ANNEXE II
I. DÉTERMINATION DE LA MATIÈRE SÈCHE
1. OBJET ET DOMAINE D'APPLICATION
Le présent chapitre décrit la méthode de référence de détermination de la teneur en matière sèche du lait.
2. DÉFINITION
«Matière sèche»: masse exprimée en pourcentage en masse, restant après la dessiccation complète spécifiée ci-après.
3. PRINCIPE
Évaporation de l'eau de la prise d'essai dans une étuve à une température de 102 ± 2 oC.
4. APPAREILLAGE ET VERRERIE
Matériel courant de laboratoire, et notamment:
|
4.1. |
Balance analytique |
|
4.2. |
Dessiccateur, pourvu d'un déshydratant efficace (par exemple gel de silice séché récemment avec indicateur hygrométrique). |
|
4.3. |
Étuve ventilée, réglable par thermostat pour opérer dans tout l'espace de travail à 102 ± 2 oC. |
|
4.4. |
Capsules à fond plat de 20 à 25 mm de hauteur, de 50 à 75 mm de diamètre, en matériau approprié, munies de couvercles bien ajustés et faciles à enlever. |
|
4.5. |
Bain d'eau bouillante |
|
4.6. |
Homogénéisateur |
5. PRÉPARATION DE L'ÉCHANTILLON POUR ESSAI
Amener l'échantillon de lait à une température de 20 à 25 oC. Mélanger soigneusement, afin d'obtenir une répartition homogène des matières grasses dans l'échantillon. Ne pas agiter trop vigoureusement, afin d'éviter la formation de mousse dans le lait ou le barattage des matières grasses. S'il est difficile de disperser la couche de crème, amener lentement de 35 à 40 oC en mélangeant soigneusement, de façon à incorporer la crème qui adhère au récipient. Refroidir rapidement l'échantillon à 20 à 25 oC.
Un homogénéisateur peut éventuellement être utilisé pour faciliter la dispersion des matières grasses.
Les résultats seront incorrects si l'échantillon contient de la matière grasse liquide apparente ou si des particules blanches, de forme irrégulière, sont visibles et adhèrent aux parois du récipient.
6. MODE OPÉRATOIRE
6.1. Préparation de la capsule
Chauffer la capsule (4.4) avec son couvercle posé à côté dans l'étuve (4.3), restée à 102 ± 2 oC, pendant 30 minutes au moins. Mettre le couvercle sur la capsule, placer immédiatement dans le dessiccateur (4.2). Laisser refroidir à température ambiante (30 minutes au moins) et peser à 0,1 mg près.
6.2. Prise d'essai
Peser rapidement dans la capsule préparée (6.1) à 0,1 mg près, trois à cinq grammes de l'échantillon pour essai (5).
6.3. Détermination
|
6.3.1. |
Sécher au préalable la capsule en la plaçant sur le bain d'eau; maintenir en ébullition pendant 30 minutes (4.5). |
|
6.3.2. |
Placer la capsule, avec son couvercle posé à côté, dans l'étuve (4.3), réglée à 102 ± 2 oC pendant deux heures. Mettre le couvercle sur la capsule et retirer de l'étuve. |
|
6.3.3. |
Laisser refroidir dans le dessiccateur (4.2) à température ambiante (30 minutes au moins) et peser à 0,1 mg près. |
|
6.3.4. |
Placer à nouveau la capsule, avec son couvercle posé à côté, dans l'étuve pendant une heure. Mettre le couvercle sur la capsule et retirer de l'étuve. Laisser refroidir pendant 30 minutes environ dans le dessiccateur et peser à 0,1 mg près. |
|
6.3.5. |
Répéter les opérations décrites au point 6.3.4 jusqu'à ce que la différence de masse entre deux pesées successives ne dépasse pas 0,5 mg. Relever la masse la plus faible. |
7. EXPRESSION DES RÉSULTATS
7.1. Calcul et formule
Calculer la matière sèche en pourcentage en masse d'après la formule:

|
WT |
= |
la matière sèche en g pour 100 g |
|
m0 |
= |
la masse, en grammes, de la capsule et du couvercle (6.1) |
|
m1 |
= |
la masse, en grammes, de la capsule, du couvercle et de la prise d'essai (6.2) |
|
m2 |
= |
la masse, en grammes, de la capsule, du couvercle et de la prise d'essai sèche (6.3.5) |
Arrondir la valeur obtenue à 0,01 % près (pourcentage en masse).
7.2. Fidélité
|
7.2.1. |
Répétabilité (r): 0,10 g de matière sèche pour 100 g de produit. |
|
7.2.2. |
Reproductibilité (R): 0,20 g de matière sèche pour 100 g de produit. |
II. DÉTERMINATION DE LA TENEUR EN MATIÈRES GRASSES
1. OBJET ET DOMAINE D'APPLICATION
Le présent chapitre décrit la méthode de référence de détermination de la teneur en matières grasses du lait entier, du lait partiellement écrémé et du lait écrémé.
2. DÉFINITION
«Teneur en matières grasses du lait»: toutes les substances déterminées par le procédé spécifié. Elle est exprimée en pourcentage en masse.
3. PRINCIPE
Extraction d'une solution ammoniaco-éthanolique d'une prise d'essai au moyen d'oxyde diéthylique et d'éther de pétrole, élimination des solvants par distillation ou évaporation puis détermination de la masse des substances extraites solubles dans l'éther de pétrole (méthode habituellement connue sous le nom de Röse-Gottlieb).
4. RÉACTIFS
Tous les réactifs doivent être de qualité analytique reconnue et ne doivent pas laisser de résidu appréciable lors de l'essai à blanc.
Pour vérifier la qualité des réactifs, effectuer une détermination comme spécifié au point 6.3. Pour le pesage, utiliser une fiole, un gobelet ou une capsule métallique vide (5.8), préparés en tant que tare comme indiqué au point 6.4 (pour pouvoir corriger l'incidence de modifications des conditions atmosphériques sur le poids obtenu). Si le résidu global, corrigé en fonction des modifications apparentes de la masse de la tare, est supérieur à 2,5 mg, déterminer le résidu des solvants en évaporant respectivement 100 ml d'oxyde diéthylique (4.4) et 100 ml d'éther de pétrole (4.5). Utiliser également une tare pour le pesage. Si le résidu est supérieur à 2,5 mg, nettoyer le solvant par distillation ou le remplacer.
|
4.1. |
«Hydroxyde d'ammonium»: solution à environ 25% (m/m) NH3. Une solution d'hydroxyde d'ammonium plus concentrée peut également être utilisée (6.5.1 et A.1.5.1). |
|
4.2. |
Éthanol, au moins 94 % (v/v). De l'éthanol dénaturé par du méthanol peut être utilisé pour autant qu'il soit certain que cela ne modifiera en rien les résultats de la détermination. |
|
4.3. |
Rouge congo ou rouge de crésol
Dissoudre 1 g de rouge congo ou de rouge de crésol dans l'eau et diluer à 100 ml. Note: L'emploi de cette solution, qui permet de mieux voir l'interface entre le solvant et la couche aqueuse, est facultatif (6.5.2). D'autres solutions aqueuses de colorants peuvent être utilisées pour autant qu'elles ne modifient pas le résultat de la détermination. |
|
4.4. |
Oxyde diéthylique, exempt de peroxydes, ne contenant pas plus de 2 mg/kg d'antioxydants et conforme aux spécifications de l'essai à blanc (6.3). |
|
4.5. |
Éther de pétrole ayant un point d'ébullition entre 30 et 60 oC. |
|
4.6 |
Mélange de solvants préparé peu de temps avant l'emploi par mélange de volumes égaux d'oxyde diéthylique (4.4) et d'éther de pétrole (4.5). |
5. APPAREILLAGE ET VERRERIE
Avertissement: Étant donné que la détermination fait intervenir des solvants volatils inflammables, l'appareillage électrique utilisé doit être conforme à la législation relative à l'emploi de ces solvants.
Matériel courant de laboratoire, et notamment:
|
5.1. |
Balance analytique |
|
5.2. |
Centrifugeuse dans laquelle les fioles ou les tubes d'extraction (5.6) peuvent être soumis à une vitesse de rotation de 500 à 600 tr/min-1, de façon à produire un champ de gravitation de 80 à 90 g à l'extrémité extérieure des fioles ou des tubes. Note: L'emploi d'une centrifugeuse est facultatif (6.5.5). |
|
5.3. |
Appareils de distillation ou d'évaporation permettant de distiller les solvants et l'éthanol des fioles ou de les évaporer des béchers et des capsules (6.5.12 et 6.5.15) à une température n'excédant pas 100 oC. |
|
5.4. |
Étuve, à chauffage électrique, munie d'ouïes de ventilation complètement ouvertes, réglables à une température de 102 ± 2 oC dans l'espace utilisé. L'étuve doit être munie d'un thermomètre convenable. |
|
5.5. |
Bain d'eau réglable à une température de 35 à 40 oC. |
|
5.6. |
Fioles d'extraction des matières grasses, type Molonnier
Note: On peut également utiliser des tubes d'extraction des matières grasses munis d'un siphon ou d'un dispositif de lavage dont le mode opératoire, différent dans ce cas, est décrit à l'appendice. Les fioles (ou les tubes) doivent être pourvus de bouchons en verre dépoli, en liège de bonne qualité ou en une autre matière inaltérable aux réactifs utilisés. Les bouchons en liège doivent être lavés à l'oxyde diéthylique (4.4), maintenus dans l'eau à 60 oC ou plus pendant au moins 15 minutes et ensuite mis à refroidir dans l'eau, de façon à en être imprégnés au moment de l'emploi. |
|
5.7. |
Portoir pour maintenir les fioles (ou les tubes) d'extraction des matières grasses (5.6). |
|
5.8. |
Flacon de lavage approprié à l'emploi du mélange de solvants (4.6). Ne pas utiliser de flacon de lavage en plastique. |
|
5.9. |
Récipients pour la récupération des matières grasses, par exemple fioles à ébullition (fioles à fond plat) ou d'Erlenmeyer d'une capacité de 125 à 250 ml ou capsules métalliques. Lorsqu'on utilise des capsules métalliques, elles doivent, de préférence, être en acier inoxydable, à fond plat, avec un bec et elles doivent avoir un diamètre de 80 à 100 mm et une hauteur d'environ 50 mm. |
|
5.10. |
Régulateurs d'ébullition, exempts de matières grasses, en porcelaine non poreuse, en carbure de silicium ou billes de verre (emploi facultatif dans le cas de capsules métalliques). |
|
5.11. |
Éprouvettes graduées de 5 et 25 ml de capacité. |
|
5.12. |
Pipettes graduées de 10 ml de capacité. |
|
5.13. |
Pinces métalliques permettant de tenir les fioles, béchers ou capsules. |
6. MODE OPÉRATOIRE
Note: L'autre mode opératoire utilisant les tubes d'extraction des matières grasses munis d'un siphon ou d'un dispositif de lavage (voir la note au point 5.6) est décrit à l'appendice.
6.1. Préparation de l'échantillon pour essai
Amener la température de l'échantillon pour essai entre 35 et 40 oC environ pendant 15 minutes en utilisant le bain d'eau (5.5) si nécessaire. Mélanger convenablement, mais doucement, l'échantillon en retournant plusieurs fois le récipient sans provoquer de mousse ou de barattage et refroidir rapidement à 20 oC environ.
6.2. Prise d'essai
Mélanger l'échantillon (6.1) en retournant doucement le récipient trois ou quatre fois et peser immédiatement, à 1 mg près, 10 à 11 grammes de l'échantillon pour essai, directement ou par différence, dans une fiole d'extraction ( 5.6).
La prise d'essai doit être placée le plus complètement possible dans le bulbe inférieur (étroit) de la fiole d'extraction.
6.3. Essai à blanc
Effectuer un essai à blanc en même temps que la détermination en utilisant le même mode opératoire et les mêmes réactifs, mais en remplaçant la prise d'essai par 10 à 11 ml d'eau.
Le changement de masse apparent du récipient de récupération des matières grasses, corrigé du changement apparent de masse du récipient de contrôle, ne doit pas être supérieur à 2,5 mg.
6.4. Préparation du récipient pour la récupération des matières grasses
Sécher un récipient (5.9) contenant quelques régulateurs d'ébullition (5.10) pour permettre une ébullition modérée au cours de l'élimination ultérieure du solvant dans l'étuve (5.4) pendant une heure. Laisser refroidir le récipient (non dans un dessiccateur, mais à l'abri de la poussière) à la température de la salle des balances (pendant une heure au moins pour les récipients en verre et pendant 30 minutes au moins pour les capsules métalliques). À l'aide de pinces (en particulier pour éviter les écarts de température), placer le récipient sur la balance et peser à 0,1 mg près.
6.5. Détermination
|
6.5.1. |
Ajouter 2 ml de solution d'hydroxyde d'ammonium (4.1) ou un volume équivalent d'une solution plus concentrée et mélanger vigoureusement avec la prise d'essai dans le bulbe étroit de la fiole. Après addition de l'hydroxyde d'ammonium, effectuer la détermination sans attendre. |
|
6.5.2. |
Ajouter 10 ml d'éthanol (4.2) et mélanger doucement, mais convenablement, en laissant le contenu de la fiole aller et venir entre les deux bulbes; éviter d'amener le liquide trop près du col de la fiole. Ajouter, éventuellement, deux gouttes de solution de rouge congo ou de rouge de cresol (4.3). |
|
6.5.3. |
Ajouter 25 ml d'oxyde diéthylique (4.4), fermer la fiole avec un bouchon en liège saturé d'eau ou avec un bouchon mouillé avec de l'eau (5.6), agiter la fiole vigoureusement, mais sans excès (afin d'éviter la formation d'émulsions persistantes) pendant une minute en position horizontale, le bulbe étroit étant en haut. De temps en temps, laisser le liquide du bulbe large passer dans le bulbe étroit. Si nécessaire, refroidir la fiole à l'eau courante puis retirer avec précaution le bouchon en liège ou le dispositif de fermeture et le rincer ainsi que le col de la fiole avec une petite quantité de mélange de solvants (4.6) en se servant du flacon de lavage (5.8), de façon à ce que les liquides de rinçage coulent dans la fiole. |
|
6.5.4. |
Ajouter 25 ml d'éther de pétrole (4.5), obturer la fiole à l'aide du bouchon en liège réhumidifié (réhumidification par trempage dans de l'eau) ou d'un autre dispositif de fermeture et agiter doucement la fiole pendant 30 secondes comme décrit au point 6.5.3. |
|
6.5.5. |
Centrifuger la fiole bouchée pendant 1 à 5 minutes à une vitesse de rotation de 500 à 600 tr/min-1 (5.2). En l'absence d'une centrifugeuse (voir la note au point 5.2), laisser la fiole bouchée reposer dans le portoir (5.7) pendant 30 minutes au moins jusqu'à ce que la couche surnageante soit claire et nettement séparée de la couche aqueuse. Si nécessaire, refroidir la fiole à l'eau courante. |
|
6.5.6. |
Enlever avec précaution le bouchon en liège ou tout autre dispositif de fermeture et le rincer ainsi que l'intérieur du col de la fiole avec un peu de mélange de solvants (4.6) de façon à ce que les liquides de rinçage coulent dans la fiole. Si l'interface se situe au-dessous du fond du col de la fiole, le faire monter à ce niveau en ajoutant doucement de l'eau par le côté de la fiole, afin de faciliter la décantation du solvant. |
|
6.5.7. |
En tenant la fiole d'extraction par le bulbe étroit, décanter avec soin le plus possible de la couche surnageante dans le récipient préparé, destiné à la récupération des matières grasses (6.4) contenant quelques régulateurs d'ébullition (5.10) dans le cas des fioles (facultatif avec les capsules métalliques), en évitant de décanter une partie quelconque de la couche aqueuse. |
|
6.5.8. |
Rincer l'extérieur du col de la fiole d'extraction avec un peu de mélange de solvants (4.6) en recueillant les liquides de rinçage dans le récipient destiné à la récupération des matières grasses et en veillant à ce que le mélange de solvants ne soit pas projeté sur l'extérieur de la fiole d'extraction. Le solvant peut, éventuellement, être éliminé totalement ou partiellement du récipient par distillation ou évaporation comme décrit au point 6.5.12. |
|
6.5.9. |
Ajouter 5 ml d'éthanol (4.2) au contenu de la fiole d'extraction en se servant de l'éthanol pour rincer l'intérieur du col de la fiole et mélanger comme décrit au point 6.5.2. |
|
6.5.10. |
Effectuer une deuxième extraction en recommençant les opérations décrites aux points 6.5.3 à 6.5.8, mais en utilisant seulement 15 ml d'oxyde diéthylique (4.4) et 15 ml d'éther de pétrole (4.5); utiliser l'oxyde diéthylique pour rincer l'intérieur du col de la fiole d'extraction. Si nécessaire, faire monter l'interface au milieu du col de la fiole, pour permettre à la décantation finale des solvants d'être aussi complète que possible. |
|
6.5.11. |
Effectuer une troisième extraction en répétant à nouveau les opérations décrites aux points 6.5.3 à 6.5.8, mais en utilisant seulement 15 ml d'oxyde diéthylique (4.4) er 15 ml d'éther de pétrole (4.5); utiliser l'oxyde diéthylique pour rincer l'intérieur du col de la fiole d'extraction. Si nécessaire, faire monter l'interface au milieu du col de la fiole d'extraction pour permettre à la décantation finale des solvants d'être aussi complète que possible. La troisième extraction peut être supprimée dans le cas du lait écrémé. |
|
6.5.12. |
Éliminer les solvants (y compris l'éthanol), aussi complètement que possible, de la fiole par distillation ou du bécher ou de la capsule par évaporation (5.3), en rinçant l'intérieur du col de la fiole avec un peu de mélange de solvants (4.6) avant de commencer la distillation. |
|
6.5.13. |
Chauffer le récipient destiné à la récupération des matières grasses (la fiole étant penchée pour permettre aux vapeurs de solvants de s'échapper) durant une heure dans l'étuve (5.4). Retirer le récipient destiné à la récupération des matières grasses de l'étuve, laisser refroidir (pas dans un dessiccateur, mais à l'abri de la poussière) à la température de la salle des balances (pendant une heure au moins pour les récipients en verre, pendant 30 minutes au moins pour les capsules métalliques) et peser à 0,1 mg près. Ne pas essuyer le récipient juste avant la pesée. Placer le récipient sur la balance à l'aide de pinces pour éviter notamment les variations de température. |
|
6.5.14. |
Répéter les opérations décrites en 5.6.13 jusqu'à ce que la masse du récipient destiné à la récupération des matières grasses diminue de 0,5 mg ou moins, ou augmente, entre deux pesées successives. Noter la masse minimale comme étant la masse du récipient de récupération des matières grasses et de la matière extraite. |
|
6.5.15. |
Ajouter 25 ml d'éther de pétrole au récipient d'extraction des matières grasses, de façon à vérifier si, oui ou non, la matière extraite est entièrement soluble. Chauffer doucement et agiter le solvant par un mouvement rotatoire jusqu'à dissolution de toutes les matières grasses. Si la matière extraite est entièrement soluble dans l'éther de pétrole, relever la masse des matières grasses comme étant la différence entre la masse finale de la fiole contenant l'extrait (6.5.14) et sa masse initiale (6.4). |
|
6.5.16. |
Si la matière extraite n'est pas entièrement soluble dans l'éther de pétrole, ou en cas de doute, extraire complètement les matières grasses du récipient par des lavages répétés avec de l'éther de pétrole chaud. Laisser déposer les matières insolubles et décanter soigneusement l'éther de pétrole sans enlever les matières insolubles. Répéter cette opération trois fois en utilisant l'éther de pétrole pour rincer l'intérieur du col du récipient. Enfin, rincer l'extérieur du sommet du récipient avec le mélange de solvants, de telle sorte que le solvant ne se répande pas à l'extérieur du récipient. Chasser les vapeurs d'éther de pétrole en chauffant le récipient pendant 1 heure dans l'étuve, laisser refroidir et peser comme décrit aux points 6.5.13 et 6.5.14. Relever la masse des matières grasses comme étant la différence entre la masse déterminée au point 6.5.14 et cette masse finale. |
7. EXPRESSION DES RÉSULTATS
7.1. Calcul et formule
Calculer la teneur en matières grasses, exprimée en pourcentage en masse, selon la formule:

où:
|
F |
= |
la teneur en matières grasses |
|
m0 |
= |
la masse, en grammes, de la prise d'essai (6.2) |
|
m1 |
= |
la masse, en grammes, du récipient destiné à la récupération des matières grasses et de la matière extraite, déterminée au point 6.5.14 |
|
m2 |
= |
la masse, en grammes, du récipient préparé pour la récupération des matières grasses ou, dans le cas de matières non dissoutes, du récipient destiné à la récupération des matières grasses et du résidu insoluble, déterminée au point 6.5.16 |
|
m3 |
= |
la masse, en grammes, du récipient de récupération des matières grasses utilisé pour l'essai à blanc (6.3.) et de la matière extraite, déterminée au point 6.5.14 |
|
m4 |
= |
la masse, en grammes, du récipient de récupération des matières grasses (6.4) utilisé pour l'essai à blanc (6.3.) ou, dans le cas des matières non dissoutes, du récipient de récupération des matières grasses et du résidu insoluble déterminée au point 6.5.16. |
Noter le résultat à 0,01 % près.
7.2. Fidélité
|
7.2.1. |
Répétabilité (r)
|
|
7.2.2. |
Reproductibilité (R)
|
III. DÉTERMINATION DE LA TENEUR EN MATIÈRE SÈCHE NON GRASSE TOTALE
1. OBJET ET DOMAINE D'APPLICATION
Le présent chapitre décrit la méthode de référence de détermination de la teneur en matière sèche non grasse totale du lait traité thermiquement.
2. DÉFINITION ET CALCUL
La teneur en matière sèche non grasse totale s'exprime en pourcentage en masse. Elle représente la différence entre la teneur en matière sèche totale (chapitre I) et la teneur en matières grasses (chapitre II).
IV. DÉTERMINATION DE LA TENEUR EN AZOTE TOTAL DU LAIT
1. OBJET ET DOMAINE D'APPLICATION
Le présent chapitre décrit la méthode de référence de détermination de la teneur en azote total du lait entier, du lait partiellement écrémé et du lait écrémé.
2. DÉFINITION
Par teneur en azote total du lait, on entend la teneur en azote, exprimée en pourcentage en masse, obtenue par application de la méthode Kjeldahl.
3. PRINCIPE
Une quantité pesée de l'échantillon de lait est minéralisée à l'aide d'acide sulfurique concentré et de sulfate de potassium en présence de sulfate de cuivre (II) faisant fonction de catalyseur, afin de convertir l'azote des composés organiques en sulfate d'ammoniaque. L'ammoniaque libéré par adjonction d'une solution d'hydroxyde de sodium est distillé et recueilli dans une solution d'acide borique. Cette solution est ensuite titrée avec une solution acide.
4. RÉACTIFS
|
4.1. |
Sulfate de potassium (K2SO4) |
|
4.2. |
Solution de sulfate de cuivre. Dissoudre 5,0 g de sulfate de cuivre (II) pentahydrate (CuSO4, 5H2O) dans de l'eau et compléter à 100 ml (à 20 oC) dans une fiole jaugée. |
|
4.3. |
Acide sulfurique, au moins 98,0% (m/m), H2SO4. |
|
4.4. |
Solution d'hydroxyde de sodium, 47% (m/m) 704 g NaOH/1 (à 20 oC). Note: On peut également utiliser une solution d'hydroxyde de sodium moins concentrée, par exemple 40 % (m/m) 572 g/1, 20 oC ou 30% (m/m) 399 g/1, 20 oC. |
|
4.5. |
Solution d'acide borique. Dissoudre 40 g d'acide borique (H3BO3) dans un litre d'eau chaude, laisser refroidir et conserver dans une bouteille en verre borosilicaté. |
|
4.6. |
Indicateur. Dissoudre 0,01 g de rouge de méthyle, 0,02 g de bleu de bromothymol et 0,06 g de vert de bromocrésol dans 100 ml d'alcool éthylique. Conserver la solution dans une bouteille bouchée brune dans un endroit frais et sombre. |
|
4.7. |
Solution volumétrique acide normalisée
C (1/2 H2SO4) ou c (HCl) = 0,05 mol/1 titrée à 0,0001 mol/1 près. |
|
4.8. |
Saccharose exempt de composés azotés |
|
4.9. |
Sel d'ammonium, pur, tel que l'ammonium (NH4)2C2O4 · H2O ou le sulfate d'ammonium (NH4)2SO4. |
|
4.10. |
Tryptophane (C11H12N2O2), phénacétine (C10H7CH2CONH2) ou mono- ou dichlorhydrate de lysine (C6H14N2O2 · HCl ou C6H14N2O2 · 2HCl). Note: Le degré de pureté des réactifs visés aux points 4.9 et 4.10 devrait être supérieur à la «qualité analytique». Pour le point 4.9 utiliser, si possible, une solution de sels d'ammonium certifiés. |
5. APPAREILLAGE ET VERRERIE
Matériel courant de laboratoire, et notamment:
|
5.1. |
Ballons Kjeldahl de 500 ml de capacité |
|
5.2. |
Régulateurs d'ébullition, par exemple billes de verre d'environ 5 mm de diamètre, granules d' Hengar et pierre ponce. |
|
5.3 |
Burette ou pipette automatique, délivrant 1,0 ml. |
|
5.4. |
Éprouvettes graduées en verre, d'une capacité de 50, 100 et 250 ml. |
|
5.5. |
Appareil de digestion (5.1) en position inclinée (à environ 45o), équipé d'un dispositif de chauffage électrique ou de brûleurs à gaz réglables pour que le contenu du ballon ne monte pas au-dessus du tiers du col, et d'un dispositif d'extraction de fumées. |
|
5.6. |
Appareil de distillation, en verre borosilicaté, auquel peut être adapté le ballon Kjeldahl (5.1), se composant d'un déflegmateur efficace relié à un réfrigérant efficace à tube intérieur droit, à la partie inférieure duquel est relié un tube d'évacuation; le tube de raccordement et le(s) bouchon(s) doivent être étanches et de préférence en néoprène. |
|
5.7. |
Pipette ou pipette automatique, délivrant 0,10 ml. |
|
5.8. |
Fioles coniques de 500 ml de capacité, graduées à 200 ml. |
|
5.9. |
Burette de 50 ml de capacité, graduée tous les 0,1 ml, avec une erreur maximale de ± 0,05 ml. |
|
5.10. |
Loupe grossissante pour une lecture précise de la burette (5.9).. |
|
5.11. |
pH-mètre |
|
5.12. |
Burette automatique |
6. MODE OPÉRATOIRE
|
6.1. |
Introduire successivement dans le ballon Kjeldahl (5.1) les régulateurs d'ébullition (5.2) (par exemple 3 billes de verre), 15 g de sulfate de potassium (4.1), 1,0 ml de solution de sulfate de cuivre (4.2), environ 5 g de lait (pesé à 0,001 g près) et 25 ml d'acide sulfurique (4.3). Utiliser l'acide pour entraîner les résidus de la solution de sulfate de cuivre, de sulfate de potassium ou de lait qui seraient restés sur le col du ballon et mélanger le contenu du ballon. Note: Étant donné que les matières organiques absorbent de l'acide sulfurique lors de l'ébullition, il est conseillé d'utiliser, pour la digestion, 30 et non 25 ml d'H2SO4 (4.3) si le lait contient plus de 5,0% (m/m) de matières grasses. Il en va de même pour l'essai à blanc. |
|
6.2. |
Chauffer chaque ballon Kjeldahl sur l'appareil de minéralisation (5.5) très doucement au début, de façon à ce que toute la mousse noire reste dans le ballon. Lorsque la mousse initiale disparaît pour faire place à de fortes fumées blanches, porter à forte ébullition (les vapeurs d'acide se condensent à mi-chemin dans le col du ballon) jusqu'à ce que les particules noires disparaissent complètement et que le contenu du ballon devienne limpide et bleu-vert pâle. Poursuivre l'ébullition plus modérément pendant au moins lh30. Veuillez noter les points suivants:
|
|
6.3. |
Lorsque les ballons Kjeldahl sont refroidis, ajouter 300 ml d'eau (voir la note) dans chacun des récipients, de façon à entraîner toutes les particules adhérant au col du ballon. Mélanger soigneusement le contenu jusqu'à dissolution des cristaux qui se sont séparés. Ajouter quelques régulateurs d'ébullition (5.2) pour obtenir une ébullition homogène. Ajouter ensuite dans chaque ballon 70 ml de solution d'hydroxyde de sodium (4.4) (voir la note) en tenant le ballon incliné, de manière à ce que la solution d'hydroxyde de sodium forme la couche inférieure dans le ballon. Veiller à ne pas en répandre sur le dessus du col. Note: Le volume combiné d'eau et de solution d'hydroxyde de sodium doit être de 370 ml pour pouvoir recueillir environ 150 ml de distillat avant que le contenu du ballon ne présente des soubresauts (6.4). Si on ajoute une plus grande quantité de solution d'hydroxyde de sodium concentrée à moins de 47 % (m/m), le volume d'eau ajouté doit être proportionnellement réduit. Ainsi, si on ajoute 85 ml de solution à 40% (m/m) ou 125 ml de solution à 30% (m/m), le volume d'eau ajouté doit être respectivement de 282 ml et de 245 ml. |
|
6.4. |
Raccorder immédiatement chaque ballon Kjeldahl à un appareil de distillation (5.6). Veiller à ce que l'extrémité du tube du réfrigérant plonge dans 50 ml de solution d'acide boriques (4.5) et 0,20 ml (5 à 6 gouttes) d'indicateur (4.6) contenus dans la fiole conique. Agiter le contenu de chaque ballon Kjeldahl pour bien mélanger et porter à ébullition tout d'abord à feu doux, pour éviter la formation excessive de mousse. Après avoir recueilli entre 100 et 125 ml de distillat, abaisser chaque fiole d'Erlenmeyer jusqu'à ce que l'extrémité du tube du réfrigérant se trouve environ 40 mm au-dessus du trait repère de 200 ml. Poursuivre chaque distillation jusqu'à ce que le contenu du ballon présente des soubresauts et couper immédiatement la source de chaleur. Enlever chaque ballon Kjeldahl et rincer l'extrémité de chaque tuyau d'évacuation du réfrigérant avec un peu d'eau en recueillant l'eau de rinçage dans une fiole d'Erlenmeyer. Veuillez noter les points suivants:
6.5 Titrer chaque distillat avec une solution volumétrique (4.7) jusqu'à pH 4,6 ± 0,1 en utilisant un ph-mètre et une burette automatique. L'addition d'un indicateur permet de contrôler si le titrage se déroule correctement. Relever les indications de chaque burette à 0,01 ml près en utilisant une loupe grossissante (5.10) pour éviter toute erreur de parallaxe. Le titrage peut être réalisé à l'aide du seul indicateur. Titrer jusqu'à ce que la couleur du distillat corresponde à celle d'une solution récemment préparée à partir de 150 ml d'eau additionnée de 50 ml de la solution d'acide borique et de 0,20 ml de l'indicateur contenus dans une fiole d'Erlenmeyer (5.8). |
|
6.6. |
Effectuer un essai à blanc conformément aux points 6.1 à 6.5 inclus en utilisant 5 ml d'eau distillée avec environ 0,1 g de saccharose (4.8) au lieu de l'échantillon de lait. Note: Pour le titrage du distillat blanc, il ne faut qu'une très faible quantité de solution volumétrique acide normalisée (4.7). |
|
6.7. |
Vérifier régulièrement l'exactitude des résultats en effectuant deux essais de récupération en suivant la procédure décrite aux points 6.1 à 6.5. |
|
6.7.1. |
S'assurer qu'il n'y a eu, au cours de la distillation, aucune perte d'azote due à une chaleur excessive ou à des fuites mécaniques en utilisant une prise d'essai composée de 0,15 g d'oxalate d'ammonium ou de sulfate d'ammonium (4.9) pesé à 0,001 g près et de 0,1 g de saccharose (4.8). Le pourcentage d'azote récupéré doit être de 99% à 100%. Des résultats inférieurs ou supérieurs sont l'indication d'une erreur de procédure et/ou d'une erreur de concentration de la solution normalisée (4.7). |
|
6.7.2. |
Vérifier que la procédure de minéralisation est capable de libérer tout l'azote protidique, en utilisant une prise d'essai composée de 0,20 g de tryptophane pur, 0,35 g de phénacétine ou 0,20 g de chlorhydrate de lysine (4.10). Tout doit être pesé à 0,001 g près. Le taux de récupération de l'azote doit être au moins de 98 à 99%. |
7. MESURES DE SÉCURITÉ
Lors de la manipulation d'acide sulfurique concentré, d'hydroxyde de sodium concentré et de ballons Kjeldahl, veiller à toujours porter un tablier de laboratoire, des lunettes de protection et des gants résistants aux acides.
Au cours de la distillation, surveiller les ballons Kjeldahl en permanence. En raison des risques, arrêter immédiatement la distillation si le contenu de la bouteille présente de violents «soubresauts». En cas de panne du système de chauffage de distillation pendant plus de deux à trois minutes, abaisser le ballon collecteur, de façon à ce que l'extrémité du réfrigérant émerge du liquide.
8. EXPRESSION DES RÉSULTATS
8.1. Calcul et formule
Calculer la teneur en azote, en grammes d'azote pour 100 g de produit à l'aide de la formule:

où:
|
N |
= |
la teneur en azote |
|
V |
= |
le volume en ml de la solution volumétrique acide normalisée utilisée pour le dosage |
|
Vo |
= |
le volume en ml de la solution volumétrique acide normalisée utilisée pour l'essai à blanc |
|
c |
= |
la concentration, en mol/1, de la solution volumétrique acide normalisée (4.7) |
|
m |
= |
la masse en g de la prise d'essai. |
Arrondir le résultat à 0,001 g près pour 100 g.
8.2. Fidélité
|
8.2.1. |
Répétabilité (r): 0,007 g par 100 g |
|
8.2.2. |
Reproductibilité (R): 0,015 g par 100 g |
9. MODE OPÉRATOIRE MODIFIÉ
|
9.1. |
Utilisation d'un bloc chauffant de minéralisation au lieu du système de minéralisation avec ballon Kjeldahl (points 5.5 et 5.1). Dans ce cas, pour repérer les pannes éventuelles, vérifier chaque point individuellement (6.7). |
|
9.2. |
Utilisation d'un système de distillation par entraînement à la vapeur au lieu d'un chauffage direct (6.4). Si l'appareillage ne permet pas d'utiliser de l'eau distillée, s'assurer que l'eau ne contient pas de matières volatiles acides ou alcalines. |
|
9.3. |
Il est possible d'utiliser une prise d'essai de 1 g de lait (semi-macro Kjeldahl) au lieu de 5 g (6.1), à condition que:
Note: L'emploi de l'une ou l'autre de ces options n'est acceptable que si la répétabilité (8.2.1) et les résultats des deux essais de contrôle de la précision (6.7) correspondent aux valeurs données dans la norme. |
V. DÉTERMINATION DE LA TENEUR EN MATIÈRES PROTÉIQUES TOTALES
1. OBJET ET DOMAINE D'APPLICATION
Le présent chapitre décrit la méthode de référence de détermination de la teneur en protéines du lait traité thermiquement (article 3 paragraphe A point 3 de la directive 85/397/CEE).
2. DÉFINITION
Teneur en matières protéiques totales: valeur obtenue en multipliant par un facteur approprié (3) la teneur en azote total, exprimée en pourcentage en masse et déterminée selon la méthode décrite au chapitre IV (3).
3. CALCUL
Teneur en matières protéiques totales du lait = 6,38 x teneur en azote total du lait en pourcentage.
VI. DÉTERMINATION DE LA MASSE VOLUMIQUE
1. OBJET ET DOMAINE D'APPLICATION
Le présent chapitre décrit la méthode de référence de détermination de la masse volumique à 20 oC du lait cru, du lait entier, du lait partiellement écrémé et du lait écrémé.
2. DÉFINITION
La masse volumique du lait est le quotient de la masse d'un certain volume de lait, à 20 oC, à ce volume.
3. PRINCIPE
La masse volumique à 20 oC est déterminée par aréométrie.
4. APPAREILLAGE ET VERRERIE
Matériel courant de laboratoire, et notamment:
|
4.1. |
Aréomètre
L'aréomètre à masse volumique est un instrument se composant d'un flotteur en verre, large et lesté à son extrémité inférieure et d'une tige cylindrique en verre, soudée à la partie supérieure du flotteur et coaxiale à celui-ci, l'extrémité supérieure de la tige étant fermée. Le flotteur en verre contient le lest (plomb, mercure, etc.) destiné à ajuster la masse de l'aréomètre. La tige comporte une échelle graduée de 1,025 à 1,035 g/ml. L'aréomètre doit être contrôlé par la méthode pycnométrique, en utilisant un pycnomètre de 100 ml environ de capacité, équipé d'un thermomètre de précision. |
|
4.2. |
Éprouvettes cylindriques (en verre ou en acier inoxydable) ayant les dimensions minimales suivantes:
|
|
4.3. |
Bain d'eau réglé à 20 ± 0,1 oC. |
|
4.4. |
Bain d'eau réglé à 40 ± 2 oC. |
|
4.5. |
Thermomètre gradué au moins par 0,5 oC. |
5. MODE OPÉRATOIRE
|
5.1. |
Mélanger l'échantillon en le retournant pour disperser les matières grasses. Placer l'échantillon dans le bain d'eau (4.4) jusqu'à ce qu'il atteigne 40 oC et le laisser à cette température pendant 5 minutes. Mélanger convenablement par retournements successifs pour obtenir une répartition homogène des matières grasses. Refroidir à 20 oC dans le deuxième bain d'eau (4.3). |
|
5.2. |
Mélanger convenablement l'échantillon en le retournant soigneusement pour éviter l'incorporation d'air. Verser le lait dans l'éprouvette cylindrique (4.2), maintenir en position inclinée pour éviter la formation de mousse ou de bulles. Utiliser suffisamment de lait pour provoquer un débordement de liquide lors de l'introduction de l'aréomètre (4.1) dans l'éprouvette. Plonger avec précaution l'aréomètre dans le lait et le laisser flotter librement jusqu'à ce qu'il atteigne son point d'équilibre. Placer l'éprouvette en position verticale. L'aréomètre doit être placé au milieu de la colonne de liquide et ne doit pas toucher les parois de l'éprouvette. |
|
5.3. |
Lorsque l'aréomètre a atteint son point d'équilibre, effectuer la lecture de la graduation à la partie supérieure du ménisque. |
|
5.4. |
Immédiatement après la lecture de l'aréomètre, introduire le thermomètre (4.5) dans l'échantillon et lire la température par 0,5 oC. La température ne doit pas différer de plus de ± 2 oC par rapport à 20 oC. |
6. CORRECTION DE LA TEMPÉRATURE
|
6.1. |
Si la température du lait à analyser n'est pas exactement de 20 oC lors de la mesure de la masse volumique, le résultat obtenu doit être corrigé en ajoutant à la masse volumique lue 0,0002 par degré Celsius au-dessus de 20 oC ou en retranchant 0,0002 par degré Celsius au-dessous de 20 oC. Cette correction est uniquement valable si la température de l'échantillon diffère de 5 oC au maximum par rapport à 20 oC. |
7. EXPRESSION DES RÉSULTATS
Le calcul de la masse volumique de l'échantillon, exprimée en g/ml de lait écrémé à 20 oC, s'effectue selon la formule suivante:
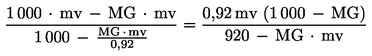
où:
|
mv |
= |
la masse volumique de l'échantillon, résultant de la lecture de l'aréomètre (5.4) en g/1 |
|
MG |
= |
la teneur en matières grasses de l'échantillon (g/1) |
|
0,92 |
= |
la densité des matières grasses. |
8. FIDÉLITÉ
|
8.1. |
Répétabilité (r): 0,0003 g/ml |
|
8.2. |
Reproductibilité (R): 0,0015 g/ml |
Appendice
(à l'annexe II)
AUTRE MODE OPÉRATOIRE AVEC TUBES D'EXTRACTION DES MATIÈRES GRASSES MUNIS D'UN SIPHON OU D'UN DISPOSITIF DE LAVAGE
A.1. MODE OPÉRATOIRE
A.1.1. Préparation de l'échantillon pour essai
Voir point 6.1.
A.1.2. Prise d'essai
Procéder comme précisé au point 6.2, mais en utilisant les tubes d'extraction des matières grasses (5.6). La prise d'essai doit être transférée aussi complètement que possible au fond du tube d'extraction.
A.1.3. Essai à blanc
Voir point 6.3.
A.1.4. Préparation du récipient destiné à la récupération des matières grasses
Voir point 6.4.
A.1.5. Détermination
|
A.1.5.1. |
Ajouter 2 ml de solution d'hydroxyde d'ammonium (4.1) ou un volume équivalent d'une solution d'hydroxyde d'ammonium plus concentrée et mélanger convenablement avec la prise d'essai prétraitée au fond du tube. Après addition de l'hydroxyde d'ammonium, effectuer la détermination sans attendre. |
|
A.1.5.2. |
Ajouter 10 ml d'éthanol (4.2) et mélanger doucement, mais convenablement, au fond du tube. Ajouter, éventuellement, deux gouttes de solution de rouge congo ou de rouge de crésol (4.3). |
|
A.1.5.3. |
Ajouter 25 ml d'oxydé diéthylique (4.4), fermer le tube avec un bouchon en liège saturé d'eau ou avec un bouchon en une autre matière mouillé avec de l'eau (5.6) et agiter vigoureusement le tube, mais pas trop fort (afin d'éviter la formation d'émulsions persistantes), par des retournements répétés pendant une minute. Si nécessaire, refroidir le tube sous l'eau courante, retirer avec précaution le bouchon en liège ou en une autre matière, le rincer ainsi que le col du tube avec une petite quantité de mélange de solvants (4.6.) en utilisant le flacon de lavage (5.8), de façon à ce que les liquides de rinçage coulent dans le tube. |
|
A.1.5.4. |
Ajouter 25 ml d'éther de pétrole (4.5), fermer le tube avec le bouchon en liège réhumidifïé (réhumidifié en le trempant dans de l'eau) ou en une autre matière et agiter soigneusement le tube pendant 30 secondes comme décrit au point A.l.5.3. |
|
A.1.5.5. |
Cetrifuger le tube fermé pendant 1 à 5 minutes à une vitesse de rotation de 500 à 600 tr/min-1 (5.2). Si l'on ne dispose pas d'une centrifugeuse (voir note au point 5.2), laisser le tube bouché reposer dans le portoir pendant au moins 30 minutes jusqu'à ce que la couche surnageante soit claire et nettement séparée de la couche aqueuse. Si nécessaire, refroidir le tube sous l'eau courante. |
|
A.1.5.6. |
Retirer avec précaution le bouchon en liège ou en une autre matière et le rincer, ainsi que le col du tube, avec une petite quantité de mélange de solvants, de sorte que les liquides de rinçage coulent dans le tube. |
|
A.1.5.7. |
Introduire un siphon ou un dispositif de lavage dans le tube et enfoncer la longue tubulure à l'intérieur jusqu'à ce que l'orifice se trouve à environ 3 mm au-dessus de l'interface des couches. La tubulure intérieure doit être parallèle à l'axe du tube d'extraction. Transvaser avec précaution la couche surnageante du tube dans le récipient préparé pour la récupération des matières grasses (6.4) et contenant quelques régulateurs d'ébullition (5.10) dans le cas de fioles (facultatif dans le cas de capsules métalliques) en évitant de transvaser une partie quelconque de la couche aqueuse. Rincer l'orifice avec une petite quantité de mélange de solvants en recueillant les liquides de rinçage dans le récipient servant à la récupération des matières grasses. |
|
A.1.5.8. |
Desserrer le dispositif du col du tube, puis le soulever légèrement et rincer la partie inférieure de sa longue tubulure interne avec un peu de mélange de solvants. Abaisser et réintroduire le dispositif et transvaser des liquides de rinçage dans le récipient de récupération des matières grasses. Rincer l'orifice externe du dispositif avec un peu de mélange de solvants, recueillir ensuite les liquides de rinçage dans le récipient. Le solvant peut éventuellement être éliminé partiellement ou totalement du récipient par distillation ou évaporation comme décrit au point 6.5.12. |
|
A.1.5.9. |
Desserrer à nouveau le dispositif du col, soulever légèrement le dispositif et ajouter 5 ml d'éthanol dans le tube en utilisant l'éthanol pour rincer la longue tubulure interne: mélanger comme décrit au point A.1.5.2. |
|
A.1.5.10. |
Effectuer une deuxième extraction en répétant les opérations décrites aux points A.1.5.3 à A.1.5.8, mais en utilisant seulement 15 ml d'oxyde diéthylique (4.4) et 15 ml d'éther de pétrole (4.5). Utiliser l'oxyde diéthylique pour rincer la longue tubulure interne pendant le retrait du dispositif du tube après la première extraction. |
|
A.1.5.11. |
Effectuer une troisième extraction en répétant à nouveau les opérations décrites aux points A.l.5.3 à A.1.5.8 en utilisant 15 ml d'oxyde diéthylique et 15 ml d'éther de pétrole et en rinçant la longue tubulure interne du dispositif comme décrit au point A.1.5.10. La troisième extraction peut être supprimée dans le cas du lait écrémé. |
|
A.1.5.12. |
Procéder comme décrit aux points 6.5.12 à 6.5.16. |