz wszystkie wyniki, |
|
— |
zastosowane kliniczne badania biochemiczne oraz wszystkie wyniki (włączając wyniki wszelkich analiz moczu), |
|
— |
wyniki badań sekcji zwłok, |
|
— |
szczegółowy opis wszystkich wyników badań histopatologicznych, |
|
— |
statystyczne ujęcie wyników, w odpowiednich przypadkach, |
|
— |
analiza wyników, |
|
— |
interpretacja wyników. |
3.2. ANALIZA I INTERPRETACJA
Zob. Wprowadzenie ogólne część B.
4. ODNIESIENIA
Zob. Wprowadzenie ogólne część B.
B.30. BADANIE TOKSYCZNOŚCI CHRONICZNEJ
1. METODA
1.1. WSTĘP
Zob. Wprowadzenie ogólne część B.
1.2. DEFINICJE
Zob. Wprowadzenie ogólne część B.
1.3. SUBSTANCJE ODNIESIENIA
Brak.
1.4. ZASADA METODY BADAWCZEJ
Substancja testowa podawana jest zwykle przez siedem dni w tygodniu, odpowiednią drogą, kilku grupom zwierząt badanych, jedna dawka dla danej grupy przez większą część długości ich życia. Podczas ekspozycji oraz po jej zakończeniu zwierzęta obserwowane są codziennie w celu wykrycia objawów toksyczności.
1.5. KRYTERIA JAKOŚCIOWE
Brak.
1.6. OPIS METODY BADAWCZEJ
1.6.1. Przygotowania
Zwierzęta podlegają warunkom przebywania i żywieniowym ustalonym do celów badania przez co najmniej 5 dni przed rozpoczęciem badania. Przed rozpoczęciem badania młode zdrowe zwierzęta są losowo przydzielane do grup badanych i kontrolnych.
1.6.2. Warunki badania
Gatunkiem preferowanym jest szczur.
W oparciu o wyniki wcześniej przeprowadzonych badań inne gatunki (gryzonie lub inne) mogą zostać wykorzystane. Należy zastosować powszechnie wykorzystywane laboratoryjne szczepy młodych, zdrowych zwierząt, a dawkowanie należy rozpocząć, jeżeli możliwe bezpośrednio po zakończeniu ssania.
W początkowej fazie badań różnice wagi ciała wykorzystywanych zwierząt nie powinny przekroczyć ± 20 % wartości średniej. W przypadku przeprowadzania badań podchronicznej toksyczności doustnej jako wstęp do długotrwałych badań do obu rodzajów badań należy wykorzystać ten sam gatunek/rasę i szczep.
W przypadku gryzoni co najmniej 40 zwierząt (20 samic i 20 samców) powinno być wykorzystanych do podania każdej z dawek i równolegle grupie kontrolnej. Samice powinny być nieciężarne i takie, które nie rodziły. Jeżeli planowane jest uśmiercenie zwierząt, ich liczba powinna zostać powiększona o liczbę zwierząt planowanych do uśmiercenia przed zakończeniem badań.
W przypadku gatunków innych niż gryzonie, akceptowana jest mniejsza liczba zwierząt, ale nie mniejsza niż cztery osobniki danej płci w danej grupie.
Należy zastosować co najmniej trzy poziomy dawek w uzupełnieniu do równoległej grupy kontrolnej. Najwyższa wielkość dawki powinna wywołać określone objawy toksyczności, nie powodując nadmiernej śmiertelności.
Najniższa dawka nie powinna wywoływać żadnych objawów toksyczności.
Dawka(-i) pośrednia powinna zostać ustalona na średnim poziomie między najwyższą a najniższą dawką.
Przy wyborze poziomu dawek należy uwzględnić dane wcześniejszych testów toksyczności i badań.
Należy zastosować zwykłą, dzienną częstotliwość ekspozycji. Jeżeli substancja chemiczna podawana jest w wodzie pitnej lub domieszana jest do pożywienia, powinna być dostępna w sposób ciągły.
Należy wykorzystać równoległą grupę kontrolną, która pod każdym względem odpowiada grupom badanym, z wyjątkiem poddania jej ekspozycji na substancję testową.
W szczególnych okolicznościach, takich jak badania inhalacyjne z użyciem aerozoli lub podawanych doustnie środków emulgujących o niepoznanej dotychczas aktywności biologicznej konieczne jest dołączenie równoległej kontrolnej grupy negatywnej. Zwierzęta z kontrolnej grupy negatywnej są traktowane w ten sam sposób co grupa badana, z wyjątkiem ekspozycji na substancję testową ani jakikolwiek nośnik.
Droga pokarmowa oraz oddechowa stanowią dwie główne drogi podawania substancji. Wybór drogi podawania substancji zależy od fizycznych i chemicznych właściwości substancji testowej i prawdopodobnych dróg ekspozycji człowieka.
Wybór drogi jaką jest stosowanie substancji na skórze przedstawia znaczne problemy natury praktycznej. Chroniczna toksyczność systemiczna, wynikająca z absorpcji przez skórę, może być normalnie wywnioskowana na podstawie wyników innego badania doustnego oraz na podstawie wiedzy z zakresu badań absorpcji przez skórę pochodzącej z wcześniejszych badań toksyczności.
W przypadku wchłaniania substancji z przewodu żołądkowo -jelitowego, oraz jeżeli droga przyjmowania pokarmu stanowi drogę ekspozycji człowieka, pożądaną drogą podawania substancji jest droga pokarmowa, jeżeli nie istnieją przeciwwskazania. Zwierzęta mogą otrzymywać substancję testową w pożywieniu, rozcieńczoną w wodzie pitnej lub podawaną w kapsułkach. Najlepiej stosować codzienne dawkowanie przez siedem dni w tygodniu, ponieważ dawkowanie na zasadzie pięciu dni w tygodniu może powodować odwracalność lub cofnięcie toksyczności w okresie nie podawania dawki, przez co może wpłynąć na wyniki i ich późniejszą analizę. Jednakże, głównie w oparciu o względy praktyczne, akceptowane jest dawkowanie na zasadzie dawkowania przez okres pięciu dni w tygodniu.
Ponieważ badania inhalacyjne przedstawiają techniczne problemy o większej złożoności niż podawanie substancji innymi drogami, niniejszym podaje się bardziej szczegółowe wskazówki dotyczące tego sposobu podawania substancji. Należy również zauważyć, iż wewnątrztchawiczne wkraplanie może stanowić poważną alternatywę w szczególnych sytuacjach.
Długoterminowe ekspozycje są zazwyczaj wzorowane na przewidywanej ekspozycji człowieka na działanie substancji. Zwierzęta poddawane są sześciogodzinnej ekspozycji po wyrównaniu stężeń komory, przez pięć dni w tygodniu (ekspozycja nieregularna), istotne w związku z ewentualnym wpływem środowiska, 22–24 godzin poddania działaniu substancji dziennie przez siedem dni w tygodniu (ciągłe działanie), z godzinną przerwą na karmienie zwierząt w ciągu dnia o podobnej porze i konserwację komory.
W obu przypadkach zwierzęta zazwyczaj poddane są działaniu stałych stężeń substancji testowej. Główna różnica między nieregularną a ciągłą ekspozycją polega na tym, że w okresie nieregularnej ekspozycji występuje przerwa 17–18 godzin, podczas której zwierzęta mogą się zregenerować w związku ze skutkami wywołanymi ekspozycją codziennej dawki, z jeszcze dłuższym okresem regeneracji w czasie przerwy weekendowej.
Wybór nieregularnego lub ciągłego dawkowania zależy od celów badań i symulacji sytuacji ekspozycji człowieka na działanie substancji. Jednakże należy wziąć pod uwagę niektóre trudności techniczne. Na przykład korzyści z ciągłej ekspozycji na symulowane warunki środowiska mogą być równoważone koniecznością pojenia oraz karmienia zwierząt podczas badania oraz potrzebą wytworzenia bardziej skomplikowanych (i pewnych) aerozoli i oparów oraz technik kontroli.
Zwierzęta powinny być badane w komorach inhalacyjnych zaprojektowanych w celu podtrzymania dynamicznego przepływu powietrza, przy co najmniej 12 wymianach powietrza na godzinę dla zapewnienia odpowiedniej zawartości tlenu oraz równomiernej dystrybucji powietrza z zawartością substancji. Komory kontrolne i badawcze powinny być identyczne pod względem konstrukcji i projektu w celu zapewnienia porównywalnych warunków badania pod każdym względem, z wyjątkiem ekspozycji na substancję testową. Niewielkie ujemne ciśnienie wewnątrz komory jest na ogół utrzymywane w celu zapobieżenia wystąpieniu przecieku substancji testowej do otoczenia. Komory powinny zminimalizować tłoczenie się badanych zwierząt. Ogólnie, w celu zapewnienia stabilności komory powietrznej ogólna liczba (objętość) badanych zwierząt nie powinna przekraczać 5 % objętości komory.
Pomiary lub kontrole powinny obejmować:
|
(i) |
przepływ powietrza: tempo przepływu powietrza przez komorę powinno być stale kontrolowane; |
|
(ii) |
stężenie: w czasie oddziaływania substancji stężenie nie powinno ulegać wahaniom wyższym niż ± 15 % średniej wartości; |
|
(iii) |
temperatura i wilgotność: dla gryzoni należy utrzymywać temperaturę na poziomie 22 ± 2 oC, i wilgotność w komorze 30–70 %, z wyjątkiem gdy woda używana jest w celu zawieszenia substancji testowej w powietrzu wewnątrz komory. Oba wskaźniki należy stale kontrolować; |
|
(iv) |
pomiary wielkości cząsteczki: należy ustalić rozkład wielkości cząstek w atmosferze komory włączając aerozole ciekłe i stałe. Cząsteczki aerozoli powinny posiadać wielkość możliwą do wchłonięcia przez badane zwierzęta. Próbki powietrza komory należy pobrać ze strefy oddechowej zwierząt. Próbka powietrza powinna być reprezentatywna dla warunków dystrybucji cząsteczek, działaniu, których poddane są zwierzęta i powinna obejmować, na zasadzie grawimetrycznej, całą ilość zawieszonego w powietrzu aerozolu nawet w przypadku, gdy duża ilość aerozolu nie jest wchłaniana. Należy przeprowadzać częste analizy wielkości cząsteczki w czasie rozwijania systemu wytwarzania w celu zapewnienia stabilności aerozolu, a następnie tak często, jak to konieczne podczas badań celem ustalenia odpowiedniego składu cząsteczek podlegających dystrybucji, których działaniu poddane są zwierzęta. |
Czas trwania podawania substancji powinien wynosić co najmniej 12 miesięcy.
1.6.3. Procedura
Należy przeprowadzić dokładne badania kliniczne, co najmniej raz dziennie. Należy przeprowadzać dodatkowe obserwacje każdego dnia podejmując odpowiednie działania w celu zminimalizowania strat w zwierzętach podczas badania, na przykład: sekcja zwłok lub schładzanie padłych zwierząt i odizolowywanie lub uśmiercanie słabych lub konających zwierząt. Należy przeprowadzić dokładne obserwacje w celu wykrycia początku powstawania skutków a także zminimalizowania strat w zwierzętach spowodowanych chorobą, autolizą lub kanibalizmem.
Objawy kliniczne, włączając zmiany neurologiczne i oczne, a także śmiertelność, powinny być odnotowane w odniesieniu do wszystkich zwierząt. Należy odnotować czas rozpoczęcia i kontynuację warunków toksyczności, włączając podejrzane nowotwory.
Należy odnotować wagę ciała indywidualnie dla każdego zwierzęcia raz w tygodniu podczas okresu pierwszych 13 tygodni okresu badania i następnie co najmniej raz na cztery tygodnie. Przyjmowanie pokarmu należy ustalać co tydzień w pierwszym okresie 13 tygodni po rozpoczęciu badań, a następnie w około trzymiesięcznych odstępach, jeżeli stan zdrowia lub zmiany wagi ciała wymuszą stosowanie innej procedury.
Badanie hematologiczne (np. badanie składu hemoglobiny, hematokrytu, całkowitej ilości krwinek czerwonych, całkowitej ilości krwinek białych, płytek krwi lub inne pomiary wskaźników krzepliwości krwi) należy przeprowadzić w ciągu trzech miesięcy, sześciu miesięcy, a następnie w sześciomiesięcznych odstępach i na zakończenie badań na próbkach krwi pobranych od gatunków innych niż gryzonie i od 10 szczurów/obojga płci. Jeżeli możliwe, próbki należy pobrać od tych samych szczurów w każdym przedziale. Ponadto należy pobrać próbki przed rozpoczęciem badań od gatunków innych niż gryzonie.
Jeżeli kliniczne obserwacje wskazują na pogorszenie stanu zdrowia zwierząt podczas badań, należy przeprowadzić obraz białokrwinkowy u zarażonych zwierząt.
Obraz białokrwinkowy należy przeprowadzić na próbkach pobranych od zwierząt z grupy najwyższego dawkowania i kontrolnych. Obrazy białokrwinkowe przeprowadzane są w grupach niższego dawkowania jedynie w przypadku wystąpienia dużej różnicy między grupą najwyższego dawkowania i kontrolnymi, lub w przypadku wystąpienia patologicznych wyników badań.
Należy pobrać próbki moczu od gatunków innych niż gryzonie i od 10 szczurów/płeć wszystkich grup, jeżeli możliwe od tych samych szczurów w tych samych odstępach czasu, w celu wykonania analizy hematologicznej. Należy dokonać następujących ustaleń dla poszczególnych zwierząt lub dla połączonych próbek/płeć/grupa gryzoni:
|
— |
wygląd: rozmiary i waga poszczególnych zwierząt, |
|
— |
białko, glukoza, ketony, krew utajona w kale (półilościowo), |
|
— |
badanie mikroskopowe szlamu (półilościowo). |
W sześciomiesięcznych odstępach i przy zakończeniu badania próbki krwi pobierane są od wszystkich gatunków innych niż gryzonie i od 10 szczurów/płeć wszystkich grup, w celu wykonania pomiarów chemii klinicznej, jeżeli możliwe od tych samych szczurów w każdym przedziale. Ponadto należy pobrać próbki przed rozpoczęciem badań od gatunków innych niż gryzonie. Z niniejszych próbek przygotowywana jest plazma krwi oraz należy dokonać następujących ustaleń:
|
— |
całkowite stężenie białka, |
|
— |
stężenie albuminy, |
|
— |
badanie funkcjonowania wątroby (takie jak działanie fosfatazy alkaicznej, działanie transaminazy pirogronianowo-glutaminowej (4) i czynność transaminazy szczawiowooctowoglutamylowej (5), transpeptydaza gammaglutamylowa, dekarboksylaza ornityny, |
|
— |
metabolizm węglowodanów, taki jak stężenie glukozy we krwi, |
|
— |
badanie funkcjonowania nerki, takie jak azotan mocznika krwi. |
Należy przeprowadzić sekcję zwłok na wszystkich zwierzętach, włączając zwierzęta martwe w czasie trwania badania lub te, które zostały uśmiercone po stwierdzeniu stanu konania. Przed uśmierceniem zwierząt należy pobrać próbki krwi od wszystkich zwierząt w celu przeprowadzenia obrazów białokrwinkowych. Wszelkie widoczne zmiany patologiczne, przewidywane nowotwory lub zmiany o podłożu nowotworowym należy zachować. Należy podjąć próbę skorelowania wyników całości obserwacji z wynikami badań mikroskopowych.
Wszystkie organy i tkanki należy zachować w celu przeprowadzenia badania histopatologicznego. Zazwyczaj dotyczy ono następujących organów i tkanek: mózg (6) (rdzeń/most, móżdżek, kora mózgowa), przysadka mózgowa, tarczyca (włączając przytarczycę), grasica, płuca (włączając tchawicę), serce, aorta, gruczoły ślinowe, wątroba (6), śledziona, nerki (6), nadnercza (6), przełyk, żołądek, dwunastnica, jelito czcze, talerz, jelito ślepe, okrężnica, odbytnica, macica, pęcherz moczowy, węzły chłonne, trzustka, gruczoły płciowe (6), dodatkowe organy genitaliów, żeńskie gruczoły malene, skóra, umięśnienie, nerwy obwodowe, rdzeń kręgowy (szyjny, śródpiersiowy, lędźwiowy), mostek wraz ze szpikiem kostnym kość udowa (włączając staw) i oczy. Napełnienie płuc i pęcherza moczowego w utrwalaczu jest optymalnym sposobem zachowania niniejszych tkanek; napełnienie płuc w badaniach inhalacyjnych jest istotna dla właściwego badania histopatologicznego. W badaniach szczególnych, takich jak badania inhalacyjne, należy dokonać badania całości dróg oddechowych, włączając nos, gardło i krtań.
W przypadku przeprowadzania innych badań klinicznych, otrzymane informacje należy udostępnić przed badaniem mikroskopowym, ponieważ mogą dostarczyć ważnych wskazówek patologowi.
Wszelkie widoczne zmiany, w szczególności nowotwory oraz inne zmiany patologiczne powstałe w jakimkolwiek organie powinny zostać poddane badaniu mikroskopowemu. Ponadto, zaleca się następujące procedury:
|
a) |
badanie mikroskopowe wszelkich zachowanych organów i tkanek wraz z pełnym opisem wszystkich wykrytych zmian:
|
|
b) |
organy lub tkanki wykazujące nieprawidłowości spowodowane lub prawdopodobnie spowodowane działaniem substancji testowej są również badane w grupach niskiego dawkowania; |
|
c) |
w przypadku gdy wyniki badań dostarczają dowodów na znaczne obniżenie normalnej długości życia zwierząt lub wywołują skutki, które mogą wpływać na reakcje toksyczności, należy zbadać następną, niższą wielkość dawki, jak opisano powyżej; |
|
d) |
informacje dotyczące występowania zmian patologicznych zazwyczaj występujących w szczepach wykorzystywanych zwierząt, w tych samych warunkach laboratoryjnych, tj. kontrola danych bazowych, są niezbędne w celu poprawnego oszacowania znaczenia obserwowanych zmian u badanych zwierząt. |
2. DANE
Dane nowinnv zostać nodsumowane w formie tabeli, wykazującej liczbę zwierząt każdej badanej grupy na początku badań, liczbę zwierząt wykazujących zmiany patologiczne oraz procent zwierząt wykazujących każdy z rodzajów zmian. Wyniki powinny zostać oszacowane właściwą metodą statystyczną. Można zastosować każdą uznaną metodę statystyczną.
3. SPRAWOZDANIE
3.1. SPRAWOZDANIE Z BADAŃ
Sprawozdanie z badań zawiera, w miarę możliwości, następujące informacje:
|
— |
gatunek lub szczep, |
|
— |
źródło, warunki środowiska, pożywienie, warunki badania. 3.1.1. Opis urządzeń badawczych: włączając projekt, typ, wymiary, źródło powietrza, system wytwarzania cząstek stałych zawieszonych w gazie i aerozoli, metoda klimatyzowania powietrza, oczyszczanie powietrza z komory i metoda umieszczania zwierząt w komorze badawczej w razie jej stosowania. Należy opisać sprzęt do pomiaru temperatury, wilgotności, w miarę potrzeb, stabilność stężenia aerozoli lub wielkości cząsteczki. 3.1.2. Dane dotyczące badania: powinny zostać umieszczone w tabeli i przedstawione jako wartość średnia wraz z pomiarem zmiennej (np. odchylenie standardowe) i powinny zawierać:
|
|
— |
poziomy dawek (charakter nośnika, jeżeli był użyty) i stężenia, |
|
— |
dane dotyczące reakcji na toksyczność ze względu na płeć i dawkę, |
|
— |
brak skutków, |
|
— |
czas zgonu podczas badań lub informacja o tym czy zwierzęta przetrwały do czasu ich zakończenia, |
|
— |
opis skutków toksyczności lub innych, |
|
— |
okres obserwacji każdego nietypowego objawu i późniejszego postępowania, |
|
— |
dane dotyczące wagi ciała i pożywienia, |
|
— |
wyniki badań oftalmologicznych, |
|
— |
zastosowane badania hematologiczne oraz wszystkie wyniki, |
|
— |
zastosowane kliniczne badania biochemiczne oraz wszystkie wyniki (włączając wyniki wszelkich analiz moczu), |
|
— |
wyniki badań sekcji zwłok, |
|
— |
szczegółowy opis wszystkich wyników badań histopatologicznych, |
|
— |
statystyczne ujęcie wyników, w odpowiednich przypadkach, |
|
— |
analiza wyników, |
|
— |
interpretacja wyników. |
3.2. ANALIZA I INTERPRETACJA
Zob. Wprowadzenie ogólne część B.
4. ODNIESIENIA
Zob. Wprowadzenie ogólne część B.
B.31. BADANIE PRZEDURODZENIOWEJ TOKSYCZNOŚCI ROZWOJOWEJ
1. METODA
Niniejsza metoda jest kopią OECD TG 414 (2001).
1.1. WPROWADZENIE
Opisana metoda określania toksyczności rozwojowej opracowana została w celu uzyskania ogólnych informacji dotyczących wpływu przedurodzeniowej ekspozycji na badane zwierzę ciężarne oraz na organizm rozwijający się in utero; może ona obejmować zarówno skutki dla matki, jak również śmierć, wady w budowie lub nieprawidłowości wzrostu płodu. Badanie niedoborów funkcjonalnych, chociaż są one ważnym elementem rozwoju, nie stanowi integralnej części tej metody. Mogą być badane w oddzielnym badaniu lub też jako dodatek do tego badania, przy wykorzystaniu metody badania neurotoksyczności rozwojowej. W celu uzyskania informacji o badaniu niedoborów funkcjonalnych i innych skutków postnatalnych należy zapoznać się z metodą badania toksyczności reprodukcji oraz badaniem neurotoksyczności rozwojowej jako metodami właściwymi.
W indywidualnych przypadkach niniejsza metoda badania może wymagać konkretnego dostosowania, uwzględniającego konkretną wiedzę, np. o fizykochemicznych lub toksykologicznych własnościach badanej substancji. Dostosowania takie są dopuszczalne, jeżeli przekonujące dowody naukowe świadczą, że doprowadzi ona do badania przynoszącego więcej informacji. W takim przypadku należy w sprawozdaniu z badań dokładnie udokumentować wspomniane dowody naukowe.
1.2. DEFINICJE
Toksykologia rozwojowa: badanie niekorzystnych skutków wywieranych na rozwijający się organizm, które mogą być wynikiem ekspozycji przed zapłodnieniem, podczas rozwoju przedurodzeniowego lub po urodzeniu, do okresu osiągnięcia dojrzałości płciowej. Główne przejawy toksyczności rozwojowej obejmują: 1) śmierć organizmu; 2) nieprawidłowość strukturalną; 3) zmianę we wzroście; i 4) niedobór funkcjonalny. Toksykologia rozwojowa była wcześniej często nazywana teratologią.
Niekorzystny skutek: jakakolwiek mająca związek z poddaniem badaniu zmiana, w stosunku do danych bazowych, która zmniejsza zdolność organizmu do przeżycia, rozmnażania lub przystosowania się do środowiska. W odniesieniu do najszerzej ujętej toksykologii rozwojowej obejmuje on każdy skutek, który stanowi przeszkodę dla normalnego rozwoju jaja płodowego, przed i po urodzeniu.
Zmiana we wzroście: zmiana w narządzie, masie ciała lub wielkości potomka.
Nieprawidłowości (anomalie): strukturalne nieprawidłowości w rozwoju, obejmujące zarówno wady rozwojowe jak i odchylenia (28).
Wada rozwojowa/znaczna nieprawidłowość: zmiana strukturalna uznana za szkodliwą dla zwierzęcia (może być również letalna), która zazwyczaj występuje rzadko.
Odchylenie/nieznaczna nieprawidłowość: zmiana strukturalna uznana za mało szkodliwą lub nieszkodliwą dla zwierzęcia; może być przejściowa i w populacji kontrolnej może występować względnie często.
Jajo płodowe: całość wytworów zapłodnionego jaja w każdym stadium rozwoju od zapłodnienia do urodzenia, wliczając w to błony pozazarodkowe, jak również sam zarodek lub płód.
Zagnieżdżenie: przyczepienie się blastocysty do nabłonka wyścielającego macicę, obejmujące również przeniknięcie blastocysty poprzez nabłonek macicy i jej zagłębienie się w śluzówce macicy.
Zarodek: wczesne stadium rozwojowe jakiegokolwiek organizmu, szczególnie rozwijający się produkt zapłodnienia jaja, po pojawieniu się długiej osi aż do stanu, w którym widoczne są wszystkie główne struktury.
Embriotoksyczność: szkodliwa dla normalnej budowy, rozwoju, wzrostu i/lub zdolności do życia zarodka.
Płód: nienarodzone potomstwo w okresie postembrionalnym.
Toksyczność dla płodu: szkodliwa dla normalnej budowy, rozwoju, wzrostu i/lub zdolności do życia płodu.
Poronienie: przedwczesne wydalenie z macicy produktów zapłodnienia: zarodka lub płodu niezdolnego do życia.
Resorpcja: jajo płodowe, które po zagnieżdżeniu się w macicy następnie obumarło i zostało lub jest resorbowane.
Wczesna resorpcja: ślad zagnieżdżenia bez widocznego zarodka/płodu.
Późna resorpcja: martwy zarodek lub płód z zewnętrznymi zmianami zwyrodnieniowymi.
NOAEL: poziom dawkowania, przy którym nie obserwuje się szkodliwych zmian oznacza najwyższą dawkę lub poziom ekspozycji przy którym nie obserwuje się szkodliwych zmian.
1.3. SUBSTANCJA ODNIESIENIA
Brak.
1.4. ZASADA METODY BADAWCZEJ
Substancja testowa podawana jest zwykle ciężarnym samicom co najmniej od zagnieżdżenia do ostatniego dnia przed planowanym uśmierceniem, które powinno przypadać jak najbliżej normalnej daty porodu bez ryzyka utraty danych wynikających z wczesnego porodu. Omawiana metoda nie ma na celu badania jedynie okresu organogenezy (np. dni 5–15 u gryzonia i dni 6–18 u królika), ale również skutki występujące w okresie przedimplantacyjnym, w stosownych przypadkach, przez cały okres ciąży do ostatniego dnia przed cesarskim cięciem. Na krótko przed cesarskim cięciem samice zostają uśmiercone, zawartość macic zbadana, a płody oceniane pod względem widocznych zewnętrznych nieprawidłowości i zmian w tkance miękkiej i kostnej.
1.5. OPIS METODY BADAŃ
1.5.1. Wybór gatunków zwierząt
Zaleca się prowadzenie badań na najbardziej odpowiednim gatunku oraz wykorzystanie gatunków i szczepów laboratoryjnych powszechnie używanych w badaniach toksyczności przeudrodzeniowej. Preferowanym gatunkiem gryzonia jest szczur, a spośród z gatunków innych niż gryzonie – królik. Jeżeli wykorzystuje się inny gatunek należy podać uzasadnienie.
1.5.2. Warunki przebywania i warunki żywieniowe
Temperatura w badawczych pomieszczeniach hodowlanych powinna wynosić 22 oC (± 3o) dla gryzoni i 18 oC (± 3o) dla królików. Chociaż wilgotność względna powinna wynosić co najmniej 30 % i pożądane jest, żeby nie przekraczała 70 % poza czasem czyszczenia pomieszczenia, celem powinno być utrzymywanie jej na poziomie 50–60 %. Oświetlenie powinno być sztuczne, w cyklu 12 godzin jasno, 12 godzin ciemno. Do żywienia można stosować konwencjonalne pasze laboratoryjne z nieograniczonym dostępem do wody pitnej.
Procedury krycia przeprowadza się w klatkach nadających się do tego celu. Chociaż pożądane jest trzymanie zwierząt krytych w oddzielnych klatkach, dopuszczalne jest również umieszczanie w klatkach w małych grupach.
1.5.3. Przygotowania
Należy wykorzystywać zdrowe zwierzęta aklimatyzowane do warunków laboratoryjnych co najmniej przez okres pięciu dni przed rozpoczęciem testu, niepoddawane wcześniej procedurom badawczym. Badane zwierzęta należy scharakteryzować ze względu na gatunek, szczep, źródło, płeć, wagę i/lub wiek. Na ile to praktycznie możliwe, zwierzęta we wszystkich grupach badanych powinny być jednakowej masy ciała i wieku. Do każdego poziomu dawek należy wykorzystać młode dojrzałe samice, które nie rodziły. Samice powinny być kryte samcami tego samego gatunku i szczepu, należy również unikać krycia pomiędzy rodzeństwem. U gryzoni dzień 0 danej ciąży jest określany w momencie wykrycia czopu nasienia i/lub spermy w pochwie; u królików dzień 0 określany jest zwykle w dniu kopulacji lub sztucznej inseminacji, jeśli zastosowano tę technikę. Pokryte samice należy przydzielić w sposób losowy do grup kontrolnych i badanych. Klatki powinny być ustawione w taki sposób, żeby zminimalizować możliwy wpływ położenia klatki. Zwierzęta są identyfikowane jednoznacznie. Pokryte samice należy przydzielić w sposób losowy do grup kontrolnych i badanych, zaś jeżeli samice są pokrywane w seriach, zwierzęta z każdej serii należy rozdzielić równomiernie pomiędzy grupy. Podobnie samice zainseminowane przez tego samego samca powinny być rozdzielone równomiernie pomiędzy grupy.
1.6. PROCEDURA
1.6.1. Liczba i płeć zwierząt
Każda grupa badana i kontrolna powinna składać się z wystarczającej liczby samic, aby znalazło się w nich około 20 samic z miejscami zagnieżdżenia, w czasie kiedy poddawane są sekcji. Grupy składające się z mniej niż 16 zwierząt z miejscami zagnieżdżenia mogą być niewłaściwe. Zgony matek nie zawsze powodują nieważność badania, pod warunkiem że nie przekraczają ok. 10 %.
1.6.2. Przygotowanie dawek
Jeżeli w celu usprawnienia dawkowania używa się nośnika lub innego dodatku, należy uwzględnić następujące charakterystyki: skutek wywierany na absorpcję, dystrybucję, metabolizm oraz na zatrzymywanie lub ekskrecję substancji testowej; skutki wywierane na chemiczne własności substancji testowej, które mogą zmienić jej charakterystykę toksyczną; należy również uwzględnić wpływ na pobieranie pokarmu lub wody, lub stan odżywienia zwierząt. Nośnik nie powinien wykazywać toksyczności rozwojowej ani wpływać na reprodukcję.
1.6.3. Dawkowanie
Zazwyczaj substancja testowa powinna być podawana codziennie od dnia zagnieżdżenia (np. w dniu 5 po kryciu) do dnia przed cięciem cesarskim. Jeżeli badania wstępne, jeśli są dostępne, nie wskazują na dużą możliwość wystąpienia strat przedimplantacyjnych, podawanie można rozciągnąć na cały okres ciąży, od dnia krycia do dnia poprzedzającego planowane uśmiercenie. Jest rzeczą powszechnie znaną, że niewłaściwe postępowanie ze zwierzętami lub stres podczas ciąży może powodować straty przedurodzeniowe. W celu zabezpieczenia się przed stratami przedurodzeniowymi, spowodowanymi przez czynniki niezwiązane z podawaniem dawek, należy unikać niekoniecznego niepokojenia i przenoszenia ciężarnych zwierząt, jak również powodowania stresów przez czynniki zewnętrzne, takie jak np. hałas.
Należy użyć co najmniej trzech grup dawkowania i jednoczesną grupę kontrolną. Zdrowe zwierzęta są losowo przydzielane do grupy kontrolnej i badanej. Poziomy dawkowania powinny być zróżnicowane w celu osiągnięcia stopniowania skutków toksycznych. Jeżeli charakter fizykochemiczny lub własności biologiczne substancji testowanej nie powodują ograniczenia, najwyższą dawkę należy dobrać w taki sposób, aby wywołać pewną toksyczność rozwojową i/lub toksyczność względem matek (objawy kliniczne lub spadek masy ciała, ale nie śmierć lub poważne cierpienie). Co najmniej jedna dawka pośrednia nie powinna dawać żadnych oznak toksyczności względem matek lub toksyczności rozwojowej. Należy zastosować sekwencję coraz mniejszych dawek, aby zademonstrować wszelkie reakcje związane z dawkowaniem i określić poziom dawkowania, przy którym nie obserwuje się szkodliwych zmian (NOAEL). Dwa do czterech interwałów dawkowania jest często optymalne dla ustalenia malejących poziomów dawki i dodanie czwartej grupy badanej jest często bardziej pożądane niż stosowanie bardzo dużych interwałów dawek (np. współczynnik większy od 10) między dawkowaniem. Chociaż celem jest ustalenie NOAEL u matek, badania, które nie ustalają tego poziomą, mogą być również do przyjęcia (1).
Poziomy dawek należy dobierać z uwzględnieniem wszelkich istniejących danych o toksyczności, jak również dodatkowych informacji dotyczących metabolizmu i toksykokinetyki. Tego rodzaju informacje mogą również pomóc w wykazaniu odpowiedniego doboru reżimu dawkowania.
Należy użyć równoczesnej grupy kontrolnej. Powinna to być grupa kontrolna poddawana pozorowanej terapii lub grupa kontrolna nośnika, jeżeli przy podawaniu substancji testowanej jest stosowany nośnik. Wszystkie grupy winny otrzymywać te same objętości substancji testowanej lub nośnika. Ze zwierzętami grup(-y) kontrolnych(-ej) powinno się obchodzić w sposób identyczny jak ze zwierzętami grupy badanej. Grupie kontrolnej nośnika należy podawać nośnik w największej stosowanej objętości (takiej jak w grupie badanej o najmniejszym poziomie dawki).
1.6.4. Badanie graniczne
Jeżeli badanie przy jednym poziomie dawki równym co najmniej 1 000 mg/dzień/kg masy ciała w podaniu doustnym, stosujący procedury opisane dla tego badania, nie wywołuje widocznych objawów zatrucia u zwierząt ciężarnych ani u ich potomstwa, i jeżeli nie należałoby spodziewać się toksyczności na podstawie istniejących danych (np. dotyczących związków strukturalnie pokrewnych i/lub powiązanych metabolicznie), wtedy pełne badanie przy zastosowaniu trzech poziomów dawkowania może być uznane za niepotrzebne. Przewidywana ekspozycja ludzi może wskazać na potrzebę zastosowania w badaniu granicznym większej dawki doustnej. W odniesieniu do innych dróg podawania, na przykład wziewnej lub stosowania na skórę, własności fizyczne i chemiczne substancji testowanej mogą często wskazywać i ograniczać najwyższy możliwy do osiągnięcia poziom ekspozycji (zastosowanie na skórę, na przykład, nie powinno powodować silnej toksyczności miejscowej).
1.6.5. Podawanie
Testowana substancja lub nośnik podawane są zazwyczaj doustnie przez intubację. Jeżeli zostanie zastosowana inna droga podawania, prowadzący badania powinien przedstawić uzasadnienie i racjonalną podstawę wyboru, mogą być również konieczne odpowiednie modyfikacje (2)(3)(4). Testowana substancja powinna być podawana każdego dnia o podobnym czasie.
Dawka dla danego zwierzęcia powinna zwykle opierać się na ostatnim określeniu jego masy ciała. Należy jednak zachować ostrożność, kiedy dostosowuje się dawki w ostatnim trymestrze ciąży. W doborze dawek należy wykorzystać dostępne dane aby zapobiec nadmiernej toksyczności względem matek. Jeśli jednak zostanie zaobserwowana nadmierna toksyczność u matek poddanych terapii, zwierzęta te należy humanitarnie uśmiercić. Jeżeli kilka ciężarnych zwierząt wykazuje oznaki nadmiernej toksyczności, należy rozważyć uśmiercenie zwierząt tej grupy dawkowania. Kiedy substancja podawana jest do żołądka, pożądane jest podawanie w jednorazowej dawce przy użyciu rurki lub odpowiedniej kaniuli dotchawicznej. Maksymalna objętość cieczy, która może być podana jednorazowo, zależy od wielkości badanego zwierzęcia. Objętość ta nie powinna przekraczać 1 ml/100 g masy ciała, oprócz roztworów wodnych, w przypadku których można użyć 2 ml/100 g masy ciała. W przypadku stosowania jako nośnika oleju kukurydzianego, objętość nie powinna przekraczać 0,4 ml/100 g masy ciała. Zmienność objętości w teście powinna być zminimalizowana przez dostosowanie stężenia, tak aby zapewnić stałą objętość dla wszystkich poziomów dawki.
1.6.6. Obserwacja matek
Obserwacje kliniczne powinny być przeprowadzane i odnotowywane co najmniej raz dziennie, najlepiej każdego dnia w tym samym czasie, biorąc pod uwagę szczytowy okres przewidywanych objawów po dawkowaniu. Stan zdrowia zwierząt powinien być odnotowywany, pod kątem śmiertelności, zachorowalności, odnośnych zmian zachowania i wszelkich objawów widocznej toksyczności.
1.6.7. Masa ciała i spożycie pożywienia
Zwierzęta należy zważyć w dniu 0 ciąży lub nie później niż w dniu 3 ciąży, jeżeli zwierzęta kryte w znanym czasie dostarczane są przez hodowcę z zewnątrz, następnie w pierwszym dniu dawkowania, potem co najmniej raz na trzy dni podczas okresu dawkowania oraz w dniu planowanego uśmiercenia.
Spożycie pożywienia odnotowuje się w odstępach trzydniowych i należy to robić równocześnie z ważeniem zwierząt.
1.6.8. Sekcja zwłok
Samice uśmierca się na jeden dzień przed przewidywaną datą porodu. Samice wykazujące objawy poronienia lub porodu przedwczesnego przed datą planowanego uśmiercenia należy uśmiercić i poddać dokładnemu badaniu oglądowemu.
W momencie uśmiercenia lub śmierci w okresie trwania badania matkę należy zbadać oglądowo w kierunku nieprawidłowości strukturalnych lub zmian patologicznych. W celu zminimalizowania błędu systematycznego najlepiej jest, jeżeli oceny matek podczas cięcia cesarskiego oraz następcze analizy płodu prowadzone są przez osoby nieświadome przynależności samicy do grupy poddanej terapii.
1.6.9. Analiza zawartości macicy
Bezpośrednio po uśmierceniu, lub tak szybko jak możliwe po śmierci, należy wyciąć macice i ustalić ciężarność. Macice, które wyglądają na nieciężarne należy poddać dalszemu badaniu (np. przy użyciu barwienia siarczkiem amonu w przypadku gryzoni, barwieniem Salewskiego lub inną odpowiednią metodą w przypadku królików) w celu potwierdzenia ich nieciężarnego statusu (5).
Należy zważyć ciężarne macice wraz z szyjką. Nie uzyskuje się wielkości mas ciężarnych macic zwierząt padłych w czasie badania.
U zwierząt ciężarnych należy określić liczbę ciałek żółtych.
Należy zbadać zawartość macicy pod kątem liczby obumarłych zarodków oraz liczby płodów zdolnych do życia. Należy określić stopień resorpcji w celu ustalenia względnego czasu śmierci jaja płodowego (zob. sekcja 1.2).
1.6.10. Badanie płodów
Należy określić płeć i ciężar ciała każdego płodu.
Każdy płód bada się w kierunku zmian zewnętrznych (6).
Płody należy zbadać pod kątem zmian w tkance miękkiej i kostnej (np. odchylenia, wady lub nieprawidłowości) (7)(8)(9)(10)(11)(12)(13)(14)(15)(16)(17)(18)(19)(20)(21)(22)(23)(24). Podział zmian w płodach na kategorie jest pożądany, ale nie jest wymagany. Jeżeli wprowadza się podział na kategorie, należy dokładnie zdefiniować kryteria każdej kategorii. Szczególną uwagę należy zwrócić na drogi rodne, które należy zbadać pod kątem zmian rozwojowych.
W przypadku gryzoni w przybliżeniu połowa każdego miotu powinna zostać wypreparowana i zbadana pod kątem zmian w tkance kostnej. Pozostała część miotu powinna zostać wypreparowana i oceniona pod kątem zmian w tkance miękkiej, z wykorzystaniem przyjętej i właściwej metody cięcia skrawków seryjnych lub z zastosowaniem techniki precyzyjnej sekcji w całości.
Dla zwierząt innych niż gryzonie, np. królików, należy zbadać wszystkie płody pod kątem zmian w tkance kostnej i miękkiej. Ciała tych płodów oceniane są poprzez precyzyjną sekcję, pod kątem zmian w tkance miękkiej, które może również obejmować procedury oceny wewnętrznej struktury serca (25). Głowy połowy zbadanych w ten sposób płodów odcina się i poddaje procedurze oceny pod kątem zmian w tkance miękkiej (wraz z oczami, mózgiem, przewodami nosowymi i językiem), stosując metody standardowego skrawania seryjnego (26) lub innej metody o podobnej czułości. Ciała tych płodów i pozostałych płodów niebadanych należy przygotować i zbadać pod kątem nieprawidłowości szkieletowych, stosując te same metody jak opisane dla gryzoni.
2. DANE
2.1. OPRACOWANIE WYNIKÓW
Dane należy podawać indywidualnie dla matek oraz ich potomstwa i podsumować w formie tabeli wykazującej, w odniesieniu do każdej grupy badanej i każdego pokolenia, liczbę zwierząt na początku badania, liczbę zwierząt padłych podczas badania lub uśmierconych ze względów humanitarnych, czas zgonu lub humanitarnego uśmiercenia zwierzęcia, liczbę ciężarnych samic, liczbę zwierząt wykazujących zaobserwowane oznaki toksyczności, z podaniem czasu ich pojawienia się, czasu trwania, ciężkości wszelkich skutków toksyczności, rodzajów obserwacji zarodków/płodów oraz wszelkich odnośnych danych miotów.
Wyniki liczbowe należy ocenić właściwą metodą statystyczną, przyjmując miot jako jednostkę w analizie danych. Należy zastosować uznaną metodę statystyczną; wyboru metod statystycznych należy dokonać w ramach projektowania badania i uzasadnić. W sprawozdaniu należy również umieścić dane dotyczące zwierząt, które nie przeżyły do terminu planowanego uśmiercenia. W odpowiednich przypadkach dane te mogą zostać włączone do średnich dla grup. W sprawozdaniu należy również zamieścić dane odnoszące się do zwierząt, które nie przeżyły do daty planowanego uśmiercenia. W stosownych przypadkach dane takie mogą być uwzględniane w średnich dla grup. Znaczenie danych uzyskanych z takich zwierząt, a tym samym ich uwzględnienie lub nieuwzględnienie w średniej (średnich), należy uzasadnić i ocenić indywidualnie dla każdego przypadku.
2.2. OCENA WYNIKÓW
Wyniki badania przeudrodzeniowej toksyczności rozwojowej należy ocenić w odniesieniu do obserwowanych skutków. Ocena obejmuje następujące informacje:
|
— |
wyniki badań matek, zarodków/płodów, w tym ocena związku lub jego braku pomiędzy ekspozycją zwierząt na badaną substancję oraz częstość i ciężkość wszelkich zaobserwowanych skutków, |
|
— |
kryteria zastosowane do wyróżnienia kategorii nieprawidłowości zewnętrznych, nieprawidłowości tkanki miękkiej i nieprawidłowości szkieletowych płodu, jeśli zastosowano kategoryzację, |
|
— |
w stosownych przypadkach, wcześniej uzyskane dane kontrolne wspomagające interpretację wyników badania, |
|
— |
liczby wykorzystane do obliczenia wszystkich danych procentowych lub wskaźników, |
|
— |
w stosownych przypadkach, odpowiednią analizę statystyczną obejmującą wystarczające informacje dotyczące metody analizy, tak aby niezależny recenzent/statystyk mógł dokonać powtórnej oceny i odtworzyć analizę. |
W każdym badaniu, które wykazało brak jakiegokolwiek skutku toksycznego, należy rozważyć dalsze badania absorpcji i biologicznej dostępności testowanej substancji.
2.3. INTERPRETACJA WYNIKÓW
Badanie przeudrodzeniowej toksyczności rozwojowej dostarcza informacji o skutkach powtarzanej w czasie ciąży ekspozycji matek na daną substancję, jak również o skutkach wywieranych na wewnątrzmaciczny rozwój ich potomstwa. Wyniki badania należy rozpatrywać w połączeniu z wynikami uzyskanymi w badaniach toksyczności podchronicznej, rozrodczej, badaniach toksokinetycznych i innych. Ponieważ nacisk położony jest zarówno na ogólną toksyczność w kategoriach punktów końcowych toksyczności względem matek, jak i toksyczności rozwojowej, wyniki badań pozwolą do pewnego stopnia rozróżnić skutki rozwojowe występujące przy nieobecności toksyczności ogólnej, od wywoływanych tylko na tych poziomach dawkowania, które są toksyczne również dla matki (27).
3. SPRAWOZDANIE
3.1. SPRAWOZDANIE Z BADAŃ
Sprawozdanie z badań musi zawierać następujące szczegółowe informacje:
|
|
Substancja testowana:
|
|
|
Nośnik (jeśli był użyty):
|
|
|
Zwierzęta badane:
|
|
|
Warunki badania:
|
|
|
Wyniki: |
|
|
Dane dotyczące toksycznej reakcji matek w rozbiciu na poziomy dawkowania powinny obejmować poniższe, ale nie powinny być do nich ograniczone:
|
|
|
Rozwojowe punkty końcowe w podziale na poziomy dawek, dla miotów z zagnieżdżeniami, obejmujące:
|
|
|
Rozwojowe punkty końcowe w podziale na poziomy dawek, dla miotów z żywymi płodami, obejmujące:
|
|
|
Dyskusja wyników. |
|
|
Wnioski. |
4. BIBLIOGRAFIA
|
(1) |
Kavlock R.J. et al. (1996) A Simulation Study of the Influence of Study Design on the Estimation of Benchmark Doses for Developmental Toxicity. Risk Analysis 16; 399–410. |
|
(2) |
Kimmel, C.A. and Francis, E.Z. (1990) Proceedings of the Workshop on the Acceptability and Interpretation of Dermal Developmental Toxicity Studies. Fundamental and Applied Toxicology 14; 386–398. |
|
(3) |
Wong, B.A., et al. (1997) Developing Specialized Inhalation Exposure Systems to Address Toxicological Problems. CIIT Activities 17; 1–8. |
|
(4) |
US Environmental Protection Agency (1985) Subpart E-Specific Organ/Tissue Toxicity, 40 CFR 798.4350; Inhalation Developmental Toxicity Study. |
|
(5) |
Salewski, E. (1964) Faerbermethode zum Makroskopischen Nachweis von Implantations Stellen am Uterusder Ratte. Naunyn-Schmeidebergs Archiv fur Pharmakologie undExperimentelle Pathologie 247: 367. |
|
(6) |
Edwards, J.A. (1968) The external Development of the Rabbit and Rat Embryo. In Advances in Teratology. D.H.M. Woolam (ed.) Vol. 3. Academic Press, NY. |
|
(7) |
Inouye, M. (1976) Differential Staining of Cartilage and Bone in Fetal Mouse Skeleton by Alcian Blue and Alizarin Red S. Congenital Anomalies 16; 171–173. |
|
(8) |
Igarashi, E. et al. (1992) Frequency Of Spontaneous Axial Skeletal Variations Detected by the Double Staining Techniquefor Ossified and Cartilaginous Skeleton in Rat Foetuses. Congenital Anomalies 32; 381–391. |
|
(9) |
Kimmel, C.A. et al. (1993) Skeletal Development Following Heat Exposure in the Rat. Teratology 47; 229–242. |
|
(10) |
Marr, M.C. et al. (1988) Comparison of Single and Double Staining for Evaluation of Skeletal Development: The Effects of Ethylene Glycol (EG) in CD Rats. Teratology 37; 476. |
|
(11) |
Barrow, M.V. and Taylor, W.J. (1969) A Rapid Method for Detecting Malformations in Rat Foetuses. Journal of Morphology 127:291–306. |
|
(12) |
Fritz, H. (1974) Prenatal Ossification in Rabbits ss Indicative of Foetal Maturity. Teratology 11; 313–320. |
|
(13) |
Gibson, J.P. et al. (1966) Use of the Rabbit in Teratogenicity Studies. Toxicology and Applied Pharmacology 9; 398–408. |
|
(14) |
Kimmel, C.A. and Wilson, J.G. (1973) Skeletal Deviation in Rats: Malformations or Variations? Teratology 8; 309–316. |
|
(15) |
Marr, M.C. et al. (1992) Developmental Stages of the CD (Sprague-Dawley) Rat Skeleton after Maternal Exposure to Ethylene Glycol. Teratology 46; 169–181. |
|
(16) |
Monie, I.W. et al. (1965) Dissection Procedures for Rat Foetuses Permitting Alizarin Red Staining of Skeleton and Histological Study of Viscera. Supplement to Teratology Workshop Manual; pp. 163–173. |
|
(17) |
Spark, C. and Dawson, A.B. (1928) The Order and Time of appearance of Centers of Ossification in the Fore and Hind Limbs of the Albino Rat, with Special Reference to the Possible Influence of the Sex Factor. American Journal of Anatomy 41; 411–445. |
|
(18) |
Staples, R.E. and Schnell, V.L. (1964) Refinements in Rapid Clearing Technique in the KOH-Alizarin Red S Method for Fetal Bone. Stain Technology 39; 61–63. |
|
(19) |
Strong, R.M. (1928) The Order Time and Rate of Ossification of the Albino Rat (Mus Norvegicus Albinus) Skeleton. American Journal of Anatomy 36; 313–355. |
|
(20) |
Stuckhardt, J.L. and Poppe, S.M. (1984) Fresh Visceral Examination of Rat and Rabbit Foetuses Used in Teratogenicity Testing. Teratogenesis, Carcinogenesis, and Mutagenesis 4; 181–188. |
|
(21) |
Walker, D.G. and Wirtschafter, Z.T. (1957) The Genesis of the Rat Skeleton. Thomas, Springfield, JL. L 216/234 EN Official Journal of the European Union 16.6.2004. |
|
(22) |
Wilson, J.G. (1965) Embryological Considerations in Teratology. In Teratology: Principles and Techniques, |
|
(23) |
Wilson J.G. and Warkany J. (eds). University of Chicago, Chicago, IL, pp 251–277. (23) Wilson, J.G. and Fraser, F.C. (eds). (1977) Handbook of Teratology, Vol. 4. Plenum, NY. |
|
(24) |
Varnagy, L. (1980) Use of Recent Fetal Bone Staining Techniques in the Evaluation of Pesticide Teratogenicity. Acta Vet. Acad. Sci. Hung. 28; 233–239. |
|
(25) |
Staples, R.E. (1974) Detection of visceral Alterations in Mammalian Foetuses. Teratology 9; 37–38. |
|
(26) |
Van Julsingha, E.B. and C.G. Bennett (1977) A Dissecting Procedure for the Detection of Anomalies in the Rabbit Foetal Head. In: Methods in Prenatal Toxicology Neubert, D., Merker, H.J. and Kwasigroch, T.E. (eds.). University of Chicago, Chicago, IL, pp. 126–144. |
|
(27) |
US Environmental Protection Agency (1991) Guidelines for Developmental Toxicity Risk Assessment. Federal Register 56; 63798–63826. |
|
(28) |
Wise, D.L. et al. (1997) Terminology of Developmental Abnormalities in Common Laboratory Mammals (Version 1) Teratology 55; 249–292. |
B.32. BADANIE RAKOTWÓRCZOŚCI
1. METODA
1.1. WSTĘP
Zob. Wprowadzenie ogólne część B.
1.2. DEFINICJE
Zob. Wprowadzenie ogólne część B.
1.3. SUBSTANCJE ODNIESIENIA
Brak.
1.4. ZASADA METODY BADAWCZEJ
Substancja testowa zwykle podawana jest siedem dni w tygodniu, odpowiednią drogą, kilku grupom zwierząt badanych, jedna dawka dla danej grupy przez większą część długości ich życia. W okresie podawania dawki testowej oraz po zakończeniu jej stosowania zwierzęta obserwowane są codziennie celem wykrycia objawów toksyczności, w szczególności po względem rozwoju nowotworów.
1.5. KRYTERIA JAKOŚCIOWE
Brak.
1.6. OPIS METODY BADAWCZEJ
Zwierzęta podlegają warunkom przebywania i żywieniowym ustalonym do celów badania przez co najmniej 5 dni przed rozpoczęciem badania. Przed rozpoczęciem badania młode zdrowe zwierzęta są losowo przydzielane do grup badanych i kontrolnych.
W oparciu o wyniki wcześniej przeprowadzonych badań inne gatunki (gryzonie lub inne) mogą zostać wykorzystane. Należy zastosować powszechnie wykorzystywane laboratoryjne szczepy młodych, zdrowych zwierząt, a dawkowanie należy rozpocząć, jeżeli możliwe, bezpośrednio po zakończeniu ssania.
W początkowej fazie badań, różnice wagi ciała wykorzystywanych zwierząt nie powinny przekroczyć ± 20 % wartości średniej. W przypadku przeprowadzania badań synchronicznej toksyczności drogą pokarmową jako wstęp do długotrwałych badań, do obu rodzajów badań należy wykorzystać ten sam gatunek/rasę i szczep.
W przypadku gryzoni, co najmniej 100 zwierząt (50 samic i 50 samców) powinno być wykorzystanych do podania każdej z dawek i tyle samo w grupie kontrolnej. Samice powinny być nieciężarne i takie, które nie rodziły. Jeżeli planowane jest uśmiercenie zwierząt, ich liczba powinna zostać powiększona o liczbę zwierząt planowanych do uśmiercenia przed zakończeniem badań.
Należy zastosować, co najmniej trzy poziomy dawek w uzupełnieniu do równoległej grupy kontrolnej. Najwyższy poziom dawki powinien wywołać objawy minimalnej toksyczności, takie jak nieznaczny spadek wagi ciała (mniej niż 10 %), nie wywołując znacznych zmian w normalnej długości życia zwierząt spowodowanej innymi skutkami niż wpływ nowotworów.
Najniższy poziom dawki nie powinien zakłócać normalnego wzrostu, rozwoju i długości życia zwierząt lub powodować jakiekolwiek objawy toksyczności. Ogółem, dawka ta nie powinna być niższa niż 10 % najwyższego poziomu dawki.
Dawka (dawki) pośrednia powinna zostać ustalona na średnim poziomie między najwyższą i najniższą dawką.
Przy wyborze poziomów dawek należy uwzględnić dane wcześniejszych testów toksyczności i badań.
Należy zastosować zwykłą, dzienną częstotliwość ekspozycji.
Jeżeli substancja chemiczna podawana jest w wodzie pitnej lub wmieszana jest do pożywienia, powinna być dostępna w sposób ciągły.
Należy wykorzystać równoległą grupę kontrolną, która pod każdym względem odpowiada grupom badanym, z wyjątkiem ekspozycji na substancję testową.
W szczególnych przypadkach, takich jak badania inhalacyjne z użyciem aerozoli lub podawanych doustnie środków emulgujących o niepoznanej dotychczas aktywności biologicznej, konieczne jest dołączenie grupy kontrolnej. Zwierzęta z kontrolnej grupy negatywnej nie są poddawane ani działaniu substancji testowej, ani jakiegokolwiek nośnika.
Droga pokarmowa, skórna oraz oddechowa stanowią trzy główne drogi podawania substancji. Wybór drogi podawania substancji zależy od fizycznych i chemicznych właściwości substancji testowej i prawdopodobnych dróg ekspozycji człowieka.
1.6.5.1. Badania doustne
W przypadku wchłaniania substancji z przewodu żołądkowo-jelitowego oraz jeżeli droga przyjmowania pokarmu stanowi drogę ekspozycji człowieka, pożądaną drogą podawania substancji jest droga pokarmowa, jeżeli nie istnieją przeciwwskazania. Zwierzęta mogą otrzymywać substancję testową w pożywieniu, rozcieńczoną w wodzie pitnej lub podawaną w kapsułkach.
Najlepiej stosować codzienne dawkowanie przez siedem dni w tygodniu ponieważ dawkowanie na zasadzie pięciu dni w tygodniu może powodować regenerację lub cofnięcie toksyczności w okresie niepodawania dawki przez co może wpłynąć na wyniki i ich późniejszą ocenę. Jednakże, głównie w oparciu o względy praktyczne, akceptowane jest dawkowanie zasadniczo przez okres pięciu dni w tygodniu.
1.6.5.2. Badania naskórne
Można wybrać drogę działania substancji testowej przez naniesienie na skórę w celu dokonania symulacji głównej drogi ekspozycji człowieka oraz jako modelowy system wywoływania uszkodzeń skóry.
1.6.5.3. Badania inhalacyjne
Ponieważ badania inhalacyjne przedstawiają techniczne problemy bardziej złożone niż w przypadku podawania substancji innymi drogami, niniejszym podaje się bardziej szczegółowe wskazówki dotyczące tego sposobu podawania substancji. Należy również zauważyć, że wewnątrztchawiczne wkraplanie może stanowić poważną alternatywę w szczególnych przypadkach.
Długoterminowa ekspozycja jest zazwyczaj wzorowana na przewidywanej ekspozycji ludzi na substancję. Zwierzęta poddawane są sześciogodzinnej ekspozycji po wyrównaniu stężeń w komorze, przez pięć dni w tygodniu (ekspozycja nieregularna), w związku z ewentualnym wpływem środowiskowym, 22–24 godzin poddania działaniu substancji dziennie przez siedem dni w tygodniu (ekspozycja ciągła), z godzinną przerwą na karmienie zwierząt w ciągu dnia o podobnej porze i konserwację komory. W obu przypadkach zwierzęta zazwyczaj poddane są działaniu stałych stężeń substancji testowej. Główna różnica między ekspozycją nieregularną a ciągłą polega na tym, że w okresie nieregularnej ekspozycji występuje przerwa 17–18 godzin, podczas której zwierzęta mogą się zregenerować w związku ze skutkami wywołanymi poddaniem działaniu codziennej dawki, z jeszcze dłuższym okresem regeneracji w czasie przerwy weekendowej.
Wybór nieregularnej lub ciągłej ekspozycji zależy od celów badań i symulacji sytuacji ekspozycji człowieka na substancję. Jednakże należy wziąć pod uwagę niektóre trudności techniczne. Na przykład korzyści z ekspozycji ciągłej na symulowane warunki środowiska mogą być równoważone koniecznością pojenia oraz karmienia zwierząt podczas badania, oraz potrzebą wytworzenia bardziej skomplikowanych (i pewnych) aerozoli i oparów oraz technik kontroli.
Zwierzęta powinny być poddane badaniu w komorach inhalacyjnych zaprojektowanych w celu podtrzymania dynamicznego przepływu powietrza przy co najmniej 12 wymianach powietrza na godzinę w celu zapewnienia odpowiedniej zawartości tlenu oraz równomiernej dystrybucji powietrza z zawartością substancji. Komory kontrolne i ekspozycyjne powinny być identyczne pod względem konstrukcji i projektu w celu zapewnienia porównywalnych warunków badania pod każdym względem z wyjątkiem poddania działaniu substancji testowej. Niewielkie ujemne ciśnienie wewnątrz komory jest na ogół utrzymywane w celu zapobieżenia wystąpienia przecieku substancji testowej do otoczenia. Komory powinny zminimalizować tłoczenie się badanych zwierząt. Ogólnie rzecz biorąc, w celu zapewnienia stabilności komory powietrznej ogólna liczba (objętość) badanych zwierząt nie powinna przekraczać 5 % objętości komory badawczej.
Pomiary lub kontrole powinny obejmować:
|
(i) |
przepływ powietrza: tempo przepływu powietrza przez komorę powinno być stale kontrolowane; |
|
(ii) |
stężenie: w czasie dziennego oddziaływania substancji testowej stężenie nie powinno ulegać wahaniom wyższym niż ± 15 % średniej wartości. Przez cały czas trwania badań codzienne stężenia powinny być utrzymywane na możliwie stałym poziomie; |
|
(iii) |
temperatura i wilgotność: dla gryzoni należy utrzymywać temperaturę na poziomie 22 ± 2 oC i wilgotność w komorze 30–70 %, z wyjątkiem gdy używana jest woda w celu zawieszenia substancji testowej w powietrzu wewnątrz komory. Oba wskaźniki należy stale kontrolować; |
|
(iv) |
pomiary wielkości cząsteczki: należy ustalić rozkład wielkości cząstek w atmosferze komory, włączając aerozole ciekłe i stałe. Cząsteczki aerozoli powinny posiadać wielkość możliwą do wchłonięcia przez badane zwierzęta. Próbki powietrza komory powinny zostać przeniesione do strefy oddychania zwierząt. Próbka powietrza powinna być reprezentatywna dla warunków dystrybucji cząsteczek, działaniu, którym poddane są zwierzęta, i powinna obejmować, na zasadzie grawimetrycznej, całą ilość zawieszonego w powietrzu aerozolu nawet w przypadku, gdy duża ilość aerozolu nie jest wchłaniana. Należy przeprowadzać częste analizy wielkość cząsteczki w czasie rozwijania systemu wytwarzania w celu zapewnienia stabilności aerozolu, a następnie, tak często jak to konieczne, podczas badań celem ustalenia odpowiedniego składu cząsteczek podlegających dystrybucji, których działaniu poddane są zwierzęta. |
Czas trwania badań rakotwórczości obejmuje większą część normalnej długości życia badanych zwierząt. Zakończenie testu powinno nastąpić w 18 miesiącu dla myszy i chomików oraz w 24 miesiącu dla szczurów; jednakże dla niektórych szczepów zwierząt o większej długowieczności i/lub współczynniku samorzutnej rakotwórczości zakończenie badań powinno nastąpić w 24 miesiącu dla myszy i chomików oraz w 30 miesiącu dla szczurów. Alternatywnie, zakończenie takich przedłużonych badań jest do przyjęcia, w przypadku gdy liczba osobników, które przetrwały w grupie najniższej wielkości dawki lub grupie kontrolnej, osiągnęła 25 %. W przypadku zakończenia badania, które wykazało widoczną różnicę reakcji ze względu na płeć, każdą płeć należy uwzględnić oddzielnie. Przypadek przedwczesnego zgonu jedynie osobników z grupy najwyższego dawkowania z przyczyn oczywistej toksyczności nie powoduje zakończenia badań, pod warunkiem że objawy toksyczności nie powodują problemów w innych grupach. W odniesieniu do negatywnych, ale akceptowalnych wyników badania dopuszcza się stratę nie więcej niż 10 % osobników danej grupy z powodu autolizy, kanibalizmu lub problemów z zarządzaniem, a liczba osobników wszystkich grup, które przetrwały badanie, jest nie mniejsza niż 50 % na 18 miesięcy dla myszy i chomików i 24 miesiące dla szczurów.
1.6.8. Procedura
Codzienne obserwacje zwierząt w klatkach powinny obejmować zmiany zachodzące na ich skórze i futrze, zmiany oczu i błony śluzowej, a także zmiany układu oddechowego, krążenia oraz autonomicznego i centralnego układu nerwowego, czynności somatomotorycznych oraz schematów zachowań.
Regularne obserwacje zwierząt są konieczne w celu zapewnienia, o ile to możliwe, że nie występują straty w zwierzętach w wyniku takich przypadków, jak kanibalizm, autoliza tkanek lub nieprawidłowe umieszczenie. Zwierzęta konające powinny być usuwane i poddawane sekcji zwłok po stwierdzeniu takiego faktu.
Należy odnotować objawy kliniczne oraz śmiertelność wszystkich zwierząt. Należy zwrócić specjalną uwagę na rozwój nowotworów; należy odnotować czas rozpoczęcia, umieszczenie, wymiary, wygląd oraz dalszy rozwój każdego bardzo widocznego lub ewidentnego nowotworu.
Należy dokonać cotygodniowych pomiarów spożycia pokarmu (oraz spożycia wody, w przypadku gdy substancja testowa podawana jest w wodzie pitnej) w okresie pierwszych 13 tygodni po rozpoczęciu badań i następnie w odstępach trzymiesięcznych, chyba że stan zdrowia lub zmiany wagi ciała spowodują inną procedurę.
Należy odnotować wagę ciała indywidualnie dla każdego zwierzęcia raz w tygodniu podczas okresu pierwszych 13 tygodni okresu badania i następnie co najmniej raz na cztery tygodnie.
1.6.8.2. Badania kliniczne
W przypadku gdy obserwacje wskazują na pogorszenie się stanu zdrowia zwierząt podczas badań, należy przeprowadzić obraz białokrwinkowy u takich zwierząt.
W 12 miesiącu badań, 18 miesiącu i przed uśmierceniem zwierząt należy pobrać wymaz krwi badanych zwierząt. Należy przeprowadzać obraz białokrwinkowy na próbkach pobranych od zwierząt z grupy najwyższego dawkowania i kontrolnych. Jeżeli te dane, w szczególności dane otrzymane przed uśmierceniem zwierząt, lub badania pochodzące z badania patologicznego wskazują na taką potrzebę, należy również przeprowadzić obrazy białokrwinkowe na kolejnej grupie (grupach) niższego dawkowania.
Należy przeprowadzić sekcję zwłok na wszystkich zwierzętach, włączając zwierzęta martwe w czasie trwania badania, lub te, które zostały uśmiercone po stwierdzeniu stanu konania. Wszelkie widoczne nowotwory lub zmiany patologiczne, lub spodziewane zmiany o podłożu nowotworowym należy zachować.
Następujące organy i tkanki należy przechować w odpowiednim środku w celu przeprowadzenia ewentualnych przyszłych badań histopatologicznych. Zazwyczaj dotyczy ono następujących organów i tkanek: mózg (włączając części rdzenia/mostu, móżdżek, kora mózgowa), przysadka mózgowa, tarczyca/przytarczyca, wszelkie tkanki grasicy, tchawica i płuca, serce, aorta, gruczoły ślinowe, wątroba, śledziona, nerki, nadnercza, trzustka, gruczoły płciowe, macica, dodatkowe organy genitaliów, skóra, przełyk, żołądek, dwunastnica, jelito czcze, talerz, jelito ślepe, okrężnica, odbytnica, macica, pęcherz moczowy, przedstawiciel węzłów chłonnych, żeńskie gruczoły malene, umięśnienie uda, nerwy obwodowe, mostek wraz ze szpikiem kostnym, kość udowa (włączając staw), rdzeń kręgowy w trzech poziomach (szyjny, śródpiersiowy, lędźwiowy) i oczy.
Napełnienie płuc i pęcherza moczowego w utrwalaczu jest optymalnym sposobem zachowania niniejszych tkanek; napełnienie płuc w badaniach inhalacyjnych jest istotne dla właściwego badania histopatologicznego. W badaniach inhalacyjnych należy zachować całość dróg oddechowych, włączając jamę nosową, gardło i krtań.
|
a) |
Pełna histopatologia powinna zostać przeprowadzona na organach i tkankach wszystkich padłych lub uśmierconych zwierząt w trakcie badania i wszystkich zwierząt grupy kontrolnej i grup najwyższego dawkowania. |
|
b) |
Wszelkie bardzo widoczne nowotwory lub zmiany patologiczne o przypuszczalnym pochodzeniu nowotworowym należy zbadać mikroskopowo we wszystkich grupach. |
|
c) |
W przypadku istotnej różnicy w częstotliwości występowania zmian nowotworowych w grupach najwyższego dawkowania i w grupach kontrolnych, należy przeprowadzić badania histopatologiczne na takim szczególnym organie i tkance w innych grupach badanych. |
|
d) |
Jeżeli ilość zwierząt pozostałych przy życiu w grupie najwyższego dawkowania jest znacznie niższa niż w grupie kontrolnej, wówczas należy przeprowadzić pełne badanie następnej grupy niższego dawkowania. |
|
e) |
W przypadku istnienia dowodów wywoływania toksyczności lub innych skutków, które mogą wpływać na reakcję nowotworową w grupie najwyższego dawkowania, należy przeprowadzić pełne badanie następnej grupy niższego dawkowania. |
2. DANE
Dane powinny zostać podsumowane w formie tabeli, wskazującej liczbę zwierząt każdej badanej grupy na początku badań, liczbę zwierząt wykazujących nowotwory wykryte podczas badania, czas wykrycia oraz liczbę zwierząt, u których wykryto nowotwory po uśmierceniu. Wyniki powinny zostać oszacowane właściwą metodą statystyczną. Można zastosować każdą uznaną metodę statystyczną.
3. SPRAWOZDAWCZOŚĆ
3.1. SPRAWOZDANIE Z BADAŃ
Sprawozdanie z badań zawiera, jeżeli możliwe, następujące informacje:
|
— |
gatunek lub szczep, źródło, warunki środowiska, pożywienie, |
|
— |
warunki badania: 3.1.1. Opis urządzeń ekspozycyjnych: włączając projekt, typ, wymiary, źródło powietrza, system wytwarzania cząstek stałych zawieszonych w gazie i aerozoli, metoda klimatyzowania powietrza, oczyszczanie powietrza z komory i metoda umieszczania zwierząt w komorze badawczej w razie jej stosowania. Należy opisać sprzęt do pomiaru temperatury, wilgotności, w miarę potrzeb, stabilność stężenia aerozoli lub wielkości cząsteczki. 3.1.2. Dane dotyczące badania: powinny zostać umieszczone w tabeli i przedstawione jako wartość średnia wraz z pomiarem zmiennej (np. odchylenie standardowe) i powinny zawierać:
|
|
— |
poziomy dawek (charakter nośnika, jeżeli był użyty) i stężenia, |
|
— |
dane dotyczące częstotliwości występowania nowotworu w odniesieniu do płci, dawki i rodzaju nowotworu, |
|
— |
czas zgonu podczas badań lub informacja o tym, czy zwierzęta przetrwały do czasu ich zakończenia, |
|
— |
dane dotyczące reakcji na toksyczność z względu na płeć i dawkę, |
|
— |
opis skutków toksyczności lub innych, |
|
— |
okres obserwacji każdego nietypowego objawu i późniejszego postępowania, |
|
— |
dane dotyczące wagi ciała i pożywienia, |
|
— |
zastosowane testy hematologiczne i wszystkie ich wyniki, |
|
— |
wyniki badań sekcji zwłok, |
|
— |
szczegółowy opis wszystkich wyników badań histopatologicznych, |
|
— |
statystyczne ujęcie wyników wraz z opisem stosowanych metod, |
|
— |
analiza wyników, |
|
— |
interpretacja wyników. |
3.2. ANALIZA I INTERPRETACJA
Zob. Wprowadzenie ogólne część B.
4. ODNIESIENIA
Zob. Wprowadzenie ogólne część B.
B.33. POŁĄCZONE BADANIE TOKSYCZNOŚCI CHRONICZNEJ/RAKOTWÓRCZOŚCI
1. METODA
1.1. WSTĘP
Zob. Wprowadzenie ogólne część B.
1.2. DEFINICJE
Zob. Wprowadzenie ogólne część B.
1.3. SUBSTANCJE ODNIESIENIA
Brak.
1.4. ZASADA METODY BADAWCZEJ
Celem połączonych badań toksyczności chronicznej/rakotwórczości jest ustalenie chronicznych i rakotwórczych skutków oddziaływania substancji na gatunki ssaków po przedłużonej ekspozycji.
W tym celu badanie rakotwórczości uzupełnione jest o co najmniej jedną satelitarną grupę badaną i satelitarną grupę kontrolną. Wielkość dawki stosowana w satelitarnej grupie najwyższego dawkowania może być wyższa niż dawka stosowana w grupie najwyższego dawkowania w badaniu rakotwórczości. W przypadku zwierząt poddanych badaniu rakotwórczości badana jest ogólna reakcja toksyczności, a także rakotwórczości. W przypadku zwierząt z badanej grupy satelitarnej badana jest ogólna reakcja toksyczności.
Substancja testowa podawana jest zwykle siedem dni w tygodniu, odpowiednią drogą, kilku grupom zwierząt badanych, jedna dawka dla danej grupy przez większą część ich życia. Podczas ekspozycji na substancję testową oraz po jej zakończeniu, zwierzęta obserwowane są codziennie celem wykrycia objawów toksyczności oraz rozwoju nowotworów.
1.5. KRYTERIA JAKOŚCIOWE
Brak.
1.6. OPIS METODY BADAWCZEJ
Zwierzęta podlegają warunkom przebywania i żywieniowym ustalonym do celów badania, przez co najmniej 5 dni przed rozpoczęciem badania. Przed rozpoczęciem badania, młode zdrowe zwierzęta są dobierane losowo i przydzielane do grup badanych i kontrolnych.
Gatunkiem pożądanym jest szczur. W oparciu o wyniki wcześniej przeprowadzonych badań inne gatunki (gryzonie lub inne) mogą zostać wykorzystane. Należy zastosować powszechnie wykorzystywane laboratoryjne szczepy młodych, zdrowych zwierząt, a dawkowanie należy rozpocząć, jeżeli możliwe, bezpośrednio po zakończeniu ssania.
W początkowej fazie badań różnice wagi ciała wykorzystywanych zwierząt nie powinny przekroczyć ± 20 % wartości średniej. W przypadku przeprowadzania testów podchroniczności doustnej jako wstępu do długotrwałych badań do obu rodzajów badań należy wykorzystać ten sam gatunek i rasę/szczep.
W przypadku gryzoni do podania każdej z dawek powinno być wykorzystanych co najmniej 100 zwierząt (50 samic i 50 samców) i tyle samo w grupie kontrolnej. Samice powinny być nieciężarne i takie, które nie rodziły. Jeżeli planowane jest uśmiercenie zwierząt, ich liczba powinna zostać powiększona o liczbę zwierząt planowanych do uśmiercenia przed zakończeniem badań.
Satelitarna grupa doświadczalna (grupy), badana w celu oszacowania patologii innych niż nowotwory, powinna zawierać 20 zwierząt każdej płci, podczas gdy satelitarna grupa kontrolna powinna zawierać 10 zwierząt każdej płci.
Do celów badania rakotwórczości należy zastosować co najmniej trzy poziomy dawek w uzupełnieniu do równoległej grupy kontrolnej. Najwyższy poziom dawki powinien wywołać objawy minimalnej toksyczności, takie jak nieznaczny spadek wagi ciała (mniej niż 10 %), nie wywołując znacznych zmian w normalnej długości życia zwierząt spowodowanej innymi skutkami niż wpływ nowotworów.
Najniższy poziom dawki nie powinien zakłócać normalnego wzrostu, rozwoju i długości życia zwierząt lub powodować jakiekolwiek objawów toksyczności. Ogółem, dawka ta nie powinna być niższa niż 10 % najwyższego poziomu dawki.
Dawka(-i) pośrednia powinna(-e) zostać ustalona(-e) na średnim poziomie między najwyższą i najniższą dawką.
Przy wyborze poziomów dawek należy uwzględnić dane wcześniejszych testów toksyczności i badań.
Do celów badania toksyczności chronicznej należy włączyć dodatkowe grupy doświadczalne i równoległą kontrolną grupę satelitarną. Najwyższa wielkość dawki dla satelitarnych zwierząt badanych powinna wywołać określone objawy toksyczności.
Należy zastosować zwykłą, dzienną częstotliwość ekspozycji.
Jeżeli substancja chemiczna podawana jest w wodzie pitnej lub wmieszana jest do pożywienia, powinna być dostępna w sposób ciągły.
Należy wykorzystać równoległą grupę kontrolną, która pod każdym względem odpowiada grupom badanym, z wyjątkiem ekspozycji na substancję testową.
W szczególnych okolicznościach, takich jak badania inhalacyjne obejmujące aerozole lub wykorzystujące emulgator niescharakteryzowanej aktywności biologicznej w badaniach drogą pokarmową, należy wykorzystać dodatkową grupę kontrolną bez ekspozycji na nośnik.
Droga pokarmowa, skóra oraz droga oddechowa stanowią trzy główne drogi podawania substancji. Wybór drogi podawania substancji zależy od fizycznych i chemicznych właściwości substancji testowej i prawdopodobnych dróg ekspozycji człowieka
1.6.5.1. Badania doustne
W przypadku wchłaniania substancji z przewodu żołądkowo-jelitowego oraz jeżeli droga pokarmowa stanowi drogę ekspozycji człowieka, preferowaną drogą podawania substancji jest droga pokarmowa, jeżeli nie istnieją przeciwwskazania. Zwierzęta mogą otrzymywać substancję testową w pożywieniu, rozcieńczoną w wodzie pitnej lub podawaną w kapsułkach.
Najlepiej stosować codzienne dawkowanie przez siedem dni w tygodniu, ponieważ dawkowanie na zasadzie pięciu dni w tygodniu może powodować regenerację lub cofnięcie toksyczności w okresie nie podawania dawki, co może wpłynąć na wyniki i ich późniejszą analizę. Jednakże, głównie w oparciu o względy praktyczne, akceptowane jest dawkowanie na zasadzie dawkowania przez okres pięciu dni w tygodniu.
1.6.5.2. Badania naskórne
Można wybrać drogę ekspozycji przez naniesienie na skórę w celu dokonania symulacji głównej drogi ekspozycji człowieka oraz jako modelowy system wywoływania uszkodzeń skóry.
1.6.5.3 Badania inhalacyjne
Ponieważ badania inhalacyjne przedstawiają problemy techniczne o większej złożoności niż podawanie substancji innymi drogami, niniejszym podaje się bardziej szczegółowe wskazówki dotyczące tego sposobu podawania substancji. Należy również zauważyć, iż wewnątrztchawiczne wkraplanie może stanowić poważną alternatywę w szczególnych sytuacjach.
Długoterminowe ekspozycje są zazwyczaj wzorowane na przewidywanej ekspozycji człowieka. Zwierzęta poddawane są sześciogodzinnej ekspozycji po wyrównaniu stężeń komory, przez pięć dni w tygodniu (ekspozycja nieregularna), istotne w związku z ewentualnym wpływem środowiskowym, 22–24 godzin poddania działaniu substancji dziennie przez siedem dni w tygodniu (ciągłe działanie), z godzinną przerwą na karmienie zwierząt w ciągu dnia o podobnej porze i konserwację komory. W obu przypadkach zwierzęta zazwyczaj poddane są działaniu stałych stężeń substancji testowej. Główna różnica między ekspozycją nieregularną i ciągłą polega na tym, że w okresie nieregularnej ekspozycji występuje przerwa 17–18 godzin, podczas której zwierzęta mogą się zregenerować w związku ze skutkami wywołanymi poddaniu działaniu codziennej dawki, z jeszcze dłuższym okresem regeneracji w czasie przerwy weekendowej.
Wybór ekspozycji nieregularnej lub ciągłej zależy od celów badań i symulacji sytuacji ekspozycji człowieka. Jednakże należy wziąć pod uwagę niektóre trudności techniczne. Na przykład, korzyści z ekspozycji ciągłej na symulowane warunki środowiska mogą być równoważone koniecznością pojenia oraz karmienia zwierząt podczas badania oraz potrzebą zastosowania bardziej skomplikowanych (i pewnych) aerozoli i oparów oraz technik kontroli.
Zwierzęta powinny być poddane badaniu w komorach inhalacyjnych zaprojektowanych w celu podtrzymania dynamicznego przepływu powietrza, przy co najmniej 12 wymianach powietrza na godzinę w celu zapewnienia odpowiedniej zawartości tlenu oraz równomiernej dystrybucji powietrza z zawartością substancji. Komory kontrolne i ekspozycyjne powinny być identyczne pod względem konstrukcji i projektu w celu zapewnienia porównywalnych warunków badania pod każdym względem, z wyjątkiem poddania działaniu substancji testowej. Niewielkie ujemne ciśnienie wewnątrz komory jest na ogół utrzymywane w celu zapobieżenia wystąpienia przecieku substancji testowej do otoczenia. Komory powinny zminimalizować tłoczenie się badanych zwierząt. Ogólnie rzecz biorąc, w celu zapewnienia stabilności komory powietrznej ogólna liczba (objętość) badanych zwierząt nie powinna przekraczać 5 % objętości komory badawczej.
Pomiary lub kontrole powinny obejmować:
|
(i) |
przepływ powietrza: tempo przepływu powietrza przez komorę powinno być stale kontrolowane; |
|
(ii) |
stężenie: w czasie dziennej ekspozycji na substancję testową stężenie nie powinno ulegać wahaniom wyższym niż ± 15 % średniej wartości. Podczas całego okresu trwania badań codzienne stężenia powinny być utrzymywane na możliwie stałym poziomie. |
|
(iii) |
temperatura i wilgotność: dla gryzoni należy utrzymywać temperaturę na poziomie 22 ± 2 oC i wilgotność w komorze 30–70 %, za wyjątkiem gdy używana jest woda w celu zawieszenia substancji testowej w powietrzu wewnątrz komory. Oba wskaźniki należy stale kontrolować; |
|
(iv) |
pomiary wielkości cząsteczki: należy ustalić rozkład wielkości cząstek w atmosferze komory, włączając aerozole ciekłe i stałe. Cząsteczki aerozoli powinny posiadać wielkość możliwą do wchłonięcia przez badane zwierzęta. Próbki powietrza komory powinny zostać przeniesione do strefy oddychania zwierząt. Próbka powietrza powinna być reprezentatywna dla cząsteczek podlegających dystrybucji, działaniu których poddane są zwierzęta i powinna obejmować, na zasadzie grawimetrycznej, całą ilość zawieszonego w powietrzu aerozolu, nawet w przypadku gdy duża ilość aerozolu nie jest wchłaniana. Należy przeprowadzać częste analizy wielkość cząsteczki w czasie rozwijania systemu wytwarzania w celu zapewnienia stabilności aerozolu, a następnie tak często, jak to konieczne, podczas badań dla ustalenia odpowiedniego składu cząsteczek podlegających dystrybucji, których działaniu poddane są zwierzęta. |
Czas trwania części badań dotyczących rakotwórczości obejmuje większą część normalnej długości życia badanych zwierząt. Zakończenie testu powinno nastąpić w 18 miesiącu dla myszy i chomików oraz w 24 miesiącu dla szczurów; jednakże dla niektórych szczepów zwierząt o większej długowieczności i/lub współczynniku samorzutnej rakotwórczości zakończenie badań powinno nastąpić w 24 miesiącu dla myszy i chomików oraz w 30 miesiącu dla szczurów. Alternatywnie, zakończenie takich przedłużonych badań jest do przyjęcia, w przypadku gdy liczba osobników, które przetrwały w grupie najniższej wielkości dawki lub grupie kontrolnej osiągnęła 25 %. W przypadku zakończenia badania, które wykazało widoczną różnicę reakcji ze względu na płeć, każdą płeć należy uwzględnić oddzielnie. Przypadek przedwczesnego zgonu jedynie osobników z grupy najwyższego dawkowania z przyczyn oczywistej toksyczności nie powoduje to zakończenia badań, pod warunkiem że objawy toksyczności nie stwarzają problemów w innych grupach. Odnośnie do negatywnych, ale akceptowalnych wyników badań nie więcej niż 10 % osobników danej grupy może zostać utracone z powodu autolizy, kanibalizmu lub problemów z zarządzaniem a liczba osobników wszystkich grup, które przetrwały badanie jest nie mniejsza niż 50 % przy 18 miesiącach dla myszy i chomików i 24 miesiącach dla szczurów.
Grupy satelitarne w ilości 20 osobników danej płci i 10 osobników danej płci z powiązanej grupy kontrolnej wykorzystywane do badania toksyczności chronicznej powinny być badane, przez co najmniej 12 miesięcy. Zwierzęta te powinny być przeznaczone do uśmiercenia w celu zbadania patologii związanych z działaniem substancji testowej, nieskomplikowanych zmianami genorologicznymi.
1.6.8. Procedura
Należy prowadzić codzienne obserwacje zwierząt w klatkach, które powinny obejmować zmiany zachodzące na ich skórze i futrze, zmiany oczu i błony śluzowej, a także zmiany układu oddechowego, krążenia oraz autonomicznego i centralnego układu nerwowego, czynności somatomotorycznych oraz schematów zachowań.
Badania kliniczne należy przeprowadzać w odpowiednich odstępach czasu na zwierzętach w satelitarnej grupie doświadczalnej (grupach).
Regularne obserwacje zwierząt są konieczne w celu zapewnienia, o ile to możliwe, że nie występują straty w zwierzętach w wyniku takich przypadków, jak kanibalizm, autoliza tkanek lub nieprawidłowe umieszczenie. Zwierzęta konające powinny być usuwane i poddawane sekcji zwłok po stwierdzeniu takiego faktu.
Należy odnotować objawy kliniczne, włączając zmiany neurologiczne i oczne, oraz śmiertelność wszystkich zwierząt. Należy zwrócić specjalną uwagę na rozwój nowotworów: należy odnotować czas rozpoczęcia, umieszczenie, wymiary, wygląd oraz dalszy rozwój każdego bardzo widocznego lub ewidentnego nowotworu; należy odnotować początek powstania oraz rozwój warunków toksyczności.
Należy dokonać cotygodniowych pomiarów spożycia pokarmu (oraz spożycia wody, w przypadku gdy substancja testowa podawana jest w wodzie pitnej) w okresie pierwszych 13 tygodni po rozpoczęciu badań i następnie w odstępach trzymiesięcznych, chyba że stan zdrowia lub zmiany wagi ciała wymuszą inną procedurę.
Należy odnotować wagę ciała indywidualnie dla każdego zwierzęcia raz w tygodniu podczas okresu pierwszych 13 tygodni okresu badania i następnie co najmniej raz na cztery tygodnie.
1.6.8.2. Badania kliniczne
Badanie hematologiczne (np. badanie składu hemoglobiny, hematokrytu, całkowitej ilości krwinek czerwonych, całkowitej ilości krwinek białych, płytek krwi lub inne pomiary wskaźników krzepliwości krwi) należy przeprowadzić w przeciągu trzech miesięcy, sześciu miesięcy, a następnie w sześciomiesięcznych odstępach i po zakończeniu badań na próbkach krwi pobranych od 10 szczurów/płeć wszystkich grup. Jeżeli możliwe, próbki należy pobrać od tych samych szczurów w każdym przedziale.
Jeżeli obserwacje zwierząt w klatkach wskazują na pogorszenie stanu zdrowia zwierząt podczas badań, należy przeprowadzić obraz białokrwinkowy u zarażonych zwierząt. Obraz białokrwinkowy należy przeprowadzić na próbkach pobranych od zwierząt z grupy najwyższego dawkowania i kontrolnych. Obrazy białokrwinkowe przeprowadzane są w grupie (grupach) niższego dawkowania jedynie w przypadku wystąpienia dużej różnicy między grupą najwyższego dawkowania a kontrolnymi lub w przypadku wystąpienia patologicznych wyników badań.
Należy pobrać próbki moczu od 10 szczurów/płeć wszystkich grup, jeżeli możliwe od tych samych szczurów w tych samych odstępach czasu, celem wykonania analizy hematologicznej. Należy dokonać następujących ustaleń dla poszczególnych zwierząt lub dla połączonych próbek/płeć/grupa gryzoni:
|
— |
wygląd: rozmiary i waga poszczególnych zwierząt, |
|
— |
białko, glukoza, ketony, krew utajona w kale (półilościowo), |
|
— |
badanie mikroskopowe szlamu (półilościowo). |
W sześciomiesięcznych odstępach i po zakończeniu badania próbki krwi pobierane są od wszystkich gatunków innych niż gryzonie i od 10 szczurów/płeć wszystkich grup, w celu wykonania pomiarów chemii klinicznej, jeżeli możliwe od tych samych szczurów w każdym przedziale. Ponadto należy pobrać próbki przed rozpoczęciem badań od gatunków innych niż gryzonie. Z próbek tych przygotowywana jest plazma krwi i należy dokonać następujących ustaleń:
|
— |
stężenie białka ogólnego, |
|
— |
stężenie albuminy, |
|
— |
badanie funkcjonowania wątroby (takie jak działanie fosfatazy alkaicznej, działanie transaminazy pirogronianowo-glutaminowej (4) i czynność transaminazy szczawiowooctowoglutamylowej (5), transpeptydaza gammaglutamylowa, dekarboksylaza ornityny, |
|
— |
metabolizm węglowodanów taki jak stężenie glukozy we krwi, |
|
— |
badanie funkcjonowania nerki takie jak azotan mocznika krwi. |
Należy przeprowadzić pełną sekcję zwłok na wszystkich zwierzętach, włączając zwierzęta martwe w czasie trwania badania lub te, które zostały uśmiercone po stwierdzeniu stanu konania. Przed uśmierceniem zwierząt należy pobrać próbki krwi od wszystkich zwierząt w celu przeprowadzenia obrazów białokrwinkowych. Wszelkie widoczne nowotwory lub zmiany o podłożu nowotworowym należy zachować. Należy podjąć próbę skorelowania wyników całości obserwacji z wynikami badań mikroskopowych.
Wszystkie organy i tkanki należy zachować w celu przeprowadzenia badania histopatologicznego. Zazwyczaj dotyczy ono następujących organów i tkanek: mózg (7) (rdzeń/most, móżdżek, kora mózgowa), przysadka mózgowa, tarczyca (włączając przytarczycę), grasica, płuca (włączając tchawicę), serce, aorta, gruczoły ślinowe, wątroba (7), śledziona, nerki (7), nadnercza (7), przełyk, żołądek, dwunastnica, jelito czcze, talerz, jelito ślepe, okrężnica, odbytnica, macica, pęcherz moczowy, węzły chłonne, trzustka, gruczoły płciowe (7), dodatkowe organy genitaliów, żeńskie gruczoły malene, skóra, umięśnienie, nerwy obwodowe, rdzeń kręgowy (szyjny, śródpiersiowy, lędźwiowy), mostek wraz ze szpikiem kostnym kość udowa (włączając staw) i oczy.
Chociaż napełnienie płuc i pęcherza moczowego w utrwalaczu jest optymalnym sposobem zachowania niniejszych tkanek, napełnienie płuc w badaniach inhalacyjnych jest wymogiem koniecznym dla właściwego badania histopatologicznego. W badaniach szczególnych, takich jak badania inhalacyjne, należy dokonać badania całości dróg oddechowych, włączając nos, gardło i krtań.
W przypadku przeprowadzania innych badań klinicznych otrzymane informacje należy udostępnić przed badaniem mikroskopowym, ponieważ mogą dostarczyć ważnych wskazówek patologowi.
W odniesieniu do części badań dotyczących toksyczności chronicznej:
Należy przeprowadzić szczegółowe badania wszystkich zachowanych organów wszystkich zwierząt z satelitarnej grupy najwyższego dawkowania i z grup kontrolnych. W przypadku wykrycia patologii związanej z działaniem substancji testowej w satelitarnej grupie najwyższego dawkowania dane organy wszystkich zwierząt z wszystkich satelitarnych grup doświadczalnych powinny zostać poddane pełnemu i szczegółowemu badaniu histologicznemu, a także organy zwierząt grup doświadczalnych w części badań dotyczących rakotwórczości w chwili ich zakończenia.
W odniesieniu do części badań dotyczących rakotwórczości:
|
a) |
pełna histopatologia powinna zostać przeprowadzona na organach i tkankach wszystkich padłych lub uśmierconych zwierząt w trakcie trwania badania i wszystkich zwierząt w grupie kontrolnej i grupach najwyższego dawkowania; |
|
b) |
wszelkie bardzo widoczne nowotwory lub zmiany patologiczne o przypuszczalnym podłożu nowotworowym pojawiające się w jakimkolwiek organie należy zbadać mikroskopowo we wszystkich grupach; |
|
c) |
w przypadku istotnej różnicy w częstotliwości występowania zmian nowotworowych w grupach najwyższego dawkowania i w grupach kontrolnych, należy przeprowadzić badania histopatologiczne na danym szczególnym organie i tkance w innych grupach badanych; |
|
d) |
jeżeli ilość zwierząt pozostałych przy życiu w grupie najwyższego dawkowania jest znacznie niższa niż w grupie kontrolnej, wówczas należy przeprowadzić pełne badanie następnej grupy niższego dawkowania; |
|
e) |
w przypadku istnienia dowodów wywołania toksyczności lub innych skutków, które mogą wpływać na reakcję nowotworową w grupie najwyższego dawkowania, należy przeprowadzić pełne badanie następnej grupy niższego dawkowania. |
2. DANE
Dane powinny zostać podsumowane w formie tabeli, wskazującej liczbę zwierząt każdej badanej grupy na początku badań, liczbę zwierząt wykazujących zmiany nowotworowe lub skutki toksyczności wykryte podczas badania, czas wykrycia oraz liczbę zwierząt, u których wykryto nowotwory po dokonaniu uśmiercenia. Wyniki powinny zostać oszacowane właściwą metodą statystyczną. Można zastosować każdą uznaną metodę statystyczną.
3. SPRAWOZDAWCZOŚĆ
3.1. SPRAWOZDANIE Z BADAŃ
Sprawozdanie z badań zawiera, jeżeli możliwe, następujące informacje:
|
— |
gatunek lub szczep, źródło, warunki środowiska, |
|
— |
pożywienie, warunki badania: 3.1.1. Opis urządzeń badawczych: włączając projekt, typ, wymiary, źródło powietrza, system wytwarzania cząstek stałych zawieszonych w gazie i aerozoli, metoda klimatyzowania powietrza, oczyszczanie powietrza z komory i metoda umieszczania zwierząt w komorze badawczej w razie jej stosowania. Należy opisać sprzęt do pomiaru temperatury, wilgotności, w miarę potrzeb, stabilność stężenia aerozoli lub wielkości cząsteczki. 3.1.2. Dane dotyczące badania: Powinny zostać umieszczone w tabeli i przedstawione jako wartość średnia wraz z pomiarem zmiennej (np. odchylenie standardowe) i powinny zawierać:
|
|
— |
poziomy dawek (charakter nośnika, jeżeli był użyty) i stężenia, |
|
— |
dane dotyczące częstotliwości występowania nowotworu w odniesieniu do płci, dawki i rodzaju nowotworu, |
|
— |
czas zgonu podczas badań lub informacja o tym czy zwierzęta przetrwały do czasu ich zakończenia, włączając grupę satelitarną, |
|
— |
dane dotyczące reakcji na toksyczność ze względu na płeć i dawkę, |
|
— |
opis skutków toksyczności lub innych, |
|
— |
czas obserwacji każdego nietypowego objawu i późniejszego postępowania, |
|
— |
wyniki badań oftalmologicznych, |
|
— |
dane dotyczące wagi ciała i pożywienia, |
|
— |
zastosowane testy hematologiczne i wszystkie ich wyniki, |
|
— |
zastosowane badanie biochemii klinicznej i wszystkie wyniki (włączając wszelką analizę moczu), |
|
— |
wyniki badań sekcji zwłok, |
|
— |
szczegółowy opis wszystkich wyników badań histopatologicznych, |
|
— |
statystyczne ujęcie wyników wraz z opisem stosowanych metod, |
|
— |
analiza wyników, |
|
— |
interpretacja wyników. |
3.2. ANALIZA I INTERPRETACJA
Zob. Wprowadzenie ogólne część B.
4. ODNIESIENIA
Zob. Wprowadzenie ogólne część B.
B.34. BADANIE TOKSYCZNOŚCI REPRODUKCJI JEDNEGO POKOLENIA
1. METODA
1.1. WSTĘP
Zob. Wprowadzenie ogólne część B.
1.2. DEFINICJE
Zob. Wprowadzenie ogólne część B.
1.3. SUBSTANCJE ODNIESIENIA
Brak.
1.4. ZASADA METODY BADAWCZEJ
Substancja testowa podawana jest w stopniowanych dawkach kilku grupom samców i samic. Samcom należy podawać dawkę w okresie wzrostu i przez co najmniej jeden pełny cykl spermatogenezy (około 56 dni u myszy i 70 dni u szczura), w celu wywołania przez substancję testową wszelkich negatywnych skutków na spermatogenezie.
Samice z pokolenia rodziców P powinny być poddane dawkowaniu przez co najmniej dwa pełne cykle rujowe w celu wywołania przez substancję testową wszelkich negatywnych skutków na cykl rajowy. Następnie zwierzęta zostają pokryte. Substancja testowa jest podawana obu płciom w okresie krycia a następnie jedynie samicom w okresie ciąży oraz przez okres ssania. W celu podania substancji przez drogi oddechowe, metoda będzie wymagać modyfikacji.
1.5. KRYTERIA JAKOŚCIOWE
Brak.
1.6. OPIS METODY BADAWCZEJ
1.6.1. Przygotowania
Przed rozpoczęciem badania młode zdrowe zwierzęta są losowo przydzielane do grup badanych i kontrolnych. Zwierzęta podlegają warunkom przebywania i żywieniowym ustalonym do celów badania, przez co najmniej 5 dni przed rozpoczęciem badania. Zalecane jest podawanie substancji testowej w pożywieniu lub w wodzie pitnej. Akceptowane są również inne drogi podawania substancji testowej. Ta sama metoda podawania substancji testowej powinna być stosowana do wszystkich zwierząt w trakcie odpowiedniego okresu badania. W przypadku użycia jakiegokolwiek nośnika lub innych dodatków w celu usprawnienia dawkowania należy upewnić się, iż nie powodują żadnych skutków toksyczności.
1.6.2. Zwierzęta badane
Wybór gatunków Pożądanymi gatunkami są szczur i mysz. Należy wykorzystać zdrowe zwierzęta, które nie były wcześniej poddawane procedurom badawczym. Nie należy stosować szczepów o niskiej płodności. Badane zwierzęta należy scharakteryzować ze względu na gatunek, szczep, płeć, wagę i/lub wiek.
W celu odpowiedniego oszacowania płodności, należy poddać badaniu zarówno samice, jak i samce. Wszystkie zwierzęta badane i kontrolne należy odstawić od ssania przed rozpoczęciem dawkowania.
Liczba i płeć
Każda grupa doświadczalna i kontrolna powinna składać się z takiej liczby zwierząt, aby znalazło się w nich około 20 samic pod koniec ciąży.
Celem jest wyprodukowanie odpowiedniej liczby ciąż i potomstwa w celu zapewnienia znaczącej oceny wpływu substancji na płodność, ciążę i zachowanie macierzyńskie w pokoleniu zwierząt rodziców P i zwierząt ssących, wzrost i rozwój potomstwa F1 w okresie od chwili poczęcia do zakończenia ssania.
Pożywienie i woda powinny być dostępne bez ograniczeń. Tuż przed porodem ciężarne samice powinny być umieszczone w klatkach porodowych lub matczynych zaopatrzonych w materiał do zrobienia gniazda.
Należy wykorzystać co najmniej trzy grupy badane i grupę kontrolną. W przypadku użycia nośnika w podawaniu substancji testowej, grupie kontrolnej należy podać nośnik o najwyższej wielkości dawki. W przypadku gdy substancja testowa zmniejsza pobieranie lub wykorzystanie pożywienia, może być niezbędne dołączenie odpowiednio żywionej grupy kontrolnej. Jeżeli dawka nie jest ograniczona fizyczno-chemicznymi lub biologicznymi właściwościami substancji, najwyższa wielkość dawki powinna wywoływać toksyczność, ale nie śmiertelność w grupie zwierząt rodziców P. Średnia dawka powinna wywoływać minimalne skutki toksyczności przypisane działaniu substancji testowej, a najniższa dawka nie powinna wywoływać żadnych niekorzystnych skutków u rodziców lub potomstwa. W przypadku substancji podawanej za pomocą dozownika lub w kapsułce dawka podawana każdemu zwierzęciu powinna być oparta na indywidualnej wadze ciała zwierzęcia i dostosowywana raz w tygodniu z uwzględnieniem zmian wagi ciała zwierzęcia. Poziom dawkowania dla samic w okresie ciąży może być oparty na wadze ciała w dniu 0 lub 6 dniu ciąży, w razie potrzeby.
W przypadku substancji o niskiej toksyczności, jeżeli poziom dawki wynoszący, co najmniej 1 000 mg/kilogram nie powoduje żadnego szkodliwego wpływu na zdolność reprodukcyjną, badania przy zastosowaniu innych poziomów dawek mogą być uznane za niekonieczne. Jeżeli badania wstępne przy wysokim poziomie dawki, z wyraźnymi dowodami toksyczności matki, nie wykazują żadnego szkodliwego wpływu na płodność, badania przy zastosowaniu innych poziomów dawek mogą być uznane za niekonieczne.
1.6.3.3. Przeprowadzenie badania
Codzienne stosowanie dawki u rodzicielskich samców P powinno rozpocząć się po przekroczeniu od pięciu do dziewięciu tygodni życia po zaprzestaniu ssania i aklimatyzacji przez okres co najmniej pięciu dni. W przypadku szczurów dawkowanie odbywa się przez okres 10 tygodni przed okresem krycia (w przypadku myszy – osiem tygodni). Samce należy uśmiercić i zbadać na końcu okresu krycia albo, alternatywnie, mogą być zachowane, podając im pożywienie wykorzystywane w badaniu w celu ewentualnej produkcji drugiego miotu, a następnie powinny być uśmiercone i zbadane w okresie poprzedzającym zakończenie badań. W przypadku rodzicielskich samic P dawkowanie powinno rozpocząć się po co najmniej pięciodniowym okresie aklimatyzacji i kontynuowane przez okres co najmniej dwóch tygodni przed rozpoczęciem krycia. Codzienne dawkowanie rodzicielskich samic P powinno być kontynuowane przez trzytygodniowy okres krycia, ciąży aż do czasu zaprzestania ssania potomstwa F1. Należy rozważyć zmodyfikowanie zasad dawkowania przy uwzględnieniu dostępnych informacji na temat substancji testowej, w szczególności pobudzania przez nią metabolizmu lub bioakumulacji.
W badaniach toksyczności reprodukcji można zastosować procedurę krycia zarówno 1:1 (jeden samiec i jedna samica), jak i 1:2 (jeden samiec i dwie samice).
W oparciu o procedurę krycia 1:1 jedną samicę należy umieścić z tym samym samcem aż do momentu pojawienia się ciąży lub przez okres trzech tygodni. Każdego dnia rano, samice należy poddać badaniu na obecność spermy lub czopów nasienia w pochwie. Dzień 0 danej ciąży jest określany w chwili wykrycia spermy lub czopu nasienia w pochwie.
Należy ustalić przyczynę wyraźnego braku płodności w odniesieniu do par, które nie zainicjują krycia.
Powyższe może obejmować takie procedury, jak ponowne, dodatkowe krycie z reproduktorami matek, mikroskopowe badanie organów rozrodczych oraz badania cyklu rujowego i cyklu spermatogenezy.
Zwierzęta poddawane dawkowaniu w okresie badań płodności są dopuszczane do miotu i wychowują swoje potomstwo do chwili zaprzestania ssania bez normalizacji miotów.
W przypadku dokonania normalizacji zaleca się następującą procedurę. Między 1 a 4 dniem po porodzie rozmiar każdego miotu może zostać dostosowany przez wyeliminowanie dodatkowych młodych w miocie, na tyle na ile to możliwe, cztery samice i cztery samce na miot.
W każdym przypadku gdy liczba młodych samców lub samic uniemożliwia zachowanie czterech osobników danej płci w każdym miocie, akceptowane jest częściowe dostosowanie (np. pięć samców i trzy samice). Nie należy przeprowadzać dostosowania w odniesieniu do miotów o liczbie osobników mniejszej niż osiem.
Przez cały okres badań każde zwierzę powinno być obserwowane co najmniej raz dziennie. Należy odnotować zmiany w zachowaniu mające związek z badaniami, objawy trudnego lub długotrwałego porodu oraz wszelkie objawy toksyczności, włączając śmiertelność. W okresie poprzedzającym krycie oraz w okresach krycia, spożycie żywności może być kontrolowane codziennie. Po porodzie oraz podczas okresu laktacji pomiary spożycia żywności (i pomiary spożycia wody, w przypadku gdy substancja testowa podawana jest w wodzie pitnej) należy sporządzać w tym samym dniu co ważenie miotu. Rodzicielskie samce i samice P należy ważyć w pierwszym dniu rozpoczęcia dawkowania i następnie w odstępach tygodniowych. Niniejsze uwagi należy odnotować indywidualnie dla każdego dorosłego zwierzęcia.
Czas trwania ciąży należy obliczać, począwszy od dnia 0. Każdy miot należy zbadać, jeżeli możliwe, natychmiast po porodzie celem ustalenia liczby oraz płci młodych, liczby martwo urodzonych, żywo urodzonych oraz obecność rażących nieprawidłowości.
Martwe młode oraz młode uśmiercone w 4 dniu należy zachować celem wykonania badań odnośnie do ewentualnych wad. Należy przeliczyć żywe młode i zważyć mioty nazajutrz po porodzie oraz w 4 i 7 dniu, a następnie w odstępach tygodniowych aż do czasu zakończenia badań, gdzie zwierzęta należy zbadać indywidualnie.
Należy odnotować obserwowalne nieprawidłowości fizyczne lub dotyczące zachowania u matek lub potomstwa.
1.6.5. Patologia
W chwili uśmiercenia lub zgonu zwierzęcia podczas badania zwierzęta pokolenia rodziców P powinny zostać poddane badaniom mikroskopowym w celu wykrycia wszelkich strukturalnych nieprawidłowości lub zmian patologicznych, ze zwróceniem szczególnej uwagi na organy systemu rozrodczego. Należy zbadać martwe lub konające młode w celu wykrycia uszkodzeń.
Jajniki, macica, szyjka macicy, pochwa, jądra, najądrza, kanaliki nasienne, gruczoł krokowy, gruczoł opuszkowo-cewkowy, przysadka mózgowa i organ (organy) docelowe wszystkich zwierząt P należy zachować celem poddania badaniu mikroskopowemu. W przypadku gdy niniejsze organy nie zostały przebadane w trakcie innych badań z wykorzystaniem wielu poziomy dawek, należy przeprowadzić ich badanie u wszystkich zwierząt grupy najwyższego dawkowania i kontrolnej, które padły w trakcie badań, w takim stopniu w jakim jest to możliwe.
Organy takich zwierząt, wykazujące objawy nieprawidłowości, powinny zostać zbadane u wszystkich zwierząt P. W tych przypadkach badanie mikroskopowe należy przeprowadzić na wszystkich tkankach wykazujących zmiany patologiczne. Jak sugerowano zgodnie z procedurami krycia, organy rozrodcze zwierząt podejrzanych o brak płodności mogą być poddane badaniu mikroskopowemu.
2. DANE
Dane powinny zostać podsumowane w formie tabeli, wskazującej liczbę zwierząt każdej badanej grupy na początku badań, liczbę płodnych samców, liczbę ciężarnych samic, rodzaje zmian i procent zwierząt wykazujących każdy rodzaj zmian.
Jeżeli możliwe, wyniki numeryczne powinny zostać oszacowane właściwą metodą statystyczną. Można zastosować każdą uznaną metodę statystyczną.
3. SPRAWOZDANIE
3.1. SPRAWOZDANIE Z BADAŃ
Sprawozdanie z badań zawiera, jeżeli możliwe, następujące informacje:
|
— |
wykorzystany gatunek/szczep, |
|
— |
dane dotyczące reakcji na toksyczność ze względu na płeć i dawkę, włączając płodność, ciążę i zdolność utrzymania się przy życiu, |
|
— |
czas zgonu podczas badań lub informacja o tym, czy zwierzęta przetrwały do czasu planowanego uśmiercenia lub zakończenia badań, |
|
— |
tabela przedstawiająca wagę każdego miotu, średnią wagę młodych i wagę poszczególnych młodych przy zakończeniu badań, |
|
— |
wpływ toksyczności lub innych skutków na reprodukcję, potomstwo i rozwój poporodowy, |
|
— |
dzień obserwacji każdego nietypowego objawu i późniejszego postępowania, |
|
— |
dane dotyczące wagi ciała dla zwierząt P, |
|
— |
wyniki badań sekcji zwłok, |
|
— |
szczegółowy opis wszystkich wyników badań mikroskopowych, |
|
— |
statystyczne ujęcie wyników, tam gdzie jest to stosowane, |
|
— |
analiza wyników, |
|
— |
interpretacja wyników. |
3.2. ANALIZA I INTERPRETACJA
Zob. Wprowadzenie ogólne część B.
4. ODNIESIENIA
Zob. Wprowadzenie ogólne część B.
B.35. DWUPOKOLENIOWE BADANIE TOKSYCZNOŚCI REPRODUKCYJNEJ
1. METODA
Niniejsza metoda jest analogiczna do OECD TG 416 (2001).
1.1. WSTĘP
Metoda badania toksyczności reprodukcji dwóch pokoleń opracowana została w celu uzyskania ogólnych informacji dotyczących skutków oddziaływania substancji testowej na integralność i działanie męskich i żeńskich układów rozrodczych, wliczając w to funkcję gruczołów rozrodczych, cykl rujowy, zachowanie kopulacyjne, zapłodnienie, ciążę, poród, laktację i zaprzestanie ssania, jak również wzrost i rozwój potomstwa. Badanie może również dostarczyć informacji dotyczących skutków substancji testowej wywieranych na zachorowalność noworodków, śmiertelności oraz wstępnych danych dotyczących przedporodowej i poporodowej toksyczności rozwojowej, oraz służyć jako wskazówka do następnych badań. Poza badaniem wzrostu i rozwoju pokolenia F1 omawiana metoda badania ma na celu ocenę integralności i działania męskich i żeńskich układów rozrodczych, jak również wzrostu i rozwoju pokolenia F2. W celu uzyskania dodatkowych informacji dotyczących toksyczności rozwojowej i niedoborów funkcjonalnych można albo włączyć do niniejszego protokołu dodatkowe segmenty badań, uwzględniające właściwe metody badania toksyczności rozwojowej i/lub neurotoksyczności rozwojowej, lub też można badać te punkty końcowe w oddzielnych badaniach z wykorzystaniem właściwych metod.
1.2 ZASADA METODY BADAWCZEJ
Substancja testowa podawana jest w stopniowanych dawkach kilku grupom samców i samic. Samcom z pokolenia rodziców P należy podawać dawkę w okresie wzrostu i przez co najmniej jeden pełny cykl spermatogenezy (około 56 dni u myszy i 70 dni u szczura) w celu wywołania przez substancję testową wszelkich negatywnych skutków wywieranych na spermatogenezę. Skutek wywierany na spermę określa się poprzez szereg parametrów spermy (np. morfologię i ruchliwość spermy) oraz w czasie preparowania tkanek i szczegółowych badań histopatologicznych. Jeśli dostępne są dane dotyczące spermatogenezy z wcześniejszego badania powtarzanego dawkowania o wystarczającym czasie trwania, np. badania 90-dniowego, nie ma potrzeby obejmować oceną samców pokolenia P. Zaleca się jednak zachowanie próbek lub cyfrowych zapisów spermy pokolenia P, aby umożliwić późniejszą ocenę. Samice z pokolenia rodziców P powinny być poddane dawkowaniu w okresie ich rozwoju i przez kilka kompletnych cyklów rujowych, w celu wykrycia wszelkich negatywnych skutków wywieranych przez substancję testową na normalny przebieg cyklu rujowego. Substancja testowa jest podawana zwierzętom rodzicielskim (P) w okresie krycia, w okresie powstałych ciąży oraz przez okres ssania ich potomstwa F1. Po zaprzestaniu ssania, należy kontynuować podawanie substancji testowej potomstwu F1 w okresie jego rozwoju do osiągnięcia dorosłości, momentu kojarzenia w pary i produkcji potomstwa F2, do chwili zaprzestania ssania potomstwa F2.
Obserwacje kliniczne i badania patologiczne są przeprowadzane na wszystkich zwierzętach w kierunku oznak toksyczności, ze szczególnym uwzględnieniem skutków wywieranych na integralność i działanie układów rozrodczych samców i samic oraz na wzrost i rozwój potomstwa.
1.3 OPIS METODY BADAWCZEJ
1.3.1 Wybór gatunków zwierząt
Szczur jest pożądanym gatunkiem zwierzęcia do badań. Jeżeli zastosowano inne gatunki, należy podać uzasadnienie, a metoda będzie wymagać odpowiednich modyfikacji. Nie należy stosować szczepów o niskiej płodności lub powszechnie znanej wysokiej częstości występowania wad rozwojowych. W momencie rozpoczęcia doświadczenia odchylenia mas ciała wśród wykorzystywanych zwierząt powinny być jak najniższe i nie powinny przekraczać 20 % średniej masy ciała u każdej z płci.
1.3.2 Warunki p rzeby w anią i warunki żywieniowe
Temperatura w badawczych pomieszczeniach hodowlanych powinna wynosić 22 oC (± 3 oC). Chociaż wilgotność względna powinna wynosić co najmniej 30 % i pożądane jest, żeby nie przekraczała 70 % poza czasem czyszczenia pomieszczenia, celem powinno być utrzymywanie jej na poziomie 50–60 %. Oświetlenie powinno być sztuczne, w cyklu 12 godzin jasno, 12 godzin ciemno. Do żywienia można stosować konwencjonalne pasze laboratoryjne z nieograniczonym dostępem do wody pitnej. Na wybór diety może mieć wpływ potrzeba zapewnienia odpowiedniej domieszki substancji testowej przy podawaniu metodą powyższą.
Zwierzęta mogą być trzymane pojedynczo lub umieszczane w klatkach w małych grupach o tej samej płci. Procedury krycia przeprowadza się w klatkach nadających się do tego celu. Po stwierdzeniu, że kopulacja nastąpiła, pokryte samice powinny być umieszczone po jednej w klatkach porodowych lub matczynych. Pokryte zwierzęta mogą być również trzymane w małych grupach i oddzielane na jeden lub dwa dni przed porodem. Tuż przed porodem należy zapewnić pokrytym samicom właściwy i określony materiał do zrobienia gniazda.
1.3.3. Przygotowanie zwierząt
Należy wykorzystać zdrowe młode zwierzęta, które były aklimatyzowane do warunków laboratoryjnych przez co najmniej 5 dni i które nie były wcześniej poddawane procedurom badawczym. Badane zwierzęta należy scharakteryzować ze względu na gatunek, szczep, źródło, płeć, wagę i/lub wiek. Należy określić stosunki pokrewieństwa w rodzeństwie wśród zwierząt w celu uniknięcia krycia rodzeństwem. Należy losowo przydzielić zwierzęta do grup kontrolnych i badanych (zaleca się dalszy podział według masy ciała). Klatki powinny być ustawione w taki sposób, żeby zminimalizować możliwy wpływ położenia klatki. Każdemu zwierzęciu powinno się nadać jednoznaczny numer identyfikacyjny. W pokoleniu P należy to zrobić przed rozpoczęciem dawkowania. W pokoleniu F1 należy to zrobić w momencie zakończenia ssania przez zwierzęta wybrane do krycia. Należy prowadzić zapisy wskazujące pochodzenie z miotu dla wszystkich wybranych zwierząt z pokolenia F1. Jeśli rozważa się indywidualne ważenie młodych lub jakiekolwiek obserwacje funkcjonalne, zaleca się ponadto indywidualną identyfikację młodych tak szybko po urodzeniu, jak to możliwe.
W momencie rozpoczęcia dawkowania zwierzęta pokolenia rodzicielskiego (P) powinny być w wieku 5–9 tygodni. Na ile to praktycznie możliwe, zwierzęta wszystkich grup badanych powinny być jednakowej masy ciała i jednego wieku.
1.4 PROCEDURA
1.4.1 Liczba i płeć zwierząt
Każda grupa badana i kontrolna powinna składać się z takiej liczby zwierząt, aby znalazło się w nich około 20 samic pod koniec ciąży. Może nie być to możliwe w przypadku substancji powodujących niepożądane skutki związane z podawaniem (np. niepłodność, nadmierną toksyczność przy najwyższym dawkowaniu). Celem jest wyprodukowanie odpowiedniej liczby ciąż w celu zapewnienia znaczącej oceny ewentualnego wpływu substancji na płodność, ciążę i zachowanie macierzyńskie w pokoleniu zwierząt rodziców P i ssących młodych, wzrost i rozwój potomstwa F1 w okresie od chwili poczęcia do osiągnięcia dojrzałości płciowej oraz na rozwój ich potomstwa (F2) do zakończenia ssania. A zatem nieosiągnięcie pożądanej liczby ciężarnych zwierząt (tj. 20) nie musi oznaczać konieczności unieważnienia badania i powinno być ocenione oddzielnie dla każdego przypadku.
1.4.2 Przygotowanie dawek
Zaleca się podawanie doustne (z pożywieniem, wodą pitną lub dozownikiem), jeżeli nie uznano za właściwą innej drogi podawania (np. naskórnej lub wziewnej).
W przypadku gdy to konieczne, substancja testowa jest rozpuszczona lub ma postać zawiesiny w odpowiednim nośniku. Zaleca się, aby – jeżeli to możliwe – najpierw było rozważone użycie wodnego roztworu/zawiesiny, następnie roztworu/emulsji w oleju (np. olej kukurydziany), a potem możliwy roztwór w innym nośnikach. W przypadku nośników niewodnych musi być znana charakterystyka toksyczna nośnika. Powinna być określona trwałość substancji testowej w nośniku.
1.4.3. Dawkowanie
Należy wykorzystać co najmniej trzy poziomy dawki i równoczesną kontrolę. Jeżeli charakter fizykochemiczny lub skutki biologiczne substancji testowej nie powodują ograniczenia, najwyższą dawkę należy dobrać w taki sposób, aby wywołać pewną toksyczność, ale nie śmierć lub poważne cierpienie. W przypadku nieoczekiwanej śmiertelności, badanie ze śmiertelnością mniejszą niż ok. 10 % zwierząt rodzicielskich (P) będzie zazwyczaj do przyjęcia. Należy zastosować sekwencję coraz mniejszych dawek, aby zademonstrować wszelkie reakcje związane z dawkowaniem i określić poziom dawkowania, przy którym nie obserwuje się niekorzystnych skutków (NOAEL). Dwa do czterech interwałów dawkowania są często optymalne dla ustalenia malejących poziomów dawki i dodanie czwartej grupy badanej jest często bardziej pożądane niż stosowanie bardzo dużych interwałów (np. współczynnik większy od 10) między dawkowaniem. Dla badań, gdzie substancja testowa podawana jest w pożywieniu, interwał dawkowania nie powinien przekraczać współczynnika 3. Poziomy dawkowania należy dobierać z uwzględnieniem wszelkich dostępnych danych o toksyczności, szczególnie z badań powtarzanego dawkowania. Należy uwzględnić wszelkie dostępne informacje o metabolizmie i kinetyce związku testowego lub odnośne materiały. Informacje te pomogą nadto w wykazaniu właściwego doboru reżimu dawkowania.
Grupa kontrolna powinna być grupą niepoddawaną dawkowaniu lub grupą kontrolną nośnika, jeśli nośnik zastosowano do podawania substancji testowej. Z wyjątkiem poddawania dawkowaniu substancji testowej, zwierzęta w grupie kontrolnej powinny być traktowane identycznie jak zwierzęta będące przedmiotem badania. Jeśli wykorzystywany jest nośnik, grupa kontrolna otrzymuje nośnik w najwyższej stosowanej objętości. Jeżeli substancja testowa jest podawana z pożywieniem i powoduje obniżenie spożycia lub wykorzystania pokarmu, można rozważyć konieczność użycia grupy kontrolnej karmionych par. Zamiast jednoczesnej grupy kontrolnej karmionych par można, alternatywnie, wykorzystać badania kontrolowane zaprojektowane w celu określenia wpływu obniżonej konsumpcji pokarmu na parametry reprodukcyjne.
Należy uwzględnić następujące charakterystyki nośnika lub innych domieszek: skutek wywierany na absorpcję, dystrybucję, metabolizm oraz na zatrzymywanie lub ekskrecję substancji testowej; skutki wywierane na chemiczne własności substancji testowej, które mogą zmienić jej charakterystykę toksyczną; należy również uwzględnić wpływ na pobieranie pokarmu lub wody, lub stan odżywienia zwierząt.
1.4.4. Test graniczny
Jeżeli badanie z podawaniem doustnym przy jednym poziomie dawki równym co najmniej 1 000 mg/kg masy ciała/dzień, lub równoważny procent w pożywieniu lub wodzie pitnej (na podstawie ustalonej masy ciała), w przypadku podawania w pożywieniu lub wodzie pitnej, stosując procedury opisane dla tego badania, nie wywołuje widocznych objawów zatrucia ani u zwierząt rodzicielskich, ani u ich potomstwa, i jeżeli nie należałoby spodziewać się toksyczności na podstawie danych dotyczących substancji strukturalnie pokrewnych i/lub powiązanych metabolicznie, wtedy pełne badanie przy użyciu trzech poziomów dawki może nie być uznane za konieczne. Ten test graniczny stosuje się z wyjątkiem przypadków, w których ekspozycja ludzi wskazuje na konieczność użycia wyższego poziomu dawki doustnej. W odniesieniu do innych dróg podawania, na przykład wziewnej lub stosowania na skórę, własności fizyczne i chemiczne substancji testowej, jak np. rozpuszczalność, mogą często wskazywać i ograniczać najwyższy możliwy do osiągnięcia poziom ekspozycji.
1.4.5. Podawanie
Zwierzętom podaje się substancję testową przez siedem dni w tygodniu. Pożądane jest podawanie doustne (z pokarmem, wodą pitną lub dozownikiem). Jeśli zastosowano inną drogę podawania, należy podać uzasadnienie; mogą też być konieczne właściwe modyfikacje. Wszystkim zwierzętom podaje się dawki tą samą metodą w czasie odpowiedniego okresu doświadczalnego. Kiedy podaje się substancję dozownikiem, powinno się to wykonywać za pomocą zgłębnika do żołądka. Objętość cieczy, która może być podana jednorazowo nie powinna przekraczać 1 ml/100 g masy ciała (0,4 ml/100 g masy ciała jest maksymalną objętością przy stosowaniu oleju kukurydzianego), z wyjątkiem roztworów wodnych można użyć 2 ml/100 g masy ciała. Z wyjątkiem substancji drażniących lub korozyjnych, które zazwyczaj wywołują zaostrzone skutki, zmienność w testowej objętości powinna być zminimalizowana poprzez skorygowanie stężenia, aby uzyskać stałą objętość na wszystkich poziomach dawki. W badaniach z wykorzystaniem dozownika młode otrzymują substancję tylko drogą pośrednią przez mleko, dopóki nie rozpocznie się dawkowanie bezpośrednie po zaprzestaniu ssania. W badaniach, gdzie podaje się substancję testową w pożywieniu lub wodzie pitnej, młode otrzymują substancję bezpośrednio, kiedy zaczynają się samodzielnie odżywiać w ostatnim tygodniu okresu laktacji.
W przypadku substancji podawanych w pożywieniu lub wodzie pitnej ważne jest zapewnienie, aby wymagane ilości substancji testowej nie kolidowały z prawidłową równowagą odżywiania lub wody. W przypadku gdy substancja testowa jest podawana w pożywieniu, może być stosowane stałe stężenie żywieniowe (ppm) albo stały poziom dawki w odniesieniu do masy ciała zwierząt; użyta opcja musi być wyszczególniona. W przypadku substancji podawanej przez dozownik, dawka powinna być podawana każdego dnia o podobnej porze, i w razie konieczności modyfikowana co najmniej raz na tydzień, żeby utrzymać stały poziom dawki w odniesieniu do masy ciała zwierzęcia. Kiedy podaje się dozownikiem dawkę przeliczoną na masę ciała, należy uwzględnić informacje dotyczące dystrybucji łożyskowej.
1.4.6. Harmonogramy badań
Codzienne stosowanie dawki u rodzicielskich samców i samic (P) powinno rozpocząć się po przekroczeniu od 5–9 tygodni życia. Codzienne dawkowanie u samców i samic F1 powinno rozpocząć się po zaprzestaniu ssania; należy pamiętać, że w przypadkach podawania substancji z pożywieniem lub wodą pitną, bezpośrednia ekspozycja młodych F1 na substancję testową może nastąpić już w okresie laktacji. U obu płci (P i F1) dawkowanie powinno się odbywać przez co najmniej 10 tygodni przed okresem krycia. Dawkowanie należy kontynuować u obu płci przez dwutygodniowy okres krycia. Samce należy humanitarnie uśmiercić, kiedy nie są już potrzebne do oceny wpływu na rozrodczość. U rodzicielskich samic P dawkowanie powinno być kontynuowane przez okres ciąży aż do czasu zaprzestania ssania potomstwa F1. Należy rozważyć zmodyfikowanie zasad dawkowania przy uwzględnieniu dostępnych informacji na temat substancji testowej, w tym dostępnych danych dotyczących toksyczności, pobudzania przez nią metabolizmu lub bioakumulacji. Dawka dla każdego zwierzęcia powinna opierać się zazwyczaj na najbardziej aktualnym indywidualnym określeniu masy ciała. Należy jednak zachować ostrożność, kiedy dostosowuje się dawki w ostatnim trymestrze ciąży.
Podawanie samcom i samicom P i F1 trwa do ich uśmiercenia. Wszystkie dorosłe samce i samice P i F1 powinny zostać humanitarnie uśmiercone, kiedy nie są już potrzebne do oceny wpływu na reprodukcję. Potomstwo F1 nie wybrane do krycia i całe potomstwo F2 powinno zostać humanitarnie uśmiercone po zaprzestaniu ssania.
1.4.7. Procedura krycia
1.4.7.1. Krycie w pokoleniu rodzicielskim (P)
W celu każdego krycia jedną samicę umieszcza się z jednym samcem z tego samego poziomu dawkowania (krycie 1:1) do momentu kiedy nastąpi kopulacja lub upłyną 2 tygodnie. Każdego dnia samice należy poddać badaniu na obecność spermy lub czopów nasienia w pochwie. Dzień 0 danej ciąży jest określany w chwili wykrycia spermy lub czopu nasienia w pochwie. Jeśli kojarzenie w parze nie uda się, można rozważyć krycie samic samcami o dowiedzionej płodności. W danych należy dokładnie zidentyfikować kojarzone pary. Należy unikać krycia pomiędzy rodzeństwem.
1.4.7.2. Krycie w potomstwie F1
W celu otrzymania pokolenia F2, do krycia w potomstwie F1 należy wybrać co najmniej jednego samca i jedną samicę, po zaprzestaniu ssania, z każdego miotu do krycia innymi młodymi osobnikami z tego samego poziomu dawkowania, ale z innego miotu. Wybór młodych z każdego miotu powinien być losowy, jeżeli nie zaobserwowano znaczących różnic w ciężarze ciała pomiędzy młodymi w miocie. W przypadku zaobserwowania takich różnic należy wybrać młode najbardziej reprezentatywne dla każdego z miotów. W sposób pragmatyczny dokonuje się tego na podstawie ciężaru ciała, ale wybór na podstawie wyglądu może być właściwszy. Młode z pokolenia F1 nie powinny być kryte zanim nie osiągną pełnej dojrzałości płciowej.
Należy ustalić przyczyny braku płodności w odniesieniu do par, które nie mają potomstwa. Powyższe może obejmować takie procedury, jak dodatkowe możliwości krycia z samcami lub samicami o dowiedzionej płodności, mikroskopowe badanie organów rozrodczych oraz badania cyklu rujowego i cyklu spermatogenezy.
1.4.7.3. Drugie krycie
W pewnych przypadkach, takich jak związane z terapią zmiany w wielkości miotu lub zaobserwowanie niejednoznacznych skutków w pierwszym kryciu, zaleca się powtórne krycie zwierząt dorosłych P i F1 w celu otrzymania drugiego miotu. Jeśli uznano za konieczne uzyskanie drugiego miotu w którymś z tych pokoleń, zwierzęta powinny być kryte po raz drugi po upływie około tygodnia od zaprzestania ssania przez ostatni miot.
1.4.7.4. Wielkość miotu
Zwierzęta są dopuszczane do miotów i wychowują swoje potomstwo do chwili zaprzestania ssania. Normalizacja wielkości miotu nie jest obowiązkowa. Kiedy dokonuje się normalizacji miotów, należy szczegółowo opisać zastosowaną metodę.
1.5. OBSERWACJE
1.5.1. Obserwacje kliniczne
Ogólne obserwacje kliniczne prowadzone są każdego dnia, a w przypadku dawkowania przez dozownik terminy podawania powinny uwzględniać szczytowy okres przewidywanych objawów po dawkowaniu. Należy odnotować zmiany w zachowaniu, objawy trudnego lub długotrwałego porodu oraz wszelkie objawy toksyczności. Dodatkowe, bardziej szczegółowe badanie każdego zwierzęcia należy przeprowadzić co najmniej raz na tydzień, przy okazji ważenia zwierząt. Dwa razy dziennie, a podczas weekendów raz dziennie tam, gdzie to konieczne, wszystkie zwierzęta są obserwowane pod kątem zachorowalności i śmiertelności.
1.5.2 Masa ciała oraz konsumpcja pożywienia/wody przez zwierzęta rodzicielskie
Zwierzęta rodzicielskie (P i F1) należy zważyć w pierwszym dniu dawkowania i następnie co najmniej raz na tydzień. Rodzicielskie samice (P and F1) należy ważyć co najmniej w dniu 0, 7, 14 i 20 lub 21 ciąży, a podczas laktacji w tych samych dniach, w których ważone są mioty, oraz w dniu, w którym uśmierca się zwierzęta. Obserwacje te należy umieścić w sprawozdaniu oddzielnie dla każdego dorosłego zwierzęcia. W okresie przed kojarzeniem i podczas ciąży, należy co najmniej raz w tygodniu mierzyć konsumpcję pokarmu. Należy mierzyć konsumpcję wody co najmniej raz w tygodniu, jeżeli substancję testową podaje się w wodzie.
1.5.3 Cykl rujowy
Długość i normalny przebieg cyklu rujowego u samic P i F1 oceniane są poprzez rozmazy śluzówki pochwy przed kryciem i nieobowiązkowo w okresie krycia, dopóki nie zostanie ustalone zaistnienie krycia. Przy uzyskiwaniu komórek pochwy/szyjki macicy należy uważać, żeby uniknąć uszkodzenia śluzówki i, w następstwie, wywołania ciąży rzekomej (1).
1.5.4 Parametry nasienia
Przy uśmiercaniu wszystkich samców P i F1 odnotowuje się masy jąder i najądrzy i po jednym z tych narządów zachowuje do badania histopatologicznego (zob. sekcja 1.5.7, 1.5.8.1). Z podgrupy co najmniej dziesięciu samców z każdej grupy samców P i F1, pozostałe jądra i najądrza należy użyć do liczenia, odpowiednio, spermatyd odpornych na homogenizację i rezerw nasienia w ogonach najądrzy. Od tych samych samców należy zebrać nasienie z ogonów najądrzy lub kanalików nasiennych do badania ruchliwości i morfologii plemników. Jeżeli zaobserwowano występowanie skutków związanych z terapią lub jeśli w innych badaniach dowiedziono możliwych skutków względem spermatogenezy, ocena nasienia powinna zostać przeprowadzona u wszystkich samców każdej grupy dawkowania; w innych przypadkach liczenie można ograniczyć do samców P i F1 grupy kontrolnej i grupy najwyższego dawkowania.
Należy określić całkowite liczby odpornych na homogenizację spermatyd jądrowych i plemników z ogona najądrzy (2)(3). Rezerwy nasienia w ogonach najądrzy można wyprowadzić ze stężenia i objętości nasienia w zawiesinie wykorzystywanej do wykonania ocen jakościowych oraz liczby plemników uzyskanych przez następne zmielenie i/lub homogenizację pozostałej tkanki ogona najądrza. Liczenia należy dokonać u podgrupy samców ze wszystkich grup dawkowania bezpośrednio po uśmierceniu zwierząt, chyba że dokonano zapisów wideo lub cyfrowych, lub kiedy uśmiercone osobniki zostają zamrożone i przeanalizowane później. W tych przypadkach grupa kontrolna i grupa wysokiego poziomu dawkowania może zostać przeanalizowana najpierw. Jeżeli nie zostaną w nich stwierdzone skutki związane z terapią (np. wpływ na liczbę plemników, ich ruchliwość, lub morfologię), nie ma potrzeby analizowania innych grup dawkowania. Kiedy w grupie najwyższego dawkowania zostaną stwierdzone skutki związane z terapią, wtedy powinna również zostać oceniona grupa z niższym poziomem dawki.
Ruchliwość plemników najądrza (lub kanalika nasiennego) powinna zostać oceniona lub zarejestrowana na taśmie wideo bezpośrednio po uśmierceniu. Nasienie należy uzyskać z minimalnymi uszkodzeniami i rozcieńczone w celu przeprowadzenia analizy ruchliwości, stosując uznane metody (4). Udział procentowy poruszających się postępowo plemników powinien zostać oceniony subiektywnie lub obiektywnie. Kiedy dokonuje się wspomaganej komputerowo analizy ruchu (5)(6)(7)(8)(9)(10), wyprowadzenie postępowej ruchliwości opiera się na zdefiniowanych przez użytkownika progach średniej prędkości wzdłuż drogi oraz wskaźniku prostości lub liniowym. Jeżeli próbki rejestrowane są na taśmie wideo (11) lub obrazy rejestruje się w inny sposób w czasie sekcji, wystarczy następnie tylko analiza grup kontrolnych i wysokiego dawkowania samców P i F1, chyba że zostaną zaobserwowane skutki związane z terapią; w tym przypadku należy przeanalizować również grupę niższego dawkowania. Jeżeli nie ma obrazu wideo lub cyfrowego, wszystkie próbki ze wszystkich grup muszą zostać przeanalizowane przy sekcji zwłok.
Należy dokonać morfologicznej oceny próbki plemników z najądrzy (lub nasieniowodu). Plemniki (co najmniej 200 na próbkę) należy badać w formie utrwalonych preparatów mokrych (12) i klasyfikować jako prawidłowe lub nieprawidłowe. Przykłady nieprawidłowości morfologicznych plemników obejmują zrośnięcie, oddzielone lub zniekształcone główki i/lub wici. Oceny dokonuje się na wybranej podgrupie samców wszystkich grup dawkowania albo bezpośrednio po uśmierceniu zwierząt albo później, opierając się na zapisie wideo lub cyfrowym. Utrwalone rozmazy mogą być również oglądane później. W tych przypadkach grupa kontrolna i grupa wysokiego poziomu dawkowania może zostać przeanalizowana najpierw. Jeżeli nie zostaną w nich stwierdzone skutki związane z terapią (np. wpływ na morfologię plemników), nie ma potrzeby analizowania innych grup dawkowania. Kiedy w grupie najwyższego dawkowania zostaną stwierdzone skutki związane z terapią, wtedy należy również przeanalizować grupy z niższym poziomem dawki.
Jeżeli którykolwiek z powyższych parametrów oceny nasienia był już wcześniej badany jako część systemowego badania toksykologicznego trwającego co najmniej 90 dni, badanie to nie musi być koniecznie powtarzane w badaniu dwupokoleniowym. Zaleca się jednak, aby próbki lub cyfrowe zapisy dotyczące nasienia pokolenia P zostały zachowane do ewentualnej późniejszej oceny, jeżeli zajdzie taka konieczność.
1.5.5. Młode
Każdy miot należy zbadać tak szybko po urodzeniu jak to możliwe (dzień laktacji 0), w celu ustalenia liczby i płci młodych, płodów urodzonych martwo, płodów urodzonych żywo, oraz obecności poważnych nieprawidłowości. Pożądane jest zbadanie młodych, które znaleziono martwe w dniu 0, pod kątem możliwych wad i przyczyny zgonu, i zachowanie ich. Żywe młode należy policzyć i zważyć pojedynczo w dniu urodzenia (dzień 0 laktacji) lub w dniu 1, oraz w dniach regularnego ważenia, np. w dniach 4, 7, 14 i 21 laktacji. Należy odnotować nieprawidłowości fizyczne lub w zachowaniu, zaobserwowane u matek lub młodych.
Rozwój fizyczny młodych powinien być odnotowany głównie w postaci przyrostów masy ciała. Inne parametry fizyczne (otwarcie uszu i oczu, wyrzynanie zębów, wzrost sierści) mogą dostarczyć dodatkowych informacji, ale dane te najlepiej jest oceniać w kontekście dojrzewania płciowego (np. wiek i masa w dniu otwarcia pochwy lub wyróżnicowania podziału żołędzi-napletka) (13). Badania funkcjonalne (np. aktywności motorycznej, funkcji sensorycznej, ontogenezy odruchu) u młodych pokolenia F1 przed i po zaprzestaniu ssania, szczególnie te, które wiążą się z dojrzewaniem płciowym są zalecane, jeżeli tego rodzaju stwierdzenia nie zostały objęte innymi badaniami. U młodych F1, które zaprzestały ssania, a są przeznaczone do krycia, należy określić wiek otwarcia pochwy i pojawienia się podziału żołędzi. Odległość między odbytem a otworem płciowym należy mierzyć w dniu 0 po urodzeniu młodych F2, jeżeli zmiany w proporcji płci w grupie lub odchylenia w dojrzewaniu płciowym wskazują na taką konieczność.
Można nie dokonywać obserwacji funkcjonalnych w grupach, które w inny sposób wykazują wyraźne oznaki kliniczne niekorzystnych skutków (np. znaczące spadki przyrostu masy itp.). Jeżeli prowadzi się obserwacje funkcjonalne, nie należy ich dokonywać na młodych wybranych do krycia.
1.5.6 Oglądowa sekcja zwłok
W chwili uśmiercenia lub zgonu zwierzęcia podczas badania, wszystkie zwierzęta pokolenia rodziców (P i F1), wszystkie młode z nieprawidłowościami zewnętrznymi lub objawami klinicznymi, jak również po jednym losowo wybranym młodym/płci/miocie, powinny zostać poddane badaniom oglądowym w celu wykrycia wszelkich strukturalnych nieprawidłowości lub zmian patologicznych, ze zwróceniem szczególnej uwagi na organy układu rozrodczego. Młode, które zostały humanitarnie uśmiercone w stanie chorobowym, oraz młode padłe, jeśli nie są zmacerowane, powinny zostać zbadane pod kątem ewentualnych wad i/lub przyczyny zgonu, i utrwalone.
Macice samic, które rodziły pierwszy raz należy zbadać w sposób nie naruszający możliwości przeprowadzenia oceny histopatologicznej na obecność i liczbę zagnieżdżonych jaj.
1.5.7. Masy narządów
Po uśmierceniu należy określić masę ciała oraz masy następujących narządów wszystkich zwierząt rodzicielskich P i F1 (narządy parzyste należy ważyć oddzielnie):
|
— |
macica, jajniki, |
|
— |
jądra, najądrza (całe oraz ogony), |
|
— |
prostata, |
|
— |
pęcherzyki nasienne wraz z gruczołami opuszkowo-cewkowymi i ich płynami oraz gruczoł krokowy (jako jedna całość), |
|
— |
mózg, wątroba, nerki, śledziona, przysadka, tarczyca, nadnercza i znane organy docelowe. |
Należy określić masy ciała przy uśmierceniu tych młodych F1 i F2, które zostały wybrane do sekcji zwłok. Należy zważyć następujące narządy z jednego losowo wybranego młodego/płci/miotu (zob. sekcja 1.5.6): mózg, śledziona i grasica.
Jeśli to możliwe, wyniki oglądowej sekcji zwłok i masy narządów powinny być oceniane w zestawieniu z innymi obserwacjami poczynionymi w innych badaniach powtarzanego dawkowania.
1.5.8. Histopatologia
1.5.8.1. Zwierzęta rodzicielskie
Należy utrwalić i przechowywać w odpowiednim środku następujące narządy i tkanki zwierząt rodzicielskich (P i F1) lub ich reprezentatywne próbki do badania histopatologicznego:
|
— |
pochwa, macica z szyjką oraz jajniki (zachowane w odpowiednim utrwalaczu), |
|
— |
jedno jądro (zachowane w utrwalaczu Bouina lub porównywalnym), jedno najądrze, kanaliki nasienne, gruczoł krokowy i gruczoł opuszkowo-cewkowy, |
|
— |
wcześniej określony organ (organy) docelowe ze wszystkich zwierząt P i F1 wybranych do kojarzenia. |
Należy wykonać pełne badanie histopatologiczne wyliczonych powyżej utrwalonych narządów i tkanek u wszystkich zwierząt grupy wysokiego dawkowania i zwierząt kontrolnych P i F1 wybranych do kojarzenia. Badanie jajników zwierząt P nie jest obowiązkowe. W grupach najmniejszego i pośredniego dawkowania należy również zbadać narządy wykazujące zmiany związane z terapią, by pomóc w wyjaśnieniu NOAEL. Ponadto należy poddać ocenie histopatologicznej organy rozrodcze zwierząt z grup najniższego i pośredniego poziomu dawkowania, podejrzanych o obniżoną płodność, tj. zwierząt, które nie kojarzyły się, nie zostały zapłodnione, nie spłodziły, albo nie urodziły zdrowego potomstwa. Należy zbadać wszystkie duże zmiany, jak atrofia lub guzy.
Należy przeprowadzić szczegółowe badania histopatologiczne jąder (np. z wykorzystaniem utrwalacza Bouina, zatapiania w parafinie i skrawania poprzecznych skrawków o grubości 4–5 μm) w celu określenia związanych z terapią skutków, takich jak zatrzymane spermatydy, braki komórek warstw zarodkowych lub ich typów, wielojądrowe komórki olbrzymie lub oddzielanie się komórek spermatogennych w światło przewodu (14). Badanie nienaruszonego najądrza powinno obejmować głowę, trzon i ogon, co można osiągnąć przez ocenę przekroju poprzecznego. Badanie najądrza powinno obejmować naciekanie leukocytami, zmiany w występowaniu poszczególnych typów komórek, typy komórek odbiegających od normy i fagocytozę plemników. W ocenie męskich narządów rozrodczych należy stosować barwienie metodą PAS i hematoksyliną.
Jajnik polaktacyjny powinien zawierać pęcherzyki rdzenne i pęcherzyki wzrastające, jak również duże ciałka żółte okresu laktacji. Badanie histopatologiczne powinno wykryć jakościowe upośledzenie populacji pęcherzyków rdzennych. U samic F1 należy przeprowadzić ilościową ocenę pęcherzyków rdzennych; liczba zwierząt, wybór przekrojów jajnika i wielkość próby przekrojów powinny mieć wielkości spełniające wymagania używanej procedury oceny statystycznej. Badanie powinno zawierać liczenie liczby pęcherzyków rdzennych, którą dla potrzeb porównania jajników zwierząt badanych i kontrolnych można połączyć z liczbą małych pęcherzyków wzrastających (15)(16)(17)(18)(19).
1.5.8.2. Młode po zaprzestaniu ssania
Tkanki i narządy docelowe ze znacznymi nieprawidłowościami ze wszystkich młodych z zewnętrznymi nieprawidłowościami lub oznakami klinicznymi, jak również z jednego losowo wybranego młodego/płci/miotu z pokoleń F1 i F2 nieprzeznaczonego do krycia, należy utrwalić i przechować w odpowiednim środku, do badań histopatologicznych. Należy określić pełną histopatologiczną charakterystykę utrwalonej tkanki ze szczególnym uwzględnieniem narządów rozrodczych.
2. DANE
2.1. OPRACOWANIE WYNIKÓW
Dane należy podawać indywidualnie i podsumować w formie tabeli wykazującej, w odniesieniu do każdej grupy badanej i każdego pokolenia, liczbę zwierząt na początku badania, liczbę zwierząt, które znaleziono martwe podczas badania lub zwierząt uśmierconych ze względów humanitarnych, czas śmierci lub humanitarnego uśmiercenia, liczbę zwierząt płodnych, liczbę ciężarnych samic, liczbę zwierząt wykazujących zaobserwowane oznaki toksyczności, z podaniem czasu ich pojawienia się, w tym czasu trwania, ciężkości wszelkich skutków toksyczności, rodzajów obserwacji zwierząt rodzicielskich i potomstwa, rodzajów zmian histopatologicznych oraz wszelkich odnośnych danych miotów.
Wyniki liczbowe należy ocenić właściwą, uznaną metodą statystyczną; wyboru metod statystycznych należy dokonać w ramach projektowania badania i uzasadnić. Użytecznym w analizie może być model statystyczny zależności reakcji od dawki. Sprawozdanie powinno zawierać wystarczające informacje dotyczące użytych metod analizy i oprogramowania komputerowego, tak aby niezależny recenzent/statystyk mógł dokonać powtórnej oceny i odtworzyć analizę.
2.2. OCENA WYNIKÓW
Wyniki uzyskane w dwupokoleniowym badaniu toksyczności reprodukcyjnej należy ocenić w kategoriach zaobserwowanych skutków, w tym ustaleń z sekcji zwłok i badań mikroskopowych. Ocena obejmuje stwierdzenie zależności – lub jej braku – pomiędzy dawką substancji testowej a obecnością lub brakiem, częstością i ciężkością nieprawidłowości, wliczając w to poważne zmiany, wskazane organy docelowe, zmienioną płodność, nieprawidłowości kliniczne, zmienione funkcje rozrodcze i funkcjonowanie miotów, zmiany masy ciała, wpływ na śmiertelność i wszelkie inne skutki toksyczne.
W ocenie wyników należy uwzględnić własności fizykochemiczne substancji testowej i (w miarę dostępności) dane toksykokinetyczne. Prawidłowo przeprowadzone badanie toksyczności reprodukcyjnej powinno dostarczyć zadowalającą ocenę poziomu niepowodującego skutków oraz dać możliwość lepszego zrozumienia niekorzystnych skutków wywieranych na reprodukcję, poród, laktację i rozwój pourodzeniowy, w tym wzrost i dojrzewanie płciowe.
2.3. INTERPRETACJA WYNIKÓW
Dwupokoleniowy test toksyczności reprodukcyjnej dostarcza informacji o skutkach powtarzanej ekspozycji na substancję, podczas wszystkich faz cyklu rozrodczego. W szczególności badanie to dostarcza informacji o parametrach rozrodu, a także o rozwoju, wzroście, dojrzewaniu i przeżywaniu młodych. Wyniki tego badania należy rozpatrywać w połączeniu z wynikami uzyskanymi w badaniach toksyczności podchronicznej, przedurodzeniowej toksyczności rozwojowej i badaniach toksykokinetycznych i innych dostępnych badaniach. Wyniki tego badania mogą być wykorzystane do oceny potrzeby dalszego badania substancji chemicznej. Ekstrapolacja wyników tego badania na człowieka jest uprawniona tylko w ograniczonym stopniu. Najlepszym sposobem ich wykorzystania jest dostarczanie informacji dotyczącej poziomów niepowodujących skutków i dopuszczalnej ekspozycji ludzi (20)(21)(22)(23).
3. SPRAWOZDAWCZOŚĆ
3.1. SPRAWOZDANIE Z BADAŃ
Sprawozdanie z badań musi zawierać następujące szczegółowe informacje:
|
|
Substancja testowa:
|
|
|
Nośnik (w stosownych przypadkach):
|
|
|
Zwierzęta badane:
|
|
|
Warunki badania:
|
|
|
Wyniki:
|
|
|
Dyskusja wyników. |
|
|
Wnioski, w tym wartości NOAEL dla skutków u matek i potomstwa. |
4 BIBLIOGRAFIA
|
(1) |
Sadleir, R.M.F.S. (1979). Cycles and Seasons, In: Reproduction in Mammals: I. Germ Cells and Fertilization, C.R. Auston and R. V. Short (eds.), Cambridge, New York. |
|
(2) |
Gray, L.E. et al., (1989). A Dose-Response Analysis of Methoxychlor-Induced Alterations of Reproductive Development and Function in the Rat. Fundamental and Applied Toxicology 12: 92–108. |
|
(3) |
Robb, G.W. et al., (1978). Daily Sperm Production and Epididymal Sperm Reserves of Pubertal and Adult Rats. Journal of Reproduction and Fertility 54: 103–107. |
|
(4) |
Klinefelter, G.R. et al., (1991). The Method of Sperm Collection Significantly Influences Sperm Motion Parameters Following Ethane Dimethanesulfonate Administration in the Rat. Reproductive Toxicology 5: 39–44. |
|
(5) |
Seed, J. et al. (1996). Methods for Assessing Sperm Motility, Morphology, and Counts in the Rat, Rabbit, and Dog: a Consensus Report. Reproductive Toxicology 10(3): 237– 244. |
|
(6) |
Chapin, R.E. et al., (1992). Methods for Assessing Rat Sperm Motility. Reproductive Toxicology 6:267–273. |
|
(7) |
Klinefelter, G.R. et al., (1992). Direct Effects of Ethane Dimethanesulphonate on Epididymal Function in Adult Rats: an In Vitro Demonstration. Journal of Andrology 13: 409–421. |
|
(8) |
Slott, V.L. et al., (1991). Rat Sperm Motility Analysis: Methodologic Considerations. Reproductive Toxicology 5: 449–458. |
|
(9) |
Slott, V.L. and Perreault, S.D., (1993). Computer-Assisted Sperm Analysis of Rodent Epididymal Sperm Motility Using the Hamilton-Thorn Motility Analyzer. In: Methods in Toxicology, Part A., Academic, Orlando, Florida, pp. 319–333. |
|
(10) |
Toth, G.P. et al. (1989). The Automated Analysis of Rat Sperm Motility Following Subchronic Epichlorhydrin Administration: Methodologic and Statistical Considerations. Journal of Andrology 10: 401–415. |
|
(11) |
Working, P.K. and M. Hurtt, (1987). Computerized Videomicrographic Analysis of Rat Sperm Motility. Journal of Andrology 8: 330–337. |
|
(12) |
Linder, R.E. et al., (1992). Endpoints of Spermatoxicity in the Rat After Short Duration Exposures to Fourteen Reproductive Toxicants. Reproductive Toxicology 6: 491–505. |
|
(13) |
Korenbrot, C.C. et al., (1977). Preputial Separation as an External Sign of Pubertal Development in the Male Rat. Biological Reproduction 17: 298–303. |
|
(14) |
Russell, L.D. et al., (1990). Histological and Histopathological Evaluation of the Testis, Cache River Press, Clearwater, Florida. |
|
(15) |
Heindel, J.J. and R.E. Chapin, (eds.) (1993). Part B. Female Reproductive Systems, Methods in Toxicology, Academic, Orlando, Florida. |
|
(16) |
Heindel, J.J. et al., (1989) Histological Assessment of Ovarian Follicle Number in Mice As a Screen of Ovarian Toxicity. In: Growth Factors and the Ovary, A.N. Hirshfield (ed.), Plenum, New York, pp. 421–426. |
|
(17) |
Manson, J.M. and Y.J. Kang, (1989). Test Methods for Assessing Female Reproductive and Developmental Toxicology. In: Principles and Methods of Toxicology, A.W. Hayes (ed.), Raven, New York. |
|
(18) |
Smith, B.J. et al., (1991). Comparison of Random and Serial Sections in Assessment of Ovarian Toxicity. Reproductive Toxicology 5:379–383. |
|
(19) |
Heindel, J.J. (1999). Oocyte Quantitation and Ovarian Histology. In: An Evaluation and Interpretation of Reproductive Endpoints for Human Health Risk Assessment, G. Daston,. and C.A. Kimmel, (eds.), ILSI Press, Washington, DC. |
|
(20) |
Thomas, J. A. (1991). Toxic Responses of the Reproductive System. In: Casarett and Doull's Toxicology, M.O. Amdur, J. Doull, and C.D. Klaassen (eds.), Pergamon, New York. |
|
(21) |
Zenick, H. and E.D. Clegg, (1989). Assessment of Male Reproductive Toxicity: A Risk Assessment Approach. In: Principles and Methods of Toxicology, A.W. Hayes (ed.), Raven Press, New York. |
|
(22) |
Palmer, A.K. (1981). In: Developmental Toxicology, Kimmel, C.A. and J. Buelke-Sam (eds.), Raven Press, New York. |
|
(23) |
Palmer, A.K. (1978). In Handbook of Teratology, Vol. 4, J.G. Wilson and F.C. Fraser (eds.), Plenum Press, New York. |
B.36. TOKSYKOKINETYKA
1. METODA
1.1. WSTĘP
Zob. Wprowadzenie ogólne część B.
1.2. DEFINICJE
Zob. Wprowadzenie ogólne część B.
1.3. SUBSTANCJE ODNIESIENIA
Brak.
1.4. ZASADA METODY BADAWCZEJ
Substancja testowa podawana jest właściwą drogą. W zależności od celu badań substancja testowa może być podawana jednorazowo lub w wielu dawkach przez wyznaczone okresy czasu jednej lub kilku grupom zwierząt badanych. Następnie, w zależności od rodzaju badań, ustalana jest ilość substancji testowej i/lub metabolitów w płynach ustrojowych, tkankach i/lub wydalinach.
Badania mogą być przeprowadzane z użyciem „nieetykietowanych” lub „etykietowanych” postaci substancji testowej. W przypadku użycia etykiety należy umieścić ją na substancji w taki sposób, aby umożliwić dostarczenie jak największej ilości informacji o przemianie badanego składnika.
1.5. KRYTERIA JAKOŚCIOWE
Brak.
1.6. OPIS METODY BADAWCZEJ
Przygotowania
Zdrowe, młode, dorosłe zwierzęta są aklimatyzowane do warunków laboratoryjnych co najmniej przez okres pięciu dni przed rozpoczęciem badania. Przed rozpoczęciem badania zwierzęta są dobierane losowo i przydzielane do grup badanych. W wyjątkowych wypadkach można wykorzystać bardzo młode, ciężarne lub niebadane zwierzęta.
Warunki badania
Zwierzęta badane
Badania toksykokinetyczne mogą być przeprowadzane na jednym lub większej ilości odpowiednich gatunków i powinny uwzględniać gatunki wykorzystywane lub mające być wykorzystanymi w innych badaniach toksykologicznych z użyciem tej samej substancji testowej. W przypadku wykorzystania gryzoni do badania, różnica wagi nie powinna przekraczać ± 20 % wagi średniej.
Liczba i płeć
Do badań absorpcji i ekskrecji początkowo należy wykorzystać cztery zwierzęta w każdej grupie dawkowania. Zachowanie preferencji w odniesieniu do płci nie jest obowiązkowe, ale w niektórych warunkach obie płcie mogą być wymagane do wykorzystania w badaniach. W przypadku różnic w reakcji danej płci na substancje testową należy poddać badaniu cztery zwierzęta każdej płci. W przypadku badań na zwierzętach innych niż gryzonie można wykorzystać mniejszą liczbę zwierząt. W przypadku badania dystrybucji w tkankach należy uwzględnić zarówno liczbę zwierząt w grupie początkowej przeznaczonych do uśmiercenia w każdym punkcie czasowym, jak i liczbę punktów czasowych do przebadania.
W przypadku badania metabolizmu wielkość grupy związana jest z potrzebami badań. W przypadku badań z użyciem wielokrotnego dawkowania i badań z wielokrotnym punktem czasowym, ustalając wielkość grupy należy wziąć pod uwagę liczbę punktów czasowych oraz planowanych uśmierceń, ale nie może składać się z mniej niż dwóch zwierząt. Wielkość grupy powinna być odpowiednia w celu odpowiedniego scharakteryzowania wchłaniania, utrzymywania się i eliminacji testowej substancji i/lub jej metabolitów (stosownie do przypadku).
Poziomy dawek
W przypadku jednorazowego dawkowania należy zastosować co najmniej dwa poziomy dawki. Należy ustalić niski poziom dawki, który nie powoduje obserwowalnych skutków toksyczności i wysoki poziom dawki stosowanie, którego może spowodować zmiany parametrów toksykokinetycznych lub pojawienie się objawów toksyczności.
W przypadku wielokrotnego podawania dawki niski poziom dawki jest zazwyczaj wystarczający, ale w niektórych okolicznościach konieczne może okazać się stosowanie wysokiego stężenia dawki.
Droga podawania substancji
Badania toksykokinetyczne należy przeprowadzić z użyciem tej samej drogi podawania i, w miarę potrzeb, tego samego nośnika co nośnik użyty lub przeznaczony do życia w innych badaniach toksyczności. Substancja testowa jest zazwyczaj podawana doustnie za pomocą dozownika lub w pożywieniu, nanoszona na skórę lub podawana drogą oddechową przez określone okresy czasu grupom zwierząt badanych. Dożylne podawanie substancji testowej może być korzystne przy ustalaniu względnej absorpcji innymi drogami. Ponadto mogą zostać dostarczone użyteczne informacje dotyczące wzorów dystrybucji wkrótce po dożylnym podaniu substancji testowej.
Należy uwzględnić możliwość ingerencji nośnika w substancję testową. Należy zwrócić uwagę na różnice w absorpcji związaną z podawaniem substancji testowej przez dozownik oraz podawaniem w pożywieniu, oraz potrzebę dokładnego określenia dawki w szczególności w przypadku podawania substancji testowej w pożywieniu.
Okres obserwacji
Wszystkie zwierzęta należy obserwować codziennie, a objawy toksyczności oraz inne istotne cechy kliniczne odnotować, włączając porę ich rozpoczęcia, stopień oraz czas trwania.
Procedura
Po przeprowadzeniu ważenia badanych zwierząt substancja testowa podawana jest właściwą drogą. Jeżeli zostanie uznane za istotne, zwierzętom można wstrzymać podawanie pożywienia przed podaniem substancji testowej.
Absorpcja
Należy oszacować stopień i zakres absorpcji podawanej substancji wykorzystując różne metody, z użyciem lub bez użycia grup odniesienia (8) na przykład przez:
|
— |
określenie ilości substancji testowej i/lub metabolitów w wydalinach, takich jak mocz, żółć, kał, wydychane powietrze i tych pozostałych w tuszy, |
|
— |
porównanie biologicznej reakcji (np. ostra toksyczność badania) między grupą badaną a kontrolną i/lub grupami odniesienia, |
|
— |
porównanie substancji wydzielanej przez nerki i/lub metabolitu w grupach badanych i grupach odniesienia, |
|
— |
określenie powierzchni zgodnie z wielkością plazmy/krzywa czasowa substancji testowej i/lub metabolitów oraz porównanie z danymi z grupy odniesienia. |
Dystrybucja
Obecnie dostępne są dwa podejścia, jedno lub oba mogą zostać wykorzystane w celu dokonania analizy wzorów dystrybucji:
|
— |
uzyskiwane są użyteczne informacje jakościowe wykorzystując autoradiograficzne techniki badania całego ciała, |
|
— |
przez uśmiercanie zwierząt w różnych okresach badania po ekspozycji na substancję testową i określeniu stężenia i ilości substancji testowej i/lub metabolitów w tkankach i organach, uzyskiwane są informacje ilościowe. |
Wydalanie
W badaniach wydalin należy pobierać mocz, kał i wydychane powietrze oraz, w niektórych sytuacjach, żółć. Należy dokonać kilkukrotnego pomiary ilości substancji testowej i/lub metabolitów w niniejszych wydalinach po ekspozycji na działanie substancji, albo do chwili wydalenia 95 % podanej dawki, albo przez okres siedmiu dni, cokolwiek nastąpi jako pierwsze.
W wyjątkowych wypadkach należy uwzględnić wydalanie substancji testowej w mleku u zwierząt w okresie laktacji.
Metabolizm
W celu ustalenia zakresu i modelu metabolizmu należy przeprowadzić analizę biologicznych próbek za pomocą odpowiednich technik. Należy wyjaśnić strukturę metabolitów i właściwe ścieżki metabolizmu w przypadku zaistnienia konieczności udzielenia odpowiedzi na pytania powstałe podczas wcześniejszych badań toksykologicznych. Pomocne może okazać się przeprowadzenie badań in vitro w celu uzyskania informacji dotyczących ścieżki metabolizmu.
Dalsze informacje dotyczące powiązań metabolizmu z toksycznością można uzyskać za pomocą badań biochemicznych, takich jak określenie wpływu na metaboliczne szlaki enzymatyczne, wyczerpanie się endogennych, niebiałkowych związków wodorosiarczkowych i wiązanie określonych związków do makromolekuł.
2. DANE
Zgodnie z rodzajem przeprowadzanych badań dane należy podsumować w formie tabeli popartej prezentacją graficzną gdzie sytuacja tego wymaga. Dla każdej badanej grupy, w odpowiednich przypadkach, należy wykazać średnie i statystyczne odchylenia pomiarów w związku z czasem, dawkowaniem, tkankami i organami. Zakres absorpcji oraz ilość i normy ekskrecji należy ustalić przy użyciu właściwych metod. W przypadku przeprowadzania badań metabolizmu, należy podać strukturę zidentyfikowanych metabolitów oraz ewentualne ścieżki przedstawionego metabolizmu.
3. SPRAWOZDAWCZOŚĆ
3.1. SPRAWOZDANIE Z BADAŃ
Zgodnie z rodzajem przeprowadzanych badań sprawozdanie z badań powinno, jeżeli możliwe, zawierać następujące informacje:
|
— |
gatunek, szczep, źródło, warunki środowiska, pożywienie, |
|
— |
charakterystyka etykietowanych materiałów, jeżeli zostały użyte, |
|
— |
wielkości dawkowania oraz stosowane odstępy czasu między dawkowaniem, |
|
— |
droga(-i) podawania substancji i użyte nośniki, |
|
— |
toksyczność oraz inne zaobserwowane objawy, |
|
— |
metody ustalania substancji testowej i/lub metabolitów w biologicznych próbkach, włączając wydychane powietrze, |
|
— |
przedstawienie pomiarów w formie tabeli w odniesieniu do płci, dawki, reżym dietetyczny, czas, tkanki i organy, |
|
— |
przedstawienie zakresu absorpcji i ekskrecji w czasie, |
|
— |
metody związane z charakterystyką i identyfikacją metabolitów w biologicznych próbkach, |
|
— |
metody pomiarów biochemicznych związanych z metabolizmem, |
|
— |
proponowane ścieżki metabolizmu, |
|
— |
analiza wyników, |
|
— |
interpretacja wyników. |
3.2. ANALIZA I INTERPRETACJA
Zob. Wprowadzenie ogólne część B.
4. ODNIESIENIA
Zob. Wprowadzenie ogólne część B.
B.37. OPÓŹNIONA NEUROTOKSYCZNOŚĆ SUBSTANCJI FOSFOROORGANICZNYCH PO DUŻEJ EKSPOZYCJI
1. METODA
1.1. WPROWADZENIE
Przy ocenie i oszacowaniu skutków toksycznych substancji ważne jest rozważenie potencjału niektórych klas substancji do wywoływania określonych typów neurotoksyczności, które mogą nie być wykryte w innych badaniach toksyczności. Zaobserwowano, że niektóre substancje fosforoorganiczne powodują opóźnioną neurotoksyczność i powinny być wzięte pod uwagę jako związki, które mogą być zaliczone do oszacowania.
Testy sortujące in vitro mogłyby być zastosowane, aby rozpoznać te substancje, które mogą powodować opóźnioną polineuropatię; jednakże negatywne wyniki badań in vitro nie są dowodem, że substancja testowana nie jest substancją neurotoksyczną
Zob. także Wprowadzenie ogólne część B.
1.2. DEFINICJE
Substancje fosforoorganiczne obejmują nienaładowane estry fosforoorganiczne, tioestry i bezwodniki pochodnych organicznych kwasu fosforowego(V), kwasu fosfonowego i kwasów fosforoamidowych i ich tio-pochodnych lub inne substancje, które mogą wywoływać opóźnioną neurotoksyczność obserwowaną czasami w tej klasie substancji.
Opóźniona neurotoksyczność jest objawem powiązanym z przedłużonym zwłocznym początkiem ataksji, obwodową aksonopatią w rdzeniu kręgowym i nerwach obwodowych, i zahamowaniem i starzeniem esterazy wywołującej neuropatię (NTE) w obojętnej tkance.
1.3. SUBSTANCJE ODNIESIENIA
Substancja odniesienia może być testowana na pozytywnej grupie kontrolnej jako środek w celu wykazania, że w warunkach testu laboratoryjnego reakcja badanego gatunku nie zmieniła się znacząco.
Przykładem szeroko stosowanego środka neurotoksycznego jest ortofosforan(V) tritoilu (CAS 78-30-8, EINECS 201-103-5, nomenklatura CAS: kwas ortofosforowy(V), ester tris(2-metylofenylowy), znany jako fosforan trikrezylu.
1.4. ZASADA METODY BADANIA
Substancja testowana jest podawana doustnie w pojedynczej dawce kurom domowym, które zostały zabezpieczone przed ostrymi skutkami cholinergicznymi, gdzie właściwe. Zwierzęta są obserwowane przez 21 dni pod kątem anomalii behawioralnych, ataksji, i paraliżu. Pomiary biochemiczne, w szczególności zahamowanie esterazy wywołującej neuropatię (NTE), przeprowadza się na kurach losowo wybranych z każdej grupy, prawidłowo 24 i 48 godzin po dawkowaniu. Dwadzieścia jeden dni po ekspozycji pozostałe kury są zabijane i przeprowadzane jest badanie histopatologiczne wybranych obojętnych tkanek.
1.5. OPIS METODY BADANIA
1.5.1. Przygotowania
Zdrowe, młode, dorosłe kury, wolne od kolidujących chorób wirusowych i leków oraz bez nieprawidłowości w sposobie chodzenia, powinny być losowo przydzielane do grupy badanej i kontrolnej oraz aklimatyzowane do warunków laboratoryjnych przez co najmniej 5 dni przed rozpoczęciem badania.
Powinno się stosować klatki i zagrody wystarczająco obszerne, aby umożliwić swobodne poruszanie się kur i niezakłóconą obserwację sposobu chodzenia.
Dawkowanie substancji testowanej powinno prawidłowo przebiegać drogą doustną, stosując aplikowanie bezpośrednio do żołądka, w kapsułkach żelatynowych lub metodą porównywalną. Ciecze powinny być podawane nierozcieńczone lub rozpuszczone w odpowiednim nośniku, takim jak olej kukurydziany; substancje stałe powinny być, w miarę możliwości, rozpuszczone, ponieważ duże dawki substancji stałych w żelatynie mogą nie być absorbowane wydajnie. W przypadku nośników niewodnych powinna być znana charakterystyka toksyczna nośnika, a jeżeli nie jest znana, powinna być określona przed testem.
1.5.2. Warunki testu
1.5.2.1. Zwierzęta badane
Zalecana jest młoda, dorosła, kura domowa nioska (Gallus gallus domesticus), w wieku 8–12 miesięcy. Powinny być wykorzystane odmiany i rasy standardowej wielkości, które prawidłowo powinny być wcześniej hodowane w warunkach umożliwiających swobodne poruszanie się.
1.5.2.2. Liczba i płeć
Oprócz grupy badanej powinna być stosowana zarówno grupa kontrolna nośnika, jak i pozytywna grupa kontrolna. Grupa kontrolna nośnika powinna być traktowana w sposób identyczny, jak grupa badana z wyjątkiem tego, że podawanie substancji testowanej jest pominięte.
W każdej grupie ptaków należy wykorzystać wystarczającą liczbę kur tak, aby co najmniej sześć ptaków można było zabić do celów oznaczenia biochemicznego (trzy w każdym z dwóch punktów czasowych) i sześć mogło przeżyć okres 21-dniowej obserwacji do celów patologii.
Pozytywna grupa kontrolna może być poddawana badaniu w tym samym czasie lub też może ona być ostatnią grupą kontrolną w historii badania. Powinna składać się z co najmniej sześciu kur, poddawanych działaniu znanego opóźnionego środka neurotoksycznego, trzy kury przeznaczone są do celów biochemii i trzy do celów patologii. Zalecane jest okresowe uaktualnianie przeszłych danych. W przypadku gdy jakiś istotny element (np. rasa, żywienie, warunki przebywania) w przebiegu badania został zmieniony przez laboratorium wykonujące, należy uzyskać nowe pozytywne dane kontrolne.
1.5.2.3. Poziomy dawki
Aby oszacować poziom dawki, który ma być zastosowany w badaniu głównym, należy przeprowadzić badanie wstępne przy użyciu odpowiedniej liczby kur i grup poziomów dawki. Typowy i nieunikniony jest pewien poziom śmiertelności w badaniu wstępnym, w celu określenia właściwej dawki do badania głównego. Jednakże aby zapobiec śmierci z powodu ostrych skutków cholinergicznych może być użyta atropina lub inny środek ochronny, o którym wiadomo, że nie koliduje z opóźnionymi reakcjami neurotoksycznymi. Rozmaite metody badania mogą być stosowane w celu oszacowania maksymalnej, niewywołującej śmierci dawki substancji testowanej (zob. metoda B.l bis). Poprzednie dane dotyczące kur lub inne dane toksykologiczne mogą być pomocne przy doborze dawki.
W głównym badaniu poziom dawki substancji testowanej powinien być możliwie najwyższy, biorąc pod uwagę wyniki wstępnego badania doboru dawki i górną dawkę graniczną wynoszącą 2 000 mg/kg masy ciała. Jakakolwiek śmiertelność, która może wystąpić, nie powinna zakłócić przeżycia 21 dni przez wystarczającą liczbę zwierząt do celów biochemii (sześć) i histologii (sześć). Powinna być użyta atropina lub inny środek ochronny, o którym wiadomo, że nie koliduje z opóźnionymi reakcjami neurotoksycznymi, w celu zapobieżenia śmierci z powodu ostrych skutków cholinergicznych.
1.5.2.4. Test graniczny
Jeżeli test przy poziomie dawki wynoszącym co najmniej 2 000 mg/kg masy ciała/dzień, z zastosowaniem procedury opisanej dla tego badania, nie powoduje widocznych objawów toksycznych i jeżeli nie należałoby spodziewać się toksyczności na podstawie danych, dotyczących substancji strukturalnie pokrewnych, wtedy badanie z zastosowaniem większej dawki może nie być konieczne. Ten test graniczny stosowany jest z wyjątkiem przypadków, w których ekspozycja ludzi wskazuje na konieczność użycia wyższego poziomu dawki.
1.5.2.5. Okres obserwacji
Okres obserwacji powinien wynosić 21 dni.
1.5.3. Procedura
Po podaniu środka ochronnego, aby zapobiec śmierci z powodu ostrych skutków cholinergicznych, substancja testowana jest podawana w pojedynczej dawce.
1.5.3.1. Ogólne obserwacje
Obserwacje powinny rozpocząć się bezpośrednio po ekspozycji. Wszystkie kury powinny być starannie obserwowane kilka razy w ciągu pierwszych 2 dni i od tego czasu co najmniej raz dziennie przez okres 21 dni lub do zaplanowanego zabicia. Wszystkie oznaki toksyczności powinny być odnotowywane, w tym czas rozpoczęcia, typ, ostrość i czas trwania anomalii behawioralnych. Ataksja powinna być mierzona według porządkowej skali klasyfikacyjnej, składającej się z co najmniej czterech poziomów, paraliż powinien być odnotowany. Co najmniej dwa razy w tygodniu wszystkie kury wybrane do celów patologii powinny być wyjmowane z klatek i poddawane okresowej wymuszonej aktywności motorycznej, takiej jak wspinanie się po drabinie, aby ułatwić obserwację minimalnych skutków toksycznych. Zwierzęta konające oraz zwierzęta przejawiające ostre cierpienie i ból powinny być usuwane, gdy zostanie to zauważone, humanitarnie zabite i poddane sekcji zwłok.
1.5.3.2. Masa ciała
Wszystkie kury powinny być ważone tuż przed podaniem substancji testowanej i od tego czasu co najmniej raz w tygodniu.
1.5.3.3. Biochemia
Sześć losowo wybranych kur z grup badanych i z grupy kontroli nośnika, i trzy kury z pozytywnej grupy kontrolnej (jeżeli grupa ta jest poddawana badaniu jednocześnie) powinny być zabite w ciągu kilku dni po dawkowaniu, mózg i lędźwiowy rdzeń kręgowy powinien być przygotowany i poddany analizie aktywności zahamowania esterazy wywołującej neuropatię. Dodatkowo może być przydatne przygotowanie i poddanie tkanki nerwu kulszowego analizie aktywności zahamowania esterazy wywołującej neuropatię. Prawidłowo, trzy ptaki grupy kontrolnej i każdej grupy badanej są zabijane po 24 godzinach i trzy po 48 godzinach, podczas gdy trzy kury z pozytywnej grupy kontrolnej powinny być zabite po 24 godzinach. Jeżeli obserwacja klinicznych oznak zatrucia (często może to być ocenione przez obserwację czasu rozpoczęcia oznak cholinergicznych) wskazuje, że środek toksyczny może być usuwany bardzo powoli, bardziej pożądane może być pobranie próbki tkanki od trzech ptaków, w każdym z dwóch terminów między 24 a 72 godziną po dawkowaniu.
Analizy acetylocholinoesterazy (AChE) mogą być również wykonane na tych próbkach, jeżeli uzna się to za właściwe. Jednakże może mieć miejsce spontaniczna reaktywacja AChE in vivo, co może prowadzić do zbyt niskiego oszacowania potencji substancji jako inhibitora AChE.
1.5.3.4. Sekcja zwłok
Sekcja zwłok wszystkich zwierząt (zabitych planowo i zabitych jako konające) powinna obejmować obserwację wyglądu mózgu i rdzenia kręgowego.
1.5.3.5. Badanie histopatologiczne
Obojętna tkanka zwierząt przeżywających okres obserwacji, nieużyta w badaniach biochemicznych, powinna być poddana badaniu mikroskopowemu. Tkanki powinny być przygotowane in situ, stosując techniki perfuzji. Odcinki powinny obejmować móżdżek (środkowy – wzdłużny poziom), rdzeń przedłużony, rdzeń kręgowy i nerwy obwodowe. Odcinki rdzenia kręgowego powinny być pobrane z górnego segmentu karkowego, okolic środkowopiersiowych i lędźwiowo-krzyżowych. Powinny być pobrane odcinki z okolic obwodowych nerwu piszczelowego i jego odgałęzienia do mięśnia brzuchatego łydki oraz nerwu kulszowego. Odcinki powinny być zabarwione odpowiednią mieliną i barwnikami charakterystycznymi dla aksonów.
2. DANE
Negatywne wyniki w punktach końcowych wybranych w tej metodzie (biochemia, histopatologia i obserwacja behawioralna) prawidłowo nie wymagałyby dalszego badania, dotyczącego opóźnionej neurotoksyczności. Niejednoznaczne lub nieprzekonujące wyniki dla tych punktów końcowych mogą wymagać dalszej oceny.
Powinny zostać dostarczone indywidualne zwierząt. Dodatkowo, wszystkie dane powinny być podsumowane w formie tabeli, przedstawiając dla każdej grupy badanej liczbę zwierząt na początku testu, liczbę zwierząt wykazujących zmiany patologiczne, skutki behawioralne i biochemiczne, typy i ostrość tych zmian patologicznych lub skutków i procent zwierząt okazujących każdy typ i ostrość zmiany patologicznej lub skutku.
Wyniki badania powinny być ocenione pod względem częstotliwości, ostrości, i korelacji skutków behawioralnych, biochemicznych i histopatologicznych oraz jakichkolwiek innych obserwowanych w grupach badanych i kontrolnych.
Wyniki liczbowe powinny być ocenione z zastosowaniem odpowiedniej i ogólnie akceptowanej metody statystycznej. Metody statystyczne powinny być wybrane podczas planowania badania.
3. SPORZĄDZANIE SPRAWOZDANIA
SPRAWOZDANIE Z TESTU
Sprawozdanie z testu obejmuje, jeżeli możliwe, następujące informacje:
|
|
Zwierzęta badane:
|
|
|
Warunki testu:
|
|
|
Wyniki:
|
|
|
Rozpatrzenie wyników |
|
|
Wnioski. |
4. BIBLIOGRAFIA
Niniejsza metoda jest analogiczna do OECD TG 418.
B.38. OPÓŹNIONA NEUROTOKSYCZNOŚĆ SUBSTANCJI FOSFOROORGANICZNYCH. 28-DNIOWE BADANIE WIELOKROTNEJ DAWKI
1. METODA
1.1. WPROWADZENIE
Przy ocenie i oszacowaniu skutków toksycznych substancji ważne jest rozważenie potencjału niektórych klas substancji do wywoływania określonych typów neurotoksyczności, które mogą nie być wykrywane podczas innych badań toksyczności. Zaobserwowano, że niektóre substancje fosforoorganiczne powodują opóźnioną neurotoksyczność i powinny być wzięte pod uwagę jako związki, które mogą być zaliczone do oszacowania.
Testy sortujące in vitro mogłyby być zastosowane w celu rozpoznania tych substancji, które mogą powodować opóźnioną polineuropatię; jednakże negatywne wyniki badań in vitro nie są dowodem na to, że substancja testowana nie jest substancją neurotoksyczną.
Ten 28-dniowy test opóźnionej neurotoksyczności dostarcza informacji, dotyczących możliwych zagrożeń zdrowia, które mogą się pojawiać się pod wpływem wielokrotnych ekspozycji przez ograniczony okres czasu. Dostarczy on informacji dotyczących reakcji na dawkę i może zapewnić oszacowanie poziomu braku obserwowanych niekorzystnych skutków, który może być przydatny przy ustalaniu kryteriów bezpieczeństwa dla ekspozycji.
Zob. także Wprowadzenie ogólne część B.
1.2. DEFINICJE
Substancje fosforoorganiczne obejmują nienaładowane estry fosforoorganiczne, tioestry i bezwodniki pochodnych organicznych kwasu fosforowego(V), kwasu fosfonowego i kwasów fosforoamidowych i ich tio-pochodnych lub inne substancje, które mogą wywoływać opóźnioną neurotoksyczność obserwowaną czasami w tej klasie substancji.
Opóźniona neurotoksyczność jest objawem związanym z przedłużonym zwłocznym początkiem ataksji, obwodową aksonopatią w rdzeniu kręgowym i nerwach obwodowych, i zahamowaniem i starzeniem esterazy wywołującej neuropatię (NTE) w obojętnej tkance.
1.3. ZASADA METODY BADANIA
Dzienne dawki substancji testowanej są podawane domowym kurom doustnie przez 28 dni. Zwierzęta są obserwowane co najmniej raz dziennie pod kątem anomalii behawioralnych, ataksji i paraliżu do 14 dnia po ostatniej dawce. Przeprowadzane są pomiary biochemiczne, w szczególności zahamowanie esterazy wywołującej neuropatię (NTE), na kurach losowo wybranych z każdej grupy, prawidłowo 24 i 48 godzin po ostatniej dawce. Dwa tygodnie po ostatniej dawce pozostałe kury są zabijane i przeprowadzane jest badanie histopatologiczne wybranych obojętnych tkanek.
1.4. OPIS METODY BADANIA
1.4.1. Przygotowania
Zdrowe, młode, dorosłe kury, wolne od kolidujących chorób wirusowych i leków oraz bez nieprawidłowości w sposobie chodzenia, powinny zostać losowo przydzielone do grupy badanej i kontrolnej oraz aklimatyzowane do warunków laboratoryjnych przez co najmniej 5 dni przed rozpoczęciem badania.
Powinny być stosowane klatki i zagrody wystarczająco obszerne, aby umożliwić swobodne poruszanie się kur i niezakłóconą obserwację sposobu chodzenia.
Dawkowanie doustne powinno być prowadzone każdego dnia, 7 dni w tygodniu, najlepiej stosując aplikowanie bezpośrednio do żołądka lub w kapsułkach żelatynowych. Ciecze powinny być podawane nierozcieńczone lub rozpuszczone w odpowiednim nośniku, takim jak olej kukurydziany; substancje stałe powinny być w miarę możliwości rozpuszczone, ponieważ duże dawki substancji stałych w kapsułkach żelatynowych mogą nie być absorbowane wydajnie. W przypadku nośników niewodnych powinna być znana charakterystyka toksyczna nośnika, a jeżeli nie jest znana, powinna być określona przed testem.
1.4.2. Warunki testu
1.4.2.1. Badane zwierzęta
Zalecana jest młoda, dorosła, domowa, kura nioska (Gallus gallus domesticus), w wieku 8–12 miesięcy. Wykorzystane powinny być odmiany i rasy standardowej wielkości, które prawidłowo powinny być wcześniej hodowane w warunkach umożliwiających swobodne poruszanie się.
1.4.2.2. Liczba i płeć
Zasadniczo, powinny być wykorzystane co najmniej trzy grupy badane i grupa kontrolna nośnika. Grupa kontrolna nośnika powinna być traktowana w sposób identyczny jak grupa badana z wyjątkiem tego, że podawanie substancji testowanej jest pominięte.
W każdej grupie ptaków należy wykorzystać wystarczającą liczbę kur, tak aby co najmniej sześć ptaków można było zabić do celów oznaczeń biochemicznych (trzy w każdym z dwóch punktów czasowych) i aby po zakończeniu działań badawczych sześć kur mogło przeżyć okres 14-dniowej obserwacji do celów patologii.
1.4.2.3. Poziomy dawki
Poziomy dawki powinny być wybierane, biorąc pod uwagę trzy wyniki testu ostrej opóźnionej neurotoksyczności i jakiekolwiek inne istniejące, osiągalne dane dotyczące toksyczności i kinetyki testowanego związku. Powinien być wybrany najwyższy poziom dawki w celu wywołania objawów zatrucia, najlepiej opóźnionej neurotoksyczności, ale nie śmierci lub widocznego cierpienia. Od tego czasu powinna być dobrana malejąca sekwencja poziomów dawki, w celu wykazania wszystkich reakcji powiązanych z dawkowaniem i braku zaobserwowania niekorzystnych objawów przy najmniejszym poziomie dawki.
1.4.2.4. Test graniczny
Jeżeli test przy poziomie dawki wynoszącym co najmniej 1 000 mg/kg masy ciała/dzień, z zastosowaniem procedury opisanej dla tego badania, nie wywołuje widocznych objawów toksycznych i jeżeli nie należałoby spodziewać się toksyczności na podstawie danych, dotyczących substancji strukturalnie pokrewnych, to badanie z zastosowaniem większej dawki może nie być konieczne. Ten test graniczny stosuje się z wyjątkiem przypadków, w których spodziewana ekspozycja ludzi wskazuje na konieczność użycia wyższego poziomu dawki.
1.4.2.5. Okres obserwacji
Wszystkie zwierzęta powinny być obserwowane co najmniej raz dziennie podczas okresu ekspozycji i 14 dni po nim, chyba że planowana jest sekcja zwłok.
1.4.3. Procedura
Substancja testowana jest dawkowana siedem dni w tygodniu przez okres 28 dni
1.4.3.1. Ogólne obserwacje
Obserwacje powinny rozpocząć się bezpośrednio po rozpoczęciu działań badawczych. Wszystkie kury powinny być starannie obserwowane co najmniej raz dziennie w każdym z 28 dni terapii i przez 14 dni po dawkowaniu lub do zaplanowanego zabicia. Wszystkie oznaki toksyczności powinny być odnotowane, w tym czas rozpoczęcia, typ, ostrość i czas trwania. Obserwacje powinny uwzględniać anomalie behawioralne, ale nie powinny być do nich ograniczone. Ataksja powinna być mierzona na porządkowej skali klasyfikacyjnej składającej się z co najmniej czterech poziomów, należy odnotować paraliż. Co najmniej dwa razy w tygodniu kury powinny być wyjęte z klatek i poddane okresowej wymuszonej aktywności motorycznej, takiej jak wspinanie się po drabinie, w celu ułatwienia obserwacji minimalnych skutków toksycznych. Zwierzęta konające, przejawiające ostre cierpienie i ból powinny być usuwane, gdy zostanie to zauważone, humanitarnie zabite i poddane sekcji zwłok.
1.4.3.2. Masa ciała
Wszystkie kury powinny być ważone tuż przed pierwszym podaniem substancji testowanej i od tego czasu co najmniej raz w tygodniu.
1.4.3.3. Biochemia
Sześć losowo wybranych kur z grup badanych i z grupy kontroli nośnika powinno zostać zabitych w ciągu kilku dni od przyjęcia ostatniej dawki. Mózg i lędźwiowy rdzeń kręgowy powinien być przygotowany i poddany analizie aktywności zahamowania esterazy (NTE) wywołującej neuropatię. Dodatkowo może być przydatne przygotowanie i poddanie tkanki nerwu kulszowego analizie aktywności zahamowania esterazy wywołującej neuropatię. Prawidłowo, trzy ptaki grupy kontrolnej i każdej grupy badanej są zabijane po 24 godzinach i trzy po 48 godzinach od ostatniej dawki. Jeżeli dane z badania ostrej toksyczności lub innych badań (np. toksykokinetycznych) wskazują, że inne terminy zabijania po końcowym dawkowaniu są bardziej pożądane niż podane, powinny być one zastosowane, a racjonalna podstawa udokumentowana.
Analizy acetylocholinoesterazy (AChE) mogą być również wykonane na tych próbkach, jeżeli jest to uznane za właściwe. Jednakże może mieć miejsce spontaniczna reaktywacja AChE in vivo i w efekcie prowadzić do zbyt niskiego oszacowania potencjału substancji jako inhibitora AChE.
1.4.3.4. Sekcja zwłok
Sekcja zwłok wszystkich zwierząt (zabitych planowo i zabitych jako konające) powinna obejmować obserwację wyglądu mózgu i rdzenia kręgowego.
1.4.3.5. Badanie histopatologiczne
Obojętna tkanka zwierząt przeżywających okres obserwacji, nie użyta w badaniach biochemicznych powinna być poddana badaniu mikroskopowemu. Tkanki powinny być przygotowane in situ, stosując techniki perfuzji. Odcinki powinny obejmować móżdżek (środkowy – wzdłużny poziom), rdzeń przedłużony, rdzeń kręgowy, i nerwy obwodowe. Odcinki rdzenia kręgowego powinny być pobrane z górnego segmentu karku, okolic środkowopiersiowych i lędźwiowo-krzyżowych. Powinny być pobrane odcinki z okolic obwodowych nerwu piszczelowego i jego odgałęzienia do mięśnia brzuchatego łydki oraz nerwu kulszowego. Odcinki powinny być zabarwione odpowiednią mieliną i barwnikami charakterystycznymi dla aksonów. Początkowo badanie mikroskopowe powinno być przeprowadzone na zakonserwowanych tkankach wszystkich zwierząt grupy kontrolnej i grupy wysokiej dawki. W przypadku gdy istnieją dowody skutków w grupie wysokiej dawki, badanie mikroskopowe powinno być również przeprowadzone na kurach z grup niskiej i pośredniej dawki.
2. DANE
Negatywne wyniki w punktach końcowych wybranych w tej metodzie (biochemia, histopatologia i obserwacja behawioralna) prawidłowo nie wymagałyby dalszego badania, dotyczącego opóźnionej neurotoksyczności. Niejednoznaczne lub nieprzekonujące wyniki dla tych punktów końcowych mogą wymagać dalszej oceny.
Powinny zostać dostarczone dane indywidualne zwierząt. Dodatkowo wszystkie dane powinny być podsumowane w formie tabeli, przedstawiając dla każdej grupy badanej liczbę zwierząt na początku testu, liczbę zwierząt wykazujących zmiany patologiczne, skutki behawioralne i biochemiczne, typy i ostrość tych zmian patologicznych lub skutków oraz procent zwierząt okazujących każdy typ i ostrość zmiany patologicznej lub skutku.
Wyniki badania powinny być ocenione pod względem częstotliwości, ostrości, i korelacji skutków behawioralnych, biochemicznych i histopatologicznych oraz jakichkolwiek innych obserwowanych w każdej z grup badanych i kontrolnych.
Wyniki liczbowe powinny być ocenione za pomocą odpowiedniej i ogólnie akceptowanej metody statystycznej. Metody statystyczne powinny być wybrane podczas planowania badania.
3. SPORZĄDZANIE SPRAWOZDANIA
SPRAWOZDANIE Z TESTU
Sprawozdanie z testu obejmuje, jeżeli możliwe, następujące informacje:
|
|
Zwierzęta badane:
|
|
|
Warunki testu:
|
|
|
Wyniki:
|
|
|
Rozpatrzenie wyników. |
|
|
Wnioski. |
4. BIBLIOGRAFIA
Niniejsza metoda jest analogiczna do OECD TG 419.
B.39. TEST POREPERACYJNEJ SYNTEZY DNA Z KOMÓRKAMI WĄTROBY SSAKÓW IN VIVO
1. METODA
Metoda ta jest powtórzeniem OECD TG 486, testu poreperacyjnej syntezy DNA z komórkami wątroby ssaków in vivo (1997).
1.1. WPROWADZENIE
Celem testu poreperacyjnej syntezy DNA z komórkami wątroby ssaków in vivo (UDS) jest zidentyfikowanie substancji badanych, które wywołują naprawę DNA w komórkach wątroby lub u zwierząt poddanych działaniu substancji (1)(2)(3)(4).
To badanie in vivo przewiduje metodę wykrywania skutków genotoksycznych substancji chemicznej w wątrobie. Zmierzony punkt końcowy jest wskaźnikiem uszkodzenia DNA oraz dalszych napraw w komórkach wątroby. Wątroba jest zazwyczaj głównym miejscem, gdzie odbywa się przemiana materii wchłanianych związków. Jest zatem właściwym miejscem pomiarów uszkodzenia DNA in vivo.
Jeżeli istnieją dowody na to, że substancja nie dotrze do tkanek docelowych, wykorzystanie tego badania nie jest właściwe.
Punkt końcowy testu poreperacyjnej syntezy DNA z komórkami wątroby ssaków in vivo mierzony jest przez oznaczenie poboru oznaczonych nukleozydów w komórkach, które nie są poddawane syntezie białka (faza S). Najpowszechniej wykorzystywaną techniką jest oznaczanie poboru wykorzystanej oznaczonej trytem tymidyny (3H-TdR) przez autoradiografię. Preferuje się wykorzystywanie wątroby szczurów do przeprowadzenia badań in vivo UDS. Tkanki inne niż wątrobowe mogą zostać wykorzystane, ale nie podlegają one tej metodzie.
Wykrycie reakcji UDS uzależnione jest od ilości zasad DNA usuniętych i zastąpionych w miejscu uszkodzenia. Z tego względu badanie UDS ma szczególną wartość w odniesieniu do wykrywania wywołanych przez substancję napraw (20–30 zasad). Odwrotnie, „reperacja krótkich fragmentów DNA” (jedna do trzech zasad) jest wykrywana ze znacznie mniejszą czułością. Co więcej, zdarzenia mutagenne mogą wynikać ze względu na nienaprawienie, niewłaściwą naprawę, niewłaściwe zastąpienie defektów DNA. Zakres reakcji UDS nie zawiera wskazania dokładności procesu naprawy. Dodatkowo możliwe jest, że mutagen reaguje z DNA, ale uszkodzenie DNA nie jest naprawione poprzez wycinek procesu naprawczego. Brak szczegółowych informacji dotyczących aktywności mutagennej dostarczonych przez badanie UDS jest wyrównywane przez potencjał wrażliwości tego punktu końcowego, ponieważ jest on mierzony w całym genomie.
Zob. także Ogólne wprowadzenie do części B.
1.2. DEFINICJE
Komórki w naprawie: względna ilość ziarnistości jądrowych (NNG) wyższa niż aktualna wartość, która ma zostać uzasadniona w laboratorium, w którym przeprowadzane są badania.
Względna ilość ziarnistości jądrowych (NNG): miara ilościowa w odniesieniu do aktywności UDS komórek w testach autoradiograficznych UDS, obliczona przez odejmowanie średniej ilości jąder cytoplazmowych w obszarach cytoplazmowych równoważnych jądru (CG) od ilości ziarnistości jądrowych (NG): NNG = NG – CG. NNG obliczeń dokonuje się dla poszczególnych komórek, a następnie łączy się dla komórek w ramach danej hodowli, w równoległych hodowlach itd.
Test poreperacyjnej syntezy DNA (UDS): synteza reperacyjna DNA po wycięciu oraz usunięciu części DNA zawierającej region uszkodzenia wywołanego substancjami chemicznymi lub czynnikami fizycznymi.
1.3. ZASAD METODY BADAWCZEJ
Badanie UDS na komórkach wątroby ssaków wskazuje syntezę naprawczą DNA po wycięciu oraz usunięciu odcinka DNA zawierającego region uszkodzenia wywołanego substancjami chemicznymi lub czynnikami fizycznymi. Badanie zazwyczaj opiera się na włączeniu 3H-TdR do DNA komórek wątroby, które posiadają niską częstotliwość komórek w fazie S cyklu komórkowego. Pobór 3H-TdR zazwyczaj jest oznaczany przez auroradiografię, ze względu na to, że ta technika nie jest tak podatna na zakłócenie przez komórki fazy S jak na przykład płynne podliczanie scyntalizacji.
1.4. OPIS METODY
1.4.1. Preparaty
1.4.1.1. Wybór gatunków zwierząt
Powszechnie wykorzystywane są szczury, chociaż każdy odpowiedni gatunek ssaków może zostać wykorzystany. Powszechnie wykorzystywane szczepy laboratoryjne młodych, zdrowych dojrzałych zwierząt powinny zostać wykorzystane. W trakcie przeprowadzania badania, zmienność masy zwierząt powinno być minimalne oraz nie powinno przekraczać ± 20 % średniej masy dla każdej z płci.
1.4.1.2. Warunki przetrzymywania oraz karmienia
Ogólne warunki określone w Ogólnym wprowadzeniu do części B są stosowane, chociaż celem w odniesieniu do wilgotności powinien być poziom 50–60 %.
1.4.1.3. Przygotowanie zwierząt
Zdrowe młode dorosłe osobniki zwierząt są losowo przypisane do grup kontrolnych oraz grup poddanych działaniu substancji. Klatki powinny zostać przygotowane w sposób minimalizujący możliwe skutki negatywne wynikające z umieszczenia w klatce. Zwierzęta są identyfikowane oraz aklimatyzowane do warunków laboratoryjnych przynajmniej przez okres pięciu dni.
1.4.1.4. Substancja badana/przygotowanie
Stała substancja badana powinna zostać rozpuszczona lub zawieszona w odpowiednich rozpuszczalnikach lub nośnikach oraz rozcieńczona, jeżeli jest to odpowiednie przed dawkowaniem zwierzętom. Płynne substancje mogą być dawkowane bezpośrednio lub rozcieńczane przed dawkowaniem. Powinny zostać wykorzystane świeżo przygotowywane substancje, chyba że dane dotyczące stabilności wskazują, iż dopuszczalne jest składowanie substancji
1.4.2. Warunki badania
1.4.2.1. Rozpuszczalnik/nośnik
Rozpuszczalnik/nośnik nie powinien wywoływać skutków toksycznych przy wykorzystanym poziomie dawek oraz w odniesieniu do niego nie powinno być spodziewane reagowanie chemiczne z substancją badaną. Jeśli inne niż dobrze znane rozpuszczalniki/nośniki są wykorzystywane, ich włączenie do doświadczenia powinno być wsparte danymi wskazującymi ich zgodność. Zaleca się, o ile to możliwe, aby w pierwszej kolejności rozważone zostało wykorzystanie wodnych rozpuszczalników/nośników.
1.4.2.2. Kontrole
Równoległe pozytywne oraz negatywne kontrole (rozpuszczalnik/nośnik) powinny zostać objęte każdą, niezależnie przeprowadzaną częścią doświadczenia. Z wyjątkiem poddawania działaniu substancji badanej, zwierzęta w grupie kontrolnej powinny być poddawane działaniom w identyczny sposób, jak zwierzęta w grupach poddanych działaniu substancji.
Pozytywnymi kontrolami powinny być substancje znane z wytwarzania UDS, jeżeli są podawane przy poziomie poddania działaniu substancji, w odniesieniu do którego oczekuje się, iż da wykrywalny wzrost powyżej tła. Pozytywne kontrole, potrzebujące aktywacji metabolicznej powinny być wykorzystywane, jeżeli są podawane przy dawkach wywołujących umiarkowane reakcje (4). Dawki mogą zostać wybrane tak, aby skutki były jasne, ale aby nie ujawniały natychmiast badającemu tożsamości zakodowanych szkiełek mikroskopowych. Przykładami pozytywnych substancji kontrolnych są:
|
Okresy pobierania próbek |
Substancje |
Nr CAS |
Nr Einecs |
|
Wczesne okresy pobierania próbek (2–4 godzin) |
N-nitrozodimetyloamina |
62-75-9 |
200-249-8 |
|
Późne okresy pobierania próbek (12-16 godzin) |
N-2-fluorenyloacetamid (2-AAF) |
53-96-3 |
200-188-6 |
Inne odpowiednie kontrolne substancje chemiczne mogą zostać wykorzystane. Dopuszczalne jest, by pozytywne kontrole były podawane drogą inną niż substancja badana.
1.5. PROCEDURA
1.5.1. Liczba oraz płeć zwierząt
Powinna zostać wykorzystana odpowiednia liczba zwierząt w celu uwzględnienia naturalnej biologicznej zmienności reakcji. Liczba zwierząt powinna przynajmniej stanowić trzy nadające się do poddania analizie osobniki na grupę. W przypadku gdy została zgromadzona znacząca baza danych, jedynie jedno zwierzę lub dwa zwierzęta są wymagane w odniesieniu do równoległych negatywnych i pozytywnych grup kontrolnych.
Jeżeli w trakcie badania istnieją dostępne dane z badań dotyczących tego samego gatunku, wykorzystując tę samą drogę poddania działaniu substancji, które wskazują, że nie istnieją zasadnicze różnice w toksyczności między płciami, badanie jednej płci będzie wystarczające. W przypadku poddawania człowieka działaniu substancji, badanie może być specyficzne co do płci, tak jak na przykład w odniesieniu do niektórych środków farmaceutycznych, badanie powinno zostać przeprowadzone z udziałem zwierząt odpowiedniej płci.
1.5.2. Harmonogram poddania działaniu substancji
Substancje badane są w ujęciu ogólnym podawane jako pojedynczy zabieg poddania działaniu substancji.
1.5.3. Poziomy dawek
Zazwyczaj wykorzystywane są przynajmniej dwa poziomy dawek. Najwyższa dawka jest określona jako dawka wywołująca oznaki toksyczności a wyższe poziomy dawek oparte na tym samym reżimie dawkowania mogłyby być zabójcze. Ogólnie niższa dawka powinna wynosić od 50 % do 25 % wysokiej dawki.
Substancje o szczególnej aktywności biologicznej przy niskich nietoksycznych dawkach (takie jak hormony i mitogeny) mogą stanowić wyjątek w odniesieniu do kryteriów ustalania dawki i powinny być oceniane na zasadzie jednostkowych przypadków. Jeżeli przeprowadzane jest studium określające zakres badania, powinno być ono przeprowadzone w tym samym laboratorium, wykorzystując ten sam reżim gatunku, szczepu, płci oraz poddawania działaniu substancji, który ma zostać wykorzystany w badaniu głównym.
Najwyższa dawka może być również określona jako dawka, która wywołuje pewne wskazania toksyczności w wątrobie (np. jądrze piknotycznym).
1.5.4. Badanie ograniczone
Jeżeli badanie w oparciu o jeden poziom dawek o przynajmniej 2 000 mg/kg masy ciała zastosowane w pojedynczym poddaniu działaniu substancji lub jako dwa zabiegi poddania działaniu substancji tego samego dnia, nie daje dostrzegalnych wyników toksycznych, oraz jeżeli nie podejrzewa się genotoksyczności w oparciu o dane dotyczące substancji powiązanych strukturalnie, wówczas przeprowadzenie pełnego badania może nie być konieczne. Spodziewa się, iż poddanie człowieka działaniu substancji może wykazać potrzebę wykorzystania większej dawki w badaniu ograniczonym.
1.5.5. Dawkowanie
Substancja badana jest zazwyczaj podawana przez zgłębnik żołądkowy lub odpowiednią rurkę intubacyjną. Inne drogi poddania działaniu substancji mogą być dopuszczalne w przypadku gdy ich wykorzystanie jest uzasadnione. Jednakże droga wewnątrzotrzewnowa nie jest zalecana ze względu na to, że mogłaby poddać wątrobę bezpośrednio działaniu substancji badanej, co jest bardziej prawdopodobne niż w przypadku poddania działaniu substancji przez krwiobieg. Maksymalna objętość płynu, jaka może być podana przez zgłębnik lub wstrzyknięta jednorazowo zależy od wielkości badanego zwierzęcia. Objętość nie powinna przekraczać 2 ml/100 g masy ciała. Wykorzystanie objętości wyższych niż te musi być uzasadnione. Z wyjątkiem podrażniających lub żrących substancji, które zazwyczaj ujawniają zwiększone skutki o dużym stężeniu, zmienności w objętości substancji badanej powinny zostać zminimalizowane przez dostosowanie stężenia w celu zapewnienia stałej objętości przy wszystkich poziomach dawek.
1.5.6. Przygotowanie komórek wątroby
Komórki wątroby przygotowywane są z poddanych działaniu substancji zwierząt zazwyczaj w 12–16 godzin po dawkowaniu. Dodatkowy wcześniejszy okres pobierania próbek (zazwyczaj 2–4 godzin po poddaniu działaniu substancji) jest generalnie niezbędny, chyba że istnieje jasna pozytywna reakcja przy 12–16 godzinach. Jednakże alternatywne okresy pobierania próbek mogą być wykorzystane, jeżeli są uzasadnione na podstawie danych toksykokinetycznych.
Krótkookresowe hodowle komórek wątrobowych ssaków są zazwyczaj ustanawiane przez przecedzanie wątroby in situ z kolagenazą i pozwalając na przytwierdzenie świeżo dysocjowanych komórek wątroby do odpowiedniej powierzchni. Komórki wątroby ze zwierząt kontroli negatywnych powinny posiadać zdolność do życia (5) wynoszącą przynajmniej 50 %.
1.5.7. Oznaczanie UDS
Świeżo oddzielone komórki wątroby ssaków są inkubowane zazwyczaj w podłożu zawierającym 3H-TdR przez odpowiedni okres czasu, np. 3–8 godzin. Na koniec okresu inkubacji podłoże powinno zostać usunięte od komórek, które mogą następnie być inkubowane w podłożu zawierającym nadmiar nieoznaczonej tymidyny w celu zmniejszenia niewłączonej radioaktywności („zimny obieg”). Następnie komórki są płukane, stabilizowane oraz suszone. W odniesieniu do okresów inkubacji bardziej przedłużonych w czasie zimny obieg może nie być konieczny. Szkiełka mikroskopowe są zanurzane w emulsji autoradiograficznej, poddawane działaniu substancji w ciemności (np. przetrzymywane w lodówce przez 7–14 dni), wytwarzane, barwione, a srebrne ziarna są podliczane. Przygotowywane są dwa lub trzy szkiełka mikroskopowe dla każdego zwierzęcia.
1.5.8. Analiza
Preparaty szkiełek mikroskopowych powinny zawierać wystarczającą liczbę komórek o normalnej morfologii w celu umożliwienia przeprowadzenia mającej znaczenie oceny UDS. Preparaty są badane mikroskopowo pod kątem oznak jawnej cytotoksyczności (np. pyknoza, zmniejszony poziom oznaczenia radioaktywności).
Szkiełka mikroskopowe powinny być kodowane przed podliczeniem ziaren. Zazwyczaj 100 komórek jest ocenianych z każdego zwierzęcia, z przynajmniej dwu szkiełek mikroskopowych; ocena mniejszej ilości niż 100 komórek/zwierzę powinna zostać uzasadniona. Ilości ziaren nie są oceniane punktowo w odniesieniu do S-fazy jądra, ale udział komórek S-fazy może zostać odnotowany.
Ilość włączenia 3H-TdR w jądrze oraz cytoplazmy normalnych pod względem morfologii komórek, jak potwierdzono przez osadzanie się srebrnych ziaren, powinna być oznaczona odpowiednią metodą.
2. DANE
2.1. PRZETWARZANIE WYNIKÓW
Powinny zostać dostarczone indywidualne szkiełka mikroskopowe oraz dane dotyczące zwierzęcia. dodatkowo wszystkie dane powinny zostać podsumowane w formie tabelarycznej. Względna ilość ziarnistości jądrowych (NNG) powinna zostać obliczona dla każdej komórki, dla każdego zwierzęcia oraz dla każdej dawki oraz czasu przez odjęcie ilości CG od ilości ziarnistości jądrowych (NG). Jeśli „komórki w naprawie” są podliczane, kryteria oznaczania „komórek w naprawie” powinny zostać uzasadnione oraz oparte na historycznych danych dotyczących kontroli negatywnej lub danych dotyczących równoległej kontroli negatywnej. Wyniki liczbowe mogą być ocenione z wykorzystaniem metod statystycznych. Jeżeli są wykorzystywane, badania statystyczne powinny zostać wybrane oraz uzasadnione przed przeprowadzeniem badania.
2.2. OCENA ORAZ INTERPRETACJA WYNIKÓW
Przykłady kryteriów w odniesieniu do pozytywnych/negatywnych reakcji obejmują:
|
pozytywne |
(i) |
wartości NNG powyżej istniejącego progu, który jest uzasadniony na podstawie historycznych danych laboratoryjnych; lub |
|
|
(ii) |
wartości NNG znacząco wyższe w porównaniu z równoległą kontrolą; |
|
negatywne |
(i) |
wartości NNG w zakresie poniżej historycznego progu kontrolnego; lub |
|
|
(ii) |
wartości NNG nieznacznie wyższe w porównaniu z równoległą kontrolą. |
Biologiczne znaczenie danych powinno zostać wzięte pod uwagę, np. parametry takie jak: zmienność międzyzwierzęca, relacja między dawką, a reakcją oraz cytotoksyczność powinny zostać uwzględnione. Metody statystyczne mogą być wykorzystane jako metody pomocnicze w ocenie wyników badania. Jednakże znaczenie statystyczne nie powinno być jedynym czynnikiem określającym w odniesieniu do reakcji pozytywnej.
Chociaż większość doświadczeń będzie dawało w jasny sposób pozytywne lub negatywne wyniki, w rzadkich przypadkach zestaw danych będzie uniemożliwiał dokonanie ostatecznej oceny dotyczącej aktywności substancji badanej. Wyniki mogą pozostać niejednoznaczne lub sporne niezależnie od tego, ile razy doświadczenie było powtarzane.
Wyniki pozytywne z testu UDS z komórkami wątroby ssaków in vivo wskazują, że substancja badana wywołuje uszkodzenia DNA w komórkach wątroby ssaków in vivo, które mogą zostać naprawione w drodze testu poreperacyjnej syntezy DNA in vitro. Wyniki negatywne wskazują, że w warunkach badawczych, badana substancja nie wywołuje uszkodzeń DNA, które są wykrywane przez to badanie.
Prawdopodobieństwo, że substancja badana dociera do głównego obiegu lub szczególnie do tkanki docelowej (np. toksyczność systemowa) powinno zostać omówione.
3. SPRAWOZDAWCZOŚĆ
SPRAWOZDANIE Z BADANIA
Sprawozdanie z badania musi zawierać następujące informacje:
|
|
Rozpuszczalnik/nośnik:
|
|
|
Badane zwierzęta:
|
|
|
Warunki badania:
|
|
|
Wyniki:
|
|
|
Omówienie wyników. |
|
|
Wnioski. |
4. BIBLIOGRAFIA
|
(1) |
Ashby, J. Lefevre, P. A., Burlinson, B. and Penman, M. G. (1985), An Assessment of the In vivo Rat. Hepatocyte DNA Repair Assay, Mutation Res., 156, pp. 1–18. |
|
(2) |
Butterworth, B. E., Ashby, J., Bermudez, E., Casciano, D., Mirsalis, J., Probst, G. and Williams, G. (19 8 7), A Protocol and Guide for the In vivo Rat Hepatocyte DNA Repair Assay, Mutation Res., 189, pp. 123–133. |
|
(3) |
Kennelly, J. C, Waters, R., Ashby, J., Lefevre, P. A., Burlinson, B., Benford, D. J., Dean, S. W. and Mitchell, I. de G. (1993), In vivo Rat Liver UDS Assay, in: Kirkland D. J. and Fox M., (eds), Supplementary Mutagenicity TestS: UKEM Recommended Procedures. UKEMS Subcommittee on Guidelines for Mutagenicity Testing. Report. Part II revised, Cambridge University Press, Cambridge, New York, Port Chester, Melbourne, Sydney, pp. 52–77. |
|
(4) |
Madle, S., Dean, S. W., Andrac, U., Brambilla. G., Burlinson, B., Doolittle, D. J., Furihata, C, Hertner, T., McQueen, C. A. and Mori, H. (19 9 3), Recommendations for the Performance of UDS Tests In vitro and In vivo, Mutation Res., 312, pp. 263–285. |
|
(5) |
Fautz, R., Hussain, B., Efstathiou, E. and Hechenberger-Freudl. C. (1993), Assessment of the Relation Between the Initial Viability and the Attachment of Freshly Isolated Rat Hepatocytes Used for the In vivo/In vitro DNA Repair Assay (UDS), Mutation Res., 291, pp. 21–27. |
|
(6) |
Mirsalis, J. C, Tyson, C. K. and Butterworth, B. E. (1982), Detection of Genotoxic Carcinogens in the In vivo/In vitro Hepalocyte DNA Repair Assay, Environ. Mutagen, 4, pp. 553–562. |
B.40. BADANIE NISZCZENIA SKÓRY METODĄ IN VITRO: TEST PRZEZSKÓRNEGO OPORU ELEKTRYCZNEGO (TER)
1. METODA
Opisywana metoda badania jest zgodna z OECD TG 430 (2004).
1.1. WSTĘP
Zniszczenie skóry oznacza powstawanie nieodwracalnych uszkodzeń tkanek skórnych w wyniku kontaktu z substancją testową (jak zdefiniowano w Globalnie Zharmonizowanym Systemie Klasyfikacji i Oznakowania Produktów Chemicznych (GHS))(1). Metoda ta umożliwia stosowanie procedury, w której ocena poziomu uszkadzania skóry nie jest przeprowadzana na żywych zwierzętach.
Do tej pory ocena zniszczenia skóry wiązała się zwykle z użyciem zwierząt laboratoryjnych (2). Potrzeba wyeliminowania bólu i cierpienia zwierząt została uwzględniona w modyfikacji metody badania B.4, umożliwiającej określenie zniszczenia skóry przy użyciu alternatywnych metod in vitro, bez zadawania zwierzętom bólu i cierpienia.
Pierwszym krokiem w kierunku zdefiniowania alternatywnych testów, które mogłyby zostać zastosowane do celów regulacyjnych, było przeprowadzenie badań prewalidacyjnych (3). Następnie przeprowadzono formalne badania walidacyjne (6)(7)(8) oceny zniszczenia skóry metodami in vitro (4)(5). Wyniki tych badań oraz inne publikacje stały się podstawą zalecenia, że do oceny niszczenia skóry in vivo można stosować następujące testy (9)(10)(11): test modelu skóry ludzkiej (zob. metoda badania B.40 bis) oraz test przezskórnego oporu elektrycznego (niniejsza metoda).
Badania walidacyjne oraz opublikowane wyniki innych badań wskazują, że test przezskórnego oporu elektrycznego (TER)(12)(13) pozwala na wiarygodne rozróżnienie znanych substancji niszczących skórę i nieniszczących skóry (5)(9).
Test przedstawiony w opisie tej metody umożliwia identyfikację związków i mieszanin chemicznych uszkadzających skórę. Ponadto w połączeniu z dowodami uzyskanymi na podstawie innych dostępnych informacji (np. pH, związek między strukturą a aktywnością, dane dotyczące człowieka i zwierząt) umożliwia identyfikację związków i mieszanin, które nie niszczą skóry (1)(2)(11)(14). Nie dostarcza informacji o podrażnieniach skóry, nie umożliwia także dzielenia substancji uszkadzających na podkategorie, jak dopuszczono w Globalnie Zharmonizowanym Systemie Klasyfikacji (GHS) (1).
Zaleca się, aby w celu pełnej oceny miejscowego działania na skórę po pojedynczej ekspozycji postępować zgodnie z sekwencyjną strategią przedstawioną w dodatku do metody badania B.4 (2) i w Globalnie Zharmonizowanym Systemie (GHS) (1). Ta strategia testowa obejmuje przeprowadzenie testów in vitro zniszczenia skóry (jak przedstawiono w opisie metody) i podrażnienia skóry przed rozważaniem przeprowadzenia badań na żywych zwierzętach.
1.2. DEFINICJE
Zniszczenie skóry in vivo : oznacza spowodowanie nieodwracalnych uszkodzeń skóry: a mianowicie widocznej martwicy skóry w warstwie naskórka i skóry właściwej w wyniku kontaktu z substancją testową w okresie do 4 godzin. Typowymi objawami zniszczenia skóry są wrzody, krwawienie, krwawiące strupy oraz, pod koniec 14-dniowego okresu obserwacji, odbarwienie skóry spowodowane zbieleniem, pojawianie się całkowitych łysin i blizn. W przypadku wątpliwych uszkodzeń należy zastosować metody histopatologiczne.
Przezskórny opór elektryczny (TER): jest miarą impedancji skóry, wartością oporu elektrycznego w kiloomach. Jest to prosta i pewna metoda ocena funkcjonowania bariery skórnej przez rejestrowanie przy użyciu mostka Wheatstone'a przechodzenia przez skórę jonów.
1.3. SUBSTANCJE ODNIESIENIA
Tabela 1
Chemiczne substancje odniesienia
|
Nazwa |
Nr EINECS |
Nr CAS |
|
|
1,2-diaminopropan |
201-155-9 |
78-90-0 |
Silne działanie niszczące |
|
Kwas akrylowy |
201-177-9 |
79-10-7 |
Silne działanie niszczące |
|
2-tert. Butylofenol |
201-807-2 |
88-18-6 |
Działanie niszczące |
|
Wodorotlenek potasu (10 %) |
215-181-3 |
1310-58-3 |
Działanie niszczące |
|
Kwas siarkowy (10 %) |
231-639-5 |
7664-93-9 |
Działanie niszczące |
|
Kwas oktanowy (kwas kaprylowy) |
204-677-5 |
124-07-02 |
Działanie niszczące |
|
4-amino-1,2,4-triazol |
209-533-5 |
584-13-4 |
Brak działania niszczącego |
|
Eugenol |
202-589-1 |
97-53-0 |
Brak działania niszczącego |
|
Bromek fenetylu |
203-130-8 |
103-63-9 |
Brak działania niszczącego |
|
Tetrachloroetylen |
204-825-9 |
27-18-4 |
Brak działania niszczącego |
|
Kwas izostearynowy |
250-178-0 |
30399-84-9 |
Brak działania niszczącego |
|
4-(metylotio)-benzaldehyd |
222-365-7 |
3446-89-7 |
Brak działania niszczącego |
Większość z wymienionych związków chemicznych pochodzi z wykazu chemikaliów przygotowanego na potrzeby międzynarodowego studium atestacyjnego ECVAM (4). Ich wybór opiera się na następujących kryteriach:
|
(i) |
taka sama liczba substancji niszczących, jak i nieniszczących; |
|
(ii) |
dostępne w handlu substancje należące do większości stosownych klas związków chemicznych; |
|
(iii) |
uwzględnienie zarówno silnie niszczących jak i słabiej niszczących substancji, aby umożliwić rozróżnianie na podstawie właściwości niszczących; |
|
(iv) |
wybór chemikaliów, które mogą być poddawane obróbce w laboratorium nie stwarzając zagrożeń innych niż zniszczenie skóry. |
1.4. ZASADA METODY BADANIA
Materiał testowy jest nakładany na okres do 24 godzin na naskórkową powierzchnię krążków skórnych w dwuczęściowym systemie testowym, w którym krążki pełnią funkcję przegrody między dwoma częściami. Krążki skóry pobierane są od szczurów w wieku 28–30 dni uśmiercanych w humanitarny sposób. Materiały uznawane są za niszczące na podstawie ich zdolności do uszkadzania struktury warstwy rogowej i zaburzania funkcji izolacyjnej, co mierzone jest spadkiem TER poniżej poziomu progowego (12). W przypadku TER u szczurów przyjęto wartość graniczną 5 kΩ; wartość tę ustalono na podstawie wielu danych dla szerokiego zakresu związków chemicznych, dla których znaczna większość wartości było albo wyraźnie wyższych (często >10 kΩ) albo znacznie niższych (często < 3 kΩ) od tej wartości (12). Generalnie materiały, które w przypadku zwierząt nie niszczą skóry, tylko ją podrażniają lub w ogóle jej nie podrażniają nie powodują spadku TER poniżej tej wartości graniczną. Co więcej, zastosowanie innych preparatów skóry lub innego sprzętu może zmienić wartość graniczną, powodując konieczność dalszej walidacji.
Etap wiązania barwnika jest włączony do procedury testowej mającej potwierdzić dodatnie wyniki TER, w tym wartości w pobliżu 5 kΩ. Etap wiązania barwnika pozwala na stwierdzenie, czy wzrost przepuszczalności jonów jest spowodowany fizycznym zniszczeniem warstwy rogowej. Wykazano, że metoda TER z wykorzystaniem skóry szczurów świadczy o potencjale niszczącym in vivo, badanym metodą badania B.4 (2) u królików. Należy zauważyć, że test in vivo na królikach jest w porównaniu z testem wycinka ludzkiej skóry wysoce konserwatywny, jeśli chodzi o potencjał niszczenia skóry (15).
1.5. OPIS METODY BADANIA
1.5.1. Zwierzęta
Szczury są gatunkiem preferowanym, ponieważ wcześniej wykazano wrażliwość ich skóry na substancje chemiczne (10). Wiek (w momencie pobierania skóry) i linia szczurów mają zasadnicze znaczenie, bowiem od tego zależy, czy mieszki włosowe są w stanie w stanie uśpienia poprzedzającym fazę wzrostu włosa u dorosłych osobników.
Włosy z grzbietu i boku ciała młodych, około 22-dniowych samców lub samic szczurów (pochodzących od linii Wistar lub podobnych), są starannie usuwane mała pincetą. Następnie zwierzęta są czyszczone przez staranne przetarcie, natomiast miejsca, z których usuwano włosy, są pokrywane roztworem antybiotyku (zawierającym na przykład streptomycynę, penicylinę, chloramfenikol i amfoterycynę w stężeniach zapewniających zahamowanie rozwoju bakterii). Zwierzęta są przemywane antybiotykiem powtórnie trzeciego lub czwartego dnia po pierwszym przemyciu i są poddawane eksperymentowi w ciągu trzech dni od drugiego przemycia, kiedy warstwa rogowa ulegnie regeneracji po usuwaniu włosów.
1.5.2. Przygotowanie krążków skóry
Zwierzęta są zabijane w sposób humanitarny, kiedy mają 28–30 dni; wiek ma tu szczególne znaczenie. Z grzbietu i boków ciała każdego zwierzęcia pobierana jest skóra, z której następnie usuwa się starannie nadmiar tłuszczu podskórnego. Pobierane są krążki skóry o średnicy około 20 mm. Przed użyciem krążków skóra może być przechowywana, jeśli wykazano, że dane dla kontroli pozytywnej i negatywnej są równoważne z danymi uzyskanymi przy użyciu świeżej skóry.
Każdy krążek skóry jest umieszczany nad jednym z końców rurki TFE (politetrafluoroetylenowej), przy czym trzeba dopilnować, aby powierzchnia naskórkowa przylegała do rurki. Na koniec rury nakładany jest gumowy pierścień, który utrzymuje skórę w odpowiedniej pozycji, a nadmiar tkanki jest odcinany. Wymiary rurki i pierścienia pokazano na rysunku 2. Połączenie między rurką PTFE a gumowym pierścieniem jest następnie starannie uszczelniane wazeliną. Rurka jest następnie mocowana sprężynowym zaciskiem wewnątrz komory receptorowej zawierającej roztwór MgSO4 (154 mM) (rysunek 1). Krążek skóry powinien być całkowicie zanurzony w roztworze MgSO4. Z jednej skóry szczura można otrzymać 10–15 krążków.
Przed rozpoczęciem testów dla każdej skóry zwierzęcej mierzy się opór elektryczny dwóch krążków skórnych, co stanowi procedurę kontroli jakości. Aby reszta krążków mogła zostać użyta do testów, wartości oporu dla obu krążków powinny być wyższe niż 10 kΩ. Jeśli wartość oporu jest mniejsza niż 10 kΩ, resztę krążków z tej skóry należy wyrzucić.
1.5.3. Nakładanie substancji testowej i kontrolnej
Aby zapewnić odpowiednią jakość testu, w każdym badaniu należy równocześnie stosować kontrolę pozytywną i negatywną. Należy używać krążków ze skóry jednego zwierzęcia. Jako substancji kontroli pozytywnej i negatywnej zaleca się używać odpowiednio 10 M kwasu solnego i wody destylowanej.
Płynne substancje testowe nakłada się równomiernie na powierzchnię naskórkową wewnątrz rurki (150 μL). Aby mieć pewność w czasie badania materiałów stałych, że pokryta jest cała powierzchnia, na krążek nakłada się równomiernie odpowiednią ilość materiału stałego. Do substancji stałej dodaje się wodę dejonizowaną (150 uL) i delikatnie potrząsa się rurką. W celu zapewnienia maksymalnego kontaktu cząstek stałych ze skórą może być konieczne albo ich podgrzanie do 30 oC, aby je stopić lub zmiękczyć, albo zmielenie, aby otrzymać granulat lub proszek.
Dla każdej substancji testowej i kontrolnej stosuje się trzy krążki skóry. Substancje testowe nakłada się na 24 godziny przy temperaturze 20–23 oC. Substancja testowa jest zmywana strumieniem wody wodociągowej o temperaturze 30 oC do momentu, aż nie da się już usunąć więcej materiału.
1.5.4. Pomiary TER
Impedancja skóry jest mierzona jako TER za pomocą niskonapięciowego mostka Wheatstone'a przy zastosowaniu prądu zmiennego (13). Ogólne dane techniczne mostka to napięcie robocze 1–3 V, prąd zmienny 50–100 Hz sinusoidalny lub prostokątny i zakres pomiaru co najmniej 0,1–30 kΩ Mostek stosowany w badaniach walidacyjnych mierzy indukcyjność, opór bierny pojemnościowy i opór do wartości odpowiednio 2 000 H, 2 000 μF oraz 2 MΩ, przy częstotliwości 100 Hz lub 1 kHz i przy połączeniach szeregowych lub równoległych. Do celów oceny poziomu niszczenia skóry TER dokonuje się pomiarów oporu przy częstotliwości 100 Hz i przy zastosowaniu połączeń szeregowych. Przed dokonaniem pomiaru oporu elektrycznego zmniejsza się napięcie skóry przez dodanie dostatecznej ilości 70 % etanolu, aby pokryć naskórek. Po paru sekundach usuwa się etanol z rurki, a tkankę uwadnia przez dodanie 3 mL roztworu MgSO4 (154 mM). W celu zmierzenia oporu krążka skóry w kΩ, po obu jego stronach są umieszczane elektrody mostka (rysunek 1). Wymiary elektrod i długość elektrod obnażonych poniżej zacisku szczękowego są pokazane na rysunku 2. W czasie pomiaru oporu zacisk przymocowany do wewnętrznej elektrody opiera się na szczycie rurki PTFE, aby mieć pewność, że w roztworze MgSO4 zanurzony jest ciągły odcinek elektrody. Elektroda zewnętrzna umieszczona jest wewnątrz części receptorowej, na jej dnie. Należy utrzymywać stałą odległość między zaciskiem szczękowym a dnem rurki PTFE (rysunek 2), ponieważ odległość ma wpływ na otrzymywane wartości oporu. W związku z tym odległość między wewnętrzną elektrodą a krążkiem skóry powinna być stała i minimalna (1–2 mm).
Jeśli zmierzona wartość oporu jest większa niż 20 kΩ, może to być spowodowane pozostałościami substancji testowej pokrywającej naskórkową powierzchnię krążka skóry. Można próbować usunąć tę warstwę, na przykład zatykając rurkę palcem w rękawiczce i potrząsając rurką przez około 10 sekund; usuwa się roztwór MgSO4 i powtarza pomiar oporu przy zastosowaniu nowej porcji MgSO4.
Na otrzymane wartości TER mogą mieć wpływ charakterystyka i wymiary aparatu testowego oraz zastosowana procedura eksperymentalna. Na podstawie danych uzyskanych przy użyciu określonej aparatury i procedury, przedstawionych w opisie tej metody, ustalono 5 kΩ próg poziomu niszczenia skóry. Jeśli warunki testu zostaną zmienione lub jeśli stosuje się inną aparaturę, zastosowanie mogą znajdować inne progi i wartości kontrolne. Tak więc konieczne jest dokonanie kalibracji metodyki i wartości progów oporu przez zbadanie szeregu wzorców wybranych spośród substancji chemicznych użytych w badaniu walidacyjnym (4) (5) lub spośród związków chemicznych podobnych do badanych substancji. Zestaw odpowiednich substancji wzorcowych zamieszczono w tabeli 1.
1.5.5. Metody wiązania barwników
Działanie niektórych materiałów nieniszczących skóry może prowadzić do zmniejszenia oporu poniżej wartości granicznej 5 kΩ, umożliwiając przechodzenie jonów przez warstwę rogową i w ten sposób redukując opór elektryczny (5). Na przykład obojętne związki organiczne oraz związki powierzchniowo czynne (w tym detergenty, emulgatory i inne surfaktanty) mogą usuwać ze skóry tłuszcz, powodując, że bariera skórna staje się bardziej przepuszczalna dla jonów. Tak więc, jeśli wartości TER substancji testowych są niższe niż lub zbliżone do 5 kΩ przy braku widocznego uszkodzenia, należy dla tkanek kontrolnych i badanych przeprowadzić test przenikania barwnika, aby stwierdzić, czy uzyskane wartości TER są wynikiem zwiększonej przepuszczalności skóry czy jej uszkodzenia (3)(5). W tym drugim przypadku, jeśli uszkodzona jest warstwa rogowa, barwnik sulforodamina B szybko przenika w głąb skóry i zabarwia leżącą głębiej tkankę. Ten szczególny barwnik nie wchodzi w reakcję z wieloma różnymi związkami chemicznymi i nie ma na niego wpływu procedura ekstrakcji opisana poniżej.
1.5.5.1. Stosowanie barwnika sulforodaminy B i jego usuwanie
Po pomiarze TER siarczan magnezu jest usuwany z rurki i skóra jest poddawana dokładnym oględzinom, aby sprawdzić, czy są na niej widoczne uszkodzenia. Jeśli nie ma oczywistych dużych uszkodzeń, na naskórkową powierzchnię każdego krążka skóry nakładany jest na dwie godziny barwnik sulforodamina B (Acid Red 52; C.I. 45100; numer EINECS 222-529-8; numer CAS 3520-42-1), 150 uL 10 % (masa/obj.), rozpuszczony w wodzie destylowanej. Krążki skóry są następnie przemywane wodą wodociągową w temperaturze nie wyższej od pokojowej przez około 10 sekund, aby usunąć wszelki zbędny/niezwiązany barwnik. Każdy krążek skóry jest starannie usuwany z rurki PTFE i umieszczany w fiolce (np. 20 mL szklanej fiolce scyntylacyjnej) zawierającej wodę dejonizowaną (8 mL). Fiolki są delikatnie wstrząsane przez 5 minut, aby usunąć cały nadmiar niezwiązanego barwnika. Procedura płukania jest powtarzana, po czym krążki skóry są wyjmowane i umieszczane w fiolkach zawierających 5 ml 30 % (masa/obj.) roztworu sodowego siarczanu dodecylu (SDS) w wodzie destylowanej i inkubowane przez noc w 60 oC.
Po inkubacji wszystkie krążki skóry są wyjmowane i usuwane, a pozostały roztwór jest wirowany przez 8 minut w temperaturze 21 oC (względna siła odśrodkowa ~ 175 × g). Próbka o objętości 1 mL supernatantu jest rozpuszczana w stosunku 1 do 5 (obj./obj) [tj. 1 mL + 4 mL] z 30 % (masa/obj.) SDS w wodzie destylowanej. Gęstość optyczną (OD) mierzy się przy 565 nm.
1.5.5.2. Obliczanie zawartości barwnika
Zawartość barwnika sulforodaminy B na krążek obliczana jest na podstawie wartości OD (5) (współczynnik molowy ekstynkcji sulforodaminy B przy 565 nm = 8,7 × 104; ciężar cząsteczkowy = 580). Zawartość barwnika jest określana dla wszystkich krążków skóry przy zastosowaniu odpowiedniej krzywej kalibracyjnej, a następnie dla wszystkich powtórzeń liczona jest średnia.
2. DANE
Wartości oporu (kΩ) i średnie zawartości barwnika (ug/krążek) dla materiału testowego, jak również dla kontroli pozytywnej i negatywnej, powinny być tam gdzie to właściwe przedstawione w formie tabelarycznej (poszczególne dane z prób i średnie ± S.D.), w tym dane dla powtórzeń/powtarzanych eksperymentów, średnie i poszczególne dane.
2.1. INTERPRETACJA WYNIKÓW
Średnie wyniki TER są uznawane, jeśli w testującym laboratorium wartości otrzymane w jednoczesnych próbach kontroli pozytywnej i negatywnej mieszczą się w zakresie dopuszczalnym dla metody. Dopuszczalne zakresy oporu dla metodyki i aparatury opisanych powyżej podano w poniższej tabeli:
|
Kontrola |
Substancja |
Zakres oporu (kΩ) |
|
Pozytywna |
10 M kwas solny |
0,5– 1,0 |
|
Negatywna |
Woda destylowana |
10–25 |
Średnie wyniki wiązania barwnika są akceptowane pod warunkiem, że wartości otrzymane w równoczesnych testach kontrolnych mieszczą się w przedziałach akceptowanych dla metody. Sugerowane dopuszczalne zakresy zawartości barwnika dla substancji kontrolnych dla metodyki i aparatury opisanych powyżej zamieszczono w poniższej tabeli:
|
Kontrola |
Substancja |
Zakres zawartości barwnika (μg/krążek) |
|
Pozytywna |
10 M kwas solny |
40–100 |
|
Negatywna |
Woda destylowana |
15–35 |
Substancja testowa uznawana jest za nieniszczącą skóry:
|
(i) |
jeśli średnia wartość TER otrzymana dla substancji testowej jest większa niż 5 kΩ lub |
|
(ii) |
średnia wartość TER jest niższa od lub równa 5 kΩ; i
|
Substancja testowa uznawana jest za niszczącą skórę:
|
(i) |
jeśli średnia wartość TER jest niższa od lub równa 5 kΩ, a krążek skóry jest niewątpliwie uszkodzony; lub |
|
(ii) |
średnia wartość TER jest niższa od lub równa 5 kΩ; i
|
3. SPRAWOZDAWCZOŚĆ
3.1. SPRAWOZDANIE DOTYCZĄCE BADANIA
Sprawozdanie z badania musi zawierać następujące informacje:
|
|
Substancje testową i kontrolną:
|
|
|
Zwierzęta testowe:
|
|
|
Warunki testu:
|
|
|
Wyniki:
|
|
|
Dyskusja wyników. |
|
|
Wnioski |
4. BIBLIOGRAFIA
|
(1) |
OECD (2001) Harmonised Integrated Classification System for Human Health and Environmental Hazards of Chemical Substances and Mixtures. OECD Series on Testing and Assessment Number 33. ENV/JM/MONO(2001)6, Paris. http ://www.olis.oecd.org/olis/2001doc.nsf/LinkTo/env-jm-mono(2001)6. |
|
(2) |
Testing Method B.4. Acute Toxicity: Dermal Irritation/Corrosion. |
|
(3) |
Botham, P.A., Chamberlain, M., Barratt, M.D., Curren, R.D., Esdaile, D.J., Gardner, J.R., Gordon, V.C., Hildebrand, B., Lewis, R.W., Liebsch, M., Logemann, P., Osborne, R., Ponec, M., Regnier, J.F., Steiling, W., Walker, A.P., and Balls, M. (1995). A prevalidation study on in vitro skin corrosivity testing. The report and recommendations of ECVAM Workshop 6. ATLA 23, 219–255. |
|
(4) |
Barratt, M.D., Brantom, P.G., Fentem, J.H., Gerner, I., Walker, A.P., and Worth, A.P. (1998). The ECVAM international validation study on in vitro tests for skin corrosivity. 1. Selection and distribution of the test chemicals. Toxic, in Vitro 12, 471–482. |
|
(5) |
Fentem, J.H., Archer, G.E.B., Balls, M., Botham, P.A., Curren, R.D., Earl, L.K., Esdaile, D.J., Holzhutter, H.-G., and Liebsch, M. (1998). The ECVAM international validation study on in vitro tests for skin corrosivity. 2. Results and evaluation by the Management Team. Toxic, in Vitro 12, 483–524. |
|
(6) |
Final Report of the OECD Workshop on Harmonization of Validation and Acceptance Criteria for Alternative Toxicological Test Methods, 62pp. |
|
(7) |
Balls, M., Blaauboer, B.J., Fentem. J.H., Bruner. L., Combes, R.D., Ekwall, B., Fielder. R.J., Guillouzo, A., Lewis, R.W., Lovell, D.P., Reinhardt, CA., Repetto, G., Sladowski. D., Spielmann, H., and Zucco, F. (1995). Practical aspects of the validation of toxicity test procedures. The report and recommendations of ECVAM workshops. ATLA 23, 129–147. |
|
(8) |
ICCVAM (Interagency Coordinating Committee on the Validation of Alternative Methods). (1997). Validation and Regulatory Acceptance of Toxicological Test Methods. NIH Publication No. 97-3981. National Institute of Environmental Health Sciences, Research Triangle Park, NC, USA. http://iccvam.niehs.nih.gov/docs/guidelines/validate.pdf |
|
(9) |
ECVAM (1998). ECVAM News & Views. ATLA 26, 275–280. |
|
(10) |
ICCVAM (Interagency Coordinating Committee on the Validation of Alternative Methods). (2002). ICCVAM evaluation of EpiDermTM, EPISKINTM (EPI-200), and the Rat Skin Transcutaneous Electrical Resistance (TER) assay: In Vitro test methods for assessing dermal corrosivity potential of chemicals. NIH Publication No. 02-4502. National Toxicology Program Interagency Center for the Evaluation of Alternative Toxicological Methods, National Institute of Environmental Health Sciences, Research Triangle Park, NC, USA. http://iccvam.niehs.nih.gov/methods/epiddocs/epis_brd.pdf. |
|
(11) |
OECD (2002) Extended Expert Consultation Meeting on The In Vitro Skin Corrosion Test Guideline Proposal, Berlin, 1st - 2nd November 2001, Secretariat's Final Summary Report, 27th March 2002, OECD ENV7EHS, available upon request from the Secretariat |
|
(12) |
Oliver, G.J.A., Pemberton, M.A., and Rhodes, C. (1986). An in vitro skin corrosivity test -modificationsand validation. Fd. Chem. Toxicol. 24, 507–512. |
|
(13) |
Botham, P.A., Hall, T.J., Dennett, R., McCall, J.C., Basketter, D.A., Whittle, E., Cheeseman, M., Esdaile, D.J., and Gardner, J. (1992). The skin corrosivity test in vitro: results of an interlaboratory trial. Toxic, in Vitro 6, 191–194. |
|
(14) |
Worth A.P., Fentem J.H., Balls M., Botham P.A., Curren RD, Earl L.K., Esdaile D.J., Liebsch M (1998). An Evaluation of the Proposed OECD Testing Strategy for Skin Corrosion. ATLA 26: 709–720. |
|
(15) |
Basketter, D.A., Chamberlain, M., Griffiths, H.A., Rowson, M., Whittle, E., York, M. (1997). The classification of skin irritants by human patch test. Fd. Chem. Toxicol. 35, 845–852. |
|
(16) |
Oliver G.J.A, Pemberton M.A and Rhodes C. (1988). An In Vitro model for identifying skin-corrosive chemicals. I. Initial Validation. Toxic. in Vitro. 2, 7–17. |
Rysunek 1
Aparatura do testu TER skóry szczura
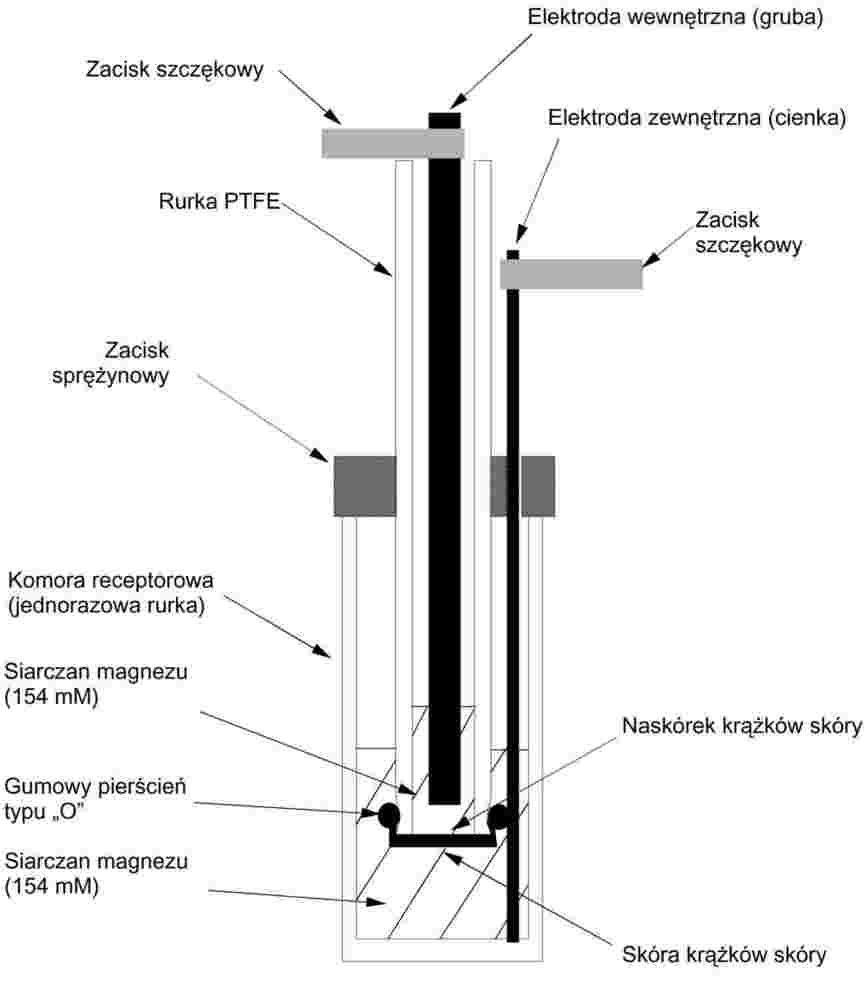
Rysunek 2
Wymiary rurek politetrafluoroetylenowej (PFTE) i receptorowej oraz stosowane elektrody

Najważniejsze elementy urządzenia przedstawionego powyżej:
|
— |
wewnętrzna średnica rurki PTFE, |
|
— |
długość elektrod w stosunku do rurki PTFE i rurki receptorowej, dobrana tak, aby elektrody nie dotykały do krążka skóry i żeby standardowa długość elektrody stykała się z roztworem MgSO4, |
|
— |
ilość roztworu MgSO4 w rurce receptorowej powinna zapewniać wysokość słupa cieczy odpowiednią do jej poziomu w rurce PFTE, jak pokazano na rysunku 1, |
|
— |
krążek skóry powinien być wystarczająco mocno przymocowany do rurki PFTE, tak aby opór elektryczny był rzeczywistą miarą właściwości skóry. |
B.40. BIS BADANIE ZNISZCZENIA SKÓRY METODĄ IN VITRO: BADANIE MODELU SKÓRY LUDZKIEJ
1. METODA
Opisywana metoda badania jest zgodna z OECD TG 431 (2004).
1.1. WSTĘP
Zniszczenie skóry oznacza powstawanie nieodwracalnych uszkodzeń tkanek skórnych w wyniku kontaktu z substancją testową (jak zdefiniowano w Globalnie Zharmonizowanym Systemie Klasyfikacji i Oznakowania Produktów Chemicznych (GHS)) (1). Niniejsza metoda badania nie wymaga wykorzystywania żywych zwierząt ani tkanek zwierzęcych do oceny stopnia niszczenia skóry.
Do tej pory ocena zniszczenia skóry wiązała się zwykle z użyciem zwierząt laboratoryjnych (2). Potrzeba wyeliminowania bólu i cierpienia zwierząt została uwzględniona przy przeglądzie metody badania B.4, umożliwiającej określenie zniszczenia skóry przy użyciu alternatywnych metod in vitro, bez zadawania zwierzętom bólu i cierpienia.
Pierwszym krokiem w kierunku określenia alternatywnych badań, które mogłyby zostać zastosowane przy badaniu niszczenia skóry do celów regulacyjnych, było przeprowadzenie badań wstępnych (3). Następnie przeprowadzono formalne badania zatwierdzające (6)(7)(8) oceny zniszczenia skóry metodami in vitro (4)(5). Wyniki tych badań oraz inne publikacje (9) stały się podstawą zalecenia, że do oceny stopnia niszczenia skóry in vivo można stosować następujące badania (10)(11)12)(13): badanie modelu skóry ludzkiej (niniejsza metoda) oraz test przezskórnego oporu elektrycznego (zob. metoda badania B.40).
Badania zatwierdzające wykazały, że badania, w których używane są modele ludzkiej skóry (3)(4)(5)(9) pozwalają na wiarygodne rozróżnienie substancji niszczących skórę i nieniszczących skóry. Protokół badania może także dostarczyć wskazówek ułatwiających odróżnienie substancji poważnie niszczących od substancji mniej niszczących.
Badanie przedstawione w opisie niniejszej metody umożliwia identyfikację związków i mieszanin chemicznych uszkadzających skórę. Ponadto w połączeniu z dowodami uzyskanymi na podstawie innych dostępnych informacji (np. pH, związek między strukturą a aktywnością, dane dotyczące ludzi i zwierząt) umożliwia identyfikację związków i mieszanin, które nie niszczą skóry (1)(2)(13)(14). Zwykle nie dostarcza wystarczających informacji o podrażnieniach skóry, nie umożliwia także dzielenia substancji niszczących na podkategorie, jak na to pozwala Globalny Zharmonizowany System Klasyfikacji (GHS) (1).
Zaleca się, aby w celu dokonania pełnej oceny miejscowego działania na skórę po pojedynczej ekspozycji postępować zgodnie z sekwencyjną strategią przedstawioną w dodatku do metody badania B.4 (2) i w Globalnie Zharmonizowanym Systemie (GHS) (1). Ta strategia badania obejmuje przeprowadzenie badań zniszczenia skóry (jak przedstawiono w opisie niniejszej metody) i podrażnienia skóry in vitro przed rozważaniem przeprowadzenia badań na żywych zwierzętach.
1.2. DEFINICJE
Zniszczenie skóry in vivo : oznacza spowodowanie nieodwracalnych uszkodzeń skóry: a mianowicie widocznej martwicy skóry w warstwie naskórka i skóry właściwej w wyniku kontaktu z substancją testową w okresie do 4 godzin. Typowymi objawami zniszczenia skóry są wrzody, krwawienie, krwawiące strupy oraz, pod koniec 14-dniowego okresu obserwacji, odbarwienie skóry spowodowane zbieleniem, pojawianie się całkowitych łysin i blizn. W przypadku wątpliwych uszkodzeń należy zastosować metody histopatologiczne.
Żywotność komórek: parametr określający całkowitą aktywność populacji komórek (np. zdolność dehydrogenaz w mitochondriach komórek do obniżenia zawartości przyżyciowego barwnika MTT), który, w zależności od pomiaru końcowego i zastosowanego schematu badania odpowiada całkowitej liczbie i/lub żywotności komórek.
1.3. SUBSTANCJE ODNIESIENIA
Tabela 1
Chemiczne substancje odniesienia
|
Nazwa |
Nr EINECS |
Nr CAS |
|
|
1,2-diaminopropan |
201-155-9 |
78-90-0 |
Silne działanie niszczące |
|
Kwas akrylowy |
201-177-9 |
79-10-7 |
Silne działanie niszczące |
|
2-tert. Butylofenol |
201-807-2 |
88-18-6 |
Działanie niszczące |
|
Wodorotlenek potasu (10 %) |
215-181-3 |
1310-58-3 |
Działanie niszczące |
|
Kwas siarkowy (10 %) |
231-639-5 |
7664-93-9 |
Działanie niszczące |
|
Kwas oktanowy (kwas kaprylowy) |
204-677-5 |
124-07-02 |
Działanie niszczące |
|
4-amino-1,2,4-triazol |
209-533-5 |
584-13-4 |
Brak działania niszczącego |
|
Eugenol |
202-589-1 |
97-53-0 |
Brak działania niszczącego |
|
Bromek fenetylu |
203-130-8 |
103-63-9 |
Brak działania niszczącego |
|
Tetrachloroetylen |
204-825-9 |
27-18-4 |
Brak działania niszczącego |
|
Kwas izostearynowy |
250-178-0 |
30399-84-9 |
Brak działania niszczącego |
|
4-(metylotio)-benzaldehyd |
222-365-7 |
3446-89-7 |
Brak działania niszczącego |
Większość z wymienionych chemikaliów pochodzi z wykazu chemikaliów przygotowanego na potrzeby międzynarodowego badania zatwierdzającego ECVAM (4). Ich wybór opiera się na następujących kryteriach:
|
(i) |
równa liczba substancji niszczących i nieniszczących; |
|
(ii) |
dostępne w handlu substancje należące do większości stosownych klas związków chemicznych; |
|
(iii) |
uwzględnienie zarówno silnie niszczących, jak i słabiej niszczących substancji, aby umożliwić rozróżnianie na podstawie właściwości niszczących; |
|
(iv) |
wybór chemikaliów, które mogą być poddawane obróbce w laboratorium nie stwarzając zagrożeń innych niż zniszczenie skóry. |
1.4. ZASADA METODY BADANIA
Materiał testowy jest nakładany miejscowo na trójwymiarowy model ludzkiej skóry, składający się co najmniej ze zrekonstruowanego naskórka z funkcjonalną warstwą rogową. Materiały niszczące są określane na podstawie ich zdolności do zmniejszania żywotności komórek, określanej na przykład przy zastosowaniu metody redukcji MTT (15), poniżej określonego poziomu w określonym czasie ekspozycji. Zasada oceny modelu ludzkiej skóry opiera się na hipotezie, że substancje niszczące są w stanie przeniknąć przez warstwę rogową na drodze dyfuzji lub w wyniku jej niszczenia i są cytotoksyczne dla leżących głębiej warstw komórek.
1.4.1. Procedura
1.4.1.1. Modele skóry ludzkiej
Modele ludzkiej skóry mogą być konstruowane lub nabyte (np. modele EpiDerm™i EPISKIN™) (16)(17)(18)(19) lub opracowywane i tworzone w laboratorium przeprowadzającym badania (20)(21). Uznaje się, że zastosowanie ludzkiej skóry podlega międzynarodowym i krajowym zastrzeżeniom i warunkom etycznym. Każdy nowy model powinien podlegać zatwierdzeniu (przynajmniej w zakresie opisanym w 1.4.1.1.2). Modele ludzkiej skóry stosowane do tego testu muszą być zgodne z następującymi kryteriami:
1.4.1.1.1.
Do tworzenia nabłonka należy użyć ludzkich keranocytów. Pod zachowującą funkcje życiowe warstwą rogową powinno znajdować się kilka warstw żywych komórek nabłonkowych. Model skóry może także zawierać element warstwy podskórnej. Warstwa rogowa powinna być wielowarstwowa, o odpowiednim profilu lipidowym, niezbędnym do wytworzenia funkcjonalnej bariery, na tyle szczelnej, aby powstrzymać szybkie przenikanie markerów cytotoksycznych. Właściwości izolacyjne modelu powinny zapobiegać przechodzeniu materiału przez warstwę rogową do żywej tkanki. Przechodzenie substancji testowej przez warstwę rogową prowadzi do niewłaściwego modelowania ekspozycji skóry. Model skóry nie powinien być skażony bakteriami (także mykoplazmami) ani grzybami.
1.4.1.1.2.
Skala żywotności jest zwykle kwantyfikowana przy użyciu MTT lub innego przekształconego metabolicznie barwnika. W tych przypadkach gęstość optyczna (OD) ekstrahowanego (rozpuszczonego) barwnika z tkanki kontroli negatywnej powinna być co najmniej 20 razy większa niż OD samego rozpuszczalnika do ekstrakcji (przegląd problematyki zamieszczono w (22)). W okresie ekspozycji testowej tkanka kontroli negatywnej powinna być stabilna w hodowli (umożliwiać podobne pomiary żywotności). Warstwa rogowa powinna być wystarczająco odporna, aby powstrzymać szybkie przenikanie niektórych cytotoksycznych markerów (np. 1 % Triron X-100). Właściwość tę można ocenić na podstawie czasu ekspozycji, potrzebnego do zmniejszenia żywotności komórek o 50 % (ET50) (np. dla modeli EpiDerm™ i EPISKIN™ czas ten wynosi > 2 godzin). Tkanka powinna charakteryzować się odtwarzalnością w czasie i jeśli to możliwe, między laboratoriami. Co więcej, powinna pozwalać na przewidywanie właściwości niszczących wzorcowych związków chemicznych (zob. tabela 1), przy stosowaniu wybranego protokołu badania.
1.4.1.2. Nakładanie substancji testowej i kontrolnej
W każdej procedurze (czasie ekspozycji), w tym w kontrolach, stosowane są dwa powtórzenia próbki tkanki. W przypadku płynnych materiałów należy równomiernie rozprowadzić na skórze dostateczną ilość substancji testowej: należy użyć co najmniej 25 uL/cm2. W przypadku materiałów stałych należy nałożyć na skórę dostateczną ilość substancji testowej, zwilżonej wodą dejonizowaną lub destylowaną, aby zapewnić dobry kontakt ze skórą. Tam, gdzie to właściwe, substancje stałe przed zastosowaniem należy zmielić na proszek. Metoda nakładania powinna być odpowiednia do substancji testowej (zob. pozycja bibliograficzna 5). Po zakończeniu okresu ekspozycji należy starannie zmyć materiał testowy ze skóry odpowiednim buforem lub 0,9 % NaCl.
Aby zapewnić odpowiednie działanie modelu, w każdym badaniu należy równocześnie stosować kontrolę pozytywną i negatywną. Jako substancji do kontroli pozytywnej zaleca się użycie kwasu octowego lodowatego lub 8N KOH. Zalecanymi substancjami w kontroli negatywnej są 0,9 % NaCl lub woda.
1.4.1.3. Pomiary żywotności komórek
Do pomiaru żywotności komórek można stosować wyłącznie sprawdzone metody ilościowe. Co więcej, miara żywotności musi być kompatybilna w użyciu z trójwymiarowym wytworem tkankowym. Wiązanie niespecyficznego barwnika nie może zaburzać pomiaru żywotności. Dlatego barwniki wiążące białka i barwniki, które nie ulegają przemianom metabolicznym (np. czerwień obojętna) są nieodpowiednie. Najczęściej stosowanym badaniem jest redukcja MTT (3-(4,5-dimetylotiazol-2-yl)-2,5 difenylo bromek tetrazolu: numer EINECS 206-069-5, numer CAS 298-93-1)), którego dokładność i powtarzalność wykazano (5), ale można zastosować także inne badania. Próbka skóry umieszczana jest na 3 godziny w roztworze MTT o odpowiednim stężeniu (np. 0,3–1 mg/mL) przy odpowiedniej temperaturze inkubacji. Wytrącony niebieski formazan jest następnie ekstrahowany przy użyciu rozpuszczalnika (izopropanol), a stężenie formazanu jest mierzone przez określenie gęstości optycznej przy długości fali między 540 a 595 nm.
Działanie chemiczne materiału testowego na barwnik przyżyciowy może przebiegać podobnie jak w metabolizmie komórkowym, prowadząc do fałszywej oceny żywotności. Wykazano, że ma to miejsce, kiedy materiał testowy nie został całkowicie usunięty ze skóry przez płukanie (9). Jeśli materiał testowy oddziałuje bezpośrednio na barwnik przyżyciowy, należy zastosować dodatkowe kontrole w celu wykrywania i korygowania zakłóceń pomiaru żywotności(9)(23).
2. DANE
Dla każdej tkanki należy przedstawić w formie tabelarycznej wartości gęstości optycznej i procentowe dane dotyczące żywotności komórek dla materiału testowego, kontroli pozytywnej i negatywnej, w tym w odpowiednich przypadkach dane z powtórzeń eksperymentu, średnie oraz poszczególne pojedyncze wartości.
2.1. INTERPRETACJA WYNIKÓW
Wartości OD otrzymane dla każdej próbki testowej mogą być wykorzystane do obliczenia procentowej żywotności w stosunku do kontroli negatywnej, dla której arbitralnie przyjmuje się 100 %. Wartość graniczna dla procentowej żywotności komórek, wyznaczająca granicę między materiałami testowymi niszczącymi skórę i nieniszczącymi skóry (lub granice między różnymi klasami niszczenia) lub statystyczne procedury stosowane do oceny wyników i identyfikacji materiałów niszczących muszą być jasno zdefiniowane i udokumentowane, przy czym należy wykazać, że są one odpowiednie. Generalnie, wartości graniczne są ustalane w trakcie optymalizacji testu, sprawdzane w fazie prewalidacyjnej i potwierdzane w badaniach walidacyjnych. Na przykład prognoza poziomu niszczenia w powiązaniu z modelem EpiDerm™ wygląda następująco (9):
Substancja testowa uznawana jest za niszczącą skórę:
|
(i) |
jeśli żywotność po trzyminutowej ekspozycji jest mniejsza niż 50 %; lub |
|
(ii) |
jeśli żywotność po trzyminutowej ekspozycji jest większa niż lub równa 50 % a żywotność po godzinnej ekspozycji jest mniejsza niż 15 %. |
Substancja testowa uznawana jest za nieniszczącą skóry:
|
(i) |
jeśli żywotność po trzyminutowej ekspozycji jest większa niż lub równa 50 %, a żywotność po godzinnej ekspozycji jest większa niż lub równa 15 %. |
3. SPRAWOZDAWCZOŚĆ
3.1. SPRAWOZDANIE Z BADANIA
Sprawozdanie z badania musi zawierać następujące informacje:
|
|
Substancja testowa i kontrolna:
|
|
|
Uzasadnienie dla zastosowanego modelu skóry i procedury. |
|
|
Warunki badania:
|
|
|
Wyniki:
|
|
|
Dyskusja wyników. |
|
|
Wnioski. |
4. BIBLIOGRAFIA
|
(1) |
OECD (2001) Harmonised Integrated Classification System for Human Health and Environmental Hazards of Chemical Substances and Mixtures. OECD Series on Testing and Assessment Number 33. ENV/JM/MONO(2001)6, Paris. http://www.olis.oecd.org/olis/2001doc.nsf/LinkTo/env-jm-mono(2001)6. |
|
(2) |
Testing Method B.4. Acute Toxicity: Dermal Irritation/Corrosion. |
|
(3) |
Botham, P.A., Chamberlain, M., Barratt, M.D., Curren, R.D., Esdaile, D.J., Gardner, J.R., Gordon, V.C., Hildebrand, B., Lewis, R.W., Liebsch, M., Logemann, P., Osborne, R., Ponec, M., Regnier, J.F., Steiling, W., Walker, A.P., and Balls, M. (1995). A prevalidation study on in vitro skin corrosivity testing. The report recommendations of ECVAM Workshop 6 ATLA 23,219–255. |
|
(4) |
Barratt, M.D., Brantom, P.G., Fentem, J.H., Gerner, I., Walker, A.P., and Worth, A.P. (1998). The ECVAM international validation study on in vitro tests for skin corrosivity. 1. Selection and distribution of the test chemicals. Toxic. In Vitro 12, 471–482. |
|
(5) |
Fentem, J.H., Archer, G.E.B., Balls, M., Botham, P.A., Curren, R.D., Earl, L.K., Esdaile, D.J., Holzhutter, H.G. and Liebsch, M. (1998). The ECVAM international validation study on in vitro tests for skin corrosivity. 2. Results and evaluation by the Management Team Toxic. In Vitro 12, 483–524. |
|
(6) |
OECD (1996). Final Report of the OECD Workshop on Harmonization of Validation and Acceptance Criteria for Alternative Toxicological Test Methods, 62pp. |
|
(7) |
Balls, M., Blaauboer, B.J., Fentem, J.H., Bruner, L., Combes, R.D., Ekwall, B., Fielder, R.J., Guillouzo, A., Lewis, R.W., Lovell, D.P., Reinhardt, CA., Repetto, G., Sladowski, D., Spielmann, H., and Zucco, F. (1995). Practical aspects of the validation of toxicity test procedures. Test report and recommendations of ECVAM workshops. ATLA 23, 129–147. |
|
(8) |
ICCVAM (Interagency Coordinating Committee on the Validation of Alternative Methods). (1997). Validation and Regulatory Acceptance of Toxicological Test Methods. NIH Publication No. 97-3981. National Institute of Environmental Health Sciences, Research Triangle Park, NC, USA. http://iccvam.niehs.nih.gov/docs/guidelines/validate.pdf. |
|
(9) |
Liebsch, M., Traue, D., Barrabas, C, Spielmann, H., Uphill, P., Wilkins, S., McPherson, J.P., Wiemann, C, Kaufmann, T., Remmele, M. and Holzhutter, H.G. (2000). The ECVAM prevalidation study on the use of EpiDerm for skin Corrosivity testing. ATLA 28, pp.371–401. |
|
(10) |
ECVAM (1998). ECVAM News & Views. ATLA 26, 275–280. |
|
(11) |
ECVAM (2000). ECVAM News & Views. ATLA 28, 365–67. |
|
(12) |
ICCVAM (Interagency Coordinating Committee on the Validation of Alternative Methods). (2002). ICCVAM evaluation of EpiDerm™, EPISKIN™ (EPI-200), and the Rat Skin Transcutaneous Electrical Resistance (TER) assay: In Vitro test methods for assessing dermal corrosivity potential of chemicals. NIH Publication No. 02-4502. National Toxicology Program Interagency Center for the Evaluation of Alternative Toxicological Methods, National Institute of Environmental Health Sciences, Research Triangle Park, NC, USA. http://iccvam.niehs.nih.gov/methods/epiddocs/epis_brd.pdf. |
|
(13) |
OECD (2002) Extended Expert Consultation Meeting on The In Vitro Skin Corrosion Test Guideline Proposal, Berlin, 1st – 2nd November 2001, Secretariat's Final Summary Report, 27th March 2002, OECD ENV/EHS, available upon request from the Secretariat |
|
(14) |
Worth A.P., Fentem J.H., Balls M., Botham P.A., Curren R.D., Earl L.K., Esdaile D.J., Liebsch M (1998). An Evaluation of the Proposed OECD Testing Strategy for Skin Corrosion. ATLA 26: 709–720. |
|
(15) |
Mosmann, T. (1983). Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity asssays. J.Immunol. Meth. 65, 55–63. |
|
(16) |
Cannon, C.L., Neal, P.J., Southee, J.A., Kubilus, J., and Klausner, M., 1994. New epidermal model for dermal irritancy testing. Toxic. In Vitro 8, 889–891. |
|
(17) |
Ponec, M., Boelsma, E., Weerheim, A., Mulder, A., Boutwstra, J., and Mommaas, M., 2000. Lipid and ultrastructural characterization of reconstructed skin models. International Journal of Pharmaceutics. 203, 211–225. |
|
(18) |
Tinois E, Gaetani Q, Gayraud B, Dupont D, Rougier A, Pouradier DX (1994). The Episkin model: Successful reconstruction of human epidermis in vitro. In In vitro Skin Toxicology. Edited by A Rougier, AM Goldberg and HI Maibach: 133–140. |
|
(19) |
Tinois E, Tiollier J, Gaucherand M, Dumas H, Tardy M, Thivolet J (1991). In vitro and post -transplantation differentiation of human keratinocytes grown on the human type IV collagen film of a bilayered dermal substitute. Experimental Cell Research 193: 310–319. |
|
(20) |
Parentau, N.L., Bilbo, P., Molte, C.J., Mason, V.S., and Rosenberg, H. (1992). The organotypic culture of human skin keratinocytes and fibroblasts to achieve form and function. Cytotechnology 9, 163–171. |
|
(21) |
Wilkins, L.M., Watson, S.R., Prosky, S.J., Meunier, S.F., Parentau, N.L. (1994). Development of a bilayered living skin construct for clinical applications. Biotechnology and Bioengineering 43/8, 747–756. |
|
(22) |
Marshall, N.J., Goodwin, C.J., Holt, S.J. (1995). A critical assessment of the use of microculture tetrazohum assays to measure cell growth and function. Growth Regulation 5, 69–84. |
|
(23) |
Fentem, J.H., Briggs, D., Chesne', C, Elliot, G.R., Harbell, J.W., Heylings, J.R., Portes, P., Rouget, R., and van de Sandt, J.J.M., and Botham, P.A. (2001). A prevalidation study on in vitro tests for acute skin irritation: results and evaluation by the Management Team. Toxic. InVItro 15, 57–93. |
B.41. BADANIE FOTOTOKSYCZNOŚCI 3T3 NRU IN VITRO
1. METODA
Opisywana metoda testowa jest zgodna z OECD TG 432 (2004).
1.1. WSTĘP
Fototoksyczność definiuje się jako toksyczny wpływ substancji, na działanie której narażony jest organizm, przy czym wpływ ten jest spowodowany lub zwiększony (widoczny przy niskim poziomie dawki) przez ekspozycję na światło albo indukowany przez naświetlanie skóry po systematycznym podawaniu substancji.
Badanie in vitro fototoksyczności 3T3 NRU jest stosowany w celu określenia potencjału fototoksycznego substancji testowej, indukowanej przez związek chemiczny wzbudzony w wyniku ekspozycji na światło. Badanie służy do oceny fotocytotoksyczności, którą to ocenę przeprowadza się na podstawie względnego zmniejszenia się żywotności komórek narażonych na działanie substancji w świetle w porównaniu z ciemnością. Istnieje duże prawdopodobieństwo, że substancje identyfikowane w tym teście po stosowaniu ogólnoustrojowym i nakładaniu na skórę lub po stosowaniu miejscowym są fototoksyczne in vivo.
Wiadome jest, że wiele rodzajów substancji chemicznych wywołuje efekt fototoksyczny (1)(2)(3)(4). Ich wspólną cechą jest zdolność do absorbowania energii światła w zakresie widma odpowiadającego widmu promieniowania słonecznego. Zgodnie z pierwszym prawem fotochemii (prawo Grotthausa-Drapera) fotoreakcja wymaga absorpcji dostatecznej liczby kwantów światła. Tak więc przed przeprowadzeniem testów biologicznych należy określić widmo absorbcyjne UV/vis substancji chemicznej według wytycznych OECD w zakresie badań 101. Stwierdzono, że jeśli molowy współczynnik ekstynkcji/absorpcji jest niższy niż 10 litrów xmo 1-1 × cm-l, to jest mało prawdopodobne, że związek jest fotoreaktywny. Takiego związku nie trzeba poddawać badaniu fototoksycznemu in vitro 3T3 NRU ani innemu testowi biologicznemu na występowanie efektu fototoksycznego. Zob. także załącznik I.
Wiarygodność i znaczenie badania fototoksyczności 3T3 NRU in vitro zostały niedawno ocenione (6)(7)(8) (9). Wykazano, że badanie fototoksyczności 3T3 NRU in vitro dostarcza informacji o efektach ostrej fototoksyczności in vivo u zwierząt i ludzi. Badanie nie jest opracowany po to, aby przewidywać inne niekorzystne skutki, do których może dojść w wyniku połączonego działania substancji chemicznych i światła, np. nie znajduje zastosowania w przypadku fotogenotoksyczności, fotoalergii i fotokarcynogenności, nie umożliwia także oceny nasilenia fototoksyczności. Ponadto badanie nie jest przeznaczone do badania pośrednich mechanizmów fototoksyczności, działania metabolitów substancji testowej ani działania mieszanin.
Podczas gdy wykorzystanie reakcji metabolicznych jest ogólnym wymogiem przy ocenie poziomu genotoksyczności i karcynogenności dla wszystkich testów in vitro, w przypadku fototoksyczności jak dotąd znaleziono tylko nieliczne przykłady sytuacji, w których substancja zaczyna działać jako fototoksyna in vivo lub in vitro dopiero po przekształceniach metabolicznych. Tak więc obecnie nie uważa się ani za konieczne, ani za naukowo uzasadnione, aby test był przeprowadzany w połączeniu z systemem aktywacji metabolicznej.
1.2. DEFINICJE
Irradiancja: intensywność światła ultrafioletowego (UV) lub widzialnego padającego na powierzchnię w W/m2 lub W/cm2.
Dawka światła: ilość (= natężenie × czas) promieniowania ultrafioletowego (UV) lub widzialnego padającego na powierzchnię, wyrażona w dżulach (= W × s) na powierzchnię, np. J/m2 lub J/cm2.
Zakresy długości fal UV: CIE (Międzynarodowa Komisja Oświetleniowa) zaleca następujące oznaczenia: UVA (315–400 nm), UVB (280–315 nm) i UVC (100–280 nm). Inne oznaczenia również mogą być stosowane; podział między UVB a UVA jest często przeprowadzany przy 320 nm, a UVA mogą być dzielone na UV-A1 i UV-A2, z granicą przy około 340 nm.
Żywotność komórek: parametr określający całkowitą aktywność populacji komórek (np. wychwyt barwnika przeżyciowego Natural Red do lizosomów komórki) który, w zależności od pomiaru końcowego i zastosowanego schematu badania, koreluje z całkowitą liczbą i/lub żywotnością komórek.
Względna żywotność komórek: żywotność komórek wyrażona w stosunku do kontroli rozpuszczalnikowych (negatywnych), których próbki były pobierane przez całą procedurę testową (+Irr lub -Irr), ale które nie były poddawane działaniu substancji testowej.
PIF (Photo Irritation Factor – współczynnik fotopodrażnienia): współczynnik uzyskany przez porównanie dwóch tak samo efektywnych stężeń cytotoksycznych (IC50) testowanej substancji, otrzymany przy braku (-Irr) i przy zastosowaniu (+Irr) niecytotoksycznej irradiacji UVA/vis.
IC50: stężenie testowanej substancji, przy której żywotność komórek jest zmniejszona o 50 %.
MPE (Mean Photo Effect – średni fotoefekt): miara uzyskana z matematycznej analizy krzywych odpowiedzi na stężenie, otrzymana przy braku (-Irr) i przy zastosowaniu (+Irr) niecytotoksycznej irradiacji UVA/vis.
Fototoksyczność: reakcja na toksyczność ostrą, pojawiająca się po pierwszej ekspozycji skóry na niektóre substancje chemiczne, a następnie ekspozycji na światło lub reakcja, która jest wywołana przez naświetlanie skóry po systematycznym, ogólnoustrojowym podawaniu substancji.
1.3. ZASADA METODY BADANIA
Badanie fototoksyczności 3T3 NRU in vitro polega na porównaniu cytotoksyczności substancji przy zastosowaniu i przy braku ekspozycji na niecytotoksyczną dawkę symulowanego światła słonecznego. Cytotoksyczność w tym teście jest wyrażona jako zależne od stężenia zmniejszanie wychwytu barwnika przyżyciowego Neutral Red, mierzone 24 godziny po wprowadzeniu substancji chemicznej i irradiacji (10). NR jest słabym barwnikiem kationowym, który łatwo przenika niedyfuzyjnie przez błony komórkowe i zbiera się wewnątrz komórek, w lizosomach. Modyfikacje powierzchni delikatnej błony lizosomów powodują kruchość lizosomów i inne zmiany, które stopniowo stają się nieodwracalne. Zmiany takie, wywołane przez działanie ksenobiotyków, prowadzą do zmniejszonego wychwytu i wiązania NR. Można więc wyróżnić w pełni żywotne, uszkodzone i martwe komórki, co stanowi podstawę testu.
Komórki Balb/c 3T3 hoduje się przez 24 godziny w celu wytworzenia monowarstw. Dwie płytki z 96 studzienkami na substancję testową są przez 1 godz. wstępnie inkubowane z substancją testową w ośmiu różnych stężeniach. Następnie jedna z dwóch płytek jest poddawana działaniu najwyższej niecytotoksycznej dawce irradiacji, podczas gdy druga płytka trzymana jest w ciemności. Na obu płytkach pożywka jest badana następnie wymieniana na pożywkę kontrolną i po następnych 24 godzinach inkubacji określa się żywotność komórek na podstawie wychwytu barwnika Neutral Red. Żywotność komórek wyrażana jest jako procent nie poddawanych działaniu substancji kontroli rozpuszczalnikowych i jest określana dla każdego stężenia testowego. W celu określenia potencjału fototoksycznego porównuje się odpowiedzi na zmienne stężenia w obecności i przy braku irradiacji, zwykle na poziomie IC50, tj. przy stężeniu zmniejszającym żywotność do 50 % w stosunku do kontroli nie poddawanej działaniu irradiacji.
1.4. OPIS METODY BADANIA
1.4.1. Przygotowania
1.4.1.1. Komórki
W badaniu walidacyjnym użyta została ustalona linia fibroplastów mysich Balb/c 3T3, klon 31, z hodowli amerykańskiej American Type Culture Collection (ATCC), Manassas, VA, USA, albo z hodowli europejskiej European Collection of Cell Cultures (ECACC), Salisbury, Wiltshire, UK i wobec tego zaleca się, aby pobierać materiał z odpowiednich banków komórek. Inne komórki lub linie komórek mogą być użyte do tej samej procedury testowej jeśli warunki hodowli są dostosowane do specyficznych potrzeb komórek, ale należy wykazać równoważność warunków.
Komórki powinny być regularnie sprawdzane pod kątem braku skażenia mykoplazmami i używane tylko, jeśli stwierdzono całkowity ich brak (11).
Ważne jest, aby regularnie sprawdzać wrażliwość komórek na UV, zgodnie z procedurą kontroli jakości przedstawioną w opisie metody. Ponieważ wrażliwość komórek na UVA może wzrastać wraz z liczbą pasaży, należy użyć komórek Balb/c 3T3 o możliwie jak najmniejszej liczbie pasaży, najlepiej poniżej stu. (Zob. część 1.4.2.2.2. i załącznik 2).
1.4.1.2. Pożywki i warunki hodowli kultury
Przy rutynowych pasażach komórek oraz w trakcie procedury testowej należy przestrzegać odpowiednich warunków inkubacji i stosować odpowiednie pożywki hodowlane, np. w przypadku komórek Balb/c 3T3 powinna to być pożywka DMEM (Dulbecco's Modified Eagle's Medium), z dodatkiem 10 % surowicy nowo urodzonych cieląt, 4 mM glutaminy, penicyliny (100IU), streptomycyny (100 μg/mL), inkubowana w wilgotnych warunkach w 37 C, 5–7,5 % CO2 w zależności od bufora (zob.część 1.4.1.4 akapit drugi). Szczególne znaczenie ma, aby warunki hodowli komórek zapewniały cykl komórek, którego okres trwania odpowiada wcześniejszym cyklom używanych komórek lub linii komórek.
1.4.1.3. Przygotowanie kultur
Komórki z zamrożonych kultur macierzystych są wysiewane do pożywki hodowlanej przy odpowiednim zagęszczeniu i następnie – zanim zostaną użyte w teście fototoksyczności 3T3 NRU in vitro – przynajmniej raz wysiane na hodowle wtórne.
Komórki używane do badania fototoksyczności są wysiewane do pożywki hodowlanej w odpowiednim zagęszczeniu, tak żeby nie dochodziło do przegęszczenia przed końcem badania, to znaczy do momentu, kiedy po 48 godzinach od wysiewania komórek oceniana jest ich żywotność. W przypadku komórek Balb/c 3T3 hodowanych na 96-studzienkowych płytkach zalecane zagęszczenie wynosi 1 × 104 komórek na studzienkę.
W przypadku każdej substancji testowej komórki są wysiewane w taki sam sposób na dwie 96-studzienkowe płytki, poddawane równocześnie całej procedurze testowej w identycznych warunkach hodowli – z wyjątkiem okresu, w którym jedna z płytek jest naświetlana (+Irr), a druga trzymana w ciemności (-Irr).
1.4.1.4. Przygotowanie substancji testowej
Substancje testowe muszą być przygotowywane na świeżo, bezpośrednio przed użyciem, chyba że istniejące dane wskazują na ich stabilność w czasie przechowywania. Zaleca się, aby wszelkie operacje z substancjami chemicznymi i wstępna obróbka komórek były wykonywane w warunkach świetlnych, które nie spowodują fotoaktywacji lub rozkładu substancji testowej przed irradiacją.
Substancje chemiczne powinny być rozpuszczane w buforowanych roztworach soli, np. w zrównoważonym roztworze soli Earla (EBSS) lub innych fizjologicznie zrównoważonych roztworach, które nie mogą zawierać składników białkowych, składników absorbujących światło (np. barwnych wskaźników pH i witamin), aby w czasie irradiacji nie dochodziło do interferencji. Ponieważ w czasie irradiacji komórki są trzymane przez około 50 minut poza inkubatorem CO2, należy uważać, aby nie doszło do alkalizacji. Jeśli stosowane są słabe bufory takie jak EBSS, można to osiągnąć przez inkubację komórek w obecności 7,5 % CO2. Jeśli komórki są inkubowane w obecności jedynie 5 % CO2, należy wybrać silniejszy bufor.
Substancje testowe o ograniczonej rozpuszczalności w wodzie powinny być rozpuszczane w odpowiednim rozpuszczalniku. Jeśli stosowany jest rozpuszczalnik, we wszystkich hodowlach musi on występować w tym samym stężeniu, tj. zarówno w kontrolach negatywnych (rozpuszczalnikowych), jak i we wszystkich stężeniach substancji testowej i nie może być przy tym stężeniu toksyczny. Stężenia substancji chemicznej należy dobierać tak, aby nie dopuścić do wytrącania się lub zmętnienia roztworów.
Zalecanymi rozpuszczalnikami są sulfotlenek dimetylu (DMSO) i etanol (ETOH). Można również użyć innych rozpuszczalników o niskiej cytotoksyczności. Przed użyciem wszystkie rozpuszczalniki powinny być ocenione pod kątem określonych właściwości, np. reakcji z substancją testową, tłumienia efektu fototoksycznego, właściwości zmiatania wolnych rodników i/lub stabilności chemicznej w rozpuszczalniku.
Jeśli nie ma to wpływu na stabilność substancji testowej można ułatwić rozpuszczanie stosując mieszanie wirowe i/lub podgrzewanie do odpowiedniej temperatury.
1.4.1.5. Warunki irradiacji
1.4.1.5.1.
W badaniach fototoksyczności wybór odpowiedniego źródła światła ma podstawowe znaczenie. Światło UVA i w widzialnych częściach widma wiąże się zwykle z fototoksycznymi reakcjami in vivo (3)(12), natomiast UVB ma z nimi mniejszy związek, ale jest wysoce cytotoksyczny; cytotoksyczność wzrasta tysiąckrotnie kiedy długość fali zmienia się z 313 do 280 nm (13). Kryteria wyboru odpowiedniego źródła światła muszą obejmować wymóg, aby źródło światła emitowało długości fal absorbowanych przez substancję testową (widmo absorpcyjne) i żeby dawka światła (osiągalna w rozsądnym czasie ekspozycji) wystarczała do wykrycia znanych substancji fotocytotoksycznych. Co więcej, zastosowane długości fal i dawki nie powinny być zbytnio szkodliwe dla systemu testowego, np. emisja ciepła (promieniowanie podczerwone).
Uważa się, że optymalnym rodzajem światła jest światło pochodzące z symulatorów światła słonecznego. Rozkład mocy irradiacji symulatora światła słonecznego wyposażonego w filtry powinien być zbliżony do widma światła dziennego podanego w (14). Jako symulatory światła słonecznego używane są zarówno łukowe lampy ksenonowe, jak i lampy (domieszkowane) rtęciowo-metalowo-halogenkowe (15). Te ostatnie mają tę zaletę, że emitują mniej ciepła i są tańsze, ale mniej doskonale niż lampy ksenonowe imitują światło słoneczne. Ponieważ wszystkie symulatory światła słonecznego emitują znaczne ilości UVB, powinny być wyposażone w odpowiednie filtry, aby zredukować wysoce toksyczne promieniowanie UVB. Ponieważ plastikowe materiały stosowane przy hodowlach komórek zawierają stabilizatory UV, widmo powinno być mierzone przez ten sam typ pokrywy płytki z 96 studzienkami, jaki jest używany w teście. Niezależnie od kroków podjętych w celu osłabienia promieniowania w niektórych częściach widma przez filtrowanie lub na skutek nieuniknionego oddziaływania sprzętu, widmo rejestrowane poniżej tych filtrów nie powinno odbiegać od widma standardowego światła dziennego (14). Przykład rozkładu widma irradiancji wyposażonego w filtry symulatora światła słonecznego stosowanego w badaniu walidacyjnym badania fotoksyczności 3T3 NRU in vitro podano w (8)(16). Zob. także załącznik 2 rysunek 1.
1.4.1.5.2
Intensywność światła (irradiancja) powinna być regularnie sprawdzana przy użyciu odpowiedniego, szerokopasmowego miernika UV przed każdym badaniem fototoksyczności. Intensywność powinna być mierzona przez tego samego typu pokrywę płytki z 96 studzienkami co stosowana w badaniu. Miernik UV musi być wykalibrowany stosownie do źródła. Należy sprawdzać sprawność miernika UV i w tym celu używać drugiego, wzorcowego licznika UV tego samego typu, zalecana jest identyczna kalibracja. W idealnym przypadku, w większych odstępach czasu, do pomiaru irradiancji spektralnej filtrowanego źródła światła i do sprawdzania szerokopasmowego miernika UV powinien być używany spektroradiometr.
Stwierdzono, że dawka 5 J/cm2 (mierzona w zakresie UVA) nie jest toksyczna dla komórek Balb/c 3T3 i jest wystarczająco silna, aby wzbudzić substancje chemiczne i spowodować ich wejście w reakcje fotoksyczne (6)(17), np. żeby uzyskać 5 J/cm2 w ciągu 50 minut irradiancja została ustalona na 1,7 mW/cm2. Zob. załacznik 2, rysunek 2. Jeśli używa się innej linii komórek albo innego źródła światła, może być konieczne przeprowadzenie kalibracji dawki irradiacji, tak aby został dobrany schemat dawek, który nie jest szkodliwy dla komórek, a zarazem jest wystarczający, aby spowodować wzbudzenie standardowych fototoksyn. Czas ekspozycji na światło wyliczany jest w następujący sposób:
|
|
1 J = 1 W sec |
1.4.2. Warunki badania
1.4.2.1. Stężenia substancji testowej
Zakresy stężeń substancji chemicznej testowanej przy naświetleniu (+Irr) i bez światła (-Irr) powinny być odpowiednio określone w eksperymentach służących wyznaczeniu zakresów dawki. Przydatne może być przeprowadzenie początkowej oceny rozpuszczalności, a następnie po 60 minutach (lub jakimkolwiek innym czasie, odpowiednim do schematu badania), jako że rozpuszczalność może znacznie się zmieniać w czasie lub w trakcie ekspozycji. Aby uniknąć toksyczności, spowodowanej niewłaściwymi warunkami hodowli lub obecnością silnie kwaśnych lub zasadowych związków chemicznych, po dodaniu badanej substancji pH hodowli komórkowych powinno mieścić się w przedziale 6,5–7,8.
Najwyższe stężenie substancji testowej powinno mieścić się w granicach fizjologicznych, tzn. należy unikać stresu pH i osmotycznego. W zależności od substancji testowej może być konieczne wzięcie pod uwagę innych właściwości fizyko-chemicznych jako czynników ograniczających najwyższe stężenie testowe. W przypadku stosunkowo trudno rozpuszczalnych substancji, które nie są toksyczne przy stężeniach do punktu nasycenia, należy testować najwyższe możliwe do osiągnięcia stężenie. Generalnie, należy unikać wytrącania substancji testowej przy jakimkolwiek stężeniu testowym. Maksymalne stężenie substancji testowej nie powinno przekraczać 1 000 g/mL; osmolarność nie powinna przekraczać 10 mmol. Należy użyć 8 stężeń substancji testowej przygotowanych zgodnie z postępem geometrycznym, ze stałym współczynnikiem rozcieńczania (zob. część 2.1 akapit drugi).
Jeśli istnieją dane wskazujące (uzyskane w toku eksperymentu określającego zakres), że substancja testowa nie jest cytotoksyczna do stężenia granicznego w eksperymencie ciemnym (-Irr), ale jest wysoce cytotoksyczna w przypadku naświetlania (+Irr), zakresy stężeń wybranych dla eksperymentu (+Irr) mogą się różnić od zakresu stężeń dla (-Irr), jeśli eksperyment ma spełnić wymaganie odpowiedniej jakości danych.
1.4.2.2. Kontrole
1.4.2.2.1.
Komórki powinny być regularnie sprawdzane (mniej więcej co piąty pasaż) pod kątem wrażliwości na źródło światła przez ocenę ich żywotności po ekspozycji na rosnące dawki światła. W ocenie należy zastosować kilka dawek irradiacji, w tym dawki, które są znacznie większe od dawek użytych w badaniu fototoksyczności 3T3 NRU. Dawki te najłatwiej skwantyfikować mierząc cześć UV widma emitowanego przez źródło światła. Komórki są wysiewane przy gęstości stosowanej w badaniu fototoksyczności 3T3 NRU in vitro i naświetlane następnego dnia. Następnie, na podstawie wychwytu barwnika Neutral Red, określana jest żywotność komórek. Należy wykazać, że ostateczna najwyższa dawka nietoksyczna (np. w badaniu walidacyjnym: 5 J/cm2 [UVA]) była wystarczająca do prawidłowego sklasyfikowania wzorcowych substancji chemicznych (tabela 1).
1.4.2.2.2.
TBadanie spełnia kryteria jakościowe, jeśli naświetlone kontrole negatywne/rozpuszczalnikowe wykazują żywotność ponad 80 % w porównaniu z nie naświetlaną kontrolą negatywną/rozpuszczalnikową.
1.4.2.2.3
Absolutna gęstość optyczna (OD540 NRU) barwnika Neutral Red wyekstrahowanego z kontroli rozpuszczalnikowych wskazuje, czy 1 × 104 komórek wysianych do każdej studzienki w ciągu dwóch dni testu mnożyło się, podwajając liczebność w normalnym czasie. Badanie spełnia kryteria akceptacji, jeśli średnia OD540 NRU kontroli nie poddanych działaniu substancji wynosi > 0,4 (tj. około dwadzieścia razy więcej niż absorbancja rozpuszczalnika w tle).
1.4.2.2.4.
Równocześnie z każdym badaniem in vitro fototoksyczności 3T3 NRU powinna być testowana znana substancja fototoksyczna. Zaleca się chlorpromazynę (CPZ). W przypadku CPZ testowanej zgodnie ze standardową procedurą, w teście fotoksyczności 3T3 NRU in vitro, określono następujące kryteria akceptacji: CPZ naświetlona (+Irr): IC50 = 0,1 do 2,0 μg/ml, CPZ nienaświetlona (-Irr): IC50 = 7,0 do 90,0 μg/mL. Współczynnik fotopodrażnienia (PIF) powinien być > 6. Należy monitorować zmiany w czasie parametrów kontroli pozytywnej.
Zamiast chloropromazyny do równoczesnych kontroli pozytywnych można użyć związków fototoksycznych, odpowiednich dla klasy chemicznej lub charakterystyki rozpuszczalności ocenianego związku chemicznego .
1.4.3 Procedura badania (6)(7)(8)(16)(17):
1.4.3.1 Pierwszy dzień:
Rozmieścić 100 μL pożywki hodowlanej w peryferyjnych studzienkach 96-studzienkowej płytki mikrotitracyjnej do hodowli tkankowych (= puste). W pozostałych studzienkach rozmieścić 100 μL zawiesiny komórek (l × 105 komórek/mL) w pożywce hodowlanej (= 1 × 104 komórek/studzienka). Dla każdej serii stężeń danej substancji testowej oraz dla kontroli rozpuszczalnikowej i kontroli dodatniej należy przygotować dwie płytki.
Inkubować komórki przez 24 godziny (zob. część 1.4.1.2), dopóki nie utworzą na wpół jednolitej monowarstwy. Okres inkubacji umożliwia komórkom odzyskanie żywotności, przyleganie i wzrost wykładniczy.
1.4.3.2 Drugi dzień:
Po inkubacji zdekantować pożywkę hodowlaną z komórek i przemyć je ostrożnie 150 μL roztworu buforowego użytego do inkubacji. Dodać 100 μL buforu zawierającego odpowiednie stężenie substancji testowej lub rozpuszczalnika (kontrola rozpuszczalnikowa). Zastosować 8 różnych stężeń substancji testowej. Inkubować komórki z substancją testową w ciemności przez 60 minut (zob. część 1.4.1.2 i 1.4.1.4 akapit drugi).
Z dwóch płytek przygotowanych do każdej serii stężeń substancji testowej i kontroli wybiera się jedną, zwykle losowo, do oznaczenia cytotoksyczności (-Irr) (tj. płytka kontrolna) i jedną (płytka eksperymentalna) do oznaczenia fototoksyczności (+Irr).
W celu przeprowadzenia ekspozycji +Irr naświetlać komórki w temperaturze pokojowej przez 50 minut przez pokrywę 96-studzienkowej płytki przy najwyższej dawce promieniowania, które nie jest toksyczne (zob. także załącznik 2). Trzymać nienaświetlone płytki (-Irr) w temperaturze pokojowej w ciemności (= czas ekspozycji na światło).
Zdekantować roztwór testowy i ostrożnie przemyć dwukrotnie 150 μL buforowanym roztworem używanym do inkubacji, ale nie zawierającym materiału testowego. Zamienić bufor na pożywkę hodowlaną i inkubować (zob. część 1.4.1.2) do następnego dnia (godz. 18–22).
1.4.3.3 Trzeci dzień:
1.4.3.3.1
Komórki powinny być sprawdzone pod kątem wzrostu, morfologii oraz kompletności monowarstwy za pomocą kontrastowego mikroskopu fazowego. Należy zanotować zmiany w morfologii komórek i wpływ na wzrost komórek.
1.4.3.3.2
Przemyć komórki 150 μL podgrzanego buforu. Usunąć roztwór do przemywania poprzez lekkie stukanie. Dodać 100 μL nośnika, zawierającego 50 μg/mL barwnika Neutral Red (NR) (chlorowodorek 3-amino-7-dimetyloamino-2-metylofenazyny, numer EINECS 209-035-8; numer CAS 553-24-2; C.I. 50040) oraz surowicę (16) i przez 3 godz. inkubować jak opisano w akapicie 1.4.1.2. Po inkubacji usunąć nośnik zawierający NR i przemyć komórki 150 μL buforu. Zdekantować i usunąć nadmiar buforu przez odsączenie lub odwirowanie.
Dodać dokładnie 150 μL roztworu desorbującego NR (świeżo przygotowanego, 49 części wody + 50 części etanolu + 1 część kwasu octowego).
Wytrząsać delikatnie płytkę do mikromiareczkowania w wytrząsarce do płytek przez 10 minut, dopóki NR nie zostanie wyekstrahowany z komórek i nie utworzy jednorodnego roztworu.
Zmierzyć gęstość optyczną ekstraktu NR w spektrofotometrze przy 540 nm, stosując próby puste jako odniesienie. Zapisać dane w elektronicznym pliku w odpowiednim formacie w celu dalszej analizy.
2. DANE
2.1. JAKOŚĆ I ILOŚĆ DANYCH
Dane testowe powinny umożliwić wiarygodną analizę odpowiedzi na stężenia, otrzymanych w obecności i przy braku irradiacji i – jeśli to możliwe – stężenia testowanej substancji, przy którym żywotność komórek jest zredukowana do 50 % (IC50). Jeśli stwierdzona została cytotoksyczność, zarówno zakres stężenia, jak i punkty przecięcia z osią krzywych dla poszczególnych stężeń powinny być ustalone w taki sposób, aby umożliwić dopasowanie krzywej do danych eksperymentalnych.
Zarówno w przypadku wyraźnie dodatnich, jak i wyraźnie ujemnych wyników (zob. część 2.3 akapit pierwszy) może wystarczyć pierwotny eksperyment, poparty jednym lub kilku wstępnymi testami określającymi zakres.
Wyniki niejednoznaczne, bliskie wartości granicznych lub niejasne powinny zostać przed dalszym testowaniem wyjaśnione (zob. także część 2.4 akapit drugi). W takim przypadku należy rozważyć modyfikację warunków eksperymentu. Do wyników eksperymentalnych, które mogą być modyfikowane, zalicza się zakres stężeń lub różnice między kolejnymi stężeniami, czas wstępnej inkubacji oraz czas ekspozycji na irradiację. W przypadku substancji, które łatwo hydrolizują może być właściwe zastosowanie krótszego czasu ekspozycji.
2.2. OCENA WYNIKÓW
Aby umożliwić ocenę danych można obliczyć współczynnik fotopodrażnienia (PIF) lub średni efekt fotoelektryczny.
Przy obliczaniu miar fotoksyczności (zob. niżej) należy wyznaczyć za pomocą odpowiedniej ciągłej krzywej odpowiedzi na stężenie (model) grupę dyskretnych wartości odpowiedzi na stężenie. Dopasowywanie krzywej do danych zwykle odbywa się za pomocą metody regresji nieliniowej (18). Aby ocenić wpływ zmienności danych na kształt dopasowanej krzywej, zaleca się zastosować metodę bootstrap.
Współczynnik fotopodrażnienia (PIF) jest obliczany według następującego wzoru:
![]()
Jeśli nie da się obliczyć IC50 w obecności lub przy braku światła, nie jest możliwe obliczenie PIF dla materiału testowego. Średni efekt fotoelektryczny (MPE) wyznacza się na podstawie porównania całych krzywych odpowiedzi na stężenie (19). Efekt ten definiowany jest jako średnia ważona reprezentatywnego zbioru wartości efektu fotoelektrycznego

Efekt fotoelektryczny PEc przy dowolnym stężeniu C jest definiowany jako iloczyn efektu odpowiedzi REc i efektu dawki DEc, tj. PEc = REc × DEc. Efekt odpowiedzi REc jest różnicą między odpowiedziami obserwowanymi przy braku światła i przy świetle, tj. REc = REc = Rc (-Irr) – Rc (+Irr). Efekt dawki oblicza się według wzoru:

gdzie C* oznacza stężenie równoważne, tj. stężenie, przy którym odpowiedź +Irr jest taka sama jak odpowiedź -Irr przy stężeniu C. Jeśli C* nie da się określić ponieważ wartości odpowiedzi na krzywej + Irr są systematycznie wyższe lub niższe niż RC (-Irr), efekt dawki jest przyjmowany za 1. Czynniki ważące są wyznaczone przez najwyższą wartość odpowiedzi, tj. Wi = MAX { Ri (+Irr), Ri (-Irr)}. Tabela stężeń Ci jest dobierana w taki sposób, że ta sama liczba punktów przypada na każdy z przedziałów stężenia, określonych przez wartości stężenia zastosowane w eksperymencie. Obliczanie MPE ogranicza się do maksymalnej wartości stężenia, przy której co najmniej jedna z dwóch krzywych wciąż wykazuje wartość odpowiedzi co najmniej 10 %. Jeśli to maksymalne stężenie jest wyższe niż najwyższe stężenie zastosowane w eksperymencie +Irr, pozostała część krzywej +Irr jest dopasowywana do wartości odpowiedzi „0”. Substancja jest klasyfikowana jako fototoksyczna, w zależności od tego, czy wartość MPE jest większa niż odpowiednio wybrany punkt odcięcia (MPEc = 0,15), czy nie.
Pakiet programów do obliczania PIF i MPE można uzyskać na stronie internetowej OECD (20).
2.3. INTERPRETACJA WYNIKÓW
Na podstawie badań walidacyjnych (8) substancja testowa z PIF < 2 lub MPE <0,1 oznacza: „brak fototoksyczności”. PIF > 2 i < 5 lub MPE > 0,1 i < 0,15 oznacza: „prawdopodobną fototoksyczność”; a PIF > 5 lub MPE > 0,15 oznacza: „fototoksyczność”.
W każdym laboratorium zaczynającym przeprowadzać to badanie, przed przebadaniem substancji testowej pod kątem fototoksyczności należy poddać testom materiały wzorcowe wymienione w tabeli 1. Wartości PIF lub MPE powinny być bliskie wartości wymienionych w tabeli 1.
Tabela 1
|
Nazwa chemiczna |
Nr EINECS |
Nr CAS |
PIF |
MPE |
Pik absorpcji |
Rozpuszczalnik (9) |
|
Amiodaron HCL |
243-293-2 |
[19774-82-4] |
> 3,25 |
0,27–0,54 |
242 nm 300 nm (prog) |
etanol |
|
Chloropromazyna HCL |
200-701-3 |
[69-09-0] |
> 14,4 |
0,33–0,63 |
309 nm |
etanol |
|
Norfloksacyna |
274-614-4 |
[70458-96-7] |
> 71,6 |
0,34–0,90 |
316 nm |
acetonitryle |
|
Antracen |
204-371-1 |
[120-12-7] |
> 18,5 |
0,19–0,81 |
356 nm |
acetonitryle |
|
Protoporfiryna IX, Disodium |
256-815-9 |
[50865-01-5] |
> 45,3 |
0,54–0,74 |
402 nm |
etanol |
|
L-Histydyna |
|
[7006-35-1] |
brak PIF |
0,05–0,10 |
211 nm |
woda |
|
Heksachlorofen |
200-733-8 |
[70-30-4] |
1,1—1,7 |
0,00–0,05 |
299 nm 317nm (prog) |
etanol |
|
Siarczan sodowy laurylu |
205-788-1 |
[151-21-3] |
1,0—1,9 |
0,00–0,05 |
brak absorpcji |
woda |
2.4. INTERPRETACJA DANYCH
Jeśli efekty fototoksyczne występują jedynie przy najwyższych stężeniach testowych (zwłaszcza w przypadku substancji testowych rozpuszczalnych w wodzie), ocena zagrożenia może wymagać uwzględnienia innych czynników. Mogą to być dane o wchłanianiu przez skórę i akumulacji substancji w skórze i/lub dane pochodzące z innych badań, np. badań in vitro na skórze ludzkiej lub zwierzęcej lub na modelach skóry.
Jeśli wykazano brak fototoksyczności (+Irr i -Irr) i jeśli słaba rozpuszczalność ograniczała wartość stężenia, jakie mogło być poddane testom, to kompatybilność substancji testowej z testem może być kwestionowana. Należy wtedy przeprowadzić testy potwierdzające przy użyciu np. innego modelu.
3. SPRAWOZDAWCZOŚĆ
SPRAWOZDANIE Z BADAŃ
Sprawozdanie z przeprowadzonego badania musi zawierać co najmniej następujące informacje:
|
|
Substancja testowa:
|
|
|
Rozpuszczalnik:
|
|
|
Komórki:
|
|
|
Warunki badania (1); inkubacja przed i po procedurze:
|
|
|
Warunki testu (2); poddawanie działaniu substancji chemicznej:
|
|
|
Warunki testu (3); irradiacja:
|
|
|
Warunki testu (4); test żywotności Neutral Red:
|
|
|
Wyniki:
|
|
|
Dyskusja o wynikach. |
|
|
Wnioski. |
4. BIBLIOGRAFIA
|
(1) |
Lovell W.W. (1993). A scheme for in vitro screening of substances for photoallergenic potential. Toxic. In Vitro 7: 95–102. |
|
(2) |
Santamaria, L. and Prino, G. (1972). List of the photodynamic substances. In Research Progress in Organic, Biological and Medicinal Chemistry-Vol. 3 part 1. North Holland Publishing Co. Amsterdam. p XI–XXXV. |
|
(3) |
Spielmann, H., Lovell, W.W., Hölzle, E., Johnson, B.E., Maurer, T., Miranda, M.A., Pape, W.J.W., Sapora, O., and Sladowski, D. (1994). The report and recommendations of ECVAM Workshop 2. ATLA, 22, 314–348. |
|
(4) |
Spikes, J.D. (1989). Photosensitization. In „The science of Photobiology” Edited by K.C. Smith. Plenum Press, New York. 2nd edition, p 79-110. |
|
(5) |
OECD (1997) Environmental Health and Safety Publications, Series on Testing and Assessment No.7 „Guidance Document On Direct Phototransformation Of Chemicals In Water” Environment Directorate, OECD, Paris. |
|
(6) |
Spielmann, H., Balls, M., Dóring, B., Holzhutter, H.G., Kalweit, S., Klecak, G., L'Eplattenier, H., Liebsch, M., Lovell, W.W., Maurer, T., Moldenhauer. F. Moore. L., Pape, W., Pfannbecker, U., Potthast, J., De Silva, O., Steiling, W., and Willshaw, A. (1994). EEC/COLIPA project on in vitro phototoxicity testing: First results obtained with a Balb/c 3T3 cell phototoxicity assay. Toxic. In Vitro 8, 793–796. |
|
(7) |
Anon (1998). Statement on the scientific validity of the 3T3 NRU PT test (an in vitro test for phototoxicity), European Commission, Joint Research Centre: ECVAM and DGXI/E/2, 3 November 1997, ATLA, 26,7–8. |
|
(8) |
Spielmann, H., Balls, M., Dupuis, J., Pape, W.J.W., Pechovitch, G. De Silva, O., Holzhutter, H.G., Clothier, R., Desolle, P., Gerberick, F., Liebsch, M., Lovell, W.W., Maurer, T., Pfannenbecker, U., Potthast, J. M., Csato, M., Sladowski, D., Steiling, W., and Brantom, P. (1998). The international EU/COLIPA In vitro phototoxicity validation study: results of phase II (blind trial), part 1: the 3T3 NRU phototoxicity test. Toxic. In Vitro 12, 305–327. |
|
(9) |
OECD (2002) Extended Expert Consultation Meeting on The In Vitro 3T3 NRU Phototoxicity Test Guideline Proposal, Berlin, 30th-31th October 2001, Secretariat's Final Summary Report, 15th March 2002, OECD ENV/EHS, available upon request from the Secretariat. |
|
(10) |
Borenfreund, E., and Puerner, J.A. (1985). Toxicity determination in vitro by morphological alterations and neutral red absorption. Toxicology Lett., 24,119–124. |
|
(11) |
Hay, R.J. (1988) The seed stock concept and quality control for cell lines. Analytical Biochemistry 171, 225–237. |
|
(12) |
Lambert L.A, Warner W.G., and Kornhauser A. (1996) Animal models for phototoxicity testing. In „Dermatotoxicology”, edited by F.N. Marzulli and H.I. Maibach. Taylor & Francis, Washington DC. 5th Edition, p 515–530. |
|
(13) |
Tyrrell R.M., Pidoux M (1987) Action spectra for human skin cells: estimates of the relative cytotoxicity of the middle ultraviolet, near ultraviolet and violet regions of sunlight on epidermal keratinocytes. Cancer Res., 47, 1825–1829. |
|
(14) |
(1993). Photography – Processed photographic colour films and paper prints – Methods for measuring image stability. |
|
(15) |
Sunscreen Testing (UV.B) TECHNICALREPORT, CIE, International Commission on Illumnation, Publication No. 90, Vienna, 1993, ISBN 3900734275 |
|
(16) |
ZEBET/ECVAM/COLIPA – Standard Operating Procedure: Final Version, 7 September, 1998. 18 pgs. |
|
(17) |
Spielmann, H., Balls, M., Dupuis, J., Pape, W.J.W., De Silva, O., Holzhutter, H.G., Gerberick, F., Liebsch, M., Lovell, W.W., and Pfannenbecker, U. (1998) A study on UV filter chemicals from Annex VII of the European Union Directive 76/768/EEC, in the in vitro 3T3 NRU phototoxicity test. ATLA 26, 679-708. |
|
(18) |
Holzhütter, H.G., and Quedenau, J. (1995) Mathematical modeling of cellular responses to external signals. J. Biol. Systems 3, 127–138. |
|
(19) |
Holzhütter, H.G. (1997). A general measure of in vitro phototoxicity derived from pairs of dose-response curves and its use for predicting the in vivo phototoxicity of chemicals. ATLA, 25, 445–462. |
|
(20) |
http://www.oecd.org/document/55/0,2340,en_2649_34377_2349687_l_l_l_l,00.html |
ZAŁĄCZNIK 1
Rola 3T3 NRU PT w sekwencyjnym podejściu do testowania fototoksyczności substancji chemicznych

ZAŁĄCZNIK 2
Rysunek 1
Rozkład mocy widma symulatora światła słonecznego wyposażonego w filtry
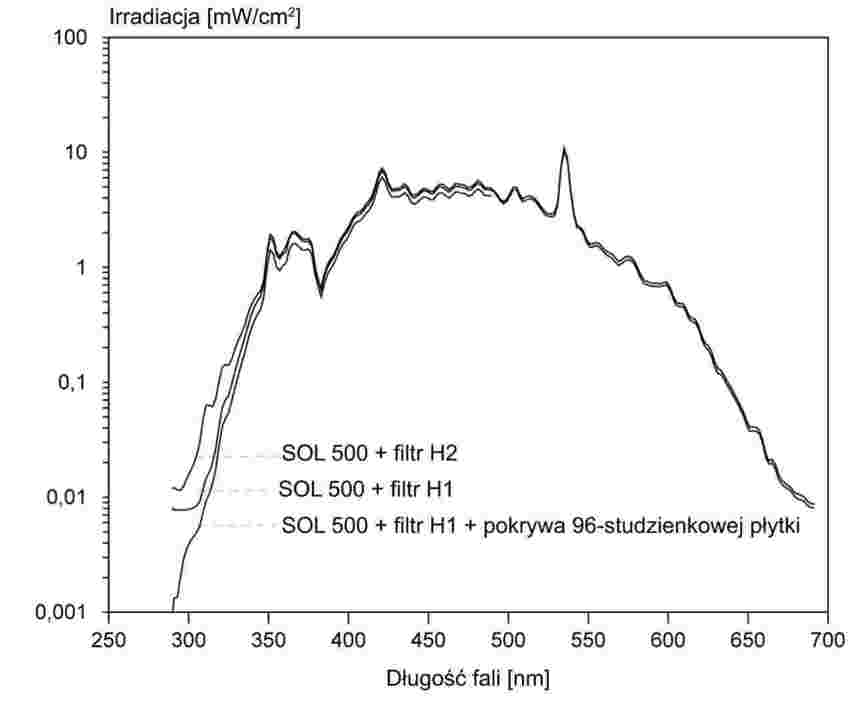
(zob. część 1.4.1.5 akapit drugi)
Na rysunku 1 podano przykład dopuszczalnego rozkładu widma irradiancji symulatora świata słonecznego wyposażonego w filtr. Opracowano na podstawie źródła halogenkowego stosowanego w badaniach walidacyjnych 3T3 NRU PT (6)(8)(17). Pokazano efekt dwóch różnych filtrów oraz dodatkowy efekt filtracyjny pokrywy 96-studzienkowej płytki do kultur komórkowych. Filtr H2 był stosowany wyłącznie w systemach testowych, które tolerują znaczną ilość UVB (model testu skórnego i test fotohemolizy krwinek czerwonych). W 3T3 NRU-PT stosowano filtr H1. Na rysunku widać, że dodatkowy efekt filtrujący pokrywy płytki występuje głównie w zakresie UVB, przy czym w widmie irradiacji wciąż jest wystarczająco dużo UVB, żeby wzbudzać substancje normalnie absorbujące promieniowanie w zakresie UVB, jak Amiodaron (zob. tabela 1).
Rysunek 2
Wrażliwość na irradiację komórek Balb/c 3T3 (mierzona w zakresie UVA)
Żywotność komórek ( % wychwytu Red Neutral w kontrolach ciemnych)
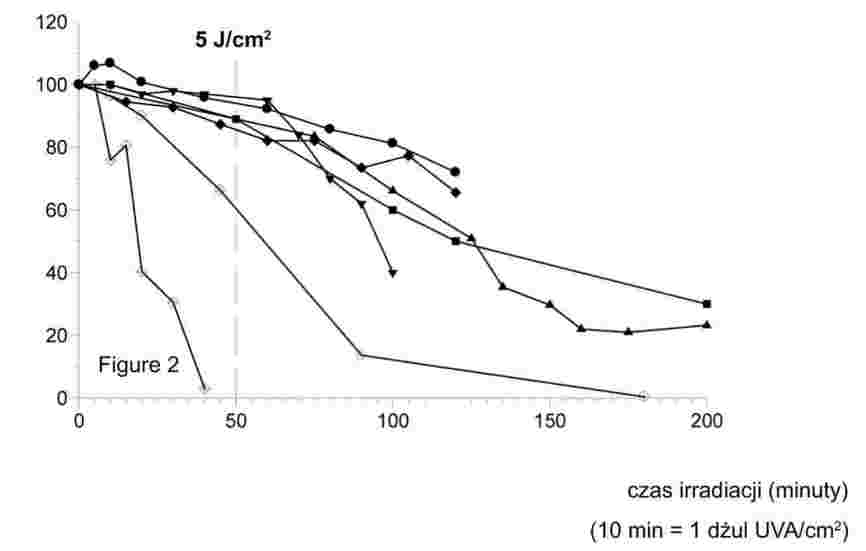
(zob. część 1.4.1.5.2 akapit drugi); 1.4.2.2.1, 1.4.2.2.2)
Wrażliwość komórek Balb/c 3T3 na irradiację przy stosowaniu symulatora światła słonecznego stosowanego w badaniach walidacyjnych testu toksyczności 3T3 NRU, mierzona w zakresie UVA. Na rysunku przedstawiono wyniki otrzymane w 7 różnych laboratoriach w badaniach przedwalidacyjnych (1). Dwie krzywe z otwartymi symbolami zostały uzyskane przy użyciu starych komórek (duża liczba pasaży), które musiały zostać zastąpione nowymi zasobami komórek; krzywe z wytłuszczonymi symbolami przedstawiają komórki z akceptowalną tolerancją na naświetlanie.
Na podstawie tych danych ustalono najwyższą niecytotoksyczną dawkę irradiacji 5 J/cm (pionowa linia przerywana). Pozioma linia przerywana pokazuje dodatkowo maksymalny akceptowalny efekt irradiacji, opisany w akapicie 1.4.2.2).
B.42. UCZULENIE SKÓRY: TEST LOKALNYCH WĘZŁÓW CHŁONNYCH
1. METODA
Niniejsza metoda jest analogiczna do OECD TG 429 (2002)
1.1. WPROWADZENIE
Test lokalnych węzłów chłonnych (LLNA) został wystarczająco potwierdzony i uznany, aby uzasadnione było jego przyjęcie jako nowej metody (1)(2)(3). Jest to druga metoda oceny potencjału uczulenia skóry przez środki chemiczne u zwierząt. Ta pierwsza metoda wykorzystuje testy na świnkach morskich, w szczególności test maksymalizacji na śwince morskiej oraz test Buehlera (4).
LLNA przedstawia alternatywną metodę określania chemikaliów uczulających skórę oraz potwierdzania, że dane środki chemiczne nie mają znaczącego potencjału do wywoływania uczulenia skóry. Nie oznacza to, że należy koniecznie we wszystkich przypadkach stosować LLNA zamiast testu na śwince morskiej, a tylko, że każdy z tych testów ma tę samą wartość merytoryczną i może zostać zastosowany jako alternatywny, i którego pozytywne lub negatywne wyniki nie wymagają już dalszego potwierdzenia.
LLNA daje pewne korzyści w odniesieniu zarówno do postępu naukowego, jak i dobrostanu zwierząt. Bada fazę indukcyjną uczulenia skóry i dostarcza danych ilościowych nadających się do oceny zależności reakcji od dawki. Szczegóły walidacji LLNA i przegląd związanej z tym bibliografii opublikowano w (5)(6)(7)(8). Trzeba ponadto zauważyć, że łagodne/umiarkowane czynniki uczulające, które zalecane są jako odpowiednie pozytywne substancje kontrolne dla metod testów na śwince morskiej, są również właściwe do stosowania w LLNA (6)(8)(9).
Będąc metodą in vivo, LLNA nie eliminuje wykorzystania zwierząt w określaniu kontaktowego działania uczulającego. Daje jednak potencjalne możliwości obniżenia liczby zwierząt potrzebnych do tego celu. Co więcej, LLNA oferuje znacznie bardziej wyrafinowany sposób wykorzystywania zwierząt do kontaktowych testów uczulenia skórnego. LLNA opiera się na rozważaniu zjawisk immunologicznych zachodzących podczas fazy indukcyjnej uczulania. W odróżnieniu od testów na śwince morskiej, test LLNA nie wymaga, żeby wywoływać reakcje nadwrażliwości skóry poprzez prowokację. Ponadto LLNA nie wymaga zastosowania adiuwanta jak wymaga tego test na śwince morskiej. Tym samym zmniejsza się niebezpieczeństwo dla zwierząt. Niezależnie od zalet LLNA w porównaniu z tradycyjnym testem na śwince morskiej należy przyjąć, że istnieją pewne ograniczenia, które mogą spowodować konieczność wykorzystania tradycyjnych testów na śwince morskiej (np. fałszywe wyniki negatywne LLNA w przypadku niektórych metali, fałszywe wyniki pozytywne w przypadku niektórych środków drażniących skórę) (10).
Zob. również Wprowadzenie do części B.
1.2. ZASADA METODY BADAWCZEJ
Podstawową zasadą u podłoża LLNA jest fakt, z czynniki uczulające wywołują pierwotną proliferację limfocytów w węźle chłonnym odprowadzającym limfę z miejsca przyłożenia środka chemicznego. Ta proliferacja jest proporcjonalna do zastosowanej dawki (oraz potencjału alergenu) i jest prostym środkiem uzyskania obiektywnego ilościowego pomiaru uczulenia. LLNA ocenia tę proliferację jako zależność reakcji od dawki, w której proliferacja w grupie badanej porównywana jest z proliferacją u zwierząt kontrolnych otrzymujących nośnik. Określa się stosunek proliferacji w grupach poddanych działaniu środka do proliferacji w grupie kontroli nośnika, nazywany wskaźnikiem stymulacji, i musi on wynosić co najmniej trzy, zanim substancję można poddać dalszym ocenom jako potencjalny czynnik uczulający skórę. Opisane metody oparte są na wykorzystaniu do pomiaru proliferacji komórek znakowania markerem promieniotwórczym. Można jednak zastosować inne punkty końcowe do oceny proliferacji, pod warunkiem że jest to odpowiednio uzasadnione i wsparte dowodami naukowymi z pełnymi cytacjami i opisem metodologii.
1.3. OPIS METODY BADAWCZEJ
1.3.1. Przygotowania
1.3.1.1. Warunki przebywania i warunki żywieniowe
Zwierzęta powinny być trzymane w pojedynczo. Temperatura w badawczych pomieszczeniach hodowlanych powinna wynosić 22 oC (± 3 oC). Chociaż wilgotność względna powinna wynosić co najmniej 30 % i pożądane jest, żeby nie przekraczała 70 %, poza czasem czyszczenia pomieszczenia, celem powinno być utrzymywanie jej na poziomie 50–60 %. Oświetlenie powinno być sztuczne, w cyklu 12 godzin jasno, 12 godzin ciemno. Do żywienia można stosować konwencjonalne pasze laboratoryjne z nieograniczonym dostępem do wody pitnej.
1.3.1.2. Przygotowanie zwierząt
Zwierzęta wybierane są losowo i znakowane w sposób umożliwiający indywidualną identyfikację (ale nie przez jakiekolwiek znaki na uszach) i trzymane w ich klatkach przez co najmniej 5 dni przed rozpoczęciem dawkowania w celu umożliwienia zaaklimatyzowania do warunków laboratoryjnych. Przed rozpoczęciem podawania bada się wszystkie zwierzęta w celu upewnienia się, że nie mają żadnych zauważalnych zmian skórnych.
1.3.2. Warunki badania
1.3.2.1. Zwierzęta doświadczalne
Mysz jest gatunkiem z wyboru do tego testu. Używa się młodych, dorosłych samic myszy szczepów CBA/Ca lub CBA/J, które nie rodziły i nie są ciężarne. Na początku doświadczenia samice powinny mieć 8–12 tygodni, a zróżnicowanie mas ciała poszczególnych zwierząt powinno być minimalne i nie przekraczać 20 % średniej masy ciała. Można wykorzystać inne szczepy i samce, jeżeli otrzymano wystarczające dowody na to, że nie istnieją znaczące specyficzne różnice w zależności od szczepu i/lub płci.
1.3.2.2 Sprawdzenie niezawodności
Kontrole pozytywne wykorzystuje się do wykazania właściwego działania testu i kompetencji laboratorium do jego udanego przeprowadzenia. Kontrola pozytywna powinna dać w teście LLNA odpowiedź pozytywną na poziom ekspozycji, przy którym oczekuje się wzrostu wskaźnika stymulacji (SI) > 3 więcej niż w kontroli negatywnej. Pozytywna dawka kontrolna powinna być dobrana w taki sposób, żeby indukcja była wyraźna, ale nie nadmierna. Preferowanymi substancjami są cynomonoheksylo aldehyd (Nr CAS 101-86-0, Nr EINECS 202-983-3) i merkaptobenzotiazol (Nr CAS 149-30-4, Nr EINECS 205-736-8). Mogą zaistnieć okoliczności, w których można użyć innych substancji spełniających powyższe kryteria, pod warunkiem że jest po temu odpowiednie uzasadnienie. Chociaż zazwyczaj w każdym teście wymagana jest pozytywna grupa kontrolna, mogą zaistnieć sytuacje, w których testujące laboratoria będą rozporządzały wcześniejszymi danymi z pozytywnych kontroli wykazującymi zadowalającą odpowiedź przez okres ponad sześciu miesięcy lub dłuższy. W takich przypadkach może być właściwym rzadsze testowanie z kontrolą pozytywną, w odstępach czasu nie większych niż 6 miesięcy. Chociaż substancja kontroli pozytywnej powinna być testowana w nośniku, o którym wiadomo, że wywołuje konsekwentną reakcję (np. aceton, oliwa z oliwek), mogą jednak zaistnieć pewne sytuacje wynikające z przepisów, w których przetestowanie w niestandardowym nośniku (o formulacji odpowiedniej klinicznie/chemicznie) może być również konieczne. W takiej sytuacji należy przetestować możliwą interakcję pozytywnej kontroli z takim niekonwencjonalnym nośnikiem.
1.3.2.3 Liczba zwierząt, poziomy dawek i wybór nośnika
W każdej grupie dawkowania powinny być użyte nie mniej niż cztery zwierzęta, z minimalną liczbą trzech stężeń substancji testowej oraz negatywną grupą kontrolną otrzymującą jedynie nośnik substancji testowej i, w stosownych przypadkach, pozytywną grupą kontrolną. W tych przypadkach, w których ma się uzyskać indywidualne wyniki dla poszczególnych zwierząt, minimalna liczba użytych zwierząt w grupie dawkowania wynosi pięć. Z wyjątkiem zastosowania substancji testowej, ze zwierzętami grup kontrolnych powinno się obchodzić w sposób identyczny jak ze zwierzętami grup badanych.
Dobór dawek i wybór nośnika powinien być oparty na zaleceniach podanych w pozycji (1) bibliografii. Dawki wybierane są z serii stężeń: 100 %, 50 %, 25 %, 10 %, 5 %, 2,5 %, 1 %, 0,5 % itd. Przy wyborze trzech kolejnych stężeń należy, w miarę dostępności, uwzględnić istniejące dane o ostrej toksyczności i podrażnieniu skóry, tak aby najwyższe stężenie maksymalizowało ekspozycję unikając ogólnoustrojowej toksyczności i nadmiernego lokalnego podrażnienia skóry (2)(11).
Wyboru nośnika należy dokonać na podstawie maksymalizowania stężeń testowych i rozpuszczalności oraz, jednocześnie, możliwości uzyskania roztworu/zawiesiny nadającej się do zastosowania substancji testowej. Zalecanymi nośnikami są, w porządku preferencji, aceton/oliwa z oliwek (w proporcji objętościowej 4:1), dimetylformamid, metylo etylo keton, glikol propylenowy i sulfotlenek dimetylowy (2)(10), ale mogą być użyte inne, jeśli przedstawi się wystarczające racjonalne uzasadnienie naukowe. W niektórych sytuacjach może być konieczne użycie, jako dodatkowej kontroli, rozpuszczalnika odpowiedniego ze względów klinicznych, lub postaci handlowej, w której substancja testowa wprowadzana jest na rynek. Szczególną uwagę należy poświęcić włączeniu substancji hydrofilowych do systemu nośnika, który nawilża skórę i nie spływa z niej natychmiast. A zatem należy unikać całkowicie wodnych roztworów.
1.3.3. Procedura testu
1.3.3.1. Harmonogram doświadczalny
Harmonogram doświadczalny badania jest następujący:
|
|
Dzień 1: Określić indywidualnie masę każdego zwierzęcia i odnotować. Rozpocząć stosowanie 25 ul właściwego roztworu lub samego nośnika, lub kontroli pozytywnej (w stosownych przypadkach), na część grzbietową każdego ucha. |
|
|
Dzień 2 i 3: Powtórzyć procedurę stosowania przeprowadzoną w dniu 1. |
|
|
Dzień 4 i 5: Bez podawania. |
|
|
Dzień 6: Zanotować masę każdego zwierzęcia. Podać w iniekcji 250ul roztworu soli fizjologicznej zbuforowanej fosforanem (PBS) zawierającego 20 uCi (7,4e + 8 Bq) 3H-metylotymidyny poprzez żyłę ogonową, wszystkim zwierzętom badanym i kontrolnym. Alternatywnie podać w iniekcji 250 uL PBS zawierających 2 uCi (7,4e + 7 Bq) 125I-jododezoksyuridiny i 10-5 M fluorodezoksyuridiny wszystkim myszom poprzez żyłę ogonową. Pięć godzin później zwierzęta zostają uśmiercone. Wycina się uszne węzły chłonne z każdego ucha i zbiera razem w PBS z każdej grupy eksperymentalnej (traktowanie zbiorcze w ujęciu grupowym); alternatywnie można wyciąć pary węzłów chłonnych z każdego zwierzęcia i zebrać razem w PBS (traktowanie zbiorcze w ujęciu osobniczym). Szczegóły i diagramy można znaleźć w załączniku I pozycji 10 bibliografii. |
1.3.3.2. Przygotowanie zawiesiny komórek
Zostaje przygotowana jedna wspólna porcja zawiesiny komórek węzła chłonnego (LNC), albo z połączonych grup dawkowania albo z obustronnych węzłów z pojedynczych osobników, przez delikatne mechaniczne rozdzielenie agregatów na siateczce ze stali nierdzewnej o 200 um oczkach. Węzły chłonne są przemywane i strącane 5 % kwasem trichlorooctowym (TCA) w 4 oC przez 18 godzin (2). Peletki są albo zawieszane ponownie w 1 ml TCA i przenoszone do fiolek scyntylacyjnych zawierających 10 ml płynu scyntylacyjnego do policzenia 3H, lub też przenoszone bezpośrednio do fiolek do liczenia impulsów gamma, w celu liczenia impulsów 125I.
1.3.3.3. Określenie proliferacji komórek (wbudowanie związku promieniotwórczego)
Wbudowanie 3H-metylotymidyny mierzone jest przez liczenie impulsów B-scyntylacji jako liczba rozpadów na minutę (DPM). Wbudowywanie 125I-jododezoksyuridiny mierzone jest jako impulsy 125I i również wyrażane jako liczba rozpadów na minutę. W zależności od użytego podejścia, wbudowanie wyrażane będzie jako DPM/grupa dawkowania (ujęcie zbiorcze) lub DPM/zwierzę (ujęcie indywidualne).
1.3.3.4. Obserwacje
1.3.3.4.1.
Zwierzęta poddaje się starannej obserwacji klinicznej raz dziennie pod kątem jakichkolwiek oznak klinicznych, czy to miejscowego podrażnienia w miejscu zastosowania, czy to ogólnoustrojowej toksyczności. Wszystkie obserwacje kliniczne są systematycznie odnotowywane w postaci indywidualnych zapisów prowadzonych dla każdego zwierzęcia.
1.3.3.4.2.
Jak podano w sekcji 1.3.3.1, indywidualne masy ciała określane są na początku testu i przy planowanym uśmierceniu zwierząt.
1.3.4 Wyliczenie wyników
Wyniki wyraża się jako wskaźnik stymulacji (SI). Jeżeli zastosowano podejście zbiorcze, SI otrzymuje się przez podzielenie zbiorczego wbudowania związku promieniotwórczego dla każdej grupy dawkowania przez wbudowanie w zbiorczej próbie grupy kontrolnej nośnika; to daje średnie SI. Jeżeli zastosowano podejście indywidualne, SI otrzymuje się przez podzielenie średniej wartości DPM/zwierzę w obrębie każdej grupy dawkowania substancji testowej i grupy pozytywnej kontroli przez średnią wartość DPM/zwierzę dla grupy kontrolnej rozpuszczalnika/nośnika. Średnie SI dla grupy kontrolnej otrzymującej nośnik wynosi zatem 1.
Wykorzystanie podejścia indywidualnego do wyliczenia SI pozwala na przeprowadzenie statystycznej analizy danych. Przez wybranie właściwej metody analizy statystycznej danych, badacz powinien zachować świadomość możliwej nierówności wariancji lub innych powiązanych problemów, które mogą spowodować konieczność transformacji danych lub przeprowadzenia nieparametrycznej analizy statystycznej. Odpowiednim podejściem do interpretacji danych jest ocena wszystkich indywidualnych danych dla grup zwierząt badanych i grup kontrolnych nośnika, i wyprowadzenie z nich najlepiej dopasowanych krzywych reakcji na dawkę, uwzględniając poziomy ufności (8)(12)(13). Jednakże badacz powinien być czujny względem możliwych „odstających” reakcji dla poszczególnych zwierząt w obrębie grupy, które mogą wymagać użycia alternatywnego pomiaru reakcji (np. mediany a nie średniej) lub wyeliminowania takiej „odstającej” reakcji.
Proces decyzyjny odnośnie do pozytywnej odpowiedzi obejmuje wskaźnik stymulacji ≥ 3 wraz z rozpatrzeniem zależności reakcji od dawki oraz, w stosownych przypadkach, istotności statystycznej (3)(6)(8)(12)(14).
Jeżeli zachodzi potrzeba objaśnienia otrzymanych wyników, należy rozważyć różne własności substancji testowej, w tym rozważyć, czy jest strukturalnie pokrewna znanym czynnikom uczulającym, czy powoduje nadmierne podrażnienie skóry, a także rozważyć charakter zaobserwowanej reakcji na dawkę. Te i inne względy omówione są szczegółowo gdzie indziej (7).
2. DANE
Dane powinny być podsumowane w formie tabelarycznej pokazującej średnie i indywidualne wartości DMP i wskaźniki stymulacji dla każdej grupy dawkowania (wliczają w to grupę kontrolną nośnika).
3. SPRAWOZDAWCZOŚĆ
3.1. SPRAWOZDANIE Z TESTU
Sprawozdanie z testu powinno zawierać następujące informacje:
|
|
Substancja testowa:
|
|
|
Nośnik:
|
|
|
Zwierzęta badane:
|
|
|
Warunki testu:
|
|
|
Sprawdzenie niezawodności:
|
|
|
Wyniki:
|
|
|
Dyskusja wyników:
|
4. BIBLIOGRAFIA
|
(1) |
Kimber, I. and Basketter, D.A. (1992). The murine local lymph node assay; collaborative studies and new directions: A commentary. Food and Chemical Toxicology 30, 165–169. |
|
(2) |
Kimber, I, Derman, R.J., Scholes E.W, and Basketter, D.A. (1994). The local lymph node assay: developments and applications. Toxicology, 93, 13–31. |
|
(3) |
Kimber, I., Hilton, J., Dearman, R.J., Gerberick, G.F., Ryan, CA., Basketter, D.A., Lea, L., House, R.V., Ladies, G.S., Loveless, S.E., Hastings, K.L. (1998). Assessment of the skin sensitisation potential of topical medicaments using the local lymph node assay: An interlaboratory exercise. Journal of Toxicology and Environmental Health, 53, 563–79. |
|
(4) |
Testing Method B.6. |
|
(5) |
Chamberlain, M. and Basketter, D.A. (1996). The local lymph node assay: status of validation. Food and Chemical Toxicology, 34, 999–1002. |
|
(6) |
Basketter, D.A., Gerberick, G.F., Kimber, I. and Loveless, S.E (1996). The local lymph node assay- A viable alternative to currently accepted skin sensitisation tests. Food and Chemical Toxicology, 34, 985–997. |
|
(7) |
Basketter, D.A., Gerberick, G.F. and Kimber, I. (1998). Strategies for identifying false positive responses in predictive sensitisation tests. Food and Chemical Toxicology. 36, 327–33. |
|
(8) |
Van Och, F.M.M, Slob, W., De Jong, W.H., Vandebriel, R.J., Van Loveren, H. (2000). A quantitative method for assessing the sensitising potency of low molecular weight chemicals using a local lymph node assay: employement of a regression method that includes determination of uncertainty margins. Toxicology, 146, 49–59. |
|
(9) |
Dearman, R.J., Hilton, J., Evans, P., Harvey, P., Basketter, D.A. and Kimber, I. (1998). Temporal stability of local lymph node assay responses to hexyl cinnamic aldehyde. Journal of Applied Toxicology, 18, 281-4. |
|
(10) |
National Institute of Environmental Health Sciences (1999). The Murine Local Lymph Node Assay: A Test Method for Assessing the Allergic Contact Dermatitis Potential of Chemicals/Compounds: The Results of an Independent Peer Review Evaluation Coordinated by the Interagency Coordinating Committee on the Validation of Alternative Methods (ICCVAM) and the National Toxicology Program Center for the Evaluation of Alternative Toxicological Methods (NICETAM). NIH Publication No: 99-4494, Research Triangle Park, N.C. (http://iccvam.niehs.nih.gov). |
|
(11) |
Testing method B.4. |
|
(12) |
Basketter, D.A., Selbie, E., Scholes, E.W. Lees, D. Kimber, I. and Botham, P.A. (1993) Results with OECD recommended positive control sensitisers in the maximisation, Buehler and local lymph node assays. Food and Chemical Toxicology, 31, 63–67. |
|
(13) |
Basketter D.A., Lea L.J., Dickens A., Briggs D., Pate I., Dearman R.J., Kimber I. (1999). A comparison of statistical approaches to the derivation of EC3 values from local lymph node assay dose responses. J. Appl. Toxicology, 19, 261–266. |
|
(14) |
Basketter DA, Blaikie L, Derman RJ, Kimber I, Ryan CA, Gerberick GF, Harvey P, Evans P, White IR and Rycroft RTG (2000). Use of local lymph node assay for the estimation of relative contact allergenic potency. Contact Dermatitis 42, 344–48. |
B.43. BADANIE NEUROTOKSYCZNOŚCI U GRYZONI
1. METODA
Ta metoda badania jest odpowiednikiem TG 424 (1997).
Ta metoda badania została zaprojektowana w celu uzyskania danych potrzebnych do potwierdzenia lub bliższego scharakteryzowania potencjalnej neurotoksyczności związków chemicznych względem dorosłych zwierząt. Można ją albo łączyć z istniejącymi metodami testowania wielokrotnej dawki, albo wykorzystywać w odrębnym badaniu. W celu uzyskania pomocy w projektowaniu badań opartych na tej metodzie zaleca się zapoznanie się z dokumentem zawierającym wytyczne OECD dotyczące strategii i metod badania neurotoksyczności (1). Jest to szczególnie ważne, jeżeli rozważa się modyfikacje obserwacji i procedur badania zalecanych do rutynowego wykorzystania tej metody. Dokument zawierający wytyczne przygotowano w celu ułatwienia wyboru innych procedur testowych do wykorzystywania w konkretnych okolicznościach.
Ocena neurotoksyczności rozwojowej nie jest tematem omawianej metody.
1.1. WPROWADZENIE
W ocenie i ewaluacji charakterystyki toksycznej substancji chemicznych ważne jest rozparzenie potencjału wywoływania skutków neurotoksycznych. Już metoda testowania ogólnoustrojowej toksyczności dawki wielokrotnej zawiera obserwacje, które mogą być badaniami sortującymi pod kątem potencjalnej neurotoksyczności. Omawiana metoda testowa może być wykorzystana do zaprojektowania badania zmierzającego do uzyskania bliższej informacji lub potwierdzenia skutków neurotoksycznych obserwowanych w badaniach ogólnoustrojowej toksyczności dawki wielokrotnej. Jednakże rozpatrywanie potencjalnej neurotoksyczności pewnych klas związków chemicznych może wskazywać, że może być ona właściwiej oceniona za pomocą omawianej metody, bez wcześniejszego wskazania potencjalnej neurotoksyczności w badaniu ogólnoustrojowej toksyczności dawki wielokrotnej. Tego rodzaju ewentualne wskazania obejmują na przykład:
|
— |
obserwacje oznak neurologicznych lub zmian neuropatologicznych w badaniach innych niż badania ogólnoustrojowej toksyczności dawki wielokrotnej, lub |
|
— |
strukturalna zależność lub inne informacje wiążące je ze znanymi substancjami neurotoksycznymi. |
Ponadto mogą być jeszcze inne przypadki, kiedy wykorzystanie omawianej metody jest właściwe; w celu uzyskania dodatkowych szczegółów, zob. (1).
Ta metoda została opracowana w taki sposób, że można ją dostosować do konkretnych potrzeb potwierdzenia specyficznej neurotoksyczności histopatologicznej i behawioralnej związku chemicznego, jak również charakteryzowania i ilościowego określania reakcji neurotoksycznej.
W przeszłości utożsamiano neurotoksyczność z neuropatią obejmującą zmiany neuropatologiczne lub dysfunkcje neurologiczne, takie jak napady, paraliż lub drżenie. Chociaż neuropatia jest ważnym objawem klinicznym neurotoksyczności, w tej chwili wiadomo już, że jest wiele innych oznak toksyczności względem układu nerwowego (np. utrata koordynacji motorycznej, niedobory czucia, dysfunkcje uczenia się i pamięci), które mogą nie ujawniać się w badaniach neuropath i innych typach badań.
Omawiana metoda badania neurotoksyczności zaprojektowana została w celu wykrywania głównych skutków neurobehawioralnych i neuropatologicznych u dorosłych gryzoni. Chociaż skutki behawioralne, nawet przy braku zmian morfologicznych mogą odzwierciedlać niekorzystny wpływ na organizm, nie wszystkie zmiany behawioralne są specyficzne dla systemu nerwowego. A zatem wszelkie obserwowane zmiany należy interpretować w powiązaniu z korelacyjnymi danymi histopatologicznymi, hematologicznymi lub biochemicznymi jak również danymi dotyczącymi innych rodzajów toksyczności ogólnoustrojowej. Testowanie potrzebne w tej metodzie do scharakteryzowania i ilościowego określenia reakcji neurotoksycznych obejmuje specyficzne procedury histopatologiczne i behawioralne, które mogą być dalej potwierdzone przez badania elektrofizjologiczne i/lub biochemiczne (1)(2)(3)(4).
Czynniki neurotoksyczne mogą oddziaływać na wiele celów w obrębie układu nerwowego i to za pośrednictwem różnych mechanizmów. Ponieważ nie ma jednego zestawu testów, który potrafiłby dokładni ocenić potencjał neurotoksyczny wszelkich substancji, może być konieczne wykorzystywanie innych testów in vivo lub in vitro specyficznych względem obserwowanego lub przewidywanego rodzaju neurotoksyczności.
Niniejsza metoda badania może być również wykorzystana w połączeniu z wytycznymi podanymi w dokumencie OECD dotyczącym strategii i metod badania neurotoksyczności (1) do zaprojektowania badań zmierzających do bliższego scharakteryzowania lub zwiększenia czułości ilościowego określania reakcji na dawkę, albo lepszej oceny poziomu dawkowania, przy którym nie obserwuje się niekorzystnych skutków lub potwierdzenia znanych, lub podejrzewanych zagrożeń związanych z danym związkiem chemicznym. Na przykład można zaprojektować badania zmierzające do zidentyfikowania i oceny mechanizmu(-ów) neurotoksycznych lub uzupełnienia danych dostępnych w ramach wykorzystania podstawowych procedur obserwacji neurobehawioralnych i neuropatologicznych. Badania takie nie muszą dublować danych, które można by uzyskać przez zastosowanie standardowych procedur zalecanych w niniejszej metodzie, jeżeli takie dane sąjuż dostępne albo nie zostały uznane za konieczne do interpretacji wyników tego badania.
Badanie neurotoksyczności, wykorzystywane odrębnie albo w połączeniu z innymi, dostarcza informacji, które mogą:
|
— |
stwierdzić, czy testowany związek chemiczny wpłynął na układ nerwowy w sposób trwały czy odwracalny, |
|
— |
przyczynić się do scharakteryzowania zmian w układzie nerwowym związanych z ekspozycją na związek chemiczny, oraz do zrozumienia mechanizmu leżącego u podłoża jego oddziaływania, |
|
— |
określić zależności reakcji od dawki i czasu, w celu ocenienia poziomu dawkowania, przy którym nie obserwuje się niekorzystnych skutków (który można wykorzystać do ustalenia kryteriów bezpieczeństwa w odniesieniu do danego związku chemicznego). |
Omawiana metoda testowa wykorzystuje doustną drogę podawania testowanej substancji. Inne drogi podawania (np. naskórna lub wziewna) mogą być właściwsze i mogą wymagać modyfikacji zalecanych procedur. Wybór drogi podawania zależy od profilu narażenia ludzi oraz dostępnych informacji w zakresie toksykologii i kinetyki.
1.2. DEFINICJE
Niekorzystny skutek: jakakolwiek mająca związek z terapią zmiana w stosunku do danych bazowych, która zmniejsza zdolność organizmu do przeżycia, rozmnażania lub przystosowania się do środowiska.
Dawka: jest ilością podawanej substancji testowej. Dawka wyrażana jest w jednostkach wagowych (g, mg) lub jako waga substancji testowej na jednostkę masy badanego zwierzęcia (np. mg/Kg), lub jako stałe stężenie pożywienia (ppm).
Dawkowanie: jest terminem ogólnym obejmującym dawkę, częstotliwość i czas trwania dawkowania.
Neurotoksyczność: jest niekorzystną zmianą struktury lub funkcji układu nerwowego wynikającą z ekspozycji na czynnik chemiczny, biologiczny lub fizyczny.
Czynnik neurotoksyczny: to jakikolwiek czynnik chemiczny, biologiczny lub fizyczny o potencjale powodowania skutków neurotoksycznych.
NOAEL: jest skrótem oznaczającym poziom, przy którym nie obserwuje się niekorzystnych skutków i jest najwyższą dawką lub poziomem ekspozycji, przy zastosowaniu którego nie obserwuje się niekorzystnych wyników terapii.
1.3. ZASADA METODY BADAWCZEJ
Testowany związek chemiczny podawany jest drogą doustną, w postaci pewnego zakresu dawek, kilku grupom gryzoni laboratoryjnych. Wymagane są zazwyczaj dawki wielokrotne, zaś reżim dawkowania może obejmować 28 dni, podchronicznej (90 dni) lub chronicznej toksyczności (1 rok lub dłużej). Procedury opisane w niniejszej metodzie badawczej mogą być również użyte w badaniu neurotoksyczności ostrej. Zwierzęta bada się w celu wykrycia lub scharakteryzowania behawioralnych i/lub neurologicznych nieprawidłowości. Zakres zachowań, na które czynniki neurotoksyczne mogą wpływać, oceniany jest w każdym okresie obserwacji. Na koniec badania podgrupę zwierząt każdej z płci poddaje się perfuzji in situ, a następnie preparuje i bada skrawki mózgu, rdzenia kręgowego i nerwów obwodowych.
Jeżeli badanie przeprowadza się jako odrębne badanie w celu sortowania pod kątem neurotoksyczności lub scharakteryzowania efektów neurotoksycznych, zwierzęta z każdej grupy, nieużyte do perfuzji i następczego badania histopatologicznego (zob. tabela 1), mogą zostać wykorzystane do konkretnych procedur neurobehawioralnych, neuropatologicznych i elektrofizjologicznych, które mogą uzupełnić dana otrzymane w standardowych badaniach wymaganych w niniejszej metodzie (1). Tego rodzaju dodatkowe procedury mogą być szczególnie użyteczne w przypadkach kiedy obserwacje empiryczne lub przewidywane skutki wskazują na jakiś szczególny rodzaj lub docelowy organ działania neurotoksycznego. Alternatywnie, pozostałe zwierzęta mogą zostać wykorzystane do ocen, jakich wymaga metoda badania toksyczności dawki wielokrotnej u gryzoni.
Jeżeli procedury powyższej metody badania łączone są z objętymi innymi metodami, należy użyć wystarczającej liczby zwierząt potrzebnej do osiągnięcia celów obu badań.
1.4. OPIS METODY BADAWCZEJ
1.4.1. Wybór gatunków zwierząt
Szczur jest pożądanym gatunkiem zwierzęcia do badań, chociaż można użyć innego gatunku gryzonia, z podaniem uzasadnienia. Należy prowadzić badania na młodych dorosłych, zdrowych osobnikach powszechnie używanych szczepów laboratoryjnych. Samice powinny być nieciężarne i takie, które nie rodziły. Dawkowanie powinno się zazwyczaj zacząć najszybciej jak to możliwe po zakończeniu ssania, najlepiej nie później niż w wieku sześciu tygodni, w każdym razie zanim zwierzęta osiągną wiek dziewięciu tygodni. Jednakże jeżeli badanie łączone jest z innym badaniem, wymagania co do wieku mogą wymagać zmodyfikowania. Na początku badania zmienność ciężaru zwierząt powinna być minimalna i nie przekraczać ± 20 % średniego ciężaru dla każdej płci. Kiedy przeprowadza się krótkotrwałe badanie z zastosowaniem dawki wielokrotnej jako wstępne badanie do badania długoterminowego, w obu badaniach należy użyć zwierząt tego samego szczepu pochodzących z tego samego źródła.
1.4.2. Warunki przebywania i warunki żywieniowe
Temperatura w badawczych pomieszczeniach hodowlanych powinna wynosić 22 oC (± 3 oC). Chociaż wilgotność względna powinna wynosić co najmniej 30 % i pożądane jest, żeby nie przekraczała 70 % poza czasem czyszczenia pomieszczenia, celem powinno być utrzymywanie jej na poziomie 50–60 %. Oświetlenie powinno być sztuczne, w cyklu 12 godzin jasno, 12 godzin ciemno. Należy zminimalizować sporadyczne głośne hałasy. Do żywienia można stosować konwencjonalne pasze laboratoryjne z nieograniczonym dostępem do wody pitnej. Na wybór diety może mieć wpływ potrzeba zapewnienia odpowiedniej domieszki substancji testowanej przy podawaniu metodą powyższą. Zwierzęta mogą być trzymane pojedynczo lub umieszczane w klatkach w małych grupach o tej samej płci.
1.4.3. Przygotowanie zwierząt
Zdrowe, młode zwierzęta są losowo przydzielane do grup badanych i kontrolnych. Klatki powinny być ustawione w taki sposób, żeby zminimalizować możliwy wpływ położenia klatki. Zwierzęta są identyfikowane jednoznacznie i przetrzymywane w klatkach przez co najmniej pięć dni przed rozpoczęciem badania, żeby umożliwić aklimatyzację do warunków laboratoryjnych.
1.4.4. Droga podawania i przygotowanie dawek
Ta metoda dotyczy konkretnie podawania doustnego substancji testowej. Podawanie może odbywać się przez rurkę, w pożywieniu, wodzie pitnej lub w kapsułkach. Można użyć innych dróg podawania (np. naskórnej lub wziewnej), ale może to wymagać modyfikacji zalecanych procedur. Wybór drogi podawania zależy od profilu narażenia ludzi oraz dostępnych informacji w zakresie toksykologii i kinetyki. Należy wskazać racjonalne uzasadnienie wyboru drogi podawania, jak również wynikające z niego modyfikacje procedury tej metody testowej.
W przypadku gdy to konieczne, substancja testowana jest rozpuszczona lub ma postać zawiesiny w odpowiednim nośniku. Zaleca się, aby – jeżeli to możliwe – najpierw było rozważone użycie wodnego roztworu/zawiesiny, następnie roztworu/emulsji w oleju (np. olej kukurydziany), a potem możliwy roztwór w innym nośniku. Musi być znana charakterystyka toksyczna nośnika. Ponadto należy uwzględnić następujące charakterystyki nośnika lub innych domieszek: skutek wywierany na absorpcję, dystrybucję, metabolizm, oraz na zatrzymywanie lub ekskrecję substancji testowej; skutki wywierane na chemiczne własności substancji testowej, które mogą zmienić jej charakterystykę toksyczną; należy również uwzględnić wpływ na pobieranie pokarmu lub wody, lub stan odżywienia zwierząt.
1.5. PROCEDURY
1.5.1. Liczba i płeć zwierząt
Kiedy badanie prowadzone jest jako odrębne, do oceny szczegółowych obserwacji klinicznych należy użyć co najmniej 20 zwierząt (10 samców i 10 samic) w każdej z grup dawkowania i kontrolnej. Co najmniej pięć samców i pięć samic należy poddać perfuzji in situ i wykorzystać do szczegółowych badań neurohistopatologicznych na zakończenie badania. W przypadkach kiedy obserwacje kliniczne pod kątem oznak skutków neurotoksycznych prowadzone są tylko w odniesieniu do ograniczonej liczby zwierząt, należy rozważyć włączenie tych zwierząt do grupy wybranych do perfuzji. Jeżeli badanie prowadzone jest w połączeniu z badaniem toksyczności dawki wielokrotnej, należy użyć odpowiedniej liczby zwierząt potrzebnej do osiągnięcia celów obu badań. Minimalne liczby zwierząt dla różnych kombinacji badań podano w tabeli 1. Jeżeli planowane jest uśmiercanie w czasie trwania badania lub rozważa się prowadzenie grup zwierząt zdrowiejących w celu obserwacji odwracalności, utrzymywania się lub opóźnionych skutków toksycznych, lub rozważa się obserwacje uzupełniające, wtedy należy zwiększyć liczbę zwierząt aby zapewnić, że wystarczy ich do celów obserwacji i badań histopatologicznych.
1.5.2. Grupy badane i grupa kontrolna
Zasadniczo, powinny być wykorzystane co najmniej trzy grupy badane i grupa kontrolna, jednak jeżeli z oceny innych danych należałoby oczekiwać braku objawów przy dawce 1 000 mg/kg masy ciała/dzień, może być przeprowadzony test graniczny. Jeżeli nie są dostępne odpowiednie dane, może być wykonane badanie wyników zakresowych, pomagające w określeniu dawek, które mają być użyte. Z wyjątkiem zastosowania substancji testowanej, ze zwierzętami grupy kontrolnej powinno się obchodzić w sposób identyczny, jak z podmiotami grupy badanej. Jeżeli przy podawaniu substancji testowanej jest stosowany nośnik, grupa kontrolna powinna otrzymać największą użytą objętość nośnika. Z wyjątkiem zastosowania substancji testowanej, ze zwierzętami grup kontrolnych powinno się obchodzić w sposób identyczny, jak ze zwierzętami grup badanych. Jeżeli przy podawaniu substancji testowanej jest stosowany nośnik, grupa kontrolna powinna otrzymać największą użytą objętość nośnika.
1.5.3. Sprawdzenie niezawodności
Laboratorium wykonujące badanie powinno przedstawić dane wykazujące jego zdolność do przeprowadzenia badania oraz czułość używanych metod. Dane te powinny udowadniać zdolność do wykrywania i ilościowego określania stosownych zmian w punktach końcowych zalecanych do obserwacji, takich jak objawy autonomiczne, reaktywność czuciowa, siła uchwytu i aktywność motoryczna. Informacje o związkach chemicznych, które powodują różne rodzaje reakcji neurotoksycznych i mogą zostać użyte jako substancje do kontroli pozytywnej można znaleźć w pozycjach bibliograficznych (2)–(9). Mogą zostać użyte wcześniej uzyskane dane jeżeli istotne aspekty procedury pozostają takie same. Zalecane jest okresowe uaktualnianie przeszłych danych. Należy uzyskać nowe dane wykazujące zachowaną czułość procedur w przypadku gdy jakiś istotny element w przebiegu badania lub procedurach został zmieniony przez laboratorium wykonujące.
1.5.4 Wybór dawki
Poziomy dawki powinny być wybierane poprzez wzięcie pod uwagę jakichkolwiek istniejących danych o toksyczności i danych kinetycznych osiągalnych dla substancji testowanej lub materiałów pokrewnych. Powinien zostać wybrany najwyższy poziom dawki, w celu wywołania skutków neurotoksycznych lub wyraźnych toksycznych skutków ogólnoustrojowych. Od tego czasu malejąca sekwencja poziomów dawki powinna być dobierana w taki sposób, aby wykazać wszelkie reakcje powiązane z dawkowaniem i brak obserwowanych objawów niekorzystnych przy najmniejszym poziomie dawki (NOAEL). W zasadzie poziomy dawek powinny być tak dobrane, aby pierwotne skutki działania toksycznego na układ nerwowy dały się odróżnić od skutków odnoszących się do toksyczności ogólnoustrojowej. Dwa do trzech interwałów są często optymalne dla ustalenia malejących poziomów dawki i dodanie czwartej grupy badanej jest często bardziej pożądane niż stosowanie bardzo dużych interwałów (np. współczynnik większy od 10) między dawkowaniem. Tam, gdzie dostępna jest racjonalna ocena ekspozycji ludzi, powinna być ona również wzięta pod uwagę.
1.5.5. Test graniczny
Jeżeli test przy jednym poziomie dawki równym co najmniej 1 000 mg/kg masy ciała/dzień, przy stosowaniu procedur opisanych dla tego badania, nie wywołuje widocznych objawów neurotoksycznych i jeżeli nie należałoby spodziewać się toksyczności na podstawie danych dotyczących substancji strukturalnie pokrewnych, wtedy pełne badanie przy użyciu trzech poziomów dawki może nie być konieczne. Ten test graniczny stosuje się z wyjątkiem przypadków, w których ekspozycja ludzi może wskazać na konieczność zastosowania w teście granicznym wyższego poziomu dawki doustnej. W odniesieniu do innych dróg podawania, na przykład wziewnej lub stosowania na skórę, własności fizyczne substancji testowanej mogą często wyznaczać najwyższy możliwy do osiągnięcia poziom ekspozycji. Dawka testu granicznego do przeprowadzenia badania doustnej toksyczności ostrej powinna wynosić co najmniej 2 000 mg/kg.
1.5.6. Podawanie dawek
Zwierzętom podaje się dawki substancji testowej raz dziennie, przez siedem dni w tygodniu, przez okres co najmniej 28 dni; stosowanie podawania przez pięć dni lub przez krótszy okres ekspozycji wymaga uzasadnienia. Kiedy substancja testowana podawana jest wprost do żołądka, powinna być podawana w jednorazowej dawce przy użyciu rurki lub odpowiedniej kaniuli dotchawicznej. Maksymalna objętość cieczy, która może być podana jednorazowo, zależy od wielkości badanych zwierząt. Objętość ta nie powinna przekraczać 1 ml/100 g masy ciała; jednakże w przypadku roztworów wodnych można rozważyć użycie do 2 ml/100 g masy ciała. Poza substancjami drażniącymi i korozyjnymi, które normalnie ujawniają zaostrzone objawy przy wyższych stężeniach, zmienność objętości w teście powinna być zminimalizowana przez dostosowanie stężenia, tak aby zapewnić stałą objętość dla wszystkich poziomów dawki.
W przypadku substancji podawanych w pożywieniu lub wodzie pitnej ważne jest zapewnienie, aby wymagane ilości substancji testowanej nie kolidowały z prawidłową równowagą odżywiania lub wody. W przypadku gdy substancja testowana jest podawana w pożywieniu, może być stosowane stałe stężenie żywieniowe (ppm) albo stały poziom dawki w odniesieniu do masy ciała zwierząt; użyta opcja musi być wyszczególniona. W przypadku substancji podawanej bezpośrednio do żołądka dawka powinna być podawana każdego dnia o podobnym czasie, i w razie konieczności modyfikowana, żeby utrzymać stały poziom dawki w odniesieniu do masy ciała zwierzęcia. W przypadku gdy badanie wielokrotnej dawki jest przeprowadzane jako badanie wstępne do badania długoterminowego, w obu badaniach powinna być stosowana podobna dieta. W badaniach toksyczności ostrej, jeżeli pojedyncza dawka nie jest możliwa, może ona być podawana w mniejszych częściach przez okres nieprzekraczający 24 godzin.
1.6. OBSERWACJE
1.6.1. Częstotliwość obserwacji i badań
W badaniach dawki wielokrotnej okres obserwacji obejmuje okres dawkowania, w badaniach toksyczności ostrej, należy przestrzegać 14-dniowego okresu poobserwacyjnego. U zwierząt w grupie satelitarnej, którą trzyma się bez ekspozycji w okresie po terapii, obserwacje powinny objąć również i ten okres.
Obserwacje należy prowadzić z wystarczającą częstotliwością by zmaksymalizować prawdopodobieństwo wykrycia nieprawidłowości w zachowaniu i/lub nieprawidłowości neurologicznych. Obserwacje powinny być przeprowadzane najlepiej każdego dnia o tej samej porze, biorąc pod uwagę szczytowy okres przewidywanych objawów po dawkowaniu. Częstotliwość obserwacji klinicznych i funkcjonalnych zestawiono w tabeli 2. Jeżeli dane kinetyczne, lub inne dane pochodzące z poprzednich badań wskazują na potrzebę wyznaczenia innych punktów czasowych, badań lub okresów poobserwacyjnych, w celu otrzymania jak największej ilości informacji, można przyjąć odmienny harmonogram. Należy przedstawić racjonalne powody zmian w harmonogramie.
1.6.1.1. Obserwacje ogólnego stanu zdrowia oraz zachorowalności/śmiertelności
Należy uważnie obserwować wszystkie zwierzęta, co najmniej raz dziennie, pod kątem ogólnego stanu zdrowia, jak również co najmniej dwa razy dziennie, pod kątem zachorowalności i śmiertelności.
1.6.1.2. Szczegółowe obserwacje kliniczne
Należy prowadzić szczegółowe obserwacje kliniczne wszystkich zwierząt wybranych do tego celu (zob. tabela 1), jeden raz przed pierwszą ekspozycją (aby uwzględnić porównanie w obrębie podmiotów), i od tego czasu w różnych odstępach czasu, w zależności od czasu trwania badania (zob. tabela 2). Szczegółowe obserwacje kliniczne w satelitarnych grupach zdrowiejących zwierząt powinny zostać przeprowadzone pod koniec okresu zdrowienia. Szczegółowe obserwacje kliniczne powinny być przeprowadzane na zewnątrz klatki macierzystej na znormalizowanej powierzchni. Powinny być one dokładnie odnotowywane przy użyciu systemów punktowania zawierających kryteria lub skale punktowe dla każdego pomiaru w obserwacjach. Kryteria zastosowanych skal powinny być wyraźnie zdefiniowane przez laboratorium testujące. Należy podjąć wysiłki, aby zagwarantować, że zmiany warunków badania są minimalne (niepowiązane systematycznie z terapią) oraz że obserwacje są przeprowadzane najlepiej przez obserwatorów nieświadomych rzeczywiście zastosowanej terapii.
Zaleca się prowadzenie obserwacji w sposób ustrukturalizowany, w którym dokładnie zdefiniowane kryteria (w tym definicja normalnego „zakresu”) stosowane są systematycznie do każdego zwierzęcia w każdym okresie obserwacji. Należy dostatecznie udokumentować „normalny zakres”. Wszelkie zaobserwowane objawy powinny być odnotowywane. Gdzie to możliwe, należy również odnotować wielkość obserwowanych objawów. Obserwacje kliniczne powinny obejmować, ale nie powinny być do nich ograniczone, zmiany w skórze, futrze, oczach, błonach śluzowych, wystąpienie wydzielin i wydalin oraz aktywność wegetatywną (np. łzawienie, podniesienie sierści, rozmiar źrenicy, niezwykła forma oddychania i/lub oddychanie przez jamę ustną, wszelkie niezwykłe oznaki oddawania moczu lub defekacji oraz odbarwiony mocz).
Należy również odnotowywać wszelkie niezwykłe reakcje w odniesieniu do postawy, poziomu aktywności (np. podwyższenie lub obniżenie poziomu eksploracji znormalizowanej powierzchni) i koordynacji ruchowej. Należy także odnotowywać zmiany w sposobie chodzenia (np. chód kaczkowaty, ataksja), postawie (np. wygięty grzbiet), i reakcji na dotyk, przenoszenie i inne bodźce ze środowiska, jak również obecność ruchów drgawkowych lub kurczowych, konwulsji lub drżenia, stereotypy (np. nadmierne czyszczenie się, niezwykłe ruchy głową, powtarzające się krążenie w kółko), lub dziwne zachowania (np. gryzienie lub nadmierne wylizywanie, samookaleczanie, chodzenie do tyłu, wydawanie dźwięków) lub agresję.
1.6.1.3. Obserwacje funkcjonalne
Podobnie jak szczegółowe obserwacje kliniczne, obserwacje funkcjonalne należy również przeprowadzić raz przed ekspozycją i później, często, u wszystkich zwierząt wybranych w tym celu (zob. tabela 1). Częstotliwość obserwacji funkcjonalnych zależy również od czasu trwania badania (zob. tabela 2). Dodatkowo, oprócz obserwacji podanych w tabeli 2, należy również przeprowadzić obserwacje funkcjonalne satelitarnej grupy zwierząt zdrowiejących, jak najbliżej terminu końcowego uśmiercenia. Badania funkcjonalne powinny obejmować reaktywność na bodźce różnego typu (np. słuchowe, wzrokowe i czucia głębokiego (5)(6)(7)), ocenę siły uchwytu (8) oraz ocenę aktywności motorycznej (9). Aktywność motoryczną należy mierzyć urządzeniem automatycznym umożliwiającym wykrywanie zarówno wzrostu jak i spadku aktywności. Jeżeli zastosowano inny zdefiniowany system, powinien on dokonywać pomiaru ilościowego, należy też wykazać jego niezawodność. Należy przetestować każde urządzenie, aby zapewnić niezawodność w czasie oraz spójność pomiędzy urządzeniami. Bliższe szczegóły procedur, które można przeprowadzać, podane są w odpowiednich pozycjach bibliografii. Jeżeli brak jest danych (np. struktura-działanie, dane epidemiologiczne, inne badania toksykologiczne) wskazujących potencjalne skutki neurotoksyczne, należy rozważyć włączenie bardziej specjalistycznych obserwacji funkcji sensorycznej, aktywności motorycznej i modelu zachowania lub uczenia się i pamięci, w celu bardziej szczegółowego zbadania tych potencjalnych skutków. Dalsze szczegóły bardziej wyspecjalizowanych obserwacji i ich zastosowania są podane w (1).
Wyjątkowo z obserwacji funkcjonalnych mogą być wyłączone zwierzęta, które ujawniają oznaki zatrucia w stopniu, który mógłby znacząco kolidować z wykonaniem obserwacji funkcjonalnej. Należy podać uzasadnienie wyłączenia zwierząt z obserwacji funkcjonalnej.
1.6.2. Masa ciała i spożycie pożywienia/wody
W badaniach trwających do 90 dni, wszystkie zwierzęta powinny być ważone co najmniej raz w tygodniu. Pomiary spożycia pożywienia (i wody, jeżeli substancja testowana podawana jest z wodą pitną) powinny być wykonywane co najmniej raz na tydzień. W badaniach długoterminowych należy ważyć zwierzęta raz na tydzień przez pierwsze 13 tygodni, później zaś co najmniej raz na 4 tygodnie. Należy dokonywać pomiarów spożycia pożywienia (i wody, jeżeli substancja testowana podawana jest w tym ośrodku) co najmniej raz na tydzień przez pierwsze 13 tygodni, a następnie w około trzymiesięcznych odstępach, jeżeli stan zdrowia lub zmiany wagi ciała wymuszą stosowanie innej procedury.
1.6.3. Oftalmologia
W przypadku badań trwających dłużej niż 28 dni badanie oftalmologiczne z wykorzystaniem oftalmoskopu lub podobnego sprzętu powinno zostać przeprowadzone przed podaniem substancji testowej oraz przed zakończeniem badań, najlepiej na wszystkich zwierzętach, ale co najmniej na zwierzętach grupy najwyższego dawkowania i grupy kontrolnej. W przypadku wykrycia zmian w oczach należy przebadać wszystkie zwierzęta. W badaniach długoterminowych należy przeprowadzić badanie oftalmologiczne również w 13 tygodniu. Nie ma potrzeby przeprowadzania badania oftalmologicznego, jeżeli takie dane są już dostępne z innych badań o podobnym okresie trwania i przy podobnych poziomach dawek.
1.6.4. Hematologia i biochemia kliniczna
Kiedy badanie neurotoksyczności prowadzone jest w połączeniu z badaniem ogólnoustrojowej toksyczności dawki wielokrotnej, należy przeprowadzić badania hematologiczne i oznaczenia w zakresie biochemii klinicznej według zasad podanych w odpowiedniej metodzie badania toksyczności ogólnoustrojowej. Próbki należy pobierać w taki sposób, żeby zminimalizować jakikolwiek potencjalny wpływ na zachowania o podłożu neurologicznym.
1.6.5. Histopatologia
Należy zaplanować badanie neuropatologiczne w celu uzupełnienia i poszerzenia obserwacji poczynionych podczas fazy in vivo badania. Tkanki pobrane od co najmniej 5 zwierząt/płeć/grupę (zob. tabela 1 i następny akapit) należy utrwalić in situ, stosując powszechnie przyjęte techniki perfuzji i utrwalania (zob. poz. bibliograficzna (3), rozdział 5, i poz. bibliograficzna (4), rozdział 50). Należy odnotować widoczne oglądowo zmiany. Jeżeli badanie jest przeprowadzane jako odrębne badanie sortujące pod kątem neurotoksyczności lub w celu scharakteryzowania skutków neurotoksycznych, można wykorzystać pozostałe zwierzęta do konkretnych procedur neurobehawioralnych (10)(11), neuropatologicznych (10)(11)(12)(13), neurochemicznych (10)(11)(14)(15) lub elektrofizjologcznych (10)(11)(16)(17), którymi można uzupełniać procedury i badania opisane powyżej; można też powiększyć o nie liczbę zwierząt badanych histopatologicznie. Tego rodzaju dodatkowe procedury są szczególnie użyteczne w przypadkach, kiedy obserwacje empiryczne lub przewidywane skutki wskazują na jakiś szczególny rodzaj lub docelowy organ działania neurotoksycznego (2)(3). Alternatywnie, można wykorzystać pozostałe zwierzęta również do rutynowych badań histopatologicznych, jakie opisano w metodzie badań dawki wielokrotnej.
Wszystkie preparaty tkanek zatopione w parafinie należy poddać ogólnej procedurze barwienia, jak np. hematoksyliną i eozyną (H&E), i zbadać pod mikroskopem. Jeżeli zaobserwuje się oznaki neuropatii obwodowej lub podejrzewa ich obecność, należy zbadać próbki tkanki nerwów obwodowych zatopione w tworzywie. Oznaki kliniczne mogą również sugerować dodatkowe lokalizacje do zbadania lub też zastosowanie specjalnych procedur barwienia. Wytyczne dotyczące dodatkowych miejsc do zbadania można znaleźć w (3)(4). Pomocne mogą być także właściwe specjalne barwniki ujawniające konkretne rodzaje zmian patologicznych (18).
Należy zbadać histologicznie reprezentatywne skrawki z centralnego i obwodowego układu nerwowego (zob. poz. bibliograficzna (3), rozdział 5, i poz. bibliograficzna (4), rozdział 50). Obszary badane powinny zwykle obejmować: przodomózgowie, centralną część móżdżku, w tym przekrój przez hipokamp, śródmózgowie, móżdżek, most, rdzeń przedłużony, oko z nerwem wzrokowym i siatkówką, rdzeń kręgowy na poziomie rozszerzenia szyjnego i lędźwiowego, zwoje korzeni grzbietowych, włókna korzeni brzusznych i grzbietowych, proksymalny nerw kulszowy, proksymalny nerw piszczelowy (w kolanie) oraz mięśniowe odgałęzienia nerwu piszczelowego. Skrawki z rdzenia kręgowego i nerwów obwodowych powinny obejmować zarówno przekroje poprzeczne jak i wzdłużne. Należy uwzględnić unaczynienie układu nerwowego. Należy również zbadać próbkę mięśnia szkieletowego, szczególnie mięśnia łydki. Szczególną uwagę należy poświęcić tym okolicom, których struktury komórkowe i włókna w centralnym i obwodowym układzie nerwowym znane są ze szczególnej podatności na działanie czynników neurotoksycznych.
W pozycjach bibliograficznych (3)(4) można znaleźć wskazówki dotyczące zmian neuropatologicznych typowych dla zmian wynikających z ekspozycji na czynnik toksyczny. Zaleca się dokonywanie badań w etapach, w których skrawki z grupy wysokiego dawkowania porównywane są najpierw ze skrawkami z grupy kontrolnej. Jeżeli nie zostaną zaobserwowane żadne zmiany neuropatologiczne w próbach z tych grup, nie jest wymagana dalsza analiza. Jeżeli zostaną zaobserwowane zmiany w grupie wysokiego dawkowania, próba każdej potencjalnie dotkniętej tkanki z grup pośredniego i niskiego dawkowania powinna zostać zakodowana i następnie zbadana sekwencyjnie.
Jeżeli w obserwacjach jakościowych zostaną zauważone dowody jakichkolwiek zmian neuropatologicznych, należy przeprowadzić badanie we wszystkich obszarach układu nerwowego wykazujących te zmiany. Skrawki z potencjalnie dotkniętych okolic, ze wszystkich grup dawkowania, należy zakodować i badać losowo bez znajomości kodu. Należy odnotować częstość i ciężkość każdej zmiany. Kiedy zostaną ocenione wszystkie grupy dawkowania, można rozkodować oznaczenia i przeprowadzić analizę statystyczną w celu oszacowania zależności reakcji od dawki. Należy opisać przykłady zmian o różnym stopniu ciężkości.
Zmiany neuropatologiczne powinny być oceniane w kontekście obserwacji i pomiarów, jak również innych danych z wcześniejszych lub jednocześnie prowadzonych badań toksyczności ogólnoustrojowej testowanej substancji.
2. DANE
2.1. OPRACOWANIE WYNIKÓW
Powinny zostać dostarczone dane indywidualne zwierząt. Dodatkowo wszystkie dane powinny być podsumowane w formie tabeli przedstawiającej dla każdej grupy badanej lub kontrolnej liczbę zwierząt na początku testu, liczbę zwierząt, które znaleziono martwe podczas testu lub uśmiercono z powodów humanitarnych i czas każdej śmierci lub humanitarnego uśmiercenia, liczbę zwierząt wykazujących oznaki toksyczności, opis obserwowanych oznak zatrucia, w tym czas rozpoczęcia, okres trwania, rodzaj i ciężkość wszelkich skutków zatrucia, liczbę zwierząt wykazujących zmiany patologiczne, typ zmian(-y).
2.2. OCENA I INTERPRETACJA WYNIKÓW
Objawy stwierdzone w badaniu należy ocenić w kategoriach częstości występowania, ciężkości i korelacji skutków neurobehawioralnych i neuropatologicznych (również neurochemicznych i elektrofizjologicznych, jeżeli włączono dodatkowe badania) oraz wszelkich innych zaobserwowanych skutków niekorzystnych. W przypadkach, gdy jest to możliwe, wyniki liczbowe powinny być ocenione za pomocą odpowiedniej i ogólnie akceptowanej metody statystycznej. Metody statystyczne powinny być wybrane podczas planowania badania.
3. SPRAWOZDAWCZOŚĆ
3.1. SPRAWOZDANIE Z BADAŃ
Sprawozdanie z badań musi zawierać następujące informacje:
|
|
Substancja testowa:
|
|
|
Nośnik (w stosownych przypadkach):
|
|
|
Zwierzęta badane:
|
|
|
Warunki testu:
|
|
|
Procedury obserwacji i testów:
|
|
|
Wyniki:
|
|
|
Dyskusja wyników:
|
|
|
Wnioski:
|
4. BIBLIOGRAFIA
|
(1) |
OECD Guidance Document on Neurotoxicity Testing Strategies and Test Methods. OECD, Paris, In Preparation. |
|
(2) |
Test Guideline for a Developmental Neurotoxicity Study, OECD Guidelines for the Testing of Chemicals. In preparation. |
|
(3) |
World Health Organization (WHO) (1986). Environmental Health Criteria document 60: Principles and Methods for the Assessment of Neurotoxicity associated with Exposure to Chemicals. |
|
(4) |
Spencer, P.S. and Schaumburg, H.H. (1980). Experimental and Clinical Neurotoxicology. Eds. Spencer, P.S. and Schaumburg, H.H. eds. Williams and Wilkins, Baltimore/London. |
|
(5) |
Tupper, D.E. and Wallace, R.B. (1980). Utility of the Neurological Examination in Rats. Acta Neurobiol. Exp., 40, 999–1003. |
|
(6) |
Gad, S.C. (1982). A Neuromuscular Screen for Use in Industrial Toxicology. J. Toxicol. Environ. Health, 9, 691–704. |
|
(7) |
Moser, V.C., McDaniel, K.M. and Phillips, P.M. (1991). Rat Strain and Stock Comparisons Using a Functional Observational Battery: Baseline Values and Effects of amitraz. Toxic. Appl. Pharmacol., 108, 267–283. |
|
(8) |
Meyer, O.A., Tilson, H.A., Byrd, W.C. and Riley, M.T. (1979). A Method for the Routine Assessment of Fore- and Hind- limb Grip Strength of Rats and Mice. Neurobehav. Toxicol., 1, 233–236. |
|
(9) |
Crofton, K.M., Haward, J.L., Moser, V.C., Gill, M.W., Reirer, L.W., Tilson, H.A. and MacPhail, R.C. (1991) Interlaboratory Comparison of Motor Activity Experiments: Implication for Neurotoxicological Assessments. Neurotoxicol. Teratol., 13, 599–609. |
|
(10) |
Tilson, H.A., and Mitchell, C.L. eds. (1992). Neurotoxicology Target Organ Toxicology Series. Raven Press, New York. |
|
(11) |
Chang, L.W., ed. (1995). Principles of Neurotoxicology. Marcel Dekker, New York. |
|
(12) |
Broxup, B. (1991). Neuopathology as a screen for Neurotoxicity Assessment. J. Amer. Coll. Toxicol., 10, 689–695. |
|
(13) |
Moser, V.C., Anthony, D.C., Sette, W.F. and MacPhail, R.C. (1992). Comparison of Subchronic Neurotoxicity of 2-Hydroxyethyl Acrylate and Acrylamide in Rats. Fund. Appl.Toxicol., 18, 343–352. |
|
(14) |
O'Callaghan, J.P. (1988). Neurotypic and Gliotypic Proteins as Biochemical Markers of Neurotoxicity. Eurotoxicol. Teratol., 10, 445–452. |
|
(15) |
O'Callaghan J.P. and Miller, D.B. (1988). Acute Exposure of the Neonatal Rat to Triethyltin Results in Persistent Changes in Neurotypic and Gliotypic Proteins. J. Pharmacol. Exp. Ther., 244, 368–378. |
|
(16) |
Fox. D.A., Lowndes, H.E. and Birkamper, G.G. (1982). Electrophysiological Techniques inNeurotoxicology. In: Nervous System Toxicology. Mitchell, CL. ed. Raven Press, New York, pp 299–335. |
|
(17) |
Johnson, B.L. (1980). Electrophysiological Methods in neurotoxicity Testing. In: Experimental and Clinical Neurotoxicology. Spencer, P.S. and Schaumburg, H.H. eds., Williams and Wilkins Co., Baltimore/London, pp. 726–742. |
|
(18) |
Bancroft, J.D. and Steven A. (1990). Theory and Pratice of Histological Techniques. Chapter 17, Neuropathological Techniques. Lowe, James and Cox, Gordon eds. Churchill Livingstone. |
Tabela 1
Minimalne liczby zwierząt w grupach potrzebne w sytuacji, kiedy badanie neurotoksyczności przeprowadzane jest odrębnie lub w połączeniu z innymi badaniami
|
|
BADANIE NEUROTOKSYCZNOŚCI PRZEPROWADZONE JAKO: |
|||
|
Odrębne badanie |
Połączone z badaniem 28-dniowym |
Połączone z badaniem 90-dniowym |
Połączone z badaniem toksyczności chronicznej |
|
|
Całkowita liczba zwierząt na grupę |
10 samców i 10 samic |
10 samców i 10 samic |
15 samców i 15 samic |
25 samców i 25 samic |
|
Liczba zwierząt wybrana do obserwacji funkcjonalnych, w tym szczegółowych obserwacji klinicznych |
10 samców i 10 samic |
10 samców i 10 samic |
10 samców i 10 samic |
10 samców i 10 samic |
|
Liczba zwierząt wybranych do perfuzji in situ i neurohistopatologii |
5 samców i 5 samic |
5 samców i 5 samic |
5 samców i 5 samic |
5 samców i 5 samic |
|
Liczba zwierząt wybranych do badań dawki wielokrotnej/obserwacji toksyczności podchronicznej/chronicznej, hematologii, biochemii klinicznej, histopatologii itp., jak wskazano to w odnośnych wytycznych |
|
5 samców i 5 samic |
||
|
Dodatkowe obserwacje w stosownych przypadkach |
5 samców i 5 samic |
|
|
|
Tabela 2
Częstotliwość obserwacji klinicznych i funkcjonalnych
|
Rodzaj obserwacji |
Czas trwania badania |
||||||||||||||||||||||||||
|
Ostra |
28-dniowe |
90-dniowe |
Chroniczna |
||||||||||||||||||||||||
|
U wszystkich zwierząt |
Ogólny stan zdrowotny |
codziennie |
codziennie |
codziennie |
codziennie |
||||||||||||||||||||||
|
Zachorowalność/śmiertelność |
dwa razy dziennie |
dwa razy dziennie |
dwa razy dziennie |
dwa razy dziennie |
|||||||||||||||||||||||
|
U zwierząt wybranych do obserwacji funkcjonalnych |
Szczegółowe obserwacje kliniczne |
|
|
|
|
||||||||||||||||||||||
|
Obserwacje funkcjonalne |
|
|
|
|
|||||||||||||||||||||||
B.44. ABSORPCJA PRZEZ SKÓRĘ: METODA IN VIVO
1. METODA
Opisywana metoda badania jest zgodna z wytyczną OECD nr 427 (2004).
1.1. WPROWADZENIE
Ekspozycja na wiele substancji chemicznych następuje głównie przez skórę, natomiast większość badań toksykologicznych na zwierzętach laboratoryjnych skupia się na doustnej drodze podawania. Badanie in vivo wchłaniania przez skórę opisane w tej instrukcji dostarcza informacji niezbędnych do ekstrapolowania wyników badań wchłaniania doustnego i wykorzystywania ich do sporządzania oceny bezpieczeństwa po ekspozycji przez skórę.
Zanim substancja dostanie się do układu krążenia, musi ona przejść przez wiele warstw komórek skóry. W przypadku większości substancji o szybkości wchłaniania decyduje warstwa rogowa, składająca się z martwych komórek. Przepuszczalność skóry zależy zarówno od lipofilności substancji chemicznej i grubości zewnętrznej warstwy naskórka, jak również czynników takich, jak masa cząsteczkowa i stężenie substancji. Ogólnie rzecz biorąc, skóra szczurów i królików jest bardziej przepuszczalna niż skóra ludzka, natomiast przepuszczalność skóry świnek morskich i małp bardziej przypomina przepuszczalność skóry ludzkiej.
Metody mierzenia wchłaniania przez skórę można podzielić na dwie kategorie: in vivo i in vitro. Metoda in vivo może dostarczyć istotnych informacji o wchłanianiu przez skórę u różnych gatunków laboratoryjnych. Metody in vitro zostały opracowane później. W metodach tych wykorzystuje się transport przez skórę zwierzęcą lub ludzką, pełnej lub niepełnej grubości, do zbiornika płynu. Metodę in vitro przedstawiono osobno w opisie innej metody badania (1). W celu doboru najwłaściwszej metody w danej sytuacji zaleca się skonsultować wytyczną OECD na temat przeprowadzania badań wchłaniania przez skórę (2), bowiem zawiera ona więcej szczegółowych informacji na temat stosowalności obu metod.
Opisywana poniżej metoda in vivo pozwala na określenie stopnia przenikania substancji testowej przez skórę do systemu. Technika ta jest powszechnie stosowana od wielu lat (3)(4)(5)(6)(7). Wprawdzie badania wchłaniania przez skórę in vitro mogą w wielu przypadkach być odpowiednie, w niektórych sytuacjach potrzebnych danych dostarczyć mogą jedynie badania in vivo.
Zalety metody in vivo polegają na tym, że stosując ją, wykorzystuje się nienaruszony fizjologicznie i metabolicznie system, używa się gatunków wspólnych dla wielu badań nad toksycznością, zaś metoda po modyfikacji może być stosowana do badań na innych gatunkach. Wadami są konieczność użycia żywych zwierząt, potrzeba stosowania materiałów znakowanych promieniotwórczo w celu uzyskania wiarygodnych wyników, trudności z określeniem wczesnej fazy wchłaniania oraz różnice w przepuszczalności skóry preferowanych gatunków (szczur) i człowieka. Skóra zwierzęca jest na ogół bardziej przepuszczalna i w związku z tym badania prowadzone z jej wykorzystaniem mogą prowadzić do przeszacowania wchłaniania przez skórę u ludzi (6)(8)(9). Na żywych zwierzętach nie należy testować substancji żrących lub korozyjnych.
1.2. DEFINICJE
Dawka niepochłonięta: jest to dawka spłukana z powierzchni skóry po ekspozycji i wszelkie substancje obecne w miejscach nieprzepuszczalnych, w tym wszelkie substancje, o których wiadomo, że wyparowały ze skóry w czasie ekspozycji.
Dawka pochłonięta (in vivo) : obejmuje substancję obecną w moczu, materiale wypłukanym z klatki, odchodach, wydychanym powietrzu (jeśli mierzono), krwi, tkankach (jeśli pobierano) oraz innych częściach ciała po usunięciu skóry w miejscu nałożenia substancji.
Dawka wchłanialna: jest masą substancji obecnej na lub w skórze po zmyciu.
1.3. ZASADA METODY BADANIA
Substancja testowa, najlepiej znakowana izotopem promieniotwórczym, nakładana jest na wystrzyżoną skórę zwierzęcia w odpowiedniej dawce lub dawkach, w takiej postaci, w jakiej jest normalnie używana. Aby zapobiec zjedzeniu przez zwierzę preparatu testowego pozostającego w kontakcie ze skórą przez określony czas, zabezpiecza się go odpowiednim opatrunkiem (nieokluzyjnym, semiokluzyjnym lub okluzyjnym). Pod koniec okresu ekspozycji opatrunek jest usuwany, a skóra czyszczona odpowiednim środkiem czyszczącym. Opatrunek i materiały użyte do czyszczenia są zachowane do analizy i zakładany jest świeży opatrunek. Przed, w trakcie i po okresie ekspozycji zwierzęta są umieszczane w oddzielnych klatkach metabolicznych. W tych okresach do celów analizy pobierane są wydaliny i wydychane powietrze. Pobieranie wydychanego powietrza nie jest konieczne, jeśli istnieje dostatecznie dużo informacji, że lotne metabolity promieniotwórcze powstają w niewielkich ilościach lub nie tworzą się wcale. Zwykle bada się kilka grup zwierząt, które poddaje się działaniu preparatu testowego. Jedna grupa jest zabijana po zakończeniu okresu ekspozycji. Inne grupy są zabijane później zgodnie z harmonogramem (2). Na koniec okresu pobierania próbek zabijane są pozostałe zwierzęta, pobierana jest krew do analizy, skóra w miejscu nałożenia substancji wycinana do analizy, a ciało poddawane badaniu w poszukiwaniu niewydalonego materiału. Próbki są badane w odpowiedni sposób i oznaczany jest poziom wchłaniania przez skórę (6)(8)(9).
1.4. OPIS METODY BADANIA
1.4.1. Dobór gatunków zwierząt
Najczęściej wykorzystuje się do tego celu szczury, ale można również użyć linii bezwłosych i gatunków, których intensywność wchłaniania przez skórę jest podobna do tej u człowieka (3)(6)(7)(8)(9). Należy użyć młodych, dorosłych zwierząt jednej płci (standardowo używa się samców) i linii powszechnie wykorzystywanych w laboratoriach. Na początku badań zróżnicowanie masy zwierząt nie powinno przekraczać ± 20 % średniej masy. Na przykład odpowiednie są samce szczurów o masie w zakresie 200–250 g, zwłaszcza o masie z górnej połowy tego zakresu.
1.4.2. Liczba oraz płeć zwierząt
Do każdego preparatu testowego i każdego zaplanowanego czasu zabijania należy użyć co najmniej czterech zwierząt jednej płci. Każda grupa zwierząt jest zabijana po różnym czasie, na przykład na koniec okresu ekspozycji (zwykle po 6 lub 24 godzinach) i następnie po kolejnych okresach (np. 48 i 72 godzinach). Jeśli są dostępne dane świadczące o istotnej różnicy między samcami i samicami w zakresie toksyczności skórnej, należy wybrać bardziej wrażliwą płeć. Jeśli brak takich danych, można użyć zwierząt dowolnej płci.
1.4.3. Warunki w pomieszczeniach dla zwierząt i pożywienie
Temperatura w pomieszczeniu ze zwierzętami doświadczalnymi powinna wynosić 22 oC (± 3 oC). Mimo że wilgotność względna powinna poza okresem sprzątania pomieszczenia wynosić co najmniej 30 % i raczej poniżej 70 %, należy starać się ją utrzymać na poziomie 50–60 %. Oświetlenie powinno być sztuczne, w cyklu 12 godzin światła, 12 godzin ciemności. Stosować można konwencjonalny pokarm laboratoryjny, który powinien być dostępny bez ograniczeń, podobnie jak woda. W czasie badań, a najlepiej także w okresie aklimatyzacji, zwierzęta są trzymane w osobnych klatkach metabolicznych. Należy zminimalizować możliwość rozsypywania pokarmu lub rozlewania wody, ponieważ może to mieć wpływ na wyniki.
1.4.4. Przygotowanie zwierząt
Zwierzęta są indywidualnie znakowane w celu umożliwienia identyfikacji osobników i trzymane w swoich klatkach przez co najmniej pięć dni przed rozpoczęciem badań w celu zapewnienia aklimatyzacji do warunków laboratoryjnych.
Po okresie aklimatyzacji i około 24 godziny przed zastosowaniem dawki na skórze każdego zwierzęcia w rejonie barków i grzbietu zostanie wystrzyżona skóra. Przepuszczalność skóry uszkodzonej jest inna niż skóry nieuszkodzonej, należy więc uważać, aby nie zranić zwierzęcia. Po wystrzyżeniu skóry i około 24 godziny przed nałożeniem substancji testowej na skórę (zob. część 1.4.7) powierzchnię skóry należy przetrzeć acetonem, aby usunąć łój. Dodatkowe przemywanie mydłem i wodą nie jest zalecane, ponieważ wszelkie resztki mydła mogą zwiększać wchłanianie substancji testowej. Aby umożliwić przeprowadzenie wiarygodnych obliczeń wchłaniania w przeliczeniu na 1 cm2 powierzchnia skóry, na którą nakładana jest substancja musi być wystarczająco duża, najlepiej co najmniej 10 cm2. Taką powierzchnię można uzyskać u szczurów o masie 200–250 g. Po przygotowaniu zwierzęta są wkładane z powrotem do klatek metabolicznych.
1.4.5. Substancja testowa
Substancja testowa jest substancją, której właściwości przenikania są poddawane badaniu. W idealnym przypadku substancja testowa powinna być znakowana izotopami promieniotwórczymi.
1.4.6. Przygotowanie do badania
Preparat substancji testowej (np. czysty, rozcieńczony lub materiał przygotowany według receptury, zawierający nakładaną na skórę substancję) powinien być taki sam (lub być realistycznym surogatem) jak substancja, na której działanie narażani są ludzie lub inne gatunki docelowe. Każda różnica między preparatem a wykorzystywaną substancją musi być uzasadniona. W razie potrzeby badaną substancję rozpuszcza się lub sporządza jej zawiesinę w odpowiednim podłożu. W przypadku podłoża innego niż woda powinna być znana charakterystyka absorpcji oraz potencjalne interakcje z substancją testową.
1.4.7. Nakładanie na skórę
Na powierzchni skóry wyznacza się miejsce nakładania o określonej powierzchni. Następnie równomiernie nakłada się na to miejsce preparat testowy. Jego ilość powinna zwykle imitować potencjalną ekspozycję u człowieka, zwykle 1–5 mg/cm2 w przypadku substancji stałej i do 10 μl/cm2 w przypadku płynu. Zastosowanie jakiejkolwiek innej ilości powinno być uzasadnione oczekiwanymi warunkami użycia, celami badania lub fizycznymi właściwościami preparatu testowego. Po nałożeniu preparatu miejsce nałożenia musi być chronione przed wylizywaniem przez zwierzę. Przykład typowego urządzenia pokazano na rysunku 1. Zazwyczaj miejsce nakładania powinno być chronione nieokluzyjnym opatrunkiem (np. przepuszczalną nylonową gazą). W przypadku dawek nieograniczonych miejsce nakładania powinno być przykryte opatrunkiem okluzyjnym. W przypadku gdy parowanie półlotnych substancji testowych zmniejsza wskaźnik odzysku substancji testowej do niedopuszczalnego poziomu (zob. także część 1.4.10 akapit pierwszy), konieczne jest zebranie wyparowanej substancji do filtra z węgla aktywowanego, pokrywającego urządzenie do nakładania (zob. rysunek 1). Ważne jest, żeby żadne urządzenie nie uszkadzało skóry, nie absorbowało substancji testowej, ani nie wchodziło z nią w reakcję. Zwierzęta wkłada się z powrotem do klatek metabolicznych, w celu zebrania wydalin.
1.4.8. Czas trwania ekspozycji i pobieranie próbek
Czas trwania ekspozycji jest czasem między nałożeniem a usunięciem preparatu przez zmycie go ze skóry. Należy zastosować odpowiedni czas ekspozycji (zwykle 6 lub 24 godziny), dostosowując go do czasu trwania ekspozycji u ludzi. Po okresie ekspozycji zwierzęta są trzymane w klatkach metabolicznych do czasu planowego uśmiercenia. Przez cały czas trwania badania zwierzęta powinny być regularnie obserwowane pod kątem oznak toksyczności lub anormalnych reakcji. Po zakończeniu okresu ekspozycji należy zaobserwować, czy na skórze narażonej na działanie substancji nie wystąpią objawy podrażnienia.
Klatki metaboliczne powinny pozwolić na oddzielne pobieranie moczu i kału przez cały czas badania. Powinny także umożliwiać pobieranie znakowanego węglem 14C dwutlenku węgla oraz lotnych związków węgla 14C, które należy poddać analizie, jeśli pojawiają się większe ilości (> 5 %). Dla każdej grupy i w trakcie każdego pobierania próbek należy osobno pobrać mocz, kał i płyny, w których znalazła się badana substancja (np. dwutlenek węgla 14C i lotne związki 14C). Jeśli jest wystarczająco dużo danych świadczących, że tworzy się niewiele lotnych promieniotwórczych metabolitów lub nie tworzą się one wcale, można stosować klatki otwarte.
Wydaliny są zbierane podczas ekspozycji, do 24 godzin po pierwszym kontakcie ze skórą, następnie codziennie do końca eksperymentu. Zazwyczaj wystarczają trzy okresy pobierania wydalin, jednak zakładane przeznaczenie preparatu testowego lub istniejące dane kinetyczne mogą wskazywać na potrzebę pobrania tego materiału w innych lub dodatkowych terminach.
Na zakończenie okresu ekspozycji urządzenie zabezpieczające jest zdejmowane z każdego zwierzęcia i zatrzymywane do osobnej analizy. Skóra w miejscu poddania działaniu substancji testowej powinna być u wszystkich zwierząt co najmniej trzykrotnie przemyta środkiem czyszczącym przy użyciu odpowiednich tamponów. Należy uważać, aby nie doprowadzić do skażenia innych części ciała. Środek czyszczący powinien być reprezentatywny dla środków stosowanych do zwykłych czynności higienicznych, np. wodny roztwór mydła. Na koniec należy wysuszyć skórę. Wszystkie tampony i płyny pozostałe po przemywaniu muszą być zachowane do analizy. Zwierzętom tworzącym grupy badane przez dłuższy okres należy przed włożeniem ich z powrotem do klatek nałożyć świeży opatrunek, chroniący miejsce poddane działaniu substancji testowej.
1.4.9. Procedury końcowe
W każdej grupie zwierzęta należy uśmiercić zgodnie z harmonogramem i pobrać krew do badań. Urządzenie ochronne lub opatrunek należy zdjąć w celu poddania analizie. Z każdego zwierzęcia powinien zostać pobrany i poddany analizie fragment skóry z miejsca nałożenia substancji oraz podobny, ostrzyżony fragment skóry, który nie był poddany działaniu substancji. Skóra z miejsca poddanego działaniu substancji testowej może zostać podzielona na frakcje, by oddzielić warstwę rogową od głębszej warstwy naskórka w celu uzyskania dokładniejszych informacji na temat rozmieszczenia substancji testowej. Określenie takiego odkładania się substancji w okresie po ekspozycji powinno dostarczyć pewnych danych na temat losu wszystkich substancji testowych w warstwie rogowej. Aby ułatwić dzielenie skóry (po ostatnim myciu skóry i uśmierceniu zwierzęcia), należy zdjąć opatrunek ochronny. Skóra z miejsca nałożenia substancji testowej, wraz z pierścieniem dookoła, jest wycinana i rozpinana na płytce. Przy zastosowaniu delikatnego nacisku na powierzchnię skóry przykleja się kawałek taśmy klejącej. Następnie odkleja się go wraz z częścią warstwy rogowej. Kolejne paski taśmy są naklejane, dopóki taśma przestanie przywierać do powierzchni skóry po usunięciu całej warstwy rogowej. Wszystkie paski taśmy pochodzące od jednego zwierzęcia mogą być umieszczone razem w jednym pojemniku, do którego dodaje się środek trawiący tkanki w celu rozpuszczenia fragmentów warstwy rogowej. Przed poddaniem reszty ciała analizie mającej określić dawkę wchłoniętą przez organizm, można pobrać wszelkie potencjalne tkanki docelowe. Ciała poszczególnych osobników należy zachować do analizy. Zwykle wystarcza analiza całkowitej zawartości. Do odrębnej analizy można pobrać określone organy (jeśli tak zalecono w innych badaniach). Do wcześniej zebranych próbek moczu należy dodać mocz obecny w pęcherzu w momencie uśmiercania zwierzęcia. Po zebraniu wydalin z klatek metabolicznych w zaplanowanym terminie uśmiercania zwierzęcia całe klatki powinny zostać umyte odpowiednim rozpuszczalnikiem. Podobnie należy poddać analizie inne urządzenia, które mogły ulec skażeniu.
1.4.10. Analiza
We wszystkich badaniach należy osiągnąć odpowiednią wartość odzysku (tj. średnio 100 ± 10 % radioaktywności). Poziom odzysku poza tym przedziałem powinien zostać uzasadniony. Wielkość zastosowanej dawki w każdej próbie powinna zostać przeanalizowana przy użyciu odpowiednio udokumentowanych procedur.
Dla każdej procedury nakładania dawki w opracowaniu statystycznym powinna się znaleźć miara wariancji dla powtórzeń.
2. DANE
W przypadku każdego zwierzęcia w każdym okresie pobierania próbek dla testowanej substancji i/lub jej metabolitów należy przeprowadzić następujące pomiary. Poza danymi indywidualnymi należy podać średnie dla danych pogrupowanych według czasów pobierania.
|
— |
ilość związana z urządzeniami ochronnymi, |
|
— |
ilość, która może zostać usunięta ze skóry, |
|
— |
ilość substancji w/na skórze, której nie można zmyć ze skóry, |
|
— |
ilość w próbce krwi, |
|
— |
ilość w wydalinach i wydychanym powietrzu (jeśli znajduje to zastosowanie), |
|
— |
ilość pozostająca w martwym ciele i wszelkich organach pobranych do oddzielnej analizy. |
Oznaczenie ilości substancji testowej i/lub metabolitów w wydalinach, wydychanym powietrzu, krwi i w martwym ciele umożliwi określenie całkowitej ilości zaabsorbowanej w każdym punkcie czasowym. Można również dokonać obliczenia ilości substancji chemicznej wchłanianej na 1 cm2 skóry poddanej działaniu substancji testowej.
3. SPORZĄDZANIE SPRAWOZDANIA
3.1. SPRAWOZDANIE Z BADANIA
Sprawozdanie z badania musi zawierać wymagania określone w protokole, w tym uzasadnienie użytego systemu testowego i powinien obejmować, co następuje:
|
|
Substancja testowa:
|
|
|
Preparat testowy:
|
|
|
Zwierzę testowe:
|
|
|
Warunki badania:
|
|
|
Wyniki:
|
|
|
Omówienie wyników. |
|
|
Wnioski. |
4. LITERATURA
|
(1) |
Testing Method B.45. Skin Absorption: In vitro Method. |
|
(2) |
OECD (2002). Guidance Document for the Conduct of Skin Absorption Studies. OECD, Paris. |
|
(3) |
ECETOC (1993) Percutaneous Absorption. European Centre for Ecotoxicology and Toxicology of Chemicals, Monograph No. 20. |
|
(4) |
Zendzian RP (1989) Skin Penetration Method suggested for Environmental Protection Agency Requirements. J. Am. Coll. Toxicol. 8(5), 829–835. |
|
(5) |
Kemppainen BW, Reifenrath WG (1990) Methods for skin absorption. CRC Press Boca Raton, FL, USA. |
|
(6) |
EPA (1992) Dermal Exposure Assessment: Principles and Applications. Exposure Assessment Group, Office of Health and Environmental Assessment. |
|
(7) |
EPA (1998) Health Effects Test Guidelines, OPPTS 870-7600, Dermal Penetration. Office of Prevention, Pedsticides and Toxic Substances. |
|
(8) |
Bronaugh R.L., Wester RC, Bucks D., Maibach H.I. and Sarason R (1990) In vivo percutaneous absorption of fragrance ingredients in reshus monkeys and humans. Fd. Chem. Toxic. 28, 369–373. |
|
(9) |
Feldman R.J. and Maibach H.I. (1970) Absorption of some organic compounds through the skin in man. J. Invest Dermatol. 54, 399–404. |
Rysunek 1
Przykład konstrukcji typowego urządzenia stosowanego do wyznaczania i zabezpieczenia miejsca stosowania substancji na skórze w trakcie badań in vivo wchłaniania przez skórę

B.45. ABSORPCJA PRZEZ SKÓRĘ: METODA IN VITRO
1. METODA
Opisywana metoda badania jest zgodna z wytyczną OECD nr 428 (2004).
1.1. WPROWADZENIE
Metoda ta została opracowana, aby dostarczyć informacji na temat wchłaniania substancji testowej nakładanej na wycinek skóry. Może być stosowana w połączeniu z metodą badania wchłaniania przez skórę in vivo (1) lub oddzielnie. Zaleca się, aby przy opracowywaniu badań opartych na tej metodzie korzystać z wytycznej OECD dotyczącej badań wchłaniania przez skórę (2). Wytyczna została opracowana, aby ułatwić wybór odpowiednich procedur in vitro, nadających się do stosowania w określonych okolicznościach, w celu zapewnienia wiarygodności wyników uzyskanych tą metodą.
Metody pomiaru wchłaniania przez skórę oraz ilości substancji dostającej się do organizmu przez skórę można podzielić na dwie kategorie: in vivo i in vitro. Metody in vivo badania wchłaniania przez skórę są dobrze opracowane i dostarczają informacji o farmakokinetyce u wielu różnych gatunków. Informacje o metodzie in vivo przedstawiono osobno w opisie innej metody badania (1). Także metody in vitro są stosowane do pomiaru wchłaniania przez skórę od wielu lat. Wprawdzie nie przeprowadzono formalnych badań walidacyjnych metod in vitro składających się na niniejszą metodę badania, jednakże specjaliści OECD zgodzili się w 1999 r., że istnieje wystarczająco dużo udokumentowanych danych na poparcie metody in vitro (3). Więcej szczegółowych informacji uzasadniających to poparcie, w tym znaczną liczbę bezpośrednich porównań metod in vitro i in vivo, zamieszczono w wytycznej (2). Istnieją monografie, w których omówiono ten temat i przedstawiono szczegółowy zakres stosowania metody in vitro (4)(5)(6)(7)(8)(9)(10)(11)(12). Metody in vitro umożliwiają pomiar rozprzestrzeniania się substancji chemicznych w głąb skóry i na jej powierzchni, a następnie do zbiornika płynu. W badaniu używa się zarówno martwej skóry, służącej wyłącznie do pomiaru dyfuzji, jak i skóry świeżej, aktywnej metabolicznie, pozwalającej na równoczesne badanie dyfuzji i metabolizmu skóry. Takie metody znalazły zastosowanie przy porównywaniu przenikania związków chemicznych zawartych w różnych substancjach chemicznych w głąb skóry i przez skórę, mogą też umożliwić uzyskanie użytecznych modeli oceny wchłaniania przezskórnego u ludzi.
Stosowanie metody in vitro może być niemożliwe we wszystkich warunkach i w przypadku wszystkich kategorii związków chemicznych. Można korzystać z metody badania in vitro do wstępnej oceny jakościowej przenikania przez skórę. W niektórych przypadkach konieczne może być na dalszym etapie przeprowadzenie badań in vivo. W celu dokładniejszego poznania sytuacji, w których metoda in vitro byłaby odpowiednia, należy sięgnąć do wytycznej (2). Dodatkowe szczegółowe informacje przydatne przy podejmowaniu decyzji zamieszczono w (3).
W opisie metody zawarto ogólne zasady pomiaru wchłaniania przez skórę i dopływu substancji testowej przy użyciu wycinka skóry. Możliwe jest użycie skóry wielu gatunków ssaków, w tym człowieka. Właściwości skóry związane z przepuszczalnością są zachowywane po wycięciu jej fragmentu z organizmu, ponieważ główną barierę przenikania stanowi martwa warstwa rogowa. Aktywny transport substancji chemicznych przez skórę nie został stwierdzony. Wykazano, że skóra ma zdolność metabolizowania niektórych związków chemicznych podczas wchłaniania przezskórnego (6), jednak proces ten nie zmniejsza ilości rzeczywiście wchłanianej dawki, choć może mieć wpływ na naturę substancji dostającej się do krwi.
1.2. DEFINICJE
Dawka niepochłonięta: jest to dawka spłukana z powierzchni skóry po ekspozycji i wszelkie substancje obecne w miejscach nieprzykrytych, w tym wszelkie substancje, o których wiadomo, że wyparowały ze skóry w czasie ekspozycji.
Dawka pochłonięta (in vitro) : masa substancji testowej docierającej do płynu receptorowego lub układu krążenia w określonym czasie.
Dawka wchłanialna (in vitro) : jest masą substancji obecnej na lub w skórze po zmyciu.
1.3. ZASADA METODY BADANIA
Substancja testowa, która może być znakowana izotopem promieniotwórczym jest nakładana na powierzchnię próbki skóry oddzielającej dwie części komory dyfuzyjnej. Substancja pozostaje na skórze w określonych warunkach, przez określony czas, zanim zostanie usunięta w trakcie odpowiedniej procedury mycia. Próbki płynu receptorowego są pobierane w odstępach czasowych w trakcie całego badania i analizowane pod kątem obecności substancji testowej i/lub metabolitów.
W przypadku stosowania systemów aktywnych metabolicznie można odpowiednimi metodami analizować metabolity substancji testowej. Na koniec eksperymentu, w razie potrzeby, rozmieszczenie substancji testowej i jej metabolitów jest wyrażane ilościowo.
W odpowiednich warunkach przedstawionych w opisie tej metody oraz w wytycznej (2), absorpcja substancji testowej w określonym czasie jest mierzona za pomocą analizy płynu receptorowego i badanej skóry. Substancja testowa pozostająca w skórze jest uznawana za zaabsorbowaną, chyba że uda się wykazać, iż poziom absorpcji można ustalić wyłącznie na podstawie wartości płynu receptorowego. Analiza innych komponentów (materiał spłukany ze skóry i pozostający w obrębie warstw skóry) umożliwia dalszą ocenę danych, w tym całkowitego rozmieszczenia substancji testowej i odzysku wyrażonego w procentach.
Aby wykazać sprawność i wiarygodność systemu testów w przeprowadzającym je laboratorium, należy dysponować wynikami odnoszącymi się do odpowiednich substancji wzorcowych, a wyniki te powinny być zgodne z publikacjami na temat wykorzystywanej metody. Wymóg ten można spełnić przeprowadzając test przy użyciu odpowiedniej substancji wzorcowej (najlepiej o lipofilności zbliżonej do lipofilności substancji testowej) równocześnie przy użyciu substancji testowej albo dostarczając odpowiednich danych historycznych dla różnych substancji wzorcowych o zróżnicowanej lipofilności (np. kofeina, kwas benzoesowy i testosteron).
1.4. OPIS METODY BADANIA
1.4.1. Komora dyfuzyjna
Komora dyfuzyjna składa się z dwóch części, donorowej i receptorowej, pomiędzy którymi umieszczona jest skóra (przykład typowego układu przedstawiono na rysunku 1). Komora powinna zapewnić szczelność wokół skóry, pozwalać na łatwe pobieranie próbek i dobre mieszanie roztworu receptorowego pozostającego w kontakcie ze spodnią częścią skóry oraz sprawną regulację temperatury komory i jej zawartości. Dopuszczalny jest zarówno układ statyczny, jak i przepływ przez komory dyfuzyjne. W czasie ekspozycji ograniczonej dawki substancji testowej części donorowe są zwykle otwarte. Jednak w przypadku stosowania nieograniczonych dawek lub przy pewnych scenariuszach stosowania dawek ograniczonych, części donorowe mogą pozostawać zamknięte.
1.4.2. Płyn receptorowy
Preferowane jest stosowanie płynu receptorowego o fizjologicznej przewodności, można jednak stosować również inne płyny, jeśli ich użycie jest uzasadnione. Należy dostarczyć precyzyjnych danych o składzie płynu receptorowego. Należy zapewnić odpowiednią rozpuszczalność substancji testowej w płynie receptorowym, tak aby nie stanowił bariery absorpcji. Ponadto płyn receptorowy nie powinien wpływać na kompletność preparatu skóry. W systemach przepływowych szybkość przepływu nie powinna hamować dyfuzji substancji testowej do płynu receptorowego. W systemie komory statycznej płyn powinien być nieustannie mieszany, a próbki powinny być pobierane regularnie. Jeśli przedmiotem badań jest metabolizm, płyn receptorowy musi zapewnić żywotność skóry przez cały czas trwania eksperymentu.
1.4.3. Preparaty skóry
Można użyć skóry ludzkiej lub zwierzęcej. Uznaje się, że zastosowanie ludzkiej skóry podlega międzynarodowym zastrzeżeniom i warunkom etycznym. Wprawdzie preferowana jest żywa skóra, można jednak użyć również skóry martwej, pod warunkiem że można wykazać kompletność wycinka. Jako błony do dyfuzji można użyć preparatu naskórka (wyizolowanego metodami enzymatycznymi, cieplnymi lub chemicznymi) lub płata skóry niepełnej grubości (zwykle o grubości 200–400 μm) przygotowanego za pomocą dermatomu. Płat skóry pełnej grubości może być użyty, jednakże należy unikać zbyt dużej grubości (ca > 1 mm), chyba że ma on służyć do oznaczenia ilości substancji chemicznej w warstwach skóry. Wybór gatunku, miejsca anatomicznego i techniki preparacji musi być uzasadniony. Wymagane są odpowiednie dane z co najmniej czterech powtórzeń na każdy preparat testowy.
1.4.4. Kompletność preparatu skóry
Właściwe przygotowanie preparatu skóry ma zasadnicze znaczenie. Nieodpowiednie postępowanie może spowodować uszkodzenie warstwy rogowej, dlatego należy sprawdzać kompletność przygotowanej próbki skóry. W przypadku badania metabolizmu skóry należy używać świeżo pobranych wycinków skóry przy zachowaniu warunków, o których wiadomo, że sprzyjają zachowaniu aktywności metabolicznej. Generalnie zaleca się użyć wycinka skóry w ciągu 24 godzin od jego pobrania, jednak dopuszczalny okres przechowywania może być różny, w zależności od systemu enzymatycznego uczestniczącego w metabolizmie oraz od temperatury przechowywania (13). Jeśli preparaty skórne były przed użyciem przechowywane, należy przedstawić dane dowodzące, że funkcja bariery została zachowana.
1.4.5. Substancja testowa
Substancja testowa jest substancją, której właściwości przenikania są poddawane badaniu. W idealnym przypadku substancja testowa powinna być znakowana izotopami promieniotwórczymi.
1.4.6. Preparat testowy
Preparat substancji testowej (np. czysty, rozcieńczony lub materiał przygotowany według receptury, zawierający substancję nakładaną na skórę) powinien być taki sam (lub być realistycznym surogatem) jak substancja, na działanie której mogą być narażeni ludzie lub inne gatunki docelowe. Każda różnica między preparatem a wykorzystywaną substancją musi być uzasadniona.
1.4.7. Stężenia i formy substancji testowych
Zwykle stosuje się więcej niż jedno stężenie substancji testowej, sięgające górnej granicy potencjalnych dawek, na które narażany jest człowiek. Należy także rozważyć poddanie testom różnych typowych form substancji.
1.4.8 Nakładanie na skórę
W normalnych warunkach człowiek jest narażany na działanie ograniczonych dawek substancji chemicznej. Z tego powodu należy nakładać testowaną substancję w sposób imitujący ekspozycję, na jaką narażany jest człowiek. Zwykle należy stosować 1–5 mg/cm2 w przypadku substancji stałej i do 10 μl/cm2 w przypadku płynów. Zastosowanie określonej ilości powinno być uzasadnione oczekiwanymi warunkami użycia, celami badania lub fizycznymi właściwościami preparatu testowego. Na przykład dawki nakładane na skórę mogą być nieograniczone tam gdzie nakładane są duże objętości na jednostkę powierzchni skóry.
1.4.9. Temperatura
Temperatura wpływa na dyfuzję bierną substancji chemicznych (i wobec tego na wchłanianie przez skórę). Komora dyfuzyjna i skóra powinny mieć stałą temperaturę, zbliżoną do normalnej temperatury skóry: 32 ± 1 oC. Różne konstrukcje komory dyfuzyjnej wymagają zastosowania różnej temperatury łaźni wodnych lub bloków grzewczych, aby mieć pewność, że receptor/skóra odpowiadają normie fizjologicznej. Wilgotność powinna mieścić się w przedziale 30–70 %.
1.4.10. Czas trwania ekspozycji i pobieranie próbek
Ekspozycja skóry na preparat testowy może trwać przez cały czas trwania eksperymentu lub przez krótszy czas (np. w celu imitacji określonego rodzaju ekspozycji, na którą narażany jest człowiek). Nadmiar preparatu testowego należy zmyć ze skóry za pomocą odpowiedniego środka czyszczącego, a spłukany materiał zebrać w celu poddania go analizie. Procedura usuwania preparatu testowego zależy od przewidywanych warunków stosowania i należy ją uzasadnić. W celu uzyskania odpowiedniej charakterystyki profilu absorpcji zwykle wymagany jest 24-godzinny okres pobierania próbek. Ponieważ po okresie dłuższym niż 24 godziny stan skóry może zacząć się pogarszać, czas pobierania próbek nie powinien zazwyczaj przekraczać 24 godzin. W przypadku substancji testowych szybko wnikających w skórę może to nie być konieczne, jednak w przypadku substancji testowych, które wnikają powoli, mogą być konieczne dłuższe okresy pobierania próbek. Częstotliwość pobierania próbek płynu receptorowego powinna umożliwić graficzne przedstawienie profilu absorpcyjnego substancji testowej.
1.4.11. Procedury końcowe
Wszystkie elementy systemu testowego powinny być poddane analizie. Należy również oznaczyć wielkość odzysku. Obejmuje to część donorową komory dyfuzyjnej, spłukiwanie powierzchni skóry, preparat skórny oraz płyn receptorowy/komorę receptorową. W niektórych przypadkach skóra może być frakcjonowana na powierzchnię eksponowaną i powierzchnię pod kryzą komory oraz na frakcje warstwy rogowej, naskórka i skóry właściwej, poddawane odrębnym analizom.
1.4.12. Analiza
We wszystkich badaniach należy osiągnąć odpowiedni poziom odzysku (celem powinna być średnia 100 ± 10 % promieniotwórczości, wszelkie odchylenia należy uzasadnić). Analizie przy użyciu odpowiedniej techniki powinna zostać poddana substancja testowa obecna w płynie receptorowym, materiale zmytym z powierzchni skóry i wypłukanym z aparatury.
2. DANE
Należy opisać wyniki analizy płynu receptorowego, rozmieszczenie testowanej substancji chemicznej w systemie testowym oraz zmiany profilu absorpcji w czasie. Jeśli ekspozycja polega na stosowaniu dawki ograniczonej, należy obliczyć ilość substancji spłukiwanej ze skóry, ilość związaną ze skórą (i w różnych warstwach skóry, jeśli jest to przedmiotem analizy) oraz ilość substancji obecnej w płynie receptorowym (współczynnik oraz ilość lub procent stosowanej dawki). Czasami możliwe jest określenie absorpcji przez skórę przy wykorzystaniu wyłącznie danych o płynie receptorowym. Jeśli jednak w momencie zakończenia badań w skórze pozostaje substancja testowa, może być konieczne uwzględnienie jej przy obliczaniu całkowitej zaabsorbowanej ilości (zob. poz. bibliograficzna (3) pkt 66). W przypadku zastosowania do ekspozycji dawki nieograniczonej, dane mogą pozwolić na obliczenie stałej przenikalności (Kp). W ostatnim przypadku procent zaabsorbowanej substancji nie ma znaczenia.
3. SPORZĄDZANIE SPRAWOZDANIA
3.1. SPRAWOZDANIE Z BADANIA
Sprawozdanie z badania musi zawierać wymagania określone w protokole, w tym uzasadnienie użycia systemu testowego, i powinno obejmować, co następuje:
|
|
Substancja testowa:
|
|
|
Preparat testowy:
|
|
|
Warunki badania:
|
|
|
Wyniki:
|
|
|
Omówienie wyników. |
|
|
Wnioski. |
4. LITERATURA
|
(1) |
Testing Method B.44. Skin Absorption: In vivo Method. |
|
(2) |
OECD (2002). Guidance Document for the Conduct of Skin Absorption Studies. OECD, Paris. |
|
(3) |
OECD (2000). Report of the Meeting of the OECD Extended Steering Committee for Percutaneous Absorption Testing, Annex 1 to ENV/JM/TG(2000)5. OECD, Paris. |
|
(4) |
Kemppainen B.W. and Reifenrath W.G. (1990). Methods for skin absorption. CRC Press, Boca Raton. |
|
(5) |
Bronaugh R.L. and Collier, S.W. (1991). Protocol for In vitro Percutaneous Absorption Studies, in In vitro Percutaneous Absorption: Principles, Fundamentals and Applications, R.L. Bronaugh and H.I. Maibach, Eds., CRC Press, Boca Raton, pp. 237–241. |
|
(6) |
Bronaugh R.L. and Maibach H.I. (1991). In vitro Percutaneous Absorption: Principles, Fundamentals and Applications. CRC Press, Boca Raton. |
|
(7) |
European Centre for Ecotoxicology and Toxicology of Chemicals (1993). Monograph No. 20, Percutaneous Absorption, ECETOC, Brussels. |
|
(8) |
Diembeck W., Beck H., Benech-Kieffer F., Courtellemont P., Dupuis J., Lovell W., Paye M., Spengler J., Steiling W (1999). Test Guidelines for In Vitro Assessment of Dermal Absorption and Percutaneous Penetration of Cosmetic Ingredients, Fd Chem Tox, 37, 191–205. |
|
(9) |
Recommended Protocol for In vitro Percutaneous Absorption Rate Studies (1996). US Federal Register, Vol. 61, No. 65. |
|
(10) |
Howes D., Guy R., Hadgraft J., Heylings J.R. et al. (1996). Methods for assessing Percutaneous absorption. ECVAM Workshop Report ATLA 24, 81 R10. |
|
(11) |
Schaefer H. and Redelmeier T.E. (1996). Skin barrier: principles of percutaneous absorption. Karger, Basel. |
|
(12) |
Roberts M.S. and Walters K.A. (1998). Dermal absorption and toxicity assessment. Marcel Dekker, New York. |
|
(13) |
Jewell, C., Heylings, J.R., Clowes, H.M. And Williams, F.M. (2000). Percutaneous absorption and metabolism of dinitrochlorobenzene in vitro. Arch Toxicol 74: 356–365. |
Rysunek 1
Przykład typowej konstrukcji statycznej komory dyfuzyjnej do badań in vitro wchłaniania przezskórnego

(1) Należy zapisać obszar zmętnienia rogówki.
(2) W przypadku kilku pomiarów w osoczu i surowicy, zwłaszcza glukozy, pożądane byłoby wstrzymanie podawania pożywienia przez całą noc. Głównym powodem takich preferencji jest fakt, że zwiększona zmienność, która nieuchronnie wynikałaby z podawania pożywienia, przyczyniałaby się do maskowania bardziej subtelnych skutków i utrudniałaby interpretację. Z drugiej strony, jednakże wstrzymanie podawania pożywienia przez całą noc może szkodzić ogólnemu metabolizmowi zwierząt i, szczególnie w badaniach żywieniowych, może naruszać dzienną ekspozycję na substancję testowaną. Jeżeli przyjęte jest wstrzymanie podawania pożywienia przez całą noc, biochemiczne oznaczenia kliniczne powinny być wykonane po przeprowadzeniu obserwacji funkcjonalnych w czwartym tygodniu badania.
(3) W odniesieniu do niektórych oznaczeń w surowicy i plazmie, najbardziej znacząco dla glukozy, zaleca się niekarmienie przez noc. Główną przyczyną tego zalecenia jest wzrost zmienności, który nieuchronnie powstaje przy nieprzestrzeganiu tego zalecenia i powoduje tendencję maskowania bardziej subtelnych efektów, co czyni interpretacje wyników trudną. Z drugiej jednak strony, niekarmienie przez noc może zakłócać ogólny metabolizm zwierząt, w szczególności w badaniach żywieniowych, może przeszkadzać w dziennej ekspozycji na działanie substancji badanej. Jeśli przyjęto metodę niekarmienia przez noc, kliniczne oznaczenia biochemiczne muszą być wykonane po wprowadzeniu do badań uwag funkcjonalnych.
(4) Obecnie znane pod nazwą aminontransferaza alaninowa surowicy.
(5) Obecnie znane pod nazwą aminotransferaza asparaginianowa surowicy.
(6) Organy te, pochodzące od dziesięciu zwierząt danej płci i grupy odnośnie gryzoni i innych gatunków, plus tarczyca (z przytarczycami), odnośnie do gatunków innych niż gryzonie, powinny zostać zważone.
(7) Organy te, pochodzące od dziesięciu zwierząt danej płci i grupy odnośnie gryzoni, powinny zostać zważone.
(8) W tej metodzie grupa odniesienia jest jedyną grupą, której substancję badaną podaje się inną drogą zapewniającą całkowitą biodostępność dawki.
(9) Rozpuszczalnik używany do mierzenia absorpcji.
|
† |
Wliczając w to pięć zwierząt wybranych do obserwacji funkcjonalnych i szczegółowych obserwacji klinicznych, stanowiących część badania neurotoksyczności |
CZĘŚĆ C: METODY USTALANIA EKOTOKSYCZNOŚCI
SPIS TREŚCI
|
C.1. |
TOKSYCZNOŚĆ OSTRA DLA RYB |
|
C.2. |
BADANIE NAGŁEGO UNIERUCHOMIENIA DAPHNIA SP. |
|
C.3. |
BADANIE INHIBICJI GLONÓW |
|
C.4. |
OZNACZENIE BIODEGRADOWALNOŚCI |
|
CZĘŚĆ I. |
ROZWAŻANIA OGÓLNE |
|
CZĘŚĆ II. |
BADANIE DOC METODĄ „DIE-AWAY” (Metoda C.4-A) |
|
CZĘŚĆ III. |
ZMODYFIKOWANE BADANIE PRZESIEWOWE OECD (Metoda C.4-B) |
|
CZĘŚĆ IV. |
BADANIE WYDZIELANIA CO2 (Metoda C.4-C) |
|
CZĘŚĆ V. |
BADANIE RESPIROMETRII MANOMETRYCZNEJ (Metoda C.4.D) |
|
CZĘŚĆ VI. |
BADANIE „ZAMKNIĘTEJ BUTLI” (Metoda C.4-E) |
|
CZĘŚĆ VII. |
BADANIE M.I.T.I. (Metoda C.4-F) |
|
C.5. |
DEGRADACJA – ZAPOTRZEBOWANIE BIOCHEMICZNE NA TLEN |
|
C.6. |
DEGRADACJA – ZAPOTRZEBOWANIE CHEMICZNE NA TLEN |
|
C.7. |
ROZKŁAD – ROZKŁAD ABIOTYCZNY: HYDROLIZA JAKO FUNKCJA PH |
|
C.8. |
TOKSYCZNOŚĆ DŻDŻOWNIC |
|
C.9. |
BIODEGRADACJA – BADANIE ZAHN-WELLENS |
|
C.10. |
BIODEGRADACJA – BADANIA SYMULACJI AKTYWOWANYCH SZLAMÓW |
|
C.11. |
BIODEGRADACJA – BADANIE HAMOWANIA ODDYCHANIA AKTYWOWANYCH SZLAMÓW |
|
C12. |
BIODEGRADACJA – ZMODYFIKOWANE BADANIE SCAS |
|
C.13. |
BIOKONCENTRACJA: BADANIE RYB W WARUNKACH PRZEPŁYWU |
|
C14. |
BADANIE ROZWOJU MŁODYCH RYB |
|
C.15. |
RYBY, KRÓTKOTERMINOWE BADANIE TOKSYCZNOŚCI NA EMBRIONIE I NARYBKU |
|
C.16. |
PSZCZOŁY MIODNE – BADANIE TOKSYCZNOŚCI OSTREJ DROGĄ POKARMOWĄ |
|
C.17. |
PSZCZOŁY MIODNE – BADANIE KONTAKTOWEJ TOKSYCZNOŚCI OSTREJ |
|
C.18. |
ADSORPCJA/DESORPCJA PRZY UŻYCIU METODY RÓWNOWAGI OKRESOWEJ |
|
C.19. |
OSZACOWANIE WSPÓŁCZYNNIKA ADSORPCJI (Koc) NA GLEBIE IW OSADZIE ŚCIEKOWYM PRZY ZASTOSOWANIU WYSOKOSPRAWNEJ CHROMATOGRAFII CIECZOWEJ (HPLC) |
|
C.20. |
BADANIE ROZRODCZOŚCI DAPHNIA MAGNA |
|
C.21. |
MIKROORGANIZMY GLEBOWE: BADANIA PRZEMIAN AZOTU |
|
C.22. |
MIKROORGANIZMY GLEBOWE: BADANIE PRZEMIAN WĘGLA |
|
C.23. |
PRZEMIANY W GLEBIE W WARUNKACH NATLENIENIA I BEZTLENOWYCH |
|
C.24. |
PRZEMIANY TLENOWE I BEZTLENOWE W UKŁADACH OSADÓW WODNYCH |
C.1. TOKSYCZNOŚĆ OSTRA DLA RYB
1. METODA
1.1. WPROWADZENIE
Celem niniejszego badania jest ustalenie ostrej toksyczności śmiertelnej substancji dla ryb w wodach słodkich. Pożądane jest posiadanie, w możliwym zakresie, informacji na temat rozpuszczalności w wodzie, ciśnienia pary, stabilności chemicznej, stałych dysocjacji i rozkładu biologicznego substancji badanej, co pomoże w wybraniu najwłaściwszej metody badania (statycznej, półstatycznej lub przepływowej), tak aby zapewnić zadowalające, stałe stężenia substancji badanej w czasie wykonywania badania.
Przy planowaniu i interpretacji wyników badania należy także wziąć pod uwagę dodatkowe informacje (np. wzór strukturalny, stopień czystości, charakter i zawartość procentowa istotnych zanieczyszczeń, obecność i ilość substancji pomocniczych oraz współczynnik podziału n-oktanol/woda).
1.2. DEFINICJE I JEDNOSTKI
Toksyczność ostra to rozpoznawalne działanie niepożądane na organizm w ciągu krótkiego okresu ekspozycji (rzędu dni) na substancję. W niniejszym badaniu toksyczność ostrą wyraża się jako medianę stężenia śmiertelnego (LC), tj. stężenie w wodzie, które powoduje śmierć 50 % badanego stada ryb podczas okresu stałej ekspozycji, który musi zostać podany.
Wszystkie stężenia substancji badanej podano jako waga na objętość (miligramy na litr). Mogą być również wyrażone jako stosunek wagi do wagi (mg/kg-1).
1.3. SUBSTANCJE ODNIESIENIA
Substancję odniesienia można zbadać w celu zademonstrowania, że w warunkach laboratoryjnego badania, reakcja badanych gatunków nie zmieniła się znacząco.
Dla niniejszego badania nie wyszczególniono żadnych substancji odniesienia.
1.4. ZASADA METODY BADANIA
Badanie graniczne można przeprowadzić przy 100 mg na litr w celu zademonstrowania że LC50 jest większy niż to stężenie.
Ryby są poddawane ekspozycji na substancje badaną dodaną do wody w danym zakresie stężeń na okres 96 godzin. Odnotowuje się liczby zgonów w odstępach co najmniej 24-godzinnych i oblicza się stężenia uśmiercające 50 % ryb (LC50) w każdym momencie obserwacji, o ile to możliwe.
1.5. KRYTERIA JAKOŚCI
Kryteria jakościowe należy odnieść do badania granicznego, jak również do pełnej metody badania.
Śmiertelność w grupach kontrolnych nie może przekraczać 10 % (lub jednej ryby, o ile stosuje się mniej niż 10) na koniec badania.
Stężenie rozpuszczonego tlenu musi być wyższa niż 60 % wartości nasycenia powietrzem przez cały czas.
Stężenia substancji badanej powinny być utrzymywane do 80 % wstępnych stężeń poprzez czas trwania badania.
Dla substancji łatwo rozpuszczalnych w podłożu badanym roztwory podatne na stabilność, to jest te które nie wykazują żadnego znacznego stopnia ulatniania, rozkładu, hydrolizy lub absorpcji, początkowe stężenie uważane jest za równoważne nominalnemu stężeniu. Należy przedstawić dowody, że utrzymano stężenia poprzez całe badanie i spełniono kryteria jakościowe.
Dla substancji, które są:
|
(i) |
słabo rozpuszczalne w podłożu badawczym; lub |
|
(ii) |
zdolne do tworzenia stabilnych emulsji lub zawiesin; lub |
|
(iii) |
niestabilne w roztworach wodnych, |
początkowe stężenie musi być pobrane jako zmierzone stężenie w roztworze (lub, jeśli to technicznie niemożliwe, zmieszone w słupie wody) na początku badania. Stężenie należy oznaczyć po okresie równowagi, lecz przed wprowadzeniem badanych ryb.
W każdym z tych przypadków, muszą być prowadzone dalsze pomiary w czasie trwania badania dla potwierdzenia rzeczywistych stężeń ekspozycji lub spełnienia kryteriów jakościowych.
pH nie może się zmieniać więcej niż o jedną jednostkę.
1.6. OPIS METODY BADANIA
Można zastosować trzy rodzaje procedury:
Badanie statyczne:
Badanie toksyczności, w którym nie zachodzi przepływ badanego roztworu. (Roztwory pozostają bez zmian w trakcie całego badania).
Badanie półstatyczne:
Badanie bez przepływu roztworu, jednak z regularnymi odnowieniami roztworów badanych z tej samej serii po wydłużonych odstępach czasu (np. co 24 godziny).
Badanie przepływowe:
Badanie toksyczności, w którym wodę stale się odnawia w komorach badawczych, przy czym badany związek chemiczny jest transportowany z wodą wykorzystywaną do odnowy podłoża badawczego.
1.6.1. Odczynniki
1.6.1.1. Roztwory substancji badanych
Przygotowuje się roztwory podstawowe o wymaganej mocy przez rozpuszczenie substancji w wodzie dejonizowanej lub w wodzie przygotowanej według opisu w ppkt 1.6.1.2.
Wybrane stężenia badane przygotowuje się przez rozcieńczanie roztworu podstawowego. W przypadku badania wysokich stężeń substancję można rozpuścić bezpośrednio w wodzie do rozcieńczania.
Substancje zwykle należy badać tylko do granicy rozpuszczalności. Dla niektórych substancji (na przykład dla substancji posiadających niską rozpuszczalność w wodzie lub o wyższym Pow lub tych tworzących raczej stabilne zawiesiny niż roztwory rzeczywiste w wodzie) jest dopuszczalne stężenie badania powyżej granicy rozpuszczalności celem zapewnienia, że uzyskano maksymalne stężenie rozpuszczenia/stabilności. Jest ważne jednakże, żeby to stężenie w żaden sposób nie zakłócało systemu badania (np. warstwa substancji na powierzchni wody zapobiegająca utlenianiu wody itp.).
Stosuje się ultradźwiękowe rozpraszanie, rozpuszczalniki organiczne, emulsyfikatory lub dyspergatory jako środki do przygotowania roztworów podstawowych substancji o niskiej rozpuszczalności lub w celu ułatwienia rozproszenia tych substancji w podłożu badawczym. Jeżeli stosuje się takie substancje pomocnicze, wszystkie badane stężenia muszą zawierać tę samą ilość substancji pomocniczej oraz dodatkowa grupa kontrolna ryb powinna być poddana ekspozycji przy tym samym stężeniu substancji pomocniczej, które użyto w badanych seriach. Stężenie takich substancji należy zminimalizować, lecz w żadnym przypadku nie może przekraczać 100 mg na litr w podłożu badawczym.
Badanie należy przeprowadzić bez regulacji pH. Jeżeli istnieją oznaki zaznaczonej zmiany pH, doradza się powtórzenie badania z odpowiednim dostosowaniem jego wartości i przedstawienie uzyskanych wtedy wyników. W takim przypadku wartość pH roztworu podstawowego należy doprowadzić do pH wody do rozcieńczenia, chyba że istnieją powody, dla których nie należy tego robić. HCl i NaOH są zalecane w tym celu. Regulację pH należy przeprowadzić w taki sposób, aby nie zmienić w istotnym stopniu stężenia substancji badanej w roztworze podstawowym. W przypadku gdyby zaszła reakcja chemiczna lub doszło do fizycznego wytrącania się osadu badanego związku z powodu ustawiania pH, należy podać ten fakt.
1.6.1.2. Woda hodowlana i woda do rozcieńczania
Można zastosować wodę do picia z sieci wodociągowej (niezanieczyszczoną przez chlor, metale ciężkie lub inne substancje w niebezpiecznym stężeniu), dobrej jakości wodę zwykłą lub wodę odtworzoną (zob. woda o całkowitej twardości pomiędzy 10 i 250 mg na litr (jako CaC03) i o pH 6,0–8,5 jest zalecana.
1.6.2. Przyrząd
Wszystkie elementy przyrządu muszą być wykonane z materiału obojętnego chemicznie:
|
— |
automatyczny układ rozcieńczający (do badania przepływowego), |
|
— |
miernik tlenu, |
|
— |
wyposażenia do oznaczania twardości wody, |
|
— |
odpowiednie urządzenia do regulacji temperatury, |
|
— |
miernik pH. |
1.6.3. Badane ryby
Ryby powinny być zdrowe i wolne od jakichkolwiek widocznych wad rozwojowych.
Stosowane gatunki należy wybrać na podstawie kryteriów praktycznych, takich jak stała dostępność w ciągu roku, łatwość utrzymania, dogodność badania, względna wrażliwość na substancje chemiczne i wszystkie czynniki ekonomiczne, bilogiczne lub środowiskowe mające posiadające jakiekolwiek znaczenie. Przy doborze gatunków ryb musi zrodzić się również przemyślenie potrzeby porównywalności uzyskanych danych do istniejących uznanych danych międzynarodowych (pozycja 1).
Wykaz zalecanych gatunków ryb do przeprowadzenia niniejszego badania podano w dodatku 2; najlepszymi gatunkami są pstrąg tęczowy i danio pręgowany.
1.6.3.1. Hodowla
Najkorzystniej, aby stosowane w badaniu ryby pochodziły z pojedynczego stada o podobnej długości i wieku. Należy je hodować przez co najmniej 12 dni w następujących warunkach:
napełnienie:
właściwe dla systemu (recyrkulacyjnego lub przepływowego) i gatunku ryb,
woda:
zob. 1.6.1.2,
światło:
12–16 godzin naświetlenia dziennie,
stężenie rozpuszczonego tlenu:
co najmniej 80 % wartości nasycenia powietrzem,
karmienie:
trzy razy tygodniowo lub codziennie przerwać 24 godzin przed rozpoczęciem badania.
1.6.3.2. Śmiertelność
Po 48-godzinnym okresie przyzwyczajania odnotowuje się śmiertelność i stosuje następujące kryteria:
|
— |
większa niż 10 % populacji w ciągu siedmiu dni: odrzucić całe stado, |
|
— |
pomiędzy 5 i 10 % populacji: okres hodowli kontynuuje się przez następnych siedem dni. Jeżeli nie stwierdzi się dodatkowych zgonów, stado można zaakceptować; w przeciwnym razie należy je odrzucić, |
|
— |
poniżej 5 % populacji: stado można zaakceptować. |
1.6.4. Adaptacja
Wszystkie ryby muszą pozostawać w kontakcie z wodą o jakości i temperaturze odpowiadającej wodzie wykorzystywanej w badaniu przez co najmniej siedem dni przed ich zastosowaniem.
1.6.5. Procedura badania
Badanie ostateczne można poprzedzić przeprowadzeniem badania służącego wyznaczeniu zakresu stężeń, aby uzyskać informacje na temat zakresu stężeń, który zostanie wykorzystany w zasadniczym badaniu.
Dodatkowo do badanych serii przeprowadza się również badanie jednej grupy kontrolnej bez badanej substancji i, jeśli stosowne, jednej grupy kontrolnej dla substancji pomocniczej.
Zależnie od właściwości fizycznych i chemicznych badanego związku należy wybrać badanie statyczne, półstatyczne lub przepływowe, tak aby zapewnić spełnienie kryteriów jakościowych.
Ryby poddaje się ekspozycji na substancję w sposób opisany poniżej:
|
— |
czas trwania: 96 godzin, |
|
— |
liczba zwierząt: co najmniej 7 na stężenie, |
|
— |
zbiorniki: o odpowiedniej pojemności w relacji do zalecanego napełnienia, |
|
— |
napełnienie: zaleca się maksymalne napełnienie 1 g na litr w przypadku badań statycznych i półstatycznych; w przypadku systemów przepływowych można zaakceptować większe napełnienie, |
|
— |
stężenie badania: co najmniej pięć stężeń różniących się stałym czynnikiem nieprzekraczającym 2,2 i tak dalece jak to możliwe, rozpiętości zakresu śmiertelności od 0 do 100 %, |
|
— |
woda: zob. 1.6.1.2, |
|
— |
oświetlenie: 12 do 16 godzin dziennie, |
|
— |
temperatura: właściwa dla gatunków (dodatek 2), jednak utrzymana w granicach ± 1 oC w przypadku każdego z badań, |
|
— |
stężenie rozpuszczonego tlenu: nie mniej niż 60 % wartości nasycenia powietrzem w wybranej temperaturze, |
|
— |
karmienie: żadne. |
Ryby kontroluje się po upływie pierwszych 2 do 4 godzin, a następnie w odstępach co najmniej 24-godzinnych. Uważa się je za martwe, gdy dotknięcie nasady ogona nie prowadzi do reakcji i nie widać ruchów oddechowych. Martwe ryby usuwa się podczas obserwacji i odnotowuje liczbę zgonów. Dokumentuje się wszelkie widoczne nieprawidłowości (np. utratę równowagi, zmiany zachowania podczas pływania, zmiany czynności oddechowej, pigmentacji itp.).
Należy wykonywać codzienne pomiary pH, rozpuszczonego tlenu i temperatury.
Badanie graniczne
Stosując procedury opisane w niniejszej metodzie badania, przeprowadza się badanie graniczne przy stężeniu 100 mg na litr w celu pokazania że LC50 jest większy niż dla danego stężenia.
Jeżeli natura badanej substancji jest taka, że nie można uzyskać stężenia 100 mg na litr w wodzie, badanie graniczne należy przeprowadzić w stężeniu równym rozpuszczalności substancji (lub w maksymalnym stężeniu tworzącym stabilną zawiesinę) w użytym podłożu (zob. także ppkt 1.6.1.1.).
Badanie graniczne należy przeprowadzić stosując 7–10 ryb, wraz z tą samą liczbą w kontroli(-ach). (Teoria rozkładu dwumianowego pokazuje, że gdy stosuje się 10 ryb o śmiertelności równej zeru, to jest 99,9 % pewności, że LC50 jest większy niż stężenie użyte w badaniu granicznym. Dla 7, 8 lub 9 ryb brak śmiertelności daje co najmniej 99 % pewności, że LC50 jest większy niż użyte stężenie).
Jeżeli występuje śmiertelność, należy przeprowadzić pełne badanie. Jeżeli obserwowane są działania subletalne, powinny być zapisywane.
2. DANE I OCENA
Dla każdego okresu czasu, gdzie zapisywano obserwacje (24, 48, 72 i 96 godzin), należy wykreślić procent śmiertelności dla każdego zalecanego okresu czasu ekspozycji w stosunku do stężenia na papierze logarytm- prawdopodobieństwo.
Jeżeli to możliwe, dla każdego czasu obserwacji, należy oszacować LC50 i granice ufności (p = 0,05), stosując standartowe procedury; wartości te należy zaokrąglić do jedności, lub najwyżej do dwóch znaczących cyfr (przykłady zaokrąglania: 170 dla 173,5; 0,13 dla 0,127; 1,2 dla 1,21).
W przypadkach gdy kąt nachylenia krzywej zależności stężenia od odpowiedzi procentowej jest zbyt stromy, aby było możliwe obliczenie LC50, wystarczy graficzna ocena szacunkowa tej wartości.
W przypadku gdy kolejne dwa stężenia w stosunku 2,2 dają jedynie umieralność 0 i 100 %, wystarczają one do wskazania zakresu, w którym znajduje się LC50.
W razie zaobserwowania, że nie da się utrzymać stabilności lub jednorodności substancji badanej, należy to zgłosić i wziąć pod uwagę przy interpretacji wyników.
3. SPORZĄDZANIE SPRAWOZDANIA
Sprawozdanie z badań zawiera w miarę możliwości następujące informacje:
|
— |
informacje na temat użytych do badania ryb (nazwa naukowa, szczep, dostawca, wszelkie wcześniejsze kontakty z innymi substancjami, rozmiary i liczba wykorzystana w przypadku każdego badanego stężenia), |
|
— |
źródło wody do rozcieńczenia i jej najważniejsze parametry chemiczne (pH, twardość, temperatura), |
|
— |
w przypadku substancji o niskiej rozpuszczalności w wodzie, metoda przygotowania roztworu podstawowego i badanego, |
|
— |
stężenie wszelkich substancji pomocniczych, |
|
— |
wykaz zastosowanych stężeń i wszelkie dostępne informacje na temat stabilności w danych stężeniach badanego związku chemicznego w roztworze badanym, |
|
— |
jeśli przeprowadzono analizy chemiczne, zastosowane metody i uzyskane wyniki, |
|
— |
wyniki badania granicznego, jeśli przeprowadzono, |
|
— |
uzasadnienie wyboru i szczegółowe dane na temat zastosowanej procedury badania (np. badanie statyczne, półstatyczne, szybkość dawkowania, szybkość przepływu, czy stosowano napowietrzanie, napełnienie rybami itp.), |
|
— |
opis urządzeń, |
|
— |
reżim oświetlenia, |
|
— |
stężenia rozpuszczonego tlenu, wartości pH i temperatury roztworów badanych co 24 godziny, |
|
— |
dowody spełnienia kryteriów jakościowych, |
|
— |
tabelę pokazującą śmiertelność skumulowaną przy każdym stężeniu i dla roztworów kontrolnych (oraz kontrolnych z substancją pomocniczą, jeśli wymagane) przy każdym z zalecanych czasów obserwacji, |
|
— |
wykres zależności stężenia od odpowiedzi procentowej pod koniec badania, |
|
— |
jeżeli możliwe, wartości LC50 w każdym z zalecanych czasów obserwacji (o granicach ufności 95 %), |
|
— |
procedury statystyczne wykorzystywane do ustalania wartości LC50, |
|
— |
uzyskane wyniki jeżeli zastosowano substancje odniesienia, |
|
— |
najwyższe stężenie badane niepowodujące zgonów w czasie trwania badania, |
|
— |
najniższe stężenie badane powodujące 100 % zgonów w czasie trwania badania. |
4. LITERATURA
|
(1) |
OECD, Paris, 1981, Test Guideline 203, Decision of the Council C(81) 30 final and updates. |
|
(2) |
AFNOR – Determination of the acute toxicity of a substance to Brachydanio rerio – Static and Flow Through methods – NFT 90-303 June 1985. |
|
(3) |
AFNOR – Determination of the acute toxicity of a substance to Salmo gairdneri – Static and Flow -Through methods – NFT 90-305 June 1985. |
|
(4) |
ISO 7346/l,/2 and/3 – Water Quality – Determination of the acute lethal toxicity of substances to a fresh water fish (Brachydanio rerio Hamilton-Buchanan – Teleostei, Cyprinidae). Part 1: Static method. Part 2: Semi-static method. Part 3: Flow-through method. |
|
(5) |
Eidgenössisches Department des Innern, Schweiz: Richtlinien fur Probenahme und Normung von Wasseruntersuchungsmethoden – Part II 1974. |
|
(6) |
DIN Testverfahren mit Wasserorganismen, 38 412 (L1) und L (15). |
|
(7) |
JIS K 0102, Acute toxicity test for fish. |
|
(8) |
NEN 6506 – Water – Bepaling van de akute toxiciteit met behulp van Poecilia reticulata, 1980. |
|
(9) |
Environmental Protection Agency, Methods for the acute toxicity tests with fish, macroinvertebrates and amphibians. The Committee on Methods for Toxicity Tests with Aquatic Organisms, Ecological Research Series EPA-660-75-009, 1975. |
|
(10) |
Environmental Protection Agency, Environmental monitoring and support laboratory, Office of Research and Development, EPA-600/4-78-012, January 1978. |
|
(11) |
Environmental Protection Agency, Toxic Substance Control, Part IV, 16 March 1979. |
|
(12) |
Standard methods for the examination of water and wastewater, fourteen edition, APHA-AWWA-WPCF, 1975. |
|
(13) |
Commission of the European Communities, Inter-laboratory test programme concerning the study of the ecotoxicity of a chemical substance with respect to the fish. EEC Study D.8368, 22 March 1979. |
|
(14) |
Verfahrensvorschlag des Umweltbundesamtes zum akuten Fisch-Test. Rudolph, P. und Boje, R. Okotoxikologie, Grundlagen fur die okotoxikologische Bewertung von Umweltchemikalien nach dem Chemikaliengesetz, ecomed 1986. |
|
(15) |
Litchfield, J. T and Wilcoxon, F., A simplified method for evaluating dose effects experiments, J. Pharm, Exp. Therap., 1949, vol. 96, 99. |
|
(16) |
Finney, D. J. Statistical Methods in Biological Assay. Griffin, Weycombe, U.K., 1978. |
|
(17) |
Sprague, J. B. Measurement of pollutant toxicity to fish. Bioassay methods for acute toxicity. Water Res., 1969, vol. 3, 793–821. |
|
(18) |
Sprague, J. B. Measurement of pollutant toxicity to fish. II Utilising and applying bioassay results. Water Res. 1970, vol. 4, 3–32. |
|
(19) |
Stephan, C. E. Methods for calculating an LC50. In Aquatic Toxicology and Hazard Evaluation (edited by F. I. Mayer and J. L. Hamelink). American Society for Testing and Materials, ASTM STP 634, 1977, 65–84. |
|
(20) |
Stephan, C. E., Busch, K. A., Smith, R., Burke, J. and Andrews, R. W A computer program for calculating an LC50. US EPA. |
Dodatek 1
Woda po regeneracji
Przykład właściwej wody rozcieńczającej
Wszystkie związki chemiczne muszą być czystości analitycznej.
Woda powinna być wodą destylowaną dobrej jakości, lub wodą dejonizowaną o przewodnictwie elektrycznym poniżej 5 μS/cm-1.
Przyrząd do destylacji wody nie może zawierać jakichkolwiek części miedzianych.
Roztwory podstawowe
|
CaCl2. 2H20 (diwodny chlorek wapnia): Rozpuścić, dopełnić wodą do jednego litra. |
11,76 g |
|
MgSO4. 7H20 (heptawodny siarczan magnezu): Rozpuścić, dopełnić wodą do jednego litra. |
4,93 g |
|
NaHC03 (wodorowęglan sodu): Rozpuścić, dopełnić wodą do jednego litra. |
2,59 g |
|
KCl (chlorek potasu): Rozpuścić, dopełnić wodą do jednego litra. |
0,23 g |
Regenerowana woda do rozcieńczania
Zmieszać po 25 ml każdego z czterech roztworów podstawowych i uzupełnić wodą do 1 litra.
Napowietrzać, aż stężenie rozpuszczonego tlenu osiągnie wartość nasycenia powietrzem.
Wartość pH powinna wynosić 7,8 ± 0,2.
W razie potrzeby należy wyregulować pH przy użyciu NaOH (wodorotlenku sodowego) lub HCl (kwasu solnego).
Tak przygotowaną wodę do rozcieńczania odstawia się na około 12 godzin, przy czym nie należy jej dalej napowietrzać.
Suma zawartości jonów Ca i Mg w tym roztworze wynosi 2,5 mmola na litr. Stosunek jonów Ca:Mg wynosi 4:1 i jonów Na:K wynosi 10:1. Całkowita zasadowość niniejszego roztworu jest 8 mmol na litr.
Wszelkie odchylenia w przygotowaniu wody do rozcieńczania nie mogą zmieniać jej składu ani właściwości.
Dodatek 2
Gatunki ryb zalecane do przeprowadzania badań
|
Zalecane gatunki |
Zalecany zakres temperatur ( oC) |
Zalecana całkowita długość badanych zwierząt (cm) |
|
Brachydanio rerio(Teleostei, Cyprinidae) (Hamilton-Buchanan) Danio pręgowany |
20 do 24 |
3,0 ± 0,5 |
|
Pimephales promelas(Teleostei, Cyprinidae) (Rafinesque) |
20 do 24 |
5,0 ± 2,0 |
|
Cyprinus carpio(Teleostei, Cyprinidae) (Linneaus 1758) Karp zwykły |
20 do 24 |
6,0 ± 2,0 |
|
Oryzias latipes(Teleostei, Poeciliidae) Cyprinodontidae (Tomminck and Schiegel 1850) Ryzowka |
20 do 24 |
3,0 ± 1,0 |
|
Poecilia reticulata (Teleostei, Poeciliidae) (Peters 1859) Gupik |
20 do 24 |
3,0 ± 1,0 |
|
Lepomis macrochirus (Teleostei, Centrarchidae) (Rafinesque Linneaus 1758) Okoń błękitnoskrzeli |
20 do 24 |
5,0 ± 2,0 |
|
Onchorhynchus mykiss (Teleostei, Salmonidae) (Walbaum 1988) Pstrąg tęczowy |
12 do 17 |
6,0 ± 2,0 |
|
Leuciscus idus (Teleostei, Cyprinidae) (Linneaus 1758) Jaz |
20 do 24 |
6,0 ± 2,0 |
Zbiór
Wymienione poniżej ryby są łatwe w hodowli i szeroko dostępne w ciągu całego roku. Nadają się one do hodowli i rozmnażania albo w gospodarstwach rybnych, albo w laboratoriach, w warunkach chroniących je od chorób i pasożytów. Ryby te dostępne są w wielu miejscach na świecie.
Dodatek 3
Przykład stężenia: procent śmiertelności
Przykład oznaczenia LC50 z użyciem papieru log-probit (logarytm-prawdopodobieństwo)

C.2. BADANIE NAGŁEGO UNIERUCHOMIENIA DAPHNIA SP.
1. METODA
Opisywana metoda badania nagłego unieruchomienia jest zgodna z OECD TG 202 (2004).
1.1. WSTĘP
Metoda ta jest stosowana w badaniu ostrej toksyczności oceniającym toksyczność w odniesieniu do rozwielitek. Istniejące metody badania były stosowane w możliwym zakresie (1)(2)(3).
1.2. DEFINICJE
W ramach tej metody stosowane są następujące definicje:
|
|
EC 50 : jest stężeniem, które według szacunków powoduje unieruchomienie 50 % rozwielitek w podanym okresie ekspozycji. Jeśli stosowana jest inna definicja, należy to zgłosić, podając jednocześnie odniesienie do niej. |
|
|
Unieruchomienie: te zwierzęta, które nie są w stanie pływać w ciągu 15 sekund po łagodnym potrząśnięciu naczyniem do badania, są uznawane za unieruchomione (nawet jeśli nadal mogą poruszać czułkami). |
1.3. ZASADA METODY BADANIA
Młode rozwielitki w wieku poniżej 24 godzin na początku badania są przez 48 godzin poddawane działaniu badanej substancji o zróżnicowanym stężeniu. Unieruchomienie jest rejestrowane po 24 oraz po 48 godzinach i porównywane z wartościami kontrolnymi. Wyniki są analizowane w celu obliczenia EC50 po 48 godz. (definicje zamieszczono w części 1.2). Nie jest konieczna ocena EC50 po 24 godz.
1.4. INFORMACJE NA TEMAT BADANEJ SUBSTANCJI
Rozpuszczalność w wodzie i ciśnienie pary badanej substancji powinny być znane, jak również powinna być dostępna niezawodna metoda określania ilościowego substancji w roztworach testowych o udokumentowanej wydajności i granicy określenia. Pomocnymi informacjami są m.in. wzór strukturalny, czystość substancji, stabilność w wodzie lub pod wpływem światła, Pow i wyniki badania podatności na rozkład biologiczny (zob. metoda C.4).
Uwaga: Instrukcje dotyczące badanych substancji, których właściwości fizykochemiczne utrudniają ich przebadanie, zamieszczono w (4).
1.5. SUBSTANCJE ODNIESIENIA
Można przeprowadzić badanie EC50 substancji odniesienia w celu sprawdzenia, czy warunki są właściwe. Zaleca się użycie do tego celu substancji toksycznych stosowanych w międzynarodowych próbach pierścieniowych (1)(5) (1). Badania z substancją odniesienia należy przeprowadzać co miesiąc, a co najmniej dwa razy do roku.
1.6. KRYTERIA JAKOŚCI
Przy uznawaniu ważności badania stosuje się następujące kryteria:
|
— |
w kontrolach, także w kontroli wykorzystującej czynnik rozcieńczający, zostało unieruchomionych nie więcej niż 10 % rozwielitek, |
|
— |
stężenie rozpuszczonego tlenu w naczyniach kontrolnych i naczyniach do badania powinno wynosić na końcu badania > 3 mg/l. |
Uwaga: W przypadku pierwszego kryterium unieruchomienie lub inne oznaki choroby lub stresu, na przykład odbarwienie, nietypowe zachowanie, takie jak utknięcie przy powierzchni, powinno wykazywać nie więcej niż 10 % kontrolnych rozwielitek.
1.7. OPIS METODY BADANIA
1.7.1. Przyrządy
Naczynia do badania i inne elementy wyposażenia mające kontakt z roztworami testowymi powinny być w całości wykonane ze szkła lub innych chemicznie obojętnych materiałów. Naczyniami do badania będą zwykle szklane probówki lub zlewki; należy je czyścić przed każdym użyciem, stosując standardowe procedury laboratoryjne. Naczynia do badania powinny być luźno przykryte, aby zmniejszyć utratę wody na skutek parowania i uniknąć dostawania się kurzu do roztworu. Substancje lotne powinny być badane w całkowicie wypełnionych i zamkniętych naczyniach, wystarczająco dużych, aby nie dopuścić do ograniczania ilości lub nadmiernego spadku poziomu tlenu (zob. część 1.6 i 1.8.3 ustęp pierwszy).
Dodatkowo należy używać niektórych lub wszystkich z następujących urządzeń: tlenomierz (z mikroelektrodą lub innym odpowiednim wyposażeniem do mierzenia poziomu tlenu w próbkach o niskiej zawartości tlenu); pehametr; odpowiedni aparat do kontroli temperatury; Sprzęt do określania całkowitego węgla organicznego (TOC); sprzęt do oznaczania chemicznego zapotrzebowania tlenu (ChZT), sprzęt do oznaczania twardości itd.
1.7.2. Badane organizmy
Zalecanym gatunkiem jest Daphnia magna Straus, ale do badania można użyć także innych, nadających się do tego celu gatunków (np. Daphnia pulex). Zwierzęta powinny być w wieku niższym niż 24 godziny na początku badania i aby zmniejszyć zróżnicowanie, zaleca się, aby nie było to potomstwo pierwszego pokolenia. Powinny one pochodzić ze zdrowej populacji (tj. nie wykazywać oznak stresu, takich jak wysoka śmiertelność, obecność samców i obecność jaj przetrwalnikowych, opóźnienia w produkcji pierwszego pokolenia, obecność odbarwionych zwierząt itd.). Wszystkie organizmy używane do określonego badania powinny pochodzić z hodowli założonych z tej samej populacji rozwielitek. Hodowane zwierzęta muszą być trzymane w warunkach hodowli (światło, temperatura, pożywka) podobnych do warunków stosowanych później w badaniu. Jeśli pożywka w hodowli rozwielitek w badaniu różni się od pożywki w rutynowych hodowlach, właściwym postępowaniem jest wprowadzenie okresu aklimatyzacji przed badaniem. Z tego powodu pokolenie rodzicielskie rozwielitek powinno być trzymane w wodzie do rozcieńczania w temperaturze do badania przez co najmniej 48 godzin przed rozpoczęciem badania.
1.7.3. Woda hodowlana i woda do rozcieńczania
Woda naturalna (powierzchniowa lub gruntowa), woda regenerowana lub odchlorowana mogą być użyte jako woda do hodowli rozwielitek i woda do rozcieńczania substancji, jeśli rozwielitki przeżyją w niej w czasie hodowli, aklimatyzacji i badania, nie wykazując oznak stresu. Jako wodę do badania można stosować każdy rodzaj wody, której charakterystyka zgodna jest z charakterystyką chemiczną wody nadającej się do rozcieńczania, jak opisano w załączniku 1. W czasie trwania badania jej jakość nie powinna się zmieniać. Woda regenerowana może być przygotowana przed dodanie do wody dejonizowanej lub destylowanej określonych ilości odczynników o czystości odpowiedniej do analizy. Przykłady wody regenerowanej podano w (1) i (6) oraz w załączniku 2. Należy pamiętać, że w przypadku badania substancji zawierającej metale należy unikać pożywek zawierających czynniki chelatujące, takie jak M4 i M7 opisane w załączniku 2. Wartość pH powinna utrzymywać się w przedziale 6–9. Dla Daphnia magna zalecana twardość powinna wynosić między 140 a 250 mg/l (CaCO3), dla innych gatunków Daphnia może być stosowana woda o mniejszej twardości. Woda do rozcieńczania może być przed użyciem do badania napowietrzona, tak aby tlen rozpuszczony osiągnął stan nasycenia.
Jeśli stosowana jest woda naturalna, jej parametry jakości powinny być sprawdzane co najmniej dwa razy do roku lub zawsze, kiedy istnieje podejrzenie, że mogły one znacznie się zmienić (zob. poprzedni ustęp i załącznik 1). Należy także sprawdzać zawartość metali ciężkich (np. Cu, Pb, Zn, Hg, Cd, Ni). Jeśli używana jest woda odchlorowana, pożądane jest przeprowadzanie codziennej analizy zawartości chloru. Jeśli woda do rozcieńczania pochodzi z wód powierzchniowych lub gruntowych, należy dokonać pomiaru przewodności i zawartości całkowitego węgla organicznego (TOC) lub chemicznego zapotrzebowania tlenu (ChZT).
1.7.4. Roztwory testowe
Roztwory testowe o wybranych stężeniach są zwykle przygotowywane przez rozcieńczenie roztworu podstawowego. Roztwory podstawowe należy przygotowywać, rozpuszczając badaną substancję w wodzie do rozcieńczania. Jeśli to tylko możliwe, należy unikać stosowania rozpuszczalników, emulgatorów czy środków dyspergujących. W niektórych przypadkach takie związki mogą być jednak potrzebne do przygotowania roztworu podstawowego o odpowiednim stężeniu. Informacje na temat odpowiednich rozpuszczalników, emulgatorów i środków dyspergujących podano w (4). W żadnym przypadku substancja badana w roztworach testowych nie powinna przekraczać granicy rozpuszczalności w wodzie do rozcieńczania.
Badanie powinno być przeprowadzone bez regulacji pH. Jeśli pH nie utrzymuje się w przedziale 6–9, można przeprowadzić drugie badanie przed dodaniem badanej substancji, dostosowując pH roztworu podstawowego do pH wody do rozcieńczania. Dostosowanie pH powinno zostać przeprowadzone w taki sposób, żeby stężenie roztworu podstawowego nie zostało istotnie zmienione i nie została spowodowana żadna reakcja chemiczna lub wytrącenie badanej substancji. Zaleca się stosowanie HCl i NaOH.
1.8. PROCEDURA
1.8.1. Warunki ekspozycji
1.8.1.1. Grupy badane i kontrolne
Naczynia do badania są napełniane odpowiednimi objętościami wody do rozcieńczania i roztworami badanych substancji. Stosunek powietrza do wody w naczyniu powinien być identyczny w grupie kontrolnej i badanej. Rozwielitki są następnie umieszczane w naczyniach do badania. Dla każdego badanego stężenia i kontroli należy użyć co najmniej 20 zwierząt podzielonych na cztery grupy po pięć zwierząt w każdej. Na każde zwierzę powinno przypadać co najmniej 2 ml roztworu badanego (tj. roztwór o objętości 10 ml dla pięciu rozwielitek na naczynie do badania). Badanie może być przeprowadzane przy zastosowaniu odnawiania semistatycznego lub przepływu przez system, jeśli badana substancja nie jest stabilna.
Oprócz właściwych serii kontrolnych należy przeprowadzić jedną serię kontrolną wody do rozcieńczania i, jeśli jest to właściwe, jedną serię kontrolną zawierającą czynnik rozpuszczający.
1.8.1.2. Badane stężenia
Jeśli nie ma danych o toksyczności badanej substancji, można przeprowadzić badanie zakresu, pozwalające określić zakres stężeń stosowanych w ostatecznych badaniach. W tym celu rozwielitki umieszcza się w szeregu roztworów o znacznie różniących się od siebie stężeniach badanej substancji. Działaniu każdego ze stężeń przez 48 godzin lub mniej powinno być poddanych pięć rozwielitek, żadne powtórzenia nie są konieczne. Okres ekspozycji może być krótszy (np. 24 godzin lub mniej), jeśli dane odpowiednie do celów badania określającego zakres stężeń można uzyskać w krótszym okresie.
Należy zastosować co najmniej pięć stężeń. Powinny one tworzyć szereg geometryczny, najlepiej o stosunku stężeń nieprzekraczającym 2,2. Jeśli zastosowano mniej niż pięć stężeń, należy przedstawić uzasadnienie. Najlepiej jeśli najwyższe badane stężenie powoduje stuprocentowe unieruchomienie, a najniższe badane stężenie nie powoduje widocznych skutków.
1.8.1.3. Warunki inkubacji
Temperatura powinna mieścić się w przedziale 18–22 oC i w żadnym badaniu nie powinna zmieniać się o więcej niż ± 1 oC. Zaleca się cykl 16 godzin oświetlenia, 8 godzin ciemności. Całkowita ciemność również jest dopuszczalna, zwłaszcza w przypadku badanych substancji niestabilnych w świetle.
W czasie badania nie należy doprowadzać powietrza do naczyń do badania. Badanie powinno być przeprowadzone bez regulacji pH. W czasie badania rozwielitki nie powinny być karmione.
1.8.1.4. Czas trwania
Czas trwania badania wynosi 48 godzin.
1.8.2. Obserwacje
W każdym naczyniu do badania należy sprawdzać obecność unieruchomionych rozwielitek po 24 i 48 godzinach od rozpoczęcia badania (definicje zamieszczono w części 1.2). Poza unieruchomieniem należy odnotować wszelkie odchylenia od normy w zachowaniu lub wyglądzie.
1.8.3. Pomiary analityczne
Zawartość tlenu rozpuszczonego i pH mierzy się na początku i na końcu badania w kontroli(-ach) i w roztworze badanej substancji o największym stężeniu. Stężenie tlenu rozpuszczonego w roztworach kontrolnych powinno spełniać kryterium ważności (zob. część 1.6). W normalnych warunkach pH nie powinno się różnić w poszczególnych badaniach o więcej niż 1,5 jednostki. Pomiary temperatury przeprowadza się zwykle w naczyniach kontrolnych i w ich otaczającym powietrzu. Temperaturę najlepiej mierzyć stale w czasie trwania badania lub przynajmniej na początku i na końcu badania.
Stężenie badanej substancji powinno być mierzone przynajmniej w roztworach badanych o najwyższym i o najniższym stężeniu, na początku i na końcu badania (4). Zaleca się, aby wyniki opierały się na mierzonych stężeniach. Jeśli jednak istnieją dane wskazujące, że stężenie badanej substancji jest skutecznie utrzymywane w granicach ± 20 % wartości nominalnej lub zmierzonej wartości początkowej przez cały czas trwania badania, to wyniki mogą opierać się na wartości nominalnej lub zmierzonej wartości początkowej.
1.9. BADANIE GRANICZNE
Stosując procedury opisane w niniejszej metodzie, można przeprowadzić badanie graniczne przy stężeniu 100 mg/l badanej substancji lub w stężeniu do granicy rozpuszczalności w środku stosowanym w badaniu (zależnie od tego, która wartość jest niższa) w celu wykazania, że EC50 jest większe niż dane stężenie. Badanie graniczne powinno być przeprowadzone przy użyciu 20 rozwielitek (najlepiej podzielonych na cztery grupy po pięć osobników) i tej samej liczby zwierząt w kontroli. Jeśli nastąpi unieruchomienie, należy przeprowadzić pełne badanie. Należy zanotować wszystkie odchylenia od normy w zachowaniu.
2. DANE
Dane powinny być podsumowane w formie tabelarycznej, dla każdej grupy badanej i kontrolnej przy każdej obserwacji należy podać liczbę użytych rozwielitek i unieruchomienie. Procent osobników unieruchomionych po 24 i 48 godzinach jest przedstawiany w funkcji stężenia badania. Dane są analizowane przy użyciu właściwych metod statystycznych (np. analiza probitowa itd.) w celu obliczenia nachylenia krzywych i EC50 przy 95 % poziomie ufności (p = 0,05) (7)(8).
W przypadku gdy standardowe metody obliczania EC50 nie mają zastosowania do otrzymanych danych, do oszacowania EC50 należy zastosować najwyższe stężenie niepowodujące unieruchomienia i najniższe stężenie powodujące 100 % unieruchomienia (wartość tę oblicza się jako średnią geometryczną tych dwóch stężeń).
3. SPRAWOZDAWCZOŚĆ
3.1. SPRAWOZDANIE Z BADANIA
Sprawozdanie z badania musi zawierać następujące dane:
Badana substancja:
|
— |
cechy fizyczne i istotne właściwości fizykochemiczne, |
|
— |
chemiczne dane identyfikacyjne, w tym czystość. |
Badany gatunek:
|
— |
źródło i gatunek Daphnia, dostawca (jeśli znany) i zastosowane warunki hodowli (w tym źródło, rodzaj i ilość pożywienia, częstotliwość karmienia). |
Warunki badania:
|
— |
opis naczyń do badania: typ naczyń, objętość roztworu, liczba rozwielitek w naczyniu do badania, liczba naczyń do badania (powtórzeń) dla każdego stężenia, |
|
— |
metody przygotowania roztworu podstawowego i badanych roztworów, w tym zastosowanie wszelkich rozpuszczalników lub środków dyspergujących, |
|
— |
szczegółowe informacje o wodzie do rozcieńczania: źródło i właściwości wody (pH, twardość, stosunek Ca/Mg, stosunek Na/K, zasadowość, przewodność itd.); skład regenerowanej wody, jeśli taka jest stosowana, |
|
— |
warunki inkubacji: temperatura, natężenie i cykliczność oświetlenia, rozpuszczony tlen, pH itd. |
Wyniki:
|
— |
liczba i procent rozwielitek, które były unieruchomione lub wykazywały jakiekolwiek objawy niekorzystnego działania substancji (w tym zachowanie odbiegające od normy) w roztworach kontrolnych i badanych przy okazji każdej obserwacji, oraz opis charakteru obserwowanych zmian, |
|
— |
wyniki i dane badania przeprowadzonego z substancja odniesienia, jeśli takie dane są dostępne, |
|
— |
nominalne stężenia badania i wynik wszystkich analiz służących określeniu stężenia badanej substancji w naczyniach do badania; należy również przedstawić informacje o wydajności stosowanej metody oraz dokładności oznaczania, |
|
— |
wyniki wszystkich pomiarów parametrów fizykochemicznych temperatury, pH i zawartości rozpuszczonego tlenu przeprowadzonych w czasie badania, |
|
— |
EC50 po 48 godzinach dla unieruchomienia wraz z przedziałami ufności i wykresy dopasowanego modelu użytego do obliczeń, nachylenia krzywych odpowiedzi na dawkę i ich błąd standardowy; procedury statystyczne użyte do określenia EC50 (dane dla unieruchomienia po 24 godzinach powinny także być przedstawione, jeżeli były rejestrowane), |
|
— |
wyjaśnienie wszelkich odstępstw od metody badania oraz określenie czy te odstępstwa miały wpływ na wyniki badań. |
4. BIBLIOGRAFIA
|
(1) |
ISO 6341. (1996). Water quality – Determination of the inhibition of the mobility of Daphnia magna Straus (Cladocera, Crustacea) – Acute toxicity test. Third edition, 1996. |
|
(2) |
EPA OPPTS 850.1010. (1996). Ecological Effects Test Guidelines – Aquatic Invertebrate Acute Toxicity Test, Freshwater Daphnids. |
|
(3) |
Environment Canada. (1996) Biological test method. Acute Lethality Test Using Daphnia spp. EPS 1/RM/11. Environment Canada, Ottawa, Ontario, Canada. |
|
(4) |
Guidance Document on Aquatic Toxicity Testing of Difficult Substances and Mixtures. OECD Environmental Health and Safety Publication. Series on Testing and Assessment. Nr. 23. Paris 2000. |
|
(5) |
Commission of the European Communities. Study D8369. (1979). Inter-laboratory Test Programme concerning the study of the ecotoxicity of a chemical substance with respect to Daphnia. |
|
(6) |
OECD Guidelines for the Testing of Chemicals. Guideline 211: Daphnia magna Reproduction Test, adopted September 1998. |
|
(7) |
Stephan C.E. (1977). Methods for calculating an LC50. In Aquatic Toxicology and Hazard Evaluation (edited by F.I. Mayer and J.L. Hamelink). ASTM STP 634 – American Society for Testing and Materials. Pp. 65–84. |
|
(8) |
Finney D.J. (1978). Statistical Methods in Biological Assay. 3rd ed. London. Griffin, Weycombe, UK. |
ZAŁĄCZNIK 1
NIEKTÓRE WŁAŚCIWOŚCI CHEMICZNE NADAJĄCEJ SIĘ DO BADAŃ WODY DO ROZCIEŃCZANIA
|
Substancja |
Stężenie |
|
Pył zawieszony |
< 20 mg/l |
|
Całkowity węgiel organiczny |
< 2 mg/l |
|
Amoniak niezjonizowany |
< 1 μg/l |
|
Chlor resztkowy |
< 10 μg/l |
|
Pestycydy fosforoorganiczne razem |
< 50 ng/l |
|
Pestycydy grupy węglowodorów chlorowanych razem plus polichlorowane bifenyle |
< 50 ng/l |
|
Całkowity chlor organiczny |
< 25 ng/1 |
ZAŁĄCZNIK 2
PRZYKŁADY WŁAŚCIWEJ REGENEROWANEJ WODY DO BADANIA
Woda do badania ISO (1)
|
Roztwory podstawowe (pojedyncza substancja) |
W celu przygotowania zregenerowanej wody dodać następujące objętości roztworów podstawowych do 1 litra wody (2) |
|
|
Substancja |
Ilość dodawana do 1 litra wody (2) |
|
|
Chlorek wapnia CaCl2, 2H2O |
11,76 g |
25 ml |
|
Siarczan magnezu MgSO4, 7H2O |
4,93 g |
25 ml |
|
Wodorowęglan sodu NaHCO3 |
2,59 g |
25 ml |
|
Chlorek potasu KC1 |
0,23 g |
25 ml |
Pożywki Elendta M7 i M4
Aklimatyzacja do pożywek Elendta M4 i M7
W niektórych laboratoriach napotkano trudności przy przenoszeniu rozwielitek do pożywek M4 i M7. Osiągnięto jednak pewne pomyślne wyniki, stosując aklimatyzację stopniową, tzn. przenosząc rozwielitki z własnej pożywki do 30 % roztworu, następnie do 60 %, a później 100 % roztworu Elendta. Konieczne może być stosowanie okresu aklimatyzacji nawet przez miesiąc.
Przygotowanie
Pierwiastki śladowe
Najpierw przygotowuje się osobne roztwory podstawowe (I) poszczególnych pierwiastków śladowych w wodzie o odpowiedniej czystości, np. dejonizowanej, destylowanej lub poddanej osmozie odwróconej. Z poszczególnych roztworów podstawowych (I) przygotowywany jest drugi, pojedynczy, roztwór podstawowy (II), który zawiera wszystkie elementy śladowe (roztwór łączony), tj.:
|
Roztwor podstawowy (pojedyncza substancja) |
Ilość dodana do wody (mg/l) |
Stężenie (odnoszące się do pożywki M4) |
W celu przygotowania kombinowanego roztworu II dodaċ następujące ilości roztworu podstawowego I do wody (ml/1) |
|
|
M4 |
M7 |
|||
|
H3 BO3 |
57 190 |
20 000-krotne |
1,0 |
0,25 |
|
MnCl2.4H2O |
7 210 |
20 000-krotne |
1,0 |
0,25 |
|
LiCl |
6 120 |
20 000-krotne |
1,0 |
0,25 |
|
RbCl |
1 420 |
20 000-krotne |
1,0 |
0,25 |
|
SrCl2.6H2O |
3 040 |
20 000-krotne |
1,0 |
0,25 |
|
NaBr |
320 |
20 000-krotne |
1,0 |
0,25 |
|
Na2MoO4.2H2O |
1 230 |
20 000-krotne |
1,0 |
0,25 |
|
CuCl2.2H2O |
335 |
20 000-krotne |
1,0 |
0,25 |
|
ZnCl2 |
260 |
20 000-krotne |
1,0 |
1,0 |
|
CoCl2.6H2O |
200 |
20 000-krotne |
1,0 |
1,0 |
|
KI |
65 |
20 000-krotne |
1,0 |
1,0 |
|
Na2 SeO3 |
43,8 |
20 000-krotne |
1,0 |
1,0 |
|
NH4VO3 |
11,5 |
20 000-krotne |
1,0 |
1,0 |
|
Na2 EDTA.2H2O |
5 000 |
2 000-krotne |
— |
— |
|
FeS04.7H2O |
1 991 |
2 000-krotne |
— |
— |
|
Zarowno roztwor Na 2 EDTA jak i FeSO4 są przygotowywane oddzielnie, zlevane razem i niezwłocznie umieszczane w autoklawie. |
||||
|
Daje to: |
||||
|
roztwor 21 Fe-EDT |
|
1 000-krotne |
20,0 |
5,0 |
Pożywki M4 i M7
Pożywki M4 i M7 są przygotowywane przy użyciu roztworu podstawowego II, makroskładników i witamin w następujący sposób:
|
|
Ilość dodana do wody (mg/l) |
Stężenie (odnoszące się do pożywki M4) |
Ilość roztworu p odstawowego II dodanego w celu otrzymania pożywkki (ml/1) |
|
|
M4 |
M7 |
|||
|
Roztwor podstawowy II (połączone pierwiastki śladowe) |
|
20-krotne |
50 |
50 |
|
Roztwory podstawowe makroskładników (pojedyncza substancja) |
|
|
|
|
|
CaCl2 • 2H2O |
293 800 |
1 000-krotne |
1,0 |
1,0 |
|
MgS04 • 7H2O |
246 600 |
2 000-krotne |
0,5 |
0,5 |
|
KCl |
58 000 |
10 000-krotne |
0,1 |
0,1 |
|
NaHCO3 |
64 800 |
1 000-krotne |
1,0 |
1,0 |
|
Na2 SiO3 • 9H2O |
50 000 |
5 000-krotne |
0,2 |
0,2 |
|
NaNO3 |
2 740 |
10 000-krotne |
0,1 |
0,1 |
|
KH2PO4 |
1 430 |
10 000-krotne |
0,1 |
0,1 |
|
K2HPO4 |
1 840 |
10 000-krotne |
0,1 |
0,1 |
|
Łączony roztwór podstawowy witamin |
— |
10 000-krotne |
0,1 |
0,1 |
|
Łączony roztwor podstawowy witamin przygotowuje się przez dodanie 3 witamin do 1 litra wody, jak przedstawiono poniżej. |
||||
|
Hydrochlorek tiaminy |
750 |
10 000-krotne |
|
|
|
Cyjanokobalami na (B12) |
10 |
10 000-krotne |
|
|
|
Biotyna |
7,5 |
10 000-krotne |
|
|
Roztwór podstawowy złożony z witamin jest przechowywany zamrożony w niewielkich alikwotach. Witaminy są dodawane do pożywki na krótko przed użyciem.
|
UWAGA |
: |
Aby uniknąć wytrącania się soli w czasie przygotowywania kompletnych pożywek, dodać alikwoty roztworów podstawowych do około 500–800 ml dejonizowanej wody, a następnie uzupełnić do 1 litra. |
|
UWAGA |
: |
Informacje o pierwszej publikacji na temat pożywki M4 można znaleźć w pracy Elendt B.P. (1990). Selenium deficiency in Crustacea; an ultrastructual approach to antennal damage in Daphnia magna Straus. Protoplasma, 154, 25-33. |
C.3. BADANIE INHIBICJI GLONÓW
1. METODA
1.1. WPROWADZENIE
Celem niniejszego badania jest oznaczenie wpływu substancji na wzrost gatunków jednokomórkowych glonów zielonych. Względnie szybkie (72 godziny) badania mogą oszacować wpływy na niektóre generacje. Niniejsza metoda może być zaadoptowana do użycia niektórych gatunków jednokomórkowych glonów, w przypadku których opis zastosowanej metody musi być przedstawiony w raporcie z badań.
Niniejsza metoda jest łatwo stosowana do substancji rozpuszczalnych w wodzie, które zgodnie z warunkami badania prawdopodobnie pozostaną w wodzie.
Metodę należy stosować dla substancji, które nie powodują bezpośredniego zakłócenia pomiaru wzrostu glonów.
Przed przystąpieniem do badania pożądane jest posiadanie, w możliwym zakresie, informacji na temat rozpuszczalności w wodzie, ciśnienia pary, stabilności chemicznej, stałych dysocjacji i rozkładu biologicznego substancji badanej.
Przy planowaniu i interpretacji wyników badania należy także wziąć pod uwagę dodatkowe informacje (np. wzór strukturalny, stopień czystości, charakter i zawartość procentowa istotnych zanieczyszczeń, obecność i ilość substancji pomocniczych oraz współczynnik podziału n-oktanol/woda).
1.2. DEFINICJE I JEDNOSTKI
Gęstość komórek: liczba komórek na mililitr.
Wzrost: wzrost gęstości komórek w okresie czasu badania.
Szybkość wzrostu: wzrost gęstości komórek na jednostkę czasu.
EC50: w niniejszej metodzie, stężenie substancji badanej, które daje 50 % zmniejszenie albo wzrostu (EbC50), albo szybkości wzrostu (ErC50) względem kontroli.
NOEC (nieobserwowany wpływ stężenia): w niniejszej metodzie – najwyższe badane stężenie, przy którym nie obserwuje się znaczącej inhibicji wzrostu względem kontroli.
Wszystkie stężenia substancji badanej podano w wadze na objętość (miligramy na litr). Mogą być także wyrażone w wadze do wagi (mg/kg-1).
1.3. SUBSTANCJE ODNIESIENIA
Można przeprowadzić badanie substancji odniesienia w celu wykazania, że reakcja badanych gatunków nie uległa istotnej zmianie w laboratoryjnych warunkach badania.
Jeżeli zastosowano substancję odniesienia, wyniki należy podać w sprawozdaniu z badania. Dichromian potasu może być użyty jako substancja odniesienia, lecz jego kolor wpływa na jakość oświetlenia i dopuszczalną intensywność dla komórek, a także na ewentualne oznaczenia spektrofotometryczne. Dichromian potasu stosowano w badaniu międzylaboratoryjnym (zob. poz. bibliograficzna (3) i dodatek 2).
1.4. ZASADA METODY BADANIA
Badanie graniczne przeprowadzane jest przy 100 mg na litr w celu zademonstrowania, że EC50 jest większe od stężenia.
Wykładniczo wzrastające kultury wybranych zielonych glonów są poddawane ekspozycji przy różnych stężeniach badanej substancji przez kilka generacji w pokreślonych warunkach.
Badane roztwory są inkubowane przez okres 72 godziny, w trakcie którego gęstość komórek w każdym z roztworów jest mierzona co najmniej co 24 godziny. Oznacza się inhibicję wzrostu w odniesieniu do kultury kontrolnej.
1.5. KRYTERIA JAKOŚCI
Do badania granicznego, jak również do pełnej metody badania należy zastosować kryteria jakościowe.
Gęstość komórek w kulturach kontrolnych musi wzrosnąć o współczynnik co najmniej 16 w ciągu okresu trzech dni.
Stężenia substancji badanej należy utrzymywać w obrębie 80 % stężeń początkowych poprzez czas odpowiadający czasowi trwania badania.
Dla substancji łatwo rozpuszczalnych w podłożu badanym, roztwory podatne na stabilność, to jest te które nie wykazują żadnego znacznego stopnia ulatniania, rozkładu, hydrolizy lub absorpcji, początkowe stężenie uważane jest za równoważne nominalnemu stężeniu. Należy przedstawić dowody, że utrzymano stężenia poprzez całe badanie i spełniono kryteria jakościowe.
Dla substancji, które są:
|
(i) |
słabo rozpuszczalne w podłożu badawczym; lub |
|
(ii) |
zdolne do tworzenia stabilnych emulsji lub zawiesin; lub |
|
(iii) |
niestabilne w roztworach wodnych, |
początkowe stężenie musi być pobrane jako zmierzone stężenie w roztworze na początku badania. Stężenie należy oznaczyć po okresie równowagi.
Znany jest fakt, że znaczne ilości badanej substancji są włączane w biomasę glonów w czasie trwania okresu badania.
Zatem w celu zademonstrowania zgodności z powyższymi kryteriami jakości, zarówno ilość substancji badanej włączonej do biomasy glonów jak i substancji w roztworze (lub jeśli to niemożliwe ze względów technicznych, zmierzone w słupie wody) muszą być wzięte pod uwagę. Jednakże jeśli oznaczenie stężenia substancji w biomasie glonów napotyka na poważne trudności techniczne, odpowiedniość z kryteriami jakościowymi, można przedstawić poprzez włączenie do procesu zbiornika badawczego z najwyższym stężeniem substancji, lecz bez glonów i pomiary stężeń w roztworze (lub, jeśli niemożliwe to technicznie, w słupie wody) na początku i na końcu okresu czasu badania.
1.6. OPIS PROCEDURY BADANIA
1.6.1. Odczynniki
1.6.1.1. Roztwory substancji badanych
Przygotowuje się roztwory podstawowe o wymaganym stężeniu przez rozpuszczenie substancji w wodzie dejonizowanej lub w wodzie przygotowanej według opisu w 1.6.1.2.
Wybrane stężenia badane są przygotowywane przez dodawanie odpowiednich podwielokrotności do prekultur glonów (zob. dodatek 1).
Substancje zwykle należy badać tylko do granicy rozpuszczalności. Dla niektórych substancji (na przykład dla substancji posiadających niską rozpuszczalność w wodzie, lub o wysokim Pow lub tych tworzących raczej stabilne zawiesiny niż roztwory rzeczywiste w wodzie) jest dopuszczalne stężenie badania powyżej granicy rozpuszczalności celem zapewnienia, że uzyskano maksymalne stężenie rozpuszczenia/stabilności. Jest ważne jednakże, żeby to stężenie w żaden sposób nie zakłócało systemu badania (np. warstwa substancji na powierzchni wody zapobiegająca utlenianiu wody itp.).
Stosuje się ultradźwiękowe rozpraszanie, rozpuszczalniki organiczne, emulsyfikatory lub dyspergatory jako środki do przygotowania roztworów podstawowych substancji o niskiej rozpuszczalności lub w celu ułatwienia rozproszenia tych substancji w podłożu badawczym. Jeżeli stosuje się takie substancje pomocnicze, wszystkie badane stężenia muszą zawierać tę samą ilość substancji pomocniczej, oraz dodatkowa grupa kontrolna powinna być poddana ekspozycji przy tym samym stężeniu substancji pomocniczej, które użyto w badanych seriach. Stężenie takich substancji należy zminimalizować, lecz w żadnym przypadku nie może przekraczać 100 mg na litr w podłożu badawczym.
Badanie należy przeprowadzić bez regulacji pH. Jeżeli istnieją oznaki zaznaczonej zmiany pH, doradza się powtórzenie badania z odpowiednim dostosowaniem jego wartości i przedstawienie uzyskanych wtedy wyników. W takim przypadku wartość pH roztworu podstawowego należy doprowadzić do pH wody do rozcieńczenia, chyba że istnieją powody, z których nie należy tego robić. Najlepsze są HCl i NaOH w tym celu. Regulację pH należy przeprowadzić w taki sposób, aby nie zmienić w istotnym stopniu stężenia substancji badanej w roztworze podstawowym. W przypadku gdyby zaszła reakcja chemiczna lub doszło do fizycznego wytrącania się osadu badanego związku z powodu ustawiania pH, należy podać ten fakt.
1.6.1.2. Podłoże badania
Woda powinna być dobrej jakości wodą destylowaną lub wodą dejonizowaną o przewodności niższej od 5 uS.cm-1. Przyrząd do destylacji nie może zawierać żadnych części wykonanych z miedzi.
Zalecane jest następujące podłoże.
Cztery roztwory podstawowe są przygotowywane zgodnie z następującą tabelą. Roztwory podstawowe są sterylizowane poprzez filtrowanie przez membranę i autoklawizowanie, i przechowywanie w ciemnym miejscu w 4 oC. Roztwór podstawowy nr 4 należy sterylizować tylko membranową filtracją. Te roztwory podstawowe rozcieńcza się do uzyskania końcowych stężeń składników pokarmowych w badanych roztworach.
|
Składnik pokarmowy |
Stężenie w roztworze podstawowym |
Końcowe stężenie w roztworze badanym |
|
Roztwór podstawowy 1: makroskładniki |
||
|
NH4Cl |
1,5 g/l |
15mg/l |
|
MgCl2.6H2O |
1,2 g/l |
12mg/l |
|
CaCl2.2H2O |
1,8 g/l |
18mg/l |
|
MgSO4.7H2O |
1,5 g/l |
15mg/l |
|
KH2PO4 |
0,16 g/l |
1,6mg/l |
|
Roztwór podstawowy 2: Fe-EDTA |
||
|
FeCl3.6H2 |
80 mg/l |
0,08 mg/l |
|
Na2EDTA.2H2O |
100 mg/l |
0,1 mg/l |
|
Roztwór podstawowy 3: pierwiastki śladowe |
||
|
H3BO3 |
185 mg/l |
0,185 mg/l |
|
MnCl2.4H2O |
415 mg/l |
0,415 mg/l |
|
ZnCl2 |
3 mg/l |
3 × 103 mg/l |
|
CoCl2.6H2O |
1,5 mg/l |
1,5 × 103 mg/l |
|
CuCl2.2H2O |
0,01 mg/l |
105 mg/l |
|
Na2 MoO4.2H2O |
7 mg/l |
7 × 103 mg/l |
|
Roztwór podstawowy 4: NaHCO3 |
||
|
NaHCO3 |
50 g/l |
50 mg/l |
Wartość pH podłoża po ustaleniu się równowagi z powietrzem wynosi około 8.
1.6.2. Przyrząd
|
— |
Zwykły sprzęt laboratoryjny. |
|
— |
Kolby do badań o stosownej objętości (np. kolby stożkowe 250 ml są odpowiednie dla objętości badanego roztworu równej 100 ml). Wszystkie kolby do badań powinny być identyczne materiałowo i wymiarowo. |
|
— |
Przyrząd do rozwoju kultur: szafka lub komora, w której temperatura wynosi 21–25 oC i jest utrzymywana z dokładnością ± 2 oC, o stałym, jednorodnym oświetleniu, dające zakres widma 400–700 nm. Jeżeli glony w kulturach kontrolnych osiągną zalecaną szybkość wzrostu, uznaje się, że warunki wzrostu, włączając intensywność oświetlenia, zostały spełnione. Zalecane jest użycie dla średniego poziomu badanych roztworów oświetlenia o intensywności w zakresie 60–120 μE.m-2.s-1 (35–70 × 1018 fotonów m-2.s-1) przy pomiarze w zakresie długości fali światła 400–700 nm, stosując odpowiedni czujnik. Dla przyrządów mierzących światło, skalibrowanych w luksach, akceptowalnym równoważnym zakresem jest 6 000–10 000 luksów. Intensywność światła może być uzyskana przez zastosowanie od czterech do siedmiu lamp fluorescencyjnych o mocy 30 W typu uniwersalnego białego (temperatura barwy około 4 300 K), w odległości 0,35 m od kultury glonów. |
|
— |
Pomiary gęstości komórek powinny być wykonywane, używając bezpośredniej metody zliczania żywych komórek, np.za pomocą mikroskopu z komorą liczącą. Jednakże można zastosować inne procedury wystarczająco czułe i wystarczająco dobrze skorelowane z gęstością komórek (np. fotometria, turbidymetria). |
1.6.3. Badane organizmy
Sugeruje się używanie takich gatunków glonów zielonych, które są gatunkami szybko wzrastającymi co jest wygodne dla kulturowania i badania. Najlepszymi gatunkami są:
|
— |
Selenastrum capricornutum, np. ATCC 22662 lub CCAP 278/4, |
|
— |
Scenedesmus subspicatus, np. 86.81 SAG, |
Uwaga:
|
ATCC |
= |
American Type Culture Collection (U.S.A.) |
|
CCAP |
= |
Culture Centre of Algae and Protozoa (U.K.) |
|
SAG |
= |
Collection of algal culture (Gottingen, F.R.G.) |
Jeżeli użyto innych gatunków, należy umieścić nazwę szczepu w sprawozdaniu.
1.6.4. Procedura badania
Zakres stężenia, w którym prawdopodobnie zajdzie wpływ, jest ustalany na podstawie wyników z badań znalezienia zakresu.
Dwa pomiary wzrostu (biomasy i szybkość wzrostu) mogą dać szeroko rozbieżne pomiary wzrostu inhibicji; obydwa należy użyć w badaniu znalezienia zakresu dla zapewnienia, że geometryczny ciąg stężeń umożliwi oszacowanie zarówno EbC50, jak i ErC50.
Początkowa gęstość komórek
Jest zalecane, aby początkowa gęstość komórek w badanych kulturach była około 104 komórek/ml dla Sehnastrum capricorn utum i Scenedesmus subspicatus. Jeśli stosuje się inne gatunki, biomasa musi być porównywalna.
Stężenia substancji badanej
Do badania przygotowuje się co najmniej pięć stężeń w seriach geometrycznych przy stosunku stężenia nieprzekraczającym 2,2. Najniższe badane stężenie powinno nie dawać obserwowalnego efektu wzrostu glonów. Najwyższe stężenie badane powinno hamować wzrost o co najmniej 50 % w stosunku do kontroli, i najkorzystniej całkowicie zatrzymać wzrost.
Repliki i kontrole
Projekt badania powinien zawierać trzy kopie badania przy każdym badanym stężeniu. Dołączane są trzy kontrole badania bez substancji badanej, oraz, jeśli stosowne, trzy kontrole zawierające substancję pomocniczą. O ile jest to uzasadnione, projekt może być zmieniony w celu zwiększenia liczby stężeń i zmniejszenia liczby replik na dane stężenie.
Przeprowadzenie badania
Badane kultury zawierające pożądane stężenia badanej substancji i pożądaną ilość inokulum glonowych są przygotowywane przez dodanie podwielokrotności roztworu podstawowego substancji badanej do odpowiednich ilości prekultur glonów (zob. dodatek 1).
Kolby z kulturami są wstrząsane i umieszczane w aparacie do hodowania kultur. Komórki glonów są utrzymywane w stanie zawiesiny poprzez wstrząsanie, wirowanie lub przepuszczaniem pęcherzyków powietrza w celu poprawy wymiany gazu z zmniejszenia zmian pH w badanych roztworach. Kultury powinny być utrzymywane w temperaturze w zakresie 21–25 oC, kontrolowanej w tolerancji ± 2 oC.
Gęstość komórkowa jest ustalana co najmniej w 24, 48 i 72 godziny po rozpoczęciu badania. Filtrowanie podłoża glonów zawierającego odpowiednie stężenia badanej substancji jest stosowane celem ustalenia tła, gdy prowadzone są pomiary gęstości komórek, inne od bezpośrednich metod zliczania.
Wartość pH jest mierzona na początku badania i w 72 godzinie.
Wartości pH w zbiornikach kontrolnych nie powinno zwykle odchylać się więcej niż 1,5 jednostki w czasie trwania badania.
Badanie substancji lotnych
Nie ma obecnie ogólnej uznanej drogi badania substancji lotnych. Jeżeli substancja jest znana z tendencji do parowania, używa się zamkniętych kolb badawczych ze zwiększoną przestrzenią głowicy. Zaproponowano zmiany do tej metody (zob. poz. bibliograficzna (4)).
Należy podjąć próby do ustalenia ilości substancji, która pozostaje w roztworze, i zwrócić ekstremalną uwagę przy interpretacji wyników badań z lotnymi związkami chemicznymi przy zastosowaniu systemów zamkniętych.
Badanie graniczne
Stosując procedury opisane w niniejszej metodzie badania, przeprowadza się badanie graniczne przy stężeniu 100 mg na litr w celu pokazania, że EC50 jest większy niż dla danego stężenia.
Jeżeli natura badanej substancji jest taka, że nie można uzyskać stężenia 100 mg na litr w badanej wodzie, badanie graniczne należy przeprowadzić w stężeniu równym rozpuszczalności substancji (lub w maksymalnym stężeniu tworzącym stabilną zawiesinę) w użytym podłożu (zob. także 1.6.1.1).
Badanie graniczne należy przeprowadzić w co najmniej potrojonej ilości, z taką samą ilością kontroli. Do badania granicznego należy wykonać dwa pomiary wzrostu (biomasy i szybkości wzrostu).
Jeżeli w badaniu granicznym znajduje się średni spadek o 25 % lub więcej biomasy albo szybkości wzrostu między badaniem granicznym i kontrolą, należy przeprowadzić pełne badanie.
2. DANE I OCENA
Zmierzone gęstości komórek w badanych kulturach i kontrolach ujmuje się w postaci stabelaryzowanej wraz ze stężeniami substancji badanej i czasami pomiarów. Wartość średnia gęstości komórek dla każdego stężenia substancji badanej oraz kontroli jest wykreślana w zależności od czasu (0–72 godz.) dla uzyskania krzywych wzrostu.
Dla ustalenia zależności stężenie/skutek należy zastosować dwa podejścia. Niektóre substancje mogą stymulować wzrost przy niskich stężeniach. Należy rozważyć tylko punkty danych wskazujących hamowanie między 0 i 100 %.
2.1. PORÓWNANIE POWIERZCHNI POD KRZYWYMI WZROSTU
Obszar między krzywymi wzrostu i poziomą linią N = N0 oblicza się zgodnie ze wzorem:

gdzie:
|
A |
= |
powierzchnia, |
|
N0 |
= |
ilość komórek/ml w czasie t (początek badania), 0, |
|
N1 |
= |
zmierzona liczba komórek/ml w czasie t1 |
|
Nn |
= |
zmierzona liczba komórek/ml w czasie t, |
|
t1 |
= |
czas pierwszego pomiaru po rozpoczęciu badania, |
|
tn |
= |
czas n-tego pomiaru po rozpoczęciu badania, |
|
n |
= |
liczba pomiarów zdjętych po rozpoczęciu badania. |
Procentowe hamowanie wzrostu komórek przy każdym stężeniu substancji badanej (IA) jest obliczane zgodnie ze wzorem:

gdzie:
|
Ac |
= |
powierzchnia między krzywą wzrostu kontroli i linią poziomą N = N0. |
|
At |
= |
powierzchnia między krzywą wzrostu przy stężeniu t i linia poziomą N = N0. |
|
IA |
= |
wartości są wykreślane na półlogarytmicznym papierze lub półlogarytmicznym probitowym papierze w relacji do odpowiadających stężeń. Jeżeli wykreśla się na papierze probitowym, punkty łączone są prostą linią albo wizualnie, albo regresją obliczeniową. |
EC50 oszacowuje się z linii regresji przez odczyt stężenia równoważnego 50 % inhibicji (IA = 50 %). Do oznaczenia niniejszej wartości jednoznacznie w odniesieniu do tej metody obliczeniowej, proponuje się użycie symbolu EbC50. Jest podstawą, że EbC50 jest dzielone przez odpowiedni okres czasu ekspozycji, np. EbC50 (0–72 godz.).
2.2. PORÓWNANIE SZYBKOŚCI WZROSTU
Średnia właściwa szybkość wzrostu (u) dla wykładniczo rosnącej kultury może być obliczona jako:
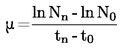
gdzie t0 jest czasem początku badania.
Alternatywnie, średnia właściwa szybkość wzrostu może być wyprowadzona z nachylenia linii regresji z wykresu N w stosunku do czasu.
Procentowe hamowanie właściwej szybkości wzrostu przy każdym stężeniu substancji badanej (Iμt) jest obliczane zgodnie ze wzorem:
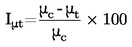
gdzie:
|
μc |
= |
średnia właściwa szybkość wzrostu dla kontroli, |
|
μt |
= |
średnia właściwa szybkość wzrostu dla stężenie badania t. |
Procentowe zmniejszenie średniej właściwej szybkości wzrostu przy każdym stężeniu substancji badanej porównane do wartości kontroli jest wykreślane w odniesieniu do logarytmu stężenia. EC50 odczytuje się z wynikowego wykresu. Dla jednoznacznego oznaczenia EC50 wyprowadzonego z niniejszej metody proponowane jest użycie symbolu ErC50. Czasy pomiaru muszą być wskazane, na przykład jeżeli wartość jest odniesiona do czasów 0 i 72 godziny, symbol staje się ErC50 (0–72 godz.).
Uwaga: właściwa szybkość wzrostu jest członem logarytmicznym, czyli mała zmiana w szybkości wzrostu prowadzi do wielkiej zmiany w biomasie. Wartości EbC i ErC są zatem porównywalne nienumerycznie.
2.3. OBLICZENIE NOEC
Stężenie bez obserwowanego działania toksycznego jest oznaczane odpowiednią procedurą statystyczną dla wielopróbkowego porównania (np. analizy wariancji i badaniem Dunnetta), stosując poszczególne replikowane wartości powierzchni spod krzywych wzrostu A (zob. 2.1) lub właściwych szybkości wzrostu μ (zob. 2.2).
3. SPORZĄDZANIE SPRAWOZDANIA
Sprawozdanie z badań zawiera w miarę możliwości następujące informacje:
|
— |
substancja badana: dane tożsamości chemicznej, |
|
— |
badane organizmy: pochodzenie, kultura laboratoryjna, numer szczepu, metoda hodowli, |
|
— |
warunki badania:
|
|
— |
wyniki:
|
4. LITERATURA
|
(1) |
OECD, Paris, 1981, Test Guideline 201, Decision of the Council C(81) 30 Final. |
|
(2) |
Umweltbundesamt, Berlin, 1984, Verfahrensvorschlag „Hemmung der Zellvermehrung bei der Grunalge Scenedesmus subspicatus”, in: Rudoiph/Boje: Okotoxikologie, ecomed, Landsberg, 1986. |
|
(3) |
ISO 8692 – Water quality – Fresh water algal wzrost inhibition test with Scenedesmus subspicatus and Selenastrum capricornutum. |
|
(4) |
S.Galassi and M.Vighi – Chemosphere, 1981, vol. 10, 1123–1126. |
Dodatek 1
Przykład procedury hodowli glonów
Uwagi ogólne
Celem hodowli glonów na podstawie niniejszej procedury jest uzyskanie kultur glonów dla badań toksyczności.
Należy użyć odpowiednich metod dla zapewnienia, że kultury glonów nie są zainfekowane bakteriami (ISO 4833). Podstawowe jest dla kultur glonów, by były wolne od pasożytów.
Wszystkie operacje powinny być prowadzone w warunkach sterylnych w celu zapobieżenia zanieczyszczeniu bakteriami i innymi glonami. Zanieczyszczone kultury należy odrzucić.
Procedury uzyskania kultur glonów
Przygotowanie roztworów składników pokarmowych (podłoża):
Podłoże przygotowuje się przez rozcieńczenie stężonych roztworów podstawowych składników pokarmowych. Dla stałego podłoża dodaje 0,8 % agaru. Używane podłoże powinno być sterylne. Sterylizacja za pomocą autoklawów prowadzi do strat amoniaku.
Kultura podstawowa:
Kultury podstawowe są kulturami małych glonów, które są regularnie przenoszone do świeżego podłoża, działają jako materiał początkowy badania. Jeżeli kultury nie są regularnie używane, tworzą pasma na nachylonych kanalikach agaru. Kultury są przenoszone do świeżego podłoża co najmniej raz na dwa miesiące.
Kultury podstawowe wzrastają w stożkowych kolbach zawierających odpowiednie podłoże (obįętość około 100 ml). Gdy glony są inkubowane w 20 oC ze stałym oświetleniem, wymagane jest cotygodniowe przenoszenie.
W czasie trwania przenoszenia ilość starej kultury jest przenoszona za pomocą sterylnych pipet do kolby ze świeżym podłożem, tak że wraz z szybkim wzrostem gatunku początkowe stężenie jest około 100 razy mniejsze niż w starej kulturze.
Szybkość wzrostu gatunku może być określona przez krzywą wzrostu. Jest wiadome, że jest możliwe oszacowanie gęstości, przy której kulturę należy przenieść do nowego podłoża. Należy to wykonać, zanim kultura osiągnie fazę martwą.
Prekultura:
Przeznaczeniem prekultury jest danie odpowiedniej ilości glonów dla inokulacji badanych kultur. Prekultura jest inkubowana w warunkach badania i stosowana, gdy wciąż wykładniczo narasta, zwykle po czasie inkubacji około trzech dni. Jeżeli kultury glonów zawierają zdeformowane lub nienormalne komórki, należy je odrzucić.
Dodatek 2
ISO 8692 – Jakość wody – badanie zahamowania wzrostu glonów słodkowodnych Scenedesmus subspicatus i Selenastrum capricornutum przedstawia następujące wyniki badania międzylaboratoryįnego między 16 laboratoriami, przy badaniu dichromianu potasu:
|
|
Średnie (mg/l) |
Zakres (mg/l) |
|
ErC50 (0–72 godz.) |
0,84 |
0,60–1,03 |
|
EbC50 (0–72 godz) |
0,53 |
0,20–0,75 |
C.4. OZNACZENIE BIODEGRADOWALNOŚCI
CZĘŚĆ I. ROZWAŻANIA OGÓLNE
I.1. WPROWADZENIE
Opisano sześć metod pozwalających na sortowanie substancji chemicznych w celu biodegradowalności w aerobowych wodnych podłożach:
|
a) |
rozpuszczalny węgiel organiczny (DOC) metoda „Die-Away” (metoda C.4-A); |
|
b) |
zmodyfikowane badanie przesiewowe OECD – DOC metoda „Die-Away” (metoda C.4-B); |
|
c) |
wydzielanie ditlenku węgla (CO2) (Zmodyfikowane badanie Sturma) (metoda C.4-C); |
|
d) |
respirometria manometryczna (metoda C.4-D); |
|
e) |
zamkniętej butli (metoda C.4-E); |
|
f) |
MITI (Ministerstwo Handlu Międzynarodowego i Przemysłu – Japonia) (Metoda C.4-F). |
Zwykłe i ogólne rozważania do wszystkich sześciu badań podano w części I metody. Punkty specyficzne dla poszczególnych metod podano w częściach II–VII. Dodatki zawierają definicje, wzory i materiał wiodący.
Ćwiczenie porównawcze międzynarodowego laboratorium OECD, przeprowadzone w 1988 r., pokazały ze metody dają zgodne wyniki. Jednakże w zależności od fizycznych charakterystyk badanych substancji jedna lub inne metody są zalecane.
I.2. WYBÓR ODPOWIEDNIEJ METODY
W celu wybrania najbardziej odpowiedniej metody podstawowe jest posiadanie informacji na temat rozpuszczalności chemicznej, prężności pary i charakterystyk adsorpcji. Powinna być znana struktura chemiczna i wzór w celu obliczenia teoretycznych wartości i/lub sprawdzenia wartości pomierzonych parametrów, na przykład ThOD, ThCO2, DOC, TÓC, COD (zob. załącznik I i II).
Badania substancji chemicznych rozpuszczalnych w wodzie w co najmniej 100 mg/l mogą być oszacowane wszystkimi metodami, pod warunkiem że są one nielotne i nieabsorbujące. Dla tych substancji chemicznych, które są słabo rozpuszczalne w wodzie, lotne lub adsorbujące, odpowiednie metody wskazano w tabeli 1. Sposób w który substancje chemiczne słabo rozpuszczalne w wodzie można przygotować opisano w załączniku III. Średnio lotne substancje chemiczne mogą być badane metodą DOC (metoda „Die-Away”), jeżeli jest wystarczająca przestrzeń na gaz w naczyniach pomiarowych (które powinny być odpowiednio zakorkowane). W tym przypadku należy ustawić kontrolę abiotyczną, by nie pozwolić na jakiekolwiek straty fizyczne.
Tabela 1
Stosowalność metod badania
|
Badanie |
Metoda analityczna |
Odpowiedniość dla substancji, które są: |
||
|
Słabo rozpuszczalne |
lotne |
adsorbujące |
||
|
DOC Die-Away |
Rozpuszczalny węgiel organiczny |
— |
— |
+/- |
|
Zmod. OECD Die-Away |
Rozpuszczalny węgiel organiczny |
— |
— |
+/- |
|
Wydzielanie CO2 |
Respirometria: wydzielanie CO2 |
+ |
— |
+ |
|
Respirometria manometryczna |
Respirometria manometryczna: zużycie tlenu |
+ |
+/- |
+ |
|
Zamkniętej butla |
Respirometria: rozpuszczony tlen |
+/- |
+ |
+ |
|
MITI |
Respirometria: zużycie tlenu |
+ |
+/- |
+ |
Wymagane są informacje na temat czystości lub względnych stosunków głównych składników badanego materiału w celu interpretacji uzyskanych wyników, szczególnie gdy wyniki są niskie lub marginalne.
Informacje na temat toksyczności badanej substancji w stosunku do bakterii (załącznik IV) są bardzo użyteczne dla wyboru odpowiednich stężeń badania i mogą być podstawowe dla prawidłowej interpretacji niskich wartości biodegradacji.
I.3. SUBSTANCJE ODNIESIENIA
W celu sprawdzenia procedury chemiczne substancje odniesienia spełniające kryteria łatwej biodegradowalności są badane przez umieszczenie w odpowiedniej kolbie równolegle do zwykłego przebiegu badania.
Odpowiednimi substancjami chemicznymi są anilina (świeżo destylowana), octan sodu i benzoesan sodu. Te chemiczne substancje odniesienia całkowicie degradują się w tych metodach, nawet gdy nie dodano przezornie inokulum.
Zasugerowano, że należy szukać chemicznej substancji odniesienia, która by była łatwo biodegradowalna, lecz wymagałaby dodania inokulum. Zaproponowano wodoroftalan potasu lecz potrzeba uzyskać więcej dowodów dla tej substancji przed jej zaakceptowaniem jako substancji odniesienia.
W badaniach respirometrycznych, związki zawierające azot oddziaływują na pobieranie tlenu z powodu procesu nitryfikacji (zob. załączniki II i V).
I.4. ZASADA METOD BADAŃ
Roztwór lub zawiesinę substancji badanej na podłożu mineralnym jest zaszczepiany i inkubowany w warunkach aerobowych w ciemności lub rozproszonym świetle. Ilość DOC w roztworze badanym powinna być utrzymywana tak mała jak to możliwe w porównaniu z ilością DOC spodziewanej w substancji badanej. Wykonuje się poprawkę na aktywność endogeniczną inokulum przez prowadzenie równoległych badań ślepych z inokulum, ale bez substancji badanej, chociaż endogeniczna aktywność komórek w obecności substancji nie tak samo dokładnie odpowiada tej w kontroli endogenicznej. Substancja odniesienia jest równolegle badana w celu sprawdzenia operacji procedury.
Ogólnie biorąc, degradacja jest wykonywana przez oznaczanie parametrów takich jak DOC, tworzenie CO2 i pobrany tlen, wykonywane są pomiary w dostatecznie częstych odstępach czasu pozwalających na identyfikację początku i końca biodegradacji. Przy zastosowaniu automatycznych respirometrów pomiar staje się ciągły. DOC jest czasem mierzony dodatkowo do innych parametrów, lecz jest to zwykle wykonywane tylko na początku i końcu badania. Stosuje się także specyficzną analizę chemiczną do oceny pierwotnej degradacji substancji badanej i do oznaczenia stężenia wszystkich utworzonych substancji pośrednich (obligatoryjne w badaniu MITI).
Zwykle badanie biegnie przez 28 dni. Badanie jednakże można zakończyć przed upływem 28 dni, to jest wtedy kiedy krzywa biodegradacji osiąga plateau dla co najmniej 3 oznaczeń. Badania można także wydłużyć ponad 28 dni, jeżeli krzywa pokazuje, że biodegradacja rozpoczęła się natomiast nie osiągnięto plateau 28 dnia.
I.5. KRYTERIA JAKOŚCI
I.5.1. Odtwarzalność
Z powodu istoty biodegradacji oraz stosowania mieszanych populacji bakteryjnych oznaczenia przeprowadzać co najmniej zdublowane.
Ze zwykłego doświadczenia wynika, iż im większe jest stężenie drobnoustrojów wstępnie dodanych do podłoża badawczego, tym mniejsze będą zmiany między replikami. Badanie obrączkowe również pokazuje, że mogą występować duże zmiany między wynikami uzyskanymi przez rożne laboratoria, lecz zwykle uzyskuje się dobrą zgodność przy zastosowaniu łatwo degradowalnych związków.
I.5.2. Ważność badania
Badanie uznaje się za ważne, jeżeli różnica ekstremów wartości replikowanych zdjętych z plateau badanej substancji pod koniec badania lub pod koniec badania 10-dniowego „okiennego” jest na koniec badania, odpowiednio, mniejsza od 20 %, oraz jeżeli procentowa degradacja substancji odniesienia osiągnęła poziom łatwej biodegradacji przez 14 dni. Jeżeli jeden z tych warunków nie jest spełniony badanie należy powtórzyć. Z powodu ostrości tych metod niskie wartości niekoniecznie oznaczają, że substancja badana nie jest biodegradowalna w warunkach środowiskowych, lecz wskazuje, że konieczne jest więcej wysiłków do ustalenia biodegradowalności.
Jeżeli w badaniu toksyczności, zawierającym zarówno substancję badaną, jak i chemiczną substancję odniesienia, zachodzi mniej niż 35 % degradacji (w oparciu o DOC) lub mniej niż 25 % (w oparciu o ThOD lub ThCO2) w ciągu 14 dni, badaną substancję można ocenić jako inhibitor (zob. także załącznik IV). Należy powtórzyć serie badań, jeżeli możliwe jest użycie niższego stężenia badanej substancji i/lub wyższego stężenia inokulum, lecz nie większego niż 30 mg ciała stałego/litr.
I.6. PROCEDURY OGÓLNE I PRZYGOTOWANIA
Ogólne warunki stosowane w badaniach podsumowano w tabeli 2. Przyrząd oraz inne warunki eksperymentalne specyficznie właściwe dla poszczególnych badań opisano dalej, pod nagłówkami dla danego badania.
Tabela 2
Warunki badania
|
Badanie |
DOC (metoda „Die-Away”) |
CO2 wydzielanie |
Respirometria manometryczna |
Zmodyfikowane OECD badanie przesiewowe |
Zamknięta butla |
MITT (I) |
|||||||||
|
Stężenie badanej substancji jako |
|
|
|
|
|
|
|||||||||
|
mg/l |
|
|
100 V |
|
2–10 |
100 |
|||||||||
|
mg DOC/l |
10–40 |
10–20 |
|
10–40 |
|
|
|||||||||
|
mg ThOD/1 |
|
|
50–100 |
|
5–10 |
|
|||||||||
|
Stężenie inokulum (komórek/l, przybliżone) |
≤ 30 mg/1 SS lub ≤ 100 ml wycieku/l (107 – 108) |
0,5 ml wtórny wyciek/1 (105) |
< 5 ml wycieku/1 (104 –106) |
30 mg/1 SS (107-108) |
|||||||||||
|
Stężenie pierwiastków w podłożu mineralnym (w mg/1) |
|
|
|
|
|
|
|||||||||
|
P |
116 |
11,6 |
29 |
||||||||||||
|
N |
1,3 |
0,13 |
1,3 |
||||||||||||
|
Na |
86 |
8,6 |
17,2 |
||||||||||||
|
K |
122 |
12,2 |
36,5 |
||||||||||||
|
Mg |
2,2 |
2,2 |
6,6 |
||||||||||||
|
Ca |
9,9 |
9,9 |
29,7 |
||||||||||||
|
Fe |
0,05-0,1 V |
0,05-0,1 |
0,15 |
||||||||||||
|
pH |
7,4±0,2 |
korzystnie 7,0 |
|||||||||||||
|
Temperatura |
22 ± 2 oC |
25 ± I oC |
|||||||||||||
|
|
|
|||||||||||||
I.6.1. Woda rozcieńczająca
Stosuje się dejonizowaną lub destylowaną wodę, wolne od inhibujących stężeń substancji toksycznych (np. jonów Cu ++). Musi zawierać nie więcej niż 10 % zawartości węgla organicznego wprowadzanego w badanym materiale. Konieczna jest woda do badań o wysokiej czystości w celu eliminacji wysokich wartości dla śłepej próby. Zanieczyszczenia mogą pochodzić z naturalnych zanieczyszczeń wody, ale również z żywic jonowymiennych oraz lizowanych materiałów bakteryjnych i glonów. Dla każdej serii badań stosuje się tylko jedną partię wody, sprawdzoną uprzednio analizą DOC. Takie sprawdzenie nie jest konieczne dla badania „zamkniętej butli”, lecz zapotrzebowanie wody na tlen musi być niskie.
I.6.2. Roztwory podstawowe składników mineralnych
Do sporządzenia roztworów badanych sporządzane są roztwory podstawowe o odpowiednich stężeniach składników mineralnych. Następujące roztwory podstawowe są używane (o różnych współczynnikach rozcieńczenia) dla metod DOC „Die-Away” zmodyfikowanego badania przesiewowego OECD, wydzielanie CO2, respirometrii manometrycznej i badania „zamkniętej butli”.
Współczynniki rozcieńczenia oraz, dla badania MITI, właściwie przygotowane podłoża mineralne podano pod nagłówkami właściwych badań.
Roztwory podstawowe:
Przygotować następujące roztwory podstawowe, stosując odczynniki czystości analitycznej.
|
a) |
Diwodoroortofosforan monopotasu, KH2PO4 |
8,50 g |
|
Monowodoroortofosforan dipotasu, K2HPO4 |
21,75 g |
|
|
Diwodny monowodoroortofosforan disodium Na2HPO4. 2 H2O |
33,40 g |
|
|
Chlorek amonu, NH4CI |
0,50 g |
|
|
Rozpuścić w wodzie i dopełnić do 1 litra. Wartość pH roztworu powinna być 7,4. |
|
|
|
b) |
Chlorek wapnia, bezwodny, CaCl2 |
27,50 g |
|
lub chlorek wapnia diwodny, CaCl2, 2 H2O |
36,40 g |
|
|
Rozpuścić w wodzie i dopełnić do 1 litra. |
|
|
|
c) |
Heptawodny siarczan magnezu, MgSO4. 7 H2O |
22,50 g |
|
Rozpuścić w wodzie i dopełnić do 1 litra. |
|
|
|
d) |
Heksawodny chlorek żelaza (III), FeCl3. 6 H2O |
0,25 g |
|
Rozpuścić w wodzie i dopełnić do 1 litra. |
|
Uwaga: aby nie przygotowywać niniejszego roztworu bezpośrednio przed użyciem, dodać kroplę stężonego kwasu solnego lub 0,4 grama soli disodowej kwasu etylenodiaminotetraoctowego (EDTA) na litr.
I.6.3. Roztwory podstawowe substancji chemicznych
Przykładowo rozpuścić 1–10 g odpowiednio badanej substancji chemicznej lub chemicznej substancji odniesienia w wodzie dejonizowanej i dopełnić do 1 litra, gdy rozpuszczalność przekracza 1 g/l. Z drugiej strony, przygotować roztwory podstawowe w podłożu mineralnym lub dodać substancje chemiczną bezpośrednio do podłoża mineralnego. Postępowanie ze słabo rozpuszczalnymi substancjami chemicznymi – zob. załącznik III, ale w badaniu MITI (metoda C.4-F) nie stosuje się ani rozpuszczalników, ani środków emulsyfikujących.
I.6.4. Materiał inokulacyjny
Inokulum może pochodzić z różnych źródeł: aktywowany osad, ścieki kanalizacyjne (niechlorowane), wody powierzchniowe i gleby lub ich mieszaniny. Dla badań DOC „Die-Away”, wydzielania CO2 i respirometrii manometrycznej, jeżeli stosowany jest aktywowany osad, powinien być pobrany z oczyszczalni ścieków i urządzeń skali laboratoryjnej przyjmujących głównie ścieki domowe. Materiał inokulacyjny z innych źródeł, okazał się dawać bardziej rozproszone wyniki. Dla zmodyfikowanego badania przesiewowego OECD i badania zamkniętej butli potrzebne jest bardziej rozcieńczone inokulum bez kłaczków osadu i zalecanym źródłem jest ściek drugiego stopnia oczyszczalnia ścieków domowych lub urządzeń skali laboratoryjnej. Dla badania MITI inokulum jest otrzymywane z mieszanych źródeł i opisane jest pod nagłówkiem tego specyficznego badania.
I.6.4.1. Inokulum z aktywowanych osadów
Zebrać świeżo pobraną próbkę aktywowanego osadu z komorą napowietrzania oczyszczalni ścieków lub urządzenia skali laboratoryjnej przerabiającego głównie ścieki domowe. Odrzucić duże cząstki, jeżeli konieczne poprzez filtracje przez siatkę o drobnym oczku i utrzymywać napowietrzanie.
Alternatywnie, zsedymentować lub odwirować (np. 1 100 rpm, 10 min) po usunięciu wszystkich dużych cząstek. Odrzucić nadsącz. Można przemyć osad w podłożu mineralnym. Zawiesić stężony osad w podłożu mineralnym dla uzyskania stężenia 3–5 g zawiesiny ciał stałych/l i napowietrzać do wymaganego momentu.
Osad powinien być pobrany z prawidłowo działającej konwencjonalnej oczyszczalni ścieków. Jeżeli pobrano osad z oczyszczalni o wysokiej szybkości oczyszczania lub podejrzewa się zawartość inhibitorów, należy go przemyć. Zsedymentować lub odwirować ponownie zawieszony osad po energicznym mieszaniu, odrzucić nadsącz i ponownie zawiesić umyty osad w dalszej objętości podłoża mineralnego. Powtarzać tę procedurę do czasu, gdy uzna się, iż jest wolny od nadmiaru substratów lub inhibitorów.
Po zakończeniu ponownego zawieszenia, lub niepoddany obróbce osad, odstawić ich próbkę do momentu jej użycia do ustalenia suchej masy ciał stałych w zawiesinie.
Dalszą alternatywą jest homogenizacja aktywowanego osadu (3–5 g zawiesiny ciał stałych/l). Rozdrobnić osad w mechanicznym rozdrabniaczu przy średniej szybkości przez 2 minuty. Zsedymentować rozdrobniony osad przez 30 minut lub dłużej, gdy trzeba, i zdekantować ciecz do użycia jako inokulum przy szybkości 10 ml/l podłoża mineralnego.
I.6.4.2. Inne źródła inokulum
Można je uzyskać ze ścieków drugiego stopnia oczyszczalni ścieków lub urządzeń skali laboratoryjnej przyjmujących głównie ścieki domowe. Zebrać świeżą próbkę i utrzymywać napowietrzaną w czasie transportu. Pozwolić osadzić się przez 1 godzinę lub przefiltrować przez grubą bibułę filtracyjną i zatrzymać zdekantowany wyciek lub jeśli wymagane przefiltrować z napowietrzaniem. Do 100 ml tego typu inokulum może być użyte na litr podłoża.
Następnym źródłem inokulum są wody powierzchniowe. W tym przypadku zebrać próbkę z odpowiedniej wody powierzchniowej, np. rzeki, jeziora i utrzymywać w napowietrzeniu przez wymagany czas. Jeżeli konieczne, zatężyć inokulum przez filtrację lub odwirowanie.
I.6.5. Wstępne sezonowanie inokulum
Materiał inokulacyjny może być wstępnie sezonowany do warunków eksperymentalnych, lecz niewstępnie adoptowany do badanej substancji. Wstępne sezonowanie składa się z napowietrzania aktywowanego osadu w podłożu mineralnym lub ścieku drugiego stopnia, przez 5–7 dni w temperaturze badania. Wstępne sezonowanie poprawia czasem precyzje metod badania przez zmniejszenie wartości dla ślepej próby. Uważane jest za niekonieczne wstępne sezonowanie inokulum dla metody MITI.
I.6.6. Kontrole abiotyczne
Gdy wymagane, sprawdzić możliwą abiotyczną degradację badanej substancji przez oznaczenie usuniętego DOC, pobieranie tlenu lub wydzielania ditlenku węgla w sterylnych kontrolach niezawierających inokulum. Sterylizować za pomocą filtracji przez membranę (0,2–0,45 mikrona) lub przez dodanie odpowiedniej toksycznej substancji o odpowiednim stężeniu. Jeżeli stosuje się filtracje membranową, pobrać próbki aseptycznie dla utrzymania sterylności. Chociaż absorpcję badanej substancji wykluczono z góry, badania, które mierzą biodegradację jako usuwanie DOC, szczególnie dla inokulum aktywowanego osadu, powinny zawierać kontrole abiotyczne, zaszczepione i intoksykowane.
I.6.7. Liczba kolb
Liczba kolb w typowym badaniu jest opisana pod nagłówkiem każdego badania.
Stosuje się następujące typy kolb:
|
— |
zawiesina badana: zawierających substancję badaną i inokulum, |
|
— |
ślepa próba inokulum: zawierające tylko inokulum, |
|
— |
kontrola procedury: zawierających substancje odniesienia i inokulum, |
|
— |
kontrola sterylności abiotycznej: sterylne, zawierających substancję badaną (zob. I.6.6), |
|
— |
kontrola adsorpcji: zawierających substancję badaną inokulum i środek sterylizujący, |
|
— |
kontrola toksyczności: zawierających substancję badaną substancje odniesienia i inokulum. |
Obowiązkowe jest, by oznaczenia w zawiesinie badanej i w ślepej próbie inokulum były wykonywane równolegle. Wskazane jest wykonywanie oznaczeń w innych kolbach również w oznaczeniach równoległych.
To jednakże nie zawsze jest możliwe. Upewnić się, że pobrano wystarczającą ilość próbek lub odczytów do oceny procentowego usunięcia DOC w 10-dniowym badaniu „okiennym”.
I.7. DANE I OCENA
W obliczeniu Dt zastosowano procent degradacji, średnie wartości zdublowanych pomiarów parametrów w obu badanych naczyniach i w ślepej próbie inokulum. Wzory są przedstawione w sekcjach poniżej właściwych badań. Przebieg degradacji jest zobrazowany graficznie i badanie 10-dniowe „okno” jest wskazane. Należy obliczyć i przedstawić w sprawozdaniu usunięcie procentowe uzyskane na koniec 10-dniowego „okna” i wartość w plateau, lub pod koniec badania, w zależności od tego, co jest odpowiednie.
W badaniach respirometrycznych związki zawierające azot mogą wpływać na pobieranie tlenu z powodu nitryfikacji (zob. załączniki II i V).
I.7.1. Degradacja zmierzona za pomocą oznaczenia DOC
Procentową degradację Dt przy każdym czasie pobierania próbki należy obliczyć oddzielnie dla kolb zawierających substancję badaną, stosując średnie wartości zdublowanych pomiarów DOC w celu oszacowania ważności badania (zob. I.5.2). Oblicza się to, stosując następujące równanie:

gdzie:
|
Dt |
= |
% degradacji w czasie t, |
|
C0 |
= |
średnie początkowe stężenie DOC w zaszczepionym podłożu kultury zawierającym substancję badaną (mg DOC/l), |
|
Ct |
= |
średnie stężenie DOC w zaszczepionym podłożu kultury zawierającym substancję badaną w czasie t (mg DOC/l), |
|
Cbo |
= |
średnie początkowe stężenie DOC w zaszczepionym podłoże mineralnym ślepej próby (mg DOC/l), |
|
Cbt |
= |
średnie stężenie DOC w zaszczepionym podłożu mineralnym ślepej próby w czasie t (mg DOC/l). |
Wszystkie stężenia są mierzone eksperymentalnie.
I.7.2. Degradacja mierzona w pojęciu właściwej analizy
Gdy dostępne są dane z właściwej analizy, można obliczyć pierwotną biodegradację z:

gdzie:
|
Dt |
= |
% degradacji w czasie t, zwykle 28 dni, |
|
Sa |
= |
ilość pozostałości substancji badanej w zaszczepionym podłożu na koniec badania (mg), |
|
Sb |
= |
ilość pozostałości substancji badanej w badaniu ślepej próby woda/podłoże, do którego dodano tylko substancję badaną (mg). |
I.7.3. Abiotyczna degradacja
Gdy stosuje się kontrolę sterylną abiotyczną, obliczyć procentową abiotyczną degradację stosując

gdzie:
|
Cs(o) |
= |
DOC stężenie w sterylnej kontroli w dniu 0, |
|
Cs(t) |
= |
DOC stężenie w sterylnej kontroli w dniu t. |
I.8. SPORZĄDZANIE SPRAWOZDANIA
W miarę możliwości sprawozdanie z badań zawiera następujące elementy:
|
— |
badana i chemiczne substancje odniesienia, i ich czystość, |
|
— |
warunki badania, |
|
— |
inokulum: natura i miejsce próbkowania(-ń), stężenie i wszystkie operacje przygotowania wstępnego, |
|
— |
proporcja i charakter odpadu przemysłowego obecnego w ścieku, jeśli znany, |
|
— |
czas trwania badania i temperatura, |
|
— |
w przypadku słabo rozpuszczalnych badanych substancji, zastosowane operacje, |
|
— |
zastosowana metoda badania; dla każdej zmiany procedury należy podać przyczyny uzasadnione naukowo i uzasadnienie, |
|
— |
arkusz danych, |
|
— |
wszystkie obserwowane zjawiska inhibicji, |
|
— |
wszystkie obserwowane abiotyczne degradacje, |
|
— |
dane właściwej analizy chemicznej, jeśli dostępne, |
|
— |
dane analityczne etapów pośrednich, jeśli dostępne, |
|
— |
wykres procentowy degradacji w stosunku do czasu dla substancji badanych i odniesienia; fazy opóźnienia, fazy degradacji, 10-dniowe „okno” oraz nachylenie wykresu należy wyraźnie wskazać (załącznik I). Jeżeli badanie odpowiadało kryteriom ważności, można użyć do wykresu średni procent degradacji w kolbach zawierających substancje badaną, |
|
— |
procentowe usunięcie po 10-dniowym „oknie” i w plateau lub na koniec badania. |
CZĘŚĆ II. BADANIE DOC METODĄ „DIE-AWAY”(Metoda C.4-A)
II.1. ZASADA METODY
Zmierzoną objętość zaszczepionego podłoża mineralnego, zawierającego znane stężenie substancji badanej (10–40 mg DOC/l), jako pojedyncze nominalne źródło węgla organicznego napowietrza się w ciemności lub w rozproszonym świetle w 22 ± 2 oC.
Postępującą degradację określa się analizą DOC w częstych przedziałach czasowych przez 28 dni. Stopień biodegradacji jest obliczany przez wyrażenie stężenia usuniętego DOC (skorygowanego do tego ze ślepej próby inokulum w kontroli) jako procent stężenia obecnego początkowo. Stopień pierwotnej biodegradacji może być także obliczony z dodatkowych analiz chemicznych wykonanych na początku i pod koniec inkubacji.
II.2. OPIS METODY
II.2.1. Przyrząd
|
a) |
Kolby stożkowe, np. 250 ml do 2 1, w zależności od objętości wymaganej dla analizy DOC. |
|
b) |
Wytrząsarka do akomodacji kolb stożkowych – albo z automatyczną kontrolą temperatury, albo używaną w stałej temperaturze pokojowej, oraz wystarczająca moc dla utrzymania aerobowych warunków we wszystkich kolbach. |
|
c) |
Przyrząd filtracyjny z odpowiednimi membranami. |
|
d) |
Analizator DOC. |
|
e) |
Przyrząd dla oznaczania rozpuszczonego tlenu. |
|
f) |
Wirówka. |
II.2.2. Przygotowanie podłoża mineralnego
W celu przygotowania roztworów podstawowych, zob. I.6.2.
Zmieszać 10 ml roztworu (a) z 800 ml wody rozcieńczającej, dodać 1 ml roztworu (b) do (d) i dopełnić do 1 l wodą rozcieńczającą.
II.2.3. Przygotowanie wstępne inokulum
Inokulum może być otrzymane z różnych źródeł: aktywowanego osadu, ścieków kanalizacyjnych, wód powierzchniowych, gleb lub z mieszaniny tu wymienionych.
Zob. I.6.4, I.6.4.1, I.6.4.2 i I.6.5.
II.2.4. Przygotowanie kolb
Jako przykład wprowadzić porcje 800 ml podłoża mineralnego do 2 1 kolb stożkowych i dodać wystarczające objętości roztworów podstawowych substancji badanej i odniesienia do oddzielnych kolb dla uzyskania stężenia równoważnika chemicznego do 10–40 mg DOC/l. Sprawdzić wartość pH i ustawić, gdy konieczne, do 7,4. Przygotować materiał inokulacyjny w kolbach z aktywowanym osadem lub innym źródłem inokuli (zob. I.6.4) do uzyskania stężenia końcowego większego niż 30 mg zawiesiny ciał stałych/litr. Przygotować także kontrole inokulum w podłożu mineralnym, lecz bez badanej substancji i chemicznej substancji odniesienia.
Jeśli wymagane, użyć jedno naczynie dla sprawdzenia możliwych działań inhibicyjnych substancji badanej przez inokulację roztworu zawierającego w podłożu mineralnym, porównywalne stężenia substancji tak badanej, jak i chemicznej substancji odniesienia.
Także, jeżeli jest to wymagane, ustawić następną sterylną kolbę dla sprawdzenia, czy badana substancja degraduje się abiotycznie, używając niezaszczepiony roztwór substancji chemicznej (zob. I.6.6).
Dodatkowo, jeżeli podejrzewa się, że badana substancja zostanie zaabsorbowana znacząco w szkle, osadzie itp., wykonać wstępną ocenę prawdopodobnego wzrostu absorpcji, a zatem przydatności badania dla substancji chemicznej (zob. tabela 1). Nastawić kolby zawierające substancję badaną, inokulum i środek sterylizujący.
Dopełnić objętość we wszystkich kolbach do 11 podłożem mineralnym i po wymieszaniu pobrać próbkę z każdej kolby do oznaczenia początkowego stężenia DOC (zob. załącznik II.4). Przykryć otwory kolb np. aluminiową folią, w taki sposób by umożliwić swobodną wymianę powietrza między kolbą i otaczającą atmosferą. Następnie włożyć naczynia do wytrząsarki celem rozpoczęcia badania.
II.2.5. Liczba kolb w typowym przebiegu
Kolby 1 i 2: Zawiesina badana
Kolby 3 i 4: Ślepa próba inokulum
Kolba 5: Kontrola procedury
Zalecane oraz gdy konieczne:
Kolba 6: Kontrola sterylności abiotycznej
Kolba 7: Kontrola adsorpcji
Kolba 8: Kontrola toksyczności
Zob. także I.6.7.
II.2.6. Przeprowadzenie badania
Poprzez całe badanie oznaczać stężenia DOC w każdej kolbie duplikatu w znanych odstępach czasu, wystarczająco często, by można było oznaczyć 10-dniowe „okno” i usunięcie procentowe na koniec 10-dniowego „okna”. Pobrać tylko minimalną objętość zawiesiny badanej konieczną dla każdego oznaczenia.
Przed pobieraniem próbek zmniejszyć straty parowania z kolb przez dodanie wody rozcieńczającej (I.6.1) w wymaganej ilości, jeśli konieczne. Zamieszać energicznie podłoże kultury przed pobraniem próbki i upewnić się, że materiał przylegający do ścianek naczynia jest rozpuszczony lub zawieszony, przed pobieraniem próbek. Przefiltrować przez filtr membranowy lub odwirować (zob. załącznik II.4) natychmiast po pobraniu próbek. Analizować odfiltrowane lub odwirowane próbki tego samego dnia, w innym razie przechować w 2–4 oC przez maksimum 48 godzin, lub w niższej temperaturze - 18 oC przez czas dłuższy.
II.3. DANE I SPORZĄDZANIE SPRAWOZDANIA
II.3.1. Obróbka wyników
Obliczyć procentową degradację w czasie t, jak podano w I.7.1 (oznaczanie DOC), i – do wyboru – według I.7.2 (właściwa analiza).
Zapisać wszystkie wyniki na dołączonych arkuszach danych.
II.3.2. Ważność wyników
Zob. I.5.2.
II.3.3. Sporządzanie sprawozdania
Zob. I.8.
II.4. ARKUSZ DANYCH
Przykład arkusza danych podano poniżej.
BADANIE DOC METODĄ „DIE-AWAY”
|
1. |
LABORATORIUM |
|
2. |
DATA ROZPOCZĘCIA BADANIA |
3. SUBSTANCJA BADANA
Nazwa:
Stężenie roztworu podstawowego: … mg/l jako substancja chemiczna.
Początkowe stężenie w podłożu, t0: … mg/l jako substancja chemiczna.
4. INOKULUM
Źródło:
Poddano traktowaniu:
Wstępne sezonowanie, jeśli wykonano:
Stężenie zawiesiny ciał stałych w mieszaninie reakcyjnej: … mg/l
5. OZNACZENIA WĘGLA
Analizator węgla:
|
|
Kolba nr |
|
DOC po n dniach (mg/1) |
||||
|
0 |
n1 |
n2 |
n3 |
nx |
|||
|
Badana substancja chemiczna plus ino-kulum |
1 |
a1 |
|
|
|
|
|
|
a2 |
|
|
|
|
|
||
|
a, średnia Ca(t) |
|
|
|
|
|
||
|
2 |
b1 |
|
|
|
|
|
|
|
b2 |
|
|
|
|
|
||
|
b, średnia Cb(t) |
|
|
|
|
|
||
|
Ślepa próba inoku-lum bez badanej substancji |
3 |
C1 |
|
|
|
|
|
|
C2 |
|
|
|
|
|
||
|
c, średnia Cc(t) |
|
|
|
|
|
||
|
4 |
d1 |
|
|
|
|
|
|
|
d2 |
|
|
|
|
|
||
|
d, średnia Cd(t) |
|
|
|
|
|
||
|
|
|
|
|
|
|
||
6. OCENA DANYCH PIERWOTNYCH
|
Kolba nr |
|
% degradacja po n dniach |
||||
|
0 |
n1 |
n2 |
n3 |
nx |
||
|
1 |
|
0 |
|
|
|
|
|
2 |
|
0 |
|
|
|
|
|
Srednia (3) |
|
0 |
|
|
|
|
Uwaga: podobne arkusze można użyć dla chemicznej substancji odniesienia i kontroli toksyczności.
7. KONTROLA ABIOTYCZNA (do wyboru)
|
|
Czas (dni) |
|
|
0 |
t |
|
|
DOC cone, (mg/l) kontrola sterylności |
Cs(o) |
Cs(t) |

8. WŁAŚCIWA ANALIZA CHEMICZNA (do wyboru)
|
|
ilość pozostałości badanej substancji na koniec badania (mg/l) |
% pierwotnej degradacji |
|
Kontrola sterylności |
Sb |
|
|
Zaszczepione podłoże badawcze |
Sa |
|
CZĘŚĆ III. ZMODYFIKOWANE BADANIE PRZESIEWOWE OECD (Metoda C.4-B)
III. 1. ZASADA METODY
Zmierzoną objętość podłoża mineralnego zawierające znane stężenie substancji badanej (10–40 mg DOC/litr) jako nominalne pojedyncze źródło węgla organicznego, zaszczepiono za pomocą 0,5 ml wycieku na litr podłoża. Mieszaninę napowietrza się w ciemności lub w rozproszonym świetle w 22 ± 2 oC.
Postępującą degradację określa się analizą DOC w częstych przedziałach czasowych przez 28 dni. Stopień biodegradacji jest obliczany przez wyrażenie stężenia usuniętego DOC (skorygowanego do tego ze ślepej próby inokulum w kontroli) jako procent stężenia obecnego początkowo. Stopień pierwotnej biodegradacji może być także obliczony z dodatkowych analiz chemicznych wykonanych na początku i pod koniec inkubacji.
III.2. OPIS METODY
III.2.1. Przyrząd
|
a) |
Kolby stożkowe, np. 250 ml do 2 1, w zależności od objętości wymaganej dla analizy DOC. |
|
b) |
Wytrząsarka do akomodacji kolb stożkowych – albo z automatyczną kontrolą temperatury, albo używaną w stałej temperaturze pokojowej, oraz wystarczająca moc dla utrzymania aerobowych warunków we wszystkich kolbach. |
|
c) |
Przyrząd filtracyjny z odpowiednimi membranami. |
|
d) |
Analizator DOC. |
|
e) |
Przyrząd dla oznaczania rozpuszczonego tlenu. |
|
f) |
Wirówka. |
III.2.2. Przygotowanie podłoża mineralnego
W celu przygotowania roztworów podstawowych, zob. I.6.2.
Zmieszać 10 ml roztworu (a) z 80 ml wody rozcieńczającej, dodać 1 ml roztworu (b) do (d) i dopełnić do 1 litra wodą rozcieńczającą.
Niniejsza metoda wykorzystuje tylko 0,5 ml wycieku/litr jako inokulum i dlatego podłoże może wymagać wzmocnienia śladowymi pierwiastkami i czynnikami wzrostu. Wykonuje się to przez dodanie 1 ml każdego z następujących roztworów, na litr końcowego podłoża:
Roztwór pierwiastków śladowych:
|
Siarczan manganu tetrawodny MnSO4. 4 H2O |
39,9 mg |
|
Kwas borowy, H3BO3 |
57,2 mg |
|
Siarczan cynku heptawodny ZnSO4. 7 H2O |
42,8 mg |
|
Heptamolibdenian amonu (NH4)6 Mo7O24 |
34,7 mg |
|
Fe-chelata (FeCl3 kwasu etylenodiaminotetraoctowego) |
100,0 mg |
|
Rozpuścić w i dopełnić do 1 000 ml wodą rozcieńczającą |
|
|
Roztwór witaminowy: |
|
|
Ekstrakt drożdży |
15,0 mg |
Rozpuścić ekstrakt drożdży w 100 ml wody. Sterylizować przez przepuszczenie przez membranę 0,2 mikrona, lub przygotować na świeżo.
III.2.3. Przygotowanie wstępne inokulum
Inokulum może być otrzymane ze ścieków drugiego stopnia oczyszczalni ścieków lub urządzeń skali laboratoryjnej przyjmujących głownie ścieki domowe (zob. I.6.4.2 i I.6.5).
Stosuje się 0,5 ml na litr podłoża mineralnego.
III.2.4. Przygotowanie kolb
Jako przykład wprowadzić porcje 800 ml podłoża mineralnego do 2-litrowych kolb stożkowych i dodać wystarczające objętości roztworów podstawowych substancji badanej i odniesienia do oddzielnych kolb dla uzyskania stężenia równoważnika chemicznego do 10–40 mg DOC/l. Sprawdzić wartość pH i ustawić, gdy konieczne, do 7,4. Zaszczepić kolby wyciekiem ściekowym 0,5 ml/litr (zob. I.6.4.2). Przygotować także kontrole inokulum w podłożu mineralnym, lecz bez badanej substancji i chemicznej substancji odniesienia.
Jeśli wymagane, użyć jedno naczynie dla sprawdzenia możliwych działań inhibicyjnych substancji badanej przez inokulację roztworu zawierającego w podłożu mineralnym, porównywalne stężenia substancji tak badanej, jak i chemicznej substancji odniesienia.
Także, jeżeli jest to wymagane, ustawić następną sterylną kolbę dla sprawdzenia, czy badana substancja degraduje się abiotycznie, używając niezaszczepiony roztwór substancji chemicznej (zob. I.6.6).
Dodatkowo, jeżeli podejrzewa się, że badana substancja zostanie zaabsorbowana znacząco w szkle, osadzie itp., wykonać wstępną ocenę prawdopodobnego wzrostu absorpcji, a zatem przydatności badania dla substancji chemicznej (zob. tabela 1). Nastawić kolby zawierające substancję badaną, inokulum i środek sterylizujący.
Dopełnić objętość we wszystkich kolbach do 1 litra podłożem mineralnym i po wymieszaniu pobrać próbkę z każdej kolby do oznaczenia początkowego stężenia DOC (zob. załącznik II.4). Przykryć otwory kolb np. aluminiową folią, w taki sposób by umożliwić swobodną wymianę powietrza między kolbą i otaczającą atmosferą. Następnie włożyć naczynia do wytrząsarki celem rozpoczęcia badania.
III.2.5. Liczba kolb w typowym przebiegu
Kolby 1 i 2: Zawiesina badana
Kolby 3 i 4: Ślepa próba inokulum
Kolba 5: Kontrola procedury
i zalecane oraz gdy konieczne:
Kolba 6: Kontrola sterylności abiotycznej
Kolba 7: Kontrola adsorpcji
Kolba 8: Kontrola toksyczności
Zob. także I.6.7.
III.2.6. Przeprowadzenie badania
Poprzez całe badanie oznaczać stężenia DOC w każdej kolbie duplikatu w znanych odstępach czasu, wystarczająco często, by można było oznaczyć 10-dniowe „okno” i usunięcie procentowe na koniec 10-dniowego „okna”. Pobrać tylko minimalną objętość zawiesiny badanej konieczna dla każdego oznaczenia.
Przed pobieraniem próbek zmniejszyć straty parowania z kolb przez dodanie wody rozcieńczającej (I.6.1) w wymaganej ilości, jeśli konieczne. Zamieszać energicznie podłoże kultury przed pobraniem próbki i upewnić się że materiał przylegający do ścianek naczynia jest rozpuszczony lub zawieszony, przed pobieraniem próbek. Przefiltrować przez filtr membranowy lub odwirować (zob. załącznik II.4) natychmiast po pobraniu próbek. Analizować odfiltrowane lub odwirowane próbki tego samego dnia, w innym razie przechować w 2–4 oC przez maksimum 48 godzin, lub w niższej temperaturze - 18 oC przez czas dłuższy.
III.3. DANE AND SPORZĄDZANIE SPRAWOZDANIA
III.3.1. Obróbka wyników
Obliczyć procentową degradację w czasie t, jak podano w I.7.1 (oznaczanie DOC), i – do wyboru – pod I.7.2. (właściwa analiza).
Zapisać wszystkie wyniki na dołączonych arkuszach danych.
III.3.2. Ważność wyników
Zob. I.5.2.
III.3.3. Sporządzanie sprawozdania
Zob. I.8.
III.4. ARKUSZ DANYCH
Przykład arkusza danych podano poniżej.
ZMODYFIKOWANE BADANIE PRZESIEWOWE OECD
|
1. |
LABORATORIUM |
|
2. |
DATA ROZPOCZĘCIA BADANIA |
3. SUBSTANCJA BADANA
Nazwa:
Stężenie roztworu podstawowego: … mg/litr jako substancja chemiczna
Początkowe stężenie w podłożu, to: … mg/litr jako substancja chemiczna
4. INOKULUM
Źródło:
Poddano traktowaniu:
Wstępne sezonowanie, jeśli wykonano:
Stężenie zawiesiny ciał stałych w mieszaninie reakcyjnej: mg/l
5. OZNACZENIA WĘGLA
Analizator wegla:
|
|
Kolba nr |
|
DOC po n dniach (mg/1) |
||||
|
0 |
n1 |
n2 |
n3 |
nx |
|||
|
Badana substancja chemiczna plus inokulum |
1 |
a1 |
|
|
|
|
|
|
a2 |
|
|
|
|
|
||
|
a, średnia Ca(t) |
|
|
|
|
|
||
|
2 |
b1 |
|
|
|
|
|
|
|
b2 |
|
|
|
|
|
||
|
b, średnia Cb(t) |
|
|
|
|
|
||
|
Ślepa próba inokulum bez badanej substancji |
3 |
C1 |
|
|
|
|
|
|
C2 |
|
|
|
|
|
||
|
c, średnia Cc(t) |
|
|
|
|
|
||
|
4 |
d1 |
|
|
|
|
|
|
|
d2 |
|
|
|
|
|
||
|
d, średnia Cd(t) |
|
|
|
|
|
||
|
|
|
|
|
|
|
||
6. OCENA DANYCH PIERWOTNYCH
|
Kolba nr |
|
% degradacja po n dniach |
||||
|
0 |
n1 |
n2 |
n3 |
nx |
||
|
1 |
|
0 |
|
|
|
|
|
2 |
|
0 |
|
|
|
|
|
Srednia (4) |
|
0 |
|
|
|
|
Uwaga: podobne arkusze można stosować dla chemicznej substancji odniesienia i kontroli toksyczności.
7. KONTROLA ABIOTYCZNA (do wyboru)
|
|
Czas (dni) |
|
|
0 |
t |
|
|
DOC cone, (mg/l) kontrola sterylności |
Cs(o) |
Cs(t) |
8. WŁAŚCIWA ANALIZA CHEMICZNA (do wyboru)
|
|
ilość pozostałości badanej substancji na koniec badania (mg/1) |
% pierwotnej degradacji |
|
Kontrola sterylności |
Sb |
|
|
Zaszczepione podłoże badawcze |
Sa |
|
CZĘŚĆ IV. BADANIE WYDZIELANIA CO2 (Metoda C.4-C)
IV.1. ZASADA METODY
Zmierzona objętość zaszczepionego podłoża mineralnego, zawierającego znane stężenie badanej substancji (10–20 mg DOG lub TOC/l) jako nominalne pojedyncze źródło węgla organicznego, jest napowietrzana przez przepuszczanie powietrza wolnego od ditlenku węgla z kontrolowaną szybkością w ciemności lub rozproszonym świetle. Degradacja jest prowadzona przez 28 dni przez oznaczanie wytwarzanego ditlenku węgla, który jest pochłaniany w wodorotlenku sodowym lub barowym i mierzony za pomocą miareczkowania pozostałego wodorotlenku lub jako nieorganiczny węgiel. Ilość ditlenku węgla wydzielonego z badanej substancji (skorygowana dla tej uzyskanej ze ślepej próby inokulum) jest wyrażana jako procent ThCO2. Stopień biodegradacji można także obliczyć z dodatkowej analizy DOG wykonanej na początku i końcu inkubacji.
IV.2. OPIS METODY
IV.2.1. Przyrząd
|
a) |
Kolby, 2–5 litra, każda zaopatrzona w rurkę napowietrzającą sięgającą blisko dna naczynia i w wylot. |
|
b) |
Mieszadła magnetyczne przy ocenie słabo rozpuszczalnych substancji chemicznych. |
|
c) |
Butle absorpcji gazu. |
|
d) |
Urządzenia do kontroli i pomiaru przepływu powietrza. |
|
e) |
Przyrząd do przemywania ditlenku węgla, dla przygotowania powietrza wolnego od ditlenku węgla; alternatywnie mieszanina tlenu i azotu wolnych od ditlenku węgla z butli gazowych, używane w prawidłowej proporcji (20 % O2: 80 % N2). |
|
f) |
Przyrząd do oznaczania ditlenku węgla – albo miareczkowaniem, albo jedną z postaci analizatora węgla nieorganicznego. |
|
g) |
Urządzenie filtracji membranowej (do wyboru). |
|
h) |
Analizator DOG (do wyboru). |
IV.2.2. Przygotowanie podłoża mineralnego
W celu przygotowania roztworów podstawowych zob. I.6.2.
Zmieszać 10 ml roztworu (a) z 800 ml wody rozcieńczającej, dodać 1 ml roztworu (b) do (d) i dopełnić do 1 l wodą rozcieńczającą.
IV.2.3. Przygotowanie wstępne inokulum
Inokulum można otrzymać z różnych źródeł: aktywowany osad; ścieki kanalizacyjne; wody powierzchniowe; gleby lub z mieszaniny tu wymienionych.
Zob. I.6.4., I.6.4.1, I.6.4.2 i I.6.5.
IV.2.4. Przygotowanie kolb
Przykładowo wskazano następujące objętości i wagi dla 5-litrowych kolb zawierających 3 litry zawiesiny. Jeżeli używa się mniejszych objętości, należy je odpowiednio zmodyfikować, ale należy zapewnić że utworzony ditlenek węgla zostanie zmierzony dokładnie.
Do każdej 5-litrowej kolby dodać 2 400 ml podłoża mineralnego. Dodać odpowiednią objętość przygotowanego aktywowanego osadu (zob. I.6.4.1 i I.6.5) dla otrzymania stężenia zawiesiny ciał stałych niewiększego niż 30 mg/l w końcowej 3-litrowej zaszczepionej mieszaninie. Alternatywnie, rozcieńczyć wpierw przygotowany osad do uzyskania zawiesiny 500–1 000 mg/l w podłożu mineralnym przed dodaniem podwielokrotności do zawartości 5-litrowej kolby, dla uzyskania stężenia 30 mg/l; zapewnia to większą dokładność. Można użyć innych źródeł inokulum (zob. I.6.4.2).
Napowietrzać te zaszczepione mieszaniny powietrzem wolnym od ditlenku węgla przez noc celem przepłukania systemu z ditlenku węgla.
Dodać badany materiał oraz substancje odniesienia oddzielnie, jako znaną objętość roztworów podstawowych, do replikowanych kolb dla uzyskania stężeń wnoszących udział przez dodane substancje chemiczne, 10 do 20 mg DOC lub TOC/l; pozostawić kilka kolb bez dodawania substancji chemicznych jako kontrole inokulum. Dodać badane słabo rozpuszczalne substancje bezpośrednio do kolb na podstawie masy lub objętości lub postępować jak opisano w załączniku III.
Jeżeli wymagane, użyć jedną kolbę do sprawdzenia możliwego działania inhibicyjnego badanej substancji przez dodanie zarówno badanej jak i substancji odniesienia, w tych samych stężeniach jak obecne w innych kolbach.
Także, jeżeli wymagane, użyć sterylnej kolby do sprawdzenia czy badana substancja jest degradowana abiotycznie przez użycie niezaszczepionego roztworu substancji chemicznej (zob. I.6.6). Sterylizować przez dodanie substancji toksycznej o odpowiednim stężeniu.
Dopełnić objętości zawiesin we wszystkich kolbach do 3l dodając podłoże mineralne uprzednio napowietrzone powietrzem wolnym od CO2. Alternatywnie, można pobrać próbki do analizy DOC (zob. załącznik II.4) i/lub właściwej analizy. Podłączyć butle absorpcyjne do wylotów powietrza kolb.
Jeżeli używany jest wodorotlenek baru, połączyć trzy butle absorpcyjne, każda zawierająca 100 ml 0,0125 M roztworu wodorotlenku baru, w seriach do kazdej 5-litrowej kolby. Roztwór musi być wolny od wytrąconych siarczanów oraz węglanów i jego stężenie musi być oznaczone bezpośrednio przed użyciem. Jeżeli używa się wodorotlenek sodu, podłączyć dwie pułapki, druga działa jako kontrola pokazania, że cały ditlenek węgla został zaabsorbowany w pierwszej. Butle absorpcyjne wyposażone w zamknięcia typu kroplówkowego są odpowiednie. Dodać 200 ml 0,05 M wodorotlenku sodu do każdej butli, która jest odpowiednia do zaabsorbowania całkowitej ilości ditlenku węgla wydzielonego po całkowitej degradacji badanej substancji. Roztwory wodorotlenku sodu, nawet świeżo przygotowane, mogą zawierać ślady węglanów; jest to korygowane przez odjęcie węglanów ze ślepej próby.
IV.2.5. Liczba kolb w typowym przebiegu
Kolby 1 i 2: Zawiesina badana
Kolby 3 i 4: Ślepa próba inokulum
Kolba 5: Kontrola procedury
oraz zalecane i gdy konieczne:
Kolba 6: Kontrola sterylności abiotycznej
Kolba 7: Kontrola toksyczności
Kolba 8: Kontrola toksyczności
Zob. także I.6.7.
IV.2.6. Przeprowadzenie badania
Rozpocząć badanie przez wpuszczenie pęcherzyków powietrza wolnego od ditlenku węgla przez zawiesiny z szybkością 30–100 ml/min. Pobierać próbki absorbenta ditlenku węgla okresowo do analiz na zawartość CO2. W trakcie pierwszych dziesięciu dni zalecane jest, by analizy były wykonywane co drugi lub trzeci dzień, a następnie co piąty, aż do 28 dnia, tak by okres 10-dniowego okna był zidentyfikowany.
W dniu 28 pobrać próbki (do wyboru) do analizy DOC i/lub właściwej analizy, pomierzyć pH zawiesin i dodać 1 ml stężonego kwasu solnego do każdej kolby; napowietrzać przez noc do wypędzenia ditlenku węgla obecnego w badanych zawiesinach. Dnia 29 wykonać analizę wydzielonego ditlenku węgla.
W dniu pomiaru CO2 rozłączyć absorber wodorotlenku baru najbliższy kolby i zmiareczkować roztwór wodorotlenku za pomocą HCl 0,05 M, stosując fenoloftaleinę jako wskaźnik. Przesunąć pozostałe absorbery o jedno miejsce bliżej kolby i umieścić nowy absorber zawierający 100 ml świeżego 0,0125 M wodorotlenku baru na dalszym końcu serii. Wykonać miareczkowania według potrzeby, na przykład gdy podstawowe strącenie jest widoczne w pierwszej pułapce i przed strąceniem w drugim, lub co najmniej co tydzień. Alternatywnie, z NaOH jako absorbentem, pobrać strzykawką małą próbkę (w zależności od charakterystyki stosowanego analizatora węgla) roztworu wodorotlenku sodu z absorbera najbliższego kolbie. Wstrzyknąć próbkę do części IC analizatora węgla do bezpośredniej analizy wydzielonego ditlenku węgla.
Analizować zawartość drugiej pułapki tylko na koniec badania dla skorygowania przenoszenia ditlenku węgla.
IV.3. DANE I SPORZĄDZANIE SPRAWOZDANIA
IV.3.1. Obróbka wyników
Ilość CO2 zatrzymana w absorberze przy miareczkowaniu jest dana przez:
mgCO2 = (100 × CB0,5 × V × CA) × 44
gdzie:
|
V |
= |
objętość HCl użytego do miareczkowania 100 ml w absorberze (ml), |
|
CB |
= |
stężenie roztworu wodorotlenku baru (M), |
|
CA |
= |
stężenie roztworu kwasu solnego (M), |
jeśli CB wynosi 0,0125 M i CA jest 0,05 M, miareczkowanie 100 ml wodorotlenku baru daje 50 ml, a waga CO2 jest dana przez:
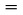
Zatem w tym przypadku dla przeliczenia objętości HCl miareczkującego na wytworzone miligramy CO2, współczynnik wynosi 1,1.
Obliczyć wagi wytworzonego CO2 z samego inokulum oraz z inokulum plus badana substancja używając odnośne wartości miareczkowania, a różnica jest wagą CO2 wytworzonego przez samą badaną substancję.
Przykładowo, jeżeli samo inokulum daje zmiareczkowane 48 ml oraz inokulum plus badana substancja daje 45 ml,
CO2 z inokulum = 1,1 × (50 – 48) = 2,2 mg
CO2 z inokulum plus badana substancja = 1,1 × (50 – 45) = 5,5 mg
Zatem waga CO2 wytworzonego z badanej substancji wynosi 3,3 mg.
Procentowa biodegradacja jest obliczana z:
![]()
lub,
![]()
3,67 istniejący współczynnik konwersji (44/12) węgla do ditlenku węgla.
Uzyskać procentową degradację po każdym przedziale czasu przez dodanie stosunków procentowych wartości ThCO2 obliczonych dla każdego z dni, do czasu w jakim zmierzono.
Dla absorberów z wodorotlenkiem sodu, obliczyć ilość wytworzonego ditlenku węgla, wyrażając jako IC (mg), przez pomnożenie stężenia IC w absorbencie przez objętość absorbenta.
Obliczyć procentową degradację z:

Obliczyć usunięcie DOC (do wyboru) jak opisano pod I.7. Zapisać te i wszystkie inne wyniki na przygotowanych arkuszach danych.
IV.3.2. Ważność wyników
Zawartość IC zawiesiny badanej substancja w podłożu mineralnym na początku badania musi być mniejsza niż 5 % wartości TC, a całkowite wydzielanie CO2 w ślepej próbie z inokulum na końcu badania nie powinna zwykle przekraczać 40 mg/l podłoża. Jeżeli uzyskuje się wartości wyższe niż 70 mg CO2/litr, dane i technika eksperymentalna muszą być krytycznie przebadane.
Zob. także I.5.2.
IV.3.3. Sporządzanie sprawozdania
Zob. I.8.
IV.4. ARKUSZ DANYCH
Przykład arkusza danych podano poniżej.
BADANIE WYDZIELANIA DITLENKU WĘGLA
|
1. |
LABORATORIUM |
|
2. |
DATA ROZPOCZĘCIA BADANIA |
3. SUBSTANCJA BADANA
Nazwa:
Stężenie roztworu podstawowego: … mg/litr jako substancja chemiczna
Wstępne stężenie. w podłożu: … mg/litr jako substancja chemiczna
Całkowity C dodany do kolby: … mg C
ThCO2: mg CO2
4. INOKULUM
Źródło:
Poddano traktowaniu:
Przygotowanie wstępne, o ile stosowano:
Stężenie zawiesiny ciał stałych w mieszaninie reakcyjnej: mg/litr
|
5. |
WYTWARZANIE DITLENKU WĘGLA I DEGRADOWALNOŚĆ Metoda: Ba(OH)/NaOH/inne |
|
Czas (dzień) |
CO2 utworzonego w badaniu (mg) |
CO2 utworzonego w ślepej próbie (mg) |
CO2 utworzony zbiorczo (mg) (badanie minus średnia ślepej próby) |
ThCO2 zbiorczo |
|||||
|
1 2 |
średnia |
3 4 |
średnia |
1 |
2 |
1 |
2 |
średnia |
|
|
0 |
|
|
|
|
|
|
|
|
|
|
n1 |
|
|
|
|
|
|
|
|
|
|
n2 |
|
|
|
|
|
|
|
|
|
|
n3 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
28 |
|
|
|
|
|
|
|
|
|
Uwaga: podobne arkusze mogą być używane dla chemicznej substancji odniesienia i kontroli toksyczności
6. ANALIZA WĘGLA (do wyboru)
Analizator węgla:
|
Czas (dzień) |
Ślepa próba mg/l |
Badana substancja chemiczna mg/l |
|
0 |
Cb(o) |
C0 |
|
28 (5) |
Cb(t) |
Ct |
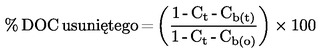
7. DEGRADACJA ABIOTYCZNA (do wyboru)

CZĘŚĆ V. BADANIE RESPIROMETRII MANOMETRYCZNEJ (Metoda C.4-D)
V.1. ZASADA METODY
Zmierzona objętość zaszczepionego podłoża mineralnego, zawierającego znane stężenie badanej substancji (100 mg/litr substancji badanej, dająca co najmniej 50–100 mg ThOD/litr) jako nominalnego pojedynczego źródła węgla organicznego, jest mieszana w zamkniętej kolbie w stałej temperaturze (± 1o C lub bliżej) do 28 dnia włącznie. Zużycie tlenu jest oznaczane albo przez pomiary ilości tlenu (wytwarzanego elektrolitycznie) wymaganego do utrzymania stałej objętości gazu w kolbie respirometru, lub przez zmianę w objętości lub ciśnieniu (lub w kombinacji dwóch) w aparacie. Wydzielany ditlenek węgla jest absorbowany w roztworze wodorotlenku potasu lub w innym odpowiednim absorbencie. Ilość tlenu pobrana przez badaną substancję (skorygowana o pobór w ślepej próbie z inokulum, biegnącej równolegle) jest wyrażana jako stosunek procentowy ThOD lub COD. Alternatywnie, pierwotna biodegradacja może być także obliczona z dodatkowych właściwych analiz wykonanych na początku i końcu inkubacji, i ostatecznie przez analizę DOC.
V.2. OPIS METODY
V.2.1. Przyrząd
|
a) |
odpowiedni respirometr; |
|
b) |
regulator temperatury, utrzymujący ± 1 oC lub lepszą; |
|
c) |
urządzenie do filtracji membranowej (do wyboru); |
|
d) |
analizator węgla (do wyboru). |
V.2.2. Przygotowanie podłoża mineralnego
W celu przygotowania roztworów podstawowych, zob. I.6.2.
Zmieszać 10 ml roztworu (a) z 800 ml wody rozcieńczającej, dodać 1 ml roztworu (b) do (d) i dopełnić do 1 litra wodą rozcieńczającą.
V.2.3. Przygotowanie i przygotowanie wstępne inokulum
Inokulum może być uzyskane z różnych źródeł: aktywowany osad; ścieki kanalizacyjne; wody powierzchniowe i gleby lub mieszanina wymienionych.
Zob. I.6.4., I.6.4.1., I.6.4.2. i I.6.5.
V.2.4. Przygotowanie kolb
Przygotować roztwory badane oraz chemicznej substancji odniesienia, w oddzielnych partiach, w podłożu mineralnym równoważnym stężeniu, zwykle 100 mg substancji chemicznej/litr (dający co najmniej 50–100 mg ThOD/litr), stosując roztwory podstawowe.
Obliczyć ThOD na podstawie tworzenia się soli amonowych, poza spodziewaną nitryfikacją, dla której należy oprzeć obliczenia na tworzeniu się azotanów (zob. załącznik II.2.)
Oznaczyć wartości pH i jeśli konieczne, ustawić na 7,4±0,2.
Substancje słabo rozpuszczalne należy dodawać na dalszym etapie (zob. poniżej).
Jeżeli ma być ustalona toksyczność badanej substancji, przygotować dalszy roztwór z podłoża mineralnego zawierającego zarówno substancję badaną jak i chemiczną substancję odniesienia o tych samych stężeniach co w poszczególnych roztworach.
Jeżeli wymagany jest pomiar fizyko-chemicznego poboru tlenu, przygotować roztwór badanej substancji zwykle 100 mg ThOD/litr który został sterylizowany dodatkiem odpowiedniej toksycznej substancji (zob. I.6.6).
Wprowadzić konieczne objętości roztworów badanej substancji i chemicznej substancji odniesienia, odpowiednio, w co najmniej zdublowanych kolbach. Dodać do następnych kolb tylko podłoże mineralne (dla kontroli inokulum) i, jeśli wymagane, zmieszane roztwory substancja badana/substancja odniesienia i roztwór sterylny.
Jeśli badana substancja chemiczna jest słabo rozpuszczalna, dodać ją bezpośrednio na niniejszym etapie na podstawie wagi lub objętości lub postępować jak opisano w dodatku III. Dodać wodorotlenek potasu, tabletki wapna sodowanego lub innego absorbenta do zbiorników absorbera CO2.
V.2.5. Liczba kolb w typowym przebiegu
Kolby 1 i 2: Zawiesina badana
Kolby 3 i 4: Ślepa próba inokulum
Kolba 5: Kontrola procedury
Zalecane i gdy konieczne:
Kolba 6: Kontrola sterylna
Kolba 7: Kontrola toksyczności
Kolba 8: Kontrola toksyczności
Zob. także I.6.7.
V.2.6. Przeprowadzenie badania
Pozwolić na uzyskanie przez naczynia żądanej temperatury i zaszczepić odpowiednie naczynia przygotowanym aktywowanym osadem lub innym źródłem inokulum by otrzymać stężenie zawiesiny ciał stałych niewiększe niż 30 mg/litr. Zestawić wyposażenie, włączyć mieszanie i sprawdzić szczelność, rozpocząć pomiar pobierania tlenu. Zwykle niewymagana jest dalsza uwaga inna niż zbieranie koniecznych danych i wykonywanie codziennych sprawdzeń czy jest utrzymywane są właściwa temperatura i stosowne mieszanie.
Obliczyć pobieranie tlenu z odczytów zbieranych w regularnych i częstych przedziałach czasowych, stosując metody podane przez producenta wyposażenia. Pod koniec inkubacji, zwykle 28-dniowej, zmierzyć pH zawartości kolb, szczególnie jeśli pobieranie tlenu jest niższe lub większe niż ThODNH4 (dla związków zawierających azot).
Jeżeli wymagane, pobrać próbki z kolb respiratora, początkowej i końcowej, do analizy DOC lub właściwej chemicznej (zob. załącznik II.4). Przy początkowym pobieraniu upewnić się, że objętość zawiesiny badanej pozostającej w kolbie jest znana. Gdy tlen zostanie pobrany przez substancję badaną zawierającą azot, oznaczyć wzrost stężenia azotynów oraz azotanów przez okres 28 dni i obliczyć poprawkę dla tlenu zużytego przez nitryfikację (załącznik V).
V.3. DANE I SPORZĄDZANIE SPRAWOZDANIA
V.3.1. Obróbka wyników
Podzielić pobór tlenu (mg) przez badaną substancję po danym czasie (skorygować o tę ze ślepej kontroli inokulum po tym samym czasie) przez wagę użytej badanej substancji. Da to BOD wyrażone jako mg tlenu/mg badanej substancji, czyli

= mg O2 na mg badanej substancji.
obliczyć procentową biodegradacje albo z:

lub postaci
![]()
Należy zauważyć że te dwie metody niekoniecznie muszą dawać te same wartości; korzystniejsze jest użycie pierwszej metody.
Dla badanych substancji zawierających azot stosuje się odpowiednie ThOD (NH4 lub N03) zgodnie z tym co jest znane lub oczekiwane o zajściu nitryfikacji (załącznik II.2). Jeżeli nitryfikacja zachodzi, lecz nie jest całkowita, obliczyć poprawkę dla tlenu zużytego przez nitryfikację ze zmian w stężeniu azotynów i azotanów (załącznik V).
Gdy wykonano alternatywne oznaczenia węgla organicznego i/lub właściwej substancji chemicznej, obliczyć procentową degradację, jak opisano pod I.7.
Zapisać wszystkie wyniki na dołączonych arkuszach danych.
V.3.2. Ważność wyników
Pobieranie tlenu w ślepej próbie z inokulum jest zwykle 20–30 mg O2/litr i powinno być niewiększe niż 60 mg/litr w ciągu 28 dni. Wartości wyższe od 60 mg/litr wymagają krytycznego sprawdzenia danych i technik eksperymentalnych. Jeżeli wartość pH jest poza zakresem 6–8,5, a zużycie tlenu przez badaną substancję jest mniejsze niż 60 %, należy powtórzyć badanie z niższym stężeniem badanej substancji.
Zob. także 1.5.2.
V.3.3. Sporządzanie sprawozdania
Zob. 1.8.
V.4. ARKUSZ DANYCH
Przykład arkusza danych podano poniżej.
BADANIE RESPIROMETRII MANOMETRYCZNEJ
|
1. |
LABORATORIUM |
|
2. |
DATA ROZPOCZĘCIA BADANIA |
3. SUBSTANCJA BADANA
Nazwa:
Stężenie roztworu podstawowego: … mg/litr
Początkowe stężenie w podłożu, C : … mg/litr
Objętość w kolbie do badań (V): ml
ThOD lub COD: mg O2/mg substancji badanej (NH4, N03)
4. INOKULUM
Źródło:
Poddano traktowaniu:
Wstępne sezonowanie, o ile wykonano:
Stężenie zawiesiny ciał stałych w mieszaninie reakcyjnej: … mg/l
5. POBÓR TLENU: BIODEGRADOWALNOŚĆ
|
|
Czas (dni) |
||||||||||||
|
0 |
|
7 |
|
14 |
|
|
21 |
|
|
28 |
|
||
|
O2 pobór (mg) badana substancja |
1 |
|
|
|
|
|
|
|
|
|
|
|
|
|
2 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
a, średnia |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
O2 pobór (mg) |
3 |
|
|
|
|
|
|
|
|
|
|
|
|
|
4 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
b, średnia |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
skorygowane BOD (mg) |
(a1- bm) |
|
|
|
|
|
|
|
|
|
|
|
|
|
(a2- bm) |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
BOD na mg badanej substancji |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
% degradacja
|
D1 (a1) |
|
|
|
|
|
|
|
|
|
|
|
|
|
D2 (a2) |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
średnia (6) |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
V = objętość podłoża w kolbie do badań |
|||||||||||||
Uwaga: Podobne arkusze używać można do chemicznej substancji odniesienia i kontroli toksyczności
6. POPRAWKA DLA NITRYFIKACJI (zob. załącznik V)
|
Dzien |
0 |
28 |
Roznica |
||
|
|
|
(N) |
||
|
— |
— |
|
||
|
|
|
(N) |
||
|
— |
— |
|
||
|
— |
— |
|
7. ANALIZA WĘGLA (alternatywna)
Analizator węgla:
|
Czas (dzień) |
Ślepa proba mg/litr |
Badana substancja chemiczna mg/litr |
|
0 |
(Cblo) |
(C0) |
|
28 (7) |
(Cblt) |
(Ct) |

8. WŁASCIWA CHEMICZNA (alternatywna)
|
Sb |
= |
stężenie w fizyko-chemicznej (sterylnej) kontroli w 28 dniu |
|
Sa |
= |
stężenie w zaszczepionej kolbie w 28 dniu |
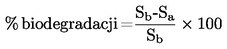
9. DEGRADACJA ABIOTYCZNA (alternatywna)
|
a |
= |
zużycie tlenu w sterylnej kolbie po 28 dniach, (mg) |

(zob. sekcje 1 i 3)

CZĘŚĆ VI. BADANIE „ZAMKNIĘTEJ BUTLI”(Metoda C.4-E)
VI. 1. ZASADA METODY BADANIA
Roztwór badanej substancji w podłożu mineralnym, zwykle 2–5 mg/litr, jest zaszczepiany relatywnie małą liczbą drobnoustrojów ze zmieszanej populacji i trzymany w całkowicie pełnej, zamkniętej butli w ciemności w stałej temperaturze. Degradacja jest oznaczana przez analizę rozpuszczonego tlenu przez okres czasu 28 dni. Ilość tlenu pobrana przez badaną substancję, skorygowany o pobór w równoległej ślepej próbie z inokulum, jest wyrażona jako stosunek procentowy ThOD lub COD.
VI.2. OPIS METODY
VI.2.1. Przyrząd
|
a) |
Butle BOD, ze szklanymi korkami, np. 250–300 ml. |
|
b) |
Łaźnia wodna lub inkubator, dla trzymania butli w stałej temperaturze (± 1 oC lub lepiej) przy wyłączonym świetle. |
|
c) |
Duże szklane butle (2–5 litrów) dla przygotowania podłoża i napełniania butli BOD. |
|
d) |
Elektroda tlenowa i miernik, lub wyposażenie oraz odczynniki dla miareczkowania Winklera. |
VI.2.2. Przygotowanie podłoża mineralnego
W celu przygotowania roztworów podstawowych zob. 1.6.2.
Zmieszać 1 (jeden) ml roztworu (a) z (d) i dopełnić do 1 litra wodą rozcieńczającą.
VI.2.3. Przygotowanie inokulum
Inokulum jest zwykle uzyskiwane ze ścieku drugiego stopnia oczyszczalni ścieków lub urządzenia skali laboratoryjnej przyjmującego głównie ścieki domowe. Alternatywnym źródłem inokulum są wody powierzchniowe. Zwykle używa się od jednej kropli (0,05 ml) do 5 ml filtratu na litr podłoża; konieczne mogą być próby dla znalezienia optymalnej objętości danego wycieku (zob. I.6.4.2 i I.6.5).
VI.2.4. Przygotowanie kolb
Silnie napowietrzyć podłoże mineralne na co najmniej 20 min. Przeprowadzić każdą serię badań z podłożem mineralnym pochodzącym z tej samej partii. Ogólnie, podłoże jest gotowe do użycia po odstaniu 20 h, w temperaturze badania. Oznaczyć stężenie rozpuszczonego tlenu do celów kontrolnych; wartość powinna wynosić około 9 mg/litr w 20 oC. Prowadzić wszystkie przenoszenia i operacje napełniania nasyconym podłożem wolnym od pęcherzyków, na przykład poprzez użycie syfonów.
Przygotować równoległe grupy butli BOD dla oznaczeń badanej i chemicznej substancji odniesienia w równoczesnych seriach doświadczalnych. Zestawić wystarczającą liczbę butli BOD, włączając ślepe próby inokulum, wykonać co najmniej zdublowane pomiary zużycia tlenu w pożądanych odstępach badawczych, na przykład po 0, 7, 14, 21 i 28 dniach. Dla zapewnienia zdolności identyfikacji 10-dniowego okna wymagana jest większa ilość butli.
Dodać całkowicie napowietrzone podłoże mineralne do dużych butli, tak by były wypełnione w około jednej trzeciej. Następnie dodać wystarczającą ilość roztworu podstawowego badanej substancji i chemicznej substancji odniesienia do oddzielnych dużych butli tak by końcowe stężenie substancji chemicznej zwykle było niewiększe niż 10 mg/litr. Nie dodawać substancji chemicznej do podłoża kontrolnego ślepej próby zawartego w następnej dużej butli.
W celu zapewnienia, że aktywność inokulum nie jest ograniczona, stężenie rozpuszczonego tlenu nie może być poniżej 0,5 mg/litr w butli BOD. To ogranicza stężenie badanej substancji do około 2 mg/litr. Jednakże dla słabo rozpuszczalnych związków i tych o niskim ThOD można użyć 5–10 mg/litr. W kilku przypadkach jest zalecane przeprowadzenie równoległych serii badań badanej substancji o dwu różnych stężeniach na przykład, 2 i 5 mg/litr. Zwykle oblicza się ThOD na podstawie tworzenia się soli amonowych, lecz jeżeli oczekiwana lub znana jest fakt, że może zajść nitryfikacja, oblicza się go na podstawie tworzenia azotanu (ThODNO3: zob. załącznik II.2). Jednakże jeśli nitryfikacja nie jest całkowita, ale zaszła, należy skorygować zmiany w stężeniu azotynu i azotanu, oznaczone analitycznie (zob. załącznik V).
Jeżeli jest do zbadania toksyczność badanej substancji (w przypadku, na przykład, gdy znaleziono wcześniejsze wartości niskiej biodegradowalności), jest konieczna inna seria butli.
Przygotować inna dużą butlę do pomieszczenia napowietrzonego podłoża mineralnego (do około jednej trzeciej jej objętości) plus badana substancja i chemiczna substancja odniesienia o końcowych stężeniach zwykle takich samych jak w tych z innych dużych butli.
Zaszczepić roztwory w dużych butlach ściekiem drugiego stopnia (jedną kroplą (0,05 ml), lub do 5 ml/litr)) lub z innego źródła, na przykład wody rzecznej (zob. I.6.4.2). Na koniec dopełnić roztwory do objętości napowietrzonym podłożem mineralnym stosując wąż wpuszczony na dno butli dla uzyskania odpowiedniego mieszania.
VI.2.5. Liczba kolb w typowym przebiegu
W typowym przebiegu stosuje się następujące butle:
|
— |
co najmniej 10 zawierających badaną substancję i inokulum (zawiesina badana), |
|
— |
co najmniej 10 zawierających tylko inokulum (ślepa próba z inokulum), |
|
— |
co najmniej 10 zawierających chemiczna substancja odniesienia i inokulum (procedura kontroli), |
|
— |
i, gdy konieczne, 6 butli zawierających badaną substancję, chemiczną substancję odniesienia i inokulum (kontrola toksyczności). Jednakże dla zapewnienia zdolności identyfikacji 10-dniowego okna konieczna może być około dwukrotnie większa ilość butli. |
VI.2.6. Przeprowadzenie badania
Rozdzielić przygotowany roztwór niezwłocznie do odpowiednich grup butli BOD za pomocą węża z dolnej ćwiartki (nie z dna) do odpowiednio dużych butli, tak aby wszystkie butle BOD zostały całkowicie napełnione. Postukać delikatnie, by usunąć wszystkie pęcherzyki powietrza. Analizować butle o zerowym czasie niezwłocznie na zawartość rozpuszczonego tlenu metodą Winkler lub elektrodowo. Zawartość butli może być zabezpieczona dla późniejszej analizy metodą Winklera przez dodanie siarczanu manganu (II) i wodorotlenku sodu (pierwszy odczynnik Winklera). Przechowywać w starannie zamkniętych, zawierających związany tlen jako brązowy uwodniony tlenek manganu (III), w ciemności w temperaturze 10–20 oC przez nie dłużej niż 24 godziny przed wykonaniem pozostałych kroków metody Winklera. Zakorkować pozostałe replikowane butle, upewniając się, że nie zawierają pęcherzyków powietrza, i inkubować w 20 oC w ciemności. Każdej serii musi towarzyszyć całkowita seria równoległa z zaszczepionymi podłożami ślepej próby. Wyjąć co najmniej zdublowane butle wszystkich serii w celu analizy rozpuszczonego tlenu w odstępach czasu (co najmniej cotygodniowo) przez 28 dni inkubacji.
Cotygodniowe pobieranie próbek powinno umożliwić ocenę stosunku procentowego usunięcia w 14-dniowym oknie, przy czym pobieranie próbek co 3–4 dni powinno umożliwić identyfikację 10-dniowego okna, wymaganego około podwójnie co większość butli.
Dla substancji badanych zawierających azot należy wykonać korekty poboru tlenu przez każdą zachodzącą nitryfikację. By to wykonać, użyć metody O2-elektrody dla oznaczania stężenia rozpuszczonego tlenu i następnie pobrać próbkę z butli BOD do analizy na azotyny i azotany. Ze wzrostu stężenia azotynów i azotanów, obliczyć użyty tlen (zob. załącznik V).
VI. 3. DANE I SPORZĄDZANIE SPRAWOZDANIA
VI.3.1. Obróbka wyników
Wpierw obliczyć BOD użyte po każdym okresie czasu przez odejmowanie zubożenia tlenu (mg/litr) ślepej próby z inokulum od tej wykazanej przez badaną substancję. Podzielić to poprawione zubożenie przez stężenie(mg/litr) badanej substancji, aby uzyskać właściwe BOD jako mg tlenu na mg badanej substancji. Obliczyć procentową biodegradowalność przez podzielenie właściwego BOD przez właściwe ThOD (obliczonego zgodnie z załącznikiem II.2) lub COD (oznaczonego przez analizę, zob. załącznik II.3), zatem:

= mg O2/mg badana substancja
![]()
lub
![]()
Należy zauważyć, że te dwie metody niekoniecznie dają te same wartości; korzystne jest użycie pierwszej metody.
Dla badanych substancji zawierających azot użyć odpowiednie ThOD (NH4 lub N03) zgodnie z tym, co jest znane lub oczekiwane o występowaniu nitryfikacji (załącznik II.2). Jeżeli nitryfikacja zachodzi lecz nie jest całkowita, obliczyć poprawkę dla tlenu zużytego przez nitryfikację ze zmiany w stężeniu azotynów i azotanów (załącznik V).
VI.3.2. Ważność wyników
Zubożenie tlenu w ślepej próbie z inokulum nie powinno przekraczać 1,5 mg rozpuszczonego tlenu/litr po 28 dniach. Wyższe wartości wymagają prześledzenia techniki eksperymentalnej. Resztkowe stężenie tlenu w badanych butlach nie powinno nigdy spaść poniżej 0,5 mg/litr. Takie niskie poziomy tlenu są ważne, gdy stosowana metoda oznaczania rozpuszczonego tlenu jest zdolna do dokładnego pomiaru takich poziomów.
Zob. także I.5.2.
VI.3.3. Sporządzanie sprawozdania
Zob. I.8.
VI.4. ARKUSZ DANYCH
Przykład arkusza danych podano poniżej.
BADANIE METODĄ „ZAMKNIĘTEJ BUTLI”
|
1. |
LABORATORIUM |
|
2. |
DATA ROZPOCZĘCIA BADANIA |
3. SUBSTANCJA BADANA
Nazwa:
Stężenie roztworu podstawowego: … mg/litr
Początkowe stężenie w butli: … mg/litr
ThOD lub COD: … mg O2/mg substancji badanej
4. INOKULUM
Źródło:
Poddano traktowaniu:
Przygotowanie wstępne, o ile stosowano:
Stężenie w mieszaninie reakcyjnej: … mg/litr
5. OZNACZANIE DO
Metoda: Winklera/elektrodowa
Analizy kolb
|
Czas inkubacji (d) |
DO mg/l) |
|||||
|
0 |
n1 |
n2 |
|
|||
|
Ślepa próba -(bez substancji chemicznej |
1 |
C1 |
|
|
|
|
|
2 |
C2 |
|
|
|
|
|
|
Średnia |
|
|
|
|
|
|
|
Badana substancja chemiczna |
1 |
a1 |
|
|
|
|
|
|
2 |
a2 |
|
|
|
|
|
Średnia |
|
|
|
|
|
|
Uwaga: Podobny arkusz może być stosowany dla substancji odniesienia i kontroli toksyczności.
6. POPRAWKA DLA NITRYFIKACJI (zob. załącznik V)
|
Czas inkubacji (d) |
0 |
n1 |
n2 |
n3 |
||
|
|
|
|
|
||
|
— |
|
|
|
||
|
— |
|
|
|
||
|
|
|
|
|
||
|
— |
|
|
|
||
|
— |
|
|
|
||
|
— |
|
|
|
7. ZUBOŻENIE DO: % DEGRADACJA
|
|
Zubożenie po n dniach (mg/litr) |
|||
|
n1 |
n2 |
n3 |
|
|
|
KOLBA 1: (mto- mtx) – (mbo – mbx) |
|
|
|
|
|
KOLBA 2: (mto- mtx) – (mbo- mbx) |
|
|
|
|
|
KOLBA 1:
|
|
|
|
|
|
KOLBA 2:
|
|
|
|
|
| % D średnia (8) =
|
|
|
|
|
|
mto |
= |
wartość w badanej kolbie w czasie 0 |
|
mtx |
= |
wartość w badanej kolbie w czasie x |
|
mbo |
= |
średnia wartość ślepej próby w czasie 0 |
|
mbx |
= |
średnia wartość ślepej próby w czasie x |
Zastosować także poprawkę dla nitryfikacji z (iii + vi) w sekcji 6.
8. ZUBOŻENIA DO W ŚLEPYCH PRÓBACH
Zużycie tlenu przez ślepą próbę: (mbo – mb28) mg/litr. Niniejsze zużycie jest istotne dla ważności naukowej badania. Powinno być mniejsze niż 1,5 mg/litr.
CZĘŚĆ VII. BADANIE M.I.T.I. (Metoda C.4-F)
VII.1. ZASADA METODY
Pobieranie tlenu trakcie mieszania roztworu lub zawiesiny badanej substancji w podłożu mineralnym, zaszczepionego specjalnie hodowanymi, nieadoptowanymi drobnoustrojami, jest mierzone automatycznie w ciągu okresu 28 dni w zaciemnionym, zamkniętym respirometrze w 25 ± 1 oC. Wydzielany ditlenek węgla jest absorbowany przez wapno sodowane. Biodegradowalność jest wyrażana jako procent pobieranego tlenu (skorygowany dla poboru w ślepej próbie) poboru teoretycznego (ThOD). Procent pierwotnej biodegradowalności jest również obliczany z dodatkowej właściwej analizy chemicznej wykonywanej na początku i pod koniec inkubacji oraz, alternatywnie, przez analizę DOC.
VII.2. OPIS METODY
VII.2.1. Przyrząd
|
a) |
Automatyczny elektrolityczny miernik BOD respirometr zwykle wyposażony w 6 butli, każda 300 ml i zaopatrzony w kubki na substancję absorbującą CO2. |
|
b) |
Stała temperatura pokojowa i/lub łaźnia wodna 25 oC ± 1 oC lub lepszej. |
|
c) |
Zespół filtracji membranowej (alternatywny). |
|
d) |
Analizator węgla (alternatywny). |
VII.2.2. Przygotowanie podłoża mineralnego
Przygotować następujące roztwory podstawowe, stosując odczynniki czystości analitycznej i wodę (I.6.1.):
|
a) |
Diwodoroortofosforan potasu, KH2P04 |
8,50 g |
|
monowodoroortofosforan dipotasu, K2HP04 |
21,75 g |
|
|
monowodoroortofosforan disodu, dodekawodny Na2HP04 12 H20 |
44,60 g |
|
|
chlorek amonu, NH4Cl |
1,70 g |
|
|
Rozpuścić w wodzie i dopełnić do 1 litra. |
|
|
|
Wartośc pH roztworu powinna wynosić 7,2. |
|
|
|
b) |
Siarczan magnezu, heptawodny MgSO4 7 H20 |
22,50 g |
|
Rozpuścić w wodzie i dopełnić do 1 litra. |
|
|
|
c) |
Chlorek wapnia bezwodny, CaCl2 |
27,50 g |
|
Rozpuścić w wodzie i dopełnić do 1 litra. |
|
|
|
d) |
Chlorek żelaza (III), heksawodny FeCl3 6 H20 |
27,50 g |
|
Rozpuścić w wodzie i dopełnić do 1 litra. |
|
Pobrać 3 ml każdego roztworu (a), (b), (c) i (d) i dopełnić do 1 litra.
VII.2.3. Przygotowanie inokulum
Zebrać świeże próbki z nie więcej niż dziesięciu miejsc, głównie z obszarów, gdzie stosowana i unieszkodliwiana jest duża rozmaitość substancji chemicznych. Z takich miejsc, jak oczyszczalnie ścieków, oczyszczalnie ścieków wodnych przemysłowych, rzek, jezior, mórz, zebrać 1 litrowe próbki osadu, gleby powierzchniowej, wody itp. i dokładnie wymieszać. Po usunięciu ciał znajdujących się na powierzchni i odstawieniu ustawić pH nadsączu na 7 ± 1 za pomocą wodorotlenku sodu lub kwasu fosforowego.
Użyć odpowiednią objętość odfiltrowanego nadsączu napełnienia zbiornika, służącego do napełniania i wylewania aktywowanego osadu, i napowietrzać roztwór około 23 1/2 godziny. Trzydzieści minut po wstrzymaniu napowietrzania odrzucić około jednej trzeciej całej objętości nadsączu i dodać równą objętość roztworu (pH 7) zawierającego po 0,1 % glukozy, peptonu i ortofosforanu potasu w celu zsedymentowania materiału i rozpocząć ponowne napowietrzanie. Powtarzać niniejszą procedurę raz dziennie. Zbiornik z osadem należy obsługiwać zgodnie z dobrą praktyką: ciecz musi być klarowna, temperaturę należy utrzymywać w 25 ± 2 oC, pH powinno wynosić 7 ± 1, osad należy prawidłowo osadzać, należy utrzymywać wystarczające napowietrzanie mieszaniny aerobowej przez cały czas, powinny być obecne pierwotniaki a aktywność osadu należy badać względem substancji odniesienia co najmniej raz na trzy miesiące. Nie używać osadu jako inokulum, przed upływem co najmniej miesiąca jego utrzymywania i po okresie dłuższym od czterech miesięcy. Następnie pobierać próbki z co najmniej 10 miejsc w regularnych odstępach czasu, raz na trzy miesiące.
W celu utrzymania świeżego i starego osadu w tej samej aktywności zmieszać odfiltrowany nadsącz aktywowanego bieżąco używanego osadu z równą objętością odfiltrowanego nadsączu z mieszaniny zebranej z dziesięciu źródeł i prowadzić kulturę połączonej cieczy, jak powyżej. Pobrać osad do użycia jako inokulum 18–24 godzin po zasileniu zbiornika.
VII.2.4. Przygotowanie kolb
Przygotować następujące sześć kolb:
Nr 1: badana substancja w wodzie rozcieńczającej w 100 mg/l
Nr 2, 3 i 4: badana substancja w podłożu mineralnym w 100 mg/l
Nr 5: chemiczna substancja odniesienia (np. anilina) w podłożu mineralnym w 100 mg/l
Nr 6: tylko podłoże mineralne
Dodać słabo rozpuszczalną badaną substancję bezpośrednio na podstawie wagi lub objętości lub postępować jak opisano w załączniku III, poza przypadkiem, gdy nie należy używać ani rozpuszczalników, ani środków emulsyfikujących. Dodać absorbent CO2 do wszystkich kolb w przygotowanych specjalnych kubkach. Ustawić pH w kolbach nr 2, 3 i 4 na 7,0.
VII.2.5. Przeprowadzenie badania
Zaszczepić kolby nr 2, 3 i 4 (zawiesina badana), nr 5 (kontrola aktywności) i nr 6 (inokulum ślepa próba) małą objętością inokulum, by uzyskać stężenie 30 mg/l zawiesiny ciał stałych. Niedodawane jest inokulum do kolby nr 1, która służy jako kontrola abiotyczna. Zestawić wyposażenie, sprawdzić szczelność, włączyć mieszadła, i rozpocząć pomiar pobierania tlenu w warunkach ciemności. Codziennie sprawdzać temperaturę, mieszadła i rejestrator kulometrycznego pobierania tlenu, notować wszystkie zmiany w kolorze zawartości kolb. Bezpośrednio odczytywać pobieranie tlenu w sześciu kolbach odpowiednią metodą, na przykład z sześciopunktowego rejestratora drukującego krzywe BOD. Na koniec inkubacji, zwykle 28 dni, zmierzyć pH zawartości kolb i oznaczyć stężenie pozostałości badanej substancji i wszystkich pośrednich, a w przypadku substancji rozpuszczalnych w wodzie, stężenie DOC (załącznik II.4). Zwrócić szczególną uwagę w przypadku lotnych substancji chemicznych. Jeżeli przewidywana jest nitryfikacja, oznaczyć jeżeli możliwe, stężenie azotynów i azotanów.
VII. 3. DANE I SPORZĄDZANIE SPRAWOZDANIA
VII.3.1. Obróbka wyników
Podzielić ilość pobranego tlenu (mg) przez badaną substancję po danym czasie (skorygować o wartość pobraną w ślepej próbie kontroli inokulum po tym samym czasie), przez wagę użytej badanej substancji. Daje to BOD wyrażone jako mg tlenu/mg badana substancja, czyli:

= mg 02/mg badana substancja.
Procentowa biodegradacja jest zatem otrzymywana z:

Dla mieszanin obliczyć ThOD z analizy składników jak dla pojedynczych związków. Użyć odpowiednie ThOD (ThODNH4 lub ThODN03) w zależności, czy nitryfikacja jest obecna czy ukończona (załącznik II.2). Jeżeli jednakże nitryfikacja zachodzi, ale nie jest całkowita, wykonać poprawkę dla tlenu zużytego w nitryfikacji, wyliczonego ze zmian stężeń azotynów i azotanów (załącznik V).
Obliczyć procentową pierwotna biodegradację ze straty właściwej macierzystej substancji chemicznej (zob. 1.7.2).

Jeżeli zaszła strata badanej substancji w kolbie nr 1 mierzącej fizyko-chemiczne usunięcie, zarejestrować to i użyć stężenia badanej substancji (Sb) po 28 dniach w tej kolbie dla obliczenia procentowej biodegradacji.
Jeżeli wykonano oznaczenia DOC (alternatywne), obliczyć ostateczną procentową biodegradację z:

jak opisano w pkt 1.7.1. Jeśli nie wystąpiła strata DOC w kolbie nr 1, mierzącej fizyko-chemiczne usunięcie, użyć stężenia DOC w niniejsze kolbie do obliczenia procentowej biodegradacji.
Zapisać wszystkie wyniki na dołączonych arkuszach danych.
VII.3.2. Ważność wyników
Pobieranie tlenu w ślepej próbie z inokulum jest zwykle 20–30 mg O2/l i powinno być nie większe niż 60 mg/l w ciągu 28 dni. Wartości wyższe od 60 mg/l wymagają krytycznego sprawdzenia danych i technik eksperymentalnych. Jeżeli wartość pH jest poza zakresem 6–8,5, a zużycie tlenu przez badaną substancję jest mniejsze niż 60 %, należy powtórzyć badanie z niższym stężeniem badanej substancji.
Zob. także I.5.2.
Jeżeli procentowa degradacja aniliny obliczona ze zużycia tlenu nie przekracza 40 % po 7 dniach i 65 % po 14 dniach, badanie uważane jest za nieważne.
VII.3.3. Sporządzanie sprawozdania
Zob.: I.8.
VII.4. ARKUSZ DANYCH
Przykład arkusza danych podano poniżej.
BADANIE MITI (I)
|
1. |
LABORATORIUM |
|
2. |
DATA ROZPOCZĘCIA BADANIA |
3. SUBSTANCJA BADANA
Nazwa:
Stężenie roztworu podstawowego: … mg/l jako substancja chemiczna
Początkowe stężenie w podłożu, CO: … mg/l jako substancja chemiczna
Objętość mieszaniny reakcyjnej, V: … ml
ThOD: … mg O2/l
4. INOKULUM
Miejsca pobierania osadu kanalizacyjnego:
|
|
||||
|
|
||||
|
|
||||
|
|
||||
|
|
Stężenie zawiesiny ciał stałych w aktywowanym osadzie po sezonowaniu z syntetycznym ściekiem = ... mg/l
Objętość aktywowanego osad na litr końcowego podłoża = ... ml
Stężenie osad końcowym podłożu = ... mg/l
5. POBÓR TLENU: BIODEGRADOWALNOŚĆ
Typ używanego respirometru:
|
|
Czas (dni) |
||||||
|
0 |
7 |
14 |
21 |
28 |
|||
|
O2 pobrany (mg) badana substancja |
a1 |
|
|
|
|
|
|
|
a2 |
|
|
|
|
|
||
|
a3 |
|
|
|
|
|
||
|
O2 pobrany (mg) ślepa proba |
b |
|
|
|
|
|
|
|
Poprawka pobranego O2 (mg) |
(a1-b) (a2-b) (a3-b) |
|
|
|
|
|
|
|
BOD na mg badanej substancji |
|
Kolba 1 |
|
|
|
|
|
|
Kolba 2 |
|
|
|
|
|
||
|
Kolba 3 |
|
|
|
|
|
||
|
% degradacja
|
|
1 |
|
|
|
|
|
|
2 |
|
|
|
|
|
||
|
3 |
|
|
|
|
|
||
|
srednia (9) |
|
|
|
|
|
||
Uwaga: podobny arkusz można używać dla substancji odniesienia.
6. ANALIZA WĘGLA (alternatywna)
Analizator węgla:
|
Kolba |
DOC |
% DOC usunięte |
Średnia |
|||
|
zmierzone |
skorygowane |
|||||
|
Woda + substancja badana |
a |
|
|
|
— |
— |
|
Osad + substancja badana |
b1 |
|
b1-c |
|
|
|
|
Osad + substancja badana |
b2 |
|
b2-c |
|
|
|
|
Osad + substancja badana |
b3 |
|
b3-c |
|
|
|
|
Ślepa próba kontrolna |
c |
|
— |
|
— |
— |
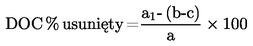
7. WŁAŚCIWE CHEMICZNE DANE ANALITYCZNE
|
|
Pozostałość badanej substancji na koniec badania |
% degradacja |
|
ślepa próba z wodą |
Sb |
|
|
zaszczepione podłoze |
Sa1 |
|
|
Sa2 |
|
|
|
Sa3 |
|

Obliczyć % degradacji odpowiednio dla kolby al i a3
8. UWAGI
Należy dołączyć, gdy dostępna, krzywą BOD w zależności od czasu.
ZAŁĄCZNIK I
SKRÓTY I DEFINICJE
|
DO |
: |
Rozpuszczony tlen (mg/l) jest stężeniem tlenu rozpuszczonego w wodnej próbce. |
|
BOD |
: |
Zapotrzebowanie biochemiczne na tlen (g) jest ilością tlenu zużytego przez drobnoustroje metabolizujące badany związek; także wyrażane jako gramy pobieranego tlenu na gramy badanego związku (zob. metoda C.5). |
|
COD |
: |
Zapotrzebowanie chemiczne na tlen (g) jest ilością tlenu zużytego w czasie trwania utleniania badanego związku gorącym zakwaszonym dichromianem; dostarcza pomiaru ilości obecnych utlenialnych ciał; także wyrażane jako gramy tlenu zużytego na gramy badanego związku (zob. metoda C.6). |
|
DOC |
: |
Rozpuszczalny węgiel organiczny jest to węgiel organiczny obecny w roztworze, lub ten przechodzący przez filtr 0,45 mikrona lub pozostaje w nadsączu po odwirowaniu przy 40 000 m/s2 (±4 000 g) dla 15 min. |
|
ThOD |
: |
Teoretyczne zapotrzebowanie na tlen (mg) jest całkowitą ilością tlenu wymaganą do całkowitego chemicznego utlenienia; jest obliczane ze wzoru cząsteczkowego (zob. załącznik II.2) i jest także wyrażane jako mg wymaganego tlenu na mg badanego związku. |
|
ThCO2 |
: |
Teoretyczny ditlenek węgla (mg) jest ilością ditlenku węgla obliczoną jako wytworzoną ze znanej lub zmierzonej zawartości węgla związku badanego, przy pełnej mineralizacji; także wyrażany jako ilość mg ditlenku węgla wydzielanego na mg badanego związku. |
|
TOC |
: |
Całkowity węgiel organiczny próbki jest sumą węgla organicznego w roztworze i w zawiesinie. |
|
IC |
: |
Węgiel nieorganiczny. |
|
TC |
: |
Całkowity węgiel, jest sumą węgla organicznego i nieorganicznego obecną w próbce. |
Pierwotna biodegradacja:
jest zmianą w chemicznej strukturze substancji, spowodowaną działaniem biologicznym, powodującą utratę specyficznych właściwości tej substancji.
Ostateczna biodegradacja (aerobowa):
jest uzyskanym poziomem degradacji, gdy badany związek zostaje całkowicie rozłożony przez drobnoustroje, objawiający się wytwarzaniem ditlenku węgla, wody, soli mineralnych i nowych mikrobowych składowych komórkowych (biomasy).
Łatwo biodegradowalne:
umowna klasyfikacja substancji chemicznych przechodzących pewne specyficzne badania przesiewowe na ostateczną biodegradowalność; te badania są tak zaostrzone, że można stwierdzić, że takie związki ulegną szybko i całkowicie biodegradacji w środowiskach wodnych w warunkach aerobowych.
Pierwotnie biodegradowalny:
klasyfikacja substancji chemicznych dla których istnieją niedwuznaczne dowody biodegradacji (pierwotnej lub ostatecznej) we wszystkich naukowo uznanych badaniach biodegradowalności.
Podatność na obróbkę:
jest uległością związków do usuwania w czasie trwania biologicznej obróbki ścieków bez zakłócania zwykłego działania procesów przetwarzania. Ogólnie łatwo biodegradowalne związki są podatne na obróbkę, lecz nie wszystkie o pierwotnej biodegradowalności. Abiotyczne procesy mogą także powodować działanie.
Czas opóźnienia:
jest czasem od inokulacji, w badaniu „die-away”, do momentu aż procent degradacji wzrośnie co najmniej 10 %. Czas opóźnienia jest często bardzo zmienny i słabo odtwarzalny:
Czas degradacji:
jest czasem od końca czasu opóźnienia do czasu uzyskania 90 % maksymalnego poziomu degradacji.
10- dniowe okno:
jest 10 dniami bezpośrednio następującymi po osiągnięciu 10 % degradacji.
ZAŁĄCZNIK II
OBLICZENIE I OZNACZENIE ODPOWIEDNICH PARAMETRÓW PODSUMOWUJĄCYCH
W zależności od wybranej metody, mogą być wymagane pewne parametry podsumowujące. Następująca sekcja opisuje wyprowadzenie tych wartości. Użycie tych parametrów jest opisane w poszczególnych metodach.
1. Zawartość węgla
Zawartość węgla jest obliczana ze znanego składu pierwiastkowego lub oznaczana poprzez analizę pierwiastkową substancji badanej.
2. Teoretyczne zapotrzebowanie tlenu (ThOD)
Teoretyczne zapotrzebowanie tlenu (ThOD) może być obliczone, jeżeli znany jest skład pierwiastkowy lub jest wyznaczony przez analizę elementarną. Dla związku:
CcHhClclNnNanaOoPpSs
lub z nitryfikacją,
 mg/mg
mg/mg
bez nitryfikacji,
 mg/mg
mg/mg
3. Zapotrzebowanie chemiczne na tlen (COD)
Zapotrzebowanie chemiczne na tlen (COD) jest oznaczane zgodnie z metodą C.6.
4. Rozpuszczalny węgiel organiczny (DOC)
Rozpuszczalny węgiel organiczny (DOC) jest według definicji węglem organicznym wszystkich substancji chemicznych i mieszanin w wodzie przechodzących przez filtr 0,45 mikrona.
Próbki z naczyń badawczych są pobierane i niezwłocznie filtrowane w aparacie filtracyjnym, stosując odpowiedni filtr membranowy. Pierwsze 20 ml (można zmniejszyć ilość, stosując mniejsze filtry) filtratu jest odrzucane. Objętości 10–20 ml lub mniej, jeżeli wstrzykuje się (objętość zależy od ilości wymaganej dla analizatora węgla), są poddawane analizie na węgiel. Stężenie DOC jest oznaczane za pomocą analizatora węgla organicznego zdolnego do dokładnych pomiarów stężenia węgla równoważnego lub niższego niż 10 % początkowego stężenia DOC użytego w badaniu.
Odfiltrowane próbki, które nie mogą być analizowane tego samego dnia, można zabezpieczyć przez przechowanie w lodówce w 2–4 oC przez 48 godz. lub niższej - 18 oC przez dłuższy czas.
Uwagi;
Filtry membranowe są często impregnowane środkami powierzchniowoczynnymi w celu hydrofilizacji. Zatem filtr może zawierać do kilku mg rozpuszczalnego węgla organicznego, który będzie zakłócał oznaczenia biodegradowalności. Usuwa się związki powierzchniowoczynne i inne rozpuszczalne związki organiczne z filtrów przez gotowanie ich w wodzie dejonizowanej przez trzy okresy czasu po jednej godzinie każdy. Filtry następnie można przechowywać w wodzie przez tydzień. Jeżeli wkłady filtrów jednorazowych są używane, każda partia musi być sprawdzona dla potwierdzenia ze nie zawiera rozpuszczalnego węgla organicznego.
W zależności od rodzaju filtra membranowego badana substancja może być zatrzymywana przez adsorpcję. Jest zatem godne polecenia upewnienie się czy badana substancja nie jest zatrzymywana przez filtr.
Odwirowanie w 40 000 m/sec2 (4 000 g) przez 15 min można stosować do zróżnicowania TOC względem DOC zamiast filtracji. Metoda nie jest pewna przy początkowym stężeniu < 10 mg DOC/l ponieważ, albo nie wszystkie bakterie są usuwane, albo węgiel jako składnik plazmy bakteryjnej jest ponownie rozpuszczany.
BIBLIOGRAFIA
|
— |
Standard Methods for the Examination of Water and Wastewater, 12th ed, Am. Pub. Hlth. Ass., Am. Wat. Poll. Control Fed., Oxygen Demand, 1965, P 65. |
|
— |
Wagner, R., Von Wasser, 1976, vol. 46, 139. |
|
— |
DIN-Entwurf 38409 Teil 41 – Deutsche Einheitsverfahren zur Wasser-, Abwasser- und Schlammunter-suchung, Summarische Wirkungs- und Stoffkenngröβen (Gruppe H). Bestimmung des Chemischen Sauerstoffbedarfs (CSB) (H 41), Normenausschuβ Wasserwesen (NAW) in DIN Deutsches Institut fur Normung e.V. |
|
— |
Gerike, P., The biodegradability testing of poorly water soluble compounds. Chemosphere, 1984, vol 13 (1), 169. |
ZAŁĄCZNIK III
OCENA BIODEGRADOWALNOŚCI SŁABO ROZPUSZCZALNYCH SUBSTANCJI
W badaniach biodegradowalności słabo rozpuszczalnych substancji należy zwrócić szczególną uwagę na następujące aspekty.
Podczas gdy jednorodne ciecze rzadko stwarzają problemy z pobieraniem ich próbek, dla materiałów w postaci ciał stałych, zalecana jest ich homogenizacja odpowiednimi metodami, aby zapobiec błędom z powodu ich niejednorodności. Należy zwrócić szczególną uwagę, gdy wymagane są reprezentatywne próbki kilkumiligramowe z mieszanin substancji chemicznych o dużej zawartości zanieczyszczeń.
W czasie badań można stosować różne formy mieszania. Należy zwrócić uwagę by używać dostatecznego mieszania tylko do utrzymania substancji chemicznej w postaci zawiesiny, w celu zapobieżenia przegrzaniu, nadmiernemu pienieniu i nadmiernym siłom poprzecznym.
Można stosować emulsyfikator dający stabilną zawiesinę substancji chemicznej. Nie może być on toksyczny dla bakterii i ulegać biodegradacji lub powodować pienienie w warunkach badania.
Do rozpuszczalników stosuje się takie same kryteria jak do emulsyfikatorów.
Nie jest zalecane używanie stałych nośników przy badaniu stałych substancji ale mogą być one odpowiednie dla substancji oleistych.
Gdy stosowane są pomocnicze substancje, takie jak emulsyfikatory, rozpuszczalniki oraz nośniki, należy przeprowadzić ślepą próbę z substancją pomocniczą.
Każde z trzech badań respirometrycznych CO2, BOD, MITI można użyć w badaniach biodegradowalności związków słabo rozpuszczalnych.
BIOBLIGRAFIA
|
— |
de Morsier, A. et al., Biodegradacja tests for słabo rozpuszczalne compounds. Chemosphere, 1987, vol. 16, 833. |
|
— |
Gerike, P., The Biodegradability testing of poorly water soluble compounds. Chemosphere, 1984, vol. 13, 169. |
ZAŁĄCZNIK IV
OCENA BIODEGRADOWALNOŚCI SUBSTANCJI CHEMICZNEJ PODEJRZEWANEJ O DZIAŁANIE TOKSYCZNE DLA INOKULUM
Jeżeli substancja chemiczna podlegająca badaniu na łatwość biodegradowalności okazuje sie być niebiodegradowalną, zalecana jest następująca procedura, jeśli pożądana jest różnica pomiędzy inhibicją a obojętnością (Reynolds et al., 1987).
Do badań biodegradacji i toksyczności należy użyć podobnych lub identycznych inokulum.
Do oceny toksyczności substancji chemicznych w badaniach łatwości biodegradacji wydaje się odpowiednie zastosowanie jednego z, lub połączonych badań: inhibicji szybkości respiracji aktywowanego osadu (Dir 88/302/EEC), BOD i/lub inhibicji wzrostu.
Aby zapobiec inhibicji z powodu toksyczności, sugeruje się, aby stężenia substancji badanej, stosowane w badaniach łatwości biodegradowalności, były mniejsze niż 1/10 wartości EC50 (lub mniejsze niż wartości EC20) otrzymywanych w badaniach toksyczności. Związki o wartości EC50 większej niż 300 mg/l nie wywierają prawdopodobnie działania toksycznego w badaniach łatwości biodegradacji.
Wartości EC50 mniejsze niż 20 mg/l prawdopodobnie stwarzają poważne problemy w następnych badaniach. Należy zastosować niskie stężenia badania, powodującego konieczność użycia surowego i czułego badania „zamkniętej butli” lub użycia znacznika 14C. Alternatywnie sezonowane inokulum pozwala użyć wyższego stężenia substancji badanej. W ostatnim przypadku gubi się jednak kryterium właściwe badaniu łatwości biodegradacji.
BIOBLIGRAFIA
Reynolds, L. et al., Evaluation of the toxicity of substances to be assessed for biodegradability. Chemosphere, 1987, vol. 16, 2259.
ZAŁĄCZNIK V
KOREKCJA POBORU TLENU Z POWODU ZAKŁÓCENIA PRZEZ NITRYFIKACJĘ
Błędy z powodu nieuwzględniania nitryfikacji w ocenie pobieranie tlenu w biodegradowalności substancji badanych niezawierających azotu są marginalne (nie większe niż 5 %), nawet jeżeli utlenianie azotu amonowego w podłożu zachodzi inaczej niż pomiędzy substancją badaną i naczyniami ślepej próby. Jednakże zawarty w badanych substancjach azot powoduje powstanie poważnych błędów.
Jeżeli zaszła nitryfikacja lecz nie jest całkowita, obserwowane pobieranie tlenu przez mieszaninę reakcyjną musi być skorygowane o ilość tlenu zużytego przy utlenieniu amoniaku do azotynów i azotanów; jeżeli zmiany w stężeniu, w czasie trwania inkubacji, azotynów i azotanów są określone następującymi równaniami:
|
2 NH4Cl + 3 O2 = 2 HNO2 + 2 HCl + 2 H2O |
(1) |
|
2 HNO2 + O2 = 2 HNO3 |
(2) |
|
ogólnie: |
|
|
2 NH4Cl + 4 O2 = 2 HNO3 + 2 HC1+ 2 H2O |
(3) |
Według równania (1) pobór tlenu przez 28 g azotu zawartego w chlorku amonu (NH4Cl) utlenianego do azotynu wyniesie 96 g, dając wskaźnik 3,43 (96/28). W ten sam sposób z równania (3) pobór tlenu przez 28 g azotu utlenionego do azotanu wynosi 28 g, dając wskaźnik 4,57 (128/28).
Ponieważ reakcje zachodzą sekwencyjnie, zostały przeprowadzone przez różne szczepy bakterii, jest możliwy wzrost lub spadek stężenia azotynów; w ostatnim przypadku utworzy się równoważne stężenie azotanów, Zatem tlen zużyty w utworzeniu azotanu jest 4,57 mnożony przez wzrost w stężeniu azotanu, podczas gdy tlen połączony z utworzeniem azotynu jest 3,43 pomnożony przez wzrost w stężeniu azotynu lub ze spadkiem w jego stężeniu strata tlenu jest -3,43 pomnożona przez spadek w stężeniu.
Oznacza to:
|
O2 zużytego w tworzeniu azotanu = 4,57 x wzrost w stężeniu azotanu |
(4) |
|
i |
|
|
O2 zużytego w tworzeniu azotynu = 3,43 x wzrost w stężeniu azotynu |
(5) |
|
i |
|
|
Strata O2 w zaniku azotynu = -3,43 x spadek w stężeniu azotanu |
(6) |
|
Tak więc |
|
|
Pobór O2 z powodu nitryfikacji = ±3,43 x zmiana stężenia azotynu +4,57 x wzrost stężenia azotanu. |
(7) |
|
I dlatego |
|
|
Pobór O2 z powodu utleniania C = całkowity obserwowany pobór – pobór z powodu nitryfikacji |
(8) |
Alternatywnie, jeżeli tylko całkowity utleniony azot jest oznaczony, pobieranie tlenu z powodu nitryfikacji, może być wzięte, w pierwszym przybliżeniu, 4,57 x wzrost w utlenionym azocie.
Wartość poprawki zużycia tlenu z powodu utleniania węgla jest następnie porównywana z ThOD NH3, jak wyliczono w załączniku II.
C.5. DEGRADACJA – ZAPOTRZEBOWANIE BIOCHEMICZNE NA TLEN
1. METODA
1.1. WPROWADZENIE
Celem metody jest pomiar zapotrzebowania biochemicznego na tlen (BOD) substancji organicznych w stanie ciekłym lub stałym.
Dane wypracowane w niniejszym badaniu odnoszą się do związków rozpuszczalnych w wodzie, jednakże związki lotne oraz te o niskiej rozpuszczalności w wodzie, mogą być również, przynajmniej z zasady badane.
Metoda jest stosowana tylko do tych badanych materiałów organicznych, które nie są inhibitorami w stosunku do bakterii w stężeniach używanych w badaniu. Jeżeli badany materiał nie jest rozpuszczalny w stężeniu badania, należy użyć specjalnych środków, takich jak użycie ultradźwięków, dla uzyskania dobrej zawiesiny badanego materiału.
Informacje na temat toksyczności substancji chemicznej są użyteczne do interpretacji niskich wyników i do wyboru odpowiednich stężeń badań.
1.2. DEFINICJA I JEDNOSTKI
BOD jest zdefiniowane jako masa rozpuszczonego tlenu pożądana przez właściwą objętość roztworu substancji dla procesu utleniania biochemicznego w opisanych warunkach.
Wyniki są wyrażane jako gramy BOD na gram badanej substancji.
1.3. SUBSTANCJE ODNIESIENIA
Pożądane jest użycie odpowiednich substancji odniesienia dla sprawdzenia aktywności inokulum.
1.4. ZASADA METODY BADANIA
Wstępnie oznaczona ilość substancji, rozpuszczona lub zawieszona w dobrze napowietrzanym odpowiednim podłożu, jest zaszczepiana drobnoustrojami i inkubowana w stałej zdefiniowanej temperaturze otoczenia w ciemności.
BOD jest oznaczane przez różnicę w zawartości rozpuszczonego tlenu na początku i na końcu badania. Czas trwania badania wynosi co najmniej 5 dni i nie więcej niż 28 dni.
Ślepa próba musi być oznaczona w równoległym badaniu niezawierającym badanej substancji.
1.5. KRYTERIA JAKOŚCI
Oznaczenie BOD nie może być uważane jako mające ważność naukową oznaczenie biodegradowalności substancji, a jedynie jako badanie przesiewowe (sortujące).
1.6. OPIS METODY BADANIA
Wstępny roztwór lub zawiesina substancji jest przygotowywana aby uzyskac stężenie BOD odpowiednie dla zastosowanej metody. Następnie BOD jest oznaczane stosując jedną z odpowiednich narodowo i międzynarodowo standaryzowanych metod.
2. DANE I OCENA
BOD zawarte w wstępnym roztworze jest obliczane zgodnie z wybraną znormalizowana metodą i przeliczane na gramy BOD na gram badanej substancji.
3. SPORZĄDZANIE SPRAWOZDANIA
Metoda stosowana musi być ustalona.
Zapotrzebowanie biochemiczne na tlen należy uśrednić z co najmniej trzech ważnych pomiarów.
Wszystkie informacje i uwagi odnośnie interpretacji wyników musza być przedstawione w sprawozdaniu, szczególnie w odniesieniu do zanieczyszczeń, postaci fizycznej, działania toksycznego i właściwości związanych z budową substancją które powodują wpływ na wyniki.
Musi być wykazane w sprawozdaniu użycie dodatków hamujących biologiczna nitryfikację.
4. LITERATURA
Wykaz standaryzowanych metod, dla przykładu:
|
|
NF T 90-103: Determination biochemical oxygen demand. |
|
|
NBN 407: Biochemical oxygen demand. |
|
|
NEN 3235 5.4: Bepaling van het biochemish zuurstofverbruik (BZV). |
|
|
The determination of biochemical oxygen demand, Methods for the examination of water and associated materials, HMSO, London. |
|
|
ISO 5815: Determination of biochemical oxygen demand after n days. |
C.6. DEGRADACJA – ZAPOTRZEBOWANIE CHEMICZNE NA TLEN
1. METODA
1.1. WPROWADZENIE
Celem metody jest pomiar zapotrzebowania chemicznego na tlen (COD) substancji organicznych w stanie ciekłym lub stałym w standardowy arbitralny sposób, w ustalonych warunkach laboratoryjnych.
Informacje na temat wzoru chemicznego są użyteczne dla prowadzenia tego badania oraz interpretacji uzyskanych wyników (np. sole halogenowe, sole żelazowe związków organicznych, związki chloroorganiczne).
1.2. DEFINICJE I JEDNOSTKI
Zapotrzebowanie chemiczne na tlen jest zmierzoną utlenialnością substancji, wyrażoną jako równoważna ilość tlenu odczynnika utleniającego zużytego przez substancje w ustalonych warunkach laboratoryjnych.
Wynik jest wyrażany w gramach COD na gram badanej substancji.
1.3. SUBSTANCJE ODNIESIENIA
Nie ma konieczności stosowania substancji odniesienia za każdym razem, gdy przeprowadzane są badania nowej substancji. Powinny one służyć głównie sprawdzeniu od czasu do czasu wydajności metody i pozwolić na porównanie wyników z innymi metodami.
1.4. ZASADA METODY BADANIA
Wstępnie oznaczona ilość substancji, rozpuszczonej lub zawieszonej w wodzie, jest utleniana przez dichromian potasu w stężonym kwasie siarkowym z siarczanem srebra jako katalizatorem, pod chłodnicą zwrotną przez dwie godziny. Pozostałość dichromiany jest oznaczana miareczkowo mianowanym roztworem siarczanu żelazoamonowego.
W przypadku substancji zawierających chlor, dodaje się siarczanu rtęci (10) celem zmniejszenia zakłócenia przez chlorki.
1.5. KRYTERIA JAKOŚCI
Z powodu arbitralnego sposobu oznaczenia, COD jest „wskaźnikiem utlenialności” i jako taki używany jest jako praktyczna metoda pomiaru związków organicznych.
Chlorki zakłócają to badanie; redukujące środki nieorganiczne lub utleniające mogą także powodować zakłócenia w oznaczaniu COD.
Niektóre związki cykliczne i wiele substancji lotnych (np. niższe kwasy tłuszczowe) nie są w pełni utleniane w niniejszym badaniu.
1.6. OPIS METODY BADANIA
Wstępny roztwór lub zawiesina substancji jest tak przygotowywana by uzyskać COD pomiędzy 250 i 600 mg na litr.
Uwagi:
W przypadku słabo rozpuszczalnych i niedających się zdyspergować substancji należy ilość drobno sproszkowanej substancji, odpowiadającej około 5 mg COD, odważyć i umieścić w aparacie doświadczalnym wraz z wodą.
Zapotrzebowanie chemiczne na tlen (COD) jest często i szczególnie w przypadku słaborozpuszczalnych substancji oznaczane korzystnie w odmianach metody, to jest w zamkniętym systemie z ciśnieniowym ekwalizerem (H. Kelkenberg, 1975). W niniejszej modyfikacji związki, które tylko z trudnością można oznaczyć metodą konwencjonalną – na przykład kwas octowy – są często z powodzeniem oznaczane. Metoda także zawodzi, jak w przypadku pirydyny. Jeżeli stężenie dichromianu potasu, jak opisano w pozycji bibliograficznej (1), wzrośnie do 0,25 N (0,0416 M), bezpośrednia doważka 5–10 mg substancji ułatwia oznaczenie COD, co jest zasadnicze dla słabo rozpuszczalnych w wodzie substancji (poz. lit. (2)).
Z drugiej strony, COD jest oznaczane, wykorzystując wszystkie odpowiednie krajowe i międzynarodowe standaryzowane metody.
2. DANE I OCENA
COD zawarte w kolbie doświadczalnej jest obliczane, stosując wybrane znormalizowane metody i przeliczane na gramy COD na gram badanej substancji.
3. SPORZĄDZANIE SPRAWOZDANIA
Zastosowana metoda odniesienia musi być ustalona.
Zapotrzebowanie chemiczne na tlen musi być średnią co najmniej z trzech pomiarów. Wszystkie informacje i uwagi odnośnie interpretacji wyników musza być przedstawione w sprawozdaniu, szczególnie w odniesieniu do zanieczyszczeń, postaci fizycznej, i właściwości związanych z budową substancji, które powodują wpływ na wyniki.
W sprawozdaniu należy przedstawić również fakt użycia siarczanu rtęci dla zminimalizowania zakłóceń pochodzących od chlorków.
4. LITERATURA
|
(1) |
Kelkenberg, H.,Z. von Wasser und Abwasserforschung, 1975, vol. 8, 146. |
|
(2) |
Gerike, P. The biodegradability testing of poorly water soluble compounds. Chemosphere, 1984, vol. 13, 169. Wykaz standaryzowanych metod, dla przykładu:
|
C.7. ROZKŁAD – ROZKŁAD ABIOTYCZNY: HYDROLIZA JAKO FUNKCJA PH
1. METODA
Niniejsza metoda badania jest równoważna metodzie OECD TG 111 (2004).
1.1. WSTĘP
Związki chemiczne mogą przedostawać się do wód powierzchniowych takimi drogami, jak: bezpośrednie stosowanie, rozpylenie poza teren upraw, odpływ, odprowadzanie wód powierzchniowych, usuwanie odpadów, ścieki przemysłowe, komunalne lub rolnicze oraz opady atmosferyczne. Związki te mogą ulegać przemianom w tych wodach w procesach chemicznych (np. hydroliza, utlenianie), fotochemicznych i/lub mikrobiologicznych. Niniejsze wytyczne opisują metodę badania laboratoryjnego mającą ocenić abiotyczne przemiany hydrolityczne związków chemicznych w układach wodnych, przy wartościach pH normalnie występujących w środowisku naturalnym (pH = 4–9) i oparte są na istniejących wytycznych (1)(2)(3)(4)(5)(6)(7).
Próby wykonuje się dla określenia: (i) szybkości hydrolizy substancji badanej w funkcji pH; oraz (ii) tożsamości lub charakteru oraz szybkości powstawania i zaniku produktów hydrolizy, na działanie których organizmy mogą być narażone. Badania takie mogą być wymagane dla związków chemicznych, które są bezpośrednio stosowane w wodzie albo które prawdopodobnie mogą się przedostawać do środowiska innymi drogami, które opisano powyżej.
1.2. DEFINICJE I JEDNOSTKI
Zob. załącznik 2.
1.3. ZAKRES STOSOWANIA METODY
Metoda ta dotyczy ogólnie substancji chemicznych (znaczonych lub nieznaczonych), dla których istnieje metoda analityczna mająca wystarczającą dokładność i czułość. Ma ona zastosowanie dla związków o niskiej lotności oraz nielotnych, o wystarczającej rozpuszczalności w wodzie. Badania nie należy przeprowadzać dla związków chemicznych, które wykazują wysoką lotność ze środowiska wodnego (np. fumiganty, rozpuszczalniki organiczne) i tym samym nie można ich utrzymać w roztworze w warunkach tego badania. Badanie może być trudne do wykonania dla substancji o minimalnej rozpuszczalności w wodzie (8).
1.4. ZASADA METODY BADANIA
Sterylne wodne roztwory buforowe o różnych wartościach pH (pH = 4, 7 i 9) poddaje się działaniu substancji badanej oraz inkubacji w ciemności w kontrolowanych warunkach laboratoryjnych (w stałych temperaturach). Po odpowiednich okresach czasu roztwory buforowe analizuje się na obecność substancji badanej i produktów hydrolizy. W przypadku substancji znaczonej (np. 14C) bilans masowy może być łatwiejszy do ustalenia.
Niniejszą metodę badania określa się jako „podejście poziomowe”, które pokazano oraz wyjaśniono w załączniku 1. Każdy „poziom” jest uruchamiany przez wyniki poprzedniego „poziomu”.
1.5. INFORMACJA O SUBSTANCJI BADANEJ
Do pomiaru szybkości hydrolizy można użyć substancji nieznaczonych albo znaczonych. Materiał znaczony jest zasadniczo preferowany przy badaniu przebiegu hydrolizy oraz przy ustalaniu bilansu masowego; jednak w przypadkach szczególnych znaczenie nie musi być absolutnie konieczne. Zaleca się znaczenie przy pomocy 14C, ale przydatne może być również użycie innych izotopów, takich jak 13C, 15N, 3H. O ile to będzie możliwe, znacznik powinien być umieszczony w najtrwalszej części cząsteczki. Na przykład, jeśli substancja badana zawiera jeden pierścień, wymagane jest znaczenie w pierścieniu; jeśli substancja badana zawiera dwa lub większą liczbę pierścieni, potrzebne mogą być osobne badania dla ustalenia losu każdego zaznaczonego pierścienia i dla uzyskania odpowiednich informacji na temat powstawania produktów hydrolizy. Czystość substancji badanej powinna wynosić co najmniej 95 %.
Przed wykonaniem testu hydrolizy należy zebrać następujące informacje na temat substancji badanej:
|
a) |
rozpuszczalność w wodzie (metoda badania A.6); |
|
b) |
rozpuszczalność w rozpuszczalnikach organicznych; |
|
c) |
prężność par (metoda badania A.4) i/lub stała w równaniu Henry'ego; |
|
d) |
współczynnik podziału n-oktanol/woda (metoda badania A.8); |
|
e) |
stała dysocjacji (pKa) (Wytyczna OECD 112) (9); |
|
f) |
szybkość fototransformacji bezpośredniej i pośredniej w wodzie, w przypadkach, których to dotyczy. |
Dostępne powinny być metody analityczne dla oznaczania ilościowego substancji badanej oraz, jeśli jest to ważne, dla identyfikacji i oznaczania ilościowego produktów hydrolizy w roztworach wodnych (zob. także sekcja 1.7.2).
1.6. SUBSTANCJE WZORCOWE
W miarę możliwości przy identyfikacji i ocenie ilościowej produktów hydrolizy metodami spektroskopowymi i chromatograficznymi albo innymi odpowiednio czułymi metodami należy korzystać z substancji wzorcowych.
1.7. KRYTERIA JAKOŚCI
1.7.1. Odzysk
Analiza co najmniej dwóch roztworów buforowych albo ich ekstraktów natychmiast po dodaniu substancji badanej daje pierwszą wskazówkę na temat powtarzalności metody analitycznej oraz jednorodności procedury aplikacyjnej dla substancji badanej. Stopnie odzysku dla kolejnych etapów eksperymentów uzyskuje się z odpowiednich bilansów masowych (jeśli używa się materiału znaczonego). Stopnie te powinny się mieścić w zakresie od 90 % do 110 % dla związków chemicznych znaczonych i nieznaczonych (7). W przypadku gdy uzyskanie tego zakresu jest technicznie trudne, odzysk wynoszący 70 % dla związków nieznaczonych jest do przyjęcia, ale należy podać uzasadnienie.
1.7.2. Powtarzalność i czułość metody analitycznej
Powtarzalność metody analitycznej albo metod analitycznych wykorzystywanych do oceny ilościowej substancji badanej i produktów hydrolizy można sprawdzić za pomocą powielanych analiz tych samych roztworów buforowych (lub ich ekstraktów) w późniejszym okresie, po zebraniu wystarczających ilości produktów hydrolizy dla oceny ilościowej.
Metoda analityczna powinna być wystarczająco czuła, aby można było ocenić ilościowo stężenia substancji badanych na poziomie 10 % lub mniej stężenia pierwotnego. Jeśli ma to zastosowanie, metody analityczne powinny także być wystarczająco czułe, aby można było oznaczyć ilościowo dowolny produkt hydrolizy stanowiący 10 % lub więcej stężenia zastosowanego (w dowolnym momencie podczas badania), do 25 % lub mniej jego najwyższego stężenia.
1.7.3. Przedziały ufności dla danych kinetycznych hydrolizy
Przedziały ufności należy obliczyć i podać dla wszystkich współczynników regresji, stałych szybkości, okresów połowicznego zaniku oraz wszelkich innych parametrów kinetycznych (np. DT50).
1.8. OPIS METODY BADANIA
1.8.1. Aparatura i wyposażenie
Badania należy wykonywać w pojemnikach szklanych (np. w probówkach lub niewielkich kolbkach), w ciemności i w warunkach sterylnych, jeśli jest to konieczne, chyba że informacje wstępne (takie jak współczynnik podziału n-oktanol/woda) wskazują, że substancja badana może przywierać do szkła. W takich przypadkach może zaistnieć potrzeba rozważenia użycia materiałów alternatywnych (takich jak teflon). Być może także uda się zmniejszyć problem przylegania do szkła przy pomocy jednej z poniższych metod:
|
— |
określenie masy substancji badanej i produktów hydrolizy zaadsorbowanych do naczynia testowego, |
|
— |
użycie łaźni ultradźwiękowej, |
|
— |
zapewnienie mycia rozpuszczalnikiem całego sprzętu szklanego dla każdego odstępu pobierania próbek, |
|
— |
używanie produktów preparowanych, |
|
— |
używanie zwiększonej ilości współrozpuszczalnika dla wprowadzania substancji badanej do układu; jeśli używa się współrozpuszczalnika, powinien to być taki materiał, który nie powoduje hydrolizy substancji badanej. |
Dla inkubacji różnych roztworów badanych zwykle potrzebne są wstrząsarki z łaźnią wodną o kontrolowanej temperaturze albo inkubatory z termostatyczną kontrolą temperatury.
Potrzebne jest standardowe wyposażenie laboratorium, obejmujące w szczególności:
|
— |
pH-metr, |
|
— |
przyrządy analityczne, takie jak wyposażenie GC, HPLC i TLC, włącznie z odpowiednimi systemami detekcji dla analizy substancji znaczonych radioaktywnie i nieznaczonych, albo odwrócona metoda rozcieńczeń izotopowych, |
|
— |
przyrządy dla potrzeb identyfikacji (np. MS, GC-MS, HPLC-MS, NMR itp.), |
|
— |
cieczowy licznik scyntylacyjny, |
|
— |
rozdzielacze dla ekstrakcji ciecz-ciecz, |
|
— |
aparatura do zatężania roztworów i ekstraktów (np. wyparka rotacyjna), |
|
— |
urządzenie do kontroli temperatury (np. łaźnia wodna). |
Odczynniki chemiczne obejmujące na przykład:
|
— |
rozpuszczalniki organiczne, cz.d.a., takie jak heksan, dichlorometan itp., |
|
— |
ciecz scyntylacyjna, |
|
— |
roztwory buforowe (szczegóły – zob. sekcja 1.8.3). |
Cały sprzęt szklany, wodę o czystości odczynnikowej oraz roztwory buforowe używane w testach hydrolizy należy poddać sterylizacji.
1.8.2. Wprowadzanie substancji badanej
Substancja badana powinna być wprowadzana jako roztwór wodny do różnych roztworów buforowych (zob. załącznik 3). Jeśli jest to konieczne dla odpowiedniego rozpuszczenia, dopuszcza się użycie niewielkich ilości rozpuszczalników mieszających się z wodą (takich jak acetonitryl, aceton, etanol) dla wprowadzenia i rozprowadzenia substancji badanej, jednak ilość ta normalnie nie powinna przekraczać 1 % obj. W przypadku jeśli rozważa się użycie wyższych stężeń rozpuszczalników (np. w przypadku słabo rozpuszczalnych substancji badanych), jest to dopuszczalne jedynie jeśli można wykazać, że rozpuszczalnik nie wywiera wpływu na hydrolizę substancji badanych.
Nie zaleca się rutynowego stosowania produktu preparowanego, gdyż nie można wykluczyć, że składniki preparatu mogą wpływać na proces hydrolizy. Jednak dla substancji badanych słabo rozpuszczalnych w wodzie albo dla substancji przywierających do szkła (zob. sekcja 1.8.1) użycie materiału preparowanego może być odpowiednią alternatywą.
Należy stosować jedno stężenie substancji badanej i nie powinno ono przekraczać 0,01 M albo połowy stężenia w stanie nasycenia (zob. załącznik 1).
1.8.3. Roztwory buforowe
Badanie hydrolizy należy wykonywać przy wartościach pH wynoszących 4, 7 oraz 9. W tym celu należy przygotować roztwory buforowe, używając chemikaliów cz.d.a. oraz wody. Pewne przydatne układy buforowe przedstawiono w załączniku 3. Należy podkreślić, że stosowany układ buforowy może wpływać na szybkość hydrolizy, i tam gdzie taki wpływ jest obserwowany należy użyć alternatywnego układu buforowego (11).
Wartość pH dla każdego roztworu buforowego należy sprawdzać wykalibrowanym pH-metrem z dokładnością do co najmniej 0,1 jednostki w wymaganej temperaturze.
1.8.4. Warunki badania
1.8.4.1. Temperatura badania
Badania dotyczące hydrolizy należy prowadzić w stałych temperaturach. Dla potrzeb ekstrapolacji ważne jest utrzymywanie temperatury z dokładnością co najmniej do +0,5 oC.
Test wstępny (poziom 1) należy wykonać w temperaturze 50 oC, jeśli hydrolityczne zachowanie się substancji badanej jest nieznane. Testy kinetyczne na wyższych poziomach należy prowadzić w minimum trzech temperaturach (włącznie z testem w 50 oC), chyba że substancja badana okaże się odporna na hydrolizę w teście początkowym. Sugerowany zakres temperatur to 10–70 oC (korzystnie z przyjęciem co najmniej jednej temperatury poniżej 25 oC), co obejmuje temperaturę podawaną w sprawozdaniu z badań, wynoszącą 25 oC, oraz większość temperatur spotykanych w warunkach polowych.
1.8.4.2. Światło i tlen
Wszystkie badania hydrolizy należy prowadzić przy użyciu odpowiedniej metody, aby uniknąć wpływów fotolitycznych. Należy podjąć wszelkie stosowne środki, aby uniknąć obecności tlenu (np. barbotując przez ciecz hel, azot albo argon, w ciągu 5 minut, przed przygotowaniem roztworu).
1.8.4.3. Czas trwania testu
Test wstępny powinien trwać 5 dni, natomiast testy na wyższych poziomach należy kontynuować, aż hydrolizie ulegnie 90 % substancji badanej albo przez okres 30 dni, zależnie od tego, co nastąpi wcześniej.
1.8.5. Wykonanie badania
1.8.5.1. Test wstępny (poziom 1)
Test wstępny wykonuje się w temperaturze 50 ±0,5 oC i przy pH = 4,0, 7,0 oraz 9,0. Jeśli po 5 dniach obserwuje się hydrolizę na poziomie mniejszym niż 10 % (t0,5 25 oC > 1 rok), substancję badaną uznaje się za trwałą hydrolitycznie i normalnie nie są wymagane żadne dodatkowe testy. Jeśli o substancji wiadomo, że nie jest ona trwała w temperaturach występujących w środowisku naturalnym (12), test wstępny nie jest wymagany. Metoda analityczna musi być wystarczająco dokładna i czuła, aby wykrywała spadek wynoszący 10 % stężenia początkowego.
1.8.5.2. Hydroliza substancji nietrwałych (poziom 2)
Test na wyższym poziomie (test zaawansowany) należy wykonywać przy wartościach pH, dla których stwierdzono w teście wstępnym, że substancja badana jest nietrwała. Roztwory substancji badanej w buforach należy termostatować w wybranych temperaturach. Przy testach na kinetykę pierwszego rzędu każdy roztwór reakcyjny należy analizować w odstępach czasu, które zapewniają co najmniej sześć równomiernie rozmieszczonych punktów pomiarowych, zwykle pomiędzy 10 % a 90 % hydrolizy substancji badanej. Należy pobierać pojedyncze próbki dla analiz powtórnych (co najmniej po dwie próbki pobierane do osobnych zbiorniczków reakcyjnych) i przeanalizować ich zawartość dla każdego z co najmniej sześciu okresów czasu (dla co najmniej dwunastu punktów zawierających dane). Pobieranie za każdym razem jednej dużej próbki roztworu badanego, z której pobiera się mniejsze ilości dla analiz powtórnych, uznaje się za niewłaściwe, gdyż procedura taka nie pozwala na analizę zmienności danych i może prowadzić do problemów z zanieczyszczaniem roztworu badanego. Na koniec badania na wyższym poziomie (to znaczy przy 90 % hydrolizy albo po 30 dniach) należy wykonywać testy potwierdzające sterylność. Jeśli jednak nie obserwuje się rozkładu (to znaczy transformacji), przeprowadzenie testów na sterylność nie uważa się za konieczne.
1.8.5.3. Identyfikacj a produktów hydrolizy (poziom 3)
Wszelkie główne produkty hydrolizy, a co najmniej te, które reprezentują > 10 % zastosowanej dawki, powinny zostać zidentyfikowane przy pomocy odpowiednich metod analitycznych.
1.8.5.4. Testy opcjonalne
Dla hydrolitycznie nietrwałej substancji badanej wymagane mogą być dodatkowe testy przy wartościach pH innych niż 4, 7 oraz 9. Na przykład dla potrzeb fizjologicznych, potrzebny może być test w warunkach bardziej kwaśnych (np. pH = 1,2) i w jednej temperaturze mającej znaczenie fizjologiczne (37 oC).
2. DANE
Ilości substancji badanej i produktów hydrolizy, jeśli takie istnieją, należy podawać w % zastosowanego stężenia początkowego oraz, tam gdzie ma to zastosowanie, w mg/l, dla każdego punktu (czasu) poboru próbek, dla każdej wartości pH oraz dla każdej temperatury. Dodatkowo, jeśli użyto znaczonej substancji badanej, należy podać bilans masowy w procentach zastosowanego stężenia początkowego.
Należy przygotować prezentację graficzną logarytmicznie przetworzonych danych dla stężeń substancji badanej w funkcji czasu. Wszelkie główne produkty hydrolizy, a przynajmniej te stanowiące > 10 % użytej dawki, powinny zostać zidentyfikowane, a ich przetworzone logarytmicznie stężenia powinny być również wykreślone w ten sam sposób, co substancja macierzysta, aby pokazać szybkości ich powstawania i zaniku.
2.1. OPRACOWANIE WYNIKÓW
Dokładniejsze oznaczenie okresów połowicznego zaniku lub wartości DT50 powinno się uzyskać stosując obliczenia na bazie odpowiedniego modelu kinetycznego. Okresy połowicznego zaniku i/lub wartości DT50 (włącznie z granicami przedziału ufności) należy podawać dla każdej wartości pH oraz wartości temperatury, wraz z opisem użytego modelu, rzędu kinetyki oraz współczynnika determinacji (r2). Jeśli jest to właściwe, obliczenia należy także stosować dla produktów hydrolizy.
W przypadku badań szybkości wykonywanych w różnych temperaturach stałe szybkości pseudo pierwszego rządu dla hydrolizy (kobs) należy przedstawić jako funkcję temperatury. Obliczenie powinno być oparte zarówno na rozdzieleniu wartości kobs na stałe szybkości dla hydrolizy katalizowanej kwasem, hydrolizy neutralnej i hydrolizy katalizowanej zasadą (odpowiednio kH, kneutral i k0H), jak i na równaniu Arrheniusa:

gdzie Ai i Bi są stałymi regresji, odpowiednio z punktu przecięcia się osią rzędnych i z nachylenia, dla najlepiej pasujących linii wygenerowanych z przeprowadzenia regresji liniowej ln ki w funkcji odwrotności temperatury absolutnej w Kelvinach (T). Przy pomocy zależności Arrheniusa dla hydrolizy katalizowanej kwasem, hydrolizy neutralnej i hydrolizy katalizowanej zasadą można wyliczyć stałe szybkości pseudo pierwszego rzędu, a tym samym okresy połowicznego zaniku, dla innych temperatur, w których bezpośrednie doświadczalne oznaczenie stałej szybkości nie jest praktycznie możliwe (10).
2.2. OCENA I INTERPRETACJA WYNIKÓW
Większość reakcji hydrolizy przebiega zgodnie z kinetyką reakcji pozornie pierwszego rzędu i dlatego okresy połowicznego zaniku są niezależne od stężenia (zob. równanie 4 w załączniku 2). Pozwala to zwykle na stosowanie wyników laboratoryjnych oznaczonych dla 10-2 do 10-3 M do warunków występujących w środowisku naturalnym (< 10-6 M) (10). Mabey i Mill (11) przedstawili kilka przykładów dobrej zgodności pomiędzy szybkościami hydrolizy zmierzonymi w wodzie czystej i w wodach naturalnych dla różnorodnych związków chemicznych, pod warunkiem że mierzy się zarówno pH, jak i temperaturę.
3. OPRACOWANIE SPRAWOZDANIA
3.1. SPRAWOZDANIE Z BADAŃ
Sprawozdanie z badań musi obejmować co najmniej następujące informacje:
Substancja badana:
|
— |
nazwa zwyczajowa, nazwa chemiczna, numer CAS, wzór strukturalny (z pokazaniem miejsca znaczenia, kiedy używa się materiału znaczonego radioaktywnie) oraz stosowne własności fizykochemiczne (zob. sekcja 1.5), |
|
— |
czystość (zanieczyszczenia) substancji badanej, |
|
— |
czystość oznaczenia znaczonego związku chemicznego oraz aktywność molowa (jeśli dotyczy), |
|
— |
roztwory buforowe, |
|
— |
daty i szczegóły dotyczące przygotowania, |
|
— |
użyte bufory i rodzaje wody, |
|
— |
molowość i pH roztworów buforowych. |
Warunki badania:
|
— |
daty wykonania badań, |
|
— |
ilość zastosowanej substancji badanej, |
|
— |
metoda i rozpuszczalniki (rodzaj i ilość) użyte dla zastosowania substancji badanej, |
|
— |
objętość inkubowanych buforowanych roztworów substancji badanej, |
|
— |
opis układu użytego do inkubacji, |
|
— |
pH i temperatura w trakcie badania, |
|
— |
czasy pobierania próbek, |
|
— |
metoda/metody ekstrakcji, |
|
— |
metody oceny ilościowej i identyfikacji substancji badanej oraz produktów jej hydrolizy w roztworach buforowych, |
|
— |
liczba powtórzeń. |
Wyniki:
|
— |
powtarzalność i czułość użytych metod analitycznych, |
|
— |
stopnie odzysku (wartości procentowe dla ważności badania podano w sekcji 1.7.1), |
|
— |
dane dla powtórzeń i wartości średnie w formie tabeli, |
|
— |
bilans masowy w trakcie i na koniec badań (jeśli użyta została znaczona substancja badana), |
|
— |
wyniki testu wstępnego, |
|
— |
omówienie i interpretacja wyników, |
|
— |
wszelkie oryginalne dane i liczby. |
Poniższe informacje są wymagane jedynie kiedy oznacza się szybkość hydrolizy:
|
— |
wykresy stężeń w funkcji czasu dla substancji badanej i, w stosownych przypadkach, dla produktów hydrolizy przy każdej wartości pH oraz temperatury, |
|
— |
tabele wyników z równania Arrheniusa dla temperatury 20 oC/25 oC, z podaniem pH, stałej szybkości [h-1 lub dzień-1], okresu połowicznego zaniku albo DT50, temperatur [ oC], włącznie z granicami dla przedziału ufności i współczynnikami korelacji (r2) albo porównywalnymi informacjami, |
|
— |
proponowany przebieg (cykl przemian w trakcie) hydrolizy. |
4. BIBLIOGRAFIA
|
(1) |
OECD (1981). Hydrolysis as a Function of pH. OECD Guideline for Testing of Chemicals Nr. 111 [OECD (1981). |
|
(2) |
US-Environmental Protection Agency (1982). 40 CFR 796,3500, Hydrolysis as a Function of pH at 25 oC. Pesticide Assessment Guidelines, Subdivision N. Chemistry: Environmental Fate. |
|
(3) |
Agriculture Canada (1987). Environmental Chemistry and Fate Guidelines for registration of pesticides in Canada. |
|
(4) |
European Union (EU) (1995). Commission Directive 95/36/EC amending Council Directive 91/414/EEC concerning the placing of plant protection products on the market. Annex V: Fate and Behaviour in the Environment. |
|
(5) |
Dutch Commission for Registration of Pesticides (1991). Application for registration of a pesticide. Section G: Behaviour of the product and its metabolites in soil, water and air. |
|
(6) |
BBA (1980). Merkblatt Nr. 55, Teil I und II: Prufung des Verhaltens von Pflanzenbehandlungsmitteln im Wasser (October 1980). |
|
(7) |
SETAC (1995). Procedures for Assessing the Environmental Fate and Ecotoxicity of Pesticides. Mark R. Lynch, Ed [SETAC (1995). |
|
(8) |
OECD (2000). Guidance document on aquatic toxicity testing of difficult substances and mixtures, OECD Environmental Health and Safety Publications Series on Testing and Assessment Nr.23 [OECD (2000). |
|
(9) |
OECD (1993). Guidelines for the Testing of Chemicals. Paris. OECD (1994 – 2000): Addenda 6-11 to Guidelines for the Testing of Chemicals [OECD (1993). |
|
(10) |
Nelson, H, Laskowski D, Thermes S, and Hendley P. (1997) Recommended changes in pesticide fate study guidelines for improving input to computer models. (Text version of oral presentation at the 14th Annual Meeting of the Society of Environmental Toxicology and Chemistry, Dallas TX, November 1993). |
|
(11) |
Mabey, W. and Mill, T. (1978). Critical review of hydrolysis of organic compounds in water under environmental conditions. J. Phys. Chem. Ref Data 7, 383–415. |
ZAŁĄCZNIK 1
Wielopoziomowy schemat badania hydrolizy

ZAŁĄCZNIK 2
Definicje i jednostki
W każdym przypadku należy stosować międzynarodowy układ jednostek miar (SI).
Substancja badana: dowolna substancja, niezależnie od tego, czy jest to związek macierzysty, czy odpowiednie produkty transformacji.
Produkty transformacji: wszystkie substancje powstające w biotycznych lub abiotycznych reakcjach transformacji substancji badanej.
Produkty hydrolizy: wszystkie substancje powstające w reakcjach transformacji hydrolitycznej substancji badanej.
Hydroliza dotyczy reakcji substancji badanej RX z wodą, z wymianą grupy X na grupę OH w centrum reakcyjnym:
|
RX + HOH → ROH + HX |
[1] |
Szybkość, z jaką stężenie RX spada w tym uproszczonym procesie, podaje się równaniem
|
szybkość = k [H2O] [RX] |
reakcja drugiego rzędu |
|
albo |
|
|
szybkość = k [RX] |
reakcja pierwszego rzędu |
zależnie od etapu decydującego o szybkości. Ponieważ woda jest obecna w dużym nadmiarze w stosunku do substancji badanej, ten typ reakcji zwykle opisuje się jako reakcję pseudopierwszego rzędu, w której obserwowaną stałą reakcji podaje zależność
|
kobs = k [H2O] |
[2] |
i można ją wyznaczyć z wyrażenia (13)
|
|
[3] |
gdzie:
t= czas,
a Co, Ct= stężenia RX dla czasu wynoszącego 0 oraz t.
Wymiar dla tej stałej to (jednostka czasu)-1, a okres połowicznego zaniku dla reakcji (czas, po którym przereaguje 50 % RX) podaje równanie
|
|
[4] |
Okres połowicznego zaniku: (t0,5) to czas potrzebny dla osiągnięcia 50 % hydrolizy substancji badanej, kiedy reakcję można opisać kinetyką pierwszego rzędu; zależy on od stężenia.
DT 50 (Disappearance Time 50 = czas zaniku 50): to czas, po którym stężenie substancji badanej zmniejsza się o 50 %; jest on różny od okresu połowicznego zaniku t0,5, kiedy reakcja nie stosuje się do kinetyki pierwszego rzędu.
Ocena k w innej temperaturze
Kiedy znane są stałe szybkości reakcji w dwóch temperaturach, stałe szybkości w innych temperaturach można wyprowadzić z równania Arrheniusa:
![]() lub
lub 
Wykres ln k w funkcji l/T jest linią prostą o nachyleniu -E/R
gdzie:
|
k |
= |
stała szybkości, mierzona w różnych temperaturach, |
|
E |
= |
energia aktywacji (kJ/mol), |
|
T |
= |
temperatura absolutna (K), |
|
R |
= |
stała gazowa (8,314 J/mol.K). |
Energię aktywacji wyliczono za pomocą analizy regresyjnej albo następującego równania:

w którym: T2 >T1
ZAŁĄCZNIK 3
Układy buforowe
A. CLARK i LUBS:
Mieszaniny buforowe według CLARKA i LUBSA (14)
|
Skład |
pH |
|
0,2 N HC1 + 0,2 N KC1 przy 20 oC |
|
|
47,5 ml HC1 + 25 ml KC1 rozcieńczyć do 100 ml |
1,0 |
|
32,25 ml HC1 + 25 ml KC1 rozcieńczyć do 100 ml |
1,2 |
|
20,75 ml HC1 + 25 ml KC1 rozcieńczyć do 100 ml |
1,4 |
|
13,15 ml HC1 + 25 ml KC1 rozcieńczyć do 100 ml |
1,6 |
|
8,3 ml HC1 + 25 ml KC1 rozcieńczyć do 100 ml |
1,8 |
|
5,3 ml HC1 + 25 ml KC1 rozcieńczyć do 100 ml |
2,0 |
|
3,35 ml HC1 + 25 ml KC1 rozcieńczyć do 100 ml |
2,2 |
|
0,1 M kwasny ftalan potasu +0,1 N HC1 przy 20 oC |
|
|
46,70 ml 0,1 N HC1 + 50 ml kwaśnego ftalanu do 100 ml |
2,2 |
|
39,60 ml 0,1 N HC1 + 50 ml kwaśnego ftalanu do 100 ml |
2,4 |
|
32,95 ml 0,1 N HC1 + 50 ml kwaśnego ftalanu do 100 ml |
2,6 |
|
26,42 ml 0,1 N HC1 + 50 ml kwaśnego ftalanu do 100 ml |
2,8 |
|
20,32 ml 0,1 N HC1 + 50 ml kwaśnego ftalanu do 100 ml |
3,0 |
|
14,70 ml 0,1 N HC1 + 50 ml kwaśnego ftalanu do 100 ml |
3,2 |
|
9,90 ml 0,1 N HC1 + 50 ml kwaśnego ftalanu do 100 ml |
3,4 |
|
5,97 ml 0,1 N HC1 + 50 ml kwaśnego ftalanu do 100 ml |
3,6 |
|
2,63 ml 0,1 N HC1 + 50 ml kwaśnego ftalanu do 100 ml |
3,8 |
|
0,1 M kwaśny ftalan potasu +0,1 N HCl przy 20 oC |
|
|
0,40 ml 0,1 N NaOH + 50 ml kwaśnego ftalanu do 100 ml |
4,0 |
|
3,70 ml 0,1 N NaOH + 50 ml kwaśnego ftalanu do 100 ml |
4,2 |
|
7,50 ml 0,1 N NaOH + 50 ml kwaśnego ftalanu do 100 ml |
4,4 |
|
12,15 ml 0,1 N NaOH + 50 ml kwaśnego ftalanu do 100 ml |
4,6 |
|
17,70 ml 0,1 N NaOH + 50 ml kwaśnego ftalanu do 100 ml |
4,8 |
|
23,85 ml 0,1 N NaOH + 50 ml kwaśnego ftalanu do 100 ml |
5,0 |
|
29,95 ml 0,1 N NaOH + 50 ml kwaśnego ftalanu do 100 ml |
5,2 |
|
35,45 ml 0,1 N NaOH + 50 ml kwaśnego ftalanu do 100 ml |
5,4 |
|
39,85 ml 0,1 N NaOH + 50 ml kwaśnego ftalanu do 100 ml |
5,6 |
|
43,00 ml 0,1 N NaOH + 50 ml kwaśnego ftalanu do 100 ml |
5,8 |
|
45,45 ml 0,1 N NaOH + 50 ml kwaśnego ftalanu do 100 ml |
6,0 |
Mieszaniny buforowe według CLARKA i LUBSA (ciąg dalszy)
|
0,1 M fosforan monopotasu +0,1 N NaOH przy 20 oC |
|
|
5,70 ml 0,1 N NaOH + 50 ml fosforanu do 100 mll |
6,0 |
|
8,60 ml 0,1 N NaOH + 50 ml fosforanu do 100 ml |
6,2 |
|
12,60 ml 0,1 N NaOH + 50 ml fosforanu do 100 m |
6,4 |
|
17,80 ml 0,1 N NaOH + 50 ml fosforanu do 100 ml |
6,6 |
|
23,45 ml 0,1 N NaOH + 50 ml fosforanu do 100 ml |
6,8 |
|
29,63 ml 0,1 N NaOH + 50 ml fosforanu do 100 ml |
7,0 |
|
35,00 ml 0,1 N NaOH + 50 ml fosforanu do 100 ml |
7,2 |
|
39,50 ml 0,1 N NaOH + 50 ml fosforanu do 100 ml |
7,4 |
|
42,80 ml 0,1 N NaOH + 50 ml fosforanu do 100 ml |
7,6 |
|
45,20 ml 0,1 N NaOH + 50 ml fosforanu do 100 ml |
7,8 |
|
46,80 ml 0,1 N NaOH + 50 ml fosforanu do 100 ml |
8,0 |
|
0,1 M H3B03 w 0,1 M KC1 + 0,1 N NaOH przy 20 oC |
|
|
2,61 ml 0,1 N NaOH + 50 ml kwasu borowego do 100 ml |
7,8 |
|
3,97 ml 0,1 N NaOH + 50 ml kwasu borowego do 100 ml |
8,0 |
|
5,90 ml 0,1 N NaOH + 50 ml kwasu borowego do 100 ml |
8,2 |
|
8,50 ml 0,1 N NaOH + 50 ml kwasu borowego do 100 ml |
8,4 |
|
12,00 ml 0,1 N NaOH + 50 ml kwasu borowego do 100 ml |
8,6 |
|
16,30 ml 0,1 N NaOH + 50 ml kwasu borowego do 100 ml |
8,8 |
|
21,30 ml 0,1 N NaOH + 50 ml kwasu borowego do 100 ml |
9,0 |
|
26,70 ml 0,1 N NaOH + 50 ml kwasu borowego do 100 ml |
9,2 |
|
32,00 ml 0,1 N NaOH + 50 ml kwasu borowego do 100 ml |
9,4 |
|
36,85 ml 0,1 N NaOH + 50 ml kwasu borowego do 100 ml |
9,6 |
|
40,80 ml 0,1 N NaOH + 50 ml kwasu borowego do 100 ml |
9,8 |
|
43,90 ml 0,1 N NaOH + 50 ml kwasu borowego do 100 ml |
10,0 |
B. KOLTHOFF i VLEESCHHOUWER:
Bufory cytrynianowe według KOLTHOFFA i VLEESCHHOUWERA
|
Sklad |
pH |
|
0,1 M cytrynian monopotasu i 0,1 N HC1 przy 18 oC (15) |
|
|
49,7 ml 0,1 N HC1 + 50 ml cytrynianu do 100 ml |
2,2 |
|
43,4 ml 0,1 N HCl+50 ml cytrynianu do 100 ml |
2,4 |
|
36,8 ml 0,1 N HCl + 50 ml cytrynianu do 100 ml |
2,6 |
|
30,2 ml 0,1 N HCl + 50 ml cytrynianu do 100 ml |
2,8 |
|
23,6 ml 0,1 N HCl+50 ml cytrynianu do 100 ml |
3,0 |
|
17,2 ml 0,1 N HCl + 50 ml cytrynianu do 100 ml |
3,2 |
|
10,7 ml 0,1 N HCl + 50 ml cytrynianu do 100 ml |
3,4 |
|
4,2 ml 0,1 N HCl + 50 ml cytrynianu do 100 ml |
3,6 |
|
0,1 M cytrynian monopotasu i 0,1 N HCl przy 18 oC (15) |
|
|
2,0 ml 0,1 N NaOH + 50 ml cytrynianu do 100 ml |
3,8 |
|
9,0 ml 0,1 N NaOH + 50 ml cytrynianu do 100 ml |
4,0 |
|
16,3 ml 0,1 N NaOH + 50 ml cytrynianu do 100 ml |
4,2 |
|
23,7 ml 0,1 N NaOH + 50 ml cytrynianu do 100 ml |
4,4 |
|
31,5 ml 0,1 N NaOH + 50 ml cytrynianu do 100 ml |
4,6 |
|
39,2 ml 0,1 N NaOH + 50 ml cytrynianu do 100 ml |
4,8 |
|
46,7 ml 0,1 N NaOH + 50 ml cytrynianu do 100 ml |
5,0 |
|
54,2 ml 0,1 N NaOH + 50 ml cytrynianu do 100 ml |
5,2 |
|
61,0 ml 0,1 N NaOH + 50 ml cytrynianu do 100 ml |
5,4 |
|
68,0 ml 0,1 N NaOH + 50 ml cytrynianu do 100 ml |
5,6 |
|
74,4 ml 0,1 N NaOH + 50 ml cytrynianu do 100 ml |
5,8 |
|
81,2 ml 0,1 N NaOH + 50 ml cytrynianu do 100 ml |
6,0 |
C. SÖRENSEN:
Mieszaniny boranowe według SÓRENSENA
|
Skład |
Sörensen 18 oC |
Valbum, pH przy |
|||
|
ml boraksu |
ml HCl/NaO H |
10 oC |
40 oC |
70 oC |
|
|
0,05 M boraksu +0,1 N HCl |
|||||
|
5,25 |
4,75 |
7,62 |
7,64 |
7,55 |
7,47 |
|
5,50 |
4,50 |
7,94 |
7,98 |
7,86 |
7,76 |
|
5,75 |
4,25 |
8,14 |
8,17 |
8,06 |
7,95 |
|
6,00 |
4,00 |
8,29 |
8,32 |
8,19 |
8,08 |
|
6,50 |
3,50 |
8,51 |
8,54 |
8,40 |
8,28 |
|
7,00 |
3,00 |
8,08 |
8,72 |
8,56 |
8,40 |
|
7,50 |
2,50 |
8,80 |
8,84 |
8,67 |
8,50 |
|
8,00 |
2,00 |
8,91 |
8,96 |
8,77 |
8,59 |
|
8,50 |
1,50 |
9,01 |
9,06 |
8,86 |
8,67 |
|
9,00 |
1,00 |
9,09 |
9,14 |
8,94 |
8,74 |
|
9,50 |
0,50 |
9,17 |
9,22 |
9,01 |
8,80 |
|
10,00 |
0,00 |
9,24 |
9,30 |
9,08 |
8,86 |
|
0,05 M boraksu +0,1 N NaOH |
|||||
|
10,0 |
0,0 |
9,24 |
9,30 |
9,08 |
8,86 |
|
9,0 |
1,0 |
9,36 |
9,42 |
9,18 |
8,94 |
|
8,0 |
2,0 |
9,50 |
9,57 |
9,30 |
9,02 |
|
7,0 |
3,0 |
9,68 |
9,76 |
9,44 |
9,12 |
|
6,0 |
4,0 |
9,97 |
10,06 |
9,67 |
9,28 |
Mieszaniny fosforanowe według SÖRENSENA
|
Skład |
pH |
|
0,0667 M fosforan monopotasu + 0,0667 M fosforan disodu przy 20 oC |
|
|
99,2 ml KH2PO4+0,8 ml Na2HPO4 |
5,0 |
|
98,4 ml KH2PO4+1,6 ml Na2HPO4 |
5,2 |
|
97,3 ml KH2PO4+2,7 ml Na2HPO4 |
5,4 |
|
95,5 ml KH2PO4+4,5 ml Na2HPO4 |
5,6 |
|
92,8 ml KH2PO4+7,2 ml Na2HPO4 |
5,8 |
|
88,9 ml KH2PO4+11,1 ml Na2HPO4 |
6,0 |
|
83,0 ml KH2PO4+17,0 ml Na2HPO4 |
6,2 |
|
75,4 ml KH2PO4+24,6 ml Na2HPO4 |
6,4 |
|
65,3 ml KH2PO4+34,7 ml Na2HPO4 |
6,6 |
|
53,4 ml KH2PO4+46,6 ml Na2HPO4 |
6,8 |
|
41,3 ml KH2PO4+58,7 ml Na2HPO4 |
7,0 |
|
29,6 ml KH2PO4+70,4 ml Na2HPO4 |
7,2 |
|
19,7 ml KH2PO4+80,3 ml Na2HPO4 |
7,4 |
|
12,8 ml KH2PO4+87,2 ml Na2HPO4 |
7,6 |
|
7,4 ml KH2PO4+92,6 ml Na2HPO4 |
7,8 |
|
3,7 ml KH2PO4+96,3 ml Na2HPO4 |
8,0 |
C.8. TOKSYCZNOŚĆ DŻDŻOWNIC
BADANIE SZTUCZNEJ GLEBY
1. METODA
1.1. WSTĘP
W tym badaniu laboratoryjnym substancja testowa dodawana jest do sztucznej gleby, w której umieszcza się dżdżownice na okres 14 dni. Po zakończeniu tego okresu (i opcjonalnie po siedmiu dniach) badane są skutki śmiertelności dżdżownic wywołane przez substancję testową. Badanie dostarcza metody na względnie krótkoterminowe badanie skutków substancji chemicznej na dżdżownice pobieranej przez skórę i układ pokarmowy.
1.2. DEFINICJA I JEDNOSTKA
LC50: Stężenie substancji testowej ocenione jako powodujące śmierć u 50 % badanych zwierząt w okresie przeprowadzania badania.
1.3. SUBSTANCJE ODNIESIENIA
Substancja odniesienia używana jest okresowo jako środek pozwalający określić, iż czułość systemu badawczego nie uległa znacznej zmianie.
Analityczny stopień chloroacetamidu zalecany jest jako substancja odniesienia.
1.4. ZASADA BADANIA
Ziemia stanowi zmienną pożywkę, dlatego też do tego badania wykorzystywana jest starannie określona sztuczna gleba. Dorosłe dżdżownice z gatunku Eisenia foetida (zob. uwaga w dodatku) utrzymywane są w ściśle określonej sztucznej glebie poddawanej działaniu różnych stężeń substancji testowej. Zawartość zbiorników rozłożona jest na tacach przez okres 14 dni (i opcjonalnie przez siedmiu dniach) po rozpoczęciu badania oraz zliczane są osobniki pozostałe przy życiu po ekspozycji na działanie każdej wielkości stężenia substancji testowej.
1.5. KRYTERIA JAKOŚCIOWE
Badanie zaprojektowano w taki sposób, aby było jak najbardziej odtwarzalne w odniesieniu do badanego substrata i organizmu. Śmiertelność w grupach kontrolnych nie może przekraczać 10 % po zakończeniu badania, w przeciwnym razie badanie uważa się za nieważne.
1.6. OPIS METODY BADAWCZEJ
1.6.1. Materiały
1.6.1.1.
Zdefiniowana sztuczna gleba używana jest jako podstawowy substrat testowy.
|
a) |
Podstawowy substrat (procenty w przeliczeniu na suchą masę)
|
|
b) |
Substrat testowy Substrat testowy zawiera substrat podstawowy, substancję testową i dejonizowaną wodę. Zawartość wody wynosi około 25–42 % suchej masy substrata podstawowego. Zawartość wody w substracie, ustalana jest przez osuszanie próbki do stałej masy przy 105 oC. Kluczowym kryterium jest nawilżanie sztucznej gleby do takiego momentu, aby nie powstały wody stojące. Należy zachować ostrożność podczas mieszania w celu równomiernego rozłożenia substancji testowej i substrata. Sposób wprowadzenia substancji testowej do substrata należy opisać. |
|
c) |
Substrat kontrolny Substrat kontrolny zawiera substrat podstawowy i wodę. W przypadku wprowadzenia dodatkowego czynnika, dodatkowy substrat kontrolny powinien zawierać tę samą ilość dodatkowego czynnika. |
1.6.1.2.
Szklane zbiorniki o pojemności około jednego litra (odpowiednio przykryte plastikowymi pokrywkami, naczynia lub folie z tworzywa sztucznego z otworami wentylacyjnymi) wypełnione pewną ilością nawilżonego lub kontrolnego substrata, równowartość 500 g suchej masy substrata.
1.6.2. Warunki badania
Zbiorniki należy przechowywać w klimatyzowanych komorach w temperaturze 20 ± 2 oC w stałym oświetleniu. Natężenie oświetlenia powinno wynosić 400–800 luksów.
Okres badania wynosi 14 dni, ale śmiertelność można fakultatywnie ocenić na siedem dni po rozpoczęciu badania.
1.6.3. Procedura badania
Stężenia substancji testowej wyrażone są jako waga substancji na suchą masę substrata podstawowego (mg/kg).
Wielkość stężeń powodująca śmiertelność 0–100 % można ustalić przez test ustalania zakresu stężeń w celu przekazania informacji o zakresie stężeń wykorzystywanym w ostatecznym badaniu.
Substancję należy zbadać przy następujących wielkościach stężeń: 1 000; 100; 10; 1; 0,1 mg substancji/kilogram badanego substrata (sucha masa).
W przypadku przeprowadzenia w pełni badania ostatecznego jedna grupa badana na daną wielkość stężenia i jedna grupa kontrolna niepoddana działaniu substancji, każda zawierająca 10 dżdżownic, powinna być wystarczająca w celu wykonania testu ustalania zakresu stężeń.
Wyniki testu ustalania zakresu stężeń wykorzystywane są w celu wybrania co najmniej pięciu wielkości stężeń uszeregowanych geometrycznie, obejmujących zakres 0–100 % śmiertelności i różniących się stałym czynnikiem nie przekraczającym 1,8.
Badania wykorzystujące te serie stężeń powinny umożliwić ustalenie wartości LC50 oraz jej granic pewności najdokładniej jak to możliwe.
W badaniu ostatecznym należy użyć co najmniej cztery grupy badane przy danej wielkości stężenia i cztery grupy kontrolne niepoddane działaniu substancji, każda zawierająca 10 dżdżownic. Wyniki takich replikujących grup podawane są jako średnia i odchylenie standardowe.
W przypadku gdy dwa równoległe stężenia o wielkości 1,8, powodują 0 % i 100 % śmiertelności, takie dwie wartości są wystarczające do oznaczenia zakresu, w którym obniża się LC50.
Badany substrat, jeżeli możliwe, nie powinien zawierać dodatkowych czynników innych niż woda. Bezpośrednio przed rozpoczęciem badania, zawiesina substancji testowej lub substancja testowa rozrzedzona w dejonizowanej wodzie lub innym rozpuszczalniku jest mieszana z podstawowym substratem testowym, lub równomiernie nad nim rozpylana za pomocą drobnego spryskiwacza chromatograficznego lub podobnego.
Jeżeli substancja testowa nie jest rozpuszczalna w wodzie, może być rozpuszczona w odpowiednim rozpuszczalniku organicznym, w możliwie jak najmniejszej ilości (np. heksan, aceton lub chloroform).
Jedynie czynniki łatwo ulatniające się mogą być użyte w celu rozpuszczenia, rozrzedzenia lub zemulgowania substancji testowej. Badany substrat należy wietrzyć przed użyciem. Należy uzupełniać ilość wyparowanej wody. Grupa kontrolna powinna zawierać tę samą ilość każdego dodawanego czynnika.
Jeżeli substancja testowa jest nierozpuszczalna, nierozrzedzalna lub nie emulguje w rozpuszczalnikach organicznych, należy zmieszać 10 g drobnoziarnistego piasku kwarcowego oraz taką ilość substancji testowej, jaka wymagana jest przy 500 g suchej masy sztucznej gleby z 490 g suchej masy badanego substrata.
W odniesieniu do każdej badanej grupy, należy umieścić ilość nawilżonego badanego substrata równą 500 g suchej masy w każdym szklanym zbiorniku wraz z 10 dżdżownicami, które przebywały przez okres 24 godzin w podobnych warunkach tj. w warunkach nawilżonego substrata podstawowego, a następnie zostały szybko umyte, a nadwyżka wody została wchłonięta w bibułę filtracyjną przed użyciem.
Zbiorniki przykrywa się perforowanymi plastikowymi pokrywkami, naczyniami lub folią w celu zapobieżenia wysychaniu substrata i utrzymuje się je w warunkach badania przez okres l4 dni.
Oszacowania należy dokonać 14 dni (opcjonalnie po siedmiu dniach) po rozpoczęciu badania. Substrat rozkładany jest na szklanej płytce lub płytce wykonanej ze stali nierdzewnej. Należy przeprowadzić badanie dżdżownic oraz ustalić liczbę osobników pozostałych przy życiu. Dżdżownice uważa się za martwe, jeżeli nie reagują na delikatne bodźce mechaniczne oddziałujące z przodu organizmu osobnika.
W przypadku gdy badanie przeprowadzane jest po siedmiu dniach, zbiornik ponownie napełniany jest substratem testowym, a osobniki pozostałe przy życiu są przenoszone na powierzchnię z tym samym substratem testowym.
1.6.4. Badane organizmy
Organizmy poddane badaniu powinny być osobnikami dorosłymi Eisenia foetida (zob. uwagi w Dodatku) (w wieku dwóch miesięcy życia z ukształtowanym siodełkiem) o wadze nawilżonego ciała 300–600 mg (metody uprawy, zob. dodatek).
2. DANE
2.1. OBRÓBKA I ANALIZA WYNIKÓW
Stężenia testowej substancji odnotowywane są w odniesieniu do procentowej ilości martwych dżdżownic.
Jeżeli dane są zadowalające, należy ustalić wartość LC50 oraz limity pewności (p = 0,05) wykorzystując standardowe metody (Litchfield i Wilcoxon, 1949, metoda równoważna). LC50 należy podać jako miligram substancji testowej na kilogram badanego substrata (sucha masa).
W przypadkach gdy nachylenie krzywej stężenia jest zbyt strome aby umożliwić obliczenie LC50, wystarczające jest graficzne oszacowanie tej wartości.
W przypadku gdy dwie kolejne wielkości stężenia o wskaźniku w wysokości 1,8 powodują jedynie śmiertelność 0 % i 100 %, te dwie wartości są wystarczające do oznaczenia granicy, przy której obniża się LC50.
3. SPRAWOZDANIE
3.1. SPRAWOZDANIE Z BADAŃ
Sprawozdanie z badań powinno, jeżeli możliwe, zawierać następujące informacje:
|
— |
stwierdzenie, że badanie zostało przeprowadzone zgodnie z wyżej wymienionymi kryteriami jakości, |
|
— |
przeprowadzone badanie (badanie zakresu wielkości stężeń i/lub badanie ostateczne), |
|
— |
dokładny opis warunków badania lub stwierdzenie, że badanie zostało przeprowadzone zgodnie z metodą; wszelkie odstępstwa od procedury metody należy odnotować, |
|
— |
dokładny opis procedury mieszania substancji testowej z podstawowym substratem testowym, |
|
— |
informacja o badanych organizmach (gatunki, wiek, średnia oraz rozpiętość wagi, warunki hodowli i utrzymywania, dostawca), |
|
— |
metoda używana w celu ustalenia LC50, |
|
— |
wyniki badania łącznie ze wszelkimi wykorzystanymi danymi, |
|
— |
opis obserwowanych objawów i zmian w zachowaniu badanych organizmów, |
|
— |
śmiertelność w grupach kontrolnych. |
|
— |
LC50 lub najwyższa wielkość stężenia testowego nie powodująca śmiertelności oraz najniższa wielkość stężenia powodująca 100 %, śmiertelności na 14 dni (oraz fakultatywnie siedem dni) po rozpoczęciu badania, |
|
— |
wykreślanie krzywej/stężenia w odniesieniu do powodowanej reakcji, |
|
— |
wyniki otrzymane po zastosowaniu substancji odniesienia, oraz czy zostały połączone z obecnym badaniem, czy na podstawie wcześniejszej kontroli jakości. |
4. BIBLIOGRAFIA
|
(1) |
OECD, Paryż, 1981, Wytyczne Badania 201, ostateczna decyzja Rady C(81) 30. |
|
(2) |
Edwards, C. A. i Lofty, J. R., 1977, Biologia Dżdżownic, Chapman i Hall, Londyn, str. 331. |
|
(3) |
Bouche, M. B., 1972, Lombriciens de France, Ecologie et Systétmatique, Institut National de la Recherche Agronomique, str. 671. |
|
(4) |
Litchfield, J. T. i Wilcoxon, F., Uproszczona metoda oceny badań skutków dawkowania. J. Pharm. Exp. Therap., tom 96, 1949, str. 99. |
|
(5) |
Komisja Wspólnot Europejskich, Rozwój standardowej metody laboratoryjnej odnośnie oceny wpływu toksyczności substancji chemicznych na dżdżownice, Sprawozdanie 8714 EUR EN, 1983. |
|
(6) |
Umweltbundesamt/Biologische Bundesanstalt fur Land- und Forstwirtschaft, Berlin, 1984, Verfahrensvorschlag „Toxizitatstest am Regenwurm Eisenia foetida in kunstlichem Boden”, in: Rudolph/Boje, Okotoxikologie, ecomed, Landsberg, 1986. |
Dodatek
Hodowla i utrzymywanie dżdżownic przed rozpoczęciem badania
W celu założenia hodowli zwierząt należy umieścić 30–50 dorosłych dżdżownic w zbiorniku hodowlanym zawierającym świeży substrat i usunąć po okresie 14 dni. Zwierzęta te można wykorzystać do hodowli kolejnych grup. Dżdżownice wylężone z kokonów wykorzystuje się do badania po osiągnięciu dojrzałości (zgodnie z zalecanymi warunkami po okresie dwóch do trzech miesięcy).
Warunki utrzymywania i hodowli
|
Komora klimatyczna |
: |
temperatura 20 ± 2 oC najlepiej stałym oświetleniem ze (natężenie 400–800 luksów). |
|
Zbiorniki hodowlane |
: |
odpowiednio płytkie zbiorniki o pojemności 10–20 1. |
|
Substrat |
: |
Eisenia foetida może być hodowana w różnych ekskrementach zwierzęcych. Zaleca się użycie mieszaniny o składzie 50 % torfu i 50 % krowiego lub końskiego obornika jako pożywki hodowlanej. Pożywka powinna zawierać wartość pH w wysokości około 6–7 (regulowanej węglanem wapnia) oraz niską przewodność elektryczną (mniej niż 6 mmhos lub 0,5 % stężenia soli). Substrat należy odpowiednio nawilżać, ale niezbyt obficie. Oprócz wyżej wspomnianej metody, można zastosować inne odpowiednie procedury. |
Uwaga: Eisenia foetida występuje w dwóch rasach, które niektórzy systematycy podzielili na dwa gatunki (Bouche, 1972). Gatunki te są podobne pod względem morfologicznym, ale jeden, Eisenia foetida foetida, posiada typowo poprzeczne paskowanie lub przedziały na segmentach, a drugi, Eisenia foetida andrei, takiego paskowania nie posiada, będąc jednocześnie różnobarwnego czerwonawego koloru. Jeżeli jest to możliwe, należy użyć Eisenia foetida andrei. Można użyć inne gatunki, jeżeli dostępna jest metodologia.
C.9. BIODEGRADACJA
BADANIE ZAHN-WELLENS
1. METODA
1.1. WSTĘP
Celem metody jest ocena wpływu całkowitej biodegradacji rozpuszczalnych w wodzie, nielotnych substancji organicznych w przypadku ich ekspozycji na stosunkowo wysokie stężenia drobnoustrojów w badaniu statycznym.
Może mieć miejsce fizyczna i chemiczna adsorpcja zawieszonych substancji stałych, co należy wziąć pod uwagę przy interpretacji wyników (zob. 3.2).
Substancje przeznaczone do badania wykorzystuje się w stężeniach odpowiadających wartościom DOC w przedziale 50–400 mg/litr lub wartościom COD w przedziale 100–1 000 mg/litr (DOC = rozwiązany węgiel organiczny; COD = chemiczne zapotrzebowanie tlenu). Te względnie wysokie stężenia posiadają zaletę wiarygodności analitycznej. Związki o właściwościach toksycznych mogą opóźnić lub zahamować proces degradacji.
W niniejszej metodzie pomiar stężenia rozwiązanego węgla organicznego lub chemicznego zapotrzebowania tlenu wykorzystywany jest w celu określenia całkowitej biodegradacji substancji testowej.
Równoczesne użycie szczególnej metody analitycznej może umożliwić ocenę podstawowej biodegradacji substancji (zanik pierwotnej budowy chemicznej).
Metodę stosuje się jedynie w odniesieniu do tych organicznych substancji testowych, które przy wielkości stężenia używanego w badaniu:
|
— |
są rozpuszczalne w wodzie w warunkach badania, |
|
— |
posiadają nieznaczne ciśnienie pary w warunkach badania, |
|
— |
nie hamują bakterii, |
|
— |
są absorbowane w badanym systemie jedynie do pewnego stopnia, |
|
— |
nie są tracone w wyniku wytwarzania piany z roztworu testowego, |
Informacje dotyczące względnych części składowych materiału testowego będą użyteczne przy interpretacji otrzymanych wyników, w szczególności w tych przypadkach, gdzie wyniki są słabe lub marginalne.
Pożądane są informacje dotyczące wpływu toksyczności substancji na drobnoustroje w celu dokonania interpretacji słabych wyników oraz wyboru właściwych wielkości stężeń.
1.2. DEFINICJE I JEDNOSTKI
Wielkość degradacji osiągniętej na końcu badania odnotowywana jest jako „Biodegradacja w badaniu Zahn-Wellens”:

gdzie:
|
DT |
= |
( %) biodegradacji w czasie T, |
|
CA |
= |
wartości DOC (lub COD) w mieszaninie testowej zmierzone trzy godziny po rozpoczęciu badania (mg/l) (DOC = rozwiązany węgiel organiczny, COD = chemiczne zapotrzebowanie tlenu), |
|
CT |
= |
wartości DOC lub COD w mieszaninie testowej w chwili pobierania próbek (mg/l), |
|
CB |
= |
wartość DOC lub COD próbki ślepej w chwili pobierania próbek (mg/l), |
|
CBA |
= |
wartość DOC lub COD próbki ślepej, mierzona trzy godziny po rozpoczęciu testu (mg/l). |
Stopień degradacji jest zaokrąglany do najbliższej pełnej wartości procentowej.
Procentową degradację stwierdza się jako procent ubytku DOC (lub COD) z substancji testowej.
Różnica między mierzoną wartością po trzech godzinach i obliczoną, lub najlepiej zmierzoną wartością początkową, może dostarczyć użytecznych informacji dotyczących wydzielania substancji (zob. 3.2, Interpretacja wyników).
1.3. SUBSTANCJE ODNIESIENIA
W niektórych przypadkach, w przypadku badania nowych substancji użyteczne mogą okazać się substancje odniesienia; jednakże nie można jeszcze zalecić szczególnych substancji odniesienia.
1.4. ZASADA METODY BADAWCZEJ
Aktywny szlam, pożywki mineralne i materiał testowy jako jedyne źródło węgla w roztworach wodnych umieszczane są razem w jednym z czterolitrowych szklanych zbiorników wyposażonych w mieszadło i napowietrzacz. Mieszanina jest mieszana i napowietrzana przy temperaturze 20–25 oC przy rozproszonym oświetleniu lub w ciemnym pomieszczeniu przez okres 28 dni. Proces degradacji jest kontrolowany przez oznaczanie wartości DOC (lub COD) w filtrowanym roztworze codziennie lub w innych właściwych, regularnych odstępach czasu. Stosunek wydzielonego DOC (lub COD) po każdym przedziale czasu do wartości, trzy godziny po rozpoczęciu, wyrażany jest jako procent biodegradacji i służy jako pomiar stopnia degradacji w danym czasie. Wynik nanoszony jest na wykres względem czasu, w celu uzyskania krzywej biodegradacji.
W przypadku użycia szczególnej metody analitycznej można dokonać pomiaru zmian w stężeniu pierwotnej molekuły w wyniku biodegradacji (podstawowa biodegradacja).
1.5. KRYTERIA JAKOŚCIOWE
Odtwarzalność niniejszego badania została pozytywnie potwierdzona w próbie pierścieniowej.
Czułość metody jest przeważnie ustalana przez zmienność próbki ślepej i, w mniejszym stopniu, przez precyzyjne oznaczenie rozwiązanego węgla organicznego i poziomu badanego związku w roztworze.
1.6. OPIS PROCEDURY BADAWCZEJ
1.6.1. Przygotowania
1.6.1.1.
Woda testowa: woda pitna o zawartości węgla organicznego < 5 mg/litr. Łączne stężenie jonów wapnia i magnezu nie może przekraczać 2,7 mola/litr; w przeciwnym razie wymagane jest odpowiednie rozcieńczenie wodą dejonizowaną lub destylowaną.
|
Kwas siarkowy, analityczny odczynnik (A.R.): |
50 g/l |
|
Roztwór wodorotlenku sodu czysty analitycznie: |
40 g/l |
|
Mineralna pożywka: rozpuszczona w jednym litrze dejonizowanej wody: |
|
|
chlorek amonowy, NH4Cl, czysty analitycznie: |
38,5 g, |
|
fosforan monosodowy diwodny, NaH2PO4.2H2O, czysty analitycznie: |
33,4 g, |
|
fosforan monopotasowy, KH2PO4, czysty analitycznie: |
8,5 g, |
|
fosforan dipotasowy, K2HPO4, czysty analitycznie: |
21,75 g. |
Mieszanina służy zarówno jako pożywka, a także jako system buforowy.
1.6.1.2.
Szklane zbiorniki o pojemności czterμech litrów (np. zbiorniki cylindryczne).
Mieszadło ze szklanym lub metalowym mieszakiem na odpowiednim trzonku (mieszak powinien obracać się około 5–10 cm ponad dnem zbiornika). W zamian można użyć magnetycznego mieszaka o długości trzonka 7–10 cm.
Szklana probówka o średnicy wewnętrznej 2–4 mm w celu doprowadzenia powietrza. Otwór probówki powinien być umieszczony około 1 cm ponad dnem zbiornika.
Wirówka (około 3 550 g).
Miernik pH.
Miernik rozpuszczonego tlenu.
Papierowe filtry.
Urządzenie do filtracji membrany.
Filtry membranowe, wielkość poru 0,45 μm. Filtry membranowe są odpowiednie w przypadku gdy potwierdzone jest, że nie przepuszczają węgla, ani nie wchłaniają substancji w fazie filtracji.
Sprzęt analityczny do określania składu węgla organicznego i chemicznego zapotrzebowania tlenu.
1.6.1.3.
Aktywny szlam pochodzący z biologicznej oczyszczalni ścieków jest oczyszczany przez (wielokrotnie) odwirowywanie lub klarowanie wodą testową (powyżej).
Aktywny szlam musi być we właściwym stanie. Taki szlam uzyskać można z właściwie pracującej oczyszczalni ścieków. W celu uzyskania różnorodnych gatunków lub szczepów bakterii można zmieszać inokulat pochodzący z różnych źródeł (np. różnych oczyszczalni ścieków, ekstraktów gleby, wód rzecznych itd.). Mieszaninę należy oczyszczać w sposób opisany powyżej.
W celu dokonania kontroli aktywnych szlamów, zob. „Kontrola funkcjonalna, poniżej”.
1.6.1.4.
Do zbiornika używanego w badaniu należy dodać 500 ml wody testowej, 2,5 ml/litr mineralnej pożywki i aktywny szlam w ilości odpowiadającej 0,2–1,0 g/litr suchej masy w ostatecznej mieszaninie. Dodać wystarczającą ilość roztworu podstawowego substancji przeznaczonej do badania, tak aby stężenie DOC w wysokości 50–400 mg/litr dało mieszaninę końcową. Odpowiednie wartości COD wynoszą 100–1 000 mg/litr. Przygotować wodę testową o ogólnej objętości jednego do czterech litrów. Wybór całkowitej ilości wody zależy od liczby próbek, mających być pobrane do określenia DOC lub COD oraz ilości koniecznej do przeprowadzenia procedury analitycznej.
Zazwyczaj ilość dwóch litrów uważana jest za wystarczającą. Zakłada się co najmniej jeden kontrolny zbiornik próbki ślepej w celu przeprowadzania badania równoległego z każdą serią; taki zbiornik zawiera jedynie aktywowany szlam i mineralną pożywkę składającą się z wody testowej o takiej samej ilości całkowitej, jak w zbiornikach używanych do badania.
1.6.2. Przeprowadzenie badania
Naczynia testowe są wytrząsane przy użyciu mieszadeł magnetycznych lub tradycyjnych (śmigłowych) w warunkach rozproszonego oświetlenia bądź w ciemności, w temperaturze 20–25 oC. Napowietrzanie jest dokonywane powietrzem pod ciśnieniem, czyszczonym za pomocą bawełnianego filtra oraz tryskawki, jeżeli konieczne. Nie należy dopuścić do osiadania szlamu, a stężenie tlenu nie może obniżyć się poniżej 2 mg/litr.
Wartość pH należy kontrolować w regularnych odstępach czasu (np. codziennie) i dostosować do poziomu pH 7–8 w razie konieczności.
Straty spowodowane parowaniem są uzupełniane tuż przed każdorazowym pobieraniem próbek wodą dejonizowaną lub destylowaną, w wymaganej ilości. Należy zaznaczyć poziom płynu na zbiorniku przed rozpoczęciem badania. Nowe znaki nanosi się po każdorazowym pobieraniu próbek (bez napowietrzania i mieszania). Pierwsze próbki zawsze pobierane są na trzy godziny po rozpoczęciu badania w celu wykrycia absorpcji materiału testowego przez aktywny szlam.
Eliminacja materiału testowego następuje po określeniu DOC lub COD dokonywanym raz dziennie lub w innych regularnych odstępach czasu. Próbki ze zbiornika testowego oraz z próbką ślepą filtrowane są przez dokładnie oczyszczony filtr papierowy. Pierwsze 5 ml przefiltrowanej substancji testowej jest odrzucane. Szlamy trudne do przefiltrowania mogą być wcześniej usunięte przez 10-minutowe odwirowywanie. Oznaczenia DOC i COD dokonuje się co najmniej dwukrotnie. Badanie przeprowadza się przez okres 28 dni.
Uwaga: Mętne próbki filtrowane są przez filtry membranowe. Filtry membranowe nie mogą uwalniać ani wchłaniać żadnego materiału organicznego.
Zbiornik zawierający znaną substancję należy prowadzić równolegle z każdą badaną serią w celu kontroli funkcjonalnej wydajności aktywnego szlamu. W tym celu przydatny jest dietylenoglikol.
Jeżeli analizy przeprowadzane są w stosunkowo krótkich odstępach czasu (np. raz dziennie), przystosowanie można łatwo rozpoznać po krzywej degradacji (zob. wykres 2). Dlatego też niniejszego badania nie należy rozpoczynać bezpośrednio przed weekendem.
W przypadku gdy dostosowanie wystąpi na końcu okresu badania, badanie możne zostać przedłużone do chwili zakończenia degradacji.
Uwaga: Jeżeli konieczna jest szersza wiedza w zakresie zachowania dostosowanego szlamu, ten sam aktywny szlam należy poddać działaniu tego samego materiału testowego zgodnie z następującą procedurą:
Należy wyłączyć mieszadło oraz napowietrzacz i umożliwić osadzenie się aktywowanego szlamu. Zebrać supernatan, nalać dwie litry wody testowej, mieszać przez 15 minut i umożliwić ponowne osadzenie się szlamu. Po ponownym zebraniu supernatanu należy użyć pozostały szlam w celu powtórzenia badania z tym samym materiałem testowym zgodnie z 1.6.1.4 i 1.6.2 powyżej. Można również wyizolować aktywny szlam przez odwirowanie zamiast osadzenia.
Dostosowany szlam można zmieszać ze świeżym szlamem w celu uzyskania stężenia o zawartości 0,2–1 g suchej masy/litr.
Zazwyczaj próbki filtrowane są przez starannie umyty filtr papierowy (do mycia należy użyć wodę dejonizowaną).
Próbki, które wciąż pozostają mętne należy filtrować przez filtry membranowe (0,45 μm).
Stężenie DOC ustala się dwukrotnie na podstawie filtrowanej próbki (pierwsze 5 ml należy odrzucić) za pomocą przyrządu TOC. Jeżeli nie można dokonać analizy filtratu w tym samym dniu, należy przechować go w chłodziarce do następnego dnia. Nie zaleca się dłuższego okresu przechowywania.
Stężenie DOC ustala się w filtratach próbki za pomocą analitycznego określenia COD na podstawie procedury ustalonej w poz. bibliograficznej (2), poniżej.
2. DANE I ANALIZA
Stężenia DOC i/lub COD ustala się co najmniej dwukrotnie w próbkach zgodnie z 1.6.2, powyżej. Degradacja w czasie T obliczana jest zgodnie ze wzorem (z definicjami) podanym zgodnie z 1.2, powyżej.
Stopień degradacji zaokrąglany jest do najbliższej wartości procentowej. Stopień degradacji uzyskany po zakończeniu badania odnotowywany jest jako „Biodegradacja w teście Zan-Wellens”.
Uwaga: W przypadku uzyskania całkowitej degradacji przed zakończeniem okresu badania oraz gdy taki wynik potwierdzony jest drugą analizą przeprowadzoną w dniu następnym, wówczas badanie może zostać zakończone.
3. SPRAWOZDANIE
3.1. SPRAWOZDANIE Z BADAŃ
Sprawozdanie z badań powinno, jeżeli możliwe, zawierać następujące informacje:
|
— |
początkowe stężenie substancji testowej, |
|
— |
wszelkie inne informacje oraz wyniki badań dotyczące substancji testowej, substancji odniesienia jeżeli zostały użyte, i próbki ślepej, |
|
— |
stężenie po trzech godzinach, |
|
— |
krzywa biodegradacji wraz z opisem, |
|
— |
data i lokalizacja pobrania próbek badanych organizmów, status dostosowania, użyte stężenie itd., |
|
— |
naukowe powody jakichkolwiek zmian procedury badania. |
3.2. INTERPRETACJA WYNIKÓW
Ubytek DOC (COD), stopniowo następujący przez okres badania obejmujący dni lub tygodnie, wskazuje, iż substancja testowa ulega biodegradacji.
Jednakże fizyko-chemiczna adsorpcja może w niektórych przypadkach odegrać istotną rolę, a sygnalizowane jest to w przypadku całkowitego lub częściowego ubytku początkowego w ciągu pierwszych trzech godzin, a różnica między grupą kontrolną i badanymi supernatanami pozostaje na niespodziewanie niskim poziomie.
Konieczne jest przeprowadzenie dalszych badań jeżeli istnieje potrzeba rozróżnienia między biodegradacją (lub częściową biodegradacją) a adsorpcją.
Można to osiągnąć na wiele sposobów, ale najbardziej przekonujące jest użycie supernatantu lub szlamu jako inokulum w teście podstawowym (najlepiej teście respirometrycznym).
Substancje testowe powodujące wysoki, nieadsorpcyjny ubytek DOC (COD) w niniejszym badaniu należy uważać za substancje wpływające biodegradacyjnie. Częściowy, nieadsorpcyjny ubytek oznacza, iż substancja chemiczna ulega biodegradacji co najmniej w pewnym stopniu. Niski lub zerowy poziom ubytku DOC (COD) może być spowodowany zahamowaniem aktywności drobnoustrojów przez substancję testową, co można również wykryć przez rozpad lub utratę szlamu, dającego mętne supernatany. Badanie należy powtórzyć, wykorzystując niskie stężenie substancji testowej.
Użycie specyficznej dla danego związku metody analitycznej lub substancji testowej znakowanej izotopem węgla 14C zwiększa czułość metody. W przypadku związków znakowanych 14C, odzyskanie 14CO2 potwierdzi, że zaszła biodegradacja.
W przypadku podawania wyników początkowej biodegradacji, jeżeli możliwe, należy wyjaśnić zmianę w budowie chemicznej, która powoduje utratę reakcji macierzystej substancji testowej.
Potwierdzenie metody analitycznej musi być dostarczone wraz z reakcją, która zaszła w próbce ślepej.
4. BIBLIOGRAFIA
|
(1) |
OECD, Paryż, 1981, Wytyczne Badania 302 B, decyzja Rady C(81) 30 wersja ostateczna. |
|
(2) |
Załącznik V C.9 Degradacja: Chemiczne zapotrzebowanie tlenu, dyrektywa Komisji 84/449/EWG (Dz.U. L 251 z 19.9.1984, s. 1). |
Dodatek
PRZYKŁAD ANALIZY
|
Związek organiczny: |
4-kwas etoksybenzoesowy |
|
Teoretyczne stężenie substancji testowej: |
600 mg/l |
|
Teoretyczne DOC: |
390 mg/l |
|
Inokulum |
Oczyszczalnia ścieków z |
|
Stężenie: |
1 gram suchej masy/litr |
|
Poziom dostosowania: |
Niedostosowany |
|
Analiza: |
Ustalenie DOC |
|
Ilość próbki: |
3 ml |
|
Substancja kontrolna: |
Dietylenoglikol |
|
Toksyczność związku chemicznego: |
Brak skutków toksyczności poniżej 1 000 mg/l Użyty test: Test probówkowy fermantacji |
|
Czas |
Substancja kontrolna |
Substancja testowa |
|||||
|
Próba ślepa DOC (16) mg/l |
DOC (16) mg/l |
DOC netto mg/l |
Degradacja % |
DOC (16) mg/l |
DOC netto mg/l |
Degradacja % |
|
|
0 |
— |
— |
300,0 |
— |
— |
390,0 |
— |
|
3 godziny |
4,0 |
298,0 |
294,0 |
2 |
371,6 |
367,6 |
6 |
|
1 dzień |
6,1 |
288,3 |
282,2 |
6 |
373,3 |
367,2 |
6 |
|
2 dni |
5,0 |
281,2 |
276,2 |
8 |
360,0 |
355,0 |
9 |
|
5 dni |
6,3 |
270,5 |
264,2 |
12 |
193,8 |
187,5 |
52 |
|
6 dni |
7,4 |
253,3 |
245,9 |
18 |
143,9 |
136,5 |
65 |
|
7 dni |
11,3 |
212,5 |
201,2 |
33 |
104,5 |
93,2 |
76 |
|
8 dni |
7,8 |
142,5 |
134,7 |
55 |
58,9 |
51,1 |
87 |
|
9 dni |
7,0 |
35,0 |
28,0 |
91 |
18,1 |
11,1 |
97 |
|
10 dni |
18,0 |
37,0 |
19,0 |
94 |
20,0 |
2,0 |
99 |
Wykres 1
Przykłady krzywych biodegradacji
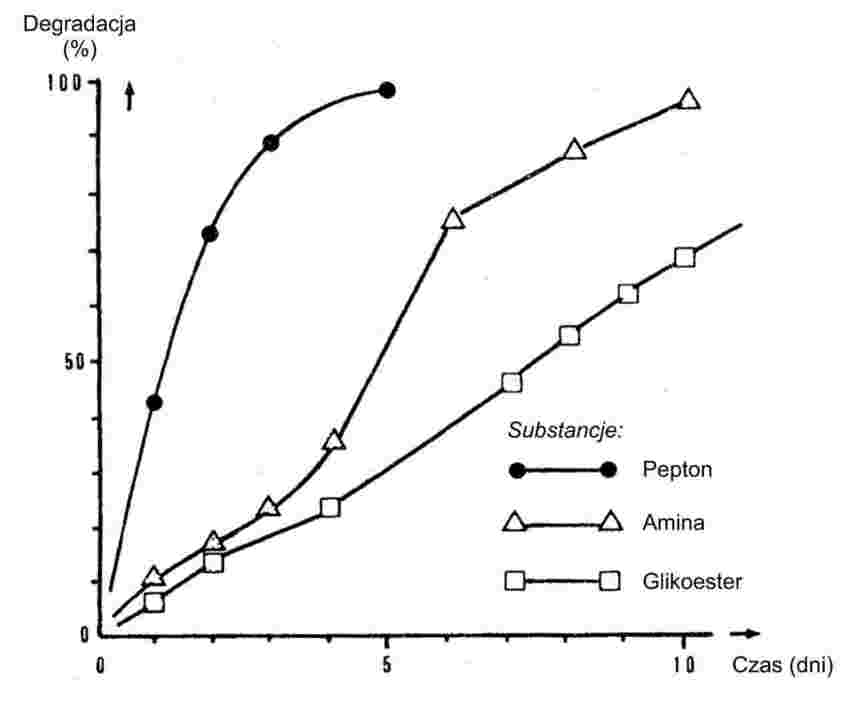
Wykres 2
Przykłady dostosowania szlamu
C.10. BIODEGRADACJA
BADANIA SYMULACJI AKTYWOWANYCH SZLAMÓW
1. METODA
1.1. WSTĘP
1.1.1. Uwagi ogólne
Metodę stosuje się jedynie w odniesieniu do tych substancji organicznych, które przy wielkości stężenia używanego w badaniu:
|
— |
są rozpuszczalne w wodzie w stopniu koniecznym do przygotowania roztworów testowych, |
|
— |
posiadają nieznaczne ciśnienie pary w warunkach badania, |
|
— |
nie hamują bakterii. |
Informacje dotyczące względnych proporcji głównych składników materiału testowego będą użyteczne przy interpretacji otrzymanych wyników, w szczególności w tych przypadkach gdzie wyniki są na niskim poziomie lub marginalne.
Pożądane są informacje dotyczące wpływu toksyczności substancji na drobnoustroje w celu dokonania interpretacji wyników o niskim poziomie oraz wyboru właściwych wielkości stężeń.
1.1.2. Ustalenie całkowitej biodegradacji (analizy DOC/COD)
Celem metody jest ustalenie całkowitej biodegradacji przez pomiar stopnia ubytku substancji i jakichkolwiek metabolitów w modelu zakładu oczyszczającego ścieki przez aktywowane szlamy przy stężeniu odpowiadającym > 12 mg DOC/litr (lub około 40 mg COD/litr); 20 mg DOC/litr wydaje się być optymalne. (DOC = rozwiązany węgiel organiczny; COD = chemiczne zapotrzebowanie tlenu).
Należy ustalić skład węgla organicznego (lub chemiczne zapotrzebowanie tlenu) materiału testowego.
1.1.3. Ustalenie początkowej biodegradacji (analizy szczególne)
Celem metody jest ustalenie początkowej biodegradacji substancji w modelu zakładu oczyszczającego ścieki przez aktywowane szlamy, przy stężeniu w wysokości około 20 mg/litr, wykorzystując szczególną metodę analityczną (można wykorzystać niższe lub wyższe stężenie, jeżeli zezwala na to metoda analityczna i stężenie toksyczności). Powyższe pozwala na oszacowanie początkowej biodegradacji substancji (zanik macierzystej budowy chemicznej).
Celem niniejszej metody nie jest ustalenie mineralizacji substancji testowej.
Wymagana jest dostępność do odpowiedniej metody analitycznej w celu ustalenia substancji testowej.
1.2. DEFINICJE I JEDNOSTKI
1.2.1. Analizy DOC/COD
Stopień ubytku substancji podawany jest przez:
|
|
[1 (a)] |
gdzie:
|
DR |
= |
procentowy stopień ubytku substancji DOC (lub COD) w danym średnim czasie retencji odnoszącym się do materiału testowego, |
|
T |
= |
stężenie materiału testowego w cieczy wpływającej do zbiornika w mg DOC/litr (lub mg COD/litr), |
|
E |
= |
stężenie DOC (lub COD) w cieczy wypływającej ze zbiornika w jednostce testowej w mg DOC/litr (lub mg COD/litr), |
|
E0 |
= |
stężenie DOC (lub COD) w cieczy wypływającej ze zbiornika w próbce ślepej w mg DOC/litr (lub mg COD/litr). |
Degradację stwierdza się jako procent ubytku DOC (lub COD) w danym czasie retencji w odniesieniu do materiału testowego.
1.2.2. Analizy szczególne
Procentowe wydzielenie testowej substancji z fazy wodnej (Rw) w danym średnim czasie retencji wyrażone jest przez:
|
|
[1 (b)] |
gdzie:
|
Cl |
= |
stężenie substancji w cieczy wpływającej do zbiornika w jednostce testowej (mg substancji/litr, ustalone na podstawie analiz szczególnych), |
|
C0 |
= |
stężenie substancji w cieczy wypływającej ze zbiornika w jednostce testowej (mg substancji/litr, ustalone na podstawie analiz szczególnych). |
1.3. SUBSTANCJE ODNIESIENIA
W niektórych przypadkach podczas badania nowych substancji użyteczne mogą okazać się substancje odniesienia; jednakże nie można jeszcze zalecić szczególnych substancji odniesienia.
1.4. ZASADA METODY BADAWCZEJ
W celu określenia całkowitej biodegradowalności należy równolegle uruchomić dwie pilotowe jednostki szlamu aktywowanego (test potwierdzający OECD lub jednostki porowatego wkładu). Substancja testowa dodawana jest do cieczy wpływającej do zbiornika (ściek syntetyczny lub wewnętrzny) jednej z jednostek, podczas gdy druga jednostka otrzymuje czysty ściek. W celu określenia podstawowej biodegradowalności, z analizą szczególną cieczy wpływającej do i wypływającej ze zbiornika, należy użyć tylko jednej jednostki.
Dokonywany jest pomiar stężeń DOC (lub COD) w cieczach wypływających ze zbiornika lub stężenia substancji ustalane są w oparciu o analizy szczególne.
Ze względu na materiał testowy nie dokonuje się pomiaru DOC, ale jedynie oznacza jego wartość.
W przypadku przeprowadzania pomiarów DOC (lub COD) zakłada się, iż różnica średnich stężeń między cieczami testowymi wypływającymi ze zbiornika a kontrolnymi cieczami wypływającymi ze zbiornika spowodowana jest brakiem degradacji materiału testowego.
W przypadku przeprowadzania szczególnych analiz można dokonać pomiaru stężenia molekuły macierzystej (biodegradacja podstawowa).
Jednostki mogą być eksploatowane zgodnie z „trybem jednostek połączonych”, zgodnie z metodą transinokulacji.
1.5. KRYTERIA JAKOŚCIOWE
Początkowe stężenie substancji zależy od rodzaju przeprowadzonej analizy i jej ograniczeń.
1.6. OPIS METODY BADAWCZEJ
1.6.1. Przygotowanie
1.6.1.1.
Wymagane są dwie jednostki tego samego rodzaju, z wyjątkiem przypadków gdy przeprowadzane są analizy szczególne. Można użyć dwa rodzaje jednostek:
Test potwierdzający OECD
Sprzęt (dodatek 11) składa się ze zbiornika (A) do magazynowania ścieków syntetycznych, pompy dozującej (B), zbiornika napowietrzającego (C), osadnika (D), powietrznego podnośnika cieczy (E) do zwracania do obiegu aktywnego szlamu i naczynia (F) do zbierania poddawanych procesowi cieczy wypływających ze zbiornika.
Zbiorniki (A) i (F) muszą być wykonane ze szkła lub odpowiedniego tworzywa sztucznego o objętości co najmniej 24 litrów. Pompa (B) musi zapewniać stały przepływ syntetycznego ścieku do napowietrzanego zbiornika; użyć można każdy odpowiedni system, po warunkiem że zapewniony zostanie przepływ i stężenie. W trakcie zwyczajnej eksploatacji wysokość osadnika (D) ustalona jest w taki sposób, aby jego objętość zawarta w napowietrzanym zbiorniku wynosiła trzy litry zmieszanego płynu. Spiekana kostka napowietrzania (G) jest zawieszana w zbiorniku (C) na wierzchołku stożka. Ilość powietrza wdmuchiwanego przez napowietrzacz może być mierzona przy użyciu przepływomierza.
Powietrzny podnośnik cieczy (E) jest umieszczony w taki sposób, że aktywny szlam z osadnika jest nieustannie i regularnie zwracany do zbiornika napowietrzającego (C).
„Porowaty wkład”
Porowaty wkład wykonany jest z płyt porowatego polietylenu (o grubości 2 mm, maksymalny rozmiar pory 95 μm), które tworzą walce o średnicy 14 cm o stożkowej podstawie 45o (rysunki 1 i 2 dodatku 2). Porowaty wkład jest zawarty w nieprzepuszczalnym zbiorniku wykonanym z odpowiedniego plastiku o średnicy wylotu 15 cm oraz wysokości 17,2 cm części walcowatej, co określa objętość (3 litrów) w naczyniu. Na szczycie wewnętrznego zbiornika znajduje się sztywna wspierająca opaska wykonana z odpowiedniego tworzywa sztucznego w taki sposób, aby powstała przestrzeń cieczy wypływającej ze zbiornika, wynosząca 0,5 cm między zbiornikiem wewnętrznym a zewnętrznym.
Porowaty wkład można zamocować u podstawy termostatycznie kontrolowanej kąpieli wodnej. Powietrze dostarczane jest do podstawy wewnętrznego zbiornika, na której umieszczone są odpowiednie dyfuzory.
Zbiorniki (A) i (E) muszą być wykonane ze szkła lub odpowiedniego tworzywa sztucznego i muszą posiadać objętość 24 litrów. Pompa (B) musi zapewniać stały przepływ syntetycznego ścieku do napowietrzanego zbiornika; użyć można każdy odpowiedni system, po warunkiem że zapewniony zostanie przepływ i stężenie.
Wymagane są zapasowe wewnętrzne porowate wkłady w celu zastąpienia jakichkolwiek naczyń, które mogą zostać zablokowane w trakcie używania; zablokowane naczynia należy oczyścić przez 24-godzinne zanurzenie w roztworze podchlorynu po gruntownym umyciu bieżącą wodą.
1.6.1.2.
Urządzenia do filtracji membranowej i filtry membranowe o rozmiarach porów 0,45 μm. Filtry membranowe są odpowiednie, jeżeli zostanie potwierdzone, że nie uwalniają węgla ani nie absorbują substancji w fazie filtracji.
1.6.1.3.
Każda odpowiednia syntetyczna pożywka lub wewnętrzne ścieki mogą zostać użyte do badania.
Przykład syntetycznej pożywki
Rozpuszczenie w każdym litrze wody bieżącej:
|
Pepton: |
160 mg, |
|
Ekstrakt z mięsa: |
110 mg, |
|
Karbamid: |
30 mg, |
|
NaCl: |
7 mg, |
|
CaCl2.2H20: |
4 mg, |
|
MgSO4.7H20: |
2 mg, |
|
K2HP04: |
28 mg. |
Ścieki wewnętrzne
Świeże ścieki należy zebrać każdego dnia z przepływowego odstojnika podstawowego w oczyszczalni ścieków, oczyszczającej w przeważającej mierze ścieki wewnętrzne.
1.6.1.4.
Należy przygotować roztwór materiału testowego, np. 1 %, celem dodania do jednostki testowej. Należy ustalić stężenie materiału w celu poznania właściwej objętości materiału mającego być dodanym do ścieków lub bezpośrednio do jednostki przez drugą pompę dla uzyskania wymaganego stężenia.
1.6.1.5.
Uwagi: W przypadku wykorzystania wewnętrznych ścieków bezcelowe byłoby użycie inokulum o niskim stężeniu bakteryjnym, ale wykorzystać można aktywowany szlam.
Zastosować można odmianę inokulum.
Podaje się trzy przykłady odpowiedniego inokulum:
|
a) |
Inokulum pochodzące z cieczy powtórnie opuszczającej zbiornik Inokulum należy uzyskać z wtórnej cieczy wypływającej ze zbiornika o dobrej jakości zebranej z oczyszczalni ścieków oczyszczającej przeważnie ścieki wewnętrzne. Ciecze wypływające ze zbiornika należy przetrzymywać w warunkach tlenowych w okresie między zebraniem, a zastosowaniem. W celu przygotowania inokulum próbka jest filtrowana przez gruboziarnisty filtr, a pierwsze 200 ml filtratu należy odrzucić. Filtrat należy przechowywać w warunkach tlenowych aż do chwili jego użycia. Inokulum należy użyć w dniu jej zebrania. Do szczepienia należy użyć co najmniej 3 ml inokulum. |
|
b) |
Inokulum łączne Inokulum z wtórnej cieczy wypływającej ze zbiornika: zob. opis powyżej. Inokulum z gleby: 100 g gleby ogrodowej (żyznej, niesterylnej) zawieszone jest w 1 000 ml bezchlorowej wody pitnej. (Gleby o znacznej frakcji iłowej, piasek lub próchnica są nieodpowiednie). Po wymieszaniu zawiesina osiada przez okres 30 minut. Supernatan filtrowany jest przez gruboziarnistą bibułę filtracyjną, pierwsze 200 ml należy odrzucić. Należy rozpocząć natychmiastowe napowietrzanie filtratu aż do chwili jego użycia. Inokulum należy użyć w dniu zebrania. Inokulum z wody powierzchniowej: Następne częściowe inokulum uzyskiwane jest z mezosaprobicznej wody powierzchniowej. Próbka filtrowana jest przez gruboziarnisty papier, pierwsze 200 ml należy odrzucić. Filtrat należy przechowywać w warunkach tlenowych aż do chwili jego użycia. Inokulum należy użyć w dniu zebrania. Równe wielkości trzech częściowych próbek inokulum są łączone i dokładnie mieszane, ostateczne inokulum uzyskiwane jest z powstałej mieszaniny. Do inokulacji należy użyć co najmniej 3 ml inokulum. |
|
c) |
Inokulum z aktywnego szlamu Jako inolukum można użyć pewnej ilości (nie więcej niż 3 litry) aktywowanego szlamu (zawieszona sucha masa do 2,5 g/litr) pobranego ze zbiornika napowietrzającego w oczyszczalni ścieków, w przeważającej mierze oczyszczającej ścieki wewnętrzne. |
1.6.2. Procedura
Badanie przeprowadzane jest w temperaturze pokojowej; temperaturę należy utrzymywać w przedziale 18–25 oC.
Odpowiednio do potrzeb, badanie można przeprowadzić w niższej temperaturze (do 10 oC); w przypadku degradacji substancji dalsze badanie nie jest konieczne. Jednakże w przypadku gdy substancja nie ulegnie degradacji, badanie należy przeprowadzić przy stałej temperaturze w przedziale 18–25 oC.
1.6.2.1.
Okres wzrostu/stabilizacji szlamu jest okresem, w którym stężenie zawieszonej suchej masy aktywnego szlamu oraz wydajność jednostek rozwija się do stanu stałego w zastosowanych warunkach działalności,
Okres eliminacji to okres, który trwa od chwili pierwszego dodania substancji testowej do czasu, gdy jej ubytek osiągnie plateau (względnie stałą wartość). Okres nie może przekroczyć sześciu miesięcy.
Okres analizy wynosi trzy tygodnie od chwili, gdy ubytek substancji testowej osiągnie względnie stałą i zazwyczaj wysoką wartość. Dla tych substancji, które nie wykazują degradacji lub wykazują znikomą degradację w pierwszych sześciu tygodniach, okres analizy przyjmuje się jako kolejne trzy tygodnie.
Początkowo należy napełnić jednostkę (jednostki) inokulum wymaganym do przeprowadzenia jednego badania i zmieszanym z cieczą wpływającą do zbiornika.
Następnie należy zainstalować napowietrzacz (i powietrzny podnośnik cieczy (E) w przypadku jednostek testu potwierdzającego OECD) oraz jednostkę dozującą (B).
Ciecz wpływająca do zbiornika bez substancji testowej musi przechodzić przez napowietrzane naczynie (C) w tempie jednego litra na godzinę albo w tempie półtorej litra na godzinę; pozwala to uzyskać średni czas retencji wynoszący trzy lub sześć godzin.
Tempo napowietrzania należy regulować w taki sposób, aby zawartość zbiornika (C) utrzymywana była stale w zawieszeniu, podczas gdy zawartość rozpuszczonego tlenu wynosi co najmniej 2 mg/litr.
Wytwarzanie piany należy powstrzymać właściwymi środkami. Nie wolno używać środków przeciwpieniących, które hamują działanie szlamu aktywowanego.
Szlam nagromadzony wokół wylotu zbiornika napowietrzającego (C) (i, w przypadku jednostek testu potwierdzającego OECD, u podstawy osadnika (D) oraz w obiegu) musi zostać ponownie dodany do zmieszanego płynu co najmniej raz dziennie przez zeszczotkowanie lub w inny właściwy sposób.
Kiedy szlamu nie udaje się oddzielić, jego gęstość można zwiększyć przez dodanie porcji 2 ml 5 % roztworu chlorku żelaza, powtarzane w razie potrzeby.
Ciecz wypływająca ze zbiornika zbierana jest w zbiorniku (E lub F) przez 20–24 godzin, a próbka pobierana jest po całkowitym wymieszaniu. Zbiornik (E lub F) należy dokładnie oczyścić.
W celu monitorowania i kontroli wydajności procesu dokonuje się pomiaru chemicznego zapotrzebowania tlenu (COD) lub rozwiązanego węgla organicznego (DOC), filtratu nagromadzonej cieczy wypływającej ze zbiornika co najmniej dwa razy w tygodniu, a także przefiltrowanej cieczy wpływającej do zbiornika (używając membrany o wielkości poru 0,45 μm, pierwsze 20 ml (około) filtratu należy odrzucić).
Obniżenie wartości COD lub DOC powinno się ustabilizować w chwili uzyskania w miarę regularnej dziennej wartości degradacji.
Zawartość suchej masy aktywowanego szlamu w zbiorniku napowietrzającym powinna być określana dwa razy w tygodniu. Jednostki mogą działać na jeden lub dwa sposoby: należy ustalić zawartość suchej masy w aktywnym szlamie dwa razy w tygodniu i – jeżeli niniejsza zawartość wyniesie więcej niż 2,5 g/litr – nadwyżka aktywnego szlamu musi zostać odrzucona albo 500 ml zmieszanego płynu usuwana jest z każdego naczynia raz dziennie w celu otrzymania średniego czasu retencji szlamu wynoszącego sześć dni.
W przypadku gdy parametry pomiarowe i szacowane (wydajność procesu (odnośnie do usuwania COD lub DOC), stężenie szlamu, oddzielanie się szlamu, mętność cieczy wypływających ze zbiorników itd.) dwóch jednostek są dostatecznie stabilne, substancję testową można wprowadzić do cieczy wpływającej do jednej z jednostek, zgodnie z 1.6.2.2.
Alternatywnie, substancję testową można dodać na początku okresu wzrostowego szlamu (1.6.2.1), w szczególności gdy szlam dodawany jest jako inokulum.
1.6.2.2.
Należy utrzymywać warunki działania w okresie eliminacji oraz należy dodawać odpowiedni roztwór podstawowy (około 1 %) materiału testowego do cieczy wpływającej do jednostki testowej w celu otrzymania pożądanego stężenia materiału testowego (w przybliżeniu 10–20 mg DOC/litr lub 40 mg COD/litr) w ścieku. Powyższe można otrzymać przez codzienne wmieszanie roztworu podstawowego do ścieków lub przez oddzielny system pompowania. Właściwe stężenie można otrzymać stopniowo. W przypadku braku wystąpienia skutków oddziaływania toksyczności substancji testowej na aktywowany szlam można również przeprowadzić badanie wyższych stężeń.
Do jednostki z próbką ślepą wprowadza się jedynie ciecz wpływającą do zbiornika bez dodanych substancji, pobierane są odpowiednie wielkości cieczy wypływających ze zbiorników do analizy i filtrowane przez filtry membranowe (0,45 μm), odrzucając (około) 20 ml pierwszego filtratu.
Przefiltrowane próbki muszą zostać przeanalizowane w tym samym dniu, w przeciwnym razie muszą być przechowane odpowiednią metodą, na przykład przez użycie 0,05 ml 1 % roztworu chlorku rtęciowego (HgCl2) dla każdego 10 ml filtratu lub przez przechowanie ich w temperaturze 2–4 oC przez okres nie dłuższy niż 24 godziny lub poniżej - 18 oC przez dłuższe okresy czasu.
Okres eliminacji, z dodatkiem substancji testowej, nie powinien przekraczać sześciu tygodni, a okres analizy nie powinien być krótszy niż trzy tygodnie, tj. około 14–20 oznaczeń powinno zostać dokonane w celu obliczenia wyników końcowych.
Tryb jednostek połączonych
Połączenie jednostek można osiągnąć przez wewnętrzną wymianę 1,5 litra zmieszanego płynu (włączając szlam) z napowietrzanego zbiornika aktywnego szlamu między dwiema jednostkami raz dziennie. W przypadku silnie absorbującego materiału testowego jedynie 1,5 litra supernatanu pobierane jest z osadnika i wlewane do zbiornika z aktywowanym szlamem w drugiej jednostce.
1.6.2.3.
Przeprowadzić można dwa rodzaje analizy w celu obserwacji zachowania substancji:
DOC i COD
Analizę stężenia DOC przeprowadza się dwukrotnie za pomocą analizatora węglowego i/lub wartości COD zgodnie z odniesieniem (2).
Szczególne analizy
Stężenia substancji testowej ustala się za pomocą odpowiedniej metody analitycznej. Jeżeli to możliwe, należy dokonać szczególnego ustalenia absorpcji substancji przez szlam.
2. DANE I OSZACOWANIE
2.1. TRYB JEDNOSTEK POŁĄCZONYCH
W przypadku wykorzystania „trybu jednostek połączonych” dzienny stopień ubytku substancji, DR, oblicza się zgodnie z 1.2.1.
Te dzienne stopnie usunięcia DR są korygowane względem DRc dla transferu materiału spowodowanego procedurą transinokulacji zgodnie z równaniem [2] dla trzygodzinnego średniego czasu retencji lub równaniem [3] dla czasu sześciogodzinnego.
|
|
[2] |
|
|
[3] |
Obliczana jest średnia serii wartości DRc, a ponadto zgodnie z równaniem [4] obliczane jest odchylenie standardowe
|
|
[4] |
gdzie:
|
SDRc |
= |
odchylenie standardowe serii wartości DRc, |
|
|
= |
średnia wartości DRc, |
|
n |
= |
liczba okrążeń |
Oddzielne wartości serii DRc są eliminowane zgodnie z odpowiednią procedurą statystyczną, np. Nalimova (6), przy 95 % stopniu prawdopodobieństwa i średnia wartość oraz odchylenie standardowe wartości DRc (wolnej od wartości oddzielnych) są obliczane ponownie.
Następnie końcowe wyniki obliczane są za pomocą równania [5], jako:
|
|
[5] |
gdzie:
|
tn-1; α |
= |
wartość tablicowa t dla n wartości par E i E0 i pewnością statystyczną P (P = 1-α), gdy wartość P = 95 % (1). |
Wynik podany jest jako średnia z granicami tolerancji o poziomie prawdopodobieństwa 95 %, odpowiednie odchylenie standardowe oraz liczba danych z zestawu wartości DRc wolnych od wartości oddzielnych oraz liczba wyników odbiegających, np.
|
DRc |
= |
98,6±2,3 % ubytku DOC, |
|
s |
= |
4,65 % ubytku DOC, |
|
n |
= |
18, |
|
x |
= |
liczba wyników odbiegających. |
2.2. TRYB JEDNOSTEK ODDZIELNYCH
Wydajność jednostek testowych można sprawdzić w następujący sposób:

Dzienna wartość usunięć może zostać graficznie zapisana w celu wykazania wszelkich tendencji, np. tendencji do aklimatyzacji.
2.2.1. Użycie ustaleń wartości COD/DOC
Dzienna wartość stopnia usunięcia DR jest obliczana zgodnie z 1.2.1.
Obliczana jest średnia serii wartości DR; ponadto jej odchylenie standardowe oblicza się zgodnie z:
|
|
[6] |
gdzie:
|
SDR |
= |
odchylenie standardowe serii wartości DRi, |
|
|
= |
średnia wartości DRi, |
|
n |
= |
liczba okrążeń. |
Oddzielne wartości serii DRc są eliminowane zgodnie z odpowiednią procedurą statystyczną, np. Nalimova (6), przy 95 % stopniu prawdopodobieństwa i średnia wartość oraz odchylenie standardowe wartości DRc (wolnej od wartości oddzielnych) są obliczane ponownie.
Końcowe wyniki obliczane są za pomocą równania [7] jako:
|
|
[7] |
gdzie:
|
tn-1; α |
= |
wartość tablicowa t dla n wartości par E i E0 i pewnością statystyczną P (P = 1-α), gdy wartość P = 95 % (1). |
Wynik podany jest jako średnia z granicami tolerancji o poziomie prawdopodobieństwa 95 %, odpowiednie odchylenie standardowe oraz liczba danych z zestawu wartości DRc wolnych od wartości oddzielnych oraz liczba wyników odbiegających, np.
|
DR |
= |
(98,6 ± 2,3) % ubytku DOC, |
|
s |
= |
4,65 % ubytku DOC, |
|
n |
= |
18, |
|
x |
= |
liczba wyników odbiegających. |
2.2.2. Wykorzystanie szczególnych analiz
Procent wydalenia substancji testowej z fazy wodnej (Rw) obliczany jest zgodnie z 1.2.2.
3. SPRAWOZDANIE
3.1. SPRAWOZDANIE Z BADAŃ
Sprawozdanie z badań powinno, jeżeli możliwe, zawierać następujące informacje:
|
— |
formularz podany w dodatku 3, zawierający warunki działań badawczych, |
|
— |
rodzaj wybranych urządzeń (test potwierdzający OECD lub porowaty wkład), |
|
— |
rodzaj wybranego trybu działania: tryb jednostek połączonych lub oddzielnych, |
|
— |
rodzaj ścieku: syntetyczny lub wewnętrzny – w przypadku ścieków wewnętrznych, data i lokalizacja próbki, |
|
— |
rodzaj inokulum, data i lokalizacja próbki, |
|
— |
deklaracja wraz z opisem metody analitycznej w przypadku przeprowadzenia analiz szczególnych, |
|
— |
wykres ubytku COD lub DOC względem czasu, włączając okres eliminacji i okres analizy, |
|
— |
analityczna regeneracja substancji testowej jako COD wartości DOC w roztworze podstawowym, |
|
— |
w przypadku przeprowadzenia szczególnych analiz, wykres procentowego ubytku substancji testowej z fazy wodnej względem czasu (okres eliminacji i okres analizy), |
|
— |
średnia wartość ubytku DOC lub COD substancji testowej i odchylenie standardowe obliczane na podstawie wyników okresu analitycznego, tj. w przypadku gdy zachodzi stałe usuwanie materiału testowego lub okres równomiernego działania, |
|
— |
wykres stężenia aktywowanego szlamu względem czasu, |
|
— |
wszelkie uwagi dotyczące aktywnego szlamu (ilość odrzuconej nadwyżki szlamu, obecność pęcznienia, FeCl3 itd.), |
|
— |
stężenie substancji używanej w badaniu, |
|
— |
wszelkie wyniki analiz przeprowadzonych na szlamie, |
|
— |
wszelkie informacje oraz wyniki badania dotyczące substancji testowej i substancji odniesienia, jeżeli została użyta, |
|
— |
naukowe powody jakichkolwiek zmian procedury. |
3.2. INTERPRETACJA WYNIKÓW
Niska wartość ubytku substancji testowej z fazy wodnej może być spowodowana zahamowaniem aktywności drobnoustrojów przez substancję testową. Powyższe można również wykazać na podstawie rozpadu i utraty szlamu, dającego mętny supernatant, i przez spadek wydajności usuwania COD (lub DOC) jednostki pilotowej.
W niektórych przypadkach istotną rolę może odegrać absorpcja fizyko-chemiczna. Różnice między biologicznym działaniem na molekułę a adsorpcją fizyko-chemiczną można ujawnić w oparciu o przeprowadzone analizy szlamu po odpowiedniej desorpcji.
Konieczne jest przeprowadzenie dalszych badań, jeżeli istnieje potrzeba rozróżnienia między biodegradacją (lub częściową biodegradacją) a adsorpcją.
Można to osiągnąć na wiele sposobów, ale najbardziej przekonujące jest użycie supernatantu lub szlamu jako inokulum w teście podstawowym (najlepiej teście respirometrycznym)
Jeżeli zaobserwowana jest wysoka wartość DOC lub COD, spowodowane jest to biodegradacją, podczas gdy przy niskich wartościach ubytku substancji biodegradacji nie można odróżnić od wydalania substancji. Na przykład jeżeli związek rozpuszczalny wykazuje wysoką stałą adsorpcję w wysokości 98 % i wskaźnik nadwyżki straty szlamu wynosi 10 % na dzień, możliwe jest wydzielenie do 40 %; przy wskaźniku nadwyżki straty szlamu w wysokości 30 % wydzielenie z powodu adsorpcji i usunięci nadwyżki szlamu może wynieść do 65 % (4).
W przypadku wykorzystania analiz szczególnych należy zwrócić uwagę na powiązanie między strukturą substancji a zastosowaną analizą szczególną. W tym przypadku obserwowane zjawisko nie można interpretować jako mineralizację substancji.
4. BIBLIOGRAFIA
|
(1) |
OECD, Paryż, 1981, Wytyczne Badania 201, decyzja Rady C(81) 30 wersja ostateczna. |
|
(2) |
Załącznik V C.9 Degradacja: Chemiczne zapotrzebowanie tlenu, dyrektywa Komisji 84/449/EWG (Dz.U. L 251 z 19.9.1984, s. 1). |
|
(3) |
Painter, H. A., King, E. F., WRC Metoda porowatego wkładu dla oznaczenia biodegradacji, Raport Techniczny TR 70, czerwiec 1978, Wodny Ośrodek Badawczy, Zjednoczone Królestwo. |
|
(4) |
Wierich, P., Gerike, P., Eliminacja związków rozpuszczalnych, opornych i adsorbujących w oczyszczalniach ścieków działających na zasadzie szlamów aktywowanych, Ekotoksykologia i Bezpieczeństwo Środowiskowe, tom 5, nr 2, czerwiec 1981, str. 161–171; |
|
(5) |
Dyrektywy Rady 82/242/EWG i 82/243/EWG, Dziennik Urzędowy Wspólnot Europejskich L 109 z 22.4.1982 (Dz.U. L 109 z 22.4.1982, s. 1), zmieniające dyrektywy Rady 73/404/EWG i 73/405/EWG w sprawie biodegradacji detergentów (Dz.U. L 347 z 17.12.1973, s. 51). |
|
(6) |
Streuli, H., Fehlerhafte Interpretation und Anwendung von Ausreißertests, insbesondere bei Ringversuchen zur Uberpriifung analytisch-chemischer Untersuchungsmethoden, Fresenius-Zeitschriftfür Analytische Chemie, 303 (1980), str. 406–408. |
Dodatek 1
Rysunek 1

Rysunek 2
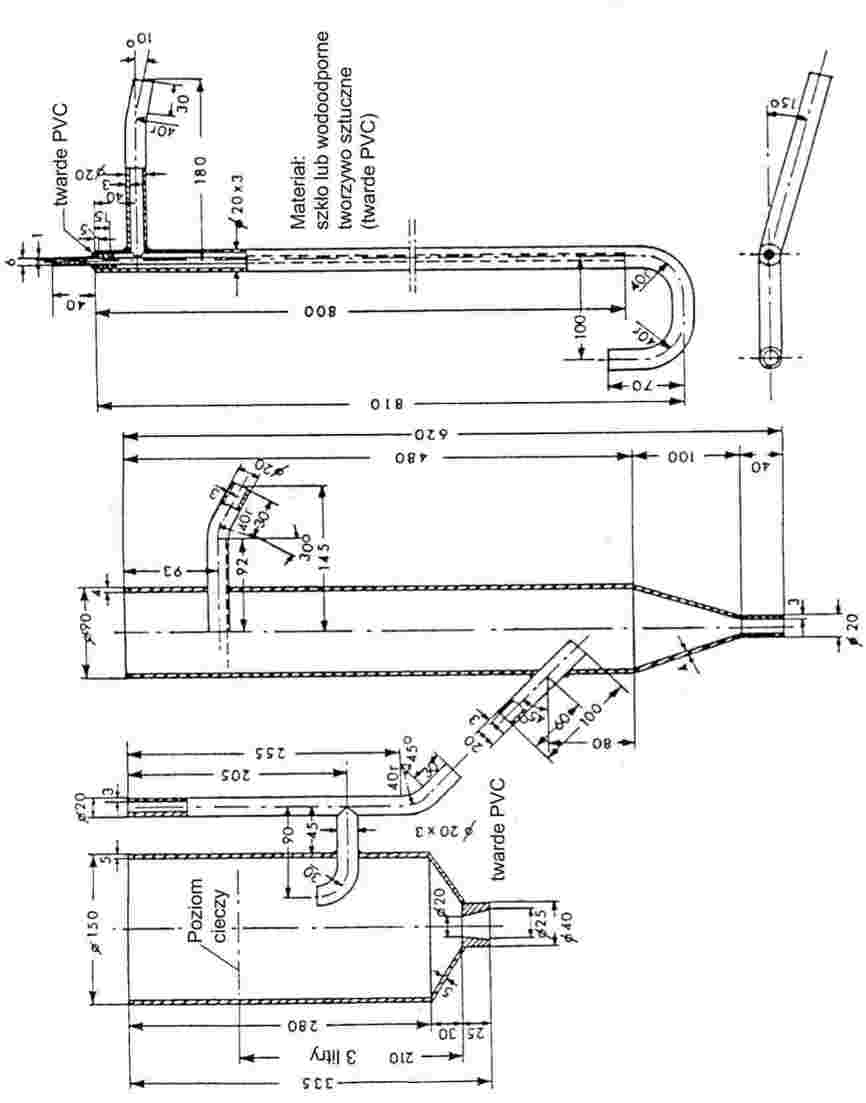
Dodatek 2
Rysunek 1
Sprzęt do określania stopnia biodegradacji

Rysunek 2
Szczegółowy opis trzylitrowego porowatego naczynia napowietrzającego
Dodatek 3
Warunki działalności w badaniu symulacji aktywnego szlamu
Kontrola w każdej grupie
Urzqdzenia
|
Test potwierdzający OECD |
|
|
Naczynie porowate |
|
Tryb działania
|
Pojedyncza jednostka |
|
|
Połączone jednostki |
|
|
Jednostki oddzielne |
|
Transinokulacja
|
Brak |
|
|
Aktywowany szlam |
|
|
Supernatan |
|
Średni czas retencji
|
Trzy godziny |
|
|
Sześć godzin |
|
Pożywka podstawowa
|
Ścieki wewnętrzne |
|
|
Ścieki syntetyczne |
|
Inokulum
|
Wtórne ciecze wypływające ze zbiornika |
|
|
Kompozyt |
|
|
Aktywny szlam |
|
Dodawanie materiału testowego
|
Od rozpoczęcia badania |
|
|
Stopniowy wzrost po |
|
|
uformowaniu się szlamu |
|
Analiza
|
Szczególna |
|
|
COD |
|
|
DOC |
|
C.11. BIODEGRADACJA
BADANIE HAMOWANIA ODDYCHANIA AKTYWOWANYCH SZLAMÓW
1. METODA
1.1. WSTĘP
Opisana metoda ocenia skutki wpływu substancji testowej na drobnoustroje, przez dokonywanie pomiaru stopnia oddychania w określonych warunkach w obecności różnych stężeń substancji testowej.
Celem tej metody jest dostarczenie bardzo szybkiej metody badań, za pomocą której można zidentyfikować związki, które mogą mieć niekorzystny wpływ na florę bakterii aerobowych oczyszczalni, oraz wskazać odpowiednie, nieinhibujące stężenia substancji testowych, które mają być użyte w testach biodegradowalności.
Badanie ustalania wielkości stężeń może poprzedzać badanie ostateczne. Dostarcza ono informacji dotyczących zakresu wielkości stężeń mających być zastosowanymi w badaniu głównym.
Do projektu badania należy włączyć dwie grupy kontrolne bez substancji testowej, jedną w chwili rozpoczęcia badania i drugą po zakończeniu serii badań. Należy skontrolować każdą partię aktywowanego szlamu, używając substancji odniesienia.
Niniejsza metoda łatwo stosuje się do substancji, które ze względu na ich rozpuszczalność w wodzie i niską lotność mogą pozostać w wodzie.
W odniesieniu do substancji o ograniczonej rozpuszczalności w badanym środku ustalenie EC50 może okazać się niemożliwe
Wyniki oparte na wchłanianiu tlenu mogą doprowadzić do błędnych wniosków w przypadku, gdy substancja testowa ma skłonność do rozszczepiania fosforylacji oksydacyjnej.
Przydatne jest posiadanie następujących informacji w celu przeprowadzenia badania:
|
— |
rozpuszczalność w wodzie, |
|
— |
ciśnienie pary, |
|
— |
wzór strukturalny, |
|
— |
czystość substancji testowej. |
Zalecenie
Aktywowany szlam może zawierać potencjalne organizmy chorobotwórcze i należy się z nim obchodzić ostrożnie.
1.2. DEFINICJE I JEDNOSTKI
Wskaźnik oddychania jest to zużycie tlenowe drobnoustrojów ściekowych w szlamie tlenowym, ogólnie wyrażane jako mg O2 na mg szlamu na godzinę.
W celu dokonania obliczeń hamującego wpływu substancji testowej przy danym stężeniu wskaźnik oddychania wyrażony jest jako procent średniej z dwóch kontrolnych wskaźników oddychania.

gdzie:
|
Rs |
= |
wskaźnik zużycia tlenu przy badanym stężeniu substancji testowej, |
|
Rc1 |
= |
wskaźnik zużycia tlenu, grupa kontrolna 1, |
|
Rc2 |
= |
wskaźnik zużycia tlenu, grupa kontrolna 2. |
W niniejszej metodzie EC50 jest stężeniem substancji testowej, przy którym wskaźnik oddychania wynosi 50 % wskaźnika wykazanego w grupie kontrolnej w warunkach opisanych w niniejszej metodzie.
1.3. SUBSTANCJE ODNIESIENIA
Zaleca się użycie 3,5-dichlorofenolu, znanego inhibitora oddychania, jako substancji odniesienia i przeprowadzenie badania w celu ustalenia EC50 każdej partii aktywowanego szlamu jako środek kontrolny w celu oznaczenia nieprawidłowości w czułości szlamu.
1.4. ZASADA METODY BADAWCZEJ
Wskaźnik oddychania aktywowanego szlamu zasilonego standardową ilością ścieku syntetycznego mierzony jest po upływie kontaktowego czasu 30 minut lub trzech godzin, lub po upływie tych okresów czasu łącznie. Dokonuje się także pomiaru wskaźnika oddychania tego samego aktywowanego szlamu w obecności różnych stężeń substancji testowej w odmiennych warunkach. Hamujący wpływ substancji testowej o danym stężeniu jest wyrażony jako procent średnich wskaźników oddychania z dwóch grup kontrolnych. Wartość EC50 obliczana jest z ustaleń różnych stężeń.
1.5. KRYTERIA JAKOŚCIOWE
Wyniki badania są ważne, jeżeli:
|
— |
wskaźniki oddychania dwóch grup kontrolnych wynoszą 15 % dla każdej z nich, |
|
— |
wartość EC50 (30 minut i/lub trzy godziny) 3,5-dichlorofenolu mieści się w zakresie 5–30 mg/litr. |
1.6. OPIS METODY BADAWCZEJ
1.6.1. Odczynniki
1.6.1.1.
Roztwory substancji testowej są przygotowywane w chwili rozpoczęcia badań z wykorzystaniem roztworu podstawowego. Stężenie roztworu podstawowego w wysokości 0,5 g/litr jest właściwe w przypadku stosowania niżej zalecanej procedury.
1.6.1.2.
Roztwór 3,5-dichlorofenolu można przygotować na przykład przez rozpuszczenie 0,5 g 3,5-dichlorofenolu w 10 ml 1 M NaOH, rozcieńczając w około 30 ml wody destylowanej, dodając podczas mieszania 0,5 M H2SO4 do punktu początkowego opadu – na koniec wymagane jest około 8 ml 0,5 M H2SO4 – a następnie należy rozcieńczyć mieszaninę wodą destylowaną do jednego litra. Wartość pH powinna wówczas wynosić 7–8.
1.6.1.3.
Syntetyczną pożywkę dla mikroflory ścieku aktywowanego otrzymuje się przez rozpuszczenie następujących ilości substancji w jednym litrze wody:
|
— |
16 g pepton, |
|
— |
11 g ekstrakt mięsa, |
|
— |
3 g mocznik, |
|
— |
0,7 g NaCl, |
|
— |
0,4 g CaCl2.2H2O, |
|
— |
0,2 g MgSO4.7H2O, |
|
— |
2,8 g K2HPO4. |
Uwaga 1: Niniejszy ściek syntetyczny jest 100-krotnym koncentratem w porównaniu z koncentratem opisanym w Raporcie technicznym OECD „Proponowana metoda określania biodegradacji środków powierzchniowo czynnych stosowanych w syntetycznych detergentach” (11 czerwca 1976 r.), z dodatkiem ortofosforanu dipotasu.
Uwaga 2: Jeżeli przygotowany środek nie zostanie niezwłocznie zastosowany, należy go przechowywać bez dostępu światła w temperaturze 0–4 oC, przez okres nie dłuższy niż jeden tydzień, w warunkach, które nie spowodują żadnej zmiany w jego składzie. Środek można również wysterylizować przed przechowaniem lub można dodać pepton i ekstrakt mięsa tuż przed rozpoczęciem badania. Przed użyciem należy go dokładnie zmieszać i dostosować pH.
1.6.2. Urządzenia
Urządzenia pomiarowe: dokładny projekt nie jest istotny. Jednakże powinna być przestrzeń u góry naczynia, a sonda powinna być ciasno dopasowana w szyjce kolby pomiarowej.
Wymagany jest zwykły sprzęt laboratoryjny, w szczególności następujące urządzenia:
|
— |
urządzenia pomiarowe, |
|
— |
urządzenie napowietrzające, |
|
— |
elektroda pH i urządzenia pomiarowe, |
|
— |
elektroda O2. |
1.6.3. Przygotowanie inokulum
Aktywowany szlam pochodzący z oczyszczalni ścieków w przeważającej mierze oczyszczającej ścieki wewnętrzne stosowany jest w badaniu jako mikrobiologiczne inokulum.
Jeżeli konieczne, po powrocie do laboratorium gruboziarniste cząsteczki można usunąć przez osadzanie przez krótki okres czasu, np. 15 minut, i przelanie górnej warstwy drobniejszej suchej masy w celu jej użycia. Alternatywnie, szlam można zmieszać przez okres kilku sekund, wykorzystując mikser.
Ponadto w przypadku podejrzenia występowania materiału hamującego szlam należy umyć bieżącą wodą lub roztworem izotonicznym. Po odwirowaniu należy przelać supernatant (procedurę powtarza się trzy razy).
Niewielką ilość szlamu należy zważyć i osuszyć. Na podstawie wyniku można obliczyć ilość mokrego szlamu, która ma być zawieszona w wodzie w celu uzyskania aktywowanego szlamu o wartości płynnej zawieszonej suchej masy wynoszącej 2–4 g/litr. Niniejszy poziom daje stężenie między 0,8 a 1,6 g/litr w badanej substancji, jeżeli przestrzegana jest niżej zalecana procedura.
Jeżeli szlamu nie można użyć w dniu jego zebrania, należy dodać 50 ml syntetycznego ścieku do każdego litra aktywowanego szlamu przygotowanego w sposób opisany powyżej; następnie należy go napowietrzać przez całą noc w temperaturze 20 ± 2 oC. Napowietrzanie należy stosować aż do chwili jego użycia w dniu następnym. Przed użyciem należy sprawdzić i dostosować pH, jeżeli jest to konieczne, do wartości pH 6–8. Płynną zawieszoną suchą masę należy ustalić w sposób określony w poprzednim ustępie.
Jeżeli wymagane jest użycie tej samej partii szlamu w kolejnych dniach badania (maksymalnie przez cztery dni), należy dodać kolejne 50 ml syntetycznej pożywki dla mikroflory ścieku aktywowanego na litr szlamu po zakończeniu każdego dnia roboczego.
1.6.4. Przeprowadzenie badania
|
Czas trwania/czas kontaktu: |
30 minut i/lub trzy godziny, podczas którego przeprowadzane jest napowietrzanie |
|
Woda: |
Woda pitna (odchlorowana w razie konieczności) |
|
Dostarczenie powietrza: |
Czyste, bezoleiste powietrze. Strumień powietrza 0,5–1 litr/minuta |
|
Aparatura pomiarowa: |
Kolba o płaskim dnie taka jak kolba BOD |
|
Tlenomierz: |
Odpowiednia elektroda tlenowa, z rejestratorem |
|
Pożywka: |
Ścieki syntetyczne (zob. powyżej) |
|
Substancja testowa: |
Substancja testowa jest świeżo przygotowywana przed rozpoczęciem badania. |
|
Substancje odniesienia: |
np. 3,5-dichlorofenol (co najmniej trzy stężenia) |
|
Kontrole: |
Zaszczepiona próbka bez substancji testowej |
|
Temperatura: |
20 ± 2 oC. |
Poniżej podano sugerowaną procedurę doświadczalną, którą można zastosować zarówno w badaniu, jak i do substancji odniesienia w trzygodzinnym czasie kontaktu.
Należy użyć kilka zbiorników (np. jednolitrowe zlewki).
Należy użyć co najmniej pięć wielkości stężeń, rozmieszczonych o stały czynnik najlepiej nieprzekraczający 3,2.
W czasie „0” 16 ml syntetycznej pożywki dla mikroflory ścieku aktywowanego uzupełniane jest wodą do objętości 300 ml. Należy dodać 200 ml inokulum bakteryjnego, a całą mieszaninę (500 ml) wlać do pierwszego zbiornika (pierwsza grupa kontrolna C1).
Zbiorniki testowe należy stale napowietrzać w celu zapewnienia, że rozpuszczony O2 nie opadnie poniżej wartości 2,5 mg/litr i że bezpośrednio przed pomiarem wskaźnika oddychania stężenie O2 wyniesie około 6,5 mg/litr.
Po „15 minutach” (15 minut stanowi bezwzględny, ale wygodny przedział czasowy) powyższa procedura jest powtarzana, z wyjątkiem 100 ml podstawowego roztworu substancji testowej, które dodaje się do 16 ml syntetycznego ścieku przed dodaniem wody do 300 ml i inokulum bakteryjnego w celu uzyskania objętości 500 ml. Następnie mieszanina ta jest wlewana do drugiego zbiornika i napowietrzana w sposób opisany powyżej. Proces powtarzany jest w 15-minutowych przedziałach czasu z różnymi wielkościami podstawowego roztworu substancji testowej celem uzyskania serii zbiorników zawierających różne stężenia substancji testowej. Na końcu przygotowuje się drugą grupę kontrolną (C2).
Po upływie trzech godzin odnotowuje się pH oraz wlewa się dobrze zmieszaną próbkę pobraną z zawartości pierwszego zbiornika do urządzeń pomiarowych i dokonuje się pomiaru wskaźnika oddychania przez czas do 10 minut.
Oznaczenie to powtarza się w odniesieniu do każdego zbiornika w 15-minutowych odstępach w taki sposób, aby czas kontaktu w każdym zbiorniku wynosił trzy godziny.
Substancja odniesienia badana jest dla każdej partii inokulum bakteryjnego według tej samej procedury.
W przypadku dokonywania pomiarów po 30 minutach kontaktu wymagany jest odmienny reżym (np. więcej niż jeden tlenomierz).
Jeżeli konieczny jest pomiar chemicznego zużycia tlenu, należy przygotować kolejne zbiorniki zawierające substancję testową, syntetyczną pożywkę dla mikroflory ścieku aktywowanego oraz wodę, ale bez aktywnego szlamu. Należy zmierzyć i odnotować zużycie tlenu po 30-minutowym okresie napowietrzania i/lub trzech godzinach (czas kontaktu).
2. DANE I OSZACOWANIE
Wskaźnik oddychania obliczany jest na podstawie historii zapisów między około 6,5 mg O2/litr a 2,5 mg O2/litr lub przez okres 10 minut, gdzie wskaźnik oddychania jest niski. Odcinek krzywej oddychania, powyżej którego następuje pomiar wskaźnika oddychania powinien być linearny.
Jeżeli wskaźniki oddychania z dwóch grup kontrolnych nie zawierają się w 15 % wartości każdego z nich lub wartość EC50 (30 minut i/lub trzy godziny) substancji odniesienia nie zawiera się w przyjętym przedziale (5–30 mg/litr dla 3,5-dichlorofenolu), badanie jest nieważne i musi zostać powtórzone.
Procent zahamowania obliczany jest dla każdej badanej wielkości stężenia (zob. 1.2). Procent zahamowania nanoszony jest na wykres względem stężenia na papierze logarytmicznym (lub papierze półlogarytmicznym) i uzyskanej wartości EC50.
95 % limit potwierdzenia dla wartości EC50 może zostać ustalony przez procedury standardowe.
3. SPRAWOZDANIE
3.1. SPRAWOZDANIE Z BADAŃ
Sprawozdanie z badań powinno, jeżeli możliwe, zawierać następujące informacje:
|
— |
dane identyfikacyjne chemicznej substancji testowej, |
|
— |
system badania: źródło, stężenie i wstępne poddanie aktywnego szlamu działaniu substancji testowej, |
|
— |
warunki badania:
|
|
— |
wyniki:
|
3.2. INTERPRETACJA DANYCH
Wartość EC50 należy interpretować jedynie jako wskazówkę do prawdopodobnej toksyczności substancji testowej zarówno dla działania na ścieki aktywowanym szlamem, jak i dla mikroorganizmów występujących w ściekach, ponieważ złożone wzajemne oddziaływania zachodzące w środowisku nie mogą być odpowiednio stymulowane podczas badania laboratoryjnego. Ponadto substancje testowe, które mogą hamująco wpływać na utlenianie amoniaku, mogą również powodować nietypowe krzywe zahamowania. W związku z tym takie krzywe należy interpretować z ostrożnością.
4. ODNIESIENIA
|
(1) |
Międzynarodowy standard ISO 8192-1986. |
|
(2) |
Broecker, B., Zahn, R., Badania wody 11, 1977, s. 165. |
|
(3) |
Brown, D., Hitz, H. R., Schaefer, L., Chemosfera 10, 1981, s. 245. |
|
(4) |
ETAD (Ekologiczne i Toksykologiczne Stowarzyszenie Przemysłu Przetwórczego Substancji Barwiących), Zalecana Metoda nr 103, również opisana przez: |
|
(5) |
Robra, B., Wasser/Abwasser 117, 1976, s. 80. |
|
(6) |
Schefer, W., Textilveredlung 6, 1977, s. 247. |
|
(7) |
OECD, Paryż, 1981, Wytyczne Badania 209, decyzja Rady C (81) 30 wersja ostateczna. |
C.12. BIODEGRADACJA
ZMODYFIKOWANE BADANIE SCAS
1. METODA
1.1. WSTĘP
Celem metody jest ocena wpływu całkowitej biodegradacji rozpuszczalnych w wodzie, nielotnych substancji organicznych, w przypadku ich ekspozycji na stosunkowo wysokie stężenia drobnoustrojów przez długi okres czasu. Żywotność drobnoustrojów utrzymywana jest przez ten okres, codziennie dodając zasiedloną pożywkę szlamową. (Odnośnie do wymogów związanych z przerwą weekendową, ścieki można przechowywać w temperaturze 4 oC. W związku z tym można użyć syntetycznych ścieków (test potwierdzający OECD).
Może mieć miejsce fizyczna i chemiczna adsorpcja zawieszonych substancji stałych, co należy wziąć pod uwagę przy interpretacji wyników (zob. 3.2).
Z powodu długiego okresu zatrzymania fazy płynnej (36 godzin) i nieregularnego dodawania odżywek badanie nie pozoruje faktycznych warunków występujących w oczyszczalni ścieków. Wyniki otrzymane z różnymi substancjami testowymi oznaczają, iż badanie wykazuje wysoki wpływ biodegradacyjny.
Warunki stworzone w badaniu wysoce sprzyjają wyborowi i/lub przystosowaniu drobnoustrojów zdolnych do degradacji badanego związku. (Procedurę można również wykorzystać w celu wytworzenia zaaklimatyzowanego inokulum mogącego być zastosowanym w innych badaniach).
W metodzie tej pomiar stężenia rozwiązanego węgla organicznego lub chemicznego zapotrzebowania tlenu wykorzystywany jest w celu określenia całkowitej biodegradacji substancji testowej. Najlepiej ustalać DOC po zakwaszeniu i oczyszczeniu niż jako różnicę Ccałkowite – Cnieorganiczne.
Równoczesne zastosowanie szczególnej metody analitycznej może umożliwić ocenę podstawowej biodegradacji substancji (zanik pierwotnej budowy chemicznej).
Metodę stosuje się jedynie w odniesieniu do tych organicznych substancji testowych, które przy wielkości stężenia użytego w badaniu:
|
— |
są rozpuszczalne w wodzie (przy co najmniej 20 ml rozpuszczonego węgla organicznego/litr), |
|
— |
posiadają nieznaczne ciśnienie pary, |
|
— |
nie hamują bakterii, |
|
— |
nie są znacznie absorbowane w badanym systemie, |
|
— |
nie są tracone w wyniku wytwarzania piany z badanego roztworu. |
Należy ustalić zawartość węgla w organicznym materiale testowym.
Informacje dotyczące względnych części składowych materiału testowego będą użyteczne przy interpretacji otrzymanych wyników, w szczególności w tych przypadkach gdzie wyniki są na niskim poziomie lub marginalne.
Informacje dotyczące wpływu toksyczności substancji na drobnoustroje mogą okazać się użyteczne w celu dokonania interpretacji słabych wyników oraz wyboru właściwych wielkości stężeń.
1.2. DEFINICJE I JEDNOSTKI
|
CT |
= |
stężenie testowego związku jako węgla organicznego obecnego w lub dodawanego do osadzonego ścieku na początku czasu napowietrzania (mg/litr), |
|
Ct |
= |
stężenie rozwiązanego węgla organicznego w supernatanie substancji testowej na końcu czasu napowietrzania (mg/litr), |
|
Cc |
= |
stężenie rozwiązanego węgla organicznego w supernatanie w grupie kontrolnej na końcu czasu napowietrzania (mg/litr). |
W metodzie tej biodegradacja określana jest jako zanik węgla organicznego. Biodegradację można wyrazić jako:
|
1. |
Procent ubytku Dda ilości substancji dodawanej codziennie:
gdzie:
|
|
2. |
Procent ubytku Dssd ilości substancji obecnej przy rozpoczęciu każdego dnia badania:
gdzie:
wskaźniki i oraz (i + 1) odnoszą się do dnia pomiaru. Zaleca się równanie 2a) w przypadku, gdy ciecz wypływająca ze zbiornika DOC różni się z dnia na dzień, podczas gdy równanie 2b) można zastosować w przypadku, gdy ciecz wypływająca ze zbiornika DOC pozostaje na względnie stałym poziomie w kolejnych dniach. |
1.3. SUBSTANCJE ODNIESIENIA
W niektórych przypadkach podczas badania nowych substancji użyteczne mogą okazać się substancje odniesienia; jednakże nie można zalecić żadnych poszczególnych substancji odniesienia.
Dane dotyczące kilku związków chemicznych przeanalizowanych za pomocą prób pierścieniowych dostarczane są głównie w celu przeprowadzenia (zob. dodatek 1) kalibracji metody odpowiednio do potrzeb i umożliwienia dokonania porównania wyników w przypadku zastosowania innej metody.
1.4. ZASADA METODY BADAWCZEJ
Aktywowany szlam, pochodzący z oczyszczalni ścieków, umieszczany jest w jednostce działającej na zasadzie szlamu aktywowanego o przepływie półstałym (SCAS). Następnie dodawany jest badany związek oraz wewnętrzne ścieki, po czym następuje napowietrzanie mieszaniny przez 23 godziny. Po 23 godzinach napowietrzanie zostaje przerwane w celu umożliwienia osadzenia się szlamu i ubytku supernatantów.
Następnie szlam pozostały w komorze napowietrzania jest mieszany z dalszą podwielokrotnością badanego związku i ścieku, a cykl jest powtarzany.
Biodegradacja ustalana jest przez określenie zawartości rozwiązanego węgla organicznego w supernatancie. Uzyskana wartość porównywana jest z wartością ustaloną dla płynu otrzymanego z probówki kontrolnej dozowanej jedynie osiadłym ściekiem.
W przypadku użycia szczególnej metody analitycznej można dokonać pomiaru zmian w stężeniu pierwotnej molekuły w wyniku biodegradacji (podstawowa biodegradacja).
1.5. KRYTERIA JAKOŚCIOWE
Reprodukcyjność metody, oparta na ubytku rozwiązanego węgla organicznego, nie została jeszcze ustalona. (W przypadku rozważania podstawowej biodegadacji bardzo dokładne dane uzyskiwane są w odniesieniu do materiałów degradowanych ekstensywnie).
Czułość metody jest przeważnie ustalana przez zmienność próbki ślepej i, w mniejszym stopniu, przez precyzję określenia rozwiązanego węgla organicznego i poziomu badanego związku w roztworze po rozpoczęciu każdego cyklu.
1.6. OPIS PROCEDURY BADAWCZEJ
1.6.1. Przygotowania
Dla każdej substancji testowej oraz grup kontrolnych składana jest odpowiednia liczba czystych napowietrzanych jednostek, ewentualnie można zastosować oryginalne, 1,5-litrowe jednostki badawcze SCAS oraz probówki doprowadzające powietrze (rysunek 1). Sprężone powietrze doprowadzane do jednostek testowych, oczyszczone przez bawełniano-wełniany filtr, nie powinno zawierać węgla organicznego oraz powinno zostać nasycone wodą w celu obniżenia jej ubytku wskutek parowania.
Próbka zmieszanego płynu, zawierająca 1–4 g zawieszonej suchej masy na litr, otrzymywana jest z oczyszczalni ścieków, oczyszczającej w głównej mierze ścieki wewnętrzne. Wymagane jest około 150 ml zmieszanego płynu dla każdej napowietrzanej jednostki.
Roztwory podstawowe substancji testowej przygotowywane są w wodzie destylowanej; wymagana wartość stężenia zazwyczaj wynosi 400 mg/litr, tak jak węgiel organiczny, którego stężenie substancji testowej wynosi 20 mg/litr węgla na początku każdego cyklu napowietrzania, jeżeli nie zachodzi biodegradacja.
Dopuszczane są wyższe stężenia, jeżeli pozwala na to wpływ toksyczności na drobnoustroje.
Skład węgla organicznego roztworów podstawowych podlega pomiarowi.
1.6.2. Warunki badania
Badanie należy przeprowadzić w temperaturze 20–25 oC.
Stosowane jest wysokie stężenie drobnoustrojów tlenowych (1–4 g/litr zawieszonej suchej masy), a skuteczny okres zatrzymania wynosi 36 godzin. Materiał zawierający węgiel w pożywce szlamowej jest rozlegle utleniany, zazwyczaj w ciągu ośmiu godzin od rozpoczęcia każdego cyklu napowietrzania. Od tego czasu szlam oddycha endogenicznie przez pozostały czas napowietrzania, w którym jedyny dostępny substrat stanowi związek testowy, jeżeli również nie ulega łatwej metabolizacji. Cechy te, połączone z codziennym szczepieniem substancją testową, w przypadku zastosowania wewnętrznego ścieku jako pożywki, zapewniają wysoce korzystne warunki zarówno dla aklimatyzacji, jak i dla wysokiego stopnia biodegradacji.
1.6.3. Przeprowadzenie badania
Próbka zmieszanego płynu pochodząca z oczyszczalni ścieków, oczyszczającej w głównej mierze ścieki wewnętrzne lub jednostki laboratoryjnej, uzyskiwana jest i utrzymywana w warunkach tlenowych aż do chwili jej użycia w laboratorium. Każda napowietrzana jednostka, a także jednostka kontrolna wypełniana jest 150 ml zmieszanego płynu (w przypadku użycia oryginalnej jednostki testowej SCAS należy pomnożyć dane wielkości przez 10) i rozpoczyna się napowietrzanie. Po 23 godzinach napowietrzanie zostaje przerwane na 45 minut, w celu umożliwienia osadzenia się szlamu. Z kolei należy otworzyć zawór każdego zbiornika i pobrać 100 ml porcję supernatanu. Próbka osiadłego ścieku wewnętrznego powinna zostać pobrana bezpośrednio przed jej użyciem i 100 ml dodawane jest do szlamu pozostającego w każdej napowietrzanej jednostce. Proces napowietrzania rozpoczyna się od nowa. W tej fazie nie są dodawane żadne materiały testowe, a jednostki zasilane są codziennie wyłącznie wewnętrznymi ściekami aż do chwili otrzymania czystego supernatanu po osadzeniu. Powyższa faza zazwyczaj trwa dwa tygodnie, podczas których rozwiązany węgiel organiczny w supernatanie zbliża się do stałej wartości na końcu każdego cyklu napowietrzania.
Po upływie tego okresu poszczególne osadzone szlamy należy zmieszać i dodać 50 ml otrzymanego w ten sposób połączonego szlamu do każdej jednostki.
95 ml osadzonego ścieku i 5 ml wody dodawane jest do jednostek kontrolnych, i 95 ml osadzonego ścieku plus 5 ml właściwego roztworu podstawowego badanego związku (400 mg/litr) dodawane jest do jednostek testowych. Napowietrzanie należy rozpocząć od nowa i kontynuować przez 23 godziny. Następnie należy umożliwić osadzenie się szlamu przez okres 45 minut oraz należy ściągnąć i przeanalizować supernatan w celu ustalenia składu rozwiązanego węgla organicznego.
Powyższą procedurę napełniania i ściągania powtarza się codziennie przez cały okres trwania badania.
Przed osadzeniem konieczne jest oczyszczenie ścian jednostek w celu zapobieżenia nagromadzania się suchej masy ponad poziom płynu. W tym celu należy użyć oddzielnego skrobaka lub szczotki dla każdej z jednostek, aby zapobiec zanieczyszczeniu krzyżowemu.
Najlepiej, aby wartość rozwiązanego węgla organicznego w supernatanie ustalana była codziennie, chociaż dopuszcza się rzadsze przeprowadzanie analiz. Przed przeprowadzeniem analiz płyny filtrowane są przez oczyszczone 0,45 μm filtry membranowe lub odwirowywane. Filtry membranowe są odpowiednie jeżeli zostanie potwierdzone, iż nie uwalniają węgla, ani nie absorbują substancji w fazie filtracji. Temperatura próbki nie może przekraczać 40 oC w chwili umieszczenia jej w wirówce laboratoryjnej.
Długość trwania badania w odniesieniu do związków wykazujących niską lub zerową biodegradację nie jest określona, ale doświadczenie sugeruje, iż taki okres powinien trwać co najmniej 12 tygodni, ale nie dłużej niż 26 tygodni.
2. DANE I OSZACOWANIE
Wartości rozwiązanego węgla organicznego w supernatanach w jednostkach testowych i jednostkach kontrolnych nanoszone są na wykres względem czasu.
W przypadku osiągnięcia biodegradacji jej poziom określony w jednostce badawczej zbliży się do poziomu w jednostce kontrolnej. Po ustaleniu stałej różnicy między dwoma poziomami w trzech kolejnych pomiarach liczba kolejnych pomiarów uzależniona jest od możliwości dokonania statystycznej analizy danych oraz należy obliczyć procentową biodegradację związku testowego (Dda lub Dssd, zob. 1.2).
3. SPRAWOZDANIE
3.1. SPRAWOZDANIE Z BADAŃ
Sprawozdanie z badań powinno, jeżeli możliwe, zawierać następujące informacje:
|
— |
wszelkie informacje dotyczące rodzaju ścieku, typu zastosowanej jednostki oraz wyniki badania dotyczące substancji testowej, substancji odniesienia, jeżeli została użyta, oraz próbki ślepej, |
|
— |
temperatura, |
|
— |
krzywa ubytku związku wraz z opisem, sposób obliczania (zob. 1.2), |
|
— |
data i lokalizacja pobrania próbki aktywowanego szlamu i ścieku, poziom przystosowania, stężenie itd., |
|
— |
naukowe powody jakichkolwiek zmian procedury badawczej, |
|
— |
data i podpis. |
3.2. INTERPRETACJA WYNIKÓW
Ponieważ substancja przeznaczona do badania niniejszą metodą nie będzie ulegać łatwej biodegradacji, jakikolwiek ubytek DOC spowodowany wyłącznie biodegradacją przebiegać będzie stopniowo w okresie dni i tygodni, z wyjątkiem takich przypadków, gdzie aklimatyzacja jest nagła i zasygnalizowana nagłym zniknięciem substancji następującym po okresie kilku tygodni.
Jednakże fizyko-chemiczna adsorpcja może w niektórych przypadkach odegrać istotną rolę; sygnalizowane jest to w chwili gdy następuje całkowity lub częściowy ubytek dodanego DOC na początku badania. To, co następuje później, zależy od czynników, takich jak stopnie adsorpcji i stężenia zawieszonej suchej masy w odrzuconej cieczy wypływającej ze zbiornika. Zwykle różnica między stężeniami DOC w supernatanach w grupie kontrolnej i w grupie badanej stopniowo wzrasta od początkowej niskiej wartości, a następnie utrzymuje się na nowym poziomie wartości w pozostałym okresie badania, jeżeli nie zachodzi aklimatyzacja.
Konieczne jest przeprowadzenie dalszych badań, jeżeli istnieje potrzeba rozróżnienia między biodegradacją (lub częściową biodegradacją) i adsorpcją. Można to osiągnąć na wiele sposobów, ale najbardziej przekonujące jest użycie supernatantu lub szlamu jako inokulum w teście podstawowym (najlepiej teście respirometrycznym).
Substancje testowe powodujące wysoki, nieadsorpcyjny ubytek DOC w niniejszym badaniu należy uważać za substancje wpływające biodegradacyjnie. Częściowy, nieadsorpcyjny ubytek oznacza, że substancja chemiczna ulega co najmniej nieznacznej biodegradacji.
Niski, lub zerowy poziom ubytku DOC może być spowodowany zahamowaniem aktywności drobnoustrojów przez substancję testową, co można również wykryć przez rozpad lub utratę szlamu, dającego mętne supernatany. Badanie należy powtórzyć, wykorzystując niskie stężenie substancji testowej.
Użycie specyficznej dla danego związku metody analitycznej lub substancji testowej znakowanej izotopem węgla 14C zwiększa czułość metody. W przypadku związków znakowanych 14C odzyskanie 14CO2 potwierdzi, że zaszła biodegradacja.
W przypadku podawania wyników z uwzględnieniem początkowej biodegradacji, jeżeli możliwe, należy wyjaśnić zmianę w budowie chemicznej, która powoduje utratę reakcji macierzystej substancji testowej.
Zatwierdzenie metody analitycznej musi być dostarczone razem z reakcją, która zaszła w próbce ślepej.
4. ODNIESIENIA
|
(1) |
OECD, Paryż, 1981, Wytyczne badania 209, decyzja Rady C (81) 30 wersja ostateczna. |
Dodatek 1
Badanie SCAS: przykładowe wyniki
|
Substancja |
CT (mg/l) |
Ct – Cc (mg/l) |
Procentowa biodegradacja, Dda |
Czas trwania badania (dni) |
|
Sulfonian 4-acetyloaminobenzenu |
17,2 |
2,0 |
85 |
40 |
|
Sulfonian tetrapropylenobenzenu |
17,3 |
8,4 |
51,4 |
40 |
|
4-nitrofenol |
16,9 |
0,8 |
95,3 |
40 |
|
Glikol dietylenowy |
16,5 |
0,2 |
98,8 |
40 |
|
Anilina |
16,9 |
1,7 |
95,9 |
40 |
|
Cyklopentanotetrakarboksylan |
17,9 |
3,2 |
81,1 |
120 |
Dodatek 2
Przykładowe urządzenia badawcze
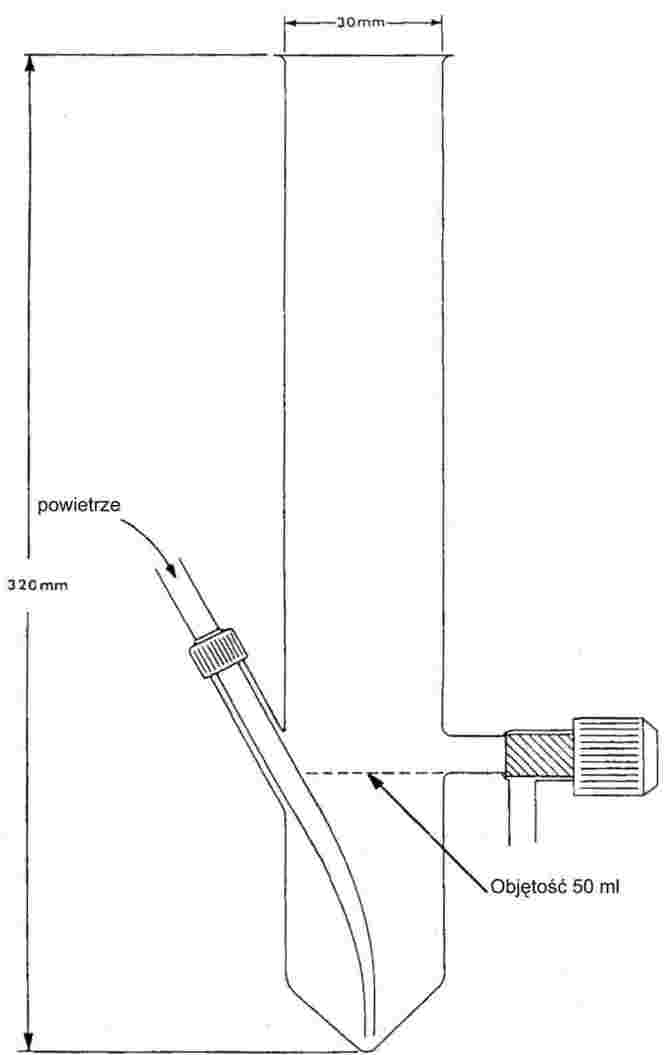
C.13. BIOKONCENTRACJA: BADANIE RYB W WARUNKACH PRZEPŁYWU
1. METODA
Ta metoda biokoncentracji jest powtórzeniem OECD WT 305 (1996).
1.1. WPROWADZENIE
Metoda ta opisuje procedurę charakteryzowania biokoncentracyjnego potencjału substancji znajdujących się w rybach w warunkach przepływu. Chociaż preferuje się bardziej wymogi obowiązujące dla badań w warunkach przepływu, dopuszczalne są wymogi obowiązujące dla badań w warunkach częściowo statycznych, pod warunkiem że spełnione są kryteria prawidłowości.
Metoda podaje wystarczające szczegóły dla przeprowadzenia badania, jednocześnie zezwalając na stosowną dowolność w dostosowywaniu procedury doświadczalnej do warunków w poszczególnych laboratoriach oraz na zróżnicowane właściwości substancji badanych. Jest ona najbardziej prawidłowo stosowana w odniesieniu do chemikaliów organicznych o wartościach log Pow między 1,5 i 6,0 (1), ale może być także stosowana do substancji superlipofilowych (o wartości log Pow > 6,0). Wstępne oszacowanie czynnika biokoncentracji (BCF), czasami oznaczanego jako KB, dla takich substancji będzie prawdopodobnie wyższe niż wartość czynnika biokoncentracji w stanie ustalonym (BCFSS), które spodziewa się uzyskać w doświadczeniach laboratoryjnych. Wstępne oszacowania czynnika biokoncentracji dla chemikaliów organicznych o wartościach log Pow wynoszących około 9,0 można uzyskać, stosując równanie Binteina i inne (2). Parametry, które charakteryzują potencjał biokoncentracji, zawierają stałą szybkość absorpcji (k1), stałą szybkość oczyszczania (k2) oraz BCFSS.
Substancje badane oznakowane radiologicznie mogą ułatwić analizę próbek wody i ryb oraz mogą być wykorzystywane do określania, czy konieczne jest wykonanie identyfikacji i kwantyfikacji degradacji. Jeżeli mierzony jest ogół pozostałości radioaktywnych (np. przez spalanie lub rozpuszczanie tkanki), BCF jest oparte na związku macierzystym, wszelkich zatrzymanych metabolitach, a także na asymilowanym węglu. Z tego względu BCFS, oparty na ogóle pozostałości radioaktywnych, nie może być bezpośrednio porównywalny z BCF uzyskiwanym w drodze szczegółowej analizy chemicznej wyłącznie związku macierzystego.
W badaniach substancji oznaczonych radiologicznie można zastosować procedury oczyszczania w celu określenia czynnika BCF w oparciu o związek macierzysty i, jeżeli uzna się to za konieczne, można opisać główne metabolity. Możliwe jest także połączenie badania metabolizmu ryb z badaniem biokoncentracji poprzez analizę i identyfikacje pozostałości w tkankach.
1.2. DEFINICJE I JEDNOSTKI
Biokoncentracja/bioakumulacja: jest wzrostem koncentracji substancji badanej w organizmie lub na nim (jego określonych tkankach), w stosunku do koncentracji substancji badanej w otaczającym środowisku.
Czynnik biokoncentracji (BCF lub KB) w każdym momencie w trakcie fazy absorpcji tego badania akumulacji jest stężeniem substancji badanej w/na rybie lub w jej określonych tkankach (Cf jako μg/g (ppm)), podzielone przez koncentrację tej substancji chemicznej w otaczającym środowisku (Cw jako μg/ml (ppm)).
Stały czynnik biokoncentracji (BCFSS lub KB) nie zmienia się znacząco w długim okresie czasu, stężenie substancji badanej w otaczającym środowisku w tym okresie czasu jest stale.
Plateau lub stan ustalony osiągany jest, wykreślając ilość substancji badanej w rybach (Cf) w zależności od momentu, kiedy krzywa staje się równoległa do osi czasu, a trzy następujące po sobie analizy Cf, wykonane na próbkach pobranych w odstępach, co najmniej dwóch dni, różnią się od siebie o ± 20 % i nie ma znacznych różnic między trzema okresami pobierania próbek. Przy wspólnej analizie próbek wymagane są przynajmniej cztery następujące po sobie analizy. W odniesieniu do substancji badanych, które są pobierane powoli, odstępy czasu powinny wynosić odpwiednio siedem dni.
Czynniki biokoncentracji, obliczane bezpośrednio ze stałych wskaźników kinetycznych (k1/k2), określane są jako czynniki koncentracji kinetycznej, BCFK.
Współczynnik podziału oktanol-woda (Pow) jest to stosunek rozpuszczalności substancji chemicznej w n-oktanolu i wodzie w równowadze (metoda A.8), wyrażany także jako Kow. Logarytm Pow wykorzystywany jest jako wskaźnik zdolności substancji chemicznej do biokoncentracji przez organizmy wodne.
Faza narażenia na działanie substancji j absorpcji jest to czas, w trakcie którego ryby narażone są na działanie badanej substancji chemicznej.
Stała szybkość absorpcji (k1) jest wartością liczbową określającą tempo wzrostu koncentracji substancji badanej w rybach i na nich (lub w nich czy ich określonych tkankach) w czasie gdy ryby wystawione są na działanie tej substancji (k1 wyraża się w dniach-1).
Faza po narażeniu na działanie substancji/oczyszczania (ubytku) jest to czas następujący po przeniesieniu ryb ze środowiska zawierającego substancje do środowiska wolnego od tej substancji, w trakcie którego badane jest oczyszczanie (lub ich określonych tkanek) z substancji (ubytek netto).
Stała szybkość oczyszczania (ubytku) (k2) jest wartością liczbową określającą tempo zmniejszania koncentracji substancji badanej w badanych rybach (lub ich określonych tkankach) po przeniesieniu badanych ryb ze środowiska zawierającego substancje badaną do środowiska wolnego od tej substancji (k2 wyrażana jest w dniach-1).
1.3. ZASADA METODY BADAWCZEJ
Badanie składa się z dwóch faz: faza narażenia na działanie substancji (absorpcja) i faza po narażeniu na działanie substancji (oczyszczania). Podczas fazy absorpcji oddzielne grupy ryb jednego gatunku narażone są na działanie substancji badanej o co najmniej dwóch różnych stężeniach. Następnie ryby przenosi się do środowiska wolnego od substancji badanej w celu przeprowadzenia fazy oczyszczania. Faza oczyszczania jest zawsze konieczna, chyba że absorpcja substancji podczas fazy absorpcji jest nieznaczna (np. czynnik BCF jest mniejszy niż 10). Stężenie substancji badanej w/na rybach (lub w ich określonych tkankach) śledzi się w obu fazach badania. Oprócz dwóch badań stężeń substancji badanej, kontrolowaną grupę ryb przetrzymuje się w identycznych warunkach, z wyjątkiem obecności substancji badanej, w celu odniesienia możliwych negatywnych skutków zaobserwowanych w badaniu biokoncentracji do odpowiedniej grupy kontrolowanej oraz w celu uzyskania bazowych stężeń substancji badanej.
Faza absorpcji trwa 28 dni, jeżeli nie wykaże się, że równowaga została osiągnięta wcześniej. Przewidywany czas trwania fazy absorpcji i czas osiągnięcia stanu ustalonego można obliczyć w oparciu o równanie podane w załączniku 3. Następnie rozpoczyna się okres oczyszczania przez przeniesienie ryb do takiego samego środowiska, ale bez substancji badanej w innym czystym naczyniu. W miarę możliwości czynnik biokoncentracji oblicza się zarówno jako stosunek (BCFSS) stężenia w rybach (Cf) i w wodzie (Cw) w pozornym stanie ustalonym, jak też jako kinetyczny czynnik biokoncentracji (BCFK), będący stosunkiem stałych szybkości absorpcji (k1) i oczyszczania (k2), uznając kinetykę reakcji pierwszego rzędu. Jeżeli kinetyka reakcji pierwszego rzędu jest w oczywisty sposób nieprzestrzegana, należy zastosować bardziej złożone wzory (załącznik 5).
Jeżeli stanu ustalonego nie osiągnie się w ciągu 28 dni, fazę absorpcji należy przedłużyć do czasu osiągnięcia stanu ustalonego lub do 60 dni, w zależności od tego, co nastąpi najpierw; następnie rozpoczyna się fazę oczyszczania.
Stała szybkość absorpcji, stała szybkość reakcji (lub stałe w przypadku gdy zastosowane są bardziej złożone wzory) oczyszczania (ubytku), czynnik biokoncentracji oraz, w miarę możliwości, przedziały ufności dla każdego z tych parametrów oblicza się w oparciu o wzór, który najlepiej opisuje mierzone stężenia substancji badanej w rybach i wodzie.
BCF wyraża się jako funkcje całkowitej mokrej masy ryb. Jednakże do szczególnych celów można wykorzystać określone tkanki lub organy (np. mięsnie, wątroba), jeżeli ryby są wystarczająco duże, lub podzielić ryby na części jadalne (filet) i niejadalne (wnętrzności). Ze względu na to, że w odniesieniu do wielu substancji organicznych istnieje wyraźny związek między zdolnością do biokoncentracji i lipofilowości, istnieje także odpowiadająca mu zależność między zawartością lipidów w rybach i zaobserwowaną biokoncentracją takich substancji. Dlatego w celu zmniejszenia tego źródła zróżnicowania w wynikach badania dla substancji o wysokiej lipofilowosci (tj. o log Pow > 3) biokoncentracja powinna być wyrażona w odniesieniu do zawartości lipidów oprócz całkowitej masy ciała.
Zawartość lipidów powinna być określana na takim samym materiale biologicznym, jaki jest wykorzystywany dla określania substancji badanej, jeżeli jest to możliwe.
1.4. INFORMACJE DOTYCZĄCE SUBSTANCJI BADANEJ
Przed przeprowadzaniem badania na biokoncentrację powinny być znane następujące informacje dotyczące substancji badanej:
|
— |
rozpuszczalność w wodzie, |
|
— |
współczynnik podziału oktanol-woda Pow (oznaczany także jako Kow ustalany za pomocą metody HPLC w A.8), |
|
— |
hydroliza, |
|
— |
fotodysocjacja w wodzie, określana w warunkach napromieniowania słonecznego lub symulowanym napromienianiu słonecznym i w warunkach napromieniania badania na biokoncentracji (3), |
|
— |
napięcie powierzchniowe (np. dla substancji, dla których nie można ustalić log Pow), |
|
— |
prężność pary, |
|
— |
biodegradalność inicjalna (jeżeli dotyczy). |
Inną wymaganą informacją jest toksyczność dla ryb, które mają być wykorzystywane w trakcie badania, najlepiej asymptotyczna LC50 (tj. niezależna od czasu). Dostępna musi być odpowiednia metoda analizy o potwierdzonej dokładności, precyzji i czułości, dla kwantyfikacji substancji badanej w roztworach badawczych i materiale biologicznym, wraz ze szczegółami dotyczącymi przygotowania i składowania próbki. Powinna być również znana analityczna granica detekcji substancji badanej zarówno w wodzie, jak i w tkankach ryb. Jeśli wykorzystywana jest substancja oznakowana 14C, powinien być znany udział radioaktywności związany z zanieczyszczeniami.
1.5. WAŻNOŚĆ BADANIA
Następujące warunki powinny mieć zastosowanie w odniesieniu do badania, aby było ono ważne:
|
— |
zmiana temperatury jest mniejsza niż ± 2 oC, |
|
— |
stężenie rozpuszczonego tlenu nie spada poniżej 60 % nasycenia, |
|
— |
stężenie substancji badanej w komorach utrzymuje się na poziomie ± 20 % średniej zmierzonych wartości w trakcie fazy absorpcji, |
|
— |
śmiertelność lub inne negatywne skutki/choroby, zarówno wśród ryb kontrolnych, jak i badanych, jest mniejsza niż 10 % po zakończeniu badania; w przypadku gdy badanie jest przedłużone o kilka tygodni lub miesięcy, śmiertelność lub inne negatywne skutki w obydwu grupach ryb powinny być niższe niż 5 % w stosunku miesięcznym i całkowicie nie powinny przekraczać 30 %. |
1.6. ZWIĄZKI ODNIESIENIA
Wykorzystanie związków odniesienia o znanej zdolności do biokoncentracji może być użyteczne przy sprawdzaniu procedury doświadczalnej, jeżeli jest to wymagane. Jednakże niemożliwe jest na razie polecenie konkretnych substancji.
1.7. OPIS METODY BADAWCZEJ
1.7.1. Aparatura
Należy zadbać, aby unikać wykorzystywania w odniesieniu do wszystkich części aparatury, materiałów, które mogą rozpuszczać się, sorbować lub ługować i wywoływać szkodliwe skutki dla ryb. Można wykorzystywać standardowe, prostokątne lub cylindryczne zbiorniki, wykonane z materiałów chemicznie obojętnych i o odpowiedniej pojemności, zgodnej ze wskaźnikiem pojemności. Wykorzystanie rur z miękkiego plastiku powinno być minimalne. Zalecane jest wykorzystywanie rur teflonowych, ze stali nierdzewnej i/lub szklanych. Doświadczenie wskazało, że dla substancji o wysokich współczynnikach absorpcji, jak np. syntetyczne pyretroidy, może być wymagane szkło silanizowane. W takich sytuacjach sprzęt będzie musiał po zostać wyrzucony po wykorzystaniu.
1.7.2. Woda
Zwykle do badania wykorzystywana jest woda naturalna, która powinna być uzyskiwana z niezanieczyszczonego źródła oraz ze źródła o jednolitej jakości. Woda rozcieńczana powinna być jakości, która pozwoli na przetrwanie wybranych gatunków ryb przez okres trwania aklimatyzacji i okres badań oraz nie będzie powodowała pojawienia się nieprawidłowego ich wyglądu lub zachowania. W idealnym przypadku powinno się wskazać, że badane gatunki mogą przeżyć, rosnąć i rozmnażać się w wodzie rozcieńczonej (np. badanie w warunkach laboratoryjnych lub na toksyczność w cyklu życia). Woda powinna być scharakteryzowana, przynajmniej przez pH, twardość, całkowitą zawartość ciał stałych, całkowitą zawartość węgla organicznego oraz w miarę możliwości amonu, azotynu oraz alkaliczności i, w odniesieniu do gatunków morskich, zasolenia. Parametry, które są ważne dla optymalnego dobrostanu ryb, są znane w pełni, jednak załącznik 1 podaje zalecane najwyższe stężenia kilku parametrów dla badanych wód słodkich i morskich.
W czasie trwania badania jakość wody powinna być stała. Wartość pH powinna mieścić się w zakresie 6,0–8,5, ale podczas danego badania powinna wynosić ±0,5 jednostek pH. W celu zapewnienia, że woda rozcieńczana nie wpłynie negatywnie na wyniki badania (np. przez zmianę właściwości substancji badanej) lub na zachowanie ryb, co pewien czas należy pobierać próbki do analizy. Powinny zostać oznaczone zawartości metali ciężkich (np. Cu, Pb, Zn, Cd, Ni), głównych anionów i kationów (np. Ca, Mg, Na, K, Cl, SO4), pestycydów (np. całkowitej zawartości pestycydów organofosforowych i organochlorowych), zawartości węgla organicznego i zawiesin, np. co trzy miesiące, w przypadku gdy wiadomo, że woda rozcieńczona ma względnie stałą jakość. Jeżeli wykazano, że woda ma stałą jakość przez okres co najmniej jednego roku, oznaczenia mogą być przeprowadzane rzadziej, a odstępy czasu między oznaczeniami wydłużone (np. co sześć miesięcy).
Naturalna zawartość cząstek, jak również całkowita zawartość węgla organicznego (TOC) wody do rozcieńczania powinna możliwie jak najniższa, w celu uniknięcia absorpcji substancji badanej do materii organicznej, która może zmniejszyć jej biodostępność (4). Najwyższa dopuszczalna wartość wynosi 5 mg/l dla cząstek stałych (masa sucha, która nie jest przepuszczana przez filtr 0,45 μm) oraz 2 mg/l dla całkowitej zawartości węgla organicznego (zob. załącznik 1). W miarę potrzeb woda powinna zostać przefiltrowana przed wykorzystaniem. Udział zawartości węgla organicznego w wodzie, z badania ryb (odchody) oraz z pozostałości pożywienia powinien być możliwie jak najniższy. W trakcie trwania badania stężenie węgla organicznego w naczyniu do badania nie powinno przekraczać stężenia węgla organicznego pochodzącego z substancji badanej i – jeżeli jest wykorzystywany – środka rozpuszczającego o więcej niż 10 mg/l (± 20 %).
1.7.3. Badane roztwory
Podstawowy roztwór substancji badanej przygotowywany jest w odpowiednim stężeniu. Roztwór podstawowy powinien być przygotowywany przez mieszanie lub wstrząsanie substancji badanej w wodzie do rozcieńczania. Nie zaleca się wykorzystywania rozpuszczalników lub dyspergatorów (czynników rozpuszczających); jednakże może to nastąpić w niektórych przypadkach, w celu wytworzenia roztworu podstawowego o odpowiednim stężeniu. Rozpuszczalniki, które mogą zostać wykorzystane, to etanol, metanol, eter monometylowy glikolu etylenowego, eter dwumetylenowy glikolu etylenowego, dwumetyloformamid i glikol trójetylenowy. Dopuszczalne dyspergatory to kremofor RH40, Tween 80, metyloceluloza 0,01 % i HCO-40. Wykorzystując czynniki łatwo ulegające biodegradacji, należy zachować ostrożność, ponieważ mogą one powodować problemy z porostem bakterii w badaniach przepływowych. Substancja badana może być oznakowana radiologicznie i powinna być o możliwie największej czystości (np. > 98 %).
W odniesieniu do badań przepływowych wymagany jest system, który nieprzerwanie dostarcza i rozcieńcza roztwór podstawowy substancji badanej (np. pompa dozująca, rozcieńczalnik proporcjonalny, sytnik), w celu dostarczenia koncentratów badanych do komór badawczych. Zezwala się na co najmniej pięciokrotną wymianę całej objętości roztworu dziennie w każdej komorze badawczej. Preferowana ma być technika przepływowa, ale w przypadkach gdy nie jest to możliwe (np. gdy zachodzi negatywne działanie na organizmy badane), dopuszczalne jest stosowanie techniki półstatycznej, pod warunkiem że spełnione są kryteria ważności. Szybkość przepływu roztworów podstawowych i wody do rozcieńczania powinny być sprawdzane 48 godzin przed badaniem, a potem co najmniej raz dziennie. W tej kontroli zawarte jest określenie szybkości przepływu przez każdą komorę badawczą i zapewnione jest, że nie różni się ona między komorami i wewnątrz nich o więcej niż 20 %.
1.7.4. Wybór gatunków
Ważnymi kryteriami w wyborze gatunków jest fakt, że są one łatwo dostępne, mogą być uzyskane w odpowiednich rozmiarach i mogą być z powodzeniem przetrzymywane w laboratorium. Inne kryteria w odniesieniu do wybierania gatunków obejmują względy rekreacyjne, handlowe i ekologiczne, jak również porównywalną wrażliwość, pomyślne wykorzystanie w przeszłości itp.
Zalecane gatunki do badania są podane w załączniku 2. Inne gatunki mogą zostać wykorzystane, ale może istnieć konieczność dostosowania procedury badawczej celu zapewnienia odpowiednich warunków dla przeprowadzenia badania. W takim przypadku powinno zostać odnotowane racjonalne uzasadnienie w odniesieniu do wyboru gatunku i metody doświadczalnej.
1.7.5. Przetrzymywanie ryb
Podstawową populacje ryb aklimatyzować przez co najmniej dwa tygodnie w wodzie w temperaturze badania i karmić w oparciu o wystarczającą dietę, tego samego rodzaju, która ma być wykorzystana w trakcie badania.
Po 48-godzinnym okresie adaptacji odnotowuje się współczynnik śmiertelności i stosuje się następujące kryteria:
|
— |
śmiertelność wyższa niż 10 % populacji w ciągu siedmiu dni: odrzucić całą partię, |
|
— |
śmiertelność między 5 i 10 % populacji w ciągu siedmiu dni: aklimatyzować przez dodatkowe siedem dni, |
|
— |
śmiertelność niższa niż 5 % populacji w ciągu siedmiu dni: zaakceptować partię – jeżeli śmiertelność jest wyższa niż 5 % w ciągu siedmiu dodatkowych dni, odrzucić całą partię. |
Zapewnić, że ryby wykorzystywane w badaniu są wolne od dostrzegalnych chorób i nieprawidłowości. Odrzucić wszystkie chore ryby. Ryby nie powinny być leczone na żadną chorobę w okresie dwóch tygodni poprzedzających badanie albo w trakcie badania.
1.8. PRZEPROWADZENIE BADANIA
1.8.1. Badanie wstępne
Może być ono użyteczne w celu przeprowadzenia wstępnego doświadczenia w celu zoptymalizowania warunków dla przeprowadzenia badania ostatecznego, np. wybór stężenia (stężeń) substancji, czasu trwania fazy absorpcji i oczyszczania.
1.8.2. Warunki narażenia na działanie substancji
1.8.2.1.
Przewidzenie czasu trwania fazy wchłaniania może zostać uzyskane w oparciu o doświadczenie praktyczne (np. z poprzedniego badania lub nagromadzenia powiązanej substancji chemicznej) lub w oparciu o pewne empiryczne związki wykorzystujące wiedzę dotyczącą albo rozpuszczalności w wodzie albo współczynnika podziału oktanol/woda substancji badanej (zob. załącznik 3).
Faza wchłaniania powinna trwać 28 dni, chyba że może zostać udowodnione, iż równowagę osiągnięto wcześniej. Jeśli stan ustalony nie został osiągnięty przed upływem 28 dni, faza absorpcji powinna zostać przedłużona, pobierając dalsze pomiary, do momentu osiągnięcia stanu ustalonego, lub też do 60 dni, w zależności od tego, który z tych dwóch okresów jest krótszy.
1.8.2.2.
Okres połowy czasu trwania fazy absorpcji jest zazwyczaj wystarczający do zmniejszenia pojawiania się (95 %) substancji w masie ciała (zob. załącznik 3 dla wyjaśnienia oszacowania). Jeżeli czas wymagany do osiągnięcia 95 % straty jest zbyt długi, co jest jednocześnie niepraktyczne, przekraczając przykładowo dwukrotnie normalny czas trwania fazy absorpcji (tzn. więcej niż 5–6 dni), może zostać wykorzystany krótszy (np. do czasu, aż stężenie substancji badanej jest niższe niż 10 % stężenia substancji w stanie ustalonym). Jednakże w odniesieniu do substancji posiadającej bardziej złożone wzory absorpcji i oczyszczania niż te, które reprezentuje jednokomorowy model ryby i które oparte są na kinetyce pierwszego rzędu, można zastosować dłuższą fazę oczyszczania, w celu określenia stałych wartości wskaźnika utraty. Okres ten jednak może być regulowany przez okres, w trakcie którego stężenie substancji badanej w rybach pozostaje na poziomie powyżej analitycznego limitu wykrywania.
1.8.2.3.
Wybrać taką liczba ryb przypadającą na badanie, aby w każdej próbce były co najmniej cztery ryby. Jeśli wymagana jest większa ilość, niezbędna będzie większa liczba ryb przypadających na badanie. Jeśli wykorzystywane są dorosłe ryby, udokumentować przed czy w doświadczeniu wykorzystano samce, samice czy też obie płci.
Jeżeli wykorzystano ryby obu płci, udokumentować różnice występujące w zawartości lipidów, aby nie miały one znaczenia dla doświadczenia przed rozpoczęciem narażenia na działanie substancji; niezbędne może być połączenie wszystkich samic i samców ryb w jednym zbiorniku.
W jakimkolwiek badaniu wybierane są ryby o podobnej masie, tak aby masa najmniejszej z nich nie była mniejsza niż dwie trzecie masy największej z ryb. Wszystkie ryby powinny należeć do tej samej klasy wiekowej i pochodzić z tego samego źródła. Ze względu na to, że masa i wiek ryb mają znaczący wpływ na wartości BCF (1), szczegóły te są odpowiednio udokumentowane. Zaleca się zważenie podpróbki z zasobów ryb przed rozpoczęciem badania, w celu ustalenia średniej masy.
1.8.2.4.
Wykorzystuje się wysoką proporcję między ilością ryb i wody w celu zminimalizowania zmniejszenia w Cw spowodowanego przez dodanie ryb na początku badania, a także w celu uniknięcia spadku stężenia rozpuszczonego tlenu. Proporcje te są stosowane również w tym celu, aby uniknąć spadków stężenia rozpuszczonego tlenu. Ważne jest, aby wskaźnik dostarczenia ryb był odpowiedni dla poszczególnych badanych gatunków. W każdym przypadku zaleca się, aby wskaźnik dostarczenia ryb wynosił 0,1–1,0 g ryb (masa mokra) na litr wody dziennie. Wysokie wskaźniki dostarczania ryb mogą być wykorzystane, jeśli wykaże się, że wymagane stężenie substancji badanej może być zachowane w ramach ograniczeń ± 20 % oraz że stężenie rozpuszczonego tlenu nie spada poniżej 60 % nasycenia.
Wybierając systemy dostarczania ryb, bierze się pod uwagę naturalne środowisko, w którym występują dane gatunki ryb. Na przykład ryby żyjące na dnie zbiorników mogą wymagać większej przestrzeni dna akwarium w odniesieniu do tej samej objętości wody niż ryby pelagiczne.
1.8.2.5.
W czasie aklimatyzacji i badania, ryby są karmione według odpowiedniej diety, o znanej całkowitej zawartości lipidów i białek, w ilości wystarczającej do utrzymania ich w stanie zdrowym i do zachowania ich masy ciała. Ryby są karmione w czasie aklimatyzacji i badania ilością pokarmu, na poziomie w przybliżeniu 1–2 % masy ich ciała dziennie; utrzymuje to stężenie lipidów u większości gatunków na w miarę stałym poziomie w czasie badania. Ilość pokarmu powinna być ponownie obliczana, na przykład raz na tydzień, w celu utrzymania stałej masy ciała i zawartości lipidów. W odniesieniu do tego obliczenia masa ciała ryb w każdej komorze badawczej może być oszacowana na postawie masy ciała ryb pobranych jako próbkę w tej komorze ostatnio. Nie ważyć ryb pozostających w komorze.
Pokarm, który nie został spożyty, oraz ekskrementy są wypłukiwane z komór badawczych wkrótce po karmieniu (od 30 minut do 1 godziny). Komory są utrzymane w miarę możliwości w czystości przez okres trwania badania tak, aby stężenie substancji organicznej utrzymane było na możliwie jak najniższym poziomie, ze względu na to, że obecność węgla organicznego może ograniczyć biodostepność substancji badanej (1).
Ze względu na to, że wiele pokarmów jest uzyskiwanych z mączki rybnej, pokarm powinien być analizowany na obecność substancji badanej. Pożądane jest także analizowanie pokarmu na obecność pestycydów i metali ciężkich.
1.8.2.6.
Fotookres trwa zwykle 12–16 godzin, a temperatura (± 2 oC) powinna być odpowiednia dla gatunków badanych (zob. załącznik 2). Powinien być znany rodzaj oraz właściwości oświetlenia. Należy również zwrócić uwagę na możliwe fototransformacje substancji badanej w warunkach napromieniowania. Unikając narażenia ryb na działanie nienaturalnych fotoproduktów, należy wykorzystywać odpowiednie oświetlenie. W niektórych przypadkach odpowiednie może być wykorzystanie filtru w celu odfiltrowania promieniowania UV poniżej 290 nm.
1.8.2.7.
Ryby są narażone na działanie przynajmniej dwóch stężeń substancji badanej w wodzie w warunkach przepływu. Zazwyczaj wybiera się wyższe (lub najwyższe) stężenie substancji badanej, aby wynosiło około 1 % jego ostrej, asymptotycznej LC50, oraz aby było przynajmniej dziesięciokrotnie wyższe od jej limitu wykrywalności w wodzie przez wykorzystywaną metodę analityczną.
Najwyższe stężenie badania można również oznaczyć przez podzielenie ostrej 96h LC50 przez odpowiedni ostry/chroniczny wskaźnik (odpowiednie wskaźniki dla niektórych substancji chemicznych wynoszą 3–100). Jeśli jest to możliwe, wybrać inne stężenie (stężenia), tak aby różniło się ono od podanego wyżej współczynnika o 10. Jeśli nie jest to możliwe ze względu na kryterium 1 % z LC50 oraz na limit analityczny, można użyć czynnika niższego od 10 albo też wziąć pod uwagę zastosowanie oznakowanej substancji badanej 14C. Żadne wykorzystane stężenie nie powinno przekraczać poziomu rozpuszczalności substancji badanej.
W przypadku gdy wykorzystuje się czynnik rozpuszczający, jego stężenie nie powinno być wyższe niż 0,1 ml/l i powinno być takie samo we wszystkich naczyniach badawczych. Wpływ stężenia, wraz z substancją badaną, na całkowitą zawartość węgla organicznego w wodzie badanej powinien być znany. Jednakże powinny zostać podjęte wszelkie wysiłki, w celu uniknięcia wykorzystania takich materiałów.
1.8.2.8.
Powinna zostać przeprowadzona jedna kontrola wody do rozcieńczania lub, jeżeli jest to stosowne, jedna kontrola obejmującą czynnik rozpuszczający powinna zostać przeprowadzona oprócz badania, pod warunkiem że ustalono, iż czynnik nie wywiera wpływu na ryby. Jeśli nie ustalono tego, powinny zostać zorganizowane obie kontrole.
1.8.3. Częstotliwość pomiarów jakości wody
W trakcie badania powinny zostać przeprowadzone pomiary rozpuszczonego tlenu, TOC, pH oraz temperatury we wszystkich zbiornikach. Całkowita twardość i zasolenie, jeśli jest to odpowiednie, powinno zostać zmierzone w czasie kontroli, natomiast jeden ze zbiorników powinien zostać zbadany przy wyższym (lub najwyższym) stężeniu. Jako minimum poziom rozpuszczonego tlenu oraz zasolenie, jeśli jest to odpowiednie powinno zostać zmierzone trzykrotnie: na początku, w połowie i pod koniec fazy absorpcji oraz raz w tygodniu w czasie fazy oczyszczania. TOC powinno zostać zmierzone na początku badania (24 godziny i 48 godzin przed rozpoczęciem fazy absorpcji), przed wpuszczeniem ryb oraz przynajmniej raz w tygodniu w czasie fazy absorpcji i oczyszczania. Temperatura powinna być mierzona codziennie, pH na początku i na końcu każdego okresu, a twardość – raz w ciągu trwania badania. Zaleca się ciągłe monitorowanie temperatury, przynajmniej jednym pojemniku.
1.8.4. Pobieranie próbek oraz analiza ryb i wody
1.8.4.1.
Próbka wody z komór badawczych do oznaczania stężenia substancji badanej, pobierana jest przed wpuszczeniem ryb, oraz w trakcie zarówno fazy absorpcji, jak i oczyszczania. Próbki wody pobierane są przed karmieniem i w tym samym czasie, co próbki ryb. W trakcie fazy absorpcji oznaczane są stężenia substancji badanych, w celu sprawdzenia ich zgodności z kryteriami ważności.
Próbki ryb są pobierane przynajmniej pięć razy podczas fazy absorpcji oraz przynajmniej cztery razy podczas fazy oczyszczania. Ze względu na to, że czasami trudno będzie obliczyć dokładne oszacowanie wartości BCF w oparciu o tą ilość próbek, zwłaszcza jeśli wskazane są inne typy kinetyki niż prosta kinetyka pierwszego rzędu, wskazane może być pobieranie próbek podczas obu faz z większą częstotliwością (zob. załącznik 4). Dodatkowe próbki są składowane i analizowane jedynie wówczas gdy wyniki badań próbek pochodzących z pierwszej serii nie są wystarczające do obliczenia wartości czynnika BCF z pożądaną dokładnością.
Przykład dopuszczalnego planu pobierania próbek podany jest w załączniku 4. Inne harmonogramy można łatwo obliczyć, wykorzystując inne przyjęte wartości Pow dla określenia czasu narażenia na działanie czynników dla 9 5 % absorpcji.
Pobieranie próbek kontynuuje się podczas fazy absorpcji do momentu, kiedy osiągnięty zostanie stan ustalony lub przez 28 dni, w zależności od tego, który z tych dwóch okresów jest krótszy. Jeśli nie osiągnięto stanu ustalonego przed w okresie 28 dni, pobieranie próbek jest kontynuowane do czasu aż osiągnięty zostanie stan ustalony lub przez 60 dni, w zależności od tego, który z tych dwóch okresów jest krótszy. Przed rozpoczęciem fazy oczyszczania ryby przenoszone są do czystych zbiorników.
1.8.4.2.
Próbki wody do analizy uzyskiwane na przykład przez ich wypłukiwanie z centralnego punktu komory badawczej. Ze względu na to, że zarówno filtracja, jak i odwirowywanie nie zawsze powoduje oddzielenie się czynnika bioniedostępnego z pierwszej substancji od czynnika biodostępnego (zwłaszcza w substancjach superlipofilowych, tzn. w tych substancjach, które mają log Pow > 5) (1)(5), próbki mogą nie być poddawane takim obróbkom.
Zamiast tego pomiary powinny być pobierane w celu utrzymania zbiorników w czystości oraz zawartość całkowita węgla organicznego powinna być monitorowana zarówno podczas fazy absorpcji, jak i oczyszczania.
Odpowiednia ilość ryb (zwykle co najmniej cztery) jest usuwana z komór badawczych podczas każdorazowego pobierania próbek. Ryby pobrane w próbkach są szybko opłukiwane wodą, plamione „na sucho”, zabijane, wykorzystując odpowiedni i humanitarny sposób, a następnie ważone.
Preferowane jest analizowanie ryby i wody natychmiast po pobraniu próbek, w celu zapobieżenia degradacji lub innym stratom oraz w celu obliczenia średnich wskaźników absorpcji i oczyszczania w trakcie postępowania badania. Natychmiastowa analiza unika również opóźnień w określaniu, kiedy plateau zostało osiągnięte.
W przypadku niepowodzenia przeprowadzenia natychmiastowej analizy, próbki są składowane, wykorzystując odpowiednią metodę. Przed rozpoczęciem badania uzyskiwane są informacje dotyczące właściwej metody składowania w odniesieniu do poszczególnych substancji badanych – np. głębokiego zamrażania, przetrzymywania w temperaturze 4 oC, czasu trwania składowania, ekstrakcji itp.
1.8.4.3.
Ze względu na to, że cała procedura uzależniona jest zasadniczo od dokładności, precyzji i czułości metody analitycznej wykorzystywanej w odniesieniu do substancji badanej, sprawdzić doświadczalnie, iż precyzja oraz powtarzalność analizy chemicznej, jak również odzyskiwanie substancji badanej zarówno z wody, jak i są wystarczające dla danej metody. Sprawdzić również, że substancja badana nie jest wykrywalna wykorzystanej wodzie do rozcieńczania.
W miarę potrzeb wartości Cw i Cf, uzyskane z badania, są korygowane w odniesieniu do wartości tłowych uzyskanych w czasie kontroli. Z próbkami ryb i wody postępuje się w międzyczasie w taki sposób, aby zminimalizować zanieczyszczenie lub straty (np. wynikające z adsorpcji przez urządzenia do pobierania próbek).
1.8.4.4.
Jeśli w badaniu wykorzystywane są substancje oznakowane radiologicznie, możliwe jest przeprowadzenie analizy w odniesieniu do całkowitego oznaczenia radiologicznego (np. macierzyste i metabolity) lub można oczyścić próbki, tak aby związek macierzysty mógł zostać poddany analizie oddzielnie. Główne metabolity mogą zostać również określone w stanie ustalonym lub pod koniec fazy pobierania, w zależności od tego, która z tych faz nastąpi szybciej. Jeśli czynnik BCF jest > 1 000 % w kategoriach całkowitych pozostałości substancji oznakowanej radiologicznie, wskazane może być, a w odniesieniu do niektórych kategorii chemikaliów, takich jak pestycydy, stanowczo zalecane zidentyfikowanie i obliczenie ilości degradatów stanowiących > 10 % całkowitej ilości pozostałości substancji w tkankach ryb w stanie ustalonym. Jeśli degradaty stanowiące > 10 % całkowitych pozostałości substancji o oznaczeniu radiologicznym w tkankach ryb są zidentyfikowane i obliczone, zaleca się również identyfikację i podanie ilości degradatów w wodzie badanej.
Stężenie substancji badanej powinno zazwyczaj być oznaczane dla każdej indywidualnie ważonej ryby. Jeśli nie jest to możliwe, można zebrać wszystkie próbki w jednym zbiorniku podczas każdego pobierania próbek, ale nie ogranicza to procedur statystycznych, które mogą mieć zastosowanie do danych. Jeżeli szczególne procedury statystyczne oraz moc statystyczna są ważnymi względami, odpowiednia liczba ryb w celu przystosowania odpowiedniej procedury zgromadzenia próbek w jednym zbiorniku powinna zostać objęta badaniem (6)(7).
BCF powinno być wyrażone zarówno jako funkcja całkowitej masy mokrej, jak i, w odniesieniu do substancji wysokolipofilowych, jako funkcja zawartości lipidów. Zawartość lipidów ryb określa się podczas każdorazowego pobierania próbek, jeśli jest to możliwe. Odpowiednie metody powinny zostać wykorzystane w celu oznaczenia zawartości lipidów (odniesienia 8 i 2 w załączniku 3). Metoda ekstrakcji chloroform/metanol może być zalecana jako metoda standardowa (9). Różne metody nie dają identycznych wartości (10), zatem ważne jest podanie szczegółów dotyczących wykorzystanej metody. Jeśli jest to możliwe, analiza dla lipidów często powinna być przeprowadzona na tym samym ekstrakcie, który wytworzony został dla potrzeb analizy na obecność substancji badanej, ze względu na to, że lipidy często muszą zostać usunięte z ekstraktu zanim zostanie on poddany analizie chromatograficznie. Zawartość lipidów w rybie (podana w mg/kg mokrej masy) pod koniec doświadczenia nie powinna różnić się od tej z początku doświadczenia o więcej niż ± 25 %. Procent zawartości ciał stałych w tkance powinien zostać także odnotowany, w celu umożliwienia przekształcenia stężenia lipidów z bazy mokrej w bazę stałą.
2. DANE
2.1. PRZETWARZANIE WYNIKÓW
Krzywa absorpcji badanej substancji uzyskiwana jest przez wykreślenie jej stężenia w/na rybie (lub w określonych tkankach) w fazie absorpcji, w danym czasie, w skali arytmetycznej. Jeśli krzywa osiągnęła plateau, to znaczy, że stała się w przybliżeniu asymptotyczna do osi czasu, stan ustalony BCFSS obliczany jest w następujący sposób:
![]()
W przypadku gdy nie został osiągnięty stan ustalony, może istnieć możliwość obliczenia BCFSS o wystarczającej precyzji dla oceny zagrożenia na podstawie „stanu ustalonego” przy 80 % (1,6/k2) lub 95 % (3,0/k2) równowagi.
Również czynnik stężenia (BCFK) jest oznaczany jako stosunek k1/k2, dwóch stałych kinetycznych pierwszego rzędu. Stała szybkość oczyszczania (k2) jest zwykle określana na podstawie krzywej oczyszczania (tzn. na podstawie wykresu spadku stężenia badanej substancji w rybie wraz z upływem czasu). Stała szybkość absorpcji (k1) wyliczana jest wówczas na podstawie k2 oraz wartości Cf, którą uzyskuje się z krzywej absorpcji (zob. także załącznik 5). Preferowaną metodą uzyskiwania BCFK oraz stałych wskaźników, k1 i k2, jest wykorzystanie metod oceny parametrów nieliniowych na komputerze (11). W przeciwnym razie dla obliczenia k1 i k2 można korzystać z metod graficznych. Jeśli krzywa oczyszczenia w wyraźny sposób nie może być uznana za krzywą procesu pierwszego rzędu, wówczas powinny zostać zastosowane modele bardziej złożone (zob. odniesienia w załączniku 3) i powinno się zasięgnąć rady u biostatystyka.
2.2. INTERPRETACJA WYNIKÓW
Wyniki powinny być interpretowane uważnie w przypadku, gdy mierzone stężenia badanych roztworów występują na poziomach zbliżonych do granicy wykrywalności metody analitycznej.
Jasno określone krzywe absorpcji i ubytku wskazują dobrej jakości dane o biostężeniu. Rozbieżność w stałych absorpcji/oczyszczenia między dwoma badanymi stężeniami powinna być niższa niż 20 %. Powinny zostać odnotowane zaobserwowane różnice dotyczące szybkości absorpcji/oczyszczenia między dwoma zastosowanymi badanymi stężeniami, podając możliwe wyjaśnienia. Ogólnie granica ufności BCFs, na podstawie dobrze zaprojektowanego podejścia badawczego, wynosi ± 20 %.
3. SPORZĄDZANIE SPRAWOZDAŃ
Sprawozdanie z badania musi zawierać następujące informacje:
3.1. BADANA SUBSTANCJA:
|
— |
własności fizyczne oraz, gdy jest to istotne, właściwości fizyczno-chemiczne, |
|
— |
dane dotyczące identyfikacji chemicznej (łącznie z zawartością węgla organicznego, w odpowiednich przypadkach), |
|
— |
w przypadku oznakowania radiologicznego, dokładne położenie znaczonego atomu (znaczonych atomów) oraz zawartość procentową radioaktywności, związanej z nieczystościami. |
3.2. BADANE GATUNKI
|
— |
nazwa naukowa, gatunek, źródło, jakakolwiek obróbka wstępna, aklimatyzacja, wiek, zakres rozmiarów itp. |
3.3. WARUNKI PRZEPROWADZANIA BADANIA:
|
— |
wykorzystana procedura badawcza (np. przepływowa lub półstatyczna), |
|
— |
rodzaj i właściwości zastosowanego oświetlenia i fotookresów, |
|
— |
projekt badania (np. numer i rozmiar komór badawczych, szybkość wymiany objętości wody, liczba powtórzeń, liczba ryb przypadających na powtórzenie, liczba stężeń badania, długość faz absorpcji i oczyszczenia, częstotliwość pobierania próbek ryb i wody), |
|
— |
metoda przygotowania roztworów podstawowych i częstotliwość ich odnawiania, (jeśli został użyty, należy podać czynnik rozpuszczający, jego stężenie oraz jego udział w zawartości węgla organicznego wody używanej do przeprowadzenia badań), |
|
— |
nominalne stężenia badania, średnia mierzonych wartości oraz ich standardowe odchylenia, odnoszące się do naczyń używanych w badaniach, oraz metoda, za pomocą której zostały one uzyskane, |
|
— |
źródło wody do rozcieńczania, opis jakiejkolwiek obróbki wstępnej, wyniki przeprowadzenia dowodu na zdolność ryb do życia w wodzie oraz właściwości wody: pH, twardość, temperatura, stężenie rozpuszczonego tlenu, poziomy pozostałościowego chloru (jeśli dokonano pomiaru), węgiel organiczny ogółem, zawiesina, stopień zasolenia środowiska zastosowanego w badaniach (jeśli występuje taka potrzeba) oraz inne wykonane pomiary, |
|
— |
jakość wody w naczyniach wykorzystane w badaniach, pH, twardość, węgiel organiczny ogółem (TOC), temperatura oraz stężenie rozpuszczonego tlenu, |
|
— |
szczegółowe informacje w sprawie karmienia (np. rodzaj karmy, jej źródło, skład – przynajmniej, jeśli to możliwe, zawartość lipidów i białka, ilość podanej karmy i częstotliwość karmienia), |
|
— |
informacje w sprawie postępowania z próbkami ryb i wody, łącznie ze szczegółami dotyczącymi przygotowania, składowania, ekstrakcji i procedur analitycznych (oraz dokładność) w odniesieniu do badanej substancji i zawartości lipidów (jeśli zmierzono). |
3.4. WYNIKI:
|
— |
wyniki z wszelkich przeprowadzonych badań wstępnych, |
|
— |
śmiertelność ryb kontrolowanych oraz ryb w każdej z komór narażonych na działanie substancji oraz wszelkie inne zaobserwowane nieprawidłowe zachowania, |
|
— |
zawartość lipidów ryb (jeśli oznaczono w momencie przeprowadzania badania), |
|
— |
krzywe (łącznie z wszystkimi zmierzonymi danymi) przedstawiające absorpcję i oczyszczenie badanej substancji chemicznej w rybie, czas poprzedzający stan ustalony, |
|
— |
Cf i Cw (razem z odchyleniami standardowymi i zakresem, jeśli jest to stosowne) dla wszystkich czasów pbierania próbek (Cf wyrażone w μg/g mokrej masy (ppm)) całego organizmu lub określonych tkanek, np. lipidy, i Cw w μg/ml (ppm). Wartości Cw dla serii kontrolnych (należy również zgłosić tło), |
|
— |
czynnik biostężenia w stanie ustalonym (BCFSS) i/lub czynnik stężenia kinetycznego (BCFK) oraz, w stosownych przypadkach, 95 % granicy ufności dla stałej szybkości absorpcji i oczyszczenia (ubytku), wyrażone w odniesieniu do całego organizmu i całkowitej zawartości lipidów jeśli dokonano pomiaru, zwierzęcia lub jego poszczególnych tkanek, granice ufności i odchylenie standardowe, jeśli są dostępne, oraz metody analizy obliczeń/danych dla każdego stężenia wykorzystanej substancji badanej, |
|
— |
w przypadku gdy wykorzystano substancje oznakowane radiologicznie oraz, jeśli jest wymagane, nagromadzenie jakichkolwiek wykrytych metabolitów może zostać przedstawione, |
|
— |
wszystko, co odbiega od normy, jeśli chodzi o badanie, wszelkie odchylenia od tych procedur oraz wszelkie inne ważne informacje; |
zminimalizować ilość wyników jako „niewykrytych w granicy wykrywalności” w drodze opracowywania metod stosowanych przed badaniem i za pomocą projektu doświadczalnego, ponieważ wyniki takie nie mogą być użyte do obliczenia stałej szybkości reakcji.
4. BIBLOGRAFIA
|
(1) |
Connell D.W. (1988). Bio accumulation behaviour of persistent chemicals with aquatic organisms. Rev. Environ. Contam. Toxicol. 102, pp. 117–156. |
|
(2) |
Bintein S., Devillers J. and Karcher W. (1993). Non-linear dependence of fish bioconcentration on noctanol/water partition coefficient. SAR and QSAR in Environmental Research, 1, pp. 29–390. |
|
(3) |
OECD, Paris (1996). Direct phototransformation of chemicals in water. Environmental Health and Safety Guidance Document Series on Testing and Assessment of Chemicals. No 3. |
|
(4) |
Kristensen P. (1991). Bioconcentration in fish: comparison of bioconcentration factors derivedfrom OECD and ASTM testing methods; influence of particulate organic matter to the bio availability of chemicals. Water Quality Institute, Denmark. |
|
(5) |
US EPA 822-R-94-002 (1994) Great Lake Water Quality Initiative Technical Support Document for the Procedure to Determine Bioaccumulation Factors. July 1994. |
|
(6) |
US FDA (Food and Drug Administration) Revision. Pesticide analytical manual, 1, 5600 Fisher's Lane, Rockville, MD 20852. Julyl975. |
|
(7) |
US EPA (1974). Section 5, A(l). Analysis of Human or animal Adipose Tissue, in Analysis of Pesticide Residues in Human and Emronmental Samples, Thompson J. F. (ed.) Research Triangle Park, N.C. 27711. |
|
(8) |
Compaan H. (1980) in „The determination of the possible effects of chemicals and wastes on the aquatic environment: degradation, toxicity, bioaccumulation”, Rozdział 2.3, Part II. Government Publishing Office, the Hague, Netherlands. |
|
(9) |
Gardner et al, (1995). Limn. & Oceanogr. 30, pp. 1099–1105. |
|
(10) |
Randall R. C, Lee H, Ozretich R.J, Lake J. L. and Pruell R.J. (1991). Evaluation of selected lipid methods for normalising pollutant bio accumulation. Envir. Toxicol. Chem. 10, pp. 1431–1436. |
|
(11) |
CEC, Bioconcentration of chemical substances in fish: the flow-through method-Ring Test Programme, 1984 to 1985. Final report Marzecl 987. Autorzy: P. Kristensen i N. Nyholm. |
|
(12) |
ASTM E-1022-84 (Reapproved 1988). Standard Practice for conducting Bioconcentration Tests with Fishes and Saltwater Bivalve Molluscs. |
ZAŁĄCZNIK 1
Właściwości chemiczne dopuszczalnej wody do rozcieńczania
|
|
Substancja |
Ograniczenie stężenia |
|
1 |
Cząstki stałe |
5 mg/l |
|
2 |
Węgiel organiczny ogółem |
2 mg/l |
|
3 |
Amoniak niejonizowany |
1 μg/l |
|
4 |
Chlor pozostały |
10 μg/l |
|
5 |
Pestycydy fosforoorganiczne |
50 ng/l |
|
6 |
Pestycydy z grupy węglowodorów chlorowanych oraz bifenyle polichlorowane |
50 ng/1 |
|
7 |
Chlor organiczny ogółem |
25 ng/1 |
|
8 |
Glin |
1 μg/l |
|
9 |
Arsen |
1 μg/l |
|
10 |
Chrom |
1 μg/l |
|
11 |
Kobalt |
1 μg/l |
|
12 |
Miedź |
1 μg/l |
|
13 |
Żelazo |
1 μg/l |
|
14 |
Ołów |
1 μg/l |
|
15 |
Nikiel |
1 μg/l |
|
16 |
Cynk |
1 μg/l |
|
17 |
Kadm |
100 ng/1 |
|
18 |
Rtęć |
100 ng/1 |
|
19 |
Srebro |
100 ng/1 |
ZAŁĄCZNIK 2
Gatunki ryb zalecane do przeprowadzenia badania
|
|
Zalecane gatunki |
Zalecany zakres temperatur badania ( oC) |
Zalecana całkowita długość badanego zwierzęcia (cm) |
|
1 |
Danio rerio (17) (Teleostei, Cyprinidae) (Hamilton-Buchanan) Danio pręgowany |
20–25 |
3,0±0,5 |
|
2 |
Pimephales promelas (Teleostei, Cyprinidae) (Rafinesque) Strzebla |
20–25 |
5,0±2,0 |
|
3 |
Cyprinus carpio (Teleostei, Cyprinidae) (Linnaeus) Karp |
20–25 |
5,0±3,0 |
|
4 |
Oryzias latipes (Teleostei, Poeciliidae) (Temminck and Schlegel) Ryżówka |
20–25 |
4,0±1,0 |
|
5 |
Poccilia reticulata (Teleostei, Poeciliidae) (Peters) Gupik |
20–25 |
3,0±1,0 |
|
6 |
Lepomis macrochirus (Teleostei, Centrarchidae) (Rafinesque) Okoń błękitnoskrzeli (Bluegill) |
20–25 |
5,0±2,0 |
|
7 |
Oncorhynchus mykiss (Teleostei, Salmonidae) (Walbaum) Pstrąg tęczowy |
13–17 |
8,0±4,0 |
|
8 |
Gasterosteus aculeatus (Teleostei, Gasterosteidae) (Linnaeus) Ciernik |
18–20 |
3,0±1,0 |
W różnych krajach wykorzystano różne gatunki występujące w wodach przybrzeżnych i gatunki morskie, na przykład:
|
Spot |
Leiostomus xanthurus |
|
Owczarz karpiowaty |
Cyprinodon variegatus |
|
Kiżucz |
Mienidia beryl |
|
Szumień mały |
Cymatogaster aggregata |
|
Złocica europejska |
Parophrys vertulus |
|
Kur rogacz |
Leptocottus armatus |
|
Ciernik trzypromienny |
Gasteroseteus aculeatus |
|
Labraks |
Dicentratracus labrax |
|
Ukleja |
Alburnus alburnus |
Gromadzenie
Ryby słodkowodne, wymienione w tabeli, są łatwe w hodowli i/lub są szeroko dostępne w ciągu roku, podczas gdy dostępność gatunków morskich i zamieszkujących wody przybrzeżne jest częściowo ograniczona do odpowiednich krajów. Nadają się one do hodowli i rozmnażania albo w gospodarstwach rybnych, albo w laboratoriach, w warunkach chroniących je od chorób i pasożytów. Ryby te dostępne są w wielu częściach świata.
ZAŁĄCZNIK 3
Przewidywanie czasu trwania faz wchłaniania i oczyszczenia
1. Przewidywanie czasu trwania fazy wchłaniania
Przed przeprowadzeniem badania, oszacować k2 i wskutek tego pewien udział procentowy czasu potrzebnego do osiągnięcia stanu ustalonego może zostać uzyskany na podstawie zależności empirycznych między k2 a n-oktanolo/wodnym współczynnikiem podziału (Pow) oraz k2 i wodną rozpuszczalnością (s).
Oszacowanie k2 (dzień-1) może zostać uzyskane na przykład z następujących zależności empirycznych (1):
|
log10 k2 = 0,414 log10(Pow) + 1,47(r2 = 0,95) |
(równanie 1) |
W odniesieniu do innych zależności – zob. poz. bibliograficzna (2).
Jeśli nie jest znany współczynnik podziału (Pow), można dokonać oszacowania (3) na podstawie wiedzy o rozpuszczalności wodnej substancji, wykorzystując wzór:
|
log10 (Pow) = 0,862 log10(s) + 0,710 (r2 = 0,994) |
(równanie 2) |
gdzie:
s = rozpuszczalność (mol/l): (n = 36).
Zależności te stosują się jedynie do substancji chemicznych o wartościach log Pow pomiędzy 2 a 6,5 (4).
Czas potrzebny do osiągnięcia pewnego udziału procentowego stanu ustalonego może zostać uzyskany przez stosowanie oszacowania k2, na podstawie ogólnego równania kinetycznego, opisującego absorpcje i oczyszczanie (kinetyka pierwszego rzędu):

lub jeśli Cw jest stałe, w następujący sposób:
|
|
(równanie 3) |
W momencie zbliżania się do stanu stacjonarnego (t → ∞) równanie 3 można zredukować (5)(6) do:
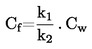 lub Cf/Cw = k1/k2
= BCF
lub Cf/Cw = k1/k2
= BCF
Wówczas k1/k2 Cw zbliża się do stężenia w rybie w „stanie ustalonym” (Cf, s).
Równanie 3 może zostać rozpisane do:
|
|
(równanie 4) |
Stosując równanie 4, czas potrzebny do osiągnięcia udziału procentowego stanu ustalonego może zostać przewidziany, jeśli k2 jest uprzednio oszacowane, wykorzystując równania 1 lub 2.
Jako wytyczna statystycznie optymalny czas trwania fazy absorpcji dla wytworzenia, statystycznie możliwy do przyjęcia danych (BCFK), jest okresem, który jest wymagany do tego, aby krzywa logarytmu stężenia substancji badanej w rybie, wykreślona w zestawieniu z czasem podanym liniowo, osiągnęła swój punkt środkowy 1 1,6/k2, lub też 80 % stanu ustalonego, ale nie więcej niż 3,0/k2 lub 95 % stanu ustalonego (7).
Czas potrzebny do osiągnięcia 80 % stanu ustalonego (równanie 4):
|
|
(równanie 5) |
Podobnie 95 % stanu ustalonego wynosi:
|
|
(równanie 6) |
Na przykład czas trwania fazy wchłaniania (up) dla substancji badanej o log Pow = 4 wynosiłby (wykorzystując równania 1, 5 i 6):
|
log10k2 = -0,414.(4) + 1,47 |
k2 = 0,652 dni-1 |
up (80 %) = 1,6/0,652, i.e. 2,45 days (59 godzin)
lub up (95 %) = 3,0/0,652, i.e. 4,60 days (110 godzin)
Podobnie, w odniesieniu do substancji badanej s = 10-5 mol/l (log(s) = -5,0) czas trwania wynosiłby (wykorzystując równania 1, 2, 5 i 6):
log10 (Pow) = 0,862 (-5,0) + 0,710 = 5,02
log10 K2 = 0,414 (5,02) + 1,47
k2 = 0,246 dni-1
up (80 %) = 1,6/0,246, tj. 6,5 dni (156 godzin)
lub up (95 %) = 3,0/0,246, tj. 12,2 dni (293 godzin)
Alternatywnie, wyrażenie:
teq = 6,54 x 103 Pow + 55,31 (godzin)
może być wykorzystane do obliczenia czasu potrzebnego do skutecznego osiągnięcia stanu ustalonego (4).
2. Przewidywanie czasu trwania fazy oczyszczenia
Przewidywanie ilości czasu potrzebnego do wyeliminowania zanieczyszczenia z organizmu do pewnej ilości udziału procentowego stężenia początkowego może zostać uzyskane na podstawie ogólnego równania, opisującego absorpcje i oczyszczanie (kinetyka pierwszego rzędu) (1)(8).
Dla fazy oczyszczania przyjmuje się Cw równe zeru. Równanie można zredukować do:
 lub
lub 
Gdzie Cf, o jest stężeniem z początku stanu oczyszczania 50 %. Oczyszczanie osiągnięte zostanie wówczas w czasie (t50):
 lub
lub 
Podobnie 95 % oczyszczania zostanie osiągnięte w następującym czasie:

Jeśli 80 % absorpcji wykorzystywane jest w odniesieniu do pierwszego okresie (1,6/k 2), a 95 % ubytku w fazie oczyszczania (3,0/k 2), wówczas faza oczyszczania jest w przybliżeniu dwa razy dłuższa niż długość fazy wchłaniania.
Jednakże ważne jest, aby odnotować, że oszacowania oparte są na założeniu, że wzory absorpcji i oczyszczania będą zgodne z kinetyką pierwszego rzędu. Jeśli w wyraźny sposób kinetyka pierwszego rzędu nie jest przestrzegana, powinny zostać wykorzystane bardziej złożone modele (np. poz. bibliograficzna (1)).
Literatura (załącznika 3)
|
(1) |
Spacie A. and Hamelink J.L. (1982) Alternative models for describing the bioconcentration of organics in fish.Toxicol. and Chem. 1, pp. 309–320. |
|
(2) |
Kristensen P. (1991) Bioconcentration in fish: comparison of BCFs derived from OecD and ASTM testing methods; influence of p articulate matter to the bio availability of chemicals. Danish Water Quality Institute. |
|
(3) |
Chiou C.T. and Schmedding (3) (1982). Partitioning of organic compounds in octanol-water systems. Environ. Sci. Technol. 16, pp. 1.4–10. |
|
(4) |
Hawker D. W. and Connell D.W. (1988). Influence of partition coefficient of lipophilic compounds on bioconcentration kinetics with fish. Wat. Res. 22 (6), pp. 701–707. |
|
(5) |
Branson D. R., Blau G.E., Alexander H. C. i Neely W. B. (19 7 5). Transactions of the American Fisheries Society, 104 (4), pp. 785–792. |
|
(6) |
Ernst W. (19 8 5). Accumulation in Aquatic organisms. W: Appraisal of tests to predict the environmental behaviour of chemicals. Ed. by Sheehman P., Korte F., Klein W. and Bourdeau P. H. Part 4.4, str. 243-255. SCOPE, 1985, John Wiley & Sons Ltd N.Y. |
|
(7) |
Reilly P.M., Bajramovic R., Blau G. E., Branson D. R. i Sauerhoff M.W. (1977). Guidelines for the optimal design of experiments to estimate parameters in first order kenetic models, Can. J. Chem. Eng. 55, pp. 614–622. |
|
(8) |
Könemann H. and Van Leeuwen K. (1980). Toxicokinetics in fish: Accumulation and Elimination of six Chlorobenzenes by Guppies. Chemosphere, 9, pp. 3–19. |
ZAŁĄCZNIK 4
Teoretyczny przykład harmonogramu pobierania próbek w odniesieniu do badań biokoncentracji substancji o log Pow = 4
|
Pobieranie próbek ryb |
Harmonogram badania próbek |
Liczba ryb przypadających na jedną próbkę |
Liczba próbek wody |
||||
|
Minimalna wymagana częstotliwość (dni) |
Dodatkowe pobieranie próbek |
||||||
|
Faza wchłaniania |
-1 0 |
|
2 (18) 2 |
Dodać 45–80 ryb |
|||
|
pierwsza |
0,3 |
0,4 |
2 (2) |
4 (4) |
|||
|
druga |
0,6 |
0,9 |
2 (2) |
4 (4) |
|||
|
trzecia |
1,2 |
1,7 |
2 (2) |
4 (4) |
|||
|
czwarta |
2,4 |
3,3 |
2 (2) |
4 (4) |
|||
|
piąta |
4,7 |
|
2 |
6 |
|||
|
Faza oczyszczania |
|
|
|
Przenieść ryby do wody wolnej od badanej substancji chemicznej |
|||
|
szósta |
5,0 |
5,3 |
|
4 (4) |
|||
|
siódma |
5,9 |
7,0 |
|
4 (4) |
|||
|
ósma |
9,3 |
11,2 |
|
4 (4) |
|||
|
dziewiąta |
14,0 |
17,5 |
|
6 (4) |
|||
|
Wartości w nawiasach są liczbami próbek (woda, ryby), które mają zostać pobrane, jeśli przeprowadzane jest dodatkowe pobieranie próbek.
|
|||||||
ZAŁĄCZNIK 5
Rozróżnianie modeli
Większość danych dotyczących biokoncentracji uznano za dość dobrze opisane za pomocą prostego modelu dwukomorowego o dwóch parametrach, jak wskazano za pomocą krzywej prostoliniowej, która zbliża się do punktów stężenia w rybach, w czasie fazy oczyszczania, gdy są one wykreślone na papierze półlogarytmicznym. (W przypadku gdy punktów tych nie można opisać za pomocą krzywej prostoliniowej, wówczas należy zastosować modele złożone, zob. na przykład Spacie i Hamelink, poz. bibliograficzna (1) w załączniku 3).
Graficzne metody oznaczania stałej szybkości oczyszczania (ubytku) k2
Wykreślić stężenie substancji badanej zawartej w każdej z próbek ryb w czasie pobierania próbek na papierze półlogarytmicznym. Nachyleniem tej linii jest k2.
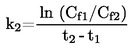
Należy zauważyć, że odchylenia od linii prostej mogą wskazywać na bardziej złożony schemat oczyszczania niż ten oparty na kinetyce pierwszego rzędu. Metoda graficzna może być zastosowana do analizowania typów oczyszczania, które są odchyleniem od kinetyki procesu pierwszego rzędu.
Graficzne metody oznaczania stałej szybkości absorpcji k1
Mając podane k2, obliczyć k1 w następujący sposób:
|
|
(równanie 1) |
Wartość Cf odczytywana jest ze środkowego punktu części gładkiej krzywej absorpcji, powstałej w oparciu o dane, gdy stężenie logarytmiczne jest wykreślone w zależności od czasu (w skali arytmetycznej).
Metoda komputerowego obliczania stałych szybkości absorpcji i oczyszczania (ubytku)
Preferowaną metodą uzyskiwania czynnika biokoncentracji oraz stałych szybkości reakcji, k1 i k2, jest wykorzystanie metod oceny parametrów nieliniowych na komputerze. Programy te przypisują wartości dla k1 i k2 w oparciu o zestaw danych dotyczących czasu sekwencyjnego stężenia i modelu:
|
|
(równanie 2) |
|
|
(równanie 3) |
gdzie: tc = czas przy końcu fazy absorpcji
Podejście to przewiduje standardowe oszacowania odchyleń k1 i k2.
Ponieważ k2 w większości przypadków może zostać oszacowane na podstawie krzywej oczyszczenia z dość wysoką dokładnością oraz ze względu na istnienie silnego związku między dwoma parametrami k1 i k2, gdy obliczane są równocześnie, wskazane może być obliczanie najpierw k2, jedynie na podstawie danych dotyczących oczyszczania, a w następnej kolejności obliczanie k1, na podstawie danych dotyczących wchłaniania, używając regresji nieliniowej.
C.14. BADANIE ROZWOJU MŁODYCH RYB
1. METODA
Niniejsza metoda wpływu toksyczności na rozwój jest kopią OECD TG 215 (2000).
1.1. WPROWADZENIE
Niniejszy badanie jest przeznaczone do oceny skutków przedłużonej ekspozycji na działanie substancji chemicznych na rozwój młodocianych ryb. Jest on oparty na metodzie, rozwiniętej i powszechnie stosowanej (1)(2) w ramach Unii Europejskiej, dla oceny działania substancji chemicznych na rozwój młodego pstrąga tęczowego (Oncorynchus mykiss) w warunkach przepływowych. Inne dobrze udokumentowane gatunki można również zastosować. Na przykład uzyskano doświadczenie z badań rozwoju danio pręgowanego (Danio rerio) (19) (3)(4) i Oryzias latipes (5)(6)(7).
Zob. także Ogólne wprowadzenie część C.
1.2. DEFINICJE
Najniższy obserwowany skutek stężenia (LOEC): jest to najmniejsze badane stężenie substancji badanej, przy którym obserwuje się znaczący skutek (przy p < 0,05) w porównaniu z doświadczeniem kontrolnym. Jednakże wszystkie badane stężenia powyżej LOEC muszą wykazywać szkodliwy skutek, równy lub większy niż ten obserwowany przy LOEC.
Nieobserwowany skutek stężenia (NOEC): jest stężeniem badanym bezpośrednio poniżej LOEC.
EC x : w niniejszej metodzie jest stężeniem badanej substancji, która powoduje x % zmienności we wskaźniku rozwoju ryb w porównaniu z doświadczeniem kontrolnym.
Wskaźnik obciążenia: jest wilgotną masą ryb na objętość wody.
Gęstość wyjściowa: jest liczbą ryb do objętości wody.
Wskaźnik szczególnego rozwoju określonej ryby: wyrażenie wskaźnika rozwoju określonej jednostki w oparciu o jej początkową wagę.
Średni zbiornikowy wskaźnik szczególnego rozwoju: wyrażenie średniego wskaźnika wzrostu populacji w zbiorniku przy jednym stężeniu.
Wskaźnik pseudoszczególnego rozwoju: wyrażenie wskaźnika określonego rozwoju w proporcji do średniej wagi początkowej populacji zbiornika.
1.3. ZASADA METODY BADANIA
Młode ryby w fazie wykładniczej rozwoju są umieszczane po zważeniu w komorach do badań i eksponowane w zakresie stężenia subletalnego substancji badanej rozpuszczonej w najlepiej strumieniem wody lub jeśli to niemożliwe, we właściwych, półstatycznych (statycznie odnawialnych) warunkach. Czas trwania badania wynosi 28 dni. Ryby są karmione codziennie. Racje żywności są oparte na początkowej wadze ryb i przeliczane po 14 dniach. Na koniec badania ryby są ważone ponownie. Skutki w odniesieniu do wskaźników rozwoju są analizowane, stosując model regresyjny w celu prognozowania stężenia, które spowoduje x % zmienności w wskaźniku rozwoju, tj. ECx (np. EC10, EC20 lub EC30). Alternatywnie, dane są porównywane z objętością w doświadczeniu kontrolnym w celu ustalenia najniższego obserwowanego skutku stężenia (LOEC), a stąd też nieobserwowanego skutku stężenia (NOEC).
1.4. INFORMACJE DOTYCZĄCE SUBSTANCJI BADANEJ
Wyniki badania ostrej toksyczności (zob. metoda badania C.1), najlepiej przeprowadzone z gatunkami wybranymi do niniejszego badania, powinny być dostępne. Oznacza to, że rozpuszczalność w wodzie i ciśnienie pary badanej substancji są znane, a wiarygodna analityczna metoda jest dostępna w odniesieniu do ujęcia ilościowego substancji w roztworach badanych, ze znaną i udokumentowaną dokładnością oraz granicą wykrywalności.
Użyteczne informacje obejmują wzór strukturalny, czystość substancji, stabilność w wodzie i na świetle, pKa, Pow oraz wyniki badania całkowitej biodegradacji (zob. metoda C.4).
1.5. WAŻNOŚĆ BADANIA
Aby badanie był ważne, stosuje się następujące warunki:
|
— |
śmiertelność w doświadczeniach kontrolnych nie może przekraczać 10 % na koniec badania, |
|
— |
średnia waga ryb w doświadczeniach kontrolnych musi wzrosnąć wystarczająco, aby pozwolić na wykrycie minimalnych zmian wskaźnika rozwoju uznanego za znaczący. Próba pierścieniowa (2) wykazała, że w odniesieniu do pstrąga tęczowego średnia waga ryb w doświadczeniach kontrolnych wzrosła co najmniej o połowę (tj. 50 %) ich średniej wagi początkowej przez ponad 28 dni; tj. waga początkowa: 1 g/ryba (= 100 %), waga końcowa po 28 dniach: ≥ 1,5 g/ryba (≥ 150 %), |
|
— |
stężenie tlenu rozpuszczonego musi wynosić co najmniej 60 % wartości nasycenia powietrzem (ASV) w trakcie badania, |
|
— |
różnica temperatury wody nie może być większa niż ± 1 oC między komorami do badań przez cały czas trwania badania i musi być utrzymywana w obrębie 2 oC zakresu temperatur określonego w odniesieniu do gatunków badanych (dodatek 1). |
1.6. OPIS METODY BADANIA
1.6.1. Aparatura
Zwykły sprzęt laboratoryjny, w szczególności następujący:
|
— |
mierniki tlenu i pH metry, |
|
— |
sprzęt do oznaczania alkaliczności i twardości wody, |
|
— |
odpowiednia aparatura do pomiaru temperatury, zalecane jest stałe monitorowanie, |
|
— |
zbiorniki wykonane z chemicznie obojętnego materiału o odpowiedniej objętości w zależności do zalecanej gęstości wyjściowej i obciążenia (zob. pkt 1.8.5 i dodatek 1), |
|
— |
waga o odpowiedniej dokładności (tj. dokładności do ±0,5 %). |
1.6.2. Woda
Każda woda, w której badane gatunki wykazują odpowiednie długoterminowe przeżycie i wzrost, może być stosowana jako woda do badań. Musi być stałej jakości w okresie trwania badania. Wartość pH wody powinna być w zakresie 6,5–8,5, lecz w trakcie danego badania musi być w zakresie ±0,5 pH. Twardość powyżej 140 mg/l (jako CaCO3) jest zalecana. W celu zapewnienia, aby rozcieńczająca woda nie miała falowego wpływu na wyniki badania (np. przez kompleksowanie substancji badanej), należy okresowo pobierać próbki wody do analizy. Co trzy miesiące na przykład powinny być wykonane pomiary metali ciężkich (takich jak Cu, Pb, Zn, Hg, Cd i Ni), głównych anionów i kationów (np. Ca, Mg, Na, K, Cl i SO4), pestycydów (np. całkowita ilość fosforo- i chloroorganicznych), całkowity węgiel organiczny i zawiesina ciał stałych, w przypadku gdy rozcieńczająca woda jest znana jako stosunkowo stała w jakości. Jeśli można przedstawić dane, że jakość wody była stała przynajmniej przez rok, oznaczenia mogą być mniej częste i przedziały poszerza się (np. co sześć miesięcy). Kilka cech chemicznych akceptowalnej wody rozcieńczającej wymieniono w dodatku 2.
1.6.3. Roztwory użyte w badaniu
Roztwory w badaniu o wybranym stężeniu są przygotowywane poprzez rozcieńczanie roztworu podstawowego.
Zalecane jest przygotowanie roztworu podstawowego przez proste wymieszanie badanej substancji w wodzie rozcieńczającej w sposób mechaniczny (np. mieszadło lub ultradźwięki). Kolumny nasyceniowe (rozpuszczające) mogą być użyte do uzyskania odpowiednio stężonego roztworu podstawowego.
W kilku przypadkach może być wymagane użycie rozpuszczalników lub dyspergatorów (środków rozpuszczających) w celu wykonania odpowiednio stężonego roztworu podstawowego. Przykładami odpowiednich rozpuszczalników są aceton, etanol, metanol, dimetylosulfotlenek, dimetyloformamid i glikol trójetylenowy. Przykładami odpowiednich dyspergatorów są Cremophor RH40, Tween 80, Metyloceluloza 0,01 % i HCO-40. Należy zachować ostrożność przy użyciu łatwo biodegradowalnych środków (np. aceton) i/lub wysokolotnych związków, gdyż mogą być przyczyną problemów z rozwojem bakterii w przepływach badań. Gdy stosuje się środki rozpuszczające, nie mogą one mieć znaczącego wpływu na wzrost ryb i widocznych negatywnych działań na młode ryby, jak odkryto w doświadczeniu kontrolnym z rozpuszczalnikiem.
Dla badań przepływowych wymagany jest system ciągłego dozowania i rozcieńczania roztworu podstawowego substancji badanej (np. pompa dozująca, rozcieńczalnik proporcjonalny, system nasycania) do przesyłania serii stężeń do komór do badań. Szybkości przepływów roztworu podstawowego i rozcieńczającej wody muszą być sprawdzane okresowo, najlepiej codziennie w trakcie trwania badania, oraz nie mogą zmieniać się więcej niż 10 % w trakcie badania. Badanie pierścieniowe (2) wykazało, że w odniesieniu do pstrąga tęczowego częstotliwość wymiany wody w trakcie trwania badania jest akceptowalna przy 6 litrów/g ryby/dzień (zob. pkt 1.8.2.2).
Dla badań półstatycznych (odnawialnych) częstotliwość odnawiania ośrodka zależy od stabilności substancji badanej, lecz zalecane jest codzienne odnawianie wody. Jeśli ze wstępnych badań stabilności (zob. pkt 1.4) wynika, że stężenie substancji badanej nie jest stabilne (tj. poza zakresem nominalnego 80–120 % lub spada poniżej 80 % zmierzonego stężenia początkowego) przez okres odnowienia, należy rozważyć celowość użycia badania przepływowego.
1.6.4. Wybór gatunków
Pstrąg tęczowy (Oncorhynchus mykiss) jest zalecanym gatunkiem do niniejszego badania, ponieważ uzyskano najwięcej doświadczeń z prób pierścieniowych z tym gatunkiem (1)(2). Można jednakże zastosować inne, dobrze udokumentowane gatunki, lecz może być niezbędne przyjęcie procedury badania w celu zapewnienia odpowiednich warunków badania. Dostępne są doświadczenia, na przykład z danio pręgowanym (Danio rerio) (3)(4) i Oryzias latipes (5)(6)(7). W takim przypadku powinno być podane racjonalne uzasadnienie wyboru i metody eksperymentalnej.
1.6.5. Przetrzymanie ryb
Badane ryby muszą być wybrane z populacji wyjściowej pojedynczo, najlepiej z tego samego tarła, przetrzymywanego co najmniej dwa tygodnie przed badaniem w warunkach jakości wody i oświetlenia zbliżonych do stosowanych w badaniu. Powinny one być karmione racją minimum 2 % wagi ciała dziennie, a najlepiej 4 % wagi ciała na dzień przez okres przetrzymania i w trakcie trwania badania.
Przez następujący od rozpoczęcia okres 48 godzin notowana jest śmiertelność i stosuje się następujące kryteria:
|
— |
śmiertelność większa niż 10 % populacji przez siedem dni: odrzucić całą partię, |
|
— |
śmiertelność między 5 % i 10 % populacji: aklimatyzacja przez siedem dodatkowych dni, jeśli więcej niż 5 % śmiertelności w ciągu tych siedmiu dni, odrzucić całą partię, |
|
— |
śmiertelność mniejsza od 5 % populacji w ciągu siedmiu dni: dopuścić partię. |
Ryby nie mogą być taktowane leczniczo przez dwa tygodnie poprzedzające badanie ani w ciągu jego trwania.
1.7. PROJEKT BADANIA
„Projekt badania” odnosi się do wyboru ilości i odstępów stężeń badanych, liczbą zbiorników dla każdego poziomu i liczbą ryb na zbiornik. W idealnym przypadku taki projekt badania powinien być wybierany z uwzględnieniem:
|
— |
celu badania, |
|
— |
metody statystycznej analizy, która będzie wykorzystana, |
|
— |
dostępność i źródła finansowania eksperymentu. |
Oświadczenie o celu badania powinno, jeśli jest to możliwe, określać statystyczną zdolność, przy której różnica danego wymiaru (np. we wskaźniku rozwoju) wymaganego jest możliwa do obserwacji lub, alternatywnie, precyzją, z jaką ECx (na przykład x = 10, 20 lub 30, a najlepiej nie mniej niż 10) ma być oszacowane. Bez tego zwięzłego określenie zakresu badania nie może być podane.
Istotne jest, aby uznać, że projekt, który jest optymalny (najlepszy z użyciem źródeł) przy użyciu jednej metody analizy statystycznej, nie jest koniecznie optymalny dla innej. Zalecany projekt dla oszacowania LOEC/NOEC dlatego nie byłby taki sam jak zalecany dla analizy metodą regresji.
W większości przypadków, analiza regresyjna jest zalecana dla analizy wariancji, z przyczyn omówionych przez Stephan i Rogers (8). Jednakże jeżeli nie znaleziono odpowiedniego modelu regresji (r2 < 0,9) powinna być zastosowana metoda NOEC/LOEC.
1.7.1. Projekt analizy regresyjnej
Ważnymi rozważaniami w projekcie badania, które mają być zanalizowane metodą regresyjną, są:
|
— |
skutek stężenia (np. EC10, 20, 30) i zakres stężeń powyżej, którego wpływ substancji badanej jest poza zainteresowaniem, powinien być koniecznie osiągnięty przez stężenie włączone do badań. Dokładność, z którą ocena wpływu stężenia jest wykonywana, jest najlepsza, gdy wpływ stężenia jest pośrodku zakresu badanych stężeń. Wstępne badanie szukania zakresu może być pomocne w wybraniu właściwych stężeń badanych, |
|
— |
w celu zapewnienia zadawalającego modelu statystycznego badanie powinno obejmować co najmniej jeden zbiornik kontrolny i pięć dodatkowych zbiorników o różnych stężeniach. Tam gdzie to jest stosowne, gdy stosowany jest środek ułatwiający rozpuszczanie, należy przeprowadzić obok serii badanej jedno doświadczenie kontrolne z tym środkiem przy najwyższym badanym stężeniu (zob. pkt 1.8.3 i 1.8.4), |
|
— |
właściwe serie geometryczne lub serie logarytmiczne (9) (zob. dodatek 3) mogą być wykorzystane. Zaleca się odstępy logarytmiczne w stężeniach badanych, |
|
— |
o ile jest dostępne więcej niż sześć zbiorników, dodatkowe zbiorniki muszą być użyte albo do zapewnienia powtórzenia lub rozdziału w poprzek zakresu stężeń, w celu zapewnienia bliższego odstępu poziomów. Oba z tych środków są w równym stopniu pożądane. |
1.7.2. Projekt dotyczący oszacowania NOEC/LOEC przy użyciu analizy wariancji (ANOVA)
Zalecane jest posiadanie skopiowanych zbiorników dla każdego stężenia i analizy statystycznej na poziomie zbiornika (10). Bez skopiowanych zbiorników nie można zastosować poprawek dla zmienności między zbiornikami, poza spodziewanymi dla poszczególnych ryb. Jednakże doświadczenie pokazało (11), że zmienność między zbiornikami jest bardzo mała w porównaniu ze zmiennością w obrębie zbiorników (tj. między rybami) w badanym przypadku. Dlatego względnie dopuszczalną alternatywą jest przeprowadzenie analizy statystycznej na poziomie poszczególnych ryb.
Zwyczajowo stosuje się co najmniej pięć stężeń badanych w serii geometrycznej ze współczynnikiem zalecanym nieprzekraczającym 3,2.
Ogólnie, gdy badania są prowadzone ze skopiowanymi zbiornikami, liczba skopiowanych zbiorników kontrolnych, a zatem i liczba ryb musi być podwojona przy każdym badaniu stężenia, które powinny być równoważnych rozmiarów (12)(13)(14). Z drugiej strony, przy nieobecności skopiowanych zbiorników, liczba ryb w grupie kontrolnej powinna być taka sama jak liczba przy każdym badaniu stężenia.
Jeśli ANOVA ma opierać się na zbiornikach, a nie na poszczególnych rybach (pociąga to albo oznaczanie poszczególnych ryb albo użycie „pseudoszczególnych” wskaźników rozwoju (zob. pkt 2.1.2), zachodzi potrzeba wystarczającego skopiowania zbiorników w celu zapewnienia oznaczenia standardowego odchylenia „stężeń w obrębie zbiorników”. To oznacza, że stopień swobody błędu w analizie wariancji musi wynosić co najmniej 5 (11). Jeśli tylko doświadczenia kontrolne są powielane zachodzi niebezpieczeństwo, że zmienność błędu będzie obciążona, ponieważ może to zwiększyć rozważaną wartość średnią wskaźnika rozwoju. Ponieważ wskaźnik rozwoju prawdopodobnie zmniejszy się ze wzrostem stężenia, będzie to powodowało przeszacowanie zmienności.
1.8. PROCEDURA
1.8.1. Wybór i ważenie ryb badanych
Istotne jest, aby zminimalizować zmienność wagi ryb na początku badania. Odpowiedni zakres wymiarowy dla różnych gatunków zalecanych do wykorzystania w tym badaniu podano w dodatku 1. W odniesieniu do całej partii ryb wykorzystanych w badaniu zakres poszczególnych wag na początku badania powinien być idealnie utrzymany w obrębie ± 10 % średniej arytmetycznej wagi, a w żadnym przypadku nie może przekraczać 25 %. Zalecane jest zważenie podpróbki ryb przed badaniem, w celu oszacowania średniej wagi.
Należy wstrzymać żywienie populacji na 24 godziny przed rozpoczęciem badania. Ryby powinny być wybrane losowo. Stosując ogólne znieczulenie (np. roztwór wodny 100 mg/l tricainy metanosulfonianu (MS 222) zneutralizowanej przez dodanie dwóch części dwuwęglanu sodu na jedną część MS 222), ryby powinny być zważone pojedynczo w stanie wilgotnym (osuszone) z dokładnością podaną w dodatku 1. Ryby, których waga jest w zamierzonym zakresie powinny być zatrzymane i następnie losowo rozdzielone do zbiorników do badań. Całkowita waga ryb w każdym zbiorniku powinna być odnotowana. Użycie znieczulenia ułatwia manipulację rybami (włączając suszenie i ważenie) może jednak powodować stres i zranienia dla młodocianych ryb, w szczególności dla gatunków o małych wymiarach. Dlatego manipulowanie młodocianymi rybami musi być wykonane z najwyższą troską, aby zapobiec stresom i zranieniom badanych zwierząt.
Ryby są ważone ponownie w 28 dniu badania (zob. pkt 1.8.6). Jednakże jeśli jest to uważane za niezbędne przeliczenie racji żywieniowej, ryby są ważone ponownie 14 dnia badania (zob. pkt 1.8.2.3). Inne metody, takie jak metoda fotograficzna, mogą być stosowane do ustalania zmiany rozmiarów ryb, na podstawie których można dostosować racje żywieniowe.
1.8.2. Warunki ekspozycji
1.8.2.1. Czas trwania
Czas trwania badania wynosi ≥ 28 dni.
1.8.2.2. Wskaźnik obciążenia i gęstości wyjściowe
Istotne jest, aby wskaźnik obciążenia i gęstość wyjściowa były właściwe dla wykorzystanych gatunków badanych (zob. dodatek 1). Jeśli gęstość wyjściowa jest za wysoka, wtedy stres z powodu zatłoczenia może prowadzić do zmniejszenia wskaźnika rozwoju i prawdopodobnie do choroby. Jeśli jest za mała, może wytwarzać zachowywanie terytorialne, które również wpływa na rozwój. W każdym przypadku wskaźnik obciążenia powinien być wystarczająco niski, aby stężenie tlenu rozpuszczonego wynosiło przynajmniej 60 % ASV i było utrzymywana bez napowietrzania. Próba pierścieniowa (2) pokazała, że w odniesieniu do pstrąga tęczowego wskaźnik obciążenia dla 16 pstrągów po 3–5 gram w 40-litrowej objętości jest dopuszczalny. Zalecana częstotliwość usuwania wody w czasie trwania badania wynosi 6 litrów/gram ryby/dzień.
1.8.2.3. Karmienie
Ryby muszą być karmione właściwym pokarmem (dodatek 1) w wystarczającej ilości dla wytworzenia akceptowalnego wskaźnika rozwoju. Należy zwrócić uwagę, aby zapobiec rozwojowi mikrobów i mętności wody. Dla pstrąga tęczowego wskaźnik 4 % ich wagi ciała dziennie prawdopodobnie spełnia te warunki (2)(15)(16)(17). Dzienna racja może być podzielona na dwie równe porcje i podawana rybom w dwóch posiłkach dziennie, w odstępie co najmniej pięciu godzin. Racje są oparte na początkowej całkowitej wadze ryb w każdym zbiorniku do badań. Jeśli ryby są ważone ponownie 14 dnia racje są wtedy przeliczane. Karmienie powinno być wstrzymane na 24 godziny przed ważeniem.
Niezjedzony pokarm i odchody ryb muszą być usuwane ze zbiorników do badań codziennie, poprzez staranne czyszczenie dna zbiorników stosując ssanie.
1.8.2.4. Światło i temperatura
Czas naświetlania i temperatura wody muszą być odpowiednie dla badanego gatunku (dodatek 1).
1.8.3. Stężenia użyte w badaniu
Zwykle wymagane jest pięć stężeń badanych, poza projektem badania (zob. pkt 1.7.2). Wcześniejsza znajomość toksyczności substancji badanej (np. z badania ostrej toksyczności i/lub ustalonego w badaniach zakresu) pomaga w doborze właściwych stężeń badanych. Najwyższe stężenie badane nie może przekraczać granicy rozpuszczalności substancji w wodzie.
W przypadku gdy środek ułatwiający rozpuszczenie towarzyszy w przygotowaniu roztworu wyjściowego jego końcowe stężenie nie może być większe niż 0,1 ml/l i zalecane jest takie same we wszystkich zbiornikach do badań (zob. pkt 1.6.3). Jednakże należy dołożyć wszelkich starań, aby uniknąć użycia takich materiałów.
1.8.4. Doświadczenia kontrolne
Liczba doświadczeń kontrolnych wody rozcieńczającej zależy od projektu badania (zob. pkt 1.7–1.7.2). Jeśli stosowany jest środek ułatwiający rozpuszczanie, powinna być przeprowadzona taka sama liczba doświadczeń kontrolnych co liczba doświadczeń kontrolnych z wodą rozcieńczającą.
1.8.5. Częstotliwość oznaczeń analitycznych i pomiarów
W czasie trwania badania stężenia substancji badanej są oznaczane w regularnych odstępach czasu (zob. poniżej).
W badaniach przepływowych wskaźniki przepływu rozcieńczalnika i roztworu wyjściowego toksykanta muszą być sprawdzane w odstępach czasu, najlepiej codziennie, i nie mogą się zmieniać więcej niż 10 % w trakcie badania. W przypadku gdy oczekuje się, że stężenie badanej substancji będzie w zakresie ± 20 % wartości nominalnej (tj. w zakresie 80–120 %; zob. pkt 1.6.2 i 1.6.3), jest zalecane, aby jako minimum wykonać analizę najmniejszego i największego stężenia substancji badanej na początku badania i w tygodniowych odstępach czasu, w okresie późniejszym. W przypadku gdy oczekuje się, że stężenie substancji badanej pozostanie w zakresie ± 20 % nominalnej wartości (na podstawie danych stabilności substancji badanej), niezbędne jest analizowanie wszystkich stężeń badanych, ale przy zastosowaniu tego samego systemu.
W półstatycznych (odnawialnych) badaniach, w przypadku gdy oczekuje się, że stężenie badanej substancji pozostanie w zakresie ± 20 % nominalnej wartości, jest zalecane, aby jako minimum analizować najniższe i najwyższe stężenia badane świeżo po przygotowaniu i zaraz przed odnowieniem, na początku badań i cotygodniowo w okresie późniejszym. Dla badań gdzie nie oczekuje się, że stężenie substancji badanej pozostanie w zakresie ± 20 % nominału, wszystkie badane stężenia muszą być analizowane, przy zastosowaniu tego samego systemu co dla substancji bardziej stabilnych.
Zalecane jest, aby wyniki oparte były na pomierzonych stężeniach. Jeżeli jednakże dostępne dowody wykazują, że stężenie badanej substancji w roztworze można z powodzeniem utrzymywać w obszarze ± 20 % nominalnego lub wstępnie zmierzonego stężenia poprzez okres badania, wtedy wyniki mogą być oparte na wartościach nominalnych lub zmierzonych.
Próbki mogą wymagać przefiltrowania (np. stosując 0,45 μm wymiar poru) lub odwirowania. Odwirowanie jest zalecaną procedurą. Jednakże jeśli badany materiał nie adsorbuje się na filtrze, dopuszczalna jest też filtracja.
W czasie trwania badania powinny być mierzone tlen rozpuszczony, pH i temperatura we wszystkich zbiornikach. Całkowita twardość, alkaliczność i zasolenie (jeśli dotyczy) muszą być mierzone w doświadczeń kontrolnych i w zbiorniku o najwyższym stężeniu. Jako minimum należy pomierzyć rozpuszczony tlen i zasolenie (jeśli dotyczy) trzy razy (na początku, w środku i pod koniec badania). W badaniach półstatycznych jest zalecane, aby rozpuszczony tlen był mierzony częściej, najlepiej przed i po każdej wymianie wody lub co najmniej raz na tydzień. Wartość pH na początku i na końcu każdej wymiany wody w badaniach statycznych odnawialnych i co najmniej cotygodniowo w badaniach przepływowych. Twardość i alkaliczność powinny być mierzone jednorazowo w każdym badaniu. Zalecane jest stałe monitorowanie temperatury w przynajmniej jednym zbiorniku do badań.
1.8.6. Obserwacje
Ważenie: pod koniec badania wszystkie ryby, które przeżyły, muszą być zważone jako masa wilgotna (osuszone) albo grupowo z badanego zbiornika albo pojedynczo. Zalecane jest ważenie ryb z badanego zbiornika, zamiast poszczególnych ryb, gdyż wymaga to indywidualnego oznaczenia ryby. W przypadku pomiarów wagi poszczególnych ryb, w celu oznaczenia wskaźnika szczególnego rozwoju określonej ryby, wybrana technika oznaczania powinna zapobiegać stresowaniu zwierząt (alternatywne do oznaczania w stanie zamrożonym jest stosowne oznaczenie ryby np. kolorową cienką linią).
Ryby powinny być sprawdzane codziennie w czasie trwania okresu badania na jakiekolwiek zewnętrzne odstępstwa od normalności (takie jak krwotoki, odbarwienia) oraz nienormalne zachowanie. Każdy przypadek śmiertelny musi być odnotowany, a martwa ryba usunięta tak szybko jak to możliwe. Martwe ryby nie są zastępowane, wskaźnik obciążenia i gęstość wyjściowa są wystarczające, aby zapobiec wpływowi na rozwój poprzez zmiany w ilości ryb na zbiornik. Jednakże zachodzi potrzeba dostosowania wskaźnika żywieniowego.
2. DANE I SPRAWOZDAWCZOŚĆ
2.1. OPRACOWANIE WYNIKÓW
Jest zalecane, aby statystyka obejmowała zarówno projekt, jak i analizę badania, ponieważ niniejsza metoda badania pozwala na uwzględnienie znacznej zmiany w projekcie doświadczenia, taką jak na przykład ilość komór do badań, liczbę stężeń badanych, liczbę ryb itd. Uwzględniając dostępne opcje w projekcie badania, nie podano tutaj informacji dotyczących procedur statystycznych.
Wskaźniki rozwoju nie powinny być obliczane w odniesieniu do zbiorników do badań, gdy śmiertelność przekracza 10 %. Jednakże wskaźnik śmiertelności powinien być wskazany w odniesieniu do wszystkich stężeń badanych.
Którakolwiek metoda jest stosowana do analizy danych, głównym pojęciem jest wskaźnik szczególnego rozwoju r między czasem t1 i czasem t2. Może to być określone na kilka sposobów zależnych od tego, czy ryby były indywidualnie oznaczane czy nie albo czy wymagana jest średnia zbiornika.

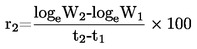
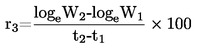
gdzie:
|
r1 |
= |
wskaźnik szczególnego rozwoju określonej ryby, |
|
r2 |
= |
średniego zbiornikowy wskaźnik szczególnego rozwoju, |
|
r3 |
= |
wskaźnik pseudoszczególnego rozwoju, |
|
w1, w2 |
= |
wagi poszczególnych ryb w czasach odpowiednio t1 i t2, |
|
logew1 |
= |
logarytm wagi określonej ryby na początku okresu badania, |
|
logew2 |
= |
logarytm wagi określonej ryby pod koniec okresu badania, |
|
loge W1 |
= |
średnia logarytmów wartości w1 w odniesieniu do ryb w zbiorniku na początku okresu badania, |
|
loge W2 |
= |
średnia logarytmów wartości w2 w odniesieniu do ryb w zbiorniku pod koniec okresu badania, |
|
t1, t2 |
= |
czas (dni) od startu do końca okresu badania |
r1, r2, r3 może być obliczony w odniesieniu do okresu 0–28 dni i, tam gdzie to jest stosowne (np. gdy pomiar wykonano w 14 dniu) w odniesieniu do okresów czasu 0–14 i 14–28 dni.
2.1.1. Regresyjna analiza wyników (modelowanie stężenie-reakcja)
Niniejsza metoda analizy dostosowuje odpowiednią matematyczną zależność między wskaźnikiem szczególnego rozwoju a stężeniem i stąd umożliwia obliczenie „ECX”, tj. każdej wymaganej wartości WE. Stosując niniejszą metodę obliczenia r dla określonej ryby (r1), nie jest konieczne i zamiast tego analiza może być oparta na średniej zbiornikowej wartości r (r2). Ta ostatnia metoda jest zalecana. Jest także bardziej właściwa w przypadku użycia najmniejszych gatunków.
Średnie zbiornikowe wskaźniki szczególnego rozwoju (r2) muszą być wykreślone graficznie w zależności od stężenia w celu sprawdzenia zależności reakcji stężenia.
Dla wyrażenia zależności między r2 a stężeniem powinien być wybrany właściwy model, a wybór musi być poparty odpowiednim uzasadnieniem.
Jeśli ilości ryb, które przeżyły w każdym zbiorniku są nierówne, wtedy proces dostosowania modelu albo prostego albo nieliniowego powinien być rozważony, aby uwzględnić nierówność rozmiarów grup.
Metoda dostosowania modelu musi umożliwiać oszacowanie czy na przykład EC20 i jego rozrzut (czy błąd standardowy czy przedział ufności) może być wyprowadzony. Wykres dopasowanego modelu powinien być przedstawiony w zależności od danych tak, aby odpowiedniość dopasowania modelu była widoczna (8)(18)(29)(20).
2.1.2. Analiza wyników dla oszacowania LOEC
Jeżeli badanie obejmuje skopiowane zbiorniki przy wszystkich poziomach stężeń, oszacowanie LOEC może być oparte na analizie wariancji (ANOVA) średniego zbiornikowego wskaźnika szczególnego rozwoju (zob. pkt 2.1), według odpowiednich metod (np. badania Dunnetta lub Williamsa (12)(13)(14)(21)) porównywania średniego r w odniesieniu do każdego stężenia ze średnią r w odniesieniu do doświadczeń kontrolnych dla określenia najniższego stężenia, dla którego ta różnica jest znacząca przy poziomie prawdopodobieństwa 0,05. Jeśli wymagane przyjęcie dla metod parametrycznych nie odpowiada nienormalnemu rozkładowi (np. badanie Shapiro-Wilka) lub heterogenicznej wariancji (badanie Bartletta), należy podjąć rozważania w celu przekształcenia danych do jednolitych wariancji przed zastosowaniem ANOVA albo przeprowadzeniem ważonego ANOVA.
Jeśli badanie nie obejmuje duplikatów zbiorników dla każdego stężenia, ANOVA oparta o zbiorniki będzie niesensytywna albo niemożliwa. W tej sytuacji dopuszczalnym kompromisem jest ANOVA na pseudoszczególnym wskaźniku rozwoju r3 dla poszczególnych ryb.
Średnia r3 dla każdego stężenia badanego może wtedy być porównywana ze średnią r3 dla doświadczeń kontrolnych. LOEC może być wtedy określona jak poprzednio. Trzeba jednak uznać, że niniejsza metoda nie umożliwia wprowadzania poprawek, ani nie zabezpiecza przed zmiennościami między zbiornikami, poza tymi, które są liczone dla zmienności między poszczególnymi rybami. Jednakże doświadczenie pokazało (8), że zmienność między zbiornikami jest bardzo mała w porównaniu z zmiennością wewnątrz zbiorników (tj. między rybami). Jeśli poszczególne ryby nie są włączone do analizy, poboczna metoda identyfikacji i uzasadnienie jej użycia życia muszą być przedstawione.
2.2. INTERPRETACJA WYNIKÓW
Wyniki muszą być interpretowane z ostrożnością w przypadku, gdy mierzone stężenie toksykanta w roztworach badanych wypada na poziomie granicy wykrywalności metody analitycznej lub w badaniach półstatycznych, gdy stężenie substancji badanej obniża się między świeżo przygotowanymi roztworami i przed wymianą.
2.3. SPRAWOZDANIE Z BADANIA
Sprawozdanie z badania musi zawierać następujące informacje:
2.3.1. Substancja użyta w badaniu:
|
— |
natura fizyczna i odpowiednie właściwości fizykochemiczne, |
|
— |
dane identyfikacji chemicznej włączając czystość i metodę analityczną kwantyfikacji substancji badanej, tam gdzie jest to stosowne. |
2.3.2. Gatunki użyte w badaniu:
|
— |
możliwie nazwa naukowa, |
|
— |
szczep, wymiar, dostawca, wszystkie przedtraktowania itp. |
2.3.3. Warunki badania:
|
— |
zastosowana procedura badania (np. półstatyczna/odnawialna, przepływowa, obciążenie, gęstość wyjściowa itp.), |
|
— |
projekt badania (np. liczba zbiorników do badań, stężenia badane i duplikatów, liczba ryb na zbiornik), |
|
— |
metoda przygotowania roztworów wyjściowych i częstotliwość odnawiania (środek ułatwiający rozpuszczanie i jego stężenie musi być podane, jeśli jest stosowany), |
|
— |
nominalne stężenia użyte w badaniu, średnie wielkości mierzonych i ich standardowe odchylenia w zbiornikach do badań metody, którymi je uzyskano, oraz dowody, że pomiary odnoszą się do stężeń substancji badanej w prawdziwym roztworze, |
|
— |
charakterystyka wody rozcieńczającej: pH, twardość, alkaliczność, temperatura, stężenie rozpuszczonego tlenu, poziom pozostałości chloru (jeśli jest zmierzony), całkowity węgiel organiczny, zawieszone cząstki stałe, zasolenie ośrodka badanego (jeśli jest zmierzony) oraz każde inne wykonane pomiary, |
|
— |
jakość wody w obrębie zbiorników do badań: pH, twardość, temperatura i stężenie rozpuszczonego tlenu, |
|
— |
szczegółowe informacje dotyczące karmienia (np. rodzaj pożywienia, źródło, ilość podawana i częstotliwość). |
2.3.4. Wyniki:
|
— |
dowody, że doświadczenia kontrolne odpowiadają kryteriom ważności dla przeżycia oraz dane śmiertelności zdarzającej się w każdym stężeniu badanym, |
|
— |
zastosowane techniki statystyczno-analityczne, statystyka oparta na duplikatach lub rybach, opracowanie danych i uzasadnienie użytych technik, |
|
— |
stabelaryzowane dane wagi średniej i indywidualnej ryb w dniach 0, 14 (jeśli są zmierzone) i 28 wartości średniej zbiornikowej lub pseudoszczególnych wskaźników rozwoju (jeśli to stosowne) w okresie 0–28 dni lub możliwie 0–14 i 14–28, |
|
— |
wyniki analizy statystycznej (tj. analizy regresyjnej lub ANOVA) zalecane w tabelarycznej i graficznej formie oraz LOEC (p = 0,05) i NOEC lub ECx z, o ile możliwe, błędami standardowymi, jeśli właściwe, |
|
— |
wypadki wszystkich niezwykłych reakcji ryb i wszystkie wizualne efekty, wytworzone przez substancję badaną. |
3. BIBLIOGRAFIA
|
(1) |
Solbe J. F. de LG (1987). Environmental Effects of Chemicals (CFM 9350 SLD). Report on a UK Ring Test of a Method for Studying the Effects of Chemicals on the Growth Rate of Fish. WRc Report No PRD 1388-M/2. |
|
(2) |
Ashley S., Mallett M. J. and Grandy N. J. (1990). EEC Ring Test of a Method for Determining the Effects of Chemicals on the Growth Rate of Fish. Final Report to the Commission of the European Communities. WRc Report No EEC 2600-M. |
|
(3) |
Crossland N. O. (1985). A method to evaluate effects of toxic chemicals on fish growth. Chemosphere, 14, pp. 1855–1870. |
|
(4) |
Nagel R., Bresh H., Caspers N., Hansen P. D., Market M., Munk R., Scholz N. and Hofte B. B. (1991). Effect of 3,4-dichloroaniline on the early life stages of the Zebrafish (Brachydanio rerio): results of a comparative laboratory study. Ecotox. Environ. Safety, 21, pp. 157–164. |
|
(5) |
Yamamoto, Tokio. (1975). Series of stock cultures in biological field. Medaka (killifish) biology and strains. Keigaku Publish. Tokio, Japan. |
|
(6) |
Holcombe, G. W., Benoit D. A., Hammermeister, D. E., Leonard, E. N. and Johnson, R. D. (1995). Acute and long-term effects of nine chemicals on the Japanese medaka (Oryzias latipes). Arch. Environ. Conta. Toxicol. 28, pp. 287–297. |
|
(7) |
Benoit, D. A., Holcombe, G. W. and Spehar, R. L. (1991). Guidelines for conducting early life toxicity tests with Japanese medaka (Oryzias latipes). Ecological Research Series EPA-600/3-91-063. US Environmental Protection Agency, Duluth, Minnesota. |
|
(8) |
Stephan C. E. and Rogers J. W. (1985). Advantages of using regression analysis to calculate results of chronic toxicity tests. Aquatic Toxicology and Hazard Assessment: Eighth Symposium, ASTM STP 891, R. C. Bahner and D. J. Hansen, eds., American Society for Testing and Materials, Philadelphia, pp. 328–338. |
|
(9) |
Environment Canada (1992). Biological test method: toxicity tests using early life stages of salmonid fish (rainbow trout, coho salmon, or atlantic salmon). Conservation and Protection, Ontario, Report EPS 1/RM/28, 81 p. |
|
(10) |
Cox D. R. (1958). Planning of experiments. Wiley Edt. |
|
(11) |
Pack S. (1991). Statistical issues concerning the design of tests for determining the effects of chemicals on the growth rate of fish. Room Document 4, OECD Ad Hoc Meeting of Experts on Aquatic Toxicology, WRc Medmenham, UK, 10–12 December 1991. |
|
(12) |
Dunnett C. W. (1955). A Multiple Comparisons Procedure for Comparing Several Treatments with a Control, J. Amer. Statist. Assoc, 50, pp. 1096–1121. |
|
(13) |
Dunnett C. W. (1964). New tables for multiple comparisons with a control. Biometrics, 20, pp. 482–491. |
|
(14) |
Williams D. A. (1971). A test for differences between treatment means when several dose levels are compared with a zero dose control. Biometrics 27, pp. 103–117. |
|
(15) |
Johnston, W. L., Atkinson, J. L., Glanville N. T. (1994). A technique using sequential feedings of different coloured food to determine food intake by individual rainbow trout, Oncorhynchus mykiss: effect of feeding level. Aquaculture 120, pp. 123–133. |
|
(16) |
Quinton, J. C. and Blake, R. W. (1990). The effect of feed cycling and ration level on the compensatory growth response in rainbow trout, Oncorhynchus mykiss. Journal of Fish Biology, 37, pp. 33–41. |
|
(17) |
Post, G. (1987). Nutrition and Nutritional Diseases of Fish. Chapter IX in Testbook of Fish Health. T.F.H. Publications, Inc. Neptune City, New Jersey, USA. 288 pp. |
|
(18) |
Bruce, R. D. and Versteeg D. J. (1992). A statistical procedure for modelling continuous toxicity data. Environ. Toxicol. Chem. 11, pp. 1485–1494. |
|
(19) |
DeGraeve, G. M., Cooney, J. M., Pollock, T. L., Reichenbach, J. H., Dean, Marcus, M. D. and McIntyre, D. O. (1989). Precision of EPA seven-day fathead minnow larval survival and growth test; intra and interlaboratory study. Report EA-6189 (American Petroleum Institute Publication, No 4468). Electric Power Research Institute, Palo Alto, CA. |
|
(20) |
Norbert-King T. J. (1988). An interpolation estimate for chronic toxicity: the ICp approach. US Environmental Protection Agency. Environmental Research Lab., Duluth, Minnesota. Tech. Rep. No 05–88 of National Effluent Toxicity Assesment Center. Sept. 1988. 12 pp. |
|
(21) |
Williams D. A. (1972). The comparison of several dose levels with a zero dose control. Biometrics 28, pp. 510–531. |
DODATEK 1
GATUNKI RYB ZALECANE DO BADANIA I ODPOWIEDNIE WARUNKI BADANIA
|
Gatunki |
Zalecana temperatura badania zakres temperatur (oC) |
Okres naświetlania (godziny) |
Zalecany zakres początkowej wagi ryby (g) |
Wymagana dokładność pomiarowa |
Wskaźnik obciążenia (g/l) |
Gęstość wyjściowa (na litr) |
Pokarm |
Czas trwania badania (dni) |
|
Zalecane gatunki: |
|
|
|
|
|
|
|
|
|
Oncorhynchus mykiss Pstrąg tęczowy |
12,5–16,0 |
12 – 16 |
1 – 5 |
100 mg |
1,2 – 2,0 |
4 |
Sucha pasza dla narybka |
≥ 28 |
|
Inne dobrze udokumentowane gatunki: |
|
|
|
|
|
|
|
|
|
Danio rerio Zebrafish |
21 – 25 |
12 – 16 |
0,050 – 0,100 |
1 mg |
0,2 – 1,0 |
5 – 10 |
Żywa żywność (Brachionus Artemia) |
≥ 28 |
|
Oryzias latipes Ricefish (Medaka) |
21 – 25 |
12 – 16 |
0,050 – 0,100 |
1 mg |
0,2–1,0 |
5 – 20 |
Żywa żywność (Brachionus Artemia) |
≥ 28 |
DODATEK 2
NIEKTÓRE CECHY CHEMICZNE DOPUSZCZONEJ WODY ROZCIEŃCZAJĄCEJ
|
Substancja |
Stężenie |
|
Cząstki stałe |
< 20 mg/l |
|
Całkowity węgiel organiczny |
< 2 mg/l |
|
Niezjonizowany amoniak |
< 1 μg/l |
|
Pozostałość chloru |
< 10 μg/l |
|
Całkowite pestycydy fosforoorganiczne |
< 50 ng/l |
|
Całkowite pestycydy chloroorganiczne plus polichlorowane bifenyle |
< 50 ng/1 |
|
Calkowity chlor organiczny |
< 25 ng/1 |
DODATEK 3
Logarytmiczne serie stężeń odpowiednich dla badania toksyczności (9)
|
Kolumna (liczba stężeń między 100 i 10 lub między 10 i 1) (20) |
||||||
|
1 |
2 |
3 |
i |
5 |
6 |
7 |
|
100 |
100 |
100 |
100 |
100 |
100 |
100 |
|
32 |
46 |
56 |
65 |
68 |
72 |
75 |
|
10 |
22 |
32 |
40 |
46 |
52 |
56 |
|
3,2 |
10 |
18 |
25 |
32 |
37 |
42 |
|
1,0 |
4,6 |
10 |
16 |
22 |
27 |
32 |
|
|
2,2 |
5,6 |
10 |
15 |
19 |
24 |
|
|
1,0 |
3,2 |
6,3 |
10 |
14 |
18 |
|
|
|
1,8 |
4,0 |
6,8 |
10 |
13 |
|
|
|
1,0 |
2,5 |
4,6 |
7,2 |
10 |
|
|
|
|
1,6 |
3,2 |
5,2 |
7,5 |
|
|
|
|
1,0 |
2,2 |
3,7 |
5,6 |
|
|
|
|
|
1,5 |
2,7 |
4,2 |
|
|
|
|
|
1,0 |
1,9 |
3,2 |
|
|
|
|
|
|
1,4 |
2,4 |
|
|
|
|
|
|
1,0 |
1,8 |
|
|
|
|
|
|
|
1,3 |
|
|
|
|
|
|
|
1,0 |
C.15. RYBY, KRÓTKOTERMINOWE BADANIE TOKSYCZNOŚCI NA EMBRIONIE I NARYBKU
1. METODA
Niniejsze badanie toksyczności krótkoterminowej jest kopią OECD TG 212 (1998).
1.1. WPROWADZENIE
Niniejsze krótkoterminowe badanie toksyczności na rybim embrionie i narybku jest krótkoterminowym badaniem, w którym etapy życia od świeżo zapłodnionego jaja do końca stadium narybka są poddane ekspozycji. W badaniu tym niewymagane jest karmienie, pod warunkiem że próba zostanie ukończona do momentu, w którym narybek są jeszcze żywione z worka żółtka.
Celem badania jest określenie śmiertelnego i, w ograniczonym zakresie, podśmiertelnego działania substancji chemicznych na szczególne stadia oraz gatunki badane. Niniejsze badanie dostarcza użytecznych informacji, tak że: a) stanowi połączenie między badaniami śmiertelnymi i podśmiertelnymi; b) jest wykorzystywane jako badanie obrazujące dla badania na pełnym wczesnym stadium życia albo dla badań przewlekłej toksyczności; i c) jest wykorzystywane dla badania gatunków w przypadku, gdy techniki gospodarki rolnej nie są wystarczająco zaawansowane do objęcia okresu zmian od stanu endogennego do egzogennego żywienia się.
Należy uwzględnić, że tylko badania obejmujące wszystkie stadia cyklu życia ryby są ogólnie zdolne do dokładnego oszacowania przewlekłej toksyczności substancji chemicznych na ryby oraz że jakiekolwiek zmniejszenie narażenia odnoszące się do etapów życia zmniejsza sensytywność i tak oszacowaną za nisko przewlekłą toksyczność. Dlatego oczekuje się, że badanie na embrionie i narybku będą mniej sensytywne niż badanie na pełnym wczesnym stadium życia, szczególnie w odniesieniu do substancji chemicznych o wysokiej lipofilności (log Pow > 4) oraz substancji chemicznych o szczególnym sposobie toksycznego działania. Jednakże mniejsze różnice w sensytywności między dwoma badaniami są oczekiwane w odniesieniu do substancji chemicznych o nieszczególnym, narkotycznym sposobie działania (1).
Przed publikacją niniejszego badania większość doświadczeń z tym badaniem embrionu i narybka było ze słodkowodną rybą Danio rerio Hamilton-Buchanan (Teleostei, Cyprinidae – zwyczajowa nazwa zebrafish). Bardziej szczegółowe informacje dotyczące przeprowadzania badań dla tego gatunku podano w dodatku 1. Nie wyklucza to użycia innych gatunków, w odniesieniu do których doświadczenie jest również dostępne (tabela 1).
1.2. DEFINICJE
Najniższy obserwowany skutek stężenia (LOEC): jest to najmniejsze badane stężenie substancji badanej, przy której obserwuje się znaczący skutek (przy p < 0,05) w porównaniu z doświadczeniem kontrolnym. Jednakże wszystkie badane stężenia powyżej LOEC muszą wykazywać szkodliwy skutek, równy lub większy niż ten obserwowany przy LOEC.
Nieobserwowany skutek stężenia (NOEC): jest stężeniem badanym bezpośrednio poniżej LOEC.
1.3. ZASADA BADANIA
Stadia embrionalne i narybka są eksponowane na zakres stężeń badanej substancji rozpuszczonej w wodzie. W granicach protokołu jest możliwy wybór między procedurami półstatyczną i przepływową. Wybór zależy od natury badanej substancji. Badanie rozpoczyna się od umieszczenia zapłodnionych jaj w komorze do badań i jest kończone zaraz przed tym, gdy worek żółtka jakiejkolwiek larwy w jakiejkolwiek z komór do badań został całkowicie zaabsorbowany lub przed śmiercią głodową rozpoczynającą się w doświadczeniach kontrolnych. Skutki śmiertelne i podśmiertne są oceniane i porównywane z wartościami kontrolnymi w celu ustalenia najniższego obserwowanego skutku stężenia, a stąd nieobserwowanego skutku stężenia. Alternatywnie, mogą być one analizowane, stosując model regresyjny w celu oszacowania stężenia powodującego dany skutek procentowy (tj. LC/ECx, gdzie x jest określony jako skutek %).
1.4. INFORMACJE DOTYCZĄCE SUBSTANCJI UŻYTEJ W BADANIU
Wyniki badania ostrej toksyczności (zob. metoda C.1) najlepiej przeprowadzonego z gatunkami wybranymi do niniejszego badania powinny być dostępne. Wyniki mogą być użyteczne w doborze odpowiedniego zakresu badanych stężeń w badaniu we wczesnym stadium życia. Rozpuszczalność w wodzie (łącznie z rozpuszczalnością w wodzie badanej) i ciśnienie pary badanej substancji powinny być znane. Powinna być dostępna wiarygodna metoda analityczna do oznaczania ilościowego substancji w roztworach badanych ze znaną i udokumentowaną dokładnością oraz granicą wykrywalności.
Informacje dotyczące substancji badanej, które są przydatne w ustaleniu warunków badania, obejmują wzór strukturalny, czystość substancji, stabilność w wodzie i na świetle, pKa, Pow i wyniki badania całkowitej biodegradacji (zob. metoda C.4).
1.5. WAŻNOŚĆ BADANIA
Aby badanie było ważne, mają zastosowanie następujące warunki:
|
— |
całkowite przeżycie zapłodnionych jaj w doświadczeniach kontrolnych, a tam gdzie to jest odpowiednie – w zbiornikach tylko rozpuszczalnikowych, musi być większe niż lub równe granicom określonym w dodatkach 2 oraz 3, |
|
— |
stężenie tlenu rozpuszczonego musi wynosić między 60 a 100 % wartości nasycenia powietrzem (ASV) w trakcie badania, |
|
— |
temperatura wody nie może się różnić więcej niż ±1,5 oC między komorami do badań lub między kolejnymi dniami w dowolnym momencie podczas trwania badania i powinno być w granicach zakresu temperatur, określonego w odniesieniu do badanych gatunków (dodatki 2 i 3). |
1.6. OPIS METODY BADANIA
1.6.1. Komory do badań
Można użyć jakiekolwiek szklane i inne obojętne chemicznie zbiorniki. Wymiary zbiorników muszą być dostatecznie duże, aby zapewnić zgodność ze wskaźnikiem obciążenia (zob. pkt 1.7.1.2). Jest zalecane, aby komory do badań były losowo rozmieszczone w obszarze badania. Losowo zaprojektowany blok z traktowaniem w każdym bloku jest zalecane do całkowicie losowo wybranego projektu, gdy występują systematyczne skutki w laboratorium, które mogą być skontrolowane poprzez wykorzystanie blokowania. Blokowanie, jeśli jest wykorzystane, powinno być uwzględnione w późniejszej analizie danych. Komory do badań powinny być ekranowane od niepożądanych zakłóceń.
1.6.2. Wybór gatunków ryb
Zalecane gatunki ryb podano w tabeli 1A. To nie wyklucza wykorzystania innych gatunków (przykłady podano w tabeli 1B), ale procedura badania musi być dostosowana w celu zapewnienia odpowiednich warunków badania. W tym przypadku należy podać racjonalne uzasadnienie dla wyboru gatunku i metody doświadczalnej.
1.6.3. Utrzymanie wylęgu ryb
Szczegółowe informacje dotyczące utrzymania wylęgu wyjściowego w zadawalających warunkach można znaleźć w OECD TG 210 (21) i w bibliografii (2)(3)(4)(5)(6).
1.6.4. Postępowanie z embrionami i larwami
Embriony i larwy poddawane są wystawianie na badanie w obrębie głównego zbiornika, w mniejszych zbiornikach wyposażonych w siatki po bokach lub na końcach, pozwalające na przepływ roztworu badanego przez zbiornik. Nieburzliwy przepływ przez te małe zbiorniki jest powodowany przez zanurzanie ich za pomocą ramienia poruszającego zbiornik w górę i w dół, ale utrzymującego zawsze organizmy w zanurzeniu; można wykorzystać także system syfonowopłuczący. Zapłodnione jaja łososiowatych ryb są podpierane na pólkach lub siatkach ze szczelinami wystarczająco dużymi, by umożliwić larwom spadanie przez nie po wylęgu. Użycie pasteryzowanych pipet jest właściwe do usunięcia embrionów i larw w badaniach półstatycznych z całkowitym odnawianiem codziennym (zob. pkt 1.6.6).
W przypadku gdy pojemniki na jaja, siatki albo oczka używano do utrzymywania jaj wewnątrz głównego zbiornika do badań, umocowania te powinny być usunięte po wylęgu larw (21), poza zachowaniem oczek zapobiegających ucieczce ryb. Jeśli zachodzi konieczność przeniesienia larw, nie można ich wystawiać na powietrze i nie wolno używać sieci do wypuszczania ryb z pojemników jaj (taka ostrożność nie jest konieczna dla trochę mniej delikatnych gatunków, np. karpia). Czas takiego przeniesienia zmienia się w zależności od gatunku i przeniesienia nie zawsze są konieczne. Dla techniki półstatycznej można stosować zlewki lub płytkie pojemniki, a jeśli konieczne – wyposażone w ekran z oczkami, nieznacznie podniesiony powyżej dna zlewki. Jeśli objętość tych pojemników jest wystarczająca do zapewnienia wymagań obciążenia (zob. 1.7.1.2), nie jest konieczne przenoszenie embrionów lub larw.
1.6.5. Woda
Każda woda, która odpowiada chemicznym cechom akceptowalnej wody rozcieńczającej, jak wymieniono w dodatku 4, i w której badane gatunki wykazują przeżycie w doświadczeniu kontrolnym, co najmniej tak dobre jak te opisane w dodatkach 2 i 3, może być stosowana jako woda do badań. Powinna być stałej jakości w okresie trwania badania. Wartość pH wody powinna pozostawać w zakresie ±0,5 jednostek pH. W celu zapewnienia, aby rozcieńczająca woda nie miała falowego wpływu na wyniki badania (np. przez kompleksowanie substancji badanej) lub negatywnego działania na przeprowadzenie wylęgu wyjściowego, należy okresowo pobierać próbki wody do analizy. Co trzy miesiące, na przykład, powinny być dokonane pomiary metali ciężkich (takich jak Cu, Pb, Zn, Hg, Cd i Ni), głównych anionów i kationów (np. Ca, Mg, Na, K, Cl i SO4), pestycydów (np. całkowita ilość fosforo- i chloroorganicznych), całkowitych węgli organicznych i zawiesin ciał stałych, w przypadku gdy rozcieńczająca woda jest znana jako stosunkowo stała w jakości. Jeśli można przedstawić dane, że jakość wody była stała przynajmniej przez rok, oznaczenia mogą być mniej częste a przedziały rozszerza się (np. co sześć miesięcy).
1.6.6. Roztwory stosowane w badaniu
Roztwory w badaniu o wybranym stężeniu są przygotowywane poprzez rozcieńczanie roztworu podstawowego.
Zalecane jest przygotowanie roztworu podstawowego przez proste wymieszanie badanej substancji w wodzie rozcieńczającej w sposób mechaniczny (np. mieszadło lub ultradźwięki). Kolumny nasyceniowe (rozpuszczające) stosuje się do uzyskania odpowiednio stężonego roztworu podstawowego. W najszerszym możliwym zakresie należy unikać stosowania rozpuszczalników lub dyspergatorów; jednakże takie związki mogą być wymagane w kilku przypadkach w celu wykonania odpowiednio stężonego roztworu podstawowego. Przykładami odpowiednich rozpuszczalników są aceton, etanol, metanol, dimetylosulfotlenek, dimetyloformamid i glikol trójetylenowy. Przykładami odpowiednich dyspergatorów są Cremophor RH40, Tween 80, metyloceluloza 0,01 % i HCO-40. Należy zachować ostrożność przy użyciu łatwo biodegradowalnych środków (np. aceton) i/lub wysokolotnych związków, gdyż mogą być przyczyną problemów z rozwojem bakterii w przepływach badań. Gdy stosuje się środki rozpuszczające, nie mogą one mieć znaczącego wpływu na przeżycie i widocznych negatywnych działań na wczesne stadium życia, jak odkryto przy doświadczeniu kontrolnym z rozpuszczalnikiem. Jednakże należy dołożyć wszelkich starań, aby zapobiec stosowaniu takich materiałów.
W odniesieniu do techniki półstatycznej mogą być prowadzone dwie różne procedury odnawiania; albo (i) nowe roztwory badane są przygotowywane w czystych zbiornikach, a jaja i larwy, które przeżyły, są delikatnie przenoszone do nowych zbiorników w małej objętości starego roztworu, zapobiegając ekspozycji na działanie powietrza; albo (ii) organizmy badane są utrzymywane w zbiornikach, o ile proporcja (co najmniej trzy czwarte) wody badanej jest zmieniona. Częstotliwość odnawiania ośrodka zależy od stabilności substancji badanej, lecz zalecane jest codzienne odnawianie wody. Jeśli z badań stabilności pierwotnej (zob. pkt 1.4) wynika, że stężenie substancji badanej nie jest stabilne (tj. poza zakresem nominalnego 80–120 % lub spada poniżej 80 % zmierzonego stężenia początkowego) ponad okres odnowienia, należy rozważyć celowość użycia badania przepływowego. W każdym przypadku należy zachować ostrożność, aby zapobiec stresom dla larw w trakcie czynności odnawiania wody.
W odniesieniu do badań przepływowych wymagany jest system ciągłego dozowania i rozcieńczania roztworu podstawowego substancji badanej (np. pompa dozująca, rozcieńczalnik proporcjonalny, system nasycania) do przesyłania serii stężeń do komór do badań. Szybkości przepływów roztworu podstawowego i rozcieńczającej wody powinny być sprawdzane okresowo, najlepiej codziennie w trakcie trwania badania, oraz nie powinny zmieniać się więcej niż 10 % w trakcie badania. Jako odpowiedni uznano wskaźnik przepływu równoważny co najmniej pięciu objętościom komory w ciągu 24 godzin (2).
1.7. PROCEDURA
Przydatne informacje dotyczące przeprowadzania na rybich embrionach i narybku badań toksyczności jest dostępna w literaturze, której kilka przykładów jest włączone do części bibliograficznej niniejszego tekstu (7)(8)(9).
1.7.1. Warunki ekspozycji
1.7.1.1. Czas trwania
Badanie powinno rozpocząć się w ciągu 30 minut po zapłodnieniu jaj. Embriony są zanurzane w roztworze badanym przed lub, tak szybko jak to możliwe, po rozpoczęciu stadium rozszczepienia tarczek zarodowych, a w każdym przypadku przed rozpoczęciem stadium gastruli. W odniesieniu do jaj uzyskanych od dostawcy handlowego niemożliwe jest rozpoczęcie badania niezwłocznie po zapłodnieniu. Ponieważ na sensytywność badania poważnie wpływa opóźnienie rozpoczęcia badania, badanie powinno być zapoczątkowane w ciągu ośmiu godzin po zapłodnieniu. Ponieważ larwy nie są karmione w czasie trwania okresu ekspozycji, badanie musi być zakończone w chwili, zanim woreczek żółtkowy jakiejkolwiek larwy w którejkolwiek z komór zostanie całkowicie zaabsorbowany lub przed początkiem śmierci głodowej w doświadczeniach kontrolnych. Czas trwania zależy od wykorzystanych gatunków. Kilka zalecanych czasów trwania podano w dodatku 2 i 3.
1.7.1.2. Obciążenie
Liczba zapłodnionych jaj na początku badania musi być wystarczająca, aby spełnić wymagania statystyczne. Powinny być one losowo rozdzielone między zabiegami, po co najmniej 30 zapłodnionych jaj, rozdzielonych równo (lub tak równo jak to możliwe, ponieważ jest trudne uzyskanie równych partii przy użyciu niektórych gatunków) między co najmniej trzy skopiowane komory do badań używane na jedno stężenie. Wskaźnik obciążenia (biomasa na objętość roztworu badanego) powinien być wystarczająco niski, aby stężenie rozpuszczonego tlenu wynosiło co najmniej 60 % ASV i mogło być utrzymane bez napowietrzania. W odniesieniu do badań przepływowych wskaźnik obciążenia nieprzekraczający 0,5 g/l na 24 godziny i nieprzekraczający 5 g/l roztworu w dowolnym momencie są zalecane (2).
1.7.1.3. Światło i temperatura
Okres naświetlania i temperatura wody badanej musi być właściwa dla badanego gatunku (dodatek 2 i 3). W celu monitorowania temperatury właściwe jest użycie dodatkowego zbiornika do badań.
1.7.2. Stężenia użyte w badaniu
Zwykle wymagane jest pięć stężeń do badań w odstępie o stały wskaźnik nieprzekraczający 3,2. Przy wyborze zakresu stężeń badanych w badaniach toksyczności ostrej należy wziąć pod uwagę krzywą zależności LC50 od okresu ekspozycji. Użycie mniej niż pięciu stężeń, na przykład w badaniach granicznych i węższego odstępu stężenia, w pewnych okolicznościach może być właściwe. Jeżeli użyto mniej niż pięciu stężeń powinno być dostarczone uzasadnienie. Stężenie substancji wyższe niż 96 godzin LC50 lub 100 mg/l, którekolwiek jest niższe, nie wymaga badania. Najwyższe stężenie badane nie może przekraczać granicy rozpuszczalności substancji w wodzie.
W przypadku gdy środek ułatwiający rozpuszczenie jest wykorzystany w przygotowaniu roztworów badanych (zob. pkt 1.6.6), jego końcowe stężenie nie może być większe niż 0,1 ml/l i zalecane jest takie same we wszystkich zbiornikach do badań.
1.7.3. Doświadczenia kontrolne
Jedno doświadczenie kontrolne z wodą rozcieńczającą (powtórzona odpowiednio), a także, jeśli jest to stosowne, jedno doświadczenie kontrolne ze środkiem ułatwiającym rozpuszczanie (powtórzone odpowiednio) powinno być dodatkowo przeprowadzone obok serii badań.
1.7.4. Częstotliwość oznaczeń analitycznych i pomiarów
W czasie trwania badania stężenia substancji badanej są oznaczane w regularnych odstępach czasu.
W półstatycznych (odnawialnych) badaniach, w przypadku gdy oczekuje się że stężenie substancji utrzyma się w zakresie ± 20 % nominalnego (tj. w zakresie 80–120 %; zob. pkt 1.4, i 1.6.6), jest zalecane, aby jako minimum analizować najniższe i najwyższe stężenia badane świeżo po przygotowaniu i zaraz przed odnowieniem, następnie w co najmniej trzech przypadkach oddzielonych równo poprzez badanie (tj. analizy powinny być wykonywane na próbce z tego samego roztworu – świeżo przygotowanym i przy wymianie).
W odniesieniu do badań, w których nie oczekuje się, że stężenie substancji badanej pozostanie w zakresie ± 20 % nominalnego (na bazie danych stabilności substancji) niezbędne jest analizowanie wszystkich badanych stężeń, świeżo po przygotowaniu i przy wymianie, zgodnie z tymi samymi warunkami (tj. w co najmniej trzech przypadkach oddzielonych równo poprzez badanie). Oznaczanie stężenia substancji badanej przed wymianą wymagane jest tylko na jednej z kopii zbiornika przy każdym stężeniu badanym. Oznaczenia powinny być dokonywane nie częściej niż co siedem dni. Zalecane jest, aby wyniki oparte były na pomierzonych stężeniach. Jeżeli jednakże dostępne dowody są zdolne wykazać, że stężenie badanej substancji w roztworze zostało z powodzeniem utrzymane w zakresie ± 20 % nominalnego lub wstępnie zmierzonego stężenia, poprzez okres badania, wtedy wyniki mogą być oparte na początkowych wartościach nominalnych lub zmierzonych.
W odniesieniu do badań przepływowych właściwe jest użycie podobnego do opisanego dla badań półstatycznych, sposobu pobierania próbek (lecz pomiar „starych” roztworów nie jest stosowany w tym przypadku). Jednakże gdy czas trwania badania jest dłuższy niż siedem dni, wskazane jest podniesienie ilości pobieranych próbek podczas pierwszego tygodnia (np. trzy zestawy pomiarów) w celu zapewnienia aby stężenia badane pozostały stałe.
Próbki mogą wymagać odwirowania lub przefiltrowania (np. stosując 0,45 μm wymiar poru). Jednakże ani odwirowanie, ani filtracja nie skutkują zawsze rozdzieleniem frakcji niebioprzyswajalnej substancji badanej od tej, która jest bioprzyswajalna, zatem próbki nie mogą być przedmiotem takiego przetwarzania.
W czasie trwania badania tlen rozpuszczony, pH oraz temperatura powinny być mierzone we wszystkich zbiornikach do badań. Całkowita twardość i zasolenie (jeśli dotyczy) powinny być mierzone w doświadczeniach kontrolnych i w zbiorniku o najwyższym stężeniu. Jako minimum należy pomierzyć rozpuszczony tlen i zasolenie (jeśli dotyczy) trzy razy (na początku, w środku i na koniec badania). W badaniach półstatycznych jest zalecane, aby rozpuszczony tlen był mierzony częściej, najlepiej przed i po każdej wymianie wody lub co najmniej raz na tydzień. Wartość pH na początku i na końcu każdej wymiany wody w badaniach statycznych odnawialnych i co najmniej cotygodniowo w badaniach przepływowych. Twardość powinna być zmierzona jednorazowo w każdym badaniu. Temperatura powinna być mierzona codziennie i zalecane jest stałe monitorowanie temperatury w przynajmniej jednym zbiorniku do badań.
1.7.5. Obserwacje
1.7.5.1. Stadium rozwoju embrionalnego
Stadium embrionalne (tj. stadium gastruli) na początku ekspozycji na działanie substancji badanej powinno być sprawdzone tak dokładnie jak to możliwe. Można tego dokonać stosując reprezentatywną próbkę jaj odpowiednio zabezpieczonych i przezroczystych. Można również uwzględnić literaturę w odniesieniu do opisu i ilustracji stadiów embrionalnych (2)(5)(10)(11).
1.7.5.2. Wylęg i przeżycie
Obserwacje w zakresie wylęgania i przeżycia powinny być dokonywane co najmniej raz dziennie, a liczby powinny być zarejestrowane. Pożądane może być częstsze dokonywanie obserwacji na początku badania (np. co 30 minut w ciągu trzech pierwszych godzin), ponieważ w kilku przypadkach czas przeżycia jest bardziej istotny od jedynie liczby zgonów (np. gdy występują skutki ostrej toksyczności). Martwe embriony i larwy powinny być usunięte natychmiast po zauważeniu, gdyż gwałtownie się rozkładają. Należy zachować najwyższą ostrożność przy usuwaniu martwych osobników, aby nie stłuc lub fizycznie uszkodzić przylegających jaj/larw, które są bardzo delikatne i wrażliwe. Kryteria dotyczące zgonu zmieniają się według stadium życia:
|
— |
w odniesieniu do jaj: szczególnie we wczesnym stadium znacząca strata przezroczystości i zmiany w zabarwieniu spowodowane przez koagulację i/lub strącania białek prowadzące do białego, nieprzezroczystego wyglądu, |
|
— |
w odniesieniu do embrionów: zanik ruchu organów i/lub uderzeń serca i/lub nieprzezroczyste odbarwienie dla gatunków, które są zwykle przezroczyste, |
|
— |
w odniesieniu do larw: nieruchomość i/lub brak ruchu oddechowego i/lub brak bicia serca i/lub białe nieprzezroczyste zabarwienie centralnego systemu nerwowego i/lub brak reakcji na bodźce mechaniczne. |
1.7.5.3. Anormalny wygląd
Liczba larw wykazujących anormalności w kształcie ciała i/lub pigmentacji, stadium absorpcji żółtko-torba powinny być rejestrowane w odpowiednich odstępach czasu zależnych od czasu trwania badania i charakteru opisanej nienormalności. Należy zauważyć, że anormalne embriony i larwy zdarzają się w naturalny sposób i w doświadczeniu(-ach) kontrolnym(-ych) może ich być rzędu kilku procent w jakimś gatunku. Anormalne zwierzęta powinny być jedynie usunięte ze zbiorników do badań i zabite.
1.7.5.4. Anormalne zachowanie
Abnormalności, np. hiperwentylacja, nieskoordynowane pływanie i nietypowy spokój powinny być zarejestrowane w odpowiednich odstępach czasu zależnych od czasu trwania badania. Skutki te, choć trudne do określenia ilościowego, gdy są obserwowane, pomagają w interpretacji danych dotyczących śmiertelności, tj. dostarczają informacji dotyczących sposobu toksycznego działania substancji.
1.7.5.5. Długość
Na koniec badania zalecany pomiar indywidualnych długości; można zastosować standardową, widełkową lub całkowitą długość. Jeśli jednakże występuje ogonowe gnicie płetwy lub nadżerka, należy zastosować długość nominalną. Ogólnie, w dobrze przebiegającej próbie współczynnik rozrzutu długości między kopiami w doświadczeniach kontrolnych powinien wynosić ≤ 20 %.
1.7.5.6. Waga
Na koniec badania, mierzona może być waga indywidualna; bardziej pożądana jest sucha waga: (24 h w 60 oC) od wilgotnej wagi (suszenie bibułą). Ogólnie, w dobrze przebiegającej próbie współczynnik rozrzutu wagi między kopiami w doświadczeniach kontrolnych powinien wynosić ≤ 20 %.
Obserwacje te wynikają z niektórych lub ze wszystkich następujących danych dostępnych dla analizy statystycznej:
|
— |
skumulowana śmiertelność, |
|
— |
liczba zdrowych larw na koniec badania, |
|
— |
czas rozpoczęcia wylęgu i czas zakończenia wylęgu (tj. 90 % wylęgu w każdej kopii), |
|
— |
ilość larw wylęgających się każdego dnia, |
|
— |
długość (i waga) zwierząt, które przeżyły na koniec badania, |
|
— |
ilość larw zdeformowanych lub o anormalnym wyglądzie, |
|
— |
ilość larw wykazujących anormalne zachowanie. |
2. DANE I SPRAWOZDAWCZOŚĆ
2.1. OPRACOWANIE WYNIKÓW
Jest zalecane, aby statystyka była włączona w projekt jak i analizę badania, ponieważ niniejsza metoda badania umożliwia uwzględnienie znacznej zmiany w projekcie doświadczenia, taką jak na przykład ilość komór do badań, liczbę stężeń badanych, liczbę zapłodnionych jaj i zmierzone parametry. Uwzględniając dostępne opcje w projekcie badania, nie podano tutaj informacji dotyczących procedur statystycznych.
Jeśli LOEC/NOECs mają być oszacowane niezbędne jest w odniesieniu do zmienności analizowanych między każdym zestawem kopii, zastosowanie analizy wariancji (ANOVA) lub procedury tablicy wielodzielczej. W celu wykonania wielokrotnego porównania między wynikami dla poszczególnych stężeń i tych dla doświadczeń kontrolnych metoda Dunnetta jest uznana za użyteczną (12)(13). Inne przydatne przykłady są także dostępne (14)(15). Rozmiar skutku wykrywalnego przy użyciu ANOVA lub innych procedur (tj. moc badania) powinna być obliczona i przedstawiona. Należy odnotować, że nie wszystkie obserwacje wymienione w pkt 1.7.5.6 są odpowiednie do analizy statystycznej przy zastosowaniu ANOVA. Na przykład skumulowana śmiertelność i liczba zdrowych larw na końcu badania są analizowane przy wykorzystaniu metod probitowych.
Jeśli LC/ECxs mają być oszacowane, odpowiednia(-e) krzywa(-e), takie jak krzywa logistyczna, muszą być dopasowane do interesujących danych, stosując metodę statystyczną taką jak najmniejszych kwadratów lub nieliniową najmniejszych kwadratów. Krzywa(-e) powinna(-y) być parametryzowana(-e) tak, aby interesujące LC/ECx oraz jego błąd standardowy były szacowane bezpośrednio. To znacznie ułatwia obliczenie granic ufności wokół LC/ECx. Pod warunkiem, że nie ma słusznych przyczyn, należy zalecać poziomy ufności dwustronne, o założonej 95 % ufności. Zalecane jest, aby procedura dopasowania dostarczała sposoby w zakresie oceniania znaczenia braku dopasowania. Stosuje się graficzne metody dla dopasowania krzywych. Analiza regresyjna jest odpowiednia w odniesieniu do wszystkich obserwacji wymienionych w pkt 1.7.5.6.
2.2. INTERPRETACJA WYNIKÓW
Wyniki powinny być interpretowane z ostrożnością w przypadku, gdy mierzone stężenie toksykanta w roztworach badanych oscyluje na poziomie granicy wykrywalności metody analitycznej. Interpretacja wyników w odniesieniu do stężeń powyżej rozpuszczalności substancji w wodzie również powinna być dokonywana z ostrożnością.
2.3. SPRAWOZDANIE Z BADANIA
Sprawozdanie z badania musi zawierać następujące informacje:
2.3.1. Substancja użyta w badaniu:
|
— |
charakter fizyczny i odpowiednie właściwości fizykochemiczne, |
|
— |
dane identyfikacji chemicznej włączając czystość i metodę analityczną kwantyfikacji substancji badanej tam gdzie to jest właściwe. |
2.3.2. Gatunki użyte w badaniu:
|
— |
nazwa naukowa, szczep, liczba rodzicielskich ryb (tj. ile samic użyto do dostarczenia żądanej ilości jaj do badań), źródło i metodę zbierania zapłodnionych jaj i dalsze postępowanie. |
2.3.3. Warunki badania:
|
— |
zastosowana procedura testowa (np. półstatyczna lub przepływowa, okres czasu od zapłodnienia do rozpoczęcia badania, obciążenie itp.), |
|
— |
okres(-y) naświetlania, |
|
— |
projekt badania (np. liczba komór do badań i kopii, liczba embrionów na kopię, stężenia badane i w kopiach, liczba ryb na zbiornik), |
|
— |
metoda przygotowania roztworów wyjściowych i częstotliwość odnawiania (środek ułatwiający rozpuszczanie i jego stężenie powinny być podane, jeśli jest zastosowany), |
|
— |
nominalne stężenia badane, średnie wielkości mierzonych i ich standardowe odchylenia w zbiornikach do badań metody, którymi je uzyskano oraz dowody, że pomiary odnoszą się do stężeń substancji badanej w roztworze, |
|
— |
cechy wody rozcieńczającej: pH, twardość, temperatura, stężenie rozpuszczonego tlenu, poziom pozostałości chloru (jeśli jest zmierzony), całkowity węgiel organiczny, zawieszone cząstki stałe, zasolenie ośrodka badanego (jeśli jest zmierzone) oraz wszelkie inne wykonane pomiary, |
|
— |
jakość wody w zbiornikach do badań: pH, twardość, temperatura i stężenie rozpuszczonego tlenu. |
2.3.4. Wyniki:
|
— |
wyniki ze wszystkich wstępnych badań stabilności badanej substancji, |
|
— |
dowody, że doświadczenia kontrolne odpowiadają kryteriom ważności w odniesieniu do przeżycia badanych gatunków (dodatki 2 i 3), |
|
— |
dane dotyczące śmiertelności/przeżycia na stadiach embrionu i larwy oraz całkowita śmiertelność/przeżycie, |
|
— |
dni do wylęgu i liczba wylęgłych, |
|
— |
dane dotyczące długości (i wagi), |
|
— |
zasięg i opis morfologicznych anormalności, jeśli istnieją, |
|
— |
zasięg i opis efektów zachowania, jeśli istnieją, |
|
— |
analiza statystyczna i opracowanie danych, |
|
— |
w odniesieniu do badań analizowanych przy pomocy ANOVA, najniższy obserwowany skutek stężenia (LOEC) przy p = 0,05 i nieobserwowany skutek stężenia (NOEC) w odniesieniu do każdej ocenianej reakcji, włączając opisanie procedur statystycznych użytych do wskazania wymiaru skutku wykrywalnego, |
|
— |
w odniesieniu do badań analizowanych przy użyciu technik regresyjnych, LC/ECx i przedziałów ufności wraz z wykresem dopasowanego modelu stosowanego dla ich wyliczenia, |
|
— |
wyjaśnienie dotyczące każdego odchylenia od niniejszej metody badania. |
3. BIBLIOGRAFIA
|
(1) |
Kristensen P. (1990). Evaluation of the Sensitivity of Short Term Fish Early Life Stage Tests in Relation to other FELS Test Methods. Final report to the Commission of the European Communities, 60 pp. June 1990. |
|
(2) |
ASTM (1988). Standard Guide for Conducting Early Life-Stage Toxicity Tests with Fishes. American Society for Testing and Materials. E 1241-88. 26 pp. |
|
(3) |
Brauhn J. L. and Schoettger R. A. (1975). Acquisition and Culture of Research Fish: Rainbow trout, Fathead minnows, Channel Catfish and Bluegills. p. 54, Ecological Research Series, EPA-660/3-75-011, Duluth, Minnesota. |
|
(4) |
Brungs W. A. and Jones B. R. (1977). Temperature Criteria for Freshwater Fish: Protocol and Procedures. p. 128, Ecological Research Series EPA-600/3-77-061, Duluth, Minnesota. |
|
(5) |
Laale H. W. (1977). The Biology and Use of the Zebrafish (Brachydanio rerio) in Fisheries Research. A Literature Review. J. Biol. 10, pp. 121–173. |
|
(6) |
Legault R. (1958). A Technique for Controlling the Time of Daily Spawning and Collecting Eggs of the Zebrafish, Brachydanio rerio (Hamilton-Buchanan) Copeia, 4, pp. 328–330. |
|
(7) |
Dave G., Damgaard B., Grande M., Martelin J. E., Rosander B. and Viktor T. (1987). Ring Test of an Embryo-larval Toxicity Test with Zebrafish (Brachydanio rerio) Using Chromium and Zinc as Toxicants. Environmental Toxicology and Chemistry, 6, pp. 61–71. |
|
(8) |
Birge J. W., Black J. A. and Westerman A. G. (1985). Short-term Fish and Amphibian Embryo-larval Tests for Determining the Effects of Toxicant Stress on Early Life Stages and Estimating Chronic Values for Single Compounds and Complex Effluents. Environmental Toxicology and Chemistry 4, pp. 807–821. |
|
(9) |
Van Leeuwen C. J., Espeldoorn A. and Mol F. (1986). Aquatic Toxicological Aspects of Dithiocarbamates and Related Compounds. III. Embryolarval Studies with Rainbow Trout (Salmo gairdneri). J. Aquatic Toxicology, 9, pp. 129–145. |
|
(10) |
Kirchen R. V. and W. R. West (1969). Teleostean Development. Carolina Tips 32(4): 1-4. Carolina Biological Supply Company. |
|
(11) |
Kirchen R. V. and W. R. West (1976). The Japanese Medaka. Its care and Development. Carolina Biological Supply Company, North Carolina. 36 pp. |
|
(12) |
Dunnett C. W. (1955). A Multiple Comparisons Procedure for Comparing Several Treatments with Control. J. Amer. Statist. Assoc, 50, pp. 1096–1121. |
|
(13) |
Dunnett C. W. (1964). New Tables for Multiple Comparisons with a Control. Biometrics, 20, pp. 482–491. |
|
(14) |
Mc Clave J. T., Sullivan J. H. and Pearson J.G. (1980). Statistical Analysis of Fish Chronic Toxicity Test Data. Proceedings of 4th Aquatic Toxicology Symposium, ASTM, Philadelphia. |
|
(15) |
Van Leeuwen C. J., Adema D. M. M. and Hermes J. (1990). Quantitative Structure-Activity Relationships for Fish Early Life Stage Toxicity. Aquatic Toxicology, 16, pp. 321–334. |
|
(16) |
Environment Canada. (1992). Toxicity Tests Using Early Life Stages of Salmonid Fish (Rainbow Trout, Coho Salmon or Atlantic Salmon). Biological Test Method Series. Report EPS 1/RM/28, December 1992, 81 pp. |
|
(17) |
Dave G. and Xiu R. (1991). Toxicity of Mercury, Nickel, Lead and Cobalt to Embryos and Larvae of Zebrafish, Brachydanio rerio. Arch. of Environmental Contamination and Toxicology, 21, pp. 126–134. |
|
(18) |
Meyer A., Bierman C. H. and Orti G. (1993). The phylogenetic position of the Zebrafish (Danio rerio), a model system in developmental biology-an invitation to the comperative methods. Proc. Royal Society of London, Series B, 252: pp. 231–236. |
|
(19) |
Ghillebaert F., Chaillou C, Deschamps F. and Roubaud P. (1995). Toxic Effects, at Three pH Levels, of Two Reference Molecules on Common Carp Embryo. Ecotoxicology and Environmental Safety 32, pp. 19–28. |
|
(20) |
US EPA, (1991). Guidelines for Culturing the Japanese Medaka, Oryzias latipes. EPA report EPA/600/3-91/064, Dec. 1991, EPA, Duluth. |
|
(21) |
US EPA, (1991). Guidelines for Conducting Early Life Stage Toxicity Tests with Japanese Medaka, (Oryzias latipes). EPA report EPA/600/3-91/063, Dec. 1991, EPA, Duluth. |
|
(22) |
De Graeve G. M., Cooney J. D., Mclntyre D. O., Poccocic T. L., Reichenbach N. G., Dean J. H. and Marcus M. D. (1991). Validity in the performance of the seven-day Fathead minnow (Pimephales promelas) larval survival and growth test: an intra- and interlaboratory study. Environ. Tox. Chem. 10, pp. 1189–1203. |
|
(23) |
Calow P. (1993). Handbook of Ecotoxicology, Blackwells, Oxford. Vol. 1, Chapter 10: Methods for spawning, culturing and conducting toxicity tests with Early Life stages of Estuarine and Marine fish. |
|
(24) |
Balon E. K. (1985). Early life history of fishes: New developmental, ecological and evolutionary perspectives, Junk Publ., Dordrecht, 280 pp. |
|
(25) |
Blaxter J. H. S. (1988). Pattern and variety in development, in: W. S. Hoar and D. J. Randall eds., Fish Physiology, Vol. XIA, Academic press, pp. 1–58. |
Tabela 1A
Gatunki ryb zalecane do badania
|
SŁODKOWODNE |
|
Oncorhynchus mykiss Pstrąg tęczowy (9)(16) |
|
Danio rerio Danio pręgowany (7)(17)(18) |
|
Cyprinus caprio Karp zwykły (8)(19) |
|
Oryzias latipes (20)(21) |
|
Pimephales promelas (8)(22) |
Tabela 1B
Przykłady innych dobrze udokumentowanych gatunków, które również zostały użyte
|
SŁODKOWODNE |
SŁONOWODNE |
|
Carassius auratus Karaś srebrzysty (8) |
Menidia peninsulae (23)(24)(25) |
|
Lepomis macrochirus (8) |
Clupea harengus Śledź (24)(25) |
|
Gadus morhua Dorsz (24)(25) |
|
|
Cyprinodon variegatus (23)(24)(25) |
DODATEK 1
WSKAZÓWKI DOTYCZĄCE PROWADZENIA BADANIA TOKSYCZNOŚCI NA EMBRIONACH I NARYBKU RYBY DANIO PRĘGOWANY (BRACHYDANIO RERIO)
WPROWADZENIE
Danio pręgowany pochodzi z wybrzeża Coromandel w Indiach, gdzie zamieszkuje szybko płynące strumienie. Jest to zwykła akwaryjna ryba z rodziny karpiowatych, a informacje na temat procedur opieki nad nią i hodowaniu można znaleźć w publikacjach informacyjnych o tropikalnych rybach. Jej biologia i badania łowisk zostały przedstawione przez Laale (1).
Ryba rzadko przekracza 45 mm długości. Ciało jest cylindryczne z 7–9 ciemnoniebieskimi poziomymi srebrzystymi pasami. Pasy te przebiegają przez ogon i płetwy odbytowe. Grzbiet jest oliwkowozielony. Samce są szczuplejsze od samic. Samice są bardziej srebrzyste i ich odwłok jest rozszerzony, szczególnie przed tarłem.
Dorosłe ryby są w stanie tolerować duże wahania temperatur, pH i twardości. Jednakże w celu uzyskania zdrowej ryby produkującej jaja dobrej jakości, powinny być zapewnione optymalne warunki.
W czasie trwania tarła samiec ściga i bodzie samicę i jak tylko jaja zostaną wyrzucone, zapładnia je. Jaja, które są przezroczyste i nieklejące, opadają na dno, gdzie są zjadane przez dorosłe ryby. Światło wpływa na tarło. Jeśli poranne światło jest odpowiednie, ryby trą się we wczesnych godzinach poprzedzających świt.
Samice produkują partie kilkuset jaj w odstępach tygodniowych.
WARUNKI RYB RODZICIELSKICH, ROZMNAŻANIE I WCZESNE STADIA ŻYCIA
Należy wybrać odpowiednią liczbę zdrowych ryb i trzymać je w odpowiedniej wodzie (np. dodatek 4) przynajmniej dwa tygodnie przed zamierzonym tarłem. Grupie ryb należy pozwolić rozmnażać się co najmniej raz, przed utworzeniem partii jaj używanych w badaniu. Gęstość zarybienia w czasie trwania tego okresu nie może przekraczać 1 gram ryby na jeden litr. Regularne zmiany wody lub użycie systemów oczyszczania umożliwia zastosowanie wyższej gęstości. Temperatura wody w zbiornikach przetrzymujących powinna być utrzymana na poziomie 25 ± 2 oC. Rybom należy dostarczyć różnorodną dietę, która składa się na przykład z handlowej suchej karmy, żywych świeżo wylęgniętych Arthemia, ochotkowatych, Daphnii, białych robaków (Enchytraeids).
Poniżej określono dwie procedury, które w praktyce doprowadziły do dostatecznej partii zdrowych, zapłodnionych jaj dla prowadzenia badania:
|
(i) |
Osiem samic i szesnaście samców jest umieszczanych w zbiorniku zawierającym 50 litrów wody rozcieńczającej, osłonięte od bezpośredniego światła i pozostawione tak długo jak to możliwe niezakłócenie przez co najmniej 48 godzin. Tackę na ikrę umieszczono na dnie akwarium, po południu dnia przed startem badania. Tacka na ikrę składa się z ramy (pleksiglas lub inny odpowiedni materiał), o 5–7 cm wysokości z 2–5 mm grubą siatką dołączoną na górze i 10–30 μm drobną siatką na dnie. Pewna liczba „drzew ikrowych” składających się z nieskręconej nylonowej liny jest dołączona do grubej siatki ramy. Po pozostawieniu ryb przez 12 godzin w ciemności włącza się słabe światło, które zapoczątkowuje tarło. Dwie do czterech godzin po tarle tacka na ikrę jest wyjmowana i jaja są zbierane. Tacka na ikrę zapobiega zjedzeniu jaj przez ryby i w tym samym czasie pozwala na łatwe zbieranie jaj. Grupa ryb powinna co najmniej raz trzeć się, przed tarłem z którego jaja są wykorzystywane do badań. |
|
(ii) |
Pięć do dziesięciu rybich samców i samica są umieszczone oddzielnie na co najmniej dwa tygodnie przed planowanym tarłem. Po 5–10 dniach odwłoki samic będą poszerzone i ich rodne brodawki widoczne. Samce nie posiadają brodawek. Tarło jest przeprowadzane w zbiornikach do tarła wyposażonych w fałszywe dno z siatki (jak wyżej). Zbiornik jest napełniany wodą rozcieńczającą, tak aby głębokość wody powyżej siatki wynosiła 5–10 cm. Jedna samica i dwa samce są umieszczane w zbiorniku na dzień przed planowanym tarłem. Temperatura wody jest stopniowo podnoszona do poziomu wyższego niż temperatura aklimatyzacji. Światło jest wyłączane i zbiornik pozostaje w możliwym zakresie w niezakłóconym stanie. Rano włącza się słabe światło, które zapoczątkowuje tarło. Po 2–4 godzinach ryby są usuwane, a jaja zbierane. Jeśli potrzebne są większe partie jaj, uzyskuje się je od jednej samicy, dostateczna ilość zbiorników do tarła zostaje podłączona równolegle. Poprzez rejestrowanie sukcesu reprodukcyjnego indywidualnych samic przed badaniem (wielkość i jakość partii) samice z najwyższym sukcesem reprodukcyjnym są wybierane do hodowli. |
Jaja powinny być przeniesione do zbiorników do badań za pomocą szklanych rurek (o wewnętrznej średnicy nie mniej niż 4 mm) zaopatrzonych w elastyczną cebulkę ssącą. Ilość wody towarzysząca jajom przy ich przenoszeniu powinna być tak niewielka, na ile to możliwe. Jaja są cięższe niż woda i opadają w rurce. Należy zachować ostrożność, aby zapobiec kontaktowi jaj (i larw) z powietrzem. Należy wykonać badania mikroskopowe próbki(-ek) z partii w celu zapewnienia, aby nie posiadały one żadnych nieregularności w pierwszym stadium rozwojowym. Dezynfekcja jaj jest niedopuszczalna.
Wskaźnik śmiertelności jaj jest najwyższy w pierwszych 24 godzinach po zapłodnieniu. Śmiertelność 5–40 % obserwowana jest często w trakcie trwania tego okresu. Jaja degenerują się jako wynik niedostatecznego zapłodnienia lub wad rozwojowych. Jakość partii jaj wydaje się zależna od samic, chociaż niektóre samice produkują konsekwentnie jaja dobrej jakości, jakiej inne nigdy nie dają. Także wskaźnik rozwoju i wskaźnik wylęgu zmienia się od jednej partii do drugiej. Zapłodnione z powodzeniem jaja i pęcherzyki żółtkowe larw przeżywają na wysokim poziomie, zwykle powyżej 90 %. Przy 25 oC jaja wylęgają sie 3–5 dni po zapłodnieniu i woreczek żółtkowy absorbuje się około 13 dni po zapłodnieniu.
Rozwój embrionalny został dobrze określony przez Hisaoka i Battle (2). Dzięki przejrzystości jaj i larw powylęgowych obserwuje się postępujący rozwój ryby oraz występowanie zniekształceń. W około cztery godziny po tarle niezapłodnione jaja są odróżnialne od zapłodnionych (3). Dla sprawdzenia tego, jaja i larwy są umieszczane w zbiornikach do badań o małej objętości i są obserwowane pod mikroskopem.
Warunki badania mającego zastosowanie do wczesnych stadiów życia są zamieszczone w dodatku 2. Optymalne wartości pH i twardości wody wynoszą odpowiednio 7,8 i 250 mg CaCO3/l.
OBLICZENIA I STATYSTYKI
Proponowane jest podejście dwuetapowe. Najpierw dane dotyczące śmiertelności, nienormalny rozwój i czas wylęgu są analizowane statystycznie. Następnie, w odniesieniu do tych stężeń, przy których nie ma skutków negatywnych żadnego z tych rejestrowanych parametrów, ocenia się statystycznie długość ciała. Niniejsze podejście jest wskazane, gdyż toksykant może zabijać selektywnie mniejsze ryby, opóźniać czas wylęgu i powodować znaczne zniekształcenia, prowadząc w ten sposób do błędnych pomiarów długości. Ponadto jest to z grubsza ta sama ilość ryb zmierzonych do poddania traktowaniu, zapewniając ważność statystyki badania.
OKREŚLENIA LC50 I EC50
Procent przeżycia jaj i larw jest obliczany i korygowany w odniesieniu do śmiertelności w doświadczeniach kontrolnych zgodnie z wzorem Abbotta (4):

gdzie,:
|
P |
= |
skorygowany % przeżycia, |
|
P' |
= |
% przeżycia obserwowanego w stężeniu do badań, |
|
C |
= |
przeżycie w doświadczeniu kontrolnym. |
Jeśli to możliwe, LC50 jest oznaczana odpowiednią metodą na końcu badania.
Jeśli włączenie morfologicznych anormalności w EC50 jest pożądane, wskazówki można znaleźć w Stephan (5).
OSZACOWANIE LOEC NOEC
Celem badania na jajach i narybku jest porównanie niezerowego stężenia z doświadczeniem kontrolnym, tj. określenie LOEC. Dlatego wielokrotne procedury porównawcze muszą być zastosowane (6)(7)(8)(9)(10).
BIBLIOGRAFIA
|
(1) |
Laale H. W. (1977). The Biology and Use of the Zebrafish (Brachydanio rerio) in Fisheries Research. A Literature Review. J. Fish Biol. 10, pp. 121–173. |
|
(2) |
Hisaoka K. K. and Battle H. I. (1958). The Normal Development Stages of the Zebrafish Brachydanio rerio (Hamilton-Buchanan) J. Morph., 102, 311 pp. |
|
(3) |
Nagel R. (1986). Untersuchungen zur Eiproduktion beim Zebrabarbling (Brachydanio rerio Hamilton-Buchanan). Journal of Applied Ichthyology, 2, pp. 173–181. |
|
(4) |
Finney D. J. (1971). Probit Analysis, 3rd ed., Cambridge University Press, Great Britain, pp. 1–333. |
|
(5) |
Stephan C. E. (1982). Increasing the Usefulness of Acute Toxicity Tests. Aquatic Toxicology and Hazard Assessment: Fifth Conference, ASTM STP 766, J. G. Pearson, R. B. Foster and W. E. Bishop, Eds., American Society for Testing and Materials, pp. 69–81. |
|
(6) |
Dunnett C. W. (1955). A Multiple Comparisons Procedure for Comparing Several Treatments with a Control. J. Amer. Statist. Assoc, 50, pp. 1096–1121. |
|
(7) |
Dunnett C. W. (1964). New Tables for Multiple Comparisons with a Control. Biometrics, 20, pp. 482–491. |
|
(8) |
Williams D. A. (1971). A Test for Differences Between Treatment Means when Several Dose Levels are Compared with a Zero Dose Control. Biometrics, 27, pp. 103–117. |
|
(9) |
Williams D. A. (1972). The Comparison of Several Dose Levels with a Zero Dose Control. Biometrics 28, pp. 519–531. |
|
(10) |
Sokal R. R. and Rohlf F. J. (1981). Biometry, the Principles and Practice of Statistics in Biological Research, W. H. Freeman and Co., San Francisco. |
DODATEK 2
WARUNKI BADANIA, CZAS TRWANIA I KRYTERIA PRZEŻYCIA W ODNIESIENIU DO ZALECANYCH GATUNKÓW
|
Gatunek |
Temperatura ( oC) |
Stopień zasolenia (0/00) |
Czas naświetlania (h) |
Czas trwania stadium (dni) |
Typowy czas trwania badania |
Przeżycie w doświadczeniu kontronym (minimum %) |
||
|
Embrion |
Narybek |
Powodzenie wylęgu |
Powylęgowe |
|||||
|
SŁODKOWODNE |
||||||||
|
Brachydanio rerio Danio pręgowany |
25 ± 1 |
— |
12–16 |
3–5 |
8–10 |
Tak szybko jak to możliwe po zapłodnieniu (wczesne stadium gastruli) do 5 dni po wylęgu (8–10 dni) |
80 |
90 |
|
Oncorhynchus mykiss Pstrąg tęczowy |
10 ± 1 (22) 12 ± l (23) |
|
0 (24) |
30–35 |
25–30 |
Tak szybko jak to możliwe po zapłodnieniu (wczesne stadium gastruli) do 20 dni po wylęgu (50–55 dni) |
66 |
70 |
|
Cyprinus carpio Zwykły karp |
21–25 |
— |
12–16 |
5 |
> 4 |
Tak szybko jak to możliwe po zapłodnieniu (wczesne stadium gastruli) do 4 dni po wylęgu (8–9 dni) |
80 |
75 |
|
Oryzias latipes |
24 ± 1 (22) 23 ± l (23) |
— |
12–16 |
8–11 |
4–8 |
Tak szybko jak to możliwe po zapłodnieniu (wczesne stadium gastruli) do 5 dni po wylęgu (13–16 dni) |
80 |
80 |
|
Pimephales promelas |
25 ±2 |
— |
16 |
4–5 |
5 |
Tak szybko jak to możliwe po zapłodnieniu (wczesne stadium gastruli) do 4 dni po wylęgu (8–9dni) |
60 |
70 |
DODATEK 3
Warunki badania, czas trwania i kryteria przeżycia w odniesieniu do innych dobrze udokumentowanych gatunków
|
Gatunki |
Temperatura ( oC) |
Stopień zasolenia (0/00) |
Czas naświetlania (godz.) |
Czas trwania stadium (dni) |
Typowy czas trwania badania embrionów i narybka |
Przeżycie w doświadczeniu kontrolnym (minimum %) |
||
|
|
|
|
|
Embrion |
Badanie narybka |
Powodzenie wylęgu |
Powylęgowe |
|
|
SŁODKOWODNE |
||||||||
|
Carassius auratus Karaś srebrzysty |
24 ± 1 |
— |
— |
3–4 |
> 4 |
Tak szybko jak to możliwe po zapłodnieniu (wczesne stadium gastruli) do 4 dni po wylęgu (7 dni) |
— |
80 |
|
Leopomis macrochirus Bassek blękitny |
21 ± 1 |
— |
16 |
3 |
> 4 |
Tak szybko jak to możliwe po zapłodnieniu (wczesne stadium gastruli) do 4 dni po wylęgu (7 dni) |
— |
75 |
|
SŁONOWODNE |
||||||||
|
Menidia peninsulae |
22–25 |
15–22 |
12 |
1,5 |
10 |
Tak szybko jak to możliwe po zapłodnieniu (wczesne stadium gastruli) do 5 dni po wylęgu (6–7 dni) |
80 |
60 |
|
Clupea harengus Śledź pospolity |
10 ± 1 |
8–15 |
12 |
20–25 |
3–5 |
Tak szybko jak to możliwe po zapłodnieniu (wczesne stadium gastruli) do 3 dni po wylęgu (23–27dni) |
60 |
80 |
|
Gadus morhua Dorsz |
5 ± 1 |
5–30 |
12 |
14–16 |
3–5 |
Tak szybko jak to możliwe po zapłodnieniu (wczesne stadium gastruli) do 3 dni po wylęgu (18 dni) |
60 |
80 |
|
Cyprinodon variegatus |
25 ± 1 |
15–30 |
12 |
— |
— |
Tak szybko jak to możliwe po zapłodnieniu (wczesne stadium gastruli) do 4/7dni po wylęgu (28 dni) |
> 75 |
80 |
DODATEK 4
NIEKTÓRE CECHY CHEMICZNE DOPUSZCZONEJ WODY ROZCIEŃCZAJĄCEJ
|
Substancja |
Stężenie |
|
Cząstki stale |
< 20 mg/l |
|
Całkowity węgiel organiczny |
< 2 mg/l |
|
Niezjonizowany amoniak |
< 1 μg/l |
|
Szczątkowy chlor |
< 10 μg/l |
|
Całkowite pestycydy fosforoorganiczne |
< 50 μg/l |
|
Całkowite pestycydy chloroorganiczne plus polichlorowane bifenyle |
< 50 ng/l |
|
Całkowity chlor organiczny |
< 25 ng/l |
C.16. PSZCZOŁY MIODNE – BADANIE TOKSYCZNOŚCI OSTREJ DROGĄ POKARMOWĄ
1. METODA
Niniejsza metoda badania toksyczności ostrej jest kopią OECD TG 213 (1998).
1.1. WPROWADZENIE
Niniejszy badanie toksyczności jest laboratoryjną metodą, zaprojektowaną do oceny ostrej toksyczności środków ochrony roślin i innych substancji chemicznych do dorosłych robotnic pszczół miodnych.
Przy oszacowaniu i ocenie cech toksyczności substancji chemicznej oznaczenie ostrej toksyczności drogą pokarmową dla pszczół miodnych jest wymagane np. gdy jest prawdopodobna ekspozycja pszczół na działanie danej substancji chemicznej. Badanie toksyczności ostrej drogą pokarmową jest przeprowadzane dla ustalenia właściwej toksyczności dla pszczół, pestycydów i innych substancji chemicznych. Wyniki niniejszego badania powinny być wykorzystane do określenia potrzeby dalszej oceny. W szczególności niniejsza metoda może być zastosowana w programach odcinkowych oceny niebezpieczeństwa pestycydów dla pszczół, w oparciu o kolejne postępy badań toksyczności laboratoryjnych do półpolowych i polowych eksperymentów (1). Pestycydy mogą być badane jako substancje aktywne (a.s.) lub jako wytworzone produkty.
Standard toksyczności powinien być zastosowany do sprawdzenia wrażliwości pszczół i precyzji procedury badania.
1.2. DEFINICJE
Ostra toksyczność drogą pokarmową: jest negatywnymi skutkami powstającymi w granicach maksymalnego okresu 96 godzin po podaniu doustnym pojedynczej dawki badanej substancji.
Dawka: jest ilością substancji badanej spożytej. Dawka jest wyrażana jako masa (μg) substancji badanej na badane zwierzę (μg/pszczoła). Rzeczywista dawka dla każdej pszczoły nie może być obliczona, ponieważ są karmione wspólnie, ale średnia dawka może być oszacowana (całkowita ilość spożytej substancji badanej/ilość badanych pszczół w jednej klatce).
LD 50 (średnia dawka śmiertelna) drogą pokarmową: jest statystycznie wyprowadzoną pojedynczą dawką substancji, która powoduje zgonu 50 % zwierząt przy podaniu drogą pokarmową. Wartość LD 50 jest wyrażona w (μg) substancji badanej na pszczołę. W odniesieniu do pestycydów substancją badaną może być substancja aktywna (a.s.) albo wytworzony produkt zawierający jedną lub więcej niż jedną substancję aktywną.
Śmiertelność: zwierzę jest rejestrowane jako martwe, gdy jest zupełnie nieruchome.
1.3. ZASADA METODY BADANIA
Dorosłe robotnice pszczół miodnych (Apis mellifera) są eksponowane w zakresie dawek substancji badanej rozproszonej w roztworze sacharozy. Następnie pszczoły są karmione tą samą dietą, wolną od substancji badanej. Śmiertelność jest rejestrowana codziennie podczas co najmniej 48 godzin i porównywana z wartościami kontrolnymi. Jeśli wskaźnik śmiertelności zwiększa się między 24 i 48 godziną, podczas gdy śmiertelność w doświadczeniu kontrolnym pozostaje na akceptowalnym poziomie, tj. < 10 %, właściwe jest rozciągnięcie czasu trwania badania do maksimum 96 godzin. Wyniki są analizowane w celu obliczenia LD50 w 24 i 48 godzinie i, w przypadku gdy badanie jest przedłużone, w 72 i 96 godzinie.
1.4. WAŻNOŚĆ BADANIA
Aby badanie było ważne, mają zastosowanie następujące warunki:
|
— |
średnia śmiertelność dla wszystkich doświadczeń kontrolnych nie może przekraczać 10 % na końcu badania, |
|
— |
LD50 normy toksyczności spełnia określony zakres. |
1.5. OPIS METODY BADANIA
1.5.1. Zbieranie pszczół
Młode dojrzałe robotnice pszczół z tej samej rasy muszą być użyte, tj. pszczoły w tym samym wieku, statusie żywieniowym itd. Pszczoły powinny być uzyskane z kolonii, właściwie odżywianej, zdrowej, tak dalece jak to możliwe wolnej od chorób, o właściwej królowej, o znanej historii i fizjologicznym statusie. Mogą być zebrane rano przed użyciem lub wieczorem przed badaniem i przechowane w warunkach badania do następnego dnia. Odpowiednie są pszczoły zebrane z ram, bez potomstwa. Należy uniknąć zbierania pszczół wczesną wiosną lub późną jesienią, gdyż podczas tego okresu mają zmienioną fizjologię. Jeżeli badania muszą być prowadzone wczesną wiosną lub późną jesienią, pszczoły umieszcza się w inkubatorze i hoduje przez tydzień na „pszczelim chlebie” (pyłek zebrany z grzebienia) i roztworze sacharozy. Pszczoły traktowane substancjami chemicznymi, takimi jak antybiotyki, produkty przeznaczone do zwalczania warrozy itd., nie powinny być używane do badań toksyczności przez cztery tygodnie od czasu końca ostatniego traktowania.
1.5.2. Pomieszczenie i warunki żywienia
Stosuje się łatwe do czyszczenia i dobrze wentylowane klatki. Należy stosować właściwe materiały, na przykład stal nierdzewną, siatkę drucianą, plastikowe lub jednorazowe drewniane klatki. Zalecane są grupy po dziesięć pszczół na klatkę. Wymiary klatek do badań muszą być właściwe do ilości pszczół, tj. zapewniać wystarczającą przestrzeń.
Pszczoły powinny być trzymane w ciemności, w pokoju doświadczalnym w temperaturze 25 ± 2 oC. Wilgotność względna, zwykle około 50–70 %, powinna być rejestrowana w czasie badania. Procedury postępowania, włączając traktowanie i obserwacje, prowadzi się w dziennym świetle. Roztwory sacharozy w wodzie o stężeniu końcowym 500 g/l (50 % wag./obj.) powinny być stosowane jako pokarm. Po podaniu próbnych dawek pokarm powinien być zapewniany ad libitum. System podawania pokarmu powinien umożliwiać rejestrowanie poboru pokarmu powinien odniesieniu do każdej klatki (zob. pkt 1.6.3.1). Może to być szklana rura (około 50 mm długości i 10 mm szerokości z otwartym zwężającym się końcem do około 2 mm średnicy).
1.5.3. Przygotowanie pszczół
Zebrane pszczoły mogą być przypadkowo rozmieszczane w klatkach do badań rozmieszczonych losowo w pokoju doświadczalnym.
Pszczoły mogą być zagłodzone przez 2 h przed rozpoczęciem badania. Zalecane jest, aby pszczoły pozbawiono pokarmu przed traktowaniem, tak aby wszystkie pszczoły miały równe warunki odnośnie do zawartości jelit na początku badania. Konające pszczoły powinny być usunięte i zastąpione zdrowymi przed rozpoczęciem badania.
1.5.4. Przygotowanie dawek
W przypadku gdy substancja badana jest związkiem mieszającym się z wodą, roztwarza się ją bezpośrednio w 50 % roztworze sacharozy. W odniesieniu do produktów technicznych i substancji o niskiej rozpuszczalności w wodzie stosuje się nośniki takie, jak organiczne rozpuszczalniki, emulgatory lub dyspergatory o niskiej toksyczności dla pszczół (np. aceton, dimetyloformamid, dimetylosulotlenek). Stężenie nośnika zależy od rozpuszczalności badanej substancji i powinno być takie same dla wszystkich badanych stężeń. Jednakże 1 % stężenie nośnika jest ogólnie właściwe i nie może być przekraczane.
Powinny być przygotowane roztwory kontrolne, tj. w przypadku gdy stosuje się rozpuszczalnik lub dyspergator do rozpuszczenia substancji badanej, należy użyć dwóch oddzielnych roztworów kontrolnych: roztwór w wodzie i roztwór sacharozy z układem rozpuszczalnik/dyspergator o stężeniu stosowanym w roztworach dozujących.
1.6. PROCEDURA
1.6.1. Grupy badane i kontrolne
Liczba dawek i testowanych kopii musi spełniać wymagania statystyczne dla oznaczenia LD50 z 95 % granicą ufności. Zwykle pięć dawek w serii geometrycznej, ze wskaźnikiem nieprzekraczającym 2,2 i obejmującym zakres dla LD50, jest wymaganych dla badania. Jednakże wskaźnik rozcieńczenia i liczba stężeń w odniesieniu do dozowania muszą być ustalone w zależności od zbocza krzywej toksyczności (dawka w zależności od śmiertelności) i uwzględnione w ramach wybranej metody statystycznej do analizy wyników. Badanie znalezienia zakresu umożliwia wybór właściwych stężeń do dozowania.
Należy użyć minimum trzech kopii grup badanych, po 10 pszczół każda, dozowanych każdym stężeniem badanym. Minimum trzy kontrolne partie, każda po 10 pszczół, powinny być dodatkowo włączone w przebieg serii badań. Partie kontrolne powinny być także włączone, gdy stosowany jest układ rozpuszczalnik/nośnik (zob. pkt 1.5.4).
1.6.2. Norma toksyczności
W badane serie należy włączyć normę toksyczności. Co najmniej trzy dawki muszą być wybrane do pokrycia wartości oczekiwanej LD50. Co najmniej trzy skopiowane klatki, każda zawierająca 10 pszczół, należy użyć dla każdej badanej dawki. Zalecaną normą toksyczności jest dimetoat, dla którego doniesienie o LD50-24 h, drogą pokarmową jest w zakresie 0,10–0,35 μg a.s./pszczoła (2). Jednakże inne normy toksyczności byłyby również dopuszczalne w przypadku, gdy można dostarczyć wystarczających danych do sprawdzenia oczekiwanej reakcji na dawkę (np. paration).
1.6.3. Ekspozycja
1.6.3.1. Podawanie dawek
Każdej badanej grupie pszczół należy dostarczyć 100–200 μl 50 % roztworu sacharozy w wodzie, zawierającego badaną substancję o właściwym stężeniu. Większa objętość jest wymagana w odniesieniu do produktów o niskiej rozpuszczalności, niskiej toksyczności lub stężeniu w składzie, ponieważ muszą być zastosowane wyższe proporcje w roztworze sacharozy. Ilość traktowanej, spożytej diety przez grupę musi być monitorowana. Zużyty podajnik (zwykle 3–4 godz.) powinien być usunięty z klatki i zastąpiony podajnikiem wypełnionym tylko roztworem sacharozy. Roztwory sacharozy są wtedy dostarczane ad libitum. W odniesieniu do niektórych związków odrzucenie wyższego stężenia dawki badanej objawia się w małej konsumpcji pokarmu lub jej braku. Po maksimum 6 godz. niespożyta traktowana dieta powinna być zastąpiona samym roztworem sacharozy. Ilość spożytej traktowanej diety musi być oszacowana (np. pomiar objętość/waga pozostałej traktowanej diety).
1.6.3.2. Czas trwania
Zalecany czas trwania badania wynosi 48 godz. po zastąpieniu roztworu badanego, roztworem samej sacharozy. Jeśli śmiertelność nadal wzrasta do więcej niż 10 % po pierwszych 24 godz., czas trwania badania powinien być przedłużony do maksimum 96 godz., zapewniając, że śmiertelność w doświadczeniu kontrolnym nie przewyższa 10 %.
1.6.4. Obserwacje
Śmiertelność jest rejestrowana w 4 godziny po rozpoczęciu badania, a w okresie późniejszym w 24 i 48 godzinie (tj. po podaniu dawki). Jeśli wymagany jest przedłużony okres obserwacji, następne oceny powinny być dokonane w 24 godzin przedziałach czasowych, do maksimum 96 godzin, pod warunkiem że śmiertelność w doświadczeniu kontrolnym nie przekroczy 10 %.
Ilości pokarmu spożytego na grupę powinna być oszacowana. Porównanie wskaźników spożycia traktowanej i nietraktowanej diety w danych 6 godzinach dostarcza informacji o strawności diety traktowanej.
Wszystkie anormalne efekty zachowania zaobserwowane w trakcie okresu badania powinny być zarejestrowane.
1.6.5. Badanie graniczne
W pewnych przypadkach (np. gdy oczekuje się, że substancja badana posiada niską toksyczność) należy przeprowadzić badanie graniczne, stosując 100 μg a.s./pszczołę w celu ukazania, że LD50 jest większa niż niniejsza wartość. Ta sama procedura powinna być zastosowana do, włączając trzy skopiowane grupy badane dla dawki badanej, odpowiednich doświadczeń kontrolnych, oceny ilości traktowanej diety spożytej oraz użycia normy toksyczności. Jeśli występuje śmiertelność, powinny być przeprowadzone pełne badania. Jeśli obserwuje się podśmiertelne skutki (zob. pkt 1.6.4), powinny być one zarejestrowane.
2. DANE I SPRAWOZDAWCZOŚĆ
2.1. DANE
Dane powinny być zestawione w formie tabelarycznej, pokazując w odniesieniu do każdej traktowanej grupy, jak również w odniesieniu do grup kontrolnych i grup norm toksyczności, ilość użytych pszczół, śmiertelność w dowolnym momencie obserwacji i liczbę pszczół o negatywnym zachowaniu. Analiza danych śmiertelności właściwymi metodami statystycznymi (np. analiza probitowa, średnia krocząca, prawdopodobieństwo dwumianu) (3)(4). Wykreślone krzywe dawka-reakcja w każdym zalecanym momencie obserwacji i obliczenia zboczy krzywych oraz średnie dawki śmiertelne (LD50) z 95 % granicami ufności. Poprawki dotyczące śmiertelności w doświadzeniach kontrolnych mogą być dokonane przy użyciu poprawki Abbotta (4)(5). W przypadku gdy traktowana dieta nie jest całkowicie spożyta, powinna być ustalona dawka substancji badanej spożytej przez grupę. LD50 powinna być wyrażona w μg substancji badanej na pszczołę.
2.2. SPRAWOZDANIE Z BADANIA
Sprawozdanie z badania musi zawierać następujące informacje:
2.2.1. Substancja użyta w badaniu:
|
— |
charakter fizyczny i stosowne właściwości fizyko-chemiczne (np. stabilność w wodzie, ciśnienie pary), |
|
— |
dane dotyczące identyfikacji chemicznej włączając wzór strukturalny, czystość (tj. w odniesieniu do pestycydów identyfikację i stężenie aktywnej(-ych) substancji). |
2.2.2. Gatunki użyte w badaniu:
|
— |
nazwa naukowa, rasa, przybliżony wiek (w tygodniach), metoda zbierania, dane dotyczące zbierania, |
|
— |
informacje dotyczące kolonii używanych do zbierania pszczół badanych, włączając stan zdrowotny, choroby wieku dorosłego, wszystkie wcześniejsze traktowania itd. |
2.2.3. Warunki badania:
|
— |
temperatura i wilgotność względna pokoju doświadczalnego, |
|
— |
warunki pomieszczenia włączając typ, wymiary i materiał klatki, |
|
— |
metody przygotowania roztworu wyjściowego i roztworów badanych (rozpuszczalnik i jego stężenia muszą być podane, gdy zostały zastosowane), |
|
— |
metoda przygotowania roztworu wyjściowego i częstotliwość odnawiania (środek ułatwiający rozpuszczanie i jego stężenia muszą być podane, gdy zostały zastosowane), |
|
— |
projekt badania, np. liczba i stężenia badane użyte, ilość doświadczeń kontrolnych w odniesieniu do każdego stężenia badanego i doświadczenia kontrolnego, liczba kopii klatek i liczba pszczół na klatkę, |
|
— |
data badania. |
2.2.4. Wyniki:
|
— |
wyniki badań wstępnego szukania zakresu, jeśli wykonano, |
|
— |
pierwotne dane: śmiertelność przy każdej dawce badanej w dowolnym momencie obserwacji, |
|
— |
wykresy krzywych dawka-reakcja na koniec badania, |
|
— |
wartość LD50 z granicami ufności 95 % w zalecanym momencie obserwacji, w odniesieniu do substancji badanej i normy toksyczności, |
|
— |
zastosowane procedury statystyczne do określenia LD50, |
|
— |
śmiertelność w doświadczeniach kontrolnych, |
|
— |
inne skutki biologiczne zaobserwowane lub pomierzone, np. anormalne zachowanie pszczół (włączając odrzucenie dawki badanej), wskaźnik spożycia diety w traktowanych i nietraktowanych grupach, |
|
— |
wszelkie odchylenia od opisanych tutaj procedur badania oraz wszelkie inne istotne informacje. |
3. BIBLIOGRAFIA
|
(1) |
EPPO/Council of Europe (1993). Decision-Making Scheme for the Environmental Risk Assessment of Plant Protection Products-Honeybees. EPPO bulletin, Vol. 23, N.1, pp. 151–165. March 1993. |
|
(2) |
Gough, H. J., McIndoe, E. C, Lewis, G. B. (1994). The use of dimethoate as a reference compound in laboratory acute toxicity tests on honeybees (Apis mellifera L.), 1981-1992. Journal of Apicultural Research 22, pp. 119–125. |
|
(3) |
Litchfield, J. T. and Wilcoxon, F. (1949). A simplified method of evaluating dose-effect experiments. Jour. Pharmacol. and Exper. Ther., 96, pp. 99–113. |
|
(4) |
Finney, D. J. (1971). Probit Analysis. 3rd ed., Cambridge, London and New York. |
|
(5) |
Abbott, W. S. (1925). A method for computing the effectiveness of an insecticide. Jour. Econ. Entomol. 18, pp. 265–267. |
C.17. PSZCZOŁY MIODNE – BADANIE KONTAKTOWEJ TOKSYCZNOŚCI OSTREJ
1. METODA
Niniejsza metoda badania kontaktowej toksyczności ostrej jest kopią OECD TG 214 (1998 r.).
1.1. WPROWADZENIE
Niniejsze badanie toksyczności jest laboratoryjną metodą, zaprojektowaną do oceny kontaktowej ostrej toksyczności środków ochrony roślin i innych substancji chemicznych do dorosłych robotnic pszczół miodnych.
Przy oszacowaniu i ocenie cech toksyczności substancji chemicznej oznaczenie ostrej toksyczności kontaktowej dla pszczół miodnych jest wymagane np. gdy jest prawdopodobna ekspozycja pszczół na działanie danej substancji chemicznej. Badanie kontaktowej toksyczności ostrej jest przeprowadzane dla ustalenia właściwej toksyczności w odniesieniu do pszczół, pestycydów i innych substancji chemicznych. Wyniki niniejszego badania powinny być wykorzystane do określenia potrzeby dalszej oceny. W szczególności niniejsza metoda może być stosowana w programach odcinkowych oceny niebezpieczeństwa pestycydów dla pszczół, w oparciu o kolejne postępy badań toksyczności laboratoryjnych do półpolowych i polowych eksperymentów (1). Pestycydy mogą być badane jako substancje aktywne (a.s.) lub jako wytworzone produkty.
Norma toksyczności powinna być zastosowana do sprawdzenia wrażliwości pszczół i precyzji procedury badania.
1.2. DEFINICJE
Ostra toksyczność drogi pokarmowej: są to negatywne skutki powstające w granicach maksymalnego okresu 96 godzin po miejscowym podaniu pojedynczej dawki substancji.
Dawka: jest ilością substancji badanej zastosowanej. Dawka jest wyrażana jako masa (μg) substancji badanej na badane zwierzę (μg/pszczołę).
LD 50 (średnia dawka śmiertelna) kontaktowa: jest statystycznie wyprowadzoną pojedyńczą dawką substancji, która powoduje zgon u 50 % zwierząt poprzez kontakt. Wartość LD50 jest wyrażona w μg substancji badanej na pszczołę. W odniesieniu do pestycydów substancją badaną może być substancja aktywna (a.s.) albo wytworzony produkt zawierający jedną lub więcej niż jedną substancję aktywną.
Śmiertelność: zwierzę jest rejestrowane jako martwe, gdy jest zupełnie nieruchome.
1.3. ZASADA METODY BADANIA
Dorosłe robotnice pszczół miodnych (Apis mellifera) są eksponowane w zakresie dawek substancji badanej rozproszonej we właściwym nośniku, przez bezpośrednie nałożenie na tułów (kropelki). Czas trwania badania wynosi 48 godz. Jeśli wskaźnik śmiertelności zwiększa się między 24 a 48 godz., podczas gdy śmiertelność w doświadzeniach kontrolnych pozostaje na akceptowalnym poziomie, tj. < 10 %, właściwe jest przedłużenie czasu trwania badania do maksimum 96 godz. Śmiertelność jest rejestrowana codziennie i porównywana z wartościami kontrolnymi .Wyniki są analizowane w celu obliczenia LD50 w 24 i 48 godz. oraz, w przypadku gdy badanie jest przedłużone, w 72 i 96 godz..
1.4. WAŻNOŚĆ BADANIA
Aby badanie było ważne, mają zastosowanie następujące warunki:
|
— |
średnia śmiertelność w odniesieniu do wszystkich doświadczeń kontrolnych nie może przekraczać 10 % na końcu badania, |
|
— |
LD50 normy toksyczności odpowiada określonemu zakresowi. |
1.5. OPIS METODY BADANIA
1.5.1. Zbieranie pszczół
Młode dojrzałe robotnice pszczół z tej samej rasy powinny być użyte, tj. pszczoły w tym samym wieku, statusie żywieniowym itd. Pszczoły powinny być uzyskane z kolonii, właściwie odżywianej, zdrowej, tak dalece jak to możliwe wolnej od chorób, o właściwej królowej, o znanej historii i fizjologicznym statusie. Muszą być zebrane rano przed użyciem lub wieczorem przed badaniem i przechowane w warunkach badania do następnego dnia. Odpowiednie są pszczoły zebrane z ram, bez potomstwa. Należy uniknąć zbierania pszczół wczesną wiosną lub późną jesienią, gdyż podczas tego okresu mają zmienioną fizjologię. Jeżeli badania muszą być prowadzone wczesną wiosną lub późną jesienią, pszczoły umieszcza się w inkubatorze i hoduje przez tydzień na „pszczelim chlebie” (pyłek zebrany z grzebienia) i roztworze sacharozy. Pszczoły traktowane substancjami chemicznymi, takimi jak antybiotyki, produkty przeznaczone do zwalczania warrozy itd., nie mogą być używane do badań toksyczności przez cztery tygodnie od chwili zakończenia ostatniego traktowania.
1.5.2. Pomieszczenie i warunki żywienia
Stosuje się łatwe do czyszczenia i dobrze wentylowane klatki. Należy stosować właściwe materiały, na przykład stal nierdzewną, siatkę drucianą, plastikowe lub jednorazowe drewniane klatki. Zalecane są grupy po dziesięć pszczół na klatkę. Wymiary klatek do badań muszą być odpowiednie do ilości pszczół, tj. zapewniać wystarczającą przestrzeń.
Pszczoły powinny być trzymane w ciemności, w pokoju doświadczalnym w temperaturze 25 ± 2 oC. Wilgotność względna, zwykle około 50–70 %, powinna być rejestrowana w czasie badania. Procedury postępowania włączając traktowanie i obserwacje prowadzi się w dziennym świetle. Roztwory sacharozy w wodzie o stężeniu końcowym 500 g/l (50 % wag./obj.) stosowane są jako pokarm podawany ad libitum w czasie trwania badania, używając podajnik pszczół. Może to być szklana rura (około 50 mm długości i 10 mm szerokości z otwartym zwężającym się końcem do około 2 mm średnicy).
1.5.3. Przygotowanie pszczół
Zebrane pszczoły są znieczulane dwutlenkiem węgla lub azotem w celu zastosowania substancji badanej. Ilość środka znieczulającego i czas ekspozycji powinien być zminimalizowany. Konające pszczoły powinny być usunięte i zastąpione zdrowymi pszczołami przed rozpoczęciem badania.
1.5.4. Przygotowanie dawek
Substancja badana ma być zastosowana jako roztwór w nośniku, tj. organicznym rozpuszczalniku lub wodnego roztworu ze środkiem zwilżającym. Z organicznych rozpuszczalników zalecany jest aceton, lecz mogą być użyte inne organiczne rozpuszczalniki o niskiej toksyczności dla pszczół (np. dwumetyloformamid, dwumetylosulfotlenek). Dla rozproszonych w wodzie określonych produktów i wysokopolarnych substancji organicznych nierozpuszczalnych w rozpuszczalnikowych organicznych nośnikach, roztwory łatwiej podawać, gdy przygotuje się słaby roztwór handlowego środka zwilżającego (np. Agral, Cittowett, Lubrol, Triton, Tween).
Powinny być przygotowane właściwe roztwory kontrolne, tj. w przypadku gdy użyto rozpuszczalnika albo dyspergator dla rozpuszczenia substancji badanej, powinny być zastosowane dwie kontrolne grupy, jedna traktowana wodą i jedna traktowana układem rozpuszczalnik/dyspergator.
1.6. PROCEDURA
1.6.1. Grupy badane i kontrolne
Liczba dawek i badanych kopii powinna odpowiadać wymaganiom statystycznym dotyczącym określenia LD50 z granicami ufności 95 %. Zwykle pięć dawek w serii geometrycznej, ze wskaźnikiem nieprzekraczającym 2,2 i obejmującym zakres dla LD50, jest wymaganych w odniesieniu do badań. Jednakże liczba dawek musi być ustalona w zależności od zbocza krzywej toksyczności (dawka w zależności od śmiertelności) i z uwzględnieniem wybranej metody statystycznej do analizy wyników. Badanie szukania zakresu umożliwia wybór właściwych stężeń do dozowania.
Należy użyć minimum trzech kopii grup badanych, po 10 pszczół każda, dozowanych każdym stężeniem badanym.
Minimum trzy kontrolne partie, każda po 10 pszczół, powinny być dodatkowo włączone w przebieg serii badań. Jeśli rozpuszczalnik organiczny lub środek zwilżający jest stosowany, trzy partie kontrolne, każda po 10 pszczół dla rozpuszczalnika i środka zwilżającego, muszą być włączone.
1.6.2. Norma toksyczności
W badane serie należy włączyć normę toksyczności. Co najmniej trzy dawki powinny być wybrane do pokrycia oczekiwanej wartości LD50. Co najmniej trzy skopiowane klatki, każda zawierająca 10 pszczół, należy użyć przy każdej badanej dawce. Zalecaną normą toksyczności jest dimetoat, dla którego oświadczone ustnie LD50-24 h, drogą pokarmową jest w zakresie 0,10–0,35 μg a.s./pszczołę (2). Jednakże inne normy toksyczności byłyby również dopuszczalne w przypadku, gdy można dostarczyć wystarczających danych do sprawdzenia oczekiwanej reakcji na dawkę (np. paration).
1.6.3. Ekspozycja
1.6.3.1. Podawanie dawek
Znieczulone pszczoły są pojedynczo poddawane zabiegowi miejscowemu podaniu dawki. Pszczoły są losowo poddawane różnym próbnym i kontrolnym dawkom. Objętość 1 μl roztworu zawierającego substancję badaną o odpowiednim stężeniu powinna być podana mikroaplikatorem do grzbietowej strony tułowia każdej pszczoły. Można użyć inne objętości, jeśli jest to uzasadnione. Po podaniu dawki pszczoły są umieszczane w klatkach do badań i żywione roztworami sacharozy.
1.6.3.2. Czas trwania
Zalecany czas trwania badania wynosi 48 godz. Jeśli śmiertelność wzrasta o więcej niż 10 % między 24 a 48 godz., czas trwania badania powinien być przedłużony do maksimum 96 godz., pod warunkiem że śmiertelność przy doświadczeniu kontrolnym nie przewyższa 10 %.
1.6.4. Obserwacje
Śmiertelność jest rejestrowana w 4 godzinie po dozowaniu, a w okresie późniejszym w 24 i 48 godzinie. Jeśli wymagany jest przedłużony okres obserwacji, dalsze oceny powinny być dokonywane w 24-godzinnych przedziałach czasowych, do maksimum 96 godzin, pod warunkiem że śmiertelność przy doświadczeniu kontrolnym nie przekracza 10 %.
Wszystkie anormalne skutki zachowywania się w czasie trwania badania powinny być zarejestrowane.
1.6.5. Badanie graniczne
W niektórych przypadkach (np. gdy oczekuje się, że substancja badana posiada niską toksyczność) można przeprowadzić badanie graniczne, stosując 100 μg a.s./pszczoła w celu ukazania, że LD50 jest większa niż niniejsza wartość. Ta sama procedura powinna być zastosowana, włączając trzy skopiowane grupy badane, w odniesieniu do dawki badanej, odpowiednich dawek kontrolnych oraz użycia normy toksyczności. Jeśli występuje śmiertelność, powinny być przeprowadzone pełne badania. Jeśli obserwuje się podśmiertelne skutki (zob. pkt 1.6.4), powinny one być zarejestrowane.
2. DANE I SPRAWOZDAWCZOŚĆ
2.1. DANE
Dane powinny być zestawione w formie tabelarycznej, pokazując w odniesieniu do każdej traktowanej grupy, jak również w odniesieniu do grup kontrolnych i grup normy toksyczności, ilość użytych pszczół, śmiertelność w każdym momencie obserwacji i liczbę pszczół o negatywnym zachowaniu. Analiza danych dotyczących śmiertelności właściwymi metodami statystycznymi (np. analiza probitowa, średnia krocząca, prawdopodobieństwo dwumianu) (3)(4). Wykreślone krzywe dawka-reakcja w każdym zalecanym momencie obserwacji (tj. 24, 48 godz. i, jeśli to stosowne, 72 i 96 godz.) i obliczenia zboczy krzywych oraz średnie dawki śmiertelne (LD50) z granicami ufności 95 %. Poprawki w odniesieniu do śmiertelności przy doświadczeniu kontrolnym powinny być wykonane przy użyciu poprawki Abbotta (4)(5). LD50 powinna być wyrażona w μg substancji badanej na pszczołę.
2.2. SPRAWOZDANIE Z BADANIA
Sprawozdanie z badania musi zawierać następujące informacje:
2.2.1. Substancja użyta w badaniu:
|
— |
charakter fizyczny i stosowne właściwości fizyko-chemiczne (np. stabilność w wodzie, ciśnienie pary), |
|
— |
dane identyfikacji chemicznej, włączając wzór strukturalny, czystość (tj. w odniesieniu do pestycydów identyfikację i stężenie aktywnej substancji). |
2.2.2. Gatunki użyte w badaniu:
|
— |
nazwa naukowa, rasa, przybliżony wiek (w tygodniach), metoda zbierania, dane dotyczące zbierania, |
|
— |
informacje dotyczące kolonii używanych do zbierania pszczół badanych, włączając stan zdrowotny, choroby wieku dorosłego, wszystkie wcześniejsze traktowania itd. |
2.2.3. Warunki badania:
|
— |
temperatura i wilgotność względna pokoju doświadczalnego, |
|
— |
warunki pomieszczenia, włączając typ, wymiary i materiał klatki, |
|
— |
metody podawania substancji badanej, np. użyty nośnik, rozpuszczalnik, objętość zastosowanego roztworu badanego, użyte znieczulenie, |
|
— |
projekt badania, np. ilość i użyte dawki badane, liczba doświadczeń kontrolnych dla próbnej dawki i pomiaru, liczba kopii klatek i liczba pszczół na klatkę, |
|
— |
data badania. |
2.2.4. Wyniki:
|
— |
wyniki badań wstępnego szukania zakresu, jeśli wykonano, |
|
— |
pierwotne dane: śmiertelność przy każdym stężeniu badanym w każdej chwili obserwacji, |
|
— |
wykresy krzywych dawka-reakcja na koniec badania, |
|
— |
wartość LD50 z 95 % limitem zaufania w każdym zalecanym momencie obserwacji, w odniesieniu do substancji badanej i normy toksyczności, |
|
— |
zastosowane procedury statystyczne do określenia LD50, |
|
— |
śmiertelność w doświadczeniach kontrolnych, |
|
— |
inne skutki biologiczne zaobserwowane lub pomierzone i wszystkie anormalne reakcje pszczół, |
|
— |
wszelkie odchylenia od opisanej tutaj metody badania i wszystkie inne istotne informacje. |
3. BIBLIOGRAFIA
|
(1) |
EPPO/Council of Europe (1993). Decision -Making Scheme for the Environmental Risk Assessment of Plant Protection Products-Honeybees. EPPO bulletin, Vol. 23, N.1, pp. 151–165. March 1993. |
|
(2) |
Gough, H. J., Mclndoe, E. C, Lewis, G. B. (1994). The use of dimethoate as a reference compound in laboratory acute toxicity tests on honeybees (Apis mellifera L.), 1981-1992. Journal of Apicultural Research 22, pp. 119–125. |
|
(3) |
Litchfield, J. T. and Wilcoxon, F. (1949). A simplified method of evaluating dose-effect experiments. Jour. Pharmacol. and Exper. Ther., 96, pp. 99–113. |
|
(4) |
Finney, D. J. (1971). Probit Analysis. 3rd ed., Cambridge, London and New York. |
|
(5) |
Abbott, W. S. (1925). A method for computing the effectiveness of an insecticide. Jour. Econ. Entomol. 18, pp. 265–267. |
C.18. ADSORPCJA/DESORPCJA PRZY UŻYCIU METODY RÓWNOWAGI OKRESOWEJ
1. METODA
Niniejsza metoda jest kopią OECD TG 106, w odniesieniu do określenia adsorpcji/desorpcji gleby przy użyciu metody równowagi okresowej (2000 r.).
1.1. WPROWADZENIE
Metoda uwzględnia próbę obrączkową i warsztat dotyczący wyboru gleby dla rozwoju badania adsorpcji (1)(2)(3)(4), a także istniejące wytyczne na poziomie krajowym (5)(6)(7)(8)(9)(10)(11).
Badania adsorpcji/desorpcji są użyteczne dla uzyskania podstawowych informacji dotyczących mobilności substancji chemicznych i ich rozłożeniu w ziemnych, wodnych i powietrznych częściach biosfery (12)(13)(14)(15)(16)(17)(18)(19)(20)(21). Informacje mogą być wykorzystywane w przewidywaniu lub szacowaniu na przykład zdolności substancji chemicznych do degradacji (22)(23), przekształcania i poboru przez organizmy (24); wymywania przez profil gleby (16)(18)(19)(21)(25)(26)(27)(28); lotności z gleby (21)(29)(30); ucieczki z powierzchni ziemi do wód naturalnych (18)(31)(32). Dane dotyczące adsorpcji są wykorzystywane do celów porównawczych i modelowania (19)(33)(34)(35).
Rozłożenie substancji chemicznej między glebę a fazę wodną jest złożonym procesem zależnym od wielu różnych czynników: charakteru chemicznego substancji (12)(36)(37)(38)(39)(40), cech gleby (4)(12)(13)(14)(41)(42)(43)(44)(45)(46)(47)(48)(49), czynników klimatycznych takich, jak opady deszczu, temperatury, światła słonecznego i wiatru. W ten sposób liczne zjawiska i mechanizmy włączają się w proces adsorpcji substancji chemicznej przez glebę i nie mogą być określone całościowo przez uproszczony model laboratoryjny, jak w niniejszej metodzie. Jednakże jeśli nawet ta próba nie może objąć środowiskowo możliwych przypadków, to dostarcza cennych informacji odnoszących się do adsorpcji substancji chemicznych w środowisku.
Zob. także Ogólne wprowadzenie.
1.2. ZAKRES
Metoda ma na celu oszacowanie zachowywania adsorpcji/desorpcji substancji w glebie. Celem jest uzyskanie wartości sorpcji, która jest użyteczna do przewidywania podziału w różnorodności warunków środowiskowych; do tego celu współczynniki adsorpcji równowagowej są ustalane w odniesieniu do substancji chemicznych w różnych glebach, jako funkcja cech gleby (np. zawartość węgla organicznego, zawartość gliny i tekstura gleby oraz pH). Należy użyć różnych typów gleby w celu objęcia jak najszerzej zdarzenia współdziałań danej substancji z glebami naturalnymi.
W niniejszej metodzie adsorpcja przedstawia proces wiązania się substancji chemicznej do powierzchni gleby; nie odróżnia między różnymi procesami adsorpcji (fizyczna i chemiczna adsorpcja) i takimi procesami, jak rozkład katalizowany powierzchniowo, masową adsorpcją i chemiczną reakcją. Adsorpcji zdarzającej się na cząstkach koloidów (średnica < 0,2 μm) wytwarzanych przez gleby nie uwzględniono.
Parametrami gleby, przypuszczalnie najistotniejszymi, w odniesieniu do gleby są: zawartość węgla organicznego (3)(4)(12)(13)(14)(41)(43)(44)(45)(46)(47)(48); zawartość gliny i tekstura gleby (3)(4)(41)(42)(43)(44)(45)(46)(47)(48) i pH w odniesieniu do zjonizowanych związków (3)(4)(42). Inne parametry gleby, które mają wpływ na absorpcję/desorpcję określonej substancji, to efektywna zdolność wymiany kationu (ECEC), zawartość amorficznego żelaza i tlenków glinu, szczególnie w odniesieniu do gleb wulkanicznych i tropikalnych (4), jak również powierzchnia właściwa (49).
Badanie jest zaprojektowane do oceny adsorpcji substancji chemicznej na różnego typu glebach o zróżnicowanym zakresie zawartości węgla organicznego, zawartości gliny i tekstury gleby oraz pH. Obejmuje to trzy warstwy:
|
Warstwa 1: |
Badania wstępne w celu ustalenia:
|
|
Warstwa 2: |
Badanie klasyfikacji: adsorpcja jest badana na pięciu różnych typach gleby za pomocą kinetyki adsorpcji w pojedynczym stężeniu oraz określenie współczynnika podziału Kd i Koc. |
|
Warstwa 3: |
Określenie izoterm adsorpcji Freundlicha dla ustalenia wpływu stężenia na zasięg adsorpcji na glebach. Badania desorpcji za pomocą kinetyki desorpcji/Freundlicha desorpcji izotermy (dodatek 1). |
1.3. DEFINICJE I JEDNOSTKI
|
Symbol |
Definicja |
Jednostki |
|
|
procent adsorpcji w czasie ti |
% |
|
Aeq |
procent adsorpcji w równowadze adsorpcji |
% |
|
|
masa substancji badanej zaadsorbowanej na glebie w czasie ti |
μg |
|
|
masa substancji badanej zaadsorbowanej na glebie podczas trwania przedziału czasowego Δti |
μg |
|
|
masa substancji badanej zaadsorbowanej na glebie w równowadze adsorpcji |
μg |
|
m0 |
masa substancji badanej w probówce na początku badania adsorpcji |
μg |
|
|
masa substancji badanej, zmierzona w podwielokrotności (
|
μg |
|
|
masa substancji w roztworze przy równowadze adsorpcji |
μg |
|
msoil |
wielkość fazy gleby, wyrażona jako sucha masa gleby |
g |
|
Cst |
stężenie masowe roztworu podstawowego substancji |
μg cm-3 |
|
C0 |
początkowe stężenie masowe roztworu badanego w kontakcie z glebą |
μg cm-3 |
|
|
stężenie masowe substancji w fazie wodnej w czasie ti, w którym dokonywana jest analiza |
μg cm-3 |
|
|
zawartość substancji zaadsorbowanej na glebie przy równowadze adsorpcji |
μg cm-1 |
|
|
stężenie masowe substancji w fazie wodnej przy równowadze adsorpcji |
μg cm-3 |
|
V0 |
początkowa objętość fazy wodnej w kontakcie z glebą, w czasie trwania badania adsorpcji |
cm3 |
|
|
objętość podwielokrotności, w której substancja badana została zmierzona |
cm3 |
|
Kd |
współczynnik podziału dla adsorpcji |
cm3 g-1 |
|
Koc |
znormalizowany współczynnik adsorpcji węgla |
cm3 g-1 |
|
Kom |
znormalizowany współczynnik ciał organicznych |
cm3 g-1 |
|
|
współczynnik adsorpcji Freundlicha |
μg1-l/n (cm3) 1/n g-1 |
|
1/n |
wykładnik Freundlicha |
|
|
|
procent desorpcji w punkcie czasu ti |
% |
|
|
procent desorpcji odpowiadający przedzialowi czasu Δti |
% |
|
Kdes |
pozorny wspolczynnik desorpcji |
μgcm-1 |
|
|
wspolczynnik desorpcji Freundlicha |
μg1-1n (cm3)1/n g-1 |
|
|
masa badanej substancji zdesorbowana z gleby w czasie tj |
μg |
|
|
masa badanej substancji zdesorbowana z gleby w czasie Δti |
μg |
|
|
masa substancji ustalona analitycznie w wodnej fazie przy równowadze desorpcyjnej |
μg |
|
|
całkowita masa badanej substancji zdesorbowanej przy równowadze desorpcyjnej |
μg |
|
|
masa substancji pozostająca zaadsorbowana na glebie po przedziale czasowym Δti |
μg |
|
|
masa substancji pozostała z równowagi adsorpcji z powodu niecałkowitego podstawienia objętości |
μg |
|
|
zawartość badanej substancji pozostająca zaadsorbowana na glebie przy równowadze desorpcyjnej |
μg g-1 |
|
|
stężenie masowe badanej substancji w fazie wodnej przy równowadze desorpcyjnej |
μg cm-3 |
|
VT |
całkowita objętość fazy wodnej w kontakcie z glebą w czasie trwania doświadczenia kinetyki desorpcji wykonanego metodą seryjną |
cm3 |
|
VR |
objętość nadsączu usuniętego z probówki po osiągnięciu równowagi adsorpcji, zastąpionego tą samą objętością roztworu 0,01 M CaCl2 |
cm3 |
|
|
Objętość podwielokrotności pobrana do celu analitycznego w czasie (i), w czasie trwania doświadczenia kinetyki adesorpcji wykonanego metodą seryjną |
cm3 |
|
|
objętość roztworu pobrana z probówki (i) dla pomiaru substancji badanej, w doświadczeniu kinetyki adesorpcji (metoda równoległa) |
cm3 |
|
|
objętość roztworu pobrana z probówki dla pomiaru substancji badanej w rownowadze desorpcji |
cm3 |
|
MB |
bilans masy |
% |
|
mE |
całkowita masa substancji badanej ekstrahowanej z gleby i ścianek zbiornika badanego w dwóch kolejnych czynnościach |
μg |
|
Vrec |
objętość nadsączu odzyskana po równowadze adsorpcji |
cm3 |
|
Pow |
współczynnik podziału oktanol/woda |
|
|
pKa |
stała dysocjacji |
|
|
Sw |
rozpuszczalność w wodzie |
g 1-1 |
1.4. ZASADA METODY BADANIA
Znane objętości roztworów badanej substancji, nieetykietowanej czy też etykietowanej izotopami o znanym stężeniu w 0,01 M CaCl2 są dodane do próbek gleby o znanej suchej masie, wcześniej zrównoważone w 0,01 M CaCl2. Mieszanina jest mieszana przez właściwy czas. Zawiesiny gleby rozdziela się następnie przez odwirowanie i, jeśli pożądane, filtrowane, a faza wodna analizowana. Ilość substancji badanej zaadsorbowana na próbce gleby jest obliczona jako różnica między ilością substancji badanej początkowo obecnej w roztworze a ilością pozostałą na koniec doświadczenia (metoda pośrednia).
Jako opcja, ilość badanej substancji zaadsorbowanej jest także bezpośrednio ustalana analizą gleby (metoda bezpośrednia). Niniejsza procedura obejmująca stopniową ekstrakcję gleby właściwym rozpuszczalnikiem, jest zalecana w przypadku, gdy różnica w stężeniu roztworu substancji nie może być dokładnie ustalona. Przykładami takich przypadków są: adsorpcja substancji badanej na ściankach zbiornika, niestabilność substancji badanej w skali czasu doświadczenia, słabej adsorpcji dającej tylko małe zmiany stężenia w roztworze i silna adsorpcja powodująca niskie stężenie, którego nie można dokładnie określić. Jeśli stosuje się substancję etykietowaną izotopowo, ekstrakcji z gleby można zapobiec za pomocą analizy fazy gleby przez spalanie i obliczenie scyntylacji na cieczowym liczniku. Jednakże cieczowy licznik scyntylacyjny jest techniką nieokreśloną, która nie może rozróżnić między macierzystymi a przekształconymi produktami; dlatego musi być używana tylko wtedy jeżeli badana substancja chemiczna jest stabilna w czasie trwania badań.
1.5. INFORMACJE DOTYCZĄCE SUB STANCJI BADANEJ
Odczynniki chemiczne powinny być czystości analitycznej. Zalecane jest użycie nieetykietowanych substancji badanych o znanym składzie i czystości co najmniej 95 % lub etykietowanych izotopami substancji badanych o znanym składzie oraz radioczystości. W przypadku wskaźników o krótkim czasie półtrwania należy zastosować poprawki na rozkład.
Przed przeprowadzeniem badania adsorpcji-desorpcji powinny być dostępne następujące informacje o badanej substancji:
|
a) |
rozpuszczalność w wodzie (A.6); |
|
b) |
ciśnienie pary (A.4) i/lub stała Henry'ego; |
|
c) |
rozkład abiotyczny: hydroliza jako funkcja pH (C.7); |
|
d) |
współczynnik podziału (A.8); |
|
e) |
szybka biodegradacja (C.4) lub aerobowe i anaerobowe przekształcenia w glebie; |
|
f) |
pKa niezjonizowanych substancji; |
|
g) |
bezpośrednia fotoliza w wodzie (tj. UV-Vis widmo absorpcji w wodzie, wydajność kwantowa) i fotodegradacja na glebie. |
1.6. MOŻLIWOŚĆ ZASTOSOWANIA BADANIA
Badanie ma zastosowanie do substancji chemicznych, dla których jest dostępna analityczna metoda z dostateczną dokładnością. Ważnym parametrem wpływającym na wiarygodność wyników, w szczególności kiedy stosowana jest metoda pośrednia, jest stabilność substancji badanej w zakresie czasu badania. Dlatego wstępnym warunkiem jest sprawdzenie stabilności we wstępnych badaniach, jeśli obserwuje się przekształcenie w skali czasu badania, zalecane jest, aby prowadzono główne badania poprzez analizę zarówno gleby jak i fazy wodnej.
Trudności w prowadzeniu niniejszego badania powstają w odniesieniu do substancji badanych, o niskiej rozpuszczalności w wodzie (Sw < 10-4 g 1-1), jak również w odniesieniu do wysoce naładowanych substancji, spowodowanych przez fakt, że stężenie w fazie wodnej nie może być zmierzone analitycznie z wystarczającą dokładnością. W tych przypadkach należy podjąć dodatkowe czynności. Wytyczne dotyczące sposobu postępowania z takimi problemami podano we właściwych punktach niniejszej metody.
Przy badaniu lotnych substancji należy zachować ostrożność by zapobiec stratom w czasie trwania badania.
1.7. OPIS METODY
1.7.1. Aparatura i odczynniki chemiczne
Zwykły sprzęt laboratoryjny, w szczególności następujące urządzenia:
|
a) |
probówki lub zbiorniki do prowadzenia doświadczeń. Istotne jest, aby te probówki i zbiorniki:
|
|
b) |
urządzenie do mieszania: podwieszany wibrator lub równoważny osprzęt, utrzymujący glebę w postaci zawiesiny; |
|
c) |
wirówka: zalecana wysokoobrotowa, na przykład siła odwirowania > 3 000 g, kontrolowana odnośnie do temperatury, zdolna do usuwania cząstek o średnicy większej niż 0,2 μm z roztworu wodnego. Pojemniki powinny być zamykane w czasie wirowania i mieszania w celu uniknięcia zmian i strat wody; w celu zminimalizowania adsorpcji na nich należy użyć nieaktywne zamknięcia na przykład pokrywki śrubowe z teflonu; |
|
d) |
fakultatywnie: urządzenie filtracyjne; filtry o porowatości 0,2 μm, sterylne, jednorazowego użytku. Należy zwrócić szczególną uwagę przy wyborze materiału filtra aby uniknąć jakichkolwiek strat na nim; w odniesieniu do słabo rozpuszczalnych substancji badanych nie zaleca się filtra z materiału organicznego; |
|
e) |
analityczne oprzyrządowanie, odpowiednie do pomiarów stężenia badanej substancji chemicznej; |
|
f) |
suszarka laboratoryjna, zdolna utrzymać temperaturę od 103 oC do 110 oC. |
1.7.2. Scharakteryzowanie i wybór gleb
Gleby powinny być scharakteryzowane przez trzy parametry uważane za w dużej mierze odpowiedzialne za zdolności adsorpcyjne: węgiel organiczny, zawartość gliny i tekstura gleby oraz pH. Jak już wspomniano (zob. zakres) inne fizykochemiczne właściwości gleby wpływają na adsorpcję/desorpcję określonych substancji i powinny być rozważone w takich przypadkach.
Metody używane do scharakteryzowania gleby są bardzo istotne oraz posiadają znaczący wpływ na wyniki. Dlatego jest zalecane, aby pH gleby było mierzone w roztworze 0,01 M CaCl2 (jest to roztwór używany w badaniach adsorpcja/desorpcja) zgodnie z odpowiednią metodą ISO (ISO-10390-1). Jest także zalecane, aby inne stosowne właściwości gleby były ustalane zgodnie ze standardowymi metodami (na przykład „Handbook of Soil Analysis” ISO); pozwala to na oparcie analizowanych danych sorpcji, na globalnie znormalizowanych parametrach gleby. Niektóre wytyczne dotyczące istniejących standardowych metod analiz gleby i jej charakterystykę podano w bibliografii (50–52). W odniesieniu do kalibracji metod badania gleby zalecane jest stosowanie gleb odniesienia.
Wytyczne dotyczące wyboru gleb do doświadczeń adsorpcja/desorpcja podano w tabeli 1. Siedem wybranych gleb pokrywa typy gleb spotykane w różnych strefach geograficznych. Dla podatnych na jonizację substancji badanych wybrane gleby powinny pokrywać szeroki zakres pH, w celu umożliwienia oceny adsorpcji substancji w jej zjonizowanych i niezjonizowanych formach. Wytyczne dotyczące tego jak wiele różnych gleb należy użyć na różnych stadiach badania podano pod 1.9 „Wykonanie badania”.
Jeśli inne typy gleb są preferowane powinny one charakteryzować się tymi samymi parametrami i powinny mieć zbliżone zmiany we właściwościach do tych opisanych w tabeli 1, nawet jeśli nie zestawiają one dokładnie kryteriów.
Tabela 1
Wytyczne dotyczące wyboru próbek gleby dla adsorpcji/desorpcji
|
Typ gleby |
Zakres pH (w 0,01 M CaCl2) |
Zawartość węgla organicznego ( %) |
Zawartość gliny ( %) |
Tekstura gleby (25) |
|
1 |
4,5–5,5 |
1,0–2,0 |
65–80 |
glina |
|
2 |
> 7,5 |
3,5–5,0 |
20–40 |
glina formierska |
|
3 |
5,5–7,0 |
1,5–3,0 |
15–25 |
mul formierski |
|
4 |
4,0–5,5 |
3,0–4,0 |
15–30 |
ił |
|
5 |
< 4,0–6,0 (26) |
< 10–15 (26) |
piasek formierski |
|
|
6 |
> 7,0 |
40–65 |
glina ił/glina |
|
|
7 |
< 4,5 |
> 10 |
< 10 |
piasek/piasek formierski |
1.7.3. Zbieranie i przechowywanie próbek gleby
1.7.3.1. Zbieranie
Nie ma specjalnych technik ani narzędzi zalecanych do pobierania próbek; technika pobierania próbek zależy od celu badania (53)(54)(55)(56)(57)(58).
Należy rozważyć:
|
a) |
szczegółowe informacje dotyczące historii pola jest konieczne, obejmuje to umiejscowienie, rodzaje wegetacji, traktowanie pestycydami i/lub nawozami, dodatki biologiczne lub przypadkowe zanieczyszczenia. Zalecenia normy ISO dotyczące pobierania próbek (ISO 10381-6) powinny zostać spełnione w odniesienia do opisu miejsca pobierania próbek; |
|
b) |
miejsce pobierania próbek musi być określone przez UTM (uniwersalne poprzeczne odwzorowanie Mercatora – europejski poziom geodezyjny) lub współrzedne geograficzne; może to umożliwić ponowne zebranie określonej gleby w przyszłości lub może pomóc w określeniu gleby w różnych systemach klasyfikacyjnych używanych w różnych krajach Zbierana powinna być gleba z poziomu A do maksymalnej głębokości do 20 cm. W szczególności w odniesieniu do gleb typu n. 7, jeśli poziom Oh jest obecny jako część gleby, która powinna być włączona przy zbieraniu próbek. |
Próbki gleby powinny być przetransportowane z użyciem pojemników i w warunkach temperatury, które zapewniają, aby początkowe właściwości gleby nie zostały zmienione w znaczący sposób.
1.7.3.2. Przechowywanie
Zalecane jest użycie tylko świeżo pobranych gleb z pola. Tylko w przypadkach, gdy jest to niemożliwe, gleba jest przechowywana w temperaturze otoczenia oraz powinna być wysuszona powietrzem. Nie ma zalecanej granicy czasu przechowywania, lecz gleby przechowywane dłużej niż trzy lata powinny być ponownie przeanalizowane przed użyciem w odniesieniu do ich zawartości węgla organicznego, pH i CEC.
1.7.3.3. Postępowanie i przygotowanie próbek gleby do badań
Gleby są suszone powietrzem w temperaturze otoczenia (zalecane 20–25 oC). Rozdrobnienie powinno być prowadzone z minimalną siłą, tak aby pierwotna tekstura gleby zmieniła się tak niewiele, jak to możliwe. Gleby przesiewa się do wielkości cząstki < 2 mm; zalecenia normy ISO dotyczące pobierania próbek gleby (ISO 10381-6) powinny być spełnione w odniesieniu do procesu przesiewania. Zalecana jest staranna homogenizacja, gdyż poprawia to odtwarzalność wyników. Zawartość wilgoci w każdej glebie jest określana z trzech podwielokrotnośći ogrzewanych w temperaturze 105 oC do zaniku występowania znaczących różnic wagi (około 12 godzin). W odniesieniu do wszystkich obliczeń masa gleby oznacza wysuszoną w suszarce suchą masę, tj. wagę gleby skorygowaną o zawartość wilgoci.
1.7.4. Przygotowanie substancji użytej w badaniu do zastosowania na glebę
Substancja badana jest rozpuszczona w roztworze 0,01 M CaCl2 w destylowanej lub dejonizowanej wodzie; roztwór CaCl2 stosuje się jako rozpuszczalnik fazy wodnej do poprawy odwirowania i zminimalizowania wymiany kationowej. Zalecane stężenie roztworu podstawowego powinno być trzy rzędy wyższe niż granica wykrywalności stosowanej metody analitycznej. Niniejszy próg gwarantuje dokładne pomiary w odniesieniu do metodologii stosowanej w niniejszej metodzie; ponadto stężenie roztworu podstawowego powinno być poniżej rozpuszczalności w wodzie badanej substancji.
Roztwór podstawowy powinien być przygotowany tuż przed zastosowaniem do próbek gleby oraz powinien być on trzymany zamknięty w ciemności w 4 oC. Czas przechowywania zależy od stabilności substancji badanej i jej stęzenia w roztworze.
Tylko w odniesieniu do słabo rozpuszczalnych substancji (Sw < 10-4 g 1-1), gdy trudno rozpuścić substancję badaną, potrzebny może być środek ułatwiający rozpuszczenie. Środek ułatwiający rozpuszczenie: a) powinien być mieszalny z wodą, jak też z metanolem lub acetonitrylem; b) jego stężenie nie powinno przekraczać 1 % całkowitej objętości roztworu podstawowego i powinno stanowić mniej niż to w roztworze substancji badanej, która wchodzi w kontakt z glebą (zalecany mniej niż 0,1 %); oraz c) nie powinien być środkiem powierzchniowo czynnym lub ulegać solwolitycznym reakcjom z badaną substancją. Użycie środka ułatwiającego rozpuszczanie powinno być przewidziane i usprawiedliwione przy przedstawianiu danych.
Inną alternatywą dla słabo rozpuszczalnych substancji jest dodanie substancji badanej do systemu badanego przez wybijanie: substancja badana jest rozpuszczona w organicznym rozpuszczalniku, jej podwielokrotność do systemu gleby a 0,01 M roztworu CaCl2 w destylowanej lub dejonizowanej wodzie. Zawartość rozpuszczalnika w fazie wodnej powinna być tak niska jak to możliwe, zwykle nie przekracza 0,1 %. Wybijanie z roztworu organicznego może ulec objętościowej nieodtwarzalności. Dlatego wprowadza się dodatkowy błąd, tak że stężenie substancji badanej i korozpuszczalnika nie jest takie samo we wszystkich badaniach.
1.8. WARUNKI WSTĘPNE PRZEPROWADZANIA BADANIA ADSORPCJI/DESORPCJI
1.8.1. Metoda analityczna
Kluczowe parametry wpływające na dokładność pomiarów sorpcji obejmują: dokładność metody analitycznej w analizie obu roztworów i faz adsorbowanych, stabilność i czystość substancji badanej, osiągnięcie równowagi sorpcji, rozpiętość zmian stężenia roztworu, stosunek gleba/roztwór i zmiany w strukturze gleby w czasie trwania procesu równowagi (35)(59–62). Niektóre przykłady dotyczące danych dokładności podano w dodatku 2.
Wiarygodność używanej metody analitycznej musi być sprawdzona w zakresie stężeń, które prawdopodobnie wynikną w trakcie badania. Eksperymentator powinien czuć się swobodnie, aby rozwijać odpowiednie metody z właściwą dokładnością, precyzją, odtwarzalnością, granicami wykrywalności i je poprawiać. Wytyczne dotyczące tego w jaki sposób należy prowadzić takie badania podano w doświadczeniu poniżej.
Właściwa objętość 0,01 M CaCl2, np. 100 cm3, jest mieszana w ciągu 4 godz. z odważką gleby, np. 20 g, o wysokiej adsorbowalności, tj. z wysoką zawartością węgla organicznego oraz gliny; te odważki i wielkości zmieniają się w zależności od potrzeb analitycznych, lecz stosunek gleba/roztwór wynoszący 1:5 jest dogodnym punktem początkowym. Mieszaninę odwirowuje się, a fazę wodną filtruje. Pewną objętość roztworu podstawowego substancji badanej dodaje się do drugiej dla uzyskania nominalnego stężenia, w zakresie stężenia, które prawdopodobnie wystąpi w czasie trwania badania. Objętość ta nie powinna przekraczać 10 % końcowej objętości fazy wodnej, w celu zmiany w najmniejszym możliwym zakresie charakteru roztworu przedrównowagowego. Roztwór poddaje się analizie.
Ślepa próba, składająca się z systemu gleba + roztwór CaCl2 (bez substancji badanej), musi być włączona w celu sprawdzenia artefaktów w metodzie analitycznej oraz dla macierzystych efektów powodowanych przez glebę.
Analityczne metody, które mogą być stosowane do pomiarów sorpcji zawierają chromatografię gazowo-cieczową (GLC), wysokiej rozdzielczości chromatografię cieczową (HPLC), spektrometrię (np. spektrometria GC/masa, spektrometria HPLC/masa) i cieczową scyntylacyjną (w odniesieniu do substancji etykietowanych izotopami). Niezależnie od używanych metod analitycznych, jest uznane za odpowiednie, jeśli wykrywalność wynosi między 90 a 110 % wartości nominalnej. W celu umożliwienia wykrycia oraz oceny po zajściu podziału granice wykrywalności metody analitycznej powinny być co najmniej dwa rzędy wielkości, poniżej stężenia nominalnego.
Cechy i granice wykrywalności metody analitycznej dostępnej do przeprowadzenia badań adsorpcji mają istotną rolę w określaniu warunków badania i całego doświadczalnego przeprowadzenia badania. Niniejsza metoda wskazuje ogólną doświadczalną drogę, dostarcza zaleceń i wytycznych dotyczących rozwiązań alternatywnych, w przypadku gdy metoda analityczna i laboratoryjne środki mogą nakładać ograniczenia.
1.8.2. Wybór optymalnych proporcji gleba/roztwór
Wybór właściwych proporcji gleby do roztworu w badaniach sorpcji zależy od współczynnika podziału Kd oraz względnego stopnia pożądanej adsorpcji. Zmiana stężenia substancji w roztworze określa statystyczną dokładność pomiaru opartego na kształcie równania adsorpcji i granicy analitycznej metodologii w wykrywaniu stężenia substancji chemicznej w roztworze. Dlatego w praktyce ogólnej jest użyteczne ustalenie kilku proporcji, w odniesieniu do których procent adsorpcji wynosi powyżej 20 %, a zalecany > 50 % (62), ponieważ należy dążyć do utrzymania stężenia substancji badanej w wodnej fazie wystarczająco wysoko, aby mogło być ono zmierzone dokładnie. Jest to szczególnie istotne w przypadku wysokiego procentu adsorpcji.
Dogodne podejście do wyboru właściwej proporcji gleba/woda jest oparte na szacowaniu wartości Kd przez badania wstępne albo przez ustalone techniki szacowania (dodatek 3). Wybór właściwej proporcji dokonywany jest w oparciu o wykres proporcji gleba/roztwór w zależności od Kd w odniesieniu do ustalonego procenta adsorpcji (rys. 1). W tym wykresie przyjęto, że równanie adsorpcji jest liniowe (28). Stosowana zależność jest uzyskiwana przez przekształcenie równania (4) dla Kd do postaci równania (1):
|
|
(1) |
lub w jego postaci logarytmicznej, przyjmując ze R = msoil/V0 i Aeq %/100 =  :
:
|
|
(2) |

Rysunek 1 przedstawia proporcje gleba/roztwór jako funkcje Kd dla różnych poziomów adsorpcji. Na przykład przy proporcji gleba/roztwór 1:5 i Kd 20 zachodzi w przybliżeniu 80 % adsorpcji. Dla uzyskania 50 % adsorpcji dla tego samego Kd należy zastosować proporcję 1:25. Takie podejście do wyboru właściwych proporcji gleba/roztwór daje badaczowi elastyczność w spełnieniu doświadczalnych wymagań.
Dziedziny, które są trudniejsze do rozwiązania, są tymi, w których substancja chemiczna jest wysoce adsorbowana lub bardzo nieznacznie. W przypadku występowania niskiej adsorpcji stosunek 1:1 gleba/roztwór jest zalecany, chociaż w odniesieniu do niektórych bardzo organicznych typów gleb, konieczne są mniejsze proporcje do uzyskania zawiesiny. Analityczna metodologia do pomiaru małych zmian stężeń w roztworach musi być stosowana z ostrożnością; inaczej pomiar adsorpcji będzie niedokładny. Z drugiej strony, bardzo wysokie współczynniki podziału Kd, dochodzące do 1:100 stosunek gleba/roztwór w celu pozostawienia znaczącej ilości substancji w roztworze. Jednakże należy zwrócić uwagę na zapewnienie dobrego mieszania oraz odpowiedniego czasu na dojście systemu do równowagi. Alternatywnym podejściem do rozwiązania takich ekstremalnych przypadków, gdy brakuje stosownej metodologii analitycznej, jest przewidywanie wartości Kd w zastosowaniu technik oceny, opartych na przykład, o wartości Pow (dodatek 3). Może to być przydatne w szczególności w odniesieniu do nisko adsorbowalnych polarnych substancji chemicznych o Pow < 20 oraz dla lipofilowych, wysoce sorpcyjnych substancji o Pow > 104.
1.9. WYKONANIE BADANIA
1.9.1. Warunki badania
Wszystkie doświadczenia są wykonywane w temperaturze otoczenia oraz, jeśli to możliwe, w stałej temperaturze między 20 a 25 oC.
Warunki odwirowania powinny umożliwić usunięcie z roztworu cząstek większych niż 0,2 μm. Ta wartość oddziela cząstki najmniejszych rozmiarów, uważanych za cząstkę stałą i jest granicą między stałą i koloidalną cząstką. Wytyczne dotyczące sposobu określania warunków odwirowania podano w dodatku 4.
Jeśli udogodnienia w zakresie odwirowania nie mogą zapewnić usunięcia cząstek większych od 0,2 μm, stosowana może być kombinacja odwirowania oraz filtracji przez filtr 0,2 μm. Filtry te powinny być wykonane z odpowiedniego obojętnego materiału, zapobiegającego stratom na nim. W każdym przypadku należy udowodnić, że nie powstają na nim jakiekolwiek straty substancji badanej podczas trwania filtracji.
1.9.2. Warstwa 1 – Badania wstępne
Cel prowadzenia badań wstępnych podano już w punkcie dotyczącym zakresu. Wytyczne dotyczące określania takiego badania wraz z doświadczeniem sugerowanym podano poniżej.
1.9.2.1. Wybór optymalnych proporcji gleba/roztwór
Używa się dwa typy gleby i trzy proporcje gleba/roztwór (sześć doświadczeń). Jeden rodzaj gleby ma wysoką zawartość węgla organicznego i gliny, druga niską zawartość węgla organicznego i gliny. Sugerowane są następujące proporcje gleby do roztworu:
|
— |
50 g gleby i 50 cm3 roztworu wodnego substancji badanej (proporcja 1/1), |
|
— |
10 g gleby i 50 cm3 roztworu wodnego substancji badanej (proporcja 1/5), |
|
— |
2 g gleby i 50 cm3 roztworu wodnego substancji badanej (proporcja 1/25). |
Minimalna ilość gleby, przy której przeprowadza się doświadczenie, zależy od możliwości laboratorium i wykonania zastosowanych metod analitycznych. Jednakże jest zalecane użycie co najmniej 1 g, a najlepiej 2 g, w celu uzyskania wiarygodnych wyników badania.
Jedna próbka kontrolna jedynie z substancją badaną w roztworze 0,01 M CaCl2 (bez gleby) podlega dokładnie tym samym kolejnym czynnościom jak badane systemy, w celu sprawdzenia stabilności badanej substancji w roztworze CaCl2 i możliwej adsorpcji na powierzchni badanego zbiornika.
Ślepa próba gleby z taką samą ilością gleby i całkowitej objętości 50 cm3 roztworu 0,01 M CaCl2 (bez substancji badanej) podlega takiej samej procedurze badania. Służy to jako dodatkowa kontrola w czasie trwania analizy mającej na celu wykrycie zakłócających substancji lub zanieczyszczonych gleb.
Wszystkie doświadczenia, łącznie z próbami ślepymi i kontrolnymi, powinny być wykonane przynajmniej z duplikatem. Całkowita ilość próbek które powinny być przygotowane dla badań mogą być obliczone w odniesieniu do metodologii, która zastanie zastosowana.
Metody w zakresie badań wstępnych i badania główne są ogólnie takie same, wyjątki są wspomniane tam, gdzie to jest odpowiednie.
Wysuszone powietrzem próbki gleby są równoważone przez wytrząsanie minimalną objętością 45 cm30,01 M CaCl2 w ciągu nocy (12 godz.) przed dniem doświadczenia. Następnie dodaje się pewną ilość roztworu podstawowego substancji badanej w celu dostosowania końcowej objętości do 50 cm3. Objętość dodanego roztworu podstawowego: a) nie powinna przekroczyć 10 % końcowej objętości 50 cm3 fazy wodnej w celu zmiany, w możliwie najmniejszym zakresie charakteru roztworu przed równowagą; oraz b) powinna zdecydowanie wpływać na wyniki początkowego stężenia substancji badanej będącej w kontakcie z glebą (C0) co najmniej dwa rzędy wielkości więcej niż granica wykrywalności metody analitycznej; niniejszy próg zabezpiecza możliwość prowadzenia dokładnych pomiarów nawet gdy zachodzi silne adsorpcja (> 90 %) i określenia późniejszych izoterm adsorpcji. Zaleca się również, jeśli to możliwe, aby początkowe stężenie substancji (C0) nie przekraczało połowy jej granicy rozpuszczalności.
Przykład sposobu obliczenia stężenia roztworu podstawowego (Cst) podano poniżej. Przyjęto granicę wykrywalności 0,01 μg cm-3 i 90 % adsorpcji; zatem zalecane wstępne stężenie substancji badanej w kontakcie z glebą powinno wynosić l μg cm-3 (dwa rzędy wielkości wyższe niż granica wykrywalności). O ile dodano zalecaną maksymalną objętość roztworu podstawowego, tj 5–45 cm30,01 M CaCl2 roztworowa równowaga (= 10 % roztworu podstawowego do 50 cm3 całkowitej objętości fazy wodnej), stężenie roztworu podstawowego powinno wynosić 10 μg cm-3; to jest trzy rzędy wielkości wyższej niż granica wykrywalności metody analitycznej.
Wartość pH fazy wodnej powinna być mierzona przed i po kontakcie z glebą, ponieważ ma to istotne znaczenie w całkowitym procesie adsorpcji, w szczególności w odniesieniu do jonizowalnych substancji.
Mieszanina jest wytrząsana do osiągnięcia równowagi adsorpcji. Czas równowagi w glebach jest wysoce zmienny w zależności od substancji chemicznej i gleby; okres 24 godzin jest ogólnie wystarczający (77). W badaniach wstępnych próbki mogą być zbierane sekwencyjnie przez 48 godzin okresu mieszania (np. w 4, 8, 24, 48 godzin). Jednakże czas analizy powinien być określony elastycznie w odniesieniu do rozkładu pracy laboratorium.
Istnieją dwie możliwości w odniesieniu do analizy substancji badanej w roztworze wodnym: a) metoda równoległa; i b) metoda seryjna. Należy pokreślić, że chociaż metoda równoległa jest doświadczalnie bardziej uciążliwa, to matematyczne opracowanie wyników jest prostsze (dodatek 5). Jednakże wybór metodologii która ma być zastosowana pozostawiona jest eksperymentatorowi, który musi rozważyć dostępne urządzania i zasoby laboratorium.
|
a) |
Metoda równoległa: przygotowuje się próbki w tej samej proporcji gleba/roztwór, w liczbie zależnej od tego w ilu przedziałach czasowych chce się zbadać kinetykę adsorpcji. Po odwirowaniu i jeśli jest to pożądane, po filtracji faza wodna z pierwszej probówki jest usuwana możliwie całkowicie i mierzona po, na przykład 4 godzinach, druga probówka po 8 godzinach, trzecia po 24 godzinach itd. |
|
b) |
Metoda seryjna: przygotowuje się tylko kopie próbek odniesieniu do każdej proporcji gleba/roztwór. W określonych przedziałach czasu mieszanina jest wirowana w celu rozdzielenia faz. Mała podwielokrotność fazy wodnej jest niezwłocznie analizowana w odniesieniu do substancji badanej; następnie doświadczenie prowadzi się nadal z oryginalną mieszaniną. Jeśli zastosowano filtrację po odwirowaniu, laboratorium powinno posiadać urządzenia umożliwiające przeprowadzenie filtracji małych wodnych podwielokrotności. Zalecane jest, aby całkowite podwielokrotności pobrane nie przekraczały 1 % całkowitej objętości roztworu, w celu nie powodowania znaczących zmian proporcji gleba/roztwór i zmniejszenia masy rozpuszczonej zdolnej do adsorpcji w czasie trwania badania. |
Procentowa adsorpcja  jest obliczana w każdym punkcie czasu (ti) na podstawie nominalnego początkowego stężenia i zmierzonego stężenia w chwili pobierania próbek (ti), skorygowana w odniesieniu do wartości ślepej próby. Wykresy
jest obliczana w każdym punkcie czasu (ti) na podstawie nominalnego początkowego stężenia i zmierzonego stężenia w chwili pobierania próbek (ti), skorygowana w odniesieniu do wartości ślepej próby. Wykresy  w zależności od czasu (rys. 1 dodatek 5) są generowane w celu oszacowania osiągnięcia plateau równowagowego (29). Wartość Kd w równowadze jest również obliczana. W oparciu o tę wartość Kd, właściwe proporcje gleba/roztwór są wybierane z rys. l, tak aby procent adsorpcji osiągał powyżej 20 %, a zalecane > 50 % (61). Wszystkie mające zastosowanie równania i zasady wykreślania podano w punkcie „Dane i sprawozdawczość” oraz w dodatku 5.
w zależności od czasu (rys. 1 dodatek 5) są generowane w celu oszacowania osiągnięcia plateau równowagowego (29). Wartość Kd w równowadze jest również obliczana. W oparciu o tę wartość Kd, właściwe proporcje gleba/roztwór są wybierane z rys. l, tak aby procent adsorpcji osiągał powyżej 20 %, a zalecane > 50 % (61). Wszystkie mające zastosowanie równania i zasady wykreślania podano w punkcie „Dane i sprawozdawczość” oraz w dodatku 5.
1.9.2.2. Określenie czasu równowagi adsorpcji i ilości badanej substancji zaadsorbowanej w równowadze
Jak już wspomniano, wykresy  lub
lub 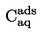 w zależności od czasu pozwalają oszacować osiągnięcie równowagi adsorpcji i ilości badanej substancji zaadsorbowanej w równowadze. Rysunki 1 i 2 w dodatku 5 wskazują przykłady takich wykresów. Czas równowagi jest czasem potrzebnym systemowi do osiągnięcia plateau.
w zależności od czasu pozwalają oszacować osiągnięcie równowagi adsorpcji i ilości badanej substancji zaadsorbowanej w równowadze. Rysunki 1 i 2 w dodatku 5 wskazują przykłady takich wykresów. Czas równowagi jest czasem potrzebnym systemowi do osiągnięcia plateau.
Jeśli przy określonej glebie nie występuje plateau, tylko stały wzrost, może być to spowodowane czynnikami zakłócającymi, takimi jak biodegradacja lub powolne rozproszenie. Biodegradacja może być wykazana przez powtórzenie doświadczenia ze sterylizowaną próbką gleby. Jeśli nie uzyskano plateau nawet w tym przypadku, eksperymentator powinien poszukiwać innych zjawisk, które mogły wpłynąć na to szczególne badanie; może to być dokonane z właściwymi zmianami warunków doświadczenia (temperatura, czas wytrząsania, proporcje gleba/roztwór). Pozostawia się do uznania ekperymentatora, czy stosować nadal procedurę badania pomimo możliwosci nieuzyskania równowagi.
1.9.2.3. Adsorpcja na powierzchni zbiornika badanego i stabilność substancji użytej w badaniu
Niektóre informacje dotyczące adsorpcji substancji badanej na powierzchni badanego zbiornika, jak również dotyczące jej stabilności są wyprowadzane poprzez analizowanie próbek kontrolnych. Jeżeli obserwuje się uszczuplenie większe niż standardowy błąd metody analitycznej, może to świadczyć o abiotycznej degradacji i/lub adsorpcji na powierzchni badanego zbiornika. Rozróżnienie między tymi dwoma zjawiskami może być uzyskane przez gruntowne mycie ścian zbiornika znaną ilością właściwego rozpuszczalnika oraz przez poddanie roztworu z mycia analizie w odniesieniu do substancji badanej. Jeśli nie obserwuje się adsorpcji na powierzchni zbiornika badanego, uszczuplenie oznacza abiotyczną niestabilność substancji badanej. Jeśli wykryto adsorpcję, konieczna jest zmiana materiału zbiornika badanego. Jednakże danych dotyczących adsorpcji na powierzchni badanego zbiornika, uzyskanych w wyniku tego doświadczenia nie można bezpośrednio ekstrapolować do doświadczenia gleba/roztwór. Obecność gleby oddziaływuje na tę adsorpcję.
Dodatkowe informacje dotyczące stabilności substancji badanej mogą być uzyskane przez określenie macierzystego bilansu masy w czasie. Oznacza to, że faza wodna, ekstrakt z gleby i ścianki zbiornika badanego są analizowane w odniesieniu do substancji badanej. Różnica między masą badanych substancji chemicznych dodanych a sumą mas badanych substancji chemicznych w fazie wodnej, ekstraktów z gleby i ścianek zbiornika badanego jest równa masie zdegradowanej i/lub ulotnionej i/lub nieekstrahowanej. W celu dokonania określenia bilansu masy równowaga adsorpcyjna powinna być osiągnięta w czasie doświadczenia.
Bilans masy wykonuje się na obu glebach i w odniesieniu do jednej proporcji gleba/roztwór na glebę, co daje uszczuplenie ponad 20 % i zalecane > 50 % w równowadze. Po zakończeniu doświadczenia znalezienia proporcji z analizą ostatniej próbki wodnej fazy po 48 godzinach, fazy są rozdzielane przez odwirowanie i, jeśli jest to pożądane, filtrowane. Faza wodna jest w najszerszym możliwym zakresie odzyskiwana oraz dodany jest odpowiedni rozpuszczalnik ekstrakcyjny (współczynnik ekstrakcji co najmniej 95 %) do gleby, aby wyekstrahować substancję badaną. Zaleca się co najmniej dwie kolejne ekstrakcje. Ilość substancji badanej w ekstrakcie z gleby i zbiornika badanego jest określana, a bilans masy oblicza się (równanie 10, Dane i sprawozdawczość). Jeżeli jest mniejszy niż 90 %, substancję badaną uznaje się za niestabilną w skali czasu badania. Jednakże badania mogą nadal być prowadzone, uwzględniając niestabilność substancji badanej, w tym przypadku zalecane jest analizowanie obu faz w badaniu głównym.
1.9.2.4. Warstwa 2 – Kinetyka adsorpcji przy jednym stężeniu substancji badanej
Stosuje się pięć typów gleb, wybranych z tabeli 1. Zaletą jest, że można włączyć niektóre lub wszystkie gleby użyte w badaniach wstępnych, jeśli to stosowne, spośród tych pięciu gleb. W tym przypadku warstwa 2 nie zostaje powtórzona w odniesieniu do gleb użytych w badaniach wstępnych.
Czas równowagi, stosunek gleba/roztwór, waga próbki gleby, objętość fazy wodnej w kontakcie z glebą oraz stężenie substancji badanej w roztworze są wybierane w oparciu o wyniki badań wstępnych. Zalecane jest, aby analiza została wykonana po około 2, 4, 6, 8 (możliwie także 10) i 24 godz. czasu kontaktu; czas mieszania może być przedłużony do maksimum 48 godz. w przypadku substancji chemicznej wymagającej dłuższego czasu równowagi w odniesieniu do wyników poszukiwania proporcji. Jednakże czas analizy należy rozważyć elastycznie.
Każde doświadczenie (jedna gleba i jeden roztwór) jest co najmniej raz kopiowane, aby umożliwić oszacowanie odchylenia wyników. W każde doświadczenie włączona jest ślepa próba. Obejmuje to glebę i roztwór 0,01 M CaCl2, bez substancji badanej, odpowiednio wagę oraz objętość identyczne do tych z doświadczenia. Kontrolna próbka tylko z substancją badaną w roztworze 0,01 M CaCl2 (bez gleby) podlega tej samej procedurze badania, służąc jako zabezpieczenie przed nieoczekiwanym.
Procent adsorpcji jest obliczany w każdym punkcie czasu  i/lub przedziale czasowym
i/lub przedziale czasowym  (wedle potrzeby) i jest wykreślany w zależności od czasu. Współczynnik podziału Kd w równowadze, jak również znormalizowany współczynnik adsorpcji węgla organicznego Koc (w odniesieniu do niepolarnych substancji chemicznych organicznych) są również obliczane.
(wedle potrzeby) i jest wykreślany w zależności od czasu. Współczynnik podziału Kd w równowadze, jak również znormalizowany współczynnik adsorpcji węgla organicznego Koc (w odniesieniu do niepolarnych substancji chemicznych organicznych) są również obliczane.
Wyniki badania adsorpcji kinetycznej
Liniowa wartość Kd jest zasadniczo dokładna aby opisać zachowanie sorpcyjne w glebie (35)(78) i przedstawia wyrażenie mobilności właściwej substancji chemicznych w glebie. Na przykład ogólnie w substancjach chemicznych o Kd < 1 cm3 g-1 są uznawane za jakościowo mobilne. Podobnie, schemat klasyfikacji mobilności oparty o wartości Koc został rozwinięty przez MacCall et al. (16). Dodatkowo, schematy klasyfikacji wypłukiwania istnieją w oparciu o zależność między Koc a DT-50 (30) (32)(79).
Również, zgodnie z badaniami analizy błędów (61), wartości Kd poniżej 0,3 cm3 g-1 nie można oszacować dokładnie z obniżenia stężenia w fazie wodnej, nawet jeżeli zastosowano najbardziej korzystny (z punktu widzenia dokładności) stosunek gleba/roztwór, tj. 1:1. Zaleca się w tym przypadku analizę obu faz, gleby i roztworu.
W odniesieniu do powyższych uwag jest zalecane, aby badania zachowania adsorpcyjnego substancji chemicznych w glebie i ich potencjalna mobilność były nadal prowadzone przez oznaczanie izoterm adsorpcji Freundlicha dla tych systemów, dla których możliwe jest oznaczenie Kd, wraz z protokołem doświadczalnym zastosowanym w niniejszej metodzie badania. Dokładne oznaczenie jest możliwe jeżeli wartość, która wynika z pomnożenia Kd i proporcji gleba/roztwór jest > 0,3, gdy pomiary są oparte na spadku stężenia w wodnej fazie (metoda pośrednia), lub > 0,1, gdy analizowane są obie fazy (metoda bezpośrednia) (61).
1.9.2.5. Warstwa 3 – Izotermy adsorpcji i kinetyka desorpcji/izotermy desorpcji
1.9.2.5.1.
Używa się pięć stężeń substancji badanej, obejmujące najlepiej dwa rzędy wielkości; przy wyborze tych stężeń powinny być uwzględnione rozpuszczalność w wodzie i wynikła stężeniowa równowaga wodną. W trakcie badań musi być zachowany ten sam stosunek gleba/roztwór. Badanie adsorpcji jest prowadzone, jak opisano powyżej, z tą tylko różnicą, że faza wodna jest analizowana tylko raz w czasie niezbędnym do osiągnięcia równowagi, jak ustalono wcześniej w warstwie 2. Stężenia równowagowe w roztworze są określone, a ilość zaadsorbowana jest obliczona z uszczuplenia substancji badanej w roztworze lub metodą bezpośrednią. Zaadsorbowana masa na jednostkę masy gleby jest wykreślana jako funkcja stężenia równowagi badanej substancji (zob. Dane i sprawozdawczość).
Wyniki z doświadczenia izoterm adsorpcji
Z matematycznych modeli adsorpcji dotąd proponowanych, izoterma Freundlicha jest jedną z najczęściej stosowanych do opisu procesów adsorpcji. Bardziej szczegółowe informacje dotyczące interpretacji i ważności modelu adsorpcji są określone w bibliografii (41)(45)(80)(81)(82).
Uwaga: Należy wspomnieć, że porównanie wartości KF (współczynnik adsorpcji Freundlicha) w odniesieniu do różnych substancji jest tylko możliwe, gdy te wartości KF są wyrażone w tych samych jednostkach (83).
1.9.2.5.2.
Celem tego doświadczenia jest zbadanie, czy substancja chemiczna jest odwracalnie czy nieodwracalnie adsorbowana w glebie. Niniejsza informacja jest istotna ponieważ proces desorpcji również ma istotne znaczenie w zachowaniu się substancji chemicznych w glebie pól. Ponadto dane w zakresie desorpcji są użyteczne jako dane wejściowe w komputerowym modelowaniu symulacji przebiegu rozpuszczania i płukania. Jeśli badania desorpcji są pożądane, jest zalecane, aby badania opisane poniżej były prowadzone na każdym systemie, dla którego dokładne oznaczenie Kd w poprzedzającym doświadczenie kinetyki adsorpcji było możliwe.
Podobnie jak w badaniach kinetyki adsorpcji, istnieją dwie możliwości wykonania doświadczenia kinetyki desorpcji: a) metoda równoległa; i b) metoda seryjna. Wybór zastosowanej metodologii pozostawia się uznaniu eksperymentatora, który musi uwzględnić dostępne urządzenia i zasoby laboratorium.
|
a) |
Metoda równoległa: w odniesieniu do każdej gleby wybranej do przeprowadzenia badań desorpcji, przygotowuje się próbki o tym samym proporcji gleba/roztwór, w liczbie zależnej od tego w ilu przedziałach czasowych chce się zbadać kinetyki desorpcji. Powinno się użyć tych samych przedziałów czasowych jak w doświadczeniu kinetyki adsorpcji; jednakże całkowity czas może być przedłużony, jeśli to właściwe, w celu osiągnięcia przez system równowagi desorpcji. W każdym doświadczeniu (jedna gleba, jeden roztwór) włącza się ślepą próbę. Obejmuje to glebę i roztwór 0,01 M CaCl2, bez substancji badanej, odpowiednio naważkę oraz objętość identyczne do tych z doświadczenia. Kontrolna próbka tylko z substancją badaną w roztworze 0,01 M CaCl2 (bez gleby) podlega tej samej procedurze badania. Wszystkie mieszaniny gleby z roztworem są mieszane do osiągnięcia równowagi adsorpcji (jak wcześniej określono w warstwie 2). Następnie fazy są rozdzielane przez odwirowanie, a fazy wodne są usuwane w najszerszym możliwym zakresie. Objętość oddzielonego roztworu jest zastępowana przez równą objętość 0,01 M CaCl2 bez substancji badanej, a nowe mieszaniny są ponownie mieszane. Faza wodna z pierwszej probówki jest usuwana całkowicie, jak to możliwe i mierzona po, na przykład 2 godz., druga probówka po 4 godzinach, trzecia po 6 godzinach itd. do osiągnięcia równowagi desorpcji. |
|
b) |
Metoda seryjna: po doświadczeniu kinetyki adsorpcji, mieszanina jest odwirowana i faza wodna jest usuwana w najszerszym możliwym zakresie. Objętość roztworu usuniętego jest zastępowana przez równą objętość 0,01 M CaCl2 bez substancji badanej. Nowa mieszanina jest mieszana do osiągnięcia równowagi desorpcji. W trakcie tego okresu czasu, w określonych przedziałach czasowych, mieszanina jest odwirowywana w celu rozdzielenia faz.. Mała podwielokrotność fazy wodnej jest niezwłocznie analizowana w odniesieniu do substancji badanej; następnie doświadczenie prowadzi się nadal z pierwotną mieszaniną. Objętość każdej poszczególnej podwielokrotności powinna być mniejsza niż 1 % całkowitej objetości. Taka sama ilość świeżego roztworu 0,01 M CaCl2 jest dodawana do mieszaniny w celu utrzymania proporcji gleby do roztworu a mieszanie jest kontynuowane do następnego przedziału czasowego. |
Procent desorpcji jest obliczany w każdym punkcie czasu (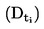 ) i/lub przedziale czasowym (
) i/lub przedziale czasowym ( ) (wedle potrzeb badania) i jest wykreślana w zależności od czasu. Współczynnik desorpcji Kdes w równowadze jest także obliczany. Wszystkie mające zastosowanie równania podano w dodatku 5 i „Dane i sprawozdawczość”.
) (wedle potrzeb badania) i jest wykreślana w zależności od czasu. Współczynnik desorpcji Kdes w równowadze jest także obliczany. Wszystkie mające zastosowanie równania podano w dodatku 5 i „Dane i sprawozdawczość”.
Wyniki z doświadczenia kinetyki desorpcji
Wspólne wykreślenie procentu desorpcji  i adsorpcji
i adsorpcji  w zależności od czasu umożliwia oszacowanie odwracalności procesu adsorpcji. Jeśli równowaga desorpcji jest osiągana nawet w obrębie podwojonego czasu równowagi adsorpcji, a całkowita desorpcja jest większa niż 75 % ilości adsorbowanej, adsorpcja jest uważana za odwracalną.
w zależności od czasu umożliwia oszacowanie odwracalności procesu adsorpcji. Jeśli równowaga desorpcji jest osiągana nawet w obrębie podwojonego czasu równowagi adsorpcji, a całkowita desorpcja jest większa niż 75 % ilości adsorbowanej, adsorpcja jest uważana za odwracalną.
1.9.2.5.3.
Izotermy desorpcji Freundlicha są określone na glebach używanych w doświadczeniu izoterm adsorpcji. Badanie desorpcji jest prowadzone jak opisano w punkcie „Kinetyka desorpcji”, z tą tylko różnicą, że faza wodna analizowana jest tylko raz, w równowadze desorpcji. Ilość substancji badanej zdesorbowanej jest obliczana. Zawartość substancji badanej pozostającej zaadsorbowaną na glebie w równowadze desorpcji jest wykreślana jako funkcja stężenia równowagi substancji badanej w roztworze.(zob. Dane i sprawozdawczość oraz dodatek 5).
2. DANE I SPRAWOZDAWCZOŚĆ
Dane analityczne są przedstawione w formie tabelarycznej (zob. dodatek 6). Podano poszczególne pomiary i obliczone średnie. Pokazano graficzne przedstawienie izoterm adsorpcji. Dokonanie obliczeń opisano poniżej.
Do celów badania uznaje się, że waga 1 cm3 roztworu wodnego wynosi 1 g. Stosunek gleba/roztwór wyrażony w jednostkach wag./wag. lub wag./obj. jest tą samą liczbą.
2.1. ADSORPCJA
Adsorpcja (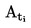 ) jest określona jako procent substancji zaadsorbowanej na glebie w proporcji do ilości na początku badania w warunkach badania. Jeśli substancja badana jest stabilna i nie adsorbuje się w znacznym stopniu na ściankach zbiornika,
) jest określona jako procent substancji zaadsorbowanej na glebie w proporcji do ilości na początku badania w warunkach badania. Jeśli substancja badana jest stabilna i nie adsorbuje się w znacznym stopniu na ściankach zbiornika, 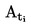 oblicza się w każdym punkcie czasu ti, zgodnie z równaniem:
oblicza się w każdym punkcie czasu ti, zgodnie z równaniem:
|
|
(3) |
gdzie:
|
|
= |
procent adsorpcji w punkcie czasu ti ( %), |
|
|
= |
masa substancji badanej zaadsorbowanej na glebie w czasie ti (μg), |
|
m0 |
= |
masa substancji badanej w probówce badanej, na początku badania (μg). |
Szczegółowe informacje dotyczące sposobu obliczenia procenta adsorpcji 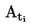 dla metody równoległej i seryjnej podano w dodatku 5.
dla metody równoległej i seryjnej podano w dodatku 5.
Współczynnik podziału Kd jest proporcją między zawartością substancji w fazie gleby i masowym stężeniem substancji w roztworze wodnym, w warunkach badania, gdy osiągnięta jest równowaga adsorpcji.
|
|
(4) |
gdzie:
|
|
= |
zawartość substancji zaadsorbowana na glebie w równowadze adsorpcji (μg g-1), |
|
|
= |
stężenie masowe substancji w fazie wodnej w równowadze adsorpcji (μg cm-3). To stężenie jest oznaczone analitycznie, uwzględniając wartości dane w ślepej próbie, |
|
|
= |
masa substancji zaadsorbowana na glebie w równowadze adsorpcji (μg), |
|
|
= |
masa substancji w roztworze w równowadze adsorpcji (μg), |
|
msoil |
= |
ilość fazy gleby, wyrażona w suchej masie gleby (g), |
|
V0 |
= |
początkowa objętość fazy wodnej w kontakcie z glebą (cm3). |
Zależność między Aeq i Kd jest określona przez:
|
|
(5) |
gdzie:
|
Aeq |
= |
procent adsorpcji w równowadze adsorpcji, %. |
Znormalizowany współczynnik adsorpcji węgla organicznego Koc wiąże współczynnik podziału Kd z zawartością węgla organicznego próbki gleby:
|
|
(6) |
gdzie:
|
% OC |
= |
procent węgla organicznego w próbce gleby (g g-1). |
Współczynnik Koc przedstawia pojedynczą wartość, która charakteryzuje podzielenie głównie niepolarnych substancji organicznych między węglem organicznym w glebie lub osadzie i wodzie. Adsorpcja tych substancji chemicznych jest skorelowana z zawartością związków organicznych sorbującego ciała stałego (7); zatem wartości Koc zależą od właściwych cech próchnicowych frakcji, które różnią się znacznie zdolnością sorpcji z powodu różnic w pochodzeniu, genezie itd.
2.1.1. Izotermy adsorpcji
Równanie izoterm Freundlicha wiąże ilość substancji badanej zaadsorbowanej ze stężeniem substancji badanej w roztworze w równowadze (równanie 8).
Dane są opracowywane jak w „Adsorpcja” i, w odniesieniu do każdej probówki badanej, jest obliczana zawartość substancji badanej zaadsorbowanej na glebie po badaniu adsorpcji ( , gdzie indziej oznaczane jako as x/m). Przyjmując, że równowaga została osiągnięta i że
, gdzie indziej oznaczane jako as x/m). Przyjmując, że równowaga została osiągnięta i że  przedstawia wartość równowagi:
przedstawia wartość równowagi:
|
|
(7) |
Równanie adsorpcji Freundlicha jest wskazane w (8):
|
|
(8) |
lub w postaci liniowej:
|
|
(9) |
gdzie:
|
|
= |
współczynnik adsorpcji Freundlicha; jego wymiarem jest cm3 g-1 tylko gdy l/n = 1; we wszystkich innych przypadkach nachylenie l/n jest wprowadzone w wymiar |
|
n |
= |
stała regresji; l/n ogólnego zakresu między 0,7–1,0, wskazując, że dane sorpcji są często lekko nieliniowe. |
Równania (8) i (9) są wykreślane i wartości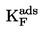 i l/n są wyliczane analizą regresyjną, używając równanie 9. Współczynnik korelacji r2 równania logarytmicznego jest również wyliczany. Przykład takich wykresów podano na rys. 2.
i l/n są wyliczane analizą regresyjną, używając równanie 9. Współczynnik korelacji r2 równania logarytmicznego jest również wyliczany. Przykład takich wykresów podano na rys. 2.
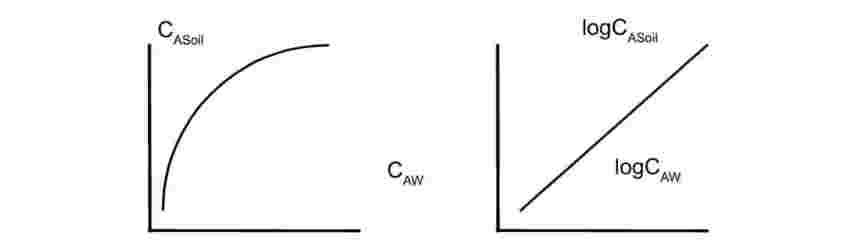
2.1.2. Bilans masy
Bilans masy (MB) określa się jako procent substancji, który może być analitycznie odzyskany po badaniu adsorpcji w zależności od nominalnej ilości substancji na początku badania.
Opracowywanie danych rozróżni, czy rozpuszczalnik jest całkowicie mieszalny z wodą. W przypadku rozpuszczalników mieszalnych z wodą opracowanie danych opisanych pod „Desorpcja” może być stosowane do określenia ilości substancji odzyskanej przez ekstrakcję rozpuszczalnikiem. Jeśli rozpuszczalnik jest słabo mieszalny z wodą, określenie odzyskanej ilości musi być dokonane.
Bilans masy MB w odniesieniu do adsorpcji jest obliczany następująco; przyjmuje się, że pojęcie (mE) odpowiada sumie badanych mas substancji chemicznych wyekstrahowanych z gleby i powierzchni zbiornika badanego, rozpuszczalnikiem organicznym:
|
|
(10) |
gdzie:
|
MB |
= |
bilans masy ( %), |
|
mE |
= |
całkowita masa substancji badanej wyekstrahowanej z gleby i ścian zbiornika badanego w dwóch krokach (μg), |
|
C0 |
= |
początkowe stężenie masowe roztworu badanego w kontakcie z glebą (μg cm-3), |
|
Vrec |
= |
objętość nadsączu odzyskanego po równowadze adsorpcyjnej (cm3). |
2.2. DESORPCJA
Desorpcję (D) określa się jako procent substancji badanej zdesorbowanej w zależności od ilości substancji uprzednio zaadsorbowanej, w warunkach badania:
|
|
(11) |
gdzie:
|
|
= |
procent desorpcji w punkcie czasu ti ( %), |
|
|
= |
masa substancji badanej zdesorbowanej z gleby w punkcie czasu ti (μg), |
|
|
= |
masa substancji badanej zaadsorbowanej na glebie w równowadze adsorpcji (μg). |
Szczegółowe informacje dotyczące sposobu obliczenia procentu desorpcji  dla metody równoległej i seryjnej podano w dodatku 5.
dla metody równoległej i seryjnej podano w dodatku 5.
Pozorny współczynnik desorpcji (Kdes) jest, w warunkach badania, proporcją między zawartością substancji pozostającą w fazie gleby a stężeniem masowym substancji desorbowanej w roztworze wodnym, gdy osiągnięta jest równowaga desorpcji:
|
|
(12) |
gdzie:
|
Kdes |
= |
współczynnik desorpcji (cm3 g-1), |
|
|
= |
całkowita masa substancji badanej zdesorbowanej z gleby w równowadze desorpcji (μg), |
|
VT |
= |
całkowita objętość fazy wodnej w kontakcie z glebą w czasie trwania badania kinetyki desorpcji (cm3). |
Wytyczne dotyczące obliczania 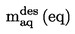 podano w dodatku 5 pod pozycją „Desorpcja”.
podano w dodatku 5 pod pozycją „Desorpcja”.
Uwaga:
Jeśli badanie adsorpcji, które było poprzedzające było wykonane metodą równoległą objętość VT w równaniu 12 uznaje się za równe V0.
2.2.1. Izotermy desorpcji
Równanie izoterm desorpcji Freundlicha wiąże zawartość substancji badanej pozostającej zaadsorbowanej na glebie, ze stężeniem substancji badanej w roztworze w równowadze desorpcji (równanie 16).
W odniesieniu do każdej probówki badanej, zawartość substancji pozostającej zaadsorbowaną na glebie, w równowadze desorpcji, jest obliczana następująco:
|
|
(13) |
 jest określone jako:
jest określone jako:
|
|
(14) |
gdzie:
|
|
= |
zawartość substancji badanej pozostającej zaadsorbowaną na glebie w równowadze desorpcji (μg g-1), |
|
|
= |
masa substancji określona analitycznie w fazie wodnej w równowadze desorpcji (μg), |
|
|
= |
masa substancji badanej pozostałej z równowagi adsorpcji z powodu niepełnego podstawienia objętości (μg), |
|
|
= |
masa substancji w roztworze w równowadze adsorpcji (μg); |
|
|
(15) |
|
|
= |
objętość roztworu pobrana z probówki do pomiaru substancji badanej, w równowadze desorpcji (cm3), |
|
Vr |
= |
objętość nadsączu usuniętego z probówki po uzyskaniu równowagi adsorpcji i zastąpieniu przez tę samą objętość roztworu 0,01 M CaCl2 (cm3). |
Równanie desorpcji Freundlicha znajduje się w (16):
|
|
(16) |
lub w postaci liniowej:
|
|
(17) |
gdzie:
|
|
= |
współczynnik desorpcji Freundlicha, |
|
n |
= |
stała regresji, |
|
|
= |
stężenie masowe substancji w fazie wodnej w równowadze desorpcji (μg cm-3). |
Równania 16 i 17 mogą być wykreślone, a wartości  i l/n są obliczane za pomocą analizy regresyjnej, stosując równanie 17.
i l/n są obliczane za pomocą analizy regresyjnej, stosując równanie 17.
Uwaga:
Jeśli wykładnik l/n adsorpcji lub desorpcji Freundlicha jest równy 1, stałe wiążące adsorpcję lub desorpcję Freundlicha (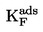 i
i  ), będą równe odpowiednio stałym równowagi adsorpcji lub desorpcji (Kd i Kdes), a wykresy Cs vs Caq będą liniowe. Jeśli wykładniki nie są równe 1, krzywe Cs vs Caq będą nieliniowe, a stałe adsorpcji i desorpcji będą się zmieniać wzdłuż izoterm.
), będą równe odpowiednio stałym równowagi adsorpcji lub desorpcji (Kd i Kdes), a wykresy Cs vs Caq będą liniowe. Jeśli wykładniki nie są równe 1, krzywe Cs vs Caq będą nieliniowe, a stałe adsorpcji i desorpcji będą się zmieniać wzdłuż izoterm.
2.2.2. Sprawozdanie z badania
Sprawozdanie z badania musi zawierać następujące informacje:
|
— |
Pełna identyfikacja użytych próbek gleby, włączając: |
|
— |
geograficzne odniesienie miejsca (szerokość geograficzna, długość geograficzna), |
|
— |
data pobierania próbek, |
|
— |
użyty rodzaj (np. gleba rolnicza, leśna itd.), |
|
— |
głębokość pobierania próbek, |
|
— |
zawartość piasek/mul/glina, |
|
— |
wartości pH (w 0,01 M CaCl2), |
|
— |
zawartość węgla organicznego, |
|
— |
zawartość substancji organicznych, |
|
— |
zawartość azotu, |
|
— |
stosunek C/N, |
|
— |
zdolność wymiany kationu (mmol/kg), |
|
— |
wszystkie informacje odnoszące się do zbierania i przechowywania próbek, |
|
— |
tam gdzie to jest stosowne, wszystkie istotne informacje dotyczące interpretacji adsorpcja/desorpcja substancji badanej, |
|
— |
bibliografia dla metod zastosowanych dla oznaczenia każdego parametru, |
|
— |
informacje dotyczące substancji badanej, jeśli to stosowne, |
|
— |
temperatura doświadczeń, |
|
— |
warunki odwirowania, |
|
— |
procedura analityczna zastosowana do analizy substancji badanej, |
|
— |
uzasadnienie w odniesieniu do każdego użycia środka ułatwiającego rozpuszczanie do przygotowania roztworu właściwego substancji badanej, |
|
— |
wyjaśnienie korekt wykonanych w obliczeniach, jeśli to odpowiednie, |
|
— |
dane zgodnie z formularzem (dodatek 6) wraz z prezentacją graficzną, |
|
— |
wszystkie informacje i obserwacje pomocne dla interpretacji wyników badania. |
3. BIBLIOGRAFIA
|
(1) |
Kukowski H. and Brümmer G., (1987). Investigations on the Adsorption and Desorption of Selected Chemicals in Soils. UBA Report 106 02 045, Part II. |
|
(2) |
Franzle O., Kuhnt G. and Vetter L., (1987). Selection of Representative Soils in the EC-Territory. UBA Report 106 02 045, Part I. |
|
(3) |
Kuhnt G. and Muntau H. (Eds.) EURO-Soils: Identification, Collection, Treatment, Characterisation. Special Publication No 1.94.60, Joint Research Centre. European Commission, ISPRA, December 1994. |
|
(4) |
OECD Test Guidelines Programme, Final Report of the OECD Workshop on Selection of Soils/Sediments, Belgirate, Italy, 18–20 January 1995 (June 1995). |
|
(5) |
US Environment Protection Agency: Pesticide Assessment Guidelines, Subdivision N, Chemistry: Environmental Fate, Series 163-1, Leaching and Adsorption/Desorption Studies, Addendum 6 on Data Reporting, 540/09-88-096, Date: 1/1988. |
|
(6) |
US Environment Protection Agency: Prevention, Pesticides and Toxic Substances, OPPTS Harmonized Test Guidelines, Series 835-Fate, Transport and Transformation Test Guidelines, OPPTS No: 835.1220 Sediment and Soil Adsorption/Desorption Isotherm. EPA No: 712-C-96-048, April 1996. |
|
(7) |
ASTM Standards, E 1195-85, Standard Test Method for Determining a Sorption Constant (Koc) for an Organic Chemical in Soil and Sediments. |
|
(8) |
Agriculture Canada: Environmental Chemistry and Fate. Guidelines for registration of pesticides in Canada, 15 July 1987. |
|
(9) |
Netherlands Commission Registration Pesticides (1995): Application for registration of a pesticide. Section G. Behaviour of the product and its metabolites in soil, water and air. |
|
(10) |
Danish National Agency of Environmental Protection (October 1988): Criteria for registration of pesticides as especially dangerous to health or especially harmful to the environment. |
|
(11) |
BBA (1990), Guidelines for the Official Testing of Plant Protection Products, Biological Research Centre for Agriculture and Forestry, Braunschweig, Germany. |
|
(12) |
Calvet R., (1989), „Evaluation of adsorption coefficients and the prediction of the mobilities of pesticides in soils”, in Methodological Aspects of the Study of Pesticide Behaviour in Soil (ed. P. Jamet), INRA, Paris, (Review). |
|
(13) |
Calvet R., (1980), „Adsorption-Desorption Phenomena” in Interactions between herbicides and the soil. (R. J. Hance ed.), Academic Press, London, pp. 83–122. |
|
(14) |
Hasset J. J., and Banwart W.L., (1989), „The sorption of nonpolar organics by soils and sediments” in Reactions and Movement of Organic Chemicals in Soils. Soil Science Society of America (S.S.S.A), Special Publication no. 22, pp. 31–44. |
|
(15) |
van Genuchten M. Th., Davidson J. M., and Wierenga P. J., (1974), „An evaluation of kinetic and equilibrium equations for the prediction of pesticide movement through porous media”. Soil Sci. Soc. Am. Proc, Vol. 38(1), pp. 29–35. |
|
(16) |
McCall P. J., Laskowski D. A., Swann R. L., and Dishburger H. J., (1981), „Measurement of sorption coefficients of organic chemicals and their use, in environmental fate analysis”, in Test Protocols for Environmental Fate and Movement of Toxicants. Proceedings of AOAC Symposium, AOAC, Washington DC. |
|
(17) |
Lambert S. M., Porter P. E., and Schieferrstein R. H., (1965), „Movement and sorption of chemicals applied to the soil”. Weeds, 13, pp. 185–190. |
|
(18) |
Rhodes R. C, Belasco I. J., and Pease H. L., (1970) „Determination of mobility and adsorption of agrochemicals in soils”. J.Agric.Food Chem., 18, pp. 524–528. |
|
(19) |
Russell M. H., (1995), „Recommended approaches to assess pesticide mobility in soil” in Environmental Behavior of Agrochemicals (ed. T. R. Roberts and P. C. Kearney). John Wiley & Sons Ltd. |
|
(20) |
Esser H. O., Hemingway R. J., Klein W., Sharp D. B., Vonk J. W. and Holland P. T., (1988), „Recommended approach to the evaluation of the environmental behavior of pesticides”, IUPAC Reports on Pesticides (24). Pure Appl. Chem., 60, pp. 901–932. |
|
(21) |
Guth J. A., Burkhard N., and D. O. Eberle, (1976), „Experimental models for studying the persistence of pesticides in soils”. Proc. BCPC Symposium: Persistence of Insecticides and Herbicides, pp. 137–157, BCPC, Surrey, UK. |
|
(22) |
Furminge C. G. L., and Osgerby J. M., (1967), „Persistence of herbicides in soil”. J. Sci. Fd Agric, 18, pp. 269–273. |
|
(23) |
Burkhard N., and Guth J. A., (1981), „Chemical hydrolysis of 2-Chloro-4,6-bis(alkylamino)-l,3,5-triazine herbicides and their breakdown in soil under the influence of adsorption”. Pestic. Sci. 12, pp. 45–52. |
|
(24) |
Guth J. A., Gerber H. R., and Schlaepfer T., (1977), „Effect of adsorption, movement and persistence on the biological availability of soil-applied pesticides”. Proc. Br. Crop Prot. Conf., 3, pp. 961–971. |
|
(25) |
Osgerby J. M., (1973), „Process affecting herbicide action in soil”. Pestic. Sci., 4, pp. 247–258. |
|
(26) |
Guth J. A., (1972), „Adsorptions- und Einwascheverhalten von Pflanzenschutzmitteln in Boden”. Schr. Reihe Ver. Wass. -Boden-Lufthyg. Berlin-Dahlem, Heft 37, pp. 143–154. |
|
(27) |
Hamaker J. W., (1975), „The interpretation of soil leaching experiments”, in Environmental Dynamics of Pesticides (eds R. Haque and V.H. freed), pp. 135–172, Plenum Press, NY. |
|
(28) |
Helling C. S., (1971), „Pesticide mobility in soils”. Soil Sci. Soc. Amer. Proc, 35, pp. 732–210. |
|
(29) |
Hamaker J. W., (1972), „Diffusion and volatilization” in Organic chemicals in the soil environment (C.A.I. Goring and J. W. Hamaker eds), Vol. I, pp. 49–143. |
|
(30) |
Burkhard N. and Guth J. A., (1981), „Rate of volatilisation of pesticides from soil surfaces; Comparison of calculated results with those determined in a laboratory model system”. Pestic. Sci. 12, pp. 37–44. |
|
(31) |
Cohen S. Z., Creeger S. M., Carsel R.F., and Enfield C.G., (1984), „Potential pesticide contamination of groundwater from agricultural uses”, in Treatment and Disposal of Pesticide Wastes, pp. 297–325, Acs Symp. Ser. 259, American Chemical Society, Washington, DC. |
|
(32) |
Gustafson D. I., (1989), „Groundwater ubiquity score: a simple method for assessing pesticide teachability”. J. Environ. Toxic. Chem., 8(4), pp. 339–357. |
|
(33) |
Leistra M., and Dekkers W. A., (1976). „Computed effects of adsorption kinetics on pesticide movement in soils”. J. of Soil Sci., 28, pp. 340–350. |
|
(34) |
Bromilov R. H., and Leistra M., (1980), „Measured and simulated behavior of aldicarb and its oxydation products in fallow soils”. Pest. Sci., 11, pp. 389–395. |
|
(35) |
Green R. E., and Karickoff S. W., (1990), „Sorption estimates for modeling”, in Pesticides in the Soil Environment: Process, Impacts and Modeling (ed. H.H. Cheng). Soil Sci. Soc. Am., Book Series no. 2, pp. 80–101, |
|
(36) |
Lambert S. M., (1967), „Functional relationship between sorption in soil and chemical structure”. J. Agri. Food Chem., 15, pp. 572–576. |
|
(37) |
Hance R. J., (1969), „An empirical relationship between chemical structure and the sorption of some herbicides by soils”. J. Agri. Food Chem., 17, pp. 667–668. |
|
(38) |
Briggs G. G. (1969), „Molecular structure of herbicides and their sorption by soils”. Nature, 223, 1288. |
|
(39) |
Briggs G. G. (1981). „Theoretical and experimental relationships between soil adsorption, octanol-water partition coefficients, water solubilities, bioconcentration factors, and the parachor”. J. Agric. Food Chem., 29, pp. 1050–1059. |
|
(40) |
Sabljic A., (1984), „Predictions of the nature and strength of soil sorption of organic polutance by molecular topology”. J. Agric. Food Chem., 32, pp. 243–246. |
|
(41) |
Bailey G. W., and White J. L., (1970), „Factors influencing the adsorption, desorption, and movement of pesticides in soil”. Residue Rev., 32, pp. 29–92. |
|
(42) |
Bailey G. W., J. L. White and Y. Rothberg., (1968), „Adsorption of organic herbicides by montomorillonite: Role of pH and chemical character of adsorbate”. Soil Sci. Soc. Amer. Proc. 32: pp. 222–234. |
|
(43) |
Karickhoff S. W., (1981), „Semi-empirical estimation of sorption of hydrophobic pollutants on natural sediments and soils”. Chemosphere 10, pp. 833–846. |
|
(44) |
Paya-Perez A., Riaz M. and Larsen B., (1989), „Soil Sorption of 6 Chlorobenzenes and 20 PCB Congeners”. Environ. Toxicol. Safety 21, pp. 1–17. |
|
(45) |
Hamaker J. W., and Thompson J. M., (1972), „Adsorption in organic chemicals” in Organic Chemicals in the Soil Environment (Goring C.A.I. and Hamaker J.W., eds), Vol I and II, Marcel Dekker, Inc., New York, NY, 1972, pp. 49–143. |
|
(46) |
Deli J., and Warren G. F., 1971, „Adsorption, desorption and leaching of diphenamid in soils”. Weed Sci. 19: pp. 67–69. |
|
(47) |
Chu-Huang Wu, Buehring N., Davinson J. M. and Santelmann, (1975), „Napropamide Adsorption, desorption and Movement in soils”. Weed Science, Vol. 23, pp. 454–457. |
|
(48) |
Haues M. H. B., Stacey M., and Thompson J. M., (1968), „Adsorption of s-triazine herbicides by soil organic preparations” in Isotopes and Radiation in Soil Organic Studies, p.75, International. Atomic Energy Agency, Vienna. |
|
(49) |
Pionke H. B., and Deangelis R. J., (1980), „Methods for distributing pesticide loss in field run-off between the solution and adsorbed phase”, CREAMS, in A Field Scale Model for Chemicals, Run-off and Erosion from Agricultural Management Systems, Chapter 19, Vol. III: Supporting Documentation, USDA Conservation Research report. |
|
(50) |
ISO Standard Compendium Environment: Soil Quality-General aspects; chemical and physical methods of analysis; biological methods of analysis. First Edition (1994). |
|
(51) |
Scheffer F., and Schachtschabel, Lehrbuch der Bodenkunde, F. Enke Verlag, Stuttgart (1982), 11th edition. |
|
(52) |
Black, Evans D. D., White J. L., Ensminger L. E., and Clark F. E., eds. „Methods of Soil Analysis”, Vol 1 and 2, American Society of Agronomy, Madison, WI, 1982. |
|
(53) |
ISO/DIS 10381-1 Soil Quality-Sampling-Part 1: Guidance on the design of sampling programmes. |
|
(54) |
ISO/DIS 10381-2 Soil Quality-Sampling-Part 2: Guidance on sampling techniques. |
|
(55) |
ISO/DIS 10381-3 Soil Quality-Sampling-Part 3: Guidance on safety of sampling. |
|
(56) |
ISO/DIS 10381-4 Soil Quality-Sampling-Part 4: Guidance on the investigation of natural and cultivated soils. |
|
(57) |
ISO/DIS 10381-5 Soil Quality-Sampling-Part 5: Guidance on the investigation of soil contamination of urban and industrial sites. |
|
(58) |
ISO 10381-6, 1993: Soil Quality-Sampling-Part 6: Guidance on the collection, handling and storage of soil for the assessment of aerobic microbial processes in the laboratory. |
|
(59) |
Green R. E., and Yamane V. K., (1970), „Precision in pesticide adsorption measurements”. Soil Sci. Am. Proc, 34, pp. 353–354. |
|
(60) |
Grover R., and Hance R. J. (1970), „Effect of ratio of soil to water on adsorption of linuron and atrazine”. Soil Sci., pp. 109–138. |
|
(61) |
Boesten, J. J. T. I, „Influence of soil/liquid ratio on the experimental error of sorption coefficients in pesticide/soil system”. Pest. Sci. 1990, 30, pp. 31–41. |
|
(62) |
Boesten, J. J. T. I. „Influence of soil/liquid ratio on the experimental error of sorption coefficients in relation to OECD guideline 106”. Proceedings of 5th international workshop on environmental behaviour of pesticides and regulatory aspects, Brussels, 26–29 April 1994. |
|
(63) |
Bastide J., Cantier J. M., et Coste C, (1980), „Comportement de substances herbicides dans le sol en fonction de leur structure chimique”. Weed Res. 21, pp. 227–231. |
|
(64) |
Brown D. S., and Flagg E. W., (1981), „Empirical prediction of organic pollutants sorption in natural sediments”. J. Environ.Qual., 10(3), pp. 382–386. |
|
(65) |
Chiou C. T., Porter P. E., and Schmedding D. W., (1983), „Partition equilibria of non-ionic organic compounds between soil organic matter and water”. Environ. Sci. Technol., 17(4), pp. 227–231. |
|
(66) |
Gerstl Z., and Mingelgrin U., (1984), „Sorption of organic substances by soils and sediments”. J. Environm. Sci. Health, B19 (3), pp. 297–312. |
|
(67) |
Vowles P. D., and Mantoura R. F. C, (1987), „Sediment-water partition coefficient and HPLC retention factors of aromatic hydrocarbons”. Chemosphere, 16(1), pp. 109–116. |
|
(68) |
Lyman W. J., Reehl W. F.and Rosenblatt D. H. (1990). Handbook of Chemical Property Estimation Methods. Environmental Behaviour of Organic Compounds. American Chemical Society, Washington DC. |
|
(69) |
Keniga E. E., and Goring, C. A. I. (1980). „Relationship between water solubility, soil sorption, octanol-water partitioning and concentration of chemicals in the biota” in Aquatic Toxicology (eds J.G. Eaton, et al.), pp.78–115, ASTM STP 707, Philadelphia. |
|
(70) |
Chiou C. T., Peters L. J., and Freed V. H., (1979), „A physical concept of soil-water equilibria for non-ionic organic compounds”. Science, Vol. 206, pp. 831–832. |
|
(71) |
Hassett J. J., Banwart W. I., Wood S. G., and Means J. C, (1981), „Sorption of/-Naphtol: implications concerning the limits of hydrophobic sorption”. Soil Sci. Soc. Am. J. 45, pp. 38–42. |
|
(72) |
Karickhoff S. W., (1981), „Semi-empirical estimation of sorption of hydrophobic pollutants on natural sediments and soils”. Chemosphere, Vol. 10(8), pp. 833–846. |
|
(73) |
Moreale A., van Bladel R., (1981), „Adsorption de 13 herbicides et insecticides par le sol. Relation solubilite-reactivite”. Revue de l'Agric, 34 (4), pp. 319–322. |
|
(74) |
Muller M., Kórdel W. (1996), „Comparison of screening methods for the determination/estimation of adsorption coefficients on soil”. Chemosphere, 32(12), pp. 2493–2504. |
|
(75) |
Kórdel W., Kotthoff G., Müller M. (1995), „HPLC-screening method for the determination of the adsorption coefficient on soil-results of a ring test”. Chemosphere 30 (7), pp. 1373–1384. |
|
(76) |
Kórdel W., Stutte J., Kotthoff G. (1993), „HPLC-screening method for the determination of the adsorption coefficient on soil-comparison of different stationary phases”. Chemosphere 27 (12), pp. 2341–2352. |
|
(77) |
Hance, R. J., (1967), „The Speed of Attainment of Sorption Equilibria in Some Systems Involving Herbicides”. Weed Research, Vol. 7, pp. 29–36. |
|
(78) |
Koskinen W. C, and Harper S. S., (1990), „The retention processes: mechanisms” in Pesticides in the Soil Environment: Processes, Impacts and Modelling (ed. H.H. Cheng). Soil Sci. Soc. Am. Book Series, No. 2, Madison, Wisconsin. |
|
(79) |
Cohen S. Z., Creeger S. M., Carsel R. F., and Enfield C. G. (1984), „Potential pesticide contamination of groundwater from agricultural uses”, in Treatment and Disposal of Pesticide Wastes, pp.297-325, ACS Symp. Ser. 259, American Chemical Society, Washington, DC. |
|
(80) |
Giles C. H., (1970), „Interpretation and use of sorption isotherms” in Sorption and Transport Processes in Soils. S.C.I. Monograph No. 37, pp. 14–32. |
|
(81) |
Giles, C. H.; McEwan J. H.; Nakhwa, S.N. and Smith, D, (1960), „Studies in adsorption: XL A system of classification of solution adsorption isotherms and its use in the diagnosis of adsorption mechanisms and in measurements of pesticides surface areas of soils”. J. Chem. Soc, pp. 3973–93. |
|
(82) |
Calvet R., Terce M., and Arvien J. C, (1980), „Adsorption des pesticides par les sols et leurs constituants: 3. Caracteristiques generates de l'adsorption”. Ann. Agron. 31: pp. 239–251. |
|
(83) |
Bedbur E., (1996), „Anomalies in the Freundlich equation”, Proc. COST 66 Workshop, Pesticides in soil and the environment, 13–15 May 1996, Stratford-upon-Avon, UK. |
|
(84) |
Guth, J. A., (1985), „Adsorption/desorption”, in Joint International Symposium, Physicochemical Properties and their Role in Environmental Hazard Assessment, July 1–3, Canterbury, UK. |
|
(85) |
Soil Texture Classification (US and FAO systems): Weed Science, 33, Suppl. 1 (1985) and Soil Sci. Soc. Amer. Proc. 26:305 (1962). |
DODATEK 1
Plan Badania
DODATEK 2
WPŁYW DOKŁADNOŚCI METODY ANALITYCZNEJ I ZMIAN STĘŻENIA NA DOKŁADNOŚĆ WYNIKÓW ADSORPCJI
Z następującej tabeli (84) jasno wynika, że kiedy różnica między masą początkową (mo = 110 μg) a masą równowagi (m = 100 μg) substancji badanej w roztworze jest bardzo mała, błąd 5 % w pomiarze stężenia w równowadze powoduje błąd 50 % w obliczeniu masy substancji zaadsorbowanej w glebie (m
= 100 μg) substancji badanej w roztworze jest bardzo mała, błąd 5 % w pomiarze stężenia w równowadze powoduje błąd 50 % w obliczeniu masy substancji zaadsorbowanej w glebie (m  ) i 52,4 % w obliczeniu Kd.
) i 52,4 % w obliczeniu Kd.
|
Ilość gleby |
msoil |
= 10 g |
|
Objętość roztworu |
V0 |
= 100 cm3 |
|
|
FOR-L_2008142PL.01044401.notes.0187.xml.jpg (μg) |
FOR-L_2008142PL.01044401.notes.0188.xml.jpg (μg cm-3) |
R |
FOR-L_2008142PL.01044401.notes.0189.xml.jpg (μg) |
FOR-L_2008142PL.01044401.notes.0190.xml.jpg (μg g-1) |
R‡ |
Kd* |
R‡ |
|
|
DLA A = 9 % |
|||||||
|
m0 = 110 μg or C0=1,100 μg/cm3 |
100 |
1,000 |
wartosc rzeczywista |
10 |
1,00 |
wartosc rzeczywista |
1 |
|
|
101 |
1,010 |
1 % |
9 |
0,90 |
10 % |
0,891 |
10,9 % |
|
|
105 |
1,050 |
5 % |
5 |
0,50 |
50 % |
0,476 |
52,4 % |
|
|
109 |
1,090 |
9 % |
1 |
0,10 |
90 % |
0,092 |
90,8 % |
|
|
|
DLA A = 55 % |
|||||||
|
m0= 110 μg or C0=1,100μg/cm3 |
50,0 |
0,500 |
wartosc rzeczywista |
60,0 |
6,00 |
wartosc rzeczywista |
12,00 |
|
|
50,5 |
0,505 |
1 % |
59,5 |
5,95 |
0,8 % |
11,78 |
1,8 % |
|
|
52,5 |
0,525 |
5 % |
57,5 |
5,75 |
4,0 % |
10,95 |
8,8 % |
|
|
55,0 |
0,550 |
10 % |
55,0 |
5,50 |
8,3 % |
10,00 |
16,7 % |
|
|
|
DLA A = 99 % |
|||||||
|
m0 = 110 μg or C0=1,100 μg/cm3 |
1,100 |
0,011 |
wartosc rzeczywista |
108,9 |
10,89 |
wartosc rzeczywista |
990 |
|
|
1,111 |
0,01111 |
1 % |
108,889 |
10,8889 |
0,01 % |
980 |
1,0 % |
|
|
1,155 |
0,01155 |
5 % |
108,845 |
10,8845 |
0,05 % |
942 |
4,8 % |
|
|
1,21 |
0,0121 |
10 % |
108,790 |
10,8790 |
0,10 % |
899 |
9,2 % |
|
gdzie:
|
|
= |
|
|
|
= |
masa substancji badanej w fazie gleby w równowadze, μg, |
|
|
= |
masa substancji badanej w fazie gleby w równowadze, μg, |
|
|
= |
zawartość substancji badanej w fazie gleby w równowadze, μg g-1, |
|
|
= |
masowe stężenie substancji badanej w fazie wodnej w równowadze, μg cm-3 |
|
R |
= |
błąd analityczny w oznaczaniu |
|
R‡ |
= |
błąd obliczony spowodowany błędem analitycznym R. |
DODATEK 3
TECHNIKI OSZACOWANIA DLA Kd
|
1. |
Techniki oszacowania pozwalają na przewidzenie Kd opartego na korelacjach z, na przykład wartością Pow (12)(39)(63–68), danymi dotyczącymi wodnej rozpuszczalności (12)(19)(21)(39)(68–73) lub danych polarności wyprowadzonych z zastosowania HPLC w fazie odwróconej (74–76). Jak wskazano w tabelach 1 i 2, istnieją Koc lub Kom obliczone tymi równaniami i następnie, pośrednio, Kd z równań:
|
|
2. |
Pojecie tych korelacji jest oparte na dwóch założeniach: 1) jest to substancja organiczna gleby, która głównie wpływa na adsorpcje substancji; i 2) współdziałania wywołane są głównie substancjami niepolarnymi. Jako wynik te korelacje: 1) nie są, lub są tylko w pewnym zakresie, stosowane do substancji polarnych, i 2) nie są stosowane w przypadkach, gdy zawartość substancji organicznych w glebie jest bardzo mała (12). Dodatkowo, chociaż odkryto zadawalające korelacje między Pow i adsorpcją (19), tego samego nie można powiedzieć o zależności między rozpuszczalnością w wodzie i zakresem adsorpcji (19)(21); tak więc badania są w istotnym stopniu sprzeczne. |
|
3. |
Niektóre przykłady korelacji między współczynnikiem adsorpcji a współczynnikiem podziału oktanol-woda, jak również rozpuszczalność w wodzie, podano odpowiednio w tabelach 1 i 2. Tabela 1 Przykłady korelacji między współczynnikiem podziału adsorpcji a współczynnikiem podziału oktanol-woda; dalsze przykłady w (12)(68)
Tabela 2 Przykłady korelacji między współczynnikiem podziału adsorpcji i rozpuszczalnością w wodzie; dalsze przykłady zob. (68)(69)
|
DODATEK 4
OBLICZENIA W ODNIESIENIU DO OKREŚLNIA WARUNKÓW WIROWANIA
|
1. |
Czas odwirowania jest podany za pomocą następującego wzoru, włączając sferyczne cząstki:
Do celów uproszczenia, wszystkie parametry są opisane w jednostkach nie-SI (g, cm). gdzie:
W ogólnej praktyce stosuje się podwojony obliczony czas w celu zapewnienia całkowitego oddzielenia. |
|
2. |
Równanie (1) może być uproszczone dalej, jeśli założymy, że lepkość (η) i gęstość (ρaq) roztworu jest równa lepkości i gęstości wody w 25 oC; zatem, η = 8,95 x 10-3 g s-1 cm-1 i ρaq = 1,0 g, cm-3. Zatem, czas wirowania jest podany za pomocą równania (2)
|
|
3. |
Z równania 2 jasno wynika, że dwa parametry są ważne dla określania warunków wirowania, tj. czas (t) szybkość (rpm), w celu uzyskania rozdzielenia cząstek o szczególnym wymiarze (w tym przypadku promień 0,1 μm): 1) gęstość gleby; oraz 2) długość mieszaniny w probówce wirówki (Rb-Rt), tj. odległość, którą przebywa cząstka z góry roztworu do dna probówki, oczywiście dla ustalonej objętości długość mieszaniny w probówce zależy od kwadratu promienia probówki. |
|
4. |
Rysunek 1 przedstawia zmiany w czasie wirowania (t) w zależności od szybkości wirowania (rpm) dla różnych gęstości gleby (ρs) (rys. la) i różnych długości mieszaniny w probówkach wirówki (rys. 2a). Z rys. la wpływ gęstości gleby wynika w sposób oczywisty, na przykład dla klasycznego wirowania przy 3000 rpm czas wirowania wynosi w przybliżeniu 240 min dla gęstości gleby 1,2 g cm3, podczas gdy jest to tylko 50 min. dla 2,0 g cm3. Podobnie, z rys. lb, dla klasycznego wirowania 3000 rpm czas wirowania wynosi w przybliżeniu 50 min dla długości mieszaniny 10 cm i tylko 7 min. dla długości 1 cm. Jednakże istotne jest odkrycie optymalnej zależności między wirowaniem które wymaga możliwie mniejszej długości i ułatwionego postępowania eksperymentatora w rozdzielaniu faz po odwirowaniu. |
|
5. |
Ponadto przy określaniu doświadczalnych warunków rozdzielania faz gleba/roztwór, istotne jest rozważenie możliwości istnienia trzeciej „pseudofazy”, koloidów. Cząstki te, o wielkości mniejszej niż 0,2 (μm, mają ważny wpływ na cały mechanizm adsorpcji substancji w zawiesinie gleby. Gdy wirowanie jest wykonywane jak opisano powyżej, koloidy pozostają w fazie wodnej i podlegają analizie wraz z wodną fazą. W ten sposób informacja o ich wpływie jest tracona. Jeśli laboratorium prowadzące badanie posiada urządzenia ultrawirujące lub ultrafiltrujące, adsorpcja/desorpcja substancji w glebie może być badana dogłębiej, włączając informacje o adsorpcji substancji na koloidach. W tym przypadku należy zastosować ultrawirowanie przy 60 000 rpm/min lub ultrafiltrację z filtrem o porowatości 100 000 daltonów w celu rozdzielenia trzech faz gleby, koloidów i roztworu. Protokół badania powinien być również odpowiednio zmodyfikowany w celu objęcia wszystkich trzech faz analizą substancji. |
Rys. 1a.
Rys. 1b.

DODATEK 5
OBLICZANIE ADSORPCJI A (%) I DESORPCJI D (%)
Schemat czasowy procedury jest następujący:
W odniesieniu do wszystkich obliczeń zakłada się, że substancja badana jest stabilna i nie adsorbuje się w sposób znaczący na ściankach naczynia.
ADSORPCJA A (A%)
a) Metoda równoległa
Procent adsorpcji jest obliczany w odniesieniu do każdej probówki badanej (i) w każdym punkcie czasu (ti), zgodnie z równaniem:
|
|
(1) (31) |
Człony niniejszego równania są obliczane następująco:
|
m0 = C0 · V0 (μg) |
(2) |
|
|
(3) |
gdzie:
|
|
= |
procent adsorpcji ( %) w punkcie czasu ti, |
|
|
= |
masa substancji badanej na glebie, w czasie ti w którym wykonywana jest analiza (μg), |
|
m0 |
= |
masa substancji badanej w probówce badanej, przy rozpoczęciu badania (μg), |
|
C0 |
= |
stężenie masowe początkowe roztworu badanego w kontakcie z glebą (μg cm-3), |
|
|
= |
stężenie masowe substancji w fazie wodnej w czasie ti w którym prowadzona jest analiza (μg cm-3); niniejsze stężenie jest ustalone analitycznie, uwzględniając wielkości określone ślepą próbą. |
|
V0 |
= |
początkowa objętość roztworu badanego w kontakcie z glebą (cm3). |
Wartości procentu adsorpcji  lub
lub  są wykreślane w zależności od czasu i czasu, po którym osiągana jest równowaga sorpcji. Przykłady takich wykresów podano odpowiednio na rys. 1 i rys.2.
są wykreślane w zależności od czasu i czasu, po którym osiągana jest równowaga sorpcji. Przykłady takich wykresów podano odpowiednio na rys. 1 i rys.2.
b) Metoda szeregowa
Następujące równania są uwzględniane, jeśli zastosowano procedurę adsorpcji, przez oznaczenie substancji badanej w małych podwielokrotnościach fazy wodnej przy właściwych przedziałach czasu.
|
— |
W ramach każdego przedziału czasu ilość substancji zaadsorbowanej na glebie jest obliczana w następujący sposób:
|
|
— |
Procent adsorpcji w każdym przedziale czasowym,
gdzie procent adsorpcji
Wartości adsorpcji |
|
— |
Przy czasie równowagi teq:
|
Parametry użyte powyżej są określone jako:
|
|
= |
masa substancji zaadsorbowana na glebie w ciągu przedziałów czasu, odpowiednio Δt1, Δt2,..., Δtn (μg), |
|
|
= |
masa substancji zmierzona w podwielokrotności |
|
|
= |
masa substancji zaadsorbowanej na glebie przy równowadze adsorpcji (μg), |
|
|
= |
masa substancji w roztworze przy równowadze adsorpcji (μg), |
|
|
= |
objętość podwielokrotności w której substancja badana jest mierzona (cm |
|
|
= |
procent adsorpcji odpowiadający przedziałowi czasowemu Δti ( %), |
|
|
= |
procent adsorpcji przy równowadze adsorpcji ( %). |
DESORPCJA D (A%)
Czas t0 rozpoczęcia doświadczenia kinetyki desorpcji, jest określany jako moment, w którym maksymalna odzyskana objętość roztworu badanej substancji (po osiągnięciu równowagi adsorpcji) jest podstawiana równą objętością roztworu 0,01 M CaCl2.
a) Metoda równoległa
W punkcie czasu ti, masa substancji badanej jest mierzona w fazie wodnej pobranej z probówki i ( ) masa zdesorbowana jest obliczana według równania:
) masa zdesorbowana jest obliczana według równania:
|
|
(13) |
Przy równowadze desorpcji ti = teq i zatem  =
= 
Masa substancji badanej zdesorbowanej podczas przedziału czasowego (Δti) jest podana za pomocą równania:
|
|
(14) |
Procent desorpcji jest obliczany:
w punkcie czasu ti z równania:
|
|
(15) |
i podczas przedziału czasowego (Δti) z równania:
|
|
(16) |
gdzie:
|
|
= |
procent desorpcji w punkcie czasu ti ( %), |
|
|
= |
procent desorpcji odpowiadający przedziałowi czasu Δti ( %), |
|
|
= |
masa substancji zdesorbowanej w punkcie czasu ti (μg), |
|
|
= |
masa zaadsorbowanej substancji badanej w ciągu przedziału czasu Δti (μg), |
|
|
= |
masa substancji badanej zmierzona analitycznie w czasie ti w objętości roztworu |
|
|
= |
masa substancji badanej pozostającej z równowagi adsorpcji wymaganej do niepełnego podstawienia objętości (μg), |
|
|
(17) |
|
|
= |
masa substancji badanej w roztworze w równowadze adsorpcji (μg), |
|
VR |
= |
objętość nadsączu zlanego z probówki po osiągnięciu równowagi adsorpcji i zastąpieniu tą samą ilością roztworu 0,01 M CaCl2 (cm3), |
|
|
= |
objętość roztworu pobranego z probówki (i) do pomiaru substancji badanej w doświadczeniu kinetyki desorpcji (cm3). |
Wielkości desorpcji  lub
lub  (zgodnie z potrzebami badań) są wykreślane w zależności od czasu i czasu po którym uzyskana równowaga desorpcji jest stała.
(zgodnie z potrzebami badań) są wykreślane w zależności od czasu i czasu po którym uzyskana równowaga desorpcji jest stała.
b) Metoda szeregowa
Następujące równania są uwzględniane, jeśli zastosowano procedurę adsorpcji, przez oznaczenie substancji badanej w małych podwielokrotnościach ( ) fazy wodnej (metoda szeregowa w 1.9. Wykonanie badania). Zakłada ona, że: a) objętość nadsączu zlanego z probówki po doświadczeniu kinetyki adsorpcji, była zastąpiona tą samą objętością roztworu 0,01 M CaCl2 (VR); i b) całkowita objętość fazy wodnej w kontakcie z glebą (VT) w czasie trwania doświadczenia pozostaje stała oraz jest podana za pomocą równania:
) fazy wodnej (metoda szeregowa w 1.9. Wykonanie badania). Zakłada ona, że: a) objętość nadsączu zlanego z probówki po doświadczeniu kinetyki adsorpcji, była zastąpiona tą samą objętością roztworu 0,01 M CaCl2 (VR); i b) całkowita objętość fazy wodnej w kontakcie z glebą (VT) w czasie trwania doświadczenia pozostaje stała oraz jest podana za pomocą równania:
|
|
(18) |
W punkcie czasu ti:
|
— |
masa substancji badanej jest mierzona w małej podwielokrotności
|
|
— |
w równowadze desorpcji ti = teq, a zatem |
|
— |
procent desorpcji
|
W przedziale czasowym (Δti):
W trakcie każdego przedziału czasu ilość substancji desorbowanej jest obliczana następująco:
|
— |
dla pierwszego przedziału czasu Δt1 = t1-t0
|
|
— |
dla drugiego przedziału czasu Δt2 = t2-t1
|
|
— |
dla n-tego przedziału czasu Δtn = tn-tn-1
|
Ostatecznie procent desorpcji w każdym przedziale czasowym,  , jest wyliczany, stosując następujące równanie:
, jest wyliczany, stosując następujące równanie:
|
|
(24) |
gdzie procent desorpcji  w punkcie czasu ti jest podany za pomocą równania:
w punkcie czasu ti jest podany za pomocą równania:
|
|
(25) |
Gdzie powyższe użyte parametry są określone jako:
|
|
= |
masa substancji zaadsorbowanej na glebie po przedziałach czasowych Δt1, Δt2,..., Δtn odpowiednio (μg); |
||
|
|
= |
masa substancji badanej zdesorbowanej podczas przedziałów czasowych Δt1, Δt2,..., Δtn odpowiednio (μg); |
||
|
|
= |
masa substancji zmierzona w podwielokrotności |
||
|
VT |
= |
całkowita objętość fazy wodnej w kontakcie z glebą, podczas trwania doświadczenia kinetycznej desorpcji, przeprowadzonego metodą szeregową (cm3), |
||
|
|
= |
masa substancji badanej pozostałej z równowagi adsorpcji wymagana do niepełnego podstawienia objętości (μg),
|
||
|
VR |
= |
objętość nadsączu usuniętego z probówki po uzyskaniu równowagi adsorpcji i zastąpiona tą samą objętością roztworu 0,01 M CaCl2 (cm3), |
||
|
|
= |
objętość podwielokrotności pobranej do celów analitycznych z probówki (i), w czasie trwania doświadczenia kinetyki desorpcji, wykonywanej metodą seryjną (cm3),
|
DODATEK 6
ADSORPCJA-DESORPCJA W GLEBACH: ARKUSZ PRZEDSTAWIANIA DANYCH
Badana substancja:
Badana gleba:
Zawartość suchej masy w glebie (105 oC, 12 godz.): … %
Temperatura: … oC
Odpowiedniość metody analitycznej
|
Zważona gleba |
g |
|
|
Gleba: sucha masa |
8 |
|
|
Objętość roztworu CaCl2 |
cm3 |
|
|
Nominalne stężenie roztworu końcowego |
μg cm-3 |
|
|
Analityczne stężenie roztworu końcowego |
μg cm-3 |
|
Zasada zastosowanej metody analitycznej:
Kalibracja metody analitycznej:
Badana substancja:
Badana gleba:
Zawartość suchej masy w glebie (105 oC, 12 godz.): … %
Temperatura: … oC
|
Zastosowana analityczna metodologia: |
Pośrednia |
|
Równoległa |
|
Szeregowa |
|
|
|
Bezpośrednia |
|
|
|
|
|
Badanie adsorpcji: próbki badane
|
|
Symbol |
Jednostki |
Czas równoważenia |
Czas równoważenia |
Czas równoważenia |
Czas równoważenia |
||||
|
Probówka nr |
|
|
|
|
|
|
|
|
|
|
|
Zważona gleba |
— |
g |
|
|
|
|
|
|
|
|
|
Gleba: sucha masa |
msoil |
g |
|
|
|
|
|
|
|
|
|
Objętość wody w zważonej glebie (obliczona) |
vws |
cm3 |
|
|
|
|
|
|
|
|
|
Objętość 0,01 M CaCl2 roztworu do zrównoważenia gleby |
|
cm3 |
|
|
|
|
|
|
|
|
|
Objętość roztworu podstawowego |
|
cm3 |
|
|
|
|
|
|
|
|
|
Całkowita objętość fazy wodnej w kontakcie z glebą |
Vo |
cm3 |
|
|
|
|
|
|
|
|
|
Początkowe stężenie badanego roztworu |
Co |
μg cm-3 |
|
|
|
|
|
|
|
|
|
Masa substancji badanej na początku badania |
m 0 |
μg |
|
|
|
|
|
|
|
|
|
Po mieszaniu i odwirowaniu |
||||||||||
|
METODA POŚREDNIA |
||||||||||
|
Metoda równoległa |
||||||||||
|
Stężenie w wodnej fazie badanej substancji, włączając ślepą próbę |
|
μg cm-3 |
|
|
|
|
|
|
|
|
|
Metoda szeregowa |
||||||||||
|
Podwielokrotna masa zmierzona substancji badanej |
|
μg |
|
|
|
|
|
|
|
|
|
METODA BEZPOŚREDNIA |
||||||||||
|
Masa badanej substancji zaadsorbowanej na glebie |
|
μg |
|
|
|
|
|
|
|
|
|
Obliczenie adsorpcji |
||||||||||
|
Adsorpcja |
|
% |
|
|
|
|
|
|
|
|
|
|
|
% |
|
|
|
|
|
|
|
|
|
Środek |
|
|
|
|
|
|
||||
|
Współczynnik adsorpcji |
Kd |
cm3 g-1 |
|
|
|
|
|
|
|
|
|
Środek |
|
|
|
|
|
|
||||
|
Współczynnik adsorpcji |
Koc |
cm3 g-1 |
|
|
|
|
|
|
|
|
|
Środek |
|
|
|
|
|
|
||||
Badana substancja:
Badana gleba:
Zawartość suchej masy w glebie (105 oC, 12 godz.): … %
Temperatura: … oC
Badanie adsorpcji: próby ślepe i kontrolne
|
|
Symbol |
Jednostki |
Ślepa |
Ślepa |
Kontrolna |
|||
|
Probówka nr |
|
|
|
|
|
|
|
|
|
Zważona gleba |
|
g |
|
|
|
|
0 |
0 |
|
Ilość wody w zważonej glebie(obliczona) |
|
cm3 |
|
|
|
|
— |
— |
|
Dodana objętość roztworu 0,01 Μ CaCl2 |
|
cm3 |
|
|
|
|
|
|
|
Dodana objętość roztworu podstawowego substancji badanej |
|
cm3 |
0 |
0 |
|
|
|
|
|
Całkowita objętość fazy wodnej (obliczona) |
|
cm3 |
|
|
|
|
— |
— |
|
Początkowe stężenie substancji badanej w fazie wodnej |
|
μg cm-3 |
|
|
|
|
|
|
|
Po mieszaniu i odwirowaniu |
||||||||
|
Stężenie w fazie wodnej |
|
μg cm3 |
|
|
|
|
|
|
Uwaga: jeśli konieczne, dodać kolumny.
Badan substancja:
Badana gleba:
Zawartość suchej masy w glebie (105 oC, 12 godz.): … %
Temperatura: … oC
Bilans masy
|
|
Symbol |
Jednostki |
|
|
|
|
|
Probówka nr |
|
|
|
|
|
|
|
Zważona gleba |
— |
g |
|
|
|
|
|
Gleba: sucha masa |
msoil |
g |
|
|
|
|
|
Objętość wody w zważonej glebie (obliczona) |
VWS |
ml |
|
|
|
|
|
Objętość roztworu 0,01 Μ CaCl2 do równoważenia gleby |
|
ml |
|
|
|
|
|
Objętość roztworu podstawowego |
|
cm3 |
|
|
|
|
|
Całkowita objętość fazy wodnej w kontakcie z glebą |
V0 |
cm3 |
|
|
|
|
|
Początkowe stężenie roztworu badanego |
Co |
μg cm-3 |
|
|
|
|
|
Czas równoważenia |
— |
h |
|
|
|
|
|
Po mieszaniu i odwirowaniu |
||||||
|
Stężenie wodnej fazy substancji badanej w równowadze adsorpcji, włączono korektę na ślepą próbę |
|
μg cm-3 |
|
|
|
|
|
Czas równoważenia |
teq |
h |
|
|
|
|
|
Pierwsze rozcieńczenie rozpuszczalnikiem |
||||||
|
Usunięta objętość fazy wodnej |
Vrec |
cm3 |
|
|
|
|
|
Dodana objętość rozpuszczalnika |
ΔV |
cm3 |
|
|
|
|
|
Pierwszasza ekstrakcja rozpuszczalnikiem |
||||||
|
Analizowany sygnał w rozpuszczalniku |
SE1 |
var. |
|
|
|
|
|
Stężenie substancji badanej w rozpuszczalniku |
CE1 |
μg cm-3 |
|
|
|
|
|
Masa substancji wyekstrahowanej z gleby i ścianek zbiornika |
ME1 |
μg |
|
|
|
|
|
Drugie rozcieńczenie rozpuszczalnikiem |
||||||
|
Usunięta objętość rozpuszczalnika |
ΔVS |
cm3 |
|
|
|
|
|
Dodana objętość rozpuszczalnika |
ΔV |
cm3 |
|
|
|
|
|
Druga ekstrakcja rozpuszczalnikiem |
||||||
|
Analizowany sygnałw fazie rozpuszczalnika |
SE2 |
var. |
|
|
|
|
|
Stężenie substancji badanej w rozpuszczalniku |
CE2 |
μg cm-3 |
|
|
|
|
|
Masa substancji wyekstrahowanej z gleby i ścianek zbiornika |
mE2 |
μg |
|
|
|
|
|
Całkowita masa substancji badanej wyekstrahowanej w dwóch kolejnych krokach |
mE |
μg |
|
|
|
|
|
Bilans masy |
MB |
% |
|
|
|
|
Badana substancja:
Badana gleba:
Zawartość suchej masy w glebie (105 oC, 12 godz.): … %
Temperatura: … oC
Izotermy adsorpcji
|
|
Symbol |
Jednostki |
|
|
|
|
|
|
|
|
|
Probówka nr |
|
|
|
|
|
|
|
|
|
|
|
Zważona gleba |
— |
g |
|
|
|
|
|
|
|
|
|
Gleba:sucha masa |
E |
g |
|
|
|
|
|
|
|
|
|
Objętość wody w zważonej glebie (obliczona) |
VWS |
cm3 |
|
|
|
|
|
|
|
|
|
Objętość roztworu 0,01 M CaCl2 do zrównoważenia gleby |
|
cm3 |
|
|
|
|
|
|
|
|
|
Objętość dodanego roztworu podstawowego |
|
cm3 |
|
|
|
|
|
|
|
|
|
Całkowita objętość fazy wodnej w kontakcie z glebą (obliczona) |
V0 |
cm3 |
|
|
|
|
|
|
|
|
|
Stężenie roztworu |
C0 |
μg cm-3 |
|
|
|
|
|
|
|
|
|
Czas równoważenia |
— |
h |
|
|
|
|
|
|
|
|
|
Po mieszaniu i odwirowaniu |
||||||||||
|
Stężenie substancji w fazie wodnej, włączono poprawkę na ślepą próbę |
|
μg cm-3 |
|
|
|
|
|
|
|
|
|
Temperatura |
|
oC |
|
|
|
|
|
|
|
|
|
Masa adsorpcji na jednostkę gleby |
|
μg g-1 |
|
|
|
|
|
|
|
|
Analiza regresyjna:
wartość KF ads:
wartość 1/n:
Współczynnik regresji r2:
Badana substancja:
Badana gleba:
Zawartość suchej masy w glebie (105 oC, 12 godz.): … %
Temperatura: … oC
|
Zastosowana analityczna |
Pośrednia Równoległa |
|
Szeregowa |
|
Metodologia: |
|
Badanie desorpcji
|
|
Symbol |
Jednostki |
Przedział czasu |
Przedział czasu |
Przedział czasu |
Przedział czasu |
|
|
Probówka nr pochodząca z kroku adsorpcji |
|
|
|
|
|
|
|
|
Masa substancji zaadsorbowanej na glebie w równowadze adsorpcji |
|
μg |
|
|
|
|
|
|
Usunięta objętość fazy wodnej, zastąpiona przez 0,01 M CaCl2 |
VR |
cm3 |
|
|
|
|
|
|
Całkowita objętość fazy wodnej w kontakcie z glebą |
PM |
V0 |
cm3 |
|
|
|
|
|
SM |
VT |
cm3 |
|
|
|
|
|
|
Masa substancji badanej pozostająca w równowadze adsorpcji wymagana do niepełnego podstawienia objętości |
|
μg |
|
|
|
|
|
|
Kinetyka desorpcji |
|||||||
|
Zmierzona masa substancji zdesorbowanej z gleby w czasie ti |
|
μg |
|
|
|
|
|
|
Objętość roztworu pobranego z probówki (i) do pomiaru substancji badanej |
PM |
vi r |
cm3 |
|
|
|
|
|
SM |
va D |
cm3 |
|
|
|
|
|
|
Masa substancji zdesorbowana z gleby w czasie ti (obliczona) |
|
μg |
|
|
|
|
|
|
Masa substancji zdesorbowana z gleby w czasie trwania przedziału czasowego Δti (obliczona) |
|
μg |
|
|
|
|
|
|
Procent desorpcji |
|||||||
|
Desorpcja w czasie ti |
Dti |
% |
|
|
|
|
|
|
Desorpcja w przedziale czasowym DΔti |
|
% |
|
|
|
|
|
|
Pozorny współczynnik desorpcji |
Kdes |
|
|
|
|
|
|
PM: metoda równoległa
SM: metoda szeregowa
C.19. OSZACOWANIE WSPÓŁCZYNNIKA ADSORPCJI (KOC) NA GLEBIE I W OSADZIE ŚCIEKOWYM PRZY ZASTOSOWANIU WYSOKOSPRAWNEJ CHROMATOGRAFII CIECZOWEJ (HPLC)
1. METODA
Niniejsza metoda jest kopią OECD TG121 (2000).
1.1. WPROWADZENIE
Zachowanie się sorpcji substancji w glebach lub osadach ściekowych może być opisane za pomocą parametrów określonych doświadczalnie w rozumieniu metody badania C.18. Ważnym parametrem jest współczynnik adsorpcji, który określa się jako stosunek między stężeniem substancji w gleba/osad ściekowy a stężeniem substancji w fazie wodnej w równowadze adsorpcji. Współczynnik adsorpcji znormalizowany do zawartości węgla organicznego gleby Koc jest użytecznym wskaźnikiem zdolności wiązania substancji chemicznych na ciałach organicznych gleby i osadu ściekowego oraz pozwalającym na dokonanie porównań między różnymi substancjami chemicznymi. Niniejszy parametr może być oszacowany poprzez korelacje rozpuszczalności wodnej i współczynnikiem podziału n-oktanol/woda (1)(2)(3)(4)(5)(6)(7).
Metoda doświadczalna opisana w niniejszym badaniu, używa HPLC dla oszacowania współczynnika adsorpcji Koc w glebie i w osadzie ściekowym (8). Oceny są wyższej wiarygodności niż te z obliczeń QSAR (9). Jako metoda oszacowania nie może w pełni zastąpić doświadczeń okresowej równowagi, zastosowanej w metodzie badanej C18. Jednakże oszacowany Koc jest użyteczny dla wyboru właściwych parametrów badania dla badań adsorpcja/desorpcja zgodnie z metodą badania C.18, poprzez wyliczenie Kd (współczynnik podziału) lub Kf (współczynnik adsorpcji Freundlicha) zgodnie z równaniem 3 (zob. pkt 1.2).
1.2. DEFINICJE
K d : Współczynnik podziału jest zdefiniowany jako stosunek stężenia równowagi C roztworu substancji badanej w systemie dwufazowym składającym się z sorbentu (gleba lub osad ściekowy) i fazy wodnej; jest to wartość bezwymiarowa, natomiast stężenia w obu fazach są wyrażone na podstawie waga/waga. W przypadku stężenia w fazie wodnej podano je na bazie waga/objętość, zatem jednostką jest ml· g-1. Kd zmienia się z właściwościami sorbentu i może być zależny od stężenia.
|
|
(1) |
gdzie:
|
Csoil |
= |
stężenie substancji badanej w glebie w równowadze (μg· g-1), |
|
Csludge |
= |
stężenie substancji badanej w osadzie w równowadze (μg· g-1), |
|
Caq |
= |
stężenie substancji badanej w fazie wodnej w równowadze (μg· g-1, μg ml-1). |
Kf: Współczynnik adsorpcji Freundlicha definiuje się jako stężenie substancji badanej w glebie lub osadzie ściekowym (x/m), gdy stężenie równowagi Caq w fazie wodnej jest równe jeden; jednostką jest μg· g-1 sorbenta. Wartość zmienia się z właściwościami sorbenta.
|
|
(2) |
gdzie:
|
x/m |
= |
ilość substancji badanej x (μg) zaadsorbowanej na ilości sorbenta m (g) w równowadze |
|
1/n |
= |
nachylenie izotermy adsorpcji Freundlicha |
|
Caq |
= |
stężenie substancji badanej w fazie wodnej w równowadze (μg . ml-1) |
At
K oc : Współczynnik podziału (Kd) lub współczynnik adsorpcji Freundlicha (Kf) znormalizowany do zawartości węgla organicznego (foc) sorbenta; w szczególności dla niezjonizowanych substancji chemicznych, jest to przybliżony wskaźnik dla zasięgu adsorpcji między substancją a sorbentem, pozwalający na dokonanie porównań między różnymi substancjami chemicznymi. W zależności od wymiarów Kd i Kf, Koc są bezwymiarowe lub mają jednostki ml . g-1 lub μg . g-1 ciała organicznego.
|
|
(3) |
Zależność między Koc i Kd nie zawsze jest liniowa i w ten sposób wartości Koc mogą różnić się od gleby do gleby, ale ich różnorodność jest znacznie zmniejszona porównując do wartości Kd lub Kf.
Współczynnik adsorpcji (Koc) jest wyprowadzony ze współczynnika zdolności (k'') przy użyciu krzywej kalibracji, wykreślając log k' w zależności od log Koc wybranych związków odniesienia.
|
|
(4) |
gdzie:
|
tR |
= |
HPLC czas retencji substancji badanej i substancji odniesienia (minuty) |
|
t0 |
= |
HPLC czas martwy (minuty) (zob. pkt 1.8.2). |
Pow: współczynnik podziału oktanol-woda jest określony jako stosunek stężenia roztworu substancji w n-oktanolu i wody; jest bezwymiarową wartością.
|
|
(5) |
1.3. SUBSTANCJE ODNIESIENIA
Wzór strukturalny, czystość, stała dysocjacji (jeśli to właściwe) powinny być znane przed zastosowaniem metody. Informacje dotyczące rozpuszczalności w wodzie i rozpuszczalnikach organicznych, współczynniku podziału oktanol-woda oraz cechy hydrolizy są przydatne.
Aby skorelować zmierzone przez HPLC dane retencji substancji badanej i jej współczynnik adsorpcji Koc, ustalana jest krzywa kalibracji log Koc w zależności od log k'. Powinno być zastosowane minimum sześć punktów odniesienia, co najmniej jeden powyżej i jeden poniżej wartości oczekiwanej substancji badanej. Dokładność metody będzie znacząco poprawiona, jeśli substancje wzorcowe są strukturalnie związane z zastosowaną substancją badaną. Jeśli takie dane nie są dostępne, jest zadaniem użytkownika wybranie właściwych substancji wzorcowych. W tym przypadku powinien zostać wybrany bardziej ogólny zestaw strukturalnie heterogenicznych substancji. Substancje stosowane i wartości Κoc są wymienione w dodatku w tabeli 1 dotyczącej osadów ściekowych i w tabeli 3 dotyczącej gleb. Wybór innych substancji kalibracyjnych powinien być uzasadniony.
1.4. ZASADA METODY BADANIA
HPLC wykonuje się na analitycznych kolumnach napełnionych handlowo dostępnym cyjanopropylem w fazie stałej zawierającej lipofilowe i polarne grupy cząsteczkowe. Umiarkowanie stacjonarna faza polarna jest oparta na stosowanej matrycy krzemionkowej:
|
-O-Si |
— CH2 — CH2 — CH2 |
-CN |
|
krzemionka |
niepolarna połowa (odstęp) |
polarna połowa |
Zasada metody badania jest podobna do metody badania A.8 (współczynnik podziału, HPLC metoda). Podczas przejścia przez kolumnę wraz z ruchomą fazą, substancja badana oddziałuje z fazą stacjonarną. W wyniku rozdzielenia między fazami ruchomą i stacjonarną badana substancja jest opóźniona. Podwójny skład fazy stacjonarnej posiadającej polarne i niepolarne powierzchnie pozwala na współdziałanie polarnych i niepolarnych grup cząsteczki w podobny sposób jak dla matryc ciał organicznych w glebie lub osadu ściekowego. Umożliwia to zależności między czasem retencji na kolumnie i współczynnikiem adsorpcji na powstających substancjach organicznych.
Wartość pH ma znaczący wpływ na zachowania sorpcyjne, w szczególności w odniesieniu do substancji polarnych. W odniesieniu do gleb rolniczych lub zbiorników oczyszczalni ścieków pH zwykle zmienia się między 5,5 a 7,5. Dla podatnych na jonizację substancji należy wykonać dwa badania zarówno z formą jonową, jak i niejonową we właściwych roztworach buforowych, ale tylko w przypadkach, gdy co najmniej 10 % związku badanego jest zdysocjowana w zakresie pH 5,5–7,5.
Ponieważ tylko zależność między retencją na kolumnie HPLC a współczynnikiem adsorpcji jest wykorzystywana do oceny, nie jest wymagana analityczna metoda ilościowa i konieczne jest określenie tylko czasu retencji. Jeśli dostępny jest odpowiedni zestaw substancji odniesienia i stosowane mogą być standardowe warunki doświadczalne, metoda stanowi szybki i skuteczny sposób oszacowania współczynnika adsorpcji Κoc.
1.5. MOŻLIWOŚĆ ZASTOSOWANIA BADANIA
Metoda HPLC ma zastosowanie do substancji chemicznych (nieetykietowanych lub etykietowanych) w odniesieniu do których jest dostępny właściwy system detakcji (np. spektrofotometer, detektor radioaktywności) i które są wystarczająco stabilne podczas czasu trwania doświadczenia. Może to być szczególnie użyteczne w odniesieniu do substancji chemicznych które są trudne do badań w innych systemach doświadczalnych (tj. substancje lotne, substancje nierozpuszczalne w wodzie w stężeniu które może być analitycznie zmierzone, substancje z wysokim powinowactwem do powierzchni systemów inkubacyjnych). Metoda jest użyteczna w odniesieniu do mieszanin dających pasma nierozuszczalnej elucji. W takim przypadku górne i dolne granice wartości log Κoc związków mieszaniny badanej powinny być określone.
Zanieczyszczenia mogą czasem powodować problemy dotyczące interpretacji wyników z HPLC, lecz mają małe znaczenie w zakresie w jakim substancja badana może analitycznie być wyraźnie określona oraz oddzielana od zanieczyszczeń.
Metoda jest uzasadniona w odniesieniu do substancji wymienionych w tabeli 1 w dodatku oraz była również stosowana do różnych innych substancji chemicznych należących do następujących chemicznych klas:
|
— |
aminy aromatyczne (np. trifluralin, 4-chloroanilina, 3,5-dinitroanilina, 4-metyloanilina, N-metyloanilina, 1-naftyloamina), |
|
— |
estry aromatycznych kwasów karboksylowych (np. metyloester kwasu benzoesowego, ester etylowy kwasu 3,5-dinitrobenzoesowego), |
|
— |
węglowodory aromatyczne (np. toluen, ksylen, etylobenzen, nitrobenzen), |
|
— |
estry kwasu aryloksyfenoksypropioniowego (np.diclofop-metylowy, fenoxaprop-etylowy, fenoxaprop-P-etylowy), |
|
— |
środki grzybobójcze benzimidazolu i imidazolu (np. carbendazim, fuberidazol, triazoxid), |
|
— |
amidy kwasu karboksylowego (np. 2-chlorobenzamid, N,N-dimetylobenzamid, 3,5-dinitrobenzamid, N-metylobenzamid, 2-nitrobenzamid, 3-nitrobenzamid), |
|
— |
chlorowane węglowodory (np. endosulfan, DDT, heksachlorobenzen, quintozen, 1,2,3-trichlorobenzen), |
|
— |
fosforoorganiczne środki owadobójcze (np. azinphos-metylowy, disulfoton, fenamiphos, isofenphos, pyrazophos, sulprofos, triazophos), |
|
— |
fenole (np. fenol, 2-nitrofenol, 4-nitrofenol, pentachlorofenol, 2,4,6-trichlorofenol, 1-naftol), |
|
— |
pochodne fenylomocznika (np. isoproturon, monolinuron, pencycuron), |
|
— |
pigmentowe barwniki (np. Acid Yellow 219, Basic Blue 41, Direct Red 81), |
|
— |
węglowodory poliaromatyczne (np. acetonaften, naftalen), |
|
— |
środki chwastobójcze 1,3,5-triazyny (np. prometryn, propazine, simazine, terbutryn), |
|
— |
pochodne triazolu (np. tebuconazole, triadimefon, tradimenol, triapenthenol). |
Metody nie stosuje się do substancji, które reagują z eluentem albo z fazą stacjonarną. Nie jest także stosowana do substancji, które oddziaływają w szczególny sposób ze składnikami nieorganicznymi (np. tworzenie grup kompleksów z minerałami gliny). Metoda nie działa z substancjami powierzchniowo czynnymi, związkami nieorganicznymi i umiarkowanymi lub mocnymi organicznymi kwasami i zasadami. Wartości log Κoc w zakresie 1,5–5,0 mogą być określone. Podatne na jonizację substancje muszą być mierzone, stosując buforowaną fazę ruchomą, należy jednak zachować ostrożność, aby zapobiec strącaniu się składników buforu lub substancji badanej.
1.6. KRYTERIA JAKOŚCIOWE
1.6.1. Dokładność
Zwykle współczynnik adsorpcji substancji badanej może być szacowany w zakresie ±0,5 log jednostki wartości określonej przez okresową metodę równowagi (zob. tabela 1 w dodatku). Wyższa dokładność może być uzyskana, jeśli substancje odniesienia zastosowane są strukturalnie związane z substancją badaną.
1.6.2. Powtarzalność
Oznaczenia powinny być prowadzone przynajmniej w dwóch kopiach. Wartości log Κoc wyprowadzone z poszczególnych pomiarów powinny mieścić się w zakresie 0,25 log jednostki.
1.6.3. Odtwarzalność
Doświadczenie uzyskane dotychczas przy zastosowaniu metody podtrzymuje jej zasadność. Badania metodą HPLC, z użyciem 48 substancji (głównie pestycydów), w odniesieniu do których były dostępne wiarygodne dane Κoc na glebach, dało współczynnik korelacji równy R = 0,95 (10)(11).
Między laboratoryjne badanie porównawcze z 11 uczestniczącymi laboratoriami przeprowadzono, aby poprawić i uwiarygodnić metodę (12). Wyniki podano w tabeli 2 dodatku.
1.7. OPISANIE METODY BADANIA
1.7.1. Wstępne oszacowanie współczynnika adsorpcji
Współczynnik podziału oktanol-woda Pow (= Κow) oraz, w pewnym zakresie, wodna rozpuszczalność może być używana jako wskaźnik dla zasięgu adsorpcji w szczególności dla niejonowych substancji, a w ten sposób może być używana do wstępnego znalezienia zakresu. Rozmaitość przydatnych korelacji opublikowano w odniesieniu do kilku grup substancji chemicznych (1)(2)(3)(4)(5)(6)(7).
1.7.2. Aparatura
Wymagane są: cieczowy chromatograf połączony z bezpulsacyjną pompą i odpowiednim urządzeniem wykrywającym. Zalecane jest użycie zaworu wtryskowego z pętlą wtryskową. Należy użyć handlowego cyjanopropylu chemicznie związanego żywicą na bazie krzemionki (np. Hypersil i Zorbax CN). Kolumna ochronna z tego samego materiału może być ustawiona między systemem wtrysku i kolumną analityczną. Kolumny od różnych dostawców mogą różnić się znacznie w wydajności rozdzielania. Jako wytyczne, powinny być osiągnięte następujące masowe proporcje podziału k': log k' > 0,0 dla log Κoc = 3,0 i log k' > 0,4 dla log Κoc = 2,0 przy użyciu metanol/woda 55/45 % jako faza ruchoma.
1.7.3. Fazy ruchome
Zbadano kilka ruchomych faz i następujące dwie są zalecane:
|
— |
metanol/woda (55/45 % obj./obj.), |
|
— |
metanol/0,01M bufor cytrynianowy pH 6,0 (55/45 % obj./obj.). |
Metanol czystości HPLC oraz destylowana woda lub bufor cytrynianowy są używane do przygotowania elucyjnego rozpuszczalnika. Mieszanina jest odgazowywana przed użyciem. Powinna być używana elucja izokratyczna. Jeśli mieszaniny metanol/woda nie są właściwe, należy wypróbować inne mieszaniny rozpuszczalnik organiczny/woda, np. etanol/woda lub acetonitryl/woda. W odniesieniu do związków jonowych zaleca się użycie buforów w celu stabilizacji pH. Należy zachować ostrożność, aby zapobiec wytrącaniu się soli i uszkodzeniu kolumny, co zdarza się przy niektórych mieszaninach faza organiczna/bufor.
Nie można używać żadnych dodatków, takich jak odczynniki z wolną parą elektronową, ponieważ wpływają na właściwości sorpcji fazy stacjonarnej. Takie zmiany w fazie stacjonarnej są nieodwracalne. Z tej przyczyny obowiązkowe jest, aby doświadczenia z użyciem dodatków prowadzić w oddzielnych kolumnach.
1.7.4. Substancje rozpuszczone
Substancje badane i substancje odniesienia powinny być rozpuszczone w fazie ruchomej.
1.8. WYKONANIE BADANIA
1.8.1. Warunki badania
Temperatura w czasie trwania pomiarów powinna być rejestrowana. Użycie segmentu kolumny z kontrolowaną temperaturą jest silnie zalecane w celu zapewnienia stałych warunków w czasie trwania procesu kalibracji, oszacowania i pomiaru substancji badanej.
1.8.2. Oznaczenie czasu martwego to
Stosuje się w odniesieniu do określenia czasu martwego dla dwóch różnych metod (zob. także pkt 1.2).
1.8.2.1. Określenie czasu martwego to za pomocą serii homologicznych
Niniejsza procedura daje wiarygodne i standaryzowane wartości to. W zakresie szczegółowych informacji zob. metoda badania A.8: współczynnik podziału (n-oktanol/woda), metoda HPLC.
1.8.2.2. Określenie czasu martwego to substancjami obojętnymi, niezatrzymywanymi przez kolumnę
Niniejsza technika jest oparta na wtrysku roztworów formamidu, mocznika lub azotanu sodowego. Pomiary powinny być wykonane co najmniej dwa razy.
1.8.3. Oznaczanie czasów retencji tR
Należy wybrać substancje odniesienia, jak opisano w pkt 1.3. Są one wstrzykiwane jako mieszaniny wzorcowe dla ustalenia ich czasów retencji, upewniając się, że czas retencji każdego wzorca nie jest zakłócony przez obecność innego wzorca. Kalibracja powinna być wykonywana w regularnych odstępach czasu, co najmniej dwa razy dziennie, w celu wyjaśnienia niespodziewanych zmian w działaniu kolumny. Dla najlepszego sposobu wstrzyknięcia kalibracyjne powinny być prowadzone przed i po wstrzyknięciu badanej substancji w celu zapewnienia, aby czas retencji nie zmieniał się. Substancje badane są wstrzykiwane oddzielnie, w ilościach tak małych, jak to możliwe (aby zapobiec przeciążeniu kolumny), a ich czasy retencji są określone.
W celu podniesienia zaufania do pomiarów, należy wykonać podwójne określenia. Wartości log Koc uzyskane z poszczególnych pomiarów powinny wchodzić w zakres 0,25 log jednostki.
1.8.4. Ocena
Masowe proporcje podziału k' są obliczane od martwego czasu do czasów retencji tR wybranych substancji odniesienia, według równania 4 (zob. pkt 1.2). Dane log k' dotyczące substancji odniesienia są wykreślane względem ich wartości log Κoc z doświadczeń równowagi wsadowej podanych w tabeli 1 i 3 dodatku. Stosując niniejszy wykres, wartość log k' badanej substancji jest zatem używana do określania wartości log Κoc. Jeśli rzeczywiste wyniki wskazują, że log Κoc substancji badanej jest poza zakresem kalibracji, badanie należy powtórzyć stosując inne, bardziej właściwe substancje odniesienia.
2. DANE I SPRAWOZDAWCZOŚĆ
Sprawozdanie musi zawierać następujące informacje:
|
— |
identyczność substancji badanej i substancji odniesienia ich czystość i wartości pKa, jeśli to stosowne, |
|
— |
opis wyposażenia i warunki działania, np. typ i wymiary kolumn analitycznych (z ochroną), sposoby wykrywania, faza ruchoma (proporcje składników i pH), zakres temperatur w czasie trwania pomiarów, |
|
— |
czas martwy i zastosowana metoda jego określenia, |
|
— |
ilość substancji badanej i substancji odniesienia wprowadzonej do kolumny, |
|
— |
czas retencji różnych związków odniesienia użytych do kalibracji, |
|
— |
szczegółowe informacje dotyczące spasowania linii regresji (log k' vs log Κoc) i wykres linii regresji, |
|
— |
dane średniej retencji i oszacowane wartości d log Koc w odniesieniu do testowanego związku, |
|
— |
chromatogramy. |
3. BIBLIOGRAFIA
|
(1) |
W. J. Lyman, W. F. Reehl, D. H. Rosenblatt (ed). (1990). Handbook of chemical property estimation methods, Chap. 4, McGraw-Hill, New York. |
|
(2) |
J. Hodson, N. A. Williams (1988). The estimation of the adsorption coefficient (Κoc) for soils by HPLC. Chemosphere, 17, 1 67. |
|
(3) |
G. G. Briggs (1981). Theoretical and experimental relationships between soil adsorption, octanol-water partition coefficients, water solubilities, bioconcentration factors, and the parachor. J. Agric. Food Chem., 29, pp. 1050–1059. |
|
(4) |
C. T. Chiou, P. E. Porter, D.W. Schmedding (1983). Partition equilibria of nonionic organic compounds between soil organic matter and water. Environ. Sci. Technol., 17, pp. 227–231. |
|
(5) |
Z. Gerstl, U. Mingelgrin (1984). Sorption of organic substances by soils and sediment. J. Environm. Sci. Health, B19, pp. 297–312. |
|
(6) |
C. T. Chiou, L. J. Peters, V. H. Freed (1979). A physical concept of soil water equilibria for nonionic organic compounds, Science, 106, pp. 831–832. |
|
(7) |
S. W. Karickhoff (1981). Semi-empirical estimation of sorption of hydrophobic pollutants on natural sediments and soils. Chemosphere, 10, pp. 833–846. |
|
(8) |
W. Kördel, D. Hennecke, M. Herrmann (1997). Application of the HPLC-screening method for the determination of the adsorption coefficient on sewage sludges. Chemosphere, 35(1/2), pp. 121–128. |
|
(9) |
M. Mueller, W. Kördel (1996). Comparison of screening methods for the estimation of adsorption coefficients on soil. Chemosphere, 32(12), pp. 2493–2504. |
|
(10) |
W. Kördel, J. Stutte, G. Kotthoff (1993). HPLC-screening method for the determination of the adsorption coefficient in soil-comparison of different stationary phases, Chemosphere, 27(12), pp. 2341–2352. |
|
(11) |
B. von Oepen, W. Kördel, W. Klein (1991). Sorption of nonpolar and polar compounds to soils: Processes, measurements and experience with the applicability of the modified OECD Guideline 106, Chemosphere, 22, pp. 285–304. |
|
(12) |
W. Kördel, G. Kotthoff, J. Müller (1995). HPLC-screening method for the determination of the adsorption coefficient on soil-results of a ring test. Chemosphere, 30(7), pp. 1373–1384. |
DODATEK
Tabela 1
Porównanie wartości Koc w odniesieniu do gleb i osadu ściekowego oraz obliczone wartości metodą rozdzielania HPLC (32), (33)
|
Substancja |
CAS No |
log Κoc osad kanalizacyjny |
log Κoc HPLC |
Δ |
log Κoc gleb |
log Κoc HPLC |
Δ |
|
Atrazyna |
1912-24-9 |
1,66 |
2,14 |
0,48 |
1,81 |
2,20 |
0,39 |
|
Linuron |
330-55-2 |
2,43 |
2,96 |
0,53 |
2,59 |
2,89 |
0,30 |
|
Fention |
55-38-9 |
3,75 |
3,58 |
0,17 |
3,31 |
3,40 |
0,09 |
|
Monuron |
150-68-5 |
1,46 |
2,21 |
0,75 |
1,99 |
2,26 |
0,27 |
|
Fenantren |
85-01-8 |
4,35 |
3,72 |
0,63 |
4,09 |
3,52 |
0,57 |
|
Fenyloester kwasu benzoesowego |
93-99-2 |
3,26 |
3,03 |
0,23 |
2,87 |
2,94 |
0,07 |
|
Benzamid |
55-21-0 |
1,60 |
1,00 |
0,60 |
1,26 |
1,25 |
0,01 |
|
4-nitrobenzamid |
619-80-7 |
1,52 |
1,49 |
0,03 |
1,93 |
1,66 |
0,27 |
|
Acetanilid |
103-84-4 |
1,52 |
1,53 |
0,01 |
1,26 |
1,69 |
0,08 |
|
Anilina |
62-53-3 |
1,74 |
1,47 |
0,27 |
2,07 |
1,64 |
0,43 |
|
2,5-dichloroanilina |
95-82-9 |
2,45 |
2,59 |
0,14 |
2,55 |
2,58 |
0,03 |
Tabela 2
Wyniki interlaboratoryjnego badania porównawczego (11 uczestniczących laboratoriów) przeprowadzonego w celu poprawy i uwiarygodnienia metody HPLC (34)
|
Substancja |
Nr CAS |
log Κoc |
Κoc |
log Κoc |
|
(OECD 106) |
(metoda HPLC) |
|||
|
Atrazine |
1912-24-9 |
1,81 |
78 ± 16 |
1,89 |
|
Monuron |
150-68-5 |
1,99 |
100 ± 8 |
2,00 |
|
Triapenthenol |
77608-88-3 |
2,37 |
292 ± 58 |
2,47 |
|
Linuron |
330-55-2 |
2,59 |
465 ± 62 |
2,67 |
|
Fenthion |
55-38-9 |
3,31 |
2062 ± 648 |
3,31 |
Tabela 3
Zalecane substancje odniesienia dla metody przeglądowej HPLC opartej o dane adsorpcji gleby
|
Substancja wzorcowa |
Nr CAS |
Log Κoc średnie wartości od równowagi wsadowej |
Liczba danych Koc |
Log S.D. |
Źródło |
|
Acetanilid |
103-84-4 |
1,25 |
4 |
0,48 |
|
|
Fenol |
108-95-2 |
1,32 |
4 |
0,70 |
|
|
2-nitrobenzamid |
610-15-1 |
1,45 |
3 |
0,90 |
|
|
N.N-dimetylobenzamid |
611-74-5 |
1,52 |
2 |
0,45 |
|
|
4-metylobenzamid |
619-55-6 |
1,78 |
3 |
1,76 |
|
|
Benzoesan metylu |
93-58-3 |
1,80 |
4 |
1,08 |
|
|
Atrazyna |
1912-24-9 |
1,81 |
3 |
1,08 |
|
|
Isoproturon |
34123-59-6 |
1,86 |
5 |
1,53 |
|
|
3-Nitrobenzamid |
645-09-0 |
1,95 |
3 |
1,31 |
|
|
Anilina |
62-53-3 |
2,07 |
4 |
1,73 |
|
|
3,5-Dinitrobenzamid |
121-81-3 |
2,31 |
3 |
1,27 |
|
|
Carbendazim |
10605-21-7 |
2,35 |
3 |
1,37 |
|
|
Triadimenol |
55219-65-3 |
2,40 |
3 |
1,85 |
|
|
Triazoxide |
72459-58-6 |
2,44 |
3 |
1,66 |
|
|
Triazophos |
24017-47-8 |
2,55 |
3 |
1,78 |
|
|
Linuron |
330-55-2 |
2,59 |
3 |
1,97 |
|
|
Naftalen |
91-20-3 |
2,75 |
4 |
2,20 |
|
|
Endosulfan-diol |
2157-19-9 |
3,02 |
5 |
2,29 |
|
|
Metiocarb |
2032-65-7 |
3,10 |
4 |
2,39 |
|
|
Barwnik Acid Yellow 219 |
63405-85-6 |
3,16 |
4 |
2,83 |
|
|
1,2,3-Trichlorobenzen |
87-61-6 |
3,16 |
4 |
1,40 |
|
|
γ-HCH |
58-89-9 |
3,23 |
5 |
2,94 |
|
|
Fention |
55-38-9 |
3,31 |
3 |
2,49 |
|
|
Barwnik Direct Red 81 |
2610-11-9 |
3,43 |
4 |
2,68 |
|
|
Pyrazophos |
13457-18-6 |
3,65 |
3 |
2,70 |
|
|
α-Endosulfan |
959-98-8 |
4,09 |
5 |
3,74 |
|
|
Diclofop metylowy |
51338-27-3 |
4,20 |
3 |
3,77 |
|
|
Fenantren |
85-01-8 |
4,09 |
4 |
3,83 |
|
|
Barwnik Basic Blue 41 (mieszany) |
26850-47-5 12270-13-2 |
4,89 |
4 |
4,46 |
|
|
ODT |
50-29-3 |
5,63 |
1 |
— |
C.20. BADANIE ROZRODCZOŚCI DAPHNIA MAGNA
1. METODA
Niniejsza metoda badania toksyczności dla rozrodczości jest kopią OECD TG 211 (1998).
1.1. WPROWADZENIE
Głównym celem badania jest ocena skutków działania substancji chemicznych na zdolność rozrodczości Daphnia magna.
1.2. DEFINICJE I JEDNOSTKI
Zwierzęta rodzicielskie: są tymi samicami Daphnia obecnymi na początku badania i których zdolność rozrodczości jest przedmiotem badań.
Potomstwo: są to młode Daphnia produkowane przez zwierzęta rodzicielskie w trakcie badania.
Najniższy obserwowany skutek stężenia (LOEC): jest to najmniejsze badane stężenie substancji badanej, przy którym obserwuje się statystycznie istotny skutek dotyczący rozrodczości i śmiertelności zwierząt macierzystych (przy p < 0,05) przy porównaniu z doświadczeniem kontrolnym, w ramach określonego czasu trwania ekspozycji na badania. Jednakże wszystkie badane stężenia powyżej LOEC muszą wykazywać szkodliwy skutek, równy lub większy niż ten obserwowany przy LOEC. Gdy te dwa warunki nie mogą być spełnione, muszą być podane wszystkie wyjaśnienia w odniesieniu do sposobu w jaki LOEC (i stąd NOEC) zostało wybrane.
Nieobserwowany skutek stężenia (NOEC): jest stężeniem badanym bezpośrednio poniżej LOEC, które, gdy jest porównane z doświadczeniem kontrolnym, nie ma statystyczni istotnego skutku (p < 0,05), w ramach określonego czasu trwania ekspozycji na badanie.
EC x : jest stężeniem substancji badanej rozpuszczonej w wodzie powodującym w x % redukcję w rozrodczości Daphnia magna w ramach okresu ekspozycji na badanie.
Wewnętrzny wskaźnik wzrostu: jest miarą wzrostu populacji, która łączy zdolność rozrodczą i określoną wiekowo śmiertelność (20)(21)(22). W populacji w stanie zrównoważonym jest równy zero. Dla rozwijających populacji będzie dodatni, a dla kurczących się populacji będzie ujemny. Jest oczywiste, że ten drugi jest niepodtrzymujący i ostatecznie prowadzi do zaniku.
Granica wykrywania: najniższe stężenie, które może być wykryte, ale nie można go ilościowo oznaczyć.
Granica oznaczania: najniższe stężenie, które można zmierzyć ilościowo.
Śmiertelność: zwierzę jest rejestrowane jako martwe, gdy jest nieruchome, tj. gdy nie jest w stanie pływać lub nie jest obserwowany ruch przydatków lub odwłoka, w ciągu 15 sekund po łagodnym poruszeniu zbiornika badanego. (Jeśli jest stosowana inna definicja, musi być to zrelacjonowane wraz z odniesieniem).
1.3. ZASADA METODY BADANIA
Młode samice Daphnia (zwierzęta rodzicielskie), w wieku poniżej 24 godzin na początku badania, są wystawiane na działanie substancji badanej dodanej do wody w zakresie stężeń. Czas trwania badania wynosi 21 dni. Na końcu badania szacowana jest całkowita ilość żyjącego potomstwa wyprodukowanego na zwierzę rodzicielskie, które przeżyło do końca badania. To oznacza, że młode wyprodukowane przez dorosłe zmarłe w czasie trwania badania są wykluczone z obliczeń. Zdolność rozrodcza zwierząt rodzicielskich może być wyrażona w inny sposób (np. liczba żyjącego potomstwa wyprodukowanego na zwierzę na dzień od pierwszego dnia zaobserwowania potomstwa), lecz powinno to być przedstawione jako dodatek do całkowitej liczby młodocianych wyprodukowanych na żywych rodziców na końcu badania. Zdolność rozrodcza zwierząt wystawianych na działanie substancji badanej jest porównywana do tej z doświadczenia kontrolnego w celu ustalenia najniższego obserwowanego skutku stężenia (LOEC), a stąd nieobserwowanego skutku stężenia (NOEC). Ponadto, w najszerszym możliwym zakresie, dane analizuje się, stosując model regresji w celu prognozowania stężenia powodującego x % zmniejszenia zdolności rozrodczej (tj. EC50, EC20 lub EC10).
Przeżycie zwierząt rodzicielskich i czas do produkcji potomstwa musi być również przedstawiony. Inne skutki związane z substancją dotyczące parametrów takich jak rozwój (np. długość) i możliwy wewnętrzny wskaźnik wzrostu może być również zbadany.
1.4. INFORMACJE DOTYCZĄCE SUBSTANCJI UŻYTEJ W BADANIU
Powinny być dostępne wyniki badania toksyczności ostrej (zob. metoda C.2, część I) wykonanej na Daphnia magna. Wyniki mogą być użyteczne przy wyborze właściwego zakresu stężeń badanych w badaniach rozrodczości. Rozpuszczalność w wodzie i ciśnienie pary substancji badanej powinny być znane, jak również powinna być dostępna niezawodna metoda określania ilościowego substancji w roztworach badanych o udokumentowanej wydajności i granicy określenia.
Informacje dotyczące substancji badanej, które mogą być użyteczne w ustalaniu warunków badania, obejmują: wzór strukturalny, czystość substancji, stabilność w świetle, stabilność w warunkach badania, pKa, Pow i wyniki badania szybkiej biodegradacji (zob. metoda C.4).
1.5. WAŻNOŚĆ BADANIA
Aby badanie było ważne, powinny być spełnione następujące kryteria wykonania doświadczeń kontrolnych:
|
— |
śmiertelność zwierząt rodzicielskich (samice Daphnia) nie przewyższa 20 % na koniec badania, |
|
— |
średnia liczba żyjącego potomstwa wyprodukowanego na zwierzę rodzicielskie przeżywające na koniec badania wynosi ≥ 60. |
1.6. OPIS METODY BADANIA
1.6.1. Aparatura
Zbiorniki do badań i inna aparatura, która wchodzi w kontakt z roztworami badanymi, powinny być wykonane całkowicie ze szkła lub innego chemicznie obojętnego materiału. Zbiornikami do badań będą zwykłe szklane zlewki.
Ponadto wymagane są wszystkie lub kilka z następujących sprzętów:
|
— |
miernik tlenu (z mikroelektrodą lub innym odpowiednim wyposażeniem do pomiaru rozpuszczonego tlenu w próbkach o małej objętości), |
|
— |
odpowiedni aparat do kontroli temperatury, |
|
— |
pH metr, |
|
— |
wyposażenie do określenia twardości wody, |
|
— |
sprzęt do określenia całkowitego węgla organicznego (TOC) w wodzie lub wyposażenie do określenia zapotrzebowania na chemiczny tlen (COD), |
|
— |
odpowiednia aparatura do kontroli parametrów światła i pomiaru intensywności oświetlenia. |
1.6.2. Organizmy użyte w badaniu
Gatunkiem wykorzystywanym w badaniu jest Daphnia magna Straus. Inne gatunki Daphnia wykorzystuje się, zapewniając, że odpowiadają właściwym kryteriom ważności (kryterium ważności dotyczące zdolności rozrodczej przy doświadczeniach kontrolnych powinny być właściwe w odniesieniu do gatunku Daphnia). Jeśli wykorzystane są inne gatunki Daphnia, muszą być one wyraźnie określone i ich wykorzystanie musi być uzasadnione.
Najlepiej, aby klon był określony za pomocą genotypowania. Badania (1) wykazały, że zdolności reprodukcyjne klonu A (pochodzącego z IRCHA we Francji) (3) konsekwentnie odpowiada kryterium ważności średniej ≥ 60 potomstwa na rodzicielskie zwierzę przeżywające, gdy jest hodowane zgodnie z warunkami opisanymi w niniejszej metodzie. Jednakże inne klony są akceptowalne, pod warunkiem że wykazano że kultura Daphnia spełnia kryteria ważności dotyczące badania.
Na początku badania zwierzęta powinny być młodsze niż 24 godziny i nie muszą być pierwszym potomstwem. Powinny one pochodzić ze zdrowej hodowli (tj. niewykazujące takich oznak stresu, jak wysoka śmiertelność, obecności samców i ephippia, opóźnień w produkcji pierwszego potomstwa, zwierząt odbarwionych itd.). Podstawowe zwierzęta muszą być utrzymywane w warunkach właściwych dla ich hodowli (światło, temperatura, ośrodek, karmienie i ilość zwierząt na jednostkę objętości), zbliżonych do tych stosowanych w badaniu. Jeśli ośrodek hodowli Daphnia używany w badaniu różni się od rutynowego dla hodowli Daphnia, jest dobrą praktyką włączyć okres aklimatyzacji przed badaniem, zwykle około trzech tygodni (tj. jedno pokolenie), aby zapobiec stresowi zwierząt rodzicielskich.
1.6.3. Ośrodek badania
Jest zalecane, aby w badaniu użyć w pełni określony ośrodek. Umożliwia to zapobieżenie użyciu dodatków (np. wodorostów, ekstraktu ziemi itd.), które trudno scharakteryzować, a w ten sposób poprawić możliwości standaryzacji między laboratoriami. Ośrodki Elendt M4 (4) i M7 (zob. dodatek 1) uznano za właściwe do tego celu. Są też akceptowane inne ośrodki odpowiednie w tym celu (np. (5)(6)) które dostarczają zdolności dla hodowli Daphnia spełnienia kryteriów ważności dotyczących badania.
Jeśli stosowane ośrodki zawierają niezdefiniowane dodatki, powinny być one wyraźnie określone, a informacja o nich powinna być zawarta w sprawozdaniu z badań dotyczącym ich składu, w szczególności w odniesieniu do zawartości węgla organicznego, mogącą wpływać na dostarczaną dietę. Jest zalecane, aby całkowity węgiel organiczny (TOC) i/lub zapotrzebowanie na tlen chemiczny (COD) w przygotowanych dodatkach ośrodka podstawowego był ustalony i dokonany szacunek wynikowego udziału TOC/COD w ośrodku badanym. Zalecane jest aby poziomy TOC w ośrodku (tj. przed dodaniem alg) były poniżej 2 mg/l (7).
W przypadku substancji badanych, zawierających metale, istotne jest aby uwzględniać, że właściwości ośrodka (np. twardość, zdolność chelatującą) mogą wpływać na toksyczność badanej substancji. Z tej przyczyny jest pożądany w pełni zdefiniowany ośrodek. Jednakże obecnie jedynymi w pełni zdefiniowanymi ośrodkami uznanymi za odpowiednie dla hodowli długoterminowej Daphnia magna są Elendt M4 i M7. Oba ośrodki zawierają związek chelatujący EDTA. Prace wykazały (2), że „oczywista toksyczność” kadmu jest ogólnie niższa, gdy badanie rozrodczości jest prowadzone w ośrodkach M4 i M7 niż w ośrodkach niezawierających EDTA. M4 i M7 dlatego nie są zalecane do badań substancji zawierających metale, a inne ośrodki zawierające znane środki chelatujące powinny być również unikane. W odniesieniu do substancji zawierających metale może być wskazane użycie alternatywnego ośrodka, takiego jak na przykład odtworzona świeża twarda woda ASTM (7), która nie zawiera EDTA, z dodatkiem ekstraktu wodorostów (8). Ta kombinacja odtworzonej świeżej twardej wody ASTM i ekstraktu wodorostów jest także odpowiednia dla długoterminowej hodowli oraz badania Daphnia magna (2), chociaż obserwuje się łagodne działanie chelatujące z powodu składnika organicznego w dodanym ekstrakcie wodorostu.
Na początku i w trakcie trwania badania stężenie rozpuszczonego tlenu powinno być powyżej 3 mg/l. Wartość pH powinna być w zakresie 6–9 i zwykle nie powinna różnić się o więcej niż 1,5 jednostki w każdym pojedynczym badaniu. Twardość powyżej 140 mg/l (jako CaCO3) jest zalecana. Badania na tym poziomie i powyżej wykazały zdolność rozrodczą w zgodności z kryteriami ważności (9)(10).
1.6.4. Roztwory użyte w badaniu
Roztwory użyte o wybranych stężeniach są zwykle przygotowywane przez rozcieńczenie roztworu podstawowego. Zalecane jest przygotowanie roztworów podstawowych przez rozpuszczenie substancji badanej w ośrodku badanym.
W niektórych przypadkach stosowanie rozpuszczalników organicznych lub dyspergatorów może być wymagane w celu uzyskania odpowiednio stężonego roztworu podstawowego, ale należy dołożyć wszelkich starań, aby zapobiec użyciu takich materiałów. Przykładami odpowiednich rozpuszczalników są aceton, etanol, metanol, dwumetyloformamid i glikol trójetylenowy. Przykładami odpowiednich dyspergatorów są Cremophor RH40, metyloceluloza 0,01 % i HCO-40. W każdym przypadku substancja badana w roztworach badanych nie powinna przekraczać granicy rozpuszczalności w ośrodku badanym.
Rozpuszczalniki są stosowane do wykonania roztworu podstawowego, który może być dozowany ściśle do wody. W zalecanym stężeniu rozpuszczalnika w końcowym ośrodku badanym (tj. < 0,1 ml/l) rozpuszczalniki wymienione powyżej nie będą toksyczne i nie mogą podnosić rozpuszczalności substancji w wodzie.
Dyspergatory mogą pomagać w dokładnej dawce i rozproszeniu. Przy zalecanym stężeniu w końcowym ośrodku badanym (< 0,1 ml/l) dyspergatory wymienione powyżej nie będą toksyczne i nie będą podnosić rozpuszczalności substancji w wodzie.
1.7. PROJEKT BADANIA
Traktowanie powinno być rozdzielone do zbiorników do badań, a wszystkie następujące czynności ze zbiornikami do badań powinny być prowadzone metodą losową. Niezastosowanie się do tego może powodować powstanie błędów systematycznych, które mogą być zinterpretowane jako wpływ stężenia. W szczególności jeśli jednostki doświadczalne są obsługiwane według traktowania lub stężenia, wtedy niektóre związane z czasem skutki, takie jak zmęczenie operatora lub inny błąd mogą prowadzić do poważniejszych skutków przy wyższych stężeniach. Ponadto, jeśli na wyniki badania prawdopodobnie wpłyną początkowe lub środowiskowe warunki, takie jak położenie w laboratorium, należy rozważyć wykonanie badania blokowego.
1.8. PROCEDURA
1.8.1. Warunki ekspozycji
1.8.1.1. Czas trwania
Czas trwania badania wynosi 21 dni.
1.8.1.2. Załadunek
Zwierzęta rodzicielskie są utrzymywane pojedynczo, jedno na zbiornik do badań, z 50–100 ml ośrodka w każdym zbiorniku.
Większe objętości czasem są niezbędne do spełnienia wymagań procedury analitycznej stosowanej do określania stężenia substancji badanej, chociaż zakładanie kopii w odniesieniu do analizy chemicznej jest też dopuszczalne. Jeśli stosuje się objętości większe niż 100 ml, racje podawane Daphnia mogą wymagać zwiększenia w celu zapewnienia odpowiedniej dostępności pożywienia i zgodności z kryteriami ważności. W odniesieniu do badań przepływowych alternatywne projekty mogą być rozważane, z przyczyn technicznych (np. cztery grupy po 10 zwierząt w większej objętości badanej) lecz wszystkie zmiany projektu badania powinny być przedstawione.
1.8.1.3. Liczba zwierząt
W odniesieniu do półstatycznych badań co najmniej 10 zwierząt jest trzymanych pojedynczo przy każdym stężeniu badanym i co najmniej 10 zwierząt trzymanych pojedynczo w seriach kontrolnych.
W odniesieniu do badań przepływowych 40 zwierząt, podzielonych na grupy po 10 zwierząt w każdym stężeniu badanym, okazało się być odpowiednie (1). Mniejsza liczba organizmów badanych może być wykorzystana, a minimum 20 zwierząt na stężenie, podzielonych w dwie lub więcej kopii z równą ilością zwierząt (np. cztery kopie każda z pięcioma daphnid. Należy zauważyć, że w odniesieniu do badań, w których zwierzęta są trzymane w grupach, nie jest możliwe wyrażenie zdolności rozrodczej jako całkowitej liczby żyjącego potomstwa wyprodukowanego na rodzicielskie zwierzę, które żyje na końcu badania, jeśli zwierzęta rodzicielskie zmarły. W tych przypadkach zdolność rozrodcza powinna być wyrażona jako „całkowita liczba żyjącego potomstwa wyprodukowanego na rodzica obecnego na początku badania”.
1.8.1.4. Karmienie
W odniesieniu do badań półstatycznych karmienie powinno być wykonywane najlepiej codziennie, lecz przynajmniej trzy razy na tydzień (tj. zgodnie z wymianami ośrodka). Odchylenia od tego (np. w odniesieniu do badań przepływowych) powinno być przedstawione.
W czasie trwania badania dieta zwierząt rodzicielskich powinno składać się z żyjących komórek glonowych jednego lub więcej z następujących typów: Chlorella sp., Selenastrum capricornutum (obecnie Pseudokirchneriella subcapitata (11)) i Scenedesmus subspicatus. Dostarczana dieta powinna być oparta na ilości węgla organicznego dostarczanego każdemu zwierzęciu rodzicielskiemu. Badania (12) wykazały, że dla Daphnia magna poziomy racji między 0,1 i 0,2 mg C/Daphnia/dzień są wystarczające do uzyskania odpowiedniej liczby potomstwa spełniającego kryteria ważności. Racje dostarczane są jako stały wskaźnik poprzez czas badania albo, jeśli to jest pożądane, stosując niski wskaźnik na początku i zwiększany w czasie trwania badania, uwzględniając rozwój zwierząt rodzicielskich. W tym przypadku racje powinny nadal pozostawać w granicach zalecanego zakresu 0,1–0,2 mg C/Daphnia/dzień przez cały czas.
Jeśli mają być stosowane środki zastępcze, takie jak ilość komórek glonowych lub absorpcja światła, do karmienia na wymaganym poziomie racji (tj. dla wygody ponieważ pomiar zawartości węgla zabiera czas), każde laboratorium musi wykonać swój własny nomogram odnoszący się do zależności środka zastępczego oraz zawartości węgla hodowli glonów (zob. dodatek 2 dotyczący zaleceń odnoszących się do wykonania nomogramu). Nomogramy powinny być sprawdzane co najmniej corocznie i częściej, gdy warunki hodowli glonów uległy zmianie. Odkryto, że absorpcja światła jest lepszą namiastką dla zawartości węgla niż ilość komórek (13).
W celu zminimalizowania objętości kultur glonów dostarczanych do ośrodka zbiorników do badań Daphnia powinny być karmione koncentratem glonów w nośniku. Stężenie glonów może być uzyskane przez odwirowanie zawiesiny w destylowanej albo dejonizowanej wodzie, z pierwotnej zawiesiny lub z ośrodka kultur Daphnia.
1.8.1.5. Światło
16 godzin światła przy intensywności nieprzekraczającej 15–20 uE • m-2 • s-1.
1.8.1.6. Temperatura
Temperatura ośrodka badanego powinna być w zakresu 18–22 oC. Jednakże w odniesieniu do jednego badania nie może, jeśli to możliwie, różnić się o więcej niż 2 oC w ramach tych granic (np. 18–20, 19–21 lub 20–22 oC). Właściwe może być użycie dodatkowego zbiornika badanego do celów monitorowania temperatury.
1.8.1.7. Napowietrzanie
W czasie trwania badania zbiorniki do badań nie mogą być napowietrzane.
1.8.2. Stężenie użyte w badaniu
Zwykle powinno być co najmniej pięć stężeń ułożonych w serii geometrycznej ze wskaźnikiem odstępu najlepiej nieprzekraczającym 3,2 oraz właściwej liczbie kopii dla każdego stężenia badanego (zob. pkt 1.8.1.3). Powinno być przedstawione uzasadnienie w przypadku zastosowania mniej niż pięciu stężeń. Nie powinny być badane substancje powyżej ich granicy rozpuszczalności w ośrodku badanym.
W określaniu zakresu stężeń należy uwzględnić do następuje:
|
(i) |
jeśli celem jest uzyskanie LOEC/NOEC, najniższe stężenie badane musi być wystarczająco niskie, aby płodność przy tym stężeniu nie była znacząco niższa niż w przy doświadczeniu kontrolnym. W innym przypadku badanie będzie musiało być powtórzone ze zmniejszonym najniższym stężeniem; |
|
(ii) |
jeśli celem jest uzyskanie LOEC/NOEC, najwyższe stężenie badane musi być wystarczająco wysokie, aby płodność przy tym stężeniu była znacząco niższa niż w doświadczeniu kontrolnym. W innym przypadku badanie będzie musiało być powtórzone ze zwiększonym najwyższym stężeniem; |
|
(iii) |
jeśli szacowane jest ECX odnoszące się do skutków dotyczących rozrodczości wskazane jest użycie wystarczającego stężenia w celu określenia ECX z właściwym poziomem wiarygodności. Jeśli szacowany jest EC50 odnoszące się do skutków dotyczących rozrodczości, wskazane jest, aby najwyższe stężenie badane było wyższe niż ten EC50. Inaczej, choć będzie jeszcze możliwe oszacowanie EC50, przedział ufności dla EC50 będzie bardzo szeroki i nie będzie możliwe zadawalające ocenienie odpowiedniości dopasowanego modelu; |
|
(iv) |
zakres stężeń badanych nie powinien obejmować żadnych stężeń, które mają statystycznie znaczący wpływ na przeżycie dorosłych, ponieważ powoduje to zmianę charakteru badania z badania prostej rozrodczości na badanie złożonej rozrodczości i śmiertelności, wymagający bardziej złożonej analizy statystycznej. |
Uprzednia wiedza dotycząca toksyczności substancji badanej (np. z badania ostrego i/lub badań szukania zakresu) powinna być pomocna w wyborze właściwych stężeń badanych.
W przypadku gdy rozpuszczalnik lub dyspergator stosowano do pomocy w przygotowaniu roztworów badanych (zob. pkt 1.6.4), ich końcowe stężenie w zbiornikach do badań nie powinno przekraczać 0,1 ml/l oraz powinno być takie samo we wszystkich zbiornikach do badań.
1.8.3. Doświadczenia kontrolne
Do serii badanej dodatkowo musi być włączona jedna seria kontrolna badanego ośrodka oraz, jeżeli to właściwe, jedna seria kontrolna zwierająca rozpuszczalnik lub dyspergator. Gdy stosowany jest rozpuszczalnik lub dyspergator ich stężenie powinno być takie samo jak używane w zbiornikach zawierających substancję badaną. Należy użyć właściwą ilość kopii (zob. pkt 1.8.1.3).
Ogólnie, w dobrze przebiegającym badaniu współczynnik zmian wokół średniej ilości żyjącego potomstwa produkowanego urodzonego na jedno rodzicielskie zwierzę w doświadczeniu(-ach) kontrolnym(-ch) powinna wynosić < 25 % oraz powinno to być przedstawione w odniesieniu do projektów badania stosującego pojedynczo trzymane zwierzęta.
1.8.4. Odnawianie ośrodka użytego w badaniu
Częstotliwość odnawiania ośrodka zależy od stabilności substancji badanej, lecz powinna następować co najmniej trzy razy na tydzień. Jeśli z wstępnego badania stabilności (zob. pkt 1.4) wynika, że stężenie substancji badanej nie jest stabilne (tj. poza zakresem 80–120 % nominalnego lub spada poniżej 80 % mierzonego stężenia początkowego) poza maksymalnym okresem odnawiania (tj. trzy dni), należy rozważyć częstszą wymianę ośrodka lub zastosowania badania przepływowego.
Gdy ośrodek jest wymieniany w badaniach półstatycznych, przygotowuje się drugą serię zbiorników do badań, a zwierzęta rodzicielskie są do nich przenoszone, na przykład szklaną pipetą o odpowiedniej średnicy. Objętość ośrodka przenoszonego z Daphnia powinna być zminimalizowana.
1.8.5. Obserwacje
Wyniki obserwacji dokonanych w trakcie trwania badania powinny być odnotowane w arkuszach danych (zob. przykłady w dodatkach 3 i 4). Jeśli inne pomiary są wymagane (zob. 1.3 i 1.8.8) dodatkowe obserwacje mogą być wymagane.
1.8.6. Potomstwo
Potomstwo wyprodukowane prze każde zwierzę rodzicielskie powinno być usunięte i policzone codziennie od pojawienia się pierwszego potomstwa, w celu powstrzymania ich przed zjedzeniem pokarmu przeznaczonego dla dorosłych. Do celów niniejszej metody potrzebne jest tylko liczenie ilości żyjącego potomstwa, lecz obecność przerwanych jaj lub martwego potomstwa powinna być także odnotowana.
1.8.7. Śmiertelność
Śmiertelność zwierząt rodzicielskich powinna być rejestrowana najlepiej codziennie, co najmniej w tych samych porach, gdy liczone jest potomstwo.
1.8.8. Inne parametry
Chociaż niniejsza metoda jest przeznaczona zasadniczo do oceny skutków dotyczących rozrodczości, możliwe jest, że inne efekty zostaną wystarczająco określone, aby umożliwić ich statystyczną analizę. Wysoce pożądane są pomiary rozwoju, ponieważ dostarczają one informacji dotyczących możliwych skutków podśmiertelnych, które mogą być bardziej użyteczne niż tylko sam pomiar rozrodczości; pomiar długości zwierząt rodzicielskich (tj. długość ciała, wyłączając kolec odbytowy) jest zalecany na końcu badania. Inne parametry do zmierzenia lub obliczenia obejmują: czas do produkcji pierwszego potomstwa (i dalszego potomstwa), liczbę i rozmiary potomstwa na zwierzę, liczbę poronień, obecność samic lub ephippię oraz wewnętrzny wskaźnik przyrostu populacji.
1.8.9. Częstotliwość analitycznych określeń i pomiarów
Stężenie tlenu, temperatura, wartości twardości i pH powinny być mierzone co najmniej raz tygodniowo, w świeżych i starych roztworach, w kontrolnych i w najwyższych stężeniach badanej substancji.
W czasie trwania badania stężenia substancji użytej w badaniu są określane w regularnych odstępach czasu.
W półstatycznych badaniach, w przypadku gdy oczekuje się, że stężenie substancji pozostanie w zakresie ± 20 % nominału (tj. w zakresie 80–120 % – zob. pkt 1.4 i 1.8.4), jest zalecane, aby jako minimum analizować najniższe i najwyższe stężenia użyte w badaniu świeżo po przygotowaniu i zaraz przed odnowieniem, po jednym w czasie trwania pierwszego tygodnia badania (tj. analizy powinny być dokonane na próbce z tego samego roztworu – gdy świeżo przygotowany i przy odnowieniu). Te określenia powinny być powtarzane przynajmniej w odstępach tygodniowych, w późniejszym czasie.
W odniesieniu do badań, w których nie oczekuje się, że stężenie substancji użytej w badaniu pozostanie w zakresie ± 20 % nominału, konieczne jest analizowanie wszystkich stężeń po świeżym przygotowaniu i przy odnowieniu. Jednakże w odniesieniu do tych badań, w których zmierzone początkowe stężenie nie jest w zakresie ± 20 % nominału, lecz gdy mogą być dostarczone wystarczające dowody wskazujące, że wstępne stężenia są powtarzalne i stabilne (tj. w zakresie 80–120 % stężenia początkowego), oznaczenia chemiczne mogą być zmniejszone w tygodniu 2 i 3 badania do najwyższego i najniższego stężenia badanego. We wszystkich przypadkach oznaczenie stężenia substancji użytej w badaniu przed odnowieniem wymaga jedynie prowadzenia na jednym kopiowanym zbiorniku dla każdego stężenia badanego.
W odniesieniu do badań przepływowych właściwe jest użycie podobnego do opisanego dla badań półstatycznych, sposobu pobierania próbek (lecz pomiar „starych” roztworów nie jest stosowany w tym przypadku). Jednakże wskazane jest zwiększenie ilości pobieranych próbek podczas pierwszego tygodnia (np. trzy zestawy pomiarów) w celu zapewnienia, aby stężenia badane pozostały stałe. W tego typu badaniach jednostka przepływu rozcieńczalnika i substancji badanej powinny być sprawdzane codziennie.
Jeśli istnieją dowody, że stężenie substancji badanej jest zadawalająco utrzymywane w granicach ± 20 % nominalnego lub zmierzonego początkowego stężenia poprzez badanie, wtedy wyniki oparte są o nominalne lub zmierzone początkowe wartości. Jeśli odchylenie od nominalnego lub zmierzonego stężenia jest większe niż ± 20 %, wyniki muszą być wyrażone w średniej ważonej czasu (zob. dodatek 5).
2. DANE I SPRAWOZDAWCZOŚĆ
2.1. OPRACOWANIE WYNIKÓW
Celem niniejszego badania jest ustalenie działania substancji użytej w badaniu na całkowitą liczbę żywego potomstwa wyprodukowanego na zwierzę rodzicielskie, żyjące na koniec badania. Całkowita liczba potomstwa na zwierzę rodzicielskie powinna być obliczona w odniesieniu do każdego badanego zbiornika (tj. kopii). Jeśli w jakimś skopiowanym zbiorniku zwierze rodzicielskie zmarło w czasie trwania badania lub okazało się samcem, to ta kopia jest wyłączona z analizy. Analiza jest wtedy oparta na zmniejszonej ilości kopii.
W odniesieniu do oszacowania LOEC, a stąd NOEC, w odniesieniu do skutków substancji chemicznych dotyczących zdolności rozrodczej, konieczne jest obliczenie średniej zdolności rozrodczej poprzez kopie dla każdego stężenia i złożonego pozostałego standardowego odchylenia poprzez użycie analizy wariancji (ANOVA). Średnia w odniesieniu do każdego stężenia musi być porównana ze średnią doświadczenia kontrolnego, stosując właściwą metodę wielokrotnego porównania. Użyteczne są badania Dunnetta lub Williamsa (14)(15)(16)(17). Koniecznie należy sprawdzić, czy założenie metody ANOVA homogeniczności odchylenia jest utrzymane. Jest zalecane, aby zostało to wykonane w sposób graficzny niż poprzez formalnie znaczące badanie (18); odpowiednią alternatywą jest przeprowadzenie badania Bartletta. Jeśli to założenie nie jest utrzymane, należy rozważyć przekształcenie danych do shomogenizowania wariancji, przed przeprowadzeniem ANOVA albo przeprowadzić ważoną ANOVA. Zakres skutków wykrywalnych przy zastosowaniu ANOVA (tj. najmniej znaczącej różnicy) powinien być obliczony i odnotowany.
Dla oszacowania stężenia, które spowoduje 50 % zmniejszenia zdolności rozrodczej (tj. EC50), odpowiednia krzywa, taka jak krzywa logistyczna powinna być dopasowana do danych, używając metodę statystyczną, taką jak najmniejszych kwadratów. Krzywa musi być sparametryzowana, tak aby EC50 i jej błąd standardowy można było obliczyć bezpośrednio. Ułatwiałoby to obliczenie granic ufności w zakresie EC50. O ile istnieją słuszne przyczyny, aby preferować różne poziomy ufności, dwustronne 95 % granice ufności powinny być podawane. Procedura spasowania powinna zasadniczo dostarczać środki oceny znaczenia braku spasowania. Może to być wykonane graficznie lub przez podzielenie pozostałej sumy kwadratów na „brak pasowania” i „czyste składniki błędu”, a przeprowadzenie badania istotności w odniesieniu do braku spasowania. Ponieważ obróbka dająca wysoką płodność prawdopodobnie zapewni większe odchylenie w ilości młodych osobników wyprodukowanych niż obróbka dająca niską płodność, muszą być podjęte rozważania do zważenia obserwowanych wartości dla odzwierciedlenia różnych wariancji w różnych opracowywanych grupach (zob. podstawowe informacje w bibliografii (18)).
W analizie danych z końcowego badania obrączkowego (2) spasowano krzywą logistyczną, posługując się następującym modelem, chociaż inny odpowiedni model może być użyty:

gdzie:
|
Y |
= |
całkowita liczba młodych osobników na zwierzę rodzicielskie żyjące na koniec badania (obliczone w odniesieniu każdego zbiornika), |
|
x |
= |
stężenie substancji, |
|
c |
= |
oczekiwana ilość młodych osobników, gdy x = 0, |
|
x0 |
= |
EC50 w populacji, |
|
b |
= |
parametr nachylenia. |
Niniejszy model jest prawdopodobnie odpowiedni w dużej ilości sytuacji, ale są badania, dla których nie jest właściwy. Ważność powyższego sugerowanego modelu powinna zostać sprawdzona. W niektórych przypadkach model hormesis, w którym niskie stężenia powodują lepsze wyniki może być właściwy (19).
Inne skutki stężeń, takie jak EC10 lub EC20, mogą również być oszacowane, chociaż bardziej pożądane jest użycie różnej parametryzacji modelu, z którego oszacowano EC50.
2.2. SPRAWOZDANIE Z BADANIA
Sprawozdanie z badania musi zawierać następujące dane:
2.2.1. Substancja użyta w badaniu:
|
— |
charakter fizyczny i stosowne właściwości fizykochemiczne, |
|
— |
dane identyfikacji chemicznej, włączając czystość. |
2.2.2. Gatunki użyte w badaniu:
|
— |
klon (czy był typowany genetycznie), dostawca lub źródło (jeśli jest znane) i warunki stosowanej hodowli. Jeśli użyto innego gatunku niż Daphnia powinno to być przedstawione i uzasadnione. |
2.2.3. Warunki badania:
|
— |
stosowana procedura badania (np. półstatyczna lub przepływowa, objętość, obciążenie w ilości Daphnia na litr), |
|
— |
czas trwania naświetlania i intensywność światła, |
|
— |
projekt badania (np. ilość kopii, ilość rodziców na kopię), |
|
— |
szczegółowe informacje dotyczące kultur stosowanego ośrodka, |
|
— |
jeśli stosowano, dodatki materiałów organicznych, włączając ich skład, źródło, metodę przygotowania, TOC/COD preparatów podstawowych, oszacowanie wynikłego TOC/COD w ośrodku badanym, |
|
— |
szczegółowe informacje o karmieniu, włączając ilość (w mg C/Daphnia/dzień) i plan (np. typ karmy, włączając dla glonów właściwą nazwę, gatunek i – jeśli znany szczep – warunki kultur), |
|
— |
metoda przygotowania roztworów podstawowych i częstotliwość odnawiania (muszą być podane stężenia rozpuszczalników i dyspergantów, jeśli je użyto). |
2.2.4. Wyniki:
|
— |
wyniki ze wszystkich wstępnych badań dotyczące stabilności substancji badanej, |
|
— |
nominalne stężenia badane i wyniki wszystkich analiz ustalających stężenia substancji badanej w zbiornikach do badań (zob. przykładowy arkusz danych w dodatku 4); zdolność wyjściową metody i granice oznaczania powinny być również przedstawione, |
|
— |
jakość wody w zbiornikach do badań (tj. pH, temperatura, stężenie rozpuszczonego tlenu, TOC i/lub COD oraz twardość, tam gdzie to ma zastosowanie) (zob. przykładowy arkusz danych w dodatku 3), |
|
— |
pełny zapis żyjącego potomstwa każdego zwierzęcia rodzicielskiego (zob. przykładowy arkusz danych w dodatku 3), |
|
— |
ilość zgonów między zwierzętami rodzicielskimi i data kiedy nastąpiły (zob. przykładowy arkusz danych w dodatku 3), |
|
— |
współczynnik rozrzutu dla kontroli płodności (oparty na całkowitej ilości żyjącego potomstwa na ilość żywych pod koniec badania zwierząt rodzicielskich), |
|
— |
wykres całkowitej liczby żyjącego potomstwa na zwierzę rodzicielskie (w odniesieniu do każdej kopi) żyjące na koniec badania w zależności od stężenia substancji badanej, |
|
— |
najniższy obserwowany skutek stężenia (LOEC) dla rozrodczości, włączając opis zastosowanych procedur statystycznych i wskazanie zakresu skutków jaki jest wykrywany oraz nie obserwowany skutek stężenia (NOEC) dla rozrodczości; gdzie stosowne, należy także przedstawić LOEC/NOEC w odniesieniu do śmiertelności zwierząt rodzicielskich, |
|
— |
tam gdzie to jest stosowne, ECX w odniesieniu do rozrodczości i przedziałów zaufania oraz wykres dopasowanego modelu użytego do obliczeń, nachylenie krzywej dawka – reakcja i jej błąd standardowy, |
|
— |
inne obserwowane skutki biologiczne lub pomiary: przedstawienie wszystkich innych biologicznych skutków zaobserwowanych lub pomierzonych (np. rozwój zwierząt rodzicielskich) włączając właściwe uzasadnienie, |
|
— |
wytłumaczenie wszystkich odchyleń od metody badania. |
3. BIBLIOGRAFIA
|
(1) |
OECD Test Guideline Programme, Report of the Workshop on the Daphnia magna Pilot Ring Test, Sheffield University, UK, 20-21 March 1993. |
|
(2) |
OECD Environmental Health and Safety Publications. Series on Testing and Assessment No. 6. Report of the Final Ring Test of the Daphnia magna Reproduction Test Paris. 1997. |
|
(3) |
Baird D. J., Barber J., Bradley M. C, Soares A. M. V M. and Calow P. (1991). A comparative study of genotype sensitivity to acute toxic stress using clones of Daphnia magna Strauss. Ecotoxicology and Environmental Safety, 21, pp. 257–265. |
|
(4) |
Elendt B. P., (1990). Selenium deficiency in Crustacea; An ultrastructural approach to antennal damage in Daphnia magna Straus. Protoplasma, 154, pp. 25–33. |
|
(5) |
EPA (1993). Methods for Measuring the Acute Toxicity of Effluents and Receiving Waters to Freshwater and Marine Organisms. (Fourth ed.). EPA/600/4-90/027F. C. I. Weber (ed), USEPA, Cincinnati, Ohio. |
|
(6) |
Vigano L., (1991) Suitability of commercially available spring waters as standard medium for culturing Daphnia magna. Bull. Environ. Contain. Toxicol., 47, pp. 775–782. |
|
(7) |
ASTM (1988). Standard Guide for Conducting Acute Toxicity Tests with Fishes, Macroinvertebrates and Amphibians. E729-88a. American Society for Testing and Materials, Philadelphia P. A. 20 pp. |
|
(8) |
Baird D. J., Soares A. M. V. M., Girling A., Barber J., Bradley M. C. and Calow P. (1989). The long term maintenance of Daphnia magna Straus for use in ecotoxicological tests; problems and prospects. In: Proceedings of the 1st European Conference on Ecotoxicology. Copenhagen 1988 (H. Lokke, H. Tyle & F. Bro-Rasmussen. Eds.), pp. 144–148. |
|
(9) |
Parkhurst B. R., Forte J. L. and Wright G. P. (1981). Reproducibility of a life-cycle toxicity test with Daphnia magna. Bull. Environ. Contam. and Toxicol., 26, pp. 1–8. |
|
(10) |
Cowgill U. M. and Milazzo D. P. (1990) The sensitivity of two cladocerans to water quality variables: salinity and hardness. Arch. Hydrobiol., 120(2), pp. 185–196. |
|
(11) |
Korshikov (1990). Pseudokirchneriella subcapitata Hindak, F-1990. Biologice Prace, 36, 209. |
|
(12) |
Sims I. R., Watson S. and Holmes D. (1993). Toward a standard Daphnia juvenile production test. Environmental Toxicology and Chemistry, 12, pp. 2053–2058. |
|
(13) |
Sims I. (1993). Measuring the growth of phytoplankton: the relationship between total organic carbon with three commonly used parameters of algal growth. Arch. Hydrobiol., 128, pp. 459–466. |
|
(14) |
Dunnett C. W., (1955). A multiple comparisons procedure for comparing several treatments with a control. J. Amer. Statist. Assoc, 50, pp. 1096–1121. |
|
(15) |
Dunnett C. W., (1964). New tables for multiple comparisons with a control. Biometrics, 20, pp. 482-491. |
|
(16) |
Williams D. A. (1971). A test for differences between treatment means when several dose levels are compared with a zero dose control. Biometrics 27, pp. 103–117. |
|
(17) |
Williams D. A. (1972). The comparison of several dose levels with a zero dose control. Biometrics, 28, pp. 510–531. |
|
(18) |
Draper N. R. and Smith H. (1981). Applied Regression Analysis, second edition, Wiley, N.Y. |
|
(19) |
Brain P. and Cousens R. (1989). An equation to describe dose responses where there is stimulation of growth at low doses. Weed Research, 29, pp. 93–96. |
|
(20) |
Wilson E. O. and Bossert, W. H. (1971). A Primer of Population Biology. Sinauer Associates Inc. Publishers. |
|
(21) |
Poole R. W. (1974). An Introduction to quantitative Ecology. McGraw-Hill Series in Population Biology, New York, pp. 532. |
|
(22) |
Meyer J. S., Ingersoll C. G., McDonald L. L. and Boyce M. S. (1986). Estimating uncertainty in population growth rates: Jackknife vs bootstrap techniques. Ecology, 67, pp. 1156–1166. |
DODATEK 1
PRZYGOTOWANIE W PEŁNI ZDEFINIOWANYCH OŚRODKÓW ELENDT M7 I M4
Aklimatyzacja do ośrodków Elendt M7 i M4
Niektóre laboratoria doświadczyły trudności w bezpośrednim przeniesieniu Daphnia do ośrodków M4 (1) i M7. Jednakże pewien sukces został osiągnięty ze stopniową aklimatyzacją, tj. zastępując własny ośrodek najpierw 30 % Elendt, następnie do 60 % Elendt, a następnie do 100 % Elendt. Okresy czasu potrzebne do aklimatyzacji mogą trwać do jednego miesiąca.
PRZYGOTOWANIE
Mikroelementy
Odrębne roztwory podstawowe (I) poszczególnych mikroelementów są uprzednio przygotowywane w wodzie o odpowiedniej czystości, np. dejonizowanej, destylowanej lub odwróconej osmozie. Z tych różnych roztworów podstawowych (I) jest przygotowywany drugi pojedynczy roztwór podstawowy (II), który zawiera wszystkie mikroelementy (połączony roztwór), np.:
|
Roztwór podstawowy I (pojedyncze substancje) |
Ilość dodana do wody (mg/l) |
Stężenie (w stosunku do ośrodka M4) (krotność) |
Do przygotowania połączonego roztworu podstawowego II dodać następującą ilość roztworu podstawowego I do wody (ml/l) |
|
|
M 4 |
M 7 |
|||
|
H3BO3 |
57 190 |
20 000 |
1,0 |
0,25 |
|
MnCl2 * 4 H2O |
7 210 |
20 000 |
1,0 |
0,25 |
|
LiCl |
6 120 |
20 000 |
1,0 |
0,25 |
|
RbCl |
1 420 |
20 000 |
1,0 |
0,25 |
|
SrCl2 * 6 H2O |
3 040 |
20 000 |
1,0 |
0,25 |
|
NaBr |
320 |
20 000 |
1,0 |
0,25 |
|
Na2MoO4 * 2 H2O |
1 260 |
20 000 |
1,0 |
0,25 |
|
CuCl2 * 2 H2O |
335 |
20 000 |
1,0 |
0,25 |
|
ZnCl2 |
260 |
20 000 |
1,0 |
1,0 |
|
CoCl2 * 6 H2O |
200 |
20 000 |
1,0 |
1,0 |
|
KI |
65 |
20 000 |
1,0 |
1,0 |
|
Na2SeO3 |
43,8 |
20 000 |
1,0 |
1,0 |
|
NH4VO3 |
11,5 |
20 000 |
1,0 |
1,0 |
|
Na2EDTA * 2 H2O |
5 000 |
2 000 |
— |
— |
|
FeSO4 * 7 H2O |
1 991 |
2 000 |
— |
— |
|
Oba roztwory Na2EDTA i FeSO2 przygotowano oddzielnie, zlane razem i niezwłocznie autoklawowane. To daje: |
||||
|
21 Fe-EDTA roztwór |
|
1 000 |
20,0 |
5,0 |
Ośrodki M4 i M7
Ośrodki M4 i M7 są przygotowane przy użyciu roztworu podstawowego II, makrosubstancje odżywcze i witaminy, jak następuje:
|
|
Ilość dodana do wody (mg/l) |
Stężenie (w stosunku do ośrodka M4) (krotność) |
Ilość roztworu podstawowego do przygotowania ośrodka (ml/1) |
|||||
|
M 4 |
M 7 |
|||||||
|
Roztwór podstawowy II połączone mikroelementy |
|
20 |
50 |
50 |
||||
|
Makro-odżywcze roztwory podstawowe (pojedyńcze substancje) |
||||||||
|
CaCl2 * 2 H2O |
293 800 |
1 000 |
1,0 |
1,0 |
||||
|
MgSO4 * 7 H2O |
246 600 |
2 000 |
0,5 |
0,5 |
||||
|
KCl |
58 000 |
10 000 |
0,1 |
0,1 |
||||
|
NaHCO3 |
64 800 |
1 000 |
1,0 |
1,0 |
||||
|
Na2SiO3 * 9 H2O |
50 000 |
5 000 |
0,2 |
0,2 |
||||
|
NaNO3 |
2 740 |
10 000 |
0,1 |
0,1 |
||||
|
KH2PO4 |
1 430 |
10 000 |
0,1 |
0,1 |
||||
|
K2HPO4 |
1 840 |
10 000 |
0,1 |
0,1 |
||||
|
Roztwór związany witamin |
— |
10 000 |
0,1 |
0,1 |
||||
|
Podstawowy roztwór związany witamin przygotowano poprzez dodanie 3 witamin do 1 litra wody jak wskazano poniżej: |
||||||||
|
Hydrochlorek tiaminy |
750 |
10 000 |
— |
— |
||||
|
Cyjanocobalamina (B12) |
10 |
10 000 |
— |
— |
||||
|
Biotyna |
7,5 |
10 000 |
— |
— |
||||
|
Wsad połączonych witamin jest przechowywany w zamrożeniu w małych podwielokrotnościach.. Witaminy są dodawane do ośrodka krótko przed użyciem.
|
||||||||
DODATEK 2
ANALIZA CAŁKOWITEGO WĘGLA ORGANICZNEGO (TOC) I WYKONANIE NOMOGRAMU ZAWARTOŚCI TOC W POKARMIE GLONOWYM
Należy uwzględnić, że zawartość węgla w pokarmie glonowym nie będzie mierzona bezpośrednio, ale z korelacji (tj. nomogramów) z pomiarami zastępczymi, takimi jak ilość komórek glonowych lub absorbancja światła.
TOC powinien być zmierzony raczej wysokotemperaturowym utlenieniem niż ultrafioletem lub metodą nadsiarczanową. (Zob. The Instrumental Determination of Total Organic Carbon, Total Oxygen Demand and Related Determinands 1979, HMSO 1980; 49 High Holborn, London WC1V 6HB).
Dla wykonania nomogramu glony powinny być oddzielone od ośrodka rozwoju poprzez odwirowanie następujące po ponownym utworzeniu zawiesiny w wodzie destylowanej. Pomiar zastępczego parametru i stężenia TOC w każdej potrojonej próbce. Destylowana woda jako ślepa próba powinna być zanalizowana, a stężenie TOC wydedukowane ze stężenia próbki glonów o danym TOC.
Nomogram musi być liniowy ponad wymagany zakres stężenia węgla. Przykłady pokazano poniżej.
Uwaga: Nie powinno to być zastosowane do konwersji; istotne jest, aby laboratorium przygotowało swoje własne nomogramy.


DODATEK 3
PRZYKŁADOWY ARKUSZ DANYCH ZAPISU WYMIANY OŚRODKA, DANYCH FIZYKO-CHEMICZNYCH MONITOROWANIA, KARMIENIA, ROZRODCZOŚCI DAPHNIA I ŚMIERTELNOŚCI DOROSŁYCH
|
Doświadczenie nr: |
Data rozpoczęcia: |
Klon: |
Ośrodek: |
Typ pokarmu |
Substancja badana: |
Stężenie nominalne |
||||||||||||||||||
|
Dzień |
0 |
1 |
2 |
3 |
4 |
5 |
6 |
7 |
8 |
9 |
10 |
11 |
12 |
13 |
14 |
15 |
16 |
17 |
18 |
19 |
20 |
21 |
|
|
|
Odnawianie ośrodka (takt) |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
PH (38) |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
nowy |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
stary |
|
|
|
O2 mg/l (38) |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
nowy |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
stary |
|
|
|
Temperatura (oC) (38) |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
nowy |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
stary |
|
|
|
Żywność dostarczona (takt) |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Nie żywe potomstwo (39) |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Całk owity |
|
zbiornik 1 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
2 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
3 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
4 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
5 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
6 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
7 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
8 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
9 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
10 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Całko wity |
|
|
Skumulowana śmiertelność dorosłych (40) |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
DODATEK 4
PRZYKŁADOWY ARKUSZ DANYCH DOTYCZĄCYCH ZAPISU WYNIKÓW ANALIZY CHEMICZNEJ
a) Pomierzone stężenia
|
Stężenie |
Próbka tydzień 1 |
Próbka tydzień 2 |
Próbka tydzień 3 |
|||
|
Świeża |
Stara |
Świeża |
Stara |
Świeża |
Stara |
|
|
|
|
|
|
|
|
|
b) Pomierzone stężenia jako procent nominału
|
Stężenie nominalne |
Próbka tydzień 1 |
Próbka tydzień 2 |
Próbka tydzień 3 |
|||
|
Świeża |
Stara |
Świeża |
Stara |
Świeża |
Stara |
|
|
|
|
|
|
|
|
|
DODATEK 5
OBLICZANIE ŚREDNIEJ WAŻONEJ CZASU
Średnia ważona czasu
Podane stężenie substancji badanej może wypaść w okresie czasu między odnawianiami ośrodka, dlatego konieczne jest, aby uważać, które stężenie powinno być wybrane jako reprezentatywne w zakresie stężeń doświadczanych przez dorosłe Daphnia. Wybór powinien być oparty na zarówno rozważaniach biologicznych, jak i statystycznych. Na przykład, jeśli sądzi się, że rozrodczość podlega doświadczalnie głównie maksymalnemu stężeniu, wtedy należy użyć maksymalnego stężenia. Jednakże jeśli skumulowane lub długoterminowe działanie substancji toksycznej jest uznawane za najważniejsze, wtedy bardziej właściwe jest średnie stężenie. W takim przypadku, stosowną średnią do zastosowania jest czasowo ważona średnia stężenia, ponieważ uwzględnia zmiany w chwilowym stężeniu czasowym.
Rysunek 1:
Przykład średniej ważonej czasu
Rysunek 1 wskazuje przykład (uproszczony) badania trwającego siedem dni z odnowieniem ośrodka w dniach 0, 2 i 4.
|
|
Cienka zygzakowata linia przedstawia stężenie w każdym punkcie czasu. Spadek stężenia ocenia się jako proces wykładniczy zanikający. |
|
|
Sześć wykreślonych punktów przedstawia pomierzone stężenia na początku i na końcu czasu każdego odnowienia. |
|
|
Gruba linia pokazuje położenie czasowo ważonej średniej. |
Średnia ważona czasu jest obliczana tak, aby powierzchnia pod średnią ważoną była równa powierzchni pod krzywą stężenia. Obliczenia w odniesieniu do powyższego przykładu zilustrowano w tabeli 1.
Tabela 1
Obliczanie średniej ważonej czasu
|
Odnowienie nr |
Dni |
Conc0 |
Conc1 |
Ln(Conc0) |
Ln(Conc1) |
Powierzchnia |
|
1 |
2 |
10,000 |
4,493 |
2,303 |
1,503 |
13,767 |
|
2 |
2 |
11,000 |
6,037 |
2,398 |
1,798 |
16,544 |
|
3 |
3 |
10,000 |
4,066 |
2,303 |
1,403 |
19,781 |
|
Suma dni: 7 |
Suma powierzchni |
50,091 |
||||
|
Średnia TW |
7,156 |
|||||
|
Dni jest ilością dni w okresie odnowienia. Conc0 jest zmierzonym stężeniem na początku każdego okresu odnowienia. Conc1 jest zmierzonym stężeniem na końcu każdego okresu odnowienia. Ln(Conc0) jest naturalnym logarytmem stężenia 0. Ln(Conc1) jest naturalnym logarytmem stężenia 1. Powierzchnia jest powierzchnią pod krzywą wykładniczą dla każdego okresu odnowienia. Jest obliczona za pomocą:
|
||||||
Średnia ważona czasu (średnia TW) jest całkowitą powierzchnią podzieloną przez sumę dni.
Oczywiście odniesieniu do badania rozrodczości Daphnia tabela musi być rozszerzona, aby objąć 21 dni.
Jest wiadome, że gdy podejmuje się obserwację tylko na początku i na końcu każdego okresu odnawiania, nie jest możliwe stwierdzenie, że proces spadku jest rzeczywiście gwałtowny. Różne krzywe powstają przy różnych obliczeniach dla „powierzchni”. Jednakże wykładniczy proces zanikający nie jest nieprawdopodobny oraz jest prawdopodobnie najlepszą krzywą do zastosowania wobec braku innych informacji.
Wymagana jest jednak uważna praca, jeśli analiza chemiczna nie odkryje jakiejś substancji na końcu okresu odnowienia. O ile możliwe jest oszacowanie jak szybko substancja zniknęła z roztworu, niemożliwe jest uzyskanie rzeczywistej powierzchni pod krzywą, a stąd niemożliwe jest uzyskanie wiarygodnej średniej ważonej czasu.
C.21. MIKROORGANIZMY GLEBOWE: BADANIA PRZEMIAN AZOTU
1. METODA
Ta metoda jest równoważna OECD TG 216 (2000).
1.1 WPROWADZENIE
Ta metoda badawcza opisuje laboratoryjną metodę opracowaną do badania długookresowych skutków jednorazowego wystawienia na działanie środków chemicznych dla aktywności mikroorganizmów glebowych związanej z przemianami azotu. To badanie jest oparte głównie na zaleceniach Europejskiej i Śródziemnomorskiej Organizacji Ochrony Roślin (1). Jednakże inne wytyczne, w tym wytyczne German Biologische Bundesanstalt (2), Amerykańskiej Agencji Ochrony Środowiska (3) SETAC (4) i Międzynarodowej Organizacji Normalizacyjnej (5), także zostały wzięte pod uwagę. W czasie warsztatów OECD na temat Selekcji Gleb/Osadów, mających miejsce w Belgirate we Włoszech w 1995 (6), ustalono ilość i rodzaj gleb stosowanych w tym badaniu. Zalecenia w sprawie pobierania, przenoszenia i przechowywania próbek gleby są oparte na wytycznych metodycznych według ISO (7) i zaleceniach z warsztatów w Belgirate. W ocenie i interpretacji toksycznych właściwości badanej substancji może być wymagane określenie skutków działania dla aktywności mikrobiologicznej, np. jeżeli są wymagane dane na temat potencjalnych efektów ubocznych działania środków ochrony upraw na mikroflorę glebową lub jeżeli spodziewane jest wystawienie mikroorganizmów na działanie związków chemicznych innych niż środki ochrony upraw. Badanie przemiany azotu jest prowadzone w celu określenia wpływu działania takich związków chemicznych na mikroflorę. Jeżeli badane są substancje agrochemiczne (np. środki ochrony upraw, nawozy, związki chemiczne stosowane w leśnictwie), to są prowadzone zarówno badania przemiany azotu, jak i węgla. Jeżeli badane są substancje nieagrochemiczne, wystarczające jest badanie przemian azotu. Jednakże, jeżeli wartości EC50 w badaniach przemiany azotu znajdą się w zakresie, w którym taki związek chemiczny uznaje się za dostępny w handlu inhibitor nitryfikacji (np. nitropiryna), można przeprowadzić badania przemian azotu w celu uzyskania dodatkowych informacji.
Gleby składają się ze składników ożywionych i nieożywionych, które występują w złożonych i heterogenicznych mieszaninach. Mikroorganizmy odgrywają istotną rolę w rozkładzie i przemianie materii organicznej w żyznych glebach, gdzie wiele gatunków przyczynia się do różnych aspektów żyzności gleby. Jakikolwiek długookresowy wpływ na procesy biochemiczne może potencjalnie zakłócić obieg substancji biogennych, a to może zmienić żyzność gleb. Przemiany węgla i azotu występują we wszystkich żyznych glebach. Pomimo że biocenozy bakteryjne różnią się w poszczególnych rodzajach gleb, szlaki przemian są w zasadzie takie same.
Ta opisana metoda badawcza jest przeznaczona do wykrywania długookresowych niepomyślnych skutków działania substancji na procesy przemiany azotu w natlenionych powierzchniowych warstwach gleb. Metoda badawcza pozwala także na określenie skutków działania substancji na przemiany węgla przez mikroflorę glebową. Tworzenie azotanów ma miejsce po degradacji wiązań węgiel-azot. Z tego powodu, jeżeli zostaną stwierdzone jednakowe tempa produkcji azotu w glebie badanej i kontrolnej, istnieje wysokie prawdopodobieństwo, że główne szlaki degradacji węgla są nienaruszone i funkcjonalne. Substrat wybrany do badań (sproszkowana mączka z lucerny) ma korzystny stosunek węgla do azotu (na ogół pomiędzy 12/1 a 16/1). Z tego powodu ograniczony zostaje deficyt węgla podczas badania, a jeżeli w wyniku działania związku chemicznego zostaną uszkodzone biocenozy bakteryjne, mogą się odtworzyć w ciągu 100 dni.
Badania, na podstawie których zastała opracowana ta metoda badawcza, były na początku przeznaczone dla substancji, dla których można było przewidzieć ich ilość będącą w kontakcie z glebą. Tak się dzieje, na przykład, w przypadku środków ochrony upraw, gdzie znana jest ich ilość podawana w terenie. Dla substancji agrochemicznych wystarczające jest badanie dwóch dawek odpowiadających ilości podanej lub przewidywanej. Substancje agrochemiczne mogą być badane jako aktywne dodatki (a.i.) lub jako gotowe produkty. Jednakże badanie nie jest ograniczone do substancji agrochemicznych. Zmieniając zarówno ilości substancji badanej podanej do gleby, jak i sposób interpretacji wyników, badania mogą być stosowane do substancji chemicznych, których przewidywana ilość przechodząca do gleby jest nieznana. Wobec tego dla substancji chemicznych innych niż agrochemiczne określa się wpływ różnych stężeń na przemiany azotu. Wyniki tych badań są wykorzystywane do przygotowania krzywej zależności dawka-odpowiedź i obliczenia wartości ECx, gdzie x zdefiniowano jako % oddziaływania.
1.2. DEFINICJE
Przemiany azotu: to ostateczna degradacja materii organicznej zawierającej azot, w wyniku działania mikroorganizmów, poprzez procesy amonifikacji i nitryfikacji, do odpowiednich nieorganicznych produktów końcowych azotanowych.
EC x (stężenie efektywne): to stężenie substancji badanej w glebie, które powoduje x procentową inhibicję przemian azotu do azotanu.
EC 50 (mediana stężenia efektywnego): stężenie substancji w glebie, które powoduje 50-procentową (50 %) inhibicję przemian azotu do azotanu.
1.3. SUBSTANCJE ODNIESIENIA
Brak.
1.4. ZASADA METODY BADANIA
Przesiana gleba jest wzbogacona sproszkowaną mączką roślinną i albo poddawana działaniu badanej substancji, albo pozostawiona nienaruszona (próbka kontrolna). Jeżeli badane są substancje agrochemiczne, zaleca się stosowanie przynajmniej dwóch stężeń i powinny one być wybrane w odniesieniu do najwyższego stężenia, jakie może występować w terenie. Po 0, 7, 14 dniach i po 28 dniach inkubacji próbki gleby badanej i kontrolnej są ekstrahowane odpowiednim rozpuszczalnikiem i oznaczane są ilości azotanów w ekstraktach. Tempo wytwarzania azotanów w próbce badanej jest porównywane z tempem w próbce kontrolnej i obliczane jest odchylenie procentowe między próbką badaną a kontrolną. Wszystkie badania są prowadzone przynajmniej przez 28 dni. Jeżeli 28 dnia różnice pomiędzy glebami badanymi a kontrolnymi będą równe lub większe niż 25 %, badania są kontynuowane przez maksymalnie 100 dni. Jeżeli badane są substancje nieagrochemiczne, do próbek gleby dodawana jest seria stężeń badanej substancji i mierzone są ilości wytworzonych azotanów w próbce badanej i kontrolnej po 28 dniach inkubacji. Wyniki badań z wieloma stężeniami są analizowane przy użyciu modelu regresyjnego i obliczane są wartości ECx (to jest EC50, EC25 i/lub EC10). Zob. definicje.
1.5. WAŻNOŚĆ BADANIA
Oceny wyników badań substancji agrochemicznych są oparte na stosunkowo niewielkich różnicach (średnia wartość ± 25 %) pomiędzy stężeniem azotanów w kontrolnych i badanych próbkach gleby, więc duże różnice próbek kontrolnych mogą prowadzić do fałszywych wyników. Z tego powodu odchylenia pomiędzy powtórzeniami próbek kontrolnych powinny być mniejsze niż ± 15 %.
1.6. OPIS METODY BADANIA
1.6.1. Aparatura
Stosowane są pojemniki z materiałów chemicznie obojętnych. Powinny one mieć odpowiednią pojemność zgodnie z procedurą stosowaną do inkubacji gleb, to jest inkubacji w masie lub jako serii pojedynczych próbek gleby (zob. sekcja 1.7.1.2). Powinno się zwrócić uwagę na minimalizację utraty wody i umożliwienie wymiany gazów podczas badania (np. pojemniki do badań mogą być przykryte perforowaną folią polietylenową). Jeżeli badane są substancje lotne, powinno się stosować pojemniki umożliwiające uszczelnienie i gazoszczelne. Powinny być takiej wielkości, aby około jednej czwartej ich objętości było wypełnione próbką gleby.
Stosowane jest następujące standardowe wyposażenie laboratoryjne:
|
— |
mieszadło: mechaniczna wytrząsarka lub urządzenie podobne, |
|
— |
wirówka (pojemność 3 000g) lub filtr (stosując papier bezazotanowy), |
|
— |
instrument o odpowiedniej czułości i powtarzalności do analizy azotanów. |
1.6.2. Wybór i ilość rodzajów gleb
Stosowany jest jeden rodzaj gleb. Zalecanymi własnościami gleby są:
|
— |
zawartość piasku: nie mniej niż 50 % i nie więcej niż 75 %, |
|
— |
pH: 5,5–7,5, |
|
— |
zawartość węgla organicznego: 0,5–1,5 %, |
|
— |
powinna zostać zmierzona mikrobiologiczna biomasa (8)(9) i jej zawartość węgla powinna wynosić przynajmniej 1 % całkowitej masy węgla organicznego w glebie. |
Przeważnie, gleby o takich własnościach stanowią najgorszy przypadek, ponieważ adsorpcja badanej substancji chemicznej jest minimalna, a jej dostępność dla mikroflory maksymalna. Wynika z tego, że badania innych gleb są na ogół niepotrzebne. Jednakże w pewnych okolicznościach, to jest gdy przewiduje się, że substancja badana będzie stosowana głównie w określonych glebach, takich jak kwaśne gleby leśne lub dla substancji chemicznych z ładunkiem elektrostatycznym, może być konieczne zastosowanie dodatkowych rodzajów gleb.
1.6.3. Pobieranie i przechowywanie próbek gleby
1.6.3.1. Pobieranie
Powinny być dostępne szczegółowe informacje na temat historii obszaru, z jakiego została pobrana badana próbka gleby. Szczegółowe informacje powinny obejmować dokładną lokalizację, szatę roślinną, daty poddania działaniu środków ochrony upraw, podanie nawozów organicznych lub nieorganicznych, dodanie materiałów biologicznych lub przypadkowe zanieczyszczenia. Obszar wybrany do pobrania gleby powinien umożliwiać długookresowe użytkowanie. Odpowiednie do tego są stałe pastwiska, pola okresowo zasiewane zbożami (z wyjątkiem kukurydzy) lub gęsto nawożone zielonką. Wybrany obszar do pobierania próbek nie powinien być poddawany działaniu produktów ochrony upraw przez co najmniej jeden rok przed pobraniem próbek. Nie powinno się również podawać żadnych nawozów organicznych przez co najmniej sześć miesięcy. Stosowanie nawozów mineralnych jest dozwolone tylko wtedy, gdy jest zgodne z wymogami upraw i próbki gleby nie powinny być pobierane przez co najmniej trzy miesiące po podaniu nawozu. Należy unikać stosowania gleb poddanych działaniu nawozów o znanym działaniu biologicznym (np. cyjanamid wapnia).
Pobierania próbek należy unikać podczas lub bezpośrednio po dłuższych okresach (przekraczających 30 dni) suszy lub zalania wodą. Dla gleb zaoranych, próbki powinny być pobierane z głębokości od 0 do 20 cm. Dla łąk (pastwisk) lub innych gleb, które nie są orane przez dłuższy czas (przynajmniej jeden sezon wegetacyjny), maksymalna głębokość pobierania próbek może być nieco większa niż 20 cm (np. do 25 cm).
Próbki gleb powinny być transportowane przy użyciu odpowiednich pojemników i w warunkach temperaturowych gwarantujących, że początkowe własności gleby nie zostaną znacząco zmienione.
1.6.3.2. Przechowywanie
Preferuje się stosowanie gleb świeżo zebranych. Jeżeli nie można uniknąć przechowywania w laboratorium, gleba może być przechowywana w ciemności w temperaturze 4 ± 2 oC przez maksymalnie trzy miesiące. Należy zapewnić warunki do napowietrzania gleby podczas jej przechowywania. Jeżeli gleby są pobierane z obszarów, które zamarzają przez przynajmniej trzy miesiące w roku, można wziąć pod uwagę przechowywanie przez sześć miesięcy w temperaturze od minus 18 oC do minus 22 oC. Mikrobiologiczna biomasa przechowywanych gleb jest mierzona przed każdym eksperymentem i zawartość węgla w biomasie powinna wynosić co najmniej 1 % całkowitej zawartości węgla organicznego w glebie (zob. sekcja 1.6.2).
1.6.4. Postępowanie i przygotowanie gleby do badań
1.6.4.1. Inkubacja wstępna
Jeżeli gleba była przechowywana (zob. sekcja 1.6.4.2), zalecana jest wstępna inkubacja przez okres 2–28 dni. Temperatura i zawartość wilgoci w glebie podczas wstępnej inkubacji powinny być podobne do tych, jakie będą w czasie badania (zob. sekcje 1.6.4.2 i 1.7.1.3).
1.6.4.2 Właściwości fizyczno-chemiczne
Z gleby usuwane są ręcznie większe obiekty (to jest kamienie, części roślin itp.) i jest przesiewana na wilgotno, bez nadmiernego wysuszenia do ziarnistości mniejszej lub równej 2 mm. Zawartość wilgoci w próbce gleby powinna zostać zwiększona przy użyciu wody destylowanej lub dejonizowanej do wartości pomiędzy 40 a 60 % maksymalnej pojemności wodnej.
1.6.4.3. Wzbogacanie substratem organicznym
Gleba powinna zostać wzbogacona odpowiednim substratem organicznym, np. mączką części zielonych lucerny (główny komponent to Medicago sativa) ze stosunkiem C/N od 12/1 do 16/1. Zalecany jest dodatek lucerny do gleby w ilości 5 g lucerny na kilogram gleby (sucha masa).
1.6.5. Przygotowanie substancji badanej do podania do gleby
Substancja badana jest zazwyczaj podawana przy użyciu nośnika. Nośnikiem może być woda (dla substancji rozpuszczalnych w wodzie) lub obojętne ciało stałe, takie jak drobnoziarnisty piasek kwarcowy (rozmiar ziaren 0,1–0,5 mm). Należy unikać nośników ciekłych innych niż woda (np. organiczne rozpuszczalniki, takie jak aceton, chloroform), ponieważ mogą niszczyć mikroflorę. Jeżeli stosowanym nośnikiem jest piasek, może on być powleczony badaną substancją rozpuszczoną lub zawieszoną w odpowiednim rozpuszczalniku. W takich przypadkach przed wymieszaniem z glebą należy usunąć rozpuszczalnik przez odparowanie. Dla zachowania optymalnego rozkładu substancji badanej w glebie zaleca się stosowanie 10 g piasku na kilogram gleby (w suchej masie). Próbki kontrolne są poddawane działaniu równoważnej ilości tylko wody i/lub piasku kwarcowego.
Jeżeli badane są lotne substancje chemiczne, powinno się unikać strat podczas poddawania działaniu i należy podjąć próbę zapewnienia jednorodnego rozkładu w glebie (np. badana substancja powinna być nastrzyknięta do gleby w kilku miejscach).
1.6.6. Stężenia stosowane w badaniach
Jeżeli badane są substancje agrochemiczne, powinno się stosować przynajmniej dwa stężenia. Niższe stężenie powinno odzwierciedlać przynajmniej maksymalną, oczekiwaną ilość dochodzącą do gleby w praktycznym zastosowaniu, natomiast wyższe stężenie powinno być wielokrotnością niższego. Stężenia substancji badanej dodanej do gleby są obliczane przy założeniu równomiernego wprowadzenia na głębokość 5 cm i gęstości nasypowej gleby 1,5. Dla substancji agrochemicznych dodawanych bezpośrednio do gleby lub dla substancji chemicznych o przewidywalnej ilości dochodzącej do gleby zalecanymi stężeniami do badań są maksymalne przewidywalne stężenia środowiskowe (Predicted Environmental Concentration – PEC) i ich pięciokrotność. Substancje, dla których przewiduje się kilkakrotne stosowanie w jednym sezonie, powinny być badane przy stężeniach wyliczonych poprzez przemnożenie PEC przez oczekiwaną maksymalną liczbę zastosowań. Wyższe badane stężenie nie powinno jednak przekraczać dziesięciokrotności pojedynczej stosowanej maksymalnej ilości. Jeżeli badane są substancje nieagrochemiczne, stosowane są ciągi geometryczne przynajmniej pięciu stężeń. Badane stężenia powinny pokryć zakres potrzebny do określenia wartości ECx.
1.7. PROWADZENIE BADANIA
1.7.1. Warunki ekspozycji
1.7.1.1. Próbki badane i kontrolne
Jeżeli badane są substancje agrochemiczne, gleba dzielona jest na trzy równe wagowo części. Dwie części są mieszane z nośnikiem zawierającym badany produkt, a trzecia jest mieszana z samym nośnikiem (próbka kontrolna). Zalecane są przynajmniej trzy powtórzenia, zarówno dla gleb poddanych działaniu badanej substancji, jak i niepoddanych. Jeżeli badane są środki niebędące substancjami agrochemicznymi, gleba dzielona jest na sześć równych wagowo części. Pięć próbek jest mieszanych z nośnikiem zawierającym substancję badaną, a szósty mieszany jest z samym nośnikiem. Zaleca się trzy powtórzenia, zarówno dla próbek badanych, jak i próbek kontrolnych. Należy zadbać o zapewnienie homogenicznego rozprowadzenia substancji badanej w poddanych jej działaniu próbkach gleby. Podczas mieszania powinno się uniknąć sklejania lub zbrylania gleby.
1.7.1.2. Inkubacja próbek gleby
Inkubacja może być prowadzona na dwa sposoby: jako zbiorcze próbki każdej gleby poddanej i niepoddanej działaniu badanej substancji albo jako seria pojedynczych podpróbek równej wielkości, poddanych i niepoddanych działaniu badanej substancji. Jednakże jeżeli badane są substancje lotne, badania powinny być prowadzone jedynie na serii pojedynczych podpróbek. Jeżeli gleby są inkubowane w masie, przygotowywane są duże ilości gleby poddanej i niepoddanej działaniu badanej substancji i podpróbki do analizy są pobierane w miarę potrzeby podczas badania. Ilość przygotowana wstępnie do każdego poddania działaniu i do kontroli zależy od wielkości podpróbek, ilości powtórzeń stosowanych do analizy oraz przewidywanej maksymalnej liczby pobieranych próbek. Gleby inkubowane w jednej objętości powinny zostać dokładnie wymieszane przed podziałem na mniejsze próbki. Jeżeli gleby są inkubowane jako seria pojedynczych próbek, każda poddana działaniu substancji badanej i kontrolna masa gleby jest dzielona na wymaganą liczbę podpróbek, które są wykorzystywane w miarę potrzeb. W eksperymentach, gdzie przewiduje się więcej niż dwa próbkowania, powinna być przygotowywana wystarczająca liczba podpróbek do wszystkich powtórzeń i próbkowań. Przynajmniej trzy powtórzone próbki badanej gleby powinny być inkubowane w warunkach napowietrzania (zob. sekcja 1.7.1.1). Podczas wszystkich badań powinny być stosowane odpowiednie pojemniki, z wystarczającą przestrzenią nad powierzchnią próbki, w celu uniknięcia rozwoju warunków beztlenowych. Jeżeli badane są substancje lotne, badanie powinno być prowadzone wyłącznie na serii pojedynczych podpróbek.
1.7.1.3. Warunki badań i czas ich trwania
Badanie jest prowadzone w ciemności w temperaturze pokojowej 20 ± 2 oC. Zawartość wilgoci w próbkach gleb podczas badania powinna wynosić 40–60 % maksymalnej pojemności wodnej (zob. sekcja 1.6.4.2) w przedziale ± 5 %. Można dodawać wodę destylowaną i dejonizowaną, stosownie do potrzeb.
Minimalny czas trwania badań wynosi 28 dni. Jeżeli badane są substancje agrochemiczne, porównywane są szybkości tworzenia azotanów próbki badanej i kontrolnej. Jeżeli 28 dnia wyniki różnią się od siebie o więcej niż 25 %, badanie jest kontynuowane aż do osiągnięcia wartości równej lub mniejszej od 25 % lub przez maksymalnie 100 dni, w zależności od tego, co nastąpi wcześniej. Dla substancji niebędących agrochemicznymi badanie przerywa się po 28 dniach. 28 dnia oznaczane są ilości azotanów w próbce badanej i kontrolnej i obliczane są wartości ECx.
1.7.2. Próbkowanie i analiza gleb
1.7.2.1. Harmonogram próbkowania gleb
Jeżeli badane są substancje agrochemiczne, próbki gleby są analizowane pod kątem azotanów w dniach 0, 7, 14 i 28. Jeżeli wymagane jest przedłużenie badania, dalsze pomiary powinny być wykonywane co 14 dni po dniu 28.
Jeżeli badane są substancje nieagrochemiczne, stosuje się przynajmniej pięć stężeń i próbki gleby są analizowane na azotany na początku (dzień 0) i na końcu okresu ekspozycji (28 dzień). Można dodać pomiary pośrednie, np. 7 dnia, jeżeli zostanie to uznane za konieczne. Dane uzyskane 28 dnia są stosowane do wyznaczenia wartości ECx dla substancji chemicznej. Jeżeli jest to pożądane, można wykorzystać wyniki z dnia 0 dla próbek kontrolnych do podania początkowej ilości azotanów w glebie.
1.7.2.2. Analizy próbek gleby
Ilość wytworzonych azotanów w każdej próbce poddanej działaniu badanej substancji i w próbce kontrolnej jest określana przy każdym próbkowaniu. Azotany są ekstrahowane z gleby przez wytrząsanie próbek z odpowiednim rozpuszczalnikiem, to jest 0,1 M roztwór chlorku potasu. Zaleca się 5 ml roztworu KCl na gram suchej masy równoważnej ilości gleby. W celu optymalizacji ekstrakcji pojemnik zawierający glebę i roztwór ekstrakcyjny nie powinien być napełniony bardziej niż do połowy. Mieszanina jest wytrząsana z szybkością 150 rpm przez 60 minut. Mieszanina jest odwirowywana lub filtrowana, a faza ciekła analizowana na azotany. Roztwory wolne od cząstek stałych mogą być przechowywane przed analizą w temperaturze minus 20 ± 5 oC przez okres do sześciu miesięcy.
2. DANE
2.1. OPRACOWANIE WYNIKÓW
Jeżeli badane są substancje agrochemiczne, powinna zostać zarejestrowana ilość wytworzonych azotanów w każdej próbce gleby i wartość średnia wszystkich powtórzeń powinna zostać przedstawiona w formie tabeli. Tempa przemian azotu należy zinterpretować przy użyciu odpowiednich i powszechnie akceptowanych metod statystycznych (np. test F, 5 % poziom istotności). Ilości wytworzonych azotanów wyrażane są w mg azotanów/kg suchej masy gleby/dzień. Tempo tworzenia azotanów w każdej próbce poddanej działaniu badanej substancji jest porównywane z próbką kontrolną i obliczane jest odchylenie procentowe od próbki kontrolnej.
Jeżeli badane są substancje niebędące agrochemicznymi, oznaczana jest ilość utworzonych azotanów w każdym powtórzeniu i do oceny wartości ECx przygotowywana jest krzywa zależności dawka-odpowiedź. Ilości azotanów (to jest mg azotanów/kg suchej masy gleby) oznaczone w badanych próbkach po 28 dniach są porównywane z wartościami określonymi dla próbki kontrolnej. Na podstawie tych danych obliczane są wielkości % inhibicji dla każdego badanego stężenia. Te wartości procentowe są wykreślane w zależności od stężenia, a następnie wykorzystuje się procedury statystyczne do obliczenia wartości ECx. Granice przedziału ufności (p = 0,95) dla obliczanej wartości ECx są również określane przy użyciu standardowych procedur (10)(11)(12).
Badane substancje zawierające wysokie ilości azotu mogą przyczyniać się do ilości azotanów wytworzonych w czasie badań. Jeżeli te substancje są badane w dużych stężeniach (np. związki chemiczne przewidziane do wielokrotnego stosowania), w badaniach trzeba uwzględnić odpowiednie próbki kontrolne (tj. gleba i substancja badana, ale bez mączki roślinnej). Dane z tych próbek kontrolnych muszą zostać uwzględnione w obliczeniach ECx.
2.2. INTERPRETACJA WYNIKÓW
Jeżeli są oceniane wyniki badań substancji agrochemicznych i różnica tempa tworzenia azotanów pomiędzy niższym stężeniem (to jest maksymalne przewidywane stężenie) a próbką kontrolną jest mniejsza lub równa 25 %, w którymkolwiek momencie pomiarów po upływie 28 dni, produkt może zostać oceniony, jako niemający długookresowego wpływu na przemiany azotu w glebie. Jeżeli oceniane są wyniki uzyskane dla substancji chemicznych innych niż agrochemiczne, stosowane są wartości EC50, EC25 i/lub EC10.
3. SPRAWOZDAWCZOŚĆ
Raport z badań musi zawierać następujące informacje:
|
|
Pełną identyfikację używanej gleby, w tym:
|
|
|
Substancja badana:
|
|
|
Substrat:
|
|
|
Warunki badania:
|
|
|
Wyniki:
|
4. BIBLIOGRAFIA
|
(1) |
EPPO (1994). Decision-Making Scheme for the Environmental Risk Assessment of Plant Protection Chemicals. Chapter 7: Soil Microflora. EPPO Bulletin 24: 1–16, 1994. |
|
(2) |
BBA (1990). Effects on the Activity of the Soil Microflora. BBA Guidelines for the Official Testing of Plant Protection Products, VI, 1–1 (2nd eds., 1990). |
|
(3) |
EPA (1987). Soil Microbial Community Toxicity Test. EPA 40 CFR Part 797.3700. Toxic Substances Control Act Test Guidelines; Proposed rule. September 28, 1987. |
|
(4) |
SETAC-Europe (1995). Procedures for assessing the environmental fate and ecotoxicity of pesticides, Ed. M.R. Lynch, Pub. SETAC-Europe, Bruxelles. |
|
(5) |
ISO/DIS 14238 (1995). Soil Quality – Determination of Nitrogen Mineralisation and Nitrification in Soils and the Influence of Chemicals on these Processes. Technical Committee ISO/TC 190/SC 4: Soil Quality - Biological Methods. |
|
(6) |
OECD (1995). Final Report of the OECD Workshop on Selection of Soils/Sediments, Belgirate, Italie, 18–20 janvier 1995. |
|
(7) |
ISO 10381-6 (1993). Soil quality – Sampling. Guidance on the collection, handling and storage of soil for the assessment of aerobic microbial processes in the laboratory. |
|
(8) |
ISO 14240-1 (1997). Soil quality – Determination of soil microbial biomass – Part 1: Substrate induced respiration method. |
|
(9) |
ISO 14240-2 (1997). Soil quality – Determination of soil microbial biomass – Part 2: Fumigation extraction method. |
|
(10) |
Litchfield, J.T. and Wilcoxon F. (1949). A simplified method of evaluating dose-effect experiments. Jour. Pharmacol. and Exper. Ther., 96, 99–113. |
|
(11) |
Finney, D.J. (1971). Probit Analysis. 3rd ed., Cambridge, London and New-York. |
|
(12) |
Finney, D.J. (1978). Statistical Methods in biological Assay. Griffin, Weycombe, UK. |
C.22. MIKROORGANIZMY GLEBOWE: BADANIE PRZEMIAN WĘGLA
1. METODA
Ta metoda jest równoważna OECD TG 217 (2000).
1.1. WPROWADZENIE
Ta metoda badawcza opisuje metodę laboratoryjną opracowaną do badania potencjalnych długoterminowych skutków jednorazowego poddania działaniu środków ochrony upraw i innych substancji chemicznych dla przemian węgla spowodowanych przez mikroorganizmy glebowe. To badanie jest oparte głównie na zaleceniach Europejskiej i Śródziemnomorskiej Organizacji Ochrony Roślin (1). Jednakże inne wytyczne, w tym wytyczne German Biologische Bundesanstalt (2), Amerykańskiej Agencji Ochrony Środowiska (3) i SETAC (4) także zostały wzięte pod uwagę. W czasie warsztatów OECD na temat Selekcji Gleb/Osadów, mających miejsce w Belgirate we Włoszech w 1995 (6), ustalono ilość i rodzaj gleb stosowanych w tym badaniu. Zalecenia w sprawie pobierania, przenoszenia i przechowywania próbek gleby są oparte na wytycznych metodycznych według ISO (6) i zaleceniach z warsztatów w Belgirate.
W ocenie i interpretacji toksycznych właściwości badanej substancji może być wymagane określenie skutków działania dla aktywności mikrobiologicznej np. jeżeli są wymagane dane na temat potencjalnych efektów ubocznych działania środków ochrony upraw dla mikroflory glebowej lub jeżeli spodziewane jest wystawienie mikroorganizmów na działanie związków chemicznych innych niż środki ochrony upraw. Badanie przemian węgla jest prowadzone w celu określenia wpływu działania takich substancji chemicznych na mikroflorę. Jeżeli badane są substancje agrochemiczne (np. środki ochrony upraw, nawozy, związki chemiczne stosowane w leśnictwie), to są prowadzone zarówno badania przemian węgla, jak i azotu. Jeżeli badane są substancje nieagrochemiczne, wystarczające jest badanie przemian azotu. Jednakże jeżeli wartości EC50 w badaniach przemian azotu znajdą się w zakresie, w którym taki związek chemiczny uznaje się za dostępny w handlu inhibitor nitryfikacji (np. nitropiryna), można przeprowadzić badania przemian azotu w celu uzyskania dodatkowych informacji.
Gleby składają się ze składników ożywionych i nieożywionych, które występują w złożonych i heterogenicznych mieszaninach. Mikroorganizmy odgrywają istotną rolę w rozkładzie i przemianie materii organicznej w żyznych glebach, gdzie wiele gatunków przyczynia się do różnych aspektów żyzności gleby. Jakikolwiek długookresowy wpływ na procesy biochemiczne może potencjalnie zakłócić obieg substancji biogennych, a to może zmienić żyzność gleb. Przemiany węgla i azotu występują we wszystkich żyznych glebach. Pomimo że biocenozy bakteryjne różnią się w poszczególnych rodzajach gleb, szlaki przemian są w zasadzie takie same.
Ta metoda badawcza jest przeznaczona do wykrywania długookresowych, niepomyślnych skutków działania substancji dla procesów przemian węgla w napowietrzonych, powierzchniowych warstwach gleb. Badanie jest czułe na zmiany wielkości i aktywności biocenoz bakteryjnych odpowiedzialnych za przemiany węgla, ponieważ poddaje te biocenozy zarówno oddziaływaniu chemicznemu, jak i deficytowi węgla. Stosowana jest piaszczysta gleba o niskiej zawartości materii organicznej. Ta gleba jest poddawana działaniu badanej substancji, a następnie jest inkubowana w warunkach pozwalających na szybki metabolizm mikrobiologiczny. W tych warunkach źródła łatwo dostępnego węgla w glebie są gwałtownie wyczerpywane. Powoduje to deficyt węgla, który zarówno zabija komórki mikrobiologiczne, jak i powoduje przejście w stan uśpienia i/lub zarodnikowania. Jeżeli badanie trwa dłużej niż 28 dni, suma wszystkich tych reakcji może zostać zmierzona w próbkach kontrolnych (niepoddanych działaniu badanej substancji) jako stopniowy spadek aktywnej metabolicznie biomasy mikrobiologicznej (7). Jeżeli biomasa w glebie ubogiej w węgiel, w warunkach badania, jest poddawana działaniu substancji chemicznej, to może ona nie wrócić do takiego samego poziomu, jak próbka kontrolna. Stąd zaburzenia spowodowane przez badaną substancję w dowolnym momencie podczas badania często trwają aż do zakończenia badania.
Badania, na podstawie których zastała opracowana ta metoda badawcza, były na początku przeznaczone dla substancji, dla których można było przewidzieć ich ilość będącą w kontakcie z glebą. Tak się dzieje, na przykład, w przypadku środków ochrony upraw, gdzie znana jest ich ilość podawana w polu. Dla produktów agrochemicznych wystarczające jest badanie dwóch dawek odpowiadających ilości podanej lub przewidywanej. Substancje agrochemiczne mogą być badane jako aktywne dodatki (a.i.) lub jako gotowe produkty. Jednakże badanie nie jest ograniczone do substancji chemicznych o przewidywalnych stężeniach środowiskowych. Zmieniając zarówno ilości substancji badanej podanej do gleby, jak i sposób interpretacji wyników, badania mogą być stosowane do substancji chemicznych, których przewidywana ilość będąca w kontakcie z glebą jest nieznana. Wobec tego dla substancji chemicznych innych niż agrochemiczne określa się wpływ różnych stężeń na przemianę węgla. Wyniki tych badań są wykorzystywane do przygotowania wykresu reakcji na dawkę i obliczenia wartości ECx, gdzie x zdefiniowano jako % oddziaływania.
1.2. DEFINICJE
Przemiany węgla: to degradacja materii organicznej w wyniku działania mikroorganizmów do końcowych, nieorganicznych produktów przemiany, jakim jest ditlenek węgla.
EC x (stężenie efektywne): to stężenie substancji badanej w glebie, które powoduje x-procentową inhibicję przemian węgla do ditlenku węgla.
EC 50 (mediana stężenia efektywnego): stężenie substancji w glebie, które powoduje 50-procentową inhibicję przemian węgla do ditlenku węgla.
1.3. SUBSTANCJE ODNIESIENIA
Brak.
1.4. ZASADA METODY BADAWCZEJ
Przesiana gleba jest albo poddawana działaniu substancji badanej, albo pozostawiona nienaruszona (próbka kontrolna). Jeżeli badane są substancje agrochemiczne, zaleca się stosowanie przynajmniej dwóch stężeń i powinny one być wybrane w odniesieniu do najwyższego stężenia, jakie może występować w terenie. Po 0, 7, 14 i 28 dniach inkubacji próbki gleb poddanych działaniu badanej substancji i próbki kontrolne są mieszane z glukozą i przez kolejne 12 godzin jest mierzone tempo respiracji indukownej glukozy. Tempo respiracji wyraża się ilością uwolnionego ditlenku węgla (mg dwutlenku węgla/kg suchej gleby/h) lub ilością pochłoniętego tlenu (mg tlenu/kg suchej gleby/h). Średnie tempo respiracji w próbce gleby poddanej działaniu badanej substancji jest porównywane z tempem respiracji dla próbki kontrolnej, a następnie oblicza się odchylenie procentowe próbki badanej od próbki kontrolnej. Wszystkie badania są prowadzone przynajmniej przez 28 dni. Jeżeli 28 dnia różnice pomiędzy glebami badanymi a kontrolnymi będą równe lub większe niż 25 %, badania kontynuuje się w odstępach 14-dniowych przez maksymalnie 100 dni. Jeżeli badane są substancje nieagrochemiczne, do próbek gleby dodawana jest seria stężeń badanej substancji i po 28 dniach mierzone są tempa respiracji indukowanej glukozą (to jest średnia ilość wytworzonego ditlenku węgla lub zużytego tlenu). Wyniki badań z serii stężeń są analizowane przy użyciu modelu regresyjnego i obliczane są wartości ECx (to jest EC50, EC25 i/lub EC10). Zob. definicje.
1.5. WAŻNOŚĆ BADANIA
Oceny wyników badań substancji agrochemicznych są oparte na stosunkowo niewielkich różnicach (średnia wartość ± 25 %) pomiędzy uwolnionym ditlenkiem węgla lub zużytym tlenem w (lub przez) kontrolnych i badanych próbkach gleby, więc duże różnice próbek kontrolnych mogą prowadzić do fałszywych wyników. Z tego powodu odchylenia pomiędzy powtórzeniami próbek kontrolnych powinny być mniejsze niż ± 15 %.
1.6. OPIS METODY BADANIA
1.6.1. Aparatura
Stosowane są pojemniki z materiałów chemicznie obojętnych. Powinny one mieć odpowiednią pojemność zgodnie z procedurą stosowaną do inkubacji gleb, to jest inkubacji w masie lub jako serii pojedynczych próbek gleby (zob. sekcja 1.7.1.2). Powinno się zwrócić uwagę na minimalizację utraty wody i umożliwienie wymiany gazów podczas badania (np. pojemniki do badań mogą być przykryte perforowaną folią polietylenową). Jeżeli badane są substancje lotne, powinno się stosować pojemniki umożliwiające uszczelnienie i gazoszczelne. Powinny być takiej wielkości, aby około jednej czwartej ich objętości było wypełnione próbką gleby.
Dla oznaczenia respiracji indukowanej glukozą, wymagany jest system inkubacyjny oraz przyrządy do pomiaru produkcji ditlenku węgla lub zużycia tlenu. Przykłady takich systemów i urządzeń można znaleźć w literaturze (8)(9)(10)(11).
1.6.2. Wybór i ilość próbek gleb
Stosowany jest jeden rodzaj gleb. Zalecanymi własnościami gleby są:
|
— |
zawartość piasku: nie mniej niż 50 % i nie więcej niż 75 %, |
|
— |
pH: 5,5–7,5, |
|
— |
zawartość węgla organicznego: 0,5–1,5 %, |
|
— |
powinna zostać zmierzona mikrobiologiczna biomasa (12)(13) i jej zawartość węgla powinna wynosić przynajmniej 1 % całkowitej masy węgla organicznego w glebie. |
Przeważnie gleby o takich własnościach stanowią najgorszy przypadek, ponieważ adsorpcja badanej substancji chemicznej jest minimalna, a jej dostępność dla mikroflory maksymalna. Wynika z tego, że badania innych gleb są na ogół niepotrzebne. Jednakże w pewnych okolicznościach, to jest gdy przewiduje się, że substancja badana będzie stosowana głównie w określonych glebach, takich jak kwaśne gleby leśne lub dla substancji chemicznych z ładunkiem elektrostatycznym, może być konieczne zastosowanie dodatkowych rodzajów gleb.
1.6.3. Pobieranie i przechowywanie próbek gleby
1.6.3.1. Pobieranie
Powinny być dostępne szczegółowe informacje na temat historii obszaru, z jakiego została pobrana badana próbka gleby. Szczegółowe informacje powinny obejmować dokładną lokalizację, szatę roślinną, daty poddania działaniu środków ochrony upraw, podanie nawozów organicznych lub nieorganicznych, dodanie materiałów biologicznych lub przypadkowe zanieczyszczenia. Obszar wybrany do pobrania gleby powinien umożliwiać długookresowe użytkowanie. Odpowiednie do tego są stałe pastwiska, pola okresowo zasiewane zbożami (z wyjątkiem kukurydzy) lub gęsto nawożone zielonką. Wybrany obszar do pobierania próbek nie powinien być poddawany działaniu produktów ochrony upraw przez co najmniej jeden rok przed pobraniem próbek. Nie powinno się również podawać żadnych nawozów organicznych przez co najmniej sześć miesięcy. Stosowanie nawozów mineralnych jest dozwolone tylko wtedy, gdy jest zgodne z wymogami upraw i próbki gleby nie powinny być pobierane przez co najmniej trzy miesiące po podaniu nawozu. Należy unikać stosowania gleb poddanych działaniu nawozów o znanym działaniu biologicznym (np. cyjanamid wapnia).
Pobierania próbek należy unikać podczas lub bezpośrednio po dłuższych okresach (przekraczających 30 dni) suszy lub zalania wodą. Dla gleb zaoranych próbki powinny być pobierane z głębokości od 0 do 20 cm. Dla łąk (pastwisk) lub innych gleb, które nie są orane przez dłuższy czas (przynajmniej jeden sezon wegetacyjny), maksymalna głębokość pobierania próbek może być nieco większa niż 20 cm (np. do 25 cm). Próbki gleb powinny być transportowane przy użyciu odpowiednich pojemników i w warunkach temperaturowych gwarantujących, że początkowe własności gleby nie zostaną znacząco zmienione.
1.6.3.2. Przechowywanie
Preferuje się stosowanie gleb świeżo zebranych. Jeżeli nie można uniknąć przechowywania w laboratorium, gleba może być przechowywana w ciemności w temperaturze 4 ± 2 oC przez maksymalnie trzy miesiące. Należy zapewnić warunki do napowietrzania gleby podczas jej przechowywania. Jeżeli gleby są pobierane z obszarów, które zamarzają przez przynajmniej trzy miesiące w roku, można wziąć pod uwagę przechowywanie przez sześć miesięcy w temperaturze minus 18 oC. Mikrobiologiczna biomasa przechowywanych gleb jest mierzona przed każdym eksperymentem i zawartość węgla w biomasie powinna wynosić co najmniej 1 % całkowitej zawartości węgla organicznego w glebie (zob. sekcja 1.6.2).
1.6.4. Postępowanie i przygotowanie gleby do badań
1.6.4.1. Inkubacja wstępna
Jeżeli gleba była przechowywana (zob. sekcje 1.6.4.2 i 1.7.1.3), zalecana jest wstępna inkubacja przez okres 2–28 dni. Temperatura i zawartość wilgoci w glebie podczas wstępnej inkubacji powinny być podobne do tych, jakie będą stosowane w czasie badania (zob. sekcje 1.6.4.2 i 1.7.1.3).
1.6.4.2. Właściwości fizyczno-chemiczne
Z gleby usuwane są ręcznie większe obiekty (to jest kamienie, części roślin itp.) i jest ona przesiewana na wilgotno, bez nadmiernego wysuszenia do ziarnistości mniejszej lub równej 2 mm. Zawartość wilgoci w próbce gleby powinna zostać zwiększona przy użyciu wody destylowanej lub dejonizowanej do wartości pomiędzy 40 a 60 % maksymalnej zdolności utrzymywania wody.
1.6.5. Przygotowanie substancji badanej do podania do gleby
Substancja badana jest zazwyczaj podawana przy użyciu nośnika. Nośnikiem może być woda (dla substancji rozpuszczalnych w wodzie) lub obojętne ciało stałe, takie jak drobnoziarnisty piasek kwarcowy (rozmiar ziaren 0,1–0,5 mm). Należy unikać nośników ciekłych innych niż woda (np. organiczne rozpuszczalniki, takie jak aceton, chloroform), ponieważ mogą niszczyć mikroflorę. Jeżeli stosowanym nośnikiem jest piasek, może on być powleczony badaną substancją rozpuszczoną lub zawieszoną w odpowiednim rozpuszczalniku. W takich przypadkach przed wymieszaniem z glebą należy usunąć rozpuszczalnik przez odparowanie. Dla zachowania optymalnego rozkładu substancji badanej w glebie, zaleca się stosowanie 10 g piasku na kilogram gleby (w suchej masie). Próbki kontrolne są poddawane działaniu równoważnej ilości tylko wody i/lub piasku kwarcowego.
Jeżeli badane są lotne substancje chemiczne, powinno się unikać strat podczas poddawania działaniu i należy podjąć próbę zapewnienia jednorodnego rozkładu w glebie (np. badana substancja powinna być nastrzyknięta do gleby w kilku miejscach).
1.6.6. Stężenia stosowane w badaniach
Jeżeli badane są środki ochrony upraw lub inne substancje chemiczne o przewidywalnym stężeniu środowiskowym, powinno się stosować przynajmniej dwa stężenia. Niższe stężenie powinno odzwierciedlać przynajmniej maksymalną, oczekiwaną ilość dochodzącą do gleby w praktycznym zastosowaniu, natomiast wyższe stężenie powinno być wielokrotnością niższego. Stężenia substancji badanej dodanej do gleby są obliczane przy założeniu równomiernego wprowadzenia na głębokość 5 cm i gęstości nasypowej gleby 1,5. Dla substancji agrochemicznych dodawanych bezpośrednio do gleby lub dla substancji chemicznych, o przewidywalnej ilości dochodzącej do gleby, zalecanymi stężeniami do badań są maksymalne przewidywalne stężenia środowiskowe (Predicted Environmental Concentration – PEC) i ich pięciokrotność. Substancje, dla których przewiduje się kilkakrotne stosowanie w jednym sezonie, powinny być badane przy stężeniach wyliczonych poprzez przemnożenie PEC przez oczekiwaną maksymalną liczbę zastosowań. Wyższe badane stężenie nie powinno jednak przekraczać dziesięciokrotności pojedynczej stosowanej maksymalnej ilości.
Jeżeli badane są substancje nieagrochemiczne, stosowane są ciągi geometryczne przynajmniej pięciu stężeń. Badane stężenia powinny pokryć zakres potrzebny do określenia wartości ECx.
1.7. PROWADZENIE BADANIA
1.7.1. Warunki ekspozycji
1.7.1.1. Próbki badane i kontrolne
Jeżeli badane są substancje agrochemiczne, gleba dzielona jest na trzy równe wagowo części. Dwie części są mieszane z nośnikiem zawierającym badany produkt, a trzecia jest mieszana z samym nośnikiem (próbka kontrolna). Zalecane są przynajmniej trzy powtórzenia, zarówno dla gleb poddanych działaniu badanej substancji, jak i niepoddanych. Jeżeli badane są środki niebędące substancjami agrochemicznymi, gleba dzielona jest na sześć równych wagowo części. Pięć próbek jest mieszanych z nośnikiem zawierającym substancję badaną, a szósty mieszany jest z samym nośnikiem. Zaleca się trzy powtórzenia, zarówno dla próbek badanych, jak i próbek kontrolnych. Należy zadbać o zapewnienie homogenicznego rozprowadzenia substancji badanej w poddanych jej działaniu próbkach gleby. Podczas mieszania powinno się uniknąć sklejania lub zbrylania gleby.
1.7.1.2. Inkubacja próbek gleby
Inkubacja może być prowadzona na dwa sposoby: jako zbiorcze próbki każdej gleby poddanej i niepoddanej działaniu badanej substancji, albo jako seria pojedynczych podpróbek równej wielkości, poddanych i niepoddanych działaniu badanej substancji. Jednakże jeżeli badane są substancje lotne, badania powinny być prowadzone jedynie na serii pojedynczych podpróbek. Jeżeli gleby są inkubowane w masie, przygotowywane są duże ilości gleby poddanej i niepoddanej działaniu badanej substancji i podpróbki do analizy są pobierane w miarę potrzeby podczas badania. Ilość przygotowana wstępnie do każdego poddania działaniu i do kontroli zależy od wielkości podpróbek, ilości powtórzeń stosowanych do analizy oraz przewidywanej maksymalnej liczby pobieranych próbek. Gleby inkubowane w jednej objętości powinny zostać dokładnie wymieszane przed podziałem na mniejsze próbki. Jeżeli gleby są inkubowane jako seria pojedynczych próbek, każda poddana działaniu substancji badanej i kontrolna masa gleby jest dzielona na wymaganą liczbę podpróbek, które są wykorzystywane w miarę potrzeb. W eksperymentach, gdzie przewiduje się więcej niż dwa próbkowania, powinna być przygotowywana wystarczająca liczba podpróbek do wszystkich powtórzeń i próbkowań. Przynajmniej trzy powtórzone próbki badanej gleby powinny być inkubowane w warunkach napowietrzania (zob. sekcja 1.7.1.1). Podczas wszystkich badań powinny być stosowane odpowiednie pojemniki, z wystarczającą przestrzenią nad powierzchnią próbki, w celu uniknięcia rozwoju warunków beztlenowych. Jeżeli badane są substancje lotne, badanie powinno być prowadzone wyłącznie na serii pojedynczych podpróbek.
1.7.1.3. Warunki badań i czas ich trwania
Badanie jest prowadzone w ciemności w temperaturze pokojowej 20 ± 2 oC. Zawartość wilgoci w próbkach gleb podczas badania powinna wynosić 40–60 % maksymalnej pojemności wodnej (zob. sekcja 1.6.4.2) w przedziale ± 5 %. Można dodawać wodę destylowaną i dejonizowaną, stosownie do potrzeb.
Minimalny czas trwania badań wynosi 28 dni. Jeżeli badane są substancje agrochemiczne, porównywane są ilości wydzielonego ditlenku węgla lub zużytego tlenu dla próbki badanej i kontrolnej. Jeżeli 28 dnia wyniki różnią się od siebie o więcej niż 25 %, badanie jest kontynuowane aż do osiągnięcia wartości równej lub mniejszej od 25 % lub przez maksymalnie 100 dni, w zależności od tego, co nastąpi wcześniej. Jeżeli badane są substancje niebędące środkami agrochemicznymi, badanie przerywa się po 28 dniach. 28 dnia oznaczane są ilości wydzielonego ditlenku węgla lub zużytego tlenu w próbce badanej i kontrolnej i obliczane są wartości ECx.
1.7.2. Próbkowanie i analiza gleb
1.7.2.1. Harmonogram próbkowania gleb
Jeżeli badane są substancje agrochemiczne, próbki gleby są analizowane pod kątem oddychania pod wpływem glukozy w dniach 0, 7, 14 i 28. Jeżeli wymagane jest przedłużenie badania, dalsze pomiary powinny być wykonywane co 14 dni po dniu 28.
Jeżeli badane są substancje nieagrochemiczne, stosuje się przynajmniej pięć stężeń i próbki gleby są analizowane na respirację wywołaną glukozą na początku (dzień 0) i na końcu okresu ekspozycji (28 dzień). Można dodać pomiary pośrednie, np. 7 dnia, jeżeli zostanie to uznane za konieczne. Dane uzyskane 28 dnia są stosowane do wyznaczenia wartości ECx dla substancji chemicznej. Jeżeli jest to pożądane, można wykorzystać wyniki z dnia 0 próbek kontrolnych do oszacowania początkowych ilości aktywnej metabolicznie biomasy mikrobiologicznej w glebie (12).
1.7.2.2. Pomiary tempa respiracji indukowanej glukozą
Tempo respiracji indukowanej glukozą w każdej próbce badanej i kontrolnej jest określane przy każdorazowym próbkowaniu. Próbki gleby są mieszane z odpowiednią ilością glukozy w celu wywołania warunków do natychmiastowej reakcji respiracyjnej. Ilość glukozy potrzebnej do wywołania maksymalnej reakcji respiracyjnej dla danej gleby może zostać wyznaczona we wstępnym badaniu z wykorzystaniem serii stężeń glukozy (14). Jednakże dla piaszczystych gleb zawierających 0,5–1,5 % węgla organicznego na ogół wystarczające jest zastosowanie 2 000 mg do 2 000 mg glukozy na kg suchej masy gleby. Glukoza może zostać roztarta z czystym piaskiem kwarcowym (10 g piasku na kg suchej masy gleby) i homogenicznie wymieszana z glebą.
Próbki gleby wzbogaconej glukozą są inkubowane w aparaturze odpowiedniej do mierzenia tempa respiracji albo w sposób ciągły, albo co godzinę bądź co dwie godziny (zob. sekcja 1.6.1) w temperaturze 20 ± 2 oC. Uwalniany ditlenek węgla lub zużyty tlen są mierzone przez kolejnych 12 godzin, a pomiary powinny rozpocząć się tak szybko, jak to możliwe, to jest w ciągu 1–2 godzin od dodania glukozy. Określa się całkowite ilości wydzielonego ditlenku węgla lub pochłoniętego tlenu w ciągu 12 godzin i wyznacza się średnią wartość tempa respiracji.
2. DANE
2.1. OPRACOWANIE WYNIKÓW
Jeżeli badane są substancje agrochemiczne, powinien zostać zapisany uwolniony ditlenek węgla lub pochłonięty tlen z każdej próbki gleby i wartość średnia wszystkich powtórzeń powinna zostać przedstawiona w formie tabeli. Wyniki powinny należy zinterpretować przy użyciu odpowiednich i powszechnie akceptowanych metod statystycznych (np. test F, 5 % poziom istotności). Tempo respiracji indukowanej glukozą jest wyrażane w mg ditlenku węgla/kg suchej masy gleby/h lub mg tlenu/kg suchej masy gleby/h. Średnie tempo tworzenia się ditlenku węgla lub średnie tempo pochłaniania tlenu dla każdej badanej próbki jest porównywane z próbką kontrolną i obliczane jest odchylenie procentowe od próbki kontrolnej.
Jeżeli badane są substancje nieagrochemiczne, oznacza się ilość wydzielonego ditlenku węgla lub pochłoniętego tlenu przez każdą próbkę i do oceny wartości ECx przygotowywana jest krzywa zależności dawka-reakcja. Tempo respiracji indukowanej glukozą (to jest mg ditlenku węgla/kg suchej masy gleby/h lub mg tlenu/kg suchej masy gleby/h) określone w badanych próbkach jest porównywane z wartością określoną dla próbki kontrolnej. Na podstawie tych danych obliczane są wielkości % inhibicji dla każdego badanego stężenia. Te wartości procentowe są wykreślane w zależności od stężenia, a następnie wykorzystuje się procedury statystyczne do obliczenia wartości ECx. Granice przedziału ufności (p = 0,95) dla obliczanej wartości ECx są również określane przy użyciu standardowych procedur (15)(16)(17).
2.2. INTERPRETACJA WYNIKÓW
Jeżeli są oceniane wyniki badań substancji agrochemicznych i różnica między tempem respiracji przy niższym stężeniu (to jest maksymalne przewidywane stężenie) a tempem respiracji próbki kontrolnej jest mniejsza lub równa 25 %, w którymkolwiek momencie pomiarów po upływie 28 dni, produkt może zostać oceniony, jako niemający długookresowego wpływu na przemiany węgla w glebie. Jeżeli oceniane są wyniki uzyskane dla substancji chemicznych innych niż agrochemiczne, stosowane są wartości EC50, EC25 i/lub EC10.
3. SPRAWOZDAWCZOŚĆ
RAPORT Z BADAŃ
Raport z badań musi zawierać następujące informacje:
|
|
Pełną identyfikację używanej gleby, w tym:
|
|
|
Substancja badana:
|
|
|
Warunki badania:
|
|
|
Wyniki:
|
4. BIBLIOGRAFIA
|
(1) |
EPPO (1994). Decision-Making Scheme for the Environmental Risk Assessment of Plant Protection Chemicals. Chapter 7: Soil Microflora. EPPO Bulletin 24: 1–16, 1994. |
|
(2) |
BBA (1990). Effects on the Activity of the Soil Microflora. BBA Guidelines for the Official Testing of Plant Protection Products, VI, 1–1 (2nd eds., 1990). |
|
(3) |
EPA (1987). Soil Microbial Community Toxicity Test. EPA 40 CFR Part 797.3700. Toxic Substances Control Act Test Guidelines; Proposed rule. September 28, 1987. |
|
(4) |
SETAC-Europe (1995). Procedures for assessing the environmental fate and ecotoxicity of pesticides, Ed. M.R. Lynch, Pub. SETAC-Europe, Brussels. |
|
(5) |
OECD (1995). Final Report of the OECD Workshop on Selection of Soils/Sediments, Belgirate, Italy, 18-20 January 1995. |
|
(6) |
ISO 10381-6 (1993). Soil quality – Sampling. Guidance on the collection, handling and storage of soil for the grade of aerobic microbial processes in the laboratory. |
|
(7) |
Anderson, J.P.E. (1987). Handling and Storage of Soils for Pesticide Experiments, in "Pesticide Effects on Soil Microflora.. Eds. L. Somerville and M.P. Greaves, Chap. 3: 45–60. |
|
(8) |
Anderson, J.P.E. (1982). Soil respiration, in "Methods of Soil Analysis – Part 2: Chemical and Microbiological Properties. Agronomy Monograph No 9. Eds. A.L. Page, R.H. Miller and D.R. Keeney. 41: 831–871. |
|
(9) |
ISO 11266-1. (1993). Soil Quality – Guidance on Laboratory Tests for Biodegradation in Soil: Part 1. Aerobic Conditions. |
|
(10) |
ISO 14239 (1997E). Soil Quality – Laboratory incubation systems for measuring the mineralization of organic chemicals in soil under aerobic conditions. |
|
(11) |
Heinemeye, r O., Insam, H., Kaiser, E.A, and Walenzik, G. (1989). Soil microbial biomass and respiration measurements; an automated technique based on infrared gas analyses. Plant and Soil, 116: 77–81. |
|
(12) |
ISO 14240-1 (1997). Soil quality – Determination of soil microbial biomass – Part 1: Substrateinduced oddychania method. |
|
(13) |
ISO 14240-2 (1997). Soil quality – Determination of soil microbial biomass – Part 2: Fumigationextraction method. |
|
(14) |
Malkomes, H.-P. (1986). Einfluß von Glukosemenge auf die Reaktion der Kurzzeit-Atmung im Boden Gegemiber Pflanzenschutzmitteln, Dargestellt am Beispiel eines Herbizide. (Influence of the Amount of Glucose Added to the Soil on the Effect of Pesticides in Short-Term Respiration, using a Herbicide as an Example). Nachrichtenbl. Deut. Pflanzenschutzd., Braunschweig, 38: 113–120. |
|
(15) |
Litchfield, J.T. and Wilcoxon, F. (1949). A simplified method of evaluating dose-effect experiments. Jour. Pharmacol. and Exper. Ther., 96, 99–113. |
|
(16) |
Finney, D.J. (1971). Probit Analysis. 3rd ed., Cambridge, London and New-York. |
|
(17) |
Finney D.J. (1978). Statistical Methods in biological Assay. Griffin, Weycombe, UK. |
C.23. PRZEMIANY W GLEBIE W WARUNKACH NATLENIENIA I BEZTLENOWYCH
1. METODA
Ta metoda badawcza jest równoważna OECD TG 307 (2002).
1.1. WPROWADZENIE
Ta metoda badawcza jest oparta na istniejących wytycznych (1)(2)(3)(4)(5)(6)(7)(8)(9). Metoda opisana w tej metodzie badawczej jest przeznaczona do oceny przemian substancji chemicznych w glebie, w warunkach natlenienia i beztlenowych. Eksperymenty są prowadzone w celu wyznaczenia: (i) szybkości przemian substancji badanej; oraz (ii) charakteru i szybkości tworzenia oraz zanikania produktów przemian, których działaniu mogą zostać poddane organizmy w glebie. Takie badania są wymagane dla substancji chemicznych, które są bezpośrednio podawane do gleby lub mogą przeniknąć do środowiska glebowego. Wyniki takich badań laboratoryjnych mogą być także wykorzystane do opracowania protokołów pobierania próbek i analizy dla związanych badań w terenie.
Badania w warunkach tlenowych i beztlenowych jednego rodzaju gleby na ogół wystarczają do oceny szlaków przemian (8)(10)(11). Tempa przemian powinny być określone przy użyciu co najmniej trzech dodatkowych rodzajów gleb (8)(10).
W czasie warsztatów OECD w sprawie wyboru gleb i osadów, które odbyły się w Belgirate we Włoszech w 1995 roku (10), uzgodniono, w szczególności, rodzaje gleb do stosowania w tym badaniu i ich liczbę. Rodzaje badanych gleb powinny być reprezentacyjne dla warunków środowiskowych, w których substancja będzie stosowana lub uwalniania. Na przykład związki chemiczne, które mogą być uwalniane w klimacie subtropikalnym lub tropikalnym, powinny być badane przy pomocy ferrazoli lub nitrozoli (system FAO). Uczestnicy warsztatów podali również zalecenia dotyczące pobierania, przenoszenia i przechowywania próbek gleby na podstawie wytycznych ISO (15). W tej metodzie uwzględniono także gleby pod mokre uprawy ryżu.
1.2. DEFINICJE
Substancja badana: jakakolwiek substancja, czy to związek macierzysty czy też odnośne produkty przemian.
Produkty przemian: wszystkie substancje powstałe w wyniku biotycznych lub abiotycznych reakcji przemian substancji badanej, w tym CO2, i produkty w pozostałościach związanych.
Pozostałości związane:„Pozostałości związane” stanowią związki w glebie, roślinach lub zwierzętach, które pozostają po ekstrakcji w materiale macierzystym w postaci substancji macierzystej lub jej metabolitu(-ów)/produktów przemiany. Metoda ekstrakcji nie może zmieniać w sposób znaczący samych związków lub struktury materiału macierzystego. Charakter wiązania można częściowo wyjaśnić przez metody ekstrakcji zmieniające materiał macierzysty i wyspecjalizowane techniki analityczne. Do dziś, na przykład, zostały w ten sposób zidentyfikowane wiązania kowalencyjne zjonizowane i sorpcyjne, a także pułapkowanie. Na ogół tworzenie się pozostałości związanych znacząco redukuje biodostępność i bioprzyswajalność (12) (zmodyfikowane w stosunku do IUPAC 1984 (13)).
Przemiany tlenowe: reakcje zachodzące w obecności tlenu cząsteczkowego (14).
Przemiany beztlenowe: reakcje zachodzące z wykluczeniem tlenu cząsteczkowego (14).
Gleba: jest mieszaniną składników chemicznych mineralnych i organicznych, te drugie zawierają związki o wysokiej zawartości węgla i azotu, i o wysokiej masie atomowej, ożywioną przez drobne (najczęściej mikro-) organizmy. Gleba może występować w dwóch stanach:
|
a) |
niezakłóconym, tak jak się rozwijała z upływem czasu, z charakterystycznymi warstwami różnych rodzajów gleb; |
|
b) |
zakłóconym, jak to ma na ogół miejsce na polach uprawnych lub kiedy próbki są pobierane przez kopanie i stosowane w tej metodzie badawczej (14). |
Mineralizacja: to całkowita degradacja związku organicznego do CO2 i H2O w warunkach tlenowych lub CH4, CO2 i H2O w warunkach beztlenowych. W kontekście tej metody badawczej, w której stosowane są związki znakowane 14C, mineralizacja oznacza ekstensywną degradację, podczas której znaczone atomy węgla są utleniane z wydzieleniem odpowiedniej ilości 14CO2 (14).
Okres półtrwania: t0,5 to czas potrzebny do przemiany 50 % substancji badanej, jeżeli przemiana może zostać opisana kinetyką pierwszego rzędu; nie zależy od stężenia.
DT 50 (czas zaniku 50): czas, w którym stężenie substancji badanej zmniejsza się o 50 %; ta wielkość różni się od okresu półtrwania t0,5, jeżeli przemiana nie może zostać opisana kinetyką pierwszego rzędu.
DT 75 (czas zaniku 75): czas, w jakim stężenie substancji badanej zmniejsza się o 75 %.
DT 90 (czas zaniku 90): czas, w jakim stężenie substancji badanej zmniejsza się o 90 %.
1.3. SUBSTANCJE ODNIESIENIA
Substancje odniesienia powinny być stosowane do charakteryzacji i/lub identyfikacji produktów przemian przy pomocy metod spektroskopowych i chromatograficznych.
1.4. ZASTOSOWANIE BADANIA
Metoda może być stosowana do wszystkich substancji chemicznych (nieznakowanych lub znakowanych izotopowo), dla których jest dostępna metoda analityczna o wystarczającej czułości i dokładności. Może być stosowana do związków słabo lotnych, nielotnych, rozpuszczalnych lub nierozpuszczalnych w wodzie. Badanie nie powinno być stosowane do substancji chemicznych o wysokiej lotności z gleby (np. fumigantów, rozpuszczalników organicznych), które nie mogą utrzymać się w glebie w warunkach eksperymentalnych badania.
1.5. INFORMACJE O SUBSTANCJI BADANEJ
Nieznakowane i znakowane izotopowo substancje badane mogą być stosowane do pomiaru tempa przemian. Materiał znakowany jest wymagany do badania szlaków przemian i do określenia bilansu masowego. Zaleca się znakowanie izotopem 14C, ale inne izotopy, takie jak 13C, 15N, 3H, 32P, także mogą być użyteczne. Na tyle, na ile jest to możliwe, znacznik powinien być umiejscowiony w najbardziej stabilnej części (częściach) cząsteczki (41). Czystość substancji badanej powinna wynosić przynajmniej 95 %.
Przed przystąpieniem do badań przemian tlenowych i beztlenowych w glebie, powinny być dostępne następujące informacje na temat substancji badanej:
|
a) |
rozpuszczalność w wodzie (metoda A.6), |
|
b) |
rozpuszczalność w rozpuszczalnikach organicznych, |
|
c) |
prężność pary (metoda A.4) i stała proporcjonalności w prawie Henry'ego, |
|
d) |
współczynnik podziału n-oktanol/woda (metoda A.8), |
|
e) |
stabilność chemiczna w ciemności (hydroliza) (metoda C.7), |
|
f) |
pKa, jeżeli molekuła jest podatna na protonowanie lub deprotonowanie (wytyczne OECD 112) (16). |
Inne przydatne informacje mogą obejmować dane na temat toksyczności substancji badanej dla mikroorganizmów w glebie (metody badawcze C.21 i C.22) (16).
Powinny być dostępne metody analityczne (w tym metody ekstrakcji i czyszczenia) do oznaczania ilościowego i identyfikacji substancji badanej i jej produktów przemian.
1.6. ZASADA METODY BADAWCZEJ
Próbki gleby są poddawane działaniu substancji badanej i inkubowane w ciemności w kolbie biometrycznej lub w układzie przepływowym, w kontrolowanych warunkach laboratoryjnych (w stałej temperaturze i wilgotności gleby). W odpowiednich odstępach czasu próbki gleby są ekstrahowane i analizowane pod kątem obecności substancji macierzystej i produktów przemian. Produkty lotne są także pobierane do analizy przy użyciu odpowiednich urządzeń do ich absorpcji. Stosując materiał znakowany izotopem 14C, można mierzyć różne stopnie mineralizacji substancji badanej poprzez pułapkowanie wydzielonego 14CO2 i można określić bilans masowy, obejmujący tworzenie pozostałości związanych w glebie.
1.7. KRYTERIA JAKOŚCIOWE
1.7.1. Odzysk
Ekstrakcja i analiza przynajmniej podwójnych próbek gleby natychmiast po dodaniu substancji badanej daje, pierwsze wskazanie co do powtarzalności metody analitycznej i jednorodności procedury podawania substancji badanej. Odzysk dla następnych etapów eksperymentu jest podawany przez odnoszący się do nich bilans masowy. Odzysk powinien mieścić się w zakresie 90–110 % dla znakowanych substancji chemicznych (8) i 70–110 % dla nieznakowanych substancji chemicznych (3).
1.7.2. Powtarzalność i czułość metody analitycznej
Powtarzalność metody analitycznej (wyłączając sprawność początkowej ekstrakcji) dla oznaczenia ilościowego substancji badanej i produktów przemian może zostać sprawdzona przez powtórną analizę tego samego ekstraktu z gleby inkubowanej wystarczająco długo, aby doszło do wytworzenia produktów przemian.
Granica wykrywalności (LOD) metody analitycznej dla badanej substancji i produktów przemian powinna wynosić co najmniej 0,01 mg × kg-1 gleby (dla substancji badanej) lub 1 % podanej dawki, w zależności od tego, co jest niższe. Granica kwantyfikacji (LOQ) także powinna zostać określona.
1.7.3. Dokładność danych przemian
Analiza regresji stężeń substancji badanej w funkcji czasu daje odpowiednie informacje o rzetelności krzywej przemian i pozwala na obliczenie granic przedziału ufności dla okresu półtrwania (w przypadku kinetyki pseudo pierwszorzędowej) lub wartości DT50 oraz w razie potrzeby wartości DT75 i DT90.
1.8. OPIS METODY
1.8.1. Wyposażenie i odczynniki
Układ inkubacyjny składa się ze statycznych systemów zamkniętych lub odpowiednich systemów przepływowych (7)(17). Przykłady odpowiedniej aparatury do przepływowej i kolby biometrycznej do inkubacji gleby są przedstawione odpowiednio na rys. 1 i 2. Obydwa systemy inkubacyjne mają swoje zalety i ograniczenia (7)(17).
Wymagane jest standardowe wyposażenie laboratoryjne, zwłaszcza następujące:
|
— |
instrumenty analityczne, takie jak aparaty do GLC, HPLC, TLC, włączając w to odpowiednie układy detekcji do analizy substancji znakowanych i nieznakowanych izotopowo lub metodę inwersji rozrzedzonych izotopów, |
|
— |
instrumenty do celów identyfikacji (np. MS, GC-MS, HPLC-MS, NMR itp.), |
|
— |
cieczowy licznik scyntylacyjny, |
|
— |
aparat do przeprowadzania utleniania do spalania materiałów radioaktywnych, |
|
— |
wirówka, |
|
— |
aparatura do ekstrakcji (na przykład wirówka rurowa do zimnej ekstrakcji i aparat ekstrakcyjny Soxhleta do ciągłej ekstrakcji pod chłodnicą zwrotną), |
|
— |
instrumenty do zatężania roztworów i ekstraktów (np. wirówka próżniowa), |
|
— |
łaźnia wodna, |
|
— |
mieszadła mechaniczne (np. zgniatarka, mikser obrotowy). |
Do stosowanych odczynników zalicza się na przykład:
|
— |
NaOH, czysty do analizy, 2 mol· dm-3, lub inne odpowiednie zasady (np. KOH, etanoloamina), |
|
— |
H2SO4, czysty do analizy, 0,05 mol· dm-3, |
|
— |
glikol etylenowy, czysty do analizy, |
|
— |
stałe materiały absorpcyjne, takie jak wapno sodowe i poliuretanowe wkłady, |
|
— |
rozpuszczalniki organiczne, czyste do analizy, takie jak aceton, metanol itp., |
|
— |
ciecz scyntylacyjna. |
1.8.2. Podawanie substancji badanej
W celu dodania i rozprowadzenia w glebie substancja badana może być rozpuszczona w wodzie (dejonizowanej lub destylowanej) lub w razie potrzeby w minimalnej ilości acetonu lub innego rozpuszczalnika organicznego (6), w jakiej substancja badana jest wystarczająco rozpuszczalna i stabilna. Jednakże ilość wybranego rozpuszczalnika nie powinna mieć znaczącego wpływu na aktywność mikrobiologiczną gleby (zob. sekcje 1.5 i 1.9.2–1.9.3). Należy unikać stosowania rozpuszczalników, które inhibitują aktywność mikrobiologiczną, takich jak: chloroform, dichlorometan i inne rozpuszczalniki fluorowcowane.
Substancja badana może być również dodana w stanie stałym, np. wymieszana z piaskiem kwarcowym (6) lub w małej podpróbce badanej gleby, która została wysuszona na powietrzu i wysterylizowana. Jeżeli substancja badana jest dodawana przy użyciu rozpuszczalnika, rozpuszczalnik powinien zostać odparowany przed dodaniem szczytowej podpróbki do właściwej, niesterylnej próbki gleby.
Dla większości substancji chemicznych, których główną drogą dostępu do gleby jest osad ściekowy/zastosowania rolnicze, substancja badana powinna zostać najpierw dodana do szlamu, który następnie jest wprowadzany do próbki gleby (zob. sekcje 1.9.2 i 1.9.3).
Stosowanie gotowych produktów nie jest rutynowo zalecane. Jednakże np. dla słabo rozpuszczalnych substancji badanych zastosowanie gotowych materiałów może być odpowiednim rozwiązaniem.
1.8.3. Gleby
1.8.3.1. Wybór gleb
Do określenia szlaków przemian, może być stosowana gleba reprezentatywna, glina piaszczysta, glina iłowa, glina, lub piasek gliniasty (zgodnie z klasyfikacją FAO i USDA (18)), o pH 5,5–8,0, zawartości węgla organicznego 0,5–2,5 % i mikrobiologicznej biomasie stanowiącej co najmniej 1 % całkowitej zawartości węgla organicznego (10).
Do badania tempa przemian powinny zostać użyte przynajmniej trzy dodatkowe gleby reprezentujące szereg istotnych gleb. Te gleby powinny mieć różną zawartość węgla organicznego, pH, zawartość gliny i mikrobiologicznej biomasy (10).
Wszystkie gleby powinny zostać scharakteryzowane, przynajmniej pod względem tekstury ( % piasku, % iłów, % gliny) (zgodnie z klasyfikacją FAO i USDA (18)), pH, pojemności wymiany kationowej, węgla organicznego, gęstości nasypowej, własności retencji wody (42) i mikrobiologicznej biomasy (tylko do badań w warunkach natlenienia). Dodatkowe informacje na temat właściwości gleby mogą być przydatne przy interpretacji wyników. Dla określenia własności gleby mogą być stosowane zalecane metody przedstawione w literaturze (19)(20)(21)(22)(23). Mikrobiologiczna biomasa powinna być oznaczona z wykorzystaniem metody respiracji wywołanej przez substrat (SIR) (25)(26) lub metodami alternatywnymi (20).
1.8.3.2. Pobieranie, przenoszenie i przechowywanie gleb
Powinny być dostępne szczegółowe informacje na temat historii obszaru, z jakiego została pobrana badana próbka gleby. Szczegółowe informacje powinny obejmować dokładną lokalizację, szatę roślinną, poddawanie działaniu substancji chemicznych, poddawanie działaniu nawozów organicznych i nieorganicznych, dodatki materiałów biologicznych lub inne zanieczyszczenia. Gleby, które były poddawane działaniu substancji badanej lub jej strukturalnych analogów w ciągu ostatnich czterech lat, nie powinny być używane do badania przemian (10)(15).
Gleba powinna być świeżo zebrana z pola (z poziomu A lub z wierzchniej warstwy o grubości 20 cm), z taką zawartością wody, jaka ułatwia przesiewanie. Dla gleb innych niż gleby z pól ryżowych, powinno się unikać pobierania próbek podczas lub bezpośrednio po długich okresach (> 30 dni) suszy, mrozu, zalewania (14). Próbki powinny być transportowane w sposób, który minimalizuje zmiany zawartości wody w glebie oraz powinny być przechowywane w ciemnym miejscu, z możliwie swobodnym dostępem powietrza. Na ogół do tego celu wystarczająca jest luźno związana torba polietylenowa.
Gleba powinna zostać przetworzona możliwie szybko po pobraniu próbki. Roślinność, większe okazy fauny glebowej oraz kamienie powinny zostać usunięte przed przesianiem gleby przez 2 mm sito, które usuwa drobne kamienie, okazy fauny i szczątki roślin. Należy unikać intensywnego suszenia i kruszenia gleby przed przesiewaniem (15).
Jeżeli zimą pobieranie próbek z pola jest trudne (gleba zamarznięta lub pokryta warstwą śniegu), można pobrać próbkę z partii gleby przechowywanej w szklarni pod pokryciem roślinnym (np. trawa lub mieszanina trawy i koniczyny). Zdecydowanie preferuje się badanie gleb świeżo pobranych z terenu, ale jeżeli zebrana i przetworzona gleba musi być przechowywana przed rozpoczęciem badań, przechowywanie musi być w odpowiednich warunkach i tylko przez ograniczony czas (4 ± 2 oC przez maksymalnie trzy miesiące), aby utrzymać aktywność mikrobiologiczną (43). Szczegółowe instrukcje pobierania, obchodzenia się i przechowywania gleby, przeznaczonej do eksperymentów z bioprzemianami, można znaleźć w (8)(10)(15)(26)(27).
Przed użyciem przetworzonej gleby w tym badaniu, powinna ona być wstępnie inkubowana, aby umożliwić kiełkowanie i usunięcie nasion, i aby przywrócić równowagę metabolizmu mikrobiologicznego następującą po przeniesieniu próbki z pola lub z warunków przechowywania do warunków inkubowania. Na ogół wystarczający jest okres wstępnej inkubacji 2–28 dni, w temperaturze i wilgotności zbliżonych do tych, jakie panują w czasie właściwego badania (15). Czas przechowywania i wstępnej inkubacji łącznie nie powinien przekraczać trzech miesięcy.
1.9. PROWADZENIE BADAŃ
1.9.1. Warunki badania
1.9.1.1. Temperatura badania
Podczas całego okresu badania gleby powinny być inkubowane w ciemności w stałej temperaturze reprezentatywnej dla warunków klimatycznych, w których wystąpi wykorzystanie lub uwolnienie substancji badanej. Zaleca się temperaturę około 20 ± 2 oC dla wszystkich substancji badanych, jakie mogą przeniknąć do gleby w klimacie umiarkowanym. Temperatura powinna być monitorowana.
Dla substancji chemicznych podawanych lub uwalnianych do gleby w chłodniejszych strefach klimatycznych (np. w krajach północnych, podczas okresów jesieni/zimy) powinno się inkubować dodatkowe próbki gleby, ale w niższej temperaturze (np. 10 ± 2 oC).
1.9.1.2. Zawartość wilgoci
Do badań przemian w warunkach natlenienia powinna zostać skorygowana zawartość wilgoci (44) i powinna być utrzymywana na poziomie pF od 2,0 do 2,5 (3). Zawartość wilgoci jest wyrażana jako masa wody na masę suchej gleby i powinna być regularnie kontrolowana (np. w odstępach dwutygodniowych) przez ważenie kolby inkubacyjnej, a straty wody powinny być kompensowane przez dodawanie wody (najlepiej sterylnie filtrowana woda z kranu). Należy uważać, by uniemożliwić lub zminimalizować straty substancji badanej i/lub produktów transformacji w wyniku ulatniania się i/lub fotodegradacji (jeśli zachodzi) podczas uzupełniania wilgoci.
Do badania przemian w warunkach beztlenowych i dla warunków pól ryżowych, gleba jest nasycana wodą przez zalewanie.
1.9.1.3. Inkubacja w warunkach tlenowych
W systemach przepływowych warunki natlenienia będą utrzymane w wyniku przerywanego przepływu cieczy lub ciągłej wentylacji nawilżonym powietrzem. W kolbach biometrycznych wymiana powietrza jest uzyskiwana przez dyfuzję.
1.9.1.4. Sterylne warunki tlenowe
Aby uzyskać informacje na temat znaczenia abiotycznej przemiany substancji badanej, próbki gleby mogą zostać wysterylizowane (metody sterylizacji zob. poz. bibliograficzne (16) i (29)), poddawane działaniu sterylnych substancji badanych (np. dodawanie roztworu przez filtr sterylny) i natleniane nawilżonym sterylnym powietrzem zgodnie z opisem w sekcji 1.9.1.3. Dla gleb pod mokre uprawy ryżu gleba i woda powinny być sterylizowane i inkubacja powinna być prowadzona jak opisano w sekcji 1.9.1.6.
1.9.1.5. Beztlenowe warunki inkubacji
Aby stworzyć i utrzymać warunki beztlenowe, gleba jest poddawana działaniu substancji badanej i inkubowana w warunkach natlenienia przez 30 dni lub jeden okres półtrwania lub DT50 (w zależności od tego, który jest krótszy), następnie jest zalewana wodą (warstwa 1–3 cm wody nad powierzchnią) i system inkubowania jest przepłukiwany strumieniem gazu obojętnego (np. azot lub argon) (45). Układ badawczy musi pozwalać na pomiary takie, jak pH, stężenie tlenu i potencjał redoks oraz zawierać urządzenie do wychwytywania produktów lotnych. Układ biometryczny musi być zamknięty i uniemożliwiać dostawanie się powietrza poprzez dyfuzję.
1.9.1.6. Warunki inkubacji mokrej uprawy ryżu
Do badania przemian w warunkach mokrej uprawy ryżu, gleba jest zalewana warstwą wody o grubości warstwy około l–5cm, a substancja badana jest dodawana do fazy wodnej (9). Zaleca się głębokość gleby przynajmniej 5 cm. System jest wentylowany powietrzem, tak jak w warunkach natlenienia. Należy monitorować i podawać pH, stężenie tlenu i potencjał redoks warstwy wody. Niezbędny jest przynajmniej dwutygodniowy okres wstępnej inkubacji przed rozpoczęciem badań przemian (zob. sekcja 1.8.3.2).
1.9.1.7. Czas trwania badań
Czas badania stopnia przemian i ich szlaków nie powinien przekraczać 120 dni (46) (3)(6)(8), ponieważ można oczekiwać, że po tym czasie spadnie aktywność mikrobiologiczna w sztucznych warunkach laboratoryjnych izolowanych od naturalnej wymiany składników. Jeżeli jest to konieczne do scharakteryzowania zaniku substancji badanej, a także tworzenia i zaniku głównych produktów przemian, badania mogą być prowadzone w dłuższym okresie czasu (np. 6 lub 12 miesięcy) (8). Powody dłuższej inkubacji powinny zostać przedstawione w sprawozdaniu z badań i powinny być dołączone pomiary biomasy sporządzone podczas i na końcu tego okresu.
1.9.2. Prowadzenie badań
Około 50–200 g gleby (w suchej masie) jest umieszczane w każdej kolbie inkubacyjnej (zob. rysunki 1 i 2 w załączniku 3), a następnie gleba jest poddawana działaniu substancji badanej jedną z metod przedstawionych w sekcji 1.8.2. Jeżeli stosowane są rozpuszczalniki organiczne do podania substancji badanej, należy je usunąć z gleby poprzez odparowanie. Następnie gleba jest dokładnie mieszana przy pomocy szpatułki i/lub poprzez wstrząsanie kolby. Jeżeli badanie jest prowadzone w warunkach mokrej uprawy ryżu, gleba z wodą powinny zostać dokładnie wymieszane po dodaniu substancji badanej. Małe podwielokrotności (np. 1 g) gleby poddanej działaniu powinny być analizowane pod kątem substancji badanej w celu sprawdzenia równomiernego rozkładu. Dla metody alternatywnej, zob. poniżej.
Stopień poddania działaniu substancji badanej powinien odpowiadać największemu stopniowi podania środka ochrony upraw zalecanemu w instrukcji stosowania i jednorodnemu wprowadzeniu na odpowiednią głębokość na polu (np. górna warstwa 10 cm (47) gleby (7)). Na przykład dla substancji chemicznych stosowanych na liście lub na glebę bez wprowadzania do niej, odpowiednia głębokość do obliczenia, ile substancji chemicznej należy dodać do kolby wynosi 2,5 cm. Dla substancji chemicznych wprowadzanych do gleby, odpowiednia głębokość jest głębokością wprowadzania określoną w instrukcji stosowania. Dla większości substancji chemicznych podawaną ilość należy oszacować na podstawie najbardziej istotnego szlaku wnikania; na przykład jeżeli główny szlak wnikania w glebę prowadzi przez osad ściekowy, substancja chemiczna powinna być dozowana do osadu w stężeniu, które odzwierciedla oczekiwane stężenie w osadzie, a ilość osadu dodawanego do gleby powinna odzwierciedlać zwykłe wprowadzanie osadu do gleby uprawnej. Jeżeli to stężenie nie jest wystarczająco wysokie do zidentyfikowania głównych produktów przemian, pomocna powinna być inkubacja oddzielnej próbki gleby zawierającej wyższe stężenie, należy jednak unikać stężeń, które po przekroczeniu pewnego poziomu wpływają na mikrobiologiczne funkcje w glebie (zob. sekcje 1.5 i 1.8.2).
Alternatywnie, można poddać działaniu substancji badanej większą partię gleby (to jest 1–2 kg), dokładnie mieszając w odpowiednim urządzeniu do mieszania, i następnie przenieść w małych porcjach 50–200 g do kolby inkubacyjnej (na przykład stosując rozdzielacz do próbek). Małe podwielokrotności (np. 1 g) partii gleby poddanej działaniu substancji badanej powinny być analizowane na jej obecność w celu sprawdzenia równomierności rozprowadzenia. Procedura ta jest preferowana, ponieważ umożliwia bardziej równomierne rozprowadzenie substancji badanej w glebie.
Także próbki gleby niepoddane działaniu substancji badanej inkubuje się w tych samych warunkach (natlenienia), jak próbki poddane jej działaniu. Próbki te stosowane są do pomiarów biomasy podczas badań i na ich koniec.
Jeżeli substancja badana podawana jest w postaci rozpuszczonej w rozpuszczalniku (rozpuszczalnikach) organicznym, próbki gleby traktowane taką samą ilością rozpuszczalnika (rozpuszczalników) są inkubowane w tych samych warunkach (natlenienia), jak próbki gleby poddanej działaniu substancji badanej. Te próbki używane do pomiarów biomasy na początku, w trakcie i na końcu badań, w celu określenia wpływu rozpuszczalnika (rozpuszczalników) na biomasę mikrobiologiczną.
Kolby zawierające glebę poddaną działaniu badanej substancji są albo podłączone do systemu przepływowego opisanego na rys. 1, albo zamykane kolumną absorpcyjną przedstawioną na rys. 2 (zob. załącznik 3).
1.9.3 Pobieranie próbek i pomiar
Dwie kolby inkubacyjne są usuwane w odpowiednich odstępach czasu i próbki gleby są ekstrahowane odpowiednimi rozpuszczalnikami o różnej polarności, a następnie analizuje się ich zawartość pod kątem substancji badanej i/lub produktów przemian. W dobrze zaprojektowanych badaniach jest wystarczająca liczba kolb, tak aby można było poświęcić dwie kolby dla każdego poboru próbek. Także, roztwory absorpcyjne i absorbenty stałe są usuwane w różnych odstępach czasu (co 7 dni przez pierwszy miesiąc, a potem w odstępach 17 dniowych) podczas i na zakończenie inkubacji każdej gleby i analizowane pod kątem produktów lotnych. Poza tym, dla próbki gleby pobranej zaraz po podaniu (próbka z dnia 0) powinno się przygotować przynajmniej 5 dodatkowych punktów pomiarowych. Odstępy czasu powinny być tak dobrane, aby można było określić profil zaniku substancji badanej i profil tworzenia i zaniku produktów przemian (np. 0, 1, 3, 7 dni; 2, 3 tygodnie; 1, 2, 3 miesiące itd.).
Jeżeli stosowane są substancje badane znakowane izotopem 14C, niewyekstrahowane izotopy promieniotwórcze będą kwantyfikowane przy pomocy spalania, a dla każdego odstępu próbkowania będzie obliczany bilans masy.
W przypadkach inkubacji w warunkach beztlenowych i w warunkach mokrej uprawy ryżu faza glebowa i faza wodna są analizowane razem na obecność substancji badanej i produktów przemian lub rozdzielane przy pomocy filtracji lub odwirowania przed ekstrakcją i analizą.
1.9.4 Badania opcjonalne
Badania w warunkach natlenienia i braku sterylności w dodatkowym zakresie temperatur i wilgotności gleby mogą być przydatne do oszacowania wpływu temperatury i wilgotności gleby na tempo przemian substancji badanej i/lub produktów przemian w glebie.
Można podjąć próbę dalszej charakteryzacji izotopów promieniotwórczych, które nie uległy ekstrakcji, stosując na przykład ekstrakcję płynem nadkrytycznym.
2. DANE
2.1. OPRACOWANIE WYNIKÓW
Ilości substancji badanej, produktów przemian, substancji lotnych (tylko w %) i nieulegających ekstrakcji powinny zostać podane jako % zastosowanego stężenia początkowego i tam, gdzie to dotyczy, jako mg· kg-1 gleby (na podstawie suchej masy gleby) dla każdego odstępu pobierania próbek. Bilans masy należy podać w procencie zastosowanego stężenia początkowego dla każdego odstępu pobierania próbek. Graficzna prezentacja stężenia substancji badanej w zależności od czasu pozwoli na oszacowanie okresu półtrwania lub DT50. Główne produkty przemian powinny zostać zidentyfikowane, a ich stężenia także powinny zostać wykreślone w zależności od czasu, w celu przedstawienia ich tempa powstawania i zaniku. Głównym produktem przemian jest każdy produkt stanowiący >10 % podanej dawki zmierzony w którymkolwiek momencie podczas badań.
Pułapkowane produkty lotne dają pewien wskaźnik odnośnie do potencjału lotności substancji badanej i jej produktów przemian pochodzących z gleby.
Przez zastosowanie obliczeń z odpowiednim modelem kinetycznym powinno się otrzymać dokładniejsze określenie okresu półtrwania lub wartości DT50 i, w miarę potrzeby, wartości DT75 i DT90. Okres półtrwania i wartości DT50 powinny być podane razem z opisem stosowanej metody, rzędowości kinetyki i współczynnikiem determinacji (r2). Preferowana jest kinetyka pierwszego rzędu, chyba że r2 < 0,7. Jeśli jest to odpowiednie, obliczenia powinny zostać także wykonane dla głównych produktów przemian. Przykłady odpowiednich modeli są opisane w pozycjach bibliograficznych 31 i 35.
W przypadku badań tempa prowadzonych w różnych temperaturach, tempo przemian powinno zostać opisane w funkcji temperatury dla zakresu temperatur eksperymentu, przy użyciu zależności Arrheniusa w postaci:
 lub
lub 
gdzie ln A i B są stałymi regresji wyznaczonymi odpowiednio z przecięcia i nachylenia najlepszej krzywej dopasowania otrzymanej z regresji liniowej ln k od l/T, k jest stałą tempa reakcji w temperaturze T, gdzie T jest wyrażone w kelwinach. Należy uważać na ograniczony zakres temperatur, w którym spełnione jest równanie Arreheniusa w przypadku przemian zależnych od aktywności mikrobiologicznej.
2.2. OCENA I INTERPRETACJA WYNIKÓW
Choć badania są prowadzone w sztucznych warunkach laboratoryjnych, wyniki pozwolą na ocenę tempa przemian substancji badanej, a także tempa powstawania i zaniku produktów przemian w warunkach panujących w terenie (36)(37).
Badanie szlaków przemian substancji badanej dostarcza informacji o sposobie, w jaki podana substancja zostaje zmieniona pod względem strukturalnym w glebie w wyniku reakcji chemicznych i mikrobiologicznych.
3. SPRAWOZDANIE
RAPORT Z BADAŃ
Sprawozdanie z badań musi zawierać:
|
|
Substancja badana:
|
|
|
Substancje odniesienia:
|
|
|
Badane gleby:
|
|
|
Warunki badań:
|
|
|
Wyniki:
|
4. BIBLIOGRAFIA
|
(1) |
US- Environmental Protection Agency (1982). Pesticide Grade Guidelines, Subdivision N. Chemistry: Environmental Fate. |
|
(2) |
Agriculture Canada (1987). Environmental Chemistry and Fate. Guidelines for registration of pesticides in Canada. |
|
(3) |
Unia Europejska (EU) (1995). Dyrektywa Komisji 95/36/EC z dnia 14 lipca 1995 zmieniająca dyrektywę Rady 91/414/EEC dotyczącą wprowadzania do obrotu środków ochrony roślin. Załącznik II, część A i załącznik III, część A: Losy i zachowanie się w środowisku. |
|
(4) |
Dutch Commission for Registration of Pesticides (1995). Application for registration of a pesticide. Section G: Behaviour of the product and its metabolites in soil, water and air. |
|
(5) |
BBA (1986). Richtlinie fur die amtliche Priifung von Pflanzenschutzmitteln, Teil IV, 4-1. Verbleib von Pflanzenschutzmitteln im Boden – Abbau, Umwandlung und Metabolismus. |
|
(6) |
ISO/DIS 11266-1 (1994). Soil Quality -Guidance on laboratory tests for biodegradation of organic chemicals in soil – Part 1: Aerobic conditions. |
|
(7) |
ISO 14239 (1997). Soil Quality . Laboratory incubation systems for measuring the mineralization of organic chemicals in soil under aerobic conditions. |
|
(8) |
SETAC (1995). Procedures for Assessing the Environmental Fate and Ecotoxicity of Pesticides. Mark R. Lynch, Ed. |
|
(9) |
MAFF – Japan 2000 – Draft Guidelines for transformation studies of pesticides in soil – Aerobic metabolism study in soil under paddy field conditions (flooded). |
|
(10) |
OECD (1995). Final Report of the OECD Workshop on Selection of Soils/Sediments. Belgirate, Italy, 18–20 January 1995. |
|
(11) |
Guth, J.A. (1980). The study of transformations. In Interactions between Herbicides and the Soil (R.J. Hance, Ed.), Academic Press, 123–157. |
|
(12) |
DFG: Pesticide Bound Residues in Soil. Wiley . VCH (1998). |
|
(13) |
T.R. Roberts: Non-extractable pesticide residue in soils and plants. Pure Appl. Chem. 56, 945–956 (IUPAC 1984). |
|
(14) |
OECD Test Guideline 304 A: Inherent Biodegradability in Soil (adopted 12 May 1981). |
|
(15) |
ISO 10381-6 (1993). Soil Quality – Sampling – Part 6: Guidance on the collection, handling and storage of soil for the grade of aerobic microbial processes in the laboratory. |
|
(16) |
Załącznik V do dyrektywy 67/548/EEC. |
|
(17) |
Guth, J.A. (1981). Experimental approaches to studying the fate of pesticides in soil. In Progress in Pesticide Biochemistry. D.H. Hutson, T.R. Roberts, Eds. J. Wiley & Sons. Vol 1, 85–114. |
|
(18) |
Soil Texture Classification (US and FAO systems): Weed Science, 33, Suppl. 1 (1985) and Soil Sci. Soc. Amer. Proc. 26:305 (1962). |
|
(19) |
Methods of Soil Analysis (1986). Part 1, Physical and Mineralogical Methods. A. Klute, Ed.) Agronomy Series No 9, 2nd Edition. |
|
(20) |
Metodas of Soil Analysis (1982). Part 2, Chemical and Microbiological Properties. A.L. Strona, R.H. Miller and D.R. Kelney, Eds. Agronomy Series No 9, 2nd Edition. |
|
(21) |
ISO Standard Compendium Environment (1994). Soil Quality – General aspects; chemical and physical methods of analysis; biological methods of analysis. First Edition. |
|
(22) |
Muckenhausen, E. (1975). Die Bodenkunde und ihre geologischen, geomorphologischen, mineralogischen und petrologischen Grundlagen. DLG-Verlag, Frankfurt, Main. |
|
(23) |
Scheffer, F., Schachtschabel, P. (1975). Lehrbuch der Bodenkunde. F. Enke Verlag, Stuttgart. |
|
(24) |
Anderson, J.P.E., Domsch, K.H. (1978) A physiological method for the quantitative measurement of microbial biomass in soils. Soil Biol. Biochem. 10, 215–221. |
|
(25) |
ISO 14240-1 and 2 (1997). Soil Quality – Determination of soil microbial biomass – Part 1: Substrate-induced respiration method. Part 2: fumigation-extraction method. |
|
(26) |
Anderson, J.P.E. (1987). Handling and storage of soils for pesticide experiments. In Pesticide Effects on Soil Microflora. L. Somerville, M.P. Greaves, Eds. Taylor & Francis, 45–60. |
|
(27) |
Kato, Yasuhiro. (1998). Mechanism of pesticide transformation in the environment: Aerobic and biotransformation of pesticides in water solution environment. Proceedings of the 16th Symposium on Environmental Science of Pesticide, 105–120. |
|
(28) |
Keuken O., Anderson J.P.E. (1996). Influence of storage on biochemical processes in soil. In Pesticides, Soil Microbiology and Soil Quality, 59-63 (SETAC-Europe). |
|
(29) |
Stenberg B., Johansson M., Pell M., Sjodahl-Svensson K, Stenstrom J., Torstensson L. (1996). Effect of freeze and cold storage of soil on microbial activities and biomass. In Pesticides, Soil Microbiology and Soil Quality, 68-69 (SETAC-Europe). |
|
(30) |
Gennari, M., Negre, M., Ambrosoli, R. (1987). Effects of ethylene oxide on soil microbial content and some chemical characteristics. Plant and Soil 102, 197–200. |
|
(31) |
Anderson, J.P.E. (1975). Einfluss von Temperatur und Feuchte auf Verdampfung, Abbau und Festlegung von Diallat im Boden. Z. PflKrankh Pflschutz, Sonderheft VII, 141–146. |
|
(32) |
Hamaker, J.W. (1976). The application of mathematical modelling to the soil persistence and accumulation of pesticides. Proc. BCPC Symposium: Persistence of Insecticides and Herbicides, 181–199. |
|
(33) |
Goring, C.A.I., Laskowski, D.A., Hamaker, J.W., Meikle, R.W. (1975). Principles of pesticide degradation in soil. In „Environmental Dynamics of Pesticides”. R. Haque and V.H. Freed, Eds., 135–172. |
|
(34) |
Timme, G., Frehse, H., Laska, V. (1986). Statistical interpretation and graphic representation of the degradational behaviour of pesticide residues. II. Pflanzenschutz – Nachrichten Bayer 39, 188–204. |
|
(35) |
Timme, G., Frehse, H. (1980). Statistical interpretation and graphic representation of the degradational behaviour of pesticide residues. I. Pflanzenschutz – Nachrichten Bayer 33, 47–60. |
|
(36) |
Gustafson D.I., Holden L.R. (1990). Non-linear pesticide dissipation in soil; a new model based on spatial variability. Environm. Sci. Technol. 24, 1032–1041. |
|
(37) |
Hurle K, Walker A. (1980). Persistence and its prediction. In Interactions between Herbicides and the Soil (R.J. Hance, Ed.), Academic Press, 83–122 |
ZAŁĄCZNIK 1
NAPIĘCIE WODY, POJEMNOŚĆ POLOWA GLEBY (FC) I POJEMNOŚĆ WODNA (WHC) (48)
|
Wysokość słupa wody (cm) |
pF (49) |
bar (50) |
Uwagi |
|
107 |
7 |
104 |
Sucha gleba |
|
1,6*104 |
4,2 |
16 |
Początek więdnięcia |
|
104 |
4 |
10 |
|
|
103 |
3 |
1 |
|
|
6*102 |
2,8 |
0,6 |
|
|
3,3*102 |
2,5 |
0,33 (51) |
Zakres pojemności polowej gleby (52) |
|
102 |
2 |
0,1 |
|
|
60 |
1,8 |
0,06 |
|
|
33 |
1,5 |
0,033 |
|
|
10 |
1 |
0,01 |
WHC (w przybliżeniu) |
|
1 |
0 |
0,001 |
Gleba nasycona wodą |
Napięcie wody jest mierzone w cm wysokości słupa wody lub w barach. Z powodu dużego zakresu napięcia kapilarnego jest wyrażane po prostu, jako wartość pF, która jest równoważna logarytmowi cm słupa wody.
Pojemność polowa jest definiowana jako ilość wody, jaka może być przechowywana pomimo grawitacji przez naturalną ziemię przez 2 dni po dłuższym okresie opadów lub po odpowiedniej irygacji. Jest określana w nienaruszonej glebie in situ w terenie. Ten pomiar nie może więc być stosowany do naruszonych, laboratoryjnych próbek gleb. Wartości FC oznaczone w naruszonych glebach mogą wykazywać większe odchylenia systematyczne.
Pojemność wodna (WHC) jest wyznaczana w laboratorium przy użyciu nienaruszonej i naruszonej gleby przez nasycanie słupa gleby wodą w wyniku ruchów kapilarnych. Jest to szczególnie użyteczne dla gleb naruszanych i może osiągać wartość do 30 % wyższą niż największa pojemność polowa8. Jest też łatwiejsza do wyznaczenia eksperymentalnie, niż rzetelne wyznaczenie wartości FC.
Uwagi
ZAŁĄCZNIK 2
ZAWARTOŚĆ WILGOCI W GLEBIE (g wody na 100 g suchej gleby) DLA RÓŻNYCH RODZAJÓW GLEB Z RÓŻNYCH KRAJÓW
|
|
|
Rodzaj gleby w danym kraju |
||
|
Wilgotność gleby |
Kraj |
|||
|
|
|
WHC (53) |
pF = 1,8 |
pF = 2,5 |
|
Piasek |
Niemcy |
28,7 |
8,8 |
3,9 |
|
Piasek gliniasty |
Niemcy |
50,4 |
17,9 |
12,1 |
|
Piasek gliniasty |
Szwajcaria |
44,0 |
35,3 |
9,2 |
|
Glina ilasta |
Szwajcaria |
72,8 |
56,6 |
28,4 |
|
Ił gliniasty |
Brazylia |
69,7 |
38,4 |
27,3 |
|
Ił gliniasty |
Japonia |
74,4 |
57,8 |
31,4 |
|
Glina piaszczysta |
Japonia |
82,4 |
59,2 |
36,0 |
|
Glina ilasta |
USA |
47,2 |
33,2 |
18,8 |
|
Glina piaszczysta |
USA |
40,4 |
25,2 |
13,3 |
ZAŁĄCZNIK 3
Rysunek 1
Przykłady aparatury przepływowej do badań związków chemicznych w glebie (54) (55)

Rysunek 2
Przykład kolby biometrycznej do badań przemian chemicznych w glebie (56)
C.24. PRZEMIANY TLENOWE I BEZTLENOWE W UKŁADACH OSADÓW WODNYCH
1. METODA
Ta metoda badawcza jest równoważna z OECD TG 308 (2002).
1.1. WPROWADZENIE
Substancje chemiczne mogą dostawać się do płytkich lub głębokich wód powierzchniowych takimi szlakami, jak bezpośrednie wprowadzenie, znoszenie wód rozpylonych, spływ, odwadnianie, usuwanie odpadów, ścieki przemysłowe, gospodarcze lub rolnicze i opady atmosferyczne. Ta metoda badawcza opisuje metodę laboratoryjną do oceny tlenowej i beztlenowej przemiany organicznych substancji chemicznych w układach osadów wodnych. Jest oparta na istniejących wytycznych (1)(2)(3)(4)(5)(6). Podczas Warsztatów OECD na temat Selekcji Gleb/Osadów, które odbyły się w Belgirate, we Włoszech w 1995 (7), uzgodniono w szczególności liczbę i rodzaj osadów do stosowania w tym badaniu. Podano również zalecenia odnoszące się do pobierania, przenoszenia i przechowywania próbek osadów, oparte na wytycznych ISO (8). Takie badania są wymagane dla substancji chemicznych, które są bezpośrednio wprowadzane do wody lub, co do których istnieje możliwość dostania się do środowiska wodnego poprzez szlaki opisane powyżej.
Warunki w naturalnych układach osadów wodnych są często tlenowe w górnej fazie wodnej. Warstwa powierzchniowa osadu może być zarówno natleniona, jak i niedotleniona, podczas gdy głębsze osady są zazwyczaj beztlenowe. Aby uwzględnić wszystkie te możliwości, w tym dokumencie opisano zarówno badania w warunkach tlenowych, jak i beztlenowych. Badanie w warunkach tlenowych symuluje natlenioną kolumnę wodną nad warstwą osadów natlenionych ze stopniowo niedotlenionym podkładem. Badanie w warunkach beztlenowych symuluje całkowicie beztlenowy układ woda-osad. Jeśli okoliczności wskazują, że jest konieczne znaczne odejście od tych zaleceń, na przykład poprzez użycie nienaruszonych rdzeni osadowych lub osadów, które mogły zostać poddane działaniu substancji badanych, dla tych celów są dostępne inne metody (9).
1.2. DEFINICJE
W każdym przypadku należy używać standardowych międzynarodowych jednostek (SI).
Substancja badana: jakakolwiek substancja, czy to związek macierzysty czy też odnośne produkty przemian.
Produkty przemian: wszystkie substancje powstałe w wyniku biotycznych lub abiotycznych reakcji przemian substancji badanej, w tym CO2 i produkty w pozostałościach związanych.
Pozostałości związane:„Pozostałości związane” stanowią związki w glebie, roślinach lub zwierzętach, które pozostają po ekstrakcji w materiale macierzystym w postaci substancji macierzystej lub jej metabolitu(-ów)/produktów przemiany. Metoda ekstrakcji nie może zmieniać w sposób znaczący samych związków lub struktury materiału macierzystego. Charakter wiązania można częściowo wyjaśnić przez metody ekstrakcji zmieniające materiał macierzysty i wyspecjalizowane techniki analityczne. Do dziś, na przykład, zostały w ten sposób zidentyfikowane wiązania kowalencyjne zjonizowane i sorpcyjne, a także pułapkowanie. Na ogół tworzenie się pozostałości związanych znacząco redukuje biodostępność i bioprzyswajalność (10) [zmodyfikowane w stosunku do IUPAC 1984 (11)].
Przemiany tlenowe: (utlenianie): reakcje zachodzące w obecności tlenu cząsteczkowego (12).
Przemiany beztlenowe: (redukcja): reakcje zachodzące z wykluczeniem tlenu cząsteczkowego (12).
Wody naturalne: są wodami powierzchniowymi otrzymanymi ze stawów, rzek, strumieni itp.
Osad: jest mieszaniną mineralnych i organicznych składników chemicznych, te drugie zawierające związki o wysokiej zawartości węgla i azotu i o dużych masach cząsteczkowych. Jest osadzony przez wodę naturalną i tworzy z tą wodą granicę rozdziału.
Mineralizacja: to całkowita degradacja związku organicznego do CO2 i H2O w warunkach tlenowych lub CH4, CO2 i H2O w warunkach beztlenowych. W kontekście tej metody badawczej, w której stosowane są związki znakowane 14C, mineralizacja oznacza ekstensywną degradację, podczas której znaczone atomy węgla są utleniane z wydzieleniem odpowiedniej ilości 14CO2
Czas półtrwania: t0,5 to czas potrzebny do przemiany 50 % substancji badanej, jeżeli przemiana może zostać opisana kinetyką pierwszego rzędu; nie zależy od stężenia.
DT 50 (Czas zaniku 50): to czas, w którym początkowe stężenie substancji badanej zmniejsza się o 50 %.
DT 75 (Czas zaniku 75): to czas, w którym początkowe stężenie substancji badanej zmniejsza się o 75 %.
DT 90 (Czas zaniku 90): to czas, w którym początkowe stężenie substancji badanej zmniejsza się o 90 %.
1.3. SUBSTANCJE ODNIESIENIA
Substancje odniesienia powinny być stosowane do identyfikacji i oznaczenia ilościowego produktów przemian metodami spektroskopowymi i chromatograficznymi.
1.4. INFORMACJE O SUBSTANCJI BADANEJ
Nieznakowane i znakowane izotopowo substancje badane mogą być użyte do pomiaru tempa przemian, chociaż preferowany jest materiał znakowany. Materiał znakowany jest wymagany do badania szlaków przemian i do określenia bilansu masowego. Zaleca się znakowanie izotopem 14C, ale inne izotopy, takie jak 13C, 15N, 3H, 32P, także mogą być użyteczne. Tak, jak to tylko możliwe, znacznik powinien być umiejscowiony w najbardziej stabilnej części(ach) cząsteczki (57). Chemiczna lub/i radiochemiczna czystość substancji testowej powinna wynosić co najmniej 95 %.
Przed rozpoczęciem badania powinny być dostępne następujące informacje o substancji badanej:
|
a) |
rozpuszczalność w wodzie (metoda A.6), |
|
b) |
rozpuszczalność w rozpuszczalnikach organicznych, |
|
c) |
prężność pary (metoda A.4) i stała proporcjonalności w prawie Henry'ego, |
|
d) |
współczynnik podziału n-oktanol/woda (metoda A.8), |
|
e) |
współczynnik adsorpcji (odpowiednio Kd, Kf, Koc) (metoda C.18), |
|
f) |
hydroliza (metoda C.7), |
|
g) |
stała dysocjacji (pKa) [OECD Wytyczna 112] (13), |
|
h) |
struktura chemiczna substancji badanej i pozycja znacznika(ów) izotopowych jeśli dotyczy. |
Uwaga: Należy podać temperaturę, w której wykonano te pomiary.
Inne przydatne informacje mogą obejmować dane na temat toksyczności substancji badanej dla mikroorganizmów, dane na temat biodegradalności gotowej i/lub nieodłącznej, oraz dane o tlenowych i beztlenowych przemianach w glebie.
Powinny być dostępne metody analityczne (w tym metody ekstrakcji i czyszczenia) do identyfikacji i oznaczania ilościowego substancji badanej i jej produktów przemian w wodzie i w osadzie (patrz sekcja 1.7.2).
1.5 ZASADA METODY BADAWCZEJ
W opisanej metodzie dla tego badania stosuje się tlenowy i beztlenowy układ osadu wodnego (zobacz załącznik 1), który umożliwia:
|
(i) |
pomiar tempa przemiany substancji badanej w układzie woda-osad; |
|
(ii) |
pomiar tempa przemiany substancji badanej w osadzie; |
|
(iii) |
pomiar tempa mineralizacji substancji badanej i/lub jej produktów przemiany (gdy stosowana jest substancja badana znakowana izotopem 14C); |
|
(iv) |
identyfikacja i oznaczenie ilościowe produktów przemiany w fazach wodnej i w osadu, w tym bilans masowy (gdy stosowana jest znakowana substancja badana); |
|
(v) |
pomiar rozdziału substancji badanej i jej produktów przemiany między dwie fazy podczas okresu inkubacji w ciemności (by uniknąć, na przykład, zakwitu glonów) w stałej temperaturze. Czasy półtrwania, wartości DT50, DT75 i DT90 są określone, tam gdzie potwierdzają to dane, ale nie powinny być ekstrapolowane daleko poza okres eksperymentalny (zob. sekcja 1.2). |
Do badań odpowiednio w warunkach tlenowych, jak i beztlenowych wymagane są conajmniej dwa osady wraz z towarzyszącymi im wodami (7). Jednakże mogą wystąpić przypadki, gdzie powinny być użyte więcej niż dwa osady wodne, na przykład, dla substancji chemicznych, które mogą być obecne w środowiskach słodkowodnych i/lub morskich.
1.6. ZASTOSOWANIE BADANIA
Metoda ogólnie może być stosowana dla substancji chemicznych (znakowanych i nieznakowanych izotopowo), dla których jest dostępna metoda analityczna o wystarczającej czułości i dokładności. Może być stosowana do związków słabo lotnych, nielotnych, rozpuszczalnych lub nierozpuszczalnych w wodzie. Badanie nie powinno być stosowane do substancji chemicznych, o wysokiej lotności z wody (np.: fumiganty, rozpuszczalniki organiczne), które nie mogą być zatrzymane w wodzie i/lub osadzie w warunkach eksperymentalnych badania.
Metoda ta została, jak dotąd, zastosowana do badania przemian substancji chemicznych w wodzie słodkiej i osadach, ale w zasadzie może być także zastosowana do układów estuaryjno-morskich. Nie nadaje się do symulacji warunków wód płynących (np. rzek) lub otwartego morza.
1.7. KRYTERIA JAKOŚCIOWE
1.7.1. Odzysk
Ekstrakcja i analiza, co najmniej podwójnych próbek wód i osadów natychmiast po dodaniu substancji badanej daje pierwsze wskazanie co do powtarzalności metody analitycznej i jednorodności procedury podawania substancji badanej. Odzysk dla następnych etapów eksperymentu jest podawany przez odnoszący się do nich bilans masowy (gdy stosowane są materiały znakowane). Odzysk powinien mieścić się w zakresie od 90 % do 110 % dla znakowanych substancji chemicznych (6) i od 70–110 % dla nieznakowanych substancji chemicznych.
1.7.2. Powtarzalność i czułość metody analitycznej
Powtarzalność metody analitycznej (z wyłączeniem początkowej wydajności ekstrakcji) dla określenia ilości substancji badanej i produktów przemian może być sprawdzona przez powtórzenie analizy tego samego ekstraktu próbek wody lub osadu, które były inkubowane wystarczająco długo, aby wytworzyły się produkty przemian.
Granica wykrywalności (LOD) metody analitycznej dla substancji badanej i dla produktów przemian powinna być na poziomie przynajmniej 0,01 mg· kg-1 wody lub osadu (jako substancji badanej) lub 1 % początkowej ilości podanej do układu badanego, w zależności od tego, co jest niższe. Granica kwantyfikacji (LOQ) również powinna zostać określona.
1.7.3. Dokładność danych przemiany
Analiza regresji stężeń substancji badanej w funkcji czasu daje właściwe informacje o dokładności krzywej przemian i pozwala na obliczenie granic przedziału ufności dla okresu półtrwania (przy założeniu kinetyki pseudo pierwszego rzędu) lub wartości DT50, oraz w razie potrzeby, wartości DT75 i DT90.
1.8. OPIS METODY
1.8.1. Badany układ i aparatura
Badanie powinno być przeprowadzone w pojemnikach szklanych (np. butlach, probówkach wirówkowych), chyba że wstępne informacje (jak współczynnik podziału n-oktanol-woda, dane sorpcyjne itp.) pokazują, że badana substancja może przylegać do szkła, w takim przypadku można rozważyć materiał alternatywny (taki jak teflon). Tam gdzie wiadomo, że substancja badana przywiera do szkła, można złagodzić ten problem przez użycie jednej lub więcej następujących metod:
|
— |
określenie masy substancji badanej i produktów przemian sorbowanych do szkła, |
|
— |
zapewnić mycie rozpuszczalnikiem całego sprzętu szklanego na końcu badania, |
|
— |
używać produktów gotowych (zobacz też sekcja 1.9.2), |
|
— |
używać zwiększonej ilości współrozpuszczalnika do dodawania substancji badanej do układu; jeśli jest użyty współrozpuszczalnik, powinien to być współrozpuszczalnik, który nie solwatuje substancji badanej. |
Przykłady typowej aparatury badawczej, np.: układy przepływowe i biometryczne są pokazane odpowiednio w załącznikach 2 i 3 (14). Inne użyteczne układy inkubacyjne są opisane w pozycji bibliograficznej 15. Konstrukcja aparatury badawczej powinna umożliwiać wymianę powietrza lub azotu i zatrzymywanie produktów lotnych. Wymiary aparatury muszą być takie, aby spełniały wymagania badania (patrz sekcja 1.9.1). Wentylację można zapewnić albo przez łagodne barbotowanie, albo przez przedmuchiwanie powietrzem lub azotem powierzchni wody. W tym drugim przypadku, wskazane łagodne mieszanie wody od góry może być wskazane dla lepszego rozprowadzenia tlenu lub azotu w wodzie. Nie powinno się używać powietrza pozbawionego CO2, ponieważ może to powodować wzrost pH wody. W każdym razie zaburzenie osadu jest niepożądane i powinno się go w miarę możliwości unikać. Lekko lotne substancje chemiczne powinny być badane w układzie biometrycznym, z łagodnym mieszaniem powierzchni wody. Można również użyć zamkniętych naczyń z fazą gazową powietrza atmosferycznego lub azotu nad roztworem, z wewnętrznym pojemniczkiem do wyłapywania produktów lotnych (16). Regularna wymiana gazów znad powierzchni roztworu jest wymagana w badaniach tlenowych, żeby skompensować zużycie tlenu przez biomasę.
Odpowiednie pułapki do wyłapywania lotnych produktów przemian obejmują, między innymi, 1 mol· dm-3 roztworu wodorotlenku potasu lub wodorotlenku sodu dla ditlenku węgla (58) i glikol etylenowy, etanolaminę lub 2 % roztwór parafiny w ksylenie dla związków organicznych. Części lotne powstające w warunkach beztlenowych, takie jak metan, mogą być wyłapywane przez, na przykład, sita molekularne. Takie części lotne mogą być spalone, na przykład do CO2 poprzez przepuszczanie gazu przez rurę kwarcową wypełnioną CuO w temperaturze 900 oC i wychwytywanie powstałego CO2 w aparacie absorpcyjnym z alkaliami (17).
Wymagane jest oprzyrządowanie laboratoryjne do analizy chemicznej substancji badanej i produktów przemian (np.: chromatografia gazowo-cieczowa (GLC), wysokociśnieniowa chromatografia cieczowa (HPLC), chromatografia cienkowarstwowa (TLC), spektroskopia masowa (MS), chromatografia gazowa sprzężona ze spektrometrią masową (GC-MS), chromatografia cieczowa sprzężona ze spektrometrią masową (LC-MS), magnetyczny rezonans jądrowy (NMR) itp.), włączając w to układy do wykrywania substancji chemicznych znakowanych i nieznakowanych radioizotopami. Jeśli jest stosowany materiał znakowany radioizotopem, będzie również potrzebny cieczowy licznik scyntylacyjny i aparat do utleniania przez spalanie (do spalania próbek osadów przed analizy promieniotwórczości).
W miarę potrzeby wymagane jest inne standardowe wyposażenie laboratoryjne do oznaczeń fizykochemicznych i biologicznych (zob. sekcja tabela 1, sekcja 1.8.2.2), szkło laboratoryjne, substancje chemiczne i odczynniki.
1.8.2. Wybór i liczba osadów wodnych.
Miejsca pobierania próbek powinny być wybierane stosownie do celu badania w dowolnej sytuacji. Przy wyborze miejsc pobierania próbek należy uwzględnić historię ewentualnych rolniczych, przemysłowych i gospodarczych wpływów na zlewnię i wody w górze rzeki. Osady nie powinny być użyte, jeśli zostały zanieczyszczone substancją badaną lub jej analogami strukturalnymi w ciągu ostatnich czterech lat.
1.8.2.1. Wybór osadów
Zazwyczaj używa się dwóch osadów do badań w warunkach tlenowych (7). Dwa wybrane osady powinny się różnić pod względem zawartości węgla organicznego i budowy. Jeden osad powinien mieć wysoką zawartość węgla organicznego (2,5–7,5 %) i budowę drobnoziarnistą, drugi osad powinien mieć niską zawartość węgla organicznego (0,5–2,5 %) i budowę gruboziarnistą. Różnica zawartości węgla organicznego powinna zazwyczaj wynosić co najmniej 2 %. „Budowa drobnoziarnista” jest zdefiniowana jako suma [glina +ił] (59) o zawartości > 50 % a „budowa gruboziarnista” jest zdefiniowana jako suma [glina + ił] o zawartości < 50 %. Różnica zawartości [glina + ił] dla tych dwóch osadów powinna zwykle wynosić co najmniej 20 %. W przypadku gdy substancja chemiczna może także dostać się do wody morskiej, przynajmniej jeden z układów woda-osad powinien być pochodzenia morskiego.
Dla badań w warunkach ściśle beztlenowych, dwa osady (wraz z towarzyszącymi wodami) powinny być pobrane ze stref beztlenowych powierzchniowych części wód (7). Obie fazy osadowa i wodna powinny być przenoszone i transportowane ostrożnie bez dostępu tlenu.
Przy wyborze osadów mogą być ważne inne parametry i powinny być rozpatrywane dla każdego przypadku indywidualnie. Na przykład zakres pH osadów byłby ważny dla badania substancji chemicznych, których przemiana i/lub sorpcja mogą być zależne od pH. Zależność sorpcji od pH może być odzwierciedlone w wartości pKa substancji badanej.
1.8.2.2. Charakterystyka próbek woda-osad
Kluczowe parametry, które muszą być zmierzone i podane w sprawozdaniu (w odniesieniu do użytej metody) zarówno dla wód, jak i osadów, oraz etap badania, na którym te parametry powinny zostać określone, są podsumowane w tabeli poniżej. Dla informacji, metody określania tych parametrów są podane w bibliografii (18)(19)(20)(21).
Dodatkowo może zachodzić potrzeba pomiaru i sprawozdania innych parametrów stosownie do indywidualnego przypadku (np.: dla wody słodkiej: cząstki zawieszone, zasadowość, twardość, przewodnictwo, NO3/PO4 (stosunek i wartości bezwzględne); dla osadów: zdolność wymiany kationowej, pojemność wodna, węglany, całkowity azot i fosfor; a dla układów morskich: zasolenie). Analiza osadów i wody na azotany, siarczany, żelazo bioprzyswajalne i ewentualnie inne akceptory elektronowe, również może być przydatna do oceny warunków redoks, szczególnie w odniesieniu do przemian beztlenowych.
Pomiar parametrów do charakteryzacji próbek woda-osad (7)(22)(23)
|
Parametr |
Etap procedury |
|||||
|
pobieranie próbek w terenie |
późniejsze przenoszenie |
początek przygotowania |
początek badania |
podczas badania |
koniec badania |
|
|
Woda |
||||||
|
Pochodzenie/źródło |
x |
|
|
|
|
|
|
Temperatura |
x |
|
|
|
|
|
|
pH |
x |
|
x |
x |
x |
x |
|
TOC |
|
|
x |
x |
|
x |
|
Stężenie O2 (60) |
x |
|
x |
x |
x |
x |
|
Potencjał redoks (60) |
|
|
x |
x |
x |
x |
|
Osad |
||||||
|
Pochodzenie/źródło |
x |
|
|
|
|
|
|
Głębokość warstwy |
x |
|
|
|
|
|
|
pH |
|
x |
x |
x |
x |
x |
|
Rozkład wielkości cząstek |
|
x |
|
|
|
|
|
TOC |
|
x |
x |
x |
|
x |
|
Biomasa mikrobiologiczn a (61) |
|
x |
|
x |
|
x |
|
Potencjał redoks (60) |
Obserwacja (kolor/zapach) |
|
x |
x |
x |
x |
1.8.3. Pobieranie, przenoszenie i przechowywanie
1.8.3.1. Pobieranie
Przy pobieraniu próbek osadu należy korzystać z projektu wytycznych ISO o pobieraniu próbek osadu dennego (8). Próbki osadu powinny być pobierane z całej górnej warstwy osadu, o grubości od 5 do 10 cm. Towarzysząca mu woda powinna być pobrana z tego samego miejsca lub lokalizacji i w tym samym czasie, co osad. Dla badań w warunkach beztlenowych osad i towarzysząca woda powinny być pobrane i transportowane bez dostępu tlenu (28) (zob. sekcja 1.8.2.1). Niektóre urządzenia do pobierania próbek są opisane w literaturze (8)(23).
1.8.3.2. Przenoszenie
Osad jest oddzielany od wody przez filtrację i przesiewany na mokro przez 2 mm sito przy użyciu nadmiaru miejscowej wody, która jest potem wylewana. Następnie, znane ilości osadów i wody są mieszane z pożądaną prędkością (zob. sekcja 1.9.1) w kolbach inkubacyjnych przez okres przystosowawczy (zob. sekcja 1.8.4). Dla badań w warunkach beztlenowych wszystkie kroki na etapie przenoszenia muszą być wykonane bez dostępu tlenu (29)(30)(31)(32)(33).
1.8.3.3. Przechowywanie
Bardzo zaleca się użycie świeżo pobranego osadu, jeśli jednak konieczne jest jego przechowywanie, osad i woda powinny być przesiane, jak opisano powyżej i przechowywane razem, zalane wodą (6–10 cm warstwy wody), w ciemności, w temperaturze 4 ± 2 oC4 przez maksymalnie 4 tygodnie (7)(8)(23). Próbki przeznaczone do badań w warunkach tlenowych powinny być przechowywane przy swobodnym dostępie powietrza (np.: w otwartych pojemnikach), natomiast próbki do badań beztlenowych, bez dostępu tlenu. Podczas transportu i magazynowania nie może zajść zamrożenie osadu i wody i wysuszenie osadu.
1.8.4. Przygotowanie próbek osad/woda do badania
Okres przystosowawczy powinien nastąpić przed dodaniem substancji badanej, gdzie każda próbka osad/woda jest umieszczana w naczyniu inkubacyjnym, do użycia w badaniu głównym, i przystosowanie powinno być prowadzone dokładnie w tych samych warunkach, co inkubacja (zob. sekcja 1.9.1). Okres przystosowawczy jest to czas potrzebny do osiągnięcia przez układ umiarkowanej stabilności, co odzwierciedla pH, stężenie tlenu w wodzie, potencjał redoks osadu i wody, oraz makroskopowa separacja faz. Okres przystosowawczy powinien zazwyczaj trwać od jednego do dwóch tygodni i nie powinien przekroczyć czterech tygodni. Wyniki oznaczeń wykonanych w tym okresie powinny zostać podane w sprawozdaniu.
1.9. PROWADZENIE BADAŃ
1.9.1. Warunki badania
Badanie powinno być wykonane w aparaturze inkubacyjnej (patrz sekcja 1.8.1) przy stosunku wody do osadu na poziomie między 3:1 a 4:1, oraz grubością warstwy osadu 2,5 cm (±0,5 cm) (62). Zaleca się minimalną ilość 50 g osadu (w suchej masie) na naczynie inkubacyjne.
Badanie powinno być wykonane w ciemności, w stałej temperaturze z przedziału 10–30 oC. Właściwa jest temperatura (20 ± 2) oC. W razie potrzeby, można rozważyć, stosownie do indywidualnego przypadku, dodatkową niższą temperaturę (np. 10 oC), zależnie od informacji wymaganych od badania. Temperatura inkubacji powinna być monitorowana i podana w sprawozdaniu.
1.9.2. Obróbka i podanie substancji badanej
Używane jest jedno badane stężenie substancji chemicznej (63). Dla środków ochrony upraw wprowadzanych bezpośrednio do części wód, należy przyjąć maksymalną dawkę na etykiecie, jako maksymalny stopień podania obliczony na podstawie pola powierzchni wody w naczyniu do badania. W innych przypadkach, użyte stężenie powinno być oparte na przewidywaniach emisji środowiskowych. Należy uważać, aby zapewnić, że zastosowane będzie odpowiednie stężenie substancji badanej, żeby scharakteryzować szlak przemian oraz powstawanie i zanik produktów przemian. Może być konieczne zastosowanie większych dawek (np.: 10 razy) w sytuacjach, w których stężenia substancji badanej są bliskie granicy wykrywalności na początku badania i/lub w których główne produkty przemian nie mogą być łatwo wykryte, jeżeli są obecne w ilości 10 % stopnia podania substancji badanej. Jednakże jeśli używa się wyższych stężeń testowych, nie powinny one mieć znaczącego niekorzystnego wpływu na aktywność mikrobiologiczną układu woda-osad. Żeby osiągnąć stałe stężenie substancji badanej w naczyniach o różnych wymiarach, można uznać za stosowne korektę ilości podanego materiału, na podstawie wysokości słupa wody w naczyniu w stosunku do głębokości wody w terenie (którą przyjmuje się jako 100 cm, ale mogą być użyte inne głębokości). Zob. załącznik 4 w celu uzyskania przykładowych obliczeń.
W idealnym przypadku substancja badana powinna być podana jako roztwór wodny do fazy wodnej układu badanego. Jeśli nie da się tego uniknąć, użycie małych ilości mieszających się z wodą rozpuszczalników (takich jak aceton, etanol) jest dopuszczalne dla podania i rozprowadzenia substancji badanej, ale nie powinno przekroczyć 1 % v/v i nie powinno mieć niekorzystnego wpływu na aktywność mikrobiologiczną układu badanego. Należy zachować ostrożność przy wytwarzaniu roztworu wodnego substancji badanej – może być właściwe użycie kolumn do wytwarzania i wstępnego mieszania, aby zapewnić kompletną homogeniczność. Po dodaniu roztworu wodnego do układu badanego zaleca się delikatne mieszanie fazy wodnej, aby możliwie najmniej zaburzyć osad.
Nie zaleca się rutynowego używania gotowych produktów, jako że składniki preparatu mogą wywierać wpływ na rozdział substancji badanej i/lub produktów przemian pomiędzy fazę wodną a osad. Jednakże dla substancji badanych o niskiej rozpuszczalności w wodzie zastosowanie produktów gotowych może być właściwym rozwiązaniem.
Liczba naczyń inkubacyjnych zależy od tego, ile razy są pobierane próbki (zob. sekcja 1.9.3). Należy uwzględnić wystarczającą liczbę układów badanych, tak żeby przy każdorazowym pobraniu próbek można było poświęcić dwa układy. Jeżeli stosuje się układy kontrolne wodnego układu osadowego, nie powinny być ona poddawane działaniu substancji badanej. Układy kontrolne mogą być użyte do oznaczenia biomasy mikrobiologicznej osadu i całkowitego węgla organicznego w wodzie i osadzie na końcu badania. Dwa z układów kontrolnych (np. jeden układ kontrolny każdego osadu wodnego) mogą być stosowane do monitorowania wymaganych parametrów w osadzie i w wodzie podczas okresu przystosowawczego (zob. tabela w sekcji 1.8.2.2). Dwa dodatkowe układy kontrolne muszą być uwzględnione w przypadku, gdy substancja badana jest podana przy pomocy rozpuszczalnika, aby zmierzyæ niekorzystne skutki dla aktywności mikrobiologicznej układu badanego.
1.9.3. Czas badania i pobieranie próbek
Eksperyment badawczy nie powinien zazwyczaj trwać dłużej niż 100 dni (6) i powinien być kontynuowany tak długo, aż zostanie określony szlak degradacji i model rozkładu woda/osad lub kiedy 90 % substancji badanej zaniknie poprzez przemiany i/lub parowanie. Liczba pobrań próbek powinna wynosić co najmniej sześć (włączając czas zerowy), z opcjonalnym wstępnym badaniem (zob. sekcja 1.9.4) stosowanym do ustalenia właściwego reżimu pobierania próbek i czasu trwania badania, chyba że dostępne są wystarczające dane o substancji badanej z poprzednich badań. Dla hydrofobowych substancji badanych mogą być konieczne dodatkowe punkty pobierania próbek podczas początkowego okresu badania, w celu określenia stopnia rozdziału między fazę wodną a osadową.
We właściwych momentach pobierania próbek całe naczynia inkubacyjne (w powtórzeniach) usuwa się do analizy. Osad i woda znad niego są analizowane osobno (64). Należy ostrożnie usunąć wodę powierzchniową, żeby jak najmniej zaburzyć osad. Ekstrakcja i scharakteryzowanie substancji badanej i produktów przemian powinno nastąpić po właściwych procedurach analitycznych. Należy uważać przy usuwaniu materiału, który mógł zostać zaadsorbowany do naczynia inkubacyjnego, albo do łączących rur stosowanych do wyłapania części lotnych.
1.9.4. Opcjonalne wstępne badanie
Jeśli czas trwania i reżim pobierania próbek nie mogą być oszacowane na podstawie innych odnośnych badań substancji badanej, można uznać za stosowne opcjonalne wstępne badanie, które powinno być przeprowadzone przy użyciu tych samych warunków badania, jakie zostały zaproponowane do badania ostatecznego. Odnośne warunki doświadczalne i wyniki z badania wstępnego, jeśli je przeprowadzono, powinny być pokrótce przedstawione w sprawozdaniu.
1.9.5. Pomiary i analizy
Przy każdym pobieraniu próbek stężenie substancji badanej i produktów przemiany w wodzie i osadzie powinno być zmierzone i podane w sprawozdaniu (jako stężenie i jako procent całości dostarczonej). Generalnie, produkty przemian wykryte przy ≥ 10 % zastosowanej radioaktywności w całym układzie woda-osad przy każdorazowym pobieraniu próbek powinny zostać zidentyfikowane, chyba że można przedstawić rozsądne uzasadnienie braku potrzeby identyfikacji. Produkty przemian, których stężenie ciągle wzrasta podczas badania, również powinny być uwzględnione do identyfikacji nawet, jeśli ich stężenia nie przekraczają granic podanych wyżej, jako że może to wskazywać na trwałość. To drugie powinno być rozpatrywane indywidualnie, stosownie do przypadku, z uzasadnieniem przedstawionym w sprawozdaniu.
Wyniki z układu wychwytującego gazy/części lotne (CO2 i inne, np.: lotne związki organiczne) powinny być podawane przy każdym pobieraniu próbek. Należy podać w sprawozdaniu tempo mineralizacji. Pozostałości w osadzie nieulegające ekstrakcji (związane) powinny być podawane przy każdym pobieraniu próbek.
2. DANE
2.1. OBRÓBKA WYNIKÓW
Całkowity bilans masy lub odzysk (zob. sekcja 1.7.1) dodanych radioizotopów ma być obliczany przy każdorazowym pobieraniu próbek. Wyniki powinny być podawane jako procent dodanych radioizotopów. Rozdział izotopów między wodę a osad należy podać jako stężenia i procenty, przy każdym pobieraniu próbek.
Czas półtrwania, DT50, i w razie potrzeby, DT75 i DT90 substancji badanej powinny być obliczone wraz z ich granicami przedziału ufności (zob. sekcja 1.7.3). Informacje o szybkości rozpraszania substancji badanej w wodzie i osadzie można otrzymać poprzez użycie odpowiednich narzędzi oceny. Mogą to być zastosowania kinetyki pseudo pierwszego rzędu, empiryczne techniki dopasowania krzywych, które stosują rozwiązania graficzne lub numeryczne i wiele bardziej złożonych ocen z wykorzystaniem, na przykład, modeli jedno- lub wieloprzedziałowych. Dalsze szczegółowe informacje można otrzymać z odnośnych publikacji (35)(36)(37).
Wszystkie podejścia mają swoje słabe i mocne strony i znacznie się różnią w swojej złożoności. Założenie kinetyki pierwszego rzędu może być zbytnim uproszczeniem procesów degradacji i rozdziału, ale tam, gdzie jest to możliwe daje termin (stała tempa lub czas półtrwania), który jest łatwo zrozumiały i o wartości w modelowaniu symulacyjnym i obliczeniach przewidywanych stężeń środowiskowych. Podejścia empiryczne lub przemiany liniowe mogą dawać lepsze dopasowanie krzywych do danych i dlatego pozwalają na lepsze oszacowanie czasów półtrwania, DT50 i, w razie potrzeby, wartości DT75 i DT90. Użycie wyprowadzonych stałych jest jednak ograniczone. Modele przedziałowe mogą wytwarzać wiele użytecznych stałych o wartości w ocenie ryzyka, które opisują tempo degradacji w różnych przedziałach i rozdział substancji chemicznej. Powinny być one również użyte do oszacowania stałych tempa dla powstawania i degradacji głównych produktów przemian. We wszystkich przypadkach wybrana metoda musi być uzasadniona i osoba badająca powinna przedstawić graficznie i/lub statystycznie zgodność wyników doświadczenia z przewidywaniami.
3. SPRAWOZDANIE
3.1 SPRAWOZDANIE Z BADANIA
Sprawozdanie musi zawierać następujące informacje:
|
|
Substancja badana:
|
|
|
Substancje odniesienia:
|
|
|
Badany osad i wody:
|
|
|
Warunki badania:
|
|
|
Wyniki:
|
4. BIBLIOGRAFIA
|
(1) |
BBA-Guidelines for the examination of plant protectors in the registration process. (1990). Part IV, Section 5-1: Degradability and fate of plant protectors in the water/sediment system. Germany. |
|
(2) |
Commission for registration of pesticides: Application for registration of a pesticide. (1991). Part G. Behaviour of the product and its metabolites in soil, water and air, Section G.2.1 (a). The Netherlands. |
|
(3) |
MAFF Pesticides Safety Directorate. (1992). Preliminary guideline for the conduct of biodegradability tests on pesticides in natural sediment/water systems. Ref No SC 9046. United-Kingdom. |
|
(4) |
Agriculture Canada: Environmental chemistry and fate. (1987). Guidelines for registration of pesticides in Canada. Aquatic (Laboratory) – Anaerobic and aerobic. Canada. pp 35–37. |
|
(5) |
US-EPA: Pesticide assessment guidelines, Subdivision N. Chemistry: Environmental fate (1982). Section 162-3, Anaerobic aquatic metabolism. |
|
(6) |
SETAC-Europe publication. (1995). Procedures for assessing the environmental fate and ecotoxicity of pesticides. Ed. Dr Mark R. Lynch. SETAC-Europe, Brussels. |
|
(7) |
OECD Test Guidelines Programme. (1995). Final Report of the OECD Workshop on Selection of Soils/sediments, Belgirate, Italy, 18–20 January 1995. |
|
(8) |
ISO/DIS 5667-12. (1994). Water quality – Sampling – Part 12: Guidance on sampling of bottom sediments. |
|
(9) |
US-EPA (1998a). Sediment/water microcosm biodegradation test. Harmonised Test Guidelines (OPPTS 835.3180). EPA 712-C-98-080. |
|
(10) |
DFG: Pesticide Bound Residues in Soil. Wiley-VCH (1998). |
|
(11) |
T.R. Roberts: Non-extractable pesticide residues in soils and plants. Pure Appl. Chem. 56, 945-956 (IUPAC 1984). |
|
(12) |
OECD Test Guideline 304A: Inherent Biodegradability in Soil (adopted 12 May 1981). |
|
(13) |
OECD (1993): Guidelines for Testing of Chemicals. Paris. OECD (1994-2000): Addenda 6-11 to Guidelines for the Testing of Chemicals. |
|
(14) |
Scholz, K., Fritz R., Anderson C. and Spiteller M. (1988) Degradation of pesticides in an aquatic model ecosystem. BCPC – Pests and Diseases, 3B-4, 149–158. |
|
(15) |
Guth, J.A. (1981). Experimental approaches to studying the fate of pesticides in soil. In Progress in Pesticide Biochemistry (D.H. Hutson, T.R. Roberts, Eds.), Vol. 1, 85–114. J. Wiley & Sons. |
|
(16) |
Madsen, T., Kristensen, P. (1997). Effects of bacterial inoculation and non-ionic surfactants on degradation of polycyclic aromatic hydrocarbons in soil. Environ. Toxicol. Chem. 16, 631–637. |
|
(17) |
Steber, J., Wierich, P. (1987). The anaerobic degradation of detergent range fatty alcohol ethoxylates. Studies with 14C-labelled model surfactants. Water Research 21, 661–667. |
|
(18) |
Black, C.A. (1965). Metodas of Soil Analysis. Agronomy Monograph No. 9. American Society of Agronomy, Madison. |
|
(19) |
APHA (1989). Standard Metodas for Examination of Water and Wastewater (17th edition). American Public Health Association, American Water Works Association and Water Pollution Control Federation, Washington D.C. |
|
(20) |
Rowell, D.L. (1994). Soil Science Metodas and Applications. Longman. |
|
(21) |
Light, T.S. (1972). Standard solution for redox potential measurements. Anal. Chemistry 44, 1038–1039. |
|
(22) |
SETAC-Europe publication (1991). Guidance document on testing procedures for pesticides in freshwater mesocosms. From the Workshop „A Meeting of Experts on Guidelines for Static Field Mesocosms Tests”, 3–4 July 1991. |
|
(23) |
SETAC-Europe publication. (1993). Guidance document on sediment toxicity tests and bioassays for freshwater and marine environments. From the Workshop On Sediment Toxicity Assessment (WOSTA), 8-10 November 1993. Eds.: I.R. Hill, P. Matthiessen and F. Heimbach. |
|
(24) |
Vink, J.P.M., van der Zee, S.E.A.T.M. (1997). Pesticide biotransformation in surface waters: multivariate analyses of environmental factors at field sites. Water Research 31, 2858–2868. |
|
(25) |
Vink, J.P.M., Schraa, G., van der Zee, S.E.A.T.M. (1999). Nutrient effects on microbial transformation of pesticides in nitrifying waters. Environ. Toxicol, 329–338. |
|
(26) |
Anderson, T.H., Domsch, K.H. (1985). Maintenance carbon requirements of actively-metabolising microbial populations under in-situ conditions. Soil Biol. Biochem. 17, 197–203. |
|
(27) |
ISO-14240-2. (1997). Soil quality – Determination of soil microbial biomass – Part 2: Fumigation-extraction method. |
|
(28) |
Beelen, P. Van and F. Van Keulen. (1990), The Kinetics of the Degradation of Chloroform and Benzene in Anaerobic Sediment from the River Rhine. Hydrobiol. Bull. 24 (1), 13–21. |
|
(29) |
Shelton, D.R. and Tiedje, J.M. (1984). General metoda for determining anaerobic biodegradation potential. App. Environ. Microbiol. 47, 850–857. |
|
(30) |
Birch, R.R., Biver, C., Campagna, R., Gledhill, W.E., Pagga, U., Steber, J., Reust, H. and Bontinck, W.J. (1989). Screening of chemicals for anaerobic biodegradation. Chemosphere 19, 1527–1550. |
|
(31) |
Pagga, U. and Beimborn, D.B. (1993). Anaerobic biodegradation tests for organic compounds. Chemoshpere 27, 1499–1509. |
|
(32) |
Nuck, B.A. and Federle, T.W. (1986). A partia test for assessing the mineralisation of 14C-radiolabelled compounds under realistic anaerobic conditions. Environ. Sci. Technol. 30, 3597–3603. |
|
(33) |
US-EPA (1998b). Anaerobic biodegradability of organic chemicals. Harmonised Test Guidelines (OPPTS 835.3400). EPA 712-C-98-090. |
|
(34) |
Sijm, Haller and Schrap (1997). Influence of storage on sediment characteristics and drying sediment on sorption coefficients of organic contaminants. Bulletin Environ. Contam. Toxicol. 58, 961–968. |
|
(35) |
Timme, G., Frehse H. and Laska V. (1986) Statistical interpretation and graphic representation of the degradational behaviour of pesticide residues II. Pflanzenschutz – Nachrichten Bayer, 39, 187–203. |
|
(36) |
Timme, G., Frehse, H. (1980) Statistical interpretation and graphic representation of the degradational behaviour of pesticide residues I. Pflanzenschutz – Nachrichten Bayer, 33, 47–60. |
|
(37) |
Carlton, R.R. and Allen, R. (1994). The use of a compartment model for evaluating the fate of pesticides in sediment/water systems. Brighton Crop Protection Conference – Pest and Diseases, pp. 1349–1354. |
ZAŁĄCZNIK 1
WYTYCZNE DLA UKLADÓW BADAWCZYCH W WARUNKACH TLENOWYCH I BEZTLENOWYCH
Układ badań w warunkach tlenowych
Układ badań w warunkach tlenowych opisany w tej metodzie badawczej składa się z natlenionej warstwy wody (typowe stężenie tlenu w zakresie 7 do 10 mg· l-1) i z warstwy osadu, natlenionej na powierzchni i beztlenowej poniżej powierzchni (typowy średni potencjał redoks (Eh) w strefie beztlenowej osadu w zakresie od - 80 do - 190 mV). Nawilżone powietrze przechodzi nad powierzchnią wody w każdym urządzeniu do inkubacji, aby utrzymać wystarczającą ilość tlenu w przestrzeni nad roztworem.
Układ badań w warunkach beztlenowych
Dla układu badań w warunkach beztlenowych procedura badawcza jest zasadniczo taka sama, jak przedstawiono dla układów tlenowych z tym, że nawilżony azot przepływa nad powierzchnią wody w każdym urządzeniu inkubacyjnym, aby utrzymać atmosferę azotową w przestrzeni nad roztworem. Osad i woda są uważane za beztlenowe, jeśli potencjał redoks (Eh) jest niższy niż - 100 mV.
W badaniu w warunkach beztlenowych ocena mineralizacji obejmuje pomiar wydzielonego ditlenku węgla i metanu.
ZAŁĄCZNIK 2
PRZYKŁAD APARATURY PRZEPŁYWOWO-GAZOWEJ

ZAŁĄCZNIK 3
PRZYKŁADY APARATURY BIOMETRYCZNEJ

ZAŁĄCZNIK 4
PRZYKŁADOWE OBLICZENIA DLA DAWKI DOSTARCZANEJ DO NACZYŃ BADAWCZYCH
|
Wewnętrzna średnica cylindra: |
= 8 cm |
|
Wysokość słupa wody bez osadu: |
=12 cm |
|
Pole powierzchni: 3,142 × 42 |
= 50,3 cm2 |
|
Stopień dostarczania: 500 g substancji badanej/ha odpowiada 5 μg/cm2 |
|
|
Dawka całkowita μg: 5 × 50,3 |
= 251,5 μg |
|
Korekta ilości w odniesieniu do głębokości 100 cm: 12 × 251,5 ÷ 100 |
= 30,18 μg |
|
Pojemność słupa wody: 50,3 × 12 |
= 603 ml |
|
Stężenie w wodzie: 30,18 ÷ 603 |
= 0,050 μg/ml lub 50 μg/l |
(1) Wyniki tych wewnętrznych badań laboratoryjnych i technicznego sprostowania w sprawie ISO 6341 dają EC50 po 24 h dichromianu potasu (K2Cr2O7) w przedziale 0,6–1,7 mg/l.
(2) Woda o odpowiedniej czystości, np. dejonizowana, destylowana lub poddana osmozie odwróconej, najlepiej mająca przewodność nieprzekraczającą 10 μS.cm-1.
(3) D1 i D2 nie należy uśredniać jeśli znacznie się różnią.
(4) D1 i D2 nie mogą być uśrednian, jeżeli znacznie się różnią.
(5) lub w końcu inkubacju
(6) D1 i D2 nie należy uśredniać jeśli się znacząco różnią.
(7) lub w końcu inkubacji
(8) Nie brać średniej jeżeli jest znacząca różnica pomiędzy replikami.
(9) Nie uśredniać przy znacznych różnicach pomiędzy replikami.
(10) Po użyciu roztwory zawierające sole rtęci należy unieszkodliwić, by zapobiec przeniesieniu rtęci do środowiska naturalnego.
(11) Mabey i Mill zalecają używanie buforów boran owych lub octanowych zamiast fosforanowych (11).
(12) Informacje takie mogą pochodzić z innych źródeł, takich jak dane dotyczące hydrolizy dla strukturalnie podobnych związków – z literatury albo ze wstępnych półilościowych testów hydrolizy dla substancji badanej, wykonanych na wcześniejszym etapie.
(13) Jeśli wykres danych w skali logarytmicznej jako funkcji czasu nie jest linią prostą (co równa się szybkości reakcji pierwszego rzędu), wówczas nie można wykorzystywać równania [3] do określania stałej szybkości hydrolizy dla substancji badanej.
(14) Wartości pH podane w tych tabelach zostały obliczone na podstawie pomiarów potencjału, przy wykorzystaniu standardowych równań Sórensena (1909). Odpowiadające im wartości pH są o 0,04 jednostki wyższe od wartości podanych w tabeli.
(15) Dodać niewielkie kryształki tymolu lub podobnej substancji aby zapobiec rozwojowi pleśni.
(16) Średnie wartości potrójnych degradacji.
(17) Meyer A, Orti G. (1993) Proc. Royal Society of London, Series B, Vol. 252, p. 231.
(18) Woda próbki po dostarczeniu co najmniej trzech objętości komór.
Wartości w nawiasach są liczbami próbek (woda, ryby), które mają zostać pobrane, jeśli przeprowadzane jest dodatkowe pobieranie próbek.
|
Uwagi |
: |
Oznaczenie k2 dla log Pow z 4,0 przed badaniem wynosi 0,652 dnia-1. Całkowita długość trwania eksperymentu ustalona jest na 3 × faza absorpcji = 3 × 4,6 dnia, np. 14 dni. W odniesieniu do oszacowania absorpcji – odnieść się do załącznika 3. |
(19) Meyer, A., Bierman, C.H. and Orti, G. (1993). The phylogenetic position of the zebrafish (Danio rerio), a model system in developmental biology: an invitation to the comparative method. Proc. R. Soc. Lond. B. 252, 231-236.
(20) Serie pięciu (lub więcej) kolejnych stężeń mogą być wybrane z kolumny. Punkt środkowy między stężeniami w kolumnie (x) są znajdowane w kolumnie (2x + 1). Zamieszczone wartości przedstawiają stężenia wyrażone w procentach objętościowo lub wagowo (mg/l lub μg/l). Wartości mogą być zwielokrotniane lub dzielone przez każdą potęgę 10, jeśli to konieczne. Kolumna 1 może być używana, jeśli była znaczna niepewność w poziomie toksyczności.
(21) OECD, Paris, 1992. Test Guideline 210, Fish, Early-life Stage Toxicity Test.
(22) Dla embrionów.
(23) Dla larw.
(24) Ciemność dla embrionów i larw do jednego tygodnia po wylęgu, z wyjątkiem, gdy są ich sprawdzane. Następnie przytłumione oświetlenie przez badanie.
(25) Zgodnie z FAO I systemem US (85).
(26) Odpowiednie zmienne powinny wykazywać wartości w ramach podanego zakresu. Jednakże jeśli występują trudności ze znalezieniem właściwego materiału gleby, wartości poniżej wskazują akceptowalne minimum.
(27) Gleby z mniej niż 0,3 % węgla organicznego mogą zakłócać korelację między organiczną zawartością a adsorpcją. Jest zalecane użycie gleb o minimalnej zawartości węgla organicznego 0,3 %.
(29) Krzywa stężenia substancji badanej fazie wodnej ( ) w zależności od czasu może być również wykorzystana do oszacowania osiągnięcia plateau równowagowego (zob. rys. 2 w dodatku 5).
) w zależności od czasu może być również wykorzystana do oszacowania osiągnięcia plateau równowagowego (zob. rys. 2 w dodatku 5).
(30) DT-50: czas degradacji dla 50 % badanej substancji.
(31) Równanie ma zastosowanie do obu metod, bezpośredniej i pośredniej. Wszystkie inne równania stosowane są tylko dla metody pośredniej.
(32) W. Kördel, D. Hennecke, M. Herrmann (1997). Application of the HPLC-screening method for the determination of the adsorption coefficient on sewage sludges. Chemosphere, 35(1/2), pp. 121–128.
(33) W. Kördel, D. Hennecke, C. Franke (1997). Determination of the adsorption-coefficients of organic substances on sewage sludges. Chemosphere, 35 (1/2), pp. 107–119.
(34) W. Kördel, G. Kotthoff, J. Müller (1995). HPLC-screening method for the determination of the adsorption coefficient on soil-results of a ring test. Chemosphere, 30(7), pp. 1373–1384.
(35) W. Kördel. Müller (1994). Bestimmung des Adsorptionskoefftiienten organischern Chemikalien mit der HPLC. UBA R & D Report No 106 01044 (1994).
(36) B.V. Oepen. W. Kördel, W. Klein (1991). Chemosphere, 22, pp. 285–304.
(37) Dane dostarczone przez przemysł.
(38) Wskazuje, który zbiornik był użyty w doświadczeniu.
(39) Zapis śmiertelności wszystkich dorosłych zwierząt jako „M” w stosownym okienku.
(40) Zapis przerw wylęgu jako „AB” w stosownym okienku.
(41) Na przykład jeżeli substancja badana zawiera jeden pierścień, wymagane jest znakowanie na tym pierścieniu; jeżeli substancja badana zawiera dwa lub więcej pierścieni, mogą być potrzebne oddzielne badania w celu oceny losu każdego znakowanego pierścienia i w celu otrzymania odpowiednich informacji na temat powstawania produktów przemian.
(42) Własności retencji wody gleby można zmierzyć jako pojemność połowa, jako pojemność wodna lub jako siła ssąca gleby (pF). Wyjaśnienia zamieszczono w załączniku 1. W sprawozdaniu z badań należy podać, czy własności retencji wody i gęstość nasypową gleb określono w próbkach z terenu niezaburzonego, czy w próbkach zaburzonych (przetworzonych).
(43) Ostatnie wyniki badań pokazują, że gleby ze stref umiarkowanych również mogą być przechowywane w temp. –20 oC przez okres dłuższy niż trzy miesiące (28)(29) bez znaczących strat aktywności mikrobiologicznej.
(44) Gleba nie powinna być ani za mokra, ani za sucha, aby utrzymać odpowiednie napowietrzenie i odżywienie mikroflory glebowej. Zawartość wilgoci zalecana dla optymalnego wzrostu mikrobiologicznego mieści się z zakresie od 40–60 % zdolności utrzymywania wody (WHC) i 0,1–0,33 bara (6). Ten drugi zakres jest równoważny zakresowi pF 2,0–2,5. Typowe zawartości wilgoci różnych rodzajów gleb są podane w załączniku 2.
(45) Warunki tlenowe dominują w glebach powierzchniowych i nawet w glebach podpowierzchniowych, jak pokazano w pracy badawczej sponsorowanej przez UE [K. Takagi et al. (1992). Microbial dirersity and activity in subsoils: Methods, field site, seasonal variation in subsoil temperatures and oxygen contents. Proc. Internat. Symp. Environment. Aspects Pesticides Microbiol., 270-277, 17–21 sierpnia 1992 r., Sigtuna, Szwecja]. Warunki beztlenowe mogą występować jedynie okazjonalnie podczas zalewania gleb po silnych opadach deszczu lub kiedy na polach ryżowych są tworzone warunki ryżowe.
(46) Badania tlenowe mogą być zakończone przed upływem 120 dni, pod warunkiem że w tym czasie jednoznacznie zostanie osiągnięta ostateczna ścieżka przemiany i ostateczna mineralizacja. Zakończenie badania jest możliwe po 120 dniach lub kiedy zostanie przemienione przynajmniej 90 % substancji badanej, ale tylko wtedy, gdy utworzy się przynajmniej 5 % C02.
(47) Obliczenie stężenia początkowego na powierzchni z wykorzystaniem następującego równania:

|
Csoil |
= |
Stężenie początkowe stężenie w glebie (mg· kg-1). |
|
A |
= |
Podana ilość [kg· ha-1]; 1 = grubość warstwy gleby z pola (m); d = gęstość nasypowa sucha gleby (kg· m-3). |
Z reguły podana ilość 1 kg· ha-1 powoduje stężenie w glebie około l mg· kg-1 w warstwie o grubości 10 cm (przy założeniu gęstości nasypowej 1 g· cm-3).
(48) Muckenhausen, E. (1975). Die Bodenkunde und ihre geologischen, geomorphologischen, mineralogischen und petrologischen Grundlagen. DLG-Verlag, Frankfurt, Main.
(49) pF = log z cm słupa wody.
(50) l bar = 105 Pa.
(51) Odpowiada przybliżonej zawartości wody 10 % w piasku, 35 % w piasku gliniastym, 45 % w glinie.
(52) Pojemność polowa nie jest stała, ale zmienia się w zakresie pF od 1,5 do 2,5.
(53) pojemność wodna (WHC).
(54) Guth, J.A. (1980). The study of transformations. In Interactions between Herbicides and the Soil (R.J. Hance, Ed.), Academic Press, 123-157.
(55) Guth, J.A. (1981). Experimental approaches to studying the fate of pesticides in soil. In Progress in Pesticide Biochemistry. D.H. Hutson, T.R. Roberts, Eds. J. Wiley & Sons. Vol 1, 85-114.
(56) Anderson, J.P.E. (1975). Einfluss von Temperatur und Feuchte auf Verdampfung, Abbau und Festlegung von Diallat im Boden. Z. PflKrankh Pflschutz, Sonderheft VII, 141-146.
(57) Na przykład, jeżeli substancja badana zawiera jeden pierścień, wymagane jest znakowanie na tym pierścieniu; jeżeli substancja badana zawiera dwa lub więcej pierścieni, mogą być potrzebne oddzielne badania w celu oceny losu każdego znakowanego pierścienia i w celu otrzymania odpowiednich informacji na temat powstawania produktów przemian.
(58) Ponieważ te zasadowe roztwory absorpcyjne pochłaniają również ditlenek węgla z wentylacji powietrzem i utworzony przez respirację w doświadczeniach tlenowych, muszą być regularnie wymieniane, aby uniknąć ich nasycenia, a tym samym utraty zdolności absorpcyjnej.
(59) [Glina i ił] jest frakcją mineralną osadu o ziarnistości < 50 μm
(60) Ostatnie badania pokazują, że pomiary stężeń tlenu w wodzie i potencjałów redoks nie mają ani mechanistycznej, ani predykcyjnej wartości, tak długo, jak zaburzony jest wzrost i rozwój populacji mikrobiologicznej w wodach powierzchniowych (24)(25). Oznaczenie zapotrzebowania na tlen biochemiczny (BOD, przy pobieraniu próbek w terenie, na początku i na końcu badania) i stężeń mikro/makro składników biogennych Ca, Mg i Mn (na początku i na końcu badania) w wodzie i pomiar całkowitego N i całkowitego P (przy pobieraniu próbek w terenie i na końcu badania) może być lepszym narzędziem do interpretacji i oceny tempa bioprzemian tlenowych i ich szlaków.
(61) Metoda tempa respiracji mikrobiologicznej (26), metoda zadymiania (27) lub zliczanie na podkładzie rozmnożeniowym (np. bakterie, promieniowce, grzyby i całe kolonie) dla badań w warunkach tlenowych; tempo metanogenezy dla badań beztlenowych.
(62) Ostatnie badania pokazały, że przechowywanie w temp. 4 oC może prowadzić do spadku zawartości węgla organicznego w osadzie, co może ewentualnie skutkować spadkiem aktywności mikrobiologicznej. (34)
(63) Badanie z drugim stężeniem może być przydatne dla substancji chemicznych, które przechodzą do wód powierzchniowych innymi szlakami wejścia, powodujących znacznie inne stężenia, pod warunkiem, że niższe stężenie może być analizowane z wystarczającą dokładnością.
(64) W przypadkach gdy może łatwo wystąpić gwałtowne ponowne utlenianie produktów przemian beztlenowych, podczas pobierania próbek i analizy należy utrzymywać warunki beztlenowe.




 (mN/m)
(mN/m)

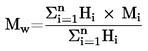


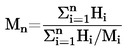



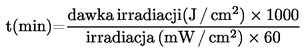






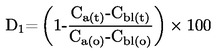










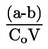
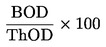

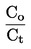

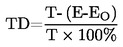



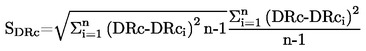

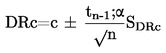




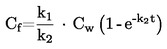
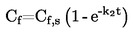

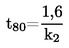






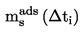
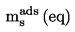

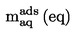
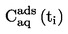






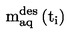

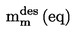


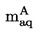


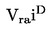


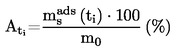
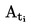




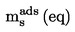



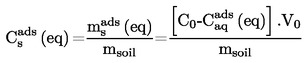

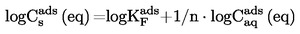
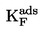
 (μg
(μg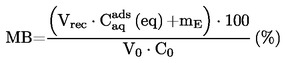







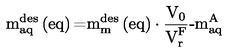



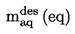



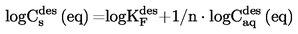
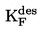



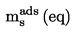



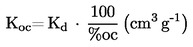


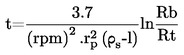





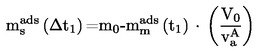


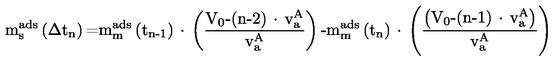
 , jest obliczany przez zastosowanie następującego równania:
, jest obliczany przez zastosowanie następującego równania:
 w punkcie czasu t
w punkcie czasu t
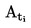 lub
lub  (w odniesieniu do potrzeb badania) są wykreślane w zależności od czasu i czasu po którym uzyskana równowaga sorpcji jest stała.
(w odniesieniu do potrzeb badania) są wykreślane w zależności od czasu i czasu po którym uzyskana równowaga sorpcji jest stała.

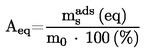


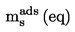
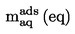


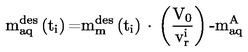
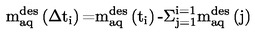




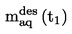


 pobranej do analizy (μg),
pobranej do analizy (μg),

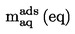


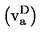 , a masa desorbowana jest obliczana według równania:
, a masa desorbowana jest obliczana według równania: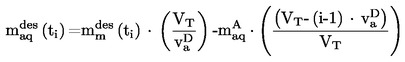
 =
=  ,
,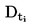 obliczany jest według równania:
obliczany jest według równania: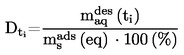






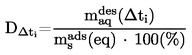



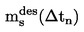
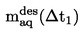





 w punktach czasu t
w punktach czasu t