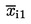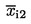|
03/Sv. 010 |
HR |
Službeni list Europske unije |
139 |
32008R0273
|
L 088/1 |
SLUŽBENI LIST EUROPSKE UNIJE |
05.03.2008. |
UREDBA KOMISIJE (EZ) br. 273/2008
od 5. ožujka 2008.
o utvrđivanju detaljnih pravila za primjenu Uredbe Vijeća (EZ) br. 1255/1999 o metodama analize i ocjenjivanja kvalitete mlijeka i mliječnih proizvoda
KOMISIJA EUROPSKIH ZAJEDNICA,
uzimajući u obzir Ugovor o osnivanju Europske zajednice,
uzimajući u obzir Uredbu Vijeća (EZ) br. 1255/1999 od 17. svibnja 1999. o zajedničkoj organizaciji tržišta mlijeka i mliječnih proizvoda (1), a posebno njezine članke 10. i 15. i članak 26. stavak 3., članak 29. stavak 1. i članak 31. stavak 4.,
budući da:
|
(1) |
Uredbom Komisije (EZ) br. 213/2001 (2) utvrđena su detaljna pravila za primjenu Uredbe Vijeća (EZ) br. 1255/1999 u pogledu metoda za analizu i ocjenjivanje kvalitete mlijeka i mliječnih proizvoda. U svjetlu tehničkog razvoja u području analitičke metodologije potrebne su znatne promjene. Radi jasnoće i učinkovitosti te s obzirom na broj i tehničku prirodu izmjena, Uredbu (EZ) br. 213/2001 treba staviti izvan snage i zamijeniti je novom uredbom. |
|
(2) |
Zahtjeve u pogledu sastava i kvalitete mlijeka i mliječnih proizvoda koji su utvrđeni u okviru mjera propisanih Uredbom (EZ) br. 1255/1999 obvezno treba provjeriti da bi se osiguralo da se dosljedno ispunjavaju. |
|
(3) |
Referentne metode za takve provjere često su metode koje objavljuju međunarodne organizacije kao što su Europski odbor za standardizaciju (CEN), Međunarodna mljekarska federacija (IDF), Međunarodna organizacija za standardizaciju (ISO) i Znanstveno društvo posvećeno analitičkoj izvrsnosti (AOAC International), koje te organizacije redovito ažuriraju. U nekim je slučajevima referentna metoda Zajednice navedena, dok u drugim slučajevima u pravilima Zajednice nema navedenih referentnih metoda. Da bi se osigurala ujednačena primjena referentnih metoda, treba sastaviti popis tih metoda i omogućiti Komisiji da ga prema potrebi prilagođava. |
|
(4) |
Primjenu rutinskih metoda ne treba isključiti. Zato treba točno navesti osnovne uvjete za njihovu primjenu. |
|
(5) |
Isto tako treba uvesti jedinstvene postupke da bi se osigurala ujednačena praksa kod ocjenjivanja rezultata analiza, senzorskog ocjenjivanja predmetnih proizvoda te kod ponovne provjere spornih rezultata. |
|
(6) |
Za neke analize trenutačno ne postoje međunarodno priznate referentne metode koje su validirane, pa nema informacija o razlikama u analitičkim rezultatima dobivenim u različitim laboratorijima. Zato na razini Zajednice treba propisati metode koje su validirane u skladu s međunarodnim pravilima i koje treba primjenjivati kao referentne metode. |
|
(7) |
Uredbom Komisije (EZ) br. 1898/2005 (3) utvrđuju se detaljna pravila za primjenu Uredbe Vijeća (EZ) br. 1255/1999 u pogledu mjera za distribuciju vrhnja, maslaca i koncentriranog maslaca na tržištu Zajednice i predviđa se sljedivost vrhnja, maslaca i koncentriranog maslaca u određenim okolnostima, da bi se osigurala pravilna konačna uporaba tih proizvoda. Sljedivost je važna za pravilno funkcioniranje sustava. Da bi se osiguralo da subjekti koji u njemu sudjeluju imaju jednak tretman, treba uvesti zajedničke metode za određivanje nekih od tih markera. |
|
(8) |
U skladu s člankom 9. Uredbe (EZ) br. 1255/1999 može se dodijeliti pomoć za privatno skladištenje sireva napravljenih od ovčjeg mlijeka. U skladu s člankom 31. te Uredbe za te se proizvode može odobriti poseban povrat. Sirevi napravljeni od ovčjeg, kozjeg i bivoljeg mlijeka smiju se iz određenih trećih zemalja uvoziti u Zajednicu po preferencijalnom režimu. S obzirom na gore navedeno, treba osigurati odgovarajuće kontrole da bi se osiguralo da se u predmetne proizvode ne dodaje kravlje mlijeko. Stoga na razini Zajednice treba utvrditi referentnu metodu za otkrivanje kravljeg mlijeka, ne dovodeći u pitanje primjenu rutinskih metoda ako one ispunjavaju određene kriterije. |
|
(9) |
U skladu s Uredbom Komisije (EEZ) br. 2921/90 od 10. listopada 1990. o potpori za proizvodnju kazeina i kazeinata iz obranog mlijeka (4) treba utvrditi odsutnost koliforma. Međunarodno prihvaćena referentna metoda za otkrivanje koliforma u mlijeku i mliječnim proizvodima je ISO 4831. Referentna metoda na razini Zajednice za otkrivanje koliforma utvrđena je na temelju navedenog standarda. |
|
(10) |
Uredbom Vijeća (EEZ) br. 2658/87 od 23. srpnja 1987. o tarifnoj i statističkoj nomenklaturi i Zajedničkoj carinskoj tarifi (5) predviđene su različite stope carine za hranu za životinje koje spadaju pod tarifni broj 2309, ovisno o njihovom sadržaju mliječnih proizvoda. Da bi se osigurala jedinstvena primjena predmetnih pravila, treba utvrditi opće priznatu metodu za analizu sadržaja laktoze čija će primjena biti obvezna u svim državama članicama. |
|
(11) |
U skladu s Uredbom (EZ) br. 1255/1999, maslac i obrano mlijeko u prahu namijenjeni za intervenciju ili, kad se radi o obranom mlijeku u prahu, namijenjeni za hranu za životinje, moraju ispunjavati određene zahtjeve u pogledu kvalitete. Treba utvrditi referentne metode kojima će se provjeravati jesu li ti zahtjevi ispunjeni. |
|
(12) |
Ovom se Uredbom neke metode uvode prvi put. Treba osigurati dovoljno dugo vremensko razdoblje od stupanja ove Uredbe na snagu da bi se laboratorijima omogućilo da te nove metode uvedu i pravilno primjenjuju. Kad god Organizacija za razvoj standarda revidira i objavi neku referentnu metodu iz Priloga I., laboratorijima treba odobriti šest mjeseci za ažuriranje njihovih analitičkih postupaka da bi ih uskladili s novim standardom. |
|
(13) |
Mjere predviđene ovom Uredbom u skladu su s mišljenjem Upravnog odbora za mlijeko i mliječne proizvode, |
DONIJELA JE OVU UREDBU:
POGLAVLJE I.
OPĆE ODREDBE
Članak 1.
Predmet i područje primjene
1. Ovom se Uredbom utvrđuju neke referentne metode za kemijsku, fizikalnu i mikrobiološku analizu te za senzorsko ocjenjivanje mlijeka i mliječnih proizvoda, koje treba koristiti u skladu s režimima predviđenim u okviru zajedničke organizacije tržišta mlijeka i mliječnih proizvoda uspostavljenim Uredbom (EZ) br. 1255/1999 te s pravilima za primjenu tih metoda.
2. Popis referentnih metoda koje se primjenjuju za analize iz stavka 1. utvrđen je u Prilogu I. ove Uredbe.
3. Komisija taj popis ažurira u skladu s postupkom utvrđenim u članku 42. Uredbe (EZ) br. 1255/1999.
Članak 2.
Rutinske metode
Za analize koje nalažu pravila Zajednice mogu se primjenjivati rutinske metode, pod uvjetom da ih se pravilno kalibrira i redovito provjerava prema referentnoj metodi. Kod usporedbe rezultata uzima se u obzir stalna pristranost, ponovljivost i obnovljivost.
U slučaju spornih rezultata, odlučujući je rezultat dobiven referentnom metodom.
O primjeni rutinskih metoda u analizama iz članka 1. države članice obavješćuju Komisiju.
POGLAVLJE II.
METODE ANALIZE
Članak 3.
Ocjenjivanje sukladnosti pošiljke sa zakonskim ograničenjima
Prilog II. ove Uredbe primjenjuje se za utvrđivanje sukladnosti sa zakonskim zahtjevima u pogledu sastava, osim za analizu markera.
Članak 4.
Senzorsko ocjenjivanje
1. Za mlijeko i mliječne proizvode, osim maslaca za javno skladištenje, kao referentnu metodu za senzorsko ocjenjivanje države članice koriste ili standard IDF 99C:1997 ili druge usporedive metode, o kojima službeno obavješćuju Komisiju.
Za provjeru rada ocjenjivača i pouzdanosti rezultata senzorskih analiza primjenjuju se postupci opisani u Prilogu III.
2. Za maslac za javno skladištenje, za provjeru rada ocjenjivača i pouzdanosti rezultata senzorskih analiza primjenjuju se postupci opisani u Prilogu III.
Postupak utvrđen u Prilogu IV. primjenjuje se kao referentna metoda za senzorsko ocjenjivanje.
Članak 5.
Markeri
1. Metoda analize utvrđena Prilogom V. koristi se kao referentna metoda za određivanje sadržaja triglicerida enantne kiseline u maslacu, maslu i vrhnju.
2. Metoda analize utvrđena u Prilogu VI. koristi se kao referentna metoda za određivanje sadržaja vanilina u koncentriranom maslacu, maslacu i vrhnju.
3. Metoda analize utvrđena u Prilogu VII. koristi se kao referentna metoda za određivanje sadržaja etil estera beta-apo-8’ karotenske kisline u koncentriranom maslacu, maslacu i vrhnju.
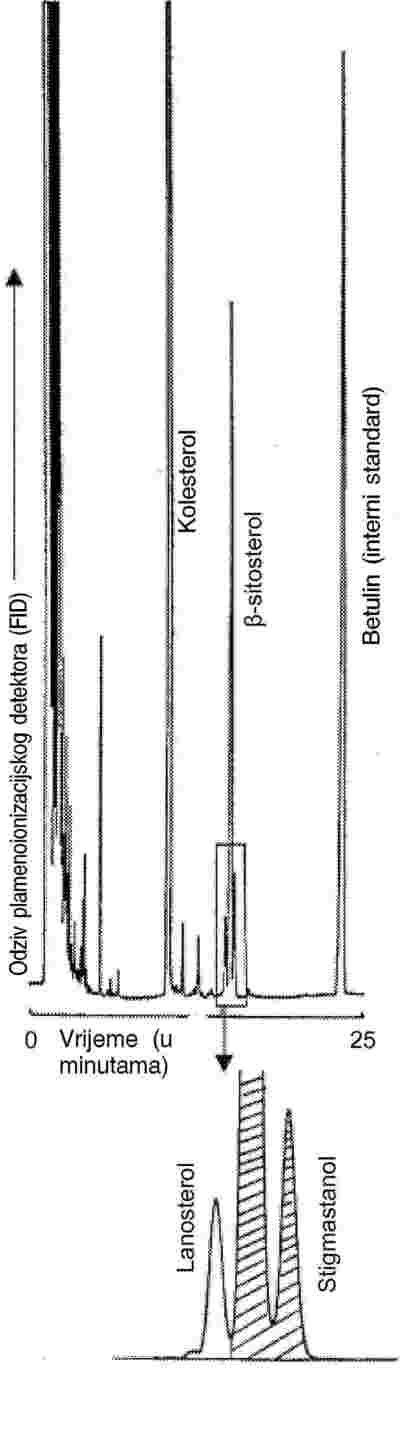
4. Metoda analize utvrđena u Prilogu VIII. koristi se kao referentna metoda za određivanje sadržaja β-sitosterola ili stigmasterola u maslacu i koncentriranom maslacu.
5. Smatra se da su koncentriranom maslacu, maslacu i vrhnju dodani markeri u skladu s relevantnim pravilima Zajednice ako su dobiveni rezultati u skladu sa specifikacijama iz točaka 10. i 11. Priloga V. i točke 8. priloga VI., VII. i VIII.
Članak 6.
Detekcija kazeina kravljeg mlijeka
1. Referentna metoda analize utvrđena u Prilogu IX. koristi se da bi se osiguralo da sir napravljen isključivo od ovčjeg, kozjeg ili bivoljeg mlijeka ne sadrži kazein kravljeg mlijeka.
Ako je udio kazeina kravljeg mlijeka u analiziranom uzorku jednak ili viši od udjela u referentnom uzorku koji sadrži 1 % kravljeg mlijeka, kako je utvrđeno u Prilogu IX., smatra se da je kazein kravljeg mlijeka prisutan.
2. Rutinske metode za detekciju kazeina kravljeg mlijeka u sirevima iz stavka 1. mogu se koristiti pod uvjetom da:
|
(a) |
granica detekcije iznosi maksimum 0,5 %; i |
|
(b) |
da nema lažno pozitivnih rezultata; i |
|
(c) |
da se kazein kravljeg mlijeka može detektirati uz potrebnu osjetljivost čak i nakon dužih razdoblja zrenja, što se može dogoditi u uobičajenim trgovinskim uvjetima. |
Ako bilo koji od gore navedenih zahtjeva nije ispunjen, koriste se referentne metode navedene u Prilogu IX.
Članak 7.
Detekcija koliforma
Koliformi u maslacu, obranom mlijeku u prahu, kazeinu i kazeinatima detektiraju se u skladu s referentnom metodom navedenom u Prilogu X.
Članak 8.
Određivanje sadržaja laktoze
Sadržaj laktoze u proizvodima koji spadaju pod oznaku KN 2309 određuje se u skladu s referentnom metodom navedenom u Prilogu XI.
Članak 9.
Detekcija slatke sirutke
1. Slatka sirutka u obranom mlijeku u prahu namijenjenom za javno skladištenje detektira se u skladu s referentnom metodom navedenom u Prilogu XII.
2. Slatka sirutka u obranom mlijeku i smjesama namijenjenim za hranu za životinje detektira se u skladu s referentnom metodom navedenom u Prilogu XII. U slučaju detekcije slatke sirutke treba primijeniti Prilog XIII.
Članak 10.
Detekcija mlaćenice
Mlaćenica u obranom mlijeku u prahu detektira se u skladu s referentnom metodom navedenom u Prilogu XIV.
Članak 11.
Detekcija antimikrobnih ostataka
Antimikrobni ostaci u obranom mlijeku u prahu detektiraju se u skladu s referentnom metodom navedenom u Prilogu XV.
Članak 12.
Određivanje sadržaja obranog mlijeka u prahu
Sadržaj obranog mlijeka u prahu u krmnim smjesama određuje se u skladu s referentnom metodom navedenom u Prilogu XVI.
Članak 13.
Detekcija škroba
Škrob u denaturiranom mliječnom prahu i krmnim smjesama detektira se u skladu s referentnom metodom navedenom u Prilogu XVII.
Članak 14.
Određivanje sadržaja vlage u vrhnju u prahu
Sadržaj vlage u vrhnju u prahu određuje se u skladu s referentnom metodom navedenom u Prilogu XVIII.
Članak 15.
Određivanje sadržaja vlage u kiseloj mlaćenici u prahu
Sadržaj vlage u vrhnju u prahu za uporabu u hrani za životinje određuje se u skladu s referentnom metodom navedenom u Prilogu XIX.
Članak 16.
Određivanje čistoće mliječne masti
Čistoća mliječne masti određuje se u skladu s referentnom metodom navedenom u Prilogu XX.
POGLAVLJE III.
OPĆE I ZAVRŠNE ODREDBE
Članak 17.
Osiguranje kvalitete
Analize se provode u laboratorijima koji imaju sustav osiguranja analitičke čistoće, koji obuhvaća i postupke interne kontrole kvalitete. Laboratoriji koji nisu akreditirani najmanje jednom godišnje sudjeluju u programima ispitivanja osposobljenosti laboratorija i njihovi rezultati ne smiju odstupati za više od 2σR (standardna devijacija obnovljivosti referentne metode) od konsenzusne vrijednosti. Detaljan opis korištenog sustava može se dobiti na uvid u predmetnom laboratoriju.
Laboratoriji koji su akreditirani u skladu sa standardima navedenim u članku 12. Uredbe (EZ) br. 882/2004 Europskog parlamenta i Vijeća od 29. travnja 2004. o službenom nadzoru koji se provodi radi provjere pridržavanja propisa o hrani i hrani za životinje te pravila o zdravlju i dobrobiti životinja (6), izuzeti su od obveze sudjelovanja u ispitivanjima osposobljenosti.
Članak 18.
Uzorkovanje i sporovi vezani uz rezultate analiza
1. Uzorkovanje se provodi u skladu s uredbom koja je relevantna za predmetni proizvod. Ako ona ne sadrži odredbe o uzorkovanju, primjenjuje se odredba iz standarda ISO 707 | IDF 50, Mlijeko i mliječni proizvodi – Smjernice za uzorkovanje.
2. Laboratorijski izvještaji o rezultatima analiza moraju sadržavati dovoljno informacija za ocjenjivanje rezultata koje se provodi u skladu s Prilogom II. i Prilogom XXI.
3. Za analize koje se traže u skladu s pravilima Zajednice uzorke obvezno treba uzimati u duplikatu.
4. Postupak opisan u Prilogu XXI. primjenjuje se u slučajevima kad operator ne prihvaća rezultate analize.
5. Ako proizvođač u roku od pet radnih dana nakon uzorkovanja može dokazati da postupak uzorkovanja nije bio ispravno proveden, uzorkovanje u slučajevima kad je to moguće treba ponoviti. Ako se uzorkovanje ne može ponoviti, pošiljka se mora prihvatiti.
Članak 19.
Prijelazno razdoblje
Ocjenjivanje sukladnosti prema Prilogu II. ove Uredbe provodi se u roku od 12 mjeseci nakon stupanja Uredbe na snagu. Države članice prema potrebi odmah izvještavaju Komisiju ako se tijekom tog razdoblja pojavi neki veći problem vezan uz postupak statističke kontrole.
Članak 20.
Stavljanje izvan snage
Uredba (EZ) br. 213/2001 stavlja se izvan snage.
Upute na Uredbu koja je stavljena izvan snage smatraju se uputama na ovu Uredbu i tumače se u skladu s korelacijskom tablicom iz Priloga XXII.
Članak 21.
Stupanje na snagu
Ova Uredba stupa na snagu trećeg dana nakon objave u Službenom listu Europske unije.
Primjenjuje se od 31. ožujka 2008.
Ova je Uredba u cijelosti obvezujuća i izravno se primjenjuje u svim državama članicama.
Sastavljeno u Bruxellesu 5. ožujka 2008.
Za Komisiju
Mariann FISCHER BOEL
Članica Komisije
(1) SL L 160, 26.6.1999., str. 48. Uredba kako je zadnje izmijenjena Uredbom (EZ) br. 1152/2007 (SL L 258, 4.10.2007., str. 3.). Uredba (EZ) br. 1255/1999 bit će 1. srpnja 2008. zamijenjena Uredbom (EZ) br. 1234/2007 (SL L 299, 16.11.2007., str. 1.).
(2) SL L 37, 7.2.2001., str. 1.
(3) SL L 308, 25.11.2005., str. 1. Uredba kako je zadnje izmijenjena Uredbom (EZ) br. 1546/2007 (SL L 337, 21.12.2007., str. 68.).
(4) SL L 279, 11.10.1990., str. 22. Uredba kako je zadnje izmijenjena Uredbom (EZ) br. 1487/2006 (SL L 278, 10.10.2006., str. 8.).
(5) SL L 256, 7.9.1987., str. 1. Uredba kako je zadnje izmijenjena Uredbom (EZ) br. 1352/2007 (SL L 303, 21.11.2007., str. 3.).
(6) SL L 165, 30.4.2004., str. 1.
PRILOG I.
(Članak 1.)
POPIS REFERENTNIH METODA
Indeks Min. = minimum, Maks. = maksimum, Prilog = prilog navedenoj uredbi, BST = bezmasna suha tvar, PB = peroksidni broj, I = izgled, O = okus, K = konzistencija, UBB = ukupan broj bakterija, Term. = broj termofilnih bakterija, DČ = država članica, IDF = Međunarodna mljekarska federacija, ISO = Međunarodna organizacija za standardizaciju, IUPAC = Međunarodna unija za čistu i primijenjenu kemiju, ADPI = Američki institut za mliječne proizvode, ZKM = zaslađeno kondenzirano mlijeko, EMV = evaporirano mlijeko ili vrhnje.
DIO A
|
Uredba Komisije |
Proizvod |
Parametar |
Granična vrijednost (1) |
Referentna metoda |
Napomena |
||||||
|
Uredba (EZ) br. 2771/1999 – javno skladište |
Nesoljeni maslac |
Mast |
Min. 82 % m/m |
ISO 17189:2003|IDF 194:2003 |
|
||||||
|
|
|
Voda |
Do 16 % m/m |
ISO 3727-1:2001|IDF 80-1:2001 |
|
||||||
|
|
|
BST |
Do 2 % m/m |
ISO 3727-2:2001|IDF 80-2:2001 |
|
||||||
|
|
|
Kiselost masti |
1,2 mmol/100 g masti |
ISO 1740:2004|IDF 6:2004 |
|
||||||
|
|
|
PB (maks.) |
0,3 meq kisika/1 000 g masti |
ISO 3976:2006|IDF 74:2006 |
Napomena 1. |
||||||
|
|
|
Koliformi |
Detekcija u 1 g nije moguća |
Prilog X. |
Napomena 3. |
||||||
|
|
|
Nemliječna mast |
Detekcija analizom triglicerida nije moguća |
Prilog XX. |
|
||||||
|
|
|
Markeri na bazi sterola |
Detekcija nije moguća, β-sitosterol ≤ 40 mg/kg |
Prilog VIII. |
|
||||||
|
|
|
Ostali markeri: |
|
|
|
||||||
|
|
|
|
Detekcija nije moguća |
Prilog VI. |
|
||||||
|
|
|
|
≤ 6 mg/kg |
Prilog VII. |
|
||||||
|
|
|
|
Detekcija nije moguća |
Prilog V. |
|
||||||
|
|
|
Senzorska svojstva |
Najmanje 4 od 5 bodova za I, O i K |
Prilog IV. |
|
||||||
|
|
|
Disperzija vode |
Najmanje 4 boda |
ISO 7586:1985|IDF 112A:1989 |
|
||||||
|
Uredba (EZ) br. 2771/1999 – Privatno skladištenje |
Nesoljeni maslac |
Mast |
Min. 82 % m/m |
ISO 17189:2003|IDF 194:2003 |
|
||||||
|
|
|
Voda |
Do 16 % m/m |
ISO 3727-1:2001|IDF 80-1:2001 |
|
||||||
|
|
|
BST |
Do 2 % m/m |
ISO 3727-2:2001|IDF 80-2:2001 |
|
||||||
|
Uredba (EZ) br. 2771/1999 – Privatno skladištenje |
Soljeni maslac |
Mast |
Min. 80 % m/m |
ISO 17189:2003|IDF 194:2003 |
|
||||||
|
|
|
Voda |
Do 16 % m/m |
ISO 3727-1:2001|IDF 80-1:2001 |
|
||||||
|
|
|
BST (bez soli) |
Do 2 % m/m |
ISO 3727-2:2001|IDF 80-2:2001 |
|
||||||
|
|
|
Sol |
Do 2 % m/m |
ISO 15648:2004|IDF 179:2004 |
|
||||||
|
Uredba (EZ) br. 1898/2005, poglavlje II. |
Nesoljeni maslac |
Mast |
Min. 82 % m/m |
ISO 17189:2003|IDF 194:2003 |
|
||||||
|
|
|
Nemliječna mast |
|
Prilog XX. |
|
||||||
|
|
|
Voda |
Do 16 % m/m |
ISO 3727-1:2001|IDF 80-1:2001 |
|
||||||
|
|
|
BST |
Do 2 % m/m |
ISO 3727-2:2001|IDF 80-2:2001 |
|
||||||
|
|
|
Markeri: |
|
|
|
||||||
|
|
|
|
Vidjeti Prilog VIII. |
Prilog VIII. |
|
||||||
|
|
|
|
Vidjeti Prilog VI. |
Prilog VI. |
|
||||||
|
|
|
|
Vidjeti Prilog VII. |
Prilog VII. |
|
||||||
|
|
|
|
Vidjeti Prilog V. |
Prilog V. |
|
||||||
|
Uredba (EZ) br. 1898/2005, poglavlje II. |
Soljeni maslac |
Mast |
Min. 80 % m/m |
ISO 17189:2003|IDF 194:2003 |
|
||||||
|
|
|
Nemliječna mast |
|
Prilog XX. |
|
||||||
|
|
|
Voda |
Do 16 % m/m |
ISO 3727-1:2001|IDF 80-1:2001 |
|
||||||
|
|
|
BST (bez soli) |
Do 2 % m/m |
ISO 3727-2:2001|IDF 80-2:2001 |
|
||||||
|
|
|
Sol |
Do 2 % m/m |
ISO 15648:2004|IDF 179:2004 |
|
||||||
|
|
|
Markeri: |
|
|
|
||||||
|
|
|
|
Vidjeti Prilog VIII. |
Prilog VIII. |
|
||||||
|
|
|
|
Vidjeti Prilog VI. |
Prilog VI. |
|
||||||
|
|
|
|
Vidjeti Prilog VII. |
Prilog VII. |
|
||||||
|
|
|
|
Vidjeti Prilog V. |
Prilog V. |
|
||||||
|
Uredba (EZ) br. 1898/2005, poglavlje II. |
Koncentrirani maslac |
Mast |
Min. 99,8 % m/m |
IDF 24:1964 |
|
||||||
|
|
|
Voda i BST |
Do 0,2 % m/m |
ISO 5536:2002|IDF 23:2002 (vlaga) IDF 24:1964 (BST) |
|
||||||
|
|
|
Kiselost masti |
1,2 mmol/100 g masti |
ISO 1740:2004|IDF 6:2004 |
|
||||||
|
|
|
PB (maks.) |
0,5 meq kisika/1 000 g masti |
ISO 3976:2006|IDF 74:2006 |
Napomena 1. |
||||||
|
|
|
Nemliječna mast |
Odsutnost |
Prilog XX. |
|
||||||
|
Okus |
Čist |
||||||||||
|
Miris |
Odsutnost stranih mirisa |
||||||||||
|
Ostalo |
Odsutnost neutralizacijskih sredstava, antioksidansa i konzervansa |
||||||||||
|
|
|
Markeri: |
|
|
|
||||||
|
Vidjeti Prilog VIII. |
Prilog VIII. |
|||||||||
|
Vidjeti Prilog VI. |
Prilog VI. |
|||||||||
|
Vidjeti Prilog VII. |
Prilog VII. |
|||||||||
|
Vidjeti Prilog V. |
Prilog V. |
|||||||||
|
Uredba (EZ) br. 1898/2005, poglavlje II. |
Vrhnje |
Mast |
Min. 35 % m/m |
ISO 2450:1999|IDF 16 C:1987 |
|
||||||
|
|
|
Nemliječna mast |
|
Prilog XX. |
|
||||||
|
|
|
Markeri: |
|
|
|
||||||
|
Vidjeti Prilog VIII. |
|
Napomena 2. |
||||||||
|
|
|
|
Vidjeti Prilog VI. |
Prilog VI. |
|
||||||
|
|
|
|
Vidjeti Prilog VII. |
|
Napomena 2. |
||||||
|
|
|
|
Vidjeti Prilog V. |
Prilog V. |
|
||||||
|
Uredba (EZ) br. 1898/2005, poglavlje III. |
Koncentrirani maslac |
Mast |
Min. 96 % m/m |
|
Napomena 2. |
||||||
|
|
|
Nemliječna mast |
|
Prilog XX. |
|
||||||
|
|
|
BST |
Do 2 % m/m |
|
Napomena 2. |
||||||
|
|
|
Markeri: |
|
|
|
||||||
|
15 g/100 kg koncentriranog maslaca |
Prilog VIII. |
|||||||||
|
|
|
|
17 g/100 kg koncentriranog maslaca |
Prilog VIII. |
|
||||||
|
|
|
|
10,34 kg/100 kg koncentriranog maslaca |
Prilog V. |
|
||||||
|
|
|
|
|
|
Napomena 2. |
||||||
|
|
|
|
|
|
Napomena 2. |
||||||
|
|
|
Lecitin (E 322) |
Do 0,5 % m/m |
|
Napomena 2. |
||||||
|
|
|
NaCl |
Do 0,75 % m/m |
ISO 15648:2004|IDF 179:2004 |
|
||||||
|
|
|
Kiselost masti |
1,2 mmol/100 g masti |
ISO 1740:2004|IDF 6:2004 |
|
||||||
|
|
|
PB (maks.) |
Do 0,5 meq. kisika/ 1 000 g masti |
ISO 3976:2006|IDF 74:2006 |
Napomena 1. |
||||||
|
|
|
Okus |
Čist |
|
|
||||||
|
|
|
Miris |
Odsutnost stranih mirisa |
|
|
||||||
|
|
|
Ostalo |
Odsutnost neutralizacijskih sredstava, antioksidansa i konzervansa |
|
|
||||||
|
Uredba (EZ) br. 1898/2005, poglavlje IV. |
Nesoljeni maslac |
Mast |
Min. 82 % m/m |
ISO 17189:2003|IDF 194:2003 |
|
||||||
|
|
|
Voda |
Do 16 % m/m |
ISO 3727-1:2001|IDF 80-1:2001 |
|
||||||
|
|
|
BST |
Do 2 % m/m |
ISO 3727-2:2001|IDF 80-2:2001 |
|
||||||
|
Uredba (EZ) br. 1898/2005, poglavlje IV. |
Soljeni maslac |
Mast |
Min. 80 % m/m |
ISO 17189:2003|IDF 194:2003 |
|
||||||
|
|
|
Voda |
Do 16 % m/m |
ISO 3727-1:2001|IDF 80-1:2001 |
|
||||||
|
|
|
BST (bez soli) |
Do 2 % m/m |
ISO 3727-2:2001|IDF 80-2:2001 |
|
||||||
|
|
|
Sol |
Do 2 % m/m |
ISO 15648:2004|IDF 179:2004 |
|
||||||
|
Članak 9. i glava II. Uredbe (EZ) br. 1255/1999 |
Sir od ovčjeg i/ili kozjeg mlijeka |
Kravlje mlijeko |
< 1 % m/m |
Prilog IX. |
|
||||||
|
Uredba (EEZ) br. 2921/90 |
Prilog I. – kiseli kazein |
Voda |
Do 12,00 % m/m |
ISO 5550:2006|IDF 78:2006 |
|
||||||
|
|
|
Mast |
Do 1,75 % m/m |
ISO 5543:2004|IDF 127:2004 |
|
||||||
|
|
|
Slobodna kiselost |
Do 0,30 ml 0,1 N otopine NaOH/g |
ISO 5547:1978|IDF 91:1979 |
|
||||||
|
Uredba (EEZ) br. 2921/90 |
Prilog I. – slatki kazein |
Voda |
Do 12,00 % m/m |
ISO 5550:2006|IDF 78:2006 |
|
||||||
|
|
|
Mast |
Do 1,00 % m/m |
ISO 5543:2004|IDF 127:2004 |
|
||||||
|
|
|
Pepeo |
Min. 7,50 % m/m |
ISO 5545:1978|IDF 90:1979 |
|
||||||
|
Uredba (EEZ) br. 2921/90 |
Prilog I. – kazeinati |
Voda |
Do 6,00 % m/m |
ISO 5550:2006|IDF 78:2006 |
|
||||||
|
|
|
Mliječne bjelančevine |
Min. 88,00 % m/m |
ISO 5549:1978|IDF 92:1979 |
|
||||||
|
|
|
Mast i pepeo |
Do 6,00 % m/m |
ISO 5543:2004|IDF 127:2004 |
|
||||||
|
|
|
Vezani pepeo |
|
ISO 5544:1978|IDF 89:1979 |
|
||||||
|
|
|
Pepeo |
|
ISO 5545:1978|IDF 90:1979 |
|
||||||
|
Uredba (EEZ) br. 2921/90 |
Prilog II. – kiseli kazein |
Voda |
Do 10,00 % m/m |
ISO 5550:2006|IDF 78:2006 |
|
||||||
|
|
|
Mast |
Do 1,50 % m/m |
ISO 5543:2004|IDF 127:2004 |
|
||||||
|
|
|
Slobodna kiselost |
Do 0,20 ml 0,1 N otopine NaOH/g |
ISO 5547:1978|IDF 91:1979 |
|
||||||
|
|
|
UBB (maks.) |
30 000/g |
ISO 4833:2003 |
Napomena 3. |
||||||
|
|
|
Koliformi |
Odsutnost u 0,1 g |
Prilog X. |
Napomena 3. |
||||||
|
|
|
Term. (maks.) |
5 000/g |
ISO 4833:2003 |
Napomene 3. i 4. |
||||||
|
Uredba (EEZ) br. 2921/90 |
Prilog II. – slatki kazein |
Voda |
Do 8,00 % m/m |
ISO 5550:2006|IDF 78:2006 |
|
||||||
|
|
|
Mast |
Do 1,00 % m/m |
ISO 5543:2004|IDF 127:2004 |
|
||||||
|
|
|
Pepeo |
Min. 7,50 % m/m |
ISO 5545:1978|IDF 90:1979 |
|
||||||
|
|
|
UBB (maks.) |
30 000/g |
ISO 4833:2003 |
Napomena 3. |
||||||
|
|
|
Koliformi |
Odsutnost u 0,1 g |
Prilog X. |
Napomena 3. |
||||||
|
|
|
Term. (maks.) |
5 000/g |
ISO 4833:2003 |
Napomene 3. i 4. |
||||||
|
Uredba (EEZ) br. 2921/90 |
Prilog II. – kazeinati |
Voda |
Do 6,00 % m/m |
ISO 5550:2006|IDF 78:2006 |
|
||||||
|
|
|
Mliječne bjelančevine |
Min. 88,00 % m/m |
ISO 5549:1978|IDF 92:1979 |
|
||||||
|
|
|
Mast i pepeo |
Do 6,00 % m/m |
ISO 5543:2004|IDF 127:2004 ISO 5544:1978|IDF 89:1979 ili ISO 5545:1978|IDF 90:1979 |
|
||||||
|
|
|
UBB (maks.) |
30 000/g |
ISO 4833:2003 |
Napomena 3. |
||||||
|
|
|
Koliformi |
Odsutnost u 0,1 g |
Prilog X. |
Napomena 3. |
||||||
|
|
|
Term. (maks.) |
5 000/g |
ISO 4833:2003 |
Napomene 3. i 4. |
||||||
|
Uredba (EEZ) br. 2921/90 |
Prilog III. – kazeinati |
Voda |
Do 6,00 % m/m |
ISO 5550:2006|IDF 78:2006 |
|
||||||
|
|
|
Mliječne bjelančevine |
Min. 85,00 % m/m |
ISO 5549:1978|IDF 92:1979 |
|
||||||
|
|
|
Mast |
Do 1,50 % m/m |
ISO 5543:2004|IDF 127:2004 |
|
||||||
|
|
|
Laktoza |
Do 1,00 % m/m |
ISO 5548:2004|IDF 106:2004 |
|
||||||
|
|
|
Pepeo |
Do 6,50 % m/m |
ISO 5544:1978|IDF 89:1979 ili ISO 5545:1978|IDF 90:1979 |
|
||||||
|
|
|
UBB (maks.) |
30 000/g |
ISO 4833:2003 |
Napomena 3. |
||||||
|
|
|
Koliformi |
Odsutnost u 0,1 g |
Prilog X. |
Napomena 3. |
||||||
|
|
|
Term. (maks.) |
5 000/g |
ISO 4833:2003 |
Napomene 3. i 4. |
||||||
|
Uredba (EZ) br. 2799/1999 |
Krmne smjese i obrano mlijeko u prahu (OMP) (za uporabu u hrani za životinje) |
Voda (kisela mlaćenica u prahu) |
Do 5 % m/m |
Prilog XIX. |
|
||||||
|
|
|
Bjelančevine |
31,4 % m/m (min.) bezmasne suhe tvari |
ISO 8968-1|2|3:2001|IDF 20-1|2|3:2001 |
|
||||||
|
|
|
Voda (OMP) |
Do 5 % m/m |
ISO 5537:2004|IDF 26:2004 |
|
||||||
|
|
|
Masti (OMP) |
Do 11 % m/m |
ISO 1736:2000|IDF 9C:1987 |
|
||||||
|
|
|
Slatka sirutka (OMP) |
Odsutnost |
Prilog XIII. |
Napomena 6. |
||||||
|
|
|
Škrob (OMP) |
Odsutnost |
Prilog XVII. |
|
||||||
|
|
|
Voda (smjese) |
Do 5 % bezmasne suhe tvari |
ISO 5537:2004|IDF 26:2004 |
|
||||||
|
|
|
Masti (smjese) |
|
Direktiva Komisije 84/4/EEZ (SL L 15, 18.1.1984., str. 29.) |
|
||||||
|
|
|
Slatka sirutka (smjese) |
Odsutnost |
Prilog XIII. |
|
||||||
|
|
|
Sadržaj OMP (u krajnjem proizvodu) |
Min. 50 % m/m |
Prilog XVI. |
|
||||||
|
|
|
Mast (u krajnjem proizvodu) |
Min. 2,5 % m/m ili 5 % m/m |
Direktiva Komisije 84/4/EEZ (SL L 15, 18.1.1984., str. 29.) |
Napomena 7. |
||||||
|
|
|
Škrob (u krajnjem proizvodu) |
Min. 2 % m/m |
Prilog XVII. |
Napomena 8. |
||||||
|
|
|
Bakar (u krajnjem proizvodu) |
25 ppm |
Direktiva Komisije 78/633/EEZ (SL L 206, 26.7.1987., str. 43.) |
|
||||||
|
Uredba (EZ) br. 214/2001 |
OMP (metoda raspršivanja) |
Mast |
Do 1,0 % m/m |
ISO 1736:2000|IDF 9C:1987 |
|
||||||
|
|
|
Bjelančevine |
31,4 % (2) m/m (min.) suhe bezmasne tvari |
ISO 8968-1/2:2001|IDF 20-1/2:2001 |
|
||||||
|
|
|
Voda |
Do 3,5 % m/m |
ISO 5537:2004|IDF 26:2004 |
|
||||||
|
|
|
Kiselost |
Do 19,5 ml, 0,1 N NaOH, 10 g bezmasne suhe tvari |
ISO 6091:1980|IDF 86:1981 |
|
||||||
|
|
|
Laktati |
Do 150 mg/100 g bezmasne suhe tvari |
ISO 8069:2005|IDF 69:2005 |
|
||||||
|
|
|
Fosfataza |
Negativan nalaz |
ISO 11816-1:2006|IDF 155-1:2006 |
|
||||||
|
|
|
Indeks netopivosti |
Do 0,5 ml na 24 °C |
ISO 8156:2005|IDF 129:2005 |
|
||||||
|
|
|
Sagorjele čestice |
Disk A ili B (15,0 mg) |
ADPI (1990) |
|
||||||
|
|
|
UBB |
40 000/g |
ISO 4833:2003 |
Napomena 3. |
||||||
|
|
|
Koliformi |
Negativan nalaz/0,1 g |
Prilog X. |
Napomena 3. |
||||||
|
|
|
Mlaćenica |
Negativan nalaz |
Prilog XIV. |
|
||||||
|
|
|
Slatka sirutka |
Negativan nalaz |
Prilog XII. |
|
||||||
|
|
|
Kisela sirutka |
Negativan nalaz |
|
Napomena 2. |
||||||
|
|
|
Antimikrobna sredstva |
|
Prilog XV. |
|
DIO B
Referentne metode navedene u dijelu B mogu se koristiti za analizu proizvoda obuhvaćenih bilo kojom od uredaba navedenih u stupcu 1.
|
Uredba Komisije |
Proizvod |
Oznaka KN |
Parametar |
Granična vrijednost |
Referentna metoda |
Primjedba |
|
Uredba (EEZ) br. 2658/87, Uredba (EZ) br. 2535/2001, Uredba (EZ) br. 1282/2006 |
Mlijeko i vrhnje koji nisu koncentrirani i ne sadrže dodani šećer ili druga sladila |
0401 |
Mast (≤ 6 % m/m) |
Granične vrijednosti jesu one koje su navedene u opisu oznake KN za određeni proizvod i koje su detaljnije utvrđene, prema potrebi, u Uredbi Komisije (EEZ) br. 3846/87 (SL L 366, 24.12.1987., str. 1.), dijelu 9. izvozne nomenklature ili u Uredbi (EZ) br. 2535/2001 (SL L 341, 22.12.2001., str. 29.) |
ISO 1211:2001|IDF 1D:1996 |
|
|
|
|
|
Mast (> 6 % m/m) |
|
ISO 2450:1999|IDF 16C:1987 |
|
|
|
Mlijeko i vrhnje koji su koncentrirani ili sadrže dodani šećer ili druga sladila |
0402 |
Mast (u tekućem obliku) |
|
ISO 1737:1999|IDF 13C:1987 |
|
|
|
|
|
Mast (u čvrstom obliku) |
|
ISO 1736:2000|IDF 9C:1987 |
|
|
|
|
|
Bjelančevine |
|
ISO 8968-1|2|3:2001|IDF 20-1|2|3:2001 |
|
|
|
|
|
Saharoza (uobičajeni sadržaj) |
|
ISO 2911:2004|IDF 35:2004 |
|
|
|
|
|
Saharoza (niski sadržaj) |
|
|
Napomena 2. |
|
|
|
|
Suha tvar (ZKM) |
|
ISO 6734:1989|IDF 15B:1991 |
|
|
|
|
|
Suha tvar (EMV) |
|
ISO 6731:1989|IDF 21B:1987 |
|
|
|
|
|
Voda (mlijeko u prahu) |
|
ISO 5537:2004/IDF 26:2004 |
|
|
|
|
|
Voda (vrhnje u prahu) |
|
Prilog XVIII. |
|
|
|
Mlaćenica, fermentirano ili zakiseljeno mlijeko i vrhnje, koncentrirani ili nekoncentrirani, s dodatkom šećera ili drugih sladila |
0403 |
Mast |
|
ISO 1211:2001|IDF 1D:1996 ISO 1736:2000|IDF 9C:1987 ISO 2450:1999|IDF 16C:1987 ISO 7208:1999|IDF 22B:1987 ISO 8262-3:2005|IDF 124-3:2005 |
|
|
|
|
|
Bjelančevine |
|
ISO 8968-1|2|3:2001|IDF 20-1|2|3:2001 |
|
|
|
|
|
Saharoza (uobičajeni sadržaj) |
|
ISO 2911:2004|IDF 35:2004 |
|
|
|
|
|
Saharoza (niski sadržaj) |
|
|
Napomena 2. |
|
|
|
|
Voda (kisela mlaćenica u prahu) |
|
Prilog XIX. |
|
|
|
|
|
Voda (slatka mlaćenica u prahu) |
|
ISO 5537:2004|IDF 26:2004 |
|
|
|
|
|
Suha tvar (drugi proizvodi) |
|
Metode odobrava nadležno tijelo |
|
|
|
Sirutka, koncentrirana ili nekoncentrirana, ili sirutka koja sadrži dodani šećer ili druga sladila; proizvodi koji se sastoje od prirodnih mliječnih sastojaka |
0404 |
Mast |
|
ISO 1736:2000|IDF 9C:1987 ISO 2450:1999|IDF 16C:1987 ISO 7208:1999|IDF 22B:1987 |
|
|
|
|
|
Bjelančevine |
|
ISO 8968-1|2|3:2001|IDF 20-1|2|3:2001 |
|
|
|
|
|
Saharoza (uobičajeni sadržaj) |
|
ISO 2911:2004|IDF 35:2004 |
|
|
|
|
|
Saharoza (niski sadržaj) |
|
|
Napomena 2. |
|
|
|
0404 90 |
Bjelančevine |
|
ISO 8968 1/2 2001|IDF 20-1/2:2001 |
|
|
|
|
|
Voda |
|
IDF 21B:1987 |
|
|
|
|
|
Suha tvar |
|
ISO 6734:1989|IDF 15B:1991 |
|
|
|
|
|
(Koncentrirani proizvodi) |
|
ISO 6731:1989|IDF 21B:1987 |
|
|
|
Maslac i druge masti dobivene od mlijeka; mliječni namazi |
0405 |
Mast (ako je ≤ 85 % m/m) |
|
ISO 17189:2003|IDF 194:2003 |
|
|
|
|
Maslac |
Voda |
|
ISO 3727-1:2001|IDF 80-1:2001 |
|
|
|
|
|
BST |
|
ISO 3727-2:2001|IDF 80-2:2001 |
|
|
|
|
|
NaCl |
|
ISO 15648:2004|IDF 179:2004 |
|
|
|
|
|
Mast (ako je > 99 % m/m) |
|
IDF 24:1964 |
|
|
|
Maslo |
|
Voda (ako je mast < 99 % m/m) |
|
ISO 5536:2002|IDF 23:2002 |
|
|
|
Sir i skuta |
0406 |
Mast |
|
ISO 1735:2004|IDF 5:2004 |
|
|
|
|
|
Suha tvar |
|
ISO 5534:2004|IDF 4:2004 |
|
|
|
|
|
Suha tvar (ricotta) |
|
ISO 2920:2004|IDF 58:2004 |
|
|
|
|
|
NaCl |
|
ISO 5943:2006|IDF 88:2006 |
|
|
|
|
|
Laktoza |
|
ISO 5765-1/2:2002|IDF 79-1/2:2002 |
|
|
Uredba (EEZ) br. 2658/87 |
Krmne smjese |
2309 |
Laktoza |
|
Prilog XI. |
|
Napomene uz popis referentnih metoda Europske unije:
Napomena 1.: Izdvajanje mliječne masti, kako je opisano u standardu ISO 1740:1991 (zaštita od svjetla).
Napomena 2.: Nije utvrđena referentna metoda. Metode koje odobrava nadležno tijelo.
Napomena 3.: Uzorak treba pripremiti u skladu sa standardom ISO 8261:2001|IDF 122:2001.
Napomena 4.: Inkubacija 48 sati na temperaturi od 55 °C, treba se pobrinuti da ne dođe do isušivanja hranjive podloge za uzgoj kulture.
Napomena 5.: % m/m BST = % m/m suhe tvari – % m/m masti.
Napomena 6.: Direktiva Komisije 84/4/EEZ.
Napomena 7.: Uredba Komisije (EZ) br. 2799/1999 (SL L 340, 31.12.1999., str. 3.-27.).
Napomena 8.: Direktiva Komisije 78/633/EEZ.
(1) Ne dovodeći u pitanje zahtjeve iz odgovarajuće Uredbe.
(2) 1. rujna 2009. minimalni sadržaj bjelančevina bio bi 34 %.
PRILOG II.
(Članak 3.)
OCJENJIVANJE SUKLADNOSTI POŠILJKE S PROPISANIM OGRANIČENJIMA
1. NAČELO
U slučajevima kad su detaljni postupci uzorkovanja utvrđeni relevantnim zakonodavstvom, primjenjuju se ti postupci. U svim drugim slučajevima uzima se uzorak koji se sastoji od najmanje 3 uzorkovne jedinice koje se nasumično uzmu iz pošiljke koja je dana na pregled. Može se pripremiti i kompozitni uzorak. Dobiveni rezultat uspoređuje se s propisanim graničnim vrijednostima na temelju izračuna 95 %-tnog intervala pouzdanosti kao dvostruke standardne devijacije, pri čemu relevantna standardna devijacija ovisi o tome (1) je li metoda potvrđena u okviru međunarodne suradnje kroz vrijednosti za σr i σR ili je (2), u slučaju interne validacije, izračunana interna obnovljivost. Ovaj interval pouzdanosti bit će tada jednak mjernoj nesigurnosti rezultata.
2. METODA SE VALIDIRA KROZ MEĐUNARODNU SURADNJU
U ovom slučaju standardna devijacija ponovljivosti σr i standardna devijacija obnovljivosti σR utvrđene su i laboratorij može dokazati sukladnost s izvedbenim karakteristikama validirane metode.
Izračunajte aritmetičku sredinu ![]() za n ponovljenih mjerenja.
za n ponovljenih mjerenja.
Izračunajte proširenu nesigurnost (k = 2)![]() kao
kao
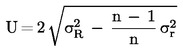
Ako se konačni rezultat mjerenja izračuna pomoću formule x = y1 + y2, x = y1 – y2, x = y1 · y2 ili x = y1 / y2, treba primijeniti uobičajene postupke za kombiniranje standardnih odstupanja u takvim slučajevima.
Pošiljka se ocjenjuje kao nesukladna s propisanom gornjom graničnom vrijednošću UL ako je
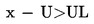 ;
;
inače se ocjenjuje kao sukladna s propisanom gornjom graničnom vrijednošću UL.
Pošiljka se ocjenjuje kao nesukladna s propisanom donjom graničnom vrijednošću LL ako je
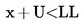 ;
;
inače se ocjenjuje kao sukladna s LL.
3. INTERNA VALIDACIJA IZRAČUNOM STANDARDNE DEVIJACIJE INTERNE OBNOVLJIVOSTI
U slučajevima kad se koriste metode koje nisu navedene u ovoj Uredbi i kad nisu utvrđene mjere za preciznost, mora se provesti interna validacija. U formuli za izračun proširene nesigurnosti U, umjesto σr i σR treba uporabiti standardnu devijaciju interne ponovljivosti sir, odnosno standardnu devijaciju interne obnovljivosti siR.
Pritom važe pravila odlučivanja u skladu s točkom 1. Međutim, ako se pošiljka ocijeni kao nesukladna s propisanim ograničenjem, mjerenja se moraju ponoviti uz primjenu metode navedene u ovoj Uredbi, a odluka se donosi u skladu s točkom 1.
PRILOG III.
(Članak 4.)
OCJENJIVANJE OCJENJIVAČA I POUZDANOST REZULTATA SENZORSKE ANALIZE
Kod primjene metode bodovanja provode se sljedeći postupci (standard IDF 99C:1997).
A. ODREĐIVANJE „INDEKSA PONOVLJIVOSTI”
Ocjenjivač ispituje najmanje 10 uzoraka slijepih proba tijekom razdoblja od 12 mjeseci. To se obično obavlja u nekoliko navrata. Rezultati za svojstva pojedinačnih proizvoda ocjenjuju se na temelju sljedeće formule:
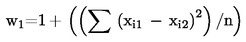
gdje je:
|
w1 |
: |
indeks ponovljivosti; |
|
xi1 |
: |
bodovi za prvo ocjenjivanje uzorka xi; |
|
xi2 |
: |
bodovi za drugo ocjenjivanje uzorka xi; |
|
n |
: |
broj uzoraka. |
Uzorci koji se ocjenjuju trebaju pokazati širok raspon kvalitete. w1 ne smije premašiti 1,5 (na ljestvici od 5 bodova).
B. ODREĐIVANJE INDEKSA ODSTUPANJA
Pomoću ovog indeksa treba provjeriti koristi li ocjenjivač istu skalu za ocjenu kvalitete kao iskusna skupina ocjenjivača. Bodovi koje postigne ocjenjivač uspoređuju se s prosječnim brojem bodova postignutim unutar skupine ocjenjivača.
Za ocjenjivanje rezultata koristi se sljedeća formula:
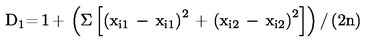
Gdje je:
|
xi1; xi2 |
: |
vidjeti odjeljak (A); |
|
|
: |
prosječan broj bodova skupine ocjenjivača za prvo, odnosno drugo ocjenjivanje uzorka xi; |
|
n |
: |
broj uzoraka (najmanje 10 u 12 mjeseci). |
Uzorci koji se ocjenjuju trebaju pokazivati širok raspon kvalitete. DI ne smije prekoračiti 1,5 (na ljestvici od 5 bodova).
Države članice moraju prijaviti sve probleme s kojima se susreću kod primjene ovog postupka.
Ako se ustanovi da su pojedinačni ocjenjivači prekoračili graničnu vrijednost 1,5 za indekse devijacije ili ponovljivosti, stručnjak/stručnjaci nadležnog tijela mora(ju) provesti jednu ili više nasumičnih naknadnih provjera uzoraka koje ti ocjenjivači ocijene u sljedećih nekoliko tjedana, ili provesti jednu ili više „praćenih” provjera zajedno s tim ocjenjivačima. Za donošenje odluke o zadržavanju njihovih usluga potrebno je pomno praćenje. Nalaze treba dokumentirati i zadržati kao dokaz o daljnjim mjerama.
C. USPOREDBA REZULTATA DOBIVENIH U RAZLIČITIM REGIJAMA DRŽAVE ČLANICE I RAZLIČITIM DRŽAVAMA ČLANICAMA
Ako je to primjenjivo, najmanje jednom godišnje organizira se ispitivanje radi usporedbe rezultata koje su dobili ocjenjivači u različitim regijama. Ako se uoče značajne razlike, treba poduzeti potrebne mjere da bi se utvrdili razlozi i da bi se došlo do usporedivih rezultata.
Države članice mogu organizirati ispitivanja za usporedbu rezultata koje su dobili njihovi vlastiti ocjenjivači i ocjenjivači u susjednim državama članicama. Značajnije razlike razlog su za detaljno istraživanje s ciljem postizanja usporedivih rezultata.
O rezultatima tih usporedbi države članice obavješćuju Komisiju.
PRILOG IV.
(Članak 4.)
SENZORSKO OCJENJIVANJE MASLACA
1. PODRUČJE PRIMJENE
Svrha je ovog postupka za senzorsko ocjenjivanje maslaca osigurati jedinstvenu metodu koja će se primjenjivati u svim državama članicama.
Detaljnije informacije potražite u važećem IDF-ovom međunarodnom standardu za mlijeko i mliječne proizvode, IDF 99 – dijelovi 1., 2., 3., o senzorskom ocjenjivanju.
2. DEFINICIJE
„Senzorsko ocjenjivanje” (procjenjivanje) znači ispitivanje svojstava proizvoda osjetilnim organima.
„Panel” znači skupinu odabranih ocjenjivača koji tijekom ocjenjivanja međusobno ne komuniciraju i ne utječu jedni na druge.
„Ocjenjivač” znači osoba odabrana zbog njezine sposobnosti da izvodi senzorsko ispitivanje. Ova vrsta ocjenjivača može imati ograničeno iskustvo.
„Stručni ocjenjivač” znači osoba s visokom razinom senzorske osjetljivosti i iskustva sa senzorskom metodologijom koja može raditi dosljedne i pouzdane senzorske procjene različitih proizvoda. Ova vrsta ocjenjivača ima dobru i dugotrajnu senzorsku memoriju.
„Bodovanje” znači senzorsko ocjenjivanje koje provodi panel primjenjujući brojčanu ljestvicu. Pritom treba koristiti nomenklaturu nedostatka.
„Klasificiranje” znači razvrstavanje proizvoda po kvaliteti, koje se obavlja na temelju bodovanja.
„Ocjenjivački list”: dokument koji se upotrebljava za zapisivanje pojedinih ocjena za svako svojstvo i konačne ocjene proizvoda. (Ovaj se dokument može koristiti i za bilježenje kemijskog sastava).
3. PROSTORIJA ZA ISPITIVANJE
Više detalja potražite u standardima ISO 8589 i ISO/DIS 22935-2 | IDF 99-2 stavak 7.
Treba poduzeti mjere opreza da na ocjenjivače u prostoriji za ocjenjivanje ne bi utjecali vanjski faktori.
U prostoriji za ocjenjivanje ne smije biti stranih mirisa te mora biti jednostavna za čišćenje. Zidovi moraju biti svijetle boje bez odsjaja.
Prostorija za ocjenjivanje i osvjetljenje u njoj moraju biti takvi da ne utječu na svojstva proizvoda koji se ocjenjuju.
Prostorija mora biti opremljena odgovarajućim uređajem za regulaciju temperature koji omogućuje održavanje stalne temperature maslaca. Dok se obavlja klasifikacija, temperatura maslaca treba biti 12 °C (± 2 °C).
4. ODABIR OCJENJIVAČA
Ocjenjivač mora poznavati proizvode od maslaca i mora biti osposobljen za senzorsko ocjenjivanje. Nadležno tijelo redovito provjerava njegovu osposobljenost (barem jedanput godišnje).
4.1. Detaljne informacije o općim zahtjevima i probirnim testovima koji se mogu primjenjivati prije službenog zapošljavanja novog ocjenjivača potražite u standardu ISO/DIS 22935-1 | IDF 99-1 stavku 4. i stavku 5.1.
Bitno je da se osposobljavanje odvija kontinuirano i da se redovito održavaju opća zasjedanja. Informacije o osposobljavanju panela potražite u standardu ISO 8586-1.
4.2. Početna obuka treba obuhvatiti sljedeće:
|
— |
općenitu teoriju i praktičnu vrijednost senzorskog ocjenjivanja, |
|
— |
metode, ljestvice i opis senzorskih dojmova, |
|
— |
otkrivanje i prepoznavanje senzorskih svojstava i specifične izraze vezane uz senzorska ispitivanja, |
|
— |
temeljnu izobrazbu o proizvodnji maslaca, |
|
— |
potvrđene reference i uzorke, koji ocjenjivaču pomažu kod prepoznavanja posebnih okusa i njihovog intenziteta u proizvodu. |
5. ZAHTJEVI U POGLEDU PANELA
Broj ocjenjivača u panelu mora biti neparan, najmanje tri. Većinu moraju činiti zaposlenici nadležnog tijela ili ovlaštene osobe koje nisu zaposlene u mliječnoj industriji.
Vođa panela odgovoran je za cijeli postupak i smije sudjelovati u radu panela.
Da bi članovi panela postigli optimalnu učinkovitost, prije ocjenjivanja treba uzeti u obzir više čimbenika:
|
— |
član panela ne smije patiti od bolesti koja bi mogla utjecati na njegov rad. U slučaju takve bolesti dotičnog člana u panelu treba zamijeniti netko drugi, |
|
— |
da bi sudjelovali u ocjenjivanju, članovi panela moraju doći na vrijeme i pobrinuti se da imaju dovoljno vremena za ocjenjivanje, |
|
— |
članovi panela ne smiju koristiti sredstva intenzivnog mirisa poput parfema, losiona poslije brijanja, dezodoransa itd. niti jesti hranu intenzivnog okusa (npr. jako začinjenu) itd., |
|
— |
pola sata prije ocjenjivanja članovi panela ne smiju pušiti, jesti niti piti bilo što osim vode. |
6. UČINKOVITOST
Svi ocjenjivači trebaju redovito sudjelovati u panelima za senzorsko ocjenjivanje radi održavanja osposobljenosti. Koliko će to biti često, ovisi o količinama i protoku maslaca, no, ako je moguće, najmanje jednom mjesečno.
Viši ocjenjivači isto tako trebaju sudjelovati u panelima svake godine, po mogućnosti najmanje jednom u tromjesečju.
7. UZORKOVANJE I PRIPREMA UZORAKA
Bitno je da identitet uzoraka tijekom ocjenjivanja bude prikriven da bi se izbjegla svaka pristranost. Uzorke treba označiti šiframa.
To treba organizirati prije ocjenjivanja. Treba utvrditi zahtijevanu temperaturu maslaca tijekom prijevoza do prostorije za ispitivanje (6 °C ± 2 °C).
Ako se senzorsko ocjenjivanje provodi u hladnjači, uzorak se uzima pomoću naprave za uzorkovanje maslaca. Ako se senzorsko ocjenjivanje provodi na nekom drugom mjestu, a ne u hladnjači, treba uzeti najmanje 500 g uzorka. Tijekom ocjenjivanja temperatura maslaca mora biti 12 °C (± 2 °C) (vidjeti: u standardu ISO/DIS 22935-2 | IDF 99-2 temperatura za ocjenjivanje maslaca je 14 °C ± 2 °C). Pod svaku cijenu treba izbjegavati veće devijacije.
8. OCJENJIVANJE VRIJEDNOSTI POJEDINAČNIH SVOJSTAVA
8.1. Senzorsko ocjenjivanje obuhvaća tri svojstva: izgled, konzistenciju i okus:
„Izgled” obuhvaća sljedeća svojstva: boju, vidljivu čistoću, odsutnost fizičkog onečišćenja, odsutnost vidljivog rasta plijesni i ujednačenost disperzije vode. Disperzija vode ispituje se u skladu sa standardom IDF 112A/1989.
„Konzinstencija” obuhvaća sljedeća svojstva: gustoću, teksturu i čvrstoću. Mazivost se može pratiti fizikalnim sredstvima ako to neka država članica želi da bi zadovoljila zahtjeve kupaca. Komisije može odlučiti o usklađivanju te metodologije u budućnosti.
„Gustoća” je pojam koji se odnosi na kohezivnost proizvoda pri konzumaciji. Obično se povezuje s čvrstoćom i mazivošću i mora biti ujednačena u cijelom proizvodu. Usko je povezana s teksturom i predstavlja sposobnost proizvoda da može stajati i nositi vlastitu težinu. Pokazuje ju otpor kod rezanja, a može se izmjeriti mehanički ili na temelju osjeta u ustima ili dodira prstom.
„Okus” je svojstvo koje se osjeća u ustima, pretežno pomoću okusnih pupoljaka na jeziku.
„Aroma” je svojstvo koje se osjeća nosom i osjetom njuha.
Značajnije odstupanje od preporučene temperature sprečava pouzdano ocjenjivanje konzistencije ili okusa. Temperatura je od ključne važnosti.
Ako je temperatura izvan preporučenih okvira, klasifikaciju maslaca treba odgoditi.
8.2. Svako se svojstvo mora senzorski ocjenjivati zasebno. Bodovanje se provodi prema tablici 1.
8.3. Bilo bi poželjno da prije početka ocjenjivanja ocjenjivači zajednički ocijene jedan ili više referentnih uzoraka s obzirom na izgled, konzistenciju i okus da bi se postigla ujednačenost.
8.4. Za prihvaćanje su potrebni sljedeći bodovi:
Vidjeti dio 7. – Nomenklatura i opis kriterija za bodovanje.
|
|
Maksimalan broj bodova |
Traženi broj bodova |
|
Izgled |
5 |
4 |
|
Konzistencija |
5 |
4 |
|
Okus/aroma |
5 |
4 |
|
— |
Ako se ne dobije zahtijevani broj bodova, potrebno je opisati nedostatak. |
|
— |
Bodovi koje svaki ocjenjivač dodijeli za svako svojstvo upisuju se u ocjenjivački list. |
|
— |
Proizvod se prihvaća ili odbija na temelju većinske odluke. |
|
— |
Ne bi se smjelo često događati da razlika ocjena za pojedino svojstvo bude veća od jednog boda (najviše jednom na 20 uzoraka). U protivnom vođa panela treba provjeriti osposobljenost njegovih članova. |
9. NADZOR
Vođa panela, koji mora biti službeni zaposlenik nadležnog tijela i može biti član panela, mora biti općenito odgovoran za čitav postupak. On/ona mora zasebno upisati bodove za svako svojstvo u ocjenjivački list i potvrditi da je proizvod prihvaćen ili odbijen.
10. NOMENKLATURA
Vidjeti priloženu tablicu 2.
11. REFERENTNA DOKUMENTACIJA
FIL-IDF 99C:1997 Senzorsko ocjenjivanje mliječnih proizvoda bodovanjem – Referentna metoda
ISO/DIS 22935 | IDF 99 Međunarodni standard za mlijeko i mliječne proizvode – Senzorska analiza – dijelovi 1. – 3.
ISO 8586-1 Senzorska analiza – Opće smjernice za odabir, osposobljavanje i praćenje ocjenjivača – dio 1.
ISO 8589 Senzorska analiza – Opće uputstvo za projektiranje prostorija za ispitivanje
FIL-IDF 112A:1989 Maslac – Određivanje vrijednosti za disperziju vode
Tablica 1.
Ocjenjivanje maslaca
|
Izgled |
Konzistencija |
Okus + miris |
||||||
|
Bodovi |
Br. (3) |
Napomene |
Bodovi (klasa kvalitete) |
Br. (3) |
Napomene |
Bodovi (klasa kvalitete) |
Br. (3) |
Napomene |
|
5 |
|
Vrlo dobar idealni tip najviša kvaliteta (ravnomjerna suhoća) |
5 |
|
Vrlo dobra idealni tip najviša kvaliteta (ravnomjerna mazivost) |
5 |
|
Vrlo dobri idealni tip najviša kvaliteta (potpuno čist, blagi miris) |
|
4 |
|
Dobar (2) bez očitih nedostataka |
4 |
17 18 |
Dobra (2) tvrda mekana |
4 |
|
Dobri (2) bez očitih nedostataka |
|
3 |
1 2 3 4 5 6 7 8 |
Prihvatljiv (manji nedostaci) razrahljen (nepovezan), vodenast neujednačene boje (dvobojan) prugast prošaran, mramorast točkast izdvajanje ulja prejako obojen slabe, otvorene teksture |
3 |
14 15 16 17 18 |
Prihvatljiva (manji nedostaci) slaba, krhka, mrvičasta mljecava, poput tijesta, masna ljepljiva tvrda mekana |
3 |
21 22 25 27 33 34 35 |
Prihvatljivi (manji nedostaci) nečisti strani okus kisli okus po kuhanom, okus po zagorenom okus po krmi sirovi, gorki preslani |
|
2 |
1 3 4 5 6 10 11 12 |
Loš (očiti nedostaci) razrahljen (nepovezan), vodenast prugast prošaran, mramorast točkast izdvajanje ulja strane tvari pljesnjiv neotopljena sol |
2 |
14 15 16 17 18 |
Loša (očiti nedostaci) slaba, krhka, mrvičasta mljecava, poput tijesta, masna ljepljiva tvrda mekana |
2 |
21 22 23 25 32 33 34 35 36 37 |
Loši (očiti nedostaci) nečisti strani okus ustajali kiseli oksidirani, metalni okus okus po krmi grubi, gorki preslani pljesnjivi, truli okus po kemikalijama |
|
1 |
1 3 4 5 6 7 9 10 11 12 |
Vrlo loš (veliki nedostaci) razrahljen (nepovezan), vodenast prugast prošaran, mramorast točkast izdvajanje ulja prejako obojen zrnat strane tvari pljesnjiv neotopljena sol |
1 |
14 15 16 17 18 |
Vrlo loša (veliki nedostaci) slaba, krhka, mrvičasta mljecava, poput tijesta, masna ljepljiva tvrda mekana |
1 |
22 24 25 26 28 29 30 31 32 34 35 36 37 38 |
Vrlo loši (veliki nedostaci) strani okus sirasti, mliječno-kiseli okus kiseli okus i miris po kvascu okus po plijesni užegli uljasti, riblji lojasti oksidirani okus, metalni okus grubi, gorki preslani pljesnjivi, truli sladni okus po kemikalijama |
Tablica 2.
Tablica nedostataka maslaca
I. Izgled
|
1. |
razrahljen (nepovezan), vodenast |
|
2. |
neujednačene boje, dvobojan |
|
3. |
prugast |
|
4. |
prošaran, mramorast |
|
5. |
točkast |
|
6. |
izdvajanje ulja |
|
7. |
prejako obojen |
|
8. |
slabe, otvorene teksture |
|
9. |
zrnat |
|
10. |
strane tvari |
|
11. |
pljesnjiv |
|
12. |
neotopljena sol |
II. Konzistencija
|
14. |
slaba, krhka, mrvičasta |
|
15. |
mljecava, poput tijesta, masna |
|
16. |
ljepljiva |
|
17. |
tvrda |
|
18. |
mekana |
III. Okus i miris
|
20. |
bez okusa |
|
21. |
nečisti (3) |
|
22. |
strani okus |
|
23. |
ustajali |
|
24. |
sirasti, mliječno-kiseli okus |
|
25. |
kiseli |
|
26. |
okus i miris po kvascu |
|
27. (a) |
kuhani okus |
|
(b) |
zagoreni okus |
|
28. |
pljesnjiv okus |
|
29. |
užegli |
|
30. |
uljasti, riblji |
|
31. |
lojasti |
|
32. (a) |
oksidirani okus |
|
(b) |
metalni okus |
|
33. |
okus po krmi |
|
34. |
grubi, gorki |
|
35. |
preslani |
|
36. |
pljesnjivi, truli |
|
37. |
sladni |
|
38. |
okus po kemikalijama |
(1) Tablica 2.
(2) Nedostaci navedeni pod „Dobar” predstavljaju samo mala odstupanja od idealnog tipa.
(3) Ovu oznaku treba koristiti što je rjeđe moguće i samo onda kad se nedostatak ne može točnije opisati.
PRILOG V.
(Članak 5.)
ODREĐIVANJE SADRŽAJA TRIGLICERIDA ENANTNE KISELINE U MASLACU, MASLU I VRHNJU PLINSKOM KROMATOGRAFSKOM ANALIZOM TRIGLICERIDA
1. PODRUČJE PRIMJENE
Ovom metodom utvrđen je način određivanja sadržaja triglicerida enantne kiseline u maslacu, maslu i vrhnju.
2. POJMOVI I DEFINICIJA
Sadržaj enantne kiseline: sadržaj triglicerida enantne kiseline određen postupkom utvrđenim u toj metodi.
Napomena: sadržaj enantne kiseline izražava se za maslo i maslac u kg po toni proizvoda, a za vrhnje u kg po toni mlijeka.
3. NAČELO
Mliječna se mast ekstrahira iz različitih proizvoda prema standardu ISO 14156 | IDF 172:2001. Kvantitativno određivanje sadržaja triglicerida enantne kiseline u ekstrahiranoj masti provodi se kapilarnom plinskom kromatografijom (GC). Rezultat dobiven za uzorak ocjenjuje se prema trigliceridima kapronske kiseline kao internom standardu.
Napomena: Tributrin se isto tako pokazao kao zadovoljavajući interni standard.
4. REAGENSI
Koristite samo reagense priznate analitičke čistoće.
4.1. N-heksan.
4.2. Standardni triglicerid kapronske kiseline, čistoće najmanje 99 %.
4.3. Standardni triglicerid enantne kiseline, čistoće najmanje 99 %.
4.4. Bezvodni natrijev sulfat (Na2SO4).
5. OPREMA
Uobičajena laboratorijska oprema, a posebno sljedeće:
5.1. Analitička vaga s točnošću od 1 mg.
5.2. Odmjerne tikvice zapremine 10 ml i 20 ml.
5.3. Epruvete za centrifugiranje zapremine 30 ml.
5.4. Rotacijski evaporator.
5.5. Sušionik koji može održavati temperaturu od 50 °C ± 5 °C.
5.6. Filtar-papir, srednje poroznosti, promjera oko 15 cm.
5.7. Oprema za plinsku kromatografiju.
5.7.1. Plinski kromatograf opremljen split/splitless ili on-column injektorom kao i plamenoionizacijskim detektorom (FID).
5.7.2. Kolona plinskog kromatografa (GC) sa stacionarnom fazom, koja je uspješno uporabljena za razdvajanje triglicerida (100 % dimetilpolisiloksan ili 5 % fenil-95 % metilpolisiloksan). Odaberite stacionarnu fazu, duljinu kolone (između 4 m i 15 m), unutarnji promjer (između 0,22 mm i 0,50 mm) i debljinu nanosa (0,12 μm ili više), uzimajući u obzir laboratorijska iskustva i injektorski sustav koji koristite. U svakom slučaju, u odabranoj koloni mora doći do potpunog razdvajanja pikova otapala i triglicerida kapronske kiseline i rezolucije pikova triglicerida kapronske i enantne kiseline po baznoj liniji. Primjeri važećih uvjeta navedeni su dalje u tekstu.
5.7.2.1. Primjer važećih uvjeta pri uporabi split injektora:
|
— |
Plin nositelj: helij, |
|
— |
Tlak u glavi kolone: 100 kPa, |
|
— |
Kolona: duljina 12 m, unutarnji promjer 0,5 mm, debljina sloja 0,1 μm, kolona od taljenog silicijevog dioksida, |
|
— |
Stacionarna faza: 100 % dimetilpolisiloksan ili 5 % fenil-95 % dimetilpolisiloksan (za ex HT5), |
|
— |
Temperatura kolone: početna temperatura od 130 °C održava se 1 minutu, povećava brzinom od 20 °C/min do 260 °C i zatim podiže brzinom od 30 °C/min do 360 °C; temperatura 360 °C održava se 10 minuta, |
|
— |
Temperatura detektora: 370 °C, |
|
— |
Temperatura injektora: 350 °C, |
|
— |
Omjer razdvajanja: 1:30, |
|
— |
Količina ubrizganog uzorka: 1 μl. |
5.7.2.2. Primjer važećih uvjeta pri uporabi on-column injektora:
|
— |
Plin nositelj: vodik (sustav sa stalnim protokom), |
|
— |
Tlak u glavi kolone: 89 kPa, |
|
— |
Kolona: duljina 4 m, unutarnji promjer 0,32 mm, debljina nanosa 0,25 μm, kolona od taljenog silicijevog dioksida, |
|
— |
Stacionarna faza: 5 % fenil, 95 % dimetilpolisiloksan, |
|
— |
Temperatura kolone: početna temperatura od 60 °C održava se 2 minute, povećava brzinom od 35 °C/min do 340 °C i održava na toj temperaturi 5 minuta, |
|
— |
Temperatura detektora: 350 °C, |
|
— |
Količina ubrizganog uzorka: 1 μl. |
5.8. Šprica za ubrizgavanje, zapremine 5 μl.
6. UZORKOVANJE
Važno je da laboratorij dobije uzorak koji je zaista reprezentativan i koji nije bio oštećen ili promijenjen tijekom prijevoza ili skladištenja.
Uzorkovanje nije dio metode utvrđene u ovom međunarodnom standardu. Preporučena metoda uzorkovanja navedena je u standardima IDF 50C:1995 ili ISO 707-1997 – Mlijeko i mliječni proizvodi – Metode uzorkovanja.
7. POSTUPAK
7.1. Priprema ispitnog uzorka i uzorka za analizu
Postupajte u skladu sa standardima ISO 14156 | IDF 172:2001.
7.1.1. Maslo, maslac
7.1.1.1. U sušioniku (5.5.) otopite 50 do 100 g ispitnog uzorka.
7.1.1.2. Stavite 0,5 do 1,0 g bezvodnog natrijevog sulfata u naborani filtar-papir.
7.1.1.3. Profiltrirajte mast kroz filtar-papir s bezvodnim natrijevim sulfatom, pri čemu se filtrat skuplja u laboratorijskoj čaši u sušioniku (5.5.). Pri dekantiranju otopljenog maslaca na filtar-papir pazite da se ne prenese serum.
7.1.2. Vrhnje
7.1.2.1. Ispitni uzorak zagrijte na temperaturu 20 °C ± 2 °C.
7.1.2.2. Uzorak dobro promućkajte ili promiješajte.
7.1.2.3. Razrijedite odgovarajuću količinu ispitnog uzorka da biste dobili uzorak za analizu od 100 ml s masenim udjelom masti od približno 4 %.
7.1.2.4. Za ekstrakciju masti iz vrhnja nastavite postupak kao kod sirovog i homogeniziranog mlijeka (vidjeti standard ISO 14156 | IDF 172:2001, stavak 8.3.).
7.1.2.5. U odmjernoj tikvici od 10 ml (5.2.) odvagnite 1 g ekstrahirane masti, s točnošću od 1 mg. Dodajte 1 ml otopine 7.2.2. Do oznake 10 ml dolijte n-heksan (4.1.) i promiješajte.
7.1.2.6. U odmjernu tikvicu od 10 ml (5.2.) stavite 1 ml otopine 7.1.1.2. i do oznake 10 ml razrijedite n-heksanom (4.1.).
7.2. Priprema kalibracijskih standarda
7.2.1. Otopite 100 mg triglicerida enantne kiseline (4.3.) u 10 ml n-heksana (4.1.).
7.2.2. Otopite 100 mg triglicerida kapronske kiseline (4.2.) u 10 ml n-heksana (4.1.).
7.2.3. U 10-mililitarsku odmjernu tikvicu (5.2.) stavite 1 ml otopine 7.2.2. Do oznake 10 ml dolijte n-heksan (4.1.).
7.2.4 U 10-mililitarsku odmjernu tikvicu (5.2.) stavite 1 ml otopine 7.2.1. i 1 ml otopine 7.2.2. Do oznake 10 ml dolijte n-heksan (4.1.).
7.2.5 U 10-mililitarsku odmjernu tikvicu (5.2.) stavite 1 ml otopine 7.2.4. i do oznake 10 ml dolijte n-heksan (4.1.).
7.3. Kromatografsko određivanje
7.3.1. Dvaput ubrizgajte po 1 μl standardne otopine 7.2.5.
7.3.2. Ubrizgajte po 1 μl otopine svakog uzorka.
Napomena: Ako se koristi on-column injektorski sustav, treba primijeniti veći stupanj razrjeđenja i na standardnu otopinu i na otopine uzoraka.
7.3.3. Postupak 7.3.1. ponovite na svaka 3 uzorka da biste obuhvatili uzorke između dva ubrizgavanja standardne otopine. Rezultati se temelje na prosječnoj srednjoj vrijednosti faktora odziva iz standardnih kromatograma.
8. IZRAČUN REZULTATA
Za svaki kromatogram povežite površine pikova koji se odnose na trigliceride enantne i kapronske kiseline.
Slijedite te upute za svaki obuhvaćeni niz tj. za seriju obuhvaćenih uzoraka; standardna otopina ubrizgana dvaput neposredno prije njih je STD1, a standardna otopina ubrizgana dvaput neposredno iza njih je STD2.
8.1. Kalibracija
8.1.1. Izračunajte faktor odziva za svaki duplikat STD1, Rf1(a) i Rf1(b)
Rf1 (a) ili (b) = (površina pika triglicerida kapronske kiseline / površina pika triglicerida enantne kiseline) × 100
Izračunajte prosječnu srednju vrijednost faktora odziva Rf1
Rf1 = (Rf1 (a) + Rf1 (b)) / 2
8.1.2. Na sličan način izračunajte prosječnu srednju vrijednost faktora odziva STD2, Rf2
8.1.3. Izračunajte prosječnu srednju vrijednost faktora odziva Rf
Rf = (Rf1 + Rf2) / 2
8.2. Ispitni uzorci
Za svaki kromatogram uzoraka dobiven između STD1 i STD2 izračunajte sadržaj enantne kiseline, C (kg/t):
C = (površina pika triglicerida enantne kiseline × Rf × 100)/(površina pika triglicerida kapronske kiseline × Wt × 1 000)
gdje je:
|
— |
Wt = masa oduzete masti (g), |
|
— |
100 = volumen razrjeđenja za uzorak, |
|
— |
1 000 = faktor konverzije (iz μg/g u kg/t). |
Za uzorke maslaca uzmite u obzir sadržaj maslaca i izračunajte korigiranu vrijednost koncentracije, Cmaslac (kg/t maslaca)
Cmaslac = Cmast × F
gdje je F sadržaj masti u maslacu.
9. PRECIZNOST
Detalji o međulaboratorijskom ispitivanju maslaca u skladu sa standardima ISO 5725-1 i ISO 5725-2 o preciznosti metode navedeni su u točki 12.
Granične vrijednosti za ponovljivost i obnovljivost izražene su za razinu vjerojatnosti od 95 % i možda neće biti primjenjive na raspon koncentracija i matrice koje se razlikuju od navedenih.
9.1. Ponovljivost
Apsolutna razlika između rezultata dvaju pojedinačnih ispitivanja, koja istom metodom na jednakom ispitnom materijalu u istom laboratoriju dobije isti analitičar koristeći istu opremu u kratkom vremenskom intervalu, ne smije u više od 5 % slučajeva biti veća od 0,35 kg/t.
9.2 Obnovljivost
Apsolutna razlika između rezultata dvaju pojedinačnih ispitivanja, koja istom metodom na jednakom ispitnom materijalu u različitim laboratorijima dobiju različiti analitičari koristeći različitu opremu, ne smije u više od 5 % slučajeva biti veća od 0,66 kg/t.
10. GRANICE DOPUŠTENOG ODSTUPANJA: DONJE GRANICE (SLUČAJ NEDOSTATNIH KOLIČINA)
10.1. Od proizvoda s dodanim markerom obvezno treba uzeti po tri uzorka radi provjere pravilnosti dodavanja
10.2. Maslac i koncentrirani maslac
10.2.1. Stupanj inkorporacije je 11 kg triglicerida enantne kiseline minimalne čistoće 95 % po toni maslaca, tj. 10,45 kg/t.
10.2.2. Rezultati analize proizvoda dobiveni na tri uzorka koriste se za provjeru stope i homogenosti inkorporacije markera, a najniži od tih rezultata uspoređuje se sa sljedećim graničnim vrijednostima:
|
— |
9,51 kg/t (95 % minimalnog stupnja inkorporacije triglicerida enantne kiseline čistoće 95 %, pojedinačno određivanje), |
|
— |
6,89 kg/t (70 % minimalnog stupnja inkorporacije triglicerida enantne kiseline čistoće 95 %, pojedinačno određivanje), |
|
— |
koncentracija markera u uzorku koji je dao najniži rezultat koristi se u vezi s interpolacijom između 9,51 kg/t i 6,89 kg/t. |
10.3. Vrhnje
10.3.1. Stupanj inkorporacije je 10 kg triglicerida enantne kiseline minimalne čistoće 95 % po toni mliječne masti, tj. 9,50 kg/t mliječne masti s dodanim markerom.
10.3.2. Rezultati analize proizvoda dobiveni na tri uzorka koriste se za provjeru stupnja homogenosti inkorporacije markera, a najniži od tih rezultata uspoređuje se sa sljedećim graničnim vrijednostima:
|
— |
8,60 kg/t (95 % minimalnog stupnja inkorporacije triglicerida enantne kiseline čistoće 95 %, pojedinačno određivanje), |
|
— |
6,23 kg/t (70 % minimalnog stupnja inkorporacije triglicerida enantne kiseline čistoće 95 %, pojedinačno određivanje), |
|
— |
koncentracija markera u uzorku koji je dao najniži rezultat koristi se u vezi s interpolacijom između 8,60 kg/t i 6,23 kg/t. |
11. GRANICE DOPUŠTENOG ODSTUPANJA: GORNJE GRANICE (SLUČAJ PREKORAČENJA KOLIČINE ZA VIŠE OD 20 %)
11.1. Od proizvoda s dodanim markerom treba uzeti po tri uzorka radi provjere pravilnosti dodavanja markera
11.2. Maslac i koncentrirani maslac
11.2.1. Rezultati analize proizvoda dobiveni na tri uzorka koriste se za provjeru stope i homogenosti inkorporacije markera, a srednja vrijednost tih rezultata uspoređuje se sa sljedećim graničnim vrijednostima:
|
— |
gornja granica iznosi 12,96 kg/t. |
11.3 Vrhnje
11.3.1. Rezultati analize proizvoda dobiveni na tri uzorka koriste se za provjeru stope i homogenosti inkorporacije markera, a srednja vrijednost tih rezultata uspoređuje se sa sljedećim graničnim vrijednostima:
|
— |
gornja granica iznosi 11,82 kg/t. |
12. DODATNI PODACI: STATISTIČKA ANALIZA REZULTATA ODREĐIVANJA TRIENANTOATA U MLIJEČNOJ MASTI ANALIZOM TRIGLICERIDA
Za određivanje sadržaja trienantoata u maslacu bila su izvedena četiri međulaboratorijska ispitivanja.
U prvom ring testu sudjelovalo je devet laboratorija, a specifikacije o metodama analize nisu bile dane.
U drugom ring testu sudjelovalo je deset laboratorija uz primjenu četiri različite metode:
|
— |
kvantifikacija metilheptanoata uz korištenje n-nonana ili metilnonanoata kao internog standarda, |
|
— |
kvantifikacija trienantoata uz korištenje trikaproata kao internog standarda, |
|
— |
kvantifikacija metilheptanoata uz korištenje kalibracijskog uzorka/mješavine, |
|
— |
kvantifikacija metilheptanoata uz korištenje kalibracijske mješavine. |
Osim toga, za analizu metilnih estera masnih kiselina (FAME) bila su primijenjena dva različita postupka metilacije (De Francesco te Christopherson i Glass).
S obzirom na dobivene rezultate, za treći ring test odabrane su dvije metode:
|
— |
kvantifikacija metilheptanoata uz korištenje n-nonana ili metilnonanoata kao internog standarda, |
|
— |
kvantifikacija trienantoata uz korištenje trikaproata kao internog standarda. |
Rezultati sedam laboratorija pokazali su da je metoda FAME proizvela veću varijabilnost pa je stoga odlučeno da se primijeni samo određivanje trienantoata kao triglicerida prema postupku kvantifikacije q za trienantoat uz korištenje trikaproata kao internog standarda. Osim toga, za analizu triglicerida treba koristiti kapilarnu kolonu.
U četvrtom ring testu radilo se na četiri uzorka (A, B, C, D) i devet laboratorija je dalo rezultate (tablice 1.-2.).
Dva su laboratorija (DE i UE) analizirala uzorke primjenjujući metodu FAME.
Zbog manjeg broja laboratorija, statistički je izračun napravljen za cijeli komplet podataka (slike 1.-2.), uključujući rezultate FAME, kao i za podatke dobivene analizom triglicerida (TG).
Testovi za ekstremne vrijednosti:
|
— |
Uzorak A. – Dixonov, Cochranov i Grubbsov test na razinama 1 i 5 % pokazali su jedan ekstremni laboratorijski rezultat. |
|
— |
Uzorak B. – Grubbsov test na razini 5 % pokazao je jedan ekstremni laboratorijski rezultat. |
|
— |
Uzorak C. – Dixonov i Grubbsov test na razinama 1 i 5 % pokazali su jedan ekstremni laboratorijski rezultat. |
|
— |
Uzorak D. – Dixonov i Grubbsov test na razinama 1 i 5 % pokazali su jedan ekstremni laboratorijski rezultat. |
Ekstremne su vrijednosti isključene iz izračuna.
Važno je napomenuti da se kod tih testova rezultati dobiveni metodom FAME nikad nisu smatrali ekstremnim vrijednostima.
Parametri preciznosti
Tablice 1. i 2. prikazuju rezultate svih laboratorija i parametre preciznosti koji su izračunani za prihvatljivi broj (8) laboratorija, no nažalost nisu dobiveni istom metodom analize.
Tablice 3. i 4. prikazuju rezultate koji su dobiveni samo metodom analize triglicerida i odgovarajuće parametre preciznosti. Prihvaćanje ovih parametara uvjetovano je malim brojem laboratorija (6).
Slike 2. i 3. prikazuju trend Sr i SR izračunan za četiri uzorka dvaju gore opisanih setova podataka.
Tablica 5. prikazuje vrijednosti Sr i SR zajedno s odgovarajućim združenim vrijednostima i sveukupne parametre r i R.
Na kraju je izračunana kritična razlika na razini vjerojatnosti od 95 %.
Tablica 1.
Statistički rezultati metoda TG + FAME*
|
Uzorak A |
|
R1 |
R2 |
Srednja vrijednost |
Br. laboratorija zadržanih nakon eliminacije ekstremnih rezultata |
8 |
|
RENNES |
FR1 |
11,0 |
11,1 |
11,1 |
Br. ekstremnih rezultata |
1 |
|
RIKILT |
NL |
11,2 |
11,2 |
11,2 |
Ekstremni rezultati |
DK |
|
ZPLA |
DE* |
11,6 |
11,8 |
11,7 |
Srednja vrijednost |
11,3 |
|
ADAS |
GB |
11,4 |
11,2 |
11,3 |
Stvarna vrijednost |
11,0 |
|
CNEVA |
FR2 |
11,4 |
11,4 |
11,4 |
Standardna devijacija ponovljivosti (Sr) |
0,09 |
|
LODI |
IT |
11,1 |
11,3 |
11,2 |
Relativna standardna devijacija ponovljivosti (RSDr%) |
0,80 |
|
EELA |
FI |
11,3 |
11,2 |
11,3 |
Ponovljivost r (95 %) |
0,26 |
|
ISPRA |
UE* |
11,0 |
11,0 |
11,0 |
Relativna ponovljivost r % |
2,24 |
|
D.V.F.A. |
DK |
13,3 |
11,8 |
12,6 |
Standardna devijacija obnovljivosti (SR) |
0,23 |
|
|
|
|
|
|
Relativna standardna devijacija obnovljivosti (RSDR%) |
2,04 |
|
|
|
|
|
|
Obnovljivost R (95 %) |
0,84 |
|
|
|
|
|
|
Relativna obnovljivost R % |
5,71 |
|
Uzorak B |
|
R1 |
R2 |
Srednja vrijednost |
Br. laboratorija zadržanih nakon eliminacije ekstremnih rezultata |
8 |
|
RENNES |
FR1 |
12,7 |
12,8 |
12,8 |
Br. ekstremnih rezultata |
1 |
|
RIKILT |
NL |
13,5 |
13,3 |
13,4 |
Ekstremni rezultati |
DK |
|
ZPLA |
DE* |
14,0 |
13,8 |
13,9 |
Srednja vrijednost |
13,4 |
|
ADAS |
GB |
13,4 |
13,5 |
13,5 |
Stvarna vrijednost |
13,5 |
|
CNEVA |
FR2 |
13,3 |
13,4 |
13,4 |
Standardna devijacija ponovljivosti (Sr) |
0,14 |
|
LODI |
IT |
13,9 |
13,5 |
13,7 |
Relativna standardna devijacija ponovljivosti (RSDr%) |
1,04 |
|
EELA |
FI |
13,4 |
13,2 |
13,3 |
Ponovljivost r (95 %) |
0,40 |
|
ISPRA |
UE* |
13,2 |
13,3 |
13,3 |
Relativna ponovljivost r % |
2,91 |
|
D.V.F.A. |
DK |
14,1 |
14,8 |
14,5 |
Standardna devijacija obnovljivosti (SR) |
0,35 |
|
|
|
|
|
|
Relativna standardna devijacija obnovljivosti (RSDR%) |
2,61 |
|
|
|
|
|
|
Obnovljivost R (95 %) |
0,99 |
|
|
|
|
|
|
Relativna obnovljivost R % |
7,31 |
Tablica 2.
Statistički rezultati metoda TG + FAME*
|
Uzorak C |
|
R1 |
R2 |
Srednja vrijednost |
Br. laboratorija zadržanih nakon eliminacije ekstremnih rezultata |
8 |
|
RENNES |
FR1 |
8,9 |
9,2 |
9,1 |
Br. ekstremnih rezultata |
1 |
|
RIKILT |
NL |
9,2 |
9,3 |
9,3 |
Ekstremni rezultati |
DK |
|
ZPLA |
DE* |
9,2 |
9,4 |
9,3 |
Srednja vrijednost |
9,3 |
|
ADAS |
GB |
9,5 |
9,3 |
9,4 |
Stvarna vrijednost |
9,3 |
|
CNEVA |
FR2 |
9,4 |
9,4 |
9,4 |
Standardna devijacija ponovljivosti (Sr) |
0,14 |
|
LODI |
IT |
9,2 |
9,5 |
9,4 |
Relativna standardna devijacija ponovljivosti (RSDr%) |
1,50 |
|
EELA |
FI |
9,4 |
9,6 |
9,5 |
Ponovljivost r (95 %) |
0,40 |
|
ISPRA |
UE* |
9,4 |
9,3 |
9,4 |
Relativna ponovljivost r % |
4,20 |
|
D.V.F.A. |
DK |
10,7 |
10,9 |
10,8 |
Standardna devijacija obnovljivosti (SR) |
0,17 |
|
|
|
|
|
|
Relativna standardna devijacija obnovljivosti (RSDR%) |
1,82 |
|
|
|
|
|
|
Obnovljivost R (95 %) |
0,47 |
|
|
|
|
|
|
Relativna obnovljivost R % |
5,10 |
|
Uzorak D |
|
R1 |
R2 |
Srednja vrijednost |
Br. laboratorija zadržanih nakon eliminacije ekstremnih rezultata |
8 |
|
RENNES |
FR1 |
1,6 |
1,6 |
1,6 |
Br. ekstremnih rezultata |
1 |
|
RIKILT |
NL |
2,1 |
2,1 |
2,1 |
Ekstremni rezultati |
DK |
|
ZPLA |
DE* |
2,3 |
2,3 |
2,3 |
Srednja vrijednost |
2,1 |
|
ADAS |
GB |
2,1 |
2,2 |
2,2 |
Stvarna vrijednost |
2,1 |
|
CNEVA |
FR2 |
2,1 |
2,1 |
2,1 |
Standardna devijacija ponovljivosti (Sr) |
0,08 |
|
LODI |
IT |
2,2 |
1,9 |
2,1 |
Relativna standardna devijacija ponovljivosti (RSDr%) |
3,81 |
|
EELA |
FI |
2,3 |
2,3 |
2,3 |
Ponovljivost r (95 %) |
0,22 |
|
ISPRA |
UE* |
2,3 |
2,3 |
2,3 |
Relativna ponovljivost r % |
10,67 |
|
D.V.F.A. |
DK |
3,4 |
2,9 |
3,2 |
Standardna devijacija obnovljivosti (SR) |
0,24 |
|
|
|
|
|
|
Relativna standardna devijacija obnovljivosti (RSDR%) |
11,43 |
|
|
|
|
|
|
Obnovljivost R (95 %) |
0,67 |
|
|
|
|
|
|
Relativna obnovljivost R % |
32,00 |
Tablica 3.
Statistički rezultati metoda TG + FAME*
|
Uzorak A |
|
R1 |
R2 |
Srednja vrijednost |
Br. laboratorija zadržanih nakon eliminacije ekstremnih rezultata |
6 |
|
RENNES |
FR1 |
11,0 |
11,1 |
11,1 |
Br. ekstremnih rezultata |
1 |
|
RIKILT |
NL |
11,2 |
11,2 |
11,2 |
Ekstremni rezultati |
DK |
|
ADAS |
GB |
11,4 |
11,2 |
11,3 |
Srednja vrijednost |
11,2 |
|
CNEVA |
FR2 |
11,4 |
11,4 |
11,4 |
Stvarna vrijednost |
11,0 |
|
LODI |
IT |
11,1 |
11,3 |
11,2 |
Standardna devijacija ponovljivosti (Sr) |
0,09 |
|
EELA |
FI |
11,3 |
11,2 |
11,3 |
Relativna standardna devijacija ponovljivosti (RSDr%) |
0,80 |
|
D.V.F.A. |
DK |
13,3 |
11,8 |
12,6 |
Ponovljivost r (95 %) |
0,25 |
|
|
|
|
|
|
Relativna ponovljivost r % |
2,24 |
|
|
|
|
|
|
Standardna devijacija obnovljivosti (SR) |
0,13 |
|
|
|
|
|
|
Relativna standardna devijacija obnovljivosti (RSDR%) |
1,16 |
|
|
|
|
|
|
Ponovljivost R (95 %) |
0,36 |
|
|
|
|
|
|
Relativna obnovljivost R % |
3,25 |
|
Uzorak B |
|
R1 |
R2 |
Srednja vrijednost |
Br. laboratorija zadržanih nakon eliminacije ekstremnih rezultata |
6 |
|
RENNES |
FR1 |
12,7 |
12,8 |
12,8 |
Br. ekstremnih rezultata |
1 |
|
RIKILT |
NL |
13,5 |
13,3 |
13,4 |
Ekstremni rezultati |
DK |
|
ADAS |
GB |
13,4 |
13,5 |
13,5 |
Srednja vrijednost |
13,3 |
|
CNEVA |
FR2 |
13,3 |
13,4 |
13,4 |
Stvarna vrijednost |
13,5 |
|
LODI |
IT |
13,9 |
13,5 |
13,7 |
Standardna devijacija ponovljivosti (Sr) |
0,15 |
|
EELA |
FI |
13,4 |
13,2 |
13,3 |
Relativna standardna devijacija ponovljivosti (RSDr%) |
1,13 |
|
D.V.F.A. |
DK |
14,1 |
14,8 |
14,5 |
Ponovljivost r (95 %) |
0,42 |
|
|
|
|
|
|
Relativna ponovljivost r % |
3,16 |
|
|
|
|
|
|
Standardna devijacija obnovljivosti (SR) |
0,33 |
|
|
|
|
|
|
Relativna standardna devijacija obnovljivosti (RSDR%) |
2,48 |
|
|
|
|
|
|
Ponovljivost R (95 %) |
0,93 |
|
|
|
|
|
|
Relativna obnovljivost R % |
6,94 |
Tablica 4.
Statistički rezultati metode TG
|
Uzorak C |
|
R1 |
R2 |
Srednja vrijednost |
Br. laboratorija zadržanih nakon eliminacije ekstremnih rezultata |
6 |
|
RENNES |
FR1 |
8,9 |
9,2 |
9,1 |
Br. ekstremnih rezultata |
1 |
|
RIKILT |
NL |
9,2 |
9,3 |
9,3 |
Ekstremni rezultati |
DK |
|
ADAS |
GB |
9,5 |
9,3 |
9,4 |
Srednja vrijednost |
9,3 |
|
CNEVA |
FR2 |
9,4 |
9,4 |
9,4 |
Stvarna vrijednost |
9,3 |
|
LODI |
IT |
9,2 |
9,5 |
9,4 |
Standardna devijacija ponovljivosti (Sr) |
0,15 |
|
EELA |
FI |
9,4 |
9,6 |
9,5 |
Relativna standardna devijacija ponovljivosti (RSDr%) |
1,61 |
|
D.V.F.A. |
DK |
10,7 |
10,9 |
10,8 |
Ponovljivost r (95 %) |
0,42 |
|
|
|
|
|
|
Relativna ponovljivost r % |
4,51 |
|
|
|
|
|
|
Standardna devijacija obnovljivosti (SR) |
0,19 |
|
|
|
|
|
|
Relativna standardna devijacija obnovljivosti (RSDR%) |
2,04 |
|
|
|
|
|
|
Obnovljivost R (95 %) |
0,53 |
|
|
|
|
|
|
Relativna obnovljivost R % |
5,71 |
|
Uzorak D |
|
R1 |
R2 |
Srednja vrijednost |
Br. laboratorija zadržanih nakon eliminacije ekstremnih rezultata |
6 |
|
RENNES |
FR1 |
1,6 |
1,6 |
1,6 |
Br. ekstremnih rezultata |
1 |
|
RIKILT |
NL |
2,1 |
2,1 |
2,1 |
Ekstremni rezultati |
DK |
|
|
|
|
|
|
Srednja vrijednost |
2,1 |
|
ADAS |
GB |
2,1 |
2,2 |
2,2 |
Stvarna vrijednost |
2,1 |
|
CNEVA |
FR2 |
2,1 |
2,1 |
2,1 |
Standardna devijacija ponovljivosti (Sr) |
0,09 |
|
LODI |
IT |
2,2 |
1,9 |
2,1 |
Relativna standardna devijacija ponovljivosti (RSDr%) |
4,29 |
|
EELA |
FI |
2,3 |
2,3 |
2,3 |
Ponovljivost r (95 %) |
0,26 |
|
D.V.F.A. |
DK |
3,4 |
2,9 |
3,2 |
Relativna ponovljivost r % |
12,01 |
|
|
|
|
|
|
Standardna devijacija obnovljivosti (SR) |
0,25 |
|
|
|
|
|
|
Relativna standardna devijacija obnovljivosti (RSDR%) |
11,90 |
|
|
|
|
|
|
Obnovljivost R (95 %) |
0,69 |
|
|
|
|
|
|
Relativna obnovljivost R % |
33,32 |
Tablica 5.
Ponovljivost i obnovljivost (metodom FAME)
|
|
Br. laboratorija |
Ekstremni rezultati |
Ponovljivost Sr (95 %) |
Obnovljivost SR (95 %) |
||
|
Uzorak A |
8 |
1 |
0,09 |
0,23 |
||
|
Uzorak B |
8 |
1 |
0,14 |
0,35 |
||
|
Uzorak C |
8 |
1 |
0,14 |
0,17 |
||
|
Uzorak D |
8 |
1 |
0,08 |
0,24 |
||
|
Objedinjena vrijednost |
|
|
0,116 |
0,256 |
||
|
|
|
|
r |
R |
||
|
Objedinjena vrijednost * 2,8 |
|
|
0,324 |
0,716 |
||
|
CrD95 = 0,40 Minimalna čistoća utvrđena za trienantoat = 95 % Minimalna granična vrijednost utvrđena za trienantoat u mliječnoj masti = 11 kg/t Uzimajući u obzir kritičnu razliku za stupanj vjerojatnosti od 95 %, srednja vrijednost dvaju rezultata nije manja od:
|
||||||
Ponovljivost i obnovljivost (bez metode FAME)
|
|
Br. laboratorija |
Ekstremni rezultati |
Ponovljivost Sr (95 %) |
Obnovljivost SR (95 %) |
||
|
Uzorak A |
6 |
1 |
0,09 |
0,13 |
||
|
Uzorak B |
6 |
1 |
0,15 |
0,33 |
||
|
Uzorak C |
6 |
1 |
0,15 |
0,19 |
||
|
Uzorak D |
6 |
1 |
0,09 |
0,25 |
||
|
Objedinjena vrijednost |
|
|
0,124 |
0,237 |
||
|
|
|
|
r |
R |
||
|
Objedinjena vrijednost * 2,8 |
|
|
0,347 |
0,663 |
||
|
CrD95 = 0,36 Minimalna čistoća utvrđena za trienantoat = 95 % Minimalna granična vrijednost utvrđena za trienantoat u mliječnoj masti = 11 kg/t Uzimajući u obzir kritičnu razliku za stupanj vjerojatnosti od 95 %, srednja vrijednost dvaju rezultata nije manja od:
|
||||||
Slika 1. (1)
Rezultati ispitivanja: uzorak A
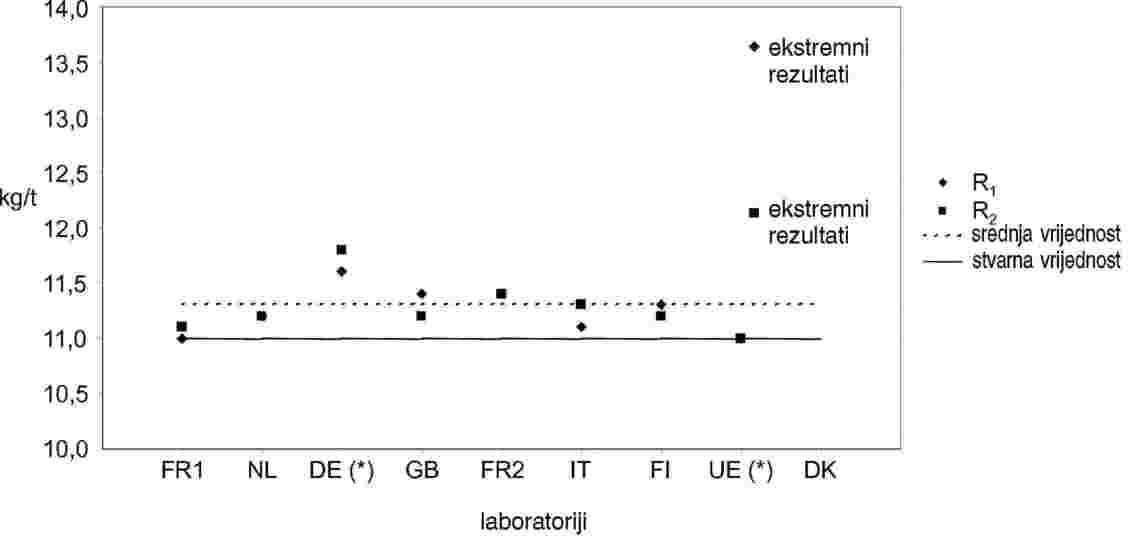
Rezultati ispitivanja: uzorak B
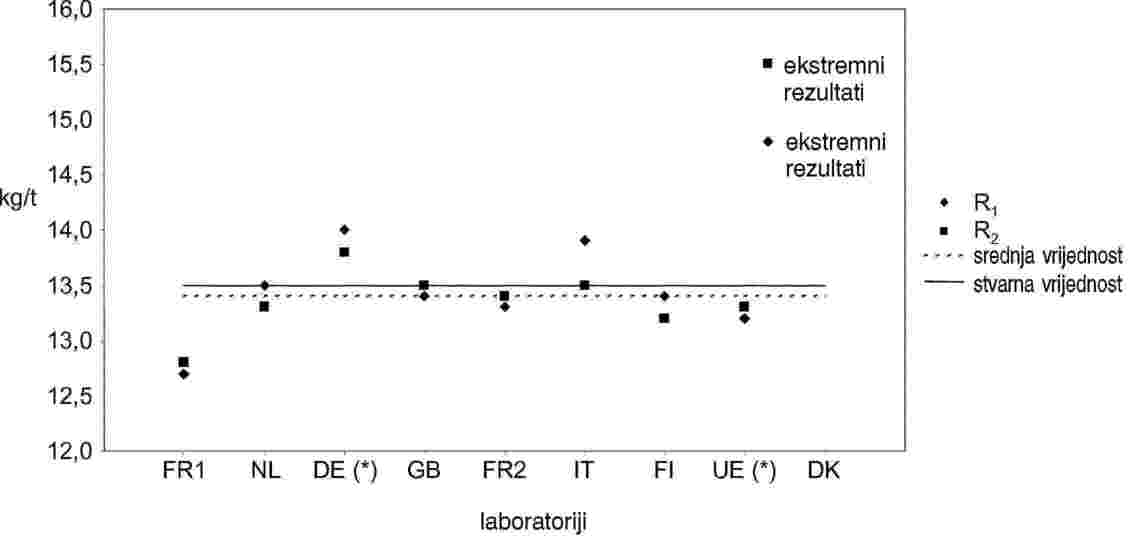
Rezultati ispitivanja: uzorak C

Rezultati ispitivanja: uzorak D

Slika 2.
Standardna devijacija ponovljivosti i obnovljivosti na različitim razinama (TG + FAME)
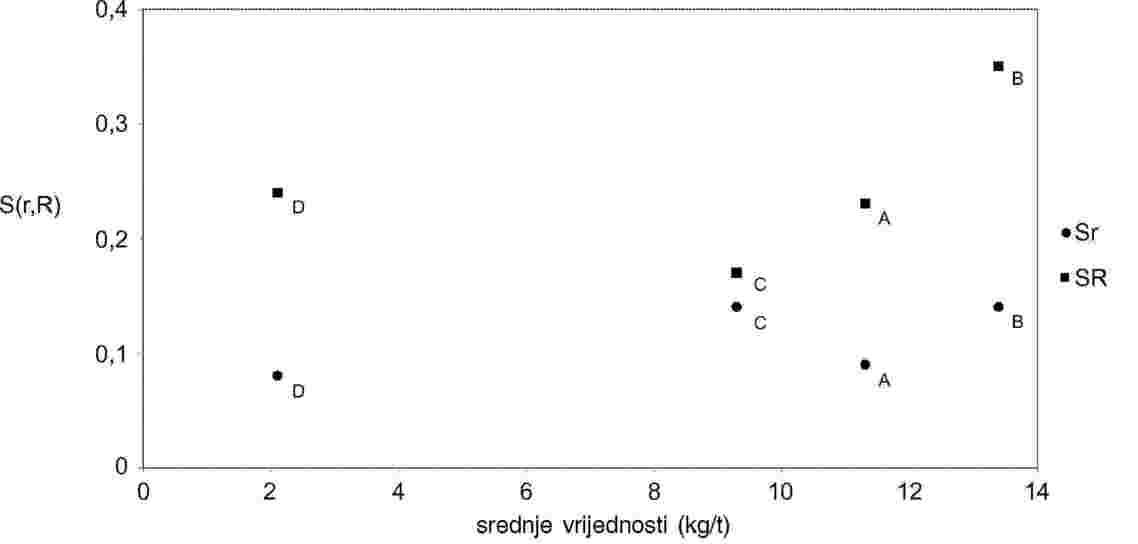
Slika 3.
Standardna devijacija ponovljivosti i obnovljivosti na različitim razinama (TG)

Slika 4.
Primjer primjene on-column injektora
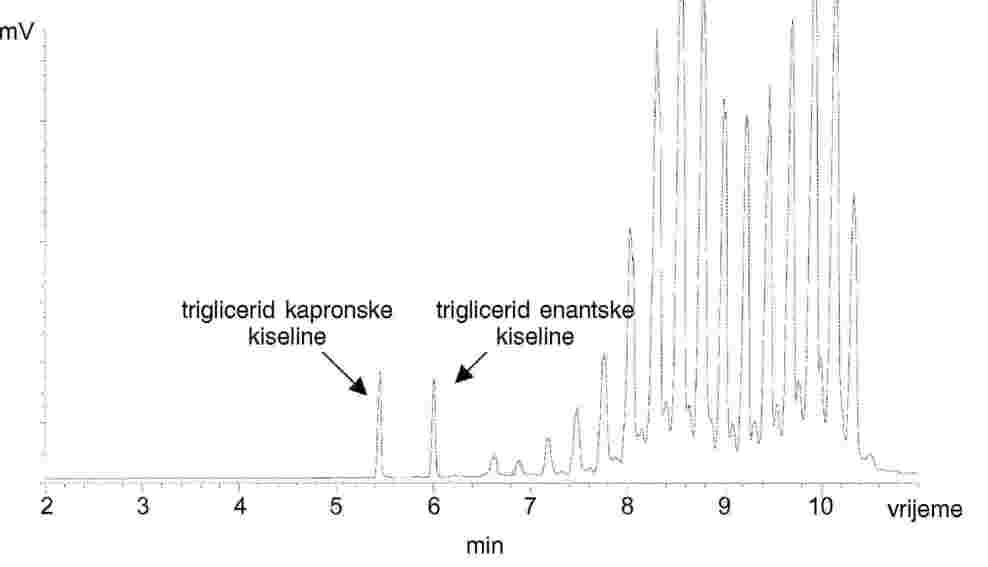
(1) = metoda FAME
PRILOG VI.
(Članak 5.)
ODREĐIVANJE SADRŽAJA VANILINA U KONCENTRIRANOM MASLACU, MASLACU ILI VRHNJU METODOM TEKUĆINSKE KROMATOGRAFIJE VISOKE DJELOTVORNOSTI
1. OPSEG I PODRUČJE PRIMJENE
U metodi je opisan postupak za kvantitativno određivanje vanilina u koncentriranom maslacu, maslacu ili vrhnju.
2. NAČELO
Ekstrakcija znane količine uzorka mješavinom izopropanola/etanola/acetonitrila (1:1:2). Precipitacija većine masti rashlađivanjem između –15 °C i –20 °C, iza čega slijedi centrifugiranje.
Nakon razrjeđivanja vodom, određivanje sadržaja vanilina metodom tekućinske kromatografije visoke djelotvornosti (HPLC).
3. OPREMA
Uobičajena laboratorijska oprema, a posebno sljedeća:
3.1. Zamrzivač koji radi unutar temperaturnog raspona od –15 °C do –20 °C.
3.2. Šprice za jednokratu uporabu, zapremine 2 ml.
3.3. Membranski mikrofiltri s porama od 0,45 μm, otporni na otopinu koja sadrži 5 % ekstrakcijske otopine (4.4.).
3.4. Sustav tekućinske kromatografije koji se sastoji od pumpe (protok 1,0 ml/min), injektora (kapacitet ubrizganja 20 μl, automatski ili ručni), UV detektora (koji radi pri 306 nm, 0,01 Å punog opsega), mjerača ili integratora i grijača kolone koji radi na 25 °C.
3.5. Analitička kolona (250 mm × 4,6 mm ID) punjena LiChrospherom RP 18 (Merck, 5 μm) ili ekvivalentnim punilom.
3.6. Predkolona (oko 20 mm × 3 mm ID) suho punjena LiChrospherom RP 18 (5 do 10 μm) ili ekvivalentnim punilom.
3.7. Centrifuga brzine 2 000 o/min.
4. REAGENSI
Svi reagensi koji se koriste moraju biti priznate analitičke čistoće.
4.1. Izopropanol
4.2. Etanol 96 % (v/v)
4.3. Acetonitril
4.4. Ekstrakcijska otopina
Izmiješajte izopropanol (4.1.), etanol (4.2.) i acetonitril (4.3.) u omjeru 1:1:2 (v/v).
Vanilin (4-hidroksi-3-metoksibenzaldehid) ≥ 98 %
4.5.1. Osnovna otopina vanilina (= 500 μg/ml)
U odmjernu tikvicu od 100 ml odvagnite oko 50 mg (CM mg) vanilina (4.5.) s točnošću od 0,1 mg, dodajte 25 ml ekstrakcijske otopine (4.4.) i dopunite vodom.
4.5.2. Standardna otopina vanilina (= 10 μg/ml)
U odmjernu tikvicu od 250 ml otpipetirajte 5 ml osnovne otopine vanilina (4.5.1.) i dopunite vodom.
4.5.3. Metanol, HPLC čistoće
4.5.4. Ledena octena kiselina
4.5.5. Voda, HPLC čistoće
4.5.6. Mobilna faza HPLC-a
U odmjernoj tikvici od 1 000 ml pomiješajte 300 ml metanola (4.5.3.) s približno 500 ml vode (4.5.5.) i 20 ml octene kiseline (4.5.4.) te dopunite vodom (4.5.5.). Mješavinu profiltrirajte kroz filtar od 0,45 μm (3.3.).
5. POSTUPAK
5.1. Priprema ispitnog uzorka
5.1.1. Maslac
Zagrijavajte uzorak dok se ne počne topiti. Izbjegavajte mjestimično pregrijavanje iznad 30 °C. Ni u kojem slučaju maslac se ne smije razdvojiti u dvije faze. Kad uzorak postane dovoljno plastičan, homogenizirajte ga mućkanjem. Prije nego što uzmete uzorak, maslac miješajte 15 s. U odmjernu tikvicu od 100 ml odvagnite oko 5 g (SM g) maslaca s točnošću od 1 mg.
5.1.2. Koncentrirani maslac
Neposredno prije uzorkovanja posudu s koncentriranim maslacem stavite u sušionik zagrijan na 40 do 50 °C dok se maslac potpuno ne rastopi. Izmiješajte maslac vrtloženjem ili miješanjem, izbjegavajući stvaranje zračnih mjehurića zbog prejakog miješanja. U odmjernu tikvicu od 100 ml odvagnite oko 4 g (SM g) koncentriranog maslaca s točnošću od 1 mg.
5.1.3. Vrhnje
Uzorak zagrijte u vodenoj kupelji ili inkubatoru na temperaturu 35 do 40 °C. Mast jednakomjerno raspršite vrtloženjem ili, prema potrebi, miješanjem. Uzorak brzo ohladite na 20 ± 2 °C. Uzorak treba izgledati homogeno; u protivnom postupak treba ponoviti. U odmjernu tikvicu od 100 ml odvagnite oko 10 g (SM g) vrhnja s točnošću od 1 mg.
5.2. Priprema ispitne otopine
U uzorak za analizu (5.1.1., 5.1.2. ili 5.1.3.) dodajte oko 75 ml ekstrakcijske otopine (4.4.), miješajte ili energično mućkajte 15 minuta i dopunite ekstrakcijskom otopinom. (4.4.). Pretočite oko 10 ml ovog ekstrakta u epruvetu s čepom. Stavite epruvetu u zamrzivač (3.1.) i ostavite oko 30 minuta. Ohlađeni ekstrakt centrifugirajte 5 minuta na oko 2 000 o/min i odmah dekantirajte. Ostavite dekantiranu otopinu da se prilagodi sobnoj temperaturi. Otpipetirajte 5 ml dekantirane otopine u odmjernu tikvicu od 100 ml i dopunite vodom. Profiltrirajte alikvot kroz membranski mikrofiltar (3.3.) koristeći špricu (3.2.). Filtrat je spreman za HPLC analizu.
5.3. Kalibracija
U odmjernu tikvicu od 100 ml otpipetirajte 5 ml standardne otopine (4.5.2.). Dodajte 5 ml ekstrakcijske otopine (4.4.) i do oznake dopunite vodom. Ova otopina sadrži 0,5 μg/ml vanilina.
5.4. Određivanje HPLC metodom
Ostavite kromatografski sustav oko 30 minuta da se stabilizira. Ubrizgajte standardnu otopinu (5.3.). Ponavljajte ovaj postupak dok razlika u površini ili visini pika između dva uzastopna ubrizgavanja ne bude iznosila manje od 2 %. Vrijeme zadržavanja vanilina u opisanim uvjetima iznosi oko 9 minuta. Analizirajte standardnu otopinu (5.3.) u duplikatu, ubrizgavanjem 20 μl. Ubrizgajte 20 μl ispitne otopine (5.2.). Odredite površinu ili visinu dobivenog pika vanilina. Nakon 10 ubrizgavanja ispitnog uzorka (5.2.) ponovite dvostruko ubrizgavanje standardne otopine (5.3.).
6. IZRAČUNAVANJE REZULTATA
Izračunajte prosječnu vrijednost površine (ili visine) (AC) pikova vanilina u vezi s dvostrukim ubrizgavanjem prije i nakon svake serije ispitnih otopina (ukupno četiri površine ili visine).
Izračunajte faktor odziva (R):
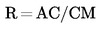
gdje je CM masa vanilina u mg (4.5.1.).
Sadržaj (mg/kg) vanilina (C) u ispitnom uzorku izračunava se prema formuli:
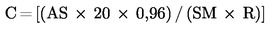
gdje je:
|
AS |
= |
površina ili visina pika vanilina u ispitnom uzorku; |
|
SM |
= |
masa ispitnog uzorka u g (5.1.1., 5.1.2. ili 5.1.3.); |
Napomena: Ako se u vrhnju analizira sadržaj vanilina, koncentracija markera mora biti izražena u mg markera/kg mliječne masti. To se radi tako da se C pomnoži sa 100/f. f je sadržaj masti u vrhnju izražen u masenom postotku (m/m);
|
20 |
= |
faktor koji uzima u obzir razrjeđenja standardnog i ispitnog uzorka; |
|
0,96 |
= |
korektivni faktor za sadržaj masti u prvom razrjeđenju ispitnog uzorka; |
Napomena: Umjesto površine pika mogu se koristiti visine pika (vidjeti 8.3.).
7. PRECIZNOST METODE
7.1. Ponovljivost (r)
Razlika između rezultata dvaju određivanja koja je u najkraćem mogućem vremenskom intervalu izvršio isti analitičar, koristeći istu opremu na jednakom ispitnom materijalu, ne smije biti veća od 16 mg/kg.
7.2. Obnovljivost (R)
Razlika između rezultata dvaju određivanja koja su izvršili analitičari u različitim laboratorijima, koristeći različitu opremu na jednakom ispitnom materijalu, ne smije biti veća od 27 mg/kg.
8. GRANICE DOPUŠTENOG ODSTUPANJA
8.1. Od uzorka proizvoda s dodanim markerom treba uzeti tri uzorka radi provjere homogenosti.
8.2. Marker dobiven iz vanilije ili sintetičkog vanilina:
8.2.1. Stopa inkorporacije za 4-hidroksi-3-metoksibenzaldehid iznosi 250 g po toni koncentriranog maslaca ili maslaca. Ako se dodaje vrhnju, stopa inkorporacije iznosi 250 g po toni mliječne masti.
8.2.2. Rezultati analize proizvoda dobiveni na tri uzorka koriste se za provjeru stope i homogenosti inkorporacije markera, a najniži od tih rezultata uspoređuje se sa sljedećim graničnim vrijednostima:
|
— |
220,8 mg/kg (95 % minimalne stope inkorporacije), |
|
— |
158,3 mg/kg (70 % minimalne stope inkorporacije). |
Koncentracija markera u uzorku koji je dao najniži rezultat koristi se u vezi s interpolacijom između 220,8 mg/kg i 158,3 mg/kg.
8.3. Marker dobiven isključivo iz zrna vanilije ili njihovih integralnih ekstrakata:
8.3.1. Stopa inkorporacije 4-hidroksi-3-metoksibenzaldehida iznosi 100 g po toni koncentriranog maslaca ili maslaca. Ako se dodaje vrhnju, stopa inkorporacije iznosi 100 g po toni mliječne masti.
8.3.2. Rezultati analize proizvoda dobiveni na tri uzorka koriste se za provjeru stope i homogenosti inkorporacije markera, a najniži od tih rezultata uspoređuje se sa sljedećim graničnim vrijednostima:
|
— |
78,3 mg/kg (95 % minimalne stope inkorporacije), |
|
— |
53,3 mg/kg (70 % minimalne stope inkorporacije). |
Koncentracija markera u uzorku koji je dao najniži rezultat koristi se u vezi s interpolacijom između 78,3 mg/kg i 53,3 mg/kg.
9. NAPOMENE
9.1. Obnavljanje dodanog vanilina na razini od 250 mg/kg masla varira od 97,0 do 103,8. Prosječni utvrđeni sadržaj iznosi 99,9 % uz standardnu devijaciju od 2,7 %.
9.2. Standardna otopina sadrži 5 % ekstrakcijske otopine koja kompenzira širenje pika uzrokovanog sadržajem od 5 % ekstrakcijske otopine u ispitnim uzorcima. Ova pojava omogućuje kvantifikaciju prema visini pika.
9.3. Analiza se temelji na linearnoj kalibracijskoj krivulji sa sjecištem u nuli.
9.4. Linearnost treba provjeriti pomoću odgovarajućih razrjeđenja standardnih otopina (4.5.2.) kad se analiza radi prvi put, a potom u redovitim vremenskim razmacima, kao i nakon izmjena ili popravaka HPLC opreme. Vanilin se može razgraditi na vanilinsku kiselinu, divanilin i druge spojeve djelovanjem intrinzičnih enzima u nepasteriziranom vrhnju ili proizvodima od njega.
PRILOG VII.
(Članak 5.)
ODREĐIVANJE ETIL ESTERA BETA-APO-8’-KAROTENSKE KISELINE U KONCENTRIRANOM MASLACU I MASLACU METODOM SPEKTROMETRIJE
1. OPSEG I PODRUČJE PRIMJENE
Ova metoda opisuje postupak za količinsko određivanje etil-estera beta-apo-8’-karotenske kiseline (apo-karotenskog estera) u koncentriranom maslacu i maslacu. Apo-karotenski ester je zbroj svih tvari prisutnih u ekstraktu uzoraka uzetih u skladu s uvjetima opisanim u metodi, koji apsorbiraju svjetlost valne duljine 440 nm.
2. NAČELO
Mliječna se mast otapa u eteru i apsorbancija se mjeri na 440 nm. Sadržaj apo-karotenskog estera utvrđuje se na temelju vanjskih standarda.
3. OPREMA
3.1. Pipete — graduirane, zapremine 0,25, 0,50, 0,75 i 1,0 ml.
3.2. Spektrofotometar — prikladan za uporabu na 440 nm (i između 447 i 449 nm), s kivetom optičkog puta duljine 1 cm.
3.3. Odmjerne tikvice, 20 ml i 100 ml.
3.4. Analitička vaga osjetljivosti 0,1 mg, koja može vagati s točnošću od 1 mg, uz mogućnost očitanja od 0,1 mg.
3.5. Sušionik, 45 °C ± 1 °C.
3.6. Filtri za brzo filtriranje bez pepela.
4. REAGENSI
Svi reagensi moraju biti priznate analitičke čistoće.
Suspenzija apo-karotenskog estera (oko 20 %)
4.1.1. Utvrdite sadržaj suspenzije na sljedeći način:
Zagrijte suspenziju na temperaturu između 45 °C i 50 °C i homogenizirajte u neotvorenoj originalnoj posudi. Odvagnite oko 400 mg u odmjernu tikvicu (100 ml), otopite u 20 ml kloroforma (4.4.) i do potrebne količine nadopunite cikloheksanom (4.5.). 5 ml ove otopine razrijedite do količine 100 ml cikloheksanom (otopina A). Razrijedite 5 ml otopine A do količine 100 ml ciklohekasnom. Izmjerite apsorbanciju na 447-449 nm (izmjerite maksimalnu vrijednost u odnosu na cikloheksan koji se koristi kao slijepa proba, pomoću kivete optičkog puta duljine 1 cm).
Sadržaj apo-karotenskog estera P (%) = (Absmaks × 40 000) / (Msusp × 2 550) ili: (Absmaks / 2 550) × (100 / 5) × (100 / 5) × (100 / Msusp)
|
Absmaks |
= |
maksimalna apsorbancija mjerene otopine |
|
Msusp |
= |
masa suspenzije (g) |
|
2 550 |
= |
referentna vrijednost Abs (1 %, 1 cm) |
|
P |
= |
čistoća (sadržaj) suspenzije (%) |
Napomena: Suspenzija apo-karotenskog estera osjetljiva je na zrak, toplinu i svjetlo. U neotvorenoj, originalnoj posudi (zapečaćenoj pod dušikom) i na hladnom mjestu može se čuvati oko 12 mjeseci. Nakon otvaranja sadržaj treba uporabiti u kratkom roku.
4.1.2. Standardna otopina apo-karotenskog estera, oko 0,2 mg/ml
Odvagnite oko 0,100 g suspenzije apo-karotenskog estera (4.1.1.) (W) s točnošću od 0,1 mg, otopite u petroleteru (4.2.), kvantitativno prenesite u odmjernu tikvicu zapremine 100 ml i do oznake nadopunite petroleterom.
Ova otopina sadrži (W × P) / 10 mg/ml apo-karotenskog estera.
Napomena: Otopina se čuva na tamnom i hladnom mjestu. Neuporabljenu otopinu bacite nakon mjesec dana.
4.2. Petroleter (40-60 °C)
4.3. Natrijev sulfat, bezvodni, zrnat, prethodno sušen dva sata na temperaturi od 102 °C
4.4. Kloroform
4.5. Cikloheksan
5. POSTUPAK
5.1. Priprema ispitnog uzorka
5.1.1. Koncentrirani maslac
Otopite uzorak u sušioniku na oko 45 °C.
5.1.2. Maslac
Otopite uzorak u sušioniku na oko 45 °C i filtrirajte reprezentativni dio pomoću filtra koji sadrži oko 10 g bezvodnog natrijevog sulfata (4.3.) u ambijentu zaštićenom od jakog prirodnog i umjetnog svjetla, na temperaturi od 45 °C. Sakupite odgovarajuću količinu mliječne masti.
5.2. Određivanje
Odvagnite oko 1 g koncentriranog maslaca (ili ekstrahirane mliječne masti (5.1.2.)) (M) s točnošću od 1 mg. Kvantitativno prenesite u odmjernu tikvicu od 20 ml (V) pomoću petroletera (4.2.), dopunite do oznake i dobro promiješajte.
Prenesite alikvot u kivetu od 1 cm i izmjerite apsorbanciju na 440 nm u odnosu na petroleter koji se koristi kao slijepa proba. Odredite koncentraciju apo-karotenskog estera u otopini prema dobivenoj standardnoj krivulji (C μ/ml).
5.3. Kalibracija
Otpipetirajte 0, 0,25, 0,5, 0,75 i 1,0 ml standardne otopine apo-karotenskog estera (4.1.2.) u 5 odmjernih tikvica od po 100 ml. Razrijedite do potrebne količine petroleterom (4.2.) i promiješajte.
Približne koncentracije otopina iznose od 0 do 2 μg/ml i precizno se računaju prema koncentraciji standardne otopine (4.1.2.) (W × P) / 10 mg/ml. Izmjerite apsorbancije na 440 nm u odnosu na petroleter koji se koristi kao slijepa proba. (4.2.).
Ucrtajte vrijednosti apsorbancije na os y, a koncentraciju apo-karotenskog estera na os x. Izračunajte jednadžbu standardne krivulje.
6. IZRAČUNAVANJE REZULTATA
6.1. Sadržaj apo-karotenskog estera, izražen u mg/kg proizvoda, računa se u:
koncentriranom maslacu: (C × V)/M
maslacu: 0,82 (C × V)/M
gdje je:
|
C |
= |
sadržaj apo-karotenskog estera, izražen u μg/ml, očitan na kalibracijskom dijagramu (5.3.); |
|
V |
= |
volumen (ml) ispitne otopine (5.2.); |
|
M |
= |
masa (g) uzorka za analizu (5.2.); |
|
0,82 |
= |
korektivni faktor za sadržaj mliječne masti u maslacu. |
7. PRECIZNOST METODE
7.1. Ponovljivost
7.1.1. Analiza maslaca
Razlika između rezultata dvaju određivanja koja je u najkraćem mogućem vremenskom intervalu izvršio isti analitičar, koristeći istu opremu na jednakom ispitnom materijalu, ne smije biti veća od 1,4 mg/kg.
7.1.2. Analiza koncentriranog maslaca
Razlika između rezultata dvaju određivanja koja je u najkraćem mogućem vremenskom intervalu izvršio isti analitičar, koristeći istu opremu na jednakom ispitnom materijalu, ne smije biti veća od 1,6 mg/kg.
7.2. Obnovljivost
7.2.1. Analiza maslaca
Razlika između rezultata dvaju određivanja koja su izvršili djelatnici u različitim laboratorijima, koristeći različitu opremu na jednakom ispitnom materijalu, ne smije biti veća od 4,7 mg/kg.
7.3. Analiza koncentriranog maslaca
Razlika između rezultata dvaju određivanja koja su izvršili djelatnici u različitim laboratorijima, koristeći različitu opremu na jednakom ispitnom materijalu, ne smije biti veća od 5,3 mg/kg.
7.4. Izvor podataka o preciznosti
Podaci o preciznosti dobiveni su pokusom izvedenim 1995., kojim je bilo obuhvaćeno 11 laboratorija i 12 uzoraka s dodanim markerima (šest slijepih proba u duplikatu) za maslac i 12 uzoraka s dodanim markerima (šest slijepih proba u duplikatu) za koncentrirani maslac.
8. GRANICE DOPUŠTENOG ODSTUPANJA
8.1. Od proizvoda s dodanim markerom obvezno treba uzeti po tri uzorka radi provjere pravilnosti dodavanja.
8.2. Maslac
8.2.1. Stopa inkorporacije za maslac, uzimajući u obzir apsorbanciju podloge, iznosi 22 mg/kg.
8.2.2. Rezultati analize proizvoda dobiveni na tri uzorka koriste se za provjeru stope i homogenosti inkorporacije markera, a najniži od tih rezultata uspoređuje se sa sljedećim graničnim vrijednostima:
|
— |
17,7 mg/kg (95 % minimalne stope inkorporacije), |
|
— |
12,2 mg/kg (70 % minimalne stope inkorporacije). |
Koncentracija markera u uzorku s najnižim rezultatom koristi se u vezi s interpolacijom između 17,7 mg/kg i 12,2 mg/kg.
8.3. Koncentrirani maslac
8.3.1. Stopa inkorporacije za koncentrirani maslac, uzimajući u obzir apsorbanciju podloge, iznosi 24 mg/kg.
Rezultati analize proizvoda dobiveni na tri uzorka koriste se za provjeru stope i homogenosti inkorporacije markera, a najniži od tih rezultata uspoređuje se sa sljedećim graničnim vrijednostima:
|
— |
19,2 mg/kg (95 % minimalne stope inkorporacije), |
|
— |
13,2 mg/kg (70 % minimalne stope inkorporacije). |
Koncentracija markera u uzorku s najnižim rezultatom koristi se u vezi s interpolacijom između 19,2 mg/kg i 13,2 mg/kg.
PRILOG VIII.
(Članak 5.)
ODREĐIVANJE SITOSTEROLA ILI STIGMASTEROLA U MASLACU ILI KONCENTRIRANOM MASLACU PUTEM KAPILARNE PLINSKE KROMATOGRAFIJE
1. OPSEG I PODRUČJE PRIMJENE
Ova metoda opisuje postupak za kvantitativno određivanje sitosterola ili stigmasterola u maslacu i koncentriranom maslacu. Sitosterolom se smatra zbroj β-sitosterola i 22-dihidro-β-sitosterola, dok se ostali sitosteroli smatraju nevažnima.
2. NAČELO
Maslac ili koncentrirani maslac saponificira se kalijevim hidroksidom u etanolskoj otopini, a tvari koje se ne mogu saponificirati ekstrahiraju se pomoću dietil etera.
Steroli se pretvaraju u trimetil-silil etere i analiziraju metodom kapilarne plinske kromatografije s obzirom na interni standard/betulin.
3. OPREMA
3.1. Tikvica za saponifikaciju od 150 ml s povratnim kondenzatorom sa spojevima od brušenog stakla.
3.2. Lijevci za odvajanje od 500 ml.
3.3. Tikvice od 250 ml.
3.4. Lijevci za izjednačavanje tlaka od 250 ml ili slično, za sakupljanje otpadnog dietil etera.
3.5. Staklena kolona, 350 mm × 20 mm, sa staklenim čepom.
3.6. Vodena kupelj ili suhi grijač s izolacijskom oblogom.
3.7. Reakcijske bočice od 2 ml.
Plinski kromatograf koji se može koristiti s kapilarnom kolonom, s razdjelnim sustavom koji se sastoji od:
3.8.1. termostatske komore za kolone, koja može održavati željenu temperaturu s točnošću od ±1 °C;
3.8.2. isparivača s podesivom temperaturom;
3.8.3. plameno-ionizacijskog detektora i pretvarača-pojačala;
3.8.4. integratora-mjerača za uporabu s pretvaračem-pojačalom (3.8.3.).
3.9. Kapilarna kolona od taljenog silicijevog dioksida potpuno obložena slojem BP1 ili ekvivalentnog materijala (ili bilo kakva druga kolona najmanje jednake rezolucije) ujednačene debljine od 0,25 μm; kolona mora biti u stanju odvojiti derivate trimetil-silila od lanosterola i sitosterola. Primjerena je kolona duljine 12 m, unutarnjeg promjera 0,2 mm.
3.10. Mikrošprica od 1 μl za plinsku kromatografiju s čvrsto fiksiranom iglom.
4. REAGENSI
Svi reagensi moraju biti priznate analitičke čistoće. Voda koja se koristi mora biti destilirana voda ili voda barem jednake čistoće.
4.1. Etanol, čistoće najmanje 95 %.
4.2. Kalijev hidroksid, 60 %-tna otopina; otopite 600 g kalijevog hidroksida (najmanje 85 %) u vodi i dopunite vodom do jedne litre.
Betulin, čistoće najmanje 99 %.
Otopine betulina u dietil eteru (4.4.).
4.3.1.1. Koncentracija otopine betulina koja se koristi za određivanje sitosterola mora iznositi 1,0 mg/ml.
4.3.1.2. Koncentracija otopine betulina koja se koristi za određivanje stigmasterola mora iznositi 0,4 mg/ml.
4.4. Dietil eter analitičke čistoće (bez peroksida ili ostataka).
4.5. Natrijev sulfat, bezvodni, zrnati, prethodno sušen dva sata na 102 °C.
4.6. Sililacijski reagens, npr. TRI-SIL (može se nabaviti od Pierce Chemical Co, kat. br. 49001) ili ekvivalentni reagens. (Važno: TRI-SIL je zapaljiv, toksičan, korozivan i potencijalno kancerogen. Osoblje laboratorija mora biti upoznato sa sigurnosnim podacima o TRI-SIL-u i poduzeti odgovarajuće mjere predostrožnosti).
4.7. Lanosterol.
Sitosterol poznate čistoće (P) od najmanje 90 %.
Napomena 1.: Čistoća standardnih materijala korištenih za kalibraciju utvrđuje se metodom normalizacije. Pretpostavlja se da su svi steroli prisutni u uzorku vidljivi na kromatogramu, da ukupna površina pikova predstavlja 100 % sastojaka sterola i da je za sve sterole odziv detektora jednak. Linearnost sustava validira se za raspon koncentracija relevantan za analizu.
4.8.1. Standardna otopina sitosterola – pripremite otopinu koja sadrži oko 0,5 mg/ml (W1) sitosterola (4.8.) u dietil eteru (4.4.) uz točnost od 0,001 mg/ml.
Stigmasterol poznate čistoće (P) ne manje od 90 %.
4.9.1. Standardna otopina stigmasterola – pripremite otopinu koja sadrži oko 0,2 mg/ml (W1) stigmasterola (4.9.) u dietil eteru (4.4.) uz točnost od 0,001 mg/ml.
4.10. Mješavina za ispitivanje rezolucije. Pripremite otopinu koja sadrži 0,05 mg/ml lanosterola (4.7.) i 0,5 mg/ml sitosterola (4.8.) u dietil eteru (4.4.).
5. METODA
5.1. Priprema standardnih otopina za kromatografiju
Interna standarnda otopina (4.3.1.) dodaje se odgovarajućoj standardnoj otopini sterola istovremeno kad i saponificiranom uzorku (vidjeti 5.2.2.).
5.1.1. Standardna kromatografska otopina sitosterola: 1 ml standardne otopine sitosterola (4.8.1.) prenesite u svaku od dvije reakcijske bočice (3.7.) i uklonite dietil eter u struji dušika. Dodajte 1 ml interne otopine (4.3.1.1.) i uklonite dietil eter u struji dušika.
5.1.2. Standardna kromatografska otopina stigmasterola: 1 ml standardne otopine stigmasterola (4.9.1.) prenesite u svaku od dvije reakcijske bočice (3.7.) i uklonite dietil eter u struji dušika. Dodajte 1 ml interne standardne otopine (4.3.1.2.) i uklonite dietil eter u struji dušika.
5.2. Priprema tvari koje se ne mogu saponificirati
5.2.1. Otopite uzorak maslaca na temperaturi do 35 °C i dobro promiješajte.
Odvagnite oko 1 g maslaca (W2) ili koncentriranog maslaca (W2) u tikvicu od 150 ml (3.1.) s točnošću od 1 mg. Dodajte 50 mg etanola (4.1.) i 10 ml otopine kalijevog hidroksida (4.2.). Namjestite povratni kondenzator i zagrijavajte 30 minuta na temperaturi od oko 75 °C. Skinite kondenzator i ohladite tikvicu približno na sobnu temperaturu.
5.2.2. Dodajte u tikvicu 1,0 ml interne standardne otopine (4.3.1.1.) ako se određuje sitosterol, ili (4.3.1.2.) ako se određuje stigmasterol. Dobro promiješajte. Sadržaj tikvice kvantitativno prenesite u lijevak za odvajanje zapremine 500 ml (3.2.) i naizmjence isperite tikvicu s 50 ml vode i 250 ml dietil etera (4.4.). Lijevak za odvajanje snažno tresite dvije minute i pričekajte da se faze odvoje. Ocijedite donji vodenasti sloj i isperite sloj etera protresanjem uz dodavanje četiri uzastopna alikvota od 100 ml vode.
Napomena 2.: Da bi se spriječilo stvaranje emulzije, prva se dva ispiranja vodom izvode lagano (10 preokretanja). Treće se ispiranje izvodi snažnim protresanjem u trajanju od 30 sekundi. Ako se emulzija ipak formira, može se odstraniti dodavanjem 5-10 ml etanola. Ako se dodaje etanol, vrlo je važno još dvaput ponoviti ispiranje vodom uz energično protresanje.
5.2.3. Bistri sloj etera bez sadržaja sapuna propustite kroz staklenu kolonu (3.5.) koja sadrži 30 g bezvodnog natrijevog sulfata (4.5.). Skupite eter u tikvicu od 250 ml (3.3.). Dodajte jedan kamenčić za vrenje i otparite gotovo do suhoga u vodenoj kupelji ili suhom grijaču s oblogom te skupite otpadna otapala.
Napomena 3.: Ako se ekstrakti uzorka potpuno posuše na previsokoj temperaturi, može doći do gubitka sterola.
5.3. Priprema trimetil-silil etera
5.3.1. Preostalu otopinu etera pretočite iz tikvice u reakcijsku bočicu od 2 ml (3.7.) s 2 ml dietil etera i uklonite eter u struji dušika. Tikvicu isperite još dva puta alikvotima od 2 ml dietil etera, svaki uz pretakanje u reakcijsku bočicu i uklanjanje etera dušikom.
5.3.2. Sililirajte uzorak dodavanjem 1 ml TRI-SIL-a (4.6.). Zatvorite reakcijsku bočicu i snažnim protresanjem otopite sadržaj. Ako otapanje nije potpuno, zagrijte na 65-70 °C. Ostavite da odstoji najmanje pet minuta prije ubrizgavanja u plinski kromatograf. Sililirajte standarde na isti način kao i uzorke. Sililirajte mješavinu za ispitivanje odvajanja (4.10.) na isti način kao i uzorke.
Napomena 4.: Sililacija se vrši u suhom okruženju. Na nepotpunu sililaciju betulina ukazuje drugi pik na kromatogramu, pored pika betulina.
Prisutnost etanola u fazi sililacije ometa sililaciju. Ta pojava može biti posljedica nedovoljnog ispiranja u fazi ekstrakcije. Ako se navedeni problem nastavi pojavljivati, može se dodati i peto ispiranje u fazi ekstrakcije, uz jako protresanje u trajanju od 30 sekundi.
5.4. Plinsko-kromatografska analiza
5.4.1. Odabir uvjeta za provođenje analize
Instalirajte plinski kromatograf prema uputama proizvođača.
Smjernice u pogledu uvjeta za provođenje analize jesu sljedeće:
|
— |
temperatura kolone: 265 °C, |
|
— |
temperatura injektora: 265 °C, |
|
— |
temperatura detektora: 300 °C, |
|
— |
brzina protoka plina nositelja: 0,6 ml/min, |
|
— |
tlak vodika: 84 kPa, |
|
— |
tlak zraka: 155 kPa, |
|
— |
razdvajanje uzorka: od 10:1 do 50:1; omjer razdvajanja mora se optimizirati u skladu s uputama proizvođača i linearnošću odziva detektora te se potom mora validirati za raspon koncentracija relevantan za analizu. Napomena 5.: Od izuzetne je važnosti redovito čišćenje uloška injektora. |
|
— |
količina ubrizgane tvari: 1 μl TMSE otopine. |
Pije početka analize ostavite sustav da se uravnoteži i postigne zadovoljavajuću stabilnost odziva.
Navedene uvjete treba varirati s obzirom na karakteristike kolone i plinskog kromatografa da bi dobiveni kromatogrami udovoljavali sljedećim zahtjevima:
|
— |
pik sitosterola na kromatogramu mora biti na odgovarajući način odvojen od pika lanosterola. Slika 1. pokazuje tipični kromatogram koji se mora dobiti za sililiziranu mješavinu za ispitivanje rezolucije (4.10.), |
|
— |
relativna vremena zadržavanja sljedećih sterola trebaju približno iznositi:
|
|
— |
vrijeme zadržavanja betulina treba iznositi približno 24 minute. |
5.4.2. Analitički postupak
Ubrizgajte 1 μl sililirane standardne otopine (stigmasterola ili sitosterola) i podesite parametre za kalibraciju integratora.
Ubrizgajte dodatni 1 μl sililirane standardne otopine radi utvrđivanja faktora odziva za betulin.
Ubrizgajte 1 μl sililirane otopine uzorka i na kromatogramu izmjerite površine pikova. Svaka kromatografska identifikacija mora biti omeđena ubrizgavanjem standarda.
Kao smjernica se može navesti da bi između dva ubrizgavanja standarda trebalo šest puta ubrizgati uzorak.
Napomena 6.: Integracija pika stigmasterola na kromatogramu treba obuhvatiti sve krajeve signala kako su definirani točkama 1., 2. i 3. na slici 2.b.
Kod procjene ukupnog sitosterola integracija pika stigmasterola na kromatogramu treba obuhvatiti i površinu pika 22-dihidro-β-sitosterola (stigmastanola) koji eluira odmah nakon sitosterola (vidjeti sliku 3.b).
6. IZRAČUNAVANJE REZULTATA
6.1. Odredite površinu pikova sterola i betulina u oba standarda koji omeđuju jednu seriju i izračunajte R1:
R1 = (prosječna površina pika sterola u standardu)/(prosječna površina pika betulina u standardu).
Odredite površinu pikova sterola (stigmasterola i sitosterola) i betulina u uzorku i izračunajte R2:
R2 = (površina pika sterola u uzorku)/(površina pika betulina u uzorku);
|
W1 |
= |
sadržaj sterola u standardu (mg) koji se nalazi u 1 ml standardne otopine (4.8.1. ili 4.9.1.); |
|
W2 |
= |
masa uzorka (g) (5.2.1.); |
|
P |
= |
čistoća standardnog sterola (4.8. ili 4.9.); |
Sadržaj sterola u uzorku (mg/kg) = ((R 2) / (R 1)) × ((W 1) / (W 2)) × P × 10.
7. PRECIZNOST METODE
7.1. Maslac
7.1.1. Ponovljivost
7.1.1.1.
Razlika između rezultata dvaju određivanja koja je u najkraćem mogućem vremenskom intervalu izvršio isti analitičar, koristeći istu opremu na jednakom ispitnom materijalu, ne smije biti veća od 19,3 mg/kg.
7.1.1.2.
Razlika između rezultata dvaju određivanja koja je u najkraćem mogućem vremenskom intervalu izvršio isti analitičar, koristeći istu opremu na jednakom ispitnom materijalu, ne smije biti veća od 23,0 mg/kg.
7.1.2. Obnovljivost
7.1.2.1.
Razlika između rezultata dvaju određivanja koja su izvršili analitičari u različitim laboratorijima, koristeći različitu opremu na jednakom ispitnom materijalu, ne smije biti veća od 31,9 mg/kg.
7.1.2.2.
Razlika između rezultata dvaju određivanja koja su izvršili analitičari u različitim laboratorijima, koristeći različitu opremu na jednakom ispitnom materijalu, ne smije biti veća od 8,7 % u odnosu na srednju vrijednost određivanja.
7.1.3. Izvor podataka o preciznosti
Podaci o preciznosti potječu iz eksperimenta provedenog 1992. kojim je bilo obuhvaćeno osam laboratorija i šest uzoraka (tri slijepe probe u duplikatu) za stigmasterol i šest uzoraka (tri slijepe probe u duplikatu) za sitosterol.
7.2. Koncentrirani maslac
7.2.1. Ponovljivost
7.2.1.1.
Razlika između rezultata dvaju određivanja koja je u najkraćem mogućem vremenskom intervalu izvršio isti analitičar, koristeći istu opremu na jednakom ispitnom materijalu, ne smije biti veća od 10,2 mg/kg.
7.2.1.2.
Razlika između rezultata dvaju određivanja koja je u najkraćem mogućem vremenskom intervalu izvršio isti analitičar, koristeći istu opremu na jednakom ispitnom materijalu, ne smije biti veća od 3,6 % u odnosu na srednju vrijednost određivanja.
7.2.2. Obnovljivost
7.2.2.1.
Razlika između rezultata dvaju određivanja koja su izvršili analitičari u različitim laboratorijima, koristeći različitu opremu na jednakom ispitnom materijalu, ne smije biti veća od 25,3 mg/kg.
7.2.2.2.
Razlika između rezultata dvaju određivanja koja su izvršili analitičari u različitim laboratorijima, koristeći različitu opremu na jednakom ispitnom materijalu, ne smije biti veća od 8,9 % u odnosu na srednju vrijednost određivanja.
7.2.3. Izvor podataka o preciznosti
Podaci o preciznosti potječu iz eksperimenta provedenog 1991. kojim je bilo obuhvaćeno devet laboratorija i šest uzoraka (tri slijepe probe u duplikatu) za stigmasterol i šest uzoraka (tri slijepe probe u duplikatu) za sitosterol.
8. GRANIČNE VRIJEDNOSTI DOPUŠTENOG ODSTUPANJA
8.1. Od proizvoda s dodanim markerom obvezno treba uzeti po tri uzorka radi provjere preciznosti.
8.2. Maslac
8.2.1. Stigmasterol
8.2.1.1. Stopa inkorporacije za stigmasterol iznosi 150 g stigmasterola minimalne čistoće 95 % po toni maslaca, odnosno 142,5 mg/kg, ili 170 g stigmasterola minimalne čistoće 85 % po toni maslaca, odnosno 144,5 mg/kg.
8.2.1.2. Rezultati analize proizvoda dobiveni na tri uzorka koriste se za provjeru stope i homogenosti inkorporacije markera, a najniži od tih rezultata uspoređuje se sa sljedećim graničnim vrijednostima:
|
— |
115,8 mg/kg (95 % minimalne stope inkorporacije za stigmasterol čistoće 95 %), |
|
— |
117,7 mg/kg (95 % minimalne stope inkorporacije za stigmasterol čistoće 85 %), |
|
— |
80,1 mg/kg (70 % minimalne stope inkorporacije za stigmasterol čistoće 95 %), |
|
— |
81,5 mg/kg (70 % minimalne stope inkorporacije za stigmasterol čistoće 85 %). |
Koncentracija markera u uzorku koji je dao najniži rezultat koristi se u vezi s interpolacijom između 115,8 mg/kg i 80,1 mg/kg, odnosno 117,7 mg/kg i 81,5 mg/kg.
8.2.2. Sitosterol
8.2.2.1. Stopa inkorporacije za sitosterol iznosi 600 g sitosterola minimalne čistoće 90 % po toni maslaca, odnosno 540 mg/kg.
8.2.2.2. Rezultati analize proizvoda dobiveni na tri uzorka koriste se za provjeru stope i homogenosti inkorporacije markera, a najniži od tih rezultata uspoređuje se sa sljedećim graničnim vrijednostima:
|
— |
482,6 mg/kg (95 % minimalne stope inkorporacije za sitosterol čistoće 90 %), |
|
— |
347,6 mg/kg (70 % minimalne stope inkorporacije za sitosterol čistoće 90 %). |
Koncentracija markera u uzorku koji je dao najniži rezultat koristi se u vezi s interpolacijom između 482,6 mg/kg i 347,6 mg/kg.
8.3. Koncentrirani maslac
8.3.1. Stigmasterol
8.3.1.1. Stopa inkorporacije za stigmasterol iznosi 150 g stigmasterola minimalne čistoće 95 % po toni koncentriranog maslaca, odnosno 142,5 mg/kg, ili 170 g stigmasterola minimalne čistoće 85 % po toni maslaca, odnosno 144,5 mg/kg.
8.3.1.2. Rezultati analize proizvoda dobiveni na tri uzorka koriste se za provjeru stope i homogenosti inkorporacije markera, a najniži od tih rezultata uspoređuje se sa sljedećim graničnim vrijednostima:
|
— |
118,5 mg/kg (95 % minimalne stope inkorporacije za stigmasterol čistoće 95 %), |
|
— |
120,4 mg/kg (95 % minimalne stope inkorporacije za stigmasterol čistoće 85 %), |
|
— |
82,9 mg/kg (70 % minimalne stope inkorporacije za stigmasterol čistoće 95 %), |
|
— |
84,3 mg/kg (70 % minimalne stope inkorporacije za stigmasterol čistoće 85 %). |
Koncentracija markera u uzorku koji je dao najniži rezultat koristi se u vezi s interpolacijom između 118,5 mg/kg i 82,9 mg/kg, odnosno 120,4 mg/kg i 84,3 mg/kg.
8.3.2. Sitosterol
8.3.2.1. Stopa inkorporacije za sitosterol iznosi 600 g sitosterola minimalne čistoće 90 % po toni maslaca, odnosno 540 mg/kg.
8.2.2.2. Rezultati analize proizvoda dobiveni na tri uzorka koriste se za provjeru stope i homogenosti inkorporacije markera, a najniži od tih rezultata uspoređuje se sa sljedećim graničnim vrijednostima:
|
— |
480,9 mg/kg (95 % minimalne stope inkorporacije za sitosterol čistoće 90 %), |
|
— |
345,9 mg/kg (70 % minimalne stope inkorporacije za sitosterol čistoće 90 %). |
Koncentracija markera u uzorku koji je dao najniži rezultat koristi se u vezi s interpolacijom između 480,9 mg/kg i 345,9 mg/kg.
Slika 1.
Kromatogram mješavine za testiranje rezolucije
Poželjna je potpuna rezolucija, odnosno pik krivulje za lanosterol trebao bi se vratiti na baznu liniju prije nego se podigne prema piku sitosterola, iako je nepotpuna rezolucija dopuštena.
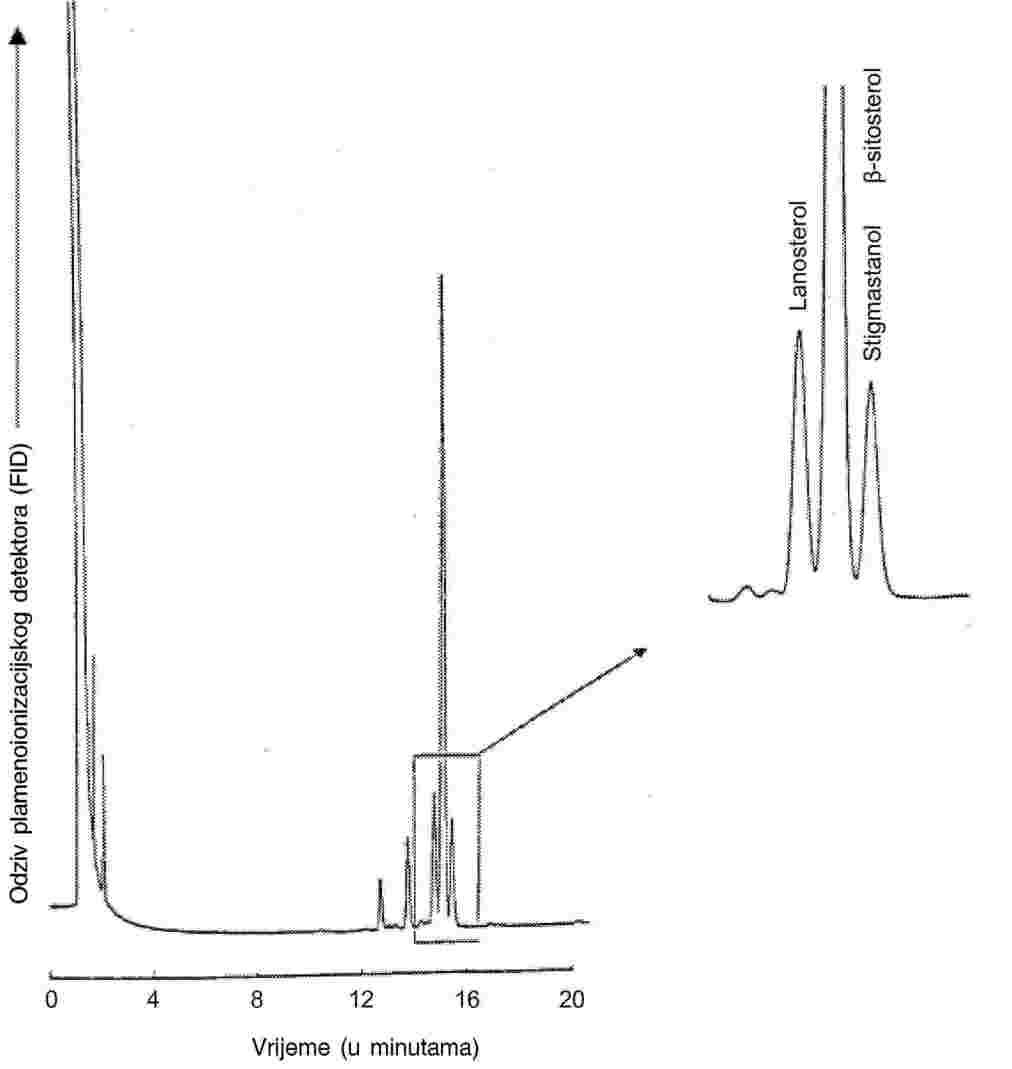
|
Slika 2.a |
Slika 2.b |
|
Standard stigmasterola |
Uzorak maslaca denaturiran stigmasterolom |
|
|
|
|
Napomena: Integracija pika stigmasterola trebala bi obuhvaćati sve krajeve signala kako su definirani točkama 1., 2. i 3. |
|
|
Slika 3.a |
Slika 3.b |
|
Standard sitosterola |
Uzorak maslaca denaturiran β-sitosterolom |
|
|
|
|
Napomena: β-sitosterol često sadrži nečistoću (definiranu kao stigmastanol) koja eluira odmah nakon β-sitosterola. Kod procjene ukupne prisutne količine β-sitosterola treba zbrojiti površine tih dvaju pikova. |
|
PRILOG IX.
(Članak 6.)
REFERENTNA METODA ZA DETEKCIJU KRAVLJEG MLIJEKA I KAZEINATA U SIREVIMA OD OVČJEG, KOZJEG ILI BIVOLJEG MLIJEKA ILI OD MJEŠAVINA OVČJEG, KOZJEG I BIVOLJEG MLIJEKA
1. PODRUČJE PRIMJENE
Detekcija kravljeg mlijeka i kazeinata u sirevima od ovčjeg, kozjeg ili bivoljeg mlijeka ili od mješavina ovčjeg, kozjeg i bivoljeg mlijeka izoelektričnim fokusiranjem γ-kazeina nakon plazminolize.
2. PODRUČJE PRIMJENE
Metoda je prikladna za osjetljivu i specifičnu detekciju sirovog i toplinski obrađenog kravljeg mlijeka i kazeinata u svježim ili zrelim sirevima od ovčjeg, kozjeg i bivoljeg mlijeka, ili od mješavina ovčjeg, kozjeg i bivoljeg mlijeka. Nije primjerena za otkrivanje patvorenja mlijeka i sira toplinski obrađenim koncentratima bjelančevina sirutke od kravljeg mlijeka.
3. NAČELO METODE
3.1. Izolacija kazeina iz sira i referentni standardi.
3.2. Otapanje izoliranih kazeina i njihovo podvrgavanje plazminskom cijepanju (EC.3.4.21.7).
3.3. Izoelektrično fokusiranje plazminski obrađenih kazeina u prisutnosti ureje i bojenje proteina.
3.4. Ocjenjivanje obojenih obrazaca γ3- i γ2-kazeina (dokaz kravljeg mlijeka) na temelju usporedbe obrasca dobivenog iz uzorka s obrascima dobivenim u istom gelu iz referentnih standarda koji sadrže 0 % i 1 % kravljeg mlijeka.
4. REAGENSI
Ako nije navedeno drukčije, moraju se koristiti kemikalije analitičke čistoće. Voda mora biti dvostruko destilirana ili ekvivalentne čistoće.
Napomena: Sljedeći detalji odnose se na laboratorijski pripravljene poliakrilamidne gelove koji sadrže ureju, dimenzija 265 × 125 × 0,25 mm. U slučajevima kad se koriste druge veličine i vrste gelova, možda treba podesiti uvjete razdvajanja.
Izoelektrično fokusiranje
4.1. Reagensi za proizvodnju poliakrilamidnih gelova koji sadrže ureju
4.1.1. Osnovna otopina gela
Otopite:
4,85 g akrilamida,
0,15 g N, N′-metilen-bis-akrilamida (BIS),
48,05 g ureje,
15,00 g glicerola (87 % w/w)
u vodi, dopunite do 100 ml i pohranite u smeđoj staklenoj bočici u hladnjak.
Napomena: Umjesto navedenih fiksnih masa neurotoksičnih akrilamida može se koristiti gotova mješavina otopina akrilamida/BIS-a, koja je dostupna na tržištu. Ako takva otopina sadrži 30 % w/v akrilamida i 0,8 % w/v BIS-a, umjesto navedenih fiksnih masa za pripravak treba upotrijebiti količinu od 16,2 ml. Rok valjanosti osnovne otopine je najviše 10 dana; ako je njena vodljivost veća od 5 μS, otopinu treba deionizirati miješajući je 30 minuta s 2 g Amberlita MB-3 i potom profiltrirati kroz membranu od 0,45 μm.
4.1.2. Otopina gela
Pripremite otopinu gela miješanjem aditiva i amfolita s osnovnom otopinom gela (vidjeti 4.1.1.).
9,0 ml osnovne otopine
24 mg β-alanina
500 μl amfolita pH 3,5-9,5 (1)
250 μl amfolita pH 5-7 (1)
250 μl amfolita pH 6-8 (1).
Otopinu gela pomiješajte i otplinjavajte dvije do tri minute u ultrazvučnoj kupelji ili vakuumu.
Napomena: Otopinu gela pripremite neposredno prije uporabe (vidjeti 6.2.).
4.1.3. Otopine katalizatora
4.1.3.1. N, N, N′ N′ – tetrametiletilendiamin (Temed).
4.1.3.2. 40 % w/v amonijev persulfat (PER):
Otopite 800 mg PER-a u vodi i dopunite do 2 ml.
Napomena: Uvijek koristite svježe pripremljenu otopinu PER-a.
4.2. Kontaktna tekućina
Kerozin ili tekući parafin
4.3. Anodna otopina
Otopite 5,77 g fosforne kiseline (85 % w/w) u vodi i razrijedite do 100 ml.
4.4. Katodna otopina
Otopite 2,00 g natrijevog hidroksida u vodi i razrijedite vodom do 100 ml.
Priprema uzorka
4.5. Reagensi za izolaciju proteina
4.5.1. Razrijeđena octena kiselina (25,0 ml ledene octene kiseline razrijedite vodom do 100 ml)
4.5.2. Diklorometan
4.5.3. Aceton
4.6. Pufer za rastapanje proteina
Otopite:
5,75 g glicerola (87 % w/w),
24,03 g ureje,
250 mg ditiotreitola
u vodi i nadopunite do 50 ml.
Napomena: Spremite pufer u hladnjak, maksimalni rok trajanja je tjedan dana.
4.7. Reagensi za plazminsko cijepanje kazeina
4.7.1. Pufer amonijevog karbonata
Titrirajte 0,2 mol/l otopine amonijevog hidrogen-karbonata (1,58 g/100 ml vode), koja sadrži 0,05 mol/l etilen-diamin-tetraoctene kiseline (EDTA, 1,46 g/100 ml), s 0,2 mol/l otopine amonijevog karbonata (1,92 g/100 ml vode), koja sadrži 0,05 mol/l kiseline EDTA do pH 8.
4.7.2. Plazmin kravljeg mlijeka (EC. 3.4.21.7), aktivnosti najmanje 5 U/ml
4.7.3. Otopina ε-aminokapronske kiseline za inhibiranje enzima
Otopite 2,624 g ε-aminokapronske kiseline (6-amino-n-heksanske kiseline) u 100 ml 40 %-tnog (v/v) etanola.
4.8. Standardi
4.8.1. Potvrđeni referentni standardi za mješavine usirenog obranog ovčjeg i kozjeg mlijeka, koje sadrže 0 % i 1 % kravljeg mlijeka, dostupni su na Institutu Komisije za referentne materijale i mjerenja, B-2440, Geel, Belgija.
4.8.2. Priprema privremenih laboratorijskih standarda za usireno bivolje mlijeko koje sadrži 0 % i 1 % kravljeg mlijeka.
Obrano se mlijeko priprema centrifugiranjem 2 500 g bivoljeg ili kravljeg sirovog mlijeka na temperaturi od 37 °C u trajanju od 20 minuta. Nakon brzog hlađenja epruvete i sadržaja na 6 do 8 °C, potpuno uklonite gornji sloj masti. Za pripremu standarda od 1 % dodajte 5,00 ml obranog kravljeg mlijeka u 495 ml obranog bivoljeg mlijeka u laboratorijskoj čaši od 1 l, podesite pH na 6,4 dodavanjem razrijeđene mliječne kiseline (10 % w/v). Podesite temperaturu na 35 °C i dodajte 100 μl telećeg sirila (aktivnost sirila 1:10 000, oko 3 000 U/ml), miješajte 1 minutu i potom ostavite posudu pokrivenu aluminijskom pločom jedan sat na temperaturi od 35 °C da bi se mogao formirati gruš. Nakon formiranja gruša, cijelo usireno mlijeko se liofilizira bez prethodne homogenizacije ili cijeđenja sirutke. Nakon liofilizacije, sitno ga sameljite da biste dobili homogeni prah. Za pripremu standarda od 0 % provodi se isti postupak uz uporabu isključivo obranog bivoljeg mlijeka. Standardi se čuvaju na temperaturi od – 20 °C.
Napomena: Prije pripreme standarda preporuča se provjeriti čistoću bivoljeg mlijeka metodom izoelektričnog fokusiranja kazeina razgrađenih djelovanjem plazmina.
Reagensi za bojenje proteina
4.9. Fiksator
Otopite 150 g triklorooctene kiseline u vodi i dopunite do 1 000 ml.
4.10. Otopina za odbojavanje
Razrijedite 500 ml metanola i 200 ml ledene octene kiseline destiliranom vodom do količine od 2 000 ml.
Napomena: Svaki dan pripremite svježu otopinu za odbojavanje; možete je pripremiti miješanjem jednakih količina osnovnih otopina 50 %-tnog (v/v) metanola i 20 %-tne (v/v) ledene octene kiseline.
4.11. Otopine za bojenje
4.11.1. Otopina za bojenje (osnovna otopina 1)
Otopite 3,0 g boje Coomassie Brilliant Blue G-250 (C.I. 42655) u 1 000 ml 90 %-tnog (v/v) metanola pomoću magnetske miješalice (oko 45 minuta), filtrirajte kroz dva naborana filtra srednje brzine.
4.11.2. Otopina za bojenje (osnovna otopina 2)
Otopite 5,0 g bakrenog sulfata pentahidrata u 1 000 ml 20 %-tne (v/v) octene kiseline.
4.11.3. Otopina za bojenje (radna otopina)
Pomiješajte po 125 ml od svake osnovne otopine (4.11.1., 4.11.2.) neposredno prije bojenja.
Napomena: Otopina za bojenje priprema se na dan kad se koristi.
5. OPREMA
5.1. Staklene ploče (265 × 125 × 4 mm); gumeni valjak (širine 15 cm); stol s nagibnom radnom površinom.
5.2. Noseća ploča za gel (265 × 125 mm).
5.3. Pokrivna ploča (280 × 125 mm). Na sve duge bridove (vidjeti sliku 1.) nanesite ljepljivu traku (280 × 6 × 0,25 mm).
5.4. Komora za elektrofokusiranje s rashladnom pločom (npr. 265 × 125 mm) i odgovarajućim električnim napajanjem (≥ 2,5 kV) ili uređajem za automatsku elektroforezu.
5.5. Cirkulacijski kriostat, termostatski reguliran na 12 ± 0,5 °C.
5.6. Centrifuga, podesiva do 3 000 g.
5.7. Elektrodne trake (duljine ≥ 265 mm).
5.8. Plastične boce s kapaljkom za anodnu i katodnu otopinu.
5.9. Aplikatori za uzorke (10 × 5 mm, od viskoze ili od filtar-papira koji slabo adsorbira bjelančevine).
5.10. Škare, skalpeli i pincete od nehrđajućeg čelika.
5.11. Posude za bojenje i odbojavanje od nehrđajućeg čelika ili stakla (npr. plitice za instrumente dimenzija 280 × 150 mm).
5.12. Podesivi štapni homogenizator (promjera osovine 10 mm), raspona brzine od 8 000 do 20 000 o/min.
5.13. Magnetska miješalica.
5.14. Ultrazvučna kupelj.
5.15. Uređaj za zavarivanje ploče.
5.16. Mikropipete od 25 μl.
5.17. Vakuumski koncentrator ili liofilizator.
5.18. Vodena kupelj, termostatski regulirana između 35 i 40 ± 1 °C s vibracijskom miješalicom.
5.19. Denzitometar za očitavanje na valnoj duljini λ = 634 nm.
6. POSTUPAK
6.1. Priprema uzoraka
6.1.1. Izolacija kazeina
Odvagnite količinu koja je ekvivalent 5 g suhe mase sira ili referentnih standarda u epruvetu za centrifugiranje od 100 ml, dodajte 60 ml destilirane vode i homogenizirajte štapnim homogenizatorom (8 000 do 10 000 o/min). Podesite pH na 4,6 pomoću razrijeđene octene kiseline (4.5.1.) i centrifugirajte (5 minuta, 3 000 g). Dekantirajte mast i sirutku, ostatak homogenizirajte na 20 000 o/min u 40 ml destilirane vode čija je pH vrijednost od 4,5 podešena pomoću razrijeđene octene kiseline (4.5.1.); dodajte 20 ml diklorometana (4.5.2.), ponovno homogenizirajte i centrifugirajte (5 minuta, 3 000 g). Špatulom uklonite sloj kazeina koji se nalazi između vodene i organske faze (vidjeti sliku 2.) i dekantirajte obje faze. Kazein ponovo homogenizirajte u 40 ml destilirane vode (vidjeti gore) i 20 ml diklorometana (4.5.2.) i centrifugirajte. Ponavljajte postupak sve dok obje faze ekstrakcije ne postanu bezbojne (dva do tri puta). Ostatak bjelančevina homogenizirajte pomoću 50 ml acetona (4.5.3.) i filtrirajte kroz naborani filtar-papir srednje brzine filtracije. Ostatak na filtru isperite dva puta, svaki put koristeći po 25 ml acetona, ostavite da se osuši na zraku ili u struji dušika te fino usitnite u tarioniku.
Napomena: Suhe izolate kazeina treba držati na temperaturi od – 20 °C.
6.1.2. Plazminsko cijepanje β-kazeina radi intenzifikacije γ-kazeina
Disperzirajte 25 mg izoliranih kazeina (6.1.1.) u 0,5 ml puferske otopine amonijevog karbonata (4.7.1.) i homogenizirajte 20 minuta, npr. ultrazvučnim postupkom. Zagrijte do 40 °C i dodajte 10 μl plazmina (4.7.2.), promiješajte i inkubirajte jedan sat na 40 °C uz neprestano tresenje. Radi inhibicije enzima dodajte 20 μl otopine ε-aminokapronske kiseline (4.7.3.) te potom dodajte 200 mg ureje u krutom obliku i 2 mg ditiotreitola.
Napomena: Radi postizanja veće simetrije fokusiranih kazeinskih vrpci, preporuča se liofiliziranje otopine nakon dodavanja ε-aminokapronske kiseline i potom otapanje ostatka u 0,5 ml pufera za otapanje bjelančevina (4.6.).
6.2. Priprema poliakrilamidnih gelova koji sadrže ureju
Pomoću nekoliko kapi vode i valjka noseću ploču za gel (5.2.) stavite na staklenu ploču (5.1.), a višak vode uklonite papirnatim ubrusom ili ručnikom. Na isti način pokrivnu ploču (5.3.) s odstojnicima (0,25 mm) stavite na drugu staklenu ploču. Položite ploču vodoravno na stol s nagibnom radnom površinom.
Dodajte 10 μl Temeda (4.1.3.1.) u pripremljenu i deaeriranu otopinu gela (4.1.2.), promiješajte i dodajte 10 μl otopine PER (4.1.3.2.), dobro izmiješajte i jednakomjerno razlijte po sredini pokrivne ploče. Stavite jedan rub noseće ploče za gel (ploča okrenuta prema dolje) na pokrivnu ploču i preostali dio polagano spuštajte tako da se između dvije plohe stvori tanki sloj gela koji će se jednakomjerno raširiti bez stvaranja mjehurića (slika 3.). Pažljivo spustite noseću ploču do kraja, koristeći pritom tanku špatulu, i na nju položite još tri staklene ploče koje će poslužiti kao utezi. Po završetku polimerizacije (oko 60 minuta), gel koji se polimerizirao na nosećoj ploči maknite zajedno s pokrivnom pločom, laganim nakretanjem staklenih ploča. Pažljivo očistite naličje noseće ploče da biste uklonili ostatake gela i ureje. Zatvorite „gel-sendvič” u cjevastu foliju i pohranite u hladnjak (najviše šest tjedana).
Napomena: Pokrivna ploča s odstojnicima može se ponovo koristiti. Poliakrilamidni gel može se razrezati na manje dijelove, što se preporuča u slučaju manjeg broja uzoraka ili ako se koristi uređaj za automatsku elektroforezu (dva gela, dimenzija 4,5 × 5 cm).
6.3. Izoelektrično fokusiranje
Termostat za hlađenje podesite na 12 °C. Kerozinom očistite stražnju stranu noseće ploče za gel i potom dodajte nekoliko kapljica kerozina (4.2.) u sredinu rashladnog bloka. Zatim na njega stavite gel-sendvič s nosećom stranom okrenutom prema dolje i povaljajte ga valjkom, pazeći da se pritom ne stvaraju mjehurići. Obrišite višak kerozina i skinite pokrivnu ploču. Elektrodne trake natopite elektrodnim otopinama (4.3., 4.4.), izrežite ih prema duljini gela i položite na predviđena mjesta (razmak između elektroda 9,5 cm).
Uvjeti za izoelektrično fokusiranje:
6.3.1. Veličina gela 265 × 125 × 0,25 mm
|
Faza |
Vrijeme (min) |
Napon (V) |
Jakost (mA) |
Snaga (W) |
Volt-sati (Vh) |
||
|
30 |
maksimalni 2 500 |
maksimalna 15 |
konstantna 4 |
oko 300 |
||
|
60 |
maksimalni 2 500 |
maksimalna 15 |
konstantna 4 |
oko 1 000 |
||
|
60 |
maksimalni 2 500 |
maksimalna 5 |
maksimalna 20 |
oko 3 000 |
||
|
40 |
maksimalni 2 500 |
maksimalna 6 |
maksimalna 20 |
oko 3 000 |
|||
|
30 |
maksimalni 2 500 |
maksimalna 7 |
maksimalna 25 |
oko 3 000 |
Napomena: Ako se promijeni debljina ili širina gela, vrijednosti za jakost i snagu treba podesiti u skladu s promjenom (npr. udvostručiti vrijednosti jakosti i snage ako se koristi gel dimenzija 265 × 125 × 0,5 mm).
6.3.2. Primjer naponskog režima za uređaj za automatsku elektroforezu (2 gela dimenzija 5,0 × 4,5 cm); elektrode bez traka nanesene izravno na gel
|
Faza |
Napon |
Jakost |
Snaga |
Temp. |
Volt-sati |
||
|
1 000 V |
10,0 mA |
3,5 W |
8 °C |
85 Vh |
||
|
250 V |
5,0 mA |
2,5 W |
8 °C |
30 Vh |
||
|
1 200 V |
10,0 mA |
3,5 W |
8 °C |
80 Vh |
||
|
1 500 V |
5,0 mA |
7,0 W |
8 °C |
570 Vh |
Postavite aplikator za uzorak u fazi 2. pri 0 Vh.
Uklonite aplikator za uzorak u fazi 2. pri 30 Vh.
6.4. Bojenje proteina
6.4.1. Fiksacija proteina
Čim isključite struju, uklonite elektrodne trake i odmah stavite gel u posudu za bojenje/odbojavanje s 200 ml fiksatora (4.9.); ostavite 15 minuta uz neprekidno mućkanje.
6.4.2. Ispiranje i bojenje ploče s gelom
Dobro ocijedite fiksator i ploču s gelom dva puta ispirite po 30 sekundi, svaki put s po 100 ml otopine za odbojavanje (4.10.). Izlijte otopinu za odbojavanje i u posudu nalijte 250 ml otopine za bojenje (4.11.3.); ostavite 45 minuta da boja djeluje, uz lagano protresanje.
6.4.3. Odbojavanje ploče s gelom
Izlijte otopinu za bojenje, ploču s gelom dva puta isperite, svaki put sa 100 ml otopine za odbojavanje (4.10.), zatim 15 minuta protresajte s 200 ml otopine za odbojavanje te ponavljajte postupak odbojavanja, najmanje dva do tri puta, dok pozadina ne postane bistra i bezbojna. Ploču s gelom isperite destiliranom vodom (2 × 2 minute) i osušite na zraku (2 do 3 sata) ili sušilom za kosu (10 do 15 minuta).
Napomena 1.: Izvršite fiksaciju, ispiranje, bojenje i odbojavanje na temperaturi od 20 °C. Ne koristite više temperature.
Napomena 2.: Ako radije primjenjujete osjetljiviju metodu bojenja srebrom (npr. Silver Staining Kit, Protein, Pharmacia Biotech, oznaka br. 17-1150-01), uzorke kazeina obrađene plazminom treba razrijediti na 5 mg/ml.
7. OCJENJIVANJE
Ocjenjivanje se provodi usporedbom struktura bjelančevina u nepoznatim uzorcima s referentnim standardima na istom gelu. Prisutnost kravljeg mlijeka u sirevima od ovčjeg, kozjeg i bivoljeg mlijeka i mješavinama ovčjeg, kozjeg i bivoljeg mlijeka utvrđuje se na temelju γ3- and γ2-kazeina, čije se izoelektrične točke nalaze između pH 6,5 i pH 7,5 (slike 4.a i b i slika 5.). Granica detekcije niža je od 0,5 %.
7.1. Vizualna procjena
Za vizualnu procjenu količine kravljeg mlijeka preporuča se izjednačiti koncentracije uzoraka i standarda radi dobivanja istih razina intenziteta ovčjih, kozjih i/ili bivoljih γ2- i γ3-kazeina (vidjeti „γ2 E, G, B” i „γ3 E, G, B” na slikama 4.a, 4.b i 5.). Nakon toga se količina kravljeg mlijeka (manje, jednako ili više od 1 %) u nepoznatom uzorku može izravno procijeniti usporedbom intenziteta kravljih γ3- i γ2-kazeina (vidjeti „γ3 C” i „γ2 C” na slikama 4.a i b te na slici 5.) s onima u referentnim standardima od 0 % i 1 % (ovčji, kozji) ili u privremenim laboratorijskim standardima (bivolji).
7.2. Denzitometrijska procjena
Za utvrđivanje omjera površina pikova kravljih i ovčjih, kozjih i/ili bivoljih kazeina γ2 i γ3 (vidjeti sliku 5.) primijenite denzitometriju (5.19.) ako je imate na raspolaganju. Usporedite tu vrijednost s omjerom površina pikova kazeina referentnog standarda od 1 % (ovčji, kozji) ili privremenog laboratorijskog standarda (bivolji) analiziranih na istom gelu.
Napomena: Ova je metoda zadovoljavajuća ako postoji jasan pozitivan signal za oba kravlja γ2- i γ3-kazeina, u referentnom standardu od 1 %, ali ne i u referentnom standardu od 0 %. U protivnom, optimizirajte postupak pažljivo slijedeći detaljne upute navedene u metodi.
Uzorak se smatra pozitivnim ako su oba kravlja γ2- i γ3-kazeina ili odgovarajući omjeri površina pikova jednaki ili veći od razine u referentnom standardu od 1 %.
8. REFERENTNA LITERATURA
|
1. |
Addeo F., Moio L., Chianese L., Stingo C., Resmini P., Berner I., Krause I., Di Luccia A., Bocca A.: Use of plasmin to increase the sensitivity of the detection of bovine milk in bovine and/or caprine cheese by gel isoelectric focusing of γ2-caseins, Milchwissenschaft 45, 1990., str. 708.-711. |
|
2. |
Addeo F., Nicolai M. A., Chianese L., Moio L., Spagna Musso S., Bocca A., Del Giovine L.: A control method to detect bovine milk in ewe and water buffalo cheese using immunoblotting, Milchwissenschaft 50, 1995., str. 83.-85. |
|
3. |
Krause I., Berner I., Klostermeyer H.: Sensitive detection of cow milk in ewe and goat milk and cheese by carrier ampholyte – and carrier ampholyte/immobilized pH gradient – isoelectric focusing of γ-caseins using plasmin as signal amplifier, v: Electrophoresis-Forum 89 (B. J. Radola, ur.), Bode-Verlag, München, 1989., str. 389.-393. |
|
4. |
Krause Ι., Belitz H.-D., Kaiser K.-P.: Nachweis von Kuhmilch in Schaf and Ziegenmilch bzw. -käse durch isoelektrische Fokussierung in harnstoffhaltigen Polyacrylamidgelen, Z. Lebensm. Unters. Forsch. 174, 1982., str. 195.-199. |
|
5. |
Radola, B. J., Ultrathin-layer isoelectric focusing in 50–100 μm polyacrylamide gels on silanised glass plates or polyester films, Electrophoresis 1, 1980., str. 43.-56. |
Slika 1.
Shematski crtež pokrivne ploče
Slika 2.
Sloj kazeina koji nakon centrifugiranja pluta između vodene i organske faze

Slika 3.
Tehnika lijevanja ultratankih poliakrilamidnih gelova poklapanjem

a = odstojnik (0,25 mm); b = pokrivna ploča (5.3.); c, e = staklene ploče (5.1.); d = otopina gela (4.1.2.); f = noseća ploča za gel (5.2.)
Slika 4.a
Izoelektrično fokusiranje kazeina obrađenih plazminom iz sireva od ovčjeg i kozjeg mlijeka koji sadrže različite količine kravljeg mlijeka
% CM = postotak kravljeg mlijeka, C = krava, E = ovca, G = koza
Prikazana je gornja polovica IEF gela.
Slika 4.b
Izoelektrično fokusiranje kazeina obrađenih plazminom iz sireva od mješavina ovčjeg, kozjeg i bivoljeg mlijeka koji sadrže različite količine kravljeg mlijeka
% CM = postotak kravljeg mlijeka; 1+ = uzorak koji sadrži 1 % kravljeg mlijeka s dodatkom čistog kravljeg kazeina u sredini traga; C = krava, E = ovca, G = koza, B = bivol
Prikazana je potpuna razdaljina razdvajanja IEF gela.
Slika 5.
Preklapanje denzitograma standarda (STD) i uzoraka sireva od mješavine ovčjeg i kozjeg mlijeka nakon izoelektričnog fokusiranja

a,b = standardi koji sadrže 0 % i 1 % kravljeg mlijeka; c-g = uzorci sireva koji sadrže 0, 1, 2, 3 i 7 % kravljeg mlijeka; C = krava, E = ovca, G = koza
Skenirana je gornja polovica IEF gela na λ = 634 nm.
(1) Proizvodi Ampholine® pH 3,5-9,5 (Pharmacia) i Resolyte® pH 5-7 i pH 6-8 (BDH, Merck) pokazali su se kao posebno primjereni za postizanje potrebnog odvajanja γ-kazeina.
(2) Aplikacija uzorka: nakon predfokusiranja (1. faza) otpipetirajte 18 μl uzorka i standardne otopine na aplikatore uzoraka (10 × 5 mm), poslažite ih na gel u razmacima od 1 mm, odnosno 5 mm uzdužno od anode te ih lagano pritisnute. Izvršite fokusiranje sukladno gore navedenim uvjetima te nakon 60 minuta fokusiranja aplikatore uzoraka pažljivo uklonite.
PRILOG X.
(Članak 7.)
REFERENTNA METODA ZA ODREĐIVANJE KOLIFORMA U MASLACU, OBRANOM MLIJEKU U PRAHU, KAZEINU I KAZEINATIMA
1. PRIPREMA UZORAKA
ISO standard 8261
2. POSTUPAK
ISO standard 4831
Uzorci koji odgovaraju 1 g maslaca, obranog mlijeka u prahu ili kazeina/kazeinata inokuliraju se u hranjivu podlogu za uzgoj kultura.
Inokuliraju se po tri epruvete za svaki uzorak.
3. REZULTATI
Ako 3 epruvete daju 3 negativna rezultata, rezultat je „sukladan”.
Ako 3 epruvete daju 2 ili 3 pozitivna rezultata, rezultat je „nesukladan”.
Ako 3 epruvete daju 2 negativna rezultata, analiza se dvaput ponavlja (s dvije epruvete).
|
— |
Ako su ta dva rezultata negativna, rezultat je „sukladan”. |
|
— |
Ako je najmanje 1 rezultat pozitivan, rezultat je „nesukladan”. |
PRILOG XI.
(Članak 8.)
ODREĐIVANJE LAKTOZE U KRMNIM SMJESAMA
1. OPSEG I POLJE PRIMJENE
Određivanje laktoze u krmnim smjesama.
2. REFERENCA
Sadržaj laktoze definira se kao maseni postotak utvrđen opisanim postupkom.
3. DEFINICIJA
Sadržaj bezvodne laktoze izražen u g na 100 g.
4. NAČELO
Krmna se smjesa rekonstituira dodavanjem vode. U razrijeđeni izvagani alikvot dodaje se Biggsova otopina da bi se istaložila mast i frakcije bjelančevinskih dijelova krmne smjese. Uzorak se filtrira (ili centrifugira), a filtrat (ili supernatant) se ubrizga u HPLC kolonu za kationsku izmjenu (olovo), pri čemu se kao mobilna faza koristi voda HPLC čistoće. Eluirana laktoza detektira se pomoću diferencijalnog refraktometra (1).
5. REAGENSI
5.1. Općenito
Ako nije drukčije navedeno, koristite samo reagense priznate analitičke čistoće i otplinjenu vodu HPLC čistoće.
5.2. Laktoza
D-laktoza monohidrat ((C12H22)O11H2O) može apsorbirati dodatnu vlagu. Prije uporabe izmjerite stvarnu količinu vode prema Karl-Fisherovoj metodi ili dodatnu vlagu odstranite tako da laktozu držite u sušioniku 8 sati na 105 °C (ovim postupkom laktoza ne gubi kristalnu vodu).
5.3 Koncentrirana Biggs/Szijartova otopina (2)
Otopite 9,10 g cinkovog acetata dihidrata (Zn(CH3COO)2.2H2O) i 5,46 g fosfovolframove kisline monohidrata (H3[P(W3O10)4.xH2O]) u približno 70 ml vode, HPLC čistoće (6.8.), u odmjernoj tikvici od 100 ml.
Dodajte 5,81 ml ledene octene kiseline (CH3COOH). Do oznake 100 ml dopunite vodom HPLC čistoće (6.8.) i promiješajte. Otopina se može čuvati na sobnoj temperaturi godinu dana.
5.4. Razrijeđena Biggs/Szijartova otopina
U odmjernoj tikvici razrijedite 25 ml koncentrirane Biggs/Szijartove otopine (5.3.) na 500 ml. Otopina se može čuvati na sobnoj temperaturi mjesec dana.
5.5. Priprema vode HPLC čistoće
Ultra čistu vodu (6.8.) filtrirajte pomoću vakuumskog sustava (6.9.). Za poboljšanje rada pumpe i postizanje stabilne bazne linije svaki dan otplinjavajte mobilnu fazu, pri čemu treba odabrati jednu od raspoloživih tehnika, kao npr. prskanje helijem, sonikacija, primjena vakuumskog ili linijskog otplinjivača.
Napomena: Za produženje vijeka kolone bitno je da sadržaj ugljičnog dioksida u eluensu bude što niži i da se spriječi njegov ponovni unos.
6. OPREMA
Uobičajena laboratorijska oprema i, posebno, sljedeće:
6.1. Kolona HPLC sa smolom za ionsku izmjenu
Punjenje kolone: 8 % križno vezanog polistiren-divinilbenzen kopolimera, na koji su vezane grupe za kationsku izmjenu u obliku olova.
Diminzije kolone: duljina 300 mm, unutarnji promjer oko 8 mm.
Mogu se koristiti i drugi promjeri, pod uvjetom da se prema njima podesi brzina protoka.
6.2. Zaštitna kolona
Zaštitna kolona je kombinacija zasebnih kationskih izmjenjivača (H+) i anionskih izmjenjivača (CO3–), koji su punjeni u kolonu od oko 30 mm × 4,6 mm (L × ID) (tj. mikrozaštitne kolone u mikrozaštitnom nosaču), povezani u nizu ili pomiješani te se sastoje od AG 50W-X4, -400 mesh (H+) i AG3-X4A, 200–400 mesh (OH–) u omjeru 35:65 (m/m), ručno punjeni u kolonu od oko 20 × 9 mm (L × ID).
6.3. Grijač kolone
Grijač koji može održavati konstantnu temperaturu od 85 °C ± 1 °C.
6.4. HPLC pumpa
Pumpa koja može proizvesti konstantnu brzinu protoka (< 0,5 % kolebanja) kod 0,2-1,0 m/min.
6.5. HPLC naprava za ubrizgavanje
Automatska naprava za uzorkovanje koja može ubrizgati 25 μL uz ponovljivost od < 0,5 %.
Alternativno se može koristiti ručna naprava (isti zahtjevi kao i za automatsku napravu za uzorkovanje).
6.6. HPLC detektor
Visoko osjetljivi detektor za refrakcijski indeks, sa šumom < 5,10–9 RI jedinica.
6.7. Integrator
Softver ili integrator za prikupljanje i obradu podataka te prikazivanje površina i visina pikova, koji se mogu pretvoriti u koncentracije laktoze.
6.8. Uređaj za pročišćavanje vode
Sustav koji može osigurati ultra čistu vodu (tipa 1), specifičnog otpora >14 MΩ.cm.
6.9. Uređaj za filtriranje otopina
Sustav koji omogućuje filtraciju vode pomoću membranskog filtra s veličinom pora 0,45 μm.
Napomena: Mnoge jedinice za pročišćavanje vode (6.8.) imaju ugrađene filtre s veličinom pora 0,45 ili 0,2 μm. Dodatna filtracija se može izbjeći ako se voda koristi izravno.
6.10. Analitička vaga
Vaga s mogućnošću očitanja od 0,1 mg.
6.11. Vodena kupelj
Vodena kupelj koja može održavati temperaturu na 40 °C (±0,5).
6.12. Centrifuga
Centrifuga koja razvija silu od najmanje 3 000 g za Eppendorfove bočice ili za ekvivalentne ili veće bočice.
6.13. Odmjerna tikvica od 50 ml
Zapremina 50 ml, klasa A.
Napomena: Mogu se koristiti tikvice drugih zapremina uzimajući u obzir faktor volumena.
6.14. Odmjerna tikvica od 100 ml
Zapremina 100 ml, klasa A.
6.15. Graduirana pipeta
Graduirana pipeta od 10 ml.
Napomena: Alternativno se može koristiti ručni pipetor zapremine 5 ml, pri čemu se količina od 5 ml reagensa dodaje dvaput (5.3.).
7. UZORKOVANJE
Važno je da laboratorij dobije uzorak uzet u skladu sa standardom ISO 707/IDF 50 (3) koji je zaista reprezentativan i nije oštećen tijekom prijevoza ili skladištenja.
8. PRIPREMA STANDARDNE OTOPINE LAKTOZE
8.1. Standard 1.
U odmjernoj tikvici od 100 ml (6.14.) otopite točno odvagnutu količinu (mogućnost očitanja 0,1 mg) od oko 50 mg monohidrata laktoze (5.2.) i do oznake dopunite vodom.
8.2. Standard 2.
U odmjernoj tikvici od 100 ml (6.14.) otopite točno odvagnutu količinu (mogućnost očitanja 0,1 mg) od oko 100 mg monohidrata laktoze (5.2.) i do oznake dopunite vodom.
Napomena: Standardne otopine mogu se čuvati maksimalno tjedan dana na oko 5 °C.
9. PRIPREMA ISPITNOG UZORKA
9.1. Rekonstitucija uzorka
Odvagnite oko 5 g praha u tikvicu od 50 ml (6.13.) i zabilježite masu s točnošću od 1 mg (W1, (11)). Dodajte 50 ml vode i zabilježite povećanje mase (W2 (11)) s točnošću od 0,01 g. Zatvorenu tikvicu stavite u vodenu kupelj (6.11.) na 30 minuta i tijekom tog razdoblja nekoliko je puta preokrenite. Nakon toga ostavite je da se ohladi na sobnu temperaturu.
9.2. Obrada uzorka
Uzmite oko 1 g te otopine i stavite je u odmjernu tikvicu od 50 ml (6.13.), zabilježite masu s točnošću od 1 mg (W3 (11)), dodajte 20 ml vode, zatim dodajte 10 ml razrijeđenog reagensa Biggs/Szijarto (5.4.) i do oznake dopunite vodom. Tijekom prvih 30 minuta 5 puta lagano preokrenite tikvicu.
Nakon jednog sata uzmite alikvot otopine i centrifugirajte (6.12.) 10 min na 3 000 g (za više g vrijeme se skraćuje u odgovarajućem omjeru). Za HPLC analizu uzmite alikvot supernatanta.
10. ODREĐIVANJE HPLC METODOM
10.1 Prethodna priprema HPLC-a
10.1.1 Postavljanje kolone i pretkolone
Postavite pretkolonu (6.2.) izvan grijača kolone (6.3.), a kolonu (6.1.) u grijač.
Napomena: Ako grijač ne sadrži cijevi za zagrijavanje eluensa, eluens mora proći kroz oko 15 cm dugu cijev od nehrđajućeg čelika u grijaču prije nego što uđe u kolonu (apsolutno je potrebno da se eluens zagrije prije ulaska u kolonu jer će inače doći do širenja pika).
10.1.2. Detektor i početni protok
Da bi se dobila stabilna bazna linija, detektor (6.6.) treba uključiti najmanje 24 sata prije početka analize. Unutarnju temperaturu detektora podesite na 35 °C. Podesite brzinu protoka na 0,2 ml/min (6.4.) i ostavite je najmanje 20 min dok je grijač kolone (6.3.) podešen na sobnu temperaturu.
10.1.3. Grijač kolone i konačna brzina protoka
Grijač kolone (6.3.) podesite na 85 °C. Kad se postigne ta temperatura, nakon 30 minuta postupno povećavajte brzinu protoka od 0,2 ml/min do 0,6 ml/min (6.4.). Pričekajte 2 sata da se sustav uravnoteži s tom brzinom protoka na 85 °C ili dok se ne postigne stabilna bazna linija.
10.1.4. Integracija
Pažljivo odaberite parametre za prikupljanje i integraciju parametara (6.7.) kao što su brzina prijenosa podataka, osjetljivost, vremenska konstanta, širina pika i prag.
Vrijeme zadržavanja laktoze je oko 11 min.
Napomena: Mnogi softveri za prikupljanje podataka (6.7.) omogućuju jednostavno mjerenje teoretskog ukupnog broja mikroorganizama. Teoretski ukupni broj standarda 1. (8.1.) treba redovito mjeriti i kolonu (6.1.) zamijeniti kad je ukupni broj mikroorganizama niži za 25 % od početne vrijednosti nove kolone.
10.1.5. Ispitivanje zaštitne kolone
Redovito provjeravajte (najmanje jednom u svakom slijedu) sposobnost zaštitne kolone (6.2.) da eliminira soli iz uzorka, ubrizgavanjem 25 μl 0,05 %-tne otopine natrijevog klorida. Kad god se pojavljuju pikovi, zaštitnu kolonu treba zamijeniti.
10.2. Primjena standarda
Na početku svake serije analiza ubrizgati 25 μl (6.5.) standarda 1. (8.1.), a potom i standarda 2. (8.2). Postupak ponavljati nakon svakih 10 do 20 uzoraka, kao i na kraju serije.
10.3. Primjena uzoraka
Ubrizgati 25 μl supernatanta (9.2.) uzorka.
11. IZRAČUNAVANJE I IZRAŽAVANJE REZULTATA
11.1. Kalibracija
Obično se za izračunavanje rezultata koriste visine pikova, međutim, ako signal sadrži previše smetnji, može se koristiti površina pikova (na utvrđivanje količine prema visini pikova manje utječu pikovi komponenti nižih koncentracija koji su djelomično, ali nedovoljno odvojeni od pika laktoze).
Softver (6.7.) treba izračunati linearnu kalibracijsku krivulju koja prolazi kroz ishodište. Provjerite eventualnu nelinearnost krivulje (očita nelinearnost vjerojatno je posljedica greške u pripremi standarda 1. (8.1.) ili 2. (8.2.), loše integracije ili, manje vjerojatno, uporabe neispravnog injektora).
Kao ulazne podatke koristite izračunane koncentracije laktoze u mg/ml standarda 1. (8.1.) i 2. (8.2.) kao bezvodne laktoze.
Kosinu (RF) kalibracijske linije definira površina/koncentracija u mg/ml.
11.2. Uzorci
Rezultat analize izražava se kao g/100 g i izračunava pomoću softvera (6.7.) ili prema sljedećoj formuli:
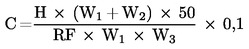
gdje je:
|
C |
: |
koncentracija laktoze u g/100 g praha; |
|
H |
: |
visina pika uzorka laktoze; |
|
RF |
: |
factor odziva (ili kosine) kalibracijskoga grafa u mV/mg/ml; |
|
W1 |
: |
masa uzorka u prahu u g (9.1.); |
|
W2 |
: |
masa vode dodane uzorku u prahu u g (9.1.); |
|
W3 |
: |
masa uzorka rekonstituirane otopine praha u g (9.2.); |
|
50 |
: |
zapremina odmjerne tikvice uporabljene u (9.2.); |
|
0,1 |
: |
konverzija rezultata u g/100 g. |
12. PRECIZNOST
Vrijednosti izvedene iz ovog međulaboratorijskog ispitivanja možda neće biti primjenjive na raspone koncentracija i matrice koji se razlikuju od zadanih. Vrijednosti za ponovljivost i obnovljivost bit će izvedene iz rezultata međulaboratorijskog ispitivanja provedenog u skladu sa standardom ISO 5725 (4).
12.1. Ponovljivost
Apsolutna razlika između rezultata dvaju nezavisnih pojedinačnih ispitivanja koja je primjenom iste metode na jednakom ispitnom materijalu u istom laboratoriju u kratkom vremenskom intervalu izvršio isti analitčar koristeći istu opremu ne smije u više od 5 % slučajeva biti veća od xxx (vrijednost se utvrđuje međulaboratorijskim ispitivanjem) (5).
12.2. Obnovljivost
Apsolutna razlika između rezultata dvaju nezavisnih pojedinačnih ispitivanja koja su primjenom iste metode na jednakom ispitnom materijalu u različitim laboratorijima izvršili različiti analitičari koristeći različitu opremu ne smije u više od 5 % slučajeva biti veća od 0,5 g/100g (vrijednost se utvrđuje međulaboratorijskim ispitivanjem).
13. REFERENTNA LITERATURA
(1) J. Koops en C. Olieman, Netherlands Milk and Dairy Journal, 39 (1985) 89.-106.
(2) D. A. Biggs en L. Szijarto, Journal of Dairy Science, 46 (1963) 1196.
(3) ISO 707 (IDF 50), Milk and milk products — Methods of sampling (Mlijeko i mliječni proizvodi – Metode uzorkovanja)
(4) ISO 5725-1, Accuracy (trueness and precision) of measurement methods and results. Part 1: General principles and definitions. (Točnost (istinitost i preciznost) mjernih metoda i rezultata. 1. dio: Opća načela i definicije)
(5) ISO 5725-2, Accuracy (trueness and precision) of measurement methods and results. Part 2: A basic method for the determination of repeatability and reproducibility of a standard measurement method. (Točnost (istinitost i preciznost) mjernih metoda i rezultata. 2. dio: Osnovna metoda određivanja ponovljivosti i obnovljivosti standardne mjerne metode)
PRILOG XII.
(Članak 9.)
DETEKCIJA SLATKE SIRUTKE U OBRANOM MLIJEKU U PRAHU ZA JAVNO SKLADIŠTENJE ODREĐIVANJEM SADRŽAJA KAZEINO-MAKROPEPTIDA METODOM TEKUĆINSKE KROMATOGRAFIJE VISOKE DJELOTVORNOSTI (HPLC)
1. OPSEG I PODRUČJE PRIMJENE
Ova metoda omogućuje detekciju slatke sirutke u obranom mlijeku u prahu za javno skladištenje određivanjem sadržaja kazeino-makropeptida.
2. REFERENCA
Međunarodni standard ISO 707 – „Mlijeko i mliječni proizvodi – Metode uzorkovanja”, u skladu sa smjernicama sadržanim u posljednjoj alineji točke (c) stavka 2. Priloga I.
3. DEFINICIJA
Sadržaj suhe tvari u slatkoj sirutki definira se kao maseni postotak kazeino-makropeptida prema opisanom postupku.
4. NAČELO
|
— |
Rekonstitucija obranog mlijeka u prahu, uklanjanje masti i bjelančevina pomoću triklorooctene kiseline, iza čega slijedi centrifugiranje ili filtracija. |
|
— |
Utvrđivanje količine kazeino-makropeptida (CMP) u supernatantu metodom tekućinske kromatografije visoke učinkovitosti (HPLC). |
|
— |
Ocjenjivanje rezultata dobivenih za uzorke na temelju usporedbe sa standardnim uzorcima koji se sastoje od obranog mlijeka u prahu sa ili bez dodatka poznatog postotka sirutke u prahu. |
5. REAGENSI
Svi korišteni reagensi moraju biti priznate analitičke čistoće. Mora se koristiti destilirana voda ili voda ekvivalentne čistoće.
5.1. Otopina triklorooctene kiseline
Otopite 240 g triklorooctene kiseline (CCl3CCOOH) u vodi i dopunite do 1 000 ml. Otopina treba biti bistra i bezbojna.
5.2. Otopina eluensa, pH 6,0
Otopite 1,74 g dikalijevog hidrogen fosfata (K2HPO4), 12,37 g kalijevog dihidrogen fosfata, (KH2PO4) i 21,41 g natrijevog sulfata (Na2SO4) u oko 700 ml vode. Prema potrebi podesiti pH na 6,0 pomoću otopine fosforne kiseline ili kalijevog hidroksida.
Dopunite vodom do 1 000 ml i homogenizirajte.
Napomena: Sastav eluensa može se prilagoditi tako da bude u skladu s potvrdom o sukladnosti sa standardima ili preporukama proizvođača materijala za punjenje kolona.
Prije uporabe profiltrirajte otopinu eluensa kroz membranski filtar s porama promjera 0,45 μm.
5.3. Otopina za ispiranje
Pomiješajte jedan volumen acetonitrila (CH3CN) i devet volumena vode. Prije uporabe profiltrirajte mješavinu kroz membranski filtar s porama promjera 0,45 μm.
Napomena: Može se koristiti bilo koja druga baktericidna otopina za ispiranje koja ne utječe na djelotvornost kolone u smislu rezolucije.
5.4. Standardni uzorci
5.4.1. Obrano mlijeko u prahu koje ispunjava uvjete te rezolucije (tj. [0]).
5.4.2. Isto obrano mlijeko u prahu kojem je dodano 5 % (m/m) slatke sirutke u prahu standardnog sastava (tj. [5]).
6. OPREMA
6.1. Analitička vaga.
6.2. Centrifuga koja može postići centrifugalnu silu od 2 200 g, s epruvetama za centrifugiranje, s čepom ili navojnim čepom, zapremine oko 50 ml.
6.3. Mehanička tresilica.
6.4. Magnetska miješalica.
6.5. Stakleni lijevci promjera oko 7 cm.
6.6. Filtar-papiri, srednje brzine filtracije, promjera oko 12,5 cm.
6.7. Staklena oprema za filtriranje s membranskim filtrom s porama promjera 0,45 μm.
6.8. Graduirane pipete od 10 ml (ISO 648, Klasa A ili ISO/R 835) ili dozirni sustav kapaciteta dobave 10,0 ml u dvije minute.
6.9. Dozirni sustav kapaciteta dobave 20,0 ml vode zagrijane na oko 50 °C.
6.10. Vodena kupelj s termostatskom regulacijom, podešena na 25 ± 0,5 °C.
Oprema za HPLC koja se sastoji od:
6.11.1. Pumpe;
6.11.2. Injektora, ručnog ili automatskog, zapremine 15 do 30 μl;
6.11.3. Dvije kolone u nizu (duljine 30 cm, unutarnjeg promjera 0,75 cm) ili ekvivalentnih kolona (npr. jednostruka TSK 2 000-SWxl, jednostruka Agilent Technologies Zorbax GF 250) i pretkolone (3 cm × 0,3 cm) napunjene s I 125 ili materijalom jednake djelotvornosti;
6.11.4. Grijača kolone s termostatom podešenim na 35 ± 1 °C;
6.11.5. UV detektora prilagodljive valne duljine koji omogućuje mjerenja na 205 nm uz osjetljivost od 0,008 Å;
6.11.6. Integratora s mogućnošću integriranja dolina-dolina.
Napomena: Rad s kolonama na sobnoj temperaturi je moguć, iako je u tim uvjetima njihova rezolucija niža. U tom slučaju temperatura ne smije varirati za više od ± 5 °C u bilo kojem rasponu analiza.
7. UZORKOVANJE
7.1. Uzorke obvezno treba uzimati u skladu s postupkom utvrđenim u međunarodnom standardu ISO 707. Međutim, države članice mogu primijeniti neku drugu metodu uzorkovanja pod uvjetom da je ona u skladu s načelima gore spomenutog standarda.
7.2. Uzorke čuvajte u uvjetima koji onemogućavaju kvarenje ili promjene u njihovom sastavu.
8. POSTUPAK
8.1. Priprema ispitnog uzorka
Stavite mlijeko u prahu u posudu dvostruko većeg volumena od volumena praha, koja ima hermetički poklopac. Posudu odmah zatvorite. Mlijeko u prahu dobro promiješajte uzastopnim preokretanjem posude.
8.2. Uzorak za analizu
Odvagnite 2,000 + 0,001 g ispitnog uzorka u epruvetu za centrifugiranje (6.2.) ili u odgovarajuću tikvicu s čepom (50 ml).
8.3. Odstranjivanje masti i bjelančevina
8.3.1. Uzorku za analizu dodajte 20,0 ml zagrijane vode (50 °C). Otopite prah protresajući ga pet minuta pomoću mehaničke tresilice (6.3.). Epruvetu stavite u vodenu kupelj (6.10.) i pustite da se temperatura uravnoteži na 25 °C.
8.3.2. U dvije minute dodajte 10,0 ml otopine triklorooctene kiseline (5.1.) uz snažno miješanje magnetskom miješalicom (6.4.). Epruvetu stavite u vodenu kupelj (6.9.) i ostavite 60 minuta.
8.3.3. Centrifugirajte (6.2.) 10 minuta na 2 200 g ili filtrirajte kroz filtar-papir (6.6.), pri čemu prvih 5 ml filtrata bacite.
8.4. Kromatografsko određivanje
8.4.1. Ubrizgajte 15 do 30 μl točno izmjerenog supernatanta ili filtrata (8.3.3.) u uređaj za HPLC (6.10.), koji radi uz brzinu protoka od 1,0 ml otopine eluensa (5.2.) u minuti.
Napomena 1.: Mogu se koristiti različite brzine protoka, ovisno o unutarnjem promjeru kolona koje se koriste ili o uputama proizvođača kolona.
Napomena 2.: Održavajte temperaturu otopine eluensa (5.2.) na 85 °C tijekom čitave kromatografske analize da bi eluens ostao otplinjen te radi sprečavanja rasta bakterija. Može se primijeniti bilo koja druga mjera opreza s istim učinkom.
Napomena 3.: Kod svakog prekida kolone isperite vodom. Nikada u njima ne ostavljajte otopinu eluensa (5.2.).
Prije svakog prekida koji traje više od 24 sata isperite kolone vodom i zatim ih najmanje tri sata perite otopinom (5.3.) uz brzinu protoka od 0,2 ml u minuti.
8.4.2. Rezultati kromatografske analize ispitnog uzorka [E] dobivaju se u obliku kromatograma kod kojeg se pikovi identificiraju prema vremenu zadržavanja (RT) kako slijedi:
|
Pik II.: |
Drugi pik kromatograma s vremenom zadržavanja (RT) od oko 12,5 minuta. |
|
Pik III.: |
Treći pik kromatograma koji odgovara CMP-u, s vremenom zadržavanja (RT) od 15,5 minuta. |
Odabir kolone ili kolona može bitno utjecati na vrijeme zadržavanja pojedinačnih pikova.
Integrator (6.11.6.) automatski izračunava površinu A svakog pika:
|
A II.: |
Površina pika II. |
|
A III.: |
Površina pika III. |
Važno je prije kvantitativne interpretacije analizirati izgled svakog kromatograma radi otkrivanja bilo kakvih nepravilnosti nastalih zbog neispravnog rada uređaja ili kolona, ili zbog podrijetla i vrste analiziranog uzorka.
U slučaju dvojbe, ponovite analizu.
8.5. Kalibracija
8.5.1. Na standardne uzorke (5.4.) točno primijenite postupak opisan od točke 8.2. do točke 8.4.2.
Koristite svježe pripremljene otopine budući da se u 8-postotnoj triklorooctenoj kiselini CMP razgrađuje. Gubitak se procjenjuje na 0,2 % na sat na temperaturi od 30 °C.
8.5.2. Prije kromatografske analize uzoraka pripremite kolone ubrizgavanjem standardnog uzorka (5.4.2.) u otopinu (8.5.1.), ponavljajući postupak sve dok se površina i vrijeme zadržavanja pika koji odgovara CMP-u ne ustale.
8.5.3. Faktore odziva R utvrdite ubrizgavanjem količine filtrata (8.5.1.) koja je jednaka količini uporabljenoj za uzorke.
9. IZRAŽAVANJE REZULTATA
9.1. Metoda za izračun i formule
9.1.1. Izračun faktora odziva R:
|
Pik II.: |
RII = 100/(AII[0]) |
gdje je:
|
RII |
= |
faktori odziva pikova II.; |
|
AII [0] |
= |
površine pikova II. standardnog uzorka [0], dobivene u 8.5.3. |
|
Pik III.: |
RIII = W/(AIII[5]) – AIII[0]) |
gdje je:
|
RIII |
= |
faktor odziva pika III.; |
|
AIII [0] i AIII [5] |
= |
površine pika III. standardnih uzoraka [0] i [5], dobivene u 8.5.3.; |
|
W |
= |
količina sirutke u standardnom uzorku [5], tj. 5. |
9.1.2. Izračun relativne površine pikova u uzorku [E]
SII[E] = RII × AII[E]
SIII[E] = RIII × AIII[E]
SIV[E] = RIV × AIV[E]
gdje je:
|
SII [E], SIII [E], SIV [E] |
= |
relativne površine pikova II., III. odnosno IV. u uzorku [E]; |
|
AII [E], AIII [E] |
= |
površine pikova II. odnosno III. u uzorku [E] dobivene u 8.4.2.; |
|
RII, RIII |
= |
faktori odziva izračunani u 9.1.1. |
9.1.3. Izračun relativnih vremena zadržavanja pika III. u uzorku [E]: RRTIII[E] = (RTIII[E])/(RTIII[5])
gdje je:
|
RRTIII [E] |
= |
relativno vrijeme zadržavanja pika III. u uzorku [E]; |
|
RTIII [E] |
= |
vrijeme zadržavanja pika III. u uzorku [E] dobiveno u 8.4.2.; |
|
RTIII [5] |
= |
vrijeme zadržavanja pika III. u kontrolnom uzorku [5] dobiveno u 8.5.3. |
9.1.4. Ispitivanja su pokazala linearnu povezanost relativnog vremena zadržavanja pika III., odnosno RRTIII [E], i postotka sirutke u prahu dodane do 10 %.
|
— |
RRTIII [E] je < 1,000 ako je sadržaj sirutke > 5 %; |
|
— |
RRTIII [E] je ≥ 1,000 ako je sadržaj sirutke ≤ 5 %. |
Dopuštena nepouzdanost za vrijednosti RRTIII iznosi ± 0,002.
U normalnim okolnostima vrijednost RRTIII [0] ne odstupa mnogo od 1,034. Ovisno o stanju kolona, navedena se vrijednost može približiti 1,000, ali uvijek mora biti viša.
9.2. Računanje postotka slatke sirutke u prahu u uzorku:
W = SIII[E] – [1, 3 + (SIII[0] – 0, 9)]
gdje je:
|
W |
= |
maseni postotak (m/m) slatke sirutke u uzorku [E]; |
|
SIII [E] |
= |
relativna površina pika III. ispitnog uzorka [E] dobivena kao u 9.1.2.; |
|
1,3 |
= |
predstavlja relativnu prosječnu površinu pika III. izraženu u gramima slatke sirutke na 100 g, utvrđenu u nepatvorenom obranom mlijeku u prahu različitog podrijetla. Ova je vrijednost dobivena eksperimentalno; |
|
SIII [0] |
= |
predstavlja relativnu površinu pika III. koja je jednaka RIII × AIII [0]. Ove su vrijednosti dobivene u 9.1.1. odnosno 8.5.3.; |
|
(SIII [0] – 0,9) |
= |
predstavlja korekciju relativne prosječne površine 1,3 ako SIII [0] ne iznosi 0,9. Eksperimentalno dobivena vrijednost relativne prosječne površine pika III. kontrolnog uzorka [0] iznosi 0,9. |
9.3. Preciznost postupka
9.3.1. Ponovljivost
Razlika između rezultata dvaju određivanja koja je izvršio isti analitičar, istovremeno ili u brzom slijedu, koristeći istu opremu na jednakom ispitnom materijalu ne smije biti veća od 0,2 % m/m.
9.3.2. Obnovljivost
Razlika između dva zasebna i neovisna rezultata dobivena u dva različita laboratorija na jednakom ispitnom materijalu ne smije biti veća od 0,4 % (m/m).
9.4. Tumačenje
9.4.1. Pretpostavlja se odsutnost sirutke ako je relativna površina pika III., SIII [E], izražena u gramima slatke sirutke na 100 g proizvoda, ≤ 2,0 + (SIII [0] – 0,9) gdje je:
|
2,0 |
= |
maksimalna vrijednost dopuštena za relativnu površinu pika III., uzimajući u obzir relativnu površinu pika III., tj. 1,3, nesigurnost zbog promjena u sastavu obranog mlijeka u prahu i obnovljivost metode (9.3.2.); |
|
(SIII [0] – 0,9) |
= |
korekcija koju treba napraviti ako je površina SIII [0] različita od 0,9 (vidjeti točku 9.2.). |
9.4.2. Ako je relativna površina pika III., SIII [E] > 2,0 + (SIII [0] – 0,9), a relativna površina pika II., SII [E] ≤ 160, utvrdite sadržaj slatke sirutke kako je navedeno u točki 9.2.
Ako je relativna površina pika III., SIII [E] > 2,0 + (SIII [0] – 0,9), a relativna površina pika II., SII [E] ≤ 160, utvrdite ukupni sadržaj bjelančevina (P %) te potom analizirajte dijagrame 1. i 2.
9.4.3.1. Podaci dobiveni analizom uzoraka nepatvorenog obranog mlijeka u prahu s visokim ukupnim sadržajem bjelančevina sabrani su u dijagramima 1. i 2.
Puna linija predstavlja lineranu regresiju za koju se koeficijenti računaju metodom najmanjih kvadrata.
Isprekidana ravna linija označava gornju graničnu vrijednost relativne površine pika III., uz vjerojatnost da u 90 % slučajeva nije prekoračena.
Jednadžbe isprekidanih ravnih linija u dijagramima 1. i 2. jesu sljedeće:
|
SIII = 0,376 P % – 10,7 |
(dijagram 1.) |
|
SIII = 0,0123 SII [E] + 0,93 |
(dijagram 2.) |
gdje je:
|
SIII |
= |
relativna površina pika III. izračunana ili prema ukupnom sadržaju bjelančevina ili prema relativnoj površini pika SII [E]; |
|
P % |
= |
ukupni sadržaj proteina izražen kao maseni postotak; |
|
SII [E] |
= |
relativna površina uzorka izračunana u točki 9.1.2. |
Ove su jednadžbe ekvivalentne vrijednosti 1,3 spomenutoj u točki 9.2.
Razlika (T1 i T2) između relativne površine SIII [E] i relativne površine SIII prikazana je sljedećom formulom: T1 = SIII[E] – [(0,376 P % – 10,7) + (SIII[0] – 0,9)]T2 = SIII[E] – [(0,0123 SII[E] + 0,93) + (SIII[0] – 0,9)].
|
Ako su T1 i/ili T2 |
jednaki ili manji od nule, prisutnost slatke sirutke nije moguće utvrditi. |
|
Ako su T1 i T2 |
veći od nule, slatka sirutka je prisutna. |
Sadržaj slatke sirutke računa se prema sljedećoj formuli: W = T2 + 0,91
gdje je:
0,91 udaljenost na okomitoj osi između pune i isprekidane ravne linije.
Obrano mlijeko u prahu
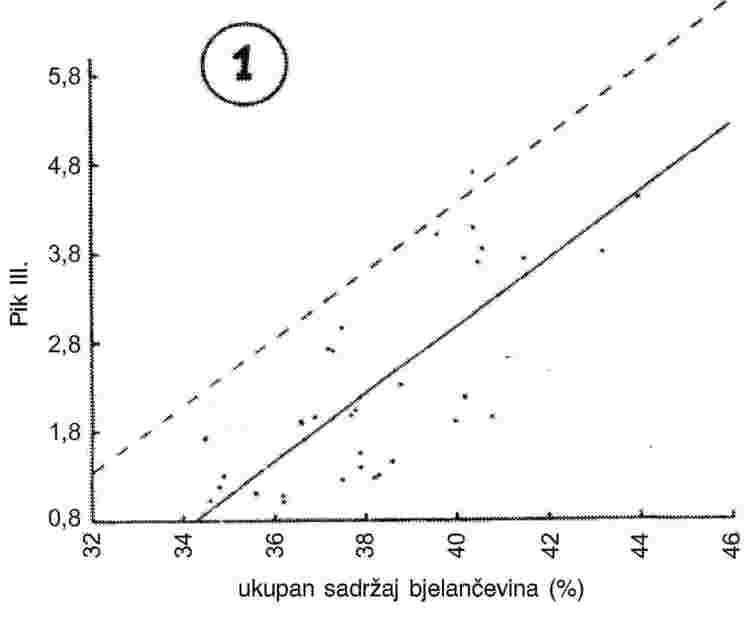
Obrano mlijeko u prahu
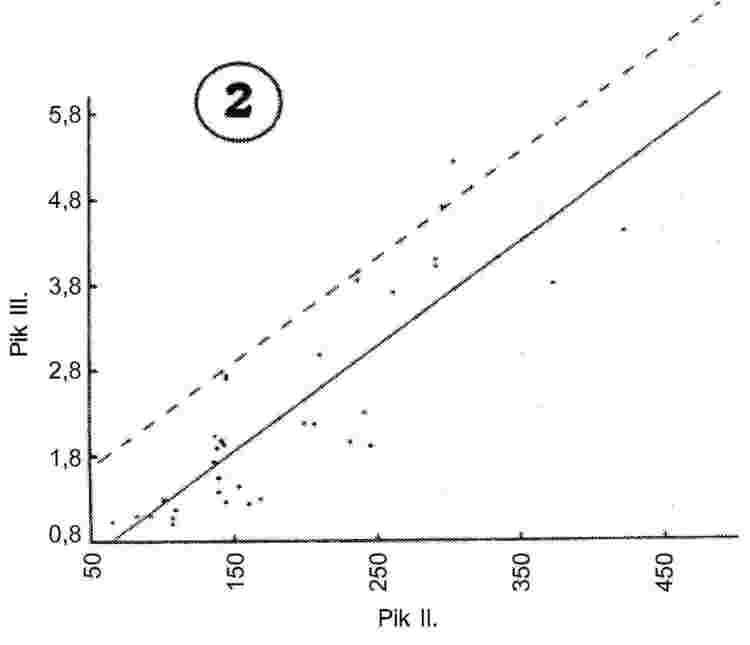
PRILOG XIII.
(Članak 9.)
ODREĐIVANJE SUHE TVARI SLATKE SIRUTKE U OBRANOM MLIJEKU U PRAHU I SMJESAMA IZ UREDBE (EZ) br. 2799/1999
1. SVRHA: DETEKCIJA DODANE SLATKE SIRUTKE U:
|
(a) |
obranom mlijeku u prahu, kako je definirano člankom 2. Uredbe (EZ) br. 2799/1999; i |
|
(b) |
smjesama, kako su definirane člankom 4. Uredbe (EZ) br. 2799/1999. |
2. REFERENCA: MEĐUNARODNI STANDARD ISO 707
3. DEFINICIJA
Udio suhe tvari slatke sirutke definira se kao maseni postotak kazeino-makropeptida prema opisanom postupku.
4. NAČELO
Udio kazeino-makropeptida određuje se u skladu s Prilogom XII. Uzorci koji pokažu pozitivne rezultate analiziraju se na prisutnost kazeino-makropeptida A metodom reverzno-fazne tekućinske kromatografije visoke djelotvornosti (HPLC postupak). Alternativno se uzorci odmah analiziraju reverzno-faznim HPLC postupkom. Dobiveni se rezultat ocjenjuje uspoređivanjem sa standardnim uzorcima koji se sastoje od obranog mlijeka u prahu sa i bez dodatka poznatog postotka sirutke u prahu. Rezultati iznad 1 % (m/m) ukazuju na prisutnost suhe tvari slatke sirutke.
5. REAGENSI
Svi reagensi moraju biti priznate analitičke čistoće. Mora se koristiti destilirana voda ili voda barem jednake čistoće. Acetonitril treba biti spektroskopske ili HPLC čistoće.
Reagensi koji su potrebni za postupak opisani su u Prilogu XII. ove Uredbe.
Reagensi za reverzno-fazni HPLC.
5.1. Otopina triklorooctene kiseline
Otopite 240 g triklorooctene kiseline (CCl3CCOOH) u vodi i dopunite do 1 000 ml. Otopina mora biti bistra i bezbojna.
5.2. Eluensi A i B
Eluens A: U odmjernu tikvicu od 1 000 ml stavite 150 ml acetonitrila (CH3CN), 20 ml izopropanola (CH3CHOHCH3) i 1,00 ml trifluorooctene kiseline (TFA, CF3COOH). Dopunite vodom do 1 000 ml.
Eluens B: U odmjernu tikvicu od 1 000 ml stavi se 550 ml acetonitrila, 20 ml izopropanola i 1,00 ml TFA. Dopunite vodom do 1 000 ml. Otopinu eulensa prije uporabe profiltrirajte kroz membranski filtar s promjerom pora od 0,45 μm.
5.3. Konzerviranje kolone
Nakon analize kolona se najprije ispere eluensom B (gradijentno ispiranje), a potom i acetonitrilom (gradijentno ispiranje u trajanju od 30 minuta). Kolona se čuva u acetonitrilu.
5.4. Standardni uzorci
5.4.1. Obrano mlijeko u prahu koje ispunjava zahtjeve za javno skladištenje (tj. [0]).
5.4.2. Isto mlijeko u prahu s dodatkom 5 % (m/m) slatke sirutke u prahu standardnog sastava (tj. [5]).
5.4.3. Isto mlijeko u prahu s dodatkom 50 % (m/m) slatke sirutke u prahu standardnog sastava (tj. [5]) (1).
6. OPREMA
Oprema potrebna za opisani postupak navedena je u Prilogu XII. ove Uredbe.
6.1. Analitička vaga
6.2. Centrifuga, neobvezna, koja može postići centrifugalnu silu od 2 200 g, opremljena epruvetama za centrifugiranje s čepom ili navojnim čepom, zapremine oko 50 ml.
6.3. Mehanička tresilica.
6.4. Magnetska miješalica.
6.5. Stakleni lijevci promjera oko 7 cm.
6.6. Filtar-papiri za srednju brzinu filtracije, promjera oko 12,5 cm.
6.7. Staklena oprema za filtriranje s membranskim filtrom s promjerom pora 0,45 μm.
6.8. Graduirane pipete koje omogućuju prijenos 10 ml (ISO 648, Klasa A ili ISO/R 835) ili dozirni sustav koji omogućuje unos 10,0 ml u dvije minute.
6.9. Dozirni sustav koji omogućuje unos 20,0 ml vode na oko 50 °C.
6.10. Vodena kupelj s termostatom, zagrijana na 25 ± 0,5 °C.
Oprema za HPLC koja se sastoji od:
6.11.1. Pumpnog sustava s binarnim gradijentom.
6.11.2. Injektora, ručnog ili automatskog, zapremine 100 μl.
6.11.3. Kolone Agilent Technologies Zorbax 300 SB-C3 (duljine 25 cm, unutarnjeg promjera 0,46 cm) ili ekvivalentne reverzno-fazne kolone sa širokim porama na bazi silicijevog dioksida.
6.11.4. Grijača kolone s termostatom podešenog na 35 ± 1 °C.
6.11.5. UV detektora promjenjive valne duljine koji omogućuje mjerenja na 210 nm (prema potrebi se mogu koristiti i veće valne duljine do 220 nm), osjetljivosti od 0,02 Å.
6.11.6. Integratora koji omogućuje integraciju na zajedničkoj baznoj liniji ili na dva minimuma („dolina–dolina”).
Napomena: Kolona se može koristiti na sobnoj temperaturi, uz uvjet da temperatura u prostoriji ne varira za više od 1 °C; u protivnom dolazi do prevelikog variranja vremena zadržavanja CMPA.
7. UZORKOVANJE
7.1. Uzorke obvezno treba uzimati u skladu s postupkom utvrđenim u međunarodnom standardu ISO 707. Međutim, države članice mogu primijeniti neku drugu metodu uzorkovanja pod uvjetom da je ona u skladu s načelima gore spomenutog standarda.
7.2. Uzorke čuvajte u uvjetima koji onemogućavaju kvarenje ili promjene u njihovom sastavu.
8. POSTUPAK
8.1. Priprema ispitnog uzorka
Stavite mlijeko u prahu u posudu dvostruko većeg volumena od volumena praha, koja ima hermetički poklopac. Posudu odmah zatvorite. Mlijeko u prahu dobro promiješajte višekratnim preokretanjem posude.
8.2. Uzorak za analizu
Odvagnite 2,00 + 0,001 g ispitnog uzorka u epruvetu za centrifugiranje (6.2.) ili u odgovarajuću tikvicu s čepom (50 ml).
Napomena: Kad se radi o smjesi, odvagnite takvu količinu ispitnog uzorka da odmašćeni uzorak za analizu iznosi 2,00 g.
8.3. Odstranjivanje masti i bjelančevina
8.3.1. Uzorku za analizu dodajte 20,0 ml zagrijane vode (50 °C). Prah otopite tresenjem pomoću mehaničke tresilice pet minuta, odnosno 30 minuta ako se radi o kiseloj mlaćenici (6.3.). Epruvetu stavite u vodenu kupelj (6.10.) i pustite da se temperatura uravnoteži na 25 °C.
8.3.2. Dodajte 10,0 ml otopine triklorooctene kiseline zagrijane na oko 25 °C (5.1.), postupno u dvije minute, uz snažno miješanje magnetskom miješalicom (6.4.). Epruvetu stavite u vodenu kupelj (6.10.) i ostavite 60 minuta.
8.3.3. Centrifugirajte (6.2.) 10 minuta na 2 200 g ili profiltrirajte kroz filtar-papir (6.6.), pri čemu prvih 5 ml filtrata bacite.
8.4. Kromatografsko određivanje
8.4.1. Izvršite HPLC analizu kako je opisana u Prilogu XII. Ako se dobije negativan rezultat, analizirani uzorak ne sadrži količine suhe tvari slatke sirutke koje je moguće detektirati. U slučaju pozitivnog rezultata treba primijeniti dolje opisani reverzno-fazni HPLC postupak. Alternativno se reverzno-fazna HPLC metoda može primijeniti odmah. Prisutnost kisele mlaćenice može kod primjene metode opisane u Prilogu XII. dovesti do pojave lažno pozitivnih rezultata. Reverzno-fazni HPLC postupak isključuje navedenu mogućnost.
8.4.2. Prije reverzno-fazne HPLC analize treba optimizirati uvjete gradijenta. Vrijeme zadržavanja od 26 ± 2 minute za CMPA optimalno je za gradijentne sustave s mrtvim volumenom od oko 6 ml (volumen od točke gdje se otopine pomiješaju do volumena petlje injektora, uključujući i njega). Optimalno vrijeme zadržavanja za gradijentne sustave s nižim mrtvim volumenom (npr. 2 ml) iznosi 22 minute.
Uzmite otopine standardnih uzoraka (5.4.) s 50 %-tnom slatkom sirutkom i bez nje.
Ubrizgajte 100 μl supernatanta ili filtrata (8.3.3.) u HPLC uređaj koji funkcionira u uvjetima referentnog gradijenta navedenim u tablici 1.
Tablica 1.
Uvjeti referentnog gradijenta za optimizaciju kromatografije
|
Vrijeme (min) |
Protok (ml/min) |
% A |
% B |
Krivulja |
|
Početno |
1,0 |
90 |
10 |
* |
|
27 |
1,0 |
60 |
40 |
linearna |
|
32 |
1,0 |
10 |
90 |
linearna |
|
37 |
1,0 |
10 |
90 |
linearna |
|
42 |
1,0 |
90 |
10 |
linearna |
Usporedba dvaju kromatograma trebala bi otkriti položaj pika CMPA.
Sastav otopine koju treba koristiti za normalni gradijent (vidjeti 8.4.3.) može se izračunati prema sljedećoj formuli: % B = 10 – 2,5 + (13,5 + (RTcmpA – 26) / 6) × 30 / 27 % B = 7,5 + (13,5 + (RTcmpA – 26) / 6) × 1,11
gdje je:
|
RTcmpA |
: |
vrijeme zadržavanja CMPA u referentnom gradijentu; |
|
10 |
: |
početni % B referentnog gradijenta; |
|
2,5 |
: |
% B u središnjoj točki minus % B na početku u normalnom gradijentu; |
|
13,5 |
: |
vrijeme u središnjoj točki referentnog gradijenta; |
|
26 |
: |
potrebno vrijeme zadržavanja CMPA; |
|
6 |
: |
omjer nagiba referentnog gradijenta i normalnog gradijenta; |
|
30 |
: |
% B na početku minus % B nakon 27 minuta u referentnom gradijentu; |
|
27 |
: |
vrijeme primjene referentnog gradijenta. |
8.4.3. Uzmite otopine ispitnih uzoraka.
Ubrizgajte 100 μl točno izmjerenog supernatanta ili filtrata (8.3.3.) u HPLC uređaj koji radi uz brzinu protoka od 1,0 ml otopine eluensa (5.2.) u minuti.
Sastav eluensa na početku analize dobiva se na način opisan u točki 8.4.2. On obično približno odgovara omjeru A:B = 76:24 (5.2.). Odmah nakon ubrizgavanja pokreće se linearni gradijent, posljedica čega je 5 % viši postotak od B nakon 27 minuta. Potom se pokreće linearni gradijent koji u pet minuta mijenja sastav eluensa u 90 % B. Taj se sastav održava pet minuta, nakon čega se pomoću linearnog gradijenta mijenja, da bi se nakon pet minuta ponovo vratio na početni sastav. Ovisno o unutarnjem volumenu pumpnog sustava, sljedeće se ubrizgavanje može izvršiti 15 minuta nakon postizanja početnih uvjeta.
Napomena 1.: Vrijeme zadržavanja CMPA trebalo bi iznositi 26 ± 2 minute. To je moguće postići mijenjanjem početnih i konačnih uvjeta prvog gradijenta. Međutim, razlika u % B između početnih i konačnih uvjeta prvog gradijenta mora ostati 5 % B.
Napomena 2.: Eluense treba otpliniti u dovoljnoj mjeri te tako otplinjeni moraju i ostati. To je od ključne važnosti za pravilan rad gradijentnog pumpnog sustava. Standardna devijacija vremena zadržavanja pika CMPA mora biti manja od 0,1 minute (n = 10).
Napomena 3.: Nakon svakih 5 uzoraka treba ubrizgati referentni uzorak (5.) te pomoću njega izračunati novi faktor odziva R (9.1.1.).
8.4.4. Rezultati kromatografske analize ispitnog uzorka (E) dobiju se u obliku kromatograma u kojem se pik CMPA utvrđuje na temelju vremena zadržavanja koje iznosi oko 26 minuta.
Integrator (6.11.6.) automatski izračunava visinu H pika CMPA. Na svakom kromatogramu treba provjeriti položaj bazne linije. Analizu ili integriranje treba ponoviti ako bazna linija nije pravilno smještena.
Napomena: Ako je pik CMPA dovoljno odvojen od drugih pikova, treba koristiti alokaciju bazne linije dolina-dolina, u suprotnom slučaju koristite padajuće okomice na baznu liniju, koje trebaju imati ishodište blizu pika CMPA (prema tome ne kad je t = 0 min!). Za standard i uzorke primjenjujte istu vrstu integracije te u slučaju zajedničke bazne linije provjerite njezinu konzistentnost za uzorke i standard.
Prije kvantitativne interpretacije od ključne je važnosti analizirati izgled svakog kromatograma radi otkrivanja bilo kakvih nepravilnosti nastalih zbog neispravnog rada uređaja ili kolona, ili zbog podrijetla i vrste analiziranog uzorka. U slučaju dvojbe ponovite analizu.
8.5. Kalibracija
8.5.1. Na standardne uzorke (od 5.4.1. do 5.4.2.) točno primijenite postupak opisan od točke 8.2. do točke 8.4.4. Koristite svježe pripremljene otopine budući da se u 8-postotnoj triklorooctenoj kiselini na sobnoj temperaturi CMP razgrađuje. Na temperaturi od 4 °C otopina ostaje stabilna 24 sata. U slučaju velike serije analiza, poželjna je uporaba ohlađenog stalka za uzorke u automatskom injektoru.
Napomena: 8.4.2. može se izostaviti ako je % B u početnim uvjetima poznat iz prethodnih analiza.
Kromatogram referentnog uzorka [5] mora biti analogan slici 1. Na toj slici piku CMPA prethode dva manja pika. Bitno je dobiti slično razdvajanje.
8.5.2. Prije kromatografskog određivanja uzoraka ubrizgajte 100 μl standardnog uzorka bez slatke sirutke [0] (5.4.1.).
Kromatogram ne smije pokazivati pik u vremenu zadržavanja pika CMPA.
8.5.3. Odredite faktore odziva R ubrizgavanjem količine filtrata (8.5.1.) jednake količini koja je korištena za uzorke.
9. IZRAŽAVANJE REZULTATA
9.1. Metoda izračuna i formule
9.1.1. Izračunavanje faktora odziva R:
Pik CMPA: R = W/H
gdje je:
|
R |
= |
faktor odziva za pik CMPA; |
|
H |
= |
visina pika CMPA; |
|
W |
= |
količina sirutke u standardnom uzorku [5]. |
9.2. Izračunavanje postotka slatke sirutke u prahu u uzorku
W(E) = R × H(E)
gdje je:
|
W(E) |
= |
postotak (m/m) slatke sirutke u uzorku (E); |
|
R |
= |
faktor odziva za pik CMPA (9.1.1.); |
|
H(E) |
= |
visina pika CMPA uzorka (E). |
Ako je W(E) veći od 1 % i ako je razlika između vremena zadržavanja i vremena standardnog uzorka [5] manja od 0,2 minute, to ukazuje na prisutnost suhe tvari slatke sirutke.
9.3. Preciznost postupka
9.3.1. Ponovljivost
Razlika između rezultata dvaju određivanja koja istodobno ili brzo jedan za drugim izvede isti analitičar koristeći istu opremu na jednakom ispitnom materijalu ne smije biti veća od 0,2 % m/m.
9.3.2. Obnovljivost
Nije određena.
9.3.3. Linearnost
Od 0 do 16 % slatke sirutke linearni se odnos treba dobiti s korelacijskim koeficijentom > 0,99.
9.4. Tumačenje
Granica od 1 % utvrđena je u skladu s odredbama točaka 9.2. i 9.4.1. Priloga XIX. Uredbi (EEZ) br. 214/2001.
Tablica 1.
Ni –4,6 standard

(1) Slatka sirutka u prahu standardnog sastava i patvoreno mlijeko u prahu nabavljaju se kod tvrtke NIZO, Kernhemseweg 2, PO Box 20 – NL-6710 BA Ede. Međutim, države članice mogu koristiti i druge prahove koji daju jednake rezultate kao prah koji proizvodi NIZO.
PRILOG XIV.
(Članak 10.)
OBRANO MLIJEKO U PRAHU: KVANTITATIVNO ODREĐIVANJE FOSFATIDILSERINA I FOSFATIDILETANOLAMINA
Metoda: reverzno-fazna HPLC metoda
1. SVRHA I PODRUČJE PRIMJENE
Ova metoda opisuje postupak količinskog određivanja fosfatidilserina (PS) i fosfatidiletanolamina (PE) u obranom mlijeku u prahu (OMP), a prikladna je za detekciju suhe tvari mlaćenice u OMP-u.
2. DEFINICIJA
Sadržaj fosfatidilserina (PS) i fosfatidiletanolamina (PE): masena frakcija određene tvari koja se utvrđuje ovdje opisanim postupkom. Rezultat se izražava u fosfatidiletanolamin dipalmitoilu (PEDP) na 100 g praha.
3. NAČELO METODE
Ekstrakcija aminofosfolipida iz rekonstituiranog mlijeka u prahu pomoću metanola. Određivanje fosfatidilserina (PS) i fosfatidiletanolamina (PE) kao derivata o-ftaldialdehida (OPA) reverzno-faznom (RP) HPLC analizom i detekcijom fluorescencije. Kvantifikacija sadržaja fosfatidilserina (PS) i fosfatidiletanolamina (PE) u ispitnom uzorku na temelju standardnog uzorka koji sadrži poznatu količinu fosfatidiletanolamin dipalmitoila (PEDP).
4. REAGENSI
Svi reagensi moraju biti priznate analitičke čistoće. Mora se koristiti destilirana voda ili voda barem jednake čistoće, osim ako je navedeno drukčije.
4.1. Standardni materijal: fosfatidiletanolamin dipalmitoil (PEDP), čistoće najmanje 99 %
Napomena: Standardni se materijal mora čuvati na temperaturi od –18 °C.
4.2. Reagensi za pripremu standardnog i ispitnog uzorka
4.2.1. Metanol, HPLC čistoće
4.2.2. Kloroform, HPLC čistoće
4.2.3. Triptamin-monohidroklorid
4.3. Reagensi za derivatizaciju o-ftaldialdehida
4.3.1. Natrijev hidroksid, vodena otopina 12 M
4.3.2. Borna kiselina, vodena otopina 0,4 M, pH vrijednost podešena na 10,0 pomoću natrijevog hidroksida (4.3.1.)
4.3.3. 2-merkaptoetanol
4.3.4. O-ftaldialdehid (OPA)
4.4. Otapala za HPLC eluaciju
4.4.1. Za pripremu otapala za eluaciju koriste se reagensi HPLC čistoće
4.4.2. Voda HPLC čistoće
4.4.3. Metanol provjerene fluorimetrijske čistoće
4.4.4. Tetrahidrofuran
4.4.5. Natrijev dihidrogen fosfat
4.4.6. Natrijev acetat
4.4.7. Octena kiselina
5. OPREMA
5.1. Analitička vaga koja može vagati s točnošću od 1 mg, uz čitljivost od 0,1 mg
5.2. Laboratorijske čaše zapremine 25 i 100 ml
5.3. Pipete za prijenos količina od 1 i 10 ml
5.4. Magnetska miješalica
5.5. Graduirane pipete za prijenos količina od 0,2, 0,5 i 5 ml
5.6. Odmjerne tikvice zapremine 10, 50 i 100 ml
5.7. Šprice zapremine 20 i 100 μl
5.8. Ultrazvučna kupelj
5.9. Centrifuga koja može raditi na 27 000 × g
5.10. Staklene bočice zapremine oko 5 ml
5.11. Menzura zapremine 25 ml
5.12. pH-metar s točnošću od 0,1 pH jedinica
Oprema za HPLC
5.13.1. Gradijentni pumpni sustav s mogućnošću rada pri brzini protoka od 1,0 ml/min i tlaku od 200 bara
5.13.2. Automatski uređaj za uzorkovanje s mogućnošću derivatizacije
5.13.3. Grijač kolona koji kolonu može održavati na temperaturi 30 °C ± 1 °C
5.13.4. Detektor fluorescencije s mogućnošću rada na 330 nm valne duljine ekscitacije i 440 nm valne duljine emisije
5.13.5. Integrator ili softver za obradu podataka koji može izmjeriti površinu pika
5.13.6. Kolona Lichrosphere – 100 (250 × 4,6 mm) ili ekvivalentna kolona ispunjena oktadecilsilanom (C 18), veličine čestica 5 μm
6. UZORKOVANJE
Uzorkovanje se provodi u skladu sa standardom ISO 707.
7. POSTUPAK
7.1. Priprema interne standardne otopine
7.1.1. Odvagnite 30,0 ± 0,1 mg triptamin-monohidroklorida (4.2.3.) u odmjernu tikvicu od 100 ml (5.6.) i dopunite do oznake metanolom (4.2.1.).
7.1.2. Otpipetirajte 1 ml (5.3.) ove otopine u odmjernu tikvicu od 10 ml (5.6.) i dopunite do oznake metanolom (4.2.1.) da biste dobili koncentraciju triptamina od 0,15 mM.
7.2. Priprema otopine ispitnog uzorka
7.2.1. Odvagnite 1,000 ± 0,001 g uzorka OMP-a u laboratorijsku čašu od 25 ml (5.2.). Pipetom (5.3.) dodajte 10 ml destilirane vode temperature 40 ± 1 °C i miješajte 30 minuta magnetskom miješalicom (5.4.) da bi se rastopile eventualne grudice.
7.2.2. Otpipetirajte 0,2 ml (5.5.) rekonstituiranog mlijeka u odmjernu tikvicu od 10 ml (5.6.), špricom (5.7.) dodajte 100 μl otopine triptamina koncentracije 0,15 mM (7.1.) i dopunite do oznake metanolom (4.2.1.). Pažljivo izmiješajte preokretanjem bočice i stavite na 15 minuta u ultrazvučnu kupelj (5.8.).
7.2.3. Centrifugirajte (5.9.) na 27 000 × g 10 minuta i skupite supernatant u staklenu bočicu (5.10.).
Napomena: Otopinu ispitnog uzorka čuvajte na temperaturi od 4 °C do izvođenja HPLC analize.
7.3. Priprema vanjske standardne otopine
7.3.1. Odvagnite 55,4 mg fosfatidiletanolamin dipalmitoila (PEDP) (4.1.) u odmjernu tikvicu od 50 ml (5.6.) i dodajte oko 25 ml kloroforma (4.2.2.) pomoću menzure (5.1.1.). Zagrijte začepljenu tikvicu na 50 °C ± 1 °C i pažljivo miješajte da se PEDP rastopi. Ohladite tikvicu na 20 °C, dopunite do oznake metanolom (4.2.1.) i izmiješajte preokretanjem tikvice.
7.3.2. Otpipetirajte 1 ml (5.3.) ove otopine u odmjernu tikvicu od 100 ml (5.6.) i dopunite do oznake metanolom (4.2.1.). Otpipetirajte 1 ml (5.3.) tako dobivene otopine u odmjernu tikvicu od 10 ml (5.6.), dodajte 100 μl (5.7.) otopine triptamina koncentracije 0,15 mM (7.1.) i dopunite do oznake metanolom (4.2.1.). Promiješajte preokretanjem tikvice.
Napomena: Otopinu referentnog uzorka do HPLC analize treba čuvati na temperaturi od 4 °C.
7.4. Priprema reagensa za derivatizaciju
Odvagnite 25,0 ± 0,1 mg o-ftaldialdehida (OPA) (4.3.4.) u odmjernu tikvicu zapremine 10 ml (5.6.), dodajte 0,5 ml (5.5.) metanola (4.2.1.) i pažljivo promiješajte da se OPA rastopi. Dopunite do oznake otopinom borne kiseline (4.3.2.) i špricom (5.7.) dodajte 20 μl 2-merkaptoetanola (4.3.3.).
Napomena: Reagens za derivatizaciju treba čuvati na 4 °C u tamnoj staklenoj bočici da bi ostao stabilan tjedan dana.
7.5. Određivanje HPLC metodom
7.5.1. Otapala za eluaciju (4.4.)
Otapalo A: Otopina 0,3 mM natrijevog dihidrogen fosfata i 3 mM otopine natrijevog acetata (pH vrijednosti podešene octenom kiselinom na 6,5 ± 0,1): metanol: tetrahidrofuran = 558:440:2 (v/v/v).
Otapalo B: metanol.
7.5.2. Preporučeni gradijent eluacije:
|
Vrijeme (min) |
Otapalo A (%) |
Otapalo B (%) |
Brzina protoka (ml/min) |
|
Početak |
40 |
60 |
0 |
|
0,1 |
40 |
60 |
0,1 |
|
5,0 |
40 |
60 |
0,1 |
|
6,0 |
40 |
60 |
1,0 |
|
6,5 |
40 |
60 |
1,0 |
|
9,0 |
36 |
64 |
1,0 |
|
10,0 |
20 |
80 |
1,0 |
|
11,5 |
16 |
84 |
1,0 |
|
12,0 |
16 |
84 |
1,0 |
|
16,0 |
10 |
90 |
1,0 |
|
19,0 |
0 |
100 |
1,0 |
|
20,0 |
0 |
100 |
1,0 |
|
21,0 |
40 |
60 |
1,0 |
|
29,0 |
40 |
60 |
1,0 |
|
30,0 |
40 |
60 |
0 |
Napomena: Da bi se postigla rezolucija prikazana na slici 1., gradijent eluacije možda treba malo modificirati.
Temperatura kolone: 30 °C.
7.5.3. Količina koja se ubrizgava: 50 μl reagensa za derivatizaciju i 50 μl otopine uzorka.
7.5.4. Uspostavljanje ravnoteže u koloni
Svakog dana pri pokretanju sustava kolonu treba ispirati 15 minuta 100 %-tnim otapalom B, a potom podesiti na A:B = 40:60 da bi se tijekom 15 minuta uspostavila ravnoteža uz brzinu protoka od 1 ml/min. Napravite slijepu probu ubrizgavanjem metanola (4.2.1.).
Napomena: Prije nego kolonu pohranite na duže vrijeme, ispirite je 30 minuta mješavinom metanol:kloroform = 80:20 (v/v).
7.5.5. Odredite sadržaj fosfatidilserina (PS) i fosfatidiletanolamina (PE) u ispitnom uzorku.
7.5.6. Izvedite niz kromatografskih analiza pazeći da vrijeme između ciklusa bude konstantno radi postizanja konstantnih vremena zadržavanja. Nakon svakih 5-10 otopina ispitnog uzorka ubrizgajte vanjsku standardnu otopinu (7.3.) radi izračuna faktora odziva 0.
Napomena: Nakon svakih 20-25 analiza neophodno je kolonu ispirati 100 %-tnim otapalom B (7.5.1.) najmanje 30 minuta.
7.6. Integracija
7.6.1. Pik fosfatidiletanolamin dipalmitoila (PEDP)
PEDP se pojavljuje kao jedan pik. Odredite površinu pika integracijom dolina-dolina.
7.6.2. Pik triptamina
Triptamin se pojavljuje kao jedan pik (slika 1.). Odredite površinu pika integracijom dolina-dolina.
7.6.3. Grupe pikova fosfatidilserina (PS) i fosfatidiletanolamina (PE)
U opisanim se uvjetima (slika 1.) PS pojavljuje kao dva glavna djelomično nerazdvojena pika, kojima prethodi jedan manji pik. PE sa javlja kao tri glavna djelomično nerazdvojena pika. Odredite ukupnu površinu svake grupe pikova postavljajući baznu liniju kako je prikazano na slici 1.
8. IZRAČUN I PRIKAZIVANJE REZULTATA
Sadržaj fosfatidilserina (PS) i fosfatidiletanolamina (PE) u ispitnom uzorku računa se pomoću sljedeće formule: C = 55,36 × ((A2)/(A1)) × ((T1)/(T2))
gdje je:
|
C |
= |
sadržaj fosfatidilserina (PS) ili fosfatidiletanolamina (PE) (mg/100 g praha) u ispitnom uzorku; |
|
A1 |
= |
površina pika fosfatidiletanolamin dipalmitoila (PEDP) u standardnoj otopini uzorka (7.3.); |
|
A2 |
= |
površina pika fosfatidilserina (PS) ili fosfatidiletanolamina (PE) u ispitnoj otopini uzorka (7.2.); |
|
T1 |
= |
površina pika triptamina u standardnoj otopini uzorka (7.3.); |
|
T2 |
= |
površina pika triptamina u ispitnoj otopini uzorka (7.2.). |
9. PRECIZNOST METODE
Napomena: Vrijednosti ponovljivosti računaju se u skladu s međunarodnim standardom IDF (1). Privremena granična vrijednost obnovljivosti izračunana je prema postupku opisanom u Prilogu III. točki B ove Uredbe.
9.1. Ponovljivost
Relativna standardna derivacija ponovljivosti, koja prikazuje varijabilnost nezavisnih analitičkih rezultata koje dobije isti analitičar koristeći istu opremu u istim uvjetima i na istom ispitnom uzorku u kratkom vremenskom intervalu, ne smije biti veća od 2 % relativno. Ako se dva određivanja izvrše pod navedenim uvjetima, relativna razlika između dvaju rezultata ne bi smjela biti veća od 6 % aritmetičke sredine rezultata.
9.2. Obnovljivost
Ako su dva određivanja izvršili analitičari u različitim laboratorijima, u različitim uvjetima, koristeći različitu opremu, na istom ispitnom uzorku, relativna razlika između dvaju rezultata ne smije biti veća od 11 % aritmetičke sredine rezultata.
10. REFERENTNA LITERATURA
10.1. Resmini P., Pellegrino L., Hogenboom J. A., Sadini V., Rampilli M., „Detection of buttermilk solids in skimmilk powder by HPLC quantification of aminophospholipids”. Sci. Tecn. Latt.-Cas., 39,395 (1988).
Slika 1.
HPLC struktura OPA-derivata fosfatidilserina (PS) i fosfatidiletanolamina (PE) u metanolnom ekstraktu rekonstituiranog obranog mlijeka u prahu. Prikazan je način integracije pikova fosfatidilserina (PS), fosfatidiletanolamina (PE) i triptamina (interni standard).
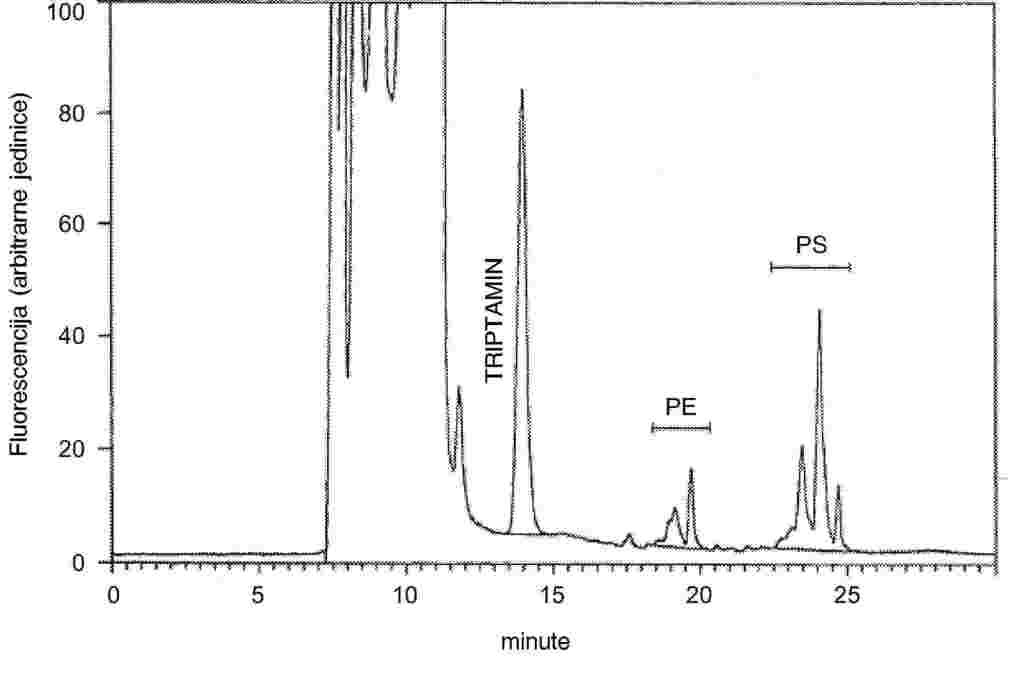
(1) Međunarodni standard IDF 135B/1991. Mlijeko i mliječni proizvodi. Značajke preciznosti metoda analize. Pregled postupka međulaboratorijske studije.
PRILOG XV.
(Članak 11.)
DETEKCIJA ANTIMIKROBNIH OSTATAKA U OBRANOM MLIJEKU U PRAHU
Primjenjuje se probirni test na bazi mikrobnih inhibitora u kojem se kao ispitni mikroorganizam koristi Geobacillus stearothermophilus var. calidolactis ATCC 10149 (jednak soju C953), koji je dovoljno osjetljiv da otkriva 4 μg benzilpenicilina u kilogramu mlijeka i 100 μg sulfadimina u kilogramu mlijeka. Na tržištu su dostupni gotovi kompleti za testiranje i može ih se koristiti ako je njihova osjetljivost na benzilpenicilin i sulfadimidin u skladu sa zahtjevima.
Za testiranje se koristi rekonstituirano obrano mlijeko u prahu (1 g praha +9 ml destilirane vode). Test se izvodi kako je opisano u standardu ISO/TS 26844:2006 – Mlijeko i mliječni proizvodi – Utvrđivanje antimikrobnih ostataka – Difuzijski test u epruveti IDF – Bilten br. 258/1991, odjeljak 1., poglavlje 2., ili prema uputama proizvođača kompleta za testiranje (1).
Pozitivni se rezultati tumače kako slijedi:
|
1. |
Prisutnost β-laktama može se potvrditi ponavljanjem testa uz dodavanje penicilinaze ispitnom sustavu (2): Negativan rezultat: inhibirajuća tvar je β-laktamski antibiotik. Pozitivan rezultat ostaje: ovim postupkom nije moguće identificirati inhibirajuću tvar, nastavite s točkom 2. |
|
2. |
Prisutnost sulfonamida može se potvrditi ponavljanjem testa uz dodavanje p-amino benzojeve kiseline testnom sustavu: Negativan rezultat: inhibirajuća tvar je sulfonamid. Pozitivan rezultat ostaje: ovim postupkom nije moguće identificirati inhibirajuću tvar, nastavite s točkom 3. |
|
3. |
Prisutnost kombinacije β-laktama i sulfonamida može se potvrditi ponavljanjem testa uz dodavanje penicilinaze + p-amino benzojeve kiseline ispitnom sustavu: Negativan rezultat: inhibirajuće tvari su β-laktamski antibiotik i sulfonamid. Pozitivan rezultat: ovim postupkom nije moguće identificirati inhibirajuću tvar. |
(1) Važna napomena: Kad se analizira obrano mlijeko u prahu, moguća je pojava lažno pozitivnih rezultata. Zbog toga je izuzetno važno provjeriti da korišteni ispitni sustav ne daje lažno pozitivne rezultate.
(2) Neki su β-laktami manje osjetljivi na β-laktamazu. U takvim se slučajevima preporuča dodatna predobrada uzorka (1 ml ispitnog uzorka s 0,3 ml koncentrata penaze 2 sata na 37 °C).
PRILOG XVI.
(Članak 12.)
KVANTITATIVNO ODREĐIVANJE OBRANOG MLIJEKA U PRAHU U KRMNIM SMJESAMA METODOM ENZIMSKE KOAGULACIJE PARA-KAZEINA
1. SVRHA
Kvantitativno određivanje obranog mlijeka u prahu u krmnim smjesama metodom enzimske koagulacije para-kazeina.
2. PODRUČJE PRIMJENE
Ova se metoda primjenjuje na krmne smjese koje sadrže barem 10 % obranog mlijeka u prahu; velike količine mlaćenice i/ili određenih nemliječnih bjelančevina mogu dovesti do smetnji.
3. NAČELO METODE
3.1. Otapanje kazeina sadržanog u krmnoj smjesi ekstrakcijom pomoću otopine natrijevog citrata.
3.2. Podešavanje koncentracije kalcijevih iona do razine potrebne za taloženje para-kazeina dodavanjem sirila.
3.3. Udio dušika u talogu para-kazeina određuje se Kjeldahlovom metodom, kako je opisana u standardu ISO 8968-2:2001/IDF 20-2:2001; količina obranog mlijeka u prahu izračunava se na temelju najmanjeg udjela kazeina od 27,5 % (vidjeti točku 8.1.).
4. REAGENSI
Korišteni reagensi moraju biti analitičke čistoće. Voda koja se koristi mora biti destilirana voda ili voda najmanje jednake čistoće. Uz iznimku sirila (4.5.), reagensi i otopine ne smiju sadržavati dušične tvari.
4.1. Trinatrijev citrat, dihidrat (1 % w/v otopine)
4.2. Kalcijev klorid (5M otopina)
Otopite 75 g of CaCl2 · 2 H2O mućkanjem u 100 ml destilirane vode (obratite pozornost na egzotermičku reakciju). Ostavite preko noći i zatim profiltrirajte. Otopinu pohranite u hladnjak.
4.3. 0,1 N natrijev hidroksid
4.4. 0,1 N klorovodična kiselina
4.5. Tekuće teleće sirilo (jakosti oko 100 IMCU/ml prema standardu ISO 11815/IDF 157). Čuvajte u hladnjaku na temperaturi od 4 do 6 °C.
4.6. Reagensi za kvantitativno određivanje dušika Kjeldahlovom metodom, kako je opisana u standardu ISO 8968-2:2001/IDF 20-2:2001.
5. OPREMA
Uobičajena laboratorijska oprema, uključujući:
5.1. Tarionik ili homogenizator;
5.2. Analitičku vagu koja može vagati s točnošću od 1 mg, uz čitljivost od 0,1 mg;
5.3. Stolnu centrifugu (500 g ili 2 000 do 3 000 o/min) s epruvetama od 50 ml i 2 000 g;
5.4. Magnetsku miješalicu s magnetima od 10 do 15 mm;
5.5. Čaše od 150 do 200 ml;
5.6. Tikvice od 250 i 500 ml;
5.7. Staklene lijevke promjera 60 do 80 mm;
5.8. Filtar-papir bez pepela za brzo filtriranje promjera 150 mm (Whatman br. 41 ili ekvivalent);
5.9. Pipete različitih nazivnih volumena;
5.10. Vodenu kupelj s termostatom zagrijanu na 37 °C ± 1 °C;
5.11. pH-metar, s preciznošću određivanja do 0,1 jedinice;
5.12. Termometri, s točnošću do 1 °C.
6. POSTUPAK
6.1. Priprema uzorka
Usitnite u tarioniku ili homogenizirajte u mlinu 10 do 20 g uzorka da biste dobili homogenu smjesu.
6.2. Otapanje mlijeka u prahu i odvajanje neotopivog ostatka
6.2.1. Odvagnite 1,000 ± 0,002 g dobro homogenizirane krmne smjese (6.1.) izravno u epruvetu za centrifugiranje od 50 ml. Dodajte 30 ml otopine trinatrijevog citrata (4.1.) prethodno zagrijane na 45 °C ± 2 °C. Miješajte barem pet minuta magnetskom miješalicom ili ručno, snažno mućkajući.
6.2.2. Centrifugirajte na 500 g (2 000 do 3 000 o/min) 10 minuta i dekantirajte bistri vodeni supernatant u čašu od 150 do 200 ml, pazeći pritom da neotopljene čestice ostanu na dnu.
6.2.3. Ostatak još dva puta ekstrahirajte ponavljajući isti postupak i dodajući dobivene ekstrakte prvom ekstraktu.
6.2.4. Ako se na površini stvori sloj ulja, hladite u hladnjaku dok se mast ne stvrdne te tvrdi sloj uklonite špatulom.
6.3. Koagulacija kazeina pomoću enzima sirila
6.3.1. Neprestano miješajući dodajte kap po kap 2 ml zasićene otopine kalcijevog klorida (4.2.) ukupnom vodenom ekstraktu (oko 100 ml). Podesite pH vrijednost na 6,4-6,5 otopinom NaOH (4.3.) ili HCl (4.4.). Držati 15 do 20 minuta u vodenoj kupelji na temperaturi od 37 °C ± 1 °C radi postizanja ravnoteže otopine soli. Ona postaje očitom kad se pojavi lagano zamućenje.
6.3.2. Prelijte tekućinu u jednu (ili dvije) epruvete za centrifugiranje i centrifugirajte na 2 000 g 10 minuta da biste uklonili istaloženi materijal. Prelijte supernatant u jednu (ili dvije) epruvete za centrifugiranje, pazeći da pritom ne prelijete i talog.
6.3.3. Supernatant ponovno zagrijte na 37 °C ± 1 °C. Miješajući ekstrakt dodajte kap po kap 0,5 ml tekućeg sirila (4.5.). Do koagulacije dolazi za dvije minute.
6.3.4. Vratite uzorak u vodenu kupelj i ostavite 15 minuta na temperaturi od 37 °C ± 1 °C. Izvadite uzorak iz vodene kupelji i miješanjem razbijte gruš. Centrifugirajte 10 minuta na 2 000 g. Filtrirajte supernatant kroz odgovarajući filtar-papir (5.8.) i taj filtar-papir sačuvajte. Talog iz epruvete za centrifugiranje uz miješanje isperite s 50 ml vode zagrijane na oko 35 °C.
Ponovno centrifugirajte 10 minuta na 2 000 g. Supernatant profiltrirajte kroz filtar-papir koji ste sačuvali od ranije.
6.4. Određivanje kazeinskog dušika
6.4.1. Nakon ispiranja, talog kvantitativno prenesite pomoću destilirane vode na filtar-papir koji ste sačuvali iz postupka opisanog u točki 6.3.4. Prenesite filtar-papir u Kjeldahlovu tikvicu. Odredite udio dušika Kjeldahlovom metodom, kako je opisana u standardu ISO 8968-2:2001/IDF 20-2:2001.
7. SLIJEPA PROBA
7.1. Slijepu probu treba redovito izvoditi mineralizacijom prema Kjeldahlovoj metodi kako je opisano u standardu ISO 8968-2:2001/IDF 20-2:2001. Pritom se koristi filtar-papir bez pepela (5.8.), koji se namoči mješavinom 90 ml (4.1.) otopine natrijevog citrata, 2 ml otopine kalcijevog klorida (4.2.), 0,5 ml tekućeg sirila (4.5.), i ispere s 3 × 15 ml destilirane vode.
7.2. Količinu kiseline korištene za slijepu probu treba oduzeti od količine kiseline (4.4.) korištene za titraciju uzroka.
8. IZRAŽAVANJE REZULTATA
8.1. Postotak obranog mlijeka u prahu u krmnoj smjesi izračunava se prema sljedećoj formuli:
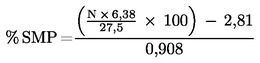
gdje:
N je postotak dušika u para-kazeinu;
27,5 je koeficijent za pretvaranje utvrđenog kazeina u postotak obranog mlijeka u prahu;
2,81 i 0,908 jesu korekcijski faktori dobiveni iz regresijske analize.
9. PRECIZNOST METODE
9.1. Ponovljivost
U barem 95 % ispitanih slučajeva razlika između rezultata dobivenih u dvije analize istog uzorka, koje je izvršio isti analitičar u istom laboratoriju, ne smije biti veća od 2,3 g obranog mlijeka u prahu u 100 g krmne smjese.
9.2. Obnovljivost
U barem 95 % ispitanih slučajeva razlika između rezultata dobivenih u dva laboratorija analizom istog uzorka ne smije biti veća od 6,5 g obranog mlijeka u prahu u 100 g krmne smjese.
10. NAPOMENE
10.1. Dodavanje visokog postotka određenih nemliječnih bjelančevina, a posebno sojinih bjelančevina, pri zagrijavanju s obranim mlijekom u prahu može dovesti do previsokih rezultata zbog zajedničkog taloženja s para-kazeinom mlijeka.
10.2. Dodavanje mlaćenice može dovesti do nešto nižih vrijednosti budući da se određuje samo nemasni dio. Dodavanje određene kisele mlaćenice može dovesti do izrazito niskih rezultata uslijed nepotpunog otapanja u otopini citrata.
10.3. Dodavanje 0,5 % ili više lecitina također može dovesti do niskih rezultata.
10.4. Dodavanje jako zagrijanog obranog mlijeka u prahu može dovesti do previsokih vrijednosti zbog zajedničkog taloženja određenih bjelančevina sirutke s para-kazeinom mlijeka.
PRILOG XVII.
(Članak 13.)
DETEKCIJA ŠKROBA U OBRANOM MLIJEKU U PRAHU, DENATURIRANOM MLIJEKU U PRAHU I KRMNIM SMJESAMA
1. PODRUČJE PRIMJENE
Ova se metoda primjenjuje za detekciju škroba koji se koristi kao marker u denaturiranom mlijeku u prahu.
Granica utvrđivanja ovom metodom iznosi približno 0,05 g škroba na 100 g uzorka.
2. NAČELO
Reakcija se temelji na reakciji koja se koristi u jodometriji:
|
— |
fiksiranje slobodnog joda u vodenoj otopini pomoću koloida; |
|
— |
apsorpcija micela škroba i stvaranje boje. |
3. REAGENSI
3.1. Otopina joda
|
— |
Jod: 1,0 g, |
|
— |
Kalijev jodid: 2,0 g, |
|
— |
Destilirana voda: 100 ml. |
|
— |
U odmjernoj tikvici od 100 ml s jednom oznakom rastopite u vodi 1,0 g joda i 2,0 g kalijevog jodida. Razrijedite vodom do oznake 100 ml i promiješajte. |
4. OPREMA
4.1. Analitička vaga
4.2. Vodena kupelj
4.3. Epruvete, 25 mm × 200 mm
5. POSTUPAK
Odvagnite 1,0 g uzorka s točnošću od 0,1 g i prenesite ga u epruvetu (4.3.).
Dodajte 20 ml destilirane vode i promućkajte da se uzorak rasprši.
Stavite u kipuću vodenu kupelj (4.2.) i ostavite 5 minuta.
Izvadite iz vodene kupelji i ohladite do sobne temperature.
Dodajte 0,5 ml otopine joda (3.1.), promućkajte i promotrite nastalo obojenje.
6. IZRAŽAVANJE REZULTATA
Plava boja ukazuje na prisutnost prirodnog škroba u uzorku.
Ako uzorak sadrži modificirani škrob, obojenje ne mora biti plavo.
7. PRIMJEDBE
Boja, intenzitet boje i izgled škroba pod mikroskopom razlikuju se ovisno o podrijetlu prirodnog škroba (npr. iz kukuruza ili krumpira) i vrsti modificiranog škroba prisutnog u uzorku.
Ako je u uzorku prisutan modificirani škrob, boja varira od ljubičaste do crvene ili smeđe, ovisno o stupnju modifikacije kristalne strukture prirodnog škroba.
PRILOG XVIII.
(Članak 14.)
ODREĐIVANJE SADRŽAJA VLAGE U VRHNJU U PRAHU
1. PODRUČJE PRIMJENE
Ovim se Prilogom utvrđuje metoda za određivanje sadržaja vlage u vrhnju u prahu.
2. POJMOVI I DEFINICIJE
U smislu ovog Priloga vrijede sljedeće definicije:
Sadržaj vlage: gubitak mase određen postupkom navedenim u ovom međunarodnom standardu.
Izražava se kao maseni postotak.
3. NAČELO
Sušenje uzorka za analizu na 102 ± 2 °C do konstantne mase i vaganje radi utvrđivanja gubitka mase.
4. OPREMA
Uobičajena laboratorijska oprema, a posebno sljedeće:
4.1. Analitička vaga prikladna za vaganje s točnošću od 1 mg, s mogućnošću očitanja 0,1 mg;
4.2. Sušionik, dobro ventiliran, koji u cijelom radnom prostoru može održavati temperaturu na 102 ± 2 °C;
4.3. Eksikator sa svježe osušenim silika gelom s higrometarskim indikatorom ili drugim djelotvornim desikantom;
4.4. Posude s ravnim dnom, dubine oko 25 mm, promjera oko 50 mm, izrađene od odgovarajućih materijala (na primjer od stakla, nehrđajućeg čelika, nikla ili aluminija) s poklopcima koji dobro naliježu i lako se skidaju;
4.5. Boce s nepropusnim čepovima za miješanje laboratorijskih uzoraka.
5. UZORKOVANJE
Važno je da laboratorij dobije uzorak koji je zaista reprezentativan i koji nije bio oštećen ili promijenjen tijekom prijevoza ili skladištenja.
Uzorkovanje nije dio metode utvrđene u ovom međunarodnom standardu. Preporučena metoda uzorkovanja navedena je u standardu ISO 707|IDF 50.
Uzorke čuvajte u uvjetima koji onemogućavaju kvarenje ili promjene u njihovom sastavu.
6. PRIPREMA ISPITNOG UZORKA
Ispitni uzorak dobro promiješajte protresanjem i preokretanjem posude (prema potrebi, nakon što prenesete sve ispitne uzorke u hermetički zatvorenu posudu dovoljne zapremine koja omogućuje takav postupak).
Ako se tim postupkom ne postigne potpuna homogenost, uzmite uzorke za analizu (za dva pojedinačna utvrđivanja) iz pripremljenog ispitnog uzorka na dva što udaljenija mjesta.
7. POSTUPAK
7.1. Priprema posude
7.1.1. Otklopljenu posudu zajedno s poklopcem (4.4.) zagrijavajte najmanje 1 sat u sušioniku (4.2.) podešenom na 102 ± 2 °C.
7.1.2. Posudu poklopite i poklopljenu prenesite u eksikator (4.3.), ostavite da se ohladi do sobne temperature i izvažite s točnošću od 1 mg, a masu zabilježite s točnošću od 0,1 mg.
7.2. Uzorak za analizu
Prenesite u posudu približno 1 do 3 g pripremljenog uzorka za analizu (6), poklopite i izvažite s točnošću od 1 mg, a masu zabilježite s točnošću od 0,1 mg.
7.3. Određivanje
7.3.1. Otklopite posudu i stavite je zajedno s poklopcem na dva sata u sušionik (4.2) podešen na 102 ± 2 °C.
7.3.2. Ponovo stavite poklopac, prenesite poklopljenu posudu u eksikator, ostavite da se ohladi do sobne temperature, izvažite s točnošću od 1 mg, a masu zabilježite s točnošću od 0,1 mg.
7.3.3. Otklopite posudu i ponovo je, zajedno s poklopcem, zagrijavajte jedan sat. Zatim ponovite postupak iz točke 7.3.2.
7.3.4. Postupak zagrijavanja i vaganja ponavljajte dok se masa posude s poklopcem ne smanji za 1 mg ili manje, ili dok se između dva uzastopna mjerenja ne poveća.
Za izračun uzmite najnižu zabilježenu masu.
8. IZRAČUN I IZRAŽAVANJE REZULTATA
8.1. Izračun
Sadržaj vlage izražen u g/100 g jednak je:
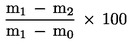
gdje je:
m 0 = masa posude i poklopca, izražena u gramima (7.1.2.);
m 1 = masa posude, poklopca i uzorka za analizu prije sušenja, izražena u gramima (7.2.);
m 2 = masa posude poklopca i uzorka za analizu nakon sušenja, izražena u gramima (7.3.4.).
Rezultat zaokružite na dvije decimale.
9. PRECIZNOST
Napomena: Vrijednosti za ponovljivost i obnovljivost izvedene su iz rezultata međulaboratorijskog ispitivanja (vidjeti Steiger, G.: Bilten IDF-a, br. 285/1993, str. 21.-28.), provedenog u skladu sa standardom IDF 135B:1991. Mlijeko i mliječni proizvodi – Obilježja preciznosti metoda analize – Prikaz postupka međulaboratorijske studije.
9.1. Ponovljivost
Apsolutna razlika između rezultata dvaju nezavisnih pojedinačnih ispitivanja koja je primjenom iste metode na jednakom ispitnom materijalu u istom laboratoriju u kratkom vremenskom intervalu izvršio isti analitičar koristeći istu opremu ne smije u više od 5 % slučajeva biti veća od 0,20 g vlage na 100 g proizvoda.
9.2. Obnovljivost
Apsolutna razlika između rezultata dvaju nezavisnih pojedinačnih ispitivanja koja su primjenom iste metode na jednakom ispitnom materijalu u različitim laboratorijima izvršili različiti analitičari koristeći različitu opremu ne smije u više od 5 % slučajeva biti veća od 0,40 g vlage na 100 g proizvoda.
10. IZVJEŠTAJ O ISPITIVANJU
U izvještaju o ispitivanju treba navesti:
|
— |
sve informacije potrebne za potpunu identifikaciju uzorka, |
|
— |
korištenu metodu uzorkovanja, ako je poznata, |
|
— |
metodu uzorkovanja korištenu u skladu s ovim međunarodnim standardom, |
|
— |
sve pojedinosti o postupku koje nisu navedene u ovom međunarodnom standardu ili se ne smatraju obveznima, zajedno s pojedinostima o svim događajima koji su mogli utjecati na rezultat(e) ispitivanja. |
Dobivene rezultate, a ako se provjeravala ponovljivost, konačni dobiveni rezultat.
PRILOG XIX.
(Članak 15.)
ODREĐIVANJE UDJELA VLAGE U KISELOJ MLAĆENICI U PRAHU
1. PODRUČJE PRIMJENE
Određivanje udjela vlage u kiseloj mlaćenici u prahu namijenjenoj za uporabu u hrani za životinje.
2. NAČELO
Uzorak se suši u vakuumu. Gubitak mase utvrđuje se vaganjem.
3. OPREMA
3.1. Analitička vaga koja može vagati s točnošću od 1 mg, uz mogućnost očitanja 0,1 mg.
3.2. Suhe posude od nehrđajućeg metala ili od stakla s poklopcem koji osigurava hermetičko zatvaranje; radna površina koja omogućuje da se 0,3 g ispitnog uzorka razmaže na 1 cm2.
3.3. Podesivi električni vakuumski sušionik opremljen uljnom pumpom i mehanizmom za dovod vrućeg zraka preko stupca koji sadrži, na primjer, kalcijev oksid ili kalcijev sulfat (kao i indikator vlage).
3.4. Eksikator s djelotvornim sredstvom za sušenje.
3.5. Sušionik s ventilacijom i termostatskom regulacijom, zagrijan na 102 ± 2 °C.
4. POSTUPAK
Posudu (3.2.) zajedno s poklopcem zagrijavajte u sušioniku (3.5.) najmanje jedan sat. Posudu poklopite i odmah prenesite u eksikator (3.4.), ostavite da se ohladi do sobne temperature i izvažite s točnošću od 1 mg, a masu zabilježite s točnošću od 0,1 mg.
Otklopite posudu i u nju stavite oko 5 g uzorka, pri čemu ga odvagnite s točnošću od 1 mg, a masu zabilježite s točnošću od 0,1 mg. Posudu s poklopcem stavite u vakuumski sušionik (3.3.) koji ste prethodno zagrijali na 83 °C. Da biste spriječili prevelik pad temperature u sušioniku, posudu u njega stavite što je moguće brže.
Podesite tlak na 100 torra (13,3 kPa) i neka se uzorak pri tom tlaku suši u struji vrućeg suhog zraka (oko četiri sata) do konstantne mase.
Vrijeme sušenja mjerite od trenutka kad sušionik ponovo dosegne temperturu od 83°C. Tlak u sušioniku pažljivo izjednačite s atmosferskim tlakom. Otvorite sušionik, na posudu odmah stavite poklopac, izvadite je iz sušionika i ostavite da se 30 do 45 minuta hladi u eksikatoru (3.4.), izvažite s točnošću od 1 mg, a masu zabilježite s točnošću od 0,1 mg. Sušite dodatnih 30 minuta u vakuumskom sušioniku (3.3.) na 83 °C i ponovo izvažite. Postupak zagrijavanja i vaganja ponavljajte dok se masa posude s poklopcem ne smanji za 1 mg ili manje, ili dok se između dva uzastopna mjerenja ne poveća. Za izračun uzmite najnižu zabilježenu masu.
5. IZRAČUNAVANJE
% vlage = (m1 – m2) / (m1 – m0) × 100 %
gdje je:
|
m0 |
masa posude i poklopca; |
|
m1 |
masa posude, poklopca i uzorka za analizu prije sušenja; |
|
m2 |
masa posude, poklopca i uzorka za analizu nakon sušenja. |
Rezultat zabilježite s točnošću od 0,1 g / 100 g.
6. PRECIZNOST
6.1. Granica ponovljivosti
Apsolutna razlika između rezultata dvaju nezavisnih pojedinačnih ispitivanja koja je primjenom iste metode na jednakom ispitnom materijalu u istom laboratoriju u kratkom vremenskom intervalu izvršio isti analitčar koristeći istu opremu ne smije u više od 5 % slučajeva biti veća od 0,4 g vode na 100 g mlaćenice u prahu.
6.2. Granica obnovljivosti
Apsolutna razlika između rezultata dvaju nezavisnih pojedinačnih ispitivanja koja su primjenom iste metode na jednakom ispitnom materijalu u različitim laboratorijima izvršili različiti analitičari koristeći različitu opremu ne smije u više od 5 % slučajeva biti veća od 0,6 g vode na 100 g kisele mlaćenice u prahu.
6.3. Izvor podataka o preciznosti
Podaci o preciznosti utvrđeni su pokusom izvedenim 1995. kojim je bilo obuhvaćeno osam laboratorija i 12 uzoraka (šest slijepih proba u duplikatu).
PRILOG XX.
(Članak 16.)
REFERENTNA METODA ZA ODREĐIVANJE ČISTOĆE MLIJEČNE MASTI PLINSKOM KROMATOGRAFSKOM ANALIZOM TRIGLICERIDA – 2. REVIZIJA
1. OPSEG I PODRUČJE PRIMJENE
U ovom je standardu opisana referentna metoda za utvrđivanje čistoće mliječne masti plinskom kromatografskom analizom triglicerida. Moguće je utvrditi prisutnost biljnih i životinjskih masti kao što su goveđi loj i svinjska mast.
Pomoću utvrđenih formula za trigliceride utvrđuje se cjelovitost mliječne masti. U osnovi se metoda primjenjuje na sirovo kravlje mlijeko ili proizvode od kravljeg mlijeka, bez obzira na prehranu, pasminu ili laktaciju. Samo iznimno visoka zastupljenost čistih biljnih ulja u prehrani životinja, kao npr. ulja iz repičinih sjemenki, može dovesti do lažnih pozitivnih rezultata. Mliječni proizvodi dobiveni od mlijeka pojedinačnih krava isto tako mogu uzrokovati lažne pozitivne rezultate.
Metoda se primjenjuje za mast ekstrahiranu iz mliječnih proizvoda za koje se podrazumijeva da sadrže čistu mliječnu mast nepromijenjenog sastava, kao npr. maslac, vrhnje, mlijeko i mlijeko u prahu. Tehnološka obrada mliječne masti, kao što je uklanjanje kolesterola ili frakcioniranje, može uzrokovati lažne pozitivne rezultate. To vrijedi i za mliječnu mast dobivenu iz obranog mlijeka ili mlaćenice. Metoda nije uvijek primjenjiva na mast ekstrahiranu iz sira jer proces zrenja može utjecati na sastav tako snažno da se dobije lažno pozitivan rezultat.
Napomena 1.: Maslačna (n-butanska) kiselina (C4) pojavljuje se isključivo u mliječnoj masti i omogućuje kvantitativnu procjenu nižih do srednjih količina mliječne masti u biljnim i životinjskim mastima. Međutim, zbog velikih varijacija C4 u rasponu prosječnog postotka masnih udjela, koji se kreće između 3,1 % i 3,8 %, teško je osigurati kvalitativne i kvantitativne podatke kad se radi o dodatku do maksimalno 20 % masenog udjela strane masti u čistoj mliječnoj masti [1].
Napomena 2.: U praksi se kvantitativni rezultati ne mogu dobiti iz sadržaja sterola biljnih masti jer ovise o uvjetima proizvodnje i obrade. Osim toga, kvalitativno određivanje stranih masti na temelju sterola je nejasno.
2. DEFINICIJA
Čistoća mliječne masti: odsutnost biljnih i životinjskih masti koja se utvrđuje prema postupku opisanom u ovom standardu.
Napomena: Čistoća se određuje pomoću S-vrijednosti koje se izračunavaju na temelju sastava triglicerida. Masene frakcije triglicerida izražavaju se u obliku postotka.
3. NAČELO METODE
Mliječna mast ekstrahirana iz mlijeka ili mliječnih proizvoda analizira se metodom plinske kromatografije pri čemu se koriste punjene ili kratke kapilarne kolone za utvrđivanje triglicerida (TG), koji se razdvajaju prema ukupnom broju ugljikovih atoma. Uvrštavanjem masenih frakcija molekula masti različitih veličina, izraženih u obliku postotka (C24 do C54, pri čemu se koriste samo parni C brojevi) u odgovarajuće trigliceridne (TG) jednadžbe, izračunavaju se S-vrijednosti. Ako S-vrijednosti premašuju granice utvrđene za čistu mliječnu mast, utvrđuje se prisutnost strane masti.
Napomena 1.: Primjerenost i jednakovrijednost punjenih i kapilarnih kolona već su ranije bile dokazane [2-4].
Napomena 2.: S-vrijednost je zbroj masenih frakcija triglicerida pomnoženih s odgovarajućim faktorima.
4. REAGENSI
Svi reagensi moraju biti priznate analitičke čistoće.
4.1. Plin nositelj: dušik ili, alternativno, helij ili vodik stupnja čistoće 99,995 %.
Standardi masti za standardiziranje standarda mliječne masti u skladu s točkom 7.3.3.
4.2.1. Standardi triglicerida, zasićeni; odgovarajući proizvodi dostupni su na tržištu.
4.2.2. Standard kolesterola.
4.3. Metanol (CH3OH), bezvodni.
4.4. n-heksan (CH3(CH2)4CH3).
4.5. n-heptan (CH3(CH2)5CH3).
4.6. Ostali plinovi: vodik, čistoće najmanje 99,995 %, bez organskih nečistoća (CnHm < 1 μl/l); sintetski zrak, bez organskih nečistoća (CnHm < 1 μl/l).
4.7. Bezvodni natrijev sulfat (Na2SO4).
5. OPREMA
Uobičajena laboratorijska oprema, a posebno sljedeće:
5.1. Visokotemperaturni plinski kromatograf
Visokotemperaturni plinski kromatograf primjeren za temperature od najmanje 400 °C, opremljen plameno-ionizacijskim detektorom (FID). Dijafragme koje se koriste u injektoru podnose visoke temperature i pokazuju vrlo nizak stupanj propuštanja. Za kapilarni plinski kromatograf (GC) koristite on-column injektor. Uvijek koristite grafitne brtve za povezivanje kolone, kao i za umetke za injektor i/ili detektor (prema potrebi).
5.2. Kolona kromatografa
5.2.1. Punjena kolona
Koristite staklenu kolonu unutarnjeg promjera 2 mm i duljine 500 mm, napunjenu stacionarnom fazom 3 % OV-1 na 125 μm do 150 μm (100 do 120 mesh) Gas ChromQ (1). Priprema, silanizacija, punjenje i kondicioniranje punjene kolone opisani su u Prilogu A.
Alternativno se može koristiti kapilarna kolona (5.2.2.).
5.2.2. Kapilarna kolona
Koristite kratku kapilarnu kolonu, npr. duljine 5 m s nepolarnom stacionarnom fazom koja može podnijeti temperature do 400 °C ili više (2). Kolonu kondicionirajte provedbom 20 analiza otopine mliječne masti (7.2.) u dva do tri dana koristeći zadane vrijednosti iz 7.3.4.2. Nakon toga faktori odziva (7.3.3.) bit će blizu 1 i niži od 1,20.
Napomena: Mogu se koristiti kolone različitih dimenzija i različita nepolarna faza otporna na visoke temperature, ako je njihova učinkovitost u skladu s tim standardom. Vidjeti također 7.3.4.2.
5.3. Kolona Extrelut zapremine 1 do 3 ml, napunjena silika gelom, koja se koristi samo za ekstrakciju mliječne masti prema 7.1.3.
5.4. Grafitne brtve koje mogu podnositi temperature od najmanje 400 °C; koriste se za povezivanje kolone plinskog kromatografa (GC) kao i za umetke za injektor i/ili detektor.
5.5. Vodena kupelj koja može održavati temperaturu od 50 °C ± 2 °C.
5.6. Sušionik koji može raditi na 50 °C ± 2 °C i 100 °C ± 2 °C.
5.7. Mikrolitarska pipeta.
5.8. Graduirana pipeta zapremine 5 ml.
5.9. Tkivica sa zaobljenim dnom zapremine 50 ml.
5.10. Erlenmeyerova tikvica nominalne zapremine 250 ml.
5.11. Lijevak.
5.12. Filtar-papir sitnih pora.
5.13. Rotacijski evaporator.
5.14. Ampule nominalne zapremine 1 ml s aluminijskim čepom ili navojnim čepom, obloženim politetrafluoroetilenom.
5.15. Šprica za ubrizgavanje s klipom koji ne ulazi u konus igle (plinska kromatogradija (GC) s punjenom kolonom).
Napomena: Šprice ove vrste omogućuju postizanje bolje ponovljivosti rezultata.
5.16. Analitička vaga koja može vagati s točnošću od 1 mg, uz mogućnost očitanja 0,1 mg.
6. UZORKOVANJE
U laboratorij treba poslati reprezentativni uzorak. Uzorak se tijekom prijevoza ili skladištenja ne smije oštetiti ili promijeniti.
Uzorkovanje nije dio metode opisane u ovom međunarodnom standardu. Preporučena metoda uzorkovanja navedena je u standardu ISO 707 | IDF 50 [5].
7. POSTUPAK
7.1. Priprema ispitnih uzoraka
Za pripremu ispitnog uzorka primijenite jednu od sljedeće tri metode ekstrakcije mliječne masti.
7.1.1. Izolacija iz maslaca ili masla
Otopite 50 do 100 g ispitnog uzorka na 50 °C u vodenoj kupelji (5.5.) ili sušioniku (5.6.). Stavite 0,5 do 1,0 g natrijevog sulfata (4.7.) u naborani filtar-papir (5.12.). Prethodno u sušioniku (5.6.) podešenom na 50 °C zagrijte Erlenmeyerovu tikvicu (5.10.) i lijevak (5.11.) s umetnutim filtar-papirom. Držeći predgrijanu tikvicu, lijevak i umetnuti filtar-papir u sušioniku, filtrirajte masni sloj otopljenog uzorka. Pazite da se pritom ne prenese nimalo seruma.
Manji ispitni uzorak smije se koristiti samo ako je količina raspoloživog ispitnog uzorka ograničena, čemu treba prilagoditi i cijeli postupak. Međutim, uzimanje manje količine uzorka za analizu povlači za sobom veći rizik od dobivanja nereprezentativnog uzorka.
Napomena 1.: Maslac se može dobiti iz vrhnja bućkanjem i temeljitim ispiranjem dobivenih zrna maslaca.
Napomena 2.: Mliječna mast dobivena postupkom iz 7.1.1. gotovo neće sadržavati fosfolipide.
7.1.2. Ekstrakcija prema Röse-Gottliebovoj gravimetrijskoj metodi
Ekstrahirajte frakciju masti iz ispitnog uzorka primjenjujući gravimetrijsku metodu opisanu u jednom od standarda ISO 1211 | IDF 001D, ISO 2450 | IDF 016C ili ISO 7328 | IDF 116A.
Napomena: Ako su u dobivenoj mliječnoj masti prisutni fosfolipidi, pik kolesterola bit će uvećan za oko 0,1 %. Utjecaj na sastav triglicerida standardiziran na 100 % uključujući kolesterol pritom je zanemariv.
7.1.3. Ekstrakcija iz mlijeka pomoću kolona punjenih silika gelom
0,7 ml ispitnog uzorka temperiranog na 20 °C unesite mikrolitarskom pipetom (5.7.) u kolonu Extrelut (5.3.) od 1 do 3 ml. Ostavite oko 5 minuta da se uzorak jednakomjerno rasporedi po silika gelu.
Za denaturiranje kompleksa bjelančevina i masti, u kolonu Extrelut graduiranom pipetom (5.8.) unesite 1,5 ml metanola (4.3.). Potom iz ispitnog uzorka pomoću 20 ml n-heksana (4.4.) ekstrahirajte frakciju masti. N-heksan treba dodavati polako, u manjim količinama. Otapalo koje istječe skupite u tikvicu sa zaobljenim dnom od 50 ml (5.9.), koja je prethodno bila osušena do konstantne poznate mase izvagane s točnošću od 1 mg i uz mogućnost očitanja 0,1 mg.
Nakon ekstrakcije ostavite kolonu da se cijedi dok se potpuno ne isprazni. Iz eluata destilacijom odvojite otapala na rotacijskom isparivaču (5.13.) pri čemu njegova vodena kupelj mora biti podešena na temperaturu između 40 i 50 °C. Nakon odvajanja otapala destilacijom osušite i potom izvažite tikvicu sa zaobljenim dnom i njezin sadržaj s točnošću od 1 mg, bilježeći masu uz točnost do 0,1 mg. Utvrdite masu ostatka masti tako da od dobivene mase oduzmete masu osušene prazne tikvice sa zaobljenim dnom.
Napomena: Ekstrakcija masti po Gerberu, Weibull–Berntropu ili Schmid–Bondzynski–Ratzlaffu ili izolacija mliječne masti pomoću deterdženata (BDI metoda) nisu prikladne metode za analizu triglicerida (TG) jer znatne količine djelomičnih glicerida ili fosfolipida mogu prijeći u masnu fazu. Zato je primjena ovog međunarodnog standarda ograničena u pogledu nekih proizvoda, posebno sira.
7.2. Priprema otopine uzorka
Za plinsku kromatografiju s punjenom kolonom pripremite 5 %-tnu (volumni udio) otopinu masti (dobivenu u skladu s točkom 7.1.) u n-heksanu (4.4.) ili n-heptanu (4.5.). Ovisno o dimenzijama kolone koristite koncentraciju od 1 % (širi unutarnji promjer 0,53 mm) ili nižu, za izravno ubrizgavanje u slučaju da koristite kapilarnu kolonu.
Ovisno o korištenoj koloni i masi masti dobivene u skladu s točkom 7.1.3. odredite količinu otapala (4.4. ili 4.5.) koju treba dodati materijalu ispitnog uzorka u tikvici, na temelju vaganja s točnošću od 1 mg i bilježenja mase s točnošću od 0,1 mg. Ostatak u cijelosti otopite.
Prenesite približno 1 ml otopine u ampulu (5.14.).
7.3. Kromatografsko određivanje triglicerida
7.3.1. Pomak bazne linije
Da bi se podizanje bazne linije svelo na minimum, kolonu treba kondicionirati kako je navedeno u točki 5.2.2. (kapilarna kolona) ili u Prilogu A.4. (punjena kolona).
Napomena: Zbog visoke temperature kolone analiza triglicerida pokazuje posebnu osjetljivost na podizanje bazne linije kod ugljikovih atoma s velikim brojevima.
7.3.2. Tehnika ubrizgavanja
7.3.2.1.
Da biste izbjegli diskriminacijske učinke i postigli bolju kvantifikaciju komponenti triglicerida s visokim vrelištem, primijenite tehniku „vruće igle”. Napunite iglu zrakom uvlačenjem otopine masti u šprici. Iglu umetnite u injektor. Prije ubrizgavanja zagrijavajte iglu oko 3 sekunde. Zatim sadržaj šprice brzo ubrizgajte.
7.3.2.2.
Kod hladnog ubrizgavanja u kolonu (7.3.4.2.) umetnite iglu u špricu i odmah ubrizgajte. Vrijeme zadržavanja igle u otvoru injektora treba biti takvo da se izbjegne širenje repa pika otapala.
Napomena: Optimalno vrijeme zadržavanja obično je oko 3 sekunde.
7.3.3. Kalibracija
7.3.3.1.
Za kalibraciju ispitnih uzoraka na početku svakog dana napravite dvije do tri analize standardizirane mliječne masti. Uporabite zadnju analizu standardizirane mliječne masti za određivanje faktora odziva Rfsi (maseni udio/površinski udio) triglicerida i kolesterola te ih primijenite na sljedeće ispitne uzorke (vidjeti 9.1.):
|
|
|
(1) |
gdje:
|
wsi |
je maseni udio, izražen kao postotak, svakog triglicerida (TG) ili kolesterola u standardiziranoj mliječnoj masti; |
|
Asi |
je numerička vrijednost površine pika svakog triglicerida (TG) ili kolesterola u standardiziranoj mliječnoj masti. |
Za dobivanje standardizirane mliječne masti s poznatim sastavom triglicerida koristite 7.3.3.2. ili 7.3.3.3.
7.3.3.2.
Najbolji način za određivanje faktora odziva svakog sastojka ispitnog uzorka uporaba je standardizirane mliječne masti s potvrđenim sastavom triglicerida (TG).
Napomena: Odgovarajući standard je CRM 519 (bezvodna mliječna mast) koji se može nabaviti u Institutu za referentne materijale i mjere (IRMM), Geel, Belgija (3).
7.3.3.3.
Pripremite oko 1 g mješavine standarda masti (vidjeti 4.2., koji sadrže najmanje zasićene trigliceride C24, C30, C36, C42, C48 i C54, kao i kolesterol; plus, po mogućnosti, C50 i C52) vaganjem s točnošću od 1 mg i bilježenjem mase s točnošću od 0,1 mg da biste dobili odgovarajući sastav triglicerida (TG), sličan mliječnoj masti.
Otopinu mješavine standarda masti više puta analizirajte u n-heksanu (4.4.) ili n-heptanu (4.5.) u skladu s točkom 7.3.4. U istom slijedu više puta analizirajte mliječnu mast prosječnog sastava.
Odredite faktore odziva triglicerida (TG) na temelju mješavine standarda masti. Privremeni faktori odziva triglicerida (TG) koji nisu prisutni u mješavini mogu se izračunati matematičkom interpolacijom. Primijenite dobivene faktore odziva na mliječnu mast da biste dobili standardizirani sastav. Tako dobivena standardizirana mliječna mast ima trajnost nekoliko godina, ako se čuva u dušiku na maksimalnoj temperaturi od –18 °C.
7.3.4. Kromatografski uvjeti
Napomena: Uporaba punjenih ili kapilarnih kolona obično za rezultat ima rezoluciju sličnu onoj na slici 1. Cijepanje triglicerida s parnim brojem obično se ne promatra i treba ga izbjegavati.
7.3.4.1.
|
(a) |
Temperaturni režim kolone: Početnu temperaturu grijača podesite na 210 °C. Održavajte tu temperaturu 1 minutu. Zatim je povećavajte brzinom od 6 °C/min do 350 °C. Tu (konačnu) temperaturu održavajte 5 minuta. |
|
(b) |
Temperatura detektora i injektora: Oba podesite na 370 °C. |
|
(c) |
Plin nositelj: Koristite dušik uz stalnu brzinu protoka od oko 40 ml/min. Točan protok plina nositelja podesite tako da C54 eluira na 341 °C. |
|
(d) |
Trajanje analize: 29,3 min. |
|
(e) |
Volumen ubrizgavanja: ubrizgajte 0,5 μl 5 %-tne (volumni udio) otopine uzorka. |
Ako se ne provode analize triglicerida (TG), održavajte početnu temperaturu grijača kako je navedeno u točki (a), temperaturu detektora i injektora kako je navedeno u točki (b), a brzinu protoka plina nositelja na stalnoj razini u skladu s točkom (c), isto tako preko noći, vikenda i praznika. To osigurava najveću učinkovitost kolone.
7.3.4.2.
|
(a) |
Temperaturni režim kolone: Početnu temperaturu grijača podesite na 80 °C. Održavajte tu temperaturu 0,5 minuta. Zatim temperaturu povećavajte brzinom od 50 °C/min do 190 °C, a zatim brzinom od 6 °C/min do 350 °C. Tu (konačnu) temperaturu održavajte 5 minuta. |
|
(b) |
Temperatura detektora: Podešena na 370 °C. |
|
(c) |
Plin nositelj: Koristite dušik uz stalnu brzinu protoka od oko 3 ml/min. |
|
(d) |
Trajanje analize: 34,4 min. |
|
(e) |
Volumen ubrizgavanja: Ubrizgajte 0,5 μl 1 %-tne (volumni udio) otopine uzorka. |
Da biste osigurali najbolju učinkovitost, zadržite ove postavke i u stanju mirovanja (vidjeti 7.3.4.1.).
Analitičke postavke navedene u točki 7.3.4.2. primjerene su za primjenu kad se koristi kolona šireg unutarnjeg promjera (ID 0,53 mm) kako je navedeno u točki 5.2.2. Ako se koristi kolona drugih dimenzija ili neka druga faza, mogu se primjenjivati drukčije postavke.
8. INTEGRACIJA, OCJENA I KONTROLA UČINKOVITOSTI ANALIZA
Ocijenite pikove kromatograma pomoću integracijskog sustava s mogućnošću crtanja i reintegracije bazne linije. Slika 1. prikazuje pravilno integrirani kromatogram, dok slika 2. prikazuje sporadičnu pogrešku u baznoj liniji koja završava iza C54, što utječe na postotke svih triglicerida (TG). Neovisno o tome, iz ocjene isključite pikove koji se pojavljuju nakon C54.
Trigliceride s neparnim acil-C brojem (2n + 1) kombinirajte s trigliceridima s prethodnim parnim brojem (2n). Nemojte uzeti u obzir niski sadržaj C56. Postotke površina preostalih triglicerida, uključujući kolesterol, pomnožite s odgovarajućim faktorima odziva standardizirane mliječne masti (posljednja kalibracija) i zajedno normalizirajte na 100 % u skladu s točkom 9.1.
Slika 1.
Primjer kromatograma triglicerida mliječne masti s pravilno postavljenom baznom linijom
Slika 2.
Primjer kromatograma triglicerida mliječne masti s nepravilno postavljenom baznom linijom

Radi kontrole mjernih uvjeta napravite usporedbu s koeficijentima varijacije (CV) različitih triglicerida, koji su izraženi kao postoci i navedeni u tablici 1., koji se temelje na 19 uzastopnih analiza istog uzorka mliječne masti.
Ako su koeficijenti varijacije (CV) znatno viši od vrijednosti navedenih u tablici 1., kromatografski uvjeti nisu odgovarajući.
Napomena: Vrijednosti navedene u tablici 1. nisu obvezne, ali su indikativne u smislu kontrole kvalitete.
Međutim, u slučaju da su vrijednosti koeficijenata varijacije (CV) veće, granice ponovljivosti i obnovljivosti navedene u klauzuli 10. usprkos tome treba poštovati.
Tablica 1.
Koeficijenti varijacije sadržaja triglicerida (19 uzastopnih analiza)
|
Triglicerid |
Koeficijent varijacije (CV) % |
|
C24 |
10,00 |
|
C26 |
2,69 |
|
C28 |
3,03 |
|
C30 |
1,76 |
|
C32 |
1,03 |
|
C34 |
0,79 |
|
C36 |
0,25 |
|
C38 |
0,42 |
|
C40 |
0,20 |
|
C42 |
0,26 |
|
C44 |
0,34 |
|
C46 |
0,37 |
|
C48 |
0,53 |
|
C50 |
0,38 |
|
C52 |
0,54 |
|
C54 |
0,60 |
9. IZRAČUN I IZRAŽAVANJE REZULTATA
9.1. Sastav triglicerida
9.1.1. Izračun
Izračunajte maseni udio svakog triglicerida (TG) (za i = C24, C26, C28, C30, C32, C34, C36, C38, C40, C42, C44, C46, C48, C50, C52 i C54) plus kolesterola, wi, izražen kao postotak, ukupnog sadržaja triglicerida (TG) u ispitnom uzorku pomoću sljedeće jednadžbe:
|
|
|
(2) |
gdje je:
|
Ai |
numerička vrijednost površine pika svakog triglicerida (TG) u ispitnom uzorku; |
|
RFsi |
faktor odziva svakog triglicerida (TG) određen kalibracijom (7.3.3.). |
9.1.2. Izražavanje rezultata ispitivanja
Izrazite rezultate na dvije decimale.
9.2. S-vrijednosti
9.2.1. Izračun
9.2.1.1. Izračunajte S-vrijednosti, izražene u postocima, tako što ćete izračunane vrijednosti wi (9.1.1.) odgovarajućih postotaka triglicerida (TG) unijeti u jednadžbe (3) do (7). Uporabite sve jednadžbe bez obzira na vrstu strane masti na koju sumnjate.
9.2.1.2. Sojino ulje, suncokretovo ulje, maslinovo ulje, repičino ulje, laneno ulje, ulje pšeničnih klica, ulje kukuruznih klica, ulje pamuka, riblje ulje
S = 2,098 3 · wC30 + 0,728 8 · wC34 + 0,692 7 · wC36 + 0,635 3 · wC38 + 3,745 2 · wC40 – 1,292 9 · wC42 + 1,354 4 · wC44 + 1,701 3 · wC46 + 2,528 3 · wC50 (3)
9.2.1.3. Kokosova mast i mast palminih koštica
S = 3,745 3 · wC32 + 1,113 4 · wC36 + 1,364 8 · wC38 + 2,154 4 · wC42 + 0,427 3 · wC44 + 0,580 9 · wC46 + 1,292 6 · wC48 + 1,030 6 · wC50 + 0,995 3 · wC52 + 1,239 6 · wC54 (4)
9.2.1.4. Palmino ulje i goveđi loj
S = 3,664 4 · wC28 + 5,229 7 · wC30 – 12,507 3 · wC32 + 4,428 5 · wC34 – 0,201 0 · wC36 + 1,279 1 · wC38 + 6,743 3 · wC40 – 4,271 4 · wC42 + 6,373 9 · wC46 (5)
9.2.1.5. Svinjska mast
S = 6,512 5 · wC26 + 1,205 2 · wC32 + 1,733 6 · wC34 + 1,755 7 · wC36 + 2,232 5 · wC42 + 2,800 6 · wC46 + 2,543 2 · wC52 + 0,989 2 · wC54 (6)
9.2.1.6. Ukupno
S = – 2,757 5 · wC26 + 6,407 7 · wC28 + 5,543 7 · wC30 – 15,324 7 · wC32 + 6,260 0 · wC34 + 8,010 8 · wC40 – 5,033 6 · wC42 + 0,635 6 · wC44 + 6,017 1 · wC46 (7)
9.2.2. Izražavanje ispitnih rezultata
Rezultate izrazite na dvije decimale.
9.3. Detekcija stranih masti
Usporedite pet S-vrijednosti iz točke 9.2.1. s odgovarajućim S-vrijednostima navedenim u tablici 2.
Ako je svih pet S-vrijednosti unutar granica navedenih u tablici 2., smatra se da je uzorak čista mliječna mast. Međutim, ako je bilo koja od S-vrijednosti veća od odgovarajuće granične vrijednosti, smatra se da uzorak sadrži strane masti.
Iako su pojedinačne jednadžbe (3) do (6) osjetljivije na neke strane masti od skupne jednadžbe (7) (vidjeti tablicu B.1.), na temelju pozitivnog rezultata dobivenog primjenom samo jedne od jednadžbi (3) do (6) ne može se izvesti zaključak o vrsti strane masti.
U Prilogu B opisan je postupak za izračunavanje sadržaja biljne ili životinjske masti u patvorenoj mliječnoj masti. Taj postupak nije validiran i samo je informativnog karaktera.
Tablica 2.
S-granice za čiste mliječne masti
|
Strana mast |
Jednadžba |
S-granice (4) |
|
Sojino ulje, suncokretovo ulje, maslinovo ulje, repičino ulje, laneno ulje, ulje pšeničnih klica, ulje kukuruznih klica, ulje pamuka, riblje ulje |
(3) |
98,05 do 101,95 |
|
Kokosova mast i mast palminih koštica |
(4) |
99,42 do 100,58 |
|
Palmino ulje i goveđi loj |
(5) |
95,90 do 104,10 |
|
Svinjska mast |
(6) |
97,96 do 102,04 |
|
Ukupno |
(7) |
95,68 do 104,32 |
10. PRECIZNOST
10.1. Međulaboratorijsko ispitivanje
Vrijednosti ponovljivosti i obnovljivosti utvrđene su na temelju jednadžbi (3) do (7) uz korištenje čiste mliječne masti i možda neće biti primjenjive na matrice koje se razlikuju od navedenih.
10.2. Ponovljivost
Apsolutna razlika između rezultata dvaju pojedinačnih ispitivanja, koje istom metodom na jednakom ispitnom materijalu u istom laboratoriju dobije isti analitičar koristeći istu opremu u kratkom vremenskom intervalu, ne smije u više od 5 % slučajeva prekoračiti granice navedene u tablici 3.
Tablica 3.
Granice ponovljivosti r za jednadžbe (3) do (7)
|
Strana mast |
Jednadžba |
r % |
|
Sojino ulje, suncokretovo ulje, maslinovo ulje, repičino ulje, laneno ulje, ulje pšeničnih klica, ulje kukuruznih klica, ulje pamuka, riblje ulje |
(3) |
0,67 |
|
Kokosova mast i mast palminih koštica |
(4) |
0,12 |
|
Palmino ulje i goveđi loj |
(5) |
1,20 |
|
Svinjska mast |
(6) |
0,58 |
|
Ukupno |
(7) |
1,49 |
10.3. Obnovljivost
Apsolutna razlika između rezultata dvaju nezavisnih pojedinačnih ispitivanja koja su primjenom iste metode na jednakom ispitnom materijalu u različitim laboratorijima izvršili različiti analitičari koristeći različitu opremu ne smije u više od 5 % slučajeva biti veća od graničnih vrijednosti navedenih u tablici 4.
Tablica 4.
Granice obnovljivosti R za jednadžbe (3) do (7)
|
Strana mast |
Jednadžba |
R % |
|
Sojino ulje, suncokretovo ulje, maslinovo ulje, repičino ulje, laneno ulje, ulje pšeničnih klica, ulje kukuruznih klica, ulje pamuka, riblje ulje |
(3) |
1,08 |
|
Kokosova mast i mast palminih koštica |
(4) |
0,40 |
|
Palmino ulje i goveđi loj |
(5) |
1,81 |
|
Svinjska mast |
(6) |
0,60 |
|
Ukupno |
(7) |
2,07 |
11. NESIGURNOST MJERENJA
Pomoću ponovljivosti r i obnovljivosti R može se izračunani proširena nesigurnost za S-vrijednost.
Uključivanjem proširene nesigurnosti (na temelju dvostrukih analiza) u S-granice iz tablice 2. dobijemo proširene S-granice kako su navedene u tablici 5.
Tablica 5.
Proširene S-granice za čiste mliječne masti koje uključuju proširenu nesigurnost
|
Strana mast |
Jednadžba |
Proširene S-granice |
|
Sojino ulje, suncokretovo ulje, maslinovo ulje, repičino ulje, laneno ulje, ulje pšeničnih klica, ulje kukuruznih klica, ulje pamuka, riblje ulje |
(3) |
97,36 do 102,64 |
|
Kokosova mast i mast palminih koštica |
(4) |
99,14 do 100,86 |
|
Palmino ulje i goveđi loj |
(5) |
94,77 do 105,23 |
|
Svinjska mast |
(6) |
97,65 do 102,35 |
|
Ukupno |
(7) |
94,42 do 105,58 |
12. IZVJEŠTAJ O ISPITIVANJU
U izvještaju o ispitivanju treba navesti:
|
— |
sve informacije potrebne za potpunu identifikaciju uzorka, |
|
— |
korištenu metodu uzorkovanja, ako je poznata, |
|
— |
metodu uzorkovanja korištenu u skladu s ovim međunarodnim standardom, |
|
— |
sve pojedinosti o postupku koje nisu navedene u ovom međunarodnom standardu ili se ne smatraju obveznima, zajedno s pojedinostima o svim događajima koji su mogli utjecati na rezultat(e) ispitivanja, |
|
— |
dobivene rezultate i, ako se provjeravala ponovljivost, konačni dobiveni rezultat. |
(1) Primjer odgovarajućeg proizvoda koji je dostupan na tržištu. Ova je informacija navedena kao pogodnost za korisnike ovog međunarodnog standarda i ne predstavlja službenu potvrdu tog proizvoda.
(2) CP-Ultimetal SimDist (5 m × 0,53 mm × 0,17 μm) primjer je odgovarajućeg proizvoda koji je dostupan na tržištu. Ova je informacija navedena kao pogodnost za korisnike ovog međunarodnog standarda i ne predstavlja službenu potvrdu tog proizvoda.
(3) Primjer odgovarajućeg proizvoda koji je dostupan na tržištu. Ova je informacija navedena kao pogodnost za korisnike ovog međunarodnog standarda i ne predstavlja službenu potvrdu tog proizvoda.
(4) Izračunato uz razinu pouzdanosti od 99 %, tako da je strana mast navedena samo ako su prekoračene granice detekcije iz odgovarajuće jednadžbe (vidjeti tablicu B.1.).
PRILOG A
(normativni)
PRIPREMA PUNJENE KOLONE
A. REAGENSI I OPREMA
A.1.1. Toluen (C6H5CH3).
A.1.2. Otopina dimetildiklorosilana [Si(CH3)2Cl2].
Otopite 50 ml dimetildiklorosilana u 283 ml toluena (A.1.1.).
A.1.3. Otopina kakao maslaca s masenim udjelom od 5 % kakao maslaca u n-heksanu (4.4.) ili n-heptanu (4.5.).
A.1.4. Stacionarna faza, 3 % OV-1 na 125 μm do 150 μm (100 do 120 mesh) Gas ChromQ (1).
Napomena: Oznaka za zrno bila je promijenjena u mikrometre u skladu sa standardom BS 410 (svi dijelovi) [6].
A.1.5. Staklena kolona unutarnjeg promjera 2 mm i duljine 500 mm, u obliku slova U.
Oprema za punjenje kolona.
A.1.6.1. Punjenje kolone, s navojnim čepovima na krajevima, s oznakom do koje se kolona može puniti potrebnom količinom stacionarne faze.
A.1.6.2. Fino sito s mrežom veličine 100 μm, s navojnim čepom prikladnim za zatvaranje staklene kolone u skladu sa slikom A.3.
A.1.6.3. Silanizirana staklena vuna, deaktivirana.
A.1.6.4. Vibrator za jednakomjerno raspoređivanje stacionarne faze tijekom punjenja.
A.1.6.5. Oprema za silaniziranje staklene površine kolone.
A.1.6.6. Woulffova boca.
A.1.6.7. Usisna pumpa za vodu.
A.2. SILANIZIRANJE (DEAKTIVACIJA STAKLENE POVRŠINE)
Nakon priključivanja Woulffove boce (A.1.6.6.) na usisnu pumpu za vodu (A.1.6.7.), uronite cijev 2. (vidjeti sliku A.1.) u otopinu dimetildiklorosilana (A.1.2.). Napunite kolonu (A.1.5.) tom otopinom, zatvaranjem zapornog ventila. Zaporni ventil ponovo otvorite i potom skinite obje cijevi. Pričvrstite kolonu na stalak. Napunite je do kraja otopinom dimetildiklorosilana (A.1.2.) koristeći pipetu.
Slika A.1.
Oprema za silaniziranje
![]()
Legenda:
|
1. |
cijev 1. |
|
2. |
cijev 2. |
|
3. |
usisna pumpa za vodu |
|
4. |
zaporni ventil |
|
5. |
staklena kolona |
|
6. |
dimetildiklorosilan i toluen |
Kolonu ostavite da odstoji 20–30 minuta. Zatim Woulffovu bocu zamijenite tikvicom za filtriranje. Kolonu ispraznite tako da je povežete s usisnom pumpom za vodu (A.1.6.7.) (vidjeti sliku A.2.). Ispražnjenu kolonu isperite najprije sa 75 ml toluena (A.1.1.), a zatim s 50 ml metanola (4.3.) uranjajući cijev 2. u otopinu. Ispranu kolonu oko 30 minuta sušite u sušioniku (5.6.) podešenom na 100 °C.
Slika A.2.
Naprava za ispiranje
![]()
Legenda:
|
1. |
cijev 1. |
|
2. |
cijev 2. |
|
3. |
usisna pumpa za vodu |
|
4. |
tikvica za filtriranje |
|
5. |
staklena kolona |
|
6. |
sredstvo za ispiranje |
A.3. PUNJENJE
Napunite kolonu koristeći opremu prikazanu na slici A.3. Kolonu za punjenje (A.1.6.1.) do oznake napunite stacionarnom fazom (A.1.4.). Niži kraj staklene kolone koju ćete puniti začepite oko 1 cm dugim čepom od silanizirane komprimirane staklene vune (A.1.6.3.). Kraj kolone zatvorite finim sitom (A.1.6.2.).
Slika A.3.
Punjenje staklene kolone

Legenda:
|
1. |
dovod dušika |
|
2. |
kolona kojom se puni i koju do oznake treba napuniti s OV-1 |
|
3. |
staklena kolona koju treba napuniti |
|
4. |
navojni čep s filtrom na koji su pritisnute staklena vuna i stacionarna faza |
Kolonu napunite pod tlakom (300 kPa i struja dušika) stacionarnom fazom. Da biste dobili ujednačeno, kontinuirano i čvrsto punjenje, duž kolone tijekom punjenja treba u oba smjera povlačiti vibrator. Nakon punjenja u drugi kraj napunjene kolone utisnite tvrdi čep od silanizirane staklene vune (A.1.6.3). Krajeve koji vire odrežite. Špatulom utisnite čep nekoliko milimetara u kolonu.
A.4. KONDICIONIRANJE
Tijekom faza (a) do (c) neka stražnji kraj kolone ne bude povezan s detektorom da bi se izbjegla kontaminacija. Napunjenu kolonu (A.3.) kondicionirajte na sljedeći način:
|
(a) |
kolonu ispirite dušikom 15 minuta, uz brzinu protoka podešenu na 40 ml/min i grijač plinskog kromatografa (GC) podešen na 50 °C; |
|
(b) |
kolonu zagrijavajte brzinom od 1 °C/min do 355 °C, uz brzinu protoka dušika podešenu na 10 ml/min; |
|
(c) |
kolonu držite na 355 °C od 12 do 15 sati; |
|
(d) |
dvaput ubrizgajte po 1 μl otopine kakao maslaca (A.1.3.) primjenjujući temperaturni režim za punjenu kolonu naveden u točki 7.3.4.1.; Napomena: Kakao maslac sastoji se gotovo isključivo od triglicerida C50 do C54 s visokim vrelištem, pa je stoga kondicioniranje kolone jednostavnije s obzirom na odgovarajuće faktore odziva. |
|
(e) |
20 puta ubrizgajte 0,5 μl otopine mliječne masti u skladu s točkom 7.2. u 2 do 3 dana koristeći postavke za punjenu kolonu navedene u točki 7.3.4.1.
|
(1) Primjer odgovarajućeg proizvoda koji je dostupan na tržištu. Ova je informacija navedena kao pogodnost za korisnike ovog međunarodnog standarda i ne znači da je proizvod potvrđen standardima ISO ili IDF.
PRILOG B
(informativan)
KVANTIFIKACIJA SADRŽAJA STRANIH MASTI
B.1. OPĆENITO
Tablica B.1. prikazuje granice detekcije za različite strane masti, izračunane uz razinu pouzdanosti od 99 %. Srednji stupac prikazuje granice detekcije pojedinačnih jednadžbi (3) do (6) s najboljim rezultatima.
Granice detekcije ukupne jednadžbe (7), prikazane u krajnjem desnom stupcu, nešto su više. U načelu, jednadžba (7) potrebna je samo za kvantifikaciju stranih masti.
Pomoću svih jednadžbi isto se tako može utvrditi prisutnost kombinacija različitih stranih masti. Varijacije sastava triglicerida (TG) pojedinačnih uzoraka iste vrste strane masti nemaju značajniji utjecaj na granice detekcije.
Ako se koriste i pojedinačne jednadžbe i skupna jednadžba, primjenjuju se granice detekcije pojedinačnih jednadžbi. Međutim, u nekim je slučajevima za kvantifikaciju potrebna S-vrijednost skupne jednadžbe (B.2.).
Tablica B.1.
99 %-tne granice detekcije stranih masti dodanih mliječnoj masti, izražene kao postoci
|
Strana mast |
Pojedinačna jednadžba % |
Ukupna jednadžba |
|
Sojino ulje |
2,1 |
4,4 |
|
Suncokretovo ulje |
2,3 |
4,8 |
|
Maslinovo ulje |
2,4 |
4,7 |
|
Kokosovo ulje |
3,5 |
4,3 |
|
Palmino ulje |
4,4 |
4,7 |
|
Mast palminih koštica |
4,6 |
5,9 |
|
Repičino ulje |
2,0 |
4,4 |
|
Laneno ulje |
2,0 |
4,0 |
|
Ulje pšeničnih klica |
2,7 |
6,4 |
|
Ulje kukuruznih klica |
2,2 |
4,5 |
|
Ulje sjemenki pamuka |
3,3 |
4,4 |
|
Svinjska mast |
2,7 |
4,7 |
|
Goveđi loj |
5,2 |
5,4 |
|
Hidrogenizirano riblje ulje |
5,4 |
6,1 |
B.2. IZRAČUN
Kvantitativno određivanje strane masti provedite samo ako je prekoračena najmanje jedna od S-granica (tablica 2. ili tablica 5.). Za dobivanje kvantitativnih podataka izračunajte maseni udio strane masti ili maseni udio mješavine stranih masti, wf, izražen u obliku postotka, u ispitnom uzorku pomoću sljedeće jednadžbe:
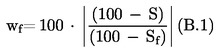
gdje:
|
S |
je rezultat dobiven uvrštavanjem podataka o trigliceridima (TG) iz mliječne masti kojima je dodana strana mast ili mješavina stranih masti u jednu od jednadžbi (3) do (7); |
|
Sf |
je konstanta, ovisna o vrsti dodane strane masti. |
Ako vrsta strane masti dodane mliječnoj masti nije poznata, koristite opću Sf -vrijednost od 7,46 (tablica B.2.). Uvijek koristite S-vrijednost dobivenu pomoću jednadžbe (7), čak i ako njezine S-vrijednosti nisu prekoračene, već su prekoračene vrijednosti iz neke druge jednadžbe.
Kad su strane masti poznate, uvrstite njihove pojedinačne Sf-vrijednosti (tablica B.2.) u jednadžbu (B.1.). Za izračun S-vrijednosti odaberite odgovarajuću jednadžbu za stranu mast među jednadžbama od (3) do (6).
Tablica B.2.
Sf-vrijednosti različitih stranih masti
|
Strana mast |
Sf |
|
Nepoznata |
7,46 |
|
Sojino ulje |
8,18 |
|
Suncokretovo ulje |
9,43 |
|
Maslinovo ulje |
12,75 |
|
Kokosovo ulje |
118,13 |
|
Palmino ulje |
7,55 |
|
Mast palminih koštica |
112,32 |
|
Repičino ulje |
3,30 |
|
Laneno ulje |
4,44 |
|
Ulje pšeničnih klica |
27,45 |
|
Ulje kukuruznih klica |
9,29 |
|
Ulje sjemenki pamuka |
41,18 |
|
Svinjska mast |
177,55 |
|
Goveđi loj |
17,56 |
|
Riblje ulje |
64,12 |
B.3. IZRAŽAVANJE REZULTATA
Rezultate izrazite na dvije decimale.
Bibliografija
|
1. |
Molkentin, J., Precht, D. Representative determination of the butyric acid content in European milk fats. Milchwissenschaft, 52, 1987., str. 82.-85. |
|
2. |
Precht, D., Molkentin, J. Quantitative triglyceride analysis using short capillary columns. Chrompack News, 4, 1993., str. 16.-17. |
|
3. |
Molkentin, J., Precht, D. Comparison of packed and capillary columns for quantitative gas chromatography of triglycerides in milk fat. Chromatographia, 39, 1994., str. 265.-270. |
|
4. |
Molkentin, J., Precht, D. Equivalence of packed and capillary GC columns with respect to suitability for foreign fat detection in butter using the triglyceride formula method. Chromatographia, 52, 2000., str. 791.-797. |
|
5. |
ISO 707|IDF 50, Mlijeko i mliječni proizvodi – upute za uzorkovanje |
|
6. |
BS 410:1988., Ispitna sita – Tehnički zahtjevi i ispitivanje |
|
7. |
Precht, D. Control of milk fat purity by gas chromatographic triglyceride analysis. Kieler Milchwirtsch. Forschungsber., 43, 1991., str. 219.-242. |
|
8. |
Precht, D., Detection of adulterated milk fat by fatty acid and triglyceride analyses, Fat Sci. Technol., 93, 1991., str. 538.-544. |
|
9. |
DIN 10336:1994, Nachweis und Bestimmung von Fremdfetten in Milchfett anhand einer gaschromatographischen Triglyceridanalyse |
|
10. |
Komisija Europskih zajednica: Consideration of results from the first, second, third, fourth, fifth and sixth EEC collaborative trial: Determination of triglycerides in milk fat; Dokumenti br. VI/2644/91, VI/8.11.91, VI/1919/92, VI 3842/92, VI/5317/92, VI/4604/93 |
|
11. |
Molkentin, J., Detection of foreign fat in milk fat from different continents by triacylglycerol analysis, Eur. J. Lipid Sci. Technol., 109, 2007., str. 505.-510. |
PRILOG XXI.
(Članak 18.)
POSTUPAK KOJI SE PRIMJENJUJE U SLUČAJEVIMA KAD SU REZULTATI ANALIZE SPORNI (KEMIJSKA ANALIZA)
1. Na zahtjev proizvođača provodi se dodatna analiza u drugom laboratoriju koji odobri nadležno tijelo, uz primjenu odgovarajuće metode, pod uvjetom da su raspoložive zapečaćene kopije uzoraka proizvoda koje je nadležno tijelo čuvalo na propisan način. Zahtjev se podnosi u roku od sedam radnih dana po objavi rezultata prve analize. Analiza se provodi unutar razdoblja od 21 radnog dana od primitka zahtjeva. Nadležno tijelo te uzorke šalje u drugi laboratorij na zahtjev i trošak proizvođača. Laboratorij mora biti ovlašten za izvođenje službenih analiza i prethodno dokazati svoju osposobljenost za izvođenje predmetne analize.
2. Proširene nesigurnosti (k = 2) srednje vrijednosti 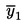
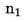 ponovljenih mjerenja u laboratoriju 1. i srednje vrijednosti
ponovljenih mjerenja u laboratoriju 1. i srednje vrijednosti 
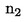 ponovljenih mjerenja u laboratoriju 2. su
ponovljenih mjerenja u laboratoriju 2. su
3. 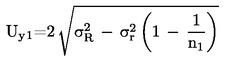 odnosno
odnosno 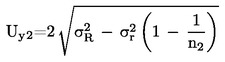 gdje je
gdje je  standardna devijacija ponovljivosti, a
standardna devijacija ponovljivosti, a ![]() standardna devijacija obnovljivosti relevantne metode. Ako se konačni rezultat y mjerenja u laboratorijima izračuna prema formuli u obliku
standardna devijacija obnovljivosti relevantne metode. Ako se konačni rezultat y mjerenja u laboratorijima izračuna prema formuli u obliku 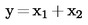 ,
, 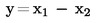 ,
, 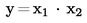 ili
ili 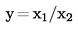 , u takvim primjerima obvezno treba primijeniti uobičajene postupke za kombiniranje standardnih devijacija da bi se dobila nesigurnost.
, u takvim primjerima obvezno treba primijeniti uobičajene postupke za kombiniranje standardnih devijacija da bi se dobila nesigurnost.
4. Da bi se provjerilo jesu li rezultati dobiveni u dva laboratorija u skladu sa standardnom devijacijom obnovljivosti ![]() metode, izračunava se proširena nesigurnost razlike
metode, izračunava se proširena nesigurnost razlike 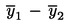 :
:
5. 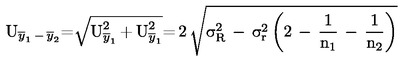 Ako apsolutna vrijednost razlike laboratorijskih srednjih vrijednosti
Ako apsolutna vrijednost razlike laboratorijskih srednjih vrijednosti  nije veća od njezine nesigurnosti
nije veća od njezine nesigurnosti 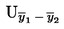 ,
,
![]() ,
,
rezultati dobiveni u dva laboratorija u skladu su sa standardnom devijacijom obnovljivosti 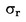 i aritmetička sredina dviju laboratorijskih srednjih vrijednosti
i aritmetička sredina dviju laboratorijskih srednjih vrijednosti
 ,
,
objavljuje se kao konačni rezultat. Njezina je proširena nesigurnost
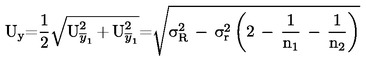 .
.
Pošiljka se odbija kao nesukladna s gornjom zakonski utvrđenom granicom UL ako je
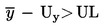 ;
;
inače se prihvaća kao sukladna s UL.
Pošiljka se odbija kao nesukladna s donjom zakonski utvrđenom granicom LL ako je
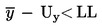 ;
;
inače se prihvaća kao sukladna s LL.
Ako je apsolutna vrijednost razlike laboratorijskih srednjih vrijednosti 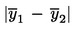 veća od njezine nesigurnosti
veća od njezine nesigurnosti 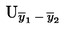 ,
,
![]() ,
,
rezultati dobiveni u dva laboratorija nisu u skladu sa standardnom devijacijom obnovljivosti.
U tom se slučaju pošiljka odbija kao nesukladna, ako druga analiza potvrdi prvu. Inače se prihvaća kao sukladna.
Nadležno tijelo mora o konačnom rezultatu što prije obavijestiti proizvođača. U slučaju odbijanja pošiljke troškove druge analize snosi proizvođač.
PRILOG XXII.
KORELACIJSKA TABLICA
|
Uredba (EZ) br. 213/2001 |
Ova Uredba |
|
Članak 1. |
Članak 1. |
|
Članak 2. |
Članak 1. |
|
Članak 3. |
Članak 2. |
|
— |
Članak 3. |
|
Članak 4. |
— |
|
Članak 5. |
— |
|
Članak 6. |
Članak 4. |
|
Članak 7. |
Članak 18. |
|
Članak 8. |
— |
|
Članak 9. |
Članak 5. |
|
Članak 10. |
Članak 6. |
|
Članak 11. |
Članak 7. |
|
Članak 12. |
Članak 8. |
|
Članak 13. |
Članak 9. |
|
Članak 14. |
Članak 10. |
|
Članak 15. |
Članak 11. |
|
Članak 16. |
Članak 12. |
|
Članak 17. |
Članak 13. |
|
— |
Članak 14. |
|
Članak 18. |
Članak 15. |
|
Članak 19. |
Članak 16. |
|
|
Članak 17. |
|
|
Članak 19. |
|
Članak 20. |
— |
|
Članak 21. |
— |
|
Članak 22. |
Članak 20. |
|
Članak 23. |
Članak 21. |