

EUR-Lex Access to European Union law
This document is an excerpt from the EUR-Lex website
Document 32009R0152
Commission Regulation (EC) No 152/2009 of 27 January 2009 laying down the methods of sampling and analysis for the official control of feed (Text with EEA relevance)
Uredba Komisije (EZ) br. 152/2009 od 27. siječnja 2009. o utvrđivanju metoda uzorkovanja i analize za službenu kontrolu hrane za životinje Tekst značajan za EGP
Uredba Komisije (EZ) br. 152/2009 od 27. siječnja 2009. o utvrđivanju metoda uzorkovanja i analize za službenu kontrolu hrane za životinje Tekst značajan za EGP
OJ L 54, 26.2.2009, p. 1–130
(BG, ES, CS, DA, DE, ET, EL, EN, FR, IT, LV, LT, HU, MT, NL, PL, PT, RO, SK, SL, FI, SV)
Special edition in Croatian: Chapter 03 Volume 006 P. 3 - 132
 In force: This act has been changed. Current consolidated version: 04/04/2024
In force: This act has been changed. Current consolidated version: 04/04/2024
|
03/Sv. 6 |
HR |
Službeni list Europske unije |
3 |
32009R0152
|
L 054/1 |
SLUŽBENI LIST EUROPSKE UNIJE |
UREDBA KOMISIJE (EZ) br. 152/2009
od 27. siječnja 2009.
o utvrđivanju metoda uzorkovanja i analize za službenu kontrolu hrane za životinje
(Tekst značajan za EGP)
KOMISIJA EUROPSKIH ZAJEDNICA,
uzimajući u obzir Ugovor o osnivanju Europske zajednice,
uzimajući u obzir Uredbu (EZ) br. 882/2004 Europskog parlamenta i Vijeća od 29. travnja 2004. o službenom nadzoru koji se provodi radi provjere pridržavanja propisa o hrani i hrani za životinje te pravila o zdravlju i dobrobiti životinja (1), a posebno njezin članak 11. stavak 4. točke (a), (b) i (c),
budući da:
|
(1) |
Sljedeći akti doneseni su za provedbu Direktive 70/373/EEZ i ostaju na snazi u skladu s člankom 61. stavkom 2. Uredbe (EZ) br. 882/2004:
|
|
(2) |
S obzirom da je Direktiva 70/373/EEZ zamijenjena Uredbom (EZ) br. 882/2004, prikladno je zamijeniti provedbene akte navedene Direktive jednom uredbom. Istodobno je potrebno prilagoditi metode s obzirom na znanstveno-tehnološki razvoj. Potrebno je staviti izvan snage metode koje više nisu valjane za njihovu predviđenu namjenu. Iako je predviđeno pravodobno ažuriranje odredaba o uzorkovanju kako bi se uzeli u obzir najnoviji rezultati razvoja u proizvodnji, skladištenju, prijevozu i trgovanju hranom za životinje, za sada je primjereno zadržati postojeće odredbe o uzorkovanju. |
|
(3) |
Zbog toga treba staviti izvan snage direktive 71/250/EEZ, 71/393/EEZ, 72/199/EEZ, 73/46/EEZ, 76/371/EEZ, 76/372/EEZ, 78/633/EEZ, 81/715/EEZ, 84/425/EEZ, 86/174/EEZ, 93/70/EEZ, 93/117/EZ, 98/64/EZ, 1999/27/EZ, 1999/76/EZ, 2000/45/EZ, 2002/70/EZ i 2003/126/EZ. |
|
(4) |
Mjere predviđene ovom Uredbom u skladu su s mišljenjem Stalnog odbora za prehrambeni lanac i zdravlje životinja, |
DONIJELA JE OVU UREDBU:
Članak 1.
Uzorkovanje za službenu kontrolu hrane za životinje u vezi s određivanjem sastojaka, dodataka i nepoželjnih tvari, osim ostataka pesticida i mikroorganizama, provodi se u skladu s metodama iz Priloga I.
Članak 2.
Priprema uzoraka za analizu i prikazivanje rezultata izvode se u skladu s metodama iz Priloga II.
Članak 3.
Analiza za službenu kontrolu hrane za životinje izvodi se pomoću metoda iz Priloga III. (Analitičke metode za kontrolu sastava hrane za životinje i krmnih smjesa), Priloga IV. (Analitičke metode za kontrolu količine dopuštenih krmnih dodataka), Priloga V. (Analitičke metode za kontrolu nepoželjnih tvari u hrani za životinje) i Priloga VI. (Analitičke metode za određivanje sastojaka životinjskog podrijetla u okviru službene kontrole hrane za životinje).
Članak 4.
Energetska vrijednost krmnih smjesa za perad izračunava se u skladu s Prilogom VII.
Članak 5.
U svrhu potvrđivanja koriste se metode analize za kontrolu nedopuštenih krmnih dodataka iz Priloga VIII.
Članak 6.
Direktive 71/250/EEZ, 71/393/EEZ, 72/199/EEZ, 73/46/EEZ, 76/371/EEZ, 76/372/EEZ, 78/633/EEZ, 81/715/EEZ, 84/425/EEZ, 86/174/EEZ, 93/70/EEZ, 93/117/EZ, 98/64/EZ, 1999/27/EZ, 1999/76/EZ, 2000/45/EZ, 2002/70/EZ i 2003/126/EZ stavljaju se izvan snage.
Upućivanja na Direktive stavljene izvan snage smatraju se upućivanjima na ovu Uredbu i tumače u skladu s korelacijskim tablicama iz Priloga IX.
Članak 7.
Ova Uredba stupa na snagu dvadesetog dana od dana objave u Službenom listu Europske unije.
Ona se primjenjuje od 26. kolovoza 2009.
Ova je Uredba u cijelosti obvezujuća i izravno se primjenjuje u svim državama članicama.
Sastavljeno u Bruxellesu 27. siječnja 2009.
Za Komisiju
Androulla VASSILIOU
Članica Komisije
(1) SL L 165, 30.4.2004., str. 1., ispravljeno u SL L 191, 28.5.2004., str. 1.
(2) SL L 155, 12.7.1971., str. 13.
(3) SL L 279, 20.12.1971., str. 7.
(4) SL L 123, 29.5.1972., str. 6.
(5) SL L 83, 30.3.1973., str. 21.
(6) SL L 102, 15.4.1976., str. 1.
(7) SL L 102, 15.4.1976., str. 8.
(8) SL L 206, 29.7.1978., str. 43.
(9) SL L 257, 10.9.1981., str. 38.
(10) SL L 238, 6.9.1984., str. 34.
(11) SL L 130, 16.5.1986., str. 53.
(12) SL L 234, 17.9.1993., str. 17.
(13) SL L 329, 30.12.1993., str. 54.
(14) SL L 257, 19.9.1998., str. 14.
(15) SL L 118, 6.5.1999., str. 36.
(16) SL L 207, 6.8.1999., str. 13.
(17) SL L 174, 13.7.2000., str. 32.
(18) SL L 209, 6.8.2002., str. 15.
(19) SL L 339, 24.12.2003., str. 78.
PRILOG I.
METODE UZORKOVANJA
1. SVRHA I PODRUČJE PRIMJENE
Uzorci za službenu kontrolu hrane za životinje uzimaju se u skladu s dolje opisanim metodama. Tako dobiveni uzorci smatraju se reprezentativnima za uzorkovane dijelove.
2. OSOBLJE KOJE UZIMA UZORKE
Uzorke uzimaju osobe koje za tu svrhu ovlašćuju države članice.
3. DEFINICIJE POJMOVA
Uzorkovani dio: količina proizvoda koja čini cjelinu i ima osobine koje se smatraju jednolikima.
Pojedinačni uzorak: količina koja se uzima iz jedne točke uzorka.
Skupni uzorak: skup pojedinačnih uzoraka uzetih iz istog uzorka.
Reducirani uzorak: reprezentativni dio skupnog uzorka dobiven postupkom smanjivanja skupnog uzorka.
Konačni uzorak: dio reduciranog uzorka ili homogeniziranog skupnog uzorka.
4. OPREMA
|
4.1. |
Oprema za uzorkovanje mora biti izrađena od materijala koji ne mogu kontaminirati proizvode namijenjene uzorkovanju. Takvu opremu države članice mogu službeno odobriti. |
4.2. Preporučena oprema za uzorkovanje krute hrane za životinje
4.2.1. Ručno uzorkovanje
|
4.2.1.1. |
Lopatica s ravnim dnom i okomitim stranicama. |
|
4.2.1.2. |
Sonda za uzorkovanje s dugim procjepom ili pregradama. Dimenzije sonde za uzorkovanje moraju odgovarati osobinama uzorka (dubina posude, veličina vreće itd.) i veličini čestica hrane za životinje. |
4.2.2. Mehaničko uzorkovanje
Za uzorkovanje hrane za životinje koja se kreće može se koristiti odobrena mehanička oprema.
4.2.3. Razdjelnik
Uređaj namijenjena za dijeljenje uzorka na približno jednake dijelove može se koristiti za uzimanje pojedinačnih uzoraka i za pripremu reduciranih i konačnih uzoraka.
5. KOLIČINSKI ZAHTJEVI
|
5.A. |
Kod kontrole tvari ili proizvoda koji su ravnomjerno raspodijeljeni u hrani za životinje. |
|
|
5.A.1. |
Uzorkovani dio Veličina uzorka mora biti takva da se od svakog njegovog sastavnog dijela može načiniti uzorak. |
|
|
5.A.2. |
Pojedinačni uzorci |
|
|
5.A.2.1. |
Hrana za životinje u rasutom stanju: |
Najmanji broj pojedinačnih uzoraka: |
|
5.A.2.1.1. |
uzorkovani dijelovi manji od 2,5 tone |
sedam |
|
5.A.2.1.2. |
uzorkovani dijelovi veći od 2,5 tone |
Kvadratni korijen iz dvadeseterokratnika broja tona u uzorkovanom dijelu (*), do najviše 40 pojedinačnih uzoraka |
|
5.A.2.2. |
Pakirana hrana za životinje: |
Najmanji broj pakovina za uzorkovanje (**): |
|
5.A.2.2.1. |
Paketi mase veće od 1 kg: |
|
|
5.A.2.2.1.1. |
uzorkovani dijelovi iz 1 do 4 pakovine |
sve pakovine |
|
5.A.2.2.1.2. |
uzorkovani dijelovi iz 5 do 16 pakovina |
četiri |
|
5.A.2.2.1.3. |
uzorkovani dijelovi iz više od 16 pakovina |
Kvadratni korijen iz broja paketa koji čine uzorkovani dio (*), do najviše 20 pakovina |
|
5.A.2.2.2. |
Pakovine mase ne veće od 1 kg |
četiri |
|
5.A.2.3. |
Tekuća ili polutekuća hrana za životinje: |
Najmanji broj spremnika za uzorkovanje (**): |
|
5.A.2.3.1. |
Spremnici zapremnine do 1 litre: |
|
|
5.A.2.3.1.1. |
uzorkovani dijelovi iz 1 do 4 spremnika |
svi spremnici |
|
5.A.2.3.1.2. |
uzorkovani dijelovi iz 5 do 16 spremnika |
četiri |
|
5.A.2.3.1.3. |
uzorkovani dijelovi iz više od 16 spremnka |
Kvadratni korijen iz broja spremnika koje čine uzorkovani dio (*), do najviše 20 spremnika |
|
5.A.2.3.2. |
Spremnici zapremnine do 1 litre |
četiri |
|
5.A.2.4. |
Krmni blokovi ili kameni za lizanje |
Najmanji broj blokova ili kamena za lizanje namijenjenih uzorkovanju (**): 1 blok ili kamen za lizanje na uzorkovani dio od 25 komada, do najviše 4 bloka ili kamena za lizanje |
|
5.A.3. |
Skupni uzorak: Zahtijeva se samo jedan skupni uzorak na uzorkovani dio. Ukupan broj pojedinačnih uzoraka koji čine skupni uzorak ne smije biti manji od: |
|
|
5.A.3.1. |
Hrana za životinje u rasutom stanju |
4 kg |
|
5.A.3.2. |
Pakirana hrana za životinje: |
|
|
5.A.3.2.1. |
Pakovine mase veće od 1 kg |
4 kg |
|
5.A.3.2.2. |
Pakovine mase do 1 kg |
masa udjela četiri izvorne pakovine |
|
5.A.3.3. |
Tekuća ili polutekuća hrana za životinje: |
|
|
5.A.3.3.1. |
Spremnici zapremnine veće od 1 litra |
četiri litre |
|
5.A.3.3.2. |
Spremnici zapremnine do od 1 litre |
zapremnina udjela četiri izvorne posude |
|
5.A.3.4. |
Krmni blokovi ili kameni za lizanje: |
|
|
5.A.3.4.1. |
pojedinačne mase veće od 1 kg |
4 kg |
|
5.A.3.4.2. |
pojedinačne mase ne veće od 1 kg |
masa četiri izvorna bloka ili kamena za lizanje |
|
5.A.4. |
Konačni uzorci Iz skupnog se uzorka prema potrebi nakon smanjivanja dobiva konačni uzorak. Potrebna je analiza najmanje jednog konačnog uzorka. Količina konačnog uzorka za analizu ne smije biti manja od: |
|
|
|
Kruta hrana za životinje |
500 g |
|
|
Tekuća ili polutekuća hrana za životinje |
500 ml |
|
5.B. |
Kod kontrole nepoželjnih tvari ili proizvoda, vjerojatno neravnomjerno raspoređenih u hrani za životinje, poput aflatoksina, ražene glavice, ricinusa i krotalarije u sirovini za dobivanje životinjske hrane (***) |
|
|
5.B.1. |
Uzorkovani dio: vidjeti točku 5.A.1. |
|
|
5.B.2. |
Pojedinačni uzorci |
|
|
5.B.2.1. |
Hrana za životinje u rasutom stanju: vidjeti točku 5.A.2.1. |
|
|
5.B.2.2. |
Pakirana hrana za životinje: |
Najmanji broj pakovina za uzorkovanje: |
|
5.B.2.2.1. |
uzorkovani dijelovi koji se sastoje od 1 do 4 pakovine |
sve pakovine |
|
5.B.2.2.2. |
uzorkovani dijelovi koji se sastoje od 5 do 16 pakovina |
4 |
|
5.B.2.2.3. |
uzorkovani dijelovi koji se sastoje od više od 16 pakovina |
Kvadratni korijen iz broja pakovina koji čine uzorkovani dio (*), do najviše 40 pakovina |
|
5.B.3. |
Skupni uzorci Broj skupnih uzoraka ovisi o veličini uzorka. Najmanji broj skupnih uzoraka po uzorkovanom dijelu, naveden je dolje. Ukupna masa pojedinačnih uzoraka koji čine svaki skupni uzorak ne smije biti manja od 4 kg |
|
|
5.B.3.1. |
Hrana za životinje u rasutom stanju |
|
|
|
Masa uzorka u tonama: |
Najmanji broj skupnih uzoraka po uzorkovanom dijelu: |
|
|
do 1 |
1 |
|
|
više od 1 do 10 |
2 |
|
|
više od 10 do 40 |
3 |
|
|
više od 40 |
4 |
|
5.B.3.2. |
Pakirana hrana za životinje |
|
|
|
Broj pakovina uzorka: |
Najmanji broj skupnih uzoraka po uzorkovanom dijelu: |
|
|
1 do 16 |
1 |
|
|
17 do 200 |
2 |
|
|
201 do 800 |
3 |
|
|
više od 800 |
4 |
|
5.B.4. |
Konačni uzorci Iz svakog se skupnog uzorka nakon smanjivanja dobiva konačni uzorak. Potrebno je napraviti analizu najmanje jednog konačnog uzorka po skupnom uzorku. Masa konačnog uzorka za analizu ne smije biti manja od 500 g. |
|
6. UPUTE ZA UZIMANJE, PRIPREMANJE I PAKIRANJE UZORAKA
6.1. Općenito
Uzorci se moraju uzimati i pripremiti u što kraćem vremenu uz pridržavanje mjera opreza kojima se sprečava promjena sastava ili kontaminacija proizvoda. Instrumenti, radne površine i posude za prihvat uzoraka moraju biti čisti i suhi.
6.2. Pojedinačni uzorci
6.2.A. Kod kontrole tvari ili proizvoda koji su ravnomjerno raspodijeljeni u hrani za životinje
Pojedinačni uzorci moraju se uzeti nasumično iz cijeloga uzorka i moraju biti približno jednake veličine.
6.2.A.1.
Uzorkovani se dio dijeli na približno jednake zamišljene dijelove. Nasumično se odabere broj dijelova koji odgovara traženom broju pojedinačnih uzoraka u skladu s točkom 5.A.2 i iz svakog od njih se uzme najmanje jedan uzorak.
Prema potrebi se uzorkovanje može izvesti kod premještanja uzorka (pri utovaru ili istovaru).
6.2.A.2.
Nakon odabira potrebnog broja paketa za uzorkovanje u skladu s točkom 5.A.2., sondom ili lopaticom uzima se dio udjela svakog paketa. Prema potrebi, uzorci se mogu uzeti nakon odvojenog pražnjenja paketa. U svakom skupnom uzorku zasebno razbiju se sve grude, prema potrebi, tako da se odvoje i zatim vrate u uzorak.
6.2.A.3.
Nakon odabira potrebnog broja posuda za uzorkovanje iz točke 5.A.2, njihov se udio prema potrebi homogenizira i iz svakog se spremnika uzima određena količina.
Pojedinačni se uzorci mogu uzimati pri pražnjenju sadržaja spremnika.
6.2.A.4.
Nakon odabira traženog broja spremnika za uzimanje uzoraka iz točke 5.A.2, uzorci se uzimaju s različitih razina.
Uzorci se mogu uzeti i za vrijeme pražnjenja udjela, ali se prva količina mora odbaciti.
U oba slučaja ukupni obujam ne smije biti manji od 10 litara.
6.2.A.5.
Nakon odabira traženog broja blokova ili kamena za lizanje namijenjenih uzorkovanju iz točke 5.A.2 uzme se dio svakog bloka ili kamena za lizanje.
6.2.B. Kod kontrole nepoželjnih tvari ili proizvoda, vjerojatno neravnomjerno raspoređenih u hrani za životinje, poput aflatoksina, ražene glavice, ricinusa i krotalarije u sirovini za dobivanje hrane za životinje
Uzorkovani dio se razdijeli na približno jednake zamišljene dijelove koji odgovaraju broju ukupnih uzoraka iz točke 5.B.3. Ako je taj broj veći od jedan, ukupan broj pojedinačnih uzoraka iz točke 5.B.2 razdijeli se približno ravnomjerno po različitim dijelovima. Zatim se uzmu uzorci približno jednake veličine (1), koji moraju biti takvi da ukupna količina u uzorcima iz svakog dijela nije manja od 4 kg, što predstavlja minimalnu količinu za svaki skupni uzorak. Pojedinačni uzorci uzeti iz različitih dijelova ne smiju se spajati u skupni uzorak.
6.3. Priprema skupnih uzoraka
6.3.A. Kod kontrole tvari ili proizvoda ravnomjerno raspodijeljenih u hrani za životinje
Pojedinačni uzorci se pomiješaju tako da tvore skupni uzorak.
6.3.B. Kod kontrole nepoželjnih tvari ili proizvoda, vjerojatno neravnomjerno raspoređenih u hrani za životinje, poput aflatoksina, ražene glavice, ricinusa i krotalarije u sirovini za dobivanje hrane za životinje
Pojedinačni uzorci iz svakog dijela uzorka se pomiješaju i pripremi se broj skupnih uzoraka utvrđen točkom 5.B.3, pri čemu treba zabilježiti izvor svakog skupnog uzorka.
6.4. Priprema konačnih uzoraka
Materijal u svakom skupnom uzorku treba pažljivo izmiješati kako bi se dobio homogenizirani uzorak (2). Prema potrebi se skupni uzorak najprije smanji na najmanje 2 kg ili 2 litre (smanjeni uzorak) mehaničkim ili automatskim razdjelnikom ili četvrtanjem.
Potom se pripremi najmanje tri konačna uzorka približno jednake veličine u skladu s količinskim zahtjevima iz točke 5.A.4 ili 5.B.4. Svaki se uzorak pohrani u prikladni spremnik. Potrebno je poduzeti sve mjere opreza kako bi se tijekom prijevoza ili skladištenja spriječila promjena sastava uzorka, kontaminacija ili onečišćenje.
6.5. Pakiranje konačnih uzoraka
Posude ili paketi moraju biti zapečaćeni i označeni etiketama (cijela etiketa mora biti uključena u pečat), tako da ih nije moguće otvoriti bez oštećenja pečata.
7. PODACI O UZORKOVANJU
O svim obavljenim uzorkovanjima moraju se voditi evidencije kako bi se svaki uzorkovani dio mogao nedvojbeno prepoznati.
8. DOSTAVLJANJE UZORAKA
Za svaki skupni uzorak treba u najkraćem mogućem roku najmanje jedan konačni uzorak dostaviti u ovlašteni laboratorij, zajedno sa svim podacima koji su potrebni osobi koja izvodi analizu.
(*) Kada dobiveni broj nije cijeli broj, zaokružuje se na sljedeći cijeli broj.
(**) Za pakovine ili spremnike udjela manjeg od 1 kg ili 1 litre, te za blokove ili kamene za lizanje pojedinačne mase ne veće od 1 kg, pojedinačni uzorak je udio pojedinačne izvorne pakovine ili spremnika, jednog bloka ili jednog kamena.
(***) Metode iz 5.A. koriste se za kontrolu aflatoksina, ražene glavice, ricinusa i krotalarije u potpunim ili dopunskim krmnim smjesama.
(1) Kod pakirane hrane za životinje, sondom ili lopaticom uzme se dio udjela paketa koji se uzorkuje, prema potrebi nakon odvojenog pražnjenja pakovine.
(2) U svakom se skupnom uzorku zasebno razbiju sve grude, prema potrebi, tako da se odvoje i zatim vrate u uzorak.
PRILOG II.
OPĆE ODREDBE O ANALITIČKIM METODAMA ZA HRANU ZA ŽIVOTINJE
A. PRIPREMA UZORAKA ZA ANALIZU
1. Svrha
U dolje navedenim postupcima opisuje se priprema konačnih uzoraka za analizu, koji se šalju nadzornim laboratorijima nakon uzorkovanja izvršenog u skladu s odredbama iz Priloga I.
Ti se uzorci moraju pripremiti tako da izvagane količine budu homogene i reprezentativne za konačne uzorke, kako je predviđeno za metode analize.
2. Mjere opreza
Postupak za pripremanje uzoraka ovisi o analitičkoj metodi koja se koristi. Stoga je vrlo važno osigurati da primijenjeni postupak za pripremanje uzorka bude primjeren analitičkoj metodi koja se koristi.
Svi potrebni postupci moraju se izvesti na način kojim se u najvećoj mogućoj mjeri sprečava kontaminacija i promjena sastava uzorka.
Mljevenje, miješanje i prosijavanje mora se vršiti se na najbrži mogući način kako bi se uzorak čim manje izlagao zraku i svjetlu. Ne smiju se koristiti mlinovi i drobilice koji bi mogli znatno zagrijati uzorak.
Preporuča se ručno mljevenje za hranu za životinje koja je posebno osjetljiva na toplinu. Treba se pobrinuti da sama oprema ne bude izvor kontaminacije elementima u tragovima.
Ako se priprema ne može obaviti bez znatne promjene udjela vlage u uzorku, određuje se udio vode prije i nakon pripreme, u skladu s metodom utvrđenoj u dijelu A Priloga III.
3. Postupak
Uzorak se prikladnim tehnikama dijeljenja, poput naizmjeničnog uzimanja uzorka lopaticom ili uzimanja uzorka pri stacionarnom ili rotacijskom miješanju, razdijeli na prikladne manje uzorke za analizu i za referentne namjene. Ne preporuča se dijeljenje uzoraka metodom stožaste hrpe i četvrtanja, budući da se na taj način mogu dobiti poduzorci s visokom pogreškom dijeljenja. Referentni se uzorak drži u primjereno čistoj i suhoj posudi s hermetičkim zatvaračem, a poduzorci mase od najmanje 100 g pripremaju se za analizu kako je navedeno u daljnjem u tekstu.
3.1. Hrana za životinje koja se može samljeti bez dodatne obrade
Ako se u analitičkim metodama ne navodi drukčije, cijeli se uzorak nakon usitnjavanja prosije kroz sito s kvadratnim očicama veličine stranice 1 mm (u skladu s preporukom ISO R565). Treba izbjegavati pretjerano usitnjavanje.
Prosijani se uzorak promiješa i prikupi u prikladno čistu i suhu posudu s hermetičkim zatvaračem. Neposredno prije vaganja količine za analizu, uzorak se ponovo promiješa.
3.2. Hrana za životinje koja se može samljeti nakon sušenja
Ako se u analitičkim metodama ne navodi drukčije, uzorak se, u skladu s postupkom za prethodno sušenje iz točke 4.3. metode za određivanje udjela vlage iz dijela A Priloga III., suši, tako da se udio vode snizi na 8 % – 12 %. Zatim se nastavlja u skladu s točkom 3.1.
3.3. Tekuća ili polutekuća hrana za životinje
Uzorak se prikupi u prikladno čistu i suhu posudu s hermetičkim zatvaračem. Neposredno prije vaganja količine za analizu, uzorak se temeljito promiješa.
3.4. Ostala hrana za životinje
Uzorci koji se ne mogu pripremiti u skladu s jednom od gore navedenih postupaka obrađuju se bilo kojim drugim postupkom kojim se osigurava da je količina uzorka odvagana za analizu homogena i reprezentativna za konačne uzorke.
4. Pohranjivanje uzoraka
Uzorci se moraju pohraniti na temperaturi koja neće promijeniti njihov sastav. Uzorci namijenjeni za analizu vitamina ili tvari posebno osjetljivih na svjetlost moraju se pohraniti u spremnicima od smeđeg stakla.
B. ODREDBE O REAGENSIMA I OPREMI, KOJI SE KORISTE U ANALITIČKIM METODAMA
|
1. |
Ako se u analitičkim metodama ne navodi drukčije, svi reagensi moraju biti analitičke čistoće (pro analisi (p.a.)). Kod analize tragova, čistoća reagensa mora se provjeriti slijepom probom. Ovisno o dobivenim rezultatima, može biti potrebno dodatno pročišćivanje reagensa. |
|
2. |
Kod svih postupaka iz analitičkih metoda koji uključuju pripremanje otopina, razrjeđivanje, ispiranje ili pranje, a kod kojih se ne navodi vrsta korištenog otapala ili razrjeđivača, koristi se voda. U pravilu, voda mora biti demineralizirana ili destilirana. U posebnim slučajevima, navedenim u analitičkim metodama, vodu treba pročistiti posebnim postupcima. |
|
3. |
S obzirom na opremu koja se uobičajeno nalazi u kontrolnim laboratorijima, u analitičkim se metodama navode samo posebni instrumenti i oprema ili oni koji zahtijevaju poseban način uporabe. Oni moraju biti čisti, posebno kod određivanja vrlo malih količina tvari. |
C. PRIMJENA ANALITIČKIH METODA I PRIKAZ REZULTATA
1. Postupak ekstrakcije
U nekoliko metoda utvrđen je poseban postupak ekstrakcije. U pravilu se, osim postupka navedenog u metodi, mogu koristiti i drugi postupci ekstrakcije, pod uvjetom da je učinkovitost korištenog postupka ekstrakcije za analiziranu matricu dokazano jednakovrijedna postupku navedenom u metodi.
2. Postupak pročišćivanja
U nekoliko metoda utvrđen je poseban postupak pročišćivanja. U pravilu se, osim postupka navedenog u metodi, mogu koristiti i drugi postupci pročišćivanja, pod uvjetom da se korištenim postupkom pročišćivanja za analiziranu matricu dokazano postižu analitički rezultati jednakovrijedni postupku navedenom u metodi.
3. Izvješćivanje o korištenoj analitičkoj metodi
Općenito je za određivanje svake tvari u hrani za životinje određena samo jedna metoda. Kada je određeno više metoda, kontrolni laboratorij u izvješću o analizi mora navesti korištenu metodu.
4. Broj postupaka određivanja
Rezultat naveden u izvješću o analizi je srednja vrijednost najmanje dvaju postupaka određivanja zadovoljavajuće ponovljivosti, izvedenih na odvojenim dijelovima uzorka.
Međutim, kod analize nepoželjnih tvari, ako je rezultat prvog određivanja znatno (> 50 %) niži od specifikacije koja se kontrolira, nisu potrebni dodatni postupci određivanja pod uvjetom da su korišteni primjereni postupci za osiguranje kvalitete.
Pri kontroli označenog udjela tvari ili sastojka, ako rezultat prvog određivanja potvrdi označeni udio, tj. ako je rezultat analize unutar prihvatljivih granica odstupanja od označenog udjela, nisu potrebni dodatni postupci određivanja pod uvjetom da su korišteni primjereni postupci za osiguranje kvalitete.
U nekim je slučajevima ta prihvatljiva granica odstupanja utvrđena propisima poput Direktive Vijeća 79/373/EEZ (1).
5. Izvješćivanje o rezultatima analize
Rezultat analize prikazuje se na način utvrđen analitičkom metodom, s primjerenim brojem značajnih znamenki, i prema potrebi se korigira s obzirom na udio vode u konačnom uzorku prije priprave.
6. Nesigurnost mjerenja i stupanj iskorištenja pri analizi nepoželjnih tvari
U vezi s nepoželjnim tvarima u smislu Direktive 2002/32/EZ, uključujući dioksine i PCB-e slične dioksinu, proizvod namijenjen za hranu za životinje smatra se da nije u skladu s najvećom dopuštenom količinom ako je rezultat analize veći od najveće dopuštene količine, uzimajući u obzir proširenu nesigurnost mjerenja i korekciju za iskorištenje. Za ocjenu usklađenosti koristi se analizirana koncentracija nakon korekcije za iskorištenje i nakon oduzimanja proširene nesigurnosti mjerenja. Taj se postupak primjenjuje samo u slučajevima kada analitička metoda omogućuje procjenu nesigurnosti mjerenja i korekciju za iskorištenje (na primjer, nije moguć u slučaju mikroskopske analize).
Rezultati analize iskazuju se na sljedeći način (ako korištena analitička metoda omogućuje ocjenu nesigurnosti mjerenja i korekciju za iskorištenje):
|
(a) |
s korekcijom za iskorištenje, pri čemu se navodi razina iskorištenja. Korekcija za iskorištenje nije potrebna ako je postotak iskorištenja između 90 % i 110 %; |
|
(b) |
kao „x +/– U”, pri čemu je x rezultat analize, a U proširena nesigurnost mjerenja uz uporabu obuhvatnog faktora 2, čime se postiže razina pouzdanosti od približno 95 %. |
Međutim, ako je rezultat analize znatno (> 50 %) niži od specifikacije koja se kontrolira i pod uvjetom da su korišteni primjereni postupci za osiguranje kvalitete, a svrha analize je samo provjera usklađenosti sa zakonskim odredbama, rezultat analize može se iskazati bez korekcije za iskorištenje i u tim se slučajevima korekcija za iskorištenje i nesigurnost mjerenja mogu izostaviti.
PRILOG III.
ANALITIČKE METODE ZA KONTROLU SASTAVA SIROVINA ZA HRANU ZA ŽIVOTINJE I KRMNIH SMJESA
A. ODREĐIVANJE VLAGE
1. Svrha i područje primjene
Ovom se metodom omogućuje određivanje udjela vlage u hrani za životinje. Kada hrana za životinje sadrži hlapljive tvari, poput organskih kiselina, treba uzeti u obzir da se pri određivanju vlage obuhvaća i znatna količina hlapljivih tvari.
Ova metoda ne obuhvaća analizu mliječnih proizvoda kao sirovina za hranu za životinje, analizu mineralnih tvari i smjesa sastavljenih uglavnom od mineralnih tvari, analizu životinjskih i biljnih masti i ulja ili analizu sjemenki uljarica i uljanog voća.
2. Načelo
Uzorak se osuši pod određenim uvjetima koji se mijenjaju ovisno o vrsti hrane za životinje. Gubitak mase utvrđuje se vaganjem. Kod krute hrane za životinje s visokim udjelom vode potrebno je prethodno sušenje.
3. Oprema
|
3.1. |
Drobilica od materijala koji ne apsorbira vlagu i lako se čisti, omogućuje brzo i ravnomjerno drobljenje bez pretjeranog zagrijavanja, u najvećoj mogućoj mjeri sprečava kontakt s vanjskim zrakom te ispunjava zahtjeve iz točaka 4.1.1 i 4.1.2 (npr. udarne ili vodom hlađene mikrodrobilice, konusni mlinovi sa sklopivim konusima, spore drobilice ili drobilice sa zupčanicima). |
|
3.2. |
Analitička vaga s preciznošću do 1 mg |
|
3.3. |
Suhe posude od nehrđajuće kovine ili stakla s poklopcima koji omogućuju hermetičko zatvaranje; radna površina koja omogućuje rasprostiranje testnog uzorka na približno 0,3 g/cm2. |
|
3.4. |
Izotermičko sušilo (± 2 °C) s električnim grijanjem i primjerenim provjetravanjem koje omogućuje brzu regulaciju temperature (1). |
|
3.5. |
Podesivo električno vakuumsko sušilo opremljeno uljnom crpkom i mehanizmom za dovod vrućeg suhog zraka ili sredstva za isušivanje (npr. kalcijevog oksida). |
|
3.6. |
Eksikator s debelom metalnom ili porculanskom perforiranom pločom koja sadrži učinkovito sredstvo za isušivanje. |
4. Postupak
|
Napomena |
: |
Postupci opisani u ovom odjeljku moraju se provesti odmah po otvaranju pakovina s uzorcima. Analiza se mora provesti najmanje dvaput zaredom. |
4.1. Priprema
4.1.1.
Uzme se najmanje 50 g uzorka. Prema potrebi se usitni ili razdijeli tako da se spriječi promjena udjela vlage (vidjeti točku 6).
4.1.2.
Uzme se najmanje 50 g uzorka. Uzorak se samelje tako da najmanje 50 % čestica prolazi kroz sito s očicama veličine 0,5 mm, a na situ s okruglim očicama veličine 1 mm ostane najviše 10 % čestica.
4.1.3.
Odvagne se približno 25 g uzorka s preciznošću od 10 mg, doda se primjerena količina bezvodnog pijeska izvagana s preciznošću od 10 mg te se miješa dok se ne dobije homogena masa.
4.2. Sušenje
4.2.1.
Izvaže se posuda (točka 3.3.) s poklopcem s preciznošću od 1 mg. U izvaganu se posudu odvagne približno 5 g uzorka s preciznošću od 1 mg i ravnomjerno raširi. Posuda se bez poklopca stavi u sušilo prethodno zagrijano na 103 °C. Posuda se stavi u sušilo što je moguće brže kako bi se spriječio prevelik pad temperature. Sušenje traje četiri sata od trenutka kada temperatura u sušilu ponovno dosegne 103 °C. Posuda se pokrije poklopcem, izvadi iz sušila, ostavi hladiti u eksikatoru (točka 3.6.) 30 do 45 minuta i izvaže s preciznošću od 1 mg.
Hrana za životinje koja se sastoji uglavnom od ulja i masti suši se u sušilu dodatnih 30 minuta na 130 °C. Razlika između dvaju vaganja ne smije biti veća od 0,1 % vlage.
4.2.2.
Izvaže se posuda (točka 3.3.) s poklopcem s preciznošću od 0,5 mg. U izvaganu se posudu odvagne približno 5 g usitnjenog uzorka s preciznošću od 1 mg i ravnomjerno raširi. Posuda se bez poklopca stavi u sušilo prethodno zagrijano na 130 °C. Posuda se stavi u sušilo što je moguće brže kako bi se spriječio prevelik pad temperature. Sušenje traje 2 sata od trenutka kada temperatura u sušilu ponovno dosegne 130 °C. Posuda se pokrije poklopcem, izvadi iz sušila, ostavi hladiti u eksikatoru (točka 3.6.) 30 do 45 minuta i izvaže s preciznošću od 1 mg.
|
4.2.3. |
Krmne smjese koje sadrže više od 4 % saharoze ili laktoze: sirovine za hranu za životinje poput rogača, hidroliziranih žitnih proizvoda, sjemena slada, sušene mljevene repe, ribe i otopine šećera; krmne smjese koje sadrže više od 25 % mineralnih soli, uključujući kristalnu vodu. Izvaže se posuda (točka 3.3.) s poklopcem s preciznošću od 0,5 mg. U izvaganu se posudu odvagne približno 5 g uzorka s preciznošću od 1 mg i ravnomjerno raširi. Posuda se bez poklopca stavi u vakuumsko sušilo (točka 3.5.) prethodno zagrijano na 80 – 85 °C. Posuda se stavi u sušilo što je moguće brže kako bi se spriječio prevelik pad temperature. Tlak se poveća na 100 torra i na tom se tlaku ostavi sušiti četiri sata na vrućem suhom zraku ili upotrebom sredstva za isušivanje (približno 300 g za 20 uzoraka). U potonjem se slučaju vakuumska crpka isključi kad se dosegne traženi tlak. Vrijeme sušenja računa se od trenutka kada temperatura u sušilu ponovno dosegne 80 – 85 °C. Oprezno se izjednači tlak u sušilu s atmosferskim tlakom. Sušilo se otvori, posuda se odmah pokrije poklopcem i izvadi iz sušila, ostavi se hladiti 30 do 45 minuta u eksikatoru (točka 3.6.) i izvaže s preciznošću od 1 mg. Suši se dodatnih 30 minuta u vakuumskom sušilu na 80 – 85 °C i ponovo važe. Razlika između dvaju vaganja ne smije biti veća od 0,1 % vlage. |
4.3. Prethodno sušenje
4.3.1.
Kruta hrana za životinje s visokim udjelom vode koja se teško drobi mora se prethodno sušiti na sljedeći način:
U prikladnu se posudu (npr. aluminijski tanjur veličine 20 × 12 cm s rubom 0,5 cm) odvagne 50 g neusitnjenog uzorka s preciznošću od 10 mg (sabijena ili zgusnuta hrana za životinje može se prema potrebi grubo razdijeliti). Ostavi se sušiti u sušilu na 60 – 70 °C, sve dok se udio vode ne smanji na 8 – 12 %. Posuda se izvadi iz sušila i jedan sat hladi bez poklopca u laboratoriju, zatim se izvaže s preciznošću od 10 mg. Hrana za životinje se odmah usitni, u skladu s točkom 4.1.1., i suši, u skladu s točkom 4.2.1. ili 4.2.3., ovisno o vrsti hrane za životinje.
4.3.2.
Zrna sa udjelom vode većim od 17 % moraju se prethodno sušiti na sljedeći način:
U prikladnu se posudu (npr. aluminijski tanjur veličine 20 × 12 cm s rubom 0,5 cm) odvagne 50 g neusitnjenog zrnja s preciznošću od 10 mg. Ostavi se sušiti 5 do 7 minuta u sušilu na 130 °C. Posuda se izvadi iz sušila i 2 sata hladi bez poklopca u laboratoriju, zatim se izvaže s preciznošću od 10 mg. Odmah se usitni, u skladu s točkom 4.1.2., i osuši, u skladu s točkom 4.2.2.
5. Izračun rezultata
Udio vode (X), iskazan kao postotni dio uzorka, izračunava se sljedećom formulom:
5.1. Sušenje bez prethodnog sušenja

pri čemu je:
|
m |
= |
početna masa uzorka u gramima, |
|
m0 |
= |
masa suhog uzorka u gramima. |
5.2. Sušenje s prethodnim sušenjem
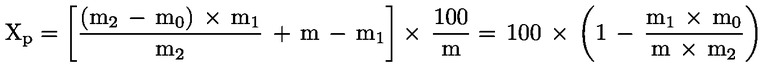
pri čemu je:
|
m |
= |
početna masa uzorka u gramima, |
|
m1 |
= |
masa uzorka nakon prethodnog sušenja u gramima, |
|
m2 |
= |
masa uzorka nakon usitnjavanja ili mljevenja u gramima, |
|
m0 |
= |
masa suhog uzorka u gramima. |
5.3. Ponovljivost
Razlika između rezultata dvaju usporednih postupaka određivanja, izvedenih na istom uzorku, ne smije prelaziti 0,2 % apsolutne vrijednosti vode.
6. Napomena
U slučaju kada je potrebno mljevenje za koje se pokaže da mijenja udio vode u proizvodu, potrebno je korigirati rezultate analize sastojaka hrane za životinje s obzirom na udio vode u uzorku u početnom stanju.
B. ODREĐIVANJE VODE U MASTIMA I ULJIMA ŽIVOTINJSKOG I BILJNOG PODRIJETLA
1. Svrha i područje primjene
Ova se metoda koristi za određivanje udjela vode i hlapljivih tvari u mastima i uljima životinjskog i biljnog podrijetla.
2. Načelo
Uzorak se osuši do konstantne mase (gubitak mase između dvaju uzastopnih vaganja ne smije prijeći 1 mg) na 103 °C. Gubitak mase utvrđuje se vaganjem.
3. Oprema
|
3.1. |
Posuda s ravnim dnom izrađena od nehrđajućeg materijala, promjera 8 do 9 cm i visine približno 3 cm. |
|
3.2. |
Termometar s ojačanom kuglicom i ekspanzijskom komorom na gornjem dijelu, baždaren od približno 80 °C do najmanje 110 °C i duljine približno 10 cm. |
|
3.3. |
Pješčana kupelj ili električna grijaća ploča |
|
3.4. |
Eksikator s učinkovitim sredstvom za isušivanje |
|
3.5. |
Analitička vaga |
4. Postupak
Odvagne se približno 20 g homogeniziranog uzorka s preciznošću od 1 mg i prenese u suhu izvaganu posudu (točka 3.1.) u kojoj se nalazi termometar (točka 3.2.). Zagrijava se na pješčanoj kupelji ili grijaćoj ploči (točka 3.3.) uz stalno miješanje termometrom, tako da temperatura dosegne 90 °C u približno 7 minuta.
Smanji se unos topline, pri čemu se nadzire učestalost podizanja mjehurića s dna posude. Temperatura ne smije prijeći 105 °C. Nastavi se miješati uz struganje dna posude sve dok ne prestane stvaranje mjehurića.
Kako bi se osiguralo potpuno uklanjanje vode, nekoliko puta se zagrije na 103 ± 2 °C uz hlađenje na 93 °C između uzastopnih zagrijavanja. Zatim se uzorak ostavi hladiti na sobnu temperaturu u eksikatoru (točka 3.4.) i izvaže. Postupak se ponavlja sve dok gubitak mase između dvaju uzastopnih vaganja ne bude manji od 2 mg.
|
Napomena |
: |
Povećanje mase uzorka nakon uzastopnog zagrijavanja upućuje na oksidaciju masti i u tom se slučaju rezultat izračunava na temelju vaganja izvedenog neposredno prije početka povećavanja mase. |
5. Izračun rezultata
Udio vode (X), iskazan kao postotni dio uzorka, izračunava se sljedećom formulom:
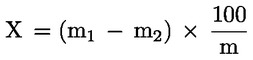
pri čemu je:
|
m |
= |
masa uzorka u gramima, |
|
m1 |
= |
masa posude sa sadržajem prije zagrijavanja, u gramima, |
|
m2 |
= |
masa posude sa sadržajem nakon zagrijavanja, u gramima. |
Rezultati manji od 0,05 % navode se kao „manji od 0,05 %”.
Ponovljivost
Razlika između rezultata dvaju usporednih postupaka određivanja, izvedenih na istom uzorku, ne smije prijeći 0,05 % apsolutne vrijednosti.
C. ODREĐIVANJE UDJELA SIROVIH BJELANČEVINA
1. Svrha i područje primjene
Ova se metoda koristi za određivanje udjela sirovih bjelančevina u hrani za životinje na temelju udjela dušika određenog Kjeldahlovom metodom.
2. Postupak
Uzorak se mineralizira sumpornom kiselinom u prisutnosti katalizatora. Kisela se otopina prevede u bazičnu dodavanjem otopine natrijevog hidroksida. Amonijak se destilira i prikupi u odmjerenoj količini sumporne kiseline, višak koje se titrira standardnom otopinom natrijevog hidroksida.
Druga je mogućnost destilacija oslobođenog amonijaka u višku otopine borne kiseline, nakon čega slijedi titriranje otopinom klorovodične ili sumporne kiseline.
3. Reagensi
|
3.1. |
Kalijev sulfat. |
|
3.2. |
Katalizator: bakrov(II) oksid CuO ili bakrov(II) sulfat pentahidrat, CuSO4 5H2O. |
|
3.3. |
Cink u zrncima. |
|
3.4. |
Sumporna kiselina, ρ20 = 1,84 g/ml. |
|
3.5 |
Sumporna kiselina, standardna volumetrijska otopina, c(H2SO4) = 0,25 mol/l. |
|
3.6. |
Sumporna kiselina, standardna volumetrijska otopina, c(H2SO4) = 0,10 mol/l. |
|
3.7. |
Sumporna kiselina, standardna volumetrijska otopina, c(H2SO4) = 0,05 mol/l. |
|
3.8. |
Indikator metil crvenilo; otopi se 300 mg metil crvenila u 100 ml etanola, σ = 95 – 96 % (v/v). |
|
3.9. |
Otopina natrijevog hidroksida (moguća uporaba tehničke čistoće) β = 40 g/100 ml (m/v: 40 %). |
|
3.10. |
Natrijev hidroksid, standardna volumetrijska otopina, c(NaOH) = 0,25 mol/l. |
|
3.11. |
Natrijev hidroksid, standardna volumetrijska otopina, c(NaOH) = 0,10 mol/l. |
|
3.12. |
Plovućac u zrnu, ispran u klorovodičnoj kiselini i kalciniran. |
|
3.13. |
Acetanilid (talište = 114 °C, udio N = 10,36 %). |
|
3.14. |
Saharoza (bez dušika). |
|
3.15. |
Borna kiselina (H3BO3). |
|
3.16. |
Otopina indikatora metilnog crvenila: otopi se 100 mg metilnog crvenila u 100 ml etanola ili metanola. |
|
3.17. |
Otopina bromkrezol zelenila: otopi se 100 mg bromkrezol zelenila u 100 ml etanola ili metanola. |
|
3.18. |
Otopina borne kiseline (10 – 40 g/l, ovisno o opremi koja se koristi). Kod metode kolorimetrijskog određivanja točke ekvivalencije, otopinama borne kiseline moraju se dodati indikatori metil crvenilo i bromkrezol zelenilo. Kod pripreme 1 litre otopine borne kiseline, prije prilagodbe volumena, doda se 7 ml otopine indikatora metil crvenila (točka 3.16.) i 10 ml otopine bromkrezol zelenila (točka 3.17.). Ovisno o vodi koja se koristi, pH vrijednost otopine borne kiseline može se razlikovati od serije do serije. Za dobivanje pozitivne slijepe probe često treba izvesti prilagodbu malim volumenom alkalija.
|
|
3.19. |
Klorovodična kiselina, standardna volumetrijska otopina, c(HCl) = 0,10 mol/l.
|
4. Oprema
Uređaj prikladna za izvođenje postupaka mineralizacije, destilacije i titracije prema Kjeldahlovoj metodi.
5. Postupak
5.1. Razgradnja
Odvagne se 1 g uzorka s preciznošću od 0,001 g i prenese u tikvicu naprave za mineralizaciju. Doda se 15 g kalijevog sulfata (točka 3.1.), prikladna količina katalizatora (točka 3.2.) (0,3 – 0,4 g bakrovog(II) oksida ili 0,9 – 1,2 g bakrovog(II) sulfat pentahidrata), 25 ml sumporne kiseline (točka 3.4.), prema potrebi nekoliko zrnaca plovućca (točka 3.12.) i promiješa.
Tikvica se prvo umjereno zagrijava i prema potrebi povremeno trese, sve dok masa potpuno ne pougljeni i nestane pjena; zatim se jače zagrijava do ravnomjernog vrenja tekućine. Zagrijavanje je primjereno ako se vrela kiselina kondenzira na stijenkama tikvice. Treba spriječiti pregrijavanje stijenki i prianjanje organskih čestica na njih.
Kada se otopina razbistri i postane svijetlozelena, ostavi se da ključa još 2 sata, a zatim se ostavi da se ohladi.
5.2. Destilacija
Oprezno se doda dovoljna količina vode kako bi se sulfati potpuno otopili. Ostavi se da se ohladi i prema potrebi doda nekoliko zrnaca cinka (točka 3.3.). Nastavlja se u skladu s točkom 5.2.1. ili 5.2.2.
5.2.1.
U tikvicu za prikupljanje naprave za destilaciju doda se točno odmjerena količina od 25 ml sumporne kiseline (točka 3.5.) ili (točka 3.7.) ovisno o pretpostavljenom udjelu dušika. Doda se nekoliko kapi indikatora metil crvenila (točka 3.8.).
Tikvica za mineralizaciju priključi se na kondenzator naprave za destilaciju i vršak kondenzatora uroni u tekućinu u tikvici za prikupljanje, najmanje do dubine od 1 cm (vidjeti napomenu pod točkom pod točkom 8.3.). U tikvicu za mineralizaciju polagano se ulije 100 ml otopine natrijevog hidroksida (točka 3.9.) bez gubitka amonijaka (vidjeti napomenu pod točkom pod točkom 8.1.). Tikvica se zagrijava do potpune destilacije amonijaka.
5.2.2.
Kod ručne titracije amonijaka iz destilata koristi se dolje navedeni postupak. Kod destilacijskih jedinica koje su u potpunosti automatizirane i obuhvaćaju titraciju amonijaka iz destilata, postupa se prema uputama proizvođača za rukovanje destilacijskom jedinicom.
Tikvica za prikupljanje s 25 – 30 ml otopine borne kiseline (točka 3.18.) postavi se ispod izlaznog otvora kondenzatora tako da ispusna cijev bude ispod površine viška otopine borne kiseline. Destilacijska se jedinica namjesti tako da isporučuje 50 ml otopine natrijevog hidroksida (točka 3.9.). Destilacijskom se jedinicom rukuje u skladu s uputama proizvođača, a oslobođeni se amonijak destilira dodavanjem otopine natrijevog hidroksida. Destilat se prikupi u otopini borne kiseline. Količina destilata (vrijeme parne destilacije) ovisi o količini dušika u uzorku. Treba se pridržavati uputa proizvođača.
|
Napomena |
: |
U poluautomatskoj destilacijskoj jedinici dodavanje viška natrijevog hidroksida i parna destilacija automatizirani su postupci. |
5.3. Titracija
Postupa se u skladu s točkom 5.3.1. ili 5.3.2.
5.3.1.
VIšak sumporne kiseline titrira se u tikvici za prikupljanje do točke ekvivalencije otopinom natrijevog hidroksida (točka 3.10. ili 3.11.), ovisno o koncentraciji korištene sumporne kiseline.
5.3.2.
Udio tikvice za prikupljanje titrira se standardnom volumetrijskom otopinom klorovodične kiseline (točka 3.19.) ili standardnom volumetrijskom otopinom sumporne kiseline (točka 3.6.), pri čemu se koristi bireta i očita se količina korištenog titranta.
Kod metode kolorimetrijskog određivanja točke ekvivalencije, točka ekvivalencije se dostiže kod prve pojave ružičastog obojenja udjela. Očita se obujam birete s preciznošću od 0,05 ml. Za pomoć kod vizualizacije točke ekvivalencije mogu se koristiti magnetna mješalica s osvjetljenjem ili fotometrijski detektor.
Ovaj se postupak može izvesti automatski upotrebom parnog destilatora s automatskom titracijom.
Kod rukovanja određenim tipom destilatora ili destilatora/titratora treba se pridržavati uputa proizvođača.
|
Napomena |
: |
Kod automatskog sustava titriranja, titracija započinje odmah po početku destilacije, a koristi se otopina borne kiseline 1 % (točka 3.18.). Kod potpuno automatizirane destilacijske jedinice, postupak automatske titracije amonijaka može se izvoditi i određivanjem točke ekvivalencije metodom potenciometrijske titracije pH. U tom se slučaju koristi automatski titrator s pH-metrom. Pravilno baždarenje pH metra izvodi se u području vrijednosti pH 4 do pH 7 uobičajenim laboratorijskim postupcima za baždarenje pH-metra. Točka ekvivalencije pH titracije dosiže se pri pH 4,6, kada je nagib titracijske krivulje najviši (točka infleksije). |
5.4. Slijepa proba
Kao potvrda da reagensi ne sadrže dušik koristi se slijepa proba (mineralizacija, destilacija i titracija), u kojoj se umjesto uzorka koristi 1 g saharoze (točka 3.14).
6. Izračun rezultata
Izračuni se izvode u skladu s točkom 6.1. ili 6.2.
6.1. Izračun za titraciju u skladu s točkom 5.3.1.
Udio sirovih bjelančevina, iskazan kao maseni postotak, izračunava se sljedećom formulom:

pri čemu je:
|
V0 |
= |
volumen (ml) NaOH (točka 3.10. ili 3.11.) korišten u slijepoj probi, |
|
V1 |
= |
volumen (ml) NaOH (točka 3.10. ili 3.11.) korišten kod titracije uzorka, |
|
c |
= |
koncentracija (mol/l) natrijevog hidroksida (točka 3.10. ili 3.11.), |
|
m |
= |
masa (g) uzorka. |
6.2. Izračun za titraciju u skladu s točkom 5.3.2.
6.2.1.
Udio sirovih bjelančevina, iskazan kao maseni postotak, izračunava se sljedećom formulom:

pri čemu je:
|
m |
= |
masa (g) uzorka, |
|
c |
= |
koncentracija (mol/l) standardne volumetrijske otopine klorovodične kiseline (točka 3.19.), |
|
V0 |
= |
volumen (ml) klorovodične kiseline korišten u slijepoj probi, |
|
V1 |
= |
volumen (ml) klorovodične kiseline korišten u uzorkovanom dijelu. |
6.2.2.
Udio sirovih bjelančevina, iskazan kao maseni postotak, izračunava se sljedećom formulom:

pri čemu je:
|
m |
= |
masa (g) uzorka, |
|
c |
= |
koncentracija (mol/l) standardne volumetrijske otopine sumporne kiseline (točka 3.6.), |
|
V0 |
= |
volumen (ml) sumporne kiseline (točka 3.6.) korišten u slijepoj probi, |
|
V1 |
= |
volumen (ml) sumporne kiseline (točka 3.6.) korišten u uzorkovanom dijelu. |
7. Provjera metode
7.1. Ponovljivost
Razlika između rezultata dvaju usporednih postupaka određivanja, izvedenih na istom uzorku, ne smije prijeći:
|
— |
0,2 % apsolutne vrijednosti za udio sirovih bjelančevina manji od 20 %, |
|
— |
1,0 % veće vrijednosti za udio sirovih bjelančevina u rasponu od 20 – 40 %, |
|
— |
0,4 % apsolutne vrijednosti za udio sirovih bjelančevina veći od 40 %. |
7.2. Točnost
Za analizu (mineralizacija, destilacija, titracija) koristi se 1,5 do 2,0 g acetanilida (točka 3.13.) u prisutnosti 1 g saharoze (točka 3.14.); za 1 g acetanilida koristi se 14,80 ml sumporne kiseline (točka 3.5.). Iskorištenje mora iznositi najmanje 99 %.
8. Napomene
|
8.1. |
Oprema može biti ručna, poluautomatska ili automatska. Ako je u radu naprave potreban prijelaz između mineralizacije i destilacije, taj se prijelaz mora izvesti bez gubitka. Ako tikvica naprave za destilaciju nije opremljena lijevkom za dokapavanje, natrijev se hidroksid dodaje neposredno prije priključivanja tikvice na kondenzator, pri čemu se tekućina ulijeva polako niz stijenke tikvice. |
|
8.2. |
Ako se udio tikvice stvrdnjava, postupak određivanja započinje se iznova, ali s količinom sumporne kiseline (točka 3.4.) većom od gore navedene. |
|
8.3. |
Kod proizvoda s niskim udjelom dušika volumen sumporne kiseline (točka 3.7.) koji se dodaje u tikvicu za prikupljanje, prema potrebi se može smanjiti na 10 ili 15 ml, a tikvica se dopuni vodom do 25 ml. |
|
8.4. |
Kod rutinske analize za određivanje sirovih bjelančevina mogu se koristiti zamjenske metode analize, iako je Kjeldahlova metoda, opisana u ovom dijelu C, referentna metoda. Za svaku matricu pojedinačno treba dokazati da su rezultati dobiveni zamjenskom metodom (npr. DUMAS) jednakovrijedni rezultatima dobivenim referentnom metodom. U izvješću o analizi treba navesti analitičku metodu korištenu za određivanje sirovih bjelančevina budući da rezultati dobiveni zamjenskom metodom, čak i nakon potvrde jednakovrijednosti, mogu odstupati od rezultata dobivenih referentnom metodom. |
D. ODREĐIVANJE UREE
1. Svrha i područje primjene
Ovom se metodom omogućuje određivanje razine uree u hrani za životinje.
2. Načelo
Uzorak se suspendira u vodi u prisutnosti tvari za bistrenje. Suspenzija se filtrira. Udio uree u filtratu određuje se nakon dodavanja 4-dimetilaminobenzaldehida (4-DMAB) mjerenjem optičke gustoće na valnoj duljini od 420 nm.
3. Reagensi
|
3.1. |
Otopina 4-dimetilaminobenzaldehida: otopi se 1,6 g 4-DMAB u 100 ml etanola 96 % i doda 10 ml klorovodične kiseline (ρ20 1,19 g/ml). Taj se reagens može čuvati najviše dva tjedna. |
|
3.2. |
Otopina Carrez I: u vodi se otopi 21,9 g cinkovog acetata Zn(CH3COO)2 2H2O i 3 g ledene octene kiseline. Dopuni se vodom do 100 ml. |
|
3.3. |
Otopina Carrez II: u vodi se otopi 10,6 g kalijevog ferocijanida K4 Fe (CN)6 3H2O. Dopuni se vodom do 100 ml. |
|
3.4. |
Aktivni ugljen koji ne apsorbira ureu (treba provjeriti). |
|
3.5. |
Urea, otopina 0,1 % (m/v). |
4. Oprema
|
4.1. |
Mješalica (rotacijska): približno 35 do 40 okr./min. |
|
4.2. |
Epruvete: 160 × 16 mm s ubrušenim čepovima. |
|
4.3. |
Spektrofotometar |
5. Postupak
5.1. Analiza uzorka
Odvagne se 2 g uzorka s preciznošću od 1 mg i prenese s 1 g aktivnog uglja (točka 3.4.) u graduiranu tikvicu obujma 500 ml. Doda se 400 ml vode i 5 ml otopine Carrez I (točka 3.2.), miješa približno 30 sekundi i doda 5 ml otopine Carrez II (točka 3.3.). Miješa se 30 minuta u mješalici. Dopuni se vodom do oznake, protrese i filtrira.
Uzme se 5 ml prozirnog bezbojnog filtrata i prenese u epruvete s ubrušenim čepovima, doda se 5 ml otopine 4-DMAB (točka 3.1.) i promiješa. Epruvete se postave u vodenu kupelj na 20 °C (+/- 4 °C). Nakon 15 minuta spektrofotometrom se izmjeri optička gustoća otopine uzorka na 420 nm. Rezultat se usporedi s otopinom reagensa iz slijepe probe.
5.2. Kalibracijska krivulja
Uzme se 1, 2, 4, 5 i 10 ml otopine uree (točka 3.5.) i prenese u odmjerne tikvice obujma 100 ml koje se dopune vodom do oznake. Iz svake se otopine odstrani 5 ml, te se u svaku doda 5 ml otopine 4-DMAB (točka 3.1.), homogenizira i izmjeri optička gustoća, kako je gore navedeno, u usporedbi s kontrolnom otopinom koja sadrži 5 ml 4-DMAB i 5 ml vode bez uree. Pripremi se kalibracijska krivulja.
6. Izračun rezultata
Količina uree u uzorku određuje se iz kalibracijske krivulje.
Rezultat se iskazuje kao postotni udio uzorka.
7. Napomene
|
7.1. |
Ako udio uree prelazi 3 %, uzorak se smanji na 1 g ili se početna otopina razrijedi tako da u 500 ml nema više od 50 mg uree. |
|
7.2. |
Ako je udio uree nizak, uzorak se povećava sve dok je filtrat proziran i bezbojan. |
|
7.3. |
Ako uzorak sadrži jednostavne dušične spojeve, poput aminokiselina, optička se gustoća mjeri na 435 nm. |
E. ODREĐIVANJE HLAPLJIVIH DUŠIKOVIH BAZA
I. MIKRODIFUZIJA
1. Svrha i područje primjene
Ova se metoda koristi za određivanje udjela hlapljivih dušikovih baza u hrani za životinje iskazanog kao amonijak.
2. Načelo
Uzorak se ekstrahira vodom, a otopina se izbistri i filtrira. Hlapljive dušične baze istiskuju se mikrodifuzijom uporabom otopine kalijevog karbonata, prikupe u otopinu borne kiseline i titriraju sumpornom kiselinom.
3. Reagensi
|
3.1. |
Trikloroctena kiselina, otopina 20 % (m/v). |
|
3.2. |
Indikator: otopi se 33 mg bromkrezol zelenila i 65 mg metilnog crvenila u 100 ml etanola 95 – 96 % (v/v). |
|
3.3. |
Otopina borne kiseline: u odmjernoj tikvici volumena od 1 litre otopi se 10 g borne kiseline u 200 ml etanola 95 – 96 % (v/v) i 700 ml vode. Doda se 10 ml indikatora (točka 3.2.). Promiješa se, a prema potrebi boja otopine promijeni u svijetlo crvenu dodavanjem otopine natrijevog hidroksida. Količina od 1 ml te otopine vezat će najviše 300 μg NH3. |
|
3.4. |
Zasićena otopina kalijevog karbonata: u 100 ml vrele vode otopi se 100 g kalijevog karbonata. Ostavi se da se ohladi, a zatim se filtrira. |
|
3.5. |
Sumporna kiselina, 0,01 mol/l. |
4. Oprema
|
4.1. |
Mješalica (rotacijska): približno 35 do 40 okr./min. |
|
4.2. |
Stakleni ili plastični Conwayevi članci (vidjeti dijagram). |
|
4.3. |
Mikrobirete graduirane s podjelom 1/100 ml. |
5. Postupak
Odvagne se 10 g uzorka s preciznošću od 1 mg i prenese sa 100 ml vode u graduiranu tikvicu obujma 200 ml. Trese se ili miješa 30 minuta u rotacijskoj mješalici. Doda se 50 ml otopine trikloroctene kiseline (točka 3.1.), dopuni vodom do oznake, snažno protrese i filtrira kroz nabrani filtar.
Pipetom se doda 1 ml otopine borne kiseline (točka 3.3.) u srednji dio Conwayevog članka i 1 ml filtrata uzorka u gornji dio članka. Djelomično se pokrije namašćenim poklopcem. U gornji dio članka brzo se nakapa 1 ml zasićene otopine kalijevog karbonata (točka 3.4.) i poklopac hermetički zatvori. Članak se oprezno okreće u vodoravnoj ravnini tako da se reagensi pomiješaju. Ostavi se najmanje četiri sata na sobnoj temperaturi ili jedan sat na temperaturi 40 °C.
Mikrobiretom (točka 4.3.) se hlapljive baze u otopini borne kiseline titriraju sumpornom kiselinom (točka 3.5.).
Slijepa se proba izvodi istim postupkom, ali bez uzorka za analizu.
6. Izračun rezultata
1 ml H2SO4 0,01 mol/l odgovara 0,34 mg amonijaka.
Rezultat analize iskazuje se kao postotni udio uzorka.
Ponovljivost
Razlika između rezultata dvaju usporednih postupaka određivanja, izvedenih na istom uzorku, ne smije prijeći:
|
— |
10 % relativne vrijednosti za udio amonijaka manji od 1,0 %, |
|
— |
0,1 % apsolutne vrijednosti za udio amonijaka 1,0 % ili veći. |
7. Napomena
Ako je udio amonijaka u uzorku veći od 0,6 % početni se filtrat razrijedi.
CONWAYEV ČLANAK
Omjer 1/1

II. DESTILACIJA
1. Svrha i područje primjene
Ova se metoda koristi za određivanje udjela hlapljivih dušikovih baza, iskazanih kao amonijak, u ribljem brašnu koje praktično ne sadrži ureu. Koristi se samo za udio amonijaka niži od 0,25 %.
2. Načelo
Uzorak se ekstrahira vodom, a otopina izbistri i filtrira. Hlapljive dušične baze se istisnu na temperaturi vrelišta dodavanjem magnezijevog oksida i prikupe u određenoj količini sumporne kiseline, višak koje se titrira otopinom natrijevog hidroksida.
3. Reagensi
|
3.1. |
Trikloroctena kiselina, otopina 20 % (m/v). |
|
3.2. |
Magnezijev oksid. |
|
3.3. |
Emulzija protiv pjenjenja (npr. silikon). |
|
3.4. |
Sumporna kiselina, 0,05 mol/l. |
|
3.5. |
Otopina natrijevog hidroksida, 0,1 mol/l. |
|
3.6. |
Otopina metilnog crvenila 0,3 % u etanolu 95 % – 96 % (v/v). |
4. Oprema
|
4.1. |
Mješalica (rotacijska): približno 35 do 40 okr./min. |
|
4.2. |
Uređaj za destilaciju po Kjeldahlu. |
5. Postupak
Odvagne se 10 g uzorka s preciznošću od 1 mg i prenese sa 100 ml vode u graduiranu tikvicu obujma 200 ml. Trese se ili miješa 30 minuta u rotacijskoj mješalici. Doda se 50 ml otopine trikloroctene kiseline (točka 3.1.), dopuni vodom do oznake, snažno protrese i filtrira kroz nabrani filtar papir.
Pipetom se prenese količina bistrog filtrata koja odgovara očekivanom sadržaju hlapljivih dušikovih baza (obično je dovoljno 100 ml). Razrijedi se na 200 ml, doda se 2 g magnezijevog oksida (točka 3.2.) i nekoliko kapljica emulzije protiv pjenjenja (točka 3.3.). Otopina mora biti alkalna pri pokusu s lakmusovim papirom; ako to nije slučaj, doda se magnezijev oksid (točka 3.2.). Postupa se u skladu s točkama 5.2. i 5.3. metode analize za određivanje udjela sirovih bjelančevina (dio C ovog Priloga).
Slijepa se proba izvodi istim postupkom, ali bez uzorka za analizu.
6. Izračun rezultata
1 ml H2SO4 0,05 mol/l odgovara 1,7 mg amonijaka.
Rezultat se iskazuje kao postotni udio uzorka.
Ponovljivost
Razlika između rezultata dvaju usporednih postupaka određivanja, izvedenih na istom uzorku, ne smije prijeći 10 % relativne vrijednosti amonijaka.
F. ODREĐIVANJE AMINOKISELINA (OSIM TRIPTOFANA)
1. Svrha i područje primjene
Ova se metoda koristi za određivanje slobodnih (sintetičkih i prirodnih) i ukupnih (vezanih na peptide i slobodnih) aminokiselina u hrani za životinje pomoću analizatora aminokiselina. Koristi se za sljedeće aminokiseline: cist(e)in, metionin, lizin, treonin, alanin, arginin, aspartatna kiselina, glutaminska kiselina, glicin, histidin, izoleucin, leucin, fenilalanin, prolin, serin, tirozin i valin.
Ovom se metodom ne mogu razlikovati soli aminokiselina niti je moguće utvrditi razliku između oblika D i L aminokiselina. Ova metoda nije primjerena za određivanje triptofana ili hidroksi-analoga aminokiselina.
2. Načelo
2.1. Slobodne aminokiseline
Slobodne aminokiseline ekstrahiraju se razrijeđenom klorovodičnom kiselinom. Koekstrahirane dušične makromolekule istalože se sulfosalicilnom kiselinom i uklone filtriranjem. Vrijednost pH filtrirane otopine podesi se na 2,20. Aminokiseline se razdvoje ionsko-izmjenjivačkom kromatografijom i odrede reakcijom s ninhidrinom fotometrijskom detekcijom na 570 nm.
2.2. Ukupne aminokiseline
Odabir postupka ovisi o ispitivanim aminokiselinama. Cist(e)in i metionin se prije hidrolize moraju oksidirati u cisteinsku kiselinu i metionin-sulfon. Tirozin se mora odrediti u hidrolizatima neoksidiranih uzoraka. Sve druge aminokiseline navedene u točki 1 mogu se odrediti u oksidiranom ili neoksidiranom uzorku.
Oksidacija se izvodi na 0 °C mješavinom peroksimravlje kiseline i fenola. Višak oksidacijskog reagensa razgradi se natrijevim disulfitom. Oksidirani ili neoksidirani uzorak se 23 sata hidrolizira klorovodičnom kiselinom (točka 3.20.). Vrijednost pH hidrolizata podesi se na 2,20. Aminokiseline se razdijele ionsko-izmjenjivačkom kromatografijom i odrede reakcijom s ninhidrinom fotometrijskom detekcijom na 570 nm (440 nm za prolin).
3. Reagensi
Obvezno se koristi dvostruko destilirana voda ili voda jednake kakvoće (provodljivost < 10 μS)
|
3.1. |
Vodikov peroksid, m (m/m) = 30 %. |
|
3.2. |
Mravlja kiselina, m (m/m) = 98 % – 100 %. |
|
3.3. |
Fenol. |
|
3.4. |
Natrijev disulfit. |
|
3.5. |
Natrijev hidroksid. |
|
3.6. |
5-sulfosalicilna kiselina dihidrat. |
|
3.7. |
Klorovodična kiselina, gustoće približno 1,18 g/ml. |
|
3.8. |
Trinatrij citrat dihidrat. |
|
3.9. |
2,2’-tiodietanol (tiodiglikol). |
|
3.10. |
Natrijev klorid. |
|
3.11. |
Ninhidrin. |
|
3.12. |
Lagani petrolej, vrelište 40 – 60 °C. |
|
3.13. |
Norleucin ili drugi spoj primjeren za uporabu kao interni standard |
|
3.14. |
Plinoviti dušik (< 10 ppm kisika). |
|
3.15. |
1-oktanol. |
|
3.16. |
Aminokiseline. |
|
3.16.1. |
Standardne tvari iz točke 1. Čisti spojevi koji ne sadrže kristalnu vodu. Prije uporabe suše se 1 tjedan pod vakuumom s P2O5 ili H2SO4. |
|
3.16.2. |
Cisteinska kiselina. |
|
3.16.3. |
Metionin-sulfon. |
|
3.17. |
Otopina natrijevog hidroksida, c = 7,5 mol/l: U vodi se otopi 300 g NaOH (točka 3.5.) i dopuni vodom do 1 litre. |
|
3.18. |
Otopina natrijevog hidroksida, c = 1 mol/l: U vodi se otopi 40 g NaOH (točka 3.5.) i dopuni vodom do 1 litre. |
|
3.19. |
Mravlja kiselina – otopina fenola: Pomiješa se 889 g mravlje kiseline (točka 3.2.) sa 111 g vode i doda 4,73 g fenola (točka 3.3.). |
|
3.20. |
Smjesa za hidrolizu, c = 6 mol HCl/l koja sadrži 1 g fenola/l: Doda se 1 g fenola (točka 3.3.) u 492 ml HCl (točka 3.7.) i dopuni vodom do 1 litre. |
|
3.21. |
Smjesa za ekstrakciju, c = 0,1 mol HCl/l koja sadrži 2 % tiodiglikola: 8,2 ml HCl (točka 3.7.) se razrijedi s približno 900 ml vode, doda se 20 ml tiodiglikola (točka 3.9.) i dopuni vodom do 1 litre (ne smiju se izravno miješati tvari iz točaka 3.7. i 3.9.). |
|
3.22. |
5-sulfosalicilna kiselina, ß = 6 %: U vodi se otopi 60 g 5-sulfosalicilne kiseline (točka 3.6.) i dopuni vodom do 1 litre. |
|
3.23. |
Oksidacijska smjesa (peroksimravlja kiselina – fenol): Pomiješa se 0,5 ml vodikovog peroksida (točka 3.1.) s 4,5 ml otopine mravlje kiseline i fenola (točka 3.19.) u maloj čaši. Inkubira se 1 sat na 20 – 30 °C kako bi se dobila peroksimravlja kiselina, zatim se hladi u ledenoj kupelji (15 min) prije dodavanja u uzorak. Upozorenje: Izbjegavati dodir s kožom i nositi zaštitnu odjeću. |
|
3.24. |
Citratni pufer, c = 0,2 mol Na+/l, pH 2,20: U približno 800 ml vode otopi se 19,61 g natrijevog citrata (točka 3.8.), 5 ml tiodiglikola (točka 3.9.), 1 g fenola (točka 3.3.) i 16,50 ml HCl (točka 3.7.). Vrijednost pH se podesi na 2,20. Dopuni se vodom do 1 litre. |
|
3.25. |
Puferi za eluiranje, pripravljeni u skladu s uputama za analizator koji se koristi (točka 4.9.). |
|
3.26. |
Reagens ninhidrin, pripravljen u skladu s uputama za analizator koji se koristi (točka 4.9.). |
|
3.27. |
Standardne otopine aminokiselina. Ove se otopine drže na temperaturi nižoj od 5 °C. |
|
3.27.1. |
Osnovna standardna otopina aminokiselina (točka 3.16.1.). c = 2,5 μmol/ml za svaku u klorovodičnoj kiselini. Može se nabaviti u slobodnoj prodaji. |
|
3.27.2. |
Osnovna standardna otopina cisteinske kiseline i metionin-sulfona, c = 1,25 μmol/ml. U odmjernoj tikvici obujma 1 l otopi se 0,2115 g cisteinske kiseline (točka 3.16.2.) i 0,2265 g metionin-sulfona (točka 3.16.3.) u citratnom puferu (točka 3.24.) i dopuni citratnim puferom do oznake. Može se pohraniti najviše 12 mjeseci na temperaturi nižoj od 5 °C. Ova se otopina ne koristi ako osnovna standardna otopina (točka 3.27.1.) sadrži cisteinsku kiselinu i metionin-sulfon. |
|
3.27.3. |
Osnovna standardna otopina internog standarda, npr. norleucina, c = 20 μmol/ml. U odmjernoj se tikvici otopi 0,6560 g norleucina (točka 3.13.) u citratnom puferu (točka 3.24.) i dopuni citratnim puferom do 250 ml. Može se pohraniti najviše 6 mjeseci na temperaturi nižoj od 5 °C. |
|
3.27.4. |
Kalibracijska otopina standardnih aminokiselina koja se koristi s hidrolizatima, c = 5 nmol/50 μl za cisteinsku kiselinu i metionin-sulfon i c = 10 nmol/50 μl za druge aminokiseline. U čaši obujma 100 ml otopi se 2,2 g natrijevog klorida (točka 3.10.) s 30 ml citratnog pufera (točka 3.24.). Doda se 4,00 ml osnovne standardne otopine aminokiseline (točka 3.27.1.), 4,00 ml osnovne standardne otopine cisteinske kiseline i metionin-sulfona (točka 3.27.2.) i 0,50 ml osnovne standardne otopine internog standarda (točka 3.27.3.) ako se isti koristi. Vrijednost pH se natrijevim hidroksidom podesi na 2,20 (točka 3.18.). Udio se kvantitativno prenese u graduiranu tikvicu obujma 50 ml, dopuni citratnim puferom (točka 3.24.) do oznake i promiješa. Pohrani se najviše 3 mjeseca na temperaturi nižoj od 5 °C. Vidjeti također napomenu pod točkom 9.1. |
|
3.27.5. |
Pripremi se kalibracijska otopina standardnih aminokiselina koja se koristi s hidrolizatima u skladu s točkom 5.3.3.1., a za ekstrakte u skladu s točkom 5.2. Kalibracijska otopina se pripremi u skladu s točkom 3.27.4., ali bez natrijevog klorida. Pohrani se najviše 3 mjeseca na temperaturi nižoj od 5 °C. |
4. Oprema
|
4.1. |
Tikvica s okruglim dnom volumena 100 ili 250 ml s povratnim hladilom. |
|
4.2. |
Staklena posuda od borosilikatnog stakla obujma 100 ml s navojnim čepom obloženim gumom/teflonom (npr. Duran, Schott) za uporabu u sušilu. |
|
4.3. |
Sušilo s prisilnom ventilacijom i regulatorom temperature točnosti veće od ± 2 °C. |
|
4.4. |
pH metar (na tri decimalna mjesta). |
|
4.5. |
Membranski filtar (0,22 μm). |
|
4.6. |
Centrifuga. |
|
4.7. |
Rotacijski vakuumski otparivač. |
|
4.8. |
Mehanička tresilica ili magnetska mješalica. |
|
4.9. |
Analizator aminokiselina ili oprema za HPLC opremljena kolonom za izmjenu iona, napravom za ninhidrin i derivatizaciju nakon kolone te fotometrijskim detektorom. Kolona se napuni smolama sulfoniranog polistirena koje mogu razdvajati aminokiseline jedne od drugih i od drugih materijala koji reagiraju na ninhidrin. Tok u puferskoj i ninhidrinskoj liniji osigurava se crpkama s tokom stabilnosti ± 0,5 % u vremenu koje obuhvaća izvođenje standardnog kalibriranja i analizu uzorka. Kod nekih analizatora aminokiselina mogu se koristiti postupci hidrolize kod kojih je koncentracija natrija u hidrolizatu c = 0,8 mol/l, a hidrolizat sadrži svu ostatnu mravlju kiselinu iz oksidacije. Drugim metodama ne postiže se zadovoljavajuće razdvajanje nekih aminokiselina ako se u hidrolizatu nalazi višak mravlje kiseline i/ili visoka koncentracija natrijevih iona. U tom se slučaju obujam kiseline nakon hidrolize i prije podešavanja pH smanjuje isparavanjem na približno 5 ml. Isparavanje se vrši pod vakuumom na najviše 40 °C. |
5. Postupak
5.1. Priprema uzorka
Uzorak se samelje tako da prolazi kroz sito veličine 0,5 mm. Uzorci s visokim udjelom vode moraju se prije mljevenja osušiti na zraku na temperaturi koja ne prelazi 50 °C ili smrzavanjem. Uzorci s visokim sadržajem masti ekstrahiraju se laganim petrolejem (točka 3.12.) prije mljevenja.
5.2. Određivanje slobodnih aminokiselina u hrani za životinje i premiksima
Odvagne se primjerena količina (1 – 5 g) pripravljenog uzorka (točka 5.1.) s preciznošću od 0,2 mg i prenese u konusnu tikvicu, te se doda 100,0 ml smjese za ekstrakciju (točka 3.21.). Smjesa se 60 minuta trese mehaničkom tresilicom ili magnetskom mješalicom (točka 4.8.). Pričeka se da talog padne na dno i pipetom prenese 10,0 ml supernatanta u čašu obujma 100 ml.
Tijekom miješanja doda se 5,0 ml otopine sulfosalicilne kiseline (točka 3.22.) i nastavi 5 minuta miješati magnetskom mješalicom. Supernatant se filtrira ili centrifugira kako bi se uklonio cjelokupni talog. U čašu obujma 100 ml izlije se 10,0 ml dobivene otopine i vrijednost pH podesi na 2,20 otopinom natrijevog hidroksida (točka 3.18.), prenese citratnim puferom (točka 3.24.) u graduiranu tikvicu primjerenog obujma i dopuni otopinom pufera (točka 3.24.) do oznake.
Ako se koristi interni standard, za svakih 100 ml konačne otopine doda se 1,00 ml internog standarda (točka 3.27.3.) i dopuni otopinom pufera (točka 3.24.) do oznake.
Nastavlja se s kromatografijom u skladu s točkom 5.4.
Ako se ekstrakti ne pregledavaju isti dan, moraju se pohraniti na temperaturi nižoj od 5 °C.
5.3. Određivanje ukupnih aminokiselina
5.3.1.
Odvagne se 0,1 – 1 g pripravljenog uzorka (točka 5.1.) s preciznošću od 0,2 mg i prenese u:
|
— |
tikvicu s okruglim dnom volumena 100 ml (točka 4.1.) za otvorenu hidrolizu (točka 5.3.2.3.) ili, |
|
— |
tikvicu s okruglim dnom volumena 250 ml (točka 4.1.) ako se zahtijeva niska koncentracija natrija (točka 5.3.3.1.) ili, |
|
— |
staklenu bocu s navojnim čepom volumena 100 ml (točka 4.2.) za zatvorenu hidrolizu (točka 5.3.2.4.). |
U odvaganom dijelu uzorka udio dušika mora iznositi približno 10 mg, a udio vode ne smije biti veći od 100 mg.
Tikvica/boca se postavi u ledenu kupelj i ohladi na 0 °C, doda se 5 ml oksidacijske smjese (točka 3.23.) i miješa staklenom lopaticom sa zakrivljenim vrhom. Tikvica/boca s lopaticom se hermetički zatvori filmom, a ledena kupelj s hermetički zatvorenom posudom stavi u hladnjak na 0 °C i ostavi 16 sati. Nakon 16 sati posuda se izvadi iz hladnjaka, a višak oksidacijskog reagensa se razgradi dodavanjem 0,84 g natrijevog disulfita (točka 3.4.).
Nastavlja se u skladu s točkom 5.3.2.1.
5.3.2.
5.3.2.1.
U oksidirani uzorak, pripravljen u skladu s točkom 5.3.1., doda se 25 ml smjese za hidrolizu (točka 3.20.), pri čemu treba brižljivo isprati sve ostatke uzorka sa stijenke posude i lopatice.
Nastavlja se u skladu s točkom 5.3.2.3. ili 5.3.2.4., ovisno o korištenom postupku hidrolize.
5.3.2.2.
U tikvicu s okruglim dnom obujma 100 ml ili 250 ml (točka 4.1.) ili u bocu s navojnim čepom obujma 100 ml (točka 4.2.) odvagne se 0,1 – 1 g pripravljenog uzorka (točka 5.1.) s preciznošću od 0,2 mg. U odvaganom dijelu uzorka udio dušika mora iznositi približno 10 mg. Oprezno se doda 25 ml smjese za hidrolizu (točka 3.20.) i pomiješa s uzorkom. Nastavlja se u skladu s točkom 5.3.2.3. ili 5.3.2.4.
5.3.2.3.
U smjesu koja se nalazi u tikvici (pripremljenu u skladu s točkom 5.3.2.1. ili 5.3.2.2.) dodaju se 3 staklene kuglice, zatim se ostavi ključati 23 sata uz stalno vrenje pod refluksom. Po završetku hidrolize kondenzator ispere se s 5 ml citratnog pufera (točka 3.24.). Tikvica se odvoji i ohladi u ledenoj kupelji.
Nastavlja se u skladu s točkom 5.3.3.
5.3.2.4.
Boca sa smjesom pripravljenom u skladu s točkom 5.3.2.1. ili 5.3.2.2. postavi se u sušilo (točka 4.3.) na temperaturi od 110 °C. Kako bi se tijekom prvog sata spriječilo povećanje tlaka i eksplozija (zbog nastanka plinovitih tvari), poklopac s navojem treba postavi na vrh posude. Posuda se ne smije zatvoriti poklopcem. Nakon jednog sata posuda se zatvori poklopcem i ostavi 23 sata u sušilu (točka 4.3.). Po završetku hidrolize boca se izvadi iz sušila, oprezno se otvori poklopac boce i boca položi u ledenu kupelj. Ostavi se da se ohladi.
Ovisno o postupku podešavanja pH (točka 5.3.3.) udio posude se citratnim puferom (točka 3.24.) kvantitativno prenese u čašu obujma 250 ml ili tikvicu s okruglim dnom obujma 250 ml.
Nastavlja se u skladu s točkom 5.3.3.
5.3.3.
Ovisno o odstupanju analizatora aminokiselina (točka 4.9.) za natrij, podešavanje pH se provodi u skladu s točkom 5.3.3.1. ili 5.3.3.2.
5.3.3.1.
Pri upotrebi analizatora aminokiselina koji zahtijevaju nisku koncentraciju natrija (kada treba smanjiti volumen kiseline) preporučuje se uporaba otopine internog osnovnog standarda (točka 3.27.3.).
U tom se slučaju hidrolizatu prije otparivanja dodaje 2,00 ml otopine internog osnovnog standarda (točka 3.27.3.).
U hidrolizat dobiven postupkom u skladu s točkom 5.3.2.3. ili 5.3.2.4. dodaje se 2 kapljice 1-oktanola (točka 3.15.).
Obujam se smanji na 5 – 10 ml rotacijskim otparivačem (točka 4.7.) pod vakuumom na 40 °C. Ako se obujam slučajno smanji na manje od 5 ml, hidrolizat se mora odbaciti i iznova započeti analizu.
Otopinom natrijevog hidroksida (točka 3.18.) vrijednost pH podesi se na 2,20 i zatim nastavi u skladu s točkom 5.3.4.
5.3.3.2.
Hidrolizati dobiveni u skladu s točkom 5.3.2.3. ili 5.3.2.4. djelomično se neutraliziraju opreznim dodavanjem, uz miješanje, 17 ml otopine natrijevog hidroksida (točka 3.17.), pri čemu se temperatura mora održavati na manje od 40 °C.
Na sobnoj temperaturi podesi se vrijednost pH na 2,20 otopinom natrijevog hidroksida (točka 3.17.) i zatim otopinom natrijevog hidroksida (točka 3.18.). Nastavlja se u skladu s točkom 5.3.4.
5.3.4.
Hidrolizat (točka 5.3.3.1. ili 5.3.3.2.) s podešenom vrijednosti pH, citratnim se puferom (točka 3.24.) kvantitativno prenese u graduiranu tikvicu obujma 200 ml i dopuni puferom (točka 3.24.) do oznake.
Ako dotada nije korišten interni standard, doda se 2,00 ml internog standarda (točka 3.27.3.) i zatim dopuni citratnim puferom (točka 3.24.) do oznake. Temeljito se promiješa.
Nastavlja se s kromatografijom (točka 5.4.).
Ako se otopine uzorka ne pregledavaju isti dan, moraju se pohraniti na temperaturi nižoj od 5 °C.
5.4. Kromatografija
Prije kromatografije temperaturu ekstrakta (točka 5.2.) ili hidrolizata (točka 5.3.4.) treba izjednačiti sa sobnom temperaturom. Smjesa se protrese i prikladna količina filtrira kroz membranski filtar 0,22 μm (točka 4.5.). Dobivena bistra otopina se podvrgne kromatografiji s izmjenom iona uporabom analizatora aminokiselina (točka 4.9.).
Ubrizgavanje se može izvesti ručno ili automatski. Važno je na kolonu za analizu standarda i uzoraka dodati istu količinu otopine (± 0,5 %) osim kada se koristi interni standard, a omjeri natrija i aminokiselina u otopinama standarda i uzorka trebaju biti što je moguće sličniji.
Općenito učestalost kalibriranja ovisi o stabilnosti reagensa ninhidrina i sustava za analizu. Standard ili uzorak se razrijede citratnim puferom (točka 3.24.) tako da površina vršaka standarda dostiže 30 – 200 % površine vršaka aminokiselina u uzorku.
Kromatografija aminokiselina neznatno se razlikuje s obzirom na vrstu korištenog analizatora i smole. Odabrani sustav mora imati mogućnost razdvajanja aminokiselina međusobno i od drugih materijala koji reagiraju na ninhidrin. U radnom području kromatografski sustav mora imati linearan odziv na promjene količina aminokiselina dodanih u kolonu.
U fazi kromatografije primjenjuju se dolje navedeni omjeri između najnižih i najviših vrijednosti kod analize ekvimolarnih otopina (aminokiselina koje se određuju). Ekvimolarna otopina mora sadržavati najmanje 30 % maksimalnog udjela svake aminokiseline koji se može točno izmjeriti sustavom za analizu aminokiselina (točka 4.9.).
Kod postupka razdvajanja treonina od serina, omjer između najniže i najviše vrijednosti za aminokiselinu s nižom vrijednosti između dviju aminokiselina koje se preklapaju na kromatogramu ne smije prijeći 2:10. (Ako se određuju samo cist(e)in, metionin, treonin i lizin, nedovoljno razdvajanje između susjednih vršaka štetno će djelovati na postupak određivanja). Za sve druge aminokiseline razdvajanje mora biti veće od 1:10.
Sustav mora jamčiti razdvajanje lizina od „lizinskih artefakata” i ornitina.
6. Izračun rezultata
Za svaku pojedinačnu aminokiselinu izmjeri se površina vršaka uzorka i standarda, a količina (X), iskazana u g aminokiseline na kg uzorka, izračunava se na sljedeći način:

Ako se koristi interni standard pomnoži se s:
|
A |
= |
površina vršaka hidrolizata ili ekstrakta |
|
B |
= |
površina vršaka standardne kalibracijske otopine |
|
C |
= |
površina vršaka hidrolizata ili ekstrakta kao internog standarda |
|
D |
= |
površina vršaka standardne kalibracijske otopine kao internog standarda |
|
M |
= |
molarna masa aminokiseline koja se određuje |
|
c |
= |
koncentracija standarda u μmol/ml |
|
m |
= |
masa uzorka u gramima (ispravljena na početnu masu ako je uzorak isušen ili odmašćen) |
|
V |
= |
ml ukupnog hidrolizata (točka 5.3.4.) ili izračunani ukupni volumen otopine ekstrakta (točka 6.1.) u ml |
Cistin i cistein se određuju kao cisteinska kiselina u hidrolizatima oksidiranog uzorka, ali se izračunavaju kao cistin (C6H12N2O4S2, M 240,30 g/mol) uporabom molarne mase 120,15 g/mol (= 0,5 × 240,30 g/mol).
Metionin se određuje kao metionin-sulfon u hidrolizatima oksidiranog uzorka, ali se izračunava kao metionin uporabom molarne mase metionina: 149,21 g/mol.
Dodani slobodni metionin određuje se nakon ekstrakcije kao metionin, a za izračun se koristi ista molekulska masa.
|
6.1. |
Ukupni volumen otopine ekstrakata (F) za određivanje slobodnih aminokiselina (točka 5.2.) izračunava se sljedećom formulom: 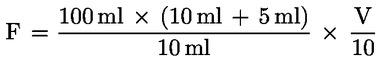
|
7. Vrednovanje metode
Metoda je provjerena u međulaboratorijskom ispitivanju na međunarodnoj razini provedenom 1990. na četiri različite vrste hrane za životinje (miješana hrana za svinje, smjesa za brojlere, proteinski koncentrat, premiks). Rezultati srednjih vrijednosti i standardnih devijacija, bez ekstremnih vrijednosti, prikazani su u tablicama u ovoj točki:
Srednje vrijednosti u g/kg
|
Referentni materijal |
Aminokiselina |
||||||
|
Treonin |
Cist(e)in |
Metionin |
Lizin |
||||
|
Miješana hrana za svinje |
6,94 n = 15 |
3,01 n = 17 |
3,27 n = 17 |
9,55 n = 13 |
|||
|
Smjesa za brojlere |
9,31 n = 16 |
3,92 n = 18 |
5,08 n = 18 |
13,93 n = 16 |
|||
|
Proteinski koncentrat |
22,32 n = 16 |
5,06 n = 17 |
12,01 n = 17 |
47,74 n = 15 |
|||
|
Premiks |
58,42 n = 16 |
— |
90,21 n = 16 |
98,03 n = 16 |
|||
|
|||||||
7.1. Ponovljivost
Ponovljivost gore navedene međulaboratorijske usporedbe, iskazana kao „unutarlaboratorijska standardna devijacija”, prikazana je u donjim tablicama:
Unutarlaboratorijska standardna devijacija (Sr), iskazana u g/kg
|
Referentni materijal |
Aminokiselina |
||||||
|
Treonin |
Cist(e)in |
Metionin |
Lizin |
||||
|
Miješana hrana za svinje |
0,13 n = 15 |
0,10 n = 17 |
0,11 n = 17 |
0,26 n = 13 |
|||
|
Smjesa za brojlere |
0,20 n = 16 |
0,11 n = 18 |
0,16 n = 18 |
0,28 n = 16 |
|||
|
Proteinski koncentrat |
0,48 n = 16 |
0,13 n = 17 |
0,27 n = 17 |
0,99 n = 15 |
|||
|
Premiks |
1,30 n = 16 |
— |
2,19 n = 16 |
2,06 n = 16 |
|||
|
|||||||
Koeficijent varijacije (%) za unutarlaboratorijsku standardnu devijaciju (Sr)
|
Referentni materijal |
Aminokiselina |
||||||
|
Treonin |
Cist(e)in |
Metionin |
Lizin |
||||
|
Miješana hrana za svinje |
1,9 n = 15 |
3,3 n = 17 |
3,4 n = 17 |
2,8 n = 13 |
|||
|
Smjesa za brojlere |
2,1 n = 16 |
2,8 n = 18 |
3,1 n = 18 |
2,1 n = 16 |
|||
|
Proteinski koncentrat |
2,7 n = 16 |
2,6 n = 17 |
2,2 n = 17 |
2,4 n = 15 |
|||
|
Premiks |
2,2 n = 16 |
— |
2,4 n = 16 |
2,1 n = 16 |
|||
|
|||||||
7.2. Ponovljivost
Rezultati gore navedene međulaboratorijske usporedbe, iskazani kao standardna devijacija između laboratorija, prikazani su u donjoj tablici:
Standardna devijacija između laboratorija (SR) u g/kg
|
Referentni materijal |
Aminokiselina |
||||||
|
Treonin |
Cist(e)in |
Metionin |
Lizin |
||||
|
Miješana hrana za svinje |
0,28 n = 15 |
0,30 n = 17 |
0,23 n = 17 |
0,30 n = 13 |
|||
|
Smjesa za brojlere |
0,48 n = 16 |
0,34 n = 18 |
0,55 n = 18 |
0,75 n = 16 |
|||
|
Proteinski koncentrat |
0,85 n = 16 |
0,62 n = 17 |
1,57 n = 17 |
1,24 n = 15 |
|||
|
Premiks |
2,49 n = 16 |
— |
6,20 n = 16 |
6,62 n = 16 |
|||
|
|||||||
Koeficijent varijacije (%) za standardnu devijaciju između laboratorija (SR)
|
Referentni materijal |
Aminokiselina |
||||||
|
Treonin |
Cist(e)in |
Metionin |
Lizin |
||||
|
Miješana hrana za svinje |
4,1 n = 15 |
9,9 n = 17 |
7,0 n = 17 |
3,2 n = 13 |
|||
|
Smjesa za brojlere |
5,2 n = 16 |
8,8 n = 18 |
10,9 n = 18 |
5,4 n = 16 |
|||
|
Proteinski koncentrat |
3,8 n = 16 |
12,3 n = 17 |
13,0 n = 17 |
3,0 n = 15 |
|||
|
Premiks |
4,3 n = 16 |
— |
6,9 n = 16 |
6,7 n = 16 |
|||
|
|||||||
8. Upotreba referentnih materijala
Pravilna primjena metode provjerava se ponovljenim mjerenjima na certificiranim referentnim materijalima, kada su dostupni. Preporučuje se izvršiti kalibraciju upotrebom certificirane otopine za kalibraciju aminokiselina.
9. Napomene
|
9.1. |
Zbog razlika između analizatora aminokiselina, kao smjernice se uzimaju konačne koncentracije otopina standardnih aminokiselina za kalibraciju (vidjeti točke 3.27.4. i 3.27.5.) i hidrolizata (vidjeti točku 5.3.4.). Za sve aminokiseline treba provjeriti područje linearnog odziva uređaja. Standardna se otopina razrijedi citratnim puferom tako da se površine vršaka nalaze u sredini područja. |
|
9.2. |
Kada se za analizu hidrolizata koristi oprema za tekućinsku kromatografiju visoke učinkovitosti, uvjeti pokusa se moraju optimizirati u skladu s preporukama proizvođača. |
|
9.3. |
Kod primjene metode za hranu za životinje koja sadrži više od 1 % klorida (koncentrat, mineralna hrana za životinje, dodaci hrani za životinje) može doći do preniske procjene udjela metionina, što zahtijeva posebnu obradu. |
G. ODREĐIVANJE TRIPTOFANA
1. Svrha i područje primjene
Ovom se metodom omogućuje određivanje ukupnog i slobodnog triptofana u hrani za životinje. Njom se ne omogućuje razlikovanje između oblika D i L.
2. Načelo
Za određivanje ukupnog triptofana uzorak se hidrolizira u bazičnim uvjetima zasićenom otopinom barijevog hidroksida i 20 sati grije na 110 °C. Nakon hidrolize dodaje se interni standard.
Za određivanje slobodnog triptofana uzorak se ekstrahira u lagano kiselim uvjetima u prisutnosti internog standarda.
Triptofan i interni standard u hidrolizatu ili ekstraktu određuju se postupkom HPLC s fluorescentnom detekcijom.
3. Reagensi
|
3.1. |
Obvezno se koristi redestilirana voda ili voda jednake kakvoće (provodljivost < 10 μS/cm). |
|
3.2. |
Standardna tvar: triptofan (čistoća/udio ≥ 99 %), osušen pod vakuumom iznad fosfornog pentoksida. |
|
3.3. |
Interna standardna tvar: α-metil-triptofan (čistoća/udio ≥ 99 %), osušen pod vakuumom iznad fosfornog pentoksida. |
|
3.4. |
Barijev hidroksid oktahidrat (treba paziti da se Ba(OH)2.8 H2O pretjerano ne izlaže zraku kako bi se izbjeglo nastajanje BaCO3 koji može remetiti postupak određivanja) (vidjeti napomenu pod točkom pod točkom 9.3.). |
|
3.5. |
Natrijev hidroksid. |
|
3.6. |
Ortofosforna kiselina, m (m/m) = 85 %. |
|
3.7. |
Klorovodična kiselina, ρ20 1,19 g/ml. |
|
3.8. |
Metanol, za HPLC. |
|
3.9. |
Lagani petrolej, raspon vrelišta 40 – 60 °C. |
|
3.10. |
Otopina natrijevog hidroksida, c = 1 mol/l: U vodi se otopi 40,0 g NaOH (točka 3.5.) i dopuni vodom do 1 litre (točka 3.1.). |
|
3.11. |
Klorovodična kiselina, c = 6 mol/l: Uzme se 492 ml HCl (točka 3.7.) i dopuni vodom do 1 litre. |
|
3.12. |
Klorovodična kiselina, c = 1 mol/l: Uzme se 82 ml HCl (točka 3.7.) i dopuni vodom do 1 litre. |
|
3.13. |
Klorovodična kiselina, c = 0,1 mol/l: Uzme se 8,2 ml HCl (točka 3.7.) i dopuni vodom do 1 litre. |
|
3.14. |
Ortofosforna kiselina, c = 0,5 mol/l: Uzme se 34 ml ortofosforne kiseline (točka 3.6.) i dopuni vodom (točka 3.1.) do 1 litre. |
|
3.15. |
Koncentrirana otopina triptofana (točka 3.2.), c = 2,50 μmol/ml: U odmjernoj tikvici obujma 500 ml otopi se 0,2553 g triptofana (točka 3.2.) u klorovodičnoj kiselini (točka 3.13.) i dopuni klorovodičnom kiselinom (točka 3.13.) do oznake. Pohrani se najviše 4 tjedna na –18 °C. |
|
3.16. |
Koncentrirana otopina internog standarda, c = 2,50 μmol/ml: U odmjernoj tikvici obujma 500 ml otopi se 0,2728 g α-metil-triptofana (točka 3.3.) u klorovodičnoj kiselini (točka 3.13.) i dopuni klorovodičnom kiselinom (točka 3.13.) do oznake. Pohrani se najviše 4 tjedna na –18 °C. |
|
3.17. |
Standardna otopina triptofana i internog standarda za kalibraciju: Pripremi se 2,00 ml koncentrirane otopine triptofana (točka 3.15.) i 2,00 ml koncentrirane otopine internog standarda (α-metil-triptofan) (točka 3.16.). Razrijedi se vodom (točka 3.1.) i metanolom (točka 3.8.) na približno jednak obujam i približno jednaku koncentraciju metanola (10 – 30 %) kao kod konačnog hidrolizata. Ta se otopina mora pripremiti svježa prije uporabe. Tijekom pripreme treba je zaštiti od izravnog sunčevog svjetla. |
|
3.18. |
Octena kiselina. |
|
3.19. |
1,1,1-triklor-2-metil-2-propanol. |
|
3.20. |
Etanolamin, m (m/m) > 98 %. |
|
3.21. |
Otopina 1 g 1,1,1-triklor-2-metil-2-propanola (točka 3.19.) u 100 ml metanola (točka 3.8.). |
|
3.22. |
Mobilna faza za HPLC: 3,00 g octene kiseline (točka 3.18.) + 900 ml vode (točka 3.1.) + 50,0 ml otopine (točka 3.21.) 1,1,1-triklor-2-metil-2-propanola (točka 3.19.) u metanolu (točka 3.8.) (1 g/100 ml). Etanolaminom (točka 3.20.) se vrijednost pH podesi na 5,00. Dopuni se vodom (točka 3.1.) do 1 000 ml. |
4. Oprema
|
4.1. |
Oprema za HPLC sa spektrofluorometrijskim detektorom. |
|
4.2. |
Kolona za tekućinsku kromatografiju, 125 mm × 4 mm C18, veličina čestica punjenja 3 μm ili jednakovrijedna. |
|
4.3. |
pH metar. |
|
4.4. |
Polipropilenska tikvica, obujma 125 ml, sa širokim vratom i navojnim čepom. |
|
4.5. |
Membranski filtar, 0,45 μm. |
|
4.6. |
Autoklav, 110 (± 2) °C, 1,4 (± 0,1) bar. |
|
4.7. |
Mehanička tresilica ili magnetska mješalica. |
|
4.8. |
Vrtložna mješalica. |
5. Postupak
5.1. Priprema uzoraka
Uzorak se samelje tako da prolazi kroz sito veličine 0,5 mm. Uzorci s visokim udjelom vode moraju se prije usitnjavanja osušiti na zraku na temperaturi koja ne prelazi 50 °C ili liofilizacijom. Uzorci s visokim sadržajem masti ekstrahiraju se laganim petrolejem (točka 3.9.) prije usitnjavanja.
5.2. Određivanje slobodnog triptofana (ekstrakt)
Odvagne se primjerena količina (1 – 5 g) pripravljenog uzorka (točka 5.1.) s preciznošću od 1 mg i prenese u konusnu tikvicu. Doda se 100,0 ml klorovodične kiseline (točka 3.13.) i 5,00 ml koncentrirane otopine internog standarda (točka 3.16.). Trese se ili miješa 60 minuta mehaničkom tresilicom ili magnetskom mješalicom (točka 4.7.). Pričeka se da talog padne na dno i pipetom prenese 10,0 ml supernatanta u čašu. Doda se 5 ml ortofosforne kiseline (točka 3.14.). Natrijevim hidroksidom (točka 3.10.) se pH vrijednost otopine podesi na 3. Doda se dovoljno metanola (točka 3.8.) kako bi u konačnom volumenu koncentracija metanola iznosila 10 – 30 %. Prenese se u graduiranu tikvicu primjerenog volumena i razrijedi vodom na volumen potreban za kromatografiju (približno jednak volumen kao kod standardne kalibracijske otopine (točka 3.17.)).
Prije ubrizgavanja na kolonu za HPLC filtrira se nekoliko ml otopine kroz membranski filtar 0,45 μm (točka 4.5.). Nastavlja se s kromatografijom u skladu s točkom 5.4.
Standardna otopina i ekstrakti se zaštite od izravnog sunčevog svjetla. Ako se ekstrakti ne mogu analizirati istog dana, mogu se pohraniti na temperaturi od 5 °C na razdoblje od najviše 3 dana.
5.3. Određivanje ukupnog triptofana (hidrolizat)
Odvagne se 0,1 – 1 g pripravljenog uzorka (točka 5.1.) s preciznošću od 0,2 mg i prenese u polipropilensku tikvicu (točka 4.4.). Udio dušika u odvaganom dijelu uzorka mora iznositi približno 10 mg. Doda se 8,4 mg barijevog hidroksid oktahidrata (točka 3.4.) i 10 ml vode. Miješa se vrtložnom mješalicom (točka 4.8.) ili magnetskom mješalicom (točka 4.7.). U smjesi se ostavi magnet obložen teflonom. Stijenke posude se isperu s 4 ml vode. Tikvica se poklopi navojnim čepom i lagano zatvori. Prenese se u autoklav (točka 4.6.) s vrelom vodom i ostavi u pari 30 – 60 minuta. Autoklav se zatvori i 20 sati autoklavira na 110 (± 2) °C.
Prije otvaranja autoklava, temperatura se snizi na malo ispod 100 °C. Kako bi se spriječila kristalizacija Ba(OH)2.8 H2O, u toplu se smjesu doda 30 ml vode temperature okoliša. Lagano se protrese ili promiješa. Doda se 2,00 ml koncentrirane otopine internog standarda (α-metil-triptofana) (točka 3.16.). Posude se 15 minuta hlade u vodenoj/ledenoj kupelji.
Zatim se doda 5 ml ortofosforne kiseline (točka 3.14.). Posuda se drži u rashladnoj kupki i uz miješanje neutralizira s HCl (točka 3.11.), a pH vrijednost se podesi na 3,0 s HCl (točka 3.12.). Doda se dovoljno metanola kako bi u konačnom obujmu koncentracija metanola iznosila 10 – 30 %. Prenese se u graduiranu tikvicu primjerenog obujma i razrijedi vodom do obujma potrebnog za kromatografiju (na primjer, 100 ml). Dodavanje metanola ne smije izazvati taloženje.
Prije ubrizgavanja na HPLC kolonu nekoliko ml otopine se filtrira kroz membranski filtar 0,45 μm (točka 4.5.). Nastavlja se s kromatografijom u skladu s točkom 5.4.
Standardna otopina i hidrolizati se zaštite od izravnog sunčevog svjetla. Ako se ekstrakti ne mogu analizirati istog dana, mogu se pohraniti na temperaturi od 5 °C na razdoblje od najviše 3 dana.
5.4. Određivanje HPLC-om
Za izokratsko se eluiranje kao smjernice predlažu sljedeći uvjeti; mogu se koristiti i drugi uvjeti ako se njima jamče jednakovrijedni rezultati (vidjeti također napomene pod točkama 9.1. i 9.2.).
|
Kolona za tekućinsku kromatografiju (točka 4.2): |
125 mm × 4 mm, C18, veličina čestica punjenja 3 μm ili jednakovrijedna |
|
Temperatura kolone: |
sobna temperatura |
|
Mobilna faza (točka 3.22.): |
3,00 g octene kiseline (točka 3.18.) + 900 ml vode (točka 3.1.) + 50,0 mlotopine točka (točka 3.21.) 1,1,1-triklor-2-metil-2-propanola (točka 3.19.) u metanolu (točka 3.8.) (1 g/100 ml). Etanolaminom (točka 3.20.) se pH vrijednost otopine podesi na 5,00. Dopuni se vodom (točka 3.1.) do 1 000 ml. |
|
Brzina protoka: |
1 ml/min |
|
Ukupno trajanje eluiranja: |
oko 34 min |
|
Valna duljina detekcije: |
ekscitacija: 280 nm, emisija: 356 nm. |
|
Volumen za ubrizgavanje: |
20 μl |
6. Izračun rezultata
Količina triptofana (X), iskazana u g na 100 g uzorka, izračunava se sljedećom formulom:

|
A |
= |
površina vrška internog standarda, standardna kalibracijska otopina (točka 3.17.) |
|
B |
= |
površina vrška triptofana, ekstrakt (točka 5.2.) ili hidrolizat (točka 5.3.) |
|
V1 |
= |
obujam, u ml (2 ml), koncentrirana otopina triptofana (točka 3.15.) dodana kalibracijskoj otopini (točka 3.17.) |
|
c |
= |
koncentracija, u μmol/ml (= 2,50), koncentrirana otopina triptofana (točka 3.15.) dodana kalibracijskoj otopini (točka 3.17.) |
|
V2 |
= |
obujam, u ml, koncentrirana otopina internog standarda (točka 3.16.) dodana pri ekstrakciji (točka 5.2.) (= 5,00 ml) ili hidrolizat (točka 5.3.) (= 2,00 ml) |
|
C |
= |
površina vrška internog standarda, ekstrakt (točka 5.2.) ili hidrolizat (točka 5.3.) |
|
D |
= |
površina vrška triptofana, standardna kalibracijska otopina (točka 3.17.) |
|
V3 |
= |
obujam, u ml (= 2,00 ml), koncentrirane otopine internog standarda (točka 3.16.), dodana standardnoj kalibracijskoj otopini (točka 3.17.) |
|
m |
= |
masa uzorka u g (korigirana na izvornu masu ako je uzorak isušen i/ili odmašćen) |
|
M |
= |
molarna masa triptofana (= 204,23 g/mol). |
7. Ponovljivost
Razlika između rezultata dvaju usporednih postupka određivanja, izvedenih na istom uzorku, ne smije prijeći 10 % u odnosu na najviši rezultat.
8. Rezultati međulaboratorijske studije
Provedena je međulaboratorijska studija na razini EZ-a (četvrta međulaboratorijska usporedba), u kojoj se u 12 laboratorija analiziralo 3 uzorka za potvrđivanje metode za hidrolizu. Svaki se uzorak analizirao pet puta. Rezultati su prikazani u donjoj tablici:
|
|
Uzorak 1 Hrana za svinje |
Uzorak 2 Hrana za svinje s dodanim L-triptofanom |
Uzorak 3 Koncentrat hrane za svinje |
|
L |
12 |
12 |
12 |
|
n |
50 |
55 |
50 |
|
Srednja vrijednost [g/kg] |
2,42 |
3,40 |
4,22 |
|
sr [g/kg] |
0,05 |
0,05 |
0,08 |
|
r [g/kg] |
0,14 |
0,14 |
0,22 |
|
CVr [%] |
1,9 |
1,6 |
1,9 |
|
SR [g/kg] |
0,15 |
0,20 |
0,09 |
|
R [g/kg] |
0,42 |
0,56 |
0,25 |
|
CVR [%] |
6,3 |
6,0 |
2,2 |
|
L |
= |
broj laboratorija koji su dostavili rezultate |
|
n |
= |
broj korištenih pojedinačnih rezultata bez ekstremnih vrijednosti (identificiranih Cochran-Dixonovim testom za ekstremne vrijednosti) |
|
sr |
= |
standardna devijacija ponovljivosti |
|
SR |
= |
standardna devijacija obnovljivosti |
|
r |
= |
ponovljivost |
|
R |
= |
obnovljivost |
|
CVr |
= |
koeficijent varijacije ponovljivosti u % |
|
CVR |
= |
koeficijent varijacije obnovljivosti u %. |
Provedena je još jedna međulaboratorijska studija na razini EZ-a (treća međulaboratorijska usporedba), u kojoj se u 13 laboratorija analiziralo 2 uzorka za potvrđivanje metode za ekstrakciju slobodnog triptofana. Svaki se uzorak analizirao pet puta. Rezultati su prikazani u donjoj tablici:
|
|
Uzorak 4 Smjesa pšenice i soje |
Uzorak 5 Smjesa pšenice i soje (= uzorak 4) s dodanim triptofanom (0,457 g/kg1) |
|
L |
12 |
12 |
|
n |
55 |
60 |
|
Srednja vrijednost [g/kg] |
0,391 |
0,931 |
|
sr [g/kg] |
0,005 |
0,012 |
|
r [g/kg] |
0,014 |
0,034 |
|
CVr [%] |
1,34 |
1,34 |
|
SR [g/kg] |
0,018 |
0,048 |
|
R [g/kg] |
0,050 |
0,134 |
|
CVR [%] |
4,71 |
5,11 |
|
L |
= |
broj laboratorija koji su dostavili rezultate |
|
n |
= |
broj korištenih pojedinačnih rezultata bez ekstremnih vrijednosti (identificiranih Cochran-Dixonovim testom za ekstremne vrijednosti) |
|
sr |
= |
standardna devijacija ponovljivosti |
|
SR |
= |
standardna devijacija obnovljivosti |
|
r |
= |
ponovljivost |
|
R |
= |
obnovljivost |
|
CVr |
= |
koeficijent varijacije ponovljivosti u % |
|
CVR |
= |
koeficijent varijacije obnovljivosti u %. |
Provedena je još jedna međulaboratorijska studija na razini EZ-a u kojoj se u 7 laboratorija analiziralo 4 uzorka za potvrđivanje triptofana za hidrolizu. Rezultati su prikazani dolje. Svaki se uzorak analizirao pet puta.
|
|
Uzorak 1 Miješana hrana za svinje (CRM 117) |
Uzorak 2 Riblje brašno s niskim udjelom masti (CRM 118) |
Uzorak 3 Sojino brašno (CRM 119) |
Uzorak 4 Obrano mlijeko u prahu (CRM 120) |
|
L |
7 |
7 |
7 |
7 |
|
n |
25 |
30 |
30 |
30 |
|
Srednja vrijednost [g/kg] |
2,064 |
8,801 |
6,882 |
5,236 |
|
sr [g/kg] |
0,021 |
0,101 |
0,089 |
0,040 |
|
r [g/kg] |
0,059 |
0,283 |
0,249 |
0,112 |
|
CVr [%] |
1,04 |
1,15 |
1,30 |
0,76 |
|
SR [g/kg] |
0,031 |
0,413 |
0,283 |
0,221 |
|
R [g/kg] |
0,087 |
1,156 |
0,792 |
0,619 |
|
CVR [%] |
1,48 |
4,69 |
4,11 |
4,22 |
|
L |
= |
broj laboratorija koji su dostavili rezultate |
|
n |
= |
broj korištenih pojedinačnih rezultata bez ekstremnih vrijednost (identificiranih Cochran-Dixonovim testom za ekstremne vrijednosti |
|
sr |
= |
standardna devijacija ponovljivosti |
|
SR |
= |
standardna devijacija obnovljivosti |
|
r |
= |
ponovljivost |
|
R |
= |
obnovljivost |
|
CVr |
= |
koeficijent varijacije ponovljivosti u % |
|
CVR |
= |
koeficijent varijacije obnovljivosti u %. |
9. Napomene
|
9.1. |
Bolje razdvajanje triptofana i α-metil-triptofana može se postići sljedećim kromatografskim uvjetima. Izokratsko eluiranje, nakon kojeg slijedi gradijentno pročišćivanje kolone:
|
||||||||||||||||||||||||||||||||||||||||||||
|
9.2. |
Postupak kromatografije se razlikuje s obzirom na vrstu HPLC-a i korišteni materijal za punjenje kolone. Odabrani sustav mora osiguravati razdvajanje bazne linije triptofana i internog standarda. Osim toga, važno je dobro odijeliti produkte razgradnje od triptofana i internog standarda. Izvede se postupak s hidrolizatima bez internog standarda za provjeru bazne linije internog standarda na nečistoće. Važno je da vrijeme eluiranja bude dovoljno za eluiranje svih produkata razgradnje, inače bi zakašnjeli vršci eluiranja mogli remetiti kasnije postupke kromatografiranja. U radnom području kromatografski sustav mora imati linearni odziv. Linearni se odziv mjeri s konstantnom (uobičajenom) koncentracijom internog standarda i različitim koncentracijama triptofana. Važno je da veličine vršaka triptofana i internog standarda budu u linearnom području sustava za HPLC/fluorescenciju. Ako su vršci triptofana i/ili internog standarda preniski ili previsoki, analizu treba ponoviti s drugom količinom uzorka i/ili drugim konačnim obujmom. |
|
9.3. |
Barijev hidroksid
S vremenom se topljivost barijevog hidroksida smanjuje. Posljedica toga je mutna otopina za HPLC, što može dovesti do niskih rezultata za triptofan. |
H. ODREĐIVANJE SIROVIH ULJA I MASTI
1. Svrha i područje primjene
Ovom se metodom omogućuje određivanje udjela sirovih ulja i masti u hrani za životinje. Ona ne obuhvaća analizu sjemenki uljarica i uljnog voća.
Primjena dolje navedenih postupaka ovisi o vrsti i sastavu hrane za životinje i razlogu za provedbu analize.
1.1. Postupak A – Sirova ulja i masti koje se ekstrahiraju izravno
Ova se metoda koristi za hranu za životinje biljnog podrijetla, osim one koja je obuhvaćena područjem primjene postupka B.
1.2. Postupak B –Ukupna sirova ulja i masti
Ova se metoda koristi za hranu za životinje životinjskog podrijetla i za sve krmne smjese. Koristi se za sve sirovine za dobivanje životinjske hrane iz kojih se bez prethodne hidrolize ne mogu potpuno ekstrahirati ulja i masti (npr. gluteni, kvasac, krumpirovi proteini i proizvodi koji se podvrgavaju postupcima poput istiskivanja, pahuljičanja i zagrijavanja).
1.3. Tumačenje rezultata
Kada je rezultat dobiven postupkom B viši od rezultata dobivenog postupkom A uvijek se kao točna vrijednost uzima rezultat dobiven postupkom B.
2. Načelo
2.1. Postupak A
Uzorak se ekstrahira laganim petrolejem. Otapalo se ukloni destilacijom, a ostatak osuši i izvaže.
2.2. Postupak B
Uzorak se uz zagrijavanje obradi klorovodičnom kiselinom. Smjesa se ohladi i filtrira. Ostatak se ispere i osuši, a zatim se provede određivanje u skladu s postupku A.
3. Reagensi
|
3.1. |
Lagani petrolej, raspon vrelišta 40 – 60 °C. Bromni broj mora biti niži od 1, a ostatak nakon isparavanja manji od 2 mg/100 ml. |
|
3.2. |
Natrijev sulfat, bezvodni. |
|
3.3. |
Klorovodična kiselina, c = 3 mol/l. |
|
3.4. |
Filtracijsko sredstvo, npr. Kieselguhr, Hyflo-supercel. |
4. Oprema
|
4.1. |
Uređaj za ekstrakciju. Ako je opremljena sifonom (uređaj Soxhlet), brzina refluksa mora biti takva da proizvodi približno 10 ciklusa na sat; ako nije opremljena sifonom, brzina povratnog toka mora iznositi približno 10 ml na minutu. |
|
4.2. |
Kartuše za ekstrakciju, bez tvari topljivih u laganom petroleju i poroznosti u skladu s zahtjevima iz točke 4.1. |
|
4.3. |
Sušilo, bilo vakuumsko, postavljeno na 75 °C ± 3 °C, ili zračno, postavljeno na 100 °C ± 3 °C. |
5. Postupak
5.1. Postupak A (vidjeti točku 8.1.)
Odvagne se 5 g uzorka s preciznošću od 1 mg, prenese u kartušu za ekstrakciju (točka 4.2.) i pokrije čepom od vate koji ne sadrži masti.
Kartuša se postavi u ekstraktor (točka 4.1.) i šest sati ekstrahira laganim petrolejem (točka 3.1.). Ekstrakt laganog petroleja se prikupi u suhu, izvaganu tikvicu koja sadrži krhotine plovućca (2).
Otapalo se ukloni destilacijom. Ostatak se osuši tako da se tikvica sat i pol drži u sušilu (točka 4.3.). Ostavi se da se ohladi u eksikatoru te se izvaže. Ponovo se suši 30 minuta kako bi se osigurala konstantna masa ulja i masti (gubitak mase između dvaju uzastopnih vaganja ne smije prijeći 1 mg).
5.2. Postupak B
Odvagne se 2,5 g uzorka s preciznošću od 1 mg (vidjeti točku 8.2.), prenese u čašu obujma 400 ml ili konusnu tikvicu obujma 300 ml i doda 100 ml klorovodične kiseline (točka 3.3.) i krhotine plovućca. Čaša se pokrije satnim staklom ili se konusna tikvica spoji s kondenzatorom s povratnim protokom. Smjesa se polagano dovede do ključanja iznad niskog plamena ili na električnoj grijaćoj ploči i tamo ostavi jedan sat. Treba paziti da se uzorak ne zalijepi za stijenke posude.
Ohladi se i doda onoliko sredstva za filtriranje (točka 3.4.) koliko je potrebno da tijekom filtriranja ne dođe do gubitka ulja i masti. Filtrira se kroz navlažen dvostruki filtar papir koji ne sadrži masti. Ostatak se ispire hladnom vodom sve dok filtrat ne postane neutralan. Treba provjeriti da filtrat ne sadrži nikakva ulja ili masti. Njihova prisutnost znači da se uzorak prije hidrolize mora ekstrahirati laganim petrolejem primjenom postupka A.
Dvostruki filtar papir na kojem je ostatak prenese se na satno staklo i sat i pol suši u sušilu (točka 4.3.) na 100 °C ± 3 °C.
Dvostruki filtar papir sa suhim ostatkom se prenese u kartušu za ekstrahiranje (točka 4.2.) i pokrije čepom od vate koji ne sadrži masti. Kartuša se postavi u ekstraktor (točka 4.1.) i postupak nastavi kako je navedeno u drugom i trećem stavku točke 5.1.
6. Prikaz rezultata
Masa ostatka iskazuje se kao postotni udio uzorka.
7. Ponovljivost
Razlika između rezultata dvaju usporednih postupaka određivanja koja na istom uzorku izvodi isti analitičar ne smije prijeći:
|
— |
0,2 % apsolutne vrijednosti za udio sirovih ulja i masti manje od 5 %, |
|
— |
4,0 % višeg rezultata za udio od 5 – 10 %, |
|
— |
0,4 % apsolutne vrijednosti za udio iznad 10 %. |
8. Napomene
|
8.1. |
Kod proizvoda s visokim udjelom ulja i masti koji se teško usitnjavaju ili nisu primjereni za pripremanje homogenog smanjenog pokusnog uzorka primjenjuje se sljedeći postupak. Odvagne se 20 g uzorka s preciznošću od 1 mg i pomiješa s 10 g ili više bezvodnog natrijevog sulfata (točka 3.2.). Ekstrahira se laganim petrolejem (točka 3.1.) u skladu s točkom 5.1. Ekstrakt se dopuni laganim petrolejem (točka 3.1.) do 500 ml i promiješa. Uzme se 50 ml otopine i prenese u malenu, suhu, izvaganu tikvicu s krhotinama plovućca. Destilacijom se ukloni otapalo, osuši se i nastavi u skladu s zadnjem stavku točke 5.1. Iz ostatka ekstrakcije koji se nalazi u tuljcu ukloni se otapalo, ostatak se usitni do veličine čestica 1 mm i vrati u tuljac za ekstrakciju (ne dodaje se natrijev sulfat), zatim se nastavi kako je navedeno u drugom i trećem stavku točke 5.1. Ucio ulja i masti kao postotni udio uzorka izračunava se sljedećom formulom: (10 m1 + m2) × 5 pri čemu je:
|
|
8.2. |
Kod proizvoda s niskim sadržajem ulja i masti pokusni se uzorak može povećati na 5 g. |
|
8.3. |
Hranu za kućne ljubimce s visokim sadržajem vode ponekad prije hidrolize i ekstrakcije trebati pomiješati s bezvodnim natrijevim sulfatom, u skladu s postupku B. |
|
8.4. |
U točki 5.2, za ispiranje ostatka nakon filtracije učinkovitije bi bilo koristiti vruću vodu umjesto hladne vode. |
|
8.5. |
Za određenu hranu za životinje možda će trebati produžiti vrijeme sušenja na više od 1,5 h. Treba izbjegavati prekomjerno sušenje zato što može dovesti do niskih rezultata. Može se koristiti i mikrovalna pećnica. |
|
8.6. |
Ako je udio sirovih ulja/masti viši od 15 %, preporučuje se prije hidrolize izvesti prethodnu ekstrakciju u skladu s postupku A i ponovnu ekstrakciju u skladu s postupku B. U određenoj mjeri, to ovisi o vrsti hrane za životinje i vrsti ulja/masti u hrani za životinje. |
I. ODREĐIVANJE SIROVE VLAKNINE
1. Svrha i područje primjene
Ovom se metodom omogućuje određivanje bezmasnih organskih tvari u hrani za životinje koje su netopljive u kiselim i baznim medijima i uobičajeno se nazivaju sirove vlaknine.
2. Načelo
Uzorak se prema potrebi odmasti i uzastopno obradi vrelim otopinama sumporne kiseline i kalijevog hidroksida u navedenim koncentracijama. Ostatak se odvaja filtracijom kroz filtar od sinteriranog stakla, ispere, osuši, izvaže i žari na 475 – 500 °C. Gubitak mase zbog žarenja odgovara udjelu sirove vlaknine u pokusnom uzorku.
3. Reagensi
|
3.1. |
Sumporna kiselina, c = 0,13 mol/l. |
|
3.2. |
Sredstvo protiv pjenjenja (npr. n-oktanol). |
|
3.3. |
Sredstvo za filtriranje (Celite 545 ili jednakovrijedno), koje se četiri sata grije na 500 °C (točka 8.6.). |
|
3.4. |
Aceton. |
|
3.5. |
Lagani petrolej s rasponom vrelišta od 40 – 60 °C. |
|
3.6. |
Klorovodična kiselina, c = 0,5 mol/l. |
|
3.7. |
Otopina kalijevog hidroksida, c = 0,23 mol/l. |
4. Oprema
|
4.1. |
Grijaća jedinica za mineralizaciju otopinama sumporne kiseline i kalijevog hidroksida, opremljena dodatkom za filtarski lončić (točka 4.2.) i ispusnom cijevi koja je preko slavine priključena na tekućinu i vakuum, po mogućnosti sa stlačenim zrakom. Svakog dana prije početka upotrebe jedinica se pet minuta zagrijava vrelom vodom. |
|
4.2. |
Stakleni filtarski lončić s filtarskom pločom od sinteriranog stakla, veličine pora 40 – 90 μm. Prije prve upotrebe treba se nekoliko minuta grijati na 500 °C i ohladiti (točka 8.6.). |
|
4.3. |
Valjak obujma najmanje 270 ml s kondenzatorom s povratnim protokom, pogodan za ključanje. |
|
4.4. |
Sušilo s termostatom |
|
4.5. |
Mufolna peć s termostatom |
|
4.6. |
Jedinica za ekstrakciju sastavljena od nosive ploče za filtarski lončić (točka 4.2.) i ispusne cijevi koja je preko slavine priključena na tekućinu i vakuum. |
|
4.7. |
Spojni prstenovi za sklapanje grijaće jedinice (točka 4.1.), lončića (točka 4.2.) i valjka (točka 4.3.) i za priključivanje jedinice za hladnu ekstrakciju (točka 4.6.) i lončića. |
5. Postupak
Odvagne se 1 g pripravljenog uzorka s preciznošću od 1 mg i prenese u lončić (točka 4.2.) (vidjeti napomene pod točkama 8.1., 8.2. i 8.3.) te se doda 1 g sredstva za filtriranje (točka 3.3.).
Najprije se sklopi grijaća jedinica (točka 4.1.) i filtarski lončić (točka 4.2.), a zatim se valjak (točka 4.3.) priključi na lončić. U sklopljeni valjak i lončić nalije se 150 ml vrele sumporne kiseline (točka 3.1) i prema potrebi nekoliko kapi sredstva protiv pjenjenja (točka 3.2.).
Tekućina se zagrijava do ključanja u 5 ± 2 minuta i ostavi snažno ključati točno 30 minuta.
Otvori se slavina na ispusnoj cijevi (točka 4.1.), sumporna kiselina se pod vakuumom filtrira kroz filtarski lončić i ostatak se triput uzastopno ispere s po 30 ml vrele vode, pri čemu treba provjeriti da se nakon svakog ispiranja ostatak filtrira do suhog.
Ispusna se slavina zatvori, u sklop valjak-lončić nalije se 150 ml vrele otopine kalijevog hidroksida (točka 3.7.) i doda se nekoliko kapi sredstva protiv pjenjenja (točka 3.2). Tekućina se zagrijava do točke ključanja u 5 ± 2 minuta i ostavi snažno ključati točno 30 minuta. Filtrira se i ponovi postupak ispiranja kako je navedeno za postupak sa sumpornom kiselinom.
Nakon konačnog ispiranja i sušenja, odspoji se lončić sa sadržajem i priključi na jedinicu za hladnu ekstrakciju (točka 4.6.). Ostatak u lončiću se pod vakuumom triput uzastopno ispere s po 25 ml acetona (točka 3.4.), pri čemu treba provjeriti da se nakon svakog ispiranja ostatak filtrira do suhog.
Lončić se osuši u sušilu na 130 °C do konstantne mase. Nakon svakog sušenja ohladi se u eksikatoru i brzo izvaže. Lončić se postavi u mufolnu peć i žari najmanje 30 minuta na 475 – 500 °C do konstantne mase (gubitak mase između dvaju uzastopnih vaganja ne smije prijeći 2 mg).
Nakon svakoga grijanja i prije vaganja, lončić se prvo ohladi u peći, a zatim u eksikatoru.
Izvodi se slijepa proba bez uzorka. Gubitak mase pri žarenju ne smije prijeći 4 mg.
6. Izračun rezultata
Udio sirovih vlakana, iskazan kao postotni udio uzorka, izračunava se sljedećom formulom:

pri čemu je:
|
m |
= |
masa uzorka u gramima, |
|
m0 |
= |
gubitak mase nakon žarenja tijekom postupka određivanja, u gramima, |
|
m1 |
= |
gubitak mase nakon žarenja tijekom slijepe probe, u gramima. |
7. Ponovljivost
Razlika između rezultata dvaju usporednih postupaka određivanja, izvedenih na istom uzorku, ne smije prijeći:
|
— |
0,6 % apsolutne vrijednosti za udio sirovih vlakana manje od 10 %, |
|
— |
6 % veće vrijednosti za udio sirovih vlakana jednak ili veći od 10 %. |
8. Napomene
|
8.1. |
Hranu za životinje koja sadrži više od 10 % sirovih masti prije analize treba odmastiti laganim petrolejem (točka 3.5.). Filtarski se lončić (točka 4.2.) sa sadržajem spoji na jedinicu za hladnu ekstrakciju (točka 4.6), a ostatak se pod vakuumom triput ispere s po 30 ml laganog petroleja, pri čemu treba provjeriti da je ostatak suh. Nakon što se lončić sa sadržajem priključi na grijaću jedinicu (točka 4.1.), nastavi se u skladu s točkom 5. |
|
8.2. |
Hranu za životinje koja sadrži masti koje se ne mogu direktno ekstrahirati laganim petrolejem (točka 3.5.), treba odmastiti u skladu s točkom 8.1. i ponovo odmastiti nakon ključanja s kiselinom. Nakon ključanja s kiselinom i naknadnog ispiranja, lončić sa sadržajem spoji se na jedinicu za hladnu ekstrakciju (točka 4.6.) i triput ispere s po 30 ml acetona i triput s po 30 ml laganog petroleja. Filtrira se pod vakuumom do suhog i nastavi u skladu s točkom 5, pri čemu se postupak započne dodavanjem kalijevog hidroksida. |
|
8.3. |
Ako hrana za životinje sadrži više od 5 % karbonata, iskazanih kao kalcijev karbonat, lončić (točka 4.2.) s izvaganim uzorkom se priključi na grijaću jedinicu (točka 4.1.). Uzorak se triput ispere s po 30 ml klorovodične kiseline (točka 3.6). Nakon svakog dodavanja, uzorak se ostavi približno jednu minutu prije filtriranja. Ispere se s 30 ml vode i nastavi u skladu s točkom 5. |
|
8.4. |
Ako se koristi uređaj u obliku stalka (nekoliko lončića priključenih na istu grijaću jedinicu), u istoj se seriji ne smiju izvesti dva postupka određivanja na istom uzorku za analizu. |
|
8.5. |
Ako se kisele i bazične otopine nakon ključanja teško filtriraju, propuše se stlačeni zrak kroz ispusnu cijev grijaće jedinice i nastavi s filtriranjem. |
|
8.6. |
Temperatura žarenja ne smije prijeći 500 °C kako bi se produljio vijek trajanja staklenih filtarskih lončića. Pri tome se mora voditi računa kako bi se spriječio pretjerani toplinski šok između ciklusa grijanja i hlađenja. |
J. ODREĐIVANJE ŠEĆERA
1. Svrha i područje primjene
Ovom se metodom omogućuje određivanje količine reducirajućih šećera i ukupnih šećera nakon inverzije, iskazanih kao glukoza ili, ovisno o slučaju, kao saharoza, s faktorom konverzije 0,95. Primjenjuje se za krmne smjese. Za drugu hranu za životinje primjenjuju se posebne metode. Prema potrebi, laktoza se određuje odvojeno i uzima u obzir pri izračunu rezultata.
2. Načelo
Šećeri se ekstrahiraju razrijeđenim etanolom; otopina se izbistri otopinama Carrez I i Carrez II. Nakon uklanjanja etanola određuju se količine prije i nakon inverzije Luff-Schoorlovom metodom.
3. Reagensi
|
3.1. |
Otopina etanola 40 % (v/v) gustoće: 0,948 g/ml na 20 °C, neutralizirana prema fenolftaleinu. |
|
3.2. |
Otopina Carrez I: u vodi se otopi 21,9 g cinkovog acetata Zn (CH3COO)2 2H2O i 3 g ledene octene kiseline. Dopuni se vodom do 100 ml. |
|
3.3. |
Otopina Carrez II: u vodi se otopi 10,6 g kalijevog ferocijanida K4Fe (CN)6 3H2O. Dopuni se vodom do 100 ml. |
|
3.4. |
Metiloranž, otopina 0,1 % (m/v). |
|
3.5. |
Klorovodična kiselina, 4 mol/l. |
|
3.6. |
Klorovodična kiselina, 0,1 mol/l. |
|
3.7. |
Otopina natrijevog hidroksida, 0,1 mol/l. |
|
3.8. |
Luff-Schoorlov reagens: Uz oprezno miješanje, u otopinu natrijevog karbonata (točka 3.8.3.) ulije se otopina limunske kiseline (točka 3.8.2.). Doda se otopina bakrovog sulfata (točka 3.8.1.) i dopuni vodom do 1 litre. Ostavi se preko noći i filtrira. Provjeri se koncentracija tako dobivenog reagensa (Cu 0,05 mol/l; Na2 CO3 1 mol/l), vidjeti zadnji stavak točke 5.4. pH vrijednost otopine je približno 9,4. |
|
3.8.1. |
Otopina bakrovog sulfata: u 100 ml vode otopi se 25 g bakrovog sulfata (Cu SO4 5H2O) koji ne sadrži željezo. |
|
3.8.2. |
Otopina limunske kiseline: u 50 ml vode otopi se 50 g limunske kiseline (C6H8O7.H2O). |
|
3.8.3. |
Otopina natrijevog karbonata: u približno 300 ml tople vode otopi se 143,8 g bezvodnog natrijevog karbonata. Ostavi se da se ohladi. |
|
3.9. |
Otopina natrijevog tiosulfata, 0,1 mol/l. |
|
3.10. |
Otopina škroba: u litru ključajuće vode doda se suspenzija 5 g topljivog škroba u 30 ml vode. Ostavi se 3 minute da vrije, zatim se ohladi i prema potrebi doda 10 mg živinog jodida kao konzervansa. |
|
3.11. |
Sumporna kiselina, 3 mol/l. |
|
3.12. |
Kalijev jodid, otopina 30 % (m/v). |
|
3.13. |
Plovućac u zrnu prokuhan u klorovodičnoj kiselini, ispran vodom i osušen. |
|
3.14. |
3-metilbutan-1-ol. |
4. Oprema
Mješalica (rotacijska): približno 35 do 40 okr./min.
5. Postupak
5.1. Ekstrakcija uzorka
Odvagne se 2,5 g uzorka s preciznošću od 1 mg i prenese u graduiranu tikvicu obujma 250 ml. Doda se 200 ml etanola (točka 3.1.) i 1 sat miješa u mješalici. Doda se 5 ml otopine Carrez I (točka 3.2.) i miješa približno 30 sekunda. Doda se 5 ml otopine Carrez II (točka 3.3.) i ponovo miješa 1 minutu. Dopuni se do oznake etanolom (točka 3.1), homogenizira i filtrira. Uzme se 200 ml filtrata i otpari približno polovica kako bi se uklonila većina etanola. Toplom vodom se ostatak nakon otparivanja kvantitativno prenese u graduiranu tikvicu obujma 200 ml, ohladi, dopuni vodom do oznake, homogenizira i prema potrebi filtrira. Ta se otopina koristi za određivanje količine reducirajućih šećera, a nakon inverzije, ukupnih šećera.
5.2. Određivanje reducirajućih šećera
Pipetom se prenese najviše 25 ml otopine koja sadrži manje od 60 mg reducirajućih šećera, iskazanih kao glukoza. Prema potrebi se dopuni destiliranom vodom do 25 ml i Luff-Schoorlovom metodom odredi udio reducirajućih šećera. Rezultat se iskazuje kao postotak glukoze sadržan u uzorku za analizu.
5.3. Određivanje ukupnih šećera nakon inverzije
Pipetom se prenese 50 ml otopine u graduiranu tikvicu obujma 100 ml. Doda se nekoliko kapi otopine metiloranža (točka 3.4.), zatim se oprezno, uz stalno miješanje, dodaje klorovodična kiselina (točka 3.5.) sve dok tekućina na postane potpuno crvena. Doda se 15 ml klorovodične kiseline (točka 3.6.), tikvica se uroni u vodenu kupelj koja snažno ključa i u njoj ostavi 30 minuta. Brzo se ohladi na približno 20 °C i doda 15 ml otopine natrijevog hidroksida (točka 3.7.). Dopuni se vodom do 100 ml i homogenizira. Pipetom se prenese najviše 25 ml otopine koja sadrži manje od 60 mg reducirajućih šećera, iskazanih kao glukoza. Prema potrebi se dopuni destiliranom vodom do 25 ml i Luff-Schoorlovom metodom odredi udio reducirajućih šećera. Rezultat se iskazuje kao postotni udio glukoze ili, prema potrebi, saharoze što se dobije množenjem s faktorom 0,95.
5.4. Titracija prema Luff-Schoorlovoj metodi
Pipetom se prenese 25 ml Luff-Schoorlovog reagensa (točka 3.8.) u Erlenmeyerovu tikvicu obujma 300 ml; doda se točno 25 ml izbistrene otopine šećera. Doda se 2 zrnca plovućca (točka 3.13.), zagrijava nad otvorenim plamenom srednje jačine, uz ručno miješanje, tako da tekućina proključa za približno dvije minute. Erlenmeyerova se tikvica odmah postavi na žičanu mrežicu presvučenu azbestom s otvorom promjera 6 cm, ispod kojeg se upali plamen. Plamen se namjesti tako da se zagrijava samo dno Erlenmeyerove tikvice. Na Erlenmeyerovu se tikvicu priključi kondenzator s povratnim protokom. Ostavi se da ključa točno 10 minuta. Odmah se ohladi u hladnoj vodi i nakon približno pet minuta titrira na sljedeći način:
Doda se 10 ml otopine kalijevog jodida (točka 3.12.) i odmah nakon toga (oprezno, zbog opasnosti od snažnog pjenjenja) 25 ml sumporne kiseline (točka 3.11.). Titrira se otopinom natrijevog tiosulfata (točka 3.9.) do pojave mutno žute boje, doda se indikator škroba (točka 3.10.) i dovrši titracija.
Jednaka se titracija izvede bez ključanja na točno izmjerenoj smjesi 25 ml Luff-Schoorlovog reagensa (točka 3.8.) i 25 ml vode, nakon dodavanja 10 ml otopine kalijevog jodida (točka 3.12.) i 25 ml sumporne kiseline (točka 3.11.).
6. Izračun rezultata
Iz tablice se odredi količina glukoze u mg koja odgovara razlici između vrijednosti obje titracije, iskazanih u mg natrijevog tiosulfata 0,1 mol/l. Rezultat se iskazuje kao postotni udio uzorka.
7. Posebni postupci
|
7.1. |
Kod hrane za životinje koja je bogata melasom i drugih vrsta hrane za životinje koje nisu posebno homogene, odvagne se 20 g uzorka i prenese s 500 ml vode u graduiranu tikvicu obujma 1 l. Miješa se 1 sat u rotacijskoj mješalici. Otopina se izbistri reagensima Carrez I (točka 3.2.) i II (točka 3.3.), u skladu s točkom 5.1., ovaj put s četverostrukom količinom svakog reagensa. Tikvica se dopuni do oznake etanolom 80 % (v/v). Homogenizira se i filtrira. Etanol se ukloni u skladu s točkom 5.1. Ako nema dekstriniranog škroba, dopuni se destiliranom vodom do oznake. |
|
7.2. |
Kod melasa i sirovina za hranu za životinje bogatih šećerom i gotovo potpuno bez škroba (rogač, sušeni rezanci šećerne repe itd.), odvagne se 5 g i prenese u graduiranu tikvicu obujma 250 ml, doda se 200 ml destilirane vode i miješa u rotacijskoj mješalici jedan sat ili prema potrebi više. Otopina se izbistri reagensima Carrez I (točka 3.2.) i II (točka 3.3.), u skladu s točkom 5.1. Dopuni se hladnom vodom do oznake, homogenizira i filtrira. Za određivanje količine ukupnih šećera nastavi se u skladu s točkom 5.3. |
8. Napomene
|
8.1. |
Za sprečavanje pjenjenja preporučuje se prije ključanja s Luff-Schoorlovim reagensom dodati (bez obzira na obujam) približno 1 ml 3-metilbutan-l-ola (točka 3.14.). |
|
8.2. |
Razlika između udjela ukupnih šećera nakon inverzije, iskazanih kao glukoza, i udjela reducirajućih šećera, iskazanih kao glukoza, pomnoženo s 0,95, daje postotni udio saharoze. |
|
8.3. |
Za određivanje udjela reducirajućih šećera, osim laktoze, mogu se koristiti dvije metode: |
|
8.3.1. |
Za približan izračun, udio se laktoze, određen drugom analitičkom metodom, pomnoži s 0,675, a dobiveni rezultat oduzme od udjela reducirajućih šećera. |
|
8.3.2. |
Za točan izračun reducirajućih šećera, osim laktoze, mora se koristiti isti uzorak za dva konačna postupka određivanja. Jedna se analiza izvodi na dijelu otopine dobivene postupkom iz točke 5.1., druga na dijelu otopine dobivene pri određivanju laktoze metodom utvrđenom za tu svrhu (nakon fermentiranja drugih vrsta šećera i bistrenja). U oba se slučaja količina šećera određuje Luff-Schoorlovom metodom i izračunava u mg glukoze. Jedna se vrijednost oduzme od druge i razlika iskazuje kao postotni udio uzorka. Primjer: Dva korištena volumena odgovaraju, za svaki postupak određivanja, uzorku od 250 mg. U prvom se slučaju potroši 17 ml otopine natrijevog tiosulfata 0,1 mol/l, što odgovara 44,2 mg glukoze; u drugom se potroši 11 ml, što odgovara 27,6 mg glukoze. Razlika je 16,6 mg glukoze. Udio reducirajućih šećera (osim laktoze), izračunan kao glukoza, dakle iznosi:
|

Tablica vrijednosti za 25 ml Luff-Schoorlovog reagensa
ml Na2 S2 O3 0,1 mol/l, dvije minute grijanja, 10 minuta ključanja
|
Na2S2O3 0,1 mol/l |
Glukoza, fruktoza, invertirani šećeri C6H12O6 |
Laktoza C12H22O11 |
Maltoza C12H22O11 |
Na2S2O3 0,1 mol/l |
|||
|
ml |
mg |
razlika |
mg |
razlika |
mg |
razlika |
ml |
|
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 |
2,4 4,8 7,2 9,7 12,2 14,7 17,2 19,8 22,4 25,0 27,6 30,3 33,0 35,7 38,5 41,3 44,2 47,1 50,0 53,0 56,0 59,1 62,2 |
2,4 2,4 2,5 2,5 2,5 2,5 2,6 2,6 2,6 2,6 2,7 2,7 2,7 2,8 2,8 2,9 2,9 2,9 3,0 3,0 3,1 3,1 |
3,6 7,3 11,0 14,7 18,4 22,1 25,8 29,5 33,2 37,0 40,8 44,6 48,4 52,2 56,0 59,9 63,8 67,7 71,7 75,7 79,8 83,9 88,0 |
3,7 3,7 3,7 3,7 3,7 3,7 3,7 3,7 3,8 3,8 3,8 3,8 3,8 3,8 3,9 3,9 3,9 4,0 4,0 4,1 4,1 4,1 |
3,9 7,8 11,7 15,6 19,6 23,5 27,5 31,5 35,5 39,5 43,5 47,5 51,6 55,7 59,8 63,9 68,0 72,2 76,5 80,9 85,4 90,0 94,6 |
3,9 3,9 3,9 4,0 3,9 4,0 4,0 4,0 4,0 4,0 4,0 4,1 4,1 4,1 4,1 4,1 4,2 4,3 4,4 4,5 4,6 4,6 |
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 |
K. ODREĐIVANJE LAKTOZE
1. Svrha i područje primjene
Ovom se metodom omogućuje određivanje razine laktoze u hrani za životinje koja sadrži više od 0,5 % laktoze.
2. Načelo
Šećeri se otope u vodi. Otopina se fermentira kvascem vrste Saccharomyces cerevisiae koji ne mijenja laktozu. Nakon bistrenja i filtriranja, udio laktoze u filtratu određuje se Luff-Schoorlovom metodom.
3. Reagensi
|
3.1. |
Suspenzija kvasca vrste Saccharomyces cerevisiae: otopi se 25 g svježeg kvasca u 100 ml vode. Suspenzija se uz držanje u hladnjaku može koristiti najviše jedan tjedan. |
|
3.2. |
Otopina Carrez I: u vodi se otopi 21,9 g cinkovog acetata Zn (CH3 COO)2 2H2O i 3 g ledene octene kiseline. Dopuni se vodom do 100 ml. |
|
3.3. |
Otopina Carrez II: u vodi se otopi 10,6 g kalijevog ferocijanida K4Fe (CN)6 3H2O. Dopuni se vodom do 100 ml. |
|
3.4. |
Luff-Schoorlov reagens: Uz oprezno miješanje, u otopinu natrijevog karbonata (točka 3.4.3.) ulije se otopina limunske kiseline (točka 3.4.2.). Doda se otopina bakrovog sulfata (točka 3.4.1.) i dopuni vodom do 1 litre. Ostavi se preko noći i filtrira. Provjeri se koncentracija tako dobivenog reagensa (Cu 0,05 mol/l, Na2 CO3 1 mol/l). pH vrijednost otopine je približno 9,4. |
|
3.4.1. |
Otopina bakrovog sulfata: u 100 ml vode otopi se 25 g bakrovog sulfata CuSO4 5H2O koji ne sadrži željezo. |
|
3.4.2. |
Otopina limunske kiseline: u 50 ml vode otopi se 50 g limunske kiseline C6H8O7.H2O. |
|
3.4.3. |
Otopina natrijevog karbonata: u približno 300 ml tople vode otopi se 143,8 g bezvodnog natrijevog karbonata. Ostavi se da se ohladi. |
|
3.5. |
Plovućac u zrnu, prokuhan u klorovodičnoj kiselini, ispran i osušen. |
|
3.6. |
Kalijev jodid, otopina 30 % (m/v). |
|
3.7. |
Sumporna kiselina, 3 mol/l. |
|
3.8. |
Otopina natrijevog tiosulfata, 0,1 mol/l. |
|
3.9. |
Otopina škroba: u litru vrele vode doda se suspenzija 5 g topljivog škroba u 30 ml vode. Ostavi se da vrije 3 minute, zatim se ohladi i prema potrebi doda 10 mg živinog jodida kao konzervansa. |
4. Oprema
Vodena kupelj s termostatom postavljenim na 38 – 40 °C
5. Postupak
Odvagne se 1 g uzorka s preciznošću od 1 mg i prenese u graduiranu tikvicu obujma 100 ml. Doda se 25 – 30 ml vode. Tikvica se potopi u ključajuću vodenu kupelj i u njoj ostavi 30 minuta, zatim se ohladi na približno 35 °C. Doda se 5 ml suspenzije kvasca (točka 3.1.) i homogenizira. Tikvica se ostavi 2 sata u vodenoj kupelji na temperaturi 38 – 40 °C. Ohladi se na približno 20 °C.
Doda se 2,5 ml otopine Carrez I (točka 3.2.) i miješa 30 sekundi, zatim 2,5 ml otopine Carrez II (točka 3.3.) i ponovo miješa 30 sekundi. Dopuni se vodom do 100 ml, promiješa i filtrira. Dio filtrata ne veći od 25 ml, koji poželjno sadrži 40 do 80 mg laktoze, pipetom se prenese u Erlenmeyerovu tikvicu obujma 300 ml. Prema potrebi se dopuni vodom do 25 ml.
Na isti se način izvodi slijepa proba s 5 ml suspenzije kvasca (točka 3.1.). Udio laktoze se određuje Luff-Schoorlovom metodom na sljedeći način: doda se točno 25 ml Luff-Schoorlovog reagensa (točka 3.4.) i 2 zrna plovućca (točka 3.5.). Uz ručno miješanje zagrijava se iznad otvorenog plamena srednje jakosti tako da tekućina proključa za približno dvije minute. Erlenmeyerova se tikvica odmah postavi na žičanu mrežicu presvučenu azbestom s otvorom promjera 6 cm, ispod kojeg se upali plamen. Plamen se namjesti tako da se zagrijava samo dno Erlenmeyerove tikvice. Na Erlenmeyerovu se tikvicu priključi kondenzator s povratnim protokom. Ostavi se da vrije točno 10 minuta. Odmah se ohladi u hladnoj vodi i nakon približno pet minuta titrira na sljedeći način:
Doda se 10 ml otopine kalijevog jodida (točka 3.6.) i odmah nakon toga (oprezno, zbog opasnosti od snažnog pjenjenja) 25 ml sumporne kiseline (točka 3.7.). Titrira se otopinom natrijevog tiosulfata (točka 3.8.) do pojave mutno žute boje, doda se indikator škroba (točka 3.9.) i dovrši titracija.
Jednaka se titracija izvede bez ključanja na točno izmjerenoj smjesi 25 ml Luff-Schoorlovog reagensa (točka 3.4.) i 25 ml vode, nakon dodavanja 10 ml otopine kalijevog jodida (točka 3.6.) i 25 ml sumporne kiseline (točka 3.7.).
6. Izračun rezultata
Iz tablice se odredi količina laktoze u mg koja odgovara razlici između vrijednosti dviju titracija, iskazanih u ml natrijevog tiosulfata 0,1 mol/l.
Rezultat se iskazuje kao postotak udjela bezvodne laktoze u uzorku.
7. Napomena
Za proizvode koji sadrže više od 40 % fermentiranog šećera koristi se više od 5 ml suspenzije kvasca (točka 3.1.).
Tablica vrijednosti za 25 ml Luff-Schoorlovog reagensa
ml Na2 S2 O3 0,1 mol/l, dvije minute grijanja, 10 minuta ključanja
|
Na2S2O3 0,1 mol/l |
Glukoza, fruktoza, invertirani šećeri C6H12O6 |
Laktoza C12H22O11 |
Maltoza C12H22O11 |
Na2S2O3 0,1 mol/l |
|||
|
ml |
mg |
razlika |
mg |
razlika |
mg |
razlika |
ml |
|
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 |
2,4 4,8 7,2 9,7 12,2 14,7 17,2 19,8 22,4 25,0 27,6 30,3 33,0 35,7 38,5 41,3 44,2 47,1 50,0 53,0 56,0 59,1 62,2 |
2,4 2,4 2,5 2,5 2,5 2,5 2,6 2,6 2,6 2,6 2,7 2,7 2,7 2,8 2,8 2,9 2,9 2,9 3,0 3,0 3,1 3,1 |
3,6 7,3 11,0 14,7 18,4 22,1 25,8 29,5 33,2 37,0 40,8 44,6 48,4 52,2 56,0 59,9 63,8 67,7 71,7 75,7 79,8 83,9 88,0 |
3,7 3,7 3,7 3,7 3,7 3,7 3,7 3,7 3,8 3,8 3,8 3,8 3,8 3,8 3,9 3,9 3,9 4,0 4,0 4,1 4,1 4,1 |
3,9 7,8 11,7 15,6 19,6 23,5 27,5 31,5 35,5 39,5 43,5 47,5 51,6 55,7 59,8 63,9 68,0 72,2 76,5 80,9 85,4 90,0 94,6 |
3,9 3,9 3,9 4,0 3,9 4,0 4,0 4,0 4,0 4,0 4,0 4,1 4,1 4,1 4,1 4,1 4,2 4,3 4,4 4,5 4,6 4,6 |
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 |
L. ODREĐIVANJE ŠKROBA
POLARIMETRIJSKA METODA
1. Svrha i područje primjene
Ovom se metodom omogućuje određivanje razine škroba i visokomolekularnih produkata razgradnje škroba u hrani za životinje, za provjeru usklađenosti s deklariranom energetskom vrijednosti (odredbe iz Priloga VII.) i Direktivom Vijeća 96/25/EZ (3).
2. Načelo
Metoda obuhvaća dva postupka određivanja. U prvom se postupku uzorak obradi razrijeđenom klorovodičnom kiselinom. Nakon bistrenja i filtriranja, polarimetrijom se izmjeri optička rotacija otopine.
U drugom se postupku uzorak ekstrahira s 40 %-tnim etanolom. Nakon zakiseljavanja filtrata klorovodičnom kiselinom, bistrenja i filtriranja, izmjeri se optička rotacija na isti način kao u prvom postupku određivanja.
Razlika između dvaju mjerenja, pomnožena s poznatim faktorom, daje udio škroba u uzorku.
3. Reagensi
|
3.1. |
Klorovodična kiselina, otopina 25 % (m/m), gustoća: 1,126 g/ml. |
|
3.2. |
Klorovodična kiselina, otopina 1,13 % (m/m). Koncentracija se mora provjeriti titracijom s otopinom natrijevog hidroksida 0,1 mol/l u prisutnosti metil crvenila 0,1 % (m/v) u etanolu 94 % (v/v). Za neutralizaciju 10 ml potrebno je 30,94 ml NaOH 0,1 mol/l. |
|
3.3. |
Otopina Carrez I: u vodi se otopi 21,9 g cinkovog acetata Zn(CH3COO)2 x 2H2O i 3 g ledene octene kiseline. Dopuni se vodom do 100 ml. |
|
3.4. |
Otopina Carrez II: u vodi se otopi 10,6 g kalijevog ferocijanida K4[Fe(CN)6] 3H2O. Dopuni se vodom do 100 ml. |
|
3.5. |
Etanol, otopina 40 % (v/v), gustoća: 0,948 g/ml na 20 °C. |
4. Oprema
|
4.1. |
Erlenmeyerova tikvica obujma 250 ml sa standardnim brušenim vratom i povratnim hladilom. |
|
4.2. |
Polarimetar ili saharimetar. |
5. Postupak
5.1. Priprema uzorka
Uzorak se usitni tako da sve čestice mogu proći kroz sito s okruglim očicama promjera 0,5 mm.
5.2. Određivanje ukupne optičke rotacije (P ili S) (vidjeti napomenu pod točkom pod točkom 7.1.)
Odvagne se 2,5 g usitnjenog uzorka s preciznošću od 1 mg i prenese u graduiranu tikvicu obujma 100 ml. Doda se 25 ml klorovodične kiseline (točka 3.2.), protrese tako da se pokusni uzorak ravnomjerno raspodijeli i doda još 25 ml klorovodične kiseline (točka 3.2.). Tikvica se potopi u ključajuću vodenu kupelj i prve 3 minute snažno i ravnomjerno trese kako bi se spriječilo nastajanje aglomerata. U vodenoj kupelji mora biti toliko vode da kada se u nju potopi tikvica kupelj ostane na točki ključanja. Tikvica se tijekom tresenja ne smije vaditi iz kupelji. Nakon točno 15 minuta, tikvica se izvadi iz kupelji, doda se 30 ml hladne vode i odmah ohladi na 20 °C.
Doda se 5 ml otopine Carrez I (točka 3.3.) i trese približno 30 sekundi. Zatim se doda 5 ml otopine Carrez II (točka 3.4.) i ponovo trese 30 sekundi. Dopuni se vodom do oznake, promiješa i filtrira. Ako filtrat nije potpuno bistar (što se rijetko događa), ponovi se postupak određivanja uz korištenje veće količine otopina Carrez I i II, na primjer 10 ml.
Polarimetrom ili saharimetrom se izmjeri optička rotacija otopine u epruveti obujma 200 ml.
5.3. Određivanje optičke rotacije (P’ ili S’) tvari topljivih u etanolu 40 %
Odvagne se 5 g uzorka s preciznošću od 1 mg, prenese u graduiranu tikvicu obujma 100 ml i doda približno 80 ml etanola (točka 3.5.) (vidjeti napomenu pod točkom 7.2.). Tikvica se ostavi 1 sat na sobnoj temperaturi; u tom se vremenskom razdoblju šest puta snažno protrese kako bi se pokusni uzorak temeljito promiješao s etanolom. Dopuni se etanolom (točka 3.5.) do oznake, promiješa i filtrira.
Pipetom se prenese 50 ml filtrata (što odgovara količini od 2,5 g uzorka) u Erlenmeyerovu tikvicu obujma 250 ml, doda se 2,1 ml klorovodične kiseline (točka 3.1.) i snažno protrese. Kondenzator s povratnim protokom se priključi na Erlenmeyerovu tikvicu i uroni u ključajuću vodenu kupelj. Nakon točno 15 minuta, Erlenmeyerova se tikvica izvadi iz kupelji, a njezin udio prenese u graduiranu tikvicu obujma 100 ml, ispere s malo hladne vode i ohladi na 20 °C.
Izbistri se otopinama Carrez I (točka 3.3.) i II (točka 3.4.), dopuni vodom do oznake, promiješa i filtrira, a zatim se izmjeri optička rotacija, kako je navedeno u drugom i trećem stavku točke 5.2.
6. Izračun rezultata
Udio škroba (%) izračunava se na sljedeći način:
6.1. Mjerenje polarimetrom Udio škroba (%)
|
P |
= |
ukupna optička rotacija u kutnim stupnjevima |
||||||||||||||
|
P’ |
= |
optička rotacija tvari topljivih u etanolu 40 % (v/v) etanolu, u kutnim stupnjevima |
||||||||||||||
|
|
= |
specifična optička rotacija čistog škroba. Brojčane vrijednosti koje se smatraju uobičajeno prihvaćenima za ovaj faktor su:
|

6.2 Mjerenje saharimetrom Udio škroba (%)

|
S |
= |
ukupna optička rotacija u stupnjevima saharimetra |
|
S’ |
= |
optička rotacija tvari topljivih u 40 %-tnom etanolu (v/v), u stupnjevima saharimetra |
|
N |
= |
masa (g) saharoze u 100 ml vode pri kojoj je optička rotacija 100 saharimetarskih stupnjeva, kada se mjeri upotrebom epruvete od 200 mm 16,29 g za francuske saharimetre 26,00 g za njemačke saharimetre 20,00 g za mješovite saharimetre. |
|
|
= |
specifična optička rotacija čistog škroba (vidjeti 6.1.) |

6.3. Ponovljivost
Razlika između rezultata dvaju usporednih postupaka određivanja, izvedenih na istom uzorku, ne smije prijeći 0,4 apsolutne vrijednosti za udio škroba niži od 40 % i 1 % relativne vrijednosti za udio škroba jednak ili veći od 40 %.
7. Napomene
|
7.1. |
Ako uzorak sadrži više od 6 % karbonata, iskazanih kao kalcijev karbonat, oni se prije određivanja ukupne optičke rotacije moraju uništiti obradom s točno potrebnom količinom razrijeđene sumporne kiseline. |
|
7.2. |
Kod proizvoda s visokim sadržajem laktoze, poput mliječnog seruma u prahu ili obranog mlijeka u prahu, nakon dodavanja 80 ml etanola (točka 3.5.) postupa se na sljedeći način. Na tikvicu se priključi kondenzator s povratnim protokom i tikvica se 30 minuta drži potopljena u vodenoj kupelji na 50 °C. Ostavi se da se ohladi i nastavi s analizom u skladu s točkom 5.3. |
|
7.3 |
Sljedeće sirovine za hranu za životinje, kada su u hrani za životinje prisutne u znatnoj količini, uzrokuju smetnje pri određivanju udjela škroba polarimetrijskom metodom, što može dovesti do neispravnih rezultata:
|
M. ODREĐIVANJE SIROVOG PEPELA
1. Svrha i područje primjene
Ovom se metodom omogućuje određivanje udjela sirovog pepela u hrani za životinje.
2. Načelo
Uzorak se spali na 550 °C; ostatak se izvaže.
3. Reagensi
Amonijev nitrat, 20 %-tna (m/v).
4. Oprema
|
4.1. |
Grijaća ploča. |
|
4.2. |
Električna mufolna peć s termostatom. |
|
4.3. |
Lončići za spaljivanje od kvarcnog stakla, porculana ili platine, pravokutni (približno 60 × 40 × 25 mm) ili okrugli (promjer: 60 do 75 mm, visina: 20 do 40 mm). |
5. Postupak
Odvagne se približno 5 g uzorka s preciznošću od 1 mg (2,5 g kod proizvoda koji lako nabubre) i prenese u lončić za spaljivanje koji se prethodno zagrije na 550 °C, ohladi i tarira. Lončić se postavi na grijaću ploču i postupno zagrijava sve dok tvar ne pougljeni. Spali se u skladu s točkom 5.1. ili 5.2.
|
5.1. |
Lončić se postavi u umjerenu mufolnu peć zagrijanu na 550 °C. Ostavi se na toj temperaturi sve do nastanka bijelog, svijetlo sivog ili crvenkastog pepela koji izgleda kao da ne sadrži ugljične čestice. Lončić se postavi u eksikator, ostavi se da se ohladi i odmah izvaže. |
|
5.2. |
Lončić se postavi u umjerenu mufolnu peć zagrijanu na 550 °C. U peći se spaljuje 3 sata. Lončić se postavi u eksikator, ostavi se da se ohladi i odmah izvaže. Ponovo se 30 minuta spaljuje kako bi se osiguralo da je masa pepela konstantna (gubitak mase između dvaju uzastopnih vaganja ne smije prijeći 1 mg). |
6. Izračun rezultata
Masa ostatka se izračuna tako da se oduzme tara.
Rezultat se iskazuje kao postotni udio uzorka.
7. Napomene
|
7.1. |
Tvari koje se teško spaljuju moraju se izložiti prvom spaljivanju u trajanju od najmanje tri sata, zatim se ohlade i dodaje im se nekoliko kapi 20 %-tne otopine amonijevog nitrata ili vode (oprezno, kako bi se spriječilo raspršivanje pepela ili nastanak gruda). Nakon sušenja u sušilu nastavi se sa spaljivanjem. Postupak se prema potrebi ponavlja, sve do potpunog spaljivanja. |
|
7.2. |
Kod tvari koje su otporne na obradu iz točke 7.1., postupa se na sljedeći način: nakon trosatnog spaljivanja, pepeo se prenese u toplu vodu i filtrira kroz mali filtar bez pepela. Filtar i njegov udio se spale u istom lončiću. Filtrat se prenese u ohlađen lončić, otpari do suhog, spali i izvaže. |
|
7.3. |
Kod ulja i masti, točno se izvaže 25 g u lončić prikladne veličine. Spaljuje se tako da se tvar zapali vrpcom od filtarnog papira bez pepela. Nakon spaljivanja navlaži se što je moguće manjom količinom vode. Osuši se i spali u skladu s točkom 5. |
N. ODREĐIVANJE PEPELA NETOPLJIVOG U KLOROVODIČNOJ KISELINI
1. Svrha i područje primjene
Ovom se metodom omogućuje određivanje razine mineralnih tvari u hrani za životinje, netopljivih u klorovodičnoj kiselini. Uzimajući u obzir prirodu uzorka mogu se primijeniti dvije metode.
|
1.1. |
Metoda A: primjenjuje se za organske sirovine za hranu za životinje i većinu krmnih smjesa. |
|
1.2. |
Metoda B: primjenjuje se za mineralne krmne smjese i krmne smjese koje, prema metodi A, sadrže više od 1 % tvari netopljivih u klorovodičnoj kiselini. |
2. Načelo
|
2.1. |
Metoda A: uzorak se spali, pepeo se otopi u vreloj klorovodičnoj kiselini, a netopljivi ostatak se filtrira i izvaže. |
|
2.2. |
Metoda B: uzorak se obradi klorovodičnom kiselinom. Otopina se filtrira, ostatak spali, a tako dobiveni pepeo obradi u skladu s metodom A. |
3. Reagensi
|
3.1. |
Klorovodična kiselina, 3 mol/l. |
|
3.2. |
Trikloroctena kiselina, otopina 20 % (m/v). |
|
3.3. |
Trikloroctena kiselina, otopina 1 % (m/v). |
4. Oprema
|
4.1. |
Grijaća ploča. |
|
4.2. |
Električna mufolna peć s termostatom. |
|
4.3. |
Lončići za spaljivanje izrađeni od kvarcnog stakla, porculana ili platine, pravokutni (približno 60 × 40 × 25 mm) ili okrugli (promjer: 60 do 75 mm, visina: 20 do 40 mm). |
5. Postupak
5.1. Metoda A
Uzorak se spali u skladu s metodom za određivanje sirovog pepela. Može se koristiti i pepeo dobiven tijekom navedene analize.
Pepeo se prenese u čašu obujma 250 ml do 400 ml sa 75 ml klorovodične kiseline (točka 3.1.). Polako se zagrije do ključanja i petnaest minuta ostavi lagano ključati. Topla otopina se filtrira kroz papir za filtriranje bez pepela, ostatak se ispire toplom vodom sve do prestanka kisele reakcije. Filtar s ostatkom i pepelom se osuši u tariranom lončiću za spaljivanje na 550 – 700 °C. Ohladi se u eksikatoru i izvaže.
5.2. Metoda B
Odvagne se 5 g uzorka s preciznošću od 1 mg i prenese u čašu obujma 250 ml do 400 ml. Doda se 25 ml vode, zatim 25 ml klorovodične kiseline (točka 3.1.), promiješa i pričeka da se reakcija umiri. Doda se još 50 ml klorovodične kiseline (točka 3.1.). Pričeka se dok potpuno ne prestane ispuštanje plina, zatim se čaša stavi u ključajuću vodenu kupelj i tamo drži 30 minuta ili prema potrebi dulje kako bi se potpuno hidrolizirao sav eventualno prisutan škrob. Udio se filtrira dok je topao kroz filtar bez pepela, a filtar se ispere s 50 ml tople vode (vidjeti napomenu pod točkom 7.). Filtar koji sadrži ostatak prenese se u lončić za spaljivanje, osuši i spali na 550 – 700 °C. Pepeo se prenese u čašu obujma 250 ml do 400 ml sa 75 ml klorovodične kiseline (točka 3.1.); nastavi se kako je opisano u drugom podstavku točke 5.1.
6. Izračun rezultata
Masa ostatka se izračuna tako da se oduzme tara. Rezultat se iskazuje kao postotni udio uzorka.
7. Napomena
Ako pri filtriranju nastupe poteškoće, analiza se ponovi, pri čemu se 50 ml klorovodične kiseline (točka 3.1.) zamijeni s 50 ml trikloroctene kiseline 20 % (točka 3.2.), a filtar ispere toplom otopinom trikloroctene kiseline 1 % (točka 3.3.).
O. ODREĐIVANJE KARBONATA
1. Svrha i područje primjene
Ovom se metodom omogućuje određivanje količine karbonata u većini hrane za životinje, koji se uobičajeno iskazuju kao kalcijev karbonat.
Međutim, u nekim se slučajevima (na primjer, kod željezovog karbonata) mora primijeniti posebna metoda.
2. Načelo
Karbonati se razlažu u klorovodičnoj kiselini; oslobođeni se ugljikov dioksid prikupi u graduiranu epruvetu, a njegov obujam usporedi s obujmom koji se pod istim uvjetima oslobađa iz poznate količine kalcijevog karbonata.
3. Reagensi
|
3.1. |
Klorovodična kiselina, gustoća 1,10 g/ml. |
|
3.2. |
Kalcijev karbonat. |
|
3.3. |
Sumporna kiselina, približno 0,05 mol/l, obojena metil crvenilom. |
4. Oprema
Scheibler-Dietrichova naprava (vidjeti sliku) ili druga jednakovrijedna naprava.
5. Postupak
Ovisno o sadržaju karbonata u uzorku odvagne se dio uzorka, kako je navedeno dalje u tekstu:
|
— |
0,5 g kod proizvoda koji sadrže 50 – 100 % karbonata, iskazanih kao kalcijev karbonat, |
|
— |
1 g kod proizvoda koji sadrže 40 – 50 % karbonata, iskazanih kao kalcijev karbonat, |
|
— |
2 do 3 g za druge proizvode. |
Odvagnuti se dio uzorka prenese u posebnu tikvicu (slika 4.) naprave, opremljenu malom epruvetom od nelomljivog materijala u kojoj se nalazi 10 ml klorovodične kiseline (točka 3.1.) te se tikvica spoji s napravom. Trosmjerni se pipac (5) okrene tako da je epruveta (1) otvorena prema van. Pomičnom epruvetom (2), napunjenom obojenom sumpornom kiselinom (točka 3.3.) i povezanom s graduiranom epruvetom (1), razina tekućine dovede se do oznake nula. Pipac (5) se okrene tako da se spoje epruvete (1) i (3), zatim se provjeri je li razina na oznaci nula.
Naginjanjem tikvice (4) klorovodična se kiselina (točka 3.1.) polako prelijeva preko uzorka. Tlak se izjednači spuštanjem epruvete (2). Tikvica (4) se trese sve dok potpuno ne prestane oslobađanje ugljikovog dioksida.
Ponovo se uspostavi tlak vraćanjem razine tekućina u epruvetama (1) i (2) na istu razinu. Rezultat se očita nakon nekoliko minuta kada se ustali obujam plina.
U istim se uvjetima izvede kontrolni pokus s 0,5 g kalcijevog karbonata (točka 3.2.).
6. Izračun rezultata
Udio karbonata, iskazanih kao kalcijev karbonat, izračunava se sljedećom formulom:

pri čemu je:
|
X |
= |
% (m/m) karbonata u uzorku, iskazanih kao kalcijev karbonat |
|
V |
= |
ml oslobođenog CO2 iz dijela uzorka |
|
V1 |
= |
ml oslobođenog CO2 iz 0,5 g CaCO3 |
|
m |
= |
masa dijela uzorka u gramima. |
7. Napomene
|
7.1. |
Kada je masa dijela uzorka veća od 2 g, prvo se u tikvicu (4) ulije 15 ml destilirane vode i promiješa prije početka pokusa. Za kontrolni se pokus koristi jednaka količina vode. |
|
7.2. |
Ako se obujam korištene naprave razlikuje od obujma Scheibler-Dietrichove naprave, potrebno je na primjeren način prilagoditi količinu dijelova uzorka i kontrolne tvari i izračun rezultata.
|

P. ODREĐIVANJE UKUPNOG FOSFORA
FOTOMETRIJSKA METODA
1. Svrha i područje primjene
Ovom se metodom omogućuje određivanje ukupnog udjela fosfora u hrani za životinje. Osobito je primjerena za analizu proizvoda s niskim sadržajem fosfora. U nekim se slučajevima (kod proizvoda bogatih fosforom) može primijeniti i gravimetrijska metoda.
2. Načelo
Uzorak se mineralizira bilo suhim sagorijevanjem (kod organske hrane za životinje) bilo digestijom kiselinom (kod mineralnih mješavina i tekuće hrane za životinje) i prenese u kiselu otopinu. Otopina se obradi molibdovanadatnim reagensom. Optička gustoća tako dobivene žute otopine mjeri se spektrofotometrom na 430 nm.
3. Reagensi
|
3.1. |
Kalcijev karbonat. |
|
3.2. |
Klorovodična kiselina, ρ20 = 1,10 g/ml (približno 6 mol/l). |
|
3.3. |
Dušična kiselina, ρ20 = 1,045 g/ml. |
|
3.4. |
Dušična kiselina, ρ20 = 1,38 do 1,42 g/ml. |
|
3.5. |
Sumporna kiselina, ρ20 = 1,84 g/ml. |
|
3.6. |
Molibdovanadatni reagens: u graduiranoj tikvici obujma 1 l pomiješa se 200 ml otopine amonijevog heptamolibdata (točka 3.6.1.), 200 ml otopine amonijevog monovanadata (točka 3.6.2.) i 134 ml dušične kiseline (točka 3.4.). Dopuni se vodom do oznake. |
|
3.6.1. |
Otopina amonijevog heptamolibdata: u vrućoj se vodi otopi 100 g amonijevog heptamolibdata ((NH4) 6Mo7O24.4H2O). Doda se 10 ml amonijaka (gustoća 0,91 g/ml) i dopuni vodom do 1 litre. |
|
3.6.2. |
Otopina amonijevog monovanadata: u 400 ml vruće vode otopi se 2,35 g amonijevog monovanadata (NH4VO3). Uz stalno miješanje polako se dodaje 20 ml razrijeđene dušične kiseline (7 ml HNO3 (točka 3.4.) + 13 ml H2O) i dopuni vodom do 1 litre. |
|
3.7 |
Standardna otopina 1 mg fosfora na ml: u vodi se otopi 4,387 g kalijevog dihidrogen fosfata (KH2PO4). Dopuni se vodom do 1 litre. |
4. Oprema
|
4.1. |
Lončići za spaljivanje od kvarcnog stakla, porculana ili platine. |
|
4.2. |
Električna mufolna peć s termostatom postavljenim na 550 °C. |
|
4.3. |
Kjeldahlova tikvica obujma 250 ml. |
|
4.4. |
Odmjerne tikvice i precizne pipete. |
|
4.5. |
Spektrofotometar |
|
4.6. |
Epruvete promjera približno 16 mm, s brušenim čepovima promjera 14,5 mm, obujma 25 – 30 ml. |
5. Postupak
5.1. Priprema otopine
Uzimajući u obzir prirodu uzorka, otopina se pripravlja u skladu s točkom 5.1.1. ili 5.1.2.
5.1.1.
Odvagne se 1 g uzorka ili više s preciznošću od 1 mg. Uzorak se prenese u Kjeldahlovu tikvicu, doda se 20 ml sumporne kiseline (točka 3.5.) i protrese kako bi se uzorak u potpunosti natopio kiselinom i spriječilo njegovo prianjanje na stijenke tikvice, zatim se zagrije i ostavi ključati 10 minuta. Ostavi se da se ohladi, doda se 2 ml dušične kiseline (točka 3.4), lagano se zagrije, malo se ohladi, doda se još malo dušične kiseline (točka 3.4.) i ponovo zagrije do ključanja. Postupak se ponavlja sve dok se ne dobije bezbojna otopina. Ohladi se, doda malo vode, tekućina se odlije u graduiranu tikvicu obujma 500 ml, a Kjeldahlova tikvica ispere vrućom vodom. Ostavi se da se ohladi, dopuni vodom do oznake, homogenizira i filtrira.
5.1.2.
Odvagne se i prenese u lončić za spaljivanje 2,5 g uzorka s preciznošću od 1 mg. Doda se 1 g kalcijevog karbonata (točka 3.1.) i miješa dok se tvari u potpunosti ne izmiješaju. Spaljuje se u pećnici pri 550 °C do nastanka bijelog ili sivog pepela (mala količina ugljena ne predstavlja problem). Pepeo se prenese u čašu obujma 250 ml. Doda se 20 ml vode i klorovodične kiseline (točka 3.2.) sve dok ne prestane pjenjenje. Doda se još 10 ml klorovodične kiseline (točka 3.2.). Čaša se stavi u pješčanu kupelj i ostavi da se otpari do suhog, kako bi kvarcni pijesak postao netopljiv. Ostatak se ponovo otopi u 10 ml dušične kiseline (točka 3.3.) i ostavi ključati 5 minuta u pješčanoj kupelji ili na grijaćoj ploči, ali se ne smije potpuno otpariti. Tekućina se odlije u graduiranu tikvicu obujma 500 ml, čaša se više puta ispere vrućom vodom. Ostavi se da se ohladi, dopuni vodom do oznake, homogenizira i filtrira.
5.2. Nastanak obojenja i mjerenje optičke gustoće
Razrijedi se alikvot filtrata iz točke 5.1.1. ili 5.1.2. do koncentracije fosfora ne veće od 40 μg/ml. U epruvetu (točka 4.6.) se prenese 10 ml te otopine i doda 10 ml molibdovanadatnog reagensa (točka 3.6.). Homogenizira se i ostavi najmanje 10 minuta na 20 °C. Spektrofotometrom se izmjeri optička gustoća na 430 nm usporedbom s otopinom dobivenom dodavanjem 10 ml molibdovanadatnog reagensa (točka 3.6.) u 10 ml vode.
5.3. Kalibracijska krivulja
Iz standardne otopine (točka 3.7.) priprave se otopine koje sadrže 5, 10, 20, 30 i 40 μg fosfora na ml. Uzme se po 10 ml od svake navedene otopine i doda 10 ml molibdovanadatnog reagensa (točka 3.6.). Homogenizira se i ostavi najmanje 10 minuta na 20 °C. Izmjeri se optička gustoća u skladu s točkom 5.2. Kalibracijska se krivulja pripremi tako da se u dijagram unesu izmjerene optičke gustoće odgovarajućih količina fosfora. Za koncentracije u rasponu od 0 – 40 μg/ml krivulja je linearna.
6. Izračun rezultata
Količina fosfora u pokusnom uzorku određuje se iz kalibracijske krivulje.
Rezultat se iskazuje kao postotni udio uzorka.
Ponovljivost
Razlika između rezultata dvaju usporednih postupaka određivanja, izvedenih na istom uzorku, ne smije prijeći:
|
— |
3 % višeg rezultata, ako je udio fosfora niži od 5 %, |
|
— |
0,15 % apsolutne vrijednosti, ako je udio fosfora 5 % ili više. |
Q. ODREĐIVANJE KLORA IZ KLORIDA
1. Svrha i područje primjene
Ovom se metodom omogućuje određivanje količine klora u kloridima topljivima u vodi koji se uobičajeno iskazuju kao natrijev klorid. Primjenjuje se za svu hranu za životinje.
2. Načelo
Kloridi se otope u vodi. Ako proizvod sadrži organske tvari, koristi se postupak bistrenja. Otopina se lagano zakiseli dušičnom kiselinom, a kloridi istalože u obliku srebrovog klorida, upotrebom otopine srebrovog nitrata. Višak srebrovog nitrata titrira se otopinom amonijevog tiocijanata po Volhardovoj metodi.
3. Reagensi
|
3.1. |
Otopina amonijevog tiocijanata, 0,1 mol/litra. |
|
3.2. |
Otopina srebrovog nitrata, 0,1 mol/l. |
|
3.3. |
Zasićena otopina amonij-željezovog sulfata (NH4)Fe(SO4)2. |
|
3.4. |
Dušična kiselina, gustoća: 1,38 g/ml. |
|
3.5. |
Dietileter. |
|
3.6. |
Aceton. |
|
3.7. |
Otopina Carrez I: u vodi se otopi 21,9 g cinkovog acetata Zn (CH3COO)2·2H2O i 3 g ledene octene kiseline. Dopuni se vodom do 100 ml. |
|
3.8. |
Otopina Carrez II: u vodi se otopi 10,6 g kalijevog ferocijanida K4Fe(CN)6·3H2O. Dopuni se vodom do 100 ml. |
|
3.9. |
Aktivni ugljen koji ne sadrži kloride niti ih apsorbira. |
4. Oprema
Mješalica (rotacijska): približno 35 do 40 okr./min.
5. Postupak
5.1. Priprema otopine
Uzimajući u obzir prirodu uzorka, otopina se pripravlja prema točki 5.1.1., 5.1.2. ili 5.1.3.
Istodobno se izvodi slijepa proba bez uzorka koji se analizira.
5.1.1.
Odvagne se najviše 10 g uzorka koji sadrži najviše 3 g klora u obliku klorida s preciznošću od 1 mg. Prenese se u graduiranu tikvicu obujma 500 ml s 400 ml vode na približno 20 °C. Miješa se 30 minuta u rotacijskoj mješalici, dopuni do oznake, homogenizira i filtrira.
5.1.2.
Odvagne se približno 5 g uzorka s preciznošću od 1 mg i prenese zajedno s 1 g aktivnog uglja u graduiranu tikvicu obujma 500 ml. Doda se 400 ml vode temperature približno 20 °C i 5 ml otopine Carrez I (točka 3.7.), miješa se 30 sekundi i doda 5 ml otopine Carrez II (točka 3.8.). Miješa se 30 minuta u rotacijskoj mješalici, dopuni do oznake, homogenizira i filtrira.
5.1.3.
Otopina se pripravi u skladu s točkom 5.1.2., ali se ne filtrira. Dekantira se (prema potrebi centrifugira), uzme se 100 ml supernatanta i prenese u graduiranu tikvicu obujma 200 ml. Pomiješa se s acetonom (točka 3.6.) i s tim otapalom dopuni do oznake, homogenizira i filtrira.
5.2. Titracija
Pipetom se u Erlenmeyerovu tikvicu prenese 25 – 100 ml filtrata (uzimajući u obzir pretpostavljeni udio klora) dobivenog postupkom iz točke 5.1.1., 5.1.2. ili 5.1.3. Alikvotni dio ne smije sadržavati više od 150 mg klora (Cl). Prema potrebi se razrijedi vodom na najmanje 50 ml, doda se 5 ml dušične kiseline (točka 3.4), 20 ml zasićene otopine amonij-željezovog sulfata (točka 3.3.) i 2 kapi otopine amonijevog tiocijanata (točka 3.1.), koji se prenese iz birete napunjene do nulte oznake. Biretom se prenese otopina srebrovog nitrata (točka 3.2.) tako da nastane 5 ml viška. Doda se 5 ml dietiletra (točka 3.5.) i dobro protrese tako da se talog zgruša. Višak srebrovog nitrata titrira se otopinom amonijevog tiocijanata (točka 3.1.) sve dok crvenkasto-smeđe obojenje ne potraje 1 minutu.
6. Izračun rezultata
Količina klora (X), iskazana kao postotni udio natrijevog klorida, izračunava se sljedećom formulom:

pri čemu je:
|
V1 |
= |
ml dodane otopine srebrovog nitrata 0,1 mol/l; |
|
V2 |
= |
ml otopine amonijevog tiocijanata 0,1 mol/l korištene za titraciju; |
|
m |
= |
masa uzorka. |
Ako se pri slijepoj probi potroši otopina srebrovog nitrata 0,1 mol/l, ta se vrijednost oduzme od obujma (V1 – V2).
7. Napomene
|
7.1 |
Titracija se može izvesti i potenciometarskom titracijom. |
|
7.2 |
Proizvodi bogati uljima i mastima prvo se odmaste dietileterom ili laganim petrolejem. |
|
7.3 |
Kod ribljeg brašna, titracija se može izvesti Mohrovom metodom. |
(1) Za sušenje žitarica, brašna, grube krupice i sitne krupice sušilo mora imati takav toplinski kapacitet da se nakon postavljanja temperature na 131 °C vraća na tu temperaturu za manje od 45 minuta nakon što se u sušilo postavi maksimalan broj testnih uzoraka za istodobno sušenje. Ventilacija mora biti takva da se, u slučaju kada se u sušilu dva sata suši najveći mogući broj uzoraka pšenice, rezultati razlikuju za manje od 0,15 % od uzoraka koji su sušeni 4 sata.
(2) Kada se ulje ili mast podvrgavaju naknadnim testovima kakvoće, krhotine plovućca zamjenjuju se staklenim kuglicama.
PRILOG IV.
ANALITIČKE METODE ZA KONTROLU RAZINE DOPUŠTENIH KRMNIH DODATAKA
A. ODREĐIVANJE VITAMINA A
1. Svrha i područje primjene
Ovom se metodom omogućuje određivanje razine vitamina A (retinola) u hrani za životinje i premiksima. Vitamin A uključuje trans-retinil alkohol i njegove cis izomere koji se određuju ovom metodom. Udio vitamina A iskazuje se u međunarodnim jedinicama (IU) na kg. Jedna IU odgovara djelovanju 0,300 μg trans-vitamin A alkohola ili 0,344 μg trans-vitamin A acetata ili 0,550 μg trans-vitamin A palmitata.
Granica kvantifikacije je 2 000 IU vitamina A/kg.
2. Načelo
Uzorak se hidrolizira otopinom etanolnog kalijevog hidroksida, a vitamin A se ekstrahira laganim petrolejem. Otapalo se ukloni otparivanjem, a ostatak otopi u metanolu i prema potrebi razrijedi do tražene koncentracije. Udio vitamina A se određuje visoko djelatnom tekućinskom kromatografijom reverznih faza (RP–HPLC) s UV ili fluorescencijskim detektorom. Odaberu se oni kromatografski parametri koji omogućuju da se trans-vitamin A alkohol i njegovi cis izomeri ne razdvoje.
3. Reagensi
|
3.1. |
Etanol, σ = 96 %. |
|
3.2. |
Lagani petrolej, raspon vrelišta 40 – 60 °C. |
|
3.3. |
Metanol. |
|
3.4. |
Otopina kalijevog hidroksida, c = 50 g/100 ml. |
|
3.5. |
Otopina natrijevog askorbata, c = 10 g/100 ml (vidjeti napomenu pod točkom pod točkom 7.7.). |
|
3.6. |
Natrijev sulfid, Na2S x H2O (x = 7 – 9). |
|
3.6.1. |
Otopina natrijevog sulfida, c = 0,5 mol/l u glicerolu, ß = 120 g/l (za x = 9) (vidjeti napomenu pod točkom pod točkom 7.8.). |
|
3.7. |
Otopina fenolftaleina, c = 2 g/100 ml u etanolu (točka 3.1.). |
|
3.8. |
2-propanol. |
|
3.9. |
Mobilna faza za HPLC: smjesa metanola (točka 3.3.) i vode, npr. 980 + 20 (v + v). Točan se omjer određuje u skladu s osobinama korištene kolone. |
|
3.10. |
Dušik, bez kisika. |
|
3.11. |
Trans-vitamin A acetat, posebne čistoće, certificiranog djelovanja, npr. 2,80 × 106 IU/g. |
|
3.11.1. |
Osnovna otopina trans-vitamin A acetata: odvagne se 50 mg acetata vitamina A (točka 3.11.) s preciznošću od 0,1 mg i prenese u graduiranu tikvicu obujma 100 ml. Otopi se u 2-propanolu (točka 3.8.) i dopuni istim otapalom do oznake. Nominalna koncentracija ove otopine iznosi 1 400 IU vitamina A na ml. Točan se udio određuje u skladu s točkom 5.6.3.1. |
|
3.12. |
Trans-vitamin A palmitat, posebne čistoće, certificiranog djelovanja, npr. 1,80 × 106 IU/g. |
|
3.12.1. |
Osnovna otopina all-trans-vitamin A palmitata:odvagne se 80 mg palmitata vitamina A (točka 3.12.) s preciznošću od 0,1 mg i prenese u graduiranu tikvicu obujma 100 ml. Otopi se u 2-propanolu (točka 3.8.) i dopuni istim otapalom do oznake. Nominalna koncentracija ove otopine je 1 400 IU vitamina A na ml. Točan se udio određuje u skladu s točkom 5.6.3.2. |
|
3.13. |
2,6-Di-tert-butil-4-metilfenol (BHT) (vidjeti napomenu pod točkom pod točkom 7.5.) |
4. Oprema
|
4.1. |
Rotacijski vakuumski otparivač. |
|
4.2. |
Laboratorijsko posuđe od smeđeg stakla. |
|
4.2.1. |
Tikvice s ravnim dnom ili konusne tikvice, obujma 500 ml, s brušenim vratom. |
|
4.2.2. |
Odmjerne tikvice s čepovima od brušenog stakla, s uskim vratom, obujma 10, 25, 100 i 500 ml. |
|
4.2.3. |
Lijevci za odjeljivanje, konusni, 1 000 ml, s čepovima od brušenog stakla. |
|
4.2.4. |
Kruškaste tikvice, 250 ml, s čepovima od brušenog stakla. |
|
4.3. |
Allihnov kondenzator, duljine omotača 300 mm sa spojem od brušenog stakla i nastavkom za dovodnu cijev plina. |
|
4.4. |
Nabrani filtar papir za odjeljivanje faza, promjera 185 mm (npr. Schleicher & Schuell 597 HY 1/2). |
|
4.5. |
Oprema za HPLC sa sustavom za ubrizgavanje. |
|
4.5.1. |
Kolona za tekućinsku kromatografiju, 250 mm × 4 mm, C18, veličina čestica punjenja 5 ili 10 μm ili jednakovrijedna (mjerilo učinkovitosti: samo jedan vršak za sve izomere retinola u uvjetima za HPLC). |
|
4.5.2. |
UV ili fluorescencijski detektor s mogućnošću promjene valne duljine. |
|
4.6. |
Spektrofotometar s kvarcnim kivetama od 10 mm. |
|
4.7. |
Vodena kupelj s magnetnom mješalicom. |
|
4.8. |
Uređaj za ekstrakciju (vidjeti sliku 1.) koja se sastoji od: |
|
4.8.1. |
Staklenog valjka obujma l litre s vratom i čepom od brušenog stakla. |
|
4.8.2. |
Uloška od brušenog stakla s bočnom ručicom i prilagodljivom cijevi koja prolazi kroz središte. Prilagodljiva cijev ima donji dio u obliku slova U i mlaznicu na suprotnom kraju tako da se gornji sloj tekućine u valjku može prenijeti u lijevak za odjeljivanje. |
5. Postupak
|
Napomena |
: |
Vitamin A je osjetljiv na UV svjetlo i oksidaciju. Svi se postupci izvode bez prisutnosti svjetla (u laboratorijskom posuđu od smeđeg stakla ili u staklenim posudama zaštićenim aluminijskom folijom) i kisika (ispire se dušikom). Tijekom ekstrakcije zrak iznad tekućine zamijeni se dušikom (povremenim popuštanjem čepa izbjegava se previsoki tlak). |
5.1. Priprema uzorka
Uzorak se samelje tako da prolazi kroz sito veličine očice 1 mm, pri čemu treba izbjegavati stvaranje topline. Mljevenje se mora izvršiti neposredno prije vaganja i saponifikacije, inače može doći do gubitka vitamina A.
5.2. Saponifikacija
Ovisno o sadržaju vitamina A, odvagne se 2 – 25 g uzorka s preciznošću od 1 mg i prenese u tikvicu s ravnim dnom ili koničnu tikvicu obujma 500 ml (točka 4.2.1.). Uz stalno vrtloženje, jedno za drugim dodaje se 130 ml etanola (točka 3.1.), približno 100 mg BHT-a (točka 3.13.), 2 ml otopine natrijevog askorbata (točka 3.5.) i 2 ml otopine natrijevog sulfida (točka 3.6.). Na tikvicu se priključi kondenzator (točka 4.3.) i tikvica potopi u vodenu kupelj s magnetnom mješalicom (točka 4.7.). Zagrije se do ključanja i 5 minuta ostavi uz refluks. Zatim se kroz kondenzator doda 25 ml otopine kalijevog hidroksida (točka 3.4.) i ostavi uz refluks (točka 4.3.) sljedećih 25 minuta, uz miješanje pod laganom strujom dušika. Zatim se kondenzator ispere s približno 20 ml vode, a udio tikvice ohladi na sobnu temperaturu.
5.3. Ekstrakcija
Otopina za saponifikaciju kvantitativno se prenese dekantiranjem u lijevak za odjeljivanje obujma 1 000 ml (točka 4.2.3.) ili u napravu za ekstrakciju (točka 4.8.), tako što se ispere s ukupno 250 ml vode. Zatim se tikvica za saponifikaciju ispere s 25 ml etanola (točka 3.1.) i 100 ml laganog petroleja (točka 3.2.), a isprane tekućine prenesu u lijevak za odjeljivanje ili uređaj za ekstrakciju. Omjer vode i etanola u pomiješanim otopinama mora biti približno 2:1. Snažno se trese 2 minute i zatim ostavi 2 minute da se slegne.
5.3.1.
Kad se slojevi razdvoje (vidjeti napomenu pod točkom pod točkom 7.3.), sloj laganog petroleja prenese se u drugi lijevak za odjeljivanje (točka 4.2.3.). Ekstrakcija se ponovi dvaput sa 100 ml laganog petroleja (točka 3.2.) i dvaput s 50 ml laganog petroleja (točka 3.2.).
Pomiješani se ekstrakti u lijevku za odjeljivanje dvaput isperu sa 100 ml vode, uz lagano vrtloženje (kako bi se spriječio nastanak emulzija), zatim uz ponavljano protresivanje s dodatnim obrocima od po 100 ml vode, sve dok voda pri dodavanju otopine fenolftaleina (točka 3.7.) ne ostane bezbojna (obično je dovoljno četiri ispiranja). Isprani se ekstrakt filtrira kroz suhi nabrani filtar za odjeljivanje faza (točka 4.4) u graduiranu tikvicu obujma 500 ml (točka 4.2.2.) kako bi se uklonila preostala voda. Lijevak za odjeljivanje i filtar se isperu s 50 ml laganog petroleja (točka 3.2.), dopuni se laganim petrolejem do oznake (točka 3.2.) i dobro promiješa.
5.3.2.
Kada se slojevi razdvoje (vidjeti napomenu pod točkom 7.3.), čep staklenog valjka (točka 4.8.1.) se zamijeni uloškom od brušenog stakla (točka 4.8.2.), a donji dio prilagodljive cijevi u obliku slova U postavi se odmah iznad razine veznog sklopa. Tlakom koji tvori dušik u bočnoj ručici, gornji se sloj laganog petroleja prenese u lijevak za odjeljivanje obujma 1 000 ml (točka 4.2.3.). U stakleni se valjak doda 100 ml laganog petroleja (točka 3.2.), začepi i dobro protrese. Pričeka se do razdvajanja slojeva, a gornji se sloj prenese u lijevak za odjeljivanje kao i prije. Postupak ekstrakcije se ponovi sa 100 ml laganog petroleja (točka 3.2.), zatim dvaput s po 50 ml laganog petroleja (točka 3.2.), a slojevi laganog petroleja dodaju u lijevak za odjeljivanje.
Pomiješani se ekstrakti laganog petroleja isperu, u skladu s točkom 5.3.1., a zatim se nastavi u skladu s navedenom točkom.
5.4. Priprema otopine uzorka za HPLC
Alikvotni dio otopine laganog petroleja (iz točke 5.3.1. ili 5.3.2.) prenese se pipetom u kruškastu tikvicu obujma 250 ml (točka 4.2.4.). Otapalo se otpari skoro do suhog u rotacijskom otparivaču (točka 4.1.) pri sniženom tlaku i temperaturi kupelji do 40 °C. Dovođenjem dušika uspostavi se atmosferski tlak (točka 3.10.), a tikvica se izvadi iz rotacijskog otparivača. Preostalo se otapalo otpari u struji dušika (točka 3.10.), a ostatak odmah otopi u poznatom volumenu (10 – 100 ml) metanola (točka 3.3.) (koncentracija vitamina A mora biti u rasponu od 5 do 30 IU/ml).
5.5. Određivanje HPLC-om
Vitamin A se razdvaja na koloni C18 s reverznim fazama (točka 4.5.1.), koncentracija se izmjeri UV detektorom (325 nm) ili fluorescencijskim detektorom (ekscitacija: 325 nm, emisija: 475 nm) (točka 4.5.2.).
Ubrizga se alikvotni dio (npr. 20 μl) otopine metanola dobivene u točki 5.4. i eluira mobilnom fazom (točka 3.9.). Izračuna se srednja vrijednost visine (površine) vršaka za više ubrizgavanja iste otopine uzorka i srednje vrijednosti visina (površina) vršaka za više ubrizgavanja kalibracijskih otopina (točka 5.6.2.).
Sljedeći uvjeti predlažu se kao smjernice; mogu se koristiti i drugi uvjeti ako se njima jamče jednakovrijedni rezultati.
|
Kolona za tekućinsku kromatografiju (točka 4.5.1.): |
250 mm × 4 mm, C18, veličina čestica punjenja 5 ili 10 μm ili jednakovrijedna |
|
Mobilna faza (točka 3.9.): |
Smjesa metanola (točka 3.3.) i vode, npr. 980 + 20 (v + v) |
|
Brzina protoka: |
1 – 2 ml/min |
|
Detektor (točka 4.5.2.): |
UV detektor (325 nm) ili fluorescencijski detektor (ekscitacija: 325 nm/emisija: 475 nm) |
5.6. Kalibracija
5.6.1.
Pipetom se prenese 20 ml osnovne otopine acetata vitamina A (točka 3.11.1.) ili 20 ml osnovne otopine palmitata vitamina A (točka 3.12.1.) u tikvicu s ravnim dnom ili konusnu tikvicu obujma 500 ml (točka 4.2.1.) i hidrolizira u skladu s točkom 5.2., ali bez dodatka BHT-a. Zatim se ekstrahira s laganim petrolejem (točka 3.2.) u skladu s točkom 5.3. i dopuni laganim petrolejem (točka 3.2.) do 500 ml. U rotacijskom se otparivaču (vidjeti 5.4.) otpari 100 ml ovog ekstrakta gotovo do suhog, strujom dušika (točka 3.10.) ukloni preostalo otapalo, a ostatak ponovno otopi u 10,0 ml metanola (točka 3.3.). Nominalna koncentracija ove otopine je 560 IU vitamina A na ml. Točan udio se određuje u skladu s točkom 5.6.3.3. Radna standardna otopina mora se pripremiti svježa prije upotrebe.
Pipetom se prenese 2,0 ml ove radne standardne otopine u graduiranu tikvicu obujma 20 ml, dopuni metanolom (točka 3.3.) do oznake i promiješa. Nominalna koncentracija takve razrijeđene radne standardne otopine iznosi 56 IU vitamina A na ml.
5.6.2.
Prenese se 1,0, 2,0, 5,0 i 10,0 ml razrijeđene radne standardne otopine u seriju graduiranih tikvica obujma 20 ml, dopuni metanolom (točka 3.3.) do oznake i promiješa. Nominalne koncentracije ovih otopina iznose 2, 8, 5, 6, 14,0 i 28,0 IU vitamina A na ml.
Više se puta ubrizga 20 μl svake kalibracijske otopine i odrede srednje vrijednosti visina (površina) vršaka. Iz srednjih se vrijednosti visina (površina) vršaka nacrta kalibracijska krivulja uzimajući u obzir rezultate UV kontrole (točka 5.6.3.3.).
5.6.3.
5.6.3.1.
Pipetom se prenese 2,0 ml osnovne otopine acetata vitamina A (točka 3.11.1.) u graduiranu tikvicu obujma 50 ml (točka 4.2.2.) i dopuni 2-propanolom (točka 3.8.) do oznake. Nominalna koncentracija ove otopine je 56 IU vitamina A na ml. Pipetom se prenese 3,0 ml ove razrijeđene otopine acetata vitamina A u graduiranu tikvicu obujma 25 ml i dopuni 2-propanolom (točka 3.8.) do oznake. Nominalna koncentracija ove otopine je 6,72 IU vitamina A na ml. Izmjeri se UV spektar ove otopine prema 2-propanolu (točka 3.8.) u spektrofotometru (točka 4.6.) između 300 i 400 nm. Maksimalna ekstinkcija mora biti između 325 i 327 nm.
Izračun udjela vitamina A:
IU vitamina A/ml = E326 × 19,0
(E1 % 1 cm za acetat vitamina A = 1 530 na 326 nm u 2-propanolu)
5.6.3.2.
Pipetom se prenese 2,0 ml osnovne otopine palmitata vitamina A (točka 3.12.1.) u graduiranu tikvicu obujma 50 ml (točka 4.2.2.) i dopuni 2-propanolom (točka 3.8.) do oznake. Nominalna koncentracija ove otopine je 56 IU vitamina A na ml. Pipetom se prenese 3,0 ml ove razrijeđene otopine palmitata vitamina A u graduiranu tikvicu obujma 25 ml i dopuni 2-propanolom (točka 3.8.) do oznake. Nominalna koncentracija ove otopine je 6,72 IU vitamina A na ml. Izmjeri se UV spektar ove otopine prema 2-propanolu (točka 3.8.) u spektrofotometru (točka 4.6.) između 300 i 400 nm. Maksimalna ekstinkcija mora biti između 325 i 327 nm.
Izračun udjela vitamina A:
IU vitamina A/ml = E326 × 19,0
(E1 % 1 cm za palmitat vitamina A = 957 na 326 nm u 2-propanolu)
5.6.3.3.
Pipetom se prenese 3,0 ml nerazrijeđene radne standardne otopine vitamina A, pripravljene u skladu s točkom 5.6.1., u graduiranu tikvicu obujma 50 ml (točka 4.2.2.) i dopuni 2-propanolom (točka 3.8.) do oznake. Pipetom se prenese 5,0 ml ove otopine u graduiranu tikvicu obujma 25 ml i dopuni 2-propanolom (točka 3.8.) do oznake. Nominalna koncentracija ove otopine je 6,72 IU vitamina A na ml. Izmjeri se UV spektar ove otopine prema 2-propanolu (točka 3.8.) u spektrofotometru (točka 4.6.) između 300 i 400 nm. Maksimalna ekstinkcija mora biti između 325 i 327 nm.
Izračun udjela vitamina A:
IU vitamina A/ml = E325 × 18,3
(E1 % 1 cm za vitamin A alkohol = 1 821 na 325 nm u 2-propanolu)
6. Izračun rezultata
Iz srednje vrijednosti visine (površine) vršaka vitamina A iz otopine uzorka, određuje se koncentracija otopine uzorka u IU/ml iz kalibracijske krivulje (točka 5.6.2.).
Udio vitamina A (w) u uzorku, iskazan u IU/kg, izračunava se sljedećom formulom:
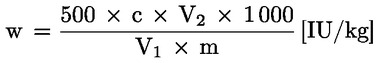
pri čemu je:
|
c |
= |
koncentracija vitamina A (točka 5.4.) u otopini uzorka, iskazana u IU/ml |
|
V1 |
= |
obujam otopine uzorka (točka 5.4.) u ml |
|
V2 |
= |
obujam alikvota korištenog u točki 5.4., u ml |
|
m |
= |
masa pokusnog uzorka u gramima |
7. Napomene
|
7.1. |
Kod uzoraka s niskom koncentracijom vitamina A može biti korisno pomiješati ekstrakte laganog petroleja iz dviju šarži za saponifikaciju (izvagana količina: 25 g) u jednu otopinu uzorka za određivanje HPLC-om. |
|
7.2. |
Masa uzorka za analizu ne smije sadržavati više od 2 g masti. |
|
7.3. |
Ako se faze ne razdvoje, doda se približno 10 ml etanola (točka 3.1.) za razbijanje emulzije. |
|
7.4. |
Kod ulja iz jetre bakalara i drugih čistih masti vrijeme trajanja saponifikacije treba produžiti na 45 – 60 minuta. |
|
7.5. |
Umjesto BHT-a može se koristiti hidrokinon. |
|
7.6. |
Izomere retinola moguće je razdvojiti uobičajenom faznom kolonom. U tom slučaju, za izračune treba zbrojiti visine (površine) vršaka svih cis i trans izomera. |
|
7.7. |
Umjesto otopine natrijevog askorbata može se koristiti približno 150 mg askorbinske kiseline. |
|
7.8. |
Umjesto otopine natrijevog sulfida može se koristiti približno 50 mg EDTA. |
|
7.9. |
Pri analizi vitamina A u nadomjescima mlijeka treba paziti na sljedeće:
Za provjeru pouzdanosti rezultata dobivenih korištenom analitičkom metodom na ovoj specifičnoj matrici (nadomjestak mlijeka) treba na dodatnom pokusnom uzorku izvesti test iskorištenja. Ako je iskorištenje manje od 80 %, rezultat analize treba korigirati za iskorištenje. |
8. Ponovljivost
Razlika između rezultata dvaju usporednih postupaka određivanja, izvedenih na istom uzorku, ne smije prijeći 15 % u odnosu na najviši rezultat.
9. Rezultati međulaboratorijske studije (1)
|
|
Premiks |
Gotove smjese hrane za stoku |
Mineralni koncentrat |
Proteinski koncentrat |
Hrana za odojke |
|
L |
13 |
12 |
13 |
12 |
13 |
|
n |
48 |
45 |
47 |
46 |
49 |
|
srednja vrijednost [IU/kg] |
17,02 × 106 |
1,21 × 106 |
537 100 |
151 800 |
18 070 |
|
Sr [IU/kg] |
0,51 × 106 |
0,039 × 106 |
22 080 |
12 280 |
682 |
|
r [IU/kg] |
1,43 × 106 |
0,109 × 106 |
61 824 |
34 384 |
1 910 |
|
CVr [%] |
3,0 |
3,5 |
4,1 |
8,1 |
3,8 |
|
SR [IU/kg] |
1,36 × 106 |
0,069 × 106 |
46 300 |
23 060 |
3 614 |
|
R [IU/kg] |
3,81 × 106 |
0,193 × 106 |
129 640 |
64 568 |
10 119 |
|
CVR [%] |
8,0 |
6,2 |
8,6 |
15 |
20 |
|
L |
= |
broj laboratorija |
|
n |
= |
broj pojedinačnih vrijednosti |
|
sr |
= |
standardna devijacija ponovljivosti |
|
SR |
= |
standardna devijacija obnovljivosti |
|
r |
= |
ponovljivost |
|
R |
= |
obnovljivost |
|
CVr |
= |
koeficijent varijacije ponovljivosti |
|
CVR |
= |
koeficijent varijacije obnovljivosti. |
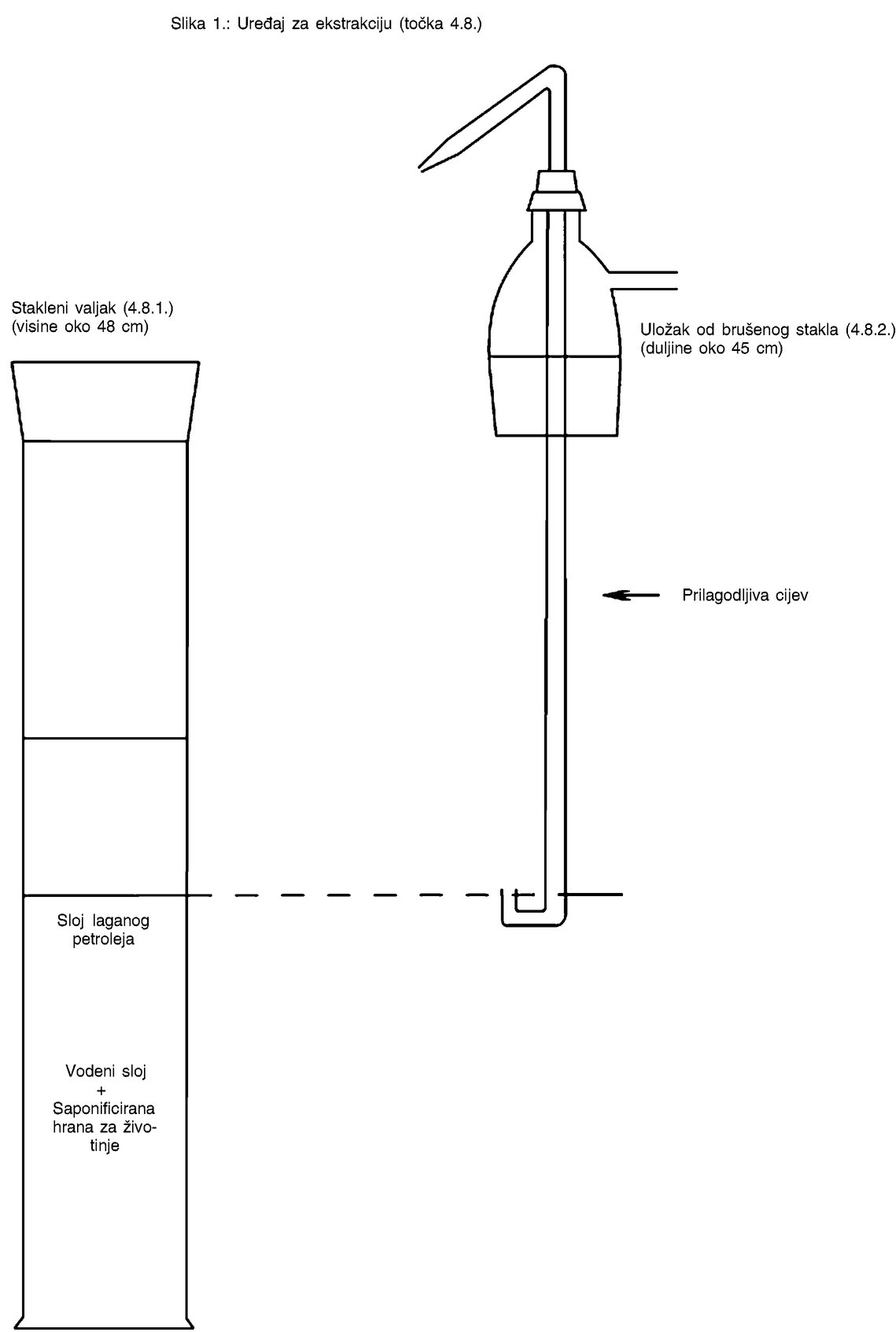
B. ODREĐIVANJE VITAMINA E
1. Svrha i područje primjene
Ovom se metodom omogućuje određivanje razine vitamina E u hrani za životinje i premiksima. Udio vitamina E iskazuje se u mg DL-α-tokoferol acetata na kg. Količina od 1 mg DL-α-tokoferol acetata odgovara količini od 0,91 mg DL-α-tokoferola (vitamina E).
Granica kvantifikacije je 2 mg vitamina E/kg. Ova se granica kvantifikacije može dosegnuti samo fluorescencijskim detektorom. Granica kvantifikacije za UV detektor iznosi 10 mg/kg.
2. Načelo
Uzorak se hidrolizira otopinom etanolnog kalijevog hidroksida, a vitamin E se ekstrahira laganim petrolejem. Otapalo se ukloni otparivanjem, a ostatak otopi u metanolu i prema potrebi razrijedi do tražene koncentracije. Udio vitamina E određuje se tekućinskom kromatografijom visoke učinkovitosti reverznih faza (RP–HPLC) i UV ili fluorescencijskim detektorom.
3. Reagensi
|
3.1. |
Etanol, σ = 96 %. |
|
3.2. |
Lagani petrolej, raspon vrelišta 40 – 60 °C. |
|
3.3. |
Metanol. |
|
3.4. |
Otopina kalijevog hidroksida, c = 50 g/100 ml. |
|
3.5. |
Otopina natrijevog askorbata, c = 10 g/100 ml (vidjeti napomenu pod točkom 7.7.). |
|
3.6. |
Natrijev sulfid, Na2S· x H2O (x = 7 – 9). |
|
3.6.1. |
Otopina natrijevog sulfida, c = 0,5 mol/l u glicerolu, β = 120 g/l (za x = 9) (vidjeti napomenu pod točkom 7.8.) |
|
3.7. |
Otopina fenolftaleina, c = 2 g/100 ml u etanolu (točka 3.1.). |
|
3.8. |
Mobilna faza za HPLC: smjesa metanola (točka 3.3.) i vode, npr. 980 + 20 (v + v). Točan se omjer određuje u skladu s osobinama korištene kolone. |
|
3.9. |
Dušik, bez kisika. |
|
3.10. |
DL-α-tokoferol acetat, posebne čistoće, certificiranog djelovanja. |
|
3.10.1. |
Osnovna otopina DL-α-tokoferol acetata: odvagne se 100 mg DL-α-tokoferol acetata (točka 3.10.) s preciznošću od 0,1 mg i prenese u graduiranu tikvicu obujma 100 ml. Otopi se u etanolu (točka 3.1.) i dopuni istim otapalom do oznake. 1 ml ove otopine sadrži 1 mg DL-α-tokoferol acetata. (Za UV kontrolu vidjeti točku 5.6.1.3., za stabilizaciju vidjeti napomenu pod točkom 7.4.). |
|
3.11. |
DL-α-tokoferol, posebne čistoće, certificiranog djelovanja. |
|
3.11.1. |
Osnovna otopina DL-α-tokoferola: odvagne se 100 mg DL-α-tokoferola (točka 3.10.) s preciznošću od 0,1 mg i prenese u graduiranu tikvicu obujma 100 ml. Otopi se u etanolu (točka 3.1.) i dopuni istim otapalom do oznake. 1 ml te otopine sadrži 1 mg DL-α-tokoferola. (Za UV kontrolu vidjeti točku 5.6.2.3., za stabilizaciju napomenu pod točkom 7.4.). |
|
3.12. |
2,6-di-tert-butil-4-metilfenol (BHT) (vidjeti napomenu pod točkom 7.5.). |
4. Oprema
|
4.1. |
Vakuumski rotacijski otparivač. |
|
4.2. |
Laboratorijsko posuđe od smeđeg stakla. |
|
4.2.1. |
Tikvice s ravnim dnom ili konusne tikvice, obujma 500 ml, s vratom od brušenog stakla. |
|
4.2.2. |
Odmjerne tikvice s ubrušenim čepovima, s uskim vratom, obujma 10 ml, 25 ml, 100 ml i 500 ml. |
|
4.2.3. |
Lijevci za odjeljivanje, konusni, obujma 1 000 ml, s ubrušenim čepovima. |
|
4.2.4. |
Kruškaste tikvice, obujma 250 ml, s ubrušenim čepovima. |
|
4.3. |
Allihnov kondenzator, duljine omotača 300 mm sa spojem od brušenog stakla i nastavkom za dovodnu cijev plina. |
|
4.4. |
Nabrani filtar papir za odjeljivanje faza, promjera 185 mm (npr. Schleicher & Schuell 597 HY 1/2). |
|
4.5. |
Oprema za HPLC sa sustavom za ubrizgavanje. |
|
4.5.1. |
Kolona za tekućinsku kromatografiju, 250 mm × 4 mm, C18, veličina čestica punjenja 5 ili 10 μm ili jednakovrijedna. |
|
4.5.2. |
UV ili fluorescencijski detektor s mogućnošću promjene valne duljine. |
|
4.6. |
Spektrofotometar s kvarcnim kivetama od 10 mm. |
|
4.7. |
Vodena kupelj s magnetnom mješalicom. |
|
4.8. |
Naprava za ekstrakciju (vidjeti sliku 1.) koja se sastoji od: |
|
4.8.1. |
Staklenog valjka obujma l litre s vratom od brušenog stakla i čepom. |
|
4.8.2. |
Uloška od brušenog stakla s bočnom ručicom i prilagodljivom cijevi koja prolazi kroz središte. Prilagodljiva cijev ima donji dio u obliku slova U i mlaznicu na suprotnom kraju tako da se gornji sloj tekućine u valjku može prenijeti u lijevak za odjeljivanje. |
5. Postupak
|
Napomena |
: |
Vitamin E osjetljiv je na (UV) svjetlo i oksidaciju. Svi se postupci izvode bez svjetla (u laboratorijskom posuđu od smeđeg stakla ili u staklenim posudama zaštićenim aluminijskom folijom) i kisika (ispire se dušikom). Tijekom ekstrakcije zrak iznad tekućine zamijeni se dušikom (povremenim popuštanjem čepa izbjegava se previsoki tlak). |
5.1. Priprema uzorka
Uzorak se samelje tako da prolazi kroz sito veličine očice 1 mm, pri čemu treba izbjegavati stvaranje topline. Mljevenje se mora izvesti neposredno prije vaganja i saponifikacije, inače može doći do gubitka vitamina E.
5.2. Saponifikacija
Ovisno o udjelu vitamina E, odvagne se 2 – 25 g uzorka s preciznošću od 0,01 g i prenese u tikvicu s ravnim dnom ili konusnu tikvicu obujma 500 ml (točka 4.2.1.). Uz stalno vrtloženje, jedno za drugim se dodaje 130 ml etanola (točka 3.1.), približno 100 mg BHT-a (točka 3.12.), 2 ml otopine natrijevog askorbata (točka 3.5.) i 2 ml otopine natrijevog sulfida (točka 3.6.). Na tikvicu se priključi kondenzator (točka 4.3.) i tikvica potopi u vodenu kupelj s magnetnom mješalicom (točka 4.7.). Zagrije se do ključanja i 5 minuta ostavi uz refluks. Kroz kondenzator (točka 4.3.) se doda 25 ml otopine kalijevog hidroksida (točka 3.4.) i ostavi uz refluks sljedećih 25 minuta, uz miješanje pod laganom strujom dušika. Zatim se kondenzator ispere s približno 20 ml vode, a udio tikvice ohladi na sobnu temperaturu.
5.3. Ekstrakcija
Otopina za saponifikaciju kvantitativno se prenese dekantiranjem u lijevak za odjeljivanje kapaciteta 1 000 ml (točka 4.2.3.) ili u napravu za ekstrakciju (točka 4.8.), tako što se ispere s ukupno 250 ml vode. Zatim se tikvica za saponifikaciju ispere s 25 ml etanola (točka 3.1.) i 100 ml laganog petroleja (točka 3.2.) i isprane tekućine prenesu u lijevak za odjeljivanje ili napravu za ekstrakciju. Omjer vode i etanola u pomiješanim otopinama mora biti približno 2:1. Snažno se trese 2 minute, zatim ostavi stajati 2.
5.3.1.
Kad se slojevi razdvoje (vidjeti napomenu pod točkom 7.3.), sloj laganog petroleja se prenese u drugi lijevak za odjeljivanje (točka 4.2.3). Ekstrakcija se ponovi dvaput sa 100 ml laganog petroleja (točka 3.2.) i dvaput s 50 ml laganog petroleja (točka 3.2.).
Pomiješani ekstrakti u lijevku za odjeljivanje isperu se dvaput sa 100 ml vode, uz lagano vrtloženje (kako bi se spriječio nastanak emulzija), zatim uz ponavljano protresivanjem s dodatnim obrocima od po 100 ml vode, sve dok voda pri dodavanju otopine fenolftaleina (točka 3.7.) ne ostane bezbojna (obično je dovoljno četiri ispiranja). Isprani se ekstrakt filtrira kroz suhi nabrani filtar za odjeljivanje faza (točka 4.4.) u graduiranu tikvicu obujma 500 ml (točka 4.2.2.) kako bi se uklonila preostala voda. Lijevak za odjeljivanje i filtar se isperu s 50 ml laganog petroleja (točka 3.2.), dopuni se laganim petrolejem (točka 3.2.) do oznake i dobro promiješa.
5.3.2.
Kada se slojevi razdvoje (vidjeti napomenu pod točkom 7.3.), zamijeni se čep staklenog valjka (točka 4.8.1.) uloškom od brušenog stakla (točka 4.8.2.), a donji dio prilagodljive cijevi u obliku slova U postavi odmah iznad razine veznog sklopa. Tlakom koji tvori dušik u bočnoj ručici gornji se sloj laganog petroleja prenese u lijevak za odjeljivanje kapaciteta 1 000 ml (točka 4.2.3.). U stakleni se valjak doda 100 ml laganog petroleja (točka 3.2.), začepi i dobro protrese. Pričeka se do razdvajanja slojeva, te se gornji sloj prenese u lijevak za odjeljivanje kao i prije. Postupak ekstrakcije se ponovi sa 100 ml laganog petroleja (točka 3.2.), zatim dvaput s 50 ml laganog petroleja (točka 3.2.), a slojevi laganog petroleja dodaju u lijevak za odjeljivanje.
Pomiješani se ekstrakti laganog petroleja isperu, u skladu s točkom 5.3.1., zatim se nastavi u skladu s uputama u navedenoj točki.
5.4. Priprema otopine uzorka za HPLC
Alikvotni dio otopine laganog petroleja (iz točke 5.3.1. ili 5.3.2.) prenese se pipetom u kruškastu tikvicu obujma 250 ml (točka 4.2.4.). Otapalo se otpari skoro do suhog u rotacijskom otparivaču (točka 4.1.) pri sniženom tlaku i temperaturi kupelji do 40 °C. Dovođenjem dušika (točka 3.9.) uspostavi se atmosferski tlak, a tikvica izvadi iz rotacijskog otparivača. Preostalo se otapalo ukloni strujom dušika (točka 3.9.), a ostatak odmah otopi u poznatom obujmu (10 – 100 ml) metanola (točka 3.3.) (koncentracija DL-α-tokoferola mora biti u području 5 μ/ml do 30 μ/ml).
5.5. Određivanje putem HPLC-a
Vitamin E se razdvaja na koloni C18 s reverznim fazama (točka 4.5.1.), a koncentracija izmjeri fluorescencijskim detektorom (ekscitacija: 295 nm, emisija: 330 nm) ili UV detektorom (292 nm) (točka 4.5.2.).
Ubrizga se alikvotni dio (npr. 20 μl) otopine metanola dobivene u točki 5.4. i eluira mobilnom fazom (točka 3.8.). Izračuna se srednja vrijednost visina (površina) vršaka za više ubrizgavanja iste otopine uzorka i srednje vrijednosti visina (površina) vršaka za više ubrizgavanja kalibracijskih otopina (točka 5.6.2.).
Sljedeći uvjeti predlažu se kao smjernice; mogu se koristiti i drugi uvjeti ako se njima jamče jednakovrijedni rezultati.
|
Kolona za tekućinsku kromatografiju (točka 4.5.1.): |
250 mm × 4 mm, C18, veličina čestica punjenja 5 ili 10 μm ili jednakovrijedna |
|
Mobilna faza (točka 3.8): |
Smjesa metanola (točka 3.3) i vode, npr. 980 + 20 (v + v) |
|
Brzina protoka: |
1 – 2 ml/min |
|
Detektor (točka 4.5.2.): |
Fluorescencijski detektor (ekscitacija: 295 nm/emisija: 330 nm) ili UV detektor (292 nm) |
5.6 Kalibracija (DL-α-tokoferol acetat ili DL-α-tokoferol)
5.6.1
5.6.1.1
Pipetom se prenese 25 ml osnovne otopine DL-α-tokoferol acetata (točka 3.10.1.) u tikvicu s ravnim dnom ili konusnu tikvicu obujma 500 ml (točka 4.2.1.) i hidrolizira, u skladu s točkom 5.2. Zatim se ekstrahira laganim petrolejem (točka 3.2.) u skladu s točkom 5.3. i laganim petrolejem dopuni do 500 ml. U rotacijskom se otparivaču (vidjeti točku 5.4.) otpari 25 ml tog ekstrakta gotovo do suhog, strujom dušika ukloni preostalo otapalo (točka 3.9.), a ostatak se ponovo otopi u 25,0 ml metanola (točka 3.3.). Nominalna koncentracija te otopine iznosi 45,5 μg DL-α-tokoferola na ml, što je jednakovrijedno 50 μg DL-α-tokoferol acetata na ml. Radna standardna otopina treba se pripremiti svježa prije upotrebe.
5.6.1.2.
Prenese se 1,0, 2,0, 4,0 i 10,0 ml radne standardne otopine u seriju graduiranih tikvica obujma 20 ml, metanolom (točka 3.3.) dopuni do oznake i promiješa. Nominalne koncentracije ovih otopina iznose 2,5, 5,0, 10,0 i 25,0 μg/ml DL-α-tokoferol acetata, tj. 2,28, 4,55, 9,10 i 22,8 μg/ml DL-α-tokoferola.
Više puta se ubrizga 20 μl svake kalibracijske otopine i odrede srednje vrijednosti visina (površina) vršaka. Iz srednjih se vrijednosti visina (površina) vršaka pripremi kalibracijska krivulja.
5.6.1.3.
Etanolom se razrijedi 5,0 ml osnovne otopine DL-α-tokoferol acetata (točka 3.10.1.) na 25,0 ml i izmjeri UV spektar te otopine prema etanolu (točka 3.1.) u spektrofotometru (točka 4.6.) između 250 i 320 nm.
Maksimalna apsorpcija je na 284 nm:
E1 % 1 cm = 43,6 na 284 nm u etanolu
Za tu se razrijeđenost mora dobiti vrijednost ekstinkcije u rasponu od 0,84 do 0,88.
5.6.2.
5.6.2.1
Pipetom se prenese 2 ml osnovne otopine DL-α-tokoferola (točka 3.11.1.) u graduiranu tikvicu obujma 50 ml, otopi u metanolu (točka 3.3.) i dopuni do oznake metanolom. Nominalna koncentracija ove otopine iznosi 40 μg DL-α-tokoferola na ml, što je jednakovrijedno 44,0 μg DL-α-tokoferol acetata na ml. Radna standardna otopina mora se pripremiti svježa prije upotrebe.
5.6.2.2.
Prenese se 1,0, 2,0, 4,0 i 10,0 ml radne standardne otopine u seriju graduiranih tikvica obujma 20 ml, metanolom (točka 3.3.) dopuni do oznake i promiješa. Nominalne koncentracije ovih otopina iznose 2,0, 4,0, 8,0 i 20,0 μg/ml DL-α-tokoferola, tj. 2,20, 4,40, 8,79 i 22,0 μg/ml DL-α-tokoferol acetata.
Više puta se ubrizga 20 μl svake kalibracijske otopine i odrede srednje vrijednosti visina (površina) vršaka. Iz srednjih se vrijednosti visina (površina) vršaka pripremi kalibracijska krivulja.
5.6.2.3.
Etanolom se razrijedi 2,0 ml osnovne otopine DL-α-tokoferola (točka 3.11.1.) na 25,0 ml te se izmjeri UV spektar ove otopine prema etanolu (točka 3.1.) u spektrofotometru (točka 4.6.) između 250 i 320 nm. Maksimalna apsorpcija je na 292 nm:
E1 % 1 cm = 75,8 na 292 nm u etanolu
Za tu se razrijeđenost mora dobiti vrijednost ekstinkcije od 0,6.
6. Izračun rezultata
Iz srednje se vrijednosti visine (površine) vršaka vitamina E iz otopine uzorka određuje koncentracija otopine uzorka u μg/ml (izračunana kao α-tokoferol acetat) iz kalibracijske krivulje (točka 5.6.1.2. ili 5.6.2.2.).
Udio vitamina E (w) u mg/kg u uzorku izračunava se sljedećom formulom:

pri čemu je:
|
c |
= |
koncentracija vitamina E (kao α-tokoferol acetata) u otopini uzorka (točka 5.4.) u μg/ml; |
|
V1 |
= |
obujam otopine uzorka (točka 5.4.) u ml; |
|
V2 |
= |
obujam alikvota korištenog u točki 5.4., u ml; |
|
m |
= |
masa pokusnog uzorka u gramima. |
7. Napomene
|
7.1. |
Kod uzoraka s niskom koncentracijom vitamina E može biti korisno pomiješati ekstrakte laganog petroleja iz dviju šarži za saponifikaciju (izvagana količina: 25 g) u jednu otopinu uzorka za određivanje HPLC-om. |
|
7.2. |
Masa uzorka za analizu ne smije sadržavati više od 2 g masti. |
|
7.3. |
Ako se faze ne razdvoje, doda se približno 10 ml etanola (točka 3.1.) za razbijanje emulzije. |
|
7.4. |
Nakon spektrofotometrijskog mjerenja otopine DL-α-tokoferol acetata ili otopine DL-α-tokoferola u skladu s točkom 5.6.1.3. ili 5.6.2.3., doda se približno 10 mg BHT-a (točka 3.12.) u otopinu (točka 3.10.1. ili 3.10.2.), a otopina pohrani u hladnjaku (može se držati najviše 4 tjedna). |
|
7.5. |
Umjesto BHT-a može se koristiti hidrokinon. |
|
7.6. |
Običnom faznom kolonom može se odijeliti α-, β-, γ- i δ-tokoferol. |
|
7.7. |
Umjesto otopine natrijevog askorbata može se koristiti približno 150 mg askorbinske kiseline. |
|
7.8. |
Umjesto otopine natrijevog sulfida može se koristiti približno 50 mg EDTA. |
|
7.9. |
Acetat vitamina E se u bazičnim uvjetima vrlo brzo hidrolizira, zato je vrlo osjetljiv na oksidaciju, posebno u prisutnosti elemenata u tragovima, poput željeza i bakra. Postupak određivanja vitamina E u premiksima s razinama višim od 5 000 mg/kg može dovesti do razgradnje vitamina E. Zbog toga se za potvrdu preporuča metoda HPLC s enzimskom razgradnjom struktura vitamina E bez alkalne saponifikacije. |
8. Ponovljivost
Razlika između rezultata dvaju usporednih postupaka određivanja, izvedenih na istom uzorku, ne smije prijeći 15 % najvišeg rezultata.
9. Rezultati međulaboratorijske studije (2)
|
|
Premiks |
Gotove smjese hrane za stoku |
Mineralni koncentrat |
Proteinski koncentrat |
Hrana za odojke |
|
L |
12 |
12 |
12 |
12 |
12 |
|
n |
48 |
48 |
48 |
48 |
48 |
|
Srednja vrijednost [mg/kg] |
17 380 |
1 187 |
926 |
315 |
61,3 |
|
Sr [mg/kg] |
384 |
45,3 |
25,2 |
13,0 |
2,3 |
|
r [mg/kg] |
1 075 |
126,8 |
70,6 |
36,4 |
6,4 |
|
CVr [%] |
2,2 |
3,8 |
2,7 |
4,1 |
3,8 |
|
SR [mg/kg] |
830 |
65,0 |
55,5 |
18,9 |
7,8 |
|
R [mg/kg] |
2 324 |
182,0 |
155,4 |
52,9 |
21,8 |
|
CVR [%] |
4,8 |
5,5 |
6,0 |
6,0 |
12,7 |
|
L |
= |
broj laboratorija |
|
n |
= |
broj pojedinačnih vrijednosti |
|
Sr |
= |
standardna devijacija ponovljivosti |
|
SR |
= |
standardna devijacija obnovljivosti |
|
r |
= |
ponovljivost |
|
R |
= |
obnovljivost |
|
CVr |
= |
koeficijent varijacije ponovljivosti |
|
CVR |
= |
koeficijent varijacije obnovljivosti. |

C. ODREĐIVANJE ŽELJEZA, BAKRA, MANGANA I CINKA U TRAGOVIMA
1. Svrha i područje primjene
Ovom se metodom omogućuje određivanje željeza, bakra, mangana i cinka u tragovima u hrani za životinje. Granice za kvantifikaciju su:
|
— |
željezo (Fe): 20 mg/kg |
|
— |
bakar (Cu): 10 mg/kg |
|
— |
mangan (Mn): 20 mg/kg |
|
— |
cink (Zn): 20 mg/kg. |
2. Načelo
Nakon razgradnje eventualno prisutnih organskih tvari uzorak se otopi u klorovodičnoj kiselini. Nakon primjerenog razrjeđivanja, atomskom se apsorpcijskom spektrometrijom određuju elementi željezo, bakar, mangan i cink.
3. Reagensi
Uvodne napomene
Za pripremanje reagensa i otopina za analizu koristi se voda bez kationa koje treba odrediti, dobivena dvostrukim destiliranjem vode u destilatoru od borosilikatnog ili kvarcnog stakla ili dvostrukom obradom ionsko-izmjenjivačkom smolom.
Reagensi moraju biti najmanje analitičke čistoće. Odsutnost elementa koji se određuje provjerava se slijepom probom. Prema potrebi se reagensi moraju dodatno pročistiti.
Umjesto dolje navedenih standardnih otopina mogu se koristiti komercijalne standardne otopine pod uvjetom da imaju jamstvo i da se prije upotrebe provjere.
|
3.1. |
Klorovodična kiselina (d: 1,19 g/ml). |
|
3.2. |
Klorovodična kiselina (6 mol/l). |
|
3.3. |
Klorovodična kiselina (0,5 mol/l). |
|
3.4. |
Fluoridna kiselina 38 – 40 % (v/v) sa sadržajem željeza (Fe) manjim od 1 mg/l i ostatkom nakon otparivanja manjim od 10 mg/l (u obliku sulfata). |
|
3.5 |
Sumporna kiselina (g: 1,84 g/ml). |
|
3.6 |
Vodikov peroksid (približno 100 volumnih dijelova kisika (30 % po masi)). |
|
3.7. |
Standardna otopina željeza (1 000 μg Fe/ml), pripravljena na sljedeći način ili jednakovrijedna komercijalno dostupna otopina: otopi se 1 g željezne žice u 200 ml klorovodične kiseline 6 mol/l (točka 3.2.), doda 16 ml vodikovog peroksida (točka 3.6.) i dopuni vodom do 1 litre. |
|
3.7.1. |
Radna standardna otopina željeza (100 μg Fe/ml), pripravljena razrjeđivanjem 1 dijela standardne otopine (točka 3.7.) s 9 dijelova vode. |
|
3.8. |
Standardna otopina bakra (1 000 μg Cu/ml), pripravljena na sljedeći način ili jednakovrijedna komercijalno dostupna otopina:
|
|
3.8.1. |
Radna standardna otopina bakra (10 μg Cu/ml), pripravljena razrjeđivanjem 1 dijela standardne otopine (točka 3.8.) s 9 dijelova vode, zatim razrjeđivanjem 1 dijela te otopine s 9 dijelova vode. |
|
3.9. |
Standardna otopina mangana (1 000 μg Mn/ml), pripravljena na sljedeći način ili jednakovrijedna komercijalno dostupna otopina:
|
|
3.9.1. |
Radna standardna otopina mangana (10 μg Mn/ml), pripravljena razrjeđivanjem 1 dijela standardne otopine (točka 3.9.) s 9 dijelova vode, zatim razrjeđivanjem 1 dijela te otopine s 9 dijelova vode. |
|
3.10. |
Standardna otopina cinka (1 000 μg Zn/ml), pripravljena na sljedeći način ili jednakovrijedna komercijalno dostupna otopina:
|
|
3.10.1. |
Radna standardna otopina cinka (10 μg Zn/ml), pripravljena razrjeđivanjem 1 dijela standardne otopine (točka 3.10.) s 9 dijelova vode, zatim razrjeđivanjem 1 dijela te otopine s 9 dijelova vode. |
|
3.11. |
Otopina lantanovoga klorida: otopi se 12 g lantanovog oksida u 150 ml vode, doda se 100 ml klorovodične kiseline 6 mol/l (točka 3.2.) i dopuni vodom do 1 litre. |
4. Oprema
|
4.1. |
Mufolna peć s regulacijom temperature i po mogućnosti zapisivačem. |
|
4.2. |
Stakleni dijelovi moraju biti od otpornog borosilikata, a preporuča se upotreba opreme koja je namijenjena isključivo određivanju elemenata u tragovima. |
|
4.3. |
Atomski apsorpcijski spektrofotometar koji ispunjava zahtjeve metode glede osjetljivosti i točnosti u potrebnom području. |
5. Postupak (3)
5.1. Uzorci koji sadrže organske tvari
5.1.1. (4)
|
5.1.1.1. |
Odvagne se 5 – 10 g uzorka s preciznošću od 0,2 mg i prenese u lončić za spaljivanje od kvarcnog stakla ili platine (vidjeti napomenu pod točkom (b)), osuši u sušilu na 105 °C i postavi u hladnu mufolnu peć (točka 4.1.). Peć se zatvori (vidjeti napomenu pod točkom (c)), a temperatura se tijekom 90 minuta postupno digne na 450 – 475 °C. Ta se temperatura zadrži 4 – 16 sati (npr. tijekom noći) kako bi se uklonile ugljične tvari, a zatim se peć otvori i ohladi (vidjeti napomenu pod točkom (d)). Pepeo se navlaži vodom i prenese u čašu obujma 250 ml. Lončić se ispere s ukupno približno 5 ml klorovodične kiseline (točka 3.1.), a udio se polako i oprezno prenese u čašu (može doći do burne reakcije zbog tvorbe CO2). U kapljicama se dodaje klorovodična kiselina (točka 3.1.), uz stalno miješanje dok ne prestane pjenjenje. Otpari se do suhog, uz povremeno miješanje staklenim štapićem. Zatim se u ostatak doda 15 ml klorovodične kiseline 6 mol/l (točka 3.2.) i približno 120 ml vode. Promiješa se staklenim štapićem, koji se ostavi u čaši, a čaša se pokrije satnim staklom. Polako se zagrijava do ključanja i ostavi ključati do potpunog otapanja pepela. Filtrira se kroz filtar koji ne sadrži pepeo i prikupi u odmjernoj tikvici obujma 250 ml. Čaša i filtar se isperu s 5 ml vruće klorovodične kiseline 6 mol/l (točka 3.2.) i dvaput vrelom vodom. Odmjerna se tikvica napuni vodom do oznake (koncentracija HCl približno 0,5 mol/l). |
|
5.1.1.2. |
Ako ostatak na filtru izgleda crn (ugljik), ponovo se stavi u peć i spali na 450 – 475 °C. To je spaljivanje, za koje treba samo nekoliko sati (približno 3 – 5 sati), gotovo kada pepeo postane bijel ili gotovo bijel. Ostatak se otopi s približno 2 ml klorovodične kiseline (točka 3.1.), otpari se do suhog i doda 5 ml klorovodične kiseline 6 mol/l (točka 3.2.). Zagrije se, otopina se filtrira u graduiranu tikvicu i dopuni vodom do oznake (koncentracija HCl približno 0,5 mol/l). Napomene:
|
5.1.2.
5.1.2.1.
Za svaki element koji se određuje pripremi se serija kalibracijskih otopina iz standardnih radnih otopina iz točaka 3.7.1., 3.8.1., 3.9.1. i 3.10.1., pri čemu koncentracija HCl u svakoj iznosi približno 0,5 mol/l (i (kod željeza, mangana i cinka) koncentracija lantanovog klorida koja odgovara 0,1 % La (m/v)).
Odabrane koncentracije elemenata u tragovima moraju biti unutar područja osjetljivosti korištenog spektrofotometra. U donjim se tablicama kao primjer navode sastavi tipičnih područja kalibracijskih otopina; međutim, ovisno o vrsti i osjetljivosti korištenog spektrofotometra možda će trebati odabrati drukčije koncentracije.
Željezo
|
μg Fe/ml |
0 |
0,5 |
1 |
2 |
3 |
4 |
5 |
|
ml radne standardne otopine (točka 3.7.1.) (1 ml = 100 μg Fe) |
0 |
0,5 |
1 |
2 |
3 |
4 |
5 |
|
ml HCl (točka 3.2.) |
7 |
7 |
7 |
7 |
7 |
7 |
7 |
|
+ 10 ml otopine lantanovog klorida (točka 3.11.) i dopuniti vodom do 100 ml |
|||||||
Bakar
|
μg Cu/ml |
0 |
0,1 |
0,2 |
0,4 |
0,6 |
0,8 |
1,0 |
|
ml radne standardne otopine (točka 3.8.1.) (1 ml = 10 μg Cu) |
0 |
1 |
2 |
4 |
6 |
8 |
10 |
|
ml HCl (točka 3.2.) |
8 |
8 |
8 |
8 |
8 |
8 |
8 |
Mangan
|
μg Mn/ml |
0 |
0,1 |
0,2 |
0,4 |
0,6 |
0,8 |
1,0 |
|
ml radne standardne otopine (točka 3.9.1) (1 ml = 10 μg Mn) |
0 |
1 |
2 |
4 |
6 |
8 |
10 |
|
ml HCl (točka 3.2) |
7 |
7 |
7 |
7 |
7 |
7 |
7 |
|
+ 10 ml otopine lantanovog klorida (točka 3.11) i dopuniti vodom do 100 ml |
|||||||
Cink
|
μg Zn/ml |
0 |
0,05 |
0,1 |
0,2 |
0,4 |
0,6 |
0,8 |
|
ml radne standardne otopine (točka 3.10.1.) (1 ml = 10 μg Zn) |
0 |
0,5 |
1 |
2 |
4 |
6 |
8 |
|
ml HCl (točka 3.2) |
7 |
7 |
7 |
7 |
7 |
7 |
7 |
|
+ 10 ml otopine lantanovog klorida (točka 3.11.) i dopuniti vodom do 100 ml |
|||||||
5.1.2.2.
Za određivanje bakra, otopina pripravljena u skladu s točkom 5.1.1. obično se može koristiti izravno. Ako je koncentraciju potrebno prilagoditi tako da bude u području kalibracijskih otopina, pipetom se prenese alikvotni dio u graduiranu tikvicu obujma 100 ml i dopuni do oznake klorovodičnom kiselinom 0,5 mol/l (točka 3.3.).
Za određivanje željeza, mangana i cinka, pipetom se u graduiranu tikvicu obujma 100 ml prenese alikvotni dio otopine pripravljene u skladu s točkom 5.1.1., doda 10 ml otopine lantanovog klorida (točka 3.11.) i dopuni do oznake klorovodičnom kiselinom 0,5 mol/l (točka 3.3.) (vidjeti također točku 8. „Napomena”).
5.1.2.3.
Slijepa proba mora obuhvaćati sve propisane korake postupka, osim što se izostavlja uzorak. Kao otopina za slijepu probu ne smije se koristiti kalibracijska otopina „0”.
5.1.2.4.
Oksidacijskim plamenom zrak-acetilen, izmjeri se atomska apsorpcija kalibracijskih otopina i otopine koju treba analizirati, na sljedećim valnim duljinama:
|
|
Fe: 248,3 nm |
|
|
Cu: 324,8 nm |
|
|
Mn: 279,5 nm |
|
|
Zn: 213,8 nm |
Svako se mjerenje izvodi četiri puta.
5.2. Mineralna hrana za životinje
Ako uzorak ne sadrži organske tvari, nije potrebno prethodno spaljivanje. Nastavlja se kako je opisano u točki 5.1.1.1. počevši od drugog stavka. Može se izostaviti otparivanje u prisutnosti fluoridne kiseline.
6. Izračun rezultata
Koncentracija elementa u tragovima u otopini koja se analizira izračuna se iz kalibracijske krivulje, a rezultat se iskazuje u miligramima elementa u tragovima na kilogram uzorka (ppm).
7. Ponovljivost
Razlika između rezultata dvaju usporednih postupaka određivanja koje na istom uzorku izvodi isti analitičar ne smije prijeći:
|
— |
5 mg/kg apsolutne vrijednosti za udio dotičnog elementa u tragovima do 50 mg/kg, |
|
— |
10 % višeg rezultata za udio dotičnog elementa u tragovima od 50 – 100 mg/kg, |
|
— |
10 mg/kg apsolutne vrijednosti za udio dotičnog elementa u tragovima od 100 – 200 mg/kg, |
|
— |
5 % višeg rezultata za udio dotičnog elementa u tragovima iznad 200 mg/kg. |
8. Napomena
Prisutnost velike količine fosfata može ometati postupak određivanje željeza, mangana i cinka. Takve se smetnje moraju korigirati dodavanjem otopine lantanovog klorida (točka 3.11.). Ako je, međutim, maseni omjer u uzorku (Ca + Mg/P) > 2, može se izostaviti dodavanje otopine lantanovog klorida (točka 3.11.) u otopinu za analizu i u kalibracijsku otopinu.
D. ODREĐIVANJE HALOFUGINONA
DL-trans-7-brom-6-klor-3-[3-(3-hidroksi-2-piperidil)acetonil]-kinazolin-4-(3H)-on hidrobromid
1. Svrha i područje primjene
Ovom se metodom omogućuje određivanje razine halofuginona u hrani za životinje. Granica kvantifikacije je 1 mg/kg.
2. Načelo
Nakon obrade vrućom vodom halofuginon se ekstrahira u obliku slobodne baze u etil acetatu, zatim razdvoji kao hidroklorid u vodenoj otopini kiseline. Ekstrakt se pročisti ionsko-izmjenjivačkom kromatografijom. Udio halofuginona određuje se tekućinskom kromatografijom visoke učinkovitosti (HPLC) reverznih faza uz upotrebu UV detektora.
3. Reagensi
|
3.1. |
Acetonitril, za HPLC. |
|
3.2. |
Smola Amberlite XAD-2. |
|
3.3. |
Amonijev acetat. |
|
3.4. |
Etil acetat. |
|
3.5. |
Ledena octena kiselina. |
|
3.6. |
Standardna tvar halofuginon (DL-trans-7-brom-6-klor-3-[3-hidroksi-2-piperidil) acetonil]-kinazolin-4-(3H)-on hidrobromid, E 764). |
|
3.6.1. |
Odvagne se 50 mg halofuginona (točka 3.6.) s preciznošću od 0,1 mg i prenese u graduiranu tikvicu obujma 500 ml, otopi u puferskoj otopini amonijevog acetata (točka 3.18.), dopuni puferskom otopinom do oznake i promiješa. Ta je otopina stabilna tri tjedna na na temperaturi od 5 °C ako se pohrani u tamnom prostoru. |
|
3.6.2. |
U seriju graduiranih tikvica obujma 100 ml prenese se 1,0, 2,0, 3,0, 4,0 i 6,0 ml osnovne standardne otopine (točka 3.6.1.). Dopuni se do oznake mobilnom fazom (točka 3.21.) i promiješa. U tim otopinama koncentracija halofuginona iznosi 1,0, 2,0, 3,0, 4,0 i 6,0 μg/ml. Ove se otopine moraju pripremiti svježe prije uporabe. |
|
3.7. |
Klorovodična kiselina (ρ20 približno 1,16 g/ml). |
|
3.8. |
Metanol. |
|
3.9. |
Srebrov nitrat. |
|
3.10. |
Natrijev askorbat. |
|
3.11. |
Natrijev karbonat. |
|
3.12. |
Natrijev klorid. |
|
3.13. |
EDTA (etilendiamintetraoctena kiselina, dinatrijeva sol). |
|
3.14. |
Voda, za HPLC. |
|
3.15. |
Otopina natrijevog karbonata, c = 10 g/100 ml. |
|
3.16. |
Otopina natrijevog karbonata zasićena natrijevim kloridom, c = 5 g/100 ml. U vodi se otopi 50 g natrijevog karbonata (točka 3.11.), razrijedi do 1 litre, zatim se dodaje natrijev klorid (točka 3.12.) do zasićenja otopine. |
|
3.17. |
Klorovodična kiselina, približno 0,1 mol/l. Vodom se razrijedi 10 ml HCl (točka 3.7.) do 1 litre. |
|
3.18. |
Puferska otopina amonijevog acetata, približno 0,25 mol/l. U vodi (točka 3.14) se otopi 19,3 g amonijevog acetata (točka 3.3.) i 30 ml octene kiseline (točka 3.5.) i razrijedi do 1 litre. |
|
3.19. |
Priprema smole Amberlite XAD-2. Vodom se ispere primjerena količina Amberlita (točka 3.2.) dok se ne uklone svi kloridni ioni, što se pokazuje pokusom sa srebrovim nitratom (točka 3.20.) u odbačenoj vodenoj fazi. Zatim se smola ispere s 50 ml metanola (točka 3.8.), metanol se odbaci, a smola pohrani u svježem metanolu. |
|
3.20. |
Otopina srebrovog nitrata, približno 0,1 mol/l. Otopi se 0,17 g srebrovog nitrata (točka 3.9.) u 10 ml vode. |
|
3.2.1. |
Mobilna faza HPLC-a. Pomiješa se 500 ml acetonitrila (točka 3.1.) s 300 ml puferske otopine amonijevog acetata (točka 3.18.) i 1 200 ml vode (točka 3.14.). Octenom kiselinom (točka 3.5.) se vrijednost pH podesi na 4,3. Filtrira se kroz filtar 0,22 μm (točka 4.8.), otopina se otplini (npr. 10 minuta u ultrazvučnoj kupelji). Ta je otopina stabilna mjesec dana ako se drži u zatvorenoj posudi u tamnom prostoru. |
4. Oprema
|
4.1. |
Ultrazvučna kupelj |
|
4.2. |
Rotacijski otparivač |
|
4.3. |
Centrifuga |
|
4.4. |
HPLC oprema s ultraljubičastim detektorom s mogućnošću promjene valne duljine ili detektorom s nizom dioda |
|
4.4.1. |
Kolona za tekućinsku kromatografiju, 300 mm × 4 mm, C18, veličina čestica punjenja 10 μm ili jednakovrijedna kolona |
|
4.5. |
Staklena kolona (300 mm × 10 mm) s filtrom od sinteriranog stakla i pipcem |
|
4.6. |
Filtri od staklenih vlakana, promjera 150 mm |
|
4.7. |
Membranski filtri, 0,45 μm |
|
4.8. |
Membranski filtri, 0,22 μm |
5. Postupak
|
Napomena |
: |
Halofuginon kao slobodna baza je nestabilan u bazičnim i etil-acetatnim otopinama. Ne smije se ostaviti u etil acetatu dulje od 30 minuta. |
5.1. Općenito
|
5.1.1. |
Analizom slijepe probe hrane za životinje treba potvrditi da nisu prisutni niti halofuginon niti interferentne tvari. |
|
5.1.2. |
Test iskorištenja izvodi se analizom slijepe probe hrane za životinje kojoj se doda količina halofuginona slična količini u uzorku. Za dobivanje koncentracije 3 mg/kg u 10 g slijepe probe hrane za životinje doda se 300 μl osnovne standardne otopine (točka 3.6.1.), promiješa i pričeka 10 minuta prije početka postupka ekstrakcije (točka 5.2.).
|
5.2. Ekstrakcija
Odvagne se 10 g pripravljenog uzorka s preciznošću od 0,1 g i prenese u epruvetu za centrifugiranje obujma 200 ml, doda se 0,5 g natrijevog askorbata (točka 3.10.), 0,5 g EDTA (točka 3.13.) i 20 ml vode i promiješa. Epruveta se 5 minuta drži u vodenoj kupelji (80 °C). Nakon hlađenja na sobnu temperaturu doda se 20 ml otopine natrijevog karbonata (točka 3.15.) i promiješa. Odmah se doda 100 ml etilacetata (točka 3.4.) i 15 sekundi snažno protrese rukom. Zatim se epruveta 3 minute drži u ultrazvučnoj kupelji (točka 4.1.) i otpusti slavina. Centrifugira se 2 minute, te se faza etilacetata dekantira kroz filtar od staklenih vlakana (točka 4.6.) u lijevak za odjeljivanje obujma 500 ml. Ponovi se ekstrakcija uzorka s dodatnih 100 ml etilacetata. Pomiješani se ekstrakti ispiru 1 minutu s 50 ml otopine natrijevog karbonata zasićene natrijevim kloridom (točka 3.16.), a vodeni se sloj odbaci.
Organski se sloj ekstrahira 1 minutu s 50 ml klorovodične kiseline (točka 3.17.). Donji se kiselinski sloj prenese u lijevak za odjeljivanje obujma 250 ml. Organski se sloj ponovo ekstrahira 1,5 minutu s dodatnih 50 ml klorovodične kiseline i pomiješa s prvim ekstraktom. Pomiješani se kiselinski ekstrakti približno 10 sekunda ispiru protresivanjem s 10 ml etilacetata (točka 3.4.).
Vodeni se sloj kvantitativno prenese u tikvicu s okruglim dnom obujma 250 ml, a organska faza se odbaci. Iz kisele se otopine u rotacijskom otparivaču (točka 4.2.) otpari sav preostali etilacetat. Temperatura vodene kupelji ne smije prijeći 40 °C. Pod vakuumom na približno 25 milibara, odstrani se sav preostali etilacetat u 5 minuta na 38 °C.
5.3. Pročišćivanje
5.3.1.
Pripremi se kolona XAD-2 za svaki ekstrakt uzorka. Metanolom (točka 3.8.) se prenese 10 g pripravljenog Amberlita (točka 3.19.) u staklenu kolonu (točka 4.5.). Na vrh posteljice od smole postavi se tampon od staklene vune. Metanol se ispusti iz kolone, a smola ispere sa 100 ml vode tako da se dotok vode zaustavi kada tekućina dosegne vrh posteljice od smole. Prije upotrebe kolona se ostavi 10 minuta kako bi se uravnotežila. Kolona se nikad ne smije isušiti.
5.3.2.
Ekstrakt (točka 5.2.) se kvantitativno prenese na vrh pripravljene kolone s Amberlitom (točka 5.3.1.) i eluira, a eluat se odbaci. Brzina eluiranja ne smije biti veća od 20 ml/min. Tikvica s okruglim dnom se ispere s 20 ml klorovodične kiseline (točka 3.17.), a udio iskoristi za ispiranje kolone sa smolom. Preostala se kiselinska otopina ukloni propuhivanjem zraka. Odbaci se tekućina od ispiranja. U kolonu se doda 100 ml metanola (točka 3.8.), 5 – 10 ml se eluira, a eluat se prikupi u tikvicu s okruglim dnom obujma 250 ml. Preostali se metanol ostavi 10 minuta kako bi se uravnotežio sa smolom te se nastavi eluiranje brzinom koja ne prelazi 20 ml/min, a eluat se prikuplja u istoj tikvici s okruglim dnom. Metanol se otpari na rotacijskom otparivaču (točka 4.2.), pri čemu temperatura vodene kupelji ne smije prijeći 40 °C. Ostatak se upotrebom mobilne faze (točka 3.21.) kvantitativno prenese u graduiranu tikvicu obujma 10 ml. Dopuni se mobilnom fazom do oznake i promiješa. Alikvot se filtrira kroz membranski filtar (točka 4.7.). Ta se otopina pohrani za određivanje putem HPLC-a (točka 5.4.).
5.4. Određivanje putem HPLC-a
5.4.1.
Sljedeći se uvjeti predlažu kao smjernice; mogu se koristiti i drugi uvjeti ako se njima jamče jednakovrijedni rezultati.
Kolona za tekućinsku kromatografiju (točka 4.4.1.)
Mobilna faza HPLC-a (točka 3.21.)
Brzina protoka: 1,5 – 2 ml/min
Valna duljina detekcije: 243 nm
Volumen za ubrizgavanje: 40 do 100 μl
Provjeri se stabilnost kromatografskog sustava višekratnim ubrizgavanjem kalibracijske otopine (točka 3.6.2) koncentracije 3,0 μg/ml, sve dok se ne dobiju konstantne visine (ili površine) vršaka i konstantna vremena retencije.
5.4.2.
Svaka se kalibracijska otopina (točka 3.6.2.) ubrizga više puta i za svaku se koncentraciju izmjere srednje vrijednosti visina (površina) vršaka. Kalibracijska krivulja se pripremi tako da se na ordinatu nanose srednje vrijednosti visina ili površina vršaka kalibracijskih otopina, a na apscisu odgovarajuće koncentracije u μg/ml.
5.4.3.
Više puta se ubrizga ekstrakt uzorka (točka 5.3.2.), pri čemu se koriste jednaki volumeni kao za kalibracijsku otopinu, a zatim se odredi srednja vrijednost visina (površina) vršaka halofuginona.
6. Izračun rezultata
Koncentracija otopine uzorka u μg/ml određuje se iz srednjih vrijednosti visina (površina) vršaka halofuginona u otopini uzorka iz kalibracijske krivulje (točka 5.4.2.).
Udio halofuginona (w) (mg/kg) u uzorku izračunava se sljedećom formulom:

pri čemu je:
|
c |
= |
koncentracija halofuginona u otopini uzorka u μg/ml, |
|
m |
= |
masa pokusnog uzorka u gramima. |
7. Vrednovanje rezultata
7.1. Identifikacija
Identifikacija analita može se potvrditi ko-kromatografijom ili upotrebom detektora s nizom dioda kojim se uspoređuje spektar ekstrakta uzorka i spektar kalibracijske otopine (točka 3.6.2.) koncentracije 6,0 μg/ml.
7.1.1.
U ekstrakt uzorka doda se primjerena količina kalibracijske otopine (točka 3.6.2.). Količina dodanog halofuginona mora biti slična procijenjenoj količini halofuginona u ekstraktu uzorka.
Dopušteno je povećanje samo visine vrška halofuginona uzimajući u obzir dodane količine i razrjeđenje ekstrakta. Širina vrška na polovici maksimalne visine mora biti unutar ± 10 % početne širine.
7.1.2.
Rezultati se ocjenjuju u skladu sa sljedećim mjerilima:
|
(a) |
valne duljine maksimalne apsorpcije spektra uzorka i spektra standarda, zabilježene na najvišoj točki vršaka kromatograma, moraju biti jednake unutar granice određene razlučivošću sustava za detekciju. Kod detektora s nizom dioda ta se vrijednost obično nalazi unutar ± 2 nm; |
|
(b) |
između 225 i 300 nm, spektar uzorka i spektar standarda, zabilježeni na najvišoj točki kromatograma, ne smiju se razlikovati za dijelove spektra unutar područja 10 – 100 % relativne apsorbance. To je mjerilo ispunjeno kada su prisutne jednake najviše vrijednosti i kada odstupanje između dva spektra ni na jednoj točki ne prelazi 15 % apsorbance standardnog analita; |
|
(c) |
između 225 i 300 nm, spektri krivulje rasta, najviše točke vrška i krivulje pada dobiveni iz ekstrakta uzorka, ne smiju se međusobno razlikovati za dijelove spektra unutar područja 10 – 100 % relativne apsorbance. To je mjerilo ispunjeno kada su prisutne jednake najviše vrijednosti i kada odstupanje između dva spektra ni na jednoj točki ne prelazi 15 % apsorbance spektra najviše točke. |
Ako nije ispunjeno jedno od navedenih mjerila, prisutnost analita nije potvrđena.
7.2. Ponovljivost
Razlika između rezultata dvaju usporednih postupaka određivanja, izvedenih na istom uzorku, ne smije prijeći 0,5 mg/kg za udio halofuginona do 3 mg/kg.
7.3. Iskorištenje
Za obogaćeni slijepi uzorak hrane za životinje iskorištenje mora iznositi najmanje 80 %.
8. Rezultati međulaboratorijske studije
Provedena je međulaboratorijska studija (5) u kojoj se u osam laboratorija analiziralo tri uzorka.
Rezultati
|
|
Uzorak A (slijepi) kod prijama |
Uzorak B (brašno) |
Uzorak C (pelete) |
||
|
|
|
Kod prijama |
Nakon 2 mjeseca |
Kod prijama |
Nakon 2 mjeseca |
|
Srednja vrijednost [mg/kg] |
ND |
2,80 |
2,42 |
2,89 |
2,45 |
|
SR [mg/kg] |
— |
0,45 |
0,43 |
0,40 |
0,42 |
|
CVR [%] |
— |
16 |
18 |
14 |
17 |
|
Rec. [%] |
|
86 |
74 |
88 |
75 |
|
ND |
= |
nije pronađeno |
|
SR |
= |
standardna devijacija obnovljivosti |
|
CVR |
= |
koeficijent varijacije obnovljivosti (%) |
|
Rec. |
= |
iskorištenje (%). |
E. ODREĐIVANJE ROBENIDINA
1,3-bis [(4-klorbenziliden)amino]gvanidin – hidroklorid
1. Svrha i područje primjene
Ovom se metodom omogućuje određivanje razine robenidina u hrani za životinje. Granica kvantifikacije je 5 mg/kg.
2. Načelo
Uzorak se ekstrahira zakiseljenim metanolom. Ekstrakt se osuši i alikvotni dio pročisti na koloni aluminijevog oksida. Robenidin se eluira metanolom iz kolone, koncentrira i dopuni mobilnom fazom do prikladnog obujma. Udio robenidina se određuje tekućinskom kromatografijom visoke učinkovitosti (HPLC) s reverznim fazama uz upotrebu UV detektora.
3. Reagensi
|
3.1. |
Metanol. |
|
3.2. |
Zakiseljeni metanol. U graduiranu tikvicu obujma 500 ml prenese se 4,0 ml klorovodične kiseline (ρ20 = 1,18 g/ml), dopuni metanolom (točka 3.1.) do oznake i promiješa. Ta se otopina mora pripremiti svježa prije uporabe. |
|
3.3. |
Acetonitril, za HPLC. |
|
3.4. |
Molekularno sito. Tip 3A, očice mreže od 8 do 12 (očice promjera 1,6 – 2,5 mm, kristalni aluminijev silikat, promjer pora 0,3 mm). |
|
3.5. |
Aluminijev oksid kiselinske aktivnosti I. stupnja, za kolonsku kromatografiju. U prikladnu se posudu prenese 100 g aluminijevog oksida i doda 2,0 ml vode. Začepi se i trese približno 20 minuta. Drži se u čvrsto zatvorenoj posudi. |
|
3.6. |
Otopina kalijevog dihidrogenfosfata, c = 0,025 mol/l. U vodi se otopi 3,40 g kalijevog dihidrogenfosfata (za HPLC) u odmjernoj tikvici obujma 1 000 ml, dopuni do oznake i promiješa. |
|
3.7. |
Otopina dinatrijevog hidrogenfosfata, c = 0,025 mol/l. U vodi se otopi 3,55 g bezvodnog dinatrijevog hidrogenfosfata (ili 4,45 g dihidrata ili 8,95 g dodekahidrata) (za HPLC) u odmjernoj tikvici obujma 1 000 ml, dopuni do oznake i promiješa. |
|
3.8. |
Mobilna faza HPLC-a. Pomiješaju se sljedeći reagensi:
Filtrira se kroz filtar 0,22 μm (točka 4.6.), otopina se zatim otplini (npr. 10 minuta u ultrazvučnoj kupelji). |
|
3.9. |
Standardna tvar. Čisti robenidin: 1,3-bis[4-klorbenziliden)amino]gvanidin – hidroklorid. |
|
3.9.1. |
Odvagne se 30 mg standardnog robenidina (točka 3.9.) s preciznošću od 0,1 mg. Otopi se u zakiseljenom metanolu (točka 3.2.) u odmjernoj tikvici obujma 100 ml, dopuni istim otapalom do oznake i promiješa. Tikvica se omota aluminijskom folijom i pohrani na tamno mjesto. |
|
3.9.2. |
U graduiranu tikvicu obujma 250 ml prenese se 10,0 ml osnovne standardne otopine robenidina (točka 3.9.1.), dopuni mobilnom fazom (točka 3.8.) do oznake i promiješa. Tikvica se omota aluminijskom folijom i pohrani na tamno mjesto. |
|
3.9.3. |
U seriju graduiranih tikvica obujma 50 ml prenese se 5,0, 10,0, 15,0, 20,0 i 25,0 ml intermedijarne standardne otopine (točka 3.9.2.). Dopuni se mobilnom fazom (točka 3.8.) do oznake i promiješa. Te otopine odgovaraju koncentracijama robenidina od 1,2, 2,4, 3,6, 4,8 i 6,0 μg/ml. Te se otopine moraju pripremiti svježe prije uporabe. |
|
3.10. |
Voda za HPLC. |
4. Oprema
|
4.1. |
Staklena kolona. Od smeđeg stakla, s pipcem i spremnikom obujma približno 150 ml, unutarnjeg promjera 10 – 15 mm, dužine 250 mm |
|
4.2. |
Mehanička tresilica ili magnetska mješalica. |
|
4.3. |
Tankoslojni rotacijski otparivač. |
|
4.4. |
Oprema za HPLC s ultraljubičastim detektorom s mogućnošću promjene valne duljine ili detektorom s nizom dioda s radnim područjem od 250 – 400 nm. |
|
4.4.1. |
Kolona za tekućinsku kromatografiju: 300 mm × 4 mm, C18, s veličinom čestica punjenja 10 μm ili jednakovrijednim. |
|
4.5. |
Filtar papir od staklenih vlakana (Whatman GF/A ili jednakovrijedan). |
|
4.6. |
Membranski filtri, 0,22 μm. |
|
4.7. |
Membranski filtri, 0,45 μm. |
5. Postupak
|
Napomena |
: |
Robenidin je osjetljiv na svjetlo. U svim se postupcima mora koristiti posuđe od smeđeg stakla. |
5.1. Općenito
|
5.1.1. |
Analizom slijepe probe hrane za životinje treba potvrditi da nije prisutan niti robenidin niti interferentne tvari. |
|
5.1.2. |
Test iskorištenja izvodi se analizom slijepe probe hrane za životinje (točka 5.1.1.) kojoj se doda količina robenidina slična količini u uzorku. Za dobivanje koncentracije 60 mg/kg prenese se 3,0 ml osnovne standardne otopine (točka 3.9.1.) u konusnu tikvicu obujma 250 ml. Otopina se otpari u struji dušika do približno 0,5 ml. Doda se 15 g slijepe probe hrane za životinje, promiješa, ostavi 10 minuta i nastavi s ekstrakcijom (točka 5.2.).
|
5.2. Ekstrakcija
Odvagne se približno 15 g uzorka s preciznošću od 0,01 g. Prenese se u konusnu tikvicu obujma 250 ml, doda 100,0 ml zakiseljenog metanola (točka 3.2.), začepi i trese jedan sat u tresilici (točka 4.2.). Otopina se filtrira kroz filtar papir od staklenih vlakana (točka 4.5.) u koničnu tikvicu obujma 150 ml. Doda se 7,5 g molekularnog sita (točka 3.4.), začepi i trese pet minuta. Odmah se filtrira kroz filtar papir od staklenih vlakana. Ova se otopina pohrani za fazu pročišćivanja (točka 5.3.).
5.3. Pročišćivanje
5.3.1.
U donji dio staklene kolone (točka 4.1.) postavi se mali čep od staklene vune i nabije staklenim štapićem. Odvagne se 11,0 g pripravljenog aluminijevog oksida (točka 3.5.) i prenese u kolonu. Pri tom treba paziti da izloženost zraku tijekom navedenog postupka bude što je moguće manja. Laganim udarcima po donjem dijelu napunjene kolone istaloži se aluminijev oksid.
5.3.2.
U kolonu se pipetom prenese 5,0 ml ekstrakta uzorka pripravljenog u (točka 5.2.). Vrh pipete se nasloni na stijenku kolone, zatim se pusti da aluminijev oksid apsorbira otopinu. Robenidin se eluira iz kolone sa 100 ml metanola (točka 3.1.) uz brzinu protoka od 2 – 3 ml/min, a eluat se prikupi u tikvicu s okruglim dnom obujma 250 ml. Otopina metanola se otpari u rotacijskom otparivaču (točka 4.3.) do suhog pri sniženom tlaku, na 40 °C. Ostatak se ponovo otopi u 3 – 4 ml mobilne faze (točka 3.8.) i prenese u graduiranu tikvicu obujma 10 ml. Tikvica se nekoliko puta ispere s obrocima od 1 – 2 ml mobilne faze, a isprana tekućina prenese u graduiranu tikvicu. Dopuni se istim otapalom do oznake i promiješa. Alikvot se filtrira kroz membranski filtar 0,45 μm (točka 4.7.). Ova se otopina pohrani za određivanje putem HPLC-a (točka 5.4.).
5.4. Određivanje putem HPLC-a
5.4.1
Sljedeći se uvjeti predlažu kao smjernice; mogu se koristiti i drugi uvjeti ako se njima jamče jednakovrijedni rezultati:
kolona za tekućinsku kromatografiju (točka 4.4.1.),
Mobilna faza HPLC (točka 3.8.),
brzina protoka: 1,5 – 2 ml/min,
valna duljina detekcije: 317 nm,
ubrizgani volumen: 20 – 50 μl.
Stabilnost kromatografskog sustava se provjerava višekratnim ubrizgavanjem kalibracijske otopine (točka 3.9.3.) koncentracije 3,6 μg/ml, sve dok se ne dobiju konstantne visine vršaka i konstantna vremena retencije.
5.4.2.
Više puta se ubrizga svaka kalibracijska otopina (točka 3.9.3.) i za svaku koncentraciju izmjere vrijednosti visina (površina) vršaka. Kalibracijska se krivulja pripremi tako da se na ordinatu nanose srednje vrijednosti visina ili površina vršaka kalibracijskih otopina, a na apscisu odgovarajuće koncentracije u μg/ml.
5.4.3.
Više puta se ubrizga ekstrakt uzorka (točka 5.3.2.), pri čemu se koriste jednaki volumeni kao za kalibracijske otopine, zatim se odredi srednja vrijednost visina (površina) vršaka robenidina.
6. Izračun rezultata
Koncentracija otopine uzorka u μg/ml određuje se iz srednje vrijednosti visina (površina) vršaka robenidina u otopini uzorka iz kalibracijske krivulje (točka 5.4.2.).
Udio robenidina (w) (mg/kg) u uzorku izračunava se sljedećom formulom:

pri čemu je:
|
c |
= |
koncentracija robenidina u otopini uzorka u μg/ml, |
|
m |
= |
masa pokusnog uzorka u gramima. |
7. Vrednovanje rezultata
7.1. Identifikacija
Identifikacija analita može se potvrditi ko-kromatografijom ili detektorom s nizom dioda kojim se uspoređuje spektar ekstrakta uzorka i spektar kalibracijske otopine (točka 3.9.3.) koncentracije 6 μg/ml.
7.1.1.
U ekstrakt uzorka doda se primjerena količina kalibracijske otopine (točka 3.9.3.). Količina dodanog robenidina mora biti slična procijenjenoj količini robenidina u ekstraktu uzorka.
Dopušteno je povećanje samo visine vrška robenidina uzimajući u obzir dodane količine i razrjeđenje ekstrakta. Širina vrška na polovici maksimalne visine mora biti unutar približno 10 % početne širine.
7.1.2.
Rezultati se ocjenjuju u skladu sa sljedećim mjerilima:
|
(a) |
valne duljine maksimalne apsorpcije spektra uzorka i spektra standarda, zabilježene na najvišoj točki vršaka kromatograma, moraju biti jednake unutar granice određene razlučivošću sustava za detekciju. Kod detektora s nizom dioda ta se vrijednost obično nalazi unutar približno 2 nm; |
|
(b) |
između 250 i 400 nm, spektar uzorka i spektar standarda, zabilježeni na najvišoj točki kromatograma, ne smiju se razlikovati za dijelove spektra unutar područja 10 – 100 % relativne apsorbance. To je mjerilo ispunjeno kada su prisutne jednake najviše vrijednosti i kada odstupanje između dva spektra ni na jednoj točki ne prelazi 15 % apsorbance standardnog analita; |
|
(c) |
između 250 i 400 nm, spektri krivulje rasta, najviše točke vrška i krivulje pada dobiveni iz ekstrakta uzorka, ne smiju se međusobno razlikovati za dijelove spektra unutar područja 10 – 100 % relativne apsorbance. To je mjerilo ispunjeno kada su prisutne jednake najviše vrijednosti i kada odstupanje između dva spektra ni na jednoj točki ne prelazi 15 % apsorbance spektra najviše točke. |
Ako nije ispunjeno jedno od navedenih mjerila, prisutnost analita nije potvrđena.
7.2. Ponovljivost
Razlika između rezultata dvaju usporednih postupaka određivanja, izvedenih na istom uzorku, ne smije prijeći 10 % vrijednosti višeg rezultata za udio robenidina viši od 15 mg/kg.
7.3. Iskorištenje
Za obogaćeni slijepi uzorak hrane za životinje iskorištenje mora iznositi najmanje 85 %.
8. Rezultati međulaboratorijske studije
Provedena je međulaboratorijska studija na razini EZ-a u kojoj se u 12 laboratorija analiziralo četiri uzorka hrane za perad i kuniće u obliku brašna i peleta. Svaki se uzorak analizirao dvaput. Rezultati su prikazani u donjoj tablici:
|
|
Hrana za perad |
Hrana za kuniće |
||
|
|
Brašno |
Pelete |
Brašno |
Pelete |
|
Srednja vrijednost [mg/kg] |
27,00 |
27,99 |
43,6 |
40,1 |
|
sr [mg/kg] |
1,46 |
1,26 |
1,44 |
1,66 |
|
CVr [%] |
5,4 |
4,5 |
3,3 |
4,1 |
|
SR [mg/kg] |
4,36 |
3,36 |
4,61 |
3,91 |
|
CVR [%] |
16,1 |
12,0 |
10,6 |
9,7 |
|
Iskorištenje [%] |
90,0 |
93,3 |
87,2 |
80,2 |
|
sr |
= |
standardna devijacija ponovljivosti, |
|
CVr |
= |
koeficijent varijacije ponovljivosti u %, |
|
SR |
= |
standardna devijacija obnovljivosti, |
|
CVR |
= |
koeficijent varijacije obnovljivosti u %. |
F. ODREĐIVANJE DIKLAZURILA
(+)-4-klorfenil [2,6-diklor-4-(2,3,4,5-tetrahidro-3,5-diokso-1,2,4-triazin-2-il)fenil] acetonitril
1. Svrha i područje primjene
Ovom se metodom omogućuje određivanje razine diklazurila u hrani za životinje i premiksima. Granica detekcije iznosi 0,1 mg/kg, a granica kvantifikacije 0,5 mg/kg.
2. Načelo
Nakon dodavanja internog standarda, uzorak se ekstrahira zakiseljenim metanolom. Kod hrane za životinje alikvotni se dio ekstrakta pročisti na kartuši C18 za ekstrakciju u krutoj fazi. Diklazuril se eluira iz kartuše smjesom zakiseljenog metanola i vode. Nakon otparivanja, ostatak se otopi u smjesi DMF/voda. Kod premiksa, ekstrakt se otpari, a ostatak otopi u smjesi DMF/voda. Udio diklazurila određuje se tekućinskom kromatografijom visoke učinkovitosti (HPLC) s reverznom fazom i ternarnim gradijentom uz upotrebu UV detektora.
3. Reagensi
|
3.1. |
Voda, za HPLC. |
|
3.2. |
Amonijev acetat. |
|
3.3. |
Tetrabutilamonijev hidrogen sulfat (TBHS). |
|
3.4. |
Acetonitril, za HPLC. |
|
3.5. |
Metanol, za HPLC. |
|
3.6. |
N,N-dimetilformamid (DMF). |
|
3.7. |
Klorovodična kiselina, ρ20 = 1,19 g/ml. |
|
3.8. |
Standardna tvar: Diklazuril II-24: (+)-4-klorfenil [2,6-diklor-4-(2,3,4,5-tetrahidro-3,5-diokso-1,2,4-triazin-2-il)fenil]acetonitril, zajamčene čistoće, E771. |
|
3.8.1. |
Odvagne se 25 mg standardne tvari diklazurila (točka 3.8.) s preciznošću od 0,1 mg i prenese u graduiranu tikvicu obujma 50 ml. Otopi se u DMF-u (točka 3.6.), tikvica se dopuni DMF-om (točka 3.6.) do oznake i promiješa. Tikvica se omota aluminijskom folijom ili se koristi tikvica od smeđeg stakla i pohrani u hladnjaku. Pri temperaturi ≤ 4 °C otopina je stabilna 1 mjesec. |
|
3.8.2. |
U graduiranu tikvicu obujma 50 ml prenese se 5,00 ml osnovne standardne otopine (točka 3.8.1.), dopuni DMF-om (točka 3.6.) do oznake i promiješa. Tikvica se omota aluminijskom folijom ili se koristi tikvica od smeđeg stakla i pohrani u hladnjaku. Na temperaturi ≤ 4 °C otopina je stabilna 1 mjesec. |
|
3.9. |
Interni standard: 2,6 diklor-α-(4-klorfenil)-4-(4,5 dihidro-3,5-diokso-1,2,4-triazin-2 (3H)– il) α-metilbenzen-acetonitril. |
|
3.9.1. |
Odvagne se 25 mg internog standarda (točka 3.9.) s preciznošću od 0,1 mg i prenese u graduiranu tikvicu obujma 50 ml. Otopi se u DMF-u (točka 3.6.), tikvica se dopuni DMF-om (točka 3.6.) do oznake i promiješa. Tikvica se omota aluminijskom folijom ili se koristi tikvica od smeđeg stakla i pohrani u hladnjaku. Na temperaturi ≤ 4 °C otopina je stabilna 1 mjesec. |
|
3.9.2. |
U graduiranu tikvicu obujma 50 ml prenese se 5,00 ml osnovne otopine internog standarda (točka 3.9.1.), dopuni DMF-om (točka 3.6.) do oznake i promiješa. Tikvica se omota aluminijskom folijom ili se koristi tikvica od smeđeg stakla i pohrani u hladnjaku. Na temperaturi ≤ 4 °C otopina je stabilna 1 mjesec. |
|
3.9.3. |
(p = nominalni udio diklazurila u premiksu, u mg/kg) Odvagne se p/10 mg internog standarda s preciznošću od 0,1 mg i prenese u graduiranu tikvicu obujma 100 ml, otopi u DMF-u (točka 3.6.) u ultrazvučnoj kupelji (točka 4.6.), dopuni DMF-om do oznake i promiješa. Tikvica se omota aluminijskom folijom ili se koristi tikvica od smeđeg stakla i pohrani u hladnjaku. Na temperaturi ≤ 4 °C otopina je stabilna 1 mjesec. |
|
3.10. |
Kalibracijska otopina, 2 μg/ml. Pipetom se prenese 2,00 ml standardne otopine diklazurila (točka 3.8.2.) i 2,00 ml otopine internog standarda (točka 3.9.2.) u graduiranu tikvicu obujma 50 ml. Doda se 16 ml DMF (točka 3.6.), tikvica se dopuni vodom do oznake i promiješa. Ova se otopina mora pripremiti svježa prije uporabe. |
|
3.11. |
Kartuša C18 za ekstrakciju u krutoj fazi, npr. Bond Elut, veličina: 1 cc, masa apsorbenta: 100 mg. |
|
3.12. |
Ekstrakcijsko otapalo: zakiseljeni metanol. Pipetom se prenese 5,0 ml klorovodične kiseline (točka 3.7.) u 1 000 ml metanola (točka 3.5.) i promiješa. |
|
3.13. |
Mobilna faza za HPLC. |
|
3.13.1. |
Eluent A: otopina amonijevog acetata i tetrabutilamonijevog hidrogen sulfata. Otopi se 5 g amonijevog acetata (točka 3.2.) i 3,4 g TBHS-a (točka 3.3.) u 1 000 ml vode (točka 3.1.) i promiješa. |
|
3.13.2. |
Eluent B: acetonitril (točka 3.4.). |
|
3.13.3. |
Eluent C: metanol (točka 3.5.). |
4. Oprema
|
4.1. |
Mehanička tresilica |
|
4.2. |
Oprema za HPLC s ternarnim gradijentom |
|
4.2.1. |
Kolona za tekućinsku kromatografiju, Hypersil ODS, veličina čestica punjenja 3 μm, 100 mm × 4,6 mm ili jednakovrijedna |
|
4.2.2. |
UV detektor s mogućnošću promjene valne duljine ili detektor s nizom dioda. |
|
4.3. |
Tankoslojni rotacijski otparivač |
|
4.4. |
Membranski filtar, 0,45 μm |
|
4.5. |
Vakuumski ventil |
|
4.6. |
Ultrazvučna kupelj |
5. Postupak
5.1. Općenito
5.1.1.
Analizom slijepe probe hrane za životinje treba potvrditi da nije prisutan niti diklazuril niti interferentne tvari. Slijepa proba hrane za životinje mora biti slična uzorku, a analizom se mora potvrditi odsutnost diklazurila ili interferentnih tvari.
5.1.2.
Test iskorištenja izvodi se analizom slijepe probe hrane za životinje kojoj se doda količina diklazurila slična količini u uzorku. Za dobivanje koncentracije 1 mg/kg doda se 0,1 ml osnovne standardne otopine (točka 3.8.1.) u 50 g slijepe probe hrane za životinje, temeljito promiješa i ostavi 10 minuta, a prije nastavka (točka 5.2.) se miješanje više puta ponovi.
Ako nije dostupna slijepa probe hrane za životinje slična uzorku (vidjeti točku 5.1.1.), test iskorištenja može se provesti metodom dodavanja standarda. U tom se slučaju uzorku za analizu doda količina diklazurila slična onoj koja se već nalazi u uzorku. Taj se uzorak analizira zajedno s uzorkom bez dodanog diklazurila, a iskorištenje se izračuna oduzimanjem.
5.2. Ekstrakcija
5.2.1.
Odvagne se približno 50 g uzorka s preciznošću od 0,01 g. Prenese se u konusnu tikvicu obujma 500 ml, doda 1,00 ml otopine internog standarda (točka 3.9.2.) i 200 ml ekstrakcijskog otapala (točka 3.12.), zatim se tikvica zatvori. Smjesa se tijekom noći protresa u tresilici (točka 4.1.). Ostavi se 10 minuta da se istaloži. U primjerenu staklenu posudu prenese se alikvot 20 ml supernatanta i razrijedi s 20 ml vode. Ta se otopina prenese u ekstrakcijsku kartušu (točka 3.11.) i filtrira pod vakuumom (točka 4.5.). Kartuša se ispere s 25 ml smjese ekstrakcijskog otapala (točka 3.12.) i vode, 65 + 35 (V + V). Prikupljene se frakcije odbace, a spojevi eluiraju s 25 ml smjese ekstrakcijskog otapala (točka 3.12.) i vode, 80 + 20 (V + V). Ova se frakcija isparava u rotacijskom otparivaču (točka 4.3) pri 60 °C dok se ne počne sušiti. Ostatak se otopi u 1,0 ml DMF-a (točka 3.6), doda se 1,5 ml vode (točka 3.1.) i promiješa. Filtrira se kroz membranski filtar (točka 4.4.). Prelazi se na postupak određivanja putem HPLC-a (točka 5.3.).
5.2.2.
Odvagne se približno 1 g uzorka s preciznošću od 0,001 g. Prenese se u konusnu tikvicu obujma 500 ml, doda se 1,00 ml otopine internog standarda (točka 3.9.3.) i 200 ml ekstrakcijskog otapala (točka 3.12.), zatim se tikvica zatvori. Smjesa se tijekom noći protresa u tresilici (točka 4.1.). Ostavi se 10 minuta da se istaloži. U primjereno veliku staklenu tikvicu s okruglim dnom prenese se alikvot (10 000/p ml, p = nominalna udio diklazurila u mg/kg u premiksu) supernatanta. Otparava se pod sniženim tlakom na 60 °C u rotacijskom otparivaču (točka 4.3.) dok se ne počne sušiti. Ostatak se ponovno otopi u 10,0 ml DMF-a (točka 3.6.), doda se 15,0 ml vode (točka 3.1.) i promiješa. Prelazi se na postupak određivanja putem HPLC-a (točka 5.3.).
5.3. Određivanje putem HPLC-a
5.3.1.
Sljedeći se uvjeti predlažu kao smjernice; mogu se koristiti i drugi uvjeti ako se njima jamče jednakovrijedni rezultati.
|
Kolona za tekućinsku kromatografiju (točka 4.2.1.): |
100 mm × 4,6 mm, Hypersil ODS, veličina čestica punjenja 3 μm ili jednakovrijedna |
|
||||||
|
Mobilna faza: |
Eluent A (točka 3.13.1.): |
vodena otopina amonijevog acetata i tetrabutil amonijevog hidrogen sulfata |
||||||
|
|
Eluent B (točka 3.13.2.): |
acetonitril |
||||||
|
|
Eluent C (točka 3.13.3.): |
metanol |
||||||
|
Način eluiranja: |
Ispiranje 10 minuta s B. |
|||||||
|
Brzina protoka: |
1,5 – 2 ml/min |
|
||||||
|
Volumen za ubrizgavanje: |
20 μl |
|
||||||
|
Valna duljina detekcije: |
280 nm |
|
||||||
Stabilnost kromatografskog sustava se provjerava višekratnim ubrizgavanjem kalibracijske otopine koncentracije 2,0 μg/ml (točka 3.10.) sve dok se ne dobiju konstantne visine vršaka i konstantna vremena retencije.
5.3.2.
Više puta se ubrizga kalibracijska otopina (točka 3.10.) i odredi srednja vrijednost visine (površine) vršaka diklazurila i internog standarda.
5.3.3.
Više puta se ubrizga 20 μl kalibracijske otopine (točka 5.2.1. ili 5.2.2.) i odredi srednja vrijednost visine (površine) vršaka diklazurila i internog standarda.
6. Izračun rezultata
6.1. Hrana za životinje
Udio diklazurila (w) (mg/kg) u uzorku izračunava se sljedećom formulom:

pri čemu je:
|
hd,s |
= |
visina (površina) vrška diklazurila u otopini uzorka (točka 5.2.1.) |
|
hi,s |
= |
visina (površina) vrška internog standarda u otopini uzorka (točka 5.2.1.) |
|
hd,c |
= |
visina (površina) vrška diklazurila u kalibracijskoj otopini (točka 3.10.) |
|
hi,c |
= |
visina (površina) vrška internog standarda u kalibracijskoj otopini (točka 3.10.) |
|
cd,c |
= |
koncentracija diklazurila u kalibracijskoj otopini u μg/ml (točka 3.10.) |
|
m |
= |
masa pokusnog uzorka u gramima |
|
V |
= |
volumen ekstrakta uzorka u skladu s točkom 5.2.1 (tj. 2,5 ml). |
6.2. Premiksi
Udio diklazurila (w) (mg/kg) u uzorku izračunava se sljedećom formulom:

pri čemu je:
|
hd,c |
= |
visina (površina) vrška diklazurila u kalibracijskoj otopini (točka 3.10.) |
|
hi,c |
= |
visina (površina) vrška internog standarda u kalibracijskoj otopini (točka 3.10.) |
|
hd,s |
= |
visina (površina) vrška diklazurila u otopini uzorka (točka 5.2.2.) |
|
hi,s |
= |
visina (površina) vrška internog standarda u otopini uzorka (točka 5.2.2.) |
|
cd,c |
= |
koncentracija diklazurila u kalibracijskoj otopini u μg/ml (točka 3.10.) |
|
m |
= |
masa pokusnog uzorka u gramima |
|
V |
= |
volumen ekstrakta uzorka u skladu s točkom 5.2.2 (tj. 25 ml) |
|
p |
= |
nominalna udio diklazurila u mg/kg u premiksu. |
7. Vrednovanje rezultata
7.1. Identifikacija
Identifikacija analita može se potvrditi ko-kromatografijom ili detektorom s nizom dioda kojim se uspoređuje spektar ekstrakta uzorka (točka 5.2.1. ili 5.2.2.) i spektar kalibracijske otopine (točka 3.10.).
7.1.1.
U ekstrakt uzorka (točka 5.2.1. ili 5.2.2.) doda se primjerena količina kalibracijske otopine (točka 3.10.). Količina dodanog diklazurila mora biti slična količini diklazurila određenoj u ekstraktu uzorka.
Dopušteno je povećanje samo visine vršaka diklazurila i internog standarda uzimajući u obzir dodane količine i razrjeđenje ekstrakta. Širina vrška na polovici visine mora biti unutar ± 10 % početne širine vrška diklazurila ili vrška internog standarda ekstrakta uzorka bez dodatka.
7.1.2.
Rezultati se ocjenjuju u skladu sa sljedećim mjerilima:
|
(a) |
Valne duljine maksimalne apsorpcije spektra uzorka i spektra standarda, zabilježene na najvišoj točki vršaka kromatograma, moraju biti jednake unutar granice određene razlučivošću sustava za detekciju. Kod detektora s nizom dioda ta se vrijednost obično nalazi unutar ± 2 nm. |
|
(b) |
Između 230 i 320 nm, spektar uzorka i spektar standarda, zabilježeni na najvišoj točki kromatograma, ne smiju se razlikovati za dijelove spektra unutar područja 10 – 100 % relativne apsorbance. To je mjerilo ispunjeno kada su prisutne jednake najviše vrijednosti i kada odstupanje između dva spektra ni na jednoj točki ne prelazi 15 % apsorbance standardnog analita. |
|
(c) |
Između 230 i 320 nm, spektri krivulje rasta, najviše točke vrška i krivulje pada dobiveni iz ekstrakta uzorka, ne smiju se međusobno razlikovati za dijelove spektra unutar područja 10 – 100 % relativne apsorbance. To je mjerilo ispunjeno kada su prisutne jednake najviše vrijednosti i kada odstupanje između dva spektra ni na jednoj točki ne prelazi 15 % apsorbance spektra najviše točke. |
Ako nije ispunjeno jedno od navedenih mjerila, prisutnost analita nije potvrđena.
7.2. Ponovljivost
Razlika između rezultata dvaju usporednih postupaka određivanja, izvedenih na istom uzorku, ne smije prijeći:
|
— |
30 % relativne vrijednosti u odnosu na najvišu vrijednost za udio diklazurila između 0,5 – 2,5 mg/kg, |
|
— |
0,75 mg/kg za udio diklazurila između 2,5 – 5 mg/kg, |
|
— |
15 % relativne vrijednosti u odnosu za najvišu vrijednost za udio diklazurila veći od 5 mg/kg. |
7.3. Iskorištenje
Za obogaćeni (slijepi) uzorak hrane za životinje iskorištenje mora iznositi najmanje 80 %.
8. Rezultati međulaboratorijske studije
Provedena je međulaboratorijska studija u kojoj se u 11 laboratorija analiziralo 5 uzoraka. Ti su se uzorci sastojali od dva premiksa; jedan je bio pomiješan s organskom matricom (O 100), a drugi s anorganskom matricom (A 100). Teoretski udio iznosi 100 mg diklazurila na kg. Tri različita proizvođača (NL) (L1/Z1/K1) proizvela su 3 mješavine hrane za perad. Teoretski udio iznosi 1 mg diklazurila na kg. Od laboratorija je zatraženo da svaki uzorak analiziraju jedanput ili dvaput. (Detaljnije informacije o navedenoj međulaboratorijskoj studiji mogu se pronaći u Journal of AOAC International, svezak 77, br. 6, 1994., str. 1 359. – 1 361.). Rezultati su prikazani u donjoj tablici.
|
|
Uzorak 1 A 100 |
Uzorak 2 O 100 |
Uzorak 3 L1 |
Uzorak 4 Z1 |
Uzorak 5 K1 |
|
L |
11 |
11 |
11 |
11 |
6 |
|
n |
19 |
18 |
19 |
19 |
12 |
|
Srednja vrijednost |
100,8 |
103,5 |
0,89 |
1,15 |
0,89 |
|
Sr (mg/kg) |
5,88 |
7,64 |
0,15 |
0,02 |
0,03 |
|
CVr (%) |
5,83 |
7,38 |
17,32 |
1,92 |
3,34 |
|
SR (mg/kg) |
7,59 |
7,64 |
0,17 |
0,11 |
0,12 |
|
CVR (%) |
7,53 |
7,38 |
18,61 |
9,67 |
13,65 |
|
Nominalni udio (mg/kg) |
100 |
100 |
1 |
1 |
1 |
|
L |
= |
broj laboratorija |
|
n |
= |
broj pojedinačnih vrijednosti |
|
Sr |
= |
standardna devijacija ponovljivosti |
|
CVr |
= |
koeficijent varijacije ponovljivosti |
|
SR |
= |
standardna devijacija obnovljivosti |
|
CVR |
= |
koeficijent varijacije obnovljivosti. |
9. Napomena
Prethodno treba dokazati da je odziv diklazurila linearan u području koncentracija koje se mjere.
G. ODREĐIVANJE LASALOCID NATRIJA
Natrijeva sol polieter monokarboksilne kiseline koju proizvodi Streptomyces lasaliensis
1. Svrha i područje primjene
Ovom se metodom omogućuje određivanje razine lasalocid natrija u hrani za životinje i premiksima. Granica detekcije iznosi 5 mg/kg, a granica kvantifikacije 10 mg/kg.
2. Načelo
Lasalocid natrij se ekstrahira iz uzorka u zakiseljeni metanol i određuje tekućinskom kromatografijom visoke učinkovitosti (HPLC) s reverznom fazom uz upotrebu spektrofluorimetrijskog detektora.
3. Reagensi
|
3.1. |
Kalijev dihidrogen fosfat (KH2PO4). |
|
3.2. |
Ortofosforna kiselina, m (m/m) = 85 %. |
|
3.3. |
Otopina ortofosforne kiseline, c = 20 %. 23,5 ml ortofosforne kiseline (točka 3.2.) razrijedi se vodom do 100 ml. |
|
3.4. |
6-metil-2-heptilamin (1,5-dimetilheksilamin), m (m/m) = 99 %. |
|
3.5. |
Metanol, za HPLC. |
|
3.6. |
Klorovodična kiselina, gustoća = 1,19 g/ml. |
|
3.7. |
Otopina fosfatnog pufera, c = 0,01 mol/l. Otopi se 1,36 g KH2PO4 (točka 3.1.) u 500 ml vode (točka 3.11.), doda se 3,5 ml ortofosforne kiseline (točka 3.2.) i 10,0 ml 6-metil-2-heptilamina (točka 3.4.). Otopinom ortofosforne kiseline (točka 3.3.) pH vrijednost se podesi na 4,0 i razrijedi vodom (točka 3.11.) do 1 000 ml. |
|
3.8. |
Zakiseljeni metanol. U graduiranu tikvicu obujma 1 000 ml prenese se 5,0 ml klorovodične kiseline (točka 3.6.), dopuni metanolom (točka 3.5.) do oznake i promiješa. Ova se otopina mora pripremiti svježa prije uporabe. |
|
3.9. |
Mobilna faza HPLC-a, otopina fosfatnog pufera i metanola 5 + 95 (V + V). Pomiješa se 5 ml otopine fosfatnog pufera (točka 3.7.) s 95 ml metanola (točka 3.5.). |
|
3.10. |
Lasalocid natrij standardna tvar, zajamčene čistoće, C34H53O8Na (natrijeva sol polieter monokarboksilne kiseline koju proizvodi Streptomyces lasaliensis), E763. |
|
3.10.1. |
Odvagne se 50 mg lasalocid natrija (točka 3.10.) s preciznošću od 0,1 mg i prenese u graduiranu tikvicu obujma 100 ml, otopi u zakiseljenom metanolu (točka 3.8.) i istim otapalom dopuni do oznake i promiješa. Ova se otopina mora pripremiti svježa prije uporabe. |
|
3.10.2. |
Pipetom se prenese 10,0 ml osnovne standardne otopine (točka 3.10.1.) u graduiranu tikvicu obujma 100 ml i zakiseljenim metanolom (točka 3.8.) dopuni do oznake i promiješa. Ova se otopina mora pripremiti svježa prije uporabe. |
|
3.10.3. |
U seriju graduiranih tikvica obujma 50 ml prenese se 1,0, 2,0, 4,0, 5,0 i 10,0 ml intermedijarne standardne otopine (točka 3.10.2.). Zakiseljenim se metanolom dopuni do oznake (točka 3.8.) i promiješa. Ove otopine odgovaraju 1,0, 2,0, 4,0, 5,0 i 10,0 μg lasalocid natrija na ml. Ove se otopine moraju pripremiti svježe prije uporabe. |
|
3.11. |
Voda, za HPLC |
4. Oprema
|
4.1. |
Ultrazvučna kupelj (ili vodena kupelj s protresanjem) s nadzorom temperature |
|
4.2. |
Membranski filtri, 0,45 μm |
|
4.3. |
Oprema za tekućinsku kromatografiju visoke učinkovitosti (HPLC) sa sustavom za ubrizgavanje primjerenim za ubrizgavanje volumena od 20 μl |
|
4.3.1. |
Kolona za tekućinsku kromatografiju 125 mm × 4 mm, reverzna faza C18, veličina čestica punjenja 5 μm ili jednakovrijedna |
|
4.3.2. |
Spektrofluorimetar s mogućnošću promjene valne duljine (ekscitacije i emisije) |
5. Postupak
5.1. Općenito
5.1.1.
Za provedbu testa iskorištenja (točka 5.1.2.) analizira se slijepa proba hrane za životinje kako bi se potvrdilo da nisu prisutni niti lasalocid natrij, niti interferentne tvari. Slijepa proba hrane za životinje mora biti slična uzorku, a analizom se mora potvrditi odsutnost lasalocid natrija i interferentnih tvari.
5.1.2.
Test iskorištenja izvodi se analizom slijepe probe hrane za životinje koja se obogaćuje dodavanjem količine lasalocid natrija slične količini u uzorku. Za dobivanje koncentracije 100 mg/kg prenese se 10,0 ml osnovne standardne otopine (točka 3.10.1.) u konusnu tikvicu obujma 250 ml i otopina otpari do približno 0,50 ml. Doda se 50 g slijepe probe hrane za životinje, temeljito promiješa i ostavi 10 minuta, a prije koraka ekstrakcije miješanje se više puta ponovi (točka 5.2.).
Ako nije dostupna slijepa probe hrane za životinje slična uzorku (vidjeti točku 5.1.1.), test iskorištenja može se provesti metodom dodavanja standarda. U tom se slučaju uzorku za analizu doda količina lasalocid natrija slična onoj koja se već nalazi u uzorku. Ovaj se uzorak analizira zajedno s uzorkom bez dodanog lasalocid natrija, a iskorištenje se izračuna oduzimanjem.
5.2. Ekstrakcija
5.2.1.
Odvagne se 5 – 10 g uzorka s preciznošću od 0,01 g i prenese u konusnu tikvicu obujma 250 ml s čepom. Pipetom se doda 100,0 ml zakiseljenog metanola (točka 3.8.). Poklopac se lagano zatvori i tikvica protrese radi disperzije uzorka. Tikvica se drži 20 minuta u ultrazvučnoj kupelji (točka 4.1.) na približno 40 °C, zatim se izvadi iz kupelji i ohladi na sobnu temperaturu. Ostavi se približno 1 sat dok se suspenzija ne istaloži, zatim se alikvotni dio filtrira kroz membranski filtar 0,45 μm (točka 4.2.) u prikladnu posudu. Prelazi se na postupak određivanja putem HPLC-a (točka 5.3.).
5.2.2.
Odvagne se približno 2 g neusitnjenog premiksa s preciznošću od 0,001 g i prenese u graduiranu tikvicu obujma 250 ml. Doda se 100,0 ml zakiseljenog metanola (točka 3.8.), tikvica se protrese radi disperzije uzorka. Tikvica se sa sadržajem drži 20 minuta u ultrazvučnoj kupelji (točka 4.1.) na približno 40 °C, zatim se izvadi iz kupelji i ohladi na sobnu temperaturu. Zakiseljenim se metanolom (točka 3.8.) razrijedi do oznake i dobro promiješa. Ostavi se 1 sat dok se suspenzija ne istaloži, zatim se alikvotni dio filtrira kroz membranski filtar 0,45 μm (točka 4.2.). Prikladan obujam čistog filtrata razrijedi se zakiseljenim metanolom (točka 3.8.), čime se dobiva konačna pokusna otopina koncentracije približno 4 μg/ml lasalocid natrija. Prelazi se na postupak određivanja putem HPLC-a (točka 5.3.).
5.3. Određivanje putem HPLC-a
5.3.1.
Sljedeći uvjeti predlažu se kao smjernice; mogu se koristiti i drugi uvjeti ako se njima jamče jednakovrijedni rezultati:
|
Kolona za tekućinsku kromatografiju (točka 4.3.1.): |
125 mm × 4 mm, povratna faza C18, veličina čestica punjenja 5 μm ili jednakovrijedna |
|
Mobilna faza (točka 3.9.): |
Smjesa otopine fosfatnog pufera (točka 3.7.) i metanola (točka 3.5.), 5 + 95 (V + V) |
|
Brzina protoka: |
1,2 ml/min |
|
Valna duljina detekcije: |
|
|
Ekscitacija: |
310 nm |
|
Emisija: |
419 nm |
|
Volumen za ubrizgavanje: |
20 μl |
Stabilnost kromatografskog sustava provjerava se višekratnim ubrizgavanjem kalibracijske otopine koncentracije 4,0 μg/ml (točka 3.10.3.) sve dok se ne postignu konstantne visine (ili površine) vršaka i konstantna vremena retencije.
5.3.2.
Više puta se ubrizga svaka kalibracijska otopina (točka 3.10.3.) i za svaku koncentraciju izmjere srednje vrijednosti visina (površina) vršaka. Kalibracijska se krivulja pripremi tako da se na ordinatu nanose srednje vrijednosti visina (površina) vršaka kalibracijskih otopina, a na apscisu odgovarajuće koncentracije u μg/ml.
5.3.3.
Više puta se ubrizgaju ekstrakti uzorka dobiveni u točki 5.2.1. ili 5.2.2., pri čemu se koristi jednak obujam kao za kalibracijsku otopinu i odrede srednje vrijednosti visina (površina) vršaka lasalocid natrija.
6. Izračun rezultata
Iz srednjih se vrijednosti visina (površina) vršaka dobivenih ubrizgavanjem otopine uzorka (točka 5.3.3.) određuje koncentracija lasalocid natrija (μg/ml) iz kalibracijske krivulje.
6.1. Hrana za životinje
Udio lasalocid natrija (w) (mg/kg) u uzorku izračunava se sljedećom formulom:

pri čemu je:
|
c |
= |
koncentracija lasalocid natrija u otopini uzorka (točka 5.2.1.) u μg/ml |
|
V1 |
= |
volumen ekstrakta uzorka u skladu s točkom 5.2.1. u ml (tj. 100) |
|
m |
= |
masa pokusnog uzorka u gramima. |
6.2. Premiksi
Udio lasalocid natrija (w) (mg/kg) u uzorku izračunava se sljedećom formulom:

pri čemu je:
|
c |
= |
koncentracija lasalocid natrija u otopini uzorka (točka 5.2.2.) u μg/ml |
|
V2 |
= |
volumen ekstrakta uzorka u skladu s točkom 5.2.2. u ml (tj. 250) |
|
f |
= |
faktor razrjeđenja u skladu s 5.2.2. |
|
m |
= |
masa pokusnog uzorka u gramima. |
7. Vrednovanje rezultata
7.1. Identifikacija
Metode koje se temelje na spektrofluorometriji manje su podložne interferencijama od metoda kod kojih se koristi UV detekcija. Identifikacija analita može se potvrditi ko-romatografijom.
7.1.1.
Ekstraktu uzorka (točka 5.2.1. ili 5.2.2.) doda se primjerena količina kalibracijske otopine (točka 3.10.3.). Količina dodanog lasalocid natrija mora biti slična količini lasalocid natrija u ekstraktu uzorka. Smije se povećati samo visina vrška lasalocid natrija uzimajući u obzir količine dodanog lasalocid natrija i razrjeđenje ekstrakta. Širina vrha, na polovici visine, mora biti unutar ± 10 % početne vrijednosti širine vrha dobivene iz ekstrakta uzorka bez dodanog lasalocid natrija.
7.2. Ponovljivost
Razlika između rezultata dvaju usporednih postupaka određivanja, izvedenih na istom uzorku, ne smije prijeći:
|
— |
15 % više vrijednosti za udio lasalocid natrija od 30 – 100 mg/kg, |
|
— |
15 mg/kg za udio lasalocid natrija od 100 – 200 mg/kg, |
|
— |
7,5 % više vrijednosti za udio lasalocid natrija veći od 200 mg/kg. |
7.3. Iskorištenje
Za obogaćeni (slijepi) uzorak hrane za životinje iskorištenje mora iznositi najmanje 80 %. Za obogaćene uzorke premiksa iskorištenje mora iznositi najmanje 90 %.
8. Rezultati međulaboratorijske studije
Provedena je međulaboratorijska studija (1) u kojoj se u 12 laboratorija analiziralo 2 premiksa (uzorci 1 i 2) i 5 uzoraka hrane za životinje (uzorci 3 – 7). Svaki se uzorak analizirao dva puta. Rezultati su prikazani u donjoj tablici:
|
|
Uzorak 1 Premiks za piliće |
Uzorak 2 Premiks za purane |
Uzorak 3 Granulat za purane |
Uzorak 4 Mrvice za piliće |
Uzorak 5 Hrana za purane |
Uzorak 6 Hrana za perad A |
Uzorak 7 Hrana za perad B |
|
L |
12 |
12 |
12 |
12 |
12 |
12 |
12 |
|
n |
23 |
23 |
23 |
23 |
23 |
23 |
23 |
|
Srednja vrijednost [mg/kg] |
5 050 |
16 200 |
76,5 |
78,4 |
92,9 |
48,3 |
32,6 |
|
sr [mg/kg] |
107 |
408 |
1,71 |
2,23 |
2,27 |
1,93 |
1,75 |
|
CVr [%] |
2,12 |
2,52 |
2,24 |
2,84 |
2,44 |
4,00 |
5,37 |
|
sR [mg/kg] |
286 |
883 |
3,85 |
7,32 |
5,29 |
3,47 |
3,49 |
|
CVR [%] |
5,66 |
5,45 |
5,03 |
9,34 |
5,69 |
7,18 |
10,70 |
|
Nominalni udio [mg/kg] |
5 000 (*) |
16 000 (*) |
80 (*) |
105 (*) |
120 (*) |
50 (**) |
35 (**) |
|
L |
= |
broj laboratorija; |
|
n |
= |
broj pojedinačnih vrijednosti |
|
sr |
= |
standardna devijacija ponovljivosti |
|
sR |
= |
standardna devijacija obnovljivosti |
|
CVr |
= |
koeficijent varijacije ponovljivosti u % |
|
CVR |
= |
koeficijent varijacije obnovljivosti u %. |
(1) Studiju je provela radna skupina za stočnu hranu pri Verband Deutscher Landwirtschaftlicher Untersuchungs- und Forschungsanstalten (VDLUFA).
(2) Studiju je provela radna skupina za stočnu hranu pri Verband Deutscher Landwirtschaftlicher Untersuchungs- und Forschungsanstalten (VDLUFA).
(3) Mogu se koristiti i druge metode za mineralizaciju ako dokazano jamče usporedive rezultate (poput, primjerice, mikrovalne mineralizacije pod tlakom).
(4) U zelenoj se krmi (svježoj ili osušenoj) mogu naći velike količine kvarca biljnog podrijetla koji može sadržavati elemente u tragovima koje treba ukloniti. Kod uzoraka takve stočne hrane treba provesti sljedeći izmijenjeni postupak. Provode se postupci iz točke 5.1.1.1. sve do filtracije. Filtarski se papir koji sadrži netopljiv ostatak dvaput ispere vrelom vodom i prenese u lončić za spaljivanje od kvarcnog stakla ili platine. Spali se u mufolnoj peći (točka 4.1.) na temperaturi nižoj od 550 °C do potpunog nestanka svih ugljičnih tvari. Ostavi se da se ohladi, doda se nekoliko kapi vode i 10 – 15 ml fluoridne kiseline (točka 3.4.), zatim se otpari do suhog na otprilike 150 °C. Ako se u ostatku i dalje nalazi kvarcni pijesak, ponovno se otopi u nekoliko mililitara fluoridne kiseline (točka 3.4.) i otpari do suhog. Doda se pet kapljica sumporne kiseline (točka 3.5.) i grije dok ne prestane izlaziti bijeli dim. Nakon dodavanja 5 ml klorovodične kiseline 6 mol/l (točka 3.2.) i oko 30 ml vode, zagrije se i otopina filtrira u graduiranu tikvicu obujma 250 ml te dopuni vodom do oznake (koncentracija HCl oko 0,5 mol/l). Nastavi se s utvrđivanjem u skladu s točkom 5.1.2.
(5) The Analyst 108, 1983., str. 1 252. - 1 256.
(1) Analyst, 1995., 120, 2 175. - 2 180.
(*) Udio koji je naveo proizvođač.
(**) Hrana za životinje pripremljena u laboratoriju.
PRILOG V.
ANALITIČKE METODE ZA KONTROLU NEPOŽELJNIH TVARI U HRANI ZA ŽIVOTINJE
A. ODREĐIVANJE SLOBODNOG I UKUPNOG GOSIPOLA
1. Svrha i područje primjene
Ovom se metodom omogućuje određivanje razine slobodnog i ukupnoga gosipola te kemijski srodnih tvari u sjemenu pamuka i u brašnu i pogači od pamukovog sjemena te u krmnim smjesama koje sadrže navedene sirovine za hranu za životinje, kada je koncentracija slobodnog i ukupnoga gosipola te kemijski srodnih tvari veća od 20 mg/kg.
2. Načelo
Gosipol se ekstrahira u prisutnosti 3-aminopropan-1-ola, bilo smjesom propan-2-ola i heksana kod određivanja slobodnoga gosipola, bilo dimetilformamidom kod određivanja ukupnoga gosipola. Gosipol se anilinom pretvori u gosipol-dianilin, čija se optička gustoća mjeri na 440 nm.
3. Reagensi
|
3.1. |
Smjesa propan-2-ol-heksana: pomiješa se 60 volumnih dijelova propan-2-ola s 40 volumnih dijelova n-heksana. |
|
3.2. |
Otapalo A: U graduiranu tikvicu obujma 1 l doda se približno 500 ml smjese propan-2-ol-heksana (točka 3.1.), 2 ml 3-aminopropan-1-ola, 8 ml ledene octene kiseline i 50 ml vode. Dopuni se do oznake smjesom propan-2-ola i heksana (točka 3.1.). Ovaj je reagens stabilan tjedan dana. |
|
3.3. |
Otapalo B: U graduiranu tikvicu obujma 100 ml pipetom se prenese 2 ml 3-aminopropan-1-ola i 10 ml ledene octene kiseline. Ohladi se na sobnu temperaturu i dopuni do oznake N,N-dimetilformamidom. Ovaj je reagens stabilan tjedan dana. |
|
3.4. |
Anilin: Ako optička gustoća u slijepom pokusu prelazi 0,022, anilin se destilira iznad cinka u prahu, pri čemu se odbaci prva i zadnja frakcija od 10 % destilata. Ako se reagens drži u hladnjaku u zatvorenoj posudi od smeđeg stakla, može se održati nekoliko mjeseci. |
|
3.5. |
Standardna otopina gosipola A: U graduiranu tikvicu obujma 250 ml doda se 27,9 mg gosipol acetata. Otopi se i dopuni otapalom A (točka 3.2.) do oznake. Pipetom se prenese 50 ml te otopine u graduiranu tikvicu obujma 250 ml i dopuni otapalom A do oznake. Koncentracija gosipola u toj otopini iznosi 0,02 mg/ml. Prije uporabe otopina se ostavi jedan sat na sobnoj temperaturi. |
|
3.6. |
Standardna otopina gosipola B: U graduiranu tikvicu obujma 50 ml doda se 27,9 mg gosipol acetata. Otopi se i dopuni otapalom B (točka 3.3.) do oznake. Koncentracija gosipola u toj otopini iznosi 0,5 mg/ml. Ako se zaštite od svjetla, standardne su otopine gosipola A i B stabilne 24 sata. |
4. Oprema
|
4.1. |
Mješalica (rotacijska): približno 35 okr./min. |
|
4.2. |
Spektrofotometar |
5. Postupak
5.1. Pokusni uzorak
Količina korištenog pokusnog uzorka ovisi o očekivanom udjelu gosipola u uzorku. Poželjno je koristiti mali pokusni uzorak i relativno velik alikvot filtrata tako da se dobije dovoljna količina gosipola za točna fotometrijska mjerenja. Kod određivanja slobodnoga gosipola u sjemenu pamuka i brašnu i pogači od pamukovog sjemena, količina pokusnog uzorka ne prelazi 1 g; za krmnu smjesu količina može biti do 5 g. U većini je primjera prikladan alikvot filtrata od 10 ml; isti sadrži 50 – 100 μg gosipola. Kod određivanja ukupnoga gosipola, pokusni uzorak iznosi 0,5 – 5 g, tako da alikvot filtrata od 2 ml sadrži 40 – 200 μg gosipola.
Analiza se provodi na sobnoj temperaturi od približno 20 °C.
5.2. Određivanje slobodnoga gosipola
Pokusni se uzorak prenese u tikvicu s brušenim vratom obujma 250 ml, čije je dno prekriveno drobljenim staklo. Pipetom se doda 50 ml otapala A (točka 3.2.), tikvica se zatvori i miješa jedan sat u mješalici. Filtrira se kroz suhi filtar i filtrat prikupi u maloj tikvici s brušenim vratom. Tijekom filtriranja lijevak se prekrije satnim staklom.
Pipetom se u svaku od dvije odmjerene tikvice obujma 25 ml (A i B) prenesu jednaki alikvoti filtrata, koji sadrže 50 – 100 μg gosipola. Prema potrebi se dopune otapalom A (točka 3.2.) do 10 ml. Tikvica (A) se do oznake dopuni smjesom propan-2-ol-heksana (točka 3.1.). Ova se otopina koristi kao referentna otopina za mjerenje otopine uzorka.
Pipetom se u svaku od dvije ostale odmjerene tikvice obujma 25 ml (C i D) prenese 10 ml otapala A (točka 3.2.). Tikvica (C) se do oznake dopuni smjesom propan-2-ol-heksana (točka 3.1.). Ova se otopina koristi kao referentna otopina za mjerenje otopine slijepe probe.
U tikvice (D) i (B) se doda po 2 ml anilina (točka 3.4.). Grije se 30 minuta nad vrelom vodenom kupelji, do obojenja. Ohladi se na sobnu temperaturu, dopuni do oznake smjesom propan-2-ol-heksana (točka 3.1.), homogenizira i ostavi jedan sat.
Optička gustoća otopine iz slijepe probe (D) odredi se usporedbom s referentnom otopinom (C), a optička gustoća otopine uzorka (B) usporedbom s referentnom otopinom (A) u spektrofotometru na 440 nm sa staklenim kivetama veličine 1 cm.
Oduzme se optička gustoća otopine slijepe probe od optičke gustoće otopine uzorka (= korigirana optička gustoća). Iz te se vrijednosti izračunava udio slobodnoga gosipola, u skladu s točkom 6.
5.3. Određivanje ukupnoga gosipola
U odmjernu se tikvicu obujma 50 ml prenese pokusni uzorak s 1 – 5 mg gosipola i doda 10 ml otapala B (točka 3.3.). Istodobno se pripremi slijepa proba, tako da se u drugu graduiranu tikvicu obujma 50 ml prenese 10 ml otapala B (točka 3.3.). Obje se tikvice griju 30 minuta iznad vrele vodene kupelji. Ohlade se na sobnu temperaturu, svaka se tikvica dopuni do oznake smjesom propan-2-ola i heksana (točka 3.1.). Homogenizira se i ostavi 10 – 15 minuta da se istaloži, zatim se filtrira u tikvicu s brušenim vratom.
U dvije se odmjerne tikvice obujma 25 ml pipetom prenese po 2 ml filtrata uzorka, a u druge dvije odmjerne tikvice obujma 25 ml po 2 ml filtrata slijepe probe. Po jedna se tikvica iz svake serije dopuni do 25 ml smjesom propan-2-ol-heksana (točka 3.1.). Ove se otopine koriste kao referentne otopine.
U svaku od preostalih dviju tikvica doda se 2 ml anilina (točka 3.4.). Zagrijavaju se 30 minuta iznad vrele vodene kupelji do nastanka obojenja. Ohladi se na sobnu temperaturu, dopuni smjesom propan-2-ola i heksana (točka 3.1.) do 25 ml, homogenizira i ostavi jedan sat.
Optička gustoća slobodnoga gosipola određuje se u skladu s točkom 5.2. Iz te se vrijednosti izračunava udio ukupnoga gosipola, u skladu s točkom 6.
6. Izračun rezultata
Rezultati se mogu izračunani bilo iz specifične optičke gustoće (točka 6.1.) ili iz kalibracijske krivulje (točka 6.2.).
6.1. Iz specifične optičke gustoće
Specifične optičke gustoće, pri opisanim uvjetima, su sljedeće:
|
Slobodni gosipol: |
|
|
Ukupni gosipol: |
|


Udio slobodnog ili ukupnoga gosipola u uzorku izračunava se sljedećom formulom:

pri čemu je:
|
E |
= |
korigirana optička gustoća određena u skladu s točkom 5.2., |
|
p |
= |
masa pokusnog uzorka u gramima, |
|
a |
= |
alikvot filtrata u mililitrima. |
6.2. Iz kalibracijske krivulje
6.2.1.
Pripreme se 2 serije po pet graduiranih tikvica obujma 25 ml. U svaku seriju tikvica pipetom se prenesu alikvoti od 2,0, 4,0, 6,0, 8,0 i 10,0 ml standardne otopine gosipola A (točka 3.5.). Otapalom A (točka 3.2.) se dopuni do 10 ml. Svaka se serija dopuni jednom odmjernom tikvicom obujma 25 ml koja sadrži samo 10 ml otapala A (točka 3.2.) (slijepa proba).
Tikvice iz prve serije (uključujući tikvicu za slijepu probu) dopune se do 25 ml smjesom propan-2-ola i heksana (točka 3.1.) (referentna serija).
U svaku tikvicu iz druge serije (uključujući tikvicu za slijepu probu) doda se 2 ml anilina (točka 3.4.). Zagrijava se 30 minuta iznad vrele vodene kupelji do nastanka obojenja. Ohladi se na sobnu temperaturu, dopuni do oznake smjesom propan-2-ol-heksana (točka 3.1.), homogenizira i ostavi jedan sat (standardna serija).
Optička gustoća otopina iz standardne serije i odgovarajućih otopina iz referentne serije odredi se u skladu s točkom 5.2. Kalibracijska se krivulja pripremi tako da se u dijagram unesu izmjerene optičke gustoće u odnosu na količine gosipola (u μg).
6.2.2.
Pripremi se šest graduiranih tikvica obujma 50 ml. U prvu se tikvicu prenese 10 ml otapala B (točka 3.3.), a u ostale 2,0, 4,0, 6,0, 8,0 i 10,0 ml standardne otopine gosipola B (točka 3.6.). Svaka se tikvica dopuni do 10 ml otapalom B (točka 3.3.). Zagrijava se 30 minuta iznad vrele vodene kupelji. Ohladi se na sobnu temperaturu, dopuni do oznake smjesom propan-2-ol-heksana (točka 3.1.) i homogenizira.
Prenese se po 2,0 ml svake od navedenih otopina u svaku od dvije serije od po šest graduiranih tikvica obujma 25 ml. Tikvice iz prve serije dopune se do 25 ml smjesom propan-2-ol-heksana (točka 3.1.) (referentna serija).
U svaku tikvicu iz druge serije doda se 2 ml anilina (točka 3.4.). Zagrijava se 30 minuta iznad vrele vodene kupelji. Ohladi se na sobnu temperaturu, dopuni do oznake smjesom propan-2-ol-heksana (točka 3.1.), homogenizira i ostavi jedan sat (standardna serija).
Optička gustoća otopina iz standardne serije i odgovarajućih otopina iz referentne serije odredi se u skladu s točkom 5.2. Kalibracijska se krivulja pripremi tako da se u dijagram unesu izmjerene optičke gustoće u odnosu na količine gosipola (u μg).
6.3. Ponovljivost
Razlika između rezultata dvaju usporednih postupaka određivanja, izvedenih na istom uzorku, ne smije prijeći:
|
— |
15 % više vrijednosti za udio gosipola manji od 500 ppm, |
|
— |
75 ppm apsolutne vrijednosti za udio gosipola od 500 – 750 ppm, |
|
— |
10 % više vrijednosti za udio gosipola veći od 750 ppm. |
(B) ODREĐIVANJE RAZINA DIOKSINA (PCDD/PCDF) I PCB-a SLIČNIH DIOKSINU
I. METODE UZORKOVANJA I TUMAČENJE REZULTATA ANALIZE
1. Svrha i područje primjene
Uzorci za službenu kontrolu razina dioksina (polikloriranih dibenzo-p-dioksina (PCDD) i polikloriranih dibenzofurana (PCDF)) te polikloriranih bifenila sličnih dioksinu (PCB) (1) u hrani za životinje uzimaju se u skladu s odredbama Priloga I. Primjenjuju se količinski zahtjevi za kontrolu tvari ili proizvoda ravnomjerno raspoređenih u hrani za životinje kako je predviđeno točkom 5.A Priloga I. Skupni uzorci dobiveni na taj način smatraju se reprezentativnim za serije i podserije iz kojih su uzeti. Sukladnost s najvišim razinama određenim Direktivom 2002/32/EZ Europskog parlamenta i Vijeća (2) utvrđuje se na temelju razina utvrđenih na laboratorijskim uzorcima.
2. Usklađenost serije ili podserije sa specifikacijom
Serija se prihvaća ako rezultat pojedinačne analize ne prelazi odgovarajuću najvišu razinu određenu Direktivom 2002/32/EZ, uzimajući u obzir nesigurnost mjerenja.
Serija nije u skladu s najvišom vrijednosti određenom Direktivom 2002/32/EZ ako rezultat analize zaokružen na više (3), potvrđen dvostrukom analizom (4), izvan razumne sumnje prelazi najvišu vrijednost uzimajući u obzir nesigurnost mjerenja.
Nesigurnost mjerenja uzima se u obzir u skladu s jednom od sljedećih pristupa:
|
— |
izračunom proširene nesigurnosti uz uporabu obuhvatnog faktora 2, čime se postiže razina pouzdanosti od 95 %. Serija nije sukladna ako je izmjerena vrijednost minus U iznad najviše vrijednosti. Kod postupka odvojenog određivanja dioksina i PCB-a sličnih dioksinu, za zbroj dioksina i PCB-a sličnih dioksinu koristi se zbroj procijenjene proširene nesigurnosti odvojenih analitičkih rezultata dioksina i PCB-a sličnih dioksinu; |
|
— |
utvrđivanjem granice odlučivanja (CCα) u skladu s Odluci Komisije 2002/657/EZ (5) (točka 3.1.2.5. Priloga – u slučaju tvari s utvrđenom dopuštenom razinom). Serija nije sukladna ako je izmjerena vrijednost jednaka ili veća od CCα. |
Sadašnja pravila tumačenja primjenjuju se na rezultat analize uzorka za službenu kontrolu. To ne utječe na prava država članica da koriste nacionalne propise za analize u obrambene ili referentne svrhe.
II. PRIPREMA UZORKA I ZAHTJEVI ZA ANALITIČKE METODE KOJE SE KORISTE KOD SLUŽBENE KONTROLE RAZINA DIOKSINA (PCDD/PCDF) I PCB-a SLIČNIH DIOKSINU
1. Svrha i područje primjene
Ovi se zahtjevi primjenjuju kada se sirovine za hranu za životinje i hrana za životinje analiziraju za određivanje količine dioksina (polikloriranih dibenzo-p-dioksina (PCDD) i polikloriranih dibenzofurana (PCDF)) te polikloriranih bifenila sličnih dioksinu (PCB).
Nadzor u pogledu prisutnosti dioksina u hrani za životinje može se provoditi strategijom temeljenom na metodi probira za odabir onih uzoraka u kojima razine dioksina ili PCB-a sličnih dioksinu prelaze značajnu vrijednost ili su od nje manje za najviše 25 %. Koncentracija dioksina u uzorcima sa značajnim razinama mora se odrediti/potvrditi potvrdnom metodom.
Orijentacijske metode su metode koje se koriste za otkrivanje prisutnosti značajnih vrijednosti dioksina i PCB-a sličnih dioksinu. Ovim se metodama može analizirati velik broj uzoraka i upotrebljavaju se za probir velikog broja uzoraka s mogućim pozitivnim rezultatima analize. Ove su metode posebno osmišljene kako bi se izbjegli lažno negativni rezultati.
Potvrdne metode su metode koje daju potpune ili dodatne informacije putem kojih se mogu identificirati i točno količinski odrediti dioksini i PCB-i slični dioksinu na razini značajne vrijednosti.
2. Kontekst primjene
Budući da uzorci iz okoliša i biološki uzorci (uključujući uzorke sirovina za hranu za životinje) općenito sadrže kompleksne smjese različitih kongenera dioksina, razvijen je koncept faktora ekvivalentne toksičnosti (TEF) kako bi se olakšala procjena opasnosti. TEF-ovi su uvedeni za iskazivanje koncentracija smjesa 2,3,7,8-supstituiranih PCDD-a i PCDF-a i određenih ne-orto i mono-orto klor-supstituiranih PCB-a s djelovanjem sličnim dioksinu u ekvivalentima toksičnosti (TEQ) 2,3,7,8-TCDD-a. Koncentracije pojedinačnih tvari u danom uzorku pomnože se s odgovarajućim TEF-om i zatim zbroje kako bi se dobila ukupna koncentracija spojeva sličnih dioksinu iskazana u ekvivalentima toksičnosti.
Isključivo u smislu ove Uredbe, prihvatljiva specifična granica kvantifikacije pojedinačnog kongenera je koncentracija analita u ekstraktu uzorka koja proizvodi odziv instrumenta na dva različita iona koji se prate s omjerom S/Š (signal/šum) 3:1 za manje osjetljiv signal i ispunjavanje temeljnih zahtjeva, kao što su npr. vrijeme retencije i omjer izotopa u skladu s postupku određivanja opisanom u EPA-inoj metodi 1613, revizija B.
3. Zahtjevi za osiguranje kvalitete kojih se treba pridržavati tijekom pripreme uzorka
Primjenjuju se opće odredbe za pripremu uzoraka za analizu iz Priloga II.
Osim toga moraju biti ispunjeni sljedeći uvjeti:
|
— |
Uzorke treba čuvati i prenositi u staklenim, aluminijskim, polipropilenskim ili polietilenskim posudama. Iz posude za uzorke treba odstraniti tragove papirne prašine. Staklene se posude ispiru otapalima koja su prethodno provjerena na prisutnost dioksina. |
|
— |
Slijepa proba vrši se tako da se provede cjelokupan analitički postupak, ali bez uzorka. |
|
— |
Masa uzorka koji se koristi za ekstrakciju mora biti dovoljna da se ispune zahtjevi glede osjetljivosti. |
4. Zahtjevi za laboratorije
|
— |
Laboratoriji moraju dokazati učinkovitost metode u području značajne vrijednosti, npr. na razini jednakoj 0,5, 1 i 2 puta značajne vrijednosti s prihvatljivim koeficijentom varijacije za ponovljene analize. Za podrobnosti o mjerilima prihvatljivosti vidjeti točku 5. |
|
— |
Granica kvantifikacije za potvrdnu metodu mora biti u rasponu jedne petine značajne vrijednosti kako bi se u području razmatrane vrijednosti osigurali prihvatljivi koeficijenti varijacije. |
|
— |
Redovite slijepe kontrole i pokusi s dodavanjem ili analize kontrolnih uzoraka (ako je dostupan, poželjan je certificirani referentni materijal) provode se kao mjere interne kontrole kvalitete. |
|
— |
Najbolji način kojim laboratorij dokazuje stručnost u provođenju određenih analiza je uspješno sudjelovanje u međulaboratorijskim studijama za ocjenu stručnosti laboratorija. Uspješno sudjelovanje u međulaboratorijskim studijama, npr. za uzorke tla ili otpadnih voda, ipak ne dokazuje nužno stručnost na području uzoraka hrane ili hrane za životinje u kojima se susreću niže razine onečišćenja. Zbog toga je obvezno stalno sudjelovanje u međulaboratorijskim studijama za određivanje dioksina i PCB-a sličnih dioksinu u mjerodavnim matricama prehrambenih proizvoda/hrane za životinje. |
|
— |
Laboratorije akreditiraju priznata tijela koja djeluju u skladu s ISO Uputama 58, čime se jamči da laboratoriji u analizama provode mjere osiguranja kvalitete. Laboratoriji se akreditiraju u skladu s normom ISO/IEC/17025. |
5. Zahtjevi za analitičke postupke za dioksine i PCB-e slične dioksinu
Osnovni zahtjevi za prihvaćanje analitičkih postupaka:
|
— |
Visoka osjetljivost i niske granice detekcije. Za PCDD-e i PCDF-e, detektabilne količine moraju biti u području pikograma TEQ (10-12 g) zbog visoke toksičnosti nekih od navedenih spojeva. Poznato je kako se PCB-i pojavljuju u većim količinama od PCDD-a i PCDF-a. Za većinu kongenera PCB-a dovoljna je osjetljivost u području nanograma (10-9 g). Međutim, za mjerenje toksičnijih kongerera PCB-a sličnih dioksinu (posebno ne-orto supstituiranih kongenera) treba postići jednaku osjetljivost kao kod PCDD-a i PCDF-a. |
|
— |
Visoka selektivnost (specifičnost). Potrebno je razlikovati između PCDD-a, PCDF-a i PCB-a sličnih dioksinu i mnogobrojnih drugih, istodobno ekstrahiranih i vjerojatno interferentnih spojeva, prisutnih u koncentracijama koje su nekoliko redova veličine veće od koncentracija predmetnih analita. Kod metoda plinske kromatografije/masene spektrometrije (GC/MS) nužno je razlikovati između različitih kongenera, na primjer između toksičnih (npr. 17 2,3,7,8-supstituiranih PCDD-a i PCDF-a i PCB-a sličnih dioksinu) i drugih kongenera. Biotestovi moraju omogućiti selektivno određivanje vrijednosti TEQ-a kao sume PCDD-a, PCDF-a i PCB-a sličnih dioksinu. |
|
— |
Visoka točnost (istinitost i preciznost). Postupak određivanja mora dati valjanu i pouzdanu procjenu prave koncentracije u uzorku. Potrebna je visoka točnost (točnost mjerenja: suglasnost između rezultata mjerenja s pravom ili dogovorenom vrijednosti mjerene veličine) kako bi se spriječilo odbacivanje rezultata analize uzorka na temelju slabe pouzdanosti procjene TEQ-a. Točnost se iskazuje kao istinitost (razlika između srednje vrijednosti koja je izmjerena za analit u certificiranom materijalu i njegove certificirane vrijednosti, iskazano kao postotak te vrijednosti) i preciznost (RSDR, relativna standardna devijacija od rezultata dobivenih pod uvjetima obnovljivosti). |
Orijentacijske metode mogu uključivati biotestove i metode GC/MS; potvrdne metode su metode plinske kromatografije visoke razlučivosti/masene spektrometrije visoke razlučivosti (HRGC/HRMS).
Za ukupnu vrijednosti TEQ-a moraju biti ispunjena sljedeća mjerila:
|
|
Orijentacijske metode |
Potvrdne metode |
|
Udio lažno negativnih rezultata |
< 1 % |
|
|
Istinitost |
|
– 20 % do + 20 % |
|
Preciznost RSDR |
< 30 % |
< 15 % |
6. Posebni zahtjevi koje moraju ispunjavati metode GC/MS koje se koriste za svrhe probira ili potvrde
|
— |
U vrednovanju metode analize, unutarnji standardi PCDD/F-ova supstituirani klorom na položaju 2,3,7,8 i obilježeni izotopom 13C i unutarnji standardi dioksinima sličnih PCB-a obilježeni izotopom 13C moraju se dodati na samom početku analitičkog postupka, npr. prije ekstrakcije. Mora se dodati najmanje po jedan kongener za svaku tetra do okta-kloriranu homolognu skupinu za PCDD/F i najmanje po jedan kongener za sve homologne skupine za PCB-e slične dioksinima (odnosno najmanje po jedan kongener za svaki izabrani ion u spektrometriji masa koja se koristi za praćenje PCDD/F-ova odnosno PCB-a sličnih dioksinu). Najbolje je koristiti, posebno za potvrdne metode, svih 17 unutarnjih standarda PCDD/F-ova supstituiranih klorom na položaju 2,3,7,8 i obilježenih izotopom 13C, te svih 12 unutarnjih standarda PCB-a sličnih dioksinima obilježenih izotopom 13C. |
|
— |
Relativne faktore odgovora treba utvrditi i za one kongenere za koje se ne dodaje analog obilježen izotopom 13C, upotrebom odgovarajuće kalibracijske otopine. |
|
— |
Za hranu biljnog i životinjskoga podrijetla koja sadrži manje od 10 % masti, unutarnji se standardi obavezno dodaju prije ekstrakcije. Za hranu životinjskoga podrijetla u kojoj je udio masti veći 10 %, unutarnji se standardi mogu dodati prije ili poslije ekstrakcije masti. Mora se provesti odgovarajuće vrednovanje učinkovitosti ekstrakcije, što ovisi o fazi u kojoj se dodaje unutarnji standard, te o tome iskazuju li se rezultati s obzirom na udio masti u uzorku ili s obzirom na cijeli uzorak. |
|
— |
Prije analize GC/MS treba dodati 1 ili 2 (surogat) standarda radi provjere iskorištenja. |
|
— |
Potrebno je kontrolirati iskorištenje. Za potvrdne metode iskorištenje pojedinačnih unutarnjih standarda mora biti između 60 % i 120 %. Prihvatljivo je manje ili veće iskorištenje za pojedinačne kongenere, a posebno za neke hepta- i okta-klorirane dibenzodioksine i dibenzofurane, pod uvjetom da je njihov doprinos vrijednosti TEQ-a manji od 10 % ukupne vrijednosti TEQ-a (dobivene na temelju zbroja PCDD/F-ova i PCB-a sličnih dioksinima). Za orijentacijske metode iskorištenje mora biti između 30 % i 140 %. |
|
— |
Dioksini se od kloriranih spojeva koji mogu interferirati u analizi, poput PCB-a koji nisu slični dioksinu te kloriranih difenil etera, odvajaju odgovarajućim kromatografskim tehnikama (najbolje pomoću kolone s florisilom, aluminijevim oksidom i/ili aktivnim ugljenom). |
|
— |
Razdvajanje izomera plinskom kromatografijom mora biti zadovoljavajuće (preklapanje signala 1,2,3,4,7,8-HxCDF i signala 1,2,3,6,7,8-HxCDF < 25 % od vrška do vrška). |
|
— |
Tetra do okta-klorirani dioksini i furani određuju se u skladu s EPA-inoj metodi 1613, revizija B, metodom izotopnog razrjeđenja uz primjenu HRGC/HRMS-a odnosno drugom metodom s jednakim kriterijima učinkovitosti. |
|
— |
Razlika između gornje i donje granice ne smije biti veća od 20 % za hranu u kojoj je zagađenje dioksinima u području oko ili iznad najviše razine. Za hranu za životinje u kojoj je zagađenost osjetno ispod najviše razine razlika može biti u rasponu od 25 % – 40 %. |
7. Orijentacijske metode analize
7.1. Uvod
Pri upotrebi orijentacijske metode, mogu se upotrijebiti različiti analitički pristupi.
Rezultati analize uzoraka uspoređuju se s rezultatima referentnog uzorka. Uzorci čiji su rezultati manji od rezultata referentnog uzorka smatraju se negativnima, a oni s većim rezultatima su potencijalno pozitivni. Zahtjevi su sljedeći:
|
— |
Uz svaku seriju uzoraka mora se analizirati jedna slijepa proba i jedan referentni uzorak koji se ekstrahiraju i analiziraju pod istovjetnim uvjetima. Rezultat referentnog uzorka mora nedvojbeno biti veći od rezultata slijepe probe. |
|
— |
Za potvrdu učinkovitosti testa u značajnom području radi provjere značajne vrijednosti koriste se dodatni referentni uzorci u kojima su količine 0,5 i 2 puta veće od značajne vrijednosti. |
|
— |
Za ispitivanje ostalih matrica potrebno je dokazati prikladnost jednog ili više referentnih uzoraka, najbolje uključivanjem uzoraka za koje je HRGC/HRMS analiza pokazala da sadrže TEQ u razini referentnoga uzorka ili tako da se slijepoj probi doda standard u toj količini. |
|
— |
Budući da se u biotestovima ne mogu koristiti unutarnji standardi, vrlo su važni testovi ponovljivosti kako bi se dobili podaci o standardnoj devijaciji unutar jedne serije ispitivanja. Koeficijent varijacije mora biti manji od 30 %. |
|
— |
U primjeni biotestova moraju se utvrditi ciljni spojevi, moguće interferencije i najveće prihvatljive količine za slijepe probe. |
Kvantitativni pristup zahtijeva seriju razrijeđenih standarda, dvostruko ili trostruko pročišćivanje i mjerenje, slijepe probe i kontrolu iskorištenja. Rezultat se može iskazati kao TEQ, čime se pretpostavlja da signali spojeva odgovaraju načelu toksične ekvivalentnosti. U tu se svrhu može koristiti TCDD (odnosno standardna smjesa dioksina/furana/PCB-a sličnih dioksinima) kako bi se dobila kalibracijska krivulja za izračun količine TEQ-a u ekstraktu i u uzorku. Rezultat za količinu TEQ-a se naknadno korigira za vrijednost slijepe probe (kako bi se isključio utjecaj nečistoća koje potječu iz korištenih otapala i kemikalija) i za iskorištenje (izračunano u kontroli kvalitete iz količine TEQ-a koja je približna odgovarajućoj toksičnoj razini). Važno je napomenuti kako na smanjenje iskorištenja dijelom može utjecati matrica uzorka i/ili razlike između vrijednosti TEF-a iz biotestova i službenih vrijednosti TEF-a koje je odredio WHO.
7.2. Zahtjevi za orijentacijske metode analize
|
— |
Za orijentaciju se mogu koristiti metode analize GC/MS i biotestovi. Za metode GC/MS primjenjuju se zahtjevi navedeni u točki 6. Posebni zahtjevi za stanične biotestove navedeni su u točki 7.3., a za biotestove u kompletu u točki 7.4. |
|
— |
Potrebno je dobiti podatke o broju lažno pozitivnih i lažno negativnih rezultata iz velikog broja uzoraka s količinama ispod ili iznad najveće dopuštene razine u odnosu na udio TEQ-a utvrđen potvrdnom analitičkom metodom. Stvarni udio lažno negativnih uzoraka mora biti manji od 1 %. Udio lažno pozitivnih uzoraka mora biti dovoljno malen da orijentacijska metoda bude uporabljiva. |
|
— |
Pozitivni rezultati se uvijek moraju potvrditi potvrdnom analitičkom metodom (HRGC/HRMS). Istom metodom potvrđuju se i uzorci iz širokog raspona TEQ vrijednosti (približno od 2 do 10 % negativnih uzoraka). Treba omogućiti uvid u odnos između rezultata dobivenih biotestom i metodom HRGC/HRMS. |
7.3. Posebni zahtjevi za stanične biotestove
|
— |
Za svaku seriju određenoga biotesta potreban je niz referentnih koncentracija TCDD-a odnosno mješavine dioksina/furana/PCB-a sličnih dioksinu (cijela krivulja odgovora na dozu pri R2 > 0,95). Međutim, za potrebe probira, kod analize uzoraka s niskim koncentracijama može se rabiti proširena krivulja za niže razine. |
|
— |
Na listi nadzora kvalitete moraju se prikazati rezultati biotesta kroz određeno neprekinuto razdoblje za referentnu koncentraciju TCDD-a (približno trostruka granica kvantifikacije). Kao druga mogućnost može se koristiti relativan odgovor referentnoga uzorka u odnosu na kalibracijski pravac za TCDD, budući da stanični odgovor može ovisiti o brojnim čimbenicima. |
|
— |
Za svaki tip referentnog materijala bilježe se i provjeravaju dijagrami kontrole kvalitete kako bi se osigurao rezultat sukladan navedenim smjernicama. |
|
— |
Posebno kod kvantitativnih izračuna, razrijeđeni se uzorak mora analizirati u linearnom području odzivne krivulje. Uzorci s odgovorom iznad linearnog područja odzivne krivulje moraju se razrijediti i ponovo testirati. Stoga valja istodobno testirati najmanje tri ili više razrjeđenja. |
|
— |
Postotak standarde devijacije ne smije biti veći od 15 % nakon trostrukog određivanja za svako od tri razrjeđenja niti smije biti veći od 30 % između tri neovisna testiranja. |
|
— |
Granica detekcije može biti tri puta veća od standardne devijacije za otopinu slijepe probe, odnosno od pozadinskog šuma. Drugi je pristup primjena odgovora višeg od pozadinskoga šuma (faktor indukcije 5x slijepa proba otapala) koji se izračunava iz kalibracijske krivulje za taj dan. Granica kvantifikacije može biti 5 – 6 puta veća od standardne devijacije za slijepu probu odnosno od pozadinskog šuma, odnosno može se primijeniti odgovor koji je neupitno veći od pozadinskoga šuma (faktor indukcije 10x slijepa proba otapala) koji se izračunava iz kalibracijske krivulje za taj dan. |
7.4. Posebni zahtjevi za biotestove u kompletu
|
— |
Potrebno je osigurati dovoljnu osjetljivost i pouzdanost biotestova u kompletu kada se primjenjuju za hranu. |
|
— |
Za pripremu uzorka treba slijediti upute proizvođača. |
|
— |
Kompleti za ispitivanje se ne smiju rabiti nakon isteka trajanja. |
|
— |
Ne smiju se rabiti materijali i sastojci namijenjeni uporabi s drugim kompletima. |
|
— |
Kompleti za ispitivanje se čuvaju na određenom rasponu temperatura te se rabe na određenoj radnoj temperaturi. |
|
— |
Granica detekcije za imunotestove određuje se kao suma srednje vrijednosti i trostruke standardne devijacije, a na temelju 10 ponovljenih analiza slijepe probe koja se dijeli s nagibom krivulje u jednadžbi linearne regresije. |
|
— |
Za laboratorijsko testiranje moraju se koristiti referentni standardi koji osiguravaju odgovor standarda u prihvatljivom području. |
8. Izvješće o rezultatima
Ako upotrijebljeni analitički postupak to omogućuje, podaci koje izvješće o ispitivanju mora sadržavati su količine pojedinačnih kongenera PCDD/F-a i PCB-a, a za koje treba navesti jesu li zaokruženi na više, na niže ili prema sredini, kako bi se u izvješće o rezultatima uključila maksimalna količina podataka i omogućilo tumačenje rezultata u skladu s posebnim zahtjevima.
U izvješće mora biti uključen udio masti u uzorku i metoda ekstrakcije masti.
Iskorištenja pojedinih unutarnjih standarda moraju biti navedena u slučaju da su izvan raspona navedenog u točki 6., u slučaju da je dobiveni rezultat veći od najveće dopuštene količine te u drugim slučajevima na poseban zahtjev.
Kako pri odluci o u skladu ssti uzorka treba uzeti u obzir mjernu nesigurnost, ovaj se parametar također mora navesti u izvješću. Rezultati analize se prikazuju kao x +/- U, gdje je x rezultat analize, a U je proširena mjerna nesigurnost uz faktor obuhvata 2, čime se dobiva razina pouzdanosti od oko 95 %. Određuju li se dioksini odvojeno od PCB-a sličnih dioksinima, tada se zbrajaju procijenjene proširene nesigurnosti za pojedinačne rezultate analiza dioksina i PCB-a sličnih dioksinima.
Ako se nesigurnost mjerenja računa iz granične količine analita CCα (kako je opisano u točki I.2. dijela B) tada se mora navesti i taj parametar.
(1) Tablica faktora ekvivalentne toksičnosti (TEF) za doksine, furane i PCB-e slične dioksinu
|
Kongeneri |
Vrijednost TEF-a |
Kongener |
Vrijednost TEF-a |
|
Dibenzo-p-dioksini („PCDD”) |
|
PCB-i slični dioksinu: |
|
|
2,3,7,8-TCDD |
1 |
|
|
|
1,2,3,7,8-PeCDD |
1 |
Ne-orto PCB-i |
|
|
1,2,3,4,7,8-HxCDD |
0,1 |
PCB 77 |
0,0001 |
|
1,2,3,6,7,8-HxCDD |
0,1 |
PCB 81 |
0,0001 |
|
1,2,3,7,8,9-HxCDD |
0,1 |
PCB 126 |
0,1 |
|
1,2,3,4,6,7,8-HpCDD |
0,01 |
PCB 169 |
0,01 |
|
OCDD |
0,0001 |
Mono-orto PCB-i |
|
|
|
|
PCB 105 |
0,0001 |
|
Dibenzofurani („PCDF”) |
|
PCB 114 |
0,0005 |
|
2,3,7,8-TCDF |
0,1 |
PCB 118 |
0,0001 |
|
1,2,3,7,8-PeCDF |
0,05 |
PCB 123 |
0,0001 |
|
2,3,4,7,8-PeCDF |
0,5 |
PCB 156 |
0,0005 |
|
1,2,3,4,7,8-HxCDF |
0,1 |
PCB 157 |
0,0005 |
|
1,2,3,6,7,8-HxCDF |
0,1 |
PCB 167 |
0,00001 |
|
1,2,3,7,8,9-HxCDF |
0,1 |
PCB 189 |
0,0001 |
|
2,3,4,6,7,8-HxCDF |
0,1 |
|
|
|
1,2,3,4,6,7,8-HpCDF |
0,01 |
|
|
|
1,2,3,4,7,8,9-HpCDF |
0,01 |
|
|
|
OCDF |
0,0001 |
|
|
|
Korištene kratice: „T” = tetra; „Pe” = penta; „Hx” =heksa; „Hp”= hepta; „O” = okta; „CDD” = klordibenzo-p-dioksin; „CDF” = klordibenzofuran; „CB” = klorbifenil. |
|||
(2) SL L 140, 30.5.2002., str. 10.
(3) Pojam „zaokruženo na više” zahtijeva uporabu granice kvantifikacije za doprinos svakog nekvantificiranog kongenera ekvivalentu toksičnosti (TEQ). Pojam „zaokruženo na niže” zahtijeva uporabu nule za doprinos svakog nekvantificiranog kongenera ekvivalentu toksičnosti (TEQ). Pojam „zaokruženo prema sredini” zahtijeva uporabu polovice granice kvantifikacije za izračun doprinosa svakog nekvantificiranog kongenera ekvivalentu toksičnosti (TEQ).
(4) Dvostruka analiza potrebna je kako bi se isključila mogućnost unutarnje križne kontaminacije ili slučajne zamjene uzoraka. Prva se analiza, uzimajući u obzir nesigurnost mjerenja, koristi za potvrdu sukladnosti.
Ako se analiza provodi u sklopu incidenta sa zagađenjem dioksinom, potvrda s dvostrukom analizom može se izostaviti ako su uzorci odabrani za analizu sljedivi do incidenta sa zagađenjem dioksinom.
PRILOG VI.
ANALITIČKE METODE ZA ODREĐIVANJE SASTOJAKA ŽIVOTINJSKOG PODRIJETLA ZA SLUŽBENU KONTROLU HRANE ZA ŽIVOTINJE
Zahtjevi za mikroskopsku detekciju, identifikaciju ili procjenu sastojaka životinjskog podrijetla u hrani za životinje
1. Cilj i područje primjene
Ovi se zahtjevi primjenjuju kada se utvrđivanje sastojaka životinjskog podrijetla (definiranih kao proizvodi iz prerade trupa ili dijelova tijela sisavaca, peradi i riba) u hrani za životinje provodi mikroskopskim pregledom u okviru usklađenog programa nadzora u području ishrane životinja u skladu s Uredbom (EZ) br. 882/2004 Europskog parlamenta i Vijeća (1). Ako se u svim službenim testovima koriste metode iz ovog Priloga, mogu se provesti i dodatni testovi koji se temelje na izmijenjenoj ili alternativnoj metodi kako bi se poboljšala detekcija određenih vrsta sastojaka životinjskog podrijetla ili za podrobnije određivanje izvora sastojaka životinjskog podrijetla. Osim toga, kod pregledavanja nekih posebnih sastojaka životinjskog podrijetla, poput plazme ili kosti u loju (vidjeti također točku 9.) može se koristiti inačica protokola pod uvjetom da se navedene analize provode dodatno uz analize predviđene usklađenim programom inspekcije.
2. Osjetljivost
Ovisno o vrsti sastojaka životinjskog podrijetla, u hrani za životinje mogu se detektirati vrlo male količine (< 0,1 %).
3. Načelo
Za identifikaciju se koristi reprezentativni uzorak, izuzet u skladu s odredbama Priloga I. te pripravljen na primjeren način. Za obradu životinjske hrane s niskim udjelom vode primjeren je sljedeći protokol. Prije obrade se hrana za životinje s udjelom vode većim od 14 % osuši (kondenzira). Posebne vrste hrane za životinje ili sirovine za hranu za životinje (npr. masti, ulja) treba namjenski obraditi (vidjeti točku 9.). Sastojci životinjskog podrijetla utvrđuju se na temelju tipičnih, mikroskopski prepoznatljivih osobina (tj. mišićnih vlakana i drugih čestica mesa, hrskavice, kosti, rogova, dlake, čekinja, krvi, perja, ljuske jaja, ribljih kosti, ljusaka). Utvrđivanje se mora izvesti i na prosijanoj frakciji (točka 6.1.) i na koncentriranom talogu (točka 6.2.) uzorka.
4. Reagensi
4.1. Sredstvo za pripremu preparata.
|
4.1.1. |
Kloral-hidrat (vodeni, 60 % m/v). |
|
4.1.2. |
Lužina (NaOH 2,5 % m/v ili KOH 2,5 % m/v) za prosijane frakcije. |
|
4.1.3. |
Parafinsko ulje ili glicerol (viskoznost: 68 – 81) za mikroskopska promatranja taloga. |
4.2. Sredstva za ispiranje.
|
4.2.1. |
Alkohol, 96 %. |
|
4.2.2. |
Aceton. |
4.3. Sredstvo za koncentriranje.
|
4.3.1. |
Tetrakloretilen (gustoća 1,62). |
4.4. Reagensi za bojenje.
|
4.4.1. |
Otopina joda/kalijevog jodida (otopi se 2 g kalijevog jodida u 100 ml vode, zatim se uz često protresivanje doda 1 g joda.). |
|
4.4.2. |
Alizarin crvenilo (otopi se 2,5 ml 1M klorovodične kiseline u 100 ml vode i toj se otopini doda 200 mg alizarin crvenila). |
|
4.4.3. |
Cistinski reagens (2 g olovnog acetata, 10 g NaOH/100 ml H2O). |
|
4.4.4. |
Otopina joda/kalijevog jodida (rastopljeno u etanolu 70 %). |
4.5. Reagens za izbjeljivanje.
|
4.5.1. |
Komercijalna otopina natrijevog hipoklorita (9,6 % aktivnog klora). |
5. Oprema i pribor
|
5.1. |
Analitička vaga (preciznost do 0,01 g, osim za koncentrirani talog: 0,001 g). |
|
5.2. |
Pribor za usitnjavanje (mlin ili mužar, posebno za hranu za životinje koja kod analize sadrži > 15 % masti). |
|
5.3. |
Sito s mrežicom kvadratnih očica širine najviše 0,50 mm. |
|
5.4. |
Lijevak za odjeljivanje ili posuda s konusnim dnom za taloženje. |
|
5.5. |
Stereomikroskop (najmanje 40x povećanje). |
|
5.6. |
Sastavljeni mikroskop (najmanje 400x povećanje), propušteno svjetlo ili polarizirano svjetlo. |
|
5.7. |
Standardno laboratorijsko posuđe od stakla. Sva se oprema mora temeljito očistiti. Lijevci za odjeljivanje i stakleno posuđe moraju se oprati u stroju za pranje posuđa. Sita se moraju očistiti četkom s tvrdim čekinjama. |
6. Postupak
Hrana za životinje u zrnu može se prethodno prosijati ako se obje frakcije analiziraju kao odvojeni uzorci.
Obradi se najmanje 50 g uzorka (prema potrebi, oprezno usitnjenog prikladnim priborom za usitnjavanje (točka 5.2.) kako bi se dobila odgovarajuća struktura). Iz usitnjenog se materijala izuzmu dva reprezentativna dijela, jedan za frakciju za prosijavanje (najmanje 5 g) (točka 6.1.) i jedan za koncentrirani talog (najmanje 5 g) (točka 6.2.). Za identifikaciju se dodatno može primijeniti bojenje reagensima za bojenje (točka 6.3.).
Za utvrđivanje prirode životinjskih bjelančevina i podrijetla čestica može se koristiti sustav potpore odlučivanju poput ARIES-a i mogu se dokumentirati referentni uzorci.
6.1. Utvrđivanje sastojaka životinjskog podrijetla u prosijanim frakcijama
Kroz sito se prosije najmanje 5 g uzorka (točka 5.3.) u dvije frakcije.
Prosijane frakcije s velikim česticama (ili reprezentativni dio frakcije) nanesu se u tankom sloju na primjerenu podlogu i sustavno pregledavaju stereomikroskopom (točka 5.5.) pod različitim povećanjima kako bi se detektirali sastojci životinjskog podrijetla.
Mikroskopski preparati iz prosijanih frakcija sa sitnim česticama sustavno se pregledavaju sastavljenim mikroskopom (točka 5.6.) pod različitim povećanjima kako bi se detektirali sastojci životinjskog podrijetla.
6.2. Utvrđivanje sastojaka životinjskog podrijetla u koncentriranom talogu
U lijevak za odjeljivanje ili posudu s konusnim dnom za taloženje prenese se najmanje 5 g (s preciznošću od 0,01 g) uzorka i obradi s najmanje 50 ml tetrakloretilena (točka 4.3.1.). Smjesa se više puta protrese ili promiješa.
|
— |
Ako se koristi zatvoreni lijevak za odjeljivanje, otopina se ostavi da odstoji dovoljno vremena (najmanje 3 minute) prije odvajanja taloga. Ponovno se protrese i otopina ostavi da odstoji najmanje 3 minute. Ponovno se odvoji talog. |
|
— |
Ako se koristi otvorena čaša, otopina se ostavi da odstoji najmanje 5 minuta prije odvajanja taloga. |
Cjelokupni talog se osuši i zatim izvaže (s točnošću na 0,001 g). Vaganje je potrebno samo kada se zahtijeva procjena. Ako se talog sastoji od velikog broja krupnijih čestica, može se prosijati kroz sito (točka 5.3.) u dvije frakcije. Osušeni se talog pregleda pod stereomikroskopom (točka 5.5.) i sastavljenim mikroskopom (točka 5.6.) kako bi se detektirali sastojci podrijetlom od kosti.
6.3. Uporaba sredstava za pripremu preparata i reagensa za bojenje
Mikroskopsko utvrđivanje sastojaka životinjskog podrijetla može se olakšati posebnim sredstvima za pripremu preparata i reagensima za bojenje.
Kloral-hidrat (točka 4.1.1.): Uz oprezno zagrijavanje stanične se strukture vide bolje zato što se škrobna zrnca želatiniziraju, a nepoželjni sastojci stanice uklone.
Lužina (točka 4.1.2.): Krmni se materijal očisti natrijevim hidroksidom ili kalijevim hidroksidom, što olakšava detekciju mišićnih vlakana, dlake i drugih keratinskih struktura.
Parafinsko ulje i glicerol (točka 4.1.3.): Sastojci podrijetlom od kosti mogu se dobro identificirati u ovom sredstvu za pripremu preparata zato što većina koštanih lakuna ostaje ispunjena zrakom i vide se kao crne rupe veličine približno 5 – 15 μm.
Otopina joda/kalijevog jodida (točka 4.4.1.): Koristi se za detekciju škroba (plavo-ljubičasto obojenje) i bjelančevina (žuto-narančasto obojenje). Prema potrebi, otopine se mogu razrijediti.
Otopina alizarin crvenila (točka 4.4.2.): Crveno-ružičasto obojenje kosti, ribljih kosti i ljusaka. Prije sušenja taloga (vidjeti točku 6.2.), cjelokupni se talog prenese u staklenu epruvetu i dvaput ispere s približno 5 ml alkohola (točka 4.2.1.) (svaki se put koristi vrtložna mješalica, a otapalo se ostavi približno 1 minutu da se slegne i zatim se odlije). Prije uporabe reagensa za bojenje, talog se izbijeli dodavanjem najmanje 1 ml otopine natrijevog hipoklorita (točka 4.5.1.). Reakcija se razvija 10 minuta. Epruveta se napuni vodom, talog se ostavi 2 – 3 minute da se slegne, te se voda i suspendirane čestice odliju. Talog se dvaput ispere s 10 ml vode (koristi se vrtložna mješalica, ostavi se da se istaloži i svaki se put odlije voda). Doda se 2 – 10 ili više kapljica (ovisno o količini ostatka) otopine alizarin crvenila. Smjesa se protrese, reakcija se razvija nekoliko sekunda. Obojeni se talog dvaput ispere s približno 5 ml alkohola (točka 4.2.1.), zatim jedanput acetonom (točka 4.2.2.) (svaki se put koristi vrtložna mješalica, a otapalo se ostavi približno 1 minutu da se slegne i zatim se odlije). Talog je tada spreman za sušenje.
Cistinski reagens (točka 4.4.3.): Uz oprezno zagrijavanje, sastojci koji sadrže cistin (dlake, perje itd.) oboje se crno-smeđe.
6.4. Pregled hrane za životinje koja može sadržavati riblje brašno
Pod sastavljenim mikroskopom pregleda se najmanje jedan mikroskopski preparat iz sitno prosijane frakcije i sitne frakcije taloga (vidjeti točke 6.1. i 6.2.).
Kada je na oznaci navedeno da sastojci sadrže riblje brašno ili kada kod početnog pregleda postoji sumnja ili se detektira riblje brašno, iz izvornog uzorka treba pregledati najmanje dva dodatna mikroskopska preparata iz sitno prosijane frakcije i sitne frakcije taloga.
7. Izračun i procjena
Države članice dužne su osigurati da se postupci opisani u ovoj točki koriste u provedbi službenih analiza za procjenu količine (a ne samo prisutnosti) sastojaka životinjskog podrijetla.
Izračun se može izvesti samo ako sastojci životinjskog podrijetla sadrže dijelove kosti.
Dijelovi kosti toplokrvnih kopnenih životinjskih vrsta (tj. sisavaca i ptica) mogu se u mikroskopskim preparatima razlučiti od različitih vrsta ribljih kosti na temelju tipičnih koštanih lakuna. Udio sastojaka životinjskog podrijetla u uzorkovanom se materijalu procjenjuje s obzirom na:
|
— |
procijenjeni udio (% mase) koštanih dijelova u koncentriranom talogu, i |
|
— |
udio (% mase) kosti u sastojcima životinjskog podrijetla. |
Procjena se mora temeljiti na najmanje tri mikroskopska preparata (ako je to moguće) i najmanje pet polja po preparatu. Koncentrirani talog u krmnim smjesama u pravilu ne sadrži samo dijelove kosti kopnenih životinja i riba, nego i druge čestice visoke specifične mase, npr. minerale, pijesak, odrvenjele dijelove biljaka i slično.
7.1. Procijenjena vrijednost postotnog udjela koštanih dijelova
% dijelova kosti kopnenih životinja = (S × c)/W
% dijelova ribljih kosti i ljusaka = (S × d)/W
(S = masa taloga (mg), c = korekcijski faktor (%) za procijenjeni udio kosti kopnenih životinja u talogu, d = korekcijski faktor (%) za procijenjeni udio dijelova ribljih kosti i ljusaka u talogu, W = masa materijala uzorka za sedimentaciju (mg)).
7.2. Procijenjena vrijednost sastojaka životinjskog podrijetla
Udio kosti u životinjskim proizvodima može se znatno razlikovati. (Udio kosti u koštanom brašnu reda je veličine 50 – 60 %, u mesnom brašnu reda veličine 20 – 30 %; u ribljem se brašnu udio kosti i ljusaka razlikuje s obzirom na kategoriju i podrijetlo ribljeg brašna, obično je reda veličine 10 – 20 %).
Ako je poznata vrsta životinjskog brašna prisutnog u uzorku, moguće je procijeniti udio:
Procijenjeni udio sastojaka proizvoda od kopnenih životinja (%) = (S × c)/(W × f) × 100
Procijenjeni udio sastojaka ribljih proizvoda (%) = (S × d)/(W × f) × 100
(S = masa taloga (mg), c = korekcijski faktor (%) za procijenjeni udio koštanih sastojaka kopnenih životinja u talogu, d = korekcijski faktor (%) za procijenjeni udio dijelova ribljih kosti i ljusaka u talogu, f = korekcijski faktor za udio kosti u sastojcima životinjskog podrijetla u pregledanom uzorku, W = masa materijala uzorka za sedimentaciju (mg)).
8. Prikaz rezultata pregleda
Izvješće sadrži najmanje podatke o prisutnosti sastojaka podrijetlom od kopnenih životinja i ribljeg brašna. O različitim se slučajevima izvješćuje na sljedeći način:
|
8.1. |
S obzirom na prisutnost sastojaka podrijetlom od kopnenih životinja:
|
|
8.2. |
i s obzirom na prisutnost ribljeg brašna:
Kada se utvrdi prisutnost sastojaka ribljeg podrijetla ili podrijetlom od kopnenih životinja, u izvješću o rezultatima pregleda može se, prema potrebi, dodatno navesti procjena količine detektiranih sastojaka (x %, < 0,1 %, 0,1 – 0,5 %, 0,5 – 5 % ili > 5 %), podrobno utvrditi vrsta kopnene životinje, ako je to moguće, te odrediti sastojke životinjskog podrijetla (mišićna vlakna, hrskavica, kosti, rogovi, dlaka, čekinje, krv, ljuske jaja, riblje kosti, ljuske). Kada se procjenjuje količina sastojaka životinjskog podrijetla, navodi se korišteni korekcijski faktor f. Kada se utvrde koštani sastojci kopnenih životinja, izvješće mora sadržavati dodatni navod: „Ne može se isključiti mogućnost da su gornji sastojci podrijetlom od sisavaca.” Takav dodatni navod nije potreban kada su dijelovi kosti kopnenih životinja točno navedeni kao dijelovi kosti peradi ili sisavaca. |
9. Neobvezni protokol za analizu masti ili ulja
Za analizu masti ili ulja može se koristiti sljedeći protokol:
|
— |
Ako je mast u krutom stanju, zagrijava se, na primjer u mikrovalnoj pećnici, dok ne postane tekuća. |
|
— |
Pipetom se prenese 40 ml masti s dna uzorka u epruvetu za centrifugiranje. |
|
— |
Centrifugira se 10 minuta na 4 000 okr./min. |
|
— |
Ako se mast skrutnula nakon centrifugiranja, ponovo se zagrijava u pećnici dok ne postane tekuća. Ponovo se centrifugira 5 minuta na 4 000 okr./min. |
|
— |
Polovica dekantirane nečistoće se prenese malom žličicom ili lopaticom u malu petrijevu zdjelicu ili na mikroskopsko stakalce za mikroskopsku identifikaciju mogućeg udjela sastojaka životinjskog podrijetla (mesnih vlakana, perja, dijelova kosti itd.). Kao sredstvo za pripremu preparata za mikroskopiju preporuča se parafinsko ulje ili glicerol, |
|
— |
Preostala nečistoća koristi se za sedimentaciju u skladu s točkom 6.2. |
(1) SL L 165, 30.4.2004., str. 1.; ispravljeno u SL L 191, 28.5.2004., str. 1.
PRILOG VII.
METODA IZRAČUNA ENERGETSKE VRIJEDNOSTI HRANE ZA PERAD
1. Metoda izračuna i prikaza energetske vrijednosti
Energetska vrijednost hrane za perad mora se izračunani prema formuli navedenoj dalje u tekstu na temelju postotka nekih analitičkih sastojaka hrane za životinje. Ta se vrijednost iskazuje u megadžulima (MJ) metaboličke energije (ME), s korekcijama za dušik, na kilogram krmne smjese:
MJ/kg ME = 0,1551 × % sirovih bjelančevina + 0,3431 × % sirovih masti + 0,1669 × % škroba + 0,1301 × % ukupnog šećera (iskazanog kao saharoza).
2. Odstupanja primjenjiva za navedene vrijednosti
Ako se službenom inspekcijom otkrije razlika (povećana ili smanjena energetska vrijednost hrane za životinje) između rezultata pregleda i navedene energetske vrijednosti, dopušteno je najmanje odstupanje od 0,4 MJ/kg ME.
3. Prikaz rezultata
Nakon primjene gore navedene formule dobiveni rezultat treba iskazati s jednim decimalnim mjestom.
4. Metode uzorkovanja i metode analize
Uzorkovanje krmnih smjesa i određivanje udjela analitičkih sastojaka navedenih u metodi izračuna moraju biti u skladu s metodama Zajednice za uzorkovanje ili analitičkim metodama Zajednice za službeni pregled hrane za životinje.
Primjenjuje se sljedeće:
|
— |
za određivanje udjela sirove masti: postupak B metode za određivanje sirovih ulja i masti iz dijela H Priloga III., |
|
— |
za određivanje udjela škroba: polarimetrijska metoda iz dijela L Priloga III. |
PRILOG VIII.
ANALITIČKE METODE ZA KONTROLU NEPROPISNE PRISUTNOSTI KRMNIH DODATAKA KOJI VIŠE NISU DOPUŠTENI U HRANI ZA ŽIVOTINJE
Važne napomene:
Za detekciju nepropisne prisutnosti krmnih dodataka koji više nisu dopušteni mogu se primjenjivati osjetljivije metode analize od analitičkih metoda navedenih u ovom Prilogu.
Za potvrdu se primjenjuju metode analize iz ovog Priloga.
A. ODREĐIVANJE METIL BENZOKVATA
7-benziloksi-6-butil-3-metoksikarbonil-4-kinolon
1. Svrha i područje primjene
Ovom se metodom omogućuje određivanje razine metil benzokvata u hrani za životinje. Granica kvantifikacije je 1 mg/kg.
2. Načelo
Metil benzokvat se iz uzorka ekstrahira otopinom metanol metansulfonske kiseline. Ekstrakt se pročisti diklormetanom ionsko-izmjenjivačkom kromatografijom i zatim ponovo diklormetanom. Udio metil benzokvata određuje se tekućinskom kromatografijom visoke učinkovitosti (HPLC) s reverznom fazom uz upotrebu UV detektora.
3. Reagensi.
|
3.1. |
Diklormetan. |
|
3.2. |
Metanol, za HPLC. |
|
3.3. |
Mobilna faza HPLC-a. Smjesa metanola (točka 3.2.) i vode (za HPLC) 75 + 25 (v + v). Filtrira se kroz filtar 0,22 μm (točka 4.5.) a otopina se otplini (npr. 10 minuta u ultrazvučnoj kupelji). |
|
3.4. |
Otopina metansulfonske kiseline, c = 2 %. Metanolom se razrijedi 20,0 ml metansulfonske kiseline do 1 000 ml (točka 3.2.). |
|
3.5. |
Otopina klorovodične kiseline, c = 10 %. Vodom se razrijedi 100 ml klorovodične kiseline (ρ20 1,18 g/ml) do 1 000 ml. |
|
3.6. |
Kationska izmjenjivačka smola Amberlite CG-120 (Na), 100 do 200 mesh. Smola se prije uporabe obradi. Suspendira se 100 g smole u 500 ml otopine klorovodične kiseline (točka 3.5.) i uz stalno miješanje zagrijava na grijaćoj ploči do ključanja. Ohladi se i kiselina odlije. Filtrira se kroz papirni filtar pod vakuumom. Smola se dvaput ispere s 500 ml vode, zatim s 250 ml metanola (točka 3.2.). Ispere se još jednom s 250 ml metanola i osuši propuhivanjem zraka kroz filtriranu pogaču. Suha se smola pohrani u zatvorenoj boci. |
|
3.7. |
Standardna tvar: čisti metil benzokvat (7-benziloksi-6-butil-3-metoksikarbonil-4-kinolon) |
|
3.7.1. |
Odvagne se 50 mg standardne tvari (točka 3.7.) s preciznošću od 0,1 mg, otopi u otopini metansulfonske kiseline (točka 3.4.) u odmjernoj tikvici obujma 100 ml, dopuni do oznake i promiješa. |
|
3.7.2. |
Prenese se 5,0 ml osnovne standardne otopine metil benzokvata (točka 3.7.1.) u graduiranu tikvicu obujma 50 ml, dopuni do oznake metanolom (točka 3.2.) i promiješa. |
|
3.7.3. |
U seriju graduiranih tikvica obujma 25 ml prenese se 1,0, 2,0, 3,0, 4,0 i 5,0 ml intermedijarne standardne otopine metil benzokvata (točka 3.7.2). Dopuni se do oznake mobilnom fazom (točka 3.3.) i promiješa. Koncentracija metil benzokvata u tim otopinama iznosi 2,0, 4,0, 6,0, 8,0 i 10,0 μg/ml. Prije uporabe ove se otopine moraju svježe pripremiti. |
4. Oprema
|
4.1. |
Laboratorijska tresilica. |
|
4.2. |
Tankoslojni rotacijski otparivač. |
|
4.3. |
Staklena kolona (250 mm × 15 mm) s pipcem i spremnikom kapaciteta približno 200 ml. |
|
4.4. |
Oprema za HPLC s ultraljubičastim detektorom s mogućnošću promjene valne duljine, ili detektorom s nizom dioda. |
|
4.4.1. |
Kolona za tekućinsku kromatografiju: 300 mm × 4 mm, C18, s veličinom čestica punjenja 10 μm ili jednakovrijedna. |
|
4.5. |
Membranski filtri, 0,22 μm. |
|
4.6. |
Membranski filtri, 0,45 μm. |
5. Postupak
5.1. Općenito
|
5.1.1. |
Analizom slijepe probe hrane za životinje provjerava se da nisu prisutni ni metil benzokvat niti interferentne tvari. |
|
5.1.2. |
Test iskorištenja provodi se analizom slijepe probe hrane za životinje kojoj se doda količina metil benzokvata slična količini u uzorku. Za dobivanje koncentracije od 15 mg/kg u 20 g slijepe probe hrane za životinje doda se 600 μl osnovne standardne otopine (točka 3.7.1.), promiješa i pričeka 10 minuta prije početka postupka ekstrakcije (točka 5.2.).
|
5.2. Ekstrakcija
Odvagne se približno 20 g pripravljenog uzorka s preciznošću od 0,01 g i prenese u konusnu tikvicu obujma 250 ml. Doda se 100,0 ml otopine metansulfonske kiseline (točka 3.4.) i mehanički protresa (točka 4.1.) 30 minuta. Otopina se filtrira kroz papirni filtar, a filtrat se spremi za postupak razdvajanja tekućina-tekućina (točka 5.3.).
5.3. Razdvajanje tekuće-tekuće
U lijevak za odjeljivanje obujma 500 ml u kojem se nalazi 100 ml otopine klorovodične kiseline (točka 3.5.) prenese se 25,0 ml filtrata dobivenog u postupku iz točke 5.2. U lijevak se doda 100 ml diklormetana (točka 3.1.) i trese jednu minutu. Pričeka se da se slojevi razdvoje te se donji (diklormetanski) sloj odlije u tikvicu s okruglim dnom obujma 500 ml. Ponovi se ekstrakcija vodene faze s 40 ml diklormetana, a dobiveni se ekstrakti pomiješaju s prvim ekstraktom u tikvici s okruglim dnom. Ekstrakt diklormetana se otpari do suhog u rotacijskom otparivaču (točka 4.2.) pri sniženom tlaku, na 40 °C. Ostatak se otopi u 20 – 25 ml metanola (točka 3.2.), tikvica se začepi i sav ekstrakt spremi za ionsko-izmjenjivačku kromatografiju (točka 5.4.).
5.4. Ionsko-izmjenjivačka kromatografija
5.4.1.
U donji dio staklene kolone postavi se mali čep od staklene vune (točka 4.3.). Pripremi se suspenzija s 5,0 g obrađene kationske izmjenjivačke smole (točka 3.6.) i 50 ml klorovodične kiseline (točka 3.5.), ulije u staklenu kolonu i pričeka da se umiri. Ukloni se višak kiseline do visine neposredno iznad površine smole, zatim se kolona ispire vodom do neutralne reakcije eluata na lakmusov papir. U kolonu se prenese 50 ml metanola (točka 3.2.) i ostavi da istekne do površine smole.
5.4.2.
Dobiveni se ekstrakt (točka 5.3.) pipetom pažljivo prenese u kolonu. Tikvica s okruglim dnom se dvaput ispere s 5 – 10 ml metanola (točka 3.2.), a isprane tekućine prenesu u kolonu. Ekstrakt se ulije do površine smole i kolona ispere s 50 ml metanola, pri čemu brzina protoka ne smije prijeći 5 ml na minutu. Eluat se odbaci. Metil benzokvat se iz kolone eluira sa 150 ml otopine metansulfonske kiseline (točka 3.4.), a eluat iz kolone prikupi u konusnu tikvicu obujma 250 ml.
5.5. Razdvajanje tekuće-tekuće
Dobiveni eluat (točka 5.4.2.) prenese se u lijevak za odjeljivanje obujma 1 l. Konusna tikvica se ispere s 5 – 10 ml metanola (točka 3.2.), a isprana tekućina pomiješa sa sadržajem u lijevku za odjeljivanje. Doda se 300 ml otopine klorovodične kiseline (točka 3.5.) i 130 ml diklormetana (točka 3.1.). Trese se 1 minutu i ostavi da se faze razdvoje. Donji (diklormetanski) sloj se odlije u tikvicu s okruglim dnom obujma 500 ml. Ekstrakcija vodene faze se ponovi dvaput sa 70 ml diklormetana, a oba se ekstrakta pomiješaju s prvim u tikvici s okruglim dnom.
Ekstrakt diklormetana se otpari do suhog u rotacijskom otparivaču (točka 4.2.) pod sniženim tlakom na 40 °C. Ostatak u tikvici se otopi u približno 5 ml metanola (točka 3.2.), a otopina prenese u graduiranu tikvicu obujma 10 ml. Tikvica s okruglim dnom se dvaput ispere s 1 – 2 ml metanola, a isprane tekućine prenesu u graduiranu tikvicu. Dopuni se metanolom do oznake i promiješa. Alikvotni dio se filtrira kroz membranski filtar (točka 4.6.). Ta se otopina pohrani za određivanje putem HPLC-a (točka 5.6.).
5.6. Određivanje putem HPLC-a
5.6.1.
Sljedeći uvjeti predlažu se kao smjernice; mogu se koristiti i drugi uvjeti ako se njima jamče jednakovrijedni rezultati:
|
— |
kolona za tekućinsku kromatografiju (točka 4.4.1.), |
|
— |
mobilna faza za HPLC-a: smjesa metanola i vode (točka 3.3.), |
|
— |
brzina protoka: 1 – 1,5 ml/min, |
|
— |
valna duljina detekcije: 265 nm, |
|
— |
volumen za ubrizgavanje: 20 – 50 μl. |
Stabilnost kromatografskog sustava se provjerava višekratnim ubrizgavanjem kalibracijske otopine koncentracije 4,0 μg/ml (točka 3.7.3.) sve dok se ne dobiju konstantne visine ili površine vršaka i konstantna vremena retencije.
5.6.2.
Više puta se ubrizga svaka kalibracijska otopina (točka 3.7.3.) i za svaku koncentraciju izmjere vrijednosti visina (površina) vršaka. Kalibracijska krivulja se pripremi tako da se na ordinatu nanose srednje vrijednosti visina ili površina vršaka kalibracijskih otopina, a na apscisu odgovarajuće koncentracije u μg/ml.
5.6.3.
Više puta se ubrizga ekstrakt uzorka (točka 5.5.), pri čemu se koriste jednaki volumeni kao za kalibracijske otopine, zatim se odredi srednja vrijednost visina (površina) vršaka metil benzokvata.
6. Izračun rezultata
Koncentracija otopine uzorka u μg/ml određuje se iz srednjih vrijednosti vršaka (površina) metil benzokvata iz kalibracijske krivulje (točka 5.6.2.).
Udio metil benzokvata (w) (mg/kg) u uzorku izračunava se sljedećom formulom:

pri čemu je:
|
c |
= |
koncentracija metil benzokvata u otopini uzorka u μg/ml. |
|
m |
= |
masa pokusnog uzorka u gramima. |
7. Vrednovanje rezultata
7.1. Identifikacija
Identifikacija analita može se potvrditi ko-kromatografijom ili detektorom s nizom dioda kojima se uspoređuje spektar ekstrakta uzorka i spektar kalibracijske otopine (točka 3.7.3.) koncentracije 10 μg/ml.
7.1.1.
Ekstraktu uzorka se doda odgovarajuća količina intermedijarne standardne otopine (točka 3.7.2.). Količina dodanog metil benzokvata mora biti slična procijenjenoj količini metil benzokvata u ekstraktu uzorka.
Može se povećati samo visina vrška metil benzokvata, uzimajući u obzir dodane količine i razrjeđenje ekstrakta. Širina vrha na polovici njegove najviše visine mora biti unutar 10 % vrijednosti prvotne širine.
7.1.2.
Rezultati se ocjenjuju u skladu sa sljedećim mjerilima:
|
(a) |
valne duljine maksimalne apsorpcije spektra uzorka i spektra standarda, zabilježene na najvišoj točki vršaka kromatograma, moraju biti jednake unutar granice određene razlučivošću sustava za detekciju. Kod detektora s nizom dioda ta se vrijednost obično nalazi unutar ± 2 nm; |
|
(b) |
između 220 i 350 nm, spektar uzorka i spektar standarda, zabilježeni na najvišoj točki kromatograma, ne smiju se razlikovati za dijelove spektra unutar područja 10 – 100 % relativne apsorbance. To je mjerilo ispunjeno kada su prisutne jednake najviše vrijednosti i kada odstupanje između dva spektra ni na jednoj točki ne prelazi 15 % apsorbance standardnog analita; |
|
(c) |
između 220 i 350 nm, spektri krivulje rasta, najviše točke vrška i krivulje pada dobiveni iz ekstrakta uzorka, ne smiju se međusobno razlikovati za dijelove spektra unutar područja 10 – 100 % relativne apsorbance. To je mjerilo ispunjeno kada su prisutne jednake najviše vrijednosti i kada odstupanje između dva spektra ni na jednoj točki ne prelazi 15 % apsorbance spektra najviše točke. |
Ako nije ispunjeno jedno od navedenih mjerila, prisutnost analita nije potvrđena.
7.2. Ponovljivost
Razlika između rezultata dvaju usporednih postupaka određivanja, izvedenih na istom uzorku, ne smije prijeći: 10 % najvišeg rezultata za udio metil benzokvata između 4 i 20 mg/kg.
7.3. Iskorištenje
Za obogaćeni slijepi uzorak hrane za životinje iskorištenje mora iznositi najmanje 90 %.
8. Rezultati međulaboratorijske studije
Provedena je analiza 5 uzoraka u 10 laboratorija. Svaki se uzorak analizirao dvaput.
|
|
Slijepa proba |
Brašno 1 |
Pelete 1 |
Brašno 2 |
Pelete 2 |
|
Srednja vrijednost |
ND |
4,50 |
4,50 |
8,90 |
8,70 |
|
[mg/kg] sr [mg/kg] |
— |
0,30 |
0,20 |
0,60 |
0,50 |
|
CVr [%] |
— |
6,70 |
4,40 |
6,70 |
5,70 |
|
sR [mg/kg] |
— |
0,40 |
0,50 |
0,90 |
1,00 |
|
CVR [%] |
— |
8,90 |
11,10 |
10,10 |
11,50 |
|
Iskorištenje [%] |
— |
92,00 |
93,00 |
92,00 |
89,00 |
|
ND |
= |
nije određeno |
|
sr |
= |
standardna devijacija ponovljivosti |
|
CVr |
= |
koeficijent varijacije ponovljivosti u % |
|
sR |
= |
standardna devijacija obnovljivosti |
|
CVR |
= |
koeficijent varijacije obnovljivosti u %. |
B. ODREĐIVANJE OLAKVINDOKSA
2-[N-2’-(hidroksietil)karbamoil]-3-metilkinoksalin-N1,N4-dioksid
1. Svrha i područje primjene
Ovom se metodom omogućuje određivanje razine olakvindoksa u hrani za životinje. Granica kvantifikacije je 5 mg/kg.
2. Načelo
Uzorak se ekstrahira smjesom vode i metanola. Udio olakvindoksa određuje se tekućinskom kromatografijom visoke učinkovitosti (HPLC) s reverznom fazom uz upotrebu UV detektora.
3. Reagensi
|
3.1. |
Metanol. |
|
3.2. |
Metanol, za HPLC. |
|
3.3. |
Voda, za HPLC. |
|
3.4. |
Mobilna faza za HPLC. Smjesa vode (točka 3.3) i metanola (točka 3.2), 900 + 100 (V + V). |
|
3.5. |
Standardna tvar: čisti olakvindoks 2-[N-2’-(hidroksietil)karbamoil]-3-metilkinoksalin-N1,N4-dioksid, E 851. |
|
3.5.1. |
Odvagne se 50 mg olakvindoksa (točka 3.5.) s preciznošću od 0,1 mg i prenese u graduiranu tikvicu obujma 200 ml, zatim se doda približno 190 ml vode. Tikvica se zatim drži 20 minuta u ultrazvučnoj kupelji (točka 4.1.). Nakon obrade ultrazvukom, otopina se zagrije do sobne temperature, dopuni vodom do oznake i promiješa. Tikvica se omota aluminijskom folijom i pohrani u hladnjaku. Ova se otopina mora svježe pripremiti svaki mjesec. |
|
3.5.2 |
Prenese se 10,0 ml osnovne standardne otopine (točka 3.5.1.) u graduiranu tikvicu obujma 100 ml, dopuni mobilnom fazom (točka 3.4.) do oznake i promiješa. Tikvica se omota aluminijskom folijom i pohrani u hladnjaku. Ova se otopina mora svježe pripremiti svaki dan. |
|
3.5.3. |
U seriju graduiranih tikvica obujma 50 ml prenese se 1,0, 2,0, 5,0, 10,0, 15,0 i 20,0 ml intermedijarne standardne otopine (točka 3.5.2.). Dopuni se mobilnom fazom (točka 3.4.) do oznake i promiješa. Tikvice se omotaju aluminijskom folijom. Ove otopine odgovaraju količini od 0,5, 1,0, 2,5, 5,0, 7,5 i 10,0 μg olakvindoksa na ml. Ove se otopine moraju svježe pripremiti svaki dan. |
4. Oprema
|
4.1. |
Ultrazvučna kupelj |
|
4.2. |
Mehanička tresilica |
|
4.3. |
Oprema za HPLC s detektorom ultraljubičastog svjetla s mogućnošću promjene valne duljine ili detektorom s nizom dioda |
|
4.3.1. |
Kolona za tekućinsku kromatografiju, 250 mm × 4 mm, C18, s veličinom čestica punjenja 10 μm ili jednakovrijedna |
|
4.4. |
Membranski filtri, 0,45 μm |
5. Postupak
|
Napomena |
: |
Olakvindoks je osjetljiv na svjetlo. Svi se postupci izvode pod prigušenim svjetlom ili u laboratorijskom posuđu od smeđeg stakla. |
5.1. Općenito
|
5.1.1. |
Analizom slijepe probe hrane za životinje potvrđuje se da nisu prisutni niti olakvindoks niti interferentne tvari. |
|
5.1.2. |
Test iskorištenja provodi se analizom slijepe probe hrane za životinje koja se obogaćuje dodavanjem količine olakvindoksa slične količini u uzorku. Kako bi se dobila koncentracija od 50 mg/kg, prenese se 10,0 ml osnovne standardne otopine (točka 3.5.1.) u konusnu tikvicu obujma 250 ml i otopina otpari do približno 0,5 ml. Doda se 50 g slijepe probe hrane za životinje, dobro promiješa i ostavi 10 minuta te ponovo promiješa nekoliko puta prije prelaska na postupak ekstrakcije (točka 5.2.).
|
5.2. Ekstrakcija
Odvagne se približno 50 g uzorka s preciznošću od 0,01 g. Prenese se u konusnu tikvicu obujma 1 000 ml, doda 100 ml metanola (točka 3.1.) i tikvica 5 minuta drži u ultrazvučnoj kupelji (točka 4.1.). Doda se 410 ml vode i ostavi daljnjih 15 minuta u ultrazvučnoj kupelji. Tikvica se izvadi iz ultrazvučne kupelji, 30 minuta protresa u tresilici (točka 4.2.) i zatim filtrira kroz nabrani filtar. Prenese se 10,0 ml filtrata u graduiranu tikvicu obujma 20 ml, dopuni vodom do oznake i promiješa. Alikvot se filtrira kroz membranski filtar (točka 4.4.) (vidjeti točku 9. Napomena). Prelazi se na postupak određivanja putem HPLC-a (točka 5.3.).
5.3. Određivanje putem HPLC-a
5.3.1.
Sljedeći se uvjeti predlažu kao smjernice; mogu se koristiti i drugi uvjeti ako se njima jamče jednakovrijedni rezultati.
|
Analitička kolona (točka 4.3.1.) |
|
|
Mobilna faza (točka 3.4.): |
smjesa vode (točka 3.3.) i metanola (točka 3.2.), 900 + 100 (V + V) |
|
Brzina protoka: |
1,5 – 2 ml/min |
|
Valna duljina detekcije: |
380 nm |
|
Volumen za ubrizgavanje: |
20 – 100 μl |
Stabilnost kromatografskog sustava se provjerava višekratnim ubrizgavanjem kalibracijske otopine (točka 3.5.3.) koncentracije 2,5 μg/ml sve dok se ne dobiju konstantne visine vršaka i konstantna vremena retencije.
5.3.2.
Više puta se ubrizga svaka kalibracijska otopina (točka 3.5.3.) i za svaku koncentraciju izmjere srednje vrijednosti visina (površina) vršaka. Kalibracijska krivulja se pripremi tako da se na ordinatu nanose srednje vrijednosti visina (površina) vršaka kalibracijskih otopina, a na apscisu odgovarajuće koncentracije u μg/ml.
5.3.3.
Više puta se ubrizga ekstrakt uzorka (točka 5. 2.), pri čemu se koriste jednaki volumeni kao za kalibracijsku otopinu, zatim se odredi srednja vrijednost visina (površina) vršaka olakvindoksa.
6. Izračun rezultata
Koncentracija otopine uzorka u μg/ml određuje se iz srednjih vrijednosti visina (površina) vršaka olakvindoksa u otopini uzorka iz kalibracijske krivulje (točka 5.3.2.).
Udio olakvindoksa (w) (mg/kg) u uzorku izračunava se sljedećom formulom:

pri čemu je:
|
c |
= |
koncentracija olakvindoksa u ekstraktu uzorka (točka 5.2.) u μg/ml |
|
m |
= |
masa pokusnog uzorka u g (točka 5.2.). |
7. Vrednovanje rezultata
7.1. Identifikacija
Identifikacija analita može se potvrditi ko-kromatografijom ili detektorom s nizom dioda kojim se uspoređuje spektar ekstrakta uzorka (točka 5.2.) i spektar kalibracijske otopine (točka 3.5.3.) koncentracije 5,0 μg/ml.
7.1.1.
U ekstrakt uzorka (točka 5.2.) doda se primjerena količina kalibracijske otopine (točka 3.5.3.). Količina dodanog olakvindoksa mora biti slična procijenjenoj količini olakvindoksa u ekstraktu uzorka.
Dopušteno je povećanje samo visine vrška olakvindoksa uzimajući u obzir dodane količine i razrjeđenje ekstrakta. Širina vrška, na polovici maksimalne visine, mora biti unutar ± 10 % početne širine vrška olakvindoksa u ekstraktu uzorka bez dodanog olakvindoksa.
7.1.2.
Rezultati se ocjenjuju u skladu sa sljedećim mjerilima:
|
(a) |
Valne duljine maksimalne apsorpcije spektra uzorka i spektra standarda, zabilježene na najvišoj točki vršaka kromatograma, moraju biti jednake unutar granice određene razlučivošću sustava za detekciju. Kod detektora s nizom dioda ta se vrijednost obično nalazi unutar ± 2 nm. |
|
(b) |
Između 220 i 400 nm, spektar uzorka i spektar standarda, zabilježeni na najvišoj točki kromatograma, ne smiju se razlikovati za dijelove spektra unutar područja 10 – 100 % relativne apsorbance. To je mjerilo ispunjeno kada su prisutne jednake najviše vrijednosti i kada odstupanje između dva spektra ni na jednoj točki ne prelazi 15 % apsorbance standardnog analita. |
|
(c) |
Između 220 i 400 nm, spektri krivulje rasta, najviše točke vrška i krivulje pada dobiveni iz ekstrakta uzorka, ne smiju se međusobno razlikovati za dijelove spektra unutar područja 10 – 100 % relativne apsorbance. To je mjerilo ispunjeno kada su prisutne jednake najviše vrijednosti i kada odstupanje između dva spektra ni na jednoj točki ne prelazi 15 % apsorbance spektra najviše točke. |
Ako nije ispunjeno jedno od navedenih mjerila, prisutnost analita nije potvrđena.
7.2. Ponovljivost
Razlika između rezultata dvaju usporednih postupaka određivanja, izvedenih na istom uzorku, ne smije prijeći 15 % najvišeg rezultata za udio olakvindoksa između 10 i 200 mg/kg.
7.3. Iskorištenje
Za obogaćeni slijepi uzorak hrane za životinje iskorištenje mora iznositi najmanje 90 %.
8. Rezultati međulaboratorijske studije
Provedena je međulaboratorijska studija na razini EZ-a u kojoj se u najviše 13 laboratorija analiziralo četiri uzorka hrane za prasad, uključujući jednu slijepu probu. Rezultati su prikazani u donjoj tablici:
|
|
Uzorak 1 |
Uzorak 2 |
Uzorak 3 |
Uzorak 4 |
|
L |
13 |
10 |
11 |
11 |
|
n |
40 |
40 |
44 |
44 |
|
Srednja vrijednost [mg/kg] |
— |
14,6 |
48,0 |
95,4 |
|
Sr [mg/kg] |
— |
0,82 |
2,05 |
6,36 |
|
SR [mg/kg] |
— |
1,62 |
4,28 |
8,42 |
|
CVr [%] |
— |
5,6 |
4,3 |
6,7 |
|
CVR [%] |
— |
11,1 |
8,9 |
8,8 |
|
Nominalni udio |
|
|
|
|
|
[mg/kg] |
— |
15 |
50 |
100 |
|
Iskorištenje % |
— |
97,3 |
96,0 |
95,4 |
|
L |
= |
broj laboratorija |
|
n |
= |
broj pojedinačnih vrijednosti |
|
Sr |
= |
standardna devijacija ponovljivosti |
|
SR |
= |
standardna devijacija obnovljivosti |
|
CVr |
= |
koeficijent varijacije ponovljivosti |
|
CVR |
= |
koeficijent varijacije obnovljivosti. |
9. Napomena
Iako ova metoda nije vrednovana za hranu za životinje koja sadrži više od 100 mg/kg olakvindoksa, mogu se dobiti zadovoljavajući rezultati uporabom manje mase uzorka i/ili razrjeđivanjem ekstrakta (točka 5.2.) tako da se dobije koncentracija u području kalibracijske krivulje (točka 5.3.2.).
C. ODREĐIVANJE AMPROLIUMA
1-[(4-amino-2-propilpirimidin-5-il)metil]-2-metil-piridinijum klorid hidroklorid
1. Svrha i područje primjene
Ovom se metodom omogućuje određivanje razine amproliuma u hrani za životinje i premiksima. Granica detekcije je 1 mg/kg, a granica kvantifikacije 5 mg/kg.
2. Načelo
Uzorak se ekstrahira smjesom metanola i vode. Nakon razrjeđivanja mobilnom fazom i membranske filtracije, udio amproliuma određuje se tekućinskom kromatografijom visoke učinkovitosti (HPLC) s izmjenom kationa uz upotrebu UV detektora.
3. Reagensi
|
3.1. |
Metanol. |
|
3.2. |
Acetonitril, za HPLC. |
|
3.3. |
Voda, za HPLC. |
|
3.4. |
Otopina natrijevog dihidrogen fosfata, c = 0,1 mol/l. U graduiranoj tikvici obujma 1 000 ml otopi se u vodi (točka 3.3.) 13,80 g natrijevog dihidrogen fosfat monohidrata, dopuni vodom (točka 3.3.) do oznake i promiješa. |
|
3.5. |
Otopina natrijevog perklorata, c = 1,6 mol/l. U odmjernoj tikvici obujma 1 000 ml otopi se u vodi (točka 3.3.) 224,74 g natrijevog perklorat monohidrata, dopuni vodom (točka 3.3.) do oznake i promiješa. |
|
3.6. |
Mobilna faza za HPLC (vidjeti napomenu pod točkom 9.1.). Smjesa acetonitrila (točka 3.2.), otopine natrijevog dihidrogen fosfata (točka 3.4.) i otopine natrijevog perklorata (točka 3.5.), 450 + 450 + 100 (v + v + v). Prije uporabe filtrira se membranskim filtrom 0,22 μm (točka 4.3.), a otopina se otplini (npr. najmanje 15 minuta u ultrazvučnoj kupelji (točka 4.4.)). |
|
3.7. |
Standardna tvar: čisti amprolium, l-[(4-amino-2-propilpirimidin-5-il)metil]-2-metil-piridinijum klorid hidroklorid, E 750 (vidjeti napomenu pod točkom 9.2). |
|
3.7.1. |
Odvagne se 50 mg amproliuma (točka 3.7.) s preciznošću od 0,1 mg i prenese u graduiranu tikvicu obujma 100 ml, otopi u 80 ml metanola (točka 3.1) i tikvicu 10 minuta drži u ultrazvučnoj kupelji (točka 4.4.). Nakon obrade ultrazvukom otopina se zagrije do sobne temperature, dopuni vodom do oznake i promiješa. Na temperaturi ≤ 4 °C otopina je stabilna 1 mjesec. |
|
3.7.2. |
Pipetom se prenese 5,0 ml osnovne standardne otopine (točka 3.7.1.) u graduiranu tikvicu obujma 50 ml, dopuni do oznake ekstrakcijskim otapalom (točka 3.8.) i promiješa. Na temperaturi ≤ 4 °C otopina je stabilna 1 mjesec. |
|
3.7.3. |
U seriju graduiranih tikvica obujma 50 ml prenese se 0,5, 1,0, i 2,0 ml intermedijarne standardne otopine (točka 3.7.2.). Dopuni se do oznake mobilnom fazom (točka 3.6.) i promiješa. Ove otopine odgovaraju koncentraciji od 0,5, 1,0 i 2,0 μg amproliuma na ml. Ove se otopine moraju pripremiti svježe prije uporabe. |
|
3.8. |
Ekstrakcijsko otapalo. Smjesa metanola (točka 3.1.) i vode 2 + 1 (v + v) |
4. Oprema
|
4.1. |
Oprema za HPLC sa sustavom za ubrizgavanje prikladnim za ubrizgavanje volumena od 100 μl. |
|
4.1.1. |
Kolona za tekućinsku kromatografiju, 125 mm × 4 mm s izmjenom kationa, punjenje Nucleosil 10 SA veličina čestica 5 ili 10 μm ili jednakovrijedno. |
|
4.1.2. |
UV detektor s mogućnošću promjene valne duljine ili detektor s nizom dioda. |
|
4.2. |
Membranski filtar, materijal PTFE, 0,45 μm. |
|
4.3. |
Membranski filtar, 0,22 μm. |
|
4.4. |
Ultrazvučna kupelj. |
|
4.5. |
Mehanička tresilica ili magnetska mješalica. |
5. Postupak
5.1. Općenito
5.1.1.
Kod primjene testa iskorištenja (točka 5.1.2.) analizom slijepe probe hrane za životinje utvrđuje se da nisu prisutni niti amprolium niti interferentne tvari. Slijepa proba hrane za životinje mora biti slične vrste kao i uzorak, a analizom se u njoj ne smiju pronaći amprolium niti interferentne tvari.
5.1.2.
Test iskorištenja provodi se analizom slijepe probe hrane za životinje u koju se doda količina amproliuma slična količini u uzorku. Kako bi se dobila koncentracija od 100 mg/kg, prenese se 10,0 ml osnovne standardne otopine (točka 3.7.1.) u konusnu tikvicu obujma 250 ml i otopina otpari do približno 0,5 ml. Doda se 50 g slijepe probe hrane za životinje, dobro promiješa i ostavi 10 minuta te ponovo promiješa nekoliko puta prije prelaska na postupak ekstrakcije (točka 5.2.).
Ako nije dostupna slijepa probe hrane za životinje slična uzorku (vidjeti točku 5.1.1.), test iskorištenja može se umjesto toga provesti metodom dodavanja standarda. U tom se slučaju uzorku za analizu doda količina amproliuma slična onoj u uzorku. Taj se uzorak analizira zajedno s uzorkom bez dodanog amproliuma, a iskorištenje se izračuna oduzimanjem.
5.2. Ekstrakcija
5.2.1.
Odvagne se 5 – 40 g uzorka, ovisno o udjelu amproliuma, s preciznošću od 0,01 g, prenese u konusnu tikvicu obujma 500 ml, te se doda 200 ml ekstrakcijskog otapala (točka 3.8.). Tikvica se stavi u ultrazvučnu kupelj (točka 4.4.) i ostavi 15 minuta. Izvadi se iz ultrazvučne kupelji i 1 sat protresa u tresilici ili miješa magnetskom mješalicom (točka 4.5.). Alikvot ekstrakta se razrijedi mobilnom fazom (točka 3.6.) do udjela amproliuma od 0,5 – 2 μg/ml i promiješa (vidjeti napomenu pod točkom 9.3.). 5 – 10 ml tako razrijeđene otopine filtrira se kroz membranski filtar (točka 4.2.). Prelazi se na postupak određivanja putem HPLC-a (točka 5.3.).
5.2.2.
Odvagne se 1 – 4 g uzorka, ovisno o udjelu amproliuma, s preciznošću od 0,001 g, prenese u konusnu tikvicu obujma 500 ml te se doda 200 ml ekstrakcijskog otapala (točka 3.8.). Tikvica se stavi u ultrazvučnu kupelj (točka 4.4.) i ostavi 15 minuta. Izvadi se iz ultrazvučne kupelji i 1 sat protresa u tresilici ili miješa magnetskom mješalicom (točka 4.5.). Alikvot ekstrakta se razrijedi mobilnom fazom (točka 3.6.) do udjela amproliuma od 0,5 – 2 μg/ml i promiješa. 5 – 10 ml tako razrijeđene otopine filtrira se kroz membranski filtar (točka 4.2.). Prelazi se na postupak određivanja putem HPLC-a (točka 5.3.).
5.3. Određivanje putem HPLC
5.3.1.
Sljedeći se uvjeti predlažu kao smjernice; mogu se koristiti i drugi uvjeti ako se njima jamče jednakovrijedni rezultati.
|
Kolona za tekućinsku kromatografiju (točka 4.1.1.): |
125 mm × 4 mm, s izmjenom kationa (punjenje Nucleosil 10 SA, veličina čestica 5 ili 10 μm ili jednakovrijedno) |
|
Mobilna faza (točka 3.6): |
Smjesa acetonitrila (točka 3.2.), otopine natrijevog dihidrogen fosfata (točka 3.4.) i otopine natrijevog perklorata (točka 3.5.), 450 + 450 + 100 (v + v + v) |
|
Brzina protoka: |
0,7 – 1 ml/min |
|
Valna duljina detekcije: |
264 nm |
|
Volumen za ubrizgavanje: |
100 μl |
Stabilnost kromatografskog sustava se provjerava višekratnim ubrizgavanjem kalibracijske otopine (točka 3.7.3.) koncentracije 1,0 μg/ml sve dok se ne dobiju konstantne visine vršaka i konstantna vremena retencije.
5.3.2.
Više puta se ubrizga svaka kalibracijska otopina (točka 3.7.3.) te se za svaku koncentraciju izmjere srednje vrijednosti visina (površina) vršaka. Kalibracijska krivulja se pripremi tako da se na ordinatu nanose srednje vrijednosti visina (površina) vršaka kalibracijskih otopina, a na apscisu odgovarajuće koncentracije u μg/ml.
5.3.3.
Više puta se ubrizga ekstrakt uzorka (točka 5.2.), pri čemu se koriste jednaki volumeni kao za kalibracijsku otopinu, zatim se odredi srednja vrijednost visina (površina) vršaka amproliuma.
6. Izračun rezultata
Koncentracija otopine uzorka u μg/ml određuje se iz srednjih vrijednosti visina (površina) vršaka amproliuma u otopini uzorka iz kalibracijske krivulje (točka 5.3.2.).
Udio amproliuma (w) (mg/kg) u uzorku izračunava se sljedećom formulom:

pri čemu je:
|
V |
= |
volumen ekstrakcijskog otapala (točka 3.8.) u ml u skladu s točkom 5.2. (tj. 200 ml) |
|
c |
= |
koncentracija amproliuma u ekstraktu uzorka (točka 5.2.) u μg/ml |
|
f |
= |
faktor razrjeđenja u skladu s točkom 5.2. |
|
m |
= |
masa pokusnog uzorka u gramima. |
7. Vrednovanje rezultata
7.1. Identifikacija
Identifikacija analita može se potvrditi ko-kromatografijom ili detektorom s nizom dioda kojim se uspoređuje spektar ekstrakta uzorka (točka 5.2.) i spektar kalibracijske otopine (točka 3.7.3.) koncentracije 2,0 μg/ml.
7.1.1.
U ekstrakt uzorka (točka 5.2.) doda se primjerena količina kalibracijske otopine (točka 3.7.3.). Količina dodanog amproliuma mora biti slična procijenjenoj količini amproliuma u ekstraktu uzorka.
Dopušteno je povećanje samo visine vrška amproliuma uzimajući u obzir dodane količine i razrjeđenje ekstrakta. Širina vrška, na polovici maksimalne visine, mora biti unutar ± 10 % početne širine amproliuma u ekstraktu uzorka bez dodanog amproliuma.
7.1.2.
Rezultati se ocjenjuju u skladu sa sljedećim mjerilima:
|
(a) |
Valne duljine maksimalne apsorpcije spektra uzorka i spektra standarda, zabilježene na najvišoj točki vršaka kromatograma, moraju biti jednake unutar granice određene razlučivošću sustava za detekciju. Kod detektora s nizom dioda ta se vrijednost obično nalazi unutar ± 2 nm. |
|
(b) |
Između 210 i 320 nm, spektar uzorka i spektar standarda, zabilježeni na najvišoj točki kromatograma, ne smiju se razlikovati za dijelove spektra unutar raspona 10 – 100 % relativne apsorbance. To je mjerilo ispunjeno kada su prisutne jednake najviše vrijednosti i kada odstupanje između dva spektra ni na jednoj točki ne prelazi 15 % apsorbance standardnog analita. |
|
(c) |
Između 210 i 320 nm, spektri krivulje rasta, najviše točke vrška i krivulje pada dobiveni iz ekstrakta uzorka, ne smiju se međusobno razlikovati za dijelove spektra unutar raspona 10 – 100 % relativne apsorbance. To je mjerilo ispunjeno kada su prisutne jednake najviše vrijednosti i kada odstupanje između dva spektra ni na jednoj točki ne prelazi 15 % apsorbance spektra najviše točke. |
Ako nije ispunjeno jedno od navedenih mjerila, prisutnost analita nije potvrđena.
7.2. Ponovljivost
Razlika između rezultata dvaju usporednih postupaka određivanja, izvedenih na istom uzorku, ne smije prijeći:
|
— |
15 % više vrijednosti za udio amproliuma od 25 – 500 mg/kg, |
|
— |
75 mg/kg za udio amproliuma od 500 – 1 000 mg/kg, |
|
— |
7,5 % više vrijednosti za udio amproliuma veći od 1 000 mg/kg. |
7.3. Iskorištenje
Kod obogaćenog (slijepog) uzorka iskorištenje iznosi najmanje 90 %.
8. Rezultati međulaboratorijske studije
Provedena je međulaboratorijska studija u kojoj se analiziralo tri uzorka hrane za perad (uzorci 1 – 3), jedan uzorak mineralne hrane za životinje (uzorak 4) i jedan uzorak premiksa (uzorak 5). Rezultati su navedeni u donjoj tablici:
|
|
Uzorak 1 (slijepa proba hrane za životinje) |
Uzorak 2 |
Uzorak 3 |
Uzorak 4 |
Uzorak 5 |
|
L |
14 |
14 |
14 |
14 |
15 |
|
n |
56 |
56 |
56 |
56 |
60 |
|
Srednja vrijednost [mg/kg] |
— |
45,5 |
188 |
5 129 |
25 140 |
|
sr [mg/kg] |
— |
2,26 |
3,57 |
178 |
550 |
|
CVr [%] |
— |
4,95 |
1,90 |
3,46 |
2,20 |
|
sR [mg/kg] |
— |
2,95 |
11,8 |
266 |
760 |
|
CVR [%] |
— |
6,47 |
6,27 |
5,19 |
3,00 |
|
Nominalni udio [mg/kg] |
— |
50 |
200 |
5 000 |
25 000 |
|
L |
= |
broj laboratorija |
|
n |
= |
broj pojedinačnih vrijednosti |
|
sr |
= |
standardna devijacija ponovljivosti |
|
CVr |
= |
koeficijent varijacije ponovljivosti |
|
sR |
= |
standardna devijacija obnovljivosti |
|
CVR |
= |
koeficijent varijacije obnovljivosti. |
9. Napomene
|
9.1. |
Ako uzorak sadrži tiamin, vršak tiamina u kromatogramu pojavljuje se malo prije vrška amproliuma. U skladu s ovom metodom, amprolium i tiamin se moraju razdvojiti. Ako se amprolium i tiamin ne razdvajaju na koloni (točka 4.1.1.) korištenoj u ovoj metodi, metanolom se zamijeni do 50 % acetonitrila u mobilnoj fazi (točka 3.6.). |
|
9.2. |
Prema Britanskoj farmakopeji, najviše točke spektra otopine amproliuma (c = 0,02 mol/l) u klorovodičnoj kiselini (c = 0,1 mol/l) nalaze se na 246 nm i 262 nm. Apsorbanca iznosi 0,84 na 246 nm i 0,80 na 262 nm. |
|
9.3. |
Ekstrakt se uvijek mora razrijediti mobilnom fazom, u suprotnom bi se vrijeme retencije vrha amproliuma moglo znatno pomaknuti zbog promjena ionske sile. |
D. ODREĐIVANJE KARBADOKSA
Metil 3-(2-kinoksalinilmetilen)karbazat N1,N4-dioksid
1. Svrha i područje primjene
Ovom se metodom omogućuje određivanje razine karbadoksa u hrani za životinje, premiksima i pripravcima. Granica detekcije iznosi 1 mg/kg. Granica kvantifikacije je 5 mg/kg.
2. Načelo
Uzorak se uravnoteži vodom i ekstrahira metanol-acetonitrilom. Kod hrane za životinje pročisti se alikvotni dio filtriranog ekstrakta na koloni iz aluminijevog oksida. Kod premiksa i pripravaka alikvotni se dio filtriranog ekstrakta razrijedi do primjerene koncentracije vodom, metanolom i acetonitrilom. Udio karbadoksa određuje se tekućinskom kromatografijom visoke učinkovitosti (HPLC) s reverznom fazom uz upotrebu UV detektora.
3. Reagensi
|
3.1. |
Metanol. |
|
3.2. |
Acetonitril, za HPLC. |
|
3.3. |
Octena kiselina, m = 100 %. |
|
3.4. |
Aluminijev oksid: neutralni, stupnja aktivnosti I. |
|
3.5. |
Metanol-acetonitril 1 + 1 (v + v). Pomiješa se 500 ml metanola (točka 3.1.) s 500 ml acetonitrila (točka 3.2.). |
|
3.6. |
Octena kiselina, σ = 10 %. Vodom se razrijedi 10 ml octene kiseline (točka 3.3.) do 100 ml. |
|
3.7. |
Natrijev acetat. |
|
3.8. |
Voda, za HPLC. |
|
3.9. |
Otopina acetatnog pufera, c = 0,01 mol/l, pH = 6,0. Otopi se 0,82 g natrijevog acetata (točka 3.7.) u 700 ml vode (točka 3.8.), vrijednost pH se octenom kiselinom (točka 3.6.) podesi na 6,0. Prenese se u graduiranu tikvicu obujma 1 000 ml, dopuni vodom (točka 3.8.) do oznake i promiješa. |
|
3.10. |
Mobilna faza za HPLC Pomiješa se 825 ml otopine acetatnog pufera (točka 3.9.) sa 175 ml acetonitrila (točka 3.2.). Filtrira se kroz filtar 0,22 μm (točka 4.5.), a otopina otplini (npr. 10 minuta u ultrazvučnoj kupelji). |
|
3.11. |
Standardna tvar Čisti karbadoks: metil 3-(2-kinoksalinilmetilen)karbazat N1,N4-dioksid, E 850 |
|
3.11.1. |
Odvagne se 25 mg standardne tvari karbadoksa (točka 3.11.) s preciznošću od 0,1 mg i prenese u graduiranu tikvicu obujma 250 ml. Otopi se u metanol-acetonitrilu (točka 3.5.) obradom u ultrazvučnoj kupelji (točka 4.7.). Nakon obrade ultrazvukom, otopina se zagrije do sobne temperature, dopuni do oznake metanol-acetonitrilom (točka 3.5.) i promiješa. Tikvica se omota aluminijskom folijom ili se koristi tikvica od smeđeg stakla i pohrani u hladnjaku. Na temperaturi ≤ 4 °C otopina je stabilna 1 mjesec. |
|
3.11.2. |
U seriju graduiranih tikvica obujma 100 ml prenese se 2,0, 5,0, 10,0 i 20,0 ml osnovne standardne otopine (točka 3.11.1.). Doda se 30 ml vode, dopuni do oznake metanol-acetonitrilom (točka 3.5.) i promiješa. Tikvice se omotaju aluminijskom folijom. Ove otopine odgovaraju koncentracijama karbadoksa od 2,0, 5,0, 10,0, i 20,0 μg/ml. Kalibracijske otopine treba pripremiti svježe prije uporabe.
|
|
3.12. |
Smjesa voda-[metanol-acetonitril] (točka 3.5.), 300 + 700 (v + v) Pomiješa se 300 ml vode sa 700 ml smjese metanola i acetonitrila (točka 3.5.). |
4. Oprema
|
4.1. |
Laboratorijska tresilica ili magnetska mješalica. |
|
4.2. |
Filtar papir od staklenih vlakana (Whatman GF/A ili jednakovrijedan). |
|
4.3. |
Staklena kolona (duljine 300 do 400 mm, unutarnji promjer približno 10 mm) sa sinteriranim staklenim filtrom i ispusnim pipcem.
|
|
4.4. |
Oprema za HPLC sa sustavom za ubrizgavanje primjerenim za ubrizgavanje volumena od 20 μl. |
|
4.4.1. |
Kolona za tekućinsku kromatografiju: 300 mm × 4 mm, C18, s veličinom čestica punjenja 10 μm ili jednakovrijedna. |
|
4.4.2. |
UV detektor s mogućnošću promjene valne duljine ili detektor s nizom dioda u području 225 do 400 nm. |
|
4.5. |
Membranski filtar, 0,22 μm. |
|
4.6. |
Membranski filtar, 0,45 μm. |
|
4.7. |
Ultrazvučna kupelj. |
5. Postupak
|
Napomena |
: |
Karbadoks je osjetljiv na svjetlo. Svi se postupci izvode pod prigušenim svjetlom ili u laboratorijskom posuđu od smeđeg stakla ili u posuđu omotanom aluminijskom folijom. |
5.1. Općenito
5.1.1.
Kod primjene testa iskorištenja (točka 5.1.2.) analizom slijepe probe hrane za životinje utvrđuje se da nisu prisutni ni karbadoks niti interferentne tvari. Slijepa proba hrane za životinje mora biti slične vrste kao i uzorak, a analizom se u njoj ne smiju pronaći karbadoks niti interferentne tvari.
5.1.2.
Test iskorištenja provodi se analizom slijepe probe hrane za životinje (točka 5.1.1.) u koju se doda količina karbadoksa slična količini u uzorku. Kako bi se dobila koncentracija od 50 mg/kg, prenese se 5,0 ml osnovne standardne otopine (točka 3.11.1.) u konusnu tikvicu obujma 200 ml. Otopina se otpari do približno 0,5 ml pod strujom dušika. Doda se 10 g slijepe probe hrane za životinje, promiješa i ostavi 10 minuta prije prelaska na postupak ekstrakcije (točka 5.2.).
Ako nije dostupna slijepa proba hrane za životinje slična uzorku (vidjeti točku 5.1.1.), test iskorištenja može se umjesto toga provesti metodom dodavanja standarda. U tom se slučaju uzorku za analizu doda količina karbadoksa slična onoj u uzorku. Taj se uzorak analizira zajedno s uzorkom bez dodanog karbadoksa, a iskorištenje se izračuna oduzimanjem.
5.2. Ekstrakcija
5.2.1.
Odvagne se 10 g uzorka s preciznošću od 0,01 g i prenese u konusnu tikvicu obujma 200 ml. Doda se 15,0 ml vode, promiješa i ostavi 5 minuta da se uravnoteži. Doda se 35,0 ml metanol-acetonitrila (točka 3.5), začepi i trese 30 minuta u tresilici ili miješa magnetnom mješalicom (točka 4.1.). Otopina se filtrira kroz filtar papir od staklenih vlakana (točka 4.2.). Ova se otopina pohrani za fazu pročišćivanja (točka 5.3.).
5.2.2.
Odvagne se 1 g neusitnjenog uzorka s preciznošću od 0,001 g i prenese u konusnu tikvicu obujma 200 ml. Doda se 15,0 ml vode, promiješa i ostavi 5 minuta da se uravnoteži. Doda se 35,0 ml metanol-acetonitrila (točka 3.5.), začepi i trese 30 minuta na tresilici ili miješa magnetnom mješalicom (točka 4.1.). Otopina se filtrira kroz filtar papir od staklenih vlakana (točka 4.2.).
Alikvot filtrata se pipetom prenese u graduiranu tikvicu obujma 50 ml. Doda se 15,0 ml vode, dopuni do oznake metanol-acetonitrilom (točka 3.5.) i promiješa. Koncentracija karbadoksa u konačnoj otopini iznosi približno 10 μg/ml. Alikvot se filtrira kroz filtar 0,45 μm (točka 4.6.).
Prelazi se na postupak određivanja putem HPLC-a (točka 5.4.).
5.2.3.
Odvagne se 0,2 g neusitnjenog uzorka s preciznošću od 0,001 g i prenese u konusnu tikvicu obujma 250 ml. Doda se 45,0 ml vode, promiješa i ostavi 5 minuta da se uravnoteži. Doda se 105,0 ml metanol-acetonitrila (točka 3.5), začepi i homogenizira. Uzorak se drži 15 minuta u ultrazvučnoj kupelji (točka 4.7.), zatim se 15 minuta protresa ili miješa (točka 4.1). Otopina se filtrira kroz filtar papir od staklenih vlakana (točka 4.2.).
Alikvot filtrata razrijedi se smjesom vode, metanola i acetonitrila (točka 3.12.) tako da konačna koncentracija karbadoksa iznosi 10 – 15 μg/ml (za 10 %-tni preparat faktor razrjeđenja je 10). Alikvot se filtrira kroz filtar 0,45 μm (točka 4.6.).
Prelazi se na postupak određivanja putem HPLC-a (točka 5.4.).
5.3. Pročišćivanje
5.3.1.
Odvagne se 4 g aluminijevog oksida (točka 3.4.) i prenese u staklenu kolonu (točka 4.3.).
5.3.2.
Kroz kolonu od aluminijevog oksida propusti se 15 ml filtriranoga ekstrakta (točka 5.2.1.), a prva 2 ml eluata se odbace. Prikupi se sljedećih 5 ml, alikvot se filtrira kroz filtar 0,45 μm (točka 4.6.).
Prelazi se na postupak određivanja putem HPLC-a (točka 5.4.).
5.4. Određivanje putem HPLC-a
5.4.1.
Sljedeći se uvjeti predlažu kao smjernice; mogu se koristiti i drugi uvjeti ako se njima jamče jednakovrijedni rezultati:
|
Kolona za tekućinsku kromatografiju (točka 4.4.1.): |
300 mm × 4 mm, C18, s veličinom čestica punjenja 10 μm ili jednakovrijedna |
|
Mobilna faza (točka 3.10.): |
Smjesa otopine acetatnog pufera (točka 3.9.) i acetonitrila (točka 3.2.), 825 + 175 (v + v) |
|
Brzina protoka: |
1,5 – 2 ml/min |
|
Valna duljina detekcije: |
365 nm |
|
Volumen za ubrizgavanje: |
20 μl |
Stabilnost kromatografskog sustava provjerava se višekratnim ubrizgavanjem kalibracijske otopine (točka 3.11.2.) koncentracije 5,0 μg/ml, sve dok se ne dobiju konstantne visine (površine) vršaka i konstantna vremena retencije.
5.4.2.
Više puta se ubrizga svaka kalibracijska otopina (točka 3.11.2.) i za svaku koncentraciju izmjere vrijednosti visina (površina) vršaka. Kalibracijska krivulja se pripremi tako da se na ordinatu nanose srednje vrijednosti visina ili površina vršaka kalibracijskih otopina, a na apscisu odgovarajuće koncentracije u μg/ml.
5.4.3.
Više puta se ubrizga ekstrakt uzorka ((točka 5.3.2.) za hranu za životinje, (točka 5.2.2.) za premikse i (točka 5.2.3.) za pripravke) i odredi srednja vrijednost visina (površina) vršaka karbadoksa.
6. Izračun rezultata
Koncentracija karbadoksa u otopini uzorka u μg/ml određuje se iz srednjih vrijednosti visina (površina) vršaka karbadoksa u otopini uzorka iz kalibracijske krivulje (točka 5.4.2.).
6.1. Hrana za životinje
Udio karbadoksa (w) (mg/kg) u uzorku izračunava se sljedećom formulom:
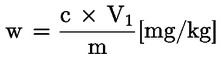
pri čemu je:
|
c |
= |
koncentracija karbadoksa u ekstraktu uzorka (točka 5.3.2.) u μg/ml |
|
V1 |
= |
volumen ekstrakta u ml (tj. 50) |
|
m |
= |
masa pokusnog uzorka u gramima. |
6.2. Premiksi i pripravci
Udio karbadoksa (w) (mg/kg) u uzorku izračunava se sljedećom formulom:

pri čemu je:
|
c |
= |
koncentracija karbadoksa u ekstraktu uzorka (točka 5.2.2. ili 5.2.3.) u μg/ml |
|
V2 |
= |
volumen ekstrakta u ml (tj. 50 za premikse; 150 za pripravke) |
|
f |
= |
faktor razrjeđenja u skladu s točkom 5.2.2. (premiksi) ili 5.2.3. (pripravci) |
|
m |
= |
masa pokusnog uzorka u gramima. |
7. Vrednovanje rezultata
7.1. Identifikacija
Identifikacija analita može se potvrditi ko-kromatografijom ili detektorom s nizom dioda kojim se uspoređuje spektar ekstrakta uzorka i spektar kalibracijske otopine (točka 3.11.2.) koncentracije 10,0 μg/ml.
7.1.1.
U ekstrakt uzorka doda se primjerena količina kalibracijske otopine (točka 3.11.2.). Količina dodanog karbadoksa mora biti slična procijenjenoj količini karbadoksa u ekstraktu uzorka.
Dopušteno je povećanje samo visine vrška karbadoksa uzimajući u obzir dodane količine i razrjeđenje ekstrakta. Širina vrška, na polovici maksimalne visine, mora biti unutar ± 10 % početne širine.
7.1.2.
Rezultati se ocjenjuju u skladu sa sljedećim mjerilima:
|
(a) |
valne duljine maksimalne apsorpcije spektra uzorka i spektra standarda, zabilježene na najvišoj točki vršaka kromatograma, moraju biti jednake unutar granice određene razlučivošću sustava za detekciju. Kod detektora s nizom dioda ta se vrijednost obično nalazi unutar ± 2 nm; |
|
(b) |
između 225 i 400 nm, spektar uzorka i spektar standarda, zabilježeni na najvišoj točki kromatograma, ne smiju se razlikovati za dijelove spektra unutar područja 10 – 100 % relativne apsorbance. To je mjerilo ispunjeno kada su prisutne jednake najviše vrijednosti i kada odstupanje između dva spektra ni na jednoj točki ne prelazi 15 % apsorbance standardnog analita; |
|
(c) |
između 225 i 400 nm, spektri krivulje rasta, najviše točke vrška i krivulje pada dobiveni iz ekstrakta uzorka, ne smiju se međusobno razlikovati za dijelove spektra unutar područja 10 – 100 % relativne apsorbance. To je mjerilo ispunjeno kada su prisutne jednake najviše vrijednosti i kada odstupanje između dva spektra ni na jednoj točki ne prelazi 15 % apsorbance spektra najviše točke. |
Ako nije ispunjeno jedno od navedenih mjerila, prisutnost analita nije potvrđena.
7.2. Ponovljivost
Razlika između rezultata dvaju usporednih postupaka određivanja, izvedenih na istom uzorku, za udio od 10 mg/kg i više ne smije prijeći 15 % vrijednosti višega rezultata.
7.3. Iskorištenje
Kod obogaćenog (slijepog) uzorka iskorištenje iznosi najmanje 90 %.
8. Rezultati međulaboratorijske studije
Provedena je međulaboratorijska studija u kojoj se u 8 laboratorija analiziralo 6 uzoraka hrane za životinje, 4 uzorka premiksa i 3 uzorka pripravaka. Svaki je uzorak analiziran dvaput. (Podrobnije informacije o navedenoj međulaboratorijskoj studiji mogu se pronaći u Journal of AOAC, svezak 71, 1988., str. 484. – 490.) Rezultati (bez ekstremnih vrijednosti) prikazani su u donjim tablicama:
Tablica 1.
Rezultati međulaboratorijske studije za hranu za životinje
|
|
Uzorak 1 |
Uzorak 2 |
Uzorak 3 |
Uzorak 4 |
Uzorak 5 |
Uzorak 6 |
|
L |
8 |
8 |
8 |
8 |
8 |
8 |
|
n |
15 |
14 |
15 |
15 |
15 |
15 |
|
Srednja vrijednost (mg/kg) |
50,0 |
47,6 |
48,2 |
49,7 |
46,9 |
49,7 |
|
Sr (mg/kg) |
2,90 |
2,69 |
1,38 |
1,55 |
1,52 |
2,12 |
|
CVr (%) |
5,8 |
5,6 |
2,9 |
3,1 |
3,2 |
4,3 |
|
SR (mg/kg) |
3,92 |
4,13 |
2,23 |
2,58 |
2,26 |
2,44 |
|
CVR (%) |
7,8 |
8,7 |
4,6 |
5,2 |
4,8 |
4,9 |
|
Nominalni udio (mg/kg) |
50,0 |
50,0 |
50,0 |
50,0 |
50,0 |
50,0 |
Tablica 2.
Rezultati međulaboratorijske studije za premikse i pripravke
|
|
Premiksi |
Pripravci |
|||||
|
A |
B |
C |
D |
A |
B |
C |
|
|
L |
7 |
7 |
7 |
7 |
8 |
8 |
8 |
|
n |
14 |
14 |
14 |
14 |
16 |
16 |
16 |
|
Srednja vrijednost (g/kg) |
8,89 |
9,29 |
9,21 |
8,76 |
94,6 |
98,1 |
104 |
|
Sr (g/kg) |
0,37 |
0,28 |
0,28 |
0,44 |
4,1 |
5,1 |
7,7 |
|
CVr (%) |
4,2 |
3,0 |
3,0 |
5,0 |
4,3 |
5,2 |
7,4 |
|
SR (g/kg) |
0,37 |
0,28 |
0,40 |
0,55 |
5,4 |
6,4 |
7,7 |
|
CVR (%) |
4,2 |
3,0 |
4,3 |
6,3 |
5,7 |
6,5 |
7,4 |
|
Nominalni udio (g/kg) |
10,0 |
10,0 |
10,0 |
10,0 |
100 |
100 |
100 |
|
L |
= |
broj laboratorija |
|
n |
= |
broj pojedinačnih vrijednosti |
|
Sr |
= |
standardna devijacija ponovljivosti |
|
CVr |
= |
koeficijent varijacije ponovljivosti |
|
SR |
= |
standardna devijacija obnovljivosti |
|
CVR |
= |
koeficijent varijacije obnovljivosti. |
PRILOG IX.
KORELACIJSKE TABLICE IZ ČLANKA 6.
1. Direktiva 71/250/EEZ
|
Direktiva 71/250/EEZ |
Ova Uredba |
|
Članak 1. prvi podstavak |
Članak 3. |
|
Članak 1. drugi podstavak |
Članak 2. |
|
Članak 2. |
— |
|
Članak 3. |
— |
|
Prilog, dio 1. |
Prilog II. |
|
Prilog, dio 2. |
— |
|
Prilog, dio 3. |
— |
|
Prilog, dio 4. |
Prilog III., dio O |
|
Prilog, dio 5. |
Prilog III., dio M |
|
Prilog, dio 6. |
Prilog III., dio N |
|
Prilog, dio 7. |
Prilog III., dio Q |
|
Prilog, dio 9. |
Prilog III., dio K |
|
Prilog, dio 10. |
— |
|
Prilog, dio 11. |
— |
|
Prilog, dio 12. |
Prilog III., dio J |
|
Prilog, dio 14. |
Prilog III., dio D |
|
Prilog, dio 16. |
— |
2. Direktiva 71/393/EEZ
|
Direktiva 71/393/EEZ |
Ova Uredba |
|
Članak 1. |
Članak 3. |
|
Članak 2. |
— |
|
Članak 3. |
— |
|
Prilog, dio I. |
Prilog III., dio A |
|
Prilog, dio II. |
Prilog III., dio E |
|
Prilog, dio III. |
Prilog III., dio P |
|
Prilog, dio IV. |
Prilog III., dio H |
3. Direktiva 72/199/EEZ
|
Direktiva 72/199/EEZ |
Ova Uredba |
|
Članak 1. |
Članak 3. |
|
Članak 2. |
— |
|
Članak 3. |
— |
|
Članak 4. |
— |
|
Prilog I., dio 1. |
Prilog III., dio L |
|
Prilog I., dio 2. |
Prilog III., dio C |
|
Prilog I., dio 3. |
— |
|
Prilog I., dio 4. |
— |
|
Prilog I., dio 5. |
Prilog V., dio A |
|
Prilog II. |
— |
4. Direktiva 73/46/EEZ
|
Direktiva 73/46/EEZ |
Ova Uredba |
|
Članak 1. |
Članak 3. |
|
Članak 3. |
— |
|
Članak 4. |
— |
|
Prilog I., dio 1. |
Prilog III., dio B |
|
Prilog I., dio 2. |
— |
|
Prilog I., dio 3. |
Prilog III., dio I |
5. Direktiva 76/371/EEZ
|
Direktiva 76/371/EEZ |
Ova Uredba |
|
Članak 1. |
Članak 1. |
|
Članak 2. |
— |
|
Članak 3. |
— |
|
Prilog |
Prilog I. |
6. Direktiva 76/372/EEZ
|
Direktiva 76/372/EEZ |
Ova Uredba |
|
Članak 1. |
— |
|
Članak 2. |
— |
|
Članak 3. |
— |
|
Prilog |
— |
7. Direktiva 78/633/EEZ
|
Direktiva 78/633/EEZ |
Ova Uredba |
|
Članak 1. |
Članak 3. |
|
Članak 2. |
— |
|
Članak 3. |
— |
|
Prilog, dio 1. |
— |
|
Prilog, dio 2. |
— |
|
Prilog, dio 3. |
Prilog IV., dio C |
8. Direktiva 81/715/EEZ
|
Direktiva 81/715/EEZ |
Ova Uredba |
|
Članak 1. |
— |
|
Članak 2. |
— |
|
Članak 3. |
— |
|
Prilog |
— |
9. Direktiva 84/425/EEZ
|
Direktiva 84/425/EEZ |
Ova Uredba |
|
Članak 1. |
— |
|
Članak 2. |
— |
|
Članak 3. |
— |
|
Prilog |
— |
10. Direktiva 86/174/EEZ
|
Direktiva 86/174/EEZ |
Ova Uredba |
|
Članak 1. |
Članak 4. |
|
Članak 2. |
— |
|
Članak 3. |
— |
|
Prilog |
Prilog VII. |
11. Direktiva 93/70/EEZ
|
Direktiva 93/70/EEZ |
Ova Uredba |
|
Članak 1. |
Članak 3. |
|
Članak 2. |
— |
|
Članak 3. |
— |
|
Prilog |
Prilog IV., dio D |
12. Direktiva 93/117/EZ
|
Direktiva 93/117/EZ |
Ova Uredba |
|
Članak 1. |
Članci 3. i 5. |
|
Članak 2. |
— |
|
Članak 3. |
— |
|
Prilog, dio 1. |
Prilog IV., dio E |
|
Prilog, dio 2. |
Prilog VIII., dio A |
13. Direktiva 98/64/EZ
|
Direktiva 98/64/EZ |
Ova Uredba |
|
Članak 1. |
Članci 3. i 5. |
|
Članak 2. |
— |
|
Članak 3. |
— |
|
Članak 4. |
— |
|
Prilog, dio A |
Prilog III., dio F |
|
Prilog, dio C |
Prilog VIII., dio B |
14. Direktiva 1999/27/EZ
|
Direktiva 1999/27/EZ |
Ova Uredba |
|
Članak 1. |
Članci 3. i 5. |
|
Članak 2. |
— |
|
Članak 3. |
— |
|
Članak 4. |
— |
|
Članak 5. |
— |
|
Članak 6. |
— |
|
Članak 7. |
— |
|
Prilog, dio A |
Prilog VIII., dio C |
|
Prilog, dio B |
Prilog IV., dio F |
|
Prilog, dio C |
Prilog VIII., dio D |
15. Direktiva 1999/76/EZ
|
Direktiva 1999/76/EZ |
Ova Uredba |
|
Članak 1. |
Članak 3. |
|
Članak 2. |
— |
|
Članak 3. |
— |
|
Članak 4. |
— |
|
Prilog |
Prilog IV., dio G |
16. Direktiva 2000/45/EZ
|
Direktiva 2000/45/EZ |
Ova Uredba |
|
Članak 1. |
Članak 3. |
|
Članak 2. |
— |
|
Članak 3. |
— |
|
Članak 4. |
— |
|
Prilog, dio A |
Prilog IV., dio A |
|
Prilog, dio B |
Prilog IV., dio B |
|
Prilog, dio C |
Prilog III., dio G |
17. Direktiva 2002/70/EZ
|
Direktiva 2002/70/EZ |
Ova Uredba |
|
Članak 1. |
Članak 1. |
|
Članak 2. |
Članci 2. i 3. |
|
Članak 3. |
— |
|
Članak 4. |
— |
|
Članak 5. |
— |
|
Prilog I. |
Prilog I. i Prilog V., dio B(I.) |
|
Prilog II. |
Prilog II. i Prilog V., dio B(II.) |
18. Direktiva 2003/126/EZ
|
Direktiva 2003/126/EZ |
Ova Uredba |
|
Članak 1. |
Članak 3. |
|
Članak 2. |
— |
|
Članak 3. |
— |
|
Članak 4. |
— |
|
Članak 5. |
— |
|
Članak 6. |
— |
|
Prilog |
Prilog VI. |