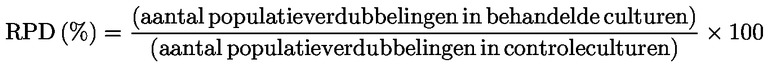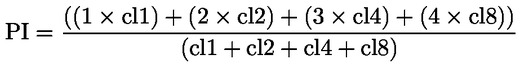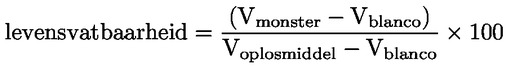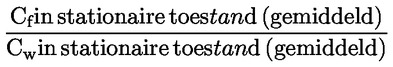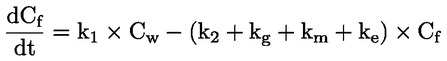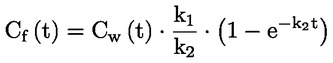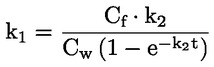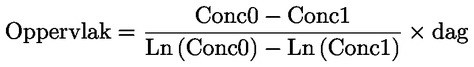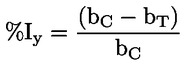ISSN 1977-0758
Publicatieblad
van de Europese Unie
L 112

Uitgave in de Nederlandse taal
Wetgeving
60e jaargang
28 april 2017
|
ISSN 1977-0758 |
||
|
Publicatieblad van de Europese Unie |
L 112 |
|
|
|
||
|
Uitgave in de Nederlandse taal |
Wetgeving |
60e jaargang |
|
|
|
|
|
(1) Voor de EER relevante tekst. |
|
NL |
Besluiten waarvan de titels mager zijn gedrukt, zijn besluiten van dagelijks beheer die in het kader van het landbouwbeleid zijn genomen en die in het algemeen een beperkte geldigheidsduur hebben. Besluiten waarvan de titels vet zijn gedrukt en die worden voorafgegaan door een sterretje, zijn alle andere besluiten. |
II Niet-wetgevingshandelingen
VERORDENINGEN
|
28.4.2017 |
NL |
Publicatieblad van de Europese Unie |
L 112/1 |
VERORDENING (EU) 2017/735 VAN DE COMMISSIE
van 14 februari 2017
tot wijziging, in verband met de aanpassing ervan aan de technische vooruitgang, van de bijlage bij Verordening (EG) nr. 440/2008 houdende vaststelling van testmethoden uit hoofde van Verordening (EG) nr. 1907/2006 van het Europees Parlement en de Raad inzake de registratie en beoordeling van en de autorisatie en beperkingen ten aanzien van chemische stoffen (REACH)
(Voor de EER relevante tekst)
DE EUROPESE COMMISSIE,
Gezien het Verdrag betreffende de werking van de Europese Unie,
Gezien Verordening (EG) nr. 1907/2006 van het Europees Parlement en de Raad van 18 december 2006 inzake de registratie en beoordeling van en de autorisatie en beperkingen ten aanzien van chemische stoffen (Reach), tot oprichting van een Europees Agentschap voor chemische stoffen, houdende wijziging van Richtlijn 1999/45/EG en houdende intrekking van Verordening (EEG) nr. 793/93 van de Raad en Verordening (EG) nr. 1488/94 van de Commissie alsmede Richtlijn 76/769/EEG van de Raad en de Richtlijnen 91/155/EEG, 93/67/EEG, 93/105/EG en 2000/21/EG van de Commissie (1), en met name artikel 13, lid 2,
Overwegende hetgeen volgt:
|
(1) |
Verordening (EG) nr. 440/2008 van de Commissie (2) bevat de testmethoden voor de bepaling van de fysisch-chemische eigenschappen, de toxiciteit en de ecotoxiciteit van chemische stoffen, die worden toegepast voor de uitvoering van Verordening (EG) nr. 1907/2006. |
|
(2) |
Verordening (EG) nr. 440/2008 moet worden bijgewerkt om daarin nieuwe en bijgewerkte testmethoden die onlangs zijn aangenomen door de Organisatie voor Economische Samenwerking en Ontwikkeling (OESO) op te nemen, teneinde rekening te houden met de technische vooruitgang en overeenkomstig Richtlijn 2010/63/EU van het Europees Parlement en de Raad (3) tot een verlaging van het aantal proefdieren te komen. De belanghebbenden zijn over het ontwerp van deze maatregel geraadpleegd. |
|
(3) |
Deze aanpassing aan de vooruitgang van de techniek betreft twintig testmethoden: één nieuwe methode voor de bepaling van een fysisch-chemische eigenschap, vijf nieuwe en één aangepaste testmethoden voor de beoordeling van de ecotoxiciteit, twee bijgewerkte testmethoden voor de beoordeling van het uiteindelijk lot en het gedrag in het milieu en vier nieuwe en zeven bijgewerkte testmethoden voor de bepaling van de effecten op de menselijke gezondheid. |
|
(4) |
De OESO evalueert haar testrichtsnoeren regelmatig om vast te stellen welke ervan in wetenschappelijk opzicht achterhaald zijn. Bij deze aanpassing aan de technische vooruitgang worden zes testmethoden geschrapt waarvoor de desbetreffende testrichtsnoeren van de OESO zijn ingetrokken. |
|
(5) |
Verordening (EG) nr. 440/2008 moet daarom dienovereenkomstig worden gewijzigd. |
|
(6) |
De in deze verordening vervatte bepalingen zijn in overeenstemming met het advies van het bij artikel 133 van Verordening (EG) nr. 1907/2006 ingestelde comité, |
HEEFT DE VOLGENDE VERORDENING VASTGESTELD:
Artikel 1
De bijlage bij Verordening (EG) nr. 440/2008 wordt gewijzigd overeenkomstig de bijlage bij deze verordening.
Artikel 2
Deze verordening treedt in werking op de twintigste dag na die van de bekendmaking ervan in het Publicatieblad van de Europese Unie.
Deze verordening is verbindend in al haar onderdelen en is rechtstreeks toepasselijk in elke lidstaat.
Gedaan te Brussel, 14 februari 2017.
Voor de Commissie
De voorzitter
Jean-Claude JUNCKER
(1) PB L 396 van 30.12.2006, blz. 1.
(2) Verordening (EG) nr. 440/2008 van de Commissie van 30 mei 2008 houdende vaststelling van testmethoden uit hoofde van Verordening (EG) nr. 1907/2006 van het Europees Parlement en de Raad inzake de registratie en beoordeling van en de autorisatie en beperkingen ten aanzien van chemische stoffen (Reach) (PB L 142 van 31.5.2008, blz. 1).
(3) Richtlijn 2010/63/EU van het Europees Parlement en de Raad van 22 september 2010 betreffende de bescherming van dieren die voor wetenschappelijke doeleinden worden gebruikt (PB L 276 van 20.10.2010, blz. 33).
BIJLAGE
De bijlage bij Verordening (EG) nr. 440/2008 wordt als volgt gewijzigd:
|
1) |
In deel A wordt het volgende hoofdstuk toegevoegd: „A.25 DISSOCIATIECONSTANTEN IN WATER (TITRATIEMETHODE — SPECTROFOTOMETRISCHE METHODE — CONDUCTOMETRISCHE METHODE) INLEIDING Deze testmethode is gelijkwaardig aan testrichtlijn (TG) 112 (1981) van de OESO. Voorwaarden
Begeleidende informatie
Kwalificerende opmerkingen
Standaarddocumenten Deze testmethode is gebaseerd op methoden die worden genoemd in de referenties onder Literatuur, en op de Preliminary Draft Guidance for Premanufacture Notices van het EPA van 18 augustus 1978. METHODE — INLEIDING, DOEL, TOEPASSINGSGEBIED, RELEVANTIE, TOEPASSING EN BEPERKINGEN VAN DE TEST De dissociatie van een stof in water is van belang bij de beoordeling van het effect van de stof op het milieu. Zij bepaalt de vorm van de stof, die op zijn beurt het gedrag en transport van de stof bepaalt. Ze kan van invloed zijn op de adsorptie van de chemische stof op bodems en sedimenten en op de absorptie in biologische cellen. Definities en eenheden Dissociatie is de omkeerbare splitsing in twee of meer chemische vormen, die ionisch kunnen zijn. Het proces wordt doorgaans beschreven door RX ⇌ R ++ X – en de concentratie-evenwichtsconstante die de reactie bepaalt, is
Voorbeeld: in het bijzondere geval waarin R waterstof is (de stof is een zuur), is de constante gelijk aan
of
Referentiestoffen Het is niet altijd nodig om bij het onderzoek van een nieuwe stof de volgende referentiestoffen te gebruiken. Ze worden vooral gegeven zodat de methode van tijd tot tijd kan worden geijkt en om de resultaten te kunnen vergelijken wanneer een andere methode wordt toegepast.
Het is nuttig een stof met meerdere pK's te hebben, zoals hieronder aangegeven in Principe van de testmethode. Een voorbeeld van een dergelijke stof is:
Principe van de testmethode Het beschreven chemische proces is in het voor het milieu relevante temperatuurbereik doorgaans slechts beperkt van de temperatuur afhankelijk. Voor de bepaling van de dissociatieconstante is een meting van de concentraties van de gedissocieerde en niet-gedissocieerde vormen van de chemische stof nodig. Op basis van de kennis van de stoichiometrie van de dissociatiereactie zoals hierboven in Definities en eenheden vermeld, kan de juiste constante worden bepaald. In het bijzondere geval dat in deze testmethode wordt beschreven, gedraagt de stof zich als een zuur of een base, en de bepaling kan het gemakkelijkst geschieden door de relatieve concentraties van de geïoniseerde en niet-geïoniseerde vormen van de stof en de pH van de oplossing te bepalen. Het verband tussen deze termen wordt hierboven gegeven in de vergelijking voor pKa in Definities en eenheden. Sommige stoffen vertonen meer dan één dissociatieconstante, waarvoor vergelijkbare vergelijkingen kunnen worden opgesteld. Enkele van de hierin beschreven methoden zijn ook geschikt voor niet-zuur/base-dissociatie. Kwaliteitscriteria Herhaalbaarheid De dissociatieconstante moet in duplo worden bepaald (minimaal drie bepalingen) tot op ± 0,1 log-eenheden. BESCHRIJVING VAN DE TESTPROCEDURES Er zijn twee basisbenaderingen om pKa te bepalen. Bij de ene wordt een bekende hoeveelheid van de stof getitreerd met standaardzuur of -base, naargelang het geval; bij de andere worden de relatieve concentratie van de geïoniseerde en niet-geïoniseerde vormen en de pH-afhankelijkheid daarvan bepaald. Voorbereidingen Op deze beginselen gebaseerde methoden kunnen worden ingedeeld als titratie-, spectrofotometrische en conductometrische procedures. Testoplossingen Voor de titratiemethode en conductometrische methode wordt de chemische stof opgelost in gedestilleerd water. Voor de spectrofotometrische en overige methoden worden bufferoplossingen gebruikt. De concentratie van de teststof mag niet hoger zijn dan het minimum van 0,01 M of, als dat lager is, de helft van de verzadigingsconcentratie, en bij het bereiden van de oplossingen wordt een zo zuiver mogelijke vorm van de stof gebruikt. Als de stof slechts matig oplosbaar is, kan zij vóór toevoeging aan de hierboven genoemde concentraties worden opgelost in een kleine hoeveelheid van een met water mengbaar oplosmiddel. Oplossingen worden met een Tyndall-kegel gecontroleerd op de aanwezigheid van emulsies, vooral als er een co-oplosmiddel is gebruikt om de oplosbaarheid te vergroten. Wanneer bufferoplossingen worden gebruikt, mag de bufferconcentratie niet hoger zijn dan 0,05 M. Testomstandigheden Temperatuur De temperatuur wordt geregeld tot op ± 1 °C of beter. De bepaling vindt bij voorkeur plaats bij 20 °C. Als een aanzienlijke temperatuurafhankelijkheid wordt vermoed, vindt de bepaling ook plaats bij ten minste twee andere temperaturen. De temperatuurintervallen zijn in dit geval 10 °C en de temperatuur wordt geregeld tot op ± 0,1 °C. Analyses De methode wordt bepaald door de aard van de stof die wordt getest. Zij moet voldoende gevoelig zijn om de verschillende vormen bij elke concentratie van de testoplossing te kunnen bepalen. Uitvoering van de test Titratiemethode De testoplossing wordt bepaald door titratie met de basische of zure standaardoplossing, naargelang hetgeen van toepassing is, waarbij na elke toevoeging van titrant de pH wordt gemeten. Er moeten ten minste 10 incrementele toevoegingen plaatsvinden vóór het equivalentiepunt. Als het evenwicht voldoende snel wordt bereikt, mag een registrerende potentiometer worden gebruikt. Voor deze methode moeten zowel de totale hoeveelheid van de stof als haar concentratie nauwkeurig bekend zijn. Er moeten voorzorgsmaatregelen worden genomen om koolstofdioxide uit te sluiten. Gedetailleerde gegevens van de procedure, voorzorgen en berekeningswijze worden gegeven in standaardtesten, bv. referenties (1), (2), (3) en (4). Spectrofotometrische methode Er wordt een golflengte gevonden waarbij de geïoniseerde en niet-geïoniseerde vormen van de stof aanmerkelijk verschillende extinctiecoëfficiënten hebben,. Het absorptiespectrum in uv/zichtbaar licht wordt verkregen van oplossingen met een constante concentratie bij pH's waarbij de stof hoofdzakelijk niet-geïoniseerd, dan wel volledig geïoniseerd, is en bij meerdere tussenliggende pH's. Dit kan worden gedaan hetzij door porties geconcentreerd(e) zuur (base) toe te voegen aan een relatief groot volume van een oplossing van de stof in een uit meerdere componenten bestaande buffer, in eerste instantie bij een hoge (lage) pH (referentie 5), of door gelijke volumes van een voorraadoplossing van de stof in bv. water of methanol toe te voegen aan constante volumes van verschillende bufferoplossingen die het gewenste pH-bereik bestrijken. Uit de pH- en extinctiewaarden bij de gekozen golflengte wordt een voldoende aantal waarden voor de pKa berekend met behulp van gegevens van ten minste 5 pH's waarbij de stof voor ten minste 10 procent en minder dan 90 procent is geïoniseerd. Nadere experimentele gegevens en de berekeningswijze zijn te vinden in referentie (1). Conductometrische methode Met behulp van een cel met een kleine, bekende celconstante wordt de geleidbaarheid van een ongeveer 0,1 M-oplossing van de stof in conductometrisch zuiver water gemeten. De geleidbaarheid van een aantal nauwkeurig gemaakte verdunningen van deze oplossing wordt ook gemeten. De concentratie wordt elke keer gehalveerd, en de reeks bestrijkt in concentratie ten minste een orde van grootte. De uiterste geleidbaarheid bij oneindige verdunning wordt gevonden door een vergelijkbaar experiment uit te voeren met het Na-zout en te extrapoleren. De mate van dissociatie kan dan met de Onsager-vergelijking worden berekend uit de geleidbaarheid van elk van de oplossingen, en daarna kan met de verdunningswet van Ostwald de dissociatieconstante worden berekend als K = α2C/(1 – α), waarin C de concentratie in mol per liter is en α de gedissocieerde fractie is. Er moeten voorzorgsmaatregelen worden genomen om CO2 uit te sluiten. Nadere experimentele details en de berekeningswijze worden gegeven in standaardteksten en in de referenties (1), (6) en (7). GEGEVENS EN RAPPORTAGE Verwerking van de resultaten Titratiemethode De pKa wordt berekend voor 10 gemeten punten op de titratiecurve. Het gemiddelde en de standaardafwijking van deze pKa-waarden worden berekend. Een grafiek van de pH uitgezet tegen het volume van de standaardbase of het standaardzuur wordt vermeld, tezamen met een weergave in tabelvorm. Spectrofotometrische methoden Er wordt een tabel gemaakt met de extinctie en pH voor elk spectrum. Er worden ten minste vijf waarden voor de pKa berekend uit de tussenliggende spectrumgegevenspunten, en ook het gemiddelde en de standaardafwijking van deze resultaten worden berekend. Conductometrische methode De equivalente geleidbaarheid Λ wordt berekend voor elke concentratie van het zuur en voor elke concentratie van een mengsel van één equivalent zuur plus 0,98 equivalent carbonaatvrij natriumhydroxide. Het zuur is in overmaat om een overmaat aan OH– als gevolg van hydrolyse te voorkomen. 1/Λ wordt in een grafiek uitgezet tegen √C, en Λo van het zout wordt gevonden door extrapolatie naar een concentratie van nul. Λo van het zuur kan worden berekend met behulp van waarden uit de literatuur voor H+ en Na+. De pKa kan worden berekend uit α = Λi /Λo en Ka = α2C/(1 – α) voor elke concentratie. Door te corrigeren voor mobiliteit en activiteit kunnen betere waarden voor Ka worden verkregen. Het gemiddelde en de standaardafwijking van de pKa-waarden moeten worden berekend. Testverslag Alle ruwe gegevens en berekende pKa-waarden worden ingediend met de berekeningsmethode (bij voorkeur in tabelvorm, zoals voorgesteld in referentie (1)) en de hierboven beschreven statistische parameters. Voor titratiemethoden worden de details van de standaardisatie van de titranten vermeld. Voor de spectrofotometrische methode worden alle spectra ingediend. Voor de conductometrische methode worden de details van de bepaling van de celconstante gerapporteerd. Er wordt informatie verstrekt over de gebruikte techniek, analysemethoden en de aard van de eventueel gebruikte buffers. De testtemperatu(u)r(en) worden gerapporteerd. LITERATUUR
|
|
2) |
In deel B wordt hoofdstuk B.5 vervangen door: „B.5 ACUTE OOGIRRITATIE/-CORROSIE INLEIDING Deze testmethode is gelijkwaardig aan testrichtlijn (TG) 405 (2012) van de OESO. De testrichtlijnen van de OESO voor het testen van chemische stoffen worden periodiek herzien om te waarborgen dat ze de beste wetenschappelijke methoden weerspiegelen. Bij eerdere herzieningen van deze testrichtlijn is bijzondere aandacht besteed aan mogelijke verbeteringen via de evaluatie van alle bestaande informatie over de teststof om onnodige tests bij proefdieren te vermijden en zodoende rekening te houden met de bezorgdheid omtrent het welzijn van dieren. In TG 405 (vastgesteld in 1981 en bijgewerkt in 1987, 2002 en 2012) is de aanbeveling opgenomen de bestaande relevante gegevens op bewijskracht te analyseren (1), alvorens de beschreven in-vivotest op acute oogirritatie/-corrosie wordt uitgevoerd. Wanneer er onvoldoende gegevens beschikbaar zijn, wordt aanbevolen deze aan te vullen door een sequentiële teststrategie te volgen (2) (3). De aanbevolen teststrategie omvat de uitvoering van gevalideerde en erkende in-vitrotests en is als supplement bij deze methode opgenomen. Voor de toepassing van Verordening (EG) nr. 1907/2006 inzake de registratie en beoordeling van en de autorisatie en beperkingen ten aanzien van chemische stoffen (REACH) (2) is in het relevante ECHA-richtsnoer (21) ook een geïntegreerde teststrategie opgenomen. Dierproeven mogen uitsluitend worden uitgevoerd als ze noodzakelijk worden geacht, nadat eerst de beschikbare alternatieve methoden zijn overwogen, en het gebruik ervan passend wordt geacht. Ten tijde van het opstellen van deze bijgewerkte testmethode zijn er gevallen bekend waarin het gebruik van deze testmethode hoe dan ook noodzakelijk is of vereist is uit hoofde van bepaalde regelgevingskaders. De laatste bijwerking richtte zich vooral op het gebruik van analgetica en anesthetica en was niet van invloed op het basisconcept en de structuur van de testrichtlijn. ICCVAM (3) en een onafhankelijk internationaal wetenschappelijk peer review panel heeft de bruikbaarheid en de beperkingen van het routinematig gebruik van lokale anesthetica, systemische analgetica en humane eindpunten tijdens in-vivotests betreffende de veiligheid ten aanzien van oogirritatie (12) beoordeeld. De beoordeling leidde tot de conclusie dat het gebruik van lokale anesthetica en systemische analgetica pijn en nood grotendeels of volledig kan voorkomen zonder dat het de uitkomst van de test beïnvloedt, en tot de aanbeveling om deze stoffen altijd te gebruiken. Deze testmethode houdt rekening met deze beoordeling. Lokale anesthetica, systemische analgetica en humane eindpunten moeten routinematig worden gebruikt tijdens het uitvoeren van in-vivotests op acute oogirritatie/-corrosie. Uitzonderingen op hun gebruik moeten worden gemotiveerd. De in deze methode beschreven verfijningen zullen pijn en nood bij dieren in de meeste testsituaties waarbij in-vivotests op de veiligheid voor ogen nog noodzakelijk zijn, aanzienlijk verminderen of voorkomen. Evenwichtige preventieve pijnbestrijding moet omvatten: i) routinematige voorbehandeling met een lokaal anestheticum (bv. proparacaïne of tetracaïne) en een systemische analgeticum (bv. buprenorfine), ii) schema voor routinematige nabehandeling met systemische analgetica (bv. buprenorfine en meloxicam), iii) geplande observatie, monitoring en registratie van dieren voor wat betreft klinische tekenen van pijn en/of nood, en iv) geplande observatie, monitoring en registratie van de aard, ernst en progressie van alle oogverwondingen. Verdere details zijn te vinden in de hieronder beschreven bijgewerkte procedures. Na toediening van de teststof mogen geen aanvullende lokale anesthetica of analgetica worden toegepast, om verstoring van het onderzoek te vermijden. Analgetica met ontstekingsremmende activiteit (bv. meloxicam) mogen niet lokaal worden toegepast, en de systemisch gebruikte doses mogen effecten op het oog niet verstoren. De definities zijn opgenomen in het aanhangsel bij deze testmethode. INLEIDENDE OVERWEGINGEN In het belang van zowel wetenschappelijke kwaliteit als het dierenwelzijn mogen in-vivotests pas worden overwogen nadat alle beschikbare gegevens over de mogelijke oogcorrosie/-irritatie door de chemische stof zijn geëvalueerd op bewijskracht. Hierbij gaat het bijvoorbeeld om de resultaten van eerdere studies bij mensen en/of proefdieren, gegevens over oogcorrosie/irritatie door een of meer qua structuur verwante stoffen of mengsels van dergelijke stoffen, gegevens waaruit blijkt dat de chemische stof sterk zuur of sterk alkalisch is (4) (5), en resultaten van gevalideerde en erkende in-vitro- of ex-vivotests op huidcorrosie en oogcorrosie/-irritatie (6) (13) (14) (15) (16) (17). De studies kunnen al vóór de bewijskrachtanalyse zijn uitgevoerd, maar dit kan ook naar aanleiding van deze analyse gebeuren. Voor bepaalde chemische stoffen kan uit een dergelijke analyse blijken dat er een in-vivo-onderzoek naar een mogelijke oogcorrosie/-irritatie door de chemische stof nodig is. In al dergelijke gevallen wordt bij voorkeur, voordat wordt overwogen om de in-vivo-oogtest te gebruiken, eerst een onderzoek naar de in vitro en/of in vivo corrosieve effecten van de chemische stof op de huid uitgevoerd en geëvalueerd in overeenstemming met de sequentiële teststrategie in testmethode B.4 (7) of de geïntegreerde teststrategie beschreven in ECHA-richtsnoer (21). Een sequentiële teststrategie, waarin de uitvoering van gevalideerde in-vitro- of ex-vivotests op oogcorrosie/-irritatie is opgenomen, is als supplement bij deze testmethode en, voor de toepassing van REACH, in ECHA-richtsnoer (21) opgenomen. Aanbevolen wordt om een dergelijke teststrategie te volgen alvorens in-vivotests uit te voeren. Voor nieuwe chemische stoffen wordt een stapsgewijze testbenadering aanbevolen om wetenschappelijk verantwoorde gegevens over corrosie/-irritatie door de chemische stof te verkrijgen. Wanneer er voor bestaande chemische stoffen onvoldoende gegevens over de huid- en oogcorrosie/-irritatie beschikbaar zijn, kan de strategie worden gebruikt om ontbrekende gegevens aan te vullen. Voor het gebruik van een andere teststrategie of -procedure of de beslissing om geen stapsgewijze testbenadering te volgen moet een motivering worden gegeven. PRINCIPE VAN DE IN-VIVOTEST Na voorbehandeling met een systemisch analgeticum en inductie van passende lokale anesthetica, wordt de te testen chemische stof in één dosis aangebracht op een van de ogen van het proefdier; het onbehandelde oog dient als controle. De mate van oogirritatie/-corrosie wordt geëvalueerd door op bepaalde tijdstippen het letsel aan de conjunctiva, de cornea en de iris in te schalen. Ook andere effecten in het oog en schadelijke systemische effecten worden beschreven om een volledige evaluatie van de effecten mogelijk te maken. De duur van de studie moet voldoende zijn om te kunnen beoordelen of de effecten reversibel of irreversibel van aard zijn. Dieren die in enig stadium van de test blijk geven van ernstige nood en/of hevige pijn of letsel vertonen dat overeenkomt met de in deze testmethode beschreven humane eindpunten (zie punt 26), worden op humane wijze gedood en de chemische stof wordt dienovereenkomstig beoordeeld. Criteria voor de beslissing om stervende en hevig lijdende dieren op humane wijze te doden zijn te vinden in een richtlijn van de OESO (8). VOORBEREIDING VAN DE IN-VIVOTEST Keuze van de diersoort Als proefdier wordt de voorkeur gegeven aan het albinokonijn, en er worden gezonde jonge volwassen dieren gebruikt. Wanneer er een andere stam of soort wordt gebruikt, moet hiervoor een motivering worden gegeven. Voorbereiding van de dieren Beide ogen van ieder voorlopig voor de test geselecteerd proefdier worden binnen 24 uur vóór het begin van de test onderzocht. Dieren die aan oogirritatie, oogaandoeningen of een reeds bestaand cornealetsel lijden, mogen niet worden gebruikt. Huisvestings- en voedingsomstandigheden De dieren worden in aparte kooien gehuisvest. De temperatuur in de proefdierruimte dient voor konijnen 20 °C (± 3 °C) te zijn. De relatieve luchtvochtigheid moet ten minste 30 % zijn en (behalve tijdens het schoonmaken van de ruimte) bij voorkeur niet hoger dan 70 % zijn, maar er moet worden gestreefd naar 50-60 %. Er moet gebruik worden gemaakt van kunstlicht met een cyclus van 12 uur licht en 12 uur donker. Een buitensporig hoge lichtintensiteit moet worden vermeden. Als voeding mag het gewone laboratoriumvoer worden gebruikt met een onbeperkte hoeveelheid drinkwater. TESTPROCEDURE Gebruik van lokale anesthetica en systemische analgetica De volgende procedures worden aanbevolen om pijn en nood in procedures voor tests betreffende de veiligheid voor de ogen te vermijden of tot een minimum te beperken. In plaats daarvan mogen ook alternatieve procedures worden gebruikt waarvan is vastgesteld dat zij pijn en nood even goed of beter voorkomen of verminderen.
Aanbrenging van de teststof De teststof wordt bij elk dier aangebracht in de conjunctivaalzak van één oog, waarbij het onderste ooglid voorzichtig van de oogbol wordt weggetrokken. Vervolgens worden de oogleden ongeveer één seconde zachtjes dichtgehouden om verlies van het materiaal te voorkomen. Het andere oog, dat niet behandeld wordt, fungeert als controle. Irrigatie De ogen van de proefdieren mogen gedurende ten minste 24 uur na het indruppelen van de teststof niet worden uitgewassen, tenzij het om een vaste stof gaat (zie punt 18) of wanneer er sprake is van onmiddellijke corrosieve of irriterende effecten. Na 24 uur mogen de ogen eventueel worden uitgewassen. Het gebruik van een satellietgroep om de invloed van het uitwassen te onderzoeken wordt niet aanbevolen, tenzij dit wetenschappelijk verantwoord is. Als er een satellietgroep nodig is, dienen hiervoor twee konijnen te worden gebruikt. De omstandigheden van het uitwassen worden zorgvuldig gedocumenteerd, bv. het tijdstip van uitwassen, de samenstelling en temperatuur van de uitwasoplossing, en duur, volume en snelheid van aanbrenging. Dosisniveau (1) Vloeibare teststoffen Bij vloeibare teststoffen wordt een dosis van 0,1 ml gebruikt. Er mogen geen sproeipompjes worden gebruikt om de chemische stof rechtstreeks in het oog te brengen. De vloeistof wordt na het sproeien opgevangen en vervolgens wordt 0,1 ml in het oog gedruppeld. (2) Vaste teststoffen Bij het testen van vaste stoffen, pasta's en vaste deeltjes wordt een hoeveelheid met een volume van 0,1 ml of een gewicht van ten hoogste 100 mg gebruikt. De teststof wordt tot een fijn poeder vermalen. Alvorens het volume te meten wordt het vaste materiaal licht ingeklonken, bijvoorbeeld door tegen de houder te tikken. Als de vaste chemische stof op het eerste observatietijdstip (1 uur na de behandeling) nog niet door fysiologische mechanismen uit het oog van het proefdier is verwijderd, kan het oog met fysiologisch zout of gedestilleerd water worden uitgewassen. (3) Aerosol-teststoffen Aanbevolen wordt alle sproeivloeistoffen en aerosolen op te vangen voordat ze in het oog worden gedruppeld. Er wordt alleen een uitzondering gemaakt voor chemische stoffen in spuitbussen onder druk, die niet kunnen worden opgevangen omdat ze verdampen. In deze gevallen wordt het oog opengehouden en wordt de teststof aan het oog toegediend door in één keer gedurende ongeveer één seconde vanaf een afstand van 10 cm recht voor het oog te spuiten. Deze afstand kan afhankelijk van de druk van de nevel en de inhoud worden aangepast. Er moet voor worden gezorgd dat het oog niet door de druk van de nevel wordt beschadigd. In bepaalde gevallen kan het nodig zijn na te gaan of een “mechanische” beschadiging van het oog door de druk van de nevel mogelijk is. De dosis uit een spuitbus kan door een simulatie als volgt worden geraamd: de chemische stof wordt door een opening die zo groot is als het oog van een konijn en recht voor het papier wordt gehouden, op een weegpapiertje gespoten. De gewichtstoename van het papier wordt gebruikt voor een benadering van de hoeveelheid die in het oog wordt gespoten. Bij vluchtige stoffen kan de dosis worden geraamd door een opvangbakje voor en na de verwijdering van de teststof te wegen. Voorlopige test (in-vivotest op oogirritatie/-corrosie met één dier) Het is zeer wenselijk dat de in-vivotest in eerste instantie met één dier wordt uitgevoerd (zie het supplement bij deze testmethode: Een sequentiële teststrategie voor oogirritatie en -corrosie). Op basis van observaties moet het mogelijk zijn de ernst en reversibiliteit vast te stellen voordat verder wordt gegaan naar een bevestigende test bij een tweede dier. Als de resultaten van deze test erop wijzen dat de chemische stof bij gebruik van de beschreven procedure corrosief of hevig irriterend is voor het oog, dienen er geen verdere tests op oogirritatie te worden uitgevoerd. Bevestigende test (in-vivotest op oogirritatie met meer dieren) Als er bij de voorlopige test geen corrosiereactie of hevige irritatie wordt waargenomen, moet de irritatie of negatieve reactie worden bevestigd met nog eens twee dieren. Als er bij de voorlopige test hevige irritatie wordt waargenomen, wordt aanbevolen de bevestigende test op sequentiële wijze bij één dier tegelijk uit te voeren en de twee andere dieren niet tegelijkertijd bloot te stellen. Als er bij het tweede dier corrosieve of hevig irriterende effecten optreden, wordt de test niet voortgezet. Als de resultaten van het tweede dier voldoende zijn om een gevarenindeling te maken, dienen geen verdere test te worden uitgevoerd. Observatieperiode De duur van de observatieperiode moet voldoende zijn om de omvang en de reversibiliteit van de waargenomen effecten volledig te kunnen beoordelen. Het experiment moet echter worden beëindigd zodra het dier tekenen van hevige pijn of ernstige nood vertoont (8). Om te bepalen of de effecten reversibel zijn, moeten de dieren normaal gesproken gedurende 21 dagen na de toediening van de teststof worden geobserveerd. Als al vóór het verstrijken van de 21 dagen wordt geconstateerd dat de effecten reversibel zijn, wordt het experiment op dat moment beëindigd. Klinische observatie en inschaling van de oogreacties De ogen worden één uur na TCA, en daarna ten minste dagelijks, uitvoerig beoordeeld op de aanwezigheid of afwezigheid van oogletsels. Dieren worden in de eerste 3 dagen meerdere keren per dag beoordeeld om ervoor te zorgen dat besluiten tot beëindiging tijdig worden genomen. Proefdieren worden gedurende de hele duur van de studie routinematig beoordeeld op klinische tekenen van pijn en/of nood (bv. herhaald krabben of wrijven van de ogen, buitensporig knipperen, buitensporig tranen) (9) (10) (11), ten minste tweemaal daags, met een minimum van 6 uur tussen de observaties, of vaker, indien nodig. Dit is nodig om i) dieren adequaat te beoordelen op tekenen van pijn en nood, teneinde onderbouwde beslissingen te kunnen nemen over de noodzaak om de dosering van analgetica te verhogen, en ii) dieren te beoordelen op tekenen van vastgestelde humane eindpunten, om onderbouwde beslissingen te kunnen nemen over de vraag of het aangewezen is om dieren op humane wijze te doden, en ervoor te zorgen dat dergelijke beslissingen tijdig worden genomen. Fluoresceïnekleuring wordt routinematig toegepast, en er wordt een spleetlampbiomicroscoop gebruikt wanneer dit passend wordt geacht (bv. beoordeling van de diepte van een verwonding bij ulceratie van de cornea) als hulpmiddel bij de detectie en meting van oogbeschadigingen, en om te beoordelen of is voldaan aan vastgestelde eindpuntcriteria voor humane euthanasie. Digitale foto's van waargenomen letsels kunnen worden verzameld als referentie en om een permanent dossier van de omvang van oogschade op te bouwen. De dieren worden niet langer dan nodig voor de test ingezet zodra er definitieve informatie is verkregen. Dieren die tekenen van hevige pijn of ernstige nood vertonen, worden onverwijld op humane wijze gedood en de chemische stof wordt dienovereenkomstig beoordeeld. Dieren die na de indruppeling de volgende oogletsels ontwikkelen, worden op humane wijze gedood (zie tabel 1 voor een beschrijving van de klassen oogletsel): perforatie van de cornea, significante ulceratie van de cornea en stafyloom; bloed in de voorste oogkamer; klasse 4 troebelheid van de cornea; afwezigheid van een lichtreflex (iris-reactie klasse 2) die gedurende 72 uur aanhoudt; ulceratie van de conjunctivae; necrose van de conjunctivae of het knipvlies; of loslatend dood weefsel. De reden hiervoor is dat dergelijk oogletsel doorgaans niet reversibel is. Het wordt tevens aanbevolen de volgende oogletsels te gebruiken als humane eindpunten om studies vóór het einde van de geplande observatieperiode van 21 dagen te beëindigen. Deze oogletsels worden beschouwd als een voorteken van hevige irritatie of corrosie en verwondingen waarvan niet wordt verwacht dat ze vóór het einde van de observatieperiode van 21 dagen volledig zijn hersteld: grote diepte van verwonding (bv. ulceratie van de cornea die dieper gaat dan de oppervlakkige lagen van het stroma), destructie van de limbus > 50 % (blijkend uit bleking van het ooglidweefsel), en ernstige ooginfectie (afscheiding van pus). Een combinatie van: vascularisatie van het corneale oppervlak (d.w.z. pannus); geen afname van de oppervlakte van fluoresceïneverkleuring in de loop van de tijd op basis van een dagelijkse beoordeling; en/of het ontbreken van re-epithelisatie 5 dagen na aanbrenging van de teststof kunnen ook worden overwogen als potentieel bruikbare criteria die van invloed zijn op het klinische besluit om de studie vroegtijdig te beëindigen. Deze bevindingen afzonderlijk zijn echter onvoldoende om vroegtijdige beëindiging van de studie te rechtvaardigen. Wanneer eenmaal ernstige oogeffecten zijn vastgesteld, dient een behandelend of gekwalificeerd dierenarts met deskundigheid op het gebied van proefdieren of personeel dat is opgeleid om klinisch letsel vast te stellen, te worden geraadpleegd voor een klinisch onderzoek om te bepalen of de combinatie van deze effecten vroegtijdige beëindiging van de studie rechtvaardigt. De score van de oogreactie (conjunctivae, cornea en iris) wordt verkregen en geregistreerd op 1, 24, 48 en 72 uur na aanbrenging van de teststof (zie tabel 1). Wanneer de dieren geen oogletsel ontwikkelen, ag de test niet eerder dan 3 dagen na de indruppeling worden beëindigd. Dieren met oogletsel dat niet ernstig is, moeten worden geobserveerd tot het letsel verdwijnt of gedurende 21 dagen; op dat tijdstip wordt de test dan afgesloten. Observaties moeten ten minste bij 1 uur, 24 uur, 48 uur, 72 uur, 7 dagen, 14 dagen en 21 dagen plaatsvinden en worden opgetekend om de toestand van de letsels en de reversibiliteit of irreversibileit ervan vast te stellen. Frequentere observaties moeten plaatsvinden als dit nodig is om vast te stellen of het proefdier uit humane overwegingen moet worden geëuthanaseerd of vanwege negatieve resultaten uit de studie moet worden verwijderd. Bij ieder onderzoek moeten de oogletsels (tabel 1) worden beoordeeld en opgetekend. Ook andere effecten in het oog (bv. pannus, verkleuring, veranderingen in de voorste kamer) en schadelijke systemische effecten worden gerapporteerd. Het onderzoek van de reacties kan worden vergemakkelijkt met behulp van een binoculaire loep, een hand-spleetlamp, een biomicroscoop of andere geschikte instrumenten. Na de registratie van de observaties na 24 uur kunnen de ogen nader worden onderzocht met behulp van fluoresceïne. De inschaling van oogreacties is per definitie subjectief. Om harmonisatie bij de inschaling van oogreacties te bevorderen en ter ondersteuning van de testlaboratoria en de personen die bij het uitvoeren en het interpreteren van de observatie betrokken zijn, moet het personeel dat de observatie uitvoert afdoende zijn opgeleid in het gebruikte scoringsysteem. GEGEVENS EN RAPPORTAGE Evaluatie van de resultaten De scores voor de oogirritatie worden in samenhang met de aard en ernst van het letsel en de vraag of dit al dan niet reversibel is, beoordeeld. De individuele scores vormen geen absolute norm voor de irriterende eigenschappen van een chemische stof, aangezien ook andere effecten van de teststof worden beoordeeld. De individuele scores moeten veeleer als referentiewaarden worden beschouwd en zijn alleen zinvol wanneer ze worden ondersteund door een volledige beschrijving en evaluatie van alle observaties. Testverslag In het testverslag moet de volgende informatie worden opgenomen:
Interpretatie van de resultaten Een extrapolatie van de resultaten van het onderzoek naar oogirritatie bij proefdieren naar de mens heeft slechts een beperkte geldigheid. In veel gevallen is het albinokonijn gevoeliger voor stoffen die oogirritatie of -corrosie veroorzaken dan de mens. Bij de interpretatie van de resultaten moet ervoor worden gezorgd dat irritatie ten gevolge van secundaire infectie wordt uitgesloten. LITERATUUR
Tabel 1 Inschaling van het oogletsel
Aanhangsel DEFINITIES Bewijskracht(proces) : het proces waarbij de sterke en zwakke punten van een verzameling gegevens worden gebruikt om tot een conclusie te komen die niet evident hoeft te zijn op basis van de afzonderlijke gegevens. Chemische stof : een stof of een mengsel. Niet-irriterend : een stof die niet is ingedeeld als voor de ogen irriterende stof van EPA-klasse I, II of III; of als voor de ogen irriterende stof van GHS-klasse 1, 2, 2A of 2B; of van EU-klasse 1 of 2 (17) (18) (19). Teststof : alle volgens deze testmethode geteste stoffen of mengsels. Trapsgewijs testen : een trapsgewijze teststrategie waarbij alle bestaande informatie over een teststof in een bepaalde volgorde wordt herbezien aan de hand van een proces op basis van bewijskracht op elke trap om te bepalen of er voldoende informatie beschikbaar is voor een beslissing tot gevarenclassificatie, vooraleer wordt overgegaan tot de volgende trap. Indien het irritatiepotentieel van een teststof kan worden bepaald op basis van de bestaande informatie, dan zijn bijkomende tests niet vereist. Indien het irritatiepotentieel van een teststof niet kan worden bepaald op basis van de bestaande informatie, dan wordt een trapsgewijze sequentiële testprocedure op dieren uitgevoerd tot een ondubbelzinnige indeling mogelijk is. Voor de ogen corrosieve stof : (a) een chemische stof die irreversibele weefselschade veroorzaakt aan de ogen; (b) een chemische stof die is ingedeeld als voor de ogen irriterende stof van GHS-klasse 1, EPA-klasse I of EU-klasse 1 (17) (18) (19). Voor de ogen hevig irriterende stof : (a) een chemische stof die weefselschade aan het oog veroorzaakt die meer dan 21 dagen na aanbrenging aanhoudt of die ernstige fysische gezichtsvermindering teweegbrengt; (b) een chemische stof die is ingedeeld als voor de ogen irriterende stof van GHS-klasse 1, EPA-klasse I of EU-klasse 1 (17) (18) (19). Voor de ogen irriterende stof : (a) een chemische stof die een reversibele wijziging in het oog teweegbrengt; (b) een chemische stof die is ingedeeld als voor de ogen irriterende stof van EPA-klasse II of III; of als voor de ogen irriterende stof van GHS-klasse 2, 2A of 2B; of van EU-klasse 2 (17) (18) (19). Zuur/alkalireserve : Voor zure preparaten is dit de hoeveelheid (g) natriumhydroxide/100 g preparaat die nodig is om een bepaalde pH te produceren. Voor alkalische preparaten is het de hoeveelheid (g) natriumhydroxide die overeenkomt met het g zwavelzuur/100 g preparaat dat nodig is om een bepaalde pH te produceren (Young et al. 1988). SUPPLEMENT BIJ TESTMETHODE B.5 (4) EEN SEQUENTIËLE TESTSTRATEGIE VOOR OOGIRRITATIE EN -CORROSIE Algemene overwegingen In het belang van verantwoorde wetenschap en het welzijn van dieren is het belangrijk dat het onnodig gebruik van dieren wordt vermeden en dat tests waarvan hevige reacties bij dieren worden verwacht, tot een minimum worden beperkt. Alle informatie over een chemische stof die relevant is voor de mogelijke oogirritatie/corrosie door die stof moet worden geëvalueerd voordat een in-vivotest wordt overwogen. Wellicht bestaat er al voldoende bewijsmateriaal om een teststof qua mogelijke oogirritatie of -corrosie in te delen zonder dat er tests bij proefdieren behoeven te worden uitgevoerd. Daarom zal het gebruik van een bewijskrachtanalyse en een sequentiële teststrategie de noodzaak van in-vivotests tot een minimum beperken, vooral als er van de chemische stof hevige reacties worden verwacht. Er wordt aanbevolen dat een bewijskrachtanalyse wordt gebruikt om bestaande informatie over oogirritatie en -corrosie door chemische stoffen te evalueren en om te bepalen of ander aanvullend onderzoek dan in-vivo-oogonderzoek moet worden uitgevoerd om te helpen bij de bepaling van deze mogelijke effecten. Wanneer nader onderzoek nodig is, wordt aanbevolen de sequentiële teststrategie te gebruiken om de relevante experimentele gegevens te verkrijgen. Voor stoffen die nog niet zijn getest, moet de sequentiële teststrategie worden gebruikt om de gegevens te vergaren die nodig zijn om de oogcorrosie/-irritatie te bepalen. De voorlopige-teststrategie die in dit supplement wordt beschreven, werd in een OESO-workshop ontwikkeld (1). Later is zij bevestigd en opgenomen in het „Harmonised Integrated Hazard Classification System for Human Health and Environmental Effects of Chemical Substances”, die de steun heeft gekregen van de 28e gezamenlijke vergadering van het Comité chemische stoffen en de Werkgroep chemische stoffen in november 1998 (2), en in 2011 is bijgewerkt door een deskundigengroep van de OESO. Hoewel deze teststrategie geen integrerend onderdeel van testmethode B.5 is, vormt zij de aanbevolen aanpak voor de bepaling van de eigenschappen qua oogirritatie/-corrosie. Deze aanpak houdt zowel een optimale gedragscode als een ethische referentie voor in-vivotests op oogirritatie/-corrosie in. De testmethode bevat een leidraad voor de uitvoering van de in-vivotest en geeft een overzicht van de factoren die vóór het overwegen van een dergelijke test in aanmerking moeten worden genomen. De sequentiële teststrategie levert een bewijskrachtbenadering voor de evaluatie van bestaande gegevens over de eigenschappen van chemische stoffen inzake oogirritatie/-corrosie en een trapsgewijze aanpak voor het vergaren van relevante gegevens over chemische stoffen waarvoor nader onderzoek nodig is of waarvoor nog geen onderzoek is uitgevoerd. De strategie omvat de uitvoering eerst van gevalideerde en erkende in-vitro- of ex-vivotests en daarna van testmethode B.4-onderzoeken onder specifieke omstandigheden (3) (4). Beschrijving van de stapsgewijze teststrategie Alvorens te beginnen met tests als onderdeel van de sequentiële teststrategie (zie figuur), moet alle beschikbare informatie worden geëvalueerd om te bepalen of in-vivo-oogonderzoek nodig is. Hoewel de evaluatie van individuele parameters (bv. een extreme pH) significante informatie kan opleveren, moet alle bestaande informatie worden geëvalueerd. Alle relevante gegevens over de effecten van de betrokken chemische stof en de qua structuur analoge verbindingen moeten bij het nemen van een beslissing over de bewijskracht worden geëvalueerd en er moet een motivering voor de beslissing worden gegeven. De nadruk moet in eerste instantie liggen op bestaande gegevens over de effecten van de chemische stof bij mens en dier en vervolgens op de resultaten van in-vitro- of ex-vivotests. Waar mogelijk moet in-vivo-onderzoek aan corrosieve chemische stoffen worden vermeden. Bij de teststrategie spelen de volgende factoren een rol:
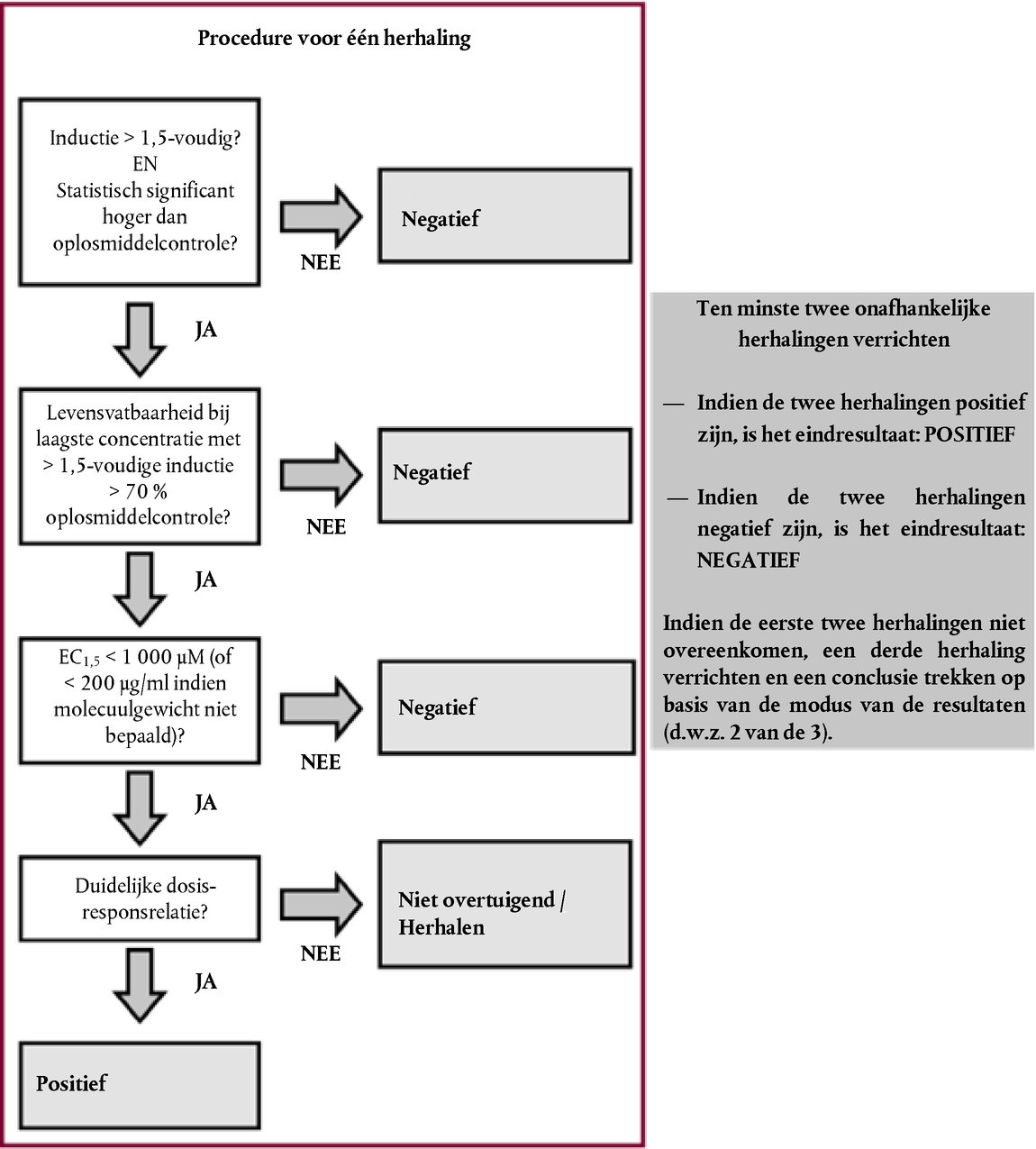
TEST- EN EVALUATIESTRATEGIE VOOR OOGIRRITATIE/-CORROSIE
LITERATUUR
|
|
3) |
In deel B wordt hoofdstuk B.10 vervangen door: „B.10 In-vitrotest op chromosoomafwijkingen in zoogdieren INLEIDING Deze testmethode is gelijkwaardig aan testrichtlijn (TG) 473 (2016) van de OESO. Zij maakt deel uit van een reeks testmethoden op het gebied van de genetische toxicologie. Er is een OESO-document opgesteld met beknopte informatie over genetisch-toxicologische tests en een overzicht van de recente wijzigingen van deze testrichtlijnen (1). De in-vitrotest op chromosoomafwijkingen is bedoeld om te bepalen welke chemische stoffen structurele chromosoomafwijkingen bij gekweekte cellen van zoogdieren veroorzaken (2) (3) (4). Er zijn twee soorten structurele afwijkingen: het chromosoomtype en het chromatidetype. Polyploïdie (met inbegrip van endoreduplicatie) kan optreden in bepalingen van chromosoomafwijkingen in vitro. Hoewel aneugenen polyploïdie kunnen induceren, wijst polyploïdie alleen niet op een aneugeen potentieel en kan het ook wijzen op een verstoring van de celcyclus of op cytotoxiciteit (5). Deze test is niet bedoeld voor het meten van aneuploïdie. Voor de opsporing van aneuploïdie wordt een in-vitromicronucleustest (6) aanbevolen. Bij de in-vitrotest op chromosoomafwijkingen kunnen culturen van permanente cellijnen of primaire celculturen van humane of knaagdierenoorsprong worden gebruikt. De gebruikte cellen moeten worden geselecteerd op basis van het groeivermogen in een cultuur, de stabiliteit van het karyotype (met inbegrip van het aantal chromosomen) en de frequentie van spontane chromosoomafwijkingen (7). Momenteel kunnen op basis van de beschikbare gegevens geen harde aanbevelingen worden gedaan, maar de gegevens wijzen erop dat het belangrijk is om bij de beoordeling van chemische gevaren rekening te houden met de p53-status, de genetische (karyotype) stabiliteit, het herstellend vermogen van het DNA en de oorsprong (knaagdier versus mens) van de voor de test geselecteerde cellen. De gebruikers van deze testmethode worden dan ook aangemoedigd om rekening te houden met de invloed van deze en andere celkenmerken op de prestaties van een cellijn bij het opsporen van de inductie van chromosoomafwijkingen, aangezien de kennis op dit gebied voortschrijdt. Gebruikte definities worden gegeven in aanhangsel 1. INLEIDENDE OVERWEGINGEN EN BEPERKINGEN Bij in vitro uitgevoerde tests moet er meestal een exogene metabole activeringsbron worden gebruikt, tenzij de cellen qua metabolisme competent zijn met betrekking tot de teststoffen. Het exogene metabole activeringssysteem kan de omstandigheden in vivo niet volledig nabootsen. Omstandigheden die zouden kunnen leiden tot artefactuele positieve resultaten, dat wil zeggen, chromosoombeschadiging niet veroorzaakt door directe interactie tussen de teststoffen en chromosomen, moeten worden vermeden. Zulke omstandigheden zijn onder andere veranderingen in de pH of de osmolaliteit (8) (9) (10), interactie met de bestanddelen van het medium (11) (12) of een sterke cytotoxiciteit (13) (14) (15) (16). Deze test wordt gebruikt voor het opsporen van chromosoomafwijkingen die het gevolg kunnen zijn van clastogene gebeurtenissen. De analyse van de inductie van chromosoomafwijkingen geschiedt met behulp van cellen in de metafase. Het is dus van groot belang dat cellen het stadium van mitose bereiken, zowel in behandelde als in onbehandelde culturen. Voor gefabriceerde nanomaterialen kunnen specifieke aanpassingen van deze testmethode noodzakelijk zijn, maar deze worden in deze testmethode niet beschreven. Voordat de testmethode wordt gebruikt op een mengsel om gegevens te genereren voor een beoogd regelgevingsdoel, moet worden nagegaan of, en zo ja, waarom deze methode betrouwbare resultaten oplevert voor dat doel. Zulke overwegingen zijn niet nodig wanneer het testen van het mengsel wettelijk vereist is. PRINCIPE VAN DE TEST Culturen van menselijke cellen of andere zoogdiercellen worden blootgesteld aan de teststof met en zonder een exogene metabole activeringsbron, tenzij cellen met een toereikende metaboliseringscapaciteit worden gebruikt (zie punt 13). Op vooraf bepaalde tijdstippen na de blootstelling van de celculturen aan de teststof worden ze met een metafasestopper (bv. colcemid of colchicine) behandeld, geoogst en gekleurd en worden de cellen in de metafase microscopisch onderzocht op de aanwezigheid van afwijkingen in het chromatidetype of het chromosoomtype. BESCHRIJVING VAN DE METHODE Voorbereidingen Cellen Er kan een verscheidenheid aan cellijnen (bv. ovariumcellen van de Chinese hamster (CHO), longcellen van de Chinese hamster V79, longcellen van de Chinese hamster (CHL)/IU, TK6) of primaire cellen, met inbegrip van lymfocyten uit perifeer bloed van de mens of andere zoogdieren, worden gebruikt (7). Voor de keuze van de gebruikte cellijnen moet een wetenschappelijke verantwoording worden gegeven. Wanneer primaire cellen worden gebruikt moet met het oog op het dierenwelzijn worden overwogen om waar mogelijk primaire cellen van menselijke oorsprong te gebruiken en te bemonsteren in overeenstemming met de ethische beginselen en regelgeving voor mensen. Menselijke lymfocyten uit perifeer bloed dienen te worden verkregen van jonge (ongeveer 18-35 jaar oud) niet-rokers zonder bekende ziekten, die voor zover bekend onlangs niet zijn blootgesteld aan genotoxische stoffen (bv. chemische stoffen, ioniserende straling) op een niveau dat de achtergrondincidentie van chromosoomafwijkingen vergroot. Dit zorgt ervoor dat de achtergrondincidentie van chromosoomafwijkingen laag en consistent is. De basisincidentie van chromosoomafwijkingen neemt toe met de leeftijd, en deze trend is bij vrouwen sterker dan bij mannen (17) (18). Indien cellen van meer dan één donor voor gebruik worden gepoold, moet het aantal donors aangegeven worden. Er moet worden aangetoond dat de cellen zich vanaf het begin van de behandeling met de teststof tot aan de bemonstering van de cellen hebben gedeeld. De celculturen worden in een fase van exponentiële celgroei (cellijnen) gehouden of worden gestimuleerd om zich te delen (primaire culturen van lymfocyten), om de cellen in verschillende stadia van de celcyclus bloot te stellen, aangezien de gevoeligheid van de verschillende celstadia voor de teststoffen mogelijk niet bekend is. De primaire cellen die met mitogene middelen moeten worden gestimuleerd om zich te delen, zijn doorgaans niet meer gesynchroniseerd tijdens de blootstelling aan de teststof (bv. menselijke lymfocyten na 48 uur mitogene stimulatie). Het gebruik van gesynchroniseerde cellen tijdens de behandeling wordt niet aangeraden, maar kan aanvaardbaar zijn, mits het naar behoren wordt gemotiveerd. Media en cultuuromstandigheden Er moet worden gezorgd voor geschikte kweekmedia en incubatieomstandigheden voor de culturen (kweekvaten, vochtige atmosfeer van 5 % CO2 indien passend, incubatietemperatuur van 37 °C). De cellijnen moeten geregeld worden gecontroleerd om na te gaan of het modale aantal chromosomen stabiel is en er geen besmetting met mycoplasma heeft plaatsgevonden (7) (19). Indien er wel een besmetting heeft plaatsgevonden of indien het modale aantal chromosomen gewijzigd is, mogen de cellen niet worden gebruikt. De normale tijd voor de celcyclus bij de in het testlaboratorium gebruikte cellijnen of primaire culturen moet worden vastgesteld en moet overeenkomen met de gepubliceerde celkenmerken (20). Prepareren van de culturen Cellijnen: cellen uit stamculturen worden met een zodanige dichtheid in een kweekmedium geënt dat de cellen in suspensies of in monolagen tot de oogsttijd exponentieel zullen blijven groeien (voor cellen die in monolagen groeien, moet bijvoorbeeld samenvloeiing worden voorkomen). Lymfocyten: met een stollingsremmer (bv. heparine) behandeld volledig bloed of geïsoleerde lymfocyten worden gekweekt (bv. gedurende 48 uur voor menselijke lymfocyten) in aanwezigheid van een mitogeen [bv. fytohemagglutinine (PHA) voor menselijke lymfocyten] om celdeling te induceren vóór blootstelling aan de teststof. Metabolische activering Exogene metabole activeringssystemen moeten worden gehanteerd bij het gebruik van cellen met ontoereikende endogene metabole capaciteit. Het meest gebruikte systeem dat als standaard wordt aanbevolen, tenzij anders wordt gemotiveerd, is een post-mitochondriale fractie aangevuld met een cofactor (S9), verkregen uit de lever van knaagdieren (doorgaans ratten) die zijn behandeld met enzym-inducerende stoffen als Aroclor 1254 (21) (22) (23) of een combinatie van fenobarbiton en β-naftoflavon (24) (25) (26) (27) (28) (29). De laatste combinatie druist niet in tegen het Verdrag van Stockholm inzake persistente organische verontreinigende stoffen (30). Bovendien is aangetoond dat zij even effectief is als Aroclor 1254 voor de inducering van oxidasen met gemengde functie (24) (25) (26) (28). De S9-fractie wordt meestal gebruikt bij een concentratie van 1 tot 2 % (v/v), maar mag worden verhoogd tot 10 % (v/v) in het uiteindelijke testmedium. Het gebruik van producten die de mitotische index verlagen, in het bijzonder calciumcomplex-vormende producten (31), moet worden vermeden tijdens de behandeling. De keuze van het type en de concentratie van het exogene metabole activeringssysteem dat of de metabole inductor die wordt gebruikt, kan worden beïnvloed door de klasse van de te testen chemische stof. Teststofbereiding Vaste teststoffen moeten in geschikte oplosmiddelen worden bereid en eventueel vóór de behandeling van de cellen worden verdund (zie punt 23). Vloeibare teststoffen kunnen direct aan het testsysteem worden toegevoegd en/of worden verdund vóór de behandeling van het testsysteem. Bij het testen van gassen of vluchtige teststoffen moeten geschikte aanpassingen van de standaardprotocollen worden gevolgd, zoals de behandeling in gesloten kweekvaten (32) (33) (34). Preparaten van de teststof moeten worden bereid vlak vóór de behandeling, tenzij met stabiliteitsgegevens is aangetoond dat bewaren van het preparaat aanvaardbaar is. Testomstandigheden Oplosmiddelen Het oplosmiddel wordt zo gekozen dat het de oplosbaarheid van de te testen chemische stoffen vergroot zonder een negatieve invloed te hebben op het verloop van de test, bv. door veranderingen in de celgroei, aantasting van de integriteit van de teststof, reacties met kweekvaten, belemmering van het metabole activeringssysteem. Het wordt aanbevolen waar mogelijk eerst het gebruik van een waterig oplosmiddel (of kweekmedium) te overwegen. Gangbare oplosmiddelen zijn bijvoorbeeld water en dimethylsulfoxide. Organische oplosmiddelen mogen in de regel de 1 % (v/v) en waterige oplosmiddelen (fysiologisch zout of water) de 10 % (v/v) in het uiteindelijke behandelingsmedium niet overschrijden. Als er andere dan gangbare oplosmiddelen worden gebruikt (bv. ethanol of aceton), moeten gegevens worden verstrekt waaruit blijkt dat ze verenigbaar zijn met de teststof en het testsysteem, en dat zij geen genetische toxiciteit vertonen bij de gebruikte concentratie. Als die gegevens ontbreken, is het belangrijk ook onbehandelde controles mee te nemen (zie aanhangsel 1) om aan te tonen dat het gekozen oplosmiddel geen schadelijke of clastogene effecten induceert. Meting van celproliferatie en cytotoxiciteit en keuze van behandelingsconcentraties Bij de bepaling van de hoogste te testen concentratie van de teststof moeten concentraties worden vermeden die kunnen leiden tot artefactuele positieve reacties, zoals reacties die bovenmatige cytotoxiciteit (zie punt 22) voortbrengen, neerslag in het cultuurmedium (zie punt 23) of uitgesproken wijzigingen in pH of osmolaliteit (zie punt 5). Indien de teststof op het ogenblik van toevoeging een uitgesproken wijziging in de pH van het medium veroorzaakt, kan de pH worden aangepast door het uiteindelijke behandelingsmedium te bufferen zodat artefactuele positieve reacties worden vermeden en passende kweekomstandigheden in stand worden gehouden. Metingen van celproliferatie worden verricht om er zeker van te zijn dat een voldoende aantal behandelde cellen tijdens de test mitose hebben ondergaan en dat de behandelingen worden uitgevoerd met de passende mate van cytotoxiciteit (zie punten 18 en 22). Cytotoxiciteit dient te worden bepaald met en zonder metabole activering in het hoofdexperiment met behulp van een passende indicatie van celdood en celgroei. Hoewel de evaluatie van cytotoxiciteit in een eerste test nuttig kan zijn om de in het hoofdexperiment te gebruiken concentraties beter te kunnen vaststellen, is een eerste test niet verplicht. Indien deze wel wordt uitgevoerd, mag deze de meting van cytotoxiciteit in het hoofdexperiment niet vervangen. De relatieve populatieverdubbeling (RPD) of de relatieve toename van de celpopulatie (RICC) zijn passende methoden voor het beoordelen van cytotoxiciteit in cytogene tests (13) (15) (35) (36) (55) (zie aanhangsel 2 voor formules). In geval van een langlopende behandeling en bemonsteringstijdstippen na het begin van de behandeling langer dan 1,5 maal de normale lengte van een celcyclus (d.w.z. in totaal langer dan 3 maal de lengte van een celcyclus), kan RPD mogelijk leiden tot een onderschatting van de cytotoxiciteit (37). Onder deze omstandigheden zou RICC een betere maat kunnen zijn of zou de evaluatie van de cytotoxiciteit na 1,5 maal de normale lengte van een celcyclus met hulp van RPD een bruikbare schatting kunnen zijn. Voor lymfocyten in primaire culturen is de mitotische index (MI) weliswaar een maat voor cytotoxische/cytostatische effecten, maar deze index is afhankelijk van het tijdstip na de behandeling waarop hij wordt gemeten, van het gebruikte mitogeen en van mogelijke ontregeling van de celcyclus. De MI is echter aanvaardbaar, omdat andere metingen van de cytotoxiciteit omslachtig en onpraktisch kunnen zijn en mogelijk niet geldig zijn voor de doelpopulatie van lymfocyten die groeit als gevolg van PHA-stimulatie. Terwijl RICC en RPD voor cellijnen en MI voor primaire culturen van lymfocyten de aanbevolen cytotoxiciteitsparameters zijn, kunnen andere indicatoren (bv. celintegriteit, apoptose, necrose, celcyclus) nuttige aanvullende informatie verschaffen. Er moeten ten minste drie testconcentraties (exclusief het oplosmiddel en positieve controles) die voldoen aan de aanvaardbaarheidscriteria (passende mate van cytotoxiciteit, aantal cellen enz.) worden geëvalueerd. Ongeacht het type cellen (cellijnen of primaire culturen van lymfocyten) kan bij elke geteste concentratie één behandelde cultuur worden gebruikt, maar ook duploculturen. Hoewel het gebruik van duploculturen raadzaam is, is het gebruik van één cultuur ook aanvaardbaar, mits voor één cultuur en duploculturen hetzelfde totale aantal cellen wordt gescoord. Het gebruik van één cultuur is in het bijzonder relevant wanneer meer dan 3 concentraties worden beoordeeld (zie punt 31). Voor de gegevensanalyse kunnen de resultaten verkregen in de onafhankelijke duploculturen bij een gegeven concentratie worden gepoold (38). Voor teststoffen die weinig of geen cytotoxiciteit vertonen, zijn intervallen tussen de testconcentraties van ongeveer een factor 2 tot 3 doorgaans passend. Wanneer er cytotoxiciteit optreedt, moeten de geselecteerde testconcentraties een interval bestrijken van een concentratie waarbij cytotoxiciteit optreedt zoals beschreven in punt 22, tot en met concentraties waarbij er matige en geen of vrijwel geen cytotoxiciteit is. Veel teststoffen vertonen een steile concentratie-responscurve, en om gegevens bij lage en matige cytotoxiciteit te verkrijgen of om in detail de dosis-responsrelatie te bestuderen zullen dichter bij elkaar liggende concentraties en/of meer dan drie concentraties moeten worden gebruikt (één cultuur of duplobepalingen), in het bijzonder in situaties waarin een herhalingsexperiment vereist is (zie punt 47). Als de maximale concentratie is gebaseerd op cytotoxiciteit, moet de hoogste concentratie zo worden gekozen dat met behulp van de aanbevolen cytotoxiciteitsparameters (d.w.z. afname van RICC en RPD voor cellijnen en verlaging van MI voor primaire culturen van lymfocyten tot 45 ± 5 % van de tegelijkertijd gemeten negatieve controle) 55 ± 5 % cytotoxiciteit wordt bereikt. Bij de interpretatie van positieve resultaten moet ervoor worden gezorgd dat deze niet alleen worden gevonden in het hogere deel van dit 55 ± 5 % cytotoxiciteitsbereik (13). Voor slecht oplosbare teststoffen die bij lagere concentraties dan de oplosbaarheidsgrens niet cytotoxisch zijn, moet de hoogste geanalyseerde concentratie troebelheid of een met het oog of met behulp van een omgekeerde microscoop aan het eind van de behandeling met de teststof zichtbaar neerslag opleveren. Zelfs indien cytotoxiciteit optreedt boven de oplosbaarheidsgrens, is het raadzaam de test uit te voeren bij slechts één concentratie die troebelheid of een zichtbaar neerslag oplevert, omdat het neerslag kan leiden tot artefactuele effecten. Bij de concentratie die neerslag oplevert, moet ervoor worden gezorgd dat het neerslag de uitvoering van de test (bv. kleuring of scoring) niet verstoort. Het kan nuttig zijn om voorafgaand aan het experiment de oplosbaarheid in het kweekmedium te bepalen. Indien geen neerslag of beperkende cytotoxiciteit wordt waargenomen, moet de hoogste testconcentratie overeenkomen met de laagste van de volgende waarden: 10 mM, 2 mg/ml of 2 μl/ml (39) (40) (41). Wanneer de samenstelling van de teststof niet vastgesteld is, bv. in geval van een stof van onbekende of variabele samenstelling, complexe reactieproducten of biologisch materiaal (UVCB) (42), een milieu-extract enz., moet de hoogste concentratie bij het ontbreken van voldoende cytotoxiciteit mogelijk hoger zijn (bv. 5 mg/ml) om de concentratie van elk van de componenten te verhogen. Er zij echter opgemerkt dat deze eisen voor geneesmiddelen voor menselijk gebruik anders kunnen zijn (43). Controles Voor elk oogsttijdstip dienen gelijktijdige negatieve controles (zie punt 15) in het experiment te worden opgenomen, waaraan uitsluitend het oplosmiddel wordt toegediend en die verder op dezelfde manier worden behandeld als de culturen met teststof. Gelijktijdige positieve controles zijn nodig om aan te tonen dat het laboratorium in staat is om onder de omstandigheden van het gebruikte testprotocol en de doeltreffendheid van het exogene metabole activeringssysteem, indien van toepassing, clastogenen te identificeren. Voorbeelden van voor positieve controle gebruikte chemische stoffen worden gegeven in tabel 1 hieronder. Er kunnen ook alternatieve positieve controlestoffen worden gebruikt, indien dat wordt gemotiveerd. Omdat in-vitrotests op genetische toxiciteit bij zoogdiercellen voldoende gestandaardiseerd zijn, mag het gebruik van positieve controles worden beperkt tot een clastogeen waarvoor metabole activering vereist is. Mits dit gelijktijdig met de niet-geactiveerde test met dezelfde behandelingsduur plaatsvindt, zal deze ene positieve-controlerespons zowel de activiteit van het metabole activeringssysteem als de gevoeligheid van het testsysteem aantonen. Een langlopende behandeling (zonder S9) moet echter haar eigen positieve controle hebben, omdat de behandelingsduur zal afwijken van de test waarbij metabole activering wordt toegepast. Elke positieve controle moet worden gebruikt bij een of meer concentraties waarvoor een reproduceerbare en detecteerbare stijging ten opzichte van het achtergrondniveau kan worden verwacht, om de gevoeligheid van het testsysteem aan te tonen (d.w.z. de effecten zijn duidelijk maar de gecodeerde objectglaasjes zijn niet onmiddellijk als zodanig herkenbaar), en de respons dient niet in het gedrang te komen door een mate van cytotoxiciteit die de in de testmethode gespecificeerde grenzen overschrijdt. Tabel 1. Aanbevolen referentiestoffen voor het beoordelen van de bekwaamheid van laboratoria en voor het selecteren van positieve controles.
PROCEDURE Behandeling met teststof Delende cellen worden zowel met als zonder metabool activeringssysteem met de teststof behandeld. Oogsttijdstippen Voor een gedegen evaluatie, die nodig is om een negatief resultaat vast te stellen, moet het experiment onder elk van de drie volgende omstandigheden worden verricht, met een kortlopende behandeling zowel met als zonder metabole activering en een langlopende behandeling zonder metabole activering (zie de punten 43, 44 en 45):
Indien een van de hierboven genoemde experimentele omstandigheden tot een positieve respons leidt, hoeven een of meer van de andere behandelingsregimes mogelijk niet te worden onderzocht. Chromosoompreparaten De celculturen worden meestal gedurende één tot drie uur vóór het oogsten behandeld met colcemid of colchicine. Elke celcultuur wordt afzonderlijk geoogst en behandeld voor het maken van chromosoompreparaten. Dit houdt in dat de cellen een hypotone behandeling krijgen, gevolgd door fixatie en kleuring. In monolagen kunnen aan het einde van de 3-6 uur durende behandeling mitotische cellen (herkenbaar aan hun ronde vorm en het feit dat ze loslaten van het oppervlak) aanwezig zijn. Omdat deze mitotische cellen gemakkelijk loskomen, kunnen ze verloren gaan wanneer het medium met de teststof wordt verwijderd. Als er aanwijzingen zijn voor een aanzienlijke toename van het aantal mitotische cellen vergeleken met de controlegroepen, wat een aanwijzing is voor het waarschijnlijk plotseling stoppen van de mitose, moeten de cellen door centrifugering worden verzameld en opnieuw aan de cultuur worden toegevoegd, opdat er geen cellen die in mitose zijn, en die gevaar voor chromosoomafwijking lopen, op het oogsttijdstip verloren gaan. Analyse Alle objectglaasjes, ook die van de positieve en negatieve controles, worden vóór de microscopische analyse op chromosoomafwijkingen onafhankelijk gecodeerd. Aangezien bij de fixatieprocedures vaak een gedeelte van de metafasecellen chromosomen verliest, moeten de gescoorde cellen een aantal centromeren bevatten dat gelijk is aan het modale aantal +/- 2. Per concentratie en controle moeten minimaal 300 goed gespreide metafasen worden gescoord om te concluderen dat een teststof duidelijk negatief is (zie punt 45). De 300 cellen moeten gelijkelijk worden verdeeld over de duplobepalingen, wanneer duploculturen worden gebruikt. Wanneer één cultuur per concentratie wordt gebruikt (zie punt 21), moeten minimaal 300 goed gespreide metafasen worden gescoord in deze ene cultuur. Het scoren van 300 cellen heeft als voordeel dat het het statistisch onderscheidingsvermogen van de test vergroot en nulwaarden bovendien zelden zullen worden waargenomen (naar verwachting slechts 5 %) (44). Het aantal te scoren metafasen kan worden verlaagd wanneer hoge aantallen cellen met chromosoomafwijkingen worden waargenomen en de teststof als duidelijk positief wordt beschouwd. Cellen met een of meer structurele chromosoomafwijkingen inclusief en exclusief hiaten moeten worden gescoord. Breuken en hiaten zijn gedefinieerd in aanhangsel 1, conform (45) (46). Afwijkingen in het chromatidetype of het chromosoomtype moeten afzonderlijk worden opgetekend en ingedeeld in subtypen (breuken, uitwisselingen). De in het laboratorium gebruikte procedures moeten ervoor zorgen dat de analyse van chromosoomafwijkingen wordt uitgevoerd door goed opgeleide scorers en zo nodig collegiaal wordt getoetst. Hoewel de test bedoeld is om structurele chromosoomafwijkingen op te sporen, is het belangrijk dat bij waarneming van polyploïdie en endoreduplicatie ook de frequenties van deze verschijnselen worden opgetekend. (Zie punt 2). Bekwaamheid van het laboratorium Om voldoende ervaring met de test vast te stellen voordat deze wordt gebruikt voor routinetests, moet het laboratorium een reeks experimenten met positieve referentiestoffen die via verschillende mechanismen werken, en met verschillende negatieve controles (met verschillende oplosmiddelen/media) hebben uitgevoerd. Deze positieve en negatieve controleresponsen moeten in overeenstemming zijn met de literatuur. Dit geldt niet voor laboratoria die ervaring hebben, d.w.z. die beschikken over referentiegegevens uit het verleden zoals omschreven in punt 37. Een selectie van positieve controlestoffen (zie tabel 1 in punt 26) moet worden onderzocht met kortlopende en langlopende behandelingen zonder metabole activering, en ook met korte behandelingen met metabole activering, teneinde bekwaamheid in het detecteren van clastogene chemische stoffen aan te tonen en de doeltreffendheid van het metabole activeringssysteem te bepalen. Er moet een bereik van concentraties van de geselecteerde chemische stoffen worden gekozen dat reproduceerbare en concentratiegerelateerde stijgingen tot boven het achtergrondniveau oplevert, om de gevoeligheid en het dynamisch bereik van het testsysteem aan te tonen. Controlegegevens uit het verleden Het laboratorium stelt vast:
Wanneer voor het eerst gegevens worden verkregen voor een verdeling van negatieve controles uit het verleden, moeten gelijktijdige negatieve controles consistent zijn met gepubliceerde controlegegevens, indien deze bestaan. Naarmate meer gegevens uit experimenten aan de controlespreiding worden toegevoegd, dienen gelijktijdige negatieve controles idealiter binnen de controlegrens van 95 % voor die spreiding te vallen (44) (47). De databank met gegevens over negatieve controles uit het verleden van het laboratorium dient aanvankelijk te worden samengesteld op basis van minimaal 10 experimenten, maar bestaat bij voorkeur uit ten minste 20 experimenten die onder vergelijkbare testomstandigheden werden uitgevoerd. Laboratoria dienen methodes voor kwaliteitscontrole, zoals regelkaarten (bv. C-kaart of X-kaart (48)), te gebruiken om na te gaan hoe variabel hun gegevens over positieve en negatieve controles zijn en om aan te tonen dat zij de methodologie “onder controle” hebben (44). Verdere aanbevelingen voor het samenstellen en gebruiken van de gegevens uit het verleden (d.w.z. criteria voor het opnemen van gegevens in en het uitsluiten ervan uit de historische gegevens en de aanvaardbaarheidscriteria voor een gegeven experiment) zijn te vinden in de literatuur (47). Bij eventuele wijzigingen in het experimentele protocol moet rekening worden gehouden met hun consistentie met de bestaande databanken met historische controlegegevens van het laboratorium. Eventuele grote inconsistenties moeten leiden tot de oprichting van een nieuwe databank met historische controlegegevens. Negatieve controlegegevens moeten bestaan uit de incidentie van cellen met chromosoomafwijkingen uit één cultuur of de som van de duploculturen zoals beschreven in punt 21. Gelijktijdige negatieve controles liggen idealiter binnen de 95 %-controlegrenzen van de verdeling van de databank met historische negatieve controlegegevens van het laboratorium (44) (47). Wanneer gegevens van gelijktijdige negatieve controles buiten de 95 %-controlegrenzen vallen, kan opneming ervan in de historische controleverdeling toch aanvaardbaar zijn, zolang deze gegevens geen extreme uitschieters zijn en er bewijs is dat het testsysteem 'onder controle' is (zie punt 37) en geen bewijs van technisch of menselijk falen. GEGEVENS EN RAPPORTAGE Presentatie van de resultaten Het percentage cellen met een of meer structurele chromosoomafwijkingen moet worden geëvalueerd. Er moet een uitgesplitst overzicht worden gegeven van de afwijkingen in het chromatidetype en chromosoomtype ingedeeld in subtypen (breuken, uitwisselingen), met de aantallen en de frequentie waarmee ze in de behandelde en controleculturen voorkomen. Hiaten worden apart opgetekend en gerapporteerd, maar meestal niet in de totale frequentie van afwijkingen opgenomen. Het percentage polyploïde cellen en/of cellen met endoreduplicatie wordt gerapporteerd wanneer dergelijke cellen worden waargenomen. Gelijktijdige metingen van de cytotoxiciteit voor alle behandelde, negatieve en positieve controleculturen bij het (de) hoofdexperiment(en) dienen te worden geregistreerd. De gegevens moeten voor elke cultuur apart worden verstrekt. Daarnaast moet een overzicht van alle gegevens in tabelvorm worden gegeven. Aanvaardbaarheidscriteria Aanvaarding van een test geschiedt aan de hand van de volgende criteria:
Beoordeling en interpretatie van resultaten Indien aan alle aanvaardbaarheidscriteria is voldaan, wordt de teststof als duidelijk positief beschouwd als in een of meer van de onderzochte experimentele omstandigheden (zie punt 28):
Wanneer aan al deze criteria is voldaan, wordt de teststof in staat geacht in gekweekte zoogdiercellen in dit testsysteem chromosoomafwijkingen te veroorzaken. Aanbevelingen voor de meest geschikte statistische methoden zijn te vinden in de literatuur (49) (50) (51). Indien aan alle aanvaardbaarheidscriteria is voldaan, wordt de teststof als duidelijk negatief beschouwd als in alle onderzochte experimentele omstandigheden (zie punt 28):
De teststof wordt dan niet niet in staat geacht in gekweekte zoogdiercellen in dit testsysteem chromosoomafwijkingen te veroorzaken. Een duidelijk positieve of negatieve reactie behoeft niet te worden bevestigd. Indien de reactie noch duidelijk negatief noch duidelijk positief is zoals hierboven beschreven, of om de biologische relevantie van een resultaat te helpen vaststellen, moeten de gegevens door een deskundige en/of nader onderzoek worden beoordeeld. Het scoren van aanvullende cellen (indien van toepassing) of het uitvoeren van een herhalingsexperiment onder mogelijk aangepaste experimentele omstandigheden (bv. concentratie-intervallen, andere omstandigheden bij metabole activering (d.w.z. S9-concentratie of S9-herkomst)) kan nuttig zijn. In zeldzame gevallen zal op basis van de gegevens, zelfs na nader onderzoek, niet kunnen worden geconcludeerd dat de resultaten positief of negatief zijn. De reactie op de teststof zal daarom als moeilijk te interpreteren worden beschouwd. Een stijging van het aantal polyploïde cellen kan erop wijzen dat de teststoffen in staat zijn mitotische processen te remmen en numerieke chromosoomafwijkingen te induceren (52). Een stijging van het aantal cellen met endoreduplicatie van chromosomen kan erop duiden dat de teststoffen in staat zijn de voortgang van de celcyclus te remmen (53) (54) (zie punt 2). De incidentie van polyploïde cellen en cellen met endoreduplicatie van chromosomen moet daarom apart worden geregistreerd. Testverslag In het testverslag moet de volgende informatie worden opgenomen:
LITERATUUR
Aanhangsel 1 DEFINITIES Afwijking van het chromatidetype : structurele chromosoombeschadiging waarbij breuken in individuele chromatiden ontstaan, eventueel gevolgd door recombinatie. Afwijking van het chromosoomtype : structurele chromosoombeschadiging waarbij breuken in beide chromatiden op dezelfde plaats ontstaan, eventueel gevolgd door recombinatie. Aneuploïdie : elke afwijking van het normale diploïde (of haploïde) aantal chromosomen met één chromosoom of meer dan één chromosoom, maar niet met (een) volledige reeks(en) chromosomen (polyploïdie). Apoptose : geprogrammeerde celdood, gekenmerkt door een reeks stappen die leiden tot de afbraak van cellen in door membraan samengehouden deeltjes die vervolgens worden geëlimineerd door fagocytose of door verwijdering. Breuk in een chromatide : discontinuïteit van één chromatide waarbij sprake is van een duidelijke fout in de uitlijning van een van de chromatiden. Celproliferatie : toename van het aantal cellen als gevolg van mitose. Chemische stof : een stof of mengsel. Clastogeen : chemische stof die structurele chromosoomafwijkingen veroorzaakt in celpopulaties of eukaryotische organismen. Concentraties : verwijzen naar uiteindelijke concentraties van de teststof in het kweekmedium. Cytotoxiciteit : Voor de bepalingen bestreken in deze testmethode met cellijnen is cytotoxiciteit gelijkgesteld aan een vermindering van de relatieve populatieverdubbeling (RPD) of relatieve toename van de celpopulatie (RICC) van de behandelde cellen vergeleken met de negatieve controle (zie punt 17 en aanhangsel 2). Voor de bepalingen bestreken in deze testmethode met primaire culturen van lymfocyten is cytotoxiciteit gelijkgesteld aan een vermindering van de mitotische index (MI) van de behandelde cellen vergeleken met de negatieve controle (zie punt 18 en aanhangsel 2). Endoreduplicatie : een proces waarbij de kern na een S-fase met DNA-replicatie niet tot mitose overgaat, maar opnieuw een S-fase begint. Het resultaat is chromosomen met 4, 8, 16, ... chromatiden. Genotoxisch : een algemene term die alle typen DNA- of chromosoombeschadiging omvat, waaronder breuken, deleties, adducten, veranderingen en bindingen van nucleotiden, herschikkingen, genmutaties, chromosoomafwijkingen en aneuploïdie. Niet alle soorten genotoxische effecten leiden tot mutaties of stabiele chromosoombeschadiging. Hiaat in een chromatide : niet-verkleurend gebied (een achromatische beschadiging) van één chromatide waarbij sprake is van een minimale fout in de uitlijning van de chromatiden. Mitose : deling van de celkern die doorgaans wordt ingedeeld in profase, prometafase, metafase, anafase en telofase. Mitotische index (MI) : de verhouding tussen het aantal cellen in de metafase en het totale aantal cellen in een populatie; deze index geeft een indicatie van de snelheid waarmee deze populatie zich voortplant. Mutageen : produceert een erfelijke wijziging van DNA-basepaarsequentie(s) in genen of van de structuur van chromosomen (chromosoomafwijkingen). Numerieke afwijking : een verandering in het aantal chromosomen ten opzichte van het normale aantal dat kenmerkend is voor de gebruikte cellen. Onbehandelde controles : culturen die geen behandeling ondergaan (d.w.z. geen teststof en geen oplosmiddel) maar gelijktijdig met en op dezelfde wijze worden verwerkt als de culturen die de teststof ontvangen. Oplosmiddelcontrole : algemene term om de controleculturen aan te duiden die uitsluitend het oplosmiddel ontvangen dat wordt gebruikt om de teststof op te lossen. p53-status : Het proteïne p53 is betrokken bij de regulering van de celcyclus, apoptose en het herstel van het DNA. Cellen met een deficiëntie aan functioneel proteïne p53, die niet in staat zijn om in antwoord op beschadigingen van het DNA de celcyclus tegen te houden of beschadigde cellen te elimineren via apoptose of andere mechanismen (bv. inductie van DNA-herstel) die verband houden met p53-functies, zouden in theorie vatbaarder moeten zijn voor genmutaties of chromosoomafwijkingen. Polyploïdie : afwijkingen in het aantal chromosomen in cellen of organismen waarbij (een) volledige reeks(en) van chromosomen betrokken is (zijn), in tegenstelling tot een afzonderlijk chromosoom of afzonderlijke chromosomen (aneuploïdie). Relatieve populatieverdubbeling (RPD) : de stijging van het aantal populatieverdubbelingen in aan een chemische stof blootgestelde culturen versus de stijging in niet-behandelde culturen, een ratio uitgedrukt als een percentage. Relatieve toename van de celpopulatie (RICC) : de stijging van het aantal cellen in aan een chemische stof blootgestelde culturen versus de stijging in niet-behandelde culturen, een ratio uitgedrukt als een percentage. S9-leverfractie : supernatant van leverhomogenaat na centrifugering bij 9 000 g, d.w.z. ruw leverextract. S9-mix : mix van de S9-leverfractie en cofactoren die nodig zijn voor de metabole enzymactiviteit. Structurele afwijking : een verandering in de chromosoomstructuur die bij microscopisch onderzoek tijdens de metafase van de celdeling kan worden waargenomen in de vorm van deleties, fragmenten en intra- of interchromosomale uitwisselingen. Teststof : alle volgens deze testmethode geteste stoffen of mengsels. Aanhangsel 2 FORMULES VOOR BEOORDELING VAN DE CYTOTOXICITEIT Mitotische index (MI):
Relatieve toename van de celpopulatie (RICC) of Relatieve populatieverdubbeling (RPD) wordt aanbevolen, aangezien beide rekening houden met het percentage van de celpopulatie dat zich gedeeld heeft.
waarbij: Populatieverdubbeling = [log (aantal cellen na behandeling ÷ aanvankelijk aantal cellen)] ÷ log 2 Voorbeeld: een RICC, of een RPD van 53 % duidt op 47 % cytotoxiciteit/cytostase en 55 % cytotoxiciteit/cytostase gemeten door MI betekent dat de werkelijke MI 45 % van de controle is. Hoe dan ook moet het aantal cellen vóór behandeling worden gemeten, en hetzelfde zijn voor behandelde en negatieve controleculturen. Terwijl RCC (d.w.z. aantal cellen in behandelde culturen / aantal cellen in controleculturen) in het verleden werd gebruikt als cytotoxiciteitsparameter, wordt dit niet langer aanbevolen, aangezien RCC de mate van cytotoxiciteit kan onderschatten. In de negatieve controleculturen moet de populatieverdubbeling verenigbaar zijn met de eis cellen na behandeling te bemonsteren op een tijdstip gelijk aan ongeveer 1,5 maal de normale lengte van een celcyclus, en de mitotische index moet hoog genoeg zijn om een voldoende aantal cellen in mitose te verkrijgen en op betrouwbare wijze een vermindering van 50 % te berekenen. |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
4) |
In deel B wordt hoofdstuk B.11 vervangen door: „B.11 Test op chromosoomafwijkingen in beenmergcellen van zoogdieren INLEIDING Deze testmethode is gelijkwaardig aan testrichtlijn (TG) 475 (2016) van de OESO. Zij maakt deel uit van een reeks testmethoden op het gebied van de genetische toxicologie. Er is een OESO-document opgesteld met beknopte informatie over genetisch-toxicologische tests en een overzicht van de recente wijzigingen van deze testrichtlijnen (1). De in-vivotest op chromosoomafwijkingen in beenmergcellen van zoogdieren is met name van belang voor de beoordeling van genotoxiciteit, omdat factoren van het in-vivometabolisme, de farmacokinetiek en de DNA-herstelprocessen actief zijn en bijdragen aan de reacties op de test, hoewel deze aspecten per soort kunnen verschillen. Een in-vivobepaling is ook geschikt om in vitro gesignaleerde genotoxiciteit nader te onderzoeken. De in-vivotest op chromosoomafwijkingen bij zoogdieren wordt gebruikt om te bepalen welke chemische stoffen structurele chromosoomafwijkingen bij beenmergcellen van dieren, meestal knaagdieren, veroorzaken (2) (3) (4) (5). Er zijn twee soorten structurele chromosoomafwijkingen: het chromosoomtype en het chromatidetype. Hoewel de meeste genotoxische door chemische stoffen geïnduceerde afwijkingen van het chromatidetype zijn, komt het chromosoomtype ook voor. Chromosoombeschadiging en soortgelijke voorvallen veroorzaken veel genetische ziekten bij de mens en het lijkt er sterk op dat deze beschadigingen en soortgelijke voorvallen, wanneer ze veranderingen in oncogenen en tumorsuppressorgenen veroorzaken, betrokken zijn bij de inductie van kanker bij de mens en bij proefdieren. Polyploïdie (met inbegrip van endoreduplicatie) kan optreden in bepalingen van chromosoomafwijkingen in vivo. Een toename van polyploïdie hoeft op zich echter niet te wijzen op een aneugeen potentieel en kan ook gewoon wijzen op een verstoring van de celcyclus of op cytotoxiciteit. Deze test is niet bedoeld voor het meten van aneuploïdie. Een in-vivomicronucleustest bij erythrocyten van zoogdieren (hoofdstuk B.12 van deze bijlage) of de in-vitromicronucleustest bij zoogdiercellen (hoofdstuk B.49 van deze bijlage) zijn respectievelijk de in-vivo- en in-vitrotests die worden aanbevolen voor de opsporing van aneuploïdie. De gebruikte definities zijn opgenomen in aanhangsel 1. INLEIDENDE OVERWEGINGEN Gewoonlijk worden in deze test knaagdieren gebruikt, maar andere soorten kunnen in sommige gevallen geschikt zijn, indien dat wetenschappelijk gerechtvaardigd is. Het doelweefsel in deze test is het beenmerg aangezien dit een sterk doorbloed weefsel is en een populatie snel delende cellen bevat die gemakkelijk kunnen worden geïsoleerd en verwerkt. De wetenschappelijke rechtvaardiging voor het gebruik van andere soorten dan ratten en muizen moet in het verslag worden opgenomen. Als andere soorten dan knaagdieren worden gebruikt, wordt aanbevolen de meting van de chromosoomafwijkingen in beenmergcellen te integreren in een andere passende toxiciteitstest. Als er aanwijzingen zijn dat de teststof(fen), of een of meer metabolieten daarvan, niet in het doelweefsel terechtkom(t)(en), is deze test mogelijk niet geschikt. Voordat de testmethode wordt gebruikt op een mengsel om gegevens te genereren voor een beoogd regelgevingsdoel, moet worden nagegaan of, en zo ja, waarom deze methode betrouwbare resultaten oplevert voor dat doel. Zulke overwegingen zijn niet nodig wanneer het testen van het mengsel wettelijk vereist is. PRINCIPE VAN DE TESTMETHODE Dieren worden aan de teststof blootgesteld via een passende wijze van blootstelling en worden op een geschikt tijdstip na de behandeling op humane wijze gedood. Voordat de dieren worden gedood, worden ze behandeld met een metafasestopper (bv. colchicine of colcemid). Vervolgens worden chromosoompreparaten van de beenmergcellen gemaakt en gekleurd, en worden de cellen in de metafase geanalyseerd op chromosoomafwijkingen. VERIFICATIE VAN DE BEKWAAMHEID VAN LABORATORIA Bekwaamheidsonderzoek Om voldoende ervaring met het uitvoeren van de bepaling vast te stellen voordat deze wordt gebruikt voor routinetests, moet het laboratorium hebben aangetoond in staat te zijn verwachte resultaten uit gepubliceerde gegevens (bv. (6)) te reproduceren voor frequenties van chromosoomafwijkingen met minimaal twee positieve controlestoffen (waaronder zwakke responsen geïnduceerd door lage doses positieve controles), zoals de stoffen vermeld in tabel 1 en met vergelijkbare medium-/oplosmiddelcontroles (zie punt 22). Bij deze experimenten moeten doses worden gebruikt die leiden tot reproduceerbare en dosisgerelateerde toenamen, en zij moeten de gevoeligheid en het dynamisch bereik van het testsysteem in het te onderzoeken weefsel (beenmerg) aantonen en gebruikmaken van de scoringmethode die binnen het laboratorium zal worden aangewend. Deze eis geldt niet voor laboratoria met ervaring, d.w.z. laboratoria die beschikken over referentiegegevens uit het verleden zoals omschreven in de punten 10-14. Controlegegevens uit het verleden Tijdens het bekwaamheidsonderzoek stelt het laboratorium vast:
Wanneer voor het eerst gegevens worden verkregen voor een verdeling van negatieve controles uit het verleden, moeten gelijktijdige negatieve controles consistent zijn met gepubliceerde controlegegevens, indien deze bestaan. Wanneer meer experimentele gegevens aan de verdeling van historische controles worden toegevoegd, moeten gelijktijdige negatieve controles idealiter binnen de 95 %-controlegrenzen van deze verdeling liggen. De databank met historische negatieve controlegegevens van het laboratorium moet statistisch robuust zijn om te waarborgen dat het laboratorium de verdeling van zijn negatieve controlegegevens kan beoordelen. Uit de literatuur komt naar voren dat daarvoor minimaal 10 experimenten nodig zijn, maar dat bij voorkeur ten minste 20 experimenten worden gebruikt die onder vergelijkbare experimentele omstandigheden zijn uitgevoerd. Laboratoria moeten methoden voor kwaliteitscontrole gebruiken, zoals controlekaarten (bv. C-kaarten of X-kaarten (7)), om vast te stellen hoe variabel hun gegevens zijn en om aan te tonen dat het laboratorium de methodologie “onder controle” heeft. Verdere aanbevelingen voor het opbouwen en gebruiken van de historische gegevens (d.w.z. criteria voor het opnemen van gegevens in en het uitsluiten ervan uit de historische gegevens en de aanvaardbaarheidscriteria voor een gegeven experiment) zijn te vinden in de literatuur (8). Wanneer het laboratorium tijdens het bekwaamheidsonderzoek (zoals beschreven in punt 9) een onvoldoende aantal experimenten voltooit om een statistisch robuuste verdeling van negatieve controles vast te stellen (zie punt 11), is het aanvaardbaar de verdeling tijdens de eerste routinetests op te bouwen. Bij deze aanpak moeten de aanbevelingen worden gevolgd die in de literatuur zijn uiteengezet (8), en de negatieve controleresultaten die in deze experimenten worden verkregen, moeten consistent blijven met gepubliceerde negatieve controlegegevens. Bij eventuele wijzigingen in het experimentele protocol moet rekening worden gehouden met hun implicaties voor de vraag of de resulterende gegevens consistent blijven met de databank met bestaande historische controlegegevens van het laboratorium. Alleen grote inconsistenties moeten leiden tot de oprichting van een nieuwe databank met historische controlegegevens, waarbij deskundigen bepalen welke controles afwijken van de eerdere verdeling (zie punt 11). Tijdens de hernieuwde opbouw is een volledige databank met negatieve controlegegevens mogelijk niet nodig om een echte test te mogen uitvoeren, mits het laboratorium kan aantonen dat zijn gelijktijdige negatieve controlewaarden consistent blijven met hetzij zijn eerdere databank hetzij de overeenkomstige gepubliceerde gegevens. Negatieve controlegegevens moeten bestaan uit de incidentie van cellen met sctructurele chromosoomafwijkingen (exclusief hiaten) in elk dier. Gelijktijdige negatieve controles liggen idealiter binnen de 95 %-controlegrenzen van de verdeling van de verzameling historische negatieve controlegegevens van het laboratorium. Wanneer gegevens van gelijktijdige negatieve controles buiten de 95 %-controlegrenzen vallen, kan opneming ervan in de historische controleverdeling toch aanvaardbaar zijn zolang deze gegevens geen extreme uitschieters zijn en er bewijs is dat het testsysteem 'onder controle' is (zie punt 11) en er geen bewijs is van technisch of menselijk falen. BESCHRIJVING VAN DE METHODE Voorbereidingen Keuze van de diersoort Er dient gebruik te worden gemaakt van gezonde jonge volwassen dieren van in het laboratorium gangbare stammen. Meestal worden ratten gebruikt, maar muizen kunnen ook geschikt zijn. Een andere geschikte zoogdiersoort kan ook in aanmerking komen, mits in het verslag een wetenschappelijke motivering wordt gegeven. Huisvestings- en voedingsomstandigheden van de dieren De temperatuur in de proefdierruimte moet voor knaagdieren 22 °C (± 3 °C) zijn. Hoewel de relatieve vochtigheid idealiter 50-60 % is, dient deze minimaal 40 % en bij voorkeur niet hoger dan 70 % te zijn, behalve bij het reinigen van de ruimte. Er moet gebruik worden gemaakt van kunstlicht met een cyclus van 12 uur licht en 12 uur donker. Als voeding mag het gewone laboratoriumvoer worden gebruikt met een onbeperkte hoeveelheid drinkwater. De keuze van het voer kan worden beïnvloed door de noodzaak om voor een afdoende mengbaarheid met de teststof te zorgen, wanneer de stof via het voer wordt toegediend. Knaagdieren moeten worden gehuisvest in kleine groepen (niet meer dan vijf dieren per kooi) van hetzelfde geslacht en dezelfde behandelingsgroep als geen agressief gedrag wordt verwacht, bij voorkeur in kooien met een stevige vloer met passende milieuverrijking. De dieren mogen uitsluitend individueel worden gehuisvest als dat wetenschappelijk gerechtvaardigd is. Voorbereiding van de dieren Normaal gesproken worden gezonde jonge volwassen dieren gebruikt (voor knaagdieren idealiter 6-10 weken oud bij het begin van de behandeling, hoewel iets oudere dieren ook aanvaardbaar zijn) en aselect ingedeeld in de behandelde en controlegroepen. De afzonderlijke dieren krijgen op humane, minimaal invasieve wijze een unieke identificatie (bv. door het aanbrengen van een ring, tag, microchip of door biometrische identificatie, maar niet door het afknippen van oren of tenen) en krijgen minimaal vijf dagen de tijd om in het laboratorium te acclimatiseren. De kooien worden zodanig geplaatst dat mogelijke effecten door de plaatsing van de kooi tot een minimum worden beperkt. Kruisverontreiniging door de positieve controle en de teststof moet worden vermeden. Bij het begin van de studie moet per geslacht het gewichtsverschil tussen de dieren zo klein mogelijk zijn en mag dit niet meer dan ± 20 % van het gemiddelde gewicht bedragen. Voorbereiding van de doses Vaste chemische teststoffen moeten worden opgelost of gesuspendeerd in geschikte oplosmiddelen of media of worden vermengd met voedsel of drinkwater voordat dit aan de dieren wordt gegeven. Vloeibare chemische teststoffen kunnen direct worden gegeven of eerst worden verdund. Voor blootstelling via inhalatie kunnen teststoffen worden toegediend als een gas, damp of een vaste/vloeibare aerosol, naargelang van de fysisch-chemische eigenschappen ervan. Er dienen verse preparaten van de teststof te worden toegepast, tenzij uit stabiliteitsgegevens blijkt dat bewaren van de preparaten aanvaardbaar is en blijkt wat de juiste bewaaromstandigheden zijn. Oplosmiddel/medium Het oplosmiddel/medium mag bij de gebruikte dosisniveaus geen toxische effecten veroorzaken en chemische reacties met de teststoffen moeten uitgesloten zijn. Als andere oplosmiddelen/media worden gebruikt dan de algemeen bekende, moet het gebruik ervan worden ondersteund met referentiegegevens die wijzen op de verenigbaarheid ervan. Het wordt aanbevolen waar mogelijk eerst het gebruik van een waterig oplosmiddel/medium te overwegen. Voorbeelden van vaak gebruikte compatibele oplosmiddelen/media zijn water, fysiologische zoutoplossing, methylcelluloseoplossing, natriumcarboxymethylcellulose-zoutoplossing, olijfolie en maïsolie. Bij het ontbreken van historische of gepubliceerde controlegegevens waaruit blijkt dat een gekozen atypisch oplosmiddel/medium geen structurele afwijkingen of andere schadelijke effecten veroorzaakt, dient een eerste onderzoek te worden uitgevoerd om vast te stellen dat de oplosmiddel/mediumcontrole aanvaardbaar is. Controles Positieve controles Normaal gesproken wordt in elke test een groep dieren opgenomen die wordt behandeld met een positieve controlestof. Hiervan kan worden afgezien wanneer het testlaboratorium aantoonbare bekwaamheid heeft in het uitvoeren van de test en een bereik van historische positieve controles heeft vastgesteld. Wanneer geen gelijktijdige positieve controlegroep wordt opgenomen, moeten in elk experiment scoringcontroles (ongekleurde en ongefixeerde preparaten) worden opgenomen. Deze kunnen worden verkregen door in de scoring van de studie passende referentiemonsters op te nemen die zijn verkregen en bewaard van een apart positieve-controle-experiment dat op gezette tijden (bv. elke 6-18 maanden) wordt verricht in het laboratorium waar de test wordt uitgevoerd; bijvoorbeeld tijdens de bekwaamheidstoetsing en daarna op gezette tijden, indien nodig. Positieve controlestoffen moeten op betrouwbare wijze leiden tot een detecteerbare stijging van de frequentie van cellen met structurele chromosoomafwijkingen ten opzichte van het spontane niveau. De doseringen van de positieve controles moeten zodanig worden gekozen dat de effecten duidelijk zijn, maar de gecodeerde monsters niet onmiddellijk als zodanig herkenbaar zijn voor de scorer. Het is aanvaardbaar dat de positieve controle langs een andere weg wordt toegediend dan de teststof, met behulp van een ander behandelingsschema, en dat bemonstering op slechts één tijdstip plaatsvindt. Tevens kan, waar dat van toepassing is, het gebruik van chemische stoffen uit een verwante chemische klasse als positieve controle worden overwogen. Voorbeelden van voor positieve controle gebruikte chemische stoffen staan vermeld in tabel 1. Tabel 1 Voorbeelden van positieve controlestoffen
Negatieve controles Dieren in een negatieve controlegroep moeten op alle bemonsteringstijdstippen worden opgenomen en ook verder op dezelfde wijze worden behandeld als de behandelingsgroepen, zij het dat zij geen teststof toegediend krijgen. Indien bij de toediening van de teststof van een oplosmiddel/medium gebruik wordt gemaakt, dient dit oplosmiddel/medium aan de controlegroep toegediend te worden. Als uit historische negatieve controlegegevens op ieder bemonsteringstijdstip voor het testlaboratorium een consistente spreiding over de dieren en consistente frequenties van cellen met structurele afwijkingen blijken, is mogelijk slechts één enkele bemonstering voor de negatieve controle nodig. Wanneer voor negatieve controles één bemonstering plaatsvindt, dient deze plaats te vinden op het eerste bemonsteringstijdstip dat in de studie wordt gebruikt. PROCEDURE Aantal en geslacht van de dieren In het algemeen is de micronucleusrespons voor mannetjes en vrouwtjes vergelijkbaar (9) en er wordt verwacht dat dit ook voor structurele chromosoomafwijkingen het geval zal zijn: de meeste studies kunnen daarom bij beide geslachten worden uitgevoerd. Gegevens waaruit blijkt dat er relevante verschillen tussen vrouwtjes en mannetjes bestaan (bv. verschillen in systemische toxiciteit, metabolisme, biologische beschikbaarheid, toxiciteit voor het beenmerg enz., met inbegrip van bv. een bereikbepalingsonderzoek), zijn een aanmoediging om beide geslachten te gebruiken. In dit geval kan het passend zijn om een onderzoek bij beide geslachten uit te voeren, bijvoorbeeld als onderdeel van een toxiciteitsonderzoek bij herhaalde toediening. Het kan passend zijn om een factoriële onderzoeksopzet te gebruiken als beide geslachten worden gebruikt. Details over de wijze waarop de gegevens bij deze onderzoeksopzet moeten worden geanalyseerd, zijn te vinden in aanhangsel 2. De vaststelling van de groepsgrootte aan het begin van het onderzoek moet erop gericht zijn minimaal 5 analyseerbare dieren van één geslacht, of van elk geslacht als beide worden gebruikt, per groep te krijgen. Wanneer de blootstelling aan een chemische stof bij de mens door het geslacht wordt bepaald, zoals dit bijvoorbeeld bij sommige geneesmiddelen het geval is, moet de test bij het desbetreffende geslacht worden uitgevoerd. Als richtsnoer voor de maximale typische behoefte aan dieren: voor een onderzoek in beenmerg op twee bemonsteringstijdstippen met drie dosisgroepen en een parallelle negatieve controlegroep, plus een positieve controlegroep (elke groep bestaande uit vijf dieren van één geslacht) zijn 45 dieren nodig. Dosisniveaus Als er een bereikbepalingsonderzoek wordt uitgevoerd omdat er nog geen bruikbare gegevens over de dosering beschikbaar zijn, moet dit in hetzelfde laboratorium gebeuren met dezelfde soort, dezelfde stam, hetzelfde geslacht en hetzelfde behandelingsschema als bij het hoofdonderzoek (10). Het onderzoek moet erop gericht zijn de maximaal verdraagbare dosis (maximum tolerated dose, MTD) vast te stellen, dat wil zeggen, de hoogste dosis die zal worden verdragen zonder aanwijzingen voor onderzoeksbeperkende toxiciteit, in verhouding tot de duur van de onderzoeksperiode (bv. door het veroorzaken van gewichtsverlies of cytotoxiciteit voor het hematopoïetisch systeem, maar geen dood of aanwijzingen voor pijn, lijden of nood die humane doding noodzakelijk maken (11)). De hoogste dosis kan ook worden gedefinieerd als een dosis die enigerlei aanwijzing van toxiciteit voor het beenmerg oplevert. Chemische stoffen die verzadiging van toxicokinetische eigenschappen vertonen, of detoxificatieprocessen induceren die kunnen leiden tot een afname van de blootstelling na langlopende behandeling, kunnen uitzonderingen zijn op de doseringscriteria en moeten per geval worden beoordeeld. Om dosisresponsinformatie te krijgen, moet een volledig onderzoek een negatieve controlegroep omvatten en minimaal drie dosisniveaus die in het algemeen een factor 2, maar niet meer dan een factor 4, uit elkaar liggen. Als in een bereikbepalingsonderzoek of op basis van bestaande gegevens geen aanwijzingen voor toxiciteit van de teststof worden gevonden, moet de hoogste dosis voor een enkele toediening 2000 mg/kg lichaamsgewicht zijn. Als de teststof echter wel toxiciteit veroorzaakt, moet de MTD de hoogste toegediende dosis zijn en moeten de gebruikte dosisniveaus bij voorkeur een bereik dekken van maximale tot weinig of geen toxiciteit. Wanneer toxiciteit voor het doelweefsel (beenmerg) bij alle geteste dosisniveaus wordt waargenomen, wordt verder onderzoek bij niet-toxische doses aanbevolen. Voor onderzoek dat ten doel heeft de kwantitatieve dosisresponsinformatie volledig te kenschetsen, kunnen aanvullende dosisgroepen nodig zijn. Voor bepaalde typen teststoffen (bv. geneesmiddelen voor menselijk gebruik) waarvoor specifieke eisen gelden, kunnen deze limieten verschillen. Limiettest Als uit doseringsbereikstudies of bestaande gegevens bij gerelateerde dierstammen blijkt dat een behandelingsregime van minstens de limietdosis (zoals hieronder beschreven) geen waarneembare toxische effecten geeft (met inbegrip van achteruitgang van de proliferatie van beenmerg of andere aanwijzingen van toxiciteit voor het doelweefsel), en als op grond van in-vitro genotoxiciteitsstudies of gegevens bij structureel verwante chemische stoffen geen genotoxiciteit wordt verwacht, dan is een volledig onderzoek met drie dosisniveaus mogelijk niet nodig, mits is aangetoond dat de teststof(fen) het doelweefsel (beenmerg) bereik(t)(en). In zulke gevallen kan één dosisniveau, gelijk aan de limietdosis, voldoende zijn. Voor een toedieningsperiode van meer dan 14 dagen is de limietdosis 1000 mg/kg lichaamsgewicht/dag. Voor een toedieningsperiode van 14 dagen of minder is de limietdosis 2000 mg/kg lichaamsgewicht/dag. Toediening van de doses Bij de opzet van een test moet rekening worden gehouden met de verwachte blootstellingsroute bij mensen. Daarom kunnen andere blootstellingsroutes zoals voeding, drinkwater, lokaal, subcutaan, intraveneus, oraal (via sonde), inhalatie, intratracheaal of implantatie worden gekozen, als deze onderbouwd worden. De route moet in elk geval zodanig worden gekozen dat adequate blootstelling van het (de) doelweefsel(s) is gewaarborgd. Intraperitoneale injectie wordt in het algemeen niet aanbevolen, omdat zij bij mensen geen beoogde blootstellingsroute is, en zij mag uitsluitend worden toegepast met een specifieke wetenschappelijke motivering. Als de teststof wordt vermengd met voedsel of drinkwater, met name in geval van één toediening, moet ervoor worden gezorgd dat de tijdsduur tussen de consumptie van voedsel en water en de bemonstering voldoende is om de effecten te kunnen detecteren (zie de punten 33-34). Het maximale vloeistofvolume dat via sonde of injectie per keer kan worden toegediend, hangt af van de grootte van het proefdier. Het volume mag normaal gesproken niet groter zijn dan 1 ml/100 g lichaamsgewicht. Bij een waterige oplossing kan echter maximaal 2 ml/100 g lichaamsgewicht worden gebruikt. Hogere volumes dan dit moeten worden onderbouwd. Behalve bij irriterende of bijtende teststoffen, die normaal gesproken bij hogere concentraties hevigere effecten hebben, moeten volumeverschillen tot een minimum worden beperkt door de concentratie aan te passen, zodat er op alle dosisniveaus hetzelfde volume in verhouding tot het lichaamsgewicht wordt toegediend. Behandelingsschema De teststoffen worden normaal gesproken in één dosis toegediend, maar de dosis kan eventueel worden gesplitst (bv. twee of meer porties op dezelfde dag met een interval van niet meer dan 2-3 uur) om de toediening van een groot volume te vergemakkelijken. In deze omstandigheden, of wanneer de teststof wordt toegediend door inhalatie, moet het bemonsteringstijdstip worden gepland op basis van het tijdstip van de laatste dosistoediening of het einde van de blootstelling. Er zijn voor deze test weinig gegevens over de geschiktheid van een protocol met herhaalde toediening beschikbaar. In omstandigheden waarin het wenselijk is om deze test te integreren met een toxiciteitsonderzoek met herhaalde toediening, moet verlies van mitotische cellen met chromosoombeschadigingen die bij toxische doses kunnen voorkomen, echter worden vermeden. Zo'n integratie is aanvaardbaar wanneer de hoogste dosis groter is dan of gelijk is aan de limietdosis (zie punt 29) en een dosisgroep voor de duur van de behandelingsperiode de limietdosis toegediend krijgt. De micronucleustest (testmethode B.12) moet worden beschouwd als de in-vivotest voor chromosoomafwijkingen die de voorkeur geniet wanneer integratie met andere onderzoeken gewenst is. Beenmergmonsters worden na een dosis in één keer op twee aparte tijdstippen genomen. Voor knaagdieren ligt het eerste bemonsteringstijdstip op 1,5 maal de normale lengte van een celcyclus (deze cyclus duurt normaal gesproken 12-18 uur) na de behandeling. Aangezien de voor de opname en het metabolisme van de teststof(fen) vereiste tijd en de effecten op de kinetiek van de celcyclus gevolgen kunnen hebben voor het optimale tijdstip om chromosoomafwijkingen te detecteren, verdient het aanbeveling 24 uur na het eerste bemonsteringstijdstip opnieuw te bemonsteren. Op het eerste bemonsteringstijdstip worden alle dosisgroepen behandeld en worden monsters genomen voor de analyse; op het (de) latere bemonsteringstijdstip(pen) hoeft echter alleen de hoogste dosis te worden toegediend. Indien het doseringsschema op grond van een wetenschappelijke onderbouwing meer dan één dag beslaat, moet in het algemeen één bemonsteringstijdstip op maximaal ongeveer 1,5 maal de normale lengte van een celcyclus na de laatste toediening worden gebruikt. Na de behandeling en voorafgaand aan de bemonstering, worden de dieren intraperitoneaal geïnjecteerd met een adequate dosis metafasestopper (bv. colcemid of colchicine) en worden de dieren na een adequate pauze bemonsterd. Voor muizen moet er ongeveer 3-5 uur worden gewacht tot de bemonstering, en voor ratten 2-5 uur. De cellen worden uit het beenmerg gehaald, opgebold, gefixeerd en gekleurd en op chromosoomafwijkingen geanalyseerd (12). Observaties Algemene klinische observaties van de proefdieren moeten ten minste eenmaal per dag plaatsvinden en klinische tekenen moeten ten minste eenmaal per dag worden geregistreerd, bij voorkeur steeds op hetzelfde tijdstip en met inachtneming van de piekperiode van de te verwachten effecten na toediening. Gedurende de toedieningsperiode moeten alle dieren ten minste tweemaal per dag worden geobserveerd op ziekteverschijnselen en sterfte. Alle dieren moeten bij aanvang van het onderzoek, tijdens onderzoeken met herhaalde toediening ten minste eenmaal per week, en bij het doden worden gewogen. In onderzoeken met een duur van ten minste één week moet het voedselverbruik ten minste wekelijks worden gemeten. Als de teststof met het drinkwater wordt toegediend, moet het waterverbruik bij elke verversing van water en ten minste wekelijks worden gemeten. Dieren die niet-letale indicatoren van excessieve toxiciteit vertonen moeten voor afloop van de testperiode op humane wijze worden gedood (11). Blootstelling van doelweefsel Op een of meer passende tijdstippen moet een bloedmonster worden genomen en moeten de plasmaspiegels van de teststoffen worden onderzocht om aan te tonen dat blootstelling van het beenmerg heeft plaatsgevonden, in situaties waar dit gerechtvaardigd is en waar geen andere blootstellingsgegevens bestaan (zie punt 44). Beenmerg en chromosoompreparaten Onmiddellijk na humane doding worden uit de dijbenen of scheenbenen van de dieren beenmergcellen verwijderd, in een hypotone oplossing gebracht en gefixeerd. De metafasecellen worden vervolgens op objectglaasjes uitgesmeerd en gekleurd met vastgestelde methoden (zie (3) (12)). Analyse Alle objectglaasjes, ook die van de positieve en negatieve controles, worden vóór de analyse onafhankelijk gecodeerd en worden gerandomiseerd, zodat de scorer de behandelingssituatie niet kent. Als maat voor de cytotoxiciteit wordt de mitotische index bepaald in ten minste 1000 cellen per dier voor alle behandelde dieren (ook de positieve controles), onbehandelde dieren voor de negatieve controle of dieren die medium/oplosmiddel toegediend hebben gekregen voor de negatieve controle. Voor elk dier worden ten minste 200 metafasen geanalyseerd op structurele chromosoomafwijkingen, inclusief en exclusief hiaten (6). Als de databank met historische negatieve controlegegevens echter uitwijst dat de gemiddelde achtergrondfrequentie van structurele chromosoomafwijkingen in het testlaboratorium <1 % is, moet worden overwogen aanvullende cellen te scoren. Afwijkingen van het chromatidetype en chromosoomtype worden apart geregistreerd en ingedeeld in subtypen (breuken, uitwisselingen). De in het laboratorium gebruikte procedures moeten ervoor zorgen dat de analyse van chromosoomafwijkingen wordt uitgevoerd door goed opgeleide scorers en zo nodig collegiaal wordt getoetst. Aangezien bij de bereiding van de objectglaasjes vaak een gedeelte van de metafasecellen breekt, waarbij chromosomen verloren gaan, mogen de gescoorde cellen niet minder dan 2n ± 2 centromeren bevatten, waarin n het haploïde aantal chromosomen voor die soort is. GEGEVENS EN RAPPORTAGE Verwerking van de resultaten Gegevens van individuele dieren moeten in tabelvorm worden gepresenteerd. De mitotische index, het aantal gescoorde metafasecellen, het aantal afwijkingen per metafasecel en het percentage cellen met een of meer structurele chromosoomafwijkingen moeten per dier worden geëvalueerd. Er moet een overzicht worden gegeven van de verschillende soorten structurele chromosoomafwijkingen met de aantallen en de frequentie waarmee ze in de behandelde en controlegroepen voorkomen. Hiaten, alsmede polyploïde cellen en cellen met endoreduplicatie van chromosomen worden apart geregistreerd. De frequentie van hiaten wordt geregistreerd, maar in het algemeen niet opgenomen in de analyse van de totale frequentie van structurele afwijkingen. Als er geen aanwijzingen zijn voor verschillen in reactie tussen de geslachten, kunnen de gegevens voor de statistische analyse worden gecombineerd. Gegevens over de toxiciteit en klinische tekenen bij dieren moeten eveneens worden gemeld. Aanvaardbaarheidscriteria Voor de aanvaardbaarheid van de test gelden de volgende criteria:
Beoordeling en interpretatie van resultaten Indien aan alle aanvaardbaarheidscriteria is voldaan, wordt een teststof als duidelijk positief beschouwd als:
Indien op een bepaald bemonsteringstijdstip alleen de hoogste dosis wordt onderzocht, wordt een teststof als duidelijk positief beschouwd als er een statistisch significante stijging vergeleken met de gelijktijdige negatieve controle is en de resultaten buiten de verdeling van de historische negatieve controlegegevens vallen (bv. op Poisson-verdeling gebaseerde 95 %-controlegrenzen). Aanbevelingen voor de passende statistische methoden zijn te vinden in de literatuur (13). Wanneer een dosisresponsanalyse wordt uitgevoerd, moeten ten minste drie behandelde dosisgroepen worden geanalyseerd. De statistische toetsen moeten het dier als de proefeenheid gebruiken. Positieve resultaten in de test op chromosoomafwijkingen geven aan dat een teststof structurele chromosoomafwijkingen in het beenmerg van de geteste soort induceert. Indien aan alle aanvaardbaarheidscriteria is voldaan, wordt een teststof als duidelijk negatief beschouwd als in alle onderzochte experimentele omstandigheden:
Aanbevelingen voor de meest passende statistische methoden zijn te vinden in de literatuur (13). Bewijs van blootstelling van het beenmerg aan een teststof kan zijn achteruitgang van de mitotische index of meting van het plasma of bloedspiegels van de teststof(fen). In geval van intraveneuze toediening is geen bewijs van blootstelling nodig. Als alternatief kunnen ADME-gegevens verkregen in een onafhankelijke studie met dezelfde toedieningsweg en dezelfde soort worden gebruikt om blootstelling van het beenmerg aan te tonen. Negatieve resultaten geven aan dat de teststof onder de testomstandigheden geen structurele chromosoomafwijkingen in het beenmerg van de onderzochte soort induceert. Een duidelijk positieve of duidelijk negatieve reactie behoeft niet te worden bevestigd. Indien de reactie noch duidelijk negatief noch duidelijk positief is en om de biologische relevantie van een resultaat te helpen vaststellen (bv. een zwakke of zeer lichte toename), moeten de gegevens door een deskundige en/of nader onderzoek van de bestaande voltooide experimenten worden beoordeeld. In sommige gevallen kan het nuttig zijn meer cellen te analyseren of een herhalingsexperiment onder aangepaste experimentele omstandigheden uit te voeren. In zeldzame gevallen zal op basis van de gegevens, zelfs na nader onderzoek, niet kunnen worden geconcludeerd dat de teststof positieve of negatieve resultaten oplevert, en zal de studie daarom als moeilijk te interpreteren worden beschouwd. De frequenties van polyploïde metafasen en metafasen met endoreduplicatie ten opzichte van het totale aantal metafasen moeten apart worden geregistreerd. Een stijging van het aantal polyploïde cellen en/of cellen met endoreduplicatie kan erop wijzen dat de teststof in staat is mitotische processen of de voortgang van de celcyclus te remmen (zie punt 3). Testverslag In het testverslag moet de volgende informatie worden opgenomen: Samenvatting
LITERATUUR
Aanhangsel 1 DEFINITIES Afwijking van het chromatidetype : structurele chromosoombeschadiging waarbij breuken in individuele chromatiden ontstaan, eventueel gevolgd door recombinatie. Afwijking van het chromosoomtype : structurele chromosoombeschadiging waarbij breuken in beide chromatiden op dezelfde plaats ontstaan, eventueel gevolgd door recombinatie. Aneuploïdie : elke afwijking van het normale diploïde (of haploïde) aantal chromosomen met één of meer chromosomen, maar niet met veelvouden van (een) volledige reeks(en) chromosomen (vgl. polyploïdie). Centromeer : het deel of de delen van een chromosoom waar tijdens de celdeling de spoeldraden aan vastzitten, zodat de verplaatsing van de dochterchromosomen naar de polen van de dochtercellen regelmatig kan verlopen. Chemische stof : een stof of mengsel. Endoreduplicatie : een proces waarbij de kern na een S-fase met DNA-replicatie niet tot mitose overgaat, maar opnieuw een S-fase begint. Het resultaat is chromosomen met 4, 8, 16, … chromatiden. Hiaat : een achromatische beschadiging die kleiner is dan de breedte van één chromatide en waarbij de fout in de uitlijning van de chromatiden minimaal is. Mitotische index : de verhouding tussen het aantal cellen in mitose en het totale aantal cellen in een populatie, die een maat is voor de proliferatie van die celpopulatie. Numerieke afwijking : een verandering in het aantal chromosomen ten opzichte van het normale aantal dat kenmerkend is voor de gebruikte dieren (aneuploïdie). Polyploïdie : een numerieke chromosoomafwijking waarbij sprake is van een verandering in het aantal van de volledige reeks chromosomen, te onderscheiden van een numerieke verandering in een deel van de reeks chromosomen (vgl. aneuploïdie). Structurele chromosoomafwijking : een verandering in de chromosoomstructuur die bij microscopisch onderzoek tijdens de metafase van de celdeling kan worden waargenomen in de vorm van deleties, fragmenten en intra- of interchromosomale uitwisselingen. Teststof : alle volgens deze testmethode geteste stoffen of mengsels. Aanhangsel 2 DE FACTORIËLE OPZET VOOR HET IDENTIFICEREN VAN VERSCHILLEN TUSSEN GESLACHTEN IN DE IN-VIVOTEST OP CHROMOSOOMAFWIJKINGEN De factoriële opzet en zijn analyse Bij deze opzet worden voor elk concentratieniveau minimaal 5 mannetjes en 5 vrouwtjes getest, wat leidt tot een opzet met minimaal 40 dieren (20 mannetjes en 20 vrouwtjes, plus relevante positieve controles). De opzet, die een van de eenvoudigere factoriële opzetten is, is gelijkwaardig aan een tweewegs variantieanalyse met geslacht en concentratieniveau als hoofdeffecten. De gegevens kunnen worden geanalyseerd met veel standaard statistische softwarepakketten, zoals SPSS, SAS, STATA, Genstat, alsook met R. De analyse verdeelt de variabiliteit in de gegevensreeks in de variabiliteit tussen de geslachten, die tussen de concentraties en die verband houdend met de interactie tussen geslacht en concentratie. Elk van de termen wordt getoetst tegen een schatting van de variabiliteit tussen de duplodieren binnen de groepen van dieren van hetzelfde geslacht die dezelfde concentratie toegediend krijgen. Volledige gegevens over de onderliggende methodologie zijn te vinden in diverse statistische standaardhandboeken (zie referenties) en in de “help”-functionaliteit van de statistische pakketten. De analyse begint met een inspectie van de interactieterm geslacht x concentratie in de ANOVA-tabel (5). Als de interactieterm niet significant is, verschaffen de gecombineerde waarden van de geslachten of van de concentratieniveaus geldige statistische toetsen tussen de niveaus op basis van de gepoolde variabiliteit binnen groepen in de ANOVA. De volgende stap in de analyse is de verdeling van de geschatte variabiliteit tussen concentraties in contrasten, die een toets op lineaire en kwadratische contrasten van de responsen over de concentratieniveaus verschaft. Wanneer de interactie tussen geslacht en concentratie significant is, kan deze term ook worden verdeeld in interactiecontrasten lineair x geslacht en kwadratisch x geslacht. Deze termen verschaffen toetsen voor de vraag of de concentratieresponsen voor de twee geslachten parallel lopen of dat de respons voor de twee geslachten gedifferentieerd is. De geschatte gepoolde variabiliteit binnen groepen kan worden gebruikt voor paarsgewijze toetsen voor het verschil tussen de gemiddelden. Deze vergelijkingen kunnen worden gemaakt tussen de gemiddelden voor de twee geslachten en tussen de gemiddelden voor de verschillende concentratieniveaus zoals voor vergelijkingen met de negatieve controleniveaus. Indien er sprake is van een significante interactie, kunnen vergelijkingen worden gemaakt tussen de gemiddelden van verschillende concentraties binnen een geslacht of tussen de gemiddelden van de geslachten bij dezelfde concentratie. Referenties Er is een groot aantal statistische handboeken waarin de theorie, opzet, methodologie, analyse en interpretatie van factoriële opzetten worden besproken, variërend van de eenvoudigste analyses met twee factoren tot de complexere vormen die worden gebruikt voor het ontwerpen van experimenten. De lijst hieronder is niet uitputtend. Enkele boeken bevatten uitgewerkte voorbeelden van vergelijkbare proefopzetten, in enkele gevallen met code voor het uitvoeren van de analyses met verschillende softwarepakketten.
|
|
5) |
In deel B wordt hoofdstuk B.12 vervangen door: „B.12 Micronucleustest bij erytrocyten van zoogdieren INLEIDING Deze testmethode is gelijkwaardig aan testrichtlijn (TG) 474 (2016) van de OESO. Zij maakt deel uit van een reeks testmethoden op het gebied van de genetische toxicologie. Er is een OESO-document opgesteld met beknopte informatie over genetisch-toxicologische tests en een overzicht van de recente wijzigingen van deze testrichtlijnen (1). De in-vivomicronucleustest bij zoogdieren is met name van belang voor de beoordeling van genotoxiciteit, omdat factoren van het in-vivometabolisme, de farmacokinetiek en de DNA-herstelprocessen actief zijn en bijdragen aan de reacties op de test, hoewel deze aspecten per soort kunnen verschillen. Een in-vivobepaling is ook geschikt om in vitro gesignaleerde genotoxiciteit nader te onderzoeken. De in-vivomicronucleustest bij zoogdieren wordt gebruikt om te bepalen of de teststof chromosoombeschadigingen of beschadiging van het mitotisch apparaat in de erytroblasten induceert. De test beoordeelt de vorming van micronuclei in erytrocyten die zijn verkregen uit het beenmerg of perifere bloedcellen van dieren, meestal knaagdieren. De micronucleustest is bedoeld om chemische stoffen te signaleren die cytogenetische schade veroorzaken waarbij micronuclei ontstaan met achtergebleven chromosoomfragmenten of volledige chromosomen. Wanneer een erytroblast uit het beenmerg zich ontwikkelt tot een onrijpe erytrocyt (ook polychromatische erytrocyt of reticulocyt genoemd), wordt de hoofdkern uitgestoten; wanneer een micronucleus is ontstaan, kan deze in het cytoplasma achterblijven. Omdat deze cellen geen hoofdkern bevatten, zijn de micronuclei gemakkelijk zichtbaar te maken of op te sporen. Een stijging van de frequentie waarmee bij behandelde dieren onrijpe erytrocyten met micronuclei worden aangetroffen, wijst op structurele of numerieke chromosoomafwijkingen. Nieuwgevormde erytrocyten met micronuclei worden geïdentificeerd en kwantitatief bepaald door kleuring gevolgd door hetzij visuele scoring met een microscoop of geautomatiseerde analyse. Het tellen van voldoende rijpe erytrocyten in het perifeer bloed of beenmerg van volwassen dieren wordt veel gemakkelijker wanneer een geautomatiseerd scoringplatform wordt gebruikt. Zulke platforms zijn aanvaardbaar als alternatief voor handmatige beoordeling (2). Uit vergelijkend onderzoek is gebleken dat deze methoden, die gebruikmaken van passende kalibratiestandaarden, een betere inter- en intralaboratoriumreproduceerbaarheid en -gevoeligheid kunnen verschaffen dan het handmatig scoren met een microscoop (3) (4). Geautomatiseerde systemen die frequenties van erytrocyten met micronuclei kunnen meten, zijn onder andere flowcytometers (5), beeldanalyseplatforms (6) (7) en laserscanningcytometers (8). Hoewel het in het kader van de test normaal gesproken niet wordt gedaan, kunnen chromosoomfragmenten aan de hand van een aantal criteria worden onderscheiden van hele chromosomen. Daartoe behoren de vaststelling van de aan- of afwezigheid van een kinetochoor- of centromeer-DNA, die beide kenmerkend zijn voor intacte chromosomen. De afwezigheid van een kinetochoor- of centromeer-DNA wijst erop dat de micronucleus uitsluitend fragmenten van chromosomen bevat, terwijl de aanwezigheid ervan wijst op verlies van chromosomen. De gebruikte definities zijn opgenomen in aanhangsel 1. INLEIDENDE OVERWEGINGEN Het beenmerg van jonge volwassen knaagdieren is het doelweefsel voor genetische schade in deze test, aangezien in dit weefsel erytrocyten worden gevormd. De meting van micronuclei in onrijpe erytrocyten in perifeer bloed is aanvaardbaar bij andere zoogdiersoorten waarvan is aangetoond dat de gevoeligheid om chemische stoffen die structurele of numerieke chromosoomafwijkingen in deze cellen veroorzaken te detecteren, afdoende is (door inductie van micronuclei in onrijpe erytrocyten) en mits een wetenschappelijke motivering wordt gegeven. De frequentie van onrijpe erytrocyten met micronuclei is het belangrijkste eindpunt. De frequentie van rijpe erytrocyten in het perifere bloed met micronuclei kan ook als eindpunt van de bepaling worden gebruikt bij diersoorten zonder sterke selectie in de milt tegen cellen met micronuclei en wanneer de dieren gedurende een periode langer dan de levensduur van de erytrocyt in de gebruikte diersoort ononderbroken worden behandeld. (bv. 4 weken of langer voor muizen). Als er aanwijzingen zijn dat de teststof(fen), of een of meer metabolieten daarvan, niet in het doelweefsel terechtkom(t)(en), is deze test mogelijk niet geschikt. Voordat de testmethode wordt gebruikt op een mengsel om gegevens te genereren voor een beoogd regelgevingsdoel, moet worden nagegaan of, en zo ja, waarom deze methode betrouwbare resultaten oplevert voor dat doel. Zulke overwegingen zijn niet nodig wanneer het testen van het mengsel wettelijk vereist is. PRINCIPE VAN DE TESTMETHODE De dieren worden langs een geschikte weg aan de teststof blootgesteld. Als beenmerg wordt gebruikt, worden de dieren op een of meer geschikte tijdstippen na de behandeling op humane wijze gedood, wordt het beenmerg verwijderd en worden er preparaten gemaakt die worden gekleurd (9) (10) (11) (12) (13) (14) (15). Wanneer perifeer bloed wordt gebruikt, wordt op een of meer geschikte tijdstippen na de behandeling bloed afgenomen en worden er preparaten gemaakt die worden gekleurd (12) (16) (17) (18). Wanneer acuut een behandeling wordt toegepast, is het belangrijk tijdstippen voor het oogsten van beenmerg of bloed te kiezen waarop de inductie van onrijpe erytrocyten met micronuclei als gevolg van de behandeling kan worden gedetecteerd. In geval van bemonstering van perifeer bloed, moet ook zoveel tijd zijn verstreken dat deze gebeurtenissen in het circulerende bloed zichtbaar worden. De preparaten worden onderzocht op de aanwezigheid van micronuclei, door visualisatie met een microscoop, beeldanalyse, flowcytometrie of laserscanningcytometrie. VERIFICATIE VAN DE BEKWAAMHEID VAN LABORATORIA Bekwaamheidsonderzoek Om voldoende ervaring met het uitvoeren van de bepaling vast te stellen voordat deze wordt gebruikt voor routinetests, moet het laboratorium hebben aangetoond in staat te zijn verwachte resultaten uit gepubliceerde gegevens (17) (19) (20) (21) (22) te reproduceren voor frequenties van micronuclei met minimaal twee positieve controlestoffen (waaronder zwakke responsen geïnduceerd door lage doses positieve controles), zoals de stoffen vermeld in tabel 1 en met vergelijkbare medium-/oplosmiddelcontroles (zie punt 26). Bij deze experimenten moeten doses worden gebruikt die leiden tot reproduceerbare en dosisgerelateerde toenamen en moeten de gevoeligheid en het dynamisch bereik van het testsysteem in het te onderzoeken weefsel (beenmerg of perifeer bloed) aantonen en gebruikmaken van de scoringmethode die binnen het laboratorium zal worden aangewend. Deze eis geldt niet voor laboratoria met ervaring, d.w.z. laboratoria die beschikken over referentiegegevens uit het verleden zoals omschreven in de punten 14-18. Controlegegevens uit het verleden Tijdens het bekwaamheidsonderzoek stelt het laboratorium vast:
Wanneer voor het eerst gegevens worden verkregen voor een verdeling van negatieve controles uit het verleden, moeten gelijktijdige negatieve controles consistent zijn met gepubliceerde controlegegevens, indien deze bestaan. Wanneer meer experimentele gegevens aan de verdeling van historische controles worden toegevoegd, moeten gelijktijdige negatieve controles idealiter binnen de 95 %-controlegrenzen van deze verdeling liggen. De databank met historische negatieve controlegegevens van het laboratorium moet statistisch robuust zijn om te waarborgen dat het laboratorium de verdeling van zijn negatieve controlegegevens kan beoordelen. Uit de literatuur komt naar voren dat daarvoor minimaal 10 experimenten nodig zijn, maar dat bij voorkeur ten minste 20 experimenten worden gebruikt die onder vergelijkbare experimentele omstandigheden zijn uitgevoerd. Laboratoria moeten methoden voor kwaliteitscontrole gebruiken, zoals controlekaarten (bv. C-kaarten of X-kaarten (23)), om vast te stellen hoe variabel hun gegevens zijn en om aan te tonen dat het laboratorium de methodologie 'onder controle' heeft. Verdere aanbevelingen voor het opbouwen en gebruiken van de historische gegevens (d.w.z. criteria voor het opnemen van gegevens in en het uitsluiten ervan uit de historische gegevens en de aanvaardbaarheidscriteria voor een gegeven experiment) zijn te vinden in de literatuur (24). Wanneer het laboratorium tijdens het bekwaamheidsonderzoek (zoals beschreven in punt 13) een onvoldoende aantal experimenten voltooit om een statistisch robuuste verdeling van negatieve controles vast te stellen (zie punt 15), is het aanvaardbaar de verdeling tijdens de eerste routinetests op te bouwen. Bij deze aanpak moeten de aanbevelingen worden gevolgd die in de literatuur zijn uiteengezet (24), en de negatieve controleresultaten die in deze experimenten worden verkregen, moeten consistent blijven met gepubliceerde negatieve controlegegevens. Bij eventuele wijzigingen in het experimentele protocol moet rekening worden gehouden met hun implicaties voor de vraag of de resulterende gegevens consistent blijven met de databank met bestaande historische controlegegevens van het laboratorium. Alleen grote inconsistenties moeten leiden tot de oprichting van een nieuwe databank met historische controlegegevens, waarbij deskundigen bepalen welke controles afwijken van de eerdere verdeling (zie punt 15). Tijdens de hernieuwde opbouw is een volledige databank met negatieve controlegegevens mogelijk niet nodig om een echte test te mogen uitvoeren, mits het laboratorium kan aantonen dat zijn gelijktijdige negatieve controlewaarden consistent blijven met hetzij zijn eerdere databank hetzij de overeenkomstige gepubliceerde gegevens. Negatieve controlegegevens moeten bestaan uit de incidentie van onrijpe erytrocyten met micronuclei in elk dier. Gelijktijdige negatieve controles liggen idealiter binnen de 95 %-controlegrenzen van de verdeling van de verzameling historische negatieve controlegegevens van het laboratorium. Wanneer gegevens van gelijktijdige negatieve controles buiten de 95 %-controlegrenzen vallen, kan opneming ervan in de historische controleverdeling toch aanvaardbaar zijn zolang deze gegevens geen extreme uitschieters zijn en er bewijs is dat het testsysteem 'onder controle' is (zie punt 15) en er geen bewijs is van technisch of menselijk falen. BESCHRIJVING VAN DE METHODE Voorbereidingen Keuze van de diersoort Er dient gebruik te worden gemaakt van gezonde jonge volwassen dieren van in het laboratorium gangbare stammen. Er kunnen muizen, ratten en andere geschikte zoogdiersoorten worden gebruikt. Wanneer perifeer bloed wordt gebruikt, moet worden vastgesteld dat verwijdering van cellen met micronuclei uit de bloedsomloop door de milt de detectie van geïnduceerde micronuclei in de gekozen soort niet in het gedrang brengt. Dit is voor perifeer bloed van muizen en ratten al aangetoond (2). De wetenschappelijke rechtvaardiging voor het gebruik van andere soorten dan ratten en muizen moet in het verslag worden opgenomen. Als andere soorten dan knaagdieren worden gebruikt, wordt aanbevolen de meting van geïnduceerde micronuclei te integreren in een andere passende toxiciteitstest. Huisvestings- en voedingsomstandigheden van de dieren De temperatuur in de proefdierruimte moet voor knaagdieren 22 °C (± 3 °C) zijn. Hoewel de relatieve vochtigheid idealiter 50-60 % is, dient deze minimaal 40 % en bij voorkeur niet hoger dan 70 % te zijn, behalve bij het reinigen van de ruimte. Er moet gebruik worden gemaakt van kunstlicht met een cyclus van 12 uur licht en 12 uur donker. Als voeding mag het gewone laboratoriumvoer worden gebruikt met een onbeperkte hoeveelheid drinkwater. De keuze van het voer kan worden beïnvloed door de noodzaak om voor een afdoende mengbaarheid met de teststof te zorgen, wanneer de stof via het voer wordt toegediend. Knaagdieren moeten worden gehuisvest in kleine groepen (niet meer dan vijf dieren per kooi) van hetzelfde geslacht en dezelfde behandelingsgroep als geen agressief gedrag wordt verwacht, bij voorkeur in kooien met een stevige vloer met passende milieuverrijking. De dieren mogen uitsluitend individueel worden gehuisvest als dat wetenschappelijk gerechtvaardigd is. Voorbereiding van de dieren Normaal gesproken worden gezonde jonge volwassen dieren gebruikt (voor knaagdieren idealiter 6-10 weken oud bij het begin van de behandeling, hoewel iets oudere dieren ook aanvaardbaar zijn) en aselect ingedeeld in de behandelde en controlegroepen. De afzonderlijke dieren krijgen op humane, minimaal invasieve wijze een unieke identificatie (bv. door het aanbrengen van een ring, tag, microchip of door biometrische identificatie, maar niet door het afknippen van oren of tenen) en krijgen minimaal vijf dagen de tijd om in het laboratorium te acclimatiseren. De kooien worden zodanig geplaatst dat mogelijke effecten door de plaatsing van de kooi tot een minimum worden beperkt. Kruisverontreiniging door de positieve controle en de teststof moet worden vermeden. Bij het begin van de studie moet per geslacht het gewichtsverschil tussen de dieren zo klein mogelijk zijn en mag dit niet meer dan ± 20 % van het gemiddelde gewicht bedragen. Voorbereiding van de doses Vaste chemische teststoffen moeten worden opgelost of gesuspendeerd in geschikte oplosmiddelen of media of worden vermengd met voedsel of drinkwater voordat dit aan de dieren wordt gegeven. Vloeibare chemische teststoffen kunnen direct worden gegeven of eerst worden verdund. Voor blootstelling via inhalatie kunnen teststoffen worden toegediend als een gas, damp of een vaste/vloeibare aerosol, naargelang van de fysisch-chemische eigenschappen ervan. Er dienen verse preparaten van de teststof te worden toegepast, tenzij uit stabiliteitsgegevens blijkt dat bewaren van de preparaten aanvaardbaar is en blijkt wat de juiste bewaaromstandigheden zijn. Testomstandigheden Oplosmiddel/medium Het oplosmiddel/medium mag bij de gebruikte dosisniveaus geen toxische effecten veroorzaken en chemische reacties met de teststoffen moeten uitgesloten zijn. Als andere oplosmiddelen/media worden gebruikt dan de algemeen bekende, moet het gebruik ervan worden ondersteund met referentiegegevens die wijzen op de verenigbaarheid ervan. Het wordt aanbevolen waar mogelijk eerst het gebruik van een waterig oplosmiddel/medium te overwegen. Voorbeelden van vaak gebruikte compatibele oplosmiddelen/media zijn water, fysiologische zoutoplossing, methylcelluloseoplossing, natriumcarboxymethylcellulose-zoutoplossing, olijfolie en maïsolie. Bij het ontbreken van historische of gepubliceerde controlegegevens waaruit blijkt dat een gekozen atypisch oplosmiddel/medium geen micronuclei en andere schadelijke effecten veroorzaakt, dient een eerste onderzoek te worden uitgevoerd om vast te stellen dat de oplosmiddel/mediumcontrole aanvaardbaar is. Controles Positieve controles Normaal gesproken wordt in elke test een groep dieren opgenomen die wordt behandeld met een positieve controlestof. Hiervan kan worden afgezien wanneer het testlaboratorium aantoonbare bekwaamheid heeft in het uitvoeren van de test en een bereik van historische positieve controles heeft vastgesteld. Wanneer geen parallelle positieve controlegroep wordt opgenomen, moeten in elk experiment scoringcontroles (ongekleurde en ongefixeerde preparaten of celsuspensiemonsters, indien van toepassing voor de scoringmethode) worden opgenomen. Deze kunnen worden verkregen door in de scoring van de studie passende referentiemonsters op te nemen die zijn verkregen en bewaard van een apart positieve controle-experiment dat op gezette tijden (bv. elke 6-18 maanden) wordt verricht; bijvoorbeeld tijdens de bekwaamheidstoetsing en daarna op gezette tijden, indien nodig. Positieve controlestoffen moeten op betrouwbare wijze leiden tot een detecteerbare stijging van de frequentie van micronuclei ten opzichte van het spontane niveau. Bij het handmatig scoren met een microscoop moeten de doseringen van de positieve controles zodanig worden gekozen dat de effecten duidelijk zijn, maar de gecodeerde monsters niet onmiddellijk als zodanig herkenbaar zijn voor de scorer. Het is aanvaardbaar dat de positieve controle langs een andere weg wordt toegediend dan de teststof, met behulp van een ander behandelingsschema, en dat bemonstering op slechts één tijdstip plaatsvindt. Tevens kan, waar dat van toepassing is, het gebruik van chemische stoffen uit een verwante chemische klasse als positieve controle worden overwogen. Voorbeelden van voor positieve controle gebruikte chemische stoffen staan vermeld in tabel 1. Tabel 1 Voorbeelden van positieve controlestoffen. Chemische stoffen en CAS RN Ethylmethaansulfonaat [CAS RN 62-50-0] Methylmethaansulfonaat [CAS RN 66-27-3] Ethylnitrosoureum [CAS RN 759-73-9] Mitomycine C [CAS RN 50-07-7] Cyclofosfamide (monohydraat) [CAS RN 50-18-0 (CAS RN 6055-19-2)] Triëthyleenmelamine [CAS RN 51-18-3] Colchicine [CAS RN 64-86-8] of vinblastine [CAS RN 865-21-4] — als aneugenen Negatieve controles Dieren in een negatieve controlegroep moeten op alle bemonsteringstijdstippen worden opgenomen en ook verder op dezelfde wijze worden behandeld als de behandelingsgroepen, zij het dat zij geen teststof toegediend krijgen. Indien bij de toediening van de teststof van een oplosmiddel/medium gebruik wordt gemaakt, dient dit oplosmiddel/medium aan de controlegroep toegediend te worden. Als uit historische negatieve controlegegevens op ieder bemonsteringstijdstip voor het testlaboratorium een consistente spreiding over de dieren en consistente frequenties van cellen met micronuclei blijken, is mogelijk slechts één enkele bemonstering voor de negatieve controle nodig. Wanneer voor negatieve controles één bemonstering plaatsvindt, dient deze plaats te vinden op het eerste bemonsteringstijdstip dat in de studie wordt gebruikt. Als perifeer bloed wordt gebruikt, kan een monster dat vóór de behandeling wordt genomen ook aanvaardbaar zijn in plaats van een gelijktijdige negatieve controle, maar alleen wanneer het gaat om een korte studie en wanneer de resultaten consistent zijn met de controlegegevens uit het verleden van het testlaboratorium. Voor ratten is aangetoond dat het nemen van kleine monsters (bv. minder dan 100 μl/dag) vóór de behandeling een minimaal effect heeft op de achtergrondfrequentie van micronuclei (25). PROCEDURE Aantal en geslacht van de dieren In het algemeen is de micronucleusrespons voor mannetjes en vrouwtjes vergelijkbaar, en de meeste studies kunnen daarom bij beide geslachten worden uitgevoerd (26). Gegevens waaruit blijkt dat er relevante verschillen tussen vrouwtjes en mannetjes bestaan (bv. verschillen in systemische toxiciteit, metabolisme, biologische beschikbaarheid, toxiciteit voor het beenmerg enz., met inbegrip van bv. in een bereikbepalingsonderzoek), zijn een aanmoediging om beide geslachten te gebruiken. In dit geval kan het passend zijn om een onderzoek bij beide geslachten uit te voeren, bijvoorbeeld als onderdeel van een toxiciteitsonderzoek bij herhaalde toediening. Het kan passend zijn om een factoriële onderzoeksopzet te gebruiken als beide geslachten worden gebruikt. Details over de wijze waarop de gegevens bij deze onderzoeksopzet moeten worden geanalyseerd, zijn te vinden in aanhangsel 2. De vaststelling van de groepsgrootte aan het begin van het onderzoek moet erop gericht zijn minimaal 5 analyseerbare dieren van één geslacht, of van elk geslacht als beide worden gebruikt, per groep te krijgen. Wanneer de blootstelling aan een chemische stof bij de mens door het geslacht wordt bepaald, zoals dit bijvoorbeeld bij sommige geneesmiddelen het geval is, moet de test bij het desbetreffende geslacht worden uitgevoerd. Als richtsnoer voor de maximale typische behoefte aan dieren: voor een onderzoek in beenmerg uitgevoerd volgens de in punt 37 vastgestelde parameters met drie dosisgroepen en parallelle negatieve en positieve controlegroepen (elke groep bestaande uit vijf dieren van één geslacht) zijn 25 tot 35 dieren nodig. Dosisniveaus Als er een bereikbepalingsonderzoek wordt uitgevoerd omdat er nog geen bruikbare gegevens over de dosering beschikbaar zijn, moet dit in hetzelfde laboratorium gebeuren met dezelfde soort, dezelfde stam, hetzelfde geslacht en hetzelfde behandelingsschema als bij het hoofdonderzoek (27). Het onderzoek moet erop gericht zijn de maximaal verdraagbare dosis (maximum tolerated dose, MTD) vast te stellen, dat wil zeggen, de hoogste dosis die zal worden verdragen zonder aanwijzingen voor onderzoeksbeperkende toxiciteit, in verhouding tot de duur van de onderzoeksperiode (bv. door het veroorzaken van gewichtsverlies of cytotoxiciteit voor het hematopoïetisch systeem, maar geen dood of aanwijzingen voor pijn, lijden of nood die humane doding noodzakelijk maken (28)). De hoogste dosis kan ook worden gedefinieerd als een dosis die enigerlei aanwijzing van toxiciteit voor het beenmerg oplevert (bv. een daling van de verhouding tussen het aantal onrijpe erytrocyten en het totale aantal erytrocyten in het beenmerg of perifeer bloed met meer dan 50 %, maar tot niet lager dan 20 % van de controlewaarde). Bij het analyseren van CD71-positieve cellen in de perifere bloedsomloop (bv. door flowcytometrie) reageert deze zeer jonge fractie onrijpe erytrocyten sneller op toxische uitdagingen dan het grotere RNA-positieve cohort onrijpe erytrocyten. Daardoor kan bij opzetten met acute blootstelling waarin de fractie CD71-positieve onrijpe erytrocyten wordt onderzocht, een hogere klaarblijkelijke toxiciteit worden gevonden dan bij opzetten die onrijpe erytrocyten identificeren op basis van RNA-gehalte. Wanneer in experimenten wordt gewerkt met niet meer dan vijf behandelingsdagen, kan het hoogste dosisniveau voor de teststoffen die toxiciteit veroorzaken, dus worden gedefinieerd als de dosis die een statistisch significante verlaging veroorzaakt in de verhouding tussen CD71-positieve onrijpe erytrocyten en het totale aantal erytrocyten, maar tot niet minder dan 5 % van de controlewaarde (29). Chemische stoffen die verzadiging van toxicokinetische eigenschappen vertonen, of detoxificatieprocessen induceren die kunnen leiden tot een afname van de blootstelling na langlopende toediening, kunnen uitzonderingen zijn op de doseringscriteria en moeten per geval worden beoordeeld. Om dosisresponsinformatie te krijgen, moet een volledig onderzoek een negatieve controlegroep omvatten en minimaal drie dosisniveaus die in het algemeen een factor 2, maar niet meer dan een factor 4, uit elkaar liggen. Als in een bereikbepalingsonderzoek of op basis van bestaande gegevens geen aanwijzingen voor toxiciteit van de teststof worden gevonden, moet de hoogste dosis voor een toedieningsperiode van 14 dagen of langer 1 000 mg/kg lichaamsgewicht/dag zijn, of voor een toedieningsperiode van minder dan 14 dagen 2 000 mg/kg lichaamsgewicht/dag. Als de teststof echter wel toxiciteit veroorzaakt, moet de MTD de hoogste toegediende dosis zijn en moeten de gebruikte dosisniveaus bij voorkeur een bereik dekken van maximale tot weinig of geen toxiciteit. Wanneer toxiciteit voor het doelweefsel (beenmerg) bij alle geteste dosisniveaus wordt waargenomen, wordt verder onderzoek bij niet-toxische doses aanbevolen. Voor onderzoek dat ten doel heeft de kwantitatieve dosisresponsinformatie volledig te kenschetsen, kunnen aanvullende dosisgroepen nodig zijn. Voor bepaalde typen teststoffen (bv. geneesmiddelen voor menselijk gebruik) waarvoor specifieke eisen gelden, kunnen deze limieten verschillen. Limiettest Als uit doseringsbereikstudies of bestaande gegevens bij gerelateerde dierstammen blijkt dat een behandelingsregime van minstens de limietdosis (zoals hieronder beschreven) geen waarneembare toxische effecten geeft (met inbegrip van achteruitgang van de proliferatie van beenmerg of andere aanwijzingen van toxiciteit voor het doelweefsel), en als op grond van in-vitro genotoxiciteitsstudies of gegevens bij structureel verwante chemische stoffen geen genotoxiciteit wordt verwacht, dan is een volledig onderzoek met drie dosisniveaus mogelijk niet nodig, mits is aangetoond dat de teststof(fen) het doelweefsel (beenmerg) bereik(t)(en). In zulke gevallen kan één dosisniveau, gelijk aan de limietdosis, voldoende zijn. Voor een toedieningsperiode van 14 dagen of meer is de limietdosis 1 000 mg/kg lichaamsgewicht/dag. Voor een toedieningsperiode van minder dan 14 dagen is de limietdosis 2 000 mg/kg lichaamsgewicht/dag. Toediening van de doses Bij de opzet van een test moet rekening worden gehouden met de verwachte blootstellingsroute bij mensen. Daarom kunnen andere blootstellingsroutes zoals voeding, drinkwater, lokaal subcutaan, intraveneus, oraal (via sonde), inhalatie, intratracheaal of implantatie worden gekozen, als deze onderbouwd worden. De route moet in elk geval zodanig worden gekozen dat adequate blootstelling van het (de) doelweefsel(s) is gewaarborgd. Intraperitoneale injectie wordt in het algemeen niet aanbevolen, omdat zij bij mensen geen beoogde blootstellingsroute is, en zij mag uitsluitend worden toegepast met een specifieke wetenschappelijke motivering. Als de teststof wordt vermengd met voedsel of drinkwater, met name in geval van één toediening, moet ervoor worden gezorgd dat de tijdsduur tussen de consumptie van voedsel en water en de bemonstering voldoende is om de effecten te kunnen detecteren (zie punt 37). Het maximale vloeistofvolume dat via sonde of injectie per keer kan worden toegediend, hangt af van de grootte van het proefdier. Het volume mag normaal gesproken niet groter zijn dan 1 ml/100 g lichaamsgewicht. Bij een waterige oplossing kan echter maximaal 2 ml/100 g lichaamsgewicht worden gebruikt. Hogere volumes dan dit moeten worden onderbouwd. Behalve bij irriterende of bijtende teststoffen, die normaal gesproken bij hogere concentraties hevigere effecten hebben, moeten volumeverschillen tot een minimum worden beperkt door de concentratie aan te passen, zodat er op alle dosisniveaus hetzelfde volume in verhouding tot het lichaamsgewicht wordt toegediend. Behandelingsschema Bij voorkeur worden 2 of meer behandelingen uitgevoerd, toegediend met tussenpozen van 24 uur, in het bijzonder wanneer deze test wordt geïntegreerd met andere toxiciteitsonderzoeken. Als alternatief kunnen doses in één keer worden toegediend, als dat wetenschappelijk gerechtvaardigd is (bv. teststoffen waarvan bekend is dat ze de celcyclus blokkeren). De doses van de teststoffen kunnen eventueel worden gesplitst (bv. twee of meer porties op dezelfde dag met een interval van niet meer dan 2-3 uur) om de toediening van een groot volume te vergemakkelijken. In deze omstandigheden, of wanneer de teststof wordt toegediend door inhalatie, moet het bemonsteringstijdstip worden gepland op basis van het tijdstip van de laatste dosistoediening of het einde van de blootstelling. De test kan bij muizen of ratten op een van de volgende drie manieren worden uitgevoerd:
Andere doserings- of bemonsteringsschema's kunnen worden gebruikt wanneer dat relevant en wetenschappelijk gerechtvaardigd is, en om integratie met andere toxiciteitstests te vergemakkelijken. Observaties Algemene klinische observaties van de proefdieren moeten ten minste eenmaal per dag plaatsvinden en klinische tekenen moeten ten minste eenmaal per dag worden geregistreerd, bij voorkeur steeds op hetzelfde tijdstip en met inachtneming van de piekperiode van de te verwachten effecten na toediening. Gedurende de toedieningsperiode moeten alle dieren ten minste tweemaal per dag worden geobserveerd op ziekteverschijnselen en sterfte. Alle dieren moeten bij aanvang van het onderzoek, tijdens onderzoeken met herhaalde toediening ten minste eenmaal per week, en bij het doden worden gewogen. In onderzoeken met een duur van ten minste één week moet het voedselverbruik ten minste wekelijks worden gemeten. Als de teststof met het drinkwater wordt toegediend, moet het waterverbruik bij elke verversing van water en ten minste wekelijks worden gemeten. Dieren die niet-letale indicatoren van excessieve toxiciteit vertonen moeten voor afloop van de testperiode op humane wijze worden gedood (28). In bepaalde situaties kan de lichaamstemperatuur van dieren worden gecontroleerd, omdat door de behandeling geïnduceerde hyper- en hypothermie een rol spelen in het veroorzaken van foutieve resultaten (32) (33) (34). Blootstelling van doelweefsel Op een of meer passende tijdstippen moet een bloedmonster worden genomen en moeten de plasmaspiegels van de teststoffen worden onderzocht om aan te tonen dat blootstelling van het beenmerg heeft plaatsgevonden, in situaties waar dit gerechtvaardigd is en waar geen andere blootstellingsgegevens bestaan (zie punt 48). Beenmerg/bloedpreparaten Beenmergcellen worden doorgaans uit de dijbenen of scheenbenen van de dieren verwijderd, onmiddellijk na humane euthanasie. Gewoonlijk worden cellen verwijderd, geprepareerd en gekleurd met vastgestelde methoden. Kleine hoeveelheden perifeer bloed kunnen volgens passende normen inzake dierenwelzijn worden verkregen met een methode waarbij het proefdier blijft leven, zoals afname van bloed uit een ader in de staart of een ander geschikt bloedvat, of door hartpunctie of monstername uit een groot bloedvat bij de doding van het dier. Voor erytrocyten uit beenmerg of perifeer bloed kunnen cellen, afhankelijk van de analysemethode, onmiddellijk supravitaal worden gekleurd (16) (17) (18) of worden er uitstrijkpreparaten gemaakt die vervolgens worden gekleurd voor microscopie of gefixeerd en passend gekleurd voor flowcytometrische analyse. Door het gebruik van een DNA-specifieke kleurstof [bv. acridine oranje (35) of Hoechst 33258 plus pyronine-Y (36)] kunnen sommige artefacten die zich bij het gebruik van niet voor DNA specifieke kleurstoffen kunnen voordoen, worden voorkomen. Dit voordeel sluit het gebruik van conventionele kleurstoffen (bv. Giemsa voor microscopische analyse) niet uit. Ook andere systemen [zoals cellulosekolommen om cellen die een kern bevatten te verwijderen (37) (38)] kunnen worden gebruikt, mits is aangetoond dat deze systemen geschikt zijn voor monsterbereiding in het laboratorium. Wanneer deze methoden van toepassing zijn, kunnen antikinetochore antilichamen (39), FISH met pancentromere DNA-proben (40) of geprepareerde in situ labeling met pancentromeer-specifieke primers, samen met de passende DNA-tegenkleuring (41) worden gebruikt om de aard (chromosoom/chromosoomfragment) van de micronuclei te identificeren, teneinde vast te stellen of het mechanisme van micronucleusinductie toe te schrijven is aan clastogene en/of aneugene activiteit. Andere methoden voor differentiatie tussen clastogenen en aneugenen kunnen worden gebruikt indien is aangetoond dat zij doeltreffend zijn. Analyse (handmatig en geautomatiseerd) Alle te analyseren objectglaasjes en monsters, ook die van positieve en negatieve controles, worden vóór willekeurig welk type analyse onafhankelijk gecodeerd en gerandomiseerd, zodat de handmatige scorer de behandelingssituatie niet kent; deze codering is niet nodig wanneer geautomatiseerde scoringsystemen worden gebruikt die niet vertrouwen op visuele inspectie en niet door hun bedieners kunnen worden beïnvloed. Voor elk dier wordt de verhouding tussen het aantal onrijpe erytrocyten en het totale aantal (onrijpe + rijpe) erytrocyten bepaald door in totaal voor het beenmerg ten minste 500 erytrocyten en voor het perifere bloed ten minste 2000 erytrocyten te tellen (42). Voor de bepaling van de frequentie waarmee onrijpe erytrocyten met micronuclei voorkomen, moeten ten minste 4000 onrijpe erytrocyten per dier worden gescoord (43). Als de databank met historische negatieve controlegegevens uitwijst dat de gemiddelde achtergrondfrequentie van onrijpe erytrocyten met micronuclei in het testlaboratorium < 0,1 % is, moet worden overwogen aanvullende cellen te scoren. Bij het analyseren van monsters mag de verhouding tussen het aantal onrijpe erytrocyten en het totale aantal erytrocyten in behandelde dieren niet lager zijn dan 20 % van de verhouding in de (medium/oplosmiddel) controles wanneer gescoord wordt met microscopie, en niet lager dan ongeveer 5 % van de verhouding in de (medium/oplosmiddel) controles wanneer CD71+ onrijpe erytrocyten gescoord worden met cytometrische methoden (zie punt 31) (29). Voorbeeld: voor een beenmergtest die wordt gescoord met microscopie, is de bovengrens voor de toxiciteit 10 % onrijpe erytrocyten als het aandeel onrijpe erytrocyten in het beenmerg in de controles 50 % is. Omdat de milt bij ratten erytrocyten met micronuclei sekwestreert en vernietigt, geniet het de voorkeur de analyse van onrijpe erytrocyten met micronuclei te beperken tot de jongste fractie om bij het analyseren van perifeer bloed van ratten een hoge gevoeligheid van de test te behouden. Wanneer geautomatiseerde analysemethoden worden gebruikt, kunnen deze meest onrijpe erytrocyten worden geïdentificeerd op basis van hun hoge RNA-gehalte, of het hoge niveau van transferrinereceptoren (CD71+) aanwezig op hun oppervlak (31). Uit een directe vergelijking van verschillende kleuringsmethoden is echter gebleken dat met verschillende methoden, ook met conventionele kleuring met acridine oranje, bevredigende resultaten kunnen worden verkregen (3) (4). GEGEVENS EN RAPPORTAGE Verwerking van de resultaten Gegevens van individuele dieren moeten in tabelvorm worden gepresenteerd. Voor elk onderzocht dier worden het aantal gescoorde onrijpe erytrocyten, het aantal onrijpe erytrocyten met micronuclei en het aandeel onrijpe erytrocyten op het totale aantal apart vermeld. Wanneer muizen ononderbroken worden behandeld gedurende een periode van ten minste 4 weken, moeten ook de gegevens over het aantal en het aandeel rijpe erytrocyten met micronuclei worden vermeld, indien deze zijn verzameld. Gegevens over de toxiciteit en klinische tekenen bij dieren moeten eveneens worden gemeld. Aanvaardbaarheidscriteria Voor de aanvaardbaarheid van de test gelden de volgende criteria:
Beoordeling en interpretatie van resultaten Indien aan alle aanvaardbaarheidscriteria is voldaan, wordt een teststof als duidelijk positief beschouwd als:
Indien op een bepaald bemonsteringstijdstip alleen de hoogste dosis wordt onderzocht, wordt een teststof als duidelijk positief beschouwd als er een statistisch significante stijging vergeleken met de gelijktijdige negatieve controle is en de resultaten buiten de verdeling van de historische negatieve controlegegevens vallen (bv. op Poisson-verdeling gebaseerde 95 %-controlegrenzen). Aanbevelingen voor de meest geschikte statistische methoden zijn te vinden in de literatuur (44) (45) (46) (47). Wanneer een dosisresponsanalyse wordt uitgevoerd, moeten ten minste drie behandelde dosisgroepen worden geanalyseerd. De statistische toetsen moeten het dier als de proefeenheid gebruiken. Positieve resultaten bij de micronucleustest wijzen erop dat een teststof micronuclei induceert die ontstaan door chromosoombeschadigingen of beschadiging van het mitotisch apparaat in de erytroblasten van de geteste soort. Indien een test werd uitgevoerd om centromeren binnen micronuclei de detecteren, is een teststof die centromeer-bevattende micronuclei oplevert (centromeer-DNA of kinetochoor, indicatief voor verlies van hele chromosomen), bewijs dat de teststof een aneugeen is. Indien aan alle aanvaardbaarheidscriteria is voldaan, wordt een teststof als duidelijk negatief beschouwd als in alle onderzochte experimentele omstandigheden:
Aanbevelingen voor de meest geschikte statistische methoden zijn te vinden in de literatuur (44) (45) (46) (47). Bewijs van blootstelling van het beenmerg aan een teststof kan zijn achteruitgang van de verhouding tussen rijpe en onrijpe erytrocyten of meting van de plasma- of bloedspiegels van de teststof. In geval van intraveneuze toediening is geen bewijs van blootstelling nodig. Als alternatief kunnen ADME-gegevens verkregen in een onafhankelijke studie met dezelfde toedieningsweg en dezelfde soort worden gebruikt om blootstelling van het beenmerg aan te tonen. Negatieve resultaten wijzen erop dat de teststof onder de testomstandigheden geen micronuclei in de onrijpe erytrocyten van de geteste soort veroorzaakt. Een duidelijk positieve of duidelijk negatieve reactie behoeft niet te worden bevestigd. Indien de reactie noch duidelijk negatief noch duidelijk positief is en om de biologische relevantie van een resultaat te helpen vaststellen (bv. een zwakke of zeer lichte toename), moeten de gegevens door een deskundige en/of nader onderzoek van de bestaande voltooide experimenten worden beoordeeld. In sommige gevallen kan het nuttig zijn meer cellen te analyseren of een herhalingsexperiment onder aangepaste experimentele omstandigheden uit te voeren. In zeldzame gevallen zal op basis van de gegevens, zelfs na nader onderzoek, niet kunnen worden geconcludeerd dat de teststof positieve of negatieve resultaten oplevert, en zal de studie daarom als moeilijk te interpreteren worden beschouwd. Testverslag In het testverslag moet de volgende informatie worden opgenomen:
LITERATUUR
Aanhangsel 1 DEFINITIES Centromeer : het deel of de delen van een chromosoom waar tijdens de celdeling de spoeldraden aan vastzitten, zodat de verplaatsing van de dochterchromosomen naar de polen van de dochtercellen regelmatig kan verlopen. Chemische stof : een stof of mengsel. Erytroblast : een vroeg ontwikkelingsstadium van een erytrocyt, onmiddellijk voorafgaand aan de onrijpe erytrocyt, waarin de cel nog een kern bevat. Kinetochoor : een structuur van eiwitten die zich op het centromeer van eukaryotische cellen vormt, die het chromosoom tijdens de mitose en meiose verbindt met polymeren van de microtubuli van de mitotische spoel, en zusterchromatiden tijdens de celdeling uit elkaar trekt. Micronucleus : kleine kern die buiten en naast de hoofdkern van cellen tijdens de telofase van de mitose (meiose) ontstaat door achtergebleven chromosoomfragmenten of volledige chromosomen. Normochromatische of rijpe erytrocyt : een geheel rijpe erytrocyt die de RNA-restanten die achterblijven na enucleatie, heeft verloren en/of die andere kortlevende celmarkers die gewoonlijk na enucleatie na de laatste deling van een erytroblast verdwijnen, heeft verloren. Polychromatische of onrijpe erytrocyt : een nieuwgevormde erytrocyt in een tussentijdse ontwikkelingsfase, die vanwege de aanwezigheid van RNA-restanten in de nieuwgevormde cel verkleurt met zowel de blauwe als de rode componenten van klassieke kleurstoffen voor bloed zoals Wright-Giemsa. Deze nieuwgevormde cellen zijn min of meer gelijk aan reticulocyten, die worden gevisualiseerd met een vitale kleurstof die de RNA-restanten doet samenklonteren tot een reticulum. Andere methoden, zoals monochromatische kleuring van RNA met fluorescente kleurstoffen of labeling van kortlevende oppervlaktemarkers zoals CD71 met fluorescente antilichamen, worden nu vaak gebruikt om de nieuwgevormde rode bloedcellen te identificeren. Polychromatische erytrocyten, reticulocyten en CD71-positieve erytrocyten zijn allemaal onrijpe erytrocyten, hoewel elk type een iets andere leeftijdsverdeling heeft. Reticulocyt : Een nieuwgevormde erytrocyt die wordt gekleurd met een vitale kleurstof die cellulaire RNA-restanten doet samenklonteren tot een karakteristiek reticulum. Reticulocyten en polychromatische erytrocyten hebben een vergelijkbare verdeling van de celleeftijd. Teststof : alle volgens deze testmethode geteste stoffen of mengsels. Aanhangsel 2 DE FACTORIËLE OPZET VOOR HET IDENTIFICEREN VAN VERSCHILLEN TUSSEN GESLACHTEN IN DE IN-VIVOTEST OP CHROMOSOOMAFWIJKINGEN De factoriële opzet en zijn analyse Bij deze opzet worden voor elk concentratieniveau minimaal 5 mannetjes en 5 vrouwtjes getest, wat leidt tot een opzet met minimaal 40 dieren (20 mannetjes en 20 vrouwtjes, plus relevante positieve controles). De opzet, die een van de eenvoudigere factoriële opzetten is, is gelijkwaardig aan een tweewegs variantieanalyse met geslacht en concentratieniveau als hoofdeffecten. De gegevens kunnen worden geanalyseerd met veel standaard statistische softwarepakketten, zoals SPSS, SAS, STATA, Genstat, alsook met R. De analyse verdeelt de variabiliteit in de gegevensreeks in de variabiliteit tussen de geslachten, die tussen de concentraties en die verband houdend met de interactie tussen geslacht en concentratie. Elk van de termen wordt getoetst tegen een schatting van de variabiliteit tussen de duplodieren binnen de groepen van dieren van hetzelfde geslacht die dezelfde concentratie toegediend krijgen. Volledige gegevens over de onderliggende methodologie zijn te vinden in diverse statistische standaardhandboeken (zie referenties) en in de 'help'-functionaliteit van de statistische pakketten. De analyse begint met een inspectie van de interactieterm geslacht x concentratie in de ANOVA-tabel (6). Als de interactieterm niet significant is, verschaffen de gecombineerde waarden van de geslachten of van de concentratieniveaus geldige statistische toetsen tussen de niveaus op basis van de gepoolde variabiliteit binnen groepen in de ANOVA. De volgende stap in de analyse is de verdeling van de geschatte variabiliteit tussen concentraties in contrasten, die een toets op lineaire en kwadratische contrasten van de responsen over de concentratieniveaus verschaft. Wanneer de interactie tussen geslacht en concentratie significant is, kan deze term ook worden verdeeld in interactiecontrasten lineair x geslacht en kwadratisch x geslacht. Deze termen verschaffen toetsen voor de vraag of de concentratieresponsen voor de twee geslachten parallel lopen of dat de respons voor de twee geslachten gedifferentieerd is. De geschatte gepoolde variabiliteit binnen groepen kan worden gebruikt voor paarsgewijze toetsen voor het verschil tussen de gemiddelden. Deze vergelijkingen kunnen worden gemaakt tussen de gemiddelden voor de twee geslachten en tussen de gemiddelden voor de verschillende concentratieniveaus zoals voor vergelijkingen met de negatieve controleniveaus. Indien er sprake is van een significante interactie, kunnen vergelijkingen worden gemaakt tussen de gemiddelden van verschillende concentraties binnen een geslacht of tussen de gemiddelden van de geslachten bij dezelfde concentratie. Referenties Er is een groot aantal statistische handboeken waarin de theorie, opzet, methodologie, analyse en interpretatie van factoriële opzetten worden besproken, variërend van de eenvoudigste analyses met twee factoren tot de complexere vormen die worden gebruikt voor het ontwerpen van experimenten. De lijst hieronder is niet uitputtend. Enkele boeken bevatten uitgewerkte voorbeelden van vergelijkbare proefopzetten, in enkele gevallen met code voor het uitvoeren van de analyses met verschillende softwarepakketten.
|
|
6) |
Deel B, hoofdstuk B.15, wordt geschrapt. |
|
7) |
Deel B, hoofdstuk B.16, wordt geschrapt. |
|
8) |
Deel B, hoofdstuk B.18, wordt geschrapt. |
|
9) |
Deel B, hoofdstuk B.19, wordt geschrapt. |
|
10) |
Deel B, hoofdstuk B.20, wordt geschrapt. |
|
11) |
Deel B, hoofdstuk B.24, wordt geschrapt. |
|
12) |
In deel B wordt hoofdstuk B.47 vervangen door: „B.47 Bovine Corneal Opacity and Permeability- (BCOP-) testmethode voor de identificatie van i) chemische stoffen die ernstig oogletsel veroorzaken en ii) chemische stoffen waarvoor classificatie voor oogirritatie of ernstig oogletsel niet nodig is INLEIDING Deze testmethode is gelijkwaardig aan testrichtlijn (TG) 437 (2013) van de OESO. De Bovine Corneal Opacity and Permeability- (BCOP-) testmethode werd in 2006 en 2010 geëvalueerd door het Interagency Coordinating Committee on the Validation of Alternative Methods (ICCVAM), samen met het Europees Centrum voor de validering van alternatieve methoden (ECVAM) en het Japans Centrum voor de validatie van alternatieve methoden (JaCVAM) (1) (2). Bij de eerste evaluatie werd de BCOP-testmethode geëvalueerd op haar bruikbaarheid voor het identificeren van chemische stoffen (stoffen en mengsels) die ernstig oogletsel veroorzaken (1). Bij de tweede evaluatie werd de BCOP-testmethode geëvalueerd op haar bruikbaarheid voor het identificeren van chemische stoffen (stoffen en mengsels) waarvoor classificatie voor oogirritatie of ernstig oogletsel niet nodig is (2). De BCOP-valideringsdatabank bevatte in totaal 113 stoffen en 100 mengsels (2) (3). Op basis van deze evaluaties en hun collegiale toetsing werd geconcludeerd dat de testmethode in staat is zowel chemische stoffen (zowel stoffen als mengsels) die ernstig oogletsel veroorzaken (categorie 1), als chemische stoffen waarvoor classificatie voor oogirritatie of ernstig oogletsel niet nodig is, zoals gedefinieerd door het mondiaal geharmoniseerd classificatie- en etiketteringssysteem voor chemische stoffen (Globally Harmonized System of Classification and Labelling of Chemicals, GHS) van de Verenigde Naties (VN) (4) en Verordening (EG) nr. 1272/2008 betreffende de indeling, etikettering en verpakking van stoffen en mengsels (CLP-verordening) (7), correct te identificeren. De testmethode werd daarom voor beide doeleinden bekrachtigd als wetenschappelijk geldig. Onder “ernstig oogletsel” wordt verstaan weefselbeschadiging in het oog of een ernstige fysieke gezichtsvermindering na het aanbrengen van een teststof op het oppervlak aan de voorzijde van het oog, die binnen 21 dagen na het aanbrengen niet volledig omkeerbaar is. Teststoffen die ernstig oogletsel veroorzaken worden ingedeeld in VN-GHS-categorie 1. Chemische stoffen die niet zijn geclassificeerd voor oogirritatie of ernstig oogletsel, zijn gedefinieerd als chemische stoffen die niet voldoen aan de voorwaarden voor indeling in VN-GHS-categorie 1 of 2 (2A of 2B), d.w.z. er wordt naar verwezen als VN-GHS Geen categorie. Deze testmethode omvat het aanbevolen gebruik en de beperkingen van de BCOP-testmethode op basis van de evaluaties ervan. De belangrijkste verschillen tussen de oorspronkelijke versie van 2009 en de bijgewerkte versie van 2013 van de OESO-testrichtlijn hebben betrekking op, maar zijn niet beperkt tot: het gebruik van de BCOP-testmethode voor de identificatie van chemische stoffen waarvoor classificatie volgens het GHS-systeem van de VN niet nodig is (punten 2 en 7); verduidelijkingen van de toepasselijkheid van de BCOP-testmethode voor het testen van alcoholen, ketonen en vaste stoffen (punten 6 en 7) en van stoffen en mengsels (punt 8); verduidelijkingen van de manier waarop oppervlakteactieve stoffen en oppervlakteactieve stoffen bevattende mengsels moeten worden getest (punt 28); bijwerkingen en verduidelijkingen met betrekking tot de positieve controles (punten 39 en 40); een bijwerking van de beslissingscriteria van de BCOP-testmethode (punt 47); een bijwerking van de acceptatiecriteria van het onderzoek (punt 48); een bijwerking van de elementen van het testverslag (punt 49); een bijwerking van aanhangsel 1 betreffende definities; de toevoeging van aanhangsel 2 betreffende de voorspellingskracht van de BCOP-testmethode onder verschillende classificatiesystemen; een bijwerking van aanhangsel 3 betreffende de lijst van stoffen voor de bekwaamheidstoetsing; en een bijwerking van aanhangsel 4 betreffende de BCOP-corneahouder (punt 1) en betreffende de opacitometer (punten 2 en 3). Momenteel wordt algemeen aangenomen dat er in de nabije toekomst geen in-vitrotest voor oogirritatie zal komen die de in-vivo-Draize-oogtest volledig zal kunnen vervangen om voorspellingen te doen voor het volledige bereik van irritatie voor verschillende klassen chemische stoffen. Strategische combinaties van meerdere alternatieve testmethoden binnen een (trapsgewijze) teststrategie kunnen de Draize-oogtest mogelijk echter wel vervangen (5). De top-downbenadering (5) is bedoeld om te worden gebruikt wanneer op basis van bestaande informatie verwacht wordt dat een chemische stof een hoog irritatiepotentieel heeft, terwijl de bottom-upbenadering (5) bedoeld is om te worden gebruikt wanneer op basis van bestaande informatie verwacht wordt dat een chemische stof onvoldoende oogirritatie zal veroorzaken om classificatie noodzakelijk te maken. De BCOP-testmethode is een in-vitrotestmethode die onder bepaalde voorwaarden en met specifieke beperkingen kan worden gebruikt voor de classificatie van gevaren voor de ogen en voor de etikettering van chemische stoffen. Hoewel de BCOP-testmethode niet geldig geacht wordt als zelfstandige vervanging van de in-vivo-oogtest bij konijnen, wordt de BCOP-testmethode aanbevolen als een eerste stap binnen een teststrategie zoals de top-downbenadering die door Scott et al. is voorgesteld (5) voor de identificatie van chemische stoffen die ernstig oogletsel veroorzaken, d.w.z. voor chemische stoffen die moeten worden ingedeeld in VN-GHS-categorie 1, zonder verdere tests (4). De BCOP-testmethode wordt ook aanbevolen voor de identificatie van chemische stoffen waarvoor classificatie voor oogirritatie of ernstig oogletsel niet nodig is, zoals gedefinieerd door het GHS van de VN (VN-GHS Geen categorie) (4), binnen een teststrategie zoals de bottom-upbenadering (5). Voor een chemische stof waarvoor met de BCOP-testmethode niet wordt voorspeld dat deze ernstig oogletsel veroorzaakt of dat classificatie voor oogirritatie/ernstig oogletsel niet nodig is, moeten echter aanvullende tests (in vitro en/of in vivo) worden uitgevoerd om tot een definitieve classificatie te komen. Deze testmethode is bedoeld om de procedures te beschrijven die worden gebruikt om het mogelijke gevaar van een teststof voor de ogen te beoordelen aan de hand van het vermogen van deze chemische stof om troebelheid en verhoogde permeabiliteit in een afgezonderde boviene cornea te veroorzaken. Toxische effecten op de cornea worden als volgt gemeten: (i) verminderde lichttransmissie (troebelheid), en (ii) verhoogde doorlating van natriumfluoresceïne (permeabiliteit). De beoordelingen van de troebelheid en permeabiliteit van de cornea na blootstelling aan een teststof worden samengevoegd om een in-vitro-irritatiescore (IVIS) af te leiden, die gebruikt wordt om het irritatieniveau van de teststof te bepalen. Definities worden gegeven in aanhangsel 1. INLEIDENDE OVERWEGINGEN EN BEPERKINGEN Deze testmethode is gebaseerd op het protocol van de BCOP-testmethode van het ICCVAM (6) (7), dat oorspronkelijk ontwikkeld werd op basis van informatie verkregen uit het protocol van het Institute for In Vitro Sciences (IIVS) en Protocol 124 van INVITTOX (8). Laatstgenoemde vertegenwoordigt het protocol dat gebruikt werd voor de door de Europese Gemeenschap gesteunde prevalideringsstudie die in 1997-1998 werd uitgevoerd. Beide protocollen waren gebaseerd op de BCOP-testmethode die voor het eerst werd beschreven door Gautheron et al. (9). De BCOP-testmethode kan worden gebruikt om chemische stoffen te identificeren die ernstig oogletsel veroorzaken zoals gedefinieerd door het GHS van de VN, d.w.z. chemische stoffen die moeten worden ingedeeld in VN-GHS-categorie 1 (4). Wanneer de BCOP-testmethode voor dit doel wordt gebruikt, heeft zij een algemene nauwkeurigheid van 79 % (150/191), een percentage vals-positieve uitslagen van 25 % (32/126) en een percentage vals-negatieve uitslagen van 14 % (9/65), vergeleken met gegevens van de in-vivo-oogtest bij konijnen die werden ingedeeld volgens het GHS-classificatiesysteem van de VN (3) (zie aanhangsel 2, tabel 1). Wanneer teststoffen binnen bepaalde chemische (d.w.z. alcoholen, ketonen) of fysische (d.w.z. vaste stoffen) klassen worden uitgesloten van de databank, heeft de BCOP-testmethode een algemene nauwkeurigheid van 85 % (111/131), een percentage vals-positieve uitslagen van 20 % (16/81) en een percentage vals-negatieve uitslagen van 8 % (4/50) voor het GHS-classificatiesysteem van de VN (3). De mogelijke tekortkomingen van de BCOP-testmethode wanneer deze wordt gebruikt voor de identificatie van chemische stoffen die ernstig oogletsel veroorzaken (VN-GHS-categorie 1), zijn gebaseerd op de waargenomen hoge percentages vals-positieve uitslagen voor alcoholen en ketonen en de waargenomen hoge percentages vals-negatieve uitslagen voor vaste stoffen in de valideringsdatabank (1) (2) (3). Omdat echter niet voor alle alcoholen en ketonen sprake is van overpredictie door de BCOP-testmethode en sommige correct zijn voorspeld als VN-GHS-categorie 1, worden deze twee organische functionele groepen niet beschouwd als groepen die buiten het toepassingsgebied van de testmethode liggen. Het is aan de gebruiker van deze testmethode om te beslissen of een mogelijke overpredictie van een alcohol of keton kan worden geaccepteerd of dat verdere tests moeten worden uitgevoerd in een bewijskrachtbenadering. Wat betreft de percentages vals-negatieve uitslagen voor vaste stoffen, zij opgemerkt dat vaste stoffen kunnen leiden tot variabele en extreme blootstellingsomstandigheden in de in-vivo-Draize-test op oogirritatie, die irrelevante voorspellingen van het werkelijke irritatiepotentieel van deze stoffen kunnen opleveren (10). Er zij ook opgemerkt dat geen van de geïdentificeerde vals-negatieve uitslagen in de ICCVAM-valideringsdatabank (2) (3), in de context van de identificatie van chemische stoffen die ernstig oogletsel veroorzaken (VN-GHS-categorie 1), heeft geleid tot een IVIS ≤ 3, dat het criterium is dat wordt gebruikt om een teststof in te delen in VN-GHS Geen categorie. Vals-negatieve uitslagen van de BCOP-testmethode zijn in deze context bovendien niet kritiek, omdat alle teststoffen met 3 < IVIS ≤ 55 vervolgens worden getest met andere afdoend gevalideerde in-vitrotests, of als laatste optie bij konijnen, afhankelijk van de wettelijke voorschriften, met een sequentiële teststrategie in een op bewijskracht gebaseerde aanpak. Aangezien sommige vaste chemische stoffen door de BCOP-testmethode correct worden voorspeld als stoffen die behoren tot VN-GHS-categorie 1, wordt deze fysische toestand evenmin beschouwd als een toestand die buiten het toepassingsgebied van de testmethode ligt. Onderzoekers zouden kunnen overwegen deze testmethode te gebruiken voor alle typen chemische stoffen, waarbij een IVIS > 55 moet worden geaccepteerd als indicatief voor een respons die ernstig oogletsel veroorzaakt waardoor de teststof zonder verder tests moet worden ingedeeld in VN-GHS-categorie 1. Positieve resultaten die verkregen werden met alcoholen of ketonen, dienen echter, zoals reeds gezegd, met voorzichtigheid te worden geïnterpreteerd vanwege het risico van overpredictie. De BCOP-testmethode kan ook worden gebruikt om chemische stoffen te identificeren die niet hoeven te worden ingedeeld voor oogirritatie of ernstig oogletsel volgens het VN-GHS-classificatiesysteem (4). Wanneer de BCOP-testmethode voor dit doel wordt gebruikt, heeft zij een algemene nauwkeurigheid van 69 % (135/196), een percentage vals-positieve uitslagen van 69 % (61/89) en een percentage vals-negatieve uitslagen van 0 % (0/107), vergeleken met gegevens van de in-vivo-oogtest bij konijnen die werden ingedeeld volgens het GHS-classificatiesysteem van de VN (3) (zie aanhangsel 2, tabel 2). Het verkregen percentage vals-negatieve uitslagen (in vivo in VN-GHS Geen categorie ingedeelde chemische stoffen die een IVIS > 3 opleveren, zie punt 47) is behoorlijk hoog, maar niet kritiek in deze context, omdat alle teststoffen met 3 < IVIS ≤ 55 vervolgens worden getest met andere afdoend gevalideerde in-vitrotests, of als laatste optie bij konijnen, afhankelijk van de wettelijke voorschriften, met een sequentiële teststrategie in een op bewijskracht gebaseerde aanpak. De BCOP-testmethode vertoont geen specifieke tekortkomingen voor het testen van alcoholen, ketonen en vaste stoffen, wanneer het doel van de test is chemische stoffen te identificeren waarvoor classificatie voor oogirritatie of ernstig oogletsel niet nodig is (VN-GHS Geen categorie) (3). Onderzoekers zouden kunnen overwegen deze testmethode te gebruiken voor alle typen chemische stoffen, waarbij een negatief resultaat (IVIS ≤ 3) moet worden geaccepteerd als een indicatie dat classificatie niet nodig is (VN-GHS Geen categorie). Aangezien de BCOP-testmethode slechts 31 % van de chemische stoffen waarvoor classificatie voor oogirritatie of ernstig oogletsel niet nodig is, correct kan identificeren, zou deze testmethode niet de eerste keuze moeten zijn om een bottom-upbenadering mee te beginnen (5) als er andere gevalideerde en erkende in-vitromethoden met een vergelijkbare hoge gevoeligheid maar een hogere specificiteit beschikbaar zijn. De BCOP-valideringsdatabank bevatte in totaal 113 stoffen en 100 mengsels (2) (3). De BCOP-testmethode wordt daarom toepasbaar geacht voor het testen van zowel stoffen als mengsels. De BCOP-testmethode wordt niet aanbevolen voor het identificeren van teststoffen die moeten worden ingedeeld als irriterend voor de ogen (VN-GHS-categorie 2 of categorie 2A) of teststoffen die moeten worden ingedeeld als licht irriterend voor de ogen (VN-GHS-categorie 2B), vanwege het aanzienlijke aantal chemische stoffen van VN-GHS-categorie 1 die te laag zijn ingedeeld in VN-GHS-categorie 2, 2A of 2B en het aanzienlijke aantal chemische stoffen van VN-GHS Geen categorie die te hoog zijn ingedeeld in VN-GHS-categorie 2, 2A of 2B (2) (3). Voor dit doel kan het nodig zijn verdere tests met een andere geschikte methode uit te voeren. Bij alle procedures met boviene ogen en cornea's dienen de op de testinrichting toepasselijke regelgeving en procedures voor de behandeling van van dieren afkomstig materiaal te worden nageleefd, met inbegrip van maar niet beperkt tot weefsels en weefselvloeistoffen. Universele voorzorgsmaatregelen voor laboratoria zijn aanbevolen (11). Hoewel de BCOP-testmethode geen rekening houdt met letsels van het ooglid en de iris, wordt wel gekeken naar effecten op het hoornvlies, die bij in-vivo-onderzoek de belangrijkste factor voor indeling zijn bij de VN-GHS-classificatie. De reversibiliteit van hoornvliesletsels op zichzelf kan niet worden beoordeeld met de BCOP-testmethode. Er is voorgesteld, op basis van studies op de ogen van konijnen, dat een beoordeling van de aanvankelijke diepte van het hoornvliesletsel kan worden gebruikt om een onderscheid te maken tussen reversibele en irreversibele effecten (12). Er is echter meer wetenschappelijke kennis nodig om te begrijpen hoe irreversibele effecten optreden die geen verband houden met de aanvankelijke diepte van het letsel. Tot slot kan de potentiële systemische toxiciteit die gepaard gaat met de blootstelling van het oog, met de BCOP-methode niet beoordeeld worden. Deze testmethode zal periodiek worden bijgewerkt wanneer nieuwe informatie en gegevens in aanmerking worden genomen. Histopathologie kan bijvoorbeeld van nut zijn wanneer een vollediger karakterisering van schade aan de cornea nodig is. Zoals beschreven in OESO-leidraad nr. 160 (13), worden gebruikers aangemoedigd cornea's te bewaren en histopathologische specimens te prepareren die kunnen worden gebruikt om een databank en beslissingscriteria op te zetten die de nauwkeurigheid van deze testmethode verder kunnen verbeteren. Elk laboratorium dat deze testmethode voor het eerst opstelt, dient gebruik te maken van de in aanhangsel 3 voorziene chemische stoffen voor bekwaamheidstoetsing. Een laboratorium kan deze chemische stoffen gebruiken om zijn technische bekwaamheden met betrekking tot het uitvoeren van de BCOP-testmethode aan te tonen, vooraleer het gegevens van de BCOP-testmethode indient voor de wettelijke gevarenclassificatie. PRINCIPE VAN DE TEST De BCOP-testmethode is een organotypisch model dat handhaving op korte termijn van de normale fysiologische en biochemische werking van de boviene cornea in vitro vereist. In deze testmethode wordt schade door de teststof beoordeeld door kwantitatieve metingen van de wijzigingen in de troebelheid en permeabiliteit van de cornea door gebruik van een opacitometer, respectievelijk een VIS-spectrofotometer. Beide metingen worden gebruikt om een IVIS te berekenen die gebruikt wordt om een in-vitro-irritatiegevarenclassificatie toe te kennen voor de voorspelling van de mogelijke in-vivo-oogirritatie door een teststof (zie beslissingscriteria in punt 48). De BCOP-testmethode gebruikt afgezonderde cornea's van de ogen van pas geslacht vee. De troebelheid van cornea's wordt kwantitatief gemeten als de hoeveelheid lichttransmissie door de cornea. De permeabiliteit wordt kwantitatief gemeten als de hoeveelheid natriumfluoresceïne die door de volledige dikte van de cornea passeert, wat gemeten wordt in het medium dat zich bevindt in de achterkamer. Teststoffen worden aangebracht op de epitheellaag van de cornea door toevoeging aan de voorkamer van de corneahouder. Aanhangsel 4 geeft een beschrijving en een tekening van een corneahouder die wordt gebruikt in de BCOP-testmethode. Corneahouders zijn verkrijgbaar in de handel of kunnen zelf worden gemaakt. Bron en ouderdom van runderogen en selectie van diersoorten Vee dat naar de slachterij wordt gebracht, wordt meestal geslacht ofwel voor menselijke consumptie ofwel voor ander commercieel gebruik. Voor gebruik in de BCOP-testmethode worden enkel de cornea's van gezonde dieren die geschikt worden geacht voor de menselijke voedselketen in aanmerking genomen. Aangezien vee een grote verscheidenheid aan gewicht vertoont, afhankelijk van ras, leeftijd en geslacht, is er geen aanbevolen gewicht van het dier op het ogenblik van de slachting. De afmetingen van de cornea's kunnen variëren als gevolg van het gebruik van de ogen van dieren van verschillende leeftijdscategorieën. Cornea's met een horizontale diameter van > 30,5 mm en een centrale corneale dikte (CCT) ≥ 1100 μm zijn meestal afkomstig van vee dat ouder is dan acht jaar, terwijl cornea's met een horizontale diameter van < 28,5 mm en een CCT < 900 μm meestal afkomstig zijn van vee dat jonger is dan vijf jaar (14). Om die reden worden doorgaans geen ogen gebruikt van vee dat ouder is dan 60 maanden. Ogen van vee jonger dan 12 maanden worden doorgaans niet gebruikt aangezien de ogen dan nog niet volgroeid zijn en de corneale dikte en diameter aanzienlijk kleiner zijn dan deze opgetekend voor ogen van volwassen vee. Het gebruik van cornea's van jonge dieren (d.w.z. 6 tot 12 maanden oud) is toegestaan aangezien hieraan voordelen verbonden zijn, zoals grotere beschikbaarheid, een nauwe leeftijdsgroep en minder gevaar met betrekking tot de blootstelling van werknemers aan boviene spongiforme encefalopathie (15). Aangezien nadere evaluatie van het effect van corneale grootte of dikte op de responsiviteit op corrosieve en irriterende chemische stoffen nuttig kan zijn, worden gebruikers aangemoedigd om de geschatte leeftijd en/of het gewicht van de dieren waarvan de cornea's in onderzoek worden gebruikt, te rapporteren. Verzameling en vervoer van ogen naar het laboratorium De ogen worden verzameld door werknemers van de slachterij. Om mechanische en andere vormen van oogschade te minimaliseren, dienen de ogen na de dood zo spoedig mogelijk verwijderd te worden en onmiddellijk na verwijdering en tijdens het vervoer gekoeld te worden. Om blootstelling van de ogen aan mogelijk irriterende chemische stoffen te voorkomen, mogen de werknemers van de slachterij geen detergent gebruiken bij het afspoelen van de kop van het dier. De ogen dienen volledig te worden ondergedompeld in gekoelde Hanks' Balanced Salt Solution (HBSS) in een houder van aangepaste grootte en naar het laboratorium te worden vervoerd op een wijze die bederf en/of bacteriële contaminatie minimaliseert. Omdat de ogen worden verzameld tijdens het slachtproces, is het mogelijk dat ze blootgesteld worden aan bloed en andere biologische materialen zoals bacteriën en andere micro-organismen. Om die reden dient te worden verzekerd dat het risico op besmetting zo klein mogelijk is (bv. door de houder met de ogen tijdens de verzameling en het vervoer op nat ijs te plaatsen en door antibiotica toe te voegen aan de HBSS die gebruikt wordt om de ogen tijdens het vervoer te bewaren [bv. penicilline aan 100 IU/ml en streptomycine aan 100 μg/ml]). Het tijdsinterval tussen de verzameling van de ogen en het gebruik van de cornea's in de BCOP-testmethode dient te worden beperkt tot een minimum (gewoonlijk verzameling en gebruik op dezelfde dag) en mag de onderzoeksresultaten niet in gevaar brengen. Deze resultaten zijn gebaseerd op de selectiecriteria voor de ogen alsook op de positieve en negatieve controleresponsen. Alle in het onderzoek gebruikte ogen dienen van dezelfde groep op een bepaalde dag verzamelde ogen te zijn. Selectiecriteria voor in de BCOP-testmethode te gebruiken ogen In het laboratorium aangekomen, worden de ogen zorgvuldig onderzocht voor gebreken zoals verhoogde troebelheid, krassen en neovascularisatie. Enkel cornea's zonder deze gebreken mogen worden gebruikt. De kwaliteit van elke cornea wordt ook op latere tijdstippen van het onderzoek beoordeeld. Cornea's met een troebelheid van meer dan zeven eenheden of equivalent voor de gebruikte opacitometer en corneahouders na een initiële equilibreerperiode van een uur, dienen te worden weggegooid (OPMERKING: de opacitometer dient te worden geijkt met troebelheidsnormen die worden gebruikt voor de vaststelling van de troebelheidseenheden, zie aanhangsel 4). Elke behandelingsgroep (teststof, tegelijkertijd gemeten negatieve en positieve controles) bevat minstens drie ogen. Drie cornea's dienen te worden gebruikt voor de negatieve controle in de BCOP-testmethode. Aangezien alle cornea's uit de oogbol worden verwijderd en in de corneale kamers worden geplaatst, bestaat het risico voor artefacten door de behandeling van de individuele corneale troebelheids- en permeabiliteitswaarden (met inbegrip van de negatieve controle). Bovendien worden de troebelheids- en permeabiliteitswaarden van de cornea's van de negatieve controle gebruikt om de troebelheids- en permeabiliteitswaarden van de met de teststof behandelde cornea's en de cornea's van de positieve controle in de IVIS-berekeningen te corrigeren. PROCEDURE Voorbereiding van de ogen Cornea's zonder gebreken worden voorzichtig losgemaakt van de harde oogrok met een rand van 2 tot 3 mm, die nodig zal zijn voor de opeenvolgende handelingen, zodat geen schade ontstaat aan het corneale epitheel en endotheel. De afzonderlijke cornea's worden in een speciaal ontworpen corneahouder geplaatst die bestaat uit een voorkamer en een achterkamer die de epitheellaag, respectievelijk de endotheellaag van de cornea raken. Beide kamers zijn overvloedig gevuld met voorverwarmde Eagle's Minimum Essential Medium (EMEM) zonder fenolrood (eerst de achterkamer) om de vorming van bellen te voorkomen. Het apparaat wordt dan geëquilibreerd op 32 ± 1 °C voor een periode van ten minste één uur om de cornea's met het medium te laten equilibreren en een normale metabolische activiteit te bereiken voor zover mogelijk (de temperatuur van het hoornvliesoppervlak in vivo is ongeveer 32 °C). Na het equilibreren wordt nieuwe voorverwarmde EMEM zonder fenolrood toegevoegd aan beide kamers en worden referentietroebelheidswaarden gemeten in elke cornea. Cornea's die macroscopische weefselschade (bv. krassen, pigmentatie, neovascularisatie) of een troebelheid van meer dan zeven eenheden of equivalent voor de gebruikte opacitometer en corneahouders vertonen, worden verwijderd. Minstens drie cornea's worden geselecteerd als cornea's voor negatieve (of oplosmiddel-) controle. De resterende cornea's worden dan onderverdeeld in een behandelings- en een positieve-controlegroep. Aangezien de warmtecapaciteit van water groter is dan die van lucht, geeft water stabielere temperatuuromstandigheden voor incubatie. Om die reden wordt het gebruik van een waterbad aanbevolen om de temperatuur van de corneahouder met inhoud op 32 ± 1 °C te houden. Luchtincubators kunnen echter ook worden gebruikt met de nodige voorzorgsmaatregelen voor behoud van de temperatuurstabiliteit (bv. door voorverwarming van de houders en media). Aanbrenging van de teststof Twee verschillende behandelingsprotocollen worden gebruikt, één voor vloeistoffen en oppervlakteactieve stoffen (vaste stoffen en vloeistoffen), en één voor niet-oppervlakteactieve vaste stoffen. Vloeistoffen worden onverdund getest. Halfvaste stoffen, pasta's en was worden gewoonlijk getest op dezelfde wijze als vloeistoffen. Zuivere oppervlakteactieve stoffen worden getest met een concentratie van 10 % g/v in een oplossing van 0,9 % natriumchloride, gedistilleerd water of een ander oplosmiddel waarvan geweten is dat het geen nadelig effect heeft op het testsysteem. Alternatieve verdunningen dienen te worden gemotiveerd. Mengsels die oppervlakteactieve stoffen bevatten, kunnen onverdund of verdund tot een passende concentratie worden getest, afhankelijk van het relevante blootstellingsscenario in vivo. De geteste concentratie dient te worden gemotiveerd. Cornea's worden blootgesteld aan vloeistoffen en oppervlakteactieve stoffen voor een periode van 10 minuten. Andere blootstellingsperioden dienen op adequate wetenschappelijke manier te worden gemotiveerd. Zie aanhangsel 1 voor een definitie van een oppervlakteactieve stof en een oppervlakteactieve stof bevattend mengsel. Niet-oppervlakteactieve stoffen worden gewoonlijk getest als oplossingen of suspensies met een concentratie van 20 % g/v in een oplossing van 0,9 % natriumchloride, gedistilleerd water of een ander oplosmiddel waarvan geweten is dat het geen nadelig effect heeft op het testsysteem. In bepaalde omstandigheden en met de juiste wetenschappelijke verklaring, kunnen vaste stoffen ook zuiver getest worden door rechtstreekse aanbrenging op het cornea-oppervlak aan de hand van de open-kamermethode (zie punt 32). Cornea's worden blootgesteld aan vaste stoffen voor een periode van vier uur, maar zoals bij vloeistoffen en oppervlakteactieve stoffen kunnen met de juiste wetenschappelijke motivering alternatieve blootstellingsperioden worden gebruikt. Verschillende behandelingsmethoden kunnen worden gebruikt afhankelijk van de fysische aard en chemische kenmerken (bv. vaste stoffen, vloeistoffen, viskeuze of niet-viskeuze vloeistoffen) van de teststof. Essentieel is dat de teststof de hele epitheellaag bedekt en dat deze chemische stof voldoende wordt verwijderd tijdens het uitwassen. Een gesloten-kamermethode wordt gewoonlijk gebruikt voor niet-viskeuze tot licht-viskeuze vloeibare teststoffen, terwijl een open-kamermethode gewoonlijk wordt gebruikt voor semi-viskeuze en viskeuze vloeibare teststoffen en voor zuiver vaste stoffen. In de gesloten-kamermethode wordt voldoende teststof (750 μl) om de epitheelzijde van de cornea te bedekken, ingebracht in de voorkamer via de doseergaten aan de bovenzijde van de kamer, waarna de gaten tijdens de blootstelling met kamerstoppen worden verzegeld. Het is belangrijk na te gaan dat elke cornea gedurende het juiste tijdsinterval is blootgesteld aan een teststof. In de open-kamermethode worden de afsluitring van het glas en het glas van de voorkamer verwijderd voor de behandeling. De controle- of teststof (750 μl of voldoende teststof om de cornea volledig te bedekken) wordt rechtstreeks op de epitheellaag van de cornea aangebracht met een micropipet. Als een teststof moeilijk te pipetteren is, kan de teststof met drukcompensatie in een plunjerpipet worden gebracht om de dosistoediening te vergemakkelijken. De pipetpunt van de plunjerpipet wordt ingebracht in de verdeelpunt van de spuit, zodat het materiaal onder druk kan worden opgeladen in de verdeelpunt. Gelijktijdig worden de spuitplunjer ingedrukt en de pipetzuiger opgetrokken. Als er zich luchtbellen vormen in de pipetpunt, dan wordt de teststof verwijderd (uitgedreven) en het proces herhaald tot de punt gevuld is zonder luchtbellen. Indien nodig, kan een gewone spuit (zonder naald) worden gebruikt, aangezien hiermee een nauwkeurige hoeveelheid teststof kan worden gemeten en de teststof gemakkelijk op de epitheellaag van de cornea kan worden aangebracht. Na de dosering wordt het glas van de voorkamer teruggeplaatst om opnieuw een gesloten systeem te creëren. Incubatie na blootstelling Na de blootstellingsperiode worden de teststof, de negatieve-controle- of de positieve-controlestof verwijderd uit de voorkamer en wordt het epitheel ten minste drie keer (of tot geen spoor van de teststof meer kan worden waargenomen) gewassen met EMEM (dat fenolrood bevat). Een medium dat fenolrood bevat, wordt voor het uitwassen gebruikt aangezien verkleuring van het fenolrood kan worden gecontroleerd om de doeltreffendheid van het uitwassen van zuurrijke of alkalische teststoffen te bepalen. De cornea's worden meer dan drie keer gewassen als het fenolrood nog steeds verkleurt (geel of purper) of als er nog teststof zichtbaar is. Zodra het medium vrij is van teststof, krijgen de cornea's een laatste uitwassing met EMEM (zonder fenolrood). EMEM (zonder fenolrood) wordt gebruikt als laatste uitwassing om te verzekeren dat alle fenolrood uit de voorkamer is verwijderd voordat de troebelheid wordt gemeten. De voorkamer wordt dan gevuld met nieuwe EMEM zonder fenolrood. Voor vloeistoffen of oppervlakteactieve stoffen worden de cornea's eerst uitgewassen en vervolgens geïncubeerd op 32 ± 1 °C voor een bijkomende periode van twee uur. Langere post-blootstellingstijd kan nuttig zijn in bepaalde omstandigheden en kan geval per geval worden beschouwd. Met vaste stoffen behandelde cornea's worden grondig uitgewassen na een blootstelling van vier uur maar hebben geen bijkomende incubatietijd nodig. Aan het einde van de incubatietijd na blootstelling voor vloeistoffen en oppervlakteactieve stoffen en aan het einde van vier uur blootstelling voor niet-oppervlakteactieve vaste stoffen, worden de troebelheid en permeabiliteit van elke cornea opgetekend. Elke cornea wordt eveneens visueel geobserveerd en relevante waarnemingen worden opgetekend (bv. loskomend weefsel, overblijvende teststof, niet-uniforme troebelheidspatronen). Deze waarnemingen kunnen belangrijk zijn aangezien ze kunnen worden weerspiegeld door variaties in de metingen van de opacitometer. Controlestoffen In elk experiment worden gelijktijdig negatieve controles of controles met oplosmiddel/medium en positieve controles uitgevoerd. Bij het testen van een vloeistof aan 100 %, wordt een gelijktijdige negatieve controle (bv. 0,9 % natriumchlorideoplossing of gedistilleerd water) in de BCOP-testmethode opgenomen, zodat niet-specifieke wijzigingen in het testsysteem kunnen worden opgespoord. Dit levert tevens een referentiekader voor de eindpunten van het onderzoek. Het verzekert ook dat de omstandigheden van het onderzoek niet op ongepaste wijze leiden tot een irriterende respons. Bij het testen van een verdunde vloeistof, oppervlakteactieve stof of vaste stof, wordt gelijktijdig een controlegroep met een oplosmiddel/medium in de BCOP-testmethode opgenomen zodat niet-specifieke wijzigingen in het testsysteem kunnen worden opgespoord; dit levert tevens een referentiekader voor de eindpunten van het onderzoek. Er kunnen enkel oplosmiddelen/media worden gebruikt waarvan geweten is dat ze geen nadelig effect op het testsysteem hebben. In elk experiment wordt een chemische stof waarvan bekend is dat deze een positieve reactie geeft, opgenomen als gelijktijdige positieve controle om de integriteit van het testsysteem te verifiëren en na te gaan dat het experiment juist is uitgevoerd. Om te verzekeren dat de variabiliteit in de positieve-controlerespons doorheen de tijd kan worden beoordeeld, mag de grootte van de irriterende respons niet buitensporig zijn. Voorbeelden van positieve controles voor vloeibare teststoffen zijn 100 % ethanol of 100 % dimethylformamide. Een voorbeeld van een positieve controle voor vaste teststoffen is een concentratie van 20 % g/v imidazool in een 0,9 % natriumchlorideoplossing. IJkstoffen zijn nuttig voor de beoordeling van potentiële oogirritatie bij onbekende chemische stoffen uit een specifieke chemische klasse of productklasse of voor de beoordeling van potentiële relatieve irritatie van een voor het oog irriterende stof binnen een reeks van irritatieresponsen. Gemeten eindpunten De troebelheid wordt gemeten door de hoeveelheid lichttransmissie door de cornea. De corneale troebelheid wordt kwantitatief gemeten met behulp van een opacitometer. De resultaten zijn troebelheidswaarden op een continue schaal. De permeabiliteit wordt bepaald door de hoeveelheid natriumfluoresceïne die alle corneale cellagen penetreert (d.w.z. van de epitheellaag aan het buitenste corneale oppervlak tot de endotheellaag aan de binnenkant). Eén ml natriumfluoresceïneoplossing (4 of 5 mg/ml voor vloeistoffen en oppervlakteactieve stoffen respectievelijk niet-oppervlakteactieve vaste stoffen) wordt toegevoegd aan de voorkamer van de corneahouder, die in verbinding staat met de epitheelzijde van de cornea, terwijl de achterkamer, die in verbinding staat met de endotheelzijde van de cornea, gevuld is met nieuwe EMEM. De houder wordt dan gedurende 90 ± 5 min op 32 ± 1 °C geïncubeerd in een horizontale positie. De hoeveelheid natriumfluoresceïne die doordringt naar de achterkamer wordt kwantitatief gemeten met behulp van UV/VIS-spectrofotometrie. Op 490 nm beoordeelde spectrofotometrische metingen worden opgetekend als optische dichtheid (OD490) of extinctiewaarden, die gemeten worden op een continue schaal. De permeabiliteitswaarden van fluoresceïne worden bepaald aan de hand van de OD490-waarden op basis van een VIS-spectrofotometer met een standaardweglengte van 1 cm. Ook kan een 96-wells-microtiterplaatlezer gebruikt worden op voorwaarde dat (i) het lineaire bereik van de plaatlezer voor de vaststelling van fluoresceïne OD490-waarden kan worden vastgesteld; en (ii) de juiste hoeveelheid fluoresceïnemonsters in de 96-wells-plaat wordt gebruikt om OD490-waarden te verkrijgen die gelijk zijn aan een standaardweglengte van 1 cm (hiervoor kan een volledige well noodzakelijk zijn [meestal 360 μl]). GEGEVENS EN RAPPORTAGE Evaluatie van de gegevens Zodra de troebelheids- en gemiddelde permeabiliteits-(OD490)-waarden gecorrigeerd zijn voor achtergrondtroebelheid en de permeabiliteits-OD490-waarden van de negatieve controle, dienen de gemiddelde troebelheids- en permeabiliteits-OD490-waarden voor elke behandelingsgroep als volgt samengevoegd te worden in een empirisch afgeleide formule om voor elke behandelingsgroep een in-vitro-irritatiescore (IVIS) te berekenen: IVIS = gemiddelde troebelheidswaarde + (15 x gemiddelde permeabiliteits-OD490-waarde) Sina et al. (16) schreef dat deze formule afgeleid was tijdens onderzoeken in eigen laboratorium en interlaboratoriumonderzoeken. De gegevens die gegenereerd werden voor een reeks van 36 verbindingen in een multilaboratoriumonderzoek werden onderworpen aan een multivariate analyse om de best passende verhouding tussen in-vivo- en in-vitrogegevens te bepalen. Wetenschappers van twee verschillende bedrijven hebben deze analyse uitgevoerd en kwamen op ongeveer identieke verhoudingen uit. De troebelheids- en permeabiliteitswaarden dienen ook onafhankelijk van elkaar beoordeeld te worden om te bepalen of een teststof corrosiviteit of hevige irritatie opwekt via slechts een van de twee eindpunten (zie beslissingscriteria). Beslissingscriteria De IVIS-drempelwaarden voor de identificatie van teststoffen als chemische stoffen die ernstig oogletsel veroorzaken (VN-GHS-categorie 1) en teststoffen waarvoor classificatie voor oogirritatie of ernstig oogletsel niet nodig is (VN-GHS Geen categorie) zijn hieronder gegeven:
Acceptatiecriteria van het onderzoek Een test wordt als aanvaardbaar beschouwd als de positieve controle een IVIS oplevert die tussen twee standaardafwijkingen van het huidige historische gemiddelde valt, dat minstens om de drie maanden wordt bijgewerkt of telkens als een aanvaardbare test wordt uitgevoerd in laboratoria waar op onregelmatige basis tests worden uitgevoerd (d.w.z. minder dan een keer per maand). De negatieve-controlerespons of controlerespons met oplosmiddel/medium dient te resulteren in troebelheids- en permeabiliteitswaarden die lager zijn dan de vastgestelde bovengrenswaarden voor achtergrondtroebelheid en permeabiliteit voor boviene cornea's die werden gebruikt voor de negatieve controle respectievelijk oplosmiddel/mediumcontrole. Eén test bestaande uit ten minste drie cornea's zou voldoende moeten zijn voor een teststof, indien de indeling ervan ondubbelzinnig is. Echter, bij grensgevallen in de eerste test moet een tweede test worden overwogen (maar deze is niet noodzakelijkerwijs vereist), evenals een derde test in geval van niet-concordante gemiddelde IVIS- resultaten tussen de eerste twee tests. In deze context wordt een resultaat in de eerste test als grensgeval beschouwd als de voorspellingen van de 3 cornea's niet concordant waren, in de zin dat:
Testverslag Het testverslag dient de volgende informatie te bevatten, indien relevant voor het voeren van het onderzoek:
LITERATUUR
Aanhangsel 1 DEFINITIES Betrouwbaarheid : indicatie van de mate waarin een testmethode, uitgevoerd volgens een vast protocol, op reproduceerbare wijze binnen één laboratorium en tussen laboratoria verspreid over de tijd kan worden uitgevoerd. De betrouwbaarheid wordt beoordeeld door berekening van intra- en interlaboratoriumreproduceerbaarheid en intralaboratoriumherhaalbaarheid. Bewijskracht(proces) : het proces waarbij de sterke en zwakke punten van verschillende gegevens worden onderzocht bij het komen tot en ondersteunen van een conclusie met betrekking tot het gevarenpotentieel van een teststof. Bottom-upbenadering : een trapsgewijze benadering die gebruikt wordt voor een chemische stof waarvan wordt vermoed dat classificatie voor oogirritatie of ernstig oogletsel niet nodig is, die begint met het onderscheiden van chemische stoffen waarvoor classificatie niet nodig is (negatief resultaat) van andere chemische stoffen (positief resultaat). Chemische stof : een stof of een mengsel. Cornea : het doorschijnende gedeelte van het voorste deel van de oogbal dat de iris en pupil bedekt en licht naar binnen toelaat. Corneale permeabiliteit : kwantitatieve meting van de schade aan het hoornvliesepitheel door bepaling van de hoeveelheid natriumfluoresceïne die door alle corneale cellagen dringt. Corneale troebelheid : meting van de mate van troebelheid van de cornea na blootstelling aan een teststof. Verhoogde corneale troebelheid is een aanwijzing voor schade aan de cornea. De troebelheid kan subjectief worden beoordeeld aan de hand van bijvoorbeeld de Draize-oogtest bij konijnen of objectief met een instrument zoals een “opacitometer”. Ernstig oogletsel : veroorzaking van weefselschade aan het oog of ernstige fysische gezichtsvermindering na aanbrenging van een teststof op de voorste ooglaag die niet volledig reversibel is binnen 21 dagen na de aanbrenging. Verwisselbaar met “Irreversibele effecten op ogen” en met “VN-GHS-categorie 1” (4). Gevaar : inherente eigenschap van een agens of situatie die nadelige effecten kan veroorzaken als een organisme, systeem of (sub)populatie aan dit agens wordt blootgesteld. Gevalideerde testmethode : een testmethode waarvoor valideringsstudies werden gedaan om de relevantie (inclusief nauwkeurigheid) en betrouwbaarheid voor een bepaald doel te bepalen. Er dient te worden opgemerkt dat een gevalideerde testmethode niet per definitie voldoende nauwkeurig en betrouwbaar is om aanvaardbaar te worden bevonden voor het voorgestelde doel. GHS (Globally Harmonized System of Classification and Labelling of Chemicals, mondiaal geharmoniseerd classificatie- en etiketteringssysteem voor chemische stoffen van de Verenigde Naties (VN)) : een systeem voor de indeling van chemische producten (stoffen en mengsels) op basis van gestandaardiseerde soorten en niveaus van gevaren van fysische aard en gevaren voor de gezondheid en het milieu, en dat de desbetreffende voorlichtingselementen bepaalt, zoals pictogrammen, signaalwoorden, gevarenaanduidingen, voorzorgsmaatregelen en veiligheidsinformatiebladen, waarmee informatie over de schadelijke effecten van de stoffen kenbaar wordt gemaakt teneinde mensen (waaronder werkgevers, werknemers, transporteurs, consumenten en spoedhulpverleners) en het milieu te beschermen (4). Ijkstof : een chemische stof die gebruikt wordt als norm voor vergelijking met een teststof. Een ijkstof moet de volgende eigenschappen hebben: (i) (een) constante en betrouwbare bron(nen); (ii) een structurele en functionele gelijkenis met de klasse chemische stoffen die getest wordt; (iii) bekende fysische-chemische kenmerken; (iv) ondersteunende gegevens over bekende effecten en (v) een bekende werkzaamheid binnen het bereik van de gewenste respons. In-vitro-irritatiescore (IVIS) : een empirisch afgeleide formule die gebruikt wordt in de BCOP-testmethode waarbij de gemiddelde troebelheid en de gemiddelde permeabiliteit voor elke behandelingsgroep gecombineerd worden in één enkele in-vitroscore voor elke behandelingsgroep. IVIS = gemiddelde troebelheidswaarde + (15 x gemiddelde permeabiliteitswaarde). Irreversibele effecten op ogen : zie “ernstig oogletsel”. Medium-/oplosmiddelcontrole : een onbehandeld monster dat alle componenten bevat van een testsysteem, met inbegrip van het oplosmiddel of medium, dat tezamen met de met de teststof behandelde monsters en andere controlemonsters verwerkt wordt om de referentierespons voor de monsters die behandeld werden met de in hetzelfde oplosmiddel of medium opgeloste teststof vast te stellen. Als dit monster getest wordt met een tegelijkertijd gemeten negatieve controle, toont het ook aan of het oplosmiddel of medium met het testsysteem interageert. Mengsel : een mengsel of een oplossing bestaande uit twee of meer stoffen waarin deze stoffen geen reactie met elkaar aangaan (4). Nauwkeurigheid : de mate van overeenstemming tussen resultaten verkregen met de testmethode en erkende referentiewaarden. Het is een maat voor de prestaties van de testmethode en één aspect van “relevantie”. Deze term en de term “concordantie”, waaronder het percentage correcte uitkomsten van een testmethode wordt verstaan, worden vaak door elkaar gebruikt. Negatieve controle : een onbehandeld monster dat alle bestanddelen van een testsysteem bevat. Dit monster wordt tezamen met de met de teststof behandelde monsters en andere controlemonsters verwerkt om te bepalen of het oplosmiddel interageert met het testsysteem. Niet ingedeeld : chemische stoffen die niet zijn geclassificeerd voor oogirritatie (VN-GHS-categorie 2, 2A of 2B) of ernstig oogletsel (VN-GHS-categorie 1). Verwisselbaar met “VN-GHS Geen categorie”. Oogirritatie : het ontstaan van veranderingen in het oog na het aanbrengen van een teststof op het oppervlak aan de voorzijde van het oog, die binnen 21 dagen na het aanbrengen volledig reversibel zijn. Verwisselbaar met “Reversibele effecten op ogen” en met “VN-GHS-categorie 2” (4). Opacitometer : een instrument dat gebruikt wordt om de “corneale troebelheid” te meten door kwantitatieve beoordeling van lichttransmissie door de cornea. Gewoonlijk bestaat dit instrument uit twee compartimenten, elk met een eigen lichtbron en fotocel. Eén compartiment wordt gebruikt voor de behandelde cornea, terwijl het andere wordt gebruikt voor de ijking en de nulinstelling van het instrument. Licht van een halogeenlamp wordt door een controlebestanddeel (lege kamer zonder vensters of vloeistof) naar een fotocel gestuurd en vergeleken met het licht dat door een experimenteel bestanddeel, dat de kamer met de cornea bevat, naar een fotocel wordt gestuurd. Het verschil in lichttransmissie van de fotocellen wordt vergeleken en er wordt een numerieke troebelheidswaarde weergegeven op een digitaal scherm. Oppervlakteactieve stof : ook genoemd “agens dat inwerkt op de oppervlakte”; dit is een stof, zoals een detergent, die de oppervlaktespanning van een vloeistof kan verlagen en het zo mogelijk maakt dat de vloeistof gaat schuimen of in vaste stoffen doordringt; ook bekend als een bevochtigingsmiddel. Oppervlakteactieve stof bevattend mengsel : in de context van deze testmethode, een mengsel dat een of meer oppervlakteactieve stoffen bevat in een uiteindelijke concentratie van > 5 %. Percentage vals-negatieve uitslagen : het percentage van alle positieve chemische stoffen dat door een testmethode onterecht als negatief wordt geïdentificeerd. Het is één indicator van de prestaties van de testmethode. Percentage vals-positieve uitslagen : het percentage van alle negatieve chemische stoffen dat door een testmethode onterecht als positief wordt geïdentificeerd. Het is één indicator van de prestaties van de testmethode. Positieve controle : een monster met alle bestanddelen van een testsysteem dat met een chemische stof behandeld werd waarvan bekend is dat deze een positieve respons opwekt. Om te verzekeren dat variabiliteit in de positieve-controlerespons doorheen de tijd kan worden beoordeeld, mag de grootte van de positieve respons niet buitensporig zijn. Reversibele effecten op ogen : zie “oogirritatie”. Stof : chemische elementen en verbindingen ervan, zoals zij voorkomen in natuurlijke toestand of bij de productie ontstaan, met inbegrip van alle additieven die nodig zijn voor het behoud van de stabiliteit van het product en alle onzuiverheden ten gevolge van het productieprocédé, doch met uitzondering van elk oplosmiddel dat kan worden afgescheiden zonder dat de stabiliteit van de stof wordt aangetast of de samenstelling ervan wordt gewijzigd (4). Top-downbenadering : een trapsgewijze benadering die gebruikt wordt voor een chemische stof waarvan wordt vermoed dat deze ernstig oogletsel veroorzaakt, die begint met het onderscheiden van chemische stoffen die ernstig oogletsel veroorzaken (positief resultaat) van andere chemische stoffen (negatief resultaat). Teststof : alle volgens deze testmethode geteste stoffen of mengsels. Trapsgewijze teststrategie : een trapsgewijze teststrategie waarbij alle bestaande informatie over een teststof in een bepaalde volgorde wordt herbezien aan de hand van een proces op basis van bewijskracht op elke trap om te bepalen of er voldoende informatie beschikbaar is voor een beslissing tot gevarenclassificatie, vooraleer wordt overgegaan tot de volgende trap. Indien het irritatiepotentieel van een teststof kan worden bepaald op basis van de bestaande informatie, dan zijn bijkomende tests niet vereist. Indien het irritatiepotentieel van een teststof niet kan worden bepaald op basis van de bestaande informatie, dan wordt een trapsgewijze sequentiële testprocedure op dieren uitgevoerd tot een ondubbelzinnige indeling mogelijk is. VN-GHS-categorie 1 : zie “ernstig oogletsel”. VN-GHS-categorie 2 : zie “oogirritatie”. VN-GHS Geen categorie : chemische stoffen die niet voldoen aan de voorwaarden voor indeling in VN-GHS-categorie 1 of 2 (2A of 2B). Verwisselbaar met “Niet ingedeeld”. Aanhangsel 2 VOORSPELLINGSKRACHT VAN DE BCOP-TESTMETHODE Tabel 1 Voorspellingskracht van de BCOP-testmethode voor de identificatie van chemische stoffen die ernstig oogletsel veroorzaken [VN-GHS-/EU-CLP-categorie 1 vs Niet categorie 1 (cat. 2 + Geen cat.); VS-EPA-klasse I vs Niet klasse I (klasse II + klasse III + klasse IV)]
Tabel 2 Voorspellingskracht van de BCOP-testmethode voor de identificatie van chemische stoffen waarvoor classificatie voor oogirritatie of ernstig oogletsel niet nodig is (“niet irriterende stoffen”) [VN-GHS/ EU-CLP Geen categorie vs Niet Geen categorie (cat. 1 + cat. 2); VS-EPA-klasse IV vs Niet klasse IV (klasse I + klasse II + klasse III)]
Aanhangsel 3 CHEMISCHE STOFFEN VOOR BEKWAAMHEIDSTOETSING VOOR DE BCOP-TESTMETHODE Vooraleer over te gaan tot routinegebruik van deze testmethode, moeten laboratoria hun technische bekwaamheid aantonen door een correcte identificatie van de gevarenclassificatie voor de ogen van de 13 chemische stoffen die in tabel 1 worden aangeraden. Deze chemische stoffen werden geselecteerd om het bereik van responsen voor gevaren voor de ogen te vertegenwoordigen op basis van resultaten in de in-vivo-oogtest bij konijnen (TG 405) (17) en het VN-GHS-classificatiesysteem (d.w.z. categorieën 1, 2A, 2B, of Niet ingedeeld) (4). Andere selectiecriteria waren dat chemische stoffen in de handel verkrijgbaar zijn, dat er in-vivo-referentiegegevens van hoge kwaliteit beschikbaar zijn, en dat er in-vitro-gegevens van hoge kwaliteit beschikbaar zijn voor de BCOP-testmethode. Er zijn referentiegegevens beschikbaar in het Streamlined Summary Document (3) en in het ICCVAM Background Review Document voor de BCOP-testmethode (2) (18). Tabel 1 Aanbevolen chemische stoffen voor het aantonen van technische bekwaamheid met de BCOP-testmethode
Aanhangsel 4 DE BCOP-CORNEAHOUDER De BCOP-corneahouders zijn gemaakt van inert materiaal (bv. polypropyleen). De houders bestaan uit twee helften (een voorste en een achterste kamer) en hebben twee gelijke cilindrische inwendige kamers. Elke kamer heeft een inhoud van ongeveer 5 ml en is afgewerkt met een glazen venster waardoor de troebelheidsmetingen worden opgetekend. Elk van de inwendige kamers heeft een diameter van 1,7 cm en is 2,2 cm diep (11). De achterste kamer is voorzien van een o-ring om lekkage te voorkomen. De cornea's worden op de o-ring van de achterste kamers geplaatst met de endotheelzijde naar beneden en de voorste kamers worden op de epitheelzijde van de cornea's geplaatst. De kamers worden op hun plaats gehouden door drie schroeven van roestvrij staal die zich bevinden aan de randen van de kamer. Aan het einde van elke kamer bevindt zich een vensterglas, dat verwijderd kan worden voor gemakkelijke toegang tot de cornea. Tussen het vensterglas en de kamer bevindt zich eveneens een o-ring om lekkage te voorkomen. Elke kamer heeft bovenaan twee openingen voor toevoeging en verwijdering van media en teststoffen. Tijdens de behandeling en de incubatieperiode worden zij afgesloten met een rubberdop. De lichttransmissie door corneahouders kan mogelijk veranderen, omdat de effecten van slijtage of opeenstapeling van residuen van bepaalde chemische stoffen op de boorgaten of op het vensterglas van de inwendige kamers de verstrooiing of reflectie van licht kunnen beïnvloeden. Hierdoor zou de referentiewaarde voor de lichttransmissie (en omgekeerd de referentiewaarde voor de troebelheidsmetingen) door de corneahouders kunnen stijgen of dalen, en dit kan zichtbaar zijn door merkbare veranderingen in de verwachte initiële corneale referentietroebelheidsmetingen in de afzonderlijke kamers (d.w.z. de initiële corneale troebelheidswaarden in specifieke afzonderlijke corneahouders kunnen stelselmatig meer dan 2 of 3 troebelheidseenheden verschillen van de verwachte referentiewaarden). Elk laboratorium zou moeten overwegen een programma vast te stellen voor het beoordelen van veranderingen in de lichttransmissie door de corneahouders, afhankelijk van de aard van de geteste verbindingen en de gebruiksfrequentie van de kamers. Om referentiewaarden vast te stellen, kunnen corneahouders voordat tot routinegebruik wordt overgegaan, worden gecontroleerd door de referentietroebelheidswaarden (of lichttransmissie) van kamers gevuld met volledig medium, zonder cornea's, te meten. De corneahouders worden daarna op gezette tijden gecontroleerd op veranderingen in de lichttransmissie tijdens gebruiksperioden. Elk laboratorium kan de frequentie voor het controleren van de corneahouders vaststellen op basis van de geteste stoffen, de gebruiksfrequentie en waarnemingen van veranderingen in de referentietroebelheidswaarden van cornea's. Indien merkbare veranderingen in de lichttransmissie door de corneahouders worden waargenomen, moeten passende reinigings- en/of polijstprocedures voor de binnenkant van de corneahouders of vervanging ervan worden overwogen. De corneahouder: Opengewerkte tekening 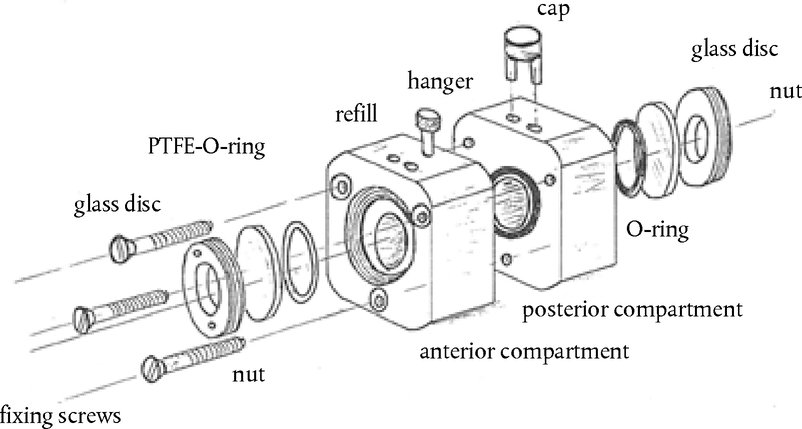
Aanhangsel 5 DE OPACITOMETER De opacitometer is een meetapparaat voor lichttransmissie. Voor bijvoorbeeld de OP-KIT-apparatuur van Electro Design (Riom, Frankrijk) die werd gebruikt in de validering van de BCOP-testmethode, wordt licht van een halogeenlamp door een controlebestanddeel (lege kamer zonder vensters of vloeistof) naar een fotocel gestuurd en vergeleken met het licht dat door een experimenteel bestanddeel, dat de kamer met de cornea bevat, naar een fotocel wordt gestuurd. Het verschil in lichttransmissie van de fotocellen wordt vergeleken en er wordt een numerieke troebelheidswaarde weergegeven op een digitaal scherm. De troebelheidseenheden worden vastgesteld. Andere typen opacitometers met een andere opzet (bv. waarvoor geen gelijktijdige metingen van de controle en experimentele compartimenten vereist zijn) kunnen worden gebruikt indien is aangetoond dat ze vergelijkbare resultaten geven als de gevalideerde apparatuur. De troebelheidsmeter dient een lineaire respons te geven met een reeks troebelheidswaarden inclusief de grenswaarden van de verschillende klassen zoals beschreven in het voorspellend model (d.w.z. tot aan de grenswaarde die de corrosiviteit/het hevig irriterend karakter bepaalt). Om lineaire en nauwkeurige waarden tot een troebelheid van 75-80 troebelheidseenheden te verzekeren, dient de opacitometer te worden gekalibreerd met een reeks kalibratiemiddelen. Kalibratiemiddelen worden in de kalibratiekamer (een corneale kamer ontworpen om de kalibratiemiddelen te bevatten) geplaatst en afgelezen op de opacitometer. De kalibratiekamer is zodanig ontworpen dat de kalibratiemiddelen op ongeveer dezelfde afstand tussen het licht en de fotocel geplaatst zijn als de cornea's tijdens de troebelheidsmetingen. De referentiewaarden en initiële ingestelde waarde hangen af van het gebruikte type apparatuur. Lineariteit van de troebelheidsmetingen moet worden verzekerd door passende (instrumentspecifieke) procedures. Voor bijvoorbeeld de OP-KIT-apparatuur van Electro Design (Riom, Frankrijk) wordt de opacitometer eerst gekalibreerd op 0 troebelheidseenheden met behulp van de kalibratiekamer zonder kalibratiemiddel. Daarna worden drie verschillende kalibratiemiddelen één voor één in de kalibratiekamer geplaatst en worden de troebelheidswaarden gemeten. Kalibratiemiddelen 1, 2 en 3 dienen te resulteren in troebelheidsmetingen die gelijk zijn aan hun vastgestelde waarden van respectievelijk 75, 150 en 225 troebelheidseenheden ± 5 %. |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
13) |
In deel B wordt hoofdstuk B.48 vervangen door: „B.48 Isolated Chicken Eye-testmethode voor de identificatie van i) chemische stoffen die ernstig oogletsel veroorzaken en ii) chemische stoffen waarvoor classificatie voor oogirritatie of ernstig oogletsel niet nodig is INLEIDING Deze testmethode is gelijkwaardig aan testrichtlijn (TG) 438 (2013) van de OESO. De Isolated Chicken Eye- (ICE-) testmethode werd in 2006 en 2010 geëvalueerd door het Interagency Coordinating Committee on the Validation of Alternative Methods (ICCVAM), samen met het Europees Centrum voor de validering van alternatieve methoden (ECVAM) en het Japans Centrum voor de validatie van alternatieve methoden (JaCVAM) (1) (2) (3). Bij de eerste evaluatie werd de ICE-testmethode bekrachtigd als een wetenschappelijk geldige testmethode voor gebruik als screeningtest voor de identificatie van chemische stoffen (stoffen en mengsels) die ernstig oogletsel veroorzaken (categorie 1) zoals gedefinieerd door het mondiaal geharmoniseerd classificatie- en etiketteringssysteem voor chemische stoffen (Globally Harmonized System of Classification and Labelling of Chemicals, GHS) van de Verenigde Naties (VN) (1) (2) (4) en Verordening (EG) nr. 1272/2008 betreffende de indeling, etikettering en verpakking van stoffen en mengsels (CLP-verordening) (12). Bij de tweede evaluatie werd de ICE-testmethode geëvalueerd op haar bruikbaarheid als screeningtest voor de identificatie van chemische stoffen die niet geclassificeerd zijn voor oogirritatie of ernstig oogletsel zoals gedefinieerd door het GHS van de VN (3) (4). Op basis van de resultaten van de valideringsstudie en de aanbevelingen van het peer review panel werd de oorspronkelijke aanbeveling gehandhaafd om de ICE-testmethode te gebruiken voor de classificatie van chemische stoffen die ernstig oogletsel veroorzaken (VN-GHS-categorie 1), omdat de beschikbare databank sinds de oorspronkelijke ICCVAM-validering ongewijzigd is gebleven. In dat stadium werden geen verdere aanbevelingen gedaan om het toepassingsgebied van de ICE-testmethode uit te breiden met andere categorieën. Met het oog op de beoordeling van de bruikbaarheid van de ICE-testmethode voor de identificatie van chemische stoffen waarvoor classificatie voor oogirritatie of ernstig oogletsel niet nodig is, werd de in-vitro- en in-vivo-gegevensverzameling die in de valideringsstudie werd gebruikt, opnieuw beoordeeld (5). Op grond van deze herbeoordeling werd geconcludeerd dat de ICE-testmethode ook kan worden gebruikt voor de identificatie van chemische stoffen waarvoor classificatie voor oogirritatie en ernstig oogletsel zoals gedefinieerd door het GHS van de VN niet nodig is (4) (5). Deze testmethode omvat de aanbevolen toepassingen en beperkingen van de ICE-testmethode op basis van deze evaluaties. De belangrijkste verschillen tussen de oorspronkelijke versie van 2009 en de bijgewerkte versie van 2013 van de testrichtlijn van de OESO hebben betrekking op, maar zijn niet beperkt tot, het gebruik van de ICE-testmethode voor de identificatie van chemische stoffen waarvoor classificatie volgens het GHS-classificatiesysteem van de VN niet nodig is, een bijwerking van de elementen van het testverslag, een bijwerking van aanhangsel 1 betreffende definities, en een bijwerking van aanhangsel 2 betreffende de stoffen voor de bekwaamheidstoetsing. Momenteel wordt algemeen aangenomen dat er in de nabije toekomst geen in-vitrotest voor oogirritatie zal komen die de in-vivo-Draize-oogtest volledig zal kunnen vervangen om voorspellingen te doen voor het volledige bereik van irritatie voor verschillende klassen chemische stoffen. Strategische combinaties van meerdere alternatieve testmethoden binnen een (trapsgewijze) teststrategie kunnen de Draize-oogtest mogelijk echter wel vervangen (6). De top-downbenadering (7) is bedoeld om te worden gebruikt wanneer op basis van bestaande informatie verwacht wordt dat een chemische stof een hoog irritatiepotentieel heeft, terwijl de bottom-upbenadering (7) bedoeld is om te worden gebruikt wanneer op basis van bestaande informatie verwacht wordt dat een chemische stof onvoldoende oogirritatie zal veroorzaken om classificatie noodzakelijk te maken. De ICE-testmethode is een in-vitrotestmethode die onder bepaalde voorwaarden en met specifieke beperkingen zoals beschreven in de punten (8) tot en met (10) kan worden gebruikt voor de classificatie van gevaren voor de ogen en de etikettering van chemische stoffen. Hoewel de ICE-testmethode niet geldig geacht wordt als zelfstandige vervanging van de in-vivo-oogtest bij konijnen, wordt de ICE-testmethode aanbevolen als een eerste stap binnen een teststrategie zoals de top-downbenadering die door Scott et al. is voorgesteld (7) voor de identificatie van chemische stoffen die ernstig oogletsel veroorzaken, d.w.z. voor chemische stoffen die moeten worden ingedeeld in VN-GHS-categorie 1, zonder verdere tests (4). De ICE-testmethode wordt ook aanbevolen voor de identificatie van chemische stoffen waarvoor classificatie voor oogirritatie of ernstig oogletsel niet nodig is, zoals gedefinieerd door het GHS van de VN (VN-GHS Geen categorie) (4), en kan derhalve worden gebruikt als eerste stap in een bottom-upbenadering van een teststrategie (7). Voor een chemische stof waarvoor met de ICE-testmethode niet wordt voorspeld dat deze ernstig oogletsel veroorzaakt of dat classificatie voor oogirritatie/ernstig oogletsel niet nodig is, moeten echter aanvullende tests (in vitro en/of in vivo) worden uitgevoerd om tot een definitieve classificatie te komen. Bovendien moeten de toepasselijke regelgevingsinstanties worden geraadpleegd voordat de ICE-testmethode wordt gebruikt in een bottom-upbenadering volgens andere classificatiesystemen dan het GHS van de VN. Deze testmethode is bedoeld om de procedures te beschrijven die worden gebruikt om het mogelijke gevaar van een teststof voor de ogen te beoordelen aan de hand van het vermogen van deze chemische stof om toxiciteit op te wekken in een verwijderd kippenoog. Toxische effecten op de cornea worden gemeten door (i) een kwalitatieve beoordeling van de troebelheid, (ii) een kwalitatieve beoordeling van de schade aan de epitheellaag op basis van aanbrenging van fluoresceïne op het oog (fluoresceïne-retentie), (iii) een kwantitatieve meting van toegenomen dikte (zwelling) en (iv) een kwalitatieve beoordeling van macroscopische morfologische schade aan het oppervlak. De beoordelingen van de corneale troebelheid, zwelling en schade na blootstelling aan een teststof worden individueel beoordeeld en daarna gecombineerd om een oogirritatieklasse te kunnen afleiden. Definities worden gegeven in aanhangsel 1. INLEIDENDE OVERWEGINGEN EN BEPERKINGEN Deze testmethode is gebaseerd op het protocol dat is voorgesteld in OESO-leidraad nr. 160 (8) en ontwikkeld werd naar aanleiding van een internationale valideringsstudie van het ICCVAM (1) (3) (9) met bijdragen van het European Centre for the Validation of Alternative Methods, het Japanese Center for the Validation of Alternative Methods en TNO Quality of Life Department of Toxicology and Applied Pharmacology (Nederland). Het protocol is gebaseerd op informatie verkregen uit gepubliceerde protocollen alsook het huidige door TNO gebruikte protocol (10) (11) (12) (13) (14). Bij de onderliggende validering van deze testmethode werd een groot aantal chemische stoffen getest en de empirische databank van de valideringsstudie bevatte 152 chemische stoffen, waarvan 72 stoffen en 80 mengsels (5). De testmethode is van toepassing op vaste stoffen, vloeistoffen, emulsies en gels. Vloeistoffen mogen waterig of niet-waterig zijn; vaste stoffen mogen al dan niet in water oplosbaar zijn. Gassen en aerosols zijn nog niet gevalideerd in een valideringsstudie. De ICE-testmethode kan worden gebruik om chemische stoffen te identificeren die ernstig oogletsel veroorzaken en die moeten worden ingedeeld in VN-GHS-categorie 1 (4). Voor het doel van dit onderzoek zijn de geïdentificeerde beperkingen van de ICE-testmethode gebaseerd op het grote aantal vals-positieve uitslagen voor alcoholen en het grote aantal vals-negatieve uitslagen voor vaste stoffen en oppervlakteactieve stoffen (1) (3) (9). In deze context (VN-GHS-categorie 1 aangemerkt als niet tot VN-GHS-categorie 1 behorend) zijn vals-negatieve uitslagen echter niet kritiek omdat alle teststoffen die een negatief resultaat geven, vervolgens worden getest met (een) andere afdoend gevalideerde in-vitrotest(s), of als laatste optie bij konijnen, afhankelijk van de wettelijke voorschriften, met een sequentiële teststrategie in een op bewijskracht gebaseerde aanpak. Voorts zij opgemerkt dat vaste stoffen kunnen leiden tot variabele en extreme blootstellingsomstandigheden in de in-vivo-Draize-test op oogirritatie, die irrelevante voorspellingen van het werkelijke irritatiepotentieel van deze stoffen kunnen opleveren (15). Onderzoekers zouden kunnen overwegen deze testmethode te gebruiken voor alle typen chemische stoffen, waarbij een positief resultaat dient te worden aangenomen als een aanwijzing van ernstig oogletsel, d.w.z. om de stof zonder verdere tests in te delen in VN-GHS-categorie 1. Positieve resultaten die verkregen werden met alcoholen dienen echter met voorzichtigheid te worden geïnterpreteerd vanwege het risico van overpredictie. Wanneer de ICE-testmethode wordt gebruikt om chemische stoffen die ernstig oogletsel veroorzaken (VN-GHS-categorie 1) te identificeren, heeft deze testmethode een algemene nauwkeurigheidsgraad van 86 % (120/140), een percentage vals-positieve uitslagen van 6 % (7/113) en een percentage vals-negatieve uitslagen van 48 % (13/27) vergeleken met gegevens van de in-vivo-oogtest bij konijnen die werden geclassificeerd volgens het GHS-classificatiesysteem van de VN (4) (5). De ICE-testmethode kan ook worden gebruik om chemische stoffen te identificeren die niet hoeven te worden ingedeeld voor oogirritatie of ernstig oogletsel volgens het VN-GHS-classificatiesysteem (4). Voor de ICE-testmethode in een bottom-upbenadering volgens andere classificatiesystemen wordt gebruikt, dienen de toepasselijke regelgevingsinstanties te worden geraadpleegd. Deze testmethode kan worden gebruikt voor alle soorten chemische stoffen, waarbij een negatief resultaat kan worden aangenomen om een chemische stof niet in te delen voor oogirritatie en ernstig oogletsel. Op basis van één resultaat van de valideringsdatabank is de voorspelling voor aangroeiwerende oplosmiddelhoudende organische verven echter mogelijk te laag (5). Wanneer de ICE-testmethode wordt gebruikt om chemische stoffen te identificeren die geen indeling voor oogirritatie en ernstig oogletsel behoeven, heeft deze testmethode een algemene nauwkeurigheidsgraad van 82 % (125/152), een percentage vals-positieve uitslagen van 33 % (26/79) en een percentage vals-negatieve uitslagen van 1 % (1/73) vergeleken met gegevens van de in-vivo-oogtest bij konijnen die werden geclassificeerd volgens het GHS-classificatiesysteem van de VN (4) (5). Wanneer teststoffen in bepaalde klassen (zoals aangroeiwerende oplosmiddelhoudende organische verven) worden uitgesloten van de databank, heeft de ICE-testmethode een nauwkeurigheidsgraad van 83 % (123/149), een percentage vals-positieve uitslagen van 33 % (26/78) en een percentage vals-negatieve uitslagen van 0 % (0/71) voor het VN-GHS-classificatiesysteem (4) (5). De ICE-testmethode wordt niet aanbevolen voor het identificeren van teststoffen die moeten worden geclassificeerd als irriterend voor de ogen (VN-GHS-categorie 2 of categorie 2A) of teststoffen die moeten worden geclassificeerd als licht irriterend voor de ogen (VN-GHS-categorie 2B), vanwege het aanzienlijke aantal chemische stoffen van VN-GHS-categorie 1 die te laag zijn ingedeeld in VN-GHS-categorie 2, 2A of 2B en het aanzienlijke aantal chemische stoffen van VN-GHS Geen categorie die te hoog zijn ingedeeld in VN-GHS-categorie 2, 2A of 2B. Voor dit doel kan het nodig zijn verdere tests met een andere geschikte methode uit te voeren. Alle procedures met kippenogen dienen de op de testinrichting toepasselijke regelgeving en procedures voor de behandeling van menselijke of van dieren afkomstige materialen na te leven, met inbegrip van maar niet beperkt tot weefsels en weefselvloeistoffen. Universele voorzorgsmaatregelen voor laboratoria zijn aanbevolen (16). Hoewel de ICE-testmethode geen rekening houdt met letsels van het ooglid en de iris zoals die in de oogirritatietestmethode bij konijnen worden beoordeeld, wordt wel gekeken naar effecten op het hoornvlies, die bij in-vivo-onderzoek de belangrijkste factor voor indeling zijn bij de VN-GHS-classificatie. Hoewel de reversibiliteit van hoornvliesletsels op zichzelf niet kan worden beoordeeld in de ICE-testmethode, werd bovendien voorgesteld, op basis van studies op de ogen van konijnen, dat een beoordeling van de aanvankelijke diepte van het hoornvliesletsel kan worden gebruikt om sommige types irreversibele effecten te identificeren (17). Zo is er in het bijzonder meer wetenschappelijke kennis nodig om te begrijpen hoe irreversibele effecten optreden die geen verband houden met de aanvankelijke diepte van het letsel. Tot slot kan de ICE-testmethode niet worden gebruikt voor een beoordeling van de mogelijkheid van met blootstelling van het oog geassocieerde systemische toxiciteit. Deze testmethode zal periodiek worden bijgewerkt wanneer nieuwe informatie en gegevens in aanmerking worden genomen. Histopathologie kan bijvoorbeeld van nut zijn wanneer een vollediger karakterisering van schade aan de cornea nodig is. Om die mogelijkheid te evalueren, worden de gebruikers aangemoedigd om ogen te bewaren en histopathologiespecimen te prepareren die kunnen worden gebruikt om een databank en beslissingscriteria te ontwikkelen die de nauwkeurigheid van deze testmethode verder kunnen verbeteren. De OESO heeft een leidraad over het gebruik van in-vitrotestmethoden voor oculaire toxiciteit uitgewerkt met gedetailleerde procedures voor het verzamelen van histopathologiespecimen en informatie over waar specimen en/of histopathologiegegevens kunnen worden ingediend (8). Elk laboratorium dat dit onderzoek voor het eerst opstelt, dient gebruik te maken van de in bijlage 2 voorziene chemische stoffen voor bekwaamheidstoetsing. Een laboratorium kan deze chemicaliën gebruiken om zijn technische bekwaamheden met betrekking tot het uitvoeren van de ICE-testmethode aan te tonen vooraleer het ICE-onderzoeksgegevens indient voor de wettelijke gevarenclassificatie. PRINCIPE VAN DE TEST De ICE-testmethode is een organotypisch model dat handhaving op korte termijn van het kippenoog in vitro vereist. In deze testmethode wordt schade door de teststof beoordeeld door vaststelling van corneale zwelling, troebelheid en fluoresceïne-retentie. Daar waar de laatste twee parameters een kwalitatieve beoordeling vergen, vereist de analyse van de corneale zwelling een kwantitatieve beoordeling. Elke meting wordt ofwel omgezet in een kwantitatieve score die gebruikt wordt voor de berekening van een algemene irritatie-index, ofwel een kwalitatieve indeling toegekend die gebruikt wordt voor de toekenning van een in-vitro-indeling van het gevaar voor de ogen, hetzij als VN-GHS-categorie 1, hetzij als niet ingedeeld volgens VN-GHS. Elk van deze resultaten kan dan worden aangewend om het potentiële ernstige oogletsel in vivo of de afwezigheid van de noodzaak van indeling als gevaarlijk voor de ogen van een teststof te voorspellen (zie beslissingscriteria). Er kan evenwel geen indeling worden gegeven voor chemische stoffen waarvan niet is voorspeld dat zij ernstig oogletsel veroorzaken of die niet zijn ingedeeld met de ICE-testmethode (zie punt 11). Bron en ouderdom van de kippenogen In het verleden werden voor dit onderzoek steeds ogen verzameld van kippen uit de slachterij die geslacht werden voor de menselijke consumptie waardoor er geen nood was aan proefdieren. Enkel ogen van gezonde dieren die geschikt zijn voor de menselijke voedselketen worden gebruikt. Hoewel een gecontroleerde studie voor de beoordeling van de ideale leeftijd van de kip nog niet werd gevoerd, was de leeftijd en het gewicht van de in deze testmethode gebruikte kippen in het verleden dat van piepkuikens van oudsher geslacht in een pluimveeslachterij (d.w.z. ongeveer 7 weken oud, 1,5 tot 2,5 kg). Verzameling en vervoer van ogen naar het laboratorium Na verdoving van de kippen, meestal met een elektrische schok, en incisie in de nek voor bloeding, dient de kop onmiddellijk verwijderd te worden. Er dient een plaatselijke bron van kippen, niet ver van het laboratorium te worden gezocht zodat de koppen snel genoeg van de slachterij naar het laboratorium kunnen worden vervoerd om bederf en/of bacteriële besmetting zoveel mogelijk te beperken. Om de naleving van de acceptatiecriteria van de test te verzekeren dient het tijdsinterval tussen de verzameling van de koppen en het moment waarop de ogen in de superfusiekamer worden geplaatst nadat zij zijn weggenomen, tot een minimum te worden beperkt (gewoonlijk maximaal twee uur). Alle in het onderzoek gebruikte ogen dienen van dezelfde groep op een bepaalde dag verzamelde ogen te zijn. Aangezien de ogen in het laboratorium ontleed worden, worden de koppen intact en op omgevingstemperatuur van de slachterij (gewoonlijk tussen 18 °C en 25 °C) vervoerd in plastic dozen, bevochtigd met in isotoon zout gedrenkte doeken. Selectiecriteria en aantal ogen gebruikt in de ICE Ogen die een hoge referentie-fluoresceïneverkleuring (d.w.z. > 0,5) of corneale troebelheidsscore (d.w.z. > 0,5) vertonen, worden na verwijdering verworpen. Elke behandelingsgroep en tegelijkertijd gemeten positieve controle bestaat uit ten minste drie ogen. De negatieve controlegroep of de oplosmiddelcontrole (indien een ander oplosmiddel dan zout gebruikt wordt) bestaat uit ten minste één oog. Wanneer vaste materialen het resultaat “GHS Geen categorie” opleveren, is een tweede cyclus met drie ogen aanbevolen om het negatieve resultaat te bevestigen of te verwerpen. PROCEDURE Voorbereiding van de ogen De oogleden worden zorgvuldig verwijderd zodat geen schade wordt berokkend aan de cornea. De corneale integriteit kan eenvoudig worden beoordeeld met een druppel van 2 % (g/v) natriumfluoresceïne die gedurende enkele seconden wordt aangebracht op het hoornvliesoppervlak en daarna uitgewassen met een isotone zoutoplossing. Met fluoresceïne behandelde ogen worden daarna onderzocht met een spleetlampmicroscoop om er zeker van te zijn dat de cornea niet beschadigd is (d.w.z. fluoresceïne-retentie en corneale troebelheidsscore ≤ 0.5). Indien het oog onbeschadigd is, wordt het voorzichtig verder verwijderd uit de schedel zonder de cornea te beschadigen. De oogbal wordt van de oogrand getrokken door het derde ooglid stevig vast te houden met een chirurgische tang terwijl de oogspier afgesneden wordt met een kromme schaar met botte punten. Het is belangrijk schade aan de cornea door buitensporige druk (d.w.z. misvorming door samendrukking) te voorkomen. Als het oog van de oogrand verwijderd is, zou een zichtbaar gedeelte van de oogzenuw nog aangehecht moeten zijn. Zodra het oog van de oogrand verwijderd is, wordt het op een absorberend verband gelegd en worden het derde ooglid en ander bindweefsel weggesneden. Het verwijderde oog wordt op een klem van roestvrij staal geplaatst met de cornea in verticale positie. De klem wordt dan overgebracht naar een kamer van het superfusieapparaat (18). De klemmen dienen zo in het superfusieapparaat te worden geplaatst dat de volledige cornea met de isotone zoutoplossing wordt bedropen (3-4 druppels per minuut of 0,1 tot 0,15 ml/min). In de kamers van het superfusieapparaat moet een temperatuur van 32 ± 1,5 °C worden gehandhaafd. Bijlage 3 bevat een diagram van een typisch superfusieapparaat met oogklemmen, dat verkrijgbaar is in de handel of zelf kan worden gemaakt. Het apparaat kan worden aangepast aan de behoeften van elk individueel laboratorium (bv. om een ander aantal ogen te houden). Nadat ze in het superfusieapparaat geplaatst zijn, worden de ogen nogmaals onderzocht met een spleetlampmicroscoop om te controleren dat er zich tijdens de dissectieprocedure geen schade heeft voorgedaan. Met behulp van de dieptemeter van de spleetlampmicroscoop wordt meteen ook de corneale dikte gemeten ter hoogte van de corneale apex. Ogen met een fluoresceïne-retentiescore van > 0,5; (ii) corneale troebelheid > 0,5; elk bijkomend teken van schade, dienen te worden vervangen. Individuele ogen die niet werden verwijderd op basis van de voorgaande criteria dienen te worden verwijderd indien de corneale dikte met meer dan 10 % afwijkt van de gemiddelde waarde van alle ogen. Gebruikers dienen zich te realiseren dat spleetlampmicroscopen verschillende resultaten voor corneale dikte kunnen opleveren naargelang van de instelling van de breedte van de spleet. De breedte van de spleet dient te worden ingesteld op 0,095 mm. Zodra alle ogen onderzocht en goedgekeurd zijn, worden de ogen gedurende ongeveer 45 tot 60 minuten geïncubeerd om ze vóór de dosering te equilibreren met het testsysteem. Na de equilibreerperiode wordt een nulreferentiemeting opgetekend voor de corneale dikte en troebelheid die zal dienen als referentie (d.w.z. tijd = 0). De fluoresceïnescore die bepaald wordt bij de ontleding wordt gebruikt als referentiemeting voor dat eindpunt. Aanbrenging van de teststof Onmiddellijk na de nulreferentiemetingen wordt het oog (in de houder) verwijderd uit het superfusieapparaat, in horizontale positie geplaatst, en wordt de teststof aangebracht op de cornea. Vloeibare teststoffen worden gewoonlijk onverdund getest, maar kunnen worden verdund indien dit nodig blijkt te zijn (bv. als onderdeel van de onderzoeksopzet). Het aanbevolen oplosmiddel voor verdunde teststoffen is fysiologische zoutoplossing. Andere oplosmiddelen mogen echter ook worden gebruikt in gecontroleerde omstandigheden maar indien aan een ander oplosmiddel dan fysiologische zoutoplossing de voorkeur wordt gegeven, dient dit te worden gemotiveerd. Vloeibare teststoffen worden zodanig aangebracht op de cornea dat het gehele oppervlak van de cornea gelijk bedekt is met de teststof; het standaardvolume is 0,03 ml. Indien mogelijk, dienen de vaste stoffen zo fijn mogelijk gemalen te worden in een vijzel of een vergelijkbaar maalinstrument. Het poeder wordt zodanig op de cornea aangebracht dat het hele oppervlak gelijk bedekt is met de teststof; de standaardhoeveelheid is 0,03 g. De teststof (vloeibaar of vast) wordt gedurende 10 seconden toegediend en dan weggespoeld met isotone zoutoplossing (ongeveer 20 ml) op omgevingstemperatuur. Het oog (in de houder) wordt daarna terug in het superfusieapparaat geplaatst in de oorspronkelijke rechtopstaande positie. Indien nodig kunnen de cornea na de toediening gedurende 10 seconden en op latere tijdstippen (bv. wanneer er residuen van de teststof op de cornea worden aangetroffen) bijkomend worden uitgewassen. Over het algemeen is de hoeveelheid zoutoplossing die extra wordt gebruikt voor het uitwassen, niet van doorslaggevend belang, maar de vaststelling dat chemische stoffen aan de cornea blijven kleven is wel belangrijk. Controlestoffen In elk experiment moeten gelijktijdig gemeten negatieve controles of controles met oplosmiddel/medium en positieve controles worden opgenomen. Bij het testen van vloeistoffen aan 100 % of vaste stoffen wordt fysiologische zoutoplossing gebruikt als tegelijkertijd gemeten negatieve controle in de ICE-testmethode om niet-specifieke veranderingen in het testsysteem op te sporen en om te garanderen dat de onderzoeksomstandigheden niet ongewenst leiden tot een irriterende respons. Bij het testen van verdunde vloeistoffen wordt een tegelijkertijd gemeten controlegroep van oplosmiddelen/media opgenomen in de testmethode om niet-specifieke veranderingen in het testsysteem op te sporen en om te garanderen dat de onderzoeksomstandigheden niet ongewenst leiden tot een irriterende respons. Zoals gesteld in punt 31, kunnen enkel oplosmiddelen/media worden gebruikt waarvan geweten is dat ze geen nadelig effect op het testsysteem hebben. Een bekende voor de ogen irriterende stof wordt in elk experiment opgenomen als gelijktijdige positieve controle om na te gaan of de juiste respons wordt opgewekt. Aangezien het ICE-onderzoek in deze testmethode wordt gebruikt om corrosieve en hevig irriterende stoffen te identificeren, is het best dat de positieve controle gebeurt met een referentiestof die een hevige respons opwekt in deze testmethode. Om te verzekeren dat de variabiliteit in de positieve-controlerespons doorheen de tijd kan worden beoordeeld, mag de grootte van de hevig irriterende respons echter niet buitensporig zijn. Voor de positieve controle dienen voldoende in-vitrogegevens te worden gegenereerd zodat een statistisch gedefinieerde aanvaardbare reeks voor de positieve controle kan worden berekend. Indien voor een bepaalde positieve controle geen gepaste historische ICE-testmethodegegevens beschikbaar zijn, dan is het mogelijk dat onderzoek dient te worden verricht om deze gegevens te verkrijgen. Voorbeelden van positieve controles voor vloeibare teststoffen zijn 10 % azijnzuur of 5 % benzalkoniumchloride. Voorbeelden van positieve controles voor vaste teststoffen zijn natriumhydroxide of imidazool. IJkstoffen zijn nuttig voor de beoordeling van potentiële oogirritatie bij onbekende chemische stoffen uit een specifieke chemische klasse of productklasse of voor de beoordeling van potentiële relatieve irritatie van een voor het oog irriterende stof binnen een reeks van irritatieresponsen. Gemeten eindpunten Behandelde cornea's worden beoordeeld vóór behandeling en op 30, 75, 120, 180 en 240 minuten (± 5 minuten) na het uitwassen na behandeling. Deze punten in de tijd leveren een geschikt aantal metingen op over een behandelingsperiode van vier uur en laten toch voldoende tijd tussen twee metingen voor de vereiste waarnemingen voor alle ogen. De eindpuntbeoordelingen zijn corneale troebelheid, zwelling, fluoresceïne-retentie en morfologische effecten (bv. kuilvorming of loskomen van de epitheellaag). Alle eindpunten, met uitzondering van de fluoresceïne-retentie (die enkel wordt bepaald vóór de behandeling en 30 minuten na blootstelling aan de teststof) worden bepaald op elk van de bovenvermelde tijdstippen. Het nemen van foto's wordt aanbevolen als documentatie van corneale troebelheid, fluoresceïne-retentie, morfologische effecten en, indien uitgevoerd, histopathologie. Na het laatste onderzoek, na vier uur, worden gebruikers aangespoord om de ogen te bewaren in het gepaste fixeermiddel (d.w.z. neutrale gebufferde formaldehyde) voor eventueel histopathologisch onderzoek (zie punt 14 en referentie (8) voor meer informatie). Corneale zwelling wordt bepaald aan de hand van de diktemetingen van de cornea met een optische pachymeter op een spleetlampmicroscoop. Zij wordt uitgedrukt als percentage en wordt berekend vanuit de corneale diktemetingen volgens de volgende formule:
Het gemiddelde percentage van corneale zwelling voor alle testogen wordt berekend voor alle waarnemingstijdstippen. Op basis van de hoogste gemiddelde score voor corneale zwelling, zoals die op elk tijdstip is waargenomen, wordt dan een algemene categoriescore toegekend aan elke teststof (zie punt 51). De corneale troebelheid wordt beoordeeld aan de hand van het gebied van de cornea waar de troebelheid het dichtst is, zoals te zien in tabel 1. De gemiddelde waarde van de corneale troebelheid voor alle testogen wordt berekend voor alle waarnemingstijdstippen. Op basis van de hoogste gemiddelde score voor corneale troebelheid, zoals die op elk tijdstip is waargenomen, wordt dan een algemene categoriescore toegekend aan elke teststof (zie punt 51). Tabel 1 Corneale troebelheidsscores
De mate van fluoresceïne-retentie wordt enkel beoordeeld op het tijdstip van de 30e minuut, zoals te zien in tabel 2. De gemiddelde fluoresceïne-retentiewaarde voor alle testogen wordt vervolgens berekend voor het tijdstip van de 30e minuut en wordt gebruikt voor de algemene categoriescore die aan elke teststof wordt gegeven (zie punt 51). Tabel 2 Fluoresceïne-retentiescores
Morfologische effecten zijn bijvoorbeeld “kuilvorming” van de corneale epitheelcellen, “loskomen” van de epitheellaag, “ongelijkmatig worden” van het corneale oppervlak en “kleven” van de teststof aan de cornea. Deze bevindingen kunnen in hevigheid variëren en kunnen eveneens gelijktijdig voorkomen. De indeling van deze bevindingen is subjectief naargelang de interpretatie van de onderzoeker. GEGEVENS EN RAPPORTAGE Evaluatie van de gegevens Resultaten van corneale troebelheid, zwelling en fluoresceïne-retentie dienen afzonderlijk te worden beoordeeld om een ICE-klasse te genereren voor elk eindpunt. De ICE-klassen voor elk eindpunt worden daarna gecombineerd om een irritatieklasse voor elke teststof te genereren. Beslissingscriteria Na beoordeling van elk eindpunt kunnen ICE-klassen worden toegekend op basis van een vooraf vastgestelde reeks. Interpretatie van corneale zwelling (tabel 3), troebelheid (tabel 4) en fluoresceïne-retentie (tabel 5) aan de hand van vier ICE-klassen gebeurt volgens onderstaande schalen. Scores voor corneale zwelling zoals te zien in tabel 3 zijn enkel van toepassing indien de dikte gemeten wordt met een spleetlampmicroscoop (bijvoorbeeld Haag-Streit BP900) met een dieptemeetapparaat nr. 1 en spleetbreedte ingesteld op 9 Tabel 3 ICE-indelingscriteria voor corneale zwelling
Tabel 4 ICE-indelingscriteria voor troebelheid
Tabel 5 ICE-indelingscriteria voor gemiddelde fluoresceïne-retentie
De in-vitroclassificatie voor een teststof wordt beoordeeld door de GHS-classificatie te nemen die overeenkomt met de combinatie van de categorieën die werden verkregen voor corneale zwelling, corneale troebelheid en fluoresceïne-retentie zoals beschreven in tabel 6. Tabel 6 Totale in-vitroclassificaties
Acceptatiecriteria van het onderzoek Een test wordt aanvaardbaar geacht als de gelijktijdige negatieve controles of controles met medium/oplosmiddel en de gelijktijdige positieve controles respectievelijk zijn geïdentificeerd als GHS niet ingedeeld en GHS categorie 1. Testverslag Het testverslag dient de volgende informatie te bevatten, indien relevant voor het voeren van het onderzoek:
LITERATUUR
Aanhangsel 1 DEFINITIES Betrouwbaarheid : indicatie van de mate waarin een testmethode, uitgevoerd volgens een vast protocol, op reproduceerbare wijze binnen één laboratorium en tussen laboratoria verspreid over de tijd kan worden uitgevoerd. De betrouwbaarheid wordt beoordeeld door berekening van intra- en interlaboratoriumreproduceerbaarheid en intralaboratoriumherhaalbaarheid. Bewijskracht(proces) : het proces waarbij de sterke en zwakke punten van verschillende gegevens worden onderzocht bij het komen tot en ondersteunen van een conclusie met betrekking tot het gevarenpotentieel van een stof. Bottom-upbenadering : een trapsgewijze benadering die gebruikt wordt voor een chemische stof waarvan wordt vermoed dat classificatie voor oogirritatie of ernstig oogletsel niet nodig is, die begint met het onderscheiden van chemische stoffen waarvoor classificatie niet nodig is (negatief resultaat) van andere chemische stoffen (positief resultaat). Chemische stof : een stof of een mengsel. Cornea : het doorschijnende gedeelte van het voorste deel van de oogbal dat de iris en pupil bedekt en licht naar binnen toelaat. Corneale troebelheid : meting van de mate van troebelheid van de cornea na blootstelling aan een teststof. Verhoogde corneale troebelheid is een aanwijzing voor schade aan de cornea. Corneale zwelling : een objectieve meting in de ICE-test van de mate van uitzetting van de cornea na blootstelling aan een teststof. Dit wordt uitgedrukt als percentage en wordt berekend vanaf de referentiedikte van de cornea (vóór dosering) en de dikte opgetekend op regelmatige intervallen na blootstelling aan de teststof in de ICE-test. De mate van corneale zwelling is een aanwijzing voor schade aan de cornea. Ernstig oogletsel : veroorzaking van weefselschade aan het oog of ernstige fysische gezichtsvermindering na aanbrenging van een teststof op de voorste ooglaag die niet volledig reversibel is binnen 21 dagen na de aanbrenging. Verwisselbaar met “Irreversibele effecten op ogen” en met “VN-GHS-categorie 1” (4). Fluoresceïne-retentie : een subjectieve meting in de ICE-test van de hoeveelheid natriumfluoresceïne die wordt weerhouden door de epitheelcellen in de cornea na blootstelling aan een teststof. De mate van fluoresceïne-retentie is een aanwijzing voor schade aan de corneale epitheellaag. Gevaar : inherente eigenschap van een agens of situatie die nadelige effecten kan veroorzaken als een organisme, systeem of (sub)populatie aan dit agens wordt blootgesteld. Gevalideerde testmethode : een testmethode waarvoor valideringsstudies werden gedaan om de relevantie (inclusief nauwkeurigheid) en betrouwbaarheid voor een bepaald doel te bepalen. Er dient te worden opgemerkt dat een gevalideerde testmethode niet per definitie voldoende nauwkeurig en betrouwbaar is om aanvaardbaar te worden bevonden voor het voorgestelde doel. GHS (Globally Harmonized System of Classification and Labelling of Chemicals, mondiaal geharmoniseerd classificatie- en etiketteringssysteem voor chemische stoffen van de Verenigde Naties (VN)) : een systeem voor de indeling van chemische producten (stoffen en mengsels) op basis van gestandaardiseerde soorten en niveaus van gevaren van fysische aard en gevaren voor de gezondheid en het milieu, en dat de desbetreffende voorlichtingselementen bepaalt, zoals pictogrammen, signaalwoorden, gevarenaanduidingen, voorzorgsmaatregelen en veiligheidsinformatiebladen, waarmee informatie over de schadelijke effecten van de stoffen kenbaar wordt gemaakt teneinde mensen (waaronder werkgevers, werknemers, transporteurs, consumenten en spoedhulpverleners) en het milieu te beschermen (4). Ijkstof : een chemische stof die gebruikt wordt als norm voor vergelijking met een teststof. Een ijkstof moet de volgende eigenschappen hebben: (i) (een) constante en betrouwbare bron(nen); (ii) een structurele en functionele gelijkenis met de klasse chemische stoffen die getest wordt; iii) bekende fysisch-chemische kenmerken; (iv) ondersteunende gegevens over bekende effecten; en (v) een bekende werkzaamheid binnen het bereik van de gewenste respons. Irreversibele effecten op ogen : zie “ernstig oogletsel” en “VN-GHS-categorie 1”. Medium-/oplosmiddelcontrole : een onbehandeld monster dat alle componenten bevat van een testsysteem, met inbegrip van het oplosmiddel of medium, dat tezamen met de met de teststof behandelde monsters en andere controlemonsters verwerkt wordt om de referentierespons voor de monsters die behandeld werden met de in hetzelfde oplosmiddel of medium opgeloste teststof vast te stellen. Als dit monster getest wordt met een tegelijkertijd gemeten negatieve controle, toont het ook aan of het oplosmiddel of medium met het testsysteem interageert. Mengsel : een mengsel of een oplossing bestaande uit twee of meer stoffen waarin deze stoffen geen reactie met elkaar aangaan (4). Nauwkeurigheid : de mate van overeenstemming tussen resultaten verkregen met de testmethode en erkende referentiewaarden. Het is een maat voor de prestaties van de testmethode en één aspect van “relevantie”. Deze term en de term “concordantie”, waaronder het percentage correcte uitkomsten van een testmethode wordt verstaan, worden vaak door elkaar gebruikt. Negatieve controle : een onbehandeld monster dat alle bestanddelen van een testsysteem bevat. Dit monster wordt tezamen met de met de teststof behandelde monsters en andere controlemonsters verwerkt om te bepalen of het oplosmiddel interageert met het testsysteem. Niet ingedeeld : stoffen die niet zijn geclassificeerd voor oogirritatie (VN-GHS-categorie 2) of ernstig oogletsel (VN-GHS-categorie 1). Verwisselbaar met “VN-GHS Geen categorie”. Oogirritatie : het ontstaan van veranderingen in het oog na het aanbrengen van een teststof op het oppervlak aan de voorzijde van het oog, die binnen 21 dagen na het aanbrengen volledig reversibel zijn. Verwisselbaar met “Reversibele effecten op ogen” en met “VN-GHS-categorie 2” (4). Oppervlakteactieve stof : ook genoemd “agens dat inwerkt op de oppervlakte”; dit is een stof, zoals een detergent, die de oppervlaktespanning van een vloeistof kan verlagen en het zo mogelijk maakt dat de vloeistof gaat schuimen of in vaste stoffen doordringt; ook bekend als een bevochtigingsmiddel. Percentage vals-negatieve uitslagen : het percentage van alle positieve chemische stoffen dat door een testmethode onterecht als negatief wordt geïdentificeerd. Het is één indicator van de prestaties van de testmethode. Percentage vals-positieve uitslagen : het percentage van alle negatieve chemische stoffen dat door een testmethode onterecht als positief wordt geïdentificeerd. Het is één indicator van de prestaties van de testmethode. Positieve controle : een monster met alle bestanddelen van een testsysteem dat met een chemische stof behandeld werd waarvan bekend is dat deze een positieve respons opwekt. Om te verzekeren dat variabiliteit in de positieve-controlerespons doorheen de tijd kan worden beoordeeld, mag de grootte van de “hevige irritatie”-respons niet buitensporig zijn. Reversibele effecten op de ogen : zie “oogirritatie” en “VN-GHS-categorie 2”. Spleetlampmicroscoop : een instrument dat wordt gebruikt om het oog rechtstreeks te onderzoeken met een binoculaire microscoop die een driedimensionaal, rechtopstaand beeld oplevert. In de ICE-testmethode wordt dit instrument gebruikt om de voorste structuren van het kippenoog te zien alsook om de corneale dikte objectief te kunnen meten met een aangehechte dieptemeter. Stof : chemische elementen en verbindingen ervan, zoals zij voorkomen in natuurlijke toestand of bij de productie ontstaan, met inbegrip van alle additieven die nodig zijn voor het behoud van de stabiliteit van het product en alle onzuiverheden ten gevolge van het productieprocédé, doch met uitzondering van elk oplosmiddel dat kan worden afgescheiden zonder dat de stabiliteit van de stof wordt aangetast of de samenstelling ervan wordt gewijzigd (4). Teststof : Alle volgens deze testmethode geteste stoffen en mengsels. Top-downbenadering : een trapsgewijze benadering die gebruikt wordt voor een chemische stof waarvan wordt vermoed dat deze ernstig oogletsel veroorzaakt, die begint met het onderscheiden van chemische stoffen die ernstig oogletsel veroorzaken (positief resultaat) van andere chemische stoffen (negatief resultaat). Trapsgewijze teststrategie : een trapsgewijze teststrategie waarbij alle bestaande informatie over een teststof in een bepaalde volgorde wordt herbezien aan de hand van een proces op basis van bewijskracht op elke trap om te bepalen of er voldoende informatie beschikbaar is voor een beslissing tot gevarenclassificatie, vooraleer wordt overgegaan tot de volgende trap. Indien het irritatiepotentieel van een teststof kan worden bepaald op basis van de bestaande informatie, dan zijn bijkomende tests niet vereist. Indien het irritatiepotentieel van een teststof niet kan worden bepaald op basis van de bestaande informatie, dan wordt een trapsgewijze sequentiële testprocedure op dieren uitgevoerd tot een ondubbelzinnige indeling mogelijk is. VN-GHS-categorie 1 : zie “ernstig oogletsel” en/of “irreversibele effecten op ogen”. VN-GHS-categorie 2 : zie “oogirritatie” en/of “reversibele effecten op het oog”. VN-GHS Geen categorie : Stoffen die niet voldoen aan de voorwaarden voor indeling in VN-GHS-categorie 1 of 2 (2A of 2B). Verwisselbaar met “Niet ingedeeld”. Aanhangsel 2 CHEMISCHE STOFFEN VOOR BEKWAAMHEIDSTOETSING VOOR DE ICE-TESTMETHODE Vooraleer over te gaan tot routinegebruik van een testmethode die deze testmethode volgt, moeten laboratoria hun technische bekwaamheid aantonen door een correcte identificatie van de gevarenclassificatie voor de ogen van de 13 chemische stoffen die in tabel 1 worden aangeraden. Deze chemische stoffen werden geselecteerd om de mogelijke responsen voor gevaren voor de ogen weer te geven, op basis van de resultaten van de in-vivo-oogtest bij konijnen (TG 405) en het VN-GHS-classificatiesysteem (bv. VN-GHS-categorieën 1, 2A, 2B of geen categorie) (4) (6). Andere selectiecriteria waren dat stoffen commercieel beschikbaar zijn, dat er in-vivoreferentiegegevens van zeer goede kwaliteit beschikbaar zijn, en dat er gegevens van zeer goede kwaliteit beschikbaar zijn van de ICE-in-vitromethode. Er zijn referentiegegevens beschikbaar in het SSD (5) en in de Background Review Documents van het ICCVAM voor de ICE-testmethode (9). Tabel 1 Aanbevolen stoffen voor het aantonen van technische bekwaamheid met ICE
Aanhangsel 3 DIAGRAMMEN VAN HET ICE-SUPERFUSIEAPPARAAT EN DE OOGKLEMMEN (Zie Burton et al. (18) voor aanvullende algemene beschrijvingen van het superfusieapparaat en de oogklem) 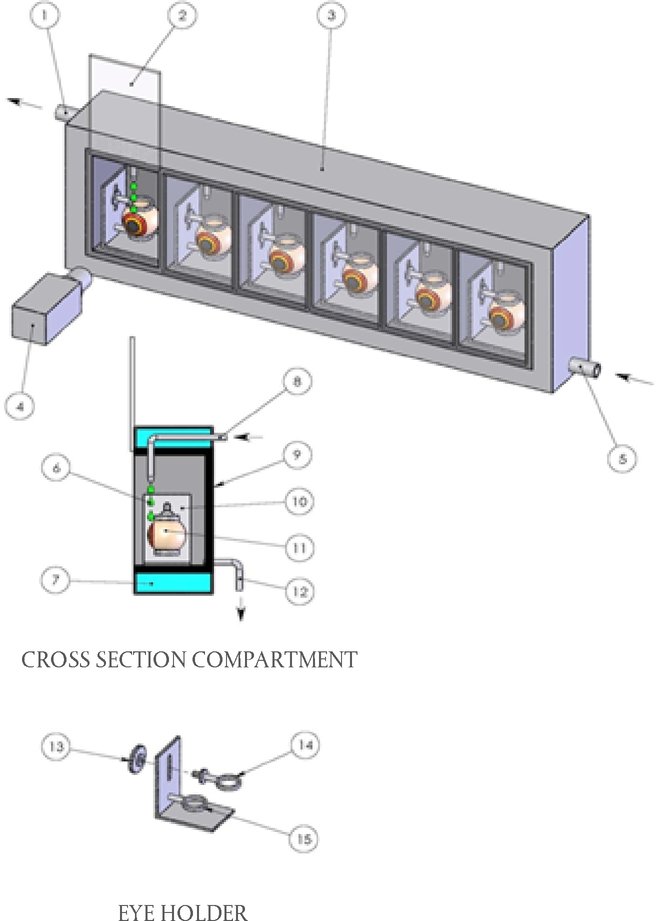
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
14) |
In deel B wordt hoofdstuk B.49 vervangen door: „B.49 In-vitromicronucleustest bij zoogdiercellen INLEIDING Deze testmethode is gelijkwaardig aan testrichtlijn (TG) 487 (2016) van de OESO. Zij maakt deel uit van een reeks testmethoden op het gebied van de genetische toxicologie. Er is een OESO-document opgesteld met beknopte informatie over genetisch-toxicologische tests en een overzicht van de recente wijzigingen van deze testrichtlijnen (1). De in-vitromicronucleustest (MNvit) is een genotoxiciteitstest waarmee micronuclei (MN) in het cytoplasma van interfasecellen worden opgespoord. Micronuclei kunnen ontstaan uit acentrische chromosoomfragmenten (d.w.z. zonder centromeer) of volledige chromosomen die tijdens het anafasestadium van de celdeling niet naar de polen kunnen migreren. De MNvit-test is dan ook een in-vitromethode die een omvattende basis biedt om het potentieel voor chromosoombeschadiging te onderzoeken in vitro, omdat zowel aneugenen als clastogenen kunnen worden opgespoord (2) (3) in cellen die tijdens of na blootstelling aan de teststof celdeling hebben ondergaan (zie punt 13 voor meer informatie). Micronuclei vertonen schade die is doorgegeven aan dochtercellen, terwijl in metafasecellen gescoorde chromosoomafwijkingen mogelijk niet doorgegeven worden. In beide gevallen zijn de veranderingen mogelijk niet verenigbaar met het overleven van de cellen. Bij deze testmethode (TM) mogen protocollen met en zonder de actinepolymerisatieremmer cytochalasine B (cytoB) worden gebruikt. De toevoeging van cytoB voor de mitose, maakt identificatie en analyse mogelijk van de micronuclei in cellen die één mitose hebben voltooid, omdat dergelijke cellen binucleair zijn (4) (5). Met deze testmethode is het ook mogelijk protocollen zonder cytokineseblokkering te gebruiken op voorwaarde dat er bewijzen zijn dat de geanalyseerde celpopulatie mitose heeft ondergaan. De MNvit-test kan niet alleen worden gebruikt om chemische stoffen te identificeren die micronuclei induceren, maar het gebruik van de immunochemische labeling van kinetochoren of hybridisatie met centromere/telomere proben (fluorescentie in situ hybridisatie (FISH)) kan ook informatie verstrekken over de mechanismen van chromosoombeschadiging en de vorming van micronuclei (6) (7) (8) (9) (10) (11) (12) (13) (14) (15) (16) (17). Deze labeling- en hybridisatieprocedures kunnen worden gebruikt wanneer sprake is van een toename van micronucleivorming en de onderzoeker wenst te bepalen of die toename het gevolg is van clastogene en/of aneugene gebeurtenissen. Omdat micronuclei in interfasecellen op relatief objectieve wijze kunnen worden beoordeeld, hoeft het laboratoriumpersoneel alleen het aantal binucleaire cellen — wanneer cytoB wordt gebruikt — en het aantal cellen met micronuclei — in alle gevallen — na te gaan. Bijgevolg kunnen de objectglaasjes vrij snel worden gescoord en kan de analyse worden geautomatiseerd. Zo wordt het in de praktijk mogelijk om niet honderden, maar duizenden cellen per behandeling te scoren, hetgeen de test krachtiger maakt. Aangezien micronuclei kunnen ontstaan uit achtergebleven chromosomen, is het tot slot mogelijk om stoffen op te sporen die aneuploïdie induceren en die moeilijk onderzocht kunnen worden in conventionele tests op chromosoomafwijkingen, bv. hoofdstuk B.10 van deze bijlage (18). Aan de hand van de MNvit-test is het zonder bijzondere technieken, zoals de in punt 4 beschreven FISH, echter niet mogelijk te differentiëren tussen stoffen die veranderingen in het aantal chromosomen en/of polyploïdie induceren en stoffen die clastogeniciteit induceren. De MNvit-test is robuust en kan worden uitgevoerd in verschillende celtypen en in de aanwezigheid of afwezigheid van cytoB. Er zijn uitgebreide gegevens beschikbaar die de validiteit van de MNvit-test met behulp van diverse celtypen (culturen van cellijnen of primaire celculturen van knaagdieren) (19) (20) (21) (22) (23) (24) (25) (26) (27) (28) (29) (30) (31) (32) (33) (34) (35) (36). Hieronder zijn met name begrepen de internationale valideringsstudies die zijn gecoördineerd door de Société Française de Toxicologie Génétique (SFTG) (19) (20) (21) (22) (23) en de verslagen van de International Workshop on Genotoxicity Testing (5) (17). De beschikbare gegevens zijn ook opnieuw beoordeeld tijdens een retrospectieve valideringsstudie naar het gewicht van de evidentie door het Europees Centrum voor de validering van alternatieve methoden (ECVAM) van de Europese Commissie, en de testmethode is door het Raadgevend wetenschappelijk comité van het ECVAM bekrachtigd als wetenschappelijk geldig (37) (38) (39). Bij de MNvit-test op zoogdiercellen kunnen culturen van cellijnen of primaire celculturen van menselijke cellen of knaagdiercellen worden gebruikt. Omdat de achtergrondfrequentie van micronuclei van invloed zal zijn op de gevoeligheid van de test, wordt aanbevolen dat celtypen met een stabiele, vastgestelde achtergrondfrequentie van micronucleusvorming worden gebruikt. De gebruikte cellen worden geselecteerd op basis van hun groeivermogen in een cultuur, de stabiliteit van het karyotype (met inbegrip van het aantal chromosomen) en de frequentie van spontane micronuclei (40). Momenteel kunnen op basis van de beschikbare gegevens geen harde aanbevelingen worden gedaan, maar de gegevens wijzen erop dat het belangrijk is om bij de beoordeling van chemische gevaren rekening te houden met de p53-status, de genetische (karyotype) stabiliteit, het herstellend vermogen van het DNA en de oorsprong (knaagdier versus mens) van de voor de test geselecteerde cellen. De gebruikers van deze testmethode worden dan ook aangemoedigd om rekening te houden met de invloed van deze en andere celkenmerken op de prestaties van een cellijn bij het opsporen van de inductie van micronuclei, aangezien de kennis op dit gebied voortschrijdt. Gebruikte definities worden gegeven in aanhangsel 1. INLEIDENDE OVERWEGINGEN EN BEPERKINGEN Bij in vitro uitgevoerde tests moet er meestal een exogene metabole activeringsbron worden gebruikt, tenzij de cellen qua metabolisme competent zijn met betrekking tot de teststoffen. Het exogene metabole activeringssysteem kan de omstandigheden in vivo niet volledig nabootsen. Omstandigheden die zouden kunnen leiden tot artefactuele positieve resultaten die de genotoxiciteit van de teststoffen niet weerspiegelen, moeten worden vermeden. Het gaat dan onder meer om veranderingen in de pH (41) (42) (43) of de osmolaliteit, wisselwerking met het celkweekmedium (44) (45) of een te sterke cytotoxiciteit (zie punt 29). Om de inductie van micronuclei te analyseren, is het van essentieel belang dat zowel in de behandelde als de onbehandelde culturen mitose heeft plaatsgevonden. Het meest informatieve stadium voor het scoren van micronuclei is in cellen die één mitose voltooid hebben tijdens of na de behandeling met de teststof. Voor gefabriceerde nanomaterialen zijn specifieke aanpassingen van deze testmethode nodig die niet in de testmethode zijn beschreven. Voordat de testmethode wordt gebruikt op een mengsel om gegevens te genereren voor een beoogd regelgevingsdoel, moet worden nagegaan of, en zo ja, waarom deze methode betrouwbare resultaten oplevert voor dat doel. Zulke overwegingen zijn niet nodig wanneer het testen van het mengsel wettelijk vereist is. PRINCIPE VAN DE TEST Culturen van menselijke cellen of andere zoogdiercellen worden blootgesteld aan de teststof met en zonder een exogene metabole activeringsbron, tenzij cellen met een toereikende metaboliseringscapaciteit worden gebruikt (zie punt 19). Tijdens of na blootstelling aan de teststof worden de cellen gekweekt gedurende een periode die lang genoeg is om ervoor te zorgen dat chromosoombeschadiging of andere effecten op de celcyclus/celdeling leiden tot de vorming van micronuclei in interfasecellen. Voor de inductie van aneuploïdie moet de teststof gewoon aanwezig zijn tijdens mitose. Geoogste en gekleurde interfasecellen worden onderzocht op de aanwezigheid van micronuclei. Idealiter mogen micronuclei alleen worden gescoord in de cellen die de mitose hebben voltooid tijdens blootstelling aan de teststof of tijdens de periode na de behandeling, als een dergelijke periode wordt toegepast. In culturen die behandeld zijn met een cytokineseblokker wordt dit eenvoudig bereikt door uitsluitend binucleaire cellen te scoren. Wanneer geen cytokineseblokker wordt gebruikt, is het belangrijk aan te tonen dat de onderzochte cellen wellicht celdeling hebben ondergaan tijdens of na de blootstelling aan de teststof, op basis van een toename van de celpopulatie. Het is belangrijk dat alle protocollen aantonen dat celproliferatie zich zowel in de controleculturen als de behandelde culturen heeft voorgedaan, en de mate van door de teststof geïnduceerde cytoxiciteit of cytostase moet worden beoordeeld in alle culturen die voor micronuclei gescoord zijn. BESCHRIJVING VAN DE METHODE Cellen Er kunnen gekweekte primaire menselijke of van andere zoogdieren afkomstige perifere bloedlymfocyten (7) (20) (46) (47) en een aantal cellijnen van knaagdieren, zoals CHO, V79, CHL/IU en L5178Y-cellen of menselijke cellijnen zoals TK6 worden gebruikt (19) (20) (21) (22) (23) (26) (27) (28) (29) (31) (33) (34) (35) (36) (zie punt 6). Andere cellijnen zoals HT29 (48), Caco-2 (49), HepaRG (50) (51), HepG2-cellen (52) (53), A549 en primaire embryocellen van de Syrische hamster (54) zijn gebruikt bij micronucleustesten, maar zijn tot nog toe niet uitgebreid gevalideerd. Het gebruik die cellijnen en celtypen dient daarom te worden verantwoord op basis van hun aangetoonde prestaties in de test, zoals beschreven in het hoofdstuk over aanvaardbaarheidscriteria. Cyto B zou van invloed kunnen zijn op de celgroei van L5178Y en wordt daarom niet aanbevolen voor deze cellijn (23). Wanneer primaire cellen worden gebruikt moet met het oog op het dierenwelzijn worden overwogen om waar mogelijk cellen van menselijke oorsprong te gebruiken en te bemonsteren in overeenstemming met de ethische beginselen en regelgeving voor mensen. Menselijke lymfocyten uit perifeer bloed dienen te worden verkregen van jonge (ongeveer 18-35 jaar oud) niet-rokers zonder bekende ziekten, die voor zover bekend onlangs niet zijn blootgesteld aan genotoxische stoffen (bv. chemische stoffen of ioniserende straling) op een niveau dat het aantal cellen met micronuclei dat op de achtergrond aanwezig is, zou verhogen. Dit zorgt ervoor dat de aanwezigheid van cellen met micronuclei op de achtergrond beperkt en constant blijft. Het aantal cellen met micronuclei dat standaard aanwezig is, neemt toe met de leeftijd, en deze trend is meer uitgesproken bij vrouwen dan bij mannen (55). Indien cellen van meer dan één donor voor gebruik worden gepoold, moet het aantal donors aangegeven worden. Er moet worden aangetoond dat de cellen zich vanaf het begin van de behandeling met de teststof tot aan de bemonstering van de cellen hebben gedeeld. De celculturen worden in een fase van exponentiële groei (cellijnen) gehouden of worden gestimuleerd om zich te delen (primaire culturen van lymfocyten), om de cellen in verschillende stadia van de celcyclus bloot te stellen, aangezien de gevoeligheid van de verschillende celstadia voor de teststoffen mogelijk niet bekend is. De primaire cellen die met mitogene middelen moeten worden gestimuleerd om zich te delen, zijn doorgaans niet meer gesynchroniseerd tijdens de blootstelling aan de teststof (bv. menselijke lymfocyten na 48 uur mitogene stimulatie). Het gebruik van gesynchroniseerde cellen tijdens de behandeling met de teststof wordt niet aangeraden, maar kan aanvaardbaar zijn, mits het naar behoren wordt gemotiveerd. Media en cultuuromstandigheden Er moet worden gezorgd voor geschikte kweekmedia en incubatieomstandigheden voor de culturen (kweekvaten, vochtige atmosfeer van 5 % CO2 indien passend, temperatuur van 37 °C). De cellijnen moeten geregeld worden gecontroleerd om na te gaan of het modale aantal chromosomen stabiel is en er geen besmetting met mycoplasma heeft plaatsgevonden. Indien er wel een besmetting heeft plaatsgevonden of indien het modale aantal chromosomen gewijzigd is, mogen de cellen niet worden gebruikt. De normale tijd voor de celcyclus bij de in het testlaboratorium gebruikte cellijnen of primaire culturen moet worden vastgesteld en moet overeenkomen met de gepubliceerde celkenmerken. Prepareren van de culturen Cellijnen: cellen uit stamculturen worden met een zodanige dichtheid in een kweekmedium geënt dat de cellen in suspensies of in monolagen tot de oogsttijd exponentieel zullen blijven groeien (voor cellen die in monolagen groeien, moet bijvoorbeeld samenvloeiing worden voorkomen). Lymfocyten: met een stollingsremmer (bv. heparine) behandeld volledig bloed of geïsoleerde lymfocyten worden gekweekt (bv. 48 uur voor menselijke lymfocyten) in aanwezigheid van een mitogeen (bv. fytohemagglutinine (PHA) voor menselijke lymfocyten) om celdeling te induceren voordat ze aan de teststof en cytoB worden blootgesteld. Metabolische activering Exogene metabole activeringssystemen moeten worden gehanteerd bij het gebruik van cellen met ontoereikende endogene metabole capaciteit. Het meest gebruikte systeem dat standaard wordt aanbevolen, tenzij een ander systeem gerechtvaardigd is, is een post-mitochondriale fractie aangevuld met een cofactor (S9), verkregen uit de lever van knaagdieren (gewoonlijk ratten) die zijn behandeld met enzyminducerende stoffen als Aroclor 1254 (56) (57) of een combinatie van fenobarbiton en b-naftoflavon (58) (59) (60). De laatste combinatie druist niet in tegen het Verdrag van Stockholm inzake persistente organische verontreinigende stoffen (61). Bovendien is aangetoond dat zij even effectief is als Aroclor 1254 voor de inducering van oxidasen met gemengde functie (58) (59) (60). De S9-fractie wordt meestal gebruikt bij een concentratie van 1 tot 2 % (v/v), maar mag worden verhoogd tot 10 % (v/v) in het uiteindelijke testmedium. Het gebruik van producten die de mitotische index verlagen, en met name van calciumcomplex-vormende producten (62), dient te worden vermeden tijdens de behandeling. De keuze van het type en de concentratie van het exogene metabole activeringssysteem dat of de metabole inductor die wordt gebruikt, kan worden beïnvloed door de klasse van de te testen chemische stof. Teststofbereiding Vaste teststoffen moeten in geschikte oplosmiddelen of media worden bereid en eventueel vóór de behandeling van de cellen worden verdund. Vloeibare teststoffen kunnen direct aan het testsysteem worden toegevoegd en/of worden verdund vóór de behandeling van het testsysteem. Bij het testen van gassen of vluchtige stoffen moeten geschikte aanpassingen van de standaardprotocollen worden gevolgd, zoals de behandeling in gesloten vaten (63) (64) (65). Preparaten van de teststof moeten worden bereid vlak vóór de behandeling, tenzij met stabiliteitsgegevens is aangetoond dat bewaren van het preparaat aanvaardbaar is. Testomstandigheden Oplosmiddelen Het oplosmiddel wordt zo gekozen dat het de oplosbaarheid van de te testen chemische stoffen vergroot zonder een negatieve invloed te hebben op het verloop van de test, bv. door veranderingen in de celgroei, aantasting van de integriteit van de teststof, reacties met kweekvaten, belemmering van het metabole activeringssysteem. Het wordt aanbevolen waar mogelijk eerst het gebruik van een waterig oplosmiddel (of kweekmedium) te overwegen. Gangbare oplosmiddelen zijn water of dimethylsulfoxide (DMSO). Over het algemeen mag de concentratie organische oplosmiddelen niet meer dan 1 % (v/v) bedragen. Als cytoB wordt opgelost in DMSO, mag de totale concentratie organisch oplosmiddel die wordt gebruikt voor zowel de teststof als cytoB niet meer dan 1 % (v/v) bedragen. Is dat niet het geval, dan moeten onbehandelde controlestoffen worden gebruikt om na te gaan of het percentage organisch oplosmiddel geen ongewenste effecten heeft. De concentratie waterige oplosmiddelen (zoutoplossing of water) mag niet meer dan 10 % (v/v) bedragen in het uiteindelijke behandelmedium. Als er andere dan de gangbare oplosmiddelen (bv. ethanol of aceton) worden gebruikt, moeten gegevens worden verstrekt waaruit blijkt dat ze verenigbaar zijn met de teststof en het testsysteem en dat zij geen genetische toxiciteit vertonen in de gebruikte concentratie. Als die gegevens ontbreken, is het belangrijk ook onbehandelde controlestoffen op te nemen (zie aanhangsel 1) en om controleoplosmiddelen te gebruiken om aan te tonen dat het gekozen oplosmiddel geen schadelijke of chromosomale effecten (zoals aneuploïdie of clastogeniciteit) induceert. Gebruik van cytoB als cytokineseblokker Een van de belangrijkste overwegingen bij de uitvoering van de MNvit-test is ervoor te zorgen dat de cellen die worden gescoord tijdens de behandeling of de incubatieperiode na de behandeling, als deze wordt toegepast, de mitose hebben voltooid. Micronucleusscoring dient daarom te worden beperkt tot cellen die tijdens of na behandeling mitose hebben ondergaan. CytoB is de stof die het meest wordt gebruikt om cytokinese te blokkeren, omdat zij de samenstelling van actine afremt en derhalve de scheiding van dochtercellen na mitose voorkomt, wat leidt tot de vorming van binucleaire cellen (6) (66) (67). Tegelijkertijd kan het effect van de teststof op de celproliferatiekinetiek worden gemeten bij het gebruik van cytoB. CytoB dient als cytokineseblokker te worden gebruikt wanneer menselijke lymfocyten worden gebruikt omdat de tijden voor de celcyclus tussen de donors variabel zullen zijn en omdat niet alle lymfocyten op PHA-stimulatie zullen reageren. CytoB is niet verplicht voor andere celtypen als kan worden vastgesteld dat zij deling hebben ondergaan zoals beschreven in punt 27. Bovendien wordt cytoB gewoonlijk niet gebruikt wanneer monsters met behulp van flowcytometriemethoden worden beoordeeld op micronuclei. De geschikte cytoB-concentratie dient door het laboratorium voor elk celtype te worden bepaald om de optimale frequentie van binucleaire cellen te verkrijgen in de culturen van de oplosmiddelcontrole en de concentratie dient aantoonbaar een goede opbrengst op te leveren van binucleaire cellen die vervolgens kunnen worden gescoord. De geschikte cytoB-concentratie is doorgaans tussen 3 en 6 μg/ml (19). Meting van celproliferatie en cytotoxiciteit en keuze van behandelingsconcentraties Bij de bepaling van de hoogste te testen concentratie van de teststof moeten concentraties worden vermeden die kunnen leiden tot artefactuele positieve reacties, zoals reacties die bovenmatige cytotoxiciteit (zie punt 29) voortbrengen, neerslag in het cultuurmedium (zie punt 30) of uitgesproken wijzigingen in pH of osmolaliteit (zie punt 9). Indien de teststof op het ogenblik van toevoeging een uitgesproken wijziging in de pH van het medium veroorzaakt, kan de pH worden aangepast door het uiteindelijke behandelingsmedium te bufferen zodat artefactuele positieve reacties worden vermeden en passende kweekomstandigheden in stand worden gehouden. Metingen van celproliferatie worden verricht om ervoor te zorgen dat voldoende behandelde cellen tijdens de test mitose hebben ondergaan en dat de behandelingen worden uitgevoerd met de passende mate van cytotoxiciteit (zie punt 29). Cytotoxiciteit dient te worden bepaald in het hoofdexperiment, met en zonder metabole activering, aan de hand van een passende indicatie van de celdood en -groei (zie punten 26 en 27). Hoewel een beoordeling van de cytotoxiciteit bij een eerste verkennende test nuttig kan zijn om nauwkeuriger te kunnen bepalen welke concentraties moeten worden gebruikt bij het hoofdexperiment, is een dergelijke eerste test niet verplicht. Indien deze wel wordt uitgevoerd, mag deze de meting van cytotoxiciteit in het hoofdexperiment niet vervangen. De behandeling van culturen met cytoB en de meting van de relatieve frequenties van mononucleaire, binucleaire en multinucleaire cellen in de cultuur verschaft een nauwkeurige methode om het effect van een behandeling op de celproliferatie en de cytotoxische of cytostatische activiteit te kwantificeren (6) en zorgt ervoor dat alleen cellen microscopisch worden gescoord die zich tijdens of na de behandeling hebben gedeeld. De cytokineseblokproliferatie-index (CBPI) (6) (27) (68) of de replicatie-index (RI) van minstens 500 cellen per cultuur (zie aanhangsel 2 voor formules) wordt aanbevolen om de cytotoxische of cytostatische activiteit van een behandeling te ramen door de waarden in de behandelde en de controleculturen te vergelijken. De beoordeling van andere cytotoxiciteitsmarkers (bv. celintegriteit, apoptose, necrose, metafasetelling, celcyclus) kan nuttige informatie opleveren, maar mag de CBPI of RI niet vervangen. In studies zonder cytoB moet worden aangetoond dat de cellen in de cultuur deling hebben ondergaan, opdat een significant deel van de gescoorde cellen deling heeft ondergaan tijdens of na de behandeling met de teststof, anders kunnen er vals-negatieve reacties worden gegenereerd. De meting van de relatieve populatieverdubbeling (RPD) of relatieve toename van de celpopulatie (RICC) wordt aanbevolen om de cytotoxische of cytostatische activiteit van een behandeling te ramen (17) (68) (69) (70) (71) (zie aanhangsel 2 voor formules). Bij verlengde bemonsteringstijden (bv. behandeling gedurende 1,5-2 maal de normale lengte van een celcyclus en oogst na nog eens 1,5-2 maal de normale lengte van een celcyclus, waardoor de bemonsteringstijden in het totaal langer zijn dan 3-4 maal de normale lengte van een celcyclus, zoals beschreven in de punten 38 en 39) kan de geraamde cytotoxiciteit te laag zijn bij RPD (71). Onder deze omstandigheden zou RICC een betere maat kunnen zijn of zou de evaluatie van de cytotoxiciteit na 1,5-2 maal de normale lengte van een celcyclus een bruikbare schatting kunnen zijn. De beoordeling van andere cytotoxiciteitsmarkers (bv. celintegriteit, apoptose, necrose, metafasetelling, proliferatie-index (PI), celcyclus, nucleoplasmatische bruggen of nucleaire knoppen) kan nuttige informatie opleveren, maar mag de RPD of RICC niet vervangen. Er moeten ten minste drie testconcentraties (exclusief het oplosmiddel en positieve controles) die voldoen aan de aanvaardbaarheidscriteria (passende mate van cytotoxiciteit, aantal cellen enz.) worden geëvalueerd. Ongeacht het type cellen (cellijnen of primaire culturen van lymfocyten) kan bij elke geteste concentratie één behandelde cultuur worden gebruikt, maar ook duploculturen. Hoewel het gebruik van duploculturen raadzaam is, is het gebruik van één cultuur ook aanvaardbaar, mits voor één cultuur en duploculturen hetzelfde totale aantal cellen wordt gescoord. Het gebruik van één cultuur is in het bijzonder relevant wanneer meer dan 3 concentraties worden beoordeeld (zie punten 44-45). Voor de gegevensanalyse kunnen de resultaten verkregen op basis van de onafhankelijke duploculturen bij een gegeven concentratie worden gepoold. Voor teststoffen die weinig of geen cytotoxiciteit vertonen, zijn intervallen tussen de testconcentraties van ongeveer een factor 2 tot 3 doorgaans passend. Wanneer er cytotoxiciteit optreedt, moeten de geselecteerde testconcentraties een interval bestrijken van een concentratie waarbij cytotoxiciteit optreedt zoals beschreven in punt 29, tot en met concentraties waarbij er matige en geen of vrijwel geen cytotoxiciteit is. Veel teststoffen vertonen een steile concentratie-responscurve, en om gegevens bij lage en matige cytotoxiciteit te verkrijgen of om in detail de dosis-responsrelatie te bestuderen zullen dichter bij elkaar liggende concentraties en/of meer dan drie concentraties moeten worden gebruikt (één cultuur of duplobepalingen), in het bijzonder in situaties waarin een herhalingsexperiment vereist is (zie punt 60). Als de maximumconcentratie is gebaseerd op de cytotoxiciteit, moet bij de hoogste concentratie worden gestreefd naar 55 ± 5 % cytotoxiciteit, gebruikmakend van de aanbevolen cytotoxiciteitsparameters (d.w.z. een verlaging bij RICC en RPD voor cellijnen wanneer geen cytoB wordt gebruikt en een verlaging bij CBPI en RI wanneer cytoB wordt gebruikt, naar 45 ± 5 % van de tegelijkertijd gemeten negatieve controle) (72). Bij de interpretatie van positieve resultaten moet ervoor worden gezorgd dat deze niet alleen worden gevonden in het hogere deel van dit 55 ± 5 % cytotoxiciteitsbereik (71). Voor slecht oplosbare teststoffen die bij lagere concentraties dan de oplosbaarheidsgrens niet cytotoxisch zijn, moet de hoogste geanalyseerde concentratie troebelheid of een met het oog of met behulp van een omgekeerde microscoop aan het eind van de behandeling met de teststof zichtbaar neerslag opleveren. Zelfs indien cytotoxiciteit optreedt boven de oplosbaarheidsgrens, is het raadzaam de test uit te voeren bij slechts één concentratie die troebelheid of een zichtbaar neerslag oplevert, omdat het neerslag kan leiden tot artefactuele effecten. Bij de concentratie die neerslag oplevert, moet ervoor worden gezorgd dat het neerslag de uitvoering van de test (bv. kleuring of scoring) niet verstoort. Het kan nuttig zijn om voorafgaand aan het experiment de oplosbaarheid in het kweekmedium te bepalen. Indien geen neerslag of beperkende cytotoxiciteit wordt waargenomen, moet de hoogste testconcentratie overeenkomen met de laagste van de volgende waarden: 10 mM, 2 mg/ml of 2 μl/ml (73) (74) (75). Wanneer de samenstelling van de teststof niet vastgesteld is, bv. in geval van een stof van onbekende of variabele samenstelling, complexe reactieproducten of biologische materialen (UVCB) (76), een milieu-extract enz., moet de hoogste concentratie bij het ontbreken van voldoende cytotoxiciteit mogelijk hoger zijn (bv. 5 mg/ml) om de concentratie van elk van de componenten te verhogen. Er zij echter opgemerkt dat deze eisen voor geneesmiddelen voor menselijk gebruik anders kunnen zijn (93). Controles Voor elk oogsttijdstip dienen gelijktijdige negatieve controles (zie punt 21) in het experiment te worden opgenomen, waaraan uitsluitend het oplosmiddel wordt toegediend en die verder op dezelfde manier worden behandeld als de culturen met teststof. Gelijktijdige positieve controles zijn nodig om aan te tonen dat het laboratorium in staat is om onder de omstandigheden van het gebruikte testprotocol en de doeltreffendheid van het exogene metabole activeringssysteem, indien van toepassing, clastogenenen aneugenen te identificeren. Voorbeelden van voor positieve controle gebruikte chemische stoffen worden gegeven in tabel 1 hieronder. Er kunnen ook alternatieve positieve controlestoffen worden gebruikt, indien dat wordt gemotiveerd. Op dit ogenblik zijn er geen aneugenen bekend die metabole activering nodig hebben voor hun genotoxische activiteit (17). Omdat in-vitrogenotoxiciteitstests op zoogdiercellen voldoende gestandaardiseerd zijn voor de korte behandelingen die gelijktijdig met en zonder metabole activering met dezelfde behandelingsduur worden toegepast, kan het gebruik van positieve controles worden beperkt tot een clastogeen dat metabole activering nodig heeft. In dat geval zal een enkele clastogene positieve controlerespons zowel de werking van het metabole activeringssysteem als het reactievermogen van het testsysteem aantonen. Een langlopende behandeling (zonder S9) moet echter haar eigen positieve controle hebben, omdat de behandelingsduur zal afwijken van de test waarbij metabole activering wordt toegepast. Als een clastogeen wordt geselecteerd als enige positieve controle voor een korte behandeling met en zonder metabole activering, dient een aneugeen te worden geselecteerd voor de langdurige behandeling zonder metabole activering. In cellen die qua metabolisme competent zijn en geen S9 nodig hebben, dienen positieve controles voor zowel clastogeniciteit als aneugeniciteit te worden gebruikt. Elke positieve controle moet worden gebruikt bij een of meer concentraties waarvoor een reproduceerbare en detecteerbare stijging ten opzichte van het achtergrondniveau kan worden verwacht, om de gevoeligheid van het testsysteem aan te tonen (d.w.z. de effecten zijn duidelijk maar de gecodeerde objectglaasjes zijn niet onmiddellijk als zodanig herkenbaar), en de respons dient niet in het gedrang te komen door een mate van cytotoxiciteit die de in deze testmethode gespecificeerde grenzen overschrijdt. Tabel 1 Aanbevolen referentiestoffen voor het beoordelen van de bekwaamheid van laboratoria en voor het selecteren van positieve controles
PROCEDURE Behandelingsschema Voor een maximale kans op detectie van een aneugeen of clastogeen dat actief is in een specifiek stadium in de celcyclus, is het belangrijk dat voldoende aantallen cellen tijdens alle stadia van hun celcyclus met de teststof worden behandeld. Alle behandelingen dienen te beginnen en te eindigen terwijl de cellen exponentieel groeien en de cellen dienen te blijven groeien tot op het moment van bemonstering. Het behandelingsschema voor cellijnen en primaire celculturen kan daarom enigszins afwijken van dat voor lymfocyten, die mitogene stimulatie nodig hebben om hun celcyclus te starten (17). Voor lymfocyten is het het meest doeltreffend om met de behandeling met de teststof te starten 44-48 uur na de PHA-stimulatie, wanneer de cellen asynchroon delen (6). Uit gepubliceerde gegevens (19) is gebleken dat de meeste aneugenen en clastogenen worden gedetecteerd na een korte behandelingsperiode van 3 tot 6 uur in aanwezigheid en in afwezigheid van S9, waarna de teststof wordt verwijderd en monsters worden genomen na een periode die overeenkomt met ongeveer 1,5 tot 2,0 maal de normale lengte van een celcyclus vanaf het begin van de behandeling (7). Voor een gedegen evaluatie, die nodig is om een negatief resultaat vast te stellen, moet het experiment onder elk van de drie volgende omstandigheden worden verricht, met een kortlopende behandeling zowel met als zonder metabole activering en een langlopende behandeling zonder metabole activering (zie de punten 56, 57 en 58):
Indien een van de hierboven genoemde experimentele omstandigheden tot een positieve respons leidt, hoeven een of meer van de andere behandelingsregimes mogelijk niet te worden onderzocht. Als geweten is of vermoed wordt dat de teststof de duur van de celcycli beïnvloedt (bv. bij het testen van nucleosideanalogen), in het bijzonder bij p53-competente cellen (35) (36) (77), kunnen de bemonsterings- of hersteltijden worden verlengd met maximaal 1,5 tot 2,0 maal de normale lengte van een celcyclus (d.w.z. tot een totaal van 3,0 tot 4,0 maal de normale lengte van een celcyclus vanaf het begin van de korte en langdurige behandelingen). Deze keuzes zijn bedoeld voor situaties waarin ongerustheid kan bestaan over mogelijke interacties tussen de teststof en cytoB. Wanneer verlengde bemonsteringstijden (d.w.z. een kweektijd van 3,0 tot 4,0 maal de normale lengte van een celcyclus) worden gehanteerd, dient ervoor te worden gezorgd dat de cellen nog steeds actief delen. Voor lymfocyten kan de exponentiële groei bijvoorbeeld afnemen 96 uur na de stimulatie en kunnen cellen in monolaagculturen confluent worden. De voorgestelde celbehandelingsschema's zijn samengevat in tabel 2. Deze algemene behandelingsschema's kunnen worden gewijzigd (en moeten worden gemotiveerd) afhankelijk van de stabiliteit of reactiviteit van de teststof of de bijzondere groeikenmerken van de gebruikte cellen. Tabel 2 Celbehandeling en oogsttijdstippen voor de MNvit-test
In monolaagculturen kunnen aan het einde van de 3-6 uur durende behandeling mitotische cellen (herkenbaar aan hun ronde vorm en het feit dat ze loslaten van het oppervlak) aanwezig zijn. Omdat deze mitotische cellen gemakkelijk loskomen, kunnen ze verloren gaan wanneer het medium met de teststof wordt verwijderd. Als er aanwijzingen zijn voor een aanzienlijke toename van het aantal mitotische cellen vergeleken met de controlegroepen, wat een aanwijzing is voor het waarschijnlijk plotseling stoppen van de mitose, moeten de cellen door centrifugering worden verzameld en opnieuw aan de cultuur worden toegevoegd, opdat er geen cellen die in mitose zijn, en die gevaar voor micronuclei/chromosoomafwijking lopen, op het oogsttijdstip verloren gaan. Oogsten van de cellen en bereiding van de objectglaasjes Elke cultuur dient afzonderlijk te worden geoogst en verwerkt. Het prepareren van de cellen kan een hypotone behandeling inhouden, maar deze stap is niet nodig als op een andere manier toereikende cellenspreiding wordt gerealiseerd. Verschillende technieken kunnen worden gebruikt bij de bereiding van de objectglaasjes op voorwaarde dat er voor het scoren hoogwaardige celpreparaten worden verkregen. Cellen met een intact celmembraan en intact cytoplasma dienen te worden bewaard om de detectie van micronuclei en (bij de methode met cytokineseblokker) een betrouwbare identificatie van binucleaire cellen mogelijk te maken. De objectglaasjes kunnen worden gekleurd met behulp van diverse methoden, zoals Giemsa of fluorescerende DNA-specifieke kleurstoffen. Door het gebruik van gepaste fluorescerende kleurstoffen (bv. acridine oranje (78) of Hoechst 33258 plus pyronine-Y (79)) kunnen sommige artefacten die zich bij het gebruik van niet voor DNA specifieke kleurstoffen kunnen voordoen, worden voorkomen. Antikinetochore antilichamen, FISH met pancentromere DNA-proben of geprepareerde in situ labeling met pancentromeer-specifieke primers, samen met de passende DNA-tegenkleuring, kunnen worden gebruikt om de inhoud (volledige chromosoom worden gekleurd, acentrische chromosoomfragmenten niet) van micronuclei te identificeren indien de mechanistische informatie over hun vorming van belang is (16) (17). Andere methoden voor differentiatie tussen clastogenen en aneugenen kunnen worden gebruikt indien is aangetoond dat zij doeltreffend zijn en indien zij zijn gevalideerd. Zo kan de meting van sub-2N-nuclei als hypodiploïde voorvallen met behulp van technieken zoals beeldanalyse, laserscanningcytometrie of flowcytometrie eveneens nuttige informatie opleveren voor bepaalde cellijnen (80) (81) (82). Ook morfologische waarnemingen van nuclei zouden aanwijzingen kunnen opleveren voor mogelijke aneuploïdie. Voorts zou ook een test voor chromosoomafwijkingen tijdens de metafase kunnen worden verricht, bij voorkeur op hetzelfde celtype en volgens een protocol met een vergelijkbare gevoeligheid, als nuttig instrument om te bepalen of micronuclei te wijten zijn aan chromosoombreuken (wetende dat chromosoomverlies niet zou worden opgemerkt in de test op chromosoomafwijkingen). Analyse Alle objectglaasjes, ook die van het oplosmiddel en de onbehandelde en (indien van toepassing) positieve controles, worden vóór de microscopische analyse onafhankelijk gecodeerd. Wanneer een automatisch scoringsysteem wordt gehanteerd, zoals flowcytometrie, laserscanningcytometrie of beeldanalyse, moeten passende technieken worden gebruikt om vertekening of verschuiving te beperken. Ongeacht het automatische platform dat wordt gebruikt om de micronuclei te tellen, CBPI, RI, RPD of RICC, dient er een gelijktijdige beoordeling plaats te vinden. In met cytoB behandelde culturen moeten de micronucleusfrequenties worden geanalyseerd in ten minste 2 000 binucleaire cellen per concentratie en controle (83), gelijk verdeeld over de culturen, indien er meerdere culturen worden gebruikt. Indien er één cultuur per dosis wordt gebruikt (zie punt 28), moeten ten minste 2 000 binucleaire cellen per cultuur (83) worden gescoord in deze ene cultuur. Als er in elke concentratie aanzienlijk minder dan 1 000 binucleaire cellen per cultuur (voor meerdere culturen), of 2 000 bij gebruik van één cultuur, beschikbaar zijn voor scoring, en als er geen aanzienlijke toename van micronuclei wordt gedetecteerd, moet de test worden herhaald met behulp van meer cellen, of met minder cytotoxische concentraties, naargelang het geval. Er moet gezorgd worden dat er geen binucleaire cellen met onregelmatige vorm worden gescoord of cellen waarvan de twee nuclei sterk in grootte verschillen; ook mogen binucleaire cellen niet worden verward met slecht verspreide multinucleaire cellen. Cellen die meer dan twee hoofdnuclei bevatten, mogen niet op micronuclei worden geanalyseerd, aangezien de basismicronucleusfrequentie in deze cellen hoger kan zijn (84). Het scoren van mononucleaire cellen is aanvaardbaar indien is aangetoond dat de teststof de cytoB-activiteit verstoort. Een nieuwe test zonder cytoB kan in dergelijke gevallen nuttig zijn. Naast binucleaire cellen ook mononucleaire cellen scoren kan nuttige informatie opleveren (85) (86), maar is niet verplicht. In niet met cytoB behandelde cellijnen moeten de micronuclei worden gescoord in ten minste 2 000 cellen per testconcentratie en controle (83), gelijk verdeeld over de culturen, indien er meerdere culturen worden gebruikt. Indien één cultuur per concentratie wordt gebruikt (zie punt 28), moeten ten minste 2 000 cellen per cultuur worden gescoord in deze ene cultuur. Als er in elke concentratie aanzienlijk minder dan 1 000 cellen per cultuur (voor meerdere culturen), of 2000 bij gebruik van één cultuur, beschikbaar zijn voor scoring, en als er geen aanzienlijke toename van micronuclei wordt gedetecteerd, moet de test worden herhaald met behulp van meer cellen, of met minder cytotoxische concentraties, naargelang het geval. Wanneer cytoB wordt gebruikt, moet een CBPI of een RI worden vastgesteld om de celproliferatie te beoordelen (zie aanhangsel 2) met behulp van ten minste 500 cellen per cultuur. Wanneer de behandelingen worden uitgevoerd zonder cytoB, is het essentieel dat er bewijs wordt geleverd dat de cellen in de cultuur hebben gedeeld, zoals besproken in punten 24-28. Bekwaamheid van het laboratorium Om voldoende ervaring met de test op te bouwen vooraleer wordt overgegaan tot routinematig gebruik, dient het laboratorium een reeks experimenten te hebben verricht met positieve controlestoffen die via verschillende mechanismen werken (ten minste één met en één zonder metabole activering en één die via aneugeen mechanisme werkt, gekozen uit de stoffen die zijn opgesomd in tabel 1) en met verschillende negatieve controles (waaronder onbehandelde culturen en verschillende oplosmiddelen/media). Deze positieve en negatieve controleresponsen moeten in overeenstemming zijn met de literatuur. Dit geldt niet voor laboratoria die ervaring hebben, d.w.z. die beschikken over referentiegegevens uit het verleden zoals omschreven in de punten 49-52. Om te bewijzen dat het laboratorium bekwaam is om clastogene en aneugene stoffen op te sporen, de doeltreffendheid van het metabole activeringssysteem te bepalen en de geschiktheid van de scoringsprocedures (visuele microscopische analyse, flowcytometrie, laserscanningcytometrie of beeldanalyse) aan te tonen, dient een selectie positieve controlestoffen (zie tabel 1) te worden onderzocht met korte en langdurige behandelingen zonder metabole activering en met een korte behandeling met metabole activering. Er moet een bereik van concentraties van de geselecteerde chemische stoffen worden gekozen dat reproduceerbare en concentratiegerelateerde stijgingen tot boven het achtergrondniveau oplevert, om de gevoeligheid en het dynamisch bereik van het testsysteem aan te tonen. Controlegegevens uit het verleden Het laboratorium stelt vast:
Wanneer voor het eerst gegevens worden vergaard voor een historische spreiding voor negatieve controles, dienen gelijktijdige negatieve controles te stroken met eventuele gepubliceerde gegevens over negatieve controles. Naarmate meer gegevens uit experimenten aan de controlespreiding worden toegevoegd, dienen gelijktijdige negatieve controles idealiter binnen de controlegrens van 95 % voor die spreiding te vallen (87) (88). De databank met gegevens over negatieve controles uit het verleden van het laboratorium dient aanvankelijk te worden samengesteld op basis van minimaal 10 experimenten, maar bestaat bij voorkeur uit ten minste 20 experimenten die onder vergelijkbare testomstandigheden werden uitgevoerd. Laboratoria dienen methoden voor kwaliteitscontrole, zoals regelkaarten (bv. C-kaart of X-kaart (88)), te gebruiken om na te gaan hoe variabel hun gegevens over positieve en negatieve controles zijn en om aan te tonen dat zij de methodologie “onder controle” hebben (83). Verdere aanbevelingen voor het samenstellen en gebruiken van de gegevens uit het verleden (d.w.z. criteria voor het opnemen van gegevens in en het uitsluiten ervan uit de historische gegevens en de aanvaardbaarheidscriteria voor een gegeven experiment) zijn te vinden in de literatuur (87). Eventuele aanpassingen van het testprotocol moeten worden bekeken vanuit het oogpunt van de samenhang van de gegevens met de bestaande historische controledatabanken van het laboratorium. Eventuele grote inconsistenties moeten leiden tot de oprichting van een nieuwe databank met historische controlegegevens. Negatieve controlegegevens moeten bestaan uit de incidentie van cellen met micronuclei uit één cultuur of de som van de duploculturen zoals beschreven in punt 28. Gelijktijdige negatieve controles liggen idealiter binnen de 95 %-controlegrenzen van de verdeling van de databank met historische negatieve controlegegevens van het laboratorium (87) (88). Wanneer gegevens van gelijktijdige negatieve controles buiten de 95 %-controlegrenzen vallen, kan opneming ervan in de historische controleverdeling toch aanvaardbaar zijn, zolang deze gegevens geen extreme uitschieters zijn en er bewijs is dat het testsysteem 'onder controle' is (zie punt 50) en geen bewijs van technisch of menselijk falen. GEGEVENS EN RAPPORTAGE Presentatie van de resultaten Als de techniek met de cytokineseblokker wordt gebruikt, worden alleen de frequenties van binucleaire cellen met micronuclei (onafhankelijk van het aantal micronuclei per cel) gebruikt bij de evaluatie van micronucleusinductie. De scoring van de aantallen cellen met een, twee of meer micronuclei kan afzonderlijk worden beschreven en kan nuttige informatie opleveren, maar is niet verplicht. Gelijktijdige metingen van de cytotoxiciteit en/of cytostase voor alle behandelde, negatieve en positieve controleculturen dienen te worden vastgesteld (16). De CBPI of RI moeten worden berekend voor alle behandelde en controleculturen als meting van celcyclusvertraging wanneer de methode met de cytokineseblokker wordt gebruikt. Als er geen cytoB wordt gebruikt, moeten de RPD of de RICC worden gebruikt (zie aanhangsel 2). De gegevens moeten voor elke cultuur apart worden verstrekt. Daarnaast moet een overzicht van alle gegevens in tabelvorm worden gegeven. Aanvaardbaarheidscriteria Aanvaarding van een test geschiedt aan de hand van de volgende criteria:
Beoordeling en interpretatie van resultaten Indien aan alle aanvaardbaarheidscriteria is voldaan, wordt de teststof als duidelijk positief beschouwd als in een of meer van de onderzochte experimentele omstandigheden (zie punten 36-39):
Wanneer aan al deze criteria is voldaan, wordt de teststof geacht chromosoombreuken en/of -winst of -verlies te kunnen veroorzaken in dit testsysteem. In de literatuur zijn eveneens aanbevelingen te vinden voor de meest geschikte statistische methoden (90) (91) (92). Indien aan alle aanvaardbaarheidscriteria is voldaan, wordt de teststof als duidelijk negatief beschouwd als in alle onderzochte experimentele omstandigheden (zie punten 36-39):
De teststof wordt in dat geval geacht geen chromosoombreuken en/of -winst of -verlies te kunnen veroorzaken in dit testsysteem. In de literatuur zijn eveneens aanbevelingen te vinden voor de meest geschikte statistische methoden (90) (91) (92). Een duidelijk positieve of negatieve reactie behoeft niet te worden bevestigd. Indien de reactie noch duidelijk negatief noch duidelijk positief is zoals hierboven beschreven, en/of om de biologische relevantie van een resultaat te helpen vaststellen, moeten de gegevens door een deskundige en/of nader onderzoek worden beoordeeld. Het scoren van aanvullende cellen (indien van toepassing) of het uitvoeren van een herhalingsexperiment onder mogelijk aangepaste experimentele omstandigheden (bv. concentratie-intervallen, andere omstandigheden bij metabole activering [d.w.z. S9-concentratie of S9-herkomst]) kan nuttig zijn. In zeldzame gevallen zal de gegevensreeks zelfs na bijkomend onderzoek geen definitief positief of negatief resultaat opleveren en moet worden besloten dat het resultaat twijfelachtig is. Teststoffen die micronuclei in de MNvit-test induceren, kunnen dit doen omdat ze chromosoombreuken, chromosoomverlies of een combinatie van beide induceren. Verdere analyse aan de hand van antikinetochore antilichamen, centromeerspecifieke in-situproben of andere methoden mogen worden gebruikt om vast te stellen of het mechanisme van micronucleusinductie te wijten is aan clastogene en/of aneugene activiteit. Testverslag In het testverslag moet de volgende informatie worden opgenomen:
LITERATUUR
Aanhangsel 1 DEFINITIES Aneugeen : elke chemische stof die of elk proces dat leidt tot aneuploïdie in cellen of organismen door de wisselwerking met componenten van de mitose en meiose in de celdelingscyclus. Aneuploïdie : elke afwijking van het normale diploïde (of haploïde) aantal chromosomen met één chromosoom of meer dan één chromosoom, maar niet met (een) volledige reeks(en) chromosomen (polyploïdie). Apoptose : geprogrammeerde celdood, gekenmerkt door een reeks stappen die leiden tot de afbraak van cellen in door membraan samengehouden deeltjes die vervolgens worden geëlimineerd door fagocytose of door verwijdering. Celproliferatie : toename van het aantal cellen als gevolg van mitose. Centromeer : DNA-plek van een chromosoom waar beide chromatiden worden samengehouden en waar beide kinetochoren zij aan zij zijn aangehecht. Chemische stof : een stof of mengsel. Concentraties : verwijzen naar uiteindelijke concentraties van de teststof in het kweekmedium. Clastogeen : elke chemische stof die of elk proces dat structurele chromosoomafwijkingen veroorzaakt in populaties van cellen of eukaryotische organismen. Cytokinese : deling van de cel onmiddellijk na de mitose, waarbij twee dochtercellen worden gevormd die elk één nucleus bevatten. Cytokineseblokproliferatie-index (CBPI) : de verhouding cellen na tweede deling in de behandelde populatie ten opzichte van de onbehandelde controlegroep (zie aanhangsel 2 voor formule). Cytostase : afremming van celgroei (zie aanhangsel 2 voor formule). Cytotoxiciteit : voor de bepalingen die aan bod komen in testmethode uitgevoerd in aanwezigheid van cytochalasine B, wordt de cytotoxiciteit geïdentificeerd als een daling van de cytokineseblokproliferatie-index (CBPI) of replicatie-index (RI) van de behandelde cellen in vergelijking met de negatieve controle (zie punt 26 en aanhangsel 2). Voor de bepalingen die aan bod komen in testmethode uitgevoerd in afwezigheid van cytochalasine B, wordt de cytotoxiciteit geïdentificeerd als een daling van de relatieve populatieverdubbeling (RPD) of relatieve toename van de celpopulatie (RICC) van de behandelde cellen in vergelijking met de negatieve controle (zie punt 27 en aanhangsel 2). Genotoxisch : een algemene term die alle typen DNA- of chromosoombeschadiging omvat, waaronder breuken, deleties, adducten, veranderingen en bindingen van nucleotiden, herschikkingen, genmutaties, chromosoomafwijkingen en aneuploïdie. Niet alle soorten genotoxische effecten leiden tot mutaties of stabiele chromosoombeschadiging. Interfasecellen : cellen die zich niet in het stadium van de mitose bevinden. Kinetochoor : een hoofdzakelijk uit eiwitten bestaande structuur die zich assembleert nabij het centromeer van een chromosoom en waar tijdens de celdeling de spoeldraden aan vastzitten, zodat de verplaatsing van de dochterchromosomen naar de polen van de dochtercellen regelmatig kan verlopen. Micronucleus : kleine kern die buiten en naast de hoofdkern van cellen tijdens de telofase van de mitose of meiose ontstaat door achtergebleven chromosoomfragmenten of volledige chromosomen. Mitose : deling van de celkern die doorgaans wordt ingedeeld in profase, prometafase, metafase, anafase en telofase. Mitotische index : de verhouding tussen het aantal cellen in de metafase en het totale aantal cellen in een populatie; deze index geeft een indicatie van de snelheid waarmee deze populatie groeit. Mutageen : produceert een erfelijke wijziging van DNA-basepaarsequentie(s) in genen of van de structuur van chromosomen (chromosoomafwijkingen). Non-disjunctie : het feit dat de chromatiden niet uit elkaar gaan en niet afzonderlijk overgaan in de dochtercellen in ontwikkeling, wat leidt tot dochtercellen met abnormale chromosoomaantallen. Onbehandelde controle : culturen die geen behandeling ondergaan (d.w.z. geen teststof en geen oplosmiddel) maar gelijktijdig met en op dezelfde wijze worden verwerkt als de culturen die de teststof ontvangen. Oplosmiddelcontrole : algemene term om de controleculturen aan te duiden die uitsluitend het oplosmiddel ontvangen dat wordt gebruikt om de teststof op te lossen. p53-status : Het proteïne p53 is betrokken bij de regulering van de celcyclus, apoptose en het herstel van het DNA. Cellen met een deficiëntie aan functioneel proteïne p53, die niet in staat zijn om in antwoord op beschadigingen van het DNA de celcyclus tegen te houden of beschadigde cellen te elimineren via apoptose of andere mechanismen (bv. inductie van DNA-herstel) die verband houden met p53-functies, zouden in theorie vatbaarder moeten zijn voor genmutaties of chromosoomafwijkingen. Polyploïdie : afwijkingen in het aantal chromosomen in cellen of organismen waarbij (een) volledige reeks(en) van chromosomen betrokken is (zijn), in tegenstelling tot een afzonderlijk chromosoom of afzonderlijke chromosomen (aneuploïdie). Proliferatie-index (PI) : methode voor cytotoxiciteitsmeting wanneer geen cytoB wordt gebruikt (zie aanhangsel 2 voor formule). Relatieve populatieverdubbeling (RPD) : methode voor cytotoxiciteitsmeting wanneer geen cytoB wordt gebruikt (zie aanhangsel 2 voor formule). Relatieve toename van de celpopulatie (RICC) : methode voor cytotoxiciteitsmeting wanneer geen cytoB wordt gebruikt (zie aanhangsel 2 voor formule). Replicatie-index (RI) : de verhouding tussen het aantal voltooide celdelingscycli in een behandelde cultuur en het aantal voltooide celdelingscycli in de onbehandelde controlecultuur, tijdens de blootstellingsperiode en het herstel (zie aanhangsel 2 voor formule). S9-leverfractie : supernatant van leverhomogenaat na centrifugering bij 9000 g, d.w.z. ruw leverextract. S9-mix : mix van de S9-leverfractie en cofactoren die nodig zijn voor de metabole enzymactiviteit. Teststof : alle volgens deze testmethode geteste stoffen of mengsels. Aanhangsel 2 FORMULES VOOR BEOORDELING VAN DE CYTOTOXICITEIT Wanneer cytoB wordt gebruikt , moet de evaluatie van de cytotoxiciteit uitgaan van de cytokineseblokproliferatie-index (CBPI) of replicatie-index (RI) (17) (69). De CBPI geeft het gemiddelde aantal nuclei per cel aan en kan worden gebruikt om de celproliferatie te berekenen. De RI geeft het relatieve aantal celcycli per cel in behandelde culturen tijdens de periode van blootstelling aan cytoB aan in vergelijking met controleculturen en kan worden gebruikt om het % cytostase te berekenen: % cytostase = 100-100{(CBPIT – 1) ÷ (CBPIC – 1)} en:
waarbij:
Zo is een CBPI van 1 (alle cellen zijn mononucleair) gelijk aan 100 % cytostase. Cytostase = 100 — RI
Zo betekent een RI van 53 % dat, in vergelijking met het aantal cellen van de controlecultuur die zich gedeeld hebben om binucleaire en multinucleaire cellen te vormen, slechts 53 % van dit aantal in de behandelde cultuur zich heeft gedeeld, d.w.z. 47 % cytostase. Wanneer geen cytoB wordt gebruikt , wordt aanbevolen om de cytotoxiciteit te beoordelen op basis van de relatieve toename van de celpopulatie (RICC) of de relatieve populatieverdubbeling (RPD) (69), aangezien beide rekening houden met het percentage van de celpopulatie dat zich gedeeld heeft.
waarbij: Populatieverdubbeling = [log (aantal cellen na behandeling ÷ aanvankelijk aantal cellen)] ÷ log 2 Zo geeft een RICC of een RPD van 53 % aan dat de cytotoxiciteit/cytostase 47 % bedraagt. Met behulp van een proliferatie-index (PI) kan de cytotoxiciteit worden beoordeeld door het aantal klonen te tellen dat bestaat uit 1 cel (cl1), 2 cellen (cl2), 3 tot 4 cellen (cl4) en 5 tot 8 cellen (cl8).
De PI is ook toegepast als waardevolle en betrouwbare cytotoxiciteitsparameter voor in vitro gekweekte cellijnen in afwezigheid van cytoB (35) (36) (37) (38) en kan als nuttige aanvullende parameter worden beschouwd. Hoe dan ook moet het aantal cellen voor behandeling hetzelfde zijn voor behandelde en negatieve controleculturen. Terwijl RCC (d.w.z. aantal cellen in behandelde culturen / aantal cellen in controleculturen) in het verleden werd gebruikt als cytotoxiciteitsparameter, wordt dit niet langer aanbevolen, aangezien RCC de mate van cytotoxiciteit kan onderschatten. Bij het gebruik van automatische scoringsystemen zoals flowcytometrie, laserscanningcytometrie of beeldanalyse kan het aantal cellen in de formule bijvoorbeeld worden vervangen door het aantal nuclei. In de negatieve controleculturen dient verdubbeling van de populatie of de replicatie-index verenigbaar te zijn met de verplichting om cellen na behandeling te bemonsteren na een periode die overeenkomt met ongeveer 1,5 tot 2,0 normale celcycli. |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
15) |
In deel B worden de volgende hoofdstukken toegevoegd: „B.59 In-chemicohuidsensibilisatie: Direct Peptide Reactivity Assay (DPRA-test) INLEIDING Deze testmethode (TM) is gelijkwaardig aan testrichtlijn (TG) 442C (2015) van de OESO. Een huidsensibiliserende stof is een stof die een allergische reactie uitlokt na contact met de huid, zoals gedefinieerd in het wereldwijd geharmoniseerd systeem voor de indeling en etikettering van chemische stoffen van de Verenigde Naties (GHS) (1) en Verordening (EG) nr. 1272/2008 van de Europese Unie betreffende de indeling, etikettering en verpakking van stoffen en mengsel (CLP-verordening) (18). Deze testmethode reikt een in-chemicoprocedure (Direct Peptide Reactivity Assay — DPRA) aan die kan worden gebruikt ter ondersteuning van de onderscheiding tussen huidsensibiliserende stoffen en niet-sensibiliserende stoffen, in overeenstemming met het VN-GHS en de CLP-verordening. Er bestaat algemene overeenstemming over de belangrijkste biologische gebeurtenissen die aan de basis liggen van huidsensibilisatie. De bestaande kennis van de chemische en biologische mechanismen die verband houden met huidsensibilisatie zijn samengevat in een Adverse Outcome Pathway (AOP) (2), van de moleculaire inleidende gebeurtenis over de tussenliggende gebeurtenissen tot het schadelijke effect, te weten allergische contactdermatitis bij mensen of contactovergevoeligheid bij knaagdieren. De moleculaire gebeurtenis in de AOP die aanleiding geeft tot huidsensibilisatie, is de covalente binding van elektrofiele stoffen aan nucleofiele centra in eiwitten in de huid. De bepaling van de huidsensibilisatie ging meestal gepaard met het gebruik van proefdieren. Bij de klassieke methoden op basis van cavia's, de maximalisatietest met cavia's (GMPT) van Magnusson-Kligman en de Buehler-test (TM B.6 (3)) worden zowel de inductie- als de uitlokkingsfase van huidsensibilisatie bestudeerd. Ook een murinetest, de lokale lymfkliertest (LLNA, TM B.42 (4)), en de twee niet-radioactieve aanpassingen ervan, LLNA: DA (TM B.50 (5)) en LLNA: BrdU-ELISA (TM B.51 (6)), waarbij telkens uitsluitend de inductierespons wordt beoordeeld, zijn gangbaar geworden aangezien zij beter zijn voor het dierenwelzijn dan tests met cavia's en het mogelijk maken om de inductiefase van huidsensibilisatie objectief te meten. Recenter zijn ook mechanistisch gebaseerde in-chemico- en in-vitrotestmethoden wetenschappelijk geldig bevonden om de potentiële huidsensibilisatie van chemische stoffen te beoordelen. Om de dierproeven die momenteel worden gebruikt, volledig te kunnen vervangen, zullen echter combinaties van methoden zonder dierproeven (in silico, in chemico, in vitro) binnen geïntegreerde test- en beoordelingsbenaderingen (Integrated Approaches to Testing and Assessment of IATA) nodig zijn, aangezien elk van de momenteel beschikbare testmethoden zonder dierproeven vanuit mechanistisch oogpunt slechts een beperkt deel van de AOP afdekt (2) (7). De DPRA wordt voorgesteld om de moleculaire gebeurtenis die aanleiding geeft tot huidsensibilisatie van de desbetreffende AOP, namelijk de eiwitreactiviteit, te bestuderen door de reactiviteit van teststoffen op synthetische modelpeptiden die lysine of cysteïne bevatten, te kwantificeren (8). Vervolgens worden de waarden voor het percentage peptide-depletie voor cysteïne en lysine gebruikt om een stof in te delen in een van vier mogelijke reactiviteitsklassen om het onderscheid tussen huidsensibiliserende stoffen en niet-sensibiliserende stoffen te staven (9). De DPRA werd geëvalueerd in een valideringsstudie onder leiding van het referentielaboratorium voor alternatieven voor dierproeven van de Europese Unie (EURL ECVAM) en in een latere onafhankelijke beoordeling door vakgenoten van het Raadgevend wetenschappelijk comité van het ECVAM (ESAC) en werd wetenschappelijk geldig bevonden (10) voor gebruik als onderdeel van een IATA ter ondersteuning van het onderscheid tussen huidsensibiliserende stoffen en niet-sensibiliserende stoffen met het oog op de gevarenindeling en etikettering. Voorbeelden van het gebruik van DPRA-gegevens in combinatie met andere informatie zijn beschreven in de literatuur (11) (12) (13) (14). Definities worden gegeven in aanhangsel 1. INLEIDENDE OVERWEGINGEN, TOEPASSELIJKHEID EN BEPERKINGEN De correlatie tussen eiwitreactiviteit en potentiële huidsensibilisatie is uitvoerig beschreven (15) (16) (17). Desondanks volstaat informatie over de eiwitreactiviteit die werd verzameld met behulp van test- en niet-testmethoden als dusdanig niet altijd om te besluiten dat een chemische stof geen huidsensibilisatie veroorzaakt, aangezien eiwitbinding slechts één belangrijke gebeurtenis is, zij het dan wel de moleculaire gebeurtenis binnen de AOP die aanleiding geeft tot huidsensibilisatie. De gegevens die deze testmethode oplevert, dienen daarom te worden bekeken in het kader van een geïntegreerde aanpak, zoals een IATA, waarin zij worden gecombineerd met andere aanvullende informatie die bijvoorbeeld is afgeleid uit in-vitrotesten waarin andere belangrijke gebeurtenissen binnen de AOP van huidsensibilisatie worden bestudeerd of uit niet-testmethoden zoals een “read-across” van chemische analogen. Deze testmethode kan in combinatie met andere aanvullende informatie worden gebruikt om het onderscheid tussen huidsensibiliserende stoffen (d.w.z. VN-GHS/CLP-categorie 1) en niet-sensibiliserende stoffen te staven in de context van IATA. Deze testmethode kan niet afzonderlijk worden gebruikt om huidsensibiliserende stoffen in te delen in de subcategorieën 1A en 1B zoals gedefinieerd door VN-GHS/CLP of om de potentie te voorspellen met het oog op besluiten inzake veiligheidsbeoordelingen. Naargelang van het regelgevingskader kan een positief resultaat van de DPRA echter wel als dusdanig worden gebruikt om een stof in te delen in VN-GHS/CLP-categorie 1. Van de DPRA-testmethode is aangetoond dat zij overdraagbaar is naar laboratoria die ervaring hebben met HLPC-analyses (high-performance liquid chromatography of hogedrukvloeistofchromatografie). De verwachte reproduceerbaarheid van voorspellingen op basis van deze testmethode bedraagt 85 % binnen laboratoria en 80 % tussen verschillende laboratoria (10). De resultaten van de valideringsstudie (18) en de gepubliceerde studies (19) geven over het algemeen aan dat de nauwkeurigheid van de DPRA bij het onderscheiden van sensibiliserende stoffen (d.w.z. VN-GHS/CLP-cat. 1) van niet-sensibiliserende stoffen 80 % (N=157) bedraagt, met een gevoeligheid van 80 % (88/109) en een specificiteit van 77 % (37/48) in vergelijking met de resultaten van LLNA-tests. Bij de DPRA is de kans op een te lage voorspelling voor chemische stoffen met een lage tot gemiddelde potentie voor huidsensibilisatie (d.w.z. VN-GHS/CLP-subcategorie 1B) hoger dan voor chemische stoffen met een hoge potentie voor huidsensibilisatie (d.w.z. VN-GHS/CLP-subcategorie 1A) (18) (19). De waarden die hier worden vermeld voor de nauwkeurigheid van de DPRA als losstaande testmethode, vormen echter slechts een indicatie, aangezien de testmethode in combinatie met andere informatiebronnen moet worden bekeken in de context van een IATA en in overeenstemming met hetgeen hierboven bepaald is in punt 9. Bovendien moet bij de beoordeling van methoden zonder dierproeven voor huidsensibilisatie rekening worden gehouden met het feit dat de LLNA-test en andere dierproeven de situatie van de doelsoort, in dit geval mensen, mogelijk niet volledig weerspiegelen. Op basis van de globale beschikbare gegevens is gebleken dat de DPRA kan worden toegepast op teststoffen die verschillende organische functionele groepen, reactiemechanismen, potenties voor huidsensibilisatie (zoals bepaald bij in-vivo-onderzoek) en fysisch-chemische eigenschappen beslaan (8) (9) (10) (19). Gecombineerd geeft deze informatie aan dat de DPRA nuttig kan zijn om het gevaar van huidsensibilisatie te bepalen. De term “teststof” wordt in deze testmethode gebruikt om te verwijzen naar datgene dat wordt getest en houdt geen verband met de toepasselijkheid van de DPRA op het testen van stoffen en/of mengsels. Deze testmethode kan niet worden toegepast om metaalverbindingen te testen, aangezien bekend is dat dergelijke verbindingen via andere mechanismen dan covalente binding reageren met eiwitten. Een teststof dient oplosbaar te zijn in een passend oplosmiddel bij een uiteindelijke concentratie van 100 mM (zie punt 18). Teststoffen die niet oplosbaar zijn bij die concentratie kunnen echter nog steeds worden getest bij een lagere oplosbare concentratie. In dat geval kan een positief resultaat nog steeds worden gebruikt om de identificatie van de teststof als huidsensibiliserende stof te ondersteunen, maar mag er aan een negatief resultaat geen definitieve conclusie inzake het gebrek aan reactiviteit worden gekoppeld. Er is momenteel slechts beperkte informatie beschikbaar over de toepasselijkheid van de DPRA op mengsels waarvan de samenstelling bekend is (18) (19). Vanuit technisch oogpunt wordt er evenwel van uitgegaan dat de DPRA kan worden toegepast op het testen van stoffen die uit meerdere componenten bestaan, en van mengsels waarvan de samenstelling bekend is (zie punt 18). Voordat deze testmethode wordt gebruikt op een mengsel om gegevens te genereren voor een beoogd regelgevingsdoel, moet worden nagegaan of, en zo ja, waarom deze methode betrouwbare resultaten oplevert voor dat doel. Zulke overwegingen zijn niet nodig wanneer het testen van het mengsel wettelijk vereist is. Het huidige voorspellingsmodel kan niet worden gebruikt voor complexe mengsels waarvan de samenstelling niet bekend is, of voor stoffen waarvan de samenstelling niet bekend of variabel is, voor complexe reactieproducten of voor biologische materialen (d.w.z. UVCB-stoffen), vanwege de vastgestelde molaire verhouding van teststof en peptide. Daarvoor zal een nieuw voorspellingsmodel op basis van een gravimetrische aanpak moeten worden ontwikkeld. Wanneer kan worden aangetoond dat de testmethode niet kan worden toegepast op andere specifieke categorieën van chemische stoffen, mag de testmethode niet voor die specifieke categorieën van chemische stoffen worden gebruikt. Deze testmethode is een in-chemicomethode die geen metabool systeem omvat. Chemische stoffen die enzymatische bioactivering nodig hebben om hun potentieel voor huidsensibilisatie uit te oefenen (zogeheten prohaptenen), kunnen met deze testmethode niet worden opgespoord. Chemische stoffen die in sensibiliserende stoffen veranderen na abiotische transformatie (zogeheten prehaptenen) zouden in sommige gevallen correct kunnen worden opgespoord met de testmethode (18). In het licht van het bovenstaande moeten negatieve resultaten verkregen met deze testmethode worden geïnterpreteerd in de context van de vermelde beperkingen en in samenhang met andere informatiebronnen in het kader van een IATA. Teststoffen die niet covalent binden aan het peptide maar de oxidatie ervan bevorderen (zogeheten cysteïnedimerisatie), kunnen mogelijk tot een te hoge inschatting van de peptidedepletie leiden, hetgeen tot vals-positieve voorspellingen en/of een onterechte toewijzing aan een hogere reactiviteitsklasse zou kunnen leiden (zie punten 29 en 30). Zoals beschreven, helpt de DPRA om huidsensibiliserende stoffen en niet-sensibiliserende stoffen van elkaar te onderscheiden. Deze testmethode kan echter ook bijdragen aan de beoordeling van de sensibiliserende potentie (11) wanneer zij wordt gebruikt in het kader van een geïntegreerde aanpak zoals een IATA. Er moet evenwel nog meer onderzoek worden verricht, bij voorkeur op basis van gegevens verzameld bij mensen, om te bepalen hoe de resultaten van de DPRA kunnen worden benut bij de beoordeling van de potentie. PRINCIPE VAN DE TEST De DPRA is een in-chemicomethode om de resterende concentratie cysteïne- of lysinebevattend peptide na 24 uur incubatie met de teststof bij 25±2,5 °C te kwantificeren. De synthetische peptiden bevatten fenylalanine om de opsporing te vergemakkelijken. De relatieve peptideconcentratie wordt gemeten met behulp van hogedrukvloeistofchromatografie (HPLC) met gradiëntelutie en uv-detectie op 220 nm. De waarden voor het percentage cysteïne- en lysinepeptidedepletie worden berekend en gebruikt in een voorspellingsmodel (zie punt 29) waarmee de teststof kan worden ingedeeld in een van de vier reactiviteitsklassen die worden gebruikt om het onderscheid tussen sensibiliserende stoffen en niet-sensibiliserende stoffen te ondersteunen. Vooraleer over te gaan tot routinegebruik van een onder deze testmethode vallende methode, dienen laboratoria eerst hun technische bekwaamheid aan te tonen met de tien stoffen die in aanhangsel 2 zijn opgesomd. PROCEDURE Deze testmethode is gebaseerd op DPRA DB-ALM-protocol nr. 154 (20), het protocol dat werd gebruikt voor de door het EURL ECVAM gecoördineerde valideringsstudie. Wanneer de methode in het laboratorium wordt ingevoerd en toegepast, wordt aanbevolen om dit protocol te volgen. Hieronder worden de belangrijkste onderdelen en procedures van de DPRA beschreven. Als er een alternatieve HPLC-opstelling wordt gebruikt, moet worden aangetoond dat die gelijkwaardig is aan de gevalideerde opstelling zoals beschreven in het DB-ALM-protocol (bv. door de stoffen voor bekwaamheidstoetsing in aanhangsel 2 te testen). Bereiding van de cysteïne of lysine bevattende peptiden Vlak voor de incubatie met de teststof moeten stamoplossingen van cysteïne (Ac-RFAACAA-COOH) en lysine (Ac-RFAAKAA-COOH) die synthetische peptiden met een zuiverheid van meer dan 85 % en bij voorkeur 90-95 % bevatten, vers worden bereid. De uiteindelijke concentratie van het cysteïnepeptide moet 0,667 mM in een fosfaatbuffer met pH 7,5 bedragen, terwijl de uiteindelijke concentratie van het lysinepeptide 0,667 mM in een ammoniumacetaatbuffer met pH 10,2 dient te zijn. De reeks experimenten van de HPLC dient zo te worden opgezet dat de HPLC-analyse minder dan 30 uur in beslag neemt. Voor de HPLC-opzet die in de valideringsstudie werd gebruikt en die in deze testmethode wordt beschreven, kunnen tot 26 analysemonsters (waaronder de teststof, de positieve controle en de nodige oplosmiddelcontroles naargelang van het aantal afzonderlijke oplosmiddelen dat bij de test wordt gebruikt, waarvan elk oplosmiddel in drievoud wordt getest) in een enkele HPLC-reeks worden meegenomen. Bij elk monster dat in dezelfde reeks wordt geanalyseerd, moeten dezelfde stamoplossingen van cysteïne- en lysinepeptide worden gebruikt. Er wordt aanbevolen om de goede oplosbaarheid van elke afzonderlijke partij peptide te controleren voor gebruik. Bereiding van de teststof De oplosbaarheid van de teststof in een passend oplosmiddel moet voorafgaand aan de test worden beoordeeld volgens de solubilisatieprocedure beschreven in het DB-ALM-protocol voor de DPRA (20). Wanneer een passend oplosmiddel wordt gebruikt, zal de teststof volledig oplossen. Aangezien de teststof bij de DPRA langdurig wordt geïncubeerd met het cysteïne- of het lysinepeptide, volstaat het om visueel te controleren of er een heldere oplossing wordt gevormd om na te gaan of de teststof (en alle componenten ervan, wanneer een stof met meerdere componenten of een mengsel wordt getest) volledig wordt opgelost. Voorbeelden van geschikte oplosmiddelen zijn acetonitril, water, een mengsel van water en acetonitril in een verhouding van 1:1, isopropanol, aceton of een mengsel van aceton en acetonitril in een verhouding van 1:1. Andere oplosmiddelen mogen worden gebruikt voor zover zij geen gevolgen hebben voor de stabiliteit van het peptide, die wordt bewaakt met behulp van referentiecontroles C (d.w.z. monsters die uitsluitend bestaan uit het peptide opgelost in een passend oplosmiddel; zie aanhangsel 3). Als een teststof niet oplosbaar is in een van deze oplosmiddelen, dient in laatste instantie te worden geprobeerd ze op te lossen in 300 μl DMSO en om de daaruit resulterende oplossing te verdunnen met 2 700 μl acetonitril. Als de teststof niet oplosbaar is dat mengsel, moet worden geprobeerd om dezelfde hoeveelheid teststof op te lossen in 1 500 μl DMSO en de resulterende oplossing te verdunnen met 1 500 μl acetonitril. De teststof dient vooraf te worden afgewogen in glazen flessen en onmiddellijk voor het testen te worden opgelost in een passend oplosmiddel om een oplossing van 100 mM te bereiden. Voor mengsels en stoffen met meerdere componenten waarvan de samenstelling bekend is, dient één enkele zuiverheid te worden bepaald door het aandeel van de verschillende componenten (behalve water) op te tellen en dient één enkel schijnbaar molecuulgewicht te worden bepaald door het afzonderlijke molecuulgewicht van elke component in het mengsel (behalve water) en het afzonderlijke aandeel daarvan in aanmerking te nemen. De aldus bepaalde zuiverheid en het schijnbare molecuulgewicht dienen vervolgens te worden gebruikt om te berekenen welk gewicht van de teststof moet worden gebruikt om 100 mM oplossing te bereiden. Voor polymeren waarvan geen bepalend molecuulgewicht kan worden vastgesteld, mag het molecuulgewicht van het monomeer (of het schijnbare molecuulgewicht van de verschillende monomeren waaruit het polymeer bestaat) worden gebruikt om 100 mM oplossing te bereiden. Bij het testen van mengsels, stoffen met meerdere componenten of polymeren waarvan de samenstelling bekend is, dient echter te worden overwogen om ook de pure chemische stof te testen. Voor vloeistoffen dient de pure chemische stof als dusdanig te worden getest, zonder voorafgaande verdunning, door ze te incuberen in een molaire verhouding van 1:10 en 1:50 met respectievelijk het cysteïne- en het lysinepeptide. Voor vaste stoffen moet de teststof worden opgelost tot de maximaal oplosbare concentratie in hetzelfde oplosmiddel dat wordt gebruikt om de schijnbare 100 mM-oplossing te bereiden. Die oplossing dient vervolgens als dusdanig te worden getest, zonder verdere verdunning, door ze te incuberen in een molaire verhouding van 1:10 en 1:50 met respectievelijk het cysteïne- en het lysinepeptide. Indien de resultaten voor de schijnbare 100 mM-oplossing en de pure chemische stof overeenkomen (reactief of niet-reactief), kan op basis daarvan een definitieve conclusie worden getrokken over het resultaat. Bereiding van de positieve controle, de referentiecontroles en de co-elutiecontroles Als positieve controle dient cinnamaldehyde (CAS 104-55-2; ≥95 % zuiver, van levensmiddelenkwaliteit) in een concentratie van 100 mM in acetonitril te worden gebruikt. Andere geschikte positieve controles, die bij voorkeur gematigde depletiewaarden opleveren, kunnen worden gebruikt als er historische gegevens beschikbaar om vergelijkbare aanvaardbaarheidscriteria voor de reeks experimenten af te leiden. Daarnaast moeten er referentiecontroles (d.w.z. monsters die enkel het peptide bevatten, opgelost in het juiste oplosmiddel) in de reeks experimenten van de HPLC worden opgenomen die worden gebruikt om de geschiktheid van het HPLC-systeem voor de analyse (referentiecontroles A) en de stabiliteit van de referentiecontroles in de loop van de tijd (referentiecontroles B) na te gaan en om te verzekeren dat het oplosmiddel dat wordt gebruikt om de teststof op te lossen, geen invloed heeft op het percentage peptidedepletie (referentiecontroles C) (zie aanhangsel 3). Voor elke chemische stof moet de juiste referentiecontrole worden gebruikt om het percentage peptidedepletie te berekenen voor die stof (zie punt 26). Daarnaast moet voor elk van de onderzochte teststoffen ook een co-elutiecontrole in de reeks experimenten worden opgenomen, waarbij voor elke geanalyseerde teststof enkel de desbetreffende stof wordt gebruikt, om eventuele co-elutie van de teststof met het lysine- of cysteïnepeptide op te sporen. Incubatie van de teststof met de cysteïne- en lysinepeptideoplossingen De cysteïne- en lysinepeptideoplossingen moeten worden geïncubeerd in glazen autosampler-flessen met de teststof in een verhouding van respectievelijk 1:10 en 1:50. Als er meteen na toevoeging van de teststofoplossing aan de peptideoplossing een neerslag wordt waargenomen, vanwege de beperkte oplosbaarheid in water van de teststof, kan niet met zekerheid worden vastgesteld hoeveel teststof in de oplossing is overgebleven om met het peptide te reageren. In dat geval kan een positief resultaat bijgevolg nog steeds worden gebruikt, maar is een negatief resultaat onzeker en dient dat resultaat met de nodige behoedzaamheid te worden geïnterpreteerd (zie ook de bepalingen in punt 11 inzake het testen van chemische stoffen die niet oplosbaar zijn tot een concentratie van 100 mM). De reactieoplossing dient voordat de HPLC-analyse wordt uitgevoerd gedurende 24±2 uur in het donker te worden bewaard op 25±2,5 °C. Elke teststof dient in drievoud te worden geanalyseerd voor beide peptiden. De monsters moeten voorafgaand aan de HPLC-analyse visueel worden gecontroleerd. Als er een neerslag of fasescheiding wordt waargenomen, kunnen de monsters op lage snelheid worden gecentrifugeerd (100-400 g) om de neerslag naar de bodem van de fles te doen zinken, aangezien grote hoeveelheden neerslag de HPLC-slangen of -kolommen kunnen verstoppen. Als na de incubatieperiode een neerslag of fasescheiding wordt waargenomen, kan de peptidedepletie te laag worden ingeschat en kan er bij een negatief resultaat geen sluitende conclusie worden getrokken over het gebrek aan reactiviteit. Opstellen van de standaardkalibratiecurve voor de HLPC Er moet voor zowel het cysteïne- als het lysinepeptide een standaardkalibratiecurve worden gegenereerd. Er dienen peptidestandaarden te worden bereid in een oplossing van 20 % of 25 % acetonitril:buffer met behulp van een fosfaatbuffer (pH 7,5) voor het cysteïnepeptide en een ammoniumacetaatbuffer (pH 10,2) voor het lysinepeptide. Aan de hand van standaarden voor de seriële verdunning van de peptidestamoplossing (0,667 mM) dienen 6 kalibratieoplossingen te worden bereid om het bereik van 0,534 tot 0,0167 mM te bestrijken. Voorts dient een blanco van de verdunningsbuffer in de standaardkalibratiecurve te worden opgenomen. Geschikte kalibratiecurven moeten een r2 > 0,99 hebben. Voorbereiding en HPLC-analyse Voordat de analyse wordt verricht, moet de geschiktheid van het HPLC-systeem worden nagegaan. De peptidedepletie wordt bewaakt via HPLC in combinatie met een uv-detector (fotodiode-arraydetector of extinctiedetector met variabele golflengte met een signaal van 220 nm). Het HPLC-systeem wordt van de juiste kolom voorzien. In HPLC-opstelling die in het gevalideerde protocol is beschreven, gaat de voorkeur uit naar een Zorbax SB-C-18 2,1 mm x 100 mm x 3,5 micron. Met deze reversed-phase-HPLC-kolom moet het hele systeem voor het experiment gedurende ten minste 2 uur op 30 °C met 50 % fase A (0,1 % (v/v) trifluorazijnzuur in water) en 50 % fase B (0,085 % (v/v) trifluorazijnzuur in acetonitril) worden geëquilibreerd. De HPLC-analyse moet worden uitgevoerd met een stroomdebiet van 0,35 ml/min en een lineaire gradiënt die het acetonnitrilpercentage in 10 minuten van 10 % naar 25 % brengt, gevolgd door een snelle verhoging van het percentage acetonitril tot 90 % om andere materialen te verwijderen. Er moeten gelijke volumes van elke standaard, elk monster en elke controle worden geïnjecteerd. De kolom dient tussen injecties gedurende 7 minuten opnieuw te worden geëquilibreerd onder de uitgangsomstandigheden. Als er een andere reversed-phase-HPLC-kolom wordt gebruikt, moeten bovenstaande opstellingsparameters mogelijk worden aangepast om een passende elutie en integratie van het cysteïne- en het lysinepeptide te garanderen. Mogelijk moet ook het injectievolume worden aangepast, dat kan variëren naargelang van het gebruikte systeem (gewoonlijk binnen het bereik van 3-10 μl). Belangrijk is ook dat als er een alternatieve HPLC-opstelling wordt gebruikt, moet worden aangetoond dat die gelijkwaardig is aan de hierboven beschreven gevalideerde opstelling (bv. door de stoffen voor bekwaamheidstoetsing in aanhangsel 2 te testen). De extinctie wordt bewaakt op 220 nm. Als een fotodiode-arraydetector wordt gebruikt, dient ook de extinctie op 258 nm te worden geregistreerd. Er zij op gewezen dat bepaalde voorraden acetonitril de stabiliteit van het peptide kunnen aantasten. Dat moet worden beoordeeld wanneer een nieuwe partij acetonitril wordt gebruikt. De ratio van de piekoppervlakte bij 220 nm en bij 258 nm kan ook als indicator voor de co-elutie worden gebruikt. Voor elk monster vormt een ratio binnen het bereik van 90 % < gemiddelde (19) oppervlakteratio van de controlemonsters <100 % een goede indicatie dat er geen co-elutie is opgetreden. Bepaalde teststoffen kunnen de oxidatie van het cysteïnepeptide bevorderen. De piek van het gedimeriseerde cysteïnepeptide kan visueel worden bewaakt. Als er dimerisatie lijkt te zijn opgetreden, dient dit te worden genoteerd, aangezien het percentage peptidedepletie te hoog kan worden ingeschat, wat aanleiding kan geven tot vals-positieve voorspellingen en/of toewijzing aan een hogere reactiviteitsklasse (zie punten 29 en 30). De HPLC-analyse voor het cysteïne- en het lysinepeptide kan gelijktijdig gebeuren (als er twee HPLC-systemen voorhanden zijn) of op verschillende dagen. Als de analyse op verschillende dagen wordt verricht, dienen alle oplossingen met teststoffen elke dag vers te worden bereid voor beide tests. De tijd van de analyse dient te worden opgenomen om te verzekeren dat de injectie van het eerste monster wordt gestart 22 tot 26 uur nadat de teststof werd gemengd met de peptideoplossing. De reeks experimenten van de HPLC dient zo te worden opgezet dat de HPLC-analyse minder dan 30 uur in beslag neemt. Voor de HLPC-opstelling die in de valideringsstudie werd gebruikt en in deze testmethode is beschreven, kunnen tot 26 analysemonsters worden meegenomen in een enkele HPLC-reeks (zie ook punt 17). Aanhangsel 3 bevat een voorbeeld van een reeks HPLC-analyses. GEGEVENS EN RAPPORTAGE Evaluatie van de gegevens De concentratie van cysteïne- en lysinepeptide wordt in elk monster fotometrisch bepaald op 220 nm door de piekoppervlakte (de oppervlakte onder de curve of AUC) van de passende pieken te meten en de concentratie peptide te berekenen met behulp van de lineaire kalibratiecurve die uit de standaarden werd afgeleid. Het percentage peptidedepletie wordt voor elk monster bepaald door de piekoppervlakte te meten en ze te delen door de gemiddelde piekoppervlakte van de relevante referentiecontroles C (zie aanhangsel 3) volgens onderstaande formule.
Aanvaardbaarheidscriteria Wil een reeks experimenten geldig zijn, dan moet aan de volgende criteria worden voldaan:
Als aan een of meerdere van deze criteria niet is voldaan, dient de reeks experimenten te worden overgedaan. Willen de resultaten voor een teststof geldig zijn, dan moet aan de volgende criteria worden voldaan:
Voorspellingsmodel De gemiddelde waarde voor het percentage cysteïne- en lysinedepletie wordt berekend voor elke teststof. Een negatieve depletie wordt bij de berekening van het gemiddelde als “0” beschouwd. Bij gebruik van het voorspellingsmodel voor cysteïne 1:10/lysine 1:50 uit tabel 1 dient de drempel van een gemiddelde peptidedepletie van 6,38 % te worden gehanteerd om het onderscheid tussen huidsensibiliserende stoffen en niet-sensibiliserende stoffen in het kader van een IATA te ondersteunen. De toepassing van het voorspellingsmodel voor het toewijzen van een teststof aan een reactiviteitsklasse (lage, matige en hoge reactiviteit) kan wellicht nuttig blijken als inbreng voor de potentiebeoordeling in het kader van een IATA. Tabel 1 Voorspellingsmodel cysteïne 1:10/lysine 1:50 (20)
In sommige gevallen zou de teststof (de stof of een of meerdere van de componenten van een stof met meerdere componenten of een mengsel) op 220 nm aanzienlijk absorbeert en dezelfde retentietijd heeft als het peptide (co-elutie). Co-elutie kan worden verholpen door de HPLS-opstelling enigszins aan te passen om het verschil in retentietijd tussen de teststof en het peptide te vergroten. Als een alternatieve HPLC-opstelling wordt gebruikt om co-elutie te voorkomen, moet worden aangetoond dat die gelijkwaardig is aan gevalideerde opstelling (bv. door de bekwaamheidsstoffen in aanhangsel 2 te testen). Wanneer co-elutie optreedt, kan de piek van het peptide niet worden geïntegreerd en kan het percentage peptidedepletie niet worden berekend. Als co-elutie van dergelijke teststoffen optreedt bij zowel het cysteïne- als het lysinepeptide, dient de analyse te worden gemeld als “niet overtuigend”. In gevallen waarin co-elutie enkel optreedt bij het lysinepeptide, kan het voorspellingsmodel voor cysteïne 1:10 uit tabel 2 worden gebruikt. Tabel 2 Voorspellingsmodel cysteïne 1:10 (22)
Het is mogelijk dat de retentietijd van de teststof en die van een van beide peptiden in sommige gevallen niet volledig overlapt. In dergelijke gevallen kunnen de waarden voor het percentage peptidedepletie worden geraamd en gebruikt binnen het voorspellingsmodel voor cysteïne 1:10/lysine 1:50, maar kan de teststof niet met de nodige nauwkeurigheid aan een reactiviteitsklasse worden toegewezen. Eén HPLC-analyse voor zowel het cysteïne- als het lysinepeptide zou voldoende moeten zijn voor een teststof, indien het resultaat ervan ondubbelzinnig is. Bij resultaten die dicht bij de drempelwaarde liggen die werd gebruikt om het onderscheid te maken tussen positieve en negatieve resultaten (d.w.z. in grensgevallen) kan het echter nodig zijn om bijkomende tests te verrichten. In gevallen waarin het gemiddelde percentage depletie tussen 3 % en 10 % bedraagt voor het voorspellingsmodel voor cysteïne 1:10/lysine 1:50 of het percentage cysteïnedepletie tussen 9 % en 17 % bedraagt voor het voorspellingsmodel voor cysteïne 1:10, moet worden overwogen om een tweede reeks experimenten uit te voeren, en eventueel ook een derde als de resultaten van de eerste twee reeksen niet overeenkomen. Testverslag In het testverslag moet de volgende informatie worden opgenomen:
LITERATUUR
Aanhangsel 1 DEFINITIES AOP (Adverse Outcome Pathway) : een reeks gebeurtenissen van de chemische structuur van een doelstof of een groep soortgelijke chemische stoffen over de moleculaire inleidende gebeurtenis tot een te onderzoeken in-vivoresultaat (2). Betrouwbaarheid : indicatie van de mate waarin een testmethode, uitgevoerd volgens een vast protocol, op reproduceerbare wijze binnen één laboratorium en tussen laboratoria verspreid over de tijd kan worden uitgevoerd. De betrouwbaarheid wordt beoordeeld door berekening van intra- en interlaboratoriumreproduceerbaarheid en intralaboratoriumherhaalbaarheid (21). Chemische stof : een stof of mengsel. Geldige testmethode : een testmethode die voldoende relevant en betrouwbaar wordt geacht voor een specifiek doel en gebaseerd is op wetenschappelijk verantwoorde beginselen. Een testmethode is nooit algemeen geldig, maar enkel met betrekking tot een specifiek doel (21). Geschiktheid van het systeem : bepaling van de prestaties van de instrumenten (bv. gevoeligheid) door een referentienorm te analyseren voordat de reeks analyses wordt uitgevoerd (22). Gevaar : inherente eigenschap van een agens of situatie die nadelige effecten kan veroorzaken als een organisme, systeem of (sub)populatie aan dit agens wordt blootgesteld. Gevoeligheid : Het percentage van alle positieve/actieve chemische stoffen dat door middel van de testmethode correct wordt ingedeeld. De gevoeligheid is een maat voor de nauwkeurigheid van een testmethode die categoriale resultaten oplevert en is een belangrijke factor voor de beoordeling van de relevantie van een testmethode (21). GHS (Globally Harmonized System of Classification and Labelling of Chemicals, mondiaal geharmoniseerd classificatie- en etiketteringssysteem voor chemische stoffen van de Verenigde Naties (VN)) : een systeem voor de indeling van chemische producten (stoffen en mengsels) op basis van gestandaardiseerde soorten en niveaus van gevaren van fysische aard en gevaren voor de gezondheid en het milieu, en dat de desbetreffende voorlichtingselementen bepaalt, zoals pictogrammen, signaalwoorden, gevarenaanduidingen, voorzorgsmaatregelen en veiligheidsinformatiebladen, waarmee informatie over de schadelijke effecten van de stoffen kenbaar wordt gemaakt teneinde mensen (waaronder werkgevers, werknemers, transporteurs, consumenten en spoedhulpverleners) en het milieu te beschermen (1). IATA (Integrated Approach to Testing and Assessment, geïntegreerde test- en beoordelingsbenadering) : een gestructureerde aanpak die wordt gebruikt voor de identificatie van gevaren (potentieel), het kenmerken van gevaren (potentie) en/of het beoordelen van de veiligheid (potentieel/potentie en blootstelling) van een chemische stof of een groep chemische stoffen, waarbinnen alle relevante gegevens strategisch worden geïntegreerd en worden afgewogen als inbreng voor regelgevingsbesluiten inzake potentiële gevaren en/of risico's en/of de noodzaak van verdere gerichte en bijgevolg minimale tests. Nauwkeurigheid : de mate van overeenstemming tussen resultaten verkregen met de testmethode en erkende referentiewaarden. Het is een maat voor de prestaties van de testmethode en één aspect van “relevantie”. Deze term en de term “concordantie”, waaronder het percentage correcte uitkomsten van een testmethode wordt verstaan, worden vaak door elkaar gebruikt (21). Kalibratiecurve : het verband tussen de responswaarde in het experiment en de analytische concentratie (ook wel standaardcurve genoemd) van een bekende stof. Mengsel : een mengsel of een oplossing bestaande uit twee of meer stoffen waarin deze stoffen geen reactie met elkaar aangaan (1). Moleculaire inleidende gebeurtenis : een door chemische stoffen geïnduceerde verstoring van een biologisch systeem op het moleculaire niveau, waarvan is vastgesteld dat zij het beginpunt is van de adverse outcome pathway. Positieve controle : een monster met alle bestanddelen van een testsysteem dat met een stof behandeld werd waarvan bekend is dat deze een positieve respons opwekt. Om te verzekeren dat variabiliteit in de positieve-controlerespons doorheen de tijd kan worden beoordeeld, mag de grootte van de positieve respons niet buitensporig zijn. Referentiecontrole : een onbehandeld monster dat alle componenten bevat van een testsysteem, met inbegrip van het oplosmiddel of medium, dat tezamen met de met de teststof behandelde monsters en andere controlemonsters verwerkt wordt om de referentierespons voor de monsters die behandeld werden met de in hetzelfde oplosmiddel of medium opgeloste teststof vast te stellen. Als dit monster getest wordt met een tegelijkertijd gemeten negatieve controle, toont het ook aan of het oplosmiddel of medium met het testsysteem interageert. Relevantie : beschrijving van het verband van de test met het te onderzoeken effect en of de test betekenis heeft en nuttig is voor een bepaald doel. Het is de mate waarin de test het te onderzoeken biologische effect correct meet of voorspelt. Relevantie omvat een beoordeling van de nauwkeurigheid (concordantie) van een testmethode (21). Reproduceerbaarheid : de overeenkomst tussen resultaten verkregen uit tests op dezelfde chemische stof met behulp van hetzelfde testprotocol (zie betrouwbaarheid) (21). Specificiteit : Het percentage van alle negatieve/niet-actieve chemische stoffen dat door middel van de testmethode correct wordt ingedeeld. De specificiteit is een maat voor de nauwkeurigheid van een testmethode die categoriale resultaten oplevert, en is een belangrijke factor bij de beoordeling van de relevantie van een testmethode (21). Stof : chemische elementen en verbindingen ervan, zoals zij voorkomen in natuurlijke toestand of bij de productie ontstaan, met inbegrip van alle additieven die nodig zijn voor het behoud van de stabiliteit van het product en alle onzuiverheden ten gevolge van het productieprocédé, doch met uitzondering van elk oplosmiddel dat kan worden afgescheiden zonder dat de stabiliteit van de stof wordt aangetast of de samenstelling ervan wordt gewijzigd (1). Stof die uit één component bestaat : een stof die wordt gekenmerkt door haar kwantitatieve samenstelling en waarin een hoofdcomponent aanwezig is in een concentratie van ten minste 80 % (w/w). Stof die uit meerdere componenten bestaat : een stof die wordt gekenmerkt door haar kwantitatieve samenstelling en waarin meer dan één hoofdcomponent aanwezig is in een concentratie van ≥ 10 % (w/w) en < 80 % (w/w). Een stof die uit meerdere componenten bestaat, is het resultaat van een fabricageproces. Het verschil tussen mengsels en stoffen die uit meerdere componenten bestaan, is dat mengsels worden verkregen door twee of meer stoffen zonder chemische reactie met elkaar te vermengen. Een stof die uit meerdere componenten bestaat, is het resultaat van een chemische reactie. Teststof : met de term “teststof” wordt verwezen naar datgene dat wordt getest. UVCB : stoffen waarvan de samenstelling niet bekend of variabel is, complexe reactieproducten of biologische materialen. Variatiecoëfficiënt : een maat voor de variabiliteit die wordt berekend voor een groep duplogegevens door de standaardafwijking te delen door het gemiddelde. Dit quotiënt kan ook worden vermenigvuldigd met 100 om het uit te drukken als een percentage. Aanhangsel 2 STOFFEN VOOR BEKWAAMHEIDSTOETSING In-chemicohuidsensibilisatie: Direct Peptide Reactivity Assay Vooraleer over te gaan tot routinegebruik van deze testmethode, dienen laboratoria hun technische bekwaamheid aan te tonen door de correcte DPRA-voorspelling te verkrijgen voor de 10 stoffen die in tabel 1 worden aangeraden en door cysteïne- en lysinedepletiewaarden te bekomen die binnen de respectieve referentiebereiken vallen voor 8 van de 10 stoffen voor elk peptide. Deze stoffen voor bekwaamheidstoetsing werden geselecteerd om alle mogelijke responsen voor huidsensibilisatiegevaar te weerspiegelen. Andere selectiecriteria waren dat stoffen commercieel beschikbaar zijn, dat er in-vivoreferentiegegevens en met de DPRA gegenereerde in-vitrogegevens van zeer goede kwaliteit beschikbaar zijn en dat zij in de door het EURL ECVAM gecoördineerde valideringsstudie werden gebruikt om de geslaagde toepassing van de testmethode in de laboratoria die aan de studie deelnamen, aan te tonen. Tabel 1 Aanbevolen stoffen voor het aantonen van technische bekwaamheid met de Direct Peptide Reactivity Assay
Aanhangsel 3 VOORBEELDEN VAN ANALYSEREEKSEN
B.60 In-vitrohuidsensibilisatie: ARE-Nrf2-luciferasetestmethode INLEIDING Deze testmethode (TM) is gelijkwaardig aan testrichtlijn (TG) 442D (2015) van de OESO. Een huidsensibiliserende stof is een stof die een allergische reactie uitlokt na contact met de huid, zoals gedefinieerd in het wereldwijd geharmoniseerd systeem voor de indeling en etikettering van chemische stoffen van de Verenigde Naties (GHS) (1) en Verordening (EG) nr. 1272/2008 van de Europese Unie betreffende de indeling, etikettering en verpakking van stoffen en mengsel (CLP-verordening) (27). Deze testmethode reikt een in-vitroprocedure (de ARE-Nrf2-luciferasetest) aan die kan worden gebruikt om het onderscheid tussen huidsensibiliserende stoffen en niet-sensibiliserende stoffen te staven, in overeenstemming met het VN-GHS (1) en de CLP-verordening. Er bestaat algemene overeenstemming over de belangrijkste biologische gebeurtenissen die aan de basis liggen van huidsensibilisatie. De bestaande kennis van de chemische en biologische mechanismen die verband houden met huidsensibilisatie zijn samengevat in een Adverse Outcome Pathway (AOP) (2), van de moleculaire gebeurtenis die er aanleiding toe geeft over de tussenliggende gebeurtenissen tot het schadelijke effect voor de gezondheid, te weten allergische contactdermatitis bij mensen of contactovergevoeligheid bij knaagdieren (2) (3). De moleculaire inleidende gebeurtenis is de covalente binding van elektrofiele stoffen aan nucleofiele centra in de eiwitten van de huid. De tweede belangrijke gebeurtenis in deze AOP vindt plaats in de keratinocyten en omvat zowel ontstekingsreacties als genexpressie die samenhangt met specifieke signaalpaden in de cellen, zoals de paden die afhankelijk zijn van het antioxidant-/elektrofiel responselement (ARE). De derde belangrijke gebeurtenis is de activering van dendritische cellen. Deze gebeurtenis wordt gewoonlijk beoordeeld aan de hand van de expressie van specifieke markers op het celoppervlak. De vierde belangrijke gebeurtenis is de proliferatie van T-cellen, die indirect wordt beoordeeld in de lokale lymfkliermurinetest (4). De bepaling van de huidsensibilisatie ging meestal gepaard met het gebruik van proefdieren. Bij de klassieke methoden op basis van cavia's, de maximalisatietest met cavia's (GMPT) van Magnusson-Kligman en de Buehler-test (TM B.6 (5)) worden zowel de inductie- als de uitlokkingsfase van huidsensibilisatie bestudeerd. Ook een murinetest, de lokale lymfkliertest (LLNA) (TM B.42 (4)), en de twee niet-radioactieve aanpassingen ervan, LLNA: DA (TM B.50 (6)) en LLNA: BrdU-ELISA (TM B.51 (7)), waarbij telkens uitsluitend de inductierespons wordt beoordeeld, zijn gangbaar geworden aangezien zij beter zijn voor het dierenwelzijn dan tests met cavia's en het mogelijk maken om de inductiefase van huidsensibilisatie objectief te meten. Recenter zijn ook mechanistisch gebaseerde in-chemico- en in-vitrotestmethoden wetenschappelijk geldig bevonden voor de beoordeling van het gevaar van huidsensibilisatie van chemische stoffen. Om de dierproeven die momenteel worden gebruikt, volledig te kunnen vervangen, zullen echter combinaties van methoden zonder dierproeven (in silico, in chemico, in vitro) binnen geïntegreerde test- en beoordelingsbenaderingen (Integrated Approaches to Testing and Assessment of IATA) nodig zijn, aangezien elk van de momenteel beschikbare testmethoden zonder dierproeven vanuit mechanistisch oogpunt slechts een beperkt deel van de AOP afdekt (2) (3). Deze testmethode (ARE-Nrf2-luciferasetest) wordt voorgesteld om de tweede belangrijke gebeurtenis zoals toegelicht in punt 2 te onderzoeken. Er zijn meldingen van huidsensibiliserende stoffen die genen induceren die worden gereguleerd door het antioxidantresponselement (ARE) (8) (9). Kleine elektrofiele stoffen zoals huidsensibiliserende stoffen kunnen inwerken op het sensoreiwit Keap1 (Kelch-like ECH-associated protein 1), bijvoorbeeld via covalente aanpassingen van het cysteïneresidu van dat eiwit, waardoor het eiwit wordt gescheiden van de transcriptiefactor Nrf2 (nuclear factor-erythroid 2-related factor 2). De afgescheiden Nrf2 kan vervolgens ARE-afhankelijke genen activeren, zoals diegenen die coderen voor fase II-ontgiftingsenzymen (8) (10) (11). Voorlopig is de KeratinoSens™-test de enige in-vitro-ARE-Nrf2-luciferasetest in deze testmethode waarvoor valideringsstudies zijn verricht (9) (12) (13), gevolgd door een onafhankelijke beoordeling door vakgenoten uitgevoerd door het referentielaboratorium voor alternatieven voor dierproeven van de Europese Unie (EURL ECVAM) (14). De KeratinoSens™-test werd wetenschappelijk geldig bevonden voor gebruik in het kader van een IATA om het onderscheid tussen huidsensibiliserende stoffen en niet-sensibiliserende stoffen te staven met het oog op de gevarenindeling en etikettering (14). Laboratoria die de testmethode willen invoeren kunnen de bij de KeratinoSens™-test gebruikte recombinante cellijn verkrijgen door een licentieovereenkomst te sluiten met de ontwikkelaar van de testmethode (15). Definities worden gegeven in aanhangsel 1. INLEIDENDE OVERWEGINGEN, TOEPASSELIJKHEID EN BEPERKINGEN Aangezien activering van het Keap1-Nrf2-ARE-pad enkel betrekking heeft op de tweede belangrijke gebeurtenis van de AOP voor huidsensibilisatie, volstaat informatie verkregen uit testmethoden die gebaseerd zijn op de activering van dat pas waarschijnlijk niet wanneer zij als dusdanig wordt gebruikt om conclusies te formuleren over het huidsensibilisatiepotentieel van chemische stoffen. De gegevens die deze testmethode oplevert, dienen daarom te worden bekeken in het kader van een geïntegreerde aanpak, zoals een IATA, waarbij zij worden gecombineerd met andere aanvullende informatie die bijvoorbeeld is afgeleid uit in-vitrotesten waarin andere belangrijke gebeurtenissen binnen de AOP van huidsensibilisatie worden bestudeerd of uit niet-testmethoden zoals een “read-across” van chemische analogen. Er zijn voorbeelden van het gebruik van ARE-Nrf2-luciferasetestmethode in combinatie met andere informatie beschreven in de literatuur (13) (16) (17) (18) (19). Deze testmethode kan worden gebruikt om het onderscheid tussen huidsensibiliserende stoffen (d.w.z. VN-GHS/CLP-categorie 1) en niet-sensibiliserende stoffen te staven in de context van IATA. Deze testmethode kan niet afzonderlijk worden gebruikt om huidsensibiliserende stoffen in te delen in de subcategorieën 1A en 1B zoals gedefinieerd door VN-GHS/CLP of om de potentie te voorspellen met het oog op besluiten inzake veiligheidsbeoordelingen. Naargelang van het regelgevingskader kan een positief resultaat echter wel afzonderlijk worden gebruikt om een stof in te delen in VN-GHS/CLP-categorie 1. Op basis van de gegevensreeks uit de valideringsstudie en interne tests die werden gebruikt voor de onafhankelijke beoordeling door vakgenoten van de testmethode bleek de KeratinoSens™-test overdraagbaar te zijn naar laboratoria met ervaring in het kweken van cellen. De verwachte reproduceerbaarheid van voorspellingen op basis van deze testmethode binnen laboratoria en tussen verschillende bedraagt 85 % (14). De nauwkeurigheid (77 % — 155/201), gevoeligheid (78 % — 71/91) en specificiteit (76 % — 84/110) van de KeratinoSens™-test bij het onderscheiden van huidsensibiliserende stoffen (d.w.z. VN GHS/CLP-cat. 1) van niet-sensibiliserende stoffen in vergelijking met de resultaten van de LLNA-tests werden berekend aan de hand van alle gegevens die bij het EURL ECVAM werden ingediend voor de evaluatie en de beoordeling door vakgenoten van de testmethode (14). Die cijfers zijn vergelijkbaar met de cijfers die onlangs werden gepubliceerd op basis van interne tests op ongeveer 145 stoffen (77 % nauwkeurigheid, 79 % gevoeligheid, 72 % specificiteit) (13). Bij de KeratinoSens™-test is de kans op een te lage voorspelling voor chemische stoffen met een lage tot gemiddelde potentie voor huidsensibilisatie (d.w.z. VN-GHS/CLP-subcategorie 1B) hoger dan voor chemische stoffen met een hoge potentie voor huidsensibilisatie (d.w.z. VN-GHS/CLP-subcategorie 1A) (13) (14). Gecombineerd geeft deze informatie een indicatie van het nut van de KeratinoSens™-test om bij te dragen aan de bepaling van het gevaar van huidsensibilisatie. De waarden die hier worden vermeld voor de nauwkeurigheid van de KeratinoSens™-test als losstaande testmethode, vormen echter slechts een indicatie, aangezien de testmethode dient te worden beschouwd in combinatie met andere informatiebronnen in de context van een IATA en in overeenstemming met hetgeen hierboven bepaald is in punt 9. Bovendien moet bij de beoordeling van methoden zonder dierproeven voor huidsensibilisatie rekening worden gehouden met het feit dat de LLNA en andere dierproeven de situatie van de doelsoort, in dit geval mensen, mogelijk niet volledig weerspiegelen. De term “teststof” wordt in deze teststof gebruikt om te verwijzen naar datgene dat wordt getest en houdt geen verband met de toepasselijkheid van de ARE-Nrf2-luciferasetestmethode op het testen van stoffen en/of mengsels. Op basis van de thans beschikbare gegevens is gebleken dat de KeratinoSens™-test van toepassing is op teststoffen die verschillende organische functionele groepen, reactiemechanismen, potenties voor huidsensibilisatie (zoals bepaald bij in-vivo-onderzoek) en fysisch-chemische eigenschappen beslaan (9) (12) (13) (14). Er werden voornamelijk stoffen die bestaan uit één component getest, hoewel er ook een beperkte hoeveelheid gegevens beschikbaar is over het testen van mengsels (20). De testmethode is vanuit technisch oogpunt evenwel toepasbaar voor het testen van stoffen die uit meerdere componenten bestaan en van mengsels. Voordat deze testmethode wordt gebruikt op een mengsel om gegevens te genereren voor een beoogd regelgevingsdoel, moet echter worden nagegaan of, en zo ja, waarom deze methode betrouwbare resultaten oplevert voor dat doel. Zulke overwegingen zijn niet nodig wanneer het testen van het mengsel wettelijk vereist is. Bovendien moet bij tests op stoffen die uit meerdere componenten bestaan of op mengsels rekening worden gehouden met mogelijke interferentie van cytotoxische componenten met de waargenomen responsen. De testmethode is van toepassing op teststoffen die oplosbaar zijn of een stabiele dispersie vormen (d.w.z. een colloïde of suspensie waarin de teststof niet bezinkt of zich van het oplosmiddel afscheidt in verschillende fasen) in water of DMSO (inclusief alle componenten van de teststof wanneer een stof met meerdere componenten of een mengsel wordt getest). Teststoffen die niet aan deze voorwaarden voldoen in de hoogste uiteindelijke vereiste concentratie van 2000 μM (zie punt 22), mogen nog steeds worden getest bij lagere concentraties. In dat geval kunnen resultaten die aan de criteria voor positiviteit voldoen zoals beschreven in punt 39, nog steeds worden gebruikt om de teststof aan te merken als huidsensibiliserende stof, terwijl een negatief resultaat verkregen bij concentraties van < 1000 μM als niet-overtuigend dient te worden beschouwd (zie voorspellingsmodel in punt 39). Over het algemeen zijn stoffen met een LogP van maximaal 5 met succes getest, terwijl bijzonder hydrofobe stoffen met een LogP van meer dan 7 buiten het bekende toepassingsbereik van de testmethode vallen (14). Voor stoffen met een LogP tussen 5 en 7 is slechts beperkte informatie beschikbaar. Negatieve resultaten dienen met de nodige voorzichtigheid te worden geïnterpreteerd, aangezien stoffen die enkel reageren met lysineresiduen, als negatief kunnen worden gedetecteerd door deze testmethode. Bovendien kunnen ook prohaptenen (d.w.z. chemische stoffen die enzymactivering nodig hebben, bijvoorbeeld via P450-enzymen) en prehaptenen (d.w.z. chemische stoffen die worden geactiveerd door auto-oxidatie), in het bijzonder met een trage oxidatiesnelheid, negatieve resultaten geven vanwege de beperkte metabole capaciteit van de gebruikte cellijn (21) en vanwege de omstandigheden bij het experiment. Teststoffen die niet huidsensibiliserend werken maar toch chemische stressoren zijn, kunnen anderzijds tot vals-positieve uitslagen leiden (14). Bovendien kunnen bijzonder cytotoxische teststoffen niet altijd betrouwbaar worden beoordeeld. Tot slot kunnen teststoffen die interfereren met het luciferase-enzym de luciferaseactiviteit in tests op celbasis verstoren, doordat zij een schijnbare remming of verhoging van de luminescentie veroorzaken (22). Zo werd bij andere reportergentests op basis van luciferase gemeld dat fyto-oestrogeenconcentraties boven 1 μM interfereerden met de luminescentiesignalen vanwege overactiviteit van het luciferasereportergen (23). De luciferase-expressie bij hoge concentraties fyto-oestrogenen of soortgelijke chemische stoffen waarvan vermoed wordt dat zij fyto-oestrogeenachtige overactivatie van het luciferasereportergen kunnen veroorzaken, dient daarom zorgvuldig te worden bestudeerd (23). In gevallen waarin kan worden aangetoond dat de testmethode niet toepasselijk is op andere specifieke categorieën van teststoffen, dient de testmethode niet te worden gebruikt voor die specifieke categorieën. De KeratinoSens™-test staaft niet alleen het onderscheid tussen huidsensibiliserende stoffen en niet-sensibiliserende stoffen, maar levert ook informatie over de concentratierespons op die kan bijdragen aan de beoordeling van de sensibiliserende potentie wanneer deze test wordt gebruikt binnen een geïntegreerde aanpak, zoals een IATA (19). Er moet echter nog meer werk worden verricht, bij voorkeur op basis van betrouwbare gegevens verzameld bij mensen, om te bepalen hoe de resultaten van de KeratinoSens™-test kunnen bijdragen aan de beoordeling van de potentie (24) en de indeling van sensibiliserende stoffen in subcategorieën volgens VN-GHS/CLP. PRINCIPE VAN DE TEST Bij de ARE-Nrf2luciferasetestmethode wordt gebruikgemaakt van een geïmmortaliseerde adherente cellijn afgeleid van menselijke HaCaT-keratinocyten die stabiel zijn getransfecteerd met een te selecteren plasmide. De cellijn bevat het luciferasegen onder transcriptionele controle door een constitutieve promotor met een ARE-element uit een gen waarvan bekend is dat het wordt gestimuleerd door contactallergenen (25) (26). Het luciferasesignaal weerspiegelt de activering van endogene Nrf2-afhankelijke genen door sensibiliserende stoffen, en er is reeds aangetoond dat het luciferasesignaal in de recombinante cellijn afhankelijk is van Nrf2 (27). Daardoor wordt een kwantitatieve meting (via luminescentiedetectie) van de inductie van het luciferasegen met behulp van gangbare lichtproducerende luciferasesubstraten mogelijk, als indicator van de activiteit van de Nrf2-transcriptiefactor in cellen die zijn blootgesteld aan elektrofiele stoffen. De teststoffen worden positief bevonden bij de KeratinoSens™-test als zij een statistisch significante inductie van de luciferaseactiviteit induceren boven een bepaalde drempel (d.w.z. > 1,5-voudige of 50 %-toename), onder een bepaalde concentratie die geen significante invloed heeft op de levensvatbaarheid van de cellen (d.w.z. lager dan 1000 μM en bij een concentratie waarbij de levensvatbaarheid van de cellen meer dan 70 % bedraagt (9) (12)). Daartoe wordt de maximale x-voudige inductie van de luciferaseactiviteit ten opzichte van de (negatieve) oplosmiddelcontrole (Imax) bepaald. Aangezien de cellen aan reeksen concentraties van de teststof worden blootgesteld, moet voorts de concentratie die nodig is voor een statistisch significante inductie van de luciferaseactiviteit, boven de drempel (d.w.z. de waarde voor EC1,5) worden geïnterpoleerd op grond van de dosis-responscurve (zie punt 32 voor de berekening). Tot slot dienen parallelle metingen van de cytotoxiciteit te worden verricht om te beoordelen of er bij subcytotoxische concentraties niveaus worden bereikt die luciferaseactiviteit induceren. Vooraleer over te gaan tot routinegebruik van de ARE-Nrf2-luciferasetest die samenhangt met deze testmethode, zouden laboratoria hun technische bekwaamheid moeten aantonen aan de hand van de tien stoffen voor bekwaamheidstoetsing die in aanhangsel 2 zijn opgesomd. Er zijn prestatienormen (PS) (28) beschikbaar om de validering van nieuwe of gewijzigde in-vitro-ARE-Nrf2-luciferasetestmethoden die vergelijkbaar zijn met de KeratinoSens™-test te vergemakkelijken en tijdige wijziging van deze testmethode met het oog op de opname van dergelijke testmethoden mogelijk te maken. De wederzijdse aanvaarding van gegevens in overeenstemming met de OESO-overeenkomst is enkel gewaarborgd voor testmethoden die zijn gevalideerd volgens de prestatienormen, voor zover die testmethoden door de OESO zijn geëvalueerd en zijn opgenomen in de overeenkomstige testrichtsnoeren. PROCEDURE Momenteel is de wetenschappelijk geldige KeratinoSens™-test de enige methode die onder deze testmethode valt (9) (12) (13) (14). De standaardwerkwijzen voor de KeratinoSens™-test zijn beschikbaar en dienen te worden gebruikt wanneer de testmethode in een laboratorium wordt toegepast en gebruikt (15). Laboratoria die de testmethode willen invoeren, kunnen de bij de KeratinoSens™-test gebruikte recombinante cellijn verkrijgen door een licentieovereenkomst te sluiten met de ontwikkelaar van de testmethode. Hieronder worden de belangrijkste onderdelen en procedures van de ARE-Nrf2-luciferasetestmethode beschreven. Bereiding van de keratinocytenculturen Er dient een transgene cellijn te worden gebruikt waarin het luciferasereportergen stabiel is ingebracht onder controle van het ARE-element (bv. de KeratinoSens™-cellijn). Bij ontvangst worden de cellen vermeerderd (bv. 2 tot 4 passages) en in bevroren toestand bewaard als homogene stam. Cellen uit deze oorspronkelijke stam kunnen worden vermeerderd tot een maximumaantal passages (25 in het geval van KeratinoSens™) en worden gebruikt voor routinetests waarbij een passend medium wordt gebruikt om de cellen in leven te houden (in het geval van KeratinoSens™ gaat het om DMEM met serum en geneticine). Voor het testen dienen de cellen 80-90 % confluent te zijn en dient ervoor te worden gezorgd dat de cellen nooit doorgroeien naar volledige confluentie. Een dag voor het testen worden de cellen geoogst en verdeeld over 96-wells-platen (10 000 cellen/well in het geval van KeratinoSens™). Er moet de nodige aandacht worden geschonken aan het voorkomen van sedimentatie van de cellen tijdens het enten om een homogene verdeling van het aantal cellen over de wells te verzekeren. Als dat niet gebeurt, kan deze stap aanleiding geven tot grote verschillen van well tot well. Voor elke herhaling worden drie duplo's gebruikt voor het meten van de luciferaseactiviteit en een parallelle duplo voor het testen van de levensvatbaarheid van de cellen. Bereiding van de teststof en de controlestoffen De teststof en controlestoffen worden bereid op de dag van de tests. Voor de KeratinoSens™-test worden de teststoffen opgelost in dimethylsulfoxide (DMSO) tot de gewenste uiteindelijke concentratie is bereikt (bv. 200 mM). De DMSO-oplossingen zijn zelfsteriliserend en behoeven dus geen steriele filtratie. Teststoffen die niet oplosbaar zijn in DMSO worden opgelost in steriel water of in een kweekmedium, waarna de oplossingen worden gesteriliseerd, bv. door filtratie. Voor een teststof waarvan het molecuulgewicht niet is vastgesteld, wordt bij de KeratinoSens™-test een stamoplossing bereid in een standaardconcentratie (40 mg/ml or 4 % (w/v)). Wanneer andere oplosmiddelen dan DMSO, water of het kweekmedium worden gebruikt, moet dat voldoende wetenschappelijk gemotiveerd worden. Op basis van de stam-DMSO-oplossingen van de teststof worden seriële verdunningen gemaakt met behulp van DMSO om 12 basisconcentraties van de te testen stof te verkrijgen (van 0,098 tot 200 mM bij de KeratinoSens™-test). Voor teststoffen die niet oplosbaar zijn in DMSO, worden de verdunningen om de basisconcentraties te verkrijgen gemaakt met behulp van steriel water of steriel kweekmedium. De basisconcentraties worden vervolgens ongeacht het gebruikte oplosmiddel 25 keer verder verdund in het serum bevattende kweekmedium en worden tot slot gebruikt voor behandeling met een bijkomende verdunningsfactor 4, zodat de uiteindelijke concentraties van de geteste chemische stof 0,98 tot 2000 μM bedragen bij de KeratinoSens™-test. Alternatieve concentraties kunnen worden gebruikt als zij worden gemotiveerd (bv. in het geval van cytotoxiciteit of slechte oplosbaarheid). De negatieve (oplosmiddel)controle die wordt gebruikt bij de KeratinoSens™-test, is DMSO (CAS-nr. 67-68-5, ≥ 99 % zuiverheid), waarvoor zes wells per plaat worden geprepareerd. Deze controle ondergaat dezelfde verdunning als beschreven voor de basisconcentraties in punt 22, zodat de uiteindelijke concentratie van de negatieve (oplosmiddel)controle 1 % bedraagt, geen gevolgen heeft voor de levensvatbaarheid van de cellen en overeenkomt met dezelfde concentratie DMSO als die in de geteste chemische stof en in de positieve controle. Voor teststoffen die niet oplosbaar zijn in DMSO, waarvoor de verdunningen met water zijn gebeurd, moet het DMSO-niveau in alle wells van de uiteindelijke testoplossing worden gecorrigeerd naar 1 %, net zoals voor de overige teststoffen en controlestoffen. De positieve controle die wordt gebruikt bij de KeratinoSens™ -test is cinnamaldehyde (CAS-nr. 14371-10-9, ≥ 98 % zuiverheid), waarvoor een reeks van 5 basisconcentraties van 0,4 tot 6,4 mM wordt bereid in DMSO (op basis van 6,4 mM stamoplossing) en verdund zoals beschreven voor de basisconcentraties in punt 22, zodat de uiteindelijke concentraties van de positieve controle 4 tot 64 μM bedragen. Andere geschikte positieve controles, die bij voorkeur gematigde EC1,5-waarden opleveren, kunnen worden gebruikt als er historische gegevens beschikbaar zijn waaruit vergelijkbare aanvaardbaarheidscriteria voor de reeks experimenten kunnen worden afgeleid. Opbrengen van de teststof en de controlestoffen Er moet voor elke teststof en positieve controlestof een experiment worden verricht om een (positieve of negatieve) voorspelling af te leiden, waarbij ten minste twee onafhankelijke herhalingen moeten plaatsvinden met elk drie duplo's (d.w.z. n=6). Wanneer de resultaten van de twee onafhankelijke herhalingen van elkaar afwijken, dient het experiment een derde keer te worden herhaald met de drie duplo's (d.w.z. n=9). Elke onafhankelijke herhaling wordt op een andere dag verricht met een verse stamoplossing van teststoffen en onafhankelijk geoogste cellen. De cellen kunnen echter wel uit dezelfde passage komen. Na het enten volgens punt 20, worden de cellen gedurende 24 uur gekweekt in de 96-wells-microtiterplaat. Het medium wordt vervolgens verwijderd en vervangen door een vers kweekmedium (150 μl kweekmedium met serum maar zonder geneticine, in het geval van KeratinoSens™) waaraan 50 μl van de 25 maal verdunde teststof en controlestoffen wordt toegevoegd. Ten minste één well per plaat dient leeg te worden gelaten (geen cellen en geen behandeling) om de achtergrondwaarden te beoordelen. Bij de KeratinoSens™-test worden de behandelde platen vervolgens gedurende ongeveer 48 uur geïncubeerd op 37±1 °C in aanwezigheid van 5 % CO2. Er moet goed op worden gelet dat vluchtige teststoffen niet verdampen en dat er geen kruisbesmetting met teststoffen tussen wells optreedt, bijvoorbeeld door de platen voor incubatie met de teststoffen af te dekken met een folie. Metingen van de luciferaseactiviteit Drie factoren zijn van kritiek belang om correcte metingen van de luminescentie te verzekeren:
Voorafgaand aan het testen dient een controle-experiment zoals gedefinieerd in aanhangsel 3 te worden verricht om na te gaan of aan die drie criteria is voldaan. Nadat de cellen bij de KeratinoSens™-test 48 uur zijn blootgesteld aan de teststof en de controlestoffen, worden zij uitgewassen met een zoutoplossing met fosfaatbuffer en wordt gedurende 20 minuten bij kamertemperatuur de relevante lysisbuffer voor de luminescentiewaarden toegevoegd aan elke well. De platen met het cellysaat worden vervolgens in de luminometer geplaatst voor de meting, die bij de KeratinoSens™-test is geprogrammeerd om: (i) het luciferasesubstraat aan elke well toe te voegen (d.w.z. 50 μl), (ii) 1 seconde te wachten en vervolgens (iii) de luciferaseactiviteit gedurende 2 seconden te integreren. Wanneer er alternatieve instellingen worden gebruikt, bv. naargelang van het gebruikte luminometermodel, moeten die worden gemotiveerd. Voorts kan ook een gloeisubstraat worden gebruikt, voor zover het kwaliteitscontrole-experiment uit aanhangsel 3 met succes wordt afgerond. Cytotoxiciteitsbeoordeling Om de levensvatbaarheid van de cellen te testen bij de KeratinoSens™-test wordt het medium na de blootstellingstijd van 48 uur vervangen door vers medium met MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-difenyltetrazoliumbromide, thiazolylblauwtetrazoliumbromide; CAS-nr. 298-93-1) en worden de cellen ongeveer 4 uur geïncubeerd op 37 °C in aanwezigheid van 5 % CO2. Vervolgens wordt het MTT-medium verwijderd en worden de cellen gedurende een nacht gelyseerd (bv. door 10 % SDS-oplossing toe te voegen aan elke well). Nadat de wells zijn geschud, wordt de extinctie gemeten op bv. 600 nm met een fotometer. GEGEVENS EN RAPPORTAGE Evaluatie van de gegevens In de KeratinoSens™-test worden de volgende parameters berekend:
Vergelijking 1:
waarbij
EC1,5 wordt berekend via lineaire interpolatie volgens vergelijking 2, en de totale EC1,5 wordt berekend als het geometrische gemiddelde van de individuele herhalingen. Vergelijking 2:
waarbij
De levensvatbaarheid wordt berekend met vergelijking 3: Vergelijking 3:
waarbij
IC50 en IC30 worden berekend via lineaire interpolatie volgens vergelijking 4, en de totale IC50 en IC30 worden berekend als het geometrische gemiddelde van de individuele herhalingen. Vergelijking 4:
waarbij
Voor elke concentratie die een > 1,5-voudige inductie van luciferaseactiviteit oplevert, wordt de statistische significantie berekend (bv. met behulp van een tweezijdige t-toets van Student), waarbij de luminescentiewaarden voor de drie duplomonsters worden vergeleken met de luminescentiewaarden in de (negatieve) oplosmiddelcontrole om te bepalen of de inductie van luciferaseactiviteit statistisch significant is (p < 0,05). De laagste concentratie die een > 1,5-voudige inductie van luciferaseactiviteit oplevert is de waarde die de EC1,5-waarde bepaalt. Voor elk geval wordt gecontroleerd of die waarde lager is dan de IC30-waarde, hetgeen erop wijst dat de daling van de levensvatbaarheid van de cellen minder dan 30 % bedraagt bij de concentratie die de EC1,5 bepaalt. Het wordt aanbevolen om de gegevens visueel te controleren aan de hand van regelkaarten. Als er geen duidelijke dosis-responscurve wordt waargenomen of als de verkregen dosis-responscurve tweefasig is (d.w.z. dat zij de drempel van 1,5 tweemaal overschrijdt), moet het experiment worden overgedaan om te controleren of dit eigen is aan de teststof of te wijten is aan een artefact van het experiment. Wanneer de tweefasige respons kan worden gereproduceerd in een onafhankelijk experiment, dient de laagste EC1,5-waarde (de concentratie waarbij de drempel van 1,5 voor het eerst wordt overschreden) te worden gemeld. In het zeldzame geval waarin een statistisch niet-significante inductie van meer dan 1,5 maal wordt waargenomen gevolgd door een hogere concentratie met een statistisch significante inductie, worden de resultaten van die herhaling enkel als geldig en positief beschouwd als de statistisch significante inductie boven de drempel van 1,5 werd verkregen bij een niet-cytotoxische concentratie. Tot slot wordt voor teststoffen die al een 1,5-voudige of hogere inductie vertonen bij de laagste testconcentratie van 0,98 μM een EC1,5-waarde van < 0,98 vastgesteld op basis van een visuele controle van de dosis-responscurve. Aanvaardbaarheidscriteria Bij gebruik van de KeratinoSens™-test dient aan de volgende aanvaardbaarheidscriteria te worden voldaan. Ten eerste dient de inductie van luciferaseactiviteit die wordt verkregen met de positieve controle, cinnamaldehyde, statistisch significant te zijn, boven de drempel van 1,5 (bv. bij gebruik van een t-toets), in ten minste een van de geteste concentraties (van 4 tot 64 μM). Ten tweede dient de EC1,5-waarde tussen twee standaardafwijkingen van het historische gemiddelde van de testinstelling te vallen (bv. tussen 7 μM en 30 μM op basis van de gegevensreeks voor validering), dat regelmatig dient te worden bijgewerkt. Daarnaast dient de gemiddelde inductie in de drie duplo's voor cinnamaldehyde bij 64 μM tussen 2 en 8 te liggen. Als niet aan dat laatste criterium is voldaan, dient de dosis-respons van cinnamaldehyde zorgvuldig te worden gecontroleerd en kunnen tests enkel worden aanvaard als er een duidelijke dosis-respons bestaat waarbij de inductie van luciferaseactiviteit in de positieve controle toeneemt als de concentratie toeneemt. Tot slot dient de gemiddelde variatiecoëfficiënt van de luminescentiewaarde voor de (negatieve) oplosmiddelcontrole DMSO minder dan 20 % te bedragen voor elke herhaling waarbij 6 wells in drievoud worden getest. Als de variatie groter is, dienen de resultaten te worden verworpen. Interpretatie van de resultaten en voorspellingsmodel Een KeratinoSens™-voorspelling wordt positief geacht als aan elk van de volgende 4 voorwaarden is voldaan in 2 van 2 of in dezelfde 2 van 3 herhalingen. Is dat niet het geval, dan wordt de KeratinoSens™-voorspelling negatief geacht (figuur 1):
Als bij een herhaling aan elk van de eerste drie voorwaarden is voldaan maar er geen duidelijke dosis-respons voor de luciferase-inductie kan worden waargenomen, dient het resultaat van die herhaling als niet-overtuigend te worden beschouwd en kunnen bijkomende tests nodig zijn (figuur 1). Voorts dient ook een negatief resultaat dat werd verkregen bij concentraties < 1 000 μM (of < 200 μg/ml voor teststoffen waarvan het molecuulgewicht niet is bepaald) als niet-overtuigend te worden beschouwd (zie punt 11). Figuur 1: voorspellingsmodel dat wordt gebruikt in de KeratinoSens™-test. Een KeratinoSens™-voorspelling dient te worden bekeken in het kader van een IATA en in overeenstemming met hetgeen is bepaald in de punten 9 en 11.
In zeldzame gevallen kunnen teststoffen die luciferaseactiviteit induceren op een niveau dat erg dicht bij het cytotoxiciteitsniveau ligt, bij niet-cytotoxische niveaus (d.w.z. wanneer de concentratie die de EC1,5 bepaalt lager is dan (<) de IC30) een positief resultaat opleveren in sommige herhalingen, terwijl dat in andere herhalingen enkel bij cytotoxische niveaus gebeurt (d.w.z. wanneer de concentratie die de EC1,5 bepaalt hoger is dan (>) de IC30). Dergelijke teststoffen worden opnieuw getest met een striktere analyse van de dosis-respons, waarbij een lagere verdunningsfactor wordt gebruikt (bv. 1,33- of Ö2-voudige (=1,41-voudige) verdunning tussen wells) om te bepalen of er inductie is opgetreden bij cytotoxische niveaus of niet (9). Testverslag In het testverslag moet de volgende informatie worden opgenomen:
LITERATUUR
Aanhangsel 1 DEFINITIES AOP (Adverse Outcome Pathway) : een reeks gebeurtenissen van de chemische structuur van een doelstof of een groep soortgelijke chemische stoffen over de moleculaire inleidende gebeurtenis tot een te onderzoeken in-vivoresultaat (2). ARE : het antioxidantresponselement (ook wel EpRE of elektrofiel responselement genoemd) is een responselement dat stroomopwaarts van de promotor van veel cytoprotectieve en fase II-genen wordt aangetroffen. Wanneer het wordt geactiveerd door Nfr2, vermindert het de transcriptionele inductie van die genen. Betrouwbaarheid : indicatie van de mate waarin een testmethode, uitgevoerd volgens een vast protocol, op reproduceerbare wijze binnen één laboratorium en tussen laboratoria verspreid over de tijd kan worden uitgevoerd. De betrouwbaarheid wordt beoordeeld door berekening van intra- en interlaboratoriumreproduceerbaarheid en intralaboratoriumherhaalbaarheid (29). Chemische stof : een stof of mengsel. EC1.5 : geïnterpoleerde concentratie voor een 1,5-voudige luciferase-inductie. Geldige testmethode : een testmethode die voldoende relevant en betrouwbaar wordt geacht voor een specifiek doel en gebaseerd is op wetenschappelijk verantwoorde beginselen. Een testmethode is nooit algemeen geldig, maar enkel met betrekking tot een specifiek doel (29). Gevaar : inherente eigenschap van een agens of situatie die nadelige effecten kan veroorzaken als een organisme, systeem of (sub)populatie aan dit agens wordt blootgesteld. Gevoeligheid : Het percentage van alle positieve/actieve chemische stoffen dat door middel van de testmethode correct wordt ingedeeld. De gevoeligheid is een maat voor de nauwkeurigheid van een testmethode die categoriale resultaten oplevert en is een belangrijke factor voor de beoordeling van de relevantie van een testmethode (29). GHS (Globally Harmonized System of Classification and Labelling of Chemicals, mondiaal geharmoniseerd classificatie- en etiketteringssysteem voor chemische stoffen van de Verenigde Naties (VN)) : een systeem voor de indeling van chemische producten (stoffen en mengsels) op basis van gestandaardiseerde soorten en niveaus van gevaren van fysische aard en gevaren voor de gezondheid en het milieu, en dat de desbetreffende voorlichtingselementen bepaalt, zoals pictogrammen, signaalwoorden, gevarenaanduidingen, voorzorgsmaatregelen en veiligheidsinformatiebladen, waarmee informatie over de schadelijke effecten van de stoffen kenbaar wordt gemaakt teneinde mensen (waaronder werkgevers, werknemers, transporteurs, consumenten en spoedhulpverleners) en het milieu te beschermen (1). IATA (Integrated Approach to Testing and Assessment, geïntegreerde test- en beoordelingsbenadering) : een gestructureerde aanpak die wordt gebruikt voor de identificatie van gevaren (potentieel), het kenmerken van gevaren (potentie) en/of het beoordelen van de veiligheid (potentieel/potentie en blootstelling) van een chemische stof of een groep chemische stoffen, waarbinnen alle relevante gegevens strategisch worden geïntegreerd en worden afgewogen als inbreng voor regelgevingsbesluiten inzake potentiële gevaren en/of risico's en/of de noodzaak van verdere gerichte en bijgevolg minimale tests. IC30 : concentratie die een daling van de levensvatbaarheid van de cellen met 30 % bewerkstelligt. IC50 : concentratie die een daling van de levensvatbaarheid van de cellen met 50 % bewerkstelligt. Imax : maximale inductiefactor van luciferaseactiviteit in vergelijking met de (negatieve) oplosmiddelcontrole gemeten bij alle concentraties van de teststof. Keap1 : Kelch-like ECH-associated protein 1 is een sensoreiwit dat de activiteit van Nrf2 kan reguleren. In niet-geïnduceerde omstandigheden werkt het Keap1-sensoreiwit in op de Nrf2-transcriptiefactor voor ubiquitinylering en proteolytische degradatie in het proteasoom. De covalente aanpassing van de reactieve cysteïneresiduen van Keap1 door kleine moleculen kan ertoe leiden dat Nrf2 zich gaat afscheiden van Keap1 (8) (10) (11). Medium-/oplosmiddelcontrole : een monster met alle bestanddelen van een testsysteem met uitzondering van de teststof, maar met inbegrip van het gebruikte oplosmiddel. Dit monster wordt gebruikt om de referentierespons vast te stellen voor de monsters die worden behandeld met de teststof opgelost in hetzelfde oplosmiddel. Mengsel : een mengsel of een oplossing bestaande uit twee of meer stoffen waarin deze stoffen geen reactie met elkaar aangaan (1). Nauwkeurigheid : de mate van overeenstemming tussen resultaten verkregen met de testmethode en erkende referentiewaarden. Het is een maat voor de prestaties van de testmethode en één aspect van “relevantie”. Deze term en de term “concordantie”, waaronder het percentage correcte uitkomsten van een testmethode wordt verstaan, worden vaak door elkaar gebruikt (29). Nrf2 : Nuclear factor (erythroid-derived 2)-like 2 is een transcriptiefactor die een rol speelt in de route van de antioxidantrespons. Als Nrf2 niet geübiquitinyleerd is, stapelt hij zich op in het cytoplasma en verplaatst hij zich naar de nucleus, waar hij in het gebied van de stroomopwaarts gelegen promotor van veel cytoprotectieve genen bindt met het ARE en zo de transcriptie van die genen start (8) (10) (11). Positieve controle : een monster met alle bestanddelen van een testsysteem dat met een stof behandeld werd waarvan bekend is dat deze een positieve respons opwekt. Om te verzekeren dat variabiliteit in de positieve-controlerespons doorheen de tijd kan worden beoordeeld, mag de grootte van de positieve respons niet buitensporig zijn. Relevantie : beschrijving van het verband van de test met het te onderzoeken effect en of de test betekenis heeft en nuttig is voor een bepaald doel. Het is de mate waarin de test het te onderzoeken biologische effect correct meet of voorspelt. Relevantie omvat een beoordeling van de nauwkeurigheid (concordantie) van een testmethode (29). Reproduceerbaarheid : de overeenkomst tussen resultaten verkregen uit tests op dezelfde chemische stof met behulp van hetzelfde testprotocol (zie betrouwbaarheid) (29). Specificiteit : het percentage van alle negatieve/niet-actieve chemische stoffen dat door middel van de testmethode correct wordt ingedeeld. De specificiteit is een maat voor de nauwkeurigheid van een testmethode die categoriale resultaten oplevert, en is een belangrijke factor bij de beoordeling van de relevantie van een testmethode (29). Stof : chemische elementen en verbindingen ervan, zoals zij voorkomen in natuurlijke toestand of bij de productie ontstaan, met inbegrip van alle additieven die nodig zijn voor het behoud van de stabiliteit van het product en alle onzuiverheden ten gevolge van het productieprocedé, doch met uitzondering van elk oplosmiddel dat kan worden afgescheiden zonder dat de stabiliteit van de stof wordt aangetast of de samenstelling ervan wordt gewijzigd (1). Stof die uit één component bestaat : een stof die wordt gekenmerkt door haar kwantitatieve samenstelling en waarin een hoofdcomponent aanwezig is in een concentratie van ten minste 80 % (w/w). Stof die uit meerdere componenten bestaat : een stof die wordt gekenmerkt door haar kwantitatieve samenstelling en waarin meer dan één hoofdcomponent aanwezig is in een concentratie van ≥ 10 % (w/w) en < 80 % (w/w). Een stof die uit meerdere componenten bestaat, is het resultaat van een fabricageproces. Het verschil tussen mengsels en stoffen die uit meerdere componenten bestaan, is dat mengsels worden verkregen door twee of meer stoffen zonder chemische reactie met elkaar te vermengen. Een stof die uit meerdere componenten bestaat, is het resultaat van een chemische reactie. Teststof : met de term “teststof” wordt verwezen naar datgene dat wordt getest. UVCB : stoffen waarvan de samenstelling niet bekend of variabel is, complexe reactieproducten of biologische materialen. Variatiecoëfficiënt : een maat voor de variabiliteit die wordt berekend voor een groep duplogegevens door de standaardafwijking te delen door het gemiddelde. Dit quotiënt kan ook worden vermenigvuldigd met 100 om het uit te drukken als een percentage. Aanhangsel 2 STOFFEN VOOR BEKWAAMHEIDSTOETSING In-vitrohuidsensibilisatie: ARE-Nrf2-luciferasetestmethode Vooraleer over te gaan tot routinegebruik van deze testmethode, dienen laboratoria hun technische bekwaamheid aan te tonen door de correcte KeratinoSens™-voorspelling te verkrijgen voor de 10 stoffen die in tabel 1 worden aangeraden en door de EC1,5- en IC50-waarden te bekomen die binnen de respectieve referentiebereiken vallen voor ten minste 8 van de 10 stoffen voor bekwaamheidstoetsing. Deze stoffen voor bekwaamheidstoetsing werden geselecteerd om alle mogelijke responsen voor huidsensibilisatiegevaar te weerspiegelen. Andere selectiecriteria waren dat stoffen commercieel beschikbaar zijn en dat er in-vivoreferentiegegevens en met de KeratinoSens™-test gegenereerde in-vitrogegevens van zeer goede kwaliteit beschikbaar zijn. Tabel 1 Aanbevolen stoffen voor het aantonen van technische bekwaamheid met de KeratinoSens™-test
Aanhangsel 3 KWALITEITSCONTROLE VAN DE LUMINESCENTIEMETINGEN Basisexperiment om optimale luminescentiemetingen te verzekeren bij de KeratinoSens™-test De volgende drie parameters zijn van kritiek belang om betrouwbare resultaten te kunnen bereiken met de luminometer:
Er wordt aanbevolen om vóór het testen te verzekeren dat de luminescentiemetingen correct zijn door een controleplaatopstelling te testen zoals hieronder beschreven (drievoudige analyse). Plaatopstelling van het eerste oefenexperiment
EGDMA= ethyleenglycoldimethacrylaat (CAS-nr.: 97-90-5) een sterk inducerende chemische stof CA= cinnamaldehyde, positieve referentie (CAS-nr.: 104-55-2) De analyse van de kwaliteitscontrole dient het volgende aan te tonen:
B.61 Fluoresceïnelekkage-testmethode voor de identificatie van voor het oog corrosieve en hevig irriterende stoffen INLEIDING Deze testmethode (TM) is gelijkwaardig aan testrichtlijn (TG) 460 (2012) van de OESO. De fluoresceïnelekkage-testmethode (FL-testmethode) is een in-vitrotestmethode die onder bepaalde voorwaarden en binnen specifieke beperkingen kan worden gebruikt om chemische stoffen (stoffen en mengsels) in te delen als voor het oog corrosieve en hevig irriterende stoffen, zoals gedefinieerd in het wereldwijd geharmoniseerd systeem voor de indeling en etikettering van chemische stoffen van de Verenigde Naties (GHS) (categorie 1), in Verordening (EG) nr. 1272/2008 van de Europese Unie betreffende de indeling, etikettering en verpakking van stoffen en mengsel (CLP-verordening) (31) (categorie 1) en door het Amerikaanse agentschap voor milieubescherming (Environmental Protection Agency of EPA) (klasse I) (1) (2). Voor de toepassing van deze testmethode worden voor de ogen hevig irriterende stoffen gedefinieerd als chemische stoffen die na aanbrenging van de teststof weefselschade aan het oog veroorzaken die niet reversibel is binnen 21 dagen of die ernstige fysische gezichtsvermindering teweegbrengt, terwijl voor het oog corrosieve stoffen chemische stoffen zijn die irreversibele weefselschade aan het oog veroorzaken. Deze chemische stoffen zijn ingedeeld als VN-GHS-categorie 1, EU-CLP-categorie 1 of VS-EPA-klasse I. Hoewel de FL-testmethode niet geldig wordt geacht om de in-vivo-oogtest bij konijnen volledig te vervangen, wordt de FL aanbevolen voor gebruik als onderdeel van een trapsgewijze teststrategie voor indeling en etikettering volgens de regelgeving. De FL wordt zo aanbevolen als eerste stap in een top-downbenadering om voor het oog corrosieve/hevig irriterende stoffen te identificeren, in het bijzonder voor specifieke soorten chemische stoffen (meer bepaald in water oplosbare stoffen en mengsels) (3) (4). Momenteel wordt algemeen aangenomen dat er in de nabije toekomst geen in-vitrotest voor oogirritatie zal komen die de in-vivotest op ogen (TM B.5 (5)) volledig kan vervangen om voorspellingen te doen voor het volledige bereik van irritatie voor verschillende klassen chemische stoffen. Strategische combinaties van meerdere alternatieve testmethoden binnen een (trapsgewijze) teststrategie kunnen de in-vivotest op ogen mogelijk echter wel vervangen (4). De top-downbenadering (4) is opgezet om te worden gebruikt wanneer op basis van bestaande informatie wordt verwacht dat een chemische stof een hoog irritatiepotentieel heeft. Op basis van het in punt 35 beschreven voorspellingsmodel kan de FL-testmethode chemische stoffen zonder bijkomende tests identificeren binnen een beperkt toepassingsgebied als voor het oog corrosieve stoffen/hevig irriterende stoffen (VN-GHS-categorie 1; EU-CLP-categorie 1; VS-EPA-klasse I). Hetzelfde wordt aangenomen voor mengsels, hoewel er geen mengsels werden gebruikt bij de validering. De FL-testmethode kan bijgevolg worden gebruikt om de oogirritatie/corrosiviteit van chemische stoffen te bepalen, volgens de trapsgewijze teststrategie van TM B.5 (5). Een chemische stof waarvoor met de FL-testmethode niet voorspeld wordt dat zij corrosief is voor de ogen of hevig irriterend is, dient echter te worden getest in een of meer aanvullende testmethoden (in vitro en/of in vivo) die geschikt zijn voor de identificatie van i) chemische stoffen die in vitro vals-negatieve uitslagen opleveren voor oogirritatie/hevige irritatie in de FL (VN-GHS-categorie 1; EU-CLP-categorie 1; VS-EPA-klasse I); ii) chemische stoffen die niet zijn ingedeeld voor corrosie/irritatie van het oog (VN GHS geen categorie; EU CLP geen categorie: VS-EPA-klasse IV); en/of iii) chemische stoffen die matig/mild irriterend zijn voor het oog (VN-GHS-categorieën 2A en 2B; EU-CLP-categorie 2; VS-EPA-klassen II en III). Het doel van deze testmethode is het beschrijven van de procedures die worden gebruikt om de mogelijke corrosie of hevige irritatie van een teststof voor de ogen te beoordelen aan de hand van het vermogen van deze stof om schade te induceren in een ondoordringbare, confluente monolaag van epitheelcellen. De integriteit van de permeabiliteit doorheen de epitheellaag is een belangrijke functie van de epitheellaag, bijvoorbeeld in de conjunctiva en de cornea. De permeabiliteit doorheen de epitheellaag wordt gereguleerd door verschillende tight junctions. Er is een verband vastgesteld tussen het verhogen van de permeabiliteit van het hoornvliesepitheel in vivo en de mate van ontsteking en oppervlakkige schade naarmate oogirritatie optreedt. Bij de FL-testmethode worden de toxische effecten na een korte blootstelling aan de teststof gemeten aan de hand van een verhoging van de hoeveelheid natriumfluoresceïne die door de epitheelmonolaag dringt in MDCK-cellen (Madin-Darby Canine Kidney-cellen) die op doorlaatbare inzetstukken zijn gekweekt. De hoeveelheid fluoresceïnelekkage die optreedt is evenredig aan de door chemische stoffen geïnduceerde schade aan de tight junctions, desmosomen en celmembranen en kan worden gebruikt om het toxisch potentieel van een teststof voor de ogen te ramen. Aanhangsel 1 bevat een diagram van op een membraan van een inzetstuk gekweekte MDCK-cellen. Definities worden gegeven in aanhangsel 2. INLEIDENDE OVERWEGINGEN EN BEPERKINGEN Deze testmethode is gebaseerd op het INVITTOX-protocol nr. 71 (6) dat werd beoordeeld in een interne valideringsstudie uitgevoerd door het Europees Centrum voor de validering van alternatieve methoden (ECVAM), in samenwerking met het Interagency Coordinating Committee on the Validation of Alternative Methods (ICCVAM) van de Verenigde Staten en het Japanese Center for the Validation of Alternative Methods (JaCVAM). De FL-testmethode wordt niet aanbevolen voor de identificatie van chemische stoffen die als matig/mild irriterend voor het oog ingedeeld dienen te worden of van chemische stoffen die niet ingedeeld dienen te worden (d.w.z. GHS-cat. 2A/2B, geen categorie; EU-CLP-cat. 2, geen categorie; VS-EPA-klasse II/III/IV), zoals aangetoond in de valideringsstudie (3) (7). De testmethode enkel toepasselijk op in water oplosbare chemische stoffen (stoffen en mengsels). De FL-testmethode voorspelt gewoonlijk nauwkeurig of in water oplosbare chemische stoffen mogelijk hevige oogirritatie kunnen veroorzaken en/of het toxische effect niet wordt beïnvloed door verdunning (7). Om een chemische stof in te delen als in water oplosbaar in het kader van een experiment, dient zij oplosbaar te zijn in een steriele Hanks' Balanced Salt Solution (HBSS) met calcium (in een concentratie van 1,0-1,8 mM) en zonder fenol rood, in een concentratie van ≥ 250 mg/ml (een dosis boven de drempelwaarde van 100 mg/ml). Als de teststof echter oplosbaar is bij een lagere concentratie dan 100 mg/ml maar al een FL-inductie van 20 % induceert bij die concentratie (dus FL20 < 100 mg/ml), kan zij nog steeds worden ingedeeld als GHS-cat. 1 of EPA-cat. I. Vanwege de vastgestelde beperkingen zijn sterke zuren en basen, celfixeermiddelen en bijzonder vluchtige chemische stoffen uitgesloten van het toepassingsgebied van deze testmethode. Deze chemische stoffen beschikken over mechanismen die niet worden gemeten door de FL-testmethode, zoals sterke stolling, verzeping of specifieke reactieve verbindingen. Andere vastgestelde beperkingen voor deze methode zijn gebaseerd op de resultaten voor het voorspellend vermogen voor gekleurde en viskeuze teststoffen (7). Er wordt gesuggereerd dat beide soorten chemische stoffen moeilijk uit de monolaag te verwijderen zijn na de korte blootstellingsperiode en dat de voorspelbaarheid van de testmethode zou kunnen worden verbeterd als er meer uitwassingen zouden worden gehanteerd. Vaste chemische stoffen die in een vloeistof in suspensie zijn gebracht, kunnen uit die vloeistof neerslaan, en de uiteindelijke concentratie voor de cellen kan moeilijk te bepalen zijn. De nauwkeurigheid van FL in het EU-, EPA- en GHS-classificatiesysteem verbetert aanzienlijk als stoffen die tot deze chemische en fysische klassen behoren, uit de databank worden verwijderd (7). Gezien het doel van deze testmethode (de identificatie van enkel die stoffen die corrosief of hevig irriterend zijn voor de ogen) zijn vals-negatieve uitslagen (zie punt 13) niet doorslaggevend, aangezien dergelijke teststoffen nadien met andere gevalideerde in-vitrotests of, naargelang van de geldende voorschriften, op konijnen worden getest met behulp van een sequentiële teststrategie in een benadering met meervoudige bewijsvoering (“weight of evidence” benadering) (5) (zie ook de punten 3 en 4). Andere vastgestelde beperkingen van de FL-testmethode zijn gebaseerd op de percentages vals-negatieve en vals-positieve uitslagen. Wanneer deze testmethode werd gebruikt als eerste stap in een top-downbenadering om in water oplosbare voor het oog corrosieve/hevig irriterende stoffen en mengsels te identificeren (VN-GHS-categorie 1; EU-CLP-categorie 1; VS-EPA-klasse I), lag het percentage vals-positieve uitslagen voor de FL-testmethode tussen 7 % (7/103; VN-GHS en EU-CLP) en 9 % (9/99; VS-EPA) en het percentage vals-negatieve uitslagen tussen 54 % (15/28; VS-EPA) en 56 % (27/48; VN-GHS en EU-CLP) in vergelijking met in-vivoresultaten. Chemische groepen die vals-positieve en/of vals-negatieve uitslagen geven bij de FL-testmethode, worden hier niet gedefinieerd. Bepaalde technische beperkingen hebben enkel betrekking op de MDCK-celcultuur. De tight junctions die de passage van het natriumfluoresceïne door de monolaag tegenhouden, worden steeds meer aangetast naarmate het aantal celpassages stijgt. Als de tight junctions niet volledig zijn gevormd, leidt dat tot een hogere FL in de niet-behandelde controle. Daarom is het belangrijk om een toelaatbare maximumlekkage in de niet-behandelde controles vast te stellen (zie punt 38: 0 % lekkage). Net zoals bij alle in-vitrotests bestaat de mogelijkheid dat cellen na verloop van tijd vervormen. Het is daarom van essentieel belang dat er een bereik voor het aantal passages voor de tests wordt vermeld. Het huidige toepassingsgebied kan in sommige gevallen worden uitgebreid, maar enkel na analyse van een ruimere gegevensreeks van onderzochte teststoffen, bij voorkeur verkregen op basis van tests (3). Deze testmethode wordt dienovereenkomstig bijgewerkt naarmate nieuwe informatie en gegevens in overweging worden genomen. Elk laboratorium dat dit onderzoek voor het eerst opstelt, dient gebruik te maken van de in bijlage 3 voorziene chemicaliën voor bekwaamheidstoetsing. Laboratoria kunnen deze chemische stoffen gebruiken om hun technische bekwaamheid met betrekking tot het uitvoeren van de FL-testmethode aan te tonen vooraleer zij FL-onderzoeksgegevens indienen voor de wettelijke gevarenclassificatie. PRINCIPE VAN DE TEST De FL-testmethode is een in-vitrotest op basis van de cytotoxiciteit en de celfunctie die wordt uitgevoerd op een confluente monolaag tubulaire MDCK CB997-epitheelcellen die op semipermeabele inzetstukken worden gekweekt en de niet-delende toestand van het hoornvliesepitheel in vivo modelleren. De MDCK-cellijn is uitvoerig beschreven en vormt tight junctions en desmosomen die vergelijkbaar zijn met die aan de apicale zijde van de epitheellagen van de conjunctiva en de cornea. In vivo voorkomen tight junctions en desmosomen dat opgeloste stoffen en vreemde materialen door het hoornvliesepitheel dringen. Het verlies van impermeabiliteit door de epitheellaag vanwege beschadigde tight junctions en desmosomen is een van de eerste gebeurtenissen die optreden bij oogirritatie door chemische stoffen. De teststof wordt aangebracht op de confluente laag cellen die op de apicale zijde van het inzetstuk is gekweekt. Gewoonlijk wordt voor een korte blootstelling van 1 min gekozen om de normale zuiveringssnelheid bij menselijke blootstelling te weerspiegelen. Een korte blootstellingsperiode biedt het voordeel dat stoffen en mengsels op waterbasis onverdund kunnen worden getest, voor zover zij na de blootstellingsperiode eenvoudig kunnen worden verwijderd. Zo zijn er meer rechtstreekse vergelijkingen tussen de chemische effecten bij mensen mogelijk. Vervolgens wordt de teststof verwijderd en wordt de niet-toxische, sterk fluorescerende kleurstof natriumfluoresceïne gedurende 30 minuten aan de apicale zijde van de monolaag aangebracht. De door de teststof veroorzaakte schade aan de tight junctions wordt bepaald aan de hand van de hoeveelheid fluoresceïne die binnen een bepaalde tijd door de cellaag lekt. De hoeveelheid natriumfluoresceïne die door de monolaag en het membraan van het inzetstuk dringt tot in een vast volume oplossing dat aanwezig is in de well (waarin het lekkende natriumfluoresceïne wordt opgevangen), wordt bepaald door de fluoresceïneconcentratie in de well te meten met een spectrofluorimeter. De hoeveelheid fluoresceïnelekkage (FL) wordt berekend onder verwijzing naar metingen van de fluorescentie-intensiteit (FI) verkregen bij twee controles: een blanco controle en een maximumlekkagecontrole. Het percentage lekkage, en bijgevolg ook de hoeveelheid schade aan de tight junctions, wordt voor alle vastgestelde concentraties van de teststof uitgedrukt in verhouding tot die controles. Vervolgens wordt de FL20 (d.w.z. de concentratie die 20 % FL veroorzaakt ten opzicht van de waarde die werd genoteerd voor de onbehandelde confluente monolaag en de inzetstukken zonder cellen) berekend. De FL20-waarde (mg/ml) wordt in het voorspellingsmodel gebruikt om voor het oog corrosieve en hevig irriterende stoffen te identificeren (zie punt 35). Recovery vormt een belangrijk onderdeel van het toxiciteitsprofiel van een teststof, dat eveneens wordt beoordeeld bij de in-vivotest op oogirritatie. Voorlopige analyses gaven aan dat gegevens over recovery (tot 72 uur na de blootstelling aan de chemische stof) het voorspellend vermogen van INVITTOX-protocol nr. 71 zouden kunnen verhogen, maar dat er bijkomende beoordelingen nodig zijn en dat aanvullende gegevens, bij voorkeur verkregen uit verdere tests, daarbij zouden kunnen helpen (6). Deze testmethode wordt dienovereenkomstig bijgewerkt naarmate nieuwe informatie en gegevens in overweging worden genomen. PROCEDURE Bereiding van de monolaag van cellen De monolaag van MDCK CB997-cellen wordt bereid met behulp van subconfluente cellen die in celkweekflessen worden gekweekt in DMEM/Nutrient Mix F12 (1x concentraat met L-glutamine, 15 mM HEPES, calcium (in een concentratie van 1,0-1,8 mM) en 10 % door warmte geïnactiveerd FCS/FBS). Belangrijk is dat alle media/oplossingen die in de loop van de FL-test worden gebruikt, calcium dienen te bevatten in een concentratie tussen 1,8 mM (200 mg/l) en 1.0 mM (111 mg/l) om de vorming van tight junctions en de integriteit ervan te verzekeren. Het bereik voor het aantal celpassages dient te worden geregeld om een gelijkmatige, reproduceerbare vorming van tight junctions te verzekeren. De cellen dienen zich bij voorkeur binnen het bereik van 3-30 passages na het ontdooien te bevinden, aangezien de functionaliteit van de cellen binnen dat passagebereik vergelijkbaar is, wat helpt om de reproduceerbaarheid van de testresultaten te verzekeren. Voordat de FL-testmethode wordt uitgevoerd, worden de cellen losgemaakt van de fles met behulp van trypsinisatie, worden ze gecentrifugeerd en wordt een passende hoeveelheid cellen op de inzetstukken geënt, die in 24-wells-platen worden geplaatst (zie aanhangsel 1). Voor het enten van de cellen dienen inzetstukken van twaalf millimeter doorsnede met een membraan van gemengde cellulose-esters, een dikte van 80-150 μm en een poriëngrootte van 0,45 μm te worden gebruikt. Bij de valideringsstudie werden Millicell-HA-inzetstukken van 12 mm gebruikt. De eigenschappen van het inzetstuk en het membraantype zijn van belang, aangezien zij van invloed kunnen zijn op de celgroei en de binding van chemische stoffen. Bepaalde types van chemische stoffen kunnen aan het membraan van het Millicell-HA-inzetstuk binden, hetgeen van invloed kan zijn op de interpretatie van de resultaten. Als er andere membranen worden gebruikt, dient de gelijkwaardigheid te worden aangetoond met behulp van chemische stoffen voor bekwaamheidstoetsing (zie aanhangsel 3). De binding van chemische stoffen aan het membraan van het inzetstuk komt vaker voor bij kationische chemische stoffen, zoals benzalkoniumchloride, die worden aangetrokken tot het geladen membraan (7). De binding van chemische stoffen aan het membraan van het inzetstuk kan de blootstellingsperiode aan de chemische stof verlengen, waardoor het toxische potentieel van de chemische stof te hoog kan worden ingeschat, maar kan ook de fluoresceïnelekkage door het inzetstuk fysiek verminderen doordat de kleurstof aan de kationische stof bindt, die op haar beurt aan het membraan van het inzetstuk is gebonden, waardoor het toxische potentieel van de chemische stof te laag kan worden ingeschat. Dit kan eenvoudig worden bewaakt door enkel het membraan bloot te stellen aan de hoogste concentratie van de geteste chemische stof en vervolgens gedurende de standaardtijd de natriumfluoresceïnekleurstof toe te voegen bij de normale concentratie (geen celcontrole). Als het natriumfluoresceïne bindt, kleurt het membraan van het inzetstuk geel nadat het testmateriaal is weggewassen. Het is dan ook van essentieel belang om de bindende eigenschappen van de teststof te kennen om het effect van de chemische stof op de cellen te kunnen interpreteren. Bij het enten van de cellen op de inzetstukken dient een confluente monolaag te ontstaan bij blootstelling aan de chemische stof. Er dienen 1,6 x 105 cellen te worden toegevoegd per inzetstuk (400 μl van een celsuspensie met een dichtheid van 4 x 105 cellen/ml). In die omstandigheden wordt gewoonlijk na 96 uur in cultuur een confluente monolaag verkregen. De inzetstukken dienen voor het enten visueel te worden gecontroleerd om ervoor te zorgen dat eventuele beschadigingen die bij de in punt 30 beschreven visuele controle worden vastgesteld, te wijten zijn aan de behandeling. De MDCK-celculturen dienen in incubatoren in een bevochtigde atmosfeer te worden bewaard bij 5 % ± 1 % CO2 en 37 ± 1 °C. De cellen mogen niet verontreinigd zijn met bacteriën, virussen, mycoplasma of schimmels. Het aanbrengen van de test- en controlestoffen Voor elke reeks experimenten dient een verse stamoplossing van de teststof te worden bereid, die binnen 30 minuten na bereiding moet worden gebruikt. De teststoffen dienen te worden bereid in HBSS met calcium (in een concentratie van 1,0-1,8 mM) en zonder fenolrood om serum-eiwitbinding te vermijden. De oplosbaarheid van de chemische stof in HBSS bij 250 mg/ml dient voor het testen te worden beoordeeld. Als de chemische stof bij die concentratie in 30 minuten een stabiele suspensie of emulsie vormt (d.w.z. dat zij haar uniformiteit behoudt en niet bezinkt en zich niet afscheidt in meerdere fasen), kan HBSS nog steeds worden gebruikt als oplosmiddel. Als wordt vastgesteld dat de chemische stof bij deze concentratie niet oplosbaar is in HBSS, moet echter worden overwogen om een andere testmethode dan FL te gebruiken. Het gebruik van lichte minerale olie als oplosmiddel wanneer de chemische stof niet oplosbaar blijkt in HBSS, dient met enige terughoudendheid te worden bekeken, aangezien er onvoldoende gegevens beschikbaar zijn om conclusies te formuleren over de prestaties van de FL-test in zulke omstandigheden. Alle te testen chemische stoffen worden bereid in steriele HBSS met calcium (in een concentratie van 1,0-1,8 mM) en zonder fenolrood op basis van de stamoplossing, in vijf vaste concentraties die verdund zijn op basis van het gewicht per volume-eenheid: 1, 25, 100, 250 mg/ml en een onverdunde of een verzadigd oplossing. Wanneer een vaste chemische stof wordt getest, dient een erg hoge concentratie van 750 mg/ml te worden opgenomen. Deze concentratie van de chemische stof moet mogelijk op de cellen worden aangebracht met behulp van een plunjerpipet. Als wordt vastgesteld dat de toxiciteit tussen 25 en 100 mg/ml ligt, dienen de volgende bijkomende concentraties in tweevoud te worden getest: 1, 25, 50, 75, 100 mg/ml. De FL20-waarde dient te worden afgeleid van die concentraties, voor zover aan de aanvaardbaarheidscriteria is voldaan. De teststoffen worden op de confluente monolagen van cellen aangebracht na verwijdering van het kweekmedium en nadat de monolagen tweemaal zijn uitgewassen met steriele, warme (37 °C) HBSS met calcium (in een concentratie van 1,0-1,8 mM) zonder fenolrood. De filters zijn vooraf visueel gecontroleerd op bestaande beschadigingen die verkeerdelijk zouden kunnen worden toegeschreven aan mogelijke incompatibiliteit met teststoffen. Er dienen in elke reeks ten minste drie duplo's te worden gebruikt voor elke concentratie van de teststof en voor de controles. Na 1 minuut blootstelling bij kamertemperatuur wordt de teststof zorgvuldig verwijderd door afzuiging, wordt de monolaag twee keer uitgewassen met steriele, warme (37 °C) HBSS met calcium (in een concentratie van 1,0-1,8 mM) zonder fenolrood en wordt de fluoresceïnelekkage onmiddellijk gemeten. In elke reeks moeten negatieve controles (NC) en positieve controles (PC) worden gebruikt om aan te tonen dat de integriteit van de monolaag (NC) en de gevoeligheid van de cellen (PC) binnen een gedefinieerd historisch aanvaardbaarheidsbereik liggen. Voor de positieve controles wordt voorgesteld om Brij 35 (CAS-nr. 9002-92-0) te gebruiken bij 100 mg/ml. Deze concentratie dient een fluoresceïnelekkage van ongeveer 30 % op te leveren (aanvaardbaar bereik fluoresceïnelekkage 20-40 %, d.w.z. schade aan de cellaag). Voor de negatieve controles wordt voorgesteld om HBSS met calcium (in een concentratie van 1,0-1,8 mM) en zonder fenolrood te gebruiken (onbehandeld, blanco controle). In elke reeks dient ook een maximumlekkagecontrole te worden opgenomen om de FL20-waarden te kunnen berekenen. De maximumlekkage wordt bepaald met behulp van een controle-inzetstuk zonder cellen. Bepaling van de fluoresceïnepermeabiliteit Onmiddellijk nadat de test- en controlestoffen zijn verwijderd, wordt 400μl 0,1 mg/ml natriumfluoresceïneoplossing (0,01 % (w/v) in HBSS met calcium [in een concentratie van 1,0-1,8 mM] en zonder fenolrood) aan de inzetstukken (bv. Millicell-HA) toegevoegd. De culturen worden gedurende 30 minuten op kamertemperatuur gehouden. Aan het eind van de incubatie met fluoresceïne worden de inzetstukken zorgvuldig uit elke well verwijderd. Elke filter wordt visueel gecontroleerd en eventuele beschadiging die mogelijk is opgetreden tijdens de behandeling, wordt genoteerd. De hoeveelheid fluoresceïne die door de monolaag en het inzetstuk is gelekt, wordt gemeten in de oplossing die in de wells is achtergebleven nadat de inzetstukken zijn verwijderd. De metingen worden verricht in een spectrofluorimeter bij een excitatie- en emissiegolflengte van respectievelijk 485 nm en 530 nm. De gevoeligheid van de spectrofluorimeter dient zo te worden ingesteld dat het numerieke verschil tussen de maximale FL (inzetstuk zonder cellen) en de minimale FL (inzetstuk met confluente monolaag behandeld met NC) zo groot mogelijk is. Vanwege de verschillen in de gebruikte spectrofluorimeter wordt aanbevolen om een gevoeligheid te hanteren die bij de maximale fluoresceïnelekkagecontrole een fluorescentie-intensiteit van > 4000 geeft. De maximale FL-waarde mag niet meer dan 9999 bedragen. De maximale intensiteit van de fluoresceïnelekkage dient binnen het lineaire bereik van de gebruikte spectrofluorimeter te vallen. Interpretatie van de resultaten en voorspellingsmodel De hoeveelheid FL is evenredig aan de door chemische stoffen geïnduceerde schade aan de tight junctions. Het percentage FL voor elke geteste concentratie van de chemische stof wordt berekend op basis van de FL-waarden die voor de teststof werden verkregen, onder verwijzing naar de FL-waarden verkregen uit de NC (waarden afkomstig van de confluente monolaag van cellen die werd behandeld met de NC) en een maximumlekkagecontrole (waarden voor de hoeveelheid FL door een inzetstuk zonder cellen). De gemiddelde maximale intensiteit van de fluoresceïnelekkage = x De gemiddelde 0 %-intensiteit van de fluoresceïnelekkage (NC) = y De gemiddelde 100 %-lekkage wordt verkregen door de gemiddelde 0 %-lekkage af te trekken van de gemiddelde maximale lekkage, d.w.z. x – y = z Het percentage lekkage voor elke vaste dosis wordt verkregen door de 0 %-lekkage af te trekken van de gemiddelde fluorescentie-intensiteit van de drie duplometingen (m) en die waarde te delen door de 100 %-lekkage, d.w.z. %FL = [(m-y) / z] x 100 %, waarbij:
Om de concentratie van de chemische stof te berekenen die 20 % FL veroorzaakt, dient de volgende vergelijking te worden gebruikt: FLD = [(A-B) / (C-B)] x (MC – MB) + MB waarbij:
Hieronder wordt de drempelwaarde van FL20 gegeven om te voorspellen of een stof voor het oog corrosief of hevig irriterend is:
De FL-testmethode wordt enkel aanbevolen voor de identificatie van in water oplosbare voor het oog corrosieve en hevig irriterende stoffen (VN-GHS-categorie 1, EU-CLP-categorie 1, VS-EPA-klasse I) (zie punten 1 en 10). Om in water oplosbare chemische stoffen (stoffen en mengsels) (3) (6) (7) in te delen als “stoffen die ernstig oogletsel veroorzaken” (VN-GHS-/EU-CLP-categorie 1) of als “voor het oog corrosieve of hevig irriterende stoffen” (VS-EPA-klasse I), dient de teststof een FL20-waarde van ≤ 100 mg/ml te induceren. Aanvaarding van de resultaten De gemiddelde waarde voor de maximale fluoresceïnelekkage (x) dient hoger dan 4 000 te zijn (zie punt 31), de gemiddelde 0 %-lekkage (y) dient gelijk te zijn aan of lager te zijn dan 300 en de gemiddelde 100 %-lekkage (z) dient tussen 3 700 en 6 000 te liggen. Een test wordt aanvaardbaar geacht als de positieve controle 20 % tot 40 % schade aan de cellaag opleverde (meting als % fluoresceïnelekkage). GEGEVENS EN RAPPORTAGE Gegevens Voor elke reeks experimenten moeten de gegevens die voor elke individuele well zijn verkregen (bv. gegevens over de waarden voor de fluorescentie-intensiteit en de berekende percentages FL voor elke teststof, inclusief de classificatie), in tabelvorm worden gerapporteerd. Daarnaast moeten voor elke reeks experimenten gemiddelden ± standaardafwijking van elke individuele meting worden vermeld. Testverslag In het testverslag moet de volgende informatie worden opgenomen:
LITERATUUR
Aanhangsel 1 DIAGRAM VAN OP EEN MEMBRAAN VAN EEN INZETSTUK GEKWEEKTE MDCK-CELLEN VOOR DE FL-TESTMETHODE Er wordt een confluente laag MDCK-cellen gekweekt op een semipermeabel membraan van een inzetstuk. De inzetstukken worden in de wells van de 24-wells-platen geplaatst. 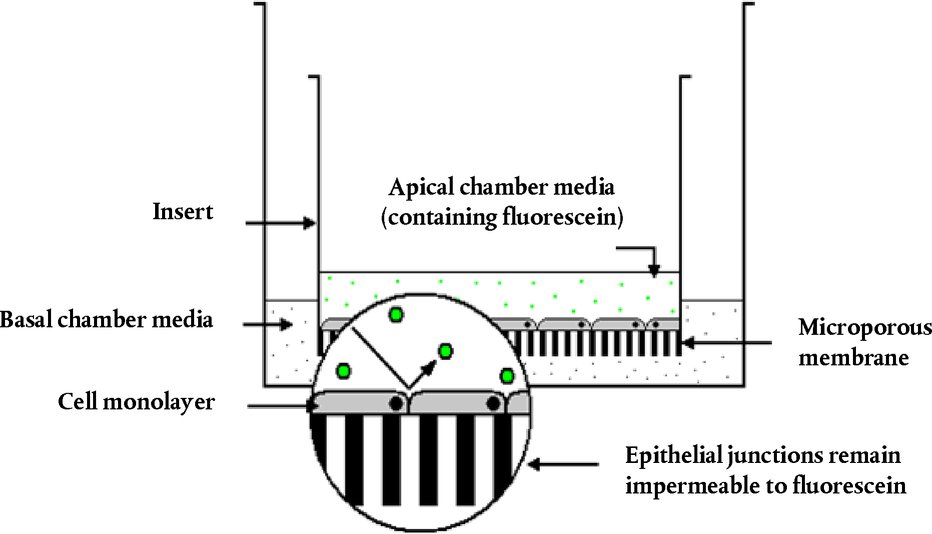
Figuur overgenomen uit: Wilkinson, P.J. (2006), Development of an in vitro model to investigate repeat ocular exposure, Ph.D. Thesis, University of Nottingham, UK. Aanhangsel 2 DEFINITIES Betrouwbaarheid : indicatie van de mate waarin een testmethode, uitgevoerd volgens een vast protocol, op reproduceerbare wijze binnen één laboratorium en tussen laboratoria verspreid over de tijd kan worden uitgevoerd. De betrouwbaarheid wordt beoordeeld door berekening van intra- en interlaboratoriumreproduceerbaarheid en intralaboratoriumherhaalbaarheid. Bewijskracht(proces) : het proces waarbij de sterke en zwakke punten van verschillende gegevens worden onderzocht bij het komen tot en ondersteunen van een conclusie met betrekking tot het gevarenpotentieel van een stof. Chemische stof : een stof of mengsel. EPA-klasse I : chemische stoffen die een corrosieve (irreversibele afbraak van oogweefsel) of corneale aandoening of irritatie veroorzaken die gedurende meer dan 21 dagen aanhoudt (2). Ernstig oogletsel : de veroorzaking van weefselschade aan het oog of ernstige fysische gezichtsvermindering na aanbrenging van een teststof op de voorste ooglaag die niet volledig reversibel is binnen 21 dagen na de aanbrenging. EU-CLP : (Verordening (EG) nr. 1272/2008 betreffende de indeling, etikettering en verpakking van stoffen en mengsels): voert in de Europese Unie (EU) het GHS-systeem van de VN uit dat chemicaliën (stoffen en mengsels) indeelt. FL20 : kan worden geraamd door de concentratie te bepalen waarbij de geteste chemische stof 20 % van de fluoresceïnelekkage door de cellaag veroorzaakt. Fluoresceïnelekkage : de hoeveelheid fluoresceïne die door de cellaag dringt, gemeten met een spectrofluorimeter. Gevaar : inherente eigenschap van een agens of situatie die nadelige effecten kan veroorzaken als een organisme, systeem of (sub)populatie aan dit agens wordt blootgesteld. Gevalideerde testmethode : een testmethode waarvoor valideringsstudies werden gedaan om de relevantie (inclusief nauwkeurigheid) en betrouwbaarheid voor een bepaald doel te bepalen. Er dient te worden opgemerkt dat een gevalideerde testmethode niet per definitie voldoende nauwkeurig en betrouwbaar is om aanvaardbaar te worden bevonden voor het voorgestelde doel (8). Gevoeligheid : het percentage van alle positieve/actieve chemische stoffen dat door middel van de test correct wordt ingedeeld. De gevoeligheid is een maat voor de nauwkeurigheid van een testmethode die categoriale resultaten oplevert en is een belangrijke factor voor de beoordeling van de relevantie van een testmethode (8). GHS (Globally Harmonized System of Classification and Labelling of Chemicals, mondiaal geharmoniseerd classificatie- en etiketteringssysteem voor chemische stoffen van de Verenigde Naties (VN)) : een systeem voor de indeling van chemische producten (stoffen en mengsels) op basis van gestandaardiseerde soorten en niveaus van gevaren van fysische aard en gevaren voor de gezondheid en het milieu, en dat de desbetreffende voorlichtingselementen bepaalt, zoals pictogrammen, signaalwoorden, gevarenaanduidingen, voorzorgsmaatregelen en veiligheidsinformatiebladen, waarmee informatie over de schadelijke effecten van de stoffen kenbaar wordt gemaakt teneinde mensen (waaronder werkgevers, werknemers, transporteurs, consumenten en spoedhulpverleners) en het milieu te beschermen. GHS-klasse 1 : veroorzaking van weefselschade aan het oog of ernstige fysische gezichtsvermindering na aanbrenging van een teststof op de voorste ooglaag die niet volledig reversibel is binnen 21 dagen na de aanbrenging. Medium-/oplosmiddelcontrole : een onbehandeld monster dat alle componenten bevat van een testsysteem, met inbegrip van het oplosmiddel of medium, dat tezamen met de met de teststof behandelde monsters en andere controlemonsters verwerkt wordt om de referentierespons voor de monsters die behandeld werden met de in hetzelfde oplosmiddel of medium opgeloste teststof vast te stellen. Als dit monster getest wordt met een tegelijkertijd gemeten negatieve controle, toont het ook aan of het oplosmiddel of medium met het testsysteem interageert. Mengsel : gebruikt in de context van het VN-GHS voor een mengsel of oplossing bestaande uit meer dan twee stoffen waarin die stoffen niet reageren. Nauwkeurigheid : de mate van overeenstemming tussen resultaten verkregen met de testmethode en erkende referentiewaarden. Het is een maat voor de prestaties van de testmethode en één aspect van “relevantie”. Deze term en de term “concordantie”, waaronder het percentage correcte uitkomsten van een testmethode wordt verstaan, worden vaak door elkaar gebruikt. Negatieve controle : een onbehandeld monster dat alle bestanddelen van een testsysteem bevat. Dit monster wordt tezamen met de met de teststof behandelde monsters en andere controlemonsters verwerkt om te bepalen of het oplosmiddel interageert met het testsysteem. Niet ingedeeld : chemische stoffen die niet zijn ingedeeld als voor de ogen irriterende stof van VN-GHS-categorie 1, 2A of 2B; EU-CLP-categorie 1 of 2; of VS-EPA-klasse I, II of III. Percentage vals-negatieve uitslagen : het percentage van alle positieve chemische stoffen dat door een testmethode onterecht als negatief wordt geïdentificeerd. Het is één indicator van de prestaties van de testmethode. Percentage vals-positieve uitslagen : het percentage van alle negatieve chemische stoffen dat door een testmethode onterecht als positief wordt geïdentificeerd. Het is één indicator van de prestaties van de testmethode. Positieve controle : een monster met alle bestanddelen van een testsysteem dat met een chemische stof behandeld werd waarvan bekend is dat deze een positieve respons opwekt. Om te verzekeren dat variabiliteit in de positieve-controlerespons doorheen de tijd kan worden beoordeeld, mag de grootte van de positieve respons niet extreem zijn. Relevantie : beschrijving van het verband van de test met het te onderzoeken effect en of de test betekenis heeft en nuttig is voor een bepaald doel. Het is de mate waarin de test het te onderzoeken biologische effect correct meet of voorspelt. Relevantie omvat een beoordeling van de nauwkeurigheid (concordantie) van een testmethode (8). Specificiteit : het percentage van alle negatieve/niet-actieve chemische stoffen dat door middel van de test correct wordt ingedeeld. Het is een maat voor de nauwkeurigheid van een testmethode die categorale resultaten oplevert, en is een belangrijke overweging bij de beoordeling van de relevantie van een testmethode. Stof : gebruikt in de context van het VN-GHS voor een chemisch element en de verbindingen ervan zoals zij voorkomen in natuurlijke toestand of bij de vervaardiging ontstaan, met inbegrip van alle additieven die nodig zijn voor het behoud van de stabiliteit van het product, en onzuiverheden ten gevolge van het toegepaste procedé, doch met uitzondering van elk oplosmiddel dat kan worden afgescheiden zonder dat de stabiliteit van de stof wordt aangetast of de samenstelling ervan wordt gewijzigd. Stoffen voor bekwaamheidstoetsing : een subgroep van de lijst van referentiestoffen die een onervaren laboratorium kan gebruiken om aan te tonen dat het bekwaam is om de gevalideerde referentietestmethode toe te passen. Teststof : alle volgens deze testmethode geteste stoffen of mengsels. Trapsgewijze teststrategie : een trapsgewijze teststrategie waarbij alle bestaande informatie over een teststof in een bepaalde volgorde wordt herbezien aan de hand van een proces op basis van bewijskracht op elke trap om te bepalen of er voldoende informatie beschikbaar is voor een beslissing tot gevarenclassificatie, vooraleer wordt overgegaan tot de volgende trap. Indien het irritatiepotentieel van een teststof kan worden bepaald op basis van de bestaande informatie, dan zijn bijkomende tests niet vereist. Indien het irritatiepotentieel van een teststof niet kan worden bepaald op basis van de bestaande informatie, dan wordt een trapsgewijze sequentiële testprocedure op dieren uitgevoerd tot een ondubbelzinnige indeling mogelijk is. Vervangende test : een test, ontworpen ter vervanging van een voor de identificatie van gevaren en/of risicobeoordeling geaccepteerde en routinematig gebruikte test, waarvan is vastgesteld dat hij, in vergelijking met de geaccepteerde test, de gezondheid van mens of dier of het milieu, naargelang het geval, gelijke of betere bescherming biedt voor alle mogelijke testsituaties en chemische stoffen. Voor de ogen corrosieve stof : (a) een chemische stof die irreversibele schade aan het oogweefsel veroorzaakt. (b) chemische stoffen die zijn ingedeeld als voor de ogen irriterende stoffen van VN-GHS-categorie 1; EU-CLP-categorie 1; of VS-EPA-klasse I. Voor de ogen hevig irriterende stof : (a) een chemische stof die na aanbrenging op het voorste oogoppervlak weefselschade aan het oog veroorzaakt die niet reversibel is binnen 21 dagen na de aanbrenging, of die ernstige fysische gezichtsvermindering teweegbrengt; (b) chemische stoffen die zijn ingedeeld als voor de ogen irriterende stoffen van VN-GHS-categorie 1; EU-CLP-categorie 1; of VS-EPA-klasse I. Voor de ogen irriterende stof : (a) een chemische stof die een reversibele wijziging in het oog teweegbrengt na aanbrenging op het voorste oogoppervlak; (b) chemische stoffen die zijn ingedeeld als voor de ogen irriterende stoffen van VN-GHS-categorie 2A of 2B; EU-CLP-categorie 2; of VS-EPA-klasse II of III. Aanhangsel 3 CHEMISCHE STOFFEN VOOR BEKWAAMHEIDSTOETSING VOOR DE FL-TESTMETHODE Vooraleer over te gaan tot routinegebruik van deze testmethode, zouden laboratoria de technische bekwaamheid moeten aantonen door een correcte identificatie van de indeling als corrosief voor de ogen van de 8 stoffen die in tabel 1 worden aangeraden. Deze chemische stoffen werden geselecteerd om de mogelijke responsen voor plaatselijke oogirritatie/-corrosie weer te geven, op basis van de resultaten in de in-vivo-oogtest bij konijnen (TG 405, TM B.5(5)) (d.w.z. categorieën 1, 2A, 2B of geen indeling volgens het VN-GHS). Aangezien het nut van de FL-test (d.w.z. uitsluitend voor het oog corrosieve/hevig irriterende stoffen te identificeren) is gevalideerd, zijn er echter slechts twee testresultaten voor indelingsdoeleinden (corrosief/hevig irriterend of niet-corrosief/niet hevig irriterend) waarvoor de bekwaamheid moet worden aangetoond. Andere selectiecriteria waren dat stoffen commercieel beschikbaar zijn, dat er in-vivoreferentiegegevens van zeer goede kwaliteit beschikbaar zijn, en dat er gegevens van zeer goede kwaliteit beschikbaar zijn van de FL-testmethode. Daarom werden de chemische stoffen voor bekwaamheidstoetsing geselecteerd uit het “Fluorescein Leakage Assay Background Review Document as an Alternative Method for Eye Irritation Testing” (8), dat werd gebruikt om de FL-testmethode met terugwerkende kracht te valideren. Tabel 1 Aanbevolen stoffen voor het aantonen van technische bekwaamheid met FL
B.62 In vivo alkalische komeettest bij zoogdieren INLEIDING Deze testmethode (TM) is gelijkwaardig aan testrichtlijn (TG) 489 (2016) van de OESO. De in vivo alkalische komeettest bij zoogdieren (gelelektroforese op cellen) (hierna verkort “de komeettest” genoemd) wordt gebruikt om breuken in de DNA-strengen te detecteren in cellen of nuclei die zijn afgezonderd uit verschillende dierlijke weefsels, gewoonlijk van knaagdieren, die zijn blootgesteld aan mogelijk genotoxische materialen. De komeettest is geëvalueerd en er zijn aanbevelingen gepubliceerd door verschillende deskundigengroepen (1) (2) (3) (4) (5) (6) (7) (8) (9) (10). Deze testmethode maakt deel uit van een reeks methoden om de genotoxiciteit te testen. Er is een OESO-document opgesteld met beknopte informatie over genetisch-toxicologische tests en een overzicht van de recente wijzigingen van deze testrichtlijnen (11). Deze komeettest is bedoeld om chemische stoffen te identificeren die schade aan het DNA veroorzaken. In alkalische omstandigheden (pH > 13) kan de komeettest zowel enkelstrengige als dubbelstrengige breuken detecteren ten gevolge van, bijvoorbeeld, een directe wisselwerking met het DNA, alkalilabiele plekken of voorbijgaande breuken van DNA-strengen ten gevolge van excisieherstel van het DNA. Deze strengbreuken kunnen worden hersteld, waarbij geen blijvende effecten ontstaan, kunnen letaal zijn voor de cel of kunnen worden vastgezet in een mutatie die tot een blijvende, levensvatbare verandering leidt. Zij kunnen ook aanleiding geven tot chromosoombeschadiging, die eveneens in verband wordt gebracht met talrijke ziekten bij de mens, waaronder kanker. Tussen 2006-2012 werd een formele validatieproef van de in-vivokomeettest bij knaagdieren uitgevoerd die werd gecoördineerd door het Japanese Center for the Validation of Alternative Methods (JaCVAM), in samenwerking met het Europees Centrum voor de validering van alternatieve methoden (ECVAM), het Interagency Coordinating Committee on the Validation of Alternative Methods (ICCVAM) en het NTP Interagency Center for the Evaluation of Alternative Toxicological Methods (NICEATM) (12). Deze testmethode omvat het aanbevolen gebruik en de beperkingen van de komeettest en is gebaseerd op het definitieve protocol (12) dat werd gebruikt bij de validatieproef en op andere relevante gepubliceerde en niet gepubliceerde gegevens (eigen gegevens van de laboratoria). De definities van kernbegrippen worden uiteengezet in aanhangsel 1. Voor deze test kunnen heel wat verschillende platforms worden gebruikt (microscoopglaasjes, geldruppels, 96-wells-platen enz.). Gemakshalve wordt in de rest van dit document de term “glaasje” gebruikt, waaronder eveneens alle overige platforms worden begrepen. INLEIDENDE OVERWEGINGEN EN BEPERKINGEN De komeettest is een methode om breuken van DNA-strengen te meten in eukaryotische cellen. Op een glaasje in agarosegel ingebedde cellen/nuclei worden met een detergent en een hoge zoutconcentratie gelyseerd. Bij deze stap wordt het cel- en nucleusmembraan verteerd en komen opgerolde DNA-lussen, ook wel nucleoïden genoemd, en DNA-fragmenten vrij. Elektroforese bij een hoge pH leidt tot structuren die op kometen lijken en die, met behulp van passende fluorescerende kleurstoffen, met een fluorescentiemicroscoop kunnen worden waargenomen. DNA-fragmenten migreren van de “kop” naar de “staart” op basis van hun grootte, en de intensiteit van de staart van de komeet in verhouding tot de totale intensiteit (kop plus staart) weerspiegelt het aantal DNA-breuken (13) (14) (15). De in vivo alkalische komeettest bij zoogdieren is in het bijzonder relevant om het genotoxische gevaar te beoordelen, aangezien de responsen op de test afhankelijk zijn van de ADME (absorptie, distributie, metabolisme en excretie) in vivo en van de DNA-herstelprocessen. Die kunnen verschillen naargelang van de diersoort, het soort weefsel en het soort DNA-beschadiging. Om aan de voorschriften op het gebied van dierenwelzijn te voldoen, in het bijzonder inzake het beperken van het gebruik van dieren (de beginselen van de drie v's — vervangen, vermijden en verfijnen), kan deze test ook worden gecombineerd met andere toxicologische studies, bv. toxiciteitsonderzoeken met herhaalde toediening (10) (16) (17), of kan het eindpunt worden gecombineerd met andere eindpunten voor genotoxiciteit, zoals in-vivomicronucleustest op erytrocyten bij zoogdieren (18) (19) (20). De komeettest wordt meestal uitgevoerd op knaagdieren, hoewel zij ook al op andere zoogdieren en niet-zoogdieren is toegepast. Het gebruik van andere diersoorten dan knaagdieren dient voor elk afzonderlijk geval wetenschappelijk en ethisch te worden gemotiveerd en het is sterk aanbevolen om de komeettest enkel te verrichten op andere soorten dan knaagdieren als onderdeel van een andere toxiciteitsstudie en niet als op zichzelf staande test. De keuze voor de blootstellingsroute en de te onderzoeken weefsels dient te worden gebaseerd op alle beschikbare/bestaande kennis over de teststoffen, zoals de beoogde/verwachte blootstellingsroute bij de mens, het metabolisme en de distributie, het potentieel voor effecten op de plek van contact, structurele waarschuwingen, andere gegevens over genotoxiciteit of toxiciteit en het doel van de studie. Waar passend dient het genotoxisch potentieel van de teststoffen daarom te worden beoordeeld in het (de) doelweefsel(s) van kankerverwekkende en/of andere toxische effecten. De test wordt ook geschikt geacht om in vitro gesignaleerde genotoxiciteit nader te onderzoeken. Wanneer redelijkerwijs kan worden verwacht dat een te onderzoeken weefsel op passende wijze kan worden blootgesteld, is het verstandig om een in-vivokomeettest op dat weefsel toe te passen. De test is het meest uitvoerig gevalideerd voor somatische weefsels van mannetjesratten, in gezamenlijke studies zoals de JaCVAM-proef (12) en in Rothfuss et al., 2010 (10). In de internationale valideringsproef van het JaCVAM werden de lever en de maag gebruikt. De keuze viel op de lever, omdat dat het orgaan is dat het actiefst is in het metabolisme van chemische stoffen en vaak ook een doelorgaan is voor carcinogeniteit. De maag werd onderzocht omdat het gewoonlijk de eerste plek van contact is voor chemische stoffen na orale blootstelling, hoewel ook andere delen van het spijsverteringskanaal, zoals de duodenum en de jejunum, kunnen worden beschouwd als weefsels die in contact komen met chemische stoffen en relevanter kunnen zijn voor mensen dan de glandulaire maag van knaagdieren. Er dient voor te worden gezorgd dat zulke weefsels niet overmatig worden blootgesteld aan hoge concentraties van de teststof (21). De techniek kan in principe worden toegepast op elk weefsel waaruit analyseerbare suspensies van afzonderlijke cellen/nuclei kunnen worden afgeleid. Eigen gegevens van verschillende laboratoria geven aan dat de techniek met succes kan worden toegepast op uiteenlopende weefsels, en er zijn talrijke publicaties te vinden waaruit blijkt dat de techniek toepasbaar is op andere organen of weefsels dan de lever of de maag, zoals de jejunum (22), de nieren (23) (24), de huid (25) (26) of de urineblaas (27) (28), de longen of bronchoalveolaire lavagecellen (relevant voor studies naar chemische stoffen die worden ingeademd) (29) (30). Voorts zijn er ook tests verricht op meerdere organen (31) (32). Hoewel het nuttig kan zijn om de genotoxische effecten op geslachtscellen te onderzoeken, dient te worden opgemerkt dat de standaard alkalische komeettest zoals beschreven in deze testmethode niet geschikt wordt geacht om breuken van DNA-strengen te meten in volwassen geslachtscellen. Aangezien er in een literatuurstudie naar het gebruik van de komeettest om de genotoxiciteit voor geslachtscellen te onderzoeken (33) hoge en variabele achtergrondniveaus voor DNA-beschadiging werden gemeld, zijn er aanpassingen van de protocollen en betere standaardiserings- en valideringsstudies nodig, alvorens de komeettest op volwassen geslachtscellen (zoals spermacellen) in de testmethode kan worden opgenomen. Bovendien is het in deze testmethode beschreven aanbevolen blootstellingsregime niet optimaal en zouden er langere blootstellingstijden of bemonsteringstijden nodig zijn om een zinvolle analyse te kunnen maken van de breuken van DNA-strengen in volwassen sperma. De bij de komeettest gemeten genotoxische effecten in testiculaire cellen in verschillende stadia van de differentiatie zijn beschreven in de literatuur (34) (35). Er zij echter op gewezen dat geslachtsklieren een combinatie van somatische en geslachtscellen bevatten. Daarom wijzen positieve resultaten in de volledige geslachtsklier (testis) niet noodzakelijk op schade aan de geslachtscellen; zij wijzen er echter wel op dat de geteste chemische stof(fen) en/of hun metabolieten tot in de geslachtsklier zijn doorgedrongen. Crosslinks kunnen niet op betrouwbare wijze worden gedetecteerd onder de gebruikelijke experimentele omstandigheden van de komeettest. In bepaalde, aangepaste experimentele omstandigheden kunnen mogelijk crosslinks tussen DNA en DNA en tussen DNA en eiwitten en andere baseveranderingen, zoals geoxideerde basen, worden gedetecteerd (23) (36) (37) (38) (39). Er moet evenwel verder onderzoek worden verricht met het oog op de adequate omschrijving van de nodige aanpassingen van de protocollen. Het detecteren van crosslinkende stoffen is dan ook niet het hoofddoel van de test zoals die hier is beschreven. De test is niet geschikt om aneugenen te detecteren, ongeacht eventuele aanpassingen. Vanwege de huidige kennisstand gelden er verschillende bijkomende beperkingen (zie aanhangsel 3) voor de in-vivokomeettest. Er wordt verwacht dat de testmethode in de toekomst zal worden geëvalueerd en indien nodig zal worden herzien in het licht van de opgedane ervaring. Voordat de testmethode wordt gebruikt op een mengsel om gegevens te genereren voor een beoogd regelgevingsdoel, moet worden nagegaan of, en zo ja, waarom deze methode betrouwbare resultaten oplevert voor dat doel. Zulke overwegingen zijn niet nodig wanneer het testen van het mengsel wettelijk vereist is. PRINCIPE VAN DE METHODE De dieren worden langs een geschikte weg aan de teststof blootgesteld. De dosering en bemonstering worden in detail beschreven in de punten 36-40. Op de gekozen bemonsteringstijdstippen worden de te onderzoeken weefsels ontleed en worden suspensies van afzonderlijke cellen/nuclei bereid (er kan een in-situperfusie worden uitgevoerd wanneer dat nuttig wordt geacht, bv. voor de lever), die in zachte agar worden ingebed om te immobiliseren op de glaasjes. De cellen/nuclei worden behandeld met lysis-buffermengsel om het cel- en/of nucleusmembraan te verwijderen en worden blootgesteld aan krachtige alkali, bv. met een pH ≥ 13, om het DNA te ontrafelen en ervoor te zorgen dat de losgekomen DNA-lussen en -fragmenten worden vrijgegeven. Het nucleaire DNA in de agar wordt vervolgens onderworpen aan elektroforese. Normaal gezien blijven niet-gefragmenteerde DNA-moleculen in de positie waarin het nucleaire DNA zich bevond in de agar, terwijl gefragmenteerd DNA en losgekomen DNA-lussen naar de anode migreren. Na elektroforese wordt het DNA zichtbaar gemaakt met behulp van een geschikte fluorescente kleurstof. De preparaten dienen te worden geanalyseerd met een microscoop en volledig of halfautomatische beeldanalysesystemen. De mate waarin het DNA tijdens de elektroforese is gemigreerd en de afstand die het daarbij heeft afgelegd, weerspiegelt de hoeveelheid en de grootte van de DNA-fragmenten. De komeettest heeft verschillende eindpunten. Het DNA-gehalte in de staart ( % DNA in de staart of % staartintensiteit) werd aanbevolen om de DNA-beschadiging te beoordelen (12) (40) (41) (42). Nadat voldoende nuclei zijn geanalyseerd, worden de gegevens met behulp van passende methoden geanalyseerd om de testresultaten te beoordelen. Er zij op gewezen dat aanpassing van verschillende aspecten van de methodologie, waaronder de bereiding van de monsters, de elektroforeseomstandigheden, de parameters voor visuele analyse (zoals de intensiteit van de kleurstof, de intensiteit van de microscooplamp en het gebruik van microscoopfilters en cameradynamica), en van de omgevingsomstandigheden (zoals de achtergrondverlichting) is onderzocht en de DNA-migratie kan beïnvloeden (43) (44) (45) (46). VERIFICATIE VAN DE BEKWAAMHEID VAN LABORATORIA Elk laboratorium dient de nodige experimentele competentie met de komeettest te ontwikkelen door aan te tonen dat het in staat is om suspensies van afzonderlijke cellen of nuclei van voldoende kwaliteit te verkrijgen voor elk doelweefsel van elke gebruikte soort. De kwaliteit van de preparaten wordt in eerste instantie beoordeeld aan de hand van het % DNA in de staart voor met een medium behandelde dieren die binnen een reproduceerbaar laag bereik vallen. De huidige gegevens suggereren dat het groepsgemiddelde voor het % DNA in de staart (op basis van het gemiddelde van de medianen — zie punt 57 voor meer informatie over deze voorwaarden) in de rattenlever bij voorkeur niet hoger mag zijn dan 6 %, hetgeen zou stroken met de waarden van de JaCVAM-valideringsproef (12) en van andere gepubliceerde en eigen gegevens. Er zijn op dit moment onvoldoende gegevens beschikbaar om aanbevelingen te doen over optimale of aanvaardbare bereiken voor andere weefsels. Dit sluit niet uit dat andere weefsels kunnen worden gebruikt, als dat wordt gemotiveerd. Het testverslag dient een passende beoordeling van de prestaties van de komeettest in die weefsels te bevatten, in verhouding tot de gepubliceerde literatuur of op basis van eigen gegevens. Ten eerste is een laag bereik van % DNA in de staart in controles wenselijk om het bereik voldoende dynamisch te houden om een positief effect te kunnen detecteren. Ten tweede dient elk laboratorium de verwachte responsen voor directe mutagenen en pro-mutagenen te kunnen reproduceren, met verschillende werkwijzen zoals voorgesteld in tabel 1 (punt 29). In voorkomend geval en mits de nodige motivering kunnen positieve stoffen worden geselecteerd, bijvoorbeeld op basis van de JaCVAM-valideringsproef (12) of van andere gepubliceerde gegevens (zie punt 9), waarbij duidelijke positieve responsen in de te onderzoeken weefsels moeten worden aangetoond. Het vermogen om zwakke effecten van bekende mutagenen, zoals EMS bij lage doses, te detecteren dient eveneens te worden aangetoond, bijvoorbeeld door dosis-responsrelaties vast te stellen met een passend aantal en een passende onderlinge afstand van de doseringen. Er moet in eerste instantie worden getracht om de bekwaamheid aan te tonen voor de vaakst gebruikte weefsels, zoals leverweefsel van knaagdieren, waarvoor vergelijkingen kunnen worden gemaakt met bestaande gegevens en verwachte resultaten (12). Tegelijkertijd kunnen gegevens van andere weefsels zoals de maag/de duodenum/de jejunum, het bloed enz. worden verzameld. Het laboratorium moet zijn bekwaamheid aantonen voor elk afzonderlijk weefsel in elke soort die het wil onderzoeken en moet eveneens aantonen dat er in dat weefsel een aanvaardbare positieve respons kan worden verkregen met een bekend mutageen (zoals EMS). Er dienen ook gegevens over mediumcontroles/negatieve controles te worden verzameld om de reproduceerbaarheid van negatieve gegevensresponsen aan te tonen en om ervoor te zorgen dat de technische aspecten van de test terdege werden gecontroleerd of om aan te geven dat er mogelijk opnieuw historische controlebereiken moeten worden vastgesteld (zie punt 22). Er zij op gewezen dat, hoewel er bij de necropsie meerdere weefsels kunnen worden verzameld voor verwerking met het oog op komeetanalyse, het laboratorium bekwaam moet zijn in het oogsten van meerdere weefsels bij één dier, om zo te verzekeren dat potentiële DNA-beschadigingen niet verloren gaan en de komeetanalyse niet in het gedrang komt. De tijd tussen de euthanasie en het verwijderen van de weefsels voor verwerking kan van kritiek belang zijn (zie punt 44). Bij het ontwikkelen van bekwaamheid in deze test dient aandacht te worden geschonken aan het dierenwelzijn. Daarom kunnen weefsels van in andere tests gebruikte dieren worden gebruikt om competenties op te bouwen in de verschillende aspecten van de test. Voorts hoeft bij het invoeren van een nieuwe testmethode in een laboratorium niet altijd een volledige studie te worden verricht en kunnen er minder dieren of testconcentraties worden gebruikt om de nodige vaardigheden te verwerven. Controlegegevens uit het verleden Het laboratorium dient in de loop van het bekwaamheidsonderzoek een historische databank samen te stellen om positieve en negatieve controlebereiken en distributies voor relevante weefsels en soorten vast te stellen. Aanbevelingen voor het samenstellen en gebruiken van de gegevens uit het verleden (zoals criteria voor opname en uitsluiting van gegevens in historische gegevens en aanvaardbaarheidscriteria voor specifieke experimenten) zijn te vinden in de literatuur (47). Verschillende weefsels en verschillende soorten, maar ook verschillende media en toedieningswegen kunnen verschillende negatieve controlewaarden voor het % DNA in de staart geven. Het is daarom belangrijk om voor elk weefsel en voor elke soort negatieve controlebereiken vast te stellen. Laboratoria moeten methoden voor kwaliteitscontrole gebruiken, zoals controlekaarten (bv. C-kaarten of X-kaarten (48)), om vast te stellen hoe variabel hun gegevens zijn en om aan te tonen dat het laboratorium de methodologie 'onder controle' heeft. Ook de keuze van passende positieve controlestoffen, dosisbereiken en experimentele omstandigheden (bv. elektroforeseomstandigheden) moet mogelijk worden geoptimaliseerd om zwakke effecten te kunnen detecteren (zie punt 17). Bij eventuele wijzigingen in het experimentele protocol moet rekening worden gehouden met hun consistentie met de bestaande databanken met historische controlegegevens van het laboratorium. Eventuele grote inconsistenties moeten leiden tot de oprichting van een nieuwe databank met historische controlegegevens. BESCHRIJVING VAN DE METHODE Voorbereidingen Keuze van de diersoort Gewoonlijk worden gangbare laboratoriumstammen van gezonde, jonge volwassen knaagdieren gebruikt (6-10 weken oud aan het begin van de behandeling, hoewel ook iets oudere dieren kunnen worden gebruikt). De keuze van de knaagdiersoort dient te worden gebaseerd op (i) de soorten die worden gebruikt in andere toxiciteitsstudies (zodat er altijd verbanden kunnen worden gelegd tussen de gegevens en om geïntegreerde studies mogelijk te maken), (ii) de soorten die tumoren ontwikkelden in een carcinogeniteitsstudie (wanneer het mechanisme van carcinogenese werd onderzocht), of (iii) de soorten waarvan het metabolisme het meest relevant is voor de mens, voor zover dat bekend is. Voor deze test worden gewoonlijk ratten gebruikt. Er kunnen evenwel andere soorten worden gebruikt als dat ethisch en wetenschappelijk gemotiveerd wordt. Huisvestings- en voedingsomstandigheden van de dieren Voor knaagdieren moet de temperatuur in de proefdierruimte idealiter 22 °C (±3 °C) zijn. De relatieve luchtvochtigheid moet ten minste 30 % en idealiter 50-60 % bedragen en mag, behalve tijdens het schoonmaken van de ruimte, bij voorkeur niet hoger zijn dan 70 %. Er moet gebruik worden gemaakt van kunstlicht met een cyclus van 12 uur licht en 12 uur donker. Als voeding mag het gewone laboratoriumvoer worden gebruikt met een onbeperkte hoeveelheid drinkwater. De keuze van het voer kan worden beïnvloed door de noodzaak om voor een afdoende mengbaarheid met de teststof te zorgen, wanneer de stof via het voer wordt toegediend. De knaagdieren moeten worden gehuisvest in kleine groepen (gewoonlijk niet meer dan vijf) van hetzelfde geslacht indien geen agressief gedrag wordt verwacht. De dieren mogen uitsluitend individueel worden gehuisvest als dat wetenschappelijk gerechtvaardigd is. Waar mogelijk moet worden gebruikgemaakt van vaste vloeren, aangezien gaasvloeren ernstige verwondingen kunnen veroorzaken (49). Er moet worden voorzien in de nodige milieuverrijking. Voorbereiding van de dieren De dieren worden willekeurig in de controle- en behandelingsgroepen ingedeeld. De dieren krijgen een unieke identificatie en krijgen voordat de test begint minimaal vijf dagen de tijd om aan de omstandigheden in het laboratorium te wennen. Voor het identificeren van de dieren dient de minst invasieve methode te worden gebruikt. Passende methoden zijn ringen, merken, microchippen en biometrische identificatie. Het afknippen van tenen of oren is in deze test niet wetenschappelijk gerechtvaardigd. De kooien moeten op zodanige wijze worden opgesteld dat eventuele plaatseffecten geminimaliseerd worden. Bij het begin van de studie moet per geslacht het gewichtsverschil tussen de dieren zo klein mogelijk zijn en mag dit niet meer dan ± 20 % bedragen. Voorbereiding van de doses Vaste chemische teststoffen moeten worden opgelost of gesuspendeerd in geschikte media of worden vermengd met voedsel of drinkwater voordat dit aan de dieren wordt gegeven. Vloeibare chemische teststoffen kunnen direct worden gegeven of eerst worden verdund. Voor blootstelling via inhalatie kunnen teststoffen worden toegediend als gas, damp of een vaste/vloeibare aerosol, naargelang van de fysisch-chemische eigenschappen ervan (50) (51). Er dienen verse preparaten van de teststof te worden toegepast, tenzij uit stabiliteitsgegevens blijkt dat bewaren van de preparaten aanvaardbaar is en blijkt wat de juiste bewaaromstandigheden zijn. Testomstandigheden Medium Het medium mag bij de gebruikte dosisvolumes geen toxische effecten veroorzaken en chemische reacties met de chemische teststoffen moeten uitgesloten zijn. Als andere media dan de algemeen bekende worden gebruikt, moet het gebruik ervan worden ondersteund met referentiegegevens die wijzen op de verenigbaarheid ervan wat de proefdieren, de toedieningsweg en het eindpunt betreft. Het wordt aanbevolen waar mogelijk eerst het gebruik van een waterig oplosmiddel/medium te overwegen. Er zij op gewezen dat sommige media (in het bijzonder viskeuze media) ontstekingen kunnen veroorzaken en de achtergrondniveaus van DNA-strengbreuken kunnen verhogen op de plek van contact, in het bijzonder bij herhaaldelijke toediening. Controles Positieve controles Op dit moment in elke test een groep van minimaal 3 analyseerbare dieren van één geslacht, of van elk geslacht als beide worden gebruikt (zie punt 32), te worden opgenomen, die behandeld is met een positieve controlestof. In de toekomst kan mogelijk voldoende bekwaamheid worden aangetoond om de noodzaak van positieve controles te beperken. Als er meerdere bemonsteringstijdstippen worden toegepast (bv. bij een protocol met enkelvoudige toediening) moeten enkel positieve controles worden opgenomen op een van de bemonsteringstijdstippen, maar moet wel voor een evenwichtige opzet worden gezorgd (zie punt 48). Gelijktijdige positieve controlestoffen hoeven niet via dezelfde weg als de teststof te worden toegediend, hoewel het bij het meten van de effecten op de plek van contact belangrijk is om dezelfde weg te gebruiken. Van de positieve controlestoffen dient te zijn aangetoond dat zij breuken van de DNA-strengen induceren in alle weefsels die voor de teststof moeten worden onderzocht. EMS is daarbij waarschijnlijk de positieve controle bij uitstek, omdat deze stof in alle onderzochte weefsels breuken van DNA-strengen heeft veroorzaakt. De doses van de voor positieve controle gebruikte chemische stoffen dienen zodanig te worden gekozen dat matige effecten worden geproduceerd waarmee de prestaties en gevoeligheid van de test kritisch worden beoordeeld en kunnen worden gebaseerd op de dosis-responscurves die het laboratorium heeft vastgesteld tijdens de bekwaamheidstoetsing. Het % DNA in de staart bij dieren die worden gebruikt voor de gelijktijdige positieve controles dient te stroken met het vooraf in het laboratorium vastgestelde bereik voor elk afzonderlijk weefsel en voor elk bemonsteringstijdstip voor die soort (zie punt 16). Voorbeelden van voor positieve controle gebruikte chemische stoffen en enkele van de doelweefsels ervan (bij knaagdieren) staan vermeld in tabel 1. Er kunnen andere stoffen worden gekozen dan die welke vermeld zijn in tabel 1, voor zover dat wetenschappelijk wordt gemotiveerd. Tabel 1 Voorbeelden van voor positieve controle gebruikte stoffen en enkele van de doelweefsels ervan Stoffen en CAS-registratienummers Ethylmethaansulfonaat (CAS RN 62-50-0) voor elk weefsel Ethylnitrosoureum (CAS RN 759-73-9) voor lever en maag, duodenum of jejunum Methylmethaansulfonaat (CAS RN 66-27-3) voor lever, maag, duodenum of jejunum, long- en bronchoalveolaire lavage- (BAL-) cellen, nier, blaas, long, testis en beenmerg/bloed N-Methyl-N'-nitro-N-nitrosoguanidine (CAS RN 70-25-7) voor maag, duodenum of jejunum 1,2-Dimethylhydrazine 2HCl (CAS RN 306-37-6) voor lever en darm N-methyl-N-nitrosoureum (CAS RN 684-93-5) voor lever, beenmerg, bloed, nier, maag, jejunum en hersenen. Negatieve controles Voor elk bemonsterinsgtijdstip en weefsel dient een groep dieren voor de negatieve controle te worden meegenomen die zijn behandeld met alleen medium en verder op dezelfde wijze zijn behandeld als de behandelingsgroepen. Het percentage DNA in de staart in dieren voor de negatieve controle dient te liggen binnen het vooraf bepaalde achtergrondbereik van het laboratorium voor elk afzonderlijk weefsel en bemonsteringstijdstip voor de betreffende soort (zie punt 16). Bij het ontbreken van historische of gepubliceerde controlegegevens waaruit blijkt dat het gekozen medium, het aantal toedieningen en de toedieningsweg geen schadelijke of genotoxische effecten induceren, dienen voorafgaand aan het volledige onderzoek voorlopige onderzoeken te worden uitgevoerd om vast te stellen dat de mediumcontrole aanvaardbaar is. PROCEDURE Aantal en geslacht van de dieren Hoewel er voor de komeettest weinig gegevens over vrouwtjes beschikbaar zijn op basis waarvan een vergelijking tussen de geslachten kan worden gemaakt, zijn andere in-vivogenotoxiciteitsresponsen voor vrouwtjes en mannetjes in het algemeen gelijk en kunnen de meeste studies dus bij beide geslachten worden uitgevoerd. Gegevens waaruit blijkt dat er relevante verschillen tussen vrouwtjes en mannetjes bestaan (bv. verschillen in systemische toxiciteit, metabolisme, biologische beschikbaarheid enz., met inbegrip van bijvoorbeeld in een bereikbepalingsonderzoek), zijn een aanmoediging om beide geslachten te gebruiken. In dit geval kan het passend zijn om een onderzoek bij beide geslachten uit te voeren, bijvoorbeeld als onderdeel van een toxiciteitsonderzoek bij herhaalde toediening. Het kan passend zijn om een factoriële onderzoeksopzet te gebruiken als beide geslachten worden gebruikt. Details over de wijze waarop de gegevens bij deze onderzoeksopzet moeten worden geanalyseerd, zijn te vinden in aanhangsel 2. De vaststelling van de groepsgrootte aan het begin van het onderzoek (en tijdens de aantoning van de bekwaamheid) moet erop gericht zijn minimaal 5 analyseerbare dieren van één geslacht, of van elk geslacht als beide worden gebruikt, per groep te krijgen (minder in de gelijktijdige positieve controlegroep — zie punt 29). Wanneer de blootstelling aan een chemische stof bij de mens door het geslacht wordt bepaald, zoals dit bijvoorbeeld bij sommige geneesmiddelen het geval is, moet de test bij het desbetreffende geslacht worden uitgevoerd. Als richtsnoer voor de maximale typische behoefte aan dieren: voor een onderzoek uitgevoerd volgens de in punt 33 vastgestelde parameters met drie dosisgroepen en parallelle negatieve en positieve controlegroepen (elke groep bestaande uit vijf dieren van één geslacht) zijn 25 tot 35 dieren nodig. BEHANDELINGSSCHEMA Dieren moeten gedurende een periode van 2 of meer dagen dagelijkse behandelingen krijgen (d.w.z. twee of meer behandelingen met een interval van ongeveer 24 uur), en monsters moeten eenmalig 2-6 uur (of op Tmax) na de laatste behandeling worden verzameld (12). Monsters van verlengde doseringsschema's (bv. dagelijkse dosistoediening gedurende 28 dagen) zijn aanvaardbaar. Er is aangetoond dat de komeettest en de micronucleustest bij erytrocyten succesvol kunnen worden gecombineerd (10) (19). Er moet echter zorgvuldig rekening worden gehouden met de logistiek die komt kijken bij de bemonstering van weefsel voor de komeetanalyse naast de vereisten voor de bemonstering van weefsel voor andere typen toxicologische beoordelingen. Oogsten 24 uur na de laatste dosistoediening, wat typisch is voor een algemeen toxiciteitsonderzoek, is in de meeste gevallen niet passend (zie punt 40 over bemonsteringstijdstippen). Het gebruik van andere behandelings- en bemonsteringsschema's moet worden gemotiveerd (zie aanhangsel 3). Er kan bijvoorbeeld één behandeling met meerdere bemonsteringen worden gebruikt; er zij echter opgemerkt dat voor een studie met één toediening meer dieren nodig zullen zijn, omdat meerdere bemonsteringstijdstippen nodig zullen zijn, maar dit kan bij gelegenheid toch de voorkeur genieten, bijvoorbeeld wanneer de teststof na herhaalde toedieningen excessieve toxiciteit veroorzaakt. Ongeacht de wijze waarop de test wordt uitgevoerd, is de test aanvaardbaar zolang de teststof leidt tot een positieve respons of, voor een negatieve studie, zolang als er direct of indirect bewijs is van blootstelling van, of toxiciteit voor, het doelweefsel of als de limietdosis is bereikt (zie punt 36). De doses van de teststoffen kunnen eventueel worden gesplitst (bv. twee porties op dezelfde dag met een interval van niet meer dan 2-3 uur) om de toediening van een groot volume te vergemakkelijken. In deze omstandigheden moet het bemonsteringstijdstip worden gepland op basis van het tijdstip van de laatste dosistoediening (zie punt 40). Dosisniveaus Als er een bereikbepalingsonderzoek wordt uitgevoerd omdat er uit andere relevante onderzoeken geen bruikbare gegevens over de dosering beschikbaar zijn, moet dit in hetzelfde laboratorium gebeuren met dezelfde soort, dezelfde stam, hetzelfde geslacht en hetzelfde behandelingsschema als bij het hoofdonderzoek en volgens de huidige benaderingen voor het uitvoeren van doseringsbereikstudies. Het onderzoek moet erop gericht zijn de maximaal verdraagbare dosis (maximum tolerated dose, MTD) vast te stellen, dat wil zeggen, de dosis die lichte toxische effecten induceert, in verhouding tot de duur van de onderzoeksperiode (bijvoorbeeld duidelijke klinische tekenen zoals afwijkend gedrag of afwijkende reacties, licht gewichtsverlies of cytotoxiciteit voor het doelweefsel), maar geen dood of aanwijzingen voor pijn, lijden of nood die humane doding noodzakelijk maken. Voor een niet-toxische teststof met een toedieningsperiode van 14 dagen of meer is de maximale (limiet)dosis 1000 mg/kg lichaamsgewicht/dag. Voor een toedieningsperiode van minder dan 14 dagen is de maximale (limiet)dosis 2000 mg/kg lichaamsgewicht/dag. Voor bepaalde typen teststoffen (bv. geneesmiddelen voor menselijk gebruik) waarvoor specifieke voorschriften gelden, kunnen deze limieten verschillen. Chemische stoffen die verzadiging van toxicokinetische eigenschappen vertonen, of detoxificatieprocessen induceren die kunnen leiden tot een afname van de blootstelling na langlopende toediening, kunnen uitzonderingen zijn op de doseringscriteria en moeten per geval worden beoordeeld. Voor zowel de acute als de sub-acute versie van de komeettest moet naast de maximale dosis (MTD, maximaal haalbare dosis, maximale blootstelling of limietdosis) een aflopende reeks van ten minste twee aanvullende dosisniveaus die niet te ver uit elkaar liggen (bij voorkeur minder dan 10) worden gekozen voor elk bemonsteringstijdstip om doseringsgerelateerde responsen aan te tonen. De gebruikte dosisniveaus moeten bij voorkeur echter ook een bereik dekken van maximale tot weinig of geen toxiciteit. Wanneer toxiciteit voor het doelweefsel bij alle geteste dosisniveaus wordt waargenomen, wordt verder onderzoek bij niet-toxische doses aanbevolen (zie punten 54-55). Voor onderzoek dat ten doel heeft de vorm van de dosisresponscurve vollediger te onderzoeken, kunnen een of meer aanvullende dosisgroepen nodig zijn. Toediening van de doses Bij de opzet van een test moet rekening worden gehouden met de verwachte blootstellingsroute bij mensen. Daarom kunnen andere blootstellingsroutes zoals voeding, drinkwater, lokaal, subcutaan, intraveneus, oraal (via sonde), inhalatie, intratracheaal of implantatie worden gekozen, als deze onderbouwd worden. De route moet in elk geval zodanig worden gekozen dat adequate blootstelling van het (de) doelweefsel(s) is gewaarborgd. Intraperitoneale injectie wordt in het algemeen niet aanbevolen, omdat zij bij mensen geen typische relevante blootstellingsroute is, en zij mag uitsluitend worden toegepast met een specifieke motivering (bv. sommige stoffen voor positieve controles, voor onderzoeksdoeleinden of voor sommige geneesmiddelen die intraperitoneaal worden toegediend). Het maximale vloeistofvolume dat via sonde of injectie per keer kan worden toegediend, hangt af van de grootte van het proefdier. Het volume mag niet groter zijn dan 1 ml/100 g lichaamsgewicht, behalve wanneer het om een waterige oplossing gaat: in dat geval mag 2 ml/100 g lichaamsgewicht worden gebruikt. Hogere volumes dan dit (mits toegestaan volgens wetgeving inzake dierenwelzijn) moeten worden onderbouwd. Indien mogelijk moeten verschillende dosisniveaus worden bereikt door de concentratie van de doseringsformule aan te passen, zodat er op alle dosisniveaus hetzelfde volume in verhouding tot het lichaamsgewicht wordt toegediend. Bemonsteringstijdstip Het bemonsteringstijdstip is een kritische variabele: het wordt bepaald door de tijd die nodig is om de teststoffen de maximale concentratie in het doelweefsel te laten bereiken en om breuken in DNA-strengen te induceren, maar zo dat deze breuken nog niet zijn verwijderd of gerepareerd en niet leiden tot celdood. De persistentie van sommige laesies die leiden tot de in de komeettest gedetecteerde breuken in DNA-strengen, kan voor sommige in vitro geteste chemische stoffen heel kort zijn (52) (53). Als dergelijke tijdelijke DNA-laesies worden vermoed, moeten dus maatregelen worden genomen om de verwijdering ervan te verminderen door ervoor te zorgen dat weefsels vroeg genoeg worden bemonsterd, mogelijk vóór de hieronder vermelde standaardtijdstippen. De optimale bemonsteringstijdstippen kunnen afhangen van de chemische stof of de toedieningsweg en kunnen leiden tot bijvoorbeeld snelle blootstelling van weefsel bij intraveneuze toediening of blootstelling via inhalatie. Indien beschikbaar, moeten de bemonsteringstijdstippen bijgevolg worden bepaald op basis van kinetische gegevens (bv. het tijdstip (Tmax) waarop de piek-plasmaconcentratie of piek-weefselconcentratie (Cmax) wordt bereikt, of op de stationaire toestand bij meerdere toedieningen). Indien kinetische gegevens niet beschikbaar zijn, kunnen de monsters bij wijze van geschikt compromis voor de meting van genotoxiciteit 2-6 uur na de laatste behandeling bij twee of meer behandelingen worden genomen, of zowel 2-6 uur als 16-26 uur na één behandeling, hoewel ervoor moet worden gezorgd dat necropsie bij alle dieren op hetzelfde tijdstip na de laatste (of enige) dosis plaatsvindt. Ook informatie over het optreden van toxische effecten in doelorganen (indien beschikbaar) kan worden gebruikt om geschikte bemonsteringstijdstippen te kiezen. Observaties Algemene klinische waarnemingen verband houdend met de gezondheid van de dieren moeten ten minste eenmaal per dag plaatsvinden en worden opgetekend, bij voorkeur steeds op hetzelfde tijdstip en met inachtneming van de piekperiode van de te verwachten effecten na toediening (54). Alle dieren moeten ten minste tweemaal per dag worden geobserveerd op ziekteverschijnselen en sterfte. Voor onderzoeken van langere duur moeten alle dieren ten minste eenmaal per week en bij afloop van de testperiode worden gewogen. Het voedselverbruik wordt bij elke wijziging in het voer en ten minste wekelijks gemeten. Als de teststof met het drinkwater wordt toegediend, moet het waterverbruik bij elke verversing van water en ten minste wekelijks worden gemeten. Dieren die niet-letale indicatoren van excessieve toxiciteit vertonen, moeten voor afloop van de testperiode worden gedood en ze worden gewoonlijk niet gebruikt voor de komeetanalyse. Weefselverzameling Omdat inductie van breuken in DNA-strengen (kometen) in bijna alle weefsels onderzocht kan worden, moet de selectie van het (de) te verzamelen weefsel(s) duidelijk worden onderbouwd en worden gebaseerd op de reden voor uitvoering van het onderzoek en eventuele bestaande gegevens over ADME, genotoxiciteit, carcinogeniteit of andere toxiciteit voor de onderzochte teststoffen. Belangrijke factoren waarmee rekening moet worden gehouden, zijn de toedieningsweg (op basis van de waarschijnlijke blootstelling bij mensen), de voorspelde weefseldistributie en -absorptie, de rol van metabolisme en het mogelijke werkingsmechanisme van de teststoffen. De lever is het weefsel dat het vaakst is onderzocht en waarvoor de meeste gegevens beschikbaar zijn. Indien geen achtergrondinformatie beschikbaar is, en als er geen specifieke weefsels van belang zijn geïdentificeerd, is bemonstering van de lever gerechtvaardigd, aangezien de lever een primaire plaats van het xenobiotisch metabolisme is en vaak sterk blootstaat aan zowel de oorspronkelijke stof(fen) als de metaboliet(en) daarvan. In sommige gevallen kan onderzoek van een plaats van direct contact heel belangrijk zijn (bijvoorbeeld de glandulaire maag, of duodenum/jejunum, voor chemische stoffen die oraal worden toegediend, of de longen voor chemische stoffen die worden geïnhaleerd). Aanvullende of alternatieve weefsels moeten worden geselecteerd op basis van de specifieke redenen waarom de test wordt uitgevoerd; het kan nuttig zijn meerdere weefsels in dezelfde dieren te onderzoeken, mits het laboratorium bekwaamheid met deze weefsels en met het gelijktijdig hanteren van meerdere weefsels heeft aangetoond. Bereiding van de monsters Voor de in de volgende punten (44-49) beschreven processen is het belangrijk dat alle oplossingen of stabiele suspensies vóór hun uiterste gebruiksdatum worden gebruikt, of vers worden bereid indien nodig. In de volgende punten worden de tijden die worden genomen (i) om elk weefsel na necropsie te verwijderen, (ii) elk weefsel te verwerken tot cel-/nuclei-suspensies, en (iii) de suspensie te verwerken en de objectglaasjes te prepareren, ook allemaal als kritische variabelen beschouwd (zie aanhangsel 1, Definities en eenheden), en bij de vaststelling van de methode en het aantonen van de bekwaamheid moeten voor elke stap tijden van aanvaardbare lengte worden vastgesteld. De dieren worden gedood op een wijze die in overeenstemming is met de geldende wetgeving inzake dierenwelzijn en de beginselen van de 3 V's (vervanging, vermindering en verfijning), op het (de) toepasselijke tijdstip(pen) na de laatste behandeling met een teststof. Het geselecteerde weefsel wordt verwijderd en in stukken gesneden, en een deel ervan wordt verzameld voor de komeettest, terwijl tegelijkertijd een gedeelte van hetzelfde deel van het weefsel moet worden uitgesneden en voor mogelijke histopathologische analyse (zie punt 55) volgens de standaardmethoden in een formaldehydeoplossing of in een geschikte fixeerstof moet worden geplaatst (12). Het weefsel voor de komeettest wordt in een maalbuffer geplaatst, voldoende uitgewassen met koude maalbuffer om resterend bloed te verwijderen, en in ijskoude maalbuffer bewaard totdat het wordt verwerkt. In-situ perfusie kan ook worden uitgevoerd, bv. voor lever of nier. In publicaties zijn allerlei methoden voor het isoleren van cellen/nuclei beschreven. Enkele daarvan zijn het fijnmalen van weefsel zoals lever en nier, het schrapen van oppervlakken van slijmvliezen in geval van het maag-darmkanaal, homogenisering en enzymatische vertering. In de valideringsstudie van het JaCVAM werden uitsluitend losse cellen onderzocht, en daarom wordt de voorkeur gegeven aan losse cellen waar het gaat om het vaststellen van de methode en om voor het aantonen van bekwaamheid naar de onderzoeksgegevens van het JaCVAM te kunnen verwijzen. Er is echter aangetoond dat het testresultaat bij gebruik van losse cellen niet essentieel verschilt van dat bij gebruik van losse nuclei (8). Ook verschillende methoden om cellen/nuclei te isoleren (d.w.z homogenisering, fijnhakken, enzymatische vertering en filtering door gaas) gaven vergelijkbare resultaten (55). Zowel geïsoleerde cellen als geïsoleerde nuclei kunnen dus worden gebruikt. Een laboratorium moet weefselspecifieke methoden voor het isoleren van afzonderlijke cellen/nuclei grondig evalueren en valideren. Zoals in punt 40 is besproken, kan de persistentie van sommige laesies die leiden tot de door de komeettest gedetecteerde breuken in DNA-strengen, heel kort zijn (52) (53). Ongeacht de methode die wordt gebruikt voor het bereiden van de cel/nuclei-suspensies, is het dus belangrijk dat weefsels zo spoedig mogelijk na doding van de dieren worden verwerkt en worden bewaard onder omstandigheden die de verwijdering van laesies verminderen (bv. door het weefsel op een lage temperatuur te houden). De celsuspensies moeten ijskoud worden bewaard tot ze gereed zijn voor gebruik, zodat kan worden aangetoond dat de variatie tussen monsters minimaal is en de positieve en negatieve controleresponsen geschikt zijn. BEREIDING VAN DE OBJECTGLAASJES De objectglaasjes dienen zo spoedig mogelijk (idealiter binnen één uur) na de bereiding van het cel/nuclei-preparaat te worden bereid, maar de temperatuur en de tijd tussen de dood van de dieren en de bereiding van de objectglaasjes dient streng te worden gecontroleerd en gevalideerd onder de omstandigheden in het laboratorium. Het volume van de celsuspensie die voor het prepareren van de objectglaasjes wordt toegevoegd aan agarose met een laag smeltpunt (gewoonlijk 0,5-1,0 %), moet zo zijn dat het percentage agarose met laag smeltpunt niet tot onder de 0,45 % daalt. De optimale celdichtheid wordt bepaald door het beeldanalysesysteem dat voor het scoren van de kometen wordt gebruikt. Lysis De lysisomstandigheden zijn ook een kritische variabele en kunnen als gevolg van specifieke typen modificaties in het DNA (bepaalde alkyleringen van DNA en base-adducten) interfereren met strengbreuken. Het wordt daarom aanbevolen de lysisomstandigheden voor alle objectglaasjes in een experiment zo constant mogelijk te houden. Wanneer de objectglaasjes eenmaal zijn bereid, moeten ze gedurende ten minste één uur (of een nacht) worden ondergedompeld in gekoelde lysisoplossing bij ongeveer 2-8 °C bij getemperde lichtomstandigheden, bijvoorbeeld geel licht (of lichtdicht), die blootstelling aan wit licht dat UV-componenten kan bevatten vermijden. Na deze incubatieperiode worden de objectglaasjes gewassen om vóór de alkalische ontwindingsstap resten detergent en zouten te verwijderen. Dit kan worden gedaan met behulp van gezuiverd water, neutralisatiebuffer of fosfaatbuffer. Elektroforesebuffer kan ook worden gebruikt. Dit houdt de alkalische omstandigheden in de elektroforesekamer in stand. Ontwinding en elektroforese De objectglaasjes dienen willekeurig te worden geplaatst op het platform van een elektroforeseapparaat van het type submarine dat zoveel elektroforeseoplossing bevat dat de oppervlakken van de objectglaasjes volledig worden bedekt (de diepte van de bedekking moet van run tot run gelijk zijn). In een ander type elektroforeseapparaten voor komeettests, d.w.z. met actieve koeling, circulatie en stroomvoorziening met een hoge capaciteit, zal een dikkere laag oplossing leiden tot hogere elektrische stromen bij gelijk voltage. Er moet voor het plaatsen van de objectglaasjes in de elektroforesetank een gebalanceerde opzet worden gebruikt, om de effecten van eventuele trends of randeffecten binnen de tank te verminderen en de variabiliteit tussen batches zo klein mogelijk te maken, d.w.z. in elke elektroforeserun dient het aantal objectglaasjes van elk dier in het onderzoek hetzelfde te zijn en dienen monsters uit de verschillende dosisgroepen, negatieve en positieve controles te worden opgenomen. De objectglaasjes moeten ten minste 20 minuten de gelegenheid krijgen om te rusten zodat het DNA kan ontwinden, en vervolgens worden onderworpen aan elektroforese onder gecontroleerde omstandigheden die de gevoeligheid en het dynamisch bereik van de test maximaliseren (d.w.z. voor negatieve en positieve controles leiden tot aanvaardbare niveaus voor het % DNA in de staart die de gevoeligheid maximaliseren). Het niveau van de DNA-migratie hangt lineair samen met de duur van de elektroforese en ook met de potentiaal (V/cm). Op basis van het JaCVAM-onderzoek kan dit 0,7 V/cm gedurende ten minste 20 minuten zijn. De duur van de elektroforese wordt als kritische variabele beschouwd, en de elektroforeseduur moet zo worden ingesteld dat het dynamisch bereik optimaal is. Langere elektroforesetijden (bv. 30 of 40 minuten om maximale gevoeligheid te verkrijgen) leiden gewoonlijk tot sterkere positieve responsen voor bekende mutagenen. Langere elektroforesetijden kunnen echter ook tot excessieve migratie in controlemonsters leiden. In elk experiment moet het voltage constant worden gehouden en de variabiliteit in de andere parameters moet binnen een smalle en voorgeschreven band liggen; in het JaCVAM-onderzoek leverde 0,7 V/cm bijvoorbeeld een beginstroom van 300 mA op. De diepte van de bufferlaag moet worden aangepast om de vereiste omstandigheden te verkrijgen en moet gedurende het hele experiment in stand worden gehouden. De stroom aan het begin en aan het einde van de elektroforeseperiode moet worden opgetekend. De optimale omstandigheden moeten daarom worden bepaald tijdens de initiële aantoning van bekwaamheid in het laboratorium dat betrokken is bij elk onderzocht weefsel. De temperatuur van de elektroforeseoplossing doorheen de ontwinding en elektroforese moet laag worden gehouden, gewoonlijk 2-10 °C (10). De temperatuur van de elektroforeseoplossing aan het begin van de ontwinding, het begin van de elektroforese en het einde van de elektroforese moet worden opgetekend. Na voltooiing van de elektroforese worden de objectglaasjes gedurende ten minste 5 minuten ondergedompeld/gewassen in de neutralisatiebuffer. Gels kunnen worden gekleurd en “vers” worden gescoord (bv. binnen 1-2 dagen) of kunnen worden gedehydrateerd voor latere scoring (bv. binnen 1-2 weken na kleuring) (56). De omstandigheden moeten echter tijdens de aantoning van bekwaamheid worden gevalideerd, en voor elk van deze omstandigheden moeten historische gegevens worden verkregen en bewaard. In het laatstgenoemde geval moeten de objectglaasjes worden gedehydrateerd door ze gedurende ten minste 5 minuten onder te dompelen in absolute ethanol, worden gedroogd aan de lucht en vervolgens bij kamertemperatuur of in een houder in een koelkast worden opgeslagen totdat ze worden gescoord. Metingsmethoden Kometen moeten kwantitatief worden gescoord met behulp van een automatisch of halfautomatisch beeldanalysesysteem. De objectglaasjes worden gekleurd met een geschikte fluorescerende kleurstof zoals SYBR Gold, Green I, propidiumjodide of ethidiumbromide, en worden gemeten bij een geschikte vergroting (bv. 200x) op een epifluorescentiemicroscoop met passende detectoren of een digitale (bv. CCD-) camera. Cellen kunnen worden geclassificeerd in drie categorieën, zoals is beschreven in de atlas van afbeeldingen van kometen (57), te weten, scoorbaar, niet-scoorbaar en “hedgehog” (zie punt 56 voor een nadere bespreking). Om artefacten te voorkomen, moet alleen voor scoorbare cellen (duidelijk afgebakende kop en staart zonder interferentie met omliggende cellen) het % DNA in de staart worden gescoord. De frequentie niet-scoorbare cellen hoeft niet te worden gerapporteerd. Omdat “hedgehogs” door het ontbreken van een duidelijk afgebakende kop niet gemakkelijk te detecteren zijn door middel van beeldanalyse, moet hun frequentie worden bepaald op basis van de visuele scoring van ten minste 150 cellen per monster (zie punt 56 voor een nadere bespreking) en apart worden gedocumenteerd. Alle objectglaasjes voor de analyse, ook die van de positieve en negatieve controles, worden onafhankelijk gecodeerd en “blind” gescoord, zodat de scorer de behandelingssituatie niet kent. Voor elk monster (per weefsel per dier) worden ten minste 150 cellen (“hedgehogs” niet inbegrepen — zie punt 56) worden geanalyseerd. Het scoren van 150 cellen per dier in ten minste 5 dieren per dosis (minder in de gelijktijdige positieve controle — zie punt 29) geeft volgens de analyse van Smith et al., 2008 (5) een voldoende statistisch onderscheidingsvermogen. Als er objectglaasjes worden gebruikt, kan dit worden gedaan door 2 of 3 objectglaasjes per monster te scoren wanneer vijf dieren per groep worden gebruikt. Er moeten meerdere gebieden van het objectglaasje worden geobserveerd bij een dichtheid die verzekert dat er geen sprake is van overlappende staarten. Scoring aan de rand van de objectglaasjes moet worden vermeden. Breuken in DNA-strengen in de komeettest kunnen worden gemeten door onafhankelijke eindpunten zoals het % DNA in de staart, de staartlengte en het staartmoment. Alle drie de metingen kunnen worden verricht als het juiste softwarematige beeldanalysesysteem wordt gebruikt. Het % DNA in de staart (ook bekend als % staartintensiteit) wordt aanbevolen voor de beoordeling en interpretatie van resultaten (12) (40) (41) (42) en is gelijk aan de DNA-fragmentintensiteit in de staart als percentage van de totale intensiteit van de cel (13). Weefselschade en cytotoxiciteit Positieve bevindingen in de komeettest zijn mogelijk niet uitsluitend te wijten aan genotoxiciteit, omdat toxiciteit voor het doelweefsel ook kan leiden tot een toename van de migratie van DNA (12) (41). Omgekeerd wordt vaak een lage tot matige cytotoxiciteit waargenomen bij bekende genotoxische stoffen (12), hetgeen laat zien dat het niet mogelijk is om enkel op basis van de komeettest DNA-migratie veroorzaakt door genotoxiciteit te onderscheiden van DNA-migratie veroorzaakt door cytotoxiciteit. Wanneer echter een toename van DNA-migratie wordt waargenomen, wordt aanbevolen een onderzoek van een of meer indicatoren van cytotoxiciteit uit te voeren, omdat dit kan helpen bij de interpretatie van de bevindingen. Een toename van de migratie van DNA met duidelijk bewijs van cytotoxiciteit moet met de nodige omzichtigheid worden geïnterpreteerd. Er zijn veel maten voor cytotoxiciteit voorgesteld. Daarvan worden histopathologische wijzigingen geacht een relevante maat voor weefseltoxiciteit te zijn. Waarnemingen zoals ontsteking, celinfiltratie, apoptotische of necrotische wijzigingen worden in verband gebracht met toenamen van DNA-migratie, maar er is, zoals de valideringsstudie van het JaCVAM (12) heeft aangetoond, geen definitieve lijst van histopathologische wijzigingen die altijd verbonden zijn aan toegenomen DNA-migratie. Wijzigingen in klinisch-chemische maten (bv. AST, ALT) kunnen ook bruikbare informatie over weefselschade verschaffen, en ook aanvullende indicatoren zoals caspase-activering, TUNEL-kleuring, annexine V-kleuring enz. kunnen in aanmerking worden genomen. Er zijn echter slechts beperkte gepubliceerde gegevens beschikbaar waarin de laatstgenoemde zijn gebruikt voor in-vivostudies, en ze zijn mogelijk niet allemaal even betrouwbaar. “Hedgehogs” (of wolken, spookcellen) zijn cellen die een microscoopbeeld laten zien dat bestaat uit een kleine of niet-bestaande kop en een grote diffuse staart, en die als zwaar beschadigde cellen worden beschouwd, hoewel de etiologie van de “hedgehogs” onzeker is (zie aanhangsel 3). Door het uiterlijk van “hedgehogs” zijn metingen van het % DNA in de staart door middel van beeldanalyse onbetrouwbaar, en daarom moeten “hedgehogs” apart worden beoordeeld. Het optreden van “hedgehogs” dient te worden opgetekend en gerapporteerd en elke relevante toename waarvan wordt gedacht dat zij te wijten is aan de teststof, moet worden onderzocht en zorgvuldig worden geïnterpreteerd. Kennis van het potentiële werkingsmechanisme van de teststoffen kan bij deze overwegingen helpen. GEGEVENS EN RAPPORTAGE Verwerking van de resultaten De experimentele eenheid is het dier en daarom moeten in tabelvorm zowel gegevens van de afzonderlijke dieren als samengevatte gegevens worden gepresenteerd. Vanwege het hiërarchische karakter van de gegevens wordt aanbevolen het mediane % DNA in de staart voor elk objectglaasje te bepalen en het gemiddelde van de mediane waarden voor elk dier te berekenen (12). Vervolgens wordt het gemiddelde van de gemiddelden voor de afzonderlijke dieren bepaald om een groepsgemiddelde te krijgen. Al deze waarden dienen in het verslag te worden opgenomen. Alternatieve benaderingen (zie punt 53) mogen worden gebruikt als ze wetenschappelijk en statistisch gerechtvaardigd zijn. De statistische analyse kan geschieden met behulp van een verscheidenheid aan benaderingen (58) (59) (60) (61). Bij de keuze van de statistische methoden die zullen worden gebruikt, moet rekening worden gehouden met de noodzaak van transformatie (bv. log of vierkantswortel) van de gegevens en/of optelling van een kleine getal (bv. 0,001) bij alle waarden (ook waarden ongelijk aan nul) om de effecten van nulwaarden voor cellen te verminderen, zoals besproken in de hierboven vermelde referenties. Gedetailleerde informatie over de analyse van interacties tussen behandeling en geslacht bij gebruik van beide geslachten en de daaropvolgende gegevensanalyse, waarbij ofwel verschillen ofwel geen verschillen worden gevonden, is opgenomen in aanhangsel 2. Gegevens over de toxiciteit en klinische tekenen moeten eveneens worden gemeld. Aanvaardbaarheidscriteria Aanvaarding van een test geschiedt aan de hand van de volgende criteria:
Beoordeling en interpretatie van resultaten Indien aan alle aanvaardbaarheidscriteria is voldaan, wordt een teststof als duidelijk positief beschouwd als:
Wanneer aan al deze criteria is voldaan, wordt de teststof in staat geacht in de onderzochte weefsels in dit testsysteem DNA-strengbreuken te veroorzaken. Zie punt 62 als slechts aan een of twee van deze criteria is voldaan. Indien aan alle aanvaardbaarheidscriteria is voldaan, wordt een teststof als duidelijk negatief beschouwd als:
De teststof wordt dan niet in staat geacht in de onderzochte weefsels in dit testsysteem DNA-strengbreuken te veroorzaken. Een duidelijk positieve of negatieve reactie behoeft niet te worden bevestigd. Indien de reactie noch duidelijk negatief noch duidelijk positief is (d.w.z. er is niet voldaan aan alle in punt 59 of punt 60 genoemde criteria) en om de biologische relevantie van een resultaat te helpen vaststellen, moeten de gegevens door een deskundige worden beoordeeld en/of moet nader onderzoek worden verricht, indien wetenschappelijk gerechtvaardigd. Het scoren van aanvullende cellen (indien van toepassing) of het uitvoeren van een herhalingsexperiment onder mogelijk geoptimaliseerde experimentele omstandigheden (bv. dosisintervallen, andere toedieningswegen, andere bemonsteringstijdstippen of andere weefsels) kan nuttig zijn. In zeldzame gevallen zal op basis van de gegevens, zelfs na nader onderzoek, niet kunnen worden geconcludeerd dat de resultaten positief of negatief zijn, en zullen ze daarom als moeilijk te interpreteren worden beschouwd. Om de biologische relevantie van een positief of moeilijk te interpreteren resultaat te beoordelen, is informatie over cytotoxiciteit voor het doelweefsel nodig (zie punten 54-55). Wanneer positieve of onduidelijke bevindingen uitsluitend worden waargenomen met duidelijk bewijs van cytotoxiciteit, wordt het onderzoek op het punt van genotoxiciteit als moeilijk te interpreteren beschouwd, tenzij er voldoende informatie is die een definitieve conclusie ondersteunt. In geval van een negatieve uitkomst van het onderzoek waarbij er bij alle geteste doses tekenen van toxiciteit zijn, kan nader onderzoek bij niet-toxische doses raadzaam zijn. Testverslag In het testverslag moet de volgende informatie worden opgenomen:
LITERATUUR
Aanhangsel 1 DEFINITIES Alkalische gelelektroforese op cellen : gevoelige techniek voor de detectie van primaire DNA-beschadigingen op het niveau van de afzonderlijke cel/nucleus. Chemische stof : een stof of een mengsel. Komeet : de vorm die nucleoïden aannemen wanneer ze worden onderworpen aan een elektroforetisch veld, vanwege de gelijkenis van deze vorm met kometen: de kop is de nucleus en de staart wordt gevormd door het DNA dat zich vanuit de nucleus verplaatst in het elektrisch veld. Kritische variabele/parameter : een protocolvariabele waarvoor een kleine wijziging een groot effect kan hebben op de conclusie van de test. Kritische variabelen kunnen weefselspecifiek zijn. Kritische variabelen mogen, vooral binnen een test, niet worden gewijzigd zonder dat zorgvuldig is nagedacht over de manier waarop de wijziging een testrespons zal beïnvloeden, bijvoorbeeld zoals blijkend uit de grootte en variabiliteit van positieve en negatieve controles. In het testverslag moeten wijzigingen in kritische variabelen die worden aangebracht tijdens de test of ten opzichte van het standaardprotocol voor het laboratorium, worden vermeld met een rechtvaardiging voor elk van de wijzigingen. Staartintensiteit of % DNA in de staart : de intensiteit van de staart van de komeet in verhouding tot de totale intensiteit (kop plus staart). Deze verhouding weerspiegelt de hoeveelheid breuken in DNA, uitgedrukt als een percentage. Teststof : alle volgens deze testmethode geteste stoffen of mengsels. UVCB : stof met een onbekende of variabele samenstelling, complexe reactieproducten en biologische materialen. Aanhangsel 2 DE FACTORIËLE OPZET VOOR HET IDENTIFICEREN VAN VERSCHILLEN TUSSEN GESLACHTEN IN DE IN-VIVOKOMEETTEST De factoriële opzet en zijn analyse Bij deze opzet worden voor elk concentratieniveau minimaal 5 mannetjes en 5 vrouwtjes getest, wat leidt tot een opzet met minimaal 40 dieren (20 mannetjes en 20 vrouwtjes, plus relevante positieve controles). De opzet, die een van de eenvoudigere factoriële opzetten is, is gelijkwaardig aan een tweewegs variantieanalyse met geslacht en concentratieniveau als hoofdeffecten. De gegevens kunnen worden geanalyseerd met veel standaard statistische softwarepakketten, zoals SPSS, SAS, STATA, Genstat, alsook met R. De analyse verdeelt de variabiliteit in de gegevensreeks in de variabiliteit tussen de geslachten, die tussen de concentraties en die verband houdend met de interactie tussen geslacht en concentratie. Elk van de termen wordt getoetst tegen een schatting van de variabiliteit tussen de duplodieren binnen de groepen van dieren van hetzelfde geslacht die dezelfde concentratie toegediend krijgen. Volledige gegevens over de onderliggende methodologie zijn te vinden in diverse statistische standaardhandboeken (zie referenties) en in de 'help'-functionaliteit van de statistische pakketten. De analyse begint met een inspectie van de interactieterm geslacht x concentratie in de ANOVA-tabel (35). Als de interactieterm niet significant is, verschaffen de gecombineerde waarden van de geslachten of van de concentratieniveaus geldige statistische toetsen tussen de niveaus op basis van de gepoolde variabiliteit binnen groepen in de ANOVA. De volgende stap in de analyse is de verdeling van de geschatte variabiliteit tussen concentraties in contrasten, die een toets op lineaire en kwadratische contrasten van de responsen over de concentratieniveaus verschaft. Wanneer de interactie tussen geslacht en concentratie significant is, kan deze term ook worden verdeeld in interactiecontrasten lineair x geslacht en kwadratisch x geslacht. Deze termen verschaffen toetsen voor de vraag of de concentratieresponsen voor de twee geslachten parallel lopen of dat de respons voor de twee geslachten gedifferentieerd is. De geschatte gepoolde variabiliteit binnen groepen kan worden gebruikt voor paarsgewijze toetsen voor het verschil tussen de gemiddelden. Deze vergelijkingen kunnen worden gemaakt tussen de gemiddelden voor de twee geslachten en tussen de gemiddelden voor de verschillende concentratieniveaus zoals voor vergelijkingen met de negatieve controleniveaus. Indien er sprake is van een significante interactie, kunnen vergelijkingen worden gemaakt tussen de gemiddelden van verschillende concentraties binnen een geslacht of tussen de gemiddelden van de geslachten bij dezelfde concentratie. Referenties Er is een groot aantal statistische handboeken waarin de theorie, opzet, methodologie, analyse en interpretatie van factoriële opzetten worden besproken, variërend van de eenvoudigste analyses met twee factoren tot de complexere vormen die worden gebruikt voor het ontwerpen van experimenten. De lijst hieronder is niet uitputtend. Enkele boeken bevatten uitgewerkte voorbeelden van vergelijkbare proefopzetten, in enkele gevallen met code voor het uitvoeren van de analyses met verschillende softwarepakketten.
Aanhangsel 3 HUIDIGE BEPERKINGEN VAN DE TEST Vanwege de huidige kennisstand gelden er voor de in-vivokomeettest verschillende beperkingen. Er wordt verwacht dat deze beperkingen zullen afnemen of nauwkeuriger zullen worden omschreven zodra er meer ervaring met de toepassing van de test is, om in een regelgevingscontext een antwoord te geven op vragen met betrekking tot de veiligheid.
Referenties
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
16) |
In deel C wordt hoofdstuk C.13 vervangen door: „C.13 Bioaccumulatie in vissen: blootstelling via water en voer INLEIDING Deze testmethode (TM) is gelijkwaardig aan testrichtlijn (TG) 305 (2012) van de OESO. Het belangrijkste doel van deze herziening van de testmethode is tweeledig. De herziening is in de eerste plaats bedoeld om een test voor bioaccumulatie via het voer (36) vast te stellen die geschikt is om het bioaccumulatiegedrag van stoffen met een zeer lage oplosbaarheid in water te bepalen. Op de tweede plaats is zij bedoeld om een testmethode op te zetten die, wanneer van toepassing, met het oog op het dierenwelzijn minder vissen gebruikt en kosteneffectiever is. In de jaren sinds de vaststelling van de geconsolideerde testmethode C.13 (1) zijn talrijke stoffen getest en is aanzienlijke ervaring opgedaan door zowel laboratoria als regelgevende autoriteiten. Dit heeft geleid tot de overtuiging dat de test minder complex kan worden gemaakt als aan specifieke criteria wordt voldaan (vgl. punt 88) en dat een getrapte aanpak mogelijk is. De ervaring leert ook dat biologische factoren zoals groei en vetgehalte van vissen grote invloed op de resultaten kunnen hebben en dat hiermee mogelijk rekening moet worden gehouden. Bovendien wordt erkend dat het testen van stoffen die zeer slecht oplosbaar zijn in water, technisch mogelijk niet haalbaar is. Voor stoffen met een zeer lage oplosbaarheid in water in het aquatische milieu is blootstelling via het water mogelijk ook van beperkt belang vergeleken met toediening via het voer. Dit heeft ertoe geleid dat een testmethode is ontwikkeld waarin vissen worden blootgesteld via hun voer (vgl. punten 7-14 en punt 97 en verder). Een validering (ringtest) van de test met blootstelling via het voer werd uitgevoerd in 2010 (51). De belangrijkste wijzigingen zijn onder meer:
Vóór de uitvoering van een van de bioaccumulatietests moet de volgende informatie over de teststof bekend zijn:
Ongeacht de gekozen blootstellingsmethode of het gekozen bemonsteringsschema, betreft deze methode een procedure voor de bepaling van het bioconcentratiegedrag van stoffen in vissen. Hoewel een doorstroomtest veruit te verkiezen is, mag eventueel ook in semistatische omstandigheden worden gewerkt, voorzover aan de validiteitscriteria wordt voldaan (vgl. punten 24 en 113). Bij blootstelling via het voer hoeft het doorstroomsysteem de concentraties van de teststof in het water niet noodzakelijk te handhaven, maar helpt het wel om adequate opgeloste zuurstofconcentraties in stand te houden, schoon water te verzekeren en invloeden van bijvoorbeeld excretieproducten te verwijderen. Ongeacht de gekozen testmethode, omvat de beschrijving van deze testmethode alle bijzonderheden die nodig zijn om de test te kunnen uitvoeren; er blijft voldoende ruimte om de proefopzet aan te passen aan de omstandigheden in het betrokken laboratorium en aan de specifieke karakteristieken van de teststof. De test voor blootstelling via het water is het meest geschikt voor toepassing op stabiele organische stoffen met een log K OW-waarde tussen 1,5 en 6,0 (13), maar mag niettemin ook worden toegepast op sterk hydrofobe stoffen (met een log K OW > 6,0), als een stabiele en volledig opgeloste concentratie van de teststof in water kan worden aangetoond. Als een stabiele concentratie van de teststof in water niet kan worden aangetoond, is een onderzoek in water niet passend en moet de stof bij vissen dus via het voer worden getest (hoewel de interpretatie en het gebruik van de resultaten van de test via het voer kunnen afhangen van het regelgevingskader). Preliminaire schattingen van de bioconcentratiefactor (BCF, soms ook genoteerd als K B) voor organische stoffen met log K OW-waarden tot ongeveer 9,0 kunnen worden verkregen door toepassing van de vergelijking van Bintein et al. (14). De preliminaire schatting van de bioconcentratiefactor voor zulke sterk hydrofobe stoffen kan hoger zijn dan de met behulp van laboratoriumexperimenten bepaalde bioconcentratiefactor in de stationaire situatie (BCFSS), vooral wanneer voor de preliminaire schatting een eenvoudig lineair model wordt gebruikt. De parameters die het bioaccumulatiepotentieel karakteriseren, omvatten de opnamesnelheidsconstante (k 1), de eliminatiesnelheidsconstanten met inbegrip van de depuratiesnelheidsconstante (k 2), de bioconcentratiefactor in de stationaire situatie (BCFSS), de kinetische bioconcentratiefactor (BCFK) en de biomagnificatiefactor (BMF) voor opnamen uit het voedsel (38). Radioactief gemerkte teststoffen kunnen de analyse van de water-, voedsel- en vismonsters vergemakkelijken en kunnen worden gebruikt om te bepalen of het noodzakelijk is eventuele metabolieten te identificeren en te kwantificeren. Indien uitsluitend de totale hoeveelheid radioactieve residuen wordt gemeten (bv. door verbranding of weefselsolubilisatie), heeft de BCF of BMF betrekking op de totale hoeveelheid van de oorspronkelijke stof, de eventueel achterblijvende metabolieten daarvan en de geassimileerde koolstof. De BCF- en BMF-waarden op basis van de totale hoeveelheid radioactieve residuen zijn derhalve mogelijk niet zonder meer vergelijkbaar met BCF- en BMF-waarden die worden verkregen door een specifieke chemische bepaling van uitsluitend de oorspronkelijke stof. Scheidingsprocedures, zoals TLC, HPLC of GC (39), kunnen worden gebruikt vóór de analyse in studies met radioactieve markers om de BCF- en BMF-waarden te bepalen op basis van de oorspronkelijke stof. Wanneer scheidingstechnieken worden toegepast, moeten de oorspronkelijke stof en de relevante metabolieten daarvan worden geïdentificeerd en gekwantificeerd (40) (vgl. punt 65) als de BCF- en BMF-waarden worden gebaseerd op de concentratie van de oorspronkelijke stof in vissen en niet op de totale hoeveelheid radioactief gemerkte residuen. Ook kan door analyse en identificatie van de residuen in de weefsels een studie van het metabolisme van de vissen of in-vivoverdelingsonderzoek worden gecombineerd met een bioaccumulatieonderzoek. De kans op metabolisme kan met geschikte instrumenten (bv. QSAR-toolbox van de OESO (15) en particuliere QSAR-programma's) worden voorspeld. Het besluit om een test met blootstelling via het water of via het voer uit te voeren en daarvoor een bepaalde opzet te gebruiken moet worden gebaseerd op de factoren in punt 3 en op het toepasselijke regelgevingskader. Voor bijvoorbeeld stoffen die een hoge log K OW hebben maar toch nog een waarneembare oplosbaarheid in water hebben met betrekking tot de gevoeligheid van de beschikbare analysetechnieken, moet in eerste instantie een test met blootstelling via het water worden overwogen. Het is echter mogelijk dat de informatie over de oplosbaarheid in water niet afdoend is voor deze hydrofobe typen stoffen, en er moet dus worden onderzocht of stabiele, meetbare opgeloste waterige concentraties kunnen worden bereid (stabiele emulsies zijn niet toegestaan) die toepasbaar zijn voor een onderzoek naar blootstelling via het water, alvorens een besluit wordt genomen over de te gebruiken testmethode (16). Het is niet mogelijk exacte bindende richtsnoeren voor de te gebruiken methode te geven op basis van drempelcriteria voor de oplosbaarheid in water en de verdelingscoëfficiënt octanol/water, omdat andere factoren (analystechnieken, afbraak, adsorptie enz.) om de hierboven genoemde redenen een aanmerkelijke invloed kunnen hebben op de toepasbaarheid van een methode. Een log K OW-waarde van meer dan 5 en een oplosbaarheid in water van minder dan ~0,01 tot 0,1 mg/l markeren het bereik van stoffen waarbij het testen door blootstelling via het water steeds moeilijker kan worden. Er moet ook rekening worden gehouden met andere factoren die van invloed kunnen zijn op de keuze van de test, met inbegrip van het potentieel van de stof om zich aan testbakken en apparaten te binden, zijn stabiliteit in waterige oplossingen versus zijn stabiliteit in visvoer (17) (18) enz. Informatie over dergelijke praktische aspecten is mogelijk beschikbaar uit andere voltooide studies in water. Nadere informatie over de beoordeling van aspecten die verband houden met de uitvoering van bioaccumulatiestudies is te vinden in de literatuur (bv. (19)). Voor stoffen waarvan de oplosbaarheid of de instandhouding van de concentraties in water alsmede de bepaling van deze concentraties geen belemmering vormen voor de verwezenlijking van een testmethode met blootstelling via het water, geniet deze methode de voorkeur om het bioconcentratiegedrag van de stof te bepalen. Er moet in alle gevallen worden nagegaan dat de via het water toe te dienen concentraties binnen het oplosbaarheidbereik in water in het testmedium liggen. Er kunnen verschillende methoden worden gebruikt om stabiele concentraties van de opgeloste teststof te handhaven, zoals het gebruik van voorraadoplossingen of passieve doseringssystemen (bv. de kolomelutiemethode), zolang kan worden aangetoond dat de concentraties stabiel kunnen worden gehouden en de testmedia niet worden gewijzigd ten opzichte van de in punt 27 aanbevolen testmedia. Voor sterk hydrofobe stoffen (log K OW > 5 en een oplosbaarheid van minder dan ~0,01-0,1 mg/l) kan testen met blootstelling via het water steeds moeilijker worden. Redenen voor beperkingen kunnen zijn dat de concentratie in het water niet op een niveau kan worden gehouden dat voldoende constant wordt geacht (bv. door sorptie aan het glas van blootstellingsvaten of door snelle opname door de vissen) of dat de toe te passen concentraties in het water zo laag zijn dat ze in hetzelfde bereik liggen als de analytische bepaalbaarheidsgrens of zelfs onder deze grens liggen (41). Voor deze zeer hydrofobe stoffen wordt de test met blootstelling via het voer aangeraden, mits de test consistent is met het toepasselijke regelgevingskader en de vereiste risicobeoordelingen. Voor oppervlakteactieve stoffen moet worden nagedacht over de vraag of de bioconcentratietest via het water haalbaar is, gezien de eigenschappen van de stoffen; anders is onderzoek via het voer waarschijnlijk geschikter. Oppervlakteactieve stoffen zijn stoffen die de grensvlakspanning tussen twee vloeistoffen verlagen. Hun amfifiele karakter (d.w.z. ze bevatten zowel een hydrofiel als een hydrofoob deel) zorgt ervoor dat ze zich ophopen op grensvlakken zoals het grensvlak tussen water en lucht, het grensvlak tussen water en voer, en glazen wanden, wat de bepaling van hun concentratie in water hindert. De test via het voer kan voor complexe mengsels met componenten met verschillende oplosbaarheidsgrenzen in water enkele blootstellingsaspecten omzeilen, in de zin dat een vergelijkbare blootstelling aan alle componenten van het mengsel waarschijnlijker is dan bij blootstelling via het water (vgl. (20)). Er zij opgemerkt dat de benadering met blootstelling via het voer een biomagnificatiefactor (BMF) voor opnamen uit het voedsel oplevert in plaats van een bioconcentratiefactor (BCF) (42). Er zijn benaderingen beschikbaar om een kinetische bioconcentratiefactor (BCFK) te schatten op basis van gegevens die in het onderzoek via het voer zijn gegenereerd (zoals besproken in aanhangsel 8), maar deze benaderingen moeten met voorzichtigheid worden gebruikt. In het algemeen veronderstellen deze benaderingen eersteordekinetiek en zijn zij uitsluitend toepasbaar op bepaalde groepen van verbindingen. Het is onwaarschijnlijk dat deze benaderingen kunnen worden toegepast voor oppervlakteactieve stoffen (zie punt 12). Een beperkte testopzet met blootstelling via het water met minder monsternamepunten om het aantal dieren en/of middelen te verlagen (vgl. punt 83 en verder) mag alleen worden toegepast op stoffen waarvoor er geen reden is te verwachten dat opname en depuratie ongeveer eersteordekinetiek zullen volgen (d.w.z. in het algemeen niet-geïoniseerde organische stoffen, vgl. punt 88). C.13 — I: Bioconcentratietest met vissen met blootstelling via het water PRINCIPE VAN DE TEST De test bestaat uit twee fasen: de blootstellingsfase (opnamefase) en de daaropvolgende depuratiefase. Gedurende de opnamefase wordt een groep vissen van dezelfde soort blootgesteld aan een of meer concentraties van de teststof, afhankelijk van de eigenschappen van de teststof (vgl. punt 49). Met de overbrenging van de vissen naar een medium dat de teststof niet bevat, wordt de depuratiefase ingeluid. Een depuratiefase is altijd vereist, tenzij de opname van de stof tijdens de opnamefase niet significant was. De concentratie van de teststof in of op de vissen (of bepaalde weefsels daarvan) wordt systematisch gedocumenteerd gedurende de beide fasen van de test. Naast de blootgestelde groep, wordt ook een controlegroep vissen gehouden onder — afgezien van de afwezigheid van de teststof — identieke omstandigheden. Op die manier kunnen de schadelijke effecten die eventueel in de bioconcentratietest worden waargenomen, worden gerelateerd aan waarnemingen op een passende controlegroep en kan een “nuleffectbepaling” van de concentraties van de teststof worden uitgevoerd. (43) In de test met blootstelling via het water duurt de opnamefase gewoonlijk 28 dagen. De fase kan worden verlengd, indien nodig (vgl. punt 18), of worden verkort als wordt aangetoond dat de stationaire toestand reeds eerder is bereikt (zie aanhangsel 1, Definities en eenheden). Een raming van de duur van de opnamefase en de tijd die nodig is om de stationaire toestand (dynamisch evenwicht) te bereiken, kan worden verkregen met behulp van de vergelijkingen in aanhangsel 5. De depuratiefase begint vervolgens wanneer de vissen niet langer worden blootgesteld aan de teststof, doordat ze worden overgebracht naar een schone bak die hetzelfde medium, maar geen teststof bevat. Zo mogelijk wordt de bioconcentratiefactor bij voorkeur berekend zowel als de verhouding van de concentratie in de vissen (C f) en in het water (C w) in de stationaire toestand (BCFSS; zie aanhangsel 1, Definities en eenheden) en als kinetische bioconcentratiefactor (BCFK; zie aanhangsel 1, Definities en eenheden), d.w.z. als de verhouding van de snelheidsconstanten k 1 (opname) en k 2 (depuratie), uitgaande van een eersteordekinetiek (44). Indien na 28 dagen nog geen stationaire toestand is bereikt, wordt de BCF berekend met behulp van de kinetische benadering (vgl. punt 38) of kan de opnamefase worden verlengd. Als dit leidt tot een onpraktisch lange opnamefase voordat de stationaire toestand wordt bereikt (vgl. punten 37 en 38, aanhangsel 5), wordt de kinetische benadering verkozen. Als alternatief kan voor zeer hydrofobe stoffen worden overwogen een onderzoek via het voer uit te voeren (45), mits de test via het voer verenigbaar is met het toepasselijke regelgevingskader. De opnamesnelheidsconstante, de depuratie- (eliminatie-)snelheidsconstante (of -constanten, indien een complexer model vereist is), de bioconcentratiefactor (stationaire toestand of kinetisch) en, zo mogelijk, een betrouwbaarheidsinterval voor elk van deze parameters worden berekend aan de hand van het model dat de gemeten concentraties van de teststof in de vissen en het water het beste beschrijft (vgl. aanhangsel 5). De toename van de massa van de vissen tijdens de test zal leiden tot een afname van de concentratie van de teststof in groeiende vissen (zogenoemde groeiverdunning) en dus zal de kinetische BCF worden onderschat als deze niet voor groei wordt gecorrigeerd (vgl. punten 72 en 73). De BCF wordt berekend aan de hand van de totale concentratie in de vis (d.w.z. per totaal versgewicht van de vis). Voor bepaalde doeleinden mogen evenwel ook specifieke weefsels of organen (bijvoorbeeld spieren, lever) worden gebruikt als de vissen groot genoeg zijn of als zij kunnen worden opgedeeld in een eetbaar (“filet”) en een niet-eetbaar (“ingewanden”) gedeelte. Aangezien er voor veel organische stoffen een duidelijk verband bestaat tussen neiging tot bioconcentratie en hydrofobiciteit, bestaat er een overeenkomstig verband tussen het vetgehalte van de in de proeven gebruikte vissen en de waargenomen bioconcentratie van die stoffen. Om deze bron van variatie in de testresultaten voor teststoffen met een hoge lipofiliciteit (d.w.z. met log K OW > 3) te verkleinen, dient de mate van bioconcentratie daarom niet alleen rechtstreeks te worden bepaald uit de studie, maar ook te worden uitgedrukt als bioconcentratie genormaliseerd naar een vis met een vetgehalte van 5 % (gebaseerd op het versgewicht van het hele lichaam). Dit is nodig om een basis te krijgen waarop resultaten voor verschillende stoffen en/of soorten proefdieren met elkaar kunnen worden vergeleken. Het cijfer van 5 % voor het vetgehalte wordt veel gebruikt, omdat dit het gemiddelde vetgehalte is van de vissen die gewoonlijk in deze testmethode worden gebruikt (21). GEGEVENS BETREFFENDE DE TESTSTOF Behalve de eigenschappen van de teststof die zijn genoemd in de Inleiding (punt 3), zijn eveneens vereist gegevens over de toxiciteit van de teststof voor de in de test gebruikte vissoort, bij voorkeur in de vorm van de asymptotische LC50 (d.w.z. tijdonafhankelijk) en/of geraamde toxiciteit van de teststof uit langlopende tests met vissen (bv. TM's C.47 (22), C.15 (23), C.14 (24)). Er dient een passende analytische methode, met bekende nauwkeurigheid, precisie en gevoeligheid, voor de kwantitatieve bepaling van de stof in de testoplossingen en in het biologisch materiaal beschikbaar te zijn, evenals een protocol voor de toebereiding en de opslag van de monsters. De analytische bepaalbaarheidsgrens van de teststof, zowel in water als in visweefsel, dient eveneens bekend te zijn. Wanneer een radioactief gemerkte teststof wordt gebruikt, dient deze van de hoogst mogelijke zuiverheid te zijn (bv. bij voorkeur > 98 %) en dient de procentuele radioactiviteit die in verband wordt gebracht met onzuiverheden bekend te zijn. GELDIGHEID VAN DE TEST Een test is uitsluitend onder de volgende voorwaarden geldig:
REFERENTIESTOFFEN Het gebruik van referentiestoffen waarvan het bioconcentratiegedrag bekend is en het metabolisme laag is, kan in sommige gevallen nuttig zijn om de experimentele procedure te toetsen (bv. wanneer een laboratorium geen eerdere ervaring met de test heeft of wanneer de experimentele omstandigheden zijn gewijzigd). BESCHRIJVING VAN DE METHODE Apparatuur Voor alle onderdelen van de installatie moet het gebruik van materialen die oplossen of sorberen, waardoor stoffen in het water worden vrijgegeven of waardoor enig ander schadelijk effect op de vissen kan worden veroorzaakt, zoveel mogelijk worden vermeden. Er kunnen normale, uit een chemisch inert materiaal vervaardigde rechthoekige of cilindervormige bakken worden gebruikt die voldoende ruimte bieden, gegeven de beoogde populatiedichtheid (aantal vissen per liter water) (vgl. punt 43). Het gebruik van slangen uit zacht plastic moet zoveel mogelijk worden vermeden. Er moeten verbindingsbuizen van polytetrafluorethyleen, roestvrij staal en/of glas worden gebruikt. De ervaring wijst uit dat het voor teststoffen met een hoge adsorptiecoëfficiënt, zoals synthetische pyretroïden, noodzakelijk kan zijn gesilaniseerd glas te gebruiken. In een dergelijk geval moet de apparatuur na gebruik worden verwijderd. Bij voorkeur worden de testsystemen vóór de introductie van de testorganismen blootgesteld aan concentraties van de teststof die in het onderzoek worden gebruikt, zolang als nodig is om het behoud van stabiele blootstellingsconcentraties aan te tonen. Water Voor de test wordt normaliter water van natuurlijke oorsprong gebruikt dat wordt verkregen uit een niet-verontreinigde bron van uniforme kwaliteit. Synthetisch water (d.w.z. gedemineraliseerd water met daaraan toegevoegd kleine hoeveelheden van specifieke nutriënten) kan echter geschikter zijn om in de loop van de tijd een uniforme kwaliteit te waarborgen. Het verdunningswater, dat wil zeggen, het water dat met de teststof vermengd wordt voordat het in de testbak wordt gebracht (vgl. punt 30), moet zodanig zijn dat de gekozen vissoort daarin gedurende de acclimatisatie en de looptijd van de test kan overleven zonder dat de specimens een abnormaal aspect of gedrag gaan vertonen. In het ideale geval wordt aangetoond dat de gekozen vissoort in het verdunningswater kan overleven, groeien en zich voortplanten (bijvoorbeeld in een laboratoriumkweek of in een toxiciteitstest die de hele levenscyclus omvat). Van het verdunningswater moeten ten minste de pH, de hardheid, het totaalgehalte aan vaste stof en het totaalgehalte aan organische koolstof (TOC (47)) worden bepaald, alsmede, zo mogelijk, het ammonium- en nitrietgehalte en de alkaliniteit en, voor mariene soorten, het zoutgehalte. De parameters die belangrijk zijn voor het optimale welzijn van de vissen zijn niet genoegzaam bekend; niettemin worden in aanhangsel 2 aanbevelingen gedaan wat betreft de maximumconcentratie van een aantal stoffen in het voor de tests gebruikte zoet en zeewater. Het verdunningswater dient gedurende de test een constante kwaliteit te vertonen. De pH-waarde dient bij aanvang van de test in het interval van 6,0 tot 8,5 te liggen en mag bovendien in de loop van de test met niet meer dan ± 0,5 pH-eenheden schommelen. Om te garanderen dat het verdunningswater de testresultaten niet noemenswaardig beïnvloedt (bijvoorbeeld door complexatie van de teststof) en geen ongunstige invloed heeft op de conditie van de vissen, moeten regelmatig monsters worden genomen en geanalyseerd, ten minste aan het begin en het einde van de test. Zware metalen (bijvoorbeeld Cu, Pb, Zn, Hg, Cd en Ni), de belangrijkste anionen en kationen (bijvoorbeeld Ca2+, Mg2+, Na+, K+, Cl- en SO4 2-), bestrijdingsmiddelen (bijvoorbeeld het totaalgehalte aan organofosforpesticiden en het totaalgehalte aan organochloorpesticiden) en het totaalgehalte aan organische koolstof en zwevende deeltjes moeten bijvoorbeeld om de drie maanden worden bepaald als van het verdunningswater bekend is dat het een relatief constante kwaliteit heeft. Indien is aangetoond dat de kwaliteit van het verdunningswater gedurende ten minste één jaar constant blijft, zijn minder frequente bepalingen en langere intervallen (bijvoorbeeld zes maanden) toelaatbaar. Het natuurlijke gehalte aan zwevende deeltjes en het totaalgehalte aan organische koolstof (TOC) van het verdunningswater dienen zo laag mogelijk te zijn om te vermijden dat de teststof aan het organisch materiaal adsorbeert waardoor de biologische beschikbaarheid ervan zou afnemen en de BCF zou worden onderschat. De maximale toelaatbare concentraties bedragen 5 mg/l voor deeltjes (droge stof, achterblijvend op een 0,45 μm filter) en 2 mg/l voor het totaal aan organische koolstof (vgl. aanhangsel 2). Desnoods moet het verdunningswater vóór gebruik worden gefilterd. Het gehalte aan organische koolstof in het testwater van de testvissen (excreta) en van de voedselresten dient zo laag mogelijk te worden gehouden (vgl. punt 46). Testoplossingen Maak een voorraadoplossing met een passende concentratie van de teststof klaar. De voorraadoplossing wordt bij voorkeur bereid door eenvoudig mengen en schudden van de teststof in het verdunningswater. Een alternatief dat in sommige gevallen passend kan zijn, is het gebruik van een doseringssysteem voor desorptie vanuit de vaste fase. Het gebruik van oplosmiddelen en dispergeermiddelen (oplosbaarheidsbevorderende agentia) verdient doorgaans geen aanbeveling (vgl. punt 25); niettemin kan het gebruik van deze stoffen aanvaardbaar zijn om een voorraadoplossing van een passende concentratie te verkrijgen, maar al het mogelijke moet worden gedaan om het gebruik van deze stoffen tot een minimum te beperken en hun kritische micelconcentratie mag niet worden overschreden (indien van toepassing). Als oplosmiddelen mogen aceton, ethanol, methanol, dimethylformamide en triëthyleenglycol worden gebruikt. Dispergeermiddelen die zijn gebruikt, zijn Tween 80, 0,01 % methylcellulose en HCO-40. De concentratie van het oplosmiddel in het uiteindelijke testmedium dient voor alle behandelingen gelijk te zijn (d.w.z. onafhankelijk van de concentratie van de teststof) en mag de overeenkomstige toxiciteitsdrempels die voor het oplosmiddel onder de testomstandigheden zijn bepaald, niet overschrijden. Het maximumniveau is een concentratie van 100 mg/l (of 0,1 ml/l). Het is onwaarschijnlijk dat een concentratie van een oplosmiddel van 100 mg/l de maximale opgeloste concentratie van de teststof die in het medium kan worden bereikt significant zal wijzigen (25). De bijdrage van het oplosmiddel en de teststof samen aan het totale organischekoolstofgehalte in het water in de testbakken moet bekend zijn. Gedurende de hele duur van de test mag het totale gehalte aan organische koolstof in de testbakken het gehalte aan organische koolstof in de teststof zelf en het oplosmiddel of het agens dat de oplosbaarheid daarvan dient te verhogen (solubisator) (48), in voorkomend geval, niet met meer dan 10 mg/l (± 20 %) overschrijden. Het gehalte aan organische stoffen kan tijdens doorstroomtests met vissen een aanmerkelijk effect hebben op de hoeveelheid vrij opgeloste teststof, vooral voor zeer lipofiele stoffen. Vaste-fase-microextractie (vgl. punt 60) kan belangrijke informatie verschaffen over de verhouding tussen gebonden en vrij opgeloste verbindingen, waarvan wordt aangenomen dat laatstgenoemde de biologisch beschikbare fractie vormen. De concentratie van de teststof dient onder de oplosbaarheidsgrens van de teststof in het testmedium te liggen, ondanks het gebruik van een oplosmiddel of solubisator. Wanneer biologisch gemakkelijk afbreekbare oplosmiddelen worden gebruikt, moet in het bijzonder worden gewaakt voor problemen die zich als gevolg van bacteriegroei in het doorstroomsysteem kunnen voordoen. Als het niet mogelijk is om zonder gebruik van een solubisator een voorraadoplossing te bereiden, moet worden nagedacht over de gepastheid van een onderzoek met blootstelling via het water in vergelijking tot blootstelling via het voer. Voor doorstroomtests is een systeem dat de voorraadoplossing van de teststof continu verdeelt en verdunt (bijvoorbeeld doseerpomp, mechanisme voor evenredige verdunning, saturatorsysteem), of een doseringssysteem voor desorptie vanuit de vaste fase vereist om ervoor te zorgen dat in de bakken met de proefdieren de gewenste testconcentratie wordt gehandhaafd. Laat bij voorkeur een verversingssnelheid van ten minste vijf bakvolumes per dag voor iedere testbak toe. Het gebruik van een doorstroomsysteem is verkieslijk, maar als dit niet mogelijk is (bijvoorbeeld omdat de gebruikte proefdieren daarvan schade ondervinden) mag een semistatisch systeem worden gebruikt op voorwaarde dat aan de validiteitscriteria wordt voldaan (vgl. punt 24). De stroomsnelheid van de voorraadoplossingen en het verdunningswater moet 48 uur vóór het begin van de test en vervolgens (gedurende de test) ten minste eenmaal per dag worden gecontroleerd. Neem in deze controle eveneens de bepaling van de stroomsnelheid door iedere testbak afzonderlijk op en zie erop toe dat de verschillen, zowel per bak als tussen de bakken onderling, niet meer dan 20 % bedragen. Keuze van de diersoort Belangrijke criteria bij de keuze van de soort zijn dat zij gemakkelijk verkrijgbaar is, dat exemplaren van de geschikte grootte beschikbaar zijn en dat de soort op een bevredigende manier in het laboratorium kan worden gehouden. Andere keuzecriteria zijn het recreatieve, commerciële of ecologische belang van de soort alsmede haar relatieve gevoeligheid, het succes waarmee zij in het verleden is gebruikt enz. In aanhangsel 3 wordt een aantal aanbevolen soorten opgesomd. Ook andere soorten mogen worden gebruikt, maar in dat geval kan het nodig zijn de testprocedure aan te passen om geschikte proefomstandigheden te creëren. Als er wordt gekozen voor een andere vissoort, moet worden aangegeven waarom er voor deze soort is gekozen en moet tevens worden gerapporteerd welke experimentele methode wordt toegepast. In het algemeen zal het gebruik van kleinere vissoorten de tijd die nodig is om een stationaire toestand te bereiken verkorten, maar mogelijk meer vissen (vismonsters) vereisen om het vetgehalte en de concentraties van de teststof in de vissen adequaat te bepalen. Bovendien kunnen verschillen in ademhalingssnelheid en metabolisme tussen jongere en oudere vissen onderlinge vergelijkingen van de resultaten van verschillende tests en testsoorten hinderen. Er zij opgemerkt dat het testen van een vissoort in een (jonge) levensfase met snelle groei de interpretatie van gegevens kan bemoeilijken. Leefomstandigheden van de vissen (relevant voor blootstelling zowel via water als via voer) Laat het visbestand gedurende ten minste twee weken acclimatiseren in water (vgl. punt 28) dat de temperatuur heeft waarbij de test zal worden uitgevoerd; verschaf continu voldoende voedsel (vgl. punt 45). Zowel het water als het voer dient van het type te zijn dat in de test gebruikt wordt. Na een aanpassingsperiode van 48 uur wordt de sterfte geregistreerd en worden de volgende criteria toegepast:
De in de tests gebruikte vissen moeten vrij zijn van waarneembare ziekten en abnormaliteiten. Zieke vissen moeten worden verwijderd. De vissen mogen in de twee weken voorafgaande aan de test of tijdens de test niet worden behandeld voor ziekten. UITVOERING VAN DE TEST Verkennende test Het verdient aanbeveling een verkennende proef uit te voeren om de proefomstandigheden bij de definitieve test (bijvoorbeeld teststofconcentraties, duur van de opname- en de depuratiefase) te optimaliseren of om te bepalen of een volledige test moet worden uitgevoerd. De opzet van de verkennende test moet zodanig zijn dat de vereiste informatie wordt verkregen. Een verkennende test kan worden overwogen als een beperkte test mogelijk voldoende is om een BCF te bepalen, of als een volledige test nodig is (vgl. punten 83-95 over de beperkte test). Blootstellingsomstandigheden Duur van de opnamefase De duur van de opnamefase kan worden geschat op basis van bestaande praktijkervaring (bijvoorbeeld gegevens uit een eerdere studie of kennis van de accumulatiesnelheid van een verwante chemische stof) of op basis van bepaalde empirische relaties, stoelend op gegevens betreffende de oplosbaarheid in water of de verdelingscoëfficiënt octanol/water van de teststof (mits de opname verloopt volgens eersteordekinetiek, vgl. aanhangsel 5). De opnamefase dient 28 dagen te duren, tenzij kan worden aangetoond dat reeds eerder een stationaire toestand wordt bereikt (zie aanhangsel 1, Definities en eenheden). Een stationaire toestand wordt bereikt wanneer in een grafiek van de concentratie van de teststof in de vissen (C f) als functie van de tijd de curve evenwijdig gaat lopen met de tijdas en drie opeenvolgende bepalingen van C f op monsters die met tussenpozen van ten minste twee dagen worden genomen, niet meer dan ± 20 % van elkaar verschillen en er bovendien in de loop van de tijd geen significante toename van C f tussen de eerste en laatste opeenvolgende bepaling is. Wanneer de analyse op samengevoegde monsters wordt uitgevoerd, zijn ten minste vier opeenvolgende bepalingen vereist. Voor teststoffen die langzaam worden opgenomen, verdient het de voorkeur om intervallen van zeven dagen te gebruiken. Indien de stationaire toestand na 28 dagen nog niet is bereikt, wordt de BCF berekend met uitsluitend de kinetische benadering, die niet vertrouwt op het al dan niet bereikt zijn van een stationaire toestand, of kan de opnamefase met 60 dagen worden verlengd, tenzij de stationaire toestand eerder wordt bereikt; gedurende de hele voortzetting van de opnamefase worden ook de metingen voortgezet. De concentratie van de teststof in de vissen aan het einde van de opnamefase moet zo hoog zijn dat k 2 betrouwbaar kan worden geschat op basis van de depuratiefase. Als er na 28 dagen geen bewijs van significante opname is, kan de test worden beëindigd. Duur van de depuratiefase Voor stoffen die eersteordekinetiek vertonen, is een tijd die half zo lang is als de opnamefase meestal voldoende voor een adequate reductie (bijvoorbeeld met 95 %) van de lichaamsconcentratie van de teststof (vgl. aanhangsel 5 voor een verklaring van deze raming). Indien de tijd die nodig is voor een vermindering met 95 % in de praktijk te lang is (bijvoorbeeld indien de depuratiefase dan meer dan twee keer zo lang als de normale duur van de opnamefase, dus meer dan 56 dagen, zou duren) mag een kortere periode worden gebruikt (bijvoorbeeld tot de concentratie van de teststof minder dan 10 % van de concentratie in de stationaire toestand bedraagt). Voor stoffen met een ingewikkelder opname- en depuratiepatroon dan kan worden beschreven met een ééncompartimentmodel dat een eersteordekinetiek vertoont, moet evenwel toch in een langere depuratiefase worden voorzien. Indien zulke ingewikkelde patronen worden waargenomen en/of verwacht, verdient het aanbeveling een biostatisticus en/of farmacokineticus te raadplegen om een juiste proefopzet te waarborgen. Wanneer de depuratieperiode wordt verlengd, kan het aantal benodigde vissen voor de monsterneming een beperkende factor worden en kunnen verschillen in groei tussen de vissen de resultaten beïnvloeden. De duur van de fase zal ook afhankelijk zijn van de tijd gedurende welke de concentratie van de teststof in de vissen boven de analytische bepaalbaarheidsgrens blijft. Aantal proefdieren Kies het aantal vissen per testconcentratie zo dat bij iedere bemonstering ten minste vier vissen per monster beschikbaar zijn. De vissen mogen uitsluitend worden gepoold wanneer analyse op basis van de afzonderlijke vissen niet haalbaar is. Als het de bedoeling is om de curve nauwkeuriger te fitten (en afgeleide parameters nauwkeurig te schatten) of als metabolismestudies noodzakelijk zijn (bv. om onderscheid te maken tussen metabolieten en de oorspronkelijke stof wanneer radioactief gemerkte teststoffen worden gebruikt), zullen meer vissen per bemonstering nodig zijn. Het vetgehalte dient te worden bepaald op hetzelfde biologisch materiaal waarop ook de concentratie van de teststof wordt bepaald. Als dit niet haalbaar is, kunnen aanvullende vissen nodig zijn (vgl. punten 56 en 57). Als volwassen (d.w.z. geslachtsrijpe) vissen worden gebruikt, mogen deze vóór de test of tijdens de test niet in een voortplantingsfase verkeren of net hebben gepaaid (d.w.z. reeds kuit of hom hebben afgezet). Tevens moet worden gerapporteerd of in het experiment mannetjes of vrouwtjes of beide worden gebruikt. Als beide geslachten worden gebruikt, moeten verschillen in groei en vetgehalte tussen de geslachten vóór aanvang van de blootstelling worden gedocumenteerd als niet-significant, in het bijzonder als wordt verwacht dat het nodig zal zijn de gegevens van mannetjes en vrouwtjes samen te voegen om aantoonbare concentraties van de teststof en/of een aantoonbaar vetgehalte te verzekeren. Selecteer voor elke test vissen met een min of meer uniform lichaamsgewicht: het gewicht van de kleinste vis mag niet minder bedragen dan twee derde van dat van de grootste. Alle vissen moeten tot dezelfde jaarklasse behoren en dezelfde oorsprong hebben. Aangezien het gewicht en de leeftijd van een vis soms een significant effect op de BCF-waarden hebben (12), moeten deze gegevens nauwkeurig worden geregistreerd. Het verdient aanbeveling kort vóór aanvang van de test een steekproef uit het visbestand te wegen om een schatting van het gemiddelde gewicht te verkrijgen (vgl. punt 61). Densiteit Gebruik een grote water/visverhouding om de vermindering van de concentratie van de teststof in het water door de introductie van de vissen bij het begin van de test zo klein mogelijk te houden en een afname van het gehalte aan opgeloste zuurstof te vermijden. Van belang is ook dat de dichtheid van de vissen op de biologische kenmerken van de gekozen soort wordt afgestemd. De aanbevolen dichtheid bedraagt normaliter 0,1-1,0 gram vis (versgewicht) per liter water per dag. Een grotere dichtheid is toelaatbaar als wordt aangetoond dat de schommelingen van de concentratie van de teststof de ± 20 %-grenzen niet overschrijden en het gehalte aan opgeloste zuurstof nooit minder bedraagt dan 60 % van het verzadigingspunt (vgl. punt 24). Houd bij de keuze van de geschikte vissendichtheid rekening met de normale biotoop van de betrokken vissoort. Zo kunnen demersale vissen bij eenzelfde watervolume in het aquarium een grotere bodemoppervlakte verlangen dan pelagische soorten. Voeding Verstrek de vissen gedurende de acclimatisatie- en de testperiode geschikt voer met een bekend vet- en totaal eiwitgehalte in een voldoende hoeveelheid om ze gezond te houden en hun lichaamsgewicht op peil te houden (enig groei is toegestaan). Geef gedurende de acclimatisatie- en de testperiode dagelijks een vaste hoeveelheid te eten, afhankelijk van de gebruikte soort, de experimentele omstandigheden en de calorische waarde van het voer (bijvoorbeeld voor regenboogforel ongeveer 1 à 2 % van hun lichaamsgewicht per dag). De dosering moet zo worden gekozen dat een snelle groei en een grote toename van het vetgehalte worden vermeden. Om de dosering constant te houden moet de hoeveelheid voer indien nodig worden herberekend, bijvoorbeeld eens per week. Voor die berekening kan het gewicht van de in iedere testbak overblijvende vissen worden geschat aan de hand van het gewicht van de vissen in de laatste uit die testbak getrokken steekproef. Het gewicht van de achterblijvende vissen zelf wordt niet bepaald. Niet opgegeten voer en uitwerpselen moeten dagelijks, korte tijd (30 minuten tot een uur) na de voedering, met behulp van een hevel uit de testbakken worden verwijderd. Die bakken moeten in de loop van de test zo schoon mogelijk worden gehouden om de concentratie van organische stoffen zo laag mogelijk te houden (vgl. punt 29), aangezien de aanwezigheid van organische koolstof de biologische beschikbaarheid van de teststof negatief kan beïnvloeden (12). Aangezien vele visvoeders uit vismeel worden vervaardigd, moet worden verzekerd dat het voer de testresultaten niet beïnvloedt en geen negatieve effecten induceert, bijvoorbeeld doordat het (sporen van) bestrijdingsmiddelen, zware metalen en/of de teststof zelf bevat. Licht en temperatuur Een fotoperiode van 12 tot 16 uur wordt aanbevolen, en de watertemperatuur (± 2 °C) moet worden afgestemd op de gekozen vissoort (vgl. aanhangsel 3). Het type belichting en de karakteristieken daarvan dienen bekend te zijn. Er dient rekening te worden gehouden met de mogelijkheid dat de teststof bij de gebruikte belichting fotochemisch in andere stoffen wordt omgezet. Door een passende belichting moet worden vermeden dat de vissen aan onnatuurlijke fotochemische omzettingsproducten worden blootgesteld. In bepaalde gevallen kan het nodig zijn de UV-straling met een golflengte van minder dan 290 nm weg te filteren. Testconcentraties De test werd oorspronkelijk opgezet voor apolaire organische stoffen. Voor dit type stoffen is blootstelling van de vissen aan één concentratie waarschijnlijk voldoende, aangezien er geen concentratie-effecten worden verwacht. Er kunnen echter twee concentraties vereist zijn voor het geldende regelgevingskader. Als andere stoffen dan apolaire organische stoffen worden getest, of als er andere aanwijzingen van mogelijke concentratieafhankelijkheid zijn, moet de test worden uitgevoerd met twee of meer concentraties. Indien slechts één concentratie wordt getest, dient een motivering voor het gebruik van één concentratie te worden gegeven (vgl. punt 79). De geteste concentratie moet zo laag zijn als praktisch en technisch mogelijk is (d.w.z. niet te dicht bij de oplosbaarheidsgrens). In sommige gevallen kan worden verwacht dat de bioconcentratie van een stof afhangt van de concentratie in het water (bv. voor metalen, waar de opname in de vis slechts gedeeltelijk kan worden gereguleerd). In dit geval moeten ten minste twee, maar bij voorkeur meer, concentraties worden getest (vgl. punt 49) die relevant zijn in het milieu. Ook voor stoffen waarvoor de geteste concentraties om praktische redenen dicht bij de oplosbaarheidsgrens moeten liggen, verdient het aanbeveling ten minste twee concentraties te testen, omdat dit inzicht in de betrouwbaarheid van de blootstellingsconcentraties kan geven. De selectie van de testconcentraties moet de in het milieu realistische concentratie, alsook de concentratie die relevant is voor het doel van de specifieke beoordeling omvatten. De concentratie(s) van de teststof moet(en) lager worden gekozen dan het niveau waarbij de teststof chronische effecten veroorzaakt of 1 % van zijn acute asymptotische LC50, binnen een band die relevant is in het milieu en ten minste een orde van grootte hoger dan de bepaalbaarheidsgrens van de stof in water voor de gebruikte analysemethode. De hoogste toegestane testconcentratie kan ook worden bepaald door de acute 96 h-LC50 te delen door een passende acuut/chronisch-verhouding (passende verhoudingen variëren bijvoorbeeld tussen ongeveer 3 voor sommige stoffen en groter dan 100 voor enkele andere). Indien een tweede concentratie wordt gebruikt, moet zij ten minste een factor tien verschillen van de hierboven genoemde concentratie. Indien dit niet mogelijk is vanwege het toxiciteitscriterium (dat de hoogste testconcentratie beperkt) en de analytische grens (die de laagste testconcentratie beperkt), kan een lagere factor dan tien worden gebruikt en moet worden overwogen een radioactief gemerkte teststof (met een zo groot mogelijkheid zuiverheid, bv. bij voorkeur > 98 %) te gebruiken. Er dient op te worden gelet dat geen van de gebruikte concentraties hoger is dan de oplosbaarheidsgrens van de teststof in de testmedia. Controles Naast de experimentele reeks dient één controlegroep te worden behandeld met verdunningswater of, in voorkomend geval (vgl. punten 30 en 31), met dat water en het oplosmiddel. Frequentie van de metingen van de waterkwaliteit In de loop van de test moeten het gehalte aan opgeloste zuurstof, de TOC, pH en temperatuur in alle test- en controlebakken worden gemeten. De totale hardheid en, voor zover relevant, het zoutgehalte moeten worden gemeten in de controlegroep(en) en in een testbak. Meet deze parameters bij de hogere (of hoogste) concentratie als twee of meer concentraties worden getest. De zuurstofconcentratie en, voor zover relevant, het zoutgehalte moeten ten minste drie keer worden gemeten tijdens de opnamefase — aan het begin, omstreeks het midden en aan het einde van die fase — en vervolgens om de week gedurende de depuratiefase. De TOC moet worden bepaald bij het begin van de opnamefase (24 uur en 48 uur vóór de vissen in de bakken worden geïntroduceerd) en vervolgens ten minste wekelijks, zowel gedurende de opname- als gedurende de depuratiefase. De temperatuur moet dagelijks worden gemeten en geregistreerd, de pH aan het begin en aan het einde van elke periode en de hardheid eenmaal in de loop van de test. Het is wenselijk de temperatuur in ten minste één bak continu te registreren. Bemonstering en analyse van de vissen en het water Bemonsteringsschema voor vissen en water Met het oog op de bepaling van de teststofconcentratie moet het water in de testbakken worden bemonsterd vóór de vissen worden geïntroduceerd en voorts zowel gedurende de opname- als gedurende de depuratiefase. Het water moet vóór de voedering en tegelijk met de vissen worden bemonsterd. Het kan nuttig zijn vaker monsters te nemen om stabiele concentraties na de introductie van de vissen te waarborgen. Gedurende de opnamefase moeten de concentraties van de teststof worden bepaald om te controleren of aan de validiteitscriteria is voldaan (punt 24). Als uit de analyse van watermonsters aan het begin van de depuratiefase blijkt dat de teststof niet wordt gedetecteerd, kan dit worden gebruikt als rechtvaardiging om test- en controlewater gedurende de resterende tijd van de depuratiefase niet te analyseren op de concentratie van de teststof. De vissen moeten ten minste vijfmaal in de loop van de opnamefase en ten minste viermaal in de loop van de depuratiefase op teststof worden bemonsterd. Omdat het in bepaalde gevallen moeilijk is op basis van dit aantal monsters een voldoende precieze schatting van de BCF-waarde te berekenen (met name wanneer er aanwijzingen bestaan dat een andere dan een simpele eersteordeopname- en depuratiekinetiek wordt gevolgd), is het raadzaam gedurende beide periodes een hogere bemonsteringsfrequentie aan te houden (vgl. aanhangsel 4). Het vetgehalte moet worden bepaald op hetzelfde biologische materiaal dat wordt gebruikt om de concentratie van de teststof te bepalen, ten minste aan het begin en aan het einde van de opnamefase en aan het einde van de depuratiefase. Als dit niet haalbaar mocht zijn, moeten ten minste drie onafhankelijke vissen worden bemonsterd om het vetgehalte op elk van deze drie tijdstippen te bepalen. Het aantal vissen per bak aan het begin van het experiment moet dienovereenkomstig worden aangepast (49). Indien in de controlevissen (d.w.z. vissen uit het visbestand) geen significante hoeveelheden van de teststof worden gedetecteerd, kunnen de controlevissen uit de test ook worden geanalyseerd op alleen het vetgehalte en kan de bepaling van de teststof in de testgroep(en) (en de daarmee samenhangende opnamesnelheidsconstante, depuratiesnelheidsconstante en BCF-waarde) worden gecorrigeerd voor wijzigingen overeenkomstig het vetgehalte van de controlegroep tijdens de test (50). Dode of zieke vissen hoeven niet te worden geanalyseerd op de teststof of hun vetgehalte. Aanhangsel 4 bevat een voorbeeld van een goed bemonsteringsschema. Uitgaande van andere hypothetische K OW-waarden ter bepaling van de blootstellingstijd die nodig is voor 95 % opname, kunnen probleemloos andere schema's worden doorgerekend (zie aanhangsel 5 voor de berekeningen). De bemonstering wordt tijdens de opnamefase voortgezet tot een stationaire toestand is bereikt (zie aanhangsel 1, Definities en eenheden) of tot de opnamefase om een andere reden wordt beëindigd (na 28 dagen of 60 dagen, vgl. punten 37 en 38). Voor de start van de depuratiefase worden de vissen naar schone bakken overgebracht. Bemonstering en monstervoorbereiding De watermonsters voor de analyse moeten bijvoorbeeld worden verkregen door afheveling uit het midden van de testbak met behulp van een slang uit inert materiaal. Noch filtratie noch centrifugatie garandeert in alle omstandigheden een volledige scheiding van de biologisch beschikbare en biologisch niet-beschikbare fractie van de teststof. Wanneer een scheidingstechniek wordt toegepast, moet in het testverslag altijd een motivering voor, of een validering van, de scheidingstechniek worden gegeven gezien de problemen met de biologische beschikbaarheid (25). Met name voor zeer hydrofobe stoffen (d.w.z. stoffen met een log K OW > 5) (12) (26), waar adsorptie aan de filermatrix of centrifugebak kan optreden, mogen deze bewerkingen niet op de monsters worden toegepast. In plaats daarvan moeten maatregelen worden getroffen om de bakken zo schoon mogelijk te houden (vgl. punt 46) en moet het totale gehalte organische koolstof (TOC) zowel gedurende de opnamefase als gedurende de depuratiefase bestendig worden gecontroleerd (vgl. punt 53). Om mogelijke problemen met verminderde biologische beschikbaarheid te voorkomen, kunnen voor slecht oplosbare en zeer hydrofobe stoffen technieken voor vaste-fase-microextractie worden gebruikt. De bemonsterde vissen worden onverwijld gedood, met behulp van de meest geschikte en humane manier (voor bepaling op de hele vis dienen geen verdere bewerkingen plaats te vinden dan de vis met water spoelen (vgl. punt 28) en droog deppen). Weeg de vis en meet zijn totale lengte (51). Voor elke vis dient het gemeten gewicht en de gemeten lengte te worden gekoppeld aan de bepaalde concentratie van de teststof (en het bepaalde vetgehalte, voor zover van toepassing), bijvoorbeeld met behulp van een unieke identificatiecode voor elke bemonsterde vis. Het verdient de voorkeur de vissen en het water onmiddellijk na de monsterneming te analyseren om afbraakprocessen en andere verliezen te vermijden. Bovendien kunnen op deze wijze naargelang de test vordert benaderde opname- en depuratiesnelheidsconstanten worden berekend. Indien de analyse meteen wordt uitgevoerd, wordt ook vermeden dat te veel tijd voorbijgaat alvorens het bereiken van het plateau (stationaire toestand) wordt geconstateerd. Indien de analyse niet onmiddellijk wordt uitgevoerd, worden de monsters op passende wijze bewaard. In dit geval moeten, vooraleer de studie wordt aangevat, gegevens worden vergaard over de geschikte wijze van bewaring voor de teststof in kwestie — bijvoorbeeld diepvriezen of bewaren bij 4 °C, wijze van extractie enz. De duur van de bewaring moet zo worden gekozen dat de stof tijdens de bewaring niet wordt afgebroken. Kwaliteit van de analysemethode Aangezien de nauwkeurigheid, de precisie en de gevoeligheid van de analysemethode ten aanzien van de teststof bepalend zijn voor de kwaliteit van de hele procedure, moet experimenteel worden gecontroleerd of de nauwkeurigheid, de precisie en de reproduceerbaarheid van de bepaling van de stof alsmede de terugvinding van de teststof in de water- en vismonsters bevredigend zijn voor de gekozen methode. Dit moet deel uitmaken van de verkennende tests. Controleer eveneens dat geen teststof aantoonbaar is in het verdunningswater. Corrigeer zo nodig de bij de test gemeten waarden van de concentratie van de teststof in het water en in de vissen aan de hand van de terugvinding en de nuleffectmetingen bij de controlegroepen. De vis- en watermonsters moeten consequent op zodanige wijze behandeld worden dat contaminatie en verliezen (bijvoorbeeld als gevolg van adsorptie aan de bemonsteringsapparatuur) zoveel mogelijk worden vermeden. Analyse van de vismonsters Indien in de test radioactief gemerkt materiaal wordt gebruikt, kan hetzij de totale hoeveelheid radioactiviteit (d.w.z. in de oorspronkelijke stof én de metabolieten daarvan) worden geanalyseerd, hetzij een zuivering worden uitgevoerd waardoor de oorspronkelijke stof afzonderlijk kan worden geanalyseerd. Indien de BCF wordt gebaseerd op de oorspronkelijke stof, moeten de belangrijkste metabolieten worden gekarakteriseerd, ten minste aan het einde van de opnamefase (vgl. punt 6). De belangrijkste metabolieten zij de metabolieten die ≥ 10 % van de totale hoeveelheid residuen in het visweefsel vertegenwoordigen, die ≥ 5 % op twee opeenvolgende bemonsteringstijdstippen vertegenwoordigen, die gedurende de hele opnamefase toenemende niveaus vertonen, en die waarvan bekend is dat ze toxicologisch van belang zijn. Indien de BCF voor de hele vis, gemeten op basis van de totale hoeveelheid radioactief gemerkte residuen, ≥ 500 is, kan het raadzaam zijn — en voor bepaalde categorieën stoffen zoals bestrijdingsmiddelen ten stelligste aan te bevelen — de metabolieten te identificeren en te kwantificeren. Kwantificering van deze metabolieten kan door sommige regelgevende autoriteiten worden geëist. Indien de afbraakproducten die 10 % of meer van de totale hoeveelheid radioactief gemerkte residuen in het visweefsel vertegenwoordigen, worden geïdentificeerd en gekwantificeerd, verdient het aanbeveling de afbraakproducten in de geanalyseerde testwatermonsters op dezelfde wijze uit te splitsen. Indien dit niet mogelijk is, moet hiervoor in het verslag een verklaring worden gegeven. De concentratie van de teststof dient normaliter voor iedere gewogen vis afzonderlijk te worden bepaald. Als zulks niet mogelijk is, mogen de gelijktijdig genomen vismonsters worden samengevoegd, maar een dergelijke samenvoeging houdt restricties in voor de statistische procedures die op de gegevens kunnen worden toegepast, dus de test moet een toereikend aantal vissen omvatten om de gewenste samenvoeging, de gewenste statistische procedure en het gewenste onderscheidingsvermogen mogelijk te maken. Referenties (27) en (28) kunnen worden gebruikt als inleiding op de relevante samenvoegingsprocedures. De BCF dient niet alleen rechtstreeks te worden bepaald uit de studie maar ook te worden uitgedrukt als BCF genormaliseerd naar een vis met een vetgehalte van 5 % (op basis van het versgewicht) (vgl. punt 21), tenzij kan worden beargumenteerd dat de teststof zich niet primair in lipiden ophoopt. Het vetgehalte van de vissen wordt zo mogelijk bij iedere bemonstering bepaald, als het kan op hetzelfde extract als datgene dat voor de bepaling van de teststof wordt gebruikt, omdat de vetten meestal toch moeten worden verwijderd alvorens het extract chromatografisch kan worden geanalyseerd. Voor de bepaling van teststoffen zijn echter vaak specifieke extractieprocedures nodig, die in tegenspraak kunnen zijn met de testmethoden voor de bepaling van het vetgehalte. In dit geval (tot geschikte niet-destructieve instrumentele methoden beschikbaar zijn) wordt aanbevolen een andere strategie te hanteren om het vetgehalte van de vissen te bepalen (vgl. punt 56). Voor de bepaling van het vetgehalte moeten geschikte methoden worden gebruikt (20). De extractietechniek met chloroform/methanol (29) kan als standaardmethode worden aanbevolen (30), maar de Smedes-methode (31) wordt als alternatieve methode aanbevolen. Deze laatste methode wordt gekenmerkt door een vergelijkbare efficiëntie van de extractie, een hoge nauwkeurigheid, het gebruik van minder toxische organische oplosmiddelen en uitvoeringsgemak. Andere methoden waarvan de nauwkeurigheid gunstig afsteekt bij die van de aanbevolen methoden mogen worden gebruikt, mits naar behoren gemotiveerd. Het is van belang dat bijzonderheden over de gebruikte methode worden verstrekt. Meting van de groei van de vissen Aan het begin van de test moeten vijf tot tien vissen uit het visbestand apart worden gewogen en moet hun totale lengte worden gemeten. Dit kunnen dezelfde vissen zijn als die voor de bepaling van het vetgehalte worden gebruikt (vgl. punt 56). Het gewicht en de lengte van de vissen uit de test- en controlegroepen die bij elke monstername worden gebruikt, moeten worden gemeten voordat het gehalte van de chemische stof of het vetgehalte wordt bepaald. De metingen van deze bemonsterde vissen kunnen worden gebruikt om het gewicht en de lengte te schatten van de vissen die in de test- en controlebakken achterblijven (vgl. punt 45). GEGEVENS EN RAPPORTAGE Verwerking van de resultaten De opnamecurve van de teststof moet worden verkregen door de concentratie in/op de vissen (of bepaalde weefsels daarvan) in de opnamefase uit te zetten tegen de tijd in een lineair coördinatenstelsel. Als de curve een plateau bereikt, d.w.z. ongeveer asymptotisch evenwijdig gaat lopen met de tijdas, wordt de BCF in de stationaire toestand (BCFSS) berekend als:
De ontwikkeling van C f kan worden beïnvloed door de groei van de vissen (vgl. punten 72 en 73). De gemiddelde blootstellingsconcentratie (Cw ) varieert in de tijd. Een tijdgewogen gemiddelde concentratie is waarschijnlijk relevanter en preciezer voor bioaccumulatiestudies, zelfs als de variatie binnen het toepasselijke geldigheidsbereik ligt (vgl. punt 24). Een tijdgewogen gemiddelde concentratie in water kan worden berekend overeenkomstig aanhangsel 5, punt 1. De kinetische bioconcentratiefactor (BCFK) wordt berekend als de verhouding k 1/k 2 van de twee kinetische eersteordesnelheidsconstanten. De snelheidsconstanten k 1 en k 2 en BCFK kunnen worden afgeleid door simultane fitting van de opname- en de depuratiefase. Als alternatief kunnen k 1 en k 2 ook sequentieel worden bepaald (zie aanhangsel 5 voor een beschrijving en vergelijking van deze methoden). De depuratiesnelheidsconstante (k 2) moet mogelijk worden gecorrigeerd voor groeiverdunning (vgl. punten 72 en 73). Indien de opname- en/of depuratiecurve duidelijk geen eersteordekinetiek vertoont, moeten complexere modellen worden gebruikt (zie de referenties in aanhangsel 5 en het ingewonnen advies van een biostatisticus en/of farmacokineticus). Gewichts- en lengtegegevens van de vissen Het versgewicht en de totale lengte van afzonderlijke vissen voor alle bemonsteringsintervallen worden voor de test- en controlegroepen tijdens de opnamefase (met inbegrip van het visbestand voor het begin van de opnamefase) en de depuratiefase in aparte tabellen geplaatst. Voor elke vis wordt het gemeten gewicht en de gemeten lengte gekoppeld aan de bepaalde concentratie van de teststof, bijvoorbeeld met behulp van een unieke identificatiecode voor elke bemonsterde vis. Voor de doeleinden van het corrigeren van kinetische BCF-waarden voor groeiverdunning geniet het gewicht de voorkeur als maat voor de groei (zie punt 73 en aanhangsel 5 voor de gebruikte methode om gegevens te corrigeren voor groeiverdunning). Correctie voor groeiverdunning en normalisatie naar vetgehalte Groei van de vissen tijdens de depuratiefase kan de concentraties van chemische stoffen in de vissen verlagen, met als effect dat de algemene depuratiesnelheidsconstante (k 2) groter is dan zij zou zijn als zij uitsluitend werd ontleend aan verwijderingsprocessen (bv. ademhaling, metabolisme, egestie). Kinetische bioconcentratiefactoren moeten worden gecorrigeerd voor groeiverdunning. Een BCFSS zal ook worden beïnvloed door groei, maar er is geen overeengekomen procedure beschikbaar om een BCFSS te corrigeren voor groei. In geval van significante groei, moet ook de BCFK, gecorrigeerd voor groei (BCFKg), worden afgeleid, aangezien deze mogelijk een relevantere maat voor de bioconcentratiefactor is. Het vetgehalte van testvissen (dat sterk samenhangt met de bioaccumulatie van hydrofiele stoffen) kan in de praktijk zoveel schommelen dat normalisatie naar een vastgesteld vetgehalte (5 % g/g) noodzakelijk is om zowel kinetische bioconcentratiefactoren als bioconcentratiefactoren in stationaire toestand op een zinvolle manier te kunnen presenteren, tenzij kan worden aangevoerd dat de teststof zich niet primair in vetten ophoopt (sommige geperfluoreerde stoffen kunnen zich bijvoorbeeld binden aan eiwitten). Vergelijkingen voor en voorbeelden van deze berekeningen zijn te vinden in aanhangsel 5. Om een kinetische BCF te corrigeren voor groeiverdunning, moet de depuratiesnelheidsconstante worden gecorrigeerd voor groei. Deze voor groei gecorrigeerde depuratiesnelheidsconstante (k 2g) wordt berekend door de groeisnelheidsconstante (k g, zoals verkregen uit de gemeten gewichtsgegevens) in mindering te brengen op de algemene depuratiesnelheidsconstante (k 2). De voor groei gecorrigeerde kinetische bioconcentratiefactor wordt vervolgens berekend door de opnamesnelheidsconstante (k 1) te delen door de voor groei gecorrigeerde depuratiesnelheidsconstante (k 2g) (vgl. aanhangsel 5). In sommige gevallen komt deze aanpak in gevaar. Zo kan de afgeleide k 2g voor zeer traag depurerende stoffen die worden getest met snel groeiende vissen bijvoorbeeld heel klein zijn, waardoor de fout in de twee snelheidsconstanten die worden gebruikt om de voor groei gecorrigeerde kinetische bioconcentratiefactor te bepalen kritisch wordt, en in sommige gevallen kunnen de schattingen van k g groter zijn dan k 2. Een alternatieve aanpak waarbij niet voor groeiverdunning hoeft te worden gecorrigeerd, maakt gebruik van depuratiegegevens met betrekking tot de massa van de teststof per vis (op basis van hele vissen) in plaats van de gebruikelijke (concentratie)gegevens met betrekking tot de massa van de teststof per massaeenheid vis. Deze aanpak kan probleemloos worden gebruikt, aangezien tests volgens deze testmethode de geregistreerde weefselconcentraties dienen te koppelen aan het gewicht van de afzonderlijke vissen. De eenvoudige procedure hiervoor is uiteengezet in aanhangsel 5. Er zij opgemerkt dat k 2 toch moet worden gerapporteerd, zelfs als deze alternatieve aanpak wordt gebruikt. Kinetische bioconcentratiefactoren en bioconcentratiefactoren in stationaire toestand moeten ook worden gerapporteerd ten opzichte van een standaardvetgehalte van de vis van 5 % (g/g), tenzij kan worden aangevoerd dat de teststof zich niet primair in vetten ophoopt. De gegevens van de concentratie in vissen, of de BCF, worden genormaliseerd volgens de verhouding tussen 5 % en het werkelijke (individuele) gemiddelde vetgehalte (in % versgewicht) (vgl. aanhangsel 5). Als de bepalingen van de chemische stof en het vetgehalte zijn uitgevoerd op dezelfde vis, moeten de genormaliseerde gegevens van het vetgehalte van de afzonderlijke vis worden gebruikt om een naar vetgehalte genormaliseerde BCF te berekenen. Indien de groei in de controlevissen en blootgestelde vissen vergelijkbaar is, kan ook alleen het vetgehalte van de controlevissen worden gebruikt om voor het vetgehalte te corrigeren (vgl. punt 56). Een methode voor het berekenen van de BCF genormaliseerd naar vetgehalte is beschreven in aanhangsel 5. Interpretatie van de resultaten De resultaten moeten met de nodige omzichtigheid worden geïnterpreteerd als de gemeten concentraties van de teststofoplossingen in de buurt liggen van de analytische aantoonbaarheidsgrens. De gemiddelde groei mag zowel in de testgroep als in de controlegroep in beginsel niet significant verschillen om toxische effecten uit te sluiten. De groeisnelheidsconstanten of de groeicurven van de twee groepen moeten worden vergeleken met behulp van een passende procedure (52). Bioconcentratiegegevens van goede kwaliteit resulteren in scherp afgetekende opname- en depuratiecurven. Voor de snelheidsconstanten moet het resultaat van een χ2 goodness-of-fit-toets een goede fit laten zien (d.w.z. klein meetfoutenpercentage (32)) voor het bioaccumulatiemodel, zodat de snelheidsconstanten als betrouwbaar kunnen worden beschouwd (vgl. aanhangsel 5). Indien meer dan één testconcentratie wordt gebruikt, behoort het verschil tussen de waarden voor de opname- c.q. de depuratieconstante als bepaald voor de testconcentraties niet meer te bedragen dan 20 % (53). Als dit niet het geval is, kan er sprake zijn van concentratieafhankelijkheid. Indien tussen de gemeten waarden van de opname- c.q. de depuratiesnelheidsconstante voor de gebruikte testconcentraties een significant verschil bestaat, moet dit worden genoteerd en moeten mogelijk verklaringen worden gesuggereerd. Bij goed opgezette studies is het 95 %-betrouwbaarheidsinterval voor de BCF's in het algemeen niet veel ruimer dan (puntschatting) ± 20 %. Indien twee of meer concentraties worden getest, worden de resultaten van beide of alle concentraties gebruikt om na te gaan of de resultaten consistent zijn en of er sprake is van concentratieafhankelijkheid. Indien slechts één concentratie wordt getest om het gebruik van dieren en/of middelen te beperken, moet een motivering voor het gebruik van slechts één concentratie worden gegeven. De resulterende BCFSS is verdacht als de BCFK significant groter is dan de BCFSS, aangezien dit een aanwijzing kan zijn dat geen stationaire toestand is bereikt of dat geen rekening is gehouden met groeiverdunning en verliesprocessen. Indien de BCFSS heel veel hoger is dan de BCFK, moet worden gecontroleerd of er fouten zijn gemaakt in de afleiding van de opname- en depuratiesnelheidsconstanten en moeten de constanten opnieuw worden berekend. Een andere fittingsprocedure kan de schatting van BCFK mogelijk verbeteren (vgl. aanhangsel 5). Testverslag Behalve de gegevens over de teststof die in punt 3 zijn genoemd, moeten in het verslag over de test de volgende gegevens worden opgenomen:
C.13 — II: Beperkte test met vissen met blootstelling via het water INLEIDING De toenemende ervaring die is opgedaan met het uitvoeren en interpreteren van de volledige test door zowel laboratoria als regelgevende instanties, laat zien dat — enkele uitzonderingen daargelaten — voor het schatten van de opname- en depuratiesnelheidsconstanten eersteordekinetiek van toepassing is. De opname- en depuratiesnelheidsconstanten kunnen derhalve worden geschat met een minimum aan monsternamepunten, en de kinetische BCF kan worden afgeleid. Het aanvankelijke doel van het onderzoeken van alternatieve opzetten voor BCF-studies was een kleine test te ontwikkelen die kan worden gebruikt als tussenstap om schattingen van de BCF op basis van K OW en QSAR's te verwerpen of te bevestigen en zo voor veel stoffen de noodzaak van een volledig onderzoek uit te sluiten, en om de kosten en het gebruik van dieren te minimaliseren door beperking van de bemonstering en van het aantal uitgevoerde analysereeksen. Terwijl in grote lijnen de opzet van de eerdere testmethode werd gevolgd om integratie van de testresultaten met bestaande BCF-gegevens mogelijk te maken en om de uitvoering van de test en de interpretatie van de gegevens te vergemakkelijken, was het doel BCF-schattingen te verschaffen die voldoende nauwkeurig en precies zijn om op basis van risicobeoordelingen besluiten te kunnen nemen. Veel overwegingen van de volledige test zijn ook van toepassing op de beperkte test, bijvoorbeeld de validiteitscriteria (vgl. punt 24) en de beëindiging van een test als aan het einde van de opnamefase geen significante opname wordt gezien (vgl. punten 16 en 38). Stoffen die in aanmerking kunnen komen voor de beperkte testopzet, moeten tot het algemene domein behoren waarvoor deze testmethode werd ontwikkeld, d.w.z. apolaire organische stoffen (vgl. punt 49). Als er enige aanwijzing is dat de te testen stof afwijkend gedrag kan vertonen (bv. een duidelijke afwijking van eersteordekinetiek), moet voor regelgevingsdoeleinden een volledige test worden uitgevoerd. Gewoonlijk duurt de beperkte test niet korter dan de standaard BCF-test, maar worden er wel minder monsters van vissen genomen (zie aanhangsel 6 voor de grondgedachte). De depuratieperiode kan voor snel depurerende stoffen echter worden verkort om te voorkomen dat de concentraties in de vissen vóór het einde van de test dalen tot onder de aantoonbaarheidsgrens/bepaalbaarheidsgrens. Een beperkte blootstellingstest met vissen met één concentratie kan worden gebruikt om vast te stellen of een volledige test noodzakelijk is, en als de resulterende gegevens die worden gebruikt voor het berekenen van de snelheidsconstanten en de BCF robuust zijn (vgl. punt 93), kan worden afgezien van de volledige test als de resulterende BCF sterk verschilt van de wettelijke waarden van belang. In sommige gevallen kan het voordelig zijn om de beperkte testopzet met meer dan één testconcentratie uit te voeren bij wijze van oriënterende test om vast te stellen of schattingen van de BCF voor een stof concentratieafhankelijk zijn. Indien uit de schattingen van de BCF op basis van de beperkte test concentratieafhankelijkheid blijkt, moet de volledige test worden uitgevoerd. Indien de schattingen van de BCF op basis van zo'n beperkte test niet concentratieafhankelijk blijken te zijn, maar de resultaten niet als afdoend worden beschouwd, kan een eventuele daaropvolgende volledige test worden uitgevoerd met één concentratie, zodat minder dieren worden gebruikt dan in een volledige test met twee (of meer) concentraties. Stoffen die voor de beperkte test in aanmerking kunnen komen moeten:
BEMONSTERINGSSCHEMA VOOR STUDIES VOLGENS DE BEPERKTE OPZET Bemonstering van de vissen De bemonstering van de vissen wordt beperkt tot vier monsternamepunten:
Bemonstering water Voor de beperkte opzet wordt water bemonsterd als in een volledige studie (vgl. punt 54) of ten minste vijf keer gelijkelijk verdeeld over de opnamefase, en wekelijks in de depuratiefase. Wijzigingen in de opzet Rekening houdend met de eigenschappen van de teststof, geldige QSAR-voorspellingen en het specifieke doel van de studie, kunnen enkele wijzigingen in de opzet van de studie worden overwogen:
Berekeningen De grondgedachte van deze aanpak is dat de bioconcentratiefactor in een volledige test kan worden bepaald als een bioconcentratiefactor in stationaire toestand (BCFSS) door de verhouding tussen de concentratie van de teststof in het weefsel van de vissen en de concentratie van de teststof in het water te berekenen, of door de kinetische bioconcentratiefactor (BCFK) te berekenen als de verhouding tussen de opnamesnelheidsconstante k 1 en de depuratiesnelheidsconstante k 2. De BCFK is ook geldig als tijdens de opname geen concentratie van de stof in stationaire toestand wordt bereikt, mits die opname- en depuratieprocessen ongeveer eersteordekinetiek vertonen. Als absoluut minimum zijn twee gegevenspunten vereist om de opname- en depuratiesnelheidsconstanten te schatten, één aan het einde van de opnamefase (d.w.z. het begin van de depuratiefase) en één aan het einde (of na een aanzienlijk deel) van de depuratiefase. Het tussenliggende monsternamepunt wordt aanbevolen ter controle van de opname- en depuratiekinetiek (57). Zie de aanhangsels 5 en 6 voor de berekeningen. Interpretatie van de resultaten Ga na dat depuratiefase langer is dan één halveringsperiode, om de geldigheid en informatieve waarde van de test te kunnen beoordelen. Ook moet de BCFKm (kinetische BCF afgeleid uit een beperkte test) worden vergeleken met de beperkte BCFSS-waarde (dat wil zeggen, de BCFSS zoals berekend aan het einde van de opnamefase, onder de aanname dat de stationaire toestand is bereikt; dit kan slechts een aanname zijn, want het aantal monsternamepunten is onvoldoende om dit aan te tonen). Indien de BCFKm kleiner is dan de beperkte BCFSS, is de beperkte BCFSS de waarde die de voorkeur geniet. Indien BCFKm kleiner is dan 70 % van de beperkte BCFSS, zijn de resultaten niet geldig en moet een volledige test worden uitgevoerd. Als de beperkte test een BCFKm oplevert in de buurt van een waarde die wettelijk van belang is, moet een volledige test worden uitgevoerd. Als het resultaat ver van een waarde van wettelijk belang ligt (ruim erboven of ruim eronder), is een volledige test mogelijk niet nodig, of kan een volledige test met één concentratie worden uitgevoerd, indien deze door het toepasselijke regelgevingskader wordt geëist. Indien na een beperkte test met één concentratie een volledige test noodzakelijk wordt bevonden, kan deze met een tweede concentratie worden uitgevoerd. Indien de resultaten consistent zijn, kan worden afgezien van een verdere volledig test met een andere concentratie, aangezien de bioconcentratie van de stof waarschijnlijk niet concentratieafhankelijk zal zijn. Indien de resultaten van de beperkte test, uitgevoerd met twee concentraties, geen concentratieafhankelijkheid laten zien, kan een volledige test met slechts één concentratie worden uitgevoerd (vgl. punt 87). Testverslag In het verslag over de beperkte test moet alle informatie worden opgenomen die verlangd wordt voor een verslag van een volledige test (vgl. punt 81), behalve de informatie die niet kan worden verstrekt (d.w.z. een curve die de tijd die nodig is om de stationaire toestand te bereiken laat zien, en de bioconcentratiefactor in de stationaire toestand; in plaats van laatstgenoemde moet de beperkte BCFss worden gegeven). In het verslag moeten tevens de motivering voor het gebruik van de beperkte test en de resulterende BCFKm worden opgenomen. C.13 — III: Bioaccumulatietest met vissen met blootstelling via het voer INLEIDING De in deze paragraaf beschreven methode moet worden gebruikt voor stoffen waarvoor de methode met blootstelling via het water niet praktisch is (bijvoorbeeld omdat geen stabiele, meetbare concentraties in het water in stand kunnen worden gehouden, of omdat het niet mogelijk is om binnen 60 dagen na de blootstelling adequate lichaamsconcentraties te bereiken; zie de eerdere paragrafen over de methode met blootstelling via het water). Men moet evenwel beseffen dat het eindpunt van deze test een biomagnificatiefactor (BMF) voor opnamen uit het voedsel zal zijn in plaats van een bioconcentratiefactor (BCF)( (58) ). In mei 2001 werd op de SETAC Europe-conferentie in Madrid een nieuwe methode gepresenteerd voor het testen van de bioaccumulatie van organische stoffen die slecht oplosbaar zijn in water (36). Dit werk bouwde voort op verschillende gerapporteerde bioaccumulatiestudies in de literatuur waarin een doseringsmethode met verrijkt voer werd gebruikt (bv. (37)). Begin 2004 werd bij een PBT-werkgroep van de EU een ontwerpprotocol (38) ingediend voor het meten van het bioaccumulatiepotentieel van organische stoffen die slecht oplosbaar zijn in water en waarvoor de standaard bioconcentratiemethode met blootstelling via het water niet praktisch is, tezamen met een ondersteunend achtergronddocument (39). Als verdere motivering voor de methode werd genoemd dat de potentiële blootstelling aan dergelijke slecht oplosbare stoffen (d.w.z. log K OW >5) in het milieu grotendeels via het voedsel kan plaatsvinden (vgl. (40) (41) (42) (43) (44)). In sommige gepubliceerde verordeningen inzake chemische stoffen wordt daarom verwezen naar blootstellingstests via het voer (59). Men moet evenwel beseffen dat blootstelling via de waterfase in de hier beschreven methode zorgvuldig wordt vermeden, zodat een uit deze testmethode verkregen BMF-waarde niet zonder meer kan worden vergeleken met een BMF-waarde uit een veldonderzoek (waarin blootstelling via het water en via het voedsel kunnen zijn gecombineerd). Deze paragraaf van de onderhavige testmethode is gebaseerd op dit protocol (38) en is een nieuwe methode die niet voorkwam in de eerdere versie van TM C.13. Deze alternatieve test maakt het mogelijk om blootstelling via het voer rechtstreeks onder gecontroleerde laboratoriumomstandigheden te onderzoeken. Potentiële onderzoekers dienen de punten 1 tot en met 14 van deze testmethode te raadplegen voor informatie over de situaties waarin de test met blootstelling via het voer kan worden verkozen boven de test met blootstelling via het water. Informatie over de verschillende overwegingen met betrekking tot stoffen wordt er uiteengezet en dient in overweging te worden genomen alvorens een test wordt uitgevoerd. Het gebruik van radioactief gemerkte stoffen kan worden overwogen, waarvoor dezelfde overwegingen gelden als voor de methode met blootstelling via het water (vgl. punten 6 en 65). De methode via het voer kan worden gebruikt om in één test meerdere stoffen te testen, mits aan bepaalde criteria wordt voldaan; hierop wordt nader ingegaan in punt 112. Voor de eenvoud wordt de methode hier beschreven voor een test waarin slechts één teststof wordt gebruikt. De test via het voer is in veel opzichten, natuurlijk uitgezonderd de blootstellingsroute, gelijk aan de methode met blootstelling via het water. Veel aspecten van de hier beschreven methode vertonen daarom een overlap met de methode met blootstelling via het water die in de vorige paragraaf is beschreven. Er worden zo veel mogelijk kruisverwijzingen naar relevante punten in de vorige paragraaf gegeven, maar een zekere mate van duplicatie is onvermijdelijk ten behoeve van de leesbaarheid en begrijpelijkheid. PRINCIPE VAN DE TEST Er kan gebruik worden gemaakt van doorstroom- of semistatische omstandigheden (vgl. punt 4); doorstroomomstandigheden worden aanbevolen om de potentiële blootstelling aan de teststof via het water als gevolg van eventuele desorptie van verrijkt voer of fecaliën te beperken. De test bestaat uit twee fasen: de opnamefase (voer verrijkt met de teststof) en de depuratiefase (schoon, onbehandeld voer) (vgl. punt 16). In de opnamefase ontvangt een ”testgroep” van vissen dagelijks een vastgesteld voer bestaande uit een commercieel visvoer van bekende samenstelling dat is verrijkt met de teststof. In het ideale geval consumeren de vissen al het aangeboden voedsel (vgl. punt 141). De vissen ontvangen vervolgens gedurende de depuratiefase het zuivere, onbehandelde commerciële visvoer. Net als bij de methode met blootstelling via het water kunnen, indien nodig, meerdere testgroepen worden gebruikt die voer krijgen dat met verschillende concentraties van de teststof is verrijkt, maar voor de meeste zeer hydrofobe organische teststoffen volstaat één testgroep (vgl. punten 49 en 107). Indien semistatische omstandigheden worden gebruikt, moeten de vissen aan het einde van de opnamefase naar een nieuw medium en/of een nieuwe testbak worden overgebracht (indien het medium en/of de gebruikte apparatuur in de opnamefase door uitloging verontreinigd is met de teststof). De concentraties van de teststof in de vissen worden in beide fasen van de test gemeten. Naast de groep vissen die het verrijkte voer krijgt (de testgroep), wordt onder identieke omstandigheden ook een controlegroep vissen gehouden die op dezelfde wijze wordt gevoederd, behalve dat het commerciële visvoer voor de controlegroep niet wordt verrijkt met de teststof. Deze controlegroep maakt het mogelijk achtergrondniveaus van de teststof in niet-blootgestelde vissen te kwantificeren en dient ter vergelijking voor eventueel met de behandeling samenhangende negatieve effecten die in de testgroep(en) worden waargenomen (60). De controlegroep maakt het ook mogelijk groeisnelheidsconstanten tussen groepen te vergelijken om te controleren dat vergelijkbare hoeveelheden van het aangeboden voedsel zijn geconsumeerd (bij het geven van verklaringen voor verschillende groeisnelheidsconstanten moet ook rekening worden gehouden met mogelijke verschillen in smakelijkheid tussen de voeders; vgl. punt 138). Het is belangrijk dat de test- en controlegroepen tijdens de opnamefase en de depuratiefase voer met een gelijke voedingswaarde krijgen. Uit de ervaring van de ontwikkelaars van de methode is gebleken dat een opnamefase van 7-14 dagen in het algemeen voldoende is (38) (39). Dit bereik zou de kosten van het uitvoeren van de test tot een minimum moeten beperken en tegelijk voor de meeste stoffen voldoende blootstelling verzekeren. In sommige gevallen kan de opnamefase echter worden verlengd (vgl. punt 127). Tijdens de opnamefase zal de concentratie van de teststof in de vissen mogelijk niet de stationaire toestand bereiken, en daarom worden de gegevensverwerking en de resultaten van deze methode gewoonlijk gebaseerd op een kinetische analyse van residuen in weefsels. (Noot: Vergelijkingen voor het schatten van de tijd die nodig is om een stationaire toestand te bereiken, kunnen hier op dezelfde wijze worden toegepast als voor de test met blootstelling via het water — zie aanhangsel 5.) De depuratiefase vangt aan wanneer de vissen voor het eerst niet-verrijkt voer krijgen, en duurt doorgaans niet meer dan 28 dagen of totdat de teststof niet meer kan worden gekwantificeerd in de hele vissen, indien dat tijdstip vroeger is. De depuratiefase kan worden verkort of worden verlengd tot meer dan 28 dagen, afhankelijk van de veranderingen in de gemeten concentraties van de chemische stof en in de grootte van de vissen in de loop van de tijd. Deze methode maakt het mogelijk de voor de stof specifieke halveringsperiode (t 1/2, uit de depuratiesnelheidsconstante k 2) te bepalen, alsmede het rendement van de assimilatie (absorptie via de darm; a), de kinetische biomagnificatiefactor voor opnamen uit het voedsel (BMFK), de voor groei gecorrigeerde kinetische biomagnificatiefactor voor opnamen uit het voedsel (BMFKg), en de voor vetgehalte gecorrigeerde (61) kinetische biomagnificatiefactor voor opnamen uit het voedsel (BMFKL) (en/of de voor groei en vetgehalte gecorrigeerde kinetische biomagnificatiefactor voor opnamen uit het voedsel, BMFKgL) voor de teststof in de vissen. Net als voor de methode met blootstelling via het water, zal de toename van de massa van de vissen tijdens de test leiden tot een afname van de concentratie van de teststof in groeiende vissen en zal de (kinetische) BMF dus worden onderschat als deze niet voor groei wordt gecorrigeerd (vgl. punten 162 en 163). Als wordt geraamd dat in de opnamefase de stationaire toestand werd bereikt, kan bovendien een indicatieve BMF in de stationaire toestand worden berekend. Er zijn benaderingen beschikbaar die het mogelijk maken een kinetische bioconcentratiefactor (BCFK) te schatten op basis van gegevens die in het onderzoek via het voer zijn gegenereerd (bv. (44) (45) (46) (47) (48)). De voor- en nadelen van deze benaderingen worden besproken in aanhangsel 8. De test is in de eerste plaats opgezet voor apolaire organische stoffen die slecht oplosbaar zijn in water en die in vissen ongeveer eersteordeopname- en depuratiekinetiek vertonen. Indien een stof wordt getest die niet ongeveer eersteordeopname- en depuratiekinetiek vertoont, moeten complexere modellen worden gebruikt (zie de referenties in aanhangsel 5) en moet advies van een biostatisticus en/of farmacokineticus worden ingewonnen. De BMF wordt normaal gesproken bepaald met behulp van een bepaling van het gehalte van de teststof in de hele vis (op basis van het versgewicht). Indien relevant voor de doelstellingen van het onderzoek, kunnen specifieke weefsels (bv. spier, lever) worden bemonsterd, als de vis kan worden opgedeeld in een eetbaar en een niet-eetbaar gedeelte (vgl. punt 21). Bovendien kan het maag-darmkanaal worden verwijderd en apart worden geanalyseerd om de bijdrage aan de concentraties in de hele vissen te bepalen voor monsternamepunten aan het einde van de opnamefase en rond het begin van de depuratiefase, of in het kader van een massabalansaanpak. Het vetgehalte van bemonsterde hele vissen moet worden gemeten zodat de concentraties kunnen worden gecorrigeerd voor vetgehalte, rekening houdend met het vetgehalte van zowel het voer als de vissen (vgl. punten 56 en 57 en aanhangsel 7). Het gewicht van de bemonsterde vissen moet worden gemeten en geregistreerd en worden gekoppeld aan de concentratie van de teststof die voor die vis is bepaald (bv. met behulp van een unieke identificatiecode voor elke bemonsterde vis), om de groei te kunnen berekenen die tijdens de test kan optreden. De totale lengte van de vissen moet eveneens worden gemeten, indien dit mogelijk is (62). Gewichtsgegevens zijn ook nodig om de BCF tijdens de depuratiefase van de test via het voer te schatten. GEGEVENS BETREFFENDE DE TESTSTOF Er dient informatie over de teststof zoals beschreven in de punten 3 en 22 beschikbaar te zijn. Gewoonlijk is een analysemethode voor concentraties van de teststof in water niet nodig; vereist zijn methoden met een geschikte gevoeligheid voor het meten van concentraties in visvoer en visweefsel. De methode kan worden gebruikt om meerdere stoffen in één test te testen. De teststoffen moeten echter met elkaar verenigbaar zijn, zodat ze niet met elkaar reageren of een andere chemische identiteit krijgen nadat ze als verrijking aan het visvoer zijn toegevoegd. Het is de bedoeling dat de gemeten resultaten voor elk van de stoffen die tezamen worden getest, niet sterk afwijkt van de resultaten die zouden zijn verkregen als voor elke teststof een afzonderlijke test was uitgevoerd. Door middel van voorafgaande analyses moet worden vastgesteld dat elke teststof kan worden teruggevonden in het meervoudig verrijkte voer en het visweefselmonster met i) hoge recovery's (bv. > 85 % van de nominale concentratie) en ii) de vereiste gevoeligheid voor de tests. De totale dosis van de samen geteste stoffen moet lager zijn dan de gecombineerde concentratie die toxische effecten zou kunnen veroorzaken (vgl. punt 51). Bij de proefopzet moet bovendien rekening worden gehouden met mogelijke negatieve effecten in vissen en het risico van interactie-effecten (bv. metabolische effecten) die verband houden met het gelijktijdig testen van meerdere stoffen. Het gelijktijdig testen van ioniseerbare stoffen moet worden vermeden. Op het punt van blootstelling is de methode ook geschikt voor complexe mengsels (vgl. punt 13, hoewel dezelfde beperkingen voor de analyse zullen gelden als voor andere methoden). GELDIGHEID VAN DE TEST Voor een valide test moet aan de volgende voorwaarden worden voldaan (vgl. punt 24):
REFERENTIESTOFFEN Als een laboratorium de test niet eerder heeft uitgevoerd of als er sprake is van materiële wijzigingen (bijvoorbeeld een verandering van visstam of leverancier, een andere vissoort, een significante verandering in de grootte van de vissen, het visvoer of de verrijkingsmethode enz.), wordt aanbevolen een technische bekwaamheidsstudie uit te voeren met behulp van een referentiestof. De referentiestof wordt vooral gebruikt om vast te stellen of de techniek voor het verrijken van het voer geschikt is om maximale homogeniteit en biologische beschikbaarheid van de teststoffen te verzekeren. Een voorbeeld van een referentiestof die is gebruikt in geval van apolaire hydrofobe stoffen, is hexachloorbenzeen (HCB), maar andere stoffen waarvoor betrouwbare gegevens over opname en biomagnificatie beschikbaar zijn, moeten worden overwogen vanwege de gevaarlijke eigenschappen van HCB (63). Indien een referentiestof is gebruikt, moeten in het verslag van de test basisgegevens over de referentiestof worden opgenomen als voor teststoffen, met inbegrip van de naam, de zuiverheid, het CAS-nummer, de structuur en toxiciteitsgegevens (indien beschikbaar) van de referentiestof (vgl. punten 3 en 22). BESCHRIJVING VAN DE METHODE Apparatuur Materialen en apparatuur moeten worden gebruikt zoals beschreven in de methode met blootstelling via het water (vgl. punt 26). Voor de test moet een doorstroom- of statisch verversingssysteem worden gebruikt dat een voldoende hoeveelheid verdunningswater naar de testbakken doet stromen. De stroomsnelheden moeten worden genoteerd. Water Het testwater moet worden gebruikt zoals beschreven in de methode met blootstelling via het water (vgl. punten 27-29). Het testmedium moet worden gekarakteriseerd als beschreven en de kwaliteit ervan moet gedurende de test constant blijven. Het natuurlijke gehalte aan zwevende deeltjes en het totaalgehalte aan organische koolstof dienen vóór aanvang van de test zo laag mogelijk te zijn (≤ 5 mg/l deeltjes; ≤ 2 mg/l totaalgehalte aan organische koolstof). TOC hoeft uitsluitend vóór de test te worden gemeten, in het kader van de karakterisering van het testwater (vgl. punt 53). Voer Het is aan te bevelen een in de handel verkrijgbaar visvoer (zwevend en/of langzaam zinkend voer in korrelvorm) te gebruiken dat is gekarakteriseerd op het punt van ten minste zijn eiwit- en vetgehalte. Het voer moet bestaan uit korrels van uniforme grootte om de efficiëntie van de blootstelling via het voer te vergroten, dat wil zeggen, dat de vis meer van het voedsel zal eten in plaats van dat hij de grotere stukken zal opeten en de kleinere zal laten liggen. De korrels moeten een grootte hebben die geschikt is voor de grootte van de vissen bij aanvang van de test (voor vissen met een totale lengte van 3 tot 7 cm kan bijvoorbeeld voer worden gebruikt met een korreldiameter van ongeveer 0,6-0,85 mm, en voor vissen met een totale lengte van 6 tot 12 cm voer met een korreldiameter van 0,85-1,2 mm). De korrelgrootte kan aan het begin van de depuratiefase worden aangepast, afhankelijk van de groei van de vissen. Een voorbeeld van een geschikte samenstelling van het voer, zoals in de handel verkrijgbaar, wordt gegeven in aanhangsel 7. Bij de ontwikkeling van deze methode zijn veelal testvoeders met een totaal vetgehalte van 15 tot 20 % (g/g) gebruikt. Visvoer met zo'n hoge vetconcentratie is in sommige gebieden mogelijk niet verkrijgbaar. In dergelijke gevallen kunnen studies met een lagere vetconcentratie in het voer worden uitgevoerd, en indien nodig kan de dosering worden aangepast om de vissen gezond te houden (op basis van voorbereidende tests). Het totale vetgehalte van de voeders voor de test- en controlegroepen moet vóór aanvang van de test en aan het einde van de opnamefase worden gemeten en geregistreerd. In het verslag van de studie moeten de bijzonderheden over de bepaling van het nutriënten-,vocht-, vezel- en asgehalte, en indien mogelijk mineralen en residuen van pesticiden (bv. “standaard” prioritaire verontreinigende stoffen), worden opgenomen die door de leverancier van het commerciële voer worden verstrekt. Wanneer het voer wordt verrijkt met teststof, moet al het mogelijk worden gedaan om te verzekeren dat het voer gedurende de test homogeen is. Bij de selectie van de concentratie van de teststof in het voer voor de testgroep moet rekening worden gehouden met de gevoeligheid van de analysetechniek, de toxiciteit (NOEC, indien bekend) en relevante fysisch-chemische gegevens van de teststof. Indien een referentiestof wordt gebruikt, wordt de referentiestof bij voorkeur toegevoegd in een concentratie van ongeveer 10 % van de concentratie van de teststof (of in elk geval zo laag als praktisch haalbaar is), rekening houdend met de gevoeligheid van de analyse (voor bijvoorbeeld hexachloorbenzeen is een concentratie van 1-100 μg/g in het voer aanvaardbaar bevonden; vgl. (47) voor meer informatie over de rendementen van de assimilatie van HCB). De teststof kan op verschillende manieren als verrijking aan het visvoer worden toegevoegd, afhankelijk van de fysische kenmerken en oplosbaarheid van de stof (zie aanhangsel 7 voor meer bijzonderheden over verrijkingsmethoden):
In enkele gevallen, bijvoorbeeld in geval van minder hydrofobe teststoffen die met grotere waarschijnlijkheid uit het voer zullen desorberen, kan het nodig zijn de bereide voederkorrels een coating te geven met een kleine hoeveelheid mais/visolie (zie punt 142). In zulke gevallen moet het controlevoer op dezelfde wijze worden behandeld en moet het uiteindelijke bereide voer worden gebruikt voor de meting van de vetconcentratie. Indien een referentiestof wordt gebruikt, moeten de resultaten van de referentiestof vergelijkbaar zijn met gegevens in de literatuur van onderzoeken die onder min of meer dezelfde omstandigheden en met een vergelijkbare dosering zijn uitgevoerd (vgl. punt 45) en moeten parameters die specifiek zijn voor de referentiestof, voldoen aan de relevante criteria in punt 113 (3e, 4e en 5e gedachtestreepje). Indien een olie of een oplosmiddel als draagvloeistof voor de teststof wordt gebruikt, moet dezelfde hoeveelheid van hetzelfde medium (exclusief de teststof) door het controlevoer worden gemengd, zodat het controlevoer gelijkwaardig blijft aan het verrijkte voer. Het is belangrijk dat de test- en controlegroepen tijdens de opnamefase en de depuratiefase voer met een gelijke voedingswaarde krijgen. Het verrijkte voer moet worden bewaard onder omstandigheden waarbij de stabiliteit van de teststof in het voermengsel behouden blijft (bv. gekoeld) en deze omstandigheden moeten worden gerapporteerd. Keuze van de vissoort In de test via het voer mogen vissoorten worden gebruikt die zijn gespecificeerd voor blootstelling via het water (vgl. punt 32 en aanhangsel 3). Vóór de publicatie van deze testmethode zijn in bioaccumulatiestudies met organische stoffen via het voer veelal regenboogforel (Oncorhynchus mykiss), karper (Cyprinus carpio) en Amerikaanse dikkop-elrits (“fathead minnow”, Pimephales promelas) gebruikt. De proefdiersoort moet een eetpatroon hebben dat leidt tot snelle consumptie van de toegediende portie voedsel om ervoor te zorgen dat factoren die van invloed zijn op de concentratie van de teststof in het voer (bv. uitloging naar het water en de mogelijkheid van blootstelling via het water), tot een minimum worden beperkt. Er moeten vissen worden gebruikt met een grootte en gewicht die binnen het aanbevolen bereik liggen (vgl. aanhangsel 3). De vissen mogen niet zo klein zijn dat analyses op individuele basis worden bemoeilijkt. Het testen van soorten in een levensfase met snelle groei kan de interpretatie van de gegevens bemoeilijken, en een hoge groeisnelheid kan de berekening van het rendement van de assimilatie beïnvloeden (64). Leefomstandigheden van de vis De criteria met betrekking tot acclimatisering, sterfte en de aanvaardbaarheid van ziekte zijn hetzelfde als voor de methode met blootstelling via het water voordat de test wordt uitgevoerd (vgl. punten 33-35). UITVOERING VAN DE TEST Voorstudie en bereikbepalingstest Voorafgaande analyses zijn noodzakelijk om terugvinding van de stof in verrijkt voer/verrijkt visweefsel aan te tonen. Een bereikbepalingstest om een geschikte concentratie in het voer te selecteren is niet altijd nodig. Om aan te tonen dat geen negatieve effecten worden waargenomen en om de smakelijkheid van het verrijkte voer, de gevoeligheid van de analysemethode voor visweefsel en voer, en de selectie van geschikte doseringen en bemonsteringsintervallen tijdens de depuratiefase enz., te beoordelen, kunnen voorafgaande voederexperimenten worden uitgevoerd, maar dit is niet verplicht. Een voorafgaande studie kan waardevol zijn om de aantallen vissen te ramen die nodig zullen zijn voor de bemonstering tijdens de depuratiefase. Dit kan leiden tot een aanzienlijke daling van het gebruikte aantal vissen, vooral voor teststoffen die bijzonder ontvankelijk zijn voor metabolisme. Blootstellingsomstandigheden Duur van de opnamefase In het algemeen is een opnamefase van 7-14 dagen voldoende, gedurende welke één groep vissen dagelijks het controlevoer krijgt en een andere groep vissen dagelijks het testvoer in een vaste dosering afhankelijk van de proefdiersoort en de experimentele omstandigheden, bv. 1-2 % van het lichaamsgewicht (versgewicht) in geval van regenboogforel. De dosering moet zo worden gekozen dat een snelle groei en een grote toename van het vetgehalte worden vermeden. Indien nodig, kan de opnamefase worden verlengd op basis van de praktische ervaring uit eerdere studies of kennis van de opname/depuratie van de teststof (of een analoge stof) in vissen. Het begin van de test is het tijdstip van de eerste verstrekking van verrijkt voer. Een experimentele dag loopt vanaf het tijdstip van de voedselverstrekking tot kort voor het tijdstip van de volgende voedselverstrekking (bv. één uur). De eerste experimentele dag van de opnamefase begint dus op het tijdstip van de eerste verstrekking van verrijkt voer en eindigt kort vóór de tweede verstrekking van verrijkt voer. In de praktijk eindigt de opnamefase kort vóór (bv. één uur) de eerste verstrekking van niet-verrijkt voer, omdat de vissen in de tussenliggende 24 uur verrijkt voer zullen blijven verteren en de teststof zullen blijven absorberen. Het is belangrijk ervoor te zorgen dat voor de analysemethode een voldoende hoge (niet-toxische) lichaamsconcentratie van de teststof wordt bereikt, zodat tijdens de depuratiefase een daling van ten minste een orde van grootte kan worden gemeten. In speciale gevallen kan een verlengde opnamefase (van maximaal 28 dagen) met aanvullende bemonstering worden gebruikt om inzicht te krijgen in de opnamekinetiek. Tijdens de opname mag de concentratie in de vissen geen stationaire toestand bereiken. Vergelijkingen voor het ramen van de tijd die nodig is om de stationaire toestand te bereiken, als indicatie voor de waarschijnlijke tijdsduur die nodig is om significante concentraties in de vissen te bereiken, kunnen hier worden toegepast zoals voor de test met blootstelling via het water (vgl. aanhangsel 5). In sommige gevallen is mogelijk bekend dat de opname van de teststof in de vissen gedurende 7-14 dagen onvoldoende zal zijn om bij de gebruikte concentratie in het voer een concentratie in de vissen te bereiken die hoog genoeg is om tijdens de depuratie een daling van ten minste een orde van grootte te bepalen, hetzij door geringe analytische gevoeligheid hetzij door een laag rendement van de assimilatie. In dergelijke gevallen kan het nuttig zijn de initiële voederfase te verlengen tot meer dan 14 dagen of moet, in het bijzonder voor zeer omzetbare stoffen, een hogere concentratie in het voer worden overwogen. Er moet echter voor worden gezorgd dat de lichaamsconcentratie tijdens de opname niet hoger wordt dan de (geschatte) chronische concentratie zonder waargenomen effecten (NOEC) in visweefsel (vgl. punt 138). Duur van de depuratiefase De depuratiefase duurt gewoonlijk niet langer dan 28 dagen en begint zodra de testgroep vissen na de opnamefase zuiver, onbehandeld voer verstrekt krijgt. De depuratie begint met de eerste verstrekking van “niet-verrijkt” voer, in plaats van onmiddellijk na de laatste verstrekking van “verrijkt” voer, aangezien de vissen in de tussenliggende 24 uur het voedsel zullen blijven verteren en de teststof zullen blijven absorberen, zoals werd opgemerkt in punt 126. Het eerste monster in de depuratiefase wordt daarom kort voor de tweede verstrekking van niet-verrijkt voer genomen. Deze depuratiefase is bedoeld om stoffen te detecteren met een potentiële halveringsperiode van niet meer dan 14 dagen, wat overeenkomst met de halveringsperiode van bioaccumulerende stoffen (65), zodat een fase van 28 dagen twee halveringsperioden van zulke stoffen bestrijkt. In geval van zeer sterk bioaccumulerende stoffen kan het nuttig zijn de depuratiefase te verlengen (indien de noodzaak daarvan is aangetoond door voorafgaande tests). Indien een stof zo traag wordt gedepureerd dat in de depuratiefase geen exacte halveringsperiode kan worden bepaald, kan de informatie voor beoordelingsdoeleinden toch voldoende zijn om te duiden op een hoog bioaccumulatieniveau. Omgekeerd, als een stof zo snel wordt gedepureerd dat geen betrouwbare tijdstip-nul-concentratie (concentratie aan het einde van de opname/begin van de depuratie, C 0,d) en k 2 kunnen worden afgeleid, kan een conservatieve schatting van k 2 worden gemaakt (vgl. aanhangsel 7). Indien analyses van vissen op eerdere momenten (bv. 7 of 14 dagen) laten zien dat de stof tot onder de bepaalbaarheidsgrens is gedepureerd voordat de volledige periode van 28 dagen is verstreken, kunnen de latere bemonsteringen worden geschrapt en kan de test worden beëindigd. In enkele gevallen zal aan het einde van de opnameperiode (of bij de tweede bemonstering in de depuratiefase) geen meetbare opname van de teststof hebben plaatsgevonden. Als kan worden aangetoond dat: i) wordt voldaan aan de geldigheidscriteria in punt 113, en ii) het gebrek aan opname niet te wijten is aan een andere tekortkoming van de test (bv. duur van de opname niet lang genoeg, gebrekkige techniek voor het verrijken van het voer met als gevolg een slechte biologische beschikbaarheid, gebrek aan gevoeligheid van de analysemethode, geen voedselverbruik door de vissen enz.), kan de studie mogelijk worden beëindigd zonder dat deze opnieuw hoeft te worden uitgevoerd met een langere opnameduur. Indien uit voorafgaand onderzoek is gebleken dat hiervan sprake kan zijn, kan het aan te bevelen zijn om als onderdeel van een “massabalansaanpak” fecaliën, indien mogelijk, te analyseren op onverteerde teststof. Aantal proefdieren Net als bij de test met blootstelling via het water moeten vissen met een min of meer uniform lichaamsgewicht en min of meer uniforme lichaamslengte worden geselecteerd, waarbij het gewicht van de kleinste vis niet minder mag bedragen dan twee derde van dat van de grootste (vgl. punten 40-42). Het totale aantal vissen voor de studie moet worden geselecteerd op basis van het bemonsteringsschema (een minimum van één monster aan het einde van de opnamefase en vier tot zes monsters gedurende de depuratiefase, maar afhankelijk van de duur van de fasen), rekening houdend met de gevoeligheid van de analysetechniek, de concentratie die waarschijnlijk aan het einde van de opnamefase wordt bereikt (op basis van eerder opgedane kennis) en de duur van de depuratiefase (indien deze kan worden geraamd op grond van eerder opgedane kennis). Bij elke bemonstering moeten vijf tot tien vissen worden bemonsterd, waarbij de groeiparameters (gewicht en totale lengte) worden gemeten voordat de concentratie van de teststof en het vetgehalte worden bepaald. Door de inherente verschillen in de grootte, groeisnelheid en fysiologie tussen de vissen en de waarschijnlijke variatie in de hoeveelheid toegediend voer die elke vis consumeert, moeten op elk bemonsteringstijdstip ten minste vijf vissen uit de testgroep en vijf vissen uit de controlegroep worden bemonsterd om de gemiddelde concentratie en de variabiliteit daarin goed te kunnen vaststellen. De variabiliteit tussen de gebruikte vissen zal waarschijnlijk meer bijdragen aan de algemene ongecontroleerde variabiliteit in de test dan de variabiliteit die inherent is aan de gebruikte analysemethoden, en rechtvaardigt in sommige gevallen derhalve het gebruik van maximaal tien vissen per monsternamepunt. Als de achtergrondconcentraties van de teststof in de controlevissen aan het begin van de depuratie echter niet meetbaar zijn, kan chemische analyse van slechts twee of drie controlevissen op uitsluitend het laatste bemonsteringstijdstip voldoende zijn, zolang op alle monsternamepunten wel het gewicht en de totale lengte van een steekproef van achterblijvende controlevissen wordt bepaald (zodanig dat uit de testgroep en controlegroep hetzelfde aantal wordt bemonsterd voor groei). De vissen moeten worden bewaard en moeten individueel worden gewogen (zelfs als het nodig blijkt de resultaten van de monsters later samen te voegen) en hun totale lengte moet worden gemeten. Voor een standaardtest met bijvoorbeeld een depuratiefase van 28 dagen met vijf depuratiemonsters komt dit neer op een totaal van 59-120 vissen uit de testgroep en 50-110 vissen uit de controlegroep, aannemende dat de analysetechniek voor de stof toelaat het vetgehalte op basis van dezelfde vissen te bepalen. Indien de bepaling van het vetgehalte niet op dezelfde vissen kan worden uitgevoerd als de bepaling van de concentratie van de teststof, en indien het ook niet haalbaar is om uitsluitend controlevissen voor de bepaling van het vetgehalte te gebruiken (vgl. punt 56), zijn 15 vissen extra nodig (drie uit het visbestand aan het begin van de test, drie uit de controlegroep en drie uit de testgroep aan het begin van de depuratie, en drie uit de controlegroep en drie uit de testgroep aan het einde van het experiment). Aanhangsel 4 bevat een voorbeeld van een bemonsteringsschema met aantallen vissen. Densiteit Er moeten even grote water/visverhoudingen worden gebruikt als voor de methode met blootstelling via het water (vgl. punten 43 en 44). Hoewel het aantal vissen per liter water in deze test niet van invloed is op de blootstellingsconcentraties, is het aan te bevelen 0,1-1,0 g vis (versgewicht) per liter water per dag te gebruiken om adequate opgeloste zuurstofconcentraties in stand te houden en stress bij de testorganismen tot een minimum te beperken. Testvoer en voeding Gedurende de acclimatiseringsperiode moet de vissen een passend voer worden verstrekt zoals hierboven beschreven (punt 117). Indien de test wordt uitgevoerd onder doorstroomomstandigheden, moet de doorstroming tijdens het voeren van de vissen worden opgeschort. Gedurende de test moet het voer voor de testgroep voldoen aan de hierboven beschreven voorwaarden (punten 116-121). Er moet niet alleen rekening worden gehouden met stofspecifieke factoren, analytische gevoeligheid, verwachte concentratie in het voer onder milieuomstandigheden en chronische toxiciteitsniveaus/lichaamsconcentratie, maar bij de selectie van de beoogde concentratie van de verrijking moet ook rekening worden gehouden met de smakelijkheid van het voedsel (zodat de vissen niet weigeren het voedsel te eten). De nominale concentratie van de verrijking met de teststof moet in het verslag worden gedocumenteerd. De ervaring heeft geleerd dat 1-1000 μg/g een praktisch werkbereik voor verrijkingsconcentraties is voor teststoffen die geen specifiek toxisch mechanisme vertonen. Voor stoffen die via een niet-specifiek mechanisme werken, mogen de niveaus van de residuen in weefsels niet hoger zijn dan 5 μmol/g vet, aangezien hogere niveaus aan residuen waarschijnlijk chronische effecten veroorzaken (19) (48) (50) (66). Voor andere stoffen dienen negatieve effecten als gevolg van geaccumuleerde blootstelling te worden voorkomen (vgl. punt 127). Dit geldt in het bijzonder indien gelijktijdig meer dan één stof wordt getest (vgl. punt 112). De aangewezen hoeveelheid teststof kan op drie manieren als verrijking aan het visvoer worden toegevoegd, zoals beschreven in punt 119 en aanhangsel 7. De gebruikte methoden en procedures voor het verrijken van het voer moeten in het verslag worden gedocumenteerd. De controlevissen krijgen onbehandeld voer verstrekt, dat een gelijke hoeveelheid niet-verrijkt oliemedium bevat als het verrijkte voer voor de opnamefase, indien olie wordt gebruikt, of dat is behandeld met “zuiver” oplosmiddel indien bij de bereiding van het voer voor de testgroep een oplosmiddelmedium wordt gebruikt. De concentratie van de teststof in het behandelde en het onbehandelde voer moet vóór het begin en aan het einde van de opnamefase ten minste in drievoud worden bepaald. Na de blootstelling aan het behandelde voer (opnamefase) krijgen de vissen (beide groepen) onbehandeld voer verstrekt (depuratiefase). De vissen krijgen een vaste rantsoen (afhankelijk van de soort; bijvoorbeeld ongeveer 1-2 % van het lichaamsgewicht (versgewicht) per dag in het geval van regenboogforel). De dosering moet zo worden gekozen dat een snelle groei en een grote toename van het vetgehalte worden vermeden. De exacte dosering die tijdens het experiment wordt ingesteld, moet worden geregistreerd. De eerste voedselverstrekking moet worden gebaseerd op de geplande metingen van het gewicht van het visbestand vlak vóór het begin van de test. De hoeveelheid voer moet bij elke bemonstering worden aangepast aan het versgewicht van de bemonsterde vissen om rekening te houden met groei tijdens het experiment. Het gewicht en de lengte van de vissen in de test- en controlebakken kan worden geraamd uit het gewicht en de totale lengte van de vissen die bij elke bemonstering worden gebruikt; de vissen die in de test- en controlebakken achterblijven, worden niet gewogen of gemeten. Het is belangrijk gedurende het experiment dezelfde vastgestelde dosering te handhaven. Bij de verstrekking van voer moet erop worden gelet dat de vissen al het aangeboden voedsel zichtbaar consumeren, om te verzekeren dat in de berekeningen de juiste ingestiesnelheden worden gebruikt. Bij de selectie van een dosering die garandeert dat alle voedsel van een dagelijkse voedselverstrekking wordt geconsumeerd, moet rekening worden gehouden met voorafgaande voederexperimenten en eerder opgedane ervaring. Indien voedsel consequent niet volledig worden opgegeten, kan het raadzaam zijn de dosis te spreiden over een extra voederperiode op elke experimentele dag (bijvoorbeeld door voedselverstrekking eenmaal daags te vervangen door verstrekking van de helft van de dosis tweemaal daags). Indien dit noodzakelijk is, moet de tweede voedselverstrekking plaatsvinden op een vast tijdstip dat zodanig is dat de tijd tot de bemonstering van de vissen maximaal is (de tijd voor de tweede voedselverstrekking wordt bijvoorbeeld bepaald op een tijdstip in de eerste helft van een experimentele dag). Hoewel de vissen het voedsel gewoonlijk snel consumeren, is het toch belangrijk ervoor te zorgen dat de teststof aan het voedsel geadsorbeerd blijft. Er moet zoveel mogelijk worden gezorgd dat de teststof niet uit het voedsel in het water wordt gedispergeerd, waardoor de vissen zowel via het water als via het voer aan concentraties van de teststof zouden kunnen worden blootgesteld. Dit kan worden bereikt door niet-opgegeten voedsel (en fecaliën) binnen één uur, maar bij voorkeur binnen 30 minuten na de voedselverstrekking uit de test- en controlebakken te verwijderen. Daarnaast kan een systeem worden gebruikt waarin het water voortdurend via een actief koolstoffilter wordt gereinigd om 'opgeloste' verontreinigende stoffen te absorberen. Doorstroomsystemen kunnen helpen om snel voedseldeeltjes en opgeloste stoffen weg te spoelen (67). In sommige gevallen kan een enigszins gewijzigde methode om voedsel te verrijken helpen om dit probleem te verminderen (zie punt 119). Licht en temperatuur Net als voor de methode met blootstelling via het water (vgl. punt 48), wordt een fotoperiode van 12 tot 16 uur aanbevolen en een temperatuur (± 2 °C) die geschikt is voor de gebruikte proefdiersoort (vgl. aanhangsel 3). Het type belichting en de karakteristieken van de belichting dienen bekend te zijn en te worden gedocumenteerd. Controles Er moet één controlegroep worden gebruikt met vissen die hetzelfde voedsel krijgen als de testgroep, maar zonder dat daarin de teststof aanwezig is. Indien een olie- of oplosmiddelmedium wordt gebruikt om het voer voor de testgroep te verrijken, moet het voedsel voor de controlegroep op precies dezelfde manier worden behandeld, maar zonder aanwezigheid van de teststof, zodat de voeders van de testgroep en controlegroep gelijkwaardig zijn (vgl. punten 121 en 139). Frequentie van de metingen van de waterkwaliteit De voorwaarden beschreven in de methode met blootstelling via het water zijn ook hier van toepassing, behalve dat de TOC uitsluitend vóór de test hoeft te worden gemeten in het kader van de karakterisering van het testwater (vgl. punt 53). Bemonstering en analyse van de vissen en het voer Analyse van de voermonsters Monsters van de test- en controlevoeders moeten ten minste vóór het begin en aan het einde van de opnamefase en ten minste in drievoud worden geanalyseerd op de concentratie van de teststof en op het vetgehalte. In het verslag moeten de analysemethoden en procedures om de homogeniteit van het voer te waarborgen worden beschreven. De teststofconcentratie in de monsters wordt bepaald met behulp van de erkende en gevalideerde methode. Er moet een voorstudie worden uitgevoerd om de bepaalbaarheidsgrens, het percentage recovery, interferenties en de analytische variabiliteit in de beoogde monstermatrix vast te stellen. Indien een radioactief gemerkt materiaal wordt getest, moeten vergelijkbare overwegingen als die voor de methode met blootstelling via het water worden gemaakt, waarbij analyse van het water wordt vervangen door analyse van het voer (vgl. punt 65). Analyse van de vissen Bij elke bemonstering van vissen worden 5-10 vissen uit de blootstellings- en controlegroepen bemonsterd (in sommige gevallen kunnen de aantallen controlevissen worden verlaagd; vgl. punt 134). De bemonsteringen moeten op elke experimentele dag op hetzelfde tijdstip (ten opzichte van het voedertijdstip) plaatsvinden en wel op een zodanig tijdstip dat de waarschijnlijkheid dat voedsel tijdens de opnamefase en het eerste begin van de depuratiefase in de darm achterblijft, tot een minimum wordt beperkt, teneinde foute bijdragen aan de totale concentraties van de teststof te voorkomen (dat wil zeggen, bemonsterde vissen moeten aan het einde van een experimentele dag worden verwijderd, in gedachten houdend dat een experimentele dag begint op het tijdstip van voedselverstrekking en eindigt op het tijdstip van de volgende voedselverstrekking, ongeveer 24 uur later; de depuratie begint bij de eerste verstrekking van niet-verrijkt voedsel; vgl. punt 128). Het eerste monster in de depuratiefase (genomen kort voor de tweede verstrekking van niet-verrijkt voer) is van belang omdat extrapolatie naar één dag vóór deze meting wordt gebruikt om de tijdstip-nul-concentratie te schatten (C 0,d, de concentratie in de vissen aan het einde van de opnamefase/begin van de depuratiefase). Facultatief kan het maag-darmkanaal van de vissen worden verwijderd en apart worden geanalyseerd aan het einde van de opnamefase en op dag 1 en dag 3 van de depuratiefase. Bij elke bemonstering moeten de vissen uit beide testbakken worden verwijderd en op dezelfde wijze worden behandeld als beschreven in de methode met blootstelling via het water (vgl. punten 61-63). Concentraties van de teststof in hele vissen (versgewicht) worden ten minste aan het einde van de opnamefase en gedurende de depuratiefase in zowel de controle- als de testgroep gemeten. Het is aan te bevelen gedurende depuratiefase vier tot zes monsternamepunten te gebruiken (bv. 1, 3, 7, 14 en 28 dagen). Facultatief kan een aanvullend monsternamepunt na 1-3 dagen opname worden opgenomen om nog dicht bij het begin van de blootstellingsperiode uit de lineaire fase van de opname voor de vissen het rendement van de assimilatie te schatten. Er zijn twee belangrijke afwijkingen van het schema: i) indien de opnamefase is verlengd om de opnamekinetiek te onderzoeken, zullen er gedurende de opnamefase aanvullende monsternamepunten zijn en zullen er dus meer vissen moeten worden opgenomen (vgl. punt 126); ii) indien de studie aan het einde van de opnamefase is beëindigd omdat er geen meetbare opname was (vgl. punt 131). De individuele vissen die worden bemonsterd, moeten worden gewogen (en hun totale lengte moet worden gemeten) zodat de groeisnelheidsconstanten kunnen worden bepaald. De concentraties van de stof in specifiek visweefsel (eetbare en niet-eetbare gedeelten) kunnen ook aan het einde van de opnamefase en op geselecteerde depuratietijdstippen worden gemeten. Indien een radioactief gemerkt materiaal wordt getest, gelden vergelijkbare overwegingen als voor de methode met blootstelling via het water, waarbij analyse van het water wordt vervangen door analyse van het voer (vgl. punt 65). Voor het periodiek gebruik van een referentiestof (vgl. punt 25) geniet het de voorkeur de concentraties in de testgroep te meten aan het einde van de opnamefase en op alle depuratietijdstippen die voor de teststof zijn gespecificeerd (hele vissen); concentraties in de controlegroep hoeven alleen aan het einde van de opnamefase te worden bepaald (hele vissen). In bepaalde situaties (bijvoorbeeld indien de analysemethoden voor de teststof en referentiestof niet verenigbaar zijn, zodat meer vissen nodig zijn om het bemonsteringsschema uit te voeren) kan als volgt een andere aanpak worden gebruikt om het vereiste aantal extra vissen tot een minimum te beperken. Concentraties van de referentiestof worden tijdens de depuratie uitsluitend gemeten op dag 1 en dag 3 en op nog twee monsternamepunten, die zodanig zijn gekozen dat voor de referentiestof betrouwbare schattingen van de tijdstip-nul-concentratie (C 0,d) en k 2 kunnen worden verkregen. Indien mogelijk, moet het vetgehalte van de afzonderlijke vissen worden bepaald bij elke bemonstering, of ten minste aan het begin en het einde van de opnamefase en aan het einde van de depuratiefase (vgl. punten 56 en 67). Afhankelijk van de analysemethode (zie punt 67 en aanhangsel 4) kunnen voor de bepaling van het vetgehalte en de concentratie van de teststof mogelijk dezelfde vissen worden gebruikt. Dit heeft de voorkeur, omdat dit het aantal vissen tot een minimum beperkt. Mocht dit niet mogelijk zijn, dan kan dezelfde aanpak worden gebruikt als hierboven is beschreven voor de methode met blootstelling via het water (zie punt 56 voor opties voor deze alternatieve vetmeting). In het verslag moet worden opgenomen welke methode werd gebruikt om het vetgehalte te kwantificeren. Kwaliteit van de analysemethode Er moeten experimentele controles worden uitgevoerd om de specificiteit, nauwkeurigheid, precisie en reproduceerbaarheid van de voor de stof specifieke analysetechniek, alsook recovery's van de teststof uit zowel het voedsel als de vissen te verzekeren. Meting van de groei van de vissen Aan het begin van de test moet een steekproef van vissen uit het visbestand worden gewogen (en moet hun totale lengte worden gemeten). Deze vissen moeten kort (bv. één uur) voor de eerste verstrekking van verrijkt voer worden bemonsterd en worden toegewezen aan experimentele dag 0. Het aantal vissen voor deze bemonstering moet ten minste hetzelfde zijn als dat voor de bemonsteringen tijdens de test. Sommige van deze vissen kunnen dezelfde zijn als gebruikt voor de bepaling van het vetgehalte vóór het begin van de opnamefase (vgl. punt 153). Op elk bemonsteringstijdstip worden de vissen eerst gewogen en wordt hun lengte gemeten. Voor elke vis dient het gemeten gewicht (en de gemeten lengte) te worden gekoppeld aan de bepaalde concentratie van de teststof (en het bepaalde vetgehalte, voor zover van toepassing), bijvoorbeeld met behulp van een unieke identificatiecode voor elke bemonsterde vis. De metingen van deze bemonsterde vissen kunnen worden gebruikt om het gewicht (en de lengte) te schatten van de vissen die in de test- en controlebakken achterblijven. Evaluatie van het experiment De sterfte moet dagelijks worden geobserveerd en geregistreerd. Er moeten aanvullende observaties voor negatieve effecten worden verricht en geregistreerd, bijvoorbeeld voor afwijkend gedrag of afwijkende pigmentatie. Vissen worden als dood beschouwd indien er geen ademhaling meer is en geen reactie op een lichte mechanische stimulus meer kan worden waargenomen. Elke dode of duidelijk stervende vis moet worden verwijderd. GEGEVENS EN RAPPORTAGE Verwerking van de resultaten De testresultaten worden gebruikt om de depuratiesnelheidsconstante (k 2) af te leiden als functie van het totale versgewicht van de vissen. De groeisnelheidsconstante, k g, op basis van de gemiddelde gewichtstoename van de vissen, wordt berekend en gebruikt om de voor groei gecorrigeerde depuratiesnelheidsconstante, k 2g, te berekenen, indien van toepassing. Daarnaast moeten het rendement van de assimilatie (a, absorptie uit de darm), de kinetische biomagnificatiefactor (BMFK) (indien nodig gecorrigeerd voor groei, BMFKg), de voor vetgehalte gecorrigeerde waarde (BMFKL of BMFKgL, indien gecorrigeerd voor groeiverdunning) en de dosering worden gerapporteerd. Indien een schatting kan worden gemaakt van de tijd die nodig is om in de opnamefase de stationaire toestand te bereiken (bv. 95 % van de stationaire toestand of t 95 = 3,0/k 2), kan ook een schatting van de BMF in stationaire toestand (BMFSS) worden opgenomen (vgl. punten 105 en 106 en aanhangsel 5), indien de t 95-waarde aangeeft dat mogelijk stationaire omstandigheden zijn bereikt. Op deze BMFSS moet dezelfde correctie voor het vetgehalte worden toegepast als op de kinetisch afgeleide BMF (BMFK), zodat een voor vetgehalte gecorrigeerde waarde, BMFSSL, wordt verkregen (er zij opgemerkt dat er geen overeengekomen procedure beschikbaar is voor het corrigeren van een BMF in stationaire toestand voor groeiverdunning). In aanhangsel 7 worden formules en voorbeeldberekeningen gegeven. Er zijn benaderingen die het haalbaar maken om een kinetische bioconcentratiefactor (BCFK) te schatten uit gegevens die in het onderzoek via het voer zijn gegenereerd. Dit wordt besproken in aanhangsel 8. Gewichts- en lengtegegevens van de vissen Het versgewicht en de lengte van afzonderlijke vissen voor alle tijdsperioden worden apart in tabellen geplaatst voor de testgroep en controlegroep en voor alle bemonsteringsdagen tijdens de opnamefase (visbestand voor het begin van de opname; controlegroep en testgroep voor einde van de opname en, indien uitgevoerd, de vroege fase (bv. dag 1-3 van de opname) en depuratiefase (bv. dagen 1, 2, 4, 7, 14 en 28, voor de controlegroep en testgroep)). Voor de doeleinden van het corrigeren voor groeiverdunning geniet het gewicht de voorkeur als maat voor de groei. Zie de punten 162 en 163 en aanhangsel 5 voor de gebruikte methode(n) voor het corrigeren van gegevens voor groeiverdunning. Gegevens over de concentratie van de teststof in de vissen Metingen van residuen van de teststof in afzonderlijke vissen (of in samengevoegde vismonsters indien metingen bij afzonderlijke vissen niet mogelijk zijn), uitgedrukt in concentratie ten opzichte van het versgewicht (g/g), worden voor de testvissen en controlevissen voor de afzonderlijke bemonsteringstijdstippen in tabellen geplaatst. Indien voor elke bemonsterde vis een bepaling van het vetgehalte is uitgevoerd, kunnen afzonderlijke voor het vetgehalte gecorrigeerde concentraties, uitgedrukt in vetconcentratie (g/g vet), worden afgeleid en in een tabel worden opgenomen.
Depuratiesnelheid en biomagnificatiefactor Om uit de gegevens de biomagnificatiefactor te berekenen, moet eerst het rendement van de assimilatie (absorptie van teststof via de darm, α) worden verkregen. Daarvoor moet vergelijking A7.1 in aanhangsel 7 worden gebruikt, waarvoor de afgeleide concentratie in de vissen op tijdstip nul van de depuratiefase (C 0,d), de (algemene) depuratiesnelheidsconstante (k 2), de concentratie in het voedsel (C voedsel), de voedselingestiesnelheidsconstante (I) en de duur van de opnameperiode (t) bekend moeten zijn. De helling en het intercept van het lineaire verband tussen de natuurlijke logaritme van de concentratie en de depuratietijd worden gerapporteerd als de algemene depuratiesnelheidsconstante (k 2 = helling) en tijdstip-nul-concentratie (C 0,d = eintercept), zoals hierboven. De afgeleide waarden moeten worden gecontroleerd op hun biologische aannemelijkheid (het rendement van de assimilatie als fractie mag bijvoorbeeld niet groter dan 1 zijn). I wordt berekend door de massa van het voer te delen door de massa van de vissen die elke dag worden gevoederd (indien ze 2 % van hun lichaamsgewicht aan voer krijgen, zal I gelijk zijn aan 0,02). Het kan echter nodig zijn de in de berekening gebruikte dosering aan te passen voor de groei van de vissen (dit kan geschieden met de bekende groeisnelheidsconstante om het gewicht van de vissen op elk tijdstip tijdens de opnamefase te schatten; vgl. aanhangsel 7). In gevallen waarin k 2 en C 0,d niet kunnen worden afgeleid omdat bijvoorbeeld de concentraties voor de tweede bemonstering tijdens de depuratie zijn gedaald tot onder de aantoonbaarheidsgrens, kan een conservatieve schatting van k 2 worden gemaakt en een “bovengrens” voor BMFk worden geschat (vgl. aanhangsel 7). Wanneer het rendement van de assimilatie (α) is verkregen, kan de biomagnificatiefactor worden berekend door α te vermenigvuldigen met de ingestiesnelheidsconstante (I) en te delen door de (algemene) depuratiesnelheidsconstante (k 2). De voor groei gecorrigeerde biomagnificatiefactor wordt op dezelfde manier berekend, maar met gebruik van de voor groei gecorrigeerde depuratiesnelheidsconstante (k 2g; vgl. punten 162 en 163). Een alternatieve schatting van het rendement van de assimilatie kan worden afgeleid als een weefselanalyse werd verricht op de vissen die werden bemonsterd in de vroege, lineaire fase van de opnamefase; vgl. punten 151 en aanhangsel 7. Deze waarde staat voor een onafhankelijke schatting van het rendement van de assimilatie voor een in wezen niet-blootgesteld organisme (d.w.z. de vissen bevinden zich ongeveer aan het begin van de opnamefase). Het rendement van de assimilatie geschat uit de depuratiegegevens wordt gewoonlijk gebruikt om de BMF af te leiden. Correctie voor vetgehalte en correctie voor groeiverdunning De groei van de vissen tijdens de depuratiefase kan de concentraties van chemische stoffen in de vissen verlagen, met als effect dat de algemene depuratiesnelheidsconstante, k2 , groter is dan zij zou zijn als zij uitsluitend werd ontleend aan verwijderingsprocessen (bv. metabolisme, egestie) (vgl. punt 72). Het vetgehalte van testvissen (dat sterk in verband wordt gebracht met de bioaccumulatie van hydrofiele stoffen) en het vetgehalte van het voer kunnen in de praktijk zoveel schommelen dat het noodzakelijk is ervoor te corrigeren om bioconcentratiefactoren op een zinvolle manier te kunnen presenteren. De biomagnificatiefactor moet worden gecorrigeerd voor groeiverdunning (net als de kinetische BCF in de methode met blootstelling via het water) en worden gecorrigeerd voor het vetgehalte van het voer in verhouding tot dat van de vissen (de vetcorrectiefactor). Vergelijkingen voor en voorbeelden van deze berekeningen zijn te vinden in respectievelijk aanhangsel 5 en aanhangsel 7. Om voor groeiverdunning te corrigeren moet de voor groei gecorrigeerde depuratiesnelheidsconstante (k 2g) worden berekend (zie aanhangsel 5 voor vergelijkingen). Deze voor groei gecorrigeerde depuratiesnelheidsconstante (k 2g) wordt vervolgens gebruikt om de voor groei gecorrigeerde biomagnificatiefactor te berekenen, als in punt 73. In sommige gevallen is deze aanpak niet mogelijk. Een alternatieve aanpak waarbij niet voor groeiverdunning hoeft te worden gecorrigeerd, maakt gebruik van depuratiegegevens met betrekking tot de massa van de teststof per vis (op basis van hele vissen) in plaats van de gebruikelijke (concentratie)gegevens met betrekking tot de massa van de teststof per massaeenheid vis. Deze aanpak kan probleemloos worden gebruikt, aangezien in tests die volgens deze methode worden uitgevoerd, de geregistreerde weefselconcentraties dienen te worden gekoppeld aan het gewicht van de afzonderlijke vissen. De eenvoudige procedure hiervoor is uiteengezet in aanhangsel 5. Er zij opgemerkt dat k 2 toch moet worden geschat en gerapporteerd, zelfs als deze alternatieve aanpak wordt gebruikt. Om voor het vetgehalte van het voer en de vissen te corrigeren wanneer het vetgehalte niet voor alle bemonsterde vissen is bepaald, worden de gemiddelde vetfracties (g/g) in de vissen en in het voer afgeleid (68). De vetcorrectiefactor (Lc ) wordt vervolgens berekend door de gemiddelde vetfractie van de vissen te delen door de gemiddelde vetfractie van het voer. De biomagnificatiefactor, al dan niet voor groei gecorrigeerd, naar gelang van hetgeen van toepassing is, wordt gedeeld door de vetcorrectiefactor om de voor vetgehalte gecorrigeerde biomagnificatiefactor te berekenen. Indien de bepalingen van de stofconcentraties en het vetgehalte bij elk monsternamepunt op dezelfde vissen werden verricht, kunnen de voor vetgehalte gecorrigeerde gegevens van weefsels voor afzonderlijke vissen worden gebruikt om rechtstreeks een voor vetgehalte gecorrigeerde BMF te berekenen (vgl. (37)). De grafiek van de voor vetgehalte gecorrigeerde concentratiegegevens levert C 0,d op op basis van vetgehalte en k 2. De wiskundige analyse kan daarna worden voortgezet met behulp van dezelfde vergelijkingen in aanhangsel 7, maar het rendement van de assimilatie (a) wordt berekend met behulp van de voedselingestiesnelheidsconstante genormaliseerd naar vetgehalte (I lipiden) en de concentratie in het voer op basis van het vetgehalte (C voedsel-lipiden). Voor vetgehalte gecorrigeerde parameters worden daarna op dezelfde manier gebruikt om BMF te berekenen (opgemerkt zij dat correctie van de groeisnelheidsconstante ook moet worden toegepast op de vetfractie in plaats van het versgewicht van de vissen om de voor vetgehalte en groei gecorrigeerde BMFKgL te berekenen). Interpretatie van de resultaten De gemiddelde groei mag zowel in de testgroep als in de controlegroep in beginsel niet significant verschillen om toxische effecten uit te sluiten. De groeisnelheidsconstanten of de groeicurven van de twee groepen moeten worden vergeleken met behulp van een passende procedure (69). Testverslag Na afloop van de studie wordt een eindverslag opgesteld met daarin informatie over de teststof, proefdiersoort en testomstandigheden zoals vermeld in punt 81 (als voor de methode met blootstelling via het water). Daarnaast is de volgende informatie vereist:
LITERATUUR
Aanhangsel 1 DEFINITIES EN EENHEDEN
LITERATUUR
Aanhangsel 2 CHEMISCHE KARAKTERISTIEKEN VAN AANVAARDBAAR VERDUNNINGSWATER
Aanhangsel 3 VOOR DE TESTS AANBEVOLEN VISSOORTEN
Minder vaak zijn ook verschillende estuariene en mariene soorten gebruikt, bijvoorbeeld:
De in de tabel genoemde zoetwatervissen zijn gemakkelijk te kweken en/of het hele jaar door vlot verkrijgbaar, maar de beschikbaarheid van de mariene en estuariene soorten verschilt van land tot land. De genoemde soorten kunnen in viskwekerijen of in het laboratorium vrij van ziekten en parasieten worden opgekweekt en tot voortplanting gebracht, zodat voor de test gezonde dieren van bekende afstamming kunnen worden gebruikt. Dergelijke vissen zijn in vele delen van de wereld verkrijgbaar. LITERATUUR
Aanhangsel 4 BEMONSTERINGSSCHEMA'S VOOR TESTS MET BLOOTSTELLING VIA VOER OF WATER 1. Theoretisch voorbeeld van een bemonsteringsschema voor een volledige bioconcentratietest met blootstelling via het water met een stof waarvoor log KOW = 4
2. Theoretisch voorbeeld van een bemonsteringsschema voor een bioaccumulatietest via het voer na een opnamefase van 10 dagen en een depuratiefase van 42 dagen
Opmerking over fase en bemonsteringstijdstippen: De opnamefase vangt aan bij de eerste verstrekking van verrijkt voer. Een experimentele dag loopt van een voedselverstrekking tot kort voor de volgende voedselverstrekking, 24 uur later. De eerste bemonstering (1 in de tabel) moet plaatsvinden kort (bv. één uur) voor de eerste voedselverstrekking. De bemonsteringen gedurende een studie vinden in het ideale geval kort voor de voedselverstrekking van de volgende dag plaats (d.w.z. ongeveer 23 uur na de voedselverstrekking van de monsternamedag). De opnamefase eindigt kort vóór de eerste verstrekking van niet-verrijkt voer, wanneer de depuratiefase begint (de vissen in de testgroep zullen in de 24 uur na de laatste verstrekking van verrijkt voer waarschijnlijk nog verrijkt voer verteren). Dit betekent dat het monster aan het einde van de opnamefase moet worden genomen kort voor de eerste verstrekking van niet-verrijkt voer en dat het eerste monster in de depuratiefase ongeveer 23 uur na de eerste verstrekking van niet-verrijkt voer moet worden genomen. Aanhangsel 5 ALGEMENE BEREKENINGEN
1. INLEIDING Het algemene aquatische bioaccumulatiemodel voor vissen kan worden beschreven in termen van opname- en eliminatieprocessen, waarbij opname uit voedsel wordt genegeerd. De differentiaalvergelijking (dC f/dt) die de snelheid beschrijft waarmee de concentratie in vissen verandert (mg·kg-1·dag-1), is (1):
waarin
Voor bioaccumulerende stoffen kan worden verwacht dat een tijdgewogen gemiddelde (TWA) de meest relevante blootstellingsconcentratie in het water (Cw ) binnen de toegestane bandbreedte is (vgl. punt 24). Het is aan te bevelen een TWA-waterconcentratie te berekenen volgens de procedure in aanhangsel 6 van TM C.20 (2). Er zij opgemerkt dat een ln-transformatie van de waterconcentratie geschikt is wanneer tussen de verversingsperioden een exponentiële daling wordt verwacht, bijvoorbeeld in geval van een semistatische opzet van de test. In een doorstroomsysteem is een ln-transformatie van de blootstellingsconcentraties mogelijk niet nodig. Indien TWA-waterconcentraties worden afgeleid, moeten deze worden gerapporteerd en in de volgende berekeningen worden gebruikt. In een standaard BCF-test met vissen kunnen de opname en depuratie worden beschreven in termen van twee processen die eersteordekinetiek vertonen:
In de stationaire toestand en ervan uitgaande dat de groei en het metabolisme verwaarloosbaar zijn (d.w.z. de waarden voor k g en k m zijn niet te onderscheiden van nul), is de opnamesnelheid gelijk aan de depuratiesnelheid. Wanneer de vergelijkingen A5.2 en A5.3 worden gecombineerd, geeft dit dus de volgende relatie:
waarin
De verhouding k 1/k 2 staat bekend als de kinetische BCF (BCFK) en moet gelijk zijn aan de BCF in de stationaire toestand (BCFSS) verkregen uit de verhouding van de concentratie in stationaire toestand in de vissen en die in het water, maar er kunnen afwijkingen optreden als het onzeker is of de stationaire toestand werd bereikt of als de kinetische BCF is gecorrigeerd voor groei. Aangezien k 1 en k 2 constanten zijn, hoeft de stationaire toestand echter niet te zijn bereikt om een BCFK af te leiden. Dit aanhangsel 5 bevat de algemene berekeningen op basis van deze eersteordevergelijkingen die nodig zijn voor zowel bioaccumulatiemethoden met blootstelling via het water als voor dergelijke tests via het voer. De paragrafen 5, 6 en 8 zijn echter uitsluitend relevant voor de methode met blootstelling via het water, maar worden hier toch opgenomen omdat ze “algemene” technieken beschrijven. De sequentiële (paragrafen 4 en 5) en simultane (paragraaf 6) methoden maken het mogelijk de opname- en depuratieconstanten te berekenen die worden gebruikt om kinetische BCF's af te leiden. De sequentiële methode voor het bepalen van k 2 (paragraaf 4) is van belang voor de methode via het voer, aangezien zij nodig is om zowel het rendement van de assimilatie als de BMF te berekenen. Aanhangsel 7 bevat bijzonderheden van de berekeningen die specifiek zijn voor de methode via het voer. 2. VOORSPELLING VAN DE DUUR VAN DE OPNAMEFASE Alvorens de test wordt uitgevoerd, kan een schatting van k 2 en derhalve van de tijd die nodig is om de stationaire toestand te bereiken, worden verkregen uit het empirische verband dat is aangetoond tussen k 2 en de n-octanol-water-verdelingscoëfficiënt (K OW) of tussen k 1 en de BCF. Men moet echter beseffen dat de vergelijkingen in deze paragraaf slechts van toepassing zijn wanneer de opname en depuratie eersteordekinetiek vertonen. Als dit duidelijk niet het geval is, verdient het aanbeveling een biostatisticus en/of farmacokineticus te raadplegen, indien ramingen van de opnamefase gewenst zijn. Een schatting van k 2 (dag-1) kan op meerdere manieren worden verkregen. In eerste instantie kan bijvoorbeeld het volgende empirische verband worden gebruikt (87):
of
W = gemiddelde gewicht van behandelde vissen (grammen versgewicht) aan het einde van de opname/begin van de depuratie (88) Voor andere gerelateerde relaties, zie (6). Het kan nuttig zijn complexere modellen voor de schatting van k2 te gebruiken wanneer het bijvoorbeeld waarschijnlijk is dat substantieel metabolisme plaatsvindt (7) (8). Naarmate de complexiteit van het model toeneemt, moeten de voorspellingen echter behoedzamer worden geïnterpreteerd. De aanwezigheid van nitro-groepen kan bijvoorbeeld wijzen op snel metabolisme, maar dit is niet altijd het geval. De gebruiker moet daarom bij de planning van een studie de resultaten van de voorspellende methode beoordelen in het licht van de chemische structuur en eventuele andere relevante informatie (bijvoorbeeld voorafgaande studies). De tijd die nodig is om een bepaald percentage van de stationaire concentratie te bereiken, kan worden verkregen door de schatting van k 2 uit de algemene kinetische vergelijking die de opname en depuratie beschrijft (eersteordekinetiek), toe te passen, ervan uitgaande dat groei en metabolisme verwaarloosbaar zijn. Indien er sprake is van substantiële groei tijdens de studie, zijn de hieronder beschreven schattingen niet betrouwbaar. In dergelijke gevallen is het beter de voor groei gecorrigeerde k 2g te gebruiken zoals verderop wordt beschreven (zie paragraaf 7 van dit aanhangsel):
of, indien C w constant is:
Bij het naderen van de stationaire toestand (t → ∞), kan vergelijking A5.10 worden vereenvoudigd (vgl. (9) (10)) tot:
of
BCF × C w is dan een benadering van de concentratie van de stof in de vissen in de stationaire toestand (C f-SS). [Opmerking: dezelfde aanpak kan worden gebruikt bij het schatten van een BMF in de stationaire toestand met de test via het voer. In dit geval wordt in de vergelijkingen hierboven BCF vervangen door BMF en C w door C voedsel, de concentratie in het voedsel.] Vergelijking A5.10 kan dan worden herschreven als:
of
Wanneer met vergelijking A5.5 of vergelijking A5.6 een preliminaire schatting van k 2 wordt gemaakt, kan met behulp van vergelijking A5.14 de tijd worden geraamd die nodig is om een bepaald percentage van de stationaire concentratie te bereiken. Als vuistregel geldt dat de statistisch optimale duur van de opnamefase voor het verkrijgen van statistisch aanvaardbare gegevens wat betreft BCFK overeenstemt met de tijd die nodig is opdat de logaritme van de concentratie van de teststof in de vissen, uitgezet tegen de niet-getransformeerde tijd, ten minste 50 % van de stationaire concentratie (d.w.z. 0,69/k 2) bereikt, maar niet meer dan 95 % van de stationaire concentratie (d.w.z. 3,0/k 2) (11). Wanneer de accumulatie een niveau van meer dan 95 % van de stationaire concentratie bereikt, wordt berekening van een BCFSS haalbaar. De tijd die nodig is om 80 % van de stationaire concentratie te bereiken is (met behulp van vergelijking A5.14):
of
Evenzo is de tijd die nodig is om 95 % van de stationaire concentratie te bereiken:
De duur van de opnamefase (d.w.z. de tijd die nodig is om voor een teststof waarvoor log K OW = 4, een bepaald percentage van de stationaire concentratie te bereiken, bv. t 80 of t 95) is (met behulp van de vergelijkingen A5.5, A5.16 en A5.17): logk2 = 1,47 – 0,414 · 4 k 2 = 0,652 dag-1
of Als alternatief kan de uitdrukking
worden gebruikt om de tijd te berekenen die nodig is voor het daadwerkelijk bereiken van de stationaire situatie (t eSS ) (12). Voor een teststof waarvoor log K OW = 4, leidt dit tot: teSS = 6,54 · 10– 3 · 104 + 55,31 = 121 uur 3. VOORSPELLING VAN DE DUUR VAN DE DEPURATIEFASE Een raming van de tijd die nodig is om de lichaamsconcentratie te laten dalen tot een bepaald percentage van de beginconcentratie, kan ook worden verkregen uit de algemene vergelijking die de opname en depuratie beschrijft (uitgaande van eersteordekinetiek, vgl. vergelijking A5.9) (1) (13). Voor de depuratiefase wordt aangenomen dat C w (of C voedsel voor de test via het voer) gelijk is aan nul. De vergelijking wordt dan gereduceerd tot
of
waarin C f,0 de concentratie is bij het begin van depuratiefase. 50 % depuratie wordt dan verkregen na t 50, die als volgt wordt berekend:
of
Zo ook wordt 95 % depuratie bereikt na
Als de opnamefase wordt afgesloten wanneer 80 % van de stationaire concentratie is bereikt (1,6/k 2) en de depuratiefase wanneer de eliminatie 95 % beloopt (3,0/k 2), bedraagt de duur van de depuratiefase ongeveer het dubbele van de duur van de opnamefase. Opgemerkt zij dat de schattingen zijn gebaseerd op de aanname dat de opname- en depuratiepatronen eersteordekinetiek vertonen. Indien het proces duidelijk geen eersteordekinetiek vertoont, zijn deze schattingen niet geldig. 4. SEQUENTIËLE METHODE: BEPALING VAN DE DEPURATIE- OF (ELIMINATIE-)SNELHEIDSCONSTANTE K 2 Er is van uitgegaan dat de meeste bioconcentratieprocessen op een “acceptabele” manier worden beschreven door een simpel twee-compartiment/twee-parametermodel, wat zich vertaalt in een lineair verband wanneer de concentratie van de stof in de vissen tijdens de depuratiefase (op logaritmische schaal) tegen de tijd wordt uitgezet.
Houd er rekening mee dat afwijkingen van een lineair verband een aanwijzing kunnen vormen voor een complex depuratiepatroon dat niet door een eersteordekinetiek kan worden beschreven. Om depuratiepatronen door te rekenen die niet door een eersteordekinetiek worden gekenmerkt, kan eveneens de grafische methode worden gebruikt. Voer een lineaire regressie uit op de natuurlijke logaritme van de concentratie als functie van de tijd om k 2 te berekenen voor meerdere (bemonsterings)tijdstippen. De helling van de regressielijn is een schatting van de depuratiesnelheidsconstante k 2 (89) . Uit het intercept kan gemakkelijk de gemiddelde concentratie in de vissen aan het begin van de depuratiefase (C 0,d; welke gelijk is aan de gemiddelde concentratie in de vissen aan het einde van de opnamefase) worden berekend (met inbegrip van onzekerheidsmarges) (89):
Vul de twee gemiddelde concentraties in de volgende vergelijking in om k 2 te berekenen wanneer slechts gegevens van twee (bemonsterings)tijdstippen beschikbaar zijn (zoals in de beperkte testopzet):
waarin ln(C f1) en ln(C f2) de natuurlijke logaritmen van de concentraties op respectievelijk de tijdstippen t 1 en t 2 zijn, en t 2 en t 1 de tijdstippen ten opzichte van het begin van de depuratie zijn waarop de twee monsters werden verzameld (90). 5. SEQUENTIËLE METHODE: BEPALING VAN DE OPNAMESNELHEIDSCONSTANTE K 1 (ALLEEN VOOR METHODE MET BLOOTSTELLING VIA HET WATER) Om een waarde voor k1 te vinden kan met een computerprogramma een reeks aan de tijd gerelateerde concentratiegegevens voor de opnamefase aan het volgende model worden gefit:
waarbij k 2 door de berekening hierboven wordt gegeven en C f(t) en C w(t) respectievelijk de concentraties in vissen en in het water op tijdstip t zijn. Gebruik de volgende formule om k 1 te berekenen wanneer slechts twee (bemonsterings)tijdstippen beschikbaar zijn (zoals in de beperkte test):
waarbij k 2 door de berekening hierboven wordt gegeven, C f de concentratie in vissen aan het begin van de depuratiefase is en C w de gemiddelde concentratie in het water tijdens de opnamefase is (91). Visuele inspectie van de resulterende hellingen k 1 en k 2 wanneer de gemeten gegevens van de bemonsteringstijdstippen in een grafiek worden uitgezet, kan worden gebruikt om de kwaliteit van de aanpassing te beoordelen. Als blijkt dat de sequentiële methode een slechte schatting voor k 1 oplevert, moet de simultane aanpak voor het berekenen van k 1 en k 2 worden toegepast (zie paragraaf 6 hierna). De resulterende hellingen moeten weer worden vergeleken met de in de grafiek uitgezette gemeten gegevens om de kwaliteit van de aanpassing visueel te beoordelen. Als de kwaliteit van de aanpassing nog steeds slecht is, kan dit een aanwijzing zijn dat het proces geen eersteordekinetiek vertoont en moeten andere, complexere modellen worden gebruikt. 6. SIMULTANE METHODE VOOR HET BEREKENEN VAN DE OPNAME- EN DEPURATIE- OF (ELIMINATIE-)SNELHEIDSCONSTANTEN (ALLEEN VOOR METHODE MET BLOOTSTELLING VIA HET WATER) Om waarden voor k 1 en k 2 te vinden kan met een computerprogramma een reeks aan de tijd gerelateerde concentratiegegevens aan het model worden gefit:
waarbij
Deze benadering verschaft rechtstreeks standaardfouten voor de schattingen van k 1 en k 2. Wanneer k 1/k 2 in vergelijking A5.25 en vergelijking A5.26 wordt vervangen door BCF (vgl. vergelijking A5.4), kunnen ook de standaardfout en het 95 %-BI van de BCF worden geschat. Dit is in het bijzonder nuttig wanneer verschillende schattingen ten gevolge van gegevenstransformaties worden vergeleken. De afhankelijke variabele (de concentratie in vissen) kan met of zonder natuurlijke logaritmische transformatie worden gefit, en de onzekerheid in de resulterende BCF kan worden geraamd. Aangezien er een sterke correlatie tussen de twee parameters k 1 en k 2 bestaat indien ze gelijktijdig worden geschat, kan het aan te bevelen zijn eerst k 2 te berekenen uit uitsluitend de depuratiegegevens (zie hierboven); k 2 kan in de meeste gevallen vrij nauwkeurig worden geschat uit de depuratiecurve. k 1 kan vervolgens met niet-lineaire regressie worden berekend uit de opnamegegevens (92). Het is aan te bevelen dezelfde gegevenstransformatie toe te passen wanneer sequentieel wordt gefit. Visuele inspectie van de resulterende hellingen wanneer de gemeten gegevens van de bemonsteringstijdstippen in een grafiek worden uitgezet, kan worden gebruikt om de kwaliteit van de aanpassing te beoordelen. Als blijkt dat de sequentiële methode een slechte schatting voor k1 oplevert, kan de simultane aanpak voor het berekenen van k 1 en k 2 worden toegepast. Het gefitte model moet weer worden vergeleken met de in de grafiek uitgezette gemeten gegevens om de kwaliteit van de aanpassing visueel te beoordelen, en de resulterende parameterschattingen voor k 1, k 2 en de resulterende BCF met hun standaardfouten en/of betrouwbaarheidsintervallen moeten worden vergeleken voor verschillende typen fits. Als de kwaliteit van de aanpassing nog steeds slecht is, kan dit een aanwijzing zijn dat het proces geen eersteordekinetiek vertoont en moeten andere, complexere modellen worden gebruikt. Een van de meest voorkomende complicaties is groei van de vissen gedurende de test. 7. CORRECTIE VOOR GROEIVERDUNNING VAN KINETISCHE BCF EN BMF In deze paragraaf wordt een standaardmethode voor het corrigeren voor groei van de vissen tijdens de test (zogenoemde 'groeiverdunning') beschreven die alleen geldig is wanneer het proces eersteordekinetiek vertoont. Als er aanwijzingen zijn dat er geen sprake is van eersteordekinetiek, verdient het aanbeveling om een biostatisticus te raadplegen voor een juiste correctie voor groeiverdunning of om de hieronder beschreven aanpak op basis van massa te gebruiken. In sommige gevallen is er bij deze methode voor het corrigeren voor groeiverdunning sprake van gebrekkige precisie of werkt de methode niet (zo kan bijvoorbeeld de afgeleide, voor groeiverdunning gecorrigeerde depuratiesnelheidsconstante, k 2g, voor zeer traag depurerende stoffen die worden getest met snel groeiende vissen heel klein zijn, waardoor de fout in de twee snelheidsconstanten die worden gebruikt om de voor groeiverdunning gecorrigeerde depuratiesnelheidsconstante te bepalen kritisch wordt, en in sommige gevallen kunnen de schattingen van k g groter zijn dan k 2). In dergelijke gevallen kan een alternatieve aanpak (d.w.z. massa-aanpak), die ook werkt wanneer het proces geen eersteordegroeikinetiek vertoont, worden gebruikt waarbij correctie niet nodig is. Deze aanpak wordt aan het einde van deze paragraaf uiteengezet. Methode voor correctie voor groei door aftrek van de groeisnelheidsconstante Voor de standaardmethode worden alle afzonderlijke gewichts- en lengtegegevens geconverteerd naar hun natuurlijke logaritme en wordt ln(gewicht) of ln(1/gewicht) voor de behandelingsgroep en controlegroep apart in een grafiek uitgezet tegen de tijd (dag). Dezelfde procedure wordt gevolgd voor de gegevens van de opnamefase en apart voor die van de depuratiefase. In het algemeen is het voor correctie voor groeiverdunning passender om de gewichtsgegevens van de hele studie te gebruiken om de groeisnelheidsconstante (k g) af te leiden, maar statistisch significante verschillen tussen de afgeleide groeisnelheidsconstanten voor de opnamefase en de depuratiefase kunnen een aanwijzing vormen dat de snelheidsconstante voor de depuratiefase moet worden gebruikt. Algemene groeisnelheden uit studies via het water voor test- en controlegroepen kunnen worden gebruikt om te controleren op het voorkomen van aan de behandeling gerelateerde effecten. Voor de natuurlijke logaritme van het gewicht van de vissen uitgezet tegen de dag (en voor de natuurlijke logaritme van 1/gewicht uitgezet tegen de dag) wordt met behulp van statistische standaardprocedures voor elke groep (test- en controlegroepen, afzonderlijke gegevens, geen gemiddelde waarden per dag) voor de hele studie, de opnamefase en de depuratiefase een lineaire kleinste-kwadratencorrelatie berekend. De varianties in de hellingen van de lijnen worden berekend en gebruikt om de statistische significantie (p = 0,05) van het verschil in de hellingen (groeisnelheidsconstanten) te schatten met de t-toets van Student (of ANOVA indien meer dan één concentratie wordt getest). Voor het corrigeren voor groei genieten gewichtsgegevens doorgaans de voorkeur. Lengtegegevens, op dezelfde manier behandeld, kunnen nuttig zijn om controle- en testgroepen te vergelijken op het punt van aan de behandeling gerelateerde effecten. Indien er in de analyse van de gewichtsgegevens geen statistisch significant verschil werd gevonden, kunnen de gegevens van de test- en controlegroepen worden samengevoegd en kan voor de studie de algemene groeisnelheidsconstante voor de vissen (k g) worden berekend als de algemene helling van het lineaire verband. Indien statistische verschillen werden waargenomen, moeten de groeisnelheidsconstanten voor elke groep vissen, en/of fase van het onderzoek, apart worden gerapporteerd. De snelheidsconstante van elke behandelde groep moet vervolgens worden gebruikt om voor die groep voor groeiverdunning te corrigeren. Indien statistische verschillen tussen de snelheidsconstanten voor de opnamefase en depuratiefase werden waargenomen, moeten de voor de depuratiefase afgeleide snelheidsconstanten worden gebruikt. De depuratiesnelheidsconstante, k 2g, wordt verkregen door de berekende groeisnelheidsconstante (k g uitgedrukt in dag-1) in mindering te brengen op de algemene depuratiesnelheidsconstante (k 2).
De voor groei gecorrigeerde kinetische BCF, genoteerd als BCFKg (of BMFKg), wordt verkregen door de opnamesnelheidsconstante te delen door de voor groei gecorrigeerde depuratiesnelheidsconstante.
De afgeleide groeisnelheidsconstante voor een studie via het voer wordt in vergelijking A7.5 gebruikt om de voor groei gecorrigeerde BMFKg te berekenen (vgl. aanhangsel 7). Methode voor correctie voor groei op basis van massa Een alternatief voor de “methode voor correctie voor groei door aftrek van de groeisnelheidsconstante” hierboven waarbij niet voor groei hoeft te worden gecorrigeerd, is de volgende. Het principe van deze alternatieve methode is dat gegevens uit de depuratiefase op basis van massa per hele vis worden gebruikt in plaats van op basis van concentratie. Converteer de weefselconcentraties uit de depuratiefase (massa van de teststof per massaeenheid vis) naar massa van de teststof per vis: plaats de concentraties en gewichten van afzonderlijke vissen naast elkaar in een tabel (bv. met een spreadsheetprogramma) en vermenigvuldig elke concentratie met het totale gewicht van de vis voor die meting, zodat voor alle bemonsteringen uit de depuratiefase een reeks massagegevens van de teststof per vis wordt verkregen. Zet de resulterende natuurlijke logaritme van de massagegevens voor de stof uit tegen de tijd voor het experiment (depuratiefase) zoals dit in het normale geval wordt gedaan. Leid voor de methode met blootstelling via het water routinematig de opnamesnelheidsconstante af (zie de paragrafen 4 en 6). Houd rekening met het feit dat de “normale” waarde van k 2 moet worden gebruikt in de vergelijkingen voor het fitten van de curve voor k 1, en leid uit de gegevens hierboven de depuratiesnelheidsconstante af. Omdat de resulterende waarde voor de depuratiesnelheidsconstante onafhankelijk is van de groei, aangezien zij is afgeleid op basis van de massa per hele vis, wordt de waarde genoteerd als k 2g en niet als k 2. 8. NORMALISATIE NAAR EEN VETGEHALTE VAN 5 % (ALLEEN VOOR METHODE MET BLOOTSTELLING VIA HET WATER) De resultaten voor de BCF's (kinetische en in stationaire toestand) uit tests met blootstelling via het water moeten ook worden gerapporteerd ten opzichte van een standaardvetgehalte van de vis van 5 % (versgewicht), tenzij kan worden aangevoerd dat de teststof zich niet primair in vetten ophoopt (sommige geperfluoreerde stoffen kunnen zich bijvoorbeeld binden aan eiwitten). De gegevens van de concentratie in de vissen, of de BCF, moeten worden geconverteerd naar een vetgehalte van 5 % op basis van het versgewicht. Indien op alle monsternamepunten voor het meten van de concentraties van de teststof en het meten van het vetgehalte dezelfde vissen werden gebruikt, moet elke afzonderlijke gemeten concentratie in de vissen worden gecorrigeerd voor het vetgehalte van de betreffende vis.
waarbij
Indien niet voor alle bemonsterde vissen een vetanalyse is uitgevoerd, wordt een gemiddelde vetwaarde gebruikt om de BCF te normaliseren. Voor de BCF in de stationaire toestand moet de gemiddelde waarde worden gebruikt die aan het einde van de opnamefase in de behandelingsgroep is geregistreerd. Voor de normalisatie van een kinetische BCF kunnen zich situaties voordoen waarin een andere aanpak gerechtvaardigd is, bijvoorbeeld als het vetgehalte tijdens de opnamefase of depuratiefase duidelijk is gewijzigd. Er moet in de regel echter een rantsoen worden gebruikt die het risico van sterke wijzigingen in het vetgehalte tot een minimum beperkt.
waarbij
LITERATUUR
Aanhangsel 6 VERGELIJKINGEN VOOR TESTS MET BLOOTSTELLING VIA HET WATER: BEPERKTE TESTOPZET De grondgedachte van deze aanpak is dat de bioconcentratiefactor in een volledige test kan worden bepaald als een bioconcentratiefactor in stationaire toestand (BCFSS) door de verhouding tussen de concentratie van de teststof in het weefsel van de vissen en de concentratie van de teststof in het water te berekenen, of door de kinetische bioconcentratiefactor (BCFK) te berekenen als de verhouding tussen de opnamesnelheidsconstante k 1 en de depuratiesnelheidsconstante k 2. De BCFK is ook geldig als tijdens de opname geen concentratie van de stof in stationaire toestand wordt bereikt, mits die opname- en depuratieprocessen ongeveer eersteordekinetiek vertonen. Als een meting van de concentratie van de stof in weefsels (C f1) wordt verricht op het tijdstip waarop de blootstelling eindigt (t 1) en de concentratie in het weefsel (C f2) opnieuw wordt gemeten nadat een bepaalde tijdsperiode is verstreken (t 2), kan de depuratiesnelheidsconstante (k 2) worden geschat met vergelijking A5.22 in aanhangsel 5. De opnamesnelheidsconstante, k 1, kan dan algebraïsch worden bepaald met vergelijking A5.23 in aanhangsel 5 (waarin C f gelijk is aan C f1 en t gelijk is aan t 1) (1). De kinetische bioconcentratiefactor voor de beperkte opzet (aangeduid als BCFKm om hem te onderscheiden van kinetische bioconcentratiefactoren die met andere methoden worden bepaald) is dus:
De concentraties of resultaten moeten worden gecorrigeerd voor groeiverdunning en worden genormaliseerd naar een vetgehalte van de vissen van 5 %, zoals is beschreven in aanhangsel 5. De beperkte BCFSS is de BCF die wordt berekend aan het einde van de opnamefase, ervan uitgaande dat de stationaire toestand is bereikt. Dit kan slechts een aanname zijn, want het aantal monsternamepunten is onvoldoende om dit aan te tonen.
waarin C f-minSS = concentratie in de vissen bij veronderstelde stationaire toestand aan het einde van de opname (mg kg-1 versgewicht), C w-minSS = concentratie in het water bij veronderstelde stationaire toestand aan het einde van de opname (mg l-1). LITERATUUR
Aanhangsel 7 VERGELIJKINGEN VOOR TESTS MET BLOOTSTELLING VIA HET VOER
1. VOORBEELD VAN HOEVEELHEDEN VAN COMPONENTEN VAN EEN GESCHIKT COMMERCIEEL VISVOER
2. VOORBEELDEN VAN METHODEN VOOR HET VERRIJKEN VAN VOER Algemene punten De controlevoeders moeten op precies dezelfde manier worden bereid als het verrijkte voer, maar zonder aanwezigheid van de teststof. Om de concentratie van het behandelde voer te controleren, moeten met een geschikte extractiemethode monsters van het toegediende voedsel in drievoud worden genomen en moet de concentratie of radioactiviteit van de teststof in de extracten worden gemeten. Er moeten hoge analytische recovery's (> 85 %) met geringe verschillen tussen monsters worden aangetoond (de concentraties van de stof in drie monsters die aan het begin van de test zijn genomen, mogen niet meer dan ± 15 % afwijken van het gemiddelde). Tijdens de test via het voer moeten op dag 0 en aan het einde van de opnamefase drie voermonsters voor analyses worden genomen om het gehalte van de teststof in het voer te bepalen. Bereiding van visvoer met een vloeibaar testmateriaal (zuiver) Er wordt een nominale doelconcentratie van de teststof in het voer van de behandelde vissen vastgesteld, bijvoorbeeld 500 μg teststof/g voer. De juiste hoeveelheid (wat betreft molaire massa of specifieke radioactiviteit) aan zuivere teststof wordt toegevoegd aan een bekende hoeveelheid visvoer in een glazen pot of kolf van een roterende verdamper. De hoeveelheid visvoer moet toereikend zijn voor de duur van de opnamefase (rekening houdend met de noodzaak om de hoeveelheden bij elke voedselverstrekking te verhogen vanwege groei van de vissen). Het visvoer/de teststof moet een nacht lang worden gemengd door trage tuimelbewegingen (bv. met een roto-rack mixer of door rotatie indien een kolf van een roterende verdamper wordt gebruikt). Het verrijkte voer moet worden bewaard onder omstandigheden waarbij de stabiliteit van de teststof in het voermengsel tot gebruik behouden blijft (bv. gekoeld). Bereiding van visvoer met mais- of visolie als medium Vaste teststoffen moeten in een vijzel tot een fijn poeder worden vermalen. Vloeibare teststoffen kunnen rechtstreeks aan de mais- of visolie worden toegevoegd. De teststof wordt opgelost in een bekende hoeveelheid mais- of visolie (bv. 5-15 ml). De gedoseerde olie wordt kwantitatief overgebracht in een kolf van geschikte grootte van een roterende verdamper. De fles die is gebruikt om de gedoseerde olie in te bereiden, moet met twee kleine aliquots olie worden gespoeld en deze moeten worden toegevoegd aan de kolf om ervoor te zorgen dat alle opgeloste teststof wordt overgebracht. Om te waarborgen dat de teststof volledig wordt opgelost/gedispergeerd in de olie (of als in de studie meer dan één teststof wordt gebruikt), wordt een microroerder toegevoegd, wordt de fles met een stop afgesloten en wordt het mengsel een nacht lang snel geschud. Aan de kolf wordt een passende hoeveelheid visvoer (gewoonlijk in korrelvorm) voor de test toegevoegd, en de inhoud van de kolf wordt gemengd tot hij homogeen is door de glazen kolf gedurende ten minste 30 minuten, maar bij voorkeur een nacht lang, voortdurend te keren. Het verrijkte voer wordt daarna onder passende omstandigheden (bv. gekoeld) bewaard om de stabiliteit van de teststof in het voer tot gebruik te waarborgen. Bereiding van visvoer met een organisch oplosmiddel Een passende hoeveelheid teststof (wat betreft molaire massa of specifieke radioactiviteit) die toereikend is om de beoogde dosis te bereiken, wordt opgelost in een geschikt organisch oplosmiddel (bv. cyclohexaan of aceton; 10-40 ml, maar indien nodig een groter volume, afhankelijk van de hoeveelheid voedsel die moet worden verrijkt). Hetzij een aliquot van deze oplossing hetzij alle oplossing (beetje bij beetje toegevoegd) wordt gemengd met de juiste hoeveelheid visvoer om in de test het vereiste nominale dosisniveau te bereiken. Het voedsel/de teststof kan worden gemengd in een mengkom van roestvrij staal, en het vers gedoseerde visvoer kan in de mengkom twee dagen (onder af en toe roeren) in een zuurkast worden geplaatst zodat het overtollige oplosmiddel kan verdampen, of in een kolf van een roterende verdamper onder voortdurend roteren worden gemengd. Het overtollige oplosmiddel kan, indien nodig, onder een stroom van lucht of stikstof worden “weggeblazen”. Er moet op worden gelet dat de teststof niet kristalliseert terwijl het oplosmiddel wordt verwijderd. Het verrijkte voer moet worden bewaard onder omstandigheden (bv. gekoeld) waarbij de stabiliteit van de teststof in het voermengsel tot gebruik behouden blijft. 3. BEREKENING VAN HET RENDEMENT VAN DE ASSIMILATIE EN DE BIOMAGNIFICATIEFACTOR Om het rendement van de assimilatie te berekenen, moet eerst de algemene depuratiesnelheidsconstante worden geschat volgens paragraaf 4 van aanhangsel 5 (met behulp van de “sequentiële methode”, d.w.z. standaard lineaire regressie) op basis van gemiddelde concentraties van de monsters uit de depuratiefase. De voedingssnelheidsconstante, I, en opnameduur, t, zijn bekende parameters in de studie. C voedsel, de gemiddelde gemeten concentratie van de teststof in het voedsel, is een gemeten variabele in de studie. C 0,d, de concentratie van de teststof in de vissen aan het einde van de opnamefase, wordt gewoonlijk afgeleid uit het intercept van een grafiek waarin de natuurlijke logaritme van de concentratie is uitgezet tegen de depuratiedag. Het rendement van de assimilatie van de teststof (a, absorptie van de teststof via de darm) wordt berekend als:
waarbij: C 0,d = afgeleide concentratie in de vissen op tijdstip nul van de depuratiefase (mg kg-1); k 2 = algemene (niet voor groei gecorrigeerde) depuratiesnelheidsconstante (dag-1), berekend volgens de vergelijkingen in aanhangsel 5, paragraaf 3; I = voedselingestiesnelheidsconstante (g voedsel g-1 vis dag-1); C voedsel = concentratie in het voedsel (mg kg-1 voedsel); t= duur van de voederperiode (dag). Het kan echter noodzakelijk zijn de in de berekening gebruikte voedingssnelheid, I, aan de groei van de vissen aan te passen om een nauwkeurig rendement van de assimilatie, a, te verkrijgen. In een test waarin vissen tijdens de opnamefase substantieel groeien (waarbij de hoeveelheden voer niet worden gecorrigeerd om een vaste voedingssnelheid te behouden), zal de effectieve voedingssnelheid lager dan de vastgestelde voedingssnelheid worden naarmate de opnamefase verstrijkt, met als gevolg een hoger 'werkelijk' rendement van de assimilatie. (Er zij opgemerkt dat dit niet van belang is voor de algemene berekening van BMF, aangezien de I-termen in vergelijking A7.1 en vergelijking A7.4 effectief tegen elkaar wegvallen.) De gemiddelde voedingssnelheid gecorrigeerd voor groeiverdunning, I g, kan op meerdere manieren worden afgeleid, maar een eenvoudige en nauwgezette manier is de bekende groeisnelheidsconstante (k g) te gebruiken om de gewichten van de testvissen op tijdstippen tijdens de opnamefase te schatten, d.w.z.:
waarbij W f(t)= gemiddelde gewicht van de vissen op opnamedag t W f,0 = gemiddelde gewicht van de vissen aan het begin van het experiment Op deze manier kan (ten minste) het gemiddelde gewicht van de vissen op de laatste blootstellingsdag (W f,end-of-uptake) worden geschat. Aangezien de voedingssnelheid was vastgesteld op basis van W f,0, kan de effectieve voedingssnelheid voor elke dag van de opname worden berekend met behulp van deze twee gewichten. De voor groei gecorrigeerde voedingssnelheid, I g (g voedsel g-1 vis dag-1), die moet worden gebruikt in plaats van I als er sprake is van snelle groei tijdens de opnamefase, kan dan worden berekend als:
Nadat het rendement van de assimilatie is verkregen, kan de BMF worden berekend door het rendement van de assimilatie te vermenigvuldigen met de voedingssnelheidsconstante I (of I g, indien deze is gebruikt om α te berekenen) en het product te delen door de algemene depuratiesnelheidsconstante k 2:
De voor groei gecorrigeerde biomagnificatiefactor moet ook op deze manier worden berekend, met behulp van de voor groei gecorrigeerde depuratiesnelheidsconstante (zoals afgeleid overeenkomstig paragraaf 7 van aanhangsel 5). Ook hier moet I g in plaats van I worden gebruikt indien I g is gebruikt om α te berekenen:
waarbij: α = rendement van de assimilatie (absorptie van de teststof via de darm); k 2 = algemene (niet voor groei gecorrigeerde) depuratiesnelheidsconstante (dag-1), berekend volgens de vergelijkingen in aanhangsel 5, paragraaf 3; k 2g = voor groei gecorrigeerde depuratiesnelheidsconstante (dag-1); I = voedselingestiesnelheidsconstante (g voedsel g-1 vis dag-1); De voor groei gecorrigeerde halveringsperiode (t 1/2) wordt als volgt berekend:
Het rendement van de assimilatie van de stof uit het voer kan ook worden geschat als tijdens de lineaire fase van de opnamefase (tussen dag 1 en dag 3) de residuen in weefsel worden bepaald. In dit geval kan het rendement van de assimilatie van de stof (α) worden bepaald als:
waarin C vis (t) = de concentratie van de teststof in de vissen op tijdstip t (mg kg-1 versgewicht). 4. CORRECTIE VOOR VETGEHALTE Indien het vetgehalte voor alle bemonsteringsintervallen bij dezelfde vissen werd gemeten als de concentratie van de teststof, moeten de afzonderlijke concentraties worden gecorrigeerd op basis van het vetgehalte en moet de natuurlijke logaritme van de voor vetgehalte gecorrigeerde concentratie worden uitgezet tegen de depuratie (dag) om C 0,d en k 2 te verkrijgen. Het rendement van de assimilatie (vergelijking A7.1) kan daarna worden berekend op basis van het vetgehalte, met behulp van C voedsel op basis van het vetgehalte (d.w.z. C voedsel wordt vermenigvuldigd met de gemiddelde vetfractie van het voedsel). Verdere berekeningen met behulp van vergelijking A7.4 en vergelijking A7.5 geven rechtstreeks de voor vetgehalte gecorrigeerde (en voor groeiverdunning gecorrigeerde) BMF. In het andere geval worden de gemiddelde vetfractie (g/g) in de vissen en in het voedsel afgeleid voor zowel de behandelings- als de controlegroep (voor voedsel en vissen in de controlegeroep wordt dit gewoonlijk gedaan op basis van gegevens die aan het begin en aan het einde van de blootstelling zijn gemeten; voor vissen in de behandelingsgroep wordt dit gewoonlijk gedaan uitsluitend op basis van gegevens die aan het einde van de blootstelling zijn gemeten). In sommige studies kan het vetgehalte in vissen duidelijk toenemen; in dergelijke gevallen is het passender om een gemiddelde vetconcentratie in de testvissen te gebruiken die is berekend uit de gemeten waarden aan het einde van de blootstelling en het einde van de depuratie. In het algemeen moeten uitsluitend gegevens van de behandelingsgroep worden gebruikt om beide vetfracties af te leiden. De vetcorrectiefactor (Lc ) wordt berekend als:
waarin L vis en L voedsel de gemiddelde vetfracties in respectievelijk vis en voedsel zijn. De vetcorrectiefactor wordt gebruikt om de voor vetgehalte gecorrigeerde biomagnificatiefactor (BMFL) te berekenen:
5. EVALUATIE VAN VERSCHILLEN TUSSEN GEMETEN TIJDSTIP-NUL-CONCENTRATIE (C 0,M) EN AFGELEIDE TIJDSTIP-NUL-CONCENTRATIE (C 0,D) De gemeten tijdstip-nul-concentratie (C 0,m) en de afgeleide tijdstip-nul-concentratie (C 0,d) moeten worden vergeleken. Indien ze min of meer gelijk zijn, ondersteunt dit het eersteordemodel dat is gebruikt om de depuratieparameters af te leiden. In sommige studies kan er een duidelijk verschil zijn tussen de afgeleide tijdstip-nul-waarde, C 0,d, en de gemiddelde gemeten tijdstip-nul-concentratie, C 0,m (zie laatste aandachtsstreepje van punt 159 van deze testmethode). Indien C 0,d heel veel lager is dan C 0,m (C 0,d << C 0,m), kan het verschil een aanwijzing vormen voor de aanwezigheid van onverteerd verrijkt voedsel in de darm. Dit kan experimenteel worden getoetst door een aparte analyse op de weggenomen darm uit te voeren indien aan het einde van de opnamefase aanvullende monsters (hele vissen) zijn genomen en bewaard. Anders kan, indien een statistisch geldige uitschietertoets die werd toegepast op de lineaire regressie voor de depuratiefase erop wijst dat het eerste monsternamepunt van de depuratie ten onrechte hoog is, het passend zijn om de lineaire regressie uit te voeren om k 2 af te leiden maar de concentratie van het eerste monsternamepunt in de depuratie weg te laten. In dergelijke gevallen kan het, als de onzekerheid in de lineaire regressie sterk verminderd is en het duidelijk is dat de depuratie ongeveer eersteordekinetiek vertoont, passend zijn om in de berekening van het rendement van de assimilatie de resulterende waarden voor C 0,d en k 2 te gebruiken. Dit moet in het verslag uitvoerig worden verantwoord. Het is ook mogelijk dat het proces in de depuratiefase geen eersteordekinetiek vertoont. Als dit waarschijnlijk is (d.w.z. als de natuurlijke logaritmen van de gegevens een kromme lijken te volgen, vergeleken met de rechte lijn in de grafiek van de lineaire regressie), zijn de berekeningen van k 2 en C 0,d waarschijnlijk niet geldig en moet het advies van een biostatisticus worden ingewonnen. Indien C 0,d heel veel hoger is dan de gemeten waarde (C 0,d >> C 0,m), kan dit een aanwijzing vormen: dat de stof heel snel werd gedepureerd (d.w.z. monsternamepunten bereikten heel vroeg in de depuratiefase de bepaalbaarheidsgrens van de analysemethode, vgl. paragraaf 6 hieronder); dat er een afwijking van eersteordedepuratiekinetiek was; dat de lineaire regressie om k 2 en C 0,d af te leiden, gebrekkig is; of dat er op een of meer bemonsteringstijdstippen een probleem met de gemeten concentraties in de studie was. In dergelijke gevallen moet de grafiek van de lineaire regressie nauwkeurig worden onderzocht op bewijs van monsters op of rond de bepaalbaarheidsgrens, op uitschieters en op een duidelijke kromming (wat een aanwijzing kan zijn dat het proces mogelijk geen eersteordekinetiek vertoont), en moet dit in het verslag naar voren worden gebracht. Elke daaropvolgende herbeoordeling van de lineaire regressie om de geschatte waarden te verbeteren, moet worden beschreven en gerechtvaardigd. Indien een duidelijke afwijking van eersteordekinetiek wordt waargenomen, zijn de berekeningen van k 2 en C 0,d waarschijnlijk niet geldig en moet het advies van een biostatisticus worden ingewonnen. 6. LEIDRAAD VOOR ZEER SNEL DEPURERENDE TESTSTOFFEN Zoals besproken in punt 129 van de testmethode, kunnen sommige stoffen zo snel depureren dat geen betrouwbare tijdstip-nul-concentratie, C 0,d, en k 2 kunnen worden afgeleid omdat de stof heel vroeg in de depuratiefase (d.w.z. vanaf het tweede monster in de depuratiefase) in feite al niet meer wordt gemeten in monsters (concentratie gerapporteerd op de bepaalbaarheidsgrens). Deze situatie werd waargenomen in de ringtest die werd uitgevoerd tot staving van deze testmethode met benzo[a]pyreen, en is gedocumenteerd in het validatierapport voor de methode. In dergelijke gevallen kan geen betrouwbare lineaire regressie worden uitgevoerd en zal lineaire regressie waarschijnlijk een onrealistisch hoge schatting van C 0,d opleveren, die leidt tot een schijnbaar rendement van de assimilatie dat veel groter is dan 1. Het is mogelijk in deze gevallen een conservatieve schatting van k 2 en een “bovengrens” voor de BMF te berekenen. Met behulp van de gegevenspunten van de depuratiefase waarvoor een concentratie werd gemeten, tot en met het eerste monsternamepunt waarvoor geen concentratie werd aangetoond (concentratie vastgesteld op de bepaalbaarheidsgrens), zal een lineaire regressie (voor de natuurlijke logaritme van de concentratiegegevens uitgezet tegen de tijd) een schatting van k 2 opleveren. Voor dit soort gevallen betreft dit waarschijnlijk slechts twee gegevenspunten (bv. bemonsteringsdagen 1 en 2 van de depuratie) en kan k 2 daarna worden geschat met behulp van vergelijking A5.22 in aanhangsel 5. Deze schatting van k2 kan worden gebruikt om een rendement van de assimilatie te schatten overeenkomstig vergelijking A7.1, waarbij de waarde van C0,d in de vergelijking wordt vervangen door de gemeten tijdstip-nul-concentratie (C0,m) in gevallen waarin C0,d duidelijk veel hoger wordt geschat dan in de test haalbaar zou zijn. Indien C0,m niet meetbaar was, moet de aantoonbaarheidsgrens in visweefsel worden gebruikt. Indien dit, in sommige gevallen, een waarde voor α groter dan 1 oplevert, wordt uitgegaan van een “worst case”, d.w.z. van een rendement van de assimilatie dat gelijk is aan 1. De maximale BMFK kan dan worden geschat met behulp van vergelijking A7.4 en moet worden opgegeven als een waarde “veel kleiner dan” (<<). Voor een studie die is uitgevoerd met een voedingssnelheid van 3 %, een halveringsperiode in de depuratiefase van minder dan 3 dagen en een α die gelijk is aan 1 (“worst case”), zal de BMFK waarschijnlijk kleiner zijn dan ongeveer 0,13. Gezien het doel van deze schatting en het feit dat waarden conservatief van aard zullen zijn, is het niet nodig ze te corrigeren voor groeiverdunning of voor het vetgehalte van de vissen of het voer. Aanhangsel 8 BENADERINGEN OM TENTATIEVE BCF'S TE SCHATTEN OP BASIS VAN GEGEVENS VERZAMELD IN DE STUDIE MET BLOOTSTELLING VIA HET VOER De methode via het voer is in deze testmethode opgenomen voor het testen van de bioaccumulatie van stoffen die in de praktijk niet met de methode met blootstelling via het water kunnen worden getest. De methode met blootstelling via het water levert een bioconcentratiefactor op, terwijl de methode via het voer rechtstreeks informatie geeft over het biomagnificatiepotentieel van voer. In veel veiligheidsvoorschriften voor chemische stoffen is informatie over de aquatische bioconcentratie vereist (bijvoorbeeld in risicobeoordelingen en het Globally Harmonization System of Classification). Het is dus nodig de in een studie via voer verkregen gegevens te gebruiken om een bioconcentratiefactor te schatten die vergelijkbaar is met de factor uit tests die volgens de methode met blootstelling via het water zijn uitgevoerd (94). In deze paragraaf worden benaderingen onderzocht die daartoe kunnen worden gevolgd en worden de tekortkomingen erkend die inherent zijn aan de schattingen. De studie via het voer meet de depuratie om een depuratiesnelheidsconstante, k 2, te verkrijgen. Indien op basis van de beschikbare gegevens een opnamesnelheidsconstante kan worden geschat voor de situatie waarin de vissen via het water werden blootgesteld aan de teststof, kan een kinetische BCF worden geschat. De schatting van een opnamesnelheidsconstante voor blootstelling aan een teststof via het water is gebaseerd op veel aannames, die allemaal bijdragen aan de onzekerheid van de schatting. Deze manier om een BCF te schatten gaat er bovendien vanuit dat de algemene depuratiesnelheid (met inbegrip van bijdragende factoren zoals verdeling in het lichaam en depuratieprocessen in afzonderlijke vissen) onafhankelijk is van de blootstellingsmethode die is gebruikt om een bepaalde lichaamsconcentratie van de teststof te genereren. De belangrijkste aannames die inherent zijn aan de aanpak van de schattingen kunnen als volgt worden samengevat. De depuratie na opname via voer is voor een gegeven stof dezelfde als de depuratie na blootstelling aan die stof via het water. De opname uit water volgt eersteordekinetiek. Afhankelijk van de gebruikte methode om de opname te schatten:
is de databank (“oefenset”) die is gebruikt om de methode voor de schatting van de opname te ontwikkelen, representatief voor de onderzochte stof. In diverse publicaties in de open literatuur zijn vergelijkingen afgeleid die de opname uit water in vissen via de kieuwen relateren aan een octanol-water-verdelingscoëfficiënt van de stof, het gewicht van vissen (1) (2) (3) (4), het volume en/of vetgehalte, permeatie/diffusie door membranen (5) (6), ventilatievolume van vissen (7) en door een fugaciteit/massabalansaanpak (8) (9) (10). Een gedetailleerde evaluatie van dergelijke methoden in deze context wordt gegeven in Crookes & Brooke (11). Een publicatie van Barber (12) die zich richt op de modellering van bioaccumulatie via opname uit het voer, is in deze context ook nuttig, aangezien zij ook ingaat op bijdragen van modellen voor de opnamesnelheid via de kieuwen. Ook een paragraaf van het achtergronddocument bij het protocol betreffende blootstelling via het voer uit 2004 (13) is gewijd aan dit aspect. De meeste van deze modellen lijken te zijn afgeleid met behulp van beperkte databanken. Voor modellen waarvoor bijzonderheden beschikbaar zijn over de databank die bij het opstellen van het model werd gebruikt, blijkt dat de gebruikte typen stoffen vaak een vergelijkbare structuur hebben of tot een vergelijkbare klasse behoren (op het punt van functionaliteit, bijvoorbeeld gechloreerde organische verbindingen). Dit vergroot de onzekerheid wanneer een model wordt gebruikt om voor een ander type stoffen een opnamesnelheidsconstante te ramen, naast de voor de test specifieke overwegingen zoals soort, temperatuur enz. In een beoordeling van de beschikbare technieken (11) werd naar voren gehaald dat niet één methode “correcter” is dan de andere. Daarom moet een duidelijke rechtvaardiging voor het gebruik van een bepaald model worden gegeven. Wanneer er meerdere methoden beschikbaar zijn waarvan het gebruik te rechtvaardigen is, kan het verstandig zijn om meerdere schattingen van k 1 (en dus van BCF) of een bereik van k 1-waarden (en BCF-waarden) volgens meerdere schattingsmethoden voor de opname te presenteren. Gezien de verschillen in de typen modellen en de gegevensverzamelingen die voor de ontwikkeling ervan werden gebruikt, is het echter niet juist een gemiddelde waarde te nemen van schattingen die op verschillende manieren zijn verkregen. Sommige onderzoekers hebben geponeerd dat dit soort BCF-schattingen moeten worden gecorrigeerd voor de biologische beschikbaarheid om rekening te houden met de adsorptie van een stof aan opgeloste organische koolstof (DOC) in omstandigheden met blootstelling via het water, teneinde de schattingen in overeenstemming te brengen met de resultaten van studies met blootstelling via het water (bv. (13) (14)). Deze correctie is mogelijk echter niet gepast, gezien de lage DOC-niveaus in een studie met blootstelling via het water voor een “worst case”-schatting (d.w.z. verhouding van biologisch beschikbare stof en stof als gemeten in de oplossing). Voor zeer hydrofobe stoffen kan de opname via de kieuwen beperkt worden door de snelheid van de passieve diffusie dicht bij het kieuwoppervlak; in dit geval pakt de correctie mogelijk dit effect aan, in plaats van hetgeen waarvoor zij was bedoeld. Het is aan te bevelen zich te richten op methoden die inputs vereisen waarvoor gemakkelijk gegevens te vinden zijn voor de stoffen die volgens de hier beschreven studie via het voer worden getest (d.w.z. log K OW, gewicht van de vis). Andere methoden waarvoor complexere inputs nodig zijn, kunnen worden toegepast, maar daarvoor kunnen aanvullende metingen in de test nodig zijn of kan gedetailleerde kennis van de teststof of de vissoort vereist zijn die mogelijk niet breed beschikbaar is. Daarnaast kan de keuze van het model ook worden beïnvloed door het niveau van de validering en het toepassingsgebied (zie (11) voor een beoordeling en vergelijking van verschillende methoden). Er moet rekening worden gehouden met het feit dat de resulterende schatting van k 1 en de geschatte BCF onzeker zijn en mogelijk samen met de afgeleide BMF en stofparameters (bv. molecuulgrootte) moeten worden onderworpen aan een op bewijskracht gebaseerde aanpak om een totaalbeeld van het bioaccumulatiepotentieel van een stof te krijgen. De interpretatie en het gebruik van deze parameters kan afhangen van het regelgevingskader. LITERATUUR
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
17) |
In deel C wordt hoofdstuk C.20 vervangen door: „C.20 Daphnia magna voortplantingstest INLEIDING Deze testmethode is gelijkwaardig aan testrichtlijn (TG) 211 (2012) van de OESO. Testrichtlijnen van de OESO worden periodiek in het licht van de wetenschappelijke vorderingen getoetst. Testrichtlijn 211 betreffende de voortplantingstest is voortgekomen uit testrichtlijn 202, deel II, Daphnia sp. voortplantingstest (1984). Er werd algemeen erkend dat gegevens van tests die zijn uitgevoerd volgens testrichtlijn 202, wisselend kunnen zijn. Dit heeft geleid tot aanzienlijke inspanningen om de oorzaken van deze variabiliteit te identificeren, teneinde een betere testmethode op te stellen. Testrichtlijn 211 is gebaseerd op de uitkomsten van deze onderzoeksactiviteiten, ringtests en valideringsstudies die zijn uitgevoerd in 1992 (1), 1994 (2) en 2008 (3). De belangrijkste verschillen tussen de oorspronkelijke versie (TG 202, 1984) en de tweede versie (TG 211, 1998) van de testrichtlijn betreffende de voortplantingstest zijn:
De gebruikte definities zijn opgenomen in aanhangsel 1. PRINCIPE VAN DE TEST Het doel van de test is in de eerste plaats het beoordelen van het effect van chemicaliën op het voortplantingsresultaat van Daphnia magna. Jonge vrouwelijke Daphnia (de moederdieren), die bij aanvang van de test nog geen 24 uur oud zijn, worden daartoe blootgesteld aan de teststof die aan het water is toegevoegd in een aantal uiteenlopende concentraties. De test duurt 21 dagen. Aan het eind van de test wordt het totale aantal levende nakomelingen beoordeeld. Het voortplantingsresultaat van de moederdieren kan ook op andere manieren worden uitgedrukt (bijvoorbeeld in het aantal levende nakomelingen dat per dag en per dier is voortgebracht vanaf de eerste dag dat er nakomelingen werden waargenomen), maar die moeten dan wel worden vermeld naast het totale aantal levende nakomelingen dat aan het eind van de test werd voortgebracht. Vanwege de specifieke opzet van de semistatische test vergeleken met andere voortplantingstestmethoden met ongewervelden, kan ook het aantal levende nakomelingen voor elk afzonderlijk moederdier worden geteld. Dit maakt het mogelijk om, anders dan bij andere voortplantingstestmethoden met ongewervelden, de nakomelingen van een moederdier dat tijdens de testperiode onopzettelijk of onbedoeld sterft, van de gegevensbeoordeling uit te sluiten. Als zich bij blootgestelde replicaties mortaliteit van moederdieren voordoet, moet daarom worden gekeken of de mortaliteit een concentratie-effectpatroon vertoont, bijvoorbeeld of een regressie van de respons uitgezet tegen de concentratie van de teststof een significant verband met een positieve helling laat zien (hiervoor kan een statistische toets zoals de trendtoets van Cochran-Armitage worden gebruikt). Indien de mortaliteit geen concentratie-effectpatroon vertoont, moeten de replicaties met mortaliteit van de moederdieren van de analyse van het testresultaat worden uitgesloten. Indien de mortaliteit wel een concentratie-effectpatroon vertoont, moet de mortaliteit van de moederdieren worden beschouwd als een effect van de teststof en mogen de replicaties niet van de analyse worden uitgesloten. Indien het moederdier tijdens de test sterft, d.w.z. ofwel onopzettelijk ten gevolge van een verkeerde behandeling of een ongeluk, ofwel onbedoeld als gevolg van een onverklaard incident dat geen verband houdt met het effect van de teststof, of indien het dier van het mannelijk geslacht blijkt te zijn, wordt de replicatie van de analyse uitgesloten (zie verder in punt 51). Het toxische effect van de teststof op het voortplantingsresultaat wordt uitgedrukt als ECx door met niet-lineaire regressie een passend model aan de gegevens te passen om de concentratie te schatten die zou leiden tot een afname van x % van het voortplantingsresultaat, of anders als de NOEC/LOEC-waarde (4). De laagste gebruikte effectconcentratie (bv. EC10) moet bij voorkeur in het bereik van de testconcentraties liggen, wat betekent dat deze waarde wordt berekend door interpolatie, en niet door extrapolatie. Het aantal overlevende moederdieren en de tijd die nodig was voor het voortbrengen van het eerste broedsel, moeten eveneens worden vermeld. Andere effecten van de teststof op parameters zoals de groei (bv. lengte), en de mogelijke intrinsieke snelheid van de populatietoename, kunnen ook worden onderzocht (zie punt 44). INFORMATIE OVER DE TESTSTOF De resultaten van een test voor acute toxiciteit (zie hoofdstuk C.2 van deze bijlage: Daphnia sp. — acute immobilisatietest) uitgevoerd met Daphnia magna kunnen bruikbaar zijn bij de selectie van een geschikt bereik van testconcentraties voor de voortplantingstests. De oplosbaarheid in water en de dampdruk van de teststof dienen bekend te zijn en er moet een betrouwbare analysemethode voor de kwantitatieve bepaling van de stof in de testoplossingen met een gerapporteerde recovery-capaciteit en bepaalbaarheidsgrens beschikbaar zijn. De informatie over de teststof die nuttig kan zijn om de testomstandigheden te bepalen, omvat bijvoorbeeld de structuurformule, zuiverheid van de chemische stof, stabiliteit in licht, stabiliteit onder de testomstandigheden, pKa, Pow en resultaten van een test op de gemakkelijke biologische afbreekbaarheid (zie hoofdstuk C.4 (Bepaling van de ”gemakkelijke“ biologische afbreekbaarheid) en hoofdstuk C.29 (Gemakkelijke biologische afbreekbaarheid — CO2 in gesloten vaten) van deze bijlage). GELDIGHEID VAN DE TEST Wil de test geldig zijn, dan moet in de controlegroep(en) aan de volgende prestatiecriteria worden voldaan:
Opmerking: hetzelfde geldigheidscriterium (20 %) kan ook worden gebruikt voor onopzettelijke en onbedoelde mortaliteit van moederdieren, zowel voor de controles als voor elk van de testconcentraties. BESCHRIJVING VAN DE METHODE Apparatuur Testvaten en andere apparaten die in aanraking komen met de testoplossingen, mogen alleen uit glas of een ander chemisch inert materiaal bestaan. Normaal gesproken zijn de testvaten glazen bekers. Daarnaast zal (een deel van) de volgende apparatuur nodig zijn:
Testorganismen De testsoort is Daphnia magna Strauss (95). De kloon moet bij voorkeur zijn geïdentificeerd door middel van bepaling van het genotype. Uit onderzoek (1) is gebleken dat de voortplantingsprestatie van kloon A (die afkomstig is van IRCHA in Frankrijk) (5) consequent voldoet aan het geldigheidscriterium van een gemiddelde van ≥ 60 levende nakomelingen per overlevend moederdier indien gekweekt onder de in deze testmethode beschreven voorwaarden. Desalniettemin zijn andere klonen aanvaard mits de Daphnia-cultuur aantoonbaar voldoet aan de geldigheidscriteria voor de test. Bij aanvang van de test mogen de dieren nog geen 24 uur oud zijn; ze mogen geen eerste nakomelingen zijn. Ze moeten afkomstig zijn van een gezonde stam (d.w.z. zonder tekenen van stress zoals hoge mortaliteit, aanwezigheid van mannelijke dieren en ephippia, vertraging bij het voortbrengen van het eerste broedsel, verkleurde dieren enz.). De stamdieren moeten onder soortgelijke kweekomstandigheden (licht, temperatuur, medium, voeding en aantal dieren per eenheid) worden gehouden als die welke van toepassing zijn tijdens de test. Indien het tijdens de test te gebruiken Daphnia-kweekmedium afwijkt van het medium voor de gangbare Daphnia-cultuur, verdient het aanbeveling een aan de test voorafgaande acclimatiseringsperiode van normaliter circa drie weken (d.w.z. één generatie aan te houden om stress bij de moederdieren te voorkomen. Testmedium Het verdient aanbeveling om bij deze test gebruik te maken van een volledig beschreven medium. Op die manier kan het gebruik van additieven vermeden worden (zoals zeewier, grondextract), die moeilijk te typeren zijn, en kan er beter gestandaardiseerd worden tussen laboratoria. De media Elendt M4 (6) en M7 (zie aanhangsel 2) zijn hiertoe geschikt gebleken. Desalniettemin zijn andere media (bijvoorbeeld (7) en (8)) aanvaardbaar mits de prestaties van de Daphnia-cultuur aantoonbaar voldoen aan de geldigheidscriteria voor de test. Indien gebruik wordt gemaakt van media met niet-beschreven additieven, moeten die additieven niet alleen duidelijk worden gespecificeerd, maar moeten er ook gegevens in het testverslag worden opgenomen over de samenstelling, in het bijzonder met betrekking tot het koolstofgehalte, aangezien dit een bijdrage kan leveren aan de voeding. Het verdient aanbeveling de totale organische koolstof (TOC) en/of het chemisch zuurstofverbruik (COD) van het stampreparaat van het organische additief te bepalen en een schatting te maken van de resulterende bijdrage aan TOC/COD in het testmedium. Voorts zijn de TOC-niveaus in het medium (d.w.z. vóór toevoeging van de algen) bij voorkeur lager dan 2 mg/l (9). In geval van teststoffen met metalen moet worden onderkend dat de eigenschappen van het testmedium (bijvoorbeeld hardheid, chelaatvormend vermogen) van invloed kunnen zijn op de toxiciteit van de teststof. Een volledig beschreven medium is dan ook wenselijk. Op dit moment zijn er echter slechts twee volledig beschreven media waarvan bekend is dat ze geschikt zijn voor langetermijnkweek van Daphnia magna: Elendt M4 en M7. Beide media bevatten de chelaatvormer EDTA. Uit werkzaamheden is naar voren gekomen (2) dat de “klaarblijkelijke toxiciteit” van cadmium in de regel lager is wanneer de voortplantingstest wordt uitgevoerd in M4- en M7-media dan bij uitvoering in media die geen EDTA bevatten. M4 en M7 worden derhalve niet aanbevolen voor metaalhoudende teststoffen, terwijl ook het gebruik van andere media die bekende chelaatvormers bevatten, vermeden dient te worden. Het kan raadzaam zijn om voor metaalhoudende chemische stoffen een ander medium te gebruiken, zoals volgens ASTM geregenereerd hard schoon water (9), dat geen EDTA bevat. Deze combinatie van volgens ASTM geregenereerd hard schoon water en zeewierextract (10) is ook geschikt voor langetermijnkweek van Daphnia magna (2). Het gehalte aan opgeloste zuurstof moet aan het begin en tijdens de test hoger zijn dan 3 mg/l. De pH moet liggen tussen 6 en 9 en normaliter mag er geen variatie van meer dan 1,5 eenheid optreden bij welke test dan ook. Een hardheid van meer dan 140 mg/l (als CaCO3) wordt aanbevolen. Uit tests op dit niveau en de hierboven beschreven tests is naar voren gekomen dat de voortplantingsprestatie voldoet aan de geldigheidscriteria (11) (12). Testoplossingen Testoplossingen van de gekozen concentraties worden gewoonlijk bereid door verdunning van een stamoplossing. Stamoplossingen moeten, indien mogelijk zonder gebruik van oplosmiddelen of dispergeermiddelen, bij voorkeur worden bereid door mengen of schudden van de teststof in het testmedium met mechanische middelen, zoals door te schudden of te roeren of met ultrasone trillingen of andere passende methoden. Bij voorkeur worden de testsystemen vóór de introductie van de testorganismen blootgesteld aan concentraties van de teststof die in het onderzoek worden gebruikt, zolang als nodig is om het behoud van stabiele blootstellingsconcentraties aan te tonen. Indien de teststof moeilijk oplosbaar is in water, moeten de procedures worden gevolgd die zijn beschreven in de OESO-leidraad voor het omgaan met moeilijke stoffen (13). Het gebruik van oplosmiddelen of dispergeermiddelen moet worden vermeden, maar kan in bepaalde gevallen toch nodig zijn om een stamoplossing van een passende concentratie voor dosering te verkrijgen. De test moet worden uitgevoerd met zowel de testconcentraties als een verdunningswatercontrole met voldoende replicaties en, indien niet te vermijden, een oplosmiddelcontrole met voldoende replicaties. In de test mogen uitsluitend oplosmiddelen of dispergeermiddelen worden gebruikt waarvoor onderzoek heeft aangetoond dat ze geen significant effect of slechts een minimaal effect op de responsvariabele hebben. Voorbeelden van geschikte oplosmiddelen (bv. aceton, ethanol, methanol, dimethylformamide en triëethyleenglycol) en dispergeermiddelen (bv. Cremophor RH40, methylcellulose 0,01 % en HCO-40) worden gegeven in (13). Wanneer een oplosmiddel of dispergeermiddel wordt gebruikt, mag de uiteindelijke concentratie ervan niet groter zijn dan 0,1 ml/l (13) en moet deze concentratie in alle testvaten, behalve de verdunningswatercontrole, dezelfde zijn. Er moet echter al het mogelijke worden gedaan om de concentratie van het oplosmiddel zo laag mogelijk te houden. PROCEDURE Blootstellingsomstandigheden Duur De test duurt 21 dagen. Densiteit De moederdieren worden afzonderlijk, d.w.z. één per testvat, in de vaten ondergebracht met 50 tot 100 ml medium in elk vat (voor Daphnia magna kan mogelijk worden volstaan met kleinere volumes, vooral voor kleinere Daphnia-soorten zoals Ceriodaphnia dubia), tenzij het gebruik van een doorstroomsysteem noodzakelijk is voor de test. Soms kunnen grotere volumes nodig zijn om te voldoen aan de eisen van de analytische procedure die wordt gebruikt voor de bepaling van de teststofconcentratie, hoewel het bundelen van replicatieonderzoeken voor chemische analyse ook is toegestaan. Bij gebruik van volumes van meer dan 100 ml dient de hoeveelheid voer die aan de Daphnia wordt gegeven, eventueel te worden verhoogd om ervoor te zorgen dat er voldoende voer aanwezig is en dat wordt voldaan aan de geldigheidscriteria. Proefdieren Bij semistatische tests ten minste tien dieren afzonderlijk gehouden bij elke testconcentratie en ten minste tien dieren afzonderlijk gehouden in de controlereeks. Wat doorstroomtests betreft, is gebleken dat een aantal van 40, in vier groepen van tien verdeelde dieren bij elke testconcentratie geschikt is (1). Een kleiner aantal testorganismen is toegestaan; een minimum van 20 dieren per concentratie verdeeld over twee of meer replicatieonderzoeken met een gelijk aantal dieren (bijvoorbeeld vier replicatieonderzoeken met elk vijf watervlooien) wordt aanbevolen. Opgemerkt dient te worden dat het bij tests waar dieren in groepen worden gehouden, niet mogelijk is nakomelingen van de statistische analyse uit te sluiten als zich na aanvang van de voortplanting onbedoelde/onopzettelijke mortaliteit van moederdieren voordoet. In die gevallen moet het voortplantingsresultaat daarom worden uitgedrukt in het “totale aantal levende nakomelingen per moederdier dat bij aanvang van de test aanwezig was”. De behandelingen moeten alle plaatsvinden in een speciaal voor de desbetreffende behandeling bestemd testvat en de testvaten moeten daarna op willekeurige wijze worden behandeld. Het nalaten hiervan kan een vertekend beeld geven dat uitgelegd zou kunnen worden als een concentratie-effect. In het bijzonder moet hierbij gedacht worden aan de mogelijkheid dat, wanneer met experimentele units wordt omgegaan in volgorde van behandeling of concentratie, bepaalde met tijd verband houdende effecten, zoals vermoeidheid bij de uitvoerder van het experiment of andere fouten, kunnen leiden tot het vaststellen van grotere effecten bij de hogere concentraties. Bovendien zou overwogen moeten worden de test af te breken als de testresultaten beïnvloed kunnen worden door een conditie waarvan reeds sprake was bij aanvang van de test of door een omgevingsconditie, zoals de plaats in het laboratorium. Voeding Bij semistatische tests moet er bij voorkeur dagelijks worden gevoerd, doch in ieder geval driemaal per week (d.w.z. naar gelang van de verandering van medium). Er moet rekening worden gehouden met de mogelijke verdunning van de blootstellingsconcentraties door toevoeging van voer en dergelijke verdunning moet zo veel mogelijk worden vermeden met goed geconcentreerde algensuspensies. Indien hiervan wordt afgeweken (bijvoorbeeld bij doorstroomtests), moet dat in het testverslag worden vermeld. Tijdens de test moet de voeding van de moederdieren bij voorkeur bestaan uit levende algencellen van ten minste een van de volgende: Chlorella sp., Pseudokirchneriella subcapitata (voorheen Selenastrum capricornutum) en Desmodesmus subspicatus (voorheen Scenedesmus subspicatus). Uitgangspunt bij de voeding moet de aan elk moederdier toegediende hoeveelheid organische koolstof (C) zijn. Uit onderzoek (14) is gebleken dat een hoeveelheid van 0,1 à 0,2 mg C/Daphnia/dag voor Daphnia magna voldoende is om een zodanig aantal levende nakomelingen te krijgen dat wordt voldaan aan de geldigheidscriteria voor de test. De voeding kan worden gegeven in een gelijkmatige, over de gehele testperiode uitgesmeerde hoeveelheid of desgewenst in een lagere hoeveelheid bij aanvang van de test, die daarna wordt opgevoerd teneinde rekening te houden met de groei van de moederdieren. Ook in dat geval moet de hoeveelheid echter altijd binnen de aanbevolen hoeveelheid van 0,1 tot 0,2 mg C/Daphnia/dag blijven. Indien er gebruik moet worden gemaakt van alternatieve middelen, zoals algencelaantal of lichtextinctie, om aan de vereiste hoeveelheid voeding te komen (d.w.z. gemakshalve, aangezien de meting van het koolstofgehalte tijdrovend is), moet elk laboratorium zijn eigen nomografie opstellen waarin het alternatieve middel wordt gerelateerd aan het koolstofgehalte van de algencultuur (zie aanhangsel 3 voor suggesties inzake het opstellen van een nomografie). Nomografieën moeten ten minste eenmaal per jaar worden gecontroleerd, doch vaker als de algencultuuromstandigheden zijn gewijzigd. Gebleken is dat lichtextinctie een beter alternatief voor het koolstofgehalte is dan het celaantal (15). Er dient een geconcentreerde algensuspensie te worden toegediend aan de Daphnia om de hoeveelheid algen-kweekmedium die in de testvaten wordt overgebracht, zoveel mogelijk te beperken. De algen kunnen worden geconcentreerd door middel van centrifugeren, gevolgd door hersuspensie in Daphnia-kweekmedium. Licht 16 uren licht met een intensiteit van ten hoogste 15-20 μE · m-2 · s-1 gemeten op het wateroppervlak van het vat. Voor lichtmeetinstrumenten die in lux gekalibreerd zijn, komt een bereik van 1000 — 1500 lux voor “cool white” licht ongeveer overeen met de aanbevolen lichtintensiteit 15-20 μE·m-2·s-1. Temperatuur De temperatuur van de testmedia moet liggen tussen 18-22 °C. Waar mogelijk mag de temperatuur, als dagelijks bereik, echter bij geen enkele test met meer dan 2 °C variëren binnen dit bereik (bijvoorbeeld 18-20, 19-21 of 20-22 °C). Voor de controle van de temperatuur kan het dienstig zijn een extra testvat te gebruiken. Beluchting De testvaten mogen niet worden belucht tijdens de test. Testopzet Bereikbepalingstest Indien nodig wordt een bereikbepalingstest uitgevoerd met bijvoorbeeld vijf teststofconcentraties en twee replicaties voor elke behandeling en controle. Aanvullende informatie, uit tests met soortgelijke chemische stoffen of uit de literatuur, over acute toxiciteit voor Daphnia en/of andere aquatische organismen kan ook nuttig zijn om de reeks concentraties voor de bereikbepalingstest vast te stellen. De duur van de bereikbepalingstest is 21 dagen of voldoende lang om effectniveaus op betrouwbare wijze te kunnen voorspellen. Aan het eind van de test wordt de voortplanting van de Daphnia beoordeeld. Het aantal moederdieren en de aanwezigheid van nakomelingen moeten worden geregistreerd. Definitieve test Normaal gesproken zijn er ten minste vijf testconcentraties, aan weerszijden van de effectieve concentratie (bv. ECx), die een meetkundige reeks vormen met een rede van bij voorkeur niet hoger dan 3,2. Er moet een passend aantal replicatieonderzoeken voor elke testconcentratie worden gebruikt (zie de punten 24-25). Als er minder dan vijf concentraties worden gebruikt, moet een motivering worden gegeven. Chemische stoffen mogen niet worden getest boven de grens van hun oplosbaarheid in het testmedium. Het is raadzaam om, alvorens het experiment uit te voeren, na te denken over het statistisch onderscheidingsvermogen van de testopzet en passende statistische methoden toe te passen (4). Bij de vaststelling van het concentratiebereik moet aandacht worden geschonken aan het volgende:
Als bij de hoogste concentratie in de bereikbepalingstest (bv. bij 10 mg/l) geen effecten worden waargenomen, of wanneer op grond van het ontbreken van toxiciteit voor andere organismen en/of een lage/geen opname wordt gedacht dat de teststof zeer waarschijnlijk een geringe/geen toxiciteit heeft, kan de voortplantingstest worden uitgevoerd als een limiettest, waarbij bijvoorbeeld 10 mg/l als testconcentratie en de controle worden gebruikt. Er moeten tien replicatieonderzoeken worden gebruikt voor zowel de behandelings- als de controlegroepen. Wanneer in een doorstroomsysteem mogelijk een limiettest moet worden uitgevoerd, kan worden volstaan met minder replicatieonderzoeken. Een limiettest biedt de mogelijkheid om aan te tonen dat er bij de limietconcentratie geen statistisch significant effect is, maar als er effecten worden waargenomen, is normaal gesproken een volledige test vereist. Controles Ter aanvulling op de testreeks moet één testmediumcontrolereeks en, indien relevant, één controlereeks met het oplosmiddel of het dispergeermiddel worden uitgevoerd. Indien gebruikt moet de oplosmiddel- of dispergeermiddelconcentratie dezelfde zijn als in de vaten met de teststof. Het juiste aantal replicatieonderzoeken moet worden verricht (zie de punten 23-24). Bij een goed uitgevoerde test moet de coëfficiënt van de variatie rond het gemiddelde aantal levende nakomelingen per moederdier in de controlegroep(en) in de regel ≤ 25 % zijn; dit moet worden vermeld voor elke testopzet waarbij gebruik wordt gemaakt van afzonderlijk gehouden dieren. Verversing van het testmedium De frequentie waarmee het medium wordt ververst, hangt af van de stabiliteit van de teststof, maar moet ten minste driemaal per week zijn. Indien uit voorafgaande stabiliteitstests (zie punt 7) blijkt dat de teststofconcentratie niet stabiel is (d.w.z. buiten het bereik van 80 tot 120 % van de nominale concentratie of dalend tot onder 80 % van de gemeten beginconcentratie) gedurende de maximale verversingsperiode (d.w.z. drie dagen), moet worden overwogen het medium vaker te verversen of gebruik te maken van een doorstroomtest. Wanneer het medium wordt ververst in semistatische tests, wordt een tweede reeks testvaten bereid waarin de moederdieren worden overgebracht door middel van bijvoorbeeld een glazen pipet met een geschikte diameter. De hoeveelheid met de Daphnia overgebracht medium moet zo klein mogelijk zijn. Observaties De resultaten van de observaties gedurende de test moeten worden vermeld op informatiebladen (zie de voorbeelden in de aanhangsels 4 en 5). Indien er nog andere metingen gedaan moeten worden (zie punt 44), zijn er mogelijk meer observaties nodig. Nakomelingen De door elk moederdier voortgebrachte nakomelingen moeten bij voorkeur dagelijks worden verwijderd en geteld vanaf het moment dat het eerste broedsel verschijnt, teneinde te voorkomen dat ze voer opeten dat voor het moederdier is bestemd. Hoewel in het kader van deze testmethode alleen het aantal levende nakomelingen moet worden geteld, moet ook de aanwezigheid van niet-uitgekomen eitjes of dode nakomelingen worden vermeld. Mortaliteit De mortaliteit onder de moederdieren moet bij voorkeur dagelijks worden geregistreerd of ten minste even vaak als de nakomelingen worden geteld. Andere parameters Hoewel deze testmethode voornamelijk bedoeld is voor de beoordeling van de effecten op het voortplantingsresultaat, kunnen er mogelijk ook andere effecten in zodanige mate worden gekwantificeerd dat statistische analyse mogelijk is. Het voortplantingsresultaat per overlevend moederdier, d.w.z. het aantal levende nakomelingen voorgebracht tijdens de testperiode per overlevend moederdier, kan worden geregistreerd. Dit kan worden vergeleken met de belangrijkste responsvariabele (voortplantingsresultaat per moederdier aan het begin van de test dat tijdens de test niet onbedoeld of onopzettelijk stierf). Als zich bij blootgestelde replicaties mortaliteit van moederdieren voordoet, moet worden gekeken of de mortaliteit een concentratie-effectpatroon vertoont, bijvoorbeeld of een regressie van de respons uitgezet tegen de concentratie van de teststof een significant verband met een positieve helling laat zien (hiervoor kan een statistische toets zoals de trendtoets van Cochran-Armitage worden gebruikt). Indien de mortaliteit geen concentratie-effectpatroon vertoont, moeten de replicaties met mortaliteit van de moederdieren van de analyse van het testresultaat worden uitgesloten. Indien de mortaliteit wel een concentratie-effectpatroon vertoont, moet de mortaliteit van de moederdieren worden beschouwd als een effect van de teststof en mogen de replicaties niet van de analyse van het testresultaat worden uitgesloten. Groeimetingen zijn zeer wenselijk aangezien zij informatie verschaffen over mogelijke subletale effecten, welke gegevens soms bruikbaarder zijn dan voortplantingsmetingen alleen; de meting van de lengte van de moederdieren (d.w.z. de lichaamslengte met uitzondering van het anale uiteinde) aan het eind van de test wordt aanbevolen. Andere parameters die kunnen worden gemeten of berekend zijn de tijd die nodig is om het eerste broedsel (en de daaropvolgende broedsels) voort te brengen, aantal en omvang van de broedsels per dier, aantal niet-uitgekomen eitjes, aanwezigheid van mannelijke neonaten (OESO, 2008) of ephippia en mogelijk de intrinsieke groeisnelheid van de populatie (zie aanhangsel 1 voor definities en aanhangsel 7 voor de bepaling van het geslacht van neonaten). Frequentie van analytische bepalingen en metingen De zuurstofconcentratie, temperatuur, hardheid en pH-waarden moeten ten minste eenmaal per week worden gemeten, in verse en oude media, in de controle(s) en in de hoogste teststofconcentratie. Tijdens de test worden de concentraties van de teststof regelmatig bepaald. Bij semistatische tests waarbij de concentratie van de teststof naar verwachting binnen ± 20 % van de nominale waarde zal blijven (d.w.z. binnen het bereik van 80 tot 120 %, zie de punten 6, 7 en 39), verdient het aanbeveling de hoogste en laagste testconcentraties in ieder geval te analyseren bij de verse bereiding ervan alsmede eenmaal bij verversing in de eerste week van de test (d.w.z. analyses moeten worden verricht op een monster van een en dezelfde oplossing — bij verse bereiding en bij verversing). Daarna moeten deze bepalingen in ieder geval wekelijks worden herhaald. Bij tests waarbij de concentratie van de teststof naar verwachting niet binnen ± 20 % van de nominale waarde zal blijven, moeten alle testconcentraties zowel bij de verse bereiding als bij verversing worden geanalyseerd. Bij tests echter waarbij de gemeten beginconcentratie van de teststof weliswaar niet binnen ± 20 % van de nominale waarde ligt, maar wel naar tevredenheid kan worden aangetoond dat de beginconcentraties herhaalbaar en stabiel zijn (d.w.z. binnen het bereik van 80 tot 120 % van de beginconcentraties), kunnen de chemische bepalingen in de weken 2 en 3 van de test worden beperkt tot de hoogste en laagste testconcentraties. In alle gevallen hoeft de bepaling van teststofconcentraties vóór de verversing slechts op één replicatieonderzoekvat bij elke testconcentratie te worden uitgevoerd. Bij doorstroomtests is een gelijksoortige monstermethode van toepassing als bij semistatische tests (met dien verstande dat de meting van oplossingen hier niet van toepassing is). Desalniettemin kan het raadzaam zijn meer monsters te nemen in de eerste week (bijvoorbeeld via drie reeksen metingen) om te bewerkstelligen dat de testconcentraties stabiel blijven. Bij dit soort testen moet de doorstromingssnelheid van het oplosmiddel en de teststof dagelijks worden gecontroleerd. Als er voldoende aanwijzingen zijn dat de concentratie van de te testen stof gedurende de gehele test en naar tevredenheid binnen ± 20 % van de nominale of gemeten beginconcentratie is gebleven, mogen de resultaten worden gebaseerd op nominale of gemeten beginwaarden. Indien de afwijking van de nominale of gemeten beginconcentratie groter is dan ± 20 %, moeten de resultaten worden uitgedrukt in tijdgewogen gemiddelde (zie leidraad voor de berekening in aanhangsel 6). GEGEVENS EN RAPPORTAGE Verwerking van de resultaten Deze test is bedoeld om het effect van de teststof op het voortplantingsresultaat te bepalen. Het totale aantal levende nakomelingen per moederdier moet per testvat worden berekend (d.w.z. via replicatieonderzoek). Daarnaast kan het voortplantingsresultaat worden berekend op basis van het aantal levende nakomelingen per overlevend moederdier. De responsvariabele die ecologisch het relevantst is, is echter het totale aantal levende nakomelingen per moederdier dat niet onopzettelijk (96) of onbedoeld (97) sterft tijdens de test. Als het moederdier onopzettelijk of onbedoeld sterft tijdens de test, of als het blijkt te gaan om een mannelijk dier, wordt het replicatieonderzoek niet meegenomen in de analyse. De analyse zal in dat geval worden gebaseerd op een lager aantal replicatieonderzoeken. Als zich bij blootgestelde replicaties mortaliteit van moederdieren voordoet, moet worden gekeken of de mortaliteit een concentratie-effectpatroon vertoont, bijvoorbeeld of een regressie van de respons uitgezet tegen de concentratie van de teststof een significant verband met een positieve helling laat zien (hiervoor kan een statistische toets zoals de trendtoets van Cochran-Armitage worden gebruikt). Indien de mortaliteit geen concentratie-effectpatroon vertoont, moeten de replicaties met mortaliteit van de moederdieren van de analyse van het testresultaat worden uitgesloten. Indien de mortaliteit wel een concentratie-effectpatroon vertoont, moet de mortaliteit van de moederdieren worden beschouwd als een effect van de teststof en mogen de replicaties niet van de analyse van het testresultaat worden uitgesloten. Samengevat, wanneer de LOEC en NOEC of ECx worden gebruikt om de effecten uit te drukken, verdient het aanbeveling het effect op de voortplanting te berekenen door de hierboven genoemde responsvariabelen beide te gebruiken, d.w.z.
en dan als eindresultaat de laagste waarde van de NOEC en LOEC of ECx te gebruiken die met gebruik van een van deze twee responsvariabelen is berekend. Alvorens de statistische analyse uit te voeren, verdient het aanbeveling een gegevenstransformatie te overwegen indien dit nodig is om te voldoen aan de eisen van een specifieke statistische toets, bijvoorbeeld ANOVA-procedures, vergelijking van de behandelingen met de controle door een t-toets van Student, Dunnett-toets, Williams-toets of stap-omlaag-trendtoets van Jonckheere-Terpstra. Als verdelingsvrije alternatieven kunnen een toets van Dunn of Mann-Whitney-toets worden overwogen. 95 %-betrouwbaarheidsintervallen worden berekend voor afzonderlijke behandelingsgemiddelden. Het aantal overlevende moederdieren in de onbehandelde controles is een geldigheidscriterium en moet worden gedocumenteerd en gerapporteerd. Ook alle andere schadelijke effecten, zoals afwijkend gedrag en toxicologisch significante bevindingen, moeten in het eindverslag worden vermeld. ECx ECx-waarden en de bijbehorende betrouwbaarheidsgrenzen worden berekend met een geschikte statistische methode (bv. logistische functie of Weibull-functie, getrimde Spearman-Karber-methode of eenvoudige interpolatie). Voor de berekening van de EC10, EC50 of een andere ECx wordt regressieanalyse uitgevoerd met de volledige gegevensreeks. NOEC/LOEC Indien het doel van een statistische analyse is de NOEC/LOEC te bepalen, moeten passende statistische methoden worden gebruikt conform document 54 van de OESO, “Current Approaches in the Statistical Analysis of Ecotoxicity Data: a Guidance to Application” (4). Negatieve effecten van de teststof, in vergelijking met de controle, worden in het algemeen onderzocht met behulp van eenzijdige hypothesetoetsing met p ≤ 0,05. Een normale verdeling van de gegevens en homogeniteit van de variantie kunnen met een passende statistische toets worden getoetst, bijvoorbeeld respectievelijk met de toets van Shapiro-Wilk en de Levene-toets (p ≤ 0,05). Er kunnen een éénwegs variantieanalyse en vervolgens toetsen voor meervoudige vergelijkingen worden uitgevoerd. Toetsen voor meervoudige vergelijkingen (bv. Dunnett-toets) of stap-omlaag-trendtoetsen (bv. Williams-toets of stap-omlaag-trendtoets van Jonckheere-Terpstra) kunnen worden gebruikt om na te gaan of er significante verschillen (p ≤ 0,05) zijn tussen de controles en de verschillende concentraties van de teststof (selectie van de aanbevolen toets overeenkomstig document 54 van de OESO (4)). Anders kunnen verdelingsvrije toetsen (bv. Bonferroni-toets volgens Holm of de Jonckheere-Terpstra-trendtoets) worden gebruikt om de NOEC en LOEC te bepalen. Limiettest Als een limiettest (vergelijking van de controle en slechts één behandeling) is uitgevoerd en er is voldaan aan de voorwaarden van parametrische toetsprocedures (normaliteit, homogeniteit), kunnen metrische responsen worden geëvalueerd met de t-toets van Student. Als niet aan die voorwaarden wordt voldaan, kan een t-toets voor ongelijke varianties (zoals de Welch-toets) of een verdelingsvrije toets, zoals de Mann-Whitney-toets, worden gebruikt. Om significante verschillen tussen de controles (controle en oplosmiddelcontrole of dispergeermiddelcontrole) vast te stellen, kunnen de replicaties van de beide controles worden getoetst als beschreven voor de limiettest. Als uit deze toetsen geen significante verschillen naar voren komen, mogen alle replicaties van de controle en oplosmiddelcontrole worden gepoold. Anders dienen alle behandelingen te worden vergeleken met de oplosmiddelcontrole. Testverslag In het testverslag moet de volgende informatie worden opgenomen:
LITERATUUR
Aanhangsel 1 DEFINITIES Voor de toepassing van deze testmethode wordt verstaan onder: Aantoonbaarheidsgrens : de laagste concentratie die wel kan worden gedetecteerd doch niet gekwantificeerd. Bepaalbaarheidsgrens : de laagste concentratie die kwantitatief meetbaar is. Chemische stof : een stof of een mengsel. Ecx : de concentratie van de in water opgeloste teststof die resulteert in een x %-vermindering van de voortplanting van Daphnia binnen een aangegeven blootstellingsperiode. Intrinsieke groeisnelheid van de populatie : een maat voor de groei van de populatie waarbij wordt uitgegaan van het voortplantingsresultaat en de leeftijdspecifieke mortaliteit (1) (2) (3). Bij stabiele populaties is die snelheid nul. Bij toenemende populaties is zij positief en bij afnemende populaties negatief. De laatstgenoemde populaties zijn uiteraard niet levensvatbaar en zullen uiteindelijk uitsterven. Lowest Observed Effect Concentration (LOEC) (concentratie waarbij het laagste effect wordt waargenomen) : de laagste testconcentratie van de stof waarbij een effect is waargenomen dat statistisch significant is voor de voortplanting en de mortaliteit van moederdieren (bij p < 0,05) in vergelijking met de controle, binnen een aangegeven blootstellingsperiode. Alle testconcentraties boven de LOEC moeten echter een schadelijk effect hebben dat gelijk is aan of groter dan de effecten die worden waargenomen bij de LOEC. Indien niet aan deze beide voorwaarden kan worden voldaan, moet uitgebreid worden toegelicht hoe de LOEC (en daarmee de NOEC) is geselecteerd. Mortaliteit : een dier wordt geregistreerd als gestorven als het immobiel is, d.w.z. als het niet tot zwemmen in staat is of als er geen beweging van aanhangsels of het postabdomen wordt waargenomen binnen 15 seconden na zachtjes bewegen van het testvat. (Gebruik van een andere definitie moet met bronvermelding worden vermeld.) Moederdieren : de vrouwelijke Daphnia die aanwezig zijn bij aanvang van de test en voorwerp zijn van de studie naar het voortplantingsresultaat. Nakomelingen : de jonge Daphnia die tijdens de test worden voortgebracht door de moederdieren. No Observed Effect Concentration (NOEC) (concentratie waarbij geen effect wordt waargenomen) : de testconcentratie direct onder de LOEC, die in vergelijking met de controle geen statistisch significant effect heeft (p < 0,05) binnen een aangegeven blootstellingsperiode. Onbedoelde mortaliteit : niet met een chemische stof samenhangende mortaliteit met onbekende oorzaak. Onopzettelijke mortaliteit : niet met een chemische stof samenhangende mortaliteit veroorzaakt door een accidentele gebeurtenis (d.w.z. een bekende oorzaak). Teststof : alle volgens deze testmethode geteste stoffen of mengsels. Voortplantingsresultaat : het aantal levende nakomelingen dat in de testperiode door moederdieren wordt voortgebracht. Literatuur
Aanhangsel 2 BEREIDING VAN VOLLEDIG BESCHREVEN MEDIA ELENDT M7 EN M4 Acclimatisering aan Elendt M7- en Elendt M4-media Sommige laboratoria hebben problemen ondervonden bij het rechtstreeks overbrengen van Daphnia in M4- (1) en M7-media. Toch is er enig succes geboekt met geleidelijke acclimatisering, d.w.z. overgang van het eigen medium naar 30 % Elendt, vervolgens naar 60 % Elendt en tot slot naar 100 % Elendt. De vereiste acclimatiseringsperiode kan soms oplopen tot een hele maand. Bereiding Spoorelementen Afzonderlijke stamoplossingen (I) van individuele spoorelementen worden eerst bereid in water met een geschikte zuiverheid, d.w.z. gedeïoniseerd, gedistilleerd of omgekeerde osmose. Uit deze verschillende stamoplossingen (I) wordt een enkelvoudige stamoplossing (II) bereid, die alle spoorelementen bevat (gecombineerde oplossing), te weten:
M4- en M7-media M4- en M7-media worden als volgt bereid met behulp van stamoplossing II, macronutriënten en vitaminen:
De gecombineerde vitaminestamoplossing wordt in bevroren toestand bewaard in kleine fracties. De vitaminen worden kort vóór gebruik aan de media toegevoegd.
Aanhangsel 3 TOTALE ORGANISCHE KOOLSTOF (TOC) ANALYSE EN OPSTELLING VAN EEN NOMOGRAFIE VOOR TOC-GEHALTE VAN ALGENVOEDING Normaal gesproken wordt het koolstofgehalte van de algenvoeding niet rechtstreeks gemeten, maar afgeleid uit correlaties (d.w.z. nomografieën) met alternatieve middelen zoals algencelaantal of lichtextinctie. De TOC moet worden gemeten via hogetemperatuuroxidatie en niet met methoden op basis van UV of persulfaat. (Zie voor advies: The Instrumental Determination of Total Organic Carbon, Total Oxygen Demand and Related Determinands 1979, HMSO 1980; 49 High Holborn, London WC1V 6HB.) Voor het opstellen van nomografieën moeten algen middels centrifugeren worden gescheiden van het groeimedium, gevolgd door hersuspensie in gedistilleerd water. De surrogaatparameter en de TOC-concentratie moet in elk monster driemaal worden gemeten. Blanco's van gedistilleerd water moeten worden geanalyseerd; de TOC-concentratie moet worden afgeleid uit die van het algenmonster. De nomografieën moeten lineair zijn over het vereiste koolstofconcentratiebereik. Voorbeelden zijn hieronder weergegeven.

Chlorella vulgaris var. viridis (CCAP 211/12). Regressie van mg/l drooggewicht op mg C/1. Gegevens van geconcentreerde suspensies van semicontinue batch-gekweekte cellen, gehersuspendeerd in gedistilleerd water. x-as: mg C/1 geconcentreerde algenvoeding y-as: mg/1 drooggewicht van geconcentreerde algenvoeding Correlatiecoëfficiënt -0,980 
Chlorella vulgaris var. viridis (CCAP 211/12). Regressie van celaantal op mg C/1. Gegevens van geconcentreerde suspensies van semicontinue batch-gekweekte cellen, gehersuspendeerd in gedistilleerd water. x-as: mg C/1 geconcentreerde algenvoeding y-as: Aantal cellen/1 geconcentreerde algenvoeding Correlatiecoëfficiënt -0,926 
Chlorella vulgaris var. viridis (CCAP 211/12). Regressie van exctinctie op mg C/1 (1 cm padlengte). Gegevens van geconcentreerde suspensies van semicontinue batch-gekweekte cellen, gehersuspendeerd in gedistilleerd water. x-as: mg C/1 geconcentreerde algenvoeding y-as: Extinctie bij 440 nm van een 1/10 verdunning van geconcentreerde algenvoeding Correlatiecoëfficiënt – 0,998 Aanhangsel 4 VOORBEELD INFORMATIEBLAD VOOR REGISTRATIE VAN MEDIUMVERVERSING, FYSISCH-CHEMISCHE CONTROLEGEGEVENS, VOEDING, DAPHNIA-VOORTPLANTING EN MORTALITEIT MOEDERDIEREN
Aanhangsel 5 VOORBEELD INFORMATIEBLAD VOOR REGISTRATIE VAN RESULTATEN CHEMISCHE ANALYSE a) Gemeten concentraties
b) Gemeten concentraties als percentage van nominale concentratie
Aanhangsel 6 BEREKENING TIJDGEWOGEN GEMIDDELDE Tijdgewogen gemiddelde Daar de concentratie van de teststof in de periode tussen de mediumverversingen kan afnemen, moet goed worden bekeken welke concentratie moet worden gekozen als representatief voor het concentratiebereik dat wordt ondergaan door de Daphnia-moederdieren. Deze keuze moet gebaseerd zijn op biologische én statistische overwegingen. Als bijvoorbeeld wordt aangenomen dat de voortplanting vóór alles wordt beïnvloed door de piekconcentratie, moet de maximumconcentratie worden gebruikt. Wanneer men er daarentegen van uitgaat dat het geaccumuleerde of langeretermijneffect van de toxische stof belangrijker is, is een gemiddelde concentratie relevanter. In dat geval is de tijdgewogen gemiddelde concentratie geschikt, aangezien daarbij rekening wordt gehouden met de variatie van de momentane concentratie over een bepaalde tijdsperiode. Figuur 1: Voorbeeld van tijdgewogen gemiddelde 
Figuur 1 laat een voorbeeld zien van een (vereenvoudigde) test over zeven dagen met mediumverversing op dag 0, 2 en 4.
Het tijdgewogen gemiddelde wordt zo berekend dat het oppervlak onder dat gemiddelde gelijk is aan het oppervlak onder de concentratiekromme. Zie onderstaande tabel 1 voor de berekening van dit voorbeeld. Tabel 1 Berekening van het tijdgewogen gemiddelde
Oppervlak: het oppervlak onder de exponentiële kromme voor elke verversingsperiode. Het wordt berekend aan de hand van de volgende formule:
Het tijdgewogen gemiddelde is het” Totaal oppervlak „gedeeld door het” Totaal dagen„. Voor de toepasbaarheid op de Daphnia-voortplantingstest moet de tabel uiteraard worden uitgebreid en betrekking hebben op een periode van 21 dagen. Het is duidelijk dat niet bevestigd kan worden dat het achteruitgangsproces inderdaad exponentieel is als de waarnemingen alleen plaatsvinden aan het begin en einde van elke verversingsperiode. Een andere kromme zou resulteren in een andere berekening van het” Oppervlak„. Hoe dan ook, het is alleszins plausibel uit te gaan van een exponentieel achteruitgangsproces en de weergegeven kromme is waarschijnlijk het beste alternatief bij gebrek aan andere gegevens. Toch is de nodige voorzichtigheid geboden als geen enkele chemische stof wordt aangetroffen bij de chemische analyse aan het eind van de verversingsperiode. Als niet ingeschat kan worden hoe snel de chemische stof uit de oplossing verdwenen is, kan er ook geen realistisch oppervlak onder de kromme worden vastgesteld, en dus ook geen aannemelijk tijdgewogen gemiddelde. Aanhangsel 7 LEIDRAAD VOOR DE BEPALING VAN HET GESLACHT VAN NEONATEN Onder veranderende omgevingscondities, zoals een korter wordende fotoperiode, temperatuur, afnemende voedselconcentratie en toenemende populatiedichtheid, kunnen neonaten van het mannelijk geslacht worden voortgebracht (Hobaek en Larson, 1990; Kleiven et al., 1992). Het voortbrengen van mannetjes is ook een bekende respons op groeiregulatoren van bepaalde insecten (Oda et al., 2005). Onder omstandigheden waarin chemische stressoren een afname van het aantal reproductieve nakomelingen van de parthenogenetische vrouwtjes veroorzaken, wordt een groter aantal mannetjes verwacht (OESO, 2008). Op basis van de beschikbare informatie kan niet worden voorspeld of de geslachtsverhouding of het voortplantingseindpunt het gevoeligst is; er zijn echter aanwijzingen (zie “Validation report” van de OESO, deel 1) dat de toename in het aantal mannetjes mogelijk minder gevoelig is dan de afname van het aantal nakomelingen. Aangezien het primaire doel van de testmethode is het aantal voortgebrachte nakomelingen te beoordelen, is observatie van de verschijning van mannetjes facultatief. Indien dit facultatieve eindpunt in een studie wordt beoordeeld, moet een aanvullend geldigheidscriterium voor de test van niet meer dan 5 % mannetjes in de controles worden gehanteerd. De meest praktische en gemakkelijkste manier om het geslacht van Daphnia te bepalen is aan de hand van hun fenotypische kenmerken, aangezien mannetjes en vrouwtjes genetisch identiek zijn en hun geslacht door het milieu wordt bepaald. Mannetjes en vrouwtjes verschillen van elkaar in de lengte en morfologie van de eerste antenne, die bij mannetjes langer is dan bij vrouwtjes (figuur 1). Dit verschil is al direct na de geboorte zichtbaar, hoewel zich tijdens de groei van de dieren nog andere secundaire geslachtskenmerken ontwikkelen (zie bijvoorbeeld figuur 2 in Olmstead en LeBlanc, 2000). Om het morfologisch geslacht waar te nemen, moeten de neonaten per proefdier met een pipet worden overgebracht in een petrischaal met testmedium. De hoeveelheid medium wordt zo klein mogelijk gehouden om beweging van de dieren tegen te gaan. De observatie van de eerste antenne kan geschieden onder een stereomicroscoop (× 10-60). Figuur 1 Mannetje (links) en vrouwtje (rechts) van D. magna van 24 uur oud. Mannetjes onderscheiden zich van vrouwtjes door de lengte en morfologie van hun eerste antenne, zoals is te zien in de cirkels (Tatarazako et al., 2004). 
REFERENTIES Hobaek A and Larson P. 1990. Sex determination in Daphnia magna. Ecology 71: 2255-2268. Kleiven O.T., Larsson P., Hobaek A. 1992. Sexual reproduction in Daphnia magna requires three stimuli. Oikos 65, 197-206. Oda S., Tatarazako N, Watanabe H., Morita M., and Iguchi T. 2005. Production of male neonates in Daphnia magna (Cladocera, Crustacea) exposed to juvenile hormones and their analogs. Chemosphere 61:1168-1174. OESO, 2008. Validation report for an enhancement of OECD TG 211 Daphnia magna reproduction test. OECD Series on Testing and Assessment, nr. 88. Organisatie voor Economische Samenwerking en Ontwikkeling, Parijs. Olmstead, A.W., LeBlanc, G.A., 2000. Effects of endocrine-active chemicals on the development characteristics of Daphnia magna. Environmental Toxicology and Chemistry 19:2107-2113. Tatarazako, N., Oda, S., Abe, R., Morita M. and Iguchi T., 2004. Development of a screening method for endocrine disruptors in crustaceans using Daphnia magna (Cladocera, Crustacea). Environmental Science 17, 439-449. |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
18) |
In deel C wordt hoofdstuk C.29, punt 66, vervangen door:
|
|
19) |
In deel C worden de volgende hoofdstukken toegevoegd: „C.47 Test op toxiciteit bij vissen in vroege levensstadia (FELS — fish early-life stage) INLEIDING
PRINCIPE VAN DE TEST
INFORMATIE OVER DE TESTSTOF
GELDIGHEID VAN DE TEST
BESCHRIJVING VAN DE METHODE Testkamers
Keuze van de soorten
Leefomstandigheden van de broedvis
Omgaan met bevruchte eieren, embryo's en larven
Water
Testoplossingen
PROCEDURE Blootstellingsomstandigheden Duur
Densiteit
Licht en temperatuur
Voeding
Testconcentraties
Controles
Frequentie van de analytische bepalingen en metingen
Waarnemingen 26. Stadium van embryonale ontwikkeling : het embryonale stadium aan het begin van de blootstelling aan de teststof moet zo nauwkeurig mogelijk worden gecontroleerd. Hiervoor kan een representatief monster van eitjes worden gebruikt die afdoende zijn gefixeerd en geklaard. 27. Uitkomen en overleven : minimaal één keer per dag moet uitkomen en overleven worden geobserveerd en de aantallen geregistreerd. Als er vroeg in de embryonale ontwikkeling (bijvoorbeeld op dag één of dag twee van de test) schimmel op eitjes wordt vastgesteld, dienen die eitjes te worden geteld en verwijderd. Dode embryo's, larven en jonge vissen moeten meteen worden verwijderd zodra ze worden opgemerkt, aangezien zij snel kunnen ontbinden en uiteen kunnen vallen door het gedrag van de overige vissen. Het verwijderen van dode organismen moet zeer voorzichtig gebeuren om beschadiging van naastgelegen eitjes/larven te voorkomen. De tekenen van dood variëren per soort en per stadium. Bijvoorbeeld:
28. Afwijkend uiterlijk : het aantal larven of jonge vissen dat een afwijkende lichaamsvorm vertoont dient op gepaste tijden te worden geregistreerd, naargelang van de duur van de test en de aard van de beschreven afwijkingen. Larven en jonge vissen met een afwijking kunnen van nature voorkomen en bij sommige vissoorten kan het aantal oplopen tot enkele procenten van de controlegroep(en). Wanneer de misvormingen en het ermee samenhangende afwijkende gedrag zo ernstig worden geacht dat het organisme er aanzienlijk onder lijdt, en het een punt heeft bereikt waarop het niet meer zal herstellen, mag het uit de test worden verwijderd. Dergelijke dieren moeten worden geëuthanaseerd en in de latere gegevensanalyse als sterfgevallen worden behandeld. Voor de meeste soorten die in deze testmethode worden aanbevolen, is de normale embryonale ontwikkeling gedocumenteerd (9) (10) (11) (12). 29. Afwijkend gedrag : afwijkingen, zoals hyperventilatie, ongecoördineerd zwemmen en atypisch(e) onbeweeglijkheid of eetgedrag moeten op gepaste tijden worden geregistreerd naargelang van de duur van de test (bv. een keer per dag voor warmwatersoorten). Deze effecten zijn moeilijk te kwantificeren, maar kunnen, als zij worden waargenomen, helpen om de sterftegegevens te interpreteren. 30. Gewicht : aan het eind van de test worden alle overlevende vissen tenminste per replicaat gewogen (waarbij het aantal dieren in het replicaat en het gemiddelde gewicht per dier worden gerapporteerd): nat gewicht — (drooggedept) geniet de voorkeur, al mogen gegevens over het droge gewicht ook worden gerapporteerd (13). 31. Lengte : aan het eind van de test worden de individuele lengtes gemeten. Het is aanbevolen om de totale lengte te meten, al kan ook de standaardlengte worden gebruikt in het geval er vinrot in de staart of vinerosie is opgetreden. Voor alle vissen in een bepaalde test moet dezelfde methode worden gebruikt. De individuele lengte kan worden gemeten met een schuifmaat, een digitale camera of een gekalibreerd micrometer-oculair. De typische minimumlengten zijn gedefinieerd in aanhangsel 2. GEGEVENS EN RAPPORTAGE Verwerking van de resultaten
Testverslag
LITERATUUR
Aanhangsel 1 DEFINITIES
Aanhangsel 2 TESTOMSTANDIGHEDEN, DUUR EN OVERLEVINGSCRITERIA VOOR AANBEVOLEN VISSOORTEN
Aanhangsel 3 RICHTSNOEREN VOOR HET VOEDEREN EN HANTEREN VAN BROED- EN TESTVISSEN VAN AANBEVOLEN SOORTEN
Aanhangsel 4 ENKELE CHEMISCHE KENMERKEN VAN GESCHIKT VERDUNNINGSWATER
Aanhangsel 5 STATISTISCHE RICHTSNOEREN VOOR NOEC-BEPALING Algemene opmerkingen De replicaatbak is de analyse-eenheid. Voor continue metingen, zoals het meten van de grootte, dient daarom een gemiddelde of mediaan te worden berekend voor het replicaat, en die replicaatwaarden zijn de gegevens die worden geanalyseerd. Het onderscheidend vermogen van de gebruikte tests moet worden aangetoond, bij voorkeur op basis van een adequate historische gegevensbank voor elk laboratorium. De grootte van het effect die kan worden gedetecteerd met een onderscheidend vermogen van 75-80 % moet voor elk eindpunt worden vermeld met de statistische toets die zal worden gebruikt. De gegevensbanken die beschikbaar zijn bij het ontwikkelen van deze testmethode, bepalen het mogelijke onderscheidende vermogen van de aanbevolen statistische toetsen. Elk afzonderlijk laboratorium moet aantonen dat het in staat is om aan deze vereiste inzake het onderscheidend vermogen te voldoen door een eigen analyse van dat vermogen te verrichten of door aan te tonen dat de variatiecoëfficiënt (VC) voor elke respons het 90e percentiel van de VC's die werden gebruikt bij de ontwikkeling van de TG, niet overschrijdt. Deze VC's zijn vermeld in tabel 1. Als er enkel gemiddelden of medianen per replicaat beschikbaar zijn, kan de VC binnen het replicaat buiten beschouwing worden gelaten.. Tabel 1 90e percentiel VC's voor geselecteerde zoetwatersoorten
Voor nagenoeg alle statistische toetsen die worden gebruikt om toxicologische onderzoeken van laboratoria te evalueren, is het de vergelijking van de dosisgroepen met de controlegroepen die van belang is. Daarom is het niet raadzaam te eisen dat er een significante ANOVA F-toets plaatsvindt voordat een toets van Dunnett of William wordt gebruikt, of een significante toets van Kruskal-Wallis voordat de toets van Jonckheere-Terpstra, Mann-Whitney of Dunn wordt gebruikt (Hochberg and Tamhane 1987, Hsu 1996, Dunnett 1955, 1964, Williams 1971, 1972, 1975, 1977, Robertson et al. 1988, Jonckheere 1954, Dunn 1964). De toets van Dunnett bevat een multipliciteitscorrectie, en de percentages vals-positieven en vals-negatieven worden nadelig beïnvloed door de F-toets als poortwachter te gebruiken. Evenzo waarborgen de stap-omlaagtoetsen van Williams en Jonckheere-Terpstra waarbij een significantieniveau van 0,05 wordt gehanteerd bij elke stap, een totaalpercentage van vals-positieven van 5 %, en dat percentage en het onderscheidend vermogen van de toetsen worden nadelig beïnvloed door de F- of de Kruskal-Wallis-toets als poortwachter te gebruiken. De toetsen van Mann-Whitney en Dunn moeten worden gecorrigeerd voor multipliciteit en daarbij wordt aanbevolen om de Bonferroni-Holm-correctie te gebruiken. De meeste aanbevelingen inzake het toetsen van hypotheses en het verifiëren van de aannamen die aan de basis liggen van deze toetsen, worden in OESO (2006) besproken, waarin ook een uitgebreide literatuurlijst is opgenomen. Behandeling van controles wanneer een oplosmiddel wordt gebruikt Als er een oplosmiddel wordt gebruikt, moeten zowel een verdunningswatercontrole als een oplosmiddelcontrole worden uitgevoerd. De twee controles dienen te worden vergeleken voor elke respons en te worden gecombineerd voor statistische analyse, als er geen significant verschil wordt vastgesteld tussen de controles. Is dat wel het geval, dan dient de oplosmiddelcontrole te worden gebruikt om de NOEC te bepalen of de ECx te ramen en wordt de watercontrole niet gebruikt. Zie de beperkingen in de geldigheidscriteria (punt 7) Voor de lengte, het gewicht, het percentage uitgekomen eitjes of sterfte onder de larven of afwijkende larven en de eerste en laatste dag van het uitkomen of het naar boven zwemmen, moet een T-toets of een Mann-Whitney-toets worden gebruikt om de verdunningswatercontrole en de oplosmiddelcontrole te vergelijken bij het significantieniveau van 0,05, waarbij alle dosisgroepen buiten beschouwing worden gelaten. De resultaten van die toetsen moeten worden gerapporteerd. Groottemetingen (lengte en gewicht) De waarden voor de lengte en het gewicht van de afzonderlijke vissen kunnen normaal of lognormaal verdeeld zijn. De gemiddelde waarden per replicaat zijn gewoonlijk normaal verdeeld vanwege de centrale limietstelling en bevestigd op basis van gegevens uit meer dan 100 ELS-onderzoeken bij drie zoetwatersoorten. Wanneer de gegevens of de historische gegevensbanken wijzen op een lognormale verdeling van de groottewaarden van de afzonderlijke vissen, kan de gemiddelde logaritme per replicaat van de waarden voor de afzonderlijke vissen worden berekend en kunnen de gegevens voor analyse de antilogaritmen van die gemiddelde replicaatlogaritmen zijn. De gegevens moeten worden beoordeeld op overeenstemming met een normale verdeling en homogeniteit van variantie. Daartoe dienen de residuen van een ANOVA-model met concentratie als enige verklarende klassevariabele te worden gebruikt. Daarbij kan worden gebruikgemaakt van een visuele bepaling op basis van puntenwolken en histogrammen of stam-en-bladgrafieken. Als alternatief kan ook een formele toets zoals die van Shapiro-Wilk of Anderson-Darling worden gebruikt. De overeenstemming met de homogeniteit van variantie kan worden beoordeeld aan de hand van een visueel onderzoek van dezelfde puntenwolk of formeel op basis van een toets van Levene. Enkel parametrische toetsen (zoals Williams en Dunnett) moeten worden beoordeeld op normaliteit en homogeniteit van variantie. Daarbij moet aandacht worden geschonken aan mogelijke uitschieters en het effect daarvan op de analyse. De uitschieterstoets van Tukey en een visueel onderzoek van de hierboven genoemde puntenwolk met residuen kunnen worden gebruikt. Er zij op gewezen dat de waarnemingen betrekking hebben op volledige replicaten. Het uitsluiten van uitschieters uit de analyse moet dus steeds zorgvuldig worden overwogen. De statistische toetsen waarbij gebruik wordt gemaakt van de kenmerken van de testopzet en de biologische verwachtingen, zijn trendtoetsen die met stapjes omlaag worden toegepast, zoals die van Williams en Jonckheere-Terpstra. In die toetsen wordt uitgegaan van een monotone concentratierespons, en de gegevens moeten worden beoordeeld op overeenstemming met die aanname. Dat kan visueel gebeuren op basis van een puntenwolk van de replicagemiddelden ten opzichte van de testconcentratie. Het is nuttig om een stuksgewijze lineaire curve over die puntenwolk te leggen waarin de concentratiegemiddelden gewogen naar monstergrootte van het replicaat met elkaar worden verbonden. Als die stuksgewijze curve sterk afwijkt van de monotoniteit, wijst dat erop dat er mogelijk andere toetsen dan trendtoetsen moeten worden gebruikt. Als alternatief kunnen formele toetsen worden gebruikt. Een eenvoudige manier om een formele toets uit te voeren is om lineaire en kwadratische contrasten van de concentratiegemiddelden te berekenen. Als het kwadratische contrast significant is en het lineaire contrast niet, dan kan dat op problemen met de monotoniteit wijzen, die nader moet worden beoordeeld aan de hand van puntenwolken. Wanneer de normaliteit of de homogeniteit van variantie een probleem kan zijn, kunnen die contrasten worden berekend op basis van op rang getransformeerde gegevens. Alternatieve werkwijzen, zoals de monotoniteitstoets van Bartholomew, kunnen worden gebruikt, maar maken het proces complexer. Figuur 2: NOEC-stroomdiagram groottemetingen (lengte en gewicht) 
Tenzij de gegevens niet stroken met de vereisten voor deze toetsen, wordt de NOEC bepaald door de toets van Williams of Jonckheere-Terpstra met stapjes omlaag toe te passen. OESO (2006) bevat meer informatie over deze procedures. Voor gegevens die niet stroken met de vereisten voor een stap-omlaagtrendtoets, kan de toets van Dunnett of van Tamhane-Dunnett (T3) worden gebruikt. Beide toetsen bevatten een correctie voor multipliciteit. In deze toetsen wordt uitgegaan van normaliteit en, in het geval van Dunnett, ook van homogeniteit van variantie. Indien niet aan die voorwaarden is voldaan, kan de niet-parametrische toets van Dunn worden gebruikt. OESO (2006) bevat meer informatie over al deze toetsen. Figuur 2 geeft een overzicht van hoe kan worden nagegaan welke test de voorkeur geniet. Aantal uitgekomen eitjes en overlevende larven Deze gegevens hebben betrekking op de percentages eitjes die uitkomen of larven die overleven in afzonderlijke replicaten. Deze percentages moeten worden beoordeeld op extrabinomiale variantie, wat gebruikelijk maar niet alomtegenwoordig is voor zulke metingen. Het stroomdiagram in figuur 3 kan helpen om de optimale toets te kiezen; zie de tekst voor een gedetailleerde beschrijving. Er zijn twee toetsen die vaak worden gebruikt: de C(α)-toets van Tarone (Tarone, 1979) en chi-kwadraattoetsen, die elk afzonderlijk op elke testconcentratie moeten worden toegepast. Als er extrabinomiale variantie wordt vastgesteld in minstens één testconcentratie, moeten methoden worden gebruikt die daarmee rekening houden. Formule 1: C (α)-toets van Tarone (Tarone 1979)
waarbij p het gemiddelde percentage voor een bepaalde concentratie is, m het aantal replicaatbakken, nj het aantal individuen in replicaat j, en xj het aantal individuen in dat replicaat die een respons vertonen, hetzij die niet uitgekomen zijn of dood zijn. Deze toets wordt afzonderlijk toegepast op elke concentratie. Deze toets kan worden gezien als een gecorrigeerde chi-kwadraattoets, maar uit beperkte simulaties van het onderscheidend vermogen uitgevoerd door Tarone is gebleken dat zij krachtiger is dan een chi-kwadraattoets. Figuur 3: NOEC-stroomdiagram voor uitgekomen eitjes en sterfte onder de larven. 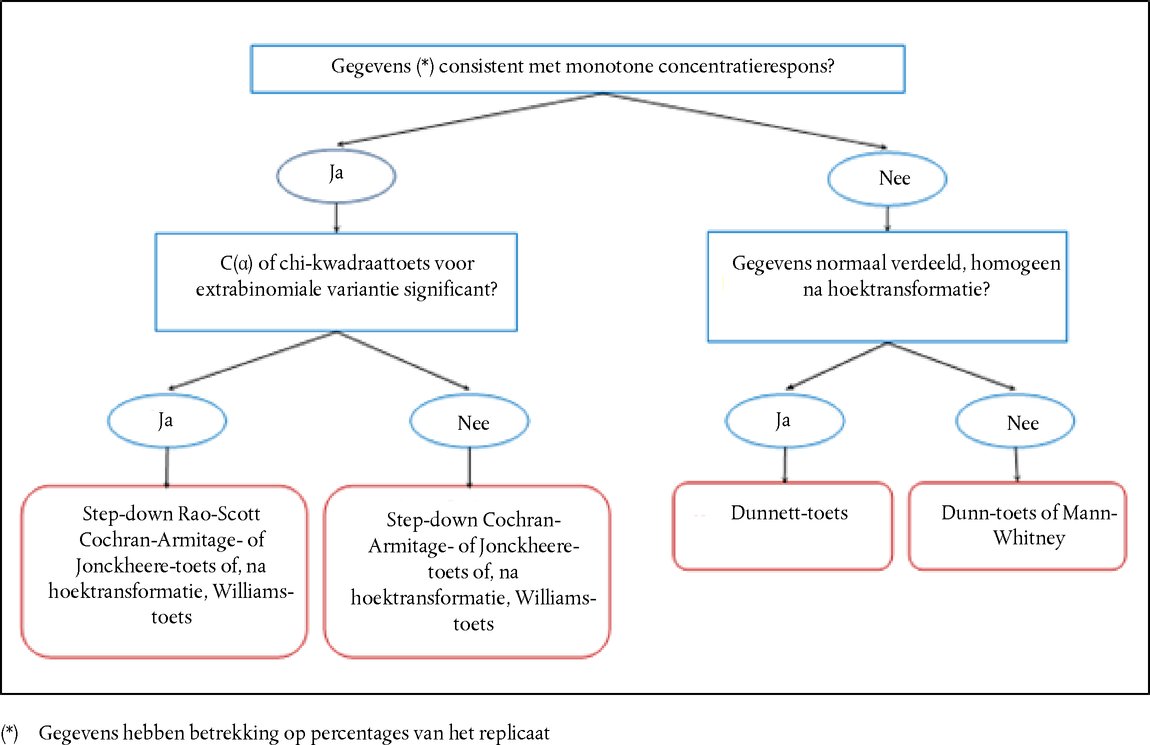
Wanneer er geen significante aanwijzingen van extrabinomiale variantie zijn, kan de stap-omlaagtoets van Cochran-Armitage worden gebruikt. Replicaten worden in deze toets buiten beschouwing gelaten, dus wanneer er dergelijke aanwijzingen zijn, wordt de correctie van Rao-Scott op de toets van Cochran-Armitage aanbevolen, die rekening houdt met replicaten, replicaatgrootte en extrabinomiale variantie. Voorbeelden van alternatieve toetsen zijn de stap-omlaagtoetsen van Williams en Jonckheere-Terpstra en de toets van Dunnett, zoals beschreven voor de groottemetingen. Deze toetsen zijn van toepassing ongeacht eventuele extrabinomiale variantie, maar hebben een iets beperkter onderscheidend vermogen (Agresti 2002, Morgan 1992, Rao and Scott 1992, 1999, Fung et al. 1994, 1996). Eerste of laatste dag van het uitkomen of naar boven zwemmen De respons is een geheel getal, dat aangeeft op welke dag van de test de aangegeven waarneming wordt waargenomen voor een bepaalde replicaatbak. Het waardenbereik is gewoonlijk erg beperkt, en vaak zijn grote delen van de waarden gelijk, waarbij bv. de eerste dag waarop eitjes uitkomen dezelfde is voor alle controlereplicaten en soms ook voor een of twee lage testconcentraties. Parametrische proeven zoals Williams en Dunnett zijn niet geschikt voor zulke gegevens. Tenzij er aanwijzingen zijn van ernstige niet-monotoniteit, is de stap-omlaagtoets van Jonckheere-Terpstra bijzonder goed in staat om effecten van de teststof te detecteren. Anders kan de proef van Dunn worden gebruikt. Afwijkingen bij de larven De respons is het aantal larven dat een of andere afwijking vertoont. Deze respons heeft vaak een lage incidentie en vertoont een aantal soortgelijke problemen als de eerste dag van het uitkomen. Soms is de concentratierespons ook grillig. Als de gegevens op zijn minst ruwweg een monotone concentratievorm volgen, is de stap-omlaagtoets van Jonckheere-Terpstra goed in staat om effecten te detecteren. Anders kan de proef van Dunn worden gebruikt. VERWIJZINGEN Agresti, A. (2002); Categorical Data Analysis, second edition, Wiley, Hoboken. Dunnett C. W. (1955); A multiple comparison procedure for comparing several treatments with a control, J. American Statistical Association 50, 1096-1121. Dunn O. J. (1964 ); Multiple Comparisons Using Rank Sums, Technometrics 6, 241-252. Dunnett C. W. (1964); New tables for multiple comparisons with a control, Biometrics 20, 482-491. Fung, K.Y., D. Krewski, J.N.K. Rao, A.J. Scott (1994); Tests for Trend in Developmental Toxicity Experiments with Correlated Binary Data, Risk Analysis 14, 639-648. Fung, K.Y, D. Krewski, R.T. Smythe (1996); A comparison of tests for trend with historical controls in carcinogen bioassay, Canadian Journal of Statistics 24, 431-454. Hochberg, Y. and A. C. Tamhane (1987); Multiple Comparison Procedures, Wiley, New York. Hsu, J.C. (1996); Multiple Comparisons: Theory and Methods; Chapman and Hall/CRC Press, Boca Raton. Jonckheere A. R. (1954); A distribution-free k-sample test against ordered alternatives, Biometrika 41, 133. Morgan, B.J.T. (1992); Analysis of Quantal Response Data, Chapman and Hall, London. OESO (2006). Current approaches in the statistical analysis of ecotoxicity data: A guidance to application. Series on Testing and Assessment, nr. 54. Organisatie voor Economische Samenwerking en Ontwikkeling, OESO, Parijs. Rao J.N.K. and Scott A.J. (1992) — A simple method for the analysis of clustered binary data, Biometrics 48, 577-585. Rao J.N.K. and Scott A.J. (1999) — A simple method for analyzing overdispersion in clustered Poisson data, Statistics in Medicine 18, 1373-1385. Robertson, T., Wright F.T. and Dykstra R.L. (1988); Order restricted statistical inference, Wiley. Tarone, R.E. (1979); Testing the goodness of fit of the Binomial distribution, Biometrika 66, 585-590. Williams D.A. (1971); A test for differences between treatment means when several dose levels are compared with a zero dose control, Biometrics 27, 103-117. Williams D.A. (1972); The comparison of several dose levels with a zero dose control, Biometrics 28, 519-531. Williams D. A. (1975); The Analysis of Binary Responses from Toxicological Experiments Involving Reproduction and Teratotlogy, Biometrics 31, 949-952. Williams D.A. (1977); Some inference procedures for monotonically ordered normal means, Biometrika 64, 9-14. Aanhangsel 6 STATISTISCHE RICHTSNOEREN VOOR REGRESSIERAMINGEN Algemene opmerkingen De waarnemingen die worden gebruikt om een model te passen zijn replicaatgemiddelden (lengte en gewicht) of percentages van het replicaat (uitgekomen eitjes en sterfte onder de larven) (OESO 2006). Gewoonlijk wordt aanbevolen om een gewogen regressie te hanteren met de monstergrootte van het replicaat als wegingsfactor. Ook andere weegschema's zijn mogelijk, zoals een weging op basis van de voorspelde gemiddelde respons of een combinatie van die laatste en een weging op basis van de monstergrootte van het replicaat. Weging op basis van reciproque waarden van de variantie binnen de concentratie van de monsters wordt niet aanbevolen (Bunke et al. 1999, Seber and Wild, 2003, Motulsky and Christopoulos 2004, Huet et al. 2003). Bij een eventuele transformatie van de responsen voorafgaand aan de analyse dient de onafhankelijkheid van de waarnemingen te worden bewaard en dienen de ECx en de betrouwbaarheidslimieten te worden uitgedrukt in de oorspronkelijke meeteenheden, en niet in getransformeerde eenheden. Een verandering met 20 % in de lengtelogaritme is bijvoorbeeld niet gelijk aan een verandering met 20 % in de lengte (Lyles et.al 2008, Draper and Smith 1999). Het stroomdiagram in figuur 4 geeft een overzicht voor ECx-ramingen. De details zijn beschreven in onderstaande tekst. Figuur 4: Stroomdiagram voor ECx, raming van de gemiddelde lengte voor het replicaat of het percentage uitgekomen eitjes of sterfte onder de larven, zie tekst voor meer informatie. 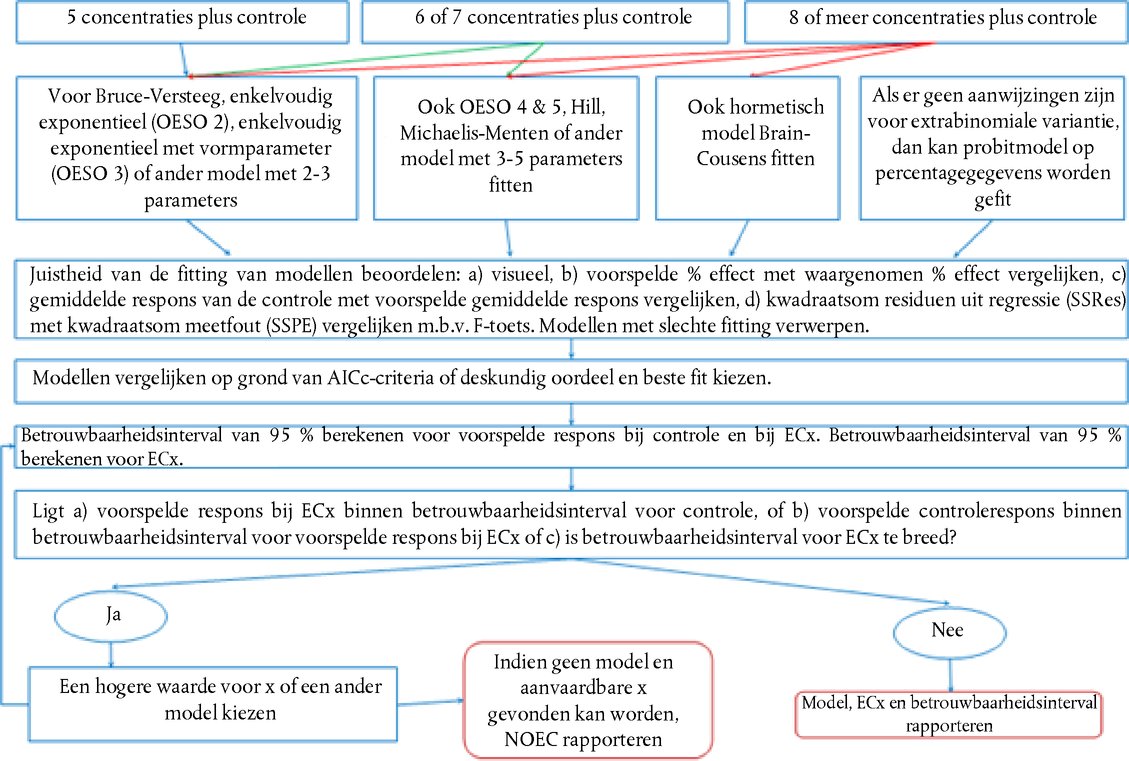
Overwegingen voor uitgekomen eitjes en sterfte onder de larven Voor het aantal uitgekomen eitjes en de sterfte onder de larven is het over het algemeen het beste om een aflopend model te passen, tenzij er een probitmodel wordt gepast, zoals hieronder beschreven. Dat wil zeggen dat het percentage eitjes dat niet uitkomt of het percentage larven dat sterft moet worden gemodelleerd. De reden daarvoor is dat ECx betrekking heeft op een concentratie waarbij er een verandering gelijk aan x % van de gemiddelde controlerespons plaatsvindt. Als 5 % van de eitjes in de controlegroep niet uitkomt en het percentage niet-uitgekomen eitjes wordt gemodelleerd, heeft EC20 betrekking op een concentratie waarbij er een verandering optreedt gelijk aan 20 % van het percentage niet-uitgekomen eitjes in de controlegroep, namelijk 5 %, en dat is een verandering van 0,2*0,05=0,01 of 1 procentpunt in een percentage niet-uitgekomen eitjes van 6 %. Een dergelijke kleine verandering kan niet op een zinvolle manier worden geraamd op basis van de beschikbare gegevens en is niet van biologisch belang. Als daarentegen het percentage eitjes dat uitkomt wordt gemodelleerd, dan zou het controlepercentage in dit voorbeeld 95 % zijn, en komt een verlaging ten opzichte van het controlegemiddelde met 20 % neer op een verandering van 0,95*0,2=0,18, dus van een percentage uitgekomen eitjes van 95 % naar een percentage uitgekomen eitjes van 77 % (= 95-18). Die effectconcentratie kan wel worden geraamd en is naar verwachting van groter belang. Bij groottemetingen is dit geen probleem, hoewel schadelijke effecten voor de grootte gewoonlijk neerkomen op een afname van de grootte. Modellen voor de grootte (lengte en gewicht) en het percentage uitgekomen eitjes of overlevende larven. Met uitzondering van het hormetische model van Brain-Cousens zijn al deze modellen beschreven en aanbevolen in OESO (2006). De zogeheten OESO 2-5 worden ook besproken voor ecotoxiciteitstests in Slob (2002). Er zijn natuurlijk nog talrijke andere modellen die nuttig kunnen zijn. In Bunke, et al. (1999) worden verschillende modellen opgesomd die hier niet zijn opgenomen, en er zijn ook talrijke modellen te vinden in de verwijzingen. Onderstaande modellen worden voorgesteld omdat zij bijzonder geschikt zouden zijn voor ecotoxiciteitstests en omdat zij gangbaar zijn. Met 5 testconcentraties plus controle
Met 6 of meer testconcentraties plus controle
Wanneer er zichtbare aanwijzingen zijn van hormese (weinig waarschijnlijk bij de percentages uitgekomen eitjes of overlevende larven, maar wordt soms waargenomen bij groottewaarnemingen)
Alternatieve modellen voor uitgekomen eitjes en sterfte onder de larven
Geschiktheid van een enkel model
Modellen vergelijken
Kwaliteit van de ECx-raming Het betrouwbaarheidsinterval (CI) voor ECx mag niet te ruim zijn. Er moet statistisch worden beoordeeld hoe ruim het betrouwbaarheidsinterval kan zijn zonder dat de ECx zijn nut verliest. Simulaties voor regressiemodellen die geschikt zijn voor het percentage uitgekomen eitjes en gegevens over de grootte wijzen uit dat ongeveer 75 % van de betrouwbaarheidsintervallen voor ECx (x=10, 20 of 30) slechts twee testconcentraties omvatten. Dat kan als algemene leidraad worden gebruikt voor wat aanvaardbaar is en als praktische leidraad voor wat haalbaar is. Verschillende auteurs vermelden dat betrouwbaarheidsintervallen moeten worden gerapporteerd voor alle modelparameters en dat ruime betrouwbaarheidsintervallen wijzen op onaanvaardbare modellen (Ott and Longnecker 2008, Alvord and Rossio 1993, Motulsky and Christopoulos 2004, Lyles et al. 2008, Seber and Wild 2003, Bunke et al. 1999, Environment Canada 2005). Het CI voor ECx (of een andere modelparameter) mag de nul niet omvatten (Motulsky and Christopoulos 2004). Dit is de regressie die gelijk is aan het minimale significante verschil dat vaak wordt genoemd in aanpakken om hypotheses te toetsen (bv. Wang et al 2000). Het betrouwbaarheidsinterval voor de gemiddelde responsen bij de LOEC dient ook het controlegemiddelde niet te omvatten. Men dient zich af te vragen of de parameterramingen wetenschappelijk plausibel zijn. Als het betrouwbaarheidsinterval voor y0 ± 20 % is, is er bijvoorbeeld geen EC10-raming plausibel. Als het model een effect van 20 % voorspelt bij concentratie C en het maximale waargenomen effect bij C en lagere concentraties 10 % bedraagt, dan is de EC20 niet plausibel (Motulsky and Christopoulos 2004, Wang et al. 2000, Environment Canada 2005). ECx hoort geen extrapolatie te vereisen buiten het bereik van positieve concentraties (Draper and Smith 1999, OESO 2006). Als algemene leidraad kan bijvoorbeeld worden gesteld dat ECx niet meer mag bedragen dan ongeveer 25 % minder dan de laagste geteste concentratie of meer dan de hoogste geteste concentratie. VERWIJZINGEN Alvord, W.G., Rossio, J.L. (1993); Determining confidence limits for drug potency in immunoassay, Journal of Immunological Methods 157, 155-163. Brain P. and Cousens R. (1989); An equation to describe dose responses where there is stimulation of growth at low doses. Weed res. 29: 93-96. Bunke, O., Droge, B. and Polzehl, J. (1999). Model selection, transformations and variance estimation in nonlinear regression. Statistics 33, 197-240. Collett, D. (2002); Modelling Binary Data, second edition, Chapman and Hall, London. Collett, D. (2003); Modelling Survival Data in Medical Research, second edition, Chapman and Hall, London. Draper, N.R. and Smith, H. (1999); Applied Regression Analysis, third edition. New York: John Wiley & Sons. Environment Canada (2005); Guidance Document on Statistical Methods for Environmental Toxicity Tests, Report EPS 1/RM/46 Huet, S., A. Bouvier, M.-A. Poursat, E. Jolivet (2003); Statistical Tools for Nonlinear Regression: A Practical Guide with S-PLUS and R Examples, Springer Series in Statistics, New York. Lyles, R. H., C. Poindexter, A. Evans, M. Brown, and C.R. Cooper (2008); Nonlinear Model-Based Estimates of IC50 for Studies Involving Continuous Therapeutic Dose-Response Data, Contemp Clin Trials. 2008 November; 29(6): 878–886. Morgan, B.J.T. (1992); Analysis of Quantal Response Data, Chapman and Hall, London. Motulsky, H., A. Christopoulos (2004); Fitting Models to Biological Data Using Linear and Nonlinear Regression: A Practical Guide to Curve Fitting, Oxford University Press, VS. O'Hara Hines, R. J. and J. F. Lawless (1993); Modelling Overdispersion in Toxicological Mortality Data Grouped over Time, Biometrics Vol. 49, blz. 107-121 OESO (2006); Current approaches in the statistical analysis of ecotoxicity data: A guidance to application. Series Testing and Assessment, nr. 54, Organisatie voor Economische Samenwerking en Ontwikkeling, OESO, Parijs. Ott, R.L., M.T. Longnecker, An Introduction to Statistical Methods and Data Analysis, sixth edition, 2008, Brooks-Cole, Belmont, CA Ratkowsky, D.A. (1993); Principles of nonlinear regression, Journal of Industrial Microbiology 12, 195-199. Seber, G.A.F., C.J. Wild, Nonlinear Regression, Wiley, 2003 Slob W. (2002); Dose-response modelling of continuous endpoints. Toxicol. Sci., 66, 298-312 Wang, Q., D.L. Denton, and R. Shukla (2000); Applications and Statistical Properties Of Minimum Significant Difference-Based Criterion Testing In a Toxicity Testing Program, Environmental Toxicology and Chemistry, Vol. 19, blz. 113–117, 2000. C.48 Test voor kortetermijneffecten op de voortplanting van vissen INLEIDING
INLEIDENDE OVERWEGINGEN EN BEPERKINGEN
PRINCIPE VAN DE TEST
AANVAARDBAARHEIDSCRITERIA VOOR DE TEST
BESCHRIJVING VAN DE METHODE Apparatuur
Water
Testoplossingen
Leefomstandigheden van de vis
Pre-blootstellingsperiode en selectie van de vissen
TESTOPZET
Selectie van testconcentraties
PROCEDURE Selectie en weging van de testvissen
Blootstellingsomstandigheden Duur
Voeding
Licht en temperatuur
Frequentie van de analytische bepalingen en metingen
Opmerkingen
Overleven
Gedrag en uiterlijk
Vruchtbaarheid
Op humane wijze doden van de vissen
Waarneming van secundaire geslachtskenmerken
Vitellogenine (VTG)
Evaluatie van de histopathologie van de geslachtsklieren
GEGEVENS EN RAPPORTAGE Evaluatie van biomerkerreacties door variantieanalyse
Rapportage van testresultaten
RICHTSNOEREN VOOR INTERPRETATIE EN AANVAARDING VAN DE TESTRESULTATEN
LITERATUUR
Aanhangsel 1 AFKORTINGEN EN DEFINITIES Bezettingsgraad : het aantal vissen per volume water. Chemische stof : een stof of mengsel Densiteit : het natte gewicht van de vissen per volume water ELISA : enzyme-linked immunosorbent assay HPG-as : hypothalamus-hypofyse-gonade-as MTC : maximale toegelaten concentratie, dat wil zeggen circa 10 % van de LC50 Teststof : alle volgens deze testmethode geteste stoffen of mengsels. VC : variatiecoëfficiënt VTG : vitellogenine is een fosfolipoglycoproteïneprecursor van eidooierproteïne die gewoonlijk voorkomt bij seksueel actieve vrouwtjes van alle ovipare soorten. Aanhangsel 2 PROEFVOORWAARDEN VOOR DE ENDOCRIENE SCREENINGTEST BIJ VISSEN
Aanhangsel 3 ENKELE CHEMISCHE KENMERKEN VAN AANVAARDBAAR VERDUNNINGSWATER
Aanhangsel 4A KUITSUBSTRAAT VOOR DE ZEBRAVIS Paaiblad: geheel glazen instrumentenschaal, bijvoorbeeld van 22 × 15 × 5,5 cm (l × b × d), bedekt met verwijderbaar roestvrijstalen gaas (met openingen van 2 mm breed). Het gaas moet de opening van de instrumentenschaal bedekken op een niveau dat onder de rand ligt. 
Op het gaas dient kuitsubstraat te worden bevestigd. Het moet de vissen een structuur bieden die ze kunnen binnengaan. Kunstmatige aquariumplanten van groen kunststofmateriaal zijn hiervoor bijvoorbeeld geschikt. (N.B. Houd rekening met mogelijke adsorptie van de teststof aan het kunststofmateriaal.) Het kunststofmateriaal moet gedurende voldoende tijd worden uitgeloogd in een ruime hoeveelheid warm water om te waarborgen dat geen chemische stoffen kunnen worden afgegeven aan het testwater. Bij het gebruik van glasmateriaal moet erop worden toegezien dat de vissen niet gewond of beklemd raken tijdens hun krachtige bewegingen. De afstand tussen het blad en de glazen wanden dient ten minste 3 cm te bedragen om te waarborgen dat het paaien niet buiten het blad plaatsvindt. De op het blad gelegde eitjes vallen door het gaas; 45-60 minuten na het begin van de belichting kunnen er monsters van worden genomen. De transparante eitjes kleven niet en kunnen gemakkelijk worden geteld met behulp van transversaal licht. Wanneer 5 vrouwtjes per bak worden gebruikt, kunnen aantallen tot 20 per dag als laag, tot 100 per dag als gemiddeld en boven de 100 per dag als hoog worden beschouwd. Zo laat mogelijk in de avond of zo vroeg mogelijk in de ochtend moet het paaiblad worden verwijderd om de eitjes te verzamelen en vervolgens het paaiblad weer aan te brengen in de testbak. De tijd tussen het verwijderen en weer aanbrengen van het blad mag niet langer dan 1 uur bedragen omdat anders de komst van het kuitsubstraat kan leiden tot afzonderlijk paren en paaien op een ongebruikelijk tijdstip. Als de situatie met zich meebrengt dat het paaiblad later moet worden aangebracht, dient dit ten minste 9 uur na het begin van de belichting te geschieden. Op zo'n laat tijdstip wordt geen paaigedrag meer uitgelokt. Aanhangsel 4B KUITSUBSTRAAT VOOR DE AMERIKAANSE DIKKOP-ELRITS In elk van de testkamers worden twee of drie gecombineerde paaibuizen en -bladen van kunststof/keramiek/glas of roestvrij staal geplaatst, bijvoorbeeld een grijze halfronde buis van 80 mm lengte op een blad met opstaande rand van 130 mm lengte (zie afbeelding). Goed geacclimatiseerde buizen van pvc of keramiek zijn aantoonbaar geschikt als kuitsubstraat (Thorpe et al., 2007). Aanbevolen wordt geschuurde buizen te gebruiken om de adhesie te verbeteren. Ook moet het blad worden afgeschermd om te voorkomen dat de vissen de gevallen eitjes bereiken, tenzij de adhesiewerking voor het gebruikte kuitsubstraat is aangetoond. 
De bladbodem is zo ontworpen dat deze alle eitjes bevat die zich niet aan het buisoppervlak hechten en anders op de bodem van de bak zouden vallen (en de eitjes die rechtstreeks op de platte kunststofbodem worden gelegd). Alle kuitsubstraten moeten voor gebruik minimaal 12 uur worden uitgeloogd in verdunningswater. Thorpe KL, Benstead R, Hutchinson TH, Tyler CR, 2007. An optimised experimental test procedure for measuring chemical effects on reproduction in the fathead minnow, Pimephales promelas. Aquatic Toxicology, 81, 90–98. Aanhangsel 5A BEOORDELING VAN SECUNDAIRE GESLACHTSKENMERKEN BIJ AMERIKAANSE DIKKOP-ELRITSEN VOOR DE OPSPORING VAN BEPAALDE ENDOCRIEN ACTIEVE CHEMISCHE STOFFEN Overzicht Potentieel belangrijke kenmerken van het fysieke voorkomen van volwassen Amerikaanse dikkop-elritsen tijdens tests met hormoonontregelaars zijn de lichaamskleur (licht/donker), kleurpatronen (aan- of afwezigheid van verticale banen), lichaamsvorm (vorm van de kop en borstregio, zwelling van de buik) en speciale secundaire geslachtskenmerken (aantal en grootte van nuptiale papillen, grootte van de nekverdikking en de legbuis). Nuptiale papillen bevinden zich op de kop (nekverdikking) van seksueel actieve mannetjes van de Amerikaanse dikkop-elrits, gewoonlijk in een tweezijdig symmetrisch patroon (Jensen et al. 2001). Controlevrouwtjes en jonge mannetjes en vrouwtjes vertonen geen papillaire ontwikkeling (Jensen et al. 2001). Er kunnen zich tot acht afzonderlijke papillen rond de ogen en tussen de neusgaten van de mannetjes bevinden. De meeste en de grootste papillen worden in twee evenwijdige lijnen direct onder de neusgaten en boven de bek aangetroffen. Veel vissen hebben groepen papillen onder de onderkaak. Die welke het dichtst bij de bek zitten, vormen meestal één paar, terwijl de reeks dichter bij de buik kan bestaan uit maximaal vier papillen. Het feitelijke aantal papillen is zelden meer dan 30 (bereik van 18-28; Jensen et al. 2001). De overheersende papillen (in aantal) zijn aanwezig als een enkele, relatief ronde structuur met een hoogte die ongeveer gelijk is aan de diameter. De meeste seksueel actieve mannetjes hebben ook ten minste een aantal papillen die zo vergroot en geprononceerd zijn dat ze niet als afzonderlijke structuren te onderscheiden zijn. Sommige soorten hormoonontregelende stoffen kunnen leiden tot abnormaal optreden van bepaalde secundaire geslachtskenmerken bij de andere sekse. Zo kunnen androgene receptoragonisten, zoals 17α-methyltestosteron of 17β-trenbolon, tot gevolg hebben dat vrouwelijke dikkop-elritsen geprononceerde nuptiale papillen ontwikkelen (Smith 1974; Ankley et al. 2001; 2003), terwijl oestrogene receptoragonisten het aantal of de grootte van nuptiale papillen bij mannetjes kunnen verkleinen (Miles-Richardson et al. 1999; Harries et al. 2000). Hierna volgt een beschrijving van de kenmerken van nuptiale papillen bij Amerikaanse dikkop-elritsen op basis van procedures die worden gebruikt in het laboratorium van het Amerikaanse Environmental Protection Agency in Duluth (Minnesota). Specifieke producten en/of apparaten kunnen worden vervangen door vergelijkbare beschikbare materialen. De beste weergave wordt bereikt met behulp van een verlicht vergrootglas of 3x vergrotende verlichte ontleedmicroscoop. Geef de vis dorsaal en met de kop naar voren (in de richting van de kijker) weer.
Tellen en beoordelen van de papillen Er zijn zes specifieke gebieden aangewezen die moeten worden beoordeeld op aanwezigheid en ontwikkeling van papillen bij volwassen Amerikaanse dikkop-elritsen. Er is een sjabloon ontwikkeld om de locatie en de hoeveelheid papillen in kaart te brengen (zie einde van dit aanhangsel). Het aantal papillen wordt genoteerd, waarbij de grootte per organisme als volgt kan worden aangegeven: 0-afwezig, 1-aanwezig, 2-vergroot en 3-geprononceerd (fig. 1). De score 0-afwezig betekent dat geen enkele papil aanwezig is. De score 1-aanwezig wordt toegekend aan elke papil die uit één punt bestaat waarvan de hoogte bijna gelijk is aan de diameter. De score 2-vergroot wordt toegekend aan weefsel dat eruitziet als een asterisk, gewoonlijk met een brede ronde basis met groeven of sporen die vanuit het midden lopen. De papilhoogte is meestal grilliger maar kan soms enigszins afgerond zijn. De score 3-geprononceerd geldt voor een tamelijk grote, afgeronde papil met een minder duidelijk gedefinieerde structuur. Soms komen deze papillen samen zodat ze één geheel vormen op één of meer gebieden (B, C en D, hierna beschreven). Kleur en vorm zijn soortgelijk aan score 2 maar af en toe tamelijk ongedifferentieerd. Toepassing van dit beoordelingssysteem leidt in het algemeen tot een totale papilscore van < 50 bij een normaal controlemannetje met 18 tot 20 papillen (Jensen et al. 2001). Figuur 1 
Het werkelijke aantal papillen kan bij sommige vissen groter zijn dan het aantal vakjes in de sjablonen voor een bepaald gebied. Als dit het geval is, kunnen extra scorecijfers worden genoteerd in of naast het kader. De sjabloon hoeft derhalve geen symmetrie te vertonen. Een aanvullende methode om papillen in kaart te brengen die paarsgewijs of verticaal verbonden langs het horizontale vlak van de bek voorkomen, is het noteren van dubbele scores voor twee papillen in één vakje. In kaart te brengen gebieden:
VERWIJZINGEN
Aanhangsel 5B BEOORDELING VAN SECUNDAIRE GESLACHTSKENMERKEN BIJ JAPANSE RIJSTVISJES VOOR DE OPSPORING VAN BEPAALDE ENDOCRIEN ACTIEVE CHEMISCHE STOFFEN Hierna volgt een beschrijving van het meten van de papillaire uitsteeksels (*10) die de secundaire geslachtskenmerken van het Japanse rijstvisje (Oryzias latipes) vormen.
Meting
Fig. 1. Diagram met verschillen naar geslacht in vorm en grootte van de anaalvin. A = mannetje; B = vrouwtje. Oka, T. B., 1931. On the processes on the fin rays of the male of Oryzias latipes and other sex characters of this fish. J. Fac. Sci., Tokyo Univ., IV, 2: 209-218. 
Fig. 2. A. Uitsteeksels op verbindingsplaten van anaalvinstralen. = verbindingsplaat; A.S. = axiale ruimte; P. = uitsteeksel. B. Distaal uiteinde van vinstraal. Actinotrichia (Act.) bevinden zich aan de punt. Oka, T. B., 1931. On the processes on the fin rays of the male of Oryzias latipes and other sex characters of this fish. J. Fac. Sci., Tokyo Univ., IV, 2: 209-218. 
Fig. 3. Foto van vis met de plaats van de snede voor het fixeren van de geslachtsklier in een andere fixeeroplossing dan 10 % neutraal gebufferde formaline. In dat geval wordt het karkas doorgesneden met een scheermes tussen de regio vóór de anaalvin en de anus (rode streep), en wordt de kopzijde van de vis in de fixeeroplossing voor de geslachtsklier en de staartzijde van de vis in de 10 % neutraal gebufferde formaline geplaatst. 
Aanhangsel 6 AANBEVOLEN PROCEDURES VOOR MONSTERNEMING TEN BEHOEVE VAN VITELLOGENINE-ANALYSE Er moet zorgvuldig worden voorkomen dat kruisbesmetting tussen VTG-monsters van mannetjes en vrouwtjes optreedt. Procedure 1A: Amerikaanse dikkop-elrits: bloedafname uit de staartader of -slagader Na anesthesering wordt de staartwortel gedeeltelijk afgesneden met een ontleedmesje en wordt bloed afgenomen uit de staartader of -slagader met een gehepariniseerd microhematocriet capillair buisje. Na de bloedafname wordt het plasma snel geïsoleerd door middel van centrifugeren gedurende 3 minuten bij 15 000 g (of gedurende 10 minuten bij 15 000 g en een temperatuur van 4 oC). Indien gewenst kan na het centrifugeren het hematocrietpercentage worden bepaald. Het plasmadeel wordt vervolgens uit het microhematocriet buisje genomen en in een centrifugebuis met 0,13 eenheden aprotinine (een proteaseremmer) bewaard bij -80 oC totdat de VTG-bepaling kan plaatsvinden. Afhankelijk van de grootte van de Amerikaanse dikkop-elrits (die sekse-afhankelijk is) lopen de af te nemen hoeveelheden plasma meestal uiteen van 5 tot 60 microliter per vis (Jensen et al. 2001). Procedure 1B: Amerikaanse dikkop-elrits: bloedafname uit het hart Als alternatief kan ook bloed worden afgenomen door middel van een hartpunctie met een gehepariniseerde injectiespuit (1000 eenheden heparine per ml). Het bloed wordt overgebracht naar eppendorfbuizen (op ijs bewaard) en vervolgens gecentrifugeerd (5 minuten bij 7 000 g en kamertemperatuur). Het plasma moet daarna worden overgebracht naar schone eppendorfbuizen (in aliquots als de hoeveelheid plasma dit mogelijk maakt) en onmiddellijk worden bevroren en bewaard bij -80 oC totdat het wordt geanalyseerd (Panter et al. 1998). Procedure 2A: Japans rijstvisje: excisie van de lever De testvissen uit de testkamer scheppen
Excisie van de lever
Na de leverexcisie is het karkas beschikbaar voor het meten van de histologie van de geslachtsklier en de secundaire geslachtskenmerken. Monsters Bewaar de uit de testvissen genomen levermonsters bij ≤ -70 oC als ze niet kort na de excisie worden gebruikt voor de voorbehandeling. Fig. 1. Net voor de borstvinnen wordt een snede aangebracht met een schaar. 
Fig. 2. De middellijn van de buik wordt met een schaar opengesneden tot ongeveer 2 mm van de craniale zijde van de anus. 
Fig. 3. De buikwanden worden gespreid met een tang om de lever en andere inwendige organen bloot te leggen. (Als alternatief kunnen de buikwanden lateraal worden vastgezet). De pijl wijst de lever aan. 
Fig. 4. De lever wordt grofweg ontleed en uitgenomen met een tang. 
Fig. 5. De ingewanden worden voorzichtig opzij getrokken met een tang. 
Fig. 6. Beide uiteinden van de ingewanden en alle mesenterische aanhechtingen worden doorgesneden met een schaar. 
Fig. 7 (vrouwtje). De procedure is identiek voor de vrouwtjesvis. 
Fig. 8. De procedure is afgerond. 
Procedure 2B: Japans rijstvisje (Oryzias latipes): voorbehandeling van de lever ten behoeve van vitellogenineanalyse Neem de fles homogenaatbuffer uit de ELISA-uitrusting en koel deze met gemalen ijs (temperatuur van de oplossing: ≤ 4 oC). Als gebruik wordt gemaakt van homogenaatbuffer van het EnBio ELISA-systeem, ontdooit u de oplossing bij kamertemperatuur en koelt u de fles vervolgens met gemalen ijs. Bereken het volume homogenaatbuffer voor de lever op basis van het gewicht van de lever (voeg 50 μl homogenaatbuffer per mg levergewicht toe). Als het gewicht van de lever bijvoorbeeld 4,5 mg is, bedraagt het volume homogenaatbuffer voor die lever 225 μl. Stel een lijst op van de volumes homogenaatbuffer voor alle levers. De lever prepareren voor de voorbehandeling
De voorbehandeling uitvoeren 1) Toevoeging van de homogenaatbuffer Controleer in de lijst het volume homogenaatbuffer dat moet worden gebruikt voor een bepaald levermonster en stel de micropipet (volumebereik: 100-1000 μl) in op het juiste volume. Bevestig een schone tip aan de micropipet. Neem de homogenaatbuffer uit de fles met reagens en voeg de buffer toe aan het microbuisje van 1,5 ml dat de lever bevat. Voeg volgens de hierboven beschreven procedure de homogenaatbuffer toe aan alle microbuisjes van 1,5 ml die een lever bevatten. Het is niet nodig de tip van de micropipet te vervangen door een nieuwe. Als de tip echter besmet of mogelijk besmet is, moet de tip wel worden vervangen. 2) Homogenisatie van de lever
3) Centrifugeren van het gesuspendeerde leverhomogenaat
4) Afname van het supernatans
Opslag van de monsters Bewaar de microbuisjes van 0,5 ml met het supernatans van het leverhomogenaat bij een temperatuur van ≤ -70 oC totdat ze voor de ELISA-test worden gebruikt. Procedure 3A: Zebravis: bloedafname uit de staartader of -slagader Na anesthesering wordt de staartwortel overdwars afgesneden en wordt bloed afgenomen uit de staartader of -slagader met een gehepariniseerd microhematocriet capillair buisje. De hoeveelheden bloed lopen uiteen van 5 tot 15 microliter, afhankelijk van de visgrootte. Er wordt een gelijke hoeveelheid aprotininebuffer (6 microgram/ml in PBS) aan het microcapillaire buisje toegevoegd en het plasma wordt van het bloed gescheiden door middel van centrifugeren (5 minuten bij 600 g). Het plasma wordt verzameld in de reageerbuisjes en bewaard bij –20 oC totdat het wordt geanalyseerd op vitellogenine of andere belangwekkende proteïnen. Procedure 3B: Zebravis: bloedafname door middel van hartpunctie Om coagulatie van het bloed en proteïneafbraak te voorkomen, worden de monsters verzameld in een fosfaatgebufferde zoutoplossing (PBS) die heparine (1 000 eenheden/ml) en de proteaseremmer aprotinine (2 TIU/ml) bevat. Als ingrediënten voor de buffer worden heparine, ammoniumzout en gevriesdroogde aprotinine aanbevolen. Voor de bloedafname wordt een injectiespuit (1 ml) met een vaste dunne naald (bijvoorbeeld Braun Omnikan-F) aanbevolen. De spuit moet vooraf gevuld zijn met de buffer (ongeveer 100 microliter) om de kleine hoeveelheden bloed volledig uit de vissen te wassen. De bloedmonsters worden afgenomen door middel van een hartpunctie. Eerst moet de vis worden geanestheseerd met MS-222 (100 mg/l). Met het juiste anesthesieniveau kan de gebruiker de hartslag van de zebravis onderscheiden. Houd tijdens de hartpunctie de zuiger van de injectiespuit onder lichte druk. De af te nemen hoeveelheden bloed lopen uiteen van 20 tot 40 microliter. Na de hartpunctie moet het mengsel van bloed en buffer in de reageerbuis worden gespoten. Het plasma wordt van het bloed gescheiden door middel van centrifuge (20 minuten bij 5 000 g) en moet bij een temperatuur van -80 oC worden bewaard totdat het nodig is voor analyse. Procedure 3C: SOP: zebravis: homogenisatie van kop en staart
Aanhangsel 7 VERRIJKTE VITELLOGENINEMONSTERS EN TESTONAFHANKELIJKE REFERENTIENORM Op elke dag dat er VTG-tests worden uitgevoerd, wordt een verrijkt monster geanalyseerd dat is gemaakt volgens een testonafhankelijke referentienorm. De VTG die wordt gebruikt voor de testonafhankelijke referentienorm is afkomstig uit een andere partij dan die welke is gebruikt om kalibratienormen samen te stellen voor de test die wordt uitgevoerd. Het verrijkte monster wordt gemaakt door toevoeging van een bekende hoeveelheid van de testonafhankelijke referentienorm aan een plasmamonster van controlemannetjes. Het monster wordt verrijkt om een VTG-concentratie te bereiken die 10 tot 100 maal zo groot is als de verwachte vitellogenineconcentratie in de controlemannetjes. Het plasmamonster van controlemannetjes dat wordt verrijkt, mag afkomstig zijn van één vis of van meerdere vissen. Een submonster van het nog niet verrijkte plasmamonster van controlemannetjes wordt ten minste in duplo geanalyseerd. Het verrijkte monster wordt eveneens ten minste in duplo geanalyseerd. De gemiddelde hoeveelheid VTG in de twee niet verrijkte plasmamonsters van controlemannetjes wordt opgeteld bij de berekende hoeveelheid VTG die is toegevoegd aan de verrijkte monsters om een verwachte concentratie te bepalen. De verhouding tussen deze verwachte concentratie en de gemeten concentratie wordt gerapporteerd samen met de resultaten van elke set tests die op die dag is uitgevoerd. Aanhangsel 8 BESLISDIAGRAM VOOR DE STATISTISCHE ANALYSE 
C.49 Acute toxiciteitstest bij visembryo's (FET, Fish Embryo Acute Toxicity Test) INLEIDING
PRINCIPE VAN DE TEST
INLEIDENDE OVERWEGINGEN
GELDIGHEID VAN DE TEST
BESCHRIJVING VAN DE METHODE
Apparatuur
Testkamers
Water en testomstandigheden
Testoplossingen
Houden van broedvissen
Bekwaamheidstoetsing
Eiproductie
Differentiatie van eitjes
PROCEDURE Blootstellingsomstandigheden
Testconcentraties
Controles
Start van de blootstelling en duur van de test
Verdeling van de eitjes over de 24-wells-platen
Waarnemingen
Analytische bepalingen
LIMIETTEST
GEGEVENS EN RAPPORTAGE Verwerking van de resultaten
Testverslag
LITERATUUR
Aanhangsel 1 DEFINITIES Apicaal eindpunt : veroorzaakt een effect op het niveau van de populatie. Blastula : een celformatie rond de dierlijke pool die een bepaald deel van de dooier bedekt. Chemische stof : een stof of mengsel. Doorstroomtest : een test waarbij de testoplossingen constant door het testsysteem stromen gedurende de blootstellingsduur. Epibolie : een massieve proliferatie van hoofdzakelijk epidermale cellen in het gastrulatiestadium van het embryo en de verplaatsing ervan van de dorsale naar de ventrale zijde, waarmee entodermale cellagen worden geïnternaliseerd tijdens een soort invaginatieproces en de dooier wordt opgenomen in het embryo. Interne plaatcontrole : een interne controle bestaande uit 4 wells met verdunningswater per 24-wells-plaat met het oog op de identificatie van potentiële verontreiniging van de platen door de fabrikant of de onderzoeker gedurende de procedure en eventuele effecten van de plaat die van invloed kunnen zijn op de resultaten van de test (bv. temperatuurgradiënt). IUPAC : International Union of Pure and Applied Chemistry. Letale-concentratiemediaan (LC50) : de concentratie van een teststof waarvan geraamd wordt dat zij binnen de testduur 50 % van de blootgestelde testorganismen doodt. Semistatische (verversings) test : een test waarbij de testoplossing regelmatig wordt ververst na een bepaalde periode (bv. elke 24 uur). SMILES : Simplified Molecular Input Line Entry Specification. Somiet : in de ontwikkeling van embryo's van gewervelde dieren zijn somieten massa's mesoderm die verdeeld zijn aan weerszijden van de neurale buis, waaruit zich uiteindelijk de dermis (dermatoom), het skeletspierweefsel (myotoom) en de wervels (sclerotoom) zullen ontwikkelen. Statische proef : een test waarbij de testoplossingen gedurende de test ongewijzigd blijven. Teststof : alle volgens deze testmethode geteste stoffen en mengsels. UVCB : stof met een onbekende of variabele samenstelling, complexe reactieproducten en biologische materialen. Water waarin de vissen worden gehouden : het water waarin de dierhouderij van de volwassen vissen plaatsvindt. Aanhangsel 2 TESTOMSTANDIGHEDEN, DUUR EN OVERLEVINGSCRITERIA VOOR AANBEVOLEN VISSOORTEN
Aanhangsel 3 NORMALE ONTWIKKELING VAN DE ZEBRAVIS BIJ 26 oC 


Aanhangsel 4 Figuur 1: Indeling van 24-wells-platen 
Figuur 2: Schematische weergave van de procedure voor de acute toxiciteitstest bij zebravisembryo's (van links naar rechts): eiproductie, verzamelen van de eitjes, pre-blootstelling meteen na bevruchting in glazen bakken, selectie van bevruchte eitjes met een omgekeerde microscoop of binoculair en verdeling van bevruchte eitjes over 24-wells-platen die zijn voorbereid met de respectieve testconcentraties/controles, n = benodigd aantal eitjes per testconcentratie/controle (hier 20), hpf = uren na bevruchting. 
Aanhangsel 5 ATLAS VAN LETALE EINDPUNTEN VOOR DE ACUTE TOXICITEITSTEST BIJ ZEBRAVISEMBRYO'S De volgende apicale eindpunten wijzen op acute toxiciteit en bijgevolg op de dood van de embryo's: coagulatie van het embryo, niet loskomen van de staart, uitblijven van somietvorming en afwezigheid van een hartslag. Om die eindpunten te illustreren werden de volgende microfoto's geselecteerd. Figuur 1 Coagulatie van het embryo: 
Onder felle verlichting vertonen gecoaguleerde zebravisembryo's verschillende ondoorzichtige inclusies. Figuur 2 Uitblijven van somietvorming: 
Hoewel de ontwikkeling van het zebravisembryo met ong. 10 uur vertraagd is, vertoont het embryo van 24 uur oud in (a) goed ontwikkelde somieten (→), terwijl het embryo in (b) geen tekenen van somietvorming vertoont (→). Hoewel het zebravisembryo een uitgesproken oedeem van de dooierzak vertoont, vertoont het embryo van 48 uur oud in (c) duidelijk gevormde somieten (→), terwijl het embryo van 96 uur oud in (d) geen tekenen van somietvorming vertoont (→). Let ook op de kromming van de ruggengraat (scoliose) en het pericardiale oedeem (*) in het embryo dat te zien is in (d). Figuur 3 Niet loskomen van de staartknop in zijaanzicht 
Figuur 4 De afwezigheid van een hartslag 
De afwezigheid van een hartslag is per definitie moeilijk te illustreren op een microfoto. De afwezigheid van een hartslag is te merken aan het feit dat het hart niet samentrekt (dubbele pijl). De onbeweeglijkheid van bloedcellen in bijvoorbeeld de aorta abdominalis (→ in tussenvoegsel) is geen indicator voor de afwezigheid van een hartslag. Let ook op het gebrek aan somietvorming in dit embryo (*, de spierweefsels zien er eerder homogeen dan gesegmenteerd uit). De observatietijd om de afwezigheid van een hartslag te registreren, dient ten minste een minuut te bedragen, met een minimale vergroting van 80×. C.50 Toxiciteitstest op Myriophyllum spicatum zonder sediment INLEIDING
PRINCIPE VAN DE TEST
INFORMATIE OVER DE TESTSTOF
GELDIGHEID VAN DE TEST
REFERENTIESTOF
BESCHRIJVING VAN DE METHODE Apparatuur
Testorganisme
Teelt
Testmedium
Testoplossingen
Dosis- en controlegroepen
Blootstelling
Testomstandigheden
Duur
Metingen en analyses
Frequentie van de metingen en analyses
Limiettest
GEGEVENS EN RAPPORTAGE Responsvariabelen
Gemiddelde specifieke groeisnelheid
Opbrengst
Verdubbelingstijd
Uitzetting van de concentratie/responscurves
Schatting van de ECx
Statistische procedures
Rapportage
LITERATUUR
Aanhangsel 1 DEFINITIES
Aanhangsel 2 GEWIJZIGD MEDIUM VAN ANDREWS VOOR STAMCULTUUR EN VOORCULTUUR Het gewijzigde medium van Andrews voor de stamcultuur en voorcultuur wordt bereid op basis van vijf afzonderlijk bereide nutriënt-stamoplossingen, waaraan 3 % sucrose wordt toegevoegd. Tabel 1 Samenstelling van nutriëntenoplossing van Andrews: (ASTM-aanduiding E 1913-04)
Stamoplossingen kunnen 6 maanden in een koelkast worden bewaard (bij 5-10 oC). Alleen stamoplossing nr. 5 heeft een beperkte houdbaarheid (twee maanden). Tabel 2 Productie van stamoplossing 3.1 voor het bereiden van stamoplossing 3
Nadat stamoplossing 3.1 (tabel 2) is bereid, moet de oplossing in ongeveer 11 ml-aliquots (bij ten minste -18 oC) worden ingevroren. De ingevroren porties zijn vijf jaar houdbaar. Om stamoplossing 3 te bereiden, ontdooit u stamoplossing 3.1, doet u 10 ml van de oplossing in een maatkolf van 1 l en voegt u ultrazuiver water toe tot aan de markering op de maatkolf. Doe om het gewijzigde medium van Andrews te bekomen ongeveer 2500 ml ultrazuiver water in een maatkolf van 5 l. Nadat er 50 ml van elke stamoplossing is toegevoegd, vult u de maatkolf voor 90 % met ultrazuiver water en brengt u de pH op 5,8. Voeg vervolgens 150 g opgeloste sucrose (3 % per 5 l) toe en vul de maatkolf tot aan de markering met ultrazuiver water. Tot slot wordt de nutriëntenoplossing in een Schott-kolf van 1 l gedaan en 20 minuten in de autoclaaf behandeld op 121 oC. De aldus geproduceerde nutriëntenoplossing kan drie maanden steriel in een koelkast worden bewaard (bij 5-10 oC). Gewijzigd medium van Andrews voor toxiciteitstest zonder sediment Het tienvoudig geconcentreerde, gewijzigde medium van Andrews dat nodig is om de testoplossingen te verkrijgen, wordt bereid op basis van de vijf nutriënt-stamoplossingen die reeds zijn vermeld in de tabellen 1 en 2, waaraan 30 % sucrose wordt toegevoegd. Doe daartoe ongeveer 100 ml ultrazuiver water in een maatkolf van 1 l. Nadat u 100 ml van elk van de stamoplossingen hebt toegevoegd, brengt u de pH op 5,8. Voeg vervolgens 30 % opgeloste sucrose (300 g per 1000 ml) toe en vul de maatkolf tot aan de markering met ultrazuiver water. Tot slot wordt de nutriëntenoplossing in een Schott-kolf van 0,5 l gedaan en 20 minuten in de autoclaaf behandeld op 121 oC. De aldus geproduceerde tienvoudig geconcentreerde gewijzigde nutriëntenoplossing kan drie maanden steriel in een koelkast worden bewaard (bij 5-10 oC). Aanhangsel 3 BEWARING VAN STAMCULTUREN In dit aanhangsel 3 wordt de stamcultuur van Myriophyllum spicatum L (103), een tweezaadlobbige waterplant uit de vederkruidfamilie beschreven. Tussen juni en augustus steken onopvallende roze-witte bloemen boven het wateroppervlak uit. De planten zijn in de grond geworteld via een systeem van robuuste rizomen en komen in het gehele noordelijke halfrond voor in eutrofe maar niet verontreinigde en meer kalkhoudende stilstaande wateren met een moddersubstraat. Myriophyllum spicatum verkiest zoet water, maar is ook te vinden in brak water. Voor de toxiciteitstest zonder sediment in laboratoriumomstandigheden zijn steriele planten nodig. Steriele planten zijn te verkrijgen bij het laboratorium voor ecotoxicologie van het Duitse Umweltbundesamt (het federale milieuagentschap van Duitsland). De testorganismen kunnen ook worden bereid uit niet-steriele planten, in overeenstemming met ASTM-aanduiding E 1913-04. Zie hieronder — uittreksel uit de ASTM Standard Guide — voor de procedure om uit het veld gehaalde Myriophyllum sibiricum te telen: „Als wordt vertrokken van uit het veld gehaalde, niet-steriele planten, moeten in het najaar winterknoppen van M. sibiricum worden verzameld. Plaats de winterknoppen in een aquarium van 20 l met 5 cm steriel sediment bedekt met kiezelzand of bijvoorbeeld Turface® en 18 l reagenswater. Belucht het aquarium en houd de temperatuur op 15 oC en de fluentiesnelheid op 200 tot 300 μmol m–2 s–1 gedurende 16 uur per dag. De plantcultuur in het aquarium kan worden gehandhaafd als reservebron van planten ingeval de steriele plantculturen worden vernietigd door een mechanische storing in de klimaatkast, verontreiniging of iets anders. De planten die in het aquarium worden geteeld, zijn niet steriel, en er kunnen geen steriele culturen worden gehouden in een cultuursysteem op basis van batches. Om de cultuur te steriliseren, worden de planten uit het aquarium gehaald en ongeveer 0,5 uur onder stromend gedeïoniseerd water gespoeld. De planten worden in minder dan 20 minuten in een laminaire-stromingskast onder aseptische omstandigheden gedesinfecteerd (tot het meeste plantenweefsel gebleekt is en enkel de groeiende top nog groen is) in een 3 % (v/v) natriumhypochlorietoplossing die 0,01 % van een geschikte oppervlakteactieve stof bevat. Schud het ontsmettingsmiddel en het plantenmateriaal. Segmenten met meerdere knopen worden naar steriele cultuurbuizen met 45 ml gesteriliseerd, gewijzigd medium van Andrews overgezet, die worden afgesloten met gewone afsluitingen voor cultuurbuizen. Er wordt slechts een plantsegment in elke testkamer geplaatst. De afsluiting wordt op het cultuurvat vastgezet met afdichtingsfolie voor laboratoria. Zodra er een steriele cultuur tot stand is gekomen, moeten plantsegmenten met meerdere knopen elke tien à twaalf dagen worden overgezet naar nieuwe testkamers met vers vloeibaar nutriëntmedium. Teelt op agarplaten heeft uitgewezen dat de planten steriel moeten zijn en achtweken steriel moeten blijven voordat de tests kunnen worden gestart.” Aangezien het gewijzigde medium van Andrews sucrose bevat (dat de groei van schimmels en bacteriën bevordert), moeten alle materialen, oplossingen en culturen onder steriele omstandigheden worden bereid. Alle vloeistoffen en uitrusting moeten voor gebruik worden gesteriliseerd. De sterilisatie gebeurt via behandeling met verhitte lucht (210 oC) gedurende 4 uur of in de autoclaaf gedurende 20 minuten op 121 oC. Daarnaast moeten alle kolven, schaaltjes, kommen enz. en andere uitrusting vlak voor gebruik op de steriele werkbank in een open vlam worden gehouden. Stamculturen kunnen onder beperkte verlichting en bij lage temperatuur (50 μE m-2 s-1, 20 ± 2 oC) geruime tijd gebruiksklaar worden bewaard. Voor Myriophyllum moet hetzelfde groeimedium worden gebruikt als voor de test, maar voor stamculturen kan een ander nutriëntrijk medium worden gebruikt. De plantsegmenten worden axenisch verdeeld over verschillende Erlenmeyers van 500 ml en/of Fernbach-kolven van 2000 ml, die gevuld worden met respectievelijk 450 en 1000 ml gewijzigd medium van Andrews. De kolven worden vervolgens axenisch afgesloten met een celluloseprop. Daarnaast is het absoluut noodzakelijk om uitrusting op de steriele werkbank vlak voor gebruik aan een grondige vlambehandeling te onderwerpen. Naargelang van hun aantal en grootte moeten de planten ongeveer elke drie weken naar een verse nutriëntenoplossing worden overgezet. Voor deze vernieuwde cultuur kunnen zowel toppen als segmenten van het middendeel van de stengel worden gebruikt. Het aantal en de grootte van de overgezette planten (of plantsegmenten) zijn afhankelijk van hoeveel planten er nodig zijn. Zo kunt u bijvoorbeeld vijf scheutsegmenten naar een Fernbach-kolf overzetten en drie scheutsegmenten naar een Erlenmeyer-kolf, elk met een lengte van 5 cm. Verwijder alle gewortelde, bloeiende, dode of anderszins verdachte delen. Figuur 1: Snijden van planten voor de stam- en voorcultuur na 3 weken teelt. 
De planten moeten worden opgekweekt in een Erlenmeyer van 500 ml en een Fernbach-kolf van 2000 ml in een koelincubator bij 20 ± 2 oC, met een constante belichting van ongeveer 100-150 μE m-2 s-1 of 6000-9000 lux (afgegeven door kamerverlichting met de kleurtemperatuur „warm wit licht”). Figuur 2: Opkweken van planten in een koelincubator met kamerverlichting. 
Er moeten chemisch zuivere (met zuur gewassen) en steriele glazen kweekvaten worden gebruikt en aseptische behandelingstechnieken worden toegepast. Bij verontreiniging van de stamcultuur door bijvoorbeeld algen, schimmels en/of bacteriën, moet een nieuwe cultuur worden bereid of moet een stamcultuur van een ander laboratorium worden gebruikt om de desbetreffende cultuur te vernieuwen. Aanhangsel 4 BEWARING VAN VOORCULTUREN EN VOORBEREIDING VAN TESTORGANISMEN VOOR HET TESTEN Voor de voorcultuur snijdt u scheuten van de stamcultuur in segmenten met elk twee kransen; doe de kransen in Fernbach-kolven gevuld met gewijzigd medium van Andrews (met 3 % sucrose). Elke kolf kan maximaal 50 scheutsegmenten bevatten. Er moet wel voor worden gezorgd dat de segmenten levenskrachtig zijn en geen wortels of zijtakken op hun knoppen hebben (zie figuur 1 in aanhangsel 3). De voorcultuurorganismen worden gedurende 14 tot 21 dagen onder steriele omstandigheden opgekweekt in een klimaatkast met een afwisselende licht-/donkercyclus van 16/8 uur. De lichtintensiteit wordt gekozen uit het bereik van 100-150 μE m-2 s-1. De temperatuur in de testvaten moet 23 ± 2 oC zijn. Aangezien het gewijzigde medium van Andrews sucrose bevat (dat de groei van algen, schimmels en bacteriën bevordert), moet het bereiden van de teststofoplossingen en het opkweken plaatsvinden onder steriele omstandigheden. Alle vloeistoffen en uitrusting moeten voor gebruik worden gesteriliseerd. De sterilisatie gebeurt via behandeling met verhitte lucht (210 oC) gedurende 4 uur of in de autoclaaf gedurende 20 minuten op 121 oC. Daarnaast moeten alle kolven, schaaltjes, kommen enz. en andere uitrusting vlak voor gebruik op de steriele werkbank in een open vlam worden gehouden. De scheuten worden axenisch verwijderd uit de kolven met de voorculturen, waarbij materiaal moet worden gekozen dat zo homogeen mogelijk is. Elke test moet ten minste 60 testorganismen omvatten (tests met acht teststofconcentraties). Neem voor de test verse zijtakken van voorculturen, kort ze in tot 2,5 cm van de voet (gemeten met een liniaal) en zet ze over naar een glas met steriel gewijzigd medium van Andrews. Deze verse zijtakken kunnen worden gebruikt voor de toxiciteitstest op Myriophyllum spicatum zonder sediment. Figuur 2: Snijden van planten uit de voorcultuur voor de sediment-vrije toxiciteitstest op Myriophyllum spicatum. 
C.51 Toxiciteitstest op Myriophyllum spicatum in water en sediment INLEIDING
PRINCIPE VAN DE TEST
INFORMATIE OVER DE TESTSTOF
GELDIGHEID VAN DE TEST
REFERENTIESTOF
BESCHRIJVING VAN DE METHODE Testapparatuur
Testorganisme
Kweken van het testorganisme
Sediment
Testmedium
Proefopzet
Teststofconcentraties en controlegroepen
Limiettest
Testoplossingen
TESTPROCEDURE
Kiemfase
Selectie van uniform plantenmateriaal
Blootstelling via de waterfase
Blootstelling via sediment
Instandhouding van het waterpeil gedurende de test
Testomstandigheden
Testduur
Metingen en analyses
Frequentie van de metingen en analyses
Analytische metingen van de teststof
EVALUATIE VAN DE GEGEVENS
Responsvariabelen
Gemiddelde specifieke groeisnelheid
Opbrengst
Uitzetting van de concentratie/responscurves
Schatting van de ECx
Statistische procedures
RAPPORTAGE
LITERATUUR
Aanhangsel 1 SAMENSTELLING MEDIUM VAN SMART EN BARKO
Aanhangsel 2 DEFINITIES Biomassa : het droog- en/of versgewicht van het levende materiaal in een populatie. In deze test is de biomassa de som van de hoofdscheut, alle zijtakken en alle wortels. Chlorose : de verandering van de kleur van het testorganisme, en in het bijzonder van de kransen, van groen naar vergeeld. ECx : de concentratie van de in het testmedium opgeloste teststof die binnen een bepaalde blootstellingsperiode (die expliciet moet worden vermeld als deze afwijkt van de volledige of normale duur van de test) leidt tot een afname van de groei van Myriophyllum spicatum met x % (bv. 50 %). Om ondubbelzinnig aan te geven of een EC-waarde gebaseerd is op de groeisnelheid of de opbrengst, worden de symbolen “ErC” voor de groeisnelheid en “EyC” voor de opbrengst gebruikt, gevolgd door de gebruikte meetvariabele, bv.ErC (lengte van de hoofdscheut). Groei : een toename van de meetvariabele, bv. de lengte van de hoofdscheut, de totale lengte van de zijtakken, de totale lengte van de scheuten, de totale lengte van de wortels, het versgewicht, het drooggewicht of het aantal kransen gedurende de testperiode. Groeisnelheid : (gemiddelde specifieke groeisnelheid): de logaritmische toename van de meetvariabele gedurende de blootstellingsperiode. Noot: De responsvariabelen die verband houden met de groeisnelheid, zijn onafhankelijk van de duur van de test voor zover het groeipatroon van niet blootgestelde controleorganismen exponentieel is. LOEC (Lowest Observed Effect Concentration) : de laagste geteste concentratie waarbij wordt waargenomen dat de stof binnen een bepaalde blootstellingstijd in vergelijking met de controle tot een statistisch significante afname van de groei (p < 0,05) leidt. Alle testconcentraties boven de LOEC moeten echter een schadelijk effect hebben dat gelijk is aan of groter dan de effecten die worden waargenomen bij de LOEC. Indien niet aan deze beide voorwaarden kan worden voldaan, moet uitgebreid worden toegelicht hoe de LOEC (en daarmee de NOEC) is geselecteerd. Meetvariabelen : alle soorten variabelen die gemeten worden om aan de hand van een of meer verschillende responsvariabelen het eindpunt van de test te beschrijven. In deze testmethode zijn de meetvariabelen de lengte van de hoofdscheut, de totale lengte van de zijtakken, de totale lengte van de scheuten, de totale lengte van de wortels, het versgewicht, het drooggewicht of het aantal kransen. Monocultuur : een cultuur met één plantensoort. Necrose : dood (d.w.z. wit of donkerbruin) weefsel van het testorganisme. NOEC (No Observed Effect Concentration) : de testconcentratie die direct onder de LOEC ligt. Opbrengst : de waarde van een meetvariabele om de biomassa uit te drukken aan het eind van de blootstellingstijd, verminderd met de waarde van de meetvariabele aan het begin van de blootstellingstijd. Noot: Wanneer het groeipatroon van niet blootgestelde organismen exponentieel is, nemen de responsvariabelen op basis van de opbrengst af in de loop van de test. Responsvariabele : een variabele voor de bepaling van de toxiciteit, met uiteenlopende berekeningsmethoden afgeleid van de gemeten variabelen voor de biomassa. Voor deze testmethode worden de responsvariabelen groeisnelheid en opbrengst afgeleid van meetvariabelen zoals lengte van de hoofdscheut, totale lengte van de scheuten, versgewicht, drooggewicht of aantal kransen. Semi-statische test (met verversing) : een test waarbij de testoplossing op specifieke tijdstippen gedurende de test periodiek wordt ververst. Statische test : een testmethode waarbij de testoplossing gedurende de test niet wordt ververst. Stof : een stof of mengsel. Testeindpunt : de algemene factor die volgens het doel van de test door de teststof zal worden veranderd in vergelijking met de controle. Het eindpunt van deze testmethode is groeiremming, die kan worden uitgedrukt door verschillende responsvariabelen die op een of meer meetvariabelen gebaseerd zijn. Testmedium : het volledige kunstmatige groeimedium waarin de testplanten gedurende de blootstelling aan de teststof groeien. De teststof wordt normaal gesproken in het medium opgelost. Teststof : alle volgens deze testmethode geteste stoffen en mengsels. UVCB : stoffen waarvan de samenstelling niet bekend of variabel is, complexe reactieproducten of biologische materialen. |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
(1) Er is geen waarde voor 20 °C beschikbaar, maar er kan worden aangenomen dat de variabiliteit van de meetresultaten groter is dan de te verwachten temperatuurafhankelijkheid
(2) Verordening (EG) nr. 1907/2006 van het Europees Parlement en de Raad van 18 december 2006 inzake de registratie en beoordeling van en de autorisatie en beperkingen ten aanzien van chemische stoffen (REACH), tot oprichting van een Europees Agentschap voor chemische stoffen, houdende wijziging van Richtlijn 1999/45/EG en houdende intrekking van Verordening (EEG) nr. 793/93 van de Raad en Verordening (EG) nr. 1488/94 van de Commissie alsmede Richtlijn 76/769/EEG van de Raad en de Richtlijnen 91/155/EEG, 93/67/EEG, 93/105/EG en 2000/21/EG van de Commissie. PB L 304 van 22.11.2007, blz. 1.
(3) US Interagency Coordinating Committee on the Validation of Alternative Methods.
(*1) Er moet worden aangegeven welk gebied van de cornea troebel is
(4) Voor het gebruik van een geïntegreerde teststrategie voor oogirritatie onder de REACH-verordening, zie ook ECHA Guidance on information requirements and chemical safety assessment, hoofdstuk R.7a: Endpoint specific guidance http://echa.europa.eu/documents/10162/13632/information_requirements_r7a_en.pdf
(5) Statistici die een modelbenadering zoals gegeneraliseerde lineaire modellen (GLM's) hanteren, pakken de analyse mogelijk op een andere, maar vergelijkbare manier aan, maar zullen niet noodzakelijkerwijs de traditionele ANOVA-tabel opstellen, die dateert uit de tijd van de algoritmische benaderingen om de statistieken te berekenen die werden ontwikkeld in het pre-computertijdperk.
(6) Statistici die een modelbenadering zoals gegeneraliseerde lineaire modellen (GLM's) hanteren, pakken de analyse mogelijk op een andere, maar vergelijkbare manier aan, maar zullen niet noodzakelijkerwijs de traditionele ANOVA-tabel opstellen, die dateert uit de tijd van de algoritmische benaderingen om de statistieken te berekenen die werden ontwikkeld in het pre-computertijdperk.
(7) Verordening (EG) nr. 1272/2008 van het Europees Parlement en de Raad van 16 december 2008 betreffende de indeling, etikettering en verpakking van stoffen en mengsels tot wijziging en intrekking van de Richtlijnen 67/548/EEG en 1999/45/EG en tot wijziging van Verordening (EG) nr. 1907/2006, PB L 353 van 31.12.2008, blz. 1.
(8) Scheikundige klassen werden toegekend aan elke teststof aan de hand van een standaard classificatieschema, gebaseerd op het classificatiesysteem National Library of Medicine Medical Subject Headings (MeSH) (beschikbaar op http//www.nlm.nih.gov/mesh).
(9) Gebaseerd op de resultaten van de in-vivo-oogtest op konijnen (OESO TG 405) (17) en aan de hand van de VN-GHS (4).
(10) Indeling in categorie 2A of 2B hangt af van de interpretatie van het VN-GHS-criterium voor het onderscheiden van deze twee categorieën, d.w.z. 1 van 3 of 2 van 3 dieren met effecten op dag 7 nodig voor indeling in categorie 2A. Het in-vivo-onderzoek omvatte 3 dieren. Alle eindpunten behalve roodheid van de conjunctiva in één dier herstelden tot een score van nul op dag 7 of eerder. Het ene dier dat op dag 7 niet volledig was hersteld, had een score van 1 (op dag 7) voor de roodheid van de conjunctiva, die volledig was hersteld op dag 10.
(11) De opgegeven afmetingen zijn gebaseerd op een corneahouder die wordt gebruikt voor koeien tussen 12 en 60 maanden oud. Indien dieren tussen 6 en 12 maanden worden gebruikt, dient de houder zodanig te worden ontworpen dat elke kamer een inhoud heeft van 4 ml en elk van de interne kamers een diameter heeft van 1,5 cm en diepte van 2,2 cm. Voor elke nieuw-ontworpen corneahouder is het zeer belangrijk dat de verhouding tussen het blootgestelde corneale oppervlak en de achterkamerinhoud dezelfde is als de verhouding in de traditionele corneahouder. Dit is noodzakelijk om te verzekeren dat de permeabiliteitswaarden correct bepaald worden voor de berekening van de IVIS aan de hand van de voorgestelde formule.
(12) Verordening (EG) nr. 1272/2008 van het Europees Parlement en de Raad van 16 december 2008 betreffende de indeling, etikettering en verpakking van stoffen en mengsels tot wijziging en intrekking van de Richtlijnen 67/548/EEG en 1999/45/EG en tot wijziging van Verordening (EG) nr. 1907/2006, PB L 353 van 31.12.2008, blz. 1.
(*2) Hoogste gemiddelde score waargenomen op elk tijdstip
(*3) Maximale gemiddelde score waargenomen op elk tijdstip (op basis van de troebelheidsscores zoals gedefinieerd in tabel 1).
(*4) Op basis van scores zoals gedefinieerd in tabel 2.
(*5) Combinaties die zelden voorkomen.
(13) Scheikundige klassen werden toegekend aan elke teststof aan de hand van een standaard classificatieschema, gebaseerd op het classificatiesysteem National Library of Medicine Medical Subject Headings (MeSH) (beschikbaar op: http//www.nlm.nih.gov/mesh).
(14) Gebaseerd op de resultaten van de in-vivo-oogtest op konijnen (OESO TG 405) en aan de hand van de VN-GHS (4) (6).
(15) Op basis van de resultaten in ICE zoals beschreven in tabel 6.
(16) Combinatie van ICE-scores met uitzondering van die scores die zijn beschreven in tabel 6 voor de identificatie van GHS Geen categorie en GHS-categorie 1 (zie tabel 6).
(17) Indeling in categorie 2A of 2B hangt af van de interpretatie van het VN-GHS-criterium voor het onderscheiden van deze twee categorieën, d.w.z. 1 van 3 of 2 van 3 dieren met effecten op dag 7 nodig voor indeling in categorie 2A. Het in-vivo-onderzoek omvatte 3 dieren. Alle eindpunten behalve roodheid van de conjunctiva in één dier herstelden tot een score van nul op dag 7 of eerder. Het ene dier dat op dag 7 niet volledig was hersteld, had een score van 1 (op dag 7) voor de roodheid van de conjunctiva, die volledig was hersteld op dag 10.
(18) Verordening (EG) nr. 1272/2008 van het Europees Parlement en de Raad van 16 december 2008 betreffende de indeling, etikettering en verpakking van stoffen en mengsels tot wijziging en intrekking van de Richtlijnen 67/548/EEG en 1999/45/EG en tot wijziging van Verordening (EG) nr. 1907/2006, PB L 353 van 31.12.2008, blz. 1.
(19) Met gemiddelde wordt in dit document het rekenkundige gemiddelde bedoeld.
(20) De cijfers verwijzen naar statistisch gegenereerde drempelwaarden en houden geen verband met de nauwkeurigheid van de meting.
(21) In het kader van een IATA en in overeenstemming met de bepalingen van punten 9 en 12 dient een DPRA-voorspelling te worden overwogen.
(22) De cijfers verwijzen naar statistisch gegenereerde drempelwaarden en houden geen verband met de nauwkeurigheid van de meting.
(23) In het kader van een IATA en in overeenstemming met de bepalingen van punten 9 en 12 dient een DPRA-voorspelling te worden overwogen.
(24) De in-vivogevaren- en (potentie)voorspellingen zijn gebaseerd op LLNA-gegevens (19). De in-vivopotentie wordt afgeleid met behulp van de criteria die zijn voorgesteld door ECETOC (23).
(25) In het kader van een IATA en in overeenstemming met de bepalingen van punten 9 en 11 dient een DPRA-voorspelling te worden overwogen.
(26) Bereiken bepaald op basis van ten minste 10 depletiewaarden gegenereerd door 6 onafhankelijke laboratoria.
(27) Verordening (EG) nr. 1272/2008 van het Europees Parlement en de Raad van 16 december 2008 betreffende de indeling, etikettering en verpakking van stoffen en mengsels tot wijziging en intrekking van de Richtlijnen 67/548/EEG en 1999/45/EG en tot wijziging van Verordening (EG) nr. 1907/2006, PB L 353 van 31.12.2008, blz. 1.
(28) De voorspellingen van de in-vivogevaren (en de potentie) zijn gebaseerd op LLNA-gegevens (13). De in-vivopotentie wordt afgeleid met behulp van de criteria die zijn voorgesteld door ECETOC (24).
(29) Een KeratinoSens™-voorspelling dient te worden bekeken in het kader van een IATA en in overeenstemming met hetgeen is bepaald in de punten 9 en 11 van deze testmethode.
(30) Op basis van de in het verleden waargenomen waarden (12).
(31) Verordening (EG) nr. 1272/2008 van het Europees Parlement en de Raad van 16 december 2008 betreffende de indeling, etikettering en verpakking van stoffen en mengsels tot wijziging en intrekking van de Richtlijnen 67/548/EEG en 1999/45/EG en tot wijziging van Verordening (EG) nr. 1907/2006, PB L 353 van 31.12.2008, blz. 1.
(32) Scheikundige klassen werden toegekend aan elke teststof aan de hand van een standaard classificatieschema, gebaseerd op het classificatiesysteem National Library of Medicine Medical Subject Headings (MeSH) (beschikbaar op: http//www.nlm.nih.gov/mesh).
(33) Gebaseerd op de resultaten van de in-vivo-oogtest op konijnen (OESO TG 405, TM B.5) en aan de hand van het VN-GHS en EU-CLP.
(34) Op basis van de resultaten verkregen met FL (INVITTOX-protocol nr. 71(6)).
(35) Statistici die een modelbenadering zoals gegeneraliseerde lineaire modellen (GLM's) hanteren, pakken de analyse mogelijk op een andere, maar vergelijkbare manier aan, maar zullen niet noodzakelijkerwijs de traditionele ANOVA-tabel opstellen, die dateert uit de tijd van de algoritmische benaderingen om de statistieken te berekenen die werden ontwikkeld in het pre-computertijdperk.
(36) Zie aanhangsel 1 voor definities en eenheden.
(37) Soms aangeduid als P OW; bepaald door een schudflesmethode in TM A.8 (4), een HPLC-methode in TM A.24 (5) en een langzaam-roerenmethode in TM A.23 (6). Voor de bepaling van log KOW wordt soms de generatorkolomtechniek gebruikt. Er is een beperkt aantal studies beschikbaar dat gebruikmaakt van deze techniek, voornamelijk voor gechloreerde bifenylen en dibenzodioxinen (bv. Li & Doucette, 1993) (3). Voor stoffen die kunnen ioniseren, moet log K OW betrekking hebben op de niet-geïoniseerde vorm.
(38) Zie aanhangsel 1 voor definities en eenheden.
(39) TLC: dunnelaagchromatografie; HPLC: hogedrukvloeistofchromatografie; GC: gaschromatografie
(40) In sommige regelgevingskaders is bepaling van de metabolieten verplicht wanneer aan bepaalde voorwaarden wordt voldaan (vgl. punt 65).
(41) In het algemeen moeten de gemeten concentraties in water in de opnamefase ten minste een orde van grootte hoger zijn dan de bepaalbaarheidsgrens, zodat in de depuratiefase van het onderzoek meer dan één halvering van de lichaamsconcentratie kan worden gemeten.
(42) Zie aanhangsel 1 voor definities en eenheden.
(43) De meeste teststoffen worden in het ideale geval niet aangetoond in het controlewater. Nuleffectbepalingen van de concentraties van de teststof mogen alleen relevant zijn voor van nature voorkomende materialen (bv. sommige metalen) en stoffen die algemeen voorkomen in het milieu.
(44) Indien het proces duidelijk geen eersteordekinetiek vertoont, moeten complexere modellen worden gebruikt (zie de referenties in aanhangsel 5) en moet het advies van een biostatisticus worden ingewonnen.
(45) De opname kan beperkt zijn door lage blootstellingsconcentraties vanwege een lage oplosbaarheid in water in de bioconcentratietest, terwijl in de test met blootstelling via het voer veel hogere blootstellingsconcentraties kunnen worden bereikt.
(46) Voor stoffen die uit meerdere componenten bestaan, UVCB's en mengsels moet bij de bepaling van de passende blootstellingsconcentraties rekening worden gehouden met de oplosbaarheid in water van elk van de relevante bestanddelen.
(47) TOC omvat organische koolstof van deeltjes en opgeloste organische koolstof, d.w.z. TOC = POC + DOC.
(48) Hoewel het doorgaans niet wordt aanbevolen, moet, wanneer een oplosmiddel of solubisator wordt gebruikt, de organische koolstof afkomstig van dit agens worden opgeteld bij de organische koolstof afkomstig van de teststof om het gehalte aan organische koolstof in de testbakken te beoordelen.
(49) Als het vetgehalte niet in dezelfde vis wordt bepaald als de teststof, moeten de vissen ten minste een vergelijkbaar gewicht hebben en, voor zover relevant, van hetzelfde geslacht zijn.
(50) Dit is alleen een geldig alternatief als de vissen in alle testgroepen worden gehouden in groepen van gelijke grootte, de vissen volgens hetzelfde patroon worden verwijderd en ze op dezelfde wijze worden gevoerd. Dit waarborgt dat de groei van de vissen in alle testgroepen vergelijkbaar is, mits de geteste concentratie lager is dan het toxisch bereik. Als de groei vergelijkbaar is, zal naar verwachting ook het vetgehalte vergelijkbaar zijn. Een andere groei in de controle wijst op een effect van de stof en maakt de studie ongeldig.
(51) Behalve het gewicht moet ook de totale lengte worden geregistreerd, omdat vergelijking van de lengtegroei tijdens de test een goede indicator is voor het optreden van negatieve effecten.
(52) Er kan een t-toets op de groeisnelheidsconstanten worden uitgevoerd om te toetsen of de groei tussen de controle- en testgroepen verschilt, of er kan een F-toets worden uitgevoerd in geval van een variantieanalyse. Indien nodig kan een F-toets of likelihood-ratio-toets worden gebruikt als hulpmiddel bij het kiezen van het juiste groeimodel (monografie 54 van de OESO, (32)).
(53) Deze percentages veronderstellen dat de analysemethoden betrouwbaar zijn en dat de halveringsperiode korter dan 14 dagen is. Als de analysemethoden minder betrouwbaar zijn of als de halveringsperiode (veel) langer is, worden deze cijfers hoger.
BI: Betrouwbaarheidsinterval (indien schatting mogelijk is)
SD: Standaarddeviatie (indien schatting mogelijk is)
(56) De beperkte test kan in feite worden gebruikt om snel metabolisme aan te tonen, wanneer bekend is dat snel metabolisme waarschijnlijk is.
(57) Wanneer slechts twee gegevenspunten worden gemeten, kunnen de betrouwbaarheidsgrenzen voor BCFKm worden geschat met behulp van bootstrapmethoden. Wanneer ook tussenliggende gegevenspunten beschikbaar zijn, kunnen betrouwbaarheidsgrenzen voor BCFKm worden berekend als in de volledige test.
(58) Zie aanhangsel 1 voor definities en eenheden.
(59) Voor de toepassing van Verordening (EG) nr. 1907/2006 van het Europees Parlement en de Raad inzake de registratie en beoordeling van en de autorisatie en beperkingen ten aanzien van chemische stoffen (REACH) (PB L 396 van 30.12.2006, blz. 1) wordt dit onderwerp behandeld in het richtsnoer voor informatie-eisen en chemische veiligheidsbeoordeling („Guidance on Information Requirements and Chemical Safety Assessment”), hoofdstukken R.7c, R.7.10.3.1, R.7.10.4.1; en figuur R7.10-2.
(60) De meeste teststoffen worden in het ideale geval niet aangetoond in het controlewater. Nuleffectbepalingen van de concentraties van de teststof mogen alleen relevant zijn voor van nature voorkomende materialen (bv. sommige metalen) en stoffen die algemeen voorkomen in het milieu.
(61) Aangezien de BMF is gedefinieerd als de verhouding tussen de concentratie van een stof in een organisme en de concentratie daarvan in het voer van het organisme in de stationaire toestand, wordt rekening gehouden met het vetgehalte door te corrigeren voor het vetgehalte in het organisme en in het voer en is het dus juister om te spreken van een „correctie”. Deze benadering verschilt van „normalisatie” naar een bepaald vetgehalte van het organisme zoals geschiedt in de bioconcentratietest met blootstelling via het water.
(62) De totale lengte moet tijdens de test ook worden geregistreerd, omdat deze een goede indicator is voor het optreden van negatieve effecten.
(63) HCB is opgenomen in de bijlagen A en C bij het Verdrag van Stockholm en in de bijlagen I en III van Verordening (EG) nr. 850/2004 betreffende persistente organische verontreinigende stoffen (PB L 158 van 30.4.2004, blz. 7).
(64) Door snelle groei tijdens de opnamefase zal de werkelijke dosering dalen tot onder de dosering die aan het begin van de blootstelling werd vastgesteld.
(65) In een studie met blootstelling via het water zou een halveringsperiode van 14 dagen overeenkomen met een BCF van ongeveer 10 000 l/kg bij gebruik van vissen van 1 g met een bijbehorende opnamesnelheid van ongeveer 500 l/kg/d (volgens de vergelijking van Sijm et al. (46)).
(66) Aangezien de werkelijke inwendige concentratie in vissen uitsluitend kunnen worden bepaald nadat de test is uitgevoerd, is een schatting van de verwachte inwendige concentratie nodig (bijvoorbeeld op basis van de geraamde BMF en de concentratie in het voer; vgl. vergelijking A5.8 in aanhangsel 5).
(67) Aanwezigheid van de teststof in het testmedium als gevolg van uitscheiding door de vissen of uitloging uit het voedsel kan mogelijk niet helemaal worden voorkomen. Een optie is daarom aan het einde van de opnameperiode de concentratie van de teststof in het water te meten, in het bijzonder als een semistatische opzet wordt gebruikt, om te helpen vaststellen of blootstelling via het water heeft plaatsgehad.
(68) Deze aanpak is specifiek voor onderzoek via het voer en wijkt af van de procedure die wordt gevolgd bij blootstelling via het water, vandaar dat er gesproken wordt van „correctie” in plaats van „normalisatie” om verwarring te voorkomen — zie ook voetnoot in punt 106.
(69) Er kan een t-toets op de groeisnelheidsconstanten worden uitgevoerd om te toetsen of de groei tussen de controle- en testgroepen verschilt, of er kan een F-toets worden uitgevoerd in geval van een variantieanalyse. Indien nodig kan een F-toets of likelihood-ratio-toets worden gebruikt als hulpmiddel bij het kiezen van het juiste groeimodel (monografie 54 van de OESO, (32)).
(70) Een methode voor het analyseren van levensmiddelen op eiwit-, vet, ruwe celstof- en asgehalte; deze informatie is gewoonlijk verkrijgbaar bij de leverancier van het voer.
BI: Betrouwbaarheidsinterval (indien schatting mogelijk is)
SD: Standaarddeviatie (indien schatting mogelijk is)
(73) Meyer et al. (1)
(74) Er zij opgemerkt dat in de test zelf gewicht wordt verkozen als maat voor de grootte en voor afleidingen van de groeisnelheidsconstante. Er wordt echter erkend dat lengte een praktischer maat is als de vissen aan het begin van een experiment op het oog moeten worden geselecteerd (d.w.z. uit het visbestand).
(75) Dit bereik voor de lengte wordt aangegeven in de Testing Methods for New Chemical Substances enz. gebaseerd op de Chemical Substances Control Law (CSCL) van Japan.
(76) De cijfers tussen haakjes zijn de aantallen extra monsters (van het water en de vissen) die worden genomen ingeval voor een aanvullende bemonstering werd geopteerd.
(77) De preliminaire, aan de test voorafgaande schatting van k 2 op basis van log K OW = 4,0 bedraagt 0,652 dag– 1. De totale duur van het experiment wordt gelijkgesteld aan 3 × t SS = 3 × 4,6 dagen, d.w.z. 14 dagen. Voor de schatting van t SS, zie aanhangsel 5.
(78) Bemonster het water nadat ten minste reeds 3 bakvolumes zijn doorgestroomd.
(79) Deze vissen worden geselecteerd uit het visbestand.
(80) Indien een grotere nauwkeurigheid of metabolismestudies noodzakelijk zijn waarvoor meer vissen nodig zijn, moeten deze in het bijzonder aan het einde van de opnamefase en depuratiefase worden bemonsterd (vgl. punt 40).
(81) Voor de bepaling van het vetgehalte zijn mogelijk ten minste 3 extra vissen nodig als het niet mogelijk is om dezelfde vissen te gebruiken als waarvoor aan het begin van de test, het einde van de opnamefase en het einde van de depuratiefase de teststofconcentratie wordt bepaald. Er zij opgemerkt dat het in veel gevallen mogelijk moet zijn uitsluitend de 3 controlevissen te gebruiken (vgl. punt 56).
(82) 3 monsters van het voer van zowel de controle als de testgroep geanalyseerd op de concentratie van de teststof en op het vetgehalte.
(83) Vissen worden zo dicht mogelijk bij het begin van de studie geselecteerd uit het visbestand; uit het visbestand moeten aan het begin van de test ten minste 3 vissen worden bemonsterd voor de bepaling van het vetgehalte.
(84) (Facultatieve) bemonstering vroeg in de opnamefase verschaft gegevens voor het berekenen van de assimilatie van de teststof uit het voer, die dan kan worden vergeleken met het rendement van de assimilatie zoals berekend uit de gegevens van de depuratiefase.
(85) Voor weefselspecifieke analyse kunnen 5 extra vissen worden bemonsterd.
(86) Voor de bepaling van het vetgehalte zijn mogelijk ten minste 3 extra vissen nodig als het niet mogelijk is om dezelfde vissen te gebruiken als waarvoor aan het begin van de test, het einde van de opnamefase en het einde van de depuratiefase de teststofconcentratie wordt bepaald. Er zij opgemerkt dat het in veel gevallen mogelijk moet zijn uitsluitend de 3 controlevissen te gebruiken (vgl. punten 56 en 153).
(87) Als bij elk empirisch verband moet worden nagegaan dat de teststof binnen het toepassingsgebied van het verband ligt.
(88) Het gewicht van de vissen aan het einde van de opnamefase kan worden geschat uit gegevens van eerder onderzoek of eerder opgedane kennis van de waarschijnlijke toename van de grootte van de soort vanaf een typisch gewicht bij aanvang van de test gedurende een opnamefase van typische duur (bijvoorbeeld 28 dagen).
(89) In de meeste programma's waarmee lineaire regressies kunnen worden uitgevoerd, worden ook standaardfouten en betrouwbaarheidsintervallen (BI's) van de schattingen gegeven, bijvoorbeeld in Microsoft Excel met de Analysis ToolPak.
(90) In tegenstelling tot de lineaire regressiemethode geeft deze formule geen standaardfout voor k2.
(91) In tegenstelling tot een lineaire fittingprocedure zal deze methode gewoonlijk geen standaardfout of betrouwbaarheidsinterval voor de geschatte k1 opleveren.
(92) Men moet beseffen dat de onzekerheid in de schatting van k2 in het bioaccumulatiemodel strikt genomen niet goed wordt gebruikt wanneer deze in wezen als een constante wordt beschouwd wanneer k1 in de sequentiële fitmethode wordt gefit. De onzekerheid in de resulterende BCF zal voor de simultane en sequentiële fitmethode daarom verschillend zijn.
(93) In sommige gebieden is mogelijk alleen visvoer verkrijgbaar met een vetconcentratie die ver onder deze bovengrens ligt. In dergelijke gevallen moeten studies met de lagere vetconcentratie in het voer zoals geleverd worden uitgevoerd, en moet de dosering worden aangepast om de vissen gezond te houden. Het vetgehalte van voer mag niet kunstmatig worden verhoogd door toevoeging van een overmaat aan olie.
(94) In het wild is de route die in aquatische milieus tot de grootste blootstelling leidt, voor zeer hydrofobe stoffen waarschijnlijk de route via ingestie, en dus is een geschatte BCF strikt genomen niet representatief voor het bioaccumulatiepotentieel van een dergelijke stof.
(95) Andere Daphnia-soorten zijn eveneens toegestaan, mits ze aan de toepasselijke geldigheidscriteria voldoen (het geldigheidscriterium met betrekking tot het voortplantingsresultaat bij de controlegroepen moet relevant zijn voor alle soorten). Indien andere Daphnia-soorten worden gebruikt, moeten deze duidelijk worden geïdentificeerd en moet hun gebruik worden verantwoord.
(96) Onopzettelijke mortaliteit: niet met een chemische stof samenhangende mortaliteit veroorzaakt door een accidentele gebeurtenis (d.w.z. een bekende oorzaak).
(97) Onbedoelde mortaliteit: niet met een chemische stof samenhangende mortaliteit met onbekende oorzaak.
(*6) Aangeven welk vat voor het experiment werd gebruikt.
(*7) Niet-uitgekomen eitjes aangeven als “AB” in het desbetreffende vakje.
(*8) Eventuele mortaliteit van moederdieren aangeven als “M” in het desbetreffende vakje.
(98) De typische minimale gemiddelde totale lengte is geen geldigheidscriterium, maar afwijkingen onder dit cijfer dienen zorgvuldig te worden onderzocht met het oog op de gevoeligheid van de test. De minimale gemiddelde totale lengte wordt afgeleid van een selectie gegevens die op het desbetreffende moment beschikbaar zijn.
(99) Naargelang van de specifieke stam van regenboogforel die wordt getest, kan het nodig zijn om andere temperaturen te hanteren. Broedvissen moeten bij dezelfde temperatuur worden gehouden als die welke voor de eitjes zal worden gebruikt. Nadat de eitjes in ontvangst zijn genomen van een commerciële kweker, is een korte aanpassingsperiode (bv. 1-2 uur) nodig om de temperatuur te testen na aankomst.
(100) Duisternis voor embryo's en larven tot een week nadat de eitjes zijn uitgekomen, behalve wanneer ze worden geïnspecteerd. Daarna zwak licht tot het eind van de test (fotoperiode van 12-16 uur)(4).
(101) Het lichtregime moet altijd constant zijn voor de desbetreffende testomstandigheden.
(102) Dit wordt voor elke test verricht tot ± 20/00.
(*9) Er dient te worden gevoederd tot het verzadigingspunt. Overtollig voedsel en fecaal materiaal moeten indien nodig worden verwijderd om te voorkomen dat er zich afvalstoffen ophopen
|
FBS |
bevroren pekelkreeftjes (frozen brine shrimp); volwassen Artemia sp |
|
BSN |
larvale pekelkreeftjes (brine shrimp nauplii); net uitgekomen |
|
BSN48 |
larvale pekelkreeftjes (brine shrimp nauplii); 48 uur oud |
(1) larven met dooierzakjes behoeven geen voedsel
(2) gefilterd uit gemengde kweek
(3) korrels van fermentatieproces
(*10) Papillaire uitsteeksels komen gewoonlijk alleen voor bij volwassen mannetjes en worden aangetroffen op de vinstralen van de tweede tot en met zevende of achtste vin, gerekend vanaf het achtereinde van de anaalvin (fig. 1 en 2). De uitsteeksels komen echter zelden voor op de eerste vinstraal vanaf het achtereinde van de anaalvin. Deze operationele standaardprocedure (Standard Operating Procedure - SOP) heeft betrekking op het meten van uitsteeksels op de eerste vinstraal (het nummer van de vinstraal verwijst in deze SOP naar de volgorde vanaf het achtereinde van de anaalvin).
(*11) Homogenisatiebuffer:
|
— |
(50 mM Tris-HCl pH 7,4; 1 % proteaseremmende cocktail (Sigma)): 12 ml Tris-HCl pH 7.4 + 120 μl proteaseremmende cocktail. |
|
— |
TRIS: TRIS-ULTRA PURE (ICN) bijvoorbeeld van Bie & Berntsen, Denemarken. |
|
— |
Proteaseremmende cocktail: van Sigma (voor zoogdierweefsel), productnummer P 8340. |
WAARSCHUWING: de homogenisatiebuffer moet worden gebruikt op de dag van samenstelling. Tijdens gebruik op ijs plaatsen.
(103) Carl von Linné (* 23 mei 1707 in Råshult /Älmhult; † 10 januari 1778 in Uppsala).
(*12) gedemineraliseerd (gedistilleerd of gedeïoniseerd) water.





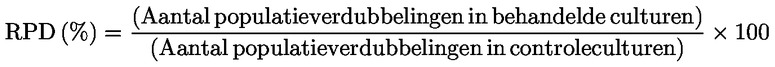

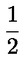 gelijk aan 0,095 mm. Gebruikers dienen zich te realiseren dat spleetlampmicroscopen verschillende resultaten voor corneale dikte kunnen opleveren naargelang van de instelling van de breedte van de spleet.
gelijk aan 0,095 mm. Gebruikers dienen zich te realiseren dat spleetlampmicroscopen verschillende resultaten voor corneale dikte kunnen opleveren naargelang van de instelling van de breedte van de spleet.