

EUR-Lex Access to European Union law
This document is an excerpt from the EUR-Lex website
Document 52016XC0726(02)
Commission Notice — The ‘Blue Guide’ on the implementation of EU products rules 2016 (Text with EEA relevance)
Commission Notice — The ‘Blue Guide’ on the implementation of EU products rules 2016 (Text with EEA relevance)
Commission Notice — The ‘Blue Guide’ on the implementation of EU products rules 2016 (Text with EEA relevance)
C/2016/1958
OJ C 272, 26.7.2016, p. 1–149
(BG, ES, CS, DA, DE, ET, EL, EN, FR, HR, IT, LV, LT, HU, MT, NL, PL, PT, RO, SK, SL, FI, SV)
|
26.7.2016 |
EN |
Official Journal of the European Union |
C 272/1 |
COMMISSION NOTICE
The ‘Blue Guide’ on the implementation of EU products rules 2016
(Text with EEA relevance)
(2016/C 272/01)
TABLE OF CONTENTS
PREFACE
IMPORTANT NOTICE
|
1. |
REGULATING THE FREE MOVEMENT OF GOODS | 5 |
|
1.1. |
A historical perspective | 5 |
|
1.1.1. |
The ‘Old Approach’ | 6 |
|
1.1.2. |
Mutual Recognition | 7 |
|
1.1.3. |
The ‘New Approach’ and the ‘Global Approach’ | 7 |
|
1.2. |
The ‘New Legislative Framework’ | 9 |
|
1.2.1. |
The concept | 9 |
|
1.2.2. |
The legal nature of the NLF acts and their relationship to other EU legislation | 10 |
|
1.2.3. |
How the system fits together | 11 |
|
1.3. |
The General Product Safety Directive | 12 |
|
1.4. |
The legislation on product liability | 12 |
|
1.5. |
Scope of the Guide | 13 |
|
2. |
WHEN DOES UNION HARMONISATION LEGISLATION ON PRODUCTS APPLY? | 15 |
|
2.1. |
Product coverage | 15 |
|
2.2. |
Making available on the market | 17 |
|
2.3. |
Placing on the market | 18 |
|
2.4. |
Products imported from countries outside the EU | 20 |
|
2.5. |
Putting into service or use (and installation) | 21 |
|
2.6. |
Simultaneous application of Union harmonisation acts | 22 |
|
2.7. |
Intended use/misuse | 23 |
|
2.8. |
Geographical application (EEA EFTA states, Overseas Countries and Territories (OCTs), Turkey) | 24 |
|
2.8.1. |
Member States and Overseas countries and territories | 24 |
|
2.8.2. |
EEA EFTA states | 25 |
|
2.8.3. |
Monaco, San Marino and Andorra | 25 |
|
2.8.4. |
Turkey | 26 |
|
2.9. |
Transitional periods in the case of new or revised EU rules | 27 |
|
2.10. |
Transitional arrangements for the EU declaration of conformity as a result of the alignment to Decision No 768/2008/EC | 27 |
|
3. |
THE ACTORS IN THE PRODUCT SUPPLY CHAIN AND THEIR OBLIGATIONS | 28 |
|
3.1. |
Manufacturer | 28 |
|
3.2. |
Authorised representative | 32 |
|
3.3. |
Importer | 33 |
|
3.4. |
Distributor | 34 |
|
3.5. |
Other intermediaries: Intermediary service providers under the E-commerce Directive | 37 |
|
3.6. |
End-user | 38 |
|
4. |
PRODUCT REQUIREMENTS | 39 |
|
4.1. |
Essential product requirements | 39 |
|
4.1.1. |
Definition of essential requirements | 39 |
|
4.1.2. |
Conformity with the essential requirements: harmonised standards | 40 |
|
4.1.3. |
Conformity with the essential requirements: other possibilities | 51 |
|
4.2. |
Traceability requirements | 51 |
|
4.2.1. |
Why does Traceability matter? | 52 |
|
4.2.2. |
Traceability provisions | 52 |
|
4.3. |
Technical documentation | 56 |
|
4.4. |
EU declaration of conformity | 57 |
|
4.5. |
Marking requirements | 58 |
|
4.5.1. |
CE marking | 58 |
|
4.5.2. |
Other mandatory markings | 64 |
|
5. |
CONFORMITY ASSESSMENT | 65 |
|
5.1. |
Modules for conformity assessment | 65 |
|
5.1.1. |
What is a conformity assessment? | 65 |
|
5.1.2. |
The modular structure of conformity assessment in Union harmonisation legislation | 65 |
|
5.1.3. |
Actors in conformity assessment — Positioning of conformity assessment in the supply chain | 66 |
|
5.1.4. |
Modules and their variants | 69 |
|
5.1.5. |
One- and two-module procedures — Procedures based on type (EU-type examination) | 69 |
|
5.1.6. |
Modules based on quality assurance | 70 |
|
5.1.7. |
Overview of modules | 70 |
|
5.1.8. |
Overview of procedures | 73 |
|
5.1.9. |
Rationale for selecting the appropriate modules | 74 |
|
5.2. |
Conformity assessment bodies | 75 |
|
5.2.1. |
Conformity assessment bodies and notified bodies | 75 |
|
5.2.2. |
Roles and responsibilities | 76 |
|
5.2.3. |
Competence of Notified bodies | 78 |
|
5.2.4. |
Coordination between notified bodies | 79 |
|
5.2.5. |
Subcontracting by notified bodies | 79 |
|
5.2.6. |
Accredited in-house bodies | 81 |
|
5.3. |
Notification | 81 |
|
5.3.1. |
Notifying authorities | 81 |
|
5.3.2. |
Notification process | 82 |
|
5.3.3. |
Publication by the commission — the NANDO website | 85 |
|
5.3.4. |
Monitoring of the competence of notified bodies — Suspension — withdrawal — appeal | 86 |
|
6. |
ACCREDITATION | 87 |
|
6.1. |
Why accreditation? | 87 |
|
6.2. |
What is accreditation? | 88 |
|
6.3. |
Scope of Accreditation | 89 |
|
6.4. |
Accreditation according to Regulation (EC) No 765/2008 | 89 |
|
6.4.1. |
National accreditation bodies | 90 |
|
6.4.2. |
Non-competition and non-commerciality of national accreditation bodies | 91 |
|
6.5. |
The European accreditation infrastructure | 92 |
|
6.5.1. |
Sectoral accreditation schemes | 92 |
|
6.5.2. |
Peer evaluation | 92 |
|
6.5.3. |
Presumption of conformity for national accreditation bodies | 93 |
|
6.5.4. |
EA's role in supporting and harmonising accreditation practice across Europe | 93 |
|
6.6. |
Cross-border accreditation | 93 |
|
6.7. |
Accreditation in the international context | 95 |
|
6.7.1. |
Cooperation between accreditation bodies | 95 |
|
6.7.2. |
The impact on trade relations in the field of conformity assessment between the EU and Third Countries | 96 |
|
7. |
MARKET SURVEILLANCE | 97 |
|
7.1. |
Why do we need market surveillance? | 98 |
|
7.2. |
controls by Market Surveillance authorities | 99 |
|
7.3. |
Control of products from third countries by customs | 101 |
|
7.4. |
Member States responsibilities | 103 |
|
7.4.1. |
National infrastructures | 103 |
|
7.4.2. |
National Market Surveillance Programmes (NMSP) and reviews of activities | 104 |
|
7.4.3. |
Public information | 105 |
|
7.4.4. |
Market surveillance procedures | 105 |
|
7.4.5. |
Corrective measures — bans — withdrawals — recalls | 107 |
|
7.4.6. |
Sanctions | 108 |
|
7.5. |
Cooperation between the Member States and the European commission | 108 |
|
7.5.1. |
safeguard mechanisms | 109 |
|
7.5.2. |
The application of safeguard mechanisms step by step | 110 |
|
7.5.3. |
Mutual assistance, administrative cooperation and exchange of information among Member States | 112 |
|
7.5.4. |
Rapid Alert System for non-food products presenting a risk | 114 |
|
7.5.5. |
ICSMS | 115 |
|
7.5.6. |
Medical devices: vigilance system | 117 |
|
8. |
FREE MOVEMENT OF PRODUCTS WITHIN THE EU | 117 |
|
8.1. |
Free movement clause | 117 |
|
8.2. |
Limits and restrictions | 118 |
|
9. |
INTERNATIONAL ASPECTS OF THE EU LEGISLATION ON PRODUCTS | 118 |
|
9.1. |
Agreements on Conformity Assessment and Acceptance (ACAAs) | 118 |
|
9.2. |
Mutual recognition agreements (MRAs) | 119 |
|
9.2.1. |
Main characteristics | 119 |
|
9.2.2. |
EU-Swiss MRA | 120 |
|
9.2.3. |
EEA EFTA States: Mutual recognition agreements and Agreements on Conformity Assessment and Acceptance | 121 |
ANNEXES
|
ANNEX I — |
EU legislation referred to in the Guide (non-exhaustive list) | 122 |
|
ANNEX II — |
Additional guidance documents | 127 |
|
ANNEX III — |
Useful Web addresses | 129 |
|
ANNEX IV — |
Conformity assessment procedures (modules from Decision No 768/2008/EC) | 130 |
|
ANNEX V — |
Relation between ISO 9001 and modules requiring a quality assurance system | 140 |
|
ANNEX VI — |
Using harmonised standards to assess the competence of Conformity Assessment Bodies | 142 |
|
ANNEX VII — |
Frequently Asked Questions on CE marking | 147 |
PREFACE
The Guide to the implementation of directives based on the New Approach and the Global Approach (the ‘Blue Guide’) was published in 2000. Since then, it has become one of the main reference documents explaining how to implement the legislation based on the New Approach, now covered by the New Legislative Framework.
Much of the 2000 edition of the ‘Blue Guide’ is still valid but it requires updating to cover new developments and to ensure the broadest possible common understanding on implementation of the New Legislative Framework (NLF) for the marketing of products. It is also necessary to take account of the changes introduced by the Lisbon Treaty (in force since 1 December 2009) with regard to the legal references and terminology applicable to EU-related documents, procedures, etc.
This new version of the Guide will therefore build on the past edition, but include new chapters, for example on the obligations of economic operators or accreditation, or completely revised chapters such as those on standardisation or market surveillance. The Guide has also been given a new title reflecting the fact that the New Legislative Framework is likely to be used, at least in part, by all types of Union harmonisation legislation and not only by the so-called ‘New Approach’ directives.
IMPORTANT NOTICE
This Guide is intended to contribute to a better understanding of EU product rules and to their more uniform and coherent application across different sectors and throughout the single market. It is addressed to the Member States and others who need to be informed of the provisions designed to ensure the free circulation of products as well as a high level of protection throughout the Union (e.g. trade and consumer associations, standardisation bodies, manufacturers, importers, distributors, conformity assessment bodies and trade unions).
This is intended purely as a guidance document — only the text of the Union harmonisation act itself has legal force. In certain cases, there may be differences between the provisions of a Union harmonisation act and the contents of this Guide, in particular where slightly divergent provisions in the individual Union harmonisation act cannot be fully described in this Guide. The binding interpretation of EU legislation is the exclusive competence of the Court of Justice of the European Union. The views expressed in this Guide cannot prejudge the position that the Commission might take before the Court of Justice. Neither the European Commission nor any person acting on behalf of the Commission is responsible for the use which might be made of the following information.
This Guide applies to the EU Member States but also to Iceland, Liechtenstein and Norway as signatories of the Agreement on the European Economic Area (EEA), as well as Turkey in certain cases. References to the Union or the single market are, accordingly, to be understood as referring to the EEA, or to the EEA market.
As this Guide reflects the state of the art at the time of its drafting, the guidance offered may be subject to later modification (1). In particular, more specific reflections are ongoing regarding various aspects of the Union legal framework applicable to online sales and this Guide is without prejudice to any future specific interpretation and guidance which may be developed on those matters.
1. REGULATING THE FREE MOVEMENT OF GOODS
1.1. A HISTORICAL PERSPECTIVE
The objectives of the first harmonisation directives focused on the elimination of barriers and on the free movement of goods in the single market. These objectives are now being complemented by a comprehensive policy geared to ensuring that only safe and otherwise compliant products find their way on to the market, in such a way that honest economic operators can benefit from a level playing field, thus promoting at the same time an effective protection of EU consumers and professional users and a competitive single EU market.
Policies and legislative techniques have evolved over the last 40 years of European integration, especially in the area of the free movement of goods, thereby contributing to the success of the Single Market today.
Historically, EU legislation for goods has progressed through four main phases:
|
— |
the traditional approach or ‘Old Approach’ with detailed texts containing all the necessary technical and administrative requirements, |
|
— |
the ‘New Approach’ developed in 1985, which restricted the content of legislation to ‘essential requirements’ leaving the technical details to European harmonised standards. This in turn led to the development of European standardisation policy to support this legislation, |
|
— |
the development of the conformity assessment instruments made necessary by the implementation of the various Union harmonisation acts, both New Approach and Old Approach, |
|
— |
the ‘New Legislative Framework’ (2) adopted in July 2008, which built on the New Approach and completed the overall legislative framework with all the necessary elements for effective conformity assessment, accreditation and market surveillance including the control of products from outside the Union. |
1.1.1. THE ‘OLD APPROACH’
The Old Approach reflected the traditional manner in which national authorities drew up technical legislation, going into great detail — usually motivated by a lack of confidence in the rigour of economic operators on issues of public health and safety. In certain sectors (e.g. legal metrology) this even led public authorities to deliver certificates of conformity themselves. The unanimity required in this field until 1986 made the adoption of such legislation very unwieldy and the continued recourse to this technique in a number of sectors is often justified for reasons of public policy (e.g. food legislation) or by international traditions and/or agreements which cannot be changed unilaterally (e.g. automobile legislation or food again).
The first attempt to break out of this situation came with the adoption of Directive 83/189/EEC (3) on 28 March 1983 setting up an information procedure between the Member States and the Commission to avoid the creation of new technical barriers to the free movement of goods which would take a long time to correct through the harmonisation process.
Under that Directive, Member States are obliged to notify draft national technical regulations to other Member States and the Commission (and national standardisation bodies (NSB) were obliged to notify draft national standards (4) to the Commission, to the European standardisation organisations (ESO) and to other national standardisation bodies). During a standstill period these technical regulations may not be adopted, leaving the Commission and the other Member States the possibility to react. In the absence of reactions within the initial standstill period of 3 months, the draft technical regulations may then be adopted. Otherwise, where objections are raised, a further 3 month standstill is imposed.
The standstill period is 12 months in the presence of a proposal for a Union harmonisation act in the area in question. However, the standstill period does not apply where a Member State is obliged to introduce technical regulations urgently to protect public health or safety, animals or plants.
1.1.2. MUTUAL RECOGNITION
Alongside legislative initiatives to prevent new barriers and promote the free movement of goods, the systematic application of the principle of mutual recognition enshrined in EU law was also pursued. National technical regulations are subject to the provisions of Articles 34 to 36 of the Treaty on the Functioning of the European Union (TFEU), which prohibit quantitative restrictions or measures having equivalent effect. Case law of the European Court of Justice, especially case 120/78 (the ‘Cassis de Dijon’ case (5)), provides the key elements for mutual recognition. The effect of this case law is as follows.
|
— |
Products lawfully manufactured or marketed in one Member State should in principle move freely throughout the Union where such products meet equivalent levels of protection to those imposed by the Member State of destination. |
|
— |
In the absence of Union harmonisation legislation, Member States are free to legislate on their territory subject to the Treaty rules on free movement of goods (Arts 34-36 TFEU). |
|
— |
Barriers to free movement which result from differences in national legislation may only be accepted if national measures:
|
To help implement these principles, the European Parliament and the Council adopted, in the 2008 goods package, Regulation (EC) No 764/2008 of 9 July 2008 laying down procedures relating to the application of certain national technical rules to products lawfully marketed in another Member State and repealing Decision 3052/95/EC (6).
However, while contributing greatly to the free movement of goods within the single market, the mutual recognition principle cannot solve all the problems and there remains, even today as underlined by the comments of the Monti Report (7), room for further harmonisation.
1.1.3. THE ‘NEW APPROACH’ AND THE ‘GLOBAL APPROACH’
The Cassis de Dijon case is well known for its important role in promoting the mutual recognition principle but it also played an immense role in modifying the EU approach to technical harmonisation on three fundamental counts:
|
— |
in stating that Member States could only justify forbidding or restricting the marketing of products from other Member States on the basis of non-conformity with ‘essential requirements’, the Court opened a reflection on the content of future harmonisation legislation: since non-respect of non-essential requirements could not justify restricting the marketing of a product, such non-essential requirements need no longer figure in EU harmonisation texts. This opened the door to the New Approach and the consequent reflection on what constitutes an essential requirement and how to formulate it in such a manner that conformity can be demonstrated, |
|
— |
in stating this principle, the Court clearly placed the onus on national authorities to demonstrate where products did not conform to essential requirements but it also begged the question of the appropriate means for demonstrating conformity in a proportionate manner, |
|
— |
by noting that Member States were obliged to accept products from other Member States except in circumscribed conditions, the Court identified a legal principle but did not produce the means to create the trust in the products that could help authorities to accept products they could not vouch for. This led to the need to develop a policy on conformity assessment. |
The New Approach legislative technique approved by the Council of Ministers on 7 May 1985 in its Resolution on a new approach to technical harmonisation and standards (8) was the logical legislative follow up to the Cassis de Dijon case. This regulatory technique established the following principles:
|
— |
legislative harmonisation should be limited to the essential requirements (preferably performance or functional requirements) that products placed on the EU market must meet if they are to benefit from free movement within the EU, |
|
— |
the technical specifications for products meeting the essential requirements set out in legislation should be laid down in harmonised standards which can be applied alongside the legislation, |
|
— |
products manufactured in compliance with harmonised standards benefit from a presumption of conformity with the corresponding essential requirements of the applicable legislation, and, in some cases, the manufacturer may benefit from a simplified conformity assessment procedure (in many instances the manufacturer's declaration of conformity, made more easily acceptable to public authorities by the existence of the product liability legislation (9)), |
|
— |
the application of harmonised or other standards remains voluntary, and the manufacturer can always apply other technical specifications to meet the requirements (but will carry the burden of demonstrating that these technical specifications answer the needs of the essential requirements, more often than not, through a process involving a third party conformity assessment body). |
The operation of Union harmonisation legislation under the New Approach requires harmonised standards to offer a guaranteed level of protection with regard to the essential requirements established by the legislation. This constitutes one of the major preoccupations of the Commission in pursuit of its policy for a strong European standardisation process and infrastructure. Regulation (EU) No 1025/2012 on European Standardisation (10) gives the Commission the possibility of inviting, after consultation with the Member States, the European standardisation organisations to draw up harmonised standards and it establishes procedures to assess and to object to harmonised standards.
Since the New Approach calls for common essential requirements to be made mandatory by legislation, this approach is appropriate only where it is possible to distinguish between essential requirements and technical specifications. Further, as the scope of such legislation is risk-related, the wide range of products covered has to be sufficiently homogeneous for common essential requirements to be applicable. The product area or hazards also have to be suitable for standardisation.
The principles of the New Approach laid the foundation for European standardisation in support of Union harmonisation legislation. The role of harmonised standards and the responsibilities of the European standardisation organisations are now defined in Regulation (EU) No 1025/2012 together with relevant Union harmonisation legislation.
The principle of reliance on standards in technical regulations has also been adopted by the World Trade Organisation (WTO). In its Agreement on Technical Barriers to Trade (TBT) it promotes the use of international standards (11).
The negotiation of the first Union harmonisation texts under the New Approach immediately highlighted the fact that the determination of essential requirements and the development of harmonised standards were not sufficient to create the necessary level of trust between Member States and that an appropriate horizontal conformity assessment policy and instruments had to be developed. This was done in parallel to the adoption of the directives (12).
Hence in 1989 and 1990 the Council adopted a Resolution on the Global Approach and Decision 90/683/EEC (updated and replaced by Decision 93/465/EEC) (13) laying down the general guidelines and detailed procedures for conformity assessment. These have now been repealed and updated by Decision No 768/2008/EC of 9 July 2008 on a common framework for the marketing of products (14).
The major thrust of these policy instruments was to develop common tools for conformity assessment across the board (for both regulated and non-regulated areas).
The policy on product standards was first developed to ensure that the standards set technical specifications to which conformity could be demonstrated. However, at the request of the Commission, CEN and Cenelec adopted the EN 45000 series of standards for the determination of the competence of third parties involved with conformity assessment. This series has since become the EN ISO/IEC 17000 harmonised series of standards. Under the New Approach directives a mechanism was set up whereby national authorities notified the third parties they designated to carry out conformity assessments based on recourse to these standards.
On the basis of ISO/IEC documentation, the Council in its Decisions developed consolidated conformity assessment procedures and the rules for their selection and use in directives (the modules). The modules are set out in a manner to favour their selection from the lightest (‘internal control of production’) for simple products or products not necessarily presenting serious risks, moving to the most comprehensive (full quality assurance with EU-design examination), where the risks are more severe or the products/technologies more complex. In order to face up to modern manufacturing processes, the modules foresee both product conformity assessment processes and quality management assessment, leaving the legislator to decide which are the most appropriate in each sector, as it is not necessarily effective to provide for individual certification for each mass produced product, for example. To reinforce the transparency of the modules and their effectiveness, at the request of the Commission, the ISO 9001 series of standards on quality assurance were harmonised at the European level and integrated into the modules. Thus, economic operators who use these tools in their voluntary quality management policies to reinforce their quality image on the market, can benefit from the use of the same tools in the regulated sectors.
These different initiatives were all geared to directly reinforcing the assessment of conformity of products prior to their marketing. Alongside these, the Commission, in close cooperation with the Member States and the national accreditation bodies, developed European cooperation in the field of accreditation in order to constitute a last level of control and reinforce the credibility of the third parties involved in carrying out product and quality assurance conformity assessment. This remained a political, rather than a legislative initiative, but it was nevertheless effective in creating the first European infrastructure in this area, and in placing European players very much in the lead in this field at international level.
These developments led to some 27 directives being adopted on the basis of New Approach elements. They are far fewer in number than traditional directives in the field of industrial products (some 700), but their wide hazard-based scope means that entire industrial sectors have benefited from free movement through this legislative technique.
1.2. THE ‘NEW LEGISLATIVE FRAMEWORK’
1.2.1. THE CONCEPT
Towards the end of the 90s the Commission started to reflect on the effective implementation of the New Approach. In 2002, a wide consultation process was launched and on 7 May 2003 the Commission adopted a Communication to the Council and European Parliament suggesting a possible revision of certain New Approach elements. This in turn led to the Council Resolution of 10 November 2003 on the Communication of the European Commission ‘Enhancing the implementation of the New Approach Directives’ (15).
The consensus on the need for the update and review was clear and strong. The major elements needing attention were also clear: overall coherence and consistency, the notification process, accreditation, the conformity assessment procedures (modules), CE marking and market surveillance (including revision of the safeguard clause procedures).
A Regulation and a Decision constituting part of the ‘Ayral goods package’ (16) were adopted by the European Parliament and the Council on 9 July 2008 (17).
Regulation (EC) No 765/2008 and Decision No 768/2008/EC brought together, in the New Legislative Framework (NLF), all the elements required for a comprehensive regulatory framework to operate effectively for the safety and compliance of industrial products with the requirements adopted to protect the various public interests and for the proper functioning of the single market.
Regulation (EC) No 765/2008 established the legal basis for accreditation and market surveillance and consolidated the meaning of the CE marking, thus filling an existing void. Decision No 768/2008/EC updated, harmonised and consolidated the various technical instruments already used in existing Union harmonisation legislation (not only in New Approach directives): definitions, criteria for the designation and notification of conformity assessment bodies, rules for the notification process, the conformity assessment procedures (modules) and the rules for their use, the safeguard mechanisms, the responsibilities of the economic operators and traceability requirements.
The NLF takes account of the existence of all the economic operators in the supply chain — manufacturers, authorised representatives, distributors and importers — and of their respective roles in relation to the product. The importer now has clear obligations in relation to the compliance of products, and where a distributor or an importer modifies a product or markets it under their own name, they become the equivalent of the manufacturer and must take on the latter's responsibilities in relation to the product.
The NLF also recognises the different facets of the responsibilities of the national authorities: the regulatory authorities, the notification authorities, those which oversee the national accreditation body, the market surveillance authorities, the authorities responsible for the control of products from third countries, etc., underlining that responsibilities depend on the activities carried out.
The NLF has changed the emphasis of EU legislation in relation to market access. Formerly the language of Union harmonisation legislation concentrated on the notion of ‘placing on the market’ which is traditional free movement of goods language, i.e. it focuses on the first making available of a product on the EU market. The NLF, recognising the existence of a single internal market, puts the emphasis on making a product available thus giving more importance to what happens after a product is first made available. This also corresponds to the logic of the putting into place of EU market surveillance provisions. The introduction of the concept of making available facilitates the tracing back of a non-compliant product to the manufacturer. It is important to note that compliance is assessed with regard to the legal requirements applicable at the time of the first making available.
The most important change brought about by the NLF to the legislative environment of the EU was the introduction of a comprehensive policy on market surveillance. This has considerably changed the balance of EU legislative provisions from being fundamentally oriented at setting product related requirements to be met when products are placed on the market to an equal emphasis on enforcement aspects during the whole life-cycle of products.
1.2.2. THE LEGAL NATURE OF THE NLF ACTS AND THEIR RELATIONSHIP TO OTHER EU LEGISLATION
1.2.2.1. Regulation (EC) No 765/2008
Regulation (EC) No 765/2008 imposes clear obligations on Member States who do not have to transpose its provisions (although many may have to take national measures to adapt their national legal framework). Its provisions are directly applicable to the Member States, to all the economic operators concerned (manufacturers, distributors, importers) and to conformity assessment bodies and accreditation bodies. Economic operators now have not only obligations but direct rights that they can enforce through the national courts against both national authorities and other economic operators for non-respect of the provisions of the Regulation.
In the presence of other EU legislation, the Regulation applies first and foremost, (a) on the basis of being directly applicable, i.e. national authorities and economic operators must apply the provisions of the Regulation as such (most of the other legislation is contained in directives) and (b) on the basis of the lex specialis rule, i.e. whenever a matter is regulated by two rules, the more specific one should be applied first.
In the absence of more specific legislation on the issues covered by its provisions, Regulation (EC) No 765/2008 will apply at the same time, with, and as a complement to, existing legislation. Where existing legislation contains similar provisions as the Regulation, the corresponding provisions will have to be examined on a one to one basis to determine which is the most specific.
In general terms, relatively few EU legislative texts contain provisions relating to accreditation, so it can be said that Regulation (EC) No 765/2008 is of general application in this area. In the area of market surveillance (including the control of products from third countries) the situation is more complex, as some Union harmonisation legislation does have various provisions relating to the issues covered by the Regulation (e.g. pharmaceuticals and medical devices legislation which provides for a specific information procedure).
1.2.2.2. Decision No 768/2008/EC
Decision No 768/2008/EC is what is referred to as a sui generis decision, meaning that it has no addressees and therefore is neither directly nor indirectly applicable. It constitutes a political commitment on the part of the three EU institutions, European Parliament, Council and Commission.
This means that for its provisions to apply in Union law, they have to be either referred to expressis verbis (expressly) in future legislation or integrated into it.
The three institutions have indeed committed themselves to adhere to and to have recourse as systematically as possible to its provisions when drawing up product related legislation. Thus, relevant future proposals are to be examined in the light of the Decision and departures from its contents, duly justified.
1.2.3. HOW THE SYSTEM FITS TOGETHER
The evolution of EU legislative techniques in this area has been progressive, tackling issues one after another, although sometimes in parallel, culminating in the adoption of the New Legislative Framework: essential or other legal requirements, product standards, standards and rules for the competence of conformity assessment bodies as well as for accreditation, standards for quality management, conformity assessment procedures, CE marking, accreditation policy, and lately market surveillance policy including the control of products from third countries.
The New Legislative Framework now constitutes a complete system bringing together all the different elements that need to be dealt with in product safety legislation in a coherent, comprehensive legislative instrument that can be used across the board in all industrial sectors, and even beyond (environmental and health policies also have recourse to a number of these elements), whenever EU legislation is required.
In this system, the legislation has to set the levels of public protection objectives of the products concerned as well as the basic safety characteristics, it should set the obligations and requirements for economic operators, it has to set-where necessary-the level of competence of the third party conformity assessment bodies who assess products or quality management systems, as well as the control mechanisms for these bodies (notification and accreditation), it must determine which are the appropriate conformity assessment processes (modules which also include the manufacturer's declaration of conformity) to be applied, and finally it must impose the appropriate market surveillance mechanisms (internal and external) to ensure that the whole legislative instrument operates in an effective and seamless manner.
All these different elements are interlinked, operate together and are complementary, forming an EU quality (18) chain. The quality of the product depends on the quality of the manufacturing, which in many instances is influenced by the quality of testing, internal or carried out by external bodies, which depends on the quality of the conformity assessment processes, which depends on the quality of the bodies which in turn depends on the quality of their controls, which depends on the quality of notification or accreditation; the entire system depending on the quality of market surveillance and controls of products from third countries.
They should all be treated in one way or another in any piece of EU product safety legislation. If one element goes missing or is weak, the strength and effectiveness of the entire ‘quality chain’ is at stake.
1.3. THE GENERAL PRODUCT SAFETY DIRECTIVE
Directive 2001/95/EC (19) on general product safety (GPSD) is intended to ensure a high level of product safety throughout the EU for consumer products that are not covered by sector-specific EU harmonisation legislation. The GPSD also complements the provisions of sector legislation in some aspects. The key provision of the GPSD is that producers are obliged to place on the market only products which are safe (20). The GPSD also provides for market surveillance provisions aimed at guaranteeing a high level of consumer health and safety protection.
The GPSD has set up the Rapid Alert System which is used for dangerous non-food products (RAPEX, Rapid Alert System) between Member States and the Commission. The Rapid Alert System ensures that the relevant authorities are rapidly informed of dangerous products. Subject to certain conditions, Rapid Alert System notifications can also be exchanged with non-EU countries. In the case of serious product risks to the health and safety of consumers in various Member States, the GPSD provides for the possibility for the Commission to take temporary Decisions on Union-wide measures, so-called ‘emergency measures’. Under certain conditions, the Commission may adopt a formal Decision (valid for 1 year, but renewable for the same period) requiring the Member States to restrict or prevent the marketing of a product posing a serious risk to the health and safety of consumers. The Rapid Alert System has been subsequently extended by Regulation (EC) No 765/2008 to apply to all harmonised industrial products irrespective of the final user (i.e. professional products) and to products posing risks to other protected interests than health and safety, for example risks to the environment.
1.4. THE LEGISLATION ON PRODUCT LIABILITY
The concept of manufacturer according to Union harmonisation legislation as shaped by the New Legislative Framework is different from that under the Directive on consumer product liability 85/374/EEC (21). In the latter case, the concept of ‘producer’ (22) covers more and different persons than the concept of ‘manufacturer’ under the New Legislative Framework.
Legal or administrative action may take place against any person in the supply or distribution chain who can be considered responsible for a non-compliant product. This may, in particular, be the case when the producer is established outside the Union. The Directive on product liability covers all movables (23) and electricity, as well as raw materials and components of final products. Services as such are currently excluded from the scope. Secondly, the Directive applies only to defective products, i.e. products not providing the safety that a person is entitled to expect. The fact that a product is not fit for the use expected is not enough. The Directive only applies if a product lacks safety. The fact that a better product is made afterwards does not render older models defective.
Liability, the responsibility to pay for damages, is placed on the producer. A producer is either a manufacturer of a finished product or a component part of a finished product, producer of any raw material, or any person who presents himself as a manufacturer (for example by affixing a trademark). Importers placing products on the Union market from third countries are all considered to be producers under the Directive on product liability. If the producer cannot be identified, each supplier of the product becomes liable, unless he informs the injured person within a reasonable time of the identity of the producer, or of the person who supplied him with the product. When several persons are liable for the same damage, they are all jointly and severally liable.
The producer must compensate damages caused by the defective product to individuals (death, personal injury) and private property (goods for private use). However, the Directive does not cover any damage to property under EUR 500 (24) for a single incident. National law may govern non-material damages (such as pain and suffering). The Directive does not cover the destruction of the defective product itself and, therefore, there is no obligation to compensate for it under the Directive on product liability. This is without prejudice to national law.
The Directive on product liability allows Member States to fix a financial ceiling for serial accidents at a minimum of EUR 70 million (25). However, most Member States have not used this possibility.
The producer is not automatically liable for damage caused by the product. The injured person, whether or not he is the buyer or user of the defective product, must claim his rights to obtain compensation. The victims will be paid only if they prove that they have suffered damage, the product was defective, and this product caused the damage. If the injured person contributes to the damage, the producer's liability may be reduced or even eliminated. However, the victims do not need to prove that the producer was negligent because the Directive on product liability is based on the principle of no-fault liability. Thus, the producer will not be exonerated even if he proves he was not negligent, if an act or omission of a third person contributes to the damage caused, if he has applied standards, or if his product has been tested. The producer will not have to pay, if he proves:
|
— |
he did not place the product on the market (for example the product was stolen), |
|
— |
the product was not defective when he placed it on the market (thus he proves that the defect was caused subsequently), |
|
— |
the product was not manufactured to be sold or distributed for economic purpose, |
|
— |
the defect was caused due to compliance with mandatory regulations issued by the public authorities (which excludes national, European and international standards) (26), |
|
— |
the state of scientific and technical knowledge at the time when the product was put on the market could not as such enable the existence of the defect to be discovered (the development risks defence) (27), or |
|
— |
where he is a subcontractor, that the defect was due either to the design of the finished product or to defective instructions given to him by the producer of the finished product. |
10 years after the product is placed on the market, the producer ceases to be liable, unless legal action is pending. Further, the victim must file an action within 3 years of the damage, the defect and the identity of the producer being known. No waivers of liability in relation to the injured person may be agreed.
The Directive on product liability does not require Member States to repeal any other legislation on liability. In this respect, the Directive's regime adds to the existing national rules on liability. It is up to the victim to choose the grounds on which to file the action.
1.5. SCOPE OF THE GUIDE
This Guide discusses non-food and non-agricultural products referred to as industrial products or products whether for use by consumers or professionals. The product-related legislation that deals with these products will be referred throughout the text indistinctly as Union harmonisation legislation, sectoral Union harmonisation legislation or Union harmonisation acts.
The New Legislative Framework consists of a set of legal documents. In particular Decision No 768/2008/EC provides for elements, which are partially or totally implemented in product-related Union harmonisation legislation addressing various public interests. The guide gives guidance for the implementation of the provisions and concepts laid down in the New Legislative Framework (28). Where there are product-specific deviations or provisions the guide refers to sectoral guides, which exist for almost all sectoral Union harmonisation legislation.
The present Guide has the ambition of explaining the different elements of the New Legislative Framework in detail and of contributing to a better overall understanding of the system so that legislation is implemented properly and is therefore effective for the protection of public interests such as health and safety, consumers, the environment and public security, as well as the proper functioning of the internal market for economic operators. Furthermore, the Guide promotes the goals of the Commission's better regulation policy by contributing to the development of more comprehensive, coherent and proportionate legislation.
Each of these chapters should be read in conjunction with the explanations set out above, in other words against the general background, and in conjunction with the other chapters, as they are all interlinked and should not be seen in isolation.
|
This guide primarily relates to the Union legislation on:
|
However, elements of this Guide might be relevant for other Union harmonisation legislation going even beyond the realm of industrial products. This is particularly true for the various definitions in the Guide as well as the chapters bearing on standardisation, conformity assessment, accreditation and market surveillance. Although, it is neither correct nor desirable to dress an exhaustive list of relevant legislation, a larger list of legislation concerned is provided in Annex I.
The Guide does not attempt to cover:
|
— |
the Directive on General Product Safety (29). The Commission services have provided specific guidance on the practical application of the GPSD (30). |
|
— |
the Union legislation on motor vehicles, construction products, REACH, and chemicals. |
2. WHEN DOES UNION HARMONISATION LEGISLATION ON PRODUCTS APPLY?
2.1. PRODUCT COVERAGE
|
Union harmonisation legislation applies to products which are intended to be placed (or put into service (31)) on the market (32). Furthermore, Union harmonisation legislation applies when the product is placed on the market (or put into service) and to any subsequent making available until the product reaches the end-user (33) (34) (35). A product still in the distribution chain falls under the obligations of the Union harmonisation legislation as long as it is a new product. (36) Once it reaches the end-user it is no longer considered a new product and the Union harmonisation legislation no longer applies (37). The end-user is not one of the economic operators who bear responsibilities under Union harmonisation legislation, i.e. any operation or transaction by the end-user involving the product is not subject to Union harmonisation legislation. However, such an operation or transaction might fall under another regulatory regime, in particular at national level.
The product must comply with the legal requirements that were in place at the time of its placing on the market (or putting into service).
Union harmonisation legislation applies to all form of supply, including distance selling and selling through electronic means. Hence, regardless of the selling technique products intended to be made available on the Union market must be in conformity with the applicable legislation.
A product intended to be placed on the Union market, offered in a catalogue or by means of electronic commerce, has to comply with Union harmonisation legislation when the catalogue or website directs its offer to the Union market and includes an ordering and shipping system (38). Where a product is not intended for the Union market or is not compliant with the applicable Union legislation, this has to be clearly indicated (e.g. by providing a visual warning).
The Union harmonisation legislation applies to newly manufactured products but also to used and second-hand products, including products resulting from the preparation for re-use of electrical or electronic waste, imported from a third country when they enter the Union market for the first time (39) (40). This applies even to used and second-hand products imported from a third country that were manufactured before the legislation became applicable (41).
Union harmonisation legislation applies to finished products. Yet, the concept of product varies between different pieces of Union harmonisation legislation. The objects covered by legislation are referred to, for instance, as products, equipment, apparatus, devices, appliances, instruments, materials, assemblies, components or safety components, units, fittings, accessories, systems or partly completed machinery. Thus, within the terms of a specific Union harmonisation act, components, spare parts or sub-assemblies may be regarded as finished products and their end-use may be the assembly or incorporation into a finished product. It is the responsibility of the manufacturer to verify whether or not the product is within the scope of a given piece of Union harmonisation legislation (42) (43).
A combination of products and parts, which each comply with applicable legislation, does not always constitute a finished product that has to comply itself as a whole with a given Union harmonisation legislation. However, in some cases, a combination of different products and parts designed or put together by the same person is considered as one finished product which has to comply with the legislation as such. In particular, the manufacturer of the combination is responsible for selecting suitable products to make up the combination, for putting the combination together in such a way that it complies with the provisions of the laws concerned, and for fulfilling all the requirements of the legislation in relation to the assembly, the EU declaration of conformity and CE marking. The fact that components or parts are CE marked does not automatically guarantee that the finished product also complies. Manufacturers must choose components and parts in such a way that the finished product itself complies. The manufacturer must verify on a case-by-case basis whether a combination of products and parts has to be considered as one finished product in relation with the scope of the relevant legislation.
A product, which has been subject to important changes or overhaul aiming to modify its original performance, purpose or type after it has been put into service, having a significant impact on its compliance with Union harmonisation legislation, must be considered as a new product. This has to be assessed on a case-by-case basis and, in particular, in view of the objective of the legislation and the type of products covered by the legislation in question. Where a rebuilt (44) or modified product is considered as a new product, it must comply with the provisions of the applicable legislation when it is made available or put into service. This has to be verified by applying the appropriate conformity assessment procedure laid down by the legislation in question. In particular, if the risk assessment leads to the conclusion that the nature of the hazard has changed or the level of risk has increased, then the modified product has to be considered as a new product, i.e. compliance of the modified product with the applicable essential requirements has to be reassessed and the person carrying out the modification has to fulfil the same requirements as an original manufacturer, for example preparation of the technical documentation, drawing up a EU declaration of conformity and affixing the CE marking on the product.
In any case, a modified product sold under the name or trademark of a natural or legal person different from the original manufacturer, should be considered as new and subject to Union harmonisation legislation. The person who carries out important changes to the product carries the responsibility for verifying whether or not it should be considered as a new product in relation to the relevant Union harmonisation legislation. If the product is to be considered as new, this person becomes the manufacturer with the corresponding obligations. Furthermore, in the case the conclusion is that it is a new product, the product has to undergo a full conformity assessment before it is made available on the market. However, the technical documentation has to be updated in as much as the modification has an impact on the requirements of the applicable legislation. It is not necessary to repeat tests and produce new documentation in relation to aspects not impacted by the modification, as long as the manufacturer has copies (or access to copies) of the original test reports for the unchanged aspects. It is up to the natural or legal person who carries out changes or has changes carried out to the product to demonstrate that not all elements of the technical documentation need to be updated.
Products which have been repaired or exchanged (for example following a defect), without changing the original performance, purpose or type, are not to be considered as new products according to Union harmonisation legislation. Thus, such products do not need to undergo conformity assessment again, whether or not the original product was placed on the market before or after the legislation entered into force. This applies even if the product has been temporarily exported to a third county for the repair operations. Such repair operations are often carried out by replacing a defective or worn item by a spare part, which is either identical, or at least similar, to the original part (for example modifications may have taken place due to technical progress, or discontinued production of the old part), by exchanging cards, components, sub-assemblies or even entire identical units. If the original performance of a product is modified (within the intended use, range of performance and maintenance originally conceived at the design stage) because the spare-parts used for its repair perform better due to technical progress, this product is not to be considered as new according to Union harmonisation legislation. Thus, maintenance operations are basically excluded from the scope of the Union harmonisation legislation. However, at the design stage of the product the intended use and maintenance must be taken into account (45).
Software updates or repairs could be assimilated to maintenance operations provided that they do not modify a product already placed on the market in such a way that compliance with the applicable requirements may be affected.
2.2. MAKING AVAILABLE ON THE MARKET
|
A product is made available on the market when supplied for distribution, consumption or use on the Union market in the course of a commercial activity, whether in return for payment or free of charge. (46) Such supply includes any offer for distribution, consumption or use on the Union market which could result in actual supply (e.g. an invitation to purchase, advertising campaigns).
Supplying a product is only considered as making available on the Union market, when the product is intended for end use on the Union market. The supply of products whether for further distribution, for incorporation into a final product, or for further processing or refinement with the aim to export the final product outside the Union market is not considered as making available. Commercial activity is understood as providing goods in a business related context. Non-profit organisations may be considered as carrying out commercial activities if they operate in such a context. This can only be appreciated on a case by case basis taking into account the regularity of the supplies, the characteristics of the product, the intentions of the supplier etc. In principle, occasional supplies by charities or hobbyists should not be considered as taking place in a business related context.
‘Use’ refers to the intended purpose of the product as defined by the manufacturer under conditions which can be reasonably foreseen. Usually, this is the end use of the product.
The central role that the concept of making available plays in Union harmonisation legislation is related to the fact that all economic operators in the supply-chain have traceability obligations and need to have an active role in ensuring that only compliant products circulate on the Union market.
The concept of making available refers to each individual product, not to a type of product, and whether it was manufactured as an individual unit or in series.
The making available of a product supposes an offer or an agreement (written or verbal) between two or more legal or natural persons for the transfer of ownership, possession or any other right (47) concerning the product in question after the stage of manufacture has taken place. The transfer does not necessarily require the physical handover of the product.
This transfer can be for payment or free of charge, and it can be based on any type of legal instrument. Thus, a transfer of a product is considered to have taken place, for instance, in the circumstances of sale, loan, hire (48), leasing and gift. Transfer of ownership implies that the product is intended to be placed at the disposal of another legal or natural person.
2.3. PLACING ON THE MARKET
|
For the purposes of Union harmonisation legislation, a product is placed on the market when it is made available for the first time on the Union market. The operation is reserved either for a manufacturer or an importer, i.e. the manufacturer and the importer are the only economic operators who place products on the market (49). When a manufacturer or an importer supplies a product to a distributor (50) or an end-user for the first time, the operation is always labelled in legal terms as ‘placing on the market’. Any subsequent operation, for instance, from a distributor to distributor or from a distributor to an end-user is defined as making available.
As for ‘making available’, the concept of placing on the market refers to each individual product, not to a type of product, and whether it was manufactured as an individual unit or in series. Consequently, even though a product model or type has been supplied before new Union harmonisation legislation laying down new mandatory requirements entered into force, individual units of the same model or type, which are placed on the market after the new requirements have become applicable, must comply with these new requirements.
Placing a product on the market requires an offer or an agreement (written or verbal) between two or more legal or natural persons for the transfer of ownership, possession or any other property right concerning the product in question after the stage of manufacture has taken place (51). This transfer could be for payment or free of charge. It does not require the physical handover of the product.
Placing on the market is considered not to take place where a product is:
|
— |
manufactured for one's own use. Some Union harmonisation legislation however covers products manufactured for own use in its scope (52) (53), |
|
— |
bought by a consumer in a third country while physically present in that country (54) and brought by the consumer into the EU for the personal use of that person, |
|
— |
transferred from the manufacturer in a third country to an authorised representative in the Union whom the manufacturer has engaged to ensure that the product complies with the Union harmonisation legislation (55), |
|
— |
introduced from a third country in the EU customs territory in transit, placed in free zones, warehouses, temporary storage or other special customs procedures (temporary admission or inward processing) (56), |
|
— |
manufactured in a Member State with a view to exporting it to a third country (this includes components supplied to a manufacturer for incorporation into a final product to be exported into a third country), |
|
— |
transferred for testing or validating pre-production units considered still in the stage of manufacture, |
|
— |
displayed or operated under controlled conditions (57) at trade fairs, exhibitions or demonstrations (58), or |
|
— |
in the stocks of the manufacturer (or the authorised representative established in the Union) or the importer, where the product is not yet made available, that is, when it is not being supplied for distribution, consumption or use, unless otherwise provided for in the applicable Union harmonisation legislation. |
Products offered for sale by online operators (59) (60) based in the EU are considered to have been placed on the Union market, regardless of who placed them on the market (the online operator, the importer, etc.). Products offered for sale online by sellers based outside the EU are considered to be placed on the Union market if sales are specifically targeted at EU consumers or other end-users. The assessment of whether or not a website located inside or outside the EU targets EU consumers has to be done on a case-by-case basis, taking into account any relevant factors such as the geographical areas to which dispatch is possible, the languages available used for the offer or for ordering, payment possibilities, etc. (61) When an online operator delivers in the EU, accepts payment by EU consumers/end-users and uses EU languages, then it can be considered that the operator has expressly chosen to supply products to EU consumers or other end-users. Online operators may offer online for sale a product type or an individual product which has been already manufactured. When the offer refers to a product type, the placing on the market will only take place after the stage of manufacture has been completed.
As the products offered for sale by an online operator are likely to be (or have already been) ordered by consumers or businesses in the EU, they are being supplied in the context of a commercial activity by way of online sales. Generally products are offered for sale online in return for payment. Nevertheless, the supply of products free of charge can also be a commercial activity (62). As for consumer to consumer (C2C) sales, these are generally not considered as commercial activities. Nevertheless the assessment of whether a C2C product is being supplied in the framework of a commercial activity has to be done on a case-by-case basis, taking into account all relevant criteria such as the regularity of the supplies, the intention of the supplier, etc. (63)
The legal consequence is that products offered for sale by online operators need to comply with all applicable EU rules when placed on the market (64). Such compliance can be physically verified by responsible authorities when the products are in their jurisdiction, at the soonest, at the customs.
In addition, products offered by online operators are generally stored in fulfilment houses located in the EU to guarantee their swift delivery to EU consumers. Accordingly, products stored in such fulfilment houses are considered to have been supplied for distribution, consumption or use in the EU market and thus placed on the EU market. When an online operator uses a fulfilment house, by shipping the products to the fulfilment house in the EU the products are in the distribution phase of the supply chain (65).
The placing on the market is the most decisive point in time concerning the application of the Union harmonised legislation (66). When made available on the market, products must be in compliance with the Union harmonisation legislation applicable at the time of placing on the market. Accordingly, new products manufactured in the Union and all products imported from third countries — whether new or used — must meet the provisions of the applicable Union harmonisation legislation when placed on the market, i.e. when made available for the first time on the Union market. Compliant products once they have been placed on the market may subsequently be made available along the delivery chain without additional considerations, even in case of revisions to the applicable legislation or the relevant harmonised standards, unless otherwise specified in the legislation.
Member States have an obligation in the framework of market surveillance to ensure that only safe and compliant products are on the market (67). Used products, which are on the Union market, are subject to free movement according to the principles laid down by Articles 34 and 36 TFEU. It must be noted that used products made available to consumers in the course of a commercial activity are subject to the GPSD.
2.4. PRODUCTS IMPORTED FROM COUNTRIES OUTSIDE THE EU
|
Union harmonisation legislation applies when the product is made available (or put into service (68)) on the Union market for the first time. It also applies to used and second-hand products imported from a third country, including products resulting from the preparation for re-use of electrical or electronic waste, when they enter the Union market for the first time, but not to such products already on the market. It applies even to used and second-hand products imported from a third country that were manufactured before the Union harmonisation legislation became applicable.
The basic principle of EU product rules is that irrespective of the origins of the products, they need to be compliant with the applicable Union harmonisation legislation if they are made available on the Union market. Products manufactured in the EU and products from non-EU countries are treated alike.
Before they can reach the end-user in the EU, products coming from countries outside the EU will be presented to customs under the release for free circulation procedure. The purpose of release for free circulation is to fulfil all import formalities so that the goods can be made available on the EU market like any product made in the EU. Therefore when products are presented to customs under the release for free circulation procedure, it can generally be considered that the goods are being placed on the EU market and so they will need to be compliant with the applicable Union harmonisation legislation. However, it may also be the case that the release for free circulation and the placing on the market do not take place at the same time. The placing on the market is the moment in which the product is supplied for distribution, consumption or use for the purposes of compliance with Union harmonisation legislation. Placing on the market can take place before the release for free circulation, for example, in the case of online sales by economic operators located outside the EU, even if the physical check of the compliance of the products can take place at the earliest when they arrive at the customs in the EU. Placing on the market can also take place after release for free circulation.
Customs authorities and market surveillance authorities have the obligation and the power, based on risk analyses, to check products arriving from third countries and intervene as appropriate before their release for free circulation, irrespective of when they are considered to be placed on the Union market. This is to prevent the release for free circulation and thus the making available in the EU territory of products which are not in compliance with the relevant Union harmonisation legislation (69).
For products imported from countries outside the EU, Union harmonisation legislation envisages a special role for the importer. The latter assumes certain obligations which to some extent mirror the obligations of manufacturers based within the EU (70).
In the case of products imported from countries outside the EU, an authorised representative may carry out a number of tasks on behalf of the manufacturer (71). If however, the authorised representative of a third country manufacturer supplies a product to a distributor or a consumer within the EU, he then no longer acts as a mere authorised representative but becomes the importer and is subject to the obligations of importers.
2.5. PUTTING INTO SERVICE OR USE (AND INSTALLATION)
|
Putting into service takes place at the moment of first use within the Union by the end user for the purposes for which it was intended (72) (73). The concept is used, for example, in the field of Lifts, Machinery, Radio equipment, Measuring instruments, Medical devices, in vitro diagnostic medical devices or products covered by the EMC or ATEX-directives, in addition to placing on the market, and results in the scope of Union harmonisation legislation being extended beyond the moment of making available of a product (74).
Where the product is put into service by an employer for use by his employees, the employer is considered as the end-user.
Member States may not prohibit, restrict or impede the putting into service of products that meet the provisions of the applicable Union harmonisation legislation (75). However, Member States are allowed to maintain and adopt, in compliance with the Treaty (in particular Articles 34 and 36 TFEU) and subject to Union harmonisation legislation, additional national provisions regarding the putting into service, installation or use, of products which are intended for the protection of workers or other users, or other products. Such national provisions may not require modifications of a product manufactured in accordance with the provisions of the applicable Union harmonisation legislation.
The need to demonstrate compliance of products at the moment of putting into service, and — if applicable — that they are correctly installed, maintained and used for the intended purpose, should be limited to products:
|
— |
which have not been placed on the market prior to their putting into service or which can be used only after an assembly, an installation or other manipulation has been carried out, or |
|
— |
whose compliance can be influenced by the distribution conditions (for example storage or transport). |
2.6. SIMULTANEOUS APPLICATION OF UNION HARMONISATION ACTS
|
Union harmonisation legislation covers a wide range of products, hazards and impacts (76), which both overlap and complement each other. As a result, the general rule is that several pieces of legislation may have to be taken into consideration for one product, since the making available or putting into service can only take place when the product complies with all applicable provisions and when the conformity assessment has been carried out in accordance with all applicable Union harmonisation legislation.
Hazards covered by the requirements of various Union harmonisation acts would typically concern different aspects that in many cases complement each other (for example the Directives relating to Electromagnetic Compatibility and Pressure Equipment cover phenomena not covered by the Directives relating to Low-voltage Equipment or Machinery). This calls for a simultaneous application of the various legislative acts. Accordingly, the product has to be designed and manufactured in accordance with all applicable Union harmonisation legislation, as well as to undergo the conformity assessment procedures according to all applicable legislation, unless otherwise provided for.
Certain Union harmonisation acts exclude from their scope products covered by other acts (77) or incorporate the essential requirements of other acts (78) which avoids simultaneous application of redundant requirements. In other instances, this is not the case and the general principle of simultaneous application still applies where the requirements of the Union harmonisation acts are complementary to each other.
Two or more Union harmonisation acts can cover the same product, hazard or impact. In such a case, the issue of overlap might be resolved by giving preference to the more specific Union harmonisation act (79). This usually requires a risk analysis of the product, or sometimes an analysis of the intended purpose of the product, which then determines the applicable legislation. In specifying the hazards relating to a product, the manufacturer may use the relevant harmonised standards applicable for the product in question.
2.7. INTENDED USE/MISUSE
|
Manufacturers have to match a level of protection corresponding to the use they prescribe to the product under the conditions of use which can be reasonably foreseen. |
Union harmonisation legislation applies in the case products made available or put into service (80) on the market are used for their intended use. Intended use means either the use for which a product is intended in accordance with the information provided by the person placing it on the market, or the ordinary use as determined by the design and construction of the product.
Usually such products are ready for use, or require only adjustments that can be performed in view of their intended use. Products are ‘ready for use’ if they can be used as intended without the insertion of additional parts. Products are also deemed ready for use if all parts from which they are to be assembled are placed on the market by one person only, or they only need to be mounted or plugged in, or they are placed on the market without the parts that are usually procured separately and inserted for the intended use (e.g. a cable for electric supply).
Manufacturers are required to match a level of protection for the users of the products which corresponds to the use that the manufacturer prescribes for the product in the product information. This is particularly relevant in the cases where a misuse of a product is at stake (81).
As far as market surveillance activities are concerned, market surveillance authorities are required to check the conformity of a product:
|
— |
in accordance with its intended purpose (as defined by the manufacturer) and |
|
— |
under the conditions of use which can be reasonably foreseen, that is when such use could result from lawful and readily predictable human behaviour. |
The consequence for manufacturers is that they have to consider the conditions of use which can be reasonably foreseen prior to placing a product on the market.
Manufacturers have to look beyond what they consider the intended use of a product and place themselves in the position of the average user of a particular product and envisage in what way they would reasonably consider to use the product (82).
It is also important that market surveillance authorities take into account that not all risks can be prevented by product design. The supervision and assistance of the intended users should be considered as part of the conditions which can be reasonably foreseen. For instance, some professional machine tools are intended for use by averagely skilled and trained workers under the supervision of their employer; the responsibility of the manufacturer cannot be engaged if such machine tools are rented by a distributor or third party service-provider for use by unskilled and untrained consumers.
In any case, the manufacturer is not obliged to expect that users will not take into consideration the lawful conditions of use of his product.
2.8. GEOGRAPHICAL APPLICATION (EEA EFTA STATES, OVERSEAS COUNTRIES AND TERRITORIES (OCTS), TURKEY)
|
2.8.1. MEMBER STATES AND OVERSEAS COUNTRIES AND TERRITORIES
The purpose of Union harmonisation legislation relating to goods adopted pursuant to Articles 114 and 115 TFEU is the establishment and functioning of the internal market for goods. Consequently, Union harmonisation legislation cannot be separated from Treaty provisions on free movement of goods and the territorial scope of application of Union harmonisation legislation should coincide with the territorial scope of application of Articles 30 and 34 to 36 TFEU.
Pursuant to Article 355 TFEU and in connection with the Article 52 of the Treaty on European Union (TEU), the Treaty and consequently the Union harmonisation legislation applies to all Member States of the European Union. Pursuant to Article 355(1) TFEU it also applies to Guadeloupe, French Guyana, Martinique, Réunion, Saint-Martin, the Azores, Madeira and the Canary Islands. Moreover, the Treaty and harmonisation legislation relating to products adopted on the basis of Articles 114 and 115 TFEU applies to certain European territories to the extent necessary to give effect to the arrangements set out in the relevant Accession Treaty (83).
However, it applies neither to Faeroe Islands, Greenland, Akrotiri and Dhekelia, nor to those overseas countries and territories having special relations with the United Kingdom of Great Britain and Northern Ireland such as Gibraltar. The Union harmonisation legislation does not apply to overseas countries and territories, in particular: New Caledonia and Dependencies, French Polynesia, French Southern and Antarctic Territories, Wallis and Futuna Islands, Saint Pierre and Miquelon, Saint-Barthélemy, Aruba, Curaçao, Sint Maarten, Caribbean Netherlands (Bonaire, Saba and Sint Eustatius), Anguilla, Cayman Islands, Falkland Islands, South Georgia and the South Sandwich Islands, Montserrat, Pitcairn, Saint Helena and Dependencies, British Antarctic Territory, British Indian Ocean Territory, Turks and Caicos Islands, British Virgin Islands, Bermuda.
2.8.2. EEA EFTA STATES
2.8.2.1. Basic elements of the Agreement on the European Economic Area
The Agreement on the European Economic Area, in force since 1 January 1994, covers all Union harmonisation legislation to which this Guide is applicable. Thus, Union harmonisation legislation covered by this Guide also applies to the so-called EEA EFTA States: Iceland, Liechtenstein and Norway.
The objective of the EEA Agreement is to establish a dynamic and homogeneous European Economic Area, based on common rules and equal conditions of competition.
Rights conferred and obligations imposed upon the Member States, or their public entities, undertakings, or individuals in relation to each other, are, according to the EEA Agreement, understood to be conferred or imposed in the same way also upon the EEA EFTA States. This ensures that the EEA EFTA States, and their economic operators, are subject to the same rights and obligations as their counterparts in the Union. For instance, the New Approach directives and other Union harmonisation legislation are implemented and applied in exactly the same way in the EEA EFTA States as in the Member States — although the safeguard clause is modified. Therefore, all guidance applicable to the Member States according to this Guide applies also to the EEA EFTA States.
For the purpose of the EEA Agreement references to the Community (now Union) or the common market in the EU/EEA acts are understood to be references to the territories of the Contracting Parties. Accordingly, a product is not only placed on the Union market, but on the EEA market (i.e. the national markets of the Member States and Iceland, Liechtenstein and Norway).
The EEA Agreement is amended on a continuous basis through decisions of the EEA Joint Committee following changes in relevant Union legislation. To arrive at and maintain a uniform interpretation and application of the Agreement, an EFTA Court and an EFTA Surveillance Authority have been established.
The EEA Agreement ensures a close cooperation between the Commission and the administration of the EEA EFTA States. The Commission seeks informal advice from experts of these States in the same way as it seeks advice from experts of the Member States. As regards the committees assisting the Commission in its work, close cooperation has been established. The EEA Council meets biannually, and the EEA Joint Parliamentary Committee and the EEA Consultative Committee regularly.
2.8.2.2. Safeguard clause procedure
The EFTA Surveillance Authority is responsible for the examination of the safeguard clause notifications from the EEA EFTA States. The Authority consults all parties concerned and exchanges information with the Commission on the proceedings of the case. The Authority transmits its decision to the EEA EFTA States and the Commission for further actions. If an EEA EFTA State does not follow the decision, the Surveillance Authority can initiate an infringement procedure.
In cases where a Member State triggers a safeguard clause, consultations between the Commission and the Surveillance Authority are envisaged. The Commission communicates its decision to the EFTA Surveillance Authority, which sends it to the EEA EFTA States for further actions. If an EEA EFTA State does not follow the decision, the Surveillance Authority can initiate an infringement procedure.
2.8.3. MONACO, SAN MARINO AND ANDORRA
Bilateral trade in products between the EU and Monaco, San Marino and Andorra, is facilitated by customs union agreements: Monaco has customs union with France and is part of the customs territory of the EU; whereas San Marino and Andorra both have a customs union agreement with the EU.
However, in order to be made available on the Union market, products from these countries must comply with the EU acquis. (84)
2.8.4. TURKEY
Turkey and the EU established a Customs Union in 1995 (Decision 1/95 of the EU-Turkey Association Council, 96/142/EC). The Customs Union Decision covers trade in manufactured products and processed agricultural products between Turkey and the EU, and entails alignment by Turkey with all EU product legislation. The Agreement aims to ensure the free movement of manufactured products and processed agricultural products between the EU and Turkey, by eliminating import controls at the EU-Turkey border on such products.
Articles 5 to 7 of the Decision provide for the elimination of measures having an effect equivalent to customs duties between the European Union and Turkey, mirroring Articles 34-36 TFEU. Pursuant to Article 66 of the Decision, its Articles 5 to 7 must, for the purposes of their implementation and application to products covered by the Customs Union Decision, be interpreted in conformity with the relevant case law of the Court of Justice, most notably the Cassis de Dijon case on mutual recognition.
As a consequence, in the sectors for which Turkey has aligned its legislation with that of the EU, a product lawfully manufactured and/or marketed in Turkey should be treated equal to a product lawfully manufactured and/or marketed in the EU and should not be subject to import controls. The same reasoning would apply in the non-harmonised sectors where Turkey has aligned its legislation with Articles 34-36 TFEU.
The Decision also requires Turkey to adopt European Union legislation on products and on quality infrastructure, notably on CE marking requirements, notified bodies, market surveillance, accreditation, standardisation, metrology and mutual recognition in the non-harmonised area.
Another Decision (Decision No 2/97 of the EC-Turkey Association Council) signed in 1997 lays down the list of the Union's legal instruments, including part of the acquis on industrial products related to the removal of technical barriers to trade and the conditions and arrangements governing their implementation by Turkey. Annex I to this Decision ensures that when Turkey adopted the legislation listed in Annex II to the Decision, the same rules and procedures would apply in the EU and Turkey for products that fall within the scope of the legislation listed in Annex II to the Decision. However, many of the legislative instruments set out in Annex II have been gradually replaced by new Union directives and regulations.
In 2006, the EU-Turkey Association Council adopted a new Decision (1/2006), providing for the designation of Turkish notified bodies and recognition of the test reports and certificates issued by such bodies in Turkey. The Parties have signed statements confirming that Turkey's legislation is equivalent to that of the EU for a number of New Approach directives and regulations.
In the non-harmonised area, the rights and obligations of economic operators supplying products to the EU market from Turkey have been laid down in the Commission's interpretative communication on ‘facilitating the access of products to the markets of other Member States: the practical application of mutual recognition’ (85).
The Turkish Accreditation Agency (TURKAK) is a member of European cooperation for Accreditation (EA) and has signed a number of mutual recognition agreements with EA. Certificates issued by Turkish conformity assessment bodies accredited by TURKAK should be deemed equivalent to those issued by conformity assessment bodies established in the EU and accredited by EU National Accreditation Bodies.
In the area of standardisation, both CEN and Cenelec granted full membership status to the Turkish Standards Institute (TSE) on 1 January 2012.
2.9. TRANSITIONAL PERIODS IN THE CASE OF NEW OR REVISED EU RULES
|
In the case of new or revised legislation, economic operators may be given additional time to adapt to the new rules which is called transitional period and correspond to the lapse of time between the entry into force of a new rule and the moment it starts applying. |
Transitional period means that the existing product rules remain applicable although new rules have been already adopted. Transitional period may be introduced by the legislator in the case EU product rules are being revised or come to replace national rules.
The aim of the transitional period is to allow manufacturers, national authorities and notified bodies to adjust gradually to the conformity assessment procedures and the essential or other legal requirements set up by a new or a revised piece of legislation, and, thus, to avert the risk of blocking production. Further, manufacturers, importers and distributors need to be given time to exercise any rights they have acquired under any pre-existing, national or EU rules, for example to sell their stocks of products manufactured in line with the pre-existing rules. Finally, the transitional period provides for extra time for the revision and adoption of harmonised standards, even though this is not a precondition for the application of Union harmonisation legislation.
Each Union harmonisation legislation providing for a transitional period sets the date for freezing the system in force. Generally, this is the date on which the legislation enters into force, but sometimes it is the date on which the legislation is adopted.
After the transitional period, products manufactured before or during this period, in line with the legislation to be repealed, may no longer be placed on the market. A product, which is placed on the market before the end of the transitional period, should be allowed to be made available on the market or put into service. (86) Nevertheless, specific Union harmonisation legislation could forbid the making available of such products if this is deemed necessary for safety reasons or other objectives of the legislation.
Products which were not placed on the market before the end of the transposition period can only be placed on the market or put into service, if they fully comply with the provisions of the new legislation (87).
According to the general rule, CE marking is an indication that products, which are subject to one or several pieces of Union harmonisation legislation providing for its affixing, conform to the provisions of all these applicable legislations. However, where one or more of these pieces of legislation allow the manufacturer, during a transitional period, to choose which arrangements to apply, the CE marking is an indication of conformity only to the legal texts applied by the manufacturer. Consequently, during a transitional period, the CE marking does not necessarily indicate that the product conforms to all applicable pieces of legislation providing for its affixing. Information concerning all Union harmonisation legislation applied by the manufacturer has to be found in the EU declaration of conformity (88).
2.10. TRANSITIONAL ARRANGEMENTS FOR THE EU DECLARATION OF CONFORMITY AS A RESULT OF THE ALIGNMENT TO DECISION NO 768/2008/EC
Union harmonisation legislation does not necessarily foresee a transitional solution for the information to be included in the EU declaration of conformity when existing legislation is replaced by a new one. This is the case for the Directives which have been revised to be aligned to the reference provisions of Decision No 768/2008/EC (89). The essential requirements in most of these Directives are not modified and there is no transitional period for referring to the old or new Directives. Furthermore, where relevant, aligned Directives specify that certificates issued under the old Directive remain valid under the new Directive. As of their entry into force, the EU Declaration of conformity will need to include the reference to the new Directives for the products placed on the market to be considered as compliant.
Union harmonisation legislation in most cases only specifies the mandatory minimum content of the EU declaration of conformity but additional useful information is generally accepted. Manufacturers may make use of this flexibility and start using the new model structure set out in the annexes to the aligned directives ahead of their entry into application. Where products comply with the requirements of both the old and new Directives, economic operators could refer to the two Directives in the EU declaration of conformity (‘old’ and aligned Directives), indicating the corresponding periods of application for each of the directives. For example, for a product in the scope of Directive 2014/30/EU, the EU declaration of conformity could contain the following statement:
‘The object of the declaration described above is in conformity with the relevant Union harmonisation legislation: Directive 2004/108/EC (until 19 April 2016) and Directive 2014/30/EU (from 20 April 2016).’
3. THE ACTORS IN THE PRODUCT SUPPLY CHAIN AND THEIR OBLIGATIONS
Union harmonisation legislation defines the manufacturer, the authorised representative, the importer and the distributor as ‘economic operators’ (90).
3.1. MANUFACTURER
|
The manufacturer is any natural or legal person who is responsible for designing or manufacturing a product and places it on the market under his own name or trademark (91). The definition contains two cumulative conditions: the person has to manufacture (or have a product manufactured) and to market the product under his own name or trademark. So, if the product is marketed under another person's name or trademark, this person will be considered as the manufacturer.
The responsibilities of the manufacturer apply also to any natural or legal person who assembles, packs, processes or labels ready-made products and places them on the market under his own name or trademark. Further, the responsibility of the manufacturer is placed on any person who changes the intended use of a product in such a way that different essential or other legal requirements will become applicable, or substantially modifies or re-builds a product (thus creating a new product), with a view to placing it on the market or for putting it into service, in those cases where the Union harmonisation legislation applicable to the product includes putting into service in its scope (92).
The manufacturer may design and manufacture the product himself. As an alternative, he may have it designed, manufactured, assembled, packed, processed or labelled with a view to placing it on the market under his own name or trademark, and thus presenting himself as a manufacturer (93). Where subcontracting takes place, the manufacturer must retain the overall control for the product and ensure that he receives all the information that is necessary to fulfil his responsibilities according to the relevant Union harmonisation act. The manufacturer who subcontracts some or all of his activities may in no circumstances discharge himself from his responsibilities, for example to an authorised representative, a distributor, a user or a subcontractor.
The manufacturer has ultimate responsibility for the conformity of the product to the applicable Union harmonisation legislation, whether he designed and manufactured the product himself or is considered as a manufacturer because the product is placed on the market under his name or trademark.
Thus, when a product is transferred to a manufacturer for further measures such as assembling, packaging, processing or labelling, when placing the product on the market, he has the sole and ultimate responsibility for ensuring the conformity of the product to the applicable legislation, and must be able to do so.
The manufacturer is responsible for designing and manufacturing the product in accordance with essential or other legal requirements laid down by the relevant Union harmonisation legislation and for carrying out conformity assessment in accordance with the procedure(s) laid down by the Union harmonisation legislation (94).
The manufacturer is obliged to understand both the design and construction of the product to be able to take the responsibility for the product being in compliance with all provisions of the relevant Union harmonisation legislation. This applies equally to situations where the manufacturer designs, manufactures, packs and labels the product himself, as to situations where some or all of these operations are carried out by a subcontractor. The manufacturer needs to have the relevant information to demonstrate compliance of the product at its disposal.
In this respect, the economic operator that places the product on the market under its name or trademark becomes automatically the manufacturer for the purposes of Union harmonisation legislation. Therefore he takes the entire responsibility for the conformity assessment (design and production) of the product, even if this has been actually done by somebody else. Furthermore he must be in the possession of all documentation and certificates necessary to demonstrate the conformity of the product, but these do not need to be under his name.
The Lifts Directive 95/16/EC defines the installer of a lift as ‘the natural or legal person who takes responsibility for the design, manufacture, installation and placing on the market of the lift and who affixes the CE marking and draws up the EC declaration of conformity’. Hence, the installer is a person who assumes responsibilities which in the context of other Union harmonisation legislation are typically assigned to the manufacturer.
Union harmonisation legislation does not require the manufacturer to be established in the European Union. Thus, when placing a product on the Union market, the responsibilities of a manufacturer are the same whether he is established outside the European Union or in a Member State.
As a general rule, when placing a product on the market the manufacturer must take all measures necessary to ensure that the manufacturing process assures compliance of the products (95) and in particular:
|
1. |
carry out the applicable conformity assessment or have it carried out, in accordance with the procedure(s) laid down by the relevant Union harmonisation legislation. Depending on the Union harmonisation act, the manufacturer may be required to submit the product to a third party (usually a notified body) to have the conformity assessment carried out, or to have a quality system approved by a notified body. In any case, the manufacturer bears full responsibility for product conformity; |
|
2. |
draw up the required technical documentation; |
|
3. |
draw up the EU declaration of conformity; |
|
4. |
accompany the product with instructions and safety information (96) (97) as required by the applicable Union harmonisation legislation (98), in a language easily understood by consumers and other end-users, as determined by the Member State concerned (99). Unless otherwise specified in specific legislation, instructions and safety information need to be provided (100), whether the product is intended for consumers or other end-users. This should include all the necessary information for the safe use of the product, to enable the consumer to assemble, install, operate, store, maintain, and dispose of the product. Instructions for assembly or installation should include the inventory parts and special skills or tools. Instructions on operation should include information for restriction of use, need for personal protective equipment, maintenance and cleaning or repair. It is for the manufacturer to determine the relevant information which should be included in the instructions and safety information for a particular product. Manufacturers have to look beyond what they consider the intended use of a product and place themselves in the position of the average user of a particular product and envisage in what way they would reasonably consider to use the product. Furthermore, a tool designed and intended to be used by professionals only might also be used by non-professionals, the design and instructions accompanied must take this possibility into account; |
|
5. |
satisfy the following traceability requirements:
|
|
6. |
affix the conformity marking (CE marking and where relevant other markings (108)) to the product in accordance with the applicable legislation; |
|
7. |
ensure that procedures are in place for series production to remain in conformity. Changes in product design or characteristics and changes in the harmonised standards or in other technical specifications by reference to which conformity of a product is declared must be adequately taken into account. The kind of action to be taken by the manufacturer depends on the nature of changes in the harmonised standards or other technical specifications, in particular whether these changes are material with regard to the coverage of the essential or other legal requirements and whether they concern the product in question. This might require for instance to update the EU Declaration of conformity, change the product design, contact the notified body (109), etc.; |
|
8. |
Where relevant, certify the product and/or the quality system. |
Under certain Union harmonisation acts, the manufacturer may be required to perform sample testing at the end of the production chain or of already marketed products in view of offering additional protection to consumers or other end-users (110) (111).
Manufacturers who consider or have reason to believe that a product which they have placed on the market is not in conformity with the applicable Union harmonisation legislation must immediately take the necessary corrective measures to bring that product into conformity, to withdraw it or recall it, if appropriate. Furthermore, where manufacturers have reason to believe that the product presents a risk to health, safety, the environment or any other public interest protected by the applicable legislation (112), they must immediately inform the competent national authorities of the Member States in which they made the product available to that effect, giving details, in particular, of the noncompliance and of any corrective measures taken. The Commission provides an IT tool, the ‘GPSD Business Application’ to facilitate practical aspects of this obligation (113).
Upon a reasoned request (114), the manufacturer has to provide the competent national authority with all the information and documentation necessary to demonstrate the conformity of a product, in a language which can be easily understood by that authority. Manufacturers must cooperate with the authority, at its request, on any action taken to eliminate the risks posed by products which they have placed on the market. Manufacturers must on request by market surveillance authorities identify any economic operator to whom they have supplied a product. They must be able to present this information for a period of 10 years after they have supplied the product.
The idea is that the national authority might accept a language they understand and which is different from the national language(s). The language chosen is subject to negotiation with the authority and could be a third language, if accepted by the authority.
In the case of a reasoned request it is sufficient for the manufacturer to provide the part of the technical documentation related to the claimed non-conformity and appropriate for demonstrating whether the issue has been dealt with by the manufacturer. Therefore, any request for translation of technical documentation should be limited to these parts of the documentation. The request may indicate a deadline for the receipt of the requested documents, depending on the Union harmonisation legislation the product is subject to. A shorter deadline can be fixed if the national authority justifies the urgency on the basis of an immediate serious risk.
If the Union harmonisation legislation covers putting into service, the natural or legal person who puts the product into service has the same responsibilities as a manufacturer who places a product on the market. He must ensure that the product complies with the Union harmonisation legislation, and that the appropriate conformity assessment procedure has been carried out (115).
Furthermore, a person who places on the Union market second-hand products from a third country, or any product not designed or manufactured for the Union market, must assume the role of the manufacturer.
Finally, if an importer or distributor modifies a product to the extent that the compliance with the applicable requirements may be affected or supplies it under his name or trademark, then he is to be considered the manufacturer and must undertake all the obligations incumbent on the manufacturer (116). Accordingly, he must ensure that the product complies with the applicable Union harmonisation legislation and that the appropriate conformity assessment procedure has been carried out (117).
3.2. AUTHORISED REPRESENTATIVE
|
Irrespectively of whether he is established in the EU or not, the manufacturer may appoint an authorised representative in the Union to act on his behalf in carrying out certain tasks. |
Whether the manufacturer is established in the EU or not, he may appoint an authorised representative in the Union to act on his behalf in carrying out certain tasks required in the applicable Union harmonisation legislation (118). A manufacturer established outside the European Union is not obliged to have an authorised representative (119).
For the purposes of Union harmonisation legislation, to be able to act on behalf of the manufacturer, the authorised representative must be established inside the Union. Commercial representatives of the manufacturer (such as authorised distributors or agents), are not to be confused with the authorised representative in the meaning of Union harmonisation legislation.
The delegation of tasks from the manufacturer to the authorised representative must be explicit and set out in writing, in particular to define the contents and limits of the representative's tasks. The tasks that may be delegated to the authorised representative according to the Union harmonisation legislation are of an administrative nature. Thus, the manufacturer may neither delegate the measures necessary to ensure that the manufacturing process assures compliance of the products nor the drawing up of technical documentation, unless otherwise provided for. Further, an authorised representative cannot modify the product on his own initiative in order to bring it into line with the applicable Union harmonisation legislation.
Where the manufacturer appoints an authorised representative, the mandate shall at least allow the authorised representative to perform the following tasks:
|
— |
keep the EU declaration of conformity and the technical documentation at the disposal of national surveillance authorities and cooperate with them at their request, |
|
— |
upon a reasoned request from a competent national authority, provide that authority with all the information and documentation necessary to demonstrate the conformity of a product, |
|
— |
cooperate with the competent national authorities, at their request, on any action taken to eliminate the risks posed by products covered by their mandate. |
Depending on the conformity assessment procedure and the Union harmonisation act in question, the authorised representative can also, for instance, be appointed to perform tasks such as:
|
— |
affix the CE marking (and where relevant other markings) and the notified body's number to the product, |
|
— |
draw up and sign the EU declaration of conformity. |
The authorised representative who is appointed by a manufacturer may be an importer or a distributor in the meaning of Union harmonisation legislation, in which case he must also to fulfil the obligations of the importer or distributor (120).
3.3. IMPORTER
|
The importer is the economic operator established in the Union who places a product from a third country on the Union market. He has important and clearly defined responsibilities under Union harmonisation legislation (121) (122). To a large extent they build on the type of responsibilities which a manufacturer based in the EU is subjected to.
The importer must ensure that the manufacturer has correctly fulfilled his obligations. The importer is not a simple re-seller of products, but has a key role to play in guaranteeing the compliance of imported products.
The importer is defined as any natural or legal person who places a product from a third country on the EU market. As a general rule, before placing a product on the market the importer must ensure:
|
1. |
that the appropriate conformity assessment procedure has been carried out by the manufacturer. If he has any doubt about the conformity of the product, he must refrain from placing it on the market. If the product has already been placed on the market, he has to take corrective actions. (123) In both cases the manufacturer might need to be contacted to clarify any doubt about the conformity of the product. |
|
2. |
that the manufacturer has drawn up the technical documentation, affixed the relevant conformity marking (e.g. CE marking), fulfilled his traceability obligations and accompanied, where relevant, the product by the instructions and safety information in a language easily understood by consumers and other end-users, as determined by the Member State concerned. (124) |
These obligations are meant to make sure that the importers are aware of their responsibility to place only compliant products on the market (125). Neither do they imply the need for importers to systematically resort to additional control procedures or (third- party) testing, nor do they preclude them from doing so.
The importer also has to:
|
— |
Indicate the following three elements: his (1) name, (2) registered trade name or trade mark and (3) the address at which he can be contacted on the product or where not possible because of the size or physical characteristics of the product or because the packaging would need to be opened, on the packaging or/and (126) on the accompanying documentation (127). By doing so, he must not impede the visibility of any safety information printed on the product or the accompanying documents. |
|
— |
Ensure that, while a product is under his responsibility, storage or transport conditions do not jeopardise its compliance with the requirements set out in the applicable legislation. |
|
— |
Keep a copy of the EU declaration of conformity for 10 years after the product has been placed on the market (128) or for the period specified in the relevant Union harmonisation act. |
|
— |
Ensure that the technical documentation can be made available to the competent national authority upon request (129). The importer has to cooperate with that authority and upon a reasoned request (130), has to provide that authority with all the information and documentation necessary to demonstrate the conformity of the product in a language which can be easily understood by that authority. The idea is that the national authority might accept a language they understand and which is different from the national language(s). The language chosen is subject to negotiation with the authority and could be a third language, if accepted by the authority. |
|
— |
In the case of a reasoned request it is sufficient for the importer to provide the part of the technical documentation related to the claimed non-conformity and appropriate for demonstrating whether the issue has been dealt with by the manufacturer. Therefore, any request for translation of technical documentation should be limited to these parts of the documentation. |
|
— |
On request by market surveillance authorities, the importer must identify any economic operator who has supplied him and to whom he has supplied the product. He must be able to present this information for a period of 10 years after he has been supplied with the product and for a period of 10 years after he has supplied the product. |
Further, under certain Union harmonisation acts, the importer, like a manufacturer, may be required to perform or have performed sample testing of products already placed on the market (131).
Equally, importers who have reason to believe that a product which they have placed on the market is not in conformity with the Union harmonisation legislation applicable, shall immediately take the corrective measures necessary to bring that product into conformity, to withdraw it or recall it, if appropriate. Furthermore, where the product presents a risk, importers shall immediately inform the competent national authorities.
The importer needs neither a mandate from the manufacturer, nor a preferential relationship with the manufacturer like the authorised representative. However, the importer must ensure, in order to fulfil his responsibilities, that a contact with the manufacturer can be established (e.g. to make the technical documentation available to the requesting authority).
The importer may wish to carry out administrative tasks on behalf of the manufacturer. In such a case, he has to be explicitly designated by the manufacturer in order to become an authorised representative.
3.4. DISTRIBUTOR
|
Along with manufacturers and importers, distributors are the third category of economic operators who are subject to specific obligations. The distributor is a natural or a legal person in the supply chain, other than the manufacturer or the importer, who makes a product available on the market.
Retailers, wholesalers and other distributors in the supply chain are not required to have a preferential relationship with the manufacturer like the authorised representative. A distributor acquires products for further distribution either from a manufacturer, from an importer, or from another distributor.
Distributor must act with due care (132) in relation to the applicable requirements (133). They have to know, for instance, which products must bear the CE marking, what information is to accompany the product (for example the EU declaration of conformity), what are the language requirements for labelling, user instructions or other accompanying documents, and what is a clear indication of the product being non-compliant. Distributors have an obligation to demonstrate to the national market surveillance authority that they have acted with due care and ensure that the manufacturer, or his authorised representative, or the person who provided him with the product has taken the measures required by the applicable Union harmonisation legislation as listed in the obligations for distributors.
Conformity assessment, drawing up and keeping the EU declaration of conformity and the technical documentation remain the responsibility of the manufacturer and/or importer in the case of products from third countries. It is not part of the distributor's obligations to check whether a product already placed on the market is still in conformity with the legal obligations that are currently applicable in case these have changed. The obligations of the distributor refer to the legislation applicable when the product was placed on the market by the manufacturer or the importer unless specific legislation provides otherwise.
The distributor must be able to identify the manufacturer, his authorised representative, the importer or the person who has provided him with the product in order to assist the market surveillance authority in its efforts to obtain the EU declaration of conformity and the necessary parts of the technical documentation. Market surveillance authorities have the possibility to address their request for the technical documentation directly to the distributor. The latter is however not expected to be in possession of the relevant documentation.
Before making a product available on the market, the distributor must verify the following formal requirements (134):
|
— |
that the product bears the required conformity marking(s) (e.g. CE marking), |
|
— |
that the product is accompanied by the relevant documents (e.g. EU declaration of conformity (135)) and by instructions and safety information (136) in a language which can be easily understood by consumers and other end-users if required by the applicable legislation, |
|
— |
that the manufacturer and importer have indicated their (1) name, (2) registered trade name or trademark and (3) the address at which they can be contacted on the product or when not possible because of the size or physical characteristics of the products, on its packaging and/or on the accompanying documentation (137), and that the product bears a type, batch or serial number or other element allowing the identification of the product. |
Distributors must not supply products that they know or should have assumed, on the basis of information in their possession and as a professional, not to be in compliance with the legislation. Further, they must cooperate with the competent authority in actions taken to avoid or minimise these risks, inform the manufacturer or the importer as well as the competent national authorities (138).
Similar obligations bind distributors once a product is made available. If they have reasonable grounds to believe that a product is not in conformity, they have to make sure that corrective measures to bring the product into conformity are taken by the manufacturer or the importer and inform the competent national authorities. Distributors have to contact the importer or manufacturer to clarify any doubt about the conformity of the product.
In addition to controlling the conformity of the product with the formal requirements, the distributor must:
|
1. |
initiate corrective measures where there is suspicion of a non-conformity (139); |
|
2. |
assist market surveillance authorities in identifying the manufacturer or importer responsible for the product; |
|
3. |
upon a reasoned request (140) from a competent authority, cooperate with that authority and provide it with all the information and documentation necessary to demonstrate the conformity of a product (141); |
|
4. |
on request by market surveillance authorities, identify any economic operator who has supplied them and to whom they have supplied the product. They must be able to present this information for a period of 10 years after they have been supplied with the product and for a period of 10 years after they have supplied the product (142). |
The distribution conditions (for example transport or storage) may have an impact on maintaining the compliance with the provisions of the applicable Union harmonisation legislation. Thus, the person in charge of the distribution conditions must take the necessary measures to protect the compliance of the product. This is to ensure that the product complies with the essential or other legal requirements at the moment of first use within the Union (143).
The distribution conditions may, in the absence of Union harmonisation legislation, be regulated to some extent on the national level in accordance with Articles 34 and 36 TFEU. National legislation that grants to members of a specific profession the exclusive right to distribute certain products is capable, insofar as it restricts sales to certain channels, of affecting the possibilities of marketing imported products. Accordingly, such legislation may constitute a measure having an effect equivalent to a quantitative restriction on imports. However, it can be justified for instance on grounds of the protection of public health, if the measure is appropriate for the purpose and does not go beyond what is necessary to achieve it (144).
Fulfilment Service Providers
Fulfilment houses (145) represent a new business model generated by e-commerce. Products offered by online operators are generally stored in fulfilment houses located in the EU to guarantee their swift delivery to EU consumers. These entities provide services to other economic operators. They store products and, further to the receipt of orders, they package the products and ship them to customers. Sometimes, they also deal with returns. There is a wide range of operating scenarios for delivering fulfilment services. Some fulfilment houses offer all of the services listed above, while others only cover them partially. Their size and scale also differ, from global operators to micro businesses.
The activities of fulfilment service providers as described above go beyond those of parcel service providers that provide clearance services, sorting, transport and delivery of parcels. The complexity of the business model they offer makes fulfilment service providers a necessary element of the supply chain and therefore they can be considered as taking part in the supply of a product and subsequently in placing it on the market. Thus, where fulfilment service providers provide services as described above which go beyond those of parcel service providers, they should be considered as distributors and should fulfil the corresponding legal responsibilities.
Taking into account the variety of fulfilment houses and the services they provide, the analysis of the economic model of some operators may conclude that they are importers or authorised representatives.
3.5. OTHER INTERMEDIARIES: INTERMEDIARY SERVICE PROVIDERS UNDER THE E-COMMERCE DIRECTIVE
The E-Commerce Directive (146) establishes the legal framework for electronic commerce in the EU. It introduces harmonised rules on issues such as the transparency and information requirements for online services providers, commercial communications or electronic contracts.
The E-commerce Directive does not cover categories of economic operators, but rather describes different categories of activities. The most relevant categories of activities, from a product safety and compliance point of view, are the hosting activities (147). Hosting activities are activities such as storing information provided by the recipient of the service, e.g. web shops and market places or platforms.
Intermediary service providers carrying out the activities described above benefit from an exemption of liability for damages or criminal sanctions related to the content provided by third parties using their networks. However, the liability exemption is not absolute. In the case of hosting activities, which are the most relevant for the product safety and compliance area, the exemption only applies if the intermediary service provider (1) has no actual knowledge or awareness about the illegal nature of the information hosted and (2) upon obtaining such knowledge or awareness of the illegal content (for instance by a ‘sufficiently precise and adequately substantiated’ notice (148)), it acts expeditiously to remove it or disable access. If they do not fulfil these conditions, they cannot be covered by the exemption and thus they can be held liable for the content they host.
Following Article 15 of the E-commerce Directive, Member States cannot impose either a general obligation on these providers to monitor the content or a general obligation to actively seek facts or circumstances indicating illegal activity. This means that national authorities cannot establish a general obligation for intermediaries to actively monitor their entire internet traffic and seek elements indicating illegal activities such as unsafe products.
The ban on requesting general monitoring, however, does not limit public authorities in establishing specific monitoring requirements, although the scope of such arrangements have to be targeted. As an example of a distinct policy area still with certain similarities, a court can order service providers to make sure that certain websites, which contain exclusively copyright-infringing content or fake products, are blocked from users of the given Member State.
In practice, this means that national authorities can contact the hosting providers who, when notified of unlawful activity, if they want to benefit from the exemption of liability, have to remove or disable the content, meaning that the unsafe/non-compliant products would no longer be accessible to EU customers through their services. Nevertheless, market surveillance authorities should base their activities on the applicable provisions of Regulation (EC) No 765/2008 and relevant Union harmonisation legislation and, thus, target, in the first place, the responsible economic operator. Market surveillance authorities should assess the most appropriate action to be taken on a case-by-case basis and with a view to the principle of the proportionality, taking into account the level of the risk, if the economic operator is identifiable, the urgency, if previously measures have been taken against given product, etc.
The term ‘content’ covers an offer of a product online (e.g. its picture, description, etc.). The term ‘illegal activity’ refers to activities covered both by criminal and administrative law. The exemption of liability refers to civil, administrative and criminal liability for all types of illegal activities initiated by third parties online, such as copyright and trademarks piracy, unfair commercial practices, etc. The Directive seeks to strike a fair balance between all interests at stake. The legal basis for notifying and requiring hosting providers to remove/disable access to illegal content is contained in the national transpositions of the E-commerce Directive. Moreover, most online intermediaries have developed their own reporting mechanisms.
3.6. END-USER
|
Union harmonisation legislation does not create obligations for the end-users of the products in their scope (149). This is the case even when there are no responsible economic operators present within the EU (for example, in the context of products sold online). Consequently, the term is not defined in that legislation. It is certain however that the term covers both professional users and consumers. The concept of ‘end use’ by a professional or a consumer is intrinsically related to the concept of ‘intended use’ (150).
Many products covered by Union harmonisation legislation are used at work. According to legislation based on Article 153 TFEU, employers have obligations as regards the use of work equipment by workers at the workplace. An employer is considered to be any natural or legal person who has an employment relationship with a worker (that is any person employed by an employer), and has responsibility for the undertaking or establishment.
According to the Directive concerning the minimum safety and health requirements for the use of work equipment by workers at work (2009/104/EC), the employer must take all measures necessary to ensure that the work equipment (for example machinery and apparatus) made available to the workers is suitable for the work carried out, and may be used by workers without impairment to their safety or health. The employer may only obtain or use work equipment that complies with the provisions of the applicable legislation at the time of its first use, or, if no other legislation is applicable or is only partially applicable, the minimum requirements laid down in Annex I to Directive 2009/104/EC. The employer must also take the necessary measures to ensure that work equipment is kept at such a level. Further, the employer has an obligation to provide information and training for workers as regards the use of work equipment.
According to the Directive concerning the minimum health and safety requirements for the use of personal protective equipment by workers at the workplace (89/656/EEC), such equipment must comply with the relevant Union provisions on design and manufacture with respect to safety and health (that is the Union harmonisation act relating to personal protective equipment). Further, the equipment must be appropriate for the risk involved, correspond to existing conditions at the workplace, take into account ergonomic requirements and the worker's state of health, fit the wearer correctly, and be compatible where more than one pieces of equipment must be used simultaneously. The employer is required, before choosing the personal protective equipment, to assess that it satisfies the requirements.
According to the Directive on the minimum safety and health requirements for work with display screen equipment (90/270/EEC), employers are obliged to perform an analysis of workstations in order to evaluate the safety and health conditions, particularly regarding possible risks to eyesight, physical problems and problems of mental stress. The Directive also lays down the minimum requirements for the display screen and other equipment.
According to the Directive on the introduction of measures to encourage improvements in the safety and health of workers at work (89/391/EEC), workers have a general responsibility to take care, as far as possible, of their own safety and health and that of other persons affected by their acts at work. In accordance with the training and the instructions given by their employer they must, for instance, make correct use of machinery, apparatus, and other means of production, and the personal protective equipment.
Directives 89/391/EEC, 2009/104/EC, 89/656/EEC and 90/270/EEC lay down minimum requirements. Therefore, Member States are allowed to adopt or retain more stringent provisions, as long as they are compatible with the TFEU. The provisions of Union harmonisation legislation must be respected and, thus, additional national provisions may neither request a modification of a product within the scope of a Union harmonisation act, nor influence the conditions of the making available on the market of such products.
4. PRODUCT REQUIREMENTS
4.1. ESSENTIAL PRODUCT REQUIREMENTS
4.1.1. DEFINITION OF ESSENTIAL REQUIREMENTS
|
A fundamental feature of a large part of Union harmonisation legislation is to limit legislative harmonisation to the essential requirements that are of public interest. These requirements deal with the protection of health and safety of users (usually consumers and workers) but may also cover other fundamental requirements (for example protection of property, scarce resources or the environment).
Essential requirements are designed to provide and ensure a high level of protection. They either arise from certain hazards associated with the product (for example physical and mechanical resistance, flammability, chemical, electrical or biological properties, hygiene, radioactivity, accuracy), or refer to the product or its performance (for example provisions regarding materials, design, construction, manufacturing process, instructions drawn up by the manufacturer), or lay down the principal protection objective (for example by means of an illustrative list). Often they are a combination of these. As a result, several Union harmonisation acts may be applicable to a given product at the same time, since the essential requirements of different Union harmonisation acts need to be applied simultaneously in order to cover all relevant public interests.
Essential requirements must be applied as a function of the hazard inherent to a given product. Therefore, manufacturers have to carry out a risk analysis to first identify all possible risks that the product may pose and determine the essential requirements applicable to the product. This analysis has to be documented and included in the technical documentation (151). In addition, the manufacturer needs to document the assessment of how he is addressing the risks identified to ensure that the product complies with the applicable essential requirements (for example, by applying harmonised standards). If only part of the harmonised standard is applied or it does not cover all applicable essential requirements, then the way applicable essential requirements not covered by it are dealt with, should be documented (152).
Essential requirements define the results to be attained, or the hazards to be dealt with, but do not specify the technical solutions for doing so. The precise technical solution may be provided by a standard or by other technical specifications or be developed in accordance with general engineering or scientific knowledge laid down in engineering and scientific literature at the discretion of the manufacturer. This flexibility allows manufacturers to choose the way to meet the requirements. It allows also that, for instance, the materials and product design may be adapted to technological progress. Accordingly, Union harmonisation legislation based on essential requirements does not necessitate regular adaptation to technical progress, since assessment of whether requirements have been met or not are based on the state of technical know-how at the moment the product is placed on the market.
The essential requirements are set out in relevant sections or annexes of a given piece of Union harmonisation legislation. Although no detailed manufacturing specifications are included in the essential requirements, the degree of detailed wording differs between different Union harmonisation acts (153). The wording is intended to be precise enough to create, on transposition into national legislation, legally binding obligations that can be enforced, and to facilitate the setting up of standardisation requests by the Commission to the European standardisation organisations in order to produce harmonised standards. They are also formulated so to enable the assessment of conformity with those requirements, even in the absence of harmonised standards or in case the manufacturer chooses not to apply them.
4.1.2. CONFORMITY WITH THE ESSENTIAL REQUIREMENTS: HARMONISED STANDARDS
|
4.1.2.1. Definition of a harmonised standard
Regulation (EU) No 1025/2012 (154) provides definitions for the terms ‘standard’, ‘national standard’, ‘European standard’, ‘harmonised standard’, and ‘international standard’.
|
— |
‘Standards’ are defined as technical specifications, adopted by a recognised standardisation body, for repeated or continuous application, with which compliance is not compulsory. |
|
— |
‘European standards’ are ‘standards’ adopted by the European standardisation organisations (ESOs) listed in Annex I to Regulation (EU) No 1025/2012 (155). |
|
— |
Taking into account the first two definitions mentioned above, ‘harmonised standards’ are ‘European standards’ adopted, upon a request made by the Commission for the application of Union harmonisation legislation. Harmonised standards maintain their status of voluntary application. |
The definition for a ‘harmonised standard’, within the context of Regulation (EU) No 1025/2012, is not restricted to harmonised standards supporting harmonised product legislation as the Regulation mainstreams the use of harmonised standards in harmonisation legislation for services in similar way as in Union harmonisation legislation for products.
4.1.2.2. Role of harmonised standards
Harmonised standards are developed and published like other European standards following the internal rules of European standardisation organisations. According to these rules, all European standards must be transposed at national level by the national standardisation bodies. This transposition means that the European standards in question must be made available as national standards in an identical way, and that all conflicting national standards must be withdrawn in a given period.
Harmonised standards are European standards to which Regulation (EU) No 1025/2012 and sectoral Union harmonisation legislation give a special meaning (156). Harmonised standards maintain their status of voluntary application (157). However it is important to note that the definition of a harmonised standard does not contain any reference to the publication of its title in the OJ. As long as a title of a harmonised standard is not published in the OJ the harmonised standard, or parts thereof, does not give presumption of conformity with the essential or other requirements it aims to cover. The Commission formally requests the European standardisation organisations to present harmonised standards by issuing a standardisation request (mandate). The role and preparation of the Commission's standardisation request to the European standardisation organisations is detailed in the Vademecum on European standardisation (158). Prior to this the Commission consults the Member States (159). Obtaining standards based on consensus within the meaning of Regulation (EU) No 1025/2012 (160) implies a wide consultation of sectoral authorities at national level. Thus, the request provides a strong indication of the expectations of public authorities.
The European standardisation organisations will formally take a position on a request from the Commission in conformity with their internal rules. Acceptance of the request and the subsequent standardisation work initiate the standstill period for national standardisation bodies as provided for in their internal rules and, in the case of harmonised standards, also in Regulation (EU) No 1025/2012.
The elaboration and adoption of harmonised standards is based on Regulation (EU) No 1025/2012 (161) and on the General Guidelines for cooperation between the European standardisation organisations and the Commission and EFTA from 28 March 2003 (162). There are a number of requirements, principles and commitments concerning standardisation, such as the participation of all interested parties (for example manufacturers, including SMEs, consumer associations, environmental stakeholders and trade unions), the role of public authorities, the quality of standards and a uniform transposition of European standards throughout the Union by national standardisation bodies.
The European standardisation organisations are responsible for identifying, in line with relevant requests, and elaborating harmonised standards within the meaning of the relevant internal market legislation and for presenting a list of adopted harmonised standards to the Commission. The technical contents of such harmonised standards are under the entire responsibility of the European standardisation organisations. Once public authorities have agreed on a request, the search for technical solutions should in principle be left to the interested parties. In certain areas, such as the environment and health and safety, the participation of public authorities on a technical level is important in the standardisation process. However, Union harmonisation legislation for products do not foresee a procedure under which public authorities would systematically verify or approve either at Union or national level the contents of harmonised standards, which have been adopted by European standardisation organisations (163). The dialogue between standardisation bodies and public authorities and, when appropriate, their participation in the standardisation process should, nevertheless, help to ensure that the terms of the standardisation request are correctly understood and public concerns are properly taken into account in the process.
The European standardisation organisations decide on the work programme for harmonised standards in line with the relevant request. They may also identify existing standards that they judge, after examination and possible revision, to meet the terms of the request, or modify existing standards in order to meet those terms. In the same way, they may identify international or national standards and adopt them as European standards, and present them to the Commission as harmonised standards. It is also possible that sometimes only certain parts or clauses of a harmonised standard support essential requirements and then only those parts or clauses will provide presumption of conformity after the references are published in the OJ.
A harmonised standard must match the relevant essential or other legal requirements of the relevant piece of legislation in line with the relevant standardisation request. A harmonised standard may contain specifications relating not only to essential requirements but dealing with other non-regulated issues. In such a case, these specifications are clearly distinguished from those covering the essential requirements. A harmonised standard does not necessarily have to cover all essential requirements but it must be always clear which requirements are ‘aimed to be covered’ (164) as otherwise a manufacturer complying with a harmonised standard, referenced in the OJ, does not know against which requirements a ‘presumption of conformity’ will apply and public authorities do not know against which essential requirements they must accept a presumption of conformity.
The relevant essential or other legal requirements aimed to be covered are usually indicated in a separate informative annex (165) to a harmonised standard. When essential requirements are covered only partially, it should be clearly indicated in the standard. In some cases the scope of a harmonised standard may also indicate the relevant requirements with sufficient clarity (e.g. when there is a clear reference to covered safety related risks). This information on the ‘aimed coverage of essential or other requirements’ given in a harmonised standard thereby determines the scope of the so-called ‘presumption of conformity with legal requirements’.
A clear distinction must be made between ‘conformity with a standard’ and ‘presumption of conformity (when applying a harmonised standard)’. ‘Conformity with a standard’ usually makes a reference to a situation where a standard is ‘fully applied’. This is for instance the case of voluntary certification against a standard. For the purposes of ‘presumption of conformity’ it is sufficient to apply only those provisions relating to the essential or other legal requirements aimed to be covered.
Harmonised standards never replace legally binding essential requirements. A specification given in a harmonised standard is not an alternative to a relevant essential or other legal requirement but only a possible technical means to comply with it. In risk related harmonisation legislation this means in particular that a manufacturer always, even when using harmonised standards, remains fully responsible for assessing all the risks of his product in order to determine which essential (or other) requirements are applicable. After this assessment a manufacturer may then choose to apply specifications given in harmonised standards to implement ‘risk reduction measures’ (166) which are specified by harmonised standards. In risk related harmonisation legislation harmonised standards most commonly provide certain means to reduce or remove risks while manufacturers remain fully responsible for risk assessment to identify applicable risks and to identify applicable essential requirements in order to select suitable harmonised standards or other specifications.
The role of harmonised standards in complying with applicable essential requirements identified by a manufacturer — a generic philosophy for cases where a manufacturer needs to identify applicable essential requirements:

Where harmonised standards fail to indicate clearly the essential requirements aimed to be covered such standards may become less useful for manufacturers as there is less legal certainty on the real ‘scope of presumption of conformity’. An unclear or incorrect indication of the essential requirements aimed to be covered may also lead, in some cases, to formal objections against harmonised standards (see point 4.1.2.5). Where a harmonised standard covers only part of the essential requirements identified as applicable by a manufacturer or only certain aspects thereof, he additionally has to use other relevant technical specifications or develop solutions in accordance with general engineering or scientific knowledge laid down in engineering and scientific literature in order to meet the essential requirements of the legislation in question. In a similar way when a manufacturer chooses not to apply all the provisions given in a harmonised standard, and which normally would provide presumption of conformity, he needs, on the basis of his own risk assessment, to indicate in his technical documentation how the compliance is reached or that relevant essential requirements are not applicable for his product.
Occasionally standards may contain errors or offer different possible readings. If a manufacturer finds such an error or uncertainty, he should first make contact with his national standardisation body to seek for clarification.
4.1.2.3. Process to harmonised standards providing presumption of conformity
The overall procedure leading to a harmonised standard giving a presumption of conformity is described in Flowchart 1.
Before a preparation of a standardisation request asking to develop harmonised standards may start there should be Union harmonisation legislation, or this legislation is under preparation (167), which foresees the use of harmonised standards as a means to comply with essential or other legal requirements, i.e. the legislator has already given the political acceptance to the harmonised standards to be developed and published within the legal framework given in Regulation (EU) No 1025/2012.
|
1. |
Planning of the Commission's standardisation request: The Commission publishes its plans on future standardisation requests in the annual Union Work Programme for European standardisation according to Article 8 of Regulation (EU) No 1025/2012. This Work Programme also identifies standardisation needs under future harmonisation legislation. |
|
2. |
Preparation of a standardisation request: The Commission prepares, according to Article 10(1) of Regulation (EU) No 1025/2012, a draft request consulting European standardisation organisations, sectoral experts of the Member States and relevant stakeholders at European level according to Articles 10(2) and 12 of Regulation (EU) No 1025/2012. |
|
3. |
Adoption and notification of a standardisation request: The Commission adopts a request as a Commission implementing decision addressed to the ESO(s) after obtaining positive opinion from the Member States in accordance with the procedure laid down in Article 22(3) of Regulation (EU) No 1025/2012. The request is then notified to the relevant European standardisation organisations. |
|
4. |
Acceptance of a standardisation request: Relevant European standardisation organisation indicates its acceptance on the request (168) within the time limit given in Article 10(3) of Regulation (EU) No 1025/2012. National standardisation bodies are obliged to respect standstill requirements according to Article 3(6) of Regulation (EU) No 1025/2012. Relevant European standardisation organisation may apply for Union financing (an action grant) on the basis of Chapter V of Regulation (EU) No 1025/2012. The Commission informs the relevant European standardisation organisation within the timeline given in Article 10(4) of Regulation (EU) No 1025/2012 about the award of a grant. |
|
5. |
Programming and agreement of a work programme: Relevant European standardisation organisation(s) elaborates a (joint) work programme in line with the relevant request and presents it to the Commission. When appropriate the Commission may inform on priorities for the standardisation work. |
|
6. |
Drafting work: Responsible technical committee of a European standardisation organisation (169) elaborates a draft European standard. European standardisation organisations follow the principles recognised by the World Trade Organisation (WTO) in the field of standardisation (coherence, transparency, openness, consensus, voluntary application and efficiency). In addition Regulation (EU) No 1025/2012 addresses directly applicable requirements on stakeholder participation and transparency of work programmes and draft standards in its Articles 3 to 5. The accepted request is one of the reference documents the responsible technical committee must follow during drafting work. According to Article 10(5) of Regulation (EU) No 1025/2012 relevant European standardisation organisation shall inform (reporting) the Commission on activities undertaken and have a suitable means (170) implemented together with the Commission in lead to assess compliance of the drafted standards with the initial request. |
|
7. |
Public Enquiry: European standardisation organisations together with national standardisation bodies organise a public enquiry where all stakeholders may submit comments through national standardisation bodies. Article 4(3) of Regulation (EU) No 1025/2012 provides a procedure if a national standardisation body receives comments indicating a possible negative impact on the internal market. |
|
8. |
Integrating comments received: Responsible technical committee considers comments received during public enquiry and prepares the final draft European standard. |
|
9. |
Formal Vote: National standardisation bodies vote on the final draft in a formal vote where national standardisation bodies have weighted votes. The final draft is adopted if there is a simple majority of the votes cast in favour and if 71,00 % or more of the weighted votes cast (abstentions not counted) are in favour. |
|
10. |
Ratification and publication of a European standard (EN): Where the voting result is positive, the relevant European standardisation organisation ratifies and publishes the European standard (EN). As in this case the relevant EN supports Union harmonisation legislation and it was drafted on the basis of a Commission request, this EN is a harmonised standard with the meaning of Article 2(1)(c) of Regulation (EU) No 1025/2012 — however it does not yet provide a presumption of conformity. |
|
11. |
Submission of references to the Commission: Relevant European standardisation organisation transmits automatically references of a relevant harmonised standard to the Commission. This information includes in particular the reference number and the title in all official languages of the EU. |
|
12. |
Verification of the conditions for publication in the OJ: According to Article 10(5) of Regulation (EU) No 1025/2012 the Commission has to verify whether the relevant harmonised standard complies with the initial request. During this verification the Commission checks in particular that the harmonised standard is covered by the relevant request and whether essential or other legal requirements ‘aimed to be covered’ are clearly indicated and covered by the standard. During this verification there is no need for a review of the technical content as the Commission does not, in general, accept the technical content or takes responsibility for it. However already during this step the Commission may also assess the adequacy of technical specifications given in a harmonised standard in satisfying corresponding essential requirements and this assessment may lead to non-publication of the references in the OJ. |
|
13. |
Publication of references in the OJ: According to Article 10(6) of Regulation (EU) No 1025/2012 the Commission publishes the references of a harmonised standard in the OJ. This publication finally initiates a presumption of conformity with essential or other legal requirements covered by the relevant harmonised standard. A presumption of conformity is usually valid from the date the publication is done in the OJ and, in most common cases (see also point 4.1.2.5), can be removed by a formal objection or when a revised version of the harmonised standard is referenced in the OJ. |
|
14. |
National transposition: National standardisation bodies are obliged to transpose the relevant European standard (171) as an identical national standard on the basis of the internal rules of the European standardisation organisations. According to Article 3(6) of Regulation (EU) No 1025/2012 they also are obliged to withdraw any national standards which are conflicting with a harmonised standard. |
|
15. |
Formal objection: According to Article 11 of Regulation (EU) No 1025/2012 (172) a Member State or the European Parliament may dispute the publication of the references of a harmonised standard in the OJ. Through this process a Member State or the European Parliament may ask the Commission to draft a Commission Decision in order to prevent or remove the presumption of conformity. A formal objection may be made as soon as the standard has been adopted and ratified (in case of CEN and Cenelec) or adopted (in case of ETSI) in accordance with the rules of the organisation concerned. |
Flowchart 1
Process to harmonised standards and presumption of conformity
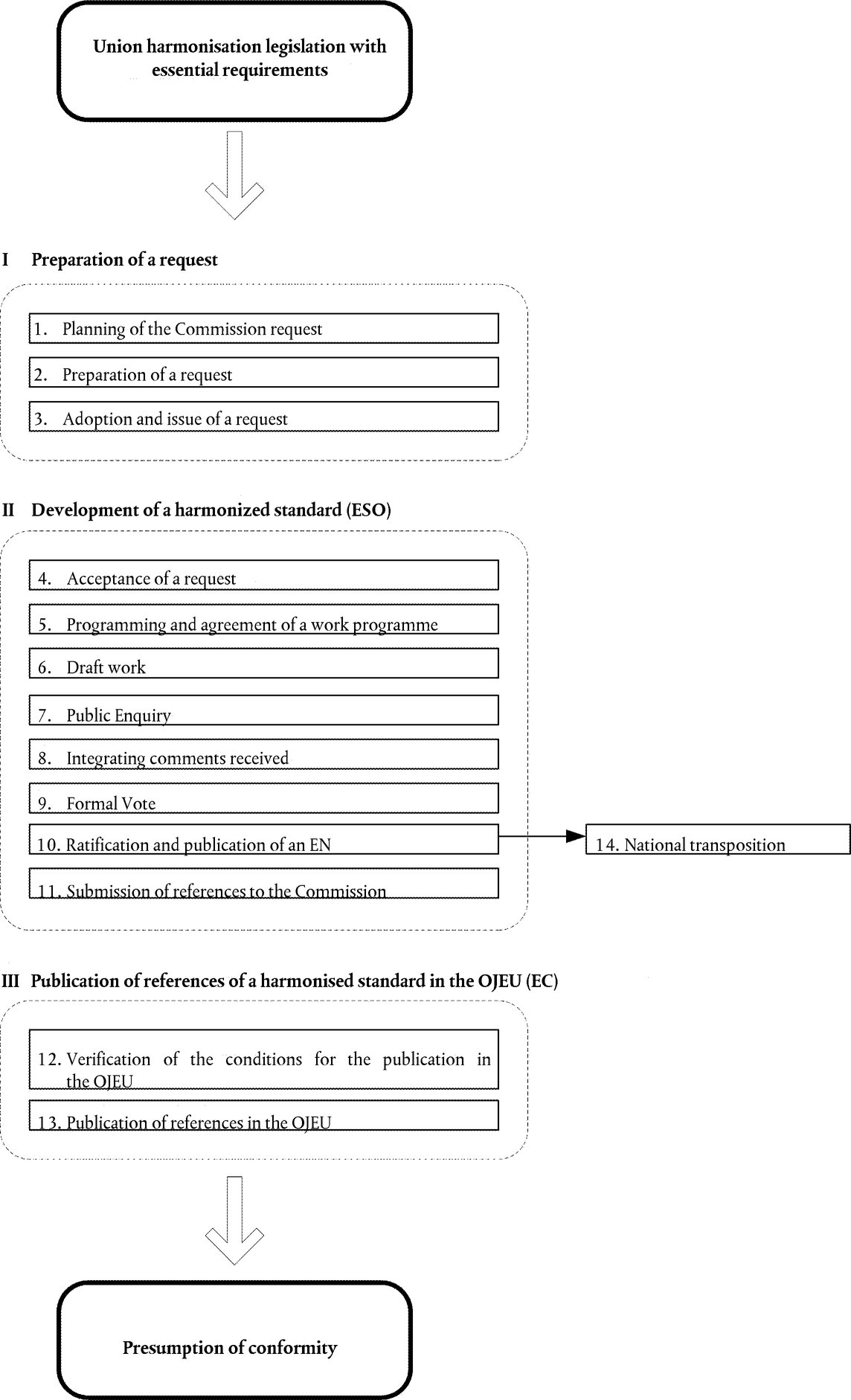
4.1.2.4. The Presumption of conformity
Harmonised standards provide a presumption of conformity with the essential requirements they aim to cover, if their references have been published in the Official Journal of the European Union. References of harmonised standards are published as Commission Communications in the C series of the OJ (173).
European standards, including harmonised standards, are often based fully or partially on international ISO or IEC standards. Sometimes, however, the presumption of conformity is possible only when applying the European version because of modifications introduced in it.
The objective of publishing the reference in the Official Journal is to set the date from which the presumption of conformity takes effect. Publication of references of harmonised standards is an administrative task for the Commission which is done without further consulting Member States or relevant sectoral Committees. It is the ultimate goal for a harmonised standard and the end of the process which started when the relevant Commission request was issued. Before the Commission publishes the references, it must however assess together with the European standardisation organisations, according to Article 10(5) of Regulation (EU) No 1025/2012, that the terms of the relevant request(s) are fulfilled and that the harmonised standard covers indeed the essential or other requirements which it claims (174) to cover.
Publication of references is not an automatic action and the Commission must perform certain checks and assessments before publication takes place. The Commission may thus refuse to publish the references or, in some cases, may set certain limitations which are published together with the references.
In a situation where a formal objection procedure has been already initiated, there is a doubt whether a harmonised standard entirely satisfies the requirements it aims to cover within the meaning of Article 11(1) of Regulation (EU) No 1025/2012. Because of this doubt, the Commission cannot publish the reference according to Article 10(6) of Regulation (EU) No 1025/2012 and a Commission Implementing Decision within the meaning of Article 11(1) must be taken.
There are also other situations where the references might not be published. The assessment according to Article 10(5) may reveal that the terms of the relevant request are not properly fulfilled or that the standard contains obvious errors. In those cases the conditions for initiating the objection procedure according to Article 11 (175) of Regulation (EU) No 1025/2012 are usually not fulfilled.
Examples of other reasons for non-publication include: the standard is not covered by the relevant standardisation request; products covered by the standard are not within the scope of relevant Union harmonisation legislation; the standard fails to indicate which legal (essential) requirements are covered (176); the standard does not cover the legal (essential) requirements which it claims to cover; the standard contains specifications which are not supporting essential requirements and without clearly separating them from specifications supporting essential requirements; the standard claims to support other legal requirements than those addressed in the request; the standard contains normative references to other specifications which are not acceptable because of their origin or lack of proper consensus building process during their adoption or normative reference are not yet accessible; other reasons because of non-application of internal rules of European standardisation organisations or disregard of the requirement given in Regulation (EU) No 1025/2012 during the preparation of a relevant harmonised standard.
In these cases the Commission ensures by non-publication a correct application of relevant Union harmonisation legislation and a coherent and proper functioning of the Single Market. Here the Commission can merely ask the relevant European standardisation organisations to correct the relevant standards making reference to the agreed requirements given in the relevant request and to other recognised and agreed principles under which these organisations should work. In some cases the Commission may consider the publication of references with a limitation, however, keeping in mind that these limitations should not overlap with reasons where a proper objection procedure should be initiated. The justification for non-publication comes from the relevant request itself but the Commission may refuse the publication also in order to protect the proper functioning of the Single Market.
The recourse to harmonised standards cited in the OJ and which give a presumption of conformity remains voluntary (177). The manufacturer can choose whether or not to refer to such harmonised standards. However, if the manufacturer chooses not to follow a harmonised standard, he has the obligation to demonstrate that his products are in conformity with essential requirements by the use of other means of his own choice (for example by means of any existing technical specifications including all other available standards). If the manufacturer applies only a part of a harmonised standard or the applicable harmonised standard does not cover entirely all applicable essential requirements, the presumption of conformity exists only to the extent the harmonised standard corresponds to the essential requirements. For that reason it is necessary that each harmonised standard contains clear and correct information on legal (essential) requirements covered.
Compliance with harmonised standards is, according to certain Union harmonisation acts, an option having effect on the applicable conformity assessment procedure, and sometimes opens the possibility for conformity assessment without the intervention of a third party or for a larger choice of procedures (178).
4.1.2.5. Withdrawal, restriction or prevention of the presumption of conformity
Regulation (EU) No 1025/2012 contains a provision according to which publication of titles of harmonised standards in the OJ can be challenged (179). This situation might arise prior to the publication of the reference of the harmonised standard in the OJ or in the case of a harmonised standard the reference of which has already been published in the OJ.
In both cases, where a Member State or the European Parliament (180) considers that a harmonised standard does not entirely satisfy the requirements which it aims to cover and which are set out in the relevant Union harmonisation legislation, it has to inform the Commission thereof. After consulting the Member States (181), the Commission can make an Implementing Decision:
|
— |
to publish, not to publish or to publish with restrictions the reference to the harmonised standard concerned in the OJ, or |
|
— |
to maintain, to maintain with restrictions or to withdraw the references to the harmonised standard concerned in or from the OJ. |
In all cases, the Commission needs to publish on its website (182) information on the harmonised standards that have been subject to such Implementing Decisions.
As part of its responsibilities and duties according to Regulation (EU) No 1025/2012 and the relevant sectoral legislation, the Commission can also draft and propose such Implementing Decisions to object to harmonised standards on its own initiative. Where a Member State has raised an action under a safeguard clause (183) against a product which complies with a harmonised standard and where such a safeguard action is considered justified, the Commission also has a responsibility to initiate an objection against the relevant harmonised standard.
The procedure to challenge a harmonised standard and its outcome does not affect its existence as a harmonised standard or as a European standard as only European standardisation organisations can make decisions on the revision or withdrawal of their deliverables. This objection procedure gives the legislator a possibility to control the presumption of conformity, i.e. the legal effect, which stems from the publication of the reference in the OJ. It may only lead to the withdrawal, restriction or prevention of such a publication in the OJ. In the two first cases, this means that the standard in question will no longer give presumption of conformity or presumption of conformity with the essential requirements is restricted. In the last case (prevention), it means that the standard will not become a harmonised standard giving presumption of conformity at all.
A harmonised standard can be challenged at any moment after its adoption by CEN, Cenelec or ETSI as a European standard.
Furthermore, the reference may be removed from the OJ by the Commission, without applying formal objection procedures, in certain exceptional cases where the relevant edition of a harmonised standard is not anymore reviewed or updated by the ESO itself and where the ESO itself does not regard it as a standard. Such cases include: the harmonised standard in question has been withdrawn by the relevant ESO without any intention to adopt a revised harmonised standard; the national standards transposing the harmonised standard are not available or valid as national standards anymore. The concept of essential requirements is based on the assumption that the harmonised standards reflect generally acknowledgeable state of the art and the ESO review standards regularly. When it is evident that a harmonised standard is not anymore recognised as a standard by the relevant ESO itself or where the standard is not anymore revised or available as a national standard such a document cannot be, as a rule, used anymore to provide presumption of conformity. The purpose of Article 11 of Regulation (EU) No 1025/2012 is to provide a procedure to challenge only valid harmonised standards, not withdrawn harmonised standards or draft harmonised standards which cannot be regarded as adopted European standards in the context of definitions given in Article 2 of Regulation (EU) No 1025/2012.
Another specific situation where the Commission may need to remove references from the OJ without formal objection relates to cases where publication in the OJ is done by a mistake or where a reference of a document which cannot be regarded as a harmonised standard is published. The latter case may include cases where the standard is not covered by a standardisation request or where the standard does not cover any essential requirements or where the standard was not correctly adopted by respective ESO following the recognised standardisation principles.
According to Regulation (EU) No 1025/2012 the Commission is obliged to inform stakeholders (184) on all pending formal objections against harmonised standards before formal decisions are taken.
4.1.2.6. Revision of harmonised standards
Harmonised standards translate the essential requirements into detailed technical specifications, methods of measurement to assess and/or declare the compliance with the essential requirements and, in some cases, numerical values to allow compliance with the essential requirements. Like any technical document, they are subject to change, or in other words to a revision.
The formal decision to revise a harmonised standard is, in principle, taken by the European standardisation organisations. This takes place on the basis of their own initiative (185), or following a standardisation request from the Commission directly or, indirectly, based on a Commission decision after a formal objection. The need for revision can result from the changes of the scope of the Union harmonisation act (such as an extension of the scope to other products or a modification of the essential requirements), from the fact that the Commission or a Member State challenges the contents of the harmonised standard, indicating that it could no longer give presumption of conformity with the essential requirements, or as a result of technological development.
When a harmonised standard is revised, the revision must be covered by a standardisation request to maintain the possibility of giving presumption of conformity. Unless the contrary can be deduced, the terms and conditions of the original request apply also for the revision of the harmonised standard. This does not exclude the possibility of a new request, in particular where the revision is related to shortcomings with respect to the essential requirements.
To give presumption of conformity, the revised standard must satisfy the general conditions according to the Union harmonisation legislation: the standard is based on a standardisation request, it is presented by the relevant European standardisation organisation to the Commission and its reference is published by the Commission in the Official Journal.
Following its internal rules, the relevant European standardisation organisation lays down for the national standardisation bodies the latest date of withdrawal of the superseded edition of the national standard — in this case for the national standards which transpose the previous edition of the harmonised standard. The transitional period during which both the withdrawn harmonised standard and the revised harmonised standard may give presumption of conformity is set by the Commission and published in the Official Journal. It is usually the time period between the date of publication of the reference of the new edition of the standard in the Official Journal and the date of withdrawal of the conflicting national standards — i.e. national standards which transpose the previous edition of the harmonised standards. It is the responsibility of the Commission to ensure that such transitional periods are of sufficient length and coherently set for all harmonised standards. After this transitional period, only the revised harmonised standard gives a presumption of conformity.
The Commission may consider that, for safety or other reasons, the superseded version of the harmonised standard must cease to give a presumption of conformity before the date of withdrawal, set by the European standardisation organisation in question, or at a later date. In such cases, the Commission fixes an earlier or later date after which the withdrawn harmonised standard will no longer give a presumption of conformity, and publishes this information in the Official Journal. If circumstances allow, the Commission may consult the Member States prior to taking a decision to reduce or extend the period during which both versions of the standard give a presumption of conformity.
Unless decided otherwise on the basis of a proposal by the Commission, the removal of the reference of a harmonised standard from the Official Journal after its revision does not automatically invalidate existing certificates issued by notified bodies; it only concerns the conformity that is conferred onto new conformity assessments that follow the new harmonised standard. Products produced according to the old certificate may still benefit from the continuing conformity with the essential requirements and may continue to be placed on the market until the end of the validity of the relevant certificates issued by notified bodies. However, manufacturers must assess the extent of the changes to the superseded version of the standard. The kind of action to be taken by the manufacturer depends on the nature of the changes in the harmonised standards, in particular whether these changes are material with regard to the coverage of the essential requirements and whether they concern the product in question. In addition, the notified body shall keep itself appraised of any changes in the generally acknowledged state of the art which indicate that the approved type may no longer comply with the applicable requirements, and shall determine whether such changes require further investigation. If so, the notified body shall inform the manufacturer accordingly. The reference of the revised harmonised standard, together with the information concerning the superseded version of the harmonised standard, and the date when the presumption of conformity of the superseded version of the standard ceases are published together in the Official Journal. It is in the interest of the manufacturer to check every publication of the list of harmonised standards and to verify within it the validity of the harmonised standards he has applied in order to assess the conformity of his product. This is particularly essential in cases where the manufacturer declares conformity himself (in case of internal production control) and where the manufacturer wants to ensure continuous presumption of conformity for the products placed on the market.
In the context of guidelines (186) agreed between the Commission and European standardisation organisations there is an expectation that all revised harmonised standards should contain specific information indicating significant changes to a revised or amended harmonised standards and this information should be made publicly available (free of charge) by the standardisation organisations.
A harmonised standard may contain normative references to other standards. Through these references those other standards or parts thereof, become indispensable for the application of a given harmonised standard. Internal rules of the European standardisation organisations are applicable when making these normative references to other standards. Because of the nature of harmonised standards undated references to other standards, where relevant clauses aim to support essential or other legal requirements, should not be normally used. Undated references may cause situations where changes in specifications contained in harmonised standards and providing presumption of conformity are uncontrolled and non-transparent — changes in normative references cannot be controlled within the meaning of Article 10(6) of the Regulation (EU) No 1025/2012 although by such changes a harmonised standard (a part of it) is de facto revised.
4.1.3. CONFORMITY WITH THE ESSENTIAL REQUIREMENTS: OTHER POSSIBILITIES
|
The application of harmonised standards is not the only means to demonstrate the conformity of a product — however only harmonised standards (187), after publication of references in the OJ, may provide an automatic presumption of conformity against essential requirements covered by such standards.
According to some Union harmonisation acts, national standards may give a presumption of conformity — as a transitional measure — insofar as there is no harmonised standard covering the same area (188). Member States may communicate to the Commission the text of those national standards, which they consider to meet the essential requirements. After consulting the Member States (189), the Commission notifies the Member States whether or not the national standard should enjoy presumption of conformity. If the opinion is positive, Member States are required to publish the references of such standards. The reference is also published in the OJ. This procedure has not been used so far in order to give full priority to the development of European standards.
The manufacturer can choose whether or not to apply and refer to harmonised standards. However, if the manufacturer chooses not to follow the harmonised standards, he has the obligation to demonstrate that his products are in conformity with essential requirements by the use of other means of his own choice that provide for the level of safety or protection of other interests required by the applicable legislation. These can be technical specifications such as national standards, European or international standards which are not harmonised, i.e. not published in the OJ or the manufacturer's own specifications. In these cases the manufacturer does not benefit from the presumption of conformity, but has to demonstrate the conformity himself. This implies that he demonstrates, in the technical file of a relevant product, in a more detailed manner how the technical specifications he uses provide conformity with the essential requirements (190).
It is important to stress that Union harmonisation legislation for products does not, as a general rule, impose the use of harmonised standards. Only essential requirements are legally binding and manufacturers may apply whatever standards and technical specifications — however only harmonised standards provide a presumption of conformity.
Furthermore, even if the manufacturer has not used harmonised standards, a change in the relevant harmonised standard could mean a change in the state of the art that implies that his product may not be compliant.
4.2. TRACEABILITY REQUIREMENTS
|
4.2.1. WHY DOES TRACEABILITY MATTER?
Traceability is the ability to trace the history of the product.
From a regulator's perspective, traceability matters because it enables effective enforcement through market surveillance via corrective measures including withdrawals and recalls. It enables unsafe or non-compliant products to be traced up the distribution chain and identifies roles and responsibilities of the economic operator throughout the chain. Traceability enables market surveillance authorities to trace products up to the factory gate and from factory to the end-user in certain cases.
From a manufacturer's perspective traceability matters because it enables effective control of the production process and suppliers before the marketing of the products, and control of their distribution chain after the placing of the product on the market. In case of non-compliance, manufacturers are able to reduce the impact of recalls or withdrawals depending on the detail of their traceability system.
4.2.2. TRACEABILITY PROVISIONS
Union harmonisation legislation is prescriptive as to the ends but not as to the means to achieve those ends. This means that Union harmonisation legislation foresees requirements for the traceability of products made available on the market, without stipulating how to achieve or implement these requirements. Union harmonisation legislation is also technology-neutral, meaning it does not prescribe the technology to be used such as printing or moulding. Manufacturers should choose the traceability system which they deem most appropriate in relation to their products and their manufacturing and distribution system.
The indication of the manufacturer's, and for imported products also the importer's, name and address on the product is a basic traceability requirement. In case of need, it allows market surveillance authorities to quickly get in contact with the economic operator responsible for the placing of an unsafe or non-compliant product on the Union market.
There is no explicit obligation that the addresses have to be preceded by the words ‘Manufactured by’, ‘Imported by’ or ‘Represented by’. This information must however not mislead the end-user and the market surveillance authorities about the place of manufacture and the address of each economic operator (191). If these words are not mentioned, market surveillance authorities will decide what the role of each economic operator is. It is then up to the economic operator to prove that he has a different role.
There is no obligation to translate into all necessary languages the words ‘manufactured by’, ‘imported by’ or ‘represented by’. These words are considered to be easily understandable in all official EU languages.
Regulation (EC) No 765/2008 setting out the requirements for accreditation and market surveillance relating to the marketing of products and Decision No 768/2008/EC on a common framework for the marketing of products establish the current practices regarding traceability by requiring specific traceability labels. The reference provisions of Decision No 768/2008/EC reflected in Union harmonisation legislation require:
|
1. |
manufacturers to indicate the following three elements: their (1) name, (2) registered trade name or registered trade mark (192) and (3) the address at which they can be contacted, on the product or, where that is not possible, on its packaging or in a document accompanying the product. The address must indicate a single point at which the manufacturer can be contacted (193); |
|
2. |
importers to indicate the following three elements: their (1) name, (2) registered trade name or registered trade mark and (3) the address at which they can be contacted, on the product or, where that is not possible, on its packaging or in a document accompanying the product (194); |
|
3. |
manufacturers to ensure that their products bear a type, batch, serial or model number or other element allowing their identification, or, where the size or nature of the product does not allow it, that the required information is provided on the packaging or in a document accompanying the product (195); and |
|
4. |
economic operators to identify any economic operator who has supplied them with a product and any economic operator to whom they have supplied a product (196). |
4.2.2.1. The requirement to indicate name and address for manufacturers
The manufacturers must indicate the following three elements: their (1) name, (2) registered trade name or registered trade mark and (3) the address at which they can be contacted on the product, or, where that is not possible, on its packaging and/or in a document accompanying the product.
The name and address must, as a rule, be affixed to the product. However, it may exceptionally be moved from the product if this rule cannot be followed. This would be justified where affixing it to the product was not possible under reasonable technical or economic conditions excluding however esthetical reasons. It is up to the manufacturer to make this assessment. This assessment has to be done according to the size or nature of the product (197). Some products, e.g. hearing aids, sensors or the like are simply too small to carry such information. In such cases the order of priority is that as a first alternative the information should be on the packaging, as a second alternative on an accompanying document, except for the cases where sectoral Union harmonisation legislation requires the information to be on both the packaging and accompanying documents.
The manufacturer has to comply with this obligation regardless of his location (within or outside the EU). This provision implies that products sold without packaging or any accompanying documents, must bear the name and address of the manufacturer on the product itself.
The address must indicate a single point at which the manufacturer can be contacted, in particular by market surveillance authorities. The legal text obliges the manufacturer to put a single contact point on the product. Only one single contact point in each product is allowed. This is not necessarily the address where the manufacturer is actually established. This address can for example be the one of the authorised representative or of the customer services.
The single contact point does not need to be in every Member State where the product is made available. The manufacturer may however put other addresses (198) provided that it is clear which one is the single contact point. The latter is then to be indicated on the product/documentation as the ‘single contact point’. The address or the country does not necessarily have to be translated into the language of the Member State where the product is made available on the market but the characters of the language used must allow identifying the origin and the name of the company.
A website is additional information, but is not enough as an address. Normally an address consists of a street and number or post-box and number and the postal code and town, but some countries might deviate from this model.
4.2.2.2. The requirement to indicate name and address for importers
Importers must also indicate the following three elements: their (1) name, (2) registered trade name or registered trade mark and (3) the address at which they can be contacted on the product, or, where that is not possible, on its packaging or in a document accompanying the product. The provision refers to an address at which they can be contacted, in particular by market surveillance authorities. This is not necessarily the address where the importer is actually established but can for example be the one of the customer services.
As a rule, the identification and the address of importer must be indicated on the product. Only where it is not possible, the identification and address of the importer may be indicated on the packaging and/or in a document accompanying the product. This may be the case when the importer would have to open the packaging to put his name and address. The additional information from the importer shall not hide the information put on the product by the manufacturer.
A website is additional information, but is not enough as contact address. Normally an address consists of a street and number or post-box and number and the postal code and town, but some countries might deviate from this model.
The product must always bear the manufacturer's name and address. Imported products must also bear the importer's name and address. Hence, in conclusion, a product normally bears one or two addresses (199):
|
— |
If the manufacturer is within the European Union, the product will bear only one (manufacturer's) address as there is no importer involved. |
|
— |
If the manufacturer (declaring himself as a manufacturer by putting his name and address on the product) is outside the EU and the products are placed on the Union market by an importer, the product will bear two addresses: the one of the manufacturer and the one of the importer. |
|
— |
If the original manufacturer is outside the EU and the importer places the product on the market under his own name or trademark or modifies the product already placed on the market (in such a way that compliance with the applicable requirements may be affected), the importer is considered the manufacturer. The only address that in this case will figure on the product (or packaging or accompanying document) is the address of the importer who is considered as the manufacturer. (200) (201) |
|
— |
If the manufacturer is within the EU (a company located in the EU declaring itself to be a manufacturer by putting its name and address on the product) although the products are manufactured outside the EU, that company is considered to be the manufacturer who places the product on the Union market, even if actual importation is done by another company. In this case there is no importer in the meaning of the importer's definition and it is sufficient to put only the manufacturer's address. |
4.2.2.3. Identification element
The product must bear a type, batch, serial or model number or other element allowing its identification. The identification must, as a rule, be affixed to the product. However, it may exceptionally be moved from the product if this rule cannot be followed. This would be justified where the size and/or the nature of the product makes the indication illegible or technically impossible (202). In such cases, the identification has to be affixed to the packaging, if it exists, and/or to the accompanying document. The identification on the product may neither be omitted nor be moved to the packaging or accompanying documents on purely aesthetic or economic grounds. It is up to the manufacturer to make this assessment.
This provision implies that if the product has no packaging or is not accompanied by any document, the identification must be on the product itself.
The requirement gives the freedom to the manufacturers to choose the element they want to use as identification of the product, as long as traceability is ensured. The identification element used must ensure a clear link to the relevant documentation that demonstrates the conformity of the specific type of product, in particular the EU declaration of conformity. This identification element of the product shall be the same as on the EU declaration of conformity. The identification element chosen by the manufacturer is also important in case of a withdrawal or recall, since all products bearing the same identification element will have to be withdrawn or recalled from the market.
In some cases, e.g. when a product consists of several parts or is an assembly of several parts, its nature does not allow for the affixing of the identification element. The identification of the product has in these cases to be affixed to the packaging (or accompanying document). In addition to the marking with an identification element on the packaging, additional marking of individual products/parts/components can be made based on the manufacturer's internal rules and ambitions to minimise the extent of a potential recall by having an advanced system for traceability of individual items (e.g. batch codes, production dates).
According to some economic operators, one way to refer to products is to use an item number (a so-called ‘SKU’-‘Stock keeping unit’) as identification. This item number can also be used as an identifier on the EU declaration of conformity together with other elements allowing traceability.
The product consists of several parts/components
Each product is enclosed in one packaging but typically some parts/components could/would also be sold in another packaging as separate parts/components or in other combinations of parts/components. Some of the parts/components in these packages may be possible to mark, while others may be too small or have a shape which does not allow the marking to be on the part. For these reasons, it is allowed to give the set/packaging an item number and to use the same item number on the EU declaration of conformity.
The main purpose of the identification element is to enable market surveillance authorities to identify an individual product and to link it to a EU declaration of conformity. If, when the market surveillance takes place, the product is still in its packaging, it will be easy to identify the element and thus ensure that the corresponding EU declaration of conformity regards the product in question. It would be more complicated to have to open the packaging and find elements on the individual items and then link these to a particular EU declaration of conformity.
The product consists of one assembled item
Also when a product consists of only one ‘item’, it is not uncommon that this item has been assembled by the manufacturer, using several parts (but it is not intended to be disassembled by the consumer). The parts composing the item (product) are often used in more than one design of products. Normally, some parts would not be large enough to bear an identification element and yet other parts might not allow marking with an identification element for technical reasons (uneven surface, spherical shaped surface, etc.). Also in this case it is allowed to affix an item number on the packaging and to use the same number on the EU declaration of conformity.
The product consists of one item which has not been assembled of several parts
This is a case where it may seem simple to mark the product itself with an identification element that is identical to the one on the EU declaration of conformity (i.e. an item number). However, the same product might be sold in combination with other products/items in a set. Since at the point of production, it is not known which of the items will be sold ‘alone’ and which will be in a packaging together with other products, it is easier to mark the item number, corresponding to the EU declaration of conformity, on the packaging. This will also facilitate market surveillance authorities to link the product to the EU declaration of conformity.
4.2.2.4. Identification of economic operators
Economic operators are obliged to keep track of the economic operators they supplied their product to or from whom they bought products for a period of 10 years. Bear in mind that the end user (consumer) is not covered by this requirement as they are not considered to be economic operators.
The way to comply with this requirement by economic operators is not prescribed by Union harmonisation legislation, but it must be noted that market surveillance authorities can ask for relevant documents, including invoices, allowing the origin of the product to be traced. Hence, it could be useful to keep invoices for a longer period than envisaged in accounting legislation to comply with the requirements on traceability.
4.3. TECHNICAL DOCUMENTATION
|
Union harmonisation legislation obliges the manufacturer to draw up technical documentation containing information to demonstrate the conformity of the product to the applicable requirements. This documentation may be part of the quality system documentation where the legislation provides for a conformity assessment procedure based on a quality system (modules D, E, H and their variants). The technical documentation must be available when the product is placed on the market, whatever its geographical origin or location (203).
The technical documentation must be kept for 10 years from the date of placing the product on the market, unless the applicable Union harmonisation legislation expressly provides for any other duration (204). This is the responsibility of the manufacturer or the authorised representative established within the Union. Since the concept of ‘placing on the market’ refers to each individual product, the time period needs to be calculated from the moment when the individual product that is covered by the technical documentation is placed on the market.
The contents of the technical documentation are laid down, in each Union harmonisation act, in accordance with the products concerned. As a rule, the documentation has to include a description of the product and of its intended use and cover the design, manufacture and operation of the product. The details included in the documentation depend on the nature of the product and on what is considered as necessary, from the technical point of view, for demonstrating the conformity of the product to the essential requirements of the relevant Union harmonisation legislation or, if the harmonised standards have been applied, to these by indicating the essential requirements covered by the standards. The requirements in Annex II to Decision No 768/2008/EC refer to the contents of the technical documentation that are relevant for proving the conformity of the product with the applicable harmonisation legislation. Furthermore, the requirement for an ‘adequate analysis and assessment of the risk(s)’ requires the manufacturer to first identify all possible risks of the product and determine the essential requirements applicable. This analysis has to be documented and included in the technical documentation. In addition, the manufacturer needs to document the assessment of how he is addressing the risks identified to ensure that the product complies with the applicable essential requirements (for example, by applying harmonised standards). If only part of the harmonised standard is applied or it does not cover all applicable essential requirements, then also the way applicable essential requirements not covered by it are dealt with should be documented in the technical documentation.
In the case where a product has been subject to re-designs and re-assessments of the conformity, the technical documentation must reflect all versions of the product; describing the changes made, how the various versions of the product can be identified and information on the various conformity assessment. This is to avoid situations where during the whole life of a product, a market surveillance authority is faced with previous versions of the product for which the version of the technical documentation it is presented with, is not applicable.
Some Union harmonisation acts require that the technical documentation is written in a language accepted by the notified body (205). In order to carry out the conformity assessment procedures requiring third-party verification in a proper way, the documentation should always be in a language understood by the notified body, even if this has not been explicitly mentioned in the Union harmonisation legislation.
4.4. EU DECLARATION OF CONFORMITY
|
Union harmonisation legislation imposes an obligation on the manufacturer to draw up and sign an EU declaration of conformity before placing a product on the market (206). The manufacturer or his authorised representative established within the Union must draw up and sign an EU declaration of conformity as part of the conformity assessment procedure provided for in the Union harmonisation legislation. The EU declaration of conformity is the document that states that that the product satisfies all the relevant requirements of the applicable legislation.
By drawing up and signing the EU declaration of conformity, the manufacturer assumes responsibility for the compliance of the product.
Just as it is the case for the technical documentation (207), the EU declaration of conformity must be kept for 10 years from the date of placing the product on the market, unless the legislation provides for any other duration (208). This is the responsibility of the manufacturer or the authorised representative established within the Union. For imported products, the importer must take on this responsibility for the DoC (209).
The contents of the EU declaration of conformity either refer to the model declaration contained in Annex III to Decision No 768/2008/EC or a model declaration directly annexed to the sectoral Union harmonisation legislation at stake. The standard EN ISO/IEC 17050-1 has been drawn up with the objective of providing the general criteria for the declaration of conformity, and it can also be used as a guidance document provided it is in line with the applicable Union harmonisation legislation. The declaration may take the form of a document, a label or equivalent, and must contain sufficient information to enable all products covered by it to be traced back to it.
The model declaration of Decision No 768/2008/EC contains:
|
1. |
A number identifying the product. This number does not need to be unique to each product. It could refer to a product, batch, type or a serial number (210). This is left to the discretion of the manufacturer (211). |
|
2. |
The name and address of the manufacturer or the authorised representative issuing the declaration. |
|
3. |
A statement that the declaration is issued under the sole responsibility of the manufacturer. |
|
4. |
The identification of the product allowing traceability. This is basically any relevant information supplementary to point 1 describing the product and allowing for its traceability. It may where relevant for the identification of the product contain an image, but unless specified as a requirement in the Union harmonisation legislation this is left to the discretion of the manufacturer. |
|
5. |
All relevant Union harmonisation legislation complied with; the referenced standards or other technical specifications (such as national technical standards and specifications) in a precise, complete and clearly defined way; this implies that the version and/or date of the relevant standard is specified. |
|
6. |
The name and identification number of the notified body when it has been involved in the conformity assessment procedure (212) (213) and the reference to the relevant certificate, if applicable. |
|
7. |
All supplementary information that may be required (for example grade, category), if applicable. |
|
8. |
The date of issue of the declaration; signature and title or an equivalent marking of authorised person (214) (215); this could be any date after the completion of the conformity assessment |
Where several pieces of Union harmonisation legislation apply to a product, the manufacturer or the authorised representative has to provide a single declaration of conformity in respect of all such Union acts (216). In order to reduce the administrative burden on economic operators and facilitate its adaptation to the modification of one of the applicable Union acts, the single declaration may be a dossier made up of relevant individual Declarations of conformity (217).
The EU declaration of conformity must be made available to the surveillance authority upon request. Moreover, Union harmonisation legislation relating to machinery, equipment in potentially explosive atmospheres, radio and terminal telecommunication equipment, measuring instruments, recreational craft, lifts, high-speed and conventional rail systems and constituents of the European Air Traffic Management network require products to be accompanied by the EU declaration of conformity.
The EU declaration of conformity must be translated into the language or languages required by the Member State in which the product is placed or made available on the market (218). Union harmonisation legislation does not necessarily specify who has the obligation to translate. Logically, this should be the manufacturer or another economic operator making the product available. The EU declaration of conformity must be signed by the manufacturer or his authorised representative. If a translation of the EU declaration of conformity has been produced by another economic operator and is not signed by the manufacturer, a copy of the original EU declaration of conformity signed by the manufacturer must also be provided together with the translated version.
4.5. MARKING REQUIREMENTS
4.5.1. CE MARKING
4.5.1.1. Definition and role of the CE marking
|
The CE marking is a key indicator (but not proof) of a product's compliance with EU legislation and enables the free movement of products within the EEA and Turkish market, whether they are manufactured in the EEA, Turkey or in another country.
Member States of the European Economic Area (EEA — EU Member States and certain EFTA countries: Iceland, Norway, Liechtenstein) are not allowed to restrict the placing on the market of CE marked products, unless such measures can be justified on the basis of evidence of the non-compliance of the product. This also applies to products made in third countries which are sold in the EEA.
CE marking does not indicate that a product was made in the European Union. The CE marking indicates conformity with the requirements laid down by the Union harmonisation text(s) in question. Therefore, it is to be considered as essential information to Member States' authorities as well as other relevant parties (for example distributors). CE marking does not serve commercial purposes, i.e. it is not a marketing tool.
CE marking is the visible consequence of a whole process comprising conformity assessment in a broad sense and indicates that a product is declared by the manufacturer as in conformity with Union harmonisation legislation.
4.5.1.2. Relationship to existing legislation
|
Regulation (EC) No 765/2008 lays down the definition, the format and the general principles governing the CE marking. Decision No 768/2008/EC provides for conformity assessment procedures that lead to its affixing.
The sectoral Union harmonisation legislation providing for the affixing of the CE marking mostly follows the principles of the Regulation (EC) No 765/2008 and Decision No 768/2008/EC.
As a general rule (219) the CE marking can be introduced in a Union legislative act as legal conformity marking if:
|
— |
the method of total harmonisation is used, which means that diverging national regulations that cover the areas as the legislative act in question are prohibited, |
|
— |
the Union harmonisation act contains conformity assessment procedures according to Decision No 768/2008/EC. |
However there is an exception to this rule.
In duly justified cases a total harmonisation piece of legislation that follows Decision No 768/2008/EC may provide for a different marking instead of the CE marking. For example the Directive on marine equipment does not provide for a CE marking, but for a specific conformity mark — the wheel mark. — The use of the wheel mark also is subject to the general principles set out in Regulation (EC) No 765/2008 and Decision No 768/2008/EC and any reference to CE marking is to be construed as a reference to the wheel mark. Similarly, for transportable pressure equipment, the ‘Pi’ marking is required instead of the CE marking..
4.5.1.3. Who must (not) affix the CE marking
|
The manufacturer, whether established inside or outside the Union, is the entity ultimately responsible for the conformity of the product with the provisions of the Union harmonisation legislation and for the affixing of the CE marking. The manufacturer may appoint an authorised representative to affix the CE marking on his behalf.
By affixing the CE marking on a product, a manufacturer is declaring, on his sole responsibility (and irrespectively of whether a third-party has been involved in the conformity assessment process), conformity with all of the legal requirements to achieve CE marking.
If the importer or distributor or another operator places products on the market under his own name or trademark or modifies them, he then takes over the manufacturer's responsibilities. This includes the responsibility for the conformity of the product and the affixing of the CE marking. In this case he must have sufficient information on the design and production of the product, as he will be assuming the legal responsibility when affixing the CE marking.
4.5.1.4. Principles of affixing the CE marking
|
The CE marking must take the form below. If the CE marking is reduced or enlarged the proportions must be respected. |

The CE marking must be affixed visibly, legibly and indelibly to the product or to its data plate. However, where this is not possible or not warranted on account of the nature of the product, it must be affixed to the packaging, if any, and/or to the accompanying documents. The CE marking may not, in principle, be affixed until the conformity assessment procedure has been completed to ensure that the product complies with all the provisions of the relevant Union harmonisation acts. This will usually be at the end of the production phase. This poses no problem if, for example, the CE marking is on a data plate that is not affixed to the product until after the final inspection. However, if (for example) the CE marking is affixed by stamping or casting, the marking can be affixed at any other stage of the production phase, provided that the conformity of the product is verified as part of the production process.
The requirement for visibility means that the CE marking must be easily accessible for all parties. It could, for instance, be affixed on the back or underside of a product. The requirement for visibility does not necessarily mean that the CE marking must be visible before opening a products' packaging because affixing the CE marking also to the packaging is only necessary in case this is explicitly required in the relevant Union acts. A minimum height of 5 mm is required to ensure that it is legible. However according to the several pieces of legislation (220) the minimum dimension of the CE marking may be waived for small devices or components.
The CE marking can take different forms (e.g. colour, solid/hollow) as long as it remains visible, legible and respects its proportions. It must also be indelible so that it cannot be removed under normal circumstances without leaving noticeable traces (for example some product standards provide for a rub test with water and petroleum spirits). Nevertheless, this does not mean that the CE marking must form an integral part of the product.
However in certain cases affixing of the CE marking to the product is impossible (for example on certain types of explosives) or not possible under reasonable technical or economic conditions. Furthermore there can be cases where the minimum dimensions for the affixing cannot be respected, or it cannot be ensured that the CE marking is visibly, legibly and indelibly affixed.
In such cases, the CE marking can be affixed to the packaging, if it exists, and/or to the accompanying document, where the Union harmonisation legislation concerned provides for such documents. The CE marking on the product may neither be omitted nor be moved to the packaging or accompanying documents on purely aesthetic grounds.
Regulation (EC) No 765/2008 and Decision 768/2008/EC lay down that the CE marking must have the dimensions, format and proportions defined in Annex II to Regulation (EC) No 765/2008 and be legible and clearly affixed. Regulation (EC) No 765/2008 and Decision No 768/2008/EC do not forbid any kind of design (e.g. ‘hollow’ design) as long as the above conditions are respected. However, electronic labelling only is not allowed.
4.5.1.5. Affixing CE marking together with the identification number of the notified body
Where a notified body is involved in the production control phase according to the applicable Union harmonisation legislation, its identification number must follow the CE marking. The manufacturer or the authorised representative affixes the identification number if the legislation so requires, under the responsibility of the notified body.
A notified body may be involved in the production phase, depending on the conformity assessment procedures applied. The CE marking must be followed by the identification number of the notified body only if it is involved in the production phase. Thus, the identification number of a notified body involved in conformity assessment in the design phase according to module B does not follow the CE marking. Sometimes several notified bodies are involved in the production phase, which is possible where more than one Union harmonisation text is applicable. In these situations several identification numbers follow the CE marking.
Thus, if the CE marking appears on products without an identification number, this means that:
|
— |
either no notified body intervened in the design or production phase (module A), |
|
— |
or upon manufacturer's choice the in-house accredited body intervened in the production phase (modules A1, A2), |
|
— |
or a notified body intervened in the design phase (module B) but no notified body intervened in the production phase, |
|
— |
(module C following module B), |
|
— |
or a notified body intervened in the design phase (module B) and upon manufacturer's choice the in-house accredited body intervened in the production phase (modules C1, C2 following module B). |
If however the CE marking appears on products with an identification number (221), this means that:
|
— |
either upon manufacturer's choice a notified body intervened in the production phase (modules A1, A2), |
|
— |
or a notified body intervened in the design phase (module B) and upon manufacturer's choice a notified body (not necessarily the same one but the one whose identification number appears) intervened in the production phase (modules C1, C2 following module B), |
|
— |
or a notified body intervened in the design phase (module B) and a notified body (not necessarily the same one but the one whose identification number appears) intervened in the production phase (modules C1, C2, D, E, F following module B), |
|
— |
or a notified body intervened in the design and production phase (modules D1, E1, F1, G1 H, H1). |
The CE marking and the identification number of the notified body do not necessarily have to be affixed within the Union. They may be affixed in a third country, for example if the product is manufactured there and the notified body carried out conformity assessment in that country in line with the relevant Union harmonisation legislation. The CE marking and the identification number can also be affixed separately, as long as they remain combined.
4.5.1.6. Which products must (not) be CE marked
|
Not all products have to be CE marked (222). The obligation to affix the CE marking extends to all products within the scope of legislative acts providing for its affixing, and which are intended for the Union market. Thus, the CE marking must be affixed:
|
— |
to all newly manufactured products that are subject to legislation providing for CE marking, whether manufactured in the Member States or in third countries, |
|
— |
to used and second-hand products imported from third countries that are subject to legislation providing for CE marking, |
|
— |
to modified products that, as new, are subject to legislation providing for CE marking and which have been modified in a way that could affect the safety or the compliance of the product with the applicable harmonisation legislation. |
In some cases, a product is deemed final for the purposes of a particular Union harmonisation act and has to be CE marked. This same product then is incorporated in another final product which is itself subject to another Union harmonisation act which also requires CE marking. This produces a situation where more than one CE marking can be found on a product (223).
Union harmonisation legislation providing in general for CE marking may exclude the application of the CE marking on certain products. As a general rule, such products are subject to free circulation, if:
|
(a) |
they are accompanied by:
|
|
(b) |
they are accompanied by an attestation of conformity in the case of components as defined in the Directive on equipment and protective systems intended for use in potentially explosive atmospheres (ATEX); |
|
(c) |
they are accompanied by a statement in the case of:
|
|
(d) |
they are accompanied by a certificate of conformity in the case of fittings referred to in the Directive relating to gas appliances; |
|
(e) |
the product bears the manufacturer's name and an indication of maximum capacity in the case of instruments not subject to conformity assessment according to the Directive relating to non-automatic weighing instruments; |
|
(f) |
the product is manufactured in accordance with sound engineering practice in the case of certain vessels referred to in the Directives relating to simple pressure vessels and pressure equipment. |
In addition, the Directive on pressure equipment entitles Member States to authorise, on their territory, the placing on the market and the putting into service by users, of pressure equipment of assemblies not bearing the CE marking, but that have been subject to a conformity assessment carried out by a user inspectorate instead of a notified body.
4.5.1.7. CE marking and other markings
|
The CE marking replaces all mandatory conformity markings having the same meaning, which existed before harmonisation took place. Such national conformity markings are incompatible with CE marking and would constitute an infringement of the applicable European legislation in question. When transposing Union harmonisation legislation, Member States are required to incorporate the CE marking into their national regulations and administrative procedures. They must also refrain from introducing any other conformity marking into their national legislation that has the same meaning as the CE marking.
However, other markings may be used as long as they contribute to the protection of public interests, are not covered by Union harmonisation legislation and their affixing does not impair the visibility, legibility and meaning of the CE marking. The affixing of additional markings (such as a protected trademark of a manufacturer or other private/national markings) is allowed to the extent that such markings do not create confusion with the CE marking. This confusion may either refer to the meaning or form of the CE marking.
In this respect, other markings additional to the CE marking need to fulfil a different function from that of the CE marking. Thus, they should provide information on conformity with objectives that are different from those to which the CE marking relates (for example environmental aspects not covered by the applicable Union harmonisation legislation).
Furthermore several Union harmonisation acts foresee additional markings that are complementary and non-overlapping to the CE marking (see under 4.5.2).
4.5.1.8. Sanctions
|
The CE marking provides the first indication that the necessary controls can be assumed to have been carried out, before the product in question is placed on the market, in order to ensure its compliance to the legislative requirements. Market surveillance authorities are entitled to proceed to additional controls for the protection of public interest. The action to be taken by market surveillance authorities should be decided on a case by case basis according to the principle of proportionality.
Member States must provide in their national legislation for appropriate measures both to prevent the abuse and misuse of CE marking, and to redress the situation if such abuse or misuse takes place. Those measures need to be effective, proportionate to the seriousness of the offence and dissuasive and may be increased if the relevant economic operator has previously committed a similar infringement. They may include withdrawal, recall of products, penalties and criminal sanctions (such as fines and imprisonment) wherever necessary.
The measures are imposed without prejudice to other measures taken where the market surveillance authorities find that a product presents a risk or does not comply with the applicable legislation. Furthermore Member States must ensure that the measures are implemented.
In this respect the affixing of the CE marking to a product that is not covered by any of the Union harmonisation legislation providing for its affixing is considered to be deceiving because consumers or users, for instance, are likely to get the impression that the product in question satisfies certain Union harmonisation legislation provisions. Competent authorities must, therefore, have at their disposal legal instruments that enable them to act against the deceptive use of CE marking. Action must be taken also against those responsible for a non-compliant product bearing the CE marking.
The affixing of markings in addition to the CE marking is subject to certain restrictions (224). The surveillance authority has to take the necessary measures to ensure that these principles are respected and, where necessary, take appropriate action.
A Member State must inform the Commission and the other Member States of its decision to restrict free movement due to incorrect affixing of the CE marking, and of its action against the person responsible for affixing the CE marking to a non- compliant product. It is then up to the other Member States to decide whether or not similar action is necessary. In the case of unduly affixed CE marking to products not subject to the CE marking requirement, Member States should inform the Commission and the other Member States about it.
4.5.2. OTHER MANDATORY MARKINGS
|
Several pieces of Union harmonisation legislation foresee additional markings that are complementary and non-overlapping to the CE marking. |
Pictograms or other markings indicating, for instance, the category of use are, according to some Union harmonisation legislation, complementary to the CE marking but do not form part of it or replace it. In general, these markings follow the same principles as the CE marking. Some examples:
|
— |
the EU energy label for energy-related products, |
|
— |
the specific marking of explosion protection required for equipment and protective systems intended for use in potentially explosive atmospheres, |
|
— |
the equipment class identifier required for radio equipment (Class 2), |
|
— |
the supplementary metrology marking required for measuring instruments and non-automatic weighing instruments. |
5. CONFORMITY ASSESSMENT
5.1. MODULES FOR CONFORMITY ASSESSMENT
5.1.1. WHAT IS A CONFORMITY ASSESSMENT?
|
Two important elements of every legislative act (either Old or New Approach) covering products are:
|
— |
the legislative requirements governing the characteristics of the products covered, |
|
— |
and the conformity assessment procedures the manufacturer carries out in order to demonstrate that a product, before it is placed on the market, conforms to these legislative requirements. |
This guide addresses conformity assessment as this is laid down under Decision No 768/2008/EC (particularly for the Union harmonisation legislation under the ‘New Approach’ and now the New Legislative Framework).
A product is subjected to conformity assessment both during the design and production phase. Conformity assessment is the responsibility of the manufacturer. Should a manufacturer subcontract design or production, he still remains responsible for the execution of conformity assessment.
Conformity assessment must not be confused with market surveillance, which consists of controls by the national market surveillance authorities after the product has been placed on the market. However both techniques are complementary and equally necessary to ensure the protection of the public interests at stake and the smooth functioning of the internal market.
The essential objective of a conformity assessment procedure is to demonstrate that products placed on the market conform to the requirements expressed in the provisions of the relevant legislation.
5.1.2. THE MODULAR STRUCTURE OF CONFORMITY ASSESSMENT IN UNION HARMONISATION LEGISLATION
|
Under Union harmonisation legislation, conformity assessment procedures are composed of one or two conformity assessment modules. As products are subjected to conformity assessment both during the design and production phase, a conformity assessment procedure covers both design and production phases; while a module may cover:
|
— |
either one of these two phases (in this case a conformity assessment procedure is composed of two modules), |
|
— |
or both (in this case a conformity assessment procedure is composed of one module). |
Decision No 768/2008/EC, lays down a ‘horizontal menu’ of conformity assessment modules and the ways procedures are built of modules.
The legislator selects from the menu of conformity assessment modules/procedures (laid down under Decision No 768/2008/EC) the most appropriate one(s) in order to address the specific needs of the concerned sector (225). The least onerous modules should be selected taking into account the type of products and hazards involved, the impact on the protection of public interests, the economic infrastructure of the given sector, the methods of production, etc., where possible a choice of inspection, certification, and/or QA modules should be provided.
Conformity assessment procedures are equivalent from a legal point of view but not technically identical in terms of methods. Their application in the sectoral legislation aims at providing high level of confidence as regards the conformity of products to the relevant essential requirements.
The intention of the modules as laid down in the Decision No 768/2008/EC is to allow for a limited number of possible procedures.
Nevertheless, the choice offered needs to be sufficiently varied in order to be applicable to the widest range of products concerned.
Union harmonisation legislation establishes conformity assessment procedures by either leaving the manufacturer no choice or by establishing a range of procedures from which the manufacturer must choose. As conformity assessment procedures in Union harmonisation legislation originate from Decision No 768/2008/EC, they remain consistent and coherent. Thus the assessment of product conformity becomes more transparent especially in cases where more than one harmonisation legislative act applies to a product.
5.1.3. ACTORS IN CONFORMITY ASSESSMENT — POSITIONING OF CONFORMITY ASSESSMENT IN THE SUPPLY CHAIN
|
Conformity assessment is a responsibility of the manufacturer. However, if required by the relevant legislation, a third party must be involved in the conformity assessment procedure.
In total there are three possibilities:
|
— |
There is no third-party involvement. This may concern the case where, according to the legislator, a declaration (accompanied by the relevant technical examinations and documentation) of the manufacturer is enough to ensure the conformity of the product(s) in question against the relevant legislative requirements. In this case the manufacturer himself carries out all required controls and checks, establishes the technical documentation and ensures the conformity of the production process. |
|
— |
Conformity assessment is performed with the involvement of an accredited in-house conformity assessment body that forms a part of the manufacturer's organisation. However this in-house body must not have any activities other than conformity assessment and must be independent from any commercial, design and production entities (see for details Article R21 of Decision No 768/2008/EC). It has to demonstrate the same technical competence and impartiality as external conformity assessment bodies, through accreditation. Whenever appropriate for a specific sector, the legislator may acknowledge the fact that manufacturers operate very well equipped testing laboratories or premises. This may be the case for new innovative complex products for which the testing know-how remains inside the manufacturers. |
|
— |
However in some other cases the legislator may consider the intervention of a third party, i.e. an external conformity assessment body, necessary. Such a body must be impartial and fully independent from the organisation or the product it assesses (see also Article R17(3) of Decision No 768/2008/EC), it cannot engage in any activity that may conflict with its independence (see also Article R21(2)(c) of Decision No 768/2008/EC) and thus it cannot have user or other interests in the product to be assessed. |
It is the responsibility of the Member States to notify those third party conformity assessment bodies within their jurisdiction that they consider technically competent to assess the compliance of products with the requirements of the Union harmonisation legislation that applies to them. In-house bodies cannot be notified but they have still to demonstrate the same technical competence as external bodies, through accreditation. Member States must also ensure that the (in-house or external) bodies permanently maintain their technical competence.
Taking the above into account, the stakeholders in a conformity assessment procedure are the following:
|
(a) |
the legislator who:
|
|
(b) |
the manufacturer who:
It must be clear that it is always the manufacturer who takes responsibility for the conformity of his products with the relevant legislative requirements. In this respect, the economic operator that places the product on the market under its name or trademark becomes automatically the manufacturer for the purposes of Union harmonisation legislation. Therefore he takes the entire responsibility for the conformity assessment (design and production) of the product, even if this has been actually done by somebody else. Furthermore he must be in the possession of all documentation and certificates necessary to demonstrate the conformity of the product, but these do not need to be under his name; |
|
(c) |
the (in-house or external) conformity assessment body that:
|
A conformity assessment body wishing to carry out conformity assessment for one or several module(s) under a given piece of Union harmonisation legislation has to be assessed according to all the requirements for the different modules it wishes to offer services for (see under §5.2.3). A body wishing to offer conformity assessment services under a Union harmonisation act has to offer services for at least one module from those indicated in the Union harmonisation act. It must be noted that there is no obligation for a body to offer services for more than one module, but it must take on the responsibility for a whole module.
The exact position of conformity assessment in the supply chain is depicted under Flowchart 2.
Flowchart 2
Conformity Assessment
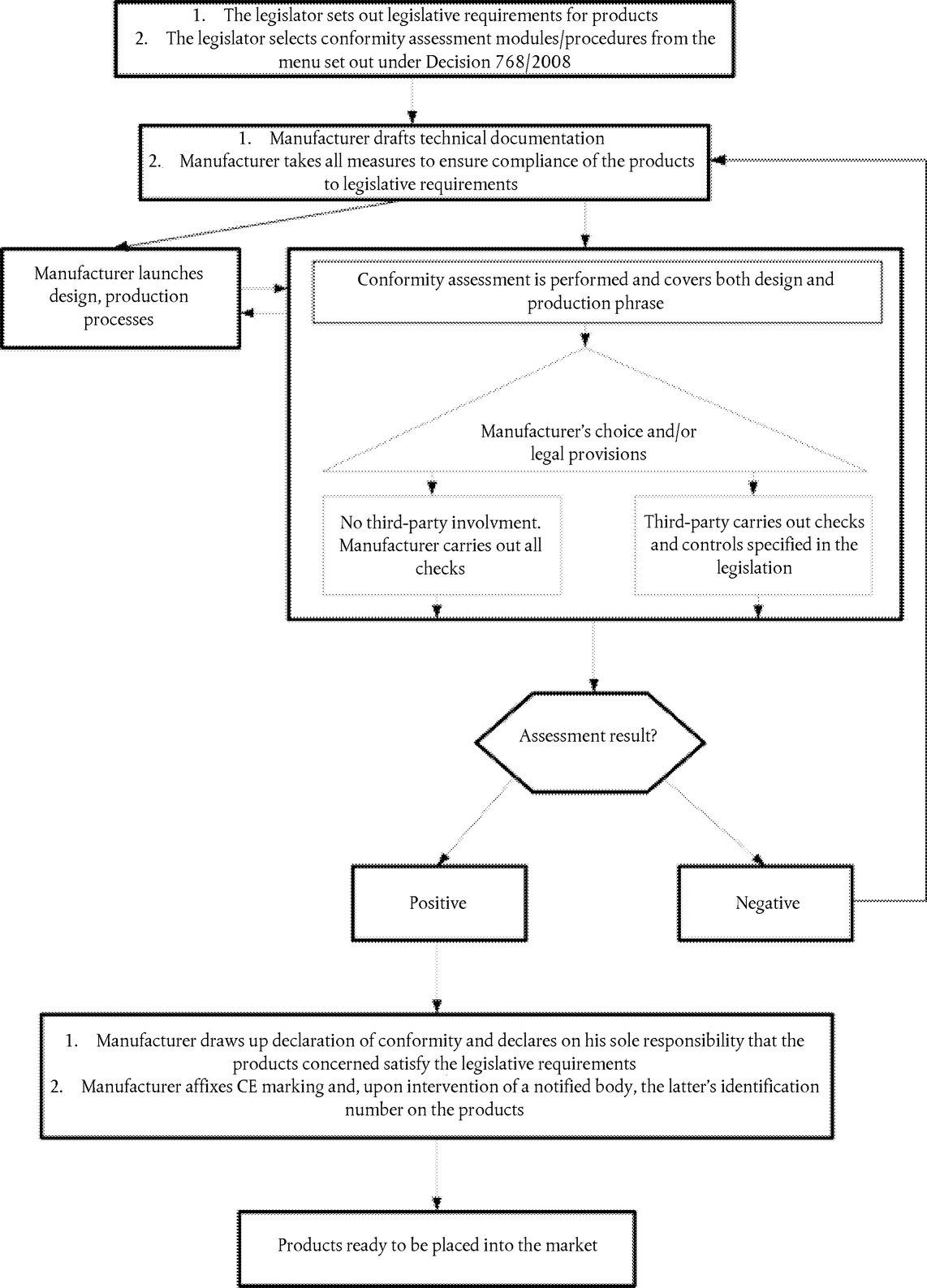
5.1.4. MODULES AND THEIR VARIANTS
|
There are eight modules. Some of them have variants. |
There are eight modules (named with the letters from A to H). They lay down the responsibilities of the manufacturer (and his authorised representative) and the degree of involvement of the in-house accredited or notified conformity assessment body. They are the components of the conformity assessment procedures laid down under Decision No 768/2008/EC, the ‘horizontal menu’.
Several modules have their variants. The reason for providing variants within modules (this applies for all variants of all modules laid down under Decision No 768/2008/EC) is to enable the necessary level of protection to be ensured for products presenting higher level of risk while avoiding the imposition of a heavier module. The idea is to minimise the burden on manufacturers to the extent possible.
5.1.5. ONE- AND TWO-MODULE PROCEDURES — PROCEDURES BASED ON TYPE (EU-TYPE EXAMINATION)
|
In some cases the conformity assessment procedure is in two steps:
|
In some cases, e.g. mass production based on a type/specimen ‘representative of the production envisaged’ and where the product in question is of complex design, the EU legislation may lay down the conformity assessment procedure in two steps:
|
— |
first the examination of conformity of the type/specimen against the relevant legal requirements (so called EU-Type examination — module B), |
|
— |
and then the determination of the conformity of the manufactured products against the approved EU-type. |
In these cases conformity assessment procedures are composed of two modules; the first module is always module B.
This method not only reduces burden and costs but is also more efficient compared to a traditional examination of the conformity products directly against the legal requirements. Once the type is approved (and this is done only once for a specific specimen), it must be checked only whether the products to be placed on the market are in conformity with the approved type.
The conformity assessment body involved under module B is not necessarily the same as the one involved in the module that is used together with module B.
In cases where there is no EU-type examination, conformity assessment procedures are composed of one two-phase (design & production) module.
The manufacturer undertaking the module (226) that is used together with module B does not need to be the same person as the one having the EU-type examination certificate under module B. Yet, that manufacturer placing then the product on the market, takes the entire responsibility for the conformity assessment (design & production) of the product. Consequently, he must be in possession of both certificates, although the EU-type examination certificate does not have to be in his name, and the full history of the product. He must have all the administrative and technical information and data, have type testing performed, manage the technical documentation related to the type testing and have batch testing performed. The above reasoning applies actually to all modules and procedures, independently of whether it is a one-phase or two-phase conformity assessment procedure. In cases where a manufacturer relies on one or more other manufacturer(s) for the design and production of the product there has to be proof that the manufacturer is fully informed about any changes in the design, production and conformity assessment of the product.
5.1.6. MODULES BASED ON QUALITY ASSURANCE
|
Some modules and their variants are based on quality assurance techniques and are derived from the EN ISO 9000 (227), EN ISO 9001 (228) standards. The modules based on quality assurance techniques (modules D, E, H and their variants) describe the elements a manufacturer must implement in his organisation in order to demonstrate that the product fulfils the essential requirements of the applicable legislation.
This means that a manufacturer is given the possibility of using an approved quality system for the purpose of demonstrating compliance with regulatory requirements. The quality system is assessed by the notified body.
A quality system implemented on the basis of the EN ISO 9000, EN ISO 9001 gives a presumption of conformity with the respective modules with regard to the provisions in the modules that these standards cover, provided that the quality system takes into consideration the specificities of the concerned products.
However, the manufacturer is free to apply other quality system models than those based on EN ISO 9001 for the purpose of complying with these modules.
In any case the manufacturer must specifically address all regulatory provisions while applying its quality system, in particular:
|
— |
The quality objectives, quality planning and quality manual must fully take on board the objective of delivering products that conform to the essential requirements. |
|
— |
The manufacturer must identify and document the essential requirements that are relevant for the product and the harmonised standards or other technical solutions that will ensure fulfilment of these requirements. |
|
— |
The identified standards or other technical solutions must be used as design input, and as verification that design output ensures that the essential requirements will be met. |
|
— |
The measures taken to control manufacturing must ensure that the products conform to the identified essential requirements. |
|
— |
Quality records, such as inspection reports and test data, calibration data, qualification reports of the personnel concerned, must be suitable to ensure the fulfilment of the applicable essential requirements. |
5.1.7. OVERVIEW OF MODULES
|
Modules |
Description |
|
A Internal production control |
Covers both design and production. The manufacturer himself ensures the conformity of the products to the legislative requirements (no EU-type examination). |
|
A1 Internal production control plus supervised product testing |
Covers both design and production. A + tests on specific aspects of the product carried out by an in-house accredited body or under the responsibility of a notified body chosen by the manufacturer. |
|
A2 Internal production control plus supervised product checks at random intervals |
Covers both design and production. A + product checks at random intervals carried out by a notified body or in-house accredited body. |
|
B EU-type examination |
Covers design. It is always followed by other modules by which the conformity of the products to the approved EU-type is demonstrated. A notified body examines the technical design and or the specimen of a type and verifies and attests that it meets the requirements of the legislative instrument that apply to it by issuing an EU-type examination certificate. There are 3 ways to carry out EU-type examination: 1) production type, 2) combination of production type and design type and 3) design type. |
|
C Conformity to EU-type based on internal production control |
Covers production and follows module B. Manufacturer must internally control its production in order to ensure product conformity against the EU-type approved under module B. |
|
C1 Conformity to EU-type based on internal production control plus supervised product testing |
Covers production and follows module B. Manufacturer must internally control its production in order to ensure product conformity against the EU-type approved under module B. C + tests on specific aspects of the product carried out by an in-house accredited body or under the responsibility of a notified body chosen by the manufacturer (*). |
|
C2 Conformity to EU-type based on internal production control plus supervised product checks at random intervals |
Covers production and follows module B. Manufacturer must internally control its production in order to ensure product conformity against the EU-type approved under module B. C + product checks at random intervals tests on specific aspects of the product carried out by a notified body or in-house accredited body. |
|
D Conformity to EU-type based on quality assurance of the production process |
Covers production and follows module B. The manufacturer operates a production (manufacturing part and inspection of final product) quality assurance system in order to ensure conformity to EU-type. The notified body assesses the quality system. |
|
D1 Quality assurance of the production process |
Covers both design and production. The manufacturer operates a production (manufacturing part and inspection of final product) quality assurance system in order to ensure conformity to legislative requirements (no EU-type, used like D without module B). The notified body assesses the production (manufacturing part and inspection of final product) quality system. |
|
E Conformity to EU-type based on product quality assurance |
Covers production and follows module B. The manufacturer operates a product quality (= production quality without the manufacturing part) assurance system for final product inspection and testing in order to ensure conformity to EU-type. A notified body assesses the quality system. The idea behind module E is similar to the one under module D: both are based on a quality system and follow module B. Their difference is that the quality system under module E aims to ensure the quality of the final product, while the quality system under module D (and D1 too) aims to ensure the quality of the whole production process (that includes the manufacturing part and the test of final product). E is thus similar to module D without the provisions relating to the manufacturing process |
|
E1 Quality assurance of final product inspection and testing |
Covers both design and production. The manufacturer operates a product quality (= production quality without the manufacturing part) assurance system for final product inspection and testing in order to ensure conformity to the legislative requirements (no module B (EU-type), used like E without module B). The notified body assesses the quality system. The idea behind module E1 is similar to the one under module D1: both are based on a quality system. Their difference is that the quality system under module E1 aims to ensure the quality of the final product, while the quality system under module D1 aims to ensure the quality of the whole production process (that includes the manufacturing part and the test of final product). E1 is thus similar to module D1 without the provisions relating to the manufacturing process. |
|
F Conformity to EU-type based on product verification |
Covers production and follows module B. The manufacturer ensures compliance of the manufactured products to approved EU-type. The notified body carries out product examinations (testing of every product or statistical checks) in order to control product conformity to EU-type. Module F is like C2 but the notified body carries out more systematic product checks. |
|
F1 Conformity based on product verification |
Covers both design and production. The manufacturer ensures compliance of the manufactured products to the legislative requirements. The notified body carries out product examinations (testing of every product or statistical checks) in order to control product conformity to the legislative requirements (no EU-type, used like F without module B) Module F1 is like A2 but the notified body carries out more detailed product checks. |
|
G Conformity based on unit verification |
Covers both design and production. The manufacturer ensures compliance of the manufactured products to the legislative requirements. The notified body verifies every individual product in order to ensure conformity to legislative requirements (no EU-type). |
|
H Conformity based on full quality assurance |
Covers both design and production. The manufacturer operates a full quality assurance system in order to ensure conformity to legislative requirements (no EU-type). The notified body assesses the quality system. |
|
H1 Conformity based on full quality assurance plus design examination |
Covers both design and production. The manufacturer operates a full quality assurance system in order to ensure conformity to legislative requirements (no EU-type). The notified body assesses the quality system and the product design and issues an EU design examination certificate. Module H1 in comparison to module H provides in addition that the notified body carries out a more detailed examination of the product design. The EU-design examination certificate must not be confused with the EU-type examination certificate of module B that attests the conformity of a specimen 'representative of the production envisaged', so that the conformity of the products may be checked against this specimen. Under EU design examination certificate of module H1, there is no such specimen. EU design examination certificate attests that the conformity of the design of the product has been checked and certified by a notified body. |
5.1.8. OVERVIEW OF PROCEDURES
The following procedures are possible:
|
— |
A — Internal production control |
|
— |
A1 — Internal production control plus supervised product testing |
|
— |
A2 — Internal production control plus supervised product checks at random intervals |
|
— |
B + C — EU-type examination (B) followed by Conformity to EU-type based on internal production control (C) |
|
— |
B + C1 — EU-type examination (B) followed by Conformity to EU-type based on internal production control plus supervised product testing (C1) |
|
— |
B + C2 — EU-type examination (B) followed by Conformity to EU-type based on internal production control plus super- vised product checks at random intervals (C2) |
|
— |
B + D — EU-type examination (B) followed by Conformity to EU-type based on quality assurance of the production pro- cess (D) |
|
— |
D1 — Quality assurance of the production process. |
|
— |
B + E — EU-type examination (B) followed by Conformity to EU-type based on product quality assurance (E) |
|
— |
E1 — Quality assurance of final product inspection and testing |
|
— |
B + F — EU-type examination (B) followed by Conformity to EU-type based on product verification (F) |
|
— |
F1 — Conformity based on product verification |
|
— |
G — Conformity based on unit verification |
|
— |
H — Conformity based on full quality assurance |
|
— |
H1 — Conformity based on full quality assurance plus design examination |

5.1.9. RATIONALE FOR SELECTING THE APPROPRIATE MODULES
|
The legislator when selecting modules for his legislative instrument should follow the following principles:
|
— |
As a general rule, products are subject to both design and production modules before placed on the market. |
|
— |
When appropriate in terms of protection of public interest, the manufacturer must be given as wide a choice of modules as possible. |
|
— |
If it is sufficient that the manufacturer carries out himself all checks in order to ensure the conformity of the products, then the legislator may select module A. This can be the case of low complexity (simple design and production mechanism) products that present a low risk for the public interest. |
|
— |
In cases of mass production based on a type/specimen and where the product in question is of complex design or presents higher risks of non-compliance for instance, the EU legislation may lay down the conformity assessment procedure in two steps: first the examination of conformity of the prototype/specimen against the relevant legal requirements (EU-type examination — module B) and then the determination of the conformity of the products against the approved EU-type (modules C and variants, D, E, F). |
|
— |
In cases where the legislator has opted for the demonstration of conformity assessment against a specimen (module B), he must examine the possibility of whether it is sufficient that the manufacturer can carry out himself all checks in order to ensure the conformity in the production phase. If this is the case, then the legislator may select module C. |
|
— |
In many cases the legislator must acknowledge that quite often, manufacturers manage very well equipped testing laboratories or premises. This is usually the case for new innovative complex products for which the testing know-how remains inside the manufacturers. In such cases the legislator may consider selecting either modules A1, A2, or, C1, C2 (the latter two, if he has opted for the demonstration of conformity assessment against a specimen — module B) that allow the use of an accredited in-house body. |
|
— |
If the demonstration of conformity of products against an approved EU-type cannot be left to the manufacturer but requires that products are supervised by a notified body during the production process, then the legislator may require from the manufacturer either to operate an approved quality system (modules D, E) or that the conformity of his products are verified be means of tests/checks (module F). In this respect, if the production mechanism is relatively ‘simple’ then the legislator may consider that it sufficient that the quality system of the manufacturer focuses only on the test of the final product without including the pure manufacturing part. If this is the case, module E is the most appropriate. |
|
— |
In the case of products of simple design but complicated production/manufacturing, the legislator may consider selecting modules D1, E1, F1 and using thus the advantages of modules D, E and F respectively, without the necessity of recurring to a more formal specimen examination (as provided under module B that precedes modules D, E, F). |
|
— |
For products, produced in small series the legislator may consider selecting module G. |
|
— |
In complex cases where it is necessary that the manufacturer must operate a full quality system covering both the design and the production phase, the legislator may opt for module H. |
|
— |
When the manufacturer operates a full quality assurance system, but the verification of the conformity of design and the issuance of EU design examination certificate by a notified body is necessary, then the legislator may select module H1. |
5.2. CONFORMITY ASSESSMENT BODIES
5.2.1. CONFORMITY ASSESSMENT BODIES AND NOTIFIED BODIES
|
Notified bodies carry out the tasks pertaining to the conformity assessment procedures referred to in the applicable technical harmonisation legislation when a third party is required. |
A conformity assessment body is a body that performs one or several elements of conformity assessment, including one or several of the following activities: calibration, testing, certification and inspection. Notified bodies are conformity assessment bodies which have been officially designated by their national authority to carry out the procedures for conformity assessment within the meaning of applicable Union harmonisation legislation when a third party is required. They are called ‘notified bodies’ under EU legislation.
Notified bodies take responsibilities in areas of public interest and, therefore, must remain accountable to the competent national authorities. To be eligible a body must be a legal entity established on the territory of a Member State and, thus, come under its jurisdiction. Member States remain free to decide whether or not to notify a body which complies with the requirements laid down in the relevant Union harmonisation legislation.
5.2.2. ROLES AND RESPONSIBILITIES
|
Although the notified body must be established on the territory of the notifying Member State, it may have activities or personnel outside the Member State, or even outside the Union. Certificates and other conformity assessment attestations are, however, always issued by and in the name of the notified body (229). Since the notified body always has to carry out its assessment functions within the jurisdiction of the designating Member State, it has to inform the notifying authority, which must be capable of ensuring the monitoring of the total body as it has to take the responsibility for its operations. If monitoring is not considered possible, the notifying authority should withdraw or limit the scope of the notification as deemed necessary.
Notified bodies must keep their national notifying authorities informed of their activities (for example concerning the conduct of conformity assessments, availability of resources, subcontracting, situations of conflicts of interest), either directly or via an authorised body (for example the national accreditation body). They must also be prepared to provide either at the request of their notifying authorities or of the Commission, all information concerning the proper implementation of the conditions under which they were notified.
Notified bodies have a general obligation to inform the notifying authority about all certificates refused, restricted, suspended or withdrawn due to safety related non-conformities and, on request, about certificates issued or other conformity assessment activities performed. In addition, notified bodies must provide other bodies notified under the same Union harmonisation legislation, carrying out similar conformity assessment activities covering the same products, with relevant information on issues relating to negative and, on request, positive conformity assessment results. Given the confidentiality requirements that notified bodies have to observe when fulfilling their tasks, the information to be shared with other notified bodies cannot concern confidential commercial information on the product. Relevant information to be exchanged on issues relating to negative conformity assessment results should thus primarily concern the refusal to issue a conformity assessment attestation identifying the product and manufacturer in question.
They must also provide the market surveillance authority and, according to some Union harmonisation legislation also the market surveillance authorities of other Member States, with relevant information for the purpose of market surveillance. Notified bodies as such are not responsible for providing the EU declaration of conformity or the technical documentation. Having said that, in line with the applicable conformity assessment procedure, they might have to keep the technical documentation as part of the technical file and provide it to the Commission or the Member States upon request (230). Further, notified bodies must provide, on request of the Commission department responsible for administering a safeguard clause, the necessary information related to the product or the conformity assessment.
Notified bodies are and must remain third parties independent of their clients and other interested parties. The legal status of bodies seeking notification, whether they are private or state-owned, is irrelevant as long as their independence, impartiality and integrity are ensured, and they are identifiable as a legal entity bearing rights and obligations.
The requirement for independence covers the whole organisation, including the board or directors, and applies also for bodies belonging to business associations or professional federations.
In order to guarantee impartiality, the notified body and its staff have to be free from any commercial, financial and other pressure that might influence their judgement. The body also has to implement procedures to ensure that its work cannot be influenced from outside. The structure of the body must safeguard its impartiality, especially if the body has activities other than those of a notified body.
Furthermore, the body must have policies and procedures that distinguish between the tasks it carries out as a notified body and any other activity in which it is engaged, and it must make this distinction clear to its customers. Accordingly, marketing material must not give any impression that assessment or other activities carried out by the body are linked with tasks described in the applicable Union harmonisation legislation.
When a conformity assessment body delivers a test report it is in its capacity as a conformity assessment body; only in its capacity as notified body can it deliver EU-type examination certificates — a certificate bearing in particular the name and identification number of the notified body. In no circumstances must the notified body issue a test report carrying its notified body number (231) in relation to tests that are not specified in the legislation, whether those tests were carried out by the body itself or by another body. Moreover, a notified body may only use its number in relation to conformity assessment activities carried out under the specific conformity assessment module that requires the intervention of a notified body, and for which it has been notified.
A notified body must require the manufacturer to take appropriate corrective measure and, if necessary suspend or withdraw a certificate it has issued, if in the course of the monitoring of conformity following the issue of the certificate, it finds that the product no longer complies (232).
In their capacity of notified bodies, they must not offer or provide additional services unless they have an added value for the conformity assessment of the product. However, notified bodies may offer any type of conformity assessment services and markings where the products are intended for the markets of third countries outside the European Union, for example in the context of Mutual Recognition Agreements (233). Such activities must be clearly separated from the activities of the body as a notified body. Notified bodies must also ensure that their activities outside the scope of technical harmonisation legislation do not compromise or diminish confidence in their competence, objectivity, impartiality or operational integrity as notified bodies. Notified bodies cannot use their notified body name and number to carry out these activities.
A notified body may not be the manufacturer, the authorised representative, a supplier or their commercial competitor, nor offer or provide (or have offered or provided) consultancy or advice to any of these parties as regards the design, construction, marketing or maintenance of the products in question. However, this does not preclude the possibility of exchanging technical information and guidance between the manufacturer, the authorised representative, suppliers and the notified body.
To safeguard impartiality and avoid conflicts of interest it is important to make a clear distinction between conformity assessment performed by notified bodies prior to placing products on the market and market surveillance. Furthermore, market surveillance authorities must carry out their duties independently, impartially and without bias. Therefore, it is to be considered as inappropriate for market surveillance authorities to be designated as notified bodies, and the necessary safeguards should be put in place to ensure the impartiality and absence of conflict of interest if a single entity is entrusted with both responsibilities (234) (235). Notified bodies must have documented procedures for the identification, review and resolution of all cases where conflict of interest is suspected or proven. The notified body should also require all staff acting on its behalf to declare any potential conflict of interest.
Notified bodies must have under their control the necessary personnel, who have sufficient knowledge and experience relating to the products and conformity assessment procedure in question, and who have appropriate training. In particular, knowledge and experience should relate to relevant regulatory requirements and enforcement policies, European and international standardisation activities, relevant technologies, production methods and verification procedures, and normal conditions of use of the product in question. The body must be in a position to manage, control and be responsible for the performance of all its resources and maintain comprehensive records concerning the suitability of all the staff it uses in particular areas, whether they are employees, employed on contract or provided by external bodies. The body must also have access to appropriate facilities and be able to test or re-test in the EU. Otherwise it will not be possible for the notifying authority to check its competence.
Notified bodies must ensure the confidentiality of all the information it obtains in the course of conformity assessment. It must make adequate arrangements to ensure that no results or other information is disclosed to any other party than the competent authority in question, and to the manufacturer or the authorised representative.
Notified bodies must have adequate insurance to cover their conformity assessment activities. The scope and overall financial value of liability insurance must correspond to the level of risk linked with the activities of the notified body. The manufacturer in particular retains, however, the overall responsibility for the conformity of the product with all the requirements of the applicable legislation, even if some stages of the conformity assessment are carried out under the responsibility of a notified body.
Notified bodies are obliged to participate in coordination activities (236). They must also take part directly or be represented in European standardisation, or otherwise ensure that they know the situation of relevant standards (237).
5.2.3. COMPETENCE OF NOTIFIED BODIES
|
The primary task of a notified body is to provide conformity assessment services on the conditions set out in the applicable Union harmonisation legislation. This is a service to the manufacturers in an area of public interest. |
Notified bodies are designated to assess conformity with the essential requirements, and to ensure consistent technical application of these requirements according to the relevant procedures in the applicable Union harmonisation legislation. The notified bodies must have appropriate facilities and technical staff that enable them to carry out technical and administrative tasks related to conformity assessment. They must also apply appropriate procedures of quality control in relation to such services provided. Manufacturers are free to choose any notified body that has been designated to carry out the conformity assessment procedure in question according to the applicable Union harmonisation legislation.
A notified body wishing to offer services according to several conformity assessment procedures must fulfil the relevant requirements for the respective tasks, and this has to be assessed according to the requirements for each different procedure in question. However, since the scope of much technical harmonisation legislation can be relatively wide and heterogeneous, a notified body does not need to be qualified to cover all products falling within the scope of that legislation, but may be notified for a defined range of products only.
Notified bodies must have appropriate structures and procedures to ensure that the conduct of conformity assessment and the issuing of certificates are subject to a review process. Relevant procedures must, in particular, cover obligations and responsibilities in relation to suspension and withdrawal of certificates, requests addressed to the manufacturer to take corrective measures, and reporting to the competent authority.
Apart from carrying out certain responsibilities in the field of public interest, notified bodies must regard themselves as rendering services to industry. Thus, they should provide relevant information to the manufacturer and the authorised representative regarding the legislation in question, apply the conformity assessment procedure without unnecessary burdens for the economic operators, and refrain from proposing additional certification or marking that has no added value for the conformity assessment of the product. The latter activities must be clearly separated from the activities of the body as a notified body. Notified bodies cannot use their notified body name and number to carry out these activities.
To avoid unnecessary burdens for economic operators and help in ensuring the protection of confidential data or intellectual property rights, the technical documentation provided to notified bodies has to be limited to that which is required solely for the purpose of assessing conformity to the legislation.
5.2.4. COORDINATION BETWEEN NOTIFIED BODIES
In recognition of the fact that notified bodies fulfil tasks delegated to them by public authorities, they are obliged to take part in coordination activities organised by the Commission. The latter, together with the Member States, ensures that coordination is organised between the notified bodies.
A coordination group of notified bodies is established for each Union harmonisation legislative act or for several related acts, and its work is limited to technical problems relating to conformity assessment in order to ensure a uniform application of the technical provisions of the applicable legislation. To that end, it should be free to define its rules of work and constitution. Each group of notified bodies has a technical secretariat and a chairman.
Generally, the groups of notified bodies are composed of representatives of notified bodies. To achieve a higher degree of efficiency in their work the groups can set up subgroups with a restricted number of participants to discuss specific technical questions. The Commission is represented in the groups. Governmental experts and representatives of the authorities directly responsible for the effective implementation of Union harmonisation legislation can participate as observers in the groups. The European standardisation organisations (CEN, Cenelec and ETSI) are represented in the groups when standards related issues arise. The groups may also invite relevant European federations other interested parties. Where the groups of notified bodies have to treat subjects of a confidential nature, the participation in meetings is restricted as deemed necessary.
If a body refuses to cooperate, the notification may be withdrawn. However, the notified bodies are not obliged to participate in meetings at European level if they keep themselves informed of, and apply the administrative decisions and documents produced by their group. The relevant working documents, meeting reports, recommendations and guidelines produced by the sectoral and inter-sectoral groups of notified bodies or their subgroups should be made available to all notified bodies forming part of those groups, whether they have taken part in the meetings or not. The information exchange and communication can be enhanced by use of a platform such as CIRCABC, hosted by the Commission.
National coordination groups are also encouraged and where those exist, notified bodies from a given Member State might be required to take part in their activities.
5.2.5. SUBCONTRACTING BY NOTIFIED BODIES
|
A notified body can only subcontract a task for which it has the competence itself. It must not be the case that a notified body subcontracts a part of the work because it does not have the required competence and knowledge.
The bodies acting as subcontractors for the notified bodies need not be notified as such. Nevertheless, the notified body must inform the Member State concerned of its intention to subcontract certain work. Consequently, the Member State may decide that it cannot take the overall responsibility as a notifying authority for such an arrangement, and withdraw or limit the scope of the notification. The notified body must keep a register of all its subcontracting activities, and update it systematically.
The body subcontracted by the notified body must be technically competent, and display independence and objectivity according to the same criteria and under the same conditions as the notified body. The Member State that has notified the body which subcontracts part of its work must be capable of ensuring effective monitoring of the competence of the body subcontracted by the notified body. Individual external auditors or specialists have to fulfil the conditions of a subcontractor.
The notified body must ensure that its subcontractors have the necessary competence and that they maintain this competence, for example by carrying out regular evaluations and by keeping itself regularly informed of the details regarding the performance of their tasks. The notified body must also be able to demonstrate the compliance of its subcontractors with the requirements laid down in the relevant Union harmonisation legislation.
Information on subcontracting activities and on the competence of the subcontractors and/or the subsidiaries must be readily available at all times, so that the notifying authority can take any necessary action, and communicate it without delay to the Commission and the other Member States on request. Compliance with the EN ISO/IEC 17000 series of standards entails a presumption of conformity of the subcontractor with most of the requirements, as is the case with the notified body itself. Where accreditation is not used to assess the competence of notified bodies, the authority should undertake on-site checks of the subcontractor to the same extent as would be provided for under accreditation.
A further condition for subcontracting is that the conformity assessment procedure can be subdivided into technical operations and assessment operations, and that the methodology used to carry out the technical operations is sufficiently precise. A notified body can subcontract strictly limited technical tasks (such as tests and examinations), as long as these can be defined as substantial and coherent parts of the technical operation. The body subcontracted by the notified body must, nevertheless, carry out substantial and coherent parts of these technical operations. The notified body staff has to be technically qualified to be able to assess the test results of subcontractors. Notified bodies must not restrict their activities to purely administrative functions.
Notified bodies may, for example, subcontract tests while continuing to assess their results and, in particular, to validate the test report in order to evaluate whether the requirements of the Union harmonisation legislation are met. Similarly, subcontracting is possible in the field of certification of quality systems provided that the notified body carries out the evaluation of the audit results. The notified body cannot under any circumstances subcontract all of its activities, as that would make the notification meaningless.
The subcontracted work must be carried out according to pre-established technical specifications setting out a detailed procedure based on objective criteria to guarantee total transparency. Where the body subcontracted by the notified body is involved in the assessment of conformity to standards, these must be used if they lay down the procedures. If this body is involved in the assessment of conformity to essential requirements, the procedure followed by the notified body itself or a procedure deemed by the notified body to be equivalent to that must be used.
The notified body must in all cases have a binding agreement with its subcontractors to ensure that its general responsibilities are fulfilled (238). Notified bodies must keep at the disposal of the notifying authority the relevant documents concerning the assessment of the qualifications of the subcontractor or the subsidiary and the work carried out by them under the relevant Union harmonisation legislation (239).
A subcontracting notified body remains responsible for all the activities covered by the notification. Subcontracting does not entail the delegation of powers or responsibilities. Certificates and other attestations of conformity are always issued in the name and under the responsibility of the notified body. Therefore, the subcontracting notified body must be competent to review the work of the subcontractor in all its elements and must take the final decision.
The conditions for subcontracting apply to any subcontractor whether or not established within the European Union. The notified body remains entirely responsible for the work carried out for it by the subcontractor.
The notified body must have appropriate facilities and staff to be able to verify the results of any tests, inspections or any other task carried out by the subcontractor. Furthermore, if accreditation is the chosen path for notification, it must cover the subsidiary companies of notified bodies to which they have recourse. Accreditation bodies must take this into account, either by properly applying the existing international guidance on cross-frontier accreditation or by specifying it in the accreditation documents. If notification is not based on accreditation, then in order to ensure the proper and consistent supervision of such subsidiaries and subcontractors, the contents of the information to be provided to the notifying authority should be further specified by aligning it to the relevant practices in accreditation.
The manufacturer can provide test reports or other elements of its technical documentation. The notified body can take these reports into account if it assumes full responsibility for the results. The notified body may accept the manufacturer's test results for the conformity assessment provided that it justifies the reason for taking account of these tests.
5.2.6. ACCREDITED IN-HOUSE BODIES (240)
Only in the cases where sectoral Union harmonisation legislation provides for it, an accredited in-house body may be used to carry out conformity assessment activities for the undertaking of which it forms a part, for the implementation of the conformity assessment procedures, modules A1, A2, C1 or C2. That body must constitute a separate and distinct part of the undertaking and must not participate in the design, production, supply, installation, use or maintenance of the products it assesses.
An accredited in-house body has to meet a number of requirements. It must be accredited in accordance with Regulation (EC) No 765/2008 (241). The body and its personnel must be identifiable within the structure of the organisation, and have reporting methods within the undertaking of which they form a part which ensure their impartiality and demonstrate it to the relevant national accreditation body. Neither the body nor its personnel may be responsible for the design, manufacture, supply, installation, operation or maintenance of the products they assess, nor may they engage in any activity that might conflict with their independence of judgment or integrity in relation to their assessment activities. An accredited in-house body can supply its services only to the undertaking of which it forms a part.
An accredited in-house body cannot be notified to the Member States or the Commission, but information concerning its accreditation must be given by the undertaking of which it forms a part, or by the national accreditation body, to the notifying authority at the request of that authority.
5.3. NOTIFICATION
5.3.1. NOTIFYING AUTHORITIES
|
A notifying authority is the governmental or public body that is tasked with designating and notifying conformity assessment bodies under Union harmonisation legislation. |
A notifying authority is the governmental or public body that is tasked with designating and notifying conformity assessment bodies under Union harmonisation legislation. Most often it is the national administration responsible for the implementation and management of the Union harmonisation act under which the body is notified. Each Member State must designate a notifying authority to be responsible for the assessment, notification and monitoring of conformity assessment bodies. The notifying authority assumes full responsibility for the competence of the bodies it notifies.
Each Member State must establish its notifying authorities in such a way that there is no conflict of interest with conformity assessment bodies. They must be organised and operated so as to safeguard the objectivity and impartiality of their activities. Each decision relating to notification of a conformity assessment body must be taken by competent persons different from those who carried out the assessment.
Further requirements on a notifying authority are that it must not offer or provide any activities that conformity assessment bodies perform, or consultancy services on a commercial or competitive basis. It must safeguard the confidentiality of the information it obtains, and it must have a sufficient number of competent personnel at its disposal for the proper performance of its tasks.
Member States must inform the Commission of their procedures for the assessment and notification of conformity assessment bodies and the monitoring of notified bodies. The Commission makes that information publicly available on its website.
5.3.2. NOTIFICATION PROCESS
|
5.3.2.1. Principles of notification
The status of Notified Body is available to conformity assessment bodies established within the European Union. Member States are responsible for the notification of notified bodies and the choice of and responsibility for notified bodies rests with national authorities. They may choose the bodies they notify from those that are established on their territory which comply with the requirements of the legislation, and which have the necessary competences to become notified. Notification is the act of the notifying authority informing the Commission and the other Member States that such a body has been designated to carry out conformity assessment according to a Union harmonisation act, and fulfils the requirements relating to notified bodies set out in that Union harmonisation act.
While designation is considered as an act of the designating authority — which may be the same body as the notifying authority — only the act of notifying the Commission and the other Member States allows a ‘designated body’ to become a ‘notified body’.
Since notification falls within the discretion of Member States, they are not obliged to notify all the bodies demonstrating technical competence. Neither are Member States obliged to notify bodies in respect of each procedure to be applied according to a specific Union harmonisation act.
Member States are free to notify a body at any time after a Union harmonisation act has been adopted. They should nevertheless take all necessary steps for notifying before the Union harmonisation act starts applying (242). This can make effective use of the transitional period provided for in the Union harmonisation act, and allow for notified bodies to be active and certificates to be granted from the date of first application of the Union harmonisation act. If on the basis of new legislation the re-notification of notified bodies is required, as soon as the Member State has transposed the necessary provisions into national law and has appointed a notifying authority for a particular Union harmonisation act, it is possible for that notifying authority to place a notification. A notified body may thus be notified under both the old and the new legislation during the transitional period, but the notification under the old legislation will expire automatically on the date of application of the new legislation, unless specific legislation provides otherwise. It must be stressed, however, that in such cases notified bodies, while they can do preparatory work, are not entitled to issue certificates before the Union harmonisation legislation starts applying unless sector legislation provides otherwise.
5.3.2.2. Assessment of conformity assessment bodies
The assessment of a conformity assessment body seeking notification determines if it is technically competent and capable of carrying out the conformity assessment procedures in question, and if it can demonstrate the necessary level of independence, impartiality and integrity.
Member States take the final responsibility for the competence of their notified bodies with respect to the other Member States and the EU institutions. They must therefore verify the competence of the bodies seeking notification, based on the criteria laid down in the applicable Union harmonisation legislation in conjunction with essential requirements and the conformity assessment procedure(s) in question. In general, the competence criteria set out in the Union harmonisation acts cover:
|
— |
availability of personnel and equipment, |
|
— |
independence and impartiality in relation to those directly or indirectly concerned with the product (such as the designer, the manufacturer, the manufacturer's authorised representative, the supplier, the assembler, the installer, the user), |
|
— |
technical competence of personnel that is relevant to the products and conformity assessment procedure in question, |
|
— |
maintenance of professional secrecy and integrity, and |
|
— |
subscription to civil liability insurance, unless that liability is covered by the state under national law. |
Notifying authorities or accreditation bodies must carry out periodic monitoring to assess the continuity of the competence of notified bodies after they are notified.
5.3.2.3. Accreditation under Regulation (EC) No 765/2008
Accreditation, performed according to the EN ISO/IEC 17000 series of standards by nationally recognised accreditation bodies that are members of the European cooperation for Accreditation (EA), is a technical assessment of the competence of the conformity assessment body seeking notification. Although it is not a requirement, it remains an important and favoured instrument for evaluating the competence and integrity of the bodies to be notified. For this reason, accreditation should be considered by national notifying authorities as the most favoured technical basis for the assessment of conformity assessment bodies so as to reduce differences in the criteria applied for notification.
Accreditation provides an authoritative statement of the competence, professional integrity and impartiality of the bodies to be notified to the Commission and the other Member States. It also entails regular monitoring and surveillance of the accredited bodies. Whenever a national accreditation body ascertains that the conformity assessment body to which it has issued an accreditation certificate is no longer competent or does not fulfil its obligations, the accreditation certificate can be withdrawn. In this case the body should be de-notified and no longer allowed to carry out conformity assessment activities under the relevant legislation.
The preference given to accreditation is based on the peer evaluation process which ensures that the accreditation body adequately supervises the conformity assessment bodies it accredits. Cases may, however arise where the national accreditation body has not been successfully peer evaluated but may nevertheless have assessed notified bodies (243). If the national accreditation body has not been peer evaluated for the specific accreditation activity in question but still evaluate the competence of a conformity assessment body for this activity, the notification of this conformity assessment body should not be considered as accredited for the purposes of EU harmonisation legislation.
If a national accreditation body was successful in a previous peer evaluation for a given activity but has been suspended at a subsequent peer evaluation, new notifications of conformity assessment bodies assessed by this national accreditation body should also be considered as unaccredited. As a principle, accreditation certificates issued up until the point of the suspension of the peer evaluation of the national accreditation body, should continue to be recognised by national authorities.
If the grounds for suspension of the national accreditation body result in serious doubts about the competence of the notified bodies, the responsible notifying authority would have to inform the Commission and other Member States of how it intends to ensure the competence of the bodies notified, and of any corrective measures taken, including the de-notification.
Although accreditation is the favoured instrument for the verification of competence of conformity assessment bodies, Member States can carry out the evaluation themselves. Following the entry into force of Regulation (EC) No 765/2008 on 1 January 2010, in such cases evidence must be given to the Commission and other Member States that the evaluated body complies with all the applicable regulatory requirements. Further, the notified body must be subject to regular surveillance similar to the practice established by the accreditation organisations.
5.3.2.4. Article 5(2) of Regulation (EC) No 765/2008
According to Article 5(2) of Regulation (EC) No 765/2008, when a Member State does not base its notification on accreditation, ‘it shall provide the Commission and the other Member States with all the documentary evidence necessary for the verification of the competence of the conformity assessment bodies it selects for the implementation of the Union harmonisation legislation in question’. (244)
In order to ensure the necessary level of confidence in the impartiality and technical competence of conformity assessment bodies and in the reports and certificates issued by them, national authorities, when carrying out the assessment without accreditation, should give detailed and comprehensive information describing how the candidate Notified Body has been assessed as qualified to carry out the tasks for which it is notified and showing that it fulfils the applicable criteria relating to notified bodies. This information, linked to a given notification, is made available to the Commission and the other Member States using the NANDO electronic notification tool.
The evaluation procedure should be based on at least the following elements:
|
— |
a formal application procedure |
|
— |
assessment against applicable requirements. |
|
— |
production of an assessment report |
|
— |
clear decision-making process |
|
— |
existence of a systematic surveillance and related sanction mechanism, providing for periodic surveillance including |
|
— |
on-site visits, in order to verify the continued fulfilment of requirements by the notified body |
|
— |
demonstration of the national authority's own technical competence for assessing conformity assessment bodies for the purpose of notification under technical harmonisation legislation. This demonstration must give equivalent assurance as the EA (245) peer evaluation system. |
|
— |
the candidate notified bodies should be made aware of general conditions, of their rights and obligations, and of the requirements relating to assessment carried out with a view to notification |
The assessment itself should consist of:
|
— |
a review of documents verifying the completeness and appropriateness from a substantial point of view with regard to conformity to the applicable requirements |
|
— |
an on-site audit to check technical and procedural aspects — such as the availability and appropriateness of facilities and equipment, the technical competence of staff, the existence of an appropriate management system — and to check other aspects demonstrating that conformity to requirements is properly implemented. The assessment must include witnessing technical activities. |
When choosing an assessment process other than formal accreditation, notifying authorities must indicate the reasons why accreditation is not chosen to back up the notification process. Moreover, notifying authorities may not outsource to the national accreditation body the assessment of unaccredited conformity assessment bodies that seek to become notified bodies, without fulfilling the whole accreditation process including the delivery of the accreditation certificate.
When accreditation is not used, the notifying authorities must perform periodical verifications to ensure the continuous competence of the notified body, in the same way as national accreditation bodies do.
5.3.2.5. Steps in the notification of a notified body
To obtain notification, a conformity assessment body submits an application for notification to the notifying authority of the Member State in which it is established. That application should be accompanied by a description of the conformity assessment activities, the conformity assessment procedures or modules and the product or products for which that body claims to be competent, as well as by an accreditation certificate, where one exists, issued by the national accreditation body attesting that the conformity assessment body fulfils the requirements laid down in the relevant harmonisation legislation.
Where the body concerned cannot provide an accreditation certificate, it must provide the notifying authority with all the documentary evidence necessary for the verification, recognition and regular monitoring of its compliance with the requirements laid down in the relevant harmonisation legislation. After verification the Member State informs the Commission and the other Member States of the details of the body.
The notification of a notified body is sent by the notifying authority to the Commission and the other Member States via NANDO (New Approach Notified and Designated Organisations), which is the electronic notification tool developed and managed by the Commission. It should include full details of the body, its conformity assessment activities, the conformity assessment procedures or modules and product or products concerned, and the relevant attestation of competence. It must also include the date set for the reassessment of the notified body by the national accreditation body or, for an unaccredited notification, the date of the next monitoring review by the notifying authority.
Where a notification is not based on an accreditation certificate, the notifying authority must provide the Commission and the other Member States with documentary evidence which demonstrates the conformity assessment body's competence, how it has been assessed, and the arrangements in place to ensure that that body will be monitored regularly and will continue to satisfy the requirements.
The notification takes effect after a notification email from NANDO has been sent to the Commission and the other Member States and published on the NANDO website. The body concerned may then perform the activities of a notified body. Under legislation that is aligned with Decision No 768/2008/EC, the notification is published following a period allowed for objections by other Member States or the Commission — 2 weeks where accreditation is used, 2 months where accreditation is not used — and then only where no such objections have been raised.
The Commission and the other Member States must be notified in similar manner of any subsequent changes to the notification that are relevant, such as a change in the scope or validity period of the notification, or changes to the details of the body itself.
5.3.3. PUBLICATION BY THE COMMISSION — THE NANDO WEBSITE
For information purposes, the Commission makes the lists of notified bodies (and other categories of conformity assessment bodies such as User Inspectorates and Recognised Third Party Organisations) publicly available on the NANDO website on its Europa server. The lists are updated as and when the notifications are published, and the website is refreshed daily to keep it up-to-date.
With its initial notification, a notified body is assigned an identity number in the NANDO system. This number is automatically generated by the system at the moment of validation of the notification in the NANDO database. A legal entity may carry only one notified body identity number, regardless of the number of Union harmonisation acts for which it is notified. Allocation of the number is a purely administrative act designed to ensure the consistent management of the lists of notified bodies, and does not confer rights or commit the Commission in any way. The numbering system in NANDO is sequential, and numbers are not re-used when a notified body is withdrawn from the list. In cases of suspension or withdrawal of a notification, the details of the notification remain in the database, and are moved to the ‘Withdrawn/Expired Notifications/NBs’ part of the website (246).
Amendments (extension or reduction) to the scope, modifications of the validity period of the notification, or cancellation of the notification are likewise notified by email to the Member States and are published on the NANDO website. The website can be searched by Union harmonisation act, by country, by notified body number or using keywords.
5.3.4. MONITORING OF THE COMPETENCE OF NOTIFIED BODIES — SUSPENSION — WITHDRAWAL — APPEAL
It is essential to ensure that notified bodies remain competent over time and that this can be made transparent to the other Member States and the Commission. Legislation at EU level clearly requires the national competent authorities to regularly monitor and assess the continuing competence of the bodies they have notified and that are listed in NANDO. The NANDO website should be transparent for these ongoing processes that back up the notification system.
All notifications of notified bodies, whether accredited or unaccredited, that are entered in the NANDO database, should be updated within a maximum period of 5 years from the date of the initial notification, or the last update, with information on the continuous monitoring of the competence of the notified body. Such updates should include the relevant new data relating to accreditation or, if the notification is unaccredited, information relating to the required monitoring of the body by the notifying authority — in particular, a report concerning the assessment process, i.e. document review, on-site assessment, description of systematic surveillance including on-site visits and demonstration of the authority's technical competence to conduct the assessment. If the notification is not updated after the 5-year period, the Commission will consider that there is reason to question the continued competence of the notified body (247) and will request the notifying Member State to provide with all information relating to the maintenance of the competence of the body concerned.
The Commission and the Member States have the responsibility to act when doubt arises about the competence of a notified body, either at the moment of notification or thereafter. Should the Commission consider, on its own initiative or after complaint, that a notified body does not comply with the requirements or fulfil its responsibilities, it will inform the national notifying authority and ask for appropriate documented evidence concerning the basis for the notification and the maintenance of the competence of the body. Should a Member State not provide such information, the Commission may bring this to the attention of the other Member States for discussion or initiate the procedure under Article 258 TFEU against the notifying Member State.
Where a notifying authority has ascertained or has been informed that a notified body no longer meets the requirements laid down in the relevant legislation, or that it is failing to fulfil its obligations, the notifying authority must — depending on the seriousness of the failure — suspend or withdraw the notification after immediately contacting the body in question. It has to immediately inform the Commission and the other Member States accordingly. The Member State must also have this information published, and inform the Commission and the other Member States following a procedure similar to that of the notification. The body in question should have the possibility to appeal against such a decision. Whether this appeal postpones the de-notification or not depends on national legislation.
Withdrawal of notification takes place when the notified body ceases to fulfil the requirements or its obligations. This may be done at the instigation of the notifying Member State, where it has received evidence regarding the failure of the notified body to meet its requirements during the periodic surveillance (done by the accreditation body or the notifying authority), or has received complaints about the notified body's competence or behaviour. It may also be a result of action by the Commission, where the latter has reason to doubt that a notified body meets or continues to meet the requirements for its notification. In such cases the Commission informs the notifying Member State accordingly and requests it to take the necessary corrective measures, including de-notification if necessary. The notifying authority must take appropriate measures. Another reason for withdrawal of a notification might be the request of the notified body itself, for instance due to planned changes in policy, organisation or ownership of the body. Withdrawal of a notification can also be the end result of an infringement procedure.
Withdrawal is the responsibility of the notifying Member State. Only the national authority is entitled to withdraw a notification. The Commission can withdraw a notified body from the NANDO list only when, at the end of an infringement procedure under Article 258 TFEU, the Court declares a Member State to be in infringement of a given Union harmonisation act and, consequently, declares a notification to be invalid. In all such cases the Commission will ensure that any sensitive information obtained in the course of its investigations is treated confidentially.
Without prejudice to sectoral specificities, the suspension or withdrawal of a notification does not affect certificates issued by the notified body up to that point, until such time as demonstration can be made that the certificates should be withdrawn. In order to ensure continuity in the event of the suspension or withdrawal of a notification, or where the notified body has ceased its activity, the notifying Member State must ensure that the files of that body are either processed by another notified body or kept available for the responsible notifying and market surveillance authorities at their request.
6. ACCREDITATION
Regulation (EC) No 765/2008 provides a legislative framework for accreditation at the national and EU levels and puts into place an overall policy with its rules, procedures and infrastructures. The reinforcement of accreditation as a means of underpinning the competence of conformity assessment bodies and hence the credibility and acceptance of certificates and other attestations, required to ensure the free movement of goods, has been a preoccupation of the Commission since the end of the 1970s. In the 1990s there was a trend towards accreditation becoming a commercial and competitive activity, hence reducing its credibility as the last level of control. The NLF however confirmed that in the EU, accreditation is a non-commercial and non-competitive public activity which is accountable to both national and European authorities.
The reinforced EU accreditation system thus put in place is in line with the standards, rules and practices of the international organisations in the field. Regulation (EC) No 765/2008 aims to ensure that accreditation serves the public interest. European Cooperation for Accreditation (EA), the European organisation of national accreditation bodies, is recognised by the Regulation, by the guidelines signed with the Member States (EFTA included) and the Commission on 1 April 2009, and benefits from a privileged relationship with the Commission through the signature of a Framework Partnership Agreement. Within this framework, the primary role of EA is to contribute to the harmonisation of European accreditation services to support the mutual recognition and acceptance of accreditation certificates throughout the Union, and to operate a rigorous peer evaluation system that controls the competence of the national accreditation bodies and the equivalence of their services.
In the field of accreditation, Regulation (EC) No 765/2008 has established a single European system which covers both the regulated domain where accreditation is required by legislation as well as for the non-regulated sphere. In the latter case, where a body voluntarily wishes to be accredited it can only go the accreditation bodies which operate under Regulation (EC) No 765/2008, thus avoiding the existence of competing systems, whichever principles they may be based on.
6.1. WHY ACCREDITATION?
|
Accreditation provides the last level of public control in a quality chain underpinning the free movement of goods in the Union. |
Regulation (EC) No 765/2008 introduced a legal framework for accreditation for the first time. Accreditation of conformity assessment bodies had previously been used in both the regulated and non-regulated domains, but it was not governed by a legal framework at European level.
The idea of regulating accreditation at European level is twofold. On the one hand a comprehensive European framework for accreditation provides the last level of public control in the European conformity assessment chain and is therefore an important element in ensuring product conformity — on the other it enhances the free movement of products and services across the EU by underpinning trust in their safety and compliance with other issues of public interest protection.
Before the entry into force of the Regulation, the lack of common rules for accreditation across Member States meant that accreditation was being used very differently, with the result that accreditation certificates were not necessarily recognised by different national authorities and market operators — leading to multiple accreditation and therefore increased cost to business and conformity assessment bodies without producing the benefits described above.
Introducing the legal framework for accreditation therefore reduced administrative burdens in the single market and enhanced public control over accreditation so that it serves as an essential tool for the functioning of the internal market.
The accreditation framework set up by the Regulation explicitly applies both to the regulated and to the voluntary spheres. This is so, because the distinction between the two can become blurred as conformity assessment bodies are active, and products are used, in both fields. A differentiation would therefore lead to unnecessary burdens for public authorities and market actors while leading to contradictions between the voluntary and regulated domains.
6.2. WHAT IS ACCREDITATION?
|
Accreditation is the attestation by a national accreditation body based on harmonised standards that a conformity assessment body has the technical competence to perform a specific conformity assessment activity. |
Accreditation is the attestation by a national accreditation body that a conformity assessment body meets the requirements set by harmonised standards and, where applicable, any additional requirements including those set out in relevant sectoral schemes, to carry out a specific conformity assessment activity.
A wide range of products are subject to third party conformity assessment. This includes unregulated products as well as products regulated at national or at EU level. For products regulated at EU level, i.e. in the harmonised area, this usually means that nationally designated conformity assessment bodies — notified bodies — test the product and issue an attestation of conformity before the product can be placed on the market.
More precisely, for there to be accreditation, there has to be an accreditable conformity assessment body (regardless of its legal personality) performing a specific conformity assessment activity.
Accreditation is the standards-based activity to ensure and attest that conformity assessment bodies have the technical competence to perform their duties as is required by the relevant regulations and standards. It assesses conformity assessment bodies' competence to perform their duties in specific fields, as accreditation is always linked to a specific scope of activity of the conformity assessment body. Operating in the public interest, accreditation assesses the technical competence, reliability and integrity of conformity assessment bodies. It does this through a process of transparent and impartial evaluation against internationally recognised standards and other requirements. Regulation (EC) No 765/2008 obliges national accreditation bodies to verify that conformity assessments are carried out in an appropriate manner and that account is taken of the size and structure of undertakings and the degree of complexity of the product technology in question and the nature of the production process.
Accreditation is based on the international standards for conformity assessment bodies that have been harmonised in the New Legislative Framework and the references of which have been published in the Official Journal of the European Union. It is the attestation by a national accreditation body that a conformity assessment body meets the requirements set by harmonised standards and where applicable any additional requirements, including those set out in relevant sectoral schemes. With Regulation (EC) No 765/2008, only national accreditation bodies are allowed to provide accreditation of conformity assessment bodies.
The reliance on harmonised standards, based on corresponding international standards, is intended to create the necessary level of transparency and confidence in the competence of conformity assessment bodies, and to ensure that the European accreditation system set up by Regulation (EC) No 765/2008 is compatible with the international accreditation system — therefore facilitating international trade.
Given the prominent role in the conformity assessment system that the Regulation has bestowed upon national accreditation bodies, accreditation bodies have to strictly follow the rules of the Regulation when assessing the competence of conformity assessment bodies. The legislator has decided to clearly limit the activities that an accreditation body may perform, keeping a tight control over their remit via the direct reference to harmonised standards. This also means that national authorities may therefore not require and should actively prevent their accreditation bodies from performing assessment services outside the full accreditation process or use conformity assessment standards that are not harmonised.
6.3. SCOPE OF ACCREDITATION
|
Accreditation is always sought and granted for a defined scope, i.e. for specific conformity assessment activities. |
Accreditation is the standards-based method of assessing and attesting the competence of conformity assessment bodies. Union policy has made use of accreditation as an instrument that is designed to create the conditions for mutual confidence because of its reliance on consensus standards. Mutual confidence can only be achieved through reliance on criteria that can be objectively verified, thus allowing transparency and comparability of conformity assessment. The relevant standards for conformity assessment bodies (248) were produced with the intention of supporting the introduction of the conformity assessment procedures set out in Union harmonisation legislation (249). These standards are conceived to cover the general competence requirements for bodies performing conformity assessment to specified requirements, irrespective of whether these are contained in regulations, standards or other technical specifications or whether such specifications are performance-based or product-specific. This concept supports the role of accreditation as a tool to facilitate the free movement of products within the internal market and has been taken over by the ISO/IEC 17000 standards at international level.
As stated in the respective clauses covering their scope, the standards specify criteria for bodies irrespective of the sector concerned. However, accreditation is always sought and granted for a defined scope, i.e. for specific conformity assessment activity and, where applicable, the types of test performed and methods used (e.g. ‘Body X is competent to undertake inspections as a type A body in the area of pressure equipment categories of Directive 97/23/EC’) and is never restricted to the mere compliance with the general 17000 standards. Therefore, accreditation on the basis of compliance with the 17000 standards always implies the need for these general criteria to be complemented and further specified by all technical specifications that are relevant for the specific technical area for which the applicant conformity assessment body seeks accreditation. Thus, accreditation implies verification of competence with regard to the current state of the art, and includes assessment on the basis of the standards for conformity assessment bodies and all relevant product- and/or technology-related regulations, standards and other specifications.
6.4. ACCREDITATION ACCORDING TO REGULATION (EC) NO 765/2008
|
6.4.1. NATIONAL ACCREDITATION BODIES
The Regulation foresees that each Member State may appoint one single national accreditation body. Only the national accreditation bodies are allowed to perform accreditation of conformity assessment bodies. No other bodies may claim to provide such services, be it according to harmonised standards or non-harmonised standards. This provision is central to the functioning of accreditation in the EU and to the framework of accreditation set up by the Regulation. Member States are not obliged to set up their own national accreditation body, should they consider it not economically viable to do so or should they not deem it useful to offer accreditation for all activities. This means that at no time more than one accreditation body may be active on the territory of a Member State for a given activity. In order to ensure transparency, Member States are therefore obliged to inform the Commission and other Member States to which national accreditation body of another Member State they are having recourse.
A list of national accreditation bodies is available online (250). National accreditation bodies have to make the activities for which they perform accreditation publicly available.
The Regulation does not prescribe the legal form a national accreditation body should take. This means that the national accreditation body may operate from within a ministry, be a governmental agency or be organised as a private company. The Regulation is, however, very clear in that accreditation is to be operated as a public authority activity and, to this effect, has to be formally recognised by the Member State.
Furthermore, the responsibilities and tasks of the national accreditation body have to be clearly distinguished from those of other national authorities. This provision aims to enhance the independence of the national accreditation body and the impartiality and objectivity of its activities. Should the national accreditation body be part of a larger public structure, such as a ministry, other departments are not allowed to influence accreditation decisions. The process of accreditation has to remain separate from other functions. It is absolutely essential to avoid a conflict of interest of the national accreditation body. This also applies to certain tasks that the national accreditation body may take on. While Decision No 768/2008/EC foresees that the national accreditation body may function as a notifying authority (251), the delegation of powers has to be clearly documented and the conditions for impartiality, namely the separation of tasks within the accreditation body ensured.
Should notification tasks be delegated to the national accreditation body, the body's obligations under the Regulation nevertheless remain applicable. This means that its task remains the assessment of the technical competence of conformity assessment bodies according to the full accreditation process and an accreditation certificate must be issued if the technical competence of the conformity assessment body has been established. The national accreditation body may not perform any other assessments that do not meet these requirements or that meet less strict requirements which would not warrant the issuing of an accreditation certificate.
In other words, if the task of notification is to be delegated to the national accreditation body, only the notification of accredited conformity assessment bodies would be possible. The notification of conformity assessment bodies whose competence has not been assessed against the full accreditation criteria will not be possible where such a delegation has been decided on. This also means that the national accreditation body would not exercise any discretion in the notification of a body — the relevant accreditation certificate would lead to an automatic notification (252).
In addition, when providing accreditation, the national accreditation body has to fulfil a number of conditions in terms of stakeholder representation, its internal management and internal controls. Decisions on assessment have to be taken by a different person to the one who carried out the assessment of the conformity assessment body. The accreditation body has to have enough competent personnel at its disposal to ensure that it can perform its tasks. Procedures have to be in place to make sure that the personnel performs adequately and is competent to carry out its tasks. Also, adequate arrangements to ensure the confidentiality of the information obtained from conformity assessment bodies have to be in place and the accreditation body is obliged not to impose unnecessary burdens on its clients. Accreditation bodies must also have a complaint handling mechanism in place.
Furthermore, the Regulation states that the national accreditation body has to have sufficient resources to fulfil its tasks; this includes on the one hand a sufficient number of competent personnel, but also special tasks such as activities for European and international accreditation cooperation and activities that are required in support of public policy and which are not self-financing. In this respect adequate participation in EA, its committees and the peer evaluation process are of foremost importance. Member States should facilitate the participation of their national accreditation bodies in this kind of activities.
In this vein, national accreditation bodies are also obliged to publish their annual audited accounts. The intentions of this provision go beyond demonstrating sound financial management, for the purposes of peer evaluation. National accreditation bodies must therefore clearly demonstrate that the guiding principles of non-commerciality and sufficient resources for ensuring its competence in all activities are respected. Bearing in mind the overall objective of the Regulation of establishing accreditation as the last level of control in the conformity assessment system, in those cases where the accreditation body is part of a larger structure, this requirement should thus be understood to be a tool to demonstrate compliance with these principles, rather than being used to create unnecessary bureaucratic burdens for Member States. Thus the accreditation bodies situated in ministerial departments must be in a position to present at least their overall budgetary and financial figures covering overall resources and their global and operational expenses; together with any financial policies that apply to them in order to be able to demonstrate that they have sufficient resources to perform their tasks adequately while safeguarding the principle of non-commerciality.
Member States have the responsibility to ensure that their national accreditation bodies meet the requirements foreseen by the Regulation on an on-going basis and to take corrective action if this should not be the case. For this reason they are to take the utmost account of the results of the peer evaluation organised by the European accreditation infrastructure.
6.4.2. NON-COMPETITION AND NON-COMMERCIALITY OF NATIONAL ACCREDITATION BODIES
The Regulation's aim of setting up a coherent framework for accreditation that establishes accreditation as the last level of control is underpinned by the principles of non-commerciality and non-competition.
For this reason, while accreditation is supposed to be a self-supporting activity, it is to be provided on a not-for-profit basis. This means that national accreditation bodies do not have the objective of maximising gains or distributing profits. They may provide their services in return for payments or receive income, but any excess revenue is to be invested in further developing their accreditation activities as long as these correspond to the overall tasks of the accreditation bodies. The primary objective of accreditation remains not to produce any gain but to fulfil a task in the public interest.
Regular excess revenues could be a signal that there is a potential to reduce the tariffs charged for accreditation and to encourage smaller conformity assessment bodies to apply for accreditation. Given the prominence that the Regulation gives to accreditation's not-for-profit character, recital 14 clarifies that accreditation is not to produce any gains to its owners or members. In the case that there should nevertheless be any gains, the situation may be corrected by the reduction of tariffs or the revenue may be reused for the further development of accreditation, so as to avoid any conflict with the not-for-profit principle of the Regulation. One could reasonably expect that any excess revenues generated by an accreditation body could also be used to support the accreditation body's involvement in the accreditation activities in the European, international or public sphere.
Regardless of the legal structure of the national accreditation body, there should thus not be a regular transfer of excess revenue to the owners or members of the national accreditation body — be they public or private. Using accreditation as another form of revenue for the state would in consequence cast serious doubts on its compliance with the Regulation's intentions concerning the not-for-profit character of accreditation
Following the same logic, accreditation is to be established as a clearly distinct activity from any conformity assessment activities. A national accreditation body is therefore not allowed to offer or provide any activities or services that a conformity assessment body offers or provides. Neither may it provide consultancy services, own shares in or otherwise have a financial interest in a conformity assessment body or compete with conformity assessment bodies, so as to avoid any kind of conflict of interest.
Furthermore, to safeguard the principle of non-commerciality the Regulation also foresees that accreditation bodies are not allowed to compete with other accreditation bodies. Within the EU, they are only to be active on the territory of their own Member State. Only in exceptional cases, specified in Article 7.1 of the Regulation (EC) No 765/2008 is cross-border accreditation foreseen. Unless these conditions are met, conformity assessment bodies have to seek accreditation with the national accreditation body of the Member State they are established in. This applies for all conformity assessment activities that take place in Europe and concern products or services that are to be placed on the market (253).
6.5. THE EUROPEAN ACCREDITATION INFRASTRUCTURE
The Regulation provides for the recognition of a European accreditation infrastructure. For the time being, this is the European cooperation for Accreditation (EA), the regional organisation of European national accreditation bodies. EA is central to the implementation of the Regulation and through the peer evaluation system is the body that has the closest oversight of the practical functioning of accreditation in Europe. The Commission and EA have concluded a framework partnership agreement on the basis of which EA carries out its tasks. One of EA's primary tasks is to operate a peer evaluation of national accreditation bodies, in line with international standards and practice, but it also contributes to the wider development, maintenance and implementation of accreditation in the EU. |
6.5.1. SECTORAL ACCREDITATION SCHEMES
At the request of the Commission, EA's tasks may include the development of sectoral accreditation schemes or the acceptance of existing schemes. A sector scheme is a scheme that is based on a relevant standard for a specific product, process, service, etc. and additional requirements that are specific to the relevant sector and/or specific legislation. Accreditation may be called upon to assess the competence of conformity assessment bodies to carry out assessments with regard to such schemes.
EA may contribute to the development of sector schemes and their corresponding evaluation criteria and peer evaluation procedures. EA may also recognise already existing schemes that lay down their evaluation criteria and peer evaluation procedures.
In the case of sector schemes that are linked to EU legislation, the Commission has to ensure that the proposed scheme meets the necessary requirements of the legislation in question in terms of the public interest expressed by this specific legislation.
6.5.2. PEER EVALUATION
One of the most important tasks of EA is the organisation of the peer evaluation system of national accreditation bodies, which is the cornerstone of the European accreditation system.
National accreditation bodies undergo peer evaluations of their systems, procedures and structures at a maximum of 4 year intervals. The aim of the peer evaluation system is to ensure consistency and equivalence of accreditation practices across Europe so that the wider market place, including the national public authorities (254), mutually recognise the services delivered by those bodies that have successfully passed the peer evaluation, and therefore accept the accreditation certificates and the attestations issued by the conformity assessment bodies accredited by them. EA provides an appropriate training system to ensure the coherence of peer evaluation activities and results across Europe. Successful peer evaluation allows a national accreditation body to sign the EA Multilateral Agreement, or to maintain signature status. Under the EA Multilateral Agreement, all signatories are obliged to recognise the equivalence of each other's accreditation systems and the equal reliability of the attestations issued by the conformity assessment bodies accredited by them.
The peer evaluation system is operated at several levels. First of all national accreditation bodies have to meet the requirements of the harmonised standard EN ISO/IEC 17011 ‘Conformity assessment — General requirements for accreditation bodies accrediting conformity assessment bodies’ and the requirements of the Regulation which are not contained in the international accreditation body standard — these are namely the principles of one national accreditation body acting as public authority, non- commerciality and non-competition.
Accreditation bodies then have to demonstrate that they are capable and competent to carry out accreditation in the different fields of conformity assessment serviced by them. These activities are themselves determined by a number of harmonised standards (such as EN ISO/IEC 17025 for testing and calibration laboratories, EN ISO/IEC 17020 for inspection bodies or EN ISO/IEC 17065 for bodies certifying products, services and processes). In addition, peer evaluators have to make sure that the accreditation body takes into account any other requirements in its assessments, which are relevant for the specific conformity assessment activities to be carried out by the bodies which they accredit. These may be the specific requirements contained in conformity assessment schemes, including European and national schemes.
6.5.3. PRESUMPTION OF CONFORMITY FOR NATIONAL ACCREDITATION BODIES
If a national accreditation body can demonstrate as a result of the peer evaluation process that it meets the requirements of the relevant harmonised standard (255), it is presumed to meet the requirements for national accreditation bodies that are outlined in Article 8 of the Regulation.
More importantly — and this is of specific importance for the regulatory sphere — if a national accreditation body has successfully undergone peer evaluation for a specific conformity assessment activity, national authorities are obliged to accept the accreditation certificates issued by this body, as well as any attestations (e.g. test or inspection reports, certificates) issued by conformity assessment bodies accredited by this accreditation body.
6.5.4. EA'S ROLE IN SUPPORTING AND HARMONISING ACCREDITATION PRACTICE ACROSS EUROPE
Following on from EA's role as the organisation that is in charge of the peer evaluation of national accreditation bodies there is a need to arrive at a coherent and equivalent approach to accreditation which then warrants the mutual recognition and acceptance of conformity assessment attestations. This means that EA has to facilitate a common approach to accreditation practice and towards the harmonised standards and the requirements that may be contained in any sector schemes. Therefore, with the involvement of all parties concerned such as stakeholders and national authorities, EA has to develop transparent guidance that its members have to respect when conducting accreditation.
6.6. CROSS-BORDER ACCREDITATION
|
The possibility of a conformity assessment body to request accreditation with a national accreditation body in another Member State is only allowed in a limited number of cases. |
According to Article 7(1) of Regulation (EC) No 765/2008, conformity assessment bodies, whether third-party or first-party/in-house, are required when requesting accreditation to do so with the national accreditation body of the Member State in which they are established. This general rule allows for exceptions: the possibility of a conformity assessment body to request accreditation with a national accreditation body in another Member State is limited to cases where
|
— |
there is no national accreditation body in its own Member State and no other national accreditation body to which recourse is had [Article 7(1)(a)], |
|
— |
the national accreditation body does not offer the requested accreditation service [Article 7(1)(b)] |
|
— |
the national accreditation body has not received a positive outcome in the peer evaluation in relation to the conformity assessment activity for which accreditation is requested, i.e. the national accreditation body is not a signatory to the EA Multilateral Agreement for the accreditation of the conformity assessment activity concerned [Article 7(1)(c)]. |
Article 7(1) of the Regulation is closely linked to and is a logical consequence of the non-competition principle.
The cross-border provision laid down in Article 7 is perceived to be very stringent and unnecessarily burdensome for multi- nationally active conformity assessment bodies having their head office in one Member State with local entities/sites established in other Member States and working under the supervision of the head office and under the same quality system and management, as implying costly duplications of assessments. The risk of suffering a competitive disadvantage compared to third-country organisations is feared. In case of a strict legal interpretation of Article 7, due to their structures, multinational conformity assessment bodies may not benefit from the advantage of one accreditation certificate sufficient for the whole territory of the EU, although avoiding multiple accreditations is one of the objectives of the Regulation.
The duplication of unnecessary assessments and burdens on multinational conformity assessment bodies should be avoided while ensuring adequate control of local entities of conformity assessment bodies. There has to be an exchange of information and effective cooperation between national accreditation bodies for assessment, re-assessment and surveillance of local sites of multinational conformity assessment bodies where necessary. Based on mutual recognition of all assessments carried out by EA members, any duplication of assessments of organisational aspects or requirements should be strictly avoided.
If necessary and on reasoned request, relevant information on carrying out accreditation against national legislative requirements of another Member State and/or requirements set out in relevant national sectoral schemes are to be provided by the local national accreditation body to the national authorities of the other Member State. National authorities of the Member States in which the local national accreditation body is established should be kept informed thereof.
The conformity assessment bodies with local sites (regardless of their legal personality), provided that the latter operate under the same global quality system and management and that the head office has the means to substantially influence and control their activities, can be considered as being only one organisation with regard to the conformity assessment activity carried out. Such a conformity assessment body is therefore allowed to request accreditation with the national accreditation body of the head office whose scope can also cover the activities performed by the local site, including those located in another Member State.
The multi-site accreditation is however only permitted under the Regulation if the accredited conformity assessment body maintains the final responsibility for the activities performed by local sites covered by the scope of the multi-site accreditation. The accreditation certificate issued by the national accreditation body where the head office is established names one legal entity — the head office — and it is this legal entity which holds the accreditation and which is responsible for the accredited activities of the conformity assessment body, including any activity performed by the local site that forms part of the scope of the accreditation. Where these local sites carry out key activities (as listed in EN ISO/IEC 17011), then the accreditation certificate (in its annexes) has to clearly identify the address of these site offices.
The local site is entitled to offer directly to the local market conformity attestations under the multisite accreditation, but only on behalf of the accredited conformity assessment body. These accredited certificates and reports are therefore issued under the accreditation, name and address of the head office without the logo of the local site. However this does not impede mentioning on the conformity assessment certificate or report the contact details of the local site issuing the certificate or report in question.
The multi-site accreditation is meant for use only by companies within the same organisation and where the head office maintains the responsibility for the activities performed and certificates/reports issued by the local sites. The responsibility has to be demonstrated on the basis of contractual or equivalent legal relationships between the head office and the local entity and internal regulations that further specify these relationships in terms of management and responsibilities.
The solution of the multi-site accreditation can be applied to all types of local entities (subsidiaries, branches, agencies, offices, etc.), regardless of their legal personality and is in principle valid for all types of conformity assessment bodies, including laboratories, inspection and certification bodies as long as they carry out clearly identified and relevant activities for the purpose of accreditation.
The multi-site accreditation solution is excluded when the above mentioned conditions are not fulfilled, i.e. the conformity assessment body cannot be considered as one organisation with regard to conformity assessment and the head office does not maintain the ultimate responsibility for the activities of the local entities. In this case the local sites being separate legal entities should apply for their own accreditation with the local national accreditation body. As a consequence it can be considered that the local entity carries out the conformity assessment service completely independently of the head office.
In case of the multi-site accreditation, initial assessment and reassessments must be carried out in close cooperation between the respective local national accreditation body and the national accreditation body of the head office taking the accreditation decision, while surveillance must be carried out in cooperation with or by the local national accreditation body. The multinational conformity assessment body must fully cooperate with the national accreditation bodies involved. Local entities cannot reject the participation of the local national accreditation body in the assessment, reassessments and surveillance process. Harmonised rules for cooperation between national accreditation bodies exist in the form of the EA cross frontier policy. Multi-site accreditation has to be managed under the EA cross frontier policy in order to guarantee the involvement of the local national accreditation body.
The multi-site accreditation does not supersede sub-contracting, which remains a viable solution in case a conformity assessment body may wish to sub-contract part of its activities to legal entities located and operating in the same or other Member States, which however do not belong to the same organisation, i.e. are not part of a multinational conformity assessment body. In this case, the subcontractor is not covered by the accreditation of the conformity assessment body. The accredited conformity assessment body may subcontract specific parts of its conformity assessment activities to a different legal entity according to the applicable conformity assessment body standard to which it is accredited and only to the extent allowed in this standard. The conformity assessment body must be able to demonstrate to the national accreditation body that the subcontracted activities are carried out in a competent and reliable manner consistent with the applicable requirements for the activities in question. The accredited conformity assessment attestation must be issued exclusively under the name and responsibility of the accredited conformity assessment body, i.e. the legal entity holding the accreditation. The contractual relationship with the client remains with the accredited conformity assessment body.
6.7. ACCREDITATION IN THE INTERNATIONAL CONTEXT
|
At international level, cooperation between accreditation bodies takes place within the International Accreditation Forum (IAF) and within the International Laboratory Accreditation Cooperation (ILAC). |
6.7.1. COOPERATION BETWEEN ACCREDITATION BODIES
Accreditation as an impartial means of assessing and conveying formal demonstration of the technical competence, impartiality and professional integrity of conformity assessment bodies is an effective quality infrastructure tool used worldwide.
At international level, cooperation between accreditation bodies takes place within two organisations: namely within the International Accreditation Forum (IAF) between accreditation bodies accrediting certification (products and management systems) bodies and within the International Laboratory Accreditation Cooperation (ILAC) between accreditation bodies accrediting laboratories and inspection bodies. Both organisations provide for multilateral mutual recognition arrangements between its accreditation body members. IAF manages a Multilateral Recognition Arrangement (MLA), while ILAC operates a Mutual Recognition Arrangement (MRA). These multilateral mutual recognition arrangements/agreements of competence at technical level between accreditation bodies have the ultimate aim to allow products and services accompanied by accredited conformity attestations to enter foreign markets without the need for re-testing or re-certification in the import country. The objective of such recognition arrangement/agreements between accreditation bodies is therefore to contribute to reinforce the acceptance of conformity assessment results.
At the regional level, to date (256), cooperation organisations between accreditation bodies have been established in:
|
— |
Europe: European cooperation for accreditation (EA) |
|
— |
America: Inter America Accreditation Cooperation (IAAC) |
|
— |
Asia — Pacific: Asia Pacific Laboratory Accreditation Cooperation (APLAC) and Pacific Accreditation Cooperation (PAC) |
|
— |
Africa: Southern African Development Community Accreditation (SADCA) |
|
— |
Africa: African Accreditation Cooperation (AFRAC) |
|
— |
Middle East: Arab Accreditation Cooperation (ARAC) |
Except for SADCA, AFRAC and ARAC which are currently developing its regional mutual recognition arrangement, the above listed cooperation organisations have agreements/arrangements in place within their region which the ILAC/IAF arrangements build upon. By granting special recognition IAF accepts the mutual recognition arrangements established within EA, IAAC and PAC: accreditation bodies being member of IAF and signatories to the EA Multilateral agreement (EA MLA) or the PAC Multilateral Recognition Arrangement (PAC MLA) are automatically accepted into the IAF MLA. ILAC accepts the mutual recognition arrangements and underlying evaluation procedures of EA, APLAC, and IAAC. Accreditation bodies which are not affiliated to any recognised regional cooperation entity may apply directly to ILAC and/or IAF for evaluation and recognition.
The requirements which the Regulation sets for accreditation bodies are in line with the globally accepted requirements laid down in the relevant international standards, although some of them can be perceived as being more rigorous. In particular
|
— |
Accreditation is carried out by one single national accreditation body appointed by its Member State (Art. 4.1) |
|
— |
Accreditation is performed as a public authority activity (Art. 4.5) |
|
— |
National accreditation bodies operates free from commercial motivations (Art. 8.1) and on a not-for- profit basis (Art. 4.7) |
|
— |
National accreditation bodies do not compete with conformity assessment bodies or among each other (Art. 6.1 and Art. 6.2) |
|
— |
Cross-border accreditation Article 7 (within the EU and EEA) |
6.7.2. THE IMPACT ON TRADE RELATIONS IN THE FIELD OF CONFORMITY ASSESSMENT BETWEEN THE EU AND THIRD COUNTRIES
The ultimate acceptance of conformity assessment attestations is decided by the public authorities in the regulatory sphere and, from an economic point of view, by industry users and consumers. The voluntary multilateral mutual recognition agreements between accreditation bodies taking place at technical level support, further develop and enhance intergovernmental trade agreements.
The requirements set out above affect the acceptance of non-European certificates and test results accredited by non-European accreditation bodies not complying with EU requirements but signatories to the ILAC/IAF MRA/MLA in the following way:
|
— |
Conformity assessment delivered in the voluntary sphere It is up to the non-European conformity assessment body operating on the European market to decide if and where to get accredited. In order to boost the acceptance of its conformity assessment attestations by the European market (industry as purchasers of conformity assessment services and ultimately consumers) the non-European conformity assessment body opting for accreditation may choose whether to resort to the service of a third country accreditation body not necessarily conforming to the new European requirements but signatory to the ILAC/IAF MRA/MLA or rather to that of an accreditation body established in the Union. Non-European conformity assessment attestations issued under accreditation by non- European accreditation bodies not fulfilling European requirements can continue to be used on the European market but only in the voluntary sphere. |
|
— |
Conformity assessment delivered in the mandatory sphere Where conformity assessment is required in regulations, national authorities of EU Member States may refuse to accept attestations of conformity issued under accreditation by non-European accreditation bodies not complying with the EU requirements even though they may be signatories to the ILAC/IAF MRA/MLA. However this refusal cannot be based on the sole argument of the non-fulfilment of the EU requirements by the third country accreditation body. The conformance to the EU requirements by the third country accreditation body is not a condition for acceptance of conformity assessment results, but such non-conformance could reinforce doubt as to the quality and value of the accreditation and therefore as to the quality and value of the accredited certificates or reports. |
However, where government-to-government Mutual recognition agreements (MRAs) between the Union and a third country in relation to conformity assessment are in place, national authorities of EU Member States will accept the test reports and certificates issued by bodies that the foreign party has designated under the MRA for assessing conformity in the categories of products or sectors covered by the MRA. The products accompanied by such conformity attestations can be exported and placed on the other party's market without undergoing additional conformity assessment procedures. Each importing party agrees, by the terms of the MRA, to recognise the conformity assessment attestations issued by agreed conformity assessment bodies of the exporting party, independently of whether accreditation has been used to back up the designation process of the conformity assessment bodies under the MRA or not, and independently of, in case accreditation is used by the non-European Party, the fulfilment by the third Party accreditation body of the EU requirements.
7. MARKET SURVEILLANCE
Under Regulation (EC) No 765/2008 national market surveillance authorities have clear obligations to proactively control products made available on the market, to organise themselves and ensure coordination between themselves at the national level and to cooperate at the EU level (257). Economic operators have the clear obligation to cooperate with the national market surveillance authorities and to take corrective action where necessary. National market surveillance authorities have the authority to take sanctions which can include the destruction of products.
Regulation (EC) No 765/2008 integrates the provisions of Regulation (EC) No 339/93 on control of products from third countries. Such controls are now part and parcel of market surveillance activities and Regulation (EC) No 765/2008 obliges national market surveillance and customs authorities to cooperate in order to ensure a seamless system. Such controls must be carried out in a non-discriminatory manner in line with the WTO rules and under the same rules and conditions as set out for internal market surveillance controls.
The European Commission has the responsibility to facilitate the exchange of information between national authorities (in relation to their national market surveillance programmes, their risk assessment methodologies, etc.) in order to ensure that market surveillance is effectively EU-wide and that Member States can pool together their means.
7.1. WHY DO WE NEED MARKET SURVEILLANCE?
|
Member States have to take appropriate measures to prevent the making available on the market and use (258) of non-compliant products. |
Market surveillance aims at ensuring that products fulfil the applicable requirements providing a high level of protection of public interests such as health and safety in general, health and safety in the workplace, protection of consumers, protection of the environment and security while ensuring that the free movement of products is not restricted to any extent greater than that which is allowed under Union harmonisation legislation or any other relevant Union rule. Market surveillance entitles citizens to an equivalent level of protection throughout the single market, regardless of the origin of the product. Further, market surveillance is important for the interest of economic operators, because it helps to eliminate unfair competition.
Market surveillance activities are not directed exclusively towards the protection of health and safety but are additionally undertaken with the aim of enforcing Union legislation which seeks also to safeguard other public interests, for example by means of regulating the accuracy of measurement, electromagnetic compatibility, energy efficiency, consumer and environment protection, following the principle of ‘high level of protection’ as laid down in Article 114(3) TFEU.
Member States must ensure effective surveillance of their market. They are required to organise and carry out the monitoring of the products made available on the market or imported. Member States have to take appropriate measures to ensure that the provisions of Regulation (EC) No 765/2008, of Directive 2001/95/EC and of the other Union harmonisation legislation, as well as non-harmonised, national legislation, in force are respected in the EU and, in particular, to prevent the making available on the market and use of non-compliant and/or unsafe products.
Market surveillance should enable unsafe products or products which otherwise do not conform to applicable requirements set out in Union harmonisation legislation to be identified and kept or taken off the market and unscrupulous or even criminal operators punished. It should also act as a powerful deterrent (259). For that purpose Member States must:
|
— |
correctly implement the provisions of the relevant legislation and allow for sanctions proportional to any infringements, |
|
— |
survey the products (whatever their origin) introduced on their market in order to ensure that they have been subjected to the necessary procedures, that the marking and documentation requirements have been respected and that they have been designed and manufactured in accordance with the Union harmonisation legislation requirements. |
In order to be effective, the market surveillance effort should be uniform across the Union. This is all the more important considering that each point of the Union's external border constitutes an access point for a great quantity of products from third countries. If market surveillance is ‘softer’ in some parts of the Union than others, weak spots are created which threaten the public interest and create unfair trade conditions. Consequently, there must be effective market surveillance along the entire length of the Union's external borders.
In order to guarantee the necessary objectivity and impartiality, market surveillance must be undertaken by the authorities of the Member States. Certain checks (e.g. tests, inspections) can be delegated to other bodies, but the official authorities must retain full responsibility for the decisions taken following these checks. Controls carried out within the framework of market surveillance may be carried out at different times during the life-cycle of a product, following its placing on the market, such as distribution, putting into use or final use. It can, therefore, be exerted in various locations, e.g. importers establishments, wholesale or retail distributors, hire companies, users, etc.
7.2. CONTROLS BY MARKET SURVEILLANCE AUTHORITIES
|
Market surveillance authorities shall check the compliance of the product with the legal requirements applicable at the moment of the placing of the market or, if relevant, putting into service.
Thus, market surveillance does not formally take place during the design and production stages, which is before the manufacturer has taken formal responsibility for the conformity of the products, usually by affixing the CE marking. However, nothing prevents market surveillance authorities and economic operators to collaborate during the design and production phase. Such collaboration may help taking preventive actions and identifying as early as possible safety and conformity issues (260).
Other exceptions to the principle that market surveillance can only take place after the manufacturer has taken formal responsibility for the products are trade fairs, exhibitions and demonstrations. Most Union harmonisation legislation allows the showing and display of non-CE marked products at trade fairs, exhibitions and demonstrations, provided that a visible sign clearly indicates that the products may not be marketed or put into service until they have been made to comply, and that adequate measures are taken during demonstrations, where appropriate, to ensure the protection of public interests. Market surveillance authorities must monitor that this obligation is respected.
For market surveillance to be efficient, resources should be concentrated where risks are likely to be higher or non-compliance more frequent, or where a particular interest can be identified. Statistics and risk assessment procedures can be used for this purpose. To be able to monitor products on the market, market surveillance authorities must have the power, competence and resources:
|
— |
to regularly visit commercial, industrial and storage premises, |
|
— |
to regularly visit, if appropriate, work places and other premises where products are put into service (261), |
|
— |
to organise random and spot checks, |
|
— |
to take samples of products, and to subject them to examination and testing, and |
|
— |
to require, upon reasoned request, all necessary information. |
The first level of control are documentary and visual checks, for example regarding the CE marking and its affixing, the availability of the EU declaration of conformity, the information accompanying the product and the correct choice of conformity assessment procedures. More profound checks may be however necessary to verify the conformity of the product, for example regarding the correct application of the conformity assessment procedure, the compliance with the applicable essential requirements, and the contents of the EU declaration of conformity.
In practice, individual market surveillance activities can focus on certain aspects of the requirements. Besides market surveillance activities that have as their explicit aim the verification of products made available on the market, other public mechanisms exist that, although not directly designed for that aim, can nevertheless have as a consequence the uncovering of non-compliance (262). Labour inspectorates that check safety at the workplace, for example, can discover that the design or construction of a machine, or personal protective equipment bearing the CE marking, is not in conformity with the applicable requirement (263).
Information on the compliance of a product at the moment when it was placed on the market can also be obtained during in-use inspections, or by analysing the factors that caused an accident. Complaints from consumers or other users about the product, or from manufacturers or distributors about unfair competition can also provide information for market surveillance purposes.
Monitoring of products made available on the market may be divided between several authorities on the national level, for example functionally or geographically. Where the same products are subject to control by more than one authority (for example customs and a sectoral authority, or local authorities), coordination between services within a Member State is necessary.
Voluntary initiatives, such as product certification or application of a quality management system, cannot be put on the same footing as market surveillance activities carried out by an authority. Still, they can contribute to the elimination of risks and non-compliances. However, market surveillance authorities must be impartial regarding all voluntary marks, labels and arrangements, and they may only be taken into consideration, in a transparent and non-discriminatory way, for the risk and compliance assessment. Accordingly, products should not be excluded from market surveillance operations even if they have been subject to voluntary certification or other voluntary initiatives.
Union harmonisation legislation provides for two different tools that enable market surveillance authorities to receive information on the product: the EU declaration of conformity and the technical documentation. These must be made available by the manufacturer, the authorised representative established within the Union or under certain circumstances by the importer (264).
Other natural or legal persons, such as distributors cannot be obliged to make these available (265). However, they are expected to assist the market surveillance authority in obtaining them. Further, the market surveillance authority may request the notified body to provide information on the conduct of conformity assessment for the product in question.
The EU declaration of conformity must be made available for the market surveillance authority without delay upon reasoned request (266). It shall accompany the product where required so by specific Union harmonisation legislation. It can be made available for surveillance purposes in each of the Member States, for instance, by means of administrative cooperation.
The technical documentation must be made available to the market surveillance authority within a reasonable period of time, in response to a reasoned request. The authority cannot request it systematically. In general, it can be requested during random checks made for market surveillance purposes, or when there are grounds for a concern that a product does not offer the level of protection required in all respects.
More detailed information (for example certificates and decisions from the notified body) can, nevertheless, be requested in cases of doubt about the conformity of the product to the applicable Union harmonisation legislation. The full technical documentation should be requested only where clearly necessary, and not, for example, when only a detail has to be checked.
This request has to be evaluated in accordance with the principle of proportionality and, thus, taking into account the need to ensure the health and safety of persons or other public interests foreseen in the applicable Union harmonisation legislation, as well as to protect the economic operators from unnecessary burden. Furthermore, failure to present the documentation in response to a reasoned request by a national market surveillance authority, within an acceptable delay, may constitute sufficient grounds for doubting the conformity of the product with the essential requirements of the applicable Union harmonisation legislation.
In the case of a reasoned request it is sufficient for the manufacturer to provide the part of the technical documentation related to the claimed non-conformity and appropriate for demonstrating whether the issue has been dealt with by the manufacturer. Therefore, the request for translation of technical documentation should be limited to these parts of the documentation. If the market surveillance authority considers a translation necessary, it must clearly indicate the part of the documentation to be translated and allow reasonable time for this to take place. No further conditions may be imposed on the translation, such as a requirement of a translator accredited or recognised by the public authorities.
National authority might accept a language they understand and which is different from the national language(s). The language chosen could be a third language, if accepted by that authority.
It must be possible to make the technical documentation available in the Union. However, it does not need to be kept inside the Union, unless otherwise provided for in the applicable Union harmonisation legislation. The requirement for making it available does not mean that the person who bears this responsibility has to store it himself (267), as long as he is capable of presenting it on request from the national authority. The name and address of the person storing the documentation does not need to be expressly mentioned on the product or on its packaging, unless otherwise specified. Further, the technical documentation can be kept and sent to market surveillance authorities in paper or electronic form, which allows it to be made available within a period of time commensurate with the risk or non-compliance in question. Member States must ensure that everyone receiving information about the contents of the technical documentation during market surveillance activities is bound to confidentiality according to principles laid down in the national legislation.
Information on procedures followed by market surveillance authorities, as well as to corrective measures and sanctions can be found in Sections 7.4.4-7.4.6.
7.3. CONTROL OF PRODUCTS FROM THIRD COUNTRIES BY CUSTOMS
Points of entry to the EU are relevant to stop non-compliant and unsafe products coming in from third countries. Being the place where all products from third countries have to pass by, they are the ideal place to stop unsafe and non-compliant products before they are released for free circulation and subsequently circulate freely within the European Union. Thus, customs have an important role in supporting market surveillance authorities in carrying out product safety and compliance controls at the external borders.
The most effective way to avoid the making available of non-conforming or unsafe imported from third countries on the Union market is to carry out adequate checks during the import control process. This requires involvement of customs and cooperation between customs and market surveillance authorities.
The authorities in charge of the control of products entering the Union market, customs or market surveillance authorities depending on the national organisational structure, are very well placed to carry out initial checks, at the first point of entry, on the safety and compliance of the imported products. There are specific guidelines for import controls in the area of product safety and compliance (268). To ensure such controls, the authorities in charge of controls of products at the external borders need an appropriate technical support in order to carry out the checks on the characteristics of the products on an adequate scale. They can perform documentary, physical or laboratory checks. They also need appropriate human and financial resources.
Regulation (EC) No 765/2008 on checks for conformity with Union harmonisation legislation in the case of products imported from third countries requires the customs authorities to be closely involved in the market surveillance activities and information systems provided for under EU and national rules. Article 27(2) of Regulation (EC) No 765/2008 foresees the obligation for cooperation between customs officers and market surveillance officers. Obligations for cooperation are also included in Article 13 of the Community Customs Code which establishes that controls performed with customs and other authorities are undertaken in close cooperation between each other. In addition, the principles of cooperation between the Member States and the Commission established in Article 24 of the Regulation are extended to authorities in charge of external controls, when relevant (Article 27(5)).
Cooperation at national level should allow for a common approach taken by customs and market surveillance authorities during the control process. This should not be hampered by the fact that various ministries and authorities may be responsible for the implementation of Regulation (EC) No 765/2008.
Customs authorities have the following responsibilities under Regulation (EC) No 765/2008:
|
— |
to suspend the release of products when there is a suspicion that the products present a serious risk to health, safety, environment or other public interest and/or do not fulfil documentation and marking requirements and/or the CE marking has been affixed in a false or misleading manner(Article 27(3)), |
|
— |
not to authorise the release for free circulation for the reasons mentioned in Article 29, |
|
— |
to authorise the release for free circulation for any product in compliance with the relevant Union harmonisation legislation and/or nor presenting risks to any public interest, |
|
— |
where the release for free circulation has been suspended, customs have to immediately notify the competent national market surveillance authority which is given 3 working days to perform a preliminary investigation of the products and to decide:
|
Customs authorities must notify their decisions to suspend release of a product to the market surveillance authorities, which in turn must be in a position to take appropriate action. Four hypotheses must be distinguished as from the moment of the notification.
|
1. |
The products in question present a serious risk If the market surveillance authority ascertains that the products present a serious risk, it must prohibit their placing on the EU market. The market surveillance authorities have to request the customs authorities to mark the commercial invoice accompanying the product, and any other relevant accompanying document, with the words ‘Dangerous product — release for free circulation not authorised — Regulation (EC) No 765/2008’ (269). Member State authorities may also decide to destroy the products or otherwise render them inoperable, where they deem it necessary and proportionate. The market surveillance authority must use in those cases the system for rapid exchange of information — RAPEX (270). As a consequence, market surveillance authorities in all Member States are informed, and they may in turn inform the national customs authorities about products imported from third countries, which display characteristics giving rise to a serious doubt as to the existence of a serious risk. This information is of particular importance for customs authorities where it involves measures banning or withdrawing from the market products imported from third countries. Feedback from market surveillance authorities on whether goods are considered as unsafe or non-compliant is crucial for customs risk management and control processes. It ensures controls can be concentrated on risky consignments, allowing for the facilitation of legitimate trade. Furthermore, when non-compliant or unsafe products are found in the internal market, it is often extremely difficult to identify how they entered the EU. Cooperation between customs and market surveillance authorities is encouraged to improve tracing in those cases. |
|
2. |
The products in question do not comply with Union harmonisation legislation In this case the market surveillance authorities must take appropriate measures, if necessary prohibiting the placing on the market under the rules in question. In cases where placing on the market is prohibited, they must ask the customs authorities to mark the commercial invoice accompanying the products, and any other relevant accompanying document, with ‘Product not in conformity — release for free circulation not authorised — Regulation (EC) No 765/2008’ (271). |
|
3. |
The products in question do not present a serious risk and cannot be considered as not conforming to the Union harmonisation legislation. In this case the products must be released for free circulation, provided that all the other conditions and formalities regarding release for free circulation are met. |
|
4. |
The customs authorities have not been notified of any action taken by the market surveillance authorities. If, within 3 working days of the suspension of release for free circulation, the market surveillance authority has not notified customs of any action taken by them, the product has to be released for free circulation provided that all the other requirements and formalities pertaining to such release have been fulfilled. |
The entire procedure from the suspension until the release for free circulation or its prohibition by customs should be completed without delay to avoid creating barriers for legitimate trade but does not necessarily have to be completed within 3 working days. The suspension of release can remain valid for the time required by the market surveillance authority to carry out appropriate checks on the products and allow them to take the final decision. Market surveillance authorities must ensure that the free movement of products is not restricted to any extent greater than that which is allowed under Union harmonisation legislation or any other relevant EU legislation. To that end market surveillance authorities perform their activities regarding products originating from third countries — including the interaction with the relevant economic operators — with the same urgency and methodologies as for products originating from within the EU.
In this case, the market surveillance authority notifies customs within these 3 working days that their final decision on the goods is pending. The release for free circulation has to remain suspended until the market surveillance authority has made a final decision. That notification empowers customs to extend the initial suspension period. The products will remain under customs supervision even if they are allowed to be stored at another place approved by customs.
7.4. MEMBER STATES RESPONSIBILITIES
|
7.4.1. NATIONAL INFRASTRUCTURES
Market surveillance is the responsibility of public authorities. This is, in particular, to guarantee the impartiality of market surveillance activities. Each Member State can decide upon the market surveillance infrastructure, for example there is no limitation on the allocation of responsibilities between authorities on a functional or geographical basis as long as surveillance is efficient and covers the whole territory. Member States organise and carry out market surveillance through the establishment of market surveillance authorities (272). Market surveillance authorities are the authorities of a Member State responsible for carrying out market surveillance on their territory. Surveillance of the market by public authorities is a fundamental element for the good implementation of Union harmonisation legislation.
Member States must ensure that the public is aware of the existence, responsibilities and identity of national market surveillance authorities, and of how those authorities may be contacted. They must also ensure that consumers and other interested parties are given an opportunity to submit complaints to the competent authorities and that these complaints are followed up appropriately.
Member States must entrust market surveillance authorities with the powers, resources and knowledge necessary for the proper performance of their tasks. This is to monitor products made available on the market and, in case of products presenting a risk or other form of non-compliance, to take appropriate action to remove the risk and enforce conformity. As regards personnel resources, the authority has to have, or have access to, a sufficient number of suitably qualified and experienced staff, with the necessary professional integrity. The market surveillance authority should also be independent, and carry out its activities in an impartial and non-discriminatory way. Further, the market surveillance authority should carry out market surveillance respecting the principle of proportionality, for example action must be in accordance with the degree of risk or non-compliance and the impact on the free circulation of products may not be more than is necessary for achieving the objectives of market surveillance.
The market surveillance authority may subcontract technical tasks (such as testing or inspection) to another body, provided that it retains the responsibility for its decisions, and provided there is no conflict of interest between the other body's conformity assessment activities carried out of behalf of economic operators and compliance assessment provided to the market surveillance authority. In doing so the market surveillance authority should exercise great care to ensure that the impartiality of the advice it receives is beyond reproach. The responsibility for any decision to be taken on the basis of such advice should reside in the market surveillance authority.
7.4.2. NATIONAL MARKET SURVEILLANCE PROGRAMMES (NMSP) AND REVIEWS OF ACTIVITIES
National authorities are obliged by Article 18(5) of the Regulation (EC) No 765/2008 to establish, implement and periodically update and communicate their NMSP (273). Programmes may be general and/or sectoral. They should ensure that the overall EU market surveillance framework is respected. Member States must also communicate the programmes to other Member States and to the Commission and make them accessible to the public via internet, without information that could hamper the effectiveness of the programme if made public. The purpose of these programmes is to allow the other countries' authorities, as well as citizens in general, to understand how, when, where and in which areas market surveillance is carried out. National programmes then contain information on activities planned to improve the general organisation of market surveillance at national level (e.g. mechanisms of coordination between different authorities, resources attributed to them, working methods, etc.) and initiatives in specific areas of intervention (e.g. product categories, risk categories, types of users, etc.) (274). Both types of information are necessary.
The Commission helped Member States by proposing common templates to lay out their programmes. The use of all relevant templates is recommended to ensure completeness of information provided. This also facilitates the comparability of national market surveillance programmes in specific product or legislation areas and makes it possible for market surveillance authorities to plan cross-border cooperation in areas of common interest.
When establishing national market surveillance programmes, market surveillance authorities should take the needs of customs into account. Programmes should take into consideration the balance between proactive and reactive control activities and any other factors which may influence enforcement priorities. Resource capabilities must be ensured at the border for this purpose.
According to Article 18(6) of the Regulation (EC) No 765/2008, the functioning of market surveillance activities needs to be periodically reviewed and assessed by Member States, at least every 4 years. The results of this assessment are then communicated to the Commission and other Member States and made available to the public (275).
7.4.3. PUBLIC INFORMATION
Considering that the aim of market surveillance is to provide a high level of protection of certain public interests, informing the public is an essential element of market surveillance. Therefore, Member States should ensure openness to the public and to interested parties and should ensure public access to the information available to the authorities on product conformity. In accordance with the principle of transparency, information available to the authorities of the Member States or the Commission relating to risks to health and safety or other public interests protected under EU harmonisation legislation posed by products should in general be available to the public, without prejudice to the restrictions required for protecting patents and other confidential business information as well as preserving personal data, and for monitoring and investigation and prosecution activities. (276)
The public should be aware of the existence, responsibilities and identity of national market surveillance authorities, and of how those authorities may be contacted. Also national market surveillance programmes and reviews of activities carried out have to be made available to the public by way of electronic communication and, where appropriate, by other means.
Among the measures that market surveillance authorities have to take, is the obligation to alert users within their territories within an adequate timeframe of hazards they have identified relating to any product so as to reduce the risk of injury or other damage particularly when the economic operator responsible fails to do so.
7.4.4. MARKET SURVEILLANCE PROCEDURES
Market surveillance is carried out through the implementation of a sequence of procedures whose aim is to ensure that an effective and consistent system of market surveillance is established across the EU. Market surveillance authorities follow these procedures when dealing with products presenting a risk to the health and safety or persons or to other aspects of public interest protection, according to Article 16(2) of Regulation (EC) No 765/2008 and in line with Articles R31 and R32 in Annex I to Decision No 768/2008/EC, and with products presenting a serious risk requiring rapid intervention, according to Articles 20 and 22 of Regulation (EC) No 765/2008.
An initial event suggesting to market surveillance authorities that a product presents a risk to the health or safety of persons or to other aspects of public interests may trigger the need for closer scrutiny of the product. It may be an accident, the reception of complaints, ex officio initiatives of market surveillance authorities (including custom authorities' control of products entering the EU) as well as information from economic operators on products presenting a risk. When there are sufficient reasons to believe that a product presents a risk, market surveillance authorities carry out an evaluation of compliance with the requirements of the relevant Union harmonisation legislation. They have to perform appropriate checks (both documentary and physical/laboratory checks, as necessary) on the characteristics of the products, duly taking into account the reports and conformity assessment certificates issued by an accredited conformity assessment body provided by the economic operators.
Market surveillance authorities carry out a risk assessment in order to verify if products present a serious risk. According to Article 20(2) of the Regulation an appropriate risk assessment ‘takes account of the nature of the hazard and the likelihood of its occurrence’ (277).
If a product presents a risk to the health or safety of persons or to other aspects of public interests, market surveillance authorities must request without delay to relevant economic operators to:
|
(a) |
take any action to bring the product into compliance with the applicable requirements laid down in the Union harmonisation legislation; and/or |
|
(b) |
withdraw the product; and/or |
|
(c) |
recall the product; and/or |
|
(d) |
stop or restrict supplying the product within a reasonable period. |
In case the risk is deemed to be ‘serious’, market surveillance authorities must adopt a rapid intervention following the specific provisions of Articles 20 and 22 of the Regulation.
The economic operators must ensure that the corrective action is taken throughout the EU. The market surveillance authorities must also inform the relevant notified body (if any) on the decision taken. In case of serious risk requiring a rapid intervention, the market surveillance authority may adopt restrictive measures without waiting for the economic operator to take corrective action to bring the product into compliance. According to Article 21 of the Regulation, the measures adopted by market surveillance authorities have to be proportionate and communicated to the relevant economic operator without delay. The market surveillance authorities must also consult the economic operator prior to the adoption of the measures and, if such consultation is not possible because of the urgency of the measures to be taken, the operator must be given the opportunity to be heard as soon as possible. The market surveillance authorities must withdraw or amend the measures taken if the economic operator demonstrates that he has taken effective action.
When non-compliance is not limited to the national territory, market surveillance authorities must inform the Commission and the other Member States about the results of the compliance evaluation and about the actions required of the economic operator or the measures adopted. In case of serious risk, market surveillance authorities notify to the Commission through the RAPEX system of any voluntary or compulsory measure according to the procedure laid down in Article 22 of the Regulation and/or Article 12 of the GPSD. In the case of products that do not present a serious risk, the Commission and the other Member States will be informed by means of the information support system indicated in Article 23 of the Regulation and/or Article 11 of the GPSD. Market surveillance authorities have to verify that adequate corrective measures have been taken. Otherwise, they adopt appropriate provisional measures, informing the Commission and the other Member States with the procedures detailed above.
In order to broaden the effectiveness of the market surveillance activity launched by the notifying Member State, the other Member States are called upon to follow up on the notification by verifying whether the same product has been made available on their territories and by adopting appropriate measures. They should inform the Commission and the other Member States according to the procedures of the initial notification.
Under Union harmonisation legislation aligned to Decision No 768/2008/EC if the Commission and the other Member States do not raise any objection within a certain period, the restrictive measures are deemed justified and must be adopted without delay by the Member States. In the case of non-compliance due to shortcomings in harmonised standards, the Commission informs the relevant standardisation bodies and brings the matter before the Committee set up under Article 22 of Regulation (EU) No 1025/2012. In light of the Committee's opinion, the Commission can decide to: (a) maintain the reference to harmonised standards in the OJ; (b) maintain with restrictions the reference to the harmonised standards in the OJ; (c) withdraw the reference to the harmonised standards in the OJ. The Commission also informs the relevant European standardisation organisation and, if necessary, requests the revision of the harmonised standards concerned.
If objections are raised, the safeguard mechanism will apply.
Additional information on the procedure allowing Member States to exchange information on measures adopted against products presenting a risk and, if appropriate, for their assessment by the European Commission is provided in Sections 7.5.1 and 7.5.2.
7.4.5. CORRECTIVE MEASURES — BANS — WITHDRAWALS — RECALLS
According to Union harmonisation legislation, Member States are required to ensure that products are made available on the market only if they comply with the applicable requirements. The latter include both the essential requirements, and a number of administrative and formal requirements. When competent national authorities discover that a product is not in compliance with the provisions of the applicable Union harmonisation legislation, they must take action to ensure it is brought into conformity or taken off the market.
The corrective action depends on the risk or non-compliance and, thus, must be in accordance with the principle of proportionality. Non-conformity to essential requirements must be considered as a substantial non-compliance, because this may lead to the product presenting a potential or actual risk to the health and safety of persons or to other aspects of public interest. In case of a serious risk, Article 20 of Regulation (EC) No 765/2008 sets out the need of prohibiting products from being made available on the market, withdrawing or recalling products.
If a product covered by Union harmonisation legislation is not CE marked, it is an indication that the product does not comply with the essential requirements or the conformity assessment procedure has not been applied and, consequently, the product may endanger the health and safety of persons or harm other public interests protected by that legislation. Only if, following further investigation, the product proves to be compliant with the essential requirements, the absence of the CE marking is to be considered as a formal non-compliance (i.e. the product does not present a risk).
Unless there are reasons to believe that the product presents a risk, there are cases where non-compliance with a number of administrative or formal requirements are defined as formal non-compliance by Union harmonisation legislation. That is the case for the incorrect affixing of the CE marking as regards, for instance, the design, size, visibility, indelibility or legibility, can usually be considered as a formal non-compliance. Examples of typically formal non-compliance could also be the situations where other conformity markings provided for in the Union harmonisation legislation are incorrectly affixed, or where the EU declaration of conformity cannot be provided for immediately or it does not accompany the product when this is mandatory, or the requirement to accompany other information provided for in sectoral Union harmonisation legislation is complied with insufficiently, or, where applicable, the identification number of the notified body has not been affixed to the CE marking.
Enforcement of conformity can be achieved by obliging the manufacturer, the authorised representative, or other responsible persons (importers, distributors), to take required measures. Corrective action can also take place if the necessary measures are taken (for example the product is modified or withdrawn from the market), either as a result of consultations carried out by the market surveillance authority or as a result of formal or informal warnings. In all cases the market surveillance authority must establish accompanying measures to ensure that conformity is enforced. PROSAFE ‘Guidelines for Businesses to manage Product Recalls & Other Corrective Actions’ (278) have been designed to assist businesses to ensure, whenever necessary, the appropriate corrective actions and follow-up once a product has been already made available on the EU market or is coming from third countries.
Actions to prohibit or restrict the placing on the market may first be temporary to allow the market surveillance authority to obtain sufficient evidence about the risk or other substantial non-compliance of the product.
In case of formal non-compliance only (i.e. without a risk), the market surveillance authority should first oblige the manufacturer, or the authorised representative, to make the product intended to be placed on the market and, if necessary, the product already on the market, comply with the provisions and to remedy the infringement within a reasonable time period. If no result can be achieved, the market surveillance authority has to, ultimately, take a further step to restrict or prohibit the placing on the market of the product and, if necessary, to ensure that it is also withdrawn or recalled from the market.
Any decision taken by national market surveillance authorities to restrict or prohibit the placing on the market or the putting into service, to withdraw or recall the products from the market must state the exact grounds on which it is based. The party concerned — in particular, the manufacturer, or the authorised representative established in the Union — must be notified. They must also be informed about remedies available under the national law in force in the Member State in question, and of the time limits to which such remedies are subjected. (279)
Unless the matter is urgent (for example the product presents a serious risk), the manufacturer, or the authorised representative established in the Union, should have an opportunity to be consulted in advance, before the competent authority takes action to restrict the free circulation of products. In practice, it should be considered as sufficient when the manufacturer or the authorised representative has been provided with an opportunity to react. (280) However, it should not delay the proceedings, if the manufacturer or the authorised representative remains passive.
The decision to restrict the free movement of a CE marked product in case of non-compliance with the essential requirements usually invokes the safeguard clause procedure. This procedure is aimed to enable the Commission to keep an overview of such measures, to consider whether or not they are justified and to ensure all Member States take similar measures in relation to the same products. A manufacturer, the authorised representative, or other economic operator may consider himself to have suffered a loss as a result of an inappropriate national measure that restricted the free movement of a product. In such a case he could be entitled to claim damages under the jurisdiction of the Member State which initiated the procedure and accordingly the Commission, at the end of a safeguard clause procedure, where the national measure is considered as non-justified. This may raise the question whether or not a liability case for incorrect implementation of EU law could take place.
7.4.6. SANCTIONS
Regulation (EC) No 765/2008 requires Member States to ensure the correct implementation of its provisions and to take appropriate action in the event of infringement. The Regulation requires penalties to be proportionate to the seriousness of the offence and constitute an effective deterrent against abuses.
It is up to the Member States to lay down and implement the mechanism for enforcing the provisions of the Regulation in their territories. According to Article 41 of the Regulation, ‘the penalties provided for shall be effective, proportionate, and dissuasive and may be increased if the relevant economic operator has previously committed similar infringement’.
In addition, Union harmonisation legislation aligned to Decision No 768/2008/EC includes as well a provision requiring Member States to lay down penalties for infringements by economic operators of that particular legislation.
Sanctions are imposed by means of fines, whose sums vary from one Member State to the other. They may also include criminal sanctions for serious infringements.
The most common legal instruments providing for sanctions are general product safety acts and/or sector specific legislation. However, in some Member States sanctions are provided in CE Marking acts, customs code or acts on conformity assessment system.
7.5. COOPERATION BETWEEN THE MEMBER STATES AND THE EUROPEAN COMMISSION
Cooperation and coordination of action among national authorities is indispensable to obtain effective and consistent surveillance of the Single Market. The EU legal framework provides a number of tools to achieve this goal. The safeguard mechanism included in Union harmonisation legislation obliges to share information about restrictive measures adopted by national authorities so that, if appropriate, follow up action can be taken by other authorities. Mutual assistance based on Regulation (EC) No 765/2008 allows authorities to enforce request of information vis-à-vis economic operators located in another Member State. Administrative cooperation groups (ADCOs), the ICSMS database, the RAPEX Rapid Alert System constitute essential tools to exchange information and optimise work sharing among authorities.
7.5.1. SAFEGUARD MECHANISMS
|
The safeguard clause procedure, based on Article 114(10) TFEU and included in most sectoral Union harmonisation legislation, authorises Member States to take restrictive measures in relation to products presenting a risk to health and safety or other aspects of public interests protection and obliges them to notify those measures to the Commission and other Member States. The safeguard clause procedure is designed to provide a means to inform all national market surveillance authorities about dangerous products, and, accordingly, to have the necessary restrictions extended to all Member States, so as to ensure an equivalent level of protection throughout the EU. Furthermore, it allows the Commission to take a position on the national measures restricting the free movement of products with a view to ensuring the functioning of the internal market.
It is to be noted that the safeguard procedure is distinct from the RAPEX Rapid Alert System procedure because of their different notification criteria and different methods of application (281).
Where, having performed an evaluation, a Member State finds that a product is non-compliant or a product is in compliance but presents a risk to the health or safety of persons or to other aspects of public interest protection, it must require the relevant economic operator to take all appropriate measures to ensure that the product concerned, when made available on the market, no longer presents that risk, to withdraw the product from the market or to recall it within a reasonable period, commensurate with the nature of the risk, as it may prescribe.
This procedure will be applicable, unless it is established that the risk does not affect a whole series of products manufactured, however limited the series, or that the risk is not due to the product itself but to its misuse, that is, when not used in accordance with their intended purpose or under conditions which can be reasonably foreseen and when not properly installed and maintained.. For an isolated error, limited to the territory of the Member State that has discovered the non-compliance, there is no need to invoke the safeguard clause, since there is no need to take action at EU level. In addition, the risk must be due to the product itself and not to its misuse.
Conformity can be enforced if the national authority requests the manufacturer or the authorised representative to take the necessary measures, or if the product is modified or voluntarily withdrawn from the market. Unless a formal decision is taken in these cases, to prohibit or restrict the making available on the market of the product or to have it withdrawn from the market, the safeguard clause procedure is not invoked. In case there is no compulsory measure; there is no need to invoke the safeguard clause (282).
However, if an economic operator does not take adequate corrective action within the period indicated by a market surveillance authority, the market surveillance authorities have to take all appropriate provisional measures to prohibit or restrict the product's being made available on their national market, to withdraw the product from that market or to recall it.
7.5.2. THE APPLICATION OF SAFEGUARD MECHANISMS STEP BY STEP
|
7.5.2.1. Compulsory restrictive measure taken
The application of the safeguard clause requires that the competent national authority takes a compulsory measure to restrict or forbid the making available on the market and, possibly, the putting into service of the product, or has it withdrawn from the market where the relevant economic operator does not take adequate corrective action himself. The contents of the decision should relate to all products belonging to the same type, batch or series. It must also have binding legal effect: it is followed by sanctions, if not respected, and can be subject to an appeals procedure. Court decisions, which restrict the free movement of CE marked product within the scope of the relevant Union harmonisation legislation, do not invoke the safeguard clause. However, where administrative proceedings initiated by the surveillance authority must be, according to the national law, confirmed by a court, such court decisions are not excluded from the safeguard clause procedure.
The findings that justify the national measure are established either by the market surveillance authority on its own initiative or based on information received from a third party (such as consumers, competitors, consumer organisations, labour inspectorates). Further, the national measure must be based on evidence (for example tests or examinations) that constitutes sufficient proof of errors in the product design or the manufacture to indicate a foreseeable potential or actual danger or other substantial non-compliance, even when the products are correctly constructed, installed, maintained and used in accordance with their intended purpose or in a reasonably foreseeable way. There is a grey zone between correct and incorrect maintenance and use, and it can be considered that, to a certain extent, products should be safe, even if maintained and used for their intended purpose in an incorrect way that can reasonably be expected. In evaluating this, the data supplied by the manufacturer on the labelling, in the instructions, in the user's manual or in promotion materials are to be taken into consideration.
The reason for taking restrictive measures may result, for instance, from differences or failures in the application of essential requirements, incorrect application of harmonised standards or shortcomings in them. The surveillance authority can add or specify other motives (for example failure to comply with good engineering practice) when invoking the safeguard clause, provided that they are directly linked with these three reasons.
Where non-compliance with harmonised standards that give a presumption of conformity is established, the manufacturer, or the authorised representative, must be requested to provide evidence about compliance with essential requirements. The decision of the competent authority to take corrective action must always be based on an established non-compliance with the essential requirements.
The measures taken by authorities have to be proportionate with the seriousness of the risk and the non-compliance of the product and have to be notified to the Commission.
7.5.2.2. Notification to the Commission
As soon as a competent national authority restricts or forbids the free movement of a product in such way that the safeguard clause is invoked, the Member State must immediately notify (283) the Commission indicating the reasons and justification for the decision.
The information has to include all available details, in particular:
|
— |
name and address of the manufacturer, the authorised representative, and in addition — if necessary — the name and address of the importer or other person responsible for making the product available on the market, |
|
— |
the data necessary for the identification of the product concerned, the origin and the supply chain of the product, |
|
— |
the nature of the risk involved and the nature of the national measures taken, |
|
— |
a reference to the Union harmonisation legislation, and in particular to the essential requirements, against which the non-compliance has been established, |
|
— |
a comprehensive assessment and evidence to justify the measure (for example harmonised standards or other technical specifications used by the authority, the test reports and identification of the testing laboratory). In particular, the market surveillance authorities must indicate whether the non-compliance is due to either:
|
|
— |
the arguments put forward by the relevant economic operator. |
If possible, the notification should also include:
|
— |
a copy of the declaration of conformity, |
|
— |
the name and number of any notified body that intervened in the conformity assessment procedure, if applicable, |
|
— |
a copy of the decision taken by the Member State authorities. |
7.5.2.3. Management of the safeguard process by the Commission
Where objections are raised against a measure taken by a Member State (284), or where the Commission considers a national measure to be contrary to Union harmonisation legislation, the Commission must without delay enter into consultation with the Member States and the relevant economic operator or operators and must evaluate the national measure. On the basis of the results of this evaluation, the Commission decides whether the national measure is justified or not.
The Commission addresses its decision to all Member States and immediately communicates it to them and the relevant economic operator or operators.
If the national measure is considered justified, all Member States must take the measures necessary to ensure that the non-compliant product is withdrawn from their market, and must inform the Commission accordingly. If the national measure is considered unjustified, the Member State concerned must withdraw the measure.
Where the national measure is considered justified and the non-compliance of the product is attributed to shortcomings in the harmonised standards, the Commission shall apply the procedure provided for in Article 11 of Regulation (EU) No 1025/2012 concerning the formal objection to harmonised standard.
Member States other than the Member State initiating the procedure must without delay inform the Commission and the other Member States of any measures adopted and of any additional information at their disposal relating to the non-compliance of the product concerned, and, in the event of disagreement with the notified national measure, of their objections. Member States must ensure that appropriate restrictive measures are taken in respect of the product concerned, such as withdrawal of the product from their market, without delay.
Where, within a certain period of time of receipt of the information, no objection has been raised by either a Member State or the Commission in respect of a provisional measure taken by a Member State, that measure should be deemed justified.
Conversely, should the Commission see no justification for the national action that invoked the safeguard clause, it will ask the Member State to withdraw its action and take immediate appropriate steps to re-establish the free movement of the products in question on its territory.
Whether the action taken by the Member State is considered justified or not, in either case, the Commission keeps the Member States informed of the progress and the results of the procedure.
Once the decision is taken by the Commission, it can be legally challenged by Member States on the basis of Article 263 TFEU. The economic operator directly concerned by the Decision may also challenge it on the basis of article 263 TFEU.
If the initiating Member State does not withdraw the measure in case of non-justification, in this case, the Commission will consider initiating the infringement procedure provided for by Article 258 TFEU.
7.5.3. MUTUAL ASSISTANCE, ADMINISTRATIVE COOPERATION AND EXCHANGE OF INFORMATION AMONG MEMBER STATES
|
The proper application of Union law depends on a smooth administrative cooperation to ensure uniform and efficient enforcement of Union legislation in all Member States. The obligation to cooperate is in line with Article 20 of the Treaty on European Union (TEU) which states that Member States must take all appropriate measures to fulfil their obligations (285), and with Article 24 of Regulation (EC) No 765/2008. Although technical harmonisation has created a single market, where products move over national borders, market surveillance is carried out on a national basis. Administrative cooperation mechanisms between nation- al surveillance authorities, therefore, need to be developed to increase the efficiency of surveillance, to minimise the effect of different surveillance practices and to reduce the overlapping of national surveillance operations. Cooperation between market surveillance authorities can also spread good surveillance practice and techniques across the Union, as it allows national authorities to compare their methods with those of other authorities, for example in the framework of comparisons and joint surveys or study visits. In addition, cooperation can be useful for exchanging views and solving practical problems.
Administrative cooperation calls for mutual trust and transparency between national surveillance authorities. Member States and the Commission need to be informed about the way enforcement of Union harmonisation legislation, in particular market surveillance of products is organised throughout the single market. This includes information about national authorities in charge of market surveillance for the different product sectors, and about national market surveillance mechanisms to clarify how monitoring of products made available on the market takes place and what corrective actions and other activities the surveillance authority is entitled to use.
Transparency is also necessary regarding the national rules on confidentiality. For the achievement of effective market surveillance in the Union, it is important that national surveillance authorities assist each other. On request, a national authority should make information available and provide other assistance. Without prior request, a national authority may consider sending to the other national authorities all relevant information concerning operations that constitute, or are likely to constitute, breaches of Union harmonisation legislation, which may have an impact on the territory of other Member States. In addition, the national authorities should communicate to the Commission any information they consider relevant, spontaneously or in response to a reasoned request from the Commission. The Commission may then communicate this information to the other national authorities when considered necessary.
Cooperation and mutual assistance according to Article 24(2) of Regulation (EC) No 765/2008 are, in particular, necessary to ensure that action can be taken against all those who are responsible for a non-compliant product being made available on the market. In some cases the authority of the Member State, where the manufacturer, the authorised representative, or other responsible person is established, needs to be contacted. This is to enforce requests of information made to these economic operators, for example to require the EU declaration of conformity or some specified details from the technical documentation, or to request information concerning the distribution chain, and not followed up by them. The Member State under whose jurisdiction the notified body operates (where applicable) needs to be contacted as well. When a national authority acts due to information it has received from another national body, it should report back to this authority on the outcome of the action.
Moreover, market surveillance would be more efficient, at the Union level, if the national surveillance authorities could agree on how to allocate their resources in such a way that a maximum number of different product types could be covered in each sector. To avoid duplication of product tests, or other investigations for market surveillance purposes, national authorities should exchange summary reports of these tests. This can be done by using the Information and Communication System for Market Surveillance (ICSMS) (286). National surveillance authorities should also consider whether or not there is a special need to carry out technical analyses or laboratory tests when another surveillance authority has already done so, and the results are available to those authorities or may at their request be placed at their disposal (287). It might also be useful to exchange results of periodic inspections on equipment in service, to the extent that they provide information on the compliance of products when they were placed on the market.
Information exchanged between national surveillance authorities has to be covered by professional confidentiality, according to the principles of the national legal system in question, and it has to enjoy the protection extended to similar information under national law. Where a Member States has rules permitting free access by persons to information held by surveillance authorities, this fact must be revealed at the time of the request to another surveillance authority, or during the exchange of information if no such request occurs. If the sending authority indicates that the information involves matters of professional or commercial confidentiality, the receiving authority should ensure that this can be provided for. Otherwise the sending authority is entitled to withhold the information. Coordination and exchange of information between national surveillance authorities need to be agreed by the parties involved and taking into account the needs of the sector concerned. The following principles could be taken into consideration, where appropriate:
|
— |
appointing a national communication point or correspondent for every sector, which would coordinate internally as appropriate, |
|
— |
agreeing about the types of cases for which the communication of surveillance information would serve a useful purpose, |
|
— |
developing a common approach to issues such as the classification of risks and hazards and their coding, |
|
— |
identifying of the details which should be communicated in each case, including the request for further information, |
|
— |
accepting the obligation to respond to enquiries within a given time scale (288), |
|
— |
transmitting information (requests and responses), as simply as possible, by email, or through a telematic system operated by the Commission (ICSMS) or an external body, and by using standard multi-language forms, |
|
— |
taking advantage of up-to-date data recording techniques so that enquiries can be easily undertaken, and |
|
— |
treating the information received in complete confidence. |
Cooperation between national administrations takes place in working groups set up under the Union harmonisation legislation. Discussions mainly focus on interpretation issues, but questions related to market surveillance and administrative cooperation are also dealt with. Administrative cooperation between national authorities carrying out market surveillance is taking place in the following sectors: measuring instruments and non-automatic weighing instruments (WELMEC), low voltage equipment (LVD ADCO), Eco-Design ADCO Group, electromagnetic compatibility (EMC administrative cooperation), machinery, medical devices (Vigilance Working Group and COEN — Compliance and Enforcement Group), PEMSAC (The Platform of European Market Surveillance Authorities for Cosmetics), Toy-ADCO (The Administrative Cooperation Group of toys), telecommunications terminal equipment (TCAM), recreational craft, personal protective equipment, ATEX equipment, Radio and Telecommunications Terminal Equipment (R&TTE), Cableways (CABLE), Energy Labelling (ENERLAB), Gas Appliances (GAD), Lifts (LIFTS), Marine Equipment (MED), Noise, Pressure equipment sector (PED/SVPD), Pyrotechnics (PYROTEC), Chemicals (REACH), Restriction of the use of certain hazardous substances (ROHS), Transportable Pressure Equipment (TPED), Labelling of tyres. There are also groups dealing with more horizontal issues such as PROSAFE (the product safety forum of Europe), the Expert Group on Internal Market for Products (IMP-MSG), a horizontal committee where, for instance, general questions related to the implementation and enforcement of Union harmonisation legislation, such as horizontal aspects of market surveillance, are discussed. The emergencies committees, set up under the GPSD, regularly discusses administrative cooperation issues of general interest.
7.5.4. RAPID ALERT SYSTEM FOR NON-FOOD PRODUCTS PRESENTING A RISK
|
The Rapid Alert System used for non-food products allows 31 participating countries (all EEA countries) and the European Commission to exchange information on products presenting a risk to health and safety or other protected interests and on the measures taken by these countries to do away with that risk. |
Article 12 of the GPSD provides a legal basis for a general and horizontal system for the rapid exchange of information on serious risks arising from the use of products (RAPEX, Rapid Alert System).
The Rapid Alert System covers consumer and professional products (289). It is applicable to non-harmonised products and products covered by the Union harmonisation legislation alike (290).
The Rapid Alert System works according to the detailed procedures laid down in Annex II to the GPSD and in the Rapid Alert System guidelines (291).
With the entry into force of Regulation (EC) No 765/2008, the scope of the Rapid Alert System system was extended to risks other than those affecting health and safety (i.e. risks for the environment and in the work place, security risks) and also to products intended for professional (as opposed to consumer) use. Member States should ensure that products which present a serious risk requiring rapid intervention, including a serious risk the effects of which are not immediate, are recalled, withdrawn or that their being made available on their market is prohibited, and that the Commission is informed without delay through Rapid Alert System under Article 22 of Regulation (EC) No 765/2008.
On 16 December 2009, the Commission adopted Decision 2010/15/EU (292) laying down the new guidelines for the management of the Rapid Alert System. Since guidelines were written before 1 January 2010 they refer explicitly only to notifications based on the GPSD. Nevertheless they are the main reference also for notifications based on Regulation (EC) No 765/2008 (see Article 22(4) therein) — professional products and risks other than health and safety.
The Rapid Alert System procedure is as follows.
|
— |
When a product (e.g. a toy, a childcare article or a household appliance) is found, for instance, to be dangerous, the competent national authority takes appropriate action to eliminate the risk. It can withdraw the product from the market, recall it from consumers or issue warnings. Economic operators can take such measures also voluntarily which has to be reported by the competent authorities as well. The National Contact Point then informs the European Commission (through IT system GRAS- Rapid Alert System (293)) about the product, the risks it poses and the measures taken by the authority or the economic operator to prevent risks and accidents. |
|
— |
The Commission disseminates the information that it receives to the National Contact Points of all other EU and EEA countries. It publishes weekly overviews of products posing a risk and the measures taken to eliminate the risks on the Commission's Rapid Alert System website (294). |
|
— |
The National Contact Points in each EU and EEA country ensure that the authorities responsible check whether the newly notified product is present on the market. If so, the authorities take measures to eliminate the risk, either by requiring that the product is withdrawn from the market, by recalling it from consumers or by issuing warnings. |
The safeguard clause procedures under the Union harmonisation legislation apply in addition to the Rapid Alert System. Accordingly, the Rapid Alert System does not necessarily have to come into play before the safeguard clause procedure is applied. However, the safeguard clause procedure has to be applied, in addition to the Rapid Alert System, when the Member State takes a decision to permanently prohibit or restrict the free movement of CE marked products on the basis of a danger or other serious risk presented by the product.
7.5.5. ICSMS
|
7.5.5.1. Role
ICSMS offers fast and efficient communication means for market surveillance authorities to exchange information within a short space of time. ICSMS allows information on non-compliant products (test results, product identification data, photographs, economic operator information, risk assessments, accident information, information on measures taken by surveillance authorities, etc.) to be quickly and efficiently shared between authorities.
The aim is not only to avoid cases where an unsafe product taken off the market in one country to be on sale for a long time in another country but mainly to have a market surveillance policy tool that allows to establish a cooperation mechanism among authorities.
While being aware of the fact that the mere reliable exchange of information is crucial for the market surveillance, it must be acknowledged that the added value of ICSMS stems from its capacity to be the platform for the implementation of the European market surveillance policy.
In this respect whenever a national authority wants to exchange information about a product under investigation with other authorities in order to share resources (e.g. for product checks), carry out common actions or consult other authorities, it must input into ICSMS the relevant information. This must be done as early as possible and certainly well before the decision to adopt measures for products found to present a risk. E.g. if a national authority cannot determine the level of the risk presented by a relevant product and carries out investigations, it must use ICSMS in order to communicate with the competent authorities of the other Member States.
ICSMS is not limited only to non-compliant products, but it gives information also regarding all products checked by authorities even if the result of the checks would be that no non-compliances have been found. This helps authorities avoiding any double (or multiple) checking of products.
Thus the ultimate role of ICSMS is to help the European Union to fulfil one of its major political objectives; i.e. to ensure reliability and coherence in the implementation and enforcement of the European legislation) in order for operators and citizens to benefit from the original intention of full access to the Internal Market.
In particular ICSMS helps market surveillance authorities to:
|
— |
proceed to quick and in-time exchange of information on market surveillance measures, |
|
— |
coordinate their activities and inspections more effectively, especially by focusing on products which have not been inspected or tested yet, |
|
— |
share resources and have thus more time to concentrate on other products which have yet to be tested, |
|
— |
carry out wide-scale market interventions wherever products of a dubious nature are concerned using the latest information and avoid thus duplicate and multiple inspections, |
|
— |
elaborate best practices, |
|
— |
ensure that market surveillance is efficient and of even rigour in all Member States and avoid thus distortion to competition, |
|
— |
establish an encyclopaedia of EU market surveillance intelligence. |
7.5.5.2. Structure
The internal area is destined for market surveillance authorities, customs authorities and the EU. It contains all information available (product description, test results, measures taken, etc.). Only ICSMS account holders may access this area.
The public area is destined for consumers, users and manufacturers. The information which is visible to the public provides only the data, which reference the product and its non-compliance and not any internal documents (i.e. information exchange between authority and importer/manufacturer).
ICSMS enables specific searches for non-compliant products. Confidentiality aspects are protected by a system of access authorisations.
Each market surveillance authority can input data about investigated products, which are not already in the database and add information (e.g. additional tests results, measures taken) to an already existing product information file.
The Commission ensures the proper functioning of ICSMS. The use of ICSMS is free of charge.
7.5.6. MEDICAL DEVICES: VIGILANCE SYSTEM
|
A specific vigilance system applies in the case of medical devices. |
Risks posed by medical devices have necessitated a comprehensive monitoring system whereby all serious product incidents will be reported (295). The medical devices vigilance system applies to all incidents which might lead to, or might have led to, the death of a patient or a user, or to a serious deterioration in their state of health, and which result from:
|
— |
any malfunction or deterioration in the characteristics or performance of a device, |
|
— |
any inadequacy in the labelling or the instructions for use, or |
|
— |
any technical or medical reason in relation to the characteristics or performance of a device, and which leads the manufacturer to systematically recall all devices of the same type. |
The manufacturer is responsible for activating the vigilance system and must, accordingly, inform the surveillance authority about incidents that invoke it. After the notification, the manufacturer is obliged to make investigations, send a report to the surveillance authority and consider, in collaboration with the authority, what action should be taken.
The manufacturer's notification is followed by an assessment carried out by the surveillance authority, if possible together with the manufacturer. After the assessment, the authority must immediately inform the Commission, and the other Member States, of the incidents for which relevant measures have been taken or are contemplated. The Commission may then take any steps to coordinate, facilitate and support measures taken by the national surveillance authorities when dealing with the same type of incidents, or, if necessary, take measures at Union level (for example envisaging the re-classification of the device). A databank containing, among other information, data obtained in accordance with the vigilance system will be set up and made accessible to the competent authorities. The vigilance system is different from the safeguard clause procedure, since it requires notification even if the manufacturer takes the necessary measures on a voluntary basis. Nevertheless, when applying the vigilance system the surveillance authority is also obliged to adopt a restrictive measure vis-à-vis non-compliant CE marked products, if the conditions for invoking the safeguard clause apply and, accordingly, notify this measure following the safeguard clause procedure. However, the vigilance system does not necessarily have to come into play before the safeguard clause procedure is applied.
8. FREE MOVEMENT OF PRODUCTS WITHIN THE EU (296)
8.1. FREE MOVEMENT CLAUSE
The objective of eliminating trade barriers among Member States and of strengthening the free movement of products is stated by a free movement clause, inserted in Union harmonisation legislation, which guarantees the free movement of products complying with the legislation. Free movement clauses are provisions inserted in EU legislative acts which expressly prevent the Member States from taking more restrictive measures on a matter, if that matter fulfils the requirements of the law in question. Therefore, Member States cannot impede the making available on the market of a product which complies with all the provisions of sectoral harmonisation legislation.
The conformity to all the obligations incumbent on manufacturers by virtue of Union harmonisation legislation is symbolised by the CE marking. Member States must presume that products bearing the CE marking comply with all the provisions of the applicable legislation providing for its affixing. Accordingly, Member States may not prohibit, restrict or impede the making available on the market in their territory of products bearing the CE marking, unless the provisions relating to CE marking are incorrectly applied.
8.2. LIMITS AND RESTRICTIONS
The Union harmonisation legislation is designed to ensure free movement of products that comply with the high level of protection laid down in the applicable legislation. Therefore, Member States may not prohibit, restrict or impede the making available of such products. However, Member States are allowed to maintain or adopt, in compliance with the Treaty (in particular Articles 34 and 36 TFEU), additional national provisions regarding the use of particular products which are intended for the protection of workers or other users, or the environment. Such national provisions may neither require modification of a product manufactured in accordance with the provisions of the applicable legislation, nor influence the conditions for making it available.
A limitation to the free movement of product might be imposed in the case of non-compliance of a product with the essential or other legal requirements. Besides that, it may be the case that products complying with the requirements of harmonised legislation present, nonetheless, a risk to the health or safety of persons or to other aspects of public interest protection. In this case, Member States must require the relevant economic operator to take corrective actions. It is possible, therefore, to limit the free movement of a product not only in case of non-compliance of the product with the requirements set up by the relevant legislation but also in the case of compliance when the essential or other requirements do not entirely cover all of the risks related to the product. (297)
9. INTERNATIONAL ASPECTS OF THE EU LEGISLATION ON PRODUCTS
In its relations with third countries the EU, among other things, endeavours to promote international trade in regulated products. Conditions for open trade include compatibility of approach, coherence of regulations and standards, transparency of rules, appropriate levels and means of regulation, impartiality in certification, compatibility of market surveillance measures and supervision practices, and an appropriate level of technical and administrative infrastructure.
Accordingly, depending on the status of the above conditions, a broad variety of measures can be applied in order to facilitate trade. The expansion of the single market of products is pursued through several international legal instruments that enable the achievement of appropriate levels of cooperation, convergence or harmonisation of legislation and thus facilitate the free movement of goods. These instruments include:
|
— |
full integration of the EEA EFTA countries in the internal market by virtue of the EEA agreement (298), |
|
— |
alignment of the legislative system and infrastructure of the Candidate countries with those of the EU |
|
— |
similar alignment by Neighbouring countries by conclusion of bilateral Agreements on conformity assessment and acceptance of industrial products (ACAAs), |
|
— |
conclusion of bilateral (inter-governmental) Mutual Recognition Agreements (MRAs) for conformity assessment, certificates and marking, which are intended to reduce the costs of testing and certification in other markets, and |
|
— |
finally, reliance on WTO Agreement on Technical Barriers to Trade (299). |
9.1. AGREEMENTS ON CONFORMITY ASSESSMENT AND ACCEPTANCE (ACAAS)
|
Agreements on Conformity Assessment and Acceptance are established between the Union and EU Neighbouring countries. |
The European Union has always been at the forefront of support for international cooperation regarding the areas of technical regulations, standards, conformity assessment and the elimination of technical barriers to trade for products. Within the framework of the European Neighbourhood Policy, the European Commission has made clear its intention to intensify cooperation with the EU's eastern and southern neighbours in the areas of trade, market access and regulatory structures.
The use of the Union system of standardisation and conformity assessment by third countries is designed to facilitate trade and market access in both directions.
Agreements on Conformity Assessment and acceptance of industrial products are intended to be established between the Union and EU Neighbouring countries (Mediterranean — Algeria, Egypt, Israel, Jordan, Lebanon, Morocco, Palestinian Authority, Tunisia, — and Eastern — Armenia, Azerbaijan, Belarus, Georgia, Moldova, Ukraine).
This mutual recognition of equivalence in technical regulation, standardisation and conformity assessment on which are based these agreements are based, operates on the basis of the EU acquis that has been transposed by the partner country, in the same way as it would apply to products placed on the market of a Member State. It allows industrial products covered by the agreements and attested as compliant with the procedures in the European Union to be placed on the market of the partner country without having to undergo any further approval procedures, and vice versa.
An ACAA requires the prior full alignment of the partner country's legal framework with EU legislation and standards and the upgrading of the implementing infrastructure in line with the model of the EU system, in relation to standardisation, accreditation, conformity assessment, metrology and market surveillance.
ACAAs consist of a framework agreement and one or more annexes, setting out the products covered, and the means adopted to extend the benefit of trade in that sector. The framework agreement provides for two mechanisms, (a) the recognition of equivalence in technical regulation, standardisation and conformity assessment for industrial products subject to equivalent regulation in Union law and the national law of the partner country, and (b) the mutual acceptance of industrial products that fulfil the requirements to be lawfully placed on the market in one of the Parties in cases where there is no European technical legislation applicable to relevant products. More sectoral annexes can be added successively.
A first ACAA entered into force in January 2013 with Israel on pharmaceutical products. At the time of writing, other Mediterranean partners are finalising the preparatory work for launching negotiations in a number of New Approach sectors (electrical products, construction materials, toys, gas appliances and pressure equipment).
9.2. MUTUAL RECOGNITION AGREEMENTS (MRAS)
|
9.2.1. MAIN CHARACTERISTICS
One of the instruments to promote international trade of regulated products is the conclusion of mutual recognition agreements (MRAs) on the basis of Article 207 and 218 TFEU. MRAs are agreements established between the Union and third countries for the purpose of mutual recognition of conformity assessment of regulated products.
MRAs are designed so that each party accepts the reports, certificates and marks that are delivered in the partner country in accordance with its own legislation. These are drawn up and issued by bodies that the other party has designated under the MRA for assessing conformity in the field(s) covered by the MRA. This can be achieved, because MRAs include all the conformity assessment requirements of the parties necessary to obtain full market access, and the products are evaluated in the country of production against the regulatory requirements of the other party. These are usually referred to as ‘Traditional MRAs’.
MRAs cover the entire territory of the parties in order to guarantee the full free movement of products certified to be in conformity, in particular in States with a federal structure. As a general rule, MRAs are limited to products that have their origin on the territory of either party.
MRAs apply to one or more categories of products or sectors falling within the regulated field (they are covered by Union harmonisation legislation in force) and, in certain cases, by non-harmonised national law. In principle MRAs should cover all the industrial products for which the regulations of at least one of the parties require third party conformity assessment.
MRAs comprise a framework agreement and sectoral annexes. The framework agreement lays down the essential principles of a traditional agreement. Sectoral annexes specify, in particular, the scope and coverage, regulatory requirements, the list of designated conformity assessment bodies, the procedures and authorities responsible for designating these bodies and, if applicable, transitional periods. More sectoral annexes can be added successively.
MRAs are not based on the necessity to mutually accept other party's standards or technical regulations, or to consider the legislation of the two parties as equivalent. They involve only the mutual acceptance of the reports, certificates and marks that are delivered in the partner country in accordance with its own legislation. However, MRAs can pave the way towards a harmonised system of standardisation and certification by the parties. Nevertheless, the two legislations are, as a rule, deemed to ensure a comparable level of protection regarding health, safety, environment or other public interests. Moreover, MRAs increase the transparency of the regulatory systems. Once established, the MRA needs to be maintained, for example by keeping lists of recognised certification bodies, and the standards or rules against which they must certify.
The benefits of the MRA arise from removal of duplicated inspection or certification. Where a product intended for two markets may still have to be assessed twice (when technical requirements or standards are different), the assessment will be cheaper when carried out by the same body. The time to market is reduced since contacts between the manufacturer and the single conformity assessment body, and a single assessment, speed up the process. Even where the underlying regulations are harmonised, for example because they refer to an international standard, the need for recognition of certificates remains, and in such cases the benefit will be clear: the product is assessed once against the commonly accepted standard instead of twice.
Currently there are MRAs into force with Australia, New Zealand, the United States, Canada, Japan and Switzerland.
The above agreements are concluded in a number of specific sectors which might vary from one country to another. More details on the agreements can be found at the following address: http://ec.europa.eu/growth/single-market/goods/international-aspects/mutual-recognition-agreements/index_en.htm. The bodies designated under MRAs figure in a dedicated part of NANDO.
9.2.2. EU-SWISS MRA
The MRA concluded with Switzerland which entered into force on 1 June 2002 (OJ L 114, 30.4.2002) is a comprehensive agreement based on the equivalence of the legislation of the EU and Switzerland (300). It covers the recognition of conformity assessments irrespective of the origin of the products, except for Chapter 15 on Medicinal products, GMP Inspection and Batch Certification. This type of MRA agreement is usually referred to as an ‘Enhanced MRA’. However, the case of Switzerland remains quite unique.
The provisions of the Agreement and the harmonisation of Swiss technical regulations with those of the EU ensure seamless market access for EU products to the Swiss market, and vice versa for Swiss products to the EU/EEA market. However, despite the MRA, there is no customs union between the EU and Switzerland.
According to the Agreement, the Swiss Accreditation Service (SAS) is a full member of European cooperation for Accreditation (EA) and is signatory of all mutual recognition agreements with the EA. In the area of standardisation, Switzerland is full member of CEN, Cenelec and ETSI and participates actively in the work of the European standardisation.
Furthermore, an EU conformity assessment body is allowed to issue certificates in the EU according to EU legislation, which are deemed equivalent to those of Switzerland. The same applies conversely to the Swiss' conformity assessment bodies. Thus, certificates issued by Swiss conformity assessment bodies accredited by SAS for product covered by the MRA are deemed equivalent to those issued by EU conformity assessment bodies.
This was only possible because, on the one hand, Switzerland has an existing technical infrastructure (e.g. the public or private institutions dealing with standards, accreditation, conformity assessment, market surveillance and consumer protection) which is equally developed and deemed equivalent to the one existing in the EU. On the other hand, Switzerland has chosen to modify its legislation in the sectors covered by the agreement, in order to align it with that of the Union. Furthermore, it has committed to maintain its legislation aligned whenever amendments to it are introduced by the Union to the applicable EU legal framework.
The so called ‘Enhanced MRA’ agreement with Switzerland currently covers twenty product sectors: Machinery, Personal Protective Equipment (PPE), Safety of Toys, Medical Devices, Gas Appliances and Boilers, Pressure Equipment, Telecommunications Terminal Equipment, Equipment and Protective Systems intended for use in potentially explosive atmosphere (ATEX), Electrical Safety and Electromagnetic Compatibility (EMC), Construction Plant and Equipment, Measuring Instruments and Pre-packages, Motor Vehicles, Agricultural and Forestry Tractors, Good Laboratory Practice (GLP), Good Manufacturing Practice (GMP) Inspection and Batch Certification, Construction products, Lifts, Biocidal Products, Cableways installations and Explosives for civil use.
A parallel MRA covering exactly the same scope has been concluded between the EEA EFTA States and Switzerland (Annex I to the EFTA Vaduz Convention which entered into force on 1 June 2002), ensuring a homogeneous market access throughout the EU internal market, the EEA and Switzerland.
9.2.3. EEA EFTA STATES: MUTUAL RECOGNITION AGREEMENTS AND AGREEMENTS ON CONFORMITY ASSESSMENT AND ACCEPTANCE
The mandate from the Council to the Commission to negotiate mutual recognition agreements and Agreements on conformity assessment and acceptance of industrial products indicated the objective that the third countries concerned will conclude with the EEA EFTA States parallel agreements equivalent to those to be concluded with the Union, and which will, possibly, have the same date for entry into force.
The system of parallel agreements formally grants the third country concerned the same market access throughout the European Economic Area for products covered by the mutual recognition agreements or Agreements on conformity assessment and acceptance of industrial products. As to the practical implementation of these agreements, common sessions of the Joint Committee meetings with the third country concerned will be arranged.
(1) On 13 February 2013, the Commission adopted a proposal for a new stand-alone Market Surveillance Regulation, bringing together all market surveillance provisions from Regulation (EC) No 765/2008, GPSD and sectoral legislation. COM(2013)75 final is available at: http://eur-lex.europa.eu/LexUriServ/LexUriServ.do?uri=COM:2013:0075:FIN:en:PDF
(2) Regulation (EC) No 765/2008 of the European Parliament and of the Council of 9 July 2008 setting out the requirements for accreditation and market surveillance relating to the marketing of products and repealing Regulation (EEC) No 339/93 and Decision No 768/2008/EC of the European Parliament and of the Council of 9 July 2008 on a common framework for the marketing of products, and repealing Council Decision 93/465/EEC.
(3) Now superseded by Directive (EU) 2015/1535 of the European Parliament and of the Council of 9 September 2015 laying down a procedure for the provision of information in the field of technical regulations and of rules on Information Society services (OJ L 241, 17.9.2015, p. 1).
(4) Since 1.1.2013 and under Regulation (EU) No 1025/2012 each national standardisation body is obliged to make its work programme publicly available and to notify the existence thereof to the other national standardisation bodies, to the European standardisation organisation and to the Commission.
(5) Judgment of the Court of Justice of 20 February 1979. — Rewe-Zentral AG v Bundesmonopolverwaltung für Branntwein. Case 120/78. European Court reports 1979, p. 649.
(6) OJ L 218, 13.8.2008, p. 21.
(7) http://ec.europa.eu/internal_market/strategy/docs/monti_report_final_10_05_2010_en.pdf
(9) For more details on the product liability legislation see Section 1.4.
(10) OJ L 316, 14.11.2012, p. 12.
(11) Article 2.4 of the WTO TBT Agreement.
(12) Initially legislation adopted under the New Approach technique was essentially in the form of Directives.
(13) Council Decision 93/465/EEC of 22 July 1993 concerning the modules for the various phases of the conformity assessment procedures and the rules for the affixing and use of the CE conformity marking, which are intended to be used in the technical harmonisation directives (OJ L 220, 30.8.1993, p. 23).
(14) OJ L 218, 13.8.2008, p. 82.
(15) OJ C 282, 25.11.2003, p. 3.
(16) So called by the European Parliament in memory of Michel Ayral, the Director in Directorate-General Enterprise and Industry, responsible for putting the Package together.
(18) The word ‘quality’ is used to designate the level of safety and other public policy objectives which are aimed by the Union harmonisation legislation. Not to be confused with the meaning of the word ‘quality’ in the commercial context allowing differentiating between different levels of product quality.
(19) OJ L 11, 15.1.2002, p. 4.
(20) Specific guidance on the practical application of the GPSD is available at: http://ec.europa.eu/consumers/safety/prod_legis/index_en.htm
(21) OJ L 210, 7.8.1985, p. 29.
(22) See Article 3 of Directive 85/374/EEC.
(23) For comparison, Union harmonisation legislation may apply to ‘movables’ such as electronic equipment, personal protective equipment, etc. or ‘not movables’ (e.g. a lift once it is integrated into immovable property).
(24) The equivalence in national currency is calculated at the exchange rate of 25 July 1985.
(25) The equivalence in national currency is calculated at the exchange rate of 25 July 1985.
(26) Accordingly, harmonised standards — although they give a presumption of conformity — do not free from liability, but they may reduce the likelihood of damage. For the use of harmonised standards and presumption of conformity, see point 4.1.2.
(27) According to the Court of Justice (case C-300/95) this refers to an objective state of knowledge, related not only to safety standards existing in a particular sector, but to any high standard the producer is presumed to be aware of and that was accessible to him. Liability for development risks exists in only two Member States.
(28) Decision No 768/2008/EC and Regulation (EC) No 765/2008.
(29) There are however references to GPSD in relation to specific situations such as second-hand products.
(30) http://ec.europa.eu/consumers/safety/prod_legis/index_en.htm
(31) Some Union harmonisation legislation covers also ‘putting into service’ (e.g. lifts) or ‘own use’ (e.g. machinery to be used by the manufacturer himself) as being equivalent to the ‘placing on the market’.
(32) Directive 2014/90/EU on marine equipment refers to the placing on board of a ship flying the flag of an EU Member State.
(33) For placing on the market, making available on the market and putting into service, see Sections 2.2, 2.3 and 2.5.
(34) Directive 1999/44/EC on certain aspects of the sale of consumer goods and associated guarantees (OJ L 171, 7.7.1999, p. 12.) is beyond the interest of this Guide. According to this directive, sellers of consumer products within the EU are obliged to guarantee the conformity of the products with a contract, for a period of 2 years after their delivery. If the products are not delivered in conformity with the sales contract, consumers can ask for the products to be repaired, replaced, and reduced in price or for the contract to be rescinded. The final seller, who is responsible to the consumer, can also hold the producer liable in their business relationship.
(35) For the Directives on medical devices, obligations apply only to the placing on the market and/or putting into service but not to any subsequent making available.
(36) See Chapter 3.4 Distributors.
(37) This is without prejudice to the level of safety or other public interest protection that a product must offer in relation to the applicable Union harmonisation legislation at the time it was placed on the market.
(38) This implies that the economic operator that offers the product has to be in a position to provide the evidence that the product complies with the applicable requirements, i.e. by providing the technical file on request of a market surveillance authority. See Chapter 2.3 for more guidance on the placing on the market of products online.
(39) Union harmonisation legislation does not prohibit the manufacture of products to meet the requirements of a non-EU Member State, if such products will not be placed and put into service in the internal market. Union harmonisation legislation does not prohibit the import of products that do not meet the requirements of the relevant Union harmonisation legislation, if such products are not intended to be placed on the market or put into service on the internal market (but, e.g. refined/processed/incorporated in the internal market) but to be exported outside the EEA.
(40) In this context the Union should be considered to mean the present Member States, where free movement of used and second-hand products takes place according to Articles 34 and 36 TFEU.
(41) Used and second-hand products supplied to consumers are covered by the GPSD and have to be safe, unless they are supplied as antiques or as products to be repaired or reconditioned prior to being used, provided that the supplier has clearly informed the person to whom he supplies the product to that effect.
(42) In some situations the responsibilities of the original manufacturer are taken over by another person, see Chapter 3.
(43) When outside the scope of relevant Union harmonisation legislation, spare parts or parts which are available and marketed separately as products intended for consumers in order to be integrated to other products, such as service parts or components intended for maintenance or repair, must nevertheless comply with the general safety requirement set out in the GPSD.
(44) Under medical devices legislation, the term ‘fully refurbished’ exists. ‘Fully refurbished’ products are assimilated to new products.
(45) For products used at the workplace the employer must take all measures necessary to ensure that work equipment is suitable and safe and that repaired machinery is no less safe than the original. See Section 3.5.
(46) See Article 2 of Regulation (EC) No 765/2008 and Article R1 of Annex I to Decision No 768/2008/EC.
(47) Excluding intellectual property rights.
(48) In case of the making available of a product taking place through renting, repeated renting of the same product does not constitute a new placing on the market. That product would need to be in compliance with the applicable Union harmonisation legislation at the time the first renting takes place.
(49) The Lifts Directive 95/16/EC uses also the concept of ‘installer’ who also places on the market.
(50) The distribution chain can also be the commercial chain of the manufacturer or the authorised representative.
(51) An offer or agreement concluded before the stage of manufacture has been finalised cannot be considered as placing on the market (e.g. an offer to manufacture a product according to certain specifications agreed by the parties to the contract, where the product will only be manufactured and delivered at a later stage).
(52) See for instance, the Directives on Machinery, Measuring Instruments, ATEX, Civil Explosives.
(53) When Union harmonisation legislation covers own use, this does not refer to the occasional manufacturing for own use by a private person in a non-commercial context.
(54) This exception does not include products which are shipped by the economic operator to the consumers in the EU, such as the case of products bought online and shipped to the EU.
(55) For authorised representative, see Section 3.2.
(56) See Council Regulation (EEC) No 2913/92 establishing the Community customs code. In accordance with this Regulation, non-Community goods placed under a suspensive customs procedure or in a free zone are subject to customs supervision and do not benefit from the free circulation in the internal market. Before benefiting from the free circulation in the internal market, these goods must be declared for release for free circulation. That entails application of commercial policy measures, completion of the other formalities laid down in respect of the importation of goods and the charging of any duties legally due.
(57) The prototype must be safe and under complete control and supervision. Controlled conditions would mean expert operators, restrictions to public contact with the product, avoiding inappropriate interaction with other neighbouring products, etc.
(58) However, in such circumstances a visible sign must clearly indicate that the product in question may not be placed on the market or put into service until it has been made to comply.
(59) As noted in the introduction, more specific reflections are ongoing regarding various aspects of the Union legal framework applicable to online sales and this Guide is without prejudice to any future specific interpretation and guidance which may be developed on those matters.
(60) An online operator is not a new category of economic operator, but it is used to refer to the classic economic operators (manufacturers, importers distributors) operating online only/mainly.
(61) Judgment of the CJEU of 12 July 2011, Case C-324/09, L'Oréal/eBay, p. 65. Although the legal context is different, this element of the judgement can be taken into account in this context.
(62) For example in the context of combined offers, Directive 2005/29/EC concerning unfair business-to-consumer commercial practices in the internal market defines ‘business-to-consumer commercial practices’ as any act, omission, course of conduct or representation, commercial communication including advertising and marketing, by a trader, directly connected with the promotion, sale or supply of a product to consumers.
(63) An element that could also be taken into account is that the Unfair Commercial Practices Directive 2005/29/EC defines a ‘trader’ as any natural or legal person who, in commercial practices covered by the Directive, is acting for purposes relating to his trade, business, craft or profession and anyone acting in the name of or on behalf of a trader. Similarly, Directive 2011/83/EU on consumer rights defines a ‘trader’ as any natural person or any legal person, irrespective of whether privately or publicly owned, who is acting, including through any other person acting in his name or on his behalf, for purposes relating to his trade, business, craft or profession in relation to contracts covered by this Directive.
(64) If products are sold online, the CE marking and any required warnings, information and labels according to applicable legislation should be indicated in that website; these should be clearly visible in its entirety before the consumer is carrying out the purchase.
(65) This explanation does not attempt to deal with the question of intermediary liability and the term ‘online operator’ used in this context may not cover such intermediaries.
(66) The design in accordance with the essential requirements of the applicable legal act, the following risk and conformity assessment, the issue of a Declaration of Conformity, the marking requirements (CE marking, name, address of the manufacturer etc.), the compilation of the technical file, must have been completed by the manufacturer at the time of placing on the market.
(67) For market surveillance, see Chapter 7.
(68) For putting into service, see Section 2.5.
(69) See articles 27 to 29 of Regulation (EC) No 765/2008.
(70) For the role of the importer, see point 3.3.
(71) Please note that in the area of medical devices, the role of the authorised representative is reinforced and he is the primary interlocutor of market surveillance authorities for products for third countries.
(72) The concept of ‘putting into service’ is not relevant for all Union harmonisation legislation. For instance, in the case of explosives, there is no ‘putting into service’.
(73) As regards lifts and equivalent products, the putting into service should be considered to take place at the moment when the first use within the Union is possible.
(74) In case there is new Union harmonisation legislation coming into force, a compliant product which is placed on the market before the end of the transitional period of the legislation to be replaced should be allowed to be put into service, unless specific legislation provides otherwise.
(75) In the case of the Directive on radio and telecommunications terminal equipment, Article 7 regulates restrictions to the putting into service. Member States may restrict the putting into service of radio equipment for reasons related to the effective and appropriate use of the radio spectrum, avoidance of harmful interference or matters related to public health
(76) E.g. energy consumption.
(77) For instance: the Directive relating to low voltage equipment is not applicable to electrical equipment for medical purposes, instead the legislation relating to medical devices will apply; the Directive relating to electromagnetic compatibility is not applicable to products covered by specific legislation that harmonises the protection requirements specified in the Directive on electromagnetic compatibility; the Directive relating to lifts is not applicable to lifts connected to machinery and intended exclusively for access to the workplace, instead the Directive relating to machinery applies; marine equipment, which is also within the scope of other directives than the Directive on marine equipment, is excluded from the application of such directives.
(78) E.g. The R&TTE Directive directly covers electromagnetic compatibility aspects and low voltage safety. In order to avoid double coverage R&TTE incorporates the essential requirements of EMCD and LVD (with no lower voltage limit) and allows a manufacturer to use some of their conformity assessment procedures. In addition harmonised standards under EMCD and LVD also have that status under the R&TTE Directive. The Lifts Directive includes relevant requirements of the Machinery Directive.
(79) For example: the Machinery Directive covers all hazards that come from machinery, including electrical hazards. However, concerning the electrical hazards of machinery, the Machinery Directive is referring to the safety objectives of the Low Voltage Directive, to be applied solely.
(80) For making available, see Section 2.2.; for putting into service, see 2.5.
(81) Please note that the Machinery Directive requires the manufacturer to take account of ‘reasonably foreseeable misuse’.
(82) Furthermore, a tool designed and intended to be used by professionals only, might eventually also be used by non-professionals; consequently the design and instructions accompanied must take this possibility into account.
(83) In the UK, these are the Channel Islands and the Isle of Man.
(84) For more details, please consult Commission Staff Working Paper on Obstacles to access by Andorra, Monaco and San Marino to the EU's Internal Market and Cooperation in other Areas (SWD(2012) 388 final) available at: http://eur-lex.europa.eu/LexUriServ/LexUriServ.do?uri=SWD:2012:0388:FIN:EN:PDF
(85) OJ C 265, 4.11.2003, p. 2.
(86) For instance, such product can still be sold legally after the transitional period if the product is on stock at the distributors' warehouse, i.e. that the product has been already placed on the market and that a change of ownership has taken place.
(87) Since the Directive on pressure equipment sets no time limit for the putting into service, products covered by this Directive can be put into service at any time without being subject to further conditions according to this Directive. For placing on the market and putting into service, see Sections 2.3 and 2.5.
(88) For the EU declaration of conformity, see Section 4.4.; for the CE marking, see Section 4.5.1.
(89) In February 2014, an ‘Alignment Package’ consisting of eight directives was adopted. The Directives of the alignment package will become applicable on 20 April 2016 and their essential requirements are not modified. It includes Directive 2014/35/EU (Low Voltage); Directive 2014/30/EU (Electromagnetic Compatibility); Directive 2014/34/EU (ATEX); Directive 2014/33/EU (Lifts); Directive 2014/29/EU (Simple Pressure Vessels); Directive 2014/32/EU (Measuring Instruments); Directive 2014/31/EU (Non-automatic Weighing Instruments); Directive 2014/28/EU (Civil Explosives). Directive 2013/29/EU (Pyrotechnic Articles) has also been aligned to Decision No 768/2008/EC and became applicable on 1 July 2015.
(90) See Article R1 (7) of Annex I to Decision No 768/2008/EC.
(91) See Article R1 (3) of Annex I to Decision No 768/2008/EC.
(92) See Article R6 of Annex I to Decision No 768/2008/EC.
(93) Those manufacturers are often referred to as ‘own brand labellers’ or ‘private labellers’.
(94) The Lifts Directive 95/16/EC uses the concept of installer to impose responsibilities on the person who makes a product operational and ready to use. The role of the installer combines elements of manufacture and putting into service and is seen as fundamental for delivering the final product.
(95) Article R2 (1) of Decision No 768/2008/EC
(96) The use of symbols according to international standards may be an alternative to written statements.
(97) In some specific cases, where several identical products are bundled and intended by the manufacturer to be sold together to the end-user or to be sold in a packaging for use in one application (e.g. installation equipment), it is sufficient to accompany the shipping unit with one set of instructions. However, if the bundle is dismantled and the different identical products sold individually, the economic operator dismantling the bundle and making available the individual products needs to make sure that a set of instructions and safety information accompanies each individual product.
(98) Not all Union harmonisation legislation requires both instructions and safety information since not all Union harmonisation legislation is safety related.
(99) The manufacturer, importer and distributor have the obligation to ensure that the product is accompanied by instructions in a language which can be easily understood by consumers and other end-users, as determined by the Member State concerned. It is for each economic operator which makes available the product in a Member State, to ensure that all the required languages are available.
(100) Unless otherwise specified in specific legislation, whilst the safety information needs to be provided on paper, it is not required that all the set of instructions is also provided on paper but they can also be on electronic or other data storage format. However, a paper version should always be available free of charge for the consumers who request it.
(101) To be understood as the last item of the product model placed on the market.
(102) For the Directives on Medical Devices, the manufacturer must indicate the place of business.
(103) This does not include esthetical reasons.
(104) Please note that some Union harmonisation legislation excludes the possibility to use the packaging to satisfy this requirement (e.g. Simple Pressure Vessels Directive)
(105) Manufacturer may add a website if they so desires. A website is additional information, but is not enough as an address. Normally an address consists of a street and number or post-box and number and the postal code and town, but some countries might deviate from this model.
(106) See Union harmonisation legislation relating to low voltage equipment, toys, machinery, non- automatic weighing instruments, active implantable medical devices, gas appliances, medical devices, potentially explosive atmospheres, recreational craft, lifts, pressure equipment, in vitro diagnostic medical devices, and radio and telecommunications terminal equipment. Further, according to the Directive on in vitro diagnostic medical devices, a manufacturer who places devices on the Union market under his own name is obliged to register in the Member State where he has his place of business.
(107) For more information on the name and address requirement, see point 4.2.2.1.
(108) E.g. ATEX marking, Class identifier in accordance with the R&TTE Directive or the supplementary metrology marking in the case of non-automatic weighing machines and measuring instruments.
(109) For the information obligations in the case of EU-type examination certificates, see Annex II to Decision No 768/2008/EC, Module B, point 7.
(110) E.g. the Directives on Simple pressure vessels and ATEX.
(111) Such sample testing should be performed when deemed appropriate with regard to the risks presented by a product, in view of protecting the health and safety of consumers (see article R2.4 of Decision No 768/2008/EC).
(112) The acceptable level of risk for the product is defined by the essential requirements set out in the applicable Union harmonisation legislation. Consequently, manufacturers must inform the competent authority where they consider or have reason to believe that the product does not comply with the applicable essential requirements.
(113) https://webgate.ec.europa.eu/gpsd-ba/index.do
(114) The reasoned request does not necessarily mean a formal decision by an authority. According to Article 19(1), paragraph 2 of Regulation (EC) No 765/2008, ‘market surveillance authorities may require economic operators to make such documentation and information available as appear to them to be necessary for the purpose of carrying out their activities’. For a request to be reasoned it is sufficient the market surveillance authority explains the context in which the information is requested (e.g. inspection on specific characteristics of the products, random checks, etc.)
(115) This is not applicable to products covered by the Union harmonisation legislation relating to toys, low voltage equipment, civil explosives and refrigeration appliances, since these directives only cover making available on the market. Further, this is not applicable to recreational craft built for own use, provided that it is not subsequently placed on the market during a period of 5 years, or to craft designed before 1950.
(116) Article R6 of Annex I to Decision No 768/2008/EC.
(117) Further, according to the Directives relating to machinery and lifts, obligations regarding the conformity assessment procedure fall to any person placing the product on the market, where neither the manufacturer nor the authorised representative or the installer of the lift fulfils these obligations.
(118) Please note that not all Union harmonisation legislation provides for an authorised representative.
(119) As an exception, according to the Directives on medical devices and in vitro diagnostic medical devices the manufacturer must designate a person who is established in the Union to be responsible for the marketing of medical devices, if he does not have a registered place of business in a Member State and he places devices on the Union market under his own name.
(120) For the obligations of the importer see Section 3.3.
(121) For the purposes of this Guide, imports are products manufactured in third countries and placed on the Union market. Products manufactured in one Member State and placed on the market in another Member State do not constitute an ‘import’ as the operation takes place within the Union internal market.
(122) The importer is not necessarily the person who transports the product, but can be the person on behalf of whom this logistic activity is performed.
(123) See the Chapter on Market Surveillance.
(124) Not all Union harmonisation legislation requires both instructions and safety information since not all Union harmonisation legislation is safety related.
(125) In light of these obligations, it is generally considered good practice for importers to: refer to the applicable EU legislation in the contract with his supplier (mentioning the obligations of manufacturers under Union law); ensure that he has access to the technical file, or ensure that the manufacturer has signed an obligation to provide the technical documentation if requested by market surveillance authorities.
(126) Depends on the applicable Union harmonisation legislation.
(127) Please note that some sectoral Union harmonisation legislation might provide for stricter requirements.
(128) To be understood as the last item of the product model placed on the market.
(129) Importers are not obliged to have a copy of the technical documentation but they shall ensure that it is made available to the relevant authorities upon request. Even if there is no explicit obligation, the importer is advised to require formal assurance in writing from the manufacturer that the documents will be made available when requested by the surveillance authority.
(130) The reasoned request does not necessarily mean a formal decision by an authority. According to Article 19(1), paragraph 2 of Regulation (EC) No 765/2008, ‘market surveillance authorities may require economic operators to make such documentation and information available as appear to them to be necessary for the purpose of carrying out their activities’. For a request to be reasoned it is sufficient the market surveillance authority explains the context in which the information is requested (e.g. inspection on specific characteristics of the products, random checks, etc.)
(131) Article R4(6) of Annex I to Decision No 768/2008/EC.
(132) Due care refers to the effort made by an ordinarily prudent or reasonable party to avoid harm to another, taking the circumstances into account. It refers to the level of judgment, care, prudence, determination, and activity that a person would reasonably be expected to do under particular circumstances.
(133) Article R5(1) of Annex I to Decision No 768/2008/EC.
(134) Article R5(2), 1st paragraph of Annex I to Decision No 768/2008/EC.
(135) Where the Union harmonisation legislation explicitly requires that the product is accompanied by the EU declaration of conformity, the distributor has to ensure that this is the case.
(136) Not all Union harmonisation legislation requires both instructions and safety information since not all Union harmonisation legislation is safety related.
(137) See the obligations of the manufacturer in point 3.1 and the obligations of the importer in point 3.3.
(138) Article R5(2), 2nd paragraph, of Annex I to Decision No 768/2008/EC.
(139) Article R5(2), 2nd paragraph and article R5(4) of Annex I to Decision No 768/2008/EC.
(140) The reasoned request does not necessarily mean a formal decision by an authority. According to Article 19(1), paragraph 2 of Regulation (EC) No 765/2008, ‘market surveillance authorities may require economic operators to make such documentation and information available as appear to them to be necessary for the purpose of carrying out their activities’. For a request to be reasoned it is sufficient the market surveillance authority explains the context in which the information is requested (e.g. inspection on specific characteristics of the products, random checks, etc.)
(141) Article R5(5) of Annex I to Decision No 768/2008/EC.
(142) Article R7(2) of Annex I to Decision No 768/2008/EC.
(143) Article R5(3) of Annex I to Decision No 768/2008/EC
(144) See Judgement of the Court: case C-271/92.
(145) As noted in the introduction, more specific reflections are ongoing regarding various aspects of the Union legal framework applicable to online sales and this Guide is without prejudice to any future specific interpretation and guidance which may be developed on those matters.
(146) Directive 2000/31/EC of the European Parliament and of the Council of 8 June 2000 on certain legal aspects of information society services, in particular electronic commerce, in the Internal Market (Directive on electronic commerce) (OJ L 178, 17.7.2000, p. 1).
(147) Other activities also described by the Directive are: (1) ‘mere conduit activities’ such as transmitting information (provided by the recipient of the service) or providing access to a communication network (e.g. internet providers) and (2) ‘caching activities’ such as making the transmission of information more efficient, e.g. duplicating a database which copies the content of the initial server for ensuring a global coverage.
(148) In Case C-324/09, L'Oréal vs. eBay, the European Court of Justice clarified that the relevant question relating to the conditions for benefiting from a liability exemption was whether eBay was aware of facts and circumstances from which the illegal activity was apparent (see paragraphs 120-123).
(149) However, Directive 2013/53/EU on Recreational craft imposes obligations on private importers.
(150) For the concept of ‘intended use’, see above Section 2.7.
(151) For the technical documentation, see point 4.3.
(152) Even where the manufacturer uses a harmonised standard (where its reference is published in the OJ and which aims to cover certain risks) to satisfy essential requirements, the risk assessment has to be carried out and he must check whether the harmonised standard covers all risks of the product. This is because it cannot be assumed that the harmonised standard covers all requirements of all legislative acts applicable to a given product (or, indeed, all the requirements of the specific act under which it has been developed) or whether the product in question introduces also other risks not considered in the harmonised standard.
(153) According to the Directive 2008/57/EC on the interoperability of the rail system each sub-system is covered by a Technical Specification of Inter-operability (TSI), which specifies the essential requirements. According to Regulation (EC) No 552/2004 on the interoperability of the European Air Traffic Management network, in case of necessity, the essential requirements are refined or complemented by implementing rules for interoperability.
(154) OJ L 316, 14.11.2012, p. 12.
(155) CEN (European Committee for Standardisation); Cenelec (European Committee for Electrotechnical Standardisation); ETSI (European Telecommunication Standards Institute).
(156) Exceptionally, harmonisation documents (HD) adopted by European standardisations organisations can also be regarded as harmonised standards (e.g. in the case of the Low Voltage Directive). The differences between European standards (EN) and harmonisation documents essentially relate to the degree of obligation on the part of the national standardisation bodies. Harmonisation documents must be implemented at national level, at least by public notification of the title and number of the document, and by the withdrawal of conflicting national standards. However, it is acceptable to retain or publish a national standard dealing with a subject covered by the harmonisation document, provided that it has technically equivalent contents. In addition, harmonisation documents allow for national divergences under special conditions, which could create some application problems if they were accepted as harmonised standards.
(157) Voluntary nature of standards makes reference to the fact that standards as such and as published by standardisation organisations are always of voluntary application. This principle is also usually applied in legislation when referencing to standards.
(158) (SWD(2015) 205 final, 27.10.2015) available at http://ec.europa.eu/growth/single-market/european-standards/vademecum/index_en.htm
(159) Following consultation of the ESOs, stakeholders and sectoral experts (the latter through the committee set up under the legislation where it exists), the Commission consults the Member State Committee established under Regulation (EU) No 1025/2012 (Standardisation Regulation) in accordance with the examination procedure of Regulation (EC) No 182/2011 (OJ L 55, 28.2.2011, p. 13).
(160) See Article 10 of Regulation (EU) No 1025/2012.
(161) In order to establish coherent principles for elaboration and adoption, including revision, of harmonised standards Vademecum on European standardisation (SWD(2015) 205 final, 27.10.2015 Part III) sets out guidelines for the execution of standardisation requests accepted by the ESOs.
(162) OJ C 91, 16.4.2003, p. 7.
(163) Still, the Commission on the basis of Regulation (EU) No 1025/2012 must verify and assess that the terms of the request are fulfilled in order to ensure the proper functioning of the Single market (see point 4.1.2.4).
(164) In reality European standardisation organisations may only declare an intention to cover certain requirements and this intention is further presumed (or removed) once a reference is published (or removed) in (from) the OJ (see points 4.1.2.4 and 4.1.2.5).
(165) European standardisation organisations usually name this annex as ‘Annex ZA, ZB or ZZ’ etc.
(166) In this context this term is understood as defined in ISO/IEC Guide 51 Safety aspects — Guidelines for their inclusion in standards, which is a generic guideline to develop standards addressing safety issues.
(167) Preparation of a request may start in parallel with the legislative process. However at the moment when a request is issued to the European standardisation organisations, there must be certainty on the legal requirements to be supported by harmonised standards
(168) This is without prejudice to their right to refuse a request.
(169) European standardisation organisation may also cooperate with other bodies which are responsible for the drafting work.
(170) Article 10(5) indicates that consensus building process according to the internal rules of European standardisation organisations is not alone as such a sufficient guarantee to assume that requirements of a request are fulfilled.
(171) The transposition of the standard is a matter for the ESOs' rules. It is usually carried out before the references of the harmonised standard are published in the OJ. However national transposition is not a precondition to get a presumption of conformity. In practise harmonised standards are usually available as transposed nationally standards while the list of harmonised standards published in the OJ and relevant Union harmonisation legislation make direct reference to original European standards.
(172) According to Article 28 of Regulation (EU) No 1025/2012 formal objection articles given in some sectoral legislation continue to be valid for some time.
(173) A Web service providing access to the latest lists of references of harmonised standards and of other European standards published in the Official Journal of the European Union (OJ) is available at: http://ec.europa.eu/growth/single-market/european-standards/harmonised-standards/index_en.htm
(174) This ‘claim’ is usually in a dedicated informative annex to a harmonised standard.
(175) In some cases a piece of sectoral legislation may still contain an article of objection. In such cases Article 11 of Regulation (EU) No 1025/2012 does not apply — see Article 28, second paragraph of Regulation (EU) No 1025/2012.
(176) A presumption of conformity would be meaningless if essential requirements covered are not known.
(177) Directive 1999/5/EC relating to telecommunications terminal equipment allows harmonised standards to be transformed into common technical regulations, compliance with which is mandatory. Regulation (EC) No 552/2004 on the interoperability of the European Air Traffic Management network requires the application of Community specifications.
(178) See the directives relating to simple pressure vessel, toys, electromagnetic compatibility, radio and telecommunications terminal equipment, machinery, lifts and recreational craft. The lack of harmonised standards may lead to the application of a specific procedure, see for instance the Pressure equipment directive (the European approval may be granted to materials which are not covered by any harmonised standard and which are intended for repeated use in the manufacture of pressure equipment).
(179) Article 11 of Regulation (EU) No 1025/2012 becomes gradually applicable after objection articles contained in sectoral legislation are removed. Meanwhile some Union harmonisation legislation may still contain specific procedures, like the Directive relating to radio and telecommunications terminal equipment provides a possibility for the Commission, in the case of shortcomings of harmonised standards, to publish in the Official Journal guidelines to the interpretation of harmonised standards, or the conditions under which compliance is possible.
(180) The European Parliament can raise this concern in cases where Article 11 of Regulation (EU) No 1025/2012 applies.
(181) In accordance with Article 11(1) and 11(4)-(5) of Regulation (EU) No 1025/2012
(182) http://ec.europa.eu/growth/single-market/european-standards/notification-system/index_en.htm
(183) For the safeguard clause, see Section 7.4
(184) http://ec.europa.eu/growth/single-market/european-standards/notification-system/index_en.htm
(185) Under the terms of their internal rules, the European standardisation organisations review their standards — whether or not initially developed on the basis of a standardisation request — at intervals not exceeding 5 years. This periodical review may lead to a confirmation (no action), a revision or a withdrawal of a relevant standard.
(186) Vademecum on European standardisation (SWD(2015) 205 final, 27.10.2015 Part III).
(187) Some Union harmonisation legislation may however give alternative ways for a presumption of conformity by means of other specifications than harmonised standards, e.g. the possibility to use the EU Ecolabel scheme in Eco-design Directive; in the in vitro diagnostic medical devices sector, compliance with so called ‘common technical specifications’ (CTS) provides presumption of conformity with the relevant essential requirements. Another example is the references to normative documents from the International Organisation of Legal Metrology (OIML) in Directive 2004/22/EC on measuring instruments.
(188) See, for instance, Directive relating to gas appliances.
(189) The Member States Committee under Regulation (EU) No 1025/2012 and, if provided for, the sectoral Committee
(190) In the case of Regulation (EC) No 552/2004 on the interoperability of the European Air Traffic Management network, if a manufacturer chooses not to follow a harmonised standard, the declaration is called declaration of suitability for use.
(191) Such confusion might occur for instance when the name of the distributor appears on the packaging while the manufacturer's name is shown on the product inside.
(192) A trade mark is a distinctive sign or indicator used by an individual, business organisation, or other legal entity to identify that the products or services to consumers with which the trade mark appears originate from a unique source, and to distinguish the products or services in question from those of other entities. A trademark is a type of intellectual property, and typically a name, word, phrase, logo, symbol, design, image, or a combination of these elements.
(193) Article R2(6) of Annex I to Decision No 768/2008/EC.
(194) Article R4(3) of Annex I to Decision No 768/2008/EC.
(195) Article R2(5) of Annex I to Decision No 768/2008/EC.
(196) Article R7 of Annex I to Decision No 768/2008/EC.
(197) See recital 25 of Decision No 768/2008/EC.
(198) For instance, an address serving as an information point for consumers and other users in the Member State where the product is made available.
(199) In the medical devices sector, the product must also bear the authorised representative's name and address.
(200) If the importer is only affixing his name and address and leaves the trademark of the original manufacturer, he remains importer. The address of importer and manufacturer will appear on the product (or packaging or accompanying documents).
(201) This is also the case if the manufacturer and importer belong to the same group of companies, and the company based in the EU importing the product into the EU assumes the full manufacturer's responsibility for the product.
(202) In the case of toys, this might be the case for toys consisting of several parts or an assembly of several parts.
(203) For placing on the market, see Section 2.3.
(204) According to the Directives relating to medical devices and in vitro diagnostic medical devices these documents must be kept for 5 years and in the case of implantable medical devices for 15 year.
(205) See the Directives relating to simple pressure vessels, machinery (for module B), non-automatic weighing instruments, active implantable medical devices, gas appliances, telecommunications terminal equipment, medical devices, potentially explosive atmospheres, lifts (for modules B, C, D, G, H), pressure equipment, in vitro diagnostic medical devices, and radio and telecommunications terminal equipment.
(206) Please note that the Machinery Directive 2006/42/EC foresees the placing on the market of ‘partly completed machinery’ to be accompanied by a so- called declaration of incorporation which is different from the EU Declaration of conformity. According to Regulation (EC) No 552/2004, constituents of the European Air Traffic Management network are accompanied either by a declaration of conformity or a declaration of suitability for use.
(207) For more information on the technical documentation, see Section 4.3.
(208) According to the Directives relating to medical devices and in vitro diagnostic medical devices the EU Declaration of Conformity must be kept for 5 years and in the case of implantable medical devices for 15 years.
(209) For responsibilities of the manufacturer, the authorised representative and the importer, see Chapter 3.
(210) The ‘number’ can be an alpha-numerical code too.
(211) In addition, whether this is expressly envisaged or not by the Union harmonisation legislation manufacturers are free to add a number identifying the EU Declaration of Conformity itself in line with EN ISO/IEC 17050-2.
(212) Not all Union harmonisation legislation requires the intervention of a notified body. For instance the Low voltage directive and the Toys directive do not.
(213) The name and address of the person who keeps the technical documentation may also be required by some pieces of Union harmonisation legislation since according to those, not only the manufacturer shall keep the technical documentation.
(214) This could be the Managing Director of the company or another representative of the company to whom this responsibility has been delegated.
(215) It is not necessary for the signatory to be domiciled in the European Union. A manufacturer established outside the Union is entitled to carry out all the conformity assessment procedures at his premises and, to sign the EU Declaration of Conformity, unless otherwise provided for in the relevant Union harmonisation legislation.
(216) Article 5 from Decision No 768/2008/EC.
(217) See for example recital 22 of Directive 2014/35/EU, or the similar recital 24 of Directive 2014/34/EU.
(218) Article R10(2) of Annex I to Decision No 768/2008/EC.
(219) Conformity assessment according to the construction products legislation does not follow Decision No 768/2008/EC although construction products legislation provides for the CE marking. The difference is that the CE marking under construction products legislation indicates the level of performance of the product and not conformity in the stricter sense as it is the case for the other legislative acts providing for the CE marking.
(220) Such as machinery, personal protective equipment, active implantable medical devices, medical devices, potentially explosives atmospheres, lifts — as regards safety components, in vitro diagnostic medical devices, radio and telecommunications terminal equipment or marine equipment.
(221) Please note that in the case where several Union harmonisation acts apply to a product and the CE marking appears with an identification number, this does not mean that the notified body intervenes in the conformity assessment process required by each of the applicable acts. Some of the applicable Union harmonisation legislation might not require the intervention of a notified body.
(222) Regulation (EC) No 552/2004 on the interoperability of the European Air Traffic Management network does not provide for CE marking.
(223) The typical example can be a computer.
(224) See under points 4.5.1.7 and 4.5.2
(225) Under the Ecodesign directive the conformity assessment procedures (to be specified in the implementing measure) is laid down in the directive itself as a rule, but in duly justified cases the modules of Decision No 768/2008/EC are prescribed.
(226) The relevant modules are modules C, C1, C2, D, E and F.
(227) Quality management systems — Fundamentals and vocabulary
(228) Quality management systems — Requirements
(*) The legislator may restrict manufacturer's choice.
(229) For subcontracting by notified bodies, see Section 5.2.5
(230) See Annex II to Decision No 768/2008/EC, Module B, point 8, 3rd paragraph.
(231) For more information on the notified body number in NANDO, see point 5.3.3.
(232) Article R27(4) of Annex I to Decision No 768/2008/EC.
(233) For Mutual Recognition Agreements, see Section 9.2.
(234) For market surveillance, see Chapter 7.
(235) Having said that it is common practice in some sectors (e.g. explosives and pyrotechnics articles) that market surveillance authorities rely on notified bodies' testing provided that there is no conflict of interests.
(236) For the coordination between notified bodies, see point 5.2.4.
(237) Article R17(11) of Annex I to Decision No 768/2008/EC
(238) For the role and responsibilities of notified bodies see point 5.2.2.
(239) Article R20(4) of Annex I to Decision No 768/2008/EC.
(240) Please note that only a limited number of Union harmonisation legislation provides for accredited in-house bodies.
(241) See Annex VI for details on the harmonised standards that in-house bodies must be accredited to, depending on the relevant module.
(242) Union harmonisation legislation aligned to Decision No 768/2008/EC includes modified provisions on notified bodies. In view of notifying bodies for the purposes of such legislation, it is essential that at least the relevant provisions concerning notified bodies (which include in particular the requirements and obligations of those bodies) are transposed into national law. In addition, notification procedures need to be communicated to the Commission and other Member States, and Member States need to appoint the notifying authority for that particular Union harmonisation legislation.
(243) Article 7 of the Regulation identifies this as a situation where a conformity assessment body may seek accreditation outside its Member State of establishment.
(244) A similar provision has been included in most Directives aligned to Decision No 768/2008/EC.
(245) On EA's role, see 6.5.2 and 6.5.4.
(246) For more information on withdrawal and de-notification, see point 5.3.4.
(247) According to Article R26 of Decision 768/2008/EC.
(248) Originally, the EN 45000 series of standards which have been revised and superseded by the EN ISO/IEC 17000 series of standards.
(249) The set of conformity assessment procedures to be used by Union harmonisation legislation was first set out in Council Decision 93/465/EEC (the so-called ‘modules Decision’).
(250) NANDO website: http://ec.europa.eu/growth/tools-databases/nando/ as well as EA website: http://www.european-accreditation.org/
(251) Article R14(2) of Annex I to Decision No 768/2008/EC
(252) Most of Union harmonisation legislation aligned with Decision No 768/2008/EC, contains a provision specifying that the notifying authority may delegate the notification tasks under certain conditions. In that case, it may entrust the notification of accredited conformity assessment bodies to a national accreditation body while the notifying authority should notify the non-accredited conformity assessment bodies (should it choose to maintain unaccredited notifications). Such a system would require good internal coordination within the Member State.
(253) See point 6.6 on cross-border accreditation.
(254) Article 11(2) of Regulation (EC) No 765/2008.
(255) ISO/IEC 17011.
(256) For latest information refer to www.ilac.org and www.iaf.nu where listings of current regional members of ILAC and IAF are available.
(257) The General Product Safety Directive also contains requirements on market surveillance. The relationship between Regulation (EC) No 765/2008 and the General Product Safety Directive is described in detail in the Working Paper of 3 March 2010 available at: http://ec.europa.eu/consumers/safety/prod_legis/docs/20100324_guidance_gspd_reg_en.pdf
(258) Subject to specific Union harmonisation legislation.
(259) According to Article 16 of Regulation (EC) No 765/2008 ‘Market surveillance shall ensure that products covered by Union harmonisation legislation which, when used in accordance with their intended purpose or under conditions which can be reasonably foreseen and when properly installed and maintained, are liable to compromise the health or safety of users, or which otherwise do not conform to applicable requirements set out in Union harmonisation legislation are withdrawn or their being made available on the market is prohibited or restricted and that the public, the Commission and the other Member States are informed accordingly. Member States shall ensure that effective measures can be taken in relation to any product category subject to Union harmonisation legislation’.
(260) In this case, the authority is expected to put in place the necessary measures (e.g. Chinese walls) to preserve objectivity and impartiality during post-marketing product checks.
(261) This is important for products (for example machinery and pressure equipment) that are directly, after being manufactured, installed and put into service at the premises of the client.
(262) According to the Directive on high-speed rail systems, each Member State authorises the putting into service of the structural subsystems in their territory. This is a systematic mechanism to monitor the compliance of subsystems and their inter-operability constituents.
(263) Member States are obliged, according to the Directive on the introduction of measures to encourage improvements in the safety and health of workers at work (89/391/EEC), to ensure adequate controls and supervision.
(264) Under Decision No 768/2008/EC, module B, Notified Bodies are required to provide, upon request from Member States, European Commission or other Notified Bodies a copy of the technical documentation.
(265) Unless the EU Declaration of Conformity is required to accompany the product, in which case the distributor should provide the market surveillance authorities with such document.
(266) The reasoned request does not necessarily mean a formal decision by an authority. According to Article 19(1), paragraph 2 of Regulation (EC) No 765/2008, ‘market surveillance authorities may require economic operators to make such documentation and information available as appear to them to be necessary for the purpose of carrying out their activities’. For a request to be reasoned it is sufficient the market surveillance authority explains the context in which the information is requested (e.g. inspection on specific characteristics of the products, random checks, etc.).
(267) For example storing the technical documentation may be delegated to the authorised representative.
(268) These guidelines are available at: http://ec.europa.eu/taxation_customs/resources/documents/common/publications/info_docs/customs/product_safety/guidelines_en.pdf
(269) If following the refusal of release for free circulation by customs the products are declared for customs-approved treatment or use other than release for free circulation, and provided the market surveillance authorities have no objections, the same wording must be added, under the same conditions, to the documents relating to that treatment or use.
(270) For RAPEX, See point 7.5.2.
(271) Also in this case, if following the refusal of release for free circulation by customs the products are declared for customs-approved treatment or use other than release for free circulation, and provided the market surveillance authorities have no objections, the same wording must be added, under the same conditions, to the documents relating to that treatment or use.
(272) A list of market surveillance authorities appointed by the Member States can be found at: http://ec.europa.eu/growth/single-market/goods/building-blocks/market-surveillance/organisation/index_en.htm
(273) A similar provision can be found in the GPSD.
(274) The public national market surveillance programmes can be consulted here: http://ec.europa.eu/growth/single-market/goods/building-blocks/market-surveillance/organisation/index_en.htm
(275) The national reviews and assessments can be found here: http://ec.europa.eu/growth/single-market/goods/building-blocks/market-surveillance/organisation/index_en.htm
(276) See General Product Safety Directive, whereas n. 24 and 35 and Article 16; see also Regulation (EC) No 765/2008, Article 19(5).
(277) See the Rapid Alert System Guidelines for a more precise definition of ‘risk’ and ‘serious risk’.
(278) http://ec.europa.eu/consumers/archive/safety/rapex/docs/corrective_action_guide_march2012.pdf
(279) See Directives relating to simple pressure vessels, toys, machinery, personal protective equipment, non-automatic weighing instruments, active implantable medical devices, gas appliances, potentially explosive atmospheres, medical devices, recreational craft, lifts refrigeration appliances, pressure equipment, ecodesign requirements for energy-related products and in vitro diagnostic medical devices.
(280) An explicit provision to consult has been included in Article 21 of Regulation (EC) No 765/2008, as well as in the Directives relating to medical devices and in vitro diagnostic medical services.
(281) The safeguard clause procedures under the Union harmonisation legislation apply independently from Rapid Alert System. Accordingly, Rapid Alert System does not necessarily have to come into play before the safeguard clause procedure is applied. However, the safeguard clause procedure has to be applied, in addition to Rapid Alert System, when a Member State takes a decision to permanently prohibit or restrict the free movement of harmonised products on the basis of a danger or other serious risk presented by the product.
(282) Even if it may not constitute a safeguard clause, the market surveillance authorities shall inform the Commission and other Member States of actions taken against non-compliant products where the non-compliance is not restricted to the national territory (see Article R31(2) of Annex I to Decision No 768/2008/EC.
(283) This notification should be made via ICSMS. A link between the ICSMS database and the GRS RAPEX IT tool will prevent double encoding of information by national authorities for the purposes respectively of the safeguard clause process and rapid alerts according to Article 22 of the Regulation (EC) No 765/2008.
(284) Union harmonisation legislation aligned to Decision No 768/2008/EC provides for a safeguard procedure which applies only in the event of disagreement between Member States over measures taken by a Member State. The aim is to ensure that proportionate and appropriate measures were taken when a non-compliant product is present in their territory and that similar approaches are taken in the different Member States.While in the past a notification of a risk of a product was notified, Commission had to open a case and elaborate an opinion, now, this burden has been removed and a safeguard case is only opened if a Member State or Commission objects to the measure taken by the notifying authority. Where the Member States and the Commission agree as to the justification of a measure taken by a Member State, no further involvement of the Commission is required, except where non-compliance can be attributed to shortcomings of a harmonised standard.
(285) An explicit obligation for administrative cooperation is laid down in the Directives relating to pressure equipment and in vitro diagnostic medical devices: Member States are required to take appropriate measures in order to encourage/ensure that the authorities responsible for implementing the Directive cooperate with each other, and provide each other (and the Commission) with information in order to assist the functioning of the Directive.
(286) For ICSMS, see point 7.5.3.
(287) See Judgement of the Court, cases 272/80 and 25/88.
(288) An information request does not infringe the right of a national authority to take whatever measures are needed to ensure compliance with Union harmonisation legislation within its jurisdiction.
(289) Under Article 22 of Regulation (EC) No 765/2008, the Rapid Alert System applies to products covered by Union harmonisation legislation.
(290) In the field of medicinal products and medical devices, there is a specific information exchange system.
(291) Adopted as Commission Decision 2010/15/EU of 16 December 2009 laying down guidelines for the management of the Community Rapid Information System ‘RAPEX’ established under Article 12 and of the notification procedure established under Article 11 of Directive 2001/95/EC (the General Product safety Directive (OJ L 22, 26.1.2010, p. 1). The Commission is in the process of drafting an EU wide Risk Assessment Methodology which builds on the RAPEX guidelines, developed within the framework of the GPSD and extends risks assessment to products that can harm the health and safety of professional users or other public interests.
(292) Decision 2010/15/EU is available at: http://ec.europa.eu/consumers/safety/rapex/docs/rapex_guid_26012010_en.pdf
(293) General Rapid Alert System for the RAPEX notifications. GRAS-RAPEX replaced RAPEX-REIS (Rapid Exchange Information System for the Rapid Alert System application and extended the scope of Rapid Alert System to professional products and to other risks than health and safety.
(294) http://ec.europa.eu/consumers/consumers_safety/safety_products/rapex/index_en.htm
(295) See Directives relating to active implantable medical devices, medical devices and in vitro diagnostic medical devices.
(296) This chapter deals only with products subject to Union harmonisation legislation. The free movement is products not subject to Union harmonisation legislation is dealt by the Guide to the application of Treaty provisions governing the free movement of goods, available at: http://ec.europa.eu/DocsRoom/documents/104
(297) A more detailed description of the procedures to be followed in case of products presenting a risk to the health or safety of persons or to other aspects of public interest protection is provided in Chapter 7.
(298) For the EEA Agreement, see point 2.8.2.
(299) The issues related to the WTO agreement are beyond the scope of the Guide.
(300) The full text of the MRA EU-Switzerland and specific provisions may be found on the homepage of the Commission: http://ec.europa.eu/growth/single-market/goods/international-aspects/mutual-recognition-agreements/index_en.htm
ANNEX I
EU LEGISLATION REFERRED TO IN THE GUIDE (NON-EXHAUSTIVE LIST)
|
Horizontal Union harmonisation Act |
Number (amendment) |
Reference in the OJ |
|
Regulation (EC) No 765/2008 of the European Parliament and of the Council of 9 July 2008 setting out the requirements for accreditation and market surveillance relating to the marketing of products and repealing Regulation (EEC) No 339/93 |
765/2008 |
|
|
Decision No 768/2008/EC of the European Parliament and of the Council of 9 July 2008 on a common framework for the marketing of products |
768/2008/EC |
|
|
Regulation (EC) No 764/2008 of the European Parliament and of the Council of 9 July 2008 laying down procedures relating to the application of certain national technical rules to products lawfully marketed in another Member State and repealing Decision No 3052/95/EC |
764/2008 |
|
|
Council Directive 85/374/EEC of 25 July 1985 on the approximation of the laws, regulations and administrative provisions of the Member States concerning liability for defective products |
85/374/EEC (1999/34/EC) |
|
|
Directive 2001/95/EC of the European Parliament and of the Council of 3 December 2001 on General Product Safety |
2001/95/EC |
|
|
Regulation (EU) No 1025/2012 of the European Parliament and of the Council of 25 October 2012 on European standardisation |
1025/2012 |
|
Sectoral Union harmonisation Act |
Number (amendment) |
Reference in the OJ |
|
Council Directive of 19 February 1973 on the harmonisation of the laws of Member States relating to electrical equipment designed for use within certain voltage limits |
73/23/EEC 93/68/ЕЕС 2006/95/EC |
|
|
Directive 2014/35/EU of the European Parliament and of the Council of 26 February 2014 on the harmonisation of the laws of the Member States relating to the making available on the market of electrical equipment designed for use within certain voltage limits (recast) |
2014/35/EU |
|
|
Directive 2009/48/EC of the European Parliament and of the Council of 18 June 2009 on the safety of toys |
2009/48/EC |
|
|
Council Directive 89/336/EEC of 3 May 1989 on the approximation of the laws of the Member States relating to electromagnetic compatibility |
89/336/EEC 92/31/EEC 93/68/ЕЕС 2004/108/EC (98/1З/ЕС) |
|
|
Directive 2014/30/EU of the European Parliament and of the Council of 26 February 2014 on the harmonisation of the laws of the Member States relating to electromagnetic compatibility (recast) |
2014/30/EU |
|
|
Directive of the European Parliament and of the Council of 22 June 1998 on the approximation of the laws of the Member States relating to machinery |
98/37/EC 98/79/EC |
|
|
Council Directive of 21 December 1989 on the approximation of the laws of the Member States relating to personal protective equipment |
89/686/EEC 93/68/ЕЕС 93/95/ЕЕС 96/58/ЕС |
|
|
Directive 2009/23/EC of the European Parliament and of the Council of 23 April 2009 on non-automatic weighing instruments |
90/384/EEC 93/68/ЕЕС 2009/23/EC |
|
|
Directive 2014/31/EU of the European Parliament and of the Council of 26 February 2014 on the harmonisation of the laws of the Member States relating to the making available on the market of non-automatic weighing instruments (recast) |
2014/31/EU |
|
|
Directive 2004/22/EC of the European Parliament and of the Council of 31 March 2004 on measuring instruments |
2004/22/EC |
|
|
Directive 2014/32/EU of the European Parliament and of the Council of 26 February 2014 on the harmonisation of the laws of the Member States relating to the making available on the market of measuring instruments (recast) |
2014/32/EU |
|
|
Council Directive 93/42/EEC of 14 June 1993 concerning medical devices |
93/42/EEC 98/79/ЕС 2000/70/EC 2001/104/EC 2007/97/EC |
|
|
Council Directive of 20 June 1990 on the approximation of the laws of the Member States relating to active implantable medical devices |
90/385/EEC 93/42/ЕЕС 93/68/ЕЕС |
|
|
Directive 98/79/EC of the European Parliament and of the Council of 27 October 1998 on in vitro diagnostic medical devices |
98/79/EC |
|
|
Council Directive 90/396/EEC of 29 June 1990 on the approximation of the laws of the Member States relating to appliances burning gaseous fuels |
90/396/EEC 93/68/ЕЕС 09/142/EC |
|
|
Council Directive of 5 April 1993 on the harmonisation of the provisions relating to the placing on the market and supervision of explosives for civil uses |
93/15/EEC |
|
|
Directive 2014/28/EU of the European Parliament and of the Council of 26 February 2014 on the harmonisation of the laws of the Member States relating to the making available on the market and supervision of explosives for civil uses (recast) |
2014/28/EU |
|
|
Directive 2007/23/EC of the European Parliament and of the Council of 23 May 2007 on the placing on the market of pyrotechnic articles |
2007/23/EC |
|
|
Directive 2013/29/EU of the European Parliament and of the Council of 12 June 2013 on the harmonisation of the laws of the Member States relating to the making available on the market of pyrotechnic articles (recast) |
2013/29/EU |
|
|
Directive 94/9/EC of the European Parliament and the Council of 23 March 1994 on the approximation of the laws of the Member States concerning equipment and protective systems intended for use in potentially explosive atmospheres |
94/9/EC |
|
|
Directive 2014/34/EU of the European Parliament and of the Council of 26 February 2014 on the harmonisation of the laws of the Member States relating to equipment and protective systems intended for use in potentially explosive atmospheres (recast) |
2014/34/EU |
|
|
Directive 94/25/EC of the European Parliament and of the Council of 16 June 1994 on the approximation of the laws, regulations and administrative provisions of the Member States relating to recreational craft |
94/25/EC 03/44/EC |
|
|
Directive 2013/53/EU of the European Parliament and of the Council of 20 November 2013 on recreational craft and personal watercraft and repealing Directive 94/25/EC |
2013/53/EU |
|
|
European Parliament and Council Directive 95/16/EC of 29 June 1995 on the approximation of the laws of the Member States relating to lifts |
95/16/EC |
|
|
Directive 2014/33/EU of the European Parliament and of the Council of 26 February 2014 on the harmonisation of the laws of the Member States relating to lifts and safety components for lifts (recast) |
2014/33/EU |
|
|
Directive 2000/9/EC of the European Parliament and of the Council of 20 March 2000 relating to cableway installations designed to carry persons |
2000/9/EC |
|
|
Directive 97/23/EC of the European Parliament and of the Council of 29 May 1997 on the approximation of the laws of the Member States concerning pressure equipment |
97/23/EC |
|
|
Directive 2014/68/EU of the European Parliament and of the Council of 15 May 2014 on the harmonisation of the laws of the Member States relating to the making available on the market of pressure equipment (recast) |
2014/68/EU |
|
|
Directive 2009/105/EC of the European Parliament and of the Council of 16 September 2009 relating to simple pressure vessels |
2009/105/EC |
|
|
Directive 2014/29/EU of the European Parliament and of the Council of 26 February 2014 on the harmonisation of the laws of the Member States relating to the making available on the market of simple pressure vessels (recast) |
2014/29/EU |
|
|
Directive 2010/35/EU of the European Parliament and of the Council of 16 June 2010 on transportable pressure equipment |
2010/35/EC |
|
|
Council Directive of 20 May 1975 on the approximation of the laws of the Member States relating to aerosol dispensers |
75/324/EEC 94/1/EC 2008/47/EC |
|
|
Directive 99/5/EC of the European Parliament and of the Council relating to radio equipment and telecommunications terminal equipment and the mutual recognition of their conformity |
99/5/EC |
|
|
Directive 2014/53/EU of the European Parliament and of the Council of 16 April 2014 on the harmonisation of the laws of the Member States relating to the making available on the market of radio equipment and repealing Directive 1999/5/EC |
2014/53/EU |
|
|
Directive 2009/125/EC of the European Parliament and of the Council of 21 October 2009 establishing a framework for the setting of ecodesign requirements for energy-related products |
2009/125/EC |
|
|
Directive 97/68/EC of the European Parliament and of the Council on the approximation of the laws of the Member States relating to measures against the emission of gaseous and particulate pollutants from internal combustion engines to be installed in non-road mobile machinery |
97/68/EC 2002/88/EC 2004/26/EC 2006/105/EC 2010/26/EU 2011/88/EU 2012/46/EU |
|
|
Directive 2000/14/EC of the European Parliament and of the Council of 8 May 2000 on the approximation of the laws of the Member States relating to the noise emission in the environment by equipment for use outdoors |
2000/14/EC 2005/88/EC 219/2009 |
|
|
Directive 2011/65/EU of the European Parliament and of the Council of 8 June 2011 on the restriction of the use of certain hazardous substances in electrical and electronic equipment (RoHS) |
2011/65/EU |
|
|
Directive 2012/19/EU of the European Parliament and of the Council of 4 July 2012 on waste electrical and electronic equipment (WEEE) |
2012/19/EU |
|
|
Council Directive 96/98/EC of 20 December 1996 on marine equipment |
96/98/EC |
|
|
Directive 2014/90/EU of the European Parliament and of the Council of 23 July 2014 on marine equipment and repealing Council Directive 96/98/EC |
2014/90/EU |
|
|
Council Directive 2008/57/EC of 17 June 2008 on the interoperability of the rail system within the Community |
2008/57/EC 2009/131/EC 2011/18/EU 2013/9/EU |
|
|
European Parliament and Council Directive 94/62/EC of 20 December 1994 on packaging and packaging waste |
94/62/EC 2004/12/EC 2005/20/EC |
|
|
Regulation (EC) No 552/2004 of the European Parliament and of the Council of 10 March 2004 on the interoperability of the European Air Traffic Management network |
552/2004 1070/2009 |
|
|
Directive 2010/30/EU of the European Parliament and of the Council of 19 May 2010 on the indication by labelling and standard product information of the consumption of energy and other resources by energy-related products |
2010/30/EU |
|
|
Regulation (EC) No 1222/2009 of the European Parliament and of the Council of 25 November 2009 on the labelling of tyres with respect to fuel efficiency and other essential parameters |
1222/2009 |
ANNEX II
ADDITIONAL GUIDANCE DOCUMENTS
|
— |
Guidance documents from the Expert Group on Toy Safety: http://ec.europa.eu/growth/sectors/toys/safety/guidance/index_en.htm |
|
— |
Measuring instruments and non-automatic weighing instruments: http://ec.europa.eu/growth/single-market/goods/building-blocks/legal-metrology/measuring-instruments/guidance-standards/index_en.htm |
|
— |
Chemicals: http://echa.europa.eu/support/guidance |
|
— |
Low Voltage Directive — Guidelines on application and recommendations: http://ec.europa.eu/growth/sectors/electrical-engineering/lvd-directive/index_en.htm |
|
— |
Electromagnetic Compatibility (EMC) — Guidance: http://ec.europa.eu/growth/sectors/electrical-engineering/emc-directive/index_en.htm |
|
— |
Radio and telecommunications terminal equipment (R&TTE) Guidance: http://ec.europa.eu/growth/sectors/electrical-engineering/rtte-directive/index_en.htm |
|
— |
Medical Devices — Interpretative documents: http://ec.europa.eu/growth/sectors/medical-devices/guidance/index_en.htm |
|
— |
Frequently Asked Questions on the Construction Products Regulation (CPR): http://ec.europa.eu/growth/sectors/construction/product-regulation/faq/index_en.htm |
|
— |
Automotive industry — Frequently asked questions: http://ec.europa.eu/growth/sectors/automotive/index_en.htm |
|
— |
RoHS 2 — Frequently asked questions: http://ec.europa.eu/environment/waste/rohs_eee/events_rohs3_en.htm |
|
— |
Pressure Equipment Directive (PED): guidelines: http://ec.europa.eu/growth/sectors/pressure-gas/pressure-equipment/guidelines/index_en.htm |
|
— |
Machinery — Guidance Documents: http://ec.europa.eu/growth/sectors/mechanical-engineering/machinery/index_en.htm |
|
— |
Directive on cableways — Guide to application: http://ec.europa.eu/growth/sectors/mechanical-engineering/cableways/index_en.htm |
|
— |
Directive on lifts — Guide to application: http://ec.europa.eu/growth/sectors/mechanical-engineering/lifts/index_en.htm |
|
— |
Directive on Personal Protective Equipment — Guide to application: http://ec.europa.eu/growth/sectors/mechanical-engineering/personal-protective-equipment/index_en.htm |
|
— |
Directive on the noise emission in the environment by equipment for use outdoors — Guide to application, publications and studies: http://ec.europa.eu/growth/sectors/mechanical-engineering/noise-emissions/index_en.htm |
|
— |
Guidelines on the application of Directive 94/9/EC of 23 March 1994 on the approximation of the laws of the Member States concerning equipment and protective systems intended for use in potentially Explosive Atmospheres (Fourth edition September 2012): http://ec.europa.eu/growth/sectors/mechanical-engineering/atex/index_en.htm |
|
— |
Healthcare industries — Frequently asked questions: http://ec.europa.eu/growth/sectors/healthcare/index_en.htm |
|
— |
Guide to the practical application of the General Product Safety Directive: http://ec.europa.eu/consumers/safety/prod_legis/index_en.htm |
|
— |
RAPEX Rapid Alert System Guidelines: http://ec.europa.eu/consumers/consumers_safety/safety_products/rapex/index_en.htm |
|
— |
European standards — General framework: http://ec.europa.eu/growth/single-market/european-standards/policy/framework/index_en.htm |
|
— |
Vademecum on European standardisation in support of Union legislation and policies (SWD(2015) 205 final, 27.10.2015): http://ec.europa.eu/growth/single-market/european-standards/vademecum/index_en.htm |
ANNEX III
USEFUL WEB ADDRESSES
|
— |
Single Market for Goods http://ec.europa.eu/growth/single-market/goods/index_en.htm |
|
— |
Internal Market for products http://ec.europa.eu/growth/single-market/goods/index_en.htm |
|
— |
European standards http://ec.europa.eu/growth/single-market/european-standards/index_en.htm |
|
— |
Rapid Alert System for non-food products posing a serious risk http://ec.europa.eu/consumers/consumers_safety/safety_products/rapex/index_en.htm |
ANNEX IV
CONFORMITY ASSESSMENT PROCEDURES (MODULES FROM DECISION NO 768/2008/EC)
|
Modules |
Manufacturer |
Manufacturer or Authorised representative |
Conformity assessment body |
||||||||||||||||||||||||||||||||||||||||
|
A (Internal production control)
|
|
|
No involvement of conformity assessment body. The manufacturer carries out himself all checks a notified body would do |
||||||||||||||||||||||||||||||||||||||||
|
A1 (Internal production control plus supervised product testing)
|
|
|
Either notified body or in-house accredited body (manufacturer's choice) (*):
|
||||||||||||||||||||||||||||||||||||||||
|
A2 (Internal production control plus supervised product checks at random intervals)
|
|
|
Either notified body or in-house accredited body(manufacturer's choice) (*):
|
||||||||||||||||||||||||||||||||||||||||
|
B (EC-type examination)
|
|
|
Notified Body
In this respect, the legislator lays down which of the following ways must be used:
|
||||||||||||||||||||||||||||||||||||||||
|
C (Conformity to type based on internal production control)
|
|
|
|
||||||||||||||||||||||||||||||||||||||||
|
C1 (Conformity to type based on internal production control plus supervised product testing)
|
|
|
Either notified body or in-house accredited body (manufacturer's choice) (*):
|
||||||||||||||||||||||||||||||||||||||||
|
C2 (Conformity to type based on internal production control plus supervised product checks at random intervals)
|
|
|
Either notified body or in-house accredited body(manufacturer's choice) (*):
|
||||||||||||||||||||||||||||||||||||||||
|
D (Conformity to EC-type based on quality assurance of the production process)
|
|
|
Notified Body
|
||||||||||||||||||||||||||||||||||||||||
|
D1 (Quality assurance of the production process)
|
|
|
Notified Body
|
||||||||||||||||||||||||||||||||||||||||
|
E (Conformity to EC-type based on product quality assurance)
|
|
|
Notified Body
|
||||||||||||||||||||||||||||||||||||||||
|
E1 (Quality assurance of final product inspection and testing)
|
|
|
Notified Body
|
||||||||||||||||||||||||||||||||||||||||
|
F (Conformity to EC-type based on product verification)
|
|
|
Notified Body
|
||||||||||||||||||||||||||||||||||||||||
|
F1 (Conformity based on product verification)
|
|
|
Notified Body
|
||||||||||||||||||||||||||||||||||||||||
|
G (Conformity based on unit verification)
|
|
|
Notified Body
|
||||||||||||||||||||||||||||||||||||||||
|
H (Conformity based on full quality assurance)
|
|
|
Notified Body
|
||||||||||||||||||||||||||||||||||||||||
|
H1 (Conformity based on full quality assurance plus design examination)
|
|
|
Notified Body
|
(*) The legislator may restrict manufacturer's choice
ANNEX V
RELATION BETWEEN ISO 9001 AND MODULES REQUIRING A QUALITY ASSURANCE SYSTEM
|
Quality requirements referred to under modules of Decision 768/2008 |
Module D |
Module D1 |
Module E |
Module E1 |
Module H |
Module H1 |
||||
|
EN ISO 9001:2008, §5.1, §5.3, §5.4, §5.5, §5.6 (without §5.6.2.b — customer feed-back) |
EN ISO 9001:2008, §5.1, §5.3, §5.4, §5.5, §5.6 (without §5.6.2.b — customer feed-back) |
EN ISO 9001:2008, §5.1, §5.3, §5.4 (without reference to §7.1), §5.5, §5.6 (without §5.6.2.b — customer feed-back) |
EN ISO 9001:2008, §5.1, §5.3, §5.4 (without reference to §7.1), §5.5, §5.6 (without §5.6.2.b — customer feed-back) |
EN ISO 9001:2008, §5.1, §5.3, §5.4, §5.5, §5.6 (without §5.6.2.b — customer feed-back) |
EN ISO 9001:2008, §5.1, §5.3, §5.4, §5.5, §5.6 (without §5.6.2.b — customer feed-back) |
||||
|
Not relevant — Module D does not cover the design phase |
Not relevant — under module D1, design issues are covered by means of technical documentation |
Not relevant — Module E does not cover the design phase |
Not relevant — under module E1, design issues are covered by means of technical documentation |
EN ISO 9001:2008, §7.3.1, §7.3.2, §7.3.3 |
EN ISO 9001:2008, §7.3.1, §7.3.2, §7.3.3 |
||||
|
Not relevant — Module D does not cover the design phase |
Not relevant — under module D1, design issues are covered by means of technical documentation |
Not relevant — Module E does not cover the design phase |
Not relevant — under module E1, design issues are covered by means of technical documentation |
EN ISO 9001:2008, §7.3.4 — §7.3.7 |
EN ISO 9001:2008, §7.3.4 — §7.3.7 |
||||
|
EN ISO 9001:2008, §7.5.1, §7.5.2, §7.5.3 |
EN ISO 9001:2008, §7.5.1, §7.5.2, §7.5.3 |
Not relevant — module E does not cover the manufacturing part |
Not relevant — module E1 does not cover the manufacturing part |
EN ISO 9001:2008, §7.5.1, §7.5.2, §7.5.3 |
EN ISO 9001:2008, §7.5.1, §7.5.2, §7.5.3 |
||||
|
EN ISO 9001:2008, §7.6, §8.2.2, §8.2.3, §8.2.4 (only first paragraph), §8.3, §8.4 (without §8.4.a -customer satisfaction), §8.5 |
EN ISO 9001:2008, §7.6, §8.2.2, §8.2.3, §8.2.4 (only first paragraph), §8.3, §8.4 (without §8.4.a -customer satisfaction), §8.5 |
EN ISO 9001:2008, §7.6, §8.2.2, §8.2.3, §8.2.4 (only first paragraph), §8.3, §8.4 (without §8.4.a -customer satisfaction), §8.5 |
EN ISO 9001:2008, §7.6, §8.2.2, §8.2.3, §8.2.4 (only first paragraph), §8.3, §8.4 (without §8.4.a -customer satisfaction), §8.5 |
EN ISO 9001:2008, §7.6, §8.2.2, §8.2.3, §8.2.4 (only first paragraph), §8.3, §8.4 (without §8.4.a -customer satisfaction), §8.5 |
EN ISO 9001:2008, §7.6, §8.2.2, §8.2.3, §8.2.4 (only first paragraph), §8.3, §8.4 (without §8.4.a -customer satisfaction), §8.5 |
||||
|
EN ISO 9001:2008, §4 |
EN ISO 9001:2008, §4 |
EN ISO 9001:2008, §4 |
EN ISO 9001:2008, §4 |
EN ISO 9001:2008, §4 |
EN ISO 9001:2008, §4 |
ANNEX VI
USING HARMONISED STANDARDS TO ASSESS THE COMPETENCE OF CONFORMITY ASSESSMENT BODIES
Please note that the annex below is indicative guidance only. It does not lay down procedures for assessing the competence of Conformity Assessment Bodies.
1. REQUIREMENTS ON CONFORMITY ASSESSMENT BODIES
A conformity assessment body wishing to be notified under an Union harmonisation act for one or several conformity assessment modules of Decision No 768/2008/EC has to be assessed in order to determine if it is technically competent to carry out the tasks required by the module(s) in question.
Equally important is the continuous surveillance of the competence of the notified body. This must be done at regular intervals and follow the practice established by the accreditation organisations.
The assessment process must determine if the conformity assessment body has adequately trained technical staff with knowledge and experience of the relevant technology, suitable facilities and equipment, policies and procedures to ensure integrity and impartiality, correct understanding of the directive, etc.
Conformity assessment includes activities such as testing (carried out by laboratories), inspection, certification, etc. Inspection and product certification can be considered similar and there is some overlapping in the definitions. They both go beyond simple testing by including tasks related to the ability to assess test results and decide on conformity. They pursue the same goal (i.e. the assessment of the conformity of a product) in slightly different ways.
Generally, inspection involves direct determination of the conformance with specifications of unique or small series products. Product certification primarily involves determination of the conformance of products manufactured in long series.
In practice, inspection may also involve professional judgement on the basis of general requirements, while product certification is done against standards or other technical specifications.
Thus different criteria apply to conformity assessment bodies depending on whether the latter are laboratories, inspection bodies or certification bodies.
2. SET OF CORE STANDARDS SETTING OUT COMPETENCE CRITERIA FOR CONFORMITY ASSESSMENT BODIES
The general criteria, independent of the sector in question, that notified bodies must fulfil in order to be positively assessed are set out in harmonised standards published in the Official Journal of the EU following mandate M417.
Sectoral legislation may, if necessary, lay down additional specific criteria related to the knowledge of the sector a body must have.
EN ISO/IEC 17025, EN ISO/IEC 17020, EN ISO/IEC 17021, EN ISO/IEC 17065 are the core standards for assessing the competence of conformity assessment bodies. EN ISO/IEC 17020, EN ISO/IEC 17065 focus on criteria for performing conformity assessment, while EN ISO/IEC 17025 tackles in more detail the testing aspect.
|
— |
EN ISO/IEC 17025 (applies to laboratories — replaces EN 45001 and ISO Guide 25) sets out the general requirements a laboratory (first-, second- and third-party and regardless of the number of personnel or the extent of the scope of activities) must meet if it is to be recognised as competent to carry out testing and/or calibration including sampling (sampling was not tackled by ISO 45001). |
These activities involve determining one or several characteristics of a product according to a defined method (can be standard, non-standard methods, laboratory-developed). Compliance of the operation of laboratories with regulatory and safety requirements is not covered by this standard.
When a laboratory does not undertake one or more of the activities covered by this International Standard, such as sampling and the design/development of new methods, the requirements of those clauses do not apply.
|
— |
EN ISO/IEC 17020 (applies to inspection bodies — replaces EN 45004). This standard specifies general criteria for the competence of impartial bodies performing inspection irrespective of the sector involved. |
Inspection involves examination of a product design, product, service, process or plant and determination of their conformity with specific requirements or, on the basis of professional judgement, with general requirements. It also specifies independence criteria. This standard does not cover testing laboratories, certification bodies or the supplier's declaration of conformity.
|
— |
EN ISO/IEC 17065 (applies to certification bodies — replaces EN 45011) specifies the general requirements that a third party operating a product certification system shall meet if it is to be recognised as competent and reliable. |
Product certification entails giving assurance that a product conforms to specified requirements such as regulations, standards or other technical specifications. A product certification system can include, e.g. type testing or examination, testing or inspection of every product or of a particular product, batch testing or inspection, design appraisal, which could be coupled with production surveillance or assessment and surveillance of the manufacturer's quality system. This standard does not cover testing laboratories, inspection bodies or the declaration of conformity. ISO/IEC 17065 means that the four eyes principle is used, that is the reviewer and decision maker is different from the evaluator.
|
— |
ISO/IEC 17021 (replacing EN 45012) contains principles and requirements for the competence and impartiality of bodies performing audit and certification of management systems of all types (e.g. quality management systems or environmental management systems). |
Bodies operating according to this standard are not obliged to offer all types of management system certification. Quality sys- tem certification involves the assessment, determination of conformity against a quality system standard and within a certain scope of activity and surveillance of the manufacturer's quality system.
3. APPROPRIATE STANDARDS RELATED TO THE COMPETENCE OF CONFORMITY ASSESSMENT BODIES FOR EVERY MODULE
The sections below describe which of the above standards are the most appropriate for the tasks in the modules as laid down in the Decision No 768/2008/EC.
3.1 Modules A1, A2, C1, C2
Under these modules the body must have the technical knowledge, experience and ability in carrying out tests. Even if the test equipment is situated with the manufacturer, requirements on the equipment's suitability, functioning, maintenance (e.g. calibration programmes) and measurement traceability must be ensured and should be considered as the responsibility of the notified body. Furthermore, if the manufacturer has not applied the relevant harmonised standards, equivalent tests must be carried out, or failing this, appropriate methods must be developed. In either case, the notified body must validate the tests used.
For A2, C2 the body must in addition be able to deal with statistical methods, sampling plan, random methods, operational characteristics, that are included in the product checking and specified by the applicable Union harmonisation legislation.
In this respect and for all these modules, as EN ISO/IEC 17025, EN ISO/IEC 17020 or EN ISO/IEC 17065 (depending if the body in question is a laboratory, inspection body, product certification body) lay down the competence and deontology criteria for performing product examination, their requirements can be considered as the most appropriate for the assessment of the bodies seeking notification for carrying out the tasks in this module.
However, if the assessment is based on EN ISO/IEC 17025 and as this standard lays down criteria only for testing/calibration without tackling the evaluation of test results by the notified body, the latter must demonstrate separately its capability of and procedures for judging and deciding, based on the results of the tests, if the essential or other legal requirements are fulfilled and/or the harmonised standards have been applied.
On the other hand if EN ISO/IEC 17020 or EN ISO/IEC 17065 are used and as these standards do not tackle criteria for testing/calibration, the requirements on testing activities as set out in EN ISO/IEC 17025 must be taken into account. In all cases the notified body must have the capability of assessing a product regardless of whether the manufacturer has applied relevant harmonised standards or not.
3.2 Module B
The notified body must determine if the product design complies with the relevant legislative requirements.
In this respect stand-alone EN ISO/IEC 17025 must be regarded as not being appropriate for the purpose of module B. The reason is that this standard tackles only pure testing issues and does not cover the important functions of module B concerning evaluation of product design, which due to its complexity (it goes well beyond the mere examination of technical documentation as in modules D1, E1, F1) requires from the notified body additional competencies (similar to modules G, H1)
The requirements in both EN ISO/IEC 17020 and EN ISO/IEC 17065 can be considered as appropriate for the assessment of bodies seeking notification for carrying out the tasks in module B, because these standards lay down the competence and deontology criteria for performing product examination and conformity assessment. However, as these standards do not tackle criteria for testing/calibration, the relevant requirements in EN ISO/IEC 17025 must always be taken into account for the testing required.
3.3 Modules D, D1, E, E1, H
The notified body assesses and decides if the manufacturer's quality system ensures that the products are in conformity with or ensure compliance with the legislative instrument that apply to them (in case of modules D1, E1, H) or with the approved EC-type (in case of modules D, E).
Thus, the requirements in EN ISO/IEC 17021 can be considered as appropriate for the assessment of the bodies seeking notification for carrying out the tasks in this module. It must be underlined that the operation of the manufacturer's quality system must ensure the conformity of the final products with the requirements of the applicable Union harmonisation legislation. Therefore, the notified body must have, in addition, adequate capability of assessing the manufacturer's ability to identify relevant product requirements and carry out the necessary inspections and tests.
EN ISO/IEC 17065 is also considered appropriate for the assessment of modules D, D1, E and E1, which assess the ability of the manufacturer's management system to ensure products are in conformity with the relevant applicable legislation and continue to conform to type. Assessments to these modules are focussed closely on the production processes and controls associated with the product(s) concerned, thus EN ISO/IEC 17065 requirements cover the product and process related aspects as well as covering the management system assessment (according to the requirements of EN ISO/IEC 17065 the management system assessment aspects have to be carried out in accordance with ISO/IEC 17021).
3.4 Modules F, F1
The notified body carries out the appropriate examinations and tests either by examination and testing of every product or by examination and testing of products on a statistical basis. Under module F1, the notified body must in addition examine the technical documentation.
In this respect and for all these modules, as EN ISO/IEC 17025, EN ISO/IEC 17020 or EN ISO/IEC 17065 (depending on whether the body in question is a laboratory, inspection body, product certification body) lay down the competence and deontology criteria for performing product examination, their requirements can be considered as the most appropriate for the assessment of the bodies seeking notification for these modules.
It must be noted that although EN ISO/IEC 17025 does not tackle the examination of product design and although module F1 also covers the design phase, this standard, even stand-alone, remains appropriate for this module: the reason is that design examination under F1 is relatively simple, and is performed only by means of examination of technical documentation and not by means of examination of any specimen or any critical parts of the design that would require additional competences from the notified body as is the case with modules B (or G -see below).
However, if the assessment is based on EN ISO/IEC 17025, and as this standard lays down criteria only for testing/calibration without tackling the evaluation of test results by the notified body, the latter must demonstrate separately its capacity and procedures for judging and deciding, based on the results of the tests, if the essential requirements are fulfilled and/or the harmonised standards have been applied.
On the other hand if EN ISO/IEC 17020 or EN ISO/IEC 17065 are used, and as these standards do not tackle criteria for testing/calibration, the requirements on testing activities as set out in EN ISO/IEC 17025 must be taken into account. In all cases the notified body must have the capability of assessing a product regardless of whether the manufacturer has applied relevant harmonised standards or not.
3.5 Module G
The notified body examines the complete individual product in both design and production phase.
In this respect stand-alone EN ISO/IEC 17025 must be regarded as not being appropriate for the purpose of module G. The reason being that this standard tackles only pure testing issues and does not cover the important functions of module G concerning evaluation of product design, which due to its complexity (it goes well beyond the mere examination of technical documentation as in modules D1, E1, F1) requires from the notified body additional competencies (similar to modules B, H1).
The requirements in both EN ISO/IEC, 17020 or EN ISO/IEC 17065 can be considered as appropriate for the assessment of bodies seeking notification for carrying out the tasks in module G, because these standards lay down the competence and deontology criteria for performing product examination and conformity assessment. However, as these standards do not tackle criteria for testing/calibration, the relevant requirements in EN ISO/IEC 17025 must be always taken into account for the testing required.
3.6 Module H1
The notified body assesses and decides if the manufacturer's quality system ensures that the products are in conformity with the legislative instrument(s) applying to them. Furthermore it examines the manufacturer's technical design specifications, including the necessary supporting evidence and the result of tests carried out by the manufacturer.
Thus, the requirements in EN ISO/IEC 17021 can be considered as appropriate for the assessment of the bodies seeking notification for this module. It must be underlined that the operation of the manufacturer's quality system must ensure the conformity of the final products with the requirements of the applicable Union harmonisation legislation. Therefore, the notified body must have, in addition, adequate capability of assessing the manufacturer's ability to identify relevant product requirements and carry out the necessary inspections and tests.
In addition, as the notified body also examines the product design in order to certify it by issuing a EC-design examination certificate, the requirements in both EN ISO/IEC 17020 or EN ISO/IEC 17065 can be considered as appropriate for the assessment of bodies seeking notification under module H1, because these standards lay down the competence and deontology criteria for performing product examination and conformity assessment. In the case of ISO/IEC 17065 testing elements are covered in that the standard requires any testing to be carried out in accordance with ISO/IEC 17025. However, as EN ISO/IEC 17020 does not tackle criteria for testing/calibration, the relevant requirements in EN ISO/IEC 17025 must be always taken into account for the testing required.
In this respect it must be noted that stand-alone EN ISO/IEC 17025 must be regarded as not being appropriate for the purpose of module H1. The reason is that this standard tackles only pure testing issues and does not cover the important functions of module H concerning evaluation of product design, which due to its complexity (it goes well beyond the mere examination of technical documentation as in modules D1, E1, F1) requires from the notified body additional competencies (similar to modules B, G).
4. SUMMARY
The table that follows reflects the desired approach to the choice of standards for the different modules.
|
Module |
EN Standard(s) applicable |
|
A1, A2 |
EN ISO/IEC 17025 (+ ability to decide on conformity), or EN ISO/IEC 17020, EN ISO/IEC 17025 to be taken into account for testing required, or EN ISO/IEC 17065, EN ISO/IEC 17025 to be taken into account for testing required |
|
B |
EN ISO/IEC 17020, EN ISO/IEC 17025 to be taken into account for testing required, or EN ISO/IEC 17065, EN 17025 to be taken into account for testing required |
|
C1, C2 |
EN ISO/IEC 17025 (+ ability to decide on conformity), or EN ISO/IEC 17020, EN ISO/IEC 17025 to be taken into account for testing required, or EN ISO/IEC 17065, EN ISO/IEC 17025 to be taken into account for testing required |
|
D, D1 |
EN ISO/IEC 17021 (+ product related knowledge) or EN ISO/IEC 17065 |
|
E, E1 |
EN ISO/IEC 17021(+ product related knowledge) or EN ISO/IEC 17065 |
|
F, F1 |
EN ISO/IEC 17025 (+ ability to decide on conformity), or EN ISO/IEC 17020, EN 17025 to be taken into account for testing required, or EN ISO/IEC 17065, EN 17025 to be taken into account for testing required |
|
G |
EN ISO/IEC 17020, EN 17025 to be taken into account for testing required, or EN ISO/IEC 17065, EN 17025 to be taken into account for testing required |
|
H |
EN ISO/IEC 17021 (+ product related knowledge) |
|
H1 |
EN ISO/IEC 17021 (+ product related knowledge) or EN ISO/IEC 17065 or EN ISO/IEC 17020, EN 17025 to be taken into account for testing required |
ANNEX VII
FREQUENTLY ASKED QUESTIONS ON CE MARKING
What does the CE marking on a product indicate?
By affixing the CE marking to a product, the manufacturer declares on his sole responsibility that the product is in conformity with the essential requirements of the applicable Union harmonisation legislation providing for its affixing and that the relevant conformity assessment procedures have been fulfilled. Products bearing the CE marking are presumed to be in compliance with the applicable Union harmonisation legislation and hence benefit from free circulation in the European Market.
Is a product affixed with the CE marking always produced in the EU?
No. The CE marking only signals that all essential requirements have been fulfilled when the product was manufactured. The CE marking is not a mark of origin, as it does not indicate that the product was manufactured in the European Union. Consequently, a product affixed with the CE marking may have been produced anywhere in the world.
Are all CE marked products tested and approved by authorities?
No. In fact, the assessment of the conformity of the products with the legislative requirements applying to them is the sole responsibility of the manufacturer. The manufacturer affixes the CE marking and drafts the EU declaration of conformity. Only products which are regarded as presenting a high risk to the public interest, e.g. pressure vessels, lifts and certain machine tools, require conformity assessment by a third party, i.e. a notified body.
Can I, as a manufacturer, affix my products with the CE marking myself?
Yes, the CE marking is always affixed by the manufacturer himself or his authorised representative after the necessary conformity assessment procedure has been performed. This means that, before being affixed with the CE marking and being placed on the market, the product must be subject to the conformity assessment procedure provided for in one or more of the applicable Union harmonisation acts. The latter establish whether the conformity assessment may be performed by the manufacturer him- self or if the intervention of a third party (the notified body) is required.
Where should the CE marking be affixed?
The marking shall be affixed either to the product or to the product's data plate. When that is not possible due to the nature of the product, the CE marking shall be affixed to the packaging and/or to any accompanying documents.
What is a manufacturer's declaration of conformity?
The EU declaration of conformity (EU DoC) is a document in which the manufacturer, or his authorised representative within the European Economic Area (EEA), indicates that the product meets all the necessary requirements of the Union harmonisation legislation applicable to the specific product. The EU DoC shall also contain the name and address of the manufacturer along with information about the product, such as the brand and serial number. The EU DoC must be signed by an individual working for the manufacturer or his authorised representative, and the employee's function shall also be indicated.
Whether a Notified Body has been involved or not, the manufacturer must draw up and sign the EE declaration of conformity.
Is CE marking mandatory, and if so; for what products?
Yes, CE marking is mandatory. However, only the products that are covered by the scope of one or more of the Union harmonisation acts providing for CE marking shall be affixed with it in order to be placed on the Union market. Examples of products that fall under Union harmonisation acts providing for CE marking are toys, electrical products, machinery, personal protective equipment and lifts. Products that are not covered by CE marking legislation shall not bear the CE marking.
Information the products that are CE marked and the Union harmonisation legislation providing for CE Marking under
http://ec.europa.eu/growth/single-market/ce-marking/index_en.htm
What is the difference between the CE marking and other markings, and can other markings be affixed on the product if there is a CE marking?
The CE marking is the only marking that indicates conformity to all the essential requirements of the Union harmonisation legislation that provide for its affixing. A product may bear additional markings provided that they do not have the same meaning as the CE marking, that they are not liable to cause confusion with the CE marking and that they do not impair the legibility and visibility of the CE marking. In this respect, other markings may be used only if they contribute to the improvement of consumer protection and are not covered by harmonisation legislation of the European Union.
Who supervises the correct use of the CE marking?
In order to guarantee the impartiality of market surveillance operations, the supervision of the CE marking is the responsibility of public authorities in the Member States in cooperation with the European Commission.
What are the sanctions for counterfeiting the CE marking?
The procedures, measures and sanctions that apply to counterfeiting of the CE marking are laid down in Member State's national administrative and penal law. Depending on the seriousness of the crime, economic operators may be liable to a fine and, in some circumstances, imprisonment. However, if the product is not regarded as an imminent safety risk, the manufacturer may be given a second opportunity to ensure that the product is in conformity with the applicable legislation before being obliged to take the product off the market.
What implications may the affixing of the CE marking have for the manufacturer/importer/distributor?
While manufacturers are responsible for ensuring product compliance and affixing the CE marking, importers and distributors also play an important role in making sure that only products complying with legislation and bearing the CE marking are placed on the market. Not only does this help to reinforce the EU's health, safety and environmental protection requirements, it also supports fair competition with all players being held accountable to the same rules.
When products are produced in third countries and the manufacturer is not represented in the EEA, importers must make sure that the products placed by them on the market comply with the applicable requirements and do not present a risk to the European public. The importer must verify that the manufacturer outside the EU has undertaken the necessary steps and that the documentation is available upon request.
Thus, importers must have an overall knowledge of the respective Union harmonisation acts and are obliged to support national authorities should problems arise. Importers should have a written assurance from the manufacturer that they will have access to the necessary documentation — such as the EU declaration of conformity and the technical documentation — and be able to provide it to national authorities, if requested. Importers should also make sure that contact with the manufacturer can always be established.
Further along in the supply chain, distributors play an important role in ensuring that only compliant products are on the market and must act with due care to ensure that their handling of the product does not adversely affect its compliance. The distributor must also have a basic knowledge of the legal requirements — including which products must bear the CE marking and the ac- companying documentation — and should be able to identify products that are clearly not in compliance.
Distributors must be able to demonstrate to national authorities that they have acted with due care and have affirmation from the manufacturer or the importer that the necessary measures have been taken. Furthermore, a distributor must be able to assist the national authority in its efforts to receive the required documentation.
If the importer or distributor markets the products under his own name, he then takes over the manufacturer's responsibilities. In this case, he must have sufficient information on the design and production of the product, as he will be assuming the legal responsibility when affixing the CE marking.
Where can I find more information?
Information about CE marking, the products that are CE marked, the Union harmonisation legislation providing for CE Marking and the steps to follow under
http://ec.europa.eu/growth/single-market/ce-marking/index_en.htm
Economic operators may contact the Enterprise Europe Network under
http://een.ec.europa.eu/