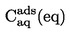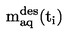ISSN 1725-2520
Den Europæiske Unions
Tidende
L 142

Dansk udgave
Retsforskrifter
51. årgang
31. maj 2008
|
ISSN 1725-2520 |
||
|
Den Europæiske Unions Tidende |
L 142 |
|
|
|
||
|
Dansk udgave |
Retsforskrifter |
51. årgang |
|
Indhold |
|
I Retsakter vedtaget i henhold til traktaterne om oprettelse af Det Europæiske Fællesskab/Euratom, hvis offentliggørelse er obligatorisk |
Side |
|
|
|
FORORDNINGER |
|
|
|
* |
Kommissionens forordning (EF) Nr. 440/2008 af 30. maj 2008 om fastlæggelse af forsøgsmetoder i henhold til Europa-Parlamentets og Rådets forordning (EF) Nr. 1907/2006 om registrering, vurdering og godkendelse af samt begrænsninger for kemikalier (REACH) ( 1 ) |
|
|
|
|
|
(1) EØS-relevant tekst |
|
DA |
De akter, hvis titel er trykt med magre typer, er løbende retsakter inden for rammerne af landbrugspolitikken og har normalt en begrænset gyldighedsperiode. Titlen på alle øvrige akter er trykt med fede typer efter en asterisk. |
I Retsakter vedtaget i henhold til traktaterne om oprettelse af Det Europæiske Fællesskab/Euratom, hvis offentliggørelse er obligatorisk
FORORDNINGER
|
31.5.2008 |
DA |
Den Europæiske Unions Tidende |
L 142/1 |
Kommissionens forordning (EF) Nr. 440/2008
af 30. maj 2008
om fastlæggelse af forsøgsmetoder i henhold til Europa-Parlamentets og Rådets forordning (EF) Nr. 1907/2006 om registrering, vurdering og godkendelse af samt begrænsninger for kemikalier (REACH)
(EØS-relevant tekst)
KOMMISSIONEN FOR DE EUROPÆISKE FÆLLESSKABER HAR —
under henvisning til traktaten om oprettelse af Det Europæiske Fællesskab,
under henvisning til Europa-Parlamentets og Rådets forordning (EF) nr. 1907/2006 af 18. december 2006 om registrering, vurdering og godkendelse af samt begrænsninger for kemikalier (REACH), om oprettelse af et europæisk kemikalieagentur og om ændring af direktiv 1999/45/EF og ophævelse af Rådets forordning (EØF) nr. 793/93 og Kommissionens forordning (EF) nr. 1488/94 samt Rådets direktiv 76/769/EØF og Kommissionens direktiv 91/155/EØF, 93/67/EØF, 93/105/EF og 2000/21/EF (1), særlig artikel 13, stk. 3, og
ud fra følgende betragtninger:
|
(1) |
I henhold til forordning (EF) nr. 1907/2006 skal der på EU-plan fastlægges forsøgsmetoder til undersøgelse af stoffer, hvis forsøg er påkrævet til fremskaffelse af oplysninger om stoffers iboende egenskaber. |
|
(2) |
I Rådets direktiv 67/548/EØF af 27. juni 1967 om tilnærmelse af lovgivning om klassificering, emballering og etikettering af farlige stoffer (2) indeholder bilag V metoderne til bestemmelse af stoffers og præparaters fysisk-kemiske egenskaber, toksicitet og økotoksicitet. Ved direktiv 2006/121/EF er bilag V til direktiv 67/548/EØF udgået med virkning fra den 1. juni 2008. |
|
(3) |
Forsøgsmetoderne i bilag V til direktiv 67/548/EØF bør indarbejdes i denne forordning. |
|
(4) |
Denne forordning udelukker ikke, at der anvendes andre testmetoder, når blot deres anvendelse er i overensstemmelse med artikel 13, stk. 3, i forordning 1907/2006. |
|
(5) |
Ved udformningen af forsøgsmetoderne bør der i fuldt omfang tages hensyn til principperne om at erstatte, begrænse og raffinere brugen af dyr i procedurer, især når der bliver rådighed over validerede metoder, som kan erstatte, begrænse eller raffinere dyreforsøg. |
|
(6) |
De i denne forordning fastsatte foranstaltninger er i overensstemmelse med udtalelse fra det udvalg, der er nedsat ved artikel 133 i forordning (EØF) nr. 1907/2006 — |
UDSTEDT FØLGENDE FORORDNING:
Artikel 1
De forsøgsmetoder, der skal anvendes i forbindelse med forordning (EF) nr. 1907/2006, er fastlagt i bilaget til nærværende forordning.
Artikel 2
Kommissionen revurderer efter behov forsøgsmetoderne i denne forordning med henblik på at erstatte, begrænse eller raffinere forsøg med hvirveldyr.
Artikel 3
Alle henvisninger til bilag V til direktiv 67/548/EØF skal betragtes som henvisninger til nærværende forordning.
Artikel 4
Denne forordning træder i kraft dagen efter offentliggørelsen i Den Europæiske Unions Tidende.
Den anvendes fra den 1. juni 2008.
Denne forordning er bindende i alle enkeltheder og gælder umiddelbart i hver medlemsstat.
Udfærdiget i Bruxelles, den 30. maj 2008.
På Kommissionens vegne
Stavros DIMAS
Medlem af Kommissionen
(1) EUT L 396 af 30.12.2006, s. 1. Berigtiget i EUT L 136 af 29.5.2007, s. 3.
(2) EFT 196 af 16.8.1967, s. 1. Senest ændret ved Europa-Parlamentets og Rådets direktiv 2006/121/EF (EUT L 396 af 30.12.2006, s. 850. Berigtiget i EUT L 136 af 29.5.2007, s. 281) — denne henvisning ajourføres, når 30. tilpasning til den tekniske udvikling er offentliggjort.
BILAG
DEL A: METODER TIL BESTEMMELSE AF FYSISK/KEMISKE EGENSKABER
INDHOLDSFORTEGNELSE
|
A.1. |
Smelte-/frysepunkt |
|
A.2. |
Kogepunkt |
|
A.3. |
Relativ massefylde |
|
A.4. |
Damptryk |
|
A.5. |
Overfladespænding |
|
A.6. |
Vandopløselighed |
|
A.8. |
Fordelingskoefficient |
|
A.9. |
Flammepunkt |
|
A.10. |
Antændelighed (faste stoffer) |
|
A.11. |
Antændelighed (gasser) |
|
A.12. |
Antændelighed (kontakt med vand) |
|
A.13. |
Væskers og faste stoffers pyroforiske egenskaber |
|
A.14. |
Eksplosive egenskaber |
|
A.15. |
Selvantændelsestemperatur for væsker og gasser |
|
A.16. |
Relativ selvantændelsestemperatur for faste stoffer |
|
A.17. |
Oxiderende egenskaber (faste stoffer) |
|
A.18. |
Polymerers antalsmiddelmolekylvægt og molekylvægtsfordeling |
|
A.19. |
Polymerers indhold af lavmolekylære bestanddele |
|
A.20. |
Polymerers opløselighed/ekstraherbarhed i vand |
|
A.21. |
Oxiderende egenskaber (væsker) |
A.1. SMELTE-/FRYSEPUNKT
1. METODE
De fleste af de beskrevne metoder er baseret på OECD-testvejledningen (1). Grundprincipperne er anført i henvisning (2) og (3).
1.1. INDLEDNING
De beskrevne metoder og apparater kan anvendes til bestemmelse af smeltepunktet for kemiske stoffer uanset renheden.
De beskrevne metoder og apparater kan anvendes til bestemmelse af smeltepunktet for kemiske stoffer uanset renheden. Valg af metode afhænger af arten af det stof, der skal undersøges. Det er således afgørende, om stoffet let, vanskeligt eller slet ikke kan pulveriseres.
For nogle stoffer er bestemmelse af fryse- eller størkningspunkt mere hensigtsmæssigt, og der er i denne metode medtaget standarder for sådanne bestemmelser.
Hvis det som følge af stoffets særlige egenskaber er umuligt at bestemme nogen af ovenstående parametre, kan det være hensigtsmæssigt at bestemme et flydepunkt.
1.2. DEFINITIONER OG ENHEDER
Smeltepunktet defineres som den temperatur, ved hvilken faseovergangen fra fast til flydende tilstand finder sted ved atmosfæretryk, og denne temperatur svarer i det ideale tilfælde til temperaturen ved frysepunktet.
Da faseovergangen for mange stoffer finder sted over et temperaturinterval, beskrives den ofte som smeltepunktsintervallet.
Enhedsomregning (K til oC)
t = T – 273,15
|
t |
: |
er temperaturen i grader celsius ( oC) |
|
T |
: |
er temperaturen i kelvin (K) |
1.3. REFERENCESTOFFER
Anvendelse af referencestoffer er ikke påkrævet, hver gang et nyt stof skal undersøges. Referencestoffer skal først og fremmest bruges til lejlighedsvis kontrol af metoden og til sammenligning med resultater opnået med andre metoder.
Der er i henvisningerne (4) angivet nogle referencestoffer.
1.4. METODENS PRINCIP
Temperaturen (temperaturintervallet) for faseovergangen fra fast til flydende tilstand eller fra flydende til fast tilstand bestemmes. I praksis bestemmes temperaturen ved begyndende smeltning/frysning og ved endelig smeltning/frysning under opvarmning/afkøling af en prøve af stoffet ved atmosfæretryk. Der er beskrevet fem typer metoder: kapillarmetoder, varmebordsmetoder, frysepunktsbestemmelser, termoanalyse og flydepunktsbestemmelse (som udviklet til mineralolier).
Det kan i nogle tilfælde være mere hensigtsmæssigt at bestemme frysepunktet end smeltepunktet.
1.4.1. Kapillarmetode
1.4.1.1. Smeltepunktsapparater med væskebad
En lille mængde af det findelte stof fyldes i et kapillarrør og pakkes tæt. Røret opvarmes sammen med et termometer, og temperaturstigningen indstilles til mindre end ca. 1 K/min under den egentlige smeltning. Temperaturen ved begyndende og endelig smeltning bestemmes.
1.4.1.2. Smeltepunktsapparater med metalblok
Som beskrevet under 1.4.1.1 bortset fra, at kapillarrøret og termometeret er anbragt i en opvarmet metalblok og kan iagttages gennem huller i blokken.
1.4.1.3. Bestemmelse med fotocelle
Prøven i kapillarrøret opvarmes automatisk i en metalcylinder. En lysstråle sendes via et hul i cylinderen gennem stoffet og ind på en nøjagtigt indstillet fotocelle. De fleste stoffers optiske egenskaber ændres under smeltning fra at være uigennemsigtige til gennemsigtige. Derved øges den lysmængde, som når frem til fotocellen, og der sendes et stopsignal til en digitalindikator, som viser temperaturen af et platinmodstandstermometer anbragt i varmekammeret. Denne metode kan ikke anvendes for visse stærkt farvede stoffer.
1.4.2. Varmeborde
1.4.2.1. Koflers varmebænk
Koflers varmebænk består af to elektrisk opvarmede metalstykker med forskellig varmeledningsevne og er udformet sådan, at dens temperaturgradient i længderetningen er næsten lineær. Varmebænken kan spænde over temperaturområdet fra 283 K til 573 K og er forsynet med en særlig anordning til temperaturaflæsning, som består af en skyder med en viser, og en fane, og som er særligt udformet til den enkelte bænk. Smeltepunktet bestemmes ved, at det pågældende stof lægges i et tyndt lag direkte på bænkens overflade, hvorefter der på få sekunder opstår en skarp skillelinje mellem flydende og fast fase. Temperaturen på skillelinjen aflæses ved at indstille viseren på denne.
1.4.2.2. Smeltemikroskop
Der anvendes en række varmeborde med mikroskop til smeltepunktsbestemmelser på meget små stofmængder. I de fleste varmeborde måles temperaturen med et følsomt termoelement, men undertiden anvendes også kviksølvtermometre. Et typisk smeltepunktsapparat, bestående af varmebord med mikroskop, er udstyret med et varmekammer indeholdende en metalplade, hvorpå prøven anbringes i en slæde. I midten af metalpladen er der et hul, hvorigennem der kan sendes lys fra mikroskopets belysningsspejl. Når apparatet er i brug, er kammeret lukket med en glasplade for at forhindre luftens adgang til prøven.
Opvarmningen af prøven reguleres med en rheostat. Kræves meget nøjagtige målinger kan der for optisk anisotrope stoffer anvendes polariseret lys.
1.4.2.3. Meniskmetoden
Denne metode anvendes kun for polyamider.
Den temperatur, ved hvilken forskydningen af en silikonoliemenisk, som er indesluttet mellem et varmebord og et dækglas hvilende på polyamid-prøveemnet, iagttages visuelt.
1.4.3. Metode til bestemmelse af frysepunkt
Prøven anbringes i en særlig type reagensglas og anbringes i et apparat til bestemmelse af frysepunkt. Prøven afkøles under stadig langsom omrøring, og temperaturen måles med passende mellemrum. Når temperaturen er konstant ved flere aflæsninger, anføres denne temperatur (korrigeret for termometerfejl) som frysepunktet.
Underafkøling skal undgås ved, at der opretholdes ligevægt mellem den faste fase og væskefasen.
1.4.4. Termoanalyse
1.4.4.1. Differential termoanalyse (DTA)
Ved denne metode registreres temperaturforskellen mellem stoffet og et referencemateriale som funktion af temperaturen, imedens stoffet og referencematerialet følger samme temperaturprogram. En faseovergang i stoffet ledsaget af en enthalpiændring viser sig ved en endoterm (smeltning) eller exoterm (frysning) afvigelse fra den registrerede grundlinje.
1.4.4.2. Differential scanning kalorimetri (DSC)
Ved denne metode registreres forskellen i den energimængde, der tilføres stoffet og et referencemateriale, som funktion af temperaturen, imedens stoffet og referencematerialet følger samme temperaturprogram. Denne forskel svarer til den energimængde, der er nødvendig for at opretholde en temperaturforskel på nul mellem stoffet og referencematerialet. En faseovergang i stoffet ledsaget af en enthalpiændring viser sig ved en endoterm (smeltning) eller exoterm (frysning) afvigelse fra den registrerede grundlinje.
1.4.5. Flydepunkt
Denne metode er udviklet til mineralolier og er egnet til olieagtige stoffer med lavt smeltepunkt.
Efter først at være opvarmet afkøles prøven med en bestemt hastighed, og dens flydeegenskaber undersøges med 3 K's mellemrum. Den laveste temperatur, ved hvilken der kan iagttages bevægelse i stoffet, registreres som flydepunktet.
1.5. KVALITETSKRITERIER
Anvendelighed og nøjagtighed for de forskellige metoder til bestemmelse af smeltepunkt/smeltepunktsinterval er angivet i følgende tabel:
TABEL: METODERNES ANVENDELIGHED
A. Kapillarmetoder
|
Målemetode |
For stoffer, som kan pulveriseres |
For stoffer, som ikke let pulveriseres |
Temperaturområde |
Skønnet (1) nøjagtighed |
Foreliggende standard |
|
Smeltepunkts-apparater med væskebad |
ja |
enkelte |
273 til 573 K |
±0,3 K |
JIS K 0064 |
|
Smeltepunkts-apparater med metalblok |
ja |
enkelte |
293 til > 573 K |
±0,5 K |
ISO 1218 (E) |
|
Bestemmelse med fotocelle |
ja |
adskillige ved hjælp af særligt udsryr |
253 til 573 K |
±0,5 K |
|
B. Varmeborde og frysemetoder
|
Målemetode |
For stoffer, som kan pulveriseres |
For stoffer, som ikke let pulveriseres |
Temperaturområde |
Skønnet (2) nøjagtigned |
Foreliggende standard |
|
Koflers varmebænk |
ja |
nej |
283 K til > 573 K |
±1,0 K |
ANSI/ASTM D 345176 |
|
Smekemikroskop |
ja |
enkelte |
273 K til > 573 K |
±0,5 K |
DIN 53736 |
|
Meniskmetode |
nej |
kun for polyamider |
293 K til > 573 K |
±0,5 K |
IS0 1218 (E) |
|
Frysepunktsmetoder |
ja |
ja |
223 K til 573 K |
±0,5 K |
f.eks. BS 4695 |
C. Termoanalyse
|
Målemetode |
For stoffer, som kan pulveriseres |
For stoffer, som ikke let pulveriseres |
Temperaturområde |
Skønnet (3) nøjagtighed |
Foreliggende standard |
|
Differential termoanalyse |
ja |
ja |
173 K til 1 273 K |
op til 600 K ±0,5 K op til 1 273 K ±2,0 K |
ASTM E 537-76 |
|
Differential scanning kalorimetri |
ja |
ja |
173 K til 1 273 K |
op til 600 K ±0,5 K op til 1 273 K ±2,0 K |
ASTM E 537-76 |
D. Flydepunkt
|
Målemetode |
For stoffer, som kan pulveriseres |
For stoffer, som ikke let pulveriseres |
Temperaturområde |
Skønnet (4) nø-jagtighcd |
Foreliggende standard |
|
Flydepunkt |
for mineralolier og olieagtige stoffer |
for mineralolier og olieagtige stoffer |
223 K til 323 K |
±0,3 K |
ASTM D 97-66 |
1.6. BESKRIVELSE AF METODERNE
Fremgangsmåden er for næsten alle undersøgelsesmetoderne beskrevet i internationale og nationale standarder (se tillægget).
1.6.1. Kapillarrørsmetoder
Fint pulveriserede stoffer udviser ved langsom opvarmning sædvanligvis et smeltningsforløb som vist på figur 1.
Figur 1

Under smeltepunktsbestemmelsen noteres temperaturerne ved begyndende og endelig smeltning.
1.6.1.1. Smeltepunktsapparater med væskebad
Figur 2 viser en type standardiseret smeltepunktsapparat (JIS K 0064). Apparatet er fremstillet af glas, og alle mål er angivet i mm.
Figur 2

Badvæske:
Der vælges en passende væske. Valget afhænger af, hvor højt et smeltepunkt der skal bestemmes, f.eks. paraffinolie for smeltepunkter op til 473 K og silikonolie for smeltepunkter op til 573 K.
For smeltepunkter over 523 K kan der anvendes en blanding af 3 dele svovlsyre og 2 dele kaliumsulfat (masseforhold). Anvendes denne blanding, skal der træffes passende sikkerhedsforholdsregler.
Termometer:
Man bør kun anvende termometre, som opfylder kravene i de følgende eller tilsvarende standarder:
ASTM E 1-71, DIN 12770, JIS K 8001.
Fremgangsmåde:
Det tørre stof pulveriseres fint i en morter og fyldes i kapillarrøret, som er lukket i den ene ende, således at fyldningshøjden er ca. 3 mm efter tæt pakning. For at opnå en ensartet pakket prøve lader man kapillarrøret falde fra en højde af ca. 700 mm gennem et lodret glasrør ned på et urglas.
Det fyldte kapillarrør anbringes i badet, således at den midterste del af termometerets kviksølvbeholder berører den del af kapillarrøret, som prøven befinder sig i. Sædvanligvis anbringes kapillarrøret i apparatet, når badets temperatur er ca. 10 K under smeltepunktet.
Badvæsken opvarmes med en temperaturstigning på ca. 3 K/min under omrøring af væsken. Når temperaturen er ca. 10 K under det forventede smeltepunkt, indstilles temperaturstigningen til højst 1 K/min.
Beregning:
Smeltepunktet beregnes således:
T = TD + 0,00016 (TD – TE) n
hvor:
|
T |
= |
korrigeret smeltepunktstemperatur i K |
|
TD |
= |
temperaturaflæsning af termometer D i K |
|
TE |
= |
temperaturaflæsning af termometer E i K |
|
n |
= |
antallet af gradinddelinger på den udestående del af termometer D's kviksøjle. |
1.6.1.2. Smeltepunktapparatar med metalblok
Apparat:
Dette består af:
|
— |
en cylindrisk metalblok, hvis øverste del er hul og danner et kammer (se figur 3) |
|
— |
en metalprop med to eller flere huller, som muliggør anbringelse af rør i metalklodsen |
|
— |
et opvarmningssystem for metalklodsen, f.eks. i form af en elektrisk modstand indbygget i blokken |
|
— |
en rheostat til regulering af energitilførslen, hvis der anvendes elektrisk opvarmning |
|
— |
fire vinduer af varmeresistent glas i kammerets sidevægge, anbragt diametralt og vinkelret på hinanden. Foran et af disse vinduer er der anbragt en lup til iagttagelse af kapillarrøret. De tre andre vinduer tjener til belysning af indersiden af kammeret ved hjælp af lamper |
|
— |
et kapillarrør af varmeresistent glas lukket i den ene ende (se 1.6.1.1). |
Termometer:
Se standarderne i 1.6.1.1. Termoelektriske måleinstrumenter med tilsvarende nøjagtighed kan også anvendes.
Figur 3

1.6.1.3. Bestemmelse med fotocelle
Apparat og fremgangsmåde:
Apparatet består af et metalkammer med et automatisk opvarmningssystem. Tre kapillarrør fyldes som i 1.6.1.1 og anbringes i ovnen.
Der er mulighed for flere lineære temperaturstigninger til kalibrering af apparatet, og temperaturstigningen indstilles elektrisk til en passende forudvalgt konstant og lineær hastighed. Skrivere viser den øjeblikkelige ovntemperatur og temperaturen af stoffet i kapillarrørene.
1.6.2. Varmeborde
1.6.2.1. Koflers varmebænk
Se tillæg.
1.6.2.2. Smeltemikroskop
Se tillæg.
1.6.2.3. Meniskmetode (polyamider)
Se tillæg.
Opvarmingshastigheden ved smeltepunktet skal være mindre end 1 K/min.
1.6.3. Metoder til bestemmelse af frysepunkt
Se tillæg.
1.6.4. Termoanalyse
1.6.4.1. Differential termoanalyse
Se tillæg.
1.6.4.2. Differential scanning kalorimetri
Se tillæg.
1.6.5. Bestemmelse af flydepunkt
Se tillæg.
2. DATA
I visse tilfælde er termometerkorrektion nødvendig.
3. RAPPORTERING
Forsøgsrapporten skal om muligt indeholde følgende oplysninger:
|
— |
anvendt metode |
|
— |
nøjagtig beskrivelse af stoffet (betegnelse og urenheder) og eventuelt indledende oprensningstrin |
|
— |
den skønnede nøjagtighed. |
Som smeltepunkt anføres gennemsnittet af mindst to målinger, der ligger inden for den skønnede nøjagtighed (jf. tabellerne).
Hvis forskellen mellem temperaturen ved begyndende og endelig smeltning ligger inden for metodens nøjagtighed, angives temperaturen ved den endelige smeltning som smeltepunktet; i modsat fald angives begge temperaturer.
Hvis stoffet dekomponerer eller sublimerer, inden smeltepunktet er nået, anføres den temperatur, hvor dette fænomen er iagttaget.
Alle oplysninger og bemærkninger af betydning for vurdering af resultaterne især vedrørende urenheder og stoffets fysiske tilstand skal anføres.
4. HENVISNINGER
|
(1) |
OECD, Paris, 1981, Test Guideline 102, Decision of the Council C(81) 30 final. |
|
(2) |
IUPAC, B. Le Neindre, B. Vodar, eds. Experimental thermodynamics, Butterworths, London 1975, vol. II, 803-834. |
|
(3) |
R. Weissberger ed.: Technique of organic Chemistry, Physical Methods of Organic Chemistry, 3rd ed., Interscience Publ., New York, 1959, vol. I, Part I, Chapter VII. |
|
(4) |
IUPAC, Physicochemical measurements: Catalogue of reference materials from national laboratories, Pure and applied chemistry, 1976, vol. 48, 505-515. |
Tillæg
Der findes supplerende tekniske oplysninger i nedenstående standarder.
1. Kapillarmetoder
1.1. Smeltepunktsapparater med væskebad
|
ASTM E 324-69 |
Standard test method for relative initial and final melting points and the melting range of organic chemicals |
|
BS 4634 |
Method for the determination of melting point and/or melting range |
|
DIN 53181 |
Bestimmung des Schmelzintervalles von Harzen nach Kapilarverfarehn |
|
JIS K 00-64 |
Testing methods for melting point of chemical products. |
1.2. Smeltepunktsapparater med metalblok
|
DIN 53736 |
Visuelle Bestimmung der Schmelztemperatur von teilkristallinen Kunststoffen |
|
ISO 1218 (E) |
Plastics — polyamides — determination of »melting point« |
2. Varmeborde
2.1. Koflers varmebænk
|
ANSI/ASTM D 3451-76 |
Standard recommended practices for testing polymeric powder coatings |
2.2. Smeltemikroskop
|
DIN 53736 |
Visuelle Bestimmung der Schmelztemperatur von teilkristallinen Kunststoffen. |
2.3. Meniskmetode (polyamider)
|
ISO 1218 (E) |
Plastics — polyamides — determination of »melting point« |
|
ANSI/ASTM D 2133-66 |
Standard specification for acetal resin injection moulding and extrusion materials |
|
NF T 51-050 |
Resines de polyamides. Determination du »point de fusion« methode du menisque |
3. Metoder til bestemmelse af frysepunkt
|
BS 4633 |
Method for the determination of crystallizing point |
|
BS 4695 |
Method for Determination of Melting Point of petroleum wax (Cooling Curve) |
|
DIN 51421 |
Bestimmung des Gefrierpunktes von Flugkraftstoffen, Ottokraftstoffen und Motorenbenzolen |
|
ISO 2207 |
Cires de petrole: determination de la temperature de figeage |
|
DIN 53175 |
Bestimmung des Erstarrungspunktes von Fettsiiuren |
|
NF T 60-114 |
Point de fusion des paraffines |
|
NF T 20-051 |
Methode de determination du point de cristallisation (point de Congelation |
|
ISO 1392 |
Method for the determination of the freezing point |
4. Termoanalyse
4.1. Differential termoanalyse
|
ASTM E 537-76 |
Standard method for assessing the thermal stability of chemicals by methods of differential thermal analysis |
|
ASTM E 473-85 |
Standard definitions of terms relating to thermal analysis |
|
ASTM E 472-86 |
Standard practice for reporting thermoanalytical data |
|
DIN 51005 |
Thermische Analyse, Begriffe |
4.2. Differential scanning kalorimetri
|
ASTM E 537-76 |
Standard method for assessing the thermal stability of chemicals by methods of differential thermal analysis |
|
ASTM E 473-85 |
Standard definitions of terms relating to thermal analysis |
|
ASTM E 472-86 |
Standard practice for reporting thermoanalytical data |
|
DIN 51005 |
Thermische Analyse, Begriffe |
5. Bestemmelse af flydepunkt
|
NBN 52014 |
Echantillonnage et analyse des produits du petrole: Point de trouble et point d'ecoulement limite — Monsterneming en ontleding van aardolieproducten: Troebelingspunt en vloeipunt |
|
ASTM D 97-66 |
Standard test method for pour point of petroleum oils |
|
ISO 3016 |
Petroleum oils — Determination of pour point |
A.2. KOGEPUNKT
1. METODE
Flertallet af de beskrevne metoder er baseret på OECD-testvejledningen (1). Grundprincipperne er anført i henvisning (2) og (3).
1.1. INDLEDNING
De her beskrevne metoder kan anvendes for alle væsker og lavtsmeltende stoffer, forudsat at der ikke indtræder nogen kemisk reaktion under kogepunktet (f.eks. autooxidation, omlejring eller nedbrydning). Metoderne kan anvendes for alle væsker uanset renheden.
De metoder, hvor der benyttes bestemmelse med fotocelle, og de, der er baseret på termoanalyse, fremhæves specielt, da de kan benyttes til bestemmelse af både smelte- og kogepunkt. Desuden kan disse målinger udføres automatisk.
Den dynamiske metode har den fordel, at den også kan benyttes til bestemmelse af damptryk, og det er ikke nødvendigt at korrigere kogepunktstemperaturen til standardtryk (101,325 kPa), da standardtrykket kan indstilles med en pressostat under målingen.
Bemærkninger:
Urenheders indflydelse på kogepunktsbestemmelsen afhænger stærkt af deres art. Hvis der i prøven er flygtige urenheder, som vil kunne få indflydelse på resultaterne, kan stoffet eventuelt renses.
1.2. DEFINITIONER OG ENHEDER
Standardkogepunktet defineres som den temperatur, ved hvilken en væskes damptryk er 101,325 kPa.
Måles kogepunktet ikke ved standardatmosfæretryk, kan damptrykkets afhængighed af temperaturen beskrives ved Clausius-Clapeyrons ligning:

hvor:
|
P |
= |
stoffets damptryk i pascal |
|
ΔHv |
= |
stoffets fordampningsvarme i J mol-1 |
|
R |
= |
gaskonstanten = 8,314 J mol-1 K-1 |
|
T |
= |
den absolutte temperatur i K |
Kogepunktet opgives i forhold til det aktuelle tryk under målingen.
Omregninger
Tryk (enhed kPa)
|
100 kPa |
= |
1 bar = 0,1 MPa (»bar« er stadig tilladt, men anbefales ikke) |
|
133 Pa |
= |
1 mm Hg = 1 Torr (enhederne »mm Hg« og »Torr« er ikke tilladt). |
|
1 atm |
= |
standardatmosfære = 101 325 Pa (»atm«-enheden er ikke tilladt). |
Temperatur (enhed: K)
t = T – 273,15
|
t |
: |
er temperaturen i grader celsius ( oC) |
|
T |
: |
er termodynamisk temperaturen i kelvin (K) |
1.3. REFERENCESTOFFER
Anvendelse af referencestoffer er ikke påkrævet, hver gang et nyt stof undersøges. Referencestoffer skal først og fremmest tjene til lejlighedsvis kontrol af metoden og til sammenligning med resultater opnået med andre metoder.
Der er i de metoder, der er anført i tillægget, angivet nogle referencestoffer.
1.4. METODENS PRINCIP
Der er fem metoder til bestemmelse af kogepunkt (kogepunktsinterval), som er baseret på måling af den temperatur, ved hvilken prøven koger, og to metoder baseret på termoanalyse.
1.4.1. Bestemmelse ved hjælp af ebulliometer
Ebullitometre blev oprindeligt udviklet til molekylvægtsbestemmelse via kogepunktsforhøjelse, men de er også velegnede til nøjagtige kogepunktsbestemmelser. Et meget enkelt apparat er beskrevet i ASTM D 1120-72 (se tillæg). I dette apparat opvarmes væsken under ligevægtsbetingelser ved atmosfæretryk, indtil den koger.
1.4.2. Dynamisk metode
Ved denne metode måles dampens fortætningstemperatur ved hjælp af et egnet termometer anbragt i tilbagesvaleren under kogning. Trykket kan varieres ved denne metode.
1.4.3. Destillationsmetode til bestemmelse af kogepunkt
Ved denne metode destilleres væsken, og dampens fortætningstemperatur samt destillatmængden måles.
1.4.4. Siwoloboffs metode
En prøve opvarmes i et prøverør, som er nedsænket i et varmebad. Et tilsmeltet kapillarrør med en luftboble i den nederste ende er nedsænket i prøverøret.
1.4.5. Bestemmelse med fotocelle
Der foretages automatisk fotoelektrisk måling af opstigende bobler under anvendelse af Siwoloboffs princip.
1.4.6. Differential termoanalyse
Ved denne metode registreres temperaturforskellen mellem stoffet og et referencemateriale som funktion af temperaturen, imedens stoffet og referencematerialet følger samme temperaturprogram. En faseovergang i stoffet ledsaget af en enthalpiændring viser sig ved en endoterm afvigelse (kogning) fra den registrerede grundlinje.
1.4.7. Differential scanning kalorimetri
Ved denne metode registreres forskellen i den energimængde, der tilføres stoffet og et referencemateriale, som funktion af temperaturen, imedens stoffet og referencematerialet følger samme temperaturprogram. Denne forskel svarer til den energimængde, der er nødvendig for at opretholde en temperaturforskel på nul mellem stoffet og referencematerialet. En faseovergang i stoffet ledsaget af en enthalpiændring viser sig ved en endoterm afvigelse (kogning) fra den registrerede grundlinje.
1.5. KVALITETSKRITERIER
Anvendelighed og nøjagtighed for de forskellige metoder til bestemmelse af kogepunkt/kogepunktsinterval er angivet i tabel 1.
Tabel 1
Sammenligning af metoderne
|
Målemetode |
Skønnet nøjagtighed |
Foreliggende standard |
|
Ebulliometer |
ASTM D 1120-72 (5) |
|
|
Dynamisk metode |
±0,5 K (op til 600 K) (6) |
|
|
Destillationsforløb (kogepunktsinterval) |
±0,5 K (op til 600 K) |
ISO/R 918, DIN 53171, BS 4591/71 |
|
Siwoloboff's metode |
± 2 K (op til 600 K) (6) |
|
|
Bestemmelse med fotocelle |
±0,3 K (ved 373 K) (6) |
|
|
Differential termoanalyse |
±0,5 K (op til 600 K) ±2,0 K (op til 1 273 K) |
ASTM E 537-76 |
|
Differential scanning kalorimetri |
±0,5 K (op til 600 K) ±2,0 K (op til 1 273 K) |
ASTM E 537-76 |
1.6. BESKRIVELSE AF METODERNE
Nogle af metodernes fremgangsmåder er beskrevet i internationale og nationale standarder (se tillæg).
1.6.1. Ebulliometer
Se tillæg.
1.6.2. Dynamisk metode
Se metode A.4 til bestemmelse af damptryk.
Den kogetemperatur, der iagttages ved et tryk på 101,325 kPa, noteres.
1.6.3. Destillationsforløb (kogepunktsinterval)
Se tillæg.
1.6.4. Siwoloboffs metode
Prøven opvarmes i et smeltepunktsapparat i et prøverør med en diameter på ca. 5 mm (figur 1).
Figur 1 viser en model af et standardiseret smelte- og kogepunktsapparat (JIS K 0064) (fremstillet af glas, alle mål i millimeter).
Figur 1
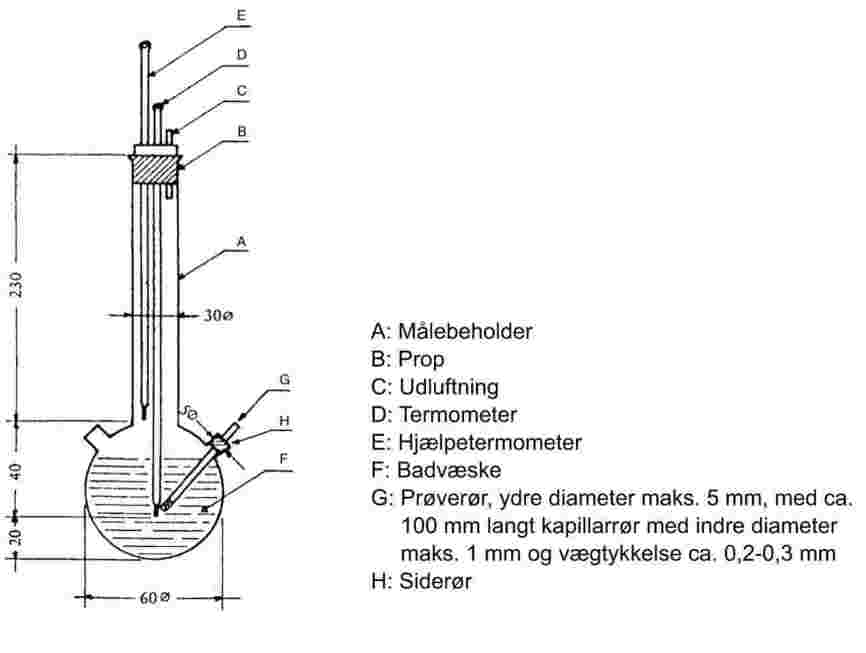
Et kapillarrør, som er tilsmeltet ca. 1 cm over den nederste ende, anbringes i prøverøret. Der fyldes så meget prøve i, at den tilsmeltede del af kapillarrøret befinder sig under væskeoverfladen. Prøverøret med kapillarrøret fastgøres til termometeret med en elastik eller fastholdes støttet fra siden (se figur 2).
|
Figur 2 Siwoloboff's princip |
Figur 3 Modificeret princip |
|
|
|
Badvæsken vælges under hensyntagen til kogepunktet. Ved en temperatur på op til 573 K kan der benyttes silikonolie. Flydende paraffin må kun anvendes op til 473 K. Badvæsken opvarmes først med en hastighed på 3 K pr. minut under omrøring. Ca. 10 K under det forventede kogepunkt sænkes varmetilførslen, så temperaturstigningen bliver mindre end 1 K pr. minut. Når temperaturen nærmer sig kogepunktet, begynder der at strømme bobler ud af kapillarrøret.
Kogepunktet er den temperatur, hvor boblestrømmen under et kortvarigt temperaturfald standser og væsken begynder at stige op i kapillarrøret. Termometeraflæsningen viser stoffets kogepunkt.
I det modificerede princip (figur 3) bestemmes kogepunktet i et smeltepunktskapillarrør. Det trækkes ud til en ca. 2 cm lang tynd spids (a), og en smule af prøven suges op heri. Den åbne ende af det tynde kapillarrør tilsmeltes, så der dannes en lille luftboble ved enden. Ved opvarmning i smeltepunktsapparatet (b) udvider luftboblen sig. Kogepunktet ligger ved den temperatur, hvor stofproppen når op i højde med badvæskens overflade (c).
1.6.5. Bestemmelse med fotocelle
Prøven opvarmes i et kapillarrør i en opvarmet metalblok.
Via passende huller i blokken sendes der en lysstråle gennem stoffet ind til en nøjagtigt kalibreret fotocelle.
Medens temperaturen i prøven stiger, undviger der enkelte bobler fra kapillarrøret. Når kogepunktet er nået, vokser antallet af bobler voldsomt. Herved ændres intensiteten af det lys, som fotocellen modtager, så der sendes et stopsignal til den indikator, der viser temperaturen af et platinmodstandstermometer, der er anbragt i blokken
Denne metode er særlig anvendelig, da den kan benyttes til bestemmelser under stuetemperatur ned til 253,15 K (– 20 oC) uden ændringer ved apparaturet. Instrumentet skal blot anbringes i et kuldebad.
1.6.6. Termoanalyse
1.6.6.1. Differential termoanalyse
Se tillæg.
1.6.6.2. Differential scanning kalorimetri
Se tillæg.
2. DATA
Ved små afvigelser fra standardtryk (maks. ± 5 kPa) korrigeres kogepunktstemperaturerne ved hjælp af Sidney-Youngs ligning:
Tn = T + (fT x Δp)
hvor
|
Δ p |
= |
(101,325 – p) [bemærk fortegnet] |
|
p |
= |
trykket målt i kPa |
|
fT |
= |
kopepunktets ændring med trykker i K/kPa |
|
T |
= |
målt kogepunkt i K |
|
Tn |
= |
kogepunkt i K, korrigeret til standardtryk |
I ovennævnte nationale og internationale standarder findes der temperaturkorrektionsfaktorer fT for mange stoffer og udtryk til tilnærmelse af dem.
Eksempelvis anføres i DIN 53171 følgende grove korrektioner for opløsningsmidler i maling:
Tabel 2
Temperaturkorrektionsfaktorer fT
|
Temperatur T |
korrektionsfaktor fT (K/kPa) |
|
323,15 |
0,26 |
|
348,15 |
0,28 |
|
373,15 |
0,31 |
|
398,15 |
0,33 |
|
423,15 |
0,35 |
|
448,15 |
0,37 |
|
473,15 |
0,39 |
|
498,15 |
0,41 |
|
523,15 |
0,44 |
|
548,15 |
0,45 |
|
573,15 |
0,47 |
3. RAPPORTERING
Forsøgsrapporten skal om muligt indeholde følgende oplysninger:
|
— |
anvendt metode |
|
— |
nøjagtig beskrivelse af stoffet (betegnelse og urenheder) og eventuel indledende rensning |
|
— |
den skønnede nøjagtighed. |
Som kogepunkt anføres gennemsnittet af mindst to målinger, der ligger inden for den skønnede nøjagtighed (jf. tabel 1).
De målte kogetemperaturer og gennemsnittet heraf skal anføres, og det tryk, hvorved målingerne er foretaget, opgives i kPa. Trykket skal helst ligge nær standardatmosfæretryk.
Alle oplysninger og bemærkninger af betydning for vurdering af resultaterne især vedrørende urenheder og stoffets fysiske tilstand skal anføres.
4. HENVISNINGER
|
(1) |
OECD, Paris, 1981, Test Guideline 103, Decision of the Council C (81) 30 final. |
|
(2) |
IUPAC, B. Le Neindre, B. Vodar, editions. Experimental thermodynamics, Butterworths, London 1975, volume II. |
|
(3) |
R. Weissberger edition: Technique of organic chemistry, Physical methods of organic chemistry, Third Edition, Interscience Publications, New York, 1959, volume I, Part I, Chapter VIII. |
Tillæg
Der findes supplerende tekniske oplysninger i nedenstående standarder
1. Ebulliometer
|
1.1. |
Smeltepunktsapparater med væskebad |
|
ASTM D 1120-72 |
Standard test method for boiling point of engine anti-freezes |
2. Destillationsforløb (kogepunktsinterval)
|
ISO/R 918 |
Test Method for Distillation (Distillation Yield and Distillation Range) |
|
BS 4349/68 |
Method for determination of distillation of petroleum products |
|
BS 4591/71 |
Method for the determination of distillation characteristics |
|
DIN 53171 |
Losungsmittel fur Anstrichstoffe, Bestimmung des Siedeverlaufes |
|
NF T 20-608 |
Distillation:determination du rendement et de l'intervalle de distillation |
3. Differential termoanalyse og differential scanning kalorimetri
|
ASTM E 537-76 |
Standard method for assessing the thermal stability of chemicals by methods of differential thermal analysis |
|
ASTM E 473-85 |
Standard definitions of terms relating to thermal analysis |
|
ASTM E 472-86 |
Standard practice for reporting thermoanalytical data |
|
DIN 51005 |
Thermische Analyse, Begriffe |
A.3. RELATIV MASSEFYLDE
1. METODE
De beskrevne metoder er baseret på OECD-testvejledningen (1). Grundprincipperne er anført i henvisning (2).
1.1. INDLEDNING
De beskrevne metoder til bestemmelse af relativ massefylde kan anvendes for faste stoffer og væsker uanset renheden. De forskellige metoder, som kan anvendes, er anført i tabel 1.
1.2. DEFINITIONER OG ENHEDER
Den relative massefylde, D4 20, af faste stoffer og væsker er forholdet mellem massen af et rumfang af det undersøgte stof bestemt ved 20 oC og massen af det samme rumfang vand bestemt ved 4 oC. Den relative massefylde er dimensionsløs.
Massefylden, P, af et stof er dets masse m divideret med dets rumfang v.
Massefylden, P, opgives i kg/m3 (SI-enheder).
1.3. REFERENCESTOFFER (1) (3)
Anvendelse af referencestoffer er ikke påkrævet, hver gang et nyt stof skal undersøges. Referencestoffer skal først og fremmest tjene til lejlighedsvis kontrol af metoden og til sammenligning med resultater opnået med andre metoder.
1.4. METODERNES PRINCIP
Der benyttes fire typer metoder.
1.4.1. Opdriftsmetoder
1.4.1.1. Aræometer (for væsker)
Der kan opnås tilstrækkelig nøjagtige og hurtige massefyldebestemmelser med aræometre, hvor massefylden af en væske udledes af flydehøjden ved aflæsning på en skala.
1.4.1.2. Hydrostatisk vægt (for væsker og faste stoffer)
Forskellen mellem massen af en prøve i henholdsvis atmosfærisk luft og en passende væske (f.eks. vand) kan anvendes til at bestemme dens massefylde.
For faste stoffer er den fundne massefylde kun repræsentativ for den bestemte prøve, som er anvendt til bestemmelsen. Til bestemmelse af væskers massefylde vejes et legeme med kendt rumfang v først i atmosfærisk luft og dernæst i væsken.
1.4.1.3. Metode med nedsænket legeme (for væsker) (4)
Ved denne metode bestemmes en væskes massefylde ud fra forskellen mellem resultaterne af vejning af væsken før og efter nedsænkning af et legeme med kendt rumfang her.
1.4.2. Pyknometermetoder
Pyknometre af forskellig udformning og med kendt rumfang kan anvendes for faste stoffer og væsker. Massefylden beregnes ud fra masseforskellen mellem det fulde og det tomme pyknometer og dets kendte rumfang.
1.4.3. Luftsammenligningspyknometer (for faste stoffer)
Massefylden af et fast stof i enhver form kan bestemmes ved stuetemperatur med et gassammenligningspyknometer. Rumfanget af en prøve af stoffet måles i atmosfærisk luft eller i en inaktiv luftart i en beholder med korrigeret rumfangsinddeling. Til beregning af massefylden foretages efter rumfangsbestemmelsen en bestemmelse af prøvens masse.
1.4.4. Oscillerende densitometer (5) (6) (7)
Massefylden af en væske kan bestemmes med et oscillerende densitometer. En mekanisk oscillator i form af et U-rør sættes i svingning ved sin resonansfrekvens, der afhænger af dens masse. Når der indføres en prøve heri, ændres oscillatorens resonansfrekvens. Apparatet kalibreres med to væsker med kendt massefylde. Disse stoffer vælges fortrinsvis således, at deres massefylde ligger i hver sin ende af det pågældende måleområde.
1.5. KVALITETSKRITERIER
Anvendeligheden af de forskellige metoder, der anvendes til bestemmelse af den relative massefylde, er opstillet i tabellen.
1.6. BESKRIVELSE AF METODERNE
Tillægget indeholder en liste over nogle standarder, hvorfra yderligere tekniske detaljer kan hentes.
Målingerne skal udføres ved 20 oC, og der foretages mindst to bestemmelser.
2. DATA
Se standarderne.
3. RAPPORTERING
Forsøgsrapporten skal om muligt indeholde følgende oplysninger:
|
— |
anvendt metode |
|
— |
nøjagtig beskrivelse af stoffet (art og urenheder) og eventuel indledende rensning. |
Den under 1.2 definerede relative massefylde  rapporteres tillige med det undersøgte stofs fysiske tilstand.
rapporteres tillige med det undersøgte stofs fysiske tilstand.
Alle oplysninger og bemærkninger af betydning for vurdering af resultaterne, især vedrørende urenheder og stoffets fysiske tilstand, skal anføres.
Tabel
Metodernes anvendelighed
|
Målemetode |
Massefylde |
Maksimal dynamisk viskositet |
Foreliggende standarder |
|||
|
fast stof |
væske |
|||||
|
|
ja |
5 Pa s |
ISO 387, ISO 649-2, NF T 20-050 |
||
|
|
|
|
|
||
|
ja |
|
|
ISO 1183 (A) |
||
|
|
ja |
5 Pa S |
ISO 901 og 758 |
||
|
|
ja |
20 Pa s |
DIN 53217 |
||
|
|
|
|
ISO 3507 |
||
|
ja |
|
|
ISO 1183 (B), NF T 20-053 |
||
|
|
ja |
500 Pa s |
ISO 758 |
||
|
ja |
|
|
DIN 55990 Teil 3, DIN 53243 |
||
|
|
ja |
5 Pa s |
|
||
4. HENVISNINGER
|
(1) |
OECD, Paris, 1981, Test Guideline 109, Decision of the Council C(81) 30 final. |
|
(2) |
R. Weissberger ed., Technique of Organic Chemistry, Physical Methods of Organic Chemistry, 3rd ed., Chapter IV, Interscience Publ., New York, 1959, vol. I, Part 1. |
|
(3) |
IUPAC, Recommended reference materials for realization of physico-chemical properties, Pure and applied chemistry, 1976, vol. 48, 508. |
|
(4) |
Wagenbreth, H., Die Tauchkugel zur Bestimmung der Dichte von Flüssigkeiten, Technisches Messen tm, 1979, vol.II, 427-430. |
|
(5) |
Leopold, H., Die digitale Messung von Flüssigkeiten, Elektronik, 1970, vol. 19, 297-302. |
|
(6) |
Baumgarten, D., Füllmengenkontrolle bei vorgepackten Erzeugnissen — Verfahren zur Dichtebestimmung bei flüssigen Produkten und ihre praktische Anwendung, Die Pharmazeutische Industrie, 1975, vol. 37, 717 - 726. |
|
(7) |
Riemann, J., Der Einsatz der digital en Dichtemessung im Brauereilaboratorium, Brauwissenschaft, 1976, vol. 9, 253-255. |
Tillæg
Der findes supplerende tekniske oplysninger i nedenstående standarder:
1. Opdriftsmetoder
1.1. Aræometer
|
DIN 12790, ISO 387 |
Hydrometer; general instructions |
|
DIN 12791 |
Part I: Density hydrometers; construction, adjustment and use Part II: Density hydrometers; standardized sizes, designation Part III: Use and test |
|
ISO 649-2 |
Laboratory glassware: Density hydrometers for general purpose |
|
NF T 20-050 |
Chemical products for industrial use — Determination of density of liquids — Areometric method |
|
DIN 12793 |
Laboratory glassware: range find hydrometers |
1.2. Hydrostatisk vægt
For faste stoffer
|
ISO 1183 |
Method A: Methods for determining the density and relative density of plastics excluding cellular plastics |
|
NF T 20-049 |
Chemical products for industrial use — Determination of the density of solids other than powders and cellular products — Hydrostatic balance method |
|
ASTM-D-792 |
Specific gravity and density of plastics by displacement |
|
DIN 53479 |
Testing of plastics and elastomers; determination of density |
For væsker
|
ISO 901 |
ISO 758 |
|
DIN 51757 |
Testing of mineral oils and related materials; determination of density |
|
ASTM D 941-55, ASTM D 1296-67 og ASTM D 1481-62 |
|
|
ASTM D 1298 |
Density, specific gravity or API gravity of crude petroleum and liquid petroleum products by hydrometer method |
|
BS 4714 |
Density, specific gravity or API gravity of crude petroleum and liquid petroleum products by hydrometer method |
1.3. Metode med nedsænket legeme
|
DIN 53217 |
Testing of paints, varnishes and similar coating materials; determination of density; immersed body method |
2. Pyknometermetoder
2.1. For væsker
|
ISO 3507 |
Pycnometers |
|
ISO 758 |
Liquid chemical products; determination of density at 20 oC |
|
DIN 12797 |
Gay-Lussac pycnometer (for non-volatile liquids which are not too viscous) |
|
DIN 12798 |
Lipkin pycnometer (for liquids with a kinematic viscosity of less than 100,10-6 m2 s-1 at 15 oC) |
|
DIN 12800 |
Sprengel pycnometer (for liquids as DIN 12798) |
|
DIN 12801 |
Reischauer pycnometer (for liquids with a kinematic viscosity of less than 100,10-6 m2 s-1 at 20 oC, applicable in particular also to hydrocarbons and aqueous solutions as well as to liquids with higher vapour pressure, approximately 1 bar at 90 oC) |
|
DIN 12806 |
Hubbard pycnometer (for viscous liquids of all types which do not have too high a vapour pressure, in particular also for paints, varnishes and bitumen) |
|
DIN 12807 |
Bingham pycnometer (for liquids, as in DIN 12801) |
|
DIN 12808 |
Jaulmes pycnometer (in particular for ethanol — water mixture) |
|
DIN 12809 |
Pycnometer with ground-in thermometer and capillary side tube (for liquids which are not too viscous) |
|
DIN 53217 |
Testing of paints, varnishes and similar products; determination of density by pycnometer |
|
DIN 51757 |
Point 7: Testing of mineral oils and related materials; determination of density |
|
ASTM D 297 |
Section 15: Rubber products — chemical analysis |
|
ASTM D 2111 |
Method C: Halogenated organic compounds |
|
BS 4699 |
Method for determination of specific gravity and density of petroleum products (graduated bicapillary pycnometer method) |
|
BS 5903 |
Method for determination of relative density and density of petroleum products by the capillary — stoppered pycnometer method |
|
NF T 20-053 |
Chemical products for industrial use — Determination of density of solids in powder and liquids — Pyknometric method |
2.2. For faste stoffer
|
ISO 1183 |
Method B: Methods for determining the density and relative density of plastics excluding cellular plastics |
|
NF T 20-053 |
Chemical products for industrial use — Determination of density of solids in powder and liquids — Pyknometric method |
|
DIN 19683 |
Determination of the density of soils |
3. Luftsammenligningspyknometer
|
DIN 55990 |
Part 3: Prüfung von Anstrichstoffen und ähnlichen Beschichrungsstoffen; Pulverlack; Bestimmung der Dichte |
|
DIN 53243 |
Anstrichstoffe; Chlorhaltige Polymere; Prüfung |
A.4. DAMPTRYK
1. METODE
Flertallet af de beskrevne metoder er baseret på OECD-testvejledningen (1). Grundprincipperne er anført i henvisning (2) og (3).
1.1. INDLEDNING
Ved udførelse af denne test er det nyttigt at have forhåndsoplysninger om stoffets struktur, smeltepunkt og kogepunkt.
Der findes ikke en enkelt metode, som kan anvendes i hele damptryksområdet. Derfor anbefales der flere forskellige metoder til måling af damptryk fra < 10-4 til 105 Pa.
Urenheder vil normalt påvirke damptrykket; denne påvirkning afhænger stærkt af urenhedens art.
Er der i prøven flygtige urenheder, der kunne tænkes at påvirke resultatet, kan man rense stoffet. Det kan også være hensigtsmæssigt at oplyse damptrykket for den tekniske kvalitet.
I nogle af de her beskrevne metoder anvendes der apparatur med metaldele; der skal tages hensyn hertil, når der udføres test med ætsende stoffer.
1.2. DEFINITIONER OG ENHEDER
Et fast eller flydende stofs damptryk defineres som mætningstrykket over stoffet. I termodynamisk ligevægt er et rent stofs damptryk kun en funktion af temperaturen.
Den SI-enhed for tryk, som anvendes, er pascal (Pa).
Nedenfor er anført nogle enheder, som tidligere har været anvendt, og deres omregningsfaktorer:
|
1 Torr (≡ 1 mm Hg) |
= 1,333 × 102 Pa |
|
1 atmosfære |
= 1,013 × 105 Pa |
|
1 bar |
= 105 Pa |
SI-enheden for temperatur er kelvin (K).
Gaskonstanten R er 8,314 J. mol-1. K-1.
Damptrykkets afhængighed af temperaturen beskrives ved Clausius-Clapeyrons ligning:
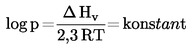
hvor:
|
p |
= |
stoffets damptryk i pascal |
|
ΔHv |
= |
stoffets fordampningsvarme i J. mol-1 |
|
R |
= |
gaskonstanten i J. mol-1. K-1 |
|
T |
= |
den absolutte temperatur i K |
1.3. REFERENCESTOFFER
Anvendelse af referencestoffer er ikke påkrævet, hver gang et nyt stof skal undersøges. Referencestoffer skal først og fremmest tjene til lejlighedsvis kontrol af metoden og til sammenligning med resultater opnået med andre metoder.
1.4. TESTMETODERNES PRINCIP
Der foreslås syv metoder til bestemmelse af damptryk, som hver kan bruges i forskellige damptryksintervaller. Ved hver metode bestemmes damptrykket ved flere forskellige temperaturer. I et begrænset temperaturinterval er logaritmen til damptrykket af et rent stof en lineær funktion af den reciprokke værdi af temperaturen.
1.4.1. Dynamisk metode
Ved den dynamiske metode måles den kogetemperatur, der svarer til et specificeret tryk.
Anbefalet område:
103 til 105 Pa.
Denne metode anbefales også til bestemmelse af kogepunkt og kan anvendes til dette formål op til 600 K.
1.4.2. Statisk metode
Ved den statiske metode bestemmes damptrykket i et lukket system, der er i termodynamisk ligevægt ved en specificeret temperatur. Metoden er egnet til faste stoffer og væsker, som består af en eller flere komponenter.
Anbefalet område:
10 til 105 Pa.
Med den fornødne omhu kan metoden også anvendes i området fra 1 til 10 Pa.
1.4.3. Isoteniskop
Denne standardiserede metode er også en statisk metode, men den kan normalt ikke anvendes til flerkomponentsystemer. Der kan søges yderligere oplysninger i ASTM D 2879-86.
Anbefalet område:
100 til 105 Pa.
1.4.4. Effusionsmetode: damptryksvægt
Den stofmængde, der forlader en celle pr. tidsenhed gennem en åbning af kendt størrelse, bestemmes under vakuum, så den mængde stof, der vender tilbage til cellen, er ubetydelig (f.eks. ved måling af den impuls, en dampstråle udøver på en følsom vægt, eller ved måling af vægttabet).
Anbefalet område:
10-3 til 1 Pa.
1.4.5. Effusionsmetode: vægttab eller opfangning af damp
Metoden er baseret på bestemmelse af den mængde teststof, der under ultravakuum strømmer ud af en Knudsen-celle (4) gennem en mikroåbning pr. tidsenhed i gasform. Massen af den udsivede damp kan konstateres enten ved at bestemme cellens vægttab eller ved at kondensere dampen ved lav temperatur og bestemme den fordampede mængde stof ved kromatografisk analyse. Damptrykket beregnes ved anvendelse af Hertz-Knudsen-relationen.
Anbefalet område:
10-3 til 1 Pa.
1.4.6. Gasmætningsmetode
Der ledes en strøm af inaktiv bæregas hen over stoffet på en sådan måde, at den mættes med dampen. Den stofmængde, en kendt bæregasmængde transporterer, kan enten måles ved opsamling i en passende fælde eller ved in-line analyse. Herudfra kan damptrykket ved en given temperatur beregnes.
Anbefalet område:
10-4 til 1 Pa.
Med den fornødne omhu kan metoden også anvendes i området fra 1 til 10 Pa.
1.4.7. Roterende kugle
I denne metode er det egentlige måleorgan en lille stålkugle, der svæver i et magnetfelt og roterer med høj hastighed. Gastrykket udledes af den trykafhængige bremsning af stålkuglen.
Anbefalet område:
10-4 til 0,5 Pa.
1.5. KVALITETSKRITERIER
I følgende tabel er de forskellige metoder til bestemmelse af damptryk sammenlignet med hensyn til anvendelse, repeterbarhed, reproducerbarhed, måleområde og foreliggende standard.
Tabel
Kvalitetskriterier
|
Målemetode |
Stoffer |
Skønnet repeterbarhed (7) |
Skønnet reproducerbarhed (7) |
Anbefalet område |
Foreliggende standard |
|||
|
faste |
væsker |
|||||||
|
lavtsmeltende |
ja |
op til 25 % |
op til 25 % |
103 Pa til 2 x 103 Pa |
— |
||
|
|
|
|
1 til 5 % |
1 til 5 % |
2 x 103 Pa til 105 Pa |
— |
||
|
ja |
ja |
5 til 10 % |
5 til 10 % |
10 Pa til 105 Pa (8) |
NFT 20-048 (5) |
||
|
ja |
ja |
5 til 10 % |
5 til 10 % |
102 Pa til 105 Pa |
ASTM-D 2879-86 |
||
|
ja |
ja |
5 til 20 % |
op til 50 % |
10-3 Pa til 1 Pa |
NFT 20-047 (6) |
||
|
ja |
ja |
10 til 30 % |
— |
10-3 Pa til 1 Pa |
— |
||
|
ja |
ja |
10 til 30 % |
op til 50 % |
10-4 Pa til 1 Pa (8) |
— |
||
|
|
|
10 til 20 % |
— |
10-4 Pa til 0,5 Pa |
— |
||
1.6. BESKRIVELSE AF METODERNE
1.6.1. Dynamisk måling
1.6.1.1. Apparatur
Måleinstrumentet består typisk af en kogebeholder med tilsluttet køler fremstillet af glas eller metal (figur 1), udstyr til måling af temperaturen og udstyr til regulering og måling af trykket. Et typisk måleudstyr som det på skitsen viste er fremstillet af varmebestandigt glas og består af fem dele:
Et stort delvis dobbeltvægget rør bestående af en slibsamling, en køler, en kølebeholder og et indløb.
Der er monteret en glascylinder med en Cottrell-pumpe på rørets kogesektion, og dens overflade er ru, så stødkogning undgås.
Temperaturen måles med en egnet temperaturføler (f.eks. modstandstermometer eller termoelement med kappe), som er ført ind i instrumentet helt til målepunktet (figur 1, nr. 5) gennem en passende åbning (f.eks. udvendigt slib).
Der foretages de fornødne tilslutninger til det udstyr, hvormed trykket reguleres og måles.
Bulben, der fungerer som stødpudevolumen, er forbundet med måleinstrumentet med et kapillarrør.
Kogebeholderen opvarmes med et varmeelement (f.eks. varmepatron), der er indført i glasapparaturet nedefra. Den nødvendige strømstyrke indstilles og reguleres via et termoelement.
Det påkrævede vakuum på mellem 102 og ca. 105 Pa skabes med en vakuumpumpe.
Der benyttes en passende ventil til regulering af trykket med luft eller nitrogen (måleområde ca. 102 til 105 Pa) og til udluftning.
Trykket måles med et manometer.
1.6.1.2. Fremgangsmåde ved målingen
Damptrykket måles ved, at prøvens kogepunkt bestemmes ved forskellige specificerede tryk mellem ca. 103 og 105 Pa. En ikke-stigende temperatur ved konstant tryk viser, at kogetemperaturen er nået. Skummende stoffers damptryk kan ikke måles ved denne metode.
Stoffet anbringes i testkammeret, der skal være rent og tørt. Det kan være vanskeligt med ikke-pulverformige faste stoffer, men det kan undertiden hjælpe at opvarme kølekappen. Når kammeret er fyldt, samles apparatet, og stoffet afgasses. Derpå indstilles der på det laveste ønskede tryk, og der tændes for opvarmningen. Samtidig tilsluttes temperaturføleren til en skriver.
Der er nået ligevægt, når der registreres konstant kogetemperatur ved konstant tryk. Det skal nøje påses, at der ikke forekommer stødkogning. Desuden skal der ske fuldstændig kondensation på svaleren. Ved bestemmelse af damptryk af lavtsmeltende faste stoffer, skal man omhyggeligt sørge for, at svaleren ikke tilstoppes.
Når ligevægtspunktet er registreret, indstilles der til et højere tryk. Der fortsættes på samme måde, indtil 105 Pa er nået (ca. 5 til 10 målepunkter i alt). Som kontrol gentages ligevægtspunkterne ved aftagende tryk.
1.6.2. Statisk måling
1.6.2.1. Apparatur
Apparaturet består af en beholder til prøven og et opvarmnings- og afkølingssystem til regulering og måling af prøvens temperatur. Apparaturet omfatter desuden instrumenter til indstilling og måling af trykket. Figur 2a og 2b viser grundprincipperne.
Testkammeret (figur 2a) er på den ene side afgrænset af en passende højvakuumventil og på den anden side forbundet med et U-rør, der indeholder en egnet manometervæske. Den anden ende af U-røret er tilsluttet til vakuumpumpen, nitrogenflasken eller udluftningsventilen og et manometer.
I stedet for U-røret kan der anvendes en trykmåler med separat viser (figur 2b).
Prøvens temperatur styres ved, at testkammeret med ventil og U-rør eller trykmåler anbringes i et bad, der holdes ved konstant temperatur ±0,2 K. Temperaturmålingerne udføres på ydersiden af testkammerets væg eller i selve testkammeret.
Der benyttes en vakuumpumpe med frysefælde til evakuering af apparatet.
I metode 2a måles stoffets damptryk indirekte ved hjælp af en nulindikator. Der tages derved hensyn til, at massefylden af væsken i U-røret ændrer sig, hvis der sker store temperaturændringer.
Følgende væsker er egnede som nulindikatorer i U-røret og vælges under hensyntagen til trykområdet og teststoffets kemiske egenskaber: siliconevæsker og phthalater. Teststoffet må ikke kunne opløses væsentligt i eller reageres med U-rørvæsken.
I manometre kan der bruges kviksølv i området fra normalt lufttryk til 102 Pa, mens siliconevæsker og phthalater er egnede fra 102 ned til 10 Pa. Varmebestandige membrankondensatormanometre kan endda benyttes under 10-1 Pa. Der findes også andre trykmålere, der kan bruges under 102 Pa.
1.6.2.2. Fremgangsmåde ved målingen
Før målingen skal alle dele af det i figur 2 viste apparat rengøres og tørres omhyggeligt.
Ved metode 2a fyldes U-røret med den valgte væske, der afgasses ved høj temperatur, inden der foretages aflæsninger.
Teststoffet anbringes i apparatet, som samles, hvorefter temperaturen sænkes tilstrækkeligt til afgasning. Temperaturen skal være så lav, at luften suges ud, men der må ikke for flerkomponentsystemers vedkommende ske nogen ændring af materialets sammensætning. Om nødvendigt kan indstillingen af ligevægt fremskyndes ved omrøring.
Prøven kan underafkøles med f.eks. flydende nitrogen (pas på kondensation i luften og pumpevæsken) eller en blanding af ethanol og tøris. Til lavtemperaturmålinger benyttes et temperaturreguleret bad forbundet med en ultrakryomat.
Idet ventilen over testbeholderen står åben, suges der i flere minutter, så luften fjernes. Derefter lukkes ventilen, og prøvens temperatur sænkes til den laveste ønskede. Om nødvendigt gentages afgasningen flere gange.
Når prøven opvarmes, stiger damptrykket, hvorved væsken i U-røret ændrer ligevægtsstilling. Som kompensation ledes der nitrogen eller luft ind i apparatet via ventilen, indtil trykindikatorvæsken atter er på nul. Det tilsvarende tryk kan aflæses på et præcisionsmanometer ved stuetemperatur. Dette tryk svarer til stoffets damptryk ved den pågældende måletemperatur.
Metode 2b er en lignende metode, men damptrykket aflæses direkte.
Damptrykkets temperaturafhængighed bestemmes med passende små intervaller (ca. 5 til 10 målepunkter) op til det ønskede maksimum. Som kontrol gentages lavtemperaturmålingerne.
En eventuel afvigelse mellem de nye målinger og den kurve, der er fundet ved stigende temperatur, kan skyldes:
|
1. |
at prøven stadig indeholder luft (f.eks. meget tyktflydende materialer) eller lavtkogende stoffer, som er frigjort under opvarmningen og kan fjernes ved sugning efterfulgt af endnu en underafkøling |
|
2. |
at afkølingstemperaturen ikke er lav nok. I så fald benyttes der flydende nitrogen som kølemiddel I tilfælde 1 og 2 må målingerne gentages |
|
3. |
at der sker en kemisk reaktion i stoffet i det undersøgte temperaturinterval (f.eks. dekomponering eller polymerisering). |
1.6.3. Isoteniskop
Henvisning (7) indeholder en fuldstændig beskrivelse af denne metode. Måleanordningens princip fremgår af figur 3. Isoteniskopet er i lighed med den under 1.6.2 beskrevne metode egnet til undersøgelse af såvel faste stoffer som væsker.
Ved måling på væsker, tjener stoffet selv som hjælpemanometervæske. Der anbringes en sådan mængde af stoffet i isoteniskopet, at kuglen og den korte arm af manometersektionen er fyldt. Isoteniskopet forbindes med et vakuumsystem, evakueres og fyldes med nitrogen. Evakueringen og rensningen af systemet gentages to gange, så alt resterende oxygen fjernes. Det fyldte isoteniskop anbringes vandret, så prøven fordeler sig i et tyndt lag i kuglen og manometersektionen (U-delen). Trykket i systemet reduceres til 133 Pa, og prøven opvarmes forsigtigt, indtil den netop koger (fjernelse af opløste fikserede gasser). Isoteniskopet anbringes nu således, at prøven atter befinder sig i kuglen og manometerets korte arm og begge er helt fyldt med væske. Der opretholdes samme tryk som ved afgasning; kuglens udtrukne spids opvarmes med en lille flamme, indtil den dannede damp udvider sig tilstrækkeligt til, at en del af prøven presses ud af kuglens øvre del og manometerarm og ud i isoteniskopets manometersektion, hvorved der opstår et dampfyldt nitrogenfrit rum.
Isoteniskopet anbringes dernæst i et bad med konstant temperatur, og nitrogentrykket indstilles til det er lig med prøvens tryk. At der er trykligevægt fremgår af isoteniskopets manometersektion. Ved ligevægt er nitrogenens damptryk lig med stoffets damptryk.
Ved måling på faste stoffer benyttes en af de i 1.6.2.1 nævnte manometervæsker, alt efter tryk- og temperaturområde. Der fyldes afgasset manometervæske ind i en udvidelse på isoteniskopets lange arm. Derefter anbringes det faste teststof i kuglen og afgasses ved høj temperatur. Dernæst hældes isoteniskopet således, at manometervæsken kan strømme ind i U-røret. Måling af damptrykket som funktion af temperaturen foregår som under 1.6.2.
1.6.4. Effusionsmetoden: damptryksvægt
1.6.4.1. Apparatur
I henvisning (1) er der beskrevet forskellige versioner af apparaturet. Det her beskrevne apparatur illustrerer det generelle princip (figur 4). Figur 4 viser apparaturets hovedkomponenter: en højvakuumbeholder af glas eller rustfrit stål, udstyr til generering og måling af vakuum og indbyggede komponenter til måling af damptryk på en vægt. Apparaturet indeholder følgende indbyggede komponenter:
|
— |
en fordampningsovn med flange og drejelig gennemføring. Fordampningsovnen er en cylindrisk beholder fremstillet i f.eks. kobber eller en godt varmeledende kemisk bestandig legering. Der kan også benyttes en glasbeholder med kobbervæg. Ovnen har en diameter på ca. 3 til 5 cm og en højde på 2 til 5 cm. Der er en til tre åbninger af forskellig størrelse til dampstrømmen. Ovnen opvarmes enten med en varmeplade underneden eller med en varmespiral omkring ydersiden. For at undgå bortledning af varme til grundpladen er varmeaggregatet fastgjort hertil med et metal med ringe varmeledningsevne (nikkel/sølv eller chromnikkelstål), f.eks. et nikkel/sølv-rør fastgjort til en drejelig gennemføring, hvis der benyttes en ovn med flere åbninger. En sådan opstilling har den fordel, at der kan indstikkes en kobberstang, så der kan køles udefra med et kuldebad |
|
— |
hvis der i ovnens kobberlåg er tre åbninger anbragt med 90o i forhold til hinanden, kan forskellige damptryksintervaller inden for det totale måleområde dækkes (åbninger med diameter mellem ca. 0,30 og 4,50 mm). De store åbninger benyttes til lave damptryk og omvendt. Ved drejning af ovnen kan den ønskede åbning eller en mellemstilling i dampstrømmen (ovnåbning — skærm — vægtskål) indstilles, så molekylestrømmen frigøres eller afbøjes af ovnåbningen og rammer vægtskålen. Stoffets temperatur måles med et termoelement eller et modstandstermometer anbragt et passende sted |
|
— |
over skærmen befinder sig vægtskålen på en meget følsom mikrovægt (se nedenfor). Vægtskålen er ca. 30 mm i diameter. Forgyldt aluminium er et egnet materiale |
|
— |
vægtskålen er omgivet af en cylindrisk kølekappe af messing eller kobber. Afhængigt af vægttypen er den forsynet med åbninger for vægtbjælken og en skærmåbning til molekylestrømmen, og den skal garantere fuldstændig kondensering af dampen på vægtskålen. Varmen bortledes til omgivelserne f.eks. med en kobberstang, der er forbundet med kølekappen. Denne stang er ført gennem grundpladen og isoleret herfra, f.eks. med et rør af chromnikkelstål. Den nedsænkes i et dewarkar med flydende nitrogen under grundpladen, eller der ledes flydende nitrogen gennem stangen. Herved holdes kølekappen på en temperatur på ca. – 120 oC. Vægtskålen køles udelukkende ved stråling, hvilket er tilfredsstillende i det pågældende trykinterval (afkøling ca. 1 time før målingerne påbegyndes) |
|
— |
vægten er anbragt over kølekappen. En højfølsom toarmet elektronisk mikrovægt (8) eller et meget følsomt drejespoleinstrument (se OECD Test Guideline 104, udgave af 12.5.1981) er eksempler på egnede vægte) |
|
— |
på grundpladen findes ligeledes de elektriske tilslutninger for termoelementer (eller modstandstermometre) og varmelegemer |
|
— |
der frembringes et vakuum i beholderen med enten en partiel vakuumpumpe eller en højvakuumpumpe (der kræves et vakuum på ca. 1 til 2 × 10–3 Pa, hvilket opnås efter 2 timers pumpning). Trykket reguleres med et egnet ioniseringsmanometer. |
1.6.4.2. Fremgangsmåde ved målingen
Teststoffet fyldes i beholderen, som lukkes med låget. Skærmen og kølekappen skubbes på plads over ovnen. Apparatet lukkes, og vakuumpumperne startes. Sluttrykket skal være ca. 10–4 Pa, før målingerne påbegyndes. Afkøling af kølekappen sættes i gang ved 10-2 Pa.
Når det krævede vakuum er nået, startes kalibreringsrækken ved den laveste temperatur. Der indstilles på den pågældende åbning i låget, og dampstrømmen passerer gennem skærmen direkte over åbningen og rammer den afkølede vægtskål. Vægtskålen skal være så stor, at hele den dampstrøm, der ledes gennem skærmen, rammer den. Dampstrømmens impuls overfører en kraft til vægtskålen, og molekylerne kondenserer på dens kolde overflade.
Impulsen og den samtidige kondensation frembringer et signal på skriveren. Ved vurdering af signalerne kan der uddrages to oplysninger:
|
1. |
Med det her beskrevne apparatur bestemmes damptrykket direkte ud fra den impuls, der overføres til vægtskålen (hertil er det ikke nødvendigt at kende molekylvægten (2)). Ved vurdering af aflæsningerne må der tages hensyn til geometriske faktorer, såsom ovnåbningen og molekylestrømmens vinkel. |
|
2. |
Kondensatets masse kan måles samtidig, og herudfra kan fordampningshastigheden beregnes. Endvidere kan damptrykket beregnes ud fra fordampningshastigheden og molekylvægten ved hjælp af Hertz' ligning (2). |

hvor
|
G |
= |
fordampningshastigheden (kg s-1 m-2) |
|
M |
= |
molekylmassen (g mol-1) |
|
T |
= |
temperaturen (K) |
|
R |
= |
gaskonstanten (Jmol-1 K -1) |
|
p |
= |
damptrykket (Pa) |
Når det ønskede vakuum er nået, påbegyndes måleserien ved den laveste ønskede måletemperatur.
Videre målinger foretages ved, at temperaturen hæves i små spring, indtil den højeste ønskede temperatur er nået. Dernæst afkøles prøven igen, og der optegnes endnu en kurve over damptrykket. Bekræfter de sidste resultater ikke de første, kan det skyldes, at stoffet dekomponerer i det undersøgte temperaturområde.
1.6.5. Effusionsmetode — vægttab
1.6.5.1. Apparatur
Et effusionsapparat består af følgende basiskomponenter:
|
— |
en beholder, der kan termostateres og evakueres, og hvori effusionscellerne placeres |
|
— |
en højvakuumpumpe (f.eks. diffusionspumpe eller turbomolekylarpumpe) med vakuummeter |
|
— |
en frysefælde med flydende nitrogen eller tøris. |
Som eksempel er der i figur 5 vist en elektrisk opvarmet vakuumbeholder af aluminium med fire effusionsceller af rustfrit stål. Den rustfrie stålfolie af ca. 0,3 mm's tykkelse har en effusionsåbning på 0,2 til 1,0 mm i diameter og er fastgjort til effusionscellen med et låg med gevind.
1.6.5.2. Fremgangsmåde ved målingen
Referencestoffet og teststoffet fyldes i hver effusionscelle, metalmembranen med åbningen fastgøres ved hjælp af låget med gevind, og hver celle vejes med en nøjagtighed på 0,1 mg. Cellen anbringes i det termostaterede apparat, som derefter evakueres til et tryk, der er mindre end en tiendedel af det forventede. Med på forhånd fastsatte tidsintervaller på mellem 5 og 30 timer lukkes der luft ind i apparatet, og effusionscellernes massetab bestemmes ved vejning.
For at sikre at resultaterne ikke påvirkes af flygtige bestanddele, vejes cellen med faste mellemrum, så det kan kontrolleres, at fordampningshastigheden er konstant over mindst to tidsintervaller.
Damptrykket p i effusionscellen er givet ved:

hvor
|
p |
= |
damptrykket (Pa) |
|
m |
= |
massen af den stofmængde, der forlader cellen i tidsrummet t (kg) |
|
t |
= |
tiden (s) |
|
A |
= |
hullets areal (m2) |
|
K |
= |
korrektionsfaktor |
|
R |
= |
gaskonstanten (Jmol-1 K -1) |
|
T |
= |
temperaturen (K) |
|
M |
= |
molekylmassen (kg mol-1) |
Korrektionsfaktoren K afhænger af forholdet mellem den cylindriske åbnings længde og radius:
|
forhold |
0,1 |
0,2 |
0,6 |
1,0 |
2,0 |
|
K |
0,952 |
0,909 |
0,771 |
0,672 |
0,514 |
Ovenstående ligning kan omskrives til:

hvor 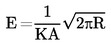 er effusionscellekonstanten.
er effusionscellekonstanten.
Denne effusionscellekonstant kan bestemmes ved hjælp af referencestoffer (2,9) ud fra følgende ligning:

hvor
|
p(r) |
= |
referencestoffets damptryk (Pa) |
|
M(r) |
= |
referencestoffets molekylmasse (kg × mol-1) |
1.6.6. Gasmætningsmetode
1.6.6.1. Apparatur
Et typisk apparat til udførelse af denne prøvning består af de komponenter, der er vist i figur 6a og beskrevet nedenfor (1).
Inert gas:
Bæregassen må ikke reagere kemisk med teststoffet. Det er normalt tilstrækkeligt at anvende nitrogen til dette formål, men undertiden kan der kræves andre gasser (10). Den anvendte gas skal være tør (se figur 6a, nr. 4: føler for relativ fugtighed).
Strømningsregulering
Der kræves et velegnet gasreguleringssystem for at sikre konstant og valgbar gennemstrømningshastighed gennem mætningskolonnen.
Fælder til opsamling af damp:
Disse afhænger af den aktuelle prøves egenskaber og af den valgte analysemetode. Dampen skal indfanges kvantitativt og i en form, som tillader efterfølgende analyse. Til visse stoffer vil fælder, der indeholder væsker såsom hexan eller ethylenglycol, være velegnede. Til andre vil der kunne anvendes faste absorptionsmidler.
Som alternativ til opsamling af damp og efterfølgende analyse kan der benyttes in-line analyseteknikker, som f.eks. chromatografi, til kvantitativ bestemmelse af den mængde materiale, som en kendt mængde bæregas fører med sig. Desuden kan prøvens massetab måles.
Varmeveksler:
Til målinger ved forskellige temperaturer kan det være nødvendigt at indsætte en varmeveksler i opstillingen.
Mætningskolonne:
Teststoffet overføres til en egnet inaktiv bærer fra en opløsning. Den coatede bærer pakkes i mætningskolonnen; dennes dimensioner og gennemstrømningshastigheden skal afpasses således, at man er sikker på, at bæregassen bliver mættet. Mætningskolonnen skal være termostateret. Ved målinger over stuetemperatur skal stykket mellem mætningskolonnen og fælderne opvarmes, så kondensation af teststoffet undgås.
For at mindske massetransport ved diffusion kan der anbringes et kapillarrør efter mætningskolonnen (figur 6b).
1.6.6.2. Fremgangsmåde ved målingen
Klargøring af mætningskolonnen:
En opløsning af teststoffet i et meget letflygtigt opløsningsmiddel blandes med en passende mængde af bæreren. Der skal være tilstrækkeligt med teststof til, at der finder mætning sted under hele testens varighed. Opløsningsmidlet afdampes fuldstændigt i luft eller i en rotationsfordamper, og efter omhyggelig blanding fyldes materialet på mætningskolonnen. Efter at prøven er termostateret, ledes der tør nitrogen gennem apparatet.
Måling:
Fælderne eller in-line detektoren forbindes med kolonnens afgangsrør, og tiden registreres. Gennemstrømningshastigheden kontrolleres i begyndelsen og med regelmæssige mellemrum under forsøget ved anvendelse af en boblemåler (eller kontinuerligt med en massestrømsmåler).
Trykket ved mætningskolonnens afgangsåbning skal måles. Dette kan ske ved:
|
a) |
enten at indsætte en trykmåler mellem mætningskolonnen og fælderne (hvilket kan være utilfredsstillende på grund af øget dødvolumen og større adsorptionsflade) |
|
b) |
eller at bestemme trykfaldet over det bestemte fældesystem, der benyttes, som funktion af gennemstrømningshastigheden i et særskilt forsøg (hvilket kan være lidet tilfredsstillende for væskefælder). |
Den tid, der kræves til opsamling af den mængde teststof, der er nødvendig til de forskellige analysemetoder, bestemmes ved forforsøg eller skønnes. Som alternativ til opsamling af stoffet til senere analyse kan der benyttes kvantitative in-line analyseteknikker (f.eks. chromatografi). Før beregning af damptrykket ved en given temperatur udføres der forforsøg til bestemmelse af den maksimale flowhastighed, hvor bæregassen mættes fuldstændigt med stofdampe. Dette sikres ved at lede bæregassen så langsomt gennem mætningskolonnen, at en lavere hastighed ikke giver større beregnet damptryk.
Den specifikke analysemetode vil afhænge af arten af teststof, der undersøges (f.eks. gaschromatografi eller gravimetri).
Den mængde stof, som et kendt rumfang bæregas fører med sig, bestemmes.
1.6.6.3. Beregning af damptryk
Damptrykket beregnes ud fra dampens massefylde W/V ved hjælp af ligningen:
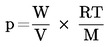
hvor:
|
P |
= |
damptrykket (Pa) |
|
W |
= |
massen af den stofmængde, der er fordampet (g) |
|
V |
= |
rumfang mættet gas (m3) |
|
R |
= |
gaskonstanten (Jmol-1 K-1) |
|
T |
= |
temperaturen (K) |
|
M |
= |
teststoffets molekylmasse (g mol-1) |
De målte rumfang skal korrigeres for temperaturforskelle mellem flowmeteret og den termostaterede mætningskolonne. Hvis flowmeteret er placeret efter dampfælden, kan det være nødvendigt at korrigere for eventuelt fordampede bestanddele fra fælden (1).
1.6.7. Roterende kugle (8, 11, 13)
1.6.7.1. Apparatur
Metoden kan udføres ved hjælp af en viskositetsmåler med roterende kugle som vist på figur 8. Figur 7 viser forsøgsopstillingen skematisk.
Måleopstillingen består typisk af et målehoved med roterende kugle anbragt i et termostateret hus (kontrolleret inden for 0,1 oC). Testbeholderen er ligeledes anbragt i et termostateret hus (kontrolleret inden for 0,01 oC), og alle opstillingens komponenter holdes ved en højere temperatur for at undgå kondensation. Systemet er ved hjælp af højvakuumventiler forbundet med en højvakuumpumpe.
Målehovedet består af en stålkugle (4 til 5 mm diameter) i et rør. Kuglen holdes stabilt svævende af et magnetfelt, sædvanligvis ved en kombination af permanentmagneter og styrespoler.
Kuglen sættes i rotation ved hjælp af roterende felter, der frembringes af spolerne. Rotationshastigheden måles med detektorspoler, der måler den svage tværmagnetisering af kuglen, der altid vil være til stede.
1.6.7.2. Fremgangsmåde ved målingen
Når kuglen har opnået en bestemt rotationshastighed v(o) (normalt omkring 400 omdrejninger pr. sekund), afbrydes energitilførslen, og gasfriktionen begynder at decelerere kuglens rotation.
Rotationshastighedstabet måles som funktion af tiden. Da friktionen i det magnetiske ophæng er ubetydelig sammenlignet med gasfriktionen, er gastrykket givet ved udtrykket:
![]()
hvor
|
|
= |
gasmolekylernes gennemsnitshastighed |
|
r |
= |
kuglens radius |
|
ρ |
= |
kuglens massefylde |
|
σ |
= |
koefficient for tangential impulsoverførsel ((ε = 1 for en ideal kugleflade |
|
t |
= |
tiden |
|
v(t) |
= |
rotationshastigheden efter tiden t |
|
v(o) |
= |
begyndelsesrotationshastigheden |
Dette udtryk kan også omskrives til
hvor tn, og tn - 1 er den tid, der kræves til et givet antal omdrejninger N. Disse tn og tn-1 følger lige efter hinanden, og tn > t n - 1.
Gasmolekylernes gennemsnitshastighed  er givet ved
er givet ved

hvor:
|
T |
= |
temperaturen |
|
R |
= |
gaskonstanten |
|
M |
= |
molekylmassen |
2. DATA
Damptrykket bestemt ved en hvilken som helst af de ovenfor beskrevne metoder bør bestemmes ved mindst to temperaturer. Tre eller flere er ønskelige i området 0 til 50 oC, så damptrykkurvens linearitet kan kontrolleres.
3. RAPPORTERING
Forsøgsrapporten skal om muligt indeholde følgende oplysninger:
|
— |
anvendt metode |
|
— |
nøjagtig beskrivelse af stoffet (betegnelse og urenheder) og eventuel indledende rensning |
|
— |
mindst to værdier for damptryk og temperatur, helst i området mellem 0 og 50 oC |
|
— |
alle rådata |
|
— |
en afbildning af log p mod 1/T |
|
— |
et skøn over damptrykket ved 20 eller 25 oC. |
Hvis der er iagttaget omdannelse (ændring i tilstandsform eller dekomponering), skal følgende oplysninger fremgå:
|
— |
ændringens art |
|
— |
den temperatur, hvor ændringen sker ved atmosfæretryk |
|
— |
damptrykket ved 10 og 20 oC under overgangstemperaturen og ved 10 og 20 oC over denne temperatur (medmindre overgangen er fra fast stof til gas). |
Alle oplysninger og bemærkninger af betydning for vurdering af resultaterne, især vedrørende urenheder og stoffets fysiske tilstand, skal anføres.
4. HENVISNINGER
|
(1) |
OECD, Paris, 1981, Test Guideline 104, Decision of the Council C(81) 30 final. |
|
(2) |
Ambrose, D. in B. Le Neindre, B. Vodar, (Eds.): Experimental Thermodynamics, Butterworths, London, 1975, Vol II. |
|
(3) |
R. Weissberger ed.: Technique of Organic Chemistry, Physical Methods of Organic Chemistry, 3rd ed. Chapter IX, Interscience Publ., New York, 1959, Vol. I, Part I. |
|
(4) |
Knudsen, M. Ann. Phys. Lpz., 1909, vol. 29, 1979; 1911, vol. 34, 593. |
|
(5) |
NF T 20-048 AFNOR (Sept. 85). Chemical products for industrial use — Determination of vapour pressure of solids and liquids within range from 10-1 to 105 Pa — Static method. |
|
(6) |
NF T 20-047 AFNOR (Sept. 85). Chemical products for industrial use — Determination of vapour pressure of solids and liquids within range from 10-3 to 1 Pa — Vapour pressure balance method. |
|
(7) |
ASTM D 2879-86, Standard test method for vapour pressure — temperature relationship and initial decomposition temperature of liquids by isoteniscope. |
|
(8) |
G. Messer, P. Rohl, G. Grosse and W. Jitschin. J. Vac. Sci. Technol.(A), 1987,'VoI. 5 (4), 2440. |
|
(9) |
Ambrose, D.; Lawrenson, I.J.; Sprake, C.H.S. J. Chem. Thermodynamics 1975, vol. 7, 1173. |
|
(10) |
B.F. Rordorf. Thermochimica Acta, 1985, vol. 85, 435. |
|
(11) |
G. Comsa, J.K. Fremerey and B. Lindenau. J. Vac. Sci. Technol., 1980, Vol. 17 (2), 642. |
|
(12) |
G. Reich. J. Vac. Sci. Technol., 1982, voI. 20 (4), 1148. |
|
(13) |
J.K. Fremerey. J. Vac. Sci. Technol.(A), 1985, Vol. 3 (3), 1715. |
Tillæg 1
Estimeringsmetode
INDLEDNING
Man kan benytte beregnede værdier for damptrykket til:
|
— |
at beslutte, hvilken af forsøgsmetoderne der er egnet |
|
— |
at foretage et skøn eller at fastsætte en grænseværdi, når man af tekniske årsager (herunder tilfælde, hvor damptrykket er meget lavt) ikke kan benytte eksperimentelle metoder |
|
— |
at konstatere, i hvilke tilfælde det er berettiget at undlade eksperimentelle målinger, fordi damptrykket sandsynligvis vil være < 10-5 Pa ved stuetemperatur. |
ESTIMERINGSMETODE
Væskers og faste stoffers damptryk kan skønnes ved hjælp af den modificerede Watson-korrelation (a). Det eneste nødvendige eksperimentelle data er kogepunktet. Metoden kan benyttes i trykintervallet fra 10-5 Pa til 10 Pa.
I »Handbook of Chemical Property Estimation Methods« (b) findes der nærmere oplysninger om metoden.
FREMGANGSMÅDE VED BEREGNINGEN
Ifølge (b) beregnes damptrykket således:
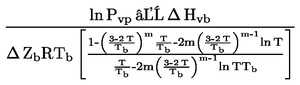
hvor
|
T |
= den pågældende temperatur |
|
Tb |
= kogepunktet |
|
Pvp |
= damptrykket ved temperaturen T |
|
ΔHvb |
= fordampningsvarmen |
|
ΔZb |
= en sammentrykkelighedsfaktor (ansættes til 0,97) |
|
m |
= en empirisk faktor, der afhænger af tilstandsformen ved den pågældende temperatur |
Yderligere haves

hvor KF er en empirisk faktor, der tager hensyn til stoffets polaritet. I henvisning (b) er der en liste med KF-faktorer for flere typer forbindelser.
Ofte forligger der data, hvor kogepunktet ved reduceret tryk er opgivet. I sådanne tilfælde kan man efter (b) beregne damptrykket således:

hvor T1 er kogepunkter ved det reducerede tryk P1.
RAPPORT
Når estimeringsmetoden benyttes, skal rapporten indeholde fyldestgørende dokumentation af beregningen.
HENVISNINGER
|
(a) |
K.M. Watson, Ind. Eng. Chem; 1943, vol. 35, 398. |
|
(b) |
W.J. Lyman, W.F. Reehl, D.H. Rosenblatt. Handbook of Chemical Property Estimation Methods, Mc Graw-Hill, 1982. |
Tillæg 2
Figur 1
Apparat til bestemmelse af damptrykskurven efter den dynamiske metode

Figur 2a
Apparat til bestemmelse af damptrykskurven efter den statiske metode (med U-rørsmanometer)

Figur 2b
Apparat til bestemmelse af damptrykskurven efter den statiske metode (med trykindikator)
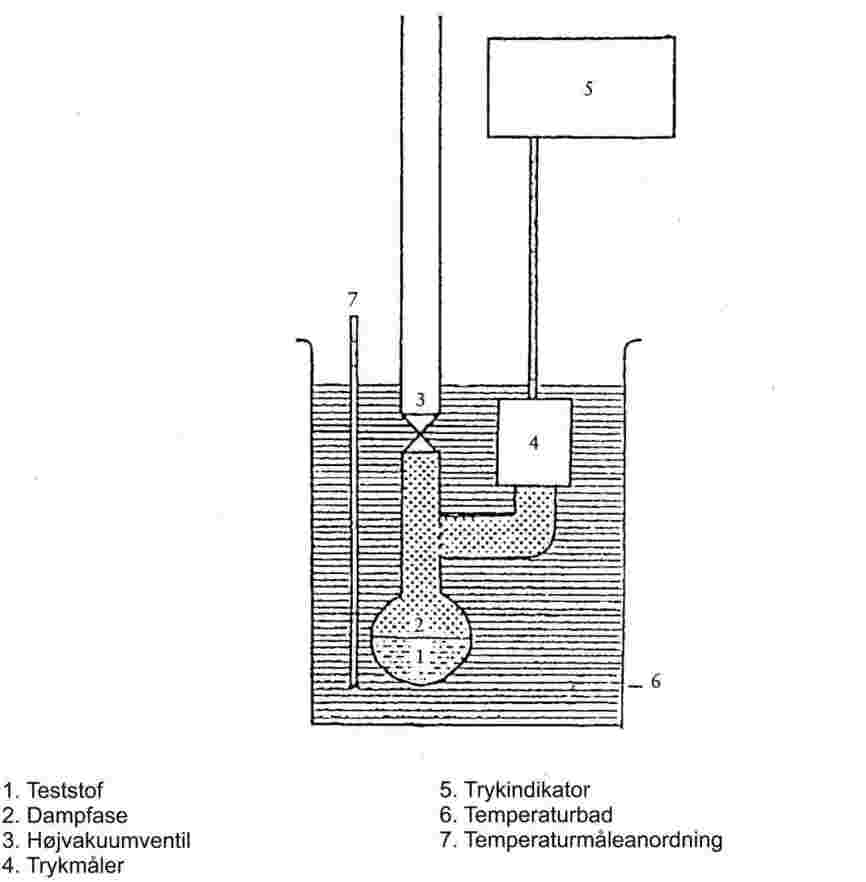
Figur 3
Isoteniskop (se reference 7)

Figur 4
Apparat til bestemmelse af damptrykskurven efter metoden med damptryksvægt

Figur 5
Eksempel på apparat til fordampning ved lavt tryk ved effusionsmetoden, med et effusionscellevolumen på 8cm3
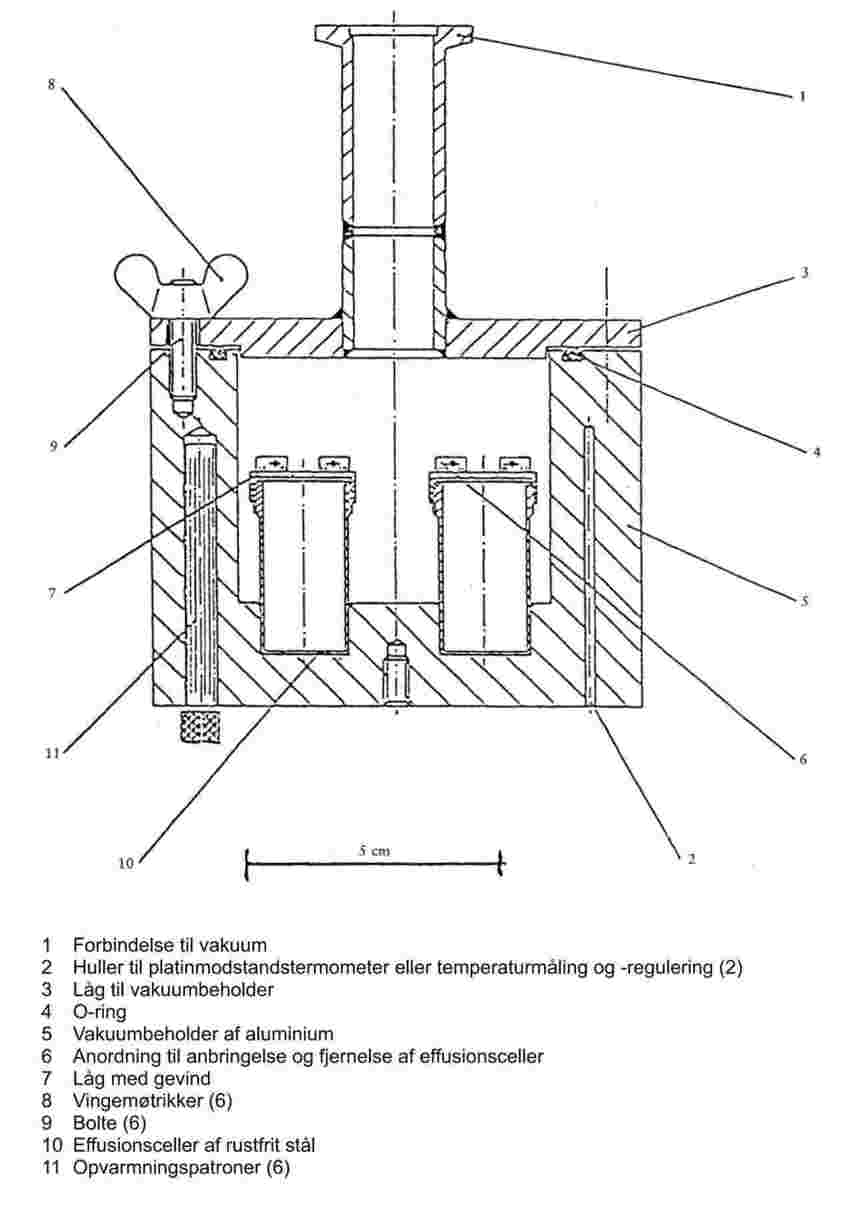
Figur 6a
Eksempel på et strømningssystem til bestemmelse af damptryk efter gasmæmingsmetoden
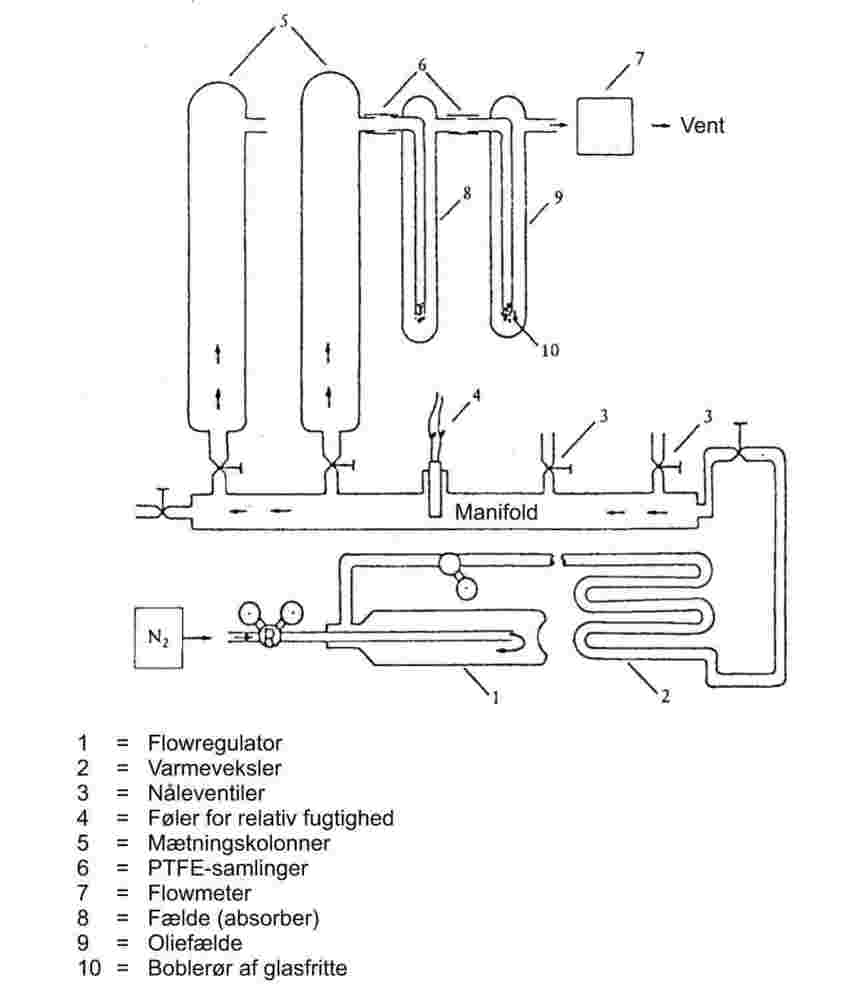
Figur 6b
Eksempel på et system til bestemmelse af damptryk efter gasmætningsmetoden med kapillarrør anbragt efter mætningskammeret

Figur 7
Eksempel på forsøgsopstilling til metoden med roterende kugle Damptryksapparat

Figur 8
Eksempel på målehoved med roterende kugle

A.5. OVERFLADESPÆNDING
1. METODE
De beskrevne metoder er baseret på OECD-testvejledningen (1). Grundprincipperne er anført i henvisning (2).
1.1. INDLEDNING
De beskrevne metoder anvendes til måling af vandige opløsningers overfladespænding.
Ved udførelsen af denne test er det nyttigt at have forhåndsoplysninger om stoffets vandopløselighed, struktur, hydrolyseegenskaber og kritiske koncentrationer for micelledannelse.
De følgende metoder kan anvendes for de fleste kemiske stoffer uanset renheden.
Måling af overfladespænding ved ringtensiometermetoden er begrænset til vandige opløsninger med en dynamisk viskositet på mindre end ca. 200 mPa s.
1.2. DEFINITIONER OG ENHEDER
Den frie overfladeenthalpi pr. overfladearealenhed betegnes som overfladespændingen.
Overfladespændingen angives i:
N/m (SI-enhed) eller
mN/m (SI-underenhed)
1 N/m = 103 dyn/cm
1 mN/m = 1 dyn/cm i det gamle cgs-system
1.3. REFERENCESTOFFER
Anvendelse af referencestoffer er ikke påkrævet, hver gang et nyt stof skal undersøges. Referencestoffer skal først og fremmest tjene til lejlighedsvis kontrol af metoden og til sammenligning med resultater opnået med andre metoder.
Referencestoffer, som dækker et bredt udvalg af overfladespændinger, er angivet i henvisning (1) og (3).
1.4. METODERNES PRINCIP
Metoderne er baseret på måling af den største kraft, der skal udøves lodret på en stigbøjle eller ring i kontakt med væskeoverfladen af en prøve i et målekar, for at denne kontakt brydes, eller den lodrette kraft, som skal udøves på en plade, hvis ene kant er i kontakt med prøvens overflade, for at den dannede hinde brydes.
Stoffer, hvis opløselighed i vand er mindst 1 mg/l, testes i vandig opløsning ved én koncentration.
1.5. KVALITETSKRITERIER
Disse metoder kan give større nøjagtighed, end der må forventes krævet ved miljøvurderinger.
1.6. BESKRIVELSE AF METODERNE
Der fremstilles en opløsning af stoffet i destilleret vand. Koncentrationen af denne opløsning skal være 90 % af stoffets mætningskoncentration i vand; er denne koncentration større end 1 g/l, benyttes der en koncentration på 1 g/l til testen. Stoffer med en opløselighed i vand på mindre end 1 mg/l behøver ikke at testes.
1.6.1. Plademetoden
Se ISO 304 og NF T 73-060 (Surface active agents — determination of surface tension by drawing up liquid films).
1.6.2. Stigbøjlemetoden
Se ISO 304 og NF T 73-060 (Surface active agents — determination of surface tension by drawing up liquid films).
1.6.3. Ringmetoden
Se ISO 304 og NF T 73-060 (Surface active agents — determination of surface tension by drawing up liquid films).
1.6.4. OECD-harmoniseret ringmetode
1.6.4.1. Apparatur
Kommercielt tilgængelige tensiometre er tilstrækkelige til denne måling. De består af følgende dele:
|
— |
bevægeligt prøvebord |
|
— |
kraftmålingssystem |
|
— |
målelegeme (ring) |
|
— |
målekar. |
1.6.4.1.1.
Det bevægelige prøvebord anvendes til understøttelse af det termostaterede målekar, som rummer den væske, der skal undersøges. Sammen med kraftmålingssystemet er det monteret på et stativ.
1.6.4.1.2.
Kraftmålingssystemet (se figuren) er placeret over prøvebordet. Usikkerheden ved kraftmålingen må ikke overstige ± 10–6 N, svarende til en usikkerhed på ±0,1 mg ved en vejning. Oftest er måleskalæn på de kommercielt tilgængelige tensiometre inddelt i mN/m, således at overfladespændingen direkte kan aflæses i mN/m med en nøjagtighed på 0,1 mN/m.
1.6.4.1.3.
Ringen er sædvanligvis fremstillet af en platin-iridium-tråd med en tykkelse på ca. 0,4 mm og en middelomkreds på 60 mm. Trådringen er ophængt vandret fra en metalstang og en trådbærearm, som danner forbindelsen til kraftmålingssystemet (se figuren).
Figur
Målelegeme
(alle dimensioner er i mm)

1.6.4.1.4.
Målekarret, som rummer testopløsningen, skal være et termostateret glaskar. Det skal have en sådan form, at temperaturen af testopløsningen og gasfasen herover er konstant under målingen, og at prøven ikke kan fordampe. Cylindriske glaskar med en indre diameter på mindst 45 mm er acceptable.
1.6.4.2. Klargøring af apparatet
1.6.4.2.1.
Glaskar rengøres omhyggeligt. Om nødvendigt vaskes de først med varm chromsvovlsyre og dernæst med en tyktflydende phosphorsyreopløsning (83 til 98 w/w % H3PO4). Herefter skylles omhyggeligt i ledningsvand og endelig vaskes i redestilleret vand til neutral reaktion. Til sidst tørres karrene, eller de skylles i noget af den prøvevæske, som skal måles.
Ringen renses først grundigt i vand, så alle vandopløselige stoffer fjernes. Derefter nedsænkes den et øjeblik i chromsvovlsyre, skylles i redestilleret vand til neutral reaktion og opvarmes til sidst et øjeblik over en methanolflamme.
Bemærk:
Forurening med stoffer, som ikke opløses eller destrueres af chromsvovlsyre eller phosphorsyre, som f.eks. silikoner, fjernes ved hjælp af et passende organisk opløsningsmiddel.
1.6.4.2.2.
Apparatet klargøres ved, at nulpunktet justeres, og apparatet indstilles til pålidelig aflæsning i mN/m.
Opstilling:
Apparatet skal stilles i vater, f.eks. ved hjælp af et vaterpas på tensiometrets fod, idet man justerer stilleskruerne.
Nulpunktsindstilling:
Efter montering af ringen i apparatet, men inden den nedsænkes i væsken, skal tensiometerviseren indstilles på nul, og man kontrollerer, at ringen er parallel med væskeoverfladen. Til dette kan væskeoverfladen anvendes som et spejl.
Kalibreringer:
Prøvekalibreringen kan udføres ved en af de følgende to metoder:
|
a) |
ved hjælp af lodder: dette er en fremgangsmåde, hvor ryttere af kendt masse mellem 0,1 og 1,0 g anbringes på ringen. Kalibreringsfaktoren Φa, som alle instrumentaflæsninger skal multipliceres med, bestemmes ifølge ligning (1).
hvor: 
|
|
b) |
ved hjælp af vand: dette er en fremgangsmåde, hvor der anvendes rent vand, hvis overfladespænding ved f.eks. 23 oC er 72,3 mN/m. Denne fremgangsmåde er hurtigere at udføre end vægtkalibreringen, men der er altid risiko for, at vandets overfladespænding er ændret ved forurening med spor af overfladeaktive stoffer. Kalibreringsfaktoren, Φb, som alle instrumentaflæsninger skal multipliceres med, bestemmes ifølge ligning (2).
hvor:
|
1.6.4.3. Testforberedelse
Der fremstilles vandige opløsninger med passende koncentrationer af de stoffer, som skal undersøges. Opløsningerne må ikke indeholde uopløst stof.
Opløsningerne holdes ved konstant temperatur (±0,5 oC). Idet en opløsnings overfladespænding ændres i målekarret med tiden, må der foretages adskillige målinger på forskellige tidspunkter, og der afsættes en kurve, der viser overfladespændingen som funktion af tiden. Når overfladespændingen ikke ændres yderligere, er ligevægtstilstanden nået.
Støv og gasformig forurening med andre stoffer forstyrrer målingen, hvorfor arbejdet bør udføres under et beskyttende dække.
1.6.5. Testbetingelser
Målingerne foretages ved ca. 20 oC, og temperaturen skal holdes inden for ±0,5 oC.
1.6.6. Udførelse af testen
De opløsninger, som skal måles, fyldes i det omhyggeligt rengjorte målekar, idet man undgår skumning. Målekarret anbringes på apparatets prøvebord. Bordet med målekarret hæves, indtil ringen er nedsænket fuldstændigt i den opløsning, som skal måles. Derefter sænkes bordet jævnt og gradvis (med en hastighed på ca. 0,5 cm/min) for at løsrive ringen fra overfladen, indtil den største kraft er nået. Det væskelag, som hænger ved ringen, må ikke fjernes fra denne. Når målingen er udført, nedsænkes ringen atter i væsken, og målingen gentages, indtil der opnås konstante værdier for overfladespændingen. Den tid, der er gået fra overførsel af opløsningen til målekarret og til selve målingen udføres, noteres ved hver bestemmelse. Aflæsningerne skal foretages ved den største kraft, som kræves til løsrivning af ringen fra væskeoverfladen.
2. DATA
For at beregne overfladespændingen skal den værdi, som er aflæst i mN/m på apparatet, først multipliceres med kalibreringsfaktoren Φa eller Φb (afhængigt af den anvendte kalibreringsmetode). Dette giver en værdi, som kun gælder tilnærmelsesvis, og som derfor kræver korrektion.
Harkins og Jordan (4) har empirisk bestemt korrektionsfaktorer for værdier af overfladespændingen, som er bestemt med ringmetoden; disse faktorer afhænger af ringens dimensioner, væskens massefylde og dens overfladespænding.
Da det er besværligt for hver enkelt måling at bestemme korrektionsfaktoren ud fra Harkins og Jordans tabeller for at beregne overfladespænding, kan man for vandige opløsninger benytte en forenklet metode, hvor de korrigerede værdier for overfladespændingen direkte aflæses i den nedenstående tabel (der anvendes interpolation til aflæsninger, som ligger mellem tabelværdierne).
Tabel
Korrektion af den målte overfladespænding
Kun for vandige opløsninger, p = 1 g/cm3
|
R |
= 9,55 mm (gennemsnitlig ringradius) |
|
r |
= 0,185 mm (ringtrådens radius) |
|
Eksperimentel værdi (mN/m) |
Korrigeret værdi (mN/m) |
|
|
kalibrering med lodder (se 1.6.4.2.2. a)) |
kalibrering med vand (se 1.6.4.2.2. b)) |
|
|
20 |
16,9 |
18,1 |
|
22 |
18,7 |
20,1 |
|
24 |
20,6 |
22,1 |
|
26 |
22,4 |
24,1 |
|
28 |
24,3 |
26,1 |
|
30 |
26,2 |
28,1 |
|
32 |
28,1 |
30,1 |
|
34 |
29,9 |
32,1 |
|
36 |
31,8 |
34,1 |
|
38 |
33,7 |
36,1 |
|
40 |
35,6 |
38,2 |
|
42 |
37,6 |
40,3 |
|
44 |
39,5 |
42,3 |
|
46 |
41,4 |
44,4 |
|
48 |
43,4 |
46,5 |
|
50 |
45,3 |
48,6 |
|
52 |
47,3 |
50,7 |
|
54 |
49,3 |
52,8 |
|
56 |
51,2 |
54,9 |
|
58 |
53,2 |
57,0 |
|
60 |
55,2 |
59,1 |
|
62 |
57,2 |
61,3 |
|
64 |
59,2 |
63,4 |
|
66 |
61,2 |
65,5 |
|
68 |
63,2 |
67,7 |
|
70 |
65,2 |
69,9 |
|
72 |
67,2 |
72,0 |
|
74 |
69,2 |
— |
|
76 |
71,2 |
— |
|
78 |
73,2 |
— |
Denne tabel er udarbejdet på basis af Harkins-Jordan korrektioner, og ligner den, der er i DIN-standarden (DIN 53914) for vand og vandige opløsninger (massefylde ρ = 1 g/cm3) og for en kommerciel tilgængelig ring med dimensionerne R = 9,55 mm (middelringradius) og r = 0,185 mm (ringtrådens radius). Tabellen angiver korrigerede værdier for overfladespændingsmålinger, der er foretaget efter kalibrering med lodder eller vand.
Alternativt kan overfladespændingen uden forudgående kalibrering beregnes efter følgende formel:
hvor:
|
F |
= |
kraften målt på dynamometeret, idet hinden brydes |
|
R |
= |
ringens radius |
|
f |
= |
korrektionsfaktoren (1) |
3. RAPPORTERING
3.1. FORSØGSRAPPORT
Forsøgsrapporten skal om muligt indeholde følgende oplysninger:
|
— |
anvendt metode |
|
— |
anvendt type vand eller opløsning |
|
— |
nøjagtig specifikation af stoffet (identitet og urenheder) |
|
— |
måleresultater: overfladespænding (aflæsning) med angivelse af både de enkelte målinger, deres middelværdi og den korrigerede middelværdi (med hensyn til apparatfaktoren og korrektionstabellen) |
|
— |
opløsningens koncentration |
|
— |
testtemperaturen |
|
— |
den anvendte opløsnings alder, og specielt den tid, der er forløbet mellem fremstillingen af opløsningen og målingen |
|
— |
beskrivelse af overfladespændingens tidsafhængighed efter overførsel til målekarret |
|
— |
alle oplysninger og bemærkninger af betydning for vurdering af resultaterne, især vedrørende urenheder og stoffets fysiske tilstandsform. |
3.2. FORTOLKNING AF RESULTATERNE
På baggrund af vands overfladespænding på 72,75 mN/m ved 20 oC anses stoffer med en overfladespænding på mindre end 60 mN/m bestemt ved denne metode for at være overfladeaktive.
4. HENVISNINGER
|
(1) |
OECD, Paris, 1981, Test Guideline 115, Decision of the Council C(81) 30 final. |
|
(2) |
R. Weissberger ed.: Technique of Organic Chemistry, Physical Methods of Organic Chemistry, 3rd ed., Interscience Publ., New York, 1959, Vol. I, Part I, Chapter XIV |
|
(3) |
Pure Appl. Chem., 1976, vol. 48, 511. |
|
(4) |
Harkins, W.D., Jordan, H.F., J. Amer. Chem. Soc., 1930, vol. 52, 1751. |
A.6. VANDOPLØSELIGHED
1. METODE
De beskrevne metoder er baseret på OECD-testvejledningen (1).
1.1. INDLEDNING
Ved udførelse af denne test er det nyttigt at have forhåndsoplysninger om stoffets strukturformel, damptryk, dissociationskonstant og hydrolyse (som funktion af pH).
Der findes ikke én enkelt metode, der dækker hele området af vandopløselighed.
De to testmetoder, der er beskrevet nedenfor, dækker hele opløselighedsområdet, men egner sig ikke til flygtige stoffer
|
— |
en, der kan anvendes til stoffer, som i det væsentlige er rene, med lav opløselighed (< 10-2 g/l), og som er stabile i vand, betegnet »kolonne-eluerings-metoden« |
|
— |
en der kan anvendes til stoffer, som i det væsentlige er rene, med større opløselighed (> 10-2 g/l), og som er stabile i vand, betegnet »kolbe-metoden«. |
De undersøgte stoffers vandopløselighed kan i betragtelig grad afhænge af tilstedeværelsen af urenheder.
1.2. DEFINITION OG ENHEDER
Et stofs opløselighed i vand angives ved mætningskoncentrationen af stoffet i vand ved en given temperatur. Opløseligheden i vand angives i enheden masse pr. rumfang opløsning. SI-enheden er kg/m3 (g/l kan også anvendes).
1.3. REFERENCESTOFFER
Anvendelse af referencestoffer er ikke påkrævet, hver gang et nyt stof skal undersøges. Referencestoffer skal først og fremmest tjene til lejlighedsvis kontrol af metoden og til sammenligning med resultater opnået med andre metoder.
1.4. PRØVNINGSMETODENS PRINCIP
Ved en simpel foreløbig test undersøges, omtrentligt hvor meget materiale og hvor lang tid der kræves til at nå mætningskoncentrationen.
1.4.1. Kolonne-eluerings-metoden
Denne metode er baseret på eluering af det undersøgte stof med vand fra en mikrokolonne, der indeholder et inert bæremateriale såsom glaskugler eller sand, belagt med overskud af teststof. Vandopløseligheden bestemmes, når koncentrationen i eluatet bliver konstant. Dette fremkommer som et koncentrationsplateau som en funktion af tiden.
1.4.2. Kolbe-metoden
Ved denne metode opløses stoffet (faste stoffer pulveriseres) i vand ved en temperatur noget over testningstemperaturen. Når mætning er opnået, afkøles blandingen til testningstemperatur og holdes der under omrøring så længe, som det er nødvendigt for at opnå ligevægt. Alternativt kan målingen foretages direkte ved testningstemperaturen, hvis det ved passende prøvetagning godtgøres, at mætningsligevægten er nået. Derefter bestemmes massekoncentrationen af stoffet i den vandige opløsning, der ikke må indeholde uopløste partikler, ved en passende analysemetode.
1.5. KVALITETSKRITERIER
1.5.1. Repeterbarhed
For kolonne-eluerings-metoden er det muligt at opnå < 30 %; for kolbe-metoden bør den være < 15 %.
1.5.2. Følsomhed
Denne afhænger af analysemetoden, men det er muligt at bestemme så lav en massekoncentration som 10-6 g/l.
1.6. BESKRIVELSE AF METODEN
1.6.1. Testbetingelser
Testen udføres helst ved 20 oC ±0,5 oC. Hvis der er mistanke om, at opløseligheden er temperaturafhængig (> 3 % pr. oC) udføres der også undersøgelser ved to andre temperaturer, der er mindst 10 oC over og under den først valgte temperatur. I så tilfælde må temperaturkontrollen være ±0,1 oC. Den valgte temperature bør holdes konstant i alle relevante dele af udstyret.
1.6.2. Indledende test
Til ca. 0,1 g af prøven (faste stoffer må være pulveriserede) i et 10 ml måleglas med glasprop sættes stigende mængder destilleret vand ved stuetemperatur, efter de i tabellen nedenfor angivne trin.
|
0,1 g stof opløselig i »x« ml vand |
0,1 |
0,5 |
1 |
2 |
10 |
100 |
> 100 |
|
omtrentlig opløselighed (g/l) |
> 1 000 |
1 000-200 |
200-100 |
100-50 |
50-20 |
10-1 |
<1 |
Efter hver tilsætning af den angivne mængde vand til blandingen omrystes der kraftigt i 10 minutter, og det undersøges visuelt, om der er noget uopløst af prøven tilbage. Hvis prøven eller dele af prøven forbliver uopløst, efter at der er tilsat 10 ml, gentages forsøget i et 100 ml måleglas med en større vandmængde. Ved lavere opløseligheder kan den tid, der kræves til opløsning af et givet stof, være betragteligt længere (der bør ventes mindst 24 timer). Den anslåede opløselighed er angivet i tabellen under det rumfang af tilsat vand, der netop forårsagede en fuldstændig opløsning af prøven. Hvis stoffet stadig er tilsyneladende uopløst, øges henstandstiden ud over de 24 timer (højst 96 timer), eller der foretages yderligere fortynding, så det kan afgøres, om det er kolonne-eluerings- eller kolbe-metoden, der skal anvendes.
1.6.3. Kolonne-eluerings-metoden
1.6.3.1. Bæremateriale, opløsningsmiddel og elueringsmiddel
Bærematerialet til kolonne-eluerings-metoden skal være inert. Mulige materialer, der kan anvendes, er glaskugler og sand. Der anvendes et passende flygtigt opløsningsmiddel af analysekvalitet til at overføre teststoffet til bærematerialet. Som opløsningsmiddel anvendes der vand, der er dobbelt destilleret i glas- eller kvartsapparatur.
Bemærk:
Vand direkte fra en organisk ionbytter må ikke anvendes.
1.6.3.2. Afsætning på bærematerialet
Ca. 600 mg bæremateriale afvejes og overføres til en 50 ml rundbundet kolbe.
En passende, afvejet mængde af prøven opløses i det valgte opløsningsmiddel. En passende mængde af denne opløsning sættes til bærematerialet. Opløsningsmidlet skal afdampes fuldstændigt, f.eks. i en rotationsinddamper; i modsat fald vil der ikke kunne opnås mætning af bærematerialet med vand som følge af fordelingseffekter på overfladen af bærematerialet.
Afsætningen på bærematerialet kan give problemer (fejlagtige resultater), hvis det undersøgte stof afsættes som en olie eller i en anden krystalstruktur. Dette problem må undersøges eksperimentelt, og resultaterne rapporteres.
Det behandlede bæremateriale henstilles til kvældning i ca. to timer i ca. 5 ml vand, hvorefter suspensionen overføres til mikrokolonnen. Alternativt kan det tørre behandlede bæremateriale hældes i mikrokolonnen, der er fyldt med vand, og derefter henstå i ca. to timer til opnåelse af ligevægt.
Testprocedure:
Elueringen af stoffet fra bærematerialet kan udføres på en af de to forskellige måder:
|
— |
med recirkulationspumpe (se figur 1) |
|
— |
med niveaubeholder (se figur 4). |
1.6.3.3. Kolonne-eluerings-metoden med recirkulationspumpe
Apparatur
Et typisk system er vist skematisk i figur 1. En egnet mikrokolonne er vist i figur 2, om end en hvilken som helst størrelse kan anvendes, forudsat at den opfylder kriterierne for reproducerbarhed og følsomhed. Kolonnen skal have et head-space på mindst fem bed-volumen vand og kunne rumme mindst fem prøver. Alternativt kan størrelsen reduceres, hvis der tilsættes opløsningsmiddel som erstatning for de første fem bed-rumfang, der fjernes med urenheder.
Kolonnen skal forbindes til en recirkulationspumpe, der kan reguleres til en strømningshastighed på ca. 25 ml/h. Pumpen forbindes med tilslutning af polytetrafluorethylen (PTFE) og/eller glas. Kolonnen og pumpen skal, når de er forbundet, give mulighed for udtagning af prøver af den eluerede væske og for indstilling af head-space-ligevægt ved atmosfæretryk. Kolonnematerialet holdes oppe med en lille (5 mm) prop af glasuld, der også fungerer som filter for partikler. Recirkulationspumpen kan f.eks. være en slangepumpe (man må være omhyggelig med ikke at få kontaminering af og/eller sorption på slangematerialet) eller en membranpumpe.
Fremgangsmåde ved målingen
Væskestrømmen gennem kolonnen startes. Det anbefales at bruge en strømningshastighed på ca. 25 ml/h (hvilket svarer til ca. ti bed-rumfang pr. time for den beskrevne kolonne). De første fem bed-rumfang (minimum) bortkastes, så vandopløselige urenheder fjernes. Derefter kører recirkulationspumpen, til der er opnået ligevægt, defineret ved at fem på hinanden følgende prøver giver koncentrationer, der ikke afviger mere end ± 30 % på tilfældig måde. Disse prøver bør være adskilt fra hinanden ved tidsintervaller, der svarer til et gennemløb af mindst ti bed-rumfang af opløsningsmiddel.
1.6.3.4. Kolonne-eluerings-metoden med niveaubeholder
Apparatur
Niveaubeholder: Forbindelsen til niveaubeholderen etableres ved at anvende et glasslibovergangsstykke, der tilsluttes med en PTFE-slange. Det anbefales, at der anvendes en gennemstrømningshastighed på ca. 25 ml/time. Successive fraktioner af eluatet opsamles og analyseres efter den valgte metode.
Fremgangsmåde ved målingen
De fraktioner fra det midterste elueringsområde, hvor koncentrationerne er konstante (± 30 %) i mindst fem på hinanden følgende prøver, anvendes til at bestemme opløseligheden i vand.
I begge tilfælde (anvendelse af recirkulationspumpe eller niveaubeholder) udføres endnu et forsøg med halvt så høj gennemstrømningshastighed som det første. Hvis resultaterne fra de to forsøg stemmer overens, er testen tilfredsstillende; hvis der er en større opløselighed ved den lavere hastighed, må halveringen af gennemstrømningshastigheden fortsættes, indtil to på hinanden følgende forsøg giver samme opløselighed.
I begge tilfælde (anvendelse af recirkulationspumpe eller niveaubeholder) kontrolleres fraktionerne for tilstedeværelse af kolloidt materiale ved hjælp af Tyndall-effekten (lysspredning). Tilstedeværelse af sådanne partikler gør resultaterne ubrugelige, og testen må gentages efter forbedring af kolonnens filtreringsformåen.
pH i alle prøver registreres. Der udføres endnu et forsøg ved samme temperatur.
1.6.4. Kolbe-metoden
1.6.4.1. Apparatur
Til kolbe-metoden kræves følgende udstyr:
|
— |
normalt laboratorieglasudstyr og -instrumentering |
|
— |
udstyr til rystning/omrøring af opløsninger ved kontrolleret konstant temperatur |
|
— |
eventuelt en centrifuge (helst termostateret) til emulsioner og |
|
— |
udstyr til analytisk bestemmelse. |
1.6.4.2. Fremgangsmåde ved målingen
Den mængde materiale, der er nødvendig for at mætte det ønskede rumfang vand, anslås ud fra den foreløbige test. Det fornødne rumfang vand vil afhænge af den analytiske metode og opløselighedsområdet. Ca. fem gange den mængde stof, der er bestemt ovenfor, afvejes i hver af tre beholdere med glasprop (f.eks. centrifugeglas eller kolber). Det valgte rumfang vand sættes til hver beholder, og beholderne lukkes helt tæt. De lukkede beholdere rystes/omrøres derefter ved 30 oC. (Der anvendes ryste- eller omrørerudstyr, som kan operere ved konstant temperatur, f.eks. magnetomrøring i et termostatbad). Efter en dag fjernes én af beholderne, og den henstilles til opnåelse af ligevægt i 24 timer ved den ønskede temperatur og lejlighedsvis omrystning. Indholdet i beholderen centrifugeres derefter ved testtemperaturen, og koncentrationen af stoffet i den klare vandige fase bestemmes med en passende analysemetode. De andre to kolber behandles på samme måde efter forudgående opnåelse af ligevægt ved 30 oC i henholdsvis to og tre dage. Hvis koncentrationsresultaterne fra mindst to af beholderne opfylder den krævede reproducerbarhed, er testen tilfredsstillende. Hvis resultaterne fra beholder 1, 2 og 3 viser en stigende tendens, gentages hele forsøget under anvendelse af længere tider til opnåelse af ligevægt.
Måleproceduren kan også udføres uden forudgående henstand ved 30 oC. Der opnås et skøn over, hvor hurtigt mætningsligevægten indstiller sig, ved udtagning af prøver, indtil omrøringstiden ikke længere indvirker på prøveopløsningens koncentration.
pH i alle prøver registreres.
1.6.5. Analyse
Der foretrækkes en stofspecifik analysemetode til disse bestemmelser, eftersom små mængder af opløselige urenheder kan forårsage store fejl i den målte opløselighed. Eksempler på sådanne metoder er gas- eller væskechromatografi, titreringsmetoder, fotometriske metoder og voltametriske metoder.
2. DATA
2.1. KOLONNE-ELUERINGS-METODEN
Middelværdien samt standardafvigelsen for mindst fem på hinanden følgende prøver taget ved mætningsplateauet udregnes for hvert forsøg. Resultaterne anføres i masseenhed pr. volumenenhed opløsning.
Gennemsnittene fra to forsøg med forskellige strømningshastigheder sammenlignes, og repeterbarheden skal være bedre end 30 %
2.2. KOLBE-METODEN
De enkelte resultater skal angives for hver af de tre kolber, og for de resultater, der skønnes at være konstante (repeterbarhed bedre end 15 %), beregnes gennemsnittet, og det angives i masseenhed pr. volumenenhed opløsning. Dette kan kræve omregning af masseenhed til volumenenhed ved hjælp af massefylden, hvis opløseligheden er meget høj (> 100 g/l).
3. RAPPORTERING
3.1. KOLONNE-ELUERINGS-METODEN
Forsøgsrapporten skal om muligt indeholde følgende oplysninger:
|
— |
resultaterne af den foreløbige test |
|
— |
nøjagtig specifikation af stoffet (identitet og urenheder) |
|
— |
koncentration, gennemstrømningshastighed og pH for hver enkelt prøve |
|
— |
middelværdi og standardafvigelse for mindst fem prøver fra mætningsplateauet fra hvert forsøg |
|
— |
gennemsnittet af to successive, acceptable forsøg |
|
— |
vandtemperaturen under mætningsprocessen |
|
— |
den anvendte analysemetode |
|
— |
arten af det anvendte bæremateriale |
|
— |
afsætning på bærematerialet |
|
— |
anvendt opløsningsmiddel |
|
— |
tegn på eventuel kemisk ustabilitet af stoffet under testen og den anvendte metode |
|
— |
alle oplysninger af betydning for vurdering af resultaterne, især vedrørende urenheder og stoffets fysiske tilstandsform. |
3.2. KOLBEMETODEN
Testrapporten skal om muligt indeholde følgende oplysninger:
|
— |
resultaterne af den foreløbige test |
|
— |
nøjagtig specifikation af stoffet (identitet og urenheder) |
|
— |
de enkelte analyseresultater og gennemsnittet, når der er bestemt mere end én værdi for hver kolbe |
|
— |
pH i hver prøve |
|
— |
gennemsnitsværdien for de forskellige kolber, der stemte overens |
|
— |
analysetemperaturen |
|
— |
den anvendte analysemetode |
|
— |
tegn på eventuel kemisk ustabilitet af stoffet under testen og den anvendte metode |
|
— |
alle oplysninger af betydning for vurdering af resultaterne, især vedrørende urenheder og stoffets fysiske tilstandsform. |
4. HENVISNINGER
|
(1) |
OECD, Paris, 1981, Test Guideline 105, Decision of the Council C(81) 30 final. |
|
(2) |
NF T 20-045 (AFNOR) (Sept. 85). Chemical products for industrial use — Determination of water solubility of solids and liquids with low solubility — Column elution method. |
|
(3) |
NF T 20-046 (AFNOR) (Sept. 85). Chemical products for industrial use — Determination of water solubility of solids and liquids with high solubility — Flask method. |
Tillæg
Figur 1
Kolonne-eluerings metoden med recirkulationspumpe

Figur 2
En typisk mikrokolonne
(alle dimensioner er i mm)
Figur 3
En typisk mikrokolonne
(alle dimensioner er i mm)
Figur 4
Kolonne-eluerings- metoden med niveau beholder
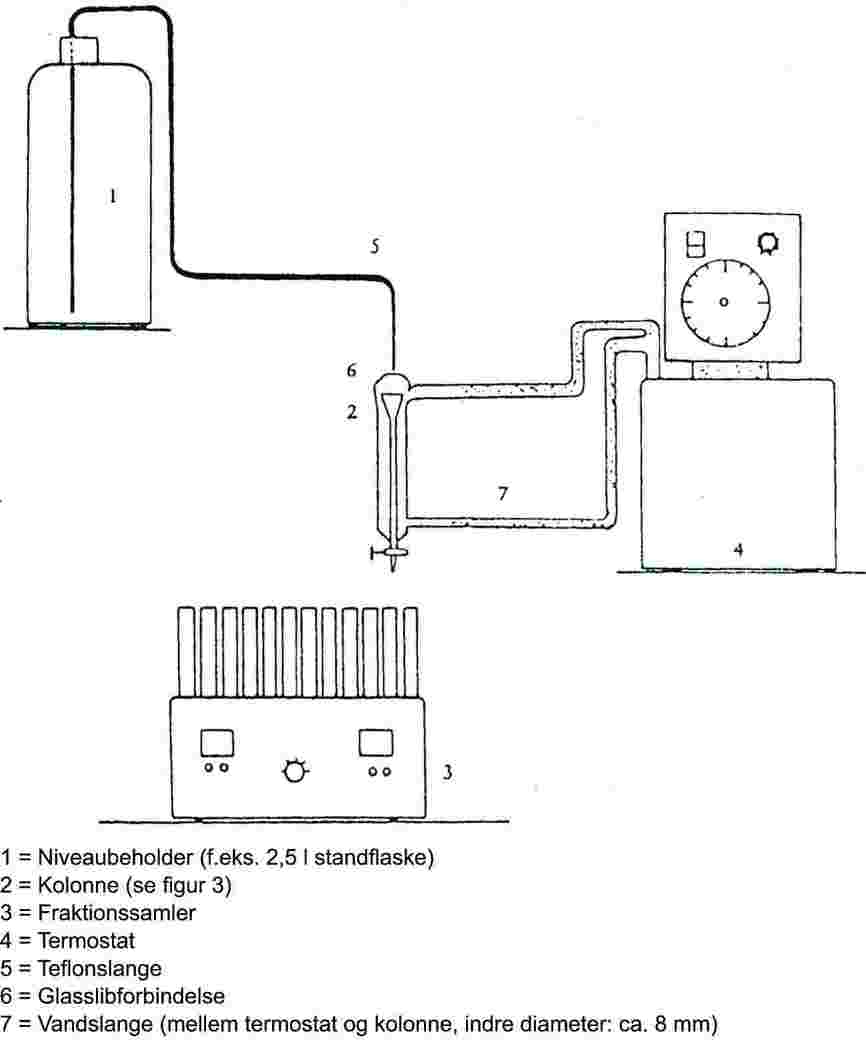
A.8. FORDELINGSKOEFFICIENT
1. METODE
Den beskrevne »rystekolbemetode« er baseret på OECD-testvejledningen (1).
1.1. INDLEDNING
Ved udførelse af denne test er det nyttigt at have forhåndsoplysninger om stoffets strukturformel, dissociationskonstant, vandopløselighed, hydrolyse, opløselighed i n-octanol og overfladespænding.
Målinger på ioniserbare stoffer udføres på den ikke-ioniserede form (fri syre eller fri base) dvs. ved anvendelse af en passende buffer med en pH-værdi på mindst 1 enhed under (fri syre) eller over (fri base) stoffets pK-værdi.
Testmetoden omfatter to særskilte fremgangsmåder — rystekolbemetoden og højtydende væskekromatografi (HPLC). Førstnævnte er brugbar, når værdien af log Pow (se definitionerne nedenfor) ligger mellem — 2 og 4, og sidstnævnte i området mellem 0 og 6. Førend nogen af fremgangsmåderne påbegyndes, foretages der er foreløbigt skøn over fordelingskoefficienten.
Rystekolbemetoden kan kun benyttes for stoffer, der i alt væsentligt er rene, og som er opløselige i vand og n-octanol. Den kan ikke anvendes til overfladeaktive materialer (for hvilke der anføres en beregnet værdi eller et skøn baseret på opløseligheden i henholdsvis n-octanol og vand).
HPLC-metoden kan ikke benyttes til stærke syrer og baser, metalkomplekser, overfladeaktive stoffer og stoffer, der reagerer med elueringsmidlet. For sådanne materialer anføres der en beregnet værdi eller et skøn baseret på opløseligheden i henholdsvis n-octanol og vand.
HPLC-metoden er mindre følsom end rystekolbemetoden over for urenheder i teststoffet. Ikke desto mindre kan urenheder i visse tilfælde vanskeliggøre fortolkningen af resultaterne på grund af usikker tilforordning af toppene. For blandinger med et uopløst bånd anføres en øvre og nedre grænse for log P.
1.2. DEFINITIONER OG ENHEDER
Fordelingkoefficienten (P) defineres som forholdet mellem ligevægtskoncentrationerne (ci) af et stof, der er opløst i et tofasesystem bestående af to stort set ublandbare opløsningsmidler. I tilfældet n-octanol og vand:

Fordelingskoefficienten (P) er derfor et forhold mellem to koncentrationer og opgives ofte som titalslogaritmen (log P).
1.3. REFERENCESTOFFER
Rystekolbemetoden
Anvendelse af referencestoffer er ikke påkrævet, hver gang et nyt stof skal undersøges. Referencestoffer skal først og fremmest tjene til lejlighedsvis kontrol af metoden og til sammenligning med resultater opnået med andre metoder.
HPLC-metoden
For at kunne korrelere målte HPLC-data for et stof med dets P-værdi må der fastlægges en kalibreringskurve for log P mod kromatografiske data, som er baseret på mindst seks referencepunkter. Det er op til brugeren at vælge de egnede referencestoffer. Så vidt muligt skal der være mindst ét referencestof med en Pow-værdi, der ligger over teststoffets, og mindst ét med en Pow-værdi under teststoffets. For værdier af log P under 4 kan kalibreringen baseres på data opnået med rystekolbemetoden. For værdier af log P over 4 kan kalibreringen baseres på validerede litteraturværdier, hvis de stemmer overens med beregnede værdier. Af hensyn til nøjagtigheden anbefales at vælge referencestoffer, der strukturmæssigt ligner teststoffet.
Der foreligger omfattende lister med værdier for log Pow for mange grupper af kemikalier (2)(3). Hvis der ikke foreligger data om fordelingskoefficienter for strukturmæssigt beslægtede stoffer, kan der bruges en mere generel kalibrering foretaget med andre referencestoffer.
Der er i tillæg 2 en liste over anbefalede referencestoffer og deres Pow-værdier
1.4. METODENS PRINCIP
1.4.1. Rystekolbemetoden
For at kunne bestemme fordelingskoefficienten må der opnås ligevægt mellem alle komponenterne i systemet, og koncentrationerne af de i de to faser opløste stoffer må bestemmes. En gennemgang af litteraturen herom har vist, at der kan benyttes en række forskellige teknikker til at løse dette problem, dvs. omhyggelig blanding af de to faser og bestemmelse af ligevægtskoncentrationen af det undersøgte stof efter adskillelse af faserne.
1.4.2. HPLC-metoden
HPLC udføres på analytiske kolonner pakket med en kommercielt tilgængelig fast fase indeholdende lange kulbrintekæder (f.eks. C8, C18) kemisk bundet til silica. Indsprøjtes der kemikalier på en sådan kolonne, vil de vandre gennem den med forskellig hastighed, da deres fordeling mellem den mobile fase og den stationære kulbrintefase ikke er den samme. Kemikalieblandinger elueres i rækkefølge efter hydrofobicitet, idet de vandopløselige kemikalier elueres først og de olieopløselige sidst, alt efter deres kulbrinte/vand-fordelingskoefficient. Derved kan sammenhængen mellem retentionstiden på en sådan kolonne (med omvendt fase) og n-octanol/vand-fordelingskøfficienten fastslås. Fordelingskoefficienten udledes af kapacitetsfaktoren k, der er givet ved udtrykket:

hvor tr er teststoffets retentionstid og to er den tid, et opløsningsmiddelmolekyle i gennemsnit bruger på at komme gennem kolonnen (kolonnens dødtid).
Der kræves ikke kvantitative analysemetoder, og det er kun nødvendigt at bestemme retentionstiderne.
1.5. KVALITETSKRITERIER
1.5.1. Repeterbarhed
Rystekolbemetoden
For at opnå en tilstrækkeligt nøjagtig fordelingskoefficient foretages der dobbeltbestemmelse under tre forskellige testbetingelser, hvor både stofmængden og forholdet mellem opløsningsmidlerne kan varieres. De fundne værdier af fordelingskoefficienten skal, når de udtrykkes som titalslogaritmer, ligge inden for et interval på ±0,3.
HPLC-metoden
For at øge tilliden til målingen foretages der dobbeltbestemmelse. Værdier af log P beregnet ud fra de enkelte målinger skal ligge inden for et interval på ±0,1.
1.5.2. Følsomhed
Rystekolbemetoden
Metodens måleområde afgrænses af analysemetodens påvisningsgrænse. Den skal tillade bestemmelse af værdier for log Pow i området fra 2 til 4 (under visse omstændigheder op til 5), når koncentrationen af opløst stof ikke er større end 0,01 mol pr. liter i nogen af faserne.
HPLC-metoden
Med HPLC-metoden kan der skønnes fordelingskoefficienter i log Pow-området fra 0 til 6.
Normalt kan en forbindelses fordelingskoefficient skønnes med en afvigelse på mindre end ± 1 fra den værdi, der opnås med rystekolbemetoden. Typiske korrelationer fremgår af henvisning (4)(5)(6)(7)(8). Der kan normalt opnås større nøjagtighed, hvis der anvendes korrelationskurver bestemt med strukturmæssigt beslægtede referencestoffer (9).
1.5.3. Specificitet
Rystekolbemetoden
Nernsts fordelingslov gælder kun for fortyndede opløsninger ved konstant temperatur, tryk og pH. Den gælder kun for et enkelt stof, der er fordelt mellem to rene opløsningsmidler. Hvis der forekommer flere opløste stoffer i den ene eller i begge faser samtidig, kan dette påvirke resultaterne.
Dissociering eller associering af de opløste molekyler resulterer i afvigelser fra Nernsts fordelingslov. At fordelingskoefficienten afhænger af opløsningens koncentration er tegn på en sådan afvigelse.
På grund af de mange involverede ligevægte, må metoden ikke anvendes på ioniserbare stoffer uden korrektion. For sådanne stoffer kan det overvejes at benytte bufferopløsninger i stedet for vand; i så fald skal bufferens pH ligge mindst 1 enhed fra stoffets pKa, og dette pH's relevans for miljøet skal overvejes
1.6. BESKRIVELSE AF METODEN
1.6.1. Indledende skøn over fordelingskoefficienten
Der kan dannes et skøn over fordelingskoefficienten enten ved beregning (se tillæg 1), hvilket foretrækkes, eller i givet fald ud fra teststoffets opløselighed i de rene opløsningsmidler (10).
1.6.2. Rystekolbemetoden
1.6.2.1. Forberedelser
n-octanol: bestemmelsen af fordelingskoefficienten udføres med reagenser af analysekvalitet.
vand: der anvendes vand, som er destilleret eller dobbeltdestilleret i glas- eller kvartsapparatur. Til ioniserbare forbindelser benyttes der bufferopløsninger i stedet for vand, hvis der er gode grunde hertil.
Bemærk:
Der må ikke bruges vand taget direkte fra en ionbytter.
1.6.2.1.1.
Før en fordelingskoefficient bestemmes, mættes hver af de faser, der indgår i opløsningsmiddelsystemet, med den anden fase ved rystning ved forsøgstemperaturen. Hertil er det praktisk at ryste to store standflasker med henholdsvis n-octanol af analysekvalitet og vand med tilstrækkelig meget af det andet opløsningsmiddel på et mekanisk rysteapparat i 24 timer, og derefter lade dem henstå så længe, at faserne skiller og der er opnået mætning.
1.6.2.1.2.
Testbeholderen skal være næsten helt fyldt af tofasesystemet. Derved hindres materialetab ved fordampning. Stofmængder og volumenforhold fastlægges på grundlag af:
|
— |
det foreløbige skøn over fordelingskoefficienten (se ovenfor) |
|
— |
den mængde teststof, der mindst kræves til analysen |
|
— |
en øvre koncentrationsgrænse på 0,01 mol/l for begge faser. |
Der udføres tre test. I den første bruges det beregnede volumenforhold mellem n-octanol og vand; i den anden divideres dette forhold med 2, og i det tredje multipliceres det med 2 (f.eks. 1:1, 1:2 og 2:1).
1.6.2.1.3.
Der fremstilles en stamopløsning i n-octanol, der på forhånd er mættet med vand. Denne stamopløsnings koncentration bestemmes nøjagtigt, inden den anvendes til bestemmelse af fordelingskoefficienten. Den opbevares under betingelser, hvor den er stabil.
1.6.2.2. Testbetingelser
Testtemperaturen skal holdes konstant (± 1 oC) og ligge mellem 20 og 25 oC.
1.6.2.3. Fremgangsmåde ved målingen
1.6.2.3.1.
For hver af testbetingelserne benyttes der to testbeholdere, der hver indeholder den nødvendige nøjagtigt afmålte mængde af de to opløsningsmidler og den nødvendige mængde stamopløsning.
n-octanolfaserne afmåles volumetrisk. Testbeholderne kan enten anbringes i et egnet rysteapparat eller rystes i hånden. Når et centrifugeglas benyttes, anbefales en hurtig 180o drejning om tværaksen af glasset, hvorved den indespærrede luft stiger op gennem de to faser. Erfaringen viser, at 50 sådanne drejninger normalt er tilstrækkeligt til, at fordelingsligevægten indtræder. For en sikkerheds skyld anbefales det at foretage 100 drejninger i løbet af 5 minutter.
1.6.2.3.2.
Blandingen kan om nødvendigt centrifugeres, så faserne adskilles. Det gøres i en laboratoriecentrifuge ved stuetemperatur; hvis der benyttes en centrifuge uden temperaturregulering, henstår centrifugeglassene i én time ved testtemperaturen til opnåelse af ligevægt før analyse.
1.6.2.4. Analyse
Til bestemmelse af fordelingskoefficienten må koncentrationen af teststoffet i begge faser bestemmes. Det kan gøres ved, at der for hver testbetingelse udtages en delprøve af hver fase fra hvert glas, som analyseres ved den valgte metode. Den samlede mængde stof i begge faser beregnes og sammenlignes med den oprindeligt tilsatte stofmængde.
Prøven af vandfasen udtages på en sådan måde, at risikoen for, at den indeholder spor af n-octanol, bliver minimal, nemlig med en glasinjektionssprøjte. Fra starten skal sprøjten være delvis fyldt med luft, som langsomt trykkes ud, medens kanylen føres ned gennem n-octanollaget. En passende mængde af vandfasen suges op i sprøjten, som hurtigt trækkes op af opløsningen, og kanylen fjernes. Sprøjtens indhold kan nu benyttes som den vandige prøve. Koncentrationen i de to adskilte faser skal helst bestemmes ved en stofspecifik metode. Eksempelvis er følgende analysemetoder egnede:
|
— |
fotometriske metoder |
|
— |
gaskromatografi |
|
— |
højtydende væskekromatografi (HPLC). |
1.6.3. HPLC-metoden
1.6.3.1. Forberedelser
Apparatur
Der kræves en væskekromatograf med pulsationsfri pumpe og passende detektionssystem. Det anbefales at bruge injektionsventil med injektionsspiral. Polære grupper i den stationære fase kan nedsætte HPLC-kolonnens ydeevne væsentligt. Derfor må stationære faser have det lavest mulige procentindhold af polære grupper (11). Der kan anvendes kommercielle mikroniserede pakkematerialer med omvendt fase eller færdigpakkede kolonner. Der kan eventuelt anbringes en beskyttelseskolonne mellem injektionssystemet og analysekolonnen.
Mobil fase
Der anvendes methanol og vand af HPLC-kvalitet til fremstilling af elueringsvæsken, som afgasses før brugen. Der benyttes isokratisk eluering. Den anvendte methanol/vand-blanding skal indeholde mindst 25 % vand. Til eluering af forbindelser med en log P på 6 på én time ved en flowhastighed på 1 ml/min vil en blanding af methanol og vand i forholdet 3:1 (v/v) typisk være tilfredsstillende. For forbindelser med en høj log P-værdi kan det være nødvendigt at nedsætte elueringstiden (også for referencestofferne) ved at benytte en mindre polær mobil fase eller en kortere kolonne.
Stoffer med meget lav opløselighed i n-octanol har tendens til at give unormalt lave værdier for log Pow med HPLC-metoden; undertiden følger sådanne forbindelsers toppe opløsningsmiddelfronten. Dette skyldes sandsynligvis, at fordelingsprocessen er for langsom til, at der opnås ligevægt inden for den tid, en HPLC-adskillelse normalt tager. I så fald kan nedsættelse af flowhastigheden og/eller mindskelse af methanol/vand-forholdet være et virkningsfuldt middel til at opnå en pålidelig værdi.
Både teststof og referencestoffer skal være opløselige i den mobile fase i en så høj koncentration, at de kan påvises. Der må kun undtagelsesvis benyttes tilsætningsstoffer til methanol/vand-blandingen, da tilsætningsstoffer vil ændre kolonnens egenskaber. Til kromatogrammer med tilsætningsstoffer skal der altid anvendes en separat kolonne af samme type. Hvis methanol/vand ikke er egnet, kan der anvendes andre blandinger af vand og organiske opløsningsmidler, f.eks. ethanol/vand eller acetonitril/vand.
For ioniserbare stoffer er elueringsvæskens pH kritisk. Det skal ligge inden for kolonnens driftsområde, som normalt er fra 2 til 8. Det anbefales at anvende buffer. Man må være omhyggelig med at undgå saltudfældning og ødelæggelse af kolonnen, hvilket kan forekomme med visse blandinger af organisk fase og buffer. Det tilrådes ikke at foretage HPLC-målinger med silicabaserede stationære faser med pH over 8, da alkalisk mobil fase kan føre til, at kolonnens ydeevne hurtigt falder.
Opløste stoffer
Referencestoffer skal være af renest mulige kvalitet. Til testnings- og kalibreringsformål opløses stofferne om muligt i den mobile fase.
Testningsbetingelser
Under målingen må temperaturen ikke variere med mere end ± 2 K.
1.6.3.2. Måling
Beregning af dødtiden to
Dødtiden to kan bestemmes enten med en homolog række (f.eks. n-alkylmethylketoner) eller med organiske stoffer, der ikke holdes tilbage (f.eks. thiourinstof eller formamid). Til beregning af dødtiden to ud fra en homolog række, indsprøjtes der mindst syv elementer af en homolog række, og retentionstiderne bestemmes. Bruttoretentionstiderne tr (n c + 1) afbildes mod tr(n c), og i regressionsligningen:
tr (n c + 1) = a + b tr (n c)
bestemmes afskæringen a og hældningskoefficienten b (nc = antallet af kulstofatomer). Dødtiden er da givet ved:
to = a/(l -b)
Kalibreringskurve
Næste trin består i at konstruere en korrelationskurve af log k mod log P for passende referencestoffer. I praksis indsprøjtes der samtidig mellem fem og ti standardreferencestoffer, hvis log P ligger i det forventede område, og retentionstiderne bestemmes, helst med en integrationsskriver, der er koblet til detektionssystemet. De tilsvarende logaritmer til kapacitetsfaktorerne, log k, beregnes og afsættes mod den log P, der er bestemt ved rystekolbemetoden. Kalibreringen foretages regelmæssigt, mindst én gang dagligt, så der kan tages højde for eventuelle ændringer i kolonnens ydeevne.
Bestemmelse af teststoffets kapacitetsfaktor
Teststoffet indsprøjtes som en så lille mængde mobil fase som mulig. Retentionstiden bestemmes (dobbeltbestemmelse), hvorved kapacitetsfaktoren k kan beregnes. Ud fra korrelationskurven over referencestofferne kan teststoffets fordelingskoefficient interpoleres. Ved meget lave og meget høje fordelingskoefficienter er det nødvendigt at ekstrapolere. I sådanne tilfælde må man være særlig opmærksom på regressionslinjens konfidensgrænser.
2. DATA
Rystekolbemetoden
De målte P-værdiers pålidelighed kan kontrolleres ved sammenligning af gennemsnittet af dobbeltbestemmelserne med det samlede gennemsnit.
3. RAPPORTERING
Forsøgsrapporten skal om muligt indeholde følgende oplysninger:
|
— |
nøjagtig beskrivelse af stoffet (betegnelse og urenheder) |
|
— |
en beregnet værdi eller et skøn baseret på de enkelte opløseligheder, hvis metoderne ikke kan anvendes (f.eks. overfladeaktivt materiale) |
|
— |
alle oplysninger og bemærkninger af betydning for vurdering af resultaterne især vedrørende urenheder og stoffets fysiske tilstand. |
For rystekolbemetoden:
|
— |
resultatet af et eventuelt indledende skøn |
|
— |
den temperatur, som bestemmelsen er foretaget ved |
|
— |
data om de analysemetoder, der er benyttet til koncentrationsbestemmelser |
|
— |
eventuel centrifugeringstid og -hastighed |
|
— |
målte koncentrationer i begge faser for hver bestemmelse (der skal således rapporteres i alt 12 koncentrationer) |
|
— |
massen af teststof, det anvendte volumen af hver fase i hver testbeholder og den samlede beregnede mængde teststof i hver fase efter indstilling af ligevægt |
|
— |
den beregnede værdi for fordelingskoefficienten (P) og gennemsnittet for hvert sæt testbetingelser samt gennemsnittet af samtlige bestemmelser. Eventuelle tegn på, at fordelingskoefficienten er koncentrationsafhængig anføres i rapporten |
|
— |
de enkelte P-værdiers standardafvigelse fra gennemsnittet |
|
— |
gennemsnitsværdien af P fra alle bestemmelser anføres tillige som titalslogaritmen |
|
— |
den beregnede teoretiske værdi af Pow, når denne værdi er bestemt eller når den målte værdi er > 104 |
|
— |
pH af det anvendte vand og af vandfasen under forsøget |
|
— |
begrundelse for anvendelse af buffere i stedet for vand, deres sammensætning, koncentration og pH, samt vandfasens pH før og efter forsøget. |
For HPLC-metoden:
|
— |
resultatet af et eventuelt foreløbigt skøn |
|
— |
test- og referencestoffer og deres renhedsgrad |
|
— |
det temperaturinterval, som bestemmelserne er foretaget ved |
|
— |
det pH, som bestemmelserne er foretaget ved |
|
— |
detaljerede oplysninger om analyse- og beskyttelseskolonne, mobil fase og detektionssystem |
|
— |
retentionsdata og litteraturværdier for log P for referencestoffer benyttet ved kalibrering |
|
— |
detaljerede oplysninger om den fittede regressionslinje (log k mod log P) |
|
— |
gennemsnitlige retentionsdata og interpoleret log P-værdi for teststoffet |
|
— |
beskrivelse af udstyr og driftsbetingelser |
|
— |
elueringsprofiler |
|
— |
den mængde test- og referencestof, der er indført i kolonnen |
|
— |
dødtiden og hvordan den er målt. |
4. HENVISNINGER
|
(1) |
OECD, Paris, 1981, Test Guideline 107, Decision of the Council C(81) 30 final. |
|
(2) |
C. Hansch and A.J. Leo, Substituent Constants for Correlation Analysis in Chemistry and Biology, John Wiley, New York 1979. |
|
(3) |
Log P and Parameter Database, A tool for the quantitative prediction of bioactivity (C. Hansch, chairman, A.J. Leo, dir.) — Available from Pomona College Medical Chemistry Project 1982, Pomona College, Claremont, California 91711. |
|
(4) |
L. Renberg, G. Sundström and K. Sundh-Nygärd, Chemosphere, 1980, vol. 80, 683. |
|
(5) |
H. Ellgehausen, C. D'Hondt and R. Fuerer, Pestic. Sci., 1981, vol. 12, 219. |
|
(6) |
B. McDuffie, Chemosphere, 1981, vol. 10, 73. |
|
(7) |
W.E. Hammers et al., J. Chromatogr., 1982, vol. 247, 1. |
|
(8) |
J.E. Haky and A.M. Young, J. Liq. Chromat., 1984, vol. 7, 675. |
|
(9) |
S. Fujisawa and E. Masuhara, J. Biomed. Mat. Res., 1981, vol. 15, 787. |
|
(10) |
O. Jubermann, Verteilen und Extrahieren, in Methoden der Organischen Chemie (Houben Weyl), Allgemeine Laboratoriumpraxis (edited by E. Muller), Georg Thieme Verlag, Stuttgart, 1958, Band I/1, 223-339. |
|
(11) |
R.F. Rekker and H.M. de Kort, Euro. J. Med. Chem., 1979, vol. 14, 479. |
|
(12) |
A. Leo, C. Hansch and D. Elkins, Partition coefficients and their uses. Chem. Rev., 1971, vol. 71, 525. |
|
(13) |
R.F. Rekker, The Hydrophobic Fragmental Constant, Elsevier, Amsterdam, 1977. |
|
(14) |
NF T 20-043 AFNOR (1985). Chemical products for industrial use — Determination of partition coefficient — Flask shaking method. |
|
(15) |
C.V. Eadsforth and P. Moser, Chemosphere, 1983, vol. 12, 1459. |
|
(16) |
A. Leo, C. Hansch and D. Elkins, Chem. Rev., 1971, vol. 71, 525. |
|
(17) |
C. Hansch, A. Leo, S.H. Unger, K.H. Kim, D. Nikaitani and E.J. Lien, J. Med. Chem., 1973, vol. 16, 1207. |
|
(18) |
W.B. Neely, D.R. Branson and G.E. Blau, Environ. Sci. Technol., 1974, vol. 8, 1113. |
|
(19) |
D.S. Brown and E.W. Flagg, J. Environ. Qual., 1981, vol. 10, 382. |
|
(20) |
J.K. Seydel and K.J. Schaper, Chemische Struktur und biologische Aktivität von Wirkstoffen, Verlag Chemie, Weinheim, New York 1979. |
|
(21) |
R. Franke, Theoretical Drug Design Methods, Elsevier, Amsterdam 1984. |
|
(22) |
Y.C. Martin, Quantitative Drug Design, Marcel Dekker, New York, Base1, 1978. |
|
(23) |
N.S. Nirrlees, S.J. Noulton, C.T. Murphy, P.J. Taylor; J. Med. Chem., 1976, vol. 19, 615. |
Tillæg 1
Metoder til beregning/estimering
INDLEDNING
I Handbook of Chemical Property Estimation Methods (a) er der en generel indføring i beregningsmetoder samt data og eksempler.
Beregnede værdier for Pow kan benyttes:
|
— |
til at afgøre, hvilken forsøgsmetode der er mest velegnet (rystekolbemetoden: log Pow — 2 til 4; HPLC-metoden: log Pow 0 til 6) |
|
— |
til at vælge de bedste forsøgsbetingelser (f.eks. referencestoffer til HPLC-metoden og volumenforholdet mellem n-octanol og vand til rystekolbemetoden) |
|
— |
som intern laboratoriekontrol for eventuelle forsøgsfejl |
|
— |
til at give et skøn over Pow i de tilfælde, hvor man af tekniske årsager ikke kan anvende eksperimentelle metoder. |
ESTIMERINGSMETODER
Foreløbigt skøn over fordelingskoefficienten
Der kan foretages et skøn over værdien af fordelingskoefficienten ved hjælp af teststoffets opløselighed i de rene opløsningsmidler: Hertil benyttes:

BEREGNINGSMETODER
Princippet bag beregningsmetoderne
Alle beregningsmetoder er baseret på formel fragmentering af molekylet i passende delstrukturer, for hvilke man har pålideligt kendskab til log Pow-bidrag. Log Pow for hele molekylet beregnes dernæst som summen af fragmentværdierne plus summen af korrektionsudtryk for intramolekylære vekselvirkninger.
Der foreligger lister over fragmentkonstanter og korrektionsudtryk (b)(c)(d)(e). Nogle af dem ajourføres regelmæssigt (b).
Kvalitetskriterier
Sædvanligvis aftager beregningsmetodens pålidelighed med, at det undersøgte stofs kompleksitet stiger. For simple molekyler med lav molekylvægt og en eller to funktionelle grupper kan der forventes en afvigelse på 0,1 til 0,3 mellem resultatet af forskellige fragmenteringsmetoder og den målte værdi for log Pow. Fejlmargenen kan være større for mere komplekse molekyler. Den afhænger af, om der foreligger fragmentkonstanter og hvor pålidelige de er, samt af, om intramolekylære vekselvirkninger (f.eks. hydrogenbindinger) erkendes, og om korrektionsudtrykkene anvendes korrekt (mindre problematisk med computerprogrammet CLOGP-3) (b). For ioniserbare forbindelsers vedkommende har det stor betydning, at ladningen eller ioniseringsgraden bedømmes korrekt.
Fremgangsmåde ved beregningen
Hanschs π-metode
Den oprindelige konstant for hydrofobe substituenter, π, som indførtes af Fujita m.fl. (f), defineres ved
πx = log Pow (PhX) – log Pow (PhH)
hvor Pow (PhX) er fordelingskoefficienten for et aromatisk derivat og Pow(PhH) er stamforbindelsens fordelingskoefficient
(f.eks. πCl = log Pow (C6H5Cl) – log Pow (C6H6) = 2,84 – 2,13 = 0,71).
Ifølge sin definition gælder π-metoden navnlig for aromatisk substitution. Der findes tabeller med π-værdier for en lang række substituenter (b)(c)(d). De benyttes til beregning af log Pow for aromatiske molekyler og delstrukturer.
Rekkers metode
Efter Rekker (g) beregnes værdien af log Pow således:

hvor fi er de forskellige molekylfragmentkonstanter og ai er den hyppighed, hvormed de forekommer i det pågældende molekyle. Korrektionsudtrykkene kan udtrykkes som et heltalsmultiplum af én enkelt konstant Cm (den såkaldte »magiske konstant«). Fragmentkonstanterne fi og Cm er bestemt på grundlag af en liste med 1 054 eksperimentelt bestemte værdier af Pow (825 forbindelser) ved hjælp af flerdimensional regressionsanalyse (c)(h). Vekselvirkningsudtrykkene bestemmes efter faste regler, der er beskrevet i henvisning (e)(h)(i).
Hansch-Leos metode
Efter Hansch og Leo (c) beregnes værdien af log Pow således:
hvor fi er de forskellige molekylfragmentkonstanter, Fj er korrektionsudtrykkene, og ai, og bj er de hertil hørende hyppigheder. Ud fra eksperimentelt bestemte værdier af Pow har man ved at prøve sig frem kunnet opstille en liste over atom- og gruppefragmentværdier og en liste over korrektionsudtryk Fj (såkaldte »faktorer«). Korrektionsudtrykkene er ordnet i flere forskellige klasser (a)(c). Det er forholdsvis kompliceret og tidkrævende at tage hensyn til alle regler og korrektionsudtryk. Der er udviklet edb-programmer hertil (b).
Kombineret metode
Beregningen af log Pow for komplekse molekyler kan forbedres betydeligt, hvis molekylet deles op i større delstrukturer, som man har pålidelige log Pow-værdier for, enten fra tabeller (b)(c) eller fra egne målinger. Sådanne fragmenter (f.eks. heterocykliske forbindelser, anthraquinon, azobenzen) kan dernæst kombineres med Hanschs π-værdier eller Rekkers eller Leos fragmentkonstanter.
Bemærkninger
|
i) |
Beregningsmetoderne kan kun anvendes på helt eller delvis ioniserede forbindelser, hvis de nødvendige korrektionsfaktorer kan tages i betragtning. |
|
ii) |
Hvis der må antages at forekomme intramolekylære hydrogenbindinger, må det hertil svarende korrektionsudtryk (ca. +0,6 til +1,0 log Pow-enheder) lægges til (a). Rumlige modeller og spektroskopiske data kan afsløre, om der kan være tale om sådanne bindinger i molekylet |
|
iii) |
Hvis flere tautomere former er mulige, benyttes den mest sandsynlige form som grundlag for beregningen. |
|
iv) |
Man skal nøje følge med i revisioner af lister over fragmentkonstanter. |
Rapport
Ved anvendelse af beregningsmetoder skal testrapporten om muligt indeholde følgende oplysninger:
|
— |
beskrivelse af stoffet (betegnelse og urenheder) |
|
— |
angivelse af alle muligheder for intramolekylære hydrogenbindinger, dissociation, ladning eller andre særlige forhold (f.eks. tautomeri) |
|
— |
beskrivelse af beregningsmetoden |
|
— |
identifikation af de anvendte datas oprindelse |
|
— |
særlige valg af fragmenter |
|
— |
udtømmende dokumentation af beregningen. |
HENVISNINGER
|
(a) |
W.J. Lyman, W.F. Reehl and D.H. Rosenblatt (ed.), Handbook of Chemical Property Estimation Methods, McGraw-Hill, New York, 1983. |
|
(b) |
Pomona College, Medicinal Chemistry Project, Claremont, California 91711, USA, Log P Database and Med. Chem. Software (Program CLOGP-3). |
|
(c) |
C. Hansch, A.J. Leo, Substituent Constants for Correlation Analysis in Chemistry and Biology, John Wiley, New York, 1979. |
|
(d) |
A. Leo, C. Hansch, D. Elkins, Chem. Rev., 1971, vol. 71, 525. |
|
(e) |
R.F. Rekker, H.M. de Kort, Eur. J. Med. Chem. — Chill. Ther. 1979, vol. 14, 479. |
|
(f) |
T. Fujita, J. Iwasa and C. Hansch, J. Amer. Chem. Soc., 1964, vol. 86, 5175. |
|
(g) |
R.F. Rekker, The Hydrophobic Fragmental Constant, Pharmacochemistry Library, Elsevier, New York, 1977, vol.1. |
|
(h) |
C.V. Eadsforth, P. Moser, Chemosphere, 1983, vol. 12, 1459. |
|
(i) |
R.A. Scherrer, ACS, American Chemical Society, Washington D.C., 1984, Symposium Series 255, p. 225. |
Tillæg 2
Anbefalede referencestoffer til HPLC-metoden
|
nr. |
referencestof |
logow |
pKa |
|
1 |
2-butanon |
0,3 |
|
|
2 |
4-acetylpyridin |
0,5 |
|
|
3 |
anilin |
0,9 |
|
|
4 |
acetanilid |
1,0 |
|
|
5 |
benzylalkohol |
1,1 |
|
|
6 |
p-methoxyphenol |
1,3 |
pKa = 10,26 |
|
7 |
phenoxyeddikesyre |
1,4 |
pKa = 3,12 |
|
8 |
phenol |
1,5 |
pKa = 9,92 |
|
9 |
2,4-dinitrophenol |
1,5 |
pKa = 3,96 |
|
10 |
benzonitril |
1,6 |
|
|
11 |
phenylacetonitril |
1,6 |
|
|
12 |
4-methylbenzylalkohol |
1,6 |
|
|
13 |
acetophenon |
1,7 |
|
|
14 |
2-nitrophenol |
1,8 |
pKa = 7,17 |
|
15 |
3-nitrobenzoesyre |
1,8 |
pKa = 3,47 |
|
16 |
4-chloranilin |
1,8 |
pKa = 4,15 |
|
17 |
nitrobenzen |
1,9 |
|
|
18 |
kanelalkohol |
1,9 |
|
|
19 |
benzoesyre |
1,9 |
pKa = 4,19 |
|
20 |
p-cresol |
1,9 |
pKa = 10,17 |
|
21 |
kanelsyre |
2,1 |
pKa = 3,89 cis 4,44 trans |
|
22 |
anisol |
2,1 |
|
|
23 |
methylbenzoat |
2,1 |
|
|
24 |
benzen |
2,1 |
|
|
25 |
3-methylbenzoesyre |
2,4 |
pKa = 4,27 |
|
16 |
4-chlorphenol |
2,4 |
pKa = 9,1 |
|
27 |
trichlorethylen |
2,4 |
|
|
28 |
atrazin |
2,6 |
|
|
29 |
ethylbenzoat |
2,6 |
|
|
30 |
2,6-dichlorbenzonitril |
2,6 |
|
|
31 |
3-chlorbenzoesyre |
2,7 |
pKa = 3,82 |
|
32 |
toluen |
2,7 |
|
|
33 |
1-naphthol |
2,7 |
pKa = 9,34 |
|
34 |
2,3-dichloranilin |
2,8 |
|
|
35 |
chlorbenzen |
2,8 |
|
|
36 |
allylphenylether |
2,9 |
|
|
37 |
brombenzen |
3,0 |
|
|
38 |
ethylbenzen |
3,2 |
|
|
39 |
benzophenon |
3,2 |
|
|
40 |
4-phenylphenol |
3,2 |
pKa = 9,54 |
|
41 |
thymol |
3,3 |
|
|
42 |
1,4-dichlorbenzen |
3,4 |
|
|
43 |
diphenylamin |
3,4 |
pKa = 0,79 |
|
44 |
naphthalen |
3,6 |
|
|
45 |
phenylbenzoat |
3,6 |
|
|
46 |
isopropylbenzen |
3,7 |
|
|
47 |
2,4,6-trichlorphenol |
3,7 |
pKa = 6 |
|
48 |
biphenyl |
4,0 |
|
|
49 |
benzylbenzoat |
4,0 |
|
|
50 |
2,4-dinitro-6-sec-butylphenol |
4,1 |
|
|
51 |
1,2,4-trichlorbenzen |
4,2 |
|
|
52 |
dodecansyre |
4,2 |
|
|
53 |
diphenylether |
4,2 |
|
|
54 |
n-butylbenzen |
4,5 |
|
|
55 |
phenanthren |
4,5 |
|
|
56 |
fluoranthen |
4,7 |
|
|
57 |
dibenzyl |
4,8 |
|
|
58 |
2,6-diphenylpyridin |
4,9 |
|
|
59 |
triphenylamin |
5,7 |
|
|
60 |
DDT |
6,2 |
|
|
Andre referencestoffer med lav log Pow |
|||
|
1 |
nikotinsyre |
-0,07 |
|
A.9. FLAMMEPUNKT
1. METODE
1.1. INDLEDNING
Ved udførelse af denne test er det nyttigt at have forhåndsoplysninger om stoffets brændbarhed. Metoden kan anvendes for væsker, hvis dampe kan antændes af en antændelseskilde. De opregnede testmetoder er kun pålidelige i de flammepunktsintervaller, der er anført ved de enkelte metoder.
Ved valg af metode skal det tages med i overvejelserne, om der kan ske en kemisk reaktion mellem stoffet og testbeholderen.
1.2. DEFINITIONER OG ENHEDER
Flammepunktet er den laveste temperatur, korrigeret til et tryk på 101,325 kPa, ved hvilken en væske under de i metoden fastsatte betingelser udvikler dampe i en sådan mængde, at de danner en brændbar blanding med luft i testbeholderen.
Enheder: oC
t = T – 273,15
(t er i oC og T er i K)
1.3. REFERENCESTOFFER
Anvendelse af referencestoffer er ikke påkrævet, hver gang et nyt stof skal undersøges. Referencestoffer skal først og fremmest tjene til lejlighedsvis kontrol af metoden og til sammenligning med resultater opnået med andre metoder.
1.4. METODENS PRINCIP
Stoffet anbringes i en testbeholder og opvarmes eller afkøles til testtemperaturen efter den fremgangsmåde, der er beskrevet i den enkelte testmetode. Der udføres antændelsesforsøg til konstatering af, om prøven antændes ved testtemperaturen eller ikke.
1.5. KVALITETSKRITERIER
1.5.1. Repeterbarhed
Repeterbarheden varierer alt efter flammepunktsområdet og den anvendte metode; højst 2 oC.
1.5.2. Følsomhed
Følsomheden afhænger af den anvendte testmetode.
1.5.3. Specificitet
Nogle af testmetoderne er begrænset til et bestemt flammepunktsinterval, og deres specificitet afhænger af andre stofegenskaber (f.eks. høj viskositet).
1.6. BESKRIVELSE AF METODEN
1.6.1. Forberedelser
Der anbringes en prøve af stoffet i testapparatet efter 1.6.3.1 og/eller 1.6.3.2.
Af sikkerhedshensyn anbefales det, at der kun anvendes en lille test, dvs. ca. 2 cm3, af energirige og giftige stoffer.
1.6.2. Testbetingelser
For så vidt som det er foreneligt med sikkerhedskravene, skal apparatet anbringes et trækfrit sted.
1.6.3. Udførelse af testen
1.6.3.1. Ligevægtsmetoden
Se ISO 1516, ISO 3680, ISO 1523 og ISO 3679.
1.6.3.2. Ikke-ligevægtsmetoden
Abel apparat:
Se BS 2000 part 170, NF M07-011 og NF T66-009.
Abel-Pensky apparat:
Se EN 57, DIN 51755 part 1 (for temperaturer fra 5 oC til 65 oC), DIN 51755 part 2 (for temperaturer under 5 oC) og NF M07-036.
Tag apparat:
Se ASTM D 56.
Pensky-Martens apparat:
See ISO 2719, EN 11, DIN 51758, ASTM D 93, BS 2000-34 og NF M07-019.
Bemærkninger:
Hvis der ved en ikke-ligevægtsmetode i 1.6.3.2 findes et flammepunkt på 0 ± 2 oC, 21 ± 2 oC eller 55 ± 2 oC, skal det bekræftes ved en ligevægtsmetode med samme apparat.
Til anmeldelsesformål kan der kun bruges metoder, der giver flammepunktstemperaturen.
Til bestemmelse af flammepunktet af tyktflydende opløsningsmiddelholdige væsker (maling, lim og lignende) må der kun benyttes apparater og testmetoder, der er egnede til bestemmelse af tyktflydende væskers flammepunkt.
Se ISO 3679, ISO 3680, ISO 1523 og DIN 53213 part 1.
|
2. |
DATA |
3. RAPPORTERING
Forsøgsrapporten skal om muligt indeholde følgende oplysninger
|
— |
nøjagtig beskrivelse af stoffet (betegnelse og urenheder) |
|
— |
angivelse af den anvendte metode samt eventuelle afvigelser herfra |
|
— |
resultaterne og supplerende bemærkninger af betydning for vurdering af dem. |
4. HENVISNINGER
Ingen.
A.10. ANTÆNDELIGHED (FASTE STOFFER)
1. METODE
1.1. INDLEDNING
Ved udførelse af denne test er det nyttigt at have forhåndsoplysninger om stoffets eventuelle eksplosive egenskaber.
Testen udføres kun på stoffer, der foreligger som pulver, granulat eller pasta.
For ikke at medtage alle stoffer, der kan antændes, men kun dem, der brænder hurtigt eller på en måde, der er særlig farlig, anses kun de stoffer, hvis forbrændingshastighed overstiger en vis værdi, for at være let antændelige.
Det kan være særlig farligt, hvis forglødning kan fortsætte gennem metalpulver, da en brand er vanskelig at slukke. Metalpulver, hvori forglødningen kan udbrede sig i et bestemt tidsrum, anses for let antændelige.
1.2. DEFINITIONER OG ENHEDER
Forbrændingstiden udtrykkes i sekunder.
1.3. REFERENCESTOFFER
Ikke specificeret.
1.4. METODENS PRINCIP
Stoffet anbringes i en ca. 250 mm lang ubrudt stribe, og der foretages en indledende screeningstest til bestemmelse af, om der efter antænding med en gasflamme sker en forbrænding, der breder sig med åben flamme eller ved glødning. Hvis forbrændingen breder sig mere end 200 mm hen ad striben i løbet af et bestemt tidsrum, udføres der et komplet testprogram til bestemmelse af forbrændingshastigheden.
1.5. KVALITETSKRITERIER
Ingen anført.
1.6. BESKRIVELSE AF METODEN
1.6.1. Indledende screeningprøve
Stoffet anbringes på en ikke-brændbar ikke-porøs dårligt varmeledende plade i en ca. 250 mm lang, 20 mm bred og 10 mm høj ubrudt stribe. En gasflamme (mindste diameter 5 mm) holdes hen til den ene ende af striben, indtil pulveret bryder i brand, dog højst 2 minutter (5 minutter for pulver af metal og metallegeringer). Det noteres, om forbrændingen breder sig 200 mm hen ad striben i løbet af testperioden på 4 minutter (40 minutter for metalpulver). Hvis stoffet ikke bryder i brand eller forbrændingen ikke med åben flamme eller ved glødning breder sig 200 mm hen ad striben i løbet af testperiodens 4 minutter (40 minutter), anses stoffet ikke for at være let antændeligt, og yderligere testning er ikke påkrævet. Hvis forbrændingen i stoffet breder sig 200 mm hen ad striben på mindre end 4 minutter eller mindre end 40 for metalpulver, gennemføres nedenstående test (1.6.2).
1.6.2. Test af forbrændingshastigheden
1.6.2.1. Forberedelse
Stoffer, der foreligger som pulver eller granulat, fyldes løst i en 250 mm lang form, der har trekantet tværsnit med indvendig højde 10 mm og bredde 20 mm. På hver side af formen i længderetningen er der anbragt en metalplade, der rager 2 mm op over det trekantede tværsnits overkant (se figuren). Derefter lader man tre gange formen falde ned på et fast underlag fra 2 cm's højde. Om nødvendigt fyldes formen op igen. Sidestykkerne fjernes, og overskydende materiale skrabes af. Der lægges en ikke-brændbar ikke-porøs dårligt varmeledende plade oven på formen, det hele vendes om, og formen fjernes.
Pasta-agtige stoffer bredes ud på en ikke-brændbar ikke-porøs dårligt varmeledende plade i en 250 mm lang pølse med et tværsnit på ca. 1 cm2.
1.6.2.2. Testbetingelser
Er stoffet følsomt over for fugt, gennemføres testen så hurtigt som muligt efter, at stoffet er taget ud af beholderen.
1.6.2.3. Udførelse af prøvningen
Volden anbringes i et stinkskab på tværs af luftstrømmen.
Lufthastigheden skal være tilstrækkelig til at forhindre, at røgen slipper ud i laboratoriet, og må ikke ændres under prøvningen. Omkring apparatet opstilles der en skærm til beskyttelse mod træk.
Volden antændes i den ene ende med en gasflamme (diameter mindst 5 mm). Når forbrændingen har forplantet sig 80 mm, måles udbredelseshastigheden over de næste 100 mm.
Testen udføres seks gange, hver gang med en ren kold plade, medmindre der observeres et positivt resultat tidligere.
2. DATA
Forbrændingstiden fra den indledende screeningstest (1.6.1) og den korteste forbrændingstid i op til seks test (1.6.2.3) er relevante for vurderingen.
3. RAPPORTERING
3.1. FORSØGSRAPPORT
Forsøgsrapporten skal om muligt indeholde følgende oplysninger:
|
— |
nøjagtig beskrivelse af stoffet (betegnelse og urenheder) |
|
— |
en nøjagtig beskrivelse af det testede stof, dets fysiske tilstandsform og dets fugtindhold |
|
— |
resultaterne af den indledende screeningstest og af testen af forbrændingshastigheden, hvis den er udført |
|
— |
alle yderligere bemærkninger af betydning for vurdering af resultaterne. |
3.2. FORTOLKNING AF RESULTATERNE
Stoffer, der foreligger i form af pulver, granulat eller pasta, anses for at være let antændelige, hvis forbrændingstiden i nogen af de test, der er udført efter 1.6.2, er mindre end 45 sekunder. Pulver af metal eller en metallegering anses for at være let antændeligt, hvis det kan antændes og flammen eller reaktionszonen forplanter sig over hele testen på 10 minutter eller derunder.
4. HENVISNINGER
NF T 20-042 (SEPT 85). Chemical products for industrial use. Determination of the flammability of solids.
Tillæg
Figur
Form og tilbehor til fremstilling af volden
(alle mal i millimeter)

A.11. ANTÆNDELIGHED (GASSER)
1. METODE
1.1. INDLEDNING
Med denne metode kan det bestemmes, om en gas blandet med luft ved stuetemperatur (ca. 20 oC) og atmosfæretryk kan antændes, og i så fald i hvilket koncentrationsområde. Blandinger med stigende koncentrationer af prøvegassen med luft påvirkes med en elektrisk gnist, og det iagttages, om der sker antændelse.
1.2. DEFINITIONER OG ENHEDER
Området for antændelighed er koncentrationsområdet mellem den nedre og den øvre eksplosionsgrænse. Nedre og øvre eksplosionsgrænse er de koncentrationsgrænser for den antændelige gas i blanding med luft, ved hvilke der ikke sker nogen flammeudbredelse.
1.3. REFERENCESTOFFER
Ikke specificeret.
1.4. METODENS PRINCIP
Koncentrationen af gassen i luft øges trinvis, og blandingen påvirkes ved hvert trin med en elektrisk gnist.
1.5. KVALITETSKRITERIER
Ikke anført.
1.6. BESKRIVELSE AF METODEN
1.6.1. Apparatur
Testbeholderen er en opretstående glascylinder med en indre diameter på mindst 50 mm og en højde på mindst 300 mm. Antændelseselektroderne er placeret 60 mm over cylinderens bund med en indbyrdes afstand på 3 til 5 mm. Cylinderen er forsynet med en trykaflastningsåbning. Apparatet skal afskærmes, så eventuelle eksplosionsskader begrænses.
Som antændelseskilde anvendes en stående induktionsgnist med en varighed på 0,5 sek. Den dannes af en højspændingstransformator med en sekundærspænding på 10 til 15 kV (største tilførte effekt 300 W). I henvisning (2) er der vist et eksempel på et egnet apparat.
1.6.2. Testbetingelser
Testen skal udføres ved stuetemperatur (ca. 20 oC).
1.6.3. Udførelse af testen
Ved hjælp af doseringspumper ledes en gas/luftblanding med kendt koncentration ind i glascylinderen. Blandingen påvirkes med en gnistudladning, og det iagttages, om der ved antændelseskilden opstår en flamme, som forplanter sig af sig selv. Gaskoncentrationen varieres i intervaller på 1 vol. %, indtil der sker antændelse som beskrevet ovenfor.
Hvis det ud fra kendskab til gassens kemiske struktur må formodes, at gassen ikke kan antændes, og sammensætningen af den støkiometriske blanding med luft kan beregnes, behøver man kun at afprøve blandinger fra 10 % under til 10 % over det støkiometriske blandingsforhold.
2. DATA
Den eneste information, der er relevant for bestemmelse af denne egenskab, er, om der forekommer flammeudbredelse.
3. RAPPORTERING
Forsøgsrapporten skal om muligt indeholde følgende oplysninger
|
— |
nøjagtig beskrivelse af stoffet (betegnelse og urenheder) |
|
— |
beskrivelse af det anvendte apparat med angivelse af dimensioner |
|
— |
den temperatur, som testen er udført ved |
|
— |
de afprøvede koncentrationer og de opnåede resultater |
|
— |
testens resultat: ikke-antændelig eller let antændelig gas |
|
— |
konkluderes det, at gassen er ikke-antændelig, anføres det koncentrationsområde, hvor prøvning med 1 % intervaller har fundet sted |
|
— |
alle oplysninger og bemærkninger, der er relevante for fortolkningen af resultaterne. |
4. HENVISNINGER
|
(1) |
NF T 20-041 (SEPT 85). Chemical products for industrial use. Determination of the flammability of gases. |
|
(2) |
W. Berthold, D. Conrad, T. Grewer, H. Grosse-Wortmann, T. Redeker und H. Schacke. »Entwicklung einer Standard-Apparatur zur Messung von Explosionsgrenzen«. Chem.-Ing.-Tech., 1984, vol. 56, 2, 126-127. |
A.12. ANTÆNDELIGHED (KONTAKT MED VAND)
1. METODE
1.1. INDLEDNING
Denne testmetode kan anvendes til bestemmelse af, om et stof ved reaktion med vand udvikler farlige mængder gas, der eventuelt er let antændelig.
Testmetoden kan anvendes til både væsker og faste stoffer. Den kan ikke anvendes til stoffer, der spontant bryder i brand, når de kommer i kontakt med luften.
1.2. DEFINITIONER OG ENHEDER
Let antændelig: stoffer, som ved kontakt med vand eller fugtig luft udvikler let antændelige gasser i farlige mængder, dvs. mindst 1 liter/kg pr. time.
1.3. METODENS PRINCIP
Testen foregår trinvis som beskrevet nedenfor; testen kan afbrydes, når der sker antænding. Hvis det vides, at stoffet ikke reagerer voldsomt med vand, kan man begynde med trin 4 (1.3.4).
1.3.1. Trin 1
Teststoffet anbringes i et kar med destilleret vand ved 20 oC, og det konstateres, om den udviklede gas bryder i brand.
1.3.2. Trin 2
Teststoffet anbringes på et stykke filtrerpapir, der flyder på overfladen i en skål med destilleret vand ved 20 oC, og det konstateres, om den udviklede gas bryder i brand. Filtrerpapiret tjener udelukkende til at holde stoffet samlet på ét sted for at øge chancerne for antændelse.
1.3.3. Trin 3
Teststoffet arrangeres i en ca. 2 cm høj bunke med en diameter på ca. 3 cm. Der tilsættes et par dråber vand til bunken, og det konstateres, om den udviklede gas bryder i brand.
1.3.4. Trin 4
Teststoffet blandes med destilleret vand ved 20 oC, og gasudviklingens hastighed måles hver time over en periode på 7 timer. Hvis hastigheden efter 7 timer er uregelmæssig eller stigende, forlænges måleperioden, dog højst til 5 dage. Testen kan afbrydes, hvis hastigheden på noget tidspunkt overstiger 1 liter/kg pr. time.
1.4. REFERENCESTOFFER
Ikke specificeret.
1.5. KVALITETSKRITERIER
Ikke anført.
1.6. BESKRIVELSE AF METODERNE
1.6.1. Trin 1
1.6.1.1. Testbetingelser
Testen udføres ved stuetemperatur (ca. 20 oC).
1.6.1.2. Udførelse af testen
En lille mængde af teststoffet (ca. 2 mm i diameter) anbringes i et kar med destilleret vand. Det noteres, i) om der udvikles gas, og ii) om gassen bryder i brand. Hvis gassen bryder i brand, behøves der ikke yderligere testning, idet stoffet anses for farligt.
1.6.2. Trin 2
1.6.2.1. Apparatur
Et stykke filtrerpapir anbringes fladt flydende på overfladen af destilleret vand i en egnet beholder, f.eks. en afdampningsskål med en diameter på 100 mm.
1.6.2.2. Testbetingelser
Testen udføres ved stuetemperatur (ca. 20 oC).
1.6.2.3. Udførelse af testen
En lille mængde af teststoffet (ca. 2 mm diameter) anbringes midt på filtrerpapiret. Det noteres, i) om der udvikles gas, og ii) om gassen bryder i brand. Hvis gassen bryder i brand, behøves der ikke yderligere testning, idet stoffet anses for farligt.
1.6.3. Trin 3
1.6.3.1. Testbetingelser
Testen udføres ved stuetemperatur (ca. 20 oC).
1.6.3.2. Udførelse af testen
Teststoffet anbringes i en ca. 2 cm høj bunke på ca. 3 cm i diameter med en fordybning i toppen. Der anbringes et par dråber vand i fordybningen, og det noteres, i) om der udvikles gas, og ii) om gassen bryder i brand. Hvis gassen bryder i brand, behøves der ikke yderligere testning, idet stoffet anses for farligt.
1.6.4. Trin 4
1.6.4.1. Apparatur
Der anvendes et apparat som vist på figuren.
1.6.4.2. Testbetingelser
Det undersøges visuelt, om beholderen med teststof indeholder pulver på under 500 μm (partikelstørrelse). Hvis pulver udgør mere end 1 % (w/w) af den samlede prøve eller prøven er smuldrende, formales hele testen til pulver inden testen, så der tages hensyn til reduktion af partikelstørrelsen under opbevaring og håndtering; i alle andre tilfælde undersøges testen i den stand, hvori den er modtaget. Testen udføres ved stuetemperatur (ca. 20 oC) og atmosfæretryk.
1.6.4.3. Udførelse af testen
Der anbringes 10-20 ml vand i apparatets tildrypningstragt og 10 g stof i den koniske kolbe. Den udviklede gasmængde kan måles ved enhver egnet metode. Tildrypningstragtens hane åbnes, så vandet løber ned i kolben, og et stopur startes. Gasudviklingen måles hver time over en periode på 7 timer. Hvis gasudviklingen er uregelmæssig i denne periode eller stiger hen mod periodens slutning, fortsættes målingerne i op til 5 døgn. Hvis gasudviklingen på noget tidspunkt kommer over 1 liter/kg pr. time, kan testen afbrydes. Testen gentages yderligere to gange.
Hvis gassens kemiske sammensætning ikke er kendt, foretages der en analyse af den. Hvis gassen indeholder let antændelige bestanddele, og det ikke vides, om hele blandingen er let antændelig, fremstilles der en blanding med samme sammensætning, som test efter metode A.11.
2. DATA
Stoffet anses for farligt, hvis
|
— |
det spontant bryder i brand på noget trin af testen eller |
|
— |
der udvikles brændbar gas i større mængde end 1 liter/kg pr. time. |
3. RAPPORTERING
Forsøgsrapporten skal om muligt indeholde følgende oplysninger
|
— |
nøjagtig beskrivelse af stoffet (betegnelse og urenheder) |
|
— |
eventuel forbehandling af teststoffet |
|
— |
resultatet af testen (trin 1, 2, 3 og 4) |
|
— |
den udviklede gas' kemiske sammensætning |
|
— |
gasudviklingshastigheden, hvis trin 4 (1.6.4) udføres |
|
— |
alle yderligere bemærkninger, der er relevante for fortolkningen af resultaterne. |
4. HENVISNINGER
|
(1) |
Recommendations on the Transport of Dangerous Goods, test and criteria, 1990, United Nations, New York. |
|
(2) |
NF T 20-040 (SEPT 85). Chemical products for industrial use. Determination of the flammability of gases formed by the hydrolysis of solid and liquid products. |
Tillæg
Figur
Apparatur
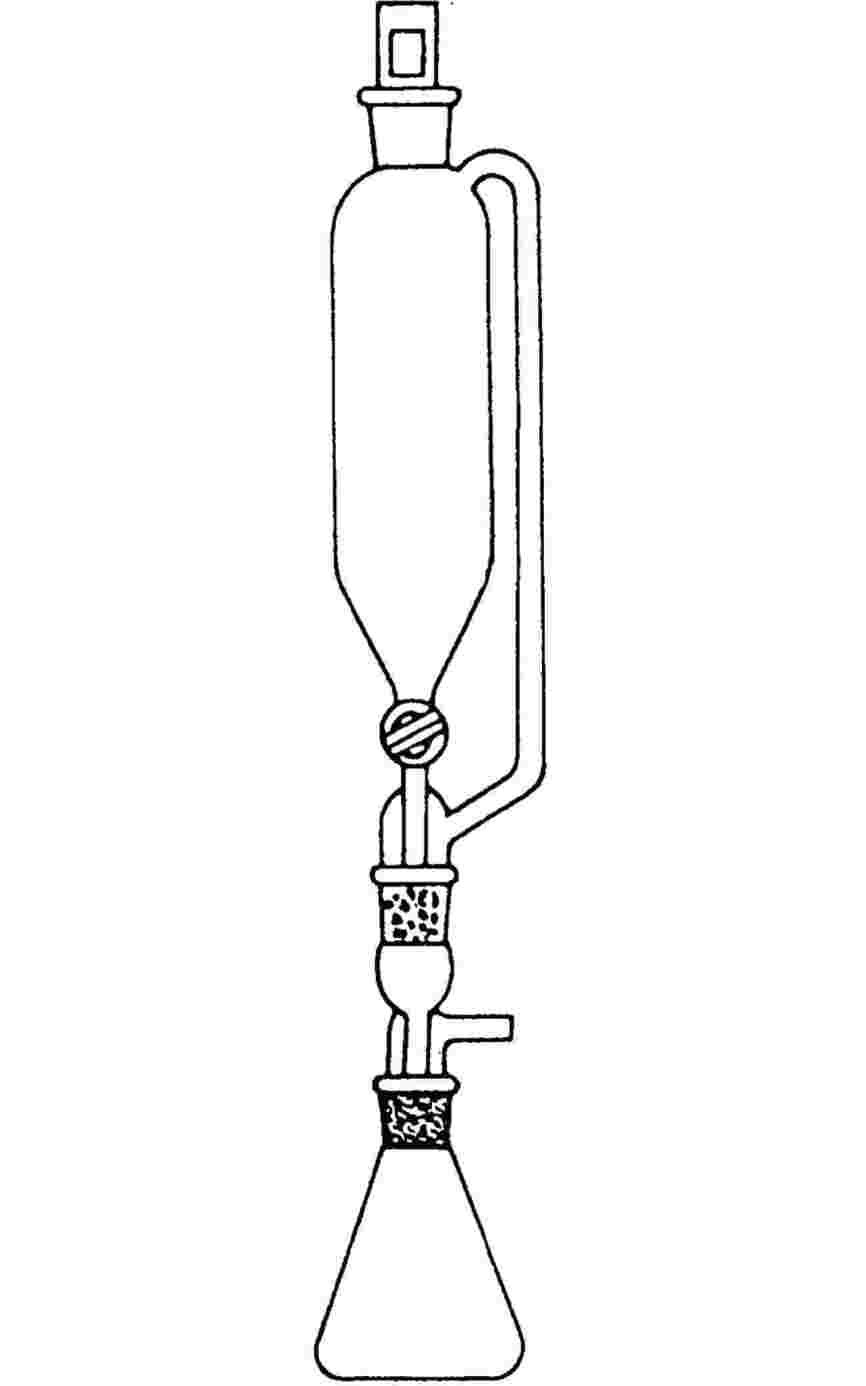
A.13 VÆSKERS OG FASTE STOFFERS PYROFORISKE EGENSKABER
1. METODE
1.1. INDLEDNING
Testmetoden kan anvendes for væsker og faste stoffer, der i små mængder spontant bryder i brand kort tid efter at være kommet i kontakt med luften ved stuetemperatur (ca. 20 oC).
Stoffer, der skal udsættes for luftens påvirkning i flere dage eller timer ved stuetemperatur, førend der sker antændelse, omfattes ikke af denne testmetode.
1.2. DEFINITIONER OG ENHEDER
Stoffer anses for at være selvantændelige, hvis de bryder i brand eller forkuller under betingelserne i 1.6.
Det kan også være påkrævet at undersøge væskers selvantændelse med metode A.15 Selvantændelsestemperatur (væsker og gasser).
1.3. REFERENCESTOFFER
Ikke specificeret.
1.4. METODENS PRINCIP
Stoffet, fast eller flydende, blandes med en inaktiv bærer og bringes i kontakt med luften ved stuetemperatur i 5 minutter. Væsker, der ikke bryder i brand, absorberes dernæst på filtrerpapir og udsættes for luftens påvirkning ved omgivende temperatur (ca. 20 oC) i 5 minutter. Flydende og faste stoffer, der bryder i brand, og væsker, der bryder i brand eller får filtrerpapiret til at forkulle, anses for at være selvantændelige.
1.5. KVALITETSKRITERIER
Repeterbarhed: på grund af den sikkerhedsmæssige betydning er et enkelt positivt resultat tilstrækkeligt til, at stoffet anses for selvantændeligt.
1.6. BESKRIVELSE AF TESTMETODEN
1.6.1. Apparatur
Der fyldes et ca. 5 mm tykt lag diatoméjord i en porcelænsskål med en diameter på ca. 10 cm ved stuetemperatur (ca. 20 oC).
Bemærk:
Diatoméjord eller et andet tilsvarende inaktivt stof, som er let at få fat i, antages at repræsentere den jord, som teststoffet kunne blive spildt på i tilfælde af uheld.
Der behøves tørt filtrerpapir til test af væsker, som ikke bryder i brand ved kontakt med luften, når de er blandet med et inaktivt bæremateriale.
1.6.2. Udførelse af testen
a) Faste stoffer i pulverform
Fra en højde af ca. 1 m hældes 1-2 cm3 af pulveret ned på en ikke-brændbar flade, og det iagttages, om stoffet bryder i brand under faldet eller inden for 5 minutter derefter.
Testen udføres seks gange, medmindre der sker antændelse.
b) Væsker
Ca. 5 cm3 af testvæsken hældes ud i den klargjorte porcelænsskål, og det iagttages, om stoffet bryder i brand, inden der er gået 5 minutter.
Hvis der ikke sker antændelse i nogen af de seks forsøg, udføres følgende forsøg:
Med en injektionssprøjte sprøjtes 0,5 ml af prøven ud på et bøjet stykke filtrerpapir, og det iagttages, om der inden for 5 minutter sker antændelse eller forkulning af filtrerpapiret. Forsøget udføres 3 gange, medmindre der sker antændelse eller forkulning.
2. DATA
2.1. BEHANDLING AF RESULTATERNE
Testen kan afbrydes, så snart et af forsøgene giver positivt resultat.
2.2. EVALUERING
Hvis stoffet bryder i brand inden 5 minutter efter, at det er tilsat til en inaktiv bærer og blevet udsat for luftens påvirkning, eller hvis en væske får et stykke filtrerpapir til at forkulle eller bryde i brand, inden 5 minutter efter at stoffet er tilsat og blevet udsat for luftens påvirkning, anses stoffet (væsken) for at være selvantændeligt.
3. RAPPORTERING
Forsøgsrapporten skal om muligt indeholde følgende oplysninger
|
— |
nøjagtig beskrivelse af stoffet (betegnelse og urenheder) |
|
— |
resultaterne af testen |
|
— |
alle yderligere bemærkninger, der er relevante for fortolkningen af resultaterne. |
4. HENVISNINGER
|
(1) |
NF T 20-039 (SEPT 85). Chemical products for industrial use. Determination of the spontaneous flammability of solids and liquids. |
|
(2) |
Recommendations on the Transport of Dangerous Goods, Test and criteria, 1990, United Nations, New York. |
A.14. EKSPLOSIVE EGENSKABER
1. METODE
1.1. INDLEDNING
Metoden indeholder et testsystem til bestemmelse af, om et fast eller pastaagtigt stof frembyder eksplosionsfare, når det udsættes for påvirkning med en flamme (varmefølsomhed) eller ved slag eller friktion (følsomhed over for mekanisk påvirkning), og om en væske frembyder eksplosionsfare, når den udsættes for påvirkning med en flamme eller ved slag.
Metoden består af tre dele:
|
a) |
en test for varmefølsomhed (1) |
|
b) |
en test for følsomhed over for slagpåvirkning (1) |
|
c) |
en test for følsomhed over for friktionspåvirkning (1). |
Metoden giver data til vurdering af sandsynligheden for, at der ved visse almindelige påvirkninger fremkaldes en eksplosion. Metoden tilsigter ikke at fastslå, om stoffet overhovedet kan bringes til at eksplodere.
Metoden er egnet til bestemmelse af, om et stof frembyder en eksplosionfare (varmefølsomhed og mekanisk følsomhed) under de særlige betingelser, der er specificeret i direktivet. Metoden er baseret på en række apparattyper, som i vidt omfang anvendes internationalt (1), og som sædvanligvis giver brugbare resultater. Det erkendes, at metoden ikke er definitiv. Der kan benyttes andet apparatur end det specificerede, forudsat at det er internationalt anerkendt, og at de fundne resultater kan korreleres til resultater opnået med det specificerede apparatur.
Testen behøver ikke at udføres, hvis det på grundlag af foreliggende termodynamiske oplysninger (f.eks. dannelsesvarme eller dekomponeringsvarme) og/eller en strukturformel, hvori visse reaktive grupper (2) ikke indgår, må anses for hævet over enhver rimelig tvivl, at stoffet er ude af stand til hurtig dekomponering ledsaget af gasudvikling eller varmefrigørelse (dvs. at stoffet ikke frembyder nogen eksplosionsfare). For væsker kræves der ikke test for friktionsfølsomhed.
1.2. DEFINITIONER OG ENHEDER
Eksplosiv:
Stoffer, som kan eksplodere ved flammepåvirkning, eller som er følsomme over for slag eller friktion (eller mere følsomme over for mekanisk påvirkning end 1,3-dinitrobenzen i et alternativt apparat).
1.3. REFERENCESTOFFER
1,3-dinitrobenzen, teknisk krystallinsk produkt, sigtet på 0,5 mm sigte, til friktions- og slagpåvirkningsmetoden.
Perhydro-1,3,5-trinitro-1,3,5-triazin (RDX, hexogen, cyclonite — CAS 121-82-4), omkrystalliseret i vandig cyclohexanon, vådsigtet på 250 μm sigte og tilbageholdt på en 150 μm sigte, tørret ved 103 ± 2 oC (i 4 timer) til anden række friktions- og slagprøvninger.
1.4. METODENS PRINCIP
Der kræves indledende testning til bestemmelse af, hvordan de tre følsomhedstest kan udføres under betryggende forhold.
1.4.1. Test for sikker håndtering (3)
Af sikkerhedshensyn foretages der før den egentlige test en indledende test bestående i, at en meget lille testmængde (ca. 10 mg) opvarmes fritliggende i en gasflamme, og påvirkes med slag i et passende apparat og ved friktion ved hjælp af en hammer mod en ambolt eller et andet friktionsapparat. Formålet er at fastslå, om stoffet er så følsomt og eksplosivt, at den foreskrevne følsomhedstest, især for varmefølsomhed, må udføres under iagttagelse af særlige forholdsregler, så operatøren ikke kan komme til skade.
1.4.2. Varmefølsomhed (virkningen af en flamme)
Metoden består i opvarmning af stoffet i et stålrør, der er lukket med en mundingsplade. Ved hjælp af mundingsplader med forskellige huldiametre kan det bestemmes, om stoffet er tilbøjeligt til at eksplodere under stærk varmepåvirkning og veldefineret indeslutningsgrad.
1.4.3. Mekanisk følsomhed (slag)
Metoden består i, at stoffet udsættes for et slag med et specificeret lod, der falder fra en specificeret højde.
1.4.4. Mekanisk følsomhed (friktion)
Metoden består i, at et fast eller pastaagtigt stof udsættes for friktion mellem standardoverflader med specificeret belastning og ved specificeret relativ bevægelse.
1.5. KVALITETSKRITERIER
Ikke anført.
1.6. BESKRIVELSE AF METODERNE
1.6.1. Varmefølsomhed (virkningen af en flamme)
1.6.1.1. Apparatur
Apparaturet består af et engangsstålrør med lukkeanordning (figur 1) anbragt i en opvarmnings- og beskyttelsesanordning. Røret er af dybtrukket stålplade (se tillægget) og har en indvendig diameter på 24 mm, en længde på 75 mm og en godstykkelse på 0,5 mm. Røret har i den åbne ende en flange, så det kan lukkes med mundingspladen ved hjælp af en todelt forskruning (møtrik og krave med gevind). Mundingspladen er trykmodstandsdygtig og har et hul i midten. Møtrikken og kraven med gevind er fremstillet i chrom-manganstål (se tillægget), som er gnistfrit op til 800 oC. Der foreligger mundingsplader med en række forskellige huldiametre, som er fremstillet i 6 mm tykt varmebestandigt stål (se tillægget).
1.6.1.2. Testbetingelser
Normalt testes stoffet i den tilstand, hvori det er modtaget, men i visse tilfælde, f.eks. hvis det foreligger i presset, støbt eller anden kompakt form, kan det være nødvendigt at knuse stoffet før testen.
For faste stoffer bestemmes den mængde materiale, der skal anvendes til testen, ved en to-trins procedure. Først fyldes der i et tareret rør 9 cm3 af stoffer, som nedstampes med en kraft på 80 N fordelt over hele rørtværsnittet. Af sikkerhedshensyn eller i tilfælde, hvor stoffet kan ændre fysisk form ved kompression, kan der benyttes andre påfyldningsmetoder; eksempelvis er stampning ikke egnet, hvis stoffet er meget friktionsfølsomt. Hvis materialet kan komprimeres, tilsættes der mere og stampes, indtil røret er fyldt til en højde af 55 mm fra overkanten. Den samlede mængde, der er brugt til fyldning af røret til denne højde, bestemmes, og der tilsættes endnu 2 portioner, som stampes med en kraft på 80 N. Der tilsættes materiale med stampning eller fjernes materiale, så røret er fyldt til 15 mm fra overkanten. Det andet trin foregår ved, at der først tilsættes og stampes en tredjedel af den samlede mængde, der blev brugt til det første trin. Der tilsættes endnu 2 portioner med stampning med 80 N, og materialets højde i røret justeres til 15 mm fra overkanten ved tilsætning eller fjernelse af materiale. Til hvert forsøg anvendes den mængde fast stof, der er fundet ved dette andet trin; påfyldningen sker i 3 lige store portioner, der hver komprimeres til 9 cm3 med den fornødne kraft. (Dette gøres lettest ved anvendelse af afstandsringe).
Væsker og geler fyldes i røret til en højde af 60 mm, idet det for gelernes vedkommende nøje påses, at der ikke dannes luftbobler. Kraven med gevind føres op om røret nedefra, den valgte mundingsplade indsættes, og møtrikken strammes efter smøring med et molybdænsulfidbaseret smøremiddel. Det er yderst vigtigt at kontrollere, at der hverken har sat sig stof mellem flangen og pladen eller i gevindet.
Opvarmningen sker med propan, der fra en industriel trykflaske med trykregulator (60-70 mbar) ledes gennem en måler og fordeles ligeligt til 4 brændere gennem en manifold (hvilket kontrolleres ved visuel inspektion af brændernes flammer). Brænderne er placeret omkring prøvekammeret som vist på figur 1. De 4 brænderes samlede forbrug er på ca. 3,2 liter propan pr. minut. Der kan anvendes andre brændstoffer og brændere, men opvarmningshastigheden skal være som specificeret i figur 3. Uanset det anvendte apparatur skal opvarmningshastigheden regelmæssigt kontrolleres med dibutylphthalatfyldte rør, som vist på figur 3.
1.6.1.3. Udførelse af testene
Hver test varer, indtil røret enten sønderdeles eller røret har været opvarmet i 5 minutter. En test, hvorved røret sønderdeles i tre eller flere fragmenter, som eventuelt hænger sammen med smalle metalstrimler som vist på figur 2, vurderes som en eksplosion. En test, der fører til færre fragmenter eller ingen fragmentering, betragtes ikke som en eksplosion.
Der udføres først tre test med en mundingsplade med et 6,0 mm hul, og opnås der ingen eksplosion, udføres endnu tre test med en mundingsplade med et 2,0 mm hul. Finder der eksplosion sted under én af disse forsøgsrækker, er yderligere testning ikke påkrævet.
1.6.1.4. Evaluering
Testens resultat anses for positivt, hvis der sker eksplosion i én af ovennævnte forsøgsrækker.
1.6.2. Mekanisk følsomhed (slag)
1.6.2.1. Apparatur (figur 4)
Hoveddelene af et typisk faldhammerapparat er en støbt stålblok med bundplade, ambolt, stander, styr, faldlodder, frigørelsesmekanisme og prøveholder. Stålambolten på 100 mm (diameter) × 70 mm (højde) er skruet fast oven på en stålblok på 230 mm (længde) × 250 mm (bredde) × 200 mm (højde) med støbt bundplade på 450 mm (længde) × 450 mm (bredde) × 60 mm (højde). En stander bestående af et sømløst trukket stålrør er fastgjort i en holder, der er fastskruet bag på stålblokken. Apparatet er forankret til en massiv betonklods på 60 × 60 × 60 cm med fire skruer, så styrene er fuldstændig lodrette og faldlodderne kan falde frit. Der findes lodder på 5 og 10 kg fremstillet i massivt stål. Loddernes slaghoved er af hærdet stål, HRC 60 til 63, og har en diameter på mindst 25 mm.
Testen er indeholdt i en slaganordning bestående af to massive stålcylindre anbragt koaksialt over hinanden i en hul cylindrisk styrering af stål. Stålcylindrene skal have en diameter på 10 (-0,003, -0,005) mm og en højde på 10 mm og have polerede endeflader, afrundede kanter (krumningsradius 0,5 mm) og en hårdhed på HRC 58 til 65. Den hule cylinder skal have en udvendig diameter på 16 mm, en poleret udboring på 10 (+0,005, +0,010) mm og en højde på 13 mm. Slaganordningen samles på en mellemambolt (diameter 26 mm, højde 26 mm) af stål og centreres med en ring, hvori der er huller til bortledning af eksplosionsprodukterne.
1.6.2.2. Testbetingelser
Testvolumenet skal være på 40 mm3 eller det volumen, der passer til et alternativt apparat. Faste stoffer testes i tør tilstand og forberedes således
|
a) |
stoffer i pulverform sigtes (0,5 mm sigte); alt hvad der passerer gennem sigten bruges til testen |
|
b) |
stoffer, der foreligger i presset, støbt eller anden kompakt form, findeles og sigtes; sigtefraktionen fra 0,5 mm til 1 mm diameter bruges til testen og skal være repræsentativ for det oprindelige stof. |
Stoffer, som normalt forekommer i pastaform skal om muligt testes i tør tilstand og under alle omstændigheder efter fjernelse af størst mulig mængde opløsningsmiddel. Ved test af væsker er der en afstand på 1 mm mellem den øverste og den nederste stålcylinder.
1.6.2.3. Udførelse af testen
Der udføres seks forsøg, hvor 10 kg's loddet falder fra en højde af 0,40 m (40 J). Forekommer der eksplosion i et af de 6 forsøg med 40 J, udføres der en ny række på seks forsøg, hvor et 5 kg's lod falder fra en højde af 0,15 m (7,5 J). I andre apparater sammenlignes prøven med det valgte referencestof ved hjælp af en fastlagt procedure (f.eks. up-and-down teknik).
1.6.2.4. Evaluering
Testresultatet anses for positivt, hvis der sker eksplosion (opflamning og knald betragtes som eksplosion) i mindst ét af forsøgene med det specificerede slagapparat, eller hvis prøven er mere følsom end 1,3-dinitrobenzen eller RDX i en alternativ slagtest.
1.6.3. Mekanisk følsomhed (friktion)
1.6.3.1. Apparatur (figur 5)
Friktionsapparatet består af en støbt bundplade af stål, hvorpå friktionsanordningen er monteret. Den består af en fastsiddende porcelænsstift og en bevægelig porcelænsplade. Porcelænspladen fastholdes i en slæde, der glider på to styr. Slæden er via en forbindelsesstang, en krumtap og et passende gear forbundet med en elmotor, således at porcelænspladen bevæges 10 mm frem og tilbage under porcelænsstiften én gang. Porcelænsstiften kan belastes f.eks. med 120 eller 360 newton.
Porcelænspladerne er flade og er fremstillet af hvidt teknisk porcelæn (ruhed 9 til 32 μm) og har dimensionerne 25 mm (længde) × 25 mm (bredde) × 5 mm (højde). Den cylindriske porcelænsstift, der ligeledes er fremstillet af hvidt teknisk porcelæn, har en længde på 15 mm, en diameter på 10 mm og ru kugleformede endeflader med en krumningsradius på 10 mm.
1.6.3.2. Testbetingelser
Prøvevolumenet skal være på 10 mm3 eller det volumen, der passer til et alternativt apparat.
Faste stoffer testes i tør tilstand og forberedes således:
|
a) |
stoffer i pulverform sigtes (0,5 mm sigte); alt hvad der passerer gennem sigten bruges til testen |
|
b) |
stoffer, der foreligger i presset, støbt eller anden kompakt form, findeles og sigtes; sigtefraktionen < 0,5 mm diameter bruges til testen. |
Stoffer, som normalt forekommer i pastaform skal om muligt testes i tør tilstand. Hvis stoffet ikke kan tørres, skal pastaen (efter fjernelse af størst mulig mængde opløsningsmiddel) testes som et 0,5 mm tykt lag med en bredde på 2 mm og en længde på 10 mm, der udlægges med en skabelon.
1.6.3.3. Udførelse af testen
Porcelænsstiften anbringes på prøven, og belastningen påsættes. Ved udførelse af testen skal skuremærkerne på porcelænspladen vende på tværs af bevægelsesretningen. Det må nøje påses, at der er tilstrækkeligt testmateriale under stiften, og at pladen bevæger sig korrekt under stiften. Til anbringelse af pastaagtige stoffer på pladen anvendes der en 0,5 mm tyk skabelon med et åbent felt på 2 × 10 mm. Porcelænspladen skal bevæge sig 10 mm frem og tilbage under porcelænsstiften i løbet af 0,44 sekunder. Hver del af porcelænspladen og -stiften må kun bruges én gang; de to ender af en stift kan bruges til to forsøg og de to flader af en plade kan hver bruges til tre forsøg.
Der udføres seks forsøg med en belastning på 360 N. Forekommer der eksplosion i et af disse seks forsøg, udføres der en ny række på seks forsøg med en belastning på 120 N. I andre apparater sammenlignes prøven med det valgte referencestof ved hjælp af en fastlagt procedure (f.eks. up-and-down teknik).
1.6.3.4. Evaluering
Testresultatet anses for positivt, hvis der sker eksplosion (knitren og/eller knald eller opflamning betragtes som eksplosion) i mindst ét af forsøgene med det specificerede friktionsapparat, eller hvis de tilsvarende kriterier i en alternativ friktionstest er opfyldt.
2. DATA
Principielt anses et stof for at være eksplosionsfarligt i direktivets forstand, hvis der ved testen for varme-, slag- eller friktionsfølsomhed opnås et positivt resultat.
3. RAPPORTERING
3.1. PRØVNINGSRAPPORTEN
Prøvningsrapporten skal om muligt indeholde følgende oplysninger:
|
— |
teststoffets betegnelse, sammensætning, renhed, fugtindhold mv. |
|
— |
prøvens fysiske tilstandsform samt oplysning om, om den er knust, findelt og/eller sigtet |
|
— |
iagttagelser under testen for varmefølsomhed (f.eks. testmasse, antal fragmenter mv.) |
|
— |
iagttagelser under testen for mekanisk følsomhed (f.eks. dannelse af væsentlige røgmængder eller fuldstændig dekomponering uden knald, flammer, gnister, knitren mv.) |
|
— |
resultaterne af hver forsøgstype |
|
— |
videnskabelig begrundelse for eventuel anvendelse af et alternativt apparat, samt godtgørelse af korrelationen mellem resultaterne med det specificerede apparat og resultaterne med et tilsvarende apparat |
|
— |
nyttige bemærkninger, såsom henvisning til forsøg med lignende produkter, som kan være relevante for korrekt fortolkning af resultaterne |
|
— |
alle yderligere bemærkninger, der er relevante for fortolkning af resultaterne. |
3.2. FORTOLKNING OG EVALUERING AF RESULTATERNE
I testrapporten skal alle resultater, der anses for ukorrekte, unormale eller ikke-repræsentative, nævnes. Hvis der skal ses bort fra nogle af resultaterne, skal dette forklares og resultaterne af supplerende eller alternativ test oplyses. Kan der ikke redegøres for et unormalt resultat, skal det tages for pålydende, og stoffet skal klassificeres i overensstemmelse hermed.
4. HENVISNINGER
|
(1) |
Recommendations on the Transport of Dangerous Goods: Tests and criteria, 1990, United Nations, New York. |
|
(2) |
Bretherick, L., Handbook of Reactive Chemical Hazards, 4th edition, Butterworths, London, ISBN 0-750-60103-5, 1990. |
|
(3) |
Koenen, H., Ide, K.H. and Swart, K.H., Explosivstoffe, 1961, vol. 3, 6-13 and 30-42. |
|
(4) |
NF T 20-038 (Sept. 85). Chemical products for industrial use — Determination of explosion risk. |
Tillæg
Eksempel på materialespecifikation for varmefølsomhedstesting (se DIN 1623)
|
(1) |
Rør: Materialespecifikation nr. 1.0336.505 g |
|
(2) |
Mundingsplade: Materialespecifikation nr. 1.4873 |
|
(3) |
Krave med gevind og møtrik: Materialespecifikation nr. 1.3817 |
Figur 1
Apparat til testing for varmefølsomhed
(alle dimensioner i mm)

Figur 2
Test for varmefølsomed
Eksempler på fragmentering

Figur 3
Kalibrering af opvarmingshastigeden ved test af varmefølsomed

Kurve over temeraturforløbet mod tiden ved opvarmning af dibutylphthalat (27 cm3 i et lukket rør (mundingslade med 1,5 mm hul) med et propanflow på 3,2 litter/minut. Temperaturen er målt med chromel/alumel-termoelement med 1 mm indkapsling i rustfrit stål, anbragt central 43 mm under rørets overkant. Mellen 135 oC og 285 oC skal opvarmningshastigheden være 185-215 K/minut.
Figur 4
Apparat til slagprøvning
(alle dimensioner i mm)

Figur 4
Fortstat

Figur 5
Apparat til friktionsfølsomhed

A.15. SELVANTÆNDELSESTEMPERATUR FOR VÆSKER OG GASSER
1. METODE
1.1. INDLEDNING
Eksplosive stoffer og stoffer, der spontant bryder i brand i kontakt med luften, underkastes ikke denne test. Testproceduren kan anvendes for gasser, væsker og dampe, som kan antændes af en hed overflade under tilstedeværelse af luft.
Selvantændelsestemperaturen kan påvirkes betydeligt i nedadgående retning af katalytiske urenheder, af overfladematerialet og af en større testbeholder.
1.2. DEFINITIONER OG ENHEDER
Tendensen til selvantændelse udtrykkes ved selvantændelsestemperaturen. Selvantændelsestemperaturen er den laveste temperatur, ved hvilken teststoffet bryder i brand i blanding med luft under de i testmetoden definerede betingelser.
1.3. REFERENCESTOFFER
I standarderne (se 1.6.3) er der nævnt referencestoffer. De skal først og fremmest tjene til lejlighedsvis kontrol af metoden og til sammenligning med resultater opnået med andre metoder.
1.4. METODENS PRINCIP
Ved metoden bestemmes den laveste temperatur af den indvendige overflade af et lukket rum, ved hvilken en gas, damp eller væske, der sprøjtes ind i rummet, bryder i brand.
1.5. KVALITETSKRITERIER
Repeterbarheden afhænger af området for selvantændelsestemperaturen og af den anvendte testmetode.
Følsomheden og specificiteten afhænger af den anvendte testmetode.
1.6. BESKRIVELSE AF METODEN
1.6.1. Apparatur
Apparaturet er beskrevet i den metode, der henvises til i 1.6.3.
1.6.2. Testbetingelser
En prøve af stoffet testes i overensstemmelse med den metode, der henvises til i 1.6.3.
1.6.3. Udførelse af testen
Se IEC 79-4, DIN 51794, ASTM-E 659-78, BS 4056 og NF T 20-037.
2. DATA
Testtemperaturen, atmosfæretrykket, den anvendte mængde prøvemateriale og tidsrummet, indtil der sker antændelse, registreres.
3. RAPPORTERING
Forsøgsrapporten skal om muligt indeholde følgende oplysninger
|
— |
nøjagtig beskrivelse af stoffet (betegnelse og urenheder) |
|
— |
den anvendte testmængde og atmosfæretrykket |
|
— |
det anvendte apparatur |
|
— |
måleresultaterne (testtemperatur, resultaterne af antændelsen og det hertil svarende tidsrum) |
|
— |
alle yderligere oplysninger, der er relevante for fortolkningen af resultaterne. |
4. HENVISNINGER
Ingen.
A.16. RELATIV SELVANTÆNDELSESTEMPERATUR FOR FASTE STOFFER
1. METODE
1.1. INDLEDNING
Eksplosive stoffer og stoffer, der spontant bryder i brand i kontakt med luften, underkastes ikke denne test.
Formålet med denne test er at fremskaffe foreløbige oplysninger om faste stoffers selvantændelighed ved høj temperatur.
Hvis den varme, der udvikles enten ved stoffets reaktion med luften eller ved eksoterm dekomponering, ikke bortledes hurtigt nok til omgivelserne, sker der en opvarmning, der fører til selvantændelse. Selvantændelse kan derfor finde sted, når varmeproduktionen sker hurtigere end varmeafgivelsen.
Testmetoden egner sig til foreløbig screening af faste stoffer. Da antændelse og forbrænding af faste stoffer er meget kompleks, bør den selvantændelsestemperatur, der bestemmes ved denne metode, kun benyttes til sammenligningsformål.
1.2. DEFINITIONER OG ENHEDER
Den selvantændelsestemperatur, der bestemmes ved denne metode, er den laveste omgivende temperatur udtrykt i oC, hvorved en bestemt mængde af et stof bryder i brand under veldefinerede betingelser.
1.3. REFERENCESTOF
Ingen.
1.4. METODENS PRINCIP
En bestemt mængde af teststoffet anbringes i en ovn ved stuetemperatur; medens ovntemperaturen hæves med 0,5 oC/min til smeltepunktet, dog højst til 400 oC, registreres temperaturen midt i prøven som funktion af tiden. I forbindelse med denne test benævnes den ovntemperatur, ved hvilken prøvens temperatur når op på 400 oC ved selvopvarmning, selvantændelsestemperaturen.
1.5. KVALITETSKRITERIER
Ingen.
1.6. BESKRIVELSE AF METODEN
1.6.1. Apparatur
1.6.1.1. Ovn
En temperaturprogrammeret laboratorieovn (volumen ca. 2 liter) med fri konvektion og eksplosionsaflastning. For at imødegå enhver eksplosionsfare må eventuelle dekomponeringsgasser ikke kunne komme i berøring med varmelegemet.
1.6.1.2. Terning af ståltrådsnet
Et stykke ståltrådsnet med maskevidden 0,045 mm skæres ud efter tegningen i figur 1. Det foldes til terninger, hvor den ene side er åben, og fastholdes med ståltråd.
1.6.1.3. Termoelementer
Egnede termoelementer.
1.6.1.4. Skriver
En to-kanals skriver, der er kalibreret op til 600 oC eller en tilsvarende spænding.
1.6.2. Testbetingelser
Stoffer testes i den form, de modtages i.
1.6.3. Udførelse af testen
Terningen fyldes med teststoffet, og der bankes let på den under tilsætning af mere af stoffet, indtil den er helt fyldt. Dernæst ophænges terningen midt i ovnen ved stuetemperatur. Det ene termoelement anbringes midt i terningen, det andet mellem terningen og ovnvæggen til måling af ovntemperaturen.
Ovntemperaturen og prøvetemperaturen registreres løbende, mens ovntemperaturen hæves til 400 oC, dog højst til stoffets smeltepunktet, med en hastighed på 0,5 oC/min.
Antændelse af stoffet vil vise sig som en meget stejl temperaturstigning i prøven sammenlignet med ovntemperaturen.
2. DATA
Den ovntemperatur, ved hvilken prøven som følge af selvopvarmning når op på en temperatur på 400 oC, er relevant for evaluering (se figur 2).
3. RAPPORTERING
Forsøgsrapporten skal om muligt indeholde følgende oplysninger:
|
— |
en beskrivelse af teststoffet |
|
— |
måleresultaterne, herunder temperatur/tid-kurven |
|
— |
alle yderligere bemærkninger, der er relevante for fortolkningen af resultaterne. |
4. HENVISNINGER
NF T 20-036 (SEPT 85). Chemical products for industrial use. Determination of the relative temperature of the spontaneous flammability of solids.
Figur 1
Tegning af 20 mm testterning

Figur 2
Typisk temperatur/tid-kurve

A.17. OXIDERENDE EGENSKABER (FASTE STOFFER)
1. METODE
1.1. INDLEDNING
Ved udførelse af denne test er det nyttigt at have forhåndsoplysninger om stoffets eventuelle eksplosive egenskaber.
Denne test kan ikke anvendes på væsker, gasser, eksplosive og yderst let antændelige stoffer samt organiske peroxider.
Det er ikke nødvendigt at udføre denne test, hvis det på grundlag af strukturformelen kan godtgøres, at stoffet med stor sandsynlighed ikke kan reagere eksotermt med et brændbart materiale.
Der udføres en indledende test til konstatering af, om den egentlige test skal udføres under iagttagelse af særlige forsigtighedsregler.
1.2. DEFINITIONER OG ENHEDER
Reaktionstid: den reaktionstid i sekunder, der kræves, for at reaktionszonen breder sig hen ad en vold, efter den fremgangsmåde, der er beskrevet i 1.6.
Reaktionshastighed: udtrykkes i millimeter pr. sekund.
Største reaktionshastighed: den største af de reaktionshastigheder, der bestemmes med blandinger indeholdende 10 til 90 vægtprocent af det oxiderende stof.
1.3. REFERENCESTOF
Som referencestof til testen og screeningstesten benyttes bariumnitrat (analysekvalitet).
Referenceblandingen er den blanding af bariumnitrat og pulveriseret cellulose, fremstillet efter 1.6, som har den største reaktionshastighed (normalt en blanding indeholdende 60 vægtprocent bariumnitrat).
1.4. METODENS PRINCIP
Af sikkerhedshensyn udføres der en indledende screeningstest. Viser denne indledende test klart, at stoffet har oxiderende egenskaber, behøves der ikke yderligere testing. Er dette ikke tilfældet, underkastes stoffet en fuldstændig test.
Ved den fuldstændige test blandes teststoffet og et veldefineret brændbart stof i forskellige forhold. Hver blanding arrangeres i en vold, der antændes i den ene ende. Den største målte reaktionshastighed sammenlignes med referenceblandingens største reaktionshastighed.
1.5. KVALITETSKRITERIER
Alle påkrævede formalings- og blandemetoder kan anvendes under den forudsætning, at den største reaktionshastighed i hver af de seks enkeltforsøg ikke afviger fra det aritmetiske gennemsnit med mere end 10 %.
1.6. BESKRIVELSE AF METODEN
1.6.1. Forberedelse
1.6.1.1. Teststof
Prøven reduceres til en partikelstørrelse på < 0,125 mm på følgende måde: Teststoffet sigtes, hvorefter den tilbageholdte fraktion formales, og denne fremgangsmåde gentages, indtil hele testmængden har passeret sigten.
Alle formalings- og sigtemetoder, der opfylder kvalitetskriterierne, kan anvendes.
Før fremstilling af blandingen tørres stoffet ved 105 oC til konstant vægt. Hvis teststoffets dekomponeringstemperatur er lavere end 105 oC, tørres stoffet ved en passende lavere temperatur.
1.6.1.2. Brændbart stof
Som brændbart stof anvendes der pulveriseret cellulose af den type, der bruges til tyndtlagskromatografi eller søjlekromatografi. En type, hvor 85 % af fibrene har en længde på mellem 0,020 og 0,075 mm, har vist sig egnet. Cellulosepulveret sigtes på en sigte med en maskevidde på 0,125 mm. Der bruges cellulose fra samme batch til hele testen.
Før fremstilling af blandingen tørres cellulosepulveret ved 105 oC til konstant vægt.
Hvis der benyttes savsmuld til den indledende test, fremstilles dette ved opsamling af den fraktion, der passerer gennem en sigte med maskevidden 1,6 mm, omhyggelig blanding og tørring ved 105 oC i 4 timer i et højst 25 mm tykt lag. Efter afkøling opbevares savsmuldet i en lufttæt beholder, der fyldes så meget som praktisk muligt, indtil der bruges, helst inden 24 timer efter tørringen.
1.6.1.3. Antændelseskilde
Som antændelseskilde anvendes der en gasflamme (diameter mindst 5 mm). Anvendes der en anden antændelseskilde (f.eks. ved test i inert atmosfære), skal dette begrundes og kilden beskrives i rapporten.
1.6.2. Udførelse af testen
Bemærk:
Blandinger af oxiderende stoffer med cellulose eller savsmuld skal betragtes som potentielt eksplosive og behandles med passende forsigtighed.
1.6.2.1. Indledende test
2 vægtdele tørret stof blandes omhyggeligt med 1 vægtdel tørret cellulose eller savsmuld, og der tildannes en lille kegleformet bunke af blandingen med en grundfladediameter på 3,5 cm og en højde på 2,5 cm, ved at fylde blandingen løst i en kegleformet form (f.eks. en laboratorietragt med tilproppet stilk).
Bunken anbringes på en kold ikke-brændbar ikke-porøs dårligt varmeledende plade. Testen udføres i stinkskab på samme måde som testen i 1.6.2.2.
Antændelseskilden bringes i berøring med bunken. Den efterfølgende reaktions voldsomhed og varighed iagttages og registreres.
Hvis reaktionen er voldsom, skal stoffet anses for oxiderende.
Hvis resultatet på nogen måde er tvetydigt, skal den nedenfor beskrevne fuldstændige udbredelsestest udføres.
1.6.2.2. Udbredelsestest
Der fremstilles en række blandinger af oxiderende stof og cellulose indeholdende fra 10 til 90 vægtprocent oxiderende stof og med intervaller på 10 vægtprocent. I grænsetilfælde anvendes der blandinger derimellem, så den største reaktionshastighed bestemmes mere nøjagtigt.
Volden tildannes ved hjælp af en 250 mm lang metalform, der har trekantet tværsnit med indvendig højde 10 mm og bredde 20 mm. På hver side af formen i længderetningen er der anbragt en metalplade, der rager 2 mm op over det trekantede tværsnits overkant (se figuren). Denne indretning fyldes løst med et lille overskud af blandingen. Efter at man har ladet formen falde ned på et fast underlag fra 2 cm's højde en gang, skrabes overskuddet af med en skråtstillet plade. Sidestykkerne fjernes, og det resterende pulver glattes ud med en valse. Der lægges en ikke-brændbar ikke-porøs dårligt varmeledende plade oven på formen, det hele vendes om, og formen fjernes.
Volden anbringes i et stinkskab på tværs af luftstrømmen.
Lufthastigheden skal være tilstrækkelig til at forhindre, at røgen slipper ud i laboratoriet, og må ikke ændres under testen. Omkring apparatet opstilles der en skærm til beskyttelse mod træk.
Da cellulose og visse teststoffer er hygroskopiske, skal testen udføres så hurtigt som muligt.
Volden antændes i den ene ende ved berøring med en flamme.
Når reaktionszonen har forplantet sig 30 mm, måles dens udbredelseshastighed over de næste 200 mm.
Testen udføres med referencestoffet og mindst én gang med hver af blandingerne af teststof med cellulose.
Hvis den fundne største reaktionshastighed er væsentligt større end referenceblandingens, kan testen afbrydes; i modsat fald gentages forsøget fem gange med hver af de tre blandinger, der giver den største reaktionshastighed.
Hvis der er mistanke om, at der er fremkommet et falsk positivt resultat, gentages testen med et inert materiale med tilsvarende partikelstørrelse i stedet for cellulosen, f.eks. kiselgur. Alternativt kan den blanding af teststof og cellulose, som har den højeste reaktionshastighed, testes i inert atmosfære (< 2 % v/v oxygen).
2. DATA
Af sikkerhedshensyn anses den største reaktionshastighed — og ikke gennemsnittet — for at være den karakteristiske oxiderende egenskab ved det undersøgte stof.
Den højeste værdi af reaktionshastigheden af en serie på seks forsøg med en given blanding er relevant for vurderingen.
Hver enkelt blandings højeste reaktionshastighed afbildes mod koncentrationen af oxiderende stof. Den største reaktionshastighed aflæses på kurven.
De seks værdier af reaktionshastigheden, som er fundet med den blanding, der har den største reaktionshastighed, må ikke afvige fra det aritmetiske gennemsnit med mere end 10 %; i modsat fald må formalings- og blandemetoderne forbedres.
Den fundne største reaktionshastighed sammenlignes med referenceblandingens største reaktionshastighed (se 1.3).
Hvis der udføres test i inert atmosfære, sammenlignes den største reaktionshastighed med referenceblandingens største reaktionshastighed bestemt i inert atmosfære.
3. RAPPORTERING
3.1. FORSØGSRAPPORT
Forsøgsrapporten skal om muligt indeholde følgende oplysninger:
|
— |
det undersøgte stofs identitet, sammensætning, renhedsgrad, vandindhold, m.v. |
|
— |
eventuel behandling af prøven (f.eks. formaling eller tørring) |
|
— |
den benyttede antændelseskilde |
|
— |
måleresultaterne |
|
— |
reaktionsforløbet (f.eks. forbrænding med flammer fra overfladen, gennembrænding af hele materialet og oplysninger om forbrændingsprodukter) |
|
— |
alle yderligere bemærkninger, der er relevante for fortolkningen af resultaterne, herunder en beskrivelse af reaktionens voldsomhed (flamme, gnister, røg, ulmen, m.v.) og omtrentlige varighed i den indledende sikkerheds/screeningstest, både af test- og referencestoffet |
|
— |
resultaterne af test med et inert stof |
|
— |
eventuelle resultater af test i inert atmosfære. |
3.2. FORTOLKNING AF RESULTATERNE
Et stof skal anses for oxiderende, hvis
|
a) |
der sker voldsom reaktion under den indledende test |
|
b) |
den største reaktionshastighed af de blandinger, der er undersøgt ved fuldstændig testning, er højere end eller lige så høj som referenceblandingens (dvs. cellulose og bariumnitrat) største reaktionshastighed. |
For at undgå falske positive resultater skal de resultater, der opnås ved test af stoffet med et inert materiale og/eller i inert atmosfære, også tages i betragtning ved fortolkningen af resultaterne.
4. HENVISNINGER
NF T 20-035 (SEPT 85). Chemical products for industrial use. Determination of the oxidizing properties of solids.
TILLÆG
Figur
Form og tilbehor til fremstilling af provolden
(alle dimensioner i mm)

A.18. POLYMERERS ANTALSMIDDELMOLEKYLVÆGT OG MOLEKYLVÆGTSFORDEL ING
1. METODE
Denne gelpermeationskromatografiske metode er en gengivelse af OECD Test Guideline 118 (1996). Grundprincipper og yderligere tekniske oplysninger findes i henvisning (1).
1.1. INDLEDNING
Da polymerer kan have vidt forskellige egenskaber, er det umuligt at beskrive én metode med nøjagtig angivelse af alle betingelser for adskillelse og evaluering, som dækker alle tænkelige særtilfælde, der kan forekomme under adskillelse af polymerer. Især er komplekse polymersystemer ofte uegnede til gelpermeationskromatografi (GPC). Er det ikke praktisk muligt at foretage GPC, kan molekylvægten bestemmes ved andre metoder (se tillægget). I så fald skal der gives en begrundelse for valget af metode og en fuldstændig beskrivelse af den.
Den beskrevne metode er baseret på DIN-standard 55672 (1). Heri findes der detaljerede oplysninger om, hvordan forsøgene udføres, og hvordan dataene evalueres. Hvis det bliver nødvendigt at ændre på forsøgsbetingelserne, skal disse ændringer begrundes. Der kan benyttes andre standarder, hvis der gives nøjagtige henvisninger dertil. I den beskrevne metode benyttes der polystyrenprøver med kendt polydispersitet til kalibrering; for visse polymerer, f.eks. vandopløselige polymerer og langkædede forgrenede polymerer, kan det være nødvendigt at ændre på dette.
1.2. DEFINITIONER OG ENHEDER
Antalsmiddelmolekylvægten Mn og vægtmiddelmolekylvægten Mw bestemmes ved følgende udtryk:
|
|
|
hvor
Hi er detektorsignalets højde over basislinjen ved retentionsvolumen Vi
Mi er molekylvægten af polymerfraktionen ved retentionsvolumen Vi
n er antallet af datapunkter.
Bredden af molekylvægtsfordelingen, som er et mål for systemets dispersitet, er givet ved forholdet Mw/Mn.
1.3. REFERENCESTOFFER
Da GPC er en relativ metode, må der foretages en kalibrering. Hertil benyttes der normalt lineære polystyrenstandarder med kendte middelmolekylvægte Mn og Mw og kendt snæver molekylvægtsfordeling. Kalibreringskurven kan kun benyttes til bestemmelse af molekylvægten af den ukendte prøve, hvis der er valgt identiske betingelser for separation af prøven og standarden.
Den sammenhæng mellem molekylvægt og elueringsvolumen, der bestemmes, gælder kun for de særlige betingelser ved det bestemte forsøg. Disse betingelser er først og fremmest temperatur, opløsningsmiddel (eller opløsningsmiddelblanding), kromatograferingsbetingelser og adskillelseskolonne eller -kolonnesystem.
Den molekylvægt, der på denne måde bestemmes for prøven, er relativ og betegnes »polystyrenækvivalent molekylvægt«. Det betyder, at molekylvægten kan afvige mere eller mindre fra den absolutte værdi, afhængigt af strukturmæssige og kemiske forskelle mellem standard og prøve. Hvis der benyttes en anden standard, f.eks. polyethylenglycol, polyethylenoxid, polymethylmethacrylat eller polyacrylsyre, skal dette begrundes.
1.4. TESTMETODENS PRINCIP
Både molekylvægtfordelingen og middelmolekylvægten af prøven (både Mn og Mw) kan bestemmes ved GPC. GPC er en særlig type væskekromatografi, hvor prøvens bestanddele adskilles efter hydrodynamisk volumen (2).
Adskillelsen sker, mens prøven passerer gennem en kolonne med porøst materiale, typisk en organisk gel. Små molekyler kan trænge ind i porerne, mens store molekyler ikke kan. På denne måde får de store molekyler en kortere vejlængde, så de elueres først. Mellemstore molekyler trænger ind i nogle af porerne og elueres senere. De mindste molekyler, hvis hydrodynamiske radius er mindre end gelens porer, kan trænge ind i alle porer og elueres sidst.
I den ideelle situation er kun molekylernes størrelse bestemmende for adskillelsen, men i praksis er det umuligt helt at undgå interferens fra absorption. Uensartet kolonnepakning og dødvolumener kan forværre dette forhold (2).
Detektoren arbejder med f.eks. brydningsindeks eller UV-absorption og giver en simpel fordelingskurve. For at få hæftet faktiske molekylvægtsværdier på kurven, må kolonnen kalibreres ved at lede polymerer med kendt molekylvægt og helst stort set samme struktur, f.eks. forskellige polystyrenstandarder, gennem den. Resultatet er typisk en gauss-kurve, undertiden trukket ud i en lille hale i den lavmolekylære ende, hvor den lodrette akse viser mængden (i vægt) af de forskellige eluerede molekyler, og den vandrette akse viser logaritmen til molekylvægten.
1.5. KVALITETSKRITERIER
Repeterbarheden (den relative standardafvigelse) bør være mindre end 0,3 % for det eluerede volumen. Den krævede repeterbarhed for analysen skal sikres ved korrektion med intern standard, hvis et kromatogram evalueres tidsafhængigt og ikke opfylder ovennævnte kriterium (1). Polydispersiteten afhænger af standardernes molekylvægt. For polystyrenstandarder er følgende værdier typiske:
|
Mp < 2 000 |
Mw/Mn < 1,20 |
|
2 000 ≤ Mp ≤ 106 |
Mw/Mn < 1,05 |
|
Mp > 106 |
Mw/Mn < 1,20 |
(Mp er standardens molekylvægt ved toppens maksimum)
1.6. BESKRIVELSE AF TESTMETODEN
1.6.1. Fremstilling af standardopløsninger af polystyren
Polystyrenstandarderne opløses ved omhyggelig blanding med det valgte elueringsmiddel. Ved fremstillingen skal der nøje tages hensyn til producentens anvisninger.
Hvilken koncentration der vælges, afhænger af forskellige faktorer, f.eks. injektionsvolumen, opløsningens viskositet og detektorens følsomhed. Det maksimale injektionsvolumen skal tilpasses til kolonnens længde, så man undgår overbelastning. Typisk er injektionsvolumen for en GPC-kolonne på 30 cm × 7,8 mm normalt mellem 40 og 100 μl til analytisk separation. Større rumfang kan tillades, men det bør ikke overstige 250 μl. Det optimale forhold mellem injektionsvolumen og koncentration må fastlægges inden den egentlige kalibrering af kolonnen.
1.6.2. Fremstilling af testopløsning
I princippet gælder der samme krav ved fremstilling af testopløsningen. Prøven opløses i et egnet opløsningsmiddel, f.eks. tetrahydrofuran (THF), ved omhyggelig omrystning. Der må under ingen omstændigheder benyttes ultralydbad til opløsningen. Om nødvendigt kan testopløsningen renses på membranfilter med en porestørrelse mellem 0,2 og 2 μm.
I den endelige rapport skal det oplyses, om der er uopløste partikler til stede, idet det kan skyldes højmolekylære bestanddele. Der benyttes en egnet metode til at bestemme den procentvise vægtmængde af de uopløste partikler. Opløsningerne benyttes inden for et døgn.
1.6.3. Apparatur
|
— |
opløsningsmiddelbeholder |
|
— |
afgasningsapparat (hvis påkrævet) |
|
— |
pumpe |
|
— |
pulsdæmper (hvis påkrævet) |
|
— |
injektionssystem |
|
— |
kromatografikolonner |
|
— |
detektor |
|
— |
flowmeter (hvis påkrævet) |
|
— |
skriver/databehandlingsenhed |
|
— |
affaldsbeholder. |
Det skal sikres, at GPC-systemet er ufølsomt over for de anvendte opløsningsmidler (f.eks. ved brug af kapillarrør af stål til THF).
1.6.4. Injektions- og opløsningsmiddeltilførselssystem
Der sættes et veldefineret volumen af prøveopløsningen på kolonnen, enten med en autosampler eller manuelt i en skarpt afgrænset zone. For hurtig tilbagetrækning eller indtrykning af stemplet i injektionssprøjten ved manuel indsprøjtning kan medføre ændringer i den iagttagne molekylvægtsfordeling. Systemet for tilførsel af opløsningsmiddel skal så vidt muligt være pulsationsfrit, helst indeholde en pulsdæmper. Flowet skal være omkring 1 ml/min.
1.6.5. Kolonne
Afhængigt af prøven karakteriseres polymeren enten på en enkelt kolonne eller på flere serieforbundne kolonner. I handelen fås en række porøse kolonnematerialer med veldefinerede egenskaber (f.eks. porestørrelse og øvre og nedre grænse). Valget af separationsgel og kolonnelængde afhænger af prøvens egenskaber (hydrodynamisk volumen og molekylvægtsfordeling) og af de specifikke adskillelsesbetingelser såsom opløsningsmiddel, temperatur og flowhastighed (1) (2) (3).
1.6.6. Teoretiske bunde
Den kolonne eller kolonnekombination, der benyttes til adskillelse, skal karakteriseres ved antallet af teoretiske bunde. Ved brug af THF som opløsningsmiddel gøres dette ved, at man sætter en opløsning af ethylbenzen eller et andet passende ikke-polært opløst stof på en kolonne af kendt længde. Antallet af teoretiske bunde er givet ved følgende udtryk:
|
|
eller |
|
hvor
|
N |
= |
er antallet af teoretiske bunde |
|
Ve |
= |
er elueringsvolumenet ved toppens maksimum |
|
W |
= |
er toppens bredde ved basislinjen |
|
W1/2 |
= |
er toppens bredde i halv højde |
1.6.7. Adskillelseseffektiviteten
Ud over antallet af teoretiske bunde, som er en størrelse, der bestemmer båndbredden, spiller også adskillelseseffektiviteten en rolle, og den bestemmes ved kalibreringskurvens stejlhed. En kolonnes adskillelseseffektivitet er givet ved følgende sammenhæng:
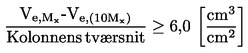
hvor
|
Ve, Mx |
= |
er elueringsvolumenet for polystyren med molekylvægten Mx |
|
Ve,(10.Mx) |
= |
er elueringsvolumenet for polystyren med en ti gange så høj molekylvægt |
Systemets adskillelsesevne defineres normalt som følger:

hvor
|
Ve1 og Ve2 |
= |
er elueringsvolumen for de to polystyrenstandarder ved toppenes maksimum |
|
W1 og W2 |
= |
er toppenes bredde ved basislinjen |
|
M1 og M2 |
= |
er molekylvægtene ved toppenes højde (normalt en forskel på en faktor 10). |
R-værdien for kolonnesystemet bør være større end 1,7 (4).
1.6.8. Opløsningsmidler
Alle opløsningsmidler skal være af høj renhed (for THF en renhedsgrad på 99,5 %). Opløsningsmiddelbeholderen (om nødvendigt med inert atmosfære) skal rumme tilstrækkelig meget til, at kolonnen kan kalibreres, og der kan analyseres flere prøver. Opløsningsmidlet skal afgasses, inden det pumpes videre til kolonnen.
1.6.9. Temperaturregulering
Temperaturen i de kritiske komponenter (injektionssløjfe, kolonner, detektor og rør og slanger) skal være konstant og passe til det valgte opløsningsmiddel.
1.6.10. Detektor
Detektoren har til formål at give et kvantitativt mål for koncentrationen af teststoffet i eluatet fra kolonnen. For at gøre den uundgåelige forbredning af toppene mindst mulig skal detektorcellens volumen være så lille som muligt. Det må ikke være større end 10 μl, undtagen for detektorer baseret på lysspredning og viskositet. Sædvanligvis benyttes der differentialrefraktometri til detektering. Hvis særlige egenskaber ved prøve eller elueringsmiddel kræver det, kan der benyttes andre detektortyper, f.eks. UV/VIS-, IR- eller viskositetsdetektor.
2. DATA OG RAPPORTERING
2.1. DATA
Der henvises til DIN-standarden (1) for så vidt angår detaljerede evalueringskriterier og krav til dataindsamling og -behandling.
Der udføres to uafhængige forsøg med hver prøve. De analyseres hver for sig.
Mn, Mw, Mw/Mn og Mp anføres for hver måling. Det skal udtrykkelig anføres, at de målte værdier er relative værdier svarende til den benyttede standards molekylvægt.
Efter at retentionsvolumen eller retentionstid (eventuelt korrigeret ved hjælp af intern standard) er bestemt, afbildes værdier af log Mp (Mp er maksimum for kalibreringsstandardens top) mod en af disse størrelser. Der kræves mindst to kalibreringspunkter i hver molekylvægtsdekade og mindst fem målepunkter for hele kurven; sidstnævnte bør spænde over prøvens formodede molekylvægt. Slutpunktet i kalibreringskurvens lavmolekylære ende fastlægges ved n-hexylbenzen eller et andet passende ikke-polært stof. Antals- og vægtmiddelmolekylvægten bestemmes i reglen ved elektronisk databehandling på grundlag af udtrykkene i punkt 1.2. Til brug for manuel behandling er der yderligere oplysninger i ASTM D 3536/91 (3).
Fordelingskurven skal forelægges i form af en tabel eller en figur (frekvens eller kumulativ sum mod log M). I en grafisk fremstilling skal en molekylær dekade normalt svare til ca. 4 cm i bredden, og den maksimale tophøjde skal være ca. 8 cm høj. I tilfælde af en integreret fordeling skal forskellen mellem 0 og 100 % på ordinaten være ca. 10 cm.
2.2. TESTRAPPORT
Testrapporten skal indeholde følgende oplysninger:
2.2.1. Teststof:
|
— |
foreliggende oplysninger om teststoffet (identitet, tilsætningsstoffer, urenheder) |
|
— |
beskrivelse af behandling af prøven, iagttagelser, problemer. |
2.2.2. Instrumenter:
|
— |
beholder til elueringsmiddel, inert gas, afgasning af elueringsmiddel, elueringsmidlets sammensætning, urenheder |
|
— |
pumpe, pulsdæmper, injektionssystem |
|
— |
separationskolonner (fabrikat, alle specifikationer for kolonnen, såsom porestørrelse og separationsmaterialets art, samt antal, længde og rækkefølge af de anvendte kolonner) |
|
— |
antal teoretiske bunde i kolonnen (eller kolonnekombinationen), adskillelseseffektivitet (systemets adskillelsesevne) |
|
— |
oplysninger om toppenes symmetri |
|
— |
kolonnetemperatur, temperaturregulering |
|
— |
detektor (måleprincip, type, cellevolumen) |
|
— |
eventuelt flowmeter (fabrikat, måleprincip) |
|
— |
dataopsamlings- og databehandlingssystem (hardware og software). |
2.2.3. Kalibrering af systemet:
|
— |
detaljeret beskrivelse af den metode, der er benyttet til fremstilling af kalibreringskurven |
|
— |
oplysninger om kvalitetskriterier for metoden (f.eks. korrelationskoefficient, sum af afvigelsers kvadrater) |
|
— |
oplysninger om alle ekstrapoleringer, forudsætninger og tilnærmelser, der er gjort under forsøget og dataevalueringen og -behandlingen |
|
— |
alle målinger, der er benyttet til fremstilling af kalibreringskurven, skal dokumenteres i en tabel, som indeholder følgende oplysninger om hvert enkelt kalibreringspunkt:
|
2.2.4. Evaluering:
|
— |
evaluering på tidsbasis: angivelse af, hvilke metoder der benyttes til at sikre den krævede reproducerbarhed (korrektionsmetode, intern standard, m.v.) |
|
— |
oplysninger om, om evalueringen er foretaget på grundlag af elueringsvolumen eller retentionstid |
|
— |
oplysninger om begrænsningerne for evalueringen, hvis en top ikke er fuldstændigt analyseret |
|
— |
beskrivelse af eventuelle udglatningsmetoder |
|
— |
procedurer for tilberedning og forbehandling af prøven |
|
— |
tilstedeværelse af eventuelle uopløste partikler |
|
— |
injektionsvolumen (μl) og injektionskoncentration (mg/ml) |
|
— |
iagttagelser, der tyder på afvigelser fra den ideelle GPC-profil |
|
— |
detaljeret beskrivelse af alle ændringer i testproceduren |
|
— |
detaljerede oplysninger om usikkerhedsintervaller |
|
— |
alle øvrige oplysninger og iagttagelser, der er relevante for fortolkningen af resultaterne. |
3. HENVISNINGER
|
(1) |
DIN 55672 (1995) Gelpermeationschromatographie (GPC) mit Tetrahydrofuran (THF) als Elutionsmittel, Teil 1. |
|
(2) |
Yau, W.W., Kirkland, J.J., and Bly, D.D. eds, (1979). Modern Size Exclusion Liquid Chromatography, J. Wiley and Sons. |
|
(3) |
ASTM D 3536-91, (1991). Standard Test Method for Molecular Weight Averages and Molecular Weight Distribution by Liquid Exclusion Chromatography (Gel Permeation Chromatography-GPC). American Society for Testing and Materials, Philadelphia, Pennsylvania. |
|
(4) |
ASTM D 5296-92, (1992). Standard Test Method for Molecular Weight Averages and Molecular Weight Distribution of Polystyrene by High Performance Size-Exclusion Chromatography. American Society for Testing and Materials, Philadelphia, Pennsylvania. |
Tillæg
Eksempler på andre metoder til bestemmelse af antalsmiddelmolekylvægt (Mn) for polymerer
Gelpermeationskromatografi er den foretrukne metode til bestemmelse af Mn, især når der er rådighed over et sæt standarder med tilnærmelsesvis samme struktur som den pågældende polymer. Hvis der er praktiske problemer med at udføre GPC, eller hvis der allerede er en formodning om, at stoffet ikke kan opfylde det fastsatte Mn-kriterium (og dette ønskes bekræftet), er der alternative metoder til rådighed, f.eks.:
1. Brug af kolligative egenskaber
|
1.1. |
Ebullioskopi/kryoskopi består i at måle et opløsningsmiddels kogepunktsforhøjelse (ebullioskopi) eller frysepunktssænkning (kryoskopi), når polymeren tilsættes. Metoden bygger på, at den opløste polymers indvirkning på væskens kogepunkt/frysepunkt afhænger af dens molekylvægt (1) (2). Anvendelsesområde: Mn < 20 000. |
|
1.2. |
Damptrykssænkning består i at måle damptrykket af en valgt referencevæske før og efter tilsætning af en kendt mængde polymer (1) (2). Anvendelsesområde: Mn < 20 000 (teoretisk; i praksis dog af begrænset værdi). |
|
1.3. |
Membranosmometri bygger på det osmotiske princip, dvs. opløsningsmiddelmolekylers naturlige tendens til at passere gennem en semipermeabel membran fra en tynd til en mere koncentreret opløsning for at etablere ligevægt. Ved testen har den tynde opløsning en koncentration på nul, mens den mere koncentrerede opløsning indeholder polymeren. Opløsningsmidlets bevægelse gennem membranen giver en trykforskel, der afhænger af polymerens koncentration og molekylvægt (1) (3) (4). Anvendelsesområde: Mn < 20 000-200 000. |
|
1.4. |
Vapour Phase Osmometry Gasfaseosmometri består i at sammenligne fordampningshastigheden i en ren opløsningsmiddelaerosol med mindst tre aerosoler, der indeholder den pågældende polymer i forskellige koncentrationer (1) (5) (6). Anvendelsesområde: Mn < 20 000. |
2. Endegruppeanalyse
For at kunne bruge denne metode skal man have kendskab både til polymerens overordnede struktur og til kædetermineringsgruppernes karakter (endegrupperne skal kunne skelnes fra hovedskelettet ved f.eks. NMR eller titrering/derivatisering). Ved bestemmelse af den molekylære koncentration af endegrupper i polymeren kan man nå frem til en værdi for molekylvægten (7) (8) (9).
Anvendelsesområde: Mn op til 50 000 (idet pålideligheden aftager).
3. Henvisninger
|
(1) |
Billmeyer, F.W. Jr., (1984). Textbook of Polymer Science, 3rd Edn., John Wiley, New York. |
|
(2) |
Glover, C.A., (1975). Absolute Colligative Property Methods. Chapter 4. In: Polymer Molecular Weights, Part I P.E. Slade, Jr. ed., Marcel Dekker, New York. |
|
(3) |
ASTM D 3750-79, (1979). Standard Practice for Determination of Number-Average Molecular Weight of Polymers by Membrane Osmometry. American Society for Testing and Materials, Philadelphia, Pennsylvania. |
|
(4) |
Coll, H. (1989). membrane Osmometry. In: Determination of Molecular Weight, A.R. Cooper ed., J. Wiley and Sons, pp. 25-52. |
|
(5) |
ASTM 3592-77, (1977). Standard Recommended Practice for Determination of Molecular Weight by Vapour Pressure, American Society for Testing and Materials, Philadelphia, Pennsylvania. |
|
(6) |
Morris, C.E.M., (1989). Vapour Pressure osmometry. In: Determinationn of Molecular Weight, A.R. Cooper ed., John Wiley and Sons. |
|
(7) |
Schröder, E., Müller, G., and Arndt, K-F., (1989). Polymer Characterisation, Carl Hanser Verlag, Munich. |
|
(8) |
Garmon, R.G., (1975). End-Group Determinations, Chapter 3 In: Polymer Molecular Weights, Part I, P.E. Slade, Jr. ed. Marcel Dekker, New York. |
|
(9) |
Amiya, S., et al. (1990). Pure and Applied Chemistry, 62, 2139-2146. |
A.19. POLYMERERS INDHOLD AF LAVMOLEKYLÆRE BESTANDDELE
1. METODE
Denne gelpermeationskromatografiske metode er en gengivelse af OECD Test Guideline 119 (1996). Grundprincipper og yderligere tekniske oplysninger findes i henvisningerne.
1.1. INDLEDNING
Da polymerer kan have vidt forskellige egenskaber, er det umuligt at beskrive én metode med nøjagtig angivelse af alle betingelser for adskillelse og evaluering, som dækker alle tænkelige særtilfælde, der kan forekomme under adskillelse af polymerer. Især er komplekse polymersystemer ofte uegnede til gelpermeationskromatografi (GPC). Er det ikke praktisk muligt at foretage GPC, kan molekylvægten bestemmes ved andre metoder (se tillægget). I så fald skal der gives en begrundelse for valget af metode og en fuldstændig beskrivelse af den.
Den beskrevne metode er baseret på DIN-standard 55672 (1). Heri findes der detaljerede oplysninger om, hvordan forsøgene udføres, og hvordan dataene evalueres. Hvis det bliver nødvendigt at ændre på forsøgsbetingelserne, skal disse ændringer begrundes. Der kan benyttes andre standarder, hvis der gives nøjagtige henvisninger dertil. I den beskrevne metode benyttes der polystrenprøver med kendt polydispersitet til kalibrering; for visse polymerer, f.eks. vandopløselige polymerer og langkædede forgrenede polymerer, kan det være nødvendigt at ændre på dette.
1.2. DEFINITIONER OG ENHEDER
Lavmolekylære bestanddele defineres arbitrært som bestanddele med en molekylvægt på mindre end 1 000 dalton.
Antalsmiddelmolekylvægten Mn og vægtmiddelmolekylvægten Mw bestemmes ved følgende udtryk:
|
|
|
hvor
|
HI |
= |
er detektorsignalets højde over basislinjen ved retentionsvolumen Vi |
|
Mi |
= |
er molekylvægten af polymerfraktionen ved retentionsvolumen Vi, og n er antallet af datapunkter |
Bredden af molekylvægtsfordelingen, som er et mål for systemets dispersitet, er givet ved forholdet Mw/Mn.
1.3. REFERENCESTOFFER
Da GPC er en relativ metode, må der foretages en kalibrering. Hertil benyttes der normalt lineære polystyrenstandarder med kendte middelmolekylvægte Mn og Mw og kendt snæver molekylvægtsfordeling. Kalibreringskurven kan kun benyttes til bestemmelse af molekylvægten af den ukendte prøve, hvis der er valgt identiske betingelser for separation af prøven og standarden.
Den sammenhæng mellem molekylvægt og elueringsvolumen, der bestemmes, gælder kun for de særlige betingelser ved det bestemte forsøg. Disse betingelser er først og fremmest temperatur, opløsningsmiddel (eller opløsningsmiddelblanding), kromatograferingsbetingelser og adskillelseskolonne eller -kolonnesystem.
Den molekylvægt, der på denne måde bestemmes for prøven, er relativ og betegnes »polystyrenækvivalent molekylvægt«. Det betyder, at molekylvægten kan afvige mere eller mindre fra den absolutte værdi, afhængigt af strukturmæssige og kemiske forskelle mellem standard og prøve. Hvis der benyttes en anden standard, f.eks. polyethylenglycol, polyethylenoxid, polymethylmethacrylat eller polyacrylsyre, skal dette begrundes.
1.4. TESTMETODENS PRINCIP
Både molekylvægtfordelingen og middelmolekylvægten af prøven (både Mw og Mn) kan bestemmes ved GPC. GPC er en særlig type væskekromatografi, hvor prøvens bestanddele adskilles efter hydrodynamisk volumen (2).
Adskillelsen sker, mens prøven passerer gennem en kolonne med porøst materiale, typisk en organisk gel. Små molekyler kan trænge ind i porerne, mens store molekyler ikke kan. På denne måde får de store molekyler en kortere vejlængde, så de elueres først. Mellemstore molekyler trænger ind i nogle af porerne og elueres senere. De mindste molekyler, hvis hydrodynamiske radius er mindre end gelens porer, kan trænge ind i alle porer og elueres sidst.
I den ideelle situation er kun molekylernes størrelse bestemmende for adskillelsen, men i praksis er det umuligt helt at undgå interferens fra absorption. Uensartet kolonnepakning og dødvolumener kan forværre dette forhold (2).
Detektoren arbejder med f.eks. brydningsindeks eller UV-absorption og giver en simpel fordelingskurve. For at få hæftet faktiske molekylvægtsværdier på kurven, må kolonnen kalibreres ved at lede polymerer med kendt molekylvægt og helst stort set samme struktur, f.eks. forskellige polystyrenstandarder, gennem den. Resultatet er typisk en gauss-kurve, undertiden trukket ud i en lille hale i den lavmolekylære ende, hvor den lodrette akse viser mængden (i vægt) af de forskellige eluerede molekyler, og den vandrette akse viser logaritmen til molekylvægten.
Af denne kurve udledes indholdet af lavmolekylære bestanddele. Beregningen er kun nøjagtig, hvis de lavmolekylære bestanddele modsvarer polymeren som helhed på massebasis.
1.5. KVALITETSKRITERIER
Repeterbarheden (den relative standardafvigelse) bør være mindre end 0,3 % for det eluerede volumen. Den krævede repeterbarhed for analysen skal sikres ved korrektion med intern standard, hvis et kromatogram evalueres tidsafhængigt og ikke opfylder ovennævnte kriterium (1). Polydispersiteten afhænger af standardernes molekylvægt. For polystyrenstandarder er følgende værdier typiske
|
Mp < 2 000 |
Mw/Mn < 1,20 |
|
2 000 ≤ Mp ≤ 106 |
Mw/Mn < 1,05 |
|
Mp > 106 |
Mw/Mn < 1,20 |
Mp et standardens molekylvægt ved toppens maksimum)
1.6. BESKRIVELSE AF TESTMETODEN
1.6.1. Fremstilling af standardopløsninger af polystyren
Polystyrenstandarderne opløses ved omhyggelig blanding med det valgte elueringsmiddel. Ved fremstillingen skal der nøje tages hensyn til producentens anvisninger.
Hvilken koncentration der vælges, afhænger af forskellige faktorer, f.eks. injektionsvolumen, opløsningens viskositet og detektorens følsomhed. Det maksimale injektionsvolumen skal tilpasses til kolonnens længde, så man undgår overbelastning. Typisk er injektionsvolumen for en GPC-kolonne på 30 cm × 7,8 mm normalt mellem 40 og 100 μl til analytisk separation. Større rumfang kan tillades, men det bør ikke overstige 250 μl. Det optimale forhold mellem injektionsvolumen og koncentration må fastlægges inden den egentlige kalibrering af kolonnen.
1.6.2. Fremstilling af testopløsning
I princippet gælder der samme krav ved fremstilling af testopløsningen. Prøven opløses i et egnet opløsningsmiddel, f.eks. tetrahydrofuran (THF) ved omhyggelig omrystning. Der må under ingen omstændigheder benyttes ultralydbad til opløsningen. Om nødvendigt kan testopløsningen renses på membranfilter med en porestørrelse mellem 0,2 og 2 μm.
I den endelige rapport skal det oplyses, om der er uopløste partikler til stede, idet det kan skyldes højmolekylære bestanddele. Der benyttes en egnet metode til at bestemme den procentvise vægtmængde af de uopløste partikler. Opløsningerne benyttes inden for et døgn.
1.6.3. Korrektion for indhold af urenheder og additiver
Det er normalt nødvendigt at korrigere indholdet af bestanddele med M < 1 000 for bidrag fra ikke-polymere bestanddele (f.eks. urenheder og/eller additiver), medmindre der allerede er målt et indhold på < 1 %. Dette gøres ved direkte analyse på polymeropløsningen eller GPC-eluatet.
Hvis eluatet efter passage af kolonnen er for fortyndet til yderligere analyse, må det koncentreres. Det kan være nødvendigt at inddampe eluatet til tørhed og genopløse det. Eluatet skal koncentreres på en sådan måde, at der med sikkerhed ikke sker ændringer i det. Behandlingen af eluatet efter GPC-trinnet vil afhænge af, hvilken analysemetode der anvendes til kvantitativ bestemmelse.
1.6.4. Apparatur
GPC-udstyr bestående af følgende komponenter:
|
— |
opløsningsmiddelbeholder |
|
— |
afgasningsapparat (hvis påkrævet) |
|
— |
pumpe |
|
— |
pulsdæmper (hvis påkrævet) |
|
— |
injektionssystem |
|
— |
kromatografikolonner |
|
— |
detektor |
|
— |
flowmeter (hvis påkrævet) |
|
— |
skriver/databehandlingsenhed |
|
— |
affaldsbeholder |
Det skal sikres, at GPC-systemet er ufølsomt over for de anvendte opløsningsmidler (f.eks. ved brug af kapillarrør af stål til THF).
1.6.5. Injektions- og opløsningsmiddeltilførselssystem
Der sættes et veldefineret volumen af prøveopløsningen på kolonnen, enten med en autosampler eller manuelt i en skarpt afgrænset zone. For hurtig tilbagetrækning eller indtrykning af stemplet i injektionssprøjten ved manuel indsprøjtning kan medføre ændringer i den iagttagne molekylvægtsfordeling. Systemet for tilførsel af opløsningsmiddel skal så vidt muligt være pulsationsfrit, helst indeholde en pulsdæmper. Flowet skal være omkring 1 ml/min.
1.6.6. Kolonne
Afhængigt af prøven karakteriseres polymeren enten på en enkelt kolonne eller på flere serieforbundne kolonner. I handelen fås en række porøse kolonnematerialer med veldefinerede egenskaber (f.eks. porestørrelse og øvre og nedre grænse). Valget af separationsgel og kolonnelængde afhænger af prøvens egenskaber (hydrodynamisk volumen og molekylvægtsfordeling) og af de specifikke adskillelsesbetingelser såsom opløsningsmiddel, temperatur og flowhastighed (1) (2) (3).
1.6.7. Teoretiske bunde
Den kolonne eller kolonnekombination, der benyttes til adskillelsen, skal karakteriseres ved antallet af teoretiske bunde. Ved brug af THF som opløsningsmiddel gøres dette ved, at man sætter en opløsning af ethylbenzen eller et anden passende ikke-polært opløst stof på en kolonne af kendt længde. Antallet af teoretiske bunde er givet ved følgende udtryk:
|
|
eller |
|
hvor,
|
N |
= |
er antallet af teoretiske bunde |
|
Ve |
= |
er elueringsvolumenet ved toppens maksimum |
|
W |
= |
er toppens bredde ved basislinjen |
|
W1/2 |
= |
er toppens bredde i halv højde |
1.6.8. Adskillelsereffektiviteten
Ud over antallet af teoretiske bunde, som er en størrelse, der bestemmer båndbredden, spiller også adskillelseseffektiviteten en rolle, og den bestemmes ved kalibreringskurvens stejlhed. En kolonnes adskillelseseffektivitet er givet ved følgende sammenhæng:

hvor
|
Ve, Mx |
= |
er elueringsvolumenet for polystyren med molekylvægten M x |
|
Ve,(10.Mx) |
= |
er elueringsvolumenet for polystyren med en ti gange så høj molekylvægt |
Systemets adskillelsesevne defineres normalt som følger:

hvor
|
Ve1 og Ve2 |
= |
er elueringsvolumen for de to polystyrenstandarder ved toppenes maksimum |
|
W1 og W2 |
= |
er toppenes bredde ved basislinjen |
|
M1 og M2 |
= |
er molekylvægtene ved toppenes højde (normalt en forskel på en faktor 10). |
R-værdien for kolonnesystemet bør være større end 1,7 (4).
1.6.9. Opløsningsmidler
Alle opløsningsmidler skal være af høj renhed (for THF en renhedsgrad på 99,5 %). Opløsningsmiddelbeholderen (om nødvendigt med inert atmosfære) skal rumme tilstrækkelig meget til, at kolonnen kan kalibreres, og der kan analyseres flere prøver. Opløsningsmidlet skal afgasses, inden det pumpes videre til kolonnen.
1.6.10. Temperaturregulering
Temperaturen i de kritiske komponenter (injektionssløjfe, kolonner, detektor og rør og slanger) skal være konstant og passe til det valgte opløsningsmiddel.
1.6.11. Detektor
Detektoren har til formål at give et kvantitativt mål for koncentrationen af teststoffet i eluatet fra kolonnen. For at gøre den uundgåelige forbredning af toppene mindst mulig skal detektorcellens volumen være så lille som muligt. Det må ikke være større end 10 μl, undtagen for detektorer baseret på lysspredning og viskositet. Sædvanligvis benyttes der differentialrefraktometri til detektering. Hvis særlige egenskaber ved prøve eller elueringsmiddel kræver det, kan der benyttes andre detektortyper, f.eks. UV/VIS-, IR-eller viskositetsdetektor.
2. DATA OG RAPPORTERING
2.1. DATA
Der henvises til DIN-standarden (1) for så vidt angår detaljerede evalueringskriterier og krav til dataindsamling og -behandling.
Der udføres to uafhængige forsøg med hver prøve. De analyseres hver for sig. Det er i alle tilfælde vigtigt også at bestemme data for blindprøver, der er behandlet på samme måde som prøven.
Det skal udtrykkelig anføres, at de målte værdier er relative værdier svarende til den benyttede standards molekylvægt.
Efter at retentionsvolumen eller retentionstid (eventuelt korrigeret ved hjælp af intern standard) er bestemt, afbildes værdier af log Mp (Mp er maksimum for kalibreringsstandardens top) mod en af disse størrelser. Der kræves mindst to kalibreringspunkter i hver molekylvægtsdekade og mindst fem målepunkter for hele kurven; sidstnævnte bør spænde over prøvens formodede molekylvægt. Slutpunktet i kalibreringskurvens lavmolekylære ende fastlægges ved n-hexylbenzen eller et andet passende ikke-polært stof. Den del af kurven, der svarer til molekylvægte under 1 000, bestemmes og korrigeres efter behov for urenheder og additiver. Elueringskurverne evalueres i reglen ved elektronisk databehandling. Til brug for manuel behandling er der yderligere oplysninger i ASTM D 3536-91 (3).
Uopløselig polymer, der måtte være tilbageholdt på kolonnen, vil efter al sandsynlighed have en molekylvægt, der er større end den opløselige fraktions, så hvis den lades ude af betragtning, vil indholdet af lavmolekylære bestanddele blive anslået for højt. I tillægget er der en vejledning i, hvordan indholdet af lavmolekylære bestanddele korrigeres for uopløselig polymer.
Fordelingskurven skal forelægges i form af en tabel eller en figur (frekvens eller kumulativ sum mod log M). I en grafisk fremstilling skal en molekylær dekade normalt svare til ca. 4 cm i bredden, og den maksimale tophøjde skal være ca. 8 cm høj. I tilfælde af en integreret fordeling skal forskellen mellem 0 og 100 % på ordinaten være ca. 10 cm.
2.2. TESTRAPPORT
Testrapporten skal indeholde følgende oplysninger:
2.2.1. Teststof:
|
— |
Foreliggende oplysninger om teststoffet (identitet, tilsætningsstoffer, urenheder) |
|
— |
Beskrivelse af behandling af prøven, iagttagelser, problemer. |
2.2.2. Instrumenter:
|
— |
Beholder til elueringsmiddel, inert gas, afgasning af elueringsmiddel, elueringsmidlets sammensætning, urenheder |
|
— |
Pumpe, pulsdæmper, injektionssystem |
|
— |
Separationskolonner (fabrikat, alle specifikationer for kolonnen, såsom porestørrelse og separationsmaterialets art, samt antal, længde og rækkefølge af de anvendte kolonner) |
|
— |
Antal teoretiske bunde i kolonnen (eller kolonnekombinationen), adskillelseseffektivitet (systemets adskillelsesevne) |
|
— |
Oplysninger om toppenes symmetri |
|
— |
Kolonnetemperatur, temperaturregulering |
|
— |
Detektor (måleprincip, type, cellevolumen) |
|
— |
Eventuelt flowmeter (fabrikat, måleprincip) |
|
— |
Dataopsamlings- og databehandlingssystem (hardware og software). |
2.2.3. Kalibrering af systemet:
|
— |
Detaljeret beskrivelse af den metode, der er benyttet til fremstilling af kalibreringskurven |
|
— |
Oplysninger om kvalitetskriterier for metoden (f.eks. korrelationskoefficient, sum af afvigelsers kvadrater |
|
— |
Oplysninger om alle ekstrapoleringer, forudsætninger og tilnærmelser, der er gjort under forsøget og dataevalueringen og -behandlingen |
|
— |
Alle målinger, der er benyttet til fremstilling af kalibreringskurven, skal dokumenteres i en tabel, som indeholder følgende oplysninger om hvert enkelt kalibreringspunkt:
|
2.2.4. Oplysninger om indholdet af lavmolekylære bestanddele:
|
— |
beskrivelse af de metoder, der er benyttet til analysen, og hvordan forsøgene er udført |
|
— |
oplysninger om det procentvise indhold (w/w) af lavmolekylære bestanddele i forhold til hele prøven |
|
— |
oplysninger om urenheder, additiver og andre ikke-polymere bestanddele i vægtprocent i forhold til hele prøven |
2.2.5. Evaluering:
|
— |
Evaluering på tidsbasis: angivelse af, hvilke metoder der benyttes til at sikre den krævede reproducerbarhed (korrektionsmetode, intern standard, m.v.) |
|
— |
Oplysninger om, om evalueringen er foretaget på grundlag af elueringsvolumen eller retentionstid |
|
— |
Oplysninger om begrænsningerne for evalueringen, hvis en top ikke er fuldstændigt analyseret |
|
— |
Beskrivelse af eventuelle udglatningsmetoder |
|
— |
Procedurer for tilberedning og forbehandling af prøven |
|
— |
Tilstedeværelse af eventuelle uopløste partikler |
|
— |
Injektionsvolumen (μl) og injektionskoncentration (mg/ml) |
|
— |
Iagttagelser, der tyder på afvigelser fra den ideelle GPC-profil |
|
— |
Detaljeret beskrivelse af alle ændringer i testproceduren |
|
— |
Detaljerede oplysninger om usikkerhedsintervaller |
|
— |
Alle øvrige oplysninger og iagttagelser, der er relevante for fortolkningen af resultaterne. |
3. HENVISNINGER
|
(1) |
DIN 55672 (1995) Gelpermeationschromatographie (GPC) mit Tetrahydrofuran (THF) als Elutionsmittel, Teil 1. |
|
(2) |
Yau, W.W., Kirkland, J.J., and Bly, D.D. eds. (1979). Modern Size Exclusion Liquid Chromatography, J. Wiley and Sons. |
|
(3) |
ASTM D 3536-91, (1991). Standard Test method for Molecular Weight Averages and Molecular Weight Distribution by Liquid Exclusion Chromatography (Gel Permeation Chromatography-GPC). American Society for Testing and Materials, Philadelphia, Pennsylvania. |
|
(4) |
ASTM D 5296-92, (1992). Standard Test method for Molecular Weight Averages and Molecular Weight Distribution of Polystyrene by High Performance Size-Exclusion Chromatography. American Society for Testing and Materials, Philadelphia, Pennsylvania. |
Tillæg
Vejledning i, hvordan indholdet af lavmolekylære bestanddele korrigeres for uopløselig polymer
Indhold af uopløselig polymer i prøven vil medføre et vist massetab under GPC-analysen. Den uopløselige polymer tilbageholdes irreversibelt på kolonnen eller prøvefiltret, mens den opløselige del af prøven passerer gennem kolonnen. Hvis tilvæksten i polymerens brydningsindeks (dn/dc) kan skønnes eller måles, kan man få et skøn over, hvor stor en masse der er gået tabt på kolonnen. I så fald foretages korrektionen ved hjælp af en ekstern kalibrering med standardmaterialer med kendt koncentration og dn/dc, så refraktometerets udslag kalibreres. I nedenstående eksempel er der benyttet en standard af poly (methylmethacrylat) (pMMA).
Ved ekstern kalibrering med henblik på analyse af acrylpolymere analyseres en pMMA-standard med kendt koncentration i tetrahydrofuran ved GPC, og de fremkomme data benyttes til bestemmelse af refraktometerkonstanten efter følgende udtryk:
K = R/(C × V × dn/dc)
hvor
|
K |
= |
er refraktometerkonstanten (i mikrovolt-sekund/ml) |
|
R |
= |
er pMMA-standardens udslag (mikrovolt-sekund) |
|
C |
= |
er pMMA-standardens koncentration (i mg/ml) |
|
V |
= |
er injektionsvolumenet (i ml), og |
|
dn/dc |
= |
er brydningsindekstilvæksten for pMMA i tetrahydrofuran (i ml/mg) |
Følgende data er typiske for en pMMA-standard:
|
R |
= |
2 937 891 |
|
C |
= |
1,07 mg/ml |
|
V |
= |
0,1 ml |
|
dn/dc |
= |
9 × 10-5 ml/mg |
Den fremkomne K-værdi på 3,05 × 1011 benyttes derefter til beregning af det teoretiske detektorudslag, hvis 100 % af polymeren elueres gennem detektoren.
A.20. POLYMERERS OPLØSELIGHED/EKSTRAHERBARHED I VAND
1. METODE
Den beskrevne metode er en gengivelse af OECD Test Guideline 120 (1997). Yderligere tekniske oplysninger findes i henvisning (1).
1.1. INDLEDNING
For visse polymerer, f.eks. emulsionspolymerer, kan det være nødvendigt at gøre yderligere forberedelser, inden den nedenfor beskrevne metode kan benyttes. Metoden kan ikke anvendes for flydende polymerer og polymerer, der kan reagere med vand under testbetingelserne.
Når metoden ikke er bekvem eller er umulig at benytte, må opløseligheden/ekstraherbarheden undersøges ved andre metoder. I så fald skal gives alle detaljer og en begrundelse for valget af metode.
1.2. REFERENCESTOFFER
Ingen.
1.3. TESTMETODENS PRINCIP
Polymerers opløselighed/ekstraherbarhed i vandigt medium bestemmes ved hjælp af kolbemetoden (jf. A.6 Vandopløselighed — kolbemetoden) med de i det følgende beskrevne ændringer.
1.4. KVALITETSKRITERIER
Ingen.
1.5. BESKRIVELSE AF TESTMETODEN
1.5.1. Udstyr
Der er brug for følgende udstyr til metoden:
|
— |
findelingsapparat, f.eks. mølle, til fremstilling af partikler med kendt størrelse |
|
— |
rysteapparat med mulighed for temperaturregulering |
|
— |
membranfilter |
|
— |
egnet analyseudstyr |
|
— |
standardsigter |
1.5.2. Prøveforberedelse
En repræsentativ prøve skal først findeles til en partikelstørrelse mellem 0,125 og 0,25 mm bestemt med passende sigter. Af hensyn til prøvens stabilitet, kan køling være påkrævet under formalingen. Materialer af gummiagtig karakter kan knuses ved flydende nitrogens temperatur (1).
Hvis det ikke er muligt at nå ned på den krævede partikelstørrelse, skal der tilstræbes den mindst mulige partikelstørrelse, og det opnåede resultat anføres. Det oplyses i rapporten, hvordan den findelte prøve har været opbevaret indtil udførelse af testen.
1.5.3. Fremgangsmåde
Der afvejes 3 prøver på hver 10 g af teststoffet i 3 glaskolber med glasprop, og der tilsættes 1 000 ml vand til hver kolbe. Hvis det ikke er praktisk muligt at håndtere 10 g af polymeren, anvendes den største mængde, der kan håndteres, og vandmængden reduceres tilsvarende.
Kolberne lukkes tæt til og omrystes ved 20 oC. Der benyttes en anordning, der kan arbejde ved konstant temperatur. Efter 24 timer centrifugeres eller filtreres indholdet af hver enkelt kolbe, og polymerkoncentrationen i den klare vandfase bestemmes med en passende analysemetode. Findes der ingen passende analysemetoder for vandfasen, kan den samlede opløselighed/ekstraherbarhed skønnes ud fra den tørre vægt af filterkagen eller bundfaldet fra centrifugeringen.
Normalt er det nødvendigt at skelne kvantitativt mellem urenheder og additiver på den ene side og lavmolekylære forbindelser på den anden side. Ved gravimetrisk bestemmelse er det ligeledes vigtigt at medtage en blindprøve uden teststof, så der tages højde for restmateriale hidrørende fra forsøgets udførelse.
Polymerers opløselighed/ekstraherbarhed i vand ved 37 oC ved pH 2 og pH 9 kan bestemmes som beskrevet ved udførelse af eksperimentet ved 20 oC. pH indstilles ved hjælp af egnede bufferopløsninger, syrer eller baser, f.eks. saltsyre, eddikesyre, natrium- eller kaliumhydroxid af analysekvalitet eller ammoniak.
Der udføres en eller to prøver, afhængigt af den benyttede analysemetode. Når der findes tilstrækkelig specifikke metoder til direkte analyse af vandfasen for polymerkomponenten, skulle én prøve som ovenfor beskrevet være tilstrækkeligt. Hvis der derimod ikke findes sådanne metoder og polymerens opløselighed/ekstraherbarhed må bestemmes indirekte ved analyse af det vandige ekstrakts indhold af organisk kulstof i alt (TOC), bør der udføres endnu en prøve. Prøven bør gentages endnu en gang, denne gang med en ti gange mindre polymermængde men samme vandmængde som ved den første prøve.
1.5.4. Analyse
1.5.4.1. Test udført med kun én prøvestørrelse
I nogle tilfælde findes der metoder til direkte analyse af polymerkomponenter i vandfasen. Alternativt kan man overveje indirekte at analysere for opløste/ekstraherede polymerkomponenter ved at bestemme den samlede mængde opløste bestanddele og korrigere for ikke-polymerspecifikke komponenter.
Analyse af vandfasen for samtlige polymerforbindelser kan foretages ved
enten en tilstrækkelig følsom metode, f.eks.
|
— |
TOC-bestemmelse ved nedbrydning til CO2 med persulfat eller dichromat efterfulgt af måling ved IR eller kemisk analyse |
|
— |
atomabsorptionsspektrometri (AAS) eller induktivt koblet plasmaemissionsspektrometri (ICP) for silicium- eller metalholdige polymerer |
|
— |
UV-absorption ellerr spektrofluorimetri for arylpolymerer |
|
— |
LC-MS for lavmolekylære prøver |
eller ved vakuuminddampning af det vandige ekstrakt til tørhed og analyse af remanensen ved spektroskopi (IR, UV m.v.) eller AAS/ICP.
Hvis det ikke er praktisk muligt at gennemføre en analyse af selve vandfasen, ekstraheres vandekstraktet med et organisk opløsningsmiddel, der ikke er blandbart med vand, f.eks. en chloreret kulbrinte. Derefter afdampes opløsningsmidlet, og remanensen analyseres som ovenfor for indhold af den pågældende polymer. Bestanddele i denne remanens, som identificeres som urenheder eller additiver, fratrækkes, når selve polymerens opløselighed/ekstraherbarhed beregnes.
Når der er forholdsvis store stofmængder til stede, kan det være nødvendigt at foretage f.eks. HPLC- eller GC-analyse af remanensen for at skelne urenheder fra monomere og monomerderivater, så det virkelige indhold af sidstnævnte kan bestemmes.
I nogle tilfælde vil simpel afdampning af det organiske opløsningsmiddel og vejning af den tørre remanens være tilstrækkeligt.
1.5.4.2. Test udført med to forskellige prøvestørrelser
Alle vandige ekstrakter analyseres for TOC.
Der foretages gravimetrisk bestemmelse af den del af prøven, der ikke er gået i opløsning. Hvis der efter centrifugering eller filtrering af de enkelte kolbers indhold stadig sidder polymerrester fast på kolbens væg, skylles kolben med filtratet, indtil der ikke længere er synlige rester i kolben. Derefter centrifugeres eller filtreres filtratet igen. Filterkagen eller bundfaldet fra centrifugeringen tørres ved 40 oC under vakuum og vejes. Tørringen fortsættes til konstant vægt.
2. DATA
2.1. TEST UDFØRT MED KUN ÉN PRØVESTØRRELSE
Resultaterne for hver enkelt kolbe og gennemsnittet for de 3 kolber anføres i masseenheder pr. volumen opløsning (typisk mg/l) eller masse pr. masse polymerprøve (typisk mg/g). Desuden anføres prøvens vægttab (beregnet som vægten af det opløste stof divideret med prøvens begyndelsesvægt). De relative standardafvigelser (RSD) beregnes. Der anføres enkelttal for den samlede stofmængde (polymer + nødvendige additiver) og for polymer alene (dvs. efter subtraktion af bidraget fra disse additiver).
2.2. TEST UDFØRT MED TO FORSKELLIGE PRØVESTØRRELSER
De enkelte TOC-værdier for de vandige ekstrakter fra de to tredobbeltforsøg og gennemsnitsværdien for hvert forsøg anføres i masseenheder pr. volumen opløsning (typisk mg C/l) eller masse pr. masse polymerprøve (typisk mg C/g).
Ingen forskel mellem resultaterne for højt og lavt forhold mellem prøve og vand er tegn på, at alle ekstraherbare komponenter faktisk er ekstraheret. I så fald er direkte analyse normalt ikke påkrævet.
Vægten af hver enkelt remanens anføres, også i procent af den oprindelige prøve. Der beregnes et gennemsnit for hvert forsøg. Det procentvise indhold af opløselige og ekstraherbare stoffer i den oprindelige prøve er givet ved forskellen mellem 100 og de fundne procenttal.
3. RAPPORTERING
3.1. TESTRAPPORT
Testrapporten skal indeholde følgende oplysninger:
3.1.1. Teststof:
|
— |
Foreliggende oplysninger om teststoffet (identitet, tilsætningsstoffer, urenheder, indhold af lavmolekylære bestanddele). |
3.1.2. Forsøgsbetingelser:
|
— |
beskrivelse af den anvendte fremgangsmåde og forsøgsbetingelserne |
|
— |
beskrivelse af analyse- og detekteringsmetoder. |
3.1.3. Resultater:
|
— |
resultaterne for opløselighed/ekstraherbarhed i mg/l; enkeltværdier og gennemsnit for ekstraktionsforsøgene i de forskellige opløsninger, opdelt på polymer og urenheder, additiver m.v. |
|
— |
resultaterne for opløselighed/ekstraherbarhed i mg/g polymer |
|
— |
TOC-værdier for vandige ekstrakter, vægt af opløst stof og beregnet procentværdi, hvis målt |
|
— |
pH af hver enkelt prøve |
|
— |
oplysninger om blindprøver |
|
— |
oplysninger om teststoffets eventuelle kemiske ustabilitet både under testproceduren og under analyseringen |
|
— |
alle oplysninger, der har betydning for fortolkning af resultaterne. |
4. HENVISNINGER
DIN 53733 (1976) Zerkleinerung von Kunststofferzeugnissen fur Prufzwecke.
A.21. OXIDERENDE EGENSKABER (VÆSKER)
1. METODE
1.1. INDLEDNING
Formålet med denne metode er måling af væskers potentiale for forøgelse af brændbare stoffers forbrændingshastighed eller -intensitet, eller deres potentiale for sammen med et brændbart stof at danne en selvantændelig blanding, når de to stoffer blandes grundigt. Metoden bygger på FN's prøve for oxiderende væsker (1) og er ækvivalent med denne. Da nærværende metode A.21 imidlertid primært er bestemt til at opfylde kravene i forordning (EF) nr. 1907/2006, kræves kun sammenligning med ét referencestof. Forventes prøvningsresultaterne anvendt til andre formål, kan sammenligning med flere referencestoffer dog være nødvendig (9).
Denne prøve kræves ikke udført, når det af strukturformlen uden rimelig tvivl fremgår, at stoffet er ude af stand til at reagere eksotermt med et brændbart materiale.
Før prøven udføres, vil det være nyttigt at råde over foreløbige oplysninger om stoffets eventuelle potentielle eksplosionsfarlighed.
Denne prøve finder ikke anvendelse på faste stoffer, gasser, eksplosive eller letantændelige stoffer samt organiske peroxider.
Denne prøve kræves ikke udført, når prøvningsresultater for prøvestoffet foreligger fra FN-prøven for oxiderende væsker (1).
1.2. DEFINITIONER OG ENHEDER
Den gennemsnitlige trykstigningstid er gennemsnitsværdien af de ved prøvningen foretagne målinger af den tid, det tager prøveblandingen at frembringe en trykstigning fra 690 kPa til 2 070 kPa over atmosfære-trykket.
1.3. REFERENCESTOF
Som referencestof anvendes 65 % (w/w) salpetersyre (analytisk kvalitet) (10).
Hvis forsøgslederen forventer, at prøveresultaterne senere kan tænkes anvendt til andre formål (9), kan også andre referencestoffer om ønsket afprøves (11).
1.4. PRØVNINGSMETODENS PRINCIP
Prøvevæsken blandes med fibercellulose i masseforholdet 1:1 og overføres til en trykbeholder. Forekommer der under blanding eller overføring selvantændelse, er yderligere prøvning unødvendig.
Hvis selvantændelse ikke opstår, gennemføres hele prøven. Blandingen opvarmes i en trykbeholder, og gennemsnitstiden for en trykstigning fra 690 kPa til 2 070 kPa over atmosfæretrykket beregnes. Resultatet sammenholdes med den gennemsnitlige trykstigningstid for blandingen af referencestof og cellulose i forholdet 1:1.
1.5. KVALITETSKRITERIER
I en serie bestående af 5 forsøg på samme stof må ingen resultater afvige mere end 30 % fra det aritmetiske gennemsnit. Resultater, som afviger over 30 % fra gennemsnittet, skal kasseres, blandings- og påfyldningsproceduren forbedres, og prøvningen gentages.
1.6. BESKRIVELSE AF METODEN
1.6.1. Forberedelse
1.6.1.1. Brændbart stof
Tørret fibercellulose med fiberlængde mellem 50 og 250 μm og gennemsnitsdiameter 25 μm anvendes som brændbart stof (12). Dette tørres til konstant vægt i et højst 25 mm tykt lag ved 105 oC i 4 timer og opbevares i exsikkator med tørremiddel, indtil det er afkølet og klart til brug. Vandindholdet i den tørrede cellulose skal være under 0,5 % på tør massebasis (13). Om nødvendigt forlænges tørretiden for at opnå dette (14). Der skal anvendes cellulose fra samme batch under hele prøven.
1.6.1.2. Apparatur
1.6.1.2.1.
Prøvningen skal udføres med en trykbeholder. Beholderen er en cylindrisk trykbeholder af stål med længde 89 mm og Ø udvendig 60 mm (se figur 1). To modstående flader af beholderen er bearbejdet (således at beholderens tværsnit er reduceret til 50 mm) for at lette fastholdelsen under montering af tændrør og udluftnings-prop. Beholderen, hvis boring er Ø 20 mm, er indvendigt falset i hver ende til en dybde af 19 mm og forsynet med gevind til isætning af 1'' britisk standardrør (BSP)eller tilsvarende metrisk størrelse. Et trykudtag i form af et siderør er skruet ind i beholderens hvælvede overflade 35 mm fra den ene ende og i en vinkel på 90o med de bearbejdede overflader. Til montering af siderøret er udført en 12 mm dyb boring med gevind til ½'' BSP (eller tilsvarende metrisk størrelse) på enden af siderøret. Om nødvendigt anvendes en pakning af inaktivt materiale for at sikre gastæt samling. Grenrøret rager 55 mm uden for trykbeholderens hus og har en boring på 6 mm. Enden af siderøret er falset og forsynet med gevind for tilslutning af tryk-transducer af membrantypen. Enhver trykmåleanordning kan anvendes, forudsat at den ikke påvirkes af de varme gasser eller nedbrydningsprodukter og reagerer på en trykstigning på 690-2 070 kPa inden for højst 5 ms.
Den ende af trykbeholderen, som er længst fra siderøret, lukkes med et tændrør, som er monteret med to elektroder, hvoraf den ene er isoleret fra rørets hus, og den anden er jordforbundet til huset. Den modsatte ende af trykbeholderen lukkes med en sprængskive (sprængningstryk ca. 2 200 kPa), som fastholdes af en låseprop med boring Ø 20 mm. Om nødvendigt anvendes til tændrøret en pakning af inaktivt materiale for at sikre gastæt samling. Et stativ (figur 2) holder enheden i korrekt stilling under brug. Stativet består sædvanligvis af en fod af blødt stål med målene 235 mm × 184 mm × 6 mm og et 185 mm langt stykke firkantet hulprofil 70 mm × 70 mm × 4 mm.
I den ene ende af hulprofilen bortskæres en del af hver af to modstående sider, således at man opnår en struktur, som har to ben med flade sider og opadtil fortsætter i et 86 mm langt stykke intakt kasseprofil. Enderne af disse flade sider skæres af i en vinkel på 60o med det vandrette plan og svejses fast på fodpladen. I den ene side af fodstykkets øverste ende foretages en 22 mm bred og 46 mm dyb udfræsning, som giver plads til siderøret, når trykbeholderen med tændrøret først sænkes ned i kasseprofilstativet. Forneden på kasseprofilens indvendige overflade fastsvejses et stykke stål med bredde 30 mm og tykkelse 6 mm, der fungerer som afstandsstykke. Til fastholdelse af trykbeholderen anvendes to 7 mm fingerskruer, som sidder i skruehuller i den modstående flade. Nedefra støttes trykbeholderen af to 12 mm brede bånd af 6 mm tykt stål, fastsvejst på sidestykkerne, som ligger op ad kasseprofilens nederste ende.
1.6.1.2.2.
Tændsystemet består af en 25 cm lang Ni/Cr ledning Ø 0,6 mm med en modstand på 3,85 ohm/m. Ledningen er spiralviklet ved hjælp af en stang Ø 5 mm og fastgjort til tændrørets elektroder. Spiralen skal være udformet på en af de i figur 3 viste måder. Afstanden fra beholderens bund til tændspolens underside skal være 20 mm. Er elektroderne ikke justerbare, skal enderne af tændtråden mellem spolen og beholderens underside være isoleret med en keramisk foring. Ledningen opvarmes af en konstant strømforsyning, som kan yde mindst 10 A.
1.6.2. Prøvens udførelse (15)
Apparatet, komplet med tryktransducer og opvarmningssystem, men uden sprængskive, anbringes under-støttet med tændrøret nedad. I et bægerglas blandes 2,5 g af prøvevæsken med 2,5 g tørret cellulose ved hjælp af en glasspatel (16). Mens blandingen finder sted, skal der være anbragt sikkerhedsskærm mellem operatøren og blandingen. Hvis der under blanding eller overføring sker selvantændelse, er yderligere prøvning unødvendig. Blandingen påfyldes trykbeholderen i små portioner ved at banke let, idet man sørger for at blandingen pakkes tæt omkring tændspolen og har god kontakt til denne. Det er vigtigt at spolen ikke forvrides under denne pakkeproces, da prøvningen ellers kan give forkerte resultater (17). Sprængskiven sættes på plads, og låseproppen skrues forsvarligt fast. Den fyldte beholder overføres til støttefoden med sprængskiven opad; støttefoden skal anbringes i et passende armeret stinkskab eller en brandcelle. Strømforsyningen tilsluttes tændrørets udvendige klemmer, og der påføres 10 A. Tiden fra sammenblandingen påbegyndes til strømmen sluttes, må ikke overstige 10 minutter.
Tryktransducerens signal registreres på et passende system, hvormed den opnåede trykprofil både kan vurderes og optegnes permanent (f.eks. en transientoptager koblet til en punktskriver). Blandingen opvarmes, indtil sprængskiven brydes, eller til der er gået mindst 60 s. Hvis sprængskiven ikke brydes, lader man blandingen afkøle, før apparatet forsigtigt adskilles, efter at der er truffet forholdsregler over for eventuelt overtryk. Der udføres 5 prøver med prøvestof og referencestof(fer). Den tid, det tager trykket at stige fra 690 kPa til 2 070 kPa over atmosfæretrykket, noteres. Den gennemsnitlige trykstigningstid beregnes.
I visse tilfælde kan et stof frembringe en trykstigning (for høj eller for lav), som skyldes kemiske reaktioner, der ikke karakteriserer stoffets oxiderende egenskaber. I så fald kan det være nødvendigt at gentage prøven med et inaktivt stof, f.eks. diatoméjord (kiselgur), i stedet for cellulose for at klarlægge reaktionens art.
2. DATA
Trykstigningstider for både prøvestof og referencestof(fer). Trykstigningstider for eventuelle prøver udført med inaktivt stof.
2.1. BEHANDLING AF RESULTATER
De gennemsnitlige trykstigningstider for prøvestof og referencestof(fer) beregnes.
Den gennemsnitlige trykstigningstid for eventuelle prøver udført med inaktivt stof beregnes.
I tabel 1 er givet nogle eksempler på resultater.
Tabel 1
Eksempler på resultater (18)
|
Stof (19) |
Gennemsnitlig trykstigningstid for en 1:1 blanding med cellulose (ms) |
|
Ammoniumdichromat, mættet vandig opløsning |
20 800 |
|
Calciumnitrat, mættet vandig opløsning |
6 700 |
|
Ferrinitrat, mættet vandig opløsning |
4 133 |
|
Lithiumperchlorat, mættet vandig opløsning |
1 686 |
|
Magnesiumperchlorat, mættet vandig opløsning |
777 |
|
Nikkelnitrat, mættet vandig opløsning |
6 250 |
|
Salpetersyre, 65 % |
4 767 (20) |
|
Perchlorsyre, 50 % |
121 (20) |
|
Perchlorsyre, 55 % |
59 |
|
Kaliumnitrat, 30 % vandig opløsning |
26 690 |
|
Sølvnitrat, mættet vandig opløsning |
|
|
Natriumchlorat, 40 % vandig opløsning |
2 555 (20) |
|
Natriumnitrat, 45 % vandig opløsning |
4 133 |
|
Inaktivt stof |
|
|
Vandxellulose |
3. RAPPORT
3.1. PRØVNINGSRAPPORT
Prøvningsrapporten skal indeholde følgende oplysninger:
|
— |
det afprøvede stofs identitet, sammensætning, renhed mv. |
|
— |
prøvestoffets koncentration |
|
— |
metode benyttet til tørring af den anvendte cellulose |
|
— |
vandindholdet i den anvendte cellulose |
|
— |
måleresultaterne |
|
— |
resultater af eventuelle prøvninger med inaktivt stof |
|
— |
beregnede gennemsnitlige trykstigningstider |
|
— |
eventuelle afvigelser fra denne metode og begrundelsen derfor |
|
— |
alle supplerende oplysninger og bemærkninger, som er relevante for fortolkningen af resultaterne. |
3.2. FORTOLKNING AF RESULTATERNE (22)
Prøvningsresultaterne bedømmes på grundlag af:
|
a) |
om blandingen af prøvestof og cellulose selvantænder, og |
|
b) |
sammenholdelse af den gennemsnitlige tid, der går til en trykstigning fra 690 kPa til 2 070 kPa, med den tilsvarende værdi for referencestoffet (-stofferne). |
Et væskeformigt stof anses for oxiderende, når:
|
a) |
en blanding af stoffet og cellulose i masseforholdet 1:1 selvantænder, eller |
|
b) |
den gennemsnitlige trykstigningstid af en blanding af prøvestoffet og cellulose i masseforholdet 1:1 højst er lig den gennemsnitlige trykstigningstid for en blanding af 65 % (w/w) vandig salpetersyre og cellulose i masseforholdet 1:1. |
For at undgå falske positive resultater må bedømmelsen af resultaterne om nødvendigt også inddrage resultaterne af prøvning af stoffet med inaktivt materiale.
4. HENVISNINGER
|
(1) |
Recommendations on the Transport of Dangerous Goods, Manual of Tests and Criteria. 3rd revised edition. UN Publication No: ST/SG/AC.10/11/Rev. 3, 1999, page 342. Test O.2: Test for oxidizing liquids. |
Fig. 1
Trykbeholder

Fig. 2
Stativ
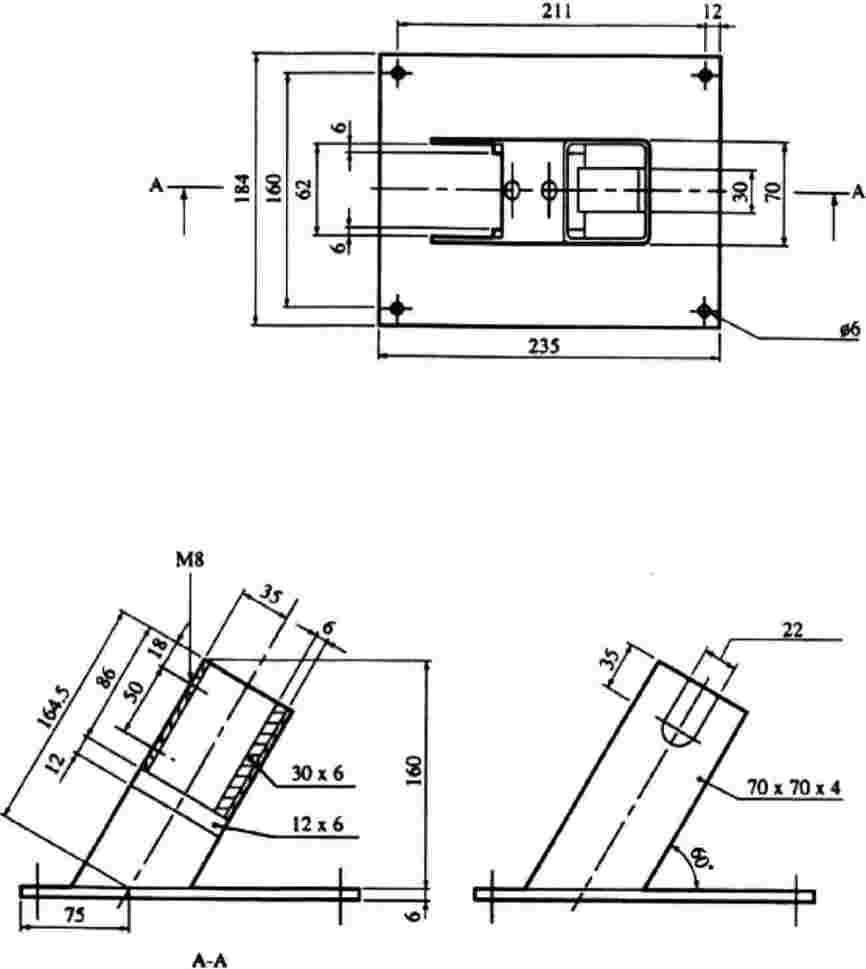
Fig. 3
Tændsystem

Note: Begge disse opstillinger kan bruges.«
(1) Afhængig af instrumenttype og stoffets renhed.
(2) Afhængig af instrumenttype og stoffets renhed.
(3) Afhxngig af instrumenttype og stoffets renhed.
(4) Afhængig af instrumenttype og stoffets renhed.
(5) Denne nøjagtighed gælder kun for det simple apparat, som er beskrevet i f.eks. ASTM D 1120-72, den kan forbedres med mere raffinerede ebulliometre.
(6) Gælder kun for rene stoffer. Anvendelse under andre betingelser skal begrundes.
(7) Afhængigt af renhedsgraden.
(8) Med den fornødne omhu kan disse metoder også anvendes i området fra 1 til 10 Pa.
(9) Som f.eks. i rammerne af FN's transportregulativer.
(10) Syren skal inden prøven titreres for at bekræfte dens styrke.
(11) F.eks. anvendes i reference (1) 50 % (w/w) perchlorsyre og 40 % (w/w) natriumchlorat.
(12) F.eks. Whatman cellulosepulver til søjlekromatografi CF 11, katalog nr. 4021 050.
(13) Bekræftet ved (f.eks.) Karl-Fisher titrering.
(14) Alternativt kan dette vandindhold også opnås (f.eks.) ved opvarmning til 105 oC under vakuum i 24 timer.
(15) Blandingen af oxiderende stoffer og cellulose skal behandles som eksplosionsfarlig og håndteres med tilbørlig forsigtighed.
(16) I praksis kan dette gøres ved at man fremstiller en 1:1 blanding af prøvevæske og cellulose i større mængde end nødvendigt til prøven og overfører 5 ±0,1 g til trykbeholderen. Blandingen skal være frisk fremstillet til hver prøvning.
(17) Navnlig må spolens naboviklinger ikke røre hinanden.
(18) Vedrørende FN-transportklassifikation henvises til ref. (1).
(19) Mættede opløsninger skal fremstilles ved 20 oC.
(20) Gennemsnitsværdi af sammenlignende laboratorieprøvninger.
(21) Det maksimale tryk 2 070 kPa ikke nået.
(22) Vedrørende fortolkning af resultater med brug af flere referencestoffer i henhold til FN's transportregulativer henvises til reference (1).
DEL B: METODER TIL BESTEMMELSE AF TOKSICITET OG ANDRE SUNDHEDSVIRKNINGER
Indholdsfortegnelse
GENEREL INDLEDNING
|
B.1.A. |
AKUT ORAL TOKSICITET — FASTDOSISMETODEN |
|
B.1.B. |
AKUT ORAL TOKSICITET — AKUT TOKSICITETSKLASSEMETODEN |
|
B.2. |
AKUT TOKSICITET (INHALATION) |
|
B.3. |
AKUT TOKSICITET (DERMAL) |
|
B.4. |
AKUT TOKSICITET: HUDIRRITATION/ÆTSNING |
|
B.5. |
AKUT TOKSICITET, ØJENIRRITATION |
|
B.6. |
HUDSENSIBILISERING |
|
B.7. |
TOKSICITET VED GENTAGEN DOSERING (28 DAGE, ORAL) |
|
B.8. |
TOKSICITET VED GENTAGEN DOSERING (28 DAGE, INHALATION) |
|
B.9. |
TOKSICITET VED GENTAGEN DOSERING (28 DAGE, DERMAL) |
|
B.10. |
MUTAGENICITET — IN VITRO-TEST FOR KROMOSOMABERRATIONER I CELLER FRA PATTEDYR |
|
B.11. |
MUTAGENICITET — IN VIVO-TEST FOR KROMOSOMABERRATIONER I KNOGLEMARV HOS PATTEDYR |
|
B.12. |
MUTAGENICITET — IN VIVO-TEST FOR MIKROKERNER I ERYTHROCYTTER HOS PATTEDYR |
|
B.13/14. |
MUTAGENICITET — TILBAGEMUTATIONSTEST MED BAKTERIER |
|
B.15. |
MUTAGENICITETSTESTNING OG SCREENING FOR KARCINOGENICITET GENMUTATION — SACCHAROMYCES CEREVISIAE |
|
B.16. |
MITOTISK REKOMBINATION — SACCHAROMYCES CEREVISIAE |
|
B.17. |
MUTAGENICITET — IN VITRO-TEST FOR GENMUTATION I PATTEDYRCELLER |
|
B.18. |
DNA-SKADE OG DNA-REPARATION — UNSCHEDULED DNA SYNTHESIS — PATTEDYRCELLER IN VITRO |
|
B.19. |
SØSTERKROMATID OMBYGNING (SCE) IN VITRO |
|
B.20. |
KØNSBUNDET RECESSIV LETAL TEST — DROSOPHILA MELANOGASTER |
|
B.21. |
CELLETRANSFORMATIONSTEST — PATTEDYRCELLER IN VITRO |
|
B.22. |
DOMINANT LETAL TEST — GNAVER |
|
B.23. |
TEST FOR KROMOSOMABERRATIONER I SPERMATOGONIER HOS PATTEDYR |
|
B.24. |
SPOTTEST — MUS |
|
B.25. |
ARVELIG TRANSLOKATION HOS MUS |
|
B.26. |
SUBKRONISK ORAL TOKSICITETSTEST — ET 90-DAGES FORSØG OVER GENTAGNE ORALE DOSERS TOKSICITET PÅ GNAVERE |
|
B.27. |
SUBKRONISK ORAL TOKSICITETSTEST — ET 90-DAGES FORSØG OVER GENTAGNE ORALE DOSERS TOKSICITET PÅ IKKE-GNAVERE |
|
B.28. |
SUBKRONISK TOKSICITET, OPTAGELSE GENNEM HUDEN — 90 DAGES GENTAGEN DOSERING PÅ HUD UNDER ANVENDELSE AF GNAVERE |
|
B.29. |
SUBKRONISK TOKSICITET, INHALATION — 90 DAGES GENTAGEN INHALATION UNDER ANVENDELSE AF GNAVERE |
|
B.30. |
UNDERSØGELSE AF KRONISK TOKSICITET |
|
B.31. |
PRÆNATAL UDVIKLINGSTOKSICITETSUNDERSØGELSE |
|
B.32. |
KARCINOGENICITETSUNDERSØGELSE |
|
B.33. |
KOMBINERET UNDERSØGELSE AF KARCINOGENICITET/KRONISK TOKSICITET |
|
B.34. |
REPRODUKTIONSTOKSICITETSUNDERSØGELSE I EN GENERATION |
|
B.35. |
REPRODUKTIONSTOKSICITETSUNDERSØGELSE I TO GENERATIONER |
|
B.36. |
TOKSIKOKINETIK |
|
B.37. |
FORSINKET NEUROTOKSICITET VED ORGANISKE PHOSPHORFORBINDELSER EFTER AKUT EKSPONERING |
|
B.38. |
FORSINKET NEUROTOKSICITET VED ORGANISKE PHOSPHORFORBINDELSER GENTAGEN DOSIS — 28 DAGE |
|
B.39. |
TEST FOR UNSCHEDULED DNA-SYNTESE (UDS) MED PATTEDYRLEVERCELLER IN VIVO |
|
B.40. |
IN VITRO-HUDÆTSNING: MÅLING AF TRANSKUTANELEKTRISK MODSTAND (TER) |
|
B.40.A. |
IN VITRO-HUDÆTSNING: TEST MED HUMAN HUDMODEL |
|
B.41. |
IN VITRO 3T3 NRU-FOTOTOKSICITETSTEST |
|
B.42. |
HUDSENSIBILISERING: ASSAY PÅ LOKALE LYMFEKNUDER |
|
B.43. |
NEUROTOKSICITETSUNDERSØGELSE I GNAVERE |
|
B.44. |
HUDABSORPTION: IN VIVO-METODE |
|
B.45. |
HUDABSORPTION: IN VITRO-METODE |
GENEREL INDLEDNING
A. BESKRIVELSE AF TESTSTOFFET
Teststoffets sammensætning, herunder de vigtigste urenheder, og dets relevante fysisk-kemiske egenskaber, herunder dets stabilitet, skal være kendt forud for enhver undersøgelse af toksicitet.
Teststoffets fysisk-kemiske egenskaber giver en række vigtige oplysninger for valget af administrationsmåde, udformningen af hver enkelt undersøgelse samt håndtering og opbevaring af teststoffet.
Forud for undersøgelsen bør der udvikles en analysemetode til kvalitativ og kvantitativ bestemmelse af stoffet (herunder vigtige urenheder, såfremt dette er muligt) i doseringsmediet og i det biologiske materiale.
Alle oplysninger om teststoffets identitet, fysisk-kemiske egenskaber, renhed og adfærd medtages i testrapporten.
B. PASNING AF DYR
Ved toksicitetstest er det afgørende, at der føres nøje kontrol med testmiljøet, og at dyrene passes korrekt.
i) Miljø
Miljøet i forsøgsstalde og -bure skal afpasses efter dyrearten. For rotter, mus og marsvin er en rumtemperatur på 22 ± 3 oC og en relativ luftfugtighed på 30-70 % passende; for kaniner bør temperaturen være på 20 ± 3 oC og den relative luftfugtighed på 30-70 %.
Nogle forsøgsteknikker er særlig følsomme over for temperaturafvigelser, og i sådanne tilfælde indeholder beskrivelsen af testmetoden nærmere oplysninger om passende forsøgsbetingelser. Ved alle undersøgelser af toksiske virkninger skal temperaturen og luftfugtigheden kontrolleres, registreres og anføres i den afsluttende forsøgsrapport.
Der bør anvendes kunstig belysning, som følger en rytme med 12 timers lys og 12 timers mørke. Oplysninger om belysningsrytmen registreres og anføres i den afsluttende forsøgsrapport.
Medmindre andet er angivet i metoden, kan dyrene opbevares i bure hver for sig eller i små grupper af samme køn, dog højst fem dyr pr. bur.
I rapporter over dyreforsøg er det vigtigt at anføre, hvilken type bure der er anvendt, og hvor mange dyr der har været i hvert bur, såvel under eksponering for det kemiske stof som i en efterfølgende observationsperiode.
ii) Fodring
Foderet skal opfylde alle de ernæringskrav, som den pågældende art af forsøgsdyr stiller. Hvis teststofferne indgives med foder, kan reaktioner mellem teststoffet og foderbestanddele medføre, at næringsværdien nedsættes. Muligheden af en sådan reaktion skal tages i betragtning, når forsøgsresultaterne fortolkes. Der kan anvendes normalt laboratoriefoder med ubegrænset forsyning af drikkevand. Valget af foder kan afhænge af, at teststoffet skal kunne iblandes foderet på passende måde, hvis teststoffet indgives med foderet.
Forureninger i foderet, som vides at influere på toksiciteten, må ikke forefindes i interfererende koncentrationer.
C. ALTERNATIV TESTNING
Den Europæiske Union har sat sig som mål at udvikle og validere alternative metoder, som kan give oplysninger på samme niveau som de nuværende dyreforsøg, men som anvender færre dyr, forårsager mindre smerte, eller hvor brugen af dyr fuldstændig kan undgås.
Efterhånden som sådanne metoder udvikles, skal de så vidt muligt tages i anvendelse i forbindelse med risikobeskrivelse og efterfølgende klassifikation og etikettering baseret på den potentielle fare ved de pågældende stoffer.
D. VURDERING OG FORTOLKNING
Ved vurdering og fortolkning af resultaterne af dyreforsøg og in vitro-undersøgelser skal der tages hensyn til, at der er grænser for, i hvilket omfang de kan ekstrapoleres direkte til mennesker, dokumentation for skadelige virkninger på mennesker, hvor sådan foreligger, skal derfor anvendes til at bekræfte testresultaterne.
E. HENVISNINGER
De fleste af disse metoder er udviklet inden for rammerne af OECD's program for retningslinjer for forsøgsvirksomhed (OECD-Test Guidelines) og bør udføres i overensstemmelse med principperne for god laboratoriepraksis, så der kan opnås størst mulig »gensidig accept af data«.
Yderligere oplysninger kan findes i henvisningerne i OECD-retningslinjerne og relevant litteratur, der er offentliggjort andetsteds.
B.1.A. AKUT ORAL TOKSICITET — FASTDOSISMETODEN
1. METODE
Denne forsøgsmetode svarer til OECD TG 420 (2001)
1.1. INDLEDNING
I traditionelle metoder til bestemmelse af akut giftighed anvendes dyrenes død som effektparameter. I 1984 foreslog British Toxicology Society en ny metode til bestemmelse af akut giftighed, baseret på administration af stoffet i en række faste dosisniveauer (1). Med denne metode undgik man at anvende dyrenes død som effektparameter, men støttede sig i stedet til observation for tydelige tegn på toksicitet ved et af en række faste dosisniveauer. Efter in vivo-validering, dels i Det Forenede Kongerige (2), dels internationalt (3), blev metoden indført som forsøgsmetode i 1992. Efterfølgende er de statistiske egenskaber af fastdosismetoden evalueret ved hjælp af matematiske modeller i en række undersøgelser (4}(5)(6). Tilsammen har in vivo-undersøgelser og modelforsøg vist, at metoden er reproducerbar, kræver færre dyr og medfører færre lidelser end traditionelle metoder, og at den gør det muligt at rangordne stofferne på tilsvarende måde som andre metoder til bestemmelse af akut giftighed.
Vejledning i valg af den mest hensigtsmæssige forsøgsmetode til et givet formål kan findes i Guidance Document on Acute Oral Toxicity Testing (7). Samme dokument indeholder desuden supplerende oplysninger om udførelse og fortolkning af forsøg efter metode B.1.A.
Undersøgelsens princip indebærer, at der i hovedundersøgelsen kun anvendes moderat toksiske doser, og at man undgår at administrere doser, som forventes at være dødelige. Ligeledes undgår man at administrere doser, der vides at medføre udtalt smerte og lidelse pga. ætsende eller stærkt irriterende virkning. Dyr, som er moribunde, har tydelige smerter eller viser tegn på svære og vedvarende lidelser, skal aflives humant og betragtes ved fortolkningen af forsøgsresultaterne på samme måde som dyr, der er døde under forsøget, Kriterierne for aflivning af moribunde eller stærkt lidende dyr samt vejledning i at fastslå forudsigelig eller forestående død er genstand for en særskilt vejledning (8).
Metoden giver oplysninger om de farlige egenskaber og gør det muligt at rangordne og klassificere stofferne efter det globale harmoniserede system (GHS) til klassificering af kemiske stoffer med akut toksisk virkning (9).
Testlaboratoriet bør inden forsøgets udførelse gennemgå alle foreliggende oplysninger om teststoffet. Sådanne oplysninger omfatter stoffets identitet og kemiske struktur, dets fysisk-kemiske egenskaber, resultaterne af andre in vitro- eller in vivo-tokstcitetsundersøgelser af stoffet, toksikologiske data for strukturelt beslægtede stoffer, samt stoffets forventede anvendelse(r). Disse oplysninger er nødvendige for til alle parters tilfredshed at godtgøre, at forsøget har betydning for beskyttelsen af menneskers sundhed, og vil desuden være nyttige ved valg af passende startdosis.
1.2. DEFINITIONER
Akut oral toksicitet: de sundhedsmæssigt uønskede virkninger, som optræder efter oral indgift af en enkelt dosis af et teststof eller efter flere doser indgivet i løbet af 24 timer.
Forsinket død: det forhold, at dyret ikke inden for 48 timer dør eller fremtræder moribund, men dør senere inden for den 14 dage lange observationsperiode.
Dosis: den mængde teststof, som indgives. Dosis angives som vægtmængde teststof pr. vægtenhed forsøgsdyr (f.eks. mg/kg).
Synlig toksicitet: et generelt udtryk, som angiver klare tegn på toksicitet efter indgift af teststoffet (eksempler herpå findes i (3)), således at der ved den næsthøjeste faste dosis enten kan forventes svære smerter og vedvarende tegn på svære lidelser, moribund status (kriterier herfor er beskrevet i Humane Endpoints Guidance Document (8)), eller størstedelen af dyrene forventes at dø.
GHS: Globally Harmonised Classification System for Chemical Substances and Mixtures. Et fælles projekt mellem OECD (sundhed og miljø), FN's ekspertudvalg vedrørende transport af farligt gods (fysisk-kemiske egenskaber) og ILO (faremærkning og -angivelse), koordineret af Interorganisation Programme for the Sound Management of Chemicals (IOMC).
Overhængende død: det forhold, at moribund tilstand eller død forventes at indtræffe inden næste planlagte observationstidspunkt. Som tegn på denne tilstand i gnavere kan nævnes kramper, sideleje, liggende stilling og tremor.
LD 50 (middel letaldosis): den statistisk beregnede enkeltdosis af et teststof, som ved oral indgift kan forventes at medføre døden hos 50 % af dyrene, LD50-værdien angives som vægtmængde teststof pr, vægtenhed kropsvægt af forsøgsdyret.
Grænsedosis: en dosis, som danner øvre grænse for testen (2 000 eller 5 000 mg/kg).
Moribund tilstand: en tilstand, hvor dyret er døende eller ikke vil overleve, selv om det behandles (se nærmere i Humane Endpoint Guidance Document (8)).
Forudsigelig død: tilstedeværelse af kliniske tegn, som antyder, at døden vil indtræffe på et kendt tidspunkt før forsøgets planlagte afslutning, f.eks. at dyret er ude af stand til at bevæge sig hen til vand eller føde (se nærmere i Humane Endpoint Guidance Document (8)).
1.3. PRINCIP FOR TESTMETODEN
Grupper af dyr af samme køn modtager trinvis stigende faste doser på 5, 50, 300 og 2 000 mg/kg (undtagelsesvis kan anvendelse af en yderligere fast dosis på 5 000 mg/kg overvejes, se punkt 1.6.2). På grundlag af en forundersøgelse vælges begyndelsesdosis som den dosis, der forventes at frembringe nogen tegn på toksicitet uden at medføre svær toksisk effekt eller dødelighed. De kliniske tegn og forhold, der er forbundet med smerte, lidelse og forestående død, er detaljeret beskrevet i en separat OECD-vejledning (8). Yderligere grupper af dyr kan behandles med højere eller lavere faste doser. Denne procedure fortsættes, indtil man har fastlagt den dosis, som medfører synlig toksicitet eller højst ét dødsfald, eller der ikke ses nogen effekt af den højeste dosis, eller der optræder dødsfald ved den laveste dosis.
1.4. BESKRIVELSE AF TESTMETODEN
1.4.1. Valg af dyreart
Den foretrukne gnaverart er rotter, men en anden gnaverart kan anvendes. Sædvanligvis anvendes hundyr (7). Baggrunden herfor er, at litteraturundersøgelser af konventionelle LD50-forsøg viser, at de to køn sædvanligvis ikke udviser stor forskel i følsomhed, men hvis der er forskel, er hundyr sædvanligvis lidt mere følsomme end handyr (10). Foreligger der imidlertid for strukturelt beslægtede stoffer oplysninger om toksikologiske eller toksikokinetiske egenskaber, som tyder på, at hanner må forventes at være mere følsomme end hunner, bør handyr anvendes. Udføres forsøget i hanner, skal fyldestgørende begrundelse herfor gives.
Der anvendes raske unge dyr af sædvanligt benyttede laboratoriestammer. Hundyrene må ikke have født og må ikke være drægtige. Hvert dyr skal ved doseringens begyndelse være mellem 8 og 12 uger gammelt, og dets vægt afvige højst ± 20 % fra gennemsnitsvægten af eventuelle tidligere behandlede dyr.
1.4.2. Opstaldning og fodring
Temperaturen i forsøgsdyrsrummet skal være 22 oC (± 3 oC). Den relative luftfugtighed skal være mindst 30 % og bør ikke være over 70 % bortset fra under rengøring af rummet, men 50-60 % bør tilstræbes, Belysningen skal være kunstig bestående af 12 timers lys og 12 timers mørke. Som foder kan anvendes sædvanligt laboratoriefoder med fri adgang til drikkevand, Dyrene i en given dosisgruppe kan være anbragt i samme bur, men antallet af dyr i buret må ikke bevirke, at det er vanskeligt at observere de enkelte dyr.
1.4.3. Klargøring af dyrene
Dyrene udtages på tilfældig måde, mærkes, så de enkelte dyr kan identificeres, og holdes i burene i mindst fem døgn forud for doseringen, så de kan akklimatisere sig til forsøgsbetingelserne.
1.4.4. Tilberedning af doser
Sædvanligvis bør teststoffet administreres i samme volumen på alle de anvendte dosisniveauer, idet man varierer koncentrationen i præparatet. Når det førdige testpræparat eller den færdige testblanding er flydende, kan anvendelse af ufortyndet teststof, dvs. med konstant koncentration, være mest relevant for den efterfølgende risikovurdering af stoffet, og nogle myndigheder stiller da også krav herom, I begge tilfælde må det maksimale volumen for indgift ikke overskrides. Den maksimale væskemængde, som kan indgives ad gangen, afhænger af forsøgsdyret størrelse, Til gnavere må rumfanget normalt ikke overskride 1 ml/100 g kropsvægt, ved vandige opløsninger kan det dog overvejes at anvende 2 ml/100 g kropsvægt. Ved formulering af doseringspræparatets anbefales det så vidt muligt at benytte en vandig opløsning/suspension/emulsion, efterfulgt (i prioriteret rækkefølge) af en opløsning/suspension/emulsion i olie (f.eks, majsolie) og derefter eventuelt en opløsning i andre vehikler. For andre vehikler end vand skal vehiklets toksiske egenskaber være kendt, Doserne skal tilberedes kort før indgift, medmindre præparatets holdbarhed inden for anvendelsesperioden kendes og er godtgjort at være tilfredsstillende.
1.5. FREMGANGSMÅDE
1.5.1. Administration of doserne
Teststoffet indgives som enkeltdosis med gastrisk sonde eller et passende intubationsrør. Kan den pågældende stofmængde ikke indgives som enkeltdosis, kan den undtagelsesvis indgives opdelt i få enkelte dele af den samlede dosis over en periode på højst 24 timer.
Dyrene skal være fastende inden doseringen (f.eks. skal rotter natten over ikke have foder, men adgang til vand; mus skal i 3-4 timer ikke have foder, men adgang til vand), Efter fasteperioden vejes dyrene, og teststoffet indgives. Når stoffet er indgivet, kan dyrene unddrages føde i yderligere 3-4 timer for rotter og 1-2 timer for mus. Når en dosis indgives delt over et vist tidsrum, kan det afhængigt af dette tidsrum være nødvendigt at give dyrene foder og adgang til vand.
1.5.2. Forundersøgelse
Forundersøgelsen har til formål at bestemme en passende startdosis i hovedundersøgelsen. Teststoffet indgives sekventielt til de enkelte dyr som angivet i skemaerne i bilag 1. Forundersøgelsen afsluttes, når det kan afgøres, hvilken startdosis der skal anvendes i hovedundersøgelsen (eller når der er et dødsfald ved den laveste faste dosis).
Startdosis til forundersøgelsen vælges blandt de faste dosisniveauer 5, 50, 300 og 2 000 mg/kg som den dosis, der forventes at medføre synlig toksicitet, om muligt baseret på dokumentation i form af in vivo- og in vitro-data for samme kemiske stof og for strukturelt beslægtede stoffer. I mangel af sådanne oplysninger anvendes en startdosis på 300 mg/kg.
Doseringerne af de enkelte dyr finder sted med et interval på mindst 24 timer. Alle dyr observeres i mindst 14 dage.
Undtagelsesvis, og kun når det kan begrundes konkret ud fra myndighedsregler, kan det overvejes at anvende et supplerende højeste fast dosisniveau på 5 000 mg/kg (se bilag 3). Af dyrevelfærdsmæssige grunde anbefales det at undgå testning i området svarende til 2 000-5 000 mg/kg i GHS kategori 5, og dette bør kun overvejes, når det er overvejende sandsynligt, at resultaterne af et sådant forsøg have direkte relevans for beskyttelsen af menneskers eller dyrs sundhed eller miljøet.
Hvis et dyr dør ved den laveste faste dosis (5 mg/kg) i forundersøgelsen, er fremgangsmåden normalt at standse forsøget og indplacere stoffet i GHS kategori 1 (som vist i bilag 1). Hvis klassificeringen kræves nærmere bekræftet, kan man dog frivilligt vælge følgende supplerende procedure: Endnu et dyr doseres med 5 mg/kg. Hvis dette dyr dør, vil det bekræfte GHS kategori 1, og undersøgelsen standses straks. Hvis dyret overlever, kan yderligere højst tre dyr behandles med 5 mg/kg. På grund af den høje dødsrisiko bør disse dyr af dyrevelfærdsmæssige grunde doseres sekventielt. Tidsrummet mellem doseringen af de enkelte dyr skal være tilstrækkeligt til at fastslå, at det foregående dyr må forventes at overleve. Indtræffer der endnu et dødsfald, skal doseringen straks indstilles, og ingen yderligere dyr doseres, Da optræden af endnu et dødsfald (uanset det testede antal dyr på ophørstidspunktet) falder ind under udfald A (to eller flere dødsfald), følger man klassificeringsreglen i bilag 2 ved fast dosis 5 mg/kg (dvs. kategori 1. hvis der er to eller flere dødsfald, og kategori 2 hvis der er højst et dødsfald), Derudover indeholder bilag 4 vejledning i, hvordan man foretager klassificeringen i EU's system, indtil det nye GHS er indført.
1.5.3. Hovedundersøgelsen
1.5.3.1. Antal dyr og dosisniveauer
De enkelte trin, der skal følges efter testning ved startdosis, er angivet i skemaerne i bilag 2. Der skal vælges en af følgende tre muligheder: enten afbrydelse af forsøget og tilordning af stoffet til den pågældende fareklasse, eller testning ved et højere fast dosisniveau, eller testning ved et lavere fast dosisniveau. Af hensyn til dyrenes beskyttelse vil et dosisniveau, der medførte død i forundersøgelsen, ikke blive gentaget i hovedundersøgelsen (se bilag 2). Erfaringen viser, at det mest sandsynlige udfald ved startdosisniveauet er, at stoffet kan klassificeres, uden at yderligere testning er nødvendig.
For hvert af de undersøgte dosisniveauer vil der normalt blive anvendt i alt fem dyr af ét køn. De fem dyr består af det dyr fra forundersøgelsen, som blev doseret ved det valgte dosisniveau, samt yderligere fire dyr (undtagen når et af hovedundersøgelsens dosisniveauer undtagelsesvis ikke indgik i forundersøgelsen).
Tidsintervallet mellem doseringerne på hvert niveau bestemmes ud fra de toksiske symptomers indsætten, varighed og sværhed. Behandlingen af dyrene ved næste dosering skal vente, indtil det kan antages, at de tidligere doserede dyr vil overleve. Det anbefales, at man om nødvendigt lader gå 3-4 dage mellem doseringerne på hvert dosisniveau for at få den nødvendige tid til at iagttage en eventuel forsinket toksisk virkning. Tidsintervallet kan korrigeres til en passende størrelse, f.eks. hvis den opnåede respons er inkonklusiv.
Overvejes det at anvende en fast højeste dosis på 5 000 mg/kg, følger man proceduren i bilag 3 (se også punkt 1.6.2).
1.5.3.2. Grænsetest
Grænsetesten anvendes fortrinsvis i situationer, hvor forsøgslederen har oplysninger om, at teststoffet må forventes at være atoksisk, dvs, kun er toksisk i doser over de grænsedoser, der er fastsat i bestemmelserne. Oplysninger om teststoffets giftighed kan fås ud fra viden om tilsvarende testede stoffer eller tilsvarende testede blandinger eller produkter, idet der tages hensyn til art og procentuelt indhold af de stoffer, der vides at være af toksikologisk betydning, Når der foreligger få eller slet ingen oplysninger om teststoffets toksicitet, eller når stoffet forventes at være toksisk, skal hovedundersøgelsen udføres.
Når den normale fremgangsmåde følges, vil en startdosis ved forundersøgelsen på 2 000 mg/kg (eller undtagelsesvis 5 000 mg/kg) efterfulgt af en dosering af fire yderligere dyr tjene som grænsetest med henblik på denne guideline.
1.6. OBSERVATIONER
Efter doseringen iagttages dyrene enkeltvis mindst én gang i de første 30 minutter, og periodisk de første 24 timer, under særlig opmærksomhed de første 4 timer, derefter dagligt, gennem i alt 14 dage, bortset fra tilfælde, hvor dyrene må tages ud af undersøgelsen og aflives humant af dyrevelfærdsmæssige grunde eller findes døde. Imidlertid bør observationsperioden ikke fastsættes ufravigeligt, Den bør fastlægges ud fra de toksiske reaktioners indsætten og restitutionsperiodens længde og kan således forlænges, når det skønnes nødvendigt. Tidspunkterne for toksiske symptomers indsætten og forsvinden er vigtige, navnlig hvis der er tendens til forsinkede toksiske symptomer (11), Alle observationer registreres systematisk, og der føres individuel protokol for hvert dyr.
Viser dyrene fortsat tegn på toksicitet, vil supplerende observationer være nødvendige. Observationerne skal omfatte forandringer i hud, pels, øjne og slimhinder, respirationssystem, kredsløb, autonomt nervesystem og centralnervesystem, samt somatomotorisk aktivitet og adfærdsmønster. Man skal være opmærksom på, om der kan iagttages tremor, kramper, spytflåd, diarré, sløvhed, søvn og koma. De principper og kriterier, der er sammenfattet i Humane Endpoints Guidance Document, skal ligeledes tages i betragtning (8). Dyr, som findes at være moribunde, og dyr, som udviser svære smerter eller vedvarende tegn på svære lidelser, skal aflives humant. Når dyr aflives af humane grunde eller findes døde, skal dødstidspunktet registreres så nøjagtigt som muligt.
1.6.1. Kropsvægt
Vægten af hvert enkelt dyr bestemmes kort før indgift af teststoffet og derefter mindst én gang om ugen. Vægtændringer beregnes og registreres. Ved forsøgets slutning vejes de overlevende dyr, som derefter aflives humant.
1.6.2. Patologi
Alle forsøgsdyr (herunder dem, der dør under forsøget eller tages ud af forsøget af dyrevelfærdsmæssige grunde) underkastes makroskopisk undersøgelse. Alle makroskopiske patologiske forandringer registreres for hvert dyr. For dyr, som overlever i 24 timer eller længere efter den første dosering, kan man endvidere overveje mikroskopisk undersøgelse af organer, som udviser makroskopiske patologiske forandringer, da dette kan give nyttige oplysninger.
2. DATA
Der skal forefindes data for de enkelte dyr. Derudover opstilles i tabelform for hver forsøgsgruppe oplysninger om antal dyr ved forsøgets begyndelse, antal dyr, som viser tegn på toksisk virkning, antal dyr, som er fundet døde under forsøget eller aflivet af humanitære grunde, dødstidspunkt for de enkelte dyr, beskrivelse af toksiske virkninger og disses tidsmæssige forløb og reversibilitet, samt obduktionsfund.
3. RAPPORTERING
3.1. FORSØGSRAPPORT
Forsøgsrapporten skal indeholde følgende oplysninger, når de er relevante:
|
|
Teststof:
|
|
|
For eventuelt vehikel:
|
|
|
For forsøgsdyrene:
|
|
|
Forsøgsbetingelser:
|
|
|
Resultater:
|
|
|
Diskussion og fortolkning af resultater. |
|
|
Konklusioner, |
4. HENVISNINGER
|
(1) |
British Toxicology Society Working Party on Toxicitiy (1984). Special report: a new approach to the classification of substances and preparations on the basis of their acute toxicity. Human Toxicol., 3, 85-92. |
|
(2) |
Van den Heuvel. M.J., Dayan. A.D. and Shillaker, R.O. (1987). Evaluation of the BTS approach to the testing of substances and preparations for their acute toxicity. Human Toxicol., 6, 279-291. |
|
(3) |
Van den Heuvel, M..J., Clark, D.G., Fielder, R.J., Koundakjian, P.P., Oliver, G.J.A., Petling, D., Tomlinson, N.J. and Walker, A.P. (1990). The international validation of a fixed-dose procedure as an alternative to the classical LD50 test. Fd. Chem. Toxicol. 28, 469-482. |
|
(4) |
Whitehead, A. and Curnow, R.N. (1992). Statistical evaluation of the fixed-dose procedure. Fd. Chem. Toxicol., 30, 313-324. |
|
(5) |
Stallard, N. and Whitehead, A. (1995). Reducing numbers in the fixed-dose procedure. Human Exptl. Toxicol. 14, 315-323. Human Exptl. Toxicol. |
|
(6) |
Stallard, N., Whitehead, A. and Ridgeway, P. (2002), Statistical evaluation of the revised fixed dose procediire.-Hum, Exp. Toxicol., 21, 183-196. |
|
(7) |
OECD (2001). Guidance Document on Acute Oral Toxicity Testing. Environmental Health and Safety Monograph Series on Testing and Assessment N. 24. Paris. |
|
(8) |
OECD (2000). Guidance Document on the Recognition, Assessment and Use of Clinical Signs as Humane Endpoints for Experimental Animals Used in Safety Evaluation. Environmental Health and Safety Monograph Series on Testing and Assesment N. 19. |
|
(9) |
OECD (1998). Harmonised Integrated Hazard Classification for Human Health and Environmental Effects of Chemical Substances as endorsed by the 28th Joint Meeting of the Chemicals Committee and the Working Party on Chemicals in November 1998, Part 2, p.ll [http://webnetl.oecd.org/oecd/pages/home/displaygeneral/0,3380,EN-documents-521-14-no-24-no-0,FF.html]. |
|
(10) |
Lipnick, R.L., Cotruvo, J.A., Hill, R.N., Bruce, R.D., Stitzel, K.A., Walker A.P., Chu, I., Goddard, M., Segal, L., Springer, J.A. and Myers, R.C. (1995). Comparison of the Up-and-Down, Conventional LD50, and Fixed-Dose Acute Toxicity Procedures. Fd. Chem. Toxicol. 33, 223-231. |
|
(11) |
Chan P.K and A.W. Haves (1994) Chapter 16 Acute Toxicity and Eye Irritation. In: Principles and Methods of Toxicology . 3rd Edition. A.W. Hayes, Editor. Raven Press, Ltd New York, USA. |
BILAG 1
FREMGANGSMÅDE VED FORUNDERSØGELSEN

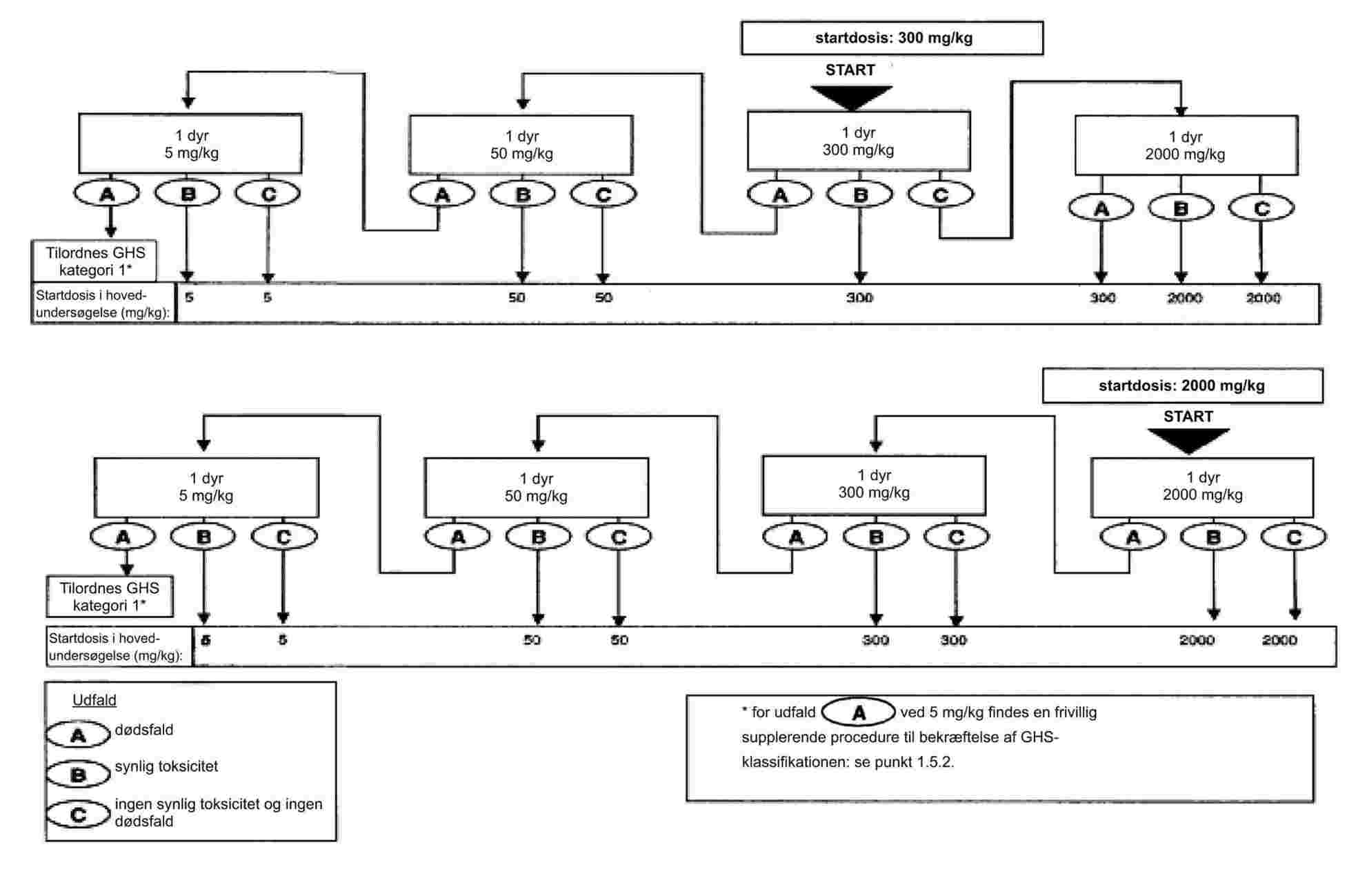
BILAG 2
FREMGANGSMÅDE VED HOVEDUNDERSØGELSEN

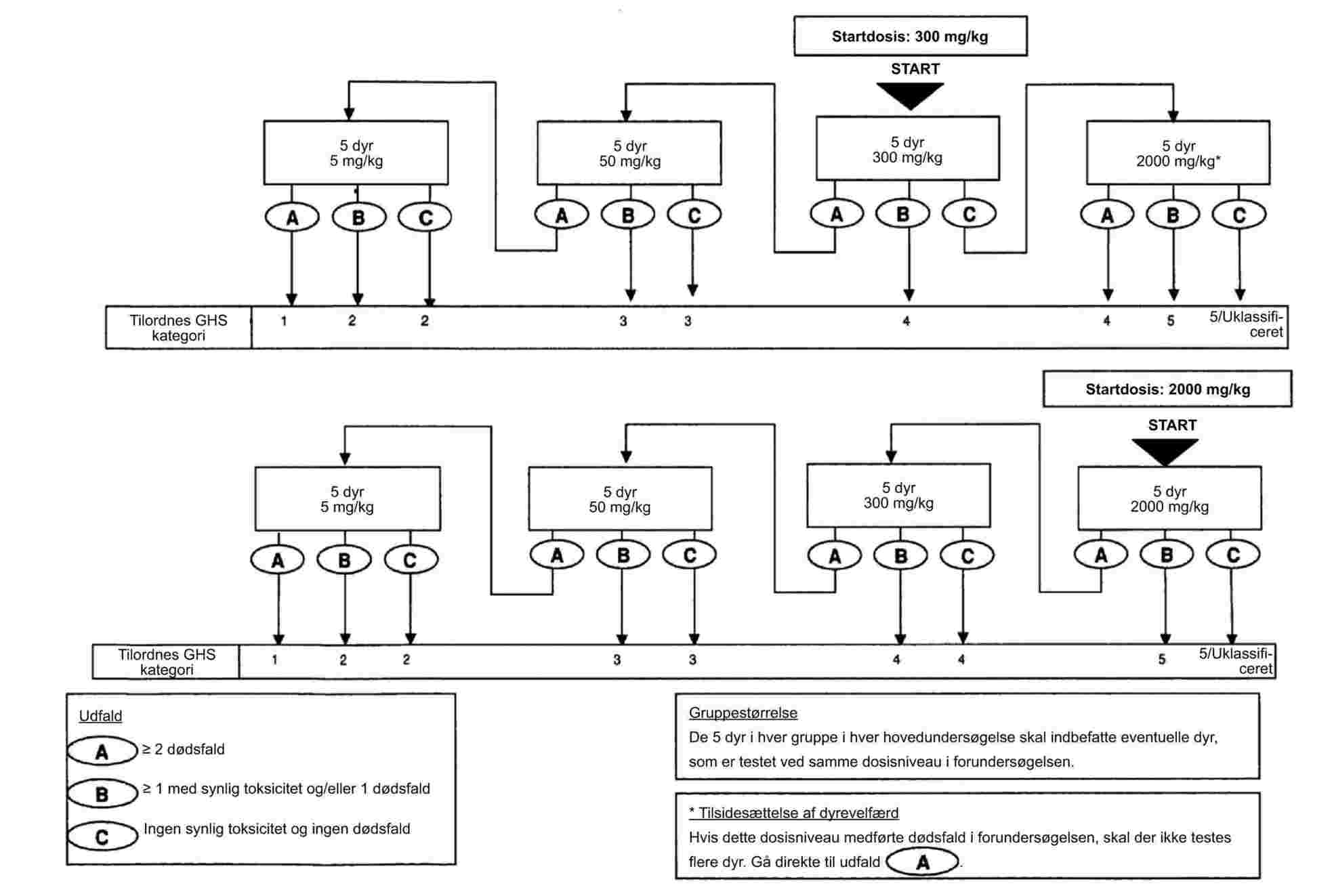
BILAG 3
KRITERIER FOR KLASSIFICERING AF TESTSTOFFER MED FORVENTET LD50-VÆRDI OVER 2 000 MG/KG, UDEN AT TESTNING ER NØDVENDIG
Kriterierne for farekategori 5 skal give mulighed for at udpege teststoffer, som udviser relativ risiko for akut gifitighed, men som under visse omstændigheder kan udgøre en fare for følsomme populationer. For disse stoffer forventes en oral eller dermal LD50 i området 2 000-5 000 mg/kg, eller tilsvarende doser for andre administrationsveje, I følgende tilfælde kan et teststof klassificeres i farekategorien defineret ved: 2 000 mg/kg < LD50 < 5 000 mg/kg (kategori 5 i GHS):
|
a) |
hvis de kan tilordnes denne kategori i henhold til enhvert af skemaerne i bilag 2 på grundlag af dødsfrekvensen |
|
b) |
hvis foreliggende pålidelige oplysninger viser, at LD50 ligger i området svarende til kategori 5, eller hvis andre dyreundersøgelser eller toksiske virkninger i mennesker viser, at der er grund til stærke sundhedsmæssige betænkeligheder |
|
c) |
ved ekstrapolation, skønsmæssig beregning eller måling af data, når der ikke er grundlag for at tilordne stoffet til en farligere klasse, og
|
TESTNING VED DOSER OVER 2 000 MG/KG
Et supplerende øvre fast dosisniveau på 5 000 mg/kg kan undtagelsesvis overvejes, når konkrete myndighedsbestemmelser gør det nødvendigt. Dyrevelfærdsmæssige grunde taler imod testning af dyr ved 5 000 mg/kg, og et sådant forsøg bør kun overvejes, når dets resultater med overvejende sandsynlighed vil få direkte relevans for beskyttelsen af dyrs eller menneskers sundhed (9),
Forundersøgelse
Reglerne i bilag 1 for anvendelse af den sekventielle metode udvides til at omfatte dosisniveauet 5 000 mg/kg. Når startdosis i forundersøgelsen er 5 000 mg/kg, vil udfald A (død) kræve testning af endnu et dyr ved 2 000 mg/kg; udfald B og C (synlig toksicitet eller ingen toksicitet) vil tillade, at 5 000 mg/kg vælges som startdosis i hovedundersøgelsen. Tilsvarende gælder, at hvis der anvendes en anden startdosis end 5 000 mg/kg, skal forsøget udvides til 5 000 mg/kg i tilfælde af udfald B eller C ved 2 000 mg/kg; fås derefter udfald A ved 5 000 mg/kg, skal startdosis i hovedundersøgelsen være 2 000 mg/kg, og fås udfald B og C, skal startdosis i hovedundersøgelsen være 5 000 mg/kg
Hovedundersøgelse
Reglerne i bilag 2 for anvendelse af den sekventielle metode udvides til at omfatte dosisniveauet 5 000 mg/kg, Når startdosis i forundersøgelsen er 5 000 mg/kg, vil udfald A (≥ 2 dødsfald) kræve testning af endnu en gruppe ved 2 000 mg/kg; udfald B (synlig toksicitet og/eller ≤ 1 dødsfald) eller C (ingen toksicitet) vil medføre, at stoffet ikke skal klassificeres i henhold til GHS. Tilsvarende gælder, at hvis der anvendes en anden startdosis end 5 000 mg/kg, vil forsøget blive udvidet til 5 000 mg/kg i tilfælde af udfald C ved 2 000 mg/kg; et efterfølgende udfald A ved 5 000 mg/kg vil placere stoffet i GHS kategori 5, mens udfald B eller C vil medføre, at stoffet ikke skal klassificeres.
BILAGÂ 4
TESTMETODE B.l a
Vejledning i klassificering efter EU-systemet i overgangsperioden indtil fuld gennemfÃ,relse af det globale harmoniserede klassifikationssystem (GHS) (fra henvisning (8))

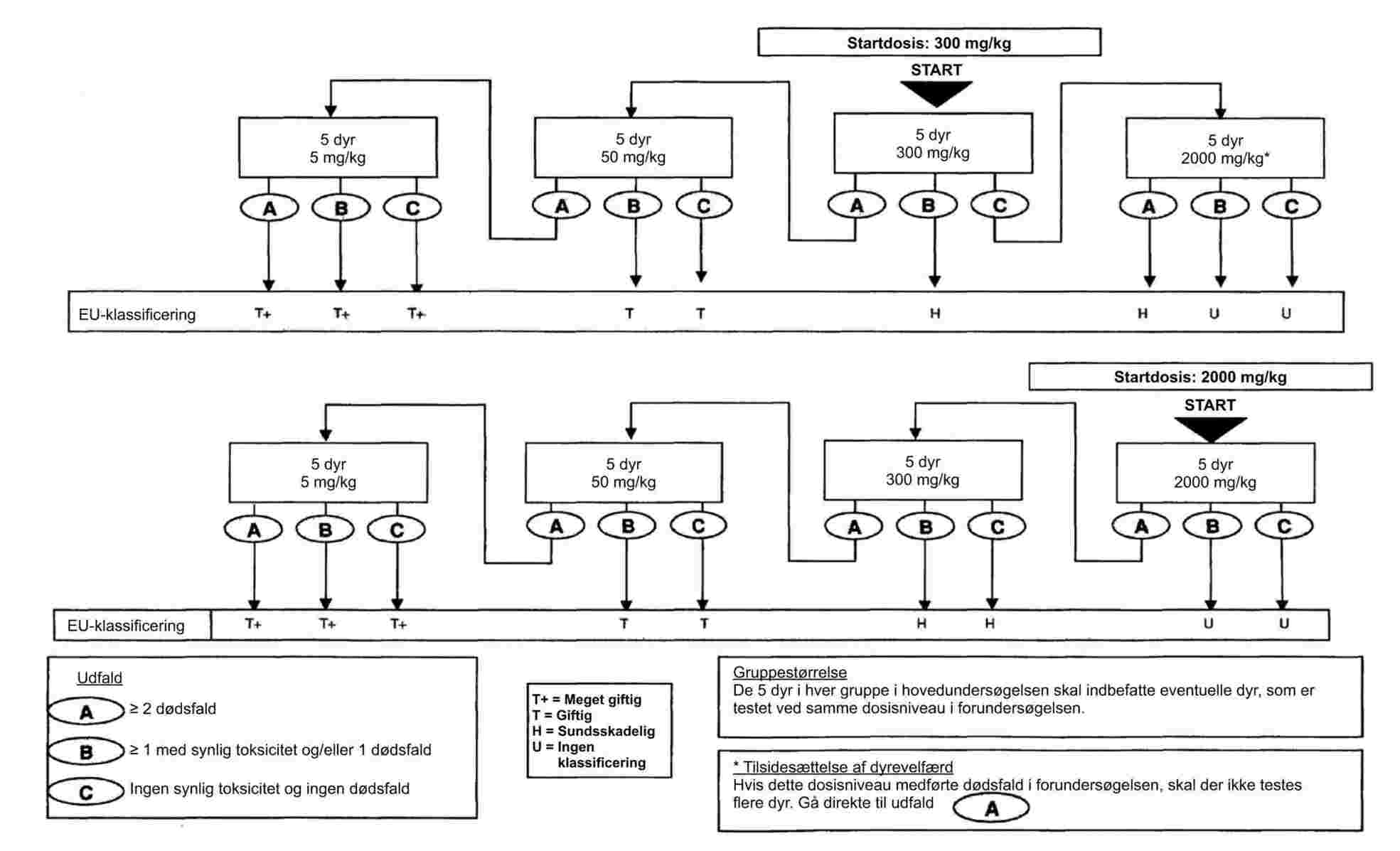
B.1.B. AKUT ORAL TOKSICITET — AKUT TOKSICITETSKLASSEMETODEN
1. METODE
Metoden svarer til OECD TG 423 (2001).
1.1. INDLEDNING
Akut toksisitetsklassemetoden (1) er en trinvis metode, hvor der anvendes tre dyr af samme køn på hvert trin. Afhængigt af dyrenes dødelighed og/eller moribunde tilstand kræves i gennemsnit 2-4 trin til vurdering af den akutte toksicitet af et teststof. Metoden er reproducerbar, kræver meget få dyr og gør det muligt at vurdere stofferne efter giftighed på tilsvarende måde som andre metoder til bestemmelse af akut toksicitet. Akut toksicitetsklassemetoden bygger på biometriske vurderinger (2)(3)(4)(5) med faste doser, der er spredt således, at et stofs giftighed kan bestemmes med henblik på klassificering og risikovurdering, Metoden blev indført i 1996 og er blevet grundigt valideret in vivo over for LD50-data fra litteraturen, både nationalt (6) og internationalt (7).
Vejledning i valg af den mest hensigtsmæssige forsøgsmetode til et givet formål kan findes i Guidance Document on Acute Oral Toxicity Testing (8). Samme dokument indeholder desuden supplerende oplysninger om udførelse og fortolkning af forsøg efter metode B.1.B.
Man behøver ikke indgive stofferne i doser, der vides at medføre udtalt smerte og lidelse på grund af ætsende eller stærkt irriterende virkning. Dyr, som er moribunde, har tydelige smerter eller viser tegn på svære og vedvarende lidelser, skal aflives humant og betragtes ved fortolkningen af forsøgsresultaterne på samme måde som dyr, der er døde under forsøget. Kriterierne for aflivning af moribunde eller stærkt lidende dyr samt vejledning i at fastslå forudsigelig eller forestående død er genstand for en særskilt vejledning (9).
Metoden giver oplysninger om de farlige egenskaber og gør det muligt at vurdere stofferne efter giftighed og klassificere stofferne efter det globale harmoniserede system (GHS) til klassificering af kemiske stoffer med akut toksisk virkning (10).
I princippet er metoden ikke beregnet til bestemmelse af en nøjagtig LD50-værdi, men giver mulighed for at fastlægge nogle eksponeringsintervaller, inden for hvilke der forventes dødelighed, idet død af en del af dyrene fortsat er testens vigtigste effektparameter. For at en LD50-værdi kan bestemmes med metoden, skal mindst to dosisniveaer resultere i større dødelighed end 0 % og lavere end 100 %. Ved at benytte en række på forhånd fastlagte doser, uafhængigt af teststoffet, og ved eksplicit at basere klassifikationen på antal dyr, der observeres i forskellige tilstande, får laboratoriet bedre mulighed for at opnå sammenhængende og repeterbare resultater.
Testlaboratoriet bør inden forsøgets udførelse gennemgå alle foreliggende oplysninger om teststoffet. Sådanne oplysninger omfatter stoffets identitet og kemiske struktur, dets fysisk-kemiske egenskaber, resultaterne af eventuelle andre in vivo og in vitro toksicitetsundersøgelser af stoffet, toksikologiske data for strukturelt beslægtede stoffer, samt stoffets forventede anvendelse(r). Disse oplysninger er nødvendige, for at det til alle parters tilfredshed kan godtgøres, at forsøget har betydning for beskyttelsen af menneskers sundhed, og vil desuden være nyttige ved valg af den bedst egnede startdosis.
1.2. DEFINITIONER
Akut oral toksicitet: de sundhedsmæssigt uønskede virkninger, som optræder efter oral indgift af en enkelt dosis af et stof eller efter flere doser indgivet i løbet af 24 timer.
Forsinket død: det forhold, at dyret ikke inden for 48 timer dør eller fremtræder moribund, men dør senere inden for den 14 dage lange observationsperiode.
Dosis: den mængde teststof, som indgives. Dosis angives som vægtmængde teststof pr. vægtenhed forsøgsdyr (f.eks. mg/kg).
GHS: Globally Harmonised Classification System for Chemical Substances and Mixtures. Et tælles projekt mellem OECD (sundhed og miljø), FN's ekspertudvalg vedrørende transport af farligt gods (fysisk-kemiske egenskaber) og ILO (faremærkning og -angivelse) koordineret af Interorganisation Programme for the Sound Management of Chemicals (IOMC).
Overhængende død: det forhold, at moribund tilstand eller død forventes at indtræffe inden næste planlagte observationstidspunkt. Som tegn på denne tilstand i gnavere kan nævnes kramper, sideleje, liggende stilling og tremor (Se nærmere i Humane End point Guidance Document (9)).
LD50 (middel oral letaldosis): den statistisk beregnede enkeltdosis af et teststof, som ved oral indgift kan forventes at medføre døden hos 50 % af dyrene. LD50-værdien angives som vægtmængde teststof pr. vægtenhed kropsvægt af forsøgsdyret (mg/kg).
Grænsedosis: en dosis, som danner øvre grænse for testen (2 000 eller 5 000 mg/kg).
Moribund tilstand: en tilstand, hvor dyret er døende eller ikke vil overleve, selv om det behandles (se nærmere i Humane Endpoint Guidance Document (9)).
Forudsigelig død: tilstedeværelse af kliniske tegn. Som antyder, at døden vil indtræffe på et kendt tidspunkt før forsøgets planlagte afslutning; f.eks. at dyret er ude af stand til at bevæge sig hen til vand eller føde (se nærmere i Humane Endpoint Guidance Document (9)).
1.3. PRINCIP FOR TESTMETODEN
Metoden bygger på en trinvis fremgangsmåde, hvor der på hvert trin anvendes det mindst mulige antal dyr, og man opnår tilstrækkelig information om teststoffets akutte giftighed til, at det kan klassificeres. Stoffet gives oralt til en gruppe forsøgsdyr ved en af de fastlagte doser. Stoffet testes ved en trinvis fremgangsmåde, hvor der på hvert trin anvendes tre dyr af samme køn (normalt hunner). Stofrelateret dødelighed (eller fravær heraf) af de behandlede dyr på det ene trin bestemmer næste trin, dvs.:
|
— |
ingen yderligere testning nødvendig |
|
— |
dosering af yderligere tre dyr med samme dosis |
|
— |
dosering af yderligere tre dyr på næste højere eller lavere dosisniveau. |
Detaljer vedrørende testmetoden er beskrevet i bilag 1. Metoden giver mulighed for vurdering af stoffets klassificering i en af de toksicitetsklasser, der er bestemt ved fast afgrænsede LD50-værdier.
1.4. BESKRIVELSE AF TESTMETODEN
1.4.1. Valg af dyreart
Den foretrukne gnaverart er rotter, men andre gnaverarter kan anvendes. Sædvanligvis anvendes hundyr (9). Baggrunden herfor er, at litteraturundersøgelser af konventionelle LD50-forsøg viser, at de to køn sædvanligvis ikke udviser stor forskel i følsomhed, men hvis der er forskel, er hundyr sædvanligvis lidt mere følsomme end handyr (11). Foreligger der imidlertid for strukturelt beslægtede stoffer toksikologiske eller toksikokinetiske oplysninger, som tyder på, at hanner må forventes at være mere følsomme end hunner, bør handyr anvendes. Udføres forsøget i hanner, skal fyldestgørende begrundelse herfor gives.
Der anvendes raske unge dyr af sædvanligt benyttede laboratoriestammer. Hundyrene må ikke have født og må ikke være drægtige. Hvert dyr skal ved doseringens begyndelse være mellem 8 og 12 uger gammelt, og dets vægt afvige højst ± 20 % fra gennemsnitsvægten af eventuelle tidligere doserede dyr.
1.4.2. Opstaldning og fodring
Temperaturen i forsøgsdyrrummet skal være 22 oC (± 3 oC). Den relative luftfugtighed skal være mindst 30 % og bør ikke være over 70 % bortset fra under rengøring af rummet, men 50-60 % bør tilstræbes. Belysningen skal være kunstig bestående af 12 timers lys og 12 timers mørke. Som foder kan anvendes sædvanligt laboratoriefoder med fri adgang til drikkevand. Dyrene i en given dosisgruppe kan være anbragt i samme bur, men antallet af dyr i buret må ikke vanskeliggøre observation af de enkelte dyr.
1.4.3. Klargøring af dyrene
Dyrene udtages på tilfældig måde, mærkes, så de enkelte dyr kan identificeres, og holdes i burene i mindst 5 døgn forud for doseringen, så de kan akklimatisere sig til forsøgsbetingelserne.
1.4.4. Tilberedning af doser
Sædvanligvis anvendes samme volumen til indgift af teststoffet på alle de anvendte dosisniveauer, idet man varierer koncentrationen i det indgivne præparat. Er det færdige produkt eller den færdige blanding flydende, kan anvendelse af ufortyndet teststof, dvs. med konstant koncentration, være mest relevant for den efterfølgende risikovurdering af stoffet, og af nogle myndigheder stilles da også krav herom. I begge tilfælde må det maksimale volumen for indgift ikke overskrides. Den maksimale væskemængde, som kan indgives ad gangen, afhænger af forsøgsdyret størrelse. Til gnavere må rumfanget normalt ikke overskride 1 ml/100 g kropsvægt. ved vandige opløsninger kan det dog overvejes at anvende 2 ml/100 g kropsvægt. Ved formulering af doseringspræparatet anbefales det så vidt muligt at benytte en vandig opløsning/suspension/emulsion, efterfulgt (i prioriteret rækkefølge) af en opløsning/suspension/emulsion i olie (f.eks. majsolie) og derefter eventuelt en opløsning i andre vehikler. For andre vehikler end vand skal vehiklets toksiske egenskaber være kendt. Doserne skal tilberedes kort før indgift, medmindre præparatets holdbarhed inden for anvendelsesperioden kendes og er godtgjort at være tilfredsstillende.
1.5. FREMGANGSMÅDE
1.5.1. Administration of doserne
Teststoffet indgives ved gavage som én enkelt dosis med gastrisk sonde eller et passende intubationsrør. Er det ikke muligt at indgive den pågældende stofmængde som en enkelt dosis, kan den undtagelsesvis undtagelsesvis indgives opdelt i få enkelte dele af den samlede dosis over en periode på højst 24 timer.
Dyrene skal være fastende inden doseringen (f.eks. skal rotter natten over ikke have foder, men adgang til vand. mus skal i 3-4 timer ikke have foder, men adgang til vand). Efter fasteperioden vejes dyrene, og teststoffet indgives, Når stoffet er indgivet, kan dyrene unddrages føde i yderligere 3-4 timer for rotter og 1-2 timer for mus. Når en dosis indgives delt over et vist tidsrum, kan det alt efter dette tidsrum være nødvendigt at give dyrene foder og adgang til vand.
1.5.2. Antal dyr og dosisniveauer
Der anvendes tre dyr til hvert trin. Som startdosis anvendes et af fire faste niveauer, 5, 50, 300 og 2 000 mg/kg kropsvægt. Som startdosis skal anvendes den, der med størst sandsynlighed forventes at fremkalde dødelighed i nogle af de doserede dyr. I skemaerne i bilag 1 beskrives den procedure, der skal følges for hver startdosis. Derudover giver bilag 4 vejledning i, hvordan man foretager klassificeringen i EU's system, indtil det nye GHS er indført.
Når de foreliggende oplysninger tyder på, at der næppe kan forventes dødelighed ved den højeste startdosis (2 000 mg/kg kropsvægt), skal der udføres en grænsetest. Når der ikke foreligger nogen oplysninger om teststoffet, anbefales 300 mg/kg kropsvægt som startdosis af dyrevelfærdsmæssige grunde.
Tidsintervallet mellem doseringerne på hvert niveau bestemmes ud fra de toksiske symptomers indsætten, varighed og sværhed. Behandlingen af dyrene ved næste dosering skal vente, indtil det er sikkert, at de tidligere doserede dyr vil overleve.
Undtagelsesvis, og kun når det kan begrundes konkret ud fra myndighedsregler, kan det overvejes at anvende et supplerende højeste fast dosisniveau på 5 000 mg/kg (se bilag 2), Af dyrevelfærdsmæssige grunde anbefales det at undgå testning i området svarende til GHS-kategori 5 (2 000-5 000 mg/kg), som kun bør overvejes, når resultaterne af et sådant forsøg med overvejende sandsynlighed ventes at få direkte relevans for beskyttelsen af dyrs eller menneskers sundhed eller miljøet.
1.5.3. Grænsetest
Grænsetesten anvendes fortrinsvis i situationer, hvor forsøgslederen har oplysninger om, at teststoffet må forventes at være atoksisk, dvs. kun er toksisk i doser over de grænsedoser, der er fastsat i bestemmelserne. Oplysninger om teststoffets toksicitet kan fås ud fra viden om tilsvarende testede stoffer eller tilsvarende testede blandinger eller produkter, idet der tages hensyn til art og procentuelt indhold af de stoffer, der vides at være af toksikologisk betydning. Når der kun foreligger få eller slet ingen oplysninger om teststoffets toksicitet, eller når stoffet forventes at være toksisk, bør hovedundersøgelsen udføres.
Der kan udføres en grænsetest ved ét dosisniveau på 2 000 mg/kg kropsvægt med seks dyr (tre dyr på hvert trin). Undtagelsesvis kan der i grænsetesten anvendes et dosisniveau på 5 000 mg/kg kropsvægt med tre dyr (se bilag 2). Hvis dette medfører teststofrelateret mortalitet, kan yderligere testning på næste lavere dosisniveau være nødvendig.
1.6 OBSERVATIONER
Efter doseringen iagttages dyrene enkeltvis mindst én gang i de første 30 minutter, og periodisk de første 24 timer, under særlig opmærksomhed de første 4 timer, derefter dagligt, gennem i alt 14 dage, bortset fra tilfælde, hvor dyrene må tages ud af undersøgelsen og aflives humant af dyrevelfærdsmæssige grunde eller findes døde. Imidlertid bør observationsperioden ikke fastsættes ufravigeligt. Den fastlægges ud fra de toksiske reaktioners indsætten og restitutionsperiodens længde og kan således forlænges, når det skønnes nødvendigt. Tidspunkterne for toksiske symptomers indsætten og forsvinden er vigtige, navnlig hvis der er tendens til forsinkede toksiske symptomer (12). Alle observationer registreres systematisk i og der føres individuel protokol for hvert dyr.
Viser dyrene fortsat tegn på toksicitet, vil supplerende observationer være nødvendige, Observationerne skal omfatte forandringer i hud, pels, øjne og slimhinder, respirationssystem, kredsløb, autonome nervesystem og centralnervesystem, samt somatomotorisk aktivitet og adfærdsmønster. Man skal være opmærksom på, om der kan iagttages tremor, kramper, spytflåd, diarré, sløvhed, søvn og coma. Ligeledes skal der tages hensyn til principperne og kriterierne i Humane Endpoints Guidance Document (9). Dyr, som findes at være moribunde, og dyr, som udviser svære smerter eller vedvarende tegn på svære lidelser, skal aflives humant. Når dyr aflives af humane grunde eller findes døde, skal dødstidspunktet registreres så nøjagtigt som muligt.
1.6.1. Kropsvægt
Vægten af hvert enkelt dyr bestemmes kort før indgift af teststoffet og derefter mindst én gang om ugen. Vægtændringer beregnes og registreres. Ved forsøgets slutning vejes de overlevende dyr, som derefter aflives humant.
1.6.2. Patologi
Alle forsøgsdyr (herunder dem, der dør under forsøget eller tages ud af forsøget af dyrevelfærdsmæssige grunde) underkastes makroskopisk undersøgelse. Alle makroskopiske patologiske forandringer registreres for hvert dyr. Endvidere kan det overvejes at foretage mikroskopisk undersøgelse af organer, som udviser makroskopiske forandringer, da dette kan give nyttige oplysninger.
2. DATA
Der skal forefindes data for de enkelte dyr. Derudover opstilles i tabelform for hver forsøgsgruppe antal dyr ved forsøgets begyndelse, antal dyr, som viser tegn på toksisk virkning, antal dyr, som er fundet døde under forsøget eller aflivet af humanitære grunde, dødstidspunkt for de enkelte dyr, beskrivelse af toksiske virkninger og disses tidsmæssige forløb og reversibilitet, samt obduktionsfund.
3. RAPPORTERING
3.1. Forsøgsrapport
Forsøgsrapporten skal indeholde følgende oplysninger, når de er relevante:
|
|
For teststoffet:
|
|
|
For eventuelt vehikel:
|
|
|
For forsøgsdyrene_
|
|
|
Forsøgsbetingelser:
|
|
|
Resultater:
|
|
|
Diskussion og fartolkning af resultater. |
|
|
Konklusioner. |
4. HENVISNINGER
|
(1) |
Roll R., Hiifer-Bosse Th. And Kayser D. (1986). New Perspectives in Acute Toxicity Testing of Chemicals. Toxicol. Lett., Suppl. 31, 86. |
|
(2) |
Roll R., Riebschlager M., Mischke U. and Kayser D. (1989). Neue Wege zur Bestimmung der akuten Toxizität van Chemikalien. Bundesgesundheitsblatt 32, 336-341. |
|
(3) |
Diener W., Sichha L., Mischke U., Kayser D. and Schlede E. (1994). The Biometric Evaluation of the Acute-Toxic-Class Method (Oral). Arch. Toxicol. 68, 559-610. |
|
(4) |
Diener W., Mischke U., Kayser D. and Schlede E. (1995). The Biometric Evaluation of the OECD Modified Version of the Acute-Toxic-Class Method (Oral). Arch. Toxicol. 69, 729-734. |
|
(5) |
Diener W., and Schlede E. (1999) Acute Toxicity Class Methods: Alterations to LD/LC50 Tests. ALTEX 16, 129-134. |
|
(6) |
Schlede E., Mischke U., Roll R. and Kayser D. (1992). A National Validation Study of the Acute-Tosic- Class Method — An Alternative to the LD50 Test. Arch. Toxicol. 66, 455-470. |
|
(7) |
Schlede E., Mischke U., Diener W. and Kayser D. (1994). The International Validation Study of the Acute-Toxic-Class Method (Oral). Arch. Toxicol. 69, 659-670. |
|
(8) |
OECD (2001) Guidance Document on Acute Oral Toxicity Testing. Environmental Health and Safety Monograph Series on Testing and Assessment N. 24. Paris. |
|
(9) |
OECD (2000) Guidance Document on the Recognition, Assessment and Use of Clinical Signs as Humane Endpoints for Experimental Animals Used in Safety Evaluation. Environmental Health and Safety Monograph Series on Testing and Assessment N 19. |
|
(10) |
OECD (1998) Harmonised Integrated Hazard Classification System For Human Health And Environmental Effects Of Chemical Substances as endorsed by the 28th Joint Meeting of the Chemicals Committee and the Working Party on Chemicals in November 1998, Part 2, p. 11 [http://webnet1.oecd.org/oecd/pages/home/displaygeneral/0,3380,EN-documents-521-14-no-24-no-0,FF.html]. |
|
(11) |
Lipnick R.L, Cotnivo. J.A, Hill R.N., Bruce R.D., Stitzel K.A., Walter A.P. Chu I: Goddard M, Segal L., Springer J.A. and Myers R.C. (1995) Comparison of the Up-and Down, Conventional LD50, and Fixed Dose Acute Toxicity Procedures. Fd. Chem. Toxicol 33, 223-231. |
|
(12) |
Chan P.K. and A.W. Hayes. (1994). Chap. 16. Acute Toxicity and Eye Irritancy. Principles und Methods of Toxicology. Third Edition. A.W. Hayes, Editor. Raven Press, Ltd, New York, USA. |
BILAG 1
FREMGANGSMÅDE, SOM SKAL FØLGES FOR HVER STARTDOSIS
GENERELLE BEMÆRKNINGER
For hver startdosis er anvist korrekt fremgangsmåde i den pågældende forsøgsplan i dette bilag.
|
— |
Bilag 1 A: Startdosis er 5 mg/kg kropsvægt |
|
— |
Bilag 1 B: Startdosis er 50 mg/kg kropsvægt |
|
— |
Bilag 1 C: Startdosis er 300 mg/kg kropsvægt |
|
— |
Bilag 1 D: Startdosis er 2 000 mg/kg kropsvægt |
Ved forsøgsgangen følges pilene, afhængigt af antallet af humant aflivede eller døde dyr.
BILAG I A
FREMGANGSMÅDE VED ANVENDELSE AF EN STARTDOSIS PÅ 5 MG/KG KROPSV ÆGT

BILAG 1 B
FREMGANGSMÅDE VED EN STARTDOSIS PÅ 50 MG/KG KROPSVÆGT

BILAG 1 C
FREMGANGSMÅDE VED EN STARTDOSIS PÅ 300 MG/KG KROPSVÆGT

B1LAG 1 D
FREMGANGSMÅDE VED EN STARTDOSIS PÅ 2 000 MG/KG KROPSVÆGT

BILAG 2
KRITERIER FOR KLASSIFICERING AF TESTSTOFFER MED FORVENTET LD50 OVER 2 000 MG/KG UDENAT TESTNING ER NØDVENDIG
Kriterierne for farekategori 5 skal gøre det muligt at udpege teststoffer, som udviser relativ lav risiko for akut giftighed, men som under visse omstændigheder kan udgøre en fare for følsomme populationer. For disse stoffer forventes en oral eller dermal LD50 i området 2 000-5 000 mg/kg, eller tilsvarende doser for andre administrationsveje. i følgende tilfælde skal et teststof klassificeres i farekategorien defineret ved: 2 000 mg/kg < LD50 < 5 000 mg/kg (kategori 5 i GHS):
|
a) |
Hvis de hører hjemme i denne kategori i henhold til enhver af testskemaerne i bilag l A-l D på grundlag af dødsfrekvensen |
|
b) |
hvis foreliggende pålidelige oplysninger viser, at LD50 ligger i området svarende til kategori 5, eller hvis andredyreundersøgelser eller toksiske virkninger i mennesker viser, at der er grund til alvorlige sundhedsmæssige betænkeligheder |
|
c) |
ved ekstrapolation, skønsmæssig ansættelse eller måling af data. når der ikke er grundlag for at tilordne stoffet til en farligere klasse, og
|
TESTNING VED DOSER OVER 2 000 MG/KG
Dyrevelfærdsmæssige hensyn taler imod testning i området 5 000 mg/kg i kategori 5 (5 000 mg/kg), og et sådant forsøg bør kun overvejes, når dets resultater med overvejende sandsynlighed må forventes at få direkte relevans for beskyttelsen af dyrs eller menneskers sundhed eller miljøet (10). Der bør ikke udføres yderligere forsøg ved højere dosisniveauer.
Er testning ved 5 000 mg/kg nødvendig, kræves kun ét trin (dvs. tre dyr). Hvis det først doserede dyr dør, fortsættes testningen ved 2 000 mg/kg efter skemaerne i bilag 1. Overlever det første dyr, doseres to dyr mere. Hvis kun ét af de tre dyr dør, er den forventede LD50-værdi over 5 000 mg/kg. Hvis begge dyr dør, fortsætter doseringen ved 2 000 mg/kg.
BILAG 3
TESTMETODE B.1 b. Vejledning i klassificering efter EU-systemet i overgangsperioden indtil fuld gennemførelse af det globale harmoniserede klassifikationssystem (GHS) (fra henvisning (8))


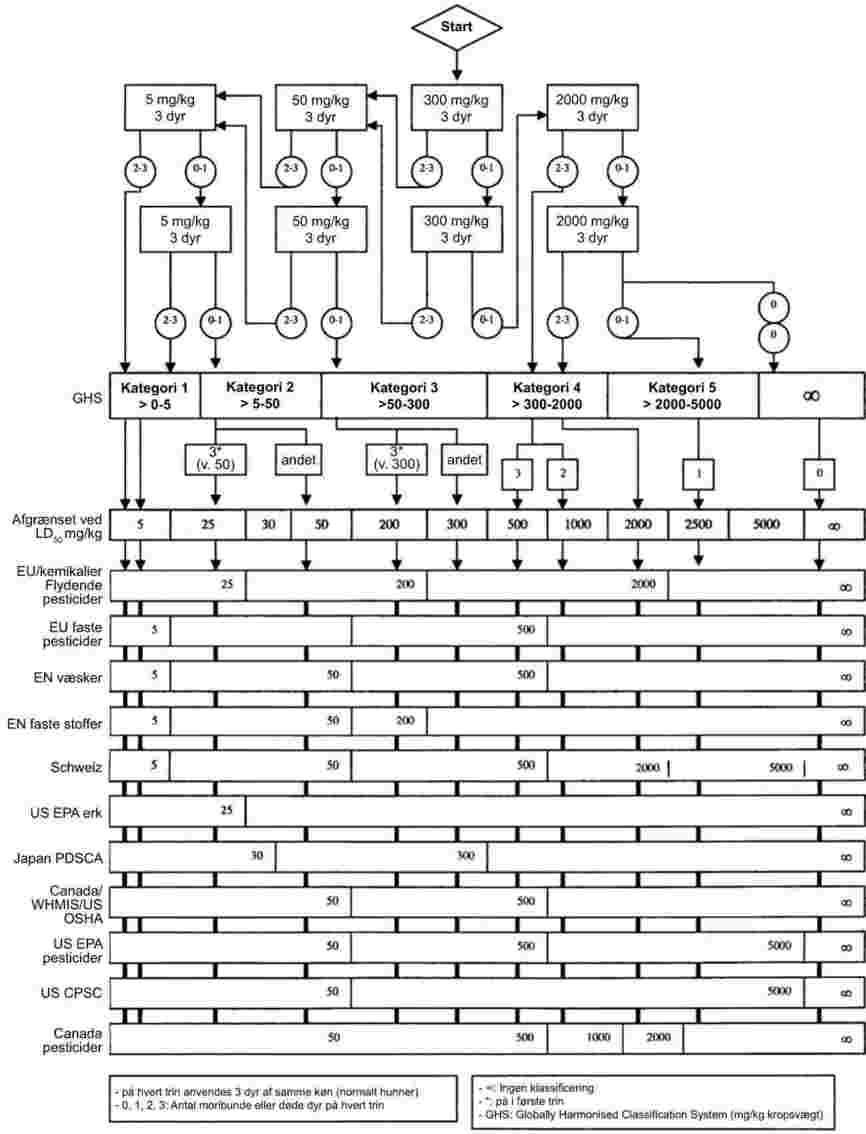

B.2. AKUT TOKSICITET (INHALATION)
1. METODE
1.1. INDLEDNING
Det er nyttigt at have forhåndsoplysninger om stoffets partikelstørrelsesfordeling, damptryk, smeltepunkt, kogepunkt, flammepunkt og eventuelle eksplosive egenskaber.
Se også den generelle indledning til del B (punkt A).
1.2. DEFINITIONER
Se den generelle indledning til del B (punkt B).
1.3. REFERENCESTOFFER
Ingen.
1.4. TESTMETODENS PRINCIP
Flere grupper forsøgsdyr udsættes i en angivet periode for teststoffet i graduerede koncentrationer, idet der anvendes en bestemt koncentration til hver enkelt gruppe. Der foretages observationer af virkningerne, herunder død. Dyr, der dør under forsøget, obduceres. Ved slutningen af forsøget aflives og obduceres overlevende dyr.
Det kan være nødvendigt, at dyr, der viser vedvarende symptomer på stærk smerte eller lidelse, aflives på human måde. Undersøgelse af stoffer på en måde, som på grund af deres ætsende eller lokalirriterende egenskaber vides at volde betydelig smerte og lidelse, er ikke påkrævet.
1.5. KVALITETSKRITERIER
Ingen.
1.6. BESKRIVELSE AF TESTMETODEN
1.6.1. Forberedelser
Dyrene holdes under forsøgsbetingelser med hensyn til miljø og fodring i mindst fem dage, inden forsøget påbegyndes. Unge, sunde, kønsmodne rotter fordeles randomiseret i det nødvendige antal grupper, inden forsøget sættes i gang. De behøver ikke udsættes for simuleret eksponering, medmindre dette indiceres af den type eksponeringsudsryr, der anvendes.
Det kan være nødvendigt at mikronisere faste teststoffer for at opnå en passende partikelstørrelse.
Om nødvendigt kan der tilsættes et egnet vehikel til teststoffet, således at der opnås en passende koncentration af teststoffet i luften. I så tilfælde skal der i forsøget indgå en kontrolgruppe, der udsættes for vehiklet. Hvis der anvendes et vehikel eller andre tilsætningsstoffer for at lette doseringen, skal det være påvist, at de ikke har nogen toksisk virkning. Her kan tidligere data benyttes.
1.6.2. Forsøgsbetingelser
1.6.2.1. Forsøgsdyr
Rotter er de foretrukne forsøgsdyr, medmindre der foreligger kontraindikationer. Der benyttes rotter af almindeligt anvendte stammer. For såvel han- som hunrotter gælder det, at vægten af de anvendte dyr ikke må variere mere end ± 20 % fra en rimelig gennemsnitsvægt ved forsøgets begyndelse.
1.6.2.2. Antal og køn
Der anvendes mindst ti gnavere (fem hunner og fem hanner) til hvert koncentrationsniveau af stoffet. Hunnerne må ikke have født og må ikke være drægtige.
Bemærk: Ved anvendelse af højere dyr end gnavere til undersøgelse af akut toksicitet skal det overvejes, om antallet af dyr kan nedsættes. Doserne skal fastlægges med omhu, og man skal bestræbe sig på ikke at benytte højere doser end moderat toksiske doser. I sådanne undersøgelser bør indgift af dødelige doser af teststoffet undgås.
1.6.2.3. Eksponeringskoncentrationer
Der anvendes et tilstrækkeligt antal dosisniveauer, mindst tre, og disse skal være valgt således, at der opnås en graduering af de toksiske virkninger og dødeligheden i testgrupperne. Der skal være tilstrækkelige data til, at der kan optegnes en dosis-virkningskurve og om muligt foretages en rimelig bestemmelse af LC50.
1.6.2.4. Grænsetest
Hvis en eksposition af fem han- og fem hundyr på 20 mg/l af et stof på gasform eller 5 mg/l af en aerosol eller et fint fordelt stof i fire timer, eller — hvis dette ikke kan lade sig gøre some følge af fysiske eller kemiske egen skaber (herunder eksplosive) ved teststoffet — den højest opnåelige koncentration ikke har teststofrelateret dødelig virkning inden for 14 dage, kan yderligere forsøg betragtes som overflødige.
1.6.2.5. Eksponeringstid
Eksponeringsperioden er fire timer.
1.6.2.6. Udstyr
Dyrene testes med inhalationsudstyr, der er udformet således, at der kan opretholdes en dynamisk luftcirkulation med et luftskifte på mindst 12 gange pr. time samtidig med et tilstrækkeligt iltindhold og jævn fordeling af eksponeringsluften. Benyttes der eksponeringskammer, skal det være således indrettet, at forsøgsdyrene sammenklumpes mindst muligt og eksponeres mest muligt for teststoffet ved inhalation. Den generelle regel for sikkerhed for stabil atmosfære er, at dyrenes totale »volumen« ikke må overstige 5 % af eksponeringskammerets volumen. Eksponeringen kan foregå gennem mund-næse, hovedet alene eller ved kamre til hele kroppen; ved anvendelse af de to første muligheder opnår man, at optagelsen af teststoffet ad andre veje minimeres.
1.6.2.7. Observationsperiode
Observationsperioden skal være mindst 14 dage. Observationsperiodens varighed bør dog ikke fastsættes rigoristisk, men bestemmes ud fra de toksiske reaktioner, hvornår disse indtræder, og restitutionsperiodens længde; den kan således forlænges, såfremt det anses for nødvendigt. Tidspunktet for, hvornår symptomer på toksicitet erkendes og forsvinder, samt dødstidspunktet er vigtige, navnlig hvis der er tendens til, at der indtræffer sene dødsfald.
1.6.3. Fremgangsmåde
Kort før eksponeringen vejes dyrene, hvorefter de udsættes for testkoncentrationen i det pågældende eksponeringsudstyr i fire timer, efter at der er opnået ligevægtskoncentration i kammeret. Ligevægten skal opnås hurtigt. Temperaturen holdes under forsøget på 22 oC ± 3 oC. Under ideelle forhold skal den relative luftfugtighed holdes på mellem 30 % og 70 %, men i visse tilfælde (f.eks. ved test af visse aerosoler) kan det være praktisk uigennemførligt. Udsivning af teststof til omgivelserne kan forhindres ved, at der i kammeret holdes et svagt undertryk (≤ 5 mm vandsøjle). Der gives hverken foder eller vand under eksponeringen. Der anvendes passende systemer til generering og kontrol af testatmosfæren. Systemet skal sikre stabile eksponeringsforhold så hurtigt som muligt. Kammeret skal være konstrueret og kunne betjenes på en sådan måde, at testatmosfæren hele tiden er ensartet fordelt i kammeret.
Der foretages målinger eller kontrol af:
|
a) |
lufthastighed (kontinuerligt) |
|
b) |
den faktiske koncentration af teststoffet målt i opholdsarealet mindst tre gange under eksponeringen (for nogle atmosfærers vedkommende, f.eks. aerosoler med høje koncentrationer, kan hyppigere kontrol være påkrævet). Koncentrationen må under eksponeringsperioden ikke variere mere end ± 15 % fra den gennemsnitlige koncentration. Det kan dog være umuligt at opnå en sådan grad af kontrol med visse aerosoler, og i så tilfælde kan større udsving accepteres. For aerosoler skal partikelstørrelsen analyseres så ofte, som det er nødvendigt (mindst én gang pr. forsøgsgruppe) |
|
c) |
temperatur og luftfugtighed, om muligt kontinuerligt. |
Under og efter eksponering foretages der systematiske observationer, som registreres for hvert enkelt dyr. Observationerne skal foretages hyppigt i løbet af den første dag. Der skal foretages en grundig klinisk undersøgelse mindst én gang hver arbejdsdag. Herudover bør dyrene dagligt observeres, så det mindst mulige antal dyr går tabt for undersøgelsen, dvs. at dyr, der findes døde, obduceres eller nedkøles, og at svage eller døende dyr isoleres eller aflives.
Inspektion af dyrene skal omfatte forandringer i hud og pels, øjne og slimhinde, derudover åndedræt og kredsløb, det autonome og centrale nervesystem, den motoriske aktivitet og adfærdsmønsteret. Der bør navnlig foretages en omhyggelig observation af respiration, rystelser, kramper, spytflåd, diarré, apati, søvn og bevidstløshed. Dødstidspunktet noteres med størst mulig nøjagtighed. Forsøgsdyrenes individuelle vægt bestemmes ugentligt og på dødstidspunktet.
De dyr, der dør under forsøget, og de, der overlever til forsøgets afslutning, obduceres med særlig opmærksomhed rettet mod eventuelle ændringer i de øvre og nedre luftveje. Alle makroskopisk-patologiske forandringer registreres. På indikation udtages der vævsprøver til histopatologisk undersøgelse.
2. DATA
Data opstilles i tabelform, som for hver gruppe viser antal dyr ved begyndelsen af forsøget, dødstidspunkt for de enkelte dyr, antal dyr, der udviser andre symptomer på toksicitet, beskrivelse af toksiske virkninger og obduktionsfund. Vægtændringer beregnes og registreres for alle dyr, der lever mere end en dag efter eksponeringen. Aflivning af dyr på human måde på grund af lidelse og smerte fremkaldt af teststoffet registreres som teststofrelaterede dødsfald, LC50 bestemmes efter en anerkendt metode. Evaluering af data bør omfatte sammenhæng, hvis en sådan findes, mellem dyrenes eksponering for teststoffet og hyppighed og grad af alle forandringer, herunder adfærdsmæssige og kliniske symptomer, makroskopisk-patologiske forandringer, ændringer i legemsvægt, dødelighed og eventuelle andre toksiske virkninger.
3. RAPPORTERING
3.1. FORSØGSRAPPORT
Forsøgsrapporten skal om muligt indeholde følgende oplysninger:
|
— |
dyreart/stamme, herkomst, miljøbetingelser, foder osv. |
|
— |
forsøgsbetingelser: beskrivelse af eksponeringsudstyret, herunder udformning, type, dimensionering, luftkilde, system til frembringelse af aerosoler, metode til konditionering af luften og den måde, hvorpå dyrene holdes i et eksponeringskammer, når et sådant benyttes. Apparatur til måling af temperatur, fugtighed og aerosolers koncentration og partikelstørrelsesfordeling skal beskrives |
Eksponeringsdata:
Disse skal opstilles i tabelform med angivelse af middelværdier og et mål for variationen (standardafvigelser) og i videst muligt omfang omfatte:
|
a) |
lufthastigheden gennem inhalationsudstyret |
|
b) |
luftens temperatur og fugtighed |
|
c) |
nominelle koncentrationer (total mængde teststof tilført inhalationsudstyret, divideret med lufrvolumen) |
|
d) |
type vehikel, hvor et sådant er anvendt |
|
e) |
faktiske koncentrationer i opholdsarealet under forsøget |
|
f) |
massemedianen af den aerodynamiske diameter (MMAD) og den geometriske standardafvigelse (GSD) |
|
g) |
periode til ligevægtsindstilling |
|
h) |
eksponeringsperiode
|
3.2. VURDERING OG FORTOLKNING
Se den generelle indledning til del B (punkt D).
4. HENVISNINGER
Se den generelle indledning til del B (punkt E).
B.3. AKUT TOKSICITET (DERMAL)
1. METODE
1.1. INDLEDNING
Se den generelle indledning til del B (punkt A).
1.2. DEFINITIONER
Se den generelle indledning til del B (punkt B).
1.3. REFERENCESTOFFER
Ingen.
1.4. TESTMETODENS PRINCIP
Teststoffet påføres huden på flere grupper af forsøgsdyr i graduerede doser, idet der anvendes en bestemt dosis til hver enkelt gruppe. Der foretages observationer af virkningerne, herunder død. Dyr, der dør under forsaget, obduceres. Ved slutningen af forsøget aflives og obduceres overlevende dyr.
Det kan være nødvendigt, at dyr, der viser vedvarende symptomer på stærk smerte eller lidelse, aflives på human måde. Undersøgelse af stoffer på en måde, som på grund af deres ætsende eller lokalirriterende egenskaber vides at volde betydelig smerte og lidelse, er ikke påkrævet.
1.5. KVALITETSKRITERIER
Ingen.
1.6. BESKRIVELSE AF TESTMETODEN
1.6.1. Forberedelser
Dyrene holdes under forsøgsbetingelser med hensyn til miljø og fodring i mindst fem dage, inden forsøget påbegyndes. Unge, sunde, kønsmodne dyr fordeles randomiseret i forsøgsgrupper, inden forsøget sættes i gang. Ca. 24 timer før forsøget påbegyndes, klippes eller barberes dyrene på ryggen. Under denne procedure må man passe på ikke at beskadige huden, da det kan ændre dens permeabilitet. Mindst 10 % af legemets overflade skal gøres klar til påføring af teststoffet. Ved testning af faste stoffer, som i givet fald kan pulveriseres, skal teststoffet fugtes tilstrækkeligt med vand eller om nødvendigt med et passende vehikel, så der sikres god kontakt med huden. Hvis der anvendes et vehikel, bør man tage hensyn til, om vehiklet har indflydelse på hudens permeabilitet for teststoffet. Flydende teststoffer påføres normalt ufortyndet.
1.6.2. Forsøgsbetingelser
1.6.2.1. Forsøgsdyr
Der kan anvendes voksne rotter eller kaniner. Der kan anvendes andre dyrearter, men i så fald skal dette være begrundet. Der benyttes dyr af almindeligt anvendte stammer. For begge køn af forsøgsdyr gælder det, at vægten af de anvendte dyr ikke må variere mere end ± 20 % fra en rimelig gennemsnitsvægt ved forsøgets begyndelse.
1.6.2.2. Antal og køn
Der anvendes mindst fem dyr (af samme køn) til hvert dosisniveau. Hunnerne må ikke have født og må ikke være drægtige. Foreligger der oplysninger om, at et bestemt køn er væsentligt mere følsomt, benyttes der dyr af dette køn.
Bemærk: Ved anvendelse af højere dyr end gnavere til undersøgelse af akut toksicitet skal det overvejes, om antallet af dyr kan nedsættes. Doserne skal fastlægges med omhu, og man skal bestræbe sig på ikke at benytte højere doser end moderat toksiske doser. I sådanne undersøgelser bør indgift af dødelige doser af teststoffet undgås.
1.6.2.3. Dosisniveauer
Der anvendes et tilstrækkeligt antal dosisniveauer, mindst tre, og disse skal være valgt således, at der opnås en graduering af de toksiske virkninger og dødeligheden i testgrupperne. Der bør ved valg af dosis tages hensyn til eventuelle hudirriterende eller ætsende virkninger. Der skal være tilstrækkelige data til, at der kan optegnes en dosis-virkningskurve og om muligt foretages en rimelig bestemmelse af LD50.
1.6.2.4. Grænsetest
Der kan med en gruppe på fem dyr af hvert køn udføres en grænsetest med en dosis på mindst 2 000 mg/kg legemsvægt efter den ovenfor beskrevne procedure. Optræder der teststofrelaterede dødsfald, kan gennemførelse af en fuldstændig undersøgelse komme i betragtning.
1.6.2.5. Observationsperiode
Observationsperioden skal være mindst 14 dage. Observationsperiodens varighed bør dog ikke fastsættes rigoristisk, men bestemmes ud fra de toksiske reaktioner, hvornår disse indtræder, og af restitutionsperiodens længde; den kan således forlænges, såfremt det anses for nødvendigt. Tidspunktet for, hvornår symptomer på toksicitet erkendes og forsvinder, samt dødstidspunktet er vigtige, navnlig hvis der er tendens til, at der indtræffer sene dødsfald.
1.6.3. Fremgangsmåde
Dyrene holdes i hver sit bur. Teststoffet påføres jævnt over et område, som svarer til ca. 10 % af den samlede legemsoverflade. Med stærkt toksiske stoffer kan der anvendes et mindre område, men stoffet bør påføres så tyndt og ensartet og over så stor en del af området som muligt.
Teststoffet skal holdes i kontakt med huden ved hjælp af en porøs gazeforbinding og en ikke-hudirriterende klæbestrimmel i en eksponeringsperiode på 24 timer. Teststedet skal desuden tildækkes på en sådan måde, at gazeforbindingen og teststoffet holdes sikkert på plads, så at dyrene ikke kan indtage teststoffet. Der kan anvendes andre foranstaltninger, som forhindrer dyrene i at indtage teststoffet; dog kan fuldstændig immobilisering ikke anbefales.
Ved eksponeringsperiodens afslutning fjernes tilbagesiddende teststof, idet der f.eks. anvendes vand eller et andet egnet middel til at rense huden.
Der foretages systematiske observationer, som registreres for hvert enkelt dyr. Observationerne skal foretages hyppigt i løbet af den første dag. Der skal foretages en grundig klinisk undersøgelse mindst én gang hver arbejdsdag. Herudover bør dyrene dagligt observeres, så det mindst mulige antal dyr går tabt for undersøgelsen, dvs. at dyr, der findes døde, obduceres eller nedkøles, og at svage eller døende dyr isoleres eller aflives.
Inspektionen af dyrene skal omfatte forandringer i pels, behandlet hud, øjne og slimhinder, derudover åndedræt og kredsløb, det autonome og centrale nervesystem, den motoriske aktivitet samt adfærdsmønsteret. Der bør foretages omhyggelig observation af rystelser, kramper, spytflåd, diarré, apati, søvn og bevidstløshed. Dødstidspunktet skal noteres så præcist som muligt. De dyr, der dør under forsøget, og de, der overlever til forsøgets afslutning, obduceres. Alle makroskopisk-patologiske forandringer registreres. På indikation udtages der vævsprøver til histopatologisk undersøgelse.
Vurdering af toksiciteten hos det modsatte køn
Efter afslutning af undersøgelsen med det ene køn testes en gruppe på fem dyr af det modsatte køn, så det konstateres, om dyr af dette køn er væsentligt mere følsomme over for teststoffet. I enkelte tilfælde kan anvendelse af færre dyr være velbegrundet. Foreligger der tilstrækkelige oplysninger om, at dyr af det køn, der er testet, er væsentligt mere følsomme, er test med dyr af det modsatte køn ikke påkrævet.
2. DATA
Data opstilles i tabelform, som for hver gruppe viser antal dyr ved begyndelsen af forsøget, dødstidspunkt for de enkelte dyr, antal dyr, der udviser andre symptomer på toksicitet, beskrivelse af toksiske virkninger og obduktionsresultater. Dyrenes individuelle vægt bestemmes og registreres, kort før teststoffet påføres, derefter ugentligt og endelig på dødstidspunktet. Vægtændringer beregnes og registreres for alle dyr, der lever mere end én dag efter påføringen af teststoffet. Aflivning af dyr på human måde på grund af lidelse og smerte fremkaldt af teststoffet registreres som teststofrelaterede dødsfald. LD50 bestemmes efter en anerkendt metode.
Evaluering af data bør omfatte sammenhæng, hvis en sådan findes, mellem dyrenes eksponering for teststof og hyppighed og grad af alle forandringer, herunder adfærdsmæssige og kliniske symptomer, makroskopisk-patologiske forandringer, ændringer i legemsvægt, dødelighed og eventuelle andre toksiske virkninger.
3. RAPPORTERING
3.1. FORSØGSRAPPORT
Forsøgsrapporten skal om muligt indeholde følgende oplysninger:
|
— |
dyreart/stamme, herkomst, miljøbetingelser, foder osv. |
|
— |
forsøgsbetingelser (herunder hvordan huden er renset og forbindingstypen; med eller uden okklusion) |
|
— |
dosisniveauer (med angivelse af typen af vehikel, hvor et sådant er anvendt, samt koncentration) |
|
— |
forsøgsdyrenes køn |
|
— |
tabelopstilling over reaktioner efter køn og dosis (dvs, antal dyr, der er døde eller aflivet under forsøget; antal dyr, der har vist symptomer på toksicitet; antal eksponerede dyr) |
|
— |
dødstidspunktet efter eksponeringen, begrundelse og kriterier for human aflivning af dyr |
|
— |
alle observationer |
|
— |
LD50 for det køn, der har været genstand for en fuldstændig undersøgelse, bestemt efter 14. dag med angivelse af beregningsmetode |
|
— |
95 % konfidensinterval for LD50 (hvor det er muligt) |
|
— |
dosis/dødelighedskurve og hældning, hvor beregningsmetoden muliggør det |
|
— |
obduktionsfund |
|
— |
eventuelle histopatologiske fund |
|
— |
resultater af forsøg med det modsatte kan |
|
— |
diskussion af resultaterne (med særlig opmærksomhed på, hvordan human aflivning af dyr under forsøget kan have indvirket på den beregnede LD50) |
|
— |
fortolkning af resultaterne. |
3.2. VURDERING OG FORTOLKNING
Se den generelle indledning til del B (punkt D).
4. HENVISNINGER
Se den generelle indledning til del B (punkt E).
B.4. AKUT TOKSICITET: HUDIRRITATION/ÆTSNING
1. METODE
Metoden er ækvivalent med OECD TG 404 (2002),
1.1. INDLEDNING
Ved udarbejdelse af denne ajourførte metode er der lagt særlig vægt på mulige forbedringer, dels vedrørende dyrevelfærd, dels vedrørende hensyntagen til alle foreliggende oplysninger om teststoffet for at undgå unødvendige forsøg med laboratoriedyr, I metoden indgår en anbefaling om, at man før iværksættelse af den beskrevne in vivo-test for ætsning/irritation fremkaldt af stoffet gennemfører en weight-of-the-evidence-analyse af foreliggende relevante data, Er de foreliggende data utilstrækkelige, kan de udbygges ved sekventiel testning (1). Den anbefalede testningsstrategi, som omfatter validerede og anerkendte in vitro-test, er givet som bilag til denne metodebeskrivelse. Når det er hensigtsmæssigt, anbefales det desuden, at man i den indledende in vivo-test anbringer de tre testlapper successivt i stedet for samtidigt på dyret.
Både den videnskabelige kvalitet og dyrenes velfærd fremmes ved, at in vivo-test ikke udføres, før alle relevante data om stoffets potentielle hudætsende eller irriterende egenskaber er vurderet ved weight-of-the-evidence-analyse. Sådanne data består af dokumentation fra foreliggende undersøgelser i mennesker og/eller laboratoriedyr, dokumentation for ætsende/irriterende virkning af et eller flere strukturelt beslægtede stoffer eller af blandinger af sådanne stoffer, data, som viser at stoffet er stærkt surt eller alkalisk (2)(3), og resultater af validerede og anerkendte in vitro- eller ex vivo-test (4)(5)(5a). En sådan analyse skulle mindske behovet for in vivo-testning for hudætsende/irriterende virkning af stoffer, som allerede er tilstrækkelig undersøgt i andre undersøgelser vedrørende disse to endepunkter.
En foretrukken testningsstrategi, som indebærer udførelse af validerede og anerkendte in vitro- eller ex vivo-test for ætsning/irritation, er medtaget som bilag til denne metodebeskrivelse, Strategien blev udviklet på en OECD-workshop (6) og enstemmigt anbefalet af deltagerne heri, og den er vedtaget som anbefalet testningsstrategi i det globalt harmoniserede system for klassificering af kemiske stoffer (GHS) (7). Skønt sekventiel testning ikke indgår som en del af testmetode B.4, anbefales det at følge denne testningsstrategi før iværksættelse af in vivo-forsøg, For nye stoffer anbefales den trinvise testningsstrategi til at indhente videnskabeligt forsvarlige oplysninger om stoffets ætsende/irriterende virkning. For eksisterende stoffer, som ikke er tilstrækkelig undersøgt for hudætsning/irritation, bør strategien anvendes til at supplere de mangelfulde data. Anvendes en anden testningsstrategi eller -metode, eller beslutter man sig for ikke at følge den trinvise testningsstrategi, bør dette begrundes.
Hvis de ætsende eller irriterende egenskaber ikke kan bestemmes ved weight-of-the-evidence-analyse i henhold til den sekventielle testningsstrategi, bør in vivo-testning overvejes (jf. bilaget).
1.2. DEFINITIONER
Hudirritation: fremkaldelse af reversibel skade på huden ved applikation af teststoffet i indtil fire timer.
Hudætsning: fremkaldelse af irreversibel skade på huden, dvs, synlig nekrose gennem epidermis og ned i dermis efter applikation af teststoffet i indtil fire timer. Typiske ætsningsreaktioner er sår, blødning, blodige skorper, efter 14 dages observation affarvning som følge af blegning af huden, hele områder med alopeci samt ardannelse. Tvivlsomme læsioner bør overvejes vurderet ved histopatologisk undersøgelse.
1.3. METODENS PRINCIP
En enkelt dosis af teststoffet påføres på huden af et forsøgsdyr; de ubehandlede hudområder af dyret tjener som kontrol. Graden af irritation/ætsning bestemmes og klassificeres med de foreskrevne intervaller og beskrives yderligere, således at virkningerne evalueres komplet. Undersøgelsen skal strække sig over tilstrækkelig lang tid til, at de iagttagne virkningers reversibilitet eller irreversibilitet kan vurderes.
Dyr, som viser tegn på stærk lidelse og/eller smerte i nogen del af forsøget, aflives humant, og teststoffet vurderes tilsvarende, Kriterier for afgørelsen om human aflivning af døende og stærkt lidende dyr kan findes i henvisning (8).
1.4. BESKRIVELSE AF TESTMETODEN
1.4.1. Forberedelse til in vivo-forsøget
1.4.1.1. Valg af dyreart
Det fortrukne forsøgsdyr er albinokaninen, og der anvendes raske unge kønsmodne dyr. Anvendes andre arter, skal der fremlægges begrundelse herfor.
1.4.1.2. Forberedelse af dyrene
Ca. 24 timer før forsøget fjernes pelsen ved tæt klipning af dyrenes ryg. Der drages omsorg for at undgå beskadigelse af huden, og kun dyr med sund, intakt hud må anvendes.
Visse kaninstammer har pletter med tæt hårvækst, som er mere eller mindre fremtrædende, afhængigt af årstiden. Sådanne områder med tæt hårvækst må ikke anvendes som testområder.
1.4.1.3. Miljøbetingelser og fodring
Dyrene holdes i hver sit bur. Temperaturen i forsøgslokalet skal være 20 oC (± 3 oC) for kaniner. Den relative luftfugtighed skal være mindst 30 % og må ikke være over 70 %, hvilket dog ikke gælder under under rengøring af lokalet, men 50-60 % bør tilstræbes. Belysningen skal være kunstig bestående af 12 timers lys og 12 timers mørke. Som foder kan anvendes sædvanligt laboratoriefoder og fri adgang til drikkevand.
1.4.2. Prøveprocedure
1.4.2.1. Applikation af teststoffet
Teststoffet påføres på et lille område (ca. 6 cm2) af huden og dækkes med en gazelap, som holdes på plads med en ikke-hudirriterende klæbestrimmel. Hvis der anvendes flydende eller pastaagtige teststoffer, kan det være nødvendigt at påføre teststoffet på gazelappen, og derefter placere lappen på huden. I ekspositionsperioden bør teststoffet holdes i løs kontakt med huden ved hiælp af en passende semiokklusiv forbinding. Hvis teststoffet påføres på gazelappen, skal denne fastgøres til huden, således at der er god kontakt og ensartet fordeling af stoffet på huden. Dyret må ikke have adgang til gazelappen og skal være forhindret i at kunne indtage eller inhalere teststoffet.
Væskeformige teststoffer anvendes sædvanligvis ufortyndet. Ved testning af faste stoffer som — hvis det er hensigtsmæssigt — kan pulveriseres, skal teststoffet fugtes tilstrækkeligt med den mindst mulige mængde vand eller om nødvendigt med et passende vehikel for at sikre god kontakt med huden. Anvendes andre vehikler end vand, må dette kun have minimal indflydelse på teststoffets hudirriterende virkning.
Ved slutningen af ekspositionsperioden, som normalt er fire timer, fjernes tilbagesiddende teststof, så vidt muligt ved hjælp af vand eller et egnet oplysningsmiddel, idet man skal undgå at påvirke det fremkomne respons eller beskadige epidermis.
1.4.2.2. Dosisniveau
Testarealet påføres 0,5 ml flydende eller 0,5 g fast eller halvflydende teststof.
1.4.2.3. Indledende test (in vivo-hudirritations/ætsningstest i ét dyr)
Det anbefales kraftigt, at in vivo-testen indledende kun udføres i ét dyr, navnlig når stoffet mistænkes for at have potentiale for ætsning. Dette er i overensstemmelse med den sekventielle testningsstrategi (jf. bilag 1).
Når et stof er bedømt som ætsende på grundlag af weight-of-the-evidence-analyse, kræves ikke yderligere dyreforsøg. For størstedelen af de stoffer, som mistænkes for at være ætsende, er yderligere in vivo-testning sædvanligvis unødvendig. I tilfælde, hvor der menes at være behov for supplering af mangelfulde data, kan en begrænset testning i dyr gennemføres med følgende fremgangsmåde: Der anbringes successivt indtil tre prøvelapper på dyret. Den første lap fjernes efter tre minutter. Ses ingen alvorlig hudirritation, anbringes endnu en prøvelap, som fjernes efter en time. Viser iagttagelserne på dette trin, at ekspositionen humant kan udvides til fire timer, anbringes en tredje lap, som fjernes efter fire timer, hvorpå responset vurderes.
Iagttages en ætsende virkning efter nogen af de tre sekventielle ekspositioner, afsluttes forsøget straks. Iagttages ingen ætsende virkning, efter at den sidste lap er fjernet, observeres dyret i 14 dage, medmindre der udvikler sig ætsning på et tidligere tidspunkt.
I tilfælde, hvor teststoffet ikke forventes at frembringe ætsning, men kan være irriterende, anbringes en enkelt lap på ét dyr i fire timer.
1.4.2.4. Verifikationstest (in vivo-hudirritationstest i flere dyr)
Er der ikke iagttaget ætsende virkning i det indledende forsøg, skal den irriterende eller negative respons verificeres i indtil to yderligere dyr, hver med én lap, ved en ekspositionsperiode på fire timer. Iagttages en irriterende virkning i det indledende forsøg, kan verifikationsforsøget udføres sekventielt eller ved eksposition af yderligere to dyr samtidigt. Er det indledende forsøg undtagelsesvis ikke udført, kan to-tre dyr behandles med en enkelt lap, som fjernes efter fire timer. Anvendes to dyr som begge udviser samme respons, er yderligere testning unødvendig. I modsat fald udføres forsøget også på det tredje dyr. Er responset usikkert, kan det være nødvendigt at evaluere resultaterne ved brug af flere dyr.
1.4.2.5. Observationsperiode
Undersøgelsen skal strække sig over tilstrækkelig lang tid til, at de iagttagne virkningers reversibilitet eller irreversibilitet kan vurderes. Forsøget skal imidlertid afsluttes, når som helst dyrene viser vedvarende tegn på stærk smerte eller lidelser. For at bestemme reversibiliteten af virkningerne skal dyrene iagttages indtil 14 dage efter fjernelse af lapperne. Iagttages reversibilitet inden 14 dage, afsluttes forsøget på dette tidspunkt.
1.4.2.6. Kliniske iagttagelser og klassificering af hudreaktioner
Alle dyr undersøges for tegn på erythem og ødem, og responset tildeles score-værdi ved 60 minutter og derefter ved 24, 48 og 72 timer efter fjernelse af lappen. Til det indledende forsøg i ét dyr undersøges testområdet desuden straks efter fjernelse af lappen. Hudreaktioner klassificeres og registreres efter inddelingen i nedenstående tabel. Er der hudskade, som ikke kan identificeres som irritation eller ætsning ved 72 timer, kan det være nødvendigt at fortsætte iagttagelsen indtil dag 14 for at fastslå reversibiliteten af virkningerne. Foruden iagttagelsen for irritation skal alle lokale toksiske virkninger, således tab af hudens fedtstofindhold, og alle systemiske bivirkninger (f.eks. kliniske symptomer på toksicitet samt kropsvægt), beskrives fuldstændigt og registreres. Til afklaring af en usikker respons bør histopatologisk undersøgelse indgå i vurderingen.
Klassificering af hudrespons er nødvendigvis subjektiv. For at give en mere ensartet klassificering af hudresponset og for at bistå prøvningslaboratorierne og dem, som foretager og fortolker iagttagelserne, må iagttagelserne foretages af personer, som er tilstrækkelig fortrolige med det anvendte score-system (se nedenstående tabel). Det kan være nyttigt at anvende en illustreret vejledning i klassificering af hudirritation og andre læsioner (9).
2. DATA
2.1. PRÆSENTATION OF RESULTATER
Undersøgelsens resultater, som sammenfattes i tabelform i den endelige testrapport, skal dække alle punkter i afsnit 3.1.
2.2. VURDERING AF RESULTATER
Score-værdierne for hudirritation vurderes i tilknytning til læsionernes art, sværhedsgrad og reversibilitet eller manglende reversibilitet. De enkelte score-værdier repræsenterer ikke en absolut standard for et stofs irritationsfremkaldende egenskaber, da andre virkninger af teststoffet ligeledes vurderes. I stedet må de enkelte score-værdier betragtes som vejledende værdier, som skal evalueres sammen med alle undersøgelsens øvrige fund.
Hudlæsionernes reversibilitet skal tages i betragtning ved bedømmelse af irritationsrespons. Hvis respons som alopeci (af et begrænset område) hyperkeratose, hyperplasi og skældannelse persisterer indtil slutningen af den 14-dags observationsperiode, anses teststoffet for irriterende.
3. RAPPORTERING
3.1. FORSØGSRAPPORT
Forsøgsrapporten skal indeholde følgende oplysninger:
|
|
Rationale for in vivo-testning: weight-of-evidence-analyse af allerede foreliggende testdata, herunder resultater af den sekventielle testningsstrategi:
|
|
|
Prøvestof:
|
|
|
Vehikel:
|
|
|
For forsøgsdyrene:
|
|
|
Omstændigheder ved forsøget:
|
|
|
Resultater:
|
4. HENVISNINGER
|
(1) |
Barratt, M.D., Castell, J.V., Chamberlain: M., Combes, R.D., Dearden, J.C., Fentem, J.H., Gerner I., Giuliani, A., Grav, T.J.B., Livingston, D.J., Provan, W.M., Rutten, F.A.J.J.L., Verhaar, H.J.M., Zbinden, P. (1995) The Integrated Use of Alternative Approaches for Predicting Toxic Hazard. ECVAM Workshop Report 8. ATLA 23, 410-429. |
|
(2) |
Young, J.R., How, M.J., Walker, A.P., Worth W.M.H. (1988) Classification as Corrosive or Irritant to Skin of Preparations Containing Acidic or Alkaline Substance Without Testing on Animals. Toxicol. In Vitro, 2, 19-26. |
|
(3) |
Worth, A.P., Fentem, J.H., Balls, M., Botham, P.A., Curren, R.D., Earl, L.K., Esdaile, D.J., Liebsch, VL (1998) Evaluation of the proposed OECD Testing Strategy for skin corrosion. ATLA 26, 709-720. |
|
(4) |
ECETOC (1990) Monograph No. 15, »Skin Irritation«, European Chemical Industry, Ecology and Toxicology Centre, Bruxelles. |
|
(5) |
Fentem, J.H., Archer, G.E.B., Balls, M. Botham, P.A., Curren, R.D., Earl, L.K., Esdaile, D.J., Holzhutter, H.G. and Liebsch, M. (1998) The ECVAM international validation study on in vitro tests for skin corrosivity. 2. Results and evaluation by the Management Team. Toxicology in Vitro 12, s. 483-524. |
|
(5a) |
Forsøgsmetode B.40 Hudætsning. |
|
(6) |
OECD (1996) OECD Test Guidelines Programme: Final Report of the OECD Workshop on Harmonization of Validation and Acceptance Criteria for Alternative Toxicological Test Methods. Afholdt i Solna, Sverige, den 22.-24. januar 1996 (http://wwwl.oecd1.org/ehs/test/background.htm). |
|
(7) |
OECD (1998) Harmonised Integrated Hazard Classification System for Human Health and Environmental Effects of Chemical Substances, som vedtaget på det 28. fælles møde mellem OECD's kemikalieudvalg og arbejdsgruppe vedrørende kemikalier, november 1998 (http://wwwl.oecd1.org/ehs/Class/HCL6.htm). |
|
(8) |
OECD (2000). Guidance Document on the Recognition. Assessment and Use of Clinical Signs as Humane Endpoints for Experimental Animals Used in Safety Evaluation. OECD Environmental Health and Satety Publitations. Series on Testing and Assessment No. 19 (http://www.oecd1.org/ehs/test/monos.htm). |
|
(9) |
EPA (1990). Atlas of Dermal Lesions, (20T-2004). United States Environmental Protection Agency, Office of Pesticides and Toxic Substances. Washington, DC, august 1990. [kan rekvireres fra OECD's sekretariat]. |
Tabel I
KLASSIFICERING AF HUDREAKTIONER
Erythem og escharadannelse
|
Intet erythem … |
0 |
|
Meget let erythem (knapt diagnosticerbart) … |
1 |
|
Veldefineret erythem … |
2 |
|
Moderat til svært erythem … |
3 |
|
Svært erythem (oksekødsfarvet) til escharadannelse, som umuliggør klassificering af erythemet … |
4 |
Maksimumværdi: 4
Ødemdannelse
|
Intet ødem … |
0 |
|
Meget let ødem (næppe synligt) … |
1 |
|
Let ødem (tydeligt afgrænset hævet område) … |
2 |
|
Moderat ødem (hævet ca. 1 mm) … |
3 |
|
Svært ødem (hævet mere end 1 mm og udstrækning ud over det eksponerede område) … |
4 |
Maksimumværdi: 4
Til afklaring af usikre responser kan histopatologisk undersøgelse foretages.
BILAG
Sekventiel testningsstrategi for hudirritation og ætsning
GENERELLE OVERVEJELSER
Af hensyn til den videnskabelige kvalitet og dyrenes velfærd er det vigtigt at undgå unødvendig brug af dyr og at minimere alle forsøg, som kan forventes at fremkalde svære reaktioner i dyr. Alle oplysninger om et stofs potentielle hudætsende og irriterende egenskaber skal vurderes, før in vivo-testning overvejes. Det kan tænkes, at der allerede foreligger tilstrækkelige vidnesbyrd til, at teststoffets hudætsende eller irriterende potentiale kan klassificeres uden laboratorieforsøg i dyr, Ved hjælp af weight-of-the-evidence-analyse og en sekventiel testningsstrategi kan behovet for in vivo-testning således nedsættes til et minimum, navnlig når stoffet forventes at fremkalde svære reaktioner.
Det anbefales, at de foreliggende oplysninger om stoffers hud irriterende og ætsende egenskaber vurderes ved weight-of-the-evidence-analyse for at afgøre, om der bortset fra in vivo-hudundersøgelser bør udføres supplerende undersøgelser til yderligere karakterisering af et sådant potentiale. Når yderligere undersøgelser er nødvendige, anbefales det at benytte den sekventielle testningsstrategi til udbygning af de relevante forsøgsdata. For ikke tidligere testede stoffer anvendes den sekventielle testningsstrategi til at udbygge det sæt data, som er nødvendigt til vurdering af stoffets potentiale for hudirritation/ætsning. Den i dette bilag beskrevne testningsstrategi er udviklet på en OECD-workshop (1) og senere bekræftet og udvidet i form af Harmonised Integrated Hazard Classification System for Human Health and Enviranmental Effects of Chemical Substanses, som vedtoges på det 28. fælles møde mellem OECD's kemikalieudvalg og arbejdsgruppe vedrørende kemikalier i november 1998 (2).
Skønt denne sekventielle testning ikke indgår som en del af testmetode B.4, er det den anbefalede strategi til bestemmelse af hudirritation/ætsning. Strategien repræsenterer både bedste praksis og en etisk standard for in vivo-testning af hudirritation/ætsning. Metoden giver vejledning i udførelse af in vivo-forsøg og sammenfatter de faktorer, som man bør tage i betragtning før et sådant forsøg påbegyndes. Strategien tilvejebringer en metode til evaluering af foreliggende oplysninger om teststoffets hudirriterende og ætsende egenskaber og en trinvis fremgangsmåde til at generere relevante data om stoffer, som har behov for yderligere undersøgelse eller ikke er blevet undersøgt, Den giver desuden anbefalinger før udførelse af validerede og anerkendte in vitro- eller ex vivo-test for hudætsning/irritation under nærmere bestemte forhold.
BESKRIVELSE AF STRATEGIEN FOR VURDERING OG TESTNING
Før man iværksætter test som led i den sekventielle testningsstrategi (vist i figuren), skal alle foreliggende oplysninger vurderes for at fastlægge nødvendigheden af in vivo-hudtest. Skønt vurdering af enkeltparametre (f.eks ekstremt pH) i sig selv kan tænkes at give væsentlige oplysninger, må alle de foreliggende oplysninger betragtes under ét. Som grundlag for en afgørelse baseret på weight-of-the-evidence må alle relevante oplysninger om virkningerne af det pågældende stof og af analoge stoffer evalueres, og et rationale for afgørelsen fremlægges. Primært må de foreliggende data om stoffets virkning på dyr og mennesker tillægges vægt, efterfulgt af resultaterne af in vitro- eller ex vivo-testning, Når det på nogen måde er muligt, bør in vivo-undersøgelser af ætsende stoffer undgås. I testningsstrategien bør følgende faktorer tages i betragtning:
Vurdering af foreliggende data fra mennesker og dyr (trin 1). Foreliggende data fra mennesker, f.eks. kliniske undersøgelser eller arbejdsmiljøundersøgelser, og case-undersøgelser fra f.eks. toksicitetsundersøgelser med enkelt eller gentagen hudeksposition bør primært tages i betragtning, da de giver oplysninger direkte om virkningerne på huden. Stoffer med kendt irriterende eller ætsende virkning og stoffer, for hvilke der foreligger klare vidnesbyrd om fravær af ætsende eller irriterende virkning, behøver ikke testes in vivo.
Analyse af struktur/aktivitet (trin 2). Resultater fra eventuelle undersøgelser af strukturelt beslægtede stoffer skal tages i betragtning. Når der for strukturelt beslægtede stoffer eller blandinger af sådanne stoffer foreligger tilstrækkelige data fra mennesker og/eller dyr til at bestemme deres hudætsende/irriterende potentiale, kan teststoffet antages at ville frembringe samme respons. I sådanne tilfælde behøver teststoffet ikke testes. Negative data fra undersøgelser af strukturelt beslægtede stoffer eller blandinger af sådanne stoffer udgør ikke tilstrækkeligt bevis for fravær af ætsende eller irriterende egenskaber af et stof i den sekventielle testningsstrategi. Potentialet for både hudætsning og hudirritation bør fastlægges ved validerede og anerkendte metoder til struktur/aktivitet analyse.
Fysisk-kemiske egenskaber og kemisk reaktivitet (trin 3). Stoffer, som udviser ekstremt pH som ≤ 2,0 og ≥ 11,5 kan have stærke lokale virkninger. Hvis et ekstremt pH er grundlaget for at udpege et stof som hudætsende, kan også dets syre/basereserve (eller bufferkapacitet) tages i betragtning (3)(4). Hvis bufferkapaciteten tyder på, at et stof muligvis ikke er hudætsende, udføres videre testning til bekræftelser heraf, fortrinsvis med brug af en valideret og anerkendt in vitro- eller ex vivo-test (jf. trin 5 og 6).
Hudtoksicitet (trin 4). Er et kemisk stof påvist meget toksisk ved optagelse gennem huden, kan in vivo-undersøgelse af dets hudirriterende/ætsende egenskaber muligvis ikke gennemføres i praksis, fordi den mængde teststof, som normalt påføres, kan overstige det stærkt toksiske niveau og følgelig vil medføre død eller svære lidelser for dyrene. Når der i forvejen er udført en hudtoksicitetsundersøgelse i albinokaniner med en dosering op til grænsen på 2 000 mg/kg kropsvægt eller højere, uden at man herved har fundet hudirritation eller ætsning, er yderligere testning for hudirritation/ætsning muligvis overflødig. Vurdering af akut hudtoksicitet på grundlag af tidligere udførte undersøgelser kræver en række overvejelser. For eksempel kan de rapporterede oplysninger om hudlæsioner være ufuldstændige. Testning og observationer kan være gjort i andre arter end kaniner, og der kan være store artsforskelle i følsomheden af responset. Endvidere kan den form, hvori teststoffet er tilført dyrene, have være uegnet til vurdering af hudirritation/ætsning (f.eks stoffets fortynding ved undersøgelse af hudtoksicitet (5)), I de tilfælde, hvor der foreligger veldesignede og veludførte hudtoksicitetsundersøgelser i kaniner, kan negative fund anses som tilstrækkelig dokumentation for, at stoffet ikke er ætsende eller irriterende.
Resultater af in vitro- eller ex vivo-undersøgelser (trin 5 og 6). Stoffer, som har udvist ætsende eller stærkt irriterende egenskaber i en valideret og anerkendt in vitro- eller ex vivo-undersøgelse (6)(7), hvis design er rettet mod vurdering af disse særlige virkninger, behøver ikke testes i dyr. Sådanne stoffer må antages at ville frembringe tilsvarende svære virkninger in vivo.
In vivo-test i kaniner (trin 7 og 8). Hvis weight-of the-evidence-analyse giver grundlag for at udføre in vivo-forsøg, bør disse begynde med en indledende test i ét dyr. Viser denne test, at stoffet ætser huden, bør der ikke udføres yderligere test. Er der ikke fundet ætsende virkning i det indledende forsøg, skal det irriterende eller negative respons verificeres i indtil to yderligere dyr med en ekspositionsperiode på fire timer. Hvis der iagttages en irriterende virkning i det indledende forsøg, kan der udføres et sekventielt verifikationsforsøg, eller yderligere to dyr kan eksponeres samtidigt.
HENVISNINGER
|
(1) |
OECD (1996), Test Guidelines Programme: Final Report on the OECD Workshop on Harmonisation of Validation and Acceptance Criteria for Alternative Toxicological Test Methods, afholdt i Solna, Sverige, den 22.-24. januar 1996 (http://www.1.oecd.org/ehs/test/background.htm). |
|
(2) |
OECD (1998). Harmonised Integrated Hazard Classification System for Human Health and Environmental Effects of Chemical Substances, som vedtaget på det 28. fælles møde mellem OECD's kemikalieudvalg og arbejdsgruppe vedrørende kemikalier, november 1998 (http://www.1oecd.org/ehs/Class/HCL6.htm). |
|
(3) |
Worth, A.P, Fentem J.H., Balls M, Botham P.A., Curren R.D, Earl L.K, Esdaile D.J., Liebsch M. (1998). An Evaluation of the Proposed OECD Testing Strategy for Skin Corrosion. ATLA 26.709-720. |
|
(4) |
Young, J.R. How, M.J., Walker, A.P., Worth. W.M.H. (1988). Classification as Corrosive or Irritant to Skin of Preparations Containing Acidic or Alkaline Substances, Without Testing on Animals. Toxic in Vitro, 2 (1) s. 19-26. |
|
(5) |
Patil, S.M., Patrick. E, Maibach, H.I. (1996) Animal, Human, and in Vitro Test Methods for Predicting Skin Irritation, in: Francis N. Marzulli and Howard I. Maibach (red.): Dermatotoxicology. Femte udgave ISBN 1-56032-356-6, kapitel 31, 411-436. |
|
(6) |
Forsøgmetode B.40. |
|
(7) |
Fentem, J.H., Archer, G.E.B., Balls, M., Botham, P.A., Curren, R.D., Earl, L.K., Esdaile, D.J., Holzhutter. H.G. and Liebsch, M. (1998) The ECVAM international validation study on in vitro tests for skin corrosivity. 2. Results and evaluation by the Management Team. Toxicology in Vitro 12, s. 483-524. |
Figur
EN STRATEGI FOR TESTNING OG VURDERING AF HUDIRRITATION/ÆTSNING

B.5. AKUT TOKSICITET, ØJENIRRITATION
1. METODE
Metoden er ækvivalent med OECD TG 405 (2002).
1.1. INDLEDNING
Ved udarbejdelse af denne ajourførte metode er der lagt særlig vægt på de forbedringer, som kan opnås ved at vurdere alle foreliggende oplysninger om teststoffet med henblik på at undgå unødvendige forsøg med laboratoriedyr og dermed komme betænkeligheder omkring dyrenes velfærd i møde. I metoden indgår en anbefaling om, at der før iværksættelse af den beskrevne in vivo-test for akut øjenirritation/ætsning udføres en weight-of-the-evidence-analyse (1) af foreliggende relevante data, Er de foreliggende data utilstrækkelige, anbefales det, at de suppleres ved sekventiel testning (2)(3). Den anbefalede testningsstrategi, som omfatter validerede og anerkendte in vitro-test, er givet som bilag til denne metodebeskrivelse, Derudover anbefales det at udføre en in vivo-test for hudirritation/ætsning med henblik på at kunne forudsige ætsning af øjet, før en in vivo-øjentest overvejes gennemført.
Både den videnskabelige kvalitet og dyrenes velfærd fremmes ved, at in vivo-test ikke udføres, før alle relevante data om stoffets potentielle øjenætsende eller -irriterende egenskaber er vurderet ved weight-of-the-evidence-analyse, Sådanne data består af dokumentation fra foreliggende undersøgelser i mennesker og/eller laboratoriedyr, dokumentation for ætsende/irriterende virkning af et eller flere strukturelt beslægtede stoffer eller af blandinger af sådanne stoffer, data, som viser at stoffet er stærkt surt eller alkalisk (4)(5), og resultater af validerede og anerkendte in vitro- eller ex vivo-test for hudætsning og -irritation (6)(6a). Undersøgelserne kan enten have været udført inden en weight-of-the-evidence-analyse eller efterfølgende som resultat heraf.
For visse stoffer kan en sådan analyse vise, at der er behov for in vivo-undersøgelser af stoffets potentiale for ætsning/irritation af øjnene, I alle sådanne tilfælde skal man, før en in vivo-øjentest påtænkes, helst først udføre en undersøgelse af stoffets virkninger på hud in vivo og vurdere denne i henhold til forsøgsmetode B.4 (7). Anvendelse af weight-of-the-evidence-analyse og sekventiel testningsstrategi skulle mindske behovet for in vivo-testning for ætsende/irriterende virkning på øjet af stoffer, som allerede er tilstrækkelig undersøgt i andre undersøgelser. Kan stoffets potentiale for ætsning eller irritation af øjet ikke bestemmes ved den sekventielle testningsstrategi, selv efter udførelse af en in vivo-undersøgelse af hudætsning og irritation, kan der udføres en in vivo-test for ætsning/irritation af øjet.
En anbefalet testningsstrategi, som indebærer udførelse af validerede og anerkendte in vitro- eller ex vivo-test for ætsning/irritation, er medtaget som bilag til denne metodebeskrivelse. Strategien blev udviklet på en OECD-workshop (8) og enstemmigt anbefalet af deltagerne heri, og den er vedtaget som anbefalet testningsstrategi i det globalt harmoniserede system for klassificering af kemiske stoffer (GHS) (9). Skønt sekventiel testning ikke indgår som en del af testmetode B.4, anbefales det at følge denne testningsstrategi, før in vivo-forsøg iværksættes, For nye stoffer anbefales den trinvise testningsstrategi til at indhente videnskabeligt forsvarlige oplysninger om stoffets ætsende/irriterende virkning. For eksisterende stoffer, som ikke er tilstrækkelig undersøgt for øjenætsning/irritation, bør man følge denne strategi for at supplere de mangelfulde data, Anvendes en anden testningsstrategi eller -metode, eller beslutter man sig for ikke at følge den trinvise testningsstrategi, bør dette begrundes.
1.2. DEFINITIONER
Øjenirritation er frembringelse af forandringer i øjet, som opstår efter påføring af et teststof på øjets anteriore overflade, og som er fuldt reversible inden for 21 dage efter påføring.
Ætsning af øjet er frembringelse af vævsbeskadigelse i øjet eller alvorlig fysisk synsnedsættelse, som opstår efter påføring af et teststof på øjets anteriore overflade, og som ikke er fuldt reversibel inden for 21 dage efter påføring.
1.3. TESTMETODENS PRINCIP
En enkelt dosis af teststoffet påføres dyrets ene øje; det ubehandlede øje tjener som kontrol. Graden af øjenirritation/ætsning vurderes ved tildeling af score-værdier til læsionerne af conjunctiva, cornea og iris med bestemte intervaller, Andre virkninger på øjet og systemiske negative virkninger beskrives ligeledes, således at man har en fuldstændig bedømmelse af virkningerne. Undersøgelsen skal strække sig over tilstrækkelig lang tid til, at virkningernes reversibilitet eller irreversibilitet kan vurderes.
Dyr, som viser tegn på stærk lidelse og/eller smerte i nogen del af forsøget, aflives humant, og teststoffet vurderes tilsvarende, Kriterier for human aflivning af døende og stærkt lidende dyr kan findes i reference (10).
1.4. BESKRIVELSE AF TESTMETODEN
1.4.1. Forberedelse til in vivo-forsøget
1.4.1.1. Valg af art
Det foretrukne forsøgsdyr er albinokaninen, og der anvendes raske unge kønsmodne dyr. Anvendes andre arter, skal der fremlægges begrundelse herfor.
1.4.1.2. Forberedelse af dyrene
På hvert af de foreløbigt udvalgte dyr undersøges begge øjne højst 24 timer før testen begynder. Dyr, som udviser øjenirritation eller øjendefekter, eller som i forvejen har beskadiget cornea, må ikke anvendes.
1.4.1.3. Miljøbetingelser og fodring
Dyrene holdes i hver sit bur. Temperaturen i forsøgslokalet skal være 20 oC (± 3 oC) for kaniner. Den relative luftfugtighed skal være mindst 30 % og helst ikke være over 70 %, hvilket dog ikke gælder under rengøring af lokalet, men 50-60 % bør tilstræbes. Belysningen skal være kunstig bestående af 12 timers lys og 12 timers mørke. Som foder kan anvendes sædvanligt laboratoriefoder og fri adgang til drikkevand.
1.4.2. Prøveprocedure
1.4.2.1. Applikation af teststoffet
Teststoffet anbringes i conjunctivalsækken af det ene øje på hvert dyr, idet det nederste øjenlåg forsigtigt trækkes lidt ud fra øjeæblet. Derefter holdes øjenlågene sammen i ca. et sekund for at forhindre, at noget af stoffet løber ud. Det ubehandlede øje fungerer som kontrol.
1.4.2.2. Skylning
I mindst 24 timer efter instillation af teststoffet må der ikke foretages skylning af forsøgsdyrenes øjne, bortset fra faststof (jf. punkt 1.4.2.3.2) eller i tilfælde af øjeblikkeligt indsættende ætsende eller irriterende virkning. Ved 24 timer kan øjet skylles, hvis det anses for hensigtsmæssigt.
Anvendelse af en satellitgruppe af dyr til undersøgelse af virkningen af skylningen kan ikke anbefales, medmindre det er videnskabeligt begrundet, Er en satellitgruppe nødvendig, skal den bestå af to kaniner. Skylningsomstændighederne skal nøje dokumenteres, således skylningstidspunkt, skyllevæskens sammensætning og temperatur samt skylningens varighed, anvendt væskevolumen og -hastighed.
1.4.2.3. Dosisniveau
1.4.2.3.1 Undersøgelse af væsker
Ved undersøgelse af flydende stoffer anvendes 0,1 ml. Der må ikke anvendes pumpespray til direkte instillation af stoffet i øjet. I stedet sprøjtes væske ud af sprayflasken og opsamles i et kar, før der instilleres 0,1 ml i øjet.
1.4.2.3.2. Undersøgelse af faste stoffer
Ved undersøgelse af faste stoffer, pastaer og fintfordelte faste stoffer anvendes et volumen på 0,1 ml eller en vægtmængde på højst 100 mg. Prøvestoffet rives til finkornet støv. Partiklernes volumen måles efter let sammenpresning, f.eks. frembragt ved små slag på målekarret. Er det faste teststof ikke forsvundet fra dyrets øje ad naturlig vej på det første observationstidspunkt 1 time efter behandlingen, kan øjet skylles med saltvand eller destilleret vand.
1.4.2.3.3. Undersøgelse af aerosoler
Det anbefales, at alle pumpesprayprodukter og aerosoler opsamles før instillation i øjet. Som eneste undtagelse gælder stoffer, som er under tryk i aerosolbeholder og ikke kan opsamles på grund af fordampning. I sådanne tilfælde holdes øjet åbent, og teststoffet administreres i øjet med et enkelt pust på ca. ét sekund fra en afstand af 10 cm direkte foran øjet. Afstanden kan variere afhængigt af trykket i aerosolbeholderen og dens indhold. Der må drages omsorg for at undgå skade på øjet som følge af trykket i spraydåsen. I visse tilfælde kan der være brug for at vurdere potentialet for »mekanisk« skade på øjet forårsaget af forstøvningens styrke.
Den afgivne dosis fra aerosolbeholderen kan vurderes ved at simulere prøven på følgende måde: Stoffet sprøjtes på vejepapir gennem en åbning af størrelse som et kaninøje placeret umiddelbart foran papiret. Papirets vægtforøgelse anvendes som omtrentligt mål for den mængde, der tilføres øjet ved forstøvning. For flygtige stoffer kan dosis vurderes ved vejning af den modtagende beholder før og efter udtagning af teststof.
1.4.2.4. Indledende test (in vivo-øjenirritations/ætsningstest i ét dyr
Som nævnt i den sekventielle teststrategi (se bilag 1), anbefales det stærkt, at in vivo-testen indledende udføres i kun ét dyr.
Viser denne test, at stoffet virker ætsende eller stærkt irriterende på øjet ved brug af den beskrevne fremgangsmåde, bør der ikke udføres yderligere test for øjenirritation.
1.4.2.5. Lokalanæstetika
Der kan anvendes lokalanæstetika efter stillingtagen i hvert enkelt tilfælde. Hvis weight-of-the-evidence-analysen viser, at stoffet har smertevoldende potentiale, eller hvis den indledende testning viser, at der vil komme en smertefuld reaktion, kan der bruges et lokalanæstetikum før instillation af teststoffet. Art, koncentration og dosering af lokalanæstetikum bør vælges omhyggeligt med henblik på at sikre, at dets anvendelse ikke medfører forskelle i reaktionen på teststoffet. Kontroløjet lokalbedøves på tilsvarende måde.
1.4.2.6. Verifikationstest (in vivo-øjenirritationstest i yderligere dyr)
Har det indledende forsøg ikke vist ætsende virkning, bør det irriterende eller negative respons verificeres i indtil to yderligere dyr. Iagttages der i det indledende forsøg en svær irriterende virkning, som tyder på, at der måske kan forventes en stærk (irreversibel) virkning i verifikationsforsøget, anbefales det at udføre dette sekventielt i ét dyr ad gangen frem for at eksponere yderligere to dyr samtidig. Udviser det andet dyr ætsning eller svær irritation, standses forsøget. Til verifikation af en svag eller moderat irritationsrespons kan det være nødvendigt at anvende yderligere dyr.
1.4.2.7. Observationsperiode
lagttagelsesperioden skal være tilstrækkelig lang til, at de iagttagne virkningers omfang og reversibilitet kan vurderes fuldt ud. Forsøget skal dog afsluttes, når som helst dyrene viser vedvarende tegn på stærk smerte eller lidelser (9). For at bestemme reversibiliteten af virkningerne skal dyrene sædvanligvis iagttages indtil 21 dage efter administration af teststoffet. Iagttages reversibilitet inden 21 dage, afsluttes forsøget på dette tidspunkt.
1.4.2.7.1. Kliniske iagttagelser og klassificering af øjenreaktioner
Øjnene undersøges 1, 24, 48 og 72 timer efter påføring af teststoffet. Dyrene skal ikke forblive længere i forsøget, end indtil dette har har givet definitive oplysninger. Dyr, som viser tegn på stærk lidelse og/eller smerte i nogen del af forsøget, skal omgående aflives humant, og teststoffet vurderes tilsvarende, Når følgende øjenlæsioner optræder efter instillation af teststoffet, skal dyret aflives humant: perforation af cornea eller nævneværdig ulceration af cornea, herunder staphylom; blod i forreste øjenkammer; uklarhed af cornea af grad 4, persisterende i 48 timer; manglende reaktion på lys (irisrespons grad 2), persisterende i 72 timer; ulceration af conjunctivaslimhinden; nekrose af conjunctivae eller blinkhinde; rynker i cornea. Begrundelsen herfor er, at sådanne læsioner sædvanligvis er irreversible.
For dyr, som ikke udvikler øjenlæsioner, kan forsøget afsluttes tidligst 3 dage efter instillation, Dyr med lette til moderate læsioner observeres, indtil læsionerne forsvinder, eller i 21 dage, på hvilket tidspunkt forsøget afsluttes, På dag 7, 14 og 21 foretages observation til bestemmelse af læsionernes status og reversibilitet eller irreversibilitet.
Graden af øjenreaktion (conjunctivae, cornea and iris) registreres ved hver undersøgelse (tabel I). Alle andre læsioner i øjet (f.eks. pannus, farvning) og systemiske negative virkning skal ligeledes rapporteres.
Undersøgelse af reaktionerne kan lettes ved brug af lup-briller, håndspaltelampe, binokulært mikroskop eller andre hensigtsmæssige hjælpemidler. Efter registrering af observationerne i 24 timer kan øjnene undersøges yderligere med fluorescein.
Klassificeringen af øjenrespons er nødvendigvis subjektiv. For at give en mere ensartet klassificering af øjenresponset og for at bistå prøvningslaboratorieme og dem, som foretager og fortolker iagttagelserne, må iagttagelserne foretages af personer, som er tilstrækkeligt fortrolige med det anvendte score-system. Klassificeringen af øjenrespons foretages blindt.
2. DATA
2.2. VURDERING AF RESULTATER
Score-værdierne for øjenirritation vurderes i tilknytning til læsionernes art, sværhedsgrad og reversibilitet eller manglende reversibilitet, De enkelte score-værdier repræsenterer ikke en absolut standard for et stofs irritationsfremkaldende egenskaber, da de også omfatter andre virkninger af teststoffet. I stedet må de enkelte score-værdier betragtes som vejledende værdier, som kun er meningsfulde, når de støttes af en fuldstændig beskrivelse og evaluering af alle observationer.
3. RAPPORTERING
3.1. FORSØGSRAPPORT
Forsøgsrapporten skal indeholde følgende oplysninger:
|
|
Rationale for in vivo-testning: weight-of-the-evidence-analyse af allerede foreliggende testdata, herunder resultater af den sekventielle testningsstrategi
|
|
|
Prøvestof:
|
|
|
Vehikel:
|
|
|
For forsøgsdyrene:
|
|
|
Resultater:
|
|
|
Diskussion af resultater. |
3.2. FORTOLKNING AF RESULTATER
Resultaterne af øjenirritationsundersøgelser i laboratoriedyr kan kun i begrænset omfang overføres til mennesker. I mange tilfælde er albinokaniner mere følsomme end mennesker for øjenirriterende eller -ætsende stoffer.
Fortolkning af data skal ske med forbehold, så man undgår at medtage irritation forårsaget af sekundær infektion.
4. HENVISNINGER
|
(1) |
Barratt, M.D., Castell, J.V., Chamberlain, M., Combes, R.D., Dearden, J.C., Fentem, J.H., Gerner, L., Giuliani, A. Gray, T.J.B., Livingston, D.J., Provan, W.M., Rutten, F.A.J.J.L., Verhaar, H.J.M., Zbinden, P. (1995) The Integrated Use of Alternative Approaches for Predicting Toxic Hazard. ECVAM Workshop Report 8. ATLA 23, 410- 429. |
|
(2) |
de Silva, O., Cottin, M., Dami, N., Roguet, R., Catroux, P., Toufic, A., Sicard, C., Dossou, K.G., Gerner, I., Schlede, E., Spielmann, H., Gupta, K.C., Hill, R.N. (1997) Evaluation of Eye Irritation Potential: Statistical Analysis and Tier Testing Strategies. Food Chem. Toxicol 35, 159-164. |
|
(3) |
Worth A.P. and Fentem J.H. (1999) A general approach for evaluating stepwise testing strategies, ATLA 27, 161-177. |
|
(4) |
Young, J.R., How, M.J., Walker, A.P., Worth W.M.H. (1988) Classification as Corrosive or Irritant to Skin of Preparations Containing Acidic or Alkaline Substance Without Testing on Animals. Toxicol. In Vitro, 2, 19-26. |
|
(5) |
Neun, D.J. (1993) Effects of Alkalinity on the Eye Irritation Potential of Solutions Prepared at a Single pH. J. Toxicol. Cut. Ocular Toxicol. 12, 227-231. |
|
(6) |
Fentem, J.H., Archer, G.E.B., Balls, M., Botham, P.A., Curren, R.D., Earl, L.K., Edsaile, D.J., Holzhutter, H.G. and Liebsch, M. (1998) The ECVAM international validation study on in vitro tests for skin corrosivity. 2. Results and evaluation by the Management Team. Toxicology in Vitro 12, s. 483-524. |
|
(6a) |
Forsøgsmetode B.40 Hudætsning. |
|
(7) |
Forsøgsmetode B.4. Akut toksicitet: Hudirritation/ætsning. |
|
(8) |
OECD (1996) OECD Test Guidelines Programme: Final Report of the OECD Workshop on Harmonization of Validation and Acceptance Criteria for Alternative Toxicological Test Methods. Afholdt i Solna, Sverige, den 22.-24. januar 1996 (http://www.oecd.org/ehs/test/background.htm). |
|
(9) |
OECD (1998) Harmonized Integrated Hazard Classification System for Human Health and Environmental Effects of Chemical Substances, som vedtaget på det 28. fælles møde mellem OECD's kemikalieudvalg og arbejdsgruppe vedrørende kemikalier, november 1998 (http://www.oecd.org/ehs/Class/HCL6.htm). |
|
(10) |
OECD (2000) Guidance Document on the Recognition, Assessment and Use of Clinical Signs as Humane Endpoints for Experimental Animals Used in Safety Evaluation. OECD Environmental Health and Safety Publications. Series on Testing and Assessment No. 19 (http://www.oecd.org/ehs/test/monos.htm). |
Tabel I
KLASSIFICERING AF ØJENSKADER
Cornea
Uklarhed: graden af uklarhed aflæses i det tætteste område (1)
|
Ingen ulceration eller uklarhed … |
0 |
|
Spredte eller diffuse uklare områder (ikke bestående i en let mattering af den normale glans); detaljer i iris klart synlige … |
1 |
|
Let skelneligt halvgennemsigtigt område; detaljerne i iris let slørede … |
2 |
|
Område med perlemorsglans; ingen detaljer i iris synlige; pupilstørrelse kan knapt skelnes … |
3 |
|
Cornea uklar; iris ikke skelnelig gennem uklarheden … |
4 |
Maksimum: 4
NOTER
Iris
|
Normal … |
0 |
|
Markant fordybede rugae, kongestion, hævelse, moderat hyperæmi omkring cornea; eller injektion; iris reagerer på lys (træg lysreaktion anses for en virkning) … |
1 |
|
Blødning, massiv ødelæggelse, eller ingen reaktion på lys … |
2 |
Maksimum: 2
Conjunctivae
Rødme (af conjunctiva palpebrae og conjunctiva bulbi; bortset fra cornea og iris)
|
Normal … |
0 |
|
Nogle blodkar hyperæmiske (injektion) … |
1 |
|
Diffus, blodrød farve; de enkelte kar ikke let skelnelige … |
2 |
|
Diffus oksekødsfarvet … |
3 |
Maksimum: 3
Chemosis
Hævelse (om øjenlåg og/eller blinkhinder)
|
Normal … |
0 |
|
Nogen unormal hævelse … |
1 |
|
Tydelig hævelse med delvis udkrængning af øjenlåg … |
2 |
|
Hævelse med øjenlåg omtrent halvt lukket … |
3 |
|
Hævelse med øjenlåg mere end halvt lukket … |
4 |
Maksimum: 4
BILAG
Sekventiel testningsstrategi for øjenirritation og ætsning
GENERELLE OVERVEJELSER
Såvel hensynet til den videnskabelige kvalitet som til dyrenes velfærd taler for at undgå unødvendig brug af dyr og at minimere alle forsøg, som kan forventes at fremkalde svære reaktioner i dyr. Alle oplysninger om et stofs potentielle øjenætsende og irriterende egenskaber skal vurderes, før in vivo-testning overvejes. Der foreligger muligvis allerede tilstrækkelige vidnesbyrd til, at teststoffets potentiale for ætsning eller irritation af øjnene kan klassificeres, uden at der behøves laboratorieforsøg udført med dyr. Ved hjælp af weight-of-the-evidence-analyse og en sekventiel testningsstrategi kan behovet for in vivo-testning således nedsættes til et minimum, navnlig når stoffet forventes at fremkalde svære reaktioner.
Det anbefales, at de foreliggende oplysninger om stoffers øjenirriterende og ætsende egenskaber vurderes ved weight-of-the-evidence-analyse for at afgøre, om der ud over in vivo-øjenundersøgelser bør udføres supplerende undersøgelser til nærmere karakterisering af et sådant potentiale. Når yderligere undersøgelser er nødvendige, anbefales det at benytte den sekventielle testningsstrategi til fastlæggelse af de relevante forsøgsdata. For ikke tidligere testede stoffer anvendes den sekventielle testningsstrategi til at fastlægge det sæt data, som er nødvendigt til vurdering af stoffets potentiale for øjenirritation/ætsning. Den i dette bilag beskrevne testningsstrategi er udviklet på en OECD-workshop (1). Den er senere blevet bekræftet og udvidet i form af Harmonised Integrated Hazard Classification System for Human Health and Environmental Effects of Chemical Substances, som vedtoges på det 28, fælles møde mellem OECD's kemikalieudvalg og arbejdsgruppe vedrørende kemikalier i november 1998 (2).
Skønt denne sekventielle testning ikke indgår som en del af testmetode B.5, er det den anbefalede strategi til bestemmelse af øjenirritation/ætsning. Strategien repræsenterer både bedste praksis og en etisk standard for in vivo-testning af øjenirritation/ætsning. Testmetoden giver vejledning i udførelse af in vivo-forsøg og sammenfatter de faktorer, som bør tages i betragtning, før et sådant forsøg påbegyndes, Strategien tilvejebringer en metode til evaluering af foreliggende oplysninger om teststoffets øjenirriterende og ætsende egenskaber og en trinvis fremgangsmåde til at generere relevante data om stoffer, som har behov for yderligere undersøgelse eller ikke er blevet undersøgt, Strategien indebærer, at der først udføres validerede og anerkendte in vivo- eller ex vivo-test, og derefter forsøgsmetode B.4, hudirritation/ætsning, alt efter omstændighederne (3)(4).
BESKRIVELSE AF DEN TRINVISE TESTNINGSSTRATEGI
Før man iværksætter test som led i den sekventielle testningsstrategi (vist i figuren), skal alle foreliggende oplysninger vurderes for at afgøre nødvendigheden af in vivo-øjentest. Skønt vurdering af enkeltparametre (f.eks. ekstremt pH) i sig selv kan tænkes at give væsentlige oplysninger, må alle de foreliggende oplysninger betragtes under ét, Som grundlag for en afgørelse baseret på weight-of-the-evidence må alle relevante oplysninger om virkningerne af det pågældende stof og af analoge stoffer evalueres, og et rationale for afgørelsen fremlægges. Primært må foreliggende oplysninger om stoffets virkning på dyr og mennesker tillægges vægt, efterfulgt af resultaterne af in vivo. eller ex vivo-testning. Når det på nogen måde er muligt, bør in vivo-undersøgelser af ætsende stoffer undgås. I testningsstrategien bør følgende faktorer tages i betragtning;
Vurdering af foreliggende data fra mennesker og dyr (trin 1). Foreliggende data fra mennesker, f.eks. kliniske undersøgelser eller arbejdsmiljøundersøgelser, case-undersøgelser og/eller data fra dyreforsøg fra f.eks. øjenstudier skal primært tages i betragtning, da de giver oplysninger direkte vedrørende virkninger på øjnene. Derefter vurderes foreliggende data fra undersøgelser af hudætsning/irritation i mennesker og/eller dyr. Stoffer med kendt ætsende eller irriterende virkning på øjet bør ikke instilleres i øjenene på dyr, hvilket ligeledes gælder stoffer, som virker ætsende eller irriterende på huden, da disse må anses for at virke ætsende og/eller irriterende også på øjnene. Stoffer, for hvilke der foreligger tilstrækkelig dokumentation for manglende ætsende og irriterende virkning fra tidligere øjenstudier, bør heller ikke testes i in vivo-øjenstudier.
Analyse af struktur/aktivitetrelationer (trin 2). Resultater fra eventuelle undersøgelser af strukturelt beslægtede stoffer skal tages i betragtning, Når der for strukturelt beslægtede stoffer eller blandinger af sådanne stoffer foreligger tilstrækkelige data fra mennesker og/eller dyr til at bestemme deres potentiale for ætsning/irritation af øjnene, kan teststoffet antages at ville frembringe samme responser. I sådanne tilfælde behøver teststoffet ikke nødvendigvis testes. Negative data fra undersøgelser af strukturelt beslægtede stoffer eller blandinger af sådanne stoffer udgør ikke tilstrækkeligt bevis for fravær af ætsende eller irriterende egenskaber af et stof i den sekventielle testningsstrategi. Potentialet for ætsende og og irriterende virkning på både hud og øje bør fastlægges ved validerede og anerkendte metoder til analyse af struktur/aktivitetrelationer.
Fysisk-kemiske og kemisk reaktivitet (trin 3). Stoffer, som udviser ekstremt pH som ≤ 2,0 eller ≤ 11,5, kan have stærke lokal virkninger. Hvis et ekstremt pH er grundlaget for at udpege et stof som ætsende eller irriterende for øjnene, kan også dets syre/basereserve (eller bufferkapacitet) tages i betragtning (5)(6). Tyder stoffets bufferkapacitet på, at det muligvis ikke virker ætsende på øjnene, bør dette bekræftes ved videre testning, fortrinsvis med brug af en valideret og anerkendt in vitro- eller ex vivo-test (jf, trin 5 og 6).
Hensyntagen til andre foreliggende oplysninger (trin 4). Alle foreliggende oplysninger om systemisk toksicitet ved påføring på huden vurderes på dette trin. Teststoffets akutte toksicitet ved optagelse gennem huden skal ligeledes tages i betragtning. Er teststoffet påvist at være meget toksisk ved optagelse gennem huden, behøver det ikke nødvendigvis testes i øjenene. Skønt der ikke nødvendigvis er sammenhæng mellem akut toksicitet ved optagelse gennem huden og øjenirritation/ætsning, kan man gå ud fra, at når et agens er meget toksisk ved optagelse gennem huden, vil det ligeledes vise sig meget toksisk ved instillation i øjet. Sådanne oplysninger kan ligeledes tages i betragtning mellem trin 2 og 3.
Resultater af in vitro- eller ex vivo-undersøgelser (trin 5 og 6). Stoffer, som har udvist ætsende eller stærkt irriterende egenskaber i en in vitro- eller ex vivo-undersøgelse (7)(8), som er valideret og anerkendt specielt til vurdering af ætsende/irriterende virkning på øjne eller hud, behøver ikke testes i dyreforsøg. Sådanne stoffer må antages at ville frembringe tilsvarende svære virkninger in vivo. Foreligger der ikke validerede og anerkendte in vitro- og/eller ex vivo-test, springes trin 5 og 6 over og fortsættes direkte med trin 7.
Vurdering af stoffets hudirriterende eller ætsende virkning in vivo (trin 7). Når ovennævnte undersøgelser ikke udgør tilstrækkelig dokumentation til at foretage en konklusiv weight-of-the-evidence-analyse af et stofs potentielle irriterende/ætsende virkning på øjet, bestemmer man først dets potentiale for in vivo-hudirritation/ætsning ved hjælp af forsøgsmetode B.4 (4) og det tilhørende bilag (9). Er stoffet påvist at forårsage ætsning eller svær hudirritation, bør det anses for øjenætsende, medmindre andre oplysninger taler for en anden konklusion. Der behøver således ikke foretages nogen in vivo-øjentest. Er stoffet ikke ætsende eller ikke stærkt hudirriterende, udføres en in vivo-øjentest.
In vivo-test i kaniner (trin 8 og 9). In vivo-øjentestning bør begynde med en indledende test i ét dyr. Viser stoffet sig i denne test ætsende eller stærkt irriterende på øjet, bør yderligere test ikke udføres. Viser testen ingen ætsende eller ingen stærkt irriterende virkninger, udføres en verifikationstest i yderligere to dyr.
HENVISNINGER
|
(1) |
OECD (1996) OECD Test Guidelines Programme: Final Report of the OECD Workshop on Harmonization of Validation and Acceptance Criteria for Alternative Toxicological Test Methods. Afholdt i Solna, Sverige, den 22.-24. januar 1996 (http://www.oecd.org/ehs/test/background.htm). |
|
(2) |
OECD (1998) Harmonized Integrated Hazard Classification System for Human Health and Environmental Effects of Chemical Substances, som vedtaget på det 28. fælles møde mellem OECD's kemikalieudvalg og arbejdsgruppe vedrørende kemikalier, november 1998 (http://www.oecd.org/ehs/Class/HCL6.htm). |
|
(3) |
Worth, A.P. and Fentem J.H. (1999). A General Approach for Evaluating Stepwise Testing Strategies, ATLA 27, 161-177. |
|
(4) |
Forsøgsmetode B.4. Akut toksicitet: hudirritation/ætsning. |
|
(5) |
Young, J.R., How, M.J., Walker, A.P., Worth W.M.H. (1988) Classification as Corrosive or Irritant to Skin of Preparations Containing Acidic or Alkaline Substance Without Testing on Animals. Toxicol. In Vitro, 2, 19-26. |
|
(6) |
Neun, D.J. (1993) Effects of Alkalinity on the Eye Irritation Potential of Solutions Prepared at a Single pH. J. Toxicol. Cut. Ocular Toxicol, 12, 227-231. |
|
(7) |
Fentem, J.H., Archer, G.E.B., Balls, M., Botham, P.A., Curren, R.D., Earl, L.K., Edsail, D.J., Holzhutter, H.G. and Liebsch, M. (1998) The ECVAM international validation study on in vitro tests for skin corrosivity. 2. Results and evaluation by the Management Team. Toxicology in Vitro 12, s. 483-524. |
|
(8) |
Forsøgsmetode B.40 Hudætsning. |
|
(9) |
Bilag til forsøgsmetode B.4: Sekventiel testningsstrategi for hudirritation og ætsning. |
Figur
En strategi for testning og vurdering af øjenirritation/ætsning
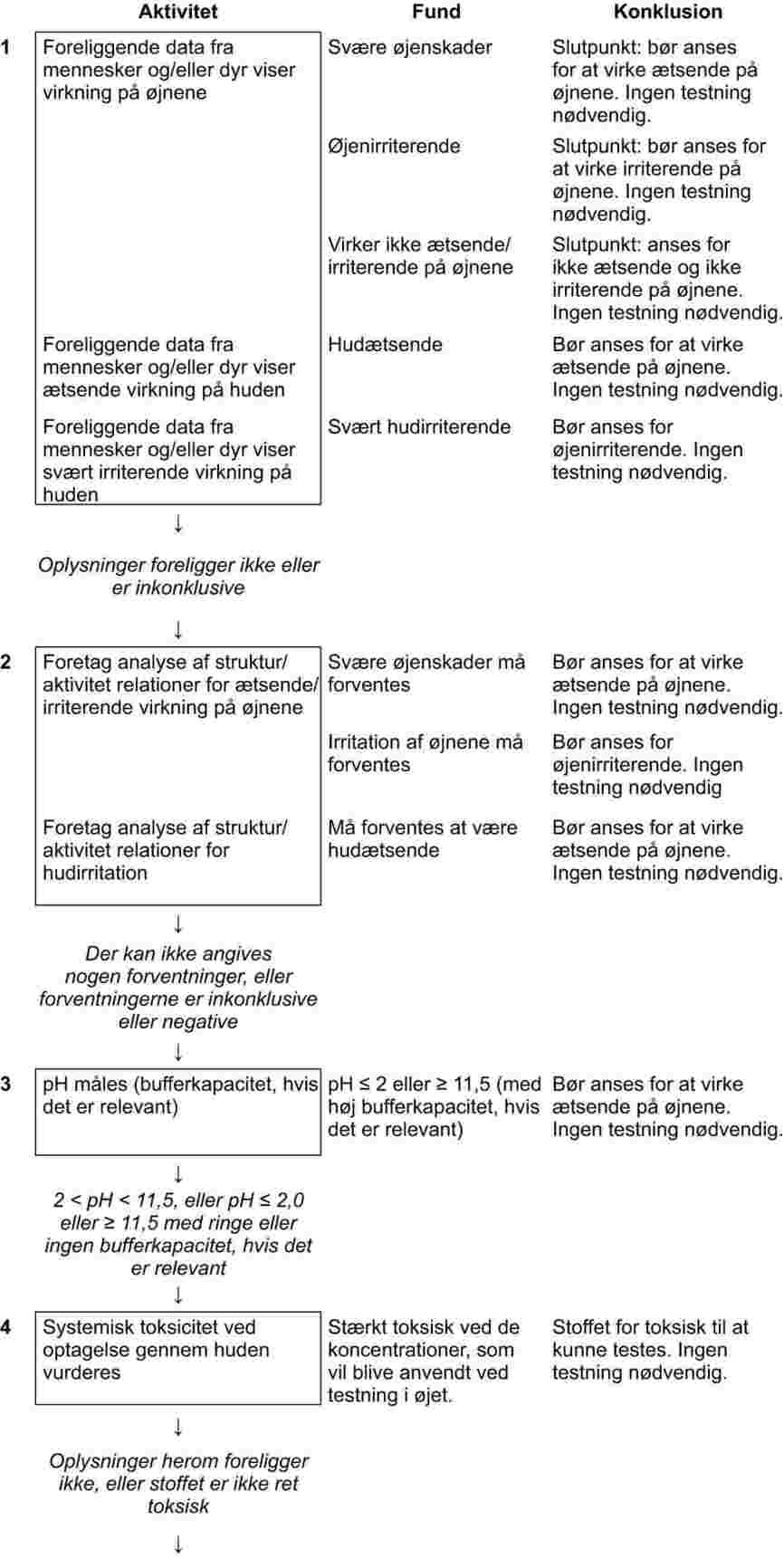

B.6. HUDSENSIBILISERING
1. METODE
1.1. INDLEDNING
Bemærkninger:
I et system for klassifikation af den toksicitet, der er relevant for folkesundheden, er det af stor betydning at have følsomme test, som kan afsløre stoffer, der kan virke hudsensibiliserende på mennesker.
Der findes ikke én enkelt testmetode, hvorved man med sikkerhed kan identificere alle stoffer, som kan virke hudsensibiliserende på mennesker, og som kan anvendes til alle stoffer.
Der skal ved valg af test tages hensyn til en række faktorer såsom stoffets fysiske egenskaber, herunder dets evne til at trænge gennem huden.
Der er udviklet to testtyper med anvendelse af marsvin: adjuvanstypen, hvor en allergisk tilstand forstærkes, når teststoffet opløses eller opslæmmes i Freunds komplette adjuvans (FCA), og typen uden adjuvans.
Test af adjuvanstypen vil normalt forudsige et stofs sandsynlige hudsensibiliserende virkning hos mennesker mere præcist end de metoder, hvor der ikke benyttes Freunds komplette adjuvans, og bliver derfor foretrukket.
Maksimeringstest på marsvin (Guinea-Pig Maximization Test — GPMT) er en meget anvendt test af adjuvanstypen. Skønt en række andre metoder kan benyttes til at undersøge et stofs potentielle hudsensibiliserende virkning, er GPMT den foretrukne metode af adjuvanstypen.
Test uden adjuvans (hvor Buehlertesten er den foretrukne) bliver for mange kemiske stofgrupper anset for at være mindre følsom.
Der kan i visse tilfælde, være gode grunde til at vælge Buehlertesten med lokal påføring i stedet for den intradermale injektion, der benyttes i maksimeringstesten på marsvin. Der skal gives en videnskabelig begrundelse for anvendelse af Buehlertesten.
I denne metode beskrives maksimeringstesten på marsvin (GPMT) og Buehlertesten. Der kan anvendes andre metoder, forudsat at de er tilstrækkeligt validerede, og at valget er videnskabeligt begrundet.
Hvis testresultatet i en af de anerkendte screeningstest er positivt, er det tilladt at betegne stoffet som potentielt sensibiliserende og det er måske ikke nødvendigt at udføre yderligere marsvineforsøg. Hvis stoffet er negativt i en af de anerkendte screeningsmetoder, skal der udføres en marsvinetest, hvor der bruges de fremgangsmåder, der er beskrevet i denne testmetode.
Se også den generelle indledning til del B.
1.2. DEFINITIONER
Hudsensibilisering: (allergisk kontaktdermatitis) er en immunologisk hudreaktion på et stof. Hos mennesker kan reaktionerne være karakteriseret ved kløen, erytem, ødem, papler, vesikler, blærer eller en kombination heraf. Andre arter kan have andre reaktioner f.eks. kun erytem og ødem.
Induktionseksponering: eksperimentel eksponering af et individ for et teststof med henblik på fremkaldelse af hypersensibel tilstand.
Induktionsperiode: tidsrum på mindst en uge efter en induktionseksponering, under hvilken en hypersensibel tilstand kan være opstået.
Provokationseksponering: eksperimentel eksponering — efter en induktionsperiode — af et tidligere behandlet individ for et teststof til konstatering af, om individet reagerer hypersensibelt.
1.3. REFERENCESTOFFER
Den anvendte forsøgsmetodes følsomhed og pålidelighed vurderes hver sjette måned med anvendelse af stoffer, der vides at have mildt til moderat hudsensibiliserende egenskaber.
I en velgennemført test bør der for mildt/moderat sensibiliserende stoffer ventes en reaktion på mindst 30 % i en adjuvanstest og mindst 15 % i en test uden adjuvans.
Følgende stoffer foretrækkes:
|
CAS-nummer |
Einecs-nummer |
Einecs-navn |
Fælles navn |
|
101-86-0 |
202-983-3 |
α-hexylkanelaldehyd |
α-hexylkanelaldehyd |
|
149-30-4 |
205-736-8 |
benzothiazol-2-thiol (mercaptobenzothiazol) |
kaptax |
|
94-09-7 |
202-303-5 |
benzocain |
norcain |
Under visse omstændigheder vil andre kontrolstoffer, der opfylder de ovennævnte kriterier, kunne anvendes, såfremt valget heraf kan begrundes.
1.4. TESTMETODENS PRINCIP
Forsøgsdyrene udsættes først for teststoffet med intradermale injektioner og/eller epidermal applikation (induktionseksponering). Efter en hvileperiode på 10-14 dage (induktionsperiode), hvor der kan udvikle sig immunrespons, udsættes dyrene for en provokationsdosis. Omfanget og graden af hudens reaktion på provokationseksponeringen i forsøgsdyrene sammenlignes med resultater fra kontroldyr, der har fået placebo-behandling i induktionsperioden og dernæst er eksponeret for provokationsdosis.
1.5. BESKRIVELSE AF TESTMETODERNE
Hvis det anses for nødvendigt at fjerne teststoffet, bør dette gøres med vand eller et passende opløsningsmiddel, uden at den fremkaldte reaktion ændres eller overhuden beskadiges.
1.5.1. Maksimeringstest på marsvin (Guinea-Pig Maximization Test — GPMT)
1.5.1.1.
Unge, sunde, kønsmodne albinomarsvin tilvænnes laboratoriebetingelserne i mindst fem dage forud for testen. Før forsøget begyndes, fordeles dyrene randomiseret i behandlingsgrupper. Behåring fjernes ved klipning, barbering eller eventuelt med kemiske midler, afhængigt af testmetode. Huden må ikke irriteres. Dyrene vejes, inden forsøget påbegyndes, og ved forsøgets afslutning.
1.5.1.2.
1.5.1.2.1. Forsøgsdyr
Der benyttes albinomarsvin af almindeligt anvendte stammer.
1.5.1.2.2. Antal og køn
Der kan anvendes handyr og/eller hundyr. Hvis der benyttes hundyr, må de ikke have født, og de må ikke være drægtige.
Forsøgsgruppen skal bestå af mindst ti dyr og kontrolgruppen af mindst fem dyr. Hvis der anvendtes mindre end tyve forsøgsmarsvin og ti kontrolmarsvin og det ikke er muligt at konkludere, at teststoffet er sensibiliserende, anbefales det stærkt at teste stoffet i yderligere dyr, så der i alt er tale om mindst tyve forsøgsdyr og ti kontroldyr.
1.5.1.2.3. Dosisniveauer
Den koncentration af teststoffet, der anvendes ved hver induktionseksponertng, må ikke forårsage systemiske gener og skal være den størst mulige, der forårsager mild til moderat hudirritation. Provokationskoncentrationen skal være den størst mulige, der ikke femkalder hudirritation. Hvis nødvendigt kan de relevante koncentrationer bestemmes ved hjælp af en pilotundersøgelse med to eller tre dyr. Det bør overvejes at bruge FCA-behand! ede dyr til dette formål.
1.5.1.3.
1.5.1.3.1. Induktion
Dag 0 — forsøgsgruppe
Tre intradermale injektioner på 0,1 ml gives parvis i skulderpartiet — hvorfra hårlaget er fjernet — således, at hvert injektionspar består af en injektion på hver side af midterlinjen.
Injektion 1: en blanding i forholdet 1:1 (volumenprocent) af FCA/vand eller fysiologisk saltopløsning.
Injektion 2: teststoffet i et passende vehikel i den udvalgte koncentration.
Injektion 3: teststoffet i den valgte koncentration i en blanding i forholdet 1:1 (volumenprocent) FCA/vand eller fysiologisk saltopløsning.
I injektion 3 opløses vandopløselige stoffer i vandfasen før de blandes med FCA. Fedtopløselige eller uopløselige stoffer opslæmmes i FCA forud for blanding med den vandige fase. Teststoffets slutkoncentration skal være den samme som i injektion 2.
Injektion 1 og 2 placeres tæt på hinanden og nærmest ved hovedet, mens injektion 3 placeres i den bageste del af testområdet.
Dag 0 — kontrolgruppe
Tre intradermale injektioner på 0,1 ml gives parvis på samme måde som for forsøgsdyrene.
Injektion 1: en blanding i forholdet 1:1 (volumenprocent) FCA/vand eller fysiologisk saltopløsning.
Injektion 2: det ufortyndede vehikel.
Injektion 3: en 50 vægtprocent formulering af vehiklet i en blanding i forholdet 1:1 (volumenprocent) af FCA/vand eller fysiologisk saltopløsning.
Dag 5-7 — forsøgsgruppe og kontrolgruppe
Hvis stoffet ikke er hudirriterende, fremkaldes der, ca. 24 timer inden den lokale induktionsapplikation, en lokal irritation ved behandling af testområdet med 0,5 ml 10 % natriumlaurylsulfat i vaseline, efter klipning og/eller barbering.
Dag 6-8 — forsøgsgruppe
Testområdet klippes. Et stykke filtrerpapir (2 × 4 cm) gennemvædes med teststoffet, der er blandet med et passende vehikel, og anbringes på testområdet og fastholdes med en okklusiv forbinding i 48 timer. Valget af vehikel skal begrundes. Faste stoffer pulveriseres og inkorporeres i et passende vehikel. Væske kan eventuelt anvendes direkte.
Dag 6-8 — kontrolgruppe
Testområdet klippes. Vehikel alene anbringes på tilsvarende måde på testområdet og fastholdes med en okklusiv forbinding i 48 timer.
1.5.1.3.2. Provokation
Dag 20-22 — forsøgs- og kontrolgruppe
Både i forsøgsgruppen og i kontrolgruppen befries dyrenes flanke for hår. På forsøgsdyrenes ene side anbringes en lap eller en kapsel med teststoffet, og, hvis det er relevant, kan der også anbringes en lap eller en kapsel med rent vehikel på den anden side. Lapperne holdes i kontakt med huden i 24 timer med en okklusiv forbinding.
1.5.1.3.3. Observation og scoring: forsøgs- og kontrolgruppe
|
— |
ca. 21 timer efter at lappen er fjernet, renses provokationsområdet og befries om nødvendigt for hår |
|
— |
ca. 3 timer senere (ca. 48 timer efter påføring af provokationsdosis) observeres hudens reaktion, som registreres efter den scoringsskala, der er vist i tillægget |
|
— |
ca. 24 timer efter denne observation foretages endnu en observation (72 timer), som registreres. |
Blindlæsning af forsøgs- og kontroldyrene anbefales.
Hvis det er nødvendige at uddybe resultaterne af den første provokationseksponering, bør det overvejes at foretage endnu en provokationseksponering (dvs. en fornyet provokation), eventuelt med en ny kontrolgruppe, ca. 1 uge efter den første eksponering. En fornyet provokation kan også foretages på den oprindelige kontrolgruppe.
Alle hudreaktioner og alle usædvanlige virkninger, herunder systemiske reaktioner, af både induktions- og provokationseksponeringen skal observeres og registreres i henhold til Magnusson/Kligmans scoringsskala (se tillægget). Andre teknikker, f.eks. histopatologisk undersøgelse og måling af hudfortykkelse, kan benyttes til afklaring af tvetydige reaktioner.
1.5.2. Buehlertest
1.5.2.1.
Unge, sunde, kønsmodne albinomarsvin tilvænnes laboratoriebetingelserne i mindst fem dage forud for testen. Før forsøget begyndes, fordeles dyrene randomiseret i behandlingsgrupper. Behåring fjernes ved klipning, barbering eller eventuelt med kemiske midler, afhængigt af testmetode. Huden må ikke irriteres. Dyrene vejes, inden forsøget påbegyndes, og ved forsøgets afslutning.
1.5.2.2.
1.5.2.2.1. Forsøgsdyr
Der benyttes albinomarsvin af almindeligt anvendte stammer.
1.5.2.2.2. Antal og køn
Der kan anvendes handyr og/eller hundyr. Hvis der benyttes hundyr, må de ikke have født, og de må ikke være drægtige.
Forsøgsgruppen skal bestå af mindst 20 dyr og kontrolgruppen af mindst 10 dyr.
1.5.2.2.3. Dosisniveauer
Der vælges en så høj koncentration af teststoffet for hver induktionseksponering, at der fremkaldes en mild, men ikke overdreven irritation. Provokationskoncentrationen bør være den størst mulige, der ikke fremkalder irritation. Hvis nødvendigt kan de relevante koncentrationer bestemmes ved hjælp af en pilotundersøgelse med to eller tre dyr.
For vandopløselige teststoffer kan der som vehikel bruges vand eller en fortyndet, ikke-irriterende opløsning af et overfladeaktivt stof. For andre teststoffer foretrækkes 80 % ethanol/vand til induktionseksponering og acetone til provokationseksponering.
1.5.2.3.
1.5.2.3.1. Induktion
Dag 0 — forsøgsgruppe
Dyrene befries for hår (klippes tæt) på den ene flanke. Testlappesystemet skal være gennemvædet med teststoffet i et egnet vehikel (valg af vehikel skal begrundes; væsker kan eventuelt anvendes direkte).
Testlappesystemet anbringes på testområdet og fastholdes i kontakt med huden med en okklusiv lap eller kapsel og en passende forbinding i seks timer.
Testlappesystemet skal være okklusivt. Et stykke gaze er passende: det kan være rundt eller firkantet, af en størrelse på ca. 4-6 cm2. For at sikre okklusion foretrækkes det, at dyrenes bevægelsesfrihed begrænses med passende midler. Hvis der anvendes bandage, kan yderligere eksponering være nødvendig.
Dag 0 — kontrolgruppe
Dyrene befries for hår (klippes tæt) på den ene flanke. Vehiklet alene anbringes på testområdet på tilsvarende måde som for forsøgsgruppen. Testlappesystemet fastholdes i kontakt med huden med en okklusiv lap eller kapsel og en passende forbinding i seks timer. Hvis det kan påvises, at der ikke er behov for en placebo-kontrolgruppe, kan en ikke forud testet kontrolgruppe (naive control group) bruges.
Dag 6-8 og 13-15 — forsøgs- og kontrolgruppen
På dag 6-8 og igen på dag 13-15 foretages samme påføring som på dag 0 på samme testområde (om nødvendigt befriet for hår).
1.5.2.3.2. Provokation
Dag 27-29 — forsøgs- og kontrolgruppe
Både i forsøgsgruppen og i kontrolgruppen befries dyrene for hår (klippes tæt) på den anden flanke. Bagest på flanken på forsøgs- og kontroldyrene anbringes en okklusiv lap eller kapsel med en passende mængde teststof i den højest mulige koncentration, der ikke fremkalder irritation.
Hvis det er relevant, anbringes desuden en okklusiv lap eller kapsel med vehikel alene forrest på flanken på såvel forsøgs- som kontroldyr. Lapper og kapsler fastholdes i kontakt med huden med en passende forbinding i seks timer.
1.5.2.3.3. Observation og scoring
|
— |
ca. 21 timer efter at lappen er fjernet, befries provokationsområdet for hår |
|
— |
ca. tre timer senere (ca. 30 timer efter påføring af provokationslap) observeres hudens reaktion, og denne registreres i henhold til den scoringsskala, der er vist i tillægget |
|
— |
ca. 24 timer efter observationen efter 30 timer (dvs. 54 timer efter påføring af provokationslap) observeres hudens reaktioner igen og registreres. |
Blindlæsning af forsøgs- og kontroldyrene anbefales.
Hvis det er nødvendigt at uddybe resultaterne af den første provokationseksponering, bør det overvejes at foretage endnu en provokationseksponering (dvs. en fornyet provokation), eventuelt med en ny kontrolgruppe, ca. en uge efter den første eksponering. En fornyet provokation kan også foretages på den oprindelige kontrolgruppe.
Alle hudreaktioner og alle usædvanlige virkninger, herunder systemiske reaktioner, af både induktions- og provokationseksponeringen skal observeres og registreres i henhold til Magnussen/Kligmans scoringsskala (se tillægget). Andre teknikker, f.eks. histopatologisk undersøgelse og måling af hudfortykkelse, kan benyttes til afklaring af tvetydige reaktioner.
2. DATA (GPMT og Buehlertest)
Data opstilles i tabelform, som angiver hudens reaktion ved hver enkelt observation af hver enkelt dyr.
3. RAPPORTERING (GPMT og Buehlertest)
Hvis der udføres en screeningtest inden marsvinetesten, skal der sammen med de resultater, der er opnået med test- og referencestofferne, gives en beskrivelse af eller henvisning til testen (f.eks. Mouse Ear Swelling Test (MEST)), herunder oplysninger om fremgangsmåde.
Forsøgsrapport (GPMT og Buehlertest)
Forsøgsrapporten skal om muligt indholde følgende oplysninger:
|
|
Forsøgsdyr:
|
|
|
Forsøgsbetingelser:
|
|
|
Resultater:
|
|
|
Diskussion af resultaterne |
|
|
Konklusioner |
4. HENVISNINGER
Denne metode svarer til TG 406 fra OECD.
Tillæg
TABEL
Magnusson/Kligmans scoringsskala til vurdering af reaktionerne på provokationslappetest
0 = ingen synlig forandring
1 = afgrænset eller pletvis erytem
2 = moderat og sammenflydende erytem
3 = kraftigt erytem og hævelse.
B.7. TOKSICITET VED GENTAGEN DOSERING (28 DAGE, ORAL)
1. METODE
1.1. INDLEDNING
Se den generelle indledning til del B.
1.2. DEFINITIONER
Se den generelle indledning til del B.
1.3. TESTMETODENS PRINCIP
Teststoffet indgives oralt dagligt i graduerede doser til grupper af forsøgsdyr, idet der gives en bestemt dosis til hver gruppe gennem en periode på 28 dage. I eksponeringsperioden observeres dyrene hver dag omhyggeligt for tegn på toksicitet. Dyr, der dør eller aflives under forsøget, obduceres, og de overlevende dyr aflives og obduceres ved forsøgets afslutning.
Med denne metode lægges der større vægt på neurologiske effekter som et specifikt endepunkt; dyrene skal underkastes omhyggelige kliniske observationer med henblik på indhentning af flest mulige oplysninger. Metoden tager sigte på at identificere kemiske stoffer med neurotoksisk potentiale, som i givet fald bør undersøges nøjere. Herudover kan metoden vise, om der er tegn på immunologiske effekter og toksicitet for kønsorganerne.
1.4. BESKRIVELSE AF TESTMETODEN
1.4.1. Forberedelser
Unge, sunde, kønsmodne dyr fordeles randomiseret i kontrol- og forsøgsgrupper. Burene bør anbringes således, at mulige virkninger som følge af burenes placering minimeres. Dyrene mærkes individuelt og tilbringer mindst fem dage i deres bure forud for forsøgets påbegyndelse, så de tilvænnes laboratoriebetingelserne.
Teststoffet indgives med mavesonde, i foderet eller i drikkevandet, afhængigt af formålet med forsøget og stoffets fysisk-kemiske egenskaber.
Om nødvendigt opløses eller opslæmmes teststoffet i et egnet vehikel. Det anbefales, at der så vidt muligt anvendes en vandig opløsning/opslæmning og, hvis dette ikke er muligt, en opløsning/emulsion i olie (f.eks. majsolie) eller eventuelt en opløsning i andre vehikler. Ikke-vandige vehiklers toksiske egenskaber skal være kendt. Teststoffets stabilitet i vehiklet bør bestemmes.
1.4.2. Forsøgsbetingelser
1.4.2.1. Forsøgsdyr
Rotten er den foretrukne gnaverart, skønt andre gnaverarter kan anvendes. Der benyttes unge, sunde rotter af almindeligt anvendte laboratoriestammer. Hundyrene må ikke have født og må ikke være drægtige. Dosering bør påbegyndes tidligst muligt efter fravænning og under alle omstændigheder, inden rotterne er ni uger gamle.
Ved forsøgets begyndelse skal vægten af de anvendte dyr variere mindst muligt og ikke mere end ± 20 % af gennemsnitsvægten af hvert køn.
Hvis en undersøgelse med gentagen oral dosis gennemføres som en forundersøgelse forud for en langtidsundersøgelse, bør der så vidt muligt anvendes dyr fra samme stamme og herkomst i begge undersøgelser.
1.4.2.2. Antal og køn
Der anvendes mindst ti dyr (fem hunner og fem hanner) ved hvert dosisniveau. Hvis der ifølge forsøgsprotokollen skal aflives dyr under forsøget, skal det oprindelige antal dyr forøges med det antal dyr, der påregnes aflivet.
Man kan samtidig udsætte en satellitgruppe på ti dyr (fem af hvert køn) for den højeste dosis i 28 dage og gennem en periode på 14 dage efter forsøget observere, om der er reversible, varige eller forsinkede toksiske virkninger. Der anvendes tillige en satellitgruppe på ti kontroldyr (fem af hvert køn).
1.4.2.3. Dosisniveauer
Der anvendes generelt mindst tre forsøgsgrupper og en kontrolgruppe. Dyrene i kontrolgruppen behandles nøjagtigt som dyrene i forsøgsgrupperne, bortset fra indgiften af teststoffet. Hvis der anvendes et vehikel til indgift af teststoffet, skal kontrolgruppen indgive vehiklet det største af de anvendte volumina.
Hvis der ud fra andre data ikke kan ventes virkninger ved en dosis på 1 000 mg/kg legemsvægt/dag, kan der gennemføres en grænsetest. Foreligger der ikke passende data, kan der udføres en »range finding« -undersøgelse til bestemmelse af de doser, der skal anvendes.
Dosisniveauerne udvælges under hensyntagen til eventuelle foreliggende data om teststoffet eller beslægtede stoffers toksicitet og (toksiko)kinetik. Den højeste dosis vælges med henblik på fremkaldelse af toksiske effekter, men ikke død eller alvorlig lidelse. Normalt vælges en faldende række dosisniveauer til påvisning af eventuelt dosisafhængigt respons og niveau, hvor skadelige effekter ikke kan observeres ved det laveste dosisniveau (NOAEL). Det optimale interval mellem to doser er ofte en faktor på 2-4, og tilføjelse af en fjerde forsøgsgruppe er ofte at foretrække frem for anvendelse af meget store intervaller (f.eks. mere end en faktor på 10) mellem dosisniveauerne.
For stoffer, der indgives med foder eller drikkevand, er det vigtigt at sikre, at de anvendte mængder af teststoffet ikke har indflydelse på den normale ernærings- eller væskebalance. Hvis teststoffet indgives med foderet, kan man enten anvende en konstant koncentration i foderet (ppm) eller en konstant dosering i forhold til dyrenes vægt; det benyttede alternativ bør specificeres. Hvis teststoffet indgives med mavesonde, gøres dette på samme tidspunkt hver dag. Doserne justeres, så de er konstant i forhold til dyrenes legemsvægt.
Hvis en undersøgelse med gentagen dosis anvendes som en forundersøgelse forud for en langtidsundersøgelse, bør foderet være det samme i begge undersøgelser.
1.4.2.4. Grænsetest
Hvis en test med et dosisniveau på mindst 1 000 mg/kg legemsvægt/dag eller, ved indgift gennem foder eller drikkevand, en tilsvarende procentdel i foderet eller drikkevandet (baseret på bestemmelse af legemsvægten), udført efter nedenstående fremgangsmåde, ikke fremkalder erkendbare toksiske virkninger, og hvis der ikke på grundlag af data fra strukturelt beslægtede stoffer kan ventes toksiske virkninger, kan en fuldstændig undersøgelse med anvendelse af tre dosisniveauer betragtes som unødvendig. I dette tilfælde bør grænsetesten anvendes, medmindre human eksponering viser, at der er behov for anvendelse af et højere dosisniveau.
1.4.2.5. Observationsperiode
Observationsperioden varer 28 dage. En eventuel satellitgruppe beregnet til opfølgningsobservationer holdes i yderligere 14 dage uden indgift af teststof, således at det kan registreres, om der er forsinkede, vedvarende eller reversible toksiske virkninger.
1.4.3. Fremgangsmåde
Dyrene indgives teststoffet dagligt syv dage om ugen i en periode på 28 dage; fem dages dosering pr. uge skal begrundes. Hvis teststoffet indgives med sonde, skal dette gøres i en enkelt dosis med anvendelse af mavesonde eller passende intubationsrør. Det maksimale væskevolumen, som kan indgives ad gangen, afhænger af forsøgsdyrets størrelse. Volumen bør ikke overstige 1 ml/100 g legemsvægt, undtagen for vandige opløsninger, hvor 2 ml/100 g legemsvægt kan anvendes. Med undtagelse af irriterende eller ætsende stoffer, som normalt vil afsløre stærkere virkninger med højere koncentrationer, bør variationen i testvolumen minimeres ved at justere koncentrationen således, at der sikres en konstant volumen ved alle dosisniveauer.
1.4.3.1. Generelle observationer
Der foretages generelle kliniske observationer mindst én gang om dagen, om muligt på det eller de samme tidspunkter hver dag og under hensyntagen til, hvornår de forventede virkninger efter dosering er stærkest. Dyrenes sundhedstilstand registreres. Dyrene observeres for sygelighed og dødelighed mindst to gange om dagen. Døende og stærkt lidende dyr fjernes, så snart de observeres, aflives humant og obduceres.
Én gang før den første eksponering (med henblik på individbaserede sammenligninger) og mindst én gang om ugen derefter foretages der indgående kliniske observationer af alle dyrene. Med henblik på disse observationer tages dyrene ud af deres egne bure og anbringes i et standardindelukke. Observationerne udføres om muligt på samme tidspunkt hver gang. De registreres omhyggeligt, om muligt med anvendelse af scoringssystemer, hvis anvendelse skal være klart defineret af forsøgslaboratoriet. Det er vigtigt at sikre, at forsøgsbetingelserne varierer minimalt, og at observationerne om muligt foretages af observatører, der ikke har kendskab til behandlingen. Observationerne bør bl.a. være rettet mod ændringer i hud, pels, øjne, slimhinder, forekomst af sekretion, ekskretion og autonom aktivitet (f.eks. tåreflod, strittende hårlag, pupilstørrelse, usædvanligt respirationsmønster). Tillige registreres ændringer i gang, holdning og reaktion på håndtering såvel som forekomsten af kloniske eller toniske bevægelser, stereotyp optræden (f.eks. overdreven soignering, stadig kredsen i cirkelbevægelser) eller mærkelig adfærd (f.eks. automutilation, baglæns gang).
I den fjerde eksponeringsuge vurderes reaktiviteten over for sensoriske stimuli af forskellig art (f.eks. auditive, visuelle og proprioceptive stimuli), gribestyrke og motorisk aktivitet. Litteraturen indeholder yderligere oplysninger om de undersøgelsesmetoder, der kan anvendes (se den generelle indledning til del B).
Funktionelle observationer i den fjerde eksponeringsuge kan udelades, hvis undersøgelsen gennemføres som en forundersøgelse til en efterfølgende undersøgelse af subkronisk toksicitet (90 dage). I så fald medtages de funktionelle observationer i den opfølgende undersøgelse. På den anden side kan data vedrørende funktionelle observationer fra undersøgelsen med gentagen dosis lette valget af dosisniveauer for en efterfølgende undersøgelse af subkronisk toksicitet.
I undtagelsestilfælde kan funktionelle observationer ligeledes udelades for grupper, der viser så tydelige tegn på toksicitet, at det i signifikant grad ville indvirke på resultaterne af den funktionelle test.
1.4.3.2. Legemsvægt og indtagelse af føde/vand
Alle dyrene vejes mindst én gang om ugen. Indtagelsen af føde og vand måles mindst én gang om ugen. Hvis teststoffet indgives via drikkevandet, bør indtagelsen af vand ligeledes måles mindst én gang om ugen.
1.4.3.3. Hæmatologi
Følgende hæmatologiske undersøgelser foretages ved forsøgsperiodens afslutning: hæmatokrit, hæmoglobinkoncentration, erythrocyttælling, total og differential leucocyttælling, blodpladetælling og en måling af koaguleringsevne/tid.
Der tages blodprøver fra et bestemt, angivet sted, lige inden eller i forbindelse med aflivning af dyrene; prøverne opbevares under passende betingelser.
1.4.3.4. Klinisk biokemi
Klinisk-biokemiske bestemmelser med henblik på undersøgelse af vigtige toksiske virkninger i væv, og navnlig i nyrer og lever, foretages med blodprøver, der udtages fra alle dyr lige inden eller i forbindelse med dyrenes aflivning (bortset fra dem, der er fundet døende og/eller som er aflivet under forsøget). Det anbefales at dyrene faster natten over forud for blodprøvetagning (2). Undersøgelser af plasma eller serum skal omfatte natrium, kalium, glukose, total kolesterol, urinstof, kreatinin, total protein og albumin, mindst to enzymer, hvis forekomst kan være tegn på hepatocellulære effekter (f.eks. alanin aminotransferase, aspartat aminotransferase, alkalisk phosphatase, gamma glutamyl transpeptidase og sorbitol dehydrogenase). Målinger af yderligere enzymer (fra lever eller andre organer) og galdesyrer kan under visse omstændigheder give nyttige oplysninger.
Følgende urinanalyser kan eventuelt udføres i forsøgets sidste uge til bestemmelse af: udseende, volumen, osmolalitet eller specifik vægtfylde, pH, protein, glukose og blod/blodceller.
Endvidere bør det overvejes at gennemføre undersøgelser af serummarkører for generelle vævsskader. Andre bestemmelser, der bør foretages, hvis teststoffets kendte egenskaber kan eller forventes at kunne indvirke på tilsvarende metaboliske funktioner, omfatter kalcium, fosfat, triglycerider ved faste, visse hormoner, methæmoglobin og cholinesterase. Disse forhold undersøges for stoffer i bestemte stofgrupper eller fra tilfælde til tilfælde.
Der er generelt brug for en fleksibel fremgangsmåde, afhængigt af dyreart og de observerede og/eller forventede effekter ved et givet stof.
Hvis der ikke foreligger passende data fra tidligere undersøgelser, bør det overvejes at bestemme hæmatologiske og klinisk biokemiske variable, inden eksponering påbegyndes.
1.4.3.5. Makroskopisk undersøgelse
Alle forsøgsdyrene underkastes en fuldstændig og tilbundsgående makroskopisk undersøgelse, der omfatter omhyggelig undersøgelse af kroppens ydre, alle åbninger samt af kraniehule, brysthule og bughule og indholdet heraf. Lever, nyrer, binyrer, testes, bitestikler, thymus, milt, hjerne og hjerte fra alle dyr befries for alt vedhængende væv og vejes hurtigst muligt efter udtagning, så udtørring undgås.
Følgende væv præserveres i det bedst egnede fikseringsmiddel både med hensyn til vævstype og den påtænkte efterfølgende histopatologiske undersøgelse: alle makroskopiske læsioner, hjerne (repræsentative regioner, herunder cerebrum, cerebellum og pons), rygmarv, mavesæk, tyktarm og tyndtarm (herunder Peyerpletter), lever, nyrer, binyrer, milt, hjerte, thymus, skjoldbruskkirtel, spiserør og lunger (præserveret ved oppumpning med fikseringsmiddel og efterfølgende immersion), gonader, sekundære kønsorganer (f.eks. uterus, prostata), urinblære, lymfeknuder (om muligt en lymfeknude, der har berøring med indgiftsvejen, og en anden lymfeknude langt fra indgiftsvejen, som kan vise systemiske virkninger), perifer nerve (ischias- eller tibianerven), om muligt tæt på musklen, og et knoglemarvssnit (eller som alternativt et nyligt præpareret knoglemarvsaspirat). De kliniske og øvrige resultater kan vise, at der er behov for at undersøge yderligere væv. Også organer, der på grundlag af teststoffets kendte egenskaber kan forventes at være målorganer, præserveres.
1.4.3.6. Histopatologisk undersøgelse
Der foretages en fuldstændig histologisk undersøgelse af de præserverede organer og væv fra alle dyr i kontrolgruppen og i højdosisgruppen. I disse undersøgelser inddrages dyr fra alle de andre dosisgrupper, hvis der i højdosisgruppen observeres eksponeringsrelaterede ændringer.
Alle makroskopiske læsioner undersøges.
Bruges der en satellitgruppe, udføres der en histopatologisk undersøgelse af væv og organer, der udviser toksisk virkning i de øvrige grupper.
2. DATA
Der tilvejebringes data for hvert enkelt dyr. Derudover opstilles alle data i tabelform, som for hver gruppe viser antallet af dyr ved begyndelsen af forsøget, antallet af dyr, der er fundet døde under forsøget eller aflivet af humane årsager, og tidspunktet for dødsfald eller human aflivning, antal dyr, der viser tegn på toksicitet, en beskrivelse af de observerede tegn på toksicitet, herunder tidspunktet for de toksiske virkningers indtræden, varigheden og styrke, antallet af dyr, der udviser læsioner, typen af læsioner, og procentdelen af dyr, der udviser hver enkelt type læsion.
Numeriske resultater vurderes om muligt med en passende og generelt accepteret statistisk metode. De statistiske metoder udvælges i forbindelse med forsøgets tilrettelæggelse.
3. RAPPORTERING
FORSØGSRAPPORT
Forsøgsrapporten skal om muligt indeholde følgende oplysninger:
|
|
Forsøgsdyr:
|
|
|
Forsøgsbetingelser:
|
|
|
Resultater:
|
|
|
Diskussion af resultaterne |
|
|
Konklusioner |
4. HENVISNINGER
Denne metode svarer til TG 407 fra OECD.
B.8. TOKSICITET VED GENTAGEN DOSERING (28 DAGE, INHALATION)
1. METODE
1.1. INDLEDNING
Det er nyttigt at have forhåndsoplysninger om stoffets partikelstørrelsesfordeling, damptryk, smeltepunkt, kogepunkt, flammepunkt og eventuelle eksplosive egenskaber.
Se også den generelle indledning til del B (punkt A).
1.2. DEFINITIONER
Se den generelle indledning til del B (punkt B).
1.3. REFERENCESTOFFER
Ingen.
1.4. TESTMETODENS PRINCIP
Flere grupper af forsøgsdyr udsættes i 28 dage dagligt i en angivet periode for teststoffet i graduerede koncentrationer, idet hver gruppe eksponeres for en bestemt koncentration.Anvendes der et vehikel til at frembringe en passende koncentration af teststoffet i luften, bør der i forsøget indgå en kontrolgruppe, der udsættes for vehiklet alene. I eksponeringsperioden observeres dyrene dagligt for symptomer på toksisk virkning. Dyr, der dør under forsøget, obduceres, og ved forsøgets afslutning aflives og obduceres de overlevende dyr.
1.5. KVALITETSKRITERIER
Ingen.
1.6. BESKRIVELSE AF TESTMETODEN
1.6.1. Forberedelser
Dyrene holdes under forsøgsbetingelser med hensyn til miljø og fodring i mindst fem dage, inden forsøget begyndes. Unge sunde dyr fordeles randomiseret i det fornødne antal grupper. Om nødvendigt kan der tilsættes et egnet vehikel til teststoffet, således at der opnås en passende koncentration af teststoffet i luften. Hvis der anvendes et vehikel eller andre tilsætningsstoffer for at lette doseringen, skal det være påvist, at de ikke har nogen toksisk virkning. Her kan tidligere data benyttes.
1.6.2. Forsøgsbetingelser
1.6.2.1. Forsøgsdyr
Rotter er de foretrukne forsøgsdyr, medmindre der foreligger kontraindikationer. Der benyttes unge sunde rotter af almindeligt anvendte stammer.
Ved forsøgets begyndelse må vægten af de anvendte dyr ikke variere mere end ± 20 % fra en rimelig gennemsnitsvægt.
1.6.2.2. Antal og køn
Der anvendes mindst ti dyr (fem hunner og fem hanner) i hver forsøgsgruppe. Hundyrene må ikke have født og må ikke være drægtige. Hvis der påregnes aflivning af dyr under forsøget, skal det oprindelige antal dyr forøges med det antal dyr, man påregner at aflive i løbet af forsøget. Man kan samtidig udsætte en satellitgruppe på ti dyr (fem af hvert køn) for den højeste koncentration i 28 dage og gennem en periode på 14 dage efter forsøget observere, om der er reversible, varige eller sent opståede toksiske virkninger. Der anvendes tillige en satellitgruppe på ti kontroldyr (fem af hvert køn).
1.6.2.3. Eksponeringskoncentrationer
Der skal anvendes mindst tre koncentrationsniveauer og en kontrolgruppe eller en vehikelkontrolgruppe (svarende til koncentrationen af vehiklet i højeste dosisniveau), såfremt der benyttes vehikel. Dyrene i kontrolgruppen behandles nøjagtigt som dyrene i forsøgsgrupperne, bortset fra inhalation af teststoffet. Den højeste koncentration bør have toksisk virkning, men kun medføre få eller ingen dødsfald. Der må ikke være tegn på toksisk virkning ved den laveste koncentration. Hvis der foreligger en anvendelig vurdering af menneskers eksponering for stoffet, skal den laveste koncentration overskride denne eksponering, Middelkoncentrationen skal helst frembringe så svage observerbare toksiske virkninger som muligt. Hvis der anvendes mere end tre koncentrationer, bør de vælges således, at der opnås graduering af de toksiske virkninger. I grupperne med lav og middel koncentration og i kontrolgrupperne skal dødeligheden holdes på et lavt niveau, så der bliver mulighed for en meningsfuld vurdering af resultaterne.
1.6.2.4. Eksponeringstid
Den daglige eksponenngsperiode er seks timer, men det kan være nødvendigt at benytte kortere eller længere perioder for at imødekomme specielle krav.
1.6.2.5. Udstyr
Dyrene testes med inhalationsapparatur, der er udformet således, at der kan opretholdes en dynamisk luftcirkulation med et luftskifte på mindst 12 gange pr. time samtidig med et tilstrækkeligt iltindhold og jævn fordeling af eksponeringsluften. Benyttes der eksponeringskammer, skal det være således indrettet, at forsøgsdyrene sammenklumpes mindst muligt og eksponeres mest muligt for teststoffet ved inhalation. Den generelle regel for sikkerhed for stabil atmosfære er, at dyrenes totale »volumen« ikke må overstige 5 % af eksponeringskammerets volumen. Eksponeringen kan foregå gennem mund-næse, hovedet alene eller ved kamre til hele kroppen; ved anvendelse af de to første muligheder opnår man, at optagelsen af teststoffet ad andre veje minimeres.
1.6.2.6. Observationsperiode
Dyrene observeres dagligt for symptomer på toksiske virkninger i hele forsøgsperioden og restitutionsperioden. Dødstidspunkt og tidspunkter for toksicitetssymptomers fremkomst og forsvinden registreres.
1.6.3. Fremgangsmåde
Dyrene eksponeres for teststoffet dagligt 5-7 dage om ugen i en periode på 28 dage. En eventuel satellitgruppe beregnet til opfølgningsobservationer holdes i yderligere 14 dage, efter at eksponering for teststoffet er ophørt, således at det kan registreres, om toksiske virkninger er reversible, eller om de vedvarer. Temperaturen under forsøget holdes på 22 oC ± 3 oC.
Under ideelle forhold skal den relative luftfugtighed holdes på mellem 30 % og 70 %, men i visse tilfælde (f.eks. ved testning af visse aerosoler) kan det være praktisk uigennemførligt. Udsivning af teststof til omgivelserne kan forhindres ved, at der i kammeret holdes et svagt undertryk (≤ 5 mm vandsøjle). Der gives hverken foder eller vand under eksponeringen.
Der anvendes et dynamisk inhalatianssystem med et passende analytisk kontrolsystem til sikring af eksponeringskoncentrationen. Det anbefales at foretage en prøvetest for at finde frem til passende eksponeringskoncentrationer. Lufthastigheden tilpasses, således at der er ensartede forhold i hele eksponeringskammeret. Systemet skal være af en sådan beskaffenhed, at der så hurtigt som muligt opnås stabile eksponeringsbetingelser.
Der foretages måling og kontrol af:
|
a) |
lufthastighed (kontinuerlig) |
|
b) |
den faktiske koncentration af teststoffet målt i opholdsarealet. Koncentrationen må under den daglige eksponeringsperiode ikke variere mere end ± 15 % fra gennemsnitskoncentrationen. Det kan være umuligt at opnå en sådan grad af kontrol med visse aerosoler, og i så tilfælde kan større udsving accepteres. Koncentrationerne bør fra dag til dag holdes så konstante som muligt under hele forsøgets forløb. For aerosoler analyseres partikelstørrelsen mindst en gang ugentligt pr. forsøgsgruppe |
|
c) |
temperatur og luftfugtighed, om muligt kontinuerligt. |
Under og efter eksponering foretages der systematiske observationer, som registreres for hvert enkelt dyr. Alk dyr observeres dagligt, og toksicitetssymptomer registreres, herunder hvornår disse symptomer optræder første gang, hvor længe de varer, og hvor kraftige de er. Inspektionen af dyrene skal omfatte forandringer i hud og pels, øjne og slimhinder, åndedræt og kredsløb, det autonome og det centrale nervesystem, den motoriske aktivitet og adfærdsmønsteret. Dyrene vejes én gang om ugen. Det anbefales, at der også holdes regnskab med den ugentlige fødeindtagelse. Dyrene observeres med jævne mellemrum, så dyr ikke går tabt for undersøgelsen på grund af f.eks. kannibalisme, autolyse af væv eller forveksling. Ved forsøgets slutning aflives og obduceres alle overlevende dyr med undtagelse af dem i satellitgruppen. Døende dyr og dyr, der viser symptomer på stærk smerte eller lidelse, fjernes, så snart de iagttages, hvorefter de aflives på human måde og obduceres.
Ved forsøgets afslutning skal alle dyr inklusive kontrolgruppen underkastes følgende undersøgelser:
|
i) |
hæmatologisk analyse omfattende mindst hæmatokrit, hæmoglobinkoncentration, erythrocyttælling, total og differential leucocyttælling og et mål for koaguleringsevnen |
|
ii) |
klinisk biokemisk blodanalyse omfattende mindst én lever- og nyrefunktionsparameter: serum alanin aminotransferase (ALAT), serum aspartat aminotransferase (ASAT), urinstof, albumin, kreatinin, total bilirubin og total serumprotein. |
Andre bestemmelser, der kan være påkrævet for en fyldestgørende toksikologisk vurdering, er calcium, phosphor, chlorid, natrium, kalium, glucose efter faste, lipidanalyser, hormoner, syre/basebalance, methæmoglobin og cholinesteraseaktivitet.
Der kan udføres yderligere klinisk biokemiske analyser, hvor det skønnes nødvendigt for en videre undersøgelse af de observerede virkninger.
1.6.3.1. Makroskopisk undersøgelse
Alle forsøgsdyrene underkastes en fuldstændig makroskopisk undersøgelse. Lever, nyrer, binyrer, lunger og testes vejes så hurtigt som muligt efter udtagning, så udtørring undgås. Væv og organer (luftveje, lever, nyrer, milt, testes, binyrer, hjerte og organer, der udviser makrokopiske læsioner eller størrelsesforandringer) opbevares i et passende medium med henblik på eventuel senere histopatologisk undersøgelse. Lunger udtages hele og intakte, vejes og behandles med et passende fixativ for at sikre, at lungestrukturen bevares.
1.6.3.2. Histopatologisk undersøgelse
Der foretages en histologisk undersøgelse af præpareret væv og organer fra dyrene i kontrolgruppen samt i den gruppe, der har været udsat for den højeste koncentration. Observeres der i gruppen med den højeste koncentration vævs- eller organdefekter, som kan tilskrives teststoffet, undersøges de samme væv og organer i grupperne med lavere koncentration. Dyrene i en eventuel satellitgruppe underkastes en histologisk undersøgelse, især de væv og organer, der udviser toksisk virkning i de øvrige grupper.
2. DATA
Data opstilles i tabelform, som for hver gruppe viser antal dyr ved begyndelsen af forsøget, og for hver type af læsion det antal dyr, der udviser den pågældende læsion.
Evaluering af samtlige observationer skal baseres på en passende statistisk metode. Alle anerkendte statistiske metoder kan anvendes.
3. RAPPORTERING
3.1. FORSØGSRAPPORT
Forsøgsrapporten skal om muligt indeholde følgende oplysninger:
|
— |
dyreart/stamme, herkomst, miljøbetingelser, foder osv. |
|
— |
forsøgsbetingelser: Beskrivelse af eksponeringsudstyret, herunder udformning, type, dimensionering, luftkilde, system til frembringelse af aerosoler, metode til konditionering af luften, behandling af udpumpet luft og den måde, hvorpå dyrene holdes i et eksponeringskammer, når et sådant benyttes. Apparatur til måling af temperatur, fugtighed og i givet fald aerosolkoncentrationens stabilitet eller partikelstørrelse skal beskrives. Eksponeringsdata: Disse skal opstilles i tabelform med angivelse af middelværdier og et mål for variationen (f.eks. standardafvigelser) og i videst muligt omfang omfatte:
|
|
— |
oplysninger om toksisk reaktion opdelt efter køn og koncentration |
|
— |
angivelse af dyrenes dødstidspunkt under forsøget eller overlevelse til forsøgets slutning |
|
— |
beskrivelse af toksiske og andre virkninger; nuleffektniveau |
|
— |
for hvert enkelt abnormitetstegn det tidspunkt, hvor det er observeret, og dets senere udvikling |
|
— |
foder- og legemsvægt |
|
— |
hæmatologiske analyser og samtlige resultater |
|
— |
klinisk-biokemiske analyser og samtlige resultater |
|
— |
obduktionsfund |
|
— |
detaljeret beskrivelse af alle histopatologiske fund |
|
— |
eventuel statistisk behandling af resultaterne |
|
— |
diskussion af resultaterne |
|
— |
fortolkning af resultaterne. |
3.2. VURDERING OG FORTOLKNING
Se den generelle indledning til del B (punkt D).
4. HENVISNINGER
Se den generelle indledning til del B (punkt E).
B.9. TOKSICITET VED GENTAGEN DOSERING (28 DAGE, DERMAL)
1. METODE
1.1. INDLEDNING
Se den generelle indledning til del B (punkt A).
1.2. DEFINITIONER
Se den generelle indledning til del B (punkt B).
1.3. REFERENCESTOFFER
Ingen.
1.4. TESTMETODENS PRINCIP
Teststoffet påføres dagligt huden i graduerede doser på grupper af forsøgsdyr, idet der anvendes en bestemt dosis til hver gruppe gennem en periode på 28 dage. I eksponeringspenoden observeres dyrene dagligt for symptomer på toksisk virkning. Dyr, der dør under forsøget, obduceres, og ved forsøgets afslutning aflives og obduceres de overlevende dyr.
1.5. KVALITETSKRITERIER
Ingen.
1.6. BESKRIVELSE AF TESTMETODEN
1.6.1. Forberedelser
Dyrene holdes under forsøgsbetingelser med hensyn til miljø og fodring i mindst fem dage, inden forsøget påbegyndes. Unge sunde dyr fordeles randomiseret i forsøgs- og kontrolgrupper, inden forsøget sættes i gang. Kort før forsøget indledes, klippes dyrene på ryggen. Såfremt dyrene barberes, bør det ske ca. 24 timer, inden forsøget indledes. Klipningen eller barberingen må som regel gentages med en uges mellemrum. Man må ved klipning og barbering passe på ikke at beskadige huden. Mindst 10 % af kroppens overflade klargøres til påføring af teststoffet. Ved afgørelse af, hvilket og hvor stort et område der skal klippes, bør der tages hensyn til dyrets vægt. Ved testning af faste stoffer, som i givet fald kan pulveriseres, skal teststoffet fugtes tilstrækkeligt med vand eller om nødvendigt med et passende vehikel, så der sikres god kontakt med huden. Flydende teststoffer påføres normalt ufortyndet. Teststoffet påføres dagligt 5-7 dage om ugen.
1.6.2. Forsøgsbetingelser
1.6.2.1. Forsøgsdyr
Der kan anvendes voksne rotter, kaniner eller marsvin. Der kan også anvendes andre dyrearter, men i så fald skal det begrundes.
Ved forsøgets begyndelse må vægten af de anvendte dyr ikke variere mere end ± 20 % fra en rimelig gennemsnitsvægt.
1.6.2.2. Antal og køn
Der anvendes mindst ti dyr (fem hunner og fem hanner) med sund hud til hvert dosisniveau. Hunnerne må ikke have født og må ikke være drægtige. Hvis der påregnes aflivning af dyr under forsøget, skal det oprindelige antal dyr forøges med det antal dyr, man påregner at aflive i løbet af forsøget. Man kan samtidig udsætte en satellitgruppe på ti dyr (fem af hvert køn) for den højeste dosis i 28 dage og gennem en periode på 14 dage efter forsøget observere, om der er reversible, varige eller forsinkede toksiske virkninger. Der anvendes tillige en satellitgruppe på ti kontroldyr (fem af hvert køn).
1.6.2.3. Dosisniveauer
Der skal anvendes mindst tre dosisniveauer samt en kontrolgruppe eller en vehikelkontrolgruppe, såfremt der benyttes vehikel. Eksponeringsperioden er på mindst seks timer dagligt. Teststoffet påføres på omtrent samme tidspunkt hver dag, og doserne justeres med jævne mellemrum (én gang eller to gange om ugen), så de er konstante i forhold til dyrenes legemsvægt. Dyrene i kontrolgruppen behandles nøjagtigt som dyrene i forsøgsgrupperne, bortset fra påføring af teststoffet. Hvis der anvendes et vehikel til at lette doseringen, skal vehikelkontrolgruppen eksponeres på samme måde som forsøgsgrupperne og påføres samme mængde vehikel som den gruppe, der modtager den højeste dosis. Den højeste dosis bør have toksisk virkning, men kun medføre få eller ingen dødsfald. Det laveste dosisniveau bør ikke have nogen toksisk virkning overhovedet. Hvis der foreligger en anvendelig vurdering af menneskers eksponering for stoffet, skal det laveste dosisniveau overskride denne eksponering. Middeldosis skal helst frembringe så svage observerbare toksiske virkninger som muligt. Hvis der anvendes mere end tre dosisniveauer, bør de vælges således, at der opnås graduering af de toksiske virkninger. I grupperne med lav og middel dosis og i kontrolgrupperne skal dødeligheden holdes på et lavt niveau, så der bliver mulighed for en meningsfuld vurdering af resultaterne.
Hvis påføring af teststoffet forårsager alvorlig hudirritation, sænkes koncentrationen, hvilket kan medføre, at andre toksiske virkninger på det højeste dosisniveau svækkes eller forsvinder. Hvis huden er blevet alvorligt beskadiget, kan det være nødvendigt at afslutte forsøget og foretage et nyt forsøg med lavere koncentrationer.
1.6.2.4. Grænsetest
Hvis der i en indledende undersøgelse med en dosis på 1 000 mg/kg legemsvægt eller en større dosis fastsat ud fra kendskab til den eksponering, mennesker kan blive udsat for, ikke konstateres nogen toksisk virkning, kan yderligere test betragtes som unødvendige.
1.6.2.5. Observationsperiode
Dyrene observeres dagligt for symptomer på toksicitet. Dødstidspunkt og tidspunkter for toksicitetssymptomers fremkomst og forsvinden registreres.
1.6.3. Fremgangsmåde
Dyrene holdes i hvert sit bur. Den ideelle dosering består i påføring af teststoffet syv dage om ugen i en periode på 28 dage. En eventuel satellitgruppe beregnet til opfølgningsobservationer holdes i yderligere 14 dage uden påføring af teststof, således at det kan registreres, om toksiske virkninger er reversible, eller om de vedvarer. Den daglige eksponeringsperiode er på mindst seks timer.
Teststoffet påføres jævnt over et område, som svarer til ca. 10 % af den samlede legemsoverflade. Med stærkt toksiske stoffer kan der anvendes et mindre område, men stoffet skal påføres så tyndt og ensartet over en så stor del af området som muligt.
Teststoffet skal holdes i kontakt med huden ved hjælp af en porøs gazeforbinding og en ikke-hudirriterende klæbestrimmel. Teststedet skal desuden tildækkes på en sådan måde, at gazeforbindingen og teststoffet holdes sikkert fast, så at dyrene ikke kan indtage teststoffet. Der kan anvendes andre foranstaltninger, som forhindrer dyrene i at indtage teststoffet; dog kan fuldstændig immobilisering ikke anbefales. Alternativt kan der benyttes en beskyttelseskrave.
Ved eksponeringsperiodens afslutning fjernes tilbagesiddende teststof, idet der f.eks. anvendes vand eller et andet egnet middel til at rense huden.
Alle dyr observeres dagligt, og toksicitetssymptomer registreres, herunder hvornår disse symptomer optræder første gang, hvor længe de varer, og hvor kraftige de er. Inspektion af dyrene skal omfatte forandringer i hud og pels, øjne og slimhinder, åndedræt og kredsløb, det autonome og centrale nervesystem, den motoriske aktivitet og adfærdsmønsteret. Dyrene vejes én gang om ugen. Det anbefales, at der også holdes regnskab med den ugentlige fødeindtagelse. Dyrene observeres med jævne mellemrum, så dyr ikke går tabt for undersøgelsen på grund af f.eks. kannibalisme, autolyse af væv eller forveksling. Ved forsøgets slutning aflives og obduceres alle overlevende dyr med undtagelse af dem i satellitgrupperne. Døende dyr og dyr, der viser symptomer på stærk smerte eller lidelse, fjernes, så snart de iagttages, hvorefter de aflives på human måde og obduceres.
Ved forsøgets afslutning skal alle dyr inklusive kontrolgruppen underkastes følgende undersøgelser:
|
1) |
hæmatologisk analyse omfattende mindst hæmatokrit, hæmoglobinkoncentration, erythrocyttælling, total og differential leucocyttælling og et mål for koaguleringsevnen |
|
2) |
klinisk biokemisk blodanalyse omfattende mindst en lever- og nyrefunktionsparameter: serum alanin aminotransferase (ALAT), serum aspartat aminotransferase (ASAT), urinstof, albumin, kreatinin, total bilirubin og total serumprotein. |
Andre bestemmelser, der kan være påkrævet for en fyldestgørende toksikologisk vurdering, er calcium, phosphor, chlorid, natrium, kalium, glucose efter faste, lipidanalyser, hormoner, syre/basebalance, methæmoglobin og cholinesteraseaktivitet.
Der kan udføres yderligere klinisk-biokemiske analyser, hvor det skønnes nødvendigt for videre undersøgelse af observerede virkninger.
1.6.4. Makroskopisk undersøgelse
Alle forsøgsdyrene underkastes en fuldstændig makroskopisk undersøgelse. Lever, nyrer, binyrer og testes vejes så hurtigt som muligt efter udtagningen, så udtørring undgås. Væv og organer, dvs. normal og eksponeret hud, lever, nyrer, milt, testes, binyrer, hjerte og målorganer (dvs. organer, der udviser makroskopiske læsioner eller forandringer i størrelse), opbevares i et passende medium med henblik på eventuel senere histopatologisk undersøgelse.
1.6.5. Histopatologisk undersøgelse
Der foretages en histologisk undersøgelse af præpareret væv og organer fra dyrene i kontrolgruppen samt i den gruppe, der har været udsat for den største dosis. Observeres der i gruppen med den højeste dosis vævs- eller organdefekter, som kan tilskrives teststoffet, undersøges de samme væv og organer i grupperne med lavere dosis. Dyrene i satellitgruppen underkastes en histologisk undersøgelse, især de væv og organer, der udviser virkning i de øvrige grupper.
2. DATA
Data opstilles i tabelform, som for hver gruppe viser antallet af dyr ved begyndelsen af forsøget, og for hver type læsion det antal dyr, der udviser den pågældende læsion.
Evaluering af samtlige observationer skal baseres på en passende statistisk metode. Alle anerkendte statistiske metoder kan anvendes.
3. RAPPORTERING
3.1. FORSØGSRAPPORT
Forsøgsrapporten skal om muligt indeholde følgende oplysninger:
|
— |
dyreart/stamme, herkomst, miljøbetingelser, foder osv. |
|
— |
forsøgsbetingelser (herunder forbindingstypen: med eller uden okklusion) |
|
— |
dosisniveauer (med angivelse af vehikel, hvor et sådant er anvendt) samt koncentration |
|
— |
nuleffektniveau, hvor det er muligt |
|
— |
oplysninger om toksiske reaktioner opdelt efter køn og dosis |
|
— |
angivelse af dyrenes dødstidspunkt under forsøget eller overlevelse til forsøgets slutning |
|
— |
toksiske og andre virkninger |
|
— |
for hvert enkelt abnormitetstegn det tidspunkt, hvor det er observeret, og dets senere udvikling |
|
— |
foder- og legemsvægt |
|
— |
hæmatologiske analyser og samtlige resultater |
|
— |
klinisk-biokemiske analyser og samtlige resultater |
|
— |
obduktionsfund |
|
— |
detaljeret beskrivelse af alle histopatologiske fund |
|
— |
eventuel statistisk behandling af resultaterne |
|
— |
diskussion af resultaterne |
|
— |
fortolkning af resultaterne. |
3.2. VURDERING OG FORTOLKNING
Se den generelle indledning til del B (punkt D).
4. HENVISNINGER
Se den generelle indledning til del B (punkt E).
B.10. MUTAGENICITET — IN VITRO-TEST FOR KROMOSOMABERRATIONER I CELLER FRA PATTEDYR
1. METODE
Denne metode er en gengivelse af OECD Test Guideline 473 In Vitro Mammalian Chromosome Aberration Test (1997).
1.1. INDLEDNING
Formålet med in vitro-testen for kromosomaberrationer er at påvise, om et stof eller andet forårsager strukturelle kromosomafvigelser i dyrkede pattedyrceller (1) (2) (3). Der er to typer strukturændringer, kromosomtypen og kromatidtypen. De fleste kemiske mutagener forårsager ændringer af kromatidtypen, men også ændringer af kromosomtypen forekommer. Ploiditetsforøgelse kan være tegn på, at et kemisk stof er i stand til at fremkalde antalsafvigelser. Den foreliggende metode er imidlertid ikke beregnet til at påvise antalsafvigelser og benyttes ikke rutinemæssigt til dette formål. Kromosommutationer og lignende er årsag til mange genetiske sygdomme hos mennesker, og meget tyder på, at kromosommutationer og lignende, som ændrer på onkogener og tumorsuppressorgener i somatiske celler, medvirker ved induktion af kræft hos mennesker og i forsøgsdyr.
I in vitro-testen for kromosomaberrationer kan der benyttes kulturer af etablerede cellelinjer, cellestammer eller primære cellekulturer. Cellerne udvælges på grundlag af deres vækstegenskaber ved dyrkning, karotypens stabilitet, kromosomtal, kromosomdiversitet og hyppighed af spontane kromosomaberrationer.
Ved udførelse af in vitro-test er det i reglen nødvendigt at benytte en exogen kilde til metabolismeaktivering. Et sådant metabolismeaktiveringssystem kan ikke fuldstændig efterligne betingelserne i et levende pattedyr. Man må omhyggeligt undgå betingelser, som fører til et positivt resultat, der ikke skyldes selve mutageniciteten, men kan være forårsaget af ændringer i pH eller osmolalitet eller høj cytotoksicitet (4) (5).
Testen benyttes til at screene for stoffer, der kan være mutagene eller carcinogene hos pattedyr. Mange af de stoffer, der giver positivt resultat med denne test, er kræftfremkaldende hos pattedyr, men der er ikke fuldstændig korrelation mellem testen og carcinogenicitet. Korrelationen afhænger af den kemiske sammensætning, og der er stadig tydeligere tegn på, at nogle carcinogener ikke påvises ved denne test, fordi de lader til at virke ved andre mekanismer end direkte beskadigelse af dna.
Se også den generelle indledning til del B.
1.2. DEFINITIONER
Aberration af kromatidtypen: beskadigelse af kromosomstrukturen i form af brud på enkeltkromatider eller brud på og sammenføjning af kromatider.
Aberration af kromosomtypen: beskadigelse af kromosomstrukturen i form af brud på — eller brud på og sammenføjning af — begge kromatider på samme sted.
Endofordobling: en proces, hvorved cellekernen efter en S-fase i dna-replikationen ikke fortsætter over i mitosen, men påbegynder endnu en S-fase. Resultatet er kromosomer med 4, 8, 16, ... kromatider.
Gap: en akromatisk beskadigelse, som er mindre end ét kromatid bred, og hvor forskydning af kromatiderne er minimal.
Mitoseindeks: forholdet mellem antallet af celler i metafase og det samlede antal celler i en population; det viser populationens formeringsgrad.
Antalsafvigelse: ændring i kromosomtallet i forhold til det for de pågældende celler normale antal.
Polyploidi: et multiplum af det haploide kromosomtal (n), som ikke er det diploide tal (dvs. 3n, 4n osv.).
Strukturel ændring: ændring i kromosomstrukturen, som kan iagttages ved mikroskopi i celledelingens metafase i form af deletioner og fragmenter, intrachange og interchange.
1.3. TESTMETODENS PRINCIP
Cellekulturer udsættes for teststoffet både med og uden metabolismeaktivering. Efter forud fastsatte tidsrum efter at være udsat for teststoffet behandles kulturerne med et metafasestandsende stof (f.eks. Colcemid® eller colchicin), hvorefter de høstes og farves; dernæst undersøges metafasecellerne mikroskopisk for forekomst af kromosomaberrationer.
1.4. BESKRIVELSE AF TESTMETODEN
1.4.1. Præparater
1.4.1.1. Celler
Der kan benyttes en række forskellige cellelinjer, -stammer og primære cellekulturer, herunder humane celler (f.eks. fibroblaster fra kinesisk hamster eller perifere blodlymfocytter fra det perifere blod hos mennesker eller andre pattedyr).
1.4.1.2. Substrater og dyrkningsforhold
Til vedligeholdelse af kulturerne benyttes der egnede dyrkningssubstrater og inkuberingsbetingelser (dyrkningsflaskcr, CO2-koncentration, temperatur og luftfugtighed). Etablerede cellelinjer og -stammer kontrolleres regelmæssigt for stabilt karakteristisk kromosomtal og mycoplasmakontaminering; i tilfælde af kontaminering anvendes kulturen ikke. Cellernes normale cyklustid og dyrkningsbetingelserne skal være kendt.
1.4.1.3. Fremstilling af kulturer
Etablerede cellelinjer og -stammer: Celler udtages fra stamkulturer og udsås i dyrkningssubstrat med en sådan tæthed, at kulturerne ikke flyder sammen inden høst, og inkuberes ved 37 oC.
Lymfocytter: fuldblod, der er behandlet med antikoaguleringsmiddel (f.eks, heparin), eller lymfocytter fra sunde individer tilsættes til dyrkningssubstratet, der indeholder et mitogen (f.eks. phytohæmagglutinin), og inkuberes ved 37 oC.
1.4.1.4. Metabolismeaktivering
Cellerne udsættes for teststoffet både med og uden et egnet metabolismeaktiveringssystem. Det mest almindeligt anvendte system er en cofaktorsuppleret postmitokondriefraktion (S9), der fremstilles af levere fra rotter, der har været behandlet med enzyminducerende stoffer såsom Aroclor 1254 (6) (7) (8) (9), eller en blanding af phenobarbiton og β-naphthoflavon (10) (11) (12).
Postmitokondriefraktionen anvendes normalt i en koncentration på 1-10 % (v/v) i det færdige testsubstrat. Et metabolismeaktiveringssystems beskaffenhed kan afhænge af, hvilken kemisk klasse teststoffet tilhører. I nogle tilfælde vil det være hensigtsmæssigt at benytte postmitokondriefraktion i mere end én koncentration.
Nogle udviklingstendenser, f.eks. genetisk konstruktion af cellelinjer, der udtrykker specifikke aktiverende enzymer, kan rumme potentiale for endogen aktivering. Valget af en bestemt cellelinje skal begrundes sagligt (f.eks. ved, at cytochrom P450-coenzymet er relevant for teststoffets metabolisme).
1.4.1.5. Teststof/testpræparat
Teststoffer i fast form opløses eller opslæmmes i passende opløsningsmidler eller bærestoffer og fortyndes passende inden behandling af cellerne. Teststoffer i væskeform kan enten tilsættes direkte til testsystemet eller fortyndes inden behandlingen. Der benyttes frisk fremstillede teststofpræparater, medmindre stabilitetsdata viser, at opbevaring er acceptabel.
1.4.2. Testbetingelser
1.4.2.1. Opløsningsmiddel/bærestof
Der må ikke være mistanke om, at opløsningsmiddel/bærestof reagerer kemisk med teststoffet, og opløsningsmiddel/bærestof må hverken hæmme cellernes overlevelse eller S9-aktiviteten. Benyttes der andre opløsningsmidler/bærestoffer end de velkendte, skal brugen af dem underbygges med kompatibilitetsdata. Det anbefales i videst muligt omfang først at forsøge at benytte et vandigt opløsningsmiddel/bærestofsystem. Ved undersøgelse af stoffer, der er ustabile i vand, skal de organiske opløsningsmidler være vandfrie. Vand kan fjernes med tilsætning af molekylsi.
1.4.2.2. Eksponeringskoncentrationer
Stoffets cytotoksicitet, dets opløselighed i testsystemet samt ændringer i pH og osmolalitet er blandt de kriterier, der skal tages hensyn til ved valg af højeste koncentration.
Cytotoksiciteten bestemmes med og uden metabolismeaktivering i hovedforsøget ved hjælp af en passende indikator for celleintegritet og -vækst, f.eks. konfluens, antal levedygtige celler eller mitoseindeks. Det kan være hensigtsmæssigt at bestemme cytotoksicitet og opløselighed ved et indledende forsøg.
Der benyttes mindst tre analyserbare koncentrationer. Hvis der er tale om cytoksicitet, skal koncentrationerne dække et interval fra største toksicitet til ringe eller ingen toksicitet; det betyder normalt, at koncentrationerne højst er adskilt med en faktor mellem 2 og √10. På højsttidspunktet skal konfluens, celleantal eller mitoseindeks være faldet signifikant ved den højeste koncentration (mere end 50 %). Mitoseindekset er kun et direkte mål for cytotoksisk/cytostatisk virkning og afhænger af, hvor lang tid der er forløbet efter behandlingen. Mitoseindeks er dog acceptabel for dyrkning i rystekolber, hvor andre toksicitetsmål er besværlige eller ubrugelige i praksis. Oplysninger om cellecyklens kinetik, såsom den gennemsnitlige generationstid (AGT), kan benyttes som supplerende information. AGT er imidlertid et overordnet gennemsnit, der ikke altid afslører forsinkede subpopulationer, og selv en lille forøgelse af den gennemsnitlige generationstid kan føre til en meget betydelig forsinkelse af tidspunktet for optimalt udbytte af aberrationer.
For stoffer, der er forholdsvis ikke-cytotoksiske, må testkoncentrationen højst være 5 μl/ml, 5 mg/ml eller 0,01 M (den laveste af de tre).
For forholdsvis uopløselige stoffer, der ikke er toksiske ved koncentrationer under opløselighedsgrænsen, skal der som højeste dosis benyttes en koncentration, der er højere end opløselighedsgrænsen i det samlede dyrkningssubstrat ved behandlingens afslutning. I nogle tilfælde (f.eks. når toksicitet kun optræder over den laveste koncentration med uopløselighed) tilrådes det at gennemføre testen ved mere end én koncentration med synlig udfældning. Det kan være hensigtsmæssigt at bedømme opløseligheden både ved behandlingens begyndelse og afslutning, eftersom opløseligheden kan ændre sig under eksponeringen i testsystemet som følge af, at der er celler S9. serum mv. til stede. Uopløselighed kan iagttages med det blotte øje. Bundfald må ikke have indflydelse på bedømmelsen.
1.4.2.3. Negative og positive kontrolprøver
I alle forsøg medtages der sideløbende både positive og negative (opløsningsmiddel eller bærestof) kontrolprøver, såvel med som uden metabolismeaktivering. Når der benyttes metabolismeaktivering, bør der som positivt kontrolkemikalie anvendes et, der kræver aktivering for at virke mutagent.
1 positive kontrolprøver benyttes der et kendt clastogen i en mængde, der forventes at give reproducerbar og påviselig forøgelse i forhold til baggrundsværdierne, hvorved testsystemets følsomhed godtgøres.
Der vælges sådanne koncentrationer i de positive kontrolprøver, at virkningerne er tydelige, uden dog at de kodede objektglas' identitet umiddelbart afsløres for den, der aflæser dem. Nedenfor er opregnet eksempler på positive kontrolstoffer:
|
Metabolismeaktivering |
Stof |
CAS nr. |
Einecs nr. |
|
Ingen exogen metabolismeaktivering |
Methylmethansulfonat |
66-27-3 |
200-625-0 |
|
Ethylmethansulfonat |
62-50-0 |
200-536-7 |
|
|
Ethylnitrosourea |
759-73-9 |
212-072-2 |
|
|
Mitomycin C |
50-07-7 |
200-008-6 |
|
|
4-Nitroquinoline-N-oxid |
56-57-5 |
200-281-1 |
|
|
Exogen metabolismeaktivering |
Benzo[a]pyrene |
50-32-8 |
200-028-5 |
|
Cyclophosphamid Cyclophosphamidmonohydrat |
50-18-0 6055-19-2 |
200-015-4 |
Der kan benyttes andre egnede stoffer til positiv kontrol. Hvis der er mulighed for det, bør der anvendes kemisk beslægtede stoffer.
Der skal i hver høst indgå negative kontrolprøver, hvor behandlingsmediet kun består af opløsningsmiddel eller bærestof, og som behandles på samme måde som de øvrige kulturer. Desuden skal der være ubehandlede kontrolprøver, medmindre der foreligger tidligere opnåede kontroldata, der viser, at det valgte opløsningsmiddel ingen skadelige eller mutagene virkninger har.
1.4.3. Fremgangsmåde
1.4.3.1. Behandling med teststof
Celler under formering behandles med teststoffet med og uden tilstedeværelse af et metabolismeaktiveringssystem. Behandling af lymfocytter påbegyndes ca. 2 døgn efter stimulering med mitogen.
|
1.4.3.2. |
Der benyttes normalt dobbeltbestemmelse ved hver koncentration, hvilket også stærkt tilrådes for negative kontrolprøver (kontrolprover med opløsningsmiddel). Hvis det på grundlag af tidligere forsøg kan godtgøres, at forskellen mellem dobbeltbestemmelser (13) (14) er minimal, kan enkeltbestemmelse ved hver koncentration accepteres. Gasformige og flygtige stoffer testes ved en egnet metode, f.eks. i forseglede dyrkningsflasker (15) (16). |
1.4.3.3. Høst af kulturer
I det første forsøg udsættes cellerne for teststoffet både med og uden metabolismeaktivering i 3-6 timer, og der udtages prøve efter et tidsrum, der svarer til ca.1,5 gange cellecykluslængden, regnet fra behandlingens begyndelse (12). Fører denne protokol til negativt resultat både med og uden aktivering, udføres endnu et forsøg, denne gang med kontinuerlig behandling indtil prøveudtagning efter et tidsrum, der svarer til ca. 1,5 gange en normal cellecyklus. Nogle kemiske stoffer er det lettere at bestemme ved behandlings/prøveudtagningstider på mere end 1,5 gange cellecyklussen. Negativt resultat med metabolismeaktivering skal bekræftes i hvert enkelt tilfælde. Anses bekræftelse af negative resultater ikke for nødvendige, skal dette begrundes.
1.4.3.4. Kromosompræparering
Cellekulturerne behandles med Colcemid® eller colchicin normalt 1-3 timer før høst. Høst og kromosompræparering skal foregå særskilt for hver enkelt kultur. Kromosompræpareringen består i hypotonisk behandling af cellerne, fiksering og farvning.
1.4.3.5. Analyse
Alle objektglas, herunder positive og negative kontrolprøver, kodes uafhængigt inden mikroskoperingen. Da fikseringen ofte medfører, at en del af metafasecellerne går i stykker, og kromosomerne går tabt, bør bedømte celler indeholde et antal centromerer svarende til kromosomtallet ± 2 for alle celletyper. Der skal bedømmes mindst 200 vel fordelte metafaser pr. koncentration og kontrolgruppe, ligeligt fordelt mellem eventuelle dobbeltprøver. Dette antal kan sættes ned, hvis der iagttages høje aberrationstal.
Selv om testens formål er at påvise ændringer i kromosomstrukturen, er det vigtigt også at notere polyploidi og endofordobling, hvis dette konstateres.
2. DATA
2.1. BEHANDLING AF RESULTATER
Da cellen er forsøgets måleenhed, bedømmes det, hvilken procentdel af cellerne der har ændret kromosomstruktur. Forskellige strukturændringer noteres med antal og hyppighed for både forsøgs- og kontrolkulturer. Gaps noteres særskilt og registreres, men medregnes normalt ikke i den samlede aberrationshyppighed.
Sideløbende målinger af cytotoksicitet for alle behandlede og negative kontrolkulturer i hovedforsøget registreres ligeledes.
Dataene præsenteres for hver enkelt kultur. Desuden sammenfattes alle data i tabelform.
Der findes ingen krav til verifikation af et klart positivt resultat. Tvetydige resultater bør afklares ved yderligere undersøgelser, helst med ændrede forsøgsbetingelser. Der er redegjort for behovet for bekræftelse af negative resultater i punkt 1.4.3.3. Ved opfølgende forsøg kan man overveje at ændre på undersøgelsens parametre, så de bedømte forsøgsbetingelser udvides. Blandt de undersøgelsesparametre, der kan ændres på, er koncentrationernes spredning og metabolismeaktiveringen.
2.2. EVALUERING OG FORTOLKNING AF RESULTATER
Der findes en række kriterier for opnåelse af et positivt resultat, f.eks. en koncentrationsafhængig forøgelse eller en reproducerbar forøgelse af antallet af celler med kromosomaberrationer. Der bør i første række ses på resultaternes biologiske relevans. Statistiske metoder kan benyttes som hjælpemiddel ved evalueringen af testresultaterne (3) (13). Statistisk signifikans bør ikke være den eneste faktor, der afgør, om resultatet bedømmes som positivt.
En forøgelse i antallet af polyploide celler kan være tegn på, at teststoffet er i stand til at inhibere mitoseprocesser og fremkaldte kromosomantalsafvigelser. En forøgelse af antallet af celler med endofordoblede kromosomer kan være tegn på, at teststoffet kan inhibere cellecyklussens forløb (17) (18).
Et teststof, der ikke opfylder ovennævnte kriterier, anses ikke for mutagent i dette system.
De fleste forsøg giver klart positivt eller negativt resultat, men i sjældne tilfælde giver dataene ikke mulighed for en endegyldig bedømmelse af teststoffets aktivitet. Resultatet kan stadig være tvetydigt eller tvivlsomt, uanset hvor mange gange forsøget gentages.
Et positivt resultat af en in vitro-test for kromosomaberrationer viser, at teststoffet fremkalder ændringer i kromosomstrukturen i dyrkede somatiske celler fra pattedyr. Et negativt resultat viser, at teststoffet under testbetingelserne ikke fremkalder ændringer i kromosomstrukturen i dyrkede somatiske celler fra pattedyr.
3. RAPPORTERING
TESTRAPPORT
Testrapporten skal indeholde følgende oplysninger:
|
|
Opløsningsmiddel/bærestof:
|
|
|
Celler:
|
|
|
Testbetingelser:
|
|
|
Resultater:
|
|
|
Diskussion af resultaterne |
|
|
Konklusioner |
4. HENVISNINGER
|
(1) |
Evans, H. J. (1976), Cytological Methods for Detecting Chemical Mutagens, in: Chemical mutagens, Principles and Methods for their Detection, Vol. 4, Hollaender. A, (ed) Plenum Press, New York and London, pp. 1-29. |
|
(2) |
Ishidate, M. Jr. and Sofuni, T. (1985), The In Vitro Chromosomal Aberration Test Using Chinese Hamster Lung (CHL) Fibroblast Cells in Culture. In: Progress in Mutation Research, Vol. 5. Ashby, J. et al., (eds) Elsevier Science Publishers, Amsterdam-New York-Oxford. pp. 427-432. |
|
(3) |
Galloway, S.M., Armstrong, M.J., Reuben, C., Colman, S, Brown, B., Cannon, C., Bloom, A.D., Nakamura, F., Ahmed, M., Duk, S., Rimpo, J., Margolin, G.H., Resnick, M.A., Andersen, G. and Zeiger, E. (1978), Chromosome aberration and sister chromatic exchanges in Chinese hamster ovary cells: Evaluation of 108 chemicals. Environs. Molec. Mutaden 10 (suppl. 10), pp. 1-175. |
|
(4) |
Scott, D., Galloway, S.M., Marshall, R.R., Ishidate, M. Jr., Brusick, D., Ashby, J. and Myhr, B.C. (1991), Genotoxicity under Extreme Culture Conditions. A report from ICPLMC Task Group 9, Mutation Res., 257, pp. 147-204. |
|
(5) |
Morita, T., Nagaki, T., Fukuda, I. and Okumura, K. (1992), Clastogenicity of low pH to Various Cultured Mammalian Cells, Mutation Res., 268. pp. 297-305. |
|
(6) |
Arnes, B.N., McCann, J,. and Yamasaki, E. (1975), Methods for Detecting Carcinogens and Mutagens with the Salmonella/Mammalian Microsome Mutagenicity Test, Mutation Res., 31, pp. 347-364. |
|
(7) |
Maron, D.M. and Arnes, B.N. (1983), Revised Methods for the Salmonella Mutagenicity Test, Mutation Res., 113, pp. 173-215. |
|
(8) |
Natarajan. A.T., Tates, A.D., van Buul, P.P.W., Meijers, M. and de Vogel, N. (1976), Cytogenetic Effects of Mutagen/Carcinogens after Activation in a Microsomal System In Vitro, J. Induction of Chromosome Aberrations and Sister Chromatid Exchange by Diethylnitrosamine (DEN) and Dimethylnitrosamine (DMN) in CHO Cells in the Presence of Rat-Liver Microsomes, Mutation Res., 37, pp. 83-90. |
|
(9) |
Matsuoka, A., Hayashi, M. and Ishidate, M. Jr. (1979), Chromosomal Aberration Tests on 29 Chemicals Combined with S9 Mix In Vitro, Mutation Res., 66, pp. 277-290. |
|
(10) |
Elliot, B.M., Combes. R.D., Elcombe, C.R., Gatehouse, D,G., Gibson, G.G., Mackay, J.M. and Wolf, R.C. (1992), Report of UK Environmental Mutagen Society Working Party. Alternatives to Aroclar 1254-induced S9 in In Vitro Genotoxicity Assays. Mutagenesis, 7, pp. 175-177. |
|
(11) |
Matsushima, T., Sawamura, M., Hara, K. and Sugimura, T. (1976), A Safe Substitute for Polychlorinated Biphenyls as an Inducer of Metabolic Activation Systems. In: de Serres, F.J., Fouts, J.R., Berid, J.R. and Philpot, R.M. (eds), In Vitro Metabolic Activalion in Mutagenesis Testing, Elsevier, North-Holland, pp. 85-88. |
|
(12) |
Galloway, S.M., Aardema, M.J., Ishidate, M. Jr., Iven, J.L., Kirkland, D.J., Morita, T., Mosesso, P., Sofuni, T. (1994), Report from Working Group on in In Vitro Tests for Chromosomal Aberrations, Mutation Res., 312, pp. 241-261. |
|
(13) |
Richardson, C., Williams, D.A., Allen, J.A., Amphlett, G., Chanter, D.O. and Phillips, B. (1989), Analysis of Data from in Vitro Cytogenetic Assays, in: Statistical Evaluation of Mulagenicity Test Data, Kirkland, D.J., (ed) Cambridge University Press, Cambridge, pp. 141-154. |
|
(14) |
Soper, K.A. and Galloway, S.M. (1994), Replicate Flasks are not Necessary for In Vitro Chromosome Aberration Assays in CHO Cells, Mutation Res., 312, pp. 139-149. |
|
(15) |
Krahn, D.F., Barsky, F.C. and McCooey, K.T. (1982), CHO/HGPRT Mutation Assay: Evaluation of Gases and Volatile Liquids. In: Tice, R.R., Costa, D.I., Schaich, K.M. (eds), Genotoxic Effects of Airborne Agents, New York, Plenum, pp. 91-103. |
|
(16) |
Zamora, P.O., Benson, J.M., Li, A.P. and Brooks, A.L. (1983), Evaluation of an Exposure System Using Cells Grown on Collagen Gels for Detecting Highly Volatile Mutagens in the CHO/HGPRT Mutation Assay, Environmental Mutagenesis, 5, pp. 795-801. |
|
(17) |
Locke-Huhle, C. (1983), Endoreduplication in Chinese hamster cells during alpha-radiation induced G2 arrest, Mulation Res., 119, pp. 403-413. |
|
(18) |
Huang, Y., Change, C. and Trosko, J.E. (1983), Aphidicolin-induced endoreduplication in Chinese hamster cells, Cancer Res., 43, pp. 1362-1364. |
B.11. MUTAGENICITET — IN VIVO-TEST FOR KROMOSOMABERRATIONER I KNOGLEMARV HOS PATTEDYR
1. METODE
Denne metode er en gengivelse af OECD Test Guideline 475 Mammalian Bone Marrow Chromosome Aberration Test (1997).
1.1. INDLEDNING
In vitro-testen for kromosomaberrationer hos pattedyr benyttes til at påvise, om et teststof forårsager strukturelle kromosomafvigelser i knoglemarvceller hos dyr, normalt gnavere (1) (2) (3) (4). Der er to typer strukturændringer, kromosomtypen og kromatidtypen. Ploiditetsforøgelse kan være tegn på, at et kemisk stof er i stand til at fremkalde antalsafvigelser. De fleste kemiske mutagener forårsager ændringer af kromosomtypen, men også ændringer af kromatidtypen forekommer. Kromosommutationer og lignende er årsag til mange genetiske sygdomme hos mennesker, og meget tyder på, at kromosommutationer og lignende, som ændrer på onkogener og tumorsuppressorgener i somatiske celler, medvirker ved induktion af kræft hos mennesker og i forsøgssystemet.
Der benyttes rutinemæssigt gnavere i denne test. Knoglemarv er målvævet i denne test, da det er et væv med høj vaskularisation og består af celler med kort cyklustid, som er lette at isolere og behandle. Metoden omfatter ikke andre arter og væv.
Denne test for kromosomaberrationer er særlig relevant for en vurdering af risikoen for mutationsfremkaldelse, da den giver mulighed for at tage sådanne faktorer som in vivo-metabolisme, farmakokinetik og DNA-reparationsprocesser i betragtning, selv om de kan variere fra art til art og fra væv til væv. En in vivo-test er ligeledes nyttig til yderligere undersøgelse af mutagene virkninger, der er påvist ved en in vitro-test.
Hvis der er grund til at tro, at teststoffet eller en reaktiv metabolit ikke vil nå frem til målvævet, er denne test ikke egnet.
Se også den generelle indledning til del B.
1.2. DEFINITIONER
Aberration af kromatidtypen: beskadigelse af kromosomstrukturen i form af brud på enkeltkromatider eller brud på og sammenføjning af kromatider.
Aberration af kromsomtypen: beskadigelse af kromosomstrukturen i form af brud på — eller brud på og sammenføjning af — begge kromatider på samme sted.
Endofordobling: en proces, hvorved cellekernen efter en S-fase i DNA-replikationen ikke fortsætter over i mitosen, men påbegynder endnu en S-fase. Resultatet er kromosomer med 4, 8, 16, ... kromatider.
Gap: en akromatisk beskadigelse, som er mindre end ét kromatid bred, og hvor misalignment af kromatiderne er minimal.
Antalsafvigelse: ændring af kromosomtallet i forhold til det for de pågældende celler normale antal.
Polyploidi: et multiplum af det haploide kromosomtal (n), som ikke er det diploide tal (dvs. 3n, 4n osv.).
Strukturel ændring: ændring i kromosomstrukturen, som kan iagttages ved mikroskopi i celledelingens metafase i form af deletioner og fragmenter, intrachange og interchange.
1.3. TESTMETODENS PRINCIP
Dyrene udsættes for teststoffet på passende måde og aflives efter passende tidsrum efter eksponeringen. Inden aflivningen behandles dyrene med et metafasestandsende stof (f.eks. colchicin eller Colcemid®). Der fremstilles derefter præparater af knoglemarvceller, som farves, og metafasecellerne undersøges for kromosomaberrationer.
1.4. BESKRIVELSE AF TESTMETODEN
1.4.1. Præparater
1.4.1.1. Valg af dyreart
Der benyttes sædvanligvis rotter, mus og kinesiske hamstere, selv om andre egnede pattedyrarter kan benyttes. Der bør anvendes almindeligt brugte laboratoriestammer af unge sunde voksne dyr. Ved undersøgelsens begyndelse bør vægtvariationen mellem dyrene være mindst mulig og ikke over ±20 % af gennemsnitsvægten for hvert køn.
1.4.1.2. Miljø og fordring
Der gælder de almindelige forhold som beskrevet i den generelle indledning til del B, blot bør der tilstræbes en fugtighed på 50-60 %.
1.4.1.3. Forberedelse af dyrene
Der udvælges tilfældigt sunde unge voksne dyr til kontrol- og behandlingsgruppen. Burene bør anbringes på en sådan måde, at deres placering får mindst mulig indvirkning. Dyrene identificeres entydigt. Dyrene tilvænnes til laboratorieforholdene i mindst fem dage.
1.4.1.4. Fremstilling uf doser
Teststoffer i fast form opløses eller opslæmmes i passende opløsningsmidler eller bærestoffer og fortyndes passende, inden de gives til dyrene. Teststoffer i væskeform kan enten gives direkte til dyrene eller fortyndes først. Der benyttes frisk fremstillede teststofpræparater, medmindre stabilitetsdata viser, at opbevaring er acceptabel.
1.4.2. Testbetingelser
1.4.2.1. Opløsningsmiddel/bærestof
Opløsningsmiddel/bærestof må ikke have toksiske virkninger ved de benyttede doser og må ikke mistænkes for at reagere kemisk med teststoffet. Benyttes der andre opløsningsmidler/bærestoffer end de velkendte, skal brugen af dem underbygges med kompatibilitetsdata. Det anbefales i videst muligt omfang først at forsøge at benytte et vandigt opløsningsmiddel/bærestofsystem.
1.4.2.2. Kontrolgrupper
I alle forsøg medtages der sideløbende både positive og negative (opløsningsmiddel eller bærestof) kontrolgrupper af begge køn. Bortset fra behandling med teststof skal dyrene i kontrolgrupperne behandles på samme måde som dyrene i behandlingsgrupperne.
Positive kontrolprøver skal frembringe strukturelle ændringer in vivo ved en dosis, der forventes at give en påviselig forøgelse i forhold til baggrundsværdierne. Der vælges sådanne doser i de positive kontrolprøver, at virkningerne er tydelige, uden dog at de kodede objektglas' identitet umiddelbart afsløres for den, der aflæser dem. Det kan accepteres, at den positive kontrol indgives på anden måde end teststoffet, og at der kun foretages én prøveudtagning. Hvis der er mulighed for det, bør der anvendes kemisk beslægtede stoffer. Nedenfor er opregnet eksempler på positive kontrolstoffer:
|
Stof |
CAS nr. |
Einecs nr. |
|
Ethylmethansulfonat |
62-50-0 |
200-536-7 |
|
Ethylnitrosourea |
759-73-9 |
212-072-2 |
|
Mitomycin C |
50-07-7 |
200-008-6 |
|
Cyclophosphamid Cyclophosphamidmonohydrat |
50-18-0 6055-19-2 |
200-015-4 |
|
Triethylenmelamin |
51-18-3 |
200-083-5 |
Der skal ved hver prøveudtagning indgå negative kontrolgrupper, som kun er behandlet med opløsningsmiddel eller bærestof, og ellers behandlet på samme måde som behandlingsgrupperne, medmindre tidligere kontroldata har vist en acceptabel variation mellem dyr og hyppighed af celler med kromosomafvigelser. Hvis der kun udtages én negativ kontrolprøve, er det mest hensigtsmæssigt at gøre det ved første prøveudtagning. Desuden skal der være ubehandlede kontrolgrupper, medmindre der foreligger tidligere opnåede eller offentliggjorte kontroldata, der viser, at det valgte opløsningsmiddel/bærestof ingen skadelige eller mutagene virkninger har.
1.5. FREMGANGSMÅDE
1.5.1. Dyrenes antal og køn
Hver behandlet gruppe og kontrolgruppe skal bestå af mindst fem analyserbare dyr af hvert køn. Hvis der på det tidspunkt, hvor undersøgelsen foretages, foreligger data fra undersøgelser med samme dyreart og med samme indgiftsmåde, som viser, at der ikke er nogen væsentlig toksicitetsforskel mellem de to køn, er test med kun ét køn tilstrækkeligt. Hvis udsættelsen af mennesker for det kemiske stof er kønsspecifik, som det f.eks. er tilfældet med visse farmaceutiske stoffer, skal testen udføres med dyr af det pågældende køn.
1.5.2. Behandlingsplan
Teststofferne indgives fortrinsvis i en enkelt dosis. De kan også indgives i to deldoser, dvs. to behandlinger samme dag med kun nogle få timers interval, hvorved det bliver lettere at indgive et større materialevolumen. Indgift på anden måde skal begrundes sagligt.
Der udtages prøver på to tidspunkter efter endagsbehandling. For gnavere udtages første prøve normalt 1,5 normal cellecykluslængde (som normalt er 12-18 timer) efter behandlingen. Da det optimale tidspunkt for påvisning af kromosomaberrationer kan afhænge af, hvor lang tid der er påkrævet for optagelse og metabolisme af teststoffet, og af teststoffets indvirkning på cellecyklussens kinetik, anbefales det at udtage endnu en prøve 24 timer efter den første. Indgives stoffet over mere end én dag, udtages der blot én prøve, når der er gået 1,5 normal cellecykluslængde efter den sidste behandling.
Inden aflivningen gives dyrene en intraperitoneal injektion med en passende dosis metafasestandsende stof (f.eks. Colcemid® eller colchicin). Der udtages prøve af forsøgsdyrene efter et passende tidsrum. For mus er dette tidsrum ca. tre-fem timer, for kinesisk hamster ca. fire-fem timer. Cellerne høstes fra knoglemarven og analyseres for kromosomaberrationer.
1.5.3. Dosisniveauer
Hvis der gennemføres en forprøve til bestemmelse af dosisinterval, fordi der ikke foreligger nogen egnede data, bør den udføres i samme laboratorium med samme art, stamme, køn og behandlingsmåde, som agtes benyttet i hovedundersøgelsen (5). Er der tale om toksicitet, benyttes der tre dosisniveauer for første prøveudtagningstidspunkt. Disse dosisniveauer skal dække et interval fra maksimal toksicitet til næsten ingen eller ingen toksicitet. Ved den efterfølgende prøveudtagning behøves kun den højeste dosis benyttet, Den højeste dosis defineres som den dosis, der fremkalder sådanne tegn på toksicitet, at højere dosis med samme indgiftsmønster må forventes at medføre død. Stoffer med specifik biologisk aktivitet ved lav ikke-toksisk dosis (f.eks. hormoner og mitogener) kan danne undtagelser fra disse dosisfastsættelseskriterier og bør evalueres individuelt. Højeste dosis kan også defineres som en dosis, der i nogen grad udviser tegn på toksicitet i knoglemarven (f.eks. mitoseindeks nedsat med mere end 50 %).
1.5.4. Grænsetest
Hvis der ved en test med én dosis på mindst 2 000 mg/kg legemsvægt indgivet på én gang eller i to omgange samme dag ikke kan iagttages nogen toksiske virkninger, og hvis man på grundlag af data om strukturmæssigt beslægtede stoffer ikke forventer gentoksicitet, kan en fuldstændig undersøgelse med tre dosisniveauer anses for unødvendig. For længerevarende undersøgelser er grænsedosis på 2 000 mg/kg legemsvægt pr. dag for behandlinger over højst 14 dage og på 1 000 mg/kg legemsvægt pr. dag for behandlinger over mere end 14 dage. Den forventede eksponering af mennesker kan foranledige, at der benyttes en højere dosis i grænsetesten.
1.5.5. Indgift af doser
Teststoffet indgives normalt ved tvangsfodring med sonde eller en passende intubationskanyle eller ved intraperitoneal injektion. Andre indgiftsmåder kan accepteres, hvis de kan begrundes. Hvor stor en væskemængde der kan indgives ad gangen ved tvangsfodring eller injektion, afhænger af forsøgsdyrenes størrelse. Mængden bør højst være på 2 ml pr. 100 g legemsvægt. Anvendelse af større rumfang skal begrundes. Bortset fra lokal-irriterende og ætsende stoffer, som normalt vil have kraftigere virkninger ved højere koncentrationer, bør testvolumenet variere så lidt som muligt, idet koncentrationen justeres, så volumenet bliver det samme ved alle dosisniveauer.
1.5.6. Præparering af kromosomer
Umiddelbart efter aflivningen udtages knoglemarven, som udsættes for hypotonisk væske og fikseres. Derefter stryges cellerne ud på objektglas og farves.
1.5.7. Analyse
Mitoseindekset bestemmes som udtryk for cytotoksiciteten i mindst 1 000 celler pr. dyr for alle behandlede dyr (inkl. positive kontroldyr) og ubehandlede (negative) kontroldyr.
For hvert dyr bør mindst 100 celler analyseres. Dette antal kan sættes ned, hvis der iagttages et stort antal aberrationer. Alle objektglas, herunder positive og negative kontrolprøver, kodes uafhængigt inden mikroskoperingen. Da præpareringen af objektglas ofte medfører, at en del af metafasecellerne går i stykker og kromosomerne går tabt, bør bedømte celler indeholde et antal centromerer svarende til kromosomtallet 2n ± 2.
2. DATA
2.1. BEHANDLING AF RESULTATER
Data for de enkelte dyr præsenteres i tabelform. Forsøgsenheden er et dyr. For hvert dyr registreres antallet af bedømte celler, antallet af aberrationer pr. celle og den procentdel af cellerne, der har kromosomaberrationer. Forskellige typer strukturelle kromosomændringer skal anføres med antal og hyppighed for behandlingsgrupper og kontrolgrupper. Gaps registreres særskilt og oplyses, men medregnes ikke generelt i den totale aberrationshyppighed. Hvis der ikke er tegn på forskellig reaktion hos de to køn, kan dataene samles med henblik på statistisk analyse.
2.2. EVALUERING OG FORTOLKNING AF RESULTATER
Der findes en række kriterier for at afgøre, om et resultat er positivt, f.eks. en dosisafhængig forøgelse af andelen af celler med kromosomaberrationer eller en tydelig forøgelse af antallet af celler med aberrationer hos en enkeltdosisgruppe på et bestemt prøveudtagningstidspunkt. Der bør i første række ses på resultaternes biologiske relevans. Statistiske metoder kan benyttes som hjælpemiddel ved evalueringen af testresultaterne (6). Statistisk signifikans bør ikke være den eneste faktor, der afgør, om resultatet bedømmes som positivt. Tvetydige resultater bør afklares ved yderligere test, helst med ændring af forsøgsbetingelserne.
En forøgelse af ploiditetsgraden kan være tegn på, at teststoffet er i stand til at fremkalde kromosomantalsafvigelser. En forøget endofordobling kan være tegn på, at teststoffet kan inhibere cellecyklussens forløb (7) (8).
Et teststof, der ikke opfylder ovennævnte kriterier, anses ikke for mutagent i dette system.
De fleste forsøg giver klart positivt eller negativt resultat, men i sjældne tilfælde giver dataene ikke mulighed for en endegyldig bedømmelse af teststoffets aktivitet. Resultatet kan stadig være tvetydigt eller tvivlsomt, uanset hvor mange gange forsøget gentages.
Et positivt resultat af en in vivo-test for kromosomaberrationer viser, at teststoffet fremkalder kromosomaberrationer i knoglemarven hos den undersøgte art. At negativt resultat viser, at teststoffet under testbetingelserne ikke fremkalder kromosomaberrationer i knoglemarven hos den undersøgte art.
Sandsynligheden for, at teststoffet eller dets metabolitter når frem til det almindelige kredsløb eller specifikt til målvævet (f.eks. systemisk toksicitet), bør diskuteres.
3. RAPPORTERING
3.1. TESTRAPPORT
Testrapporten skal indeholde følgende oplysninger:
|
|
Opløsningsmiddel/bærestof:
|
|
|
Forsøgsdyr:
|
|
|
Testbetingelser:
|
|
|
Resultater:
|
|
|
Diskussion af resultaterne |
|
|
Konklusioner |
4. HENVISNINGER
|
(1) |
Adler, L.D. (1984), Cytogenetic Tests in Mammals, in: Mutagenicity Testing: a Practical Approach. S. Venitt and J.M. Parry (eds.). IRL Press, Oxford. Washington D.C., pp. 27 5-306. |
|
(2) |
Preston. R.J., Dean, B.J., Galloway, S., Holden, H. McFee, A.F. and Shelby, M. (1987), Mammalian In Vivo Cytogenetic Assays: Analysis of Chromosome Aberrations in Bone Marrow Cells, Mutation Res., 189, pp. 157-165. |
|
(3) |
Richold, M., Chandly, A., Ashby, J., Gatehouse D.G., Bootman, J. and Henderson, L. (1990), In Vivo Cytogenetic Assays, in: D.J. Kirkland, (ed.), Basic Mutagenicity Tests, UKEMS Recommended Procedures. UKEMS Sub-Committee on Guidelines for Mutagenicity Testing Report. Part I revised, Cambridge University Press, Cambridge, New Cork, Port Chester, Melbourne, Sydney, pp. 115-141. |
|
(4) |
Tice, R.R. Hayashi, M., MacGregor, J.T., Anderson, D., Blakey, D.H., Holden, H.E., Kirch-Volders, M., Oleson Jr., F.B., Paccierotti, F., Preston, R.J., Romagna F., Shimada, H., Sutou, S. and Vannier, B. (1994), Report from the Working Group on the In Vivo Mammalian Bone Marrow Chromosomal Aberration Test, Mutation Res., 312, pp. 305-312. |
|
(5) |
Fielder, R.J., Alleen, J.A., Boobis, A.R., Botham, P.A., Doe, J., Esdaile, D.J., Gatehouse, D.G., Hodson-Walker, G., Morton, D.B., Kirkland, D.J. and Richold, M. (1992), Report of British Toxicology Society/UK, Environmental Mutagen Society Working Group: Dose setting in in Vivo Mutagenicity Assays, Mutagenesis, 7, pp. 313-319. |
|
(6) |
Lovell, D.P., Andersen, D., Albanese, R., Amphlett, G.E., Clare, G., Ferguson, R., Richold, M., Papworth, D.G. and Savage, J.R.K. (1989), Statistical Analysis of In Vivo Cytogenetic Assays, in: UKEMS Sub-Committee on Guidelines for Mutagenicity Testing. Report Part III. Statistical Evaluation of Mutagenicity Test Data. D.J. Kirkland, (ed.) Cambridge University Press, Cambridge, pp. 184-232. |
|
(7) |
Locke-Huhle, C. (1983), Endoreduplication in Chinese hamster cells during alpha-radiation-induced G2 arrest, Mutation Res. 119, pp. 403-413. |
|
(8) |
Huang. Y., Change, C. and Trosko, J.E. (1983), Aphidicolin-induced endoreduplication in Chinese hamster cells, Cancer Res., 43, pp. 1363-1 364. |
B.12. MUTAGENICITET — IN VIVO-TEST FOR MIKROKERNER I ERYTHROCYTTER HOS PATTEDYR
1. METODE
Denne metode er en gengivelse af OECD Test Guideline 474 Mammalian Erythrocyte Micronucleus Test (1997).
1.1. INDLEDNING
In vivo-testen for mikrokerner hos pattedyr benyttes til påvisning af skader, som teststoffet har forvoldt på kromosomer eller mitoseapparatet i erythroblaster, ved analyse af erythrocytter i prøver fra knoglemarv og/eller det perifere blod hos dyr, sædvanligvis gnavere.
Formålet med mikrokernetesten er at påvise stoffer, der forvolder cytogenetiske skader, som medfører dannelse af mikrokerner indeholdende tiloversblevne kromosomfragmenter eller hele kromosomer.
Når en erythroblast i knoglemarven udvikler sig til en polykromatisk erythrocyt, udstødes cellekernen, og en eventuel dannet mikrokerne kan blive tilbage i det ellers kernefri cytoplasma. I disse celler er mikrokerner let synlige, da de ellers ikke har nogen kerne. Forøget forekomst af polykromatiske erythrocytter med mikrokerne hos behandlede dyr er tegn på inducerede kromosomskader.
I denne test benyttes der rutinemæssigt knoglemarv hos gnavere, da der i dette væv produceres polykromatiske erythrocytter. Måling af umodne (polykromatiske) erythrocytter med mikrokeme i det perifere blod kan også accepteres i andre arter, hvor det er vist, at milten ikke kan fjerne erythrocytter med mikrokerner, eller at deres følsomhed over for stoffer, der forårsager strukturelle eller antalsmæssige kromosomaberrationer, er tilstrækkelig. Mikrokerner kan genkendes på en række kriterier, bl.a. om der er en DNA-centromer til stede. Det vigtigste endpoint er hyppigheden af umodne (polykromatiske) erythrocyter med mikrokerne. Det antal modne (normokromatiske) erythrocytter i det perifere blod, som indeholder mikrokerner, ud af et givet antal modne erythrocytter kan også benyttes som endpoint for testen, når dyrene behandles kontinuerligt i mindst fire uger.
Denne in vivo-test for mikrokerner hos pattedyr er særlig relevant for vurdering af den reelle mutagenitetsfare, idet den tager højde for sådanne faktorer som in vivo-metabolisme, farmakokinetik og DNA-reparationsprocesser, selv om disse kan variere fra art til art, væv til væv og genetisk endpoint til genetisk endpoint. En in vivo-test er ligeledes nyttig til yderligere undersøgelse af mutagene virkninger, der er påvist ved en in vitro-test.
Hvis der er grund til at tro, at teststoffet eller en reaktiv metabolit ikke vil nå frem til målvævet, er denne test ikke egnet.
Se også den generelle indledning til del B.
1.2. DEFINITIONER
Centromer: region(er) af et kromosom, hvortil tentrådene er påhæftet under celledeling, således at datterkromosomerne kan ordnes i forhold til dattercellernes poler.
Mikrokerner: små ekstrakerner, der er adskilt fra cellens hovedkerne, og som dannes under mitosens telofase (meiose) ud fra efterladte kromosomfragmenter eller hele kromosomer.
Normochromatisk erythrocyt: moden erythrocyt. der mangler ribosomer og kan skelnes fra umodne polykromatiske erythrocytter ved ribosomselektiv farvning.
Polychromatisk erythocyt: umoden erythrocyt på et udviklingsmellemstadium, som stadig indeholder ribosomer og derfor kan skelnes fra modne normokromatiske erythrocytter ved ribosomselektiv farvning.
1.3. TESTMETODENS PRINCIP
Dyrene udsættes for teststoffet på passende måde. Hvis der benyttes knoglemarv, aflives dyrene på et passende tidspunkt efter behandlingen, hvorefter knoglemarven udtages, og der fremstilles præparater og farves (1) (2) (3) (4) (5) (6) (7). Hvis der benyttes perifert blod, tages der blodprøver på et passende tidspunkt efter behandlingen, hvorefter der fremstilles udstrygningspræparater og farves (4) (8) (9) (10). I undersøgelser med perifert blod bør der gå så kort tid som muligt mellem sidste eksponering og cellehøst. Præparaterne analyseres for tilstedeværelse af mikrokerner.
1.4. BESKRIVELSE AE TESTMETODEN
1.4.1. Præparater
1.4.1.1. Valg af dyreart
Der anbefales mus eller rotter, hvis der bruges knoglemarv, selv om andre egnede pattedyrarter kan benyttes. Bruges der perifert blod, anbefales mus. Dog kan enhver egnet pattedyrart benyttes, hvis dens milt ikke kan fjerne erythrocytter med mikrokerne, eller hvis den er vist at være tilstrækkelig følsom til at påvise stoffer, der forårsager strukturelle eller antalsmæssige kromosomaberrationer. Der bør anvendes almindeligt brugte laboratoriestammer af unge sunde dyr. Ved undersøgelsens begyndelse bør vægtvariationen mellem dyrene være mindst mulig og ikke over ± 20 % af gennemsnitsvægten for hvert køn.
1.4.1.2. Miljø og fodring
Der gælder de almindelige forhold som beskrevet i den generelle indledning til del B, blot bør der tilstræbes en fugtighed på 50-60 %.
1.4.1.3. Forberedelse af dyrene
Der udvælges tilfældigt unge sunde voksne dyr til kontrol- og behandlingsgruppen. Dyrene identificeres entydigt. Dyrene tilvænnes til laboratorieforholdene i mindst fem dage. Burene bør anbringes på en sådan måde, at deres placering får mindst mulig indvirkning.
1.4.1.4. Fremstilling af doser
Teststoffer i fast form opløses eller opslæmmes i passende opløsningsmidler eller bærestoffer og fortyndes passende, inden de gives til dyrene. Teststoffer i væskeform kan enten gives direkte til dyrene eller fortyndes først. Der benyttes frisk fremstillede teststofpræparater, medmindre stabilitetsdata viser, at opbevaring er acceptabel.
1.4.2. Testbetingelser
1.4.2.1. Opløsningsmiddel/bærestof
Opløsningsmiddel/bærestof må ikke have toksiske virkninger ved de benyttede doser og må ikke mistænkes for at reagere kemisk med teststoffet. Benyttes der andre opløsningsmidler/bærestoffer end de velkendte, skal brugen af dem underbygges med kompatibilitetsdata. Det anbefales i videst muligt omfang først at forsøge at benytte et vandigt opløsningsmiddel/bærestofsystem.
1.4.2.2. Kontrolgrupper
I alle forsøg medtages der sideløbende både positive og negative (opløsningsmiddel eller bærestof) kontrolgrupper af begge køn. Bortset fra behandling med teststof skal dyrene i kontrolgrupperne behandles på samme måde som dyrene i behandlingsgrupperne.
Positive kontrolprøver skal frembringe mikrokerner in vivo ved en dosis, der forventes at give en påviselig forøgelse i forhold til baggrundsværdierne. Der vælges sådanne doser i de positive kontrolprøver, at virkningerne er tydelige, uden dog at de kodede objektglas' identitet umiddelbart afsløres for den, der aflæser dem. Det kan accepteres, at den positive kontrol indgives på anden måde end teststoffet, og at der kun foretages én prøveudtagning. Hvis der er mulighed for det, bør der anvendes kemisk beslægtede stoffer. Nedenfor er opregnet eksempler på positive kontrolstoffer:
|
Stof |
CAS nr. |
Einecs nr. |
|
Ethylmethansulfonat |
62-50-0 |
200-536-7 |
|
N-ethyl-N-nitrosourea |
759-73-9 |
212-072-2 |
|
Mitomycin C |
50-07-7 |
200-008-6 |
|
Cyclophosphamid Cyclophosphamidmonohydrat |
50-18-0 6055-19-2 |
200-015-4 |
|
Triethylenmelamin |
51-18-3 |
200-083-5 |
Der skal ved hver prøveudtagning indgå negative kontroldyr, som kun er behandlet med opløsningsmiddel eller bærestof og ellers behandlet på samme måde som behandlingsgrupperne, medmindre tidligere kontroldata har vist en acceptabel variation mellem dyr indbyrdes og acceptabel hyppighed af celler med mikrokerner. Hvis der kun udtages én negativ kontrolprøve, er det mest hensigtsmæssigt at gøre det ved første prøveudtagning. Desuden skal der være ubehandlede kontrolgrupper, medmindre der foreligger tidligere opnåede eller offentliggjorte kontroldata, der viser, at det valgte opløsningsmiddel ingen skadelige eller mutagene virkninger har.
Hvis der benyttes perifert blod, kan en forbehandlingsprøve også accepteres som sideløbende negativ kontrolprøve, men kun i de korte undersøgelser (f.eks. 1-3 behandlinger), når de opnåede resultater ligger inden for det forventede interval for den forudgående kontrolprøve.
1.5. FREMGANGSMÅDE
1.5.1. Dyrenes antal og køn
Hver behandlet gruppe og kontrolgruppe skal bestå af mindst fem analyserbare dyr af hvert køn (11). Hvis der på det tidspunkt, hvor undersøgelsen foretages, foreligger data fra undersøgelser med samme dyreart og med samme indgiftsmåde, som viser, at der ikke er nogen væsentlig toksicitetsforskel mellem de to køn, er test med kun ét køn tilstrækkelig. Hvis udsættelsen af mennesker for det kemiske stof er kønsspecifik, som det f.eks. er tilfældet med visse farmaceutiske stoffer, skal testen udføres med dyr af det pågældende køn.
1.5.2. Behandlingsplan
Der kan ikke anbefales nogen standardbehandlingsplan (dvs. 1, 2 eller flere behandlinger med 24 timers mellemrum). Prøver fra længdervarende indgiftsplaner er acceptable, når blot der i undersøgelsen er påvist en positiv virkning eller — for en negativ undersøgelse — der er påvist toksicitet, eller grænsedosis har været anvendt, og når indgiften er fortsat indtil prøveudtagningen. Prøvestoffet kan også indgives i to deldoser, dvs. to behandlinger samme dag med kun nogle få timers interval, hvorved det bliver lettere at indgive et større materialevolumen.
Testen kan udføres på følgende to måder:
|
a) |
Dyrene behandles med teststoffet én gang, Der udtages prøver af knoglemarv mindst to gange, første gang tidligst 24 timer efter behandlingen, med passende intervaller mellem prøverne, sidste prøve dog senest 48 timer efter behandlingen. Prøveudtagning tidligere end 24 timer efter behandlingen skal begrundes. Der udtages prøver af perifert blod mindst to gange, tidligst 36 timer efter behandlingen og med passende intervaller efter den første prøve, sidste prøve dog senest efter 72 timer. Når der i en prøve er konstateret positiv reaktion, er yderligere prøveudtagning ikke nødvendig. |
|
b) |
Benyttes der to eller flere daglige behandlinger (f.eks. to eller flere behandlinger med 24 timers mellemrum) udtages der for knoglemarvs vedkommende prøver én gang mellem 18 og 24 timer efter den sidste behandling, og for perifert blods vedkommende én gang mellem 36 og 48 timer efter den sidste behandling (12). |
Derudover kan der udtages prøver på andre tidspunkter, hvis det er relevant.
1.5.3. Dosisniveauer
Hvis der gennemføres en forprøve til bestemmelse af dosisinterval, fordi der ikke foreligger nogen egnede data, bør den udføres i samme laboratorium med samme art, stamme, køn og behandlingsmåde som agtes benyttet i hovedundersøgelsen (13). Er der tale om toksicitet, benyttes der tre dosisniveauer for første prøveudtagningstidspunkt. Disse dosisniveauer skal dække et interval fra maksimal toksicitet til næsten ingen eller ingen toksicitet. Ved den efterfølgende prøveudtagning behøves kun den højeste dosis benyttet. Den højeste dosis defineres som den dosis, der fremkalder sådanne tegn på toksicitet, at højere dosis med samme indgiftsmønster må forventes at medføre død. Stoffer med specifik biologisk aktivitet ved lav ikke-toksisk dosis (f.eks. hormoner og mitogener) kan danne undtagelser fra disse dosisfastsættelseskriterier og bør evalueres individuelt. Højeste dosis kan også defineres som en dosis, der i nogen grad udviser tegn på toksicitet i knoglemarven (f.eks. en nedsættelse af andelen af umodne erythrocytter i forhold til totale erythrocytter i knoglemarv eller perifert blod).
1.5.4. Grænsetest
Hvis der ved en test med én dosis på mindst 2 000 mg p. kg legemsvægt indgivet på én gang eller i to omgange samme dag ikke kan iagttages nogen toksiske virkninger, og hvis man på grundlag af data om strukturnæssigt beslægtede stoffer ikke forventer gentoksicitet, kan en fuldstændig undersøgelse med tre dosisniveauer anses for unødvendig, for længerevarende undersøgelser er grænsedosis på 2 000 mg pr. kg legemsvægt pr. dag for behandlinger over højst 14 dage og på 1 000 mg pr. kg legemsvægt pr. dag for behandlinger over mere end 14 dage. Den forventede eksponering af mennesker kan foranledige, at der benyttes en højere dosis i grænsetesten.
1.5.5. Indgift af doser
Teststoffet indgives normalt ved tvangsfodring med sonde eller en passende intubationskanyle eller ved intraperitoneal injektion. Andre indgiftsmåder kan accepteres, hvis de kan begrundes. Hvor stor en væskemængde, der kan indgives ad gangen ved tvangsfodring eller injektion, afhænger af forsøgsdyrenes størrelse. Mængden bør højst være på 2 ml pr. 100 g legemsvægt. Anvendelse af større rumfang skal begrundes. Bortset fra lokalirriterende og ætsende stoffer, som normalt vil have kraftigere virkninger ved højere koncentrationer, bør testvolumenet variere så lidt som muligt, idet koncentrationen justeres, så volumenet bliver det samme ved alle dosisniveauer.
1.5.6. Præparering af knoglemarv/blod
Knoglemarvceller udtages normalt fra lårben eller skinneben umiddelbart efter aflivningen. Sædvanligvis udtages cellerne fra lårben eller skinneben, hvorefter de præpareres og farves med velkendte metoder. Perifert blod udtages fra halevenen eller et andet egnet blodkar. Blodlegemerne farves straks supravitalt (8) (9) (10), eller der fremstilles udstrygningspræparater, der derefter farves. Ved at bruge en DNA-specifik farvning (f.eks. acridinorange (14) eller Hoechst 33258 plus pyronin-Y (15)) kan man undgå nogle af de artifakter, der optræder ved ikke-DNA-specifik farvning. Denne fordel udelukker ikke, at der benyttes konventionelle farvestoffer (f.eks. Giemsa). Supplerende systemer (f.eks. cellulosekolonner til fjernelse af kerneindeholdende celler (16)) kan også benyttes, forudsat at de er påvist at fungere tilfredsstillende ved præparering af mikrokerner i laboratoriet.
1.5.7. Analyse
For hvert dyr bestemmes andelen af umodne erythrocytter i forhold til det samlede antal (umodne og modne) erythrocytter, idet der tælles mindst 200 erythrocytter i alt for knoglemarv og 1 000 erythrocytter for perifert blod (17). Alle objektglas, herunder positive og negative kontrolprøver, kodes uafhængigt inden mikroskoperingen. For hvert dyr bedømmes mindst 2 000 umodne erythrocytter for forekomst af umodne etythrocytter med risikokerner. Der kan opnås yderligere information ved bedømmelse af modne erythrocytter for indhold af mikrokerner. Ved analyse af objektglas må andelen af umodne erythrocytter i forhold til totale erythrocytter ikke være mindre end 20 % af kontrolværdien. Når dyrene behandles kontinuerligt i fire uger eller derover, kan også mindst 2 000 modne erythrocytter pr. dyr bedømmes for forekomst af mikrokerner. Systemer til automatisk analyse (billedanalyse og flow-cytometri af celleopslæmninger) er acceptable som alternativer til manuel evaluering, hvis de er behørigt begrundet og valideret.
2. DATA
2.1. BEHANDLING AF RESULTATER
Data for de enkelte dyr præsenteres i tabelform. Forsøgsenheden er et dyr. For hvert dyr registreres antallet af bedømte umodne erythrocytter, antallet af umodne erythrocytter med mikrokerne og antallet af umodne erythrocytter i forhold til det samlede antal erythrocytter. Hvis dyrene behandles kontinuerligt i mere end fire uger, anføres også dataene for modne erythrocytter, hvis de foreligger. Andelen af umodne erythrocytter i forhold til totale erythrocytter oplyses for hvert dyr, og, hvis det skønnes hensigtsmæssigt, procentdelen af erythrocytter med mikrokerne. Hvis der ikke er tegn på forskellig reaktion hos de to køn, kan dataene samles med henblik på statistisk analyse.
2.2. EVALUERING OG FORTOLKNING AF RESULTATER
Der findes en række kriterier for at afgøre, om et resultat er positivt, f.eks. en dosisafhængig forøgelse af antallet af celler med mikrokerner eller en tydelig forøgelse af antallet af celler med mikrokerner hos en enkeltdosisgruppe på et bestemt prøveudtagningstidspunkt. Der bør i første række ses på resultaternes biologiske relevans. Statistiske metoder kan benyttes som hjælpemiddel ved evalueringen af testresultaterne (18) (19). Statistisk signifikans bør ikke være den eneste faktor, der afgør, om resultatet bedømmes som positivt. Tvetydige resultater bør afklares ved yderligere test, helst med ændring af forsøgsbetingelserne.
Et teststof, der ikke opfylder ovennævnte kriterier, anses ikke for mutagent i dette system.
De fleste forsøg giver klart positivt eller negativt resultat, men i sjældne tilfælde giver dataene ikke mulighed for en endegyldig bedømmelse af teststoffets aktivitet. Resultatet kan stadig være tvetydigt eller tvivlsomt, uanset hvor mange gange forsøget gentages.
Et positivt resultat af mikrokernetesten viser, at teststoffet fremkalder mikrokerner, som følge af beskadigelse af kromosomer eller mitoseapparat hos erythroblaster i den undersøgte art. Et negativt resultat viser, at teststoffet under testbetingelserne ikke fremkalder mikrokerner hos umodne erythrocytter i den undersøgte art.
Sandsynligheden for, at teststoffet eller dets metabolitter når frem til det almindelige kredsløb eller specifikt til målvævet (f.eks. systemisk toksicitet), bør diskuteres.
3. RAPPORTERING
TESTRAPPORT
Testrapporten skal indeholde følgende oplysninger:
|
|
Opløsningsmiddel/bærestof:
|
|
|
Forsøgsdyr:
|
|
|
Testbetingelser:
|
|
|
Resultater:
|
|
|
— Diskussion af resultaterne |
|
|
— Konklusioner |
4. HENVISNINGER
|
(1) |
Heddle, J.A. (1973), A Rapid In Vivo Test for Chromosomal Damage, Mutation Res., 18, pp. 187-190. |
|
(2) |
Schmid, W. (1975), The Micronucleus Test, Mulation Res., 31, pp. 9-15. |
|
(3) |
Heddle, J.A., Salamone, M.F., Hite, M, Kirkhart, B., Mavournin, K., MacGregor, J.G. and Newell, G.W. (1983), The Induction of Micronuclei as a Measure of Genotoxicity, Mutation Res. 123, pp. 61-118. |
|
(4) |
Mavournin, K.H., Blakey, D.H., Cimino, M.C, Salamone, M.F. and Heddie, J.A. (1990), The In Vivo Micronucleus Assay in Mammalian Bone Marrow and Peripheral Blood. A report of the U.S. Environmental Protection Agency Gene-Tox Program, Mutation Res., 239, pp. 29-80. |
|
(5) |
MacGregor, J.T. Schlegel, R. Choy. W.N., and Wehr, C.M. (1983), Micronuclei in Circulating Erythrocytes: A Rapid Screen for Chromosomal Damage During Routine Toxicity Testing in Mice, in: Developments in Science and Practice of Toxicology, cd. A.W. Hayes, R.C. Schnell and T.S. Miya, Elsevier, Amsterdam, pp., 555-558. |
|
(6) |
MacGregor, J.T., Heddle, J.A., Hite. M., Margolin, G.H., Ramel, C, Salamone, M.F., Tice, R.R. and Wild, D. (1987), Guidelines for the Conduct of Micronucleus Assays in Mammalian Bone Marrow Erythrocytes, Mutation Res., 189, pp. 103-112. |
|
(7) |
MacGregor, J.T., Wehr, C.M., Henika, P.R. and Shelby, M.E. (1990), The in vivo Erythrocyte Micronucleus Test: Measurement at Steady State Increases Assay Efficiency and Permits Integration with Toxicity Studies, Fund. Appl. Toxicol. 14, pp. 513-522. |
|
(8) |
Hayashi, M., Morita, T., Kodama, Y., Sofuni, T. and Ishidate, M. Jr. (1990), The Micronucleus Assay with Mouse Peripheral Blood Reticulocytes Using Acridine Orange-Coated Slides, Mutation Res. 245, pp. 245-249. |
|
(9) |
The Collaborative Study Group for the Micronucleus Test (1992). Micronucleus Test with Mouse Peripheral Blood Erythrocytes by Acridine Orange Supravital Staining: The Summary Report of the 5th Collaborative Study by CSGMT/JEMMS. MMS, Mutation Res., 278, pp. 83-98. |
|
(10) |
The Collaborative Study Group for the Micronucleus Test (CSGMT/JEMMS, MMS: The Mammalian Mutagenesis Study Group of the Environmental Mutagen Society of Japan) (1995). Protocol recommended for the short-term mouse peripheral blood micronucleus test, Mutagenesis, 10, pp. 15 3-159. |
|
(11) |
Hayashi, M., Tice, R.R., MacGregor, J.T., Anderson, D., Blackey, D.H., Kirsch-Volders, M., Oleson, Jr. F.B., Pacchierotti, F., Romagna, F., Shimada, H., Sutou, S. and Vannier, B. (1994), In Vivo, Rodent Erythrocyte Micronucleus Assay, Mutation Res., 312. pp. 293-304. |
|
(12) |
Higashikuni, N. and Sutou, S. (1995), An optimal, generalised sampling time of 30 ± 6 h after double dosing in the mouse peripheral blood micronucleus test, Mutagenesis, 10, pp. 313-319. |
|
(13) |
Fielder, R.J., Allen, J.A., Boobis, A.R.. Botham, P.A., Doe, J., Esdaile, D.J., Gatehouse, D.G., Hodson-Walker, G., Morton, D.B., Kirkland, D.J. and Rochold, M. (1992), Report of British Toxicology Society/UK Environmental Mutagen Society Working Group: Dose Setting in In Vivo Mutagenicity Assays, Mutagenesis. 7, pp. 313-319. |
|
(14) |
Hayashi, M., Sofuni, T, and Ishidate, M. Jr. (1983), An Application of Acridine Orange Fluorescent Staining to the Micronucleus Test, Mutation Res., 120, pp. 241-247. |
|
(15) |
MacGregor, J.T., Wehr, C.M. and Langlois, R.G. (1983), A Simple Fluorescent Staining Procedure for Micronuclei and RNA in Eriythrocytes Using Hoechst 33258 and Pyronin Y, Mutation Res. 120, pp. 269-275. |
|
(16) |
Romagna, F. and Staniforth, C.D. (1989), The automated bone marrow micronucleus test, Mutation Res., 213, pp. 91-104. |
|
(17) |
Gollapudi, B. and McFadden, L.G. (1995), Sample size for the estimation of polychromatic to normochromatic erythrocyte ratio in the bone marrow micronucleus test, Mutation Res. 347, pp. 97-99. |
|
(18) |
Richold, M., Ashby, J., Bootman, J., Chandley, A., Gatehouse, D.G. and Henderson, L. (1990), In Vivo Cytogenetics Assay, in: D.J. Kirkland (ed.), Basic Mutagenicity tests. UKEMS Recommended Procedures, UKEMS Sub-Committe on Guidelines for Mutagenicity Testing Report, Part 1, revised, Cambridge University Press, Cambridge, New York, Port Chester, Melbourne, Sydney, pp. 115-141. |
|
(19) |
Lovell, D.P., Anderson, D., Albanese, R., Amphlett, G. E., Clare, G., Ferguson, R., Richold, M., Papworth, D.G. and Savage, J.R.K. (1989), Statistical Analysis of In Vivo Cytogenetic Assays, in: D. J. Kirkland (ed.), Statistical Evaluation of Mutagenicity Test Data. UKEMS Sub-Committee on Guidelines for Mutagenicity Testing. Report, Part III, Cambridge University Press, Cambridge, New York, Port Chester, Melbourne, Sydney, pp. 184-232. |
B.13/14. MUTAGENICITET — TILBAGEMUTATIONSTEST MED BAKTERIER
1. METODE
Denne metode er en gengivelse af OECD Test Guideline 471 Bacterial Reverse Mutation Test (1997).
1.1. INDLEDNING
I tilbagemutationstesten med bakterier benyttes der aminosyrekrævede stammer af Salmonella typhimurium og Escherichia coli til at påvise punktmutationer med substitution, addition eller deletion af et eller nogle få dna-basepar (1) (2) (3). Princippet i denne tilbagemutationstest med bakterier er, at den påviser mutationer, som tilbagemuterer mutationer i teststammerne og dermed sætter bakterien i stand til at syntetisere en essentiel aminosyre. Revertantbakterierne påvises ved deres evne til at vokse uden den aminosyre, som den oprindelige teststamme kræver.
Punktmutationer er årsagen til mange genetiske sygdomme hos mennesker, og der er meget, der tyder på, at punktmutationer i onkogener og tumorsuppressorgener i somatiske celler spiller en rolle for dannelse af tumorer hos mennesker og i forsøgsdyr. Tilbagemutationstesten med bakterier er hurtig, billig og forholdsvis let at udføre. Mange teststammer har forskellige egenskaber, der gør dem mere følsomme til påvisning af mutationer, bl.a. responsive dna-sekvenser på reversionsstedet, høj cellepermeabilitet for store molekyler og manglende dna-reparationssystemer eller forstærkede fejlbehæftede dna-reparationsprocesser. Teststammernes specificitet kan give nyttige oplysninger om, hvilke typer mutationer et gentoksisk stof inducerer. Der findes en meget stor database med resultater for en lang række strukturer med tilbagemutationstest med bakterier, og der er udviklet veletablerede metodologier for test af kemikalier med forskellige fysiske og kemiske egenskaber, herunder flygtige forbindelser.
Se også den generelle indledning til del B.
1.2. DEFINITIONER
En tilbagemutationstest med enten Salmonella typhimurium eller Escherichia coli påviser mutation i en aminosyrekrævende stamme (histidin eller tryptophan) til en stamme, der er uafhængig af tilførsel af aminosyren udefra.
Mutagener for baseparsubstitution er stoffer, der forårsager en baseændring i dna'et. I en tilbagemutationstest kan denne ændring ske samme sted som den oprindelige mutation eller et andet sted i bakteriens genom.
Læserammemutagener er stoffer, der medfører insertion eller deletion af et eller flere basepar i dna'et, således at RNA'ets læseramme ændres.
1.5. INDLEDENDE OVERVEJELSER
I tilbagemutationstesten med bakterier benyttes der prokaroytiske celler, som adskiller sig fra pattedyrceller med hensyn til f.eks. optagelse, metabolisme, kromosomstruktur og dna-reparationsprocesser. In vitro-test kræver normalt en exogen kilde til metabolismeaktivering. In vitro-metabolismeaktiveringssystemer kan ikke fuldstændig efterligne in vivo-forholdene i pattedyr. Testen vil derfor ikke give direkte oplysninger om et stofs mutagene og carcinogene potentiale i pattedyr.
Tilbagemutationstesten med bakterier anvendes generelt til en indledende screening for gentoksiske virkninger og, især, punktmutationsinducerende aktivitet. Det er ved hjælp af en omfattende database påvist, at mange kemiske stoffer, som giver positivt resultat i denne test, også har mutagen virkning i andre test. Der findes mutagene stolfer, der ikke er påvist ved denne test; årsagen hertil kan tilskrives det påviste endpoints specifikke karakter, forskelle i metabolismeaktivering og forskelle i biotilgængelighed. På den anden side kan faktorer, der øger tilbagemutationstestens følsomhed, føre til et for højt skøn over stoffets mutagene aktivitet.
Tilbagemutationstesten med bakterier kan være uegnet til evaluering af bestemte klasser af kemiske stoffer, f.eks. stærkt baktericide forbindelser (f.eks. visse antibiotika) og forbindelser, der antages (eller vides) at gribe specifikt ind i pattedyrcellers replikationssystem (f.eks. visse topoisomeraseinhibitorer og visse nucleosidanaloger). I sådanne tilfælde kan mutationstest i pattedyr være mere velegnede.
Selv om mange af de stoffer, der er positive i denne test, er kræftfremkaldende hos pattedyr, er korrelationen ikke fuldstændig. Den afhænger af den kemiske klasse, og der findes carcinogener, der ikke påvises ved denne test, fordi de virker via andre ikke-gentoksiske mekanismer eller mekanismer, der ikke findes i bakterieceller.
1.4. TESTMETODENS PRINCIP
Opslæmninger af bakterieceller udsættes for teststoffet med og uden et exogent metabolismeaktiveringssystem. Ved pladeinkorporeringsmetoden blandes disse opslæmninger med en topagar og hældes ud på et minimalsubstrat. Ved præinkubationsmetoden inkuberes behandlingsblandingen, hvorefter den blandes med topagar inden udhældning på minimalsubstrat. For begge metoders vedkommende tælles revertantkolonierne efter to-tre dages inkubation, og der sammenlignes med antallet af spontane revertantkolonier på kontrolpladerne med opløsningsmiddel.
Der er beskrevet en række fremgangsmåder for udførelse af tilbagemutationstesten med bakterier, Blandt de mest almindeligt benyttede er pladeinkorporeringsmetoden (1) (2) (3) (4), præinkubationsmetoden (2) (3) (5) (6) (7) (8), fluktuationsmetoden (9) (10) og suspensionsmetoden (11). Der er beskrevet modifikationer til test af gasser og dampe (12).
De i denne metode beskrevne fremgangsmåder vedrører især pladeinkorporeringsmetoden og præinkubationsmetoden. De er begge acceptable til udførelse af forsøgene både med og uden metabolismeaktivering. Nogle stoffer, bl.a. kortkædede alifatiske nitrosaminer, divalente metaller, aldehyder, azofarvestoffer og diazoforbindelser, pyrollizidinalkaloider, allylforbindelser og nitroforbindelser (3), kan mest effektivt påvises ved præinkubationsmetoden. Det er også kendt, at visse klasser af mutagener ikke altid påvises ved standardmetoder som f.eks. pladeinkorporeringsmetoden og præinkubationsmetoden. Sådanne tilfælde bør betragtes som »særtilfælde«, og det anbefales kraftigt at anvende alternative metoder til påvisning i disse tilfælde. Følgende »særtilfælde« er konstateret (samt eksempler på fremgangsmåder, der kan benyttes til påvisning): azofarvestoffer og diazoforbindelser (3) (5) (6) (13), gasser og flygtige stoffer (12) (14) (15) (16) og glycosider (17) (18), Afvigelser fra standardmetoden skal begrundes sagligt.
1.5. BESKRIVELSE AF TESTMETODEN
1.5.1. Præparater
1.5.1.1. Bakterier
Friske bakteriekulturer fremdyrkes til den sidste del af den eksponentielle fase eller til begyndelsen af den stationære fase (ca. 109 celler pr. ml). Kulturer sidst i den stationære fase bør ikke benyttes. Det er vigtigt, at de kulturer, der benyttes i forsøgene, har et højt indhold af levedygtige bakterier. Titeren kan godtgøres enten ved hjælp af tidligere kontroldata for vækstkurver eller ved bestemmelse af antallet af levedygtige celler i det enkelte forsøg ved udpladning.
Som inkubationstemperatur anbefales 37 oC.
Der skal benyttes mindst fem bakteriestammer, deriblandt fire stammer af S. typhimurium (TA1535; TA1537 eller TA97a eller TA97; TA98; og TA100), som er påvist at være pålidelige, og hvis respons er reproducerbar mellem laboratorier. Disse fire S. typhimurium-stammer har GC-basepar på de primære reversionssted, og det vides, at de ikke altid påviser visse oxiderende mutagener, tværbindende stoffer og hydraziner. Sådanne stoffer kan påvises med E. coli WP2-stammer eller S. typhimurium TA102 (19), som har AT-basepar på det primære reversionssted. Derfor kan følgende kombination af stammer anbefales:
|
— |
S. typhimurium TA 1535, og |
|
— |
S. typhimurium TA 1537, TA97 eller TA97a, og |
|
— |
S. typhimurium TA98, og |
|
— |
S. typhimurium TA100, og |
|
— |
E. coli WP2 uvrA, eller E. coli WP2 uvrA (pKM101) eller S. typhimurium TA102. |
For at kunne påvise tværbindende mutagener kan det foretrækkes at vælge TA102 eller at tilføje en DNA-reparationsdygtig stamme af E. coli (f.eks. E. coli WP2 eller E. coli WP2 (pKM101)).
Der benyttes anerkendte metoder til stamkulturfremstilling, markørverifikation og opbevaring. For hver enkelt frosset kulturpræparat påvises den aminosyrebetingede vækst (histidin for S. typhimurium-stammer og tryptophan for E. coli-stammer). Andre fænotypiske egenskaber skal kontrolleres på lignende måde, således tilstedeværende eller manglende R-faktorplasmider, når det er relevant (dvs. ampicillinresistens hos TA98-stammen, TA-100- og TA97a- eller TA97-stammcn, WP2 uvrA-stammen og WP2 uvrA (pKM101)-stammen, samt ampicillin- og tetracyclinresistens hos TA102-stammen), tilstedeværelse af karakteristiske mutationer (dvs. rfa-mutation hos S. typhimurium ved hjælp af følsomhed over for krystalviolet og uvrA-mutation hos E. coli eller uvrB-mutation hos S. typhimurium ved hjælp af følsomhed over for ultraviolet lys) (2) (3). Stammerne skal ligeledes give kimtal for spontane revertanter inden for den hyppighed, der må forventes ud fra laboratoriets hidtidige kontroldata, og helst inden for den hyppighed, der er angivet i litteraturen.
1.5.1.2. Substrat
Der benyttes en egnet minimalagar (f.eks. med Vogel-Bonner minimalsubstrat E og glucose) og en topagar med histidin og biotin eller tryptophan, så der tillades nogle få celledelinger (1) (2) (9).
1.5.1.3. Metabolismeaktivering
Bakterierne udsættes for teststoffet både med og uden et egnet metabolismeaktiveringssystem. Det mest almindeligt anvendte system er en cofaktorsuppleret postmitokondriefraktion (S9), der fremstilles af levere fra rotter, der har været behandlet med enzyminducerede stoffer såsom Aroclor 1254 (1) (2) eller en blanding af phenobarbiton og β-naphthoflavon (18) (20) (21). Postmitokondriefraktionen anvendes normalt i en koncentration på 5-30 % (v/v) i S9-blandingen. Valg af metabolismeaktiveringssystem og dets beskaffenhed kan afhænge af, hvilken kemisk klasse teststoffet tilhører. I nogle tilfælde vil det være hensigtsmæssigt at benytte postmitokondriefraktion i mere end én koncentration. For azofarvestoffer og diazoforbindelser kan et reduktivt metabolismeaktiveringssystem være mere velegnet (6) (13).
1.5.1.4. Teststof/testpræparat
Teststoffer i fast form opløses eller opslæmmes i passende opløsningsmidler eller bærestoffer og fortyndes passende inden behandling af bakterierne. Teststoffer i væskeform kan enten tilsættes direkte til testsystemet eller fortyndes inden behandlingen. Der benyttes frisk fremstillede teststofpræparater, medmindre stabilitetsdata viser, at opbevaring er acceptabel.
Der må ikke være mistanke om, at opløsningsmiddel/bærestof reagerer kemisk med teststoffet, og opløsningsmiddel/bærestof skal være kompatibel med bakteriernes overlevelse og S9-aktiviteten (22). Benyttes der andre opløsningsmidler/bærestoffer end de velkendte, skal brugen af dem underbygges med kompatibilitetsdata. Det anbefales i videst muligt omfang først at forsøge at benytte et vandigt opløsningsmiddel/bærestofsystem. Ved undersøgelse af stoffer, der er ustabile i vand, skal de organiske opløsningsmidler være vandfrie.
1.5.2. Testbetingelser
1.5.2.1. Teststammer (se punkt 1.5.1.1)
1.5.2.2. Eksponeringskoncentration
Stoffets cytotoksicitet og dets opløselighed i den endelige testblanding er blandt de kriterier, der skal tages hensyn til ved valg af højeste koncentration.
Det kan være hensigtsmæssigt at bestemme toksicitet og opløselighed ved et indledende forsøg. Cytotoksicitet kan påvises ved et fald i antallet af revertantkolonier, ved en opklaring eller hæmning af baggrundsvæksten eller ved graden af overlevelse i de behandlede kulturer. Et stofs cytotoksicitet kan ændres ved, at der er metabolismeaktiveringssystemer til stede. Uopløselighed bedømmes ved, at der med det blotte øje kan iagttages udfældning i slutblandingen under de faktiske forsøgsbetingelser.
Der anbefales en maksimal testkoncentration af opløselige ikke-cytotoksiske stoffer på 5 mg/plade eller 5 μl/plade. For ikke-cytotoksiske stoffer, der ikke er opløselige med 5 mg/plade eller 5 μl/plade, skal en eller flere af de undersøgte koncentrationer være uopløselig i slutblandingen. Teststoffer, der er cytotoksiske allerede under 5 mg/plade eller 5 μl/plade. testes op til den cytotoksiske koncentration. Bundfald må ikke have indflydelse på bedømmelsen.
Der benyttes mindst fem forskellige analyserbare koncentrationer af teststoffer med en afstand på ca. en halv logaritme (√10) mellem koncentrationerne i det indledende forsøg. Ved undersøgelse af en koncentration/respons-sammenhæng kan mindre intervaller være hensigtsmæssigt. Test ved koncentrationer over 5 mg/plade eller 5 μl/plade kan tages under overvejelse, når bedømmelsen vedrører stoffer med et betydeligt indhold af potentielt mutagene urenheder.
1.5.2.3. Negative og positive kontrolprøver
I alle forsøg medtages der sideløbende både positive og negative (opløsningsmiddel eller bærestof) stammespecifikke kontrolprøver, såvel med som uden metabolismeaktivering. Der vælges en sådan koncentration i den positive kontrol, at det enkelte forsøg effektivitet godtgøres.
Ved forsøg med metabolismeaktivering udvælges positive kontrolreferencestoffer ud fra den valgte type bakteriestamme.
Nedenfor er der eksempler på stoffer, som er egnede som positive kontrolstoffer i forsøg med metabolismeaktivering:
|
CAS nr. |
Einecs nr. |
Stof |
|
781-43-1 |
212-308-4 |
9,10-dimethylanthracen |
|
57-97-6 |
200-359-5 |
7,1 2-dimethylbenz[a]anthracen |
|
50-32-8 |
200-028-5 |
benzo[a]pyren |
|
613-13-8 |
210-330-9 |
2-aminoanthracen |
|
50-18-0 |
|
cyclophosphamid |
|
6055-19-2 |
200-015-4 |
cyclophosphamidmonohydrat |
Følgende stof er egnet som positivt kontrolstof ved reduktiv metabolismeaktivering:
|
CAS nr. |
Einecs nr. |
Stof |
|
573-58-0 |
209-358-4 |
Congo-rødt |
2-aminoanthracen bør ikke anvendes som eneste indikator for, om .S9-blandingen er effektiv. Hvis der benyttes 2-aminoanthracen, skal hver batch af S9 tillige karakteriseres med et mutagen, der kræver metabolismeaktivering med mikrosomenzymer, f.eks, benzo[a]pyren eller dimethylbenzanthracen.
Nedenfor er der eksempler på stoffer til brug som stammespecifikke positive kontrolstoffer i forsøg uden exogen metabolismeaktivering:
|
CAS nr. |
Einecs nr. |
Stof |
Stamme |
|
26628-22-8 |
247-852-1 |
natriumazid |
TA 1535 og TA 100 |
|
607-57-8 |
210-138-5 |
2-nitrofluoren |
TA 98 |
|
90-45-9 |
201-995-6 |
9-aminoacridin |
TA 1537, TA 97 og TA 97a |
|
17070-45-0 |
241-129-4 |
ICR 191 |
TA 1537, TA 97 og TA 97a |
|
80-15-9 |
201-254-7 |
cumenbydroperoxid |
TA 102 |
|
50-07-7 |
200-008-6 |
mitomycin C |
WP2 uvrA og TA 102 |
|
70-25-7 |
200-730-1 |
N-ethyl-N-nitro-N-nitrosoguanidin |
WP2, WP2 uvrA og WP2 uvrA (pKM101) |
|
56-57-5 |
200-281-1 |
4-nitroquinolin-l-oxid |
WP2, WP2 uvrA og WP2 uvrA (pKM101) |
|
3688-53-7 |
|
furylfuramid (AF2) |
plasmidholdige stammer |
Der kan benyttes andre egnede referencestoffer til positiv kontrol. Hvis der er mulighed for det, bør der anvendes kemisk beslægtede stoffer.
Der skal indgå negative kontrolprøver, der kun består af opløsningsmiddel eller bærestof uden teststof, og som behandles på samme måde som de øvrige testgrupper. Desuden skal der være ubehandlede kontrolprøver, medmindre der foreligger tidligere opnåede kontroldata, der viser, at det valgte opløsningsmiddel ingen skadelige eller mutagene virkninger har.
1.5.3. Fremgangsmåde
Ved pladeinkorporeringsmetoden (1) (2) (3) (4) uden metabolismeaktivering blandes normalt 0,05 ml eller 0,1 ml testopløsning, 0,1 ml frisk bakteriekultur (indeholdende ca. 108 levedygtige celler) og 0,5 ml steril buffer med 2,0 ml topagar. Ved forsøg med metabolismeaktivering blandes normalt ca. 0,5 ml metabolismeaktiveringsblanding, indeholdende en passende mængde postmitokondriefraktion (5-30 % (v/v) i metabolismeaktiveringsblandingen), med topagaren (2,0 ml), bakterierne og teststoffet/testopløsningen. Indholdet i de enkelte glas blandes og hældes ud over overfladen på en minimalagarplade. Topagaren henstilles til størkning inden inkubering.
Ved præinkubationsmetoden (2) (3) (5) (6) forinkuberes teststof/testopløsning sammen med teststammen (indeholdende ca. 108 levedygtige celler) og steril buffer eller metabolismeaktiveringssystemet (0,5 ml) normalt i 20 minutter ved 30-37 oC, inden der blandes med topagaren, og blandingen hældes ud på overfladen af en minimalagarplade. Normalt blandes 0,05 ml eller 0,1 ml teststof/testopløsning, 0,1 ml bakteriekultur og 0,5 ml S9-blanding eller steril buffer med 2,0 ml topagar. Glassene bør under forinkuberingen beluftes i rysteapparat.
Til forsvarlig bestemmelse af variationen udføres der tredobbelt udpladning ved hvert dosisniveau. Dobbelt udpladning kan accepteres, hvis den begrundes sagligt. Tab af en plade af og til gør ikke nødvendigvis testen ubrugelig.
Gasformige og flygtige stoffer testes ved en egnet metode, f.eks. i forseglede dyrkningsflasker (12) (14) (15) (16).
1.5.4. Inkubering
Alle plader i hvert forsøg inkuberes ved 37 oC i 48-72 timer. Efter inkuberingen tælles antallet af revertantkolonier pr. plade.
2. DATA
2.1. BEHANDLING AF RESULTATER
Dataene forelægges som antallet af revertantkolonier pr. plade. Også antallet af revertantkolonier på både negative (opløsningsmiddelkontrol og en eventuel ubehandlet kontrol) og positive kontrolplader oplyses. Tallene for hver enkelt plade, gennemsnittet af revertantkolonier pr. plade og standardafvigelsen opgives for teststoffet og for positive og negative kontrolplader (ubehandlede og/eller med opløsningsmiddel).
Der findes ingen krav til verifikation af et klart positivt resultat. Tvetydige resultater bør afklares ved yderligere undersøgelser, helst med ændrede forsøgsbetingelser. Negative resultater bekræftes efter en vurdering af de enkelte tilfælde. Anses bekræftelse af negative resultater ikke for nødvendige, skal dette begrundes. Ved opfølgende forsøg kan man overveje at ændre på undersøgelsens parametre, så de bedømte forsøgsbetingelser udvides. Blandt de undersøgelsesparametre, der kan ændres på, er koncentrationernes spredning, behandlingsmetoden (pladeinkorporering eller væske-præinkubation) og metabolismeaktiveringen.
2.2. EVALUERING OG FORTOLKNING AF RESULTATER
Der findes en række kriterier for, om der er opnået et positivt resultat, f.eks. en koncentrationsafhængig forøgelse af antallet af revertantkolonier pr. plade i det undersøgte interval og/eller en reproducerbar forøgelse af antallet af revertantkolonier pr. plade ved en eller flere koncentrationer, for mindst én stamme med eller uden metabolismeaktivering (23). Der bør i første række ses på resultaternes biologiske relevans. Statistiske metoder kan benyttes som hjælpemiddel ved evalueringen af testresultaterne (24). Statistisk signifikans bør dog ikke være den eneste faktor, der afgør, om resultatet bedømmes som positivt.
Et teststof, der ikke opfylder ovennævnte kriterier, anses ikke for mutagent i denne test.
De fleste forsøg giver klart positivt eller negativt resultat, men i sjældne tilfælde giver dataene ikke mulighed for en endegyldig bedømmelse af teststoffets aktivitet. Resultatet kan stadig være tvetydigt eller tvivlsomt, uanset hvor mange gange forsøget gentages.
Et positivt resultat af en tilbagemutationstest med bakterier viser, at teststoffet fremkalder punktmutationer ved basesubstitution eller læserammeforskydning i genomet hos Salmonella typhimurium og/eller Escherichia coli. Et negativt resultat viser, at teststoffet under testbetingelserne ikke er mutagent i den undersøge organisme.
3. RAPPORTERING
TESTRAPPORT
Testrapporten skal indeholde følgende oplysninger:
|
|
Opløsningsmiddel/bærestof:
|
|
|
Stammer:
|
|
|
Testbetingelser:
|
|
|
Resultater:
|
|
|
Diskussion af resultaterne |
|
|
Konklusioner |
4. HENVISNINGER
|
(1) |
Ames, B.N., McCann, J. and Yamasaki E. (1975), Methods for Detecting Carcinogens and Mutagens with the Salmonella/Mammalian-Microsome Mutagenicity Test, Mutation Res., 31, pp. 347-364. |
|
(2) |
Maron, D.M. and Arnes, B.N. (1983), Revised Methods for the Salmonella Mutagenicity Test, Mutation Res. 113, pp. 173-215. |
|
(3) |
Gatehouse, D., Haworth, S., Cebula, T., Gocke, E., Kier, L., Matsushima, T., Melcion, C, Nohmi, T., Venitt. S. and Zeiger, E. (1994), Recommendations for the Performance of Bacterial Mutation Assays, Mutation Res., 312, pp. 217-233. |
|
(4) |
Kier, L.D., Brusick, D.J., Auletta, A.E., Von Halle, E.S., Brown, M.M., Simmon, V.F., Dunkel, V., McCann, J., Mortelmans. K., Prival, M., Rao, T.K. and Ray V. (1986). The Salmonella typhimurium/Mammalian Microsomal Assay: A Report of the U.S. Environmental Protection Agency Gene-Tox Program, Mutation Res., 168, pp. 69-240. |
|
(5) |
Yahagi, T., Degawa, M., Seino, Y.Y., Matsushima, T., Nagao, M., Sugimura, T. and Hashimoto. Y. (1975), Mutagenicity of Carcinogen Azo Dyes and their Derivatives, Cancer Letters, 1, pp. 91-96. |
|
(6) |
Matsushima, M., Sugimura, T., Nagao, M., Yahagi, T., Shirai, A. and Sawamura, M. (1980), Factors Modulating Mutagenicity Microbial Tests, in: Short-term Test Systems for Detecting Carcinogens, ed. Norpoth K.H. and Garner, R.C., Springer, Berlin-Heidelberg-New York, pp. 273-285. |
|
(7) |
Gatehouse. D.G. Rowland. I.R., Wilcox, P., Callender, R.D. and Foster R. (1980), Bacterial Mutation Assays, in: Basic Mutagenicity Tests: UKEMS Part 1 Revised, ed. D.J. Kirkland, Cambridge University Press, pp. 13-61. |
|
(8) |
Aeschacher, H.U., Wolleb, U. and Porchet, L. (198 7), Liquid Preincubation Mutagenicity Test for Foods, J. food Safety. 8. pp. 167-177. |
|
(9) |
Green, M.H.L., Muriel, W.J. and Bridges, B.A. (1976), Use of a simplified fluctuation test to detect low levels of mutagens, Mutations Res., 38, pp. 33-42. |
|
(10) |
Hubbard, S.A., Green, M.H.L., Gatehouse, D. and J.W. Bridges (1984), The Fluctuation Test in Bacteria, in: Handbook of Mutagenicity Test Procedures, 2nd Edition, ed. Kilbey, B. J., Legator, M., Nichols, \V. and Ramel, C, Elsevier, Amsterdam-New York-Oxford, pp. 141-161. |
|
(11) |
Thompson, E.D. and Melampy, P.J. (1981), An Examination of the Quantitative Suspension Assay for Mutagenesis with Strains of Salmonella typhimurium, Environmental Mutagenesis, 3, pp. 453-465. |
|
(12) |
Araki, A., Noguchi, T., Kato, F. and T. Matsushima (1994), Improved Method for Mutagenicity Testing of Gaseous Compounds by Using a Gas Sampling Bag, Mutation Res., 307, pp. 335-344. |
|
(13) |
Prival, M.]., Bell, S.J., Mitchell, V.D., Reipert, M.D. and Vaughan, V.L. (1984), Mutagenicity of Benzidinc and Benzidine-Congener Dyes and Selected Monoazo Dyes in a Modified Salmonella Assay, Mutation Res., 136, pp. 33-47. |
|
(14) |
Zeiger, E., Anderson B.E., Haworth, S., Lawlor, T. and Mortelmans, K. (1992), Salmonella Mutagenicity Tests. V. Results from the Testing of 311 Chemicals, Environ. Mol. Mutagen. 19, pp. 2-141. |
|
(15) |
Simmon, V., Kauhanen K. and Tardiff, R.G. (1977), Mutagenic Activity of Chemicals Identified in Drinking Water, in Progress in Genetic Toxicology. D. Scott, B. Bridges and F. Sobels (eds.) Elsevier, Amsterdam, pp. 249-258. |
|
(16) |
Hughes, T.J., Simmons, D.M., Monteith, I.,G. and Claxton, L.D, (1987), Vaporisation Technique to Measure Mutagenic Activity of Volatile Organic Chemicals in the Ames/Salmonella Assay, Environmental Mutagenesis. 9, pp. 421-441. |
|
(17) |
Matsushima, T., Matsumoto, A. Shirai, M.. Sawamura, M. and Sugimura T. (1979), Mutagenicity of the Naturally Occurring Carcinogen Cycasin and Synthetic Methylazoxy Methane Conjugates in Salomonella typhimurium. Cancer Res., 39, pp. 3780-3782. |
|
(18) |
Tamura, G., Gold, C, Ferro-Luzzi, A. and Arnes, B. N. (1980), Fecalase: A Model for Activation of Dictary Glycosides to Mutagens by Intestinal Flora, Proc. Natl. Acad. Sci. USA, 77, pp. 4961-4965. |
|
(19) |
Wilcox, P., Naidoo, A., Wcdd, D. J. and Gatehouse, D. G. (1990), Comparison of Salmonella lyphimurium TA 102 with Escherichia coli WP2 Tester strains, Mutagenesis, 5, pp. 285-291. |
|
(20) |
Matsushima, T., Sawamura, M., Hara, K. and Sugimura T. (1976), A Safe Substitute for Polychlorinated Biphenyls as an Inducer or Metabolic Activation Systems, in: In Vitro Metabolic Activation in Mutagenesis Testing, eds. F. J. de Serres et al. Elsevier, North Holland, pp. 85-88. |
|
(21) |
Elliot, B.M., Combes, R.D., Elcombe, C.R., Tatehouse, D.G., Gibson, G.G., Mackay, J.M. and Wolf, R.C. (1992), Alternatives to Aroclor 1254-induced S9 in in viiro Genotoxicity Assays, Mutagenesis, 7, pp. 175-177. |
|
(22) |
Maron, D., Katzenellenbogen, J. and Arnes, B.N. (1981), Compatibility of Organic Solvents with the Salmonella/Microsome Test, Mutation Res., 88, pp. 343-350. |
|
(23) |
Claxton, L.D., Allen J., Auletta, A., Mortelmans, K., Nestmann, E. and Zeiger, E. (1987), Guide for the Salmonella typhimurium/Mammalian Microsome Tests for Bacterial Mutagenicity, Mutation Res., 189, pp. 83-91. |
|
(24) |
Mahon, G.A.T. Green, M.H.L, Middleton, B., Mitchell, I., Robinson, W.D. and Tweats, D.J. (1989), Analysis of Data from Microbial Colony Assays, in: UKEMS Sub-Committee on Guidelines for Mutagenicity Testing, Part II. Statistical Evaluation of Mutagenicity Test Data, ed. Kirkland, D.J., Cambridge University Press, pp. 28-65. |
B.15. MUTAGENICITETSTESTNING OG SCREENING FOR KARCINOGENICITET GENMUTATION — SACCHAROMYCES CEREVlSIAE
1. METODE
1.1. INDLEDNING
Se den generelle indledning til del B.
1.2. DEFINITIONER
Se den generelle indledning til del B.
1.3. REFERENCESTOFFER
Ingen.
1.4. PRINCIP FOR TESTMETODEN
En række forskellige haploide og diploide stammer af gæren Saccharomyces cerevisiae kan anvendes ved undersøgelse af genmutationer, som induceres af kemiske stoffer med og uden metabolisk aktivering.
Man har anvendt fremadmutationssystemer med haploide stammer f.eks, ved bestemmelse af mutation fra røde, adeninafhængige mutanter (ade-1, ade-2) til dobbelt adeninafhængige hvide mutanter og selektive systemer som induktion af canavanin-resistens og cycloheximid-resistens.
Det bedst validerede tilbagemutationssystem bygger på anvendelse af den haploide stamme XV185-14C, der er bærer af ochre nonsensemutationerne ade 2-1, arg 4-17, lys 1-1 og trp 5-48, og som muterer tilbage med baseparsubstitutionsmutagener, der inducerer punktmutationer eller ochre suppressormutationer. XV185-14C er også bærer af his 1-7 markøren, som er en missense-mutation, der hovedsagelig muterer tilbage ved mutationer i andre loci, og markøren hom 3-10, der muterer tilbage med frameshift-mutagener.
Den eneste bredt anvendte diploide stamme af S. cerevisiae er D7, der er homozygot i ilv 1-92.
1.5. KVALITETSKRITERIER
Ingen.
1.6. BESKRIVELSE AF TESTMETODEN
Forberedelser
Opløsninger af teststof og kontrol fremstilles umiddelbart inden testen under anvendelse af et passende vehikel. Hvis der anvendes organiske kemiske forbindelser, som er uopløselige i vand, kan organiske opløsningsmidler som ætanol, acetone eller dimetylsulfoxid (DMSO) bruges i koncentrationer på op til 2 % v/v. Koncentrationen af vehiklet bør ikke medføre nogen ændring i cellevitalitet og vækstkarakteristika.
Cellerne eksponeres for teststoffet både med og uden tilsætning af et passende eksogent metabolisk aktiveringssystem.
Det hyppigst anvendte system består af en co-faktorberiget postmitochondrisk fraktion af lever fra gnavere, som er forbehandlede med enzyminducerende stoffer. Andre dyrearter, væv, postmitochondriske fraktioner eller fremgangsmåder kan også være egnede til metabolisk aktivering.
Forsøgsbetingelser
Den haploide stamme XV 185-14C og den diploide stamme D7 er de hyppigst anvendte i genmutationsundersøgelser. Også andre stammer kan være egnede.
Ved bestemmelse af overlevelsesgrad og antal mutanter anvendes passende kulturmedier.
Sideløbende med testkulturerne dyrkes positive kontrolkulturer, ubehandlede kontrolkulturer og kontrolkulturer med det anvendte opløsningsmiddel. Der anvendes egnede positive kontrolstoffer for hvert enkelt genetisk endpoint.
Der anvendes mindst fem forskellige koncentrationer af teststoffet med passende intervaller mellem koncentrationerne. Når der testes toksiske stoffer, bør den højeste koncentration ikke reducere overlevelsesgraden til mindre end 5 til 10 %. Stoffer, som er relativt uopløselige i vand, testes under anvendelse af passende fremgangsmåder op til opløselighedsgrænsen. Den højeste koncentration af ikke-toksiske teststoffer, som er frit opløselige i vand, fastlægges fra stof til stof.
Pladerne inkuberes i fire til syv dage mellem 28 og 30 oC i mørke.
Der anvendes subkulturer med spontane mutationsfrekvenser, som ligger inden for de almindeligt accepterede områder.
Der anvendes mindst tre parallelle plader for hver koncentration ved bestemmelsen af prototrofer, som er fremkommet ved genmutation, og af cellernes levedygtighed. Når man tester markører, der som hom 3-10 har en lav mutationsfrekvens, må antallet af plader forøges, således at der kan tilvejebringes en statistisk relevant mængde data.
Fremgangsmåde
Stammer af S. cerevisiae eksponeres normalt ved test i væske, hvortil der anvendes enten celler i stationærfase eller celler i vækstfase. Indledende undersøgelser bør udføres med celler i vækstfase, 1-5 × 107 celler pr. ml eksponeres for teststoffet i op til 18 timer ved 28 til 37 oC under omrystning. Hvis det er nødvendigt, tilsættes en passende mængde metabolisk aktiveringssystem under eksponeringen. Efter eksponeringen centrifugeres og vaskes cellerne, hvorefter de udsås på et egnet kulturmedium. Når inkubationen er slut, registreres induktionen af genmutationer og antal overlevende på hver plade. Hvis den første undersøgelse er negativ, bør en anden udføres under anvendelse af celler i stationærfase. Hvis den første undersøgelse er positiv, skal det efterprøves i et passende uafhængigt forsøg.
2. DATA
Data opstilles i tabelform. Antal optalte kolonier, antal mutanter, overlevelsesfrekvens samt mutationsfrekvens angives. Alle resultater efterprøves i et uafhængigt forsøg, Evalueringen af data baseres på passende statistiske metoder.
3. RAPPORTERING
3.1. FORSØGSRAPPORT
Forsøgsrapporten skal om muligt indeholde følgende:
|
— |
den anvendte stamme |
|
— |
forsøgsbetingelser: celler i stationær fase eller vækstfase, mediernes sammensætning, inkubationstemperatur og -periode, metabolisk aktiveringssystem |
|
— |
eksponeringsbetingelser: eksponeringskoncentrationer, fremgangsmåde og varighed, eksponeringstemperatur, positive og negative kontroller |
|
— |
antal optalte kolonier, antal mutanter, overlevelses- og mutationsfrekvens, om muligt dosis-responsforhold, statistisk evaluering af data |
|
— |
diskussion af resultaterne |
|
— |
fortolkning af resultaterne. |
3.2. EVALUERING OG FORTOLKNING
Se den generelle indledning til del B.
4. HENVISNINGER
Se den generelle indledning til del B.
B.16. MITOTISK REKOMBINATION — SACCHAROMYCES CEREVISIAE
1. METODE
1.1. INDLEDNING
Se den generelle indledning til del B.
1.2. DEFINITIONER
Se den generelle indledning til del B.
1.3. REFERENCESTOFFER
Ingen.
1.4. PRINCIP FOR TESTMETODEN
Der kan hos Saccharomyces cerevisiae konstateres mitotisk rekombination mellem gener (eller mere generelt mellem et gen og dets centromer) og inden for det samme gen. Den førstnævnte form kaldes mitotisk overkrydsning og frembringer reciprokke typer, mens den sidstnævnte som oftest ikke er reciprok og kaldes genbytning. Overkrydsning bestemmes almindeligvis ved frembringelse af recessive homozygote kolonier eller sektorer i en heterozygot stamme, mens genbytning bestemmes ved frembringelse af prototrofe revertanter i en auxotrof heteroallel stamme, som bærer to forskellige, defekte alleler af det samme gen. De hyppigst anvendte stammer ved undersøgelse af mitotisk genbytning er D4 (med heteroallele ade 2 og trp 5), BZ34 (med heteroallel arg 4), D7 (med heteroallel trp 5) og JD1 (med heteroallele his 4 og trp 5). Mitotisk overkrydsning, som frembringer røde og lyserøde sektorer, kan undersøges i D5 og D7 (der også kan bruges til bestemmelse af mitotisk genbytning og tilbagemutation af ilv 1-92), idet begge stammer bærer heteroallele komplementære alleler til ade 2.
1.5. KVALITETSKRITERIER
Ingen.
1.6. BESKRIVELSE AF TESTMETODEN
Forberedelser
Opløsninger af teststof og kontrol- eller referencestoffer fremstilles umiddelbart inden testen under anvendelse af et passende opløsningsmiddel. Hvis der anvendes organiske kemiske forbindelser, som er uopløselige i vand, kan organiske opløsningsmidler som ætanol, acetone eller dimetylsulfoxid (DMSO) bruges i koncentrationer på op til 2 % v/v. Koncentrationen af vehiklet bør ikke medføre nogen ændring i cellevitalitet og vækstkarakteristika.
Cellerne eksponeres for teststof både med og uden tilsætning af et passende eksogent metabolisk aktiveringssystem. Det hyppigst anvendte system består af en co-faktorberiget postmitochondrisk fraktion af lever fra gnavere, som er forbehandlede med enzyminducerende stoffer. Andre dyrearter, væv, postmitochondriske fraktioner eller fremgangsmåder kan også være egnede til metabolisk aktivering.
Forsøgsbetingelser
De hyppigst anvendte stammer er de diploide D4, D5, D7 og JD1. Også andre stammer kan være egnede.
Ved bestemmelse af overlevelsesgrad og mitotisk rekombinationsfrekvens anvendes passende kulturmedier.
Sideløbende med testkulturerne dyrkes positive kontrolkulturer, ubehandlede kontrolkulturer og kontrolkulturer med det anvendte opløsningsmiddel. Der anvendes egnede positive kontrolstoffer for hvert enkelt genetisk endpoint.
Der anvendes mindst fem forskellige koncentrationer af teststoffet med passende intervaller mellem koncentrationerne. Herunder må man tage hensyn til faktorer som cytotoksicitet og opløselighed. De laveste koncentrationer bør ikke have nogen indvirkning på cellernes levedygtighed. Når der testes toksiske stoffer, bør den højeste koncentration ikke reducere overlevelsesgraden til mindre end 5 til 10 %. Stoffer, som er relativt uopløselige i vand, testes under anvendelse af passende fremgangsmåder op til opløselighedsgrænsen. Den højeste koncentration af ikke-toksiske teststoffer, som er frit opløselige i vand, fastlægges fra test til test.
Celler i stationær fase eller vækstfase kan eksponeres for teststoffet i op til 18 timer. Benyttes der lange eksponeringsperioder, bør kulturerne dog undersøges mikroskopisk for sporedannelse, idet disses tilstedeværelse gør testen værdiløs.
Pladerne inkuberes i fire til syv dage mellem 28 og 30 oC i mørke. Plader, som anvendes til bestemmelse af røde og lyserøde homozygote sektorer, der er frembragt ved mitotisk overkrydsning, opbevares i køleskab (omkring 4 oC) i yderligere en til to dage inden optælling for at muliggøre udvikling af de rene pigmenterede kolonier.
Der anvendes subkulturer med spontane mitotiske frekvenser, som ligger inden for de almindeligt accepterede områder.
Der anvendes mindst tre parallelle plader for hver koncentration ved bestemmelsen af prototrofer, som er fremkommet ved mitotisk genbytning, og af cellernes levedygtighed. Når man tester recessive homozygoter, som er frembragt ved mitotisk overkrydsning, forøges antallet af plader, således at der opnås et passende antal kolonier.
Fremgangsmåde
Stammer af S. cerevisiae eksponeres normalt ved test i væske, hvortil der anvendes enten celler i stationær fase eller celler i vækstfase. Indledende undersøgelser bør udføres med celler i vækstfase. 1-5 × 107 celler pr. ml eksponeres for teststoffet i op til 18 timer ved 28 til 37 oC under omrystning. Under eksponeringen tilsættes en passende mængde metabolisk aktiveringssystem.
Efter eksponeringen centrifugeres og vaskes cellerne, hvorefter de udsås på et egnet kulturmedium. Når inkubationen er slut, registreres induktionen af mitotisk rekombination og antal overlevende på hver plade.
Hvis den første undersøgelse er negativ, bør en anden udføres under anvendelse af celler i stationær fase. Hvis den første undersøgelse er positiv, skal det efterprøves i et passende uafhængigt forsøg.
2. DATA
Data opstilles i tabelform. Antal optalte kolonier, antal rekombinanter, overlevelsesgrad samt rekombinationsfrekvens angives.
Alle resultater efterprøves i et uafhængigt forsøg.
Evalueringen af data baseres på passende statistiske metoder.
3. RAPPORTERING
3.1. FORSØGSRAPPORT
Forsøgsrapporten skal om muligt indeholde følgende:
|
— |
den anvendte stamme |
|
— |
forsøgsbetingelser: celler i stationær fase eller vækstfase, mediernes sammensætning, inkubationstemperatur og -periode, metabolisk aktiveringssystem |
|
— |
eksponeringsbetingelser: eksponeringskoncentrationer, fremgangsmåde og varighed, eksponeringstemperatur, positive og negative kontroller |
|
— |
antal optalte kolonier, antal rekombinanter, overlevelses- og rekombinationsfrekvens, om muligt dosis/responsforhold, statistisk evaluering af data |
|
— |
diskussion af resultaterne |
|
— |
fortolkning af resultaterne. |
3.2. EVALUERING OG FORTOLKNING
Se den generelle indledning til del B.
4. HENVISNINGER
Se den generelle indledning til del B.
B.17. MUTAGENICITET — IN VITRO-TEST FOR GENMUTATION I PATTEDYRCELLER
1. METODE
Denne metode er en gengivelse af OECD Test Guideline 476 In Vitro Mammalian Cell Gene Mutation Test (1997).
1.1. INDLEDNING
In vitro-testen for genmutation i pattedyrceller kan benyttes til at påvise genmutationer, som induceres af kemiske stoffer. Blandt egnede cellelinjer er muselymfomceller L5178Y, cellelinjerne CHO, CHO-AS52 og V79 fra kinesisk hamster og humane lymfoblastceller TK6 (1). I disse cellelinjer benyttes som genetiske end-points almindeligvis bestemmelse af mutationer ved thymidin-kinase (TK), hypoxantin-guanin-fosforibosyl-transferase (HPRT) og et transgen af xanthin-guanin-fosforibosyl-transferase (XPRT). I TK-, HPRT- og XPRT-mutationstestene påvises forskellige spektre af genetiske hændelser. Med TK og XPRT kan der på grund af deres autosomale placering påvises genetiske hændelser (f.eks. større deletioner), der ikke kan påvises ved HPRT-locus'et på X-kromosomer (2) (3) (4) (5) (6).
I in vitro-testen for genmutation i pattedyrceller kan der benyttes kulturer af etablerede cellelinjer eller cellestammer. Cellerne udvælges på grundlag af deres vækstegenskaber ved dyrkning og den spontane mutationsfrekvens' stabilitet.
Normalt kræver en in vitro-test, at der benyttes en exogen metabolismeaktivering. Et sådant metabolismeaktiveringssystem kan ikke fuldstændigt efterligne forholdene i pattedyr in vivo. Man må omhyggeligt undgå forhold, der fører til resultater, der ikke afspejler egentlig mutagenicitet. Positive resultater, der ikke afspejler egentlig mutagenicitet, kan være forårsaget af ændringer i pH eller osmolalitet eller høj cytotoksicitet (7).
Testen benyttes til screening for stoffer, der kan være mutagene eller carcinogene hos pattedyr. Mange af de stoffer, der er positive ved denne test, er carcinogene hos pattedyr, men korrelationen mellem denne test og carcinogenicitet er ikke fuldstændig. Den afhænger af den kemiske klasse, og stadig mere tyder på, at der findes carcinogener, der ikke påvises ved denne test, fordi de tilsyneladende virker gennem andre ikke-gentoksiske mekanismer eller mekanismer, der ikke findes i bakterieceller (6).
Se også den generelle indledning til del B.
1.2. DEFINITIONER
Fremadmutation: En genmutation fra forældretypen til mutanten, som medfører ændring i eller tab af det kodede proteins enzymaktivitet.
Basesubstitutionsmutagen: Et stof, der fremkalder substitution af et eller flere DNA-basepar.
Læserammemutagen: Et stof, der fremkalder addition eller deletion af et eller flere basepar i DNA-molekylet.
Fænotypeekspressionsperiode: Et tidsrum, hvorunder uændrede genprodukter gradvis forsvinder fra nylig muterede celler.
Mutanthyppighed: Antallet af observerede mutantceller divideret med antallet af levedygtige celler.
Relativ total vækst: Forøgelsen af celleantallet i et tidsrum, sammenlignet med en kontrolpopulation af celler: beregnes som produktet af suspensionsvæksten i forhold til den negative kontrol og klondannelseseffektiviteten i forhold til den negative kontrol.
Relativ suspensionsvækst: Forøgelsen af celleantallet i ekspressionsperioden i forhold til den negative kontrol.
Levedygtighed: De behandlede cellers klondannelseseffektivitet på tidspunktet for selektiv udpladning efter ekspressionsperioden.
Overlevelse: De behandlede cellers klondannelseseffektivitet ved udpladningen efter behandlingsperioden; overlevelsen udtrykkes normalt i forhold til overlevelsen i kontrolcellepopulationen.
1.3. TESTMETODENS PRINCIP
Celler, som mangler thymidinkinase TK på grund af fremadmutationen TK+/– → TK–/–, er resistente over for de cytotoksiske virkninger af pyridinanalogen trifluorthymidin (TFT). Celler, som har thymidinkinase, er følsomme over for TFT, som inhiberer cellemetabolismen og standser den videre celledeling. Det betyder, at mutantceller kan formere sig under tilstedeværelse af TFT, mens normale celler, der indeholder thymidinkinase, ikke kan. På tilsvarende måde kan celler, der mangler HPRT eller XPRT, udvælges ved deres modstandsdygtighed over for henholdsvis 6-thioguanin (TG) og 8-azaguanin (AG). Hvis der i en af genmutationstestene med pattedyrceller testes et stof, der er baseanalogt med det selektive stof eller beslægtet hermed, må der først ses nøje på teststoffets egenskaber. Eksempelvis bør enhver mistanke om, at teststoffet har selektiv toksicitet over for mutantceller og ikke-mutantceller, undersøges. Det vil sige, at det selektive system/stofs egnethed skal bekræftes, når der testes stoffer, der kemisk er beslægtet med det selektive stof (8).
Celler i suspensionskultur eller enkeltlagskultur udsættes for teststoffet både med og uden metabolismeaktivering i et passende tidsrum, hvorefter der ved dyrkning i subkultur påvises cytotoksicitet og gives mulighed for fænotypeekspression, inden mutanterne udvælges (9) (10) (11) (12) (13). Cytotoksiciteten bestemmes sædvanligvis ved at måle kulturernes relative klondannelseseffektivitet (overlevelse) eller deres relative totale vækst efter behandlingsperioden. De behandlede kulturer holdes så lang tid i vækstmediet, afhængigt af hvert valgt locus og celletype, at fænotypeekspressionen af inducerede mutationer bliver næsten optimal. Mutanthyppigheden bestemmes ved udsåning af et kendt antal celler i et medium, der indeholder det selektive stof, til påvisning af mutantceller og i et medium uden selektivt stof til bestemmelse af klondannelseseffektiviteten (levedygtigheden). Efter en passende inkuberingstid tælles kolonierne. Mutanthyppigheden afledes af antallet af mutantkolonier i det selektive medium og antallet af kolonier i det ikke-selektive medium.
1.4. BESKRIVELSE AF TESTMETODEN
1.4.1. Præparater
1.4.1.1. Celler
En lang række cellelinjer lader sig anvende i denne test, herunder subkloner af L5178Y, CHO, CHO-AS52, V-79 og TK6-celler. For de celletyper, der benyttes i denne test, bør der være påvist følsomhed over for kemiske mutagener, høj klondannelseseffektivitet og stabil spontan mutationshyppighed. Cellerne bør kontrolleres for forurening med mycoplasma og bør ikke anvendes, hvis de er forurenet.
Testen bør tilrettelægges med forud fastlagt følsomhed og styrke. Antallet af celler, kulturer og koncentrationer af teststof bør afspejle disse valgte parametre (14). Det mindste antal levedygtige celler, der overlever behandling og benyttes i testens faser, baseres på den spontane mutationshyppighed. Som rettesnor kan der anvendes et celleantal, der er mindst ti gange den reciprokke spontane mutationshyppighed. Det anbefales dog, at der mindst anvendes106 celler. Der skal foreligge tilstrækkelige historiske data om cellesystemet til at dokumentere, at testen har konstant høj effektivitet.
1.4.1.2. Medier og dyrkningsbetingelser
Der benyttes egnede dyrkningsmedier og inkuberingsbetingelser (dyrkningsbeholdere. temperatur, CO2-koncentration, luftfugtighed). Medier vælges efter, hvilke selektive systemer og celletyper der benyttes i testen. Det er især vigtigt at vælge sådanne dyrkningsbetingelser, at både mutantcellers og ikke-mutantcellers vækst og kolonidannelse bliver optimal under ekspressionsperioden.
1.4.1.3. Fremstilling af kulturer
Cellerne opformeres fra stamkulturer, udsås i dyrkningsmedium og inkuberes ved 37 oC. Det kan være nødvendigt at rense kulturerne for allerede-eksisterende mutanter, inden de benyttes i denne test.
1.4.1.4. Metabolismeaktivering
Cellerne udsættes for teststoffet både med og uden et egnet metabolismeaktiveringssystem. Det mest almindeligt anvendte system er en cofaktorsuppleret postmitokondriefraktion (S9), der fremstilles af levere fra rotter, der har været behandlet med enzyminducerende stoffer såsom Aroclor 1254 (15) (16) (17) (18) eller en blanding af phenobarbiton og β-naphthoflavon (19) (20).
Postmitokondriefraktionen anvendes normalt i en koncentration på 1-10 % (v/v) i det færdige testsubstrat. Valget af metabolismeaktiveringssystem og dets beskaffenhed kan afhænge af, hvilken kemisk klasse teststoffet tilhører. I nogle tilfælde vil det være hensigtsmæssigt at benytte postmitokondriefraktion i mere end én koncentration.
Nogle udviklingstendenser, f.eks. genetisk konstruktion af cellelinjer, der udtrykker specifikke aktiverende enzymer, kan rumme potentiale for endogen aktivering. Valget af en bestemt cellelinje skal begrundes sagligt (f.eks. ved, at cytochrom P450-coenzymet er relevant for teststoffets metabolisme).
1.4.1.5. Teststof/testpræparat
Teststoffer i fast form opløses eller opslæmmes i passende opløsningsmidler eller bærestoffer og fortyndes passende inden behandling af cellerne. Teststoffer i væskeform kan enten tilsættes direkte til testsystemet eller fortyndes inden behandlingen. Der benyttes frisk fremstillede teststofpræparater, medmindre stabilitetsdata viser, at opbevaring er acceptabel.
1.4.2. Testbetingelser
1.4.2.1. Opløsningsmiddel/bærestof
Der må ikke være mistanke om. at opløsningsmiddel/bærestof reagerer kemisk med teststoffet, og opløsningsmiddel/bærestof må hverken hæmme cellernes overlevelse eller S9-aktiviteten. Benyttes der andre opløsningsmiddel/bærestoffer end de velkendte, skal brugen af dem underbygges med kompatibilitetsdata. Det anbefales i videst muligt omfang først at forsøge at benytte et vandigt opløsningsmiddel/bærestofsystem. Ved undersøgelse af stoffer, der er ustabile i vand, skal de organiske opløsningsmidler være vandfrie. Vand kan fjernes ved tilsætning af molekylsi.
1.4.2.2. Eksponeringskoncentrationer
Stoffets cytotoksicitet, dets opløselighed i testsystemet samt ændringer i pH og osmolalitet er blandt de kriterier, der skal tages hensyn til ved valg af højeste koncentration.
Cytotoksiciteten bestemmes med og uden metabolismeaktivering i hovedforsøget ved hjælp af en passende indikator for celleintegritet og -vækst, f.eks. relativ klondannelseseffektivitet (overlevelse) eller relativ total vækst. Det kan være hensigtsmæssigt at bestemme cytotoksicitet og opløselighed ved et indledende forsøg.
Der benyttes mindst fire analyserbare koncentrationer. Hvis der er tale om cytotoksicitet, skal koncentrationerne dække et interval fra største toksicitet til ringe eller ingen toksicitet; det betyder normalt, at koncentrationerne højst er adskilt med en faktor mellem 2 og √10. Hvis den højeste koncentration er baseret på cytotoksicitet, bør den føre til en relativ overlevelse (relativ klondannelseseffektivitet) eller relativ total vækst på ca. 10-20 % (ikke under 10 %). For stoffer, der er forholdsvis ikke-cytotoksiske, må testkoncentrationen højst være 5 mg/ml, 5 μl/ml eller 0,01 M (den laveste af de tre).
Forholdsvis uopløselige stoffer testes op til eller over opløselighedsgrænsen ved dyrkningsbetingelserne. Tegn på uopløselighed bør konstateres i det endelige behandlingsmedium, som cellerne udsættes for. Det kan være hensigtsmæssigt at bedømme opløseligheden både ved behandlingens begyndelse og afslutning, eftersom opløseligheden kan ændre sig under eksponeringen i testsystemet som følge af, at der er celler, S9, serum mv. til stede. Uopløselighed kan iagttages med det blotte øje. Bundfald må ikke have indflydelse på bedømmelsen.
1.4.2.3. Kontrolprøver
I alle forsøg medtages der sideløbende både positive og negative (opløsningsmiddel eller bærestof) kontrolprøver, såvel med som uden metabolismeaktivering, Når der benyttes metabolismeaktivering, bør der som positivt kontrolkemikalie anvendes et, der kræver aktivering for at virke mutagent.
Nedenfor er opregnet eksempler på positive kontrolstoffer.
|
Metabolismeaktivering |
Locus |
Stof |
CAS nr. |
Einecs nr. |
|
Ingen exogen metabolismeaktivering |
HPRT |
Ethylmethansulfonat |
62-50-0 |
200-536-7 |
|
Ethylnitrosourea |
759-73-9 |
212-072-2 |
||
|
TK (små og store kolonier) |
Methylmethansulfonat |
66-27-3 |
200-625-0 |
|
|
XPRT |
Ethylmethansulfonat |
62-50-0 |
200-536-7 |
|
|
Ethylnitrosourea |
759-73-9 |
212-072-2 |
||
|
Exogen metabolismeaktivering |
HFRT |
3-methylcholanthren |
56-49-5 |
200-276-4 |
|
N-nitrosodimethylamin |
62-75-9 |
200-549-8 |
||
|
7,1 2-dimethylbenzanthracen |
57-97-6 |
200-359-5 |
||
|
TK (små og store kolonier) |
Cyclophosphamid |
50-18-0 |
200-015-4 |
|
|
Cyclophosphamidmonohydrat |
6055-19-2 |
|
||
|
Benzo[a]pyren |
50-32-8 |
200-028-5 |
||
|
3-methylcholanthren |
56-49-5 |
200-276-5 |
||
|
XPRT |
N-nitrosodimethylamin (ved højt indhold af S9) |
62-75-9 |
200-549-8 |
|
|
Benzo[a]pyren |
50-32-8 |
200-028-5 |
Der kan benyttes andre egnede stoffer til positiv kontrol, f.eks. kan et laboratorium, der har en historisk database over 5-brom-2'-deoxyuridin (CAS nr. 59-14-3, Einecs nr. 200-415-9), også benytte dette referencestof. Hvis der er mulighed for det, bør der anvendes kemisk beslægtede stoffer.
Der skal indgå negative kontrolprøver, hvor behandlingsmediet kun består af opløsningsmiddel eller bærestof, og som behandles på samme måde som de øvrige kulturer. Desuden skal der være ubehandlede kontrolprøver, medmindre der foreligger tidligere opnåede kontroldata, der viser, at det valgte opløsningsmiddel ingen skadelige eller mutagene virkninger har.
1.4.3. Fremgangsmåde
1.4.3.1. Behandiing med teststof
Celler under formering udsættes for teststoffet både med og uden metabolismeaktivering. Eksponeringen skal vare en passende tid (normalt har tre-fem timer den fornødne virkning). Eksponeringen kan strække sig over én eller flere cellecyklusser.
Ved hver undersøgt koncentration benyttes der enten en eller to behandlede kulturer. Benyttes der kun én kultur, øges antallet af koncentrationer, således at der bliver tilstrækkeligt mange kulturer til analyse (dvs. mindst otte analyserbare koncentrationer). Der bør anvendes to negative kontrolkulturer (opløsningsmiddel).
Gasformige og flygtige stoffer testes ved en egnet metode, f.eks. i forseglede dyrkningsflasker (21) (22).
1.4.3.2. Måling af overlevelse, levedygtighed og mutanthyppighed
Efter eksponeringen vaskes cellerne og dyrkes med henblik på at bestemme overlevelsen og give mulighed for ekspression af mutantfænotypen. Måling af cytotoksicitet ved bestemmelse af kulturernes relative klondannelseseffektivitet (overlevelse) eller relative totale vækst påbegyndes normalt efter behandlingsperioden.
Hvert locus har et fast mindste tidsrum, som kræves til nær optimal fænotypeekspression af nyligt inducerede mutanter (HPRT og XPRT kræver mindst seks-otte dage og TK mindst to dage). Cellerne dyrkes i medier med og uden selektivt stof med henblik på bestemmelse af henholdsvis antallet af mutanter og klondannelseseffektiviteten. Måling af levedygtigheden (der benyttes til beregning af mutanthyppigheden) påbegyndes efter ekspressionstiden ved udpladning på ikke-selektivt medium.
Hvis teststoffet reagerer positivt i L5178Y TK+/–-testen, bestemmes kolonistørrelsen i mindst en af testkulturerne (den højeste positive koncentration) og i de positive og negative kontrolkulturer. Hvis teststoffet reagerer negativt i L517SY TK+/–-testen, bestemmes kolonistørrelsen i de positive og negative kontrolkulturer. Også ved undersøgelser med TK6TK+/– kan kolonistørrelsen bestemmes.
2. DATA
2.1. BEHANDLING AF RESULTATER
Dataene skal omfatte bestemmelse af cytotoksicitet og levedygtighed, kimtælling og mutanthyppighed for de behandlede kulturer og kontrolkulturerne. I tilfælde af positiv reaktion i L5178Y TK+/–-testen, bedømmes kolonierne ud fra kriterier om små og store kolonier ved mindst én koncentration af teststoffet (højeste positive koncentration) og med den negative og positive kontrol. Den molekylære og cytogenetiske natur af mutanter i både store og små kolonier er udforsket detaljeret (23) (24). I TK+/–-testen bedømmes kolonierne efter kriterier for normalt voksende (store) og langsomt voksende (små) kolonier (25). De mutantceller, der har de mest alvorlige genetiske skader, har længere fordoblingstider og danner derfor mindre kolonier. Sådanne skader ligger typisk fra tab af hele genet til karyotypisk synlige kromosomaberrationer. Induktion af mutanter i små kolonier har været tilskrevet kemikalier, der fremkalder kromosomaberrationer, der kan ses med det blotte øje (26). Mindre alvorligt berørte mutantceller vokser med omtrent samme hastighed som modercellerne og danner store kolonier.
Overlevelsen (den relative klondannelseseffektivitet) eller den relative totale vækst skal oplyses. Mutanthyppigheden opgives som antallet af mutantceller i forhold til antallet af overlevende celler.
Der anføres data for hver enkelt kultur. Derudover sammenfattes alle data i tabelform.
Der findes ingen krav til verifikation af et klart positivt resultat. Tvetydige resultater bør afklares ved yderligere undersøgelser, helst med ændrede forsøgsbetingelser. Negative resultater bekræftes efter en vurdering af de enkelte tilfælde. Anses bekræftelse af negative resultater ikke for nødvendigt, skal dette begrundes. Ved forsøg til opfølgning af tvetydige eller negative resultater bør man overveje at ændre på undersøgelsens parametre, så de bedømte forsøgsbetingelser udvides. Blandt de undersøgelsesparametre. der kan ændres på, er koncentrationernes spredning og metabolismeaktiveringen.
2.2. EVALUERING OG FORTOLKNING Af RESULTATER
Der findes en række kriterier for, om der er opnået et positivt resultat, f.eks. en koncentrationsafhængig eller reproducerbar forøgelse af mutanthyppigheden. Der bør i første række ses på resultaternes biologiske relevans. Statistiske metoder kan benyttes som hjælpemiddel ved evalueringen af testresultaterne. Statistisk signifikans bør ikke være den eneste faktor, der afgør, om resultatet bedømmes som positivt.
Et teststof, der ikke opfylder ovennævnte kriterier, anses ikke for mutagent i denne test.
De fleste forsøg giver klart positivt eller negativt resultat, men i sjældne tilfælde giver dataene ikke mulighed for en endegyldig bedømmelse af teststoffets aktivitet. Resultatet kan stadig være tvetydigt eller tvivlsomt, uanset hvor mange gange forsøget gentages.
Et positivt resultat af en in vitro-test for genmutation i pattedyrceller viser, at teststoffet fremkalder genmutationer i de benyttede dyrkede pattedyrceller. En positiv koncentrationsafhængig reproducerbar respons er mest sigende. Et negativt resultat viser, at teststoffet under testbetingelserne ikke fremkalder genmutationer i de benyttede dyrkede pattedyrceller.
3. RAPPORTERING
TESTRAPPORT
Testrapporten skal indeholde følgende oplysninger:
|
|
Opløsningsmiddel/bærestof:
|
|
|
Celler:
|
|
|
Testbetingelser:
|
|
|
Resultater:
|
|
|
Diskussion af resultaterne |
|
|
Konklusioner |
4. HENVISNINGER
|
(1) |
Moore, M.M., DeMarini, D.M., DeSerres, F.J. and Tindall, K.R. (eds.) (1987), Banbury Report 28: Mammalian Cell Mutagenesis, Cold Spring Harbor Laboratory, New York. |
|
(2) |
Chu, E.H.Y. and Malling, H.V. (1968), Mammalian Cell Genetics. II.. Chemical Induction of Specific Locus Mutations in Chinese Hamster Cells In Vitro, Proc. Natl. Acad. Sri. USA, 61, pp. 1306-1312. |
|
(3) |
Liber, H.L. and Thilly, W.G. (1982), Mutation Assay at the Thymidine Kinase Locus in Diploid Human Lymphoblasts, Mutation Res., 94, pp. 467-485. |
|
(4) |
Moore, M.M., Harington-Brock, K., Doerr, C.L. and Dearfield, K.L. (1989), Differential Mutant Quantitation at the Mouse Lymphoma TK and CHO HGPRT Loci, Mutagenesis, 4, pp. 394-403. |
|
(5) |
Aaron, C. S. and Stankowski, Jr.L.F. (1989), Comparison of the AS52/XPRT and the CHO/HPRT Assays: Evaluation of Six Drug Candidates, Mutation Res., 223, pp. 121-128. |
|
(6) |
Aaron, C.S., Bolcsfoldi, G., Glatt, H.R., Moore, M, Nishi, Y., Stankowski, Jr.L.F., Theiss, J. and Thompson, E. (1994), Mammalian Cell Gene Mutation Assays Working Group Report. Report of the International Workshop on Standardisation of Genotoxicity Test Procedures. Mutation Res.. 312, pp. 235-239. |
|
(7) |
Scott. D., Galloway, S.M., Marshall, R.R., Ishidate, M., Brusick, D., Ashby, J. and Myhr, B.C. (1991), Genotoxicity Under Extreme Culture Conditions. A report from ICPEMC Task Group 9, Mutation Res., 257, pp. 147-204. |
|
(8) |
Clive, D., McCuen, R. Spector, J.F.S., Piper, C. and Mavournin, K.H. (1983), Specific Gene Mutations in L5178Y Cells in Culture. A Report of the U.S. Environmental Protection Agency Gene-Tox Program, Mutation Res., 115, pp. 225-251. |
|
(9) |
Li, A.P., Gupta, R.S., Heflich, R.H. and Wasson, J.S. (1988), A Review and Analysis of the Chinese Hamster Ovary/Hypoxanthine Guanine Phosphoribosyl Transferase System to Determine the Mutagenicity of Chemical Agents: A Report of Phase III of the U.S. Environment Protection Agency Gene-Tox Program. Mutation Res., 196, pp. 17-36. |
|
(10) |
Li. A.P., Carver, J.H., Choy, W.N., Hsie, A.W., Gupta, R.S., Loveday, K.S., O'Neill, J.P., Riddle, J.C, Stankowski, L.F. Jr. and Yang, L.L. (1987), A Guide for the Performance of the Chinese Hamster Ovary Cell/Hypoxanthine-Cuanine Phosphoribosyl Transferase Gene Mutation Assay, Mutation Res., 189, pp. 135-141. |
|
(11) |
Liber, H.I., Yandell, D.W. and Little, J.B. (1989). A Comparison of Mutation Induction at the TK and HPRT Lod in Human Lymphoblastoid Cells: Quantitative Differences are Due to an Additional Class of Mutations at the Autosomal TK Locus, Mutation Res. 216, pp. 9-17. |
|
(12) |
Stankowski, L.F. Jr., Tindall, K.R. and Hsie, A.W. (1986), Quantitative and Molecular Analyses of Ethyl Methanosulphonate — and ICR 191-Induced Molecular Analyses of Ethyl Methanosulphonate — and 1CR 191-Induced Mutation in AS52 Cells, Mutation Res., 160, pp. 133-147. |
|
(13) |
Turner, N.T., Batson, A.G. and Clive, D. (1984), Procedures for the L5178Y/TK+/– — TK+/– Mouse Lymphoma Cell Mutagenicity Assay, in: Kilbey, B.J. et al (eds.) Handbook of Mutagenicity Test Procedures, Elsevier Science Publishers, New York, pp. 239-268. |
|
(14) |
Arlett, C.F., Smith, D.M., Clarke, G.M, Green, M.H.L., Cole, J., McGregor, D.B. and Asquith, J.C. (1989). Mammalian Cell Gene Mutation Assays Based upon Colony Formation, in: Statistical Evaluation of Mutagenicity Test Data, Kirkland D.J., ed., Cambridge University Press, pp. 66-101. |
|
(15) |
Abbondandolo, A., Bonatti, S., Corti, G., Fiorio, R., Loprieno, N. and Mazzaccaro, A. (1977), Induction of 6-Thioguanine-Resistant Mutants in V79 Chinese Hamster Cells by Mouse-Liver Microsome-Activated Dimethylnitrosamine, Mutation Res., 46, pp. 36 5-37 3. |
|
(16) |
Arnes, B.N., McCann, J. and Yamasaki, E. (1975), Methods for Detecting Carcinogens and Mutagens with the Salmonella/Mammalian-Microsome Mutagenicity Test, Mutation Res. 31, pp. 347-364. |
|
(17) |
Clive, D., Johnson, K.O., Spector, J.F.S., Batson, A.G. and Brown M.M.M. (1979), Validation and Characterisation of the L5178Y/TK+/- Mouse Lymphoma Mutagen Assay System, Mutat Res., 59, pp. 61-108. |
|
(18) |
Maron, D.M. and Arnes, B.N. (1983), Revised Methods for the Salmonella Mutagenicity Test, Mutation Res., 113, pp. 173-215. |
|
(19) |
Elliott, B.M., Combes, R.D., Elcombe, C.R., Gatehouse, D.C, Gibson, G.G., Mackay, J.M. and Wolf, R.C. (1992), Alternatives to Aroclor 1254-Induced S9 in In Vitro Genotoxicity Assays, Mutagenesis, 7, pp. 175-177. |
|
(20) |
Matsushima, T., Sawamura, M., Hara, K. and Sugimura, T. (1976). A Safe Substitute for Polychlorinated Biphenyls as an Inducer of Metabolic Activation Systems, in In Vitro Metabolic Activation in Mutagenesis Testing, de Serres, F.J., Fouts, J.R., Bend, J.R. and Philpot. R.M. (eds.) Elsevier, North-Holland, pp. 85-88. |
|
(21) |
Krahn, D.F., Barsky, F.C. and McCooey, K.T. (1982), CHO/HGPRT Mutation Assay: Evaluation of Gases and Volatile Liquids, In: Tice, R.R., Costa, D.L, Schaich, K.M, (eds.), Genotoxic Effects of Airborne Agents, New York, Plenum, pp. 91-103. |
|
(22) |
Zamora, P.O., Benson, J.M., Li, A.P. and Brooks, A.L. (1983), Evaluation of an Exposure System Using Cells Grown on Collagen Gels for Detecting Highly Volatile Mutagens in the CHO/HGPRT Mutation Assay, Environmental Mutagenesis, 5, pp. 795-801. |
|
(23) |
Applegate, M.L., Moore, M.M., Broder, C.B., Burrell, A. and Hozier, J.C. (1990), Molecular Dissection of Mutations at the Heterozygous Thymidine Kinase Locus in Mouse Lymphoma Cells, Proc. Natl. Acad. Sti. USA. 87, pp, 51-55. |
|
(24) |
Moore, M.M., Clive, D., Hozier, J.C. Howard, B.E., Batson, A.G., Turner, N.T. and Sawyer, J. (1985), Analysis of Trifluorothymidine-Resistant (TFTr) Mutants of L5178Y/TK+/- Mouse Lymphoma Cells, Mutation Res. 151, pp. 161-174. |
|
(25) |
Yandell, D.W., Dryja, T.P. and I.ittle, J.B. (1990), Molecular Genetic Analysis of Recessive Mutations at a Heterozygous Autosomal Locus in Human Cells, Mutation Res., 229, pp. 89-102. |
|
(26) |
Moore, M.M. and Doerr, C.L. (1990), Comparison of Chromosome Aberration Frequency and Small-Colony TK-Deficient Mutant Frequency in L5178Y/TK+/- — 3.7.2C Mouse Lymphoma Cells, Mutagenesis 5, pp.609-614. |
B.18. DNA-SKADE OG DNA-REPARATION — UNSCHEDULED DNA SYNTHESIS — PATTEDYRCELLER IN VITRO
1. METODE
1.1. INDLEDNING
Se den generelle indledning til del B.
1.2. DEFINITIONER
Se den generelle indledning til del B.
1.3. REFERENCESTOFFER
Ingen.
1.4. PRINCIP FOR TESTMETODEN
Ved Unscheduled DNA Synthesis (UDS) test undersøges dna-reparationen efter bortskæring og fjernelse af skader, som er induceret af kemiske eller fysiske agentia. Testen bygger på inkorporering af tritiummærket thymidin (3H-TdR) i pattedyrcellers dna, når disse celler ikke er i S-fase. Optagelsen af 3H-TdR kan måles ved autoradiografi eller væskescintillationstælling (LSC) af dna fra de eksponerede celler. Medmindre der anvendes primærkulturer af rottehepatocytter, eksponeres dyrkede pattedyrceller for teststoffet med og uden et eksogen metabolisk aktiveringssystem. UDS-test kan også foretages i in vivo-systemer.
1.5. KVALITETSKRITERIER
Ingen.
1.6. BESKRIVELSE AF TESTMETODEN
Forberedelser
Teststof samt kontrol- og referencestoffer tilsættes kulturmediet eller opløses eller suspenderes i egnede vehikler og tilsættes derefter kulturmediet inden testen. Den derved opnåede vehikelkoncentration bør ikke påvirke cellernes levedygtighed.
Der kan anvendes primærkulturer af rottehepatocytter, humane lymfocytter eller etablerede cellelinjer (f.eks. humane diploide fibroblaster).
Cellerne eksponeres for teststoffet med og uden et passende metabolisk aktiveringssystem.
Forsøgsbetingelser
Ved autoradiografisk påvisning af UDS anvendes mindst to og ved LSC seks kulturer (eller mindre hvis det er videnskabeligt begrundet) for hvert eksperimentelt punkt.
Der foretages sideløbende både positiv og negativ (ubehandlet og/eller vehikel) kontrol med og uden metabolisk aktivering i hvert forsøg.
Som positiv kontrol ved anvendelse af rottehepatocytter kan f.eks. benyttes 7,12-DMBA (7,12-dimetylbenzanthracen) eller 2-AAF (2-acetylaminofluoren). Som positiv kontrol ved anvendelse af etablerede cellelinjer kan f.eks. benyttes 4-NQO (4-nitroquinolin-N-oxid) både i forbindelse med autoradiografisk registrering og LSC uden metabolisk aktivering. Når der anvendes metaboliske aktiveringssystemer kan f.eks. N-dimetylnitrosamin benyttes til positiv kontrol.
Der anvendes en række forskellige koncentrationer af teststoffet over et interval, som dækker alle påvirkninger. Den højeste koncentration bør have en vis cytotoksisk virkning. Stoffer, som er relativt uopløselige i vand, testes op til opløselighedsgrænsen. Den højeste koncentration af ikke-toksiske teststoffer, som er frit opløselige i vand, fastlægges fra test til test.
Der benyttes passende vækstmedier, CO2-koncentration, temperatur og fugtighed til opretholdelse af kulturerne. Det undersøges regelmæssigt, om etablerede cellelinjer er kontamineret med mycoplasma.
Der anvendes ikke metaboliske aktiveringssystemer i forbindelse med primærkulturer af hepatocytter. Etablerede cellelinjer og lymfocytter eksponeres for teststoffer både med og uden et passende metabolisk aktiveringssystem.
Fremgangsmåde
Etablerede cellelinjer fremstilles på basis af stamkulturer (f.eks. ved trypsinering eller afrystning) og udsås med passende tæthed i dyrkningsskåle, hvorefter de inkuberes ved 37 oC.
Primære rottehepatocytkulturer etableres, når hepatocytter umiddelbart efter isolering anbringes i et passende medium og får lov til at hæfte sig til den voksende overflade.
Humane lymfocytkulturer dyrkes under anvendelse af dertil egnede teknikker.
Primærkulturer af rottehepatocytter
Rottehepatocytter eksponeres for teststoffet kort tid efter isolering i et passende tidsrum i et medium, som indeholder 3H-TdR. Efter eksponeringen fjernes cellerne fra mediet, hvorefter de vaskes, fikseres og tørres. Pladerne dyppes i en autoradiografisk emulsion (alternativt kan »stripping« -film anvendes) og eksponeres, fremkaldes, farves og tælles.
Etablerede cellelinjer og lymfocytter
Autoradiografisk teknik: Cellekulturerne eksponeres først for teststoffet i et passende tidsrum og derefter for 3H-TdR. Eksponeringsperioderne afhænger af stofferne, de metaboliske aktiveringssystemer og celletyperne. Eksponeringen for 3H-TdR bør enten foregå samtidig med eksponeringen for teststoffet eller få minutter efter, således at UDS måles på det tidspunkt, hvor aktiviteten er størst. Valget mellem de to fremgangsmåder afhænger af risikoen for interaktion mellem teststoffet og 3H-TdR. Det er muligt at skelne mellem UDS og semi-konservativ dna replication, idet den sidstnævnte proces inhiberes ved anvendelse af f.eks. argininfattige medier, et lavt serumindhold eller tilsætning af hydroxyurinstof til kulturmediet.
LSC-måling af UDS: Inden eksponering for teststoffet blokeres cellernes overgang til S-fasen som tidligere beskrevet. Derefter eksponeres cellerne på samme måde som ved den autoradiografiske teknik. Umiddelbart efter inkubationen ekstraheres cellernes dna og det totale dna-indhold samt mængden af inkorporeret 3H-TdR måles.
Når de nævnte teknikker anvendes på humane lymfocytter, er det unødvendigt at hæmme den semi-konservative dna replication, med mindre der er tale om stimulerede kulturer.
Analyse
Ved måling af UDS i dyrkede celler medregnes kerner i S-fasen ikke. Der tælles mindst 50 celler pr. koncentration. Objektglassene kodes før tællingen. På hvert glas tælles en række forskellige tilfældigt valgte områder med stor indbyrdes afstand. Mængden af inkorporeret 3H-TdR i cytoplasmaet bestemmes ved tælling i tre områder på størrelse med kernen i hver optalt celles cytoplasma.
Ved LSC-måling af UDS anvendes et passende antal kulturer pr. koncentration og pr. kontrolforsøg.
Alle resultater efterprøves i et uafhængigt forsøg.
2. DATA
Data opstilles i tabelform.
2.1. AUTORADIOGRAFISK MÅLING
Mængden af inkorporeret 3H-TdR i cytoplasmaet og antallet af korn, som registreres i cellekerneområdet, anføres separat.
Fordelingen af mængden af inkorporeret 3H-TdR i cytoplasmaet og antallet af korn pr. kerne kan beskrives ved hjælp af middel, median og modus.
2.2. LSC-MÅLING
Ved LSC-måling angives 3H-TdR indlejringen som dpm/μg dna. Indlejringsfordelingen kan beskrives ved hjælp af middel dpm/μg dna med tilhørende standardafgivelse.
Evalueringen af data baseres på passende statistiske metoder.
3. RAPPORTERING
3.1. FORSØGSRAPPORT
Forsøgsrapporten skal om muligt indeholde følgende:
|
— |
de anvendte celler, tæthed og antal passager på eksponeringstidspunktet, antal cellekulturer |
|
— |
metoder til opretholdelse af cellekulturer, herunder medium, temperatur og CO2-koncentration |
|
— |
teststof, vehikel, koncentrationer og begrundelse for valget af de anvendte koncentrationer |
|
— |
detaljerede oplysninger om de metaboliske aktiveringssystemer |
|
— |
eksponeringsplan |
|
— |
positiv og negativ kontrol |
|
— |
den anvendte autoradiografiske teknik |
|
— |
fremgangsmåde ved blokering af cellernes overgang til S-fasen |
|
— |
fremgangsmåde ved ekstration af dna og bestemmelse af det totale dna-indhold ved LSC-målingen |
|
— |
om muligt dosis-responsforhold |
|
— |
statistisk evaluering |
|
— |
diskussion af resultaterne |
|
— |
fortolkning af resultaterne. |
3.2. EVALUERING OG FORTOLKNING
Se den generelle indledning til del B.
4. HENVISNINGER
Se den generelle indledning til del B.
B.19. SØSTERKROMATID OMBYTNING (SCE) IN VITRO
1. METODE
1.1. INDLEDNING
Se den generelle indledning til del B.
1.2. DEFINITIONER
Se den generelle indledning til del B.
1.3. REFERENCESTOFFER
Ingen.
1.4. PRINCIP FOR TESTMETODEN
Søsterkromatid ombytning (SCE) test er en korttidstest, som anvendes til påvisning af reciprok udveksling af DNA mellem de to søsterkromatider i et kromosom under replikation. Ved SCE sker der en udveksling af dna replikationsprodukter ved tilsyneladende homologe loci. Udvekslingsprocessen bygger antageligt på brud og genforening af dna-kæder, men dens molekylære grundlag er næsten ukendt. For at kunne påvise SCE er det nødvendigt at differentialmærke søsterkromatiderne, hvilket kan gøres ved at inkorporere bromdeoxyuridin (BrdU) i kromosomernes dna gennem to cellecykler.
Pattedyrceller eksponeres in vitro for teststoffet med og uden et eksogent metabolisk aktiveringssystem fra pattedyr, hvis dette skønnes relevant, og dyrkes gennem to cellecykler i et medium, som indholder BrdU. Cellerne behandles med en tentrådsinhibitor (f.eks. colchicin) med henblik på at akkumulere celler på et metafaselignende trin i mitosen (c-metafase), hvorefter de høstes og præpareres til kromosomanalyse.
1.5. KVALITETSKRITERIER
Ingen.
1.6. BESKRIVELSE AF TESTMETODEN
Forberedelser
|
— |
Der kan anvendes primærkulturer (humane lymfocytter) eller etablerede cellelinjer (f.eks. Chinese Hamster Ovary celler). Ved anvendelse af cellelinjer bør det undersøges, om kulturen er kontamineret med mycoplasma. |
|
— |
Der anvendes egnede kulturmedier og inkubationsbetingelser (temperatur, dyrkningsskåle, CO2-koncentration og fugtighed). |
|
— |
Teststoffet tilsættes kulturmediet eller opløses eller suspenderes i et egnet vehikel, inden cellerne eksponeres. Den derved opnåede koncentration af vehikel i testkulturen bør ikke påvirke cellernes levedygtighed eller vækstrate og en mulig påvirkning af SCE-frekvensen kontrolleres ved dyrkning af en kontrolkultur i opløsningsmidlet. |
|
— |
Cellerne eksponeres for teststoffet både med og uden et eksogent metabolisk aktiveringssystem fra pattedyr. Når der benyttes celletyper med endogen metabolisk aktivering, bør aktiveringsform og -hastighed være egnet til den kemiske stofklasse, der testes. |
Forsøgsbetingelser
Der anvendes mindst to parallelle kulturer for hvert eksperimentelt punkt.
Som positiv kontrol anvendes i hvert enkelt forsøg både en direkte virkende kemisk forbindelse og en forbindelse, som kræver metabolisk aktivering. Desuden foretages vehikelkontrol.
Følgende kemiske forbindelser kan f.eks. anvendes til positiv kontrol:
|
— |
direkte virkende forbindelse:
|
|
— |
indirekte virkende forbindelse:
|
Når det skønnes relevant, bør der foretages supplerende positiv kontrol med anvendelse af en kemisk forbindelse af samme kemiske stofklasse som teststoffet.
Der anvendes mindst tre koncentrationer af teststoffet med passende intervaller mellem koncentrationerne. Den højeste koncentration bør have en tydelig toksisk virkning, men den må ikke forhindre den nødvendige celledeling. Stoffer, som er relativt uopløselige i vand, testes under anvendelse af passende fremgangsmåder op til opløselighedsgrænsen. Den højeste koncentration af ikke-toksiske teststoffer, som er frit opløselige i vand, fastlægges fra test til test.
Fremgangsmåde
Cellelinjer etableres på basis af stamkulturer (f.eks. ved trypsinering eller afrystning) og udsås med passende tæthed i dyrkningsskåle, hvorefter de inkuberes ved 37 oC. Når der er tale om monolagskulturer, justeres antallet af celler pr. kulturskål, således at kulturerne på høsttidspunktet højst dækker 50 % af vækstarealet. Cellerne kan også dyrkes i suspension. Humane lymfocytkulturer dyrkes under anvendelse af dertil egnede teknikker på basis af hepariniseret blod og inkuberes ved 37 oC.
Celler i eksponentiel vækstfase eksponeres for teststoffet i et passende tidsrum. Som oftest vil en eller to timer være tilstrækkeligt, men i nogle tilfælde kan eksponeringsperioden udvides til at dække to fulde cellecykler. Celler, hvis endogene metaboliske aktivering er utilstrækkelig, eksponeres både med og uden tilsætning af et passende metabolisk aktiveringssystem. Efter eksponeringen vaskes cellerne frie for teststof og dyrkes gennem to cellecykler i et medium, som indeholder BrdU. Alternativt kan cellerne eksponeres for teststoffet og BrdU samtidigt gennem en periode svarende til to cellecykler.
Humane lymfocytter eksponeres, mens de befinder sig i semisynkron tilstand.
Cellerne analyseres under deres anden deling efter eksponeringen, hvilket sikrer, at eksponeringsperioden dækker størstedelen af de følsomme faser i cellecyclus. Alle kulturer, som er tilsat BrdU, opbevares og manipuleres i mørke eller svagt lys fra glødelamper frem til cellerne høstes for at minimere fotolysen af dna, som indeholder BrdU.
Cellekulturerne behandles med en tentrådsinhibitor (f.eks. colchicin) en til fire timer, før de høstes. Høst og kromosompræparering foregår særskilt for hver enkelt kultur.
Ved kromosompræpareringen anvendes cytogenetiske standardteknikker. Farvning på objektglas med henblik på at påvise SCE kan foretages på en række forskellige måder (f.eks. fluorescens plus Giemsafarvning).
Antallet af celler, som underkastes analyse, baseres på den spontane SCE-frekvens i kontrolforsøg. Normalt undersøges mindst 25 velpræparerede metafaser pr. kultur for SCE. Objektglassene kodes før analysen. Ved analyse af humane lymfocytter medtages kun metafaser med 46 centromerer. Ved analyse af etablerede cellelinjer medtages kun metafaser med det modale antal centromerer ± 2. Det anføres, hvorvidt ombytning i mærkningen omkring centromeret betragtes som SCE. Resultaterne efterprøves i et uafhængigt forsøg.
2. DATA
Data opstilles i tabelform. Antal SCE pr. metafase og antal SCE pr. kromosom anføres separat for alle eksponerede kulturer og kontrolkulturer.
Evalueringen af data baseres på passende statistiske metoder.
3. RAPPORTERING
3.1. FORSØGSRAPPORT
Forsøgsrapporten skal om muligt indeholde følgende:
|
— |
de anvendte celler, metoder til opretholdelse af cellekulturer |
|
— |
forsøgsbetingelser: mediernes sammensætning, CO2-koncentration, teststofkoncentration, det anvendte vehikel, inkubationstemperatur, eksponeringsperiode, den anvendte tentrådsinhibitor plus oplysninger om koncentration og behandlingens varighed, det anvendte metaboliske aktiveringssystem, positiv og negativ kontrol |
|
— |
antal cellekulturer pr. eksperimentelt punkt |
|
— |
detaljerede oplysninger om objektpræpareringsteknikken |
|
— |
antal analyserede metafaser (separate oplysninger for hver enkelt kultur) |
|
— |
gennemsnitligt antal SCE pr. celle og pr. kromosom (data angives separat for hver kultur) |
|
— |
SCE-registreringskriterier |
|
— |
begrundelse for valget af doser |
|
— |
om muligt dosis-responsforhold |
|
— |
statistisk evaluering |
|
— |
diskussion af resultaterne |
|
— |
fortolkning af resultaterne. |
3.2. EVALUERING OG FORTOLKNING
Se den generelle indledning til del B.
4. HENVISNINGER
Se den generelle indledning til del B.
B.20. KØNSBUNDET RECESSIV LETAL TEST — DROSOPHILA MELANOGASTER
1. METODE
1.1. INDLEDNING
Se den generelle indledning til del B.
1.2. DEFINITIONER
Se den generelle indledning til del B.
1.3. REFERENCESTOFFER
Ingen.
1.4. PRINCIP FOR TESTMETODEN
Ved kønsbundet recessiv letal test (SLRL) under anvendelse af Drosophila melanogaster undersøges forekomsten af både punktmutationer og mindre tabsmutationer hos insektets afkom. Der er tale om en fremadmutationstest, hvorved det er muligt at screene ca. 800 loci på X-kromosomet med henblik på registrering af mutationer. Dette antal udgør ca. 80 % af alle X-kromosomets loci. X-kromosomet udgør omkring en femtedel af den samlede haploide kromosommængde.
Mutationer i Drosophila melanogasters X-kromosom kommer fænotypisk til udtryk hos hanner, som er bærere af det muterede gen. Når mutationen under hemizygote betingelser er letal, konstateres dette som fravær af en af de to typer hanner, der normalt findes i en heterozygot huns afkom. I SLRL-testen udnyttes dette forhold ved hjælp af specielt arrangerede kromosomer med særlige markører.
1.5 KVALITETSKRITERIER
Ingen.
1.6. BESKRIVELSE AF TESTMETODEN
Forberedelser
Der kan anvendes hanner af veldefinerede vildtypestammer og hunner fra Muller-5-stammen. Hunner fra andre stammer med passende mærkning og multipelt inverterede X-kromosomer kan også anvendes.
Teststoffet opløses i vand. Stoffer, som er uopløselige i vand, kan opløses eller suspenderes i egnede bærestoffer (f.eks, en blanding af ætanol og Tween-60 eller -80) og derefter fortyndes i vand eller saltvand inden indgift. Dimetylsulfoxid bør ikke benyttes som bærestof.
Testen udformes således, at testfølsomhed og -specificitet er fastlagt på forhånd. Den spontane mutationsfrekvens, som registreres i kontrolforsøg, vil være af stor betydning for antallet af eksponerede kromosomer, der skal analyseres.
Eksponeringen kan foregå ved oral indgivelse, injektion eller ved udsættelse for gasser eller dampe. Teststoffet kan eventuelt indgives i en sukkeropløsning. Om nødvendigt kan det opløses i en 0,7 % NaCl-opløsning og injiceres i thorax eller abdomen.
Der foretages negativ (vehikel) og positiv kontrol. Hvis der foreligger egnede historiske kontroldata fra laboratorieforsøg, er det imidlertid ikke nødvendigt at foretage sideløbende kontrol.
Der anvendes tre eksponeringskoncentrationer. Til en foreløbig vurdering kan anvendes en enkelt koncentration, som enten kan være den højeste koncentration, insekterne kan tåle, eller en koncentration, som forårsager nogen grad af toksisk virkning. Ved ikke-toksiske stoffer eksponeres for de størst mulige koncentrationer.
Vildtypehanner (tre til fem dage gamle) eksponeres for teststoffet og parres enkeltvis med flere jomfruelige hunner fra Muller-5-stammen eller fra andre stammer med passende markører (multipelt inverterede X-kromosomer). Hunnerne udskiftes med nye jomfruelige hunner hver anden eller hver tredje dag, indtil hele kønscellecyklus er dækket. Hunnernes afkom registreres med henblik på konstatering af letale effekter, som svarer til eksponeringens virkning på henholdsvis modne sædceller, spermatider i sene stadier eller middelstadier, spermatider i tidlige stadier, spermatocytter og spermatogonier.
Heterozygote F1-hunner fra de ovennævnte krydsninger parres individuelt (dvs. en hun pr. glas) med deres brødre. I F2-generationen registreres hver bestand med henblik på konstatering af fravær af vildtypehanner. Hvis en bestand er afkom af en F1-hun, som synes at bære et letalt gen i det parentale X-kromosom (dvs. der optræder ingen hanner med det eksponerede kromosom), testes døtre af denne hun med samme genotype med henblik på konstatering af, om den letale effekt gentages i næste generation.
2. DATA
Data opstilles i tabelform. Antal testede X-kromosomer, antal ufrugtbare hanner og antal letale kromosomer anføres for hver eksponeringskoncentration og for hver enkelt af de eksponerede hanners parringsperioder. Ligeledes anføres antal klynger af forskellig størrelse pr. han. Alle resultater efterprøves i et uafhængigt forsøg.
Evalueringen af kønsbunden recessiv letal test baseres på passende statistiske metoder. I tilfælde af ophobning af generationer med recessive letale gener, som stammer fra en enkelt han, registreres disse og evalueres på et passende statistisk grundlag.
3. RAPPORTERING
3.1. FORSØGSRAPPORT
Forsøgsrapporten skal om muligt indeholde følgende:
|
— |
stammer: de anvendte Drosophila-stammer, insekternes alder, antal eksponerede hanner, antal sterile hanner, antal etablerede F2-bestande, antal F2-bestande uden afkom, antal kromosomer med letale gener på hvert kønscellestadium |
|
— |
kriterier for fastlæggelse af størrelsen af de eksponerede grupper |
|
— |
forsøgsbetingelser: detaljeret beskrivelse af eksponerings- og stikprøvetagningsplan, eksponeringskoncentrationer, toksicitetsdata, negativ (opløsningsmiddel) og positiv kontrol, såfremt der foretages kontrol |
|
— |
kriterier for registrering af letale mutationer |
|
— |
om muligt dosis-responsforhold |
|
— |
statistisk evaluering |
|
— |
diskussion af resultaterne |
|
— |
fortolkning af resultaterne. |
3.2. EVALUERING OG FORTOLKNING
Se den generelle indledning til del B.
4. HENVISNINGER
Se den generelle indledning til del B.
B.21. CELLETRANSFORMATIONSTEST — PATTEDYRCELLER IN VITRO
1. METODE
1.1. INDLEDNING
Se den generelle indledning til del B.
1.2. DEFINITIONER
Se den generelle indledning til del B.
1.3. REFERENCESTOFFER
Ingen.
1.4. PRINCIPPER FOR TESTMETODEN
Kulturer af pattedyrceller kan anvendes til påvisning in vitro af fænotypiske forandringer, som induceres af kemiske forbindelser, der forbindes med maligne transformationer in vivo. Hyppigt anvendte celletyper er C3H10T1/2, 3T3, SHE, Fischer-rotte, og testene bygger på forandringer i cellemorfologien, vækstevnen eller forankringsevnen i blød agar. Der findes også mindre hyppigt anvendte systemer, som bruges til undersøgelse af andre fysiologiske eller morfologiske forandringer i celler som følge af eksponering for karcinogene kemiske forbindelser. Der er ikke beskrevet nogen direkte virkningsmekanistisk forbindelse mellem cancer og de genetiske endpoints, der anvendes i disse in vitro test. Nogle af testsystemerne kan anvendes til påvisning af tumorpromotorer. Den cytotoksiske effekt kan bestemmes ved måling af teststoffets påvirkning af evnen til at danne kolonier (kloningsstyrken) eller af kulturernes vækstrater. Formålet med at måle den cytotoksiske effekt er at konstatere, om eksponeringen for teststoffet er toksikologisk relevant, men kan ikke benyttes til beregning af transformationsfrekvensen i alle test, eftersom nogle kræver lang inkubationstid og/eller overførsel til nye plader.
1.5. KVALITETSKRITERIER
Ingen.
1.6. BESKRIVELSE AF TESTMETODEN
Forberedelser
Der kan anvendes en række forskellige cellelinjer eller primærceller, afhængigt af hvilken transformationstest der foretages. Forsøgslederen må sikre, at de anvendte celler undergår relevante fænotypiske forandringer efter eksponering for kendte karcinogener, og forsøgslederens eget laboratorium må have påvist og dokumenteret den anvendte tests validitet og pålidelighed.
Der anvendes medier og forsøgsbetingelser i overensstemmelse med den anvendte transformationstest.
Teststoffet tilsættes kulturmediet eller opløses eller suspenderes i et egnet vehikel, inden cellerne eksponeres. Den derved opnåede vehikelkoncentration bør ikke påvirke cellernes levedygtighed, vækstrate eller transformationsfrekvens.
Cellerne eksponeres for teststoffet både med og uden et eksogent metabolisk aktiveringssystem fra pattedyr. Når der benyttes celler med endogen metabolisk aktivering, bør det være påvist, at aktiveringsformen egner sig til den form for kemisk forbindelse, der testes.
Forsøgsbetingelser
Som positiv kontrol anvendes i hvert enkelt forsøg både en direkte virkende kemisk forbindelse og en forbindelse, som kræver metabolisk aktivering. Desuden foretages negativ (vehikel) kontrol.
Følgende kemiske forbindelser kan f.eks. anvendes til positiv kontrol:
|
— |
direkte virkende forbindelser:
|
|
— |
forbindelser som kræver metabolisk aktivering:
|
Når det skønnes relevant, bør der foretages supplerende positiv kontrol med anvendelse af en forbindelse af samme kemiske stofklasse som teststoffet.
Der anvendes en række forskellige koncentrationer af teststoffet, således at der frembringes koncentrationsafhængige toksiske virkninger. Den højeste koncentration bør medføre en lav overlevelsesgrad, og overlevelsesgraden ved den laveste koncentration bør være af samme størrelsesorden som ved det negative kontrolforsøg. Stoffer, som er relativt uopløselige i vand, testes under anvendelse af passende fremgangsmåder op til opløselighedsgrænsen. Den højeste koncentration af ikke-toksiske teststoffer, som er frit opløselige i vand, fastlægges fra test til test.
Fremgangsmåde
Cellerne eksponeres i en passende periode afhængig af det anvendte testsystem. Dette kan indebære gentagne doseringer med udskiftning af medium (og om nødvendigt ny metabolisk aktivering), hvis der er tale om langvarig eksponering. Celler, hvis endogene metaboliske aktivering er utilstrækkelig, testes både med og uden et passende metabolisk aktiveringssystem. Efter eksponeringen vaskes cellerne frie for teststof, hvorefter de dyrkes med henblik på at lade den transformerede fænotype, testen bygger på, komme til udtryk og med henblik på bestemmelse af transformationsfrekvensen. Alle resultater efterprøves i et uafhængigt forsøg.
2. DATA
Data opstilles i tabelform og kan foreligge i forskellige former afhængigt af den anvendte test. Der kan f.eks. være tale om optælling på plader, registrering af positive plader eller antal transformerede celler. Når det er relevant, udtrykkes overlevelsesgraden som en procentdel af overlevelsen i kontrolforsøgene, og transformationsfrekvensen udtrykkes som antal transformerede celler pr. overlevende. Evalueringen af data baseres på passende statistiske metoder.
3. RAPPORTERING
3.1. FORSØGSRAPPORT
Forsøgsrapporten skal om muligt indeholde følgende:
|
— |
den anvendte celletype, antal cellekulturer, metoder til opretholdelse af cellekulturer |
|
— |
forsøgsbetingelser: teststofkoncentration, anvendt vehikel, inkubationstemperatur, inkubationstid, eksponeringens varighed og frekvens, celletæthed under eksponeringen, det anvendte eksogene metaboliske aktiveringssystem, positive og negative kontroller, specifikation af den fænotype testen bygger på, det anvendte selektive system (hvis et sådant er anvendt), begrundelse for valget af doser |
|
— |
metoder til registrering af levedygtige og transformerede celler |
|
— |
statistisk evaluering |
|
— |
diskussion af resultaterne |
|
— |
fortolkning af resultaterne. |
3.2. EVALUERING OG FORTOLKNING
Se den generelle indledning til del B.
4. HENVISNINGER
Se den generelle indledning til del B.
B.22. DOMINANT LETAL TEST — GNAVER
1. METODE
1.1. INDLEDNING
Se den generelle indledning til del B.
1.2. DEFINITIONER
Se den generelle indledning til del B.
1.3. REFERENCESTOFFER
Ingen.
1.4. PRINCIP FOR TESTMETODEN
Dominante letale virkninger forårsager, at fostret dør. Nå der ved eksponering for kemiske forbindelser induceres dominante letale mutationer, er dette et tegn pi, at forsøgsdyreartens kønscellevæv er blevet afficeret. Der er bred enighed om, at dominante letale gener opstår på grund af kromosomskader (strukturelle og numeriske anomalier). Hvis eksponerede hunners afkom dør på det embryonale stade, kan dette også skyldes toksiske virkninger.
Normalt eksponeres hanner for teststoffet, hvorefter de parres med ueksponerede jomfruelige hunner. Kønscellernes forskellige stadier kan undersøges separat ved anvendelse af på hinanden følgende parringsperioder. Forøgelsen af antallet af døde implantater pr. hun i den eksponerede gruppe i forhold til antallet af døde implantater pr. hun i kontrolgruppen er udtryk for den letale virkning efter implantation. Den letale virkning før implantation kan vurderes på grundlag af optælling af corpora lutea eller ved sammenligning af det totale antal implantater pr. hun i de eksponerede grupper og kontrolgruppen. Den totale dominante letale virkning er summen af de letale virkninger før og efter implantation. Beregningen af den totale dominante letale virkning baseres på sammenligning af antallet af levende implantater pr. hun i forsøgsgruppen og antallet af levende implantater pr. hun i kontrolgruppen. Fald i antallet af implantater i bestemte tidsperioder kan skyldes celledrab (af spermatocytter og/eller spermatogonier).
1.5. KVALITETSKRITERIER
Ingen.
1.6. BESKRIVELSE AF TESTMETODEN
Forberedelser
Hvis det er muligt, opløses eller suspenderes teststoffet i fysiologisk saltvand. Kemiske forbindelser, som er uopløselige i vand, opløses eller suspenderes i egnede vehikler. Det anvendte vehikel bør ikke interferere med teststoffet eller have nogen toksisk virkning. Testopløsningen anvendes umiddelbart efter fremstillingen.
Forsøgsbetingelser
Normalt gives hele dosis på en gang. Der kan også anvendes dosering ad flere gange, når der er toksikologisk begrundelse herfor. Sædvanligvis indgives teststoffet ved oral intubering eller intraperitoneal injektion. Der kan også benyttes andre former for indgift.
Rotter eller mus anbefales som forsøgsdyr. Sunde og fuldt kønsmodne dyr fordeles randomiseret i forsøgs- og kontrolgrupper.
Der anvendes et passende antal hanner under hensyntagen til den spontane variation i den biologiske egenskab, der danner grundlag for testen. Det valgte antal baseres på den på forhånd fastlagte testfølsomhed og signifikansstyrke. I et typisk forsøg vil antallet af hanner være tilstrækkeligt til, at mellem 30 og 50 hunner bliver drægtige i hver parringsperiode.
Normalt foretages sideløbende positiv og negativ (vehikel) kontrol i hvert forsøg. Når der foreligger acceptable positive kontrolresultater fra nyligt foretagne forsøg i samme laboratorium, kan disse dog anvendes i stedet for sideløbende positiv kontrol. Positive kontrolstoffer anvendes i tilstrækkeligt lave doser (f.eks. MMS, i.p., 10 mg/kg) til at vise testens følsomhed.
Der anvendes normalt tre forskellige dosisniveauer. Den største dosis bør forårsage tegn på toksisk virkning eller nedsætte de eksponerede dyrs fertilitet. I nogle tilfælde vil et enkelt højt dosisniveau være tilstrækkeligt.
Når der er tale om ikke-toksiske stoffer, testes en enkeltdosis på 5 g/kg eller gentagen indgift af 1 g/kg/dag.
Fremgangsmåde
Der kan anvendes flere forskellige doseringsskemaer. Normalt gives hele dosis på en gang, men andre skemaer lader sig også anvende.
Hannerne parres efter doseringen individuelt med en eller to ikke-doserede jomfruelige hunner i på hinanden følgende perioder af passende længde. Hunnerne placeres hos hannerne i en periode svarende til mindst en ægcyklus, eller indtil parringen har fundet sted, hvilket konstateres ved tilstedeværelse af sperma i vagina eller en vaginalprop.
Antallet af parringer efter doseringen fastlægges på grundlag af doseringsskemaet, som skal sikre, at alle kønscellestadier er repræsenteret efter doseringen.
Hunner aflives i anden halvdel af drægtighedsperioden, og uterus' indhold undersøges med henblik på bestemmelse af antallet af levende og døde implantater. Ovarierne undersøges med henblik på bestemmelse af antallet af corpora lutea.
2. DATA
Data opstilles i tabelform med angivelse af antal hanner samt antal drægtige og ikke drægtige hunner. Resultaterne af hver enkelt parring, herunder identificering af hver enkelt han og hun anføres individuelt. For hver hun anføres parringsuge, hannens dosis og antallet af levende og døde implantater.
Beregningen af den totale dominante letale virkning baseres på sammenligning af antallet af levende implantater pr. hun i forsøgsgruppen og antallet af levende implantater pr. hun i kontrolgruppen. Forholdet mellem døde og levende implantater i forsøgsgruppen sammenlignes med forholdet i kontrolgruppen med henblik på bestemmelse af den letale virkning efter implantation.
Hvis data registreres som tidlige og sene dødsfald, skal dette fremgå af tabellerne. Hvis der foretages vurdering af den letale virkning før implantation, anføres denne. Den letale virkning før implantation kan beregnes som forskellen mellem antallet af corpora lutea og antallet af implantater eller som et lavere gennemsnitligt antal implantationer pr. uterus i forhold til kontrolparringerne.
Evalueringen af data baseres på passende statistiske metoder.
3. RAPPORTERING
3.1. FORSØGSRAPPORT
Forsøgsrapporten skal om muligt indeholde følgende:
|
— |
dyreart, stamme, de anvendte dyrs alder og vægt, antal dyr af hvert køn i forsøgs- og kontrolgrupperne |
|
— |
teststof, bærestof, de anvendte dosisniveauer og begrundelse for valget af disse, negativ og positiv kontrol, toksicitetsdata |
|
— |
administrationsmåde og doseringens varighed |
|
— |
parringsskema |
|
— |
metode til konstatering af, om parring har fundet sted |
|
— |
aflivningstidspunkt |
|
— |
kriterier for registrering af dominante letale gener |
|
— |
om muligt dosis-responsforhold |
|
— |
statistisk evaluering |
|
— |
diskussion af resultaterne |
|
— |
fortolkning af resultaterne. |
3.2. EVALUERING OG FORTOLKNING
Se den generelle indledning til del B.
4. HENVISNINGER
Se den generelle indledning til del B.
B.23. TEST FOR KROMOSOMABERRATIONER I SPERMATOGONIER HOS PATTEDYR
1. METODE
Denne metode er en gengivelse af OECD Test Guideline 48 3 Mammalian Spermatogonial Chromosome Aberration Test (1997).
1.1. INDLEDNING
Formålet med in vivo-testen for kromosomaberrationer i spermatogonier hos pattedyr er et påvise, om et stof forårsager strukturelle kromosomafvigelser i spermatogonier fra pattedyr (1) (2) (3) (4) (5). Der er to typer strukturændringer, kromosomtypen og kromatidtypen. De fleste kemiske mutagener forårsager ændringer af kromatidtypen, men også ændringer af kromosomtypen forekommer. Den foreliggende metode er ikke beregnet til at påvise antalsafvigelser og benyttes ikke rutinemæssigt til det formål. Kromosommutationer og lignende er årsag til mange genetiske sygdomme hos mennesker.
Ved denne test måles kromosomhændelser i spermatogonier, og den forventes derfor at kunne forudsige induktion af arvelige mutationer i kimceller.
Der benyttes rutinemæssigt gnavere i denne test. Ved denne cytogenetiske in vivo-test påvises kromosomaberrationer ved spermatogoniers mitose. Metoden omfatter ikke andre målceller.
For at påvise aberrationer af kromatidtypen i spermatogonier må den første mitotiske celledeling efter behandlingen undersøges, inden eventuelle skader går tabt ved efterfølgende celledelinger. Der kan indhentes yderligere information fra behandlede spermatogonstamceller ved meiotisk kromosomanalyse for aberrationer af kromosomtypen ved diakinese-metafase I, når de behandlede celler bliver til spermatocytter.
Denne in vivo-test er tilrettelagt til at vise, om stoffer, der er mutagene over for somatiske celler, også er aktive i kimceller. Derudover er spermatogontesten relevant for en vurdering af den reelle mutagenicitetsfare, idet der tages hensyn til sådanne faktorer som in vivo-metabolisme, farmakokinetik og dna-reparationsprocesser.
I testiklerne er der flere generationer af spermatogonier til stede, som udviser forskellig følsomhed over for kemisk behandling. På denne måde repræsenterer de påviste aberrationer den samlede reaktion fra behandlede spermatogonpopulationer, hvor resultatet domineres af de mere talrige differentierede spermatogonier. Forskellige generationer af spermatogonier er, alt efter deres placering i testiklerne, i varierende grad i berøring med det almindelige kredsløb på grund af den fysiske og fysiologiske Sertoli-cellebarriere og blod-testikelbarrieren.
Hvis der er grund til at tro, at teststoffet eller en reaktiv metabolit ikke vil nå frem til målvævet, er denne test ikke egnet.
Se også den generelle indledning til del B.
1.2. DEFINITIONER
Aberration af kromatidtypen: beskadigelse af kromosomstrukturen i form af brud på enkeltkromatider eller brud på og sammenføjning af kromatider.
Aberration af kromosomtypen: beskadigelse af kromosomstrukturen i form af brud på — eller brud på og sammenføjning af — begge kromatider på samme sted.
Gap: en kromatisk beskadigelse, som er mindre end ét kromatid bred, og hvor misalignment af kromatiderne er minimal.
Antalsafvigelse: ændring i kromosomtallet i forhold til det for de pågældende dyr normale antal.
Polyploidi: et multiplum af det haploide kromosomtal (n), som ikke er det diploide tal (dvs. 3n, 4n osv.).
Strukturel ændring: ændring i kromosomstrukturen, som kan iagttages ved mikroskopi i celledelingens metafase i form af deletioner, intrachange og interchange.
1.3. TESTMETODENS PRINCIP
Dyrene udsættes for teststoffet på passende måde og aflives efter passende tidsrum efter eksponeringen. Inden aflivningen behandles dyrene med et metafasestandsende stof (f.eks. colchicin eller Colcemid®). Der fremstilles derefter præparater af kimceller, som farves, og metafasecellerne undersøges for kromosomaberrationer.
1.4. BESKRIVELSE AE TESTMETODEN
1.4.1. Præparater
1.4.1.1. Valg af dyreart
Der benyttes sædvanligvis hanmus og -hamstere, selv om hanner af andre egnede pattedyrarter kan benyttes. Der bør anvendes almindeligt brugte laboratoriestammer af unge sunde voksne dyr. Ved undersøgelsens begyndelse bør vægtvariationen mellem dyrene være mindst mulig og ikke over ± 20 % af gennemsnitsvægten.
1.4.1.2. Miljø og fodring
Der gælder de almindelige forhold som beskrevet i den generelle indledning til del B, blot bør der tilstræbes en fugtighed på 50-60 %.
1.4.1.3. Forberedelse af dyrene
Der udvælges tilfældigt sunde unge voksne handyr til kontrol- og behandlingsgruppen. Burene bør anbringes på en sådan måde, at deres placering får mindst mulig indvirkning. Dyrene identificeres entydigt. Dyrene tilvænnes laboratorieforholdene i mindst fem dage.
1.4.1.4. Fremstilling af doser
Teststoffer i fast form opløses eller opslæmmes i passende opløsningsmidler eller bærestoffer og fortyndes passende, inden de gives til dyrene. Teststoffer i væskeform kan enten gives direkte til dyrene eller fortyndes først. Der benyttes frisk fremstillede teststofpræparater, medmindre stabilitetsdata viser, at opbevaring er acceptabel.
1.4.2. Testbetingelser
1.4.2.1. Opløsningsmiddel/bærestof
Opløsningsmiddel/bærestof må ikke have toksiske virkninger ved de benyttede doser og må ikke mistænkes for at reagere kemisk med teststoffet. Benyttes der andre opløsningsmidler/bærestoffer end de velkendte, skal brugen af dem underbygges med kompatibilitetsdata. Det anbefales i videst muligt omfang først at forsøge at benytte et vandigt opløsningsmiddel/bærestofsystem.
1.4.2.2. Kontrolgrupper
I alle forsøg medtages der sideløbende både positive og negative (opløsningsmiddel eller bærestof) kontrolgrupper. Bortset fra behandling med teststof skal dyrene i kontrolgrupperne behandles på samme måde som dyrene i behandlingsgrupperne.
Positive kontrolprøver skal frembringe strukturelle ændringer i spermatogonier in vivo ved en dosis, der forventes at give en påviselig forøgelse i forhold til baggrundsværdierne.
Der vælges sådanne doser i de positive kontrolprøver, at virkningerne er tydelige, uden dog at de kodede objektglas' identitet umiddelbart afsløres for den, der aflæser dem. Det kan accepteres, at den positive kontrol indgives på anden måde end teststoffet, og at der kun foretages én prøveudtagning. Hvis der er mulighed for det, bør der anvendes kemisk beslægtede stoffer. Nedenfor er opregnet eksempler på positive kontrolstoffer:
|
Stof |
CAS nr. |
Einecs nr. |
|
Cyclophosphamid Cyclophosphamidmonohydrat |
50-18-0 6055-19-2 |
200-015-4 |
|
Cyclohexylamin |
108-91-8 |
203-629-0 |
|
Mitomycin C |
50-07-7 |
200-008-6 |
|
Monomer acrylamid |
79-06-1 |
201-173-7 |
|
Triethylenmelamin |
51-18-3 |
200-083-5 |
Der skal ved hver prøveudtagning indgå negative kontroldyr, som kun er behandlet med opløsningsmiddel eller bærestof og ellers behandlet på samme måde som behandlingsgrupperne, medmindre tidligere kontroldata har vist en acceptabel variation mellem dyr indbyrdes og acceptabel hyppighed af celler med kromosomafvigelser. Desuden skal der være ubehandlede kontrolgrupper, medmindre der foreligger tidligere opnåede eller offentliggjorte kontroldata, der viser, at det valgte opløsningsmiddel ingen skadelige eller mutagene virkninger har.
1.5. FREMGANGSMÅDE
1.5.1. Dyrenes antal
Hver behandlet gruppe og kontrolgruppe skal bestå af mindst fem analyserbare handyr.
1.5.2. Behandlingsplan
Teststofferne indgives fortrinsvis ad én eller to gange (dvs. som én behandling eller to behandlinger). De kan også indgives i to deldoser, dvs. to behandlinger samme dag med kun nogle få timers interval, hvorved det bliver lettere at indgive et større materialevolumen. Indgift på anden måde skal begrundes sagligt.
I gruppen med højeste dosis udtages der prøver på to tidspunkter efter behandling. Da teststoffet kan indvirke på cellecyklussens kinetik, udtages der er tidlig og en sen prøve, nemlig ca. 24 timer og ca. 48 timer efter behandlingen. For alle andre doser end den højeste dosis udtages en prøve 24 timer eller 1,5 gange cellecyklus efter behandlingen, medmindre et andet prøveudtagningstidspunkt vides at være bedre egnet til påvisning af virkninger (6).
Der kan desuden udtages prøver på andre tidspunkter. For kemikalier, der kan fremkalde kromosom-lagging eller have S-uafhængige virkninger, kan tidligere prøveudtagning være hensigtsmæssigt (1).
Det vurderes i det enkelte tilfælde, om en behandlingsplan med gentagen indgift er velegnet. Ved behandling med gentagen indgift aflives dyrene 24 timer (1,5 cellecykluslængde) efter sidste behandling. Der kan udtages yderligere prøver, hvis det er hensigtsmæssigt.
Inden aflivningen gives dyrene en intraperitoneal injektion med en passende dosis metafasestandsende stof (f.eks. Colcemid® eller colchicin). Der udtages prøver af forsøgsdyr efter et passende tidsrum. For mus er dette tidsrum ca. tre-fem timer, for hamster ca. fire-fem timer.
1.5.3. Dosisniveauer
Hvis der gennemføres en forprøve til bestemmelse af dosisinterval, fordi der ikke foreligger nogen egnede data, bør den udføres i samme laboratorium med samme art, stamme og behandlingsmåde, som agtes benyttet i hovedundersøgelsen (7). Er der tale om toksicitet, benyttes der tre dosisniveauer for første prøveudtagningstidspunkt. Disse dosisniveauer skal dække et interval fra maksimal toksicitet til næsten ingen eller ingen toksicitet. Ved den efterfølgende prøveudtagning behøves kun den højeste dosis benyttet. Den højeste dosis defineres som den dosis, der fremkalder sådanne tegn på toksicitet, at højere dosis med samme indgiftsmønster må forventes at medføre død.
Stoffer med specifik biologisk aktivitet ved lav ikke-toksisk dosis (f.eks. hormoner og mitogener) kan danne undtagelser fra disse dosisfastsættelseskriterier og bør evalueres individuelt. Højeste dosis kan også defineres som en dosis, der i nogen grad udviser tegn på toksicitet i spermatogonier (f.eks. et lavere forhold mellem spermatogonmitoser og første og anden meiosemetafaser: faldet må ikke være større end 50 %).
1.5.4. Grænsetest
Hvis der ved en test med én dosis på mindst 2 000 mg pr. kg legemsvægt indgivet ved én eller i to behandlinger på samme dag, ikke kan iagttages nogen toksiske virkninger, og hvis man på grundlag af data om strukturnæssigt beslægtede stoffer ikke forventer gentoksicitet, kan en fuldstændig undersøgelse med tre dosisniveauer anses for unødvendig. Den forventede eksponering af mennesker kan foranledige, at der benyttes en højere dosis i grænsetesten.
1.5.5. Indgift af doser
Teststoffet indgives normalt ved tvangsfodring med sonde eller en passende intubationskanyle eller ved intraperitoneal injektion. Andre indgiftsmåder kan accepteres, hvis de kan begrundes. Hvor stor en væskemængde, der kan indgives ad gangen ved tvangsfodring eller injektion, afhænger af forsøgsdyrenes størrelse. Mængden bør højst være på 2 ml pr. 100 g legemsvægt. Anvendelse af større rumfang skal begrundes. Bortset fra lokalirriterende og ætsende stoffer, som normalt vil have kraftigere virkninger ved højere koncentrationer, bør testvolumenet variere så lidt som muligt, idet koncentrationen justeres, så volumenet bliver det samme ved alle dosisniveauer.
1.5.6. Præparering af kromosomer
Umiddelbart efter aflivningen udtages der celleopslæmninger fra en eller begge testikler, som udsættes for hyptonisk væske og fikseres. Derefter stryges cellerne ud på objektglas og farves.
1.5.7. Analyse
For hvert dyr bør mindst 100 godt fordelte metafaseceller analyseres (dvs. mindst 500 metafaseceller pr. gruppe). Dette antal kan sættes ned, hvis der iagttages et stort antal aberrationer. Alle objektglas, herunder positive og negative kontrolprøver, kodes uafhængigt inden mikroskoperingen. Da fikseringen ofte medfører, at en del af metafasecellerne går i stykker, og kromosomerne går tabt, bør bedømte celler indeholde et antal centromerer svarende til kromosomtallet 2n ± 2.
2. DATA
2.1. BEHANDLING AF RESULTATER
Data for de enkelte dyr præsenteres i tabelform. Forsøgsenheden er et dyr. For hvert dyr registreres antallet af celler med strukturelle kromosomaberrationer, og antallet af kromosomaberrationer pr. celle bedømmes. Forskellige typer strukturelle kromosomaberrationer skal anføres med antal og hyppighed for behandlingsgrupper og kontrolgrupper. Gaps registreres særskilt og oplyses, men medregnes ikke generelt i den totale aberrationshyppighed.
Hvis der iagttages såvel mitose som meiose, bestemmes forholdet mellem spermatogonmitoser og første og anden meiosemetafase som et mål for cytotoksiciteten for alle behandlede dyr og negative kontroldyr i en samlet prøve på 100 celler under deling for hvert dyr. Hvis der kun iagttages mitose, bestemmes mitoseindekset i mindst 1 000 celler for hvert dyr.
2.2. EVALUERING OG FORTOLKNING AF RESULTATER
Der findes en række kriterier for at afgøre, om et resultat er positivt, f.eks. en dosisafhængig forøgelse af andelen af celler med kromosomaberrationer eller en tydelig forøgelse af antallet af celler med aberrationer hos en enkeltdosisgruppe på et bestemt prøveudtagningstidspunkt. Der bør i første række ses på resultaternes biologiske relevans. Statistiske metoder kan benyttes som hjælpemiddel ved evalueringen af testresultaterne (8). Statistisk signifikans bør ikke være den eneste faktor, der afgør, om resultatet bedømmes som positivt. Tvetydige resultater bør afklares ved yderligere test, helst med ændring af forsøgsbetingelserne.
Et teststof, der ikke opfylder ovennævnte kriterier, anses ikke for mutagent i dette system.
De fleste forsøg giver klart positivt eller negativt resultat, men i sjældne tilfælde giver dataene ikke mulighed for en endegyldig bedømmelse af teststoffets aktivitet. Resultatet kan stadig være tvetydigt eller tvivlsomt, uanset hvor mange gange forsøget gentages.
Et positivt resultat af en in vivo-test for kromosomaberrationer i spermatogonier hos pattedyr viser, at teststoffet fremkalder strukturelle kromosomaberrationer i kimceller hos den undersøgte art. Et negativt resultat viser, at teststoffet under testbetingelserne ikke fremkalder strukturelle kromosomaberrationer i kimceller hos den undersøgte art.
Sandsynligheden for, at teststoffet eller dets metabolitter når frem til målvævet, bør diskuteres.
3. RAPPORTERING
TESTRAPPORT
Testrapporten skal indeholde følgende oplysninger:
|
|
Opløsningsmiddel/bærestof:
|
|
|
Forsøgsdyr:
|
|
|
Testbetingelser:
|
|
|
Resultater:
|
|
|
Diskussion af resultaterne |
|
|
Konklusioner |
4. HENVISNINGER
|
(1) |
Adler, I.D. (1986), Clastogenic Potential in Mouse Spermatogonia of Chemical Mutagens Related to their Cell-Cycle Specifications. In: Genetic Toxicology of Environmental Chemicals, Part B: Genetic Effects and Applied Mutagenesis, Ramel. C, Lambert B., and Magnusson, J. (eds.) Liss, New York, pp. 477-484. |
|
(2) |
Adler. I.D. (1984), Cytogenic tests in Mammals. In: Mutagenicity Testing: a Practical Approach, ed. S. Venitt and J.M. Parry, IRL. Press, Oxford, Washington DC, pp. 275-306. |
|
(3) |
Evans, E.P., Breckon, G. and Ford, C.E. (1964), An Air-Drying Method for Meiotic Preparations from Mammalian Testes, Cytogenetics and Cell Genetics. 3. pp. 289-294. |
|
(4) |
Richold, M., Ashby, J., Chandley, A., Gatehouse, D.G. and Henderson L. (1990), In Vivo Cytogenetic Assays. In: D.J. Kirkland (ed.), Basic Mutagenicity Tests, UKEMS Recommended Procedures. UKEMS Subcommittee on Guidelines for Mutagenicity Testing. Report. Part 1 revised, Cambridge University Press, Cambridge, New York, Port Chester, Melboume, Sydney, pp. 115-141. |
|
(5) |
Yamamoto, K. and Kikuchi, Y. (1978), A New Method for Preparation of Mammalian Spermatogonial Chromosomes. Mutation Res. 52. pp. 207-209. |
|
(6) |
Adler, I.D., Shelby, M.D., Bootman, J., Favor, J., Generoso, W., Pacchierotti, F., Shibuya, T, and Tanaka, N. (1994), International Workshop on Standardisation of Genotoxicity Test Procedures. Summary- Report of the Working Group on Mammalian Germ Cell Tests, Mutation Res., 312, pp. 313-318. |
|
(7) |
Fielder, R.J., Allen, J.A., Boobis, A.R., Botham, P.A., Doe, J., Esdaile, D.J., Gatehouse, D.G., Hodson-Walker, G., Morton, D.B., Kirkland D.J. and Richold, M. (1992), Report of British Toxicology Society/UK Environmental Mutagen Society Working group: Dose setting in In Vivo Mutagenicity Assays, Mutagenesis. 7, pp. 313-319. |
|
(8) |
Lovell, D.P., Anderson, D., Albanese, R., Amphlett, G.E., Clare, G., Ferguson, R., Richold, M., Papworth, D.G. and Savage, J.R.K. (1989), Statistical Analysis of In Vivo Cytogenetic Assays, in: D.J. Kirkland (ed.), Statistical Evaluation of Mutagenicity Test Data. UKEMS SubCommittee on Guidelines for Mutagenicity Testing, report, Part III, Cambridge University Press, Cambridge, New York, Port Chester, Melbourne, Sydney, pp. 184-232. |
B.24. SPOTTEST — MUS
1. METODE
1.1. INDLEDNING
Se den generelle indledning til del B.
1.2. DEFINITIONER
Se den generelle indledning til del B.
1.3. REFERENCESTOFFER
Ingen.
1.4. PRINCIPPER FOR TESTMETODEN
Spottesten er en in vivo-test i mus, hvor musefostre på det embryonale stade eksponeres for kemiske forbindelser. Målcellerne i fostrene er melanoblasterne, og målgenerne er de gener, der bestemmer pelshårenes pigmentering. Fostrene er heterozygote i et antal af disse pelsfarvegener. Ved mutation eller tab af det dominante allel i en melanoblast ved en række genetiske hændelser kommer den recessive fænotype til udtryk i de celler, der dannes fra denne stamcelle, hvilket fører til anderledes farvede pletter i musens pels. Mængden af afkom med sådanne pletter, dvs. mutationer, registreres og sammenlignes med den tilsvarende mængde blandt afkom af hunmus, der kun har været eksponeret for opløsningsmidlet. Ved spottesten i mus konstateres formodede somatiske mutationer i fosterceller.
1.5. KVALITETSKRITERIER
Ingen.
1.6. BESKRIVELSE AF TESTMETODEN
Forberedelser
Teststoffet opløses eller suspenderes sà vidt muligt i isotonisk saltvand. Kemiske forbindelser, som er uopløselige i vand, opløses eller suspenderes i passende vehikler. Det anvendte vehikel må ikke indvirke på teststoffet eller forårsage toksiske effekter. Fremstillingen af teststofpræparationen foretages umiddelbart inden anvendelsen.
Mus af stammen T-stock (nonagouti, a/a; chinchilla, pink eye, cchp/cchp; brown, b/b; dilute, short ear, d se/d se; piebald spotting, s/s) parres med enten stammen HT-stock (pallid, nonagouti, brachypody, pa a bp/pa a bp; leaden fuzzy, In fz/ln fz; pearl pe/pe) eller C57 BL (nonagouti, a/a). Andre egnede krydsninger som NMRI (nonagouti, a/a; albino, c/c) og DBA (nonagouti, a/a; brown, b/b; dilute, d/d) kan anvendes, under forudsætning af, at afkommet er nonagouti.
Der anvendes et antal drægtige hunner, som er tilstrækkeligt stort til, at der produceres en passende mængde afkom for hver af de anvendte doser. Holdstørrelsen kan anslås på grundlag af antallet af observerede pletter hos de eksponerede mus og kontroldatas størrelsesorden. Et resultat kan først accepteres som negativt, hvis mindst 300 unger af hunner, som har været eksponeret for den største dosis, er undersøgt.
Der foretages sideløbende kontrol med mus, som kun eksponeres for vehikel (negativ kontrol). Historiske kontroldata fra samme laboratorium kan, under forudsætning af at der er tale om homogene data, indregnes med henblik på at forøge testens sensitivitet. Der bør foreligge positive kontroldata fra nyligt foretagne forsøg i samme laboratorium med en kemisk forbindelse, som vides at være mutagen i denne test, i tilfælde af at der ikke konstateres nogen mutagen virkning af teststoffet.
Sædvanligvis indgives teststoffet ved oral intubering eller intraperitoneal injektion i de drægtige hunner. Inhalation og andre administrationsmåder kan anvendes, hvis de er hensigtsmæssige.
Der anvendes mindst to doseringer, hvoraf den ene skal medføre tegn på toksisk virkning eller reduceret kuldstørrelse. Ikke-toksiske stoffer testes under anvendelse af så store doser som praktisk muligt.
Fremgangsmåde
Normalt gives forsøgsdyrene hele dosis på en gang på drægtighedsperiodens dag 8, 9 eller 10, idet dag 1 er den dag, vaginalproppen observeres første gang. Disse dage svarer til 7,25, 8,25 og 9,25 dage efter befrugtningen. Gentagne doseringer kan foretages på disse dage.
Afkommet blindkodes og eventuelle pigmenterede pletter registreres mellem tre og fire uger efter fødslen. Der skelnes mellem tre typer pletter:
|
a) |
hvide pletter inden for en afstand af 5 mm fra den ventrale midtlinje; disse pletter antages at være et resultat af celledrab (WMVS) |
|
b) |
gule agouti-agtige pletter på eller omkring pattevorter, kønsåbninger, strube, axiller, lyske og midt i panden; disse pletter antages at være et resultat af fejldifferentiering (MDS) |
|
c) |
pigmenterede og hvide pletter tilfældigt fordelt i pelsen; disse pletter antages at være et resultat af somatiske mutationer (RS). |
Alle tre typer registreres, men kun den sidstnævnte, RS, et genetisk relevant. Hvis der er problemer med at skelne mellem MDS og RS, kan der benyttes fluorescensmikroskopi af hårprøver.
Synlige morfologiske abnormiteter hos afkommet registreres også.
2. DATA
Det samlede antal undersøgte unger og antal unger med en eller flere pletter, som antages at være et resultat af somatisk mutation, anføres. Data fra forsøgsgrupper og negativ kontrolgruppe sammenlignes ved hjælp af egnede statistiske metoder. Data anføres også pr. kuld.
3. RAPPORTERING
3.1. FORSØGSRAPPORTEN
Forsøgsrapporten skal om muligt indeholde følgende:
|
— |
de anvendte stammer |
|
— |
antal drægtige hunner i forsøgs- og kontrolgrupperne |
|
— |
den gennemsnitlige kuldstørrelse i forsøgs- og kontrolgrupperne ved fødslen, og når ungerne er vænnet fra |
|
— |
dosisniveauer |
|
— |
opløsningsmiddel |
|
— |
den dag i drægtighedsperioden, hvor doseringen finder sted |
|
— |
administreringsform |
|
— |
det samlede antal undersøgte unger og antal unger med WMVS, MDS og RS i forsøgs- og kontrolgrupperne |
|
— |
makroskopiske morfologiske abnormiteter |
|
— |
om muligt dosis-responsforhold for RS |
|
— |
statistisk evaluering |
|
— |
diskussion af resultaterne |
|
— |
fortolkning af resultaterne. |
3.2. EVALUERING OG FORTOLKNING
Se den generelle indledning til del B.
4. HENVISNINGER
Se den generelle indledning til del B.
B.25. ARVELIG TRANSLOKATION HOS MUS
1. METODE
1.1. INDLEDNING
Se den generelle indledning til del B.
1.2. DEFINITIONER
Se den generelle indledning til del B.
1.3. REFERENCESTOFFER
Ingen.
1.4. PRINCIP FOR TESTMETODEN
Ved test af arvelig translokation hos mus konstateres strukturelle og numeriske kromosomforandringer i pattedyrskønsceller udtrykt i førstegenerations afkom. Kromosomforandringerne, der måles, er reciprokke translokationer, og, når afkommet også omfatter hundyr, tab af et X-kromosom. Bærere af translokationer og XO-hunner har nedsat fertilitet, hvilket udnyttes ved udvælgelsen af F1-afkom med henblik på cytogenetisk analyse. Visse typer translokationer (X-autosome og c-t-typen) forårsager fuldstændig sterilitet. Translokationer konstateres cytogenetisk i meiosens diakinese-metafase I hos hanner, som enten kan være af F1-generationen eller sønner af F1-hunner. XO-hunner identificeres cytogenetisk ved, at kromosomtallet i knoglemarvmitoser kun er 39.
1.5. KVALITETSKRITERIER
Ingen.
1.6. BESKRIVELSE AF TESTMETODEN
Forberedelser
Teststoffet opløses i isotonisk saltvand. Uopløselige stoffer opløses eller suspenderes i egnede vehikler. Teststofopløsning anvendes umiddelbart efter fremstillingen. Hvis der anvendes et vehikel med henblik på at lette doseringen, må det ikke interferere med teststoffet eller have nogen toksisk virkning.
Sædvanligvis indgives teststoffet ved oral intubering eller intraperitoneal injektion. Andre administrationsmåder kan være hensigtsmæssige.
Som forsøgsdyr benyttes mus, idet disse giver de letteste betingelser med hensyn til avl og cytologisk verifikation. Der kræves ikke nogen bestemt stamme. Hvis det påregnes at teste fertiliteten, bør den anvendte stammes gennemsnitlige kuldstørrelse være over otte og relativt konstant.
Der anvendes sunde kønsmodne dyr.
Det nødvendige antal dyr afhænger af den spontane translokationsfrekvens og den induktionsfrekvens, som er nødvendig for at opnå et positivt resultat.
Normalt gennemføres testen ved undersøgelse af F1-hanner. Der undersøges mindst 500 F1-hanner pr. dosis. Hvis der tillige anvendes F1-hunner, undersøges 300 af hvert køn.
Adækvate kontroldata, stammende fra sideløbende og historiske kontrolforsøg, skal være tilgængelige. Når der foreligger acceptable positive kontrolresultater fra undersøgelser foretaget for nyligt i det samme laboratorium, kan disse anvendes i stedet for en sideløbende positiv kontrol.
Der testes en dosis, normalt den største dosis med minimal toksisk virkning, men uden indflydelse på reproduktion eller overlevelse. Hvis dosis-responsforholdet skal bestemmes, anvendes yderligere to mindre doser. Ikke-toksiske stoffer testes under anvendelse af så store doser som praktisk muligt.
Fremgangsmåde
Der kan anvendes to forskellige eksponeringsskemaer. I det hyppigst anvendte skema gives hele dosis på en gang. Teststoffet kan også gives syv dage om ugen i 35 dage. Antallet af parringer efter eksponeringen afhænger af eksponermgsskemaet og skal være stort nok til at sikre, at alle eksponerede kønscellestadier kan undersøges. Efter parringen placeres hunnerne i hver sit bur, Ved fødslen registreres dato, kuldstørrelse og afkommets køn. Alle hanner blandt afkommet vænnes fra og hunnerne frasorteres, medmindre de medtages i forsøget.
Der anvendes en af to mulige metoder:
|
— |
fertilitetsundersøgelse af F1-generationen med efterfølgende verifikation af mulige translokationsbærere ved hjælp af cytogenetisk analyse |
|
— |
cytogenetisk analyse af alle F1-hanner uden forudgående udvælgelse på grundlag af fertilitetsundersøgelser. |
|
a) |
Fertilitetsundersøgelse Nedsat fertilitet hos F1-generationens medlemmer kan konstateres ved observation af kuldstørrelsen og/eller analyse af de parrede hundyrs uterusindhold. Forinden fastlægges der kriterier for normal og nedsat fertilitet hos den anvendte stamme. Observation af kuldstørrelse: De F1-hanner, som skal undersøges, placeres i hver sit bur sammen med hunner fra samme forsøg eller fra bestanden. Fra og med 18 dage efter parringen iagttages burene dagligt. Kuldstørrelse og køn i F2-generationen registreres ved fødslen, hvorefter kuldene fjernes. Hvis der testes F1-hunner, bevares de små F2-kuld med henblik på yderligere undersøgelser. Konstatering af, om hunner er translokationsbærere, forgår ved cytogenetisk analyse af translokation hos en vilkårlig han blandt deres afkom. XO-hunner identificeres gennem ændring af forholdet mellem hanner og hunner i deres afkom fra 1:1 til 1:2. Ved en sekventielt ordnet fremgangsmåde elimineres normale F1-dyr fra den videre undersøgelse, hvis størreisen af det første F2-kuld er lig med eller højere end en på forhånd fastsat normalværdi. I modsat fald observeres det andet eller tredje F2-kuld. F1-dyr, der ikke kan klassificeres som normale efter observation af op til tre F2-kuld underkastes enten yderligere undersøgelse ved analyse af de parrede hunners uterusindhold eller ved direkte cytogenetisk analyse. Analyse af uterusindholdet: Den nedsatte kuldstørrelse hos translokationsbærere skyldes, at fostrene dør på det embryonale stade, så et stort antal døde implantater tyder på translokation hos forsøgsdyret. De F1-hanner, som skal undersøges, parres hver især med to til tre hunner. Ved daglig undersøgelse mellem kl. 10 og 12 om formiddagen af forekomsten af vaginalprop, konstateres det, om befrugtning har fundet sted. Hunnerne aflives 14 til 16 dage senere, og antallet af levende og døde implantater i uteri registreres. |
|
b) |
Cytogenetisk analyse Der fremstilles testikelpræparater ved lufttørringsteknik. Translokationsbærere identificeres på grundlag af multivalente konfigurationer i primære spermatocytters diakinese-metafase I. Hvis der observeres mindst to celler med multivalente konfigurationer betragtes det som bevist, at forsøgsdyret er translokationsbærer. Når der ikke gennemføres udvælgelse på basis af fertilitetsundersøgelser, undersøges alle F1-hanner cytogenetisk. Der foretages mikroskopisk undersøgelse af mindst 25 diakinese-metafaser I pr. han. F1-hanner med små testikler og defekt meiose, som standser før diakinesen, samt F1-hunner, som formodes at være XO-bærere, underkastes undersøgelse af mitotiske metafaser i spermatogonier eller knoglemarv. Hvis der observeres usædvanlig lange og/eller korte kromosomer i ti celler, betragtes det som påvist, at der er tale om en translokation, som gør hanner sterile (c-t-typen). Visse X-autosome translokationer, som gør hanner sterile, kan kun identificeres ved båndanalyse af mitosekromosomer. Hvis der observeres 39 kromosomer i ti mitoser, betragtes det som påvist, at der er tale om en XO-hun. |
2. DATA
Data opstilles i tabelform.
Den gennemsnitlige kuldstørrelse og kønsfordeling i forældregenerationens afkom ved fødslen og ved afvænningen angives for hver parringsperiode.
Med henblik på vurdering af F1-dyrenes fertilitet anføres den gennemsnitlige kuldstørrelse efter de normale parringer og hver enkelt kuldstørrelse efter parring af F1-translokationsbærere. Efter analyse af uterusindhold anføres det gennemsnitlige antal levende og døde implantater efter normale parringer og antallet af levende og døde implantater efter hver enkelt parring af F1-translokationsbærere.
Efter cytogenetisk analyse af diakinese-metafaser I anføres antallet af multivalente konfigurationer og det totale antal undersøgte celler for hver enkelt translokationsbærer.
Det samlede antal parringer og parringsperiodernes længde anføres for sterile F1-dyr. Der gives detaljerede oplysninger om testikelvægt og cytogenetisk analyse.
Den gennemsnitlige kuldstørrelse, F2-afkommets kønsfordeling og resultaterne af den cytogenetiske analyse anføres for XO-hunner.
Hvis mulige F1-translokationsbærere udvælges ved fertilitetsundersøgelser, skal tabellerne indeholde oplysninger om, hvor mange af de udvalgte dyr der senere viste sig at være heterozygote efter translokation.
Data fra negative og positive kontrolforsøg rapporteres på lignende måde.
3. RAPPORTERING
3.1. FORSØGSRAPPORT
Forsøgsrapporten skal om muligt indeholde følgende:
|
— |
stamme, dyrenes alder, de doserede dyrs vægt |
|
— |
antal af forældredyr af hvert køn i forsøgs- og kontrolgrupper |
|
— |
forsøgsbetingelser, detaljeret beskrivelse af dyrenes behandling, dosisniveau, opløsningsmidler, parringsskema |
|
— |
afkommets antal og køn for hver enkelt hun, antal og køn af afkom, som opdrættes med henblik på translokationsanalyse |
|
— |
translokationsanalytiske kriterier og analysetidspunkt |
|
— |
antal translokationsbærere samt detaljeret beskrivelse af disse, herunder avlingsdata og oplysninger om uterusindhold, hvis sådanne foreligger |
|
— |
cytogenetisk fremgangsmåde og detaljerede oplysninger om den mikroskopiske analyse, helst vedlagt billeder |
|
— |
statistisk evaluering |
|
— |
diskussion af resultaterne |
|
— |
fortolkning af resultaterne. |
3.2. EVALUERING OG FORTOLKNING
Se den generelle indledning til del B.
4. HENVISNINGER
Se den generelle indledning til del B.
B.26. SUBKRONISK ORAL TOKSICITETSTEST — ET 90-DAGES FORSØG OVER GENTAGNE ORALE DOSERS TOKSICITET PÅ GNAVERE
1. METODE
Denne metode til udførelse af en subkronisk oral toksicitetstest svarer til OECD TG 408 (1998).
1.1. INTRODUKTION
Efter at en akut eller gentagen 28-dages toksicitetstest er udført med henblik på primær vurdering af et kemisk stofs toksicitet, kan man udføre en subkronisk oral toksicitetstest, hvor man anvender gentagne doser til bestemmelse og evaluering af stoffets toksiske egenskaber. Undersøgelsen på 90 dage skaffer viden om mulige trusler mod sundheden, som kan opstå ved gentagen belastning i en længere periode, som dækker afvænning, modning og den første del af voksenlivet. Undersøgelsen skaffer viden om større toksiske virkninger, peger på, hvilke organer der rammes og muligheden for akkumulation; den kan give et overslag over det belastningsniveau, der kan være skadeligt, uden at det bemærkes, som igen kan anvendes til valg af dosis til undersøgelser af kronisk effekt og til fastlæggelse af sikkerhedskriterier for, hvad mennesker kan tåle.
Metoden lægger yderligere vægt på virkninger på neurologiske områder og peger på virkninger på det immunologiske og reproduktive system. Nødvendigheden af omhyggelig klinisk observation af dyrene for at opnå størst mulig viden bliver ligeledes understreget. Undersøgelsen skulle muliggøre identifikation af kemikalier med neurotoksiske virkninger og virkninger på det immunologiske og reproduktive system, som kan føre til yderligere dybdegående undersøgelser.
Se også den generelle indledning til del B.
1.2. DEFINITIONER
Dosis: Mængden af indgivet testkemikalium. Dosis angives som vægt (g, mg) eller som vægt af testkemikalium i forhold til dyrets vægt (f.eks. mg/kg) eller en konstant del af føden (ppm).
Dosering: Er et generelt udtryk for dosis, hyppighed af indgift og varigheden af indgiften.
NOAEL: Er en forkortelse for det niveau, der ikke medfører observeret skadelig virkning, og er det højeste dosisniveau, hvor ingen negative virkninger, der kan tilskrives behandlingen, er fundet.
1.3. TESTMETODENS PRINCIP
Testkemikaliet administreres peroralt dagligt i forskellige doser til forskellige grupper af forsøgsdyr, idet hver gruppe får en bestemt dosis i en periode på 90 dage. I denne periode observeres dyrene nøje for tegn på forgiftning. Dyr, der dør eller aflives, mens testen står på, bliver obduceret, og ved undersøgelsens afslutning bliver resten af dyrene aflivet og obduceret.
1.4. BESKRIVELSE AF TESTMETODEN
1.4.1. Forberedelse af dyrene
Der anvendes sunde dyr, som er tilvænnet laboratorieforholdene i mindst fem dage, og som ikke før har været anvendt til eksperimenter. Dyrene beskrives hvad angår art, stamme, hvorfra de kommer (kilde), køn, vægt og/eller alder. Dyrenes randomiseres til henholdsvis kontrol og behandlingsgruppe. Bure skal anbringes, så deres placering får mindst mulig betydning. Hvert dyr tildeles et eget, særligt identifikationsnummer.
1.4.2. Fremstilling af doser
Stoffet, der skal undersøges, indgives gennem sonde eller i føden eller drikkevandet. Måden at indgive på afhænger af undersøgelsens formål og testmaterialets fysisk-kemiske egenskaber.
Om nødvendigt opløses eller opslæmmes stoffet i et passende medie. Hvor muligt anbefales det først at forsøge at anvende vandige opløsninger eller opslæmninger; dernæst overvejes opløsninger eller opslæmninger i olie (f.eks. majsolie) og først derefter andre opløsnings-/opslæmningsmidler. For andre midler end vand bør de toksiske egenskaber kendes. Testkemikaliets stabilitet i forbindelse med indgiften bør fastslås.
1.4.3. Testomstændighederne
1.4.3.1. Forsøgsdyrene
Den foretrukne art er rotten, men andre gnaverarter, f.eks. mus, kan anvendes. Almindeligt anvendte laboratoriestammer af unge, sunde, voksne dyr bør anvendes. Hundyrene skal ikke have haft unger og ikke være gravide. Indgiften bør begynde snarest efter fravænning, og i hvert fald før dyrene er ni uger gamle. Ved forsøgets begyndelse bør den indbyrdes vægtforskel være minimal og ikke overstige ± 20 % af gennemsnittet for hvert køn. Når forsøget foretages som forløber for en længerevarende toksicitetsstudie, bør man i begge forsøg anvende samme stamme og fra samme kilde.
1.4.3.2. Antal og køn
Mindst 20 dyr (10 af hvert køn) bør anvendes for hvert dosisniveau. Hvis man planlægger at aflive nogle dyr undervejs i forsøget, skal antallet af dyr, der påbegynder forsøget, forøges tilsvarende. Ud fra tidligere opnået viden om kemikaliet eller nære analoge stoffer bør man overveje en yderligere »satellit«-gruppe på ti dyr (fem af hvert køn) i kontrollen og i gruppen med den højeste dosis, således at man efter forsøgsperioden kan observere evt. reversibilitet eller vedvarende følger af behandlingen. Varigheden af denne observationsperiode afhænger af, hvilke virkninger man vil følge.
1.4.3.3. Dosisstørrelse
Mindst tre dosisstørrelser og en sideløbende kontrol bør anvendes, undtagen i tilfælde af en begrænset test (se 1.4.3.4). Dosisstørrelsen kan afhænge af tidligere tilsvarende forsøg eller forsøg med henblik på opsøgning af dosisgrænser og skal tage hensyn til kendte toksikologiske og toksokinetiske data for testkemikaliet eller tilsvarende stoffer. Medmindre kemikaliets fysisk-kemiske natur eller biologiske effekt sætter grænser, bør den højeste dosisstørrelse vælges med henblik på toksisk virkning, men ikke på død eller alvorlig lidelse. Faldende dosisstørrelse bør vælges med henblik på at påvise en eventuel dosisrelateret virkning og det niveau, der ikke medfører observeret skadelig virkning (NOAEL). To til firedobbelte intervaller er ofte bedst til fastlæggelse af faldende dosisstørrelser, og tilføjelse af en fjerde test-gruppe er ofte at foretrække frem for brug af meget store intervaller (f.eks. mere end en faktor på 6-10) mellem dosisstørrelserne.
Kontrolgruppen skal være en ubehandlet gruppe eller en gruppe, der får det eventuelle medie, i hvilket de behandlede dyr får kemikaliet. Bortset fra behandlingen skal dyrene i kontrolgruppen behandles nøjagtigt som dyrene i testgruppen. Hvis kemikaliet indgives i et medie, skal kontrolgruppen have mediet i den størst anvendte mængde. Hvis testkemikaliet indgives via foderet, og hvis dette medfører reduceret fødeindtagelse, kan det være nyttigt med en gruppe, der parallelfodres mængdemæssigt, således at et eventuelt vægttab kan tilskrives, at foderet har en dårlig smag, respektive virker toksisk.
Man bør overveje mediets eller eventuelle additivers egenskaber med hensyn til absorption, distribution, stofskifte eller retention af testkemikalium; virkningen på testkemikaliets kemiske egenskaber, som igen kan have indflydelse på dets toksiske egenskaber; og indflydelsen på føde- eller vandindtag eller dyrenes ernæringsmæssige tilstand.
1.4.3.4. Grænsetest
Et totalforsøg med tre dosisstørrelser behøves næppe udført, hvis man ikke forventer nogen toksisk effekt ud fra kendskab til strukturelt beslægtede stoffer, og hvis der ikke ses nogen skadelig virkning ved forsøg som beskrevet ovenfor med enkelt dosis svarende til mindst 1 000 mg/kg legemsvægt/dag. Grænsetest er konklusive, undtagen hvis belastning af mennesker indikerer nødvendigheden af anvendelse af en større dosis.
1.5. FREMGANGSMÅDE
1.5.1. Dosisadministration
Dyrene får testkemikaliet syv gange ugentlig hver uge i 90 dage. Ethvert andet regime, f.eks. fem gange ugentlig, skal begrundes. Hvis testkemikaliet indgives med sonde, skal det gøres i en enkelt dosis, med anvendelse af en mavesonde eller passende intubationsrør. Det maksimale væskevolumen, der kan tilføres ad gangen, afhænger af dyrets størrelse. Volumen bør ikke overstige 1 ml/100 g kropsvægt, undtaget vandige opløsninger, hvor man kan indgive 2 ml/100 g kropsvægt. Undtaget irritative og ætsende stoffer, som normalt vil have øget generende effekt ved større koncentrationer, bør variationen i testvolumen begrænses ved justering af koncentrationen af testkemikaliet for at sikre et konstant volumen ved alle dosisstørrelser.
Det er vigtigt at sikre, at de kvantiteter af kemikalier, som indtages via føden eller drikkevandet, ikke får indflydelse på den normale ernæring eller vandbalancen. Når testkemikaliet tilføres med føden, kan man tilføre det enten som en konstant del af føden (ppm) eller en konstant dosisstørrelse i forhold til dyrets vægt; det, man vælger, skal specificeres. Tilføres kemikaliet via sonde, bør det gøres på samme tidspunkt hver dag, og dosisstørrelse skal rettes til, så den udgør en konstant størrelse i forhold til dyrets vægt. Hvis et 90-dages forsøg foretages som forløber for et langtids kronisk-toksicitetsforsøg, skal der i begge forsøg gives samme fødesammensætning.
1.5.2. Observationer
Observationsperioden skal mindst vare 90 dage. Dyrene i satellitgruppen, hvor man vil foretage »follow-up« -observationer, skal beholdes i en passende periode uden behandling, med henblik på konstatering af fortsat eller ophør af toksisk virkning.
Almindelig klinisk observation bør foretages mindst en gang om dagen, fortrinsvis på samme tidspunkt(er), idet man tager i betragtning, hvornår maksimal effekt kan forventes efter indgift. Dyrenes kliniske tilstand bør noteres. Mindst to gange daglig, sædvanligvis morgen og aften, undersøges alle dyrene for tegn på morbiditet eller mortalitet.
I hvert fald før første belastning (for at tage højde for individuelle forskelle) og en gang ugentlig derefter bør detaljerede kliniske undersøgelse foretages af alle dyrene. Disse undersøgelser skal foretages uden for burene, fortrinsvis på et standardareal og på samme tid i hvert tilfælde. De skal noteres omhyggeligt, fortrinsvis ved anvendelse af et scoringssystem, eksplicit defineret af undersøgelseslaboratoriet. Man bør bestræbe sig på, at variationerne i observationsbetingelserne er mindst mulige. Blandt de fund, der skal noteres, (men ikke som de eneste) er ændringer i hud, pels, øjne, slimhinder, forekomst af sekretion og ekskreter og autonom aktivitet (f.eks. tåreflåd, hårrejsning, pupilstørrelse, usædvanligt respirationsmønster). Forandringer i gang, stilling og respons på håndtering såvel som tilstedeværelse af toniske og kloniske bevægelser, stereotypi (f.eks. overdreven pudsning, repetitive cirkelbevægelser) eller bizar opførsel (f.eks. selvmutilation, baglæns gang) skal også noteres (1).
Oftalmologisk undersøgelse ved hjælp af oftalmoskop eller andet, passende udstyr bør udføres, før første gang testkemikaliet tilføres og ved afslutning af forsøget, fortrinsvis på alle dyrene, men i hvert fald på dem, der fik højeste dosis og på kontrollerne. Hvis der opdages øjenforandringer, skal alle dyrene undersøges.
Hen imod slutningen af belastningsperioden og i hvert fald ikke tidligere end uge 11 foretages undersøgelser af reaktion på sensoriske stimuli af forskellig slags (1) (f.eks. auditoriske, visuelle og proprioceptive stimuli) (2), (3), (4), vurdering af gribestyrke (5) og af motorisk aktivitet (6). Nærmere detaljer af de procedurer, der kan foretages, findes i de respektive referencer. Andre, alternative fremgangsmåder end de refererede kan imidlertid også anvendes.
Funktionsundersøgelser kan udelades nær forsøgets afslutning, hvis tilsvarende data kan findes i andre tilsvarende forsøg og de daglige kliniske undersøgelser ikke påviste funktionelle mangler.
Undtagelsesvis kan funktionsundersøgelser udelades på grupper, der på anden vis viser tegn på forgiftning i en grad, der vil have afgørende indflydelse på funktionsundersøgelserne.
1.5.2.1. Kropsvægt og føde/vandindtagelse
Alle dyr skal vejes mindst en gang ugentlig. Mængden af fødeindtagelse skal beregnes mindst en gang ugentlig. Hvis testkemikaliet administreres via drikkevandet, skal vandindtagelsen beregnes mindst en gang ugentlig. Vandforbruget bør også tages i betragtning, når belastningen foretages via føden eller via sonde, da drikkeaktiviteten kan ændres herved.
1.5.2.2. Hæmatologi og klinisk biokemi
Stedet, hvor blodprøver tages, bør angives, og de bør gemmes på passende vis. Ved forsøgets afslutning tages der prøver lige før eller i forbindelse med aflivningen af dyrene.
Følgende hæmatologiske undersøgelser bør foretages ved forsøgets afslutning, og når blodprøver bliver indsamlet undervejs: hæmatokrit, hæmoglobinkoncentrationen, erytrocyttal, leukocyttal og differentialtælling, trombocyttal og koagulationstid/koagulationsevnen.
Klinisk biokemiske analyser, som kan vise toksisk virkning på væv, specielt på lever og nyrer, skal udføres på blodprøver taget fra alle dyrene lige før eller i forbindelse med aflivningen (undtaget er de dyr, der findes moribunde og/eller aflives undervejs i forsøget). På samme måde, som gælder for hæmatologiske prøver, undersøges også blodprøver taget undervejs. Det anbefales at tage fastende blodprøver (3). Analyser på serum og plasma bør inkludere: natrium, kalium, glukose, total kolesterol, carbamid, BUN, kreatinin, total protein og albumin, og mere end to enzymer, der belyser den hepatocellulære effekt (f.eks. alanin aminotransferase, aspartat aminotransferase, alkalisk fosfatase, gamma glutamyl transpeptidase og sorbitol dehydrogenase). Derudover kan inkluderes måling af andre enzymer (fra lever eller andetsteds), galdesure salte, som kan give nyttig information i visse tilfælde.
I den sidste uge af forsøget kan eventuelt følgende urinanalyser udføres, idet timediuresen måles: udseende, volumen, osmolalitet eller vægtfylde, pH, protein, glukose og blod eller blodceller.
Yderligere bør man overveje at måle serummarkører for generel vævsskade. Andre analyser, som bør udføres, hvis testkemikaliets egenskaber vides eller formodes at afficere beslægtede metaboliske markører, inkluderer calcium, fosfor, fastende triglycerider, specielle hormoner, methæmoglobin og kolinesterase. Disse bør måles i forbindelse med kemikalier af særlige typer eller fra tilfælde til tilfælde.
Almindeligvis må man have en fleksibel indstilling ud fra dyrearten og de observerede og/eller forventede virkninger af et givet kemikalium.
Hvis der ikke er tilstrækkeligt kendskab til de biokemiske eller hæmatologiske udgangsværdier, bør man overveje om disse analyser skal foretages, før belastningen begynder, men det anbefales almindeligvis ikke (7).
1.5.2.3. Makroskopisk obduktion
Alle dyrene, som indgår i forsøget, skal have udført en fuldstændig, detaljeret makroskopisk obduktion, hvilket inkluderer omhyggelig undersøgelse af alle overflader, alle åbninger, og kraniets, brystkassens og abdomens kaviteter og deres indhold. Lever, nyrer, binyrer, testes, bitestikler, uterus, ovarier, thymus, milt, hjerne og hjerte skal på alle dyr renses for andet vedhængende væv som behørigt (undtaget er de dyr, der findes moribunde og/eller aflives undervejs i forsøget) og vejes snarest muligt efter dissektionen for at undgå udtørring.
Følgende væv skal præserveres ved hjælp af det mest passende fiksationsmedie både med henblik på vævstypen og den følgende histopatologiske undersøgelse: alle større læsioner, hjernen (repræsentative regioner, inklusive cerebrum, cerebellum og medulla/pons), rygmarven (fra tre niveauer: cervikal, midt-torakal og lumbal), hypofyse, tyreoidea og paratyreoidea, tymus, øsofagus, spytkirtlerne, mavesækken, tynd- og tyktarm, (inklusive de Peyerske plaques) lever, pankreas, nyrer, binyrer, milt, hjerte, luftrør og lunger (bevaret ved indblæsning med fiksativ og påfølgende neddypning), aorta, gonader, uterus, sekundære kønsorganer, brystkirtlerne fra hundyrene, prostata, urinblære, galdeblære (mus), lymfeknuder (helst en lymfeknude, som dækker belastningsvejen, og en anden langt derfra for at dække den systemiske virkning), perifer nerve (ischiadicus eller tibialis) helst nær ved musklen, en sektion af knoglemarven (og/eller frisk knoglemarvsaspirat), hud og øjne (hvis oftalmologisk undersøgelse viste ændringer), Kliniske og andre fund kan foranledige undersøgelse af andet væv. Også særlige organer, der sandsynligvis kan være mål for testkemikaliets kendte virkninger, bør præserveres.
1.5.2.4. Histopatologi
En komplet histopatologisk undersøgelse skal udføres af de præserverede organer og væv på alle dyrene i kontrol- og højdosisgrupperne. Undersøgelserne skal omfatte alle dyrene i de andre grupper, også hvis der observeres ændringer i højdosisgruppen, der kan tilskrives belastningen.
Alle større læsioner skal undersøges.
Hvis der anvendes en »satellit«-gruppe, skal der på dem udføres histopatologisk undersøgelse af væv og organer, på hvilke der var vist ændringer i den belastede gruppe.
2. DATA OG RAPPORTERING
2.1. DATA
Alle data skal anføres. Yderligere skal alle data opsummeres i tabeller, der for hver testgruppe viser antal dyr ved forsøgets begyndelse, antal dyr fundne døde i løbet af testen eller aflivede af humane grunde og tidspunktet for deres død, det antal dyr, der udviser tegn på forgiftning, en beskrivelse af disse tegn inklusive tidspunktet for deres begyndelse, deres varighed og forgiftningsgraden; det antal dyr, der har læsioner, typen af læsioner og den procentdel af dyrene, der udviser de forskellige typer af læsion.
Hvor det er muligt, skal tallene evalueres ved hjælp af en passende og accepteret statistisk metode. Den statistiske metode og de data, der skal analyseres med den, skal vælges under planlægningen af forsøget.
2.2. TESTRAPPORT
Testrapporten skal inkludere følgende informationer:
2.2.1. Testkemikalium:
|
— |
fysiske egenskaber, renhedsgrad og fysisk-kemiske egenskaber |
|
— |
identifikationsdata |
|
— |
indgivelsesmåde, som begrundes, hvis der ikke er tale om drikkevand. |
2.2.2. Dyreart:
|
— |
anvendte art og stamme |
|
— |
antal dyr, deres alder og køn |
|
— |
kilde, opbevaringsforhold, føde osv. |
|
— |
de enkelte dyrs vægt ved forsøgets begyndelse. |
2.2.3. Forsøgsbetingelser:
|
— |
rationale for valg af dosismængde |
|
— |
detaljer vedrørende testkemikaliets form/opblanding i føden, resulterende koncentration, stabilitet og homogenitet |
|
— |
detaljer vedrørende indgift af testkemikaliet |
|
— |
sande dosis (mg/kg kropsvægt/dag) og omregningsfaktor fra testkemikaliets koncentration i føde/vand (ppm) til sand dosis, hvis muligt |
|
— |
detaljer vedrørende kvalitet af vand og føde. |
2.2.4. Resultater:
|
— |
kropsvægt og ændringer i kropsvægt |
|
— |
fødeindtagelse og indtagelse af vand, om muligt |
|
— |
data vedrørende forgiftning sat over for køn og dosis, inklusive forgiftningstegnene |
|
— |
art, alvor og varighed af det klinisk observerede (om det observerede er reversibelt) |
|
— |
resultater af oftalmologisk undersøgelse |
|
— |
vurdering, hvis muligt, af sanseaktiviteter, gribestyrke og motorisk aktivitet |
|
— |
hæmatologiske målinger med relevante udgangsværdier |
|
— |
klinisk-biokemiske målinger med relevante udgangsværdier |
|
— |
slutvægt, organernes vægt og organ/kropsvægtforholdene |
|
— |
sektionsfund |
|
— |
en detaljeret beskrivelse af de histopatologiske fund |
|
— |
data for absorption, hvis de forefindes |
|
— |
statistisk behandling af resultaterne, hvor rimeligt. |
Diskussion af resultater
Konklusioner
3. HENVISNINGER
|
(1) |
IPCS (1986). Principles and Methods for the Assessment of Neurotoxicity Associated with Exposure to Chemicals. Environmental Health Criteria Document No 60. |
|
(2) |
Tupper, D.E., Wallace, R.B. (1980). Utility of the Neurologie Examination in Rats. Acta Neurobiol. Exp., 40, pp. 999-1003. |
|
(3) |
Gad. S.C. (1982). A Neuromuscular Screen for Use in lndustrial Toxicology. J. Toxicol. Environ. Health, 9, pp. 691-704. |
|
(4) |
Moser. V.C, Mc Daniel, K.M. Phillips, P.M. (1991). Rat Strain and Stock Comparisons Using a Functional Observational Battery: Baseline Values and Effects of Amitraz. Toxicol. Appl. Pharmacol., 108, pp. 267-283. |
|
(5) |
Meyer O.A., Tilson H.A., Byrd W.C, Riley M.T. (1979). A Method for the Routine Assessment of Fore- and Hind- limb grip Strength of Rats and Mice. Neurobehav. Toxivol., 1, pp. 233-236. |
|
(6) |
Crofton K.M., Howard J.L, Moser V.C, Gill M.W., Reiter L.W., Tilson H.A., MacPhail R.C. (1991). Interlaboratory Comparison of Motor Activity Experiments: Implication for Neurotoxicological Assessments. Neurotoxicol. Teratol., 13, pp. 599-609. |
|
(7) |
Weingand K., Brown G., Hall R. et al. (1996). Harmonisation of Animal Clinic Pathology Testing in Toxicity and Safety Studies, Fundam. & Appl. Toxicol., 29, pp. 198-201. |
B.27. SUBKRONISK ORAL TOKSICITETSTEST — ET 90-DAGES FORSØG OVER GENTAGNE ORALE DOSERS TOKSICITET PÅ IKKE-GNAVERE
1. METODE
Denne metode til udførelse af en subkronisk oral toksictetstest svarer til OECD TG 409 (1998).
1.1. INTRODUKTION
Efter at en akut eller gentagen 28-dages toksicitetstest er udført med henblik på primær vurdering af et kemisk stofs toksicitet, kan man udføre en subkronisk oral toksicitetstest, hvor man anvender gentagne doser til bestemmelse og evaluering af stoffets toksiske egenskaber. Undersøgelsen på 90 dage skaffer viden om mulige trusler mod sundheden, som kan opstå ved gentagen belastning i en periode med hurtig vækst, modning og den første del af voksenlivet. Undersøgelsen skaffer viden om større toksiske virkninger, peger på, hvilke organer der rammes og muligheden for akkumulation; den kan give et overslag over det belastningsniveau, der kan være skadeligt, uden at det bemærkes, som igen kan anvendes til valg af dosis til undersøgelser af kronisk effekt og til fastlæggelse af sikkerhedskriterier for, hvad mennesker kan tåle.
Undersøgelsesmetoder muliggør, at man på dyrearter, der ikke er gnavere, finder frem til negative virkninger ved belastning med kemikalier, og den bør kun bruges:
|
— |
hvor virkninger i andre undersøgelser peger på nødvendigheden af afklaring/karakterisering i en anden dyreart end gnavere, eller |
|
— |
hvor toksiko-kinetiske undersøgelser peger på, at anvendelse af en speciel dyreart, der ikke er gnaver, er det mest relevante valg blandt laboratoriedyr, eller |
|
— |
hvor andre specielle grunde retfærdiggør anvendelsen af dyrearter, der ikke er gnavere. |
Se ligeledes den generelle indledning til del B.
1.2. DEFINITIONER
Dosis: Mængden af indgivet testkemikalium. Dosis angives som vægt (g, mg) eller som vægt af testkemikalium i forhold til dyrets vægt (f.eks. mg/kg) eller en konstant del af føden (ppm).
Dosering: Er et generelt udtryk for dosis, hyppighed af indgift og varigheden af indgiften.
NOAEL: Er en forkortelse for det niveau, der ikke medfører observeret skadelig virkning, og er det højeste dosisniveau, hvor ingen negative virkninger, der kan tilskrives behandlingen, er fundet.
1.3. TESTMETODENS PRINCIP
Testkemikaliet administreres peroralt dagligt i forskellige doser til forskellige grupper af forsøgsdyr, idet hver gruppe får en bestemt dosis i en periode på 90 dage. I denne periode observeres dyrene nøje for tegn på forgiftning. Dyr, der dør eller aflives, mens testen står på, bliver obduceret og ved undersøgelsens afslutning bliver resten af dyrene aflivet og obduceret.
1.4. BESKRIVELSE AF TESTMETODEN
1.4.1. Valg af dyreart
Den hyppigst anvendte ikke-gnaverdyreart er hunden, som bør være af en veldefineret race; beagle-hunde er ofte anvendt. Af andre arter bruges f.eks. også grise og minigrise. Primater anbefales ikke, og brug af dem skal forsvares. Der skal anvendes unge, sunde dyr, og for hunde bør belastningen begynde fortrinsvis ved 4- til 6-månedersalderen og ikke senere end ved 9-månedersalderen. Når der er tale om et primærforsøg efterfulgt af et langtidsforsøg med kronisk toksicitet, bør man anvende samme dyreart i begge forsøg.
1.4.2. Forberedelse af dyrene
Der anvendes sunde dyr, som er tilvænnet laboratorieforholdene og som ikke før har været anvendt til eksperimenter. Varigheden af tilvænning afhænger af den valgte dyreart og kilden. Mindst fem dage for hunde og grise, der er opdrættet til formålet og hører til i en stedlig koloni, men mindst to uger for disse dyr, hvis de kommer udefra. Dyrene beskrives hvad angår art, stamme, hvorfra de kommer (kilde), køn, vægt og/eller alder. Dyrene randomiseres til henholdsvis kontrol og behandlingsgruppe. Bure skal anbringes, så deres placering får mindst mulig betydning. Hvert dyr tildeles et eget, særligt identifikationsnummer.
1.4.3. Fremstilling af doser
Stoffet, der skal undersøges, kan indgives via føden eller drikkevandet, ved hjælp af sonde eller i kapsler. Måden at indgive på gennem munden afhænger af undersøgelsens formål og testmaterialets fysisk-kemiske egenskaber.
Om nødvendigt opløses eller opslæmmes stoffet i et passende medie. Hvor muligt anbefales det først at forsøge at anvende vandige opløsninger eller opslæmninger; dernæst overvejes opløsninger eller opslæmninger i olie (f.eks. majsolie) og først derefter andre opløsnings-/opslæmningsmidler. For andre midler end vand bør de toksiske egenskaber kendes. Testkemikaliets stabilitet i forbindelse med indgiften bør fastslås.
1.5. FREMGANGSMÅDE
1.5.1. Antal dyr og deres køn
Mindst otte dyr (fire af hvert køn) bør anvendes for hvert dosisniveau. Hvis man planlægger at aflive nogle dyr undervejs i forsøget, skal antallet af dyr, der påbegynder forsøget, forøges tilsvarende. Antallet af dyr ved forsøgets afslutning må være tilstrækkelig stort, så evalueringen af den toksiske virkning er mulig. Ud fra tidligere opnået viden om kemikaliet eller nære analoge stoffer bør man overveje en yderligere »satellit«-gruppe på otte dyr (fire af hvert køn) i kontrollen og i gruppen med den højeste dosis, således at man efter forsøgsperioden kan observere evt. reversibilitet eller vedvarende følger af behandlingen. Varigheden af denne observationsperiode afhænger af, hvilke virkninger man vil følge.
1.5.2. Dosisstørrelse
Mindst tre dosisstørrelser og en sideløbende kontrol bør anvendes, undtagen i tilfælde af en begrænset test (se 1.5.3). Dosisstørrelsen kan afhænge af tidligere tilsvarende forsøg eller forsøg med henblik på opsøgning af dosisgrænser og skal tage hensyn til kendte toksikologiske og toksokinetiske data for testkemikaliet eller tilsvarende stoffer. Medmindre kemikaliets fysisk-kemiske natur eller biologiske effekt sætter grænser, bør den højeste dosisstørrelse vælges med henblik på toksisk virkning, men ikke på død eller alvorlig lidelse. Faldende dosisstørrelse bør vælges med henblik på at påvise en eventuel dosisrelateret virkning og det niveau, der ikke medfører observeret skadelig virkning (NOAEL). To til firedobbelte intervaller er ofte bedst til fastlæggelse af faldende dosisstørrelser, og tilføjelse af en fjerde testgruppe er ofte at foretrække frem for brug af meget store intervaller (f.eks. mere end en faktor på 6-10) mellem dosisstørrelserne.
Kontrolgruppen skal være en ubehandlet gruppe eller en gruppe, der får det eventuelle medie, i hvilket de behandlede dyr får kemikaliet. Bortset fra behandlingen skal dyrene i kontrolgruppen behandles nøjagtigt som dyrene i testgruppen. Hvis kemikaliet indgives i et medie, skal kontrolgruppen have mediet i den størst anvendte mængde. Hvis testkemikaliet indgives via foderet, og hvis dette medfører reduceret fødeindtagelse, kan det være nyttigt med en gruppe, der parallelfodres mængdemæssigt, således at et eventuelt vægttab kan tilskrives, at foderet har en dårlig smag, respektive virker toksisk.
Man bør overveje mediets eller eventuelle additivers egenskaber med hensyn til absorption, distribution, stofskifte eller retention af testkemikalium; virkningen på testkemikaliets kemiske egenskaber, som igen kan have indflydelse på dets toksiske egenskaber; og indflydelsen på føde- eller vandindtag eller dyrenes ernæringsmæssige tilstand.
1.5.3. Grænse-test
Et totalforsøg med tre dosisstørrelser behøves næppe udført, hvis man ikke forventer nogen toksisk effekt ud fra kendskab til strukturelt beslægtede stoffer, og hvis der ikke ses nogen skadelig virkning ved forsøg som beskrevet ovenfor med enkelt dosis svarende til mindst 1 000 mg/kg legemsvægt/dag. Grænsetest er konklusive, undtagen hvis belastning af mennesker indikerer nødvendigheden af anvendelse af en større dosis.
1.5.4. Dosisadministration
Dyrene får test-kemikaliet syv gange ugentlig hver uge i 90 dage. Ethvert andet regime, f.eks. fem gange ugentlig, skal begrundes. Hvis testkemikaliet indgives med sonde, skal det gøres i en enkelt dosis, med anvendelse af en mavesonde eller passende intubationsrør. Det maksimale væskevolumen, der kan tilføres ad gangen, afhænger af dyrets størrelse. Normalt bør volumen holdes så lavt som muligt. Undtaget irritative og ætsende stoffer, som normalt vil have øget generende effekt ved større koncentrationer, bør variationen i testvolumen begrænses ved justering af koncentrationen af testkemikaliet for at sikre et konstant volumen ved alle dosisstørrelser.
Det er vigtigt at sikre, at de kvantiteter af kemikalier, som indtages via føden eller drikkevandet, ikke får indflydelse på den normale ernæring eller vandbalancen. Når testkemikaliet tilføres med føden, kan man tilføre det enten som en konstant del af føden (ppm) eller en konstant dosisstørrelse i forhold til dyrets vægt; det, man vælger, skal specificeres. Tilføres kemikaliet via sonde eller kapsel, bør det gøres på samme tidspunkt hver dag, og dosisstørrelse skal rettes til, så den udgør en konstant størrelse i forhold til dyrets vægt. Hvis et 90-dages forsøg foretages som forløber for et langtids kronisk-toksicitetsforsøg, skal der i begge forsøg gives samme fødesammensætning.
1.5.5. Observationer
Observationsperioden skal mindst vare 90 dage. Dyrene i »satellit«-gruppen, hvor man vil foretage »follow-up« -observationer, skal beholdes i en passende periode uden behandling, med henblik på konstatering af fortsat eller ophør af toksisk virkning.
Almindelig klinisk observation bør foretages mindst en gang om dagen, fortrinsvis på samme tidspunkt(er), idet man tager i betragtning, hvornår maksimal effekt kan forventes efter indgift. Dyrenes kliniske tilstand bør noteres. Mindst to gange daglig, sædvanligvis morgen og aften, undersøges alle dyrene for tegn på morbiditet eller mortalitet.
I hvert fald før første belastning (for at tage højde for individuelle forskelle) og en gang ugentlig derefter bør detaljerede kliniske undersøgelse foretages af alle dyrene. Disse undersøgelser skal foretages, hvor det er praktisk muligt, uden for burene, på et standardareal og fortrinsvis på samme tid i hvert tilfælde. Man bør bestræbe sig på. at variationerne i observationsbetingelserne er mindst mulige. Blandt de fund, der skal noteres (men ikke som de eneste), er ændringer i hud, pels, øjne, slimhinder, forekomst af sekretion og ekskreter og autonom aktivitet (f.eks. tåreflåd, hårrejsning, pupilstørrelse, usædvanligt respirationsmønster). Forandringer i gang, stilling og respons på håndtering såvel som tilstedeværelse af toniske og kloniske bevægelser, stereotypi (f.eks. overdreven pudsning, repetitive cirkelbevægelser) eller bizar opførsel skal også noteres.
Oftalmologisk undersøgelse ved hjælp af oftalmoskop eller andet passende udstyr bør udføres, før test-kemikaliet tilføres første gang og ved afslutning af forsøget, fortrinsvis på alle dyrene, men i hvert fald på dem, der fik højeste dosis og på kontrollerne. Hvis der opdages øjenforandringer. der kan tilskrives behandlingen, skal alle dyrene undersøges.
1.5.5.1. Kropsvægt og føde/vandindtagelse
Alle dyr skal vejes mindst en gang ugentlig. Mængden af fødeindtagelse skal beregnes mindst en gang ugentlig. Hvis test-kemikaliet administreres via drikkevandet, skal vandindtagelsen beregnes mindst en gang ugentlig. Vandforbruget bør også tages i betragtning, når belastningen foretages via føden eller via sonde, da drikkeaktiviteten kan ændres herved.
1.5.5.2. Hæmalologi og klinisk biokemi
Stedet, hvor blodprøver tages, bør angives, og de bør gemmes på passende vis. Ved forsøgets afslutning tages der prøver lige før eller i forbindelse med aflivningen af dyrene.
Hæmatologi, inkluderende hæmatokrit, hæmoglobinkoncentration, tælling af røde og hvide blodlegemer, differentialtælling, trombocyttælling og et mål for koagulationsevnen, som f.eks. koagulationstid, protrombintid eller tromboplastintid, bør måles før forsøget påbegyndes, siden hver måned eller midtvejs i forsøget og endelig ved forsøgets afslutning.
Klinisk kemiske målinger med henblik på undersøgelse af toksisk virkning på væv og specielt på nyrer og lever skal foretages på blodprøver taget fra alle dyrene ved forsøgets begyndelse, siden enten hver måned eller midtvejs i forsøget og til sidst ved forsøgets afslutning. Man bør måle elektrolytbalancen, kulhydratstofskiftet og lever- og nyrefunktionen. Valg af særlige målinger vil afhænge af testkemikaliets virkningsmåde. Dyrene bør faste, afhængig af dyrearten, før blodprøvetagningen. Det foreslås, at man måler calcium, fosfor, klorid, kalium, natrium, fastende glukose, alanin aminotransferase, aspartat aminotransferase, ornitin decarboksylase, gammaglutamyl transpeptidase, carbamid, albumin, serumkreatinin, total bilirubin og total serumprotein.
Urinanalyser bør udføres i hvert fald ved begyndelsen, midtvejs og ved afslutningen af forsøget, med opsamling af timediuresen. Analyserne inkluderer udseende, volumen, osmolalitet eller vægtfylde, pH, protein, glukose og blod/blodceller. Andre parametre kan inkluderes, hvor man finder det nødvendigt at udvide undersøgelsen af en eller flere observerede virkninger.
Herudover bør man overveje måling af markører for generel vævsskade. Andre målinger, som kan være nødvendige for en tilstrækkelig toksikologisk vurdering, inkluderer analyser af lipider, hormoner, syre/base-balancen, methæmoglobin, og hæmning af kolinesterasen. Hvor det skønnes nødvendigt, kan andre biokemiske målinger inddrages for at udvide undersøgelsen af observerede virkninger. Disse bør måles i forbindelse med kemikalier af særlige typer eller fra tilfælde til tilfælde.
Almindeligvis må man have en fleksibel indstilling ud fra dyrearten og de observerede og/eller forventede virkninger af et givet kemikalium.
1.5.5.3. Makroskopisk obduktion
Alle dyr, som indgår i forsøget, skal have udført en fuldstændig, detaljeret makroskopisk obduktion, hvilket inkluderer omhyggelig undersøgelse af alle overflader, alle åbninger, og kraniets, brystkassens og abdomens kaviteter og deres indhold. Lever med galdeblære, nyrer, binyrer, testes, bitestikler, ovarier, uterus, tyreoidea med paratyreoideae. thymus, milt hjerne og hjerte skal på alle dyr renses for andet vedhængende væv som behørigt (undtaget er de dyr, der findes moribunde og/eller aflives undervejs i forsøget) og vejes snarest muligt efter dissektionen for at undgå udtørring.
Følgende væv skal præserveres ved hjælp af det mest passende fiksationsmedie både med henblik på vævstypen og den følgende histopatologiske undersøgelse: alle større læsioner, hjernen (repræsentative regioner, inklusive cerebrum, cerebellum og medulla/pons), rygmarven (fra tre niveauer: cervikal, midt-torakal og lumbal), hypofyse, øjne. tyreoidea, paratyreoideae, tymus, øsofagus, spytkirtlerne, mavesækken, tynd- og tyktarm, (inklusive de Peyerske plaques) lever, galdeblære, pankreas, nyrer, binyrer, milt, hjerte, luftrør og lunger, aorta, gonader, uterus, sekundære kønsorganer, brystkirtlerne fra hundyrene, prostata, urinblære, lymfeknuder (helst en lymfeknude, som dækker belastningsvejen, og en anden langt derfra for at dække den systemiske virkning), perifer nerve (ischiadicus eller tibialis) helst nær ved musklen, en sektion af knoglemarven (og/eller frisk knoglemarvsaspirat), hud. Kliniske og andre fund kan foranledige undersøgelse af andet væv. Også særlige organer, der sandsynligvis kan være mål for testkemikaliets kendte virkninger, bør præserveres.
1.5.5.4. Histopatologi
En komplet histopatologisk undersøgelse skal udføres af de præserverede organer og væv på i hvert fald alle dyrene i kontrol- og højdosisgrupperne. Undersøgelsen skal omfatte alle dyrene i de andre gruppe også, hvis der observeres ændringer i højdosisgruppen, der kan tilskrives belastningen.
Alle større læsioner skal undersøges.
Hvis der anvendes en »satellit« -gruppe, skal der på dem udføres histopatologisk undersøgelse af væv og organer, på hvilke der var vist ændringer i den belastede gruppe.
2. DATA OG RAPPORTERING
2.1. DATA
Alle data skal anføres. Yderligere skal alle data opsummeres i tabeller, der for hver testgruppe viser antal dyr ved forsøgets begyndelse, antal dyr fundne døde i løbet af testen eller aflivede af humane grunde og tidspunktet for deres død, det antal dyr, der udviser tegn på forgiftning, en beskrivelse af disse tegn inklusive tidspunktet for deres begyndelse, deres varighed og forgiftningsgraden: det antal dyr. der har læsioner, typen af læsioner og den procentdel af dyrene, der udviser de forskellige typer af læsion.
Hvor det er muligt, skal tallene evalueres ved hjælp af en passende og accepteret statistisk metode. Den statistiske metode og de data, der skal analyseres med den, skal vælges under planlægningen af forsøget.
2.2. TESTRAPPORT
Testrapporten skal inkludere følgende informationer:
2.2.1. Testkemikalium:
|
— |
fysiske egenskaber, renhedsgrad og fysisk-kemiske egenskaber |
|
— |
identifikationsdata |
|
— |
indgivelsesmåde, som begrundes, hvis der ikke er tale om drikkevand. |
2.2.2. Dyreart:
|
— |
anvendte art og stamme |
|
— |
antal dyr, deres alder og køn |
|
— |
kilde, opbevaringsforhold, føde osv. |
|
— |
de enkelte dyrs vægt ved forsøgets begyndelse. |
2.2.3. Forsøgsbetingelser:
|
— |
rationale for valg af dosismængde |
|
— |
detaljer vedrørende test-kemikaliets form/opblanding i føden, resulterende koncentration, stabilitet og homogenitet |
|
— |
detaljer vedrørende indgift af testkemikaliet |
|
— |
sande dosis (mg/kg kropsvægt/dag) og omregningsfaktor fra testkemikaliets koncentration i føde/vand (ppm) til sand dosis, hvis muligt |
|
— |
detaljer vedrørende kvalitet af vand og føde. |
2.2.4. Resultater:
|
— |
kropsvægt og ændringer i kropsvægt |
|
— |
fødeindtagelse og indtagelse af vand, om muligt |
|
— |
data vedrørende forgiftning sat over for køn og dosis, inklusive forgiftningstegnene |
|
— |
art, alvor og varighed af det klinisk observerede (om det observerede er reversibelt) |
|
— |
oftalmologisk undersøgelse |
|
— |
hæmatologiske målinger med relevante udgangsværdier |
|
— |
klinisk-biokemiske målinger med relevante udgangsværdier |
|
— |
slutvægt, organernes vægt og organ/kropsvægtforholdene |
|
— |
sektionsfund |
|
— |
en detaljeret beskrivelse af de histopatologiske fund |
|
— |
data for absorption, hvis de forefindes |
|
— |
statistisk behandling af resultaterne, hvor rimeligt |
Diskussion af resultater
Konklusioner
B.28. SUBKRONISK TOKSICITET, OPTAGELSE GENNEM HUDEN — 90 DAGES GENTAGEN DOSERING PÅ HUD UNDER ANVENDELSE AF GNAVERE
1. METODE
1.1. INDLEDNING
Se den generelle indledning til del B.
1.2. DEFINITIONER
Se den generelle indledning til del B.
1.3. REFERENCESTOFFER
Ingen.
1.4. PRINCIP FOR TESTMETODEN
Teststoffet påføres dagligt huden på grupper af forsøgsdyr, idet der gives en bestemt gradueret dosis til hver gruppe gennem en periode på 90 dage. Dyrene observeres dagligt i forsøgsperioden. Eventuelle tegn på toksicitet registreres. De dyr, der dør under forsøget eller aflives ved afslutningen af forsøget, obduceres.
1.5. KVALITETSKRITERIER
Ingen.
1.6. BESKRIVELSE AF TESTMETODEN
1.6.1. Forberedelser
Dyrene holdes under forsøgsbetingelserne med hensyn til miljø og fodring i mindst fem dage inden forsøget påbegyndes. Unge, sunde, dyr fordeles randomiseret i grupper. Kort før forsøget indledes, klippes pelsen på ryggen på forsøgsdyrene. Såfremt pelsen barberes bort, bør det ske ca. 24 timer, inden forsøget indledes. Klipningen eller barberingen må som regel gentages med en uges mellemrum. Man må ved klipning eller barbering passe på ikke at beskadige huden. Mindst 10 % af dyrets overflade skal kunne anvendes til påføring af teststoffet. Ved afgørelse af, hvilket og hvor stort et område, der skal klippes, bør der tages hensyn til dyrets vægt. Ved testning af faste stoffer, som — hvis det er hensigtsmæssigt — kan pulveriseres, skal teststoffet fugtes tilstrækkeligt med vand eller om nødvendigt med et passende vehikel for at sikre god kontakt med huden. Flydende teststoffer påføres normalt ufortyndet. Der skal anvendes daglig applikation fem til syv dage ugentligt.
1.6.2. Forsøgsbetingelser
1.6.2.1.
Der kan anvendes voksne rotter eller kaniner. Der kan anvendes andre dyrearter, men i så fald skal dette være begrundet. Der benyttes dyr af almindeligt anvendte stammer, For begge køn af forsøgsdyr gælder det, at variationen i deres vægt ikke må overskride ± 20 % af en rimelig gennemsnitsvægt. Hvis en undersøgelse af subkronisk toksicitet udføres som et forstudium til en langtidsundersøgelse, bør samme dyreart og stamme anvendes i begge undersøgelser.
1.6.2.2.
Der anvendes mindst 20 dyr (10 hunner og 10 hanner) med sund hud til hvert dosisniveau. Hunnerne må ikke have født og må ikke være drægtige. Hvis der påregnes aflivning af dyr under forsøget, skal det oprindelige antal dyr forøges med det antal dyr, man påregner at aflive i løbet af forsøget. Man kan samtidig udsætte en satellitgruppe på 20 dyr (10 af hvert køn) for den højeste koncentration i 90 dage og gennem en periode på 28 dage efter forsøget observere, om der er reversible, varige eller sent opståede toksiske virkninger.
1.6.2.3.
Der skal anvendes mindst tre dosisniveauer samt en kontrolgruppe eller en vehikelkontrolgruppe, såfremt der benyttes vehikel. Eksponeringsperioden er på mindst seks timer dagligt. Teststoffet påføres på omtrent samme tidspunkt hver dag, og doserne justeres med jævne mellemrum (én gang eller to gange om ugen), så de er konstante i forhold til dyrenes legemsvægt. Dyrene i kontrolgruppen behandles nøjagtigt som dyrene i forsøgsgrupperne, bortset fra påføring af teststoffet. Hvis der anvendes et vehikel til at lette doseringen, skal vehikelkontrolgruppen eksponeres på samme måde som forsøgsgrupperne og påføres samme mængde vehikel som den gruppe, der modtager den højeste dosis. Den højeste dosis bør have toksisk virkning, men kun medføre få eller ingen dødsfald. Det laveste dosisniveau bør ikke have nogen toksisk virkning overhovedet. Hvis der foreligger en anvendelig vurdering af menneskers eksponering for stoffet, skal det laveste dosisniveau overskride denne eksponering. Middeldosis skal helst frembringe så svage observerbare toksiske virkninger som muligt. Hvis der anvendes mere end tre dosisniveauer, bør de vælges således, at der opnås graduering af de toksiske virkninger. I grupperne med lav og middel dosis og i kontrolgrupperne skal dødeligheden holdes på et lavt niveau, så der bliver mulighed for en meningsfuld vurdering af resultaterne.
Hvis teststoffet forårsager alvorlig hudirritation, bør koncentrationen sænkes, og der kan derved ske en reduktion eller et fuldstændigt bortfald af de øvrige toksiske effekter på den højeste dosis. Hvis huden er blevet alvorligt beskadiget, kan det være nødvendigt at afbryde forsøget og foretage et nyt forsøg med en lavere koncentration.
1.6.3.
Hvis et dosisniveau på 1 000 mg/kg legemsvægt eller et højere dosisniveau fastsat ud fra kendskabet til den eventuelle eksponering, mennesker udsættes for, ikke giver nogen toksisk virkning, kan yderligere forsøg betragtes som unødvendige.
1.6.4.
Dyrene observeres dagligt, og det registreres, om der optræder tegn på toksicitet. Dødstidspunkt og tidspunkt for toksicitetstegns optræden og forsvinden skal registreres.
1.6.5. Fremgangsmåde
Dyrene skal holdes i hvert sit bur. Den ideelle doseringer påføring af teststoffet syv dage om ugen i en periode på 90 dage.
Eventuelle satellitgrupper beregnet til efterfølgende observationer bør holdes under observation i yderligere 28 dage, efter at påføringen af teststoffet er afsluttet, således at man har mulighed for at registrere, om toksiske virkninger er reversible eller om de persisterer. Ekspositionsperioden skal være seks timer dagligt.
Teststoffet påføres jævnt over et område, som svarer til ca. 10 % af den samlede kropsoverflade. Med stærkt toksiske stoffer kan der anvendes et mindre område, men stoffet bør påføres så tyndt og ensartet og over så stor en del af området som muligt.
Teststoffet holdes i kontakt med huden ved hjælp af en porøs gazeforbinding og en ikke-hudirriterende klæbestrimmel. Teststedet skal desuden dækkes på en sådan måde, at gazeforbindingen og teststoffet holdes sikkert fast, så det sikres, at dyrene ikke indtager teststoffet. Der kan anvendes andre foranstaltninger, som forhindrer dyrene i at indtage teststoffet, men man bør ikke immobilisere dem fuldstændigt.
Ved eksponeringsperiodens slutning fjernes eventuelt tilbagesiddende teststof, idet der anvendes vand eller en anden egnet metode til at rense huden.
Alle dyr observeres dagligt, og det registreres, om der optræder tegn på toksicitet, hvornår disse tegn optræder første gang, hvor længe de varer, og hvor kraftige de er. Inspektion af dyrene skal omfatte forandringer i hud og pels, øjne og slimhinder, åndedræt og kredsløb, det autonome og centrale nervesystem, den motoriske aktivitet og adfærdsmønstret. Der holdes ugentligt regnskab med foderindtagelsen, og dyrene vejes en gang om ugen. Dyrene observeres med jævne mellemrum med henblik på, at det mindst mulige antal dyr går tabt for undersøgelsen på grund af f.eks. kannibalisme, autolyse af væv eller forveksling. Ved forsøgets afslutning aflives og obduceres alle tilbageværende dyr med undtagelse af satellitgrupperne. Døende dyr skal fjernes og obduceres, så snart de registreres.
Alle dyr inklusive kontrolgruppen underkastes normalt følgende undersøgelser:
|
a) |
Oftalmologisk undersøgelse med anvendelse af et oftalmoskop eller lignende passende udstyr skal foretages inden påføring af teststoffet og ved undersøgelsens afslutning. Det må foretrækkes, at alle dyr undersøges, men sker dette ikke, undersøges mindst den gruppe, der påføres den højeste dosis og kontrolgruppen. Hvis der observeres forandringer i øjnene, skal alle dyrene undersøges. |
|
b) |
Hæmatologiske undersøgelser omfattende hæmatokrit, hæmoglobinkoncentration, erytrocyttælling, total og differential leukocyttælling, måling af koaguleringsevne f.eks. koagulationstid, protrombintid, tromboplastintid eller blodpladetælling skal foretages ved forsøgets afslutning. |
|
c) |
Klinisk biokemisk analyse af blod skal foretages ved afslutningen af forsøget. Relevante analyseområder for alle forsøg er elektrolytbalance, kulhydratstofskifte, lever- og nyrefunktion. Valg af specifikke test afhænger af observationer af teststoffets virkning. Følgende bestemmelser kan foreslås: kalcium, fosfor, klorid, natrium, kalium, fasteglukose (fasteperioden vil afhænge af dyrearten), serum alanin aminotransferase (ALAT), serum aspartat aminotransferase (ASAT), ornitin decarboxylase, gamma glutamyl transpeptidase, urinstof, albumin, kreatinin, total bilirubin og total serumprotein. Andre bestemmelser, der kan være påkrævede i en adækvat toksikologisk vurdering, omfatter lipidanalyser, hormonanalyser, syre/basebalance, methæmoglobin, cholinesteraseaktivitet. Der kan anvendes yderligere klinisk-biokemisk analyse, hvor det skønnes nødvendigt for en videre undersøgelse af de observerede virkninger. |
|
d) |
Rutinemæssig urinanalyse er ikke påkrævet, medmindre den indiceres af forventede eller observerede toksiske virkninger. |
Hvis historiske basaldata er inadækvate, skal man gøre sig overvejelser over anvendelsen af hæmatologiske og klinisk biokemiske parametre, inden forsøget begynder.
Alle forsøgsdyrene skal underkastes en fuldstændig makroskopisk vurdering, der omfatter undersøgelse af kropsoverflade, alle kropsåbninger og kranie-, thorax- og abdominalhulerne og deres indhold. Lever, nyrer, binyrer og testikler vejes så hurtigt som muligt efter dissektion for at undgå udtørring, Følgende væv og organer skal opbevares i et passende medium med henblik på mulig senere histopatologisk undersøgelse: alle væv og organer, som udviser læsioner, hjerne, herunder snit af medulla/pons, den cerebellare og cerebrale cortex, hypofyse, skjoldbrusk/biskjoldbruskkirtel, thymusvæv, (trakea), lunger, hjerte, aorta, spytkirtler, lever, milt, nyrer, binyrer, bugspytkirtel, kønskirtler, uterus, assesoriske kønskirtler, galdeblære (hvis den findes), spiserør, mavesæk, duodenum, jejunum, ileum, coecum, tyktarm, endetarm, urinblære, en repræsentativ lymfeknude, (kvindelig brystkirtel), (lårmuskulatur), perifer nerve, (øjne), (sternum med benmarv), (femur inklusive ledflade), (rygmarv i tre forskellige snit — cervikalt, midt for thorax og lumbalt) og (exorbitale tårekirtler). Væv, der er nævnt i parentes, behøver kun undersøges, hvis der er tale om toksicitetstegn eller kan sættes i forbindelse med målorganer.
|
a) |
Der skal foretages en fuldstændig histopatologisk undersøgelse af organer og væv fra dyrene i kontrolgruppen og i den gruppe, der har fået påført den højeste dosis. |
|
b) |
Alle alvorlige læsioner skal undersøges. |
|
c) |
Målorganer skal også undersøges i øvrige doserede grupper. |
|
d) |
Når der anvendes rotter, undersøges lungerne fra dyrene i den lavest doserede og de middeldoserede grupper histopatologisk med henblik på konstatering af tegn på infektion, idet dette giver en egnet vurdering af dyrenes helbredstilstand. Der er ikke rutinemæssigt behov for yderligere histopatologiske undersøgelser af dyrene i disse grupper, men organer, som viser tegn på læsioner i den gruppe, der har fået påført den højeste dosis, skal altid undersøges. |
|
e) |
Dyrene i en eventuel satellitgruppe skal underkastes en histopatologisk undersøgelse af de væv og organer, der udviser toksisk påvirkning i de øvrige grupper. |
2. DATA
Data opstilles i tabelform, som for hver gruppe viser antal dyr ved begyndelsen af forsøget, antal dyr med læsioner og for hver type af læsioner den procentdel dyr, der udviser den pågældende læsion, Evaluering af resultaterne skal baseres på en passende statistisk metode. Alle anerkendte statistiske metoder kan anvendes.
3. RAPPORTERING
3.1. FORSØGSRAPPORT
Forsøgsrapporten skal om muligt indeholde følgende:
|
— |
dyreart, stamme, herkomst, miljøbetingelser, foder osv. |
|
— |
forsøgsbetingelser |
|
— |
dosisniveauer (med angivelse af vehikel, hvor et sådant er anvendt) samt koncentration |
|
— |
oplysninger om toksisk reaktion inddelt efter køn og dosis |
|
— |
ikke toksisk dosis, hvis en sådan er konstateret |
|
— |
dødstidspunkt under forsøget eller overlevelse af forsøgsdyr til forsøgets slutning |
|
— |
toksiske eller andre virkninger |
|
— |
observationstidspunkt for hvert enkelt abnormitetstegn og den senere udvikling af abnormiteten |
|
— |
data for foder og vægt |
|
— |
oftalmologiske fund |
|
— |
hæmatologiske undersøgelser og samtlige resultater |
|
— |
klinisk biokemiske analyser og samtlige resultater (herunder resultater af urinanalyse) |
|
— |
obduktionsfund |
|
— |
detaljeret beskrivelse af alle histopatologiske fund |
|
— |
eventuel statistisk behandling af resultaterne |
|
— |
diskussion af resultaterne |
|
— |
fortolkning af resultaterne. |
3.2. VURDERING OG FORTOLKNING
Se den generelle indledning til del B.
4. HENVISNINGER
Se den generelle indledning til del B.
B.29. SUBKRONISK TOKSICITET, INHALATION — 90 DAGES GENTAGEN INHALATION UNDER ANVENDELSE AF GNAVERE
1. METODE
1.1. INDLEDNING
Se den generelle indledning til del B.
1.2. DEFINITIONER
Se den generelle indledning til del B.
1.3. REFERENCESTOFFER
Ingen.
1.4. PRINCIP FOR TESTMETODEN
Flere grupper af forsøgsdyr eksponeres dagligt i en fastlagt periode for teststoffet, idet hver gruppe eksponeres for en bestemt koncentration i en periode på 90 dage. Anvendes der et vehikel til at frembringe en passende koncentration af teststoffet, bør der i forsøget indgå en kontrolgruppe, der udsættes for vehiklet alene. Dyrene observeres dagligt for toksicitetstegn. Dyr, der dør under forsøget, eller som aflives ved forsøgets slutning, obduceres.
1.5. KVALITETSKRITERIER
Ingen.
1.6. BESKRIVELSE AF TESTMETODEN
1.6.1. Forberedelser
Dyrene holdes under forsøgsbetingelser med hensyn til miljø og fodring i mindst fem dage, før forsøget påbegyndes. Unge, sunde, dyr fordeles randomiseret i det nødvendige antal grupper. Der kan anvendes et vehikel til at frembringe en passende koncentration af teststoffet i luften. Hvis der anvendes et vehikel eller andre tilsætningsstoffer med henblik på at lette doseringen, skal det være påvist, at de ikke har nogen toksisk virkning. Her kan tidligere opsamlede data benyttes.
1.6.2. Forsøgsbetingelser
Rotter er den foretrukne dyreart, medmindre der foreligger kontraindikationer. Der benyttes unge, sunde rotter af almindeligt anvendte laboratoriestammer. Ved forsøgets begyndelse må vægten af de anvendte dyr ikke variere mere end ± 20 % af gennemsnitsvægten. Hvis en undersøgelse af subkronisk toksicitet ved inhalation udføres som et forstudium til en langtidsundersøgelse, bør samme dyreart og stamme anvendes i begge undersøgelser.
Der anvendes mindst 20 dyr (10 hun- og 10 handyr) til hver dosisniveau. Hundyrene må ikke have født og må ikke være drægtige. Hvis der påregnes aflivning af dyr under forsøget, bør det oprindelige antal dyr forøges med det antal dyr, man påregner at aflive i løbet af forsøget. Man kan samtidig eksponere en satellitgruppe på 20 dyr (10 af hvert køn) for det højeste koncentrationsniveau gennem 90 dage og observere, hvorvidt virkninger i en periode på 28 dage efter forsøget er reversible, persisterende eller optræder forsinket.
Der skal anvendes mindst tre forskellige koncentrationsniveauer samt en kontrolgruppe eller en vehikelkontrolgruppe (svarende til den højeste koncentration af vehikel), såfremt der benyttes et vehikel. Dyrene i kontrolgruppen behandles nøjagtigt som dyrene i forsøgsgrupperne bortset fra eksponering for teststoffet. Den højeste koncentration bør have toksisk effekt, men ikke forårsage dødelighed eller kun en meget begrænset dødelighed. Hvis der foreligger en anvendelig vurdering af menneskers eksponering for stoffet, bør den laveste koncentration overskride denne eksponering. Ideelt set bør middelkoncentrationen frembringe de mindste observerbare toksiske effekter. Hvis der anvendes mere end en middelkoncentration, bør de vælges således, at der opnås en graduering af den toksiske virkning. I den lavt doserede og middeldoserede gruppe og i kontrolgruppen bør dødeligheden holdes på et lavt niveau for at give mulighed for en meningsfuld vurdering af resultaterne.
Den daglige eksponeringsperiode er seks timer, efter at koncentrationerne i kamrene er ækvilibreret. Andre perioder kan anvendes, hvis undersøgelsen skal imødekomme særlige krav.
Dyrene skal testes med inhalationsapparatur, der er således udformet, at der kan opretholdes en dynamisk luftgennemstrømning på mindst 12 luftskift pr. time, samtidig med at der sikres et adækvat iltindhold og en jævnt fordelt ekspositionsluft. Benyttes der et kammer, bør det være således indrettet, at der undgås sammenklumpning af forsøgsdyrene og sikres en så stor inhalation af teststoffet som muligt. Den generelle regel er, at hvis der skal sikres en stabil atmosfære, må dyrenes totale »volumen« ikke overstige 5 % af ekspositionskammerets volumen. Eksposition kan foregå gennem mund-næse, hovedet alene eller ved kamre til hele kroppen. Ved anvendelse af de to første muligheder opnår man, at indtagelse af teststoffet ad andre veje reduceres.
Alle dyr observeres dagligt for tegn på toksicitet under hele forsøget og under restitutionsperioden. Dødstidspunkt og toksicitetstegns optræden og forsvinden registreres.
1.6.3. Fremgangsmåde
Dyrene eksponeres for teststoffet dagligt i fem til syv dage om ugen i en periode på 90 dage. Eventuelle satellitgrupper beregnet til længerevarende observationer skal holdes under observation i yderligere 28 dage, efter at eksponering for teststoffet er afsluttet, således at man har mulighed for at registrere, om en toksisk virkning er reversibel eller persisterer. Temperaturen under forsøget bør holdes på 22 oC ± 3 oC. Under ideelle forhold skal den relative luftfugtighed holdes på mellem 30 % og 70 %, men i visse tilfælde (f.eks. ved testning af aerosoler) kan dette være praktisk uigennemførligt. Der gives hverken foder eller vand under ekspositionen.
Der anvendes et dynamisk inhalationssystem med et passende analytisk kontrolsystem til sikring af ekspositionskoncentration. Det anbefales at foretage en prøvetest for at finde frem til passende ekspositionskoncentrationer. Lufthastigheden tilpasses, således at der er ensartede forhold i hele ekspositionskammeret. Systemet bør være af en sådan beskaffenhed, at der så hurtigt som muligt opnås stabile ekspositionsbetingelser.
Der skal foretages måling og kontrol af:
|
a) |
Lufthastighed (kontinuerlig). |
|
b) |
Den faktiske koncentration af teststoffet målt i vejrtrækningszonen. Koncentrationen må under den daglige eksponeringsperiode ikke variere mere end ± 15 % fra gennemsnitskoncentrationen. Det kan imidlertid være umuligt at opnå en sådan grad af kontrol med støvpartikler og aerosoler, og i så tilfælde kan større udsving accepteres. Koncentrationerne bør fra dag til dag holdes så konstante som muligt under hele forsøgets forløb. Under indkøring af udstyret bør der foretages analyser af partikelstørrelse for at fastslå aerosolkoncentrationernes stabilitet. Under eksponering skal der foretages analyser så ofte, som det er nødvendigt for at fastslå, om partikelstørrelsen er jævnt fordelt. |
|
c) |
Temperatur og luftfugtighed. |
|
d) |
Under og efter eksposition foretages der systematiske observationer, som registreres for hvert enkelt dyr. Alle dyr inspiceres dagligt, og det registreres, om der optræder tegn på toksicitet, hvornår disse tegn optræder første gang, hvor længe de varer, og hvor kraftige de er. Inspektionen af dyrene skal omfatte forandringer i hud og pels, øjne og slimhinder, åndedræt og kredsløb, det autonome og centrale nervesystem, den motoriske aktivitet og adfærdsmønstret. Der holdes ugentligt regnskab med foderindtagelsen, og dyrene vejes en gang om ugen. Dyrene observeres med jævne mellemrum med henblik på, at det mindst mulige antal dyr går tabt for undersøgelsen på grund af f.eks. kannibalisme, autolyse af væv eller forveksling. Ved forsøgets afslutning aflives og obduceres alle tilbageværende dyr med undtagelse af satellitgruppen. Døende dyr bør fjernes og obduceres, så snart de registreres. |
Alle dyr inklusive kontrolgruppen skal normalt underkastes følgende undersøgelser:
|
a) |
Oftalmologisk undersøgelse med anvendelse af et oftalmoskop eller lignende passende udstyr skal foretages inden eksposition for teststoffet og ved undersøgelsens afslutning. Det må foretrækkes, at alle dyr undersøges, men sker dette ikke, undersøges mindst den gruppe, der eksponeres for den højeste dosis og kontrolgruppen. Hvis der observeres forandringer i øjnene, bør alle dyrene undersøges. |
|
b) |
Hæmatologiske undersøgelser omfattende hæmotokrit, hæmoglobinkoncentration, erytrocyttælling; total og differential leukocyttælling, måling af koaguleringsevne f.eks. koagulationstid, protrombintid, tromboplastintid eller blodpladetælling skal foretages ved forsøgets afslutning. |
|
c) |
Klinisk-biokemisk analyse af blod skal foretages ved afslutningen af forsøget. Relevante analyseområder for alle forsøg er elektrolytbalance, kulhydratstofskifte, lever- og nyrefunktion. Valg af specifikke test afhænger af observationer af teststoffets virkning. Følgende bestemmelser kan foreslås: kalcium, fosfor, klorid, natrium, kalium, fasteglukose (fasteperioden vil afhænge af dyrearten), serum alanin aminotransferase (ALAT), serum aspartat aminotransferase (ASAT), ornitin, decarboxylase, gamma glutamyl transpeptidase, urinstof, albumin, blodkreatinin, total bilirubin og total serumprotein. Andre bestemmelser, der kan være påkrævede i en adækvat toksikologisk vurdering omfatter lipidanalyser, hormonanalyser, syre/basebalance, methæmoglobin, cholinesteraseaktivitet. Der kan anvendes yderligere klinisk-biokemisk analyse, hvor det skønnes nødvendigt for en videre undersøgelse af de observerede virkninger. |
|
d) |
Rutinemæssig urinanalyse er ikke påkrævet, medmindre den indiceres af forventede eller observerede toksiske virkninger. Hvis historiske basaldata er inadækvate, bør man gøre sig overvejelser over bestemmelse af hæmatologiske og klinisk-biokemiske parametre, inden forsøget begynder. |
Makroskopisk undersøgelse
Alle forsøgsdyrene skal underkastes en fuldstændig makroskopisk vurdering, der omfatter undersøgelse af kropsoverflade, alle kropsåbninger og kranie-, thorax- og abdominalhulerne og deres indhold. Lever, nyrer, binyrer og testikler vejes så hurtigt som muligt efter dissektionen for at undgå udtørring. Følgende væv og organer skal opbevares i et passende medium med henblik på mulig senere histopatologisk undersøgelse: alle væv og organer, som udviser læsioner, lunger, som bør udtages intakt, vejes og behandles med et passende fiksativ, således at lungestrukturen bevares (perfusion med fiksativet betragtes som en effektiv fremgangsmåde), væv fra næse og naso-farynx, hjerne, herunder snit af medulla/pons, den cerebellare og cerebrale cortex, hypofyse, skjoldbrusk/biskjoldbruskkirtel, thymusvæv, trakea, hjerte, aorta, spytkirtler, lever, milt, nyrer, binyrer, bugspytkirtel, kønskirtler, uterus, (assesoriske kønskirtler), (hud), galdeblære (hvis en sådan forefindes), spiserør, mavesæk, duodenum, jejunum, ileum, coecum, tyktarm, endetarm, urinblære, en repræsentativ lymfeknude, (kvindelig brystkirtel), (lårmuskulatur), perifer nerve, (øjne), sternum med benmarv, (femur inklusive ledflade), (rygmarv i tre forskellige snit — cervikalt midt for thorax og lumbalt) og (exorbitale tårekirtler). Væv, der er nævnt i parentes, behøver kun undersøges, hvis der er tale om toksicitetstegn eller kan sættes i forbindelser til målorganer.
|
a) |
Der skal foretages en fuldstændig histopatologisk undersøgelse af luftvejene og andre organer og væv fra dyrene i kontrolgruppen og i den gruppe, der eksponeres for den højeste dosis. |
|
b) |
Alle alvorlige læsioner skal undersøges. |
|
c) |
Målorganer skal også undersøges i øvrige doserede grupper. |
|
d) |
Lungerne fra dyrene i den lavest doserede og de middeldoserede grupper skal underkastes histopatologisk undersøgelse, eftersom dette giver en egnet vurdering af dyrenes helbredstilstand. Der er ikke rutinemæssigt behov for yderligere histopatologiske undersøgelser af dyrene i disse grupper, men organer, som viser tegn på læsioner i den gruppe, der eksponeres for den højeste dosis, skal altid undersøges. |
|
e) |
Dyrene i en eventuel satellitgruppe skal underkastes en histopatologisk undersøgelse af de væv og organer, der udviser toksisk påvirkning i de øvrige grupper. |
2. DATA
Data opstilles i tabelform, som for hver gruppe viser antal dyr ved begyndelsen af forsøget, antal dyr med læsioner og for hver type af læsioner den procentdel dyr, der udviser den pågældende læsion. Evaluering af resultaterne skal baseres på en passende statistisk metode. Alle anerkendte statistiske metoder kan anvendes.
3. RAPPORTERING
3.1. FORSØGSRAPPORT
Forsøgsrapporten skal om muligt indeholde følgende:
|
— |
dyreart, stamme, herkomst, miljøbetingelser, foder osv. |
|
— |
forsøgsbetingelser: Beskrivelse af ekspositionsapparatur, herunder udformning, type, dimensionering, luftkilde, system til frembringelse af partikler og aerosoler, metode til konditionering af luften, behandling af udpumpet luft og den måde, hvorpå dyrene holdes i et ekspositionskammer, når et sådant benyttes. Apparaturet til måling af temperatur, fugtighed og eventuelle partikulære aerosolers koncentration og partikelstørrelse beskrives. Ekspositionsdata: Disse skal opstilles i tabelform og angive middelværdier og variation (f.eks. standardafvigelser) og bør omfatte:
|
|
— |
oplysning om toksisk reaktion inddelt efter køn og dosis |
|
— |
ikke toksisk dosis, hvis en sådan er konstateret |
|
— |
dødstidspunkt under forsøget eller overlevelse til forsøgets slutning |
|
— |
toksiske eller andre virkninger |
|
— |
observationstidspunkt for hvert enkelt abnormitetstegn og den senere udvikling af abnormiteten |
|
— |
data for foder og vægt |
|
— |
oftalmologiske fund |
|
— |
hæmatologiske undersøgelser og samtlige resultater |
|
— |
klinisk-biokemiske analyser og samtlige resultater (herunder resultater af urinanalyse) |
|
— |
obduktionsfund |
|
— |
detaljeret beskrivelse af alle histopatologiske fund |
|
— |
eventuel statistisk behandling af resultaterne |
|
— |
diskussion af restultaterne |
|
— |
fortolkning af resultaterne. |
3.2. VURDERING OG FORTOLKNING
Se den generelle indledning til del B.
4. HENVISNINGER
Se den generelle indledning til del B.
B.30. UNDERSØGELSE AF KRONISK TOKSICITET
1. METODE
1.1. INDLEDNING
Se den generelle indledning til del B.
1.2. DEFINITIONER
Se den generelle indledning til del B.
1.3. REFERENCESTOFFER
Ingen.
1.4. PRINCIP FOR TESTMETODEN
Flere grupper af forsøgsdyr doseres normalt syv dage om ugen med teststof på passende måde. Der gives en dosis pr. gruppe i størstedelen af dyrenes levetid. Under og efter dosering med teststoffet observeres dyrene dagligt, og eventuelle tegn på toksicitet registreres.
1.5. KVALITETSKRITERIER
Ingen.
1.6. BESKRIVELSE AF TESTMETODEN
Forberedelser
Dyrene holdes under forsøgsbetingelser med hensyn til miljø og fodring i mindst fem dage, inden forsøget påbegyndes. Unge, sunde, dyr fordeles randomiseret i test- og kontrolgrupper.
Forsøgsbetingelser
Rotten er det foretrukne forsøgsdyr.
Andre dyrearter (gnavere eller ikke-gnavere) kan anvendes, hvis tidligere gennemførte undersøgelser giver grundlag herfor. Der bør benyttes unge, sunde dyr af almindeligt anvendte laboratoriestammer, og doseringen bør indledes så hurtigt som muligt, efter at dyrene er vænnet fra.
Ved forsøgets begyndelse må vægten af de anvendte dyr ikke variere mere end ± 20 % af gennemsnitsvægten. Hvis en undersøgelse af subkronisk toksicitet ved oral indgift udføres som et forstudium til en langtidsundersøgelse, bør samme dyreart og stamme anvendes i begge undersøgelser.
Hvis forsøgsdyrene er gnavere, anvendes mindst 40 dyr (20 hun- og 20 handyr) på hvert dosisniveau samt en kontrolgruppe. Hundyrene må ikke have født og må ikke være drægtige. Hvis der påregnes aflivning af dyr under forsøget, bør det oprindelige antal forøges med det antal dyr, man påregner at aflive i løbet af forsøget.
Ved benyttelse af andre dyr end gnavere kan der anvendes et mindre antal, dog mindst fire af hvert køn pr. gruppe.
Der anvendes mindst tre forskellige dosisniveauer ud over kontrolgruppen. Det højeste dosisniveau bør have tydelig toksisk effekt uden dog at forårsage voldsom dødelighed.
Det laveste dosisniveau bor ikke have nogen toksisk effekt overhovedet.
Middeldosis (evt. flere) bør ligge midtvejs mellem det højeste og det laveste dosisniveau.
Ved valget af dosisniveauer bør der tages hensyn til data fra tidligere toksicitetsundersøgelser.
Dyrene doseres normalt med teststoffet hver dag. Hvis stoffet administreres via drikkevandet eller med foderet, skal det være kontinuert tilgængeligt.
Der skal anvendes en kontrolgruppe, som på alle måder er identisk med forsøgsgrupperne bortset fra dosering med teststof.
Under særlige omstændigheder som f.eks. i inhalationsundersøgelser, hvor der benyttes aerosoler, eller hvis der benyttes en emulgator med ukendt biologisk virkning i undersøgelser med oral indgift, anvendes også en negativ kontrolgruppe. Den negative kontrolgruppe behandles som dyrene i forsøgsgrupperne, bortset fra at den ikke eksponeres hverken for teststoffet eller vehiklet.
De to hovedformer for administration af teststof er oral indgift og inhalation. Valget af administrationsform afhænger af teststoffets fysiske og kemiske karakteristika og den sandsynlige måde, hvorpå mennesker eksponeres for stoffet.
Der er betydelige praktiske problemer forbundet med at påføre teststoffet på huden. Hvis kronisk systemisk toksicitet er et resultat af perkutan absorption, kan dette normalt udledes af resultaterne af test med oral indgivelse, kombineret med et kendskab til graden af perkutan absorption på grundlag af tidligere perkutane toksicitetstest.
Hvis teststoffet absorberes i mave-tarmkanalen, og hvis mennesker kan udsættes for indtagelse gennem munden, anvendes oral indgift, medmindre der foreligger kontraindikationer. Dyrene indgives teststoffet med foderet, opløst i drikkevandet eller i kapsler. Den ideelle dosering er, at dyrene får indgivet teststoffet syv dage om ugen. Hvis indgift indskrænkes til fem dage om ugen, kan der ske tilbagevenden til normal, eller der kan optræde abstinenssymptomer imellem eksponeringsperioderne, hvilket kan indvirke på resultaterne og den senere vurdering. Med udgangspunkt i praktiske overvejelser kan det accepteres, at dyrene indgives teststoffet fem dage om ugen.
Inhalationsundersøgelser giver mere komplekse tekniske problemer end andre former for administration af teststof. I det følgende gives derfor en mere detaljeret vejledning i gennemførelse af disse undersøgelser. Man bør også være opmærksom på, at intratrakeal instillation under særlige omstændigheder kan være et egnet alternativ.
Forsøg med langtidseksposition udformes normalt på grundlag af erfaringer fra arbejdspladser, således at dyrene eksponeres fem dage om ugen seks timer dagligt, efter at koncentrationen i kammeret er ækvilibreret (intermitterende eksposition), eller der benyttes eksponering på grundlag af erfaringer fra miljøet, dvs. syv dage om ugen og 22 til 24 timer pr. dag (kontinuert eksposition) med ca. en time om dagen på et bestemt klokkeslet til fodring af dyrene og rengøring af kammeret.
I begge tilfælde eksponeres dyrene normalt for fastlagte koncentrationer af teststof. En vigtig forskel mellem intermitterende og kontinuert eksponering, som man må være opmærksom på, er, at der ved den førstnævnte metode er en periode på 17 til 18 timer, hvor dyrene kan overvinde virkningerne af den daglige eksposition og en endnu længere periode i weekenderne.
Valget mellem intermitterende og kontinuert eksponering afhænger af undersøgelsens formål og den menneskelige eksponering, der simuleres. Der er imidlertid en række tekniske vanskeligheder at tage i betragtning. F.eks. kan fordelene ved kontinuert eksponering, når det drejer sig om at simulere miljøbetingelser, falde væk på grund af behovet for vanding og fodring i ekspositionsperioden og behovet for mere komplicerede (og pålidelige) teknikker til generering og kontrol af aerosoler og dampe.
Dyrene testes i ekspositionskamre, der er således udformet, at der kan opretholdes en dynamisk luftstrøm på mindst 12 luftskift pr. time, samtidig med at der sikres et adækvat iltindhold og en jævnt fordelt ekspositionsluft. Kontrolkamre og ekspositionskamre bør være identiske med hensyn til konstruktion og udformning, så det sikres, at ekspositionsbetingelserne er sammenlignelige på alle måder bortset fra eksponering fra teststoffet. Der holdes normalt et let undertryk i kammeret for at forhindre, at teststoffet lækker ud i det omgivende rum. Kammeret indrettes således, at der undgås trængsel mellem forsøgsdyrene. Den generelle regel er, at hvis der skal sikres en stabil rumatmosfære, må dyrenes totale volumen ikke overstige 5 % af ekspositionskammerets volumen.
Der skal foretages måling og kontrol af:
|
i) |
Luftgennemstrømning: det må foretrækkes, at luftstrømmen gennem kammeret kontrolleres konstant. |
|
ii) |
Koncentrationen må i den daglige eksponeringsperiode ikke variere mere end ± 15 % af den gennemsnitlige værdi. Under hele forsøget bør koncentrationen fra dag til dag holdes så konstant som praktisk muligt. |
|
iii) |
Temperatur og fugtighed: ved anvendelse af gnavere holdes temperaturen på 22 oC (± 2 oC) og fugtigheden i kammeret mellem 30 og 70 %, undtagen når der anvendes vand til suspension af teststoffet i kammerets atmosfære. Det må foretrækkes, at både temperatur og fugtighed kontrolleres konstant. |
|
iv) |
Måling af partikelstørrelse: Hvis der anvendes atmosfærer med flydende eller faste aerosoler i kammeret, bør der foretages en beregning af partikelstørrelsens distribution. Aerosolpartiklerne skal være respirable for de anvendte forsøgsdyr. Der tages prøver af kammerets atmosfære i dyrenes vejrtrækningszone. Luftprøverne skal være repræsentative for den partikeldistribution, dyrene eksponeres for, og det skal gennem gravimetrisk analyse være muligt på grundlag af disse luftprøver at beskrive hele aerosolen, også selv om meget af den ikke er respirabel. Under indkøring af udstyret bør der foretages hyppige analyser af partikelstørrelsen for at sikre aerosolkoncentrationernes stabilitet. Under eksponering foretages analyser så ofte, som det er nødvendigt for en adækvat bestemmelse af, om den partikeldistribution, dyrene eksponeres for, er konstant. |
Ekspositionen bør vare i mindst 12 måneder.
Fremgangsmåde
Der foretages en grundig klinisk undersøgelse mindst en gang om dagen. Herudover bør dyrene med jævne mellemrum inspiceres, for at det mindst mulige antal dyr går tabt for undersøgelsen. Det vil sige, døde dyr obduceres eller nedfryses, og svage eller døende dyr isoleres eller aflives. Der foretages omhyggelig registrering af alle tegn på toksicitet, hvornår disse tegn optræder første gang, og hvor længe de varer, og det sikres, at det mindst mulige antal dyr går tabt for undersøgelsen på grund af sygdomme, autolyse af væv eller kannibalisme.
Hos alle dyr foretages registrering af kliniske symptomer, herunder neurologiske forandringer, forandringer i øjne og mortalitet. Toksicitetstegns optræden og udvikling og mistanke om forekomst af tumorer skal registreres.
Dyrene vejes enkeltvis en gang om ugen i de første 13 uger af forsøgsperioden og mindst en gang hver fjerde uge derefter. Foderindtagelse bestemmes en gang om ugen i de første 13 uger og derefter ca. hver tredje måned, medmindre andet indiceres af helbredstilstanden eller ændringer i legemsvægten.
Hæmatologisk undersøgelse (f.eks. hæmoglobinkoncentration, hæmatokrit, erytrocyttælling, total leukocyttælling, blodpladetælling og målinger af koaguleringsevnen) udføres efter tre måneder, seks måneder og derefter ca. hver sjette måned samt ved forsøgets afslutning på blodprøver, som ved andre dyr end gnavere tages på hvert enkelt dyr og på rotter på ti dyr pr. køn i alle grupper. Hvis det er muligt, bør prøverne tages fra de samme rotter hver gang, Fra andre dyr end gnavere bør der tages en prøve, inden forsøget påbegyndes.
Hvis kliniske observationer tyder på, at dyrenes helbredstilstand er blevet dårligere under forsøget, foretages en differential tælling på prøver fra de pågældende dyr.
Der foretages en differential tælling på prøver fra dyr i den gruppe, som har været eksponeret for den højeste dosis og fra dyr i kontrolgruppen. Der udføres kun differential tælling på prøver fra dyr i den gruppe, der har fået den næsthøjeste dosis, hvis der er stor forskel på den højest doserede gruppe og kontrolgruppen, eller hvis den patologiske undersøgelse indicerer det.
Der indsamles urinprøver til analyse fra alle andre dyr end gnavere og fra ti rotter pr. køn i alle grupper om muligt fra de samme rotter hver gang med samme intervaller som de ovenfor nævnte blodprøver. Følgende undersøgelser gennemføres på prøver enten fra enkelte dyr eller for rotters vedkommende på en samlet urinprøve/køn/gruppe:
|
— |
udseende: rumfang og vægtfylde for hvert enkelt dyr |
|
— |
protein, glukose, ketonstof, hæmaturi (semikvantitativt), og |
|
— |
sedimentmikroskopi (semikvantitativt). |
Der udtages blodprøver til klinisk-kemisk undersøgelse med ca. seks måneders mellemrum under forsøget og ved forsøgets afslutning fra hvert enkelt dyr, såfremt det drejer sig om andre aner end gnavere, og fra ti rotter/køn i alle grupper så vidt muligt fra de samme rotter hver gang. Fra andre dyr end gnavere skal der tages en prøve, inden forsøget påbegyndes. Plasma fra prøverne underkastes følgende undersøgelser:
|
— |
total serumprotein |
|
— |
albumin |
|
— |
leverfunktionsundersøgelser (f.eks. alkalisk fosfataseaktivitet, serum alanin aminotransferase (ALAT) og serum aspartat aminotransferase (ASAT)), gamma glutamyl transpeptidase, ornitin decarboxylase |
|
— |
kulhydratstofskifte, f.eks. fasteglukose |
|
— |
nyrefunktionsundersøgelser f.eks. urinstof. |
Alle dyr inklusive de dyr, som er døde under forsøget, eller som blev aflivet, fordi de var døende, underkastes en fuldstændig makroskopisk vurdering. Før aflivning tages blodprøver af alle dyr til differential tælling. Alle makroskopisk synlige læsioner, tumorer eller læsioner, som mistænkes for at være tumorer, opbevares. Makroskopiske observationer korreleres så vidt muligt med mikroskopiske fund.
Alle væv og organer bør opbevares med henblik på mikroskopisk undersøgelse. Det drejer sig normalt om følgende væv og organer: hjerne (4) (medulla/pons, det cerebellare og cerebrale cortex), hypofyse, skjoldbruskkirtel (inklusive biskjoldbruskkirtel), thymusvæv, lunger (inklusive trakea), hjerte, aorta, spytkirtler, lever (4), milt, nyrer (4), binyrer (4), spiserør, mavesæk, duodenum, jejunum, ileum, coecum, tyktarm, endetarm, uterus, urinblære, lymfeknuder, bygspytkirtel, kønskirtler (4), assesoriske kønskirtler, kvindelig brystkirtel, hud, muskulatur, perifer nerve, rygmarv (cervikalt, thorakalt, lumbalt), sternum med benmarv og femur (inklusive led) og øjne. Den optimale præparering af lunger og urinblærer er inflation med et fiksativ og i forbindelse med inhalationsundersøgelser er det centralt for de histopatologiske undersøgelser, at lungerne inflateres. I særlige undersøgelser som inhalationsundersøgelser bør de samlede luftveje undersøges, herunder næse, farynx og larynx.
Hvis der gennemføres andre kliniske undersøgelser, bør resultaterne af disse foreligge, før der foretages mikroskopiske undersøgelser, fordi de kan være af afgørende værdi for patologen.
Alle makroskopisk synlige forandringer af et hvilket som helst organ, især tumorer og andre læsioner, skal undersøges mikroskopisk. Det anbefales yderligere at foretage:
|
a) |
Mikroskopisk undersøgelse af alle præparerede organer og væv med fuldstændig beskrivelse af alle fundne læsioner hos
|
|
b) |
Organer eller væv, som udviser abnormiteter, der skyldes eller kan skyldes teststoffet, undersøges også hos de lavere doserede grupper. |
|
c) |
Såfremt forsøgsresultaterne viser betydelige ændringer i dyrenes normale levetid eller induktioner af effekter, der kan have betydning for en toksisk respons, undersøges den gruppe, der eksponeres for den næsthøjeste dosis, efter ovenstående retningslinjer. |
|
d) |
En korrekt vurdering af betydningen af de forandringer, der observeres hos de eksponerede dyr, forudsætter kendskab til den normalt forekommende incidens af læsioner i den anvendte dyrestamme (under samme laboratoriebetingelser, dvs. på grundlag af tidligere opsamlede data). |
2. DATA
Data opstilles i tabelform, som for hver gruppe viser antal dyr ved begyndelsen af forsøget, antal dyr med læsioner og for hver type af læsioner den procentdel dyr, der udviser den pågældende læsion. Evaluering af resultaterne skal baseres på en passende statistisk metode. Alle anerkendte statistiske metoder kan anvendes.
3. RAPPORTERING
3.1. FORSØGSRAPPORT
Forsøgsrapporten skal om muligt indeholde følgende:
|
— |
dyreart, stamme, herkomst, miljøbetingelser, foder osv. |
|
— |
forsøgsbetingelser: Beskrivelse af ekspositionsapparatur herunder udformning, type, dimensionering, luftkilde, system til frembringelse af partikler og aerosoler, metode til konditionering af luften, behandling af udpumpet luft og den måde, hvorpå dyrene holdes i et ekspositionskammer, når et sådan benyttes. Apparatur til måling af temperatur, fugtighed og eventuelle partikulære aerosolers koncentration og partikelstørrelse beskrives. Ekspositionsdata: Disse skal opstilles i tabelform og angive middelværdier og variation (f.eks. standardafvigelser) og bør omfatte:
|
|
— |
dosisniveauer (inklusive vehikel, hvor et sådant er anvendt) og koncentrationer |
|
— |
oplysning om toksisk reaktion inddelt efter køn og dosis |
|
— |
minimal virkningsfuld dosis |
|
— |
dødstidspunkt under forsøget eller overlevelse til forsøgets slutning |
|
— |
beskrivelse af toksiske eller andre virkninger |
|
— |
observationstidspunkt for hvert enkelt abnormitetstegn og den senere udvikling af abnormiteten |
|
— |
data for foder og vægt |
|
— |
oftalmologiske fund |
|
— |
hæmatologiske undersøgelser og samtlige resultater |
|
— |
klinisk-biokemiske analyser og samtlige resultater (herunder resultater af urinanalyse) |
|
— |
obduktionsfund |
|
— |
detaljeret beskrivelse af alle histopatologiske fund |
|
— |
statistisk behandling af resultaterne |
|
— |
diskussion af resultaterne |
|
— |
fortolkning af resultaterne. |
3.2. VURDERING OG FORTOLKNING
Se den generelle indledning til del B.
4. HENVISNINGER
Se den generelle indledning til del B.
B.31. PRÆNATAL UDVIKLINGSTOKSICITETSUNDERSØGELSE
1. METODE
Metoden er gengivet efter OECD TG (2001).
1.1. INDLEDNING
Formålet med denne metode til udviklingstoksicitetsundersøgelse er at skaffe generel information om virkningerne af prænatal eksponering på det drægtige forsøgsdyr og på organismen, mens den udvikles in utero; heri kan også indgå en vurdering af maternel påvirkning såvel som død, strukturabnormiteter og ændret vækst af fosteret. Skønt funktionelle mangler er en vigtig del af udviklingen, indgår de ikke i denne forsøgsmetode. Funktionelle mangler kan i stedet undersøges i et særskilt forsøg eller ved at denne undersøgelse suppleres med anvendelse af metoden til undersøgelse af udviklingsneurotoksicitet. Til undersøgelse for funktionelle mangler og andre postnatale effekter kan anvendes metoderne fra reproduktionstoksicitetsundersøgelse i to generationer og udviklingsneurotoksicitetsundersøgelse.
Denne metode kan kræve individuel tilpasning på grundlag af konkret viden om f.eks. teststoffets fysisk-kemiske eller toksikologiske egenskaber. En sådan tilpasning kan godtages, når det på overbevisende måde videnskabeligt dokumenteres, at tilpasningen vil gøre forsøget mere informativt. I sådanne tilfælde skal denne videnskabelige dokumentation være nøje beskrevet i forsøgsrapporten.
1.2. DEFINITIONER
Udviklingstoksikologi: Undersøgelse af de ugunstige virkninger på organismens udvikling, som kan forårsages af eksposition før konceptionen, under den prænatale udvikling og posnatalt i tiden indtil kønsmodning. Hovedmanifestationerne af toksisk virkning på udviklingen er 1) organismens død, 2) strukturabnormiteter, 3) ændret vækst og 4) funktionelle mangler. Udviklingstoksikologi er tidligere hovedsagelig blevet betegnet teratologi.
Negativ virkning: Enhver sådan behandlingsrelateret afvigelse fra grundlinjen, som forringer en organismes overlevelsesevne, reproduktionsevne eller tilpasningsevne til omgivelserne. Udviklingstoksikologi omfatter i bredeste forstand enhver virkning, som interfererer med den normale udvikling af conceptus både før og efter fødslen.
Ændret vækst: Ændring i afkommets organ- eller kropsvægt eller -størrelse.
Forandringer (abnormiteter): Strukturelle forandringer i udviklingen, herunder både misdannelser og afvigelser (28).
Misdannelse/væsentlig abnormitet: Strukturforandring, som anses for skadelig for dyret (og kan være dødelig), og som sædvanligvis er sjælden.
Afvigelse/mindre abnormitet: Strukturændring, som anses for at have ingen eller ringe skadelig virkning for dyret; kan være forbigående og kan optræde forholdsvis hyppigt i kontrolpopulationen.
Conceptus: Det samlede produkt af et befrugtet æg på ethvert stadium af udviklingen fra befrugtning til fødsel, herunder ekstraembryonale membraner samt selve embryo eller føtus.
Implantation (nidation): Fasthæftning af blastocysten til det indvendige epitel i uterus, herunder penetration af uterusepitelet og indlejring i endometriet.
Embryo: Organismens tidlige stadium eller udviklingsstadium, specielt det fremvoksende produkt af æggets befrugtning, efter at længdeaksen viser sig, og indtil alle hovedstrukturer er til stede.
Embryotoksisk: Skadelig for den normale struktur, udvikling, vækst og eller levedygtighed af et embryo.
Føtus: Det ufødte afkom i den postembryonale periode.
Føtotoksicitet: Skadevirkning på den normale struktur, udvikling, vækst og eller levedygtighed af et føtus.
Abort: Præmatur udstødelse fra uterus af konceptionsprodukter, dvs. af embryonet eller af et ikke levedygtigt føtus.
Resorption: Et conceptus, som efter at have være implanteret i uterus er dødt og er ved at blive resorberet eller er blevet det.
Tidlig resorption: Tegn på implantation uden genkendeligt embryo/føtus.
Sen resorption: Et dødt embryo eller føtus med ydre degenerative forandringer.
NOAEL: forkortelse for »no-observed-adverse-effect level«, dvs, den højeste dosis eller det højeste eksponeringsniveau, hvor der ikke observeres behandlingsrelaterede negative virkninger.
1.3. REFERENCESTOF
Intet.
1.4. FORSØGSMETODENS PRINCIP
Teststoffet administreres normalt til drægtige dyr, i det mindste fra implantationsdagen til én dag før planlagt aflivning, som bør finde sted så kort som muligt før det forventede fødselstidspunkt, uden at der er risiko for tab af data som følge af for tidlig fødsel. Det er ikke hensigten, at metoden alene skal omhandle organogeneseperioden (f.eks. dag 5-15 hos gnavere og dag 6-18 hos kaniner), men, når det er hensigtsmæssigt, også virkninger fra præimplantationsperioden gennem hele drægtighedsperioden indtil dagen før kejsersnit. Kort inden kejsersnittet aflives hundyrene, uterusindholdet undersøges, og føtus bedømmes for synlige udvendige abnormiteter og bløddels- og skeletforandringer.
1.5. BESKRIVELSE AF FORSØGSMETODEN
1.5.1. Valg af dyreart
Det anbefales, at forsøget udføres i den mest relevante dyreart, og at de anvendte laboratoriedyr er af arter og stammer, som sædvanligvis benyttes til teratogenicitetsforsøg. Den foretrukne art gnavere er rotter, og den foretrukne art ikke-gnavere er kaniner. Anvendes andre dyrearter, bør dette begrundes.
1.5.2. Omgivende miljø og fodring af dyrene
Temperaturen i forsøgslokalet skal være 22 oC (± 3 oC) for gnavere og 18 oC (± 3 oC) for kaniner, Den relative luftfugtighed skal være mindst 30 % og helst ikke over 70 % undtagen under rengøring af rummet, men 50-60 % bør tilstræbes. Belysningen skal være kunstig bestående af 12 timers lys og 12 timers mørke. Som foder kan anvendes sædvanligt laboratoriefoder med fri adgang til drikkevand,
Parring bør finde sted i bure egnede til formålet. Par bør fortrinsvis holdes for sig, men gruppevis anbringelse af få dyr kan også godtages.
1.5.3. Forberedelse af dyrene
Der anvendes raske dyr, som er akklimatiseret til laboratoriemiljøet i mindst fem døgn og ikke tidligere er indgået i forsøg. Forsøgsdyrene skal være karakteriseret med hensyn til art, stamme, oprindelse, køn, vægt og/eller alder. Dyrene i alle forsøgsgrupper skal så vidt muligt være af ensartet vægt og alder, På hvert dosisniveau skal anvendes unge voksne hundyr, som ikke tidligere har født. Hunnerne parres med hanner af samme art og stamme, idet parring af søskendedyr undgås. For gnavere er drægtighedsperiodens dag 0 den dag, hvor der observeres en vaginal prop og/eller semen; for kaniner er dag 0 sædvanligvis dagen for coitus eller for kunstig insemination, hvis denne teknik anvendes. De parrede hunner fordeles uden skævhed mellem kontrol- og behandlingsgruppeme. Burene arrangeres således, at eventuelle virkninger af burenes placering minimeres. Hvert dyr tilordnes et unikt identifikationsnummer. Parrede hunner fordeles uden skævhed mellem kontrol- og behandlingsgrupperne, og hvis hunnerne parres holdvis, skal dyrene i hvert hold fordeles ligeligt mellem grupperne. Tilsvarende skal hunner, som insemineres af samme han, fordeles jævnt mellem grupperne.
1.6. FREMGANGSMÅDE
1.6.1. Dyrenes antal og køn
I hver behandlings- og kontrolgruppe skal der være tilstrækkeligt med hunner til, at der ved obduktion fås ca. 20 hundyr med implantationssteder. Hvis der færre end 16 dyr med implantationssteder i en gruppe, kan det blive anset for utilstrækkeligt. Mortalitet af moderdyrene gør ikke nødvendigvis undersøgelsens resultater ugyldige, forudsat at den ikke er over ca. 10 %.
1.6.2. Tilberedning af doser
Anvendes et vehikel eller andet tilsætningsstof, skal følgende egenskaber tages i betragtning: Indvirkningen på teststoffets absorption, fordeling, metabolisme og retention eller udskillelse; eventuel sådan indvirkning på teststoffets kemiske egenskaber, som kan ændre dets toksiske egenskaber, samt indvirkningen på dyrenes føde- og vandindtagelse eller ernæringstilstand, Vehiklet må hverken være udviklingstoksisk eller påvirke reproduktionen.
1.6.3. Dosering
Normalt administreres teststoffet dagligt fra implantationstidspunktet (f.eks. dag 5 efter parring) til dagen inden det planlagte kejsersnit. Hvis eventuelle indledende undersøgelser ikke tyder på et stort potentiale for præimplantationstab, kan behandlingen udvides til at omfatte hele drægtighedsperioden fra parringen til dagen før den planlagte aflivning. Det er velkendt, at uhensigtsmæssig behandling eller stress i drægtighedsperioden kan medføre prænatalt tab. For at sikre mod prænatalt tab som følge af ikke-behandlingsrelaterede faktorer skal man undgå unødvendig håndtering af drægtige dyr samt stress forårsaget af ydre faktorer som støj.
Der skal være mindst tre dosisgrupper og en sideløbende kontrolgruppe. Raske dyr skal fordeles uden skævhed mellem kontrol- og behandlingsgrupperne. Dosisniveauerne bør vælges med sådanne intervaller, at der frembringes en gradueret toksisk effekt. Medmindre doseringen må begrænses pga. teststoffets fysisk-kemiske eller biologiske egenskaber, vælges den højeste dosering således, at den har en vis udviklingstoksisk effekt og/eller toksisk effekt på moderdyret (kliniske symptomer eller vægttab), men uden at den forårsager død eller alvorlige lidelser. Der skal være mindst ét mellemniveau, som fremkalder det laveste observerbare niveau af toksisk effekt. Den laveste dosering må hverken medføre tegn på materiel toksicitet eller udviklingstoksicitet. Der vælges en aftagende række dosisniveauer. som giver mulighed for at påvise en eventuel dosisafhængig respons samt det niveau, som ikke medfører tegn på skadelig effekt (NOAEL). Ved fastlæggelse af den aftagende række dosisniveauer svarer de optimale intervaller ofte til en faktor to til fire, og i mange tilfælde må tilføjelse af en fjerde behandlingsgruppe foretrækkes frem for meget store intervaller (f.eks. større end en faktor 10) mellem dosisniveauerne. Skønt målet er at fastlægge en maternel NOAEL, kan undersøgelser, hvor en sådan dosis ikke fastlægges, også være acceptable (1).
Doseringen vælges under hensyntagen til eventuelle foreliggende toksicitetsdata samt supplerende oplysninger om metabolisme og toksikokinetik af teststof eller beslægtede stoffer. Sådanne oplysninger kan desuden være med til at godtgøre det hensigtsmæssige i den valgte dosering.
Der skal være en sideløbende kontrolgruppe. Kontrolgruppen skal være ubehandlet eller behandles med vehikel, hvis et sådant anvendes til administration af teststoffet. Alle grupper skal modtage samme volumen af enten teststof eller bærestof. Dyrene i kontrolgruppen (-grupperne) skal håndteres på samme måde som behandlingsgruppen. Kontrolgrupper på vehikel skal modtage dette i den største mængde, det anvendes i (som til behandlingsgruppen på laveste dosis).
1.6.4. Grænseprøve
Såfremt et oralt testdosisniveau på mindst 1 000 mg/kg kropsvægt/døgn med de til dette forsøg beskrevne metoder ikke frembringer observerbar toksisk virkning i de drægtige dyr eller deres afkom, og såfremt foreliggende data (f.eks. fra strukturelt og/eller metabolisk beslægtede forbindelser) ikke giver anledning til at forvente nogen virkning, skal et komplet forsøg med tre dosisniveauer ikke absolut anses for nødvendigt. Den påregnede eksponering af mennesker kan tilsige, at der anvendes et højere oralt dosisniveau end det. der er anvendt i grænseforsøget. For andre administrationsmåder — f.eks. inhalation eller dermal applikation — kan teststoffets fysisk-kemiske egenskaber ofte være bestemmende for og begrænse det maksimale opnåelige ekspositionsniveau (f.eks. må dermal applikation ikke forårsage alvorlige lokale toksiske virkninger).
1.6.5 Administration af doser
Teststoffet administreres sædvanligvis oralt med sonde. Anvendes anden administrationsvej, skal forsøgslederen begrunde valget, og passende modifikationer kan være nødvendige (2)(3)(4). Teststoffet bør administreres på omtrent samme tidspunkt hver dag.
Doseringen til det enkelte dyr bør normalt baseres på den seneste bestemmelse af dyrets vægt. I drægtighedsperiodens sidste trimester må justering af doseringen dog ske med varsomhed. Doseringen bør på grundlag af foreliggende data vælges, så man undgår kraftig maternel toksicitet, Konstateres der kraftig toksisk effekt i de behandlede moderdyr, skal disse aflives af humane grunde. Viser flere drægtige dyr tegn på stærk toksicitet, bør man overvejs at indstille forsøget for den pågældende dosisgruppe. Administreres stoffet med sonde, bør dette fortrinsvis ske som enkeltdosis ved hjælp af gastrisk sonde eller et passende intubationsrør. Den maksimale væskemængde, som kan administreres ad gangen, afhænger af forsøgsdyrets størrelse. Dette volumen må højst være 1 ml/100 g kropsvægt, bortset fra vandige opløsninger, af hvilke der kan anvendes 2 ml/100 g kropsvægt. Anvendes majsolie som vehikel, må volumenet ikke overstige 0,4 ml/100 g kropsvægt. Variationer i testvolumen skal ved justering af koncentrationerne minimeres, så der sikres et konstant volumen for alle dosisgrupper.
1.6.6. Iagttagelse af moderdyrene
Klinisk observation skal foretages og registreres mindst én gang dagligt, fortrinsvis på samme tidspunkt(er) hver dag, idet man retter sig efter tidspunktet for den forventede maksimale effekt af indgivelsen. Dyrenes tilstand registreres, herunder dødelighed, moribund tilstand, væsentlige adfærdsændringer og alle tegn på åbenbar toksicitet.
1.6.7. Kropsvægt og fødeindtagelse
Dyrene vejes på drægtighedsperiodens dag 0 eller, for tidsparrede dyr leveret fra en ekstern opdrætter, på den første dag i doseringsperioden, og derefter mindst hver tredje dag i doseringsperioden samt på dagen for den planlagte aflivning.
Fødeindtagelsen registreres med tre dages mellemrum, hvilket skal falde sammen med kropsvægtbestemmelsen.
1.6.8. Post-mortem undersøgelse
Hunnerne aflives ét døgn for den forventede fødsel. Hunner, som inden den planlagte aflivning viser tegn på abort eller for tidlig fødsel, aflives og underkastes grundig makroskopisk undersøgelse.
Ved forsøgets slutning eller ved indtræden af død under forsøget undersøges moderdyret makroskopisk for eventuelle strukturabnormiteter eller patologiske forandringer. Vurderingen af hundyrene under kejsersnittet og de efterfølgende undersøgelser af føtus bør fortrinsvis ske uden kendskab til behandlingsgruppen for at minimere skævhed.
1.6.9. Undersøgelse af uterusindholdet
Umiddelbart efter undersøgelsens afslutning eller snarest muligt efter dødens indtræden udtages uterus, og dyrenes drægtighedsstatus fastslås. Uteri, som fremtræder ikke-gravide, undersøges nærmere (f.eks. med ammoniumsulfidfarvning til gnavere og Salewski-farvning som en velegnet alternativ metode til kaniner) med henblik på bekræftelse af den ikke-gravide status (5).
De gravide uteri inklusive cervix vejes. Vægten af den gravide uterus skal ikke bestemmes for dyr, som findes døde i løbet af undersøgelsen.
Antal corpora lutea hos drægtige dyr bestemmes.
Uterusindholdet undersøges for antal døde embryoner eller føtus samt levende fostre, Resorptionsgraden beskrives, for at det relative dødstidpunkt for conceptus skønsvis kan fastsættes (se punkt 1.2).
1.6.10. Undersøgelse af føtus
For hvert føtus bestemmes køn og kropsvægt.
Hvert føtus undersøges for ydre forandringer (6).
Føtus undersøges for skelet- og bløddelsforandringer (f.eks. variationer, misdannelser og abnormiteter) (7)(8)(9)(10)(11)(12)(13)(14)(15)(16)(17)(18)(19)(20)(21)(22)(23)(24). Det må foretrækkes, at føtale forandringer kategoriseres, men dette er ikke et krav. Har kategorisering fundet sted, skal det klart angives, efter hvilke kriterier hver kategori er fastlagt. Særlig opmærksomhed skal udvises over for reproduktionssystemet, som skal undersøges for tegn på forandringer i udviklingen.
For gnavere skal ca, halvdelen af hvert kuld præpareres og undersøges for skeletforandringer. Den resterende del præpareres og undersøges for bløddelsforandringer med anerkendte eller egnede metoder til seriesnit eller omhyggelig makroskopisk dissektion.
For ikke-gnavere, f.eks. kaniner, skal alle fostre undersøges for både bløddels- og skeletforandringer. Disse fostre vurderes for bløddelsforandringer ved omhyggelig dissektion. herunder også metoder til nærmere vurdering af hjertets indvendige struktur (25). Hovedet på halvdelen af de således undersøgte fostre fjernes og præpareres til undersøgelse af bløddelsforandringer (herunder øjne, hjerne, næsesvælg og tunge), idet der anvendes sædvanlige seriesnitmetoder (26) eller en metode med tilsvarende følsomhed. Kroppen af disse fostre og de resterende intakte fostre behandles og undersøges for skeletforandringer med anvendelse af de samme metoder som beskrevet for gnavere.
2. DATA
2.1. BEHANDLING AF RESULTATER
Data rapporteres for hver enkelt moderdyr og dets afkom og sammenfattes i tabelform med angivelse, for hver forsøgsgruppe, af antal dyr ved forsøgets begyndelse, antal dyr, som er fundet døde under forsøget eller aflivet af humanitære grunde, tidspunkt for eventuel død eller humanitær aflivning, antal drægtige hundyr, antal dyr, som viser tegn på toksisk virkning, beskrivelse af iagttagne toksicitetstegn, herunder begyndelsestidspunkt, varighed og sværhed af alle toksiske virkninger, arten af de fund, der er gjort på embryo/føtus, samt alle relevante data vedrørende afkommet.
Talresultaterne evalueres med en passende statistisk metode, hvor kuldet benyttes som enhed for dataanalysen, Der anvendes en almindelig anerkendt statistisk metode; valg af statistiske metoder skal indgå som en del af undersøgelsens design og skal begrundes. Data fra dyr, som ikke overlever indtil den planlagte aflivning, skal ligeledes rapporteres. Disse data kan, når det er relevant, indgå i gruppegennemsnittet. Relevansen af data for sådanne dyr og dermed om de skal indgå i et gruppegennemsnit eller ikke, skal begrundes og vurderes på individuel basis.
2.2. VURDERING AF RESULTATER
Fundene i den prænatale udviklingstoksikologiske undersøgelse skal vurderes med hensyn til de iagttagne effekter. I vurderingen skal indgå følgende oplysninger:
|
— |
maternelle og embryonale/føtale testresultater, herunder vurdering af sammenhæng, eller mangel herpå, mellem dyrenes udsættelse for teststoffet og forekomst og sværhedsgrad af alle fund |
|
— |
de anvendte kriterier for inddeling af føtale eksterne, bløddels- og skeletforandringer, såfremt inddeling er foretaget |
|
— |
når det er hensigtsmæssigt, historiske kontroldata, som kan være nyttige ved tolkning af undersøgelsesresultaterne |
|
— |
de tal, der ligger til grund for beregning af alle procenttal og indekser |
|
— |
når det er hensigtsmæssigt, fyldestgørende statistisk analyse af undersøgelsesresultaterne, med tilstrækkelige oplysninger om analysemetoden til, at en uafhængig rapportør/statistiker kan revurdere og eftergøre analysen. |
I enhver undersøgelse, som viser fravær af toksiske virkninger, skal nødvendigheden af supplerende undersøgelser til bestemmelse af teststoffets absorption og biotilgængelighed diskuteres.
2.3. FORTOLKNING AF RESULTATER
En prænatal udviklingstoksicitetsundenøgelse giver oplysninger om virkningerne af gentagen eksponering for et stof på moderdyrene og på den intrauterine udvikling af afkommet. Undersøgelsens resultater skal fortolkes i sammenhæng med fundene fra subkroniske, reproduktions-, toksikokinetiske og andre undersøgelser. Da der lægges vægt både på almen toksisk respons i form af maternel toksicitet og på udviklingstoksikologiske endpoints, vil undersøgelsens resultater i nogen grad gøre det muligt at skelne mellem de udviklingsmæssige effekter, som ikke er ledsaget af en almen toksisk virkning, og dem, som kun påføres på niveauer, som også er toksiske for moderdyret (27).
3. RAPPORTERING
3.1. FORSØGSRAPPORT
Forsøgsrapporten skal indeholde følgende specifikke oplysninger:
|
|
For teststoffet:
|
|
|
For eventuelt vehikel:
|
|
|
For forsøgsdyrene:
|
|
|
For forsøgsbetingelserne:
|
|
|
Resultater: |
|
|
Data vedrørende materiel toksisk respons for hver dosisgruppe, inklusive, men ikke begrænset til:
|
|
|
Udviklingsmæssige endpoints for kuld med implantater, herunder:
|
|
|
Udviklingsmæssige endpoints for kuld med levende fostre, herunder:
|
|
|
Diskussion af resultaterne. |
|
|
Konklusioner. |
4. HENVISNINGER
|
(1) |
Kavlock R.J. et al. (1996) A Simulation Study of the Inf luence of Study Design on the Estimation of Benchmark Doses for Developmental Toxicity. Risk Analysis 16; 399-410. |
|
(2) |
Kimmel, C.A. and Francis, E.Z. (1990) Proceedings of the Workshop on the Acceptability and Interpretation of Dermal Developmental Toxicity Studies. Fundamental and Applied Toxicologv 14; 386-398. |
|
(3) |
Wong, B.A., et al. (1997) Developing Specialized Inhalation Exposure Systems to Address Toxicological Problems. CIIT Activities 17; 1-8. |
|
(4) |
US Environmental Protection Agency (1985) Subpart E-Speciflc Organ/Tissue Toxicity, 40 CFR 798.4350: Inhalation Developmental Toxicity Study. |
|
(5) |
Salewski, E. (1964) Färbermethode zum Makroskopischen Nachweis von Implantations Stellen am Uterus der Ratte. Naunyn-Schmeidebergs Archiv fur Pharmakologie und Experimenielle Pathologie 247:367. |
|
(6) |
Edwards, J.A. (1968) The external Development of the Rabbit and Rat Embryo. In Advances in Teratology. D.H.M. Woolam (ed.) Vol. 3. Academic Press, NY. |
|
(7) |
Inouye, M. (1976) Ditferential Staining of Cartilage and Bone in Fetal Mouse Skeleton by Alcian Blue and Alizarin Red S. Cengenital Anomalies 16; 171-173. |
|
(8) |
Igarashi, E. et al. (1992) Frequency Of Spontaneous Axial Skeletal Variations Detected by the Double Staining Technique for Ossified and Cartilaginous Skeleton in Rat Foetuses. Congenital Anomalies 32; :381-391. |
|
(9) |
Kimmel. C.A. et al. (1993) Skeletal Development Following Heat Exposure in the Rat. Teratology 47:229-242. |
|
(10) |
Marr, M.C. et al. (1988) Comparison of Single and Double Staining for Evaluation of Skeletal Development: The Effects of Ethylene Glycol (EG) in CD Rats. Teratology 37; 476. |
|
(11) |
Barrow, M.V. and Taylor, W.J. (1969) A Rapid Method for Detecting Malformations in Rat Foetuses. Journal of Morphology 127:291-306. |
|
(12) |
Fritz, H. (1974) Prenatal Ossification in Rabbits ss Indicative of Foetal Maturity. Teratology 11; 313-320. |
|
(13) |
Gibson, J.P. et al. (1966) Use of the Rabbit in Teratogenicity Studies. Toxicology and Applied Pharmacology 9; :398-408. |
|
(14) |
Kimmel, C.A. and Wilson, J.G. (1973) Skeletal Deviation in Rats: Malformations or Variations? Teratology 8: 309-316. |
|
(15) |
Marr, M.C. et al. (1992) Developmental Stages of the CD (Sprague-Dawley) Rat Skeleton after Maternal Exposure to Ethylene Glycol. Teratology 46: 169-181. |
|
(16) |
Monie, I.W. et al. (1965) Dissection Procedures for Rat Foetuses Permitting Alizarin Red Staining of Skeleton and Histological Study of Viscera. Supplement to Teratology Workshop Manual, pp. 163-173. |
|
(17) |
Spark, C. and Dawson, A.B. (1928) The Order and Time of appearance of Centers of Ossification in the Fore and Hind Limbs of the Albino Rat, with Special Reference to the Possible Influence of the Sex Factor. American Journal of Anatomy 41; 411-445. |
|
(18) |
Staples, R.E. and Schnell, V.L. (1964) Refinements in Rapid Clearing Technique in the KOH-Alizarin Red S Method for Fetal Bone. Slam Technology 39; 61-63. |
|
(19) |
Strong, R.M. (1928) The Order Time and Rate of Ossification of the Albino Rat (Mus Norvegicus Albinus) Skeleton. American Journal of Anatomy 36; 313-355. |
|
(20) |
Stuckhardt, J.L. and Poppe, S.M. (1984) Fresh Visceral Examination of Rat and Rabbit Foetuses Used in Teratogenicity Testing. Teratogenesis, Carcinogenesis, and Mutagenesis 4; 181-188. |
|
(21) |
Walker, D.G. and Wirtschafter, Z.T. (1957) The Genesis of the Rat Skeleton. Thomas, Springfield, II. |
|
(22) |
Wilson. J.G. (1965) Embryological Considerations in Teratology. In Teratology: Principles and Techniques, Wilson J.G. and Warkany J. (eds). University of Chicago, Chicago, IL, pp 251-277. |
|
(23) |
Wilson, J.G. and Fraser, F.C. (eds). (1977) Handbook of Teratology, Vol. 4. Plenum, NY. |
|
(24) |
Varnagy, L. (1980) Use of Recent Fetal Bone Staining Techniques in the Evaluation of Pesticide Teratogenicity, Acta Vet. Acad. Sci. Hung. 28; 233-239. |
|
(25) |
Staples, R.E. (1974) Detection of visceral Alterations in Mammalian Foetuses. Teratology 9; 37-38. |
|
(26) |
Van Julsingha, F.B. and CG. Bennett (1977) A Dissseting Procedure for the Detection of Anomalies in the Rabbit Foetal Head. In: Methods in Prenatal Toxicology Neubert, D., Merker, H.J. and Kwasigroch, T.E. (eds.). University of Chicago, Chicago, II, pp, 126-144. |
|
(27) |
US Environmental Protection Agency (1991) Guidelines for Developmental Toxicity Risk Assessment, Federal Register 56; 63798-63826. |
|
(28) |
Wise, D.L. et al. (1997) Terminology of Developmental Abnormalities in Common Laboratory Mammals (Version 1) Teratology 55; 249-292. |
B.32. KARCINOGENICITETSUNDERSØGELSE
1. METODE
1.1. INDLEDNING
Se den generelle indledning til del B.
1.2. DEFINITIONER
Se den generelle indledning til del B.
1.3. REFERENCESTOFFER
Ingen.
1.4. PRINCIP FOR TESTMETODEN
Flere grupper af forsøgsdyr doseres normalt syv dage om ugen med teststof på passende måde. Der gives en dosis pr. gruppe i størstedelen af dyrenes levetid. Under og efter dosering med teststoffet observeres dyrene dagligt, og eventuelle tegn på toksicitet, især udvikling af tumorer, registreres.
1.5. KVALITETSKRITERIER
Ingen.
1.6. BESKRIVELSE AF TESTMETODEN
Dyrene holdes under forsøgsbetingelser med hensyn til miljø og fodring i mindst fem dage, inden forsøget påbegyndes. Unge, sunde, dyr fordeles randomiseret i test- og kontrolgrupper.
Rotten er det foretrukne forsøgsdyr. Andre dyrearter (gnavere eller ikke-gnavere) kan anvendes, hvis tidligere gennemførte undersøgelser giver grundlag herfor. Der bør benyttes unge, sunde dyr af almindeligt anvendte laboratoriestammer, og doseringen bør indledes så hurtigt som muligt, efter at dyrene er vænnet fra.
Ved forsøgets begyndelse må vægten af de anvendte dyr ikke variere mere end ± 20 % af gennemsnitsvægten. Hvis en undersøgelse af subkronisk toksicitet ved oral indgift udføres som et forstudium til en langtidsundersøgelse, bør samme dyreart og stamme anvendes i begge undersøgelser.
Hvis der anvendes gnavere, skal der anvendes mindst 100 dyr (50 hun- og 50 handyr) til hvert dosisniveau samt en kontrolgruppe. Hundyrene må ikke have født og må ikke være drægtige. Hvis der påregnes aflivning af dyr under forsøget, bør det oprindelige antal forøges med det antal dyr, man påregner at aflive i løbet af forsøget.
Der anvendes mindst tre forskellige dosisniveauer ud over kontrolgruppen. Det højeste dosisniveau bør fremkalde minimale toksicitetstegn f.eks. en let formindskelse af legemsvægten (mindre end 10 %), uden at der er tale om væsentlige ændringer af den normale levetid undtagen som følge af tumorer.
Den mindste dosis bør ikke påvirke dyrenes normale vækst, udvikling og levetid eller have nogen toksisk effekt overhovedet. I almindelighed bør denne dosis ikke være mindre end 10 % af den største dosis.
Middeldosis (evt. flere) bør ligge midtvejs mellem det højeste og laveste dosisniveau.
Ved valg af dosisstørrelser bør der tages hensyn til data fra tidligere toksicitetsundersøgelser.
Dyrene doseres normalt med teststoffet hver dag.
Hvis stoffet administreres via drikkevandet eller med foderet, bør det være kontinuert tilgængeligt.
Der skal anvendes en kontrolgruppe, som på alle måder er identisk med forsøgsgrupperne bortset fra dosering med teststoffet.
Under særlige omstændigheder som f.eks. inhalationsundersøgelser, hvor der benyttes aerosoler, eller hvis der benyttes en emulgator med ukendt biologisk virkning i undersøgelser med oral indgift, anvendes en negativ kontrolgruppe, som ikke doseres hverken med teststoffet eller vehiklet.
De tre hovedformer for administration af teststof er oral indgift, påføring på huden og inhalation, Valget af administrationsform afhænger af teststoffets fysiske og kemiske karakteristika og den sandsynlige måde, hvorpå mennesker ekponeres for stoffet.
1.6.5.1. Oral indgift
Hvis teststoffet absorberes i mave-tarmkanalen, og hvis mennesker kan udsættes for indtagelse gennem munden, anvendes oral indgift, medmindre der foreligger kontraindikationer. Dyrene indgives teststoffet med foderet, opløst i drikkevandet eller i kapsler.
Den ideelle dosering er, at dyrene får indgivet teststoffet syv dage om ugen. Hvis indgift indskrænkes til fem dage om ugen, kan der ske tilbagevenden til normal, eller der kan optræde abstinenssymptomer imellem eksponeringsperioderne, hvilket kan indvirke på resultaterne og den senere vurdering. Med udgangspunkt i praktiske overvejelser kan det accepteres, at dyrene indgives teststoffet fem dage om ugen.
1.6.5.2. Påføring på huden
Kutan påføring ved hudpensling kan vælges som model ved induktion af hudlæsioner med henblik på at simulere en vigtig human ekspositionsform.
1.6.5.3. Inhalation
Inhalationsundersøgelser giver mere komplekse tekniske problemer end andre former for administrering af teststof, i det følgende gives derfor en mere detaljeret vejledning i gennemførelse af disse undersøgelser. Man bør også være opmærksom på, at intratrakeal instillation under særlige omstændigheder kan være et egnet alternativ.
Forsøg med langtidseksposition udformes normalt på grundlag af erfaringer fra arbejdspladser, således at dyrene eksponeres fem dage om ugen seks timer dagligt, efter at koncentrationen i kammeret er ækvilibreret (intermitterende eksposition), eller der benyttes eksponering på grundlag af erfaringer fra miljøet, dvs. syv dage om ugen og 22 til 24 timer pr. dag (kontinuert eksposition) med ca. en time om dagen på et bestemt klokkeslæt til fodring af dyrene og rengøring af kammeret. I begge tilfælde eksponeres dyrene normalt for fastlagte koncentrationer af teststof. En vigtig forskel mellem intermitterende og kontinuert eksposition, som man må være opmærksom på, er, at der ved den førstnævnte metode er en periode på 17 til 18 timer, hvor dyrene kan overvinde virkningerne af en daglig eksposition og en endnu længere periode i weekenderne.
Valget mellem intermitterende og kontinuert eksposition afhænger af undersøgelsens formål og den menneskelige eksponering, der simuleres. Der er imidlertid en række tekniske vanskeligheder at tage i betragtning. F.eks. kan fordelene ved kontinuert eksponering, når det drejer sig om at simulere miljøbetingelser, falde væk på grund af behovet for vanding og fodring i ekspositionsperioden og for mere komplicerede (og pålidelige) teknikker til generering og kontrol af aerosoler og dampe.
Dyrene testes i ekspositionskamre, der er således udformet, at der kan opretholdes en dynamisk luftgennemstrømning på mindst 12 luftskift pr. time, samtidig med at der sikres et adækvat iltindhold og en jævnt fordelt ekspositionsluft. Kontrolkamre og ekspositionskamre bør være identiske med hensyn til konstruktion og udformning, så det sikres, at ekspositionsbetingelserne er sammenlignelige på alle måder bortset fra eksponering for teststoffet. Der holdes normalt et let undertryk i kammeret for at forhindre, at teststoffet lækker ud i det omgivende rum. Kammeret indrettes, således at der undgås trængsel mellem forsøgsdyrene. Den generelle regel er, at hvis der skal sikres en stabil rumatmosfære, må dyrenes totale volumen ikke overstige 5 % af ekspositionskammerets volumen.
Der skal foretages måling og kontrol af:
|
i) |
Luftgennemstrømning: Det må foretrækkes, at luftstrømmen gennem kammeret kontrolleres kontinuerligt. |
|
ii) |
Koncentrationen må i den daglige eksponeringsperiode ikke variere mere end ± 15 % af den gennemsnitlige værdi. Under hele forsøget skal koncentrationen fra dag til dag holdes så konstant som praktisk muligt. |
|
iii) |
Temperatur og fugtighed: Ved anvendelse af gnavere holdes temperaturen på 22 oC (± 2 oC) og fugtigheden i kammeret mellem 30 og 70 %, undtagen når der anvendes vand til suspension af teststof i kammerets atmosfære. Det må foretrækkes, at både temperatur og fugtighed kontrolleres konstant. |
|
iv) |
Måling af partikelstørrelse: Hvis der anvendes atmosfære med flydende eller faste aerosoler i kammeret, bør der foretages en beregning af partikelstørrelsens distribution. Aerosolpartiklerne skal være respirable for de anvendte forsøgsdyr. Der tages prøver af kammerets atmosfære i dyrenes vejrtrækningszone. Luftprøverne skal være repræsentative for den partikeldistribution, dyrene eksponeres for, og det skal gennem gravimetrisk analyse være muligt på grundlag af disse luftprøver at beskrive hele aerosolen, også selv om meget af den ikke er respirabel. Under indkøring af udstyret bør der foretages hyppige analyser af partikelstørrelsen for at sikre aerosolkoncentrationernes stabilitet. Under eksponering foretages analyser så ofte, som det er nødvendigt for en adækvat bestemmelse af, om den panikeldistribution, dyrene eksponeres for, er konstant. |
En karcinogenicitetsundersøgelse omfatter størstedelen af forsøgsdyrenes normale levetid. Hvis der anvendes mus og hamstre bør forsøget vare 18 måneder, og hvis der anvendes rotter 24 måneder. Ved anvendelse af visse dyrestammer med længere levetid og/eller en lav spontan tumorincidens bør forsøget vare 24 måneder med mus og hamstre og 30 måneder med rotter. Det kan dog accepteres, at en sådan forlænget undersøgelse afsluttes, når antallet af overlevende dyr i den gruppe, der eksponeres for den mindste dosis, eller kontrolgruppen er nede på 25 %. Hvis der er en åbenlys forskel i respons afhængig af dyrenes køn, bør hvert køn betragtes som et forsøg for sig, og forsøgets længde indrettes herefter. Hvis der kun optræder præmature dødsfald i den gruppe, som eksponeres for den højeste dosis, og hvis dette klart skyldes toksiske effekter, er det ikke nødvendigt at afslutte undersøgelsen, under forudsætning af at de toksiske effekter ikke skaber problemer i de andre grupper. Hvis et negativt testresultat skal kunne accepteres, må højst 10 % af hver gruppe falde uden for undersøgelsen på grund af autolyse af væv, kannibalisme eller forveksling, og overlevelsesprocenten i alle grupper må ikke være under 50 % efter 18 måneders forløb med anvendelse af mus og hamstre og 24 måneder med anvendelse af rotter.
Fremgangsmåde
Observation af dyrene skal omfatte forandringer i hud og pels, øjne og slimhinder, åndedræt og kredsløb, det autonome og centrale nervesystem, den motoriske aktivitet og adfærdsmønstret.
Dyrene inspiceres regelmæssigt for at det mindst mulige antal dyr går tabt for undersøgelsen på grund af f.eks. kannibalisme, autolyse af væv eller forveksling. Døende dyr fjernes og obduceres, når de observeres.
For alle dyrs vedkommende registreres kliniske symptomer og mortalitet. Der foretages en nøje registrering af udviklingen af tumorer. Ved alle makroskopisk synlige eller palpable tumorer registreres symptomer, lokalisering, dimensioner, fremtræden og udvikling.
Foderindtagelse (og vandforbrug, hvis teststof indgives med drikkevandet) bestemmes en gang om ugen i de første 13 uger og derefter ca. hver tredje måned, medmindre andet indiceres af helbredstilstanden eller ændringer i legemsvægten.
Dyrene vejes enkeltvis en gang om ugen i de første 13 uger af forsøgsperioden og mindst en gang hver fjerde uge derefter.
Klinisk undersøgelse
Hvis observationer af dyrene tyder på, at deres helbredstilstand er blevet dårligere under forsøget, foretages en differential tælling på blodprøver fra de pågældende dyr.
Efter hhv. 12 og 18 måneders varighed samt umiddelbart før aflivning tages blod fra hvert enkelt dyr til udstrygningspræparater. Der foretages en differential tælling på prøver fra dyrene i den gruppe, som har været doseret med den højeste dosis, og fra dyrene i kontrolgruppen. Hvis resultaterne af disse analyser, især af de prøver, som tages umiddelbart inden aflivning, eller resultaterne af den patologiske undersøgelse indicerer det, udføres differential tælling på prøver fra dyr i den gruppe, der har fået den næsthøjeste dosis.
Alle dyr inklusive de dyr, som er døde under forsøget, eller som blev aflivet, fordi de var døende, underkastes en fuldstændig makroskopisk vurdering. Alle makroskopisk synlige rumorer eller læsioner, som mistænkes for at være tumorer, opbevares.
Følgende væv og organer opbevares i et passende medium med henblik på senere histopatologisk undersøgelse: hjerne, (herunder snit af medulla/pons, den cerebellare og cerebrale cortex), hypofyse, skjoldbrusk/biskjoldbruskkirtel, thymusvæv, trakea, lunger, hjerte, aorta, spytkirtler, lever, milt, nyrer, binyrer, bugspytkirtel, kønskirtler, uterus, assesoriske kønskirtler, hud, spiserør, mavesæk, duodenum, jejunum, ileum, coecum, tyktarm, endetarm, urinblære, en repræsentativ lymfeknude, kvindelig brystkirtel, lårmuskulatur, perifer nerve, sternum med benmarv, femur (inklusive ledflade), rygmarv i tre snit (cervikalt, midt for thorax og lumbalt) samt øjne.
Den optimale præparering af lunger og urinblære er inflation med et fiksativ. I forbindelse med inhalationsundersøgelser er det centralt for de histopatologiske undersøgelser, at lungerne inflateres. I inhalationsundersøgelser bør de samlede luftveje undersøges, herunder næse, farynx og larynx.
|
a) |
Der skal foretages en fuldstændig histopatologisk undersøgelse af organer og væv fra dyr, som dør eller aflives under forsøget, samt dyrene i kontrolgruppen og i den gruppe, der er blevet doseret med den højeste dosis. |
|
b) |
Alle makroskopisk synlige tumorer eller læsioner, som mistænkes for at være tumorer, undersøges i alle grupper. |
|
c) |
Hvis der er signifikant forskel på de neoplastiske læsioners incidens i den højest doserede gruppe og kontrolgruppen, skal de relevante organer eller væv undersøges histopatologisk i de øvrige grupper. |
|
d) |
Hvis overlevelse i den højest doserede gruppe er væsentlig lavere end i kontrolgruppen, underkastes den gruppe, der er doseret med den næsthøjeste dosis, en fuldstændig histopatologisk undersøgelse. |
|
e) |
Hvis undersøgelsen af den højest doserede gruppe giver grund til at formode, at der er tale om induktion af toksiske eller andre effekter, som kan have betydning for et neoplastisk respons, underkastes den gruppe, der eksponeres for den næsthøjeste dosis, en fuldstændig histopatologisk undersøgelse. |
2. DATA
Data opstilles i tabelform, som for hver gruppe viser antal dyr ved begyndelsen af forsøget, antal dyr med tumorer, som er registreret under forsøget, registreringstidspunkt og antal dyr, hos hvem der er fundet tumorer efter aflivning. Evaluering af resultaterne skal baseres på en passende statistisk metode. Alle anerkendte statistiske metoder kan anvendes.
3. RAPPORTERING
3.1. FORSØGSRAPPORT
Forsøgsrapporten skal om muligt indeholde følgende:
|
— |
dyreart, stamme, herkomst, miljøbetingelser, foder osv. |
|
— |
forsøgsbetingelser: Beskrivelse af ekspositionsapparatur herunder udformning, type, dimensionering, luftkilde, system til frembringelse af partikler og aerosoler, metode til konditionering af luften, behandling af udpumpet luft og den måde, hvorpå dyrene holdes i et ekspositionskammer, når et sådant benyttes. Apparatur til måling af temperatur, fugtighed og eventuelle partikulære aerosolers koncentration og partikelstørrelse beskrives. Ekspositionsdata: Disse skal opstilles i tabelform og angive middelværdier og variation (f.eks. standardafvigelser) og bør omfatte:
|
|
— |
doser (inklusive vehikel, hvor et sådant er anvendt) og koncentrationer |
|
— |
oplysning om tumorincidens inddelt efter køn, dosis og tumortype |
|
— |
dødstidspunkt under forsøget eller overlevelse til forsøgets slutning |
|
— |
oplysning om toksisk reaktion inddelt efter køn og dosis |
|
— |
toksisk eller anden virkning |
|
— |
observationstidspunkt for hvert enkelt abnormitetstegn og den senere udvikling af abnormiteten |
|
— |
data for foder og vægt |
|
— |
resultaterne af de hæmatologiske analyser |
|
— |
obduktionsfund |
|
— |
detaljeret beskrivelse af alle histopatologiske fund |
|
— |
statistisk behandling af resultaterne og beskrivelse af den anvendte metoder |
|
— |
diskussion af resultaterne |
|
— |
fortolkning af resultaterne. |
3.2. VURDERING OG FORTOLKNING
Sen den generelle indledning til del B.
4. HENVISNINGER
Se den generelle indledning til del B.
B.33. KOMBINERET UNDERSØGELSE AF KARCINOGENICITET/KRONISK TOKSICITET
1. METODE
1.1. INDLEDNING
Se den generelle indledning til del B.
1.2. DEFINITIONER
Se den generelle indledning til del B.
1.3. REFERENCESTOFFER
Ingen.
1.4. PRINCIP FOR TESTMETODEN
Formålet med en kombineret undersøgelse af kronisk toksicitet/karcinogenicitet er at bestemme et stofs kroniske og karcinogene virkninger på pattedyr efter langvarig eksponering.
Med henblik herpå suppleres karcinogenicitetsundersøgelsen med mindst en satellitgruppe, der eksponeres for teststoffet, og en kontrolsatellitgruppe. Den dosis, som den højst doserede satellitgruppe eksponeres for, kan være større end den største dosis, der anvendes i karcinogenicitetsundersøgelsen. Dyrene i karcinogenicitetsundersøgelsen undersøges både for toksisk respons og for karcinogen respons. Dyrene i den eksponerede satellitgruppe undersøges for toksisk respons.
Flere grupper af forsøgsdyr doseres normalt syv dage om ugen med teststof på passende måde. Der gives en dosis pr. gruppe i størstedelen af dyrenes levetid. Under og efter doseringen af teststoffet observeres dyrene dagligt, og eventuelle tegn på toksicitet og udvikling af tumorer registreres.
1.5. KVALITETSKRITERIER
Ingen.
1.6. BESKRIVELSE AF TESTMETODEN
Dyrene holdes under forsøgsbetingelser med hensyn til miljø og fodring i mindst fem dage, inden forsøget påbegyndes. Unge, sunde, kønsmodne dyr fordeles randomiseret i test- og kontrolgrupper.
Rotten er det foretrukne forsøgsdyr. Andre dyrearter (gnavere eller ikke-gnavere) kan anvendes, hvis tidligere gennemførte undersøgelser giver grundlag herfor. Der bør benyttes unge, sunde dyr af almindeligt anvendte laboratoriestammer, og doseringen bør indledes så hurtigt som muligt, efter at dyrene er vænnet fra.
Ved forsøgets begyndelse må vægten af de anvendte dyr ikke variere mere end ± 20 % af gennemsnitsvægten. Hvis en undersøgelse af subkronisk toksicitet udføres som et forstudium til en langtidsundersøgelse, bør samme dyreart og stamme anvendes i begge undersøgelser.
Hvis der anvendes gnavere, skal der anvendes mindst 100 dyr (50 hun- og 50 handyr) til hvert dosisniveau samt en kontrolgruppe. Hundyrene må ikke have født og må ikke være drægtige. Hvis der påregnes aflivning af dyr under forsøget, bør det oprindelige antal forøges med det antal dyr, man påregner at aflive i løbet af forsøget.
Den (de) doserede satellitgruppe(r), der anvendes til bedømmelse af andre patologiske virkninger end tumorer, skal omfatte 20 dyr af hvert køn, mens kontrolsatellitgruppen skal omfatte 10 dyr af hvert køn.
Til testning af karcinogenicitet anvendes mindst tre forskellige dosisniveauer ud over kontrolgruppen. Det højeste dosisniveau bør fremkalde minimale toksicitetstegn f.eks. en let formindskelse af legemsvægten (mindre end 10 %), uden at der er tale om væsentlige ændringer af den normale levetid undtagen som følge af tumorer.
Den mindste dosis bør ikke påvirke dyrenes normale vækst, udvikling og levetid eller have nogen toksisk effekt overhovedet. I almindelighed bør denne dosis ikke være mindre end 10 % af den største dosis.
Middeldosis (evt. flere) bør ligge midtvejs mellem det højeste og laveste dosisniveau.
Ved valg af dosisstørrelser bør der tages hensyn til data fra tidligere toksicitetsundersøgelser.
Med henblik på testning af kronisk toksicitet inkluderes flere forsøgsgrupper og en dertil hørende kontrolsatellitgruppe i undersøgelsen, Den højeste dosis, som dyrene i satellitgrupperne eksponeres for, bør være så høj, at den giver tydelige tegn på toksicitet.
Dyrene doseres normalt med teststoffet hver dag.
Hvis stoffet administreres via drikkevandet eller med foderet, bør det være kontinuert tilgængeligt.
Der skal anvendes en kontrolgruppe, som på alle måder er identisk med forsøgsgrupperne bortset fra dosering med teststoffet.
Under særlige omstændigheder som f.eks, inhalationsundersøgelser, hvor der benyttes aerosoler, eller hvis der benyttes en emulgator med ukendt biologisk virkning i undersøgelser med oral indgift, anvendes en negativ kontrolgruppe, som ikke doseres hverken med teststoffet eller vehiklet.
De tre hovedformer for administration af teststof er oral indgift, påføring på huden og inhalation. Valget af administrationsform afhænger af teststoffets fysiske og kemiske karakteristika og den sandsynlige måde, hvorpå mennesker eksponeres for stoffet.
Oral indgift
Hvis teststoffet absorberes i mave-tarmkanalen, og hvis mennesker kan udsættes for indtagelse gennem munden, anvendes oral indgift, medmindre der foreligger kontraindikationer. Dyrene indgives teststoffet med foderet, opløst i drikkevandet eller i kapsler.
Den ideelle dosering er, at dyrene får indgivet teststoffet syv dage om ugen. Hvis indgift indskrænkes til fem dage om ugen, kan der ske tilbagevenden til normal, eller der kan optræde abstinenssymptomer imellem eksponeringsperioderne, hvilket kan indvirke på resultaterne og den senere vurdering. Med udgangspunkt i praktiske overvejelser, kan det accepteres, at dyrene indgives teststoffet fem dage om ugen.
Påføring på huden
Kutan påføring ved hudpensling kan vælges som model ved induktion af hudlæsioner med henblik på at simulere en vigtig human ekspositionsform.
Inhalation
Inhalationsundersøgelser giver mere komplekse tekniske problemer end andre former for administrering af teststof. I det følgende gives derfor en mere detaljeret vejledning i gennemførelse af disse undersøgelser. Man bør også være opmærksom på, at intratrakeal instillation under særlige omstændigheder kan være et egnet alternativ.
Forsøg med langtidseksposition udformes normalt på grundlag af erfaringer fra arbejdspladser, således at dyrene eksponeres fem dage om ugen seks timer dagligt, efter at koncentrationen i kammeret er ækvilibreret (intermitterende eksposition), eller der benyttes eksponering på grundlag af erfaringer fra miljøet, dvs. syv dage om ugen og 22 til 24 timer pr. dag (kontinuert eksposition) med ca. en time om dagen på et bestemt klokkeslæt til fodring af dyrene og rengøring af kammeret. I begge tilfælde eksponeres dyrene normalt for fastlagte koncentrationer af teststof. En vigtig forskel mellem intermitterende og kontinuert eksposition, som man må være opmærksom på, er, at der ved den førstnævnte metode er en periode på 17 til 18 timer, hvor dyrene kan overvinde virkningerne af den daglige eksposition og en endnu længere periode i weekenderne.
Valget mellem intermitterende og kontinuert eksposition afhænger af undersøgelsens formål og den menneskelige eksponering, der simuleres. Der er imidlertid en række tekniske vanskeligheder at tage i betragtning. F.eks. kan fordelene ved kontinuert eksponering, når det drejer sig om at simulere miljøbetingelser, falde væk på grund af behovet for vanding og fodring i ekspositionsperioden og for mere komplicerede (og pålidelige) teknikker til generering og kontrol af aerosoler og dampe.
Dyrene testes i ekspositionskamre, der er således udformet, at der kan opretholdes en dynamisk luftgennemstrømning på mindst 12 luftskift pr. time, samtidig med at der sikres et adækvat iltindhold og en jævnt fordelt ekspositionsluft. Kontrolkamre og ekspositionskamre bør være identisk med hensyn til konstruktion og udformning, så det sikres, at ekspositionsbetingelserne er sammenlignelige på alle måder bortset fra eksponering for teststoffet. Der holdes normalt et let undertryk i kammeret for at forhindre, at teststoffet lækker ud i det omgivende rum. Kammeret indrettes, således at der undgås trængsel mellem forsøgsdyrene Den generelle regel er, at hvis der skal sikres en stabil rumatmosfære, må dyrenes totale volumen ikke overstige 5 % af ekspositionskammerets volumen.
Der skal foretages måling og kontrol af:
|
i) |
Luftgennemstrømning: Det må foretrækkes, at luftstrømmen gennem kammeret kontrolleres kontinuerligt. |
|
ii) |
Koncentrationen må i den daglige eksponeringsperiode ikke variere mere end ± 15 % af den gennemsnitlige værdi. Under hele forsøget skal koncentrationen fra dag til dag holdes så konstant som praktisk muligt. |
|
iii) |
Temperatur og fugtighed: Ved anvendelse af gnavere holdes temperaturen på 22 oC (± 2 oC) og fugtigheden i kammeret mellem 30 og 70 %, undtagen når der anvendes vand til suspension af teststof i kammerets atmosfære. Det må foretrækkes, at både temperatur og fugtighed kontrolleres konstant. |
|
iv) |
Måling af partikelstørrelse: Hvis der anvendes atmosfære med flydende eller faste aerosoler i kammeret, bør der foretages en beregning af partikelstørrelsens distribution. Aerosolpartiklerne skal være respirable for de anvendte forsøgsdyr. Der tages prøver af kammerets atmosfære i dyrenes vejrtrækningszone. Luftprøverne skal være repræsentative for den partikeldistribution, dyrene eksponeres for, og det skal gennem gravimetrisk analyse være muligt på grundlag af disse luftprøver at beskrive hele aerosolen, også selv om meget af den ikke er respirabel. Under indkøring af udstyret bør der foretages hyppige analyser af partikelstørrelsen for at sikre aerosolkoncentrationernes stabilitet. Under eksponering foretages analyser så ofte, som det er nødvendigt for en adækvat bestemmelse af, om den panikeldistribution, dyrene eksponeres for, er konstant. |
Varigheden af den del af undersøgelsen, der drejer sig om karcinogenicitet, omfatter størstedelen af forsøgsdyrenes normale levetid, Hvis der anvendes mus og hamstre, bør forsøget vare 18 måneder, og hvis der anvendes rotter 24 måneder. Ved anvendelse af visse dyrestammer med længere levetid og/eller en lav spontan tumorincidens, bør forsøget vare 24 måneder med mus og hamstre og 30 måneder med rotter. Det kan dog accepteres, at en sådan forlænget undersøgelse afsluttes, når antallet af overlevende dyr i den gruppe, der doseres på det laveste dosisniveau, eller kontrolgruppen er nede på 25 %. Hvis der er åbenlys forskel i respons afhængig af dyrenes køn, bør hvert køn betragtes som et forsøg for sig, og forsøgets varighed indrettes herefter. Hvis der kun optræder præmature dødsfald i den gruppe, som doseres på det højeste dosisniveau, og hvis dette klart skyldes toksiske effekter, er det ikke nødvendigt at afslutte undersøgelsen, under forudsætning af at de toksiske effekter ikke skaber problemer i de andre grupper. Hvis et negativt testresultat skal kunne accepteres, må højst 10 % af hver gruppe falde uden for undersøgelsen på grund af autolyse af væv, kannibalisme eller forveksling, og overlevelsesprocenten i alle grupper må ikke være under 50 % efter 18 måneders forløb ved anvendelse af mus og hamstre og 24 måneder ved anvendelse af rotter.
Satellitgrupperne, som består af 20 doserede dyr pr. køn og en kontrolgruppe på 10 dyr pr. køn, og som anvendes til undersøgelse af kronisk toksicitet, holdes under forsøgsbetingelser i mindst 12 måneder. Tidspunktet for aflivning af disse dyr bør fastsættes ud fra en vurdering af patologiske fund relateret til teststof, og således at eventuelle geriatriske forandringer ikke griber forstyrrende ind.
Fremgangsmåde
Dyrene inspiceres dagligt for forandringer i hud og pels, øjne og slimhinder, åndedræt og kredsløb, det autonome og centrale nervesystem, den motoriske aktivitet og adfærdsmønster.
Dyrene i den (de) doserede satellitgruppe(r) skal undersøges klinisk med passende mellemrum.
Dyrene inspiceres regelmæssigt, for at det mindst mulige antal dyr går tabt for undersøgelsen på grund af f.eks. kannibalisme, autolyse af væv eller forveksling. Døende dyr fjernes og obduceres, når de observeres.
Kliniske symptomer, herunder neurologiske forandringer og forandringer i øjne samt mortalitet, registreres for alle dyr. Der foretages en nøje registrering af udviklingen af tumorer. De første symptomer og udvikling af toksicitetstegn registreres. Ved alle makroskopisk synlige eller palpable tumorer registreres de første symptomer, lokalisering, dimension, fremtræden og udvikling.
Foderindtagelse (og vandforbrug, hvis teststoffet indgives via drikkevandet) bestemmes en gang om ugen i de første 13 uger og derefter ca. hver tredje måned, medmindre andet indiceres af helbredstilstanden eller ændringer i legemsvægten.
Dyrene vejes enkeltvis en gang om ugen i de første 13 uger af forsøgsperioden, og mindst en gang hver fjerde uge derefter.
Kliniske undersøgelser
Hæmarologisk undersøgelse (f.eks. hæmoglobinkoncentration, hæmatokrit, erytrocyttælling, total leukocyttælling, blodpladetælling og målinger af koaguleringsevnen) udføres efter tre måneder, seks måneder og derefter ca. hver sjette måned samt ved forsøgets afslutning på blodprøver fra ti rotter pr. køn i alle grupper. Hvis det er muligt, skal prøverne tages fra de samme rotter hver gang.
Hvis kliniske observationer af dyrene tyder på, at deres helbredstilstand er blevet dårligere under forsøget, foretages en differential tælling på prøver fra de pågældende dyr. Der foretages en differential tælling på prøver fra dyrene i den gruppe, som har været doseret med den højeste dosis, og fra dyrene i kontrolgruppen. Der udføres kun differential tælling på prøver fra dyrene i den gruppe, der har fået den næsthøjeste dosis, hvis der er stor forskel på den højest doserede gruppe og kontrolgruppen, eller hvis den patologiske undersøgelse indicerer det.
Der indsamles urinprøver til analyse fra ti rotter pr. køn i alle grupper om muligt fra de samme rotter hver gang og med samme intervaller som de ovenfor nævnte blodprøver. Følgende undersøgelser gennemføres på prøver enten fra enkelte dyr eller for rotters vedkommende på en samlet urinprøve/køn/gruppe:
|
— |
udseende: rumfang og vægtfylde for hvert enkelt dyr |
|
— |
protein, glukose, ketonstof, hæmaturi (semikvantitativt), og |
|
— |
sedimentmikroskopi (semikvantitativt). |
Der udtages blodprøver til klinisk-kemisk undersøgelse med ca. seks måneders mellemrum og ved forsøgets afslutning fra hvert enkelt dyr, såfremt det drejer sig om andre arter end gnavere, og fra ti rotter/køn i alle grupper så vidt muligt fra de samme rotter hver gang. Fra andre dyr end gnavere skal der tages en prøve inden forsøget påbegyndes. Plasma fra prøverne underkastes følgende undersøgelser:
|
— |
total serumprotein |
|
— |
albumin |
|
— |
leverfunktionsundersøgelser (f.eks. alkalisk fosfataseaktivitet, serum alanin aninotransferase (ALAT) og serum aspartat aminotransferase (ASAT)), gamma glutamyl transpeptidase, ornitin decarboxylase |
|
— |
kulhydratstofskifte f.eks. fasteglukose |
|
— |
nyrefunktionsundersøgelser f.eks. urinstof. |
Alle dyr inklusive de dyr, som er døde under forsøget, eller som blev aflivet, fordi de var døende, underkastes en fuldstændig makroskopisk vurdering. Før aflivning tages blodprøver af alle dyr til differential tælling. Alle makroskopisk synlige læsioner, tumorer, som mistænkes for at være tumorer, opbevares. Makroskopiske observationer korreleres så vidt muligt med mikroskopiske fund.
Alle væv og organer bør opbevares med henblik på mikroskopisk undersøgelse. Det drejer sig normalt om følgende væv og organer: hjerne (5) (medulla/pons, det cerebellare og cerebrale cortex), hypofyse, skjoldbruskkirtel (inklusive biskjoldbruskkirtel), thymusvæv, lunger (inklusive trakea), hjerte, aorta, spytkirtler, lever (5), milt, nyrer (5), binyrer (5), spiserør, mavesæk, duodenum, jejunum, ileum, coecum, tyktarm, uterus, urinblære, lymfeknuder, bugspytkirtel, kønskirtler (5), assesoriske kønskirtler, kvindelig brystkirtel, hud, muskulatur, perifer nerve, rygmarv (cervikalt, thorakalt, lumbalt), sternum med benmarv og femur (inklusive led) og øjne.
Den optimale præparering af lunger og urinblære er inflation med et fiksativ, og i forbindelse med inhalationsundersøgelser er det centralt for de histopatologiske undersøgelser, at lungerne inflateres. I særlige undersøgelser som inhalationsundersøgelser bør de samlede luftveje undersøges, herunder næse, farynx og larynx.
Hvis der gennemføres andre kliniske undersøgelser, bør resultaterne af disse foreligge, før der foretages mikroskopiske undersøgelser, fordi de kan være af afgørende værdi for patologen.
For den del af undersøgelsen, der drejer sig om kronisk toksicitet:
Der bar foretages en detaljeret undersøgelse af alle opbevarede organer fra dyr i den højest doserede satellitgrupe og kontrolgruppen. Hvis der findes patologiske tegn, som kan relateres til teststoffet i den højest doserede satellitgruppe, underkastes målorganerne hos dyrene i de øvrige doserede satellitgrupper samt de doserede grupper i den del af undersøgelsen, der drejer sig om karcinogenicitet, en fuldstændig og detaljeret histologisk undersøgelse ved forsøgets afslutning.
For den del af undersøgelsen, der drejer sig om karcinogenicitet:
|
a) |
Der skal foretages en fuldstændig histopatologisk undersøgelse af organer og væv fra de dyr, som dør eller aflives under forsøget, samt dyrene i kontrolgruppen og i den gruppe, der er blevet doseret med den højeste dosis. |
|
b) |
Alle makroskopisk synlige tumorer eller læsioner, som mistænkes for at være tumorer, undersøges. |
|
c) |
Hvis der er signifikant forskel på de neoplastiske læsioners incidens i den højest doserede gruppe og kontrolgrupper, skal de relevante organer og væv undersøges histopatologisk i de øvrige grupper. |
|
d) |
Hvis overlevende i den højest doserede gruppe er væsentlig lavere end i kontrolgruppen, underkastes den gruppe, der er doseret med den næsthøjeste dosis, en fuldstændig histopatologisk undersøgelse. |
|
e) |
Hvis undersøgelsen af den højest doserede gruppe giver grund til at formode, at der er tale om induktion af toksiske eller andre effekter, som kan have betydning for et neoplastisk respons, underkastes den gruppe, der eksponeres for den næsthøjeste dosis, en fuldstændig histopatologisk undersøgelse. |
2. DATA
Data opstilles i tabelform, som for hver gruppe viser antal dyr ved begyndelsen af forsøget, antal dyr med tumorer eller toksisk reaktion, som er registreret under forsøget, registreringstidspunkt og antal dyr, hos hvem der er fundet tumorer efter aflivning. Evaluering af resultaterne skal baseres på en passende statistisk metode. Alle anerkendte statistiske metoder kan anvendes.
3. RAPPORTERING
3.1. FORSØGSRAPPORT
Forsøgsrapporten skal om kuligt indeholde følgende.
|
— |
fyreart, stamme, herkomst, miljøbetingelser, foder osv. |
|
— |
forsøgsbetingelser: Beskrivelse af ekspositionsapparatur herunder udformning, type, dimensionering, luftkilde, system til frembringelse af partikler og aerosoler, metode til konditionering af luften, behandling af udpumpet luft og den måde, hvorpå dyrene holdes i et ekspositionskammer, når et sådant benyttes. Apparatur til måling af temperatur, fugtighed og eventuelle partikulære aerosolers koncentrationer og partikelstørrelse beskrives. Ekspositionsdata: Disse skal opstilles i tabelform og angive middelværdier og variation (f.eks. standardafvigelser) og bør omfatte:
|
|
— |
dosisniveauer (inklusive vehikel, hvor et sådant er anvendt) og koncentrationer |
|
— |
oplysning om tumorincidens inddelt efter køn, dosis og tumortype |
|
— |
dødstidspunkt under forsøget eller overlevelse til forsøgets afslutning, inklusive satellitgruppen |
|
— |
oplysning om toksisk respons inddelt efter køn og dosis |
|
— |
toksisk eller anden virkning |
|
— |
observationstidspunkt for hvert enkelt abnormitetstegn og den senere udvikling af abnormiteten |
|
— |
oftalmologiske fund |
|
— |
data for foder og vægt |
|
— |
hæmatologiske analyser og samtlige resultater |
|
— |
resultaterne af de klinisk-biokemiske analyser (herunder urinanalyse) |
|
— |
obduktionsfund |
|
— |
detaljeret beskrivelse af alle histopatologiske fund |
|
— |
statistisk behandling af resultaterne og beskrivelse af den anvendte metode |
|
— |
diskussion af resultaterne |
|
— |
fortolkning af resultaterne. |
3.2. VURDERING OG FORTOLKNING
Se den generelle indledning til del B.
4. HENVISNINGER
Se den generelle indledning til del B.
B.34. REPRODUKTIONSTOKSICITETSUNDERSØGELSE I EN GENERATION
1. METODE
1.1. INDLEDNING
Se den generelle indledning til del B.
1.2. DEFINITIONER
Se den generelle indledning til del B.
1.3. REFERENCESTOFFER
Ingen.
1.4. PRINCIP FOR TESTMETODEN
Flere grupper af han- og hundyr doseres med teststoffet, idet der gives en dosis pr. gruppe. Handyr doseres under væksten og under mindst en fuldstændig spermiedannelsescyklus (ca. 56 dage for mus og 70 dage for rotter) med henblik på at fremkalde sundhedsmæssige uønskede virkninger, teststoffet måtte have på spermatogenesen.
Hundyr i F0-generationen doseres i mindst to fuldstændige østrale perioder for at fremkalde de skadelige virkninger, teststoffet eventuelt måtte have på brunsten. Derefter parres dyrene. Begge køn doseres med teststof i parringsperioden, og derefter doseres kun hundyrene i drægtighedsperioden og laktationsperioden. Hvis dosering med teststof foregår ved inhalation, kræves ændringer af metoden.
1.5. KVALITETSKRITERIER
Ingen.
1.6. BESKRIVELSE AF TESTMETODEN
Forberedelser
Unge, sunde, kønsmodne dyr fordeles randomiseret i grupper. Dyrene holdes under forsøgsbetingelser med hensyn til miljø og fodring i mindst fem dage, inden forsøget påbegyndes. Det anbefales, at teststoffet administreres med foderet eller drikkevandet. Andre former for administration kan også accepteres. Alle dyr skal doseres på samme måde i hele forsøgsperioden. Hvis der anvendes et vehikel eller andre tilsætningsstoffer for at lette doseringen, skal det være påvist, at de ikke har nogen toksisk virkning. Dyrene skal doseres med teststoffet syv dage om ugen.
Valg af dyreart
Rotten eller musen er de foretrukne forsøgsdyr. Stammen bør ikke have en lav fertilitet. Der benyttes sunde dyr, som ikke tidligere har været anvendt i forsøg. Forsøgsdyrene bør være velbeskrevne med hensyn til art, stamme, køn, vægt og/eller alder.
For at få en adækvat vurdering af fertilitet skal både han- og hundyr undersøges. Alle dyr i forsøgsgrupper og kontrolgrupper skal være fravænnet, før dosering påbegyndes.
Antal og køn
Hver enkelt forsøgs- og kontrolgruppe skal indeholde tilstrækkelig mange dyr, til at der kan findes ca. 20 drægtige hundyr, som er nær ved eller på fødetidspunktet.
Formålet hermed er at sikre, at der produceres tilstrækkelig meget afkom, til at der kan foretages en meningsfuld vurdering af teststoffets potentielle virkning på fertilitet, drægtighed og moderdyrenes adfærd i F0-generationen samt dieadfærd, vækst og udvikling i F1-generationen fra undfangelse til dyrene er vænnet fra.
Foder og vand gives ad libitum. Når fødetidspunktet nærmer sig, placeres de drægtige hundyr i hvert sit bur, idet der benyttes fødebure eller yngelbure, og dyrene kan eventuelt forsynes med materiale til redebygning.
Der anvendes mindst tre forskellige dosisniveauer samt en kontrolgruppe. Hvis der anvendes et vehikel, doseres kontrolgruppen med samme mængde vehikel som den gruppe, der modtager den højeste dosis. Hvis et teststof fører til formindsket foderindtagelse eller interfererer med absorption og stofskifte, kan det være nødvendigt at anvende en kontrolgruppe, som er matchet med hensyn til foder. Medmindre der optræder begrænsninger som følge af teststoffets fysisk-kemiske natur eller biologiske egenskaber, bør den højeste dosis forårsage toksisk virkning, men ikke dødsfald i forældre (F0)-generationen. Middeldosis (evt. flere) bør fremkalde de mindste konstaterbare toksiske virkninger af teststoffet, og den laveste dosis bør ikke have nogen toksisk effekt hverken på forældregenerationen eller afkommet, Hvis stoffet indgives med mavesonde eller i kapsel, bør den dosis, som hvert enkelt dyr modtager, udregnes på grundlag af dyrets legemsvægt og ugentligt tilpasses ændringer i denne legemsvægt. Dosering af drægtige hundyr kan baseres på legemsvægten på dag 0 eller 6, hvis man ønsker det.
Hvis der ikke konstateres nogen virkning på reproduktionen ved en dosis på mindst 1 000 mg/kg af et lavtoksisk stof, kan yderligere testning betragtes som unødvendig. Hvis en indledende undersøgelse, hvor kun den højeste dosis benyttes, klart viser toksisk virkning på moderdyret, men ikke har nogen skadelig virkning på fertiliteten, kan yderligere testning betragtes som unødvendig.
Fremgangsmåde
Daglig dosering af forældre (F0)-generationen indledes, når dyrene er mellem fem og ni uger gamle, og efter at de er vænnet fra og akklimatiseret i mindst fem dage. Hvis der anvendes rotter, fortsættes doseringen i ti uger før parringsperioden (ved mus otte uger). Handyrene skal enten aflives og undersøges efter parringsperioden, eller man kan fortsætte med at indgive dem teststoffet frem til en mulig produktion af et andet kuld, hvorefter de aflives og undersøges, et stykke tid før undersøgelsen afsluttes. Dosering af forældregenerationens hundyr indledes efter mindst fem dages akklimatisering og fortsætter mindst to uger før parring. Daglig dosering af F0-hundyrene fortsættes gennem den tre uger lange parringsperiode, drægtighedsperioden og op til F1-generationen er vænnet fra. Man bør overveje eventuelle ændringer i forsøgsplanen, hvis der foreligger informationer om teststoffet fra specielle stofskifteundersøgelser eller undersøgelse af akkumulation.
Der kan anvendes parring i forholdet 1:1 (et handyr og et hundyr) eller 1:2 (et handyr og to hundyr) i undersøgelser af reproduktionstoksicitet.
Anvendes 1:1 parring, anbringes et hundyr med ethandyr, indtil der indtræder drægtighed, eller tre uger er gået, Hver morgen undersøges hundyrene for tilstedeværelse af sperma eller vaginalprop. Drægtighedsperiodens dag 0 fastlægges under hensyntagen til spermatogenesen som den dag, hvor der observeres vaginalprop eller sperma.
De par, som ikke parrer sig, bør undersøges for fastlæggelse af årsagen til den tilsyneladende ufrugtbarhed.
Dette kan gøres ved, at dyrene f.eks. får lejlighed til at parre sig med han- eller hundyr, hvis frugtbarhed er påvist, ved mikroskopisk undersøgelse af reproduktionsorganerne og ved undersøgelse af den østrale periode eller spermatogenesen.
De dyr, som doseres i fertilitetsundersøgelsen, bør have lov til at yngle normalt og pleje deres afkom frem til afvænning uden standardisering.
Hvis der gennemføres standardisering, foreslås følgende fremgangsmåde. Mellem dag 1 og 4 efter fødslen, standardiseres hvert kuld, ved at overskydende unger fjernes, således at hvert kuld kommer så tæt som muligt på at bestå af fire handyr og fire hundyr,
Hvis det på grund af antallet af han- og hundyr ikke er muligt at standardisere hvert kuld, så det indeholder fire af hvert køn, kan delvis standardisering accepteres (f.eks. fem handyr og tre hundyr). Der foretages ikke standardisering af kuld på mindre end otte unger.
Dyrene inspiceres mindst en gang om dagen under hele forsøget. Relevante adfærdsændringer, tegn på vanskelig eller forlænget fødsel og alle toksicitetstegn herunder mortalitet, registreres. Før og under parringsperioden registreres foderindtagelsen ugentligt. I drægtighedsperioden kan foderindtagelsen (og vandindtagelsen, hvis teststoffet administreres heri) registreres dagligt. Efter fødslen og i dieperioden måles foderindtagelsen på de samme dage, som kuldene vejes. Han- og hundyr af F0-generationen vejes den dag, doseringen indledes og derefter en gang om ugen. Disse observationer bar rapporteres individuelt for hvert enkelt udvokset dyr.
Drægtighedsperioden beregnes fra dag 0. Hvert kuld undersøges så hurtigt som muligt efter fødslen for at fastslå ungernes antal og køn, antal dødfødte og levendefødte og forekomst af makroskopisk synlige abnormiteter.
Døde unger og unger, som aflives på dag 4, skal opbevares og undersøges for mulige defekter. Levende unger tælles, og kuldet vejes morgenen efter fødslen, på dag 4 og dag 7, og derefter en gang om ugen indtil undersøgelsens afslutning, hvor dyrene skal vejes individuelt.
Fysiske eller adfærdsmæssige abnormiteter, som observeres hos hundyrene eller afkommet skal registreres.
Patologisk undersøgelse
Fo-generationsdyr, som aflives efter eller dør under forsøget, undersøges makroskopisk med henblik på konstatering af strukturelle abnormiteter eller patologiske forandringer, idet opmærksomheden særlig rettes mod reproduktionssystemet. Døde eller døende unger undersøges for defekter.
Ovarier, uterus, cervix, vagina, testikler, bitestikler, sædblærer, prostata, accessoriske kønskirtler, hypofyse og målorgan(er) fra alle dyr i F0-generationen præpareres med henblik på mikroskopisk undersøgelse. Skulle det undtagelsesvist forekomme, at disse organer endnu ikke har været underkastet undersøgelse i andre forsøg med multipel dosering, undersøges organerne fra dyrene i den gruppe, der har modtaget den højeste dosis, og i kontrolgruppen samt de dyr, som er døde under forsøget, mikroskopisk, hvis det er praktisk muligt.
Hvis der konstateres abnormiteter i organer fra disse dyr, undersøges disse organer også hos alle andre dyr i F0-generationen. Der foretages i så fald mikroskopisk undersøgelse af alle væv, som udviser makroskopisk patologiske forandringer. Som anført i afsnittet parring kan reproduktionsorganerne fra dyr, som formodes at være ufrugtbare, også underkastes mikroskopisk undersøgelse.
2. DATA
Data opstilles i tabelform, som for hver gruppe viser antal dyr ved begyndelsen af forsøget, antal frugtbare handyr, antal drægtige hundyr, typer af forandringer og den procentdel dyr, som udviser hver enkelt type forandring.
Evaluering af resultaterne baseres så vidt muligt på en passende statistisk metode. Enhver anerkendt statistisk metode kan anvendes.
3. RAPPORTERING
3.1. FORSØGSRAPPORT
Forsøgsrapporten skal om muligt indeholde følgende:
|
— |
anvendt dyreart/stamme |
|
— |
oplysning om toksisk respons inddelt efter køn og dosis, herunder fertilitet, drægtighed og afkommets levedygtighed |
|
— |
dødstidspunkt under forsøget eller overlevelse til forsøgets afslutning eller planlagt aflivningstidspunkt |
|
— |
tabel med angivelse af vægten af hvert enkelt kuld, ungernes gennemsnitlige vægt og hver enkelt unges vægt ved forsøgets afslutning |
|
— |
toksisk eller anden virkning på reproduktion, afkom, postnatal vækst |
|
— |
observationstidspunkt for hvert enkelt abnormitetstegn og den senere udvikling af abnormiteten |
|
— |
data for foder- og legemsvægt for Fo-generationen |
|
— |
obduktionsfund |
|
— |
detaljeret beskrivelse af alle mikroskopiske fund |
|
— |
eventuel statistisk behandling af resultaterne |
|
— |
diskussion af resultaterne |
|
— |
fortolkning af resultaterne. |
3.2. VURDERING OG FORTOLKNING
Se den generelle indledning til del B.
4. HENVISNINGER
Se den generelle indledning til del B.
B.35. REPRODUKTIONSTOKSICITETSUNDERSØGELSE I TO GENERATIONER
1. METODE
Metoden er gengivet efter OECD TG 416 (2001).
1.1. INDLEDNING
Formålet med denne metode til reproduktionsundersøgelse i to generationer er at skaffe generelle oplysninger om et teststofs effekter på forplantningssystemets integritet og funktion hos han- og hundyr, herunder gonadefunktion, brunstcyklus, parringsadfærd, konception, drægtighed, fødsel, laktation og fravænning samt afkommets vækst og udvikling. Undersøgelsen kan desuden give oplysninger om teststoffets effekter på den neonatale morbiditet og mortalitet samt foreløbige data om præ- og postnatal udviklingstoksicitet, foruden at tjene som vejledning for efterfølgende forsøg. Ud over at undersøge Fl-generationens vækst og udvikling har metoden til formål at vurdere forplantningssystemets integritet og funktion hos han- og hundyr samt vækst og udvikling i F2-generationen. For at skaffe supplerende viden om udviklingstoksicitet og funktionelle mangler kan man enten supplere denne protokol med forsøgsafsnit, som alt efter behov er baseret på metoder til undersøgelse af udviklingstoksicitet og/eller udviklingsneurotoksicitet, eller gøre disse endpoints til genstand for særskilte forsøg med egnede forsøgsmetoder.
1.2. FORSØGSMETODENS PRINCIP
Teststoffet administreres i graduerede doseringer til flere grupper hanner og hunner. Hannerne i P-generationen (F0-generationen) modtager stoffet i vækstperioden og i mindst én hel spermatogenesecyklus (ca. 56 døgn hos mus og 70 døgn hos rotter) for at fremkalde eventuelle negative virkninger på spermatogenesen. Effekterne på sperma bestemmes ved en række spermaparametre (f.eks. spermiemorfologi og -motilitet) og ved fremstilling af vævspræparater til detaljeret histopatologisk undersøgelse. Hvis der foreligger data vedrørende spermatogenesen fra et tidligere forsøg med gentagen dosering af tilstrækkelig varighed, f.eks. et 90-dages forsøg, behøver hannerne i P-generationen ikke indgå i vurderingen. Det anbefales dog at opbevare prøver eller digitale optagelser af spermier fra P-generationen med henblik på senere vurdering. Hunner i P-generationen doseres i vækstperioden og gennem flere hele brunstcyklusser for at bestemme eventuelle negative virkninger af teststoffet på den normale brunstcyklus, Teststoffet administreres til forældredyrene (P) i parringsperioden, gennem den resulterende drægtighedsperiode og gennem fravænningsperioden af afkommet F1, Fra fravænningstidspunktet administreres stoffet til F1-afkommet under dettes opvækst og ind i voksenalderen, parringsperioden og frembringelse af en F2-generation, indtil F2-generationen er fravænnet.
Alle dyr iagttages klinisk og undersøges patologisk for tegn på toksicitet med særlig vægt på integritet og funktion af forplantningssystemet hos han- og hundyr samt afkommets vækst og udvikling.
1.3 BESKRIVELSE AF FORSØGSMETODEN
1.3.1. Valg af dyreart
Rotten er den foretrukne art til forsøget. Anvendes andre arter, bør dette begrundes, og passende modifikationer vil være nødvendige. Der bør ikke anvendes stammer med ringe frugtbarhed eller kendt høj forekomst af udviklingsdefekter. Ved forsøgets begyndelse skal vægtvariationen mellem de anvendte dyr være mindst mulig og for hvert køn ikke over 20 % af gennemsnitsvægten.
1.3.2. Miljøbetingelser og fodring
Temperaturen i forsøgsdyrrummet skal være 22 oC (± 3 oC). Den relative luftfugtighed skal være mindst 30 % og bør ikke overstige 70 % bortset fra under rengøring af rummet, men 50-60 % bør tilstræbes. Belysningen skal være kunstig bestående af 12 timers lys og 12 timers mørke. Som foder kan anvendes sædvanligt laboratoriefoder og fri adgang til drikkevand. Valget af foder kan afhænge af nødvendigheden af at sikre passende iblanding af teststoffet, når denne administrationsmåde anvendes.
Dyrene kan enten anbringes enkeltvis eller i bure med små grupper af samme køn. Parring bør finde sted i bure egnede til formålet. Efter tegn på kopulation anbringes de parrede hunner enkeltvis i fødebure eller yngelbure. De parrede rotter kan også holdes i små grupper og adskilles en eller to dage før fødslen. Parrede dyr skal forsynes med egnet og nærmere bestemt redemateriale, når fødselstidspunktet nærmer sig.
1.3.3. Forberedelse af dyrene
Der anvendes raske unge dyr. som er akklimatiseret til laboratoriemiljøet i mindst fem døgn og ikke tidligere er indgået i forsøg, Forsøgsdyrene skal være karakteriseret med hensyn til art, stamme, oprindelse, køn, vægt og/eller alder. Eventuelle søskenderelationer mellem dyrene skal være kendt, så parring af søskende undgås. Dyrene fordeles randomiseret i forsøgs- og kontrolgrupper (stratificering efter kropsvægt anbefales). Burene skal arrangeres således, at eventuelle virkninger af burenes placering er mindst mulig. Hvert dyr tilordnes et unikt identifikationsnummer, For P-generationen skal dette ske inden doseringen begynder, For F1-generationen skal dette ske ved fravænningen for dyr, som udvælges til parring. For alle udvalgte F1-dyr registreres, hvilket kuld de er fra. Når det overvejes at benytte individuel vejning af unger eller foretage funktionstest, anbefales, at ungerne identificeres individuelt snarest efter fødslen.
Forældredyr (P) skal være 5 til 9 uger gamle, når doseringen påbegyndes. Dyrene i alle forsøgsgrupper skal så vidt muligt være af ensartet vægt og alder.
1.4. FREMGANGSMÅDE
1.4.1. Dyrenes antal og køn
I hver behandlings- og kontrolgruppe skal der være tilstrækkeligt med dyr til, at gruppen ved eller nær fødselstidspunktet består af helst mindst 20 drægtige hunner, For stoffer, som er årsag til uønskede behandlingsrelaterede virkninger (f.eks. sterilitet, stærk toksicitet i den høje dosering), kan dette eventuelt være umuligt. Formålet er at producere tilstrækkeligt med afkom til at sikre en meningsfuld vurdering af teststoffets potentiale for påvirkning af fertilitet, drægtighed og moderdyrenes adfærd og diegivning, Fl-afkommets vækst og udvikling fra undfangelsen til fuld kønsmodning, og udviklingen af dettes afkom (F2), frem til dyrene er vænnet fra. Hvis det ikke er lykkedes at frembringe det ønskede antal drægtige dyr (dvs. 20), er undersøgelsen ikke dermed nødvendigvis ugyldig, og der bør finde en konkret vurdering sted i det enkelte tilfælde.
1.4.2. Tilberedning af doser
Det anbefales, at teststoffet administreres oralt (i foderet, i drikkevandet eller med sonde), medmindre en anden administrationsvej (f.eks. dermal applikation eller inhalation) anses for mere velegnet.
Om nødvendigt opløses eller opslæmmes teststoffet i et egnet vehikel. Det anbefales, at man som første valg vurderer muligheden af at benytte en vandig opløsning/suspension, derefter en opløsning/suspension i olie (f.eks. majsolie), og først derefter eventuel opløsning i andre vehikler. For andre vehikler end vand må vehiklets toksiske egenskaber være kendt. Teststoffets stabilitet i vehiklet skal bestemmes.
1.4.3. Dosering
Der skal være mindst tre dosisgrupper og en sideløbende kontrolgruppe. Medmindre der optræder begrænsninger som følge af teststoffets fysisk-kemiske natur eller biologiske egenskaber, skal den højeste dosering vælges således, at den giver toksisk effekt, men ikke død eller alvorlige lidelser. I tilfælde af uventet dødelighed er forsøg med en dødelighed på under ca. 10 % i forældregenerationen (P) normalt stadig acceptable. Der vælges en aftagende række dosisniveauer, som giver mulighed for at påvise en eventuel dosisafhængig respons samt det niveau, hvor ingen skadelig effekt iagttages (NOAEL). Ved fastlæggelse af en sådan aftagende række dosisniveauer vil det optimale interval ofte svare til en faktor to til fire, og i mange tilfælde må det foretrækkes at tilføje en fjerde behandlingsgruppe frem for at operere med meget store intervaller (f.eks. større end en faktor 10) mellem dosisniveauerne. I undersøgelser med indgift i foderet bør dosisintervallet ikke være større end svarende til en faktor 3. Doseringen vælges under hensyntagen til eventuelle foreliggende toksicitetsdata, især resultaterne af forsøg med gentagen dosering. Eventuelle foreliggende oplysninger om metabolisme og kinetik af teststoffet eller beslægtede stoffer skal ligeledes tages i betragtning. Sådanne oplysninger kan desuden være med til at godtgøre det hensigtsmæssige i den valgte dosering.
Kontrolgruppen skal være ubehandlet eller behandles med vehikel, hvis et sådant anvendes til administration af teststoffet. Bortset fra behandling med teststof skal dyrene i kontrolgruppen (-grupperne) håndteres på samme måde som behandlingsgruppen. Anvendes vehikel, skal dyrene i kontrolgruppen modtage vehiklet i det største anvendte volumen. Hvis et teststof administreret i foderet medfører reduceret fødeindtagelse eller -udnyttelse, kan parvis fodring sammen med kontrolgruppen være nødvendig. I stedet for parvis fodring sammen med kontrolgruppen kan anvendes data fra kontrollerede undersøgelser, som er udformet til vurdering af virkningerne af nedsat fødeindtagelse på reproduktionsparametrene.
For vehiklet og andre tilsætningsstoffer skal følgende egenskaber tages i betragtning: Dets effekt på teststoffets absorption, fordeling, metabolisme og retention, dets eventuelle indvirkning på teststoffets kemiske egenskaber med deraf følgende ændring af dettes toksiske egenskaber, og dets indvirkning på dyrenes føde- og vandindtagelse og ernæringstilstand.
1.4.4. Grænsetest
Såfremt et oralt enkeltdosisniveau på mindst 1 000 mg/kg kropsvægt/døgn eller en ækvivalent procentdel tilsat foderet eller drikkevandet med de til dette forsøg beskrevne metoder hverken frembringer observerbar toksisk virkning i forældredyrene eller deres afkom, og såfremt foreliggende data fra strukturelt og/eller metabolisk beslægtede forbindelser ikke giver anledning til at forvente nogen virkning, er et komplet forsøg med flere dosisniveauer ikke nødvendigvis påkrævet, Grænsetesten skal være gældende, medmindre den påregnede udsættelse af mennesker tilsiger, at der anvendes et højere oralt dosisniveau. For andre administrationsmåder — f.eks. inhalation eller dermal applikation — kan teststoffets fysisk-kemiske egenskaber, f.eks. opløseligheden, ofte være bestemmende for og begrænsende for det maksimale opnåelige ekspositionsniveau.
1.4.5. Administration af doser
Dyrene modtager teststoffet syv dage om ugen. Oral administration (foder, drikkevand eller sonde) er at foretrække. Anvendes anden administrationsvej, bør valget begrundes, og passende modifikationer kan være nødvendige. Alle dyr skal modtage stoffet på samme måde i den pågældende forsøgsperiode. Administreres teststoffet med sonde, skal en gastrisk sonde anvendes. Der må ikke på én gang gives et væskevolumen over 1 ml/l00 g kropsvægt (for majsolie ikke over 0,4 ml/100 g kropsvægt), bortset fra vandige opløsninger, som kan gives i mængder på 2 ml/100 g kropsvægt, Hvis der ikke er tale om lokalirriterende eller ætsende stoffer, hvis effekt sædvanligvis er værre ved højere koncentration, skal variabiliteten af testvolumenet minimeres gennem justering af koncentrationen, således at der sikres et konstant volumen ved alle dosisniveauer. I forsøg med indgift gennem sonde vil ungerne normalt kun få teststoffet indirekte gennem mælken, indtil den direkte dosering begynder ved fravænning. I forsøg med indgift gennem foder eller drikkevand vil ungerne desuden modtage teststoffet direkte, når de begynder selv at æde i den sidste del af laktationsperioden.
For stoffer administreret i foderet eller drikkevandet er det vigtigt at sikre, at det pågældende teststof i den anvendte mængde ikke griber ind i den normale ernærings- eller vandbalance, Ved indgift af teststoffet i foderet kan stoffet enten gives ved konstant koncentration i foderet (ppm) eller ved et konstant dosisniveau i forhold til dyrets kropsvægt; det skal angives, hvilken mulighed der er valgt. Ved indgift med sonde skal dosis gives pa tilsvarende tidspunkt hver dag og skal justeres mindst en gang om ugen, så dosisniveauet holdes konstant i forhold til kropsvægten. Ved justering af doseringen via sonde på grundlag af vægten skal kendskabet til fordelingsforholdene i placenta tages betragtning,
1.4.6. Forsøgsplaner
Den daglige indgift til hanner og hunner i forældregenerationen (P) begynder, når dyrene er 5 til 9 uger gamle. Den daglige dosering af hanner og hunner i F1-generationen begynder ved fravænningen; man bør have for øje. at ved administration af teststoffet i foder eller drikkevand kan ungerne i F1-gruppen blive direkte eksponeret for teststoffet allerede i laktationsperioden. For begge køn (P og F1) skal doseringen fortsætte i mindst 10 uger før parringsperioden. Doseringen fortsætter hos begge køn i den to uger lange parringsperiode. Når der ikke længere er brug for handyrene til vurdering af reproduktionstoksiske virkninger, aflives de af humane grunde og undersøges. For hunner i forældregenerationen (P) fortsætter doseringen gennem hele drægtighedspeiioden og indtil fravænning af afkommet i F1-generationen. Ændringer i doseringsplanen må overvejes ud fra de foreliggende oplysninger om teststoffet, herunder foreliggende toksicitetsdata, induktion af metabolisering samt bioakkumulering. Doseringen til det enkelte dyr bør normalt baseres på den seneste individuelle bestemmelse af dyrets vægt. I drægtighedsperiodens sidste trimester må dog udvises forsigtighed med justering af doseringen.
Behandlingen af hanner og hunner i P- og F1-generationen skal fortsætte til forsøgets afslutning, Af humane grunde aflives alle voksne hanner og hunner i P- og F1-generationen, når der ikke længere er brug for dem til vurdering af reproduktionstoksiske virkninger. Af humane grunde aflives de dyr i F1 generationen, som ikke udvælges til parring, og alle dyr i F2-generationen.
1.4.7. Parringsmetode
1.4.7.1. Parring af forældregenerationen (P)
Til hver parring anbringes hver hun sammen med én han på samme dosisniveau (1:1 parring), indtil kopulation finder sted eller der er gået to uger. Hunnerne undersøges hver dag for tilstedeværelse af sperma eller vaginalprop. Drægtighedsperiodens dag 0 er den dag, hvor der findes en vaginalprop eller sperma. Lykkes parring ikke, kan fornyet parring af hunnerne med afprøvede hanner fra samme gruppe overvejes. De parrede dyr skal være tydeligt identificeret i data. Parring af søskende skal undgås.
1.4.7.2. Parring af F1
Til parring af Fl-afkommet vælges mindst én han og én hun fra hvert kuld ved afvænningen til parring med andre unger på samme dosisniveau, men fra et andet kuld, med henblik på frembringelse af F2-generationen. Valg af unger fra hvert kuld skal ske tilfældigt, når der ikke iagttages væsentlige forskelle i kropsvægt eller udseende mellem magerne fra kuldene. Iagttages sådanne forskelle, vælges den bedste repræsentant for hvert kuld. Det mest pragmatiske er at dette sker på vægtbasis, men det kan være mere hensigtsmæssigt at lade det ske på basis af udseendet. F1-afkommet må ikke parres, før de er fuldt kønsmodne.
Par uden afkom vurderes med henblik på bestemmelse af den tilsyneladende årsag til steriliteten. Dette kan bestå i at give mulighed for en ekstra parring med afprøvede fædre eller mødre, makroskopisk undersøgelse af kønsorganerne og undersøgelse af brustcyklus eller spermatogenese.
1.4.7.3. Anden parring
I visse tilfælde, således ved behandlingsrelaterede ændringer i kuldstørrelse eller tvivlsom effekt af første parring, anbefales det, at de voksne dyr i P- eller F1-generationen parres igen, så de får endnu et kuld unger. Det anbefales, at de hanner eller hunner, som ikke har frembragt afkom, parres igen med påvist yngledygtige dyr af det modsatte køn. Hvis det skønnes nødvendigt at producere et ekstra kuld i nogen af generationerne, skal dyrene parres igen ca. en uge efter fravænning af sidste kuld.
1.4.7.4. Kuldstørrelse
Dyrene skal yngle på normal måde og pleje afkommet indtil fravænning. Standardisering af kuldstørrelserne er frivillig. Anvendes standardisering, skal metoden beskrives i detaljer.
1.5. OBSERVATIONER
1.5.1. Klinisk observation
Dyrene observeres dagligt klinisk, og ved sondeindgift foretages denne observation under hensyntagen til tidspunktet for den forventede maksimale virkning efter administrationen. Adfærdsændringer, tegn på vanskelig eller langstrakt fødsel og alle tegn på toksicitet registreres. En yderligere, mere detaljeret undersøgelse af hvert dyr foretages mindst en gang ugentligt, hvilket passende kan ske i forbindelse med vejning af dyret. Alle dyr iagttages to gange dagligt for morbiditet ug mortalitet, i weekenden én gang dagligt når det er hensigtsmæssigt.
1.5.2. Forældredyrenes kropsvægt og føde- og vandindtagelse
Forældredyrene (P og F1) vejes den første dag de modtager stoffet og derefter mindst en gang om ugen. Hunnerne i forældregenerationerne (P og F1) vejes som minimum på dag 0, 7, 14, og 20 eller 21 i drægtighedsperioden, i laktationsperioden på de dage, hvor kuldene vejes, samt på aflivningsdagen. Disse iagttagelser rapporteres individuelt for hvert voksent dyr. I tiden før parring og i drægtighedsperioden skal fødeindtagelsen måles mindst en gang om ugen. Vandindtagelsen skal måles mindst en gang om ugen, hvis teststoffet gives i vandet.
1.5.3. Brunstcyklus
Hos hunnerne i P- og F1-generationen vurderes brunstcyklussens længde og normale forløb med vaginalt smear før parring samt ikke obligatorisk i parringsperioden, indtil der findes tegn på parring, Udtagning af celleprøve fra vagina/cervix skal ske forsigtigt for at undgå forstyrrelse af slimhinden med efterfølgende induktion af pseudodrægtighed (1).
1.5.4. Spermaparametre
For alle hanner i P- og F1 generationen registreres vægten af testes og epididymides ved forsøgets slutning, og det ene af hvert organ udtages til histopatologisk undersøgelse (se punkt 1.5.7, 1.5.8.1). Fra en delmængde bestående af mindst 10 hanner af hver gruppe hanner i P- og F1-generationen anvendes de øvrige testes og epididymides til tælling af henholdsvis homogeniseringsresistente spermatider og spermiebeholdning i cauda epididymidis. For samme delmængde af hanner opsamles sperma fra cauda epididymidis eller vas deferens til undersøgelse af spermiemotilitet og spermiemorfologi. Hvis der iagttages behandlingsrelaterede virkninger eller der i andre undersøgelser er fundet tegn på en mulig påvirkning af spermatogenesen, skal spermaundersøgelse finde sted hos alle hanner i hver dosisgruppe; ellers kan optællingen indskrænkes til kontrolgruppen og hannerne i højdosisgrupperne i P og F1.
Det samlede antal homogeniseringsresistente testikulære spermatider og spermier i cauda epididymidis opgøres (2)(3). Spermiebeholdningen i cauda kan beregnes af spermiekoncentration og -volumen i den suspension, der benyttes til den kvalitative vurdering og antallet af spermier, som opsamles ved findeling og/eller homogenisering af det øvrige caudavæv. Optællingen på den valgte delmængde af hanner af alle dosisgrupper foretages straks efter aflivning af dyrene, medmindre prøverne fryses og analyseres senere, I disse tilfælde kan kontroldyrene og højdosisgruppen analyseres først. Hvis der ikke optræder behandlingsrelaterede virkninger (på f.eks, spermietal, -motilitet og -morfologi), behøver de andre dosisgrupper ikke analyseres. Findes der behandlingsrelaterede virkninger i højdosisgruppen, skal grupperne på lavere dosis også analyseres.
Motiliteten af spermier i epididymis (eller ductus deferens) bedømmes eller optages på videobånd straks efter aflivning. Udtagning af af spermier skal ske på en måde, så skaden er mindst muligt, og spermierne fortyndes med henblik på motilitetsanalyse med acceptable metoder (4). Procentdelen af fremadbevægelige (progressivt motile) spermier bestemmes enten subjektivt eller objektivt. Foretages der computerassisteret motilitetsanalyse (5)(6)(7)(8)(9)(10) er beregningen af progressiv motilitet baseret på brugerdefinerede tærskelværdier for gennemsnitsvejlængde, hastighed og retlinjethed (lineært indeks). Hvis der optages videobånd af prøverne (11) eller billederne på anden måde registreres på obduktionstidspunktet, kan den efterfølgende analyse af handyr i P- og F1-generationen indskrænkes til kontrol- og højdosisgrupppen, medmindre der konstateres behandlingsrelaterede virkninger, i hvilket tilfælde også grupperne på de lavere doseringer skal bedømmes. I mangel af videooptagelse eller digitalt billede skal alle prøver fra alle behandlingsgrupper analyseres ved obduktionen.
Der foretages en morfologisk bedømmelse af en spermaprøve fra epididymis (eller vas deferens), Spermier (mindst 200 i hver prøve) undersøges som fikserede, våde præparater (12) og klassificeres som enten normale eller anormale. Som eksempel på morfologiske spermieabnormiteter kan nævnes sammensmeltning, isoleret hoved og misdannet hoved og/eller hale. Optællingen finder sted på den udvalgte delmængde af hanner fra alle grupper enten straks efter aflivning af dyrene eller senere baseret på videobånd og digitale optagelser, Smears kan, når først de er fikseret, undersøges senere. I disse tilfælde kan kontroldyrene og højdosisgruppen analyseres først. Hvis der ikke optræder behandlingsrelaterede virkninger (f.eks. virkninger på spermiemorfologien), behøver de andre dosisgrupper ikke analyseres. Når der konstateres behandlingsrelaterede virkninger i højdosisgruppen, skal grupperne på lavere dosering også analyseres.
Hvis nogen af ovennævnte parametre for spermieevaluering allerede er undersøgt som led i en systemisk toksicitetsundersøgelse af mindst 90 dages varighed, behøver de ikke nødvendigvis gentages i to generationers forsøget. Det anbefales dog, at prøver eller digitale optagelser af spermier fra P-generationen opbevares for at give mulighed for senere vurdering, hvis dette viser sig nødvendigt.
1.5.5. Afkom
Hvert kuld undersøges hurtigst muligt efter fødslen (dag 0 i laktationsperioden) til bestemmelse af ungernes antal og køn. dødfødte, levendefødte og tilstedeværelse af alvorlige abnormiteter. Unger, som findes døde på dag 0 bør, hvis de ikke er opløst, helst undersøges for eventuelle defekter og dødsårsag og præserveres. Levende unger tælles og vejes enkeltvis ved fødslen (dag 0 i laktationsperioden) eller på dag 1, og derefter på de regelmæssige vejningsdage, dvs, på dag 4, 7, 14 og 21 i laktationsperioden. Fysiske eller adfærdsmæssige abnormiteter iagttaget hos moderdyr eller afkom skal registreres.
Afkommets fysiske udvikling skal registreres, hovedsagelig som stigning i kropsvægt, Andre fysiske parametre (f.eks åbning af ører og øjne, tandfrembrud, hårvækst) kan give supplerende oplysninger, men evalueringen deraf bør fortrinsvis finde sted sammen med data vedrørende kønsmodning (f.eks. alder og kropsvægt på tidspunktet for åbning af vagina eller for den balano-præputiale separation) (13). Det anbefales at udføre funktionelle undersøgelser (f.eks udvikling i motorisk aktivitet, sansefunktioner, reflekser) på F1-generationen før og/eller efter afvænning, medmindre sådanne undersøgelser indgår i særskilte forsøg: Især gælder dette undersøgelser relateret til kønsmodningen. For de fravænnede unger i F1-generationen, som er udvalgt til parring, bestemmes alderen på tidspunktet for åbning af vagina og separation af præputium. På ungerne i F2-generationen måles den anogenitale afstand ved dag 0 efter fødslen, når dette tilsiges af forandringer i Figenerationens kønssammensætning eller tidspunktet for kønsmodning.
Funktionsundersøgelser kan udelades for grupper, som på anden måde viser tydelige tegn på negative påvirkninger (f.eks. væsentligt nedsat tilvækst osv.). Foretages der funktionsundersøgelser, skal disse ikke omfatte de unger, der er udvalgt til parring.
1.5.6 Makroskopisk undersøgelse ved obduktion
Ved forsøgets slutning eller ved indtræden afdød under undersøgelsen undersøges alle forældredyr (P og Fl), alle unger med ydre abnormiteter eller kliniske symptomer samt en (et) tilfældigt udvalgt unge/køn/kuld fra både Fl- og F2-generationen makroskopisk for eventuelle strukturabnormiteter og patologiske forandringer. Særligt kønsorganerne skal undersøges. Unger, som aflives af humane grunde i moribund tilstand samt døde unger, når de ikke er opløst, undersøges for eventuelle defekter og/eller dødsårsag og præserveres.
På alle førstegangsfødende hunner undersøges uterus på en måde, som ikke vanskeliggør histopatologisk bedømmelse, for tilstedeværelse af og antal implantationssteder.
1.5.7. Organvægt
Ved forsøgets afslutning bestemmes kropsvægt og vægten af følgende organer på alle forældredyr i P-generationen og F1-generationen (parrede organer vejes enkeltvis):
|
— |
uterus, ovarier |
|
— |
testes, epididymides (total og cauda) |
|
— |
prostata |
|
— |
vesiculae seminales med prostata anterior (coagulating glands) og disses væskeindhold samt prostata (som én samlet enhed) |
|
— |
hjerne, lever, nyrer, milt, hypofyse, skjoldbruskkirtel og binyrer samt kendte målorganer. |
Kropsvægten ved forsøgets slutning bestemmes for under i F1- og F2-generationen, som udvælges til obduktion, og følgende organer fra den (det) ene tilfældigt udvalgte unge/køn/kuld (se punkt 1.5.6) vejes: hjerne, milt og thymus.
Resultaterne af den makroskopiske obduktion og organvejningen sammenholdes, når det er praktisk muligt, med observationer fra andre forsøg med gentagen dosering.
1.5.8. Histopatologi
1.5.8.1. Forældredyr
Følgende organer og væv fra forældredyr (P og F1) eller repræsentative prøver deraf fikseres og opbevares i et passende medium til histopatologisk undersøgelse:
|
— |
vagina, uterus med cervix og ovarier (præserveret i hensigtsmæssigt fiksativ) |
|
— |
én testis (præserveret i Bouins fiksativ eller tilsvarende), én epididymis, vesiculae seminales, prostata og prostata anterior (coagulating gland) |
|
— |
tidligere udpegede målorgan(er) fra alle de dyr i P- og F1-generationen, som er udvalgt til parring. |
Af ovennævnte præserverede organer og væv foretages fuldstændig histopatologisk undersøgelse for alle de dyr i højdosis- og kontrolgrupperne i P- og F1-generationen, som er udvalgt til parring. Undersøgelse af ovarierne fra dyrene i P-generationen er ikke obligatorisk. De organer fra lav- og mellemdosisgrupperne, som udviser behandlingsrelaterede forandringer, skal ligeledes undersøges som led i fastsættelsen af NOAEL. Derudover skal den histopatologiske undersøgelse omfatte kønsorganerne fra de dyr i lav- og mellemdosisgruppen, som mistænkes for nedsat fertilitet, fordi de f.eks. ikke har været i stand til at parre sig, undfange, besvangre eller føde rask afkom, eller hvis brunstcyklus, spermietal -motilitet eller -morfologi var påvirket. Alle makroskopiske forandringer som atrofi eller tumorer undersøges.
Detaljeret histopatologisk undersøgelse af testes (f.eks. med Bouins fiksativ, paraffinfiksering og tværsnit af 4-5μm's tykkelse) skal foretages til påvisning af behandlingsrelaterede effekter som manglende frigivelse af spermatider, manglende kimcellelag eller -typer, multinukleære kæmpeceller eller vandring af spermatogene celler ind i lumen (14). Undersøgelse af den intakte epididymis skal omfatte caput, corpus og cauda, hvilket kan ske ved vurdering af et længdesnit. Epididymis skal gennemgås med henblik på leukocytinfiltration, ændret forekomst af celletyper, afvigende celletyper, og fagocytose af spermier. Til undersøgelse af handyrenes kønsorganer kan anvendes PAS- og hæmatoxylinfarvning.
Ovariet i postlaktationsperioden skal indeholde primordial- og voksende follikler foruden de store corpora lutea fra laktationsperioden. Den histopatologiske undersøgelse skal kunne påvise en eventuel kvalitativ forringelse af bestanden af primordialfollikler. For hundyr i F1-generationen foretages en kvantitativ vurdering af primordialfolliklerne; antal dyr, valg af ovariesnit og størrelsen af prøvesnit skal være statistisk tilpasset den anvendte vurderingsmetode. Undersøgelsen skal omfatte en optælling af primordialfollikler, hvilket kan kombineres med optælling af små voksende follikler, med henblik på sammenligning af ovarierne i behandlingsgmppe og kontrolgruppe (15)(16)(17)(18)(19).
1.5.8.2. Fravænnede unger
Makroskopisk abnormt væv og målorganer fra alle unger med ydre abnormiteter eller kliniske symptomer samt fra den/det ene tilfældigt udvalgte unge/køn/kuld fra både F1- og F2-generationen, som ikke er udvalgt til parring, fikseres og opbevares i et passende medium til histopatologisk undersøgelse. Det præserverede væv skal gennemgå fuld histopatologisk karakterisering med særlig vægt på kønsorganerne.
2. DATA
2.1. BEHANDLING AF RESULTATER
Data rapporteres for hver forsøgsgruppe og generation og sammenfattes i tabelform med angivelse, for hver forsøgsgruppe og hver generation, af antal dyr ved forsøgets begyndelse, antal dyr, som er fundet døde under forsøget eller aflivet af humanitære grunde, tidspunkt for eventuel død eller humanitær aflivning, antal fertiele dyr, antal drægtige hundyr, antal dyr, som viser tegn på toksisk virkning, beskrivelse af iagttagne toksicitetstegn, herunder begyndelsestidspunkt, varighed og sværhed af alle de toksiske virkninger, arten af de fund, der er gjort på forældredyr og afkom, arten af de histopatologiske forandringer, og alle relevante data om kuldene.
Til bedømmelse af talmateriale anvendes en hensigtsmæssig, almindelig anerkendt statistisk metode; valg af statistiske metoder skal indgå som en del af undersøgelsens design og skal begrundes. Statistiske modeller for dosis-respons-sammenhæng kan være nyttige til dataanalyse. Rapporten skal indeholde tilstrækkelige oplysninger om analysemetoden til, at en uafhængig rapportør/statistiker kan revurdere og gentage analysen.
2.2. VURDERING AF RESULTATER
Fundene i denne reproduktionsundersøgelse i to generationer vurderes med hensyn til de iagttagne effekter, herunder obduktionsfund og mikroskopiske fund. Vurderingen skal omfatte sammenhæng, eller mangel herpå, mellem dyrenes udsættelse for teststoffet og tilstedeværelse eller fravær, incidens og sværhedsgrad af abnormiteter, herunder makroskopiske forandringer, identificerede målorganer, påvirkning af fertiliteten, unormale kliniske fund, påvirkning af reproduktions- og ynglepræstationer, kropsvægtforandringer, indvirkning på mortaliteten og eventuelle toksiske effekter. Prøvestoffets fysisk-kemiske egenskaber og eventuelle foreliggende toksikokinetiske data skal indgå i vurderingen af forsøgets resultater.
En korrekt udført reproduktions-toksicitetsundersøgelse skal give et tilfredsstillende skøn over nulvirkningsniveauet og indsigt i negative virkninger på reproduktion, fødsel, laktation og postnatal udvikling, herunder vækst og kønsmodning.
2.3. FORTOLKNING AF RESULTATER
En reproduktionstoksicitetsundersøgelse i to generationer giver oplysninger om virkningerne af gentagen eksponering for et stof i alle faser af reproduktionscyklussen. Navnlig giver undersøgelsen oplysninger om reproduktionsparametre og om afkommets udvikling, vækst, kønsmodning og overlevelse. Undersøgelsens resultater skal fortolkes i sammenhæng med fundene i forsøgene vedrørende subkronisk toksicitet, prænatal udviklingstoksicitet, toksikokinetik og andre foreliggende undersøgelser. Undersøgelsens resultater kan anvendes til at vurdere behovet for yderligere undersøgelse af et kemisk stof. Undersøgelsens resultater kan i begrænset omfang overføres til mennesker. Resultaterne kan bedst anvendes til at give oplysninger om nulvirkningsniveau og tilladelig eksponering af mennesker (20)(21)(22)(23).
3. RAPPORTERING
FORSØGSRAPPORT
Forsøgsrapporten skal indeholde følgende oplysninger:
|
|
For teststoffet:
|
|
|
For eventuel vehikel:
|
|
|
For forsøgsdyrene:
|
|
|
For forsøgsbetingelserne:
|
|
|
For resultaterne:
|
|
|
Diskussion af resultater. |
|
|
Konklusioner, herunder NOAEL (højeste niveau uden tegn på skadelig effekt) for moderdyr og afkom. |
4. HENVISNINGER
|
(1) |
Sadleir, R.M.F.S. (1979). Cycles and Seasons, In: Reproduction in Mummals: I. Germ Cells and Fertilization, C.R. Auston and R.V. Short (eds.), Cambridge, New York. |
|
(2) |
Gray, L.E. et al, (1989). A Dose-Response Analysis of Methoxychlor-lnduced Alterations of Reproductive Development and Function in the Rat. Fundamental and Applied Toxicology 12:92-108. |
|
(3) |
Robb, G.W. et al., (1978). Daily Sperm Production and Epididymal Sperm Reserves of Pubertal and Adult Rats. Journal of Reproduction und Fertility 54:103-107. |
|
(4) |
Kimefelter, G.R. et al., (1991). The Method of Sperm Collection Significantly Influences Sperm Motion Parameters Following Ethane Dimethanesulfonate Administration in the Rat. Reproductive Toxicology 5:39 44. |
|
(5) |
Seed. J. et al. (1996). Methods for Assessing Sperm Motility, Morphology, and Counts in the Rat, Rabbit, and Dog: a Consensus Report. Reproductive Toxicology 10(3):237-244. |
|
(6) |
Chapin, R.E. et al. (1992).Methods for Assessing Rat Sperm Motility. Reproductive Toxicology 6:267-273. |
|
(7) |
Klinefelter, G.R. et al., (1992). Direct Effects of Ethane Dimethanesulphonate on Epididymal Function in Adult Rats: an In Vitro Demonstration. Journal of Andrology 13:409-421. |
|
(8) |
Slott, V.L. et al., (1991). Rat Sperm Motility Analysis: Methodologie Considerations. Reproductive Toxicology 5:449-458. |
|
(9) |
Slott, V.L. and Perreault, S.D., (1993). Computer-Assisted Sperm Analysis of Rodent Epididymal Sperm Motility Using the Hamilton-Thorn Motility Analyzer. In: Methods in Toxicology, Part A., Academic, Orlando, Florida. pp. 319-333. |
|
(10) |
Toth. G.P. et al. (1989). The Automated Analysis of Rat Sperm Motility Following Subchronic Epichlorhydrin Administration: Methodologie and Statistical Considerations. Journal of Andrology 10:401-415. |
|
(11) |
Working, P.K. and M. Hurtt, (1987). Computerized Videomicrographie Analysis of Rat Sperm Motility. Journal of Andrology 8:330-337. |
|
(12) |
Linder, R.E. et al., (1992). Endpoints of Spermatoxicity in the Rat After Short Duration Exposures to Fourteen Reproductive Toxicants. Reproductive Toxicology 6:491-505. |
|
(13) |
Korenbrot, C.C. et al., (1977). Preputial Separation as an External Sign of Pubertal Development in the Male Rat. Biological Reproduction 17:298303. |
|
(14) |
Russell, L.D. et al., (1990). Histological and Histopathological Evaluation of the Testis, Cache River Press, Clearwater, Florida. |
|
(15) |
Heindel, J.J. and R.E. Chapin, (eds.) (1993). Part B. Female Reproductive Systems, Methods in Toxicology, Academic, Orlando, Florida. |
|
(16) |
Heindel, J.J. et al., (1989) Histological Assessment of Ovarian Follicle Number in Mice As a Screen of Ovarian Toxicity. In: Growth Factors and the Ovary, A.N. Hirshfield (ed.), Plenum, New York, pp. 421-426. |
|
(17) |
Manson, J.M. and Y.J.Kang, (1989). Test Methods for Assessing Female Reproductive and Developmental Toxicology. In: Principles and Methods of Toxicology, A.W. Hayes (ed.), Raven, New York. |
|
(18) |
Smith, B.J. et al,. (1991). Comparison of Random and Serial Sections in Assessment of Ovarian Toxicity. Reproductive Toxicology 5:379-383. |
|
(19) |
Heindel, J.J. (1999). Oocyte Quantitation and Ovarian Histology. In: An Evaluation and Interpretation of Reproductive Endpoints for Human Health Risk Assessment, G. Daston,. and C.A. Kimmel, (eds.), ILSI Press, Washington, DC. |
|
(20) |
Thomas, J. A. (1991). Toxic Responses of the Reproductive System. In: Casarett and Doull's Toxicology, M.O. Amdur, .I. Doull, and CD. Klaassen (eds.), Pergamon, New York. |
|
(21) |
Zenick, H. and E.D. Clegg, (1989). Assessment of Male Reproductive Toxicity: A Risk Assessment Approach. In: Principles and Methods of Toxicology, A.W. Hayes (ed.). Raven Press, New York. |
|
(22) |
Palmer, A.K. (1981). In: Developmental Toxicology, Kimmel, C.A. and J. Buelke-Sam (eds.), Raven Press, New York. |
|
(23) |
Palmer, A.K. (1978). In Handbook of Teratology, Vol. 4, J.G. Wilson and F.C. Fraser (eds.), Plenum Press, New York. |
B.36. TOKSIKOKINETIK
1. METODE
1.1. INDLEDNING
Se den generelle indledning til del B.
1.2. DEFINITIONER
Se den generelle indledning til del B.
1.3. REFERENCESTOFFER
Ingen.
1.4. PRINCIP FOR TESTMETODEN
Teststoffet indgives på passende måde. Afhængigt af undersøgelsens formål kan teststoffet gives på en gang eller ad flere gange fordelt over fastlagte perioder, og der kan benyttes en eller flere grupper af forsøgsdyr. Afhængigt af undersøgelsens art foretages derefter bestemmelse af teststoffet og/eller dets metabolitter i kropsvæsker, væv og/eller excreta.
Ved undersøgelsen kan anvendes både »mærket« og »umærket« teststof. Hvis der benyttes mærkning, skal denne være i en sådan position i teststoffet, at det giver flest muligt oplysninger om teststoffets omdannelse i organismen.
1.5. KVALITETSKRITERIER
Ingen.
1.6. BESKRIVELSE AF TESTMETODEN
Forberedelser
Sunde, unge, voksne dyr akklimatiseres til laboratoriets undersøgelsesbetingelser i mindst fem dage, inden forsøget påbegyndes. Forud for undersøgelsen randomiseres dyrene og fordeles i grupper. Under særlige omstændigheder kan der anvendes ganske unge, drægtige eller forbehandlede dyr.
Forsøgsbetingelser
Forsøgsdyr
Ved toksikokinetiske undersøgelser kan anvendes en eller flere egnede dyrearter, og der må tages hensyn til, hvilke dyrearter man har brugt eller har til hensigt at bruge i andre toksikologiske undersøgelser af det samme teststof. Hvis der anvendes gnavere, må dyrenes vægt ved undersøgelsens begyndelse ikke variere mere end ± 20 % af gennemsnitsvægten.
Antal og køn
Ved undersøgelse af optagelse og udskillelse anvendes fire dyr til hver dosis ved forsøgets begyndelse. Der stilles ikke krav vedrørende dyrenes køn — det kan dog under visse omstændigheder være nødvendigt at anvende begge køn. Hvis der er tale om kønsbetingede forskelle i teststoffets virkning, anvendes fire dyr af hvert køn. I forsøg med andre dyr end gnavere kan der benyttes et mindre antal dyr. Ved undersøgelse af fordelingen i væv fastlægges den størrelse, forsøgsgrupperne skal have ved forsøgets begyndelse, under hensyntagen til både hvor mange dyr, der skal aflives på forud fastlagte tidspunkter, og hvor mange sådanne tidspunkter forsøget skal omfatte.
Ved undersøgelse af metaholismen fastlægges gruppestørrelsen efter behov. Ved undersøgelse med flere doser og flere tidsafhængige delundersøgelser fastlægges gruppestørrelsen under hensyntagen til påregnet aflivning og antal undersøgelsestidspunkter, idet der dog ikke må anvendes mindre end to dyr pr. gruppe. Grupperne skal være tilstrækkeligt store til at muliggøre en acceptabel beskrivelse af absorption, koncentrationsniveauer og udskillelsesforhold for teststoffet og/eller dets metabolitter.
Dosisniveauer
Når teststoffet gives på en gang anvendes mindst to forskellige doser. Der anvendes en lav dosis, hvor der ikke observeres nogen toksisk effekt, og en høj dosis hvor der kan være tale om ændringer i de toksikokinetiske parametre eller om toksiske effekter.
Hvis teststoffet indgives gentagne gange, er det normalt tilstrækkeligt at anvende den lave dosis, men under visse omstændigheder kan det være nødvendigt også at anvende en høj dosis.
Administrationsmåde
Ved toksikokinetiske undersøgelser indgives teststoffet på samme måde og om muligt under anvendelse af samme vehikel som i allerede foretagne eller planlagte toksikologiske undersøgelser af samme teststof. Sædvanligvis indgives teststoffet enten oralt med mavesonde eller via foderet, ved påføring på huden eller ved inhalation i på forhånd fastlagte tidsrum. Intravenøs indgift kan være nyttig ved bestemmelse af den relative absorption ved andre administrationsmåder. Dertil kommer, at der kan tilvejebringes nyttige oplysninger om fordelingsmønstret kort tid efter intravenøs indgift.
Der bør tages hensyn til muligheden for, at vehiklet interfererer med teststoffet. Samtidig må man være opmærksom på, at der kan være forskelle i absorptionen afhængigt af, om teststoffet indgives med mavesonde eller via foderet, og at der derfor er behov for en nøjagtig bestemmelse af størrelsen af dosis, især når teststoffet indgives via foderet.
Observationsperiode
Alle dyr observeres dagligt, og toksicitetstegn og andre relevante kliniske symptomer registreres, idet det blandt andet registreres, hvornår de optræder første gang, hvor alvorlige de er, og hvor længe de varer.
Procedure
After weighing test animals, the test substance is administered by an appropriate route. If considered relevant, animals may be fasted before the test substance is administered.
Absorption
Den mængde af det indgivne stof, der optages, og den hastighed, hvormed optagelsen foregår, kan vurderes ved hjælp af forskellige metoder med og uden referencegrupper (6), f.eks.:
|
— |
bestemmelse af mængden af teststof og/eller metabolitter i excreta som urin, galde, fæces og udåndingsluft og den tilbageværende mængde i kroppen efter aflivning |
|
— |
sammenligning af biologisk respons (f.eks, i undersøgelser af akut toksicitet) i forsøgs-, kontrol- og/eller referencegrupper |
|
— |
sammenligning af mængden af teststof og/eller metabolitter, som udskiltes gennem nyrerne i forsøgs- og referencegrupperne |
|
— |
bestemmelse af arealet under kurven over plasmakoncentrationen af teststoffet og/eller dets metabolitter som funktion af tiden og sammenligning med data fra en referencegruppe. |
Fordeling
Der er i dag mulighed for at analysere fordelingsmønstrene på to måder, og man kan benytte den ene eller begge fremgangsmåder:
|
— |
der kan tilvejebringes nyttige kvalitative oplysninger ved anvendelse af helkropsautoradiografiske teknikker |
|
— |
kvantitative oplysninger tilvejebringes ved aflivning af dyr på forskellige tidspunkter efter eksponeringen og fastlæggelse af mængden og koncentrationen af teststof og/eller metabolitter i væv og organer. |
Udskillelse
Ved undersøgelse af udskillelsen opsamles urin, fæces og udåndingsluft samt i visse tilfælde galde. Mængden af teststof og/eller metabolitter i disse excreta måles på flere tidspunkter efter eksponeringen, indtil ca. 95 % af dosis er udskilt, dog højst i syv dage.
I specielle tilfælde kan det være nødvendigt at tage hensyn til udskillelse af teststof i mælken fra diegivende forsøgsdyr.
Metabolisme
Biologiske prøver analyseres under anvendelse af dertil egnede metoder med henblik på at bestemme metabolismens omfang og mønster. Hvor der er behov for at besvare spørgsmål som følge af tidligere toksikologiske undersøgelser, opklares metabolitternes struktur, og der fremsættes forslag til beskrivelse af sandsynlige metabolismeveje. Undersøgelser in vitro kan være en hjælp til fremskaffelse af oplysninger om metabolismens forløb.
Yderligere oplysninger om sammenhængen mellem metabolisme og toksisk effekt kan indhentes gennem biokemiske undersøgelser ved f.eks. at bestemme virkningen på metaboliserende enzymsystemer, reduktion i mængden af endogene non-protein sulfhydrylforbindelser og teststoffets binding til makromolekyler.
2. DATA
Data opstilles i tabelform og støttes, når det er muligt, af grafiske fremstillinger, alt afhængigt af undersøgelsestypen. Når det er muligt, anføres middelværdi og statistisk variation af alle målinger i relation til tid, doser, væv og organer for samtlige forsøgsgruppers vedkommende. Den absorberede og udskilte mængde samt udskillelseshastigheden bestemmes med dertil egnede metoder. Ved metabolismeundersøgelser oplyses de identificerede metabolitters struktur, og sandsynlige metabolismeveje fremlægges.
3. RAPPORTERING
3.1. FORSØGSRAPPORT
Forsøgsrapporten skal om muligt indeholde følgende:
|
— |
dyreart, stamme, herkomst, miljøbetingelser, foder |
|
— |
beskrivelse af mærkede stoffer, når sådanne er anvendt |
|
— |
dosisniveauer og anvendte intervaller |
|
— |
administrationsmåde og eventuelle vehikler |
|
— |
toksisk og anden virkning |
|
— |
metoder til bestemmelse af teststof og/eller metabolitter i biologiske prøver, herunder udåndingsluft |
|
— |
måleresultater opstillet i tabelform efter køn, doser, andre testparametre, tid, væv og organer |
|
— |
den absorberede og udskilte mængde som en funktion af tiden |
|
— |
metoder til karakterisering og identificering af metabolitter i biologiske prøver |
|
— |
biokemiske målemetoder anvendt i forbindelse med metabolisme |
|
— |
forslag til metabolismeveje |
|
— |
diskussion af resultaterne |
|
— |
fortolkning af resultaterne. |
3.2. VURDERING OG FORTOLKNING
Se de generelle oplysninger til del B.
4. HENVISNINGER
Se de generelle oplysninger til del B.
B.37. FORSINKET NEUROTOKSICITET VED ORGANISKE PHOSPHORFORBINDELSER EFTER AKUT EKSPONERING
1. METODE
1.1. INDLEDNING
Ved vurdering af stoffers toksiske virkninger er det vigtigt at undersøge visse stofgruppers mulighed for at fremkalde specifikke former for neurotoksicitet, som i givet fald ikke kan påvises i andre toksicitetsundersøgelser. Visse organiske phosphorforbindelser har vist sig at forårsage forsinket neurotoksicitet og bør undersøges med henblik herpå.
Der kan anvendes in vitro-screeningtest til identificering af stoffer, som kan forårsage forsinket polyneuropati; negative resultater af in vitro-undersøgelser kan imidlertid ikke bruges som dokumentation for, at teststoffet ikke er neurotoksisk.
Se den generelle indledning til del B.
1.2. DEFINITIONER
Organiske phosphorforbindelser omfatter uladede estere, thioestere og anhydrider af organiske phosphorsyrer, phosphonsyrer og phosphoramidsyrer eller tilsvarende phosphosthiosyrer, phosphonthiosyrer og phosphorthioamidsyrer eller andre stoffer, der kan forårsage den forsinkede neurotoksicitet, der til tider forekommer for denne stofgruppe.
Forsinket neurotoksicitet er et syndrom, der er knyttet til langvarig, forsinket opståen af ataxi, distal axonopati i rygmarv og perifere nerver og inhibition og aldring af neurotoksisk esterase (NTE) i nervevæv.
1.3. REFERENCESTOFFER
Et referencestof kan testes med en positiv kontrolgruppe som middel til at påvise, at den undersøgte arts reaktion ikke har ændret sig signifikant under laboratorieforsøgsbetingelserne.
Et eksempel på et meget anvendt neurotoksisk stof er tri-o-tolylphosphat (CAS nr. 78-30-8, Einecs nr. 201-103-5, CAS-nomenklatur: phosphorsyre, tris(2-methylphenyl)ester), også kendt som tris-o-cresylphosphat.
1.4. TESTMETODENS PRINCIP
Teststoffet indgives oralt i en enkelt dosis til tamhøner, som om nødvendigt har været beskyttet mod akutte kolinerge effekter. Dyrene observeres i 21 dage for unormal adfærd, ataxi og paralyse. Der foretages biokemiske målinger, navnlig inhibition af neurotoksisk esterase, på tilfældigt udvalgte høner fra hver gruppe, normalt 24 og 48 timer efter indgift. 21 dage efter eksponering aflives de resterende høner, og der foretages en histopatologisk undersøgelse af udvalgt nervevæv.
1.5. BESKRIVELSE AF TESTMETODEN
1.5.1. Forberedelser
Unge, sunde kønsmodne høner, der er fri for virussygdomme og medicinering, der kan interferere med undersøgelsen, og hvis gang er normal, fordeles randomiseret i forsøgs- og kontrolgrupper og tilvænnes laboratoriebetingelserne i mindst fem dage inden undersøgelsens påbegyndelse.
Der anvendes bure eller indelukker, der giver hønsene fri bevægelighed, og som tillader uhindret observation af dyrenes gang.
Teststoffet bør normalt indgives oralt med mavesonde, gelatinekapsler eller en tilsvarende metode. Væsker kan indgives ufortyndet eller opløst i et passende vehikel, som f.eks. majsolie; faste stoffer bør om muligt opløses, eftersom store doser faste stoffer i gelatinekapsler ikke altid absorberes effektivt. For ikke-vandige vehikler skal vehiklets toksiske egenskaber kendes eller i givet fald bestemmes inden forsøget.
1.5.2. Forsøgsbetingelser
1.5.2.1. Forsøgsdyr
Unge, kønsmodne læggehøner (Gallus gallus domesticus) på otte til tolv måneder anbefales. Der anvendes racer og stammer af standardstørrelse, og hønsene bør normalt være opdrættet under betingelser, som tillader bevægelsesfrihed.
1.5.2.2. Antal og køn
Ud over forsøgsgruppen anvendes både en vehikelkontrolgruppe og en positiv kontrolgruppe. Vehikelkontrolgruppen behandles på samme måde som forsøgsgruppen, med undtagelse af administration af teststoffet.
Der anvendes et tilstrækkeligt antal høner i hver gruppe, så mindst seks høns kan aflives med henblik på biokemisk bestemmelse (tre på hvert af de to valgte tidspunkter) og seks kan overleve den 21 dage lange observationsperiode med henblik på patologisk undersøgelse.
Den positive kontrolgruppe kan undersøges samtidig eller kan være en nylig historisk kontrolgruppe. Gruppen indeholder mindst seks høner, der behandles med et kendt neurotoksisk stof med forsinket virkning, tre høner til biokemisk undersøgelse og tre høner til patologisk undersøgelse. Regelmæssig ajourføring af historiske data anbefales. Der tilvejebringes nye data om positiv kontrol, hvis det pågældende laboratorium har ændret væsentlige elementer (f.eks. stamme, foder, laboratoriemiljø) i forsøgets gennemførelse.
1.5.2.3. Dosisniveauer
Der gennemføres en forundersøgelse med et passende antal høner og dosisniveaugrupper med henblik på at fastsætte det dosisniveau, der skal anvendes i hovedundersøgelsen. En vis dødelighed er normalt nødvendig i denne forundersøgelse til fastsættelse af en passende dosis til hovedundersøgelsen. For at forhindre dødsfald som følge af akutte kolinerge effekter kan imidlertid atropin eller et andet beskyttende middel, der vides ikke at interferere med forsinkede neurotoksiske reaktioner, anvendes. En række forskellige testmetoder kan anvendes til at vurdere den højeste ikke dødelige dosis af teststofferne (se metode B.l.A). Historiske data fra undersøgelser af høner eller andre toksikologiske oplysninger kan også anvendes som støtte ved valg af dosis.
Dosisniveauet for teststoffet i hovedundersøgelsen bør være så højt som muligt under hensyntagen til resultaterne af forundersøgelsen og den øvre dosisgrænse på 2 000 mg/kg legemsvægt. Eventuel dødelighed må ikke forhindre, at et tilstrækkeligt antal dyr overlever med henblik på biokemisk undersøgelse (seks) og histologisk undersøgelse (seks) efter 21 dage. Atropin eller et andet beskyttende middel, der vides ikke at interferere med forsinkede neurotoksiske raktioner, anvendes for at forhindre dødsfald som følge af akutte kolinerge effekter.
1.5.2.4. Grænsetest
Hvis et forsøg udført efter de her beskrevne retningslinjer med et dosisniveau på mindst 2 000 mg/kg legemsvægt/dag ikke fremkalder observerbare toksiske virkninger, og hvis der ikke kan ventes toksisk virkning på grundlag af data fra strukturelt beslægtede stoffer, kan en undersøgelse med anvendelse af en højere dosis betragtes som unødvendig. Grænsetesten er kun relevant, hvis human eksponering indicerer, at der er behov for anvendelse af et højere dosisniveau.
1.5.2.5. Observationsperiode
Observationsperioden varer 21 dage.
1.5.3. Fremgangsmåde
Efter indgift af et beskyttende middel til forhindring af dødsfald som følge af akut kolinerg effekt indgives teststoffet i en enkelt dosis.
1.5.3.1. Generelle observationer
Observationsperioden starter umiddelbart efter eksponering. Alle hønerne observeres omhyggeligt flere gange i løbet af de første to dage og derefter mindst en gang dagligt i en periode på 21 dage eller indtil det fastlagte aflivningstidspunkt, Alle tegn på toksicitet registreres, herunder tidspunktet for indtræden af unormal adfærd og dennes type, grad og varighed. Ataxi måles efter en talskala med mindst fire niveauer, og paralyse registreres. Mindst to gange om ugen udtages de høner, der er udvalgt til patologisk undersøgelse, af burene og underkastes en periode med tvungen motorisk aktivitet, som f.eks. stigeklatring, med henblik på at lette observationen af minimale toksiske virkninger. Døende dyr og dyr, der viser symptomer på stærk smerte eller lidelse, fjernes, så snart de iagttages, hvorefter de aflives på human måde og obduceres.
1.5.3.2. Legemsvægt
Alle hønerne vejes lige inden indgift af teststoffet og mindst én gang om ugen derefter.
1.5.3.3. Biokemisk undersøgelse
Seks vilkårligt udvalgte høner fra hver forsøgs- og vehikelkontrolgruppe og tre høner fra den positive kontrolgruppe (hvis denne guppe undersøges samtidig) aflives inden for få dage efter indgift, og hjernen og den lumbale rygmarv præpareres og undersøges for inhibition af neurotoksisk esterase. Normalt aflives tre haner fra vehikelkontrolgruppen og fra hver forsøgsgruppe efter 24 timer og tre efter 48 timer, mens de tre høner fra den positive kontrolgruppe aflives efter 24 timer. Hvis observation af kliniske tegn på forgiftning (dette kan ofte vurderes ved observation af tidspunktet for indtræden af kolinerge symptomer) indicerer, at det toksiske stof udskiltes meget langsomt, kan det være bedre at udtage væv fra tre høner på hvert af to andre tidspunkter mellem 24 timer og 72 timer efter administration.
Hvis det skønnes hensigtsmæssigt, kan der også foretages analyser af acetylcholinesterase (AChE) på disse prøver. Spontan reaktivering af AChE kan imidlertid indtræde in vivo og således føre til, at stoffets AChE-inhiberende aktivitet undervurderes.
1.5.3.4. Makroskopisk undersøgelse
Den makroskopiske undersøgelse af alle dyrene (aflivet efter forsøgets afslutning eller under forsøget, fordi de var døende) omfatter observation af udseende af hjerne og rygmarv.
1.5.3.5. Histopatologisk undersøgelse
Der foretages en mikroskopisk undersøgelse af nervevæv fra dyr, der overlever observationsperioden, og som ikke er anvendt til de biokemiske undersøgelser. Væv fikseres in situ ved anvendelse af perfusionsmetoder. Snittene bør omfatte cerebellum (længdesnit langs midterlinjen), medulla oblongata, rygmarv og de perifere nerver. Rygmarvssnit tages fra det øvre cervikale segment, midt i brysthulen og det lumbosakrale område. Der tages snit af det distale område af tibianerven og dens udløbere til musculus gastrocnemius og af ischiasnerven. Snittene farves med passende myelin- og axonspecifikke farver.
2. DATA
Hvis resultaterne for de udvalgte endepunkter i denne metode (biokemi, histopatologi og adfærdsobservation) er negative, kræves der normalt ikke yderligere undersøgelse for forsinket neurotoksicitet. Ved tvetydige eller inkonklusive resultater for disse endepunkter kan yderligere vurdering være nødvendig.
Der tilvejebringes individuelle data. Herudover opstilles alle data i tabelform, der for hver forsøgsgruppe viser antallet af dyr ved forsøgets start, antallet af dyr, der udviser læsioner, adfærdsmæssige eller biokemiske virkninger, typen og graden af disse læsioner eller virkninger og procentdelen af dyr, der udviser hver enkelt type eller grad af læsion eller virkning.
Resultaterne af denne undersøgelse vurderes med hensyn til incidensen og sværhedsgraden af og korrelationen mellem adfærdsmæssige, biokemiske og histopatologiske virkninger og eventuelle andre observerede virkninger i forsøgs- og kontrolgrupper.
Resultater i talform vurderes med passende og generelt accepterede statistiske metoder. De anvendte statistiske metoder udvælges i forbindelse med forsøgets udformning.
3. RAPPORTERING
FORSØGSRAPPORT
Forsøgsrapporten skal om muligt indeholde følgende oplysninger:
|
|
Forsøgsdyr:
|
|
|
Forsøgsbetingelser
|
|
|
Resultater:
|
|
|
Diskussion af resultaterne |
|
|
Konklusioner |
4. HENVISNINGER
Denne metode svarer til TG 418 fra OECD.
B.38. FORSINKET NEUROTOKSICITET VED ORGANISKE PHOSPHORFORBINDELSER GENTAGEN DOSIS — 28 DAGE
1. METODE
1.1. INDLEDNING
Ved vurdering af stoffers toksiske virkninger er det vigtigt at undersøge visse stofgruppers mulighed for at fremkalde specifikke former for neurotoksicitet, som i givet fald ikke kan påvises i andre toksicitetsundersøgelser. Visse organiske phosphorforbindelser har vist sig at forårsage forsinket neurotoksicitet og bør undersøges med henblik herpå.
Der kan anvendes in vitro-screeningtest til identificering af stoffer, som kan forårsage forsinket polyneuropati; negative resultater af in vitro-undersøgelser kan imidlertid ikke bruges som dokumentation for, at teststoffet ikke er neurotoksisk.
Denne 28 dages test for forsinket neurotoksicitet giver oplysninger om mulige sundhedsrisici som følge af gentagen eksponering i et begrænset tidsrum. Testen giver oplysninger om dosis/respons og kan danne grundlag for en vurdering af et nul-effekt-niveau (NOAEL), som kan anvendes ved fastsættelse af sikkerhedskriterier for eksponering.
Se den generelle indledning til del B.
1.2. DEFINITIONER
Organiske phosphorforbindelser omfatter uladede estere, thioestere og anhydrider af organiske phosphorsyrer, phosphonsyrer og phosphoramidsyrer eller tilsvarende phosphorthiosyrer, phosphonthiosyrer og phosphorthioamidsyrer eller andre stoffer, der kan forsårsage den forsinkede neurotoksicitet, der til tider forekommer for denne stofgruppe.
Forsinket neurotoksicitet er et syndrom, der er knyttet til langvarig, forsinket opståen af ataxi, distal axonopati i rygmarv og perifere nerver og inhibition og aldring af neurotoksisk esterase (NTE) i nervevæv.
1.3. TESTMETODENS PRINCIP
Daglige doser af teststoffet indgives oralt til høner i 28 dage. Dyrene observeres mindst en gang dagligt for unormal adfærd, ataxi og paralyse indtil 14 dage efter sidste dosis. Der foretages biokemiske målinger, navnlig inhibition af neurotoksisk esterase (NTE), på tilfældigt udvalgte høner fra hver gruppe, normalt 24 og 48 timer efter sidste dosis. To uger efter sidste dosis aflives de resterende høner, og der foretages en histopatologisk undersøgelse af udvalgt nervevæv.
1.4. BESKRIVELSE AF TESTMETODEN
1.4.1. Forberedelser
Unge, sunde kønsmodne høner, der er fri for virussygdomme og medicinering, der kan interferere med undersøgelsen, og hvis gang er normal, fordeles randomiseret i forsøgs- og kontrolgrupper og tilvænnes laboratoriebetingelserne i mindst fem dage inden undersøgelsens påbegyndelse.
Der anvendes bure eller indelukker, der giver hønsene fri bevægelighed, og som tillader uhindret observation af dyrenes gang.
Teststoffet bør normalt indgives oralt med mavesonde, gelatinekapsler eller en tilsvarende metode. Væsker kan indgives ufortyndet eller opløst i et passende vehikel, som f.eks. majsolie; faste stoffer bør om muligt opløses, eftersom store doser faste stoffer i gelatinekapsler ikke altid absorberes effektivt. For ikke-vandige vehikler skal vehiklets toksiske egenskaber kendes eller i givet fald bestemmes inden forsøget.
1.4.2. Forsøgsbetingelser
1.4.2.1. Forsøgsdyr
Unge, kønsmodne høner (Gallus gallus domesticus) på otte til tolv måneder anbefales. Der anvendes racer og stammer af standardstørrelse, og hønerne bør normalt være opdrættet under betingelser, som tillader bevægelsesfrihed.
1.4.2.2. Antal og køn
Der anvendes generelt mindst tre forsøgsgrupper og en vehikelkontrolgruppe. Vehikelkontrolgruppen behandles på samme måde som forsøgsgruppen, med undtagelse af administration af teststoffet.
Der anvendes et tilstrækkeligt antal høner i hver gruppe, så mindst seks høner kan aflives med henblik på biokemisk bestemmelse (tre på hvert af de to valgte tidspunkter) og seks kan overleve den 14 dage lange observationsperiode efter eksponering med henblik på patologisk undersøgelse.
1.4.2.3. Dosisniveauer
Dosisniveauerne vælges under hensyntagen til resultaterne af en test for forsinket neurotoksicitet efter akut eksponering og eventuelle andre foreliggende data vedrørende teststoffets toksicitet eller kinetik. Det højeste dosisniveau vælges med henblik på at inducere toksiske virkninger, om muligt forsinket neurotoksicitet, men ikke død eller tydelig lidelse. Herefter vælges en række dosisniveauer i faldende størrelse til påvisning af dosisrelaterede reaktioner og fravær af erkendbare virkninger (NOAEL) ved det laveste dosisniveau.
1.4.2.4. Grænsetest
Hvis et forsøg udført efter de her beskrevne retningslinjer med et dosisniveau på mindst 1 000 mg/kg legemsvægt/dag ikke fremkalder observerbare toksiske virkninger, og hvis der ikke kan ventes toksiske virkninger på grundlag af data fra strukturelt beslægtede stoffer, kan en undersøgelse med anvendelse af en højere dosis betragtes som unødvendig. Grænsetesten er kun relevant, hvis human eksponering indicerer, at der er behov for anvendelse af et højere dosisniveau.
1.4.2.5. Observationsperiode
Dyrene observeres mindst en gang dagligt i eksponeringsperioden og 14 dage efter, medmindre de skal obduceres.
1.4.3. Fremgangsmåde
Dyrene indgives teststoffet syv dage om ugen i en periode på 28 dage.
1.4.3.1. Generelle observationer
Observationsperioden starter umiddelbart efter eksponering. Alle hønerne observeres omhyggeligt mindst en gang daglig i de 28 dage og i 14 dage efter eksponering, eller indtil det fastlagte aflivningstidspunkt. Alle tegn på toksicitet registreres, herunder tidspunktet for indtræden af unormal adfærd og dennes type, grad og varighed. Observationerne omfatter, men er ikke begrænset til, unormal adfærd. Ataxi måles efter en talskala med mindst fire niveauer, og paralyse registreres. Mindst to gange om ugen udtages hønerne af burene og underkastes en periode med tvungen motorisk aktivitet, som f.eks. stigeklatring, med henblik på at lette observationen af minimale toksiske virkninger. Døende dyr og dyr, der viser symptomer på stærk smerte eller lidelse, fjernes, så snart de iagttages, hvorefter de aflives på human måde og obduceres.
1.4.3.2. Legemsvægt
Alle hønsene vejes lige inden indgift af teststoffet og mindst én gang om ugen derefter.
1.4.3.3. Biokemisk undersøgelse
Seks vilkårligt udvalgte høner fra hver forsøgs- og vehikelkontrolgruppe aflives få dage efter indgift af sidste dosis, og hjernen og den lumbale rygmarv præpareres og undersøges for inhibition af neurotoksisk esterase, Derudover kan det være nyttigt at præparere og undersøge ischiasnervevæv for inhibition af neurotoksisk esterase. Normalt aflives tre høner fra kontrolgruppen og fra hver forsøgsgruppe 24 timer og tre 48 timer efter sidste dosis. Hvis data fra undersøgelsen for akut toksicitet eller andre undersøgelser (f.eks. toksikokinetik) indicerer, at andre tidspunkter for aflivning efter sidste dosis er bedre end de her nævnte, anvendes disse med angivelse af begrundelsen herfor.
Hvis det skønnes hensigtsmæssigt, kan der også foretages analyser af acetylcholinesterase (AChE) på disse prøver. Spontan reaktivering af AChE kan imidlertid indtræde in vivo og således føre til, at stoffets AChE-inhiberende aktivitet undervurderes.
1.4.3.4. Makroskopisk undersøgelse
Den makroskopiske undersøgelse af alle dyrene (aflivet efter forsøgets afslutning eller under forsøget, fordi de var døende) omfatter observation af hjerne og rygmarv.
1.4.3.5. Histopatologisk undersøgelse
Der foretages en mikroskopisk undersøgelse af nervevæv fra dyr, der overlever observationsperioden, og som ikke er anvendt til de biokemiske undersøgelser. Væv fikseres in situ ved anvendelse af perfusionsmetoder. Snittene bør omfatte cerebellum (længdesnit langs midterlinjen) medulla oblongata, rygmarv og de perifere nerver. Rygmarvssnit tages fra det øvre cervikale segment, midt i brysthulen og det lumbosakrale område. Der tages snit af det distale område af tibianerven og dens udløbere til musculus gastrocnemius og ischiasnerven. Snittene farves med passende myelin- og axonspecifikke farver. Først foretages der en mikroskopisk undersøgelse af præserveret væv fra alle dyrene i kontrolgruppen og højdosisgruppen. Hvis der er tegn på virkninger i højdosisgruppen, foretages der også en mikroskopisk undersøgelse af høner fra mellem- og lavdosisgrupperne.
2. DATA
Hvis resultaterne for de udvalgte endepunkter i denne metode (biokemi, histopatologi og adfærdsobservation) er negative, kræves der normalt ikke yderligere undersøgelse for forsinket neurotoksicitet Ved tvetydige eller inkonklusive resultater for disse endepunkter kan yderligere vurdering være nødvendig.
Der tilvejebringes individuelle data. Herudover opstilles alle data i tabelform, der for hver forsøgsgruppe viser antallet af dyr ved forsøgets start, antallet af dyr, der udviser læsioner, adfærdsmæssige eller biokemiske virkninger, typen og graden af disse læsioner eller virkninger og procentdelen af dyr, der udviser hver enkelt type eller grad af læsion eller virkning.
Resultaterne af denne undersøgelse vurderes med hensyn til incidensen og sværhedsgraden og af korrelationen mellem adfærdsmæssige, biokemiske og histopatologiske virkninger og eventuelle andre observerede virkninger i forsøgs- og kontrolgrupper.
Resultater i talform vurderes med passende og generelt accepterede statistiske metoder. De anvendte statistiske metoder udvælges i forbindelse med forsøgets udformning.
3. RAPPORTERING
FORSØGSRAPPORT
Forsøgsrapporten skal om muligt indeholde følgende oplysninger:
|
|
Forsøgsdyr:
|
|
|
Forsøgsbetingelser:
|
|
|
Resultater
|
|
|
Diskussion af resultaterne |
|
|
Konklusioner |
4. HENVISNINGER
Denne metode svarer til TG 419 fra OECD.
B.39. TEST FOR UNSCHEDULED DNA-SYNTESE (UDS) MED PATTEDYRLEVERCELLER IN VIVO
1. METODE
Denne metode er en gengivelse af OECD Test Guideline 486 Unscheduled DNA Synthesis (UDS) Test with Mammalian Liver Cells In Vivo (1997).
1.1. INDLEDNING
Formalet med testen for unscheduled DNA-syntese (UDS) med pattedyrleverceller in vivo er at påvise, om et teststof inducerer dna-reparation i levercellerne hos de behandlede dyr (1) (2) (3) (4).
Denne in vivo-test indeholder en metode til undersøgelse af kemikaliers gentoksiske virkninger i leveren. Det målte endpoint er tegn på dna-beskadigelse og deraf følgende reparation i leverceller. Leveren er normalt det sted, hvor absorberede stoffer først og fremmest metaboliseres. Den er derfor et egnet sted til in vivo-måling af dna-beskadigelse.
Hvis der er grund til at tro, at teststoffet ikke vil nå frem til målvævet, er denne test ikke egnet.
Endpoint for unscheduled DNA-syntese (UDS) måles ved at bestemme optagelsen af mærkede nukleosider i celler, hvor der ikke foregår planmæssig dna-syntese (S-fase). Den mest benyttede teknik består i at bestemme optagelsen af tritiummærket thymidin (3H-TdR) ved autoradiografi. Der bør fortrinsvis benyttes rottelevere i UDS-test in vivo. Der kan benyttes andet væv end levervæv, men det er ikke omfattet af denne metode.
Påvisningen af et UDS-respons afhænger af, hvor mange dna-baser der er skåret ud og erstattet på det beskadigede sted. Derfor er UDS-testen særlig værdifuld til påvisning af stoffremkaldt »longpatch repair« (20-30 baser). Til gengæld er følsomheden over for »shortpatch repair« (1-3 baser) meget lavere. Endvidere kan der optræde mutagene hændelser som følge af manglende reparation, forkert reparation eller forkert replikation af dna-skader. UDS-metodens reaktion er ikke noget mål for, i hvor høj grad reparationsprocessen er lykkedes. Ydermere er det muligt, at et mutagen reagerer med dna, men at dna-beskadigelsen ikke repareres ved en udskæringsreparationsproces. De beskedne specifikke oplysninger om mutagen aktivitet, som UDS-testen giver, opvejes af, at dens endpoint potentielt har høj følsomhed, eftersom det måles i hele genomet.
Se også den generelle indledning til del B.
1.2. DEFINITIONER
Celler under reparation: et NNG-tal, der er højere end en forud fastsat værdi, som det laboratorium, der udfører testen, skal begrunde.
Nettokernekorn (NNG): kvantitativt mål for cellers UDS-aktivitet ved en autoradiografisk UDS-test; tallet beregnes ved at trække gennemsnitsantallet af cytoplastmakorn i kerneækvivalente områder af cytoplasmaet (CG) fra antallet af kernekorn (NG), dvs. NNG = NG-CG. NNG-tallet beregnes for den enkelte celle, hvorefter tallene samles i puljer for celler i samme kultur, for parallelle kulturer osv.
Unscheduled DNA-syntese (UDS): dna-reparationssyntese efter udskæring og fjernelse af en dna-streng, der indeholder en region, der er beskadiget af kemiske stoffer eller fysiske påvirkninger.
1.3. TESTMETODENS PRINCIP
UDS-testen med pattedyrleverceller in vivo viser dna-reparationssyntese efter udskæring og fjernelse af en dna-streng, der indeholder en region, der er beskadiget af kemiske stoffer eller fysiske påvirkninger. Testen baseres sædvanligvis på indsætning af 3H-TdR i dna'et i leverceller, hvor der er lav forekomst af celler i cellecyklussens S-fase. Optagelse af 3H-TdR bestemmes normalt ved autoradiografi, eftersom denne teknik, i modsætning til f.eks. væskescintillation, ikke er følsom over for interferens fra celler i S-fase.
1.4. BESKRIVELSE AF TESTMETODEN
1.4.1. Præparater
1.4.1.1. Valg af dyreart
Der benyttes sædvanligvis rotter, selv om andre egnede pattedyrarter kan benyttes. Der bør anvendes almindeligt brugte laboratoriestammer af unge sunde voksne dyr. Ved undersøgelsens begyndelse bør vægtvariationen mellem dyrene være mindst mulig og ikke over ± 20 % af gennemsnitsvægten for hvert køn.
1.4.1.2. Miljø og fodring
Der gælder de almindelige forhold som beskrevet i den generelle indledning til del B, blot bør der tilstræbes en fugtighed på 50-60 %.
1.4.1.3. Forberedelse af dyrene
Der udvælges tilfældigt sunde unge voksne dyr til kontrol- og behandlingsgruppen. Burene bør anbringes på en sådan måde, at deres placering får mindst mulig indvirkning. Dyrene identificeres entydigt og holdes i deres bure i mindst fem dage, inden undersøgelsen påbegyndes, så de tilvænnes laboratorieforholdene.
1.4.1.4. Teststof/testpræparat
Teststoffer i fast form opløses eller opslæmmes i passende opløsningsmidler eller bærestoffer og fortyndes passende, inden de gives til dyrene. Teststoffer i væskeform kan enten gives direkte til dyrene eller fortyndes først. Der benyttes frisk fremstillede teststofpræparater, medmindre stabilitetsdata viser, at opbevaring er acceptabel.
1.4.2. Testbetingelser
1.4.2.1. Opløsningsmiddel/bærestof
Opløsningsmiddel/bærestof må ikke have toksiske virkninger ved de benyttede doser og må ikke mistænkes for at reagere kemisk med teststoffet. Benyttes der andre opløsningsmidler/bærestoffer end de velkendte, skal brugen af dem underbygges med kompatibilitetsdata. Det anbefales i videst muligt omfang først at forsøge at benytte et vandigt opløsningsmiddel/bærestofsystem.
1.4.2.2. Kontrolgrupper
I hver individuelt gennemført del af forsøget medtages der sideløbende både positive og negative (opløsningsmiddel eller bærestof) kontrolgrupper. Bortset fra behandling med teststof skal dyrene i kontrolgrupperne behandles på samme måde som dyrene i behandlingsgrupperne.
Positive kontrolstoffer bør være stoffer, der vides at fremkalde UDS, når de indgives i en dosis, der forventes at give en påviselig forøgelse i forhold til baggrundsværdierne. Positive kontrolstoffer, der kræver metabolismeaktivering, bør benyttes i doser, der frembringer moderat reaktion (4). Doserne kan vælges på en sådan måde, at virkningerne er tydelige, uden dog at de kodede objektglas' identitet umiddelbart afsløres for den, der aflæser dem. Nedenfor er opregnet eksempler på positive kontrolstoffer:
|
Prøveudtagningstidspunkt |
Stof |
CAS nr. |
Einecs nr. |
|
Tidlig prøveudtagning (2-4 timer) |
N-nitrosodimethylamin |
62-75-9 |
200-249-8 |
|
Sen prøveudtagning |
N-2-fluorenylacetamid |
53-96-3 |
200-188-6 |
Der kan anvendes andre egnede positive kontrolstoffer. Det kan accepteres, at den positive kontrol indgives på anden måde end teststoffet.
1.5. FREMGANGSMÅDE
1.5.1. Dyrenes antal og køn
Der anvendes tilstrækkelig mange dyr til, at der er taget hensyn til den naturlige variation i reaktionerne på testen. Der skal være mindst tre analyserbare dyr i hver gruppe. Er der indsamlet en betydelig historisk database, kræves der kun et eller to dyr i sideløbende negative og positive kontrolgrupper.
Hvis der på det tidspunkt, hvor undersøgelsen foretages, foreligger data fra undersøgelser med samme dyreart og med samme indgiftsmåde, som viser, at der ikke er nogen væsentlig toksicitetsforskel mellem de to køn, er test med kun et køn, helst hanner, tilstrækkeligt. Hvis udsættelsen af mennesker for det kemiske stof er kønsspecifik, som det f.eks. er tilfældet med visse farmaceutiske stoffer, skal testen udføres med dyr af det pågældende køn.
1.5.2. Behandlingsplan
Teststofferne indgives normalt i en enkelt behandling.
1.5.3. Dosisniveauer
Normalt benyttes der to dosisniveauer. Den højeste dosis defineres som den dosis, der fremkalder sådanne tegn på toksicitet, at højere dosis med samme indgiftsmønster må forventes at medføre død. Normalt bør laveste dosis være mellem 50 % og 25 % af den højeste dosis.
Stoffer med specifik biologisk aktivitet ved lav ikke-toksisk dosis (f.eks. hormoner og mitogener) kan danne undtagelser fra disse dosisfastsættelseskriterier og bør evalueres individuelt. Hvis der gennemføres en forprøve til bestemmelse af dosisinterval, fordi der ikke foreligger nogen egnede data, bør den udføres i samme laboratorium med samme art, stamme, køn og behandlingsmåde, som agtes benyttet i hovedundersøgelsen.
Højeste dosis kan også defineres som en dosis, der i nogen grad udviser tegn på toksicitet i leveren (f.eks. pyknotiske kerner).
1.5.4. Grænsetest
Hvis der ved en test med én dosis på mindst 2 000 mg pr. kg legemsvægt indgivet på én gang eller i to omgange samme dag ikke kan iagttages nogen toksiske virkninger, og hvis man på grundlag af data om strukturmæssigt beslægtede stoffer ikke forventer gentoksicitet, er en fuldstændig undersøgelse muligvis ikke nødvendig. Den forventede eksponering af mennesker kan foranledige, at der benyttes en højere dosis i grænsetesten.
1.5.5. Indgift af doser
Teststoffet indgives normalt ved tvangsfodring med sonde eller en passende intubationskanyle. Andre indgiftsmåder kan accepteres, hvis de kan begrundes. Intraperitoneal injektion anbefales ikke, da leveren derved kan udsættes for teststoffet direkte og ikke via kredsløbet. Hvor stor en væskemængde, der kan indgives ad gangen ved tvangsfodring eller injektion, afhænger af forsøgsdyrenes størrelse. Mængden bør højst være på 2 ml pr. 100 g legemsvægt. Anvendelse af større rumfang skal begrundes. Bortset fra lokalirriterende og ætsende stoffer, som normalt vil have kraftigere virkninger ved højere koncentrationer, bør testvolumenet variere så lidt som muligt, idet koncentrationen justeres, så volumenet bliver det samme ved alle dosisniveauer.
1.5.6. Præparering af leverceller
Leverceller fra behandlede dyr præpareres normalt 12-16 timer efter indgiften. Medmindre reaktionen på dette tidspunkt er tydeligt positiv, er desuden yderligere en tidlig prøveudtagning (normalt to-fire timer efter indgiften) sædvanligvis påkrævet. Andre tidspunkter for prøveudtagning kan benyttes, når de kan begrundes med toksikokinetiske data.
Korttidskulturer af pattedyrleverceller etableres sædvanligvis ved perfusion af leveren in situ med collagenase, idet man lader frisk dissocierede leverceller sætte sig fast på en egnet flade. Leverceller fra negative kontroldyr skal have en levedygtighed (5) på mindst 50 %.
1.5.7. Bestemmelse af UDS
Nyligt isolerede pattedyrleverceller inkuberes normalt i et medium, der indeholder 3H-TdR i et passende tidsrum, f.eks. tre-otte timer. Når inkubationstiden er afsluttet, fjernes mediet fra cellerne, som dernæst kan inkuberes i et medium, der indeholder overskud af umærket thymidin, så ikke-indbygget radioaktivitet (»cold chase«) bliver mindst mulig. Cellerne skylles, fikseres og tørres. Ved længere inkuberingstider er »cold chase« ikke altid nødvendigt. Objektglassene dyppes i autoradiografiemulsion, eksponeres i mørke (f.eks. i køleskab i 7-14 dage), fremkaldes og farves, og de eksponerede sølvkorn tælles. Der fremstilles to-tre objektglas pr. dyr.
1.5.8. Analyse
De præparede objektglas skal indeholde tilstrækkelig mange celler med normal morfologi til, at der kan foretages en meningsfyldt vurdering af UDS. Præparaterne undersøges mikroskopisk for tegn på åbenlys cytotoksicitet (f.eks. pyknose eller nedsat radioaktiv mærkning).
Objektglassene kodes, inden kornene tælles. Normalt bedømmes der 100 celler fra hvert dyr på mindst to objektglas; bedømmelse af mindre end 100 celler pr. dyr skal begrundes. Der tælles ikke korn for celler i S-fase, men andelen af celler i S-fase kan registreres.
Den mængde 3H-TdR, der indbygges i kerner og cytoplasma i morfologisk normale celler, og som giver sig udtryk i afsættelse af sølvkorn, bestemmes med egnede metoder.
Der foretages korntælling over kernerne (kernekorn, NG) og kerneækvivalente områder over cytoplasmaet (cytoplasmakorn, CG). CG-tallene fremkommer enten ved at tage de kraftigst mærkede cytoplasmaområder eller ved at tage gennemsnittet af to-tre tilfældigt foretagne tællinger af cytoplasmakorn i nærheden af kernerne. Der kan benyttes andre tællemetoder (f.eks. helcelletælling), hvis de kan begrundes (6).
2. DATA
2.1. BEHANDLING AF RESULTATER
Dataene præsenteres for hvert enkelt objektglas og hvert enkelt dyr. Desuden sammenfattes alle data i tabel form. For hver celle, hvert dyr og hver dosis og tidspunkt beregnes tallet for nettokernekorn (NNG) ved at trække CG-tallene fra NG-tallene. Hvis der tælles »celler under reparation«, skal de kriterier, der benyttes til definering af »celler under reparation«, begrundes og underbygges med tidligere og sideløbende negative kontroldata. Talresultater kan evalueres ved statistiske metoder. Eventuelt benyttede statistiske test skal vælges og begrundes, inden undersøgelsen gennemføres.
2.2. EVALUERING OG FORTOLKNING AF RESULTATER
Nedenfor følger nogle eksempler på kriterier for positiv/negativ reaktion:
|
positiv |
(i) |
NNG-værdier, der er højere end en forud fastsat tærskelsværdi, som er begrundet med udgangspunkt i laboratoriets tidligere data eller |
|
|
(ii) |
NNG-værdier, der er signifikant større end sideløbende kontrol |
|
negativ |
(i) |
NNG-værdier, der er inden for eller under en kontroltærskelværdi, der bygger på tidligere resultater eller |
|
|
(ii) |
NNG-værdier, der ikke er signifikant større end sideløbende kontrol |
Dataenes biologiske relevans skal tages i betragtning, f.eks. sådanne parametre som variation mellem dyr, sammenhæng mellem dosis og respons og cytotoksicitet. Statistiske metoder kan benyttes som hjælpemiddel ved evalueringen af testresultaterne. Statistisk signifikans bor ikke være den eneste faktor, der afgør, om resultatet bedømmes som positivt.
De fleste forsøg giver klart positivt eller negativt resultat, men i sjældne tilfælde giver dataene ikke mulighed for en endegyldig bedømmelse af teststoffets aktivitet. Resultatet kan stadig være tvetydigt eller tvivlsomt, uanset hvor mange gange forsøget gentages.
Et positivt resultat af en test for unscheduled DNA-syntese (UDS) med pattedyrleverceller in vivo viser, at teststoffet inducerer dna-skader i pattedyrleverceller in vivo, som kan repareres ved unscheduled DNA-syntese in vivo. Et negativt resultat viser, at teststoffet under testbetingelserne ikke inducerer dna-skader, der kan påvises ved denne test.
Sandsynligheden for, at teststoffet eller dets metabolitter når frem til målvævet (f.eks. systemisk toksicitet), bør diskuteres.
3. RAPPORTERING
TESTRAPPORT
Testrapporten skal indeholde følgende oplysninger:
|
|
Opløsningsmiddel/bærestof:
|
|
|
Forsøgsdyr:
|
|
|
Testbetingelser:
|
|
|
Resultater:
|
|
|
Diskussion af resultaterne |
|
|
Konklusioner |
4. HENVISNINGER
|
(1) |
Ashby, J., Lefevre, P.A., Burlinson, B. and Penman, M.G. (1985), An Assessment of the In Vivo Rat Hepatocyte DNA Repair Assay, Mutation Res., 156, pp. 1-18. |
|
(2) |
Butterworth, B.E., Ashby, J., Bermudez, H., Casciano, D., Mirsalis, J., Probst G. and Williams, G. (1987), A Protocol and Guide for the In Vivo Rat Hepatocyte DNA-Repair Assay, Mutation Res., 189, pp. 12-133. |
|
(3) |
Kennelly, J.C, Waters, R., Ashby, J., Lefevre, P.A., Burlinson B., Benford, D.J., Dean, S.W. and Mitchell I. de G. (1993), In Vivo Rat Liver UDS Assay, in: Kirkland D.J. and Fox, M., (eds.), Supplementary Mutagenicity Tests: UKEM Recommended Procedures. UKEMS Subcommittee on Guidelines for Mutagenicity Testing Report. Part II revised, Cambridge University Press, Cambridge, New York, Port Chester, Melbourne, Sydney, pp. 52-77. |
|
(4) |
Madle, S., Dean, S.W., Andrae, U., Brambilla, G., Burlinson, B., Doolittle, D.J., Furihata, C, Hertner, T., McQueen, C.A. and Mori, H. (1993), Recommendations for the Performance of UDS Tests In Vitro and In Vivo, Mutations Res.. 312, pp. 263-285. |
|
(5) |
Fautz, R., Hussain, B., Efstathiou, E. and Hechenberger-Freudl, C. (1993), Assessment of the Relation Between the Initital Viability and the Attachment of Freshly Isolated Rat Hepatocytes Used for the In Vivo/InVitro DNA Repair Assay (UDS), Mutation Res., 291, pp. 21-27. |
|
(6) |
Mirsalis, J.C., Tyson, C.K. and Butterworth, B.L. (1982), Detection of Genotoxic Carcinogens in the In Vivo/ln Vitro Hepatocyte DNA Repair Assay, Environ Mutagen, 4, pp. 553-562. |
B.40. IN VITRO-HUDÆTSNING: MÅLING AF TRANSKUTAN ELEKTRISK MODSTAND (TER)
1. METODE
Denne testmetode er ækvivalent med OECD TG 430 (2004).
1.1. INDLEDNING
Hudætsning defineres som fremkomsten af irreversibel vævsødelæggelse i huden efter påføring af teststof (jf. definitionen i Globally Harmonised System for the Classification and Labelling of Chemical Substances and Mixtures (GHS)) (1). Med denne metode opnås der en procedure, hvor ætsning kan vurderes uden brug af levende dyr.
Vurderingen af hudætsning har typisk omfattet brug af laboratoriedyr (2). Betænkelighederne ved de smerter og lidelser, der er forbundet med denne fremgangsmåde, er omhandlet i revisionen af testmetode B.4, som muliggør bestemmelse af hudætsning ved hjælp af alternative in vitro-metoder, så dyrene undgår smerter og lidelser.
Første trin mod definitionen af alternative test, der kan anvendes til test af hudætsning, og som kan anvendes i lovgivningsøjemed, var gennemførelsen af prævalideringsundersøgelser (3). Herefter blev der gennemført (6)(7)(8) en formel valideringsundersøgelse af in vitro-metoder til vurdering af hudætsning (4)(5). På baggrund af resultatet af disse undersøgelser og anden offentliggjort litteratur henstilles det, at følgende test anvendes til vurdering af in vivo-hudætsning (9)(10)(11): test med human hudmodel (se testmetode B.40.A) og test til måling af transkutan elektrisk modstand (denne metode).
I henhold til valideringsundersøgelserne og andre offentliggjorte undersøgelser kan målingen af transkutan elektrisk modstand (TER) i rottehud (12)(13) på pålidelig vis skelne mellem stoffer, der er hudætsende, og stoffer, der ikke er hudætsende.
Den test, der er beskrevet i denne metode, muliggør identifikation af ætsende kemiske stoffer og blandinger. Den muliggør endvidere identifikation af stoffer og blandinger, der ikke er hudætsende, med støtte af væsentlig dokumentation om bestemmelse ved hjælp af andre eksisterende oplysninger (f.eks. pH, forholdet mellem struktur og aktivitet samt human- og/eller dyredata) (1)(2)(11)(14). Den giver ikke tilstrækkelige oplysninger om hudirritation og muliggør heller ikke underinddeling af ætsende stoffer som tilladt i henhold til GHS-systemet (Globally Harmonised Classification System) (1).
For fuldt ud at kunne vurdere de lokale virkninger på huden efter en enkelt dermal eksponering anbefales det at følge den sekventielle teststrategi, der fremgår af testmetode B.4 (2) og GHS-systemet (Globally Harmonised System) (1). Denne teststrategi omfatter gennemførelse af in vitro-test for hudætsning (som beskrevet i forbindelse med denne metode) og hudirritation, inden test i levende dyr overvejes.
1.2. DEFINITION
In vivo-hudætsning: er fremkomsten af irreversibel ødelæggelse af huden, nemlig synlig nekrose gennem epidermis til dermis, efter applikation af et teststof i op til fire timer. Typiske eksempler på ætsende reaktioner er ulcera, blødning, blodige sår og ved slutningen af observationsperioden på 14 dage misfarvning som følge af afblegning af huden, hele områder af alopecia og ar. Histopatologisk undersøgelse bør overvejes for at evaluere tvivlsomme læsioner.
Transkutan elektrisk modstand (TER): er et mål for hudens elektriske impedans som en modstand i kiloohm. En simpel og robust metode til vurdering af barrierefunktion, hvor passagen af ioner gennem hud registreres ved hjælp af en Wheatstone-målebro.
1.3. REFERENCESTOFFER
Tabel 1
Referencestoffer
|
Navn |
EINECS nr. |
CAS nr. |
|
|
1,2-diaminopropan |
201-155-9 |
78-90-0 |
Stærkt ætsende |
|
acrylsyre |
201-177-9 |
79-10-7 |
Stærkt ætsende |
|
2-tert-butylphenol |
201-807-2 |
88-18-6 |
Ætsende |
|
kaliumhydroxid (10 %) |
215-181-3 |
1310-58-3 |
Ætsende |
|
svovlsyre (10 %) |
231-639-5 |
7664-93-9 |
Ætsende |
|
octansyre (kaprylsyre) |
204-677-5 |
124-07-02 |
Ætsende |
|
4-amino-1,2,4-triazol |
209-533-5 |
584-13-4 |
Ikke ætsende |
|
eugenol |
202-589-1 |
97-53-0 |
Ikke ætsende |
|
phenethylbromid |
203-130-8 |
103-63-9 |
Ikke ætsende |
|
tetrachlorethylen |
204-825-9 |
27-18-4 |
Ikke ætsende |
|
isostearinsyre |
250-178-0 |
30399-84-9 |
Ikke ætsende |
|
4-(methylthio)-benzaldehyd |
222-365-7 |
3446-89-7 |
Ikke ætsende |
De fleste af de anførte kemiske stoffer er hentet fra listen over kemiske stoffer, der var udvalgt til den internationale ECVAM-valideringsundersøgelse (4). De er udvalgt ud fra følgende kriterier:
|
i) |
der skal være et lige stort antal ætsende og ikke-ætsende stoffer |
|
ii) |
der skal medtages stoffer, som findes i handelen, og som dækker de fleste af de relevante kemiske grupper |
|
iii) |
der skal indgå stærkt ætsende og mindre ætsende stoffer for at muliggøre skelnen mellem ætsningsstyrke |
|
iv) |
de udvalgte kemiske stoffer skal kunne håndteres på et laboratorium, uden at der opstår andre alvorlige risici end risikoen for ætsning. |
1.4. TESTMETODENS PRINCIP
Testmaterialet påføres i op til 24 timer på den epidermale overflade af hudlapper i et testsystem med to rum, hvor hudlapperne fungerer som adskillelse mellem de to rum. Hudlapperne tages fra 28-30 dage gamle rotter, der er aflivet på en human måde. Ætsende stoffer identificeres ved deres evne til at beskadige stratum corneum og svække dets barrierefunktion, hvilket kan måles som en reduktion i den naturlige TER under en tærskelværdi (12). For rottehud-TER er der valgt en tærskelværdi på 5 kΩ på baggrund af omfattende data om en lang række kemiske stoffer, hvor langt de fleste værdier var enten klart langt over (ofte > 10 kΩ) eller langt under (ofte < 3 kΩ) denne værdi (12). Stoffer, som ikke er ætsende hos dyr, men som er irriterende eller ikke-irriterende, medfører generelt ikke en TER under denne tærskelværdi. Brugen af andre hudpræparater eller andet apparatur kan endvidere ændre tærskelværdien og nødvendiggøre yderligere validering.
Testproceduren omfatter et farvebindingstrin til bekræftende test af positive resultater i TER, herunder værdier omkring 5 kΩ. Farvebindingstrinnet viser, om stigningen i gennemtrængeligheden for ioner skyldes fysisk ødelæggelse af stratum corneum. TER-metoden med rottehud har vist sig at kunne forudsige in vivo-ætsningen hos kaniner vurderet i henhold til testmetode B.4 (2). Det bør bemærkes, at in vivo-kanintesten er særdeles konservativ med hensyn til hudætsning og hudirritation sammenlignet med lappeprøven med human hud (15).
1.5. BESKRIVELSE AF TESTMETODEN
1.5.1. Dyr
Rotter er valgt, fordi deres huds følsomhed over for kemiske stoffer i denne test tidligere er påvist (10). Rottens alder (når hudprøven tages) og stamme er særligt vigtig for at sikre, at hårfolliklerne er i hvile, inden væksten af voksenhår begynder.
Hårene på ryggen og flanken af unge (ca. 22 dage gamle) han- eller hunrotter af (Wistar-stammen eller lignende stamme) fjernes omhyggeligt med en lille dyresaks. Dyrene vaskes dernæst omhyggeligt, idet der gnubbes, og det pågældende hudområde holdes neddyppet i en opløsning af antibiotika, som f.eks. indeholder streptomycin, penicillin, chloramphenicol og amphotericin i koncentrationer, der er høje nok til at hæmme bakterievækst. Dyrene vaskes igen med antibiotika på tredje- og fjerdedagen efter den første vask og skal anvendes inden tre dage efter den anden vask, når stratum corneum har regenereret sig efter fjernelsen af hår.
1.5.2. Fremstilling af hudlapper
Dyrene aflives på human måde, når de er 28-30 dage gamle. Alderen er kritisk. Ryghuden fjernes fra hvert dyr, og overflødigt fedtvæv fjernes omhyggeligt, idet det skrælles af huden. Hudlapper med en diameter på ca. 20 mm udtages. Huden kan gemmes, inden lapperne anvendes, hvis det er påvist, at positive og negative kontroldata svarer til de data, der opnås med frisk hud.
Hver hudlap anbringes for enden af et rør af PTFE (polytetrafluorethylen), idet det sikres, at den epidermale overflade er i kontakt med røret. En O-ring af gummi sættes tætsluttende omkring rørets ende for at holde huden på plads, og overflødigt væv klippes af. Rørets og O-ringens dimensioner fremgår af figur 2. O-ringen af gummi forsegles derefter omhyggeligt til enden af PTFE-røret med vaseline. Røret fastholdes ved hjælp af fjederklemmer i et ydre kammer, som indeholder en MgSO4-opløsning (154 mM) (figur 1). Hudlappen skal være fuldstændigt dækket af MgSO4-opløsningen. Der kan fremstilles op til 10-15 hudlapper af huden fra en enkelt rotte.
Inden testen begynder, måles den elektriske modstand i to hudlapper for at kontrollere den enkelte dyrehuds kvalitet. For begge hudlapper skal modstanden være større end 10 kΩ, hvis resten af lapperne skal anvendes i testen. Hvis modstanden er mindre end 10 kΩ, kasseres resten af lapperne fra den pågældende hud.
1.5.3. Påføring af test- og kontrolstoffer
Samtidige positive og negative kontrolværdier skal anvendes i hver undersøgelse for at sikre, at forsøgsmodellen fungerer korrekt. Der skal anvendes hudlapper fra et enkelt dyr. Som positive og negative kontrolstoffer foreslås henholdsvis 10 M saltsyre og destilleret vand.
Flydende teststoffer (150 μl) påføres på den epidermale overflade inde i røret. Ved faste teststoffer skal tilstrækkeligt af stoffet påføres hudlappen, så hele overfladen er dækket af stoffet. Der tilsættes deioniseret vand (150 μl) oven på stoffet, og røret rystes forsigtigt. For at sikre maksimal kontakt med huden kan det være nødvendigt at opvarme visse faste stoffer til 30 oC, så de smelter eller blødgøres, eller formale dem til granulat eller pulver.
Der bruges tre hudlapper til hvert test- og kontrolstof. Teststofferne skal sidde på huden i 24 timer ved 20-23 oC. Teststoffet fjernes under rindende ledningsvand med en temperatur på højst 30 oC, indtil der ikke kan fjernes mere af stoffet.
1.5.4. TER-målinger
Hudens impedans måles, idet TER måles ved hjælp af en Wheatstone-lavvoltvekselstrømsmålebro (13). De generelle specifikationer for broen er en driftsspænding på 1-3 volt, en sinusformet eller rektangulært formet vekselstrøm på 50-1 000 Hz og et måleområde på mindst 0,1-30 kΩ. Den målebro, der anvendes i valideringsundersøgelsen, måler induktans, kapacitet og modstand op til værdier på henholdsvis 2 000 H, 2 000 μF og 2 MΩ ved frekvenser på 100 Hz eller 1 kHz ved hjælp af serier eller parallelle værdier. I forbindelse med TER-ætsningstesten registreres målingerne af modstand ved en frekvens på 100 Hz og ved hjælp af serieværdier. Før den elektriske modstand måles, reduceres hudens overfladespænding ved tilsætning af 70 % ethanol i så tilstrækkelig mængde, at hele hudens overflade dækkes. Efter nogle få sekunder fjernes ethanolen, og vævet fugtes ved tilsætning af 3 ml MgSO4-opløsning (154 mM). Elektroderne fra målebroen anbringes på hver sin side af hudlappen for at måle modstanden i kΩ/hudlap (figur 1). Elektrodedimensioner og længden af elektroderne under krokodillenæbbene ses i figur 2. Krokodillenæbbet på den indre elektrode hviler på toppen af PTFE-røret under modstandsmålingen, hvorved man sikrer sig, at et konstant stykke af elektroden er nede i MgSO4-opløsningen. Den ydre elektrode er anbragt inde i det ydre kammer, så den rører ved kammerbunden. Afstanden mellem fjederklemmen og bunden af PTFE-røret skal holdes konstant (figur 2), da den har indflydelse på den målte modstand. Det betyder, at afstanden mellem den indre elektrode og hudlappen skal være konstant og så lille som mulig (1-2 mm).
Hvis den målte modstand er større end 20 kΩ, kan dette skyldes, at rester af teststoffet ligger som en belægning på hudlappens overflade. Man kan forsøge at fjerne denne belægning ved at holde en tommelfinger iført en gummitut for PTFE-røret og ryste røret i ca. 10 sekunder. Derefter kasseres MgSO4-opløsningen, og målingen gentages med en ny MgSO4-opløsning.
Testapparaturets og forsøgsprocedurens egenskaber og dimensioner kan have indflydelse på de opnåede TER-værdier. Tærskelværdien for ætsning, 5 kΩ, blev bestemt med det apparatur og den fremgangsmåde, der er beskrevet i denne metode. Hvis testbetingelserne ændres, kan der blive tale om andre tærskel- og kontrolværdier. Det anbefales derfor, at metoden og tærskelværdien for modstand kalibreres ved at teste en række referencestandarder, der vælges blandt de kemikalier, som blev anvendt i valideringsundersøgelsen (4) (5), eller blandt de kemiske grupper, der svarer til de undersøgte kemiske stoffer. Tabel 1 viser et sæt relevante referencestoffer.
1.5.5. Farvebindingsmetoder
Eksponering for visse ikke-ætsende stoffer kan bevirke, at modstanden falder til under tærskelværdien på 5 kΩ, hvorved ioner kan passere gennem stratum corneum, hvilket sænker den elektriske modstand (5). Neutrale organiske stoffer og kemikalier med overfladeaktive egenskaber (herunder rengøringsmidler, emulgatorer og andre overfladeaktive stoffer) kan fjerne hudfedt og gøre barrieren gennemtrængelig for ioner. Hvis TER-værdierne for teststofferne er mindre end eller omkring 5 kΩ, og der ikke forekommer synlige ødelæggelser, skal farvestoffets evne til at trænge ned i kontrolvævet og det behandlede væv vurderes med henblik på at afgøre, om de opnåede TER-værdier skyldes øget hudgennemtrængelighed eller hudætsning (3)(5). I sidstnævnte tilfælde, hvor stratum corneum er ødelagt, trænger farvestoffet sulforhodamin B efter påføring på hudoverfladen hurtigt ned og farver det underliggende væv. Netop dette farvestof er stabilt i forbindelse med en lang række kemiske stoffer og påvirkes ikke af den ekstraktionsprocedure, der er beskrevet i det følgende.
1.5.5.1. Påføring og fjernelse af farvestoffet sulforhodamin B
Efter TER-undersøgelsen fjernes magnesiumsulfatopløsningen fra røret, og huden undersøges omhyggeligt for synlige ødelæggelser. Hvis der ikke forekommer synlig større ødelæggelse, påføres 150 μl af en 10 % (w/v)-opløsning af sulforhodamin B (Acid Red 52; C.I. 45100, EINECS nr. 222-529-8, CAS nr. 3520-42-1) i destilleret vand på den epidermale overflade af hver hudlap i 2 timer. Disse hudlapper vaskes derefter under vandhanen i ca. 10 sekunder med vand, der ikke er varmere end stuetemperatur, for at fjerne overskydende/ikke-bundet farvestof. Hver hudlap fjernes omhyggeligt fra PTFE-røret og anbringes i et glas (f.eks. et 20 ml-glas til scintillationsmålinger) indeholdende deioniseret vand (8 ml). Glasset rystes forsigtigt i 5 minutter for at fjerne yderligere ikke-bundet farvestof. Denne skylning gentages, hvorefter hudlapperne fjernes og anbringes i glas, der indeholder 5 ml af en 30 % (w/v)-opløsning af natriumdodecylsulfat (SDS) i destilleret vand. Glassene inkuberes ved 60 oC til næste dag.
Efter inkubationen fjernes og kasseres hudlapperne, og den tilbageværende opløsning centrifugeres i 8 minutter ved 21 oC (relativ centrifugalkraft ~ 175 × g). 1 ml af supernatanten fortyndes 5 gange (v/v) (dvs. 1 ml + 4 ml) med en 30 % (w/v) SDS-opløsning i destilleret vand. Opløsningens ekstinktion/optical density (OD) måles ved 565 nm.
1.5.5.2. Beregning af farveindhold
Mængden af sulforhodamin B pr. hudlap beregnes ud fra OD-værdierne (5) (sulforhodamin B's molære ekstinktionskoefficient ved 565 nm = 8,7 × l04 ; molekylvægten = 580). Farveindholdet bestemmes for hver hudlap ved hjælp af en speciel kalibreringskurve, hvorefter et gennemsnitligt farveindhold beregnes for flergangsbestemmelser.
2. DATA
De målte resultater for modstand (kΩ) og eventuelt gennemsnitligt farveindhold (μg/hudlap) i teststoffet og i positive og negative kontroller skal fremlægges i tabelform, herunder også data fra flergangsbestemmelser/gentagne undersøgelser, gennemsnitsværdier og individuelle værdier.
2.1. FORTOLKNING AF RESULTATER
De gennemsnitlige TER-resultater godkendes, hvis de samtidige positive og negative kontrolværdier falder inden for de acceptable intervaller for metoden i testlaboratoriet. De acceptable modstandsintervaller for den metode og det apparatur, der er beskrevet ovenfor, er anført i følgende tabel:
|
Kontrol |
Stof |
Modstandsinterval (kΩ) |
|
Positiv |
10M saltsyre |
0,5-1,0 |
|
Negativ |
Destilleret vand |
10-25 |
De gennemsnitlige resultater for farvebinding godkendes, hvis de samtidige positive og negative kontrolværdier falder inden for de acceptable intervaller for metoden. De foreslåede acceptable intervaller for farveindhold for de kontrolstoffer, den metode og det apparatur, der er beskrevet ovenfor, er anført i følgende tabel:
|
Kontrol |
Stof |
Interval af farveindhold (μg/hudlap) |
|
Positiv |
10M saltsyre |
40-100 |
|
Negativ |
Destilleret vand |
15-35 |
Teststoffet betragtes ikke som værende hudætsende:
|
i) |
hvis den gennemsnitlige TER-værdi for teststoffet er større end 5 kΩ, eller |
|
ii) |
hvis den gennemsnitlige TER-værdi er mindre end eller lig med 5 kΩ, og
|
Teststoffet betragtes som værende hudætsende:
|
i) |
hvis den gennemsnitlige TER-værdi er mindre end eller lig med 5 kΩ, og hudlappen er synligt beskadiget, eller |
|
ii) |
hvis den gennemsnitlige TER-værdi er mindre end eller lig med 5 kΩ, og
|
3. RAPPORTERING
3.1. TESTRAPPORT
Testrapporten skal indeholde følgende oplysninger:
|
|
Test- og kontrolstoffer:
|
|
|
Forsøgsdyr:
|
|
|
Testbetingelser:
|
|
|
Resultater:
|
|
|
Diskussion af resultater |
|
|
Konklusioner |
4. HENVISNINGER
|
(1) |
OECD (2001) Harmonised Integrated Classification System for Human Health and Environmental Hazards of Chemical Substances and Mixtures. OECD Series on Testing and Assessment Number 33. ENV/JM/MONO(2001)6, Paris. http://www.olis.oecd.org/olis/2001doc.nsf/LinkTo/env-jm-mono(2001)6. |
|
(2) |
Testmetode B.4. Akut toksicitet: hudirritation/ætsning. |
|
(3) |
Botham, P.A., Chamberlain, M., Barratt, M.D., Curren, R.D., Esdaile, D.J., Gardner, J.R., Gordon, V.C., Hildebrand, B., Lewis, R.W., Liebsch, M., Logemann, P., Osborne, R., Ponec, M., Regnier, J.F., Steiling, W., Walker, A.P., and Balls, M. (1995). A prevalidation study on in vitro skin corrosivity testing. The report and recommendations of ECVAM Workshop 6. ATLA 23, 219-255. |
|
(4) |
Barratt, M.D., Brantom, P.G., Fentem, J.H., Gerner, I., Walker, A.P., and Worth, A.P. (1998). The ECVAM international validation study on in vitro tests for skin corrosivity. 1. Selection and distribution of the test chemicals. Toxic. in Vitro 12, 471-482. |
|
(5) |
Fentem, J.H., Archer, G.E.B., Balls, M., Botham, P.A., Curren, R.D., Earl, L.K., Esdaile, D.J., Holzhutter, H.-G., and Liebsch, M. (1998). The ECVAM international validation study on in vitro tests for skin corrosivity. 2. Results and evaluation by the Management Team. Toxic. in Vitro 12, 483- 524. |
|
(6) |
OECD (1996). Final Report of the OECD Workshop on Harmonization of Validation and Acceptance Criteria for Alternative Toxicological Test Methods, 62 pp. |
|
(7) |
Balls, M., Blaauboer, B.J., Fentem. J.H., Bruner. L., Combes, R.D., Ekwall, B., Fielder. R.J., Guillouzo, A., Lewis, R.W., Lovell, D.P., Reinhardt, C.A., Repetto, G., Sladowski. D., Spielmann, H., and Zucco, F. (1995). Practical aspects of the validation of toxicity test procedures. The report and recommendations of ECVAM workshops. ATLA 23, 129-147. |
|
(8) |
ICCVAM (Interagency Coordinating Committee on the Validation of Alternative Methods). (1997). Validation and Regulatory Acceptance of Toxicological Test Methods. NIH Publication No. 97-3981. National Institute of Environmental Health Sciences, Research Triangle Park, NC, USA. http://iccvam.niehs.nih.gov/docs/guidelines/validate.pdf. |
|
(9) |
ECVAM (1998). ECVAM News & Views. ATLA 26, 275-280. |
|
(10) |
ICCVAM (Interagency Coordinating Committee on the Validation of Alternative Methods). (2002). ICCVAM evaluation of EpiDermTM, EPISKINTM (EPI-200), and the Rat Skin Transcutaneous Electrical Resistance (TER) assay: In Vitro test methods for assessing dermal corrosivity potential of chemicals. NIH Publication No. 02-4502. National Toxicology Program Interagency Center for the Evaluation of Alternative Toxicological Methods, National Institute of Environmental Health Sciences, Research Triangle Park, NC, USA. http://iccvam.niehs.nih.gov/methods/epiddocs/episbrd.pdf. |
|
(11) |
OECD (2002) Extended Expert Consultation Meeting on The In Vitro Skin Corrosion Test Guideline Proposal, Berlin, 1st-2nd November 2001, Secretariat's Final Summary Report, 27* March 2002, OECD ENV/EHS, available upon request from the Secretariat. |
|
(12) |
Oliver, G.J.A., Pemberton, M.A., and Rhodes, C. (1986). An in vitro skin corrosivity test — modifications and validation. Fd. Chem. Toxicol. 24, 507-512. |
|
(13) |
Botham, P.A., Hall, T.J., Dennett, R., McCall, J.C., Basketter, D.A., Whittle, E., Cheeseman, M., Esdaile, D.J., and Gardner, J. (1992). The skin corrosivity test in vitro: results of an interlaboratory trial. Toxic. in Vitro 6, 191-194. |
|
(14) |
Worth A.P., Fentem J.H., Balls M., Botham P.A., Curren R.D., Earl L.K., Esdaile D.J., Liebsch M. (1998). An Evaluation of the Proposed OECD Testing Strategy for Skin Corrosion. ATLA 26: 709-720. |
|
(15) |
Basketter, D.A., Chamberlain, M., Griffiths, H.A., Rowson, M., Whittle, E., York, M. (1997). The classification of skin irritants by human patch test. Fd. Chem. Toxicol. 35, 845-852. |
|
(16) |
Oliver G.J.A, Pemberton M.A and Rhodes C. (1988). An In Vitro model for identifying skin-corrosive chemicals. I. Initial Validation. Toxic. in Vitro. 2, 7-17. |
Figur 1
Apparatur til TER-test med rottehud

Figur 2
Dimensioner på anvendte PFTE-rør, ydre rør og elektroder
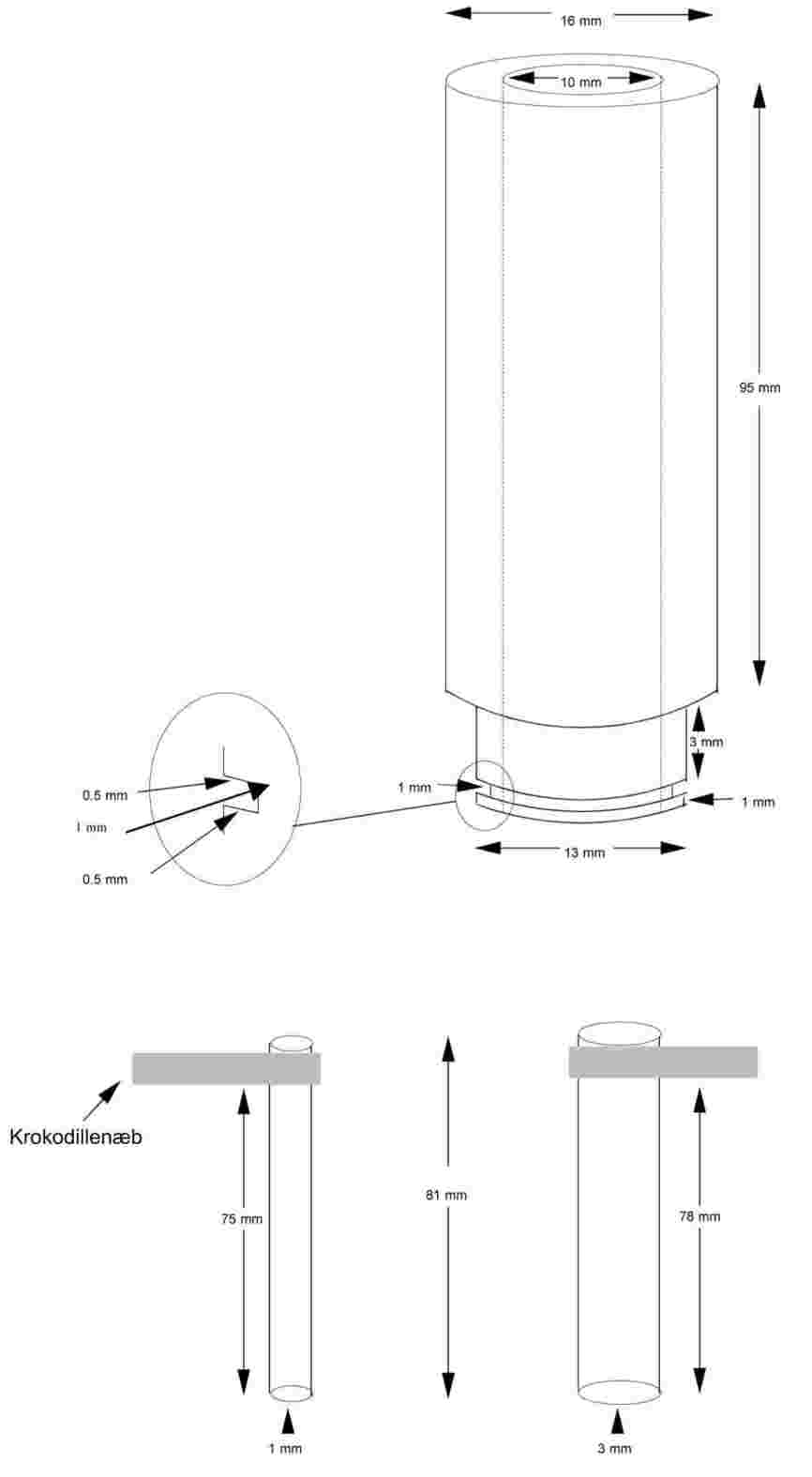
Kritiske faktorer for det ovenfor viste apparatur:
|
— |
PTFE-rørets indvendige diameter |
|
— |
længden af elektroderne i forhold til PTFE-røret og det ydre rør, så hudlappen ikke berøres af elektroderne, og så en standardlængde af elektroden er i kontakt med MgSO4-opløsningen |
|
— |
mængden af MgSO4-opløsning i det ydre rør bør sikre en væskedybde i forhold til niveauet i PFTE-røret som vist i figur 1 |
|
— |
hudlappen bør være tilstrækkeligt fastgjort til PFTE-røret, så den elektriske måling er et sandt mål for hudens egenskaber. |
B.40.A. IN VITRO-HUDÆTSNING: TEST MED HUMAN HUDMODEL
1. METODE
Denne testmetode er ækvivalent med OECD TG 431 (2004).
1.1. INDLEDNING
Hudætsning defineres som fremkomsten af irreversibel vævsødelæggelse i huden efter påføring af teststof (jf. definitionen i Globally Harmonised System for the Classification and Labelling of Chemical Substances and Mixtures (GHS)) (1). Denne testmetode kræver ikke brug af levende dyr eller dyrevæv til vurdering af hudætsning.
Vurderingen af hudætsning har typisk omfattet brug af laboratoriedyr (2). Betænkelighederne ved de smerter og lidelser, der er forbundet med denne fremgangsmåde, er omhandlet i revisionen af testmetode B.4, som muliggør bestemmelse af hudætsning ved hjælp af alternative in vitro-metoder, så dyrene undgår smerter og lidelser.
Første trin mod definitionen af alternative test, der kan anvendes til test af hudætsning, og som kan anvendes i lovgivningsøjemed, var gennemførelsen af prævalideringsundersøgelser (3). Herefter blev der gennemført (6)(7)(8) en formel valideringsundersøgelse af in vitro-metoder til vurdering af hudætsning (4)(5). På baggrund af resultatet af disse undersøgelser og anden offentliggjort litteratur (9) henstilles det, at følgende test anvendes til vurdering af in vivo-hudætsning (10)(11)12)(13): test med human hudmodel (denne metode) og test til måling af transkutan elektrisk modstand (se testmetode B.40).
I henhold til valideringsundersøgelserne kan test baseret på humane hudmodeller (3)(4)(5)(9) på pålidelig vis skelne mellem stoffer, der er hudætsende, og stoffer, der ikke er hudætsende. Med forsøgsprotokollen var det endvidere muligt at skelne mellem stærkt hudætsende og mindre stærkt hudætsende stoffer.
Den test, der er beskrevet i denne metode, muliggør identifikation af ætsende kemiske stoffer og blandinger. Den muliggør endvidere identifikation af stoffer og blandinger, der ikke er hudætsende, med støtte af væsentlig dokumentation om bestemmelse ved hjælp af andre eksisterende oplysninger (f.eks. pH, forholdet mellem struktur og aktivitet samt human- og/eller dyredata) (1)(2)(13)(14). Den giver normalt ikke tilstrækkelige oplysninger om hudirritation og muliggør heller ikke underinddeling af ætsende stoffer som tilladt i henhold til GHS-systemet (Globally Harmonised Classification System) (1).
For fuldt ud at kunne vurdere de lokale virkninger på huden efter en enkelt dermal eksponering anbefales det at følge den sekventielle teststrategi, der fremgår af testmetode B.4 (2) og GHS-systemet (Globally Harmonised System) (1). Denne teststrategi omfatter gennemførelse af in vitro-test for hudætsning (som beskrevet i forbindelse med denne metode) og hudirritation, inden test i levende dyr overvejes.
1.2. DEFINITIONER
In vivo-hudætsning: er fremkomsten af irreversibel ødelæggelse af huden, nemlig synlig nekrose gennem epidermis til dermis, efter applikation af et teststof i op til fire timer. Typiske eksempler på ætsende reaktioner er ulcera, blødning, blodige sår og ved slutningen af observationsperioden på 14 dage misfarvning som følge af afblegning af huden, hele områder af alopecia og ar. Histopatologisk undersøgelse bør overvejes for at evaluere tvivlsomme læsioner.
Cellernes levedygtighed: er en parameter, der måler den samlede aktivitet i en cellepopulation (f.eks. evnen hos cellulær mitokondrial dehydrogenase til at reducere det livsvigtige farvestof MTT), og som, afhængigt af det valgte endpoint og testens udformning, korrelerer med det totale antal celler og/eller deres levedygtighed.
1.3. REFERENCESTOFFER
Tabel 1
Referencestoffer
|
Navn |
EINECS nr. |
CAS nr. |
|
|
1,2-diaminopropan |
201-155-9 |
78-90-0 |
Stærkt ætsende |
|
acrylsyre |
201-177-9 |
79-10-7 |
Stærkt ætsende |
|
2-tert-butylphenol |
201-807-2 |
88-18-6 |
Ætsende |
|
kaliumhydroxid (10 %) |
215-181-3 |
1310-58-3 |
Ætsende |
|
svovlsyre (10 %) |
231-639-5 |
7664-93-9 |
Ætsende |
|
octansyre (kaprylsyre) |
204-677-5 |
124-07-02 |
Ætsende |
|
4-amino-1,2,4-triazol |
209-533-5 |
584-13-4 |
Ikke ætsende |
|
eugenol |
202-589-1 |
97-53-0 |
Ikke ætsende |
|
phenethylbromid |
203-130-8 |
103-63-9 |
Ikke ætsende |
|
tetrachlorethylen |
204-825-9 |
27-18-4 |
Ikke ætsende |
|
isostearinsyre |
250-178-0 |
30399-84-9 |
Ikke ætsende |
|
4-(methylthio)-benzaldehyd |
222-365-7 |
3446-89-7 |
Ikke ætsende |
De fleste af de anførte kemiske stoffer er hentet fra listen over kemiske stoffer, der var udvalgt til den internationale ECVAM-valideringsundersøgelse (4). De er udvalgt ud fra følgende kriterier:
|
i) |
der skal være et lige stort antal ætsende og ikke-ætsende stoffer |
|
ii) |
der skal medtages stoffer, som findes i handelen, og som dækker de fleste af de relevante kemiske grupper |
|
iii) |
der skal indgå stærkt ætsende og mindre ætsende stoffer for at muliggøre skelnen mellem ætsningsstyrke |
|
iv) |
de udvalgte kemiske stoffer skal kunne håndteres på et laboratorium, uden at der opstår andre alvorlige risici end risikoen for ætsning. |
1.4. TESTMETODENS PRINCIP
Teststoffet påføres på en tredimensional human hudmodel, som mindst omfatter en rekonstrueret epidermis med et fungerende stratum corneum. Ætsende stoffer identificeres ved deres evne til at bringe cellers levedygtighed (f.eks. påvist med MTT-reduktionstesten (15)) ned under en fastlagt tærskelværdi efter en bestemt eksponeringstid. Testmetoden med forsøgsopstilling med human hud er baseret på den hypotese, at kemikalier er ætsende, når de er i stand til at trænge gennem stratum corneum ved diffusion eller erosion og er cytotoksiske for de underliggende cellelag.
1.4.1. Procedure
1.4.1.1. Humane hudmodeller
Humane hudmodeller kan konstrueres eller købes i handelen (f.eks. EpiDerm™-og EPISKIN™-modellerne) (16)(17)(18)(19), eller de kan udvikles eller konstrueres i testlaboratoriet (20)(21). Det anerkendes, at brugen af human hud er underlagt nationale og internationale etiske hensyn og betingelser. En ny model bør valideres (som minimum i det omfang, der er beskrevet i 1.4.1.1.2). Humane hudmodeller, der anvendes i forbindelse med denne test, skal overholde følgende:
1.4.1.1.1.
Humane keratinocytter skal anvendes til at konstruere epithel. Der skal være flere lag af levedygtige epithelceller til stede under et fungerende stratum corneum. Hudmodellen kan også have lag med en stromal komponent. Stratum corneum skal være flerlaget og have den nødvendige fedtprofil til at udgøre en effektiv barriere, der kan modstå hurtig gennemtrængning af cytotoksiske markører. Modellens fysiske udformning skal forhindre, at stof passerer uden om stratum corneum til det levedygtige væv. Passage af kemiske teststoffer uden om stratum corneum vil forringe modelleringen af hudens eksponering. Hudmodellen skal være fri for kontamination med bakterier (herunder mykoplasma) og svampe.
1.4.1.1.2.
Graden af levedygtighed kvantificeres normalt ved hjælp MTT eller andre livsvigtige farvestoffer, der omdannes metabolisk. I disse tilfælde skal absorbansen (optical density (OD)) i det ekstraherede (opløste) farvestof fra den negative kontrol være mindst 20 gange større end absorbansen i ekstraktionsmidlet alene (se oversigten i (22)). Vævet i den negative kontrol skal have stabil kultur (levere enslydende levedygtighedsmålinger) i hele testeksponeringsperioden. Stratum corneum skal være tilstrækkeligt robust til at modstå hurtig gennemtrængning af visse cytotoksiske kemiske markørstoffer (f.eks. 1 % Triton X-100). Denne egenskab kan bedømmes ud fra den eksponeringstid, der er nødvendig for at reducere cellelevedygtigheden med 50 % (ET50) (for the EpiDerm™- og EPISKIN™-modellen er det f.eks. > 2 timer). Vævet skal give reproducerbarhed over længere tid og helst mellem laboratorier. Det skal endvidere kunne forudsige referencestoffernes ætsningsstyrke (se tabel 1), når det anvendes i forbindelse med den valgte forsøgsprotokol.
1.4.1.2. Påføring af test- og kontrolstoffer
Der anvendes to vævsprøver til hver behandling (eksponeringstid), inkl. kontroller. Ved flydende stoffer skal der påføres tilstrækkeligt teststof til, at hudoverfladen dækkes (mindst 25 μl/cm2). Ved faste stoffer skal der påføres tilstrækkeligt stof til, at huden dækkes med et jævnt lag. Stofferne skal fugtes med deioniseret eller destilleret vand for at sikre god hudkontakt. Om nødvendigt skal de males til et pulver, før de påføres. Påføringsmetoden skal være velegnet til det pågældende teststof (se f.eks. henvisning (5)). Efter eksponeringsperioden skal teststoffet omhyggeligt vaskes af hudoverfladen med en buffer eller 0,9 % NaCl.
Samtidige positive og negative kontrolværdier skal anvendes i hver undersøgelse for at sikre, at forsøgsmodellen fungerer korrekt. Som positivt kontrolstof foreslås iseddikesyre eller 8N KOH. Som negativt kontrolstof foreslås 0,9 % NaCl eller vand.
1.4.1.3. Måling af cellelevedygtighed
Enhver kvantitativ, godkendt metode kan anvendes til at måle cellernes levedygtighed. Målingen af levedygtighed skal endvidere kunne anvendes sammen med en tredimensional hudmodel. Ikke-specifik farvebinding må ikke interferere med levedygtighedsmålingen. Proteinbindende farvestoffer og farvestoffer, som ikke undergår metabolisk omdannelse (f.eks. Neutral Red), kan derfor ikke anvendes. Den hyppigst anvendte måling er MTT-reduktionstesten [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazoliumbromid, Thiazolyl Blue: EINECS nr. 206-069-5, CAS nr. 298-93-1], som har vist sig at give nøjagtige og reproducerbare resultater (5), men andre kan anvendes. Hudlappen lægges i en MTT-opløsning af passende koncentration (f.eks. 0,3-1 mg/ml) ved en passende inkubationstemperatur i tre timer. Det udfældede blå formazan ekstraheres ved hjælp af et opløsningsmiddel (isopropanol), og formazankoncentrationen beregnes ud fra måling af absorbans ved en bølgelængde mellem 540 og 595 nm.
Teststoffets kemiske reaktion med det livsvigtige farvestof kan efterligne den metaboliske reaktion i cellerne, hvilket resulterer i en falsk levedygtighedsmåling. Dette har vist sig at ske, når et sådant teststof ikke fjernes fuldstændigt fra huden ved skylning (9). Hvis teststoffet reagerer direkte med det levedygtige farvestof, skal der anvendes yderligere kontroller til at spore og korrigere for teststoffernes interferens med levedygtighedsmålingen (9)(23).
2. DATA
For hver hudlap skal der for teststof og for positive og negative kontroller fremlægges absorbansværdier og beregnede tal for procentuel cellelevedygtighed i tabelform, herunder data fra flergangsbestemmelser/gentagne bestemmelser, gennemsnitsværdier og enkeltværdier.
2.1. FORTOLKNING AF RESULTATER
De absorbansværdier, som måles ved hver undersøgelse, anvendes til beregning af den procentuelle levedygtighed i forhold til en negativ kontrol, som vilkårligt sættes til 100 %. Den procentuelle tærskelværdi af cellelevedygtighed, som adskiller ætsende fra ikke-ætsende stoffer (eller som skelner mellem forskellige grader af ætseevne), skal klart defineres, dokumenteres og påvises at være hensigtsmæssig. Generelt bliver disse tærskelværdier fastlagt under testoptimeringen, afprøvet i prævalideringsfasen og bekræftet ved en valideringsundersøgelse. Følgende benyttes f.eks. ved forudsigelse af ætsningsevne i forbindelse med EpiDerm™ -modellen (9):
Teststoffet betragtes som værende hudætsende:
|
i) |
hvis levedygtigheden efter tre minutters eksponering er mindre end 50 %, eller |
|
ii) |
hvis levedygtigheden efter tre minutters eksponering er større end eller lig med 50 %, og levedygtigheden efter en times eksponering er mindre end 15 %. |
Teststoffet betragtes ikke som værende hudætsende:
|
i) |
hvis levedygtigheden efter tre minutters eksponering er større end eller lig med 50 %, og levedygtigheden efter en times eksponering er større end eller lig med 15 %. |
3. RAPPORTERING
3.1. TESTRAPPORT
Testrapporten skal indeholde følgende oplysninger:
|
|
Test- og kontrolstof:
|
|
|
Begrundelse for valg af hudmodel og forsøgsprotokol. |
|
|
Testbetingelser:
|
|
|
Resultater:
|
|
|
Diskussion af resultater. |
|
|
Konklusion |
4. HENVISNINGER
|
(1) |
OECD (2001) Harmonised Integrated Classification System for Human Health and Environmental Hazards of Chemical Substances and Mixtures. OECD Series on Testing and Assessment Number 33. ENV/JM/MONO(2001)6, Paris. http://www.olis.oecd.org/olis/2001doc.nsf/LinkTo/env-jm-mono(2001)6. |
|
(2) |
Testmetode B.4. Akut toksicitet: hudirritation/ætsning. |
|
(3) |
Botham, P.A., Chamberlain, M., Barratt, M.D., Curren, R.D., Esdaile, D.J., Gardner, J.R., Gordon, V.C., Hildebrand, B., Lewis, R.W., Liebsch, M., Logemann, P., Osborne, R., Ponec, M., Regnier, J.F., Steiling, W., Walker, A.P., and Balls, M. (1995). A prevalidation study on in vitro skin corrosivity testing. The report recommendations of ECVAM Workshop 6 ATLA 23, 219-255. |
|
(4) |
Barratt, M.D., Brantom, P.G., Fentem, J.H., Gerner, L, Walker, A.P., and Worth, A.P. (1998). The ECVAM international validation study on in vitro tests for skin corrosivity. 1. Selection and distribution of the test chemicals. Toxic. In Vitro 12, 471-482. |
|
(5) |
Fentem, J.H., Archer, G.E.B., Balls, M., Botham, P.A., Curren, R.D., Earl, L.K., Esdaile, D.J., Holzhutter, H.G. and Liebsch, M. (1998). The ECVAM international validation study on in vitro tests for skin corrosivity. 2. Results and evaluation by the Management Team Toxic In Vitro 12, 483-524. |
|
(6) |
OECD (1996). Final Report of the OECD Workshop on Harmonization of Validation and Acceptance Criteria for Alternative Toxicological Test Methods, 62 pp. |
|
(7) |
Balls, M., Blaauboer, B.J., Fentem, J.H., Bruner, L., Combes, R.D., Ekwall, B., Fielder, R.J., Guillouzo, A., Lewis, R.W., Lovell, D.P., Reinhardt, C.A., Repetto, G., Sladowski, D., Spielmann, H., and Zucco, F. (1995). Practical aspects of the validation of toxicity test procedures. Test report and recommendations of ECVAM workshops. ATLA 23, 129-147. |
|
(8) |
ICCVAM (Interagency Coordinating Committee on the Validation of Alternative Methods). (1997). Validation and Regulatory Acceptance of Toxicological Test Methods. NIH Publication No. 97-3981. National Institute of Environmental Health Sciences, Research Triangle Park, NC, USA. http://iccvam.niehs.nih.gov/docs/guidelines/validate.pdf. |
|
(9) |
Liebsch, M., Traue, D., Barrabas, C, Spielmann, H., Uphill, P., Wilkins, S., McPherson, J.P., Wiemann, C, Kaufmann, T., Remmele, M. and Holzhutter, H.G. (2000). The ECVAM prevalidation study on the use of EpiDerm for skin Corrosivity testing. ATLA 28, pp. 371-401. |
|
(10) |
ECVAM (1998). ECVAM News & Views. ATLA 26, 275-280. |
|
(11) |
ECVAM (2000). ECVAM News & Views. ATLA 28, 365-67. |
|
(12) |
ICCVAM (Interagency Coordinating Committee on the Validation of Alternative Methods). (2002). ICCVAM evaluation of EpiDerm™, EPISKIN™ (EPI-200), and the Rat Skin Transcutaneous Electrical Resistance (TER) assay: In Vitro test methods for assessing dermal corrosivity potential of chemicals. NIH Publication No. 02-4502. National Toxicology Program Interagency Center for the Evaluation of Alternative Toxicological Methods, National Institute of Environmental Health Sciences, Research Triangle Park, NC, USA. http://iccvam.niehs.nih.gov/methods/epiddocs/epis_brd.pdf. |
|
(13) |
OECD (2002) Extended Expert Consultation Meeting on The In Vitro Skin Corrosion Test Guideline Proposal, Berlin, 1st-2nd November 2001, Secretariat's Final Summary Report, 27th March 2002, OECD ENV/EHS, available upon request from the Secretariat. |
|
(14) |
Worth AP, Fentem JH, Balls M, Botham PA, Curren RD, Earl LK, Esdaile DJ, Liebsch M (1998). An Evaluation of the Proposed OECD Testing Strategy for Skin Corrosion. ATLA 26: 709-720. |
|
(15) |
Mosmann, T. (1983). Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity asssays. J.Immunol. Meth. 65, 55-63. |
|
(16) |
Cannon, C.L., Neal, P.J., Southee, J.A., Kubilus, J., and Klausner, M., 1994. New epidermal model for dermal irritancy testing. Toxic. In Vitro 8, 889-891. |
|
(17) |
Ponec, M., Boelsma, E., Weerheim, A., Mulder, A., Boutwstra, J., and Mommaas, M., 2000. Lipid and ultrastructural characterization of reconstructed skin models. International Journal of Pharmaceutics. 203, 211-225. |
|
(18) |
Tinois E., Gaetani Q., Gayraud B., Dupont D., Rougier A., Pouradier D.X. (1994). The Episkin model: Successful reconstruction of human epidermis in vitro. In In vitro Skin Toxicology. Edited by A Rougier, AM Goldberg and HI Maibach: 133-140. |
|
(19) |
Tinois E., Tiollier J., Gaucherand M., Dumas H., Tardy M., Thivolet J. (1991). In vitro and post -transplantation differentiation of humankeratinocytes grown on the human type IV collagen film of a bilayereddermal substitute. Experimental Cell Research 193: 310-319. |
|
(20) |
Parentau, N.L., Bilbo, P., Molte, C.J., Mason, V.S., and Rosenberg, H.(1992). The organotypic culture of human skin keratinocytes and fibroblasts to achieve form and function. Cytotechnology 9, 163-171. |
|
(21) |
Wilkins, L.M., Watson, S.R., Prosky, S.J., Meunier, S.F., Parentau, N.L. (1994). Development of a bilayered living skin construct for clinical applications. Biotechnology and Bioengineering 43/8, 747-756. |
|
(22) |
Marshall, N.J., Goodwin, C.J., Holt, S.J. (1995). A critical assessment of the use of microculture tetrazolium assays to measure cell growth and function. Growth Regulation 5, 69-84. |
|
(23) |
Fentem, J.H., Briggs, D., Chesne', C, Elliot, G.R., Harbell, J.W., Heylings, J.R., Portes, P., Rouget, R., and van de Sandt, J.J.M., and Botham, P.A. (2001). A prevalidation study on in vitro tests for acute skin irritation: results and evaluation by the Management Team. Toxic. In VItro 15, 57-93. |
B.41. IN VITRO 3T3 NRU-FOTOTOKSICITETSTEST
1. METODE
Denne metode er ækvivalent med OECD TG 432 (2004).
1.1. INDLEDNING
Fototoksicitet defineres som det toksiske respons på et stof, der påføres kroppen, og som enten er blevet fremkaldt af eller forstærket af (kan konstateres ved lavere doser) eksponering for lys, eller som er forårsaget af bestråling af huden efter systemisk indgift af et stof.
In vitro 3T3 NRU-fototoksicitetstesten anvendes til at afdække det fototoksiske potentiale i et testmateriale, når det exciteres ved lyspåvirkning. Testen evaluerer fotocytotoksicitet ud fra den relative reduktion i levedygtigheden af de celler, der udsættes for det kemiske stof under lyspåvirkning, sammenlignet med samme situation uden lys. De stoffer, som identificeres ved denne test, er sandsynligvis fototoksiske in vivo efter systemisk påføring og fordeling på huden eller efter lokal påføring.
Der er rapporteret om mange typer kemiske stoffer, der fremkalder fototoksiske virkninger (1)(2)(3)(4). Et fælles træk er deres evne til at absorbere lysenergi inden for sollysets spektrum. Ifølge fotokemiens grundlov (Grotthaus-Draper-loven) forudsætter fotoreaktion en tilstrækkelig optaget lysmængde. Derfor skal man, inden man overvejer en biologisk testning, bestemme testkemikaliets absorptionsspektrum for UV/synligt lys i henhold til OECD Test Guideline 101. Det er påstået, at det kemiske stof sandsynligvis ikke er fotoreaktivt, hvis den molære ekstinktions- og absorptionskoefficient er mindre end 10 liter × mol-1 × cm-1. Et kemisk stof af denne type behøver ikke nødvendigvis at blive testet i in vitro 3T3 NRU-fototoksicitetstesten eller i nogen anden biologisk test for skadelige fotokemiske virkninger (1)(5). Se også bilag 1.
Pålideligheden og relevansen af in vitro 3T3 NRU-fototoksicitetstesten er blevet evalueret for nylig (6)(7)(8)(9). In vitro 3T3 NRU-fototoksicitetstesten har vist sig at kunne forudsige akutte fototoksicitetsvirkninger på dyr og mennesker in vivo. Testen er ikke beregnet til at forudsige andre skadelige virkninger, der måtte opstå fra samtidig påvirkning fra et kemisk stof og lys. Eksempelvis påviser den ikke fotogenotoksicitet, fotoallergi eller fotocarcinogenicitet, og den er heller ikke beregnet til vurdering af fototoksisk styrke. Testen er endvidere ikke beregnet til at påvise indirekte mekanismer ved fototoksicitet, virkninger af stofskifteprodukter på testmaterialet eller virkninger af blandinger.
Hvor anvendelsen af metaboliserende systemer er et generelt krav til alle in vitro-testninger til forudsigelse af genotoksisk og carcinogent potentiale, er der indtil nu i forbindelse med fototoksikologi kun få tilfælde, hvor der kræves metabolisk transformation for at få det kemiske stof til at reagere som fototoksin in vivo eller in vitro. Det anses derfor hverken som nødvendigt eller videnskabeligt begrundet at gennemføre nærværende testning med et metabolisk aktiverings system.
1.2. DEFINITIONER
Strålingsintensitet: intensiteten af ultraviolet (UV) eller synligt lys, der falder på en overflade. Intensiteten måles i W/m2 eller mW/cm2.
Lysdosis: mængden (= intensitet × tid) af ultraviolet (UV) eller synligt lys, der falder på en overflade, udtrykt i joule (= W × s) pr. overfladeareal, f.eks. J/m2 eller J/cm2.
UV-lys, bølgelængdeområder: følgende betegnelser er anbefalet af CIE (Den Internationale Belysningskommission): UVA (315-400 nm), UVB (280-315 nm) og UVC (100-280 nm). Der anvendes også andre betegnelser. Grænsen mellem UVB og UVA sættes ofte ved 320 nm, og UVA kan inddeles i UV-A1 og UV-A2, hvor grænsen ligger ved ca. 340 nm.
Cellernes levedygtighed: parameter, der måler den samlede aktivitet i en cellepopulation (f.eks. optagelse af det livsvigtige farvestof Neutral Red i cellelysosomer), hvilket, afhængigt af det målte endepunkt og det anvendte testdesign, korrelerer med det samlede celleantal og/eller cellevitaliteten.
Relativ cellelevedygtighed: cellelevedygtighed udtrykt i forhold til negative kontroller (opløsningsmiddel), der har været gennem hele testforløbet (enten +Irr eller –Irr), og som ikke er behandlet med kemiske teststoffer.
PIF (fotoirritationsfaktor): faktor, der beregnes ved sammenligning mellem to lige effektive cytotoksiske koncentrationer (IC50) af det kemiske teststof ved fravær af (–Irr) og ved tilstedeværelse af (+Irr) i en ikke-cytotoksisk bestråling med UVA/synligt lys.
IC50: koncentrationen af det kemiske teststof, der reducerer cellelevedygtigheden med 50 %.
MPE (gennemsnitlig lyseffekt): måling, der er afledt af en matematisk analyse af de koncentrationsresponskurver, der fremkommer ved fravær af (–Irr) og ved tilstedeværelse af (+Irr) i en ikke-cytotoksisk bestråling med UVA/synligt lys.
Fototoksicitet: akut toksisk respons, der fremkommer efter den første eksponering af huden for visse kemiske stoffer og efterfølgende eksponering for lys, eller som er fremkaldt på samme måde ved hudbestråling efter systemisk indgift af et kemisk stof.
1.3. TESTMETODENS PRINCIP
In vitro 3T3 NRU-fototoksicitetstesten er baseret på en sammenligning mellem cytotoksiciteten af et kemisk stof, når det afprøves med og uden eksponering for en ikke-cytotoksisk dosis af simuleret sollys. Cytotoksicitet er i denne testning udtrykt som en koncentrationsafhængig reduktion af optagelsen af det livsvigtige farvestof Neutral Red som målt 24 timer efter behandling med det kemiske teststof og bestråling (10). NR er et svagt kationisk farvestof, der let gennemtrænger cellemembraner ved ikke-diffusion og akkumuleres intracellulært i lysosomer. Forandringer i overfladen i den sensitive lysosommembran fører til lysosomal ustabilitet og andre forandringer, der gradvist bliver uoprettelige. Den type forandringer, der forårsages af miljøfremmede stoffer, medfører et fald i optagelsen og bindingen af NR. Det er således muligt at skelne mellem levedygtige, beskadigede og døde celler, hvilket er grundlaget for denne test.
Balb/c 3T3-celler dyrkes i 24 timer, så der dannes encellede lag. To 96-huls plader for hvert kemisk teststof præinkuberes med otte forskellige koncentrationer af teststoffet i 1 time. Derefter eksponeres den ene af de to plader for den højeste ikke-cytotoksiske strålingsdose, mens den anden plade holdes i mørke. På begge plader erstattes behandlingsmediet derefter af kulturmedium, og efter endnu 24 timers inkubation bestemmes cellelevedygtigheden ved Neutral Red-optagelsen. Cellelevedygtigheden udtrykkes som en procentdel i forhold til ubehandlede kontrolkulturer med opløsningsmiddel og beregnes for hver testkoncentration. For at forudsige det fototoksiske potentiale sammenholdes koncentrationerne for samme respons med og uden stråling, sædvanligvis på IC50-niveau, dvs. den koncentration, der reducerer cellelevedygtigheden til 50 % sammenlignet med de ubehandlede kontroller.
1.4. BESKRIVELSE AF TESTMETODEN
1.4.1. Forberedelser
1.4.1.1. Celler
I valideringsundersøgelserne blev der anvendt en permanent muse-fibroblast cellelinje, Balb/c 3T3, klon 31, enten fra American Type Culture Collection (ATCC), Manassas, VA, USA, eller fra European Collection of Cell Cultures (ECACC), Salisbury, Wiltshire, Det Forenede Kongerige, og det anbefales derfor at anskaffe materiale fra en velanskrevet cellebank. Hvis der blot kan påvises ækvivalens, kan der anvendes andre celler eller cellelinjer til samme testprocedure, hvis kulturforholdene er tilpasset cellernes specifikke behov.
Celler bør regelmæssigt undersøges for, at de ikke indeholder mycoplasmasmitte, og de bør kun anvendes, hvis de er absolut smittefrie (11).
Det er vigtigt, at cellernes UV-følsomhed undersøges regelmæssigt i henhold til den procedure for kvalitetskontrol, som er beskrevet i denne metode. Eftersom cellers UVA-følsomhed kan stige med antallet af cellegenerationer, bør der anvendes Balb/c 3T3 celler med lavest muligt antal, helst mindre end 100 (se punkt 1.4.2.2.2 og bilag 2).
1.4.1.2. Medier og dyrkningsbetingelser
Der bør anvendes korrekte dyrkningsmedie- og inkubationsforhold til rutinemæssige celledyrkninger under testproceduren. For Balb/c 3T3-celler er de korrekte forhold f.eks. DMEM (Dulbecco's Modified Eagle's Medium) suppleret med 10 % serum fra nyfødte kalve, 4 mM glutamin, penicillin (100 IU), streptomycin (100 g/ml) og inkubation i fugtig atmosfære ved 37 oC, 5-7,5 % CO2, afhængigt af buffer (se punkt 1.4.1.4, andet afsnit). Der er især vigtigt, at dyrkningsforholdene for cellekulturen tillader en cellecyklustid inden for cellernes eller cellelinjernes normale historiske område.
1.4.1.3. Forberedelse af kulturerne
Celler fra nedfrosne kulturer podes i kulturmedier ved passende tæthed og dyrkes i subkultur mindst en gang, inden de anvendes i in vitro 3T3 NRU-fototoksicitetstesten.
Celler, der anvendes til fototoksicitetstesten, podes i et kulturmedium med passende tæthed, så kulturerne ikke løber sammen ved testens afslutning, dvs. cellelevedygtigheden bestemmes 48 timer efter podning af cellerne. For Balb/c 3T3-celler, der dyrkes på 96-huls plader, er den anbefalede podningstæthed for cellerne 1 × 104 celler pr. hul.
For hver test podes kemikalieceller ensartet i to separate 96-huls plader, der begge sideløbende gennemfører hele testproceduren under identiske kulturforhold med undtagelse af den tid, hvor den ene af pladerne bestråles (+Irr), og den anden holdes i mørke (–Irr).
1.4.1.4. Forberedelse af teststof
Teststoffer skal være friske og forberedes umiddelbart inden brug, medmindre data påviser, at de er stabile ved opbevaring. Det anbefales, at enhver form for kemisk håndtering og den indledende behandling af cellerne udføres under lysforhold, der udelukker fotoaktivering eller nedbrydning af teststoffet inden bestråling.
Testkemikalierne skal være opløst i buffer-saltopløsninger, f.eks. Earle's Balanced Salt Solution (EBSS), eller andre fysiologiske buffer-opløsninger, der skal være fri for proteinkomponenter og lysabsorberende komponenter (f.eks. pH-indikatorfarver og vitaminer) for at undgå interferens under bestråling. Da celler under bestråling holdes uden for CO2-inkubatoren i ca. 50 minutter, skal man være særligt påpasselig for at undgå alkalisering. Hvis der anvendes svage buffere som f.eks. EBSS, kan alkalisering undgås ved at inkubere cellerne ved 7,5 % CO2. Hvis cellerne inkuberes ved kun 5 % CO2, skal der vælges en stærkere buffer.
Testkemikalier med begrænset opløselighed i vand skal opløses i et passende opløsningsmiddel. Hvis der anvendes opløsningsmiddel, skal det forefindes i et konstant volumen i alle kulturer, dvs. i de negative (opløsningsmiddel)kontroller og i alle koncentrationer af testkemikaliet, og det skal være ikke-cytotoksisk ved den pågældende koncentration. Der skal vælges en sådan koncentration af testkemikaliet, at man undgår udfældning og uklare opløsninger.
Dimethylsulfoxid (DMSO) og ethanol (EtOH) er de anbefalede opløsningsmidler. Andre opløsningsmidler med lav cytotoksicitet kan også være velegnede. Inden brug skal alle opløsningsmidler vurderes for specifikke egenskaber f.eks. reaktion med testkemikaliet, quenching af den fototoksiske effekt, evne til at opfange radikaler og/eller kemisk stabilitet i opløsningen.
Vortexblanding og/eller ultralydbehandling og/eller opvarmning til passende temperaturer kan anvendes for at fremme opløsningen, medmindre dette kan påvirke testkemikaliets stabilitet.
1.4.1.5. Betingelser for bestråling
1.4.1.5.1.
Valg af den rette lyskilde og filtre er en afgørende faktor i fototoksicitetstestning. Lys i UVA-området og det synlige område forbindes almindeligvis med fototoksiske reaktioner in vivo (3)(12), hvorimod UVB ofte er mindre relevant, men kraftigt cytotoksisk. Cytotoksiciteten stiger 1 000 gange, når bølgelængden går fra 313 til 280 nm (13). Kriterierne for valg af passende lyskilde skal bl.a. omfatte krav om, at lyskilden udsender bølgelængder, der absorberes af testkemikaliet (absorptionsspektrum), og at lysmængden (der kan opnås inden for en acceptabel eksponeringstid) skal være tilstrækkelig til bestemmelse af kendte fotocytotoksiske kemiske stoffer. Endvidere skal bølgelængder og de anvendte doser ikke påføre testsystemet unødige skader, f.eks. som følge af varmestråling (det infrarøde område).
Simulering af sollys med solsimulatorer anses for at være den optimale kunstlyskilde. Fordelingen af bestråling s styrken i den filtrerede solsimulator skal ligge tæt på fordelingen i udendørs dagslys, som det fremgår af (14). Både xenonlysbuer og (doterede) kviksølvmetalhalogenidlysbuer anvendes som solsimulatorer (15). Sidstnævnte har den fordel, at de udsender mindre varme og er billigere, men ligheden med sollys er knap så perfekt som xenonlysbuernes. Eftersom alle solsimulatorer udsender væsentlige mængder UVB, skal de være behørigt filtrerede for at dæmpe de stærkt cytotoksiske UVB-bølgelængder. Da plastmaterialer til cellekulturer indeholder UV-stabilisatorer, skal spektret måles gennem den samme type 96-huls pladelåg, som anvendes ved testningen. Uanset hvilke forholdsregler der er taget for at dæmpe dele af spektret ved filtrering eller som følge af uundgåelige filtereffekter fra udstyret, må det spektrum, der måles på under disse filtre, ikke afvige fra standard udendørs dagslys (14). Et eksempel på den spektrale strålingsintensitetsfordeling i den filtrerede solsimulator, der anvendes i valideringsundersøgelsen i in vitro 3T3 NRU-fototoksicitetstesten, fremgår af (8)(16). Se også bilag 2, figur 1.
1.4.1.5.2.
Lysintensiteten kontrolleres regelmæssigt før hver fototoksicitetstest ved hjælp af et bredbåndet UV-meter. Intensiteten måles gennem den samme type 96-huls pladelåg, som anvendes ved testen. UV-meteret skal være kalibreret til kilden. UV-meterets ydelse kontrolleres, og til dette formål anbefales det at anvende et ekstra UV-meter af samme type og med samme kalibrering som reference. Det optimale ville være, med lidt længere mellemrum, at anvende et spektroradiometer til måling af den spektrale intensitet af den filtrerede lyskilde og til kontrol af kalibreringen af det bredbåndede UV-meter.
En dosis på 5 J/cm2 (målt i UVA-området) er blevet bestemt til at være ikke-cytotoksisk for Balb/c 3T3-celler og tilstrækkelig kraftig til at få kemiske stoffer til at udløse fototoksiske reaktioner (6) (17). For at nå op på 5 J/cm2 inden for en periode på 50 min blev intensiteten f.eks. justeret til 1,7 mW/cm2. Se bilag 2, figur 2. Hvis der anvendes en anden cellelinje eller en anden lyskilde, skal strålingsdosen eventuelt kalibreres, så der vælges en dosering, der ikke er skadelig for cellerne, men samtidig tilstrækkelig til at udløse standardfototoksiner. Lyseksponeringens varighed beregnes på følgende måde:
|
|
(1J = 1 Wsek) |
1.4.2. Forsøgsbetingelser
1.4.2.1. Teststoffets koncentrationer
Koncentrationsområdet for et kemisk stof, der testes henholdsvis med (+Irr) og uden (–Irr) lys, skal nøje fastlægges ud fra eksperimenter til bestemmelse af dosisområde. Det kan være nyttigt at vurdere opløseligheden fra starten og ved 60 min (eller ved den behandlingstid, der skal anvendes), idet opløseligheden kan ændre sig efterhånden eller i løbet af eksponeringstiden. For at undgå toksicitet fremkaldt af forkerte kulturforhold eller af stærkt sure eller alkaliske kemikalier skal pH-værdien i cellekulturer, der er tilsat testkemikalier, ligge i området 6,5-7,8.
Den højeste koncentration af teststoffet skal ligge inden for fysiologiske testforhold. F.eks. skal belastende osmotiske og pH-mæssige forhold undgås. Afhængigt af teststof kan det være nødvendigt at overveje andre fysisk-kemiske egenskaber som faktorer til fastlæggelse af den højeste testkoncentration. Med hensyn til relativt opløselige stoffer, der ikke er toksiske ved koncentrationer op til mætningspunktet, skal den højest mulige koncentration testes. Generelt skal man undgå udfældning af teststoffet ved enhver testkoncentration. Den maksimale koncentration af et teststof bør ikke overstige 1 000 μg/ml. Osmolalitet bør ikke overstige 10 mM. Der skal anvendes en geometrisk fortyndingsrække bestående af otte teststofkoncentrationer med en konstant fortyndingsfaktor (se punkt 2.1, andet afsnit).
Hvis der findes oplysninger (fra et eksperiment til områdebestemmelse) om, at testkemikaliet ikke er cytotoksisk op til grænsekoncentrationen i mørkeeksperimentet (–Irr), men at det er stærkt cytotoksisk, når det bestråles (+Irr), kan man for at overholde kvalitetskravene til relevante data vøæge andre koncentrationsområder for (+Irr)-eksperimentet end dem, der er valgt for (–Irr)-eksperimentet.
1.4.2.2. Kontrolkulturer
1.4.2.2.1.
Celler skal kontrolleres regelmæssigt (ca. ved hver femte cellegeneration) for følsomhed over for lyskilden ved at vurdere deres levedygtighed efter at være eksponeret for stigende doser af bestråling. Til denne vurdering skal der anvendes adskillige bestrålingsdoser og også niveauer, der er væsentligt højere end de værdier, der anvendes ved 3T3 NRU-fototoksicitetstest. Disse doser kvantificeres lettest ved at måle UV-dele af lyskilden. Celler podes med den tæthed, der er anvendt i in vitro 3T3 NRU-fototoksicitetstesten, og bestråles den følgende dag. Cellelevedygtigheden bestemmes en dag senere ud fra Neutral Red-optagelsen. Det skal påvises, at den heraf følgende højeste ikke-cytotoksiske dosis (f.eks. i valideringsundersøgelsen: 5 J/cm2 [UVA]) var tilstrækkelig til at klassificere referencekemikalierne (tabel 1) korrekt.
1.4.2.2.2.
Testen overholder kvalitetskriterierne, hvis de bestrålede negative/opløsningsmiddelholdige kontrolkulturer udviser en levedygtighed på mere end 80 % sammenlignet med ikke-bestrålede negative/opløsningsmiddelholdige kontroller.
1.4.2.2.3.
Den absolutte absorbans (optical density) (OD 540 NRU ) for Neutral Red ekstraheret fra kontrolkulturer med opløsningsmiddel indikerer, om de 1 × 104 celler, der er podet pr. hul, har vokset med normal fordoblingstid i løbet af testens to dage. En testning opfylder de accepterede kriterier, hvis middelværdien af OD 540 NRU for de ubehandlede kontrolkulturer er ≥ 0,4 (dvs. ca. 20 gange baggrundsopløsningsmidlets absorbans).
1.4.2.2.4.
Et velkendt fototoksisk kemisk stof skal undersøges sideløbende med hver in vitro 3T3 NRU-fototoksicitetstest. Klorpromazin (CPZ) kan anbefales hertil. For CPZ, der testes ifølge standardprotokollen for in vitro 3T3 NRU-fototoksicitetstesten, er der defineret følgende godkendelseskriterier for afprøvningen: bestrålet CPZ (+Irr): IC50 = 0,1 til 2,0 g/ml, ikke-bestrålet CPZ (–Irr): IC50 = 7,0 til 90,0 g/ml. Fotoirritationsfaktoren (PIF) skal være > 6. Den positive kontrols historiske ydelse skal overvåges.
Andre fototoksiske kemikalier, der er velegnede i forhold til den kemiske klasse eller opløselighedskarakteristikken af det kemiske stof, der skal evalueres, kan anvendes som sideløbende positiv kontrol i stedet for klorpromazin.
1.4.3. Testprocedure (6)(7)(8)(16)(17)
1.4.3.1. Første dag
Der anbringes 100 μl kulturmedium i de ydre huller i en 96-huls mikrotiterplade til vævskultur (= negative kontroller). I de øvrige huller tilsættes 100 μl af en cellesuspension med 1 × 105 celler/ml i kulturmedium (= 1 × 104 celler/hul). Der skal klargøres to plader for hver serie af individuelle teststofkoncentrationer samt til opløsningsmiddelkontrollen og den positive kontrol.
Cellerne inkuberes i 24 timer (se punkt 1.4.1.2), indtil de danner et halvt sammenflydende encellet lag. Denne inkubationsperiode tillader cellegendannelse, adhæsion og eksponentiel vækst.
1.4.3.2. Anden dag
Efter inkubation dekanteres kulturmediet fra cellerne, som vaskes omhyggeligt med 150 μl af det bufferopløsningsmiddel, der blev anvendt til inkubationen. Der tilsættes 100 μl af bufferen med den korrekte koncentration af teststoffet eller opløsningsmidlet (kontrolkultur med opløsningsmiddel). Der benyttes otte forskellige koncentrationer af teststoffet. Cellerne med teststoffet inkuberes i mørke i 60 minutter (se punkt 1.4.1.2 og punkt 1.4.1.4, andet afsnit).
Blandt de to plader, der er klargjort til hver serie koncentrationer af teststof og kontrolmateriale, udvælges der en tilfældig plade til bestemmelse af cytotoksicitet (–Irr), dvs. kontrolpladen, og desuden en behandlingsplade til bestemmelse af fotocytotoksicitet (+Irr).
+Irr-eksponeringen gennemføres ved at bestråle cellerne ved stuetemperatur i 50 minutter gennem låget på 96-huls pladen med den størst mulige strålingsdosis, der ikke er cytotoksisk (se også bilag 2). Ikke-bestrålede plader (–Irr) opbevares ved stuetemperatur i en mørk kasse i 50 minutter (= lyseksponeringstiden).
Testopløsningen dekanteres fra, og der vaskes to gange omhyggeligt med 150 μl af den bufferopløsning, der anvendes til inkubation, men som ikke indeholder teststoffet. Bufferen erstattes med kulturmediet, og der inkuberes til næste dag (18-22 timer). Se punkt 1.4.1.2.
1.4.3.3. Tredjedag:
1.4.3.3.1
Med et fasekontrast-mikroskop undersøges cellerne med hensyn til vækst, morfologi og enkeltlagets integritet. Ændringer i cellemorfologi og påvirkninger af cellevækst registreres.
1.4.3.3.2.
Cellerne vaskes med 150 μl forvarmet buffer. Vaskeopløsningen fjernes ved at banke let på pladen. Der tilsættes 100 μl af en 50 μg/ml Neutral Red (NR) (3-amino-7-dimethylamino-2-methylphenazinhydrochlorid, EINECS nr. 209-035-8; CAS nr. 553-24-2; C.I. 50040) til dyrkningsmediet uden serum (16), og der inkuberes som beskrevet i punkt 1.4.1.2 i tre timer. Efter inkubation fjernes Neutral Red-mediet, og cellerne vaskes med 150 μl buffer. Efter dekantering fjernes overskydende buffer med sugende papir eller ved centrifugering.
Der tilsættes nøjagtig 150 μl Neutral Red-desorptionsopløsning (frisk fremstillet af 49 dele vand + 50 dele ethanol + 1 del eddikesyre).
Mikrotiterpladen rystes forsigtigt på en mikrotiterpladeshaker i 10 minutter, indtil al Neutral Red er ekstraheret fra cellerne, og der er dannet en homogen opløsning.
OD af Neutral Red-ekstraktet måles ved 540 nm i et spektrofotometer med blindprøver som reference. Resultaterne gemmes i et passende filformat med henblik på senere analyse.
2. DATA
2.1. DATAKVALITET OG -MÆNGDE
Resultaterne af undersøgelsen skal give mulighed for en relevant analyse af koncentration/respons fremkommet med og uden bestråling og, hvis det er muligt, af den koncentration af kemisk stof, hvor cellelevedygtigheden reduceres til 50 % (IC50). Hvis der påvises cytotoksicitet, skal både koncentrationsområdet og enkeltkoncentrationerne fastsættes på en sådan måde, at der kan tilpasses en kurve til forsøgsdataene.
Hvis resultaterne er enten klart positive eller klart negative (se punkt 2.3, første afsnit), vil første forsøg, der understøttes af et eller flere foreløbige eksperimenter til bestemmelse af dosisområde, være tilstrækkeligt.
Usikre og tvetydige resultater skal kontrolleres ved at gentage undersøgelsen (se også punkt 2.4, andet afsnit). I disse tilfælde kan det være påkrævet at ændre forsøgsbetingelserne for at opnå et klart resultat. Forsøgsbetingelser, der kan ændres, er bl.a. koncentrationsområde, inkubationstid før bestråling og eksponeringstid. En kortere eksponeringstid kan være relevant for stoffer, der er ustabile i vandig opløsning.
2.2. EVALUERING AF RESULTATERNE
Til evaluering af resultaterne kan der beregnes en fotoirritationsfaktor (PIF) eller en gennemsnitlig lyseffekt (MPE).
Ved beregning af parametrene for fotocytotoksicitet (se nedenfor) skal de spredte koncentration-responsværdier tilnærmes en passende kontinuerlig koncentration-responskurve (model). Kurven tilnærmes normalt til dataene ved hjælp af en ikke-lineær regressionsmetode (18). For at kunne vurdere virkningen af dataafvigelser på den tilpassede kurve anbefales det at gennemføre en bootstrap-proces.
Fotoirritationsfaktoren (PIF) beregnes ved hjælp af følgende formel:
![]()
Hvis der ikke kan beregnes en IC50 med eller uden lys, kan der ikke fastlægges en PIF for testmaterialet. Den gennemsnitlige lyseffekt (MPE) er baseret på en sammenligning af de færdige koncentrationsresponskurver (19). Den defineres som et vægtet gennemsnit af repræsentative sæt af lyseffektværdier:

Lyseffekten PEC ved en given koncentration C defineres som produktet af responseffekt REC og dosiseffekt DEC dvs. PEC = REC × DEC. Responseffekt REC er forskellen mellem den konstaterede respons med og uden lys, dvs. REC = Rc (–Irr) – Rc (+Irr). Dosiseffekten beregnes ved hjælp af:

hvor C* repræsenterer ækvivalenskoncentrationen, dvs. den koncentration, hvor et +Irr respons svarer til et –Irr respons ved koncentration C. Hvis C* ikke kan bestemmes, fordi responsværdierne i +Irr kurven systematisk er højere eller lavere end RC (–Irr), sættes dosiseffekten til 1. Vægtningsfaktorerne wi fremkommer ved den højeste responsværdi, dvs. wi = MAX {Ri (–Irr), Ri (-Irr) }. Koncentrationstabellen C; udvælges, så der er lige mange punkter i hvert af de koncentrationsintervaller, som de koncentrationsværdier, der anvendes i eksperimentet, afgrænser. Beregningen af MPE er begrænset til den højeste koncentrationsværdi, hvor mindst en af de to kurver udviser en responsværdi på mindst 10 %. Hvis denne højeste koncentration er højere end den højeste koncentration, der er blevet anvendt i +Irr-forsøget, sættes den resterende del af +Irr-kurven til responsværdien »0«. Afhængigt af om MPE-værdien er større end en korrekt udvalgt skæringsværdi (MPEC = 0,15) eller ej, klassificeres stoffet som fototoksisk.
En softwarepakke til beregning af PIF og MPE kan bestilles hos (20).
2.3. FORTOLKNING AF RESULTATER
På grundlag af valideringsundersøgelsen (8) vil et teststof med en PIF < 2 eller en MPE < 0,1 give resultatet: »ingen fototoksicitet«. En PIF > 2 men < 5 eller en MPE > 0,1 men < 0,15 vil give resultatet: »sandsynlig fototoksicitet«, og en PIF > 5 eller en MPE > 0,15 vil give resultatet: »fototoksicitet«.
Alle laboratorier, der starter brug af denne test op, skal undersøge de referencematerialer, der er opstillet i tabel 1, inden de begynder at teste stoffer for fototoksicitet. PIF- eller MPE-værdier skal ligge tæt op ad værdierne i tabel 1.
Tabel 1
|
Kemisk betegnelse |
EINECS nr. |
CAS nr. |
PIF |
MPE |
Absorptionsmaksima |
Opløsningsmiddel (7) |
|
Amiodaron HCl |
243-293-2 |
[19774-82-4] |
> 3,25 |
0,27-0,54 |
242 nm 300 nm (skulder) |
ethanol |
|
Klorpromazin HCl |
200-701-3 |
[69-09-0] |
> 14,4 |
0,33-0,63 |
309 nm |
ethanol |
|
Norfloxacin |
274-614-4 |
[70458-96-7] |
> 71,6 |
0,34-0,90 |
316 nm |
acetonitril |
|
Anthracen |
204-371-1 |
[120-12-7] |
> 18,5 |
0,19-0,81 |
356 nm |
acetonitril |
|
Protoporphyrin IX, dinatrium |
256-815-9 |
[50865-01-5] |
> 45,3 |
0,54-0,74 |
402 nm |
ethanol |
|
L-Histidin |
|
[7006-35-1] |
ingen PIF |
0,05-0,10 |
211 nm |
vand |
|
Hexachlorophen |
200-733-8 |
[70-30-4] |
1,1-1,7 |
0,00-0,05 |
299 nm 317 nm (skulder) |
ethanol |
|
Natriumlaurylsulfat |
205-788-1 |
[151-21-3] |
1,0-1,9 |
0,00-0,05 |
ingen absorption |
vand |
2.4. FORTOLKNING AF DATA
Hvis der kun kan konstateres fototoksiske virkninger ved den højeste testkoncentration (specielt i forbindelse med vandopløselige teststoffer), skal der indgå andre overvejelser i risikovurderingen. Det kan f.eks. være data om hudabsorption og ophobning af det kemiske stof i huden og/eller data fra andre test, f.eks. test af det kemiske stof in vitro i dyrehud eller human hud eller hudmodeller.
Hvis der ikke påvises nogen toksicitet (+Irr og –Irr), og hvis dårlig opløselighed har begrænset de mulige koncentrationer, kan man betvivle testens egnethed til det pågældende stof, og der bør gennemføres bekræftende undersøgelser baseret på en anden model.
3. RAPPORTERING
TESTRAPPORT
Testrapporten skal mindst indeholde følgende oplysninger:
|
|
Teststof:
|
|
|
Opløsningsmiddel:
|
|
|
Celler:
|
|
|
Testbetingelser (1) ved inkubation før og efter eksponering for kemikaliet:
|
|
|
Testbetingelser (2) ved eksponering for kemikaliet:
|
|
|
Testbetingelser (3) ved bestråling:
|
|
|
Testbetingelser (4) ved Neutral Red-levedygtighedstest:
|
|
|
Resultater:
|
|
|
Diskussion af resultater |
|
|
Konklusioner |
4. HENVISNINGER
|
(1) |
Lovell W.W. (1993). A scheme for in vitro screening of substances for photoallergenic potential. Toxic. In Vitro 7: 95-102. |
|
(2) |
Santamaria, L. and Prino, G. (1972). List of the photodynamic substances. In »Research Progress in Organic, Biological and Medicinal Chemistry« Vol. 3 part 1. North Holland Publishing Co. Amsterdam. p XI-XXXV. |
|
(3) |
Spielmann, H., Lovell, W.W., Hölzle, E., Johnson, B.E., Maurer, T., Miranda, M.A., Pape, W.J.W., Sapora, O., and Sladowski, D. (1994). In vitro phototoxicity testing: The report and recommendations of ECVAM Workshop 2. ATLA, 22, 314-348. |
|
(4) |
Spikes, J.D. (1989). Photosensitization. In »The science of Photobiology« Edited by K.C. Smith. Plenum Press, New York. 2nd edition, p 79-110. |
|
(5) |
OECD (1997) Environmental Health and Safety Publications, Series on Testing and Assessment No.7 »Guidance Document On Direct Phototransformation Of Chemicals In Water« Environment Directorate, OECD, Paris. |
|
(6) |
Spielmann, H., Balls, M., Doring, B., Holzhiitter, H.G., Kalweit, S., Klecak, G., L'Eplattenier, H., Liebsch, M., Lovell, W.W., Maurer, T., Moldenhauer. F. Moore. L., Pape, W., Pfannbecker, U., Potthast, J., De Silva, O., Steiling, W., and Willshaw, A. (1994). EEC/COLIPA project on in vitro phototoxicity testing: First results obtained with a Balb/c 3T3 cell phototoxicity assay. Toxic. In Vitro 8, 793-796. |
|
(7) |
Anon (1998). Statement on the scientific validity of the 3T3 NRU PT test (an in vitro test for phototoxicity), European Commission, Joint Research Centre: ECVAM and DGXI/E/2, 3 November 1997, ATLA, 26, 7-8. |
|
(8) |
Spielmann, H., Balls, M., Dupuis, J., Pape, W.J.W., Pechovitch, G. De Silva, O., Holzhutter, H.G., Clothier, R., Desolle, P., Gerberick, F., Liebsch, M., Lovell, W.W., Maurer, T., Pfannenbecker, U., Potthast, J. M., Csato, M., Sladowski, D., Steiling, W., and Brantom, P. (1998). The international EU/COLIPA/n vitro phototoxicity validation study: results of phase II (blind trial), part 1: the 3T3 NRU phototoxicity test. Toxic. In Vitro 12, 305-327. |
|
(9) |
OECD (2002) Extended Expert Consultation Meeting on The In Vitro 3T3 NRU Phototoxicity Test Guideline Proposal, Berlin, 3Oth-31th October 2001, Secretariat's Final Summary Report, 15th March 2002, OECD ENV/EHS, available upon request from the Secretariat. |
|
(10) |
Borenfreund, E., and Puerner, J.A. (1985). Toxicity determination in vitro by morphological alterations and neutral red absorption. Toxicology Lett, 24, 119-124. |
|
(11) |
Hay, R.J. (1988) The seed stock concept and quality control for cell lines. Analytical Biochemistry 171, 225-237. |
|
(12) |
Lambert L.A, Warner W.G., and Kornhauser A. (1996) Animal models for phototoxicity testing. In »Dermatotoxicology«, edited by F.N. Marzulli and H.I. Maibach. Taylor & Francis, Washington DC. 5th Edition, p 515-530. |
|
(13) |
Tyrrell R.M., Pidoux M (1987) Action spectra for human skin cells: estimates of the relative cytotoxicity of the middle ultraviolet, near ultraviolet and violet regions of sunlight on epidermal keratinocytes. Cancer Res., 47, 1825-1829. |
|
(14) |
ISO 10977. (1993). Photography — Processed photographie colour films and paper prints — Methods for measuring image stability. |
|
(15) |
Sunscreen Testing (UV.B) TECHNICALREPORT, CIE, International Commission on Illumnation, Publication No. 90, Vienna, 1993, ISBN 3 900 734 275. |
|
(16) |
ZEBET/ECVAM/COLIPA — Standard Operating Procedure: In Vitro 3T3 NRU Phototoxicity Test. Final Version, 7 September, 1998. 18 pgs. |
|
(17) |
Spielmann, H., Balls, M., Dupuis, J., Pape, W.J.W., De Silva, O., Holzhütter, H.G., Gerberick, F., Liebsch, M., Lovell, W.W., and Pfannenbecker, U. (1998) A study on UV filter chemicals from Annex VII of the European Union Directive 76/768/EEC, in the in vitro 3T3 NRU phototoxicity test. ATLA 26, 679-708. |
|
(18) |
Holzhütter, H.G., and Quedenau, J. (1995) Mathematical modeling of cellular responses to external signals. J. Biol. Systems 3, 127-138. |
|
(19) |
Holzhütter, H.G. (1997). A general measure of in vitro phototoxicity derived from pairs of dose-response curves and its use for predicting the in vivo phototoxicity of chemicals. ATLA, 25, 445-462. |
|
(20) |
http://www.oecd.org/document/55/0,2340,en_2649_34377_2349687_1_1_1_1,00.html |
BILAG 1
3T3 NRU PT's rolle i forhold til de forskellige trin i fototoksicitetstest af kemikalier

BILAG 2
Figur 1
Spektraleffektfordeling i en filtreret solsimulator

(Se punkt 1.4.1.5, andet afsnit)
Figur 1 viser et eksempel på en acceptabel spektralintensitetsfordeling i en filtreret solsimulator. Målingerne er fra den doterede metalhalogenidkilde, der anvendes i valideringstesten i 3T3 NRU PT (6)(8)(17). Kurverne viser to forskellige filtres virkning og den supplerende filtereffekt af låget på en 96-huls cellekulturplade. H2-filter blev kun anvendt i forbindelse med testsystemer, der tolererer en større mængde UVB (test med hudmodel og fotohæmolysetest af røde blodlegemer). I 3T3 NRU-PT blev H1-filteret anvendt. Af figuren fremgår, at supplerende filtreringseffekt fra pladelåget hovedsagelig konstateres i UVB-området, hvor det tillader tilstrækkeligt UVB i bestrålingsspektret til at excitere kemikalier, der typisk absorberer i UVB-området, som f.eks. Amiodaron (se tabel 1).
Figur 2
Balb/c 3T3-cellers bestrålingsfølsomhed (målt i UVA-området)
Cellelevedygtighed ( % Neutral Red-optagelse i mørkekontroller)

(se punkt 1.4.1.5.2, andet afsnit, samt punkt 1.4.2.2.1 og 1.4.2.2.2)
Balb/c 3T3-cellers følsomhed for bestråling med solsimulatoren under valideringstesten af 3T3 NRU-fototoksicitetstesten målt i UVA-området. Figuren viser de resultater, man kom frem til i syv forskellige laboratorier forud for valideringsundersøgelsen (1). De to kurver med åbne symboler repræsenterer gamle celler (stort antal cellegenerationer), der måtte udskiftes med nye celler, og kurverne med udfyldte symboler viser celler med acceptabel bestrålingstolerance.
På baggrund af disse data blev den højeste ikke-cytotoksiske bestrålingsdosis på 5 J/cm2 fastlagt (lodrette stiplede linje). Den vandrette stiplede linje viser den maksimale acceptable bestrålingseffekt, der omtales i punkt 1.4.2.2.
B.42. HUDSENSIBILISERING: ASSAY PÅ LOKALE LYMFEKNUDER
1. METODE
Denne forsøgsmetode er ækvivalent med OECD TG 429 (2002)
1.1. INDLEDNING
Assay på lokale lymfeknuder (LLNA) er tilstrækkeligt valideret og anerkendt til, at det berettiger, at den indføres som ny metode (1)(2)(3). Der er tale om endnu en metode til vurdering af kemiske stoffers potentiale for hudsensibilisering i dyr. I den anden metode (B.6) anvendes marsvin, specielt bør nævnes Maksimeringstest på marsvin (Guinea Pig Maximization Test — GPMT) og Buehler-testen (4).
LLNA er en alternativ metode til at udpege hudsensibiliserende kemiske stoffer og til at fastslå, om et kemisk stof er uden nævneværdigt hudsensibiliserende potentiale. Dette indebærer ikke, at LLNA i alle tilfælde bør benyttes i stedet for marsvinetesten, men snarere, at dette assay er lige så nyttigt og kan anvendes som et alternativ, for hvilket det gælder, at positive og negative resultater sædvanligvis ikke kræver yderligere bekræftelse.
LLNA har visse fordele, både i videnskabelig henseende og hvad angår dyrs velfærd. Metoden består i undersøgelse af induktionsfasen i hudsensibilisering og giver kvantitative data, som er velegnede til at vurdere dosis/respons-forholdet, En validering af LLNA og en oversigt over det tilknyttede arbejde findes i litteraturen (5)(6)(7)(8). Det skal endvidere bemærkes, at de let til moderat sensibiliserende stoffer, der anbefales som velegnede positive kontrolstoffer ved marsvineforsøg, også er egnede til brug i forbindelse med LLNA (6)(8)(9).
LLNA er en in vivo-metode og eliminerer således ikke brugen af dyr til vurdering af kontaktsensibiliserende aktivitet. Den giver imidlertid mulighed for at mindske det nødvendige antal dyr til dette formål. Desuden repræsenterer LLNA en væsentligt mere sofistikeret måde at anvende dyr til kontaktsensibiliseringstest på. LLNA er baseret på immunologiske hændelser, som udløses af kemiske stoffer i induktionsfasen af sensibiliseringen. I modsætning til marsvinetest kræver LLNA ikke, at der fremkaldes challenge-inducerede dermale hypersensivitetsreaktioner. Desuden kræver LLNA ikke anvendelse af et adjuvans, således som det er tilfældet for Maksimeringstesten på marsvin. LLNA mindsker således dyrenes lidelser. Trods LLNA's fordele frem for traditionelle marsvinetest må man have for øje, at metoden har visse begrænsninger, som kan gøre traditionelle marsvinetest nødvendige (således forekommer falske negative fund i LLNA for visse metaller og falske positive fund for visse hudirriterende stoffer) (10).
Der henvises desuden til indledningens del B.
1.2. METODENS PRINCIP
Det tilgrundliggende princip for LLNA består i, at det sensibiliserende agens inducerer en primær lymfocytproliferation i den lymfeknude, som dræner påføringsstedet for det kemiske stof. Denne proliferation er proportional med den påførte dosis (og med allergenets styrke) og gør det let at få et objektivt, kvantitativt mål for sensibiliseringen. I LLXA-testen vurderes denne proliferation som en dosis-respons-forhold, hvor proliferationen i forsøgsgrupperne sammenholdes med proliferationen i en kontrolgruppe, der er behandlet med vehiklet. Forholdet mellem proliferation i behandlingsgrupperne og proliferationen i kontrolgruppen kaldes stimulationsindekset og skal være mindst tre, før et prøvestof kan vurderes nærmere som et potentielt hudsensibiliserende agens. De her beskrevne metoder bygger på anvendelse af radioaktiv mærkning til måling af celleproliferationen. Andre endepunkter til bedømmelse af proliferationen kan imidlertid anvendes, forudsat at dette begrundes og dokumenteres videnskabeligt forsvarligt med fuldstændige litteraturhenvisninger og beskrivelse af metoden.
1.3. BESKRIVELSE AF TESTMETODEN
1.3.1. Forberedelser
1.3.1.1. Miljøbetingelser og fodring
Dyrene holdes i hver sit bur. Temperaturen i forsøgsdyrenes rum skal være 22 oC (± 3 oC). Den relative luftfugtighed skal være mindst 30 % og bør ikke være over 70 % bortset fra under rengøring af rummet, men 50-60 % bør tilstræbes. Belysningen skal være kunstig bestående af 12 timers lys og 12 timers mørke. Som foder kan anvendes sædvanligt laboratoriefoder og fri adgang til drikkevand.
1.3.1.2. Forberedelse af dyrene
Dyrene udtages på tilfældig måde, mærkes, så hvert dyr kan identificeres (dog ikke ved nogen form for mærkning af øret) og holdes i deres bure i mindst fem døgn forud for doseringen, for at de kan akklimatisere sig til forsøgsbetingelserne. Før behandlingen påbegyndes, undersøges alle dyr for at sikre, at de er uden observerbare hudlæsioner.
1.3.2. Forsøgsbetingelser
1.3.2.1. Forsøgsdyr
Mus må foretrækkes til denne test, Der anvendes unge voksne hunmus af stamme CBA/Ca eller CBA/J, som ikke har født og ikke er drægtige. Ved forsøgets begyndelse skal dyrene være 8-12 uger gamle, og vægtvariationen mellem de anvendte dyr være mindst mulig og må ikke overstige 20 % af gennemsnitsvægten. Andre stammer og handyr kan anvendes, når der er genereret tilstrækkelige data til at dokumentere, at der ikke er væsentlige stamme- og/eller kønsspecifikke forskelle i LLNA-respons.
1.3.2.2. Pålidelighedskontrol
I forsøget anvendes positive kontroller til at godtgøre, at assayets præstationer og laboratoriets kompetence er tilstrækkelige til at assayet bliver vellykket. Den positive kontrol skal fremkalde en positiv LLNA-respons ved det eksponeringsniveau, som forventes at give en stigning i stimulationsindekset (SI) som er mere end > 3 større end i den negative kontrolgruppe. Dosis til den positive kontrol skal vælges således, at der er tydelig, men ikke overdreven induktion, De foretrukne stoffer hertil er hexylkanelaldehyd (CAS nr. 101-86-0, Einecs nr. 202-983-3) og mercaptobenzothiazol (CAS nr. 149-30-4, Einecs nr. 205-736-8). Der kan forekomme omstændigheder, hvor man kan anvende andre kontrolstoffer, som opfylder ovenstående kriterier, forudsat at dette er velbegrundet. Sædvanligvis vil en positiv kontrolgruppe i hvert assay være nødvendig, men undertiden råder prøvningslaboratorierne over historiske positive kontroldata, som viser ensartet tilfredsstillende respons gennem en seks måneders periode eller længere. Under sådanne omstændigheder kan mindre hyppig testning med positive kontroller være på sin plads med intervaller på højst seks måneder. Skønt det positive kontrolstof bør afprøves i et vehikel, som vides at fremkalde ensartet respons (f.eks. acetone, olivenolie), kan det i visse godkendelsessituationer desuden være nødvendigt med testning i andre vehikler end standardvehiklet (med en klinisk-kemisk relevant formulering). I sådanne tilfælde bør en mulig interaktion mellem et positivt kontrolstof og det pågældende ukonventionelle vehikel undersøges.
1.3.2.3. Antal dyr, dosisniveauer og valg af vehikel
Der anvendes mindst fire dyr i hver dosisgruppe, mindst tre forskellige koncentrationer af prøvestoffet, en negativ kontrolgruppe, som udelukkende behandles med det til prøvestoffet anvendte vehikel, samt, når det er hensigtsmæssigt, en positiv kontrolgruppe. Når data skal indsamles fra de enkelte dyr, anvendes mindst fem dyr i hver dosisgruppe. Bortset fra behandlingen med prøvestof behandles dyrene i kontrolgrupperne på samme måde som dyrene i behandlingsgrupperne.
Dosering og valg af vehikel baseres på anbefalingerne i henvisning (1). Dosisniveauerne vælges af koncentrationsserier på 100 %, 50 %, 25 %, 10 %, 5 %, 2,5 %, 1 %, 0,5 % osv. Foreliggende data vedrørende akut toksicitet og hudirritation tages i betragtning, således at der vælges tre på hinanden følgende koncentrationer, hvoraf den højeste er den. der giver størst mulig eksponering uden systemisk toksicitet eller for stærk hudirritation (2)(11).
Vehiklet vælges således, at testkoncentrationer og opløselighed maksimeres, samtidig med al den fremstillede opløsning/suspension er egnet til påføring af prøvestoffet. De anbefalede vehikler er i prioriteret rækkefølge acetone/olivenolie (4:1 v/v), dimethylformamid. methylethylketon, propylenglycol og dimethylsulphoxid (2)(10), men andre kan anvendes, hvis der fremlægges et fyldestgørende videnskabeligt rationale derfor. I visse situationer kan det som yderligere kontrol være nødvendigt med et klinisk relevant opløsningsmiddel eller den formulering, hvori prøvestoffet markedsføres. Man må specielt være opmærksom på, at vehikelsystemet bør indeholde hydrofile stoffer, som befugter huden og ikke straks løber af. Vehikler kun bestående af vand bør således undgås.
1.3.3. Forsøgets udførelse
1.3.3.1. Forsøgsplan
Forsøgsplanen for assayet er følgende:
|
|
Dag 1 Hvert dyrs vægt bestemmes og registreres. Dorsum af hvert øre påføres åbent 25 μl af den pågældende prøvestofkoncentration, af vehiklet alene eller af det positive kontrolstof. |
|
|
Dag 2 og 3 Påføringen fra dag 1 gentages. |
|
|
Dag 4 og 5 Ingen behandling. |
|
|
Dag 6 Hvert dyrs vægt registreres. 250 μl saltvand med phosphatbuffer indeholdende 20 μCi (7,4 e + 8 Bq)3H-methylthymidin injiceres i alle testdyr og kontroldyr gennem halevenen. Alternativt injiceres 250 μL saltvand med phosphatbuffer indeholdende 2 μCi (7,4 e + 7 Bq) 125I-ioddeoxyuridin og 10-5 M fluordeoxyuridin i alle dyr gennem halevenen. Fem timer senere aflives dyrene. De drænende aurikulære lymfeknuder fra hvert øre eksciseres og pooles i saltvand med phosphatbuffer for hver forsøgsgruppe (metode med poolet behandlingsgruppe); alternativt kan hvert par lymfeknuder fra hvert enkelt dyr eksciseres og pooles i saltvand med phosphatbuffer for hvert dyr (enkeltdyrsmetode). Detaljer og diagrammer vedrørende identifikation og dissektion af lymfeknuder findes i bilag I til henvisning (10). |
1.3.3.2. Fremstilling af cellesuspensioner
En enkeltcellesuspension af lymfeknudeceller (LNC) enten fra de poolede behandlingsgrupper eller bilateralt fra de enkelte dyr fremstilles ved forsigtig mekanisk disaggregering gennem 200 μm trådnet af rustfrit stål. Lymfeknudecellerne vaskes to gange med overskud af saltvand med phosphatbuffer og fældes med 5 % trichloreddikesyre (TCA) ved 4 oC i 18 timer (2). De dannede pellets genopslæmmes enten i 1 ml TCA og overføres til scintillationsglas indeholdende 10 ml scintillatorvæske til 3H-tælling, eller overføres direkte i gamma-tælleglas til tælling af 125l.
1.3.3.3. Bestemmelse af celleproliferationen (optagen radioaktivitet)
Optagelsen af 3H-methylthymidin måles ved β-scintillationstælling som antal disintegrationer pr. minut (DPM). Optagelsen af 125l-ioddeoxyuridin måles ved 125l-tælling og angives også som DPM. Afhængigt af den valgte metode angives optagelsen som DPM/behandlingsgruppe (poolet metode) eller DPM/dyr (individuel metode).
1.3.3.4. Bemærkninger
1.3.3.4.1.
Dyrene observeres en gang dagligt for alle kliniske tegn på lokalirritation på påføringsstedet og systemisk toksicitet. Alle observationer registreres systematisk i enkeltjournaler for hvert dyr.
1.3.3.4.2.
Som angivet i 1.3.3.1 måles kropsvægten af hvert dyr ved forsøgets begyndelse og ved den planmæssige aflivning af dyrene.
1.3.4 Beregning af resultater
Resultaterne angives som stimulationsindeks (SI), Ved den poolede metode fås SI ved division af den poolede optagelse af radioaktivitet for hver behandlingsgruppe med den poolede optagelse for kontrolgruppen på vehikel: Resultatet er den gennemsnitlige SI-værdi. Ved enkeltdyrsmetoden fås Sl-værdien ved at dividere den gennemsnitlige DPM/dyr fra hver stof-testgruppe og den positive kontrolgruppe med den gennemsnitlige DPM/dyr fra opløsningsmiddel/vehikel-kontrolgruppen. Gennemsnitsværdien for de vehikelbehandlede kontroldyr er således 1.
Ved at bruge enkeltdyrsmetoden til beregning af SI får man mulighed for en statistisk analyse af data. Ved valg af statistisk analysemetode bør undersøgeren være opmærksom på mulige forskelle i varians eller andre tilknyttede problemer, som kan kræve transformation af data eller anvendelse af ikke-parametrisk statistisk analyse. En hensigtsmæssig metode til fortolkning af data er at vurdere alle individuelle data for behandlede dyr og kontroldyr på vehikel og på grundlag heraf optegne den bedst tilpassede dosis-responskurve, idet der tages hensyn til konfidensgrænserne (8)(12)(13). Undersøgeren må dog være på vagt over for mulige »vildskud« i form af responsværdier for enkeltdyr, som falder uden for gruppen og kan gøre det nødvendigt at benytte et alternativt mål for responsen (f.eks. medianværdi i stedet for gennemsnit) eller frasortering af vildskuddet.
Afgørelsen vedrørende en positiv respons omfatter et stimulationsindeks ≥ 3 foruden en gennemgang af dosis-respons og, når det er hensigtsmæssigt, statistisk signifikans (3)(6)(8)(12)(14).
Hvis det er nødvendigt at klargøre de opnåede resultater, bør forskellige egenskaber af prøvestoffet tages i betragtning, herunder om det er strukturelt beslægtet med kendte hudsensibiliserende stoffer, om det medfører kraftig hudirritation og arten af den iagttagne dosis-respons. Disse og andre overvejelser er der mere indgående redegjort for andetsteds (7).
2. DATA
Data sammenfattes i tabelform med angivelse af gennemsnitlige og individuelle DPM-værdier og stimulationsindeks for hver dosisgruppe (herunder også kontrolgruppen på vehikel).
3. RAPPORTERING
3.1. FORSØGSRAPPORT
Forsøgsrapporten skal indeholde følgende oplysninger:
|
|
Prøvestof:
|
|
|
Vehikel:
|
|
|
For forsøgsdyrene:
|
|
|
Omstændigheder ved forsøget:
|
|
|
Pålidelighedskontrol:
|
|
|
Resultater:
|
|
|
Behandling af resultater:
|
4. HENVISNINGER
|
(1) |
Kimber, I. and Basketter, D.A. (1992). The murine local lymph node assay; collaborative studies and new directions: A commentary. Food and Chemical Toxicology 30, 165-169. |
|
(2) |
Kimber, I, Derman, R.J., Scholes E.W, and Basketter, D.A. (1994). The local lymph node assay: developments and applications. Toxicology, 93, 13-31. |
|
(3) |
Kimber, I., Hilton, J., Dearman, R.J., Gerberick, G.F., Ryan, C,A., Basketter, DA. Lea, L,. House, R,V. Ladies, G.S., Loveless, S.E., Hastings, K.L. (1998). Assessment of the skin sensitisation potential of topical medicaments using the local lymph node assay: An interlaboratory exercise Journal of Toxicology and Environmental Health, 53, 563-79. |
|
(4) |
Testing Method B.6. |
|
(5) |
2 Chamberlain, M. and Basketter, D.A. (1996) The local lymph node assay: status of validation. Food and Chemical Toxicology, 34, 999-1002. |
|
(6) |
Basketter, D.A., Gerberick, G.F., Kimber. I. and Loveless, S.E (1996). The local lymph node assay- A viable alternative to currently accepted skin sensitisation tests. Food and Chemical Toxicology, 34, 985-997. |
|
(7) |
Basketter, D.A., Gerberick, G.F. and Kimber, I. (1998). Strategies for identifying false positive responses in predictive sensitisation tests. Food and Chemical Toxicology. 36, 327-33. |
|
(8) |
Van Och, F.M.M, Slob, W., De Jong, W.H., Vandebriel, R.J., Van Loveren, H. (2000). A quantitative method for assessing the sensitising potency of low molecular weight chemicals using a local lymph node assay: employement of a regression method that includes determination of uncertainty margins. Toxicology, 146, 49-59. |
|
(9) |
Dearman, R.J., Hilton, J., Evans, P., Harvey, P., Basketter, D.A. and Kimber, I. (1998). Temporal stability of local lymph node assay responses to hexyl cinnamic aldehyde. Journal of Applied Toxicology, 18, 281-4. |
|
(10) |
National Institute of Environmental Health Sciences (1999). The Murine Local Lymph Node Assay: A Test Method for Assessing the Allergic Contact Dermatitis Potential of Chemicals/Compounds: The Results of an Independent Peer Review Evaluation Coordinated by the Interagency Coordinating Committee on the Validation of Alternative Methods (ICCVAM) and the National Toxicology Program Center for the Evaluation of Alternative Toxicological Methods (NICETAM). NIH Publication No: 99-4494, Research Triangle Park, N.C. (http://iccvam.niehs.nih.gov). |
|
(11) |
Testing method B.4. |
|
(12) |
Basketter, D.A., Selbie, E., Scholes, E.W. Lees, D. Kimber, I. and Botham, P.A. (1993) Results with OECD recommended positive control sensitisers in the maximisation, Buehler and local lymph node assays. Food and Chemical Toxicology, 31, 63-67. |
|
(13) |
Basketter D.A., Lea L.J., Dickens A., Briggs D., Pate I., Dearman R.J., Kimber I. (1999). A comparison of statistical approaches to the derivation of EC3 values from local lymph node assay dose responses. J. Appl. Toxicology, 19, 261-266. |
|
(14) |
Basketter DA, Blaikie L, Derman RJ, Kimber I, Ryan CA, Gerberick GF, Harvey P, Evans P, White IR and Rycroft RTG (2000), Use of local lymph node assay for the estimation of relative contact allergenic potency. Contact Dermatitis 42 ,344-48. |
B.43. NEUROTOKSICITETSUNDERSØGELSE I GNAVERE
1. METODE
Metoden svarer til OECD TG 424 (1997).
Denne forsøgsmetode har til formål at give den nødvendige information for at kunne bekræfte eller nærmere karakterisere af den potentielle neurotoksicitet af kemiske stoffer i voksne dyr. Den kan enten kombineres med eksisterende metoder til toksicitetsundersøgelse ved gentagen indgift eller kan udføres som en separat undersøgelse. Det anbefales at konsultere OECD Guidance Document on Neurotoxicity Testing Strategies and Methods (1) med henblik på design af undersøgelser baseret på denne testmetode. Navnlig er dette vigtigt, hvis man overvejer at ændre på de observationer og testprocedurer, som anbefales til rutinemæssig brug af metoden. Det nævnte Guidance Document er udarbejdet med henblik på at lette valget af andre testprocedurer til brug under givne omstændigheder.
Vurdering af udviklingsneurotoksicitet er ikke omfattet af denne metode.
1.1. INDLEDNING
Ved vurdering af kemiske stoffers toksiske egenskaber er det vigtigt, at potentialet for neurotoksiske virkninger tages i betragtning. I metoden til systemisk toksicitetsundersøgelse med gentagen indgift indgår allerede screening for neurotoksisk potentiale. Metoden kan benyttes til at fastlægge design for en undersøgelse, der supplerer eller bekræfter de neurotoksiske virkninger, som er set i undersøgelserne for systemisk toksicitet ved gentagen indgift. Når man betragter det neurotoksiske potentiale af visse kategorier af kemiske stoffer, kan disse tænkes at ville blive vurderet mere korrekt, hvis denne metode anvendes, uden at man på forhånd har fået nogen indikation af stoffernes neurotoksiske potentiale fra forsøg med systemisk toksicitet ved gentagen indgift. Sådanne betragtninger kan f.eks. bestå i.
|
— |
observation af neurologiske symptomer eller neuropatologiske forandringer i andre toksicitetsundersøgelser end forsøg med gentagen indgift, eller |
|
— |
strukturelt slægtskab eller andre oplysninger, der kæder stofferne sammen med kendte neurotoksiske stoffer. |
Derudover kan det i andre tilfælde være hensigtsmæssig at benytte denne metode; for nærmere enkeltheder henvises til (1).
Denne metode er udviklet således, at den kan tilpasses med henblik på at bekræfte givne histopatologiske og adfærdsmæssige neurotoksiske egenskaber ved et kemisk stof foruden at karakterisere og kvantificere den neurotoksiske respons.
Tidligere blev neurotoksicitet anset for at være ensbetydende med neuropati ledsaget af neuropatologiske forandringer eller af neurologisk dysfunktion, som f.eks. kramper, lammelse eller tremor. Skønt neuropati er en vigtig manifestation af neurotoksicitet, er det nu klart, at der er mange symptomer på toksisk effekt på nervesystemet (f.eks. nedsat motorisk koordination, nedsat sansefunktion, indlærings- og hukommelsesdysfunktion), som ikke nødvendigvis afspejler sig i neuropati eller i andre typer forsøg.
Denne neurotoksicitets-forsøgsmetode er udformet med henblik på at finde væsentlige neuroadfærdsmæssige og neuropatologiske virkninger i voksne gnavere. Skønt adfærdspåvirkninger, også uden morfologiske forandringer, kan afspejle en negativ påvirkning af organismen, er det ikke alle adfærdsændringer, der er specifikke for nervesystemet. Derfor bør eventuelle observerede forandringer vurderes i sammenhæng med sideløbende histopatologiske, hæmotologiske og biokemiske data foruden data vedrørende andre former for systemisk toksicitet. I de test, der anvendes i denne metode til karakterisering og kvantificering af neurotoksisk respons, indgår specifikke histopatologiske og adfærdsmæssige metoder, som yderligere kan understøttes gennem elektrofysiologiske og/eller biokemiske undersøgelser (1)(2)(3)(4).
Neurotoksiske stoffer kan virke på en række steder i nervesystemet og ved forskellige mekanismer. Da der ikke findes nogen enkelt række test, der giver mulighed for tilbundsgående vurdering af det neurotoksiske potentiale af alle stoffer, kan det være nødvendigt at anvende andre in vivo- eller in vitro-test, som er specifikt rettet mod den observerede eller forventede form for neurotoksisk virkning.
Denne testmetode kan desuden, i forbindelse med den vejledning, der gives i OECD's Guidance Document on Neurotoxicity Testing Strategies and Methods (1), anvendes tit nærmere karakterisering eller forbedring af følsomheden af kvantificeringen af dosis-responsforholdet med henblik på at kunne opstille et bedre skøn over No Observed Adverse Effect Level (NOAEL), eller på at underbygge kendte eller formodede farer ved det kemiske stof. For eksempel kan der udformes undersøgelser til fastlæggelse og vurdering af neurotoksiske mekanismer eller supplering af de oplysninger, der allerede kan fås ved hjælp af grundlæggende neuroadfærdsmæssige og neuropatologiske observationsmetoder. Sådanne undersøgelser behøver ikke dublere de data, der fås ved brug af de i denne metode anbefalede standardmetoder, når sådanne data allerede foreligger og ikke anses for nødvendige til fortolkning af undersøgelsens resultater.
Denne neurotoksikologiske undersøgelse, anvendt alene eller i kombination, giver oplysninger, som kan:
|
— |
fastslå, om nervesystemet påvirkes permanent eller reversibelt af det testede kemiske stof |
|
— |
bidrage til karakterisering af de nervesystemforandringer, der er forbundet med udsættelse for det kemiske stof, og til forståelsen af den underliggende mekanisme |
|
— |
fastlægge dosis- og tidsrespons-forholdet med henblik på bestemmelse af et skønnet niveau, der er uden observerede sundhedsmæssigt uønskede effekter, (og som kan danne grundlag for fastsættelse af sikkerhedsniveau for det kemiske stof). |
I metoden administreres teststoffet oralt. Andre administrationsveje (f.eks. ved dermal applikation eller inhalation) kan være mere velegnede og kan kræve tilpasning af den anbefalede fremgangsmåde. Overvejelser vedrørende valg af administrationsvej bygger på stoffets profil med hensyn til eksponering af mennesker og på foreliggende toksikologiske og kinetiske oplysninger.
1.2. DEFINITIONER
Sundhedsmæssigt uønsket virkning: enhver sådan behandlingsrelateret afvigelse fra den normale variation, som forringer en organismes overlevelsesevne, reproduktionsevne eller tilpasningsevne til omgivelserne.
Dosis: den mængde teststof, som indgives. Dosis angives som vægtmængde (g, mg) teststof pr. vægtenhed forsøgsdyr (f.eks. mg/kg), eller som fast koncentration i føden (ppm).
Dosering: et generelt udtryk, som indbefatter dosis, doseringens hyppighed og anvendelsens varighed.
Neurotoksicitet: en negativ ændring i nervesystemets struktur eller funktion som følge af udsættelse for et kemisk, biologisk eller fysisk agens.
Neurotoksisk agens: ethvert kemisk, biologisk eller fysisk agens med potentiale til at fremkalde neurotoksicitet.
NOAEL: forkortelse for »no-observed-adverse-effect level«, dvs. det højeste dosisniveau, hvor der ikke observeres behandlingsrelaterede uønskede virkninger.
1.3. PRINCIP FOR TESTMETODEN
Teststoffet administreres oralt ved forskellige doser til forskellige grupper af gnavere. Normalt kræves gentagen dosering, og dosisregimet kan være 28 dages indgift, subkronisk (90 dage) eller kronisk (1 år eller længere). Procedurerne i denne testmetode kan ligeledes anvendes til et forsøg for akut neurotoksicitet. Dyrene testes, således at man kan påvise og karakterisere anomalier af adfærdsmæssig eller neurologisk art. I hver observationsperiode vurderes adfærden på en række punkter, som kan tænkes påvirket af neurotoksiske stoffer. Ved testens slutning bliver en undergruppe af dyr af hvert køn fra hver gruppe perfunderet in situ, og af hjerne, rygmarv og perifere nerver fremstilles snit, som undersøges.
Når undersøgelsen fungerer som enkeltstående undersøgelse til screening for neurotoksicitet eller til karakterisering af neurotoksiske virkninger, kan de dyr i hver gruppe, som ikke er anvendt til perfusion og efterfølgende histopatologisk undersøgelse (se tabel 1), anvendes til specifikke neuroadfærdsmæssige, neuropatolotiske, neurokemiske eller elektrofysiologiske procedurer, som kan supplere de data, man har fået ved hjælp af standardundersøgelserne i denne metode (1). Sådanne supplerende procedurer kan være særlig nyttige, når empiriske observationer eller forventede virkninger peger på neurotoksicitet af specifik art eller med et specifikt mål. Alternativt kan de tilbageværende dyr anvendes til vurderinger som f.eks. dem, der foreskrives i metoder til toksicitetsundersøgelser med gentagen indgift i gnavere.
Når procedurerne i denne forsøgsmetode kombineres med procedurerne i andre forsøgsmetoder, skal antallet af dyr være tilstrækkeligt til at opfylde kravene om observationer i begge undersøgelser.
1.4. BESKRIVELSE AF FORSØGSMETODEN
1.4.1. Valg af dyreart
Den foretrukne gnaverart er rotter, emn en anden gnaverart kan anvendes, når begrundelse herfor gives. Der anvendes unge raske dyr af sædvanligt benyttede laboratoriestammer. Hundyrene må ikke have født og må ikke være drægtige. Doseringen skal normalt begynde snarest muligt efter fravænning, fortrinsvis senest ved seks ugers alderen, og under alle omstændigheder før ni ugers alderen. Når denne undersøgelse kombineres med andre undersøgelser, kan det dog være nødvendigt at tilpasse dette alderskrav. Ved forsøgets begyndelse må vægtvariationen mellem de anvendte dyr ikke være over 20 % af gennemsnitsvægten for hvert køn. Når en kortvarig toksicitetsundersøgelse med gentagen dosering udføres som indledning til en langvarig undersøgelse, skal de anvendte dyr i de to undersøgelser være af samme stamme og oprindelse.
1.4.2. Opstaldning og fodring
Temperaturen i forsøgsdyrrummet skal være 22 oC (± 3 oC). Den relative luftfugtighed skal være mindst 30 % og ikke over 70 % bortset fra under rengøring af rummet, men 50-60 % bør tilstræbes. Belysningen skal være kunstig bestående af 12 timers lys og 12 timers mørke. Høj intermitterende støj skal holdes på et minimum. Som foder kan anvendes sædvanligt laboratoriefoder med fri adgang til drikkevand. Ved valg af foder kan der tages hensyn til nødvendigheden af at sikre passende iblanding af teststoffet, når denne administrationsmåde anvendes. Dyrene kan enten anbringes enkeltvis eller i bure med små grupper af samme køn.
1.4.3. Klargøring af dyrene
Unge raske dyr tilordnes behandlings- og kontrolgrupper på tilfældig måde. Burene arrangeres, så at eventuelle virkninger af burenes placering minimeres, Dyrene mærkes entydigt, så de enkelte dyr kan identificeres, og holdes i burene i mindst fem døgn forud for doseringen, så de kan akklimatisere sig til forsøgsbetingelserne.
1.4.4. Indgiftsvej og fremstilling af doserne
Metoden vedrører specifikt oral indgift af teststoffet. Oral indgift kan ske ved gavage, tilsætning til foder eller drikkevand, eller gennem kapsler. Der kan anvendes andre adrninistrationsveje (f.eks. dermal applikation eller inhalation), men i så fald kan det blive nødvendigt at tilpasse den anbefalede fremgangsmåde. I overvejelser vedrørende valg af administrationsvej indgår stoffets profil med hensyn til eksponering af mennesker og foreliggende toksikologiske og kinetiske oplysninger. Den valgte administrationsvej og deraf følgende ændringer af procedurerne i denne forsøgsmetode skal begrundes.
Om nødvendigt opløses eller opslæmmes teststoffet i et egnet vehikel. Det anbefales, at man som første valg vurderer muligheden af at benytte en vandig opløsning/suspension, efterfulgt af en opløsning/suspension i olie (f.eks majsolie), og derefter eventuel opløsning i andre vehikler. Vehiklets toksiske egenskaber skal være kendt. Derudover skal følgende egenskaber ved vehiklet tages i betragtning: dets effekt på teststoffets absorption, fordeling, metabolisme og retention med deraf følgende eventuel ændring af dettes toksiske egenskaber, og dets indvirkning på dyrenes føde- og vandindtagelse og ernæringstilstand.
1.5. PROCEDURER
1.5.1. Dyrenes antal og køn
Når undersøgelsen udføres som enkeltstående undersøgelse, skal der anvendes mindst 20 dyr (10 hunner og 10 hanner) i hver dosis- og kontrolgruppe til detaljeret klinisk og funktionsmæssig observation. Af disse 10 hanner og 10 hunner udvælges mindst fem hanner og fem hunner, som perfunderes in situ og gennemgår detaljeret neurohistopatologisk undersøgelse ved forsøgets afslutning. Når kun et begrænset antal dyr i en given dosisgruppe observeres for tegn på neurotoksiske virkninger, skal det overvejes at lade de pågældende dyr indgå blandt dem, der udvælges til perfusion. Når undersøgelsen udføres i kombination med en toksicitetsundersøgelse med gentagen dosering, skal antallet af dyr være tilstrækkeligt til at opfylde målene i begge undersøgelser. Det minimale antal dyr i hver gruppe ved forskellige kombinationer af undersøgelser er givet i tabel 1. Hvis der planlægges interimsaflivninger eller restitutionsgrupper til observation for toksiske virkningers reversibilitet, persistens eler forsinkede indtræden efter behandling, eller hvis supplerende observationer overvejes, skal antal dyr øges, så der er tilstrækkeligt med dyr til rådighed for observation og histopatologisk undersøgelse.
1.5.2. Behandlings- og kontrolgruppe
Sædvanligvis skal der være mindst tre dosisgrupper og en kontrolgruppe, men hvis andre data ikke giver anledning til at forvente virkning af en gentagen dosis på 1 000 mg/kg kropsvægt/dag, kan en grænsetest udføres. Foreligger der ikke egnede data, kan der udføres et dosistitreringsforsøg for at lette fastlæggelsen af de doser, der skal anvendes. Bortset fra behandling med teststof skal dyrene i kontrolgruppen håndteres på samme måde som dyrene i behandlingsgruppen. Anvendes vehikel til administration af teststoffet, skal dyrene i kontrolgruppen modtage vehiklet i den største anvendte volumen.
1.5.3. Kvalitetskontrol
Det laboratorium, som udfører undersøgelsen, skal fremlægge data, som godtgør laboratoriets egnethed til at udføre undersøgelsen og følsomheden af de anvendte metoder. Sådanne data skal dokumentere evnen til at påvise og, når det er hensigtsmæssigt, kvantificere ændringer i de forskellige effektparametre, som anbefales observeret, således autonome symptomer, sensorisk reaktionsformåen, gribestyrke af lemmerne og motorisk aktivitet. Oplysninger om kemiske stoffer, som fremkalder forskellige former for neurotoksisk respons og kan anvendes som positive kontrolstoffer, findes i henvisning (2) til (9). Historiske data kan anvendes, hvis den eksperimentelle metode i alt væsentligt er den samme. Jævnlig ajourføring af historiske data anbefales. Når forsøgslaboratoriet på væsentlige punkter ændrer udførelsen af forsøget eller procedurerne, skal der forelægges nye data, som godtgør metodernes fortsatte følsomhed.
1.5.4. Valg af dosis
Dosisniveauet vælges under hensyntagen til eventuelle foreliggende toksicitets- og kinetikdata for teststoffet og beslægtede stoffer. Det højeste dosisniveau vælges med henblik på at fremkalde neurotoksisk effekt eller tydelig systemisk toksisk effekt. Derefter vælges en aftagende række dosisniveauer, som giver mulighed for at påvise en eventuel dosisafhængig respons, og som laveste dosisniveau et niveau, hvor ingen skadelig effekt iagttages (NOAEL). I princippet skal dosisniveauerne fastsættes således, at de primære toksiske virkninger på nervesystemet kan skelnes fra virknigner knyttet til den systemiske toksicitet. I mange tilfælde vil to til tre intervaller være optimalt, og ofte må det foretrækkes at tilføje en fjerde behandlingsgruppe frem for at operere med meget store intervaller (f.eks. større end en faktor 10) mellem dosisniveauerne. Når der foreligger et rimeligt skøn over den humane eksponering, må også dette tages i betragtning.
1.5.5. Grænsetest
Når et dosisniveau på mindst 1 000 mg/kg kropsvægt/døgn med de beskrevne metoder ikke fremkalder observerbar neurotoksisk virkning, og når heller ikke foreliggende data fra strukturelt og/eller metabolisk beslægtede forbindelser giver anledning til at forvente nogen virkning, er et komplet forsøg med tre dosisniveauer ikke nødvendigvis påkrævet. Den forventede eksponering af mennesker kan betyde, at der anvendes et højere oralt dosisniveau i grænseforsøget. For andre indgiftsmåder — f.eks. inhalation eller dermal applikation — kan teststoffets fysisk-kemiske egenskaber ofte være bestemmende for det maksimale opnåelige eksponiringsniveau. Til brug for en oral akut undersøgelse bør dosis ved grænsetesten være mindst2 000 mg/kg.
1.5.6 Administration af doserne
Dyrene doseres med teststoffet dagligt syv dage om ugen i en periode af mindst 28 dage; bruges fem-dages dosisregime eller kortere eksponeringstid, skal dette begrundes, Administreres teststoffet ved gavage, skal det ske som enkeltdosis med gastrisk sonde eller et passende intubationsrør. Den maksimale væskemængde, som kan indgives ad gangen, afhænger af forsøgsdyret størrelse. Dette volumen må højst være 1 ml/100 g kropsvægt. Ved vandige opløsninger kan det dog overvejes at anvende indtil 2 ml/100 g kropsvægt. Bortset fra lokalirriterende eller ætsende stoffer, hvis effekt sædvanligvis forstærkes ved højere koncentration, skal variation af testvolumenet minimeres ved at justere koncentrationen, så volumenet er konstant ved alle dosisniveauer.
For teststoffer, som administreres i foderet eller drikkevandet, er det vigtigt at sikre, at det pågældende stof i den anvendte mængde ikke griber ind i den normale ernærings- eller vandbalance. Ved indgift af teststoffet i foderet kan stoffet enten gives ved konstant koncentration i foderet (ppm) eller ved et konstant dosisniveau i forhold til dyrets kropsvægt; den anvendte mulighed skal specificeres. Ved indgift med sonde skal dosis gives på tilsvarende tidspunkt hver dag og efter behov justeres, så dosisniveauet holdes konstant i forhold til kropsvægten. Når der som indledning til en langtidsundersøgelse udføres et korttids-toksicitetsforsøg med gentagen dosering, skal der i de to undersøgelser anvendes ensartet foder. Når det i et akut toksicitetsforsøg ikke er muligt at indgive den pågældende stofmængde som en enkelt dosis, kan den undtagelsesvis administreres delt inden for en periode af højst 24 timer.
1.6. OBSERVATION
1.6.1. Hyppighed af observationer og test
I forsøg med gentagen dosering skal observationsperioden omfatte doseringsperioden. I akutte undersøgelser skal iagttages en 14-dages efterbehandlingsperiode. For dyr i satellitgrupper, som holdes uden eksponering i en efterbehandlingsperiode, skal observationerne ligeledes omfatte denne periode.
Observationerne skal finde sted tilstrækkelig hyppigt til at maksimere sandsynligheden for at påvise eventuelle adfærdsmæssige og/eller neurologiske anomalier. Observationerne skal fortrinsvis finde sted på samme tidspunkt hver dag, idet man retter sig efter tidspunktet for den forventede maksimale effekt af indgiften. I tabel 2 vises hyppigheden af de kliniske observationer og funktionsprøver. Hvis kinetiske eller andre data fra tidligere undersøgelser viser, at der bør benyttes andre tidspunkter til observationer, test eller post-observationsperioder, skal der benyttes en alternativ plan med henblik på at skaffe flest mulige oplysninger. Eventuelle ændringer af planen skal begrundes.
1.6.1.1. Observationer af almindelig sundhedstilstand og mortalitet/morbiditet
Alle dyr observeres nøje mindst en gang dagligt med hensyn til deres almindelige helbredstilstand og to gange dagligt for morbiditet og mortalitet.
1.6.1.2. Detaljerede kliniske observationer
Der foretages detaljerede kliniske observationer af alle dyr, som er udvalgt til dette formål (se tabel 1) én gang inden første eksponering (for at sammenligne gentagne observationer af samme individ) og derefter med forskellige intervaller, alt efter undersøgelsens varighed (se tabel 2). Ved restitutionsperiodens slutning foretages detaljerede kliniske observationer af satellit-restitutionsgrupperne. Detaljerede kliniske observationer finder sted uden for opholdsburet på en »open field«. Observationerne registreres nøje med brug af bedømmelsessystemer, der omfatter kriterier eller bedømmelsesskalaer for hver af de til observationerne hørende målinger. De anvendte kriterier eller skalaer skal være tydeligt fastlagt af testlaboratoriet. Der skal drages omsorg for at sikre, at variationerne i forsøgsbetingelserne er mindst mulige (ikke systematisk behandlingsrelaterede) og at observationerne foretages af faguddannede observatører, som er uden viden om den pågældende behandling.
Det anbefales, at observationerne udføres på struktureret måde, således at veldefinerede kriterier (herunder definition af »normalområdet«) systematisk finder anvendelse på hvert dyr på hvert observationstidspunkt. »Normalområdet« skal være veldokumenteret. Alle observerede tegn skal registreres. Også størrelsesordenen af de observerede tegn registreres, mår det er muligt. De kliniske observationer skal omfatte, men er ikke begrænset til, ændringer i hud, skin, øjne, slimhinder, optræden af sekretion og ekskretion samt autonom aktivitet (f.eks. tåreflåd, pilorektion, pupilstørrelse, usædvanligt respirationsmønster og/eller mundrespiration, eventuelle symptomer vedrørende vandladning eller defækation, samt farveforandringer af urinen).
Eventuel usædvanlig respons med hensyn til kropsstilling, aktivitetsniveau (f.eks nedsat eller øget udforskning af »open field«) og koordinering af bevægelser registreres ligeledes. Ændringer i gangen (f.eks vralten, ataksi), stilling (f.eks. pukkelrygget) og reaktion på håndtering samt optræden af kloniske eller toniske bevægelser, kramper eller tremor, stereotypier (f.eks. overdreven pleje af pels, usædvanlige bevægelser af hovedet, gentagne cirkelbevægelser) eller bizar adfærd (bidning eller overdreven slikning, selvmutilering, baglæns gang, vokalisation) eller aggression protokolleres.
1.6.1.3. Funktionsprøver
I lighed med de detaljerede kliniske observationer skal funktionsprøver udføres én gang før eksponering og derefter hyppigt for alle dyr, der er udvalgt til dette formål (se tabel 1). Hyppigheden af funktionsprøverne fastsættes ligeledes i henhold til undersøgelsens varighed (se tabel 2). Ud over de i tabel 2 angivne observationsperioder foretages observation af satellit-restitutionsgrupper så tæt som muligt på tidspunktet for den afsluttende aflivning. Funktionsprøverne skal omfatte sensorisk reaktionsformåen på forskellige stimuli (f.eks auditive, visuelle og proprioceptive (5)(6)(7)), bedømmelse af lemmernes gribestyrke (8) samt bedømmelse af motorisk aktivitet (9). Den motoriske aktivitet måles med en automatiseret anordning, som kan registrere både fald og stigning i aktivitet. Anvendes et andet system, skal dette være kvantitativt, og dets følsomhed og pålidelighed skal være godtgjort. Hver anordning skal afprøves for at sikre varig pålidelighed af og overensstemmelse mellem anordningerne. Nærmere detaljer om de fremgangsmåder, der kan benyttes, er givet i de pågældende litteraturhenvisninger. Hvis der ikke er data, som peger på potentielle neurotoksiske virkninger (f.eks. vedrørende struktur-aktivitet, epidemiologiske data, andre toksikologiundersøgelser), må man overveje mere specialiserede test af sensoriske og motoriske funktioner eller indlæring og hukommelse for at undersøge disse mulige virkninger i nærmere detaljer. Nærmere oplysninger om specialiserede test og deres anvendelse er givet i (1).
Undtagelsesvis kan man i den pågældende test udelade dyr, som viser tegn på toksicitet af et omfang, som vil forstyrre funktionstesten væsentligt. Elimination af dyr fra en funktionstest skal begrundes.
1.6.2. Kropsvægt og foder-/vandindtagelse
I undersøgelser af indtil 90 dages varighed skal alle dyr vejes mindst én gang om ugen, og ligeledes mindst én gang om ugen måles foderindtagelsen (vandindtagelsen, når teststoffet administreres i vandet). I langtidsundersøgelser vejes alle dyr mindst én gang om ugen i de første 13 uger, derefter mindst én gang hver fjerde uge. Foderindtagelsen (vandindtagelsen, når teststoffet indgives i vandet) måles mindst én gang om ugen de første 13 uger, derefter ca. hver tredje måned, medmindre dyrenes sundhedstilstand eller ændringer i kropsvægt tilsiger noget andet.
1.6.3. Oftalmologi
I undersøgelser af længere varighed end 28 dage foretages oftalmologisk undersøgelse med oftalmoskop eller tilsvarende egnet instrument inden indgift af teststoffet og ved undersøgelsens slutning, helst på alle dyr, men i det mindste på dyr i højdosis- og kontrolgrupperne. Hvis der findes øjenforandringer eller de kliniske symptomer viser, at der er behov derfor, skal alle dyr undersøges. I langtidsundersøgelser skal der endvidere foretages en oftalmologisk undersøgelse ved 13 uger. Oftalmologisk undersøgelse behøver ikke udføres, når sådanne data allerede foreligger fra andre undersøgelser med tilsvarende varighed og dosisniveau.
1.6.4. Hæmatologiske og klinisk-biokemiske prøver
Når neurotoksicitetsundersøgelsen udføres i kombination med en systemisk toksicitetsundersøgelse med gentagen dosering, foretages hæmatologisk undersøgelse og klinisk-biokemiske bestemmelser som angivet i den pågældende metode til systemiske toksicitetsforsøg. Prøvetagning skal ske således, at en eventuel påvirkning af neuroadfærd bliver mindst mulig.
1.6.5. Histopatologi
Den neuropatologiske undersøgelse skal være udformet, så den supplerer og udvider de observationer, der er gjort i undersøgelsens in vivo-fase. Væv fra mindst fem dyr/køn/gruppe (se tabel 1 og næste afsnit) fikseres in situ med almindeligt anerkendte perfusions- og fiksationsteknikker (se henvisning (3), kapitel 5, og henvisning (4), kapitel 50). Alle makroskopiske forandringer protokolleres. Når undersøgelsen udføres som enkeltstående screening for neurotoksicitet eller til karakterisering af neurotoksiske virkninger, kan de resterende dyr enten bruges til specifikke neuroadfærdsmæssige (10)(11), neuropatologiske (10)(11)(12)(13), neurokemiske (10)(11)(14)(15) eller elektrofysiologiske (10)(11)(16)(17) procedurer, som kan supplere de her beskrevne procedurer og undersøgelser, eller til at øge det antal dyr, som undersøges histopatologisk. Sådanne supplerende procedurer kan være særligt nyttige, når empiriske observationer eller forventede virkninger peger på neurotoksicitet af specifik art eller med specifikt målorgan (2)(3). Alternativt kan de resterende dyr også bruges til rutinemæssig patologisk bedømmelse som beskrevet i metoden til undersøgelser med gentagen dosering.
Alle paraffinindstøbte vævsprøver farves med en all-round farvemetode som hematoxylin-eosin og undersøges mikroskopisk. Hvis der observeres eller formodes at være tegn på perifer neuropati. skal plastindstøbte prøver af perifert nervevæv undersøges. Kliniske symptomer kan også give anledning til, at der vælges supplerende prøvetagningssteder eller benyttes specialfarvning. Vejledning om supplerende prøvetagningssteder kan findes i (3)(4). Passende specialfarvning til påvisning af bestemte typer patologiske forandringer kan ligeledes være nyttig (18).
Repræsentative snit af centralnervesystem og det perifere nervesystem undersøges histologisk (se henvisning (3), kapitel 5 og henvisning (4), kapitel 50). De undersøgte områder skal normalt omfatte forhjerne, den centrale del af cerebrum, herunder et snit gennem hippocampus, midterhjernen, cerebellum, pons, medulla oblongata, øjet med nervus opticus og retina, rygmarven med intumescentia cervicalis og lumbalis, de dorsale rodganglier, fibre fra de dorsale og ventrale rødder, den proksimale del af nervus sciaticus, den proksimale del af nervus tibialis (ved knæene) og lægmuskelgrenene af nervus tibialis. Der skal være både tværsnit og længdesnit af rygmarv og perifere nerver. Man skal være opmærksom på nervesystemets kar. En prøve af skeletmuskel, helst lægmusklen, skal ligeledes undersøges. Især skal man være opmærksom på celle- og fiberstruktur i de dele af centralnervesystem og det perifere nervesystem, der særligt påvirkes af neurotoksiske stoffer.
Vejledning i de neuropatologiske forandringer, der typisk resulterer af toksisk påvirkning, findes i henvisning (3)(4). Trinvis undersøgelse af vævsprøverne anbefales, således at snittene fra højdosisgrupperne først sammenlignes med dem fra kontrolgrupperne. Ses ingen neuropatologiske forandringer i prøverne fra disse grupper, er efterfølgende analyse unødvendig. Er der neuropatologiske forandringer i højdosisgruppen, skal prøver fra hvert af de potentielt berørte væv fra mellem- og lavdosisgrupperne kodes og undersøges sekventielt.
Findes der ved den kvalitative undersøgelse tegn på neuropatologiske forandringer, udføres endnu en undersøgelse af alle de dele af nervesystemet, som udviser sådanne forandringer. Snit fra alle dosisgrupper fra hver af de potentielt berørte regioner kodes og undersøges vilkårligt uden kendskab til koden. For hver forandring registreres dens frekvens og sværhed. Når alle regioner fra alle dosisgrupper er blevet bedømt, kan koden brydes og statistisk analyse foretages til bedømmelse af dosis-respons-forholdet. For hver forandring skal der gives eksempler på beskrivelse af forskellige sværhedsgrader.
De neuropatologiske fund bedømmes i sammenhæng med de adfærdsmæssige observationer og målinger samt øvrige data fra forudgående og sideløbende toksicitetsundersøgelser af teststoffet.
2. DATA
2.1. BEHANDLING AF RESULTATER
Der skal forefindes data for de enkelte dyr. Derudover sammenfattes alle data i tabelform med angivelse, for hver forsøgs- og kontrolgruppe, af antal dyr ved forsøgets begyndelse, antal dyr, som er fundet døde under forsøget eller aflivet af humanitære grunde, tidspunkt for eventuel død eller humanitær aflivning, antal dyr, som viser tegn på toksisk virkning, beskrivelse af de observerede toksicitetstegn, herunder begyndelsestidspunkt, varighed, art og sværhed af alle toksiske virkninger, antal dyr, som udviser forandringer, herunder forandringernes art og sværhed.
2.2. EVALUERING OG FORTOLKNING AF RESULTATER
Undersøgelsens fund bedømmes med hensyn til forekomst, sværhed og korrelation mellem neuroadfærdsmæssige og neuropatologiske virkninger (foruden neurokemiske og elektrofysiologiske virkninger, når supplerende undersøgelser indgår) samt eventuelle andre observerede negative virkninger. Når det er muligt, bedømmes resultaterne ved en egnet og almindeligt anerkendt statistisk metode. Valg af statistisk metode skal finde sted i forbindelse med fastlæggelsen af undersøgelsens design.
3. RAPPORTERING
3.1. FORSØGSRAPPORT
Forsøgsrapporten skal indeholde følgende oplysninger:
|
|
For teststoffet:
|
|
|
For eventuelt vehikel:
|
|
|
For forsøgsdyrene:
|
|
|
For forsøgsbetingelserne:
|
|
|
For observationer og testprocedurer:
|
|
|
Resultater:
|
|
|
Diskussion af resultaterne:
|
|
|
Konklusioner:
|
4. HENVISNINGER
|
(1) |
OECD Giudance Document on Neorutoxicity Testing Strategies and Test Methods. OECD, Paris, In Preparation. |
|
(2) |
Test Guidline for a Developmental Neurotoxicity Study, OECD Guidelines for the Testing of Chemicals. In preparation. |
|
(3) |
World Health Organization (WHO) (1986). Environmental Health Criteria document 60: Principles and Methods for the Assessment of Neurotoxicity associated with Exposure to Chemicals. |
|
(4) |
Spencer, P.S. and Schaumburg, H.H. (1980). Experimental and Clinical Neurotoxicology. Eds. Spencer, P.S. and Schaumburg, H.H. eds, Williams and Wilkins, Battimore/London. |
|
(5) |
Tupper, D.E. and Wallace, R.B. (1980), Utility of the Neurological Examination in Rats. Acta Neurobiol. Exp., 40, 999-1003. |
|
(6) |
Gad, S.C. (1982). A Neuromuscular Screen for Use in Industrial Toxicology. J. Toxicol. Environ. Health, 9, 691-704. |
|
(7) |
Moser, V.C., McDaniel. K.M. and Phillips, P.M. (1991). Rat Strain and Stock Comparisons Using a Functional Observational Battery: Baseline Values and Effects of amitraz. Toxic, Appl, Pharmacol., 108, 267-283. |
|
(8) |
8. Meyer, O.A., Tilson, H.A., Byrd, W.C. and Riley, M.T. (1979). A Method for the Routine Assessment of Fore- and Hind-limb Grip Strength of Rats and Mice. Neurobehav. Toxicol., 1, 233-236. |
|
(9) |
9. Crofton, K.M., Haward, J.L., Moser, V.C., Gill, M.W., Reirer, L.W., Tilson, H.A. and MacPhail, R.C. (1991) Interlaboratory Comparison of Motor Activity Experiments: Implication for Neurotoxicological Assessments. Neurotoxicol. Teratol., 13, 599-609. |
|
(10) |
Tilson, H.A., and Mitchell, C.L. eds. (1992). Neurotoxicology Target Organ Toxicology Series. Raven Press, New York. |
|
(11) |
Chang, L.W., ed. (1995). Principles of Neurotoxicology. Marcel Dekker, New York. |
|
(12) |
Broxup, B. (1991). Neuopathology as a screen for Neurotoxicity Assessment. J. Amer. Coll. Toxicol., 10, 689-695. |
|
(13) |
Moser, V.C., Anthony, D.C., Sette, W.F. and MacPhail, R.C. (1992). Comparison of Subchronic Neurotoxicity of 2-Hydroxyethyl Acrylate and Acrylamide in Rats. Fund. Appl.Toxicol., 18, 343-352. |
|
(14) |
O'Callaghan, J.P. (1988). Neurotypic and Gliotypic Proteins as Biochemical Markers of Neurotoxicity. Eurotoxicol. Teratol., 10, 445-452. |
|
(15) |
O'Callaghan J.P. and Miller, D.B. (1988). Acute Exposure of the Neonatal Rat to Triethyltin Results in Persistent Changes in Neurotypic and Gliotypic Proteins. J. Pharmacol. Exp. Ther., 244, 368-378. |
|
(16) |
Fox. D.A., Lowndes, H.E. and Birkamper, G.G. (1982). Electrophysiological Techniques in Neurotoxicology. In: Nervous System Toxicology. Mitchell, C.L. ed. Raven Press, New York, pp 299-335. |
|
(17) |
Johnson, B.L. (1980). Electrophysiological Methods in neurotoxicity Testing. In: Experimental and Clinical Neurotoxicology. Spencer, P.S. and Schaumburg, H.H. eds., Williams and Wilkins Co.,. Baltimore/London, pp. 726-742. |
|
(18) |
Bancroft, J.D. and Steven A. (1990). Theory and Pratice of Histological Techniques. Chapter 17, Neuropathological Techniques. Lowe, James and Cox, Gordon eds. Churchill Livingstone. |
Tabel 1
Mindste antal dyr pr. gruppe, når neuroloksicitetsundersøgelsen udføres separat eller i kombination med andre undersøgelser
|
|
Neurotoksicitetsundersøgelse udført som: |
|||
|
Separat undersøgelse |
Kombineret med 28-dages forsøg |
Kombineret med 90-dages forsøg |
Kombineret med det kroniske toksicitetsforsøg |
|
|
Samlet antal dyr pr. gruppe |
10 hanner og 10 hunner |
10 hanner og 10 hunner |
15 hanner og 15 hunner |
25 hanner og 25 hunner |
|
Antal dyr udvalgt til funktionstestning, herunder detaljerede kliniske observationer |
10 hanner og 10 hunner |
10 hanner og 10 hunner |
10 hanner og 10 hunner |
10 hanner og 10 hunner |
|
Antal dyr udvalgt til perfusion in situ og neurohistopatologisk undersøgelse |
5 hanner og 5 hunner |
5 hanner og 5 hunner |
5 hanner og 5 hunner |
5 hanner og 5 hunner |
|
Antal dyr udvalgt til observation af toksicitet ved gentagen dosering/subkronisk/kronisk forsøg, hæmatologisk undersøgelse, klinisk-biokemisk undersøgelse, histopatologisk undersøgelse osv. som angivet i de respektive Guidelines |
|
5 hanner og 5 hunner |
||
|
Eventuelle supplerende observationer |
5 hanner og 5 hunner |
|
|
|
Tabel 2
Hyppighed af kliniske observationer og funktionstest
|
Type observationer |
Undersøgelsens varighed |
||||||||||||||||||||||||||
|
Akut |
28-dages |
90-dages |
Kronisk |
||||||||||||||||||||||||
|
I alle dyr |
Generel sundhedstilstand |
dagligt |
dagligt |
dagligt |
dagligt |
||||||||||||||||||||||
|
Mortalitet/morbiditet |
To gange dagligt |
To gange dagligt |
To gange dagligt |
To gange dagligt |
|||||||||||||||||||||||
|
I dyr, som er udvalgt til observation af funktioner |
Detaljerede kliniske observationer |
|
|
|
|
||||||||||||||||||||||
|
Funktionsprøver |
|
|
|
|
|||||||||||||||||||||||
B.44. HUDABSORPTION: IN VIVO-METODE
1. METODE
Denne testmetode er ækvivalent med OECD TG 427 (2004).
1.1. INDLEDNING
Skønt det for mange kemiske stoffers vedkommende hovedsagelig er gennem huden, at eksponering forekommer, anvendes der oral indgift i de fleste toksikologiske undersøgelser i forsøgsdyr. I denne guideline beskrives en in vivo-undersøgelsesmetode for perkutan absorption, der danner det bindeled, man behøver for at ekstrapolere fra orale undersøgelser og foretage en sikkerhedsvurdering efter eksponering af huden.
For at få adgang til kredsløbet må et stof trænge gennem mange cellelag i huden. For de fleste stoffers vedkommende er det hastighedsbestemmende lag stratum corneum, der består af døde celler. Hudens permeabilitet afhænger dels af det kemiske stofs lipofile egenskaber, dels af tykkelsen af det yderste lag af epidermis, og dels faktorer som stoffets molekylvægt og koncentration. Hud fra rotter og kaniner er sædvanligvis mere permeabel end hud fra mennesker, mens permeabiliteten af hud fra marsvin og aber bedre svarer til menneskets.
Metoder til måling af perkutan absorption kan inddeles i to kategorier: in vivo og in vitro. In vivo-metoderne er gode til at give oplysninger om hudabsorptionen i forskellige laboratoriedyrearter. I den senere tid er der udviklet in vitro-metoder. Med dem undersøges transporten gennem dyre- eller menneskehud i hel eller partiel tykkelse til et væskereservoir. In vitro-metoden er beskrevet i en særskilt testmetode (1). Med henblik på valg af den bedst egnede metode under de givne betingelser anbefales det at støtte sig til OECD's Guidance Document for the Conduct of Skin Absorption Studies (2), der nærmere beskriver in vivo- og in vitro-metodernes anvendelighed.
Med den her beskrevne in vivo-metode kan prøvestoffets penetration gennem huden til det systemiske rum bestemmes. Teknikken har fundet udbredt anvendelse i en længere årrække (3)(4)(5)(6)(7). Skønt in vitro-undersøgelser af perkutan absorption i mange tilfælde kan være hensigtsmæssige, kan der forekomme situationer, hvor kun et in vivo-forsøg giver de nødvendige oplysninger.
Fordelene ved in vivo-metoden er, at der anvendes et fysiologisk og metabolisk intakt system, at dyrearten er den samme som i mange toksicitetsundersøgelser, og at metoden kan tilpasses andre dyrearter. Ulemperne er, at der anvendes levende dyr, at der må bruges radioaktivt mærket stof for lettere at skaffe pålidelige resultater, at den tidlige absorptionsfase er vanskelig at bestemme, og at permeabiliteten af hud fra den foretrukne art (rotte) afviger fra den menneskelige huds. Hud fra dyr er sædvanligvis lettere permeabel, hvilket kan medføre overvurdering af den perkutane absorption i mennesker (6)(8)(9). Ætsende stoffer bør ikke testes i levende dyr.
1.2. DEFINITIONER
Uabsorberet dosis: den del af dosis, der vaskes af hudoverfladen efter eksponering, og den, der findes på den ikke lukkede afdækning, samt den del, der påvises at forflygtiges fra huden under eksponeringen.
Absorberet dosis (in vivo): den del af dosis, der er til stede i urin, vaskevand fra bure, fæces, udåndingsluft (hvis den måles), blod, væv (hvis dette opsamles) og resten af kadaveret efter fjernelse af huden fra påføringsstedet.
Absorberbar dosis, den del af dosis, der er til stede på eller i huden efter afvaskning.
1.3. TESTMETODENS PRINCIP
Teststoffet, helst radioaktivt mærket, påføres den glatragede hud på dyrene i en eller flere passende doseringer i form af en repræsentativ, ibrugværende formulering. Testpræparatet efterlades i kontakt med huden i et fast tidsrum under en passende afdækning (ikke tillukket, delvis tillukket eller helt tillukket), der forhindrer, at dyret indtager testpræparatet. Når eksponeringstiden er gået, fjernes afdækningen, huden renses med passende rengøringsmiddel, afdækning og rengøringsmaterialer opbevares til analyse, og der anlægges en ny afdækning. Før, under og efter eksponeringsperioden holdes dyrene i individuelle metabolismebure, og ekskreter og udåndingsluft fra de pågældende perioder opsamles til analyse. Opsamling af udåndingsluft kan undlades, når der foreligger fyldestgørende oplysninger om, at der slet ikke eller kun i ringe omfang dannes radioaktive metabolitter. Normalt er der i hver undersøgelse flere grupper dyr, der eksponeres for testpræparatet. Én gruppe aflives ved udløb af eksponeringsperioden. De øvrige grupper aflives efterfølgende efter en fast tidsplan (2). Ved udløb af prøvetagningsperioden aflives de resterende dyr, blod opsamles til analyse, påføringsstedet fjernes til analyse, og kadaveret analyseres for eventuelt ikke udskilt stof. Prøvernes indhold bestemmes med egnede metoder, og graden af perkutan absorption beregnes (6)(8)(9).
1.4. BESKRIVELSE AF METODEN
1.4.1. Valg af dyreart
Den mest anvendte art er rotten, men der kan også anvendes ubehårede stammer eller arter med en hudabsorptionsrate bedre svarende til menneskets (3)(6)(7)(8)(9). Der benyttes unge voksne, raske dyr af samme køn (handyr er standardvalget) af sædvanligt anvendte laboratoriestammer. Ved testens begyndelse må intet dyr afvige mere end ± 20 % fra gennemsnitsvægten. Som eksempel på egnede dyr kan nævnes hanrotter på 200 g–250 g, navnlig i øverste halvdel af dette område.
1.4.2. Dyrenes antal og køn
Der skal være en gruppe på mindst fire dyr af samme køn for hvert testpræparat og hvert planlagt sluttidspunkt. Hver gruppe dyr aflives efter forskellige tidsrum, f.eks. ved afslutningen af eksponeringsperioden (typisk 6 eller 24 timer) og efterfølgende (f.eks. 48 og 72 timer). Er der påvist væsentlig forskel i hudtoksicitet mellem hanner og hunner, vælges det følsomste køn. I modsat fald vælges enten det ene eller det andet køn.
1.4.3. Miljøbetingelser og fodring
Temperaturen i forsøgslokalet skal være 22 oC (± 3 oC). Den relative luftfugtighed skal være mindst 30 % og helst ikke over 70 % bortset fra under rengøring af lokalet, men 50-60 % skal tilsigtes. Belysningen skal være kunstig og bestå af 12 timers lys og 12 timers mørke. Der anvendes sædvanligt laboratoriefoder og fri adgang til drikkevand. Under forsøget, og helst også under akklimatiseringen, skal dyrene være anbragt enkeltvis i metabolismebure. Risikoen for spild af foder og vand skal nedsættes til det mindst mulige, da man ellers vil få upålidelige resultater.
1.4.4. Forberedelse af dyrene
Dyrene mærkes, så de kan identificeres enkeltvis, og holdes i burene i mindst fem døgn forud for forsøgets begyndelse, for at de kan akklimatisere sig til laboratorieomgivelserne.
Efter akklimatiseringsperioden og ca. 24 timer inden dosering glatrages et hudområde omkring skuldre og ryg på hvert dyr. Dette skal ske forsigtigt, så skrabning af huden undgås, da beskadiget hud har andre permeabilitetsegenskaber end intakt hud. Efter glatragning og ca. 24 timer før påføring af prøvestoffet på huden (se punkt 1.4.7) aftørres huden med acetone for at fjerne talg. Supplerende vask med sæbe og vand frarådes, da eventuelle sæberester kan fremme absorptionen af teststoffet. Arealet skal have en størrelse, der muliggør en pålidelig beregning af den absorberede mængde teststof pr. cm2 hud, helst mindst 10 cm2. Denne arealstørrelse er praktisk mulig på rotter med kropsvægt 200-250 g. Efter forberedelse sættes dyrene tilbage i metabolismeburene.
1.4.5. Teststof
Teststoffet er den enhed, hvis penetrationsegenskaber skal undersøges. Ideelt bør teststoffet være radioaktivt mærket.
1.4.6. Testpræparat
Testpræparatet (f.eks. ufortyndet, fortyndet eller formuleret materiale, der indeholder teststoffet og påføres huden) skal være det samme (eller en realistisk erstatning herfor) som det, mennesker eller andre potentielle målarter kan blive eksponeret for. Enhver afvigelse fra det »ibrugværende« præparat skal begrundes. Om nødvendigt opløses eller opslæmmes teststoffet i et egnet bærestof. For andre bærestoffer end vand skal dets absorptionsegenskaber og potentielle vekselvirkning med teststoffet være kendt.
1.4.7. Påføring på huden
Et påføringssted med et nærmere bestemt overfladeareal afgrænses på hudoverfladen. En kendt mængde testpræparat fordeles derefter jævnt på stedet. Sædvanligvis anvendes en mængde svarende til den potentielle eksponering af mennesker, typisk 1-5 mg/cm2 for faste stoffer og indtil 10 μl/cm2 for væsker. Eventuelle andre mængder skal begrundes i de forventede anvendelsesbetingelser, forsøgets målsætning eller testpræparatets fysiske egenskaber. Efter påføring beskyttes det behandlede område mod vartning. Figur 1 viser et eksempel på en typisk anordning. Sædvanligvis tildækkes påføringsstedet med en ikke lukket afdækning (f.eks. permeabel nylongaze). Ved påføring af ubegrænsede doser bør påføringsstedet dog være tillukket. Når genfindingen af et halvflygtigt på grund af fordampning mindskes uacceptabelt (se også punkt 1.4.10, første afsnit), skal det fordampede stof opfanges i et aktivt kulfilter, der dækker påføringsanordningen (se figur 1). Det er vigtigt at eventuelle sådanne anordninger ikke beskadiger huden, absorberer testpræparatet eller reagerer med dette. Dyrene sættes tilbage i hvert sit metabolismebur med henblik på opsamling af ekskreter.
1.4.8. Varighed af eksponering og prøvetagning
Eksponeringens varighed er tidsrummet mellem påføring af testpræparatet og fjernelse af dette ved afvaskning af huden. Der vælges en eksponeringsperiode (typisk 6 eller 24 timer), som er relevant i forhold til forventede eksponeringsvarighed for mennesker. Efter eksponeringsperioden holdes dyrene i metabolismeburene indtil den planmæssige afslutning. Dyrene observeres for tegn på toksicitet/unormale reaktioner med regelmæssige mellemrum under hele forsøget. Ved afslutningen af eksponeringsperioden observeres den behandlede hud for synlige tegn på irritation.
Metabolismeburene skal muliggøre separat opsamling af urin og fæces under hele undersøgelsen. Desuden skal de muliggøre opsamling af 14C-carbondioxid og flygtige 14C-carbonforbindelser, som analyseres hvis de dannes i en mængde af over 5 %. Urin, fæces og udskilte væsker (f.eks. 14C-carbondioxid og flygtige C-forbindelser) opsamles individuelt fra hver gruppe på hvert prøvetagningstidspunkt. Åbne bure kan anvendes, hvis det er tilstrækkeligt dokumenteret, at der kun i ringe omfang eller slet ikke dannes radioaktive metabolitter.
Ekskreter opsamles i eksponeringsperioden indtil 24 timer efter den indledende hudkontakt og derefter dagligt til forsøgets slutning. Normalt vil tre opsamlingstidspunkter for ekskreter være tilstrækkeligt, men testpræparatets påtænkte formål eller foreliggende kinetiske data kan tilsige, at andre tidspunkter vil være mere hensigtsmæssige, eller at de må suppleres.
Ved eksponeringsperiodens slutning aftages beskyttelsesanordningen fra hvert dyr og opbevares separat med henblik på analyse. Den behandlede hud fra alle dyr vaskes mindst tre gange med rengøringsmiddel og passende servietter. Der skal vises omhu for at undgå kontaminering af andre dele af kroppen. Rengøringsmidlet skal være repræsentativt for sædvanlig hygiejnepraksis, f.eks. sæbevand. Endelig skal huden tørres. Alle servietter og alt vaskevand opbevares med henblik på analyse. På dyr i grupperne svarende til efterfølgende tidspunkter anlægges en frisk afdækning til at beskytte det behandlede sted, hvorefter dyrene sættes tilbage i hvert sit bur.
1.4.9. Afsluttende procedurer
For hver gruppe aflives de enkelte dyr på det fastsatte tidspunkt, og blod opsamles til analyse. Beskyttelsesanordning eller afdækning fjernes med henblik på analyse. Huden fra påføringsstedet og et tilsvarende areal af ikke-doseret, glatraget hud fjernes fra hvert dyr til separat analyse. For at skaffe flere oplysninger om teststoffets fordeling kan påføringsstedet spaltes, så stratum corneum adskilles fra det underliggende epidermis. Fastlæggelse af denne fordelings tidsmæssige forløb efter eksponeringsperioden vil give et fingerpeg om teststoffets skæbne i stratum corneum. For at lette spaltningen af huden (efter den endelige hudvask og aflivning af dyret) fjernes hver afdækning. Huden fra påføringsstedet eksciseres fra rotten sammen med det ringformede parti af omgivende hud og hæftes op på en plade. Et stykke tape lægges på hudoverfladen med et let tryk, og tapen fjernes sammen med en del af stratum corneum. Der fortsættes med flere tapestykker, indtil tapen ikke længere hænger fast i hudoverfladen, når hele stratum corneum er fjernet. Alle tapestykker fra hvert dyr kan lægges i en enkelt beholder, som derefter tilsættes et vævsopløsende middel for at opløse stratum corneum. Alle potentielle målvæv kan fjernes med henblik på separat bestemmelse, før resten af kadaveret analyseres for absorberet dosis. Kadaveret af hvert enkelt dyr opbevares til analyse. Sædvanligvis vil analyse af det totale indhold være tilstrækkelig. Målorganer kan udtages til separat analyse (hvis det tilsiges af andre undersøgelser). Blærens indhold af urin på det planmæssige aflivningstidspunkt hældes sammen med den foregående urinopsamling. Efter at ekskreterne fra metabolismeburene på det planmæssige aflivningstidspunkt er opsamlet, vaskes burene og deres opsamlingsanordning med et passende opløsningsmiddel. Andet potentielt kontamineret udstyr analyseres ligeledes.
1.4.10. Analyse
I alle undersøgelser skal opnås tilstrækkelig genfinding (dvs. gennemsnitligt 100 ± 10 % af radioaktiviteten). Er genfindingen uden for dette område, skal det begrundes. Den tilførte mængde dosis i hver prøve bestemmes med behørigt validerede metoder.
I de statistiske overvejelser skal indgå et mål for variansen mellem gentagelserne for hver påføring.
2. DATA
Følgende målinger foretages for hvert dyr på hvert prøvetagningstidspunkt af teststoffet og/eller dets metabolitter. Foruden individuelle data angives gennemsnitsværdier for hvert prøvetagningstidspunkt.
|
— |
den mængde, der er knyttet til beskyttelsesanordningerne |
|
— |
den mængde der kan fjernes fra huden |
|
— |
den mængde i eller på huden, der ikke kan vaskes af huden |
|
— |
mængde i de udtagne blodprøver |
|
— |
mængde i ekskreter og udåndingsluft (hvis relevant) |
|
— |
resterende mængde i kadaveret og eventuelle organer, der er udtaget til separat analyse. |
Ud fra mængden af teststof og/eller metabolitter i ekskreter, udåndingsluft, blod og kadaver kan den totale absorberede mængde på hvert tidspunkt bestemmes. Desuden kan man beregne mængde teststof absorberet pr. cm2 hud, der er eksponeret for teststoffet i eksponeringsperioden.
3. RAPPORTERING
3.1. TESTRAPPORT
Testrapporten skal dække de krav, der er fastsat i protokollen, herunder en begrundelse for det anvendte testsystem, og skal omfatte følgende:
|
|
Teststof:
|
|
|
Testpræparat:
|
|
|
Forsøgsdyr:
|
|
|
Forsøgsbetingelser:
|
|
|
Resultater:
|
|
|
Diskussion af resultater |
|
|
Konklusioner |
4. HENVISNINGER
|
(1) |
Testing Method B.45. Skin Absorption: In vitro Method. |
|
(2) |
OECD (2002). Guidance Document for the Conduct of Skin Absorption Studies. OECD, Paris. |
|
(3) |
ECETOC (1993) Percutaneous Absorption. European Centre for Ecotoxicology and Toxicology of Chemicals, Monograph No. 20. |
|
(4) |
Zendzian RP (1989) Skin Penetration Method suggested for Environmental Protection Agency Requirements. J. Am. Coll. Toxicol. 8(5), 829-835. |
|
(5) |
Kemppainen BW, Reifenrath WG (1990) Methods for skin absorption. CRC Press Boca Raton, FL, USA. |
|
(6) |
EPA (1992) Dermal Exposure Assessment: Principles and Applications. Exposure Assessment Group, Office of Health and Environmental Assessment. |
|
(7) |
EPA (1998) Health Effects Test Guidelines, OPPTS 870-7600, Dermal Penetration. Office of Prevention, Pedsticides and Toxic Substances. |
|
(8) |
Bronaugh RL, Wester RC, Bucks D, Maibach HI and Sarason R (1990) In vivo percutaneous absorption of fragrance ingredients in reshus monkeys and humans. Fd. Chem. Toxic. 28, 369-373. |
|
(9) |
Feldman RJ and Maibach HI (1970) Absorption of some organic compounds through the skin in man. J. Invest Dermatol. 54, 399-404. |
Figur 1
Eksempel på typisk anordning til afgrænsning og beskyttelse af påføringsstedet på huden ved in vivo-undersøgelser af perkutan absorption

B.45. HUDABSORPTION: IN VITRO-METODE
1. METODE
Denne testmetode er ækvivalent med OECD TG 428 (2004).
1.1. INDLEDNING
Metodens formål er undersøgelse af teststoffers absorption ved påføring af disse på eksciseret hud. Den kan enten kombineres med metoden til hudabsorption in vivo (1) eller anvendes separat. Vedrørende udformningen af undersøgelser baseret på denne testmetode anbefales det at konsultere OECD's vejledning »Guidance Document for the Conduct of Skin Absorption Studies« (2). OECD's vejledning er udarbejdet for at lette valget af egnede in vitro-metoder til anvendelse under nærmere bestemte omstændigheder og derigennem sikre pålideligheden af de resultater, der fås ved brug af metoden.
Metoder til måling af hudabsorption og afgivelse til huden kan inddeles i to kategorier: in vivo og in vitro. In vivo-metoder til hudabsorptionsundersøgelser er veletablerede og giver farmakokinetiske oplysninger i en række dyrearter. En in vivo-metode er beskrevet særskilt i en anden testmetode (1). Også in vitro-metoder er i mange år blevet anvendt til måling af hudabsorption. Skønt der ikke er udført formelle valideringsundersøgelser af de in vitro-metoder, der er omfattet af denne testmetode, var OECD-eksperter i 1999 enige om, at in vitro-metoden (3) var tilstrækkeligt underbygget med evaluerede data. I vejledningen (2) underbygges metoden yderligere, bl.a. med et betydeligt antal direkte sammenligninger af in vitro- og in vivo-metoder. Emnet er gennemgået i en række monografier, hvor der detaljeret redegøres for baggrunden for anvendelse af en in vitro-metode (4)(5)(6)(7)(8)(9)(10)(11)(12). Med in vitro-metoder måles diffusionen af kemiske stoffer ind i og tværs gennem hud til en væskebeholder, idet der enten kan bruges ikke-levende hud og således alene måles diffusion, eller frisk, metabolisk aktiv hud til måling af både diffusion og hudmetabolisme. Sådanne metoder har især fundet anvendelse til screening af kemiske stoffers indtrængen i og optagelse gennem hud fra forskellige formuleringer og kan desuden være nyttige som modeller ved vurdering af perkutan absorption i mennesker.
In vitro-metoden er ikke nødvendigvis anvendelig på alle situationer eller på alle klasser af kemiske stoffer. In vitro-metoden kan være brugbar til indledende kvalitativ evaluering af hudpenetration. I visse tilfælde kan det være nødvendigt efterfølgende at supplere med in vivo-data. Med henblik på nærmere fastlæggelse af situationer, hvor in vitro-metoden vil være egnet, henvises til vejledningen (2). Mere detaljerede oplysninger til støtte for denne afgørelse findes i (3).
I beskrivelsen af denne metode redegøres for generelle principper for måling af teststoffers absorption af og afgivelse til eksciseret hud. Der kan anvendes hud fra mange pattedyrarter, herunder mennesker. Hudens permeabilitetsegenskaber bevares efter ekscision fra kroppen, da den vigtigste diffusionsbarriere er det ikke-levende stratum corneum; der ikke er fundet aktiv transport af kemiske stoffer gennem huden. Skønt huden er påvist at kunne metabolisere visse kemiske stoffer under perkutan absorption (6), er processen er ikke hastighedsbegrænsende for den faktisk absorberede dosis, selv om den kan påvirke arten af de stoffer, der træder ind i blodet.
1.2. DEFINITIONER
Uabsorberet dosis: den del af dosis, der vaskes af hudoverfladen efter eksponering, og den, der findes på den ikke lukkede afdækning, samt den del, der påvises at forflygtiges fra huden under eksponeringen.
Absorberet dosis (in vitro): massen af teststof, der tilføres receptorvæsken eller det systemiske kredsløb inden for et nærmere fastlagt tidsrum.
Absorberbar dosis (in vitro): den del af dosis, der er til stede på eller i huden efter afvaskning.
1.3. TESTMETODENS PRINCIP
Teststoffet, som kan være radioaktivt mærket, påføres på overfladen af en hudprøve, der danner adskillelse mellem de to kamre i en diffusionscelle. Stoffet efterlades på huden i et fastlagt tidsrum under nærmere angivne betingelser, hvorefter det fjernes med en passende rensningsmetode. Der udtages prøver af receptorvæsken på tidspunkter fordelt over hele forsøget, og prøverne analyseres for teststoffet og/eller dets metabolitter.
Anvendes metabolisk aktive systemer, kan teststoffets metabolitter analyseres med egnede metoder. Ved forsøgets slutning bestemmes den kvantitative fordeling af teststoffet og dets metabolitter, når det er relevant.
Under passende betingelser, der er beskrevet i denne metode og i vejledningen (2), måles teststoffets absorption i løbet af et givet tidsrum ved analyse af receptorvæsken og den behandlede hud. Teststof, der er tilbageblevet i huden, anses for absorberet, medmindre det kan vises at absorptionen kan bestemmes alene ud fra værdier for receptorvæsken. Efter analyse af de øvrige komponenter (materiale vasket af huden og materiale tilbageholdt i hudlagene) kan der foretages en nærmere vurdering af data, herunder teststoffets totale fordeling og genfindingsprocent.
For at godtgøre testsystemets ydeevne og pålidelighed, således som det anvendes i det pågældende laboratorium, skal der for relevante referencestoffer foreligge resultater, som udviser overensstemmelse med offentliggjort litteratur om den anvendte metode. Dette krav kan tænkes opfyldt ved test af et passende referencestof (fortrinsvis med lipofile egenskaber, der ligner teststoffets) sideløbende med teststoffet eller ved fremlæggelse af fyldestgørende historiske data for en række referencestoffer med forskellige lipofile egenskaber (f.eks. koffein, benzoesyre og testosteron).
1.4. BESKRIVELSE AF TESTMETODEN
1.4.1. Diffusionscelle
En diffusionscelle består af et donorkammer og et receptorkammer, hvorimellem huden anbringes (en typisk udformning er vist i figur 1). Cellen skal slutte tæt omkring huden og skal lette prøvetagning fra og opblanding af receptoropløsningen, som er i kontakt med undersiden af huden, og skal derudover give gode muligheder for temperaturregulering af cellen og dens indhold. Både statiske diffusionsceller og gennemstrømningsceller kan godtages. Donorkammeret er sædvanligvis utillukket under eksponering for en endelig dosis af testpræparatet, men kan være tillukket ved ubegrænsede doser og ved visse scenarier med endelig dosis.
1.4.2. Receptorvæske
En receptorvæske af fysiologisk karakter må foretrækkes, men også væske af anden art kan anvendes, når dette begrundes. Receptorvæskens nøjagtige sammensætning skal angives. Det skal godtgøres, at teststoffet er tilstrækkelig opløseligt i receptorvæsken, så denne ikke virker som en barriere for absorptionen. Desuden må receptorvæsken ikke påvirke hudpræparatets integritet. I gennemstrømningssystemer må gennemstrømningshastigheden ikke være sådan, at den hæmmer teststoffets diffusion til receptorvæsken. I systemer med statisk celle skal væsken være under konstant omrøring, og der skal regelmæssigt tages prøver af væsken. Undersøges metabolismen, skal receptorvæsken kunne holde huden levende under hele forsøget.
1.4.3. Hudpræparater
Hud fra mennesker eller dyr kan anvendes. Det anerkendes, at brugen af human hud er underlagt nationale og internationale etiske hensyn og betingelser. Levende hud må foretrækkes, men også ikke-levende hud kan anvendes, forudsat at hudens integritet kan godtgøres. Såvel membraner bestående af epidermis (fraspaltet ad enzymatisk, termisk eller kemisk vej) som delhudspræparater (tykkelse typisk 200-400 μm) fremstillet med dermatom, kan godtages. Hud i fuld tykkelse kan anvendes, men for stor tykkelse (> ca. 1 mm) må undgås, medmindre det specielt er nødvendigt til bestemmelse af teststoffet i hudens forskellige lag. Den valgte art, anatomiske placering og præparationsteknik skal begrundes. Der kræves acceptable data fra mindst fire gentagelser for hvert testpræparat.
1.4.4. Hudpræparatets integritet
Korrekt præparation af huden er afgørende. Ved uhensigtsmæssig håndtering kan stratum corneum blive beskadiget, hvorfor integriteten af den præparerede hud må kontrolleres. Undersøges hudmetabolisme, skal huden anvendes snarest muligt efter excisionen og ved betingelser, der vides at støtte den metaboliske aktivitet. Som hovedregel skal nyeksciseret hud anvendes inden for 24 timer, men den acceptable opbevaringsperiode kan afhænge af det enzymsystem, der deltager i metaboliseringen, samt af opbevaringstemperaturen (13). For hudpræparater, der har været opbevaret inden brug, skal der forelægges dokumentation for, at barrierefunktionen er bevaret.
1.4.5. Teststof
Teststoffet er den enhed, hvis penetrationsegenskaber skal undersøges. Ideelt bør teststoffet være radioaktivt mærket.
1.4.6. Testpræparat
Testpræparatet (f.eks. ufortyndet, fortyndet eller formuleret materiale, der indeholder teststoffet og påføres huden) skal være det samme (eller en realistisk erstatning herfor) som det, mennesker eller andre potentielle målarter kan blive eksponeret for. Enhver afvigelse fra det »ibrugværende« præparat skal begrundes.
1.4.7. Teststoffets koncentration og formuleringer
Normalt anvendes flere koncentrationer af teststoffet, således at overgrænsen for potentiel eksponering af mennesker er inkluderet. Tilsvarende må det overvejes at teste en række typiske formuleringer.
1.4.8. Påføring på huden
Når mennesker eksponeres for kemiske stoffer, er der sædvanligvis tale om endelige doser. For at påføringen skal modsvare forholdene ved eksponering af mennesker, skal der således normalt anvendes 1-5 mg/cm2 hud for faste stoffer og indtil 10 μl/cm2 for væsker. Mængden skal være begrundet i de forventede anvendelsesbetingelser, undersøgelsens målsætning eller testpræparatets fysiske egenskaber. F.eks. kan påføringen på hudoverfladen svare til »ubegrænset«, når der anvendes et stort volumen pr. arealenhed.
1.4.9. Temperatur
Kemiske stoffers passive diffusion (og dermed deres hudabsorption) er temperaturafhængig. Diffusionskammeret og huden skal holdes ved konstant temperatur tæt på den normale hudtemperatur på 32 ± 1 oC. Forskellige cellekonstruktioner kræver forskellig temperatur af vandbad eller varmeplade for at sikre, at receptor/hud er i det fysiologiske normalområde. Fugtigheden skal helst være mellem 30 og 70 %.
1.4.10. Varighed af eksponering og prøvetagning
Eksponeringen af huden for testpræparatet kan strække sig over hele forsøgets varighed eller over kortere tid (med henblik på efterligning af en given type human eksponering). Overskydende testpræparat vaskes af huden med et passende rengøringsmiddel, og vaskevandet opsamles til analyse. Fremgangsmåden ved fjernelse af testpræparatet bør afhænge af de forventede anvendelsesbetingelser og skal begrundes. Normalt kræves en prøvetagningsperiode på 24 timer for at få en fyldestgørende karakterisering af absorptionsprofilen. Da hudens integritet kan begynde at forringes efter 24 timer, bør prøvetagningstidspunkterne normalt ikke strække sig ud over 24 timer. Så lang en periode er ikke nødvendigvis påkrævet for teststoffer, der hurtigt trænger gennem huden, men til testning af langsomt penetrerende teststoffer kan længere tid behøves. Prøvetagning fra receptorvæsken skal ske med en frekvens, der gør det muligt at optegne en kurve over teststoffets absorptionsprofil.
1.4.11. Afsluttende procedurer
Alle testsystemets komponenter analyseres, og genfindingsgraden bestemmes. Det gælder donorkammer, skyllevand til hudoverflade, hudpræparat og receptorvæske/-kammer. I nogle tilfælde kan huden opdeles i det eksponerede hudareal og hudarealet under celleflangen, samt i fraktioner bestående af stratum corneum, epidermis og dermis, som analyseres hver for sig.
1.4.12. Analyse
I alle undersøgelser skal opnås tilstrækkelig genfinding (målet bør være et gennemsnit på 100 ± 10 % af radioaktiviteten, og eventuel afvigelse herfra skal begrundes). Teststofindhold i receptorvæske, hudpræparat, vaskevand fra hudoverfladen og skyllevand fra apparaturet bestemmes med en egnet metode.
2. DATA
Der redegøres for analysen af receptorvæsken, teststoffets fordeling i testsystemet, og absorptionsprofilens sammenhæng med tiden. Når der eksponeres ved betingelser svarende til en begrænset dosis, beregnes mængde vasket af huden, mængde bundet til huden (og til de forskellige hudlag, hvis disse analyseres) samt mængde i receptorvæsken (hastighed og mængde eller procentdel af påført dosis). Hudabsorption kan undertiden udtrykkes alene med receptorvæskedata. Når der er teststof tilbage i huden ved slutningen af undersøgelsen, kan det dog være nødvendigt at medregne det i den samlede absorberede mængde (se punkt 66 i henvisning (3)). Når der eksponeres med ubegrænset dosis, kan data gøre det muligt at beregne en permeabilitetskonstant (Kp). I så fald er den absorberede procentdel ikke relevant.
3. RAPPORTERING
3.1. TESTRAPPORT
Testrapporten skal dække de krav, der er fastsat i protokollen, herunder en begrundelse for det anvendte testsystem, og skal omfatte følgende:
|
|
Teststof:
|
|
|
Testpræparat:
|
|
|
Forsøgsbetingelser:
|
|
|
Resultater:
|
|
|
Diskussion af resultaterne |
|
|
Konklusioner |
4. REFERENCER
|
(1) |
Testmetode B.44. Hudabsorption: in vivo-metode. |
|
(2) |
OECD (2002). Guidance Document for the Conduct of Skin Absorption Studies. OECD, Paris. |
|
(3) |
OECD (2000). Report of the Meeting of the OECD Extended Steering Committee for Percutaneous Absorption Testing, Annex 1 to ENV/JM/TG(2000)5. OECD, Paris. |
|
(4) |
Kemppainen B.W. and Reifenrath W.G. (1990). Methods for skin absorption. CRC Press, Boca Raton. |
|
(5) |
Bronaugh RL and Collier, SW. (1991). Protocol for In vitro Percutaneous Absorption Studies, in In vitro Percutaneous Absorption: Principles, Fundamentals and Applications, RL Bronaugh and HI Maibach, Eds., CRC Press, Boca Raton, pp. 237-241. |
|
(6) |
Bronaugh RL and Maibach HI. (1991). In vitro Percutaneous Absorption: Principles, Fundamentals and Applications. CRC Press, Boca Raton. |
|
(7) |
European Centre for Ecotoxicology and Toxicology of Chemicals (1993). Monograph No. 20, Percutaneous Absorption, ECETOC, Brussels. |
|
(8) |
Diembeck W, Beck H, Benech-Kieffer F, Courtellemont P, Dupuis J, Lovell W, Paye M, Spengler J, Steiling W (1999). Test Guidelines for In Vitro Assessment of Dermal Absorption and Percutaneous Penetration of Cosmetic Ingredients, Fd ChemTox, 37, 191-205. |
|
(9) |
Recommended Protocol for In vitro Percutaneous Absorption Rate Studies (1996). US Federal Register, Vol. 61, No. 65. |
|
(10) |
Howes D, Guy R, Hadgraft J, Heylings JR et al. (1996). Methods for assessing Percutaneous absorption. ECVAM Workshop Report ATLA 24, 81 R10. |
|
(11) |
Schaefer H and Redelmeier TE. (1996). Skin barrier: principles of percutaneous absorption. Karger, Basel. |
|
(12) |
Roberts MS and Walters KA. (1998). Dermal absorption and toxicity assessment. Marcel Dekker, New York. |
|
(13) |
Jewell, C, Heylings, J.R., Clowes, H.M. And Williams, F.M. (2000). Percutaneous absorption and metabolism of dinitrochlorobenzene in vitro. Arch Toxicol 74: 356-365. |
Figur 1
Eksempel på typisk udformning af statisk diffusionscelle til in vitro-undersøgelse af perkutan absorption
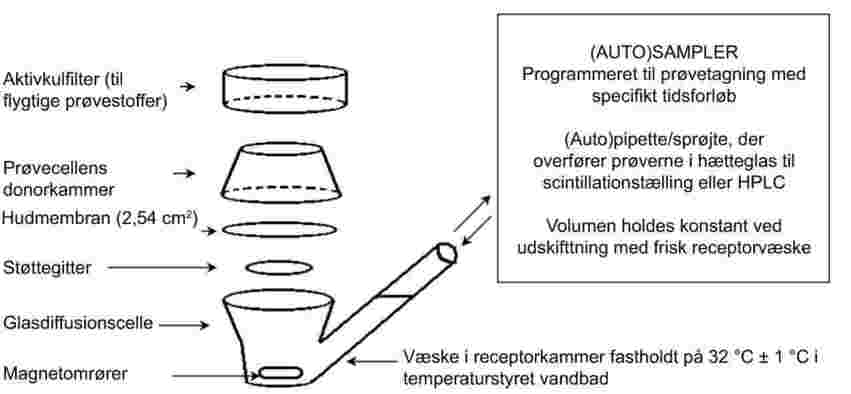
(1) Området med corneauklarhed skal angives.
(2) For en række målinger i serum og plasmaer skal dyrene helst have fastet natten over. Den vigtigste årsag hertil er, al den øgede variation i resultaterne, som manglende faste uundgåeligt medfører, vil kunne skjule de mindre tydelige effekter og gøre fortolkningen vanskelig. På den anden side kan faste indvirke på dyrenes generelle stofskifte og vil, navnlig i ernæringsundersøgelser, forstyrre den daglige eksponering for teststoffet. Hvis der benyttes faste, bør analyserne udføres efter de funktionelle observationer i forsøgets fjerde uge.
(3) Til en del analyser på serum og plasma, mest udtalt for glukose, foretrækkes fastende blodprøver. Hovedårsagen er den øgede variation, som uundgåeligt følger ikke-faste, og som vil kunne maskere små effekter og derved gøre tolkningen vanskelig. På den anden side vil faste kunne influere på dyrenes almindelige stofskifte, specielt i forsøg, hvor fødeindtagelse står centralt, og derved forstyrre den daglige belastning med testkemikaliet. Hvis blodprøver tages fastende, skal de tages efter udførelsen af de funktionelle observationer.
(4) Disse organer udtages og vejes fra ti dyr pr. køn pr. gruppe, når det drejer sig om gnavere og fra alle andre dyr end gnavere. For andre dyrearter end gnavere tages desuden skjoldbruskkirtel (med biskjoldbruskkirtel), som også vejes.
(5) Disse organer udtages og vejes fra ti dyr pr. køn pr. gruppe, når det drejer sig om gnavere.
(6) I den her beskrevne metode forstås ved referencegruppe en gruppe, som indgives teststoffet under anvendelse af en anden administreringsform, som sikrer, at hele dosis kan optages.
(7) Opløsningsmiddel anvendt til måling af absorption.
(8) Heri indgår fem dyr, som er udvalgt til funktionstestning og detaljerede kliniske observationer som led i neurotoksicitetsundersøgelsen.
DEL C: METODER TIL BESTEMMELSE AF ØKOTOKSICITET
INDHOLDSFORTEGNELSE
|
C.1. |
AKUT TOKSICITET FOR FISK |
|
C.2. |
DAPHNIA SP. AKUT IMMOBILISATIONSTEST |
|
C.3. |
VÆKSTHÆMNINGSTEST FOR ALGER |
|
C.4. |
BESTEMMELSE AF »LET« BIONEDBRYDELIGHED |
|
DEL I. |
GENERELLE BETRAGTNINGER |
|
DEL II. |
DOC-ELIMINERING (METODE C.4-A) |
|
DEL III. |
MODIFICERET OECD-SCREENINGTEST (METODE C.4-B) |
|
DEL IV. |
CO2-UDVIKLING (METODE C.4-C) |
|
DEL V. |
MANOMETRISK RESPIROMETRI (METODE C.4-D) |
|
DEL VI. |
CLOSED BOTTLE TEST (METODE C.4-E) |
|
DEL VII. |
MITI-TEST (METODE C.4-F) |
|
C.5. |
NEDBRYDNING — BIOKEMISK OXYGENFORBRUG |
|
C.6. |
NEDBRYDNING — KEMISK OXYGENFORBRUG |
|
C.7. |
NEDBRYDNING — ABIOTISK NEDBRYDNING: HYDROLYSE SOM FUNKTION AF pH |
|
C.8. |
TOKSICITET FOR REGNORME — TEST I SYNTETISK JORD |
|
C.9. |
BIOLOGISK NEDBRYDNING — ZAHN-WELLENS TEST |
|
C.10. |
BIOLOGISK NEDBRYDNING — AKTIVERET SLAM — SIMULATIONSTEST |
|
C.11. |
BIOLOGISK NEDBRYDNING — AKTIVERET SLAM — RESPIRATIONSHÆMNINGSTEST |
|
C.12. |
BIOLOGISK NEDBRYDNING — MODIFICERET SCAS-TEST |
|
C.13. |
BIOKONCENTRERING: GENNEMSTRØMNINGSTEST MED FISK |
|
C.14. |
VÆKSTTEST PÅ FISKEYNGEL |
|
C.15. |
FISK, KORTTIDSTOKSICITETSTEST PÅ FISKEEMBRYONER OG BLOMMESÆKYNGEL |
|
C.16. |
HONNINGBIER — AKUT ORAL TOKSICITETSTEST |
|
C.17. |
HONNINGBIER — AKUT KONTAKTTOKSICITETSTEST |
|
C.18. |
ADSORPTION/DESORPTION MED EN BATCH-LIGEVÆGTMETODE |
|
C.19. |
BESTEMMELSE AF ADSORPTIONSKOEFFICIENTEN (KOC ) I JORD OG I KLOAKSLAM MED HPLC (HIGH PERFORMANCE LIQUID CHROMATOGRAPHY) |
|
C.20. |
DAPHNIA MAGNA FORMERINGSTEST |
|
C.21. |
JORDMIKROORGANISMER: KVÆLSTOFOMDANNELSESTEST |
|
C.22. |
JORDMIKROORGANISMER: KULSTOFOMDANNELSESTEST |
|
C.23. |
AEROB OG ANAEROB OMDANNELSE I JORD |
|
C.24. |
AEROB OG ANAEROB OMDANNELSE I AKVATISKE SEDIMENTSYSTEMER |
C.1. AKUT TOKSICITET FOR FISK
1. METODE
1.1. INDLEDNING
Formålet med denne test er at bestemme et stofs akutte dødelige toksicitet for ferskvandsfisk. Det er ønskeligt at have flest mulige oplysninger om stoffets vandopløselighed, damptryk, kemiske stabilitet, dissociationskonstanter og bionedbrydelighed, når man skal vælge, med hvilken testmetode (statisk, semistatisk eller gennemløb) der mest hensigtsmæssigt kan opnås tilfredsstillende konstante koncentrationer af teststoffet under testens forløb.
Øvrige oplysninger (f.eks. strukturformel, renhedsgrad, art og procentdel af betydende urenheder, tilstedeværelse og mængde af tilsætningsstoffer, samt n-octanol/vand-fordelingskoefficienten) bør tages i betragtning både ved planlægningen af testen og ved fortolkningen af testresultaterne.
1.2. DEFINITIONER OG ENHEDER
Akut toksicitet er erkendbar skadelig virkning, der fremkaldes hos en organisme inden for kort tid (dage) ved eksponering for et stof. I denne test udtrykkes akut toksicitet som middel dødelig koncentration (LC50), dvs, den koncentration i vand, som dræber 50 % af fiskene i en forsøgsgruppe under kontinuerlig eksponering i en vis periode, som skal angives.
Alle koncentrationer af teststoffet er angivet i vægt pr. volumenenhed (mg/l). De kan også udtrykkes i vægtforhold (mg/kg).
1.3. REFERENCESTOFFER
Der kan anvendes et referencestof til at godtgøre, at forsøgsdyrenes følsomhed ikke har ændret sig væsentligt under forsøgsbetingelserne i laboratorier.
Der er ikke specificeret nogen referencestoffer til denne test.
1.4. TESTMETODENS PRINCIP
Der kan udføres en grænsetest med 100 mg/l til konstatering af, om LC50 er større end denne koncentration.
Fiskene udsættes for teststoffet i vand i en række koncentrationer i 96 timer. Dødeligheden registreres med højst 24 timers mellemrum, og man beregner om muligt de koncentrationer, der på hvert observationstidspunkt dræber 50 % af fiskene (LC50).
1.5. KVALITETSKRITERIER
Kvalitetskriterierne gælder for såvel grænsetesten som den komplette testmetode.
Dødeligheden i kontrolgruppen må ikke overstige 10 % (eller én fisk, hvis der benyttes mindre end ti fisk) ved testens afslutning.
Koncentrationen af opløst oxygen skal være højere end 60 % af mætning gennem hele testen.
Koncentrationen af teststoffet skal være op til 80 % af startkoncentrationen gennem hele testen.
For stoffer, der let opløses i testmediet og giver stabile opløsninger, dvs. stoffer, der ikke i væsentlig grad fordamper, nedbrydes, hydrolyseres eller adsorberes, kan startkoncentrationen antages at være lig med den nominelle koncentration. Det skal godtgøres, at koncentrationen har været opretholdt gennem hele testen, og at kvalitetskriterierne er opfyldt.
For stoffer, som
|
i) |
er tungt opløselige i testmediet eller |
|
ii) |
kan danne stabile emulsioner eller dispersioner eller |
|
iii) |
er ustabile i vandig opløsning, |
skal der som værdi for startkoncentrationen anvendes den koncentration, som er målt i opløsningen (eller hvis dette ikke er teknisk muligt, målt i vandsøjlen) ved testens begyndelse. Koncentrationen bestemmes efter, at der opnået ligevægt, men førend testfisk kommes i.
I alle disse tilfælde skal yderligere målinger foretages i løbet af testen for at sikre, at koncentrationen har været opretholdt eller kvalitetskriterierne har været opfyldt.
pH må ikke variere med mere end 1 enhed.
1.6. BESKRIVELSE AF TESTMETODEN
Der kan anvendes tre typer procedurer:
Statisk test:
Toksicitetstest, hvor der ikke foregår gennemløb af testopløsning. (Opløsningerne udskiftes ikke under testens forløb).
Semi-statisk test:
Test uden gennemløb af testopløsning, men hvor hele testopløsningen udskiftes med regelmæssige lange mellemrum (f.eks. hver 24. time).
Gennemstrømningstest:
Toksicitetstest, hvor vandet i testkamrene løbende udskiftes, og hvor teststoffet transporteres med udskiftnings-vandet.
1.6.1. Reagenser
1.6.1.1. Teststofopløsninger
Der fremstilles stamopløsninger af den påkrævede styrke ved opløsning af stoffet i deioniseret vand eller vand som beskrevet i 1.6.1.2.
De valgte testkoncentrationer fremstilles ved fortynding af stamopløsningen, Hvis der testes høje koncentrationer, kan stoffet opløses direkte i det vand, der ellers anvendes til fortynding.
Stoffer testes normalt kun op til opløselighedsgrænsen, For visse stoffer (f.eks. stoffer, som er tungt opløselige i vand, som har en høj Pow-værdi, eller som danner en stabil dispersion og ikke en ægte opløsning i vand) kan det accepteres, at der anvendes en testkoncentration, der er større end opløseligheden, så det sikres, at den højeste opløste/stabile koncentration er nået. Det er dog vigtigt, at denne koncentration ikke på anden måde forstyrrer testsystemet (f.eks. dannelse af en hinde af stoffet på vandoverfladen, så vandet ikke iltes, osv.).
Der kan tages ultralyd, organiske opløsningsmidler, emulgatorer eller dispergeringsmidler til hjælp til fremstilling af stamopløsninger af stoffer, der er tungt opløselige i vand, og til dispergering af sådanne stoffer i testmediet. Hvis der anvendes hjælpestoffer, skal alle testkoncentrationer indeholde samme mængde hjælpestof, og der anvendes en ekstra kontrolgruppe, der udsættes for samme koncentration af hjælpestoffet, som er anvendt i testserien. Sådanne hjælpestoffer anvendes i den lavest mulige koncentration, der i hvert tilfælde ikke må være større end 100 mg/l i testmediet.
Testen gennemføres uden justering af pH, Hvis der konstateres en væsentlig pH-ændring, anbefales det at gentage testen med justeret pH, og disse resultater rapporteres. I så fald justeres stamopløsningens pH til samme værdi som i det vand, der anvendes til fortynding, medmindre der er særlige grunde til at undlade dette. Til justering af pH foretrækkes HCl og NaOH. Justering af pH skal foregå på en sådan måde, at koncentrationen af teststof i stamopløsningen ikke ændres væsentligt. Hvis teststoffet reagerer kemisk eller udfældes ved justeringen, rapporteres dette.
1.6.1.2. Vand til opbevaring og fortynding
Der kan benyttes drikkevand (ikke forurenet med potentielt skadelige koncentrationer af chlor, tungmetaller eller andre stoffer), råvand af egnet kvalitet eller kunstigt fremstillet ferskvand (se tillæg 1). Der foretrækkes vand med en total hårdhed mellem 10 og 250 mg/l (som CaCO3) og med et pH mellem 6,0 og 8,5.
1.6.2. Apparatur
Alt apparatur skal være af kemisk inert materiale.
|
— |
automatisk fortyndingssystem (for gennemstrømningstest) |
|
— |
oxygenmåleudstyr |
|
— |
udstyr til bestemmelse af vandets hårdhed |
|
— |
passende apparatur til temperaturkontrol |
|
— |
pH-meter. |
1.6.3. Testfisk
Fiskene skal være i god helbredstilstand og uden synlige misdannelser.
Arterne udvælges på grundlag af praktiske kriterier såsom, om de er til rådighed hele året, hvor lette de er at holde, om de er egnede til testning, og hvor følsomme de er, samt andre økonomiske, biologiske og økologiske faktorer af betydning. Hensynet til dataenes sammenlignelighed og den allerede gennemførte internationale harmonisering (henvisning (1)) bør ligeledes tages i betragtning ved valg af fiskearterne.
Tillæg 2 indeholder en liste over de fiskearter, der anbefales til gennemførelse af denne test; zebrafisk og regnbueørred foretrækkes.
1.6.3.1. Opbevaring
Testfisk skal fortrinsvis komme fra en enkelt bestand og være af samme længde og alder. Fiskene skal holdes i mindst 12 dage under følgende betingelser:
Belastning:
Passende for systemet (recirkulation eller gennemstrømning) og fiskearten.
Vand:
Se 1.6.1.2.
Lys:
12 til 16 timers lys dagligt.
Koncentration af opløst oxygen:
Mindst 80 % mætning.
Fodring:
Dagligt eller tre gange ugentligt, sidste gang 24 timer før testen påbegyndes.
1.6.3.2. Dødelighed
Efter en 48 timers tilpasningsperiode registreres dødeligheden, og følgende kriterier anvendes;
|
— |
større end 10 % af populationen på syv dage: Hele partiet kasseres |
|
— |
mellem 5 og 10 % af populationen: Observationsperioden forlænges med yderligere syv dage. Hvis der ikke sker flere dødsfald, accepteres partiet; i modsat fald må det kasseres |
|
— |
mindre end 5 % af populationen: Partiet accepteres. |
1.6.4. Tilvænning
Alle fisk skal udsættes for vand af den kvalitet og med den temperatur, som vil blive anvendt i testen, i mindst syv dage, før de tages i anvendelse.
1.6.5. Testens udførelse
Forud for den endelige test kan der udføres en indledende test med det formål at skaffe oplysninger om, hvilke koncentrationer der skal anvendes i den endelige test.
Ud over testserien medtages en kontrolgruppe uden teststof og i givet fald en kontrolgruppe med hjælpestof.
Der vælges en statisk test, en semi-statisk test eller en gennemstrømningstest alt efter teststoffets fysiske og kemiske egenskaber, idet den bedst egnede til opfyldelse af kvalitetskriterierne vælges.
Fiskene udsættes for stoffet, som beskrevet nedenfor:
|
— |
varighed: 96 timer |
|
— |
antal dyr: mindst 7 pr, koncentration |
|
— |
kar: af passende størrelse i forhold til den anbefalede belastning |
|
— |
belastning: For statiske og semi-statiske test anbefales 1 g/l som den maksimale belastning; for gennemstrømningstest kan større belastning accepteres |
|
— |
testkoncentration: mindst fem koncentrationer med et konstant indbyrdes forhold på ikke over 2,2, som så vidt muligt dækker hele dødelighedsintervallet fra 0 til 100 % |
|
— |
vand: se 1.6.1.2 |
|
— |
lys: 12 til 16 timers lys dagligt |
|
— |
temperatur: passende for arten (tillæg 2) men inden for ± 1 oC i den enkelte test |
|
— |
koncentration af opløst oxygen: ikke mindre end 60 % af mætning ved den valgte temperatur |
|
— |
fodring: ingen. |
Fiskene inspiceres efter de første 2 til 4 timer og derefter med højst 24 timers mellemrum. Fisk betragtes som døde, hvis berøring af haleroden ikke giver nogen reaktion og der ikke er synlige respirationsbevægelser. Døde fisk fjernes, når de observeres, og dødeligheden registreres. Synlige abnormaliteter (f.eks. tab af ligevægt, ændringer i svømmeadfærd, respirationsbevægelser og pigmentering) registreres.
pH, opløst oxygen og temperatur måles dagligt.
Grænsetest
En grænsetest med 100 mg/l til konstatering af, om LC50 er større end denne koncentration kan anvendes, hvis den fremgangsmåde, som er anført i denne testmetode, benyttes,
Hvis stoffet er af en sådan art, at der ikke kan opnås en koncentration på 100 mg/l i testvandet, udføres grænsetesten ved stoffets mætningskoncentration (eller den højeste koncentration, ved hvilken der dannes en stabil dispersion) i det anvendte medium (se også 1.6.1.1).
Grænsetesten udføres med 7-10 fisk og en eller to kontrolgrupper af samme størrelse. (Ifølge binomialteorien er der ved anvendelse af 10 fisk 99,9 % sikkerhed for, at LC50 er større end den anvendte koncentration, hvis dødeligheden er nul. Med 7, 8 eller 9 fisk giver en dødelighed på nul 99 % sikkerhed for, at LC50 er større end den anvendte koncentration).
Hvis der sker dødsfald, må der gennemføres en komplet undersøgelse. Eventuelle subletale virkninger registreres.
2. DATA OG EVALUERING
For hvert af de tidspunkter, hvor der er foretaget observationer (efter 24, 48, 72 og 96 timer), afsættes den procentvise dødelighed mod koncentrationen på logaritmisk sandsynlighedspapir.
For hvert observartionstidspunkt skønnes om muligt LC50 og konfidensgrænserne (p = 0,05) ved hjælp af standardprocedurer; disse værdier afrundes til ét, eller højst to betydende cifre (eksempler på afrunding til to cifre: 173,5 til 170; 0,127 til 0,13; 1,21 til 1,2).
I de tilfælde, hvor hældningen af koncentration/responsprocentkurven er for stejl til, at LC50 kan beregnes, er et skøn ud fra kurven tilstrækkeligt.
Hvis to på hinanden følgende koncentrationer med et indbyrdes forhold på 2,2 giver en dødelighed på 0 og 100 %, er disse to værdier tilstrækkelige til angivelse af, i hvilket interval LC50 ligger.
Hvis det bemærkes, at teststoffet ikke kan holdes stabilt eller homogent, skal dette anføres, og resultaterne må fortolkes med forsigtighed.
3. RAPPORTERING
Forsøgsrapporten skal om muligt indeholde følgende oplysninger:
|
— |
oplysninger om testfisk (videnskabeligt navn, stamme, leverandør, eventuel forbehandling, størrelse samt hvor mange der er anvendt ved hver restkoncentration) |
|
— |
hvor det vand, der er anvendt til fortynding, kommer fra og dets vigtigste kemiske egenskaber (pH, hårdhed og temperatur) |
|
— |
for stoffer, der er tungtopløselige i vand, hvilken metode der er anvendt til fremstilling af stam- og testopløsning |
|
— |
koncentrationen af eventuelle hjælpestoffer |
|
— |
en liste over de anvendte koncentrationer og alle foreliggende oplysninger om testkemikaliets stabilitet i testopløsningen ved disse koncentrationer |
|
— |
metoder og resultater, hvis der er udført kemiske analyser |
|
— |
resultaterne af en eventuel grænsetest |
|
— |
begrundelse for og detaljer vedrørende den valgte testprocedure (f.eks. statisk, semi-statisk, doseringshastighed, gennemstrømningshastighed, beluftning, antal fisk, osv.) |
|
— |
beskrivelse af testudstyret |
|
— |
lysforhold |
|
— |
koncentrationen af opløst oxygen, pH og temperatur i testopløsningerne hver 24. time |
|
— |
godtgørelse af, at kvalitetskriterierne er opfyldt |
|
— |
en tabel med den kumulative dødelighed for hver koncentration og for kontrolgruppen (samt i givet fald for kontrolgruppen med hjælpestof) på alle de anbefalede observationstidspunkter |
|
— |
afbildning af koncentration/responsprocentkurve ved testens afslutning |
|
— |
om muligt LC50-værdier på alle de anbefalede observationstidspunkter (med 95 % konfidensgrænser) |
|
— |
hvilke statistiske metoder der er anvendt til bestemmelse af LC50 |
|
— |
hvis der er benyttet et referencestof, de fundne resultater |
|
— |
højeste testkoncentration, der ikke forårsager nogen dødsfald inden for testperioden |
|
— |
laveste testkoncentration, der forårsager 100 % dødelighed inden for testperioden. |
4. HENVISNINGER
|
(1) |
OECD, Paris, 1981, Test Guideline 203, Decision of the Council C(81) 30 final and updates. |
|
(2) |
AFNOR — Determination of the acute toxicity of a substance to Brachydanio rerio — Static and Flow Through methods — NFT 90-303 June 1985. |
|
(3) |
AFNOR — Determination of the acute toxidty of a substance to Salmo gairdneri — Static and Flow Through methods — NFT 90-305 June 1985. |
|
(4) |
ISO 7346/1,/2 and/3 — Water Quality — Determination of the acute lethal toxicity of substances to a fresh water fish (Brachydanio rerio Harailton-Buchanan — Teleostei, Cyprinidae). Part 1: Static method. Part 2: Semi-static method. Part 3: Flow-through method. |
|
(5) |
Eidgenössisches Department des Innern, Schweiz: Richtlinien fur Probenahme und Normung von Wasseruntersuchungsmethoden — Part II 1974. |
|
(6) |
DIN Testverfahren mit Wasserorganismen, 38 412 (11) und 1 (15). |
|
(7) |
JIS K 0102, Acute toxicity test for fish. |
|
(8) |
NEN 6506 — Water — Bepaling van de akute toxiciteit met behulp van Poecilia recticulata, 1980. |
|
(9) |
Environmental Protection Agency. Methods for the acute toxicity tests with fish, macroinvertebrates and amphibians, The Committee on Methods for Toxidty Tests with Aquatic Organisms, Ecological Research Series EPA-660-75-009, 1975. |
|
(10) |
Environmental Protection Agency, Environmental monitoring and support laboratory, Office of Research and Development, EPA-600/4-78-012, January 1978. |
|
(11) |
Environmental Protection Agency, Toxic Substance Control, Part IV, 16 March 1979. |
|
(12) |
Standard methods for the examination of water and wastewater, 14th edition, APHA-AWWA-WPCF, 1975. |
|
(13) |
Commission of the European Communities, Inter-Laboratory test programme concerning the study of the ecotoxicity of a chemical substance with respect to the fish. EEC Study D.8368, 22 March 1979. |
|
(14) |
Verfahrenvorschlag des Umweltbundesamtes zum akuten Fisch-Test. Rudolph, P, und Boje, R. Okotoxikologie, Grundlagen fur die ökotoxikologische Bewertung von Umwekchemikalien nach dem Chemikaliengesetz, ecomed 1986. |
|
(15) |
Litchfield, J.T. and Wilcoxon, F., A simplified method for evaluating dose effects experiments, J. Pharm, Exp. Therap., 1949, vol. 96, 99. |
|
(16) |
Finney, D.J., Statistical Methods in Biological Assay. Griffin, Weycombe, U.K., 1978. |
|
(17) |
Sprague, J.B. Measurement of pollutant toxicity to fish. Bioassay methods for acute toxicity. Water Res., 1969, vol. 3, 793-821. |
|
(18) |
Sprague, J.B. Measurement of pollutant toxicity to fish. II Utilising and applying bioassay results. Water Res., 1970, vol. 4, 3-32. |
|
(19) |
Stephan, C.E. Methods for calculating an LC50. In Aquatic Toxicology and Hazard Evaluation (edited by F.I. Mayer and J.L. Hamelinck). American Society for Testing and Materials. ASTM STP 634, 1977, 65-84. |
|
(20) |
Stephan, C.E., Busch, K.A., Smith, R., Burke, J. and Andrews, R.W. A computer program for calculating an LC50. US EPA. |
Tillæg 1
Kunstigt fremstillet ferskvand
Eksempel på vand egnet til fortynding
Alle kemikalier skal være af analysekvalitet.
Vandet skal være destilleret vand af god kvalitet eller deioniseret vand med en ledningsevne på mindre end 5 μScm-1.
Vanddestillationsapparatet må ikke indeholde dele af kobber.
Stamopløsninger
|
CaCl2 — 2H2O (calciumchlorid dihydrat): opløses i vand og fortyndes til 1 liter |
11,76 g |
|
MgSO4 — 7H2O (magnesiumsulfat heptahydrat): opløses i vand og fortyndes til 1 liter |
4,93 g |
|
NaHCO3 (natriumhydrogencarbonat): opløses i vand og fortyndes til 1 liter |
2,59 g |
|
KC1 (kaliumchlorid): opløses i vand og fortyndes til 1 liter |
0,23 g |
Kunstigt fremstillet ferskvand til fortynding
25 ml af hver af de fire stamopløsninger blandes, og der fortyndes til 1 liter med vand.
Opløsningen beluftes, indtil koncentrationen af opløst oxygen svarer til værdien ved mætning.
pH skal være 7,8 ± 0,2.
Om nødvendigt justeres pH med NaOH (natriumhydroxid) eller HC1 (saltsyre).
Det således fremstillede vand til fortynding henstår i ca. 12 timer og beluftes ikke yderligere.
Summen af Ca- og Mg-ioner i denne opløsning er 2,5 mmol/l. Forholdet mellem Ca- og Mg-ioner er 4:1 og mellem Na- og K-ioner 10:1. Opløsningens totale alkalinitet er 0,8 mmol/l.
Afvigelser ved fremstillingen af fortyndingsvandet må ikke medføre ændringer i vandets sammensætning og egenskaber.
Tillæg 2
Fiskearter, der anbefales til testning
|
Anbefalede aner |
Anbefalet temperaturinterval ( oC) |
Anbefalet længde af testdyr (cm) |
|
Bracbydanio rerio (Teleostei, Cyprinidae) (Hamilton-Buchanan) Zebrafisk |
20 til 24 |
3,0±0,5 |
|
Pimephales promelas (Teleostei, Cyprinidae) (Rafinesque) Fathead minnow |
20 til 24 |
5,0±2,5 |
|
Cyprinus carpio (Teleostei, Cyprinidae) (Linnaeus 1758) Karpe |
20 til 24 |
6,0±2,0 |
|
Oryzias latipes (Teleostei, Poeciliiae) Cyprinodontidae(Tomminck og Schlegel 1850) japansk risfisk |
20 til 24 |
3,0±1,0 |
|
Poecilia reticulata (Teleostei, Poeciliidae) (Peters 1859) Guppy |
20 til 24 |
3,0±1,0 |
|
Lepomis macrochirus (Teleostei, Centrarchidae) (Rafinesque Linnaeus 1758) |
20 til 24 |
5,0±2,0 |
|
Onchorhynchus mykiss (Teleostei, Salmonidae) (Walbaum 1988) Regenbueørred |
12 til 17 |
6,0±2,0 |
|
Leuciscus idus (Teleostei, Cyprinidae) (Linnaeus 1758) Guldfisk |
20 til 24 |
6,0±2,0 |
Opdrætsbetingelser
De ovenfor nævnte fisk er lette at opdrætte og/eller let tilgængelige hele året rundt. De kan opdrættes enten i dambrug eller i laboratoriet under sygdoms- og parasitfrie forhold, sådan at forsøgsdyrene er sunde og af kendt afstamning. Disse fisk er tilgængelige i store dele af verden.
Tillæg 3
Eksempel på kurve over koncentration mod dødelighed
Eksempel på bestemmelse af LC50 på log/sandsynlighedspapir

C.2. DAPHNIA SP. AKUT IMMOBILISATIONSTEST
1. METODE
Den her beskrevne akutte immobilisationstest er ækvivalent med OECD TG 202 (2004).
1.1. INDLEDNING
Metoden beskriver en akut toksicitetstest til vurdering af kemiske stoffers virkning på dafnier. Så vidt muligt er anvendt eksisterende testmetoder (1)(2)(3).
1.2. DEFINITIONER
I forbindelse med denne metode anvendes følgende definitioner:
|
|
EC50: er den koncentration, der er fundet at immobilisere 50 % af dafnierne i løbet af en nærmere angivet eksponeringsperiode. Benyttes en anden definition, skal dette angives tillige med den tilhørende henvisning. |
|
|
Immobilisering: Dyr, der ikke er i stand til at svømme inden for 15 sekunder efter forsigtig omrystning af testkarret, anses for at være immobiliseret (uanset om de stadig kan bevæge antennerne). |
1.3. TESTMETODENS PRINCIP
Unge dafnier, som ved testens begyndelse er mindre end 24 timer gamle, eksponeres for forskellige koncentrationer af prøvestoffet i et tidsrum af 48 timer. Ved 24 og 48 timer registreres immobiliseringen, og der sammenholdes med kontrolværdier. Resultaterne analyseres med henblik på beregning af EC50 ved 48h (se definitioner i punkt 1.2). Bestemmelse af EC50 ved 24h er ikke obligatorisk.
1.4. OPLYSNINGER OM PRØVESTOFFET
Prøvestoffets vandopløselighed og damptryk skal være kendt, og til kvantitativ bestemmelse af stoffet i prøveopløsningerne skal der foreligge en pålidelig analysemetode med dokumenteret genfindingsgrad og bestemmelsesgrænse. Nyttige oplysninger om stoffet er dets strukturformel, renhed, stabilitet i vand og over for lys, Pow samt resultater af en nedbrydelighedstest (se metode C.4).
Anm.: For stoffer, der er vanskelige at teste som følge af deres fysiske-kemiske egenskaber, er der vejledning i (4).
1.5. REFERENCESTOFFER
Der kan testes et referencestof for EC50 for at sikre, at forsøgsbetingelserne er pålidelige. Hertil anbefales det at benytte de giftstoffer, der anvendes i internationale ringtest (1) (1)(5). Test med referencestof bør helst finde sted hver måned og mindst to gange årligt.
1.6. KVALITETSKRITERIER
Følgende præstationskriterier tjener til at afgøre om en test er gyldig:
|
— |
i kontrolprøverne, herunder kontrollen indeholdende opløsningsmidlet, må højst 10 % af dafnierne blive immobiliseret |
|
— |
koncentrationen af opløst oxygen skal ved testens afslutning være ≥ 3 mg/l i kontrol- og testkar. |
Anm.: Med henblik på det første kriterium må højst 10 % af kontroldafnierne udvise immobilisering eller andre tegn på sygdom eller stress såsom affarvning eller usædvanlig adfærd som f.eks. fastsidden i vandoverfladen.
1.7. BESKRIVELSE AF TESTMETODEN
1.7.1. Apparatur
Testkar og andet apparatur, der kommer i berøring med prøveopløsningerne, skal være udført helt i glas eller andet kemisk inaktivt materiale. Som testkar anvendes normalt reagensglas eller bægerglas; hver gang disse anvendes, skal de forinden være rengjort med sædvanlige laboratoriemetoder. Testkarrene skal være løst tildækket for at mindske vandtab ved fordampning og for at undgå, at der kommer støv i opløsningerne. Flygtige stoffer testes i helt fyldte lukkede kar, der er store nok til at forhindre at oxygenkoncentrationen bliver en begrænsende faktor eller for lav (se punkt 1.6 og punkt 1.8.3, første afsnit).
Derudover anvendes der et eller flere af følgende udstyr: oxygenmeter (med mikroelektrode eller andet egnet udstyr til måling af opløst oxygen i prøver med lille volumen); pH-meter; egnet apparatur til temperaturregulering; udstyr til bestemmelse af total mængde organisk kulstof (TOC); udstyr til bestemmelse af kemisk iltforbrug (COD); udstyr til bestemmelse af hårdhed osv.
1.7.2. Testorganisme
Daphnia magna Straus er den foretrukne art, men andre egnede Daphnia-arter kan anvendes (f.eks. Daphnia pulex). Ved testens begyndelse skal dyrene være under 24 timer gamle, og for at mindske variabiliteten anbefales det stærkt, at man ikke anvender første kuld afkom. Dyrene skal hidrøre fra en sund bestand (dvs. fri for tegn på stress som høj dødelighed, tilstedeværelse af hanner og ephippia, forsinket frembringelse af første kuld, affarvede dyr osv.). Alle organismer, der anvendes i en given test, skal udgå fra kulturer etableret fra samme bestand af dafnier. Dyrene i bestanden skal holdes under vækstbetingelser (lys, temperatur, medium) svarende til de i testen anvendte. Skal der til testen benyttes et andet vækstmedium end de sædvanligvis anvendte vækstmedier til dafnier, er det god praksis at anvende en akklimatiseringsperiode inden testen. Denne består i, at dafnier fra kuldet opholder sig i fortyndingsvand ved testtemperaturen i mindst 48 timer, inden testen begynder.
1.7.3. Opbevarings- og fortyndingsvand
Som opbevarings- og fortyndingsvand kan accepteres råvand (overflade- eller grundvand), rekonstitueret vand eller afkloret ledningsvand, såfremt dafnier kan overleve i vandet i et tidsrum svarende til dyrknings-, akklimatiserings- og testperioden uden at vise tegn på stress. Vand er egnet som testvand, når det er i overensstemmelse med de kemiske karakteristika for fortyndingsvand i bilag 1. Vandets kvalitet skal være konstant under hele testen. Rekonstitueret vand kan fremstilles ved, at deioniseret eller destilleret vand tilsættes nærmere bestemte mængder reagenser af anerkendt analysekvalitet. Eksempler på rekonstitueret vand er givet i (1) og (6) og i bilag 2. Det skal bemærkes, at metalholdige stoffer ikke bør testes med medier indeholdende kendte chelaterende stoffer som M4- og M7-medierne i bilag 2. pH skal være mellem 6 og 9. Til Daphnia magna anbefales en hårdhed på mellem 140 og 250 mg/l (som CaCO3), men mindre hårdhed kan også være hensigtsmæssig til andre arter af Daphnia. Fortyndingsvandet kan luftes inden dets anvendelse i testen, så det er mættet med opløst oxygen.
Anvendes råvand, skal kvalitetsparametrene måles mindst to gange årligt, samt hver gang der er formodning om, at de kan have ændret sig væsentligt (se foregående afsnit og bilag 1). Måling af tungmetaller (f.eks. Cu, Pb, Zn, Hg, Cd, Ni) skal ligeledes foretages. Anvendes afkloret ledningsvand, er daglig chlorbestemmelse ønskelig. Hidrører fortyndingsvandet fra overflade- eller grundvand, skal dets ledningsevne og totale organiske kulstofindhold (TOC) eller kemiske iltforbrug (COD) bestemmes.
1.7.4. Testopløsninger
Sædvanligvis fremstilles testopløsningerne i de valgte koncentrationer ved fortynding af en stamopløsning. Stamopløsninger skal fortrinsvis fremstilles ved opløsning af prøvestoffet i fortyndingsvandet. Brug af opløsningsmidler, emulgatorer og dispergeringsmidler skal så vidt muligt undgås. I visse tilfælde kan sådanne forbindelser dog være nødvendige for at få en tilpas koncentreret stamopløsning. Vejledning om egnede opløsningsmidler, emulgatorer og dispergeringsmidler er givet i (4). I alle tilfælde må indholdet af prøvestof i testopløsningerne ikke overstige stoffets opløselighed i fortyndingsvandet.
Testen skal udføres uden justering af pH. Hvis pH ikke holder sig i området 6-9, kan der efterfølgende foretages endnu en test, hvor stamopløsningens pH er justeret til pH af fortyndingsvandet, før prøvestof tilsættes. pH-justeringen skal ske således, at stamopløsningens koncentration ikke ændres nævneværdigt, og så det ikke forårsager en kemisk reaktion eller udfældning af prøvestoffet. HCl og NaOH må foretrækkes.
1.8. FREMGANGSMÅDE
1.8.1. Eksponeringsbetingelser
1.8.1.1. Testgrupper og kontrolgrupper
Testkarrene påfyldes det korrekte rumfang fortyndingsvand og prøvestofopløsning. Luft/vand-volumenforholdet i karret skal være ens i test- og kontrolgruppen. Dafnierne overføres derefter til testkarrene. For hver testkoncentration og til kontrol skal anvendes mindst 20 dyr, helst opdelt i fire grupper på hver fem dyr. Der skal være mindst 2 ml testopløsning for hvert dyr (altså 10 ml til fem dafnier pr. testkar). Er prøvestofkoncentrationen ikke stabil, kan testen udføres i et system med halvstatisk udskiftning eller et gennemstrømningssystem.
Foruden behandlingsserien testes én kontrolserie med fortyndingsvand og én kontrolserie indeholdende et eventuelt opløsningsmiddel.
1.8.1.2. Testkoncentrationer
Der kan udføres et forberedende forsøg til fastlæggelse af koncentrationsområdet for den endelige test, medmindre oplysninger om prøvestoffets toksicitet allerede foreligger. Herunder eksponeres dafnierne for en række vidt forskellige koncentrationer af prøvestoffet. Fem dafnier eksponeres for hver testkoncentration i op til 48 timer, og flerdobbeltbestemmelse er ikke nødvendigt. Eksponeringsperioden kan afkortes (f.eks. til 24 timer eller derunder), hvis egnede data i det forberedende forsøg kan opnås på kortere tid.
Der skal anvendes mindst fem testkoncentrationer. Disse vælges som en kvotientrække med en kvotient på helst ikke over 2,2. Anvendes færre end fem koncentrationer, skal dette begrundes. Den højeste testede koncentration skal helst medføre 100 % immobilisering, mens den laveste helst ikke må medføre nogen observerbar effekt.
1.8.1.3. Inkuberingsbetingelser
Temperaturen skal være mellem 18 oC og 22 oC og skal i den enkelte test være konstant inden for ± 1 oC. En cyklus bestående af 16 timers lys og 8 timers mørke anbefales. Fuldstændigt mørke er også acceptabelt, specielt for prøvestoffer, der er ustabile i lys.
Testkarrene må ikke beluftes under testen. Testen udføres uden justering af pH. Dafnierne må ikke fodres under testen.
1.8.1.4. Varighed
Testens varighed er 48 timer.
1.8.2. Bemærkninger
Hvert testkar kontrolleres for immobiliserede dafnier 24 og 48 timer efter testens begyndelse. (se definitionerne i punkt 1.2). Ud over immobilitet rapporteres om al unormal adfærd eller fremtræden.
1.8.3. Analytiske målinger
Ved testens begyndelse og slutning måles opløst oxygen og pH i kontrolprøve(r) og i den højeste prøvestofkoncentration. Koncentrationen af opløst oxygen i kontrollerne skal være i overensstemmelse med validitetskriteriet (se punkt 1.6). pH må normalt ikke variere mere end 1,5 enhed i nogen enkelt test. Temperaturen måles sædvanligvis i kontrolkarrene eller i den omgivende luft og skal helst registreres kontinuerligt under testen eller i det mindste ved testens begyndelse og slutning.
Prøvestofkoncentrationen måles i det mindste ved den højeste og laveste prøvestofkoncentration ved testens begyndelse og slutning (4). Det anbefales at resultaterne baseres på de målte koncentrationer. Hvis prøvestofkoncentrationen kan dokumenteres at have været holdt tilfredsstillende inden for ± 20 % af den nominelle eller målte begyndelseskoncentration under hele testen, kan resultaterne dog baseres på de nominelle eller målte begyndelsesværdier.
1.9. GRÆNSETEST
Med den i denne metode beskrevne fremgangsmåde kan udføres en grænsetest ved en prøvestofkoncentration på 100 mg/l, dog ikke højere end stoffets opløselighed i testmediet, med henblik på at godtgøre at EC50 er større end denne koncentration. Til grænsetesten anvendes 20 dafnier (helst opdelt i fire grupper på hver fem dyr), og samme antal kontroldyr. Hvis der forekommer immobilisering, udføres en fuld undersøgelse. Al observeret unormal adfærd registreres.
2. DATA
Data sammenfattes i tabeller, der for hver behandlings- og kontrolgruppe viser det anvendte antal dafnier og immobiliserede dyr ved hver observation. Der optegnes en kurve over procentdel immobiliserede dyr ved 24 timer og 48 timer som funktion af prøvestofkoncentrationen. Der anvendes passende statistiske metoder (f.eks. probabalistisk analyse osv.) til beregning af kurvehældninger og EC50 med 95 % konfidensintervaller (p = 0,05) (7) (8).
Når standardmetoder til beregning af EC50 ikke kan anvendes på de målte data, benyttes den højeste koncentration, der ikke medfører immobilitet, og den laveste koncentration, der medfører 100 % immobilitet, til tilnærmet beregning af EC50 (idet denne sættes til det geometriske gennemsnit af de to koncentrationer).
3. RAPPORTERING
3.1. TESTRAPPORT
Testrapporten skal indeholde følgende:
Teststof:
|
— |
fysisk form og, når det er relevant, fysiske-kemiske egenskaber |
|
— |
kemiske identifikationsdata, herunder renhed. |
Den testede dyreart:
|
— |
kilde til og art af Daphnia, leverandør af kilden (hvis den kendes) og anvendte vækstbetingelser (herunder foderets oprindelse, art og mængde samt fodringshyppighed). |
Testbetingelser:
|
— |
beskrivelse af testkarrene: karrenes art, opløsningens volumen, antal dafnier pr. testkar, antal testkar (gentagelser) pr. koncentration |
|
— |
metoder til fremstilling af stam- og testopløsninger herunder eventuelle opløsnings- og dispergeringsmidler, samt anvendte koncentrationer |
|
— |
oplysninger om fortyndingsvandet: oprindelse og kvalitetskarakteristika (pH, hårdhed, Ca/Mg-forhold, Na/K-forhold, alkalinitet, ledningsevne osv.); hvis rekonstitueret vand anvendes, dettes sammensætning |
|
— |
inkubationsbetingelser: temperatur, lysintensitet og -periodicitet, opløst oxygen, pH osv. |
Resultater:
|
— |
antal og procentdel dafnier, der er blevet immobiliseret eller udviser negativ påvirkning (herunder unormal opførsel) i kontroldyrene og i hver behandlingsgruppe, på hvert observationstidspunkt, samt beskrivelse af arten af de iagttagne virkninger |
|
— |
resultater og dato for test udført med referencestof, hvis de foreligger |
|
— |
nominelle prøvekoncentrationer og resultat af alle analyser til bestemmelse af prøvestofkoncentrationen i testkarrene; metodens genfindingsgrad og bestemmelsesgrænse skal ligeledes rapporteres |
|
— |
alle fysiske-kemiske målinger af temperatur, pH og opløst oxygen, som er foretaget under testen |
|
— |
EC50 ved 48 h for immobilisering, med konfidensintervaller og kurver over den tilpassede model, der anvendt til beregning heraf, dosis-responskurvers hældning og middelfejlen herpå; statistiske metoder, der er anvendt til bestemmelse af EC50; (tilsvarende data vedrørende immobilisering ved 24 h skal ligeledes rapporteres, når de er målt) |
|
— |
forklaring vedrørende eventuel afvigelse fra testmetoden, og hvorvidt afvigelsen påvirkede testresultaterne. |
4. HENVISNINGER
|
(1) |
ISO 6341. (1996). Water quality — Determination of the inhibition of the mobility of Daphnia magna Straus (Cladocera, Crustacea) — Acute toxicity test. Third edition, 1996. |
|
(2) |
EPA OPPTS 850.1010. (1996). Ecological Effects Test Guidelines — Aquatic Invertebrate Acute Toxicity Test, Freshwater Daphnids. |
|
(3) |
Environment Canada. (1996) Biological test method. Acute Lethality Test Using Daphnia spp. EPS 1/RM/1 1. Environment Canada, Ottawa, Ontario, Canada. |
|
(4) |
Guidance Document on Aquatic Toxicity Testing of Difficult Substances and Mixtures. OECD Environmental Health and Safety Publication. Series on Testing and Assessment. No. 23. Paris 2000. |
|
(5) |
Commission of the European Communities. Study D8369. (1979). Inter-laboratory Test Programme concerning the study of the ecotoxicity of a chemical substance with respect to Daphnia. |
|
(6) |
OECD Guidelines for the Testing of Chemicals. Guideline 211: Daphnia magna Reproduction Test, adopted September 1998. |
|
(7) |
Stephan C.E. (1977). Methods for calculating an LC50. In Aquatic Toxicology and Hazard Evaluation (edited by F.I. Mayer and J.L. Hamelink). ASTM STP 634 — American Society for Testing and Materials. Pp. 65-84. |
|
(8) |
Finney D.J. (1978). Statistical Methods in Biological Assay. 3rd ed. London. Griffin, Weycombe, UK. |
Bilag 1
NOGLE KEMISKE KARAKTERISTIKA AF ACCEPTABELT FORTYNDINGSVAND
|
Stof |
Koncentration |
|
Partikler |
< 20 mg/l |
|
Total mængde organisk kulstof (TOC) |
< 2 mg/l |
|
Ikke-ioniseret ammoniak |
< 1 μg/l |
|
Chloroverskud |
< 10 μg/l |
|
Total mængde organiske fosforpesticider |
< 50 ng/l |
|
Total mængde organiske chlorpesticider plus polychlorerede biphenyler |
< 50 ng/l |
|
Total mængde organisk chlor |
< 25 ng/l |
Bilag 2
EKSEMPLER PÅ VELEGNET REKONSTITUERET TESTVAND
ISO-testvand (1)
|
Stamopløsninger (enkeltstof) |
Rekonstitueret vand fremstilles ved tilsætning af følgende rumfang stamopløsninger til 1 liter vand (2) |
|
|
Stof |
Mængde tilsat til 1 liter vand (2) |
|
|
Calciumchlorid CaCl2, 2H2O |
11,76 g |
25 ml |
|
Magnesiumsulfat MgSO4, 7H2O |
4,93 g |
25 ml |
|
Natriumbicarbonat NaHCO3 |
2,59 g |
25 ml |
|
Kaliumchlorid KCl |
0,23 g |
25 ml |
Elendt medium M7 og M4
Akklimatisering til Elendt medium M4 og M7
I nogle laboratorier har det medført problemer at overføre dafnier direkte til M4- og M7-medium. Dog er med noget held anvendt gradvis akklimatisering, dvs. flytning fra eget medium til 30 % Elendt efterfulgt af 60 % Elendt og derefter 100 % Elendt. En akklimatiseringsperiode på helt op til en måned kan være nødvendig.
Fremstilling
Mikronæringsstoffer
Først fremstilles der separate stamopløsninger (I) af hvert mikronæringsstof i vand af passende renhed, f.eks. deioniseret, destilleret eller behandlet ved omvendt osmose. Af disse forskellige stamopløsninger (I) fremstilles én enkelt stamopløsning (II) indeholdende alle mikronæringsstoffer (kombineret opløsning), dvs.:
|
Stamopløsning(er) I (enkeltstof) |
Mængde tilsat til vand (mg/l) |
Koncentration (i forhold til medium M4) |
Til fremstilling af den kombinerede stamopløsning II tilsættes følgende mængde stamopløsning I til vand (ml/l) |
|
|
M4 |
M7 |
|||
|
H3BO3 |
57 190 |
20 000 gange |
1,0 |
0,25 |
|
MnCl2.4H2O |
7 210 |
20 000 gange |
1,0 |
0,25 |
|
LiCl |
6 120 |
20 000 gange |
1,0 |
0,25 |
|
RbCl |
1 420 |
20 000 gange |
1,0 |
0,25 |
|
SrCl2.6H2O |
3 040 |
20 000 gange |
1,0 |
0,25 |
|
NaBr |
320 |
20 000 gange |
1,0 |
0,25 |
|
Na2 MoO4.2H2O |
1 230 |
20 000 gange |
1,0 |
0,25 |
|
CuCl2.2H2O |
335 |
20 000 gange |
1,0 |
0,25 |
|
ZnCl2 |
260 |
20 000 gange |
1,0 |
1,0 |
|
CoCl2.6H2O |
200 |
20 000 gange |
1,0 |
1,0 |
|
Kl |
65 |
20 000 gange |
1,0 |
1,0 |
|
Na2 SeO3 |
43,8 |
20 000 gange |
1,0 |
1,0 |
|
NH4VO3 |
11,5 |
20 000 gange |
1,0 |
1,0 |
|
Na2 EDTA.2H2O |
5 000 |
2 000 gange |
— |
— |
|
FeSO4.7H2O |
1 991 |
2 000 gange |
— |
— |
|
De to opløsninger af Na2 EDTA og FeSO4 fremstilles hver for sig, hældes sammen og autoklaveres straks. |
||||
|
Derved fås: |
||||
|
2 l Fe-EDTA opløsning |
|
1 000 gange |
20,0 |
5,0 |
M4- og M7-medium
M4- og M7-medierne fremstilles af stamopløsning II, makronæringsstoffer og vitaminer på følgende måde:
|
|
Mængde tilsat til vand (mg/l) |
Koncentration (i forhold til medium M4) |
Mængde stamopløsning II tilsat til fremstilling afmediet (ml/l) |
|
|
M4 |
M7 |
|||
|
Stamopløsning II (kombinerede sporstoffer) |
|
20 gange |
50 |
50 |
|
Stamopløsninger af makronæringsstoffer (enkeltstof) |
|
|
|
|
|
CaCl2 • 2H20 |
293 800 |
1 000 gange |
1,0 |
1,0 |
|
MgSO4 • 7H2O |
246 600 |
2 000 gange |
0,5 |
0,5 |
|
KCl |
58 000 |
10 000 gange |
0,1 |
0,1 |
|
NaHCO3 |
64 800 |
1 000 gange |
1,0 |
1,0 |
|
Na2 SiO3 • 9H2O |
50 000 |
5 000 gange |
0,2 |
0,2 |
|
NaNO3 |
2 740 |
10 000 gange |
0,1 |
0,1 |
|
KH2PO4 |
1 430 |
10 000 gange |
0,1 |
0,1 |
|
K2HPO4 |
1 840 |
10 000 gange |
0,1 |
0,1 |
|
Kombineret vitamin-stamopløsning |
— |
10 000 gange |
0,1 |
0,1 |
|
Den kombinerede vitamin-stamopløsning fremstilles ved tilsætning af de tre vitaminer til 1 liter vand som vist nedenfor: |
||||
|
Thiaminhydrochlorid |
750 |
10 000 gange |
|
|
|
Cyanocobalamin (B12) |
10 |
10 000 gange |
|
|
|
Biotin |
7,5 |
10 000 gange |
|
|
Den kombinerede vitamin-stamopløsning opbevares nedfrosset i små aliquoter. Vitaminer tilsættes til medierne kort inden disse anvendes.
|
NB |
: |
For at undgå saltudfældning ved fremstilling af de komplette medier tilsætter man først aliquoterne af stamopløsningerne til 500-800 ml deioniseret vand, hvorefter der fyldes op til 1 liter. |
|
NB |
: |
M4-mediet blev første gang beskrevet i Elendt, B. P. (1990): Selenium deficiency in crustacea; an ultrastructual approach to antennal damage in Daphnia magna Straus. Protoplasma, 154, 25-33. |
C.3. VÆKSTHÆMN1NGSTEST FOR ALGER
1. METODE
1.1. INDLEDNING
Formålet med denne test er at bestemme et stofs virkning på væksten af en encellet grønalgeart. Med relativt korte test (72 timer) kan virkningerne vurderes over flere generationer, Da metoden kan tilpasses til brug med adskillige encellede algearter, må forsøgsrapporten indeholde en beskrivelse af den metode, der er anvendt.
Metoden er lettest at anvende for vandopløselige stoffer, som under forsøgsbetingelserne kan forventes at forblive i vandet.
Metoden kan benyttes til stoffer, der ikke direkte interfererer med måling af algevækst.
Det er ønskeligt at have flest mulige oplysninger om teststoffets vandopløselighed, damptryk, kemiske stabilitet, dissociationskonstanter og bionedbrydelighed, førend testen påbegyndes.
Øvrige oplysninger (f.eks. strukturformel, renhedsgrad, art og procentdel af betydende urenheder, tilstedeværelse og mængde af tilsætningsstoffer, samt n-octanol/vand-fordelingskoefficienten) bør tages i betragtning både ved planlægningen af testen og ved fortolkningen af dens resultater.
1.2. DEFINITIONER OG ENHEDER
Celletæthed: antal celler pr. milliliter.
Vækst: forøgelsen i celletætheden i testperioden.
Væksthastighed: forøgelsen i celletætheden pr, tidsenhed.
EC50: i denne metode den koncentration af teststoffet, der medfører, at enten væksten (EbC50) eller væksthastigheden (E7C50) nedsættes med 50 % i forhold til kontrolkulturen.
NOEC (no observed effect concentration): i denne metode den højeste koncentration, hvor der ikke iagttages nogen signifikant væksthæmning i forhold til kontrolkulturen.
Alle koncentrationer af teststoffet er angivet i vægt pr, volumenenhed (mg/l). De kan også udtrykkes i vægtforhold (mg/kg).
1.3. REFERENCESTOFFER
Der kan anvendes et referencestof til at godtgøre, at forsøgsorganismernes følsomhed ikke har ændret sig væsentligt under forsøgsbetingelserne i laboratoriet.
Hvis der benyttes et referencestof, skal resultaterne anføres i forsøgsrapporten. Kaliumdichromat kan anvendes som referencestof, men dets farve kan indvirke dels på kvaliteten og styrken af det lys, som cellerne har til rådighed, dels på en eventuel spektrofotometrisk bestemmelse. Der blev benyttet kaliumdichromat i en international ringtest (se henvisning (3) og tillæg 2).
1.4. TESTMETODENS PRINCIP
Der kan udføres en grænsetest med 100 mg/l til konstatering af, om EC50 er større end denne koncentration.
Kulturer af en udvalgt grønalge i den eksponentielle vækstfase udsættes for forskellige koncentrationer af teststoffet gennem flere generationer under veldefinerede forhold.
Testopløsningerne inkuberes i 72 timer, og i denne periode måles celletætheden mindst hver 24. time. Væksthæmningen i forhold til en kontrolkultur bestemmes.
1.5. KVALITETSKRITERIER
Kvalitetskriterierne gælder for såvel grænsetesten som den komplette testmetode.
Celletætheden i kontrolkulturerne skal være vokset med en faktor på mindst 16 i løbet af tre dage.
Koncentrationen af teststoffet skal ligge over 80 % af startkoncentrationen i et tidsrum, der svarer til testens varighed.
For stoffer, der let opløses i testmediet og giver stabile opløsninger, dvs. stoffer, der ikke i væsentlig grad fordamper, nedbrydes, hydrolyseres eller adsorberes, kan startkoncentrationen antages at være lig med den nominelle koncentration. Det skal godtgøres, at koncentrationen har været opretholdt gennem hele testen, og at kvalitetskriterierne er opfyldt.
For stoffer, som
|
i) |
er tungt opløselige i testmediet eller |
|
ii) |
kan danne stabile emulsioner eller dispersioner eller |
|
iii) |
er ustabile i vandig opløsning, |
skal der som værdi for startkoncentrationen anvendes den koncentration, som er målt ved testens begyndelse. Koncentrationen bestemmes efter, at der opnået ligevægt.
I alle disse tilfælde skal yderligere målinger foretages i løbet af testen for at sikre at koncentrationen har været opretholdt eller kvalitetskriterierne har været opfyldt.
Det er velkendt, at en væsentlig mængde teststof vil være indeholdt i algebiomassen ved testens afslutning. Derfor burde både den del af teststoffet, der er indeholdt i biomassen, og den del, der er i opløsning (eller hvis det ikke er teknisk muligt, målt i vandsøjlen), tages i betragtning ved godtgørelse af, at ovennævnte kvalitetskriterier er opfyldt. Da det kan volde betydelige tekniske problemer at bestemme teststofkoncentrationen i algebiomassen, kan opfyldelse af kvalitetskriterierne godtgøres ved, at der anvendes en testbeholder med den højeste stofkoncentration men uden alger, hvori koncentrationen i opløsningen (eller hvis det ikke er teknisk muligt, i vandsøjlen) måles ved testperiodens begyndelse og slutning.
1.6. BESKRIVELSE AF TESTMETODEN
1.6.1. Reagenser
1.6.1.1. Teststofopløsninger
Der fremstilles stamopløsninger af den påkrævede styrke ved opløsning af stoffet i deioniseret vand eller vand som beskrevet i 1.6.1.2.
De valgte testkoncentrationer fremstilles ved tilsætning af passende teststofmængder til algeforkulturer (se tillæg 1).
Stoffer testes normalt kun op til opløselighedsgrænsen. For visse stoffer (f.eks. stoffer, som er tungt opløselige i vand, som har en høj Pow-værdi, eller som danner en stabil dispersion og ikke en ægte opløsning) kan det accepteres, at der anvendes en testkoncentration, der et større end opløseligheden, så det sikres, at den højeste opløste/stabile koncentration er nået. Det er dog vigtigt, at denne koncentration ikke på anden måde forstyrrer testsystemet (f.eks. dannelse af en hinde af stoffet på vandoverfladen, så vandet ikke iltes osv.).
Der kan tages ultralyd, organiske opløsningsmidler, emulgatorer eller dispergeringsmidler til hjælp til fremstilling af stamopløsninger af stoffer, der er tungt opløselige i vand, og til dispergering af sådanne stoffer i testmediet. Hvis der anvendes hjælpestoffer, skal alle testkoncentrationer indeholde samme mængde hjælpestof, og der anvendes en ekstra kontrolkultur, der udsættes for samme koncentration af hjælpestoffet, som der er anvendt i testserien. Sådanne hjælpestoffer anvendes i den lavest mulige koncentration, der i hvert tilfælde ikke må være større end 100 mg/l i testmediet.
Testen gennemføres uden justering af pH. Hvis der konstateres en væsentlig pH-ændring, anbefales det at gentage testen med justeret pH, og disse resultater rapporteres. I så fald justeres stamopløsningens pH til samme værdi som i det vand, der anvendes til fortynding, medmindre der er særlige grunde til at undlade dette. Til justering af pH foretrækkes HCl og NaOH. Justering af pH skal foregå på en sådan måde, at koncentrationen af teststof i stamopløsningen ikke ændres væsentligt. Hvis teststoffet reagerer kemisk eller udfældes ved justeringen, rapporteres dette.
1.6.1.2. Testmedium
Vandet skal være destilleret vand af god kvalitet eller deioniseret vand med en ledningsevne på mindre end 5 μS.cm-1. Vanddestillationsapparater må ikke indeholde dele af kobber.
Følgende medium anbefales.
Der fremstilles fire stamopløsninger ifølge nedenstående tabel. Stamopløsningerne steriliseres ved autoklavering eller membranfiltrering, hvorefter de opbevares ved 4 oC beskyttet mod lys. Stamopløsning nr. 4 må kun steriliseres ved membranfiltrering. Testopløsningernes endelige næringsstofkoncentration opnås ved fortynding af stamopløsningerne.
|
Næringsstof |
Koncentration i stamopløsning |
Slutkoncentration i testopløsning |
|
Sumopløsning 1: hovednæringsstoffer |
||
|
NH4C1 |
1,5 g/l |
15 mg/l |
|
MgCl2-6H2O |
1,2 g/l |
12 mg/l |
|
CaCl2-2H2O |
1,8 g/l |
18 mg/l |
|
MgS04 7H20 |
1,5 g/l |
15 mg/l |
|
KH2 PO4 |
0,16 g/l |
1,6 mg/l |
|
Stamopløsning 2: Fe-EDTA |
||
|
FeCl3 6H20 |
80 mg/l |
0,08 mg/l |
|
Na2EDTA-2H2O |
100 mg/l |
0,1 mg/l |
|
Stamopløsning 3: sporstoffer |
||
|
H3BO3 |
185 mg/l |
0,185 mg/l |
|
MnCl2 4H2O |
415 mg/l |
0,415 mg/l |
|
ZnCl2 |
3 mg/l |
3 × l0-3 mg/l |
|
CoCl2-6H2O |
1,5 mg/l |
1,5 × 10-3 mg/l |
|
CuCl2-2H2O |
0,01 mg/l |
10-5 mg/l |
|
Na2MoO4-2H2O |
7 mg/l |
7 × 10-3 mg/l |
|
Stamopløsning 4: NaHCO 3 |
||
|
NaHCO3 |
50 g/l |
50 mg/l |
Når mediet er kommet i ligevægt med luft, er dets pH ca. 8.
1.6.2. Apparatur
|
— |
Sædvanligt laboratorieudstyr. |
|
— |
Testkolber af passende volumen (f.eks. er 250 ml konisk kolbe passende til et testopløsningsvolumen på 100 ml). Alle testkolber skal være identiske, både med hensyn til materiale og dimensioner. |
|
— |
Dyrkningsappararur: kammer, hvori en temperatur mellem 21 og 25 oC kan holdes inden for ± 2 oC, og som har kontinuerlig ensartet belysning i området 400-700 nm. Vækstbetingelserne, herunder lysstyrken, anses for at have været tilfredsstillende, hvis kontrolkulturerne når op på den anbefalede væksthastighed. Det anbefales at anvende en lysintensitet, som er på 60-120 μE m-2 .S -1 (35-70 × 1018 fotoner m-2.s-1), når den måles i området 400-700 nm med en egnet modtager. For lysmåleudstyr, der er kalibreret i lux, er det tilsvarende passende niveau på 6 000-10 000 lux. Denne lysintensitet kan opnås ved hjælp af 4-7 lysstofrør på 30 W af hvid standardtype (farvetemperatur ca. 4 000 K) anbragt i en afstand af 35 cm fra algekulturen. |
|
— |
Målinger af celletætheden foretages ved direkte tælling af levende celler, f.eks. ved hjælp af et mikroskop med tællekammer. Andre metoder (fotometri, turbidimetri m.fl.) kan dog benyttes, hvis de er tilstrækkelig følsomme og det påvises, at de korrelerer tilstrækkelig godt med celletætheden. |
1.6.3. Testorganismer
Det foreslås, at der anvendes en hurtigtvoksende grønalgeart, der er egnet til dyrkning og test. Følgende arter foretrækkes;
|
— |
Selenastrum capricornutum, f.eks. ATCC 22662 eller CCAP 278/4 |
|
— |
Scenedesmus subsptcatus, f.eks. 86.81 SAG. |
Bemærk:
|
ATCC |
= |
American Type Culture Collection (USA) |
|
CCAP |
= |
Culture Centre of Algae and Protozoa (UK) |
|
SAG |
= |
Collection of algal culture (Göttingen, Tyskland) |
Ved anvendelse af andre arter skal den benyttede stamme anføres i rapporten.
1.6.4. Testens udførelse
Ved hjælp af indledende test bestemmes det koncentrationsområde, hvor der kan forventes at indtræde virkninger.
De to mål for væksten (biomasse og væksthastighed) kan føre til vidt forskellige resultater for væksthæmning; de skal begge anvendes i de indledende test, så det sikres, at både EbC50 og ErC50 kan skønnes ud fra den kvotientrække, der vælges for koncentrationerne.
Celletætheden ved forsøgets begyndelse
Den anbefalede celletæthed i prøvekulturerne ved forsøgets begyndelse er ca. 104 celler/ml for Selenastrum capricornutum og Scenedesmus subspicatus. Ved anvendelse af andre arter skal biomasser være af samme størrelsesorden.
Koncentrationer af teststoffet
Til forsøget anvendes der mindst fem koncentrationer, der danner en kvotientrække med en kvotient på højst 2,2. Den laveste koncentration må ikke have nogen synlig virkning på algernes vækst. Den højeste koncentration skal hæmme væksten med mindst 50 % i forhold til kontrolkulturen og helst standse væksten helt.
Gentagelser og kontrolkulturer
Forsøgsplanen skal omfatte tredobbelt bestemmelse ved hver koncentration. Der medtages desuden tre kontrolkulturer uden teststof og i givet fald tre kontrolkulturer med hjælpestof. Hvis det er begrundet, kan forsøgsplanen ændres ved, at antallet af koncentrationer øges og antallet af bestemmelser pr. koncentration nedsættes.
Udførelse af testen
Der fremstilles testkulturer med den ønskede teststofkoncentration og den ønskede mængde algeinoculum ved tilsætning af stamopløsning af teststoffet til en passende mængde algeforkultur (se tillæg 1).
Testkolberne rystes og anbringes i dyrkningsapparaturet. Algecellerne holdes i suspension ved rystning, omrøring eller gennembobling med luft, således at der bliver bedre luftudveksling og mindre pH-variationer i testopløsningerne. Kulturerne holdes ved en temperatur mellem 21 og 25 oC, der holdes konstant inden for ± 2 oC.
Celletætheden i hver kolbe bestemmes mindst tre gange, nemlig 24, 48 og 72 timer efter forsøgets begyndelse. Når celletætheden måles på anden måde end ved direkte tælling, anvendes der filtreret algemedium med den pågældende teststofkoncentration til bestemmelse af baggrundsværdien.
pH måles ved forsøgets begyndelse og efter 72 timer.
Kontrolkulturernes pH må normalt ikke afvige med mere end 1,5 i løbet af forsøget.
Test af flygtige stoffer
Der findes i dag ingen generelt accepteret metode til test af flygtige stoffer. Hvis et stof vides at have tendens til at fordampe, kan der benyttes lukkede testkolber med ekstra luftrum over væsken. Muligheden for CO2-mangel skal tages i betragtning, når dette ekstra luftrum beregnes. Der er foreslåer varianter af denne metode (se henvisning (4)).
Den mængde stof, der forbliver i opløsning, skal forsøges bestemt, og det tilrådes, at der udvises den største forsigtighed ved fortolkning af resultater af test, der er udført med flygtige kemikalier i lukkede kolber.
Grænsetest
Der kan under anvendelse af den i denne test anførte fremgangsmåde udføres en grænsetest med 100 mg/l til konstatering af, om EC50 er større end denne koncentration.
Hvis stoffet er af en sådan art, at der ikke kan opnås en koncentration på 100 mg/l i testvandet, udføres grænsetesten ved stoffets mætningskoncentration (eller den højeste koncentration, ved hvilken der dannes en stabil dispersion) i det anvendte medium (se også 1.6.1.1).
Grænsetesten udføres mindst som en tredobbeltbestemmelse og med kontrolkulturer af samme størrelse. Begge mål for væksten (biomasse og væksthastighed) benyttes i grænsetesten.
Hvis biomassen eller væksthastigheden er mindst 25 % lavere i grænsetesten end i kontrolkulturen, må der gennemføres en komplet undersøgelse.
2. DATA OG EVALUERING
De celletætheder, der er målt i test- og kontrolkulturerne, opstilles i en tabel sammen med teststoffets koncentrationer og måletidspunkterne, Der optegnes vækstkurver for hver teststofkoncentration og for kontrolkulturerne ved afsætning af den gennemsnitlige celletæthed mod tiden (0-72 timer).
De to nedenstående metoder kan benyttes til bestemmelse af sammenhængen mellem koncentration og virkning. Nogle stoffer kan virke vækstfremmende ved lave koncentrationer, Kun datapunkter, hvor hæmningen er mellem 0 og 100 %, tages i betragtning.
2.1. SAMMENLIGNING AF AREALER UNDER VÆKSTKURVERNE
Arealet mellem vækstkurverne og den vandrette linje N = No kan beregnes ved følgende formel:
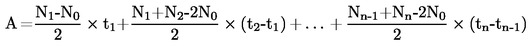
hvor
|
A |
= |
arealet |
|
N0 |
= |
antallet af celler pr. ml på tidspunktet t0 (testens begyndelse) |
|
N1 |
= |
antallet af celler pr. ml på tidspunktet t1 |
|
Nn |
= |
antallet af celler pr. ml på tidspunktet tn |
|
t1 |
= |
tidspunktet for første måling efter testens begyndelse |
|
tn |
= |
tidspunktet for n'te måling efter testens begyndelse |
|
n |
= |
det antal målinger, der er foretaget efter testens begyndelse. |
Den procentvise hæmning af cellevæksten beregnes ved hver teststofkoncentration (IA) efter følgende formel:

hvor
|
Ac |
= |
arealet mellem den vandrette linje N = No og vækstkurven for kontrolkulturen |
|
At |
= |
arealet mellem den vandrette linje N = NO og vækstkurven ved koncentrationen t. |
|
IA |
= |
værdierne afsættes på semilogaritmisk papir eller semilogaritmisk sandsynlighedspapir mod de tilsvarende koncentrationer. Benyttes der sandsynlighedspapir, indtegnes den bedste rette linje, enten på øjemål eller ved regressionsberegning. |
Ud fra regressionslinjen skønnes EC50 ved aflæsning af den koncentration, der svarer til 50 % inhibering (IA = 50 %). Som utvetydig betegnelse for den værdi, der er fundet ved denne beregningsmetode, foreslås det at benytte EbC50. Det er vigtigt, at den pågældende eksponeringstid anføres sammen med EbC50, f.eks. EbC50 (0-72 timer).
2.2. SAMMENLIGNING AF VÆKSTRATER (SPECIFIKKE VÆKSTHASTIGHEDER)
Den gennemsnitlige specifikke væksthastighed (μ) for kulturer i den eksponentielle vækstfase kan beregnes ved
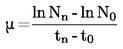
hvor t0 er tidspunktet for forsøgets begyndelse.
Alternativt kan den gennemsnitlige specifikke væksthastighed afledes af hældningen af regressionslinjen i en afbildning af ln N mod tiden.
Den procentvise hæmning af den gennemsnitlige specifikke væksthastighed beregnes for hver teststofkoncentration (Iμt) ved hjælp af følgende formel:

hvor
|
μc |
= |
kontrolkulturens gennemsnitlige specifikke væksthastighed (= gennemsnitlige vækstrate) |
|
μt |
= |
den gennemsnitlige specifikke væksthastighed ved testkoncentrationen t. |
Den procentvise nedsættelse i den gennemsnitlige specifikke væksthastighed ved hver teststofkoncentration i forhold til kontrolværdien afbildes mod logaritmen til koncentrationen. EC50 kan aflæses af den fremkomne kurve. Som utvetydig betegnelse for den EC50-værdi, der er fundet ved denne beregningsmetode, foreslås det at benytte ErC50. Måletidspunkterne skal anføres, dvs. at betegnelsen bliver f.eks. ErC50 (0-72 timer), hvis værdierne vedrører tidspunkterne 0 og 72 timer.
Bemærk: Specifik væksthastighed er et logaritmisk udtryk, og små ændringer i vækstraten (= den specifikke væksthastighed) kan medføre store ændringer i biomassen. Man kan derfor ikke sammenligne talværdierne for EbC og ErC direkte.
2.3. BEREGNING AF NOEC-VÆRDIEN
Den koncentration, hvor der ikke kan iagttages nogen virkning, (NOEC-værdien) bestemmes ved hjælp af en egnet statistisk metode til sammenligning af flere prøver (f.eks. variansanalyse eller Dunnetts test), som anvendes på de enkelte værdier af arealerne under vækstkurverne A (se 2.1) eller af de specifikke væksthastigheder μ (se 2.2).
3. RAPPORTERING
Forsøgsrapporten skal om muligt indeholde følgende oplysninger:
|
— |
teststof: kemisk betegnelse |
|
— |
testorganismer: oprindelse, laboratoriekultur, stammenummer og dyrkningsmetode |
|
— |
testbetingelser:
|
|
— |
resultater:
|
4. HENVISNINGER
|
(1) |
OECD, Paris, 1981, Test Guideline 201, Decision of thc Council C(81) 30 final. |
|
(2) |
Umweltbundesamt. Berlin, 1984, Verfahrensvorschlag »Hemmung der Zellvermehrung bei der Grünalge Scenedesmus subspicatus«, in: Rudolph/Boje: Okotoxikologie, ecomed, Landsberg, 1986. |
|
(3) |
ISO 8692 — Water Quality — Fresh water algal growth inhibition test with Scenedesmus subspicatus and Selenastrum capricornurum. |
|
(4) |
S.Galassi and M.Vighi — Chemosphere, 1981, vol. 10, 1123-1126. |
Tillæg 1
Eksempel på fremgangsmåde ved dyrkning af alger
Almindelige bemærkninger
Formålet med dyrkning efter følgende fremgangsmåde er at fremskaffe algekulturer til toksicitetstest.
Der træffes foranstaltninger til at sikre, at algekulturerne ikke inficeres med bakterier (ISO 4833). Det er ønskeligt at have axeniske kulturer, men det er absolut nødvendigt, at kulturerne kun består af én algeart.
Alle operationer foretages under sterile forhold, så kontaminering med bakterier og andre alger undgås. Kontaminerede kulturer kasseres.
Fremgangsmåde til fremskaffelse af algekulturer
Fremstilling af næringsopløsninger (substrater)
Substratet kan fremstilles ved fortynding af koncentrerede næringsstofopløsninger. Til faste substrater tilsættes 0,8 % agar. Der anvendes kun sterile substrater. Ved autoklavering kan der ske tab af NH3.
Stamkultur
Stamkulturerne er små algekulturer, som regelmæssigt overføres til frisk substrat, og som benyttes som udgangsmateriale til testen. Hvis disse kulturer ikke anvendes regelmæssigt, spredes de ud på skrå agarrør. Sådanne kulturer overføres til frisk substrat mindst hver anden måned.
Stamkulturerne dyrkes i koniske kolber med et egnet substrat (volumen ca. 100 ml). Dyrkes algerne ved 20 oC med kontinuerlig belysning, er overførsel én gang ugentligt påkrævet.
Ved overførslen overføres en mængde af den »gamle« kultur med en steril pipette til en kolbe med frisk substrat, så der med de hurtigtvoksende arter er en startkoncentration på ca. 1/100 af koncentrationen i den gamle kultur.
En arts væksthastighed kan bestemmes ud fra vækstkurven. Hvis denne kendes, kan man skønne, ved hvilken tæthed kulturen må overføres til et nyt substrat. Dette skal gøres, før kulturen når dødsfasen.
Forkultur
Forkulturen har til formål at give en passende algemængde til podning af testkulturerne. Forkulturer inkuberes under forsøgsbetingelserne og tages i anvendelse, medens væksten stadig er eksponentiel, normalt efter ca. tre dage. Algekulturer, der indeholder deforme eller abnorme celler, kasseres.
Tillæg 2
I ISO 8692 — Water Qualiry — Fresh water algal growth inhibition test with Scenedesmus subspicatus and Selenastrum capricornutum er nedenstående resultater af en sammenlignende laboratorietest med kaliumdichromat udført på 16 laboratorier:
|
|
Gennemsnit (mg/l) |
Interval (mg/1) |
|
ErC50 (0-72 h) |
0,84 |
– 0,60–1,03 |
|
EbC50 (0-72 h) |
0,53 |
– 0,20–0,75 |
C.4. BESTEMMELSE AF »LET« BIONEDBRYDELIGHED
DEL I. GENERELLE BETRAGTNINGER
I.1. INDLEDNING
Der er beskrevet seks metoder til screening af kemikalier for let bionedbrydelighed i aerobt vandigt miljø:
|
a) |
Eliminering af opløst organisk kulstof (DOC) (metode C.4-A) |
|
b) |
Modificeret OECD-screeningtest — eliminering af DOC (metode C.4-B) |
|
c) |
Kuldioxidudvikling (modificeret Sturm-test) (metode C.4-C) |
|
d) |
Manometrisk respirometri (metode C.4-D) |
|
e) |
Closed Bottle Test (metode C.4-E) |
|
f) |
MITI-test (Ministry of International Trade and Industry — Japan) (metode C.4-F) |
Del I indeholder generelle betragtninger og betragtninger, der gælder for alle seks test. I del II-VII findes de punkter, der er specifikke for de enkelte test. Tillæggene indeholder definitioner, formler og vejledende oplysninger.
I 1988 gennemførte OECD en ringtest, som viste, at metoderne giver indbyrdes overensstemmende resultater. Imidlertid kan en bestemt metode foretrækkes, alt efter det undersøgte stofs fysiske egenskaber.
I.2. VALG AF METODE
For at kunne vælge den bedst egnede metode er det afgørende at have kendskab til kemikaliets opløselighed, damptryk og adsorptionsegenskaber. Den kemiske struktur eller formel bør kendes for at kunne beregne den teoretiske værdi og/eller kontrollere målte værdier af parametre som f.eks. ThOD, ThCO2, DOC, TOC og COD (se bilag I og II).
Testkemikalier, der er opløselige i vand med mindst 100 mg/l og ikke-flygtige og ikke-adsorberende, kan vurderes ved alle metoderne. Af tabel 1 fremgår, hvilke metoder der er egnede for kemikalier, der er tungtopløselige i vand, flygtige eller adsorberende. I bilag III er det beskrevet, hvordan man kan bære sig ad med tungt vandopløselige og flygtige kemikalier. Kemiske stoffer med moderat flygtighed kan testes i DOC-henfaldsmetoden, hvis der er tilstrækkelig luftrum i testbeholderen (som skal tillukkes passende). I dette tilfælde skal der benyttes en abiotisk kontrolkolbe, som tillader ethvert fysisk tab.
Tabel 1
Testmetodernes anvendelsesområder
|
Test |
Analysemetode |
Egnet til stoffer, der er: |
||
|
tungt opløselige |
flygtige |
adsorberende |
||
|
DOC-eliminering |
Opløst organisk kulstof |
– |
– |
± |
|
Mod. OECD DOC-elim. |
Opløst organisk kulstof |
– |
– |
± |
|
CO2-udvikling |
Respirometri: CO2-udvikling |
+ |
– |
+ |
|
Manometrisk respirometri |
Manometrisk respirometri: oxygenforbrug |
+ |
± |
+ |
|
Closed Bottle |
Respirometri: opløst oxygen |
± |
+ |
+ |
|
MITI |
Respirometri: oxygenforbrug |
+ |
± |
+ |
Oplysninger om testmaterialets renhed eller forholdet mellem dets hovedtebstanddele er nødvendige for fortolkning af de fundne resultater, især hvis de er små eller usikre.
Oplysninger om testkemikaliets toksicitet over for bakterier (bilag IV) kan være til nytte ved valg af passende testkoncentrationer og kan være afgørende for korrekt fortolkning af lave værdier for bionedbrydning.
I.3. REFERENCESTOFFER
Til kontrol af fremgangsmåden benyttes der referencestoffer, som opfylder kriterierne for let bionedbrydelighed, og som testes i en separat kolbe parallelt med de øvrige prøver.
Anilin (frisk destilleret), natriumacetat og natriumbenzoat er egnede kemikalier. Alle disse referencekemikalier nedbrydes ved disse metoder, selv når der ikke tilsættes inoculum.
Det har været fremført, at man burde finde frem til et referencekemikalie, der er let nedbrydeligt men kræver tilsætning af inoculum, selv i Closed Bottle-metoden. Kaliumhydrogenphthalat har været foreslået, men der mangler endnu konkret bevis med dette stof, før det kan accepteres som et referencestof.
I metoder, der er baseret på respirometri, kan nitrogenholdige forbindelser påvirke oxygenoptagelsen på grund af nitrifikation (se bilag II og V).
I.4. TESTMETODERNES PRINCIP
Teststoffet opløses eller opslæmmes i et uorganisk medium, inoculeres og inkuberes under aerobe forhold i mørke eller dæmpet lys. Den del af DOC i opløsningen, der stammer fra inoculum, skal være så lavt som muligt i forhold til den del, der stammer fra teststoffet. Der tages hensyn til inoculums endogene aktivitet ved at køre parallelle blindprøver med inoculum men uden teststof i opløsningen, selvom cellernes endogene aktivitet under tilstedeværelse af stoffet ikke præcis svarer til denne i den endogene kontrol. Et referencestof benyttes parallelt for at kontrollere, hvorledes procedurerne fungerer.
I reglen følges nedbrydningen ved bestemmelse af parametre såsom DOC, CO2-dannelse og oxygenoptagelse, og der foretages målinger med tilstrækkeligt hyppige intervaller, således at begyndelsen og afslutningen af bionedbrydningen kan identificeres. Med automatiske respirometre sker målingen kontinuerligt. Undertiden måles både DOC og en anden parameter, men det er normalt kun tilfældet ved testens begyndelse og slutning. Der kan også anvendes specifik kemisk analyse til bedømmelse af den primære nedbrydning af teststoffet og til bestemmelse af koncentrationen af eventuelle mellemprodukter (obligatorisk i MITI-testen).
Normalt løber testen over 28 dage. Når bionedbrydningskurven har nået et plateau ved mindst tre målinger, kan testen dog afbrydes. Hvis kurven viser, at bionedbrydningen er begyndt men ikke har nået et plateau inden de 28 dage, kan testen forlænges ud over denne periode.
I.5. KVALITETSKRITERIER
I.5.1. Reproducerbarhed
Som følge af bionedbrydningens natur og den blandede bakteriepopulation i inoculum, foretages der mindst dobbeltbestemmelse.
Den almindelige erfaring er, at variationen mellem gentagelser bliver mindre, desto større startkoncentrationen af mikroorganismer i testmediet er. Ringtest har desuden vist, at der kan være betydelige forskelle mellem de resultater, forskellige laboratorier når frem til, men der opnås generelt god overensstemmelse med let bionedbrydelige forbindelser.
I.5.2. Testens gyldighed
En test anses for gyldig, hvis forskellene mellem de to yderste værdier for teststoffets forsvinden ved en multibestemmelse i plateau-fasen, ved afslutningen af testen eller ved afslutningen af 10 døgns-vinduet er mindre end 20 %, og hvis den procentvise nedbrydning af referencestoffet har nået niveauet for let bionedbrydelighed inden for 14 dage. Hvis ikke begge betingelser er opfyldt, gentages testen. Da metoderne er ret strikse, betyder lave værdier ikke nødvendigvis, at stoffet ikke er bionedbrydeligt under miljøforhold, men at der kræves yderligere arbejde til bestemmelse af bionedbrydeligheden.
Er nedbrydningen i en toksicitetstest med både teststof og referencekemikalie mindre end 35 % (baseret på DOC) eller 25 % (baseret på ThOD eller ThCO2) på 14 dage, må testkemikaliet antages at være hæmmende (se også bilag IV). Testrækken gentages, om muligt med lavere teststofkoncentration og/eller højere inoculumkoncentration, dog højst 30 mg tørstof pr. liter.
I.6. GENERELLE PROCEDURER
De generelle betingelser for testene er sammenfattet i tabel 2. Apparatur og forsøgsomstændigheder, der er specifikke for en bestemt test, er beskrevet senere under den pågældende test.
Tabel 2
Testbetingelser
|
Test |
DOC-eliminering |
CO2-udvikling |
Manometrisk respirometri |
Modificeret OECD-screening |
Closed Bottle |
MITI(I) |
|||||||||
|
Teststofkoncentration |
|
|
|
|
|
|
|||||||||
|
i mg/t |
|
|
100 |
|
2-10 |
100 |
|||||||||
|
mg DOC/l |
10-40 |
10-20 |
|
10-40 |
|
|
|||||||||
|
mg ThOD/l |
|
|
50-100 |
|
5-10 |
|
|||||||||
|
Inoculumkoncentration (omtrentligt antal celler pr. liter) |
≤ 30 mg/l TS eller ≤ 100 ml spildev and pr. l (107-108) |
0,5 ml sekundært spildevand pr. 1 (105) |
< 5 ml spildevand pr. l (104-106) |
30 mg/l TS (107-108) |
|||||||||||
|
Grundstofkoncentration i uorganisk medium (i mg/l) |
|
|
|
|
|
|
|||||||||
|
P |
116 |
11,6 |
29 |
||||||||||||
|
N |
1,3 |
0,13 |
1,3 |
||||||||||||
|
Na |
86 |
8,6 |
17,2 |
||||||||||||
|
K |
122 |
12,2 |
36,5 |
||||||||||||
|
Mg |
2,2 |
2,2 |
6,6 |
||||||||||||
|
Ca |
9,9 |
9,9 |
29,7 |
||||||||||||
|
Fe |
0,05-0,1 |
0,05-0,1 |
0,15 |
||||||||||||
|
pH |
7,4±0,2 |
helst 7,0 |
|||||||||||||
|
Temperatur |
22 ± 2 oC |
25 ± 1 oC |
|||||||||||||
|
|
|
|||||||||||||
I.6.1. Vand
Det deioniserede eller destillerede vand, der benyttes, må hverken indeholde toksiske stoffer (f.eks. Cu++-ioner) i hæmmende mængder eller organisk kulstof svarende til mere end 10 % af testmaterialets organiske kulstof. Denne høje renhed er nødvendig for at undgik høje blindværdier. Forureninger kan stamme fra naturlige urenheder og fra ionbytterharpikser og lyseret materiale fra bakterier og alger. Til hver testserie anvendes kun én bestemt batch af vand, som i forvejen er kontrolleret ved DOC-analyse. En sådan kontrol er ikke nødvendig for Closed Bottle-testen, men oxygenforbruget fra vandet skal være lavt.
I.6.2. Stamopløsninger af uorganiske medium
Der fremstilles stamopløsninger af uorganiske medier med passende koncentrationer af salte til brug ved fremstilling af testopløsningerne. Følgende stamopløsninger kan (med forskellige fortyndingsforhold) anvendes til DOC-eliminering, modificeret OECD-screening, CO2-udvikling, manometrisk respirometri og Closed Bottle-test.
Fortyndingsforholdene og for MITI-testens vedkommende fremstilling af det specifikke uorganiske medium, er anført under de enkelte metoder.
Stamopløsninger:
Følgende stamopløsninger fremstilles ud fra reagenser af analysekvalitet:
|
a) |
monokaliumdihydrogenorthophosphat, KH2PO4 |
8,50 g |
|
dikaliummonohydrogenorthophosphat, K2HPO4 |
21,75 g |
|
|
dinatriummonohydrogenorthophosphat dihydrat, Na2HPO4 2H2O |
33,40 g |
|
|
ammoniumchlorid, NH4Cl |
0,50 g |
|
|
Opløses i vand og fortyndes til 1 liter. Opløsningens pH skal være 7,4. |
|
|
|
b) |
calciumchlorid, vandfrit, CaCl2 |
27,50 g |
|
eller calciumchlorid dihydrat, CaCl2 2H2O |
36,40 g |
|
|
Opløses i vand og fortyndes til 1 liter. |
|
|
|
c) |
magnesiumsulfat heptahydrat, MgSO4 7H2O |
22,50 g |
|
Opløses i vand og fortyndes til 1 liter. |
|
|
|
d) |
jern(III)chlorid hexahydrat, FeCl3 6H20 |
0,25 g |
|
Opløses i vand og fortyndes til 1 liter. |
|
Anmærkning: For at undgå at skulle fremstille denne opløsning umiddelbart før brugen tilsættes der 1 dråbe koncentreret HCl eller 0,40 g ethylendiamintetraeddikesyre dinatriumsalt (EDTA) pr. liter.
I.6.3. Stamopløsninger af kemikalier
Hvis opløseligheden er større end 1 g/l opløses f.eks. 1-10 g af test- eller referencekemikalie i deioniseret vand, og der fortyndes til 1 liter. I modsat fald fremstilles stamopløsningen i det uorganiske medium, eller kemikaliet tilsættes direkte til det uorganiske medium. Jf. bilag III for håndtering af mindre opløselige kemikalier; dog må der i MITI-testen (metode C.4-F) hverken anvendes opløsningsmidler eller emulgatorer.
I.6.4. Inoculum
Inoculum kan stamme fra en række forskellige kilder, f.eks. aktivt slam, spildevandsafløb (ikke chloreret), overfladevand og -jord eller en blanding af disse. Aktivt slam til DOC-eliminering, CO2-udvikling og manometrisk respirometri skal udtages fra et rensningsanlæg eller et laboratorieanlæg, der først og fremmest behandler husholdningsspildevand. Inoculum fra andre kilder har vist sig at føre til større spredning af resultaterne. Til den modificerede OECD-screening og Closed Bottle-testen kræves der et mere fortyndet inoculum uden slampartikler, og som kilde foretrækkes sekundært afløbsvand fra et rensningsanlæg for husholdningsspildevand eller et anlæg i laboratorieskala. Til MITI-testen fremstilles inoculum fra en blanding af kilder, hvilket er beskrevet under denne specifikke test.
I.6.4.1. Inoculum fra aktivt slam
Der udtages en frisk prøve af aktivt slam fra beluftningstanken ved et rensningsanlæg eller et laboratorieanlæg, der fortrinsvis behandler husholdningsspildevand. Om nødvendigt fjern grove partikler fra ved filtrering gennem en finmasket sigte og hold herefter slammet aerobt.
Alternativt kan slammet henstå til bundfældning eller centrifugeres (f.eks. ved 1 100 g i 10 min), efter at grove partikler er fjernet. Supernatanten fjernes. Slammet kan vaskes i uorganisk medium. Det koncentrerede slam opslæmmes til en koncentration på 3-5 g opslæmmet tørstof pr. liter, hvorefter det beluftes indtil anvendelse.
Slammet skal tages fra et ordentligt fungerende normalt anlæg. Slammet vaskes, hvis det er nødvendigt at tage det fra et stærkt belastet rensningsanlæg, eller et renseanlæg, som menes at indeholde hæmmende stoffer. Det genopslæmmede slam henstår til bundfældning eller centrifugeres efter grundig blanding, supernatanten fjernes, og det vaskede slam genopslæmmes i uorganisk medium. Denne fremgangsmåde gentages, indtil slammet menes at være fri for overskud af substrat eller hæmmende stoffer.
Lige før brugen udtages der en prøve af det fuldstændigt genopslæmmede slam eller af det ubehandlede slam, til bestemmelse af mængden af opslæmmet tørstof.
Et andet alternativ er at homogenisere aktivt slam (3-5 g opslæmmet tørstof pr. liter). Slammet behandles i en mekanisk blender i 2 min ved middel hastighed. Det homogeniserede slam henstår til bundfældning i 30 min eller om nødvendigt længere, og væsken dekanteres fra til brug som inoculum i en mængde på 10 ml pr. liter uorganisk medium.
I.6.4.2. Andre inoculumkilder
Det kan fremstilles af sekundært afløbsvand fra et rensningsanlæg eller et laboratorieanlæg, der overvejende behandler husholdningsspildevand. Der udtages en frisk prøve, der opbevares under aerobe forhold under transporten. Den henstår til bundfældning i 1 time eller filtreres gennem et groft.papirfilter, og denne væske opbevares under aerobe forhold indtil brugen. Op til 100 ml af denne inoculumtype kan bruges pr. liter medium.
Overfladevand er en yderligere inoculumkilde. I dette tilfælde udtages en prøve af egnet overfladevand, f.eks, flod- eller søvand, som opbevares under aerobe forhold indtil anvendelsen. Om nødvendigt kan inoculum koncentreres ved filtrering eller centrifugering.
I.6.5. Forkonditionering af inoculum
De fremstillede inocula kan forkonditioneres til forsøgsbetingelserne, men må ikke tilvænnes til testkemikaliet. Forkonditioneringen består i beluftning af aktivt slam i uorganisk medium eller af sekundært afløbsvand i 5-7 dage ved stuetemperatur. Undertiden gør forkonditionering metoderne mere præcise, idet blindværdierne reduceres. Det anses for unødvendigt at forkonditionere MITI-inoculum.
I.6.6. Abiotiske kontrolprøver
Om nødvendig: foretages der kontrol af eventuel abiotisk nedbrydning af teststoffet ved bestemmelse af DOC-tab, oxygenoptagelse og kuldioxidudvikling i sterile kontrolprøver, der ikke indeholder inoculum. Der steriliseres ved membranfiltrering (0,2-0,45 μm); eller ved tilsætning af et passende toksisk stof i passende koncentration. Hvis der benyttes membranfiltrering, skal prøverne tages aseptisk for at opretholde steriliteten. Med mindre adsorption af teststoffet i forvejen er blevet udelukket, skal test, som måler bionedbrydning som DOC-fjernelse, inkludere en abiotisk kontrol, som inoculeres og forgiftes. Dette gælder særligt for inocula med aktiveret slam.
I.6.7. Antal kolber
For hver test er antallet af kolber i en typisk forsøgsrække beskrevet under den enkelte metode.
Følgende typer af kolber kan bruges:
|
— |
testopslæmning: indeholder teststof og inoculum |
|
— |
inoculumblind: indeholder kun inoculum |
|
— |
procedurekontrol: indeholder referencestof og inoculum |
|
— |
abiotisk steril kontrol: sterile, indeholder teststof (I.6.6) |
|
— |
adsorptionskontrol: indeholder teststof, inoculum og steriliserende stof |
|
— |
toksicitetskontrol: indeholder teststof, referencestof og inoculum. |
Bestemmelser i testopslæmning og inoculumblind skal obligatorisk foretages sideløbende. Det anbefales ligeledes at foretage sideløbende bestemmelser i den anden kolbe.
Det er dog ikke altid muligt. Man må sørge for, at der udtages tilstrækkeligt med prøver eller foretages tilstrækkeligt mange aflæsninger til, at den procentvise eliminering i 10-dages-vinduet kan vurderes.
I.7. DATA OG EVALUERING
Ved beregning af den procentvise nedbrydning Dt anvendes gennemsnitsværdierne af dobbeltbestemmelserne af parameteren i testopslæmningen og inoculumblindprøven. Formlerne er givet i afsnittene om de enkelte testmetoder. Nedbrydningens forløb vises grafisk med angivelse af 10-dages-vinduet. Den procentvise eliminering ved afslutningen af 10-dages-vinduet og ved plateauet eller testens afslutning beregnes og rapporteres.
I de respirometriske test kan nitrogenholdige forbindelser indvirke på oxygenoptagelsen som følge af nitrifikation (se bilag II og V).
I.7.1. Nedbrydning målt ved bestemmelse af DOC
Den procentuelle nedbrydning (Dt) på ethvert prøvetagningstidspunkt skal beregnes særskildt for kolberne, som indeholder teststof, idet gennemsnitsværdierne af dobbeltbestemmelseme af DOC benyttes for at kunne bedømme pålideligheden af testen (se I.5.2). Den beregnes ved at bruge følgende udtryk:

hvor:
|
Dt |
= |
% nedbrydning til tidspunktet t |
|
Co |
= |
gennemsnitlig startkoncentration af DOC i kulturmediet med teststof og inoculum (mg DOC/l) |
|
Ct |
= |
gennemsnitskoncentration af DOC i kulturmediet med teststof og inoculum til tidspunktet t (mg DOC/l) |
|
Cbo |
= |
gennemsnitlig startkoncentration af DOC i blindprøve med uorganisk medium og inoculum (mg DOC/l) |
|
Cbt |
= |
gennemsnitskoncentration af DOC i blindprøve med uorganisk medium og inoculum til tidspunktet t (mg DOC/l). |
Alle koncentrationer bestemmes eksperimentelt.
I.7.2. Nedbrydning målt ved specifik analyse
Når der foreligger specifikke analysedata, beregnes den primære bionedbrydning ved udtrykket:

hvor
|
Dt |
= |
% nedbrydning til tidspunktet t, normalt 28 dage |
|
Sa |
= |
restmængde teststof i medium med inoculum ved testens afslutning (mg) |
|
Sb |
= |
restmængde teststof i blindprøve kun med vand/medium og teststof (mg) |
I.7.3. Abiotisk nedbrydning
Når en abiotisk kontrol bruges, beregnes den procentuelle abiotiske nedbrydning ved at bruge

hvor
|
CS(0) |
= |
DOC-koncentrationen, sterilkontrollen på dag 0 |
|
Cs(t) |
= |
DOC-koncentrationen, sterilkontrollen på dag t |
I.8. RAPPORTERING
Testrapporten skal om muligt indeholde følgende oplysninger:
|
— |
test- og referencekemikalier og deres renhed |
|
— |
testbetingelser |
|
— |
inoculum: art og udtagningssted(er), koncentration og eventuel forkonditionering |
|
— |
art og andel af industrispildevand i afløbsvandet, hvis kendt |
|
— |
testens varighed og temperatur |
|
— |
for tungt opløselige testkemikalier, den foretagne behandling |
|
— |
anvendt testmetode; ændringer i fremgangsmåden skal begrundes videnskabeligt |
|
— |
forsøgsresultater i skema |
|
— |
eventuelt iagttagne hæmningsfænomener |
|
— |
eventuelt iagttagen abiotisk nedbrydning |
|
— |
eventuelle data fra specifikke kemiske analyser |
|
— |
om muligt analytiske data vedrørende nedbrydningsprodukter |
|
— |
afbildning af den procentvise nedbrydning mod tiden for både test- og referencestof med tydelig angivelse af lag-periode, nedbrydningstid, 10-dages-vindue og hældning (se bilag I). Hvis der i testen benyttes pålidelighedskriterier kan gennemsnittet af nedbrydningsprocenterne i kolberne indeholdende teststoffet benyttes til fremstilling af grafen |
|
— |
procentvis eliminering efter 10-dages-vinduet og ved enten plateau eller testens afslutning. |
DEL II. DOC-ELIMINERING (Metode C.4-A)
II.1. METODENS PRINCIP
En afmålt mængde uorganisk medium indeholdende inoculum og, som den nominelle eneste organiske kulstofkilde, teststof i kendt koncentration (10-40 mg/l) beluftes i mørke eller dæmpet lys ved 22 ± 2 oC.
Nedbrydningen følges ved hyppig DOC-analyse i en periode på 28 dage. Bionedbrydningsgraden beregnes ved at udtrykke koncentrationen af fjernet DOC (korrigeret for koncentrationen i inoculumblindprøven) i procent af startkoncentrationen af DOC. Den primære bionedbrydning kan også beregnes ud fra supplerende kemisk analyse ved inkubationsperiodens begyndelse og afslutning.
II.2. BESKRIVELSE AF METODEN
II.2.1. Apparatur
|
a) |
Koniske kolber, f.eks. 250 ml op til 2 1, afhængigt af det volumen, der kræves til DOC-analyse |
|
b) |
Rysteapparat til de koniske kolber, enten med automatisk temperaturkontrol eller anbragt i et lokale med konstant temperatur, og tilstrækkelig kraftigt til, at forholdene holdes aerobe i alle kolberne |
|
c) |
Filtreringsapparatur med passende membraner |
|
d) |
DOC-analysator |
|
e) |
Apparatur til bestemmelse af opløst oxygen |
|
f) |
Centrifuge. |
II.2.2. Fremstilling af uorganisk medium
Se I.6.2 om fremstilling af stamopløsning.
10 ml af opløsning (a) blandes med 800 ml fortyndingsvand, og af opløsning (b), (c) og (d) tilsættes der 1 ml af hver, hvorefter der fyldes op til 1 l med fortyndingsvand.
II.2.3. Fremstilling og forkonditionering af inoculum
Inoculum kan stamme fra en række forskellige kilder: aktivt slam, spildevandsafløb, overfladevand og -jord eller en blanding af disse.
Se I.6.4., I.6.4.1., I.6.4.2. og I.6.5.
II.2.4. Klargøring af kolberne
Eksempelvis kan der overføres 800 ml portioner af uorganisk medium til koniske 2-liters kolber, hvorefter der tilsættes så meget af stamopløsningerne af test- og referencestof til de forskellige kolber, at kemikaliekoncentrationen bliver på 10-40 mg DOC pr. liter. Kontroller pH-værdier og juster om nødvendigt til 7,4. Der tilsættes inoculum fra aktivt slam eller en anden kilde (se I.6.4) til en slutkoncentration på højst 30 mg opslæmmet tørstof pr. liter. Der klargøres også inoculumkontrolprøver i uorganisk medium uden test- og referencekemikalier.
Om nødvendigt kan der medtages en kolbe til kontrol af. om testkemikaliet har en hæmmende virkning, ved tilsætning af omtrent lige store mængder test- og referencekemikalie til uorganisk medium indeholdende inoculum.
Der kan også om nødvendigt inkluderes en steril kolbe. dvs. en kolbe uden inoculum, til undersøgelse af, om testkemikaliet nedbrydes abiotisk (se I.6.6.).
Hvis testkemikaliet mistænkes for i væsentlig grad at kunne adsorberes til glas, slampartikler eller lign., foretages desuden en indledende vurdering af denne adsorption og dermed af testens egnethed for det pågældende testkemikalie (se tabel 1). Benyt en flaske indeholdende teststoffet, inoculum og det steriliserende stof.
Alle kolber fyldes op til 1 liter med uorganisk medium, og efter blanding udtages der en prøve af hver kolbe til bestemmelse af startkoncentrationen af DOC (se bilag II, punkt 4). Kolbernes åbning tildækkes, f.eks. med aluminiumfolie, på en sådan måde, at luften frit kan passere ind og ud af kolberne. Dernæst startes testen ved, at kolberne anbringes i rysteapparatet.
II.2.5. Antal kolber i en typisk testrække
Kolbe 1 og 2: testopslæmning
Kolbe 3 og 4: inoculumblind
Kolbe 5: procedurekontrol
anbefalet og om nødvendigt:
Kolbe 6: abiotisk steril kontrol
Kolbe 7: adsorptionskontrol
Kolbe 8: toksicitetskontrol
Se I.6.7.
II.2.6. Gennemførelse af testen
Under testens forløb foretages der dobbeltbestemmelse af DOC-koncentrationen i alle kolberne med bestemte tidsintervaller og så ofte, at både i 10-dages-vinduets start og den procentvise fjernelse ved 10-dages-vinduets afslutning kan konstateres. Der udtages kun netop så meget af testopslæmningen, som er nødvendigt til analysen.
Inden prøveudtagningen kompenseres der for eventuelt fordampningstab fra kolberne ved tilsætning af vand (I.6.1). Inden prøven udtages, blandes kulturmediet omhyggeligt, idet det påses, at materiale på kolbens sider først bringes i opløsning eller opslæmning. Der centrifugeres eller filtreres på membranfilter (se bilag II, punkt 4) straks efter prøveudtagningen. Den filtrerede eller centrifugerede prøve analyseres samme dag; ellers kan den opbevares i højst 48 timer ved 2-4 oC eller i længere tid ved højst – 18 oC.
II.3. DATA OG RAPPORTERING
II.3.1. Behandling af resultaterne
Den procentvise nedbrydning til tidspunktet t beregnes som anført under I.7.1 (DOC-bestemmelse) og eventuelt som under I.7.2 (specifik analyse).
Alle resultater noteres på de tilhørende skemaer.
II.3.2. Resultaternes gyldighed
Se I.5.2.
II.3.3. Rapportering
Se I.8.
II.4. RESULTATSKEMA
Nedenfor er vist et eksempel på et resultatskema.
DOC-ELIMINERING
|
1. |
LABORATORIUM |
|
2. |
DATO VED TESTENS BEGYNDELSE |
3. TESTSTOF
Navn:
Stamopløsningens koncentration: … mg/l stof
Begyndelseskoncentration i mediet, t0: … mg/l stof
4. INOCULUM
Kilde:
Behandling:
Eventuel forkonditionering:
Koncentrationen af opslæmmet tørstof i reaktionsblandingen: … mg/l
5. KULSTOFBESTEMMELSER
Kulstofanalysator:
|
|
Kolbe nr. |
|
DOC efter n dage (mg/l) |
||||
|
0 |
n1 |
n2 |
n3 |
nx |
|||
|
Testkemikalie plus inoculum |
1 |
a1 |
|
|
|
|
|
|
a2 |
|
|
|
|
|
||
|
a, gnsn. Ca(t) |
|
|
|
|
|
||
|
2 |
b1 |
|
|
|
|
|
|
|
b2 |
|
|
|
|
|
||
|
b, gnsn. Cb(t) |
|
|
|
|
|
||
|
Inoculumblindprøve uden testkemikalie |
3 |
c1 |
|
|
|
|
|
|
c2 |
|
|
|
|
|
||
|
c, gnsn.Cc(t) |
|
|
|
|
|
||
|
4 |
d1 |
|
|
|
|
|
|
|
d2 |
|
|
|
|
|
||
|
d, gnsn. Cd(t) |
|
|
|
|
|
||
|
|
|
|
|
|
|
||
6. VURDERING AF RÅDATA
|
Kolbe nr. |
|
% nedbrydning efter n dage |
||||
|
0 |
n1 |
n2 |
n3 |
nx |
||
|
1 |
|
0 |
|
|
|
|
|
2 |
|
0 |
|
|
|
|
|
Gnsn. (3) |
|
0 |
|
|
|
|
Anmærkning: Der kan benyttes tilsvarende formler for referencekemikaliet og toksicitetskontrolprøverne.
7. ABIOTISK KONTROL (frivillig)
|
|
Tid (dage) |
|
|
0 |
t |
|
|
DOC-koncentration (mg/l) i sterilkontrol |
Cs(0) |
Cs(t) |

8. SPECIFIK KEMISK ANALYSE (frivillig)
|
|
Restmængde af teststof ved afslutning af test (mg/l) |
% primær nedbrydning |
|
Sterilkontrol |
Sb |
|
|
Inoculerer testmedium |
Sa |
|
DEL III. MODIFICERET 0ECD-SCREENINGTEST (Metode C.4-B)
III.1. METODENS PRINCIP
Til en afmålt mængde uorganisk medium indeholdende teststof i kendt koncentration (10-40 mg/l) som den nominelle eneste organisk kulstofkilde, tilsættes der 0,5 ml spildevandsinoculum pr. liter. Blandingen beluftes i mørke eller dæmpet lys ved 22 ± 2 oC.
Nedbrydningen følges ved hyppig DOC-analyse i en periode på 28 dage. Bionedbrydningsgraden beregnes ved at udtrykke koncentrationen af fjernet DOC (korrigere' for koncentrationen i inoculumblindprøven) i procent af startkoncentrationen af DOC. Den primære bionedbrydning kan også beregnes ud fra supplerende kemisk analyse ved inkubationsperiodens begyndelse og afslutning.
III.2. BESKRIVELSE AF METODEN
III.2.1. Apparatur
|
a) |
Koniske kolber, f.eks. 250 ml op til 2 1, afhængigt af det volumen, der kræves til DOC-analyse |
|
b) |
Rysteapparat til de koniske kolber, enten med automatisk temperaturkontrol eller anbragt i et lokale med konstant temperatur, og tilstrækkelig kraftigt til, at forholdene holdes aerobe i alle kolberne |
|
c) |
Filtreringsapparatur med passende membraner |
|
d) |
DOC-analysator |
|
e) |
Apparatur til bestemmelse af opløst oxygen |
|
f) |
Centrifuge. |
III.2.2. Fremstilling af uorganisk medium
Se I.6.2 om fremstilling af stamopløsning.
10 ml af opløsning (a) blandes med 800 ml fortyndingsvand, og af opløsning (b), (c) og (d) tilsættes der 1 ml af hver, hvorefter der fyldes op til 1 l med fortyndingsvand.
I denne metode anvendes der kun 0,5 ml spildevand pr. liter som.inoculum, og derfor kan det være nødvendigt at tilsætte yderligere sporstoffer og vækstfaktorer til mediet. Dette gøres ved tilsætning af 1 ml af hver af følgende opløsninger pr, liter medium (slutvolumen).
Opløsning af sporstoffer:
|
Mangansulfat tetrahydrat, MnSO4 4H,0 |
39,9 mg |
|
Borsyre, H3BO4 |
57,2 mg |
|
Zinksulfat heptahydrat, ZnSO4 7H2O |
42,8 mg |
|
Ammoniumheptamolybdat (NH4)6Mo7O24 |
34,7 mg |
|
Jernchelat (FeCl3 ethylendiamintetraeddikesyre) |
100,0 mg |
|
opløses i vand og fortyndes til 1 000 ml. |
|
|
Vitaminopløsning: |
|
|
Gærekstrakt |
15,0 mg |
opløses i 100 ml vand. Der steriliseres ved filtrering igennem 0,2 μm membranfilter, hvis opløsningen friskfremstilles hver gang.
III.2.3. Fremstilling og forkonditionering af inoculum
Inokulum tages fra det sekundære afløb fra et spildevandsanlæg eller et laboratorieanlæg, som fortrinsvis modtager husholdningsspildevand. Se I.6.4.2 og I.6.5.
0,5 ml pr. liter uorganisk næringsstofopløsning benyttes.
III.2.4. Klargøring af kolberne
Eksempelvis kan der overføres 800 ml portioner af uorganisk medium til koniske 2-liters kolber, hvorefter der tilsættes så meget af stamopløsningerne af test- og referencestof til de forskellige kolber, at kemikaliekoncentrationen bliver på 10-40 mg DOC pr, liter. Kontroller pH-værdier og juster om nødvendigt til 7,4. Der tilsættes 0,5 ml spildevand pr. liter (se I.6.4.2.) til kolberne som inoculum. Der klargøres også inoculumkontrolprøver i uorganisk medium uden test- og referencekemikalier.
Om nødvendigt kan der medtages en kolbe til kontrol af, om testkemikaliet virker hæmmende; der tilsættes omtrent lige store mængder test- og referencekemikalie til uorganisk medium indeholdende inoculum.
Der kan også om nødvendigt inkluderes en steril kolbe, dvs. en kolbe uden inoculum, til kontrol af, om testkemikaliet nedbrydes abiotisk (se I.6.6).
Hvis testkemikaliet mistænkes for i væsentlig grad at kunne adsorberes til glas, slampartikler eller lign., foretages desuden en indledende vurdering af denne adsorption og dermed af testens egnethed for det pågældende testkemikalie (se tabel 1). Benyt en kolbe indeholdende teststoffet, inoculum og det steriliserende stof.
Alle kolber fyldes op til 1 liter med uorganisk medium, og efter blanding udtages der en prøve af hver kolbe til bestemmelse af startkoncentrationen af DOC (se tillæg II, punkt 4). Kolbernes åbning tildækkes, f.eks. med aluminiumfolie, på en sådan måde, at luften frit kan passere ind og ud af kolberne, Dernæst startes testen ved, at kolberne anbringes i rysteapparatet,
III.2.5. Antal kolber i en typisk testrække
Kolbe 1 og 2: testopslæmning
Kolbe 3 og 4: inoculumblind
Kolbe 5: procedurekontrol
anbefalet og om nødvendigt:
Kolbe 6: abiotisk steril kontrol
Kolbe 7: adsorptionskontrol
Kolbe 8: toksicitetskontrol
Se I.6.7.
III.2.6. Gennemførelse af testen
Under testens forløb foretages der dobbeltbestemmelse af DOC-koncentrationen i alle kolberne med bestemte tidsintervaller og så ofte, at både 10-dages vinduets start og den procentvise fjernelse ved 10-dages-vinduets afslutning kan konstateres. Der udtages kun netop så meget af testopslæmningen, som er nødvendig til analysen.
Inden prøveudtagningen kompenseres der for eventuelt fordampningstab fra kolberne ved tilsætning af fortyndingsvand (I.6.1.). Inden prøven udtages, blandes kulturmediet omhyggeligt, idet materiale på kolbens sider først bringes i opløsning eller opslæmning. Der centrifugeres eller filtreres på membranfilter (se bilag II.4.) straks efter prøveudtagningen. Den filtrerede eller centrifugerede prøve analyseres samme dag; ellers kan den opbevares i højst 48 timer ved 2-4 oC eller i længere tid ved højst – 18 oC.
ΙII.3. DATA OG RAPPORTERING
III.3.1. Behandling af resultaterne
Den procentvise nedbrydning til tidspunktet t beregnes som anført under I.7.1. (DOC-bestemmelse) og eventuelt som under I.7.2. (specifik analyse).
Alle resultater noteres på de tilhørende skemaer.
III.3.2. Resultaternes gyldighed
Se I.5.2.
III.3.3. Rapportering
Se I.8.
IIΙ.4. RESULTATSKEMA
Nedenfor er vist et eksempel på et resultatskema.
MODIFICERET OECD-SCREENINGTEST
|
1. |
LABORATORIUM |
|
2. |
DATO VED TESTENS BEGYNDELSE |
3. TESTSTOF
Navn:
Stamopløsningens koncentration: … mg/l stof
Begyndelseskoncentration i mediet, t0: … mg/l stof
4. INOCULUM
Kilde:
Behandling:
Eventuel forkonditionering:
Koncentrationen af opslæmmet tørstof i reaktionsblandingen: … mg/l
5. KULSTOFBESTEMMELSER
Kulstofanalysator:
|
|
Kolbe nr. |
|
DOC efter n dage (mg/l) |
||||
|
0 |
n1 |
n2 |
n3 |
nx |
|||
|
Testkemikalie plus inokulum |
1 |
a1 |
|
|
|
|
|
|
a2 |
|
|
|
|
|
||
|
a, gnsn. Ca(t) |
|
|
|
|
|
||
|
2 |
b1 |
|
|
|
|
|
|
|
b2 |
|
|
|
|
|
||
|
b, gnsn. Cb(t) |
|
|
|
|
|
||
|
Inoculumblindprøve uden testkemikalie |
3 |
C1 |
|
|
|
|
|
|
C2 |
|
|
|
|
|
||
|
C, gnsn. Cc(t) |
|
|
|
|
|
||
|
4 |
d1 |
|
|
|
|
|
|
|
d2 |
|
|
|
|
|
||
|
d, gnsn. Cd(t) |
|
|
|
|
|
||
|
|
|
|
|
|
|
||
6. VURDERING AF RÅDATA
|
Kolbe nr. |
|
% nedbrydning efter n dage |
||||
|
0 |
n1 |
n2 |
n3 |
nx |
||
|
1 |
|
0 |
|
|
|
|
|
2 |
|
0 |
|
|
|
|
|
Gnsn. (4) |
|
0 |
|
|
|
|
Anmærkning: Der kan benyttes tilsvarende formler for referencekemikaliet og toksicitetskontrolprøverne.
7. ABIOTISK KONTROL (frivillig)
|
|
Tid (dage) |
|
|
0 |
t |
|
|
DOC-koncentration (mg/l) i sterilkontrol |
Cs(o) |
C s(t) |

8. SPECIFIK KEMISK ANALYSE (frivillig)
|
|
Restmængde af teststof ved afslutning af test |
% primær nedbrydning |
|
Sterilkontrol |
Sb |
|
|
Inoculeret testmedium |
Sa |
|
DEL IV. CO2-UDVIKLING (Metode C.4-C)
IV.1. METODENS PRINCIP
En afmålt mængde uorganisk medium indeholdende inoculum og, som den nominelle eneste organiske kulstofkilde, teststof i kendt koncentration (10-20 mg DOC eller TOC/1) beluftes ved gennemledning af kuldioxidfri luft med kontrolleret hastighed i mørke eller i diffust lys. Nedbrydningen følges gennem 28 dage ved bestemmelse af den dannede mængde kuldioxid, idet den opsamles i barium- eller natriumhydroxid og måles enten ved titrering af det resterende hydroxid eller som uorganisk kulstof. Den mængde kuldioxid, som er udviklet af testkemikaliet (korrigeret for inoculumblindværdien), udtrykkes i procent af ThCO2. Graden af bionedbrydning kan også beregnes ud fra supplerende DOC-analyser ved inkubationsperiodens begyndelse og afslutning.
IV.2. BESKRIVELSE AF METODEN
IV.2.1. Apparatur
|
a) |
Kolber, 2-5 l, alle forsynet med beluftningsrør, der næsten når ned til bunden af beholderen, og afgangsåbning |
|
b) |
Magnetomrørere, når der testes tungtopløselige kemikalier |
|
c) |
Gasabsorptionsflasker |
|
d) |
Udstyr til styring og måling af luftgennemstrømning |
|
e) |
Apparatur til udvaskning af kuldioxid, til fremstilling af kuldioxidfri luft; alternativt kan der benyttes CO2-fri oxygen og CO2-fri nitrogen på stålflaske i det rette blandingsforhold (20 % O2 og 80 % N2) |
|
f) |
Udstyr til bestemmelse af kuldioxid, enten titrimetrisk eller ved en eller anden form for analysator for uorganisk kulstof |
|
g) |
Membranfiltreringsapparatur (valgfrit) |
|
h) |
DOC-analysator (valgfri). |
IV.2.2. Fremstilling af uorganisk medium
Se I.6.2 om fremstilling af sumopløsning.
10 ml af opløsning (a) blandes med 800 ml fortyndingsvand, og af opløsning (b), (c) og (d) tilsættes der 1 ml af hver, hvorefter der fyldes op til 1 l med fortyndingsvand.
IV.2.3. Fremstilling og forkonditionering af inoculum
Inoculum kan stamme fra en række forskellige kilder: aktivt slam, spildevandsafløb, overfladevand og -jord eller en blanding af disse.
Se I.6.4., I.6.4.1., I.6.4.2. og I.6.5.
IV .2.4. Klargøring af kolberne
Følgende volumen- og vægtangivelser gælder som eksempel for 5-liters kolber med 3 l opslæmning. Anvendes der mindre rumfang, tilpasses værdierne, dog skal den dannede mængde kuldioxid kunne bestemmes nøjagtigt.
Der overføres 2 400 ml uorganisk medium til hver 5-liters kolbe. Der tilsættes så meget klargjort aktivt slam (se I.6.4.1 og I.6.5), at koncentrationen af opslæmmet tørstof bliver højst 30 mg/l i de 3 liter slutblanding. Alternativt kan det klargjorte slam først fortyndes med uorganisk medium til en opslæmning indeholdende 500-1 000 mg/l, hvorefter en del heraf tilsættes til 5-liters kolbens indhold til en koncentration på 30 mg/l; dette giver større nøjagtighed. Der kan benyttes andre inoculumkilder (se I.6.4.2).
Disse blandinger beluftes natten over med CO2-fri luft, så al kuldioxiden drives ud af systemet.
Test- og referencestof tilsættes hver for sig som v.h.a. kendte mængder af stamopløsninger til replikakolberne, så der opnås en koncentration på 10-20 mg DOC eller TOC pr. liter; til nogle af kolberne tilsættes der ingen kemikalier, og de fungerer som inoculumkontrolprøver. Tungtopløselige teststoffer tilsættes direkte til kolberne på vægt- eller volumenbasis, eller håndteres som beskrevet i bilag III.
Om nødvendigt kan der medtages en kolbe til kontrol af, om testkemikaliet virker hæmmende; der tilsættes både test- og referencekemikalie til samme koncentration som i de andre kolber.
Der kan også om nødvendigt inkluderes en steril kolbe, dvs. en kolbe uden inoculum, til kontrol af, om testkemikaliet nedbrydes abiotisk (se I.6.6}. Steriliser ved tilsætning af et giftigt stof i en passende koncentration.
1 alle kolberne fyldes der op til et opslætnningsvolumen på 3 l ved tilsætning af uorganisk medium, der i forvejen er gennemluftet med CO2-fri luft. Der kan eventuelt udtages prøver til DOC-analyse (se bilag II.4) og/eller specifik analyse. Absorptionsflaskerne forbindes til kolbernes afgangsåbning.
Hvis der anvendes bariumhydroxid, serieforbindes der 3 absorptionsflasker med hver 100 ml 0,0125 M bariumhydroxidopløsning til hver 5-liters kolbe. Opløsningen skal være fri for bundfald af sulfat og carbonat, og dens styrke skal bestemmes umiddelbart før brugen. Anvendes der natriumhydroxid forbindes der 2 fælder, hvor den sidste fungerer som kontrol af, at al kuldioxiden er absorberet i den første. Absorptionsflasker med serumflaskelukker er egnede. Der anbringes 200 ml 0,05 M natriumhydroxid i hver flaske, hvilket er tilstrækkeligt til at absorbere hele den mængde kuldioxid, der udvikles, når testkemikaliet nedbrydes fuldstændigt. Selv en friskfremstillet natriumhydroxidopløsning vil altid indeholde spor af carbonat; dette korrigeres der for ved subtraktion af blindprøvens carbonatindhold.
IV.2.5. Antal kolber i en typisk testrække
Kolbe 1 og 2: testopslæmning
Kolbe 3 og 4: inoculumblind
Kolbe 5: procedurekontrol
anbefalet og om nødvendigt:
Kolbe 6: abiotisk steril kontrol
Kolbe 7: toksicitetskontrol
Se I.6.7.
IV.2.6. Gennemførelse af testen
Testen indledes ved gennembobling af opslæmningerne med 30-100 ml CO2-fri luft pr. minut. Der udtages regelmæssigt prøver af kuldioxidabsorbenten til analyse for CO2-indhold. Det anbefales, at der i de første 10 dage foretages analyser hver anden eller tredje dag og derefter hver femte dag indtil den 28. dag, således at 10-dages-vinduet kan identificeres.
På den 28. dag udtages der (eventuelt) prøver til DOC-analyse og/eller specifik analyse, opslæmningernes pH måles, og der tilsættes 1 ml koncentreret saltsyre til hver kolbe; kolberne beluftes natten over, så al kuldioxid drives ud af testopslæmningerne. På den 29. dag foretages den sidste analyse af udviklet kuldioxid.
På de dage, hvor CO2 bestemmes, fjernes den bariumhydroxidabsorptionsflaske, der er nærmest kolben, og indholdet titreres med 0,05 M HCl med phenolphthalein som indikator. De øvrige absorptionsflasker rykkes én plads nærmere kolben, og der anbringes en ny flaske indeholdende 100 ml friskfremstillet 0,0125 M bariumhydroxyd bagest i serien. Titreringerne foretages efter behov, f.eks. når der er betragteligt bundfald i den første flaske og intet synligt i den anden, dog mindst én gang ugentligt. Anvendes der NaOH som absorptionsmiddel, udtages der med en injektionssprøjte en lille prøve (afhængigt af den anvendte kulstofanalysator) af natriumhydroxidopløsningen i den flaske, der er nærmest ved kolben. Prøven sprøjtes ind i IC-delen af kulstofanalysatoren til direkte analyse af den udviklede kuldioxid.
Indholdet af den anden absorptionsflaske analyseres først ved testens afslutning, så der kan korrigeres for eventuel overførsel af kuldioxid.
IV.3. DATA OG RAPPORTERING
IV.3.1. Behandling af resultaterne
Den absorberede CO2-mængde er efter titrering givet ved:
mg CO2 = [(100 × CB – 0,5 × V × CA)] × 44
hvor:
|
V |
= |
den mængde HCl, der forbruges til titrering af de 100 ml absorptionsmiddel (ml) |
|
CB |
= |
bariumhydroxidopløsningens koncentration (M) |
|
CA |
= |
saltsyreopløsningens koncentration (M). |
Hvis CB = 0,0125 M og CA = 0,05 M, skal der anvendes 50 ml til titrering af 100 ml bariumhydroxid, og CO2-mængden er givet ved:

I dette tilfælde skal der altså anvendes en faktor på 1,1 ved omregning fra det rumfang HCl, der medgår til titrering, til det antal mg CO2, der er udviklet.
Den vægtmængde CO2, der dannes af inoculum alene og af inoculum plus testkemikalie, beregnes ud fra de pågældende titreringsværdier, og forskellen er den vægtmængde CO2, som testkemikaliet alene udvikler.
Hvis f.eks. inoculum alene kræver 48 ml til titreringen og inoculum plus testkemikalie kræver 45 ml, fås
CO2 fra inoculum = 1,1 × (50-48) = 2,2 rng
CO2 fra inoculum plus testkemikalie = 1,1 × (50-45) = 5,5 mg
og den vægtmængde CO2, som testkemikaliet har udviklet, bliver altså 3,3 mg.
Den procentvise bionedbrydning beregnes ved:
![]()
eller
![]()
idet 3,67 er omregningsfaktoren (44/12) fra kulstof til kuldioxid.
Den procentvise nedbrydning efter et givet tidsrum beregnes ved summering af de enkelte procentdele af ThCO2, der er målt indtil det pågældende tidspunkt.
Anvendes der natriumhydroxid som absorptionsmiddel, beregnes den udviklede CO2- mængde, udtrykt som IC (mg), ved multiplikation af absorptionsmidlets IC- koncentration med dets volumen.
Den procentvise nedbrydning beregnes ved:
![]()
Det fjernede DOC beregnes (eventuelt) som beskrevet under I.7. Dette og alle øvrige resultater noteres på de tilhørende skemaer.
IV.3.2. Resultaternes gyldighed
IC-indholdet i testkemikalieopslæmningen i uorganisk medium ved testens begyndelse skal være mindre end 5 % af TC, og den udviklede CO2-mængde i inoculumblindprøven må ved testens afslutning normalt ikke overstige 40 mg pr. liter medium. Hvis der findes værdier på over 70 mg CO2 pr. liter, gennemgås data og forsøgsteknikker kritisk.
Se også I.5.2.
IV.3.3. Rapportering
Se I.8.
IV.4. RESULTATSKEMA
Nedenfor er vist et eksempel på et resultatskema.
KULDIOXIDUDVIKLING
|
1. |
LABORATORIUM |
|
2. |
DATO VED TESTENS BEGYNDELSE |
3. TESTSTOF
Navn:
Stamopløsningens koncentration: … mg/l stof
Begyndelseskoncentration i mediet: … mg/l stof
Total C tilsat til kolben: … mg C
ThCO2: … mgCO2
4. INOCULUM
Kilde:
Behandling:
Eventuel forkonditionering:
Koncentrationen af opslæmmet tørstof i reaktionsblandingen: … mg/l
5. KULDIOXIDUDVIKLING OG NEDBRYDELIGHED
Metode: Ba(OH)2/NaOH/andet
|
Tid (dage) |
udviklet CO2 test (mg) |
udviklet CO2 blind (mg) |
udviklet CO2 kumuleret (mg) (test minus blindprøve) |
% ThCO2 kumuleret |
|||||
|
1 2 |
gnsn. |
3 4 |
gnsn. |
1 |
2 |
1 |
2 |
gnsn. |
|
|
0 |
|
|
|
|
|
|
|
|
|
|
n1 |
|
|
|
|
|
|
|
|
|
|
n2 |
|
|
|
|
|
|
|
|
|
|
n3 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
28 |
|
|
|
|
|
|
|
|
|
Anmærkning: Der kan benyttes en tilsvarende opstilling for referencekemikaliet og toksicitetskontrolprøverne.
6. KULSTOFANALYSE (valgfri)
Kulstofanalysator:
|
Tid (dage) |
Blindprøve mg/l |
Testkemikalie mg/l |
|
0 |
Cb(o) |
Co |
|
28 (5) |
Cb(t) |
Ct |

7. ΑΒΙOTISΚ NEDBRYDNING (valgfri)

DEL V. MANOMETRISK RESPIROMETRI (Metode C.4-D)
V.1. METODENS PRINCIP
En afmålt mængde uorganisk medium indeholdende inoculum og, som den nominelle eneste organiske kulstofkilde, teststof i kendt koncentration (100 mg teststof pr. liter svarende til mindst 50-100 mg ThOD pr. l) omrøres i en lukket kolbe ved konstant temperatur (± 1 oC eller mindre) i op til 28 dage, Oxygenforbruget bestemmes enten ved måling af den mængde oxygen (fremstillet elektrolytisk), der kræves til opretholdelse af konstant gasvolumen i respirometerkolben, eller ud fra volumen- eller trykændringen (eller en kombination af dem) i apparatet. Udviklet kuldioxid absorberes i en kaliumhydroxidopløsning eller andet egnet absorptionsmiddel. Den oxygenmængde, testkemikaliet har optaget (korrigeret for den parallelle inoculumblindprøves optagelse), udtrykkes som procent af ThOD eller COD. Frivilligt kan primær bionedbrydning også beregnes ved supplerende specifik analyse ved begyndelsen og slutningen af inkuberingen, og fuldstændig bionedbrydning ved analyse af DOC.
V.2. BESKRIVELSE AF METODEN
V.2.1. Apparatur
|
a) |
Egnet respirometer |
|
b) |
Temperaturregulering inden for ± 1 oC eller mindre |
|
c) |
Membranfiltreringsapparatur (valgfrit) |
|
d) |
Kulstofanalysator (valgfri). |
V.2.2. Fremstilling af uorganisk medium
Se I.6.2. om fremstilling af stamopløsning.
10 ml af opløsning (a) blandes med 800 ml fortyndingsvand, og af opløsning (b), (c) og (d) tilsættes der 1 ml af hver, hvorefter der fyldes op til 1 l med fortyndingsvand.
V.2.3. Fremstilling og forkonditionering af inoculum
Inoculum kan stamme fra en række forskellige kilder: aktivt slam, spildevandsafløb, overfladevand og -jord eller en blanding af disse.
Se I.6.4., I.6.4.1., I.6.4.2. og I.6.5.
V.2.4. Klargøring af kolberne
Ud fra stamopløsningerne fremstilles der særskilte opløsninger af test- og referencestof i uorganisk medium med en kemikaliekoncentration på normalt 100 mg/l (svarende til mindst 50-100 mg ThOD pr. l).
ThOD beregnes ud fra dannelsen af ammoniumsalte, medmindre der forventes nitrifikation; i så fald baseres beregningen på nitratdannelsen (se bilag II.2).
pH måles og justeres om nødvendigt til 7,4 ± 0,2.
Tungtopløselige stoffer tilsættes på et senere tidspunkt (se nedenfor).
Hvis teststoffets toksicitet skal bestemmes, fremstilles endnu en opløsning i uorganisk medium, som indeholder både test- og referencestof i samme koncentration som i de enkelte opløsninger.
Hvis bestemmelse af den fysisk-kemiske oxygenoptagelse er påkrævet, fremstilles der en opløsning af teststof i en koncentration på normalt 100 mg ThOD pr. l som er blevet steriliseret ved tilsætningen af et passende giftigt stof (se I.6.6.).
Den nødvendige mængde af henholdsvis test- og referencestofopløsningerne overføres til mindst to kolber (dobbeltbestemmelse). Der klargøres yderligere kolber med uorganisk medium alene (inoculumblindprøver) og hvis nødvendigt med den blandede test- og referencestofopløsning og med den sterile opløsning.
Hvis teststoffet er tungtopløseligt, tilsættes det direkte på dette tidspunkt, enten på vægtbasis eller volumenbasis, eller ved håndtering som beskrevet i bilag III. Der tilsættes kaliumhydroxid, natriumhydroxidperler eller et andet absorptionsmiddel til CO2-absorbtionskamrene.
V.2.5. Antal kolber i en typisk testrække
Kolbe 1 og 2: testopslæmning
Kolbe 3 og 4: inoculumblind
Kolbe 5: procedurekontrol
anbefalet og om nødvendigt:
Kolbe 6: abiotisk steril kontrol
Kolbe 7: toksicitetskontrol
Se I.6.7.
V.2.6. Gennemførelse af testen
Beholderne henstår, indtil de har nået den ønskede temperatur, og der tilsættes klargjort aktivt slam eller et andet inoculum til de pågældende beholdere, så der opnås en koncentration af opslæmmet tørstof på højst 30 mg/l. Apparaturet samles, omrøringen sættes i gang, der kontrolleres for lufttæthed, og oxygenoptagelsesmålingen påbegyndes. Normalt kræves der ikke anden pasning end de nødvendige aflæsninger og daglig kontrol af, at temperaturen er korrekt og omrøringen tilstrækkelig.
Oxygenoptagelsen beregnes på grundlag af hyppige regelmæssige aflæsninger, foretaget efter apparaturfabrikantens anvisninger. Ved inkubationsperiodens afslutning, normalt 28 dage, måles pH i kolberne, navnlig hvis oxygenoptagelsen er meget lav eller større end ThODNH4 (for nitrogenholdige forbindelser).
Om nødvendigt udtages der før og efter inkuberingen prøver fra respirometerkolberne til DOC-analyse eller specifik kemisk analyse (se bilag II.4). Man skal sørge for, at rumfanget af den testopslæmning, der er tilbage i kolben efter den første prøveudtagning, er kendt. Hvis teststoffet er N-holdigt, bestemmes koncentrationsforøgelsen af nitrit og nitrat over 28 dage, og korrektionen for oxygenforbruget ved nitrifikation beregnes (bilag V).
V.3. DATA OG RAPPORTERING
V.3.1. Behandling af resultaterne
Testkemikaliets oxygenoptagelse (mg) efter et givet tidsrum (korrigeret for inoculumblindprøvens oxygenoptagelse i samme tidsrum) divideres med vægten af den anvendte teststofmængde. Herved fremkommer BOD udtrvkt i mg oxygen pr. mg testsrof. dvs.

= mg O2 pr. mg teststof.
Den procentvise bionedbrydning beregnes enten ved:

eller
![]()
Det bemærkes, at de to metoder ikke nødvendigvis giver samme resultat; førstnævnte metode bør foretrækkes.
Der benyttes den ThOD (NH4 eller NO3), der svarer til det foreliggende kendskab til nitrifikation (bilag II.2). Sker der ufuldstændig nitrifikation beregnes korrektionen for oxygenforbruget til nitrifikationen ud fra ændringerne i nitrit- og nitratkoncentrationen (bilag V).
Hvis det vælges at foretage bestemmelser af organisk kulstof og/eller af et specifikt kemikalie, beregnes den procentvise nedbrydning som beskrevet under punkt I.7.
Alle resultater noteres på de tilhørende skemaer.
V.3.2. Resultaternes gyldighed
Inoculumblindprøvens oxygenoptagelse er normalt 20-30 mg O2/l på 28 dage og bør ikke være større end 60 mg/l. Findes der værdier over 60 mg/l, må data og forsøgsteknikker gennemgås kritisk. Hvis pH ligger uden for intervallet 6-8,5 og teststoffets oxygenoptagelse er mindre end 60 %, gentages testen med lavere teststofkoncentration.
Se også I.5.2.
V.3.3. Rapportering
Se I.8.
V.4. RESULTATSKEMA
Nedenfor er vist et eksempel på et resultatskema.
MANOMETRISK RESPIROMETRI
|
1. |
LABORATORIUM |
|
2. |
DATO VED TESTENS BEGYNDELSE |
3. TESTSTOF
Navn:
Stamopløsningens koncentration: … mg/l
Begyndelseskoncentration i mediet, Co: … mg/l
Volumen i testkolbe (V): … ml
ThOD eller COD: … mg O2/mg teststof (NH4, NO3)
4. INOCULUM
Kilde:
Behandling:
Eventuel forkonditionering:
Koncentrationen af opslæmmet tørstof: reaktionsblandingen: … mg/l
5. OXYGENOPTAGELSE: BIONEDBRYDELIGHED
|
|
Tid (dage) |
||||||||||||
|
0 |
|
7 |
|
14 |
|
|
21 |
|
|
28 |
|
||
|
O2 opt. (mg) teststof |
1 |
|
|
|
|
|
|
|
|
|
|
|
|
|
2 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
a, gnsn. |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
O2 opt. (mg) blindprøve |
3 |
|
|
|
|
|
|
|
|
|
|
|
|
|
4 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
b, gnsn. |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Korrigeret BOD (mg) |
(a1 – bm) |
|
|
|
|
|
|
|
|
|
|
|
|
|
(a2 – bm) |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
BOD pr. mg teststof |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
% nedbrydning edbrydning
|
D1 (a1) |
|
|
|
|
|
|
|
|
|
|
|
|
|
D2 (a2) |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Gnsn. (6) |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
V = volumen af medium i testkolben. |
|||||||||||||
Anmærkning: Der kan anvendes en tilsvarende opstilling for reference kemi kaliet og toksicitetskontrolprøveme.
6. KORREKTION FOR NITRIFIKATION (se bilag V)
|
Dag |
0 |
28 |
forskel |
||
|
|
|
(N) |
||
|
— |
— |
|
||
|
|
|
(N) |
||
|
— |
— |
|
||
|
— |
— |
|
7. KULSTOFANALYSE (valgfri)
Kulstofanalysator:
|
Tid (dage) |
Blindprøve mg/l |
Testkemikalie mg/l |
|
0 |
(Cblo) |
(Co) |
|
28 (7) |
(Cblt) |
(Ct) |
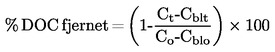
8. SPECIFIK KEMIKALIEBESTEMMELSE (valgfri)
|
Sb |
= |
koncentrationen af testkemikalie i steril kontrolkolbe efter 28 dage |
|
Sa |
= |
koncentrationen af testkemikalie i kolbe med inoculum efter 28 dage |

9. ABIOTISK NEDBRYDNING (valgfri)
|
a |
= |
oxygenforbruget i sterile kolber efter 28 dage (mg) |

(se afsnit 1 og 3)

DEL VI. CLOSED BOTTLE TEST (metode C.4-E)
VI.1. TESTMETODENS PRINCIP
Til en sædvanligvis 2-5 mg/l opløsning af teststoffet i uorganisk medium tilsættes der inoculum bestående af et forholdvis lille antal mikroorganismer fra en blandet population, og opløsningen opbevares i mørke ved konstant temperatur i helt fyldte lukkede kolber. Nedbrydningen følges gennem 28 dage ved analyse af opløst oxygen. Den oxygenmængde, som teststoffet har forbrugt, korrigeres for den oxygenmængde, der er forbrugt i den parallelle inoculumblindprøve, og udtrykkes i procent af ThOD eller COD.
VI.2. BESKRIVELSE AF METODEN
VI.2.1. Apparatur
|
a) |
BOD-kolber med glasprop, f.eks. 250-300 ml |
|
b) |
vandbad eller inkuberingsapparat, som kan holde kolberne ved konstant temperatur (± 1 oC eller mindre) i mørke |
|
c) |
store glasflasker (2-5 1) til fremstilling af medium og opfyldning af BOD-kolberne |
|
d) |
oxygenelektrode og -måler eller udstyr og reagenser til Winkler-titrering. |
VI.2.2. Fremstilling af uorganisk medium
Se I.6.2 om fremstilling af stamopløsning.
1 ml af opløsning (a), (b), (c) og (d) fortyndes til 1 liter med vand.
VI.2.3. Fremstilling af inoculum
Inoculum tages normalt fra det sekundære afløb fra et spildevandsanlæg eller et laboratorieanlæg, som fortrinsvis modtager husholdningspildevand. En alternativ inoculumkilde er overfladevand. Brug normalt fra en dråbe (0,05 ml) til 5 ml filtrat pr. liter medium. Afprøvninger kan være nødvendige for at klarlægge det optimale volumen for et givet afløb (se I.6.4.2 og I.6.5).
VI.2.4. Klargøring af kolberne
Det uorganiske medium beluftes kraftigt i mindst 20 minutter. En given forsøgsrække udføres med uorganisk medium fra samme batch. Normalt er mediet klar til brug efter henstand i 20 timer ved testtemperaturen. Som kontrol bestemmes koncentrationen af opløst oxygen; værdien skal ligge omkring 9 mg/l ved 20 oC. Al overføring og opfyldning med det luftmættede medium udføres, uden at der dannes bobler, f.eks. med en hævert.
Der klargøres parallelle grupper af BOD-kolber til samtidige forsøg med test- og referencestof. Der samles så mange BOD-kolber, inklusive inoculumblindprøver, at der mindst kan foretages dobbeltbestemmelse af oxygenforbruget med den ønskede hyppighed, f.eks. efter 0, 7, 14, 21 og 28 dage. Det kan være nødvendigt at anvende flere kolber, hvis man vil være sikker på at kunne identificere 10-dages-vinduet.
De store kolber fyldes en tredjedel op med fuldt beluftet uorganisk medium. Dernæst tilsættes der så meget stamopløsning af test- og referencestof til forskellige kolber, at deres slutkoncentration normalt ikke bliver større end 10 mg/l. Til en særskilt kolbe med kontrolmedium tilsættes der ingen kemikalier.
For at være sikker på, at inoculumaktiviteten ikke begrænses, må koncentrationen af opløst oxygen ikke falde til under 0,5 mg/l i BOD-kolberne, hvilket begrænser testkemikaliets koncentration til ca. 2 mg/l. Af tungt nedbrydelige stoffer og stoffer med lavt ThOD kan der dog anvendes 5-10 mg/l. I visse tilfælde tilrådes det at køre parallelle serier med teststof i to forskellige koncentrationer, f.eks. 2 og 5 mg/l. Normalt beregnes ThOD på basis af dannelsen af ammoniumsalte, men hvis der forventes eller vides at ske nitrifikation, beregnes værdien på grundlag af nitratdannelsen (ThODNO3: se bilag II.2). Sker der ufuldstændig nitrifikation korrigeres der for de ændringer i nitrit- og nitratkoncentrationen, der er bestemt ved analyse (se bilag V).
Endnu en serie kolber er påkrævet, hvis teststoffets toksicitet skal undersøges (f.eks. hvis der tidligere er fundet en lav bionedbrydelighedsværdi).
Hertil klargøres der endnu en stor kolbe med beluftet uorganisk medium (ca. en tredjedel fuld) plus teststof og referencestof normalt i samme slutkoncentrationer som i de andre kolber.
Der tilsættes inoculum til kolberne i form af sekundært afløbsvand (fra 1 dråbe (ca. 0,05 ml) til 5 ml pr. liter) eller f.eks. flodvand (se I.6.4.2). Endelig fyldes der op med beluftet uorganisk medium ved hjælp af en slange, der når helt ned til bunden, så det bliver tilstrækkelig omrøring.
VI.2.5. Antal kolber i en typisk testrække
I en rypisk testrække anvendes følgende antal kolber:
|
— |
mindst 10 med teststof og inoculum (restopslæmning) |
|
— |
mindst 10 kun med inoculum (inoculumblindprøve) |
|
— |
mindst 10 med referencestof og inoculum (procedurekontrol) |
|
— |
og i givet fald 6 kolber med teststof, referencestof og inoculum (toksicitetskontrol). Til sikker identifikation af 10-dages-vinduet kræves der dog det dobbelte antal kolber. |
VI.2.6. Gennemførelse af testen
De fremstillede opløsninger fordeles straks i de pågældende grupper af BOD-kolber, idet de suges op fra den nederste fjerdedel af den passende store kolbe (ikke bunden) med en slange, og således at alle BOD-kolberne fyldes helt. Eventuelle luftbobler fjernes ved en let banken på kolbens side. Starttidskolberne analyseres straks for opløst oxygen enten ved Winklers metode eller med elektrode. Indholdet i kolberne kan opbevares til senere analyse efter Winklers metode, hvis der straks tilsættes mangan(II)sulfat og natriumhydroxid (det første Winkler- reagens). De omhyggeligt tilproppede kolber, hvor oxygenet er fikseret som brunt hydratiseret mangan(III)oxid, opbevares i mørke ved 10-20 oC i højst 24 timer, inden Winkler-testen gøres færdig. De resterende kolber tiilproppes, idet man sørger for, at der ikke indesluttes nogen luftbobler, og de inkuberes ved 20 oC i mørke. Parallelt med hver serie skal der være en fuldstændig serie til bestemmelse af inoculumblindværdien. Under de 28 dages inkubering udtages der med bestemte mellemrum (mindst en gang om ugen) mindst 2 kolber af hver serie til analyse for opløst oxygen.
Med ugentlig prøveudtagning skulle den procentvise fjernelse kunne bedømmes i et 14-dages-vindue, mens der ved prøveudtagning hver tredje til fjerde dag, hvilket kræver omtrent det dobbelte antal kolber, bliver mulighed for identificering af 10-dages-vinduet.
For N-holdige stoffers vedkommende må der korrigeres for oxygenforbrug ved eventuel nitrifikation. Hertil anvendes der en O2-elektrode til bestemmelse af koncentrationen af opløst oxygen, hvorefter der udtages en prøve fra BOD-kolben til analyse for nitrit og nitrat. Den forbrugte oxygenmængde beregnes på grundlag af forøgelsen i nitrit- og nitratkoncentrationen (se bilag V).
V.3. DATA OG RAPPORTERING
VI.3.1. Behandling af resultaterne
Først beregnes BOD efter hvert tidsinterval ved at trække oxygentabet (mg O2 pr. l) i inoculumblindprøven fra oxygentabet i kolben med teststof. Dette korrigerede oxygentab divideres med teststoffets koncentration (mg/1), hvorved den specifikke BOD fremkommer i mg oxygen pr. mg teststof. Den procentvise bionedbrydelighed beregnes ved at dividere den specifikke BOD med den specifikke ThOD (beregnet som anført i bilag II.2) eller COD (bestemt ved analyse, se bilag II.3), dvs.:

= mg O2 pr. mg teststof.
![]()
eller
![]()
Det bemærkes, at de to metoder ikke nødvendigvis giver samme resultat; førstnævnte metode foretrækkes.
Der benyttes den ThOD (NH4 eller ΝΟ3), der svarer til det foreliggende kendskab til nitrifikation (bilag II.2). Sker der ufuldstændig nitrifikation beregnes korrektionen for oxygenforbruget til nitrifikationen ud fra ændringerne i nitrit- og nitratkoncentrationen (bilag V).
VI.3.2. Resultaternes gyldighed
Inoculumblindprøvens oxygentab bør ikke være større end 1,5 mg opløst oxygen pr. liter efter 28 dage. Findes der højere værdier, må forsøgsteknikkerne gennemgås kritisk. Restkoncentrationen af oxygen i testkolberne må ikke falde til under 0,5 mg/l på noget tidspunkt. Målinger af så lavt oxygenindhold er kun gyldige, hvis den anvendte målemetode for opløst oxygen tillader nøjagtig måling af så lave indhold.
Se også I.5.2.
VI.3.3. Rapportering
Se I.8.
VI.4. RESULTATSKEMA
Nedenfor er vist et eksempel på et resultatskema.
CLOSED BOTTLE TEST
|
1. |
LABORATORIUM |
|
2. |
DATO VED TESTENS BEGYNDELSE |
3. TESTSTOF
Navn:
Stamopløsningens koncentration: … mg/l
Begyndelseskoncentration i kolbe: … mg/l
ThOD eller COD: … mg O2/mg teststof
4. INOCULUM
Kilde:
Behandling:
Eventuel forkonditionering:
Koncentrationen i reaktionsblandingen: … ml/l
5. DO-BESTEMMELSE
Metode: Winkler/elektrode
Kolbeanalyser
|
Inkuberingstid (dage) |
DO (mg/l) |
|||||
|
0 |
n1 |
n2 |
|
|||
|
Blindprøve (uden teststof) |
1 |
C1 |
|
|
|
|
|
2 |
C2 |
|
|
|
|
|
|
Gennemsnit |
|
|
|
|
|
|
|
Teststof |
1 |
a1 |
|
|
|
|
|
|
2 |
a2 |
|
|
|
|
|
Gennemsnit |
|
|
|
|
|
|
Anmærkning: Der kan anvendes en tilsvarende opstilling for referencekemikaliet og toksicitetskontrolprøverne
6. KORREKTION FOR NITRIFIKATION (se bilag V)
|
Inkuberingstid (dage) |
0 |
n1 |
n2 |
n3 |
||
|
|
|
|
|
||
|
— |
|
|
|
||
|
— |
|
|
|
||
|
|
|
|
|
||
|
— |
|
|
|
||
|
— |
|
|
|
||
|
— |
|
|
|
7. TAB AF DO: % NEDBRYDNING
|
|
Tab efter n dage (mg/l) |
|||
|
n1 |
n2 |
n3 |
|
|
|
Kolbe 1: (mto – mtx) – (mbo – mbx) |
|
|
|
|
|
Kolbe 2: (mto – mtx) – (mbo – mbx) |
|
|
|
|
|
Kolbe 1:
|
|
|
|
|
|
Kolbe 2:
|
|
|
|
|
| % D gnsn. (8)
|
|
|
|
|
|
mto |
= |
værdi i testkolbe til tid 0 |
|
mtx |
= |
værdi i testkolbe til tid x |
|
mbo |
= |
gennemsnitsblindværdi til tid 0 |
|
mbx |
= |
gennemsnitsblindværdi til tid x |
Der foretages desuden korrektion for nitrifikation som beregnet i punkt 6.
8. BLINDPRØVER FOR TAB AF DO
Oxygenforbrug i blindprøve: (mbo – mb28) mg/l. Dette forbrug har betydning for testens gyldighed og må højst være 1,5 mg/l.
DEL VII. ΜITI-TEST (Metode C.4-F)
VII.1. METODENS PRINCIP
Oxygenoptagelsen i en omrørt opløsning eller opslæmning af teststoffet i uorganisk medium, som er tilsat inoculum i form af særligt dyrkede ikke-tilvænnede mikroorganismer, måles automatisk gennem en periode på 28 dage i et mørklagt lukket respirometer ved 25 ± 1 oC. Den udviklede kuldioxid absorberes i natriumhydroxid. Bionedbrydeligheden udtrykkes som den optagne oxygenmængde (korrigeret for blindprøvens optagelse) i procent af den teoretiske oxygenoptagelse (ThOD). Også den procentvise primære bionedbrydelighed beregnes, nemlig på grundlag af supplerende specifikke kemiske analyser ved inkuberingens begyndelse og slutning og eventuelt ved DOC-analyse.
VII.2. BESKRIVELSE AF METODEN
VII.2.1. Apparatur
|
a) |
Automatisk elektrolytisk BOD-meter eller respirometer, normalt med 6 kolber på hver 300 ml og med skåle til CO2-absorptionsmiddel |
|
b) |
Lokale med konstant temperatur og/eller vandbad på 25 ± 1 oC eller mindre |
|
c) |
Membranfiltreringsudstyr (valgfrit) |
|
d) |
Kulstofanalysator (valgfri). |
VII.2.2. Fremstilling af uorganisk medium
Følgende stamopløsninger fremstilles ud fra reagenser af analysekvalitet og vand (I.6.I):
|
a) |
monokaliumdihydrogenorthophosphat, KH2PO4 |
8,50 g |
|
dikaliummonohydrogenorthophosphat, K2HPO4 |
21,75 g |
|
|
dinatriummonohydrogenorthophosphat dodecahydrat, Na2HPO4 12 H2O |
44,60 g |
|
|
ammoniumchlorid, NH4Cl |
1,70 g |
|
|
Opløses i vand og fortyndes til 1 liter. |
|
|
|
Opløsningens pH skal være 7,2. |
|
|
|
b) |
magnesiumsulfat heptahydrat, MgSO4 7 H2O |
22,50 g |
|
Opløses i vand og fortyndes til 1 liter. |
|
|
|
c) |
calciumchlorid, vandfrit, CaCl2 |
27,50 g |
|
Opløses 1 vand og fortyndes til 1 liter. |
|
|
|
d) |
jern(III)chlorid hexahydrat, FcCl3 6 H2O |
0,25 g |
|
Opløses i vand og fortyndes til 1 liter. |
|
Der udtages 3 ml af opløsning a), b), c) og d), som fortyndes til 1 liter.
VII.2.3. Fremstilling af inoculum
Der indsamles friske prøver fra mindst 10 lokaliteter, hovedsagelig områder, hvor der anvendes og udledes forskellige kemikalier. Fra sådanne lokaliteter som spildevandsanlæg, vandløb, søer og havområder indsamles 1-liters prøver af slam, overfladejord, vand, mv., og det hele blandes grundigt. Materiale, der flyder ovenpå, fjernes, og efter henstand indstilles supernatantens pH til 7 ± 1 med natriumhydroxid og phosphorsyre.
Der benyttes en passende mængde filtrat til at fylde en »fill-and-draw« aktiv slambeholder, og væsken beluftes i ca. 23 1/2 time. 30 minutter efter at beluftningen er standset, borthældes ca. 1/3 af det samlede supernatantvolumen, og der tilsættes en lige så stor mængde af en opløsning (med pH 7), der indeholder 0,1 % glucose, 0,1 % pepton og 0,1 % monokaliumorthophosphat, og beluftningen genoptages. Dette gentages hver dag. Slamenheden drives efter god praksis: Afløbsvandet skal være klart, temperaturen skal holdes på 25 ± 2 oC, pH skal være 7 ± 1, slam skal bundfældes godt, tilstrækkelig beluftning skal opretholdes for at holde blandingen aerob hele tiden, protozoer skal være til stede, og slamaktiviteten skal testes mod referencestoffet mindst hver tredje måned. Slammet må tidligst anvendes som inoculum efter en måneds drift og ikke senere end efter fire måneders drift. Derefter udtages der regelmæssigt prøver fra mindst 10 lokaliteter hver tredje måned.
For at holde det friske og det gamle slam på samme aktivitet, blandes den filtrerede supernatant fra aktivt slam i brug med en lige så stor mængde filtreret supernatant fra en frisk indsamlet blanding fra 10 kilder, og denne væske opdyrkes som ovenfor nævnt. 18-24 timer efter at enheden er fyldt op, kan der udtages slam til brug som inoculum.
VII.2.4. Klargøring af kolber
Der klargøres 6 kolber som følger:
nr. 1: teststof i fortyndingsvand, 100 mg/l
nr. 2, 3 og 4: teststof fortyndet med uorganisk medium til 100 mg/l
nr. 5: referencestof (f.eks. anilin) fortyndet med uorganisk medium til 100 mg/l
nr. 6: rent uorganisk medium
Tungtopløselige teststoffer tilsættes direkte på vægt- eller volumenbasis eller håndteres som beskrevet i bilag III, bortset fra, at der hverken må benyttes opløsningsmidler eller emulgatorer. Der anbringes CO2-absorptionsmiddel i alle kolber i den særlige skål. pH i kolbe nr. 2, 3 og 4 justeres til 7,0.
VII.2.5. Gennemførelse af testen
Der tilsættes en lille mængde inoculum til kolbe nr. 2, 3 og 4 (testopslæmninger), nr. 5 (aktivitetskontrol) og nr. 6 (inoculumblindprøve), således at koncentrationen af opslæmmet tørstof bliver på 30 mg/l. Der tilsættes ikke inoculum til kolbe nr. 1, der tjener som abiotisk kontrol. Udstyret samles, der kontrolleres for lufttæthed, omrørerne startes, og oxygenoptagelsesmålingen startes i mørke. Temperaturen, omrøringen og den coulometriske oxygenoptagelsesskriver kontrolleres dagligt, og eventuelle farveændringer i kolberne noteres. Oxygenoptagelsen i de 6 kolber aflæses direkte v.h.a. en passende metode, f.eks. på sekskanalsskriveren, som giver en BOD-kurve. Ved inkuberingens afslutning, normalt efter 28 dage, måles pH i kolberne, og koncentrationen af resterende teststof og af eventuelle mellemprodukter og, når der er tale om vandopløselige stoffer, eventuelt DOC-koncentrationen bestemmes (bilag II.4). Der må udvises særlig omhu, når der er tale om flygtige kemikalier. Forventes der nitrifikation, bestemmes om muligt nitrat- og nitritkoncentrationen.
VII.3. DATA OG RAPPORTERING
VII.3.1. Behandling af resultaterne
Teststoffets oxygenfarbrug (mg) efter et givet tidsrum, korrigeret for oxygenoptagelsen i inoculumblindprøven efter samme tidsrum, divideres med den anvendte vægtmængde teststof. Herved fremkommer BOD udtrykt som mg oxygen pr. mg teststof, dvs.

= mg O2 pr. mg teststof.
Den procentvise bionedbrydning fremkommer af:
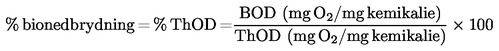
For blandinger beregnes ThOD på samme måde som for enkeltstoffer, nemlig på grundlag af grundstofanalysen. Den ThOD, der benyttes, (ThODNH4 eller ThODNO3) afhænger af, om der slet ikke sker nitrifikation eller der sker fuldstændig nitrifikation (bilag II.2). Sker der ufuldstændig nitrifikation beregnes korrektionen for oxygenforbruget til nitrifikationen ud fra ændringerne i nitrit- og nitratkoncentrationen (bilag V).
Den procentvise primære bionedbrydning beregnes ud fra den mængde af det specifikke (oprindelige) kemikalie, der forsvinder (se I.7.2).

Er der forsvundet teststof i kolbe nr. 1, som måler fysisk og kemisk nedbrydning, anføres dette, og teststoffets koncentration (Sb) i denne kolbe efter 28 dage benyttes til beregning af den procentvise bionedbrydning.
Foretages der (valgfrie) bestemmelser af DOC, beregnes den procentvise fuldstændige bionedbrydning ved:

som anført under punkt I.7.1. Er der forsvundet teststof (DOC) i kolbe nr. 1, som måler fysisk og kemisk nedbrydning, benyttes DOC-koncentrationen i denne kolbe til beregning af den procentvise bionedbrydning.
Alle resultater noteres på de tilhørende skemaer.
VII.3.2. Resultaternes gyldighed
Inoculumblindprøvens oxygenforbrug er normalt 20-30 mg O2/l på 28 dage og bør ikke være større end 60 mg/l. Findes der værdier over 60 mg/l, må data og forsøgsteknikker gennemgås kritisk. Hvis pH ligger uden for intervallet 6-8,5 og teststoffets oxygenforbrug er mindre end 60 %, gentages testen med lavere teststofkoncentration.
Se også I.5.2.
Hvis den procentvise nedbrydning af anilin beregnet ud fra oxygenforbrug ikke er større end 40 % efter 7 dage og 65 % efter 14 dage, anses testen for ugyldig.
VII.3.3. Rapportering
Se I.8.
VII.4. RESULTATSKEMA
Nedenfor er vist et eksempel på et resultatskema.
MITI (I) TEST
|
1. |
LABORATORIUM |
|
2. |
DATO VED TESTENS BEGYNDELSE |
3. TESTSTOF
Navn:
Stamopløsningens koncentration: … mg/l stof
Begyndelseskoncentration i kolbe, Co: … mg/l stof
Reaktions blandingens volumen, V: … ml
ThOD eller COD: … mg O2/l
4. INOCULUM
Lokaliteter for prøveudtagning:
|
|
||||
|
|
||||
|
|
||||
|
|
||||
|
|
Koncentration af opslæmmet tørstof i det aktive slam efter akklimatisering med syntetisk spildevand: … mg/l
Rumfang af aktivt slam pr. liter slutmedium: … ml
Slamkoncentration i slutmedium: mg/l
5. OXYGENOPTAGELSE: BIONEDBRYDEL IGHED
Respirometertype:
|
|
Tid (dage) |
||||||
|
0 |
7 |
14 |
21 |
28 |
|||
|
O2 optag, (mg) teststof |
a1 |
|
|
|
|
|
|
|
a2 |
|
|
|
|
|
||
|
a3 |
|
|
|
|
|
||
|
O2 optag, (mg) blindprøve |
b |
|
|
|
|
|
|
|
Korrigeret O2 opt. (mg) |
(a1-b) (a2-b) (a3-b) |
|
|
|
|
|
|
|
BOD pr. mg teststof |
|
Kolbe 1 |
|
|
|
|
|
|
Kolbe 2 |
|
|
|
|
|
||
|
Kolbe 3 |
|
|
|
|
|
||
|
% nedbrydning
|
|
2 3 |
|
|
|
|
|
|
2 |
|
|
|
|
|
||
|
3 |
|
|
|
|
|
||
|
gnsn. (9) |
|
|
|
|
|
||
Anmærkning: Der kan anvendes en tilsvarende opstilling for referencekemikaliet og toksicitetskontrolprøverne.
6. KULSTOFANALYSE (valgfri)
Kulstofanalysator:
|
Kolbe |
DOC |
% DOC fjernet |
Gennemsnit |
|||
|
Målt |
Korrigeret |
|||||
|
Vand + teststof |
a |
|
|
|
|
|
|
Slam + teststof |
b1 |
|
b1-c |
|
|
|
|
Slam + teststof |
b2 |
|
b2-c |
|
|
|
|
Slam + teststof |
b3 |
|
b3-c |
|
|
|
|
Kontrolblind |
c |
|
— |
|
— |
— |

7. DATA FRA SPECIFIK KEMISK ANALYSE
|
|
Restmængde af teststof ved testens afslutning |
% nedbrydning |
|
blindprøve med vand |
Sb |
|
|
medium med inoculum |
Sa1 |
|
|
Sa2 |
|
|
|
Sa3 |
|

% nedbrydning beregnes for kolbe a1, a2 og a3.
8. BEMÆRKNINGER
En kurve over BOD mod tiden skal vedlægges, hvis den foreligger.
Bilag I
FORKORTELSER OG DEFINITIONER
|
DO |
: |
Opløst oxygen (mg/l) er koncentrationen af opløst oxygen i en vandig prøve. |
|
BOD |
: |
Biokemisk oxygenforbrug (g) er den mængde oxygen, som mikroorganismer forbruger under metabolisme af teststoffet; udtrykkes også i g optaget oxygen pr. g teststof (se metode C.6). |
|
COD |
: |
Kemisk oxygenforbrug (g) er den mængde oxygen, der forbruges ved oxidation af teststoffet med varm dichromat i sur væske; det er et mål for den tilstedeværende mængde oxiderbart stof; udtrykkes også i g forbrugt oxygen pr. g teststof (se metode C.6). |
|
DOC |
: |
Opløst organisk kulstof er den mængde organisk kulstof, der er til stede i opløsning, passerer gennem et 0,45 μm filter, eller forbliver i centrifugatet efter centrifugering ved 40 000 ms-2± (4 000 g) i 15 minutter. |
|
ThOD |
: |
Teoretisk oxygenforbrug (mg) er den samlede mængde oxygen, der kræves til fuldstændig oxidation af et kemisk stof; det beregnes på grundlag af bruttoformlen (se bilag II, punkt 2) og udtrykkes også i mg oxygen pr. mg teststof. |
|
ThCO2 |
: |
Teoretisk kuldioxid (mg) er den mængde kuldioxid, der ifølge beregninger på grundlag af teststoffets kendte eller målte kulstofindhold skulle dannes ved fuldstændig nedbrydning af teststoffet; den udtrykkes også i mg udviklet kuldioxid pr. mg teststof. |
|
TOC |
: |
En prøves totale indhold af organisk kulstof er summen af organisk kulstof i opløsning og i opslæmning. |
|
IC |
: |
Uorganisk kulstof |
|
TC |
: |
En prøves totale kulstofindhold er summen af organisk og uorganisk kulstof. |
Primær bionedbrydning:
er en sådan ændring af et stofs kemiske struktur, fremkaldt ad biologisk vej, at stoffet mister sine specifikke egenskaber.
Fuldstændig bionedbrydning (aerob):
er den nedbrydningsgrad, der er nået, når mikroorganismer har opbrugt et teststof fuldstændigt under samtidig dannelse af kuldioxid, vand, uorganiske salte og nye cellebestanddele (biomasse).
Let bionedbrydelig:
kendetegner en arbitrær klasse af kemikalier, der har klaret visse specifikke screeningstest for fuldstændig bionedbrydning; disse test er så krævende, at sådanne stoffer i vandigt miljø under aerobe forhold hurtigt vil blive fuldstændigt nedbrudt.
Inherent (potentielt) bionedbrydelig:
karakteriserer en klasse af kemikalier, som ved enhver anerkendt bionedbrydelighedstest klart har vist sig at kunne omdannes ved bionedbrydning (primær eller fuldstændig).
Behandlingsmulighed:
er muligheden for at fjerne et stof ved biologisk spildevandsrensning uden at spildevandsrensningsprocessernes normale forløb forstyrres. Generelt er alle let bionedbrydelige, men ikke alle inherent bionedbrydelige, stoffer mulige at behandle. Abiotiske processer kan også spille ind.
Lag-periode:
er det tidsrum, der i en elimineringstest forløber fra inokuleringen, indtil nedbrydningsprocenten er mindst 10 %. Lag-perioden er ofte stærkt varierende og vanskeligt reproducerbar.
Nedbrydnings tid:
er tidsrummet mellem lagperiodens afslutning og det tidspunkt, hvor nedbrydningen har nået 90 % af sin maksimale værdi.
10-dages-vindue:
er den periode på 10 dage, der begynder, når nedbrydningen er nået op på 10 %.
Bilag II
BEREGNING OG BESTEMMELSE AF PASSENDE HOVEDPARAMETRE
Der vil afhængigt af den valgte metode være behov for forskellige hovedparametre. I dette afsnit beskrives, hvordan disse værdier udledes. Parametrenes anvendelse er beskrevet i de enkelte metoder.
1. Kulstofindhold
Kulstofindholdet kan enten beregnes ud fra teststoffets grundstofsammensætning elier bestemmes ved elementaranalyse.
2. Teoretisk oxygenforbrug (ThOD)
Det teoretiske oxygenforbrug (ThOD) kan beregnes, hvis grundstofsammensætningen er kendt, og ellers bestemmes ved elementaranalyse. For forbindelsen:
CcHhClclNnNanaOoPpSs
er det uden nitrifikation
 mg/mg
mg/mg
og med nitrifikation
 mg/mg
mg/mg
3. Kemisk oxygenforbrug (COD)
Det kemiske oxygenforbrug (COD) bestemmes efter metode C.6.
4. Opløst organisk kulstof (DOC)
Opløst organisk kulstof (DOC) defineres som den organiske kulstofmængde i et kemikalie eller en kemikalie-blanding, som i vandig opløsning kan passere gennem et 0,45 μm filter.
Der udtages prøver fra testbeholderne, som straks filtreres gennem filtreringsudstyret, der er forsynet med et passende membranfilter. De første 20 ml af filtratet bortkastes (ved små filtre kan denne mængde reduceres). Der opsamles en mængde på 10-20 ml til kulstofanalyse (ved indsprøjtning mindre, afhængigt af den anvendte kulstofanalysator). DOC-koncentrationen bestemmes ved hjælp af en organisk kulstofanalysator, som tillader nøjagtig bestemmelse af en kulstofkoncentration på mindre end 10 % af startkoncentrationen af DOC i testen.
Kan filtrerede prøver ikke analyseres samme arbejdsdag, kan de opbevares i højst 48 timer i køleskab ved 2-4 oC eller i længere tid ved – 18 oC.
Anmærkninger:
Membranfiltre er ofte behandlet med overfladeaktive stoffer, så de bliver mere hydrofile. De kan derfor indeholde op til flere mg opløseligt organisk kulstof, som vil gribe forstyrrende ind i bionedbrydehghedsbestemmelsen. De overfladeaktive stoffer og andre opløselige organiske stoffer fjernes fra filtrene ved kogning i demineraliseret vand i 3 gange 1 time. Derefter kan filtrene opbevares i vand i op til 1 uge. Benyttes der engangsfiltre, må hvert parti kontrolleres for afgivelse af opløseligt organisk kulstof.
På visse typer membranfiltre kan der ske adsorption af teststoffet. Det tilrådes derfor at kontrollere, at teststoffet ikke tilbageholdes på filteret.
Centrifugering ved 40 000 ms~2 (4 000 g) i 15 minutter kan træde i stedet for filtrering, når der skal skelnes mellem TOC og DOC. Metoden er ikke pålidelig ved en startkoncentration på < 10 mg DOC pr. l, da enten ikke alle bakterier fjernes eller noget af kulstoffet i bakteriernes cytoplasma genopløses.
BIBLIOGRAFI
|
— |
Standard Methods for the Examination of Water and Wastewater, 12th ed., Am. Pub. Hlth. Ass., Am. Wat. Poll. Control Fed., Oxygen Demand, 1965, Ρ 65. |
|
— |
Wagner, R. Von Wasser, 1976, Vol. 46, 139. |
|
— |
DIN-Entwurf 38409 Teil 41 — Deutsche Einheitsverfahren zur Wasser-, Abwasser- und Schlammuntersuchung, Summarische Wirkungs- und Stoffkenngrößen (Gruppe H). Bestimmung des Chemischen Sauerstoffbedarfs (CSB) (H 41), Normenausschüß Wasserwesen (NAW) in DIN Deutsches Institut fur Normung e.V. |
|
— |
Gerike, P. The biodegradability testing of poorly water soluble compounds. Chemosphere, 1984, Vol. 13 (1), 169. |
Bilag IIΙ
VURDERING AF TUNGTOPLØSELIGE STOFFERS BIONEDBRYDELIGHED
Nedenstående forhold kræver særlig opmærksomhed ved test af tungtopløselige stoffers bionedbrydelighed.
Homogene væsker vil sjældent volde problemer ved prøveudtagningen, hvorimod det anbefales at homogenisere faste materialer på passende måde, således at fejl som følge af inhomogenitet undgås. Der må udvises særlig omhu, når der skal udtages en repræsentativ prøve på nogle få mg af blandinger af kemikalier og af stoffer, der indeholder store mængder urenheder.
Under testen kan der anvendes forskellige former for omrøring. Omrøringen skal blot være så kraftig, at kemikaliet holdes dispergeret, og overophedning, stærk skumning og voldsomme forskydningskræfter skal undgås.
Der kan benyttes en emulgator tii at frembringe en stabil dispersion af kemikaliet. Emulgatoren må ikke være toksisk for bakterier og må hverken kunne bionedbrydes eller vare skumdannende under testbetingelserne.
Der gælder samme kriterier for opløsningsmidler som for emulgatorer.
Det frarådes at benytte faste bærematerialer til faste teststoffer, hvorimod de kan være egnede til olieagtige stoffer.
Når der anvendes hjælpestoffer såsom emulgatorer, opløsningsmidler og bærematerialer, udføres der en blindprøve med hjælpestoffet.
Alle tre respirometertest, CO2, BOD og MITI, kan benyttes til undersøgelse af tungtopløselige stoffers bionedbrydelighed.
BIBLIOGRAFI
|
— |
de Morsier, A. et al. Biodegradation tests for poorly-soluble compounds. Chemosphere, 1987, Vol. 16, 833. |
|
— |
Gerike, P. The biodegradability testing of poorly water soluble compounds. Chemosphere, 1984, Vol. 13, 169. |
Bilag IV
VURDERING AF BIONEDBRYDELIGHEDEN AF KEMISKE STOFFER, DER MISTÆNKES FOR AT VÆRE TOKSISKE FOR INOCULUM
Hvis et kemisk stof, som underkastes en test for let bionedbrydelighed, ikke synes at være bionedbrydeligt, tilrådes det at følge nedenstående fremgangsmåde, hvis der ønskes skelnet mellem hæmning og manglende nedbrydelighed (Reynolds et al., 1987).
Der anvendes samme eller tilsvarende inoculum til toksicitetstest som til bionedbrydningstest.
For at kunne vurdere toksiciteten af kemikalier, der undersøges i test for let bionedbrydelighed, kan man anvende en af følgende metoder eller en kombination af dem: Hæmning af slammets respirationshastighed (aktiveret slam — respirationshæmningstest — direktiv 88/302/EØF); BOD og/eller væksthæmningstest.
Hvis hæmning som følge af toksicitet skal undgås, anbefales det at benytte teststofkoncentrationer i test for let bionedbrydelighed, som er højst 1/10 af de EC50-værdier (eller højst de EC20-værdicr), der er fundet ved toksicitetstest. Stoffer, hvis EC50 er sterre end 300 mg/l, vil næppe have toksiske virkninger i test for let bionedbrydelighed.
En EC50 på mindre end 20 mg/l vil sandsynligvis skabe betydelige vanskeligheder for den efterfølgende testning. Testkoncentrationerne skal være lave, hvilket kræver anvendelse af den strenge og følsomme Closed Bottle-test eller af 14C-mærket materiale. Alternativt kan der ved tilvænning af inoculum blive mulighed for at benytte højere teststofkoncentrationer. I sidstnævnte tilfælde mister testen for let bionedbrydelighed dog sin specifikke karakter.
BIBLIOGRAFI
Reynolds, L. et al. Evaluation of the toxicity of substances to be assessed for biodegradability. Chemosphere, 1987, Vol. 16, 2259.
Bilag V
KORREKTION AF NITRIFIKATIONENS INTERFERENS PÅ OXYGENOPTAGELSEN
Fejl ved at undlade at tage hensyn til nitrifikation ved vurdering af bionedbrydeligheden af teststoffer, der ikke indeholder N, er minimal (højst 5 %), selv om oxidationen af mediets indhold af ammonium-N svinger vilkårligt fra testprøve til blindprøve. For teststoffer, der indeholder N, kan der imidlertid opstå væsentlige fejl.
Hvis der sker ufuldstændig nitrifikation, kan reaktionsblandingens iagttagne oxygenoptagelse korrigeres for den oxygenmængde, der er medgået til oxidation af ammonium til nitrit og nitrat, hvis ændringerne i nitrit- og nitratkoncentrationen under inkuberingen bestemmes under hensyntagen til følgende reaktionsligninger:
|
2 NH4Cl + 3 O2 = 2 HNO2 + 2 HCl + 2 H2O |
(1) |
|
2 HNO2 + O2 = 2 HNO3 |
(2) |
|
brutto: |
|
|
2 NH4Cl + 4 O2 = 2 HNO3 + 2 HCl + 2 H2O |
(3) |
Ud fra ligning (1) bliver oxygenoptagelsen ved oxidation af 28 g nitrogen i form af ammoniumchlorid (NH4Cl) til nitrit 96 g, dvs. en faktor på 3,43 (96/28). På tilsvarende måde bliver oxygenoptagelsen ved oxidation af 28 g nitrogen til nitrat (3) 128 g, dvs. en faktor på 4,57 (128/28).
Da reaktionerne er sekventielle, idet de finder sted p.g.a. hver deres bakteriearter, kan nitritkoncentrationen stige eller falde; i sidstnævnte tilfælde vil der blive dannet en ækvivalent mængde nitrat. Derfor bliver oxygenforbruget ved nitratdannelsen 4,57 gange forøgelsen i nitratkoncentrationen, hvorimod den oxygenmængde, der er involveret i nitritdannelsen, er 3,43 gange forøgelsen i nitritkoncentrationen, eller ved et fald i nitritkoncentrationen, et oxygentab på —3,43 gange koncentrationsfaldet.
Det vil sige, at
|
O2-forbrug ved nitratdannelse = 4,57 × N-nitratkoncentrationsforøgelsen |
(4) |
|
og |
|
|
O2-forbrug ved nitritdannelse = 3,43 × N-nitritkoncentrationsforøgelsen |
(5) |
|
og |
|
|
O2-tab ved nitritomdannelse = – 3,43 × N-nitratkoncentrationsfaldet |
(6) |
|
Derved bliver |
|
|
O2-forbrug til nitrifikation = ±3,43 × N-nitritkoncentrationsændringen +4,57 × N-nitratkoncentrationsforøgelsen |
(7) |
|
og dermed |
|
|
O2-forbrug til C-oxidation = samlet iagttaget forbrug — forbrug til nitrifikation |
(8). |
Alternativt kan oxygenoptagelsen som følge af nitrifikation som en første tilnærmelse sættes til 4,57 × tilvæksten i oxideret N, hvis kun total oxyderet Ν bestemmes.
Den korrigerede værdi for oxygenforbruget til C-oxidation sammenlignes dernæst med ThODNH4 beregnet efter bilag II.
C.5. NEDBRYDNING — BIOKEMISK OXYGENFORBRUG
1. METODE
1.1. INDLEDNING
Formålet med metoden er måling af det biokemiske oxygenforbrug (BOD) for faste eller flydende organiske stoffer.
Data, som opnås med denne test, vedrører vandopløselige stoffer; dog kan, i det mindste i princippet, flygtige stoffer samt stoffer med lav vandopløselighed også testes.
Denne metode kan kun anvendes på organiske testmaterialer, der ikke virker hæmmende for bakterier ved den koncentration, som anvendes ved testen. Hvis testmaterialet ikke lader sig opløse ved testkoncentrationen, kan det være nødvendigt at anvende særlige foranstaltninger, såsom ultralyddispersion for at opnå god dispersion af testmaterialet.
Oplysninger om kemikaliets toksicitet kan være nyttige for fortolkningen af lave resultater og ved valget af passende testkoncentrationer.
1.2. DEFINITIONER OG ENHEDER
BOD defineres som den mængde opløst oxygen, der kræves til biokemisk oxidation af en nærmere angivet mængde opløsning af stoffet under foreskrevne betingelser.
Resultatet udtrykkes som g BOD pr. g undersøgt stof.
1.3. REFERENCESTOFFER
Brug af et egnet referencestof til kontrol af inoculums aktivitet er ønskelig.
1.4. TESTMETODENS PRINCIP
En afmålt mængde stof, opløst eller dispergeret i et egnet og godt gennemluftet medium, podes med mikroorganismer og inkuberes ved konstant kendt temperatur i mørke.
BOD bestemmes som forskellen mellem indholdet af opløst oxygen ved forsøgets begyndelse og ved forsøgets afslutning. Forsøgets varighed skal være mindst fem dage og ikke over 28 dage.
Der udføres parallelt en blindprøve uden indhold af reststof.
1.5. KVALITETSKRITERIER
En BOD-bestemmelse kan ikke betragtes som en gyldig bestemmelse af et stofs bionedbrydelighed, men kun som en screeningtest.
1.6. BESKRIVELSE AF TESTMETODEN
En foreløbig opløsning eller dispersion af stoffet fremstilles for at opnå en BOD-koncentration, som er forenelig med den anvendte metode. BOD bestemmes derpå efter en hvilken som helst egnet national eller international standardmetode.
2. DATA OG EVALUERING
BOD-indholdet i opløsningen beregnes efter den valgte standardmetode og omregnes til g BOD pr. g stof.
3. RAPPORTERING
Den anvendte metode skal anføres.
Det biokemiske oxygenforbrug skal være gennemsnittet af mindst tre gyldige målinger.
Alle oplysninger og bemærkninger af betydning for fortolkningen skal rapporteres, navnlig med hensyn til stoffets egen sammensætning, urenheder, fysiske tilstand og toksiske virkninger, som vil kunne påvirke resultaterne.
Eventuel brug af tilsætningsstof til at hindre biologisk nitrifikation skal rapporteres.
4. HENVISNINGER
Liste over standardiserede metoder, f.eks.
|
|
NF Τ 90-103: Determination of the biochemical oxygen demand. |
|
|
NBN 407: Biochemical oxygen demand. |
|
|
NEN 3235 5.4: Bepaling van het biochemisch zuurstofverbruik (BZV). |
|
|
The determination of biochemical oxygen demand, Methods for the examination of water and associated materials, HMSO, London. |
|
|
ISO 5815: Determination of biochemical oxygen demand after n days. |
C.6. NEDBRYDNING — KEMISK OXYGENFORBRUG
1. METODE
1.1. INDLEDNING
Formålet med denne metode er at måle det kemiske oxygenforbrug (COD) for faste og flydende organiske stoffer på en vilkårlig standardiseret måde under bestemte laboratoriebetingelser.
Oplysninger om stoffets formel vil være nyttige ved gennemførelse af testen og for fortolkningen af de opnåede resultater (f.eks. halogensalte, organiske jern(II)salte, organiske chlorforbindelser).
1.2. DEFINITIONER OG ENHEDER
Det kemiske oxygenforbrug er et mål for et stofs evne til at kunne oxideres, og det udtrykkes som den ækvivalente mængde oxygen i det oxiderende reagens, der forbruges af stoffet under bestemte laboratoriebetingelser.
Resultatet udtrykkes som g COD pr. g undersøgt stof.
1.3. REFERENCESTOFFER
Anvendelse af referencestoffer er ikke påkrævet, hver gang et nyt stof skal undersøges. Referencestoffer skal først og fremmest tjene til lejlighedsvis kalibrering af metoden og til sammenligning med resultater opnået med andre metoder.
1.4. TESTMETODENS PRINCIP
En afmålt mængde af stoffet, opløst eller dispergeret i vand, oxideres med kaliumdichromat i stærkt svovlsur opløsning med sølvsulfat som katalysator under tilbagesvaling i to timer. Den uforbrugte mængde dichromat bestemmes ved titrering med en standardopløsning af ferroammoniumsulfat.
I tilfælde af chlorholdige stoffer tilsættes mercurisulfat (10) for at nedsætte interferens fra chlorid.
1.5. KVALITETSKRITERIER
Da metoden har en vis vilkårlig karakter, må COD betragtes som en »oxiderbarhedsindikator«, der er et praktisk mål for indholdet af organisk stof.
Chlorid kan forstyrre målingen; også uorganiske reducerende eller oxiderende stoffer kan forstyrre COD-bestemmelsen.
Visse cykliske forbindelser og mange flygtige stoffer (f.eks. lavere fedtsyrer) oxideres ikke fuldstændigt ved denne metode.
1.6. BESKRIVELSE AF TESTMETODEN
Der fremstilles en foreløbig opløsning eller dispersion af stoffet, således at der opnås en COD-værdi mellem 250 og 600 mg pr. liter.
Bemærkning:
I tilfælde af vanskeligt opløselige og ikke dispergerbare stoffer kan der afvejes en mængde fint pulveriseret eller væskeformigt stof svarende til ca. 5 mg COD, som overføres til forsøgsopstillingen med vand.
Det kemiske oxygenforbrug kan ofte, og især for stoffer med ringe opløselighed, med fordel bestemmes med en variant af metoden, nemlig et lukket system med trykudligning (H. Kelkenberg, 1975). Med denne modifikation kan der ofte med held foretages kvantitativ bestemmelse af forbindelser, der kun vanskeligt kan bestemmes ved konventionelle metoder, f.eks. eddikesyre. Metoden duer dog ikke til pyridin. Forøgelse af kaliumdichromatkoncentrationen til 0,25 Ν (0,0416 M) som anført i henvisning (1) kan lette den direkte afvejning af 5-10 mg stof, hvilket er en betingelse for COD-bestemmelse på stoffer med ringe vandopløselighed (henvisning (2)).
I øvrigt bestemmes COD derefter ved hjælp af en egnet national eller international standardmetode.
2. DATA OG EVALUERING
COD-indholdet i forsøgskolben beregnes i henhold til den valgte standardmetode og omregnes til g COD pr. g teststof.
3. RAPPORTERING
Den anvendte referencemetode skal anføres.
Det kemiske oxygenforbrug skal være et gennemsnit af mindst tre målinger. Alle oplysninger og bemærkninger af betydning for fortolkningen af resultaterne skal rapporteres, navnlig med hensyn til stoffets egen sammensætning, urenheder og fysiske tilstand (hvis de kendes), som vil kunne påvirke resultaterne.
Anvendelse af mercurisulfat til formindskelse af interferens fra chlorid skal rapporteres.
4. HENVISNINGER
|
(1) |
Kelkenberg, H. Z, von Wasser und Abwasserforschung, 1975, vol. 8, 146. |
|
(2) |
Gerike, P. The biodegradability testing of poorly water soluble compounds. Chemosphere, 1984, vol. 13, 169. Liste over standardiserede metoder, f.eks.:
|
C.7. NEDBRYDNING — ABIOTISK NEDBRYDNING: HYDROLYSE SOM FUNKTION AF pH
1. METODE
Denne testmetode er ækvivalent med OECD TG 111 (2004).
1.1. INDLEDNING
Kemiske stoffer kan tilføres overfladevande ved direkte udledning, vinddrift, afstrømning, afvanding, affaldsbortskaffelse, udledning fra industri, husholdninger eller landbrug samt ved afsætning fra atmosfæren og kan i sådanne vande omdannes ved kemiske (f.eks. hydrolyse, oxidation), fotokemiske og/eller mikrobielle processer. I denne vejledning beskrives en laboratoriemetode til vurdering af abiotisk hydrolytisk omdannelse af kemiske stoffer i akvatiske systemer ved de i miljøet normalt forekommende pH-værdier (pH 4-9), baseret på eksisterende guidelines (1)(2)(3)(4)(5)(6)(7).
Formålet med forsøgene er at bestemme i) hydrolysehastigheden af prøvestoffet som funktion af pH og ii) identitet eller art af samt dannelses- og nedbrydningshastighed for hydrolyseprodukter, som organismer kan blive udsat for. Sådanne undersøgelser kan være påkrævede for kemiske stoffer, der direkte tilføres vand eller må forventes at nå miljøet ad de andre veje beskrevet ovenfor.
1.2. DEFINITIONER OG ENHEDER
Se bilag 2.
1.3. METODENS ANVENDELSESOMRÅDE
Metoden finder anvendelse på alle kemiske stoffer (umærkede eller mærkede), for hvilke der foreligger en tilstrækkelig nøjagtig og følsom analysemetode. Den finder anvendelse på tungtflygtige og ikke-flygtige forbindelser med tilstrækkelig vandopløselighed. Metoden bør ikke anvendes på stoffer, der er letflygtige fra vand (f.eks. rygningsmidler eller organiske opløsningsmidler) og således ikke kan holdes i opløsning under de her beskrevne forsøgsbetingelser. Den kan være vanskelig at anvende på stoffer med meget ringe vandopløselighed (8).
1.4. TESTMETODENS PRINCIP
Sterile vandige bufferopløsninger med forskellige pH-værdier (pH 4, 7 og 9) behandles med prøvestoffet og inkuberes i mørke under kontrollerede laboratoriebetingelser (ved konstant temperatur). Efter passende tidsintervaller analyseres bufferopløsningerne for prøvestoffet og for hydrolyseprodukter. Anvendelse af mærket prøvestof (f.eks. 14C) letter opstilling af en massebalance.
I metoden anvendes en trinvis fremgangsmåde som illustreret og forklaret i bilag 1. Hvert trin udløses af resultaterne af det foregående trin.
1.5. OPLYSNINGER OM PRØVESTOFFET
Til måling af hydrolysehastighed kan anvendes umærket eller mærket prøvestof. Til undersøgelse af hydrolysevejen og opstilling af massebalance er mærket stof sædvanligvis at foretrække; i særlige tilfælde er mærkning dog ikke absolut nødvendig. 14C-mærkning anbefales, men andre isotoper, såsom 13C, 15N eller 3H, kan ligeledes være nyttige. Mærket bør så vidt muligt placeres i molekylets mest stabile del(e). Indeholder prøvestoffet således én ring, skal denne ring være mærket; indeholder stoffet to eller flere ringe, kan det være nødvendigt med separate undersøgelser til fastlæggelse af skæbnen af hver mærket ring og til at skaffe tilstrækkelige oplysninger om dannelsen af hydrolyseprodukter. Prøvestoffets renhed skal være mindst 95 %.
Før udførelse af et hydrolyseforsøg bør følgende oplysninger om prøvestoffet foreligge:
|
a) |
vandopløselighed (testmetode A.6) |
|
b) |
opløselighed i organiske opløsningsmidler |
|
c) |
damptryk (testmetode A.4) og/eller Henrys lovkonstant |
|
d) |
fordelingskoefficient n-oktanol/vand (testmetode A.8) |
|
e) |
dissociationskonstant (pKa) (OECD Guideline 112) (9) |
|
f) |
når det er hensigtsmæssigt, direkte og indirekte fototransformationshastighed i vand. |
Der bør foreligge analysemetoder til kvantitativ bestemmelse afprøvestoffet og, hvis relevant, identifikation og kvantitativ bestemmelse af hydrolyseprodukter i vandige opløsninger (se også punkt 1.7.2).
1.6. REFERENCESTOFFER
Når det er muligt, bør der anvendes referencestoffer til identifikation og kvantitativ bestemmelse af hydrolyseprodukter ved spektroskopiske og kromatografiske metoder eller andre metoder med tilstrækkelig følsomhed.
1.7. KVALITETSKRITERIER
1.7.1. Genfinding
Analyse af mindst dobbelte bufferopløsninger eller af ekstrakter deraf efter tilsætning af prøvestoffet giver et første fingerpeg om analysemetodens repeterbarhed og om ensartetheden af tilsætningen af prøvestoffet. Genfindingsgraden for senere forsøgsstadier fås af de respektive massebalancer (når mærket stof anvendes). Genfindingen bør være mellem 90 % og 110 % for mærkede og umærkede kemiske stoffer (7). Hvis det er teknisk vanskeligt at nå dette område, er en genfindingsgrad på 70 % for umærkede kemiske stoffer acceptabel, men skal begrundes.
1.7.2. Analysemetodens repeterbarhed og følsomhed
Repeterbarheden af de(n) analysemetode(r), der senere anvendes til kvantificering af prøvestoffet og hydrolyseprodukter, kan kontrolleres ved dobbeltanalyse af de samme bufferopløsninger (eller af ekstrakter deraf), efter at der er dannet en tilstrækkelig mængde hydrolyseprodukter til kvantitativ bestemmelse.
Analysemetoden skal være tilstrækkelig følsom til at kvantificere prøvestofkoncentrationer ned til 10 % eller derunder af begyndelseskoncentrationen. Hvis det er relevant, skal analysemetoderne desuden være tilstrækkelig følsomme til kvantificering af eventuelle hydrolyseprodukter, der udgør 10 % eller mere af den tilsatte mængde (til et vilkårligt tidspunkt under undersøgelsen) ned til 25 % eller derunder af deres spidskoncentration.
1.7.3. Konfidensintervaller for hydrolysekinetikdata
Der beregnes og angives konfidensintervaller for alle regressionskoefficienter, hastighedskonstanter, halveringstider og eventuelle andre kinetikparametre (f.eks. DT50).
1.8. BESKRIVELSE AF TESTMETODEN
1.8.1. Udstyr og apparatur
Undersøgelsen udføres i glasbeholdere (f.eks. reagensglas eller små kolber), om nødvendigt i mørke og under sterile betingelser, medmindre foreløbige oplysninger (som f.eks. n-oktanol-vand fordelingskoefficienten) viser, at prøvestoffet kan adhærere til glas. I sådanne tilfælde kan det være nødvendigt at overveje alternative materialer (som f.eks. Teflon). Problemet med adhæsion til glas kan desuden løses ved en eller flere af følgende metoder:
|
— |
bestemmelse af den masse prøvestof og hydrolyseprodukter, der er sorberet til prøvebeholderen |
|
— |
anvendelse af ultralydsbad |
|
— |
vask af alt glasapparatur i opløsningsmiddel ved hvert prøvetagningsinterval |
|
— |
anvendelse af færdigformuleret produkt |
|
— |
anvendelse af ekstra hjælpefortynder ved tilsætning af prøvestof til systemet; som hjælpefortynder må undgås stoffer der hydrolyserer prøvestoffet. |
Til inkubering af de forskellige prøveopløsninger skal normalt anvendes temperaturstyrede vandbade eller termostaterede inkubatorer.
Sædvanligt laboratorieudstyr skal være til rådighed, herunder specielt følgende:
|
— |
pH-meter |
|
— |
analyseapparatur som f.eks. GC, HPLC og TLC med passende detektionssystem til analyse af radioaktivt mærkede eller umærkede stoffer eller til omvendt isotopfortyndingsmetode |
|
— |
instrumenter til identifikation (f.eks. MS, GC-MS, HPLC-MS, NMR, mv.) |
|
— |
væskescintillationstæller |
|
— |
skilletragte til væske-væske ekstraktion |
|
— |
udstyr til koncentrering af opløsninger og ekstrakter (f.eks. rotationsfordamper) |
|
— |
temperaturreguleringsanordning (f.eks. vandbad). |
Kemiske reagenser, f.eks. følgende:
|
— |
organiske opløsningsmidler af analysekvalitet, således hexan, dichlormethan mv. |
|
— |
scintillationsvæske |
|
— |
bufferopløsninger (som nærmere angivet i pkt. 1.8.3). |
Alt glasudstyr, vand af analysekvalitet og bufferopløsninger, som anvendes i hydrolyseforsøgene, skal være steriliseret.
1.8.2. Tilsætning af prøvestoffet
Prøvestoffet tilsættes som vandig opløsning til de forskellige bufferopløsninger (se bilag 3). Om nødvendigt kan man for at få opløst tilstrækkeligt af stoffet tilsætte lidt vandopløseligt opløsningsmiddel (f.eks. acetonitril, acetone, ethanol) når prøvestoffet tilsættes og fordeles, men normalt ikke i større mængde end 1 % v/v. Anvendelse af opløsningsmidler i højere koncentration (f.eks. til tungtopløselige prøvestoffer) er kun tilladt, hvis det godtgøres at opløsningsmidlet er uden indflydelse på hydrolysen afprøvestoffet.
Rutinemæssig brug af formuleret produkt kan ikke anbefales, da det ikke kan udelukkes at hjælpestoffer i formuleringen vil påvirke hydrolyseprocessen. For prøvestoffer der er tungtopløselige i vand, og for stoffer der adhærerer til glas (se 1.8.1) kan anvendelse af det formulerede stof dog være et hensigtsmæssigt alternativ.
Prøvestoffet bør anvendes i én koncentration, der ikke bør være over 0,01 M eller det halve af mætningskoncentrationen (se bilag 1).
1.8.3. Bufferopløsninger
Hydrolyseforsøget udføres ved pH 4, 7 og 9. Vand og kemikalier anvendt til fremstilling af sådanne bufferopløsninger skal være af analysekvalitet. I bilag 3 beskrives nogle nyttige buffersystemer. Man må være opmærksom på, at det anvendte buffersystem kan influere på hydrolysehastigheden, og er dette tilfældet, må der vælges et andet buffersystem (11).
pH af hver bufferopløsning skal kontrolleres med et kalibreret pH-meter med en nøjagtighed på mindst 0,1 ved den foreskrevne temperatur.
1.8.4. Forsøgsbetingelser
1.8.4.1. Forsøgstemperatur
Hydrolyseforsøgene skal udføres ved konstant temperatur. Til brug ved ekstrapolation er det vigtigt, at temperaturen holdes konstant inden for ±0,5 oC.
Der bør udføres et indledende forsøg (trin 1) ved en temperatur på 50 oC, hvis prøvestoffets hydrolyseegenskaber er ukendte. Kinetiske forsøg på efterfølgende trin udføres ved mindst tre temperaturer (herunder forsøget ved 50 oC), medmindre prøvestoffet ved trin 1-forsøget er fundet stabilt over for hydrolyse. Det foreslås at anvende et temperaturområde på 10-70 oC (idet mindst én temperatur bør være under 25 oC), der således dækker rapporteringstemperaturen på 25 oC og størstedelen af det temperaturområde, der forekommer i marken.
1.8.4.2. Lys og oxygen
Alle hydrolyseforsøg skal udføres med en metode, der bevirker at fotolytiske effekter undgås. Der skal træffes alle nødvendige forholdsregler til undgåelse af oxygen (f.eks. ved gennembobling med helium, nitrogen eller argon i 5 minutter, før opløsningen fremstilles).
1.8.4.3. Forsøgets varighed
Forprøven strækker sig over 5 døgn, hvorimod de efterfølgende forsøg fortsætter indtil prøvestoffet er 90 % hydrolyseret, dog højst 30 døgn.
1.8.5. Forsøgets udførelse
1.8.5.1. Forprøve (trin 1)
Forprøven udføres ved 50 ± 0,5 oC og pH 4,0, 7,0 og 9,0. Iagttages mindre end 10 % hydrolyse efter 5 døgn (t0,525 oC > 1 år), anses prøvestoffet for hydrolytisk stabilt, og yderligere forsøg kræves normalt ikke. Vides stoffet at være ustabilt ved miljømæssigt relevante temperaturer (12), er forprøve unødvendig. Analysemetoden skal være tilstrækkelig præcis og følsom til at påvise en reduktion på 10 % af begyndelseskoncentrationen.
1.8.5.2. Hydrolyse af ustabile stoffer (trin 2)
Forsøget på næste trin (avanceret) udføres ved de pH-værdier, hvor prøvestoffet i ovennævnte forprøve er fundet ustabilt. De bufrede opløsninger af prøvestof termostateres ved de valgte temperaturer. Med henblik på fastlæggelse af førsteordens-kinetik analyseres hver reaktionsopløsning med tidsintervaller svarende til mindst seks adskilte datapunkter, normalt svarende til mellem 10 % og 90 % hydrolyse af prøvestoffet. De enkelte gentagelsesprøver (mindst dobbeltprøver, der opbevares i separate reaktionsbeholdere) udtages, og indholdet analyseres på hvert af mindst seks prøvetagningstidspunkter (svarende til mindst tolv datapunkter med gentagelser). Det anses for uhensigtsmæssigt at anvende en enkelt bulkprøve og heraf udtage de enkelte aliquoter af prøveopløsningen på hvert prøvetagningstidspunkt, da der derved ikke tages hensyn til analysevariationen, og da kontamination af prøveopløsningen kan være et problem. Sterilitetskontrol udføres ved afslutningen af det efterfølgende trin (dvs. ved 90 % hydrolyse eller efter 30 døgn). Iagttages ingen nedbrydning (dvs. omdannelse), anses sterilitskontrol dog for unødvendig.
1.8.5.3. Identifikation af hydrolyseprodukter (trin 3)
De vigtigste hydrolyseprodukter, i det mindste dem, der udgør > 10 % af den tilførte dosis, identificeres med egnede analysemetoder.
1.8.5.4. Ikke-obligatoriske forsøg
For hydrolytisk ustabile prøvestoffer kan supplerende forsøg ved andre pH-værdier end 4, 7 og 9 være nødvendige. Det kan således være nødvendigt at foretage et forsøg ved større surhedsgrad (f.eks. pH 1,2) ved en enkelt fysiologisk relevant temperatur (37 oC).
2. DATA
Mængden af prøvestof og af eventuelle hydrolyseprodukter angives i % af den anvendte begyndelseskoncentration og, når det er hensigtsmæssigt, i mg/l for hvert prøvetagningsinterval og for hver pH-værdi og forsøgstemperatur. Anvendes mærket prøvestof, skal der desuden opstilles en massebalance, angivet i procent af den anvendte begyndelseskoncentration.
De logaritmisk transformerede prøvestofkoncentrationsdata afbildes grafisk som funktion af tiden. Alle vigtige hydrolyseprodukter, i det mindste dem der udgør ≥ 10 % af den anvendte dosis, identificeres, og deres logaritmisk transformerede koncentration afsættes som for moderstoffet til påvisning af deres dannelses- og nedbrydningshastighed.
2.1. BEHANDLING AF RESULTATER
Der skal foretages en nøjagtigere bestemmelse af halveringstider eller DT50-værdier ved passende kinetiske modelberegninger. For hvert pH og hver temperatur angives halveringstid og/eller DT50 (med konfidensinterval) sammen med en beskrivelse af den anvendte model, den kinetiske orden og determinationskoefficienten (r2). Samme beregninger foretages desuden for hydrolyseprodukterne, når det er relevant.
For kinetikforsøg udført ved forskellige temperaturer angives hastighedskonstanterne for hydrolysen svarende til en pseudoførsteordensreaktion (kobs) som funktion af temperaturen. Beregningen baseres på opdeling af kobs i hastighedskonstanter for syrekatalyseret, neutral og basekatalyseret hydrolyse (hhv. kH, kneutral og kOh) og på Arrhenius-ligningen:
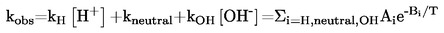
hvor Ai og Bi er regressionskonstanterne beregnet af hhv. akseafskæring og hældning af de bedst tilnærmede linjer, der genereres ved lineær regression af ln ki mod den reciprokke absolutte temperatur i kelvin (T). Ved at anvende Arrhenius-relationen på sur, neutral og basekatalyseret hydrolyse kan pseudoførsteordens hastighedskonstanter og dermed halveringstider beregnes for andre temperaturer, for hvilke det ikke er praktisk muligt at bestemme hastighedskonstanten direkte eksperimentelt (10).
2.2. EVALUERING OG FORTOLKNING AF RESULTATER
De fleste hydrolysereaktioner følger en tilsyneladende førsteordens kinetik, hvorfor halveringstiden er koncentrationsuafhængig (jf. ligning 4 i bilag 2). Derfor kan laboratorieresultater opnået i området 10-2 til 10-3 M sædvanligvis anvendes på virkelige forhold (10-6 M) (10). Mabey og Mill (11) har rapporteret om flere eksempler på god overensstemmelse mellem hydrolysehastigheder målt i rent vand og råvand for forskellige kemiske stoffer, forudsat at pH og temperatur måles.
3. RAPPORTERING
3.1. TESTRAPPORT
Testrapporten skal mindst indeholde følgende oplysninger:
Teststof:
|
— |
trivialnavn, kemisk navn, CAS-nummer, strukturformel (med angivelse af mærkets placering, når radioaktivt mærket stof anvendes) og relevante fysiske/kemiske egenskaber (se punkt 1.5) |
|
— |
renhed (urenheder) afprøvestoffet |
|
— |
for mærkede stoffer, renhed af radioaktiv mærkning samt molær aktivitet (hvis relevant). |
|
— |
Bufferopløsninger: |
|
— |
fremstillingsdato og nærmere oplysninger om fremstillingen |
|
— |
anvendte buffere og vand |
|
— |
bufferopløsningernes molaritet og pH. |
Forsøgsbetingelser:
|
— |
dato for forsøgenes udførelse |
|
— |
anvendt mængde prøvestof |
|
— |
tilsætning afprøvestoffet: fremgangsmåde og anvendte opløsningsmidler (type og mængde) |
|
— |
volumen bufret prøvestofopløsning, som er inkuberet |
|
— |
beskrivelse af det anvendte inkubationssystem |
|
— |
pH og temperatur under forsøget |
|
— |
prøvetagningstidspunkter |
|
— |
ekstraktionsmetode(r) |
|
— |
metoder til kvantitativ bestemmelse og identifikation afprøvestoffet og dets hydrolyseprodukter i bufferopløsningerne |
|
— |
antal gentagelser af bestemmelsen. |
Resultater:
|
— |
de anvendte analysemetoders repeterbarhed og følsomhed |
|
— |
genfinding (procentangivelser for en gyldig undersøgelse er givet i punkt 1.7.1) |
|
— |
resultater af gentagne bestemmelser og gennemsnitsværdier, opstillet i tabel |
|
— |
ved anvendelse afmærket prøvestof, massebalance før, under og efter forsøgene |
|
— |
resultater af forprøven |
|
— |
diskussion og fortolkning af resultaterne |
|
— |
alle oprindelige data og talværdier. |
Følgende oplysninger er kun nødvendige, når hydrolysehastigheden bestemmes:
|
— |
koncentrationstidskurver over prøvestof og relevante hydrolyseprodukter, for hver pH-værdi og temperatur |
|
— |
tabeller over beregningsresultater baseret på Arrhenius-ligningen for temperaturen 20 oC/25 oC, sammen med pH, hastighedskonstant (h-1 eller døgn-1), halveringstid eller DT50, temperatur ( oC) herunder konfidensintervaller og korrelationskoefficienter (r2), eller tilsvarende oplysninger |
|
— |
formodet hydrolysevej. |
4. HENVISNINGER
|
(1) |
OECD (1981). Hydrolysis as a Function of pH. OECD Guideline for Testing of Chemicals Nr. 111, vedtaget 12. maj 1981. |
|
(2) |
US-Environmental Protection Agency (1982). 40 CFR 796.3500, Hydrolysis as a Function of pH at 25 oC. Pesticide Assessment Guidelines, Subdivision N. Chemistry: Environmental Fate. |
|
(3) |
Agriculture Canada (1987). Environmental Chemistry and Fate Guidelines for registration of pesticides in Canada. |
|
(4) |
Den Europæiske Union (EU) (1995). Kommissionens direktiv 95/36/EF om ændring af Rådets direktiv 91/414/EØF om markedsføring af plantebeskyttelsesmidler. Bilag V: Skæbne og opførsel i miljøet. |
|
(5) |
Dutch Commission for Registration of Pesticides (1991). Application for registration of a pesticide. Section G: Behaviour of the product and its metabolites in soil, water and air. |
|
(6) |
BBA (1980). Merkblatt Nr. 55, Teil I und II: Prüfung des Verhaltens von Pflanzenbehandlungsmitteln im Wasser (oktober 1980). |
|
(7) |
SETAC (1995). Procedures for Assessing the Environmental Fate and Ecotoxicity of Pesticides. Mark R. Lynch (red.). |
|
(8) |
OECD (2000). Guidance document on aquatic toxicity testing of difficult substances and mixtures, OECD Environmental Health and Safety Publications Series on Testing and Assessment Nr. 23. |
|
(9) |
OECD (1993). Guidelines for the Testing of Chemicals. Paris. OECD (1994 — 2000): Addenda 6-11 to Guidelines for the Testing of Chemicals. |
|
(10) |
Nelson, H, Laskowski D, Thermes S, and Hendley P. (1997) Recommended changes in pesticide fate study guidelines for improving input to computer models. (Skriftlig version af mundtlig præsentation ved 14. årsmøde i Society of Environmental Toxicology and Chemistry, Dallas TX, november 1993). |
|
(11) |
Mabey, W. and Mill, T. (1978). Critical review of hydrolysis of organic compounds in water under environmental conditions. J. Phys. Chem. Ref. Data 7, 383-415. |
Bilag 1
Trinvis forsøgsplan for hydrolyse
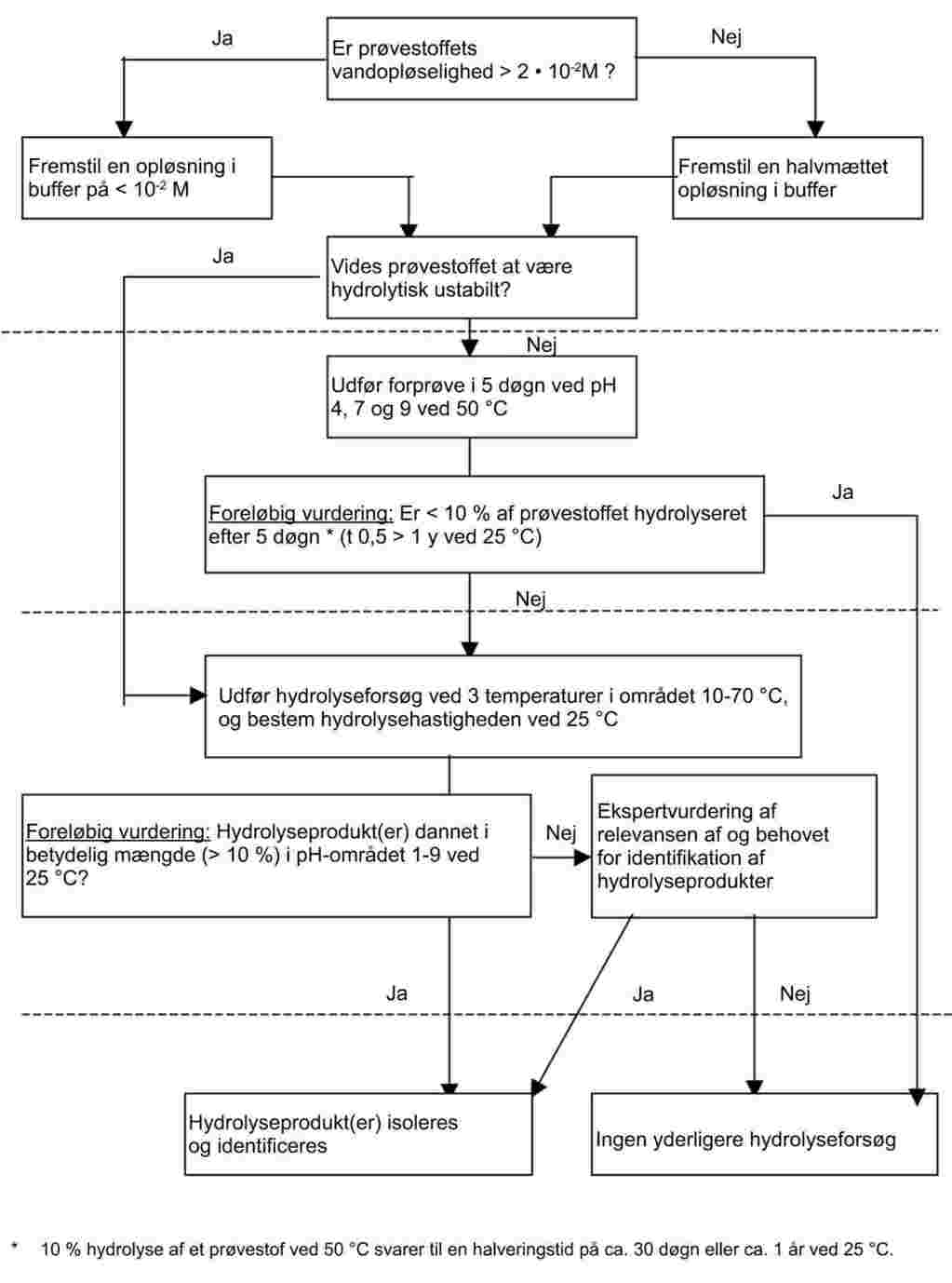
Bilag 2
Definitioner og enheder
SI-enheder skal anvendes i alle tilfælde.
Teststof: ethvert stof, hvad enten det er moderstoffet eller relevante omdannelsesprodukter.
Omdannelsesprodukter: alle stoffer hidrørende fra biotisk eller abiotisk omdannelse af prøvestoffet.
Hydrolyseprodukter: alle stoffer hidrørende fra hydrolytisk omdannelse af prøvestoffet.
Ved hydrolyse forstås reaktionen af et prøvestof RX med vand, resulterende i nettoudskiftning af gruppen X med OH på reaktions stedet:
|
RX + HOH → ROH +HX |
[1] |
I denne forenklede proces er den hastighed, hvormed koncentrationen af RX aftager, givet ved
|
hastighed = k [H2O] [RX] |
andenordens reaktion |
|
eller |
|
|
hastighed = k [RX] |
førsteordens reaktion |
afhængigt af det hastighedsbestemmende trin. Da vand er til stede i stort overskud i forhold til prøvestoffet, beskrives denne type reaktion sædvanligvis som en pseudoførsteordens reaktion, hvor den iagttagne hastighedskonstant er givet ved relationen
|
kobs = k[H2O] |
[2] |
og kan bestemmes af udtrykket (13)
|
|
[3] |
hvor
t= tiden
og Co, Ct= koncentration af RX til tiden 0 og t.
Denne konstant har dimensionen (tid)-1, og halveringstiden for reaktionen (den tid, det tager, før 50 % af RX har reageret) er givet ved
|
|
[4] |
Halveringstid: (t0,5) er den tid, der går, før 50 % prøvestoffet er hydrolyseret, når reaktionen kan beskrives ved førsteordens kinetik; den er uafhængig af koncentrationen.
DT 50 (Disappearance Time 50): er den tid, i løbet af hvilken koncentrationen afprøvestoffet mindskes med 50 %; den er forskellig fra halveringstiden t0,5, når reaktionen ikke følger førsteordens kinetik.
Skønsmæssig beregning af k ved forskellige temperaturer
Når hastighedskonstanterne kendes for to temperaturer, kan hastighedskonstanterne for andre temperaturer beregnes ved hjælp af Arrhenius-ligningen:
![]() eller
eller 
Ved afbildning af ln k mod 1/T fås en ret linje med hældning –E/R,
hvor:
|
k |
= |
hastighedskonstanten, målt ved forskellige temperaturer |
|
Ε |
= |
aktiveringsenergi (kJ/mol) |
|
Τ |
= |
absolut temperatur (K) |
|
R |
= |
gaskonstanten (8,314 J/mol.K) |
Aktiveringsenergien er beregnet ved regressionsanalyse eller af følgende udtryk:

hvor: T2 > T1.
Bilag 3
Buffersystemer
A. CLARK OG LUBS:
Bufferblandinger efter CLARK og LUBS (14)
|
Sammensætning |
pH |
|
0,2 N HCl og 0,2 N KCl ved 20 oC |
|
|
47,5 ml HCl + 25 ml KCl fortyndet til 100 ml |
1,0 |
|
32,25 ml HCl + 25 ml KCl fortyndet til 100 ml |
1,2 |
|
20,75 ml HCl + 25 ml KCl fortyndet til 100 ml |
1,4 |
|
13,15 ml HCl + 25 ml KCl fortyndet til 100 ml |
1,6 |
|
8,3 ml HCl + 25 ml KCl fortyndet til 100 ml |
1,8 |
|
5,3 ml HCl + 25 ml KCl fortyndet til 100 ml |
2,0 |
|
3,35 ml HCl + 25 ml KCl fortyndet til 100 ml |
2,2 |
|
0,1 M kaliumbiphthalat +0,1 N HCl ved 20 oC |
|
|
46,70 ml 0,1 N HCl + 50 ml biphthalat til 100 ml |
2,2 |
|
39,60 ml 0,1 N HCl + 50 ml biphthalat til 100 ml |
2,4 |
|
32,95 ml 0,1 N HCl + 50 ml biphthalat til 100 ml |
2,6 |
|
26,42 ml 0,1 N HCl + 50 ml biphthalat til 100 ml |
2,8 |
|
20,32 ml 0,1 N HCl + 50 ml biphthalat til 100 ml |
3,0 |
|
14,70 ml 0,1 N HCl + 50 ml biphthalat til 100 ml |
3,2 |
|
9,90 ml 0,1 N HCl + 50 ml biphthalat til 100 ml |
3,4 |
|
5,97 ml 0,1 N HCl + 50 ml biphthalat til 100 ml |
3,6 |
|
2,63 ml 0,1 N HCl + 50 ml biphthalat til 100 ml |
3,8 |
|
0,1 M kaliumbiphthalat +0,1 N NaOH ved 20 oC |
|
|
0,40 ml 0,1 N NaOH + 50 ml biphthalat til 100 ml |
4,0 |
|
3,70 ml 0,1 N NaOH + 50 ml biphthalat til 100 ml |
4,2 |
|
7,50 ml 0,1 N NaOH + 50 ml biphthalat til 100 ml |
4,4 |
|
12,15 ml 0,1 N NaOH + 50 ml biphthalat til 100 ml |
4,6 |
|
17,70 ml 0,1 N NaOH + 50 ml biphthalat til 100 ml |
4,8 |
|
23,85 ml 0,1 N NaOH + 50 ml biphthalat til 100 ml |
5,0 |
|
29,95 ml 0,1 N NaOH + 50 ml biphthalat til 100 ml |
5,2 |
|
35,45 ml 0,1 N NaOH + 50 ml biphthalat til 100 ml |
5,4 |
|
39,85 ml 0,1 N NaOH + 50 ml biphthalat til 100 ml |
5,6 |
|
43,00 ml 0,1 N NaOH + 50 ml biphthalat til 100 ml |
5,8 |
|
45,45 ml 0,1 N NaOH + 50 ml biphthalat til 100 ml |
6,0 |
Bufferblandinger efter CLARK og LUBS (fortsat)
|
0,1 M monokaliumphosphat +0,1 N NaOH ved 20 oC |
|
|
5,70 ml 0,1 N NaOH + 50 ml phosphat til 100 ml |
6,0 |
|
8,60 ml 0,1 N NaOH + 50 ml phosphat til 100 ml |
6,2 |
|
12,60 ml 0,1 N NaOH + 50 ml phosphat til 100 ml |
6,4 |
|
17,80 ml 0,1 N NaOH + 50 ml phosphat til 100 ml |
6,6 |
|
23,45 ml 0,1 N NaOH + 50 ml phosphat til 100 ml |
6,8 |
|
29,63 ml 0,1 N NaOH + 50 ml phosphat til 100 ml |
7,0 |
|
35,00 ml 0,1 N NaOH + 50 ml phosphat til 100 ml |
7,2 |
|
39,50 ml 0,1 N NaOH + 50 ml phosphat til 100 ml |
7,4 |
|
42,80 ml 0,1 N NaOH + 50 ml phosphat til 100 ml |
7,6 |
|
45,20 ml 0,1 N NaOH + 50 ml phosphat til 100 ml |
7,8 |
|
46,80 ml 0,1 N NaOH + 50 ml phosphat til 100 ml |
8,0 |
|
0,1 M H 3 B0 3 i 0,1 M KCl +0,1 N NaOH ved 20 oC |
|
|
2,61 ml 0,1 N NaOH + 50 ml borsyre til 100 ml |
7,8 |
|
3,97 ml 0,1 N NaOH + 50 ml borsyre til 100 ml |
8,0 |
|
5,90 ml 0,1 N NaOH + 50 ml borsyre til 100 ml |
8,2 |
|
8,50 ml 0,1 N NaOH + 50 ml borsyre til 100 ml |
8,4 |
|
12,00 ml 0,1 N NaOH + 50 ml borsyre til 100 ml |
8,6 |
|
16,30 ml 0,1 N NaOH + 50 ml borsyre til 100 ml |
8,8 |
|
21,30 ml 0,1 N NaOH + 50 ml borsyre til 100 ml |
9,0 |
|
26,70 ml 0,1 N NaOH + 50 ml borsyre til 100 ml |
9,2 |
|
32,00 ml 0,1 N NaOH + 50 ml borsyre til 100 ml |
9,4 |
|
36,85 ml 0,1 N NaOH + 50 ml borsyre til 100 ml |
9,6 |
|
40,80 ml 0,1 N NaOH + 50 ml borsyre til 100 ml |
9,8 |
|
43,90 ml 0,1 N NaOH + 50 ml borsyre til 100 ml |
10,0 |
B. KOLTHOFF OG VLEESCHHOUWER:
Citratbuffere efter KOLTHOFF og VLEESCHHOUWER
|
Sammensætning |
pH |
|
0,1 M monokaliumcitrat og 0,1 N HCl ved 18 oC (15) |
|
|
49,7 ml 0,1 N HCl + 50 ml citrat til 100 ml |
2,2 |
|
43,4 ml 0,1 N HCl + 50 ml citrat til 100 ml |
2,4 |
|
36,8 ml 0,1 N HCl + 50 ml citrat til 100 ml |
2,6 |
|
30,2 ml 0,1 N HCl + 50 ml citrat til 100 ml |
2,8 |
|
23,6 ml 0,1 N HCl + 50 ml citrat til 100 ml |
3,0 |
|
17,2 ml 0,1 N HCl + 50 ml citrat til 100 ml |
3,2 |
|
10,7 ml 0,1 N HCl + 50 ml citrat til 100 ml |
3,4 |
|
4,2 ml 0,1 N HCl + 50 ml citrat til 100 ml |
3,6 |
|
0,1 M monokaliumcitrat og 0,1 N NaOH ved 18 oC (15) |
|
|
2,0 ml 0,1 N NaOH + 50 ml citrat til 100 ml |
3,8 |
|
9,0 ml 0,1 N NaOH + 50 ml citrat til 100 ml |
4,0 |
|
16,3 ml 0,1 N NaOH + 50 ml citrat til 100 ml |
4,2 |
|
23,7 ml 0,1 N NaOH + 50 ml citrat til 100 ml |
4,4 |
|
31,5 ml 0,1 N NaOH + 50 ml citrat til 100 ml |
4,6 |
|
39,2 ml 0,1 N NaOH + 50 ml citrat til 100 ml |
4,8 |
|
46,7 ml 0,1 N NaOH + 50 ml citrat til 100 ml |
5,0 |
|
54,2 ml 0,1 N NaOH + 50 ml citrat til 100 ml |
5,2 |
|
61,0 ml 0,1 N NaOH + 50 ml citrat til 100 ml |
5,4 |
|
68,0 ml 0,1 N NaOH + 50 ml citrat til 100 ml |
5,6 |
|
74,4 ml 0,1 N NaOH + 50 ml citrat til 100 ml |
5,8 |
|
81,2 ml 0,1 N NaOH + 50 ml citrat til 100 ml |
6,0 |
C. SØRENSEN:
Boratblandinger efter SØRENSEN
|
Sammensætning |
Sørensen 18 oC |
Walbum, pH ved |
|||
|
ml Borax |
ml HCl/NaOH |
10 oC |
40 oC |
70 oC |
|
|
0,05 M borax +0,1 N HCl |
|||||
|
5,25 |
4,75 |
7,62 |
7,64 |
7,55 |
7,47 |
|
5,50 |
4,50 |
7,94 |
7,98 |
7,86 |
7,76 |
|
5,75 |
4,25 |
8,14 |
8,17 |
8,06 |
7,95 |
|
6,00 |
4,00 |
8,29 |
8,32 |
8,19 |
8,08 |
|
6,50 |
3,50 |
8,51 |
8,54 |
8,40 |
8,28 |
|
7,00 |
3,00 |
8,08 |
8,72 |
8,56 |
8,40 |
|
7,50 |
2,50 |
8,80 |
8,84 |
8,67 |
8,50 |
|
8,00 |
2,00 |
8,91 |
8,96 |
8,77 |
8,59 |
|
8,50 |
1,50 |
9,01 |
9,06 |
8,86 |
8,67 |
|
9,00 |
1,00 |
9,09 |
9,14 |
8,94 |
8,74 |
|
9,50 |
0,50 |
9,17 |
9,22 |
9,01 |
8,80 |
|
10,00 |
0,00 |
9,24 |
9,30 |
9,08 |
8,86 |
|
0,05 M borax+0,1 N NaOH |
|||||
|
10,0 |
0,0 |
9,24 |
9,30 |
9,08 |
8,86 |
|
9,0 |
1,0 |
9,36 |
9,42 |
9,18 |
8,94 |
|
8,0 |
2,0 |
9,50 |
9,57 |
9,30 |
9,02 |
|
7,0 |
3,0 |
9,68 |
9,76 |
9,44 |
9,12 |
|
6,0 |
4,0 |
9,97 |
10,06 |
9,67 |
9,28 |
Phosphatblandinger efter SØRENSEN
|
Sammensætning |
pH |
|
0,0667 M monokaliumphosphat + 0,0667 M dinatriumphosphat ved 20 oC |
|
|
99,2 ml KH2PO4 + 0,8 ml Na2HPO4 |
5,0 |
|
98,4 ml KH2PO4 + 1,6 ml Na2HPO4 |
5,2 |
|
97,3 ml KH2PO4 + 2,7 ml Na2HPO4 |
5,4 |
|
95,5 ml KH2PO4 + 4,5 ml Na2HPO4 |
5,6 |
|
92,8 ml KH2PO4 + 7,2 ml Na2HPO4 |
5,8 |
|
88,9 ml KH2PO4 + 11,1 ml Na2HPO4 |
6,0 |
|
83,0 ml KH2PO4 + 17,0 ml Na2HPO4 |
6,2 |
|
75,4 ml KH2PO4 + 24,6 ml Na2HPO4 |
6,4 |
|
65,3 ml KH2PO4 + 34,7 ml Na2HPO4 |
6,6 |
|
53,4 ml KH2PO4 + 46,6 ml Na2HPO4 |
6,8 |
|
41,3 ml KH2PO4 + 58,7 ml Na2HPO4 |
7,0 |
|
29,6 ml KH2PO4 + 70,4 ml Na2HPO4 |
7,2 |
|
19,7 ml KH2PO4 + 80,3 ml Na2HPO4 |
7,4 |
|
12,8 ml KH2PO4 + 87,2 ml Na2HPO4 |
7,6 |
|
7,4 ml KH2PO4 + 92,6 ml Na2HPO4 |
7,8 |
|
3,7 ml KH2PO4 + 96,3 ml Na2HPO4 |
8,0 |
C.8. TOKSICITET FOR REGNORME
TEST I SYNTETISK JORD
1. METODE
1.1. INDLEDNING
I denne laboratorietest tilsættes teststoffet til syntetisk jord, hvori der anbringes orme i 14 dage. Efter denne periode (og eventuelt efter syv dage) undersøges stoffets dødelige virkning på regnormene. Testen giver en metode til i løbet af relativt kort tid at foretage en screening af kemikaliers virkning på regnorme ved optagelse gennem huden eller føden.
1.2. DEFINITION OG ENHED
LC50: Koncentration af et stof, som kan forventes at forårsage 50 % af testdyrenes død i løbet af testperioden.
1.3. REFERENCESTOF
Der anvendes regelmæssigt et referencestof til påvisning af, at testsystemets følsomhed ikke har ændret sig signifikant.
Som referencestof anbefales analytisk rent chloracetamid.
1.4. TESTMETODENS PRINCIP
Jord er et variabelt medium, så der anvendes en omhyggeligt defineret syntetisk lerjord til denne test. Der holdes voksne regnorme af arten Eisenia foetida (se anmærkningen i tillægget) i en veldefineret syntetisk jord, som er behandlet med forskellige koncentrationer af teststoffet. Beholdernes indhold spredes på en bakke 14 dage (og eventuelt syv dage) efter forsøgets indledning, og der optælles for hver koncentration, hvor mange regnorme der har overlevet.
1.5. KVALITETSKRITERIER
Testen er beregnet til at være så reproducerbar som muligt med hensyn til testsubstrat og organisme. Dødeligheden hos kontroldyrene må ikke overstige 10 % ved testens afslutning; ellers er testen ugyldig.
1.6. BESKRIVELSE AF TESTMETODEN
1.6.1. Materialer
1.6.1.1.
Der anvendes veldefineret syntetisk jord som grundsubstrat.
|
a) |
Grundsubstrat (vægtprocent tørstof)
|
|
b) |
Testsubstrat Testsubstratet består af grundsubstrat, teststof og deioniseret vand. Vandindholdet svarer til ca. 25 til 42 vægtprocent af grundsubstratets tørstof. Substratets vandindhold bestemmes ved tørring af en prøve til konstant vægt ved 105 oC. Hovedkriteriet er, at den syntetiske jord er befugtet så meget, at der ikke er noget stående vand. Blandingen foretages omhyggeligt, således at der bliver en jævn fordeling af teststoffet og substratet. Metoden for tilsætning af teststoffet til substratet skal rapporteres. |
|
c) |
Kontrolsubstrat Kontrolsubstratet bestir af grundsubstrat og vand. Hvis der anvendes et tilsætningsstof, skal et ekstra kontrolsubstrat indeholde samme mængde af tilsætningsstoffet. |
1.6.1.2.
Glasbeholdere med er rumfang på ca. en liter (hensigtsmæssigt tildækket med plastlåg, skåle eller plastfolie med ventilationshuller), fyldt med en mængde vådt test- eller kontrolsubstrat svarende til 500 g substrattørstof.
1.6.2. Testbetingelser
Beholderne opbevares i klimakamre ved en temperatur pi 20 ± 2 oC og i konstant lys; Lysstyrken bør være 400 til 800 lux.
Testperioden er på 14 dage, men dødeligheden kan eventuelt vurderes syv dage efter testens indledning.
1.6.3. Fremgangsmåde
Koncentrationerne af teststof udtrykkes som stoffets vægt i forhold til grundsubstratets tørstofvægt (mg/kg).
Det koncentrationsområde, der netop forårsager dødelighed fra 0 til 100 % kan bestemmes ved en forprøve, der giver viden om de koncentrationsintervaller, der skal anvendes ved den endelige test.
Stoffet bør testes i følgende koncentrationer: 1 000, 100, 10, 1 og 0,1 mg/stof/kg testsubstrat (tørstofvægt).
Hvis der skal foretages en fuldstændig endelig test, vil én testgruppe pr. koncentration og én til den ubehandlede kontrolgruppe med hver ti orme være tilstrækkelig til forprøven.
Resultaterne af forprøven anvendes til at vælge mindst fem koncentrationer i en kvotientrække, der går fra 0 til 100 % dødelighed, og hvor den konstante faktor ikke må være over 1,8.
Ved test med disse koncentrationsrækker vil LC50-værdien og dens konfidensgrænser kunne skønnes med størst mulig nøjagtighed.
Ved den endelige test anvendes der mindst fire testgrupper pr. koncentration og fire ubehandlede kontrolgrupper med hver ti orme. Resultaterne af disse gentagne grupper angives som en middelværdi og en standardafvigelse.
Hvis to på hinanden følgende koncentrationer med et forhold på 1,8 kun giver 0 og 100 % dødelighed, er disse to værdier tilstrækkelige som angivelse for LC50-intervallet.
Når det er muligt, tilberedes teststoffet uden anden tilsætning end vand. Umiddelbart før testens indledning blandes en emulsion eller dispersion af teststoffet i deioniseret vand eller andet opløsningsmiddel med grundsubstratet eller sprøjtes jævnt ud over dette med en fin kromatografisprøjte eller lignende.
Hvis teststoffet ikke er opløseligt i vand, kan det opløses i den mindst mulige mængde af et passende organisk opløsningsmiddel (f.eks. hexan, acetone eller chloroform).
Kun midler, der let fordamper, må bruges til at opløse, dispergere eller emulgere teststoffet. Testsubstratet beluftes inden brug, Den fordampede vandmængde erstattes. Kontrolsubstratet må indeholde eventuelle tilsætningsstoffer i samme mængde.
Hvis teststoffet ikke kan opløses, dispergeres eller emulgeres i organiske opløsningsmidler, blandes en blanding af finsand af formalet kvarts og den til behandling af 500 g tørstofvægt syntetisk jord nødvendige mængde teststof med 490 g tørstofvægt testsubstrat.
For hver testgruppe anbringes en mængde vådt testsubstrat svarende til 500 g tørstofvægt i hver glasbeholder, og ti regnorme, der i 24 timer er blevet tilvænnet i et lignende vådt grundsubstrat og derefter hurtigt afskyllet og afdryppet på filtrerpapir før brugen, anbringes på testsubstratets overflade.
Beholderne tildækkes med perforerede plastlåg, skåle eller folie for at forhindre, at substratet indtørrer, og de opbevares under testbetingelser i 14 dage.
Vurderingerne foretages 14 dage (og eventuelt syv dage) efter testens indledning. Substratet spredes på en plade af glas eller rustfrit stål. Regnormene undersøges, og antallet af overlevende regnorme bestemmes. Regnorme anses for døde, hvis de ikke reagerer på en let, mekanisk påvirkning af forender.
I tilfælde af at undersøgelsen foretages efter syv dage, fyldes substratet atter i beholderen, og de overlevende regnorme genanbringes på den samme testsubstratoverflade.
1.6.4. Testorganismer
Testorganismerne bør være voksne Eisenia foetida (se anmærkningen i tillægget) (mindst to måneder gamle med bælte) af en vægt (våd) på 300 til 600 mg. (Med hensyn til opdrætningsmetode henvises til tillægget).
2. DATA
2.1. BEHANDLING OG VURDERING AF RESULTATERNE
De anvendte teststofkoncentrationer rapporteres med henvisning til den procentvise dødelighed for hver koncentration.
Når de opnåede data er dækkende, kan LC50 og dens konfidensgrænser (p = 0,05) bestemmes ved brug af standardmetoder (Litchfield and Wilcoxon, 1949, eller tilsvarende metode). LC50 angives som mg teststof pr, kg testsubstrat (tørstofvægt).
I de tilfælde, hvor koncentrationskurvens hældning er for stejl til, at LC50 kan beregnes, er en grafisk bestemmelse af denne værdi tilstrækkelig.
Hvis to på hinanden følgende koncentrationer med et forhold på 1,8 kun giver 0 og 100 % dødelighed, er disse to værdier tilstrækkelige som angivelse for LC50-intervallet.
3. RAPPORTERING
3.1. TESTRAPPORT
Testrapporten skal om muligt indeholde følgende:
|
— |
angivelse af, at testen er foretaget i overensstemmelse med ovenstående kvalitetskriterier |
|
— |
angivelse af den foretagne test (forprøve og/eller endelig test) |
|
— |
nøje beskrivelse af testbetingelserne eller angivelse af, at testen er foretaget i overensstemmelse med metoden; eventuelle afvigelser skal rapporteres |
|
— |
nøje beskrivelse af, hvorledes teststoffet er blevet iblandet i grundsubstratet |
|
— |
oplysninger om testorganismer (art, alder, gennemsnitsvægt samt minimums- og maksimumsvægt, betingelserne for avl og opdrætning samt leverandør) |
|
— |
angivelse af den anvendte metode til bestemmelse af LC50 |
|
— |
testresultaterne, herunder alle anvendte data |
|
— |
beskrivelse af observerede symptomer eller adfærdsændringer hos testorganismerne |
|
— |
dødelighed i kontrolgrupperne |
|
— |
LC50 eller højeste testkoncentration uden dødelighed og laveste testkoncentration med en dødelighed på 100 % 14 dage (og eventuelt syv dage) efter testens indledning |
|
— |
afbildning af koncentrationsresponskurven |
|
— |
resultater, der er opnået med referencestoffet enten i forbindelse med den pågældende test eller ved tidligere kvalitetskontrolundersøgelser. |
4. HENVISNINGER
|
(1) |
OECD, Paris, 1981, Test Guideline 207, Decision of the Council C(81) 30 Final. |
|
(2) |
Edwards, C. A. and Lofty, 1977. Biology of Earthworms. London: Chapman and Hall, 331 pp. |
|
(3) |
Bouche, M. B. 1972. Lombriciens de France, Écologie et Systématique. Publ. Institut National de la Recherche Agronomique, 671 pp. |
|
(4) |
Litchfield, J. T.and Wilcoxon J., 1949. A simplified method of evaluaring dose-effect experiments J. Pharm. Exp. Therap., vol. 96, 99-113. |
|
(5) |
EEC 1983. Development of a standardized labotatory method for assessing the toxicity of chemical substances to earthworms, Report EUR. 8714 EN. |
|
(6) |
Umweltbundesamt/Biologische Bundesanstalt für Land- und Forstwirtschaft, Berlin, 1984, Verfahrensvorschlag Toxizitätstest am Regenwurm Eisenia foetida in künstlichem Boden, in: Rudolph/Boje: Ökotoxikologie, ecomed, Landsberg, 1986. |
Tillæg
Avl og opdrætning af ormene før test
Med henblik på avl anbringes 30 til 50 voksne orme i en avlekasse med frisk substrat og fjernes efter 14 dage. Disse dyr kan bruges til avl af senere grupper. De regnorme, der klækkes af kokonerne, anvendes til test, når de er kønsmodne (under de foreskrevne betingelser efter to til tre måneders forløb).
Avls- og opdrætningsbetingelser
|
Klimakammer: |
: |
temperatur 20 ± 2 oC, helst med konstant lys (lysintensitet 400 til 800 lux). |
|
Avlekasser: |
: |
Egnede lave beholdere med et rumfang på 10 til 20 l. |
|
Substrat: |
: |
Eisenia foetida kan opdrættes i forskellige dyreekskrementer. Som avlsmedium kan anbefales en blanding af lige dele tørv og ko- eller hestegødning. Substratet må have en pH-værdi på ca. 6 til 7 (justeret med calciumcarbonat) og lav ionledningsevne (under 6 mmhos eller 0,5 % saltkoncentration). Substratet bør være fugtigt, men ikke for vådt. Andre egnede fremgangsmåder ud over ovennævnte metode kan benyttes. |
Anmærkning: Der findes to racer af Eisenia foetida, som nogle systematikere har klassificeret som arter (Bouche, 1972). De er morfologisk set omtrent ens, men den ene, Eisenia foetida foetida, har typisk tværgående striber på leddene, hvad den anden, Eisenia foetida andrei, ikke har, og dennes farve er rødbroget. Så vidt muligt bør Eisenia foetida andrei anvendes. Andre arter kan anvendes, hvis den fornødne metodik er til rådighed.
C.9. BIOLOGISK NEDBRYDNING
ZAHN-WELLENS TEST
1. METODE
1.1. INDLEDNING
Metoden har til formål at vurdere vandopløselige ikke flygtige organiske stoffers potentielle fuldstændige bionedbrydelighed, når de skal udsættes for forholdsvis høje koncentrationer af mikroorganismer under en statisk test.
Der kan forekomme fysisk/kemisk adsorption på de opslæmmede artikler, og der må tages hensyn hertil, når resultaterne fortolkes (jf. 3.2).
De stoffer, der skal undersøges, anvendes i koncentrationer svarende til DOC-værdier i området 50 til 400 mg/l eller COD-vædier i området 100 til 1 000 mg/l (DOC = opløst organisk kulstof; COD = kemisk iltforbrug). Disse forholdsvis høje koncentrationer har den fordel, at de i analytisk henseende er pålidelige. Forbindelser med toksiske egenskaber kan hæmme eller hindre nedbrydningsprocessen.
Ved denne metode anvendes måling af koncentrationer af opløst organisk kulstof eller af det kemiske iltforbrug til bedømmelse af teststoffets fuldstændige biologiske nedbrydning.
Samtidig anvendelse af en specifik analysemetode kan muliggøre vurdering af stoffets primære biologiske nedbrydning (dvs. den oprindelige kemiske forbindelses forsvinden).
Metoden finder kun anvendelse pi de organiske teststoffer, som ved koncentrationen under testen:
|
— |
er opløselige i vand under forsøgsbetingelserne |
|
— |
har forsvindende lavt damptryk under forsøgsbetingelserne |
|
— |
ikke hæmmer bakterier |
|
— |
kun adsorberes i testsystemet i begrænset omfang |
|
— |
ikke forsvinder fra testopløsningen som følge af skumning. |
Oplysninger om mængdeforholdet mellem teststoffets hovedbestanddele vil være til nytte ved fortolkning af resultaterne, især hvis disse er lave eller usikre.
Ved fortolkningen af lave resultater og ved valget af passende testkoncentrationer er det ønskeligt at have oplysninger om stoffets giftvirkning over for mikroorganismer.
1.2. DEFINITIONER OG ENHEDER
Nedbrydningsgraden ved testens afslutning anføres således: Biologisk nedbrydelighed i Zahn-Wellens test:
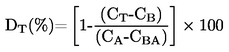
hvor:
|
DT |
= |
biologisk nedbrydning ( %) ved tidspunktet T |
|
CA |
= |
DOC (eller COD)-værdier i forsøgsblandingen, målt tre timer efter igangsætning af testen (mg/l). (DOC = opløst organisk kulstof, COD = kemisk iltforbrug) |
|
CT |
= |
DOC- eller COD-værdier i forsøgsblandingen ved tidspunktet for prøveudtagning (mg/l) |
|
CB |
= |
DOC- eller COD-værdi for blindprøven ved tidspunktet for prøveudtagning (mg/l) |
|
CBA |
= |
DOC- eller COD-værdi for blindprøven, målt tre timer efter igangsætning af testen (mg/l). |
Nedbrydningsgraden afrundes til nærmeste hele procent.
Den procentvise nedbrydning er angiver som den procentvise DOC- (eller COD-) fjernelse af teststoffet.
Forskellen mellem den værdi, der er målt efter tre timer, og den beregnede, eller helst målte, startværdi kan give nyttige oplysninger om stoffets eliminering (jf. 3.2 »Fortolkning af resultater«).
1.3. REFERENCESTOFFER
I visse tilfælde kan referencestoffer være nyttige ved undersøgelse af nye stoffer; der kan dog endnu ikke anbefales bestemte referencestoffer.
1.4. TESTMETODENS PRINCIP
Aktiveret slam, uorganiske næringsstoffer og teststoffet, der er den eneste kulstofkilde i vandig opløsning, anbringes sammen i en glasbeholder på 1 til 4 liter, som er forsynet med omrøring og beluftning. Blandingen omrøres og beluftes ved 20 til 25 oC under indirekte belysning eller mørke i op til 28 dage. Nedbrydningsprocessen følges ved bestemmelse af DOC- (eller COD-) værdier i den filtrerede opløsning en gang dagligt eller regelmæssigt med et andet passende mellemrum. Forholdet mellem den eliminerede DOC (eller COD) efter hvert tidsrum og værdier tre timer efter starten udtrykkes som procentvis biologisk nedbrydning og er et mål for nedbrydningsgraden på dette tidspunkt. Resultatet afbildes mod tiden, hvorved kurven over biologisk nedbrydning fremkommer.
Hvis der anvendes en specifik analysemetode, kan ændringer i det oprindelige stofs koncentration som følge af biologisk nedbrydning måles (primær bionedbrydelighed).
1.5. KVALITETSKRITERIER
Det er ved ringtest godtgjort, at denne metodes reproducerbarhed er tilfredsstillende.
Metodens følsomhed afhænger hovedsagelig af blindprøvens variationer og i mindre grad af nøjagtigheden ved bestemmelsen af opløst organisk kulstof og teststofkoncentrationen i væsken.
1.6. BESKRIVELSE AF FREMGANGSMÅDEN
1.6.1. Forberedelser
1.6.1.1.
vand: drikkevand med mindre end 5 mg organisk kulstof pr. liter. Koncentrationen af calcium- og magnesiumioner må ikke overstige 2,7 mmol/l tilsammen; i modsat fald må der fortyndes passende med deioniseret eller destilleret vand
|
svovlsyre, analyseren (A.R.): |
50 g/l |
|
natriumhydroxidopløsning, A.R.: |
40 g/l |
|
opløsning af uorganiske næringsstoffer: i 1 1 deioniseret vand opløses: |
|
|
Ammoniumchlorid, NH4Cl, A.R.: |
38,5 g |
|
Natriumdihydrogenphosphat, NaH2PO4. 2H2O, A.R.: |
33,4 g |
|
Kaliumdihydrogenphosphat, KH2PO4, A.R.: |
8,5 g |
|
Dikaliummonohydrogenphosphat, K2HPO4, A.R.: |
21,75 g |
Blandingen fungerer dels som næringsstofkilde, dels som buffersystem.
1.6.1.2.
glasbeholdere på 1 til 4 (f.eks. cylindriske beholdere)
omrører med rørehoved af glas eller metal på en passende aksel (omrøreren bør rotere 5 til 10 cm over beholderens bund). Der kan i stedet anvendes en magnetomrører med en 7 til 10 cm lang magnetpind
glasrør med indvendig diameter på 2 til 4 mm til beluftning. Rørets åbning bør befinde sig ca. 1 cm over beholderens bund
centrifuge (ca. 3 550 g)
pH-meter
apparat til måling af opløst ilt
papirfiltre
apparatur til membranfiltrering
membranfiltre med porestørrelse 0,45 μm. Membranfiltre er kun egnede, hvis der er vished for, at de hverken afgiver kulstof eller absorberer stoffet under filtreringen
analyseudstyr ti! bestemmelse af indholdet af organisk kulstof og det kemiske iltforbrug.
1.6.1.3.
Aktiveret slam fra et biologisk rensningsanlæg vaskes (flere gange) med vand (jf. ovenfor) ved centrifugering eller dekantering.
Det aktiverede slam skal være i passende stand. Sådant slam kan fås fra et velfungerende anlæg til spildevandsrensning. For at få så mange forskellige bakteriestammer som muligt kan det være bedre at blande inocula fra forskellige kilder (f.eks. forskellige rensningsanlæg, jordekstrakter, flodvand osv.).
Det aktiverede slams aktivitet kontrolleres, jf. »egnethedskontrol«.
1.6.1.4.
Til testbeholderen sættes 500 ml vand, 2,5 ml opløsning af uorganiske næringsstoffer pr. liter samt en mængde aktiveret slam, der svarer til 0,2 til 1,0 g tørstof pr. liter færdigblanding. Der tilsættes en så stor mængde stamopløsning af teststoffer, at den færdige blanding får en DOC-koncentration på 50 til 400 mg/l. Det tilsvarende COD-interval er 100 til 1 000 mg/l. Der fyldes op til i alt 1 til 4 l med vand. Valg af det samlede volumen afhænger af det antal prøver, der skal udtages til DOC- eller COD-bestemmelser, og det nødvendige analysevolumen.
Normalt kan et volumen på 2 l betragtes som tilstrækkeligt. Mindst én kontrolbeholder (blindprøve) sættes i gang samtidig med hver testserie; den indeholder kun aktiveret slam og opløsning af uorganiske næringsstoffer fortyndet med vand til samme totalvolumen som i testbeholderne.
1.6.2. Testens gennemførelse
Testbeholderne omrøres med magnetomrørere eller propelomrørere under indirekte belysning eller i mørke ved 20 til 25 oC. Beluftning sker med komprimeret luft, der om nødvendigt renses med et vatfilter og en vaskeflaske. Det må påses, at slammet ikke bundfælder, og at iltkoncentrationen ikke falder til under 2 mg/l.
pH-værdien kontrolleres regelmæssigt (f.eks. dagligt) og indstilles om nødvendigt til pH 7 til 8.
Lige før prøveudtagningen kompenseres der for tab ved fordampning ved tilsætning af deioniseret eller destilleret vand. Det kan f.eks. anbefales at afmærke væskehøjden i beholderen, før testen startes. Der sættes nye mærker efter hver prøveudtagning (uden beluftning og omrøring). De første-prøver udtages altid tre timer efter testens start, således at det kan konstateres, om testmaterialet adsorberes på det aktiverede slam.
Elimineringen af testmaterialet følges ved DOC- eller COD-bestemmelser dagligt eller regelmæssigt med et andet interval. Prøverne fra testbeholderen og blindforsøget filtreres gennem et omhyggeligt vasket papirfilter. De første 5 ml filtrat bortkastes. Slam, der er vanskeligt at filtrere, kan fjernes ved først at centrifugere i ti min. Der foretages mindst dobbeltbestemmelse af DOC og COD. Testens løber i op til 28 dage.
Anmærkning: Prøver, der stadig er uklare, filtreres gennem et membranfilter. Membranfiltrene må hverken afgive eller adsorbere organisk materiale.
I hver testserie bør indgå en beholder, der indeholder et kendt stof, således at man kan kontrollere egnetheden af det aktiverede slam. Til dette formål er diethylenglycol fundet egnet.
Hvis der foretages analyser med forholdsvis korte mellemrum (f.eks, dagligt), vil adaptation klart fremgå af nedbrydningskurven (jf. figur 2). Testen bør derfor ikke sættes i gang umiddelbart før weekenden.
Hvis der forekommer adaptation ved slutningen af perioden, kan testen fortsættes, indtil nedbrydningen er afsluttet.
Anmærkning: Hvis der er behov for større viden om det adapterede slams opførsel, udsattes det samme aktiverede slam endnu engang for det samme testmateriale efter følgende fremgangsmåde:
Omrøringen og beluftningen standses, og man lader det aktiverede slam bundfælde sig. Den ovenstående væske fjernes, der fyldes op til 2 l med vand og omrøres i 15 minutter, og blandingen henstår til ny bundfældning. Efter fjernelse af den ovenstående væske anvendes det tilbageblevne slam til gentagelse af testen med det samme materiale som under punkt 1.6.1.4 og 1.6.2. Det aktiverede slam kan isoleres ved centrifugering i stedet for bundfældning.
Det adapterede slam kan eventuelt blandes med frisk slam, så man når op på i alt 0,2 til 1,0 g tørstof pr. liter.
Normalt filtreres prøver gennem et omhyggeligt vasket papirfilter (til vask anvendes deioniseret vand).
Prøver, der stadig er uklare, filtreres gennem et membranfilter (0,45 μm).
Der foretages dobbeltbestemmelse af DOC-koncentrationen i prøvefiltraterne (de første 5 ml bortkastes) med TOC-instrumentet. Hvis filtratet ikke kan analyseres-samme dag, skal det opbevares i køleskab til næste dag. Længere tids opbevaring kan ikke anbefales.
COD-koncentrationen bestemmes i prøvefiltraterne med et COD-analyseudstyr efter den fremgangsmåde, der er beskrevet i henvisning (2) nedenfor.
2. DATA OG VURDERING
Der foretages mindst dobbeltbestemmelse af DOC- og COD-koncentrationerne i prøverne ifølge 1.6.2. Nedbrydningen ved tidspunktet T beregnes med den formel (med definitioner), der anført under 1.2.
Nedbrydningsgraden afrundes til nærmeste hele procent. Den nedbrydning, der opnås ved slutningen af testen, anføres som »den biologiske nedbrydelighed ved Zahn-Wellens test«.
Anmærkning: Hvis der opnås fuldstændig nedbrydning før testperioden er udløbet og dette resultat bekræftes ved endnu en analyse den følgende dag, kan testen afsluttes.
3. RAPPORTERING
3.1. TESTRAPPORT
Testrapporten skal om muligt indeholde følgende:
|
— |
stoffets begyndelseskoncentration |
|
— |
alle andre oplysninger og forsøgsresultaterne vedrørende teststoffet, et eventuelt referencestof og blindforsøget |
|
— |
koncentrationen efter tre timer |
|
— |
kurve over biologisk nedbrydning med forklaring |
|
— |
hvornår og hvor prøveorganismerne er udtaget, adaptionsgrad, anvendt koncentration osv. |
|
— |
fyldestgørende begrundelse for alle afvigelser fra fremgangsmåden. |
3.2. FORTOLKNING AF RESULTATER
Gradvis fjernelse af DOC (COD) i løbet af dage eller uger viser, at teststoffet nedbrydes biologisk.
Fysisk/kemisk adsorbtion kan dog i visse tilfælde spille en vis rolle, og et tegn herpå er, at der inden for de første tre timer sker fuldstændig eller delvis fjernelse, og at forskellen mellem supernatantvæsken fra kontrolforsøget og prøven bliver liggende på et uventet lavt niveau.
Det er nødvendigt at foretage yderligere testning, hvis der skal skelnes mellem biologisk nedbrydning (eller delvis biologisk nedbrydning) og adsorption.
Dette kan gøres på flere måder, mest troværdigt ved at anvende supernatantvæsken som inoculum i en basissat test (helst en respirometrisk test).
Teststoffer, der giver høj fjernelse af DOC (COD), som ikke skyldes adsorption, ved denne test, bør betragtes som potentielt biologisk nedbrydelige. Delvis fjernelse, som ikke skyldes adsorption, betyder, at kemikaliet i hvert fald i et vist omfang nedbrydes biologisk. Ringe eller ingen fjernelse af DOC (COD) kan skyldes, at teststoffet hæmmer mikroorganismerne; dette kan undertiden også vise sig ved, at slammet opløses og forsvinder, hvorved supernatantvæsken bliver uklar. Testen bør gentages med en lavere koncentration af teststof.
Anvendelse af en stofspecifik analysemetode eller 14C-mærket teststof kan give større følsomhed. Hvis der anvendes l4C-mærket teststof, vil påvisning af 14CO2 bekræfte, at der er tale om biologisk nedbrydning.
Dersom resultaterne anføres som primær biologisk nedbrydning, bør der om muligt anføres en forklaring pi den kemiske strukturændring, som fører til, at analyseresultaterne for det oprindelige teststof falder.
Valget af analysemetode skal underbygges, og analyseresultaterne for blindprøven skal anføres.
4. HENVISNINGER
|
(1) |
OECD, Paris, 1981 Test Guideline 302 B. Decision of the Council C(81) 30 Final. |
|
(2) |
Bilag V C.9 Nedbrydning: Kemisk iltforbrug. Kommissionens direktiv 84/449/EØF, De Europæiske Fællesskabers Tidende, L 251 af 19.9.1984. |
Tillæg
EKSEMPLER PÅ EVALUERING
|
Organisk forbindelse: |
4-ethoxybenzoesyre |
|
Teoretisk testkoncentration: |
600 mg/l |
|
Teoretisk DOC: |
390 mg/l |
|
Inoculum |
rensningsanlægget ... |
|
Koncentration: |
1 g tørstof pr. liter |
|
Eventuel adaptation: |
ikke adapteret |
|
Analyse: |
DOC-bestemmelse |
|
Testmængde: |
3 ml |
|
Kontrolstof: |
diethylenglycol |
|
Stoffets toksicitet |
ingen giftvirkning under 1 000 mg/l anvendt prøve: fermeteringsglas |
|
Tidspunkt |
Kontrolstof |
Teststof |
|||||
|
Blindprøve DOC (16) mg/l |
DOC (16) mg/l |
DOC netto mg/1 |
Nedbrydning % |
DOC (16) mg/l |
DOC netto mg/l |
Nedbrydning % |
|
|
0 |
— |
— |
300,0 |
— |
— |
390,0 |
— |
|
3 timer |
4,0 |
298,0 |
294,0 |
2 |
371,6 |
367,6 |
6 |
|
1 dag |
6,1 |
288,3 |
282,2 |
6 |
373,3 |
367,2 |
6 |
|
2 dage |
5,0 |
281,2 |
276,2 |
8 |
360,0 |
355,0 |
9 |
|
5 dage |
6,3 |
270,5 |
264,2 |
12 |
193,8 |
187,5 |
52 |
|
6 dage |
7,4 |
253,3 |
245,9 |
18 |
143,9 |
136,5 |
65 |
|
7 dage |
11,3 |
212,5 |
201,2 |
33 |
104,5 |
93,2 |
76 |
|
8 dage |
7,8 |
142,5 |
134,7 |
55 |
58,9 |
51,1 |
87 |
|
9 dage |
7,0 |
35,0 |
28,0 |
91 |
18,1 |
11,1 |
97 |
|
10 dage |
18,0 |
37,0 |
19,0 |
94 |
20,0 |
2,0 |
99 |
Figur 1
Eksempler på kurver over biologisk nedbrydning

Figur 2
Eksempler på slamadaptation

C.10. BIOLOGISK NEDBRYDNING
AKTIVERET SLAM — SIMULATIONSTEST
1. METODE
1.1. INDLEDNING
1.1.1. Generelle bemærkninger
Metoden er kun anvendelig for organiske teststoffer, der ved den benyttede testkoncentration:
|
— |
er opløselige i vand i det omfang, det er nødvendigt for tilberedning af testopløsningerne |
|
— |
har et ubetydeligt damptryk under testbetingelserne |
|
— |
ikke er hæmmende for bakterier. |
Oplysninger om de relative mængder af teststoffets hovedbestanddele vil være nyttige for fortolkningen af de opnåede resultater, især i de tilfælde, hvor resultaterne er lave eller marginale.
Oplysninger om stoffets toksicitet for mikroorganismerne kan være nyttige for fortolkningen af lave resultater og ved valget af egnede testkoncentrationer.
1.1.2. Bestemmelse af fuldstændig bionedbrydelighed (DOC/COD-analyse)
Formålet med metoden er at bestemme den fuldstændige bionedbrydelighed ved måling af fjernelsen af stoffet og eventuelle metabolitter i en aktivslamanlægsmodel ved en koncentration svarende til 12 mg DOC/l (eller ca. 40 mg COD/l); 20 mg DOC/l synes at være optimal. (DOC = dissolved organic carbon, dvs. opløst organisk kulstof; COD = chemical oxygen demand, dvs. kemisk oxygenforbrug).
Teststoffets indhold af organisk kulstof (eller dets kemiske oxygenforbrug) skal fastlægges.
1.1.3. Bestemmelse af primær bionedbrydelighed (specifik analyse)
Formålet med metoden er at bestemme den primære bionedbrydelighed af et stof i en aktivslamanlægsmodel ved en koncentration på ca. 20 mg/l ved anvendelse af en specifik analysemetode (der kan benyttes lavere eller højere koncentration, hvis analysemetoden og toksicitetshensyn tillader dette). Herved kan stoffets primære bionedbrydelighed bedømmes (dvs. forsvinden af den oprindelige kemiske forbindelse).
Formålet med denne metode er ikke at bestemme teststoffets mineralisering.
Der må foreligge en passende analysemetode til bestemmelse af teststoffet.
1.2. DEFINITIONER OG ENHEDER
1.2.1. DOC/COD-analyse
Nedbrydningen er defineret som den del af teststoffet, der fjernes, efter følgende formel:
|
|
[1 a)] |
hvor:
|
DR. |
= |
procent DOC (eller COD) fjernet i den givne middelopholdstid med hensyn til teststoffet |
|
T |
= |
teststofkoncentrationen i indløbet i mg DOC/l (eller mg COD/l) |
|
E |
= |
DOC-koncentration (eller COD-koncentration) i testenhedens udløb i mg DOC/l (eller mg COD/l) |
|
Eo |
= |
DOC-koncentration (eller COD-koncentration) i blindprøveenhedens udløb i mg DOC/l (eller COD/l). |
Nedbrydningen angives som den procentdel DOC (eller COD), der fjernes i en given retentionstid med hensyn til teststoffet.
1.2.2. Specifik analyse
Den procentdel teststof, der elimineres fra den vandige fase (Rw) i den givne middelopholdstid, fremgår af følgende formel:
|
|
[1 b)] |
hvor:
|
Cl |
= |
stofkoncentrationen: i testenhedens indløb (mg stof/l, bestemt ved specifik analyse) |
|
Co |
= |
stofkoncentrationen i testenhedens udløb (mg stof/l, bestemt ved specifik analyse). |
1.3. REFERENCESTOFFER
I nogle tilfælde, hvor et nyt stof undersøges, kan referencestoffer være nyttige; der kan endnu ikke anbefales specifikke referencestoffer.
1.4. TESTMETODENS PRINCIP
Til bestemmelse af den endelige bionedbrydelighed benyttes der parallelt-to aktiv slam pilotenheder (OECD »Confirmatory test-enheder« eller »Porous pot-enheder«). Teststoffet tilsættes til den ene enheds indløb (syntetisk-spildevand eller husholdningsspildevand), medens den anden enhed kun tilføres spildevand. Til bestemmelse af primær bionedbrydning med specifik analyse i indløb og udløb benyttes der kun en enhed.
DOC-koncentrationerne (eller COD-koncentrationerne) måles i udløbene, eller stofkoncentrationerne bestemmes ved specifik analyse.
DOC, kom skyldes teststoffet, måles ikke, men angives blot.
Når der foretages DOC-målinger (eller COD-målinger), antages forskellene mellem test- og kontroludløbenes gennemsnitkoncentrationer at skyldes ikke-nedbrudt teststof.
Når der foretages specifikke analyser, kan ændringen i stamforbindelsens koncentration måles (primær bionedbrydning).
Der kan arbjedes med enhederne efter »koblingsmetoden«, hvor der foretages transinokulering.
1.5. KVALITETSKRITERIER
Stoffets startkoncentration afhænger af, hvilken type analyse der foretages og dens begrænsniger.
1.6. BESKRIVELSE AF TESTMETODEN
1.6.1. Forberedelser
1.6.1.1.
Der kræves to enheder af samme type, undtagen når der foretages specifikke analyser. Der er to typer, der kan benyttes:
OECD Confirmatory test
Udstyret (tillæg 1) består af holdetank (A) til syntetisk spildevand, doseringspumpe (B), luftningstank (C), separator (sedimenteringstank) (D), slampumpe (E) til recirkulering af aktiveret slam samt tank (F) til opsamling af det behandlede spildevand.
Tank (A) og (F) skal være af glas eller egnet plast og skal mindst kunne rumme 24 l. Pumpen (B) skal give et konstant flow af syntetisk spildevand til luftningstanken; ethvert egnet system kan benyttes, forudsat at indløbsflow og koncentration er sikret. Normalt er separatorens højde (D) indstillet således, at luftningstanken indeholder 3 l blandet væske. En sintret luftningsklods (G) er ophængt i tank (C) over keglens spids. Den luftmængde, der blæses gennem luftningstanken, kan kontrolleres ved hjælp af et flowmeter.
Slampumpen (E) er indstillet således, at de aktiverede slam fra separatoren kontinuerligt og regelmæssigt recirkuleres til luftningstanken (C).
Porous pot:
Den porøse krukke laves af porøs polyethylenplade (2 mm tyk, maksimal porestørrelse 95 μm), der formes til cylindre med en diameter på 14 cm og en konisk bund, der danner en vinkel på 45o med siderne (figur 1 og 2 i tillæg 2). Den porøse krukke anbringes i en uigennemtrængelig beholder af egnet plastic med en diameter på 15 cm og et udtag i 17,2 cm højde på den cylindriske del, som bestemmer, hvor meget krukken kan rumme (3 l). Der er en stiv støttering af egnet-plast omkring den indre beholders øverste kant, således at der er 0,5 cm plads til udløbsvand mellem den indre og den ydre beholder.
De porøse krukker kan opstilles på bunden af et termostatstyret vandbad. Den indre beholder tilføres luft i bunden, hvorpå der er anbragt egnede luftningsklodser.
Beholder (A) og (E) skal være af glas eller egnet plast og mindst kunne rumme 24 l. Pumpen (B) skal give et konstant flow af syntetisk spildevand til luftningstanken; ethvert egnet system kan benyttes, forudsat at indløbsflow og koncentration er sikret.
Der er behov for ekstra indre porøse krukker til erstatning af krukker, der måtte blive tilstoppet; tilstoppede krukker renses ved 24 timers neddypning i en hypochloritopløsning efterfulgt af grundig afskylning i ledningsvand.
1.6.1.2.
Membranfiltreringsapparatur og membranfiltre med en porestørrelse pi 0,45 μm. Membranfiltre er egnede, hvis det sikres, at de hverken afgiver kulstof eller adsorberer teststoffet under filtreringen.
1.6.1.3.
Der kan anvendes enten syntetisk spildevand eller husholdningsspildevand.
Eksempel på syntetisk spildevand
Pr. liter ledningsvand opløses:
|
pepton: |
160 mg |
|
kødekstrakt: |
110 mg |
|
urea: |
30 mg |
|
NaCl: |
7 mg |
|
CaCl2.2H2O: |
4 mg |
|
MgSO4.7H2O: |
2 mg |
|
K2HPO4: |
28 mg. |
Husholdningsspildevand:
Husholdningsspildevandet indsamles frisk hver dag fra overløbet fra primærsedimenteringstanken i et rensningsanlæg, der overvejende behandler husholdningsspildevand.
1.6.1.4.
Der fremstilles en opløsning af teststoffet (f.eks. 1 %) for tilsætning til testenheden. Stofkoncentrationen skal bestemmes, så det vides, hvilken mængde der skal tilsættes spildevandet eller direkte i enheden via en anden pumpe for at give den krævede testkoncentration.
1.6.1.5.
Anmærkning: Hvis der anvendes husholdningsspildevand, tjener det ikke noget formål at anvende et inoculum med lav bakteriekoncentration, men der kan bruges aktiveret slam.
Der kan anvendes forskellige former for inoculum.
Her er tre eksempler på et egnet inoculum:
|
a) |
Inoculum fra sekundært spildevand Inoculum laves af sekundært spildevand af god kvalitet fra et rensningsanlæg, der overvejende behandler husholdningsspildevand, Prøven skal holdes under aerobe forhold indtil brugen. Som forberedelse af inoculum filtreres prøven gennem et groft filter, idet de første 200 ml bortkastes. Filtratet holdes under aerobe forhold indtil brugen. Inoculum skal anvendes samme dag, som prøven er taget. Der skal bruges mindst 3 ml til inokulering. |
|
b) |
Blandet inoculum Inoculum fra sekundært spildevand: Se beskrivelsen ovenfor. Inoculum fra jord: 100 g havejord (fertil, ikke steril) opslæmmes i 1 000 ml chlorfrit drikkevand (jord med et ekstremt højt indhold af ler, sand eller humus er ikke egnet). Efter omrøring henstår suspensionen i 30 minuttet for sedimentering. Supernatanten filtreres gennem et groft filtrerpapir, idet de første 200 ml bortkastes. Beluftning af filtratet begyndes straks og fortsættes indtil brugen. Inoculum skal anvendes samme dag, som prøven er taget. Inoculum fra overfladevand: Der tages endnu en del til inoculum fra et mesosaprobt overfladevand. Prøven filtreres gennem et groft filtrepapir, idet de første 200 ml bortkastes. Filtratet holdes under aerobe forhold indtil brugen. Inoculum skal anvendes samme dag, som prøven er taget. Lige dele af de tre inoculumprøver blandes godt sammen, og det endelige inoculum tages fra denne blanding. Der skal bruges mindst 3 ml til inokulering. |
|
c) |
Inoculum af aktiveret slam Der kan som inoculum bruges en mængde (hejst 3 l) aktiveret slam (indhold af suspenderede faste bestanddele på op til 2,5 g/l) udtaget fra luftningstanken i et anlæg, der overvejende behandler husholdningsspildevand. |
1.6.2. Fremgangsmåde
Testen foretages ved stuetemperatur; temperaturen skal være mellem 18 og 25 oC.
Hvis det er hensigtsmæssigt, kan testen foretages ved en lavere temperatur (ned til 10 oC): hvis stoffet nedbrydes, kræves der normalt ikke noget yderligere. Hvis stoffet imidlertid ikke nedbrydes, må testen foretages ved en konstant temperatur på mellem 18 og 25 oC.
1.6.2.1.
Slamdannelses-/stabiliseringsperioden er den periode, hvori koncentrationen af de suspenderede faste bestanddele af det aktiverede slam og enhedernes ydeevne bliver stationær under de anvendte driftsbetingelser.
Indkøringsperioden er den periode, der går fra det tidspunkt, hvor teststoffet første gang tilsættes, indtil det tidspunkt, hvor dets fjernelse har nået et plateau (relativt konstant værdi). Denne periode må højst være på seks uger.
Vurderingsperioden er en tre-ugersperiode, tre uger fra det tidspunkt, hvor teststoffets fjernelse når en relativt konstant og sædvanligvis høj værdi. For stoffer, der viser ringe eller ingen nedbrydning i de første seks uger, regnes vurderingsperioden for de følgende tre uger.
Man indleder med at fylde den eller de enheder, der er nødvendige til én test, med inoculum blandet med tilløb.
Beluftningen (og luftpumpen (E), hvis der er tale om OECD Confirmatory test-enheder) og doseringspumpen (B) sættes derefter i gang.
Tilløb uden teststof skal passere gennem luftningstanken (C) med en hastighed på enten 1 l i timen eller 0,5 l i timen; det giver en middelopholdstid på enten tre eller seks timer.
Beluftningshastigheden reguleres, således at tankens (C) indhold konstant holdes i suspension, medens indholdet af opløst ilt er mindst 2 mg/l.
Man må med egnede midler forhindre skumdannelse. Der må ikke anvendes skumdæmpende midler, der hæmmer det aktiverede slam.
Det slam, der har samlet sig rundt om toppen af luftningstanken (C) (og for OECD Confirmatory test-enheders vedkommende i bunden af sedimenteringstanken (D) og i kredsløbet), skal mindst en gang om dagen føres tilbage til den blandede væske ved afbørstning eller på anden passende vis.
Hvis slammet ikke sedimenterer, kan dets massefylde øges ved tilsætning af 2 ml-portioner af en 5 % ferri-chloridopløsning; dette gentages om fornødent.
Udløbet opsamles i tank (E) eller (F) i 20 til 24 timer, og der udtages en prøve efter grundig blanding. Tanken (E eller F) må omhyggeligt rengøres.
For at kunne overvåge og kontrollere, at processen er effektiv, måles mindst to gange om ugen det kemiske oxygenforbrug (COD) eller indholdet af opløst organisk kulstof (DOC) både i filtratet af det akkumulerede udløb og i det filtrerede indløb (der bruges en membran med en porestørrelse på 0,45 μm, idet de første (ca.) 20 ml af filtratet bortkastes.
COD- eller DOC-reduktionen skulle flade ud, når der er opnået en nogenlunde regelmæssig daglig nedbrydning.
Tørstofindholdet i det aktiverede slam i luftningstanken bestemmes mindst to gang: om ugen (i g/l). Man kan benytte enhederne på to forskellige måder: enten bestemmes det aktiverede slams tørstofindhold to gange ugentligt, og hvis det er over 2,5 g/l, bortkastes det overskydende aktiverede slam, eller der fjernes dagligt 500 ml blandet væske fra hver krukke, således at slammet får en middelopholdstid på seks dage.
Når de målte og skønnede parametre (processens effektivitet (med hensyn til COD- eller DOC-fjernelse), slamkoncentration, slamsedimenteringsegenskaber, udløbenes uklarhed osv.) for de to enheder er tilstrækkeligt stabile, kan teststoffet tilsættes indløbet til den ene af enhederne (i henhold til 1.6.2.2).
Det er også muligt at tilsætte teststoffet ved begyndelsen af slamdannelsesperioden (1.6.2.1), især hvis slammet tilsættes som inoculum.
1.6.2.2.
Arbejdsbetingelserne er de samme som for indkøringsperioden, og der tilsættes så meget stamopløsning (ca. 1 %) af teststoffet til testenhedens indløb, at den ønskede teststofkoncentration (ca. 10 til 20 mg DOC/l eller 40 mg COD/l) i spildevandet opnås. Det kan ske ved, at stamopløsningen blandes i spildevandet dagligt eller ved hjælp af et særskilt pumpesystem. Denne koncentration kan opnås gradvis. Hvis teststoffet ikke har nogen toksiske virkninger på det aktiverede slam, kan også højere koncentrationer testes.
Blindprøveenheden tilføres kun indløb uden tilsatte stoffer. Der udtages passende mængder af udløbene til analyse, og de filtreres gennem membranfiltre (0,45 μm), idet de første (ca.) 20 ml filtrat bortkastes.
De filtrerede prøver skal analyseres samme dag eller konserveres ved en egnet metode, f.eks. ved brug af 0,05 ml af en 1 % mercurichloridopløsning (HgCl2) for hver 10 ml filtrat eller ved opbevaring ved 2 til 4o C i indtil 24 timer eller under – 18o C i en længere periode.
Indkøringstiden med tilsætning af teststoffet bør ikke være på over seks uger, og vurderingsperioden bør ikke være på under tre uger, dvs. at der foreligger 14 til 20 bestemmelser til beregning af det endelige resultat.
Koblingsmetoden:
Enhederne kobles ved at ombytte 1,5 l blandet væske (inkl. slam) mellem de to enheders luftningskamre en gang om dagen. Hvis der er tale om stærkt adsorberende teststoffer, tages der kun 1,5 l supernatant fra sedimenteringstankene, som hældes i den anden enheds aktivslamtank.
1.6.2.3.
Der kan foretages to former for analyse for at følge, hvad der sker med stoffet:
DOC og COD
Der foretages dobbeltbestemmelse af DOC-koncentrationerne med kulstofanalysatoren og/eller af COD-værdierne ifølge henvisning (2).
Specifik analyse:
Teststofkoncentrationerne bestemmes ved en egnet analysemetode. Når det er muligt, foretages der en særlig bestemmelse af det stof, der er adsorberet på slammet.
2. DATA OG VURDERING
2.1. KOBLINGSMETODEN
Når koblingsmetoden anvendes, udregnes den dagligt fjernelse, DR, ifølge 1.2.1.
De daglige værdier for fjernelse, DR, korrigeres for den materialeoverførsel, der skyldes transinokuleringen, og benævnes DRc; der benyttes ligning [2] for en tre timers eller ligning [3] for en seks timers middelopholdstid.
|
|
[2] |
|
|
[3] |
Gennemsnittet af DRc-værdierne udregnes samt standardafvigelsen ifølge ligning [4].
|
|
[4] |
hvor:
|
SDRc |
= |
DRc-værdiernes standardafvigelse |
|
|
= |
gennemsnit af DRc-værdierne |
|
n |
= |
antal bestemmelser. |
Outliers i DRc-værdierne elimineres ved en egnet statistisk fremgangsmåde, f.eks. Nalimov (henvisning (6)), på 95 % sandsynlighedsniveauet, og gennemsnit og standardafvigelse for DRc-datasættet uden outliers udregnes igen.
Slutresultatet udregnes derefter med ligning [5] som:
|
|
[5] |
hvor:
|
tn-1;α |
= |
tabelværdien af t for n værdipar af E og E0 og ved en statistisk konfidens på P (P = 1-α), hvor P sættes til 95 % (henvisning (1)). |
Resultatet angives som gennemsnittet med tolerancer på 95 % sandsynlighedsniveauet, den tilsvarende standardafvigelse og antallet af data i DRc-datasættet uden outliers samt antallet af outliers, f.eks.:
|
DRc |
= |
98,6±2,3 % DOC-fjernelse |
|
s |
= |
4,65 % DOC-fjernelse |
|
n |
= |
18 |
|
X |
= |
antal outliers. |
2.2. METODE UDEN KOBLING
Enhedernes ydeevne kan kontrolleres på følgende måde:

Den daglige fjernelse kan afbildes grafisk, således at det fremgår, om der er nogen tendenser til f.eks. tilvænning.
2.2.1. Benyttelse af COD/DOC-bestemmelser
Den daglige procent fjernelse, % DR, udregnes ifølge 1.2.1.
Gennemsnittet af DR- værdierne udregnes samt standardafvigelsen ifølge:
|
|
[6] |
hvor:
|
SDR |
= |
DRi-værdiernes standardafvigelse |
|
|
= |
gennemsnit i DRi-værdierne |
|
n |
= |
antal bestemmelser. |
Outliers i DR-værdierne elimineres ved en egnet statistisk fremgangsmåde, f.eks. Nalimov (henvisning (6)), på 95 % sandsynlighedsniveauet, og gennemsnit og standardafvigelse for DR-datasættet uden outliers udregnes igen.
Slutresultatet udregnes derefter med ligning [7] som:
|
|
[7] |
hvor:
|
tn-1;α |
= |
tabelværdi af t for n værdipar af E og E0 og ved en statistisk konfidens på P(P = 1-α), hvor P sættes til 95 % (henvisning (1)). |
Resultatet angives som gennemsnittet med tolerancer på 95 % sandsynlighedsniveauet, den tilsvarende standardafvigelse og antallet af data i DR-datasættet uden outliers samt antallet af outliers, f.eks.:
|
DR |
= |
(98,6±2,3) % DOC-fjernelse |
|
s |
= |
4,65 % DOC-fjernelse |
|
n |
= |
18 |
|
x |
= |
antal outliers. |
2.2.2. Benyttelse af specifik analyse
Den procentdel teststof, der elimineres fra den vandige fase (Rw), udregnes ifølge 1.2.2.
3. RAPPORTERING
3.1. TESTRAPPORT
Testrapporten skal om mulige indeholde følgende:
|
— |
det i tillæg 3 gengivne skema, der viser arbejdsbetingelserne for testen |
|
— |
det valgte apparatur (OECD Confirmatory test eller porous pot) |
|
— |
den valgte arbejdsmetode: koblingsmetode eller ej |
|
— |
form for spildevand: syntetisk spildevand eller husholdningsspildevand. Hvis der er tale om husholdningsspildevand anføres dato og sted for prøveudtagning |
|
— |
anvendt inoculum med dato og sted for prøvens udtagning |
|
— |
angivelse af analysemetoden, hvis der er foretaget specifikke analyser, og en beskrivelse af den |
|
— |
afbildning af COD- eller DOC-fjernelse mod tiden, herunder indkørings- og vurderingsperiode |
|
— |
analytisk genfinding af teststoffet som COD eller DOC i stamopløsningen |
|
— |
afbildning af den procentvise fjernelse af teststof fra den vandige fase mod tiden (indkøringsperiode og vurderingsperiode), hvis der er foretaget specifikke analyser |
|
— |
den gennemsnitlige fjernelse af DOC, COD eller teststof samt standardafvigelse udregnes af resultaterne fra vurderingsperioden, dvs. når der er en stabil fjernelse af reststof |
|
— |
afbildning af aktivslamkoncentration mod tiden |
|
— |
eventuelle bemærkninger vedrørende det aktiverede slam (bortkastet overskydende slam, forekomst af klumpdannelse FeCl3 osv.) |
|
— |
teststofkoncentration benyttet ved testen |
|
— |
eventuelle resultater af analyser af slammet |
|
— |
alle oplysninger og forsøgsresultater vedrørende teststoffet og referencestoffer, hvis sådanne er anvendt |
|
— |
videnskabelige grunde til eventuelle ændringer af fremgangsmåden. |
3.2. FORTOLKNING AF RESULTATERNE
Hvis den procent teststof, der fjernes fra den vandige fase, er lav, kan de skyldes, at teststoffet hæmmer mikroorganismer. Også lysis og tab af slam, som giver uklar supernatant, samt nedsat effektivitet med hensyn til fjernelse af COD (eller DOC) kan afsløre dette.
Undertiden kan fysisk-kemisk adsorption spille en rolle. Forskelle mellem biologisk virkning på molekylet og fysisk-kemisk adsorption kan afsløres ved analyse af slammet efter passende desorption.
Der kræves yderligere test, hvis der skal skelnes mellem bionedbrydning (eller delvis bionedbrydning) og adsorption.
Det kan gøres på flere forskellige måder, men den mest overbevisende er brug af supernatanten som inoculum i en basis-sæt test (helst respirometrisk test)
Hvis det observeres, at den procentvise fjernelse af DOC eller COD er høj, skyldes dette bionedbrydning, hvorimod der ved lave fjernelsesprocenter ikke kan skelnes mellem bionedbrydning og eliminering. Som eksempel kan nævnes, at hvis et opløseligt stof udviser en høj adsorptionskonstant på 98 %, og der bortkastes 10 % overskydende slam pr. dag, er en eliminering på op til 40 % mulig; hvis der bortkastes 30 % overskydende slam, kan eliminering på grund af adsorption på og fjernelse sammen med overskydende slam komme op på 65 % (henvisning (4)).
Når der benyttes specifik analyse, må man være opmærksom på forholdet mellem stoffets struktur og den benyttede specifikke analyse. I dette tilfælde kan det observerede fænomen ikke fortolkes som en mineralisering af stoffet.
4. HENVISNINGER
|
(1) |
OECD, Paris, Test Guideline 303 A, Decision of the Council C(81) 30 Final. |
|
(2) |
Bilag V C9 Nedbrydningstest — kemisk oxygenforbrug Kommissionens direktive 84/449/EØF, De Europæiske Fællesskabers Tidende, L 251 af 19.9.1984. |
|
(3) |
Painter, H, A, and King, E. F., WRC Porous-Pot method for assessing biodegradability. Technical Report TR70. June 1978, Water Research Center, UK. |
|
(4) |
Wierich, P. and Gerike, P., The Fate of Soluble, Recalcitrant, and Adsorbing Compounds in Activated Sludge Plants — Ecotoxicology and Environmental Safety, Vol 5, no 2, June 1981, p. 161, 171. |
|
(5) |
Rådets direktiv 82/242/EØF og 82/243/EØF, De Europæiske Fællesskabers Tidende L 109 af 22.4.1982) om ændring af direktiv 73/404/EØF og 73/405/EØF: vaske- og rengøringsmidlers bionedbrydelighed, De Europæiske Fællesskabers Tidende L 347 af 17.12.1973). |
|
(6) |
H. Streuli, Fehierhafte Interpretation und Anwendung von Ausreissertests, insbesundere bei Ringversuchen zur Überprüfung analytisch-chemischer Untersuchungsmethoden, Fesenius-Zeitschrift für Analytische Chemie 303 (1980) 406–408. |
Tillæg 1
Figur 1

Figur 2

Figur 1
Udstyr til bedømmelse af bionedbrydelighed
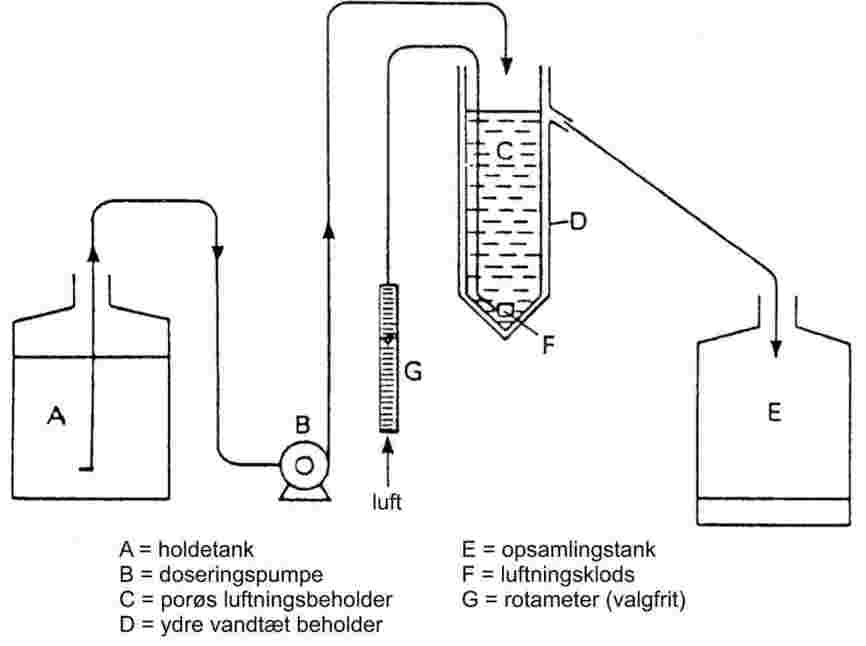
Figur 2
Tre-liters pores luftningskrukke

Tillæg 3
Arbejdsbetingelser for simulationstest for aktiveret slam
Der sættes et kryds i hver gruppe
Apparatur
|
OECD Confirmatory |
|
|
Porous Pot |
|
Arbejdsmetode
|
enkelt enhed |
|
|
koblede enheder |
|
|
ikke-koblede enheder |
|
Transinokulering
|
ingen |
|
|
aktiveret slam |
|
|
supernatant |
|
Middelopholdstid
|
tre timer |
|
|
seks timer |
|
Basisnæringsstof
|
husholdningsspildevand |
|
|
syntetisk spildevand |
|
Inoculum
|
sekundært spildevand |
|
|
blandet inoculum |
|
|
aktiveret slam |
|
Tilsætning af teststof
|
fra starten |
|
|
trinvis forøgelse |
|
|
efter slamdannelse |
|
Analyse
|
specifik |
|
|
COD |
|
|
DOC |
|
C.11. BIOLOGISK NEDBRYDNING
AKTIVERET SLAM — RESPIRATIONSHÆMNINGSTEST
1. METODE
1.1. INDLEDNING
Den beskrevne metode vurderer virkningen af et teststof på mikroorganismer ved måling af respirationshastigheden under definerede betingelser ved forskellige koncentrationer af teststoffet.
Formålet med metoden er at give en hurtig screening-metode til identifikation af stoffer, der kan påvirke et aerobt mikrobielt rensningsanlæg uheldigt, og at indikere passende ikke-hæmmende koncentrationer af teststoffer til anvendelse i bionedbrydelighedstest.
Inden den endelige test kan der udføres en forprøve. Den giver oplysninger om det koncentrationsinterval, som skal anvendes i hovedtesten.
I testen indgår to kontroltest, én i begyndelsen og én i slutningen af testrækken, Desuden bør hvert parti aktiveret slam også kontrolleres ved brug af et referencestof.
Metoden kan lettest benyttes for stoffer, der på grund af deres opløselighed i vand og ringe flygrighed sandsynligvis vil forblive i vandet.
Det kan for stoffer med begrænset opløselighed i testmediet vise sig umuligt at bestemme EC50.
Resultater, der er baseret på oxygenoptagelse, kan føre til fejlslutninger, hvis teststoffet har tendens til at afkoble oxidativ phosphorylering.
Det er nyttigt at have følgende oplysninger, når testen skal foretages:
|
— |
vandopløselighed |
|
— |
damptryk |
|
— |
strukturformel |
|
— |
teststoffets renhed. |
Anbefaling
Aktiveret slam kan indeholde potentielt patogene organismer og skal håndteres med forsigtighed.
1.2. DEFINITIONER OG ENHEDER
Respirationshastigheden er oxygenforbruget hos mikroorganismer i aerobt spildevandsslam og udtrykkes almindeligvis i mg O2 pr. mg slam pr. time.
Til beregning af et teststofs hæmmende virkning i en bestemt koncentration udtrykkes respirationshastigheden som en procentdel af gennemsnittet af to kontrolrespirationshasrigheder:
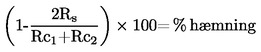
hvor:
|
Rs |
= |
oxygenforbruget ved testkoncetrationen af teststoffet |
|
RC1 |
= |
oxygenforbruget, kontrol 1 |
|
RC2 |
= |
oxygenforbruget, kontrol 2. |
I forbindelse med denne metode er EC50 den koncentration af teststoffet, hvorved respirationen er 50 % af den, som kontroltesten viser under de i denne metode beskrevne betingelser.
1.3. REFERENCESTOFFER
Det anbefales at bruge 3,5-dichlorphenol, som vides at hæmme respiration, som referencestof og at teste det for EC50 for hvert parti aktiveret slam for at kontrollere, at slammets følsomhed ikke er unormal.
1.4. TESTMETODENS PRINCIP
Respirationshastigheden i aktiveret slam tilført en standardmængde syntetisk spildevand måles efter en kontakttid på 30 minutter og/eller tre timer. Respirationshastigheden i det samme aktiverede slam ved forskellige koncentrationer af teststoffet under i øvrigt identiske betingelser måles også. Teststoffets hæmmende virkning ved en bestemt koncentration udtrykkes som en procentdel af middelrespirationshastigheden i to kontroltest. Der beregnes en EC50-værdi ud fra bestemmelser ved forskellige koncentrationer.
1.5. KVALITETSKRITERIER
Testresultaterne er gyldige, hvis:
|
— |
respirationshastigheden i de to kontroltest ligger inden for 15 % af hinanden |
|
— |
EC50 (30 minutter og/eller tre timer) for 3,.5-dichlorphenol ligger inden for det accepterede område på 5 til 30 mg/l. |
1.6. BESKRIVELSE AF TESTMETODEN
1.6.1. Reagenser
1.6.1.1.
Teststofopløsningerne frisklaves ved undersøgelsens indledning ved brug af en stamopløsning. En stamopløsningskoncentration på 0,5 g/l er egnet, hvis nedenstående anbefalede fremgangsmåde følges.
1.6.1.2.
Der fremstilles som eksempel en 3,5-dichlorphenolopløsning ved at opløse 0,5 g 3,5-dichlorphenol i 10 ml 1 M NaOH og fortynde med destilleret vand til ca. 30 ml; derefter tilsættes under omrøring 0,5 M H2SO4 til det punkt, hvor udfældning begynder — der kræves ca. 8 ml 0,5 M H2SO4 — og endelig fortyndes blandingen med destilleret vand til 1 l. pH skulle derefter være omkring 7 til 8.
1.6.1.3.
Der kan tillaves syntetisk spildevand ved opløsning af følgende mængder stof i 1 l vand:
|
— |
16 g pepton |
|
— |
11 g kødekstrakt |
|
— |
3 g urea |
|
— |
0,7 g NaCl |
|
— |
0,4 g CaCl2.2H2O |
|
— |
0,2 g MgSO4.7H20 |
|
— |
2,8 g K2HPO4. |
Note 1: Dette syntetiske spildevand er 100 gange så koncentreret som det, der er beskrevet i OECD Technical Report »Proposed method for the determination of the biodegradability of surfactants used in synthetic detergents« af 11. juni 1976 med tilsætning af dikaliumhydrogenphosphat.
Note 2: Hvis det fremstillede medium ikke anvendes straks, skal det opbevares i mørke ved 0 til 4 oC, dog ikke længere end en uge og under forhold som ikke giver anledning til ændring i sammensætningen. Medium kan også steriliseres forud for oplagringen, eller der kan tilsættes pepton og kødekstrakt, kort tid før testen udføres. Umiddelbart før brug blandes omhyggeligt og pH justeres.
1.6.2. Apparatur
Måleapparatur: Det er ikke afgørende, hvordan apparaturet nærmere er udformet. Der må imidlertid ikke være luftmellemrum mellem væske og prop, og sonden skal slutte tæt til kolbens hals.
Der kræves normalt laboratorieudstyr og især følgende:
|
— |
måleapparatur |
|
— |
beluftningsapparatur |
|
— |
pH-elektrode og -måleudstyr |
|
— |
O2-elektrode. |
1.6.3. Forberedelse af inoculum
Som mikrobielt inoculum til testen anvendes aktiveret slam fra et rensningsanlæg, der overvejende behandler husholdningsspildevand.
På laboratoriet kan grovere praktikler om nødvendigt fjernes ved kort henstand, f.eks. 15 minutter, efterfulgt af dekantering af det øverste lag med finere slampartikler. Alternativt kan slammet anvendes efter blanding i få sekunder i blender.
Hertil kommer, at slammet bør vaskes med vand (fra hanen) eller en isotonisk opløsning, hvis hæmmende stoffer kan forudses at være til stede. Efter centrifugering dekanteres supernatanten fra, hvilket gentages tre gange.
En mindre mængde af slammet afvejes og tørres. Herudfra kan beregnes den mængde af vådt slam, som skal suspenderes i vand for at opnå et aktivt slam med et indhold af faste bestanddele suspenderet i en blandet væske på 2 til 4 g/l. Dette svarer til en koncentration på 0,8 til 1,6 g/l i testmediet, hvis den nedenfor anbefalede procedure følges.
Hvis slammet ikke kan anvendes den dag, det er indsamlet, tilsættes der 50 ml syntetisk spildevand til hver liter aktiveret slam, der er fremstillet som ovenfor beskrevet; slammet beluftes derefter natten over ved 20 ± 2 oC. Det holdes derefter beluftet, indtil det anvendes den følgende dag. Inden anvendelsen kontrolleres pH, som om nødvendigt bufferes til pH 6 til 8. De faste bestanddele, der er suspenderet i den blandede væske, bestemmes som beskrevet i foregående afsnit.
Hvis der er behov for at anvende det samme parti slam de efterfølgende dage (højst fire dage), tilsættes yderligere 50 ml syntetisk spildevand ved hver arbejdsdags slutning.
1.6.4. Fremgangsmåde
|
Varighed/kontakttid: |
30 minutter og/eller tre timer under beluftning |
|
Vand: |
Drikkevand (om nødvendigt befriet for chlor) |
|
Lufttilførsel: |
Ren, oliefri luft. Luftgennemstrømning 0,5 til 1,0 l/min. |
|
Måleapparatur: |
Fladbundet kolbe som f.eks. en BOD-kolbe (se figur 1) |
|
Oxygenmåler: |
Oxygenelektrode med tilhørende skriver |
|
Næringsopløsning: |
Syntetisk spildevand (se ovenfor) |
|
Teststof: |
Testopløsningen laves frisk ved testens begyndelse |
|
Referencestof: |
F.eks. 3,5-dichlorphenol (mindst tre koncentrationer) |
|
Kontroltest: |
Inokuleret prøve uden teststof |
|
Temperatur: |
20 ± 2 oC. |
Nedenfor gives forslag til en eksperimentel procedure, der kan følges for både testen og referencestoffet i tre-timers kontaktperioden:
Der anvendes flere beholdere (f.eks. et-liters bægerglas).
Der benyttes mindst fem koncentrationer med et fast koncentrationsforhold på helst ikke over 3,2.
På tidspunktet »0« fortyndes 16 ml af det syntetiske spildevand med vand til 300 ml. Der tilsættes 200 ml af det mikrobielle inoculum, og hele blandingen (500 ml) hældes i en første beholder (første kontrol C1).
Testbeholderen skal gennemluftes kontinuerligt for at sikre, at opløst ilt (O2) ikke falder under 2,5 mg/l, og at iltkoncentrationen er i størrelsesorden 6,5 mg/l umiddelbart før målingen af respirationshastigheden.
På tidspunkt »15 minutter« (15 minutter er et vilkårligt, men bekvemt interval) gentages ovenstående proces, bortset fra at der tilsættes 100 ml teststofstamopløsning til de 16 ml syntetisk spildevand, før der tilsættes vand ti! 300 ml og mikrobielt inoculum til i alt 500 ml. Denne blanding hældes dernæst et andet bægerglas og beluftes som omhandler ovenfor. Denne proces gentages med 15 minutters intervaller med forskellige mængder teststofstamopløsning, således at der bliver en række beholdere med forskellige koncentrationer af teststoffet. Til slut fremstilles en anden kontrol (C2).
Efter tre timers forløb måles pH, og efter god blanding hældes en del af den første beholders indhold over i måleapparaturet, og respirationshastigheden måles over en periode pi indtil ti minutter.
Denne bestemmelse gentages for hver beholders indhold med 15 minutters intervaller, således at kontakttiden for hver beholder bliver tre timer.
Referencestoffet testes med hver parti mikrobielt inoculum på samme måde.
Et andet system (f.eks. mere end en oxygenmåler) vil være nødvendigt, når der skal foretages målinger efter 30 minutters kontakt.
Hvis der kræves måling af det kemiske oxygenforbrug, må der fremstilles andre beholdere, som indeholder teststof, syntetisk spildevand og vand, men ikke aktiveret slam. Oxygenforbruget måles og registreres efter en beluftningstid på 30 minutter og/eller tre timer (kontakttid).
2. DATA OG VURDERING
Respirationshastigheden beregnes ud fra skriverkurven som mg O2/l × h mellem ca. 6,5 mg O2/l og 2,5 mg O2/l, eller over en ti minutters periode, hvis respirationshastigheden er lav. Den del af respirationskurven, som respirationshastigheden måles over, skal være lineær.
Hvis respirationshastighedeme i de to kontroltest ikke ligger inden for 15 % af hinanden, eller hvis referencestoffets EC50 (30 minutter og/eller tre timer) ikke ligger inden for det accepterede område (5 til 30 mg/l for 3,5-dichlorphenol), er testen ugyldig og må gentages.
Hæmningsprocenten beregnes ved hver testkoncentration (se 1.2). Hæmningsprocenten aftegnes mod koncentration på semilogaritmisk papir (eller probit), og der udledes en EC50-værdi.
95 %-konfidensgrænserne for EC50-værdierne kan bestemmes ved hjælp af standardprocedurer.
3. RAPPORTERING
3.1. TESTRAPPORT
Testrapporten skal om muligt indeholde følgende:
|
— |
teststof: data for kemisk identifikation |
|
— |
testsystem: kilde, koncentration og eventuel forbehandling af det aktiverede slam |
|
— |
testbetingelser:
|
|
— |
resultater:
|
3.2. FORTOLKNING AF DATA
EC50 værdien må kun betragtes som vejledende for teststoffets sandsynlige toksicitet for enten spildevandsbehandling med aktiveret slam eller mikroorganismer i spildevand, da de komplekse vekselvirkninger, der forekommet i miljøet, ikke kan simuleres nøjagtigt i en laboratorietest. Hertil kommer, at teststoffer, som har en hæmmende effekt på ammoniakoxidation, også kan give anledning til atypiske hæmningskurver. Sådanne kurver må fortolkes med forsigtighed.
4. HENVISNINGER
|
(1) |
International Standard ISO/8192 — 1986. |
|
(2) |
Broecker, B. and Zahn, R., Water Research 11, 1997, s. 165. |
|
(3) |
Brown, D., Hitz, H. R. and Schaefer, L., Chemosphere 10, s. 245 (1981). |
|
(4) |
ETAD (Ecological and Toxicological Association of Dyestuffs Manufacturing Industries), Recommended Method No. 3 03, også beskrevet af: |
|
(5) |
B. Robra, Wasser/Abwasser 117, s. 80 (1976). |
|
(6) |
W. Schefer, Textilveredlung 6, s. 247 (1977). |
|
(7) |
OECD, Paris, 1981, Test Guideline 209. Decision of the Council C(81) 30 Final. |
C.12. BIOLOGISK NEDBRYDNING
MODIFICERET SCAS-TEST
1. METODE
1.1. INDLEDNING
Formålet med denne metode er at bedømme den potentielle fuldstændige bionedbrydelighed af vandopløselige, ikke-flygtige organiske stoffer, når disse udsættes for ret høje koncentrationer af mikroorganismer over et længere tidsrum. I dette tidsrum holdes mikroorganismerne i live ved en daglig tilførsel af næring bestående af bundfældet spildevand. (I forbindelse med weekenden kan spildevandet opbevares ved 4 oC, eller det syntetiske spildevand til OECD's confirmatory test kan anvendes).
Der kan finde fysisk-kemisk adsorption sted på de suspenderede faste stoffer, og der skal tages hensyn hertil ved fortolkningen af resultaterne (jf. 3.2).
På grund af den flydende fases lange opholdstid (36 timer), og da der med mellemrum tilføres næringsstoffer, simulerer testen ikke de normale vilkår i et spildevandsanlæg. De resultater, der er opnået med forskellige teststoffer, viser, at denne metode har en høj bionedbrydningsevne.
Delbetingelser, der skabes i denne metode, letter i høj grad selektion og/eller adaptation af mikroorganismer, der er i stand til at nedbryde teststoffet. (Denne fremgangsmåde kan ligeledes anvendes til at at fremstille akklimatiserede inocula til brug i andre test).
I denne test vurderes teststoffernes fuldstændige bionedbrydelighed ved måling af koncentrationen af opløst organisk kulstof (DOC). Det bør foretrækkes at bestemme koncentrationen af opløst organisk kulstof efter oprensning af den sure opløsning snarere end som forskellen mellem total C og uorganisk C.
Ved samtidig brug af en specifik analysemetode kan teststoffets primære nedbrydning (nedbrydning af en oprindelige kemiske struktur) vurderes.
Denne metode kan kan anvendes på organiske teststoffer, som ved den i analysen anvendte koncentration:
|
— |
er opløselige i vand (mindst 20 mg opløst organisk kulstof/l) |
|
— |
har et ubetydeligt damptryk |
|
— |
ikke er hæmmende over for bakterier |
|
— |
ikke adsorberes væsentligt i testsystemet |
|
— |
ikke går tabt ved, at testopløsningen skummer. |
Teststoffets indhold af organisk kulstof skal være bekendt.
Oplysninger om de relative mængder af teststoffets hovedkomponenter vil være nyttige for fortolkningen af de opnåede resultater, specielt i de tilfælde, hvor resultaterne er lave eller marginale.
Oplysninger om stoffets toksicitet for mikroorganismer kan være nyttige for fortolkningen for lidet signifikante resultater og for valg af en egnet testkoncentration.
1.2. DEFINITIONER OG ENHEDER
|
CT |
= |
koncentration af organisk kulstof i teststoffet, til stede i eller tilsat det bundfældende spildevand ved begyndelsen af beluftningsperioden (mg/l) |
|
Ct |
= |
koncentrationen af opløst organisk kulstof i supernatanten ved afslutningen af beluftningsperioden (mg/l) |
|
Cc |
= |
koncentrationen af opløst organisk kulstof i kontrollens supernatant ved afslutningen af beluftningsperioden (mg/l). |
Bionedbrydeligheden defineres i denne test som det organiske kulstofs orsvinden. Bionedbrydningen kan udtrykkes som:
|
1. |
Den procentvise forsvinden Dda af den mængde af stoffet, der tilføres dagligt:
hvor:
|
|
2. |
Den procentvise forsvinden Dssd af den mængde stof, der er til stede ved hver dags begyndelse:
hvor:
Indekserne i og (i+ 1) henviser til testdagen. Ligning [2(a)] bør anvendes, hvis spildevandets indhold af opløst organisk kulstof svinger fra dag til dag, medens ligning [2(b)] kan anvendes, når mængden af opløst organisk kulstof er forholdsvis konstant fra dag til dag. |
1.3. REFERENCESTOFFER
I visse tilfælde kan referencestoffer være nyttige ved undersøgelsen af et nyt stof. Der kan imidlertid ikke anbefales noget bestemt referencestof.
Der angives oplysninger om flere forskellige stoffer, der er evalueret i ringtest (jf. tillæg 1), først og fremmest for at testmetoden af og til kan kalibreres, men også for at resultaterne kan sammenlignes, hvis der anvendes en anden metode.
1.4. TESTMETODENS PRINCIP
Aktiveret slam fra et biologisk rensningsanlæg placeres i en semi-kontinuerlig aktiveret slamenhed (semi-continous activated sludge — SCAS). Teststoffet og bundfældet husholdningsspildevand tilføres, og blandingen beluftes i 23 timer. Beluftningen indstilles derefter, slammet gives tid til at bundfælde sig, og supernatantvæsken fjernes.
Det tilbageblevne slam i beluftningskammeret iblandes dernæst en yderligere mængde af teststoffet og spildevandet, hvorefter cyklussen begynder forfra.
Bionedbrydning konstateres ved at bestemme indholdet af opløst organisk kulstof i supernatantvæsken. Dette resultat sammenlignes med resultatet af analysen af væsken fra en kontrolenhed, som kun er tilført bundfældet spildevand.
Såfremt der anvendes en specifik analysmetode, kan ændringer i koncentrationen af det oprindelige stof som følge af bionedbrydningen måles (primær bionedbrydelighed).
1.5. KVALITETSKRITERIER
Denne metodes reporducerbarhed, baseret på fjernelse af opløst organisk kulstof, er endnu ikke fastslået. (For så vidt angår primær bionedbrydning, er der opnået meget nøjagtige data for let nedbrydelige stoffer).
Metodens følsomhed afhænger i vid udstrækning af variabiliteten i blindprøven og i mindre grad af den nøjagtighed, hvormed indholdet af opløst organisk kulstof kan bestemmes, eller af væskens indhold af teststof ved påbegyndelsen af hver enkelt cyklus.
1.6. BESKRIVELSE AF TESTPROCEDUREN
1.6.1. Forberedelse
Et tilstrækkeligt antal rene beluftningsenheder (i stedet kan den oprindelige 1,5 l SCAS-prøveenhed anvendes) og lufttilførselsslanger (figur 1) for hvert enkelt teststof og til kontrollen klargøres. Den komprimerede luft, der tilføres testenhederne, renses med er vatfilter og bør være fri for organisk kulstof og være forudmættet med vand, så fordampningstab reduceres.
Fra et aktiveret slamanlæg, der primært behandler husholdningsspildevand, tages en prøve af blandet væske, indeholdende 1 til 4 g suspenderet fast stof pr. liter. Til hver beluftningsenhed kræves ca. 150 ml af den blandede væske.
Stamopløsninger af teststoffet tilberedes i destilleret vand. Den normalt krævede koncentration er 400 mg organisk kulstof pr. liter hvilket giver en koncentration af teststoffet på 20 mg/l ved påbegyndelsen af hver beluftningscyklus, såfremt der ikke finder bionedbrydning sted.
Der; kan anvendes højere koncentrationer, hvis toksiciteten over for mikroorganismerne tillader det.
Slamopløsningernes indhold af organisk kulstof måles.
1.6.2. Testbetingelser
Testen bar foretages ved 20 til 25 oC.
Der anvendes en høj koncentration af aerobe mikroorganismer (fra 1 til 4 g suspenderet fast stof pr. liter), og den effektive opholdstid er 36 timer. Det kulstofholdige materiale i det tilførte spildevand iltes grundigt, sædvanligvis inden otte timer efter påbegyndelsen af hver beluftningscyklus. Derefter respirerer slammet endogent i resten af beluftningsperioden, hvorunder det eneste tilgængelige substrat er teststoffet, medmindre også dette metaboliseres hurtigt. Disse forhold, kombineret med daglig genpodning af prøven, når husholdningsspildevand anvendes som medium, giver uhyre gunstige betingelser for såvel akklimatisering som høj grad af bionedbrydning.
1.6.3. Gennemførelsen af testen
Der fremstilles en prøve af blandet væske fra en laboratorieenhed, der overvejende behandler husholdningsspildevand eller fra et egnet aktiveret slamanlæg, og prøven opbevares under aerobe betingelser indtil anvendelsen i laboratoriet. Hver beluftningsenhed samt kontrolenheden påfyldes 150 ml (hvis den oprindelige SCAS-test anvendes, multipliceres de anførte volumener med 10) blandet væske, og beluftningen påbegyndes. Efter 23 timers forløb indstilles beluftningen, og slammet bundfældes i 45 minutter. Hanen på hver kolbe åbnes successivt, og der udtages 100 ml af supernatantvæsken. Der klargøres en prøve bundfældet husholdningsspildevand umiddelbart før brug, og 100 ml heraf tilsættes det tilbageblevne slam i hver beluftningsenhed. Beluftningen påbegyndes på ny. På dette stadium tilsættes intet teststof. Enhederne tilsættes dagligt husholdningsspildevand, men kun indtil det tidspunkt, hvor bundfældningen resulterer i en klar supernatant væske. Dette tager sædvanligvis op til to uger, hvorefter det opløste organiske kulstof i supernatantvæsken ved afslutningen af hver beluftningscyklus nærmer sig en konstant værdi.
Ved afslutningen af dette tidsrum blandes de forskellige bundfældede slamprøver, og 50 ml af denne blandede slamprøve tilsættes til hver enhed.
Der tilsættes 95 ml bundfældet spildevand og 5 ml vand til kontrolenhederne, og 95 ml bundfældet spildevand plus 5 ml af den pågældende stamopløsning af teststoffet (400 mg/l) til testenhederne. Beluftningen påbegyndes atter og fortsættes i 23 timer. Slammet bundfældes i 45 minutter, og supernatantvæsken fjernes og analyseres for opløst organisk kulstof.
Ovenstående påfyldnings- og aftapningsprocedure gentages dagligt under hele testens forløb.
Inden bundfældningen kan det være nødvendigt at rense enhedernes vægge for at forhindre akkumulering af faste stoffer over væskens niveau. For at undgå krydskontaminering anvendes der en separat skraber eller børste for hver enkelt enhed.
Ideelt set bør indholdet af opløst organisk kulstof i supernatantvæskerne bestemmes dagligt, selv om mindre hyppige analyser kan tillades. Inden analysen filtreres væskerne gennem vaskede membranfiltre med en porestørrelse på 0,45 μm, eller væskerne centrifugeres. Membranfiltre er egnede, hvis der er sikkerhed for, at de hverken afgiver kulstof eller adsorberer stoffet under filtreringen. Prøvens temperatur må ikke overstige 40 oC, så længe den er i centrifugen.
Testens varighed er ubestemt for stoffer, der udviser ringe eller ingen bionedbrydning. Erfaringerne viser dog, at den generelt bør vare mindst 12 uger, men ikke længere end 26 uger.
2. DATA OG EVALUERING
Værdierne for indholdet af opløst kulstof i supernatant væskerne i testenhederne og i kontrolenhederne afbildes mod tiden i et koordinatsystem.
Efterhånden som nedbrydningen skrider frem, vil niveauet i testenheden nærme sig niveauet i kontrolenheden. Når forskellen mellem de ro niveauer er konstant i tre på hinanden følgende målinger, gennemføres der et tilstrækkeligt antal yderligere målinger til en statistisk behandling af dataene, og den procentvise bionedbrydning af teststoffet beregnes (Dda eller Dssd, jf. 1.2).
3. RAPPORTERING
3.1. TESTRAPPORT
Testrapporten skal om muligt indeholde følgende:
|
— |
fuldstændige oplysninger om spildevandets art, om hvilken type enhed der er anvendt samt om forsøgsresultaterne vedrerende teststoffet, referencestoffet, hvis et sådan er anvendt, og blindprøven |
|
— |
temperatur |
|
— |
kurve for det organiske kulstofs forsvinden, med beskrivelse af beregningsmetode (jf. 1.2) |
|
— |
dato og sted for prøveudtagning af aktiveret slam og spildevand, status vedrørende adaptation, koncentration osv. |
|
— |
videnskabelige grunde til eventuelle ændringer i testproceduren |
|
— |
underskrift og dato. |
3.2. FORTOLKNING AF RESULTATERNE
Eftersom det stof, der kan testes med denne metode, ikke vil være let bionedbrydeligt, vil enhver forsvinden af opløst organisk kulstof, som udelukkende skyldes bionedbrydning, normalt forløbe gradvis over dage eller uger, undtagen i sådanne tilfælde, hvor akklimatiseringen er pludselig, hvilket fremgår af en abrupt forsvinden efter nogle uger.
Fysisk-kemisk adsorption kan imidlertid nogle gange spille en betydelig rolle. Dette er tilfældet, når der ved begyndelsen indtræder en fuldstændig eller delvis forsvinden af det tilsatte opløste organiske kulstof. Hvad der derefter sker, afhænger af forskellige faktorer, såsom adsorptionsgraden og koncentrationen af suspenderet fast stof i det bortkastede spildevand. Forskellen mellem koncentrationen af opløst organisk kulstof i kontrol- og testenhedernes supernatantvæsker øges sædvanligvis gradvis fra den først indtrufne værdi, og denne forskel forbliver dernæst på den nye værdi under resten af forsøget, medmindre der finder akklimatisering sted.
Når der skal skelnes mellem bionedbrydning (eller delvis bionedbrydning) og adsorption, er yderligere analyser påkrævet. Dette kan gøres på flere forskellige måder, men den mest pålidelige metode er at anvende supernatantvæsken, eller slam, som podestof i en basissættest (helst en respirometrisk test).
Teststoffet, der resulterer i omfattende, ikke adsorptiv forsvinden af opløst organisk kulstof, bør i denne test betragtes som potentielt bionedbrydeligt. Delvis ikke-adsorptiv forsvinden tyder på, at stoffet i hvert fald er bionedbrydeligt til en vis grad.
Ringe eller ingen forsvinden af opløst organisk kulstof kan skyldes teststoffets hæmning af mikroorganismerne. Dette kan ligeledes afsløres af lysis og tab af slam, hvilket giver uklare supernatantvæsker. Testen bør i så fald gentages med anvendelse af en lavere koncentration af teststoffet.
Anvendelsen af en specifik analysemetode eller C-14-mærket teststof kan give større følsomhed. Anvendes der et C-14-teststof, vil genfinding af 14CO2 bekræfte, at der har fundet bionedbrydning sted.
Såfremt der ligeledes angives resultater for primær bionedbrydning, bør der om muligt gives en forklaring på den forandring i den kemiske struktur, der medfører, at det oprindelige teststof ikke kan påvises.
Valideringen af analysemetoden skal anføres sammen med værdierne for blindprøvemediet.
4. HENVISNINGER
|
(1) |
OECD, Paris, 1981, Test Guideline 302 A. Decision of the Council C(81) 30 Final. |
Tillæg 1
SCAS-test: eksempel på resultater
|
Stof |
CT (mg/l) |
Ct–Cc (mg/l) |
Procent bionedbrydning Dda |
Testens varighed (dage) |
|
4-acetylaminobenzensulfonat |
17,2 |
2,0 |
85 |
40 |
|
Tetrapropylonbenzensulfonat |
17,3 |
8,4 |
51,4 |
40 |
|
4-nitrophenol |
16,9 |
0,8 |
95,3 |
40 |
|
Diethylen glycol |
16,5 |
0,2 |
98,8 |
40 |
|
Anilin |
16,9 |
1,7 |
95,9 |
40 |
|
Cyclopentantetracarboxylat |
17,9 |
3,2 |
81,1 |
120 |
Tillæg 2
Eksempel på apparatur

C.13. BIOKONCENTRERING: GENNEMSTRØMNINGSTEST MED FISK
1. METODE
Denne biokoncentreringsmetode er en gengivelse af OECD Test Guideline 305 (1996).
1.1. INDLEDNING
I denne metode beskrives en fremgangsmåde for karakterisering af stoffers biokoncentreringspotentiale i fisk under gennemstrømning. Test ved gennemstrømning er langt at foretrække, men også semistatiske systemer tillades, forudsat at gyldighedskriterierne er opfyldt.
Testen er så detaljeret beskrevet, at den kan udføres, samtidig med at der er tilstrækkelig frihed til at tilpasse tilrettelægningen af forsøgene til særlige forhold i laboratorierne og teststoffernes særlige egenskaber. Metoden er bedst til stabile organiske kemiske stoffer med en log Pow.-værdi mellem 1,5 og 6,0 (1), men den kan også anvendes for ekstremt lipofile stoffer (med log Pow > 6,0). Den på forhånd skønnede biokoncentreringsfaktor (BCF), undertiden benævnt KB, for sådanne ekstremt lipofile stoffer vil normalt være højere end den værdi af ligevægtsbiokoncentreringsfaktoren (BCFSS), man må forvente at finde ved laboratorieforsøg. Forhåndsskøn over biokoncentreringsfaktoren for organiske stoffer med log Pow-værdier op til ca. 9,0 kan beregnes efter udtrykket i Bintein et al (2). De parametre, der karakteriserer biokoncentreringspotentialet, er hastighedskonstanten for optagelse (k1), hastighedskonstanten for udskillelse (k2) og BCFSS.
Radioaktivt mærkede teststoffer kan gøre analyse af vand- og fiskeprøver lettere og kan benyttes til at afgøre, om der skal foretages identifikation og kvantitativ bestemmelse af nedbrydningsprodukter. Hvis den samlede mængde radioaktivt restmateriale måles (f.eks. ved forbrænding eller opløsning af væv), vil BCF blive baseret på den oprindelige forbindelse, eventuelle tilbageholdte metabolitter og assimileret kulstof. BCF-værdier, der baseres på det samlede indhold af radioaktivt materiale, kan derfor ikke altid sammenlignes direkte med en BCF-værdi, der er bestemt ved specifik kemisk analyse for den oprindelige forbindelse alene.
Ved undersøgelser med radioaktivt mærkede stoffer kan der benyttes oprensningsprocedurer til bestemmelse af BCF baseret på den oprindelige forbindelse, og de vigtigste metabolitter kan om nødvendigt karakteriseres. Man kan også kombinere en undersøgelse af metabolisme i fisk med en biokoncentreringsundersøgelse ved at analysere og identificere stofrester i væv.
1.2. DEFINITIONER OG ENHEDER
Ved biokoncentrering/bioakkumulering forstås en koncentrationsforøgelse af teststoffet i eller på en organisme (eller specificerede væv deraf) i forhold til koncentrationen af teststoffet i det omgivende medium.
Ved biokoncentreringsfaktoren (BCF eller KB) på et givet tidspunkt under optagelsesfasen af akkumuleringstesten forstås koncentrationen af teststoffet i/på fiskene eller specificerede væv deraf (Cf i μg/g (ppm)) divideret med koncentrationen af det kemiske stof i det omgivende medium (Cw i μg/ml (ppm)).
Ligevægtsbiokoncentreringsfaktoren (BCFSS eller KB) ændrer sig ikke væsentligt over et længere tidsrum, idet koncentrationen af teststoffet i det angivende medium er konstant i dette tidsrum.
Man taler om et plateau eller en ligevægtstilstand i afbildningen af teststoffet i fisk (Cf) mod tiden, når kurven bliver parallel med tidsaksen, når tre på hinanden følgende analyser af Cf i prøver, der er udtaget med mindst to dages mellemrum, ligger inden for ± 20 % af hinanden, og når der ikke er nogen signifikante forskelle mellem de tre prøveudtagningsperioder. Ved pooling af prøver til analyse, kræves der mindst fire på hinanden følgende analyser. For teststoffer, der optages langsomt, vil et interval på syv dage være mere passende.
En biokoncentreringsfaktor, der er beregnet direkte ud fra kinetiske hastighedskonstanter (k1/k2), betegnes den kinetiske koncentreringsfaktor, BCFk.
Ved octanol-vand fordelingskoefficienten (Pow) forstås forholdet mellem et kemisk stofs opløselighed i n-octanol og vand ved ligevægt (metode A.8), også betegnet Kow. Logaritmen til Pow benyttes som rettesnor for et kemisk stofs potentiale for biokoncentrering i vandorganismer.
Ved eksponeringsfasen eller optagelsesfasen forstås det tidsrum, hvori fiskene udsættes for teststoffet.
Ved hastighedskonstant for optagelse (k1) forstås den talværdi, der angiver hastigheden for koncentrationsforøgelsen af teststoffet i eller på fiskene (eller specificerede væv deraf), når fiskene udsættes for stoffet (k1 udtrykkes i dag-1).
Ved udskillelsesfasen forstås det tidsrum efter overførsel af forsøgsfiskene fra et medium med teststoffet i til et medium uden stoffet, hvor man undersøger udskillelsen (eller nettoafgivelsen) af stoffet fra forsøgsfiskene (eller specificerede væv deraf).
Ved hastighedskonstant for udskillelse (k2) forstås den talværdi, der angiver hastigheden for koncentrationsfaldet af teststoffet i forsøgsfiskene (eller specificerede væv deraf), efter at de er overført fra et medium med teststoffet i til et medium uden dette stof (k2 udtrykkes i dag-1).
1.3. TESTMETODENS PRINCIP
Testen falder i to faser, en eksponeringsfase (optagelsesfase) og en udskillelsesfase. I optagelsesfasen udsættes separate grupper af fisk af én art for mindst to koncentrationer af teststoffet. De overføres dernæst til et medium uden teststoffet, hvor udskillelsesfasen forløber. Der kræves altid en udskillelsesfase, undtagen hvis optagelsen af stoffet under eksponeringsfasen har været ubetydelig (f.eks.hvis BCF er mindre end 10). Under begge testens faser følges koncentrationen af stoffet i eller på fiskene (eller specificerede væv deraf). Der holdes samtidig en kontrolgruppe af fisk under samme betingelser, bortset fra at der ikke er noget teststof til stede, så man kan sammenholde eventuelle skadevirkninger i biokoncentreringstesten med en tilsvarende kontrolgruppe og bestemme baggrundskoncentrationer af teststof.
Optagelsesfasen varer i 28 dage, medmindre det godtgøres, at der inden da er indtrådt ligevægt. Ud fra ligningerne i bilag 3 kan man forudsige, hvor lang tid optagelsesfasen vil vare, og hvor lang tid der vil gå, inden der er ligevægt. Udskillelsesperioden indledes dernæst ved, at fiskene overføres til en ren beholder med samme medium, men uden teststof. Når det er muligt, beregnes biokoncentreringsfaktoren både som forholdet (BCFSS) mellem koncentrationen i fiskene (Cf) og i vandet (CW) ved ligevægt, og som kinetisk biokoncentreringsfaktor (BCFK) som forholdet mellem hastighedskonstanterne for optagelse (k1) og udskillelse (k2), idet kinetikken antages at være af første orden. Hvis det er åbenbart, at kinetikken ikke er af første orden, må der benyttes mere komplekse modeller (bilag 5).
Hvis der ikke nås ligevægt inden 28 dage, forlænges optagelsesfasen, indtil der er opnået ligevægt, dog højst 60 dage: Derefter påbegyndes udskillelsesfasen.
Hastighedskonstanten for optagelse, hastighedskonstanten for udskillelse (eller konstanterne, hvis der er tale om mere komplekse modeller), biokoncentreringsfaktoren og så vidt muligt konfidensgrænser for hver af disse parametre beregnes ud fra den model, der bedst beskriver de målte koncentrationer af teststof i fisk og vand.
BCF udtrykkes som en funktion af fiskenes samlede vægt. I særlige tilfælde kan dog bestemte væv eller organer (f.eks. muskelvæv eller lever) anvendes, hvis fiskene er tilstrækkelig store, eller fiskene kan opdeles i en spiselig fraktion (filet) og en ikke-spiselig fraktion (indvolde). Da der for mange organiske stoffer er en tydelig sammenhæng mellem biokoncentreringspotentialet og stoffets lipofile karakter, er der en tilsvarende sammenhæng mellem forsøgsfiskenes fedtindhold og stoffernes observerede biokoncentrering. For at imødegå sådanne udsving i forsøgsresultater for stærkt lipofile stoffer (dvs. log Pow > 3) bør biokoncentreringen udtrykkes både i forhold til fedtindholdet og i forhold til den samlede legemsvægt.
Fedtindholdet skal om muligt bestemmes på det samme biologiske materiale, som benyttes til bestemmelse af teststoffets koncentration.
1.4. OPLYSNINGER OM TESTSTOFFET
Inden biokoncentreringstesten udføres, skal følgende oplysninger om teststoffet foreligge:
|
— |
opløselighed i vand |
|
— |
octanol-vand fordelingskoefficient Pow (betegnes også Kow, bestemt ved en HPLC-metode i A.8) |
|
— |
hydrolyse |
|
— |
fotokemisk omdannelse i vand bestemt ved bestråling med sollys og simuleret sollys under samme bestrålingsforhold som ved testen for biokoncentrering (3) |
|
— |
overfladespænding (dvs. for stoffer, hvis Pow ikke kan bestemmes) |
|
— |
damptryk |
|
— |
umiddelbar bionedbrydelighed (når det er relevant) |
Blandt andre nødvendige oplysninger er toksiciteten over for den fiskeart, der benyttes til testen, helst den asymptotiske (dvs. tidsuafhængige) LC50. Der skal foreligge en passende analysemetode med kendt nøjagtighed, præcision og følsomhed til kvantitativ bestemmelse af teststoffet i opløsninger og biologisk materiale og oplysninger om, hvordan prøver forberedes og opbevares. Hvis der bruges 14C-mærkede teststoffer, skal den procentdel af radioaktiviteten, der skyldes urenheder, være kendt.
1.5. TESTENS GYLDIGHED
Følgende betingelser skal være til stede, for at testen betragtes som gyldig:
|
— |
temperaturudsving må ikke være større end ± 2 oC |
|
— |
koncentrationen af opløst oxygen må ikke komme under 60 % af mætning |
|
— |
koncentrationen af teststoffet i kamrene skal ligge inden for ± 20 % af gennemsnittet af de målte værdier under eksponeringsfasen |
|
— |
dødeligheden og andre skadevirkninger/sygdomme må ved testens afslutning højst være 10 % både i kontrolgruppen og i de eksponerede fisk: Hvis testen strækker sig over flere uger eller måneder, må dødsfald og andre skadevirkninger i begge grupper højst være 5 % om måneden og højst 30 % i alt. |
1.6. REFERENCESTOFFER
Det kan være nyttigt at anvende referencestoffer med kendt biokoncentreringspotentiale til kontrol af forsøgsmetoden, hvis dette er påkrævet. Der kan dog endnu ikke anbefales nogen bestemte stoffer.
1.7. BESKRIVELSE AF TESTMETODEN
1.7.1. Apparatur
Det skal omhyggeligt undgås, at der i nogen dele af udstyret anvendes materialer, der kan opløses eller adsorbere, absorbere eller afgive stoffer og have en skadelig virkning på fiskene. Der kan benyttes rektangulære eller cylindriske standardkar af kemisk inert materiale og passende størrelse i forhold til mængden af fisk. Bløde plastslanger bør benyttes mindst muligt. Der skal helst bruges rør og slanger af teflon (R), rustfrit stål eller glas. Erfaringerne har vist, at det til stoffer med høj adsorptionskoefficient såsom syntetiske pyrethroider kan være nødvendigt at bruge silancoatet glas. I sådanne tilfælde må udstyret kasseres efter brug.
1.7.2. Vand
Til testen benyttes sædvanligvis råvand, som skal være af ensartet kvalitet og komme fra en ikke-forurenet kilde. Vand til fortynding skal være af en sådan kvalitet, at den valgte fiskeart kan overleve akklimatiserings- og testperioden, uden at deres udseende eller adfærd bliver unormal. Allerhelst bør det godtgøres, at fiskearten kan overleve, vokse og formere sig i fortyndingsvandet (f.eks. i laboratoriekultur eller en toksicitetstest over en hel livscyklus). Vandet skal beskrives i hvert fald ved sit pH, hårdhed, tørstof i alt og organisk kulstof i alt, og gerne også indholdet af ammonium, nitrit og nitrat, samt for saltvandsfisk saltholdigheden. Selv om det er velkendt, hvilke parametre der er vigtige for fisks velfærd, er der i bilag 1 anført anbefalede maksimumkoncentrationer for en række parametre for fersk- og saltvand til forsøg.
Vandet skal være af konstant kvalitet under hele forsøget. Dets pH skal være mellem 6 og 8,5, men for en given test skal det ligge konstant inden for ±0,5. For at sikre, at fortyndingsvandet ikke ubehørigt påvirker forsøgsresultatet (f.eks. ved kompleksdannelse med teststoffet) eller indvirker negativt på fiskebestanden, udtages der regelmæssigt prøver til analyse. Indholdet af tungmetaller (f.eks. Cu, Pb, Zn, Hg, Cd og Ni), vigtige anioner og kationer (f.eks. Ca, Mg, Na, K, Cl og SO4) pesticider (f.eks. organiske phosphorforbindelser i alt og organiske chlorforbindelser i alt), organisk kulstof i alt og opslæmmet tørstof bør bestemmes f.eks. hver tredje måned, når fortyndingsvandet vides at være af forholdsvis konstant kvalitet. Hvis vandkvaliteten kan påvises at have været konstant i mindst et år, kan hyppigheden sættes ned (til f.eks. hvert halve år).
Både det naturlige partikelindhold og indholdet af organisk kulstof i alt (TOC) i fortyndingsvandet skal være så lavt som muligt, så man undgår absorption af teststoffet ti! organisk materiale, hvilket kan nedsætte dets biotilgængelighed (4). Den højeste acceptable værdi er 5 mg/l for partikler (tørstof, der ikke passerer et 0,45 μm filter) og 2 mg/l for organisk kulstof i alt (se bilag 1). Om nødvendigt må vandet filtreres inden brugen. Bidraget til indholdet af organisk kulstof i vandet fra testfiskene (ekskrementer) og føderester skal holdes så lavt som muligt. Under hele testen må koncentrationen af organisk kulstof i testkarret ikke overstige koncentrationen af organisk kulstof hidrørende fra teststoffet og et eventuelt opløsende stof med mere end 10 mg/l (± 20 %).
1.7.3. Testopløsninger
Der fremstilles en stamopløsning af teststoffet med en passende koncentration. Den skal helst fremstilles ved simpel blanding eller omrystning af teststoffet med vand. Brug af opløsningsmidler eller dispergeringsmidler (opløsende stoffer) anbefales ikke, men det kan i visse tilfælde være nødvendigt for at opnå en passende koncentreret stamopløsning. Som opløsningsmiddel kan der benyttes ethanol, methanol, ethylenglycolmonomethylether, ethylenglycoldimethylether, dimethylformamid eller triethylenglycol. Som dispergeringsmiddel kan benyttes Cremophor RH40, Tween 80, 0,01 % methylcellulose eller HCO-40. Der bør udvises forsigtighed med brug af let bionedbrydelige midler, da de kan skabe problemer med bakterievækst i test med gennemstrømning. Teststoffet kan være radioaktivt mærket og skal være af størst mulig renhed (f.eks. helst > 98 %).
Til forsøg med gennemstrømning kræves der er system, der kontinuerligt leverer og fortynder en stamopløsning af teststoffet (f.eks. doseringspumpe, proportionalfortynder, mætningssystem), til tilførsel af teststoffet ti! kamrene. Der tilstræbes en udskiftning på mindst fem gange dagligt pr. testkammer. Gennemstrømningsmetoden bør foretrækkes, men kan det ikke lade sig gøre (f.eks. hvis testorganismerne tager skade), kan der bruges en semistatisk teknik, forudsat at gyldighedskriterierne er opfyldt. Flowhastighederne for stamopløsning og fortyndingsvand kontrolleres 48 timer før testens begyndelse og derefter mindst dagligt under testen. Ved denne kontrol måles flowet i hvert testkammer, og det sikres, at det ikke varierer med mere end 20 %, hverken inden for hvert kammer eller mellem kamrene.
1.7.4. Valg af fiskeart
Blandt de vigtige kriterier ved valg af fiskearten er, at den skal være let at få fat i, kunne fås i passende størrelser og let kunne holdes i laboratoriet. Andre kriterier for valg af fiskeart kan være dens rekreative, kommercielle eller økologiske betydning, dens følsomhed i forhold til andre arter og gode erfaringer med den.
I bilag 2 er anført nogle fiskearter, som anbefales til test. Der kan anvendes andre arter, men testmetoden må i så fald tilpasses, så testbetingelserne bliver passende. Der skal da anføres en begrundelse for valg af art og testmetode.
1.7.5. Opbevaring af fisk
Stampopulationen af fisk akklimatiseres i mindst to uger i vand ved testtemperaturen og gives hele tiden tilstrækkeligt af det foder, som skal benyttes under testen.
Efter en 48-timers tilpasningsperiode noteres dødeligheden, og der anvendes følgende kriterier:
|
— |
dødelighed over 10 % efer syv dage: Hele partiet forkastes |
|
— |
dødelighed mellem 5 % og 10 % efter syv dage: Akklimatiseringen fortsættes i endnu syv dage |
|
— |
dødelighed under 5 % efter syv dage: Partiet accepteres; hvis dødeligheden er over 5 % efter endnu syv dage, forkastes hele partiet. |
Det sikres, at ingen af fiskene til testen har synlige sygdomme eller misdannelser. Syge fisk kasseres. Fisk må ikke behandles mod sygdomme hverken under testen eller i to uger forud for testen.
1.8. TESTENS UDFØRELSE
1.8.1. Indledende test
Det kan være nyttigt at gennemføre et indledende eksperiment med henblik på at optimere testbetingelserne for den endelige test, f.eks. valg af koncentration(er) af teststoffet og længden af eksponerings- og udskillelsesfasen.
1.8.2. Eksponeringsbetingelser
1.8.2.1.
Forudsigelse af optagelsesfasens længde kan ske på grundlag af praktiske erfaringer (f.eks. fra en tidligere undersøgelse eller et andet kemisk stof, der akkumuleringsmæssigt ligner) eller ved hjælp af forskellige empiriske sammenhænge ud fra kendskab til stoffets opløselighed i vand eller dets octanol/vand fordelingskoefficient (se bilag 3).
Optagelsesfasen bør vare 28 dage, medmindre det kan godtgøres, at der inden da er indtrådt ligevægt. Er der ikke opnået ligevægt efter 28 dage, forlænges optagelsesfasen, og målingerne fortsættes, indtil der er nået ligevægt, dog højst 60 dage.
1.8.2.2.
Normalt er en periode på halvdelen af optagelsesfasen tilstrækkeligt til at opnå et passende fald (f.eks. 95 %) i fiskenes belastning med stoffet (se redegørelsen for skønnet i bilag 3). Hvis der skal bruges urealistisk lang tid til at opnå et fald på 95 %, f.eks. dobbelt så lang tid som en normal optagelsesfase (dvs. mere end 56 dage), kan tidsrummet sættes ned (dvs. indtil koncentrationen af teststoffet er mindre end 10 % af ligevægtskoncentrationen). For stoffer med mere komplekse optagelses- og udskillelsesmønstre end det, hvor fisken betragtes som en helhed, og hvor der gælder førsteordenskinetik, må der dog benyttes en længere udskillelsesfase til bestemmelse af hastighedskonstanterne. Periodens længde kan f.eks. bestemmes af, hvor længe koncentrationen af teststoffet i fiskene er højere end påvisningsgrænsen.
1.8.2.3.
Der udvælges så mange fisk pr. testkoncentration, at der mindst er fire fisk til rådighed pr. prøve ved hver prøveudtagning. Ønskes der større statistisk styrke, må antallet af fisk pr. prøve øges.
Hvis der benyttes voksne fisk, anføres i rapporten, om der er brugt hanner, hunner eller begge køn i eksperimentet. Hvis begge køn er anvendt, skal det dokumenteres, at der inden eksponeringen ikke er nogen signifikant forskel i fedtindhold mellem kønnene; pooling af alle hanner og hunner kan være påkrævet.
Til en given test udvælges der fisk af samme vægt, således at den mindste ikke vejer mindre end to tredjedele af den største. Alle fisk skal være af samme årgang og komme fra samme kilde. Da fiskenes alder og vægt undertiden synes at indvirke signifikant på BCF-værdierne (1), skal disse oplysninger noteres nøjagtigt. Det anbefales, at man inden testen danner sig et skøn over gennemsnitsvægten ved at veje en delprøve af fiskebestanden.
1.8.2.4.
Der benyttes et højt forhold mellem vand og fisk, så faldet i Cw som følge af overførsel af fiskene ved testens begyndelse bliver så lille som muligt, og for at undgå, at koncentrationen af opløst oxygen falder. Det er vigtigt, at belastningen afpasses efter den anvendte fiskeart. I alle tilfælde anbefales normalt en belastning på 0,1-1,0 g fisk (våd vægt) pr. liter vand pr. dag. Belastningen kan sættes op, hvis det godtgøres, at den nødvendige koncentration af teststof kan fastholdes inden for ± 20 %, og at koncentrationen af opløst oxygen ikke kommer under 60 % af mætning.
Der skal tages hensyn til fiskeartens normale levevilkår ved valget af belastning. Eksempelvis kan bundfisk kræve større bundareal i akvariet for samme vandrumfang end pelagiske fiskearter.
1.8.2.5.
Under akklimatisering og test gives fiskene passende foder med kendt fedt- og proteinindhold i en sådan mængde, at de forbliver sunde og bevarer deres legemsvægt. Fiskene fodres dagligt under akklimatisering og test med en mængde svarende til ca. 1-2 % af legemsvægten; på denne måde holdes fedtkoncentrationen i de fleste fiskearter nogenlunde konstant under testen. Fodermængden beregnes på ny f.eks. en gang om ugen, så legemsvægt og fedtindhold holdes konstant. Til denne beregning kan man skønne vægten af fiskene i de enkelte kamre ud fra vægten af de fisk, der senest er taget op fra det pågældende kammer. De fisk, der bliver tilbage i kamrene, må ikke vejes.
Hver dag kort efter fodringen (30 min-l time) suges overskydende foder og ekskrementer op fra testkamrene. Kamrene holdes så rene som muligt under testen, så koncentrationen af organisk kulstof er så lav som mulig, eftersom tilstedeværende organisk kulstof kan nedsætte teststoffets biotilgængelighed (1).
Da meget foder er fremstillet af fiskemel, må det analyseres for teststoffet. Foderet bør tillige analyseres for pesticider og tungmetaller.
1.8.2.6.
Lysperioden er normalt 12-16 timer, og temperaturen (± 2 oC) skal være afpasset efter testfiskearten (se bilag 2). Belysningens type og karakteristika skal være kendt. Man bør være opmærksom på en eventuel fotokemisk omdannelse af teststoffet ved belysningsforholdene under undersøgelsen. Der skal benyttes en passende belysning, hvor det undgås, at fiskene udsættes for kunstigt fremkaldte fotokemiske omdannelsesprodukter. Det kan i nogle tilfælde være hensigtsmæssigt at afblænde UV-stråling under 290 nm med et filter.
1.8.2.7.
Fiskene udsættes under gennemstrømning for mindst to koncentrationer af teststoffet i vand. Normalt sættes den højeste koncentration af teststoffet til ca. 1 % af den akutte asymptotiske LC50 og mindst ti gange højere end påvisningsgrænsen i vand ved den anvendte analysemetode.
De højeste testkoncentration kan også fastsættes ved division af den akutte 96 h LC50 med et passende akut/kronisk-forhold (for nogle kemiske stoffer vil et passende forhold ligge fra ca 3 op til 100). Om muligt vælges den eller de andre testkoncentrationer, således at den (de) er en faktor ti lavere. Kan det ikke lade sig gøre på grund af 1 % af LC50 -kriteriet og analysegrænsen, kan der benyttes en mindre faktor, eller man kan overveje at benytte 14C-mærket teststof. Der må ikke benyttes koncentrationer højere end teststoffets opløselighed.
Hvis der benyttes et opløsende stof, må koncentrationen ikke være større end 0,1 ml/1, og den skal være den samme i alle testkar. Dette stofs og teststoffets bidrag til det samlede indhold af organisk kulstof i testvandet skal være kendt. Det skal dog så vidt muligt undgås at benytte sådanne materialer.
1.8.2.8.
Der skal ud over testrækken være en kontrolgruppe med fortyndingsvand eller en kontrolgruppe med et eventuelt opløsende stof, forudsat at det er fastslået, at det opløsende stof ikke har nogen virkning på fisk. Er det ikke tilfældet, skal begge kontrolgrupper forefindes.
1.8.3. Hyppighed af målinger af vandkvaliteten
Under testen måles opløst oxygen, TOC, pH og temperatur i alle kar. I kontrolkarrene og karret med den højeste koncentration måles vandets samlede hårdhed og saltholdighed, hvis det er relevant. Som minimum måles opløst oxygen og eventuel saltholdighed tre gange — ved begyndelsen, midt i og ved slutningen af optagelsesperioden — og en gang ugentligt i udskillelsesperioden. TOC måles ved testens begyndelse (24 h og 48 h før optagelsesfasen begynder), før fiskene overføres, og mindst en gang ugentligt under både optagelses- og udskillelsesfasen. Temperaturen måles dagligt, pH ved begyndelsen og slutningen af hver periode, og hårdheden én gang ved hver test. Temperaturen skal helst følges kontinuerligt i mindst ét at karrene.
1.8.4. Prøveudtagning og analyse af fisk og vand
1.8.4.1.
Der udtages prøver af vandet fra testkamrene med henblik på bestemmelse af teststofkoncentrationen, både inden fiskene overføres og under optagelses- og udskillelsesfasen. Som minimum udtages der prøver af vandet samtidig med prøver af fisk og før fodring. Under optagelsesfasen bestemmes teststofkoncentrationen til kontrol af, at gyldighedskriterierne er opfyldt.
Der udtages prøver af fisk mindst fem gange under optagelsesfasen og mindst fire gange under udskillelsesfasen. Da det i nogle tilfælde vil være vanskeligt at beregne et rimelig præcist skøn over BCF-værdien ud fra dette prøveantal, især hvis det formodes, at udskillelsen ikke følger en simpel førsteordenskinetik, kan det anbefales at øge prøveudtagningshyppigheden i begge perioder (se bilag 4). De ekstra prøver opbevares og analyseres kun, hvis resultaterne af den første analyserunde viser sig utilstrækkelig til beregning af BCF med den ønskede præcision.
I bilag 4 er der vist et eksempel på en acceptabel prøveudtagningsplan. Der kan let opstilles andre planer efter beregning af eksponeringstider for 95 % optagelse ud fra andre antagne Pow-værdier.
Prøveudtagningen fortsættes under optagelsesfasen, indtil der er opnået ligevægt, dog højst 28 dage. Er der ikke nået ligevægt inden for 28 dage, fortsættes prøveudtagningen, indtil der er opnået ligevægt, dog højst 60 dage. Før udskillelsesfasen overføres fiskene til rene kar.
1.8.4.2.
Vandprøver til analyse kan f.eks. udtages ved opsugning gennem et rør af inert materiale fra et sted midt i testkammeret. Da hverken filtrering eller centrifugering synes at kunne adskille den ikke-biotilgængelige fraktion af teststoffet fra den biotilgængelige (især for ekstremt lipofile stoffer, dvs. stoffer med log Pow > 5) (1) (5) i alle tilfælde, må prøverne ikke behandles på denne måde.
I stedet må man sørge for at holde karrene så rene som muligt, og indholdet af organisk kulstof i alt følges under såvel optagelses- som udskillelsesfasen.
Der udtages et passende antal fisk (normalt mindst fire) fra testkarrene ved hver prøveudtagning. De udtagne fisk skylles hurtigt med vand, duppes tørre og aflives med det samme på den mest velegnede og humane måde, hvorefter de vejes.
Fiske- og vandprøver skal helst analyseres umiddelbart efter udtagningen, så man undgår nedbrydning eller andre former for tab, og så der løbende under testen kan beregnes omtrentlige optagelses- og udskillelseshastigheder. Ved at foretage analyserne med det samme opdager man straks, når et plateau er nået.
Foretages analyserne ikke med det samme, opbevares prøverne hensigtsmæssigt. Inden undersøgelsen påbegyndes, indhentes der oplysninger om, hvordan det pågældende teststof bør opbevares, f.eks. dybfrysning, henstand ved 4 oC, opbevaringstid og ekstraktion.
1.8.4.3.
Da hele proceduren i vid udstrækning afhænger af analysemetodens nøjagtighed, præcision og følsomhed, kontrolleres det eksperimentelt, at den kemiske analyse giver tilfredsstillende præcision, reproducerbarhed og genfinding af teststoffet i både vand og fisk for den valgte metode. Det kontrolleres tillige, at teststoffet ikke kan påvises i det anvendte fortyndingsvand.
Om nødvendigt korrigeres de under testen funde Cw- og Cf-værdier for genfinding og baggrundsværdier i kontrolgrupperne. Fiske- og vandprøver håndteres hele tiden på en sådan måde, at kontaminering og tab (f.eks. ved adsorption i prøveudtagningsudstyret) er mindst mulige.
1.8.4.4.
Hvis der i testen anvendes radioaktivt mærket materiale, kan man analysere for det samlede indhold af radioaktivitet (dvs. det oprindelige stof og dets metabolitter), eller prøverne kan oprenses, så den oprindelige forbindelse kan analyseres særskilt. Også de vigtigste metabolitter kan karakteriseres ved ligevægtstilstanden eller ved afslutningen af optagelsesfasen, hvis den nås først. Hvis BCF udtrykt som samlet mængde radioaktivt mærket materiale er ≥ 1 000 % tilrådes det — for visse stoffers, f.eks. pesticiders, vedkommende henstilles det kraftigt — at nedbrydningsprodukter, der udgør ≥ 10 % af restindholdet i alt i fiskevævet ved ligevægt, identificeres og bestemmes kvantitativt. Hvis nedbrydningsprodukter, der udgør ≥ 10 % af radioaktivt mærket restindhold i alt i fiskevævet, identificeres og bestemmes kvantitativt, anbefales det at gøre det samme for nedbrydningsprodukter i testvandet.
Normalt skal koncentrationen af teststoffet bestemmes i hver enkelt fisk efter vejning. Er det ikke muligt, kan prøverne fra hver prøveudtagning pooles, men pooling indskrænker mulighederne for statistisk behandling af dataene. Hvis en bestemt statistisk metode og styrke er vigtige hensyn, bør testen omfatte tilstrækkeligt mange fisk til, at den ønskede pooling og styrke tilgodeses (6) (7).
BCF udtrykkes både som funktion af samlet våd vægt og — for stærkt lipofile stoffer — som funktion af fedtindholdet. Fiskenes fedtindhold bestemmes om muligt ved hver prøveudtagning. Bestemmelsen af fedtindholdet skal bestemmes ved hjælp af egnede metoder (henvisning (8) og (2) i bilag 3). Som standardmetode kan ekstraktion med chloroform/methanol anbefales (9). Da de forskellige metoder ikke giver identiske værdier (10), er det vigtigt at give nærmere oplysninger om, hvilken metode der er benyttet. Når det er muligt, skal lipidanalysen foretages på samme ekstrakt, som er benyttet til analyse for teststoffet, eftersom lipiderne ofte må fjernes fra ekstraktet, inden det kan analyseres ved gaskromatografi. Forskellen mellem fiskenes fedtindhold (i mg pr. kg våd vægt) ved begyndelsen af eksperimentet og ved slutningen må ikke være større end ± 25 %. Også vævets tørstofindhold skal oplyses, så lipidkoncentrationen kan omregnes fra våd basis til tørstofbasis.
2. DATA
2.1. BEHANDLING AF RESULTATER
Teststoffets optagelseskurve fås ved at afsætte koncentrationen i/på fiskene (eller bestemt væv) under optagelsesfasen mod tiden (lineære akser). Hvis kurven viser et plateau, dvs. at den er omtrent parallel med tidsaksen, beregnes ligevægtsværdien BCFSS ved hjælp af udtrykket
![]()
Hvis der ikke opnås ligevægt, kan der beregnes en BCFSS-værdi, der er tilstrækkelig nøjagtig til risikovurderingsformål, ud fra en »stationær tilstand« ved 80 % (1,6/k2) eller 95 % (3,0/k2) af ligevægt.
Også biokoncentreringsfaktoren BCFk beregnes som forholdet k1/k2, de to førsteordens hastighedskonstanter. Hastighedskonstanten for udskillelse (k2) bestemmes normalt ud fra udskillelseskurven (dvs. en afbildning af faldet i teststoffets koncentration i fiskene mod tiden). Dernæst beregnes hastighedskonstanten for optagelse (k1) på grundlag af k2 og en værdi af Cf, som udledes af optagelseskurven (se også bilag 5). Til at finde BCFk og hastighedskonstanterne k1 og k2 foretrækkes ikke-lineære parameterestimeringsmetoder på computer (11). Ellers kan der benyttes grafiske metoder til beregning af k1 og k2. Hvis udskillelseskurven tydeligvis ikke er af første orden, må der tages mere komplekse modeller i brug (se henvisningerne i bilag 3) og søges biostatistisk bistand.
2.2. FORTOLKNING AF RESULTATER
Hvis der i prøveopløsninger måles koncentrationer nær analysemetodens påvisningsgrænse, skal resultaterne fortolkes med forsigtighed.
Skarpe optagelses- og udskillelseskurver er tegn på biokoncentreringsdata af høj kvalitet. Forskellen mellem optagelses- og udskillelseskonstanterne ved to testkoncentrationer bør være mindre end 20 %, Hvis der iagttages signifikante forskelle i optagelses- og udskillelseskonstanter mellem de to anvendte testkoncentrationer, skal dette noteres og forsøges forklaret. Normalt ligger konfidensgrænserne for BCF-værdier i vel tilrettelagte undersøgelser omkring ± 20 %.
3. RAPPORTERING
Testrapporten skal indeholde følgende oplysninger:
3.1. TESTSTOF
|
— |
dets fysiske tilstandsform og relevante fysiske og kemiske egenskaber |
|
— |
data til kemisk identifikation (herunder et eventuelt indhold af organisk kulstof) |
|
— |
hvis stoffet er radioaktivt mærket, de mærkede atomers nøjagtige position og den procentdel af radioaktiviteten, der skyldes urenheder. |
3.2. TESTART
|
— |
dens videnskabelige navn, stamme, kilde, eventuel forbehandling, akklimatisering, alder, størrelse mv. |
3.3. TESTBETINGELSER
|
— |
testprocedure (f.eks. gennemstrømning eller semistatisk) |
|
— |
belysningskildens art og karakteristika og belysningsperiodernes varighed |
|
— |
testens tilrettelægning (f.eks. testkamrenes antal og størrelse, vandudskiftningsrate, antal gentagelser, antal fisk pr. gentagelse, antal testkoncentrationer, optagelses- og udskillelsesfasens længde samt udtagningshyppighed for fiske- og vandprøver) |
|
— |
metode til fremstilling af stamopløsninger og fornyelseshyppighed (hvis der er benyttet et opløsende stof, oplyses dets koncentration og dets bidrag til testvandets indhold af organisk kulstof) |
|
— |
de nominelle testkoncentrationer, gennemsnittet af de målte værdier og deres standardafvigelser i testkarrene samt hvilken metode der er benyttet til bestemmelsen |
|
— |
kilde til fortyndingsvand, beskrivelse af en eventuel forbehandling, resultater af forsøg til bekræftelse af, at fisk kan leve i vandet, samt vandets parametre: pH, hårdhed, temperatur, koncentration af opløst oxygen, restchlorindhold (hvis bestemt), organisk kulstof i alt, opslæmmet tørstof, testmediets saltholdighed (hvis relevant) samt alle andre foretagne målinger |
|
— |
kvaliteten af vandet i testkarrene, pH, hårdhed, TOC, temperatur og koncentration af opløst oxygen |
|
— |
detaljerede oplysninger om fodring (f.eks. foderets type, kilde og sammensætning (i hvert fald fedtog proteinindhold) samt fodermængde og fodringshyppighed) |
|
— |
oplysninger om, hvordan fiske- og vandprøver er behandlet, herunder forberedelse, opbevaring, ekstraktion og analysemetoder (og præcision) for teststof og fedtindhold (hvis bestemt). |
3.4. RESULTATER
|
— |
resultater af eventuelle indledende undersøgelser |
|
— |
dødelighed for kontrolfiskene og fiskene i de enkelte testkamre samt eventuelt iagttaget unormal adfærd |
|
— |
fiskenes fedtindhold (hvis bestemt i forbindelse med testen) |
|
— |
kurver (og alle måledata), der viser optagelse og udskillelse af teststoffet i fiskene, samt tiden til ligevægt |
|
— |
Cf og Cw (med standardafvigelse og interval, hvis det er relevant) for alle udtagne prøver (Cf anføres i μg pr. g våd vægt (ppm), enten legemsvægt eller vægt af et bestemt væv, f.eks. fedtvæv, og Cw μg/ml (ppm)); Cw-værdier for kontrolgruppen (også baggrundsværdier oplyses) |
|
— |
ligevægtsbiokoncentreringsfaktoren (BCFSS) og/eller den kinetiske koncentreringsfaktor (BCFk) og i relevante tilfælde 95 % konfidensgrænser for hastighedskonstanter for optagelse og udskillelse (alle udtrykt i forhold til fiskenes legemsvægt eller fiskenes eller de specificerede vævs fedtindhold), konfidensgrænser og standardafvigelse (hvis de foreligger samt beregnings- og dataanalysemetoder for hver koncentration af teststof |
|
— |
hvis der er benyttet radioaktivt mærkede stoffer, kan en eventuel akkumulering af påviste metabolitter anføres, hvis det er påkrævet |
|
— |
alle usædvanlige omstændigheder ved testen, eventuelle afvigelser fra proceduren samt alle andre relevante oplysninger. |
Resultater som »ikke påvist ved påvisningsgrænsen« bør ved forudgående metodeudvikling og god tilrettelæggelse af forsøget være indskrænket til et minimum, da sådanne resultater ikke kan benyttes til beregning af hastighedskonstanter.
4. HENVISNINGER
|
(1) |
Connell D.W. (1988). Bioaccumulation behaviour of persistent chemicals with aquatic organisms. Rev. Environ. Contam. Toxicol. 102, pp 117-156. |
|
(2) |
Bintein S., Devillers J. and Karcher W. (1993). Nonlinear dependence of fish bioconcentration on n-octanol/water partition coefficient. SAR and QSAR in Environmental Research, 1, 29-390. |
|
(3) |
OECD, Paris (1996). Direct Phototransformation of chemicals in water. Environmental Health and Safety Guidance Document Series on Testing and Assessment of Chemicals No 3. |
|
(4) |
Kristensen P. (1991) Bioconcentration in fish: Comparison of bioconcentration factors derived from OECD and ASTM testing methods; influence of particulate organic matter to the bioavailability of chemicals. Water Quality Institute, Denmark. |
|
(5) |
US EPA 822-R-94-002 (1994) Great Lake Water Quality Initiative Technical Support Doc. for the Procedure to Determine Bioaccumulation Factors. July 1994. |
|
(6) |
US FDA (Food and Drug Administration) Revision. Pesticide analytical manual, 1, 5600 Fisher's Lane, Rockville, MD 20852, July 1975. |
|
(7) |
US EPA (1974). Section 5, A(l) Analysis of Human Adipose Tissue, in Analysis of Pesticide Residues in Human and Environmental Samples, Thompson J.F. (ed.) Research Triangle Park, N.C. 27711. |
|
(8) |
Compaan H. (1980) in »The determination of the possible effects of chemicals and wastes on the aquatic environment: degradation, toxicity, bioaccumulation«, Ch. 2.3, Part II. Government Publisching Office, The Hague, The Netherlands. |
|
(9) |
Gardner et al, (1995) Limn. & Oceanogr. 30, 1099-1105. |
|
(10) |
Randall R.C, Lee H., Ozretich R.J., Lake J.L. and Pruell R.J. (1991). Evaluation of selected lipid methods for normalising pollutant bioaccumulation. Envir. Toxicol. Chem. 10, pp 1431-1436. |
|
(11) |
CEC, Bioconcentration of chemical substances in fish: the flow-through method-Ring Test Programme, 1984-1985. Final report March 1987. Authors: P. Kristensen and N. Nyholm. |
|
(12) |
ASTME E-1022-84 (Reapproved 1988) Standard Practice for conducting Bioconcentration Tests with Fishes and Saltwater Bivalve Molluscs. |
Bilag 1
Kemiske egenskaber af acceptabelt vand til fortynding
|
|
Stof |
Koncentrationsgrænse |
|
1 |
Partikler |
5 mg/l |
|
2 |
Organisk kulstof i alt |
2 mg/l |
|
3 |
Ikke-ioniseret ammoniak |
1 μg/l |
|
4 |
Restchlor |
10 μg/l |
|
5 |
Organophosphorpesticider i alt |
50 ng/l |
|
6 |
Organochlorpesticider i alt plus polychlorerede biphenyler |
50 ng/l |
|
7 |
Organisk chlor i alt |
25 ng/l |
|
8 |
Aluminium |
1 μg/l |
|
9 |
Arsen |
1 μg/l |
|
10 |
Chrom |
1 μg/l |
|
11 |
Cobalt |
1 μg/l |
|
12 |
Kobber |
1 μg/l |
|
13 |
Jern |
1 μg/l |
|
14 |
Bly |
l μg/l |
|
15 |
Nikkel |
1 μg/l |
|
16 |
Zink |
1 μg/l |
|
17 |
Cadmium |
100 ng/l |
|
18 |
Kviksølv |
100 ng/l |
|
19 |
Sølv |
100 ng/l |
Bilag 2
Anbefalede testfiskearter
|
|
Anbefalet art |
Anbefalet testtemperaturinterval ( oC) |
Anbefalet størrelse af testfisk (længde i cm) |
|
1 |
Danio rerio (17) (Teleostei, Cyprinidae) (Hamilton-Buchanan) zebrafisk |
20-25 |
3,0±0,5 |
|
2 |
Pimephales promelas (Teleostei, Cyprinidae) (Rafinesque) Fathead minnow |
20-25 |
5,0±2,0 |
|
3 |
Cyprinus carpio (Teleostei, Cyprinidae) (Linnaeus) Karpe |
20-25 |
5,0±3,0 |
|
4 |
Oryzias latipes (Teleostei, Poeciliidae) (Temminck and Schlegel) japansk risfisk |
20-25 |
4,0±1,0 |
|
5 |
Poecilia reticulata (Teleostei, Poeciliidae) (Peters) guppy |
20-25 |
3,0±1,0 |
|
6 |
Lepomis macrochirus (Teleostei, Centrarchidae) (Rafinesque) |
20-25 |
5,0±2,0 |
|
7 |
Oncorhynchus mykiss (Teleostei, Salmonidae) (Walbaum) regnbueørred |
13-17 |
8,0±4,0 |
|
8 |
Gasterosteus aculeatus (Teleostei, Gasterosteidae) (Linnaeus) trepigget hundestejle |
18-20 |
3,0±1,0 |
I forskellige lande benyttes der en række brakvands- og saltvandsarter, f.eks.
|
Trommefisk |
Leiostomus xanthurus |
|
Sheepshead minnow |
Cyprinodon variegatus |
|
Stribefisk |
Menidia beryllina |
|
Shiner perch |
Cymatogaster aggregata |
|
English sole |
Parophrys vetulus |
|
Staghjorn sculpin |
Leptocottus armatus |
|
Trepigget hundestejle |
Gasterosteus aculeatus |
|
Havaborre |
Dicentracus labrax |
|
Løje |
Alburnus alburnus |
Samling
De i ovenstående tabel opregnede ferskvandsfisk er lette at opdrætte og/eller fremskaffe hele året, mens saltvands- og brakvandsarterne til dels kun findes i bestemte lande. De kan opdrættes enten i fiskefarme eller i laboratoriet under kontrollerede forhold med hensyn til sygdomme og parasitter, så testdyrene er sunde og af kendt afstamning. Disse fisk kan fås over det meste af verden.
Bilag 3
Forudsigelse af længden af optagelsesfasen og udskillelsesfasen
1. Forudsigelse af optagelsesfasens længde
Før testen udføres, kan man ud fra en empirisk sammenhæng mellem k2 og n-octanol/vand fordelingskoefficienten (Pow) eller k2 og opløseligheden i vand (s) foretage et skøn over k2 og dermed en procentdel af den tid, der vil være påkrævet til at nå ligevægt.
Eksempelvis kan k2 (dag -1) skønnes ved hjælp af følgende empiriske udtryk (1):
|
log10k2 = – 0,414 log10(Pow) + 1,47 (r2 = 0,95) |
[ligning 1] |
Andre udtryk, se henvisning (2).
Hvis fordelingskoefficienten ikke er kendt, kan denne skønnes (3) ud fra stoffets opløselighed i vand (s) ifølge udtrykket:
|
log10(Pow) = 0,862 log10(s) + 0,710 (r2 = 0,994) |
[ligning 2] |
hvor
s = opløseligheden (mol/l) : (n = 36)
Disse sammenhænge gælder kun for stoffer med log Pow-værdier mellem 2 og 6,5 (4).
Den tid, der går til en bestemt procent af ligevægt, kan beregnes ud fra det generelle udtryk, der beskriver optagelses- og udskillelseskinetikken (førsteordenskinetik) ved at anvende k2-skønnet:

eller hvis Cw er konstant:
|
|
[ligning 3] |
Når der er næsten ligevægt (t → ∞), kan ligning 3 reduceres til (5) (6):
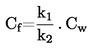 eller Cf/Cw = k1/k2
= BCF
eller Cf/Cw = k1/k2
= BCF
Da er k1/k2 . Cw en tilnærmelse til koncentrationen i fiskene ved »ligevægt« (Cf,s).
Ligning 3 kan omskrives til:
|
|
[ligning 4] |
Ved at anvende ligning 4 kan man forudsige, hvor lang tid der går indtil en vis procent af ligevægt er nået, når k2 allerede er skønnet ved hjælp af ligning 1 eller 2.
Som rettesnor kan anføres, at den statistisk optimale længde af optagelsesfasen, når der ønskes statistisk acceptable data (BCFk), er den tid, der går, indtil kurven over logaritmen til koncentrationen af teststoffet i fiskene mod tiden har nået sit midtpunkt, dvs. ved 1,6/k2 svarende til 80 % ligevægt, men ikke over 3,0/k2 svarende til 95 % ligevægt (7).
Den tid, der går indtil 80 % ligevægt, er (ligning 4):
|
|
[ligning 5] |
Tilsvarende for 95 % ligevægt:
|
|
[ligning 6] |
For eksempel vil optagelsesfasens længde (up) for et teststof med Pow = 4 være (ligning 1, 5 og 6) :
|
log10k2 = – 0,414.(4) + 1,47 |
k2 = 0,652 dag-1 |
up (80 %) = 1,6/0,652, i.e. 2,45 dag (59 timer)
eller up (95 %) = 3,0/0,652, i.e. 4,60 dag (110 timer)
Tilsvarende vil optagelsesfasens for et teststof med s = 10-5 mol/l (log(s) = – 5,0) være (ligning 1, 2, 5 og 6)
log10 (Pow) = 0,862 (– 5,0) + 0,710 = 5,02
log10 k2 = 0,414 (5,02) + 1,47
k2 = 0,246 dag-1
up (80 %) = 1,6/0,246, i.e. 6,5 dage (156 timer)
eller up (95 %) = 3,0/0,246, i.e. 12,2 dage (293 timer)
Alternativt kan undtrykket :
teq = 6,54 × 10-3 Pow + 55,31 (timer)
benyttes til beregning af, hvor lang tid der går til faktisk ligevægt (4).
2. Forudsigelse af udskillelsesfasens længde
Ud fra det generelle udtryk, der beskriver optagelses- og udskillelseskinetikken (førsteordenskinetik), kan man ligeledes forudsige, hvor lang tid der kræves for, at legemsbelastningen falder til en vis procentdel af begyndelseskoncentrationen (1) (8).
I udskillelsesfasen antages Cw at være nul. Udtrykket kan da reduceres til:
 eller
eller 
hvor Cf,o er koncentrationen ved udskillelsesperiodens begyndelse. 50 % udskillelse vil da være nået til tidspunktet (t50):
 eller
eller 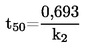
Tilsvarende vil 95 % udskillelse være nået ved:

Hvis der i den første periode benyttes 80 % optagelse (1,6/k2), og der i udskillelsesfasen benyttes 95 % tab (3,0/k2), bliver udskillelsesfasen omtrent dobbelt så lang som optagelsesfasen.
Det er vigtigt at bemærke, at skønnene er baseret på antagelsen om førsteordenskinetik for optagelse og udskillelse. Er denne forudsætning tydeligvis ikke opfyldt, må der tages mere komplekse modeller i brug (f.eks. henvisning 1).
Henvisning (til bilag 3)
|
(1) |
Spacie, A. and Hamelink, J.L. (1982) Alternative models for describing the bioconcentration of organics in fish. Environ. Toxicol. and Chem. 1, pp 309-320. |
|
(2) |
Kristensen, P, (1991) Bioconcentration in fish: comparison of BCF's derived from OECD and ASTM testing methods; influence of particulate matter to the bioavailability of chemicals. Danish Water Quality Institute. |
|
(3) |
Chiou, C.T. and Schmedding, D.W. (1982) Partitioning of organic compounds in octanol-water systems. Environ. Sci. Technol. 16 (1), pp. 4-10. |
|
(4) |
Hawker, D.W. and Connell, D.W. (1988) Influence of partition coefficient of lipophilic compounds on bioconcentration kinetics with fish. Wat. Res. 22 (6), pp 701-707. |
|
(5) |
Branson, D.R., Blau, G.E., Alexander, H.C. and Neely, W.B. (1975) Transactions of the American Fisheries Society, 104 (4), pp 785-792. |
|
(6) |
Ernst, W. (1985) Accumulation in aquatic organisms. In: Appraisal of tests to predict the environmental behaviour of chemicals. Sheehman, P., Korte, F., Klein, W. and Bourdeau P.H. (red) Part 4.4, pp 243-255. SCOPE, 1985, John Wiley & Sons Ltd. N.Y. |
|
(7) |
Reilly, P.M., Bajramovic, R, Blau, G.E., Branson, D.R. and Sauerhof, f M.W. (1977) Guidelines for the optimal design of experiments to estimate parameters in first order kenetic models, Can. J. Chem. Eng. 55, pp 614-622. |
|
(8) |
Könemann, H. and Van Leeuwen, K, (1980) Toxicokinetics in fish: Accumulation and elimination of six Chlorobenzenes by Guppies. Chemosphere, 9, pp 3-19. |
Bilag 4
Teoretisk eksempel på prøveudtagningsplan for biokoncentreringstest af stoffer med log Pow = 4
|
Fiskeprøve |
Tidspunkt for prøveudtagningen |
Antal vandprøver |
Antal fisk pr. prøve |
||||
|
Mindste hyppighed (dage) |
Supplerende prøve |
||||||
|
Optagelsesfasen |
– 1 0 |
|
2 (18) 2 |
Tilførsel af 45-80 fisk |
|||
|
1. |
0,3 |
0,4 |
2 (2) |
4 (4) |
|||
|
2. |
0,6 |
0,9 |
2 (2) |
4 (4) |
|||
|
3. |
1,2 |
1,7 |
2 (2) |
4 (4) |
|||
|
4. |
2,4 |
3,3 |
2 (2) |
4 (4) |
|||
|
5. |
4,7 |
|
2 |
6 |
|||
|
Udskillelsesfasen |
|
|
|
Fiskene overføres til vand uden teststof |
|||
|
6. |
5,0 |
5,3 |
|
4 (4) |
|||
|
7. |
5,9 |
7,0 |
|
4 (4) |
|||
|
8. |
9,3 |
11,2 |
|
4 (4) |
|||
|
9. |
14,0 |
17,5 |
|
6 (4) |
|||
|
Tallene i parentes angiver, hvor mange prøver (vand, fisk) der skal udtages, hvis der foretages supplerende prøveudtagning.
|
|||||||
Bilag 5
Modelvalg
De fleste biokoncentreringsdata antages at kunne beskrives »rimelig« godt ved en simpel model med to dele (»compartments«) og to parametre, hvilket fremgår af den bedste retlinede kurve gennem punkterne for koncentrationen i fisk under udskilielsesfasen, når de afsættes på semilogaritmisk papir. (Hvis punkterne ikke kan beskrives ved en ret linje, må der benyttes mere komplekse modeller, se f.eks. Spacie og Hamelinck, henvisning (1) til bilag 3).
Grafisk metode til bestemmelse af hastighedskonstanten for udskillelse, k2
Koncentrationen af teststoffet i hver fiskeprøve afbildes mod tiden på semilogaritmisk papir. Linjen har hældningen k2.
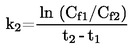

Det skal bemærkes, at afvigelser fra en ret linje kan tyde på, at udskillelsens kinetik er mere kompleks end første orden. Der kan benyttes en grafisk metode til løsning af udskillelse, der ikke har af førsteordens kinetik.
Grafisk metode til bestemmelse af hastighedskonstanten for optagelse, k1
På grundlag af k2 beregnes k1 efter udtrykket
|
|
(ligning 1) |
Værdien af Cf aflæses ved midtpunktet af den glatte optagelseskurve fra afbildningen af logaritmen til koncentrationen mod tiden (lineært).
Computermetode til bestemmelse af hastighedskonstanterne for optagelse og udskillelse
Til bestemmelse af biokoncentreringsfaktoren og hastighedskonstanterne k1 og k2 foretrækkes det, at der benyttes ikke-lineære estimeringsmetoder på computer. Sådanne programmer finder værdier for k1 og k2 ud fra et sæt sekventielle data for koncentration og tid og følgende model:
|
|
(ligning 2) |
|
|
(ligning 3) |
hvor tc er tidspunktet for afslutningen af optagelsesfasen.
Denne metode giver skøn over standardafvigelser for k1 og k2.
Eftersom k2 i de fleste tilfælde kan skønnes ud fra udskillelseskurven med forholdsvis stor præcision, og der er en stærk korrelation mellem de to parametre k1 og k2, hvis skønnet samtidigt, er det undertiden bedst først at beregne k2, ud fra udskillelsesdataene alene og derefter at beregne kl ud fra optagelsesdataene ved hjælp af ikke-lineær regression.
C.14. VÆKSTTEST PÅ FISKEYNGEL
1. METODE
Denne væksttoksicitetstestmetode svarer til OECD TG 215 (2000).
1.1. INDLEDNING
Formålet med denne test er at vurdere virkningerne på væksten hos fiskeyngel, som gennem længere tid eksponeres for kemikalier. Testen er baseret på en metode, som er udviklet og ringtestet (1)(3) inden for den Europæiske Union med henblik på at vurdere virkningerne af kemikalier på væksten hos yngel af regnbueørred (Oncorhynchus mykiss) under gennemstrømningsbetingelser. Andre veldokumenterede aner kan anvendes. For eksempel har man erfaring fra væksttest med zebrafisk (Danio rerio) (19) (3)(4) og riskarpe (medaka, Oryzias latipes) (5)(6)(7).
Der henvises også til den generelle indledning til del C.
1.2. DEFINITIONER
Laveste koncentration med observeret effekt (LOEC) er den laveste koncentration af et testkemikalie, som i testen har vist sig at have en signifikant virkning (p < 0,05) i sammenligning med en kontrolgruppe. Dog skal alle testkoncentrationer over LOEC have en skadevirkning, som er lig med eller større end de virkninger, der er observeret ved LOEC.
Koncentration uden observeret effekt (NOEC) er testkoncentrationen umiddelbart under LOEC.
EC X er ved denne testmetode den koncentration af testkemikaliet, som forårsager en variation i fiskenes tilvækst på x % i sammenligning med kontrolgrupperne.
Belastningsgraden er fiskenes vådvægt pr. vandvolumen.
Bestandtætheden er antallet af fisk pr. vandvolumen.
Enkeltfisk-specifik tilvækst er et udtryk for en enkelt fisks tilvækst på grundlag af fiskens startvægt.
Beholdergennemsnits-specifik tilvækst er et udtryk for den gennemsnitlige tilvækst for en beholderpopulation ved én koncentration.
Pseudo-specifik tilvækst er et udtryk for den enkelte fisks tilvækst i sammenligning med beholderpopulationens gennemsnitlige startvægt.
1.3. TESTMETODENS PRINCIP
Fiskeyngel i den eskponentielle vækstfase vejes og anbringes derefter i testbeholdere og eksponeres for en række subletale koncentrationer af testkemikaliet opløst i vand, fortrinsvis ved gennemstrømning eller, hvis dette ikke er muligt, under passende semistatiske betingelser (statisk-udskiftning). Testens varighed er 28 dage. Fiskene fodres dagligt. Foderrationen baseres på fiskenes startvægt og kan genberegnes efter 14 dage. Ved testens afslutning vejes fiskene igen. Virkningerne på tilvæksten analyseres ved anvendelse af en regressionsmodel for at beregne den koncentration, som ville forårsage en variation i tilvæksten på x %, dvs. ECX (f.eks. EC10, EC20 eller EC30). Alternativt kan dataene sammenlignes med kontrolværdierne for at bestemme den laveste koncentration med observeret effekt (LOEC) og derudfra koncentrationen uden observeret effekt (NOEC).
1.4. INFORMATION OM TESTKEMIKALIET
Der bør foreligge resultater fra en akut-toksidtetstest (se testmetode C.1), fortrinsvis udført på den art, som er valgt til denne test. Dette indebærer, at testkemikaliets vandopløselighed og damptryk er kendt, og at der foreligger en pålidelig analysemetode til kvantitativ bestemmelse af kemikaliet i testopløsningerne med kendt og rapporteret nøjagtighed, samt en detektionsgrænse.
Nyttige oplysninger omfatter strukturformlen, kemikaliets renhed, vand- og lysstabilitet, pKa, Pow samt resultater fra en test for umiddelbar biologisk nedbrydelighed (se testmetode C.4).
1.5. TESTENS VALIDITET
Testens validitet forudsætter følgende:
|
— |
mortaliteten i kontrolgruppen(-erne) må ikke overstige 10 % ved testens afslutning |
|
— |
den gennemsnitlige vægt af fiskene i kontrolgruppen(-erne) skal være øget tilstrækkeligt til, at detektion af den minimale variation i tilvæksten kan betragtes som signifikant. En ringtest (2) har vist, at for regnbueørreders vedkommende skal gennemsnitsvægten for fiskene i kontrolgrupperne i løbet af 28 dage være øget med mindst det halve (dvs. 50 %) af deres gennemsnitlige startvægt; f.eks. startvægt: 1 g/fisk (= 100 %), slutvægt efter 28 dage: ≥ 1,5 g/fisk (≥ 150 %) |
|
— |
koncentrationen af opløst ilt skal gennem hele testperioden have været mindst 60 % af værdien for luftmætning (ASV) |
|
— |
vandtemperaturen må ikke på noget tidspunkt i løbet af testen variere med mere end 1 oC mellem testbeholdeme og skal holdes inden for 2 oC omkring de temperaturintervaller, der er specificeret for de arter, der anvendes i testen (tillæg 1). |
1.6. BESKRIVELSE AF TESTMETODEN
1.6.1. Apparatur
Normalt laboratorieudstyr og i særdeleshed følgende:
|
— |
ilt- og pH-måleudstyr |
|
— |
udstyr til bestemmelse af vandets hårdhed og alkalinitet |
|
— |
egnet apparatur til temperaturkontrol og fortrinsvis kontinuerlig overvågning |
|
— |
beholdere fremstillet af et kemisk inaktivt materiale og med passende kapacitet i forhold til den anbefalede belastningsgrad og bestandtæthed (se 1.8.5 og tillæg 1) |
|
— |
vægt med tilstrækkelig nøjagtighed (dvs. nøjagtighed på ±0,5 %). |
1.6.2. Vand
Som testvand kan anvendes vand af enhver beskaffenhed, hvori den art, der anvendes i testen, udviser passende langtidsoverlevelse og vækst. Vandets kvalitet skal være ensartet i hele testperioden. Vandets pH skal ligge inden for området fra 6,5 til 8,5, men bør i løbet af en given test ligge inden for et område på ±0,5 pH-enheder. En hårdhedsgrad på over 140 mg/l (som CaCO3) anbefales. For at sikre, at fortyndingsvandet ikke får urimelig indflydelse på testresultatet (f.eks. ved dannelse af et kompleks med testkemikaliet), skal der regelmæssigt udtages prøver til analyse. Målinger af tungmetaller (f.eks. Cu, Pb, Zn, Hg, Cd og Ni), de vigtigste anioner og kationer (f.eks. Ca, Mg, Na, K, Cl og SO4, pesticider (f.eks. samlet mængde phosphor- og chlorholdige organiske pesticider), samlet mængde organisk kulstof og opslæmmet tørstof skal foretages, f.eks. hver tredje måned, såfremt man ved, at fortyndingsvandet har en relativt konstant kvalitet. Hvis vandkvaliteten påviseligt har været konstant gennem mindst ét år, kan analyserne foretages mindre hyppigt og med længere intervaller (f.eks. hver sjette måned). I tillæg 2 er anført nogle kemiske karakteristika for acceptabelt fortyndingsvand.
1.6.3. Testopløsninger
Testopløsninger med de valgte koncentrationer fremstilles ved fortynding af en stamopløsning.
Stamopløsningen bør fortrinsvis fremstilles ved mekanisk opblanding eller omrystning af testkemikaliet i fortyndingsvandet (f.eks. ved omrøring eller ultralydbehandling). Mætningssøjler (opløselighedssøjler) kan anvendes for at opnå en passende koncentration i stamopløsningen.
Anvendelse af opløsningsmidler eller dispergeringsmidler (opløselighedsfremmere) kan i nogle tilfælde være nødvendig for at fremstille en stamopløsning med en passende koncentration. Eksempler på egnede opløsningsmidler er acetone, ethanol, methanol, dimethylsulfoxid, dimethylformamid og triethylenglycol. Eksempler på egnede dispergeringsmidler er Cremophor RH40, Tween 80, methylcellulose 0,01 % og HCO-40. Man skal udvise forsigtighed ved anvendelse af stoffer, der er umiddelbart biologisk nedbrydelige (f.eks. acetone) og/eller meget flygtige, idet de ved gennemstrømnmgstest kan give anledning til problemer med bakteriebelægninger. Når der anvendes en opløselighedsfremmer, må dette ikke have nogen signifikant virkning på fiskenes vækst eller synlig negativ virkning på fiskeyngelen, hvilket kan afsløres ved en kontroltest, hvor kun opløsningsmidlet anvendes.
Til gennemstrømningstest kræves et system, som kontinuerligt doserer og fortynder en stamopløsning af testkemikaliet (f.eks. udmålingspumpe, proportionalfortynder, mætningssystem), så der afgives en række forskellige koncentrationer til testbeholderne. Stamopløsningernes og fortyndingsvandets strømningshastigheder skal i løbet af testen kontrolleres regelmæssigt, fortrinsvis dagligt, og må ikke variere mere end 10 % gennem hele testforløbet. En ringtest (2) har vist, at en vandudskiftningshyppighed på 6 l pr. g fisk pr. døgn i løbet af testen er passende for regnbueørreds vedkommende (se 1.8.2.2).
Til semistatiske (udskiftnings-) test vil medieudskiftningshyppigheden være afhængig af testkemikaliets stabilitet; men det anbefales at udskifte vandet hver dag. Hvis det på baggrund af præliminære stabilitetstest (se 1.4) har vist sig, at testkemikaliets koncentration ikke er stabil (dvs. ligger uden for området på 80-120 % af den nominelle værdi, eller under 80 % af den målte startkoncentration) i udskiftningstidsrummet, bør man overveje at anvende en gennemstrømningstest.
1.6.4. Valg af fiskeart
Regnbueørred (Oncorhynchus mykiss) er den anbefalede art til denne test, idet man har mest erfaring fra ringtest med denne art (1)(2). Andre veldokumenterede arter kan dog anvendes; men testproceduren må eventuelt tilpasses, så man opnår de bedst egnede testbetingelser. For eksempel har man også erfaring med zebrafisk (Danio rerio) (3)(4) og riskarpe (medaka, Oryzias latipes) (5)(6)(7). En begrundelse for valget af art og testmetode skal i så fald anføres.
1.6.5. Opbevaring af fiskene
Testfiskene skal udvælges fra en population af en enkelt bestand, fortrinsvis fra samme gydning, som i mindst to uger forud for testen holdes under vandkvalitets- og lysbetingelser, som svarer til de i testen anvendte. Fiskene skal fodres med en minimumsration på 2 % af kropsvægten pr. dag, og fortrinsvis 4 % af kropsvægten pr. dag, gennem hele opbevaringsperioden og under testen.
Efter en akklimatiseringsperiode på 48 timer registreres mortaliteten, og følgende kriterier bringes i anvendelse:
|
— |
mortalitet på mere end 10 % af populationen på syv dage: Hele fiskegruppen kasseres |
|
— |
mortalitet på mellem 5 % og 10 % af populationen: akklimatisering i yderligere syv dage, og hvis mortaliteten er over 5 % i løbet af de følgende syv dage, kasseres hele fiskegruppen |
|
— |
mortalitet på mindre end 5 % af populationen på syv dage: Fiskegruppen godkendes. |
I de to uger forud for testen eller under testen bør fiskene ikke behandles for sygdomme.
1.7. TESTDESIGN
»Testdesignet« vedrører valget af testkoncentrationernes antal og intervaller, antal beholdere med hvert koncentrationsniveau samt antal fisk pr. beholder. Optimalt bør testedesignet vælges under hensyntagen til:
|
— |
undersøgelsens formål |
|
— |
den anvendte statistiske analysemetode |
|
— |
forsøgsressourcernes tilgængelighed og pris. |
Beskrivelsen af formålet bør, om muligt, angive den statistiske opløsningsgrad, ved hvilken en given forskels-størrelse (f.eks. i tilvækst) skal detekteres, eller alternativt den nøjagtighed, hvormed ECx (f.eks. hvor x = 10, 20 eller 30, og fortrinsvis ikke mindre end 10) skal beregnes. Dette er en forudsætning for, at undersøgelsens omfang kan fastlægges med sikkerhed.
Det er vigtigt at forstå, at et design, som er optimalt (giver den bedste udnyttelse af ressourcerne) til brug i forbindelse med én statistisk analysemetode, ikke nødvendigvis er optimalt ved en anden metode. Det design, der anbefales til vurdering af LOEC/NOEC, vil derfor ikke være det samme som det, der anbefales til brug ved regressionsanalyse.
I de fleste tilfælde foretrækkes regressionsanalyse frem for variansanalyse, af årsager, som Stephan og Rogers (8) har gjort rede for. Hvor der imidlertid ikke foreligger nogen egnet regressionsmodel, bør (r2 < 0,9) NOEC/LOEC anvendes.
1.7.1. Design for regressionsanalyse
Følgende punkter er vigtige at tage i betragtning, når en test skal designes til regressionsanalyse:
|
— |
Testen må nødvendigvis omfatte et interval af koncentrationer, der ligger omkring den virksomme koncentration (f.eks. EC10,20,30) og det koncentrationsområde, hvor man er interesseret i testkemikaliets virkning. Beregningen af de virksomme koncentrationer vil være mest nøjagtig, når den virksomme koncentration befinder sig midt i testkoncentrationsområdet. En præliminær test til bestemmelse af dette område kan være nyttig, når det skal afgøres, hvilke testkoncentrationer der er passende. |
|
— |
For at man skal kunne opstille en fyldestgørende statistisk model, skal testen omfatte mindst én kontrolbeholder og fem yderligere beholdere med forskellige koncentrationer. Når der anvendes en opløselighedsfremmer, og hvor det er aktuelt, bør der i tillæg til testrækken udføres en test på én kontrolbeholder indeholdende opløselighedsfremmeren i den højeste testkoncentration (se 1.8.3 og 1.8.4). |
|
— |
En passende geometrisk eller logaritmisk progression (9) (se tillæg 3) kan anvendes. Logaritmiske progressioner mellem testkoncentrationerne er at foretrække. |
|
— |
Hvis man har mere end seks beholdere til rådighed, bør de ekstra beholdere enten anvendes til replikation eller fordeles inden for koncentrationsområdet, så der bliver mulighed for mindre intervaller mellem niveauerne. Disse to muligheder er lige ønskelige. |
1.7.2. Design for vurdering af NOEC/LOEC ved anvendelse af variansanalyse (ANOVA)
Fortrinsvis bør der være replikatbeholdere for hver enkelt koncentration, og statistiske analyser bør foretages på beholderniveau (10). Hvis man ikke har replikatbeholdere, kan man ikke tage højde for variabiliteten mellem beholdere, ud over hvad der må tilskrives enkeltfisk. Man har imidlertid erfaret (11), at variabiliteten mellem beholdere har været meget lille i sammenligning med variabiliteten inden for beholderen (dvs. mellem enkeltfisk) i det undersøgte tilfælde. Det er derfor et relativt acceptabelt alternativ at udføre statistisk analyse på enkeltfiskniveau.
Normalt anvendes mindst fem testkoncentrationer i en geometrisk progression med en faktor, som ikke overstiger 3.2.
Når testene udføres med replikatbeholdere, bør antallet af replikatkontrolbeholdere, og dermed antallet af fisk, almindeligvis være det dobbelte for hver af testkoncentrationerne, som bør omfatte samme antal (12)(13)(14). Hvis man derimod ikke har replikatbeholdere, bør der være lige så mange fisk i kontrolgruppen som i hver af restkoncentrationerne.
Hvis ANOVA skal baseres på beholdere i stedet for enkeltfisk (hvilket vil indebære enten, at hver enkelt fisk skal mærkes, eller anvendelse af »pseudo«-specifik tilvækst (se 2.1.2)), vil det være nødvendigt med tilstrækkeligt mange replikatbeholdere til, at standardafvigelsen for »beholdere inden for koncentrationerne« kan bestemmes. Dette betyder, at fejlfrihedsgraden i variansanalysen bør være mindst fem (10). Hvis kun kontrolgrupperne replikeres, vil der være en risiko for skævhed i fejlvariabiliteten, idet denne kan stige, når middelværdien for den pågældende tilvækst stiger. Eftersom det er sandsynligt, at tilvæksten vil falde, når koncentrationen øges, vil dette give en tilbøjelighed til overvurdering af variabiliteten.
1.8. FREMGANGSMÅDE
1.8.1. Udvælgelse og vejning af testfisk
Det er vigtig: a: minimere variationen i fiskenes vægt ved testens begyndelse. Egnede størrelser for de forskellige fiskearter, der anbefales til brug i denne test. er angivet i tillæg 1. For hele gruppen af fisk, der bruges i testen, gælder det, at området for individuel vægt ved testens begyndelse ideelt bør holdes inden for ± 10 % af gennemsnitsvægten og må under ingen omstændigheder overstige 25 %. Det anbefales at veje en delprøve af fisk inden testen for at beregne gennemsnitsvægten.
Stampopulationen bør ikke fodres i 24 timer forud for testens begyndelse. Derpå bør fiskene vælges tilfældigt. Ved anvendelse af et totalbedøvende middel (f.eks. en vandig opløsning af 100 mg/l tricainmethansulfonat (MS 222) neutraliseret ved tilsætning af to dele natriumbicarbonat pr. del MS 222) bør fiskene vejes enkeltvis som vådvægt (duppes tørre) med den nøjagtighed, som er anført i tillæg 1. De fisk, hvis vægt ligger inden for det ønskede område, tages fra og fordeles derpå tilfældigt mellem testbeholderne. Den samlede vådvægt af fisk i hver testbeholder registreres. Både anvendelsen af bedøvelsesmidlet og håndteringen af fiskene (herunder aftørring og vejning) kan give anledning til stress og beskadigelser af fiskeyngelen, i særdeleshed hvor det drejer sig om de arter, der er små af størrelse. Derfor skal håndteringen af fiskeyngelen foretages med den yderste forsigtighed for at undgå at stresse og beskadige testfiskene.
Fiskene vejes igen på testens 28. dag (se 1.8.6). Hvis det imidlertid skønnes nødvendigt at genberegne foderrationen, kan fiskene vejes igen på testens 14. dag (se 1.8.2.3). Andre metoder, som f.eks. en fotografisk metode, vil kunne anvendes for at bestemme ændringer i fiskenes størrelse, og foderrationerne vil så kunne justeres ud fra dette.
1.8.2. Eksponeringsbetingelser
1.8.2.1. Varighed
Testens varighed er ≥ 28 dage.
1.8.2.2. Belastningsgrad og bestandtæthed
Det er vigtigt, at belastningsgraden og bestandtætheden er passende for den fiskeart, der bruges i testen (se tillæg 1). Hvis bestandtætheden er for stor, vil der opstå stress på grund af trængsel, hvilket vil føre til reduceret tilvækst og øget risiko for sygdomme. Hvis bestandtætheden er for lav, kan det medføre territorialadfærd, hvilket også vil kunne indvirke på væksten. Under alle omstændigheder skal belastningsgraden være så lav, at der kan opretholdes en koncentration af opløst ilt på mindst 60 % ASV uden beluftning. For regnbueørreders vedkommende har man ved en ringtest (2) påvist, at en belastningsgrad på 16 ørreder a 3-5 g i et volumen på 40 liter er acceptabel. Den anbefalede hyppighed for udskiftning af vandet under testen er 6 l pr. g fisk pr. dag.
1.8.2.3. Fodring
Fiskene skal fodres med et passende foder (tillæg 1) og tilstrækkeligt ofte til at frembringe en acceptabel tilvækst. Man må udvise omhu for at undgå mikrobiel vækst og urenheder i vandet. For regnbueørreders vedkommende vil en fodertilførsel på 4 % af kropsvægten pr. dag som regel opfylde disse betingelser (2)(15)(16)(17). Den daglige foderration kan deles i to lige store portioner, som gives fiskene ved to daglige fodringer med mindst fem timers mellemrum. Rationen baseres på fiskenes samlede startvægt i hver enkelt testbeholder. Hvis fiskene vejes igen på testens 14. dag, genberegnes rationen ud fra denne vægt. Fiskene bør ikke fodres i 24 timer inden vejningen.
Foderrester og fækalmateriale skal hver dag fjernes fra testbeholderne ved omhyggelig rengøring af bunden i hver enkelt beholder ved hjælp af opsugning.
1.8.2.4. Lys og temperatur
Lysperioderne og vandtemperaturen skal passe til den art, der bruges i testen (tillæg 1).
1.8.3. Testkoncentrationer
Normalt kræves, uanset testens design, fem koncentrationer af testkemikaliet (se 1.7.2). Forudgående kendskab til testkemikaliets toksicitet (f.eks. fra akut-testning og eller fra »range finding tests«) vil være nyttigt ved udvælgelsen af passende testkoncentrationer. Begrundelse skal anføres, hvis der anvendes færre end fem koncentrationer. Den højeste testkoncentration må ikke overstige kemikaliets opløselighedsgrænse i vand.
Når der anvendes en opløselighedfremmer som hjælpemiddel ved fremstillingen af stamopløsninger, må dens slutkoncentration ikke være højere end 0,1 ml/l og bør fortrinsvis være den samme i alle testbeholdere (se 1.6.3). Anvendelse af sådanne stoffer skal imidlertid så vidt muligt undgås.
1.8.4. Kontrolprøver
Antallet af fortyndingsvand-kontrolprøver afhænger af testens design (se 1.7-1.7.2). Hvis der anvendes en opløselighedsfremmer, skal der desuden foretages det samme antal opløselighedsfremmer-kontrolprøver som fortyndingsvand-kontrolprøver.
1.8.5. Hyppighed af analyser og målinger
Under testen måles koncentrationerne af testkemikaliet med regelmæssige mellemrum (se nedenfor).
Ved gennemstrørnningstest skal strømningshastighederne for fortyndingsmidlet og giftstofstamopløsningen kontrolleres regelmæssigt, fortrinsvis dagligt, og de må ikke udvise en variation på mere end 10 % gennem hele testperioden. Hvis testkemikaliets koncentrationer forventes at ligge inden for ± 20 % af de nominelle værdier (dvs. inden for området fra 80 til 120 % — se 1.6.2 og 1.6.3), anbefales det som et minimum, at den højeste og den laveste testkoncentration analyseres ved testens begyndelse og hver uge derefter. Ved test, hvor testkemikaliets koncentration ikke forventes at holde sig inden for ± 20 % af de nominelle værdier (baseret på oplysninger om testkemikaliets stabilitet), vil det være nødvendigt at analysere samtlige testkoncentrationer, men efter samme retningslinjer.
Ved semistatiske (udskiftnings-) test, hvor testkemikaliets koncentration forventes at holde sig inden for ± 20 % af de nominelle værdier, anbefales det som et minimum, at den højeste og den laveste testkoncentration analyseres, når de lige er fremstillet og umiddelbart inden udskiftning, ved undersøgelsens begyndelse og hver uge derefter. Ved test, hvor testkemikaliets koncentration ikke forventes at holde sig inden for ± 20 % af de nominelle værdier, skal samtlige testkoncentrationer analyseres efter samme retningslinjer som for mere stabile kemikalier.
Det anbefales, at resultaterne baseres på målte koncentrationer. Hvis det imidlertid kan påvises, at testkemikaliets koncentration i tilstrækkelig grad har holdt sig inden for ± 20 % af de nominelle værdier eller den målte startkoncentration gennem hele testen, kan resultaterne baseres på de nominelle eller de målte værdier.
Det kan være nødvendigt at centrifugere eller filtrere prøverne (f.eks. med et filter med en porestørrelse på 0,45 μm). Det anbefales at centrifugere prøverne; men hvis testmaterialet ikke adsorberer på filteret, kan også filtrering accepteres.
Under testen skal opløst ilt, pH og temperatur måles i samtlige testbeholdere. Samlet hårdhedsgrad, alkalinitet og saltindhold (hvis dette er aktuelt) skal måles i kontrolbeholderne og i én beholder med den højeste koncentration. Som et minimum skal opløst ilt og saltindhold (hvis aktuelt) måles tre gange (ved testens begyndelse, midte og afslutning). Ved semistatiske test anbefales, at opløst ilt måles hyppigere, fortrinsvis før og efter hver vandudskiftning og mindst én gang om ugen. pH skal måles ved begyndelsen og afslutningen af hver vandudskiftning ved statiske udskiftningstest og mindst én gang ugentligt ved gennemstrømningstest. Hårdhedsgrad og alkalinitet skal måles én gang i hver test. Temperaturen skal fortrinsvis overvåges kontinuerligt i mindst én testbeholder.
1.8.6. Observationer
Vægt: Ved testens afslutning skal samtlige overlevende fisk vejes som vådvægt (duppes tørre), enten gruppevis pr. testbeholder eller enkeltvis. Vejning af fisk pr. testbeholder foretrækkes frem for individuel vejning, som kræver, at hver enkelt fisk skal mærkes. Hvis fiskene skal vejes enkeltvis med henblik på bestemmelse af enkeltfisk-specifik tilvækst, skal man vælge en mærkningsmåde, som undgår at stresse fiskene (der er egnede alternativer til frysemærkning, f.eks. anvendelse af tynd farvet fiskesnøre).
Fiskene skal i løbet af testperioden undersøges dagligt for eventuelle ydre abnormiteter (såsom blødninger eller misfarvning), og unormal adfærd registreres. Eventuelle mortaliteter skal registreres, og døde fisk skal fjernes hurtigst muligt. Døde fisk erstattes ikke. idet belastningsgraden og bestandtætheden er tilstrækkelige til. at indvirkninger på væksten som følge af ændringer i fiskeantallet pr. beholder undgås. Fodermængden skal imidlertid justeres.
2. DATA OG RAPPORTERING
2.1. BEHANDLING AF RESULTATERNE
Det anbefales, at en statistiker inddrages i både design og analyse af testen, eftersom denne testmetode tillader betydelige variationer i forsøgets design, som f.eks. antal testbeholdere, antal testkoncentrationer, antal fisk osv. I betragtning af valgmulighederne med hensyn til testens design vil der ikke blive givet nogen vejledning i statistiske procedurer her.
Tilvæksten bør ikke beregnes for testbeholdere, hvor mortaliteten overstiger 10 %. Mortalitetsprocenten skal imidlertid anføres for samtlige testkoncentrationers vedkommende.
Uanset hvilken metode, der anvendes til dataanalysen, er det centrale koncept den specifikke tilvækst r mellem tidspunktet t1 og tidspunktet t2. Denne kan defineres på flere måder afhængigt af, hvorvidt fiskene er mærket individuelt, eller hvorvidt der ønskes et beholdergennemsnit.



hvor:
|
r1 |
= |
enkeltfisk-specifik tilvækst |
|
r2 |
= |
beholdergennemsnitsspecifik tilvækst |
|
r3 |
= |
»pseudo«-specifik tilvækst |
|
w1, w2 |
= |
en bestemt fisks vægt på henholdsvis tidspunkt t1 og t2 |
|
loge w1 |
= |
logaritmen til en enkelt fisks vægt ved testperiodens begyndelse |
|
loge w2 |
= |
logaritmen til en enkelt fisks vægt ved testperiodens afslutning |
|
loge W1 |
= |
gennemsnit af logaritmerne til værdierne w1 for fiskene i beholderen ved testperiodens begyndelse |
|
loge W2 |
= |
gennemsnit af logaritmerne til værdierne w2 for fiskene i beholderen ved testperiodens afslutning |
|
t1, t2 |
= |
tidspunktet (dag) for testperiodens begyndelse og afslutning |
r1, r2, r3 kan udregnes for tidsrummet mellem 0 og 28 dage og, hvor det er aktuelt (dvs. når der er foretaget måling på dag 14), for tidsrummene mellem 0 og 14 samt mellem 14 og 28 dage.
2.1.1. Resultatanalyse ved regression (koncentrationsrespons-model)
Denne analysemetode fører frem til en passende matematisk sammenhæng mellem den specifikke tilvækst og koncentrationen og gør det således mulig: at beregne »ECx«, dvs. en hvilken som helst ønsket EC-værdi. Ved anvendelse af denne metode er udregningen af r for enkeltfisk (r1) ikke nødvendig, og analysen kan i stedet baseres på r-beholdergennemsnitsværdien (r2). Sidstnævnte metode foretrækkes. Den er også mest hensigtsmæssig, hvor den mindste fiskeart bruges.
Den beholdergennemsnitsspecifikke tilvækst (r2) bør plottes grafisk som funktion af koncentrationen, for at forholdet mellem koncentration og respons kan undersøges.
Når forholdet mellem r2 og koncentration skal vises, bør man vælge en passende model, og valget skal begrundes på hensigtsmæssig vis.
Hvis der ikke er lige mange overlevende fisk i de enkelte beholdere, bør modeltilpasningsmetoden vægtes, hvad enten denne er simpel eller nonlineær, for at tage højde for grupper af uens størrelse.
Modeltilpasningsmetoden skal give mulighed for, at en beregning af f.eks. EC20 og dennes fordeling (enten standardfejl eller konfidensinterval) kan udledes. Grafen for den tilpassede model bør vises i forhold til dataene, så modeltilpasningens tilstrækkelighed tydeligt fremgår (8)(18)(19)(20).
2.1.2. Analyse af resultaterne for beregningen af LOEC
Hvis testen har omfattet replikation af samtlige beholdere ved samtlige koncentrationsniveauer, vil beregningen af LOEC kunne baseres på en variansanalyse (ANOVA) af den beholdergennemsnitsspecifikke tilvækst (se 2.1), efterfulgt af en egnet metode (f.eks. Dunnetts eller Williams' test (12)(13)(14){21)) til sammenligning af det gennemsnitlige r for hver enkelt koncentration med det gennemsnitlige r for kontrolgrupperne, for at identificere den laveste koncentration, ved hvilken denne forskel er signifikant på et sandsynlighedsniveau på 0,05. Hvis de nødvendige forudsætninger for parametriske metoder ikke er opfyldt — ikke-normal fordeling (f.eks. Shapiro-Wilks test) eller heterogen varians (Bartletts test) — bør man overveje at transformere dataene for at homogenisere varianserne, inden ANOVA foretages, eller at foretage en vægtet ANOVA.
Hvis testen ikke har omfattet replikation af beholdere ved hver enkelt koncentration, vil en ANOVA, der baseres på beholdere, være ikke-udslagsgivende eller umulig. I denne situation vil det være et acceptabelt kompromis at basere ANOVA på den »pseudo«-specifikke tilvækst r3 for enkeltfisk.
Det gennemsnitlige r3 for hver enkelt testkoncentration kan så sammenlignes med det gennemsnitlige r3 for kontrolgrupperne. Derpå kan LOEC identificeres som tidligere beskrevet. Man må forstå, at denne metode hverken tager hensyn til eller yder beskyttelse mod variabilitet mellem beholdere, ud over hvad der kan tilskrives variabiliteten mellem enkeltfisk. Erfaringerne har imidlertid vist (8), at variabilitet mellem beholdere har været meget lille sammenlignet med variabiliteten inden for de enkelte beholdere (dvs. mellem fisk). Hvis analysen ikke omfatter enkeltfisk, skal det med begrundelse anføres, hvilken metode der er benyttet til identifikation af ekstreme værdier.
2.2. TOLKNING AF RESULTATERNE
Resultaterne skal tolkes med forsigtighed, hvor den målte koncentration af giftstof i testopløsningeme ligger nær analysemetodens detektionsgrænse, eller ved semistatiske test, hvor testkemikaliets koncentration i den nyligt fremstillede opløsning falder inden udskiftningen.
2.3. TESTRAPPORTEN
Testrapporten skal indeholde følgende oplysninger:
2.3.1. Testkemikaliet:
|
— |
fysisk beskaffenhed og relevante fysisk-kemiske egenskaber |
|
— |
kemiske identifikationsdata, herunder renhedsgrad og eventuelt analysemetode til kvantitativ bestemmelse af testkemikaliet. |
2.3.2. Fiskeart(er), der indgår i testen:
|
— |
videnskabeligt navn, og eventuelt |
|
— |
stamme, størrelse, leverandør, evt. forudgående behandling osv. |
2.3.3. Testbetingelser:
|
— |
anvendt testprocedure (f.eks. semistatisk/udskiftning. gennemstrømning, belastning, bestandtæthed osv.) |
|
— |
testdesign (f.eks. antal testbeholdere, testkoncentrationer og replikater, antal fisk pr. beholder) |
|
— |
metode til fremstilling af stamopløsmnger samt udskiftningshyppighed (eventuelt anvendt opløselighedsfremmer og dennes koncentration skal anføres) |
|
— |
de nominelle testkoncentrationer, de målte værdiers gennemsnit og standardafvigelser i testbeholderne, metoden, hvorved de er opnået, samt dokumentation for, at målingerne refererer til testkemikaliets faktiske koncentration i opløsning |
|
— |
fortyndingsvandets karakteristika: pH, hårdhedsgrad, alkalinitet, temperatur, koncentration af opløst ilt, restindhold af chlor (hvis målt), samlet mængde organisk kulstof, opslæmmet tørstof, testmediets saltindhold (hvis målt) og samtlige andre udførte målinger |
|
— |
vandkvaliteten i testbeholderne: pH, hårdhedsgrad, temperatur og koncentration af opløst ilt |
|
— |
detaljerede oplysninger om fodring (f.eks. fodertype(r), -kilde, -mængde og fodringshyppighed). |
2.3.4. Resultater:
|
— |
dokumentation for, at kontrolgrupperne opfylder validitetskritenet for overlevelse, samt data vedrørende mortaliteten ved de enkelte testkoncentrationer |
|
— |
anvendte statistiske analyseteknikker, statistikker baseret på replikater eller fisk, behandling af data og begrundelse for de anvendte teknikker |
|
— |
data i tabelform vedrørende fiskenes individuelle og gennemsnitlige vægt på dag 0, 14 (hvis aktuelt) og 28, værdier for beholdergennemsnits- eller pseudospecifik tilvækst (alt efter hvad der er aktuelt) i tidsrum met fra dag 0 til 28 eller eventuelt 0-14 og 14-28 |
|
— |
resultater af den statistiske analyse (dvs. regressionsanalyse eller ANOVA), fortrinsvis i tabelform og grafisk form, og LOEC (p = 0,05) og NOEC eller ECx, eventuelt med standardfejl, alt efter hvad der er aktuelt |
|
— |
forekomst af eventuelle usædvanlige reaktioner hos fiskene samt eventuelle synlige virkninger frembragt af testkemikaliet. |
3. HENVISNINGER
|
(1) |
Solbe, J.F. de LG (1987). Environmental Effects of Chemicals (CFM 9350 SLD). Report on a UK Ring Test of a Method for Studying the Effects of Chemicals on the Growth rate of Fish. WRc Report No. PRD 1388-M/2. |
|
(2) |
Ashley, S., Mallett, M.J. and Grandy, N.J. (1990). EEC Ring Test of a Method for Determining the Effects of Chemicals on the Growth Rate of Fish. Final Report to the Commission of the European Communities. WRc Report No EEC 2600-M. |
|
(3) |
Crossland, N.O. (1985). A method to evaluate effects of toxic chemicals on fish growth. Chemosphere, 14, pp 1855-1870. |
|
(4) |
Nagel, R., Bresh, H., Caspers, N., Hansen, P.D., Market, M., Munk, R., Scholz, N. and Höfte, B.B. (1991). Effect of 3,4-dichloroaniline on the early life stages of the Zebrafish (Brachydanio rerio): results of a comparative laboratory study. Ecotox. Environ. Safety, 21, pp 157-164. |
|
(5) |
Yamamoto, Tokio. (1975). Series of stock cultures in biological field. Medaka (killifish) biology and strains. Keigaku Publish. Tokio, Japan. |
|
(6) |
Holcombe, G.W., Benoit, D.A., Hammermeister, D.E., Leonard, E.N. and Johnson, R.D. (1995). Acute and long-term effects of nine chemicals on the Japanese medaka (Oryzias latipes).Arch. Environ. Conta. Toxicol. 28, pp 287-297. |
|
(7) |
Benoit, D.A., Holcombe, G.W. and Spehar, R.L. (1991). Guidelines for conducting early life toxicity tests with Japanese medaka (Oryzias latipes). Ecological Research Series EPA-600/3-91-063. U. S. Environmental Protection Agency, Duluth, Minesota. |
|
(8) |
Stephan, C.E. and Rogers, J.W. (1985). Advantages of using regression analysis to calculate results of chronic toxicity tests. Aquatic Toxicology and Hazard Assessment: Eighth Symposium, ASTM STP 891, R.C. Bahner and D.J. Hansen, Eds., American Society for Testing and Materials, Philadelphia, pp 328-338. |
|
(9) |
Environment Canada (1992). Biological test method: toxicity tests using early life stages of salmonid fish (rainbow trout, coho salmon, or atlantic salmon). Conservation and Protection, Ontario, Report EPS 1/RM/28, 81 p. |
|
(10) |
Cox, D.R. (1958). Planning of experiments. Wiley Edt. |
|
(11) |
Pack, S. (1991). Statistical issues concerning the design of tests for determining the effects of chemicals on the growth rate offish. Room Document 4, OECD Ad Hoc Meeting of Experts on Aquatic Toxicology, WRc Medmenham, UK, 10-12 December 1991. |
|
(12) |
Dunnett, C.W. (1955). A Multiple Comparisons Procedure for Comparing Several Treatments with a Control, J. Amer. Statist. Assoc., 50, pp 1096-1121. |
|
(13) |
Dunnett, C.W. (1964). New tables for multiple comparisons with a control. Biometrics, 20, pp 482-491. |
|
(14) |
Williams, D.A. (1971). A test for differences between treatment means when several dose levels are compared with a zero dose control. Biometrics 27, pp 103-117. |
|
(15) |
Johnston, W.L., Atkinson, J.L., Glanville, N.T. (1994). A technique using sequential feedings of different coloured food to determine food intake by individual rainbow trout, Oncorhynchus mykiss: effect of feeding level. Aquaculture 120, 123-133. |
|
(16) |
Quinton, J. C. and Blake, R.W. (1990). The effect of feed cycling and ration level on the compensatory growth response in rainbow trout, Oncorhynchus mykiss. Journal of Fish Biology, 37, 33-41. |
|
(17) |
Post, G. (1987). Nutrition and Nutritional Diseases of Fish. Chapter IX in Testbook of Fish Health. T.F.H. Publications, Inc. Neptune City, New Jersey, USA. 288 P. |
|
(18) |
Bruce, R.D. and Versteeg, D.J. (1992). A statistical procedure for modelling continuous toxicity data. Environ. Toxicol. Chem. 11, 1485-1494. |
|
(19) |
DeGraeve, G.M., Cooney, J.M., Pollock, T.L., Reichenbach, J.H., Dean, Marcus, M.D. and McIntyre, D.O. (1989). Precision of EPA seven-day fathead minnow larval survival and growth test; intra and interlaboratory study. report EA-6189 (American Petroleum Institute Publication, n. 4468). Electric Power Research Institute, Palo alto, CA. |
|
(20) |
Norbert-King, T.J. (1988). An interpolation estimate for chronic toxicity: the ICp approach. US Environmental Protection Agency. Environmental Research Lab., Duluth, Minesota. Tech. Rep. No 05-88 of National Effluent Toxicity Assesment Center. Sept. 1988. 12 pp. |
|
(21) |
Williams, D.A. (1972). The comparison of several dose levels with a zero dose control. Biometrics 28, pp 510-531. |
Tillæg 1
FISKEARTER ANBEFALET TIL TESTNING, OG EGNEDE TESTBETINGELSER
|
Arter |
Anbefalet område for testtemperatur ( oC) |
Lysperiode (timer) |
Anbefalet område for fiskenes startvægt (g) |
Påkrævet målingsnøjagtighed |
Belastningsgrad (g/l) |
Bestandtæthed {pr. liter) |
Foder |
Testens varighed (dage) |
|
Anbefalet art: |
|
|
|
|
|
|
|
|
|
Oncorhynchuss mykiss Regnbueørred |
12,5-16,0 |
12-16 |
1-5 |
Til nærmeste 100 mg |
1,2-2,0 |
4 |
Mærkevaretørfoder til laksefiskeyngel |
≥ 28 |
|
Andre veldokumenterede arter: |
|
|
|
|
|
|
|
|
|
Danio rerio Zebrafisk |
21-25 |
12-16 |
0,050-0,100 |
Til nærmeste 1 mg |
0,2-1,0 |
5-10 |
Levende foder (Brachionus Artemia) |
≥ 28 |
|
Oryzias latipes Riskarpe (Medaka) |
21-25 |
12-16 |
0,050-0,100 |
Til nærmeste 1 mg |
0,2-1,0 |
5-20 |
Levende foder (Brachionus Artemia) |
≥ 28 |
Tillæg 2
NOGLE KEMISKE KARAKTERISTIKA FOR ACCEPTABELT FORTYNDINGSVAND
|
Stof |
Koncentrationer |
|
Partikler |
< 20 mg/l |
|
Total organisk kulstof |
< 2 mg/l |
|
Ammoniak |
< 1 μg/l |
|
Restchlor |
< 10 μg/l |
|
Total phosphorholdige organiske pesticider |
< 50 ng/l |
|
Total chlorholdige organiske pesticider plus polychlorerede biphenyler |
< 50 ng/1 |
|
Total organisk chlor |
< 25 ng/1 |
Tillæg 3
Logaritmiske rækker af koncentrationer, der er egnede til toksicitetstest (9)
|
Søjle (antal koncentrationer mellem 100 og 10, eller mellem 10 og 1) (20) |
||||||
|
1 |
2 |
3 |
4 |
5 |
6 |
7 |
|
100 |
100 |
100 |
100 |
100 |
100 |
100 |
|
32 |
46 |
56 |
63 |
68 |
72 |
75 |
|
10 |
22 |
32 |
40 |
46 |
52 |
56 |
|
3,2 |
10 |
18 |
25 |
32 |
37 |
42 |
|
1,0 |
4,6 |
10 |
16 |
22 |
27 |
32 |
|
|
2,2 |
5,6 |
10 |
15 |
19 |
24 |
|
|
1,0 |
3,2 |
6,3 |
10 |
14 |
18 |
|
|
|
1,8 |
4,0 |
6,8 |
10 |
13 |
|
|
|
1,0 |
2,5 |
4,6 |
7,2 |
10 |
|
|
|
|
1,6 |
3,2 |
5,2 |
7,5 |
|
|
|
|
1,0 |
2,2 |
3,7 |
5,6 |
|
|
|
|
|
1,5 |
2,7 |
4,2 |
|
|
|
|
|
1,0 |
1,9 |
3,2 |
|
|
|
|
|
|
1,4 |
2,4 |
|
|
|
|
|
|
1,0 |
1,8 |
|
|
|
|
|
|
|
1,3 |
|
|
|
|
|
|
|
1,0 |
C.15. FISK, KORTTIDSTOKSICITETSTEST UDFØRT PÅ FISKEEMBRYONER OG BLOMMESÆKYNGEL
1. METODE
Denne korttids toksicitetstestmetode svarer til OECD TG 212 (1998).
1.1. INDLEDNING
Denne korttidstoksicitetstest udført på fiskeembryoner og blommesækyngel er en korttidstest, i hvilken det nybefrugtede æg er udsat for giftstof indtil slutningen af blommesækstadiet. Der skal ikke fodres i denne test, og testen skal således afsluttes, medens yngelen stadig får sin næring fra blommesækken.
Testens formål er at finde letale og til en vis grad subletale virkninger af kemikalier på specielle stadier og arter. Denne test kunne skaffe nyttig viden, idet den kunne a) danne en bro mellem letale og subletale test, b) blive anvendt som en screeningtest enten for en test på alle tidlige livsstadier eller for en test for kronisk toksicitet og c) bruges til at teste arter, hvor dyrkningsteknikkerne ikke er tilstrækkeligt gode til at dække overgangsperioden mellem endogen og eksogen fodring.
Man bør erindre sig, at kun test, der omfatter alle stadier i fisks livscyklus, kan påregnes at give en præcis vurdering af kemikaliers kroniske toksicitet over for fisk, og at enhver begrænsning i påvirkning inden for livsstadierne kan reducere følsomheden og således undervurdere den kroniske toksicitet. Man må derfor forvente, at testen på fiske-embryoner og blommesækyngel har mindre følsomhed end en test, der omfatter alle tidlige livsstadier, specielt med hensyn til meget lipofile kemikalier (log Pow > 4) og til kemikalier med en særlig type toksisk virkning. Imidlertid må man forvente mindre forskelle i følsomhed mellem de to test med hensyn til kemikalier, der har en ikke-specifik, narkotisk virkningsmåde (1).
Forud for offentliggørelsen af denne test udført på fiskeembryoner og blommesækyngel havde man størst erfaring med anvendelsen af ferskvandsfisken Danio rerio Hamilton-Buchanan (Teleostei, Cyprinidae — almindeligt navn: zebrafisk). Mere detaljerede retningslinjer for resultater med denne art er derfor anført i tillæg 1. Dette udelukker ikke brugen af andre arter, for hvilke der ligeledes foreligger erfaringer (tabel 1A og 1B).
1.2. DEFINITIONER
Laveste koncentration med statistisk sikkert observeret effekt (LOEC) er den laveste kemikaliekoncentration, der i testen har vist sig at resultere i en statistisk signifikant virkning (p < 0,05), sammenlignet med kontrollen. Alle testkoncentrationer over LOEC skal forårsage skadelige virkninger på samme eller højere niveau end observeret ved LOEC.
Koncentration uden statistisk sikkert observeret effekt (NOEC) er testkoncentrationen lige under LOEC.
1.3. TESTPRINCIPPET
Fiskeembryoner og blommesækyngel udsættes for en række koncentrationer af testkemikaliet opløst i vand. Protokollen tillader valg mellem en semistatisk og en gennemstrømningsprocedure. Valget afhænger af testkemikaliets natur. Testen begynder med, at man anbringer befrugtede æg i forsøgsbeholderne, og den slutter, lige før blommesækken er blevet fuldstændig resorberet af en hvilken som helst af fiskelarverne i en hvilken som helst forsøgsbeholder, eller før dødsfald indtræffer i kontrolbeholderne på grund af sult. Letale og subletale virkninger måles og sammenlignes med kontrolværdierne for at fastslå den laveste koncentration med statistisk observeret effekt og ud derfra den koncentration, der ikke giver nogen statistisk sikker virkning. Alternativt kan man analysere resultaterne ved hjælp af en regressionsmodel for at finde frem til den koncentration, der ville give en bestemt procentisk effekt (dvs. LC/ECx, hvor x er en defineret procentisk virkning).
1.4. KENDSKAB TIL TESTKEMIKALIET
Resultater fra en akut toksicitetstest (se metode C.1), fortrinsvis udført på arten valgt til denne test. bør foreligge. De kan være nyttige med henblik på valg af passende område af testkoncentrationer i testen udført på tidlige livsstadier. Vandopløselighed, (inklusive opløseligheden i testvandet) og testkemikaliets damptryk bør kendes. En pålidelig analyse til kvantificering af kemikaliet i testopløsningerne med kendt og publiceret »accuracy« og nederste målegrænse bør foreligge.
Informationer om testkemikaliet, nyttige for testbetingelserne, inkluderer strukturformel, renhedsgrad, lysstabilitet, stabilitet under testens betingelser, pKa, Pow og resultater fra en test for spontan biologisk nedbrydelighed (se metode C.4).
1.5. TESTENS VALIDITET
En tests validitet forudsætter:
|
— |
den generelle overlevelse af befrugtede æg i kontrollerne, og, hvor relevant, i kar med rent opløsningsmiddel skal være større end eller lig med grænserne definerede i tillæg 2 og 3 |
|
— |
koncentrationen af opløst ilt skal være mellem 60 og 100 % af værdien for mætning med luft (ASV) i hele testperioden |
|
— |
vandtemperaturen må ikke variere mere end ±1,5 oC imellem forsøgsbeholderne eller fra dag til dag på noget tidspunkt i testen, og den skal ligge i det temperaturområde, som gælder for den anvendte art. (tillæg 2 og 3). |
1.6. BESKRIVELSE AF TESTMETODEN
1.6.1. Forsøgsbeholdeme
Enhver type glas eller andet inaktivt materiale kan anvendes. Forsøgsbeholdernes dimensioner skal være store nok til at klare kravene til belastningsraten (se 1.7.1.2) Det anbefales at anbringe forsøgsbeholdeme randomiseret. Et randomiseret design med grupper af forsøgsbeholdere, hvor samme behandling forefindes i hver gruppe, bør foretrækkes frem for et helt randomiseret system, når der er systematiske virkninger i laboratoriet, som kan kontrolleres ved gruppering. I den påfølgende dataanalyse bør der tages hensyn til en eventuel gruppering. Forsøgsbeholderne bør skånes for unødvendige forstyrrelser.
1.6.2. Valg af fiskeart
Anbefalede fiskearter ses i tabel 1A. Dette udelukker ikke anvendelsen af andre arter (eksempler ses i tabel 1B), men testproceduren må muligvis ændres for at opnå passende testbetingelser. Brug af andre arter og undersøgelsesmetode skal i så fald begrundes.
1.6.3. Opbevaring af fisk til yngel
Detaljer vedrørende pasning af fisk til yngel under tilfredsstillende omstændigheder kan findes i OECD TG 210 (21) og i henvisning (2)(3)(4)(5)(6).
1.6.4. Håndtering af embryoner og fiskelarver
Embryoner og fiskelarver kan, mens de udsættes for testkemikaliet, opholde sig i mindre kar med netvægge eller i rør med net for enderne med disse mindre kar placerede i forsøgskarret, således at testopløsningen kan flyde igennem. Man kan bevirke et ikke-turbulent flow gennem de mindre kar ved at lade dem hænge ned fra en arm, som bevæger karrene op og ned, dog altid med organismerne under overfladen; man kan også anvende et system med gennemskylning ved hjælp af overtryk. Befrugtede lakseæg kan anbringes på stativer eller net med masker tilstrækkeligt store til at lade larverne falde igennem efter udklækning. Pasteurpipetter er anvendelige til at flytte embryoner og larver i de semistatiske test i forbindelse med den daglige totale udskiftning af testopløsningen (se 1.6.6).
I de tilfælde, hvor man har anvendt beholdere, riste eller netværk til at holde æg inde i forøgskarret, skal disse fjernes, når æggene klækkes (1), idet man dog beholder det netværk, som hindrer fiskene i at forsvinde. Hvis det er nødvendigt at flytte fiskelarver, må de ikke udsættes for luft, og net bør ikke bruges til at fjerne fisk fra beholdere (fra en sådan advarsel kan nok undtages mindre sarte arter, som f.eks. karpen). Tidspunktet for en sådan overførsel varierer med arten og behøves ikke i alle tilfælde. Ved brug af den semistatiske teknik kan man anvende bægerglas eller lave beholdere, eventuelt, hvis nødvendigt, forsynet med et netværk løftet en smule op over bægerglassets bund. Hvis beholderne er store nok til at opfylde kravene til belastningsraten (se 1.7.1.2) kan overflytning af embryoner eller larver eventuelt undgås.
1.6.5. Vand
Til brug i testen kan anvendes vand, der er i overensstemmelse med de kemiske karakteristika for acceptabelt fortyndingsvand, som beskrevet i tillæg 4 og i hvilket fiskearten i kontrolbeholderne har en overlevelse mindst lige så god som beskrevet i tillæg 2 og 3. Kvaliteten må ikke ændre sig i løbet af testperioden. pH skal holde sig inden for ±0,5 pH-enheder. For at sikre, at vand til fortynding ikke får urimelig indflydelse på testresultaterne (f.eks. ved at danne kompleks med testkemikaliet) eller ved at indvirke uheldigt på de præstationer, man forventer af fisk til yngel, bør man regelmæssigt udtage prøver til analyse. Målinger af tungmetaller (f.eks. Cu, Pb, Zn, Hg, Cd og Ni), væsentlige kationer og anioner (f.eks. Ca, Mg, Na, K, Cl og So4), pesticider (f.eks. totale organofosfor- og totale organochlorinopesticider), totalt organisk kulstof og opslæmmede faste stoffer bør f.eks. foretages hver tredje måned, hvis man ved, at fortyndingsvandet har en relativt konstant kvalitet. Hvis vandkvaliteten påviseligt har været konstant over mindst et år, kan analyserne foretages mindre hyppigt og med større intervaller (f.eks. hver sjette måned).
1.6.6. Testopløsning
Testopløsninger med den valgte koncentration fremstilles ved fortynding af en stamopløsning.
Stamopløsningen bør fortrinsvis fremstilles ved opblanding eller oprystning af testkemikaliet i opløsningsvandet ad mekanisk vej (f.eks. omrøring og ultralydbehandling). Saturation colums (solubility columns) kan anvendes til fremstilling af en passende koncentreret stamopløsning. Videst muligt bør brugen af opløsningsmidler eller dispergeringsmidler undgås; imidlertid kan den type stoffer være nødvendige til fremstilling af en passende koncentreret stamopløsning. Som eksempler på anvendelige opløsningsmidler kan nævnes acetone, ethanol, methanol, dimethylformamid og triethyleneglycol. Som eksempel på en anvendelige dispergenser kan nævnes Cremophor RH40, Tween 80, methylcellulose 0,01 % og HCO-40. Stor omhu bør udvises, når man anvender stoffer, der er let biologisk nedbrydelige (f.eks. acetone) og/eller let fordampelige, da de kan medføre problemer med bakterielle belægninger i gennemstrømningstestene, Når et opløsningsmiddel anvendes, må det ikke have nogen signifikant virkning på overlevelsen eller synlig uheldig virkning på de tidlige livsstadier, som det kan kontrolleres i en test med kun opløsningsmiddel. Alle anstrengelser bør imidlertid gøres for at undgå brugen af sådanne stoffer.
I den semistatiske teknik kan man følge to forskellige procedurer ved udskiftning af testopløsningen; enten i) fremstilles nye testopløsninger i rene forsøgsbeholdere og overlevende æg og larver flyttes forsigtigt inde i små volumina af den gamle opløsning til det nye forsøgsbeholdere, idet man undgår at udsætte dem for luft, eller ii) testorganismerne bibeholdes i forsøgsbeholderne, medens mindst tre fjerdedele af testvandet udskiftes. Hvor hyppigt det skal gøres, afhænger af testkemikaliets stabilitet, men en daglig udskiftning anbefales. Hvis tidligere stabilitetsforsøg (se 1.4) har vist, at testkemikaliets koncentration ikke er stabil (dvs. falder uden for 80-120 % af de nominelle grænser eller under 80 % af den målte begyndelseskoncentration) i løbet af udskiftningsperioden, bør man overveje at anvende en test med gennemstrømning. Under alle omstændigheder skal man være omhyggelig med ikke at stresse larverne, når vandet fornyes.
Til gennemstrømningstestene bruges et system, som kontinuerligt afmåler og fortynder en stamopløsning med test-kemikaliet (f.eks. peristaltpumper, proportional diluter, saturator system) for at yde en række forskellige koncentrationer til forsøgsbeholderne. Med mellemrum, fortrinsvis dagligt, kontrolleres flow-hastigheden fra stamopløsningen og vandfortyndingen, og de bør ikke variere mere end 10 % i testforløbet. En flow-hastighed svarende til volumenet af mindst fem målekamre pr. døgn er fundet passende (2).
1.7. FREMGANGSMÅDE
Nyttig viden om, hvad testen med fiskeembryoner og blommesækyngel kan præstere, kan findes i litteraturen, og eksempler herpå kan findes i henvisning (7)(8)(9).
1.7.1. Omstændigheder ved belastningen
1.7.1.1. Varighed
Helst skal testen begynde mindre end 30 minutter efter befrugtningen. Embryonerne sænkes ned i testopløsningen før eller så snart som muligt efter at blastocysten har delt sig. og i hvert fald for begyndelsen af gastrula-stadiet. Med æg, der leveres udefra, kar. man ikke altid begynde testen umiddelbart efter befrugtningen. Da testens følsomhed kan blive alvorligt påvirket af en forsinket teststart, bør testen påbegyndes senest otte timer efter befrugtningen. Da fiskelarverne ikke fodres i testperioden, afsluttes testen, lige før blommesækken er resorberet fuldstændigt i en hvilken som helst larve i en hvilken som helst forsøgsbeholder, og før sultedøden indtræffer i kontrollerne. Varigheden afhænger af den anvendte art. Nogle anbefalede tider ses i tillæg 2 og 3.
1.7.1.2. Belastning
Antallet af befrugtede æg ved testens begyndelse skal kunne opfylde de statistiske krav. Æggene skal fordeles tilfældigt mellem de forskellige behandlinger og mindst 30 befrugtede æg, ligeligt fordelt, til mindst tre replikate testbeholdere pr. koncentration af testkemikalium (så vidt gørligt, det kan være vanskeligt med nogle arter at opnå lige store mængder). Belastningsraten (biomasse per volumen af testopløsningen) bør være så lav, så den opløste iltkoncentration på mindst 60 % ASV kan vedligeholdes uden beluftning. Til gennemstrømningstest anbefales en belastningsrate på ikke over 0,5 g/l per døgn og ikke over 5 g/l opløsning på noget tidspunkt (2).
1.7.1.3. Lys og temperatur
Perioderne med lys og testvandets temperatur skal passe til den valgte art (tillæg 2 og 3). For at holde temperaturen konstant kan det være nødvendigt med endnu et forsøgskar.
1.7.2. Testkoncentrationer
Normalt kræves fem koncentrationer af testkemikaliet med en afstandsfaktor, som ikke overstiger 3,2. Kurven fra de akutte toksicitetstest, som knytter LC50 til testperioden, bør tages i betragtning, når man vælger området for testkoncentrationerne. Brug af færre end fem koncentrationer, for eksempel i en grænsetest, og et snævrere interval mellem koncentrationerne kan være rimeligt under nogle omstændigheder. Begrundelse skal anføres, hvis færre end fem koncentrationer anvendes. Det er ikke nødvendigt at teste koncentrationer højere end svarende til 96 timer LC50 eller 100 mg/l, afhængigt af hvilken af disse, der er lavest. Kemikalier bør ikke testes i koncentrationer, der overstiger deres opløselighed i testvandet.
Når der anvendes et opløsningsmiddel som hjælp ved fremstillingen af testopløsninger (se 1.6,6), bør dets endelige koncentration i forsøgsbeholderen ikke overstige 0,1 ml/l og den skal være den samme i alle forsøgsbeholdere.
1.7.3. Kontroller
Sammen med testserien skal udføres en kontrol af fortyndingsvandet (replikate bestemmelser, hvis nødvendigt) og ligeledes, hvis relevant, en kontrol, som indeholder opløsningsmiddel (replikate bestemmelser, hvis nødvendigt).
1.7.4. Hyppighed af analyser og målinger
I testperioden måles koncentrationen af testkemikaliet med regelmæssige mellemrum.
I semistatiske test, hvor koncentrationen af testkemikaliet forventes at holdes inden for 20 % af det nominelle (dvs. inden for 80-120 %; se 1.4 og 1.6.6), anbefales det, som et minimum, at den højeste og laveste testkoncentration måles, når den er frisk fremstillet og umiddelbart før udskiftning ved mindst tre lejligheder jævnt fordelt over test-perioden (dvs. analyserne skal udføres på en prøve fra den samme opløsning, når den er frisk fremstillet og ved dens anvendelse ved udskiftning).
I test, hvor koncentrationen af testkemikaliet ikke forventes at holde sig inden for 20 % af den nominelle (baseret på kemikaliets stabilitetsdata), er det nødvendig; at analysere alle testkoncentrationer, når de er frisk fremstillede og ved udskiftning, efter samme system (dvs. ved mindst tre lejligheder jævnt fordelt over testperioden). Målingen af koncentrationen af testkemikaliet før udskiftning er kun nødvendigt på en af beholderne i replikate bestemmelser for hver testkoncentration. Målingerne skal foretages mindst hver uge. Det anbefales, at resultaterne baseres på målte koncentrationer. Hvis det imidlertid kan vises, at testkemikaliets koncentration testen igennem i tilstrækkelig grad har været holdt inden for ± 20 % af den nominelle eller målte begyndelseskoncentration, kan resultaterne baseres på nominelle eller målte begyndelsesværdier.
Hvad angår gennemstrømningstest er det passende med et prøvetagningsregime som for de semistatiske test (men måling af udskiftede opløsninger er ikke mulig i disse tilfælde). Hvis imidlertid testen varer mere end en uge. er det tilrådeligt at øge antallet af prøvetagninger i den første uge (f.eks. til tre) for at sikre, at test-koncentrationcme holder sig stabile.
Det kan være nødvendigt at centrifugere eller filtrere (f.eks. med et filter med 0,45 μm porestørrelse). Da imidlertid hverken centrifugering eller filtrering altid synes at skille den biotilgængelige fra den ikke-biotilgængelige del af testkemikaliet, behøver prøver ikke at gennemgå disse behandlinger.
1 løbet af testen skal opløst ilt, pH og temperatur måles i alle testbeholderne. Hårdhedsgrad og saltindhold (hvis relevant) bør måles i kontrollerne og i en beholder med den højeste koncentration. Som et minimum skal opløst ilt og saltindhold (hvis relevant) måles tre gange (i begyndelsen, midt i og ved afslutningen af testen). I de semistatiske test anbefales det at måle ilt hyppigere, fortrinsvis før og efter hver vandudskiftning og mindst en gang om ugen. pH skal måles før og efter vandudskiftning i de semistatiske test og mindst en gang ugentlig i gennemstrømningstestene. I hver test skal hårdhedsgraden måles en gang. Temperaturen skal måles daglig og helst være monitoreret kontinuerligt i mindst en test-beholder.
1.7.5. Observationer
1.7.5.1. Fosterets udviklingstrin
Fosterstadiet (dvs. gastrula-stadiet) bør fastslås så sikkert som muligt ved testens begyndelse, hvor fosteret udsættes for testkemikaliet. Dette kan gøres ved undersøgelse af en repræsentativ samling æg, som på passende vis præpareres og godkendes. I litteraturen kan man finde beskrivelser og illustrationer af fosterstadier (2)(5)(10)(11).
1.7.5.2. Udklækning og overlevelse
Udklækning og overlevelse observeres mindst en gang om dagen og noteres. I begyndelsen af testen kan det være ønskeligt at foretage hyppigere observationer (f.eks. hver halve time i løbet af de første tre timer), da overlevelsestid i nogle tilfælde kan være mere relevant end antallet af dødsfald alene (f.eks. når der er akutte toksiske virkninger). Døde embryoner og larver skal fjernes med det samme, eftersom de kan henfalde hurtigt. Meget stor omhu skal udvises, når døde individer fjernes, så nærtliggende æg eller larver ikke bliver ramt eller fysisk beskadigede, da de er meget sarte og følsomme. Kriterier for død varierer efter stadium:
|
— |
for æg: særligt i de tidlige stadier et tydeligt tab af gennemsigtighed og ændring i farve forårsaget af koagulation og/eller bundfældelse af proteiner, hvilket medfører en hvid uklarhed |
|
— |
for embryoner: manglende bevægelighed og/eller hjerteslag og/eller uklar misfarvning i arter, hvor embryonerne almindeligvis er gennemskinnelige |
|
— |
for larver: manglende bevægelighed og/eller manglende respirationsbevægelser og/eller manglende hjerteslag og/eller hvid uklar farvning af centralnervesystemet og/eller manglende reaktion på mekaniske stimuli. |
1.7.5.3. Abnormt udseende
Antallet af larver, der udviser abnorm kropsform og/eller pigmentering, og tidspunkt for resorption af blommesækken skal noteres med passende mellemrum afhængigt af testens varighed og den beskrevne abnormitet. Det skal bemærkes, at abnorme embryoner og larver optræder almindeligt og kan opgøres til adskillige procent i kontrollerne hos visse arter. Abnorme dyr skal kun fjernes fra beholderne, når de er døde.
1.7.5.4. Abnorm opførsel
Abnorm opførsel, f.eks. hyperventilation, ukoordineret svømning og atypisk ubevægelighed, skal noteres med passende intervaller afhængigt af testens varighed. Disse virkninger kan, når de opdages, selvom de er vanskelige at kvantitere, hjælpe med til at tolke mortalitetsdata, dvs. skaffe viden om, hvorledes kemikaliets toksiske virkning udspiller sig.
1.7.5.5. Længde
Ved testens afslutning anbefales måling af individernes forskellige længde: Standard, fork eller totallængden kan anvendes. Hvis imidlertid der forekommer finneråd (svamp) ved halefinnen eller erosion af finner, anvendes standardlængden. Almindeligvis vil i en vellykket testvariationskoefficient for længden i replikate bestemmelser ligge på ≤ 20 %.
1.7.5.6. Vægt
Ved testens afslutning kan foretages vejning af dyrene: Tørvægt (24 timer ved 60 oC) bør foretrækkes frem for vådvægt (aftørret). Almindeligvis vil i en vellykket test variationskoefficienten på vægten i replikate bestemmelser ligge på ≤ 20 %.
Disse observationer vil medføre, at nogle eller alle nedenstående data kan gøres til genstand for statistisk analyse:
|
— |
kumulativ mortalitet |
|
— |
antal sunde larver ved afslutning af testen |
|
— |
begyndelses- og sluttidspunkt for klækning (dvs. 90 % klækning i hvert replikat) |
|
— |
det daglige antal larver, der klækkes |
|
— |
længde (og vægt) af overlevende dyr ved testens afslutning |
|
— |
antal larver, der er deforme eller har abnormt udseende |
|
— |
antal larver, der opfører sig abnormt. |
2. DATA OG RAPPORTERING
2.1. BEHANDLING AF RESULTATER
Det anbefales, at en statistiker involveres både i design og i analyse af testen, eftersom metoden tillader betragtelige variationer i tilrettelæggelse af eksperimentet, som f.eks. antallet af forsøgsbeholdere, antal testkoncentrationer, begyndelsesantal af befrugtede æg og de målte parametre. I betragtning af valgmulighederne i testdesign, vil her ikke blive vejledt i statistiske procedurer.
Hvis LOEC/NOECs skal beregnes, er det, for at beregne variationen inden for hvert sæt af replikater, nødvendigt at bruge variansanalyse (ANOVA) eller at anvende kontingenstabeller. Dunnets metode kan være nyttig til at foretage multiple sammenligninger mellem resultater fra individuelle koncentrationer og kontrollerne (12)(13). Andre nyttige eksempler kan også findes (14)(15). Størrelsen af den målbare effekt ved anvendelse af ANOVA eller andre procedurer (dvs. styrken af testen) bør udregnes og angives. Det bør bemærkes, at ikke alle observationer listet i 1.7.5.6 kan analyseres ved hjælp af ANOVA. F.eks. bør kumuleret død og antallet af sunde larver analyseres ved hjælp af probitmetoder.
Hvis LC/ECx'er skal beregnes, bør (en) passende kurve(r), f.eks. den logistiske kurve, fittes til de ønskede data ved hjælp af en statistisk metode som f.eks. mindste kvadraters metode eller ikke-lineære mindste kvadraters metode. Kurverne bør parametriseres, så den ønskede LC/ECx og dens standard error kan udregnes direkte. Dette vil gøre beregningen af sikkerhedsgrænserne for LC/ECx meget nemmere. Medmindre der er gode grunde til at foretrække andre sikkerhedsgrænser, bør tosidede 95 %-sikkerhedsgrænser angives. Fitningsmetoden skulle helst give mulighed for beregning af en signifikans for mangel på fit. Grafiske metoder for fitning af kurver kan anvendes. Regressionsanalyse kan anvendes på alle observationer listet i 1.7.5.6.
2.2. TOLKNING AF RESULTATER
Hvor den målte koncentration af giftstoffet i testopløsningen ligger nær analysemetodens detektionsgrænse, skal resultaterne tolkes med forsigtighed. Tolkning af resultater, hvor koncentrationerne ligger over stoffets vandopløselighed. skal også gøres med forsigtighed.
2.3. TESTRAPPORTEN
Testrapporten bør indeholde følgende information:
2.3.1. Test-kemikaliet:
|
— |
dets fysiske natur og relevante fysisk-kemiske egenskaber |
|
— |
kemiske identitets-data, heri renhedsgrad og, hvor nødvendigt, den analytiske metode til kvantificering af stoffet. |
2.3.2. Arten, anvendt i testen:
|
— |
dens videnskabelige navn, herkomst, antal forældrefisk (dvs. hvor mange hundyr blev brugt for at fremskaffe det ønskede antal æg til testen), kilde og metode til indsamling af befrugtede æg og behandlingen af dem. |
2.3.3. Forsøgsbetingelser:
|
— |
anvendt fremgangsmåde (f.eks.. semistatisk eller gennemstrømsforsøg, tidsforløb fra befrugtning til begyndelsen af testen, belastning osv.) |
|
— |
lysperiode(r) |
|
— |
testdesign (f.eks. antal forsøgsbeholdere og replikater, antal embryoner pr. replikat |
|
— |
fremstillingsmetode til stamopløsning og hyppighed af udskiftning (hvis der er anvendt opløsningsmiddel, skal dets koncentration anføres) |
|
— |
den nominelle testkoncentration, de målte værdier, gennemsnit og standarddeviationer for forsøgskarrene og metoden, hvorved de er opnået, og, hvis testkemikaliet er opløseligt i vand i koncentrationer, der ligger under de undersøgte, skal der skaffes bevis for, at målingerne refererer til testkemikaliet i opløsning |
|
— |
karakteristik af fortyndningsvandet: pH, hårdhedsgrad, temperatur, koncentration af opløst ilt, niveau for residual-chlor (hvis målt), total organisk kulstof, opslæmmede substanser, saltindhold i testmediet (hvis målt) og enhver anden ting, som er målt |
|
— |
vandkvalitet i forsøgskarrene: pH. hårdhedsgrad, temperatur og koncentration af opløst ilt. |
2.3.4. Resultater:
|
— |
resultater fra ethvert præliminært forsøg med testkemikaliets stabilitet |
|
— |
bevis på, at testdyrene gennemsnitligt har overlevet i kontrollerne svarende til den accepterede standard (tillæg 2 og 3) |
|
— |
data vedrørende mortalitet/overlevelse på embryon- og larvestadierne og den totale mortalitet/overlevelse |
|
— |
antal dage til klækningen og antal klækninger |
|
— |
data vedrørende længde (og vægt) |
|
— |
morfologiske abnormiteter beskrives, hvis de forekommer |
|
— |
abnorm opførsel beskrives, hvis den forekommer |
|
— |
statistisk analyse og behandling af data |
|
— |
vedrørende test, hvor ANOVA er anvendt, anføres laveste observerede effekt-koncentration (LOEC) ved p = 0,05 og den koncentration, der var uden effekt (NOEC) ved hver måling, inklusive de statistiske procedurer og en angivelse af, hvilken størrelse effekt der kunne påvises |
|
— |
i forbindelse med test, hvor regressionsteknik er anvendt, anføres LC/ECx og sikkerhedsgrænser og en graf med den fitningsmetode, der blev brugt til beregningen |
|
— |
redegørelse for enhver afvigelse fra denne testmetode. |
3. HENVISNINGER
|
(1) |
Kristensen P, (1990). Evaluation of the Sensitivity of Short Term Fish Early Life Stage Tests in Relation to other FELS Test Methods. Final report to the Commission of the European Communities, 60 pp. June 1990. |
|
(2) |
ASTM (1988). Standard Guide for Conducting Early Life-Stage Toxicity Tests with Fishes. American Society for Testing and Materials. E 1241-88. 26 pp. |
|
(3) |
Brauhn, J.L. and Schoettger, R.A. (1975). Acquisition and Culture of Research Fish: Rainbow trout, Fathead minnows, Channel Catfish and Bluegills. p. 54, Ecological Research Series, EPA-660/3-75-011, Duluth, Minnesota. |
|
(4) |
Brungs, W.A. and Jones, B.R. (1977). Temperature Criteria for Freshwater Fish: Protocol and Procedures, p. 128, Ecological Research Series EPA-600/3-77-061, Duluth, Minnesota. |
|
(5) |
Laale, H.W. (1977). The Biology and Use of the Zebrafish (Brachydanio rerio) in Fisheries Research. A Literature Review. J. Biol. 10, pp. 121-173. |
|
(6) |
Legault, R. (1958). A Technique for Controlling the Time of Daily Spawning and Collecting Eggs of the Zebrafish, Bmchydanio rerio (Hamilton-Buchanan) Copeia, 4, pp. 328-330. |
|
(7) |
Dave, G., Damgaard, B., Grande, M., Martelin, J.E., Rosander, B. and Viktor, T. (1987). Ring Test of an Embryo-larval Toxicity Test with Zebrafish (Brachydanio rerio) Using Chromium and Zinc as Toxicants. Environmental Toxicology and Chemistry, 6, pp. 61-71. |
|
(8) |
Birge, J.W., Black, J.A. and Westerman, A.G. (1985). Short-term Fish and Amphibian Embryo-larval Tests for Determining the Effects of Toxicant Stress on Early Life Stages and Estimating Chronic Values for Single Compounds and Complex Effluents. Environmental Toxicology and Chemistry 4, pp. 807-821. |
|
(9) |
Van Leeuwen, C.J., Espeldoom, A. and Mol, F, (1986). Aquatic Toxicological Aspects of Dithiocarbamates and Related Compounds. III. Embryolarval Studies with Rainbow Trout {Salmo gairdneri), J. Aquatic Toxicology, 9, pp. 129-145. |
|
(10) |
Kirchen, R.V. and W.R. West (1969). Teleostean Development. Carolina Tips 32(4): 1-4. Carolina Biological Supply Company. |
|
(11) |
Kirchen, R.V. and W.R. West (1976). The Japanese Medaka. Its care and Development. Carolina Biological Supply Company, North Carolina. 36 pp. |
|
(12) |
Dunnett, C.W. (1955). A Multiple Comparisons Procedure for Comparing Several Treatments with Control. J. Amer. Statist. Assoc, 50, pp. 1096-1121. |
|
(13) |
Dunnett, C.W. (1964). New Tables for Multiple Comparisons with a Control. Biometrics, 20, pp. 482-491. |
|
(14) |
Mc Clave, J.T., Sullivan, J.H. and Pearson, J.G. (1980). Statistical Analysis of Fish Chronic Toxicity Test Data. Proceedings of 4th Aquatic Toxicology Symposium, ASTM, Philadelphia. |
|
(15) |
Van Leeuwen, C.J., Adema, D.M.M. and Henries, J. (1990). Quantitative Structure-Activity Relationships for Fish Early Life Stage Toxicity. Aquatic Toxicology, 16, pp. 321-334. |
|
(16) |
Environment Canada. (1992). Toxicity Tests Using Early Life Stages of Salmonid Fish (Rainbow Trout, Coho Salmon or Atlantic Salmon). Biological Test Method Series. Report EPS l/RM/28, December 1992, 81 pp. |
|
(17) |
Dave, G. and Xiu, R. (1991). Toxicity of Mercury, Nickel, Lead and Cobalt to Embryos and Larvae of Zebrafish, Brachydanio rerio. Arch. of Environmental Contamination and Toxicology, 21. pp. 126-134. |
|
(18) |
Meyer, A., Bierman, C.H. and Orti, G. (1993). The phylogenetic position of the Zebrafish (Danio rerio), a model system in developmental biology — an invitation to the comperative methods. Proc. Royal Society of London, Series B, 25 2: pp. 231-236. |
|
(19) |
Ghillebaert, F., Chaillou, C., Deschamps, F. and Roubaud, P. (1995). Toxic Effects, at Three pH Levels, of Two Reference Molecules on Common Carp Embryo. Ecotoxicology and Environmental Safety 32, pp. 19-28. |
|
(20) |
US EPA, (1991). Guidelines for Culturing the Japanese Medaka, Oryzias latipes. EPA report EPA/600/3-91/064, Dec. 1991, EPA, Duluth. |
|
(21) |
US EPA, (1991). Guidelines for Conducting Early Life Stage Toxicity Tests with Japanese Medaka, (Oryzias latipes). EPA report EPA/600/3-91/063, Dec. 1991, EPA, Duluth. |
|
(22) |
De Graeve, G.M, Cooney, J.D., Mclntyre, D.O., Poccocic, T.L, Reichenbach, N.G., Dean, J.H. and Marcus, M.D. (1991). Validity in the performance of the seven-day Fathead minnow (Pimephales promelas) larval survival and growth test: an intra- and interlaboratory study. Environ. Tox. Chem. 10, pp. 1189-1203. |
|
(23) |
Calow, P, (1993). Handbook of Ecotoxicology, Blackwells, Oxford. Vol. 1, Chapter 10: Methods for spawning, culturing and conducting toxicity tests with Early Life stages of Estuarine and Marine fish. |
|
(24) |
Balon, E. K. (198 5). Early life history of fishes: New developmental, ecological and evolutionary perspectives, Junk Publ., Dordrecht, 280 pp. |
|
(25) |
Blaxter, J.H.S. (1988). Pattern and variety in development, in: W.S. Hoar and D. J. Randall eds., Fish Physiology, Vol. XIA, Academic press, pp. 1-58. |
Tabel 1A
Fiskearter, som er anbefalet til testning
|
Ferskvand |
|
Oncorhynchus mykiss Regnbueørred (9) (16) |
|
Danio rerio Zebrafisk (7) (17) (18) |
|
Cyprinus caprio Alm. karpe (8) (19) |
|
Oryzias latipes Japanese ricefish/Medaka (20) (21) |
|
Pimephales promelas Fathead minnow (8) (22) |
Tabel 1B
Eksempler på andre veldokumenterede arter, som også har været anvendt
|
Ferskvand |
Saltvand |
|
Carassius auralus Guldfisk (S) |
Menidia peninsulae Tidewater silverside (23) (24) (25) |
|
Lepomis macrochirus Bluegill (8) |
Clupea harengus Sild (24) (25) |
|
Gadus morhua Torsk (24) (25) |
|
|
Cyprinodon variegatus Sheepshead minnow (23) (24) (25) |
Tillæg 1
VEJLEDNING I UDFØRELSE AF TOKSICITETSTEST PÅ EMBRYONER OG BLOMMESÆKYNGEL FRA ZEBRAFISK (BRACHYDANIO RERIO)
INDLEDNING
Zebrafisken kommer oprindeligt fra Coromandelkysten i Indien, hvor den opholder sig i hurtigtrindende strømme. Den er en almindelig akvariefisk af karpefamilien, og oplysninger om fremgangsmåde i behandlingen og dyrkningen af den kan findes i standardreferenceværker om tropiske fisk. Dens biologi og anvendelse i fiskeriundersøgelser findes i et review af Laale (1).
Fisken bliver sjældent længere end 45 cm. Kroppen er cylindrisk med 7-9 mørkeblå horisontale striber. Disse striber løber ud i de caudale og anale finner. Ryggen er olivengrøn. Hannerne er slankere end hunnerne. Hunnerne er mere sølvskinnende, og abdomen er udspilet, særligt lige før æglægning.
Voksne fisk tåler store svingninger i temperatur, pH og vandhårdhed. For at sikre sig sunde fisk, som lægger æg af god kvalitet, bør man sørge for optimale betingelser.
Under æglægningen forfølger hannen hunnen og skubber til hende med snuden, og når æggene er kommet ud, bliver de befrugtede. Æggene, som er gennemsigtige og ikke-klæbende, synker til bunds, hvor de kan ædes af forældrene. Lys har indflydelse på æglægningen. Hvis lyset om morgenen er tilstrækkeligt, vil fiskene almindeligvis lægge æg i de tidlige timer efter morgengry.
En hun kan producere adskillige hundrede af æg ad gangen med ugers mellemrum.
FORÆLDREFISK, REPRODUKTION OG TIDLIGE LIVSSTADIER
Udvælg et passende antal sunde fisk og hold dem i passende vand (f.eks. tillæg 4) i mindst to uger før den ønskede æglægning. Fiskene bør formere sig mindst en gang, før de producerer den samling æg, der skal bruges i testen. Fisketætheden bør ikke overstige 1 gram fisk pr liter. Regelmæssig fornyelse af vand eller anvendelse af rensningsanlæg kan tillade større fisketæthed. Temperaturen i tankene bør holdes på 25 ± 2 oC. Fiskene bør fodres med en varieret kost f.eks. af passende kommercielt tørfoder, levende nyklækkede Arthemia, chironomider, dafnier, hvide orm (Enchytraeider).
Nedenfor er skitseret to fremgangsmåder, der i praksis har ydet en tilstrækkelig mængde sunde, befrugtede æg til brug i en test:
|
i) |
8 hunner og 16 hanner placeres i et akvarium med 50 liter fortyndingsvand, skærmet mod direkte lys og holdt så uforstyrret som muligt i mindst 48 timer. En bakke til ægopsamling anbringes i bunden af akvariet om eftermiddagen, dagen før testen begynder. Ægopsamlingsbakken består af en ramme (plexiglas eller andet passende materiale), 5-7 cm høj med et groft 2-5 mm net fæstnet til kanten og et 10-30 μm fint net på bunden. Et passende antal »æglægnings-træer«, lavet af optrevlet nylonreb, sættes fast på det grove net. Fiskene efterlades i mørke i 12 timer, hvorefter et svagt lys tændes, hvilket vil føre til æglægning. To til fire timer efter æglægningen fjernes bakken, og æggene samles. Bakken vil hindre fiskene i at æde æggene og letter samtidig indsamlingen af æggene. Disse fisk skal have lagt æg mindst en gang før den, hvorfra æggene anvendes til testning. |
|
ii) |
Fra 5 til 10 han- og hunfisk holdes adskilt hver for sig i mindst to uger før den planlagte æglægning. Efter 5-10 dage vil hunnernes abdomen være udspilet og deres papillae genitales synlige. Sådanne har hanfisk ikke. Æglægning foregår i et æglægningsakvarium, forsynet med en falsk netværksbund (som ovenfor). Akvariet fyldes med fortyndingsvand, så der står 5-10 cm vand over nettet. En hun og to hanner anbringes i akvariet dagen før den planlagte æglægning. Vandtemperaturen øges gradvist til en grad over akklimatisationstemperaturen. Lyset slukkes, og akvariet holdes så uforstyrret som muligt. Næste morgen tændes et svagt lys, som får æglægningen til at gå i gang. 2-4 timer efter fjernes fiskene, og æggene indsamles. Hvis man har brug for flere æg, end en enkelt hun kan levere, opstilles flere akvarier parallelt. Ved at notere sig formeringens størrelse og kvalitet for hver hun før testen, kan man udvælge sig de bedste hunner til formering. |
Æggene flyttes til forsøgsakvariet ved hjælp af glasrør med en indre diameter ikke mindre end 4 mm og forsynede med en sugeballon. Mængden af vand, der flyttes med æggene ved flytningen, skal være så ringe som muligt. Æggene er tungere end vand og synker selv ud af røret. Omhu skal udvises, så æg (og larver) ikke kommer i kontakt med luften. Ved hjælp af mikroskopiske undersøgelser af prøver fra æggene sikrer man sig, at de første stadier ikke udviser irregulære former. Det er ikke tilladt at desinficere æggene.
Mortaliteten blandt æggene er størst inden for de første 24 timer efter befrugtningen. Ofte ses en mortalitet på 5-40 %. Æg degenerer på grund af befrugtning, der ikke lykkes, eller udviklingsfejl. Æggenes kvalitet synes at bero på hunfisken, da nogle hunner hele tiden producerer gode ægkvaliteter, mens andre aldrig gør det. Udviklingshastigheden og klækningshastigheden varierer fra et kuld til et andet. Hvor befrugtningen lykkes, og hvor blommesækynglen klarer sig godt, overlever normalt over 90 %. Ved 25 oC klækkes æggene 3-5 dage efter befrugtningen, og blommesækken resorberes ca. 13 dage efter befrugtningen.
Fosterudviklingen er vel beskrevet af Hisaoka og Battle (2). På grund af æggenes og de udklækkede larvers gennemsigtighed kan fiskenes udvikling følges, og tilstedeværelsen af malformationer observeres. Omkring 4 timer efter æglægningen kan de ikke-befrugtede æg skelnes fra de befrugtede (3). Til denne undersøgelse bliver æg og larver anbragt i små undersøgelseskar og set på under mikroskop.
Testbetingelserne, som vedrører de tidlige livsstadier, findes i tillæg 2. De optimale værdier for pH og vandhårdhed er 7,8 og 250 mg CaCO3/l, respektive.
BEREGNINGER OG STATISTIK
Der foreslås beregninger i to tempi. Først foretages statistisk analyse af data vedrørende mortalitet, unormal udvikling og klækningstidspunkt. Derefter evalueres kropslængden statistisk for de koncentrationer, hvor ingen af ovennævnte negative virkninger kunne ses. Denne fremgangsmåde tilrådes, eftersom giftstoffet kan tænkes selektivt at dræbe mindre fisk, forsinke klækningstidspunktet og forårsage grove malformationer og på den måde føre til længdemålinger med bias. Ydermere vil der være stort set samme antal fisk at måle pr. undersøgelse, hvilket sikrer teststatistikkens validitet.
LC50 OG EC50-BESTEMMELSER
Procentdelen af overlevende æg og larver beregnes og korrigeres for mortalitet i kontrollerne efter Abbotts formel (4):

hvor
|
P |
= |
= korrigeret % overlevelse |
|
P' |
= |
= % observeret overlevelse i testkoncentrationen |
|
C |
= |
= % overlevelse i kontrollen |
Hvis man ønsker at inkludere morfologiske abnormiteter i EC50-statistikken, kan man finde vejledning i Stephan (5).
ESTIMERING AF LOEC OG NOEC
Et af formålene med æg og blommesækyngelstestene er at sammenligne koncentrationerne over nul med kontrolgruppens, dvs. at bestemme LOEC. Derfor bør multiple sammenligningsprocedurer anvendes (6)(7)(8)(9)(10).
HENVISNINGER
|
(1) |
Laale, H.W. (1977). The Biology and Use of the Zebrafish (Brachydanio raid) in Fisheries Research. A Literature Review. J. Fish Biol. 10, pp. 121-173. |
|
(2) |
Hisaoka, K.K. and Battle, H.I. (1958). The Normal Development Stages of the Zebrafish Brachydanio rerio (Hamilton-Buchanan) J. Morph., 102, 311 pp. |
|
(3) |
Nagel, R. (1986). Untersuchungen zur Eiproduktion beim Zebrabårbling (Brachydanio rerio Hamilton-Buchanan). Journal of Applied Ichthyology, 2, pp. 173-181. |
|
(4) |
Finney, D.J. (1971). Probit Analysis, 3rd ed., Cambridge University Press, Great Britain, pp. 1-333. |
|
(5) |
Stephan, C.E. (198 2). Increasing the Usefulness of Acute Toxicity Tests. Aquatic Toxicology and Hazard ssessment: Fifth Conference, ASTM STP 766, J.G. Pearson, R.B. Foster and W.E. Bishop, Eds., American Society for Testing and Materials, pp. 69-81. |
|
(6) |
Dunnett, C.W. (1955). A Multiple Comparisons Procedure for Comparing Several Treatments with a Control. J. Amer. Statist. Assoc, 50, pp. 1096-1121. |
|
(7) |
Dunnett, C.W, (1964). New Tables for Multiple Comparisons with a Control. Biometrics, 20, pp. 482-491. |
|
(8) |
Williams, D.A. (1971). A Test for Differences Between Treatment Means when Several Dose Levels are Compared with a Zero Dose Control. Biometrics, 27, pp. 103-117. |
|
(9) |
Williams, D.A. (1972). The Comparison of Several Dose Levels with a Zero Dose Control. Biometrics 28, pp. 519-531. |
|
(10) |
Sokal, R.R. and Rohlf, F.J. (1981). Biometry, the Principles and Practice of Statistics in Biological Research, W. H. Freeman and Co., San Francisco. |
Tillæg 2
TESTBETINGELSER, VARIGHED OG OVERLEVELSESKRITERIER FOR DE ANBEFALEDE ARTER
|
Art |
Temperatur (0C) |
Saltindhold (0/00) |
Lysperiode (timer) |
Varighed af stadier (dage) |
Typisk testvarighed |
Overlevelseskontrol (minimum %) |
||
|
Embryoner |
Blommesækyngel |
Klækningssucces |
Efter klækning |
|||||
|
FERSKVAND |
||||||||
|
Brachydanio rerio Zebrafisk |
25 ± 1 |
— |
12-16 |
3-5 |
8-10 |
Så hurtigt som muligt efter befrugtning (tidl. gastrula-stadium) til 5 dage efter klækning (8-10 dage) |
80 |
90 |
|
Oncorhynchus mykiss Regnbueørred |
10 ± 1 (22) 12 ± 1 (23) |
— |
0 (24) |
30-35 |
25-30 |
Så hurtigt som muligt efter befrugtning (tidl. gastrula-stadium) til 20 dage efter klækning (50-5 5 dage) |
66 |
70 |
|
Cyprinus carpio Alm. karpe |
21-25 |
— |
12-16 |
5 |
> 4 |
Så hurtigt som muligt efter befrugtning (tidl. gastrula-stadium) til 4 dage efter klækning (8-9 dage) |
80 |
75 |
|
Oryzias latipes Japanese ricefish/Medaka |
24 ± 1 (22) 23 ± 1 (23) |
— |
12-16 |
8-11 |
4-8 |
Så hurtigt som muligt efter befrugtning (tidl. gastruia-sta-dium) til 5 dage efter klækning (13-16 dage) |
80 |
80 |
|
Pimephales promelas Fathead minnow |
25 ± 2 |
— |
16 |
4-5 |
5 |
Så hurtigt som muligt efter befrugtning (tidl. gastrula-sta-dium) til 4 dage efter klækning (8-9 dage) |
60 |
70 |
TILLÆG 3
Veldokumenterede testbetingelser, varighed og overlevelseskriterier for andre brugbare arter
|
Art |
Temperatur ( oC) |
Saltindhold (0/00) |
Lysperiode (timer) |
Varighed af stadier (dage) |
Typisk testvarighed for embryoner og blommesækyngel |
Overlevelse kontrol (minimum %) |
||
|
|
|
|
|
Embryoner |
Blommesækyngel |
Klækningssucces |
Efter klækning |
|
|
FERSKVAND |
||||||||
|
Carassius auratus Guldfisk |
24 ± 1 |
— |
— |
3-4 |
> 4 |
Så hurtigt som muligt efter befrugtning (tidl. gastrula-stadium) til 4 dage efter klækning (7 dage) |
— |
80 |
|
Leopomis macrochirus Blugill sunfish |
21 ± 1 |
— |
16 |
3 |
> 4 |
Så hurtigt som muligt efter befrugtning (tidl. gastrula-stadium) til 4 dage efter klækning (7 dage) |
— |
75 |
|
SALTVAND |
||||||||
|
Menidia peninsulae Tidewater silverside |
22-25 |
15-22 |
12 |
1,5 |
10 |
Så hurtigt som muligt efter befrugtning (tidl. gastrula-stadium) til 5 dage efter klækning (6-7 dage) |
80 |
60 |
|
Clupea harengus Sild |
10 ± 1 |
8-15 |
12 |
20-25 |
3-5 |
Så hurtigt som muligt efter befrugtning (tidl. gastrula-stadium) til 3 dage efter klækning (23-27 dage) |
60 |
80 |
|
Gadus morhua Torsk |
5 ± 1 |
5-30 |
12 |
14-16 |
3-5 |
Så hurtigt som muligt efter befrugtning (tidl. gastrula-stadium) til 3 dage efter klækning (18 dage) |
60 |
80 |
|
Cyprinodon variegatus Sheepshead minnow |
25 ± 1 |
15-30 |
12 |
— |
— |
Så hurtigt som muligt efter befrugtning (tidl. gastrula-sta-dium) til 4-7 dage efter klækning (28 dage) |
> 75 |
80 |
TILLÆG 4
NOGLE KEMISKE KARAKTERISTIKA FOR ACCEPTABELT FORTYNDINGSVAND
|
Substans |
Koncentration |
|
Faste stoffer |
< 20 mg/l |
|
Totalt organisk kulstof |
< 2 mg/l |
|
Ikke-ioniseret ammonium |
< 1 μg/l |
|
Residualt chlor |
< 10 μg/l |
|
Total organophosphor-pesticider |
< 50 ng/l |
|
Total organochlorino-pcsticider + polychlor-biphenyler |
< 50 ng/l |
|
Total organisk chlor |
< 25 ng/1 |
C.16. HONNINGBIER — AKUT ORAL TOKSICITETSTEST
1. METODE
Denne korttids toksicitetstestmetode svarer til OECD TG 213 (1998).
1.1. INDLEDNING
Denne toksicitetstest er en laboratoriemetode beregnet til at fastlægge den orale akutte toksicitet af plantebeskyttelsesmidler og andre kemikalier på voksne arbejdsbier.
I forbindelse med fastlæggelse og evaluering af stoffers toksiske egenskaber kan det være nødvendigt at fastslå den akutte orale toksicitet over for honningbier, f.eks. når det er sandsynligt, at bier bliver udsat for stoffet. Den akutte orale toksicitetstest udføres for at fastslå den iboende toksicitet af pesticider og andre kemikalier over for bier. Resultaterne af testen bør anvendes for at fastslå en eventuel nødvendighed af yderligere undersøgelser. Særligt kan metoden anvendes i programmer med henblik på at vurdere de risici, pesticider udgør for bier, baseret på en trinvis progression fra laboratorietoksicitetstest til tilnærmede feltforsøg og rene feltforsøg (l). Pesticider kan testes som aktive substanser (a.s.) eller som færdiggjorte produkter.
En toksisk standard bør medtages til at fastslå biernes følsomhed og testprocedurens præcision.
1.2. DEFINITIONER
Akut oral toksicitet: er den skadelige effekt, som indtræffer inden for maksimum 96 timer efter en oral indtagelse af en enkelt dosis af testkemikaliet.
Dosis: er den mængde af test-kemikaliet, som er indtaget. Dosis udtrykkes som masse (mg) af testkemikalium per test-dyr (mg/bi). Den reelle dosis, som hver bi får, kan ikke beregnes, da bierne fodres kollektivt, men en gennemsnitlig dosis kan skønnes (den totale mængde testkemikalium indtaget/antal test-bier i et bur).
LD 50 (Mediane letale dosis) oral: er en statistisk beregnet enkeltdosis af et kemikalium, der kan forårsage død af 50 % af dyrene, indtaget oralt. LD50-værdien udtrykkes i mg testkemikalium pr. bi. Hvad pesticider angår, kan testkemikaliet være enten en aktiv substans (a.s.) eller et færdiggjort produkt, indeholdende et eller flere aktive kemikalier.
Mortalitet: et dyr regnes for dødt, når det er totalt immobilt.
1.3. TESTMETODENS PRINCIP
Voksne arbejdshonningbier (Apis mellifera) udsættes for en række doser af testkemikaliet i en rørsukkeropløsning. Derefter får bierne samme føde, men uden testkemikaliet. Mortaliteten noteres daglig i mindst 48 timer og sammenlignes med kontrolværdierne. Hvis mortaliteten øges mellem 24 og 48 timer, mens kontrolværdierne holder sig på det accepterede niveau, dvs. ≤ 10 %, bør man forlænge testperioden til de maksimale 96 timer. Resultaterne analyseres for at beregne LD50 ved 24 timer og 48 timer og, hvis forsøget forlænges, ved 72 og 96 timer.
1.4. TESTENS VALIDITET
For at testen skal være valid, gælder
|
— |
at den gennemsnitlige mortalitet for alle kontroller skal være 10 % eller mindre ved forsøgets afslutning |
|
— |
LD50 for den toksiske standard falder inden for det forventede. |
1.5. BESKRIVELSE AF TESTMETODEN
1.5.1. Indsamling af bier
Unge. voksne bier af samme race bør anvendes, dvs. bier på samme alder, i samme foderstand osv. Bierne hentes fra tilstrækkeligt fodrede, sunde, så vidt muligt sygdomsfri kolonier, som er dronningret. Deres fortid og fysiologiske status bør kendes. De kan indsamles om morgenen eller om aftenen før brugen og holdes under testbetingelserne til næste dag. Bier fra rammer uden yngel kan anvendes. Indsamling i det tidlige forår eller sene efterår bør undgås, da bierne har ændret fysiologi på den tid. Hvis testen skal udføres i det tidlige forår eller sene efterår, kan bierne holdes i en inkubator i en uge og fodres med »bi-brød« (pollen indsamlet fra vokskagen) og rørsukkeropløsning. Bier, der har været behandlet med kemiske stoffer, såsom antibiotika, anti-varroaprodukter osv., bør ikke bruges til toksicitetsundersøgelser før fire uger efter slutningen af behandlingen.
1.5.2. Opbevaring og fodringsbetingelser
Der anvendes bure, der er rene og godt ventilerede. Alt passende materiale kan anvendes, såsom rustfrit stål, metalnet, plastik eller engangstræbure, osv. Det er bedst med ti bier pr bur. Størrelsen af testburene skal passe til antallet af bier, dvs. der skal være for tilstrækkelig plads.
Bierne holdes i mørke i forsøgsrummet ved en temperatur på 25 ± 2 oC. Den relative fugtighed, normalt omkring 50-70 %, skal registreres under hele forsøget. Håndtering, inklusive behandling og observation kan forgå ved (dags)lys. Som føde anvendes vandig rørsukkeropløsning med en slutkoncentration på 500 g/l (50 % w/v). Efter at testdosis er givet, forsynes bierne med føde ad libitum. Fordringssystemet bør tillade registrering af fødeindtagelse for hvert bur (se 1.6.3.1). Et glasrør, ca. 50 mm langt med 10 mm lysning, med den åbne ende indsnævret til 2 mm, kan anvendes.
1.5.3. Forberedelse af bierne
De indsamlede bier fordeles tilfældigt i burene, som placeres tilfældigt i forsøgsrummet.
Bierne må sulte op til to timer før forsøgets begyndelse. Det anbefales, at bierne sulter før behandlingen, så at alle bier står lige med hensyn til tarmindhold ved forsøgets begyndelse. Moribunde bier skal kasseres og erstattes med sunde bier, før forsøget indledes.
1.5.4. Fremstilling af doser
Hvor testkemikaliet er kan blandes i vand. kan det dispenseres direkte i en 50 % rørsukkeropløsning. Der kan i forbindelse med tekniske produkter og stoffer med lav vandopløselighed anvendes hjælpestoffer såsom organiske opløsningsmidler, blødgøringsmidler eller dispergenser med lav toksicitet over for bier (f.eks. acetone, dimethylformamid, dimethylsulfoxid). Hjælpestoffets koncentration afhænger af teststoffets opløselighed og bør være den samme i alle koncentrationer, der undersøges. Imidlertid er en koncentration af hjælpestoffet på 1 % normalt passende og bør ikke overskrides.
Tilsvarende kontrolopløsninger bør fremstilles, dvs. hvor opløsningsmiddel eller dispergens bliver anvendt til at gøre test-kemikaliet opløseligt, bør der anvendes to adskilte kontrolgrupper: en til opløsning i vand og en til rørsukkeropløsning med opløsningsmiddel/hjælpestof med samme koncentration som i forsøgsopløsningerne.
1.6. FREMGANGSMÅDE
1.6.1. Test- og kontrolgrupper
Antallet af doser og replikater bør svare til de statistiske krav for bestemmelse af LD50 med 95 % sikkerhedsgrænser. Normalt kræves dertil fem doser i en geometrisk serie med en faktor, der ikke overstiger 2,2, og som dækker området for LD50. Fortyndingsfaktor og antal dosiskoncentrationer bør imidlertid beregnes ud fra toksiciteteskurvens hældning (dosis over for mortalitet), idet man tager hensyn til den statistiske metode, hvormed man har valgt til at analysere resultaterne. En test til fastlæggelse af dosisområdet kan være til hjælp for fastlæggelse af dosiskoncentrationerne.
Mindst tre testgrupper, hver på ti bier, bør udsættes for hver testkoncentration. Mindst tre kontrolgrupper, hver på ti bier, deltager parallelt med testserien. Kontrolgrupper skal også inkluderes til belysning af de opløsningsmidler og hjælpestoffer, der anvendes (se 1.5.4).
1.6.2. Toksisk standard
En toksisk standard (dvs. et kendt toksisk stof) skal inkluderes i testserien. Af den bør udvælges mindst tre doser for at dække den forventede LD50-værdi. Mindst tre bure. hver med ti bier. skal udsættes for hver testdosis. Den foretrukne toksiske standard er dimethoat, for hvilket det angives, at den orale LD50-24 timer befinder sig i størrelsesordenen 0,10-0,35 mg aktive substanser/bi (2). Andre toksiske standarder kan naturligvis anvendes, hvis tilstrækkelige data kan fremskaffes til belysning af forventet dosisrespons (f.eks. parathion).
1.6.3. Belastning
1.6.3.1. Administration af doser
Hver test-gruppe bier skal forsynes med 100-200 ml 50 % vandig rørsukkeropløsning, indeholdende testkemikaliet i den rigtige koncentration. Større volumen kræves til kemikalier, der er tungt opløselige, har lav toksicitet eller lav koncentration i præparatet, da der skal bruges større mængder i rørsukkeropløsningen. Mængden af behandlet føde, der indtages, skal monitoreres. Når den er indtaget (almindeligvis efter 3-4 timer), skal fodringssystemet fjernes fra buret og udskiftes med et nyt, som kun indeholder rørsukkeropløsning, og som tilføres ad libitum. Nogle stoffer kan i højere doser medføre, at der indtages kun lidt eller ingen føde. Efter højst 6 timer skal resten af den behandlede føde fjernes og erstattes med ren rørsukkeropløsning. Mængden af behandlet føde, der er indtaget, skal beregnes (f.eks. via måling af volumen/vægt af resterende behandlet føde).
1.6.3.2. Varighed
Testen varer fortrinsvis til 48 timer efter at testopløsningen er blevet erstattet med ren rørsukkeropløsning. Hvis mortaliteten fortsætter med at stige med mere end 10 % efter de første 24 timer, bør testvarigheden udvides til maksimalt 96 timer, under forudsætning af, at mortaliteten blandt kontroldyrene ikke overstiger 10 %.
1.6.4. Observationer
Mortaliteten registreres 4 timer efter testens start og derefter 24 og 48 timer efter (dvs. efter givet dosis). Hvis en længere observationsperiode er nødvendig, registreres yderligere hver 24. time op til 96 timer, under forudsætning af, at mortaliteten blandt kontroldyrene ikke overstiger 10 %.
Den mængde føde, der indtages af hver gruppe, skal vurderes. Sammenligning af den hastighed, hvormed den behandlede og den ikke behandlede føde indtages inden for 6 timer, kan skaffe viden om den behandlede fødes smagskvalitet.
Enhver unormal opførsel observeret blandt bierne i forsøgsperioden skal registreres.
1.6.5. Grænsetest
I visse tilfælde (f.eks. hvis et testkemikalium forventes at besidde lav toksicitet) kan man udføre en grænsetest, hvor man anvender 100 mg aktiv substans/bi, for at vise, at LD50 er større end denne værdi. Samme fremgangsmåde skal anvendes, inklusive tre replikate testgrupper til testdosis, de relevante kontroller, vurderingen af indtaget behandlet føde og brugen af toksisk standard. Hvis dødsfald indtræder, skal der udføres et komplet forsøg. Hvis subletale virkninger observeres (se 1.6.4), skal de registreres.
2. DATA OG RAPPORTERING
2.1. DATA
Data skal opsummeres i tabelform, der viser, for hver behandlet gruppe, kontrolgrupper og grupper med toksisk standard, antallet af anvendte bier, mortaliteten for hvert observationstidspunkt og bier med usædvanlig opførsel. Analyser mortaliteten ved hjælp af passende statistiske metoder (f.eks. probitanalyser, vægtede gennemsnit, binominal sandsynlighed) (3)(4), Plot dosisresponskurver for hver tidspunkt, hvor observationer anbefales, og beregn kurvehældninger og den mediane letale dosis, LD5o med 95 % sikkerhedsgrænser. Korrektioner for kontrolleret mortalitet kan foretages v.h.a. Abbotts korrektion (4) (5). Hvis den behandlede føde ikke er totalt indtaget, skal dosis af testkemikalium indtaget pr. gruppe bestemmes. LD50 skal udtrykkes som mg testkemikalium pr. bi.
2.2. TESTRAPPORT
Testrapporten skal indeholde følgende informationer:
2.2.1. Test-kemikalium:
|
— |
fysiske egenskaber og relevante fysisk-kermske egenskaber (f.eks. stabilitet i vand, damptryk) |
|
— |
data for kemisk identifikation, inklusive strukturformel, renhedsgrad (dvs. for pesticider: identitet og koncentration af aktive substans(er)). |
2.2.2. Testdyr
|
— |
videnskabeligt navn, race, omtrentlige alder (i uger), indsamlingsmetode, dato for indsamling |
|
— |
informationer om kolonier, der er anvendt til indsamlingen af testbier, inklusive sundhed, voksensygdomme, tidligere behandling osv. |
2.2.3. Testomstændigheder
|
— |
temperatur og forsøgsrummets relative fugtighedsgrad |
|
— |
opbevaringsbetingelser, inklusive burets type, størrelse og materiale |
|
— |
fremgangsmåde ved fremstilling af stamopløsning og testopløsning (eventuelt opløsningsmiddel og dets koncentration skal angives) |
|
— |
testdesign, dvs. antal testkoncentrationer og deres størrelse, antal kontroller; for hver testkoncentration og kontrol angives antal replikater og antal bier pr. bur |
|
— |
dato for testen. |
2.2.4. Resultater:
|
— |
resultater af eventuelle forsøg med koncentrationsområder |
|
— |
rådata: mortaliteten for hver koncentration ved hvert observationstidspunkt |
|
— |
graf af dosisresponskurve ved forsøgets slutning |
|
— |
LD50-værdier med 95 %-sikkerhedsgrænser for hvert anbefalet observationstidspunkt, for testkemikalium og toksisk standard |
|
— |
statistisk procedure for fastlæggelse af LD50 |
|
— |
mortaliteten blandt kontroldyrene |
|
— |
andre biologiske effekter oberverede eller målte, f.eks. abnorm opførsel blandt bierne (inklusive manglende indtagelse af testdosis), konsumptionshastighed både hvad angår den behandlede og den ubehandlede gruppe |
|
— |
enhver fravigelse af testfremgangsmåden, som her er beskrevet, samt enhver anden relevant oplysning. |
3. HENVISNINGER
|
(1) |
EPPO/Council of Europe (1993). Decision-Making Scheme for the Environmental Risk Assessment of Plant Protection Products — Honeybees. EPPO Bulletin, Vol. 23, N.1. pp. 151-165. March 1993. |
|
(2) |
Gough, H.J., McIndoe, E.C., Lewis, G.B. (1994). The use of dimethoate as a reference compound in laboratory acute toxicity tests on honeybees (Apis mellifera L) 1981-1992. Journal of Apicultural Research, 22, pp. 119-125. |
|
(3) |
Litchfield, J.T. and Wilcoxon, F. (1949). A simplified method of evaluating dose-effect experiments. Jour. Pharmacol. and Exper. Ther., 96, pp. 99-113. |
|
(4) |
Finney, D.J. (1971). Probit Analysis. 3rd ed., Cambridge, London and New York. |
|
(5) |
Abbott, W.S. (1925). A method for computing the effectiveness of an insecticide. Jour. Econ. Entomol, 18, pp. 265-267. |
C.17. HONNINGBIER — AKUT KONTAKTTOKSICITETSTEST
1. METHODE
Denne akut toksicitets-testmetode svarer til OECD TG 214 (1998).
1.1. INDLEDNING
Denne toksicitetstestmetode er en laboratoriemetode, beregnet til at vurdere den akutte toksicitet over for voksne arbejdshonningbier ved kontakt med plantebeskyttelsesmidler og andre kemikalier.
Ved vurderingen og evalueringen af kemikaliers toksiske karakteristika kan det være ønskværdigt at bestemme den akutte kontakttoksicitet over for honningbier, f.eks. når det er sandsynligt, at bier kan blive udsat for de pågældende kemikalier. Den akutte toksicitetstest udføres for at bestemme pesticiders og andre kemikaliers naturlige toksicitet over for bier. Resultaterne af denne test er tænkt anvendt til at fastslå behovet for yderligere evaluering. Specielt kan metoden bruges i programmer, der evaluerer den fare, som pesticider betyder for bier, baseret på en trinvis progression fra laboratorietoksicitetstest til tilnærmede feltforsøg og rene feltforsøg (1). Pesticider kan testes som aktive substanser (a.s.) eller som færdiggjorte produkter.
En toksisk standard bør medtages til at fastslå biernes følsomhed og testprocedurens præcision.
1.2. DEFINITIONER
Akut kontakt toksicitet: er den skadelige effekt, som indtræffer inden for maksimum 96 timer efter en lokal applikation af en enkelt dosis af et kemikalium.
Dosis: er den mængde af test-kemikaliet, som er påført. Dosis udtrykkes som masse (mg) af testkemikalium per test-dyr (mg/bi). Den reelle dosis, som hver bi får, kan ikke beregnes, da bierne fodres kollektivt, men en gennemsnitlig dosis kan skønnes (den totale mængde test-kemikalium indtaget/antal test-bier i et bur).
LD 50 (Mediane letale dosis) kontakt: er en statistisk beregnet enkeltdosis af et kemikalium, der kan forårsage død af 50 % af dyrene, når det administreres via kontakt. LD50-værdien udtrykkes i mg testkemikalium pr. bi. Hvad pesticider angår, kan testkemikaliet være enten en aktiv substans (a.s.) eller et færdiggjort produkt, indeholdende et eller flere aktive kemikalier.
Mortalitet: et dyr regnes for dødt, når det er totalt immobilt.
1.3. TESTMETODENS PRINCIP
Voksne arbejdshonningbier (Apis mellifera) udsættes for en serie forskellige doser af testkemikaliet, opløst i et passende medie, gennem en direkte applikation på thorax (små dråber). Forsøget varer 48 timer. Hvis mortaliteten øges mellem 24 og 48 timer, mens kontrolværdierne holder sig på det accepterede niveau, dvs. < 10 %, bør man forlænge testperioden til de maksimale 96 timer. Resultaterne analyseres for at beregne LD50 ved 24 timer og 48 timer og, hvis forsøget forlænges, ved 72 og 96 timer.
1.4. TESTENS VALIDITET
For at testen skal være valid, gælder
|
— |
at den gennemsnitlige mortalitet for alle kontroller skal være 10 % eller mindre ved forsøgets afslutning |
|
— |
LD50 for den toksiske standard falder inden for det forventede. |
1.5. BESKRIVELSE AF TESTMETODEN
1.5.1. Indsamling af bier
Unge, voksne bier af samme race bør anvendes, dvs. bier på samme alder, i samme foderstand, race osv. Bierne hentes fra tilstrækkeligt fodrede, sunde, så vidt muligt sygdomsfri kolonier, som er dronningret. Deres fortid og fysiologiske status bør kendes. De kan indsamles aftenen for brugen og holdes under testbetingelserne til næste dag. Bier fra rammer uden yngel kan anvendes. Indsamling i det tidlige forår eller sene efterår bør undgås, da bierne har ændret fysiologi på den tid. Hvis testen skal udføres i det tidlige forår eller sene efterår, kan bierne holdes i en inkubator i en uge og fodres med »bi-brød« (pollen indsamlet fra vokskagen) og rørsukkeropløsning. Bier, der har været behandlet med kemiske stoffer, såsom antibiotika, anti-varroaprodukter osv., bør ikke bruges til toksicitetsundersøgelser før fire uger efter slutningen af behandlingen.
1.5.2. Opbevaring og fodringsbetingelser
Der anvendes bure, der er rene og godt ventilerede. Alt passende materiale kan anvendes, såsom rustfrit stål, metalnet, plastik eller engangstræbure, osv. Størrelsen af testburene skal passe til antallet af bier, dvs. der skal være tilstrækkelig plads. Det er bedst med ti bier pr. bur.
Bierne holdes i mørke i forsøgsrummet ved en temperatur på 25 ± 2 oC. Den relative fugtighed, normalt omkring 50-70 %, skal registreres under hele forsøget. Håndtering, inklusive behandling og observation kan forgå ved (dags)lys. Som føde anvendes vandig rørsukkeropløsning med en slutkoncentration på 500 g/l (50 % w/v) og tilbydes ad libitum under forsøget, idet man anvender et fodringssystem, der kan være et glasrør (ca. 50 mm langt og med en lysning på 10 mm og med den åbne ende indsnævret til 2 mm i diameter).
1.5.3. Forberedelse af bierne
De indsamlede bier kan bedøves med carbondioxid eller nitrogen, når testkemikaliet skal appliceres. Mængden af anæstetikum og bedøvelsestiden bør være mindst mulig. Moribunde bier skal kasseres og erstattes af sunde bier, før testen påbegyndes.
1.5.4. Fremstilling af doser
Testkemikaliet skal appliceres som en opløsning i et medium, dvs. et organisk opløsningsmiddel eller en vandig opløsning med overfladeaktive stoffer. Som organisk opløsningsmiddel foretrækkes acetone, men andre organiske opløsningsmidler med lav toksicitet over for bier kan bruges (f.eks. dimethylformamid, dimethylsulfoxid). Vandopløselige færdiggjorte produkter og højpolære organiske substanser, som ikke er opløselige i organiske opløsningsmidler, kan være lettere at påføre, hvis de tilberedes i en svag opløsning af kommercielle overfladeaktive stoffer (f.eks.. Agral, Cittowet, Lubrol, Triton, Tween).
Tilsvarende kontrolopløsninger bør fremstilles, dvs. hvor opløsningsmiddel eller dispergens bliver anvendt til at gøre testkemikaliet opløseligt, bør der anvendes to adskilte kontrolgrupper, en behandlet med vand og en med opløsningsmiddel/dispergent.
1.6. FREMGANGSMÅDE
1.6.1. Test- og kontrolgrupper
Antallet af doser og replikater bør svare til de statistiske krav for bestemmelse af LD50med 95 % sikkerhedsgrænser. Normalt kræves dertil fem doser i en geometrisk serie med en faktor, der ikke overstiger 2,2, og som dækker området for LD50. Antallet af doser bør imidlertid beregnes ud fra toksiciteteskurvens hældning (dosis over for mortalitet), idet man tager hensyn til den statistiske metode, hvormed man har valgt til at analysere resultaterne. En test til fastlæggelse af dosisområdet kan være til hjælp for fastlæggelse af dosiskoncentrationerne.
Mindst tre testgrupper, hver på ti bier, bør udsættes for hver testkoncentration.
Mindst tre kontrolgrupper, hver på ti bier, deltager parallelt med testserien. Hvis der er anvendt et organisk opløsningsmiddel eller et overfladeaktivt stof, skal der inkluderes yderligere tre kontrolgrupper på ti bier hver for opløsningsmidlet eller det overfladeaktive stof.
1.6.2. Toksisk standard
En toksisk standard skal inkluderes i testserien. Af den bør udvælges mindst tre doser for at dække den forventede LD50-værdi. Mindst tre bure, hver med ti bier, skal udsættes for hver testdosis. Den foretrukne toksiske standard er dimethoat, for hvilket det angives, at kontakt-LD50-24 timer befinder sig i størrelsesordenen 0,10-0,30 mg aktive substanser/bi (2). Andre toksiske standarder kan naturligvis anvendes, hvis tilstrækkelige data kan fremskaffes til belysning af forventet dosis-respons (f.eks. parathion).
1.6.3. Belastning
1.6.3.1. Administration af doser
Bedøvede bier behandles individuelt med lokal applikation. Bierne randomiseres til de forskellige testdoser og kontroller. 1 ml af opløsningen, der indeholder testkemikaliet i den passende koncentration, anbringes med en mikroapplikator på dorsalsiden af thorax af bien. Andre volumina kan anvendes, hvis der er grund til det. Efter applikationen anbringes bierne i testburene og forsynes med rørsukkeropløsning.
1.6.3.2. Varighed
Forsøgets varighed er almindeligvis 48 timer. Hvis mortaliteten øger mere end 10 % mellem 24 og 48 timer, skal forsøgsvarigheden forøges til op til 96 timer, under forudsætning af, at mortaliteten blandt kontrollerne ikke overstiger 10 %.
1.6.4. Observationer
Mortaliteten registreres 4, 24 og 48 timer efter applikationen. Hvis det er nødvendigt med en forlænget observationsperiode, skal observationerne foretages med 24-timers intervaller, op til 96 timer, under forudsætning af, at kontrolmortaliteten ikke overstiger 10 %.
Enhver unormal opførsel som observeres i testperioden skal registreres.
1.6.5. Grænsetest
I visse tilfælde (f.eks. hvis et testkemikalium forventes at besidde lav toksicitet) kan man udføre en grænsetest, hvor man anvender 100 mg aktiv substans/bi, for at vise, at LD50 er større end denne værdi. Samme fremgangsmåde skal anvendes, inklusive tre replikate testgrupper til testdosis, de relevante kontroller og brugen af toksisk standard. Hvis dødsfald indtræder, skal der udføres et komplet forsøg. Hvis subletale virkninger observeres (se 1.6.4), skal de rapporteres.
2. DATA OG RAPPORTERING
2.1. DATA
Data skal opsummeres i tabelform, der viser, for hver behandlet gruppe, kontrolgrupper og grupper med toksisk standard, antallet af anvendte bier, mortaliteten for hvert observationstidspunkt og bier med usædvanlig opførsel. Analyser mortaliteten ved hjælp af passende statistiske metoder (f.eks. probitanalyser, vægtede gennemsnit, binominal sandsynlighed) (3)(4). Plot dosisresponskurver for hver tidspunkt, hvor observationer anbefales, (dvs. efter 24, 48 og, hvis relevant. 72 og 96 timer) og beregn kurvehældninger og den mediane letale dosis, LD50, med 95 %-sikkerhedsgrænser. Korrektioner for kontrolleret mortalitet kan foretages v.h.a. Abbotts korrektion (4)(5). LD50 skal udtrykkes som mg testkemikalium pr. bi.
2.2. TESTRAPPORT
Testrapporten skal indeholde følgende informationer:
2.2.1. Testkemikalium:
|
— |
fysiske egenskaber og relevante fysisk-kemiske egenskaber (f.eks. stabilitet i vand, damptryk) |
|
— |
data for kemisk identifikation, inklusive struktur-formel, renhedsgrad (dvs. for pesticider: identitet og koncentration af aktive substans(er)). |
2.2.2. Testdyr
|
— |
videnskabeligt navn. race. omtrentlige alder (i uger), indsamlingsmetode, dato for indsamling |
|
— |
informationer om kolonier, der er anvendt til indsamlingen af testbier, inklusive sundhed, voksensygdomme, tidligere behandling osv. |
2.2.3. Testomstændigheder
|
— |
temperatur og forsøgsrummets relative fugtighedsgrad |
|
— |
opbevaringsbetingelser, inklusive burets type, størrelse og materiale |
|
— |
fremgangsmåde ved administration af testkemikalium (det anvendte opløsningsmedium, volumen af testopløsning, det anvendte anæstetikum |
|
— |
testdesign, f.eks. antal og størrelse af testdoser, antal kontroller; for hver testdosis og kontrol angives antal replikater og antal bier pr. bur |
|
— |
dato for testen. |
2.2.4. Resultater:
|
— |
resultater af eventuelle forsøg med koncentrationsområder |
|
— |
rådata: mortaliteten for hver koncentration ved hvert observationstidspunkt |
|
— |
graf af dosisresponskurve ved forsøgets slutning |
|
— |
LD50-værdier med 95 %-sikkerhedsgrænser for hvert anbefalet observationstidspunkt, for testkemikalium og toksisk standard |
|
— |
statistisk procedure for fastlæggelse af LD50 |
|
— |
mortaliteten blandt kontroldyrene |
|
— |
andre biologiske effekter, observerede eller målte, og abnorm opførsel blandt bierne |
|
— |
enhver fravigelse af testfremgangsmåden, som her er beskrevet, samt enhver anden relevant oplysning. |
3. HENVISNINGER
|
(1) |
EPPO/Council of Europe (1993). Decision-Making Scheme for the Environmental Risk Assessment of Plant Protection Products — Honeybees. EPPO bulletin, Vol. 23, N.l, pp. 151-165. March 1993. |
|
(2) |
Gough, H.J., McIndoe, E.C., Lewis, G.B. (1994). The use of dimethoate as a reference compound in laboratory acute toxicity tests on honeybees (Apis mellifera L.), 1981-1992. Journal of Apicultural Research 22, pp. 119-125. |
|
(3) |
Litchfield, J.T. and Wilcoxon, F. (1949). A simplified method of evaluating dose-effect experiments. Jour. Pharmacol. and Exper. Ther., 96, pp. 99-113. |
|
(4) |
Finney, D.J. (1971), Probit Analysis. 3rd ed., Cambridge, London and New York. |
|
(5) |
Abbott, W.S. (1925). A method for computing the effectiveness of an insecticide. Jour. Econ. Entomol. 18, pp. 265-267. |
C.18. ADSORPTION/DESORPTION MED EN BATCH-LIGEVÆGTSMETODE
1. METODE
Metoden er gengivet efter OECD TG 106: »Determination of Soil Adsorption/Desorption, using a Batch Equili-brium Method« (2000).
1.1. INDLEDNING
I metoden er taget hensyn til en ringtest og en workshop om udvælgelse af jord med henblik på udvikling af en adsorptionstest (1) (2) (3) (4) samt eksisterende guidelines på nationalt plan (5) (6) (7) (8) (9) (10) (11).
Adsorptions-/desorptionsundersøgelser kan skaffe vigtige oplysninger om kemiske stoffers mobilitet og fordeling i biosfærens jord-, vand- og luftcompartments (12) (13) (14) (15) (16) (17) (18) (19) (20) (21). Sådanne oplysninger kan anvendes til opstilling af prognoser eller overslagsberegninger f.eks. vedrørende muligheden for kemisk nedbrydning (22) (23), omdannelse og optagelse i organismer (24), udvaskning gennem jordbundsprofilen (16) (18) (19) (21) (25) (26) (27) (28), fordampning fra jorden (21) (29) (30) og afstrømning fra jordoverfladen til naturlige vande (18) (31) (32). Adsorptionsdata kan anvendes til at drage sammenligninger og opstille modeller (19) (33) (34) (35).
Et kemisk stofs fordeling mellem jord- og vandfase er en kompleks proces, som bestemmes af en række forskellige faktorer: stoffets kemiske egenskaber (12) (36) (37) (38) (39) (40), jordens karakteristika (4) (12) (13), (14) (41) (42) (43) (44) (45) (46) (47) (48) (49) samt klimatiske faktorer som nedbør, temperatur, sollys og vind. De mange fænomener og mekanismer, som medvirker ved adsorption af et kemisk stof til jord, kan således ikke fastlægges fuldt ud ved en forenklet laboratoriemodel som nærværende metode. Men uanset at denne tentativt opstillede metode ikke kan dække alle tilfælde, der kan forekomme i miljøet, giver den tilstrækkelige oplysninger om den miljømæssige relevans af adsorptionen af et kemisk stof.
Se også den generelle indledning.
1.2. OMRÅDE
Metodens formål er at gøre det muligt at foretage en skønsmæssig beregning af et stofs adsorption/desorption i jord. Målet er at nå frem til en sorptionsværdi, med hvilken fordelingen under forskellige miljøforhold kan forudsiges; til dette formål bestemmes ligevægtsadsorptionskoefficienter for et kemisk stof på forskellige jordtyper som funktion af jordens egenskaber (f.eks. dens indhold af organisk kulstof, lerindholdet samt jordens struktur og pH). Det er nødvendigt at anvende forskellige jordtyper for at få så bred dækning som muligt af vekselvirkningen mellem et givet stof og naturligt forekommende jordtyper.
I denne metode forstås ved adsorption den proces, hvorved et kemisk stof bindes til overfladen af forskellige jordtyper; der skelnes her ikke mellem forskellige adsorptionsprocesser (fysisk og kemisk adsorption) og processer som overfladekatalyseret nedbrydning, masseadsorption og kemiske reaktioner. Den adsorption, som finder sted på kolloide partikler (diameter < 0,2 μm) dannet af jordarterne, tages ikke i betragtning.
De jordparametre, som anses for at have størst betydning for adsorptionen, er: organisk kulstofindhold (3) (4) (12) (13) (14) (41) (43) (44) (45) (46) (47) (48), lerindhold og jordstruktur (3) (4) (41) (42) (43) (44) (45) (46) (47) (48) samt for ioniserbare forbindelser pH-værdien (3)(4)(42). Andre jordparametre, der kan have indflydelse på adsorptionen/desorptionen af et stof, er effektiv kationudvekslingskapacitet (ECEC), indhold af amorfe jern- og aluminiumoxider, specielt for vulkanske og tropiske jordarter (4), samt virksom overflade (49).
Testen er udviklet med henblik på vurdering af et kemisk stofs adsorption på forskellige jordtyper med varierende organisk kulstofindhold, lerindhold og jordstruktur samt pH. Testen omfatter tre trin:
|
Trin 1: |
Indledende undersøgelse til bestemmelse af:
|
|
Trin 2: |
Screeningtest: adsorptionen i fem forskellige jordtyper undersøges adsorptionskinetisk ved én enkelt koncentration og bestemmelse af fordelingskoefficienterne Kd og Koc. |
|
Trin 3: |
Bestemmelse af Freundlich-adsorptionsisotermer til fastlæggelse af koncentrationens indflydelse på adsorptionen på jord. Undersøgelse af desorption ved hjælp af desorptionskinetik/Freundlich desorptionsisotermer (tillæg 1). |
1.3. DEFINITIONER OG ENHEDER
|
Symbol |
Definition |
Enhed |
|
|
adsorptionsprocent til tiden ti |
% |
|
Aeq |
adsorptionsprocent ved adsorptionsligevægt |
% |
|
|
masse prøvestof adsorberet til jorden til tiden ti |
μg |
|
|
masse prøvestof adsorberet på jorden i løbet af tidsintervallet Δti |
μg |
|
|
masse prøvestof adsorberet på jorden ved adsorptionsligevægt |
μg |
|
m0 |
masse prøvestof i prøveglasset ved adsorptionstestens begyndelse |
μg |
|
|
masse prøvestof målt i prøve (
|
μg |
|
|
masse af stoffet i opløsningen ved adsorptionsligevægt |
μg |
|
msoil |
mængde jordfase, angivet som tør masse jord |
g |
|
Cst |
massekoncentration af stamopløsningen af stoffet |
μg cm-3 |
|
C0 |
initial massekoncentration af prøveopløsning i berøring med jorden |
μg cm-3 |
|
|
massekoncentration af stoffet i den vandige fase til tidspunktet ti for analysens udførelse |
μg cm-3 |
|
|
indhold af stof adsorberet på jorden ved adsoprtionsligevægt |
μg g-1 |
|
|
massekoncentration af stoffet i vandig fase ved adsorptionsligevægt |
μg cm-1 |
|
Vo |
initialt volumen af vandig fase i kontakt med jorden under adsorptionstesten |
cm3 |
|
|
volumen af den prøve, hvro prøvestoffet måles |
cm3 |
|
Kd |
fordelingskoefficient for adsorption |
cm3 g-1 |
|
Koc |
normaliseret adsorptionskoefficient for organisk kulstof |
cm3 g-1 |
|
Kum |
normaliseret adsorptionskoefficient for organisk kulstof |
cm3 g-1 |
|
|
Freudlich adsorptionskoefficient |
μg-1-l/n (cm3) l/n g-1 |
|
l/n |
Freudlich-eksponent |
|
|
|
desorptionsprocent til tidspunktet ti |
% |
|
|
desorptionsprocent til tidsintervallet Δti |
% |
|
Kdes |
tilsyneladende desorptionskoefficient |
cm3 g-1 |
|
|
Freudlich desorptionskoefficient |
μg 1 -l/n (cm3) l/n g-1 |
|
|
masse af prøvestoffe desorbere fra jorden i lobet af tiden ti |
μg |
|
|
masse af prøvestoff desorberet fra jorden i lobet af tidsintervallet Δti |
μg |
|
|
masse af stof. som er bestemt analytisk i den vandige fase ved desorptionsligevægt |
μg |
|
|
samlet masse af prøvestof desorberet ved desorptionsligevægt |
μg |
|
|
masse af prøvestof. som stadig er adsorberet på jorden efter tidsintervallet Δti |
μg |
|
|
masse af stof. som er tilbage efter indtræden af adsorptionsligevægt som følge af ufuldstændig volumenudskiftning |
μg |
|
|
indhold af prøvestof, som stadig er adsorberet til jorden ved desorptionsligevægt |
μg g-1 |
|
|
massekoncentration af prøvestof i vandig fase ved desorptionsligevægt |
μg cm-3 |
|
VT |
samlet rumfang af vandig fase i kontakt med jorden under desorptionskinetikforsøget undført med den sekventielle metode |
cm3 |
|
VR |
rumfang supermatant, som efter indtræden af adsorptionsligevægt er fjernet fra prøveglasset og erstattet af samme rumfang 0,01 M CaCl2-opløsning |
cm3 |
|
|
rumfang af prøve udtaget til analyse fra tidspunktet (i) under desorptionskinetikforsøget udført med den sekventielle metode |
cm3 |
|
|
rumfang af opløsning udtaget af prøveglas (i) til bestemmelse af prøvestof i desorptionskinetkforsøget (parallelle metode) |
cm3 |
|
|
rumfang af opløsning udtaget af prøveglas (i) til bestemmelse af prøvestof, ved desorptionsligevægt |
cm3 |
|
MB |
massebalance |
% |
|
mE |
samlet masse prøvestof ekstraheret fra jord og prøveglassets vægge i to trin |
μg |
|
Vrec |
rumfang af supermatant, som er opsamlet efter indtræden af adsorptionsligevægt |
cm3 |
|
Pow |
oktanol/vand fordelingskoefficient |
|
|
pKa |
dissociationskonstant |
|
|
Sw |
vandopløselighed |
g l-1 |
1.4. TESTMETODENS PRINCIP
Kendte rumfang af en opløsning af umærket eller radioaktivt mærket prøvestof med kendt koncentration i 0,01 M CaCl2 tilsættes til jordprøver, hvis tørre vægt kendes, og som i forvejen er ekvilibreret i 0,01 M CaCl2. Blandingen omrystes et passende stykke tid. Jordsuspensionerne adskilles derefter ved centrifugering og om ønsket filtrering, og den vandige fase analyseres. Den mængde prøvestof, som er adsorberet på jordprøven, beregnes som forskellen mellem den mængde prøvestof, som i begyndelsen er til stede i oplosningen og den mængde, der er tilbage ved forsøgets slutning (indirekte metode).
Om ønsket kan man vælge at bestemme den adsorberede mængde prøvestof direkte ved analyse af jorden (direkte metode). Denne metode, som indebærer trinvis ekstraktion af jorden med passende opløsningsmiddel, anbefales i tilfælde, hvor koncentrationsforskellen af stoffet i opløsningen ikke kan bestemmes nøjagtigt. Som eksempel på sådanne tilfælde kan nævnes: Prøvestoffet adsorberes på overfladen af prøveglasset, prøvestoffet er ustabilt inden for forsøgets tidsramme, adsorptionen er enten så svag, at der kun fås lille koncentrationsændring i stoffet, eller så stærk, at koncentrationen bliver for lille til at kunne bestemmes nøjagtigt. Anvendes radiomærket stof, kan ekstraktion af jorden undgås ved analyse af jordfasen ved forbrænding og væskescintillationstælling. Væskescintillationstælling er imidlertid en uspecifik teknik, som ikke gør det muligt at skelne mellem moderstof og omdannelsesprodukter; den bør derfor kun anvendes, hvis prøvestoffet er stabilt i hele undersøgelsens varighed.
1.5. OPLYSNINGER OM PRØVESTOFFET
Kemiske reagenser skal være af analytisk kvalitet. Det anbefales at anvende umærket prøvestof med kendt sammensætning og helst mindst 95 % renhed eller radioaktivt mærket prøvestof af kendt sammensætning og radioaktiv renhed. Anvendes radioaktive tracerstoffer med kort halveringstid, skal der korrigeres for henfald.
Før man udfører en test for adsorption-desorption, skal der foreligge følgende oplysninger om prøvestoffet:
|
a) |
vandopløselighed (A.6) |
|
b) |
damptryk (A.4) og/eller Henrys lovkonstant |
|
c) |
abiotisk nedbrydning: hydrolyse som funktion af pH (C.7) |
|
d) |
fordelingskoefficient (A.8) |
|
e) |
umiddelbar biologisk nedbrydelighed (C.4) eller aerob og anaerob omdannelse i jord |
|
f) |
pKa for ioniserbare stoffer |
|
g) |
direkte fotolyse i vand (dvs. UV-Vis absorptionsspektrum i vand, kvanteudbytte) og fotonedbrydning på jord. |
1.6. PRØVENS ANVENDELIGHED
Prøven kan anvendes på kemiske stoffer, for hvilke en analysemetode med tilstrækkelig nøjagtighed er til rådighed. En vigtig parameter, som kan have betydning for resultaternes pålidelighed, navnlig ved anvendelse af den indirekte metode, er prøvestoffets stabilitet inden for det tidsrum, testen strækker sig over. Det er således en forudsætning, at stabiliteten efterprøves i en indledende undersøgelse; iagttages der en omdannelse i løbet af den tid, testen strækker sig over, anbefales hovedundersøgelsen udført med analyse af både jord- og vandfase.
Man kan komme ud for vanskeligheder med at udføre testen for prøvestoffer med ringe vandopløselighed (Sw < 10-4 g l-1), såvel som for stærkt ladede stoffer som følge af, at koncentrationen i den vandige fase ikke kan bestemmes analytisk med tilstrækkelig nøjagtighed. I sådanne tilfælde må der træffes supplerende foranstaltninger. Vejledning i, hvordan sådanne problemer gribes an. er givet i de pågældende afsnit i denne metode.
Ved testning af flygtige stoffer må der drages omsorg for at undgå tab under behandlingen.
1.7. BESKRIVELSE AF METODEN
1.7.1. Apparatur og kemiske reagenser
Standardlaboratorieudstyr, navnlig følgende:
|
a) |
Glas eller beholdere til udførelse af forsøgene. Det er vigtigt, at disse glas eller beholdere:
|
|
b) |
Til omrystning anvendes overhead-rystebord eller tilsvarende; rystebordet skal holde jorden opslæmmet under omrystningen. |
|
c) |
Centrifuge: helst af højhastighedstypen, f.eks centrifugalkraft > 3 000 g, temperaturkontrolleret, skal kunne fjerne partikler med diameter over 0,2 μm fra vandige opløsninger. Beholderne skal være tilproppet under omrystning og centrifugering for at undgå tab ved fordampning og væskespild; for at mindske adsorptionen på propperne bør deaktiverede propper anvendes, f.eks. teflonbelagte skruepropper. |
|
d) |
Valgfrit: filtreringsapparat, sterile engangsfiltre med 0,2 μm porestørrelse. Filtermaterialet bør udvælges særligt nøje med henblik på at undgå ethvert tab af prøvestof på filtrene; til tungtopløselige prøvestoffer kan filtre af organisk materiale ikke anbefales. |
|
e) |
Analyseudstyr, som er velegnet til måling af prøvestoffets koncentration. |
|
f) |
Laboratorievarmeskab, som kan holde temperaturer fra 103 oC til 110 oC. |
1.7.2. Karakterisering og udvælgelse af jord
Jorden bør være karakteriseret ved tre parametre, som i hovedsagen anses for bestemmende for adsorptionskapaciteten: organisk kulstofindhold, lerindhold og jordstruktur, samt pH. Som allerede nævnt (jf. »Område«) kan andre fysisk-kemiske jordegenskaber have betydning for et givet stofs adsorption/desorption og bør i så fald tages i betragtning.
Det har meget stor betydning, hvilke metoder der anvendes til karakterisering af jorden, og disse kan have stor indflydelse på resultaterne. Det anbefales derfor, at jordens pH måles i en opløsning af 0,01 M CaCl2 (dvs. den opløsning, der benyttes ved undersøgelse af adsorption/desorption) i henhold til den pågældende ISO-metode (ISO-10390-1). Ligeledes anbefales, at de øvrige relevante jordegenskaber bestemmes efter standardmetoder (ISO Handbook of soil analysis); derved kan analysen af sorptionsdata baseres på globalt standardiserede jordparametre. Nogen vejledning vedrørende foreliggende standardmetoder til jordanalyse og -karakterisering er givet i henvisning (50-52). Til kalibrering af metoderne til jordbundsanalyse anbefales brug af referencejordtyper.
Vejledning i udvælgelse af jord til adsorptions-/desorptionsforsøgene er givet i tabel 1. De syv udvalgte jordtyper er dem, man finder i tempererede geografiske områder. For ioniserbare stoffer bør de udvalgte jordtyper dække et bredt pH-område for at give mulighed for vurdering af adsorptionen af stoffet, når dette er henholdsvis ioniseret og uioniseret. Vejledning i, hvor mange forskellige jordtyper der skal undersøges i de forskellige trin af testen, findes under 1.9. »Testens præstationer«.
Foretrækker man andre jordtyper, bør de være karakteriseret ved de samme parametre og have tilsvarende variation i egenskaber som dem, der er beskrevet i tabel 1, også selv om de ikke modsvarer kriterierne nøjagtigt.
Tabel 1
Vejledning i valg af jordprøver til adsorption-desorption
|
Jordtype |
pH-område (i 0,01 M CaCl2) |
Organisk kulstofindhold ( %) |
Lerindhold ( %) |
Jordstruktur (25) |
|
1 |
4,5-5,5 |
1,0-2,0 |
65-80 |
clay |
|
2 |
> 7,5 |
3,5-5,0 |
20-40 |
clay loam |
|
3 |
5,5-7,0 |
1,5-3,0 |
15-25 |
silt loam |
|
4 |
4,0-5,5 |
3,0-4,0 |
15-30 |
loam |
|
5 |
< 4,0-6,0 (26) |
< 10-15 (26) |
loamy sand |
|
|
6 |
> 7,0 |
40-65 |
clay loam/clay |
|
|
7 |
|< 4,5 |
> 10 |
< 10 |
sand/loamy sand |
1.7.3. Indsamling og opbevaring af jordprøver
1.7.3.1. Indsamling
Der anbefales ikke bestemte prøveudtagningsmetoder eller -redskaber; prøveudtagningsmetoden afhænger af undersøgelsens formål (53)(54)(55)(56)(57){58).
Følgende bør tages i betragtning:
|
a) |
Udførlige oplysninger om områdets historie er nødvendige, herunder beliggenhed, plantedække, behandling med pesticider og/eller gødning, biologisk tilvækst eller tilfældig forurening. Anbefalingerne i ISO-standarden for jordprøvetagning (ISO 10381-6) bør følges med hensyn til beskrivelse af prøvetagningsstedet. |
|
b) |
Prøvetagningsstedet skal være fastlagt ved UTM (universel transversal Mercator-projektion/europæisk horisontal datum) eller geografiske koordinater; dette skulle gøre det muligt at genfinde en bestemt jordtype i fremtiden og kan være en hjælp ved bestemmelse af jorden efter forskellige klassifikationssystemer, som anvendes i andre lande. Endvidere må kun indsamles A-horisont til en maksimal dybde af 20 cm. Specielt gælder for jordtype n.7, at hvis en Oh-horisont indgår i jorden, bør den medtages ved prøveindsamlingen. |
Jordprøverne bør transporteres i sådanne beholdere og under sådanne temperaturforhold, at der med sikkerhed ikke sker nævneværdige ændringer i jordens oprindelige egenskaber.
1.7.3.2. Opbevaring
Der bør fortrinsvis anvendes jord, som er frisk optaget fra marken. Kun når dette ikke er muligt, bør jorden opbevares ved rumtemperatur i lufttørret tilstand. Der anbefales ingen grænse for opbevaringstiden, men har jorden været opbevaret i over tre år, bør den før anvendelse genanalyseres for organisk kulstofindhold, pH og kationbytningskapacitet.
1.7.3.3. Håndtering og klargøring af jordprøver til testen
Jordprøverne lufttørres ved rumtemperatur (fortrinsvis ved 20-25 oC). Disaggregering bør foretages med mindst mulig kraft, således at den oprindelige jordstruktur ændres mindst muligt. Jordens sigtes til en partikelstørrelse ≤ 2 mm; ved sigtningen følges anbefalingerne i ISO-standarden for jordprøvetagning (ISO 10381-6). Skånsom homogenisering anbefales, da reproducerbarheden af resultaterne derved øges. Fugtindholdet i hver jordtype bestemmes på tre prøver ved opvarmning til 105 oC, indtil vægten ikke længere ændrer sig væsentligt (ca. 12 h). I alle beregninger forstås ved masse af jord den ovntørrede masse, dvs. jordens vægt korrigeret for fugtindhold.
1.7.4. Klargøring af prøvestof, som skal tilføres jorden
Prøvestoffet opløses i 0,01 M CaCl2 i destilleret eller deioniseret vand; CaCl2-opløsningen anvendes som vandig fase for at forbedre centrifugeringen og nedsætte kationbytningen til det mindst mulige. Stamopløsningens koncentration bør fortrinsvis være tre størrelsesordener over detektionsgrænsen for den anvendte analysemetode. Denne grænse garanterer nøjagtige målinger med hensyn til de her anviste metoder; endvidere bør stamopløsningens koncentration være lavere end prøvestoffets opløselighed i vand.
Stamopløsningen bør fortrinsvis fremstilles umiddelbart før stoffet tilføres jordprøverne og bør opbevares tillukket og mørkt ved 4 oC. Opbevaringstiden afhænger af prøvestoffets stabilitet og dets koncentration i opløsningen.
Kun ved tungtopløselige stoffer (Sw < 10-4 g l-1) kan det være nødvendigt at anvende et passende opløsningsmiddel, når prøvestoffet er vanskeligt at bringe i opløsning. For dette opløsningsmiddel gælder; a) det bør være blandbart med vand, som f.eks. methanol eller acetonitril; b) dets koncentration bør ikke være større end svarende til 1 % af stamopløsningens samlede rumfang og bør være mindre i den opløsning af prøvestof, som kommer i kontakt med jorden (helst under 0,1 %), og c) det bør ikke være overfladeaktivt eller indgå i lyolytiske reaktioner med prøvestoffet. Brug af opløsningsmiddel bør være angivet og begrundet i rapporteringen af data.
Et andet alternativ ved tungtopløselige stoffer er at tilsætte prøvestoffet til prøvesystemet ved spiking: Prøvestoffet opløses i et organisk opløsningsmiddel, og en lille del heraf tilsættes systemet bestående af jord og 0,01 molær opløsning af CaCl2 i destilleret eller deioniseret vand. Indholdet af organisk opløsningsmiddel i den vandige fase skal holdes så lavt som muligt, normalt ikke over 0,1 %. Spiking fra en organisk opløsning kan være behæftet med manglende reproducerbarhed af volumen. Derved kan der indføres en ekstra fejlkilde i forsøget, da koncentrationen af prøvestof og hjælpeopløsningsmiddel ikke vil være den samme i alle test.
1.8. BETINGELSER FOR UDFØRELSE AF ADSORPTIONS-/DESORPTIONSTESTEN
1.8.1. Analysemetode
De vigtigste parametre, som kan påvirke nøjagtigheden af sorptionsmålingerne, er nøjagtigheden af analysemetoden anvendt til både opløsningsfase og adsorberet fase, prøvestoffets stabilitet og renhed, om sorptionsligevægt opnås, hvor meget opløsningens koncentration ændrer sig, forholdet jord/opløsning og ændringer i jordstrukturen under ekvilibreringen (35)(59-62). Nogle eksempler med relevans for nøjagtigbedsproblematikken er givet i tillæg 2.
Pålideligheden af den anvendte analysemetode skal være kontrolleret i det koncentrationsområde, som forventes at optræde under testen. Eksperimentatoren bør ikke afholde sig fra selv at udvikle en velegnet metode med passende nøjagtighed, præcision, reproducerbarhed, detektionsgrænser og genfindingsgrad. Nedenfor er givet en forsøgsvejledning, som viser udførelsen af en sådan test.
Et passende rumfang 0,01 M CaCl2, f.eks. 100 cm3, omrystes i 4 h med en afvejet mængde jord, f.eks. 20 g, med stor adsoptionsevne, dvs. stort organisk kulstofindhold; der kan afviges fra de angivne vægtmængder og rumfang afhængigt af, hvad hensynet til analysen kræver, men et forhold jord/opløsning på 1:5 vil være et passende udgangspunkt. Blandingen centrifugeres, og den vandige fase kan eventuelt filtreres. Til den vandige fase tilsættes et bestemt rumfang af stamopløsningen af prøvestof, således at den nominelle koncentration når inden for det koncentrationsområde, der forventes at optræde under testen. Dette rumfang bør højst udgøre 10 % af den vandige fases endelige rumfang, for at forekvilibreringsopløsningens beskaffenhed skal ændres mindst muligt. Opløsningen analyseres.
Én blindprøve bestående af systemets jord + CaCl2-opløsning (uden prøvestof) skal indgå i analysen, for at man kan kontrollere, om analysemetoden medfører artefakter, og om jorden giver matrixvirkninger.
Til sorptionsmåling kan anvendes gas-væskekromatografi (GLC), high-performance væskekromatografi (HPLC), spektrometri (f.eks. GC/massespektrometri, HPLC/massespektrometri) og væskescintillationstælling (ved radioaktivt mærket prøvestof). Uanset den anvendte analysemetode anses det for passende, hvis der genfindes mellem 90 % og 110 % af den nominelle værdi. For at give mulighed for detektion og evaluering efter at adskillelse af faserne har fundet sted, bør analysemetodens detektionsgrænser være mindst to størrelsesordener under den nominelle koncentration.
Karakteristika og detektionsgrænser for den analysemetode, der er til rådighed for adsorptionsundersøgelserne, spiller en vigtig rolle ved fastlæggelse af testbetingelserne og testens samlede præstationer. I denne metode angives en generel forsøgsgang, medens der gives anvisninger og vejledning for alternative løsninger, hvor analysemetode og laboratoriefaciliteter kan sætte begrænsninger.
1.8.2. Valg af optimalt jord/opløsning-forhold
Et passende jord til opløsning forhold for sorptionsundersøgelser vælges ud fra fordelingskoefficienten Kd og den ønskede relative adsorptionsgrad. Den statistiske nøjagtighed, med hvilken stoffets koncentration i opløsningen lader sig bestemme, er givet ved koncentrationsændringen for stoffet i opløsningen, afhængigt af adsorptionsligningens form og analysemetodens grænseværdi. I praksis er det derfor sædvanligvis nyttigt at bestemme sig for nogle få faste forhold, som giver en adsorptionsprocent på over 20 %, helst også > 50 % (62), samtidig med at man sørger for, at prøvestoffets koncentration i den vandige fase er høj nok til at kunne måles nøjagtigt. Dette er særlig vigtigt ved høje adsorptionsprocenter.
En bekvem måde at vælge passende jord/vandforhold på er at basere det på en skønnet Kd-værdi, enten fra de indledende forsøg eller fra tilnærmet beregning med en anerkendt metode (tillæg 3). Derefter kan et passende forhold jord/opløsning vælges ved afsætte en kurve over dette forhold som funktion af Kd for fastlagte adsorptionsprocenter (figur1). Ved optegning af kurven antages adsorptionsligningen lineær (28). Det pågældende forhold findes ved at omordne ligning 4 for Kd i samme form som ligning 1:
|
|
(1) |
eller i logaritmisk form, idet det forudsættes, at R = msoil/V0 og Aeq %/100 = 
|
|
(2) |

Figur 1 viser det ønskede jord/opløsningsforhold som funktion af Kd ved forskellige adsorptionsgrader. F.eks. fås med et forhold jord/opløsning på 1:5 og Kd = 20 en adsorption på ca. 80 %. For at få 50 % adsorption ved samme Kd-værdi skal forholdet være 1:25. Denne måde at vælge passende jord/opløsningsforhold på giver undersøgeren den fleksibilitet, som er nødvendig under forsøgsomstændigheder.
Vanskeligere at håndtere er de områder, hvor det kemiske stof udviser enten meget høj eller meget lav adsorption. I tilfælde af lav adsorption anbefales et jord/opløsningsforhold på 1:1, dog kan det for visse meget organiske jordtyper være nødvendigt med et lavere forhold for at få en vællinglignende konsistens. Der må vises omhu med analysemetoden ved måling af små ændringer i opløsningens koncentration; ellers vil adsorptionsmålingen blive unøjagtig. Hvis fordelingskoefficienten Kd på den anden side er meget høj, kan man gå op til et forhold jord/opløsning på 1:100 for at en væsentlig del af det kemiske stof skal forblive i opløsningen. Der må dog sørges omhyggeligt for god opblanding, og man må give systemet tilstrækkelig tid til at ekvilibreres, En alternativ fremgangsmåde er at forudsige Kd-værdien med teknikker til skønsvis beregning, f.eks. baseret på Pow-værdier (tillæg 3), Især kan dette være nyttigt for lavt adsorberede/polære kemiske stoffer med Pow < 20 og for lipofile/stærkt sorptive kemiske stoffer med Pow > 104.
1.9. TESTENS PRÆSTATIONER
1.9.1. Testbetingelser
Alle forsøg udføres ved rumtemperatur, og om muligt ved konstant temperatur mellem 20 oC og 25 oC.
Centrifugeringsbetingelserne skal gøre det muligt at fjerne partikler større end 0,2 μm fra opløsningen. Denne værdi medtager den mindste partikel, der betragtes som en faststofpartikel, og danner overgangen mellem faste og kolloide partikler. Vejledning for fastlæggelse af centrifugeringsbetingelserne findes i tillæg 4.
Kan centrifugeringsfaciliteterne ikke garantere fjernelse af partikler over 0,2 μm, kan man eventuelt kombinere centrifugeringen med filtrering med filtre med 0,2 μm porestørrelse. Sådanne filtre skal være fremstillet af passende inaktivt materiale for at undgå tab af prøvestof på dem. Under alle omstændigheder skal det være godtgjort, at filtreringen ikke medfører tab af prøvestof.
1.9.2. Trin 1 — Indledende undersøgelse
Formålet med den indledende undersøgelse er beskrevet i afsnittet »Område«. Vejledning for opsætning af en sådan test gives i forbindelse med det nedenfor foreslåede forsøg.
1.9.2.1. Valg af optimalt forhold jord/opløsning
Der anvendes to jordtyper og tre forskellige jord/opløsningforhold (seks enkeltforsøg). Den ene jordtype har højt organisk kulstofindhold og lavt lerindhold, medens den anden har lavt organisk kulstofindhold og højt lerindhold. Der foreslås følgende forhold:
|
— |
50 g jord til 50 cm3 vandig opløsning af prøvestoffet (forhold 1:1) |
|
— |
10 g jord til 50 cm3 vandig opløsning af prøvestoffet (forhold 1:5) |
|
— |
2 g jord til 50 cm3 vandig opløsning af prøvestoffet (forhold 1:25). |
Den mindste mængde jord, som forsøget kan gennemføres på, afhænger af laboratoriefaciliteterne og præstationerne af de anvendte analysemetoder. Dog anbefales det at anvende mindst 1 g, og helst 2 g for at få pålidelige resultater af testen.
Én kontrolprøve, alene indeholdende prøvestof i 0,01 M CaCl2-opløsning (ingen jord), underkastes nøjagtig de samme procedurer som prøvesystemerne for at kontrollere prøvestoffets stabilitet i CaCl2-opløsning og dets eventuelle adsorption på overfladen af prøvekarrene.
For hver jordtype gennemføres samme testprocedure på en blindprøve med samme jordmængde og et totalt rumfang på 50 cm30,01 M CaCl2-opløsning (uden prøvestof). Blindprøven tjener til baggrundskontrol under analysen med henblik på at opdage forstyrrende stoffer eller forurenet jord.
Alle forsøgene, herunder kontrol- og blindforsøg, skal udføres mindst som dobbeltforsøg. Det samlede antal prøver, som skal fremstilles til forsøget, kan beregnes ud fra den metode, der skal anvendes.
Sædvanligvis benyttes samme metoder ved forundersøgelsen og hovedundersøgelsen; undtagelser herfra nævnes i givet fald.
Dagen før forsøgsdagen ekvilibreres de lufttørrede jordprøver ved omrystning med et mindsterumfang på 45 cm30,01 M CaCl2 natten over (12 h). Derefter tilsættes et bestemt rumfang af stamopløsningen af prøve-stoffet, således at det endelige rumfang justeres til 50 cm3. Dette tilsatte rumfang stamopløsning: a) må ikke være over 10 % af det endelige rumfang på 50 cm3 af den vandige fase for ikke at ændre beskaffenheden af forekvilibreringsopløsningen mere end nødvendigt, og b) bør fortrinsvis resultere i, at den prøveopløsning, som er i kontakt med jorden (C0), har en begyndelseskoncentration mindst to størrelsesordener over detektionsgrænsen for analysemetoden; denne grænse sikrer, at der kan foretages nøjagtige målinger, selv når der optræder stærk adsorption (> 90 %), og giver mulighed for senere bestemmelse af adsorptionsisotermerne. Ligeledes anbefales det, at begyndelseskoncentrationen af det undersøgte stof (C0) ikke er større end halvdelen af dets opløselighed.
Nedenfor er givet et eksempel på, hvordan stamopløsningens koncentration (Cst) beregnes. Det antages, at detektionsgrænsen er 0,01 μg cm-3, og at adsorptionen er 90 %; begyndelseskoncentrationen af prøvestoffet, som er i kontakt med jorden, bør derfor helst være 1 μg cm-3 (to størrelsesordener over detektionsgrænsen). Når det forudsættes, at der tilsættes det anbefalede maksimale rumfang stamopløsning, dvs. 5 cm3 til 45 cm30,01 M CaCl2 ekvilibreringsopløsning (= 10 % stamopløsning i 50 cm3 samlet vandigt rumfang), bør koncentrationen af stamopløsningen være 10 μg cm-3, altså tre størrelsesordener over detektionsgrænsen for analysemetoden.
pH af den vandige fase bør måles før og efter kontakt med jorden, da den spiller en vigtig rolle i hele adsorptionsprocessen, navnlig for ioniserbare stoffer.
Blandingen omrystes godt, indtil adsorptionsligevægt er nået. Ligevægtstiden i jord er stærkt varierende og afhænger af det kemiske stof og af jorden; et tidsrum på 24 h er sædvanligvis tilstrækkeligt (77). I forundersøgelsen kan prøverne udtages sekventielt gennem en omrystningsperiode på 48 h (f.eks. 4. 8, 24, 48 h). Analysetidspunkterne bør dog opfattes fleksibelt under hensyntagen til laboratoriets arbejdsplan.
For analyse af prøvestoffet i den vandige opløsning er der to valgmuligheder: a) den parallelle metode og b) den sekventielle metode. Det må understreges, at skønt den parallelle metode er forsøgsmæssigt mere langstrakt, er den matematiske behandling af resultaterne enklere (tillæg 5). Hvilken metode man vil vælge, er imidlertid op til eksperimentatoren, som må tage hensyn til de forhåndenværende laboratoriefaciliteter og ressourcer.
|
a) |
Den parallelle metode: Der fremstilles prøver med ens jordopløsningforhold, idet antal prøver skal være lig det antal tidsintervaller, hvori adsorptionskinetikken ønskes undersøgt. Efter centrifugering og (om ønsket) filtrering opsamles den vandige fase i første prøveglas så fuldstændig som muligt og måles efter f.eks. 4 h, væsken i det andet prøveglas efter 8 h, i det tredje prøveglas efter 24 h osv. |
|
b) |
Den sekventielle metode: Der fremstilles kun en dobbeltprøve for hvert forhold jordopløsning. Ved fastlagte tidsintervaller centrifugeres blandingen for at skille faserne. En lille prøve af den vandige fase analyseres straks for prøvestof; derefter fortsætter forsøget med den oprindelige blanding. Filtreres der efter centrifugeringen, bør laboratoriet have de nødvendige faciliteter til filtrering af små vandige prøver. Det anbefales, at det samlede rumfang af de udtagne prøver ikke udgør over 1 % af opløsningens samlede rumfang, således at man ikke ændrer forholdet jordopløsning væsentligt og nedsætter massen af opløst stof, som er til rådighed for adsorption under testen. |
Adsorptionsprocenten 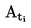 beregnes for hvert tidspunkt (ti) på grundlag af den nominelle begyndelseskoncentration og den målte koncentration på prøvetagningstidspunktet (ti), korrigeret for blindværdien. Grafer over som
beregnes for hvert tidspunkt (ti) på grundlag af den nominelle begyndelseskoncentration og den målte koncentration på prøvetagningstidspunktet (ti), korrigeret for blindværdien. Grafer over som  -funktion af tiden (Figur 1, tillæg 5) optegnes for at vurdere, om der er nået et ligevægtsplateau (29). Kd-værdien ved ligevægt beregnes ligeledes. På grundlag af denne Kd-værdi vælges passende forhold jord/opløsning i figur 1, således at adsorptionsprocenten kommer over 20 %, og helst > 50 % (61). Alle de pågældende ligninger og principper for grafisk afbildning er givet i afsnittet om data og rapportering i tillæg 5.
-funktion af tiden (Figur 1, tillæg 5) optegnes for at vurdere, om der er nået et ligevægtsplateau (29). Kd-værdien ved ligevægt beregnes ligeledes. På grundlag af denne Kd-værdi vælges passende forhold jord/opløsning i figur 1, således at adsorptionsprocenten kommer over 20 %, og helst > 50 % (61). Alle de pågældende ligninger og principper for grafisk afbildning er givet i afsnittet om data og rapportering i tillæg 5.
1.9.2.2. Bestemmelse af ekvilibreringstid for adsorptionen og mængde prøvestof adsorberet ved ligevægt
Som nævnt giver grafer over  eller
eller  som funktion af tiden mulighed for vurdere, om der er nået adsorptionsligevægt, samt den adsorberede mængde prøvestof ved ligevægt. Figur 1 og 2 i tillæg 5 viser eksempler på sådanne afbildninger. Ekvilibreringstiden er den tid, systemet behøver for at nå et plateau.
som funktion af tiden mulighed for vurdere, om der er nået adsorptionsligevægt, samt den adsorberede mængde prøvestof ved ligevægt. Figur 1 og 2 i tillæg 5 viser eksempler på sådanne afbildninger. Ekvilibreringstiden er den tid, systemet behøver for at nå et plateau.
Hvis der med en given jord ikke nås et plateau, men fås en konstant stigende værdi, kan det skyldes komplicerende faktorer som biologisk nedbrydning eller langsom diffusion. Biologisk nedbrydning kan påvises ved, at forsøget gentages med en steriliseret prøve af jorden. Hvis der heller ikke i dette tilfælde nås et plateau, bør eksperimentatoren søge efter andre fænomener, som kan tænkes at gøre sig gældende i netop hans undersøgelser; dette kan ske gennem passende ændringer af forsøgsbetingelserne (temperatur, omrystningstid, jord/opløsningforhold). Det er op til eksperimentatoren at afgøre, om testen skal fortsættes, uanset at det måske ikke lykkes at nå ligevægt.
1.9.2.3. Adsorption af prøvestof på overfladen af prøvebeholderne og prøvestoffets stabilitet
Ved at analysere kontrolprøverne kan man skaffe sig en del oplysninger om prøvestoffets adsorption på overfladen af prøvebeholderne samt dets stabilitet. Hvis der iagttages en udtynding, som er større end svarende til middelfejlen på analysemetoden, kan der være tale om abiotisk nedbrydning og/eller adsorption på overfladen af prøveglassene. Man vil kunne skelne mellem disse to fænomener ved at vaske prøveglassets vægge grundigt med et kendt volumen af et passende opløsningsmiddel og derefter analysere vaskeopløsningen for prøvestoffet. Hvis der ikke konstateres adsorption af prøvestof på overfladen af prøveglassene, er udtyndingen et bevis på abiotisk ustabilitet af prøvestoffet. Hvis der findes adsorption, må der anvendes prøveglas af andet materiale. De af dette forsøg afledte oplysninger om adsorption af prøvestof på overfladen af prøveglassene kan imidlertid ikke overføres direkte til jord/opløsningforsøget. Tilstedeværelsen af jord vil indvirke på denne adsorption.
Supplerende oplysninger om prøvestoffets stabilitet kan fås ved bestemmelse af massebalancen for moderstoffet i tid. Dette indebærer, at den vandige fase, ekstrakterne af jord samt prøvekammerets vægge analyseres for prøvestof. Forskellen mellem massen af tilsat prøvestof og summen af masserne af prøvestof i den vandige fase, ekstrakterne fra jord og fra prøveglassets vægge er lig den masse, som er nedbrudt og/eller fordampet og/eller ikke ekstraheret. For at massebalancen kan beregnes, bør der være nået adsorptionsligevægt inden for forsøgsperioden.
Massebalancen beregnes for begge jordprøver og for ét jord/opløsningforhold for hver jordtype, som giver en udtynding over 20 % og helst > 50 % ved ligevægt. Når forsøget til fastlæggelse at forholdet jord/opløsning er fuldført med analyse af den sidste prøve af den vandige fase efter 48 h, adskilles faserne ved centrifugering og, om ønsket, filtrering. Der opsamles så meget som muligt af den vandige fase, og til jorden tilsættes et passende ekstraktionsopløsningsmiddel (ekstraktionskoefficient mindst 95 %) for at ekstrahere prøvestoffet. Det anbefales at foretage mindst to på hinanden følgende ekstraktioner. Mængden af prøvestof i ekstrakterne fra jord og prøveglas bestemmes, og massebalancen beregnes (ligning 10, data og rapportering). Hvis den er mindre end 90 %, regnes prøvestoffet for ustabilt inden for testperioden. Undersøgelserne kan dog i så tilfælde fortsættes, idet der tages hensyn til prøvestoffets ustabilitet; det anbefales da at analysere begge faser i hovedundersøgelsen.
1.9.2.4. Trin 2 — Adsorptionskinetik ved én prøvestofkoncentration
Der anvendes fem jordtyper, udvalgt fra tabel 1. Det vil være en fordel, hvis nogle af eller alle de anvendte jordtyper fra forundersøgelsen i givet fald er med blandt disse fem jordtyper. Derved undgår man at gentage trin 2 for de jordtyper, der er anvendt i forundersøgelsen.
Ekvilibreringstiden, forholdet jord/opløsning, jordprøvens vægt, rumfanget af vandig fase i berøring med jorden og prøvestoffets koncentration i opløsningen vælges på grundlag af resultaterne af forundersøgelsen. Analyse bør helst finde sted efter ca. 2, 4, 6, 8 (eventuelt også 10) og 24 h kontakttid: omrystningstiden kan strækkes til højst 48 h i tilfælde af, at prøvestoffet kræver længere ekvilibreringstid ud fra de resultater, der danner grundlag for fastlæggelse af opblandingsforholdet. Analysetidspunkterne bør dog opfattes fleksibelt.
Hvert forsøg (ét med jord og ét med opløsning) gennemføres mindst som dobbeltforsøg, for at resultaternes overensstemmelse kan vurderes. I hvert forsøg skal indgå én blindprøve. Denne består af jord og 0,01 M CaCl2 opløsning uden stofprøve og med henholdsvis vægt og rumfang identisk med forsøgets. En kontrolprøve, alene indeholdende prøvestof i 0,01 M CaCl2-opløsning (uden jord) underkastes samme testprocedure for at sikre mod det uventede.
Adsorptionsprocenten beregnes for hvert tidspunkt  og/eller tidsinterval
og/eller tidsinterval  (afhængigt af behov) og afsættes mod tiden. Desuden beregnes fordelingskoefficienten Kd ved ligevægt og den normaliserede adsorptionskoefficient Koc for organisk kulstof (for upolære organiske stoffer).
(afhængigt af behov) og afsættes mod tiden. Desuden beregnes fordelingskoefficienten Kd ved ligevægt og den normaliserede adsorptionskoefficient Koc for organisk kulstof (for upolære organiske stoffer).
Resultaterne af adsorptionskinetikforsøget
Den lineære Kd-værdi giver sædvanligvis en nøjagtig beskrivelse af sorptionsforholdene i jord (35)(78) og er et udtryk for den iboende mobilitet af kemiske stoffer i jord. For eksempel anses kemiske stoffer med Kd ≤ 1 cm3 g–1 sædvanligvis for kvalitativt mobile. Tilsvarende har MacCall et al. (16) opstillet et system til opdeling efter mobilitet, baseret på Koc-værdier. Endvidere findes der klassifikationssystemer for udvaskning, baseret på sammenhængen mellem Koc og DT-50 (30) (32)(79).
1 henhold til fejlanalyseundersøgelser (61) kan Kd-værdier under 0,3 cm3 g-1 desuden ikke beregnes nøjagtigt ud fra et fald i koncentrationen i den vandige fase, selv når der anvendes det (fra en nøjagtighedssynsvinkel) gunstigste forhold jord/opløsning, dvs. 1:1. I så tilfælde anbefales det analysere begge faser — jord og opløsning.
Vedrørende ovenstående bemærkninger anbefales det. at undersøgelsen af adsorptionsforholdene for et kemisk stof i jord og dets potentielle mobilitet fortsættes med bestemmelse af Freundlich-adsorptionsisotermer for disse systemer, for hvilke Kd kan bestemmes nøjagtigt ved brug af forsøgsprotokollen i denne testmetode. Nøjagtig bestemmelse er mulig, såfremt multiplikation af Kd med forholdet jord/opløsning giver en værdi > 0,3, når målingerne baseres på koncentrationsfaldet i den vandige fase (indirekte metode), hhv. > 0,1 når begge faser analyseres (direkte metode) (61).
1.9.2.5. Trin 3 — Adsorptionsisotermer og desorptionskinetik/desorptionsisotermer
1.9.2.5.1.
Der anvendes fem prøvestofkoncentrationer, helst således at de spænder over to størrelsesordener; valg af disse koncentrationer bør ske på grundlag af vandopløseligheden og den resulterende ligevægtskoncentration i vand. For hver jordtype bør anvendes samme forhold jord/opløsning i hele undersøgelsen. Adsorptionstesten udføres som ovenfor beskrevet, blot med den forskel, at den vandige fase kun analyseres én gang efter forløb af den nødvendige tid til indtræden af ligevægt som tidligere bestemt i trin 2. Ligevægtskoncentrationerne i opløsningen bestemmes, og den adsorberede mængde beregnes enten ud fra svindet af prøvestof i opløsningen eller ved hjælp af den direkte metode. Den adsorberede masse pr. masseenhed jord afsættes som funktion af ligevægtskoncentrationen af prøvestoffet (se »Data og rapportering«).
Resultater af bestemmelsen af adsorptionsisotermer
Af de matematiske adsorptionsmodeller, som hidtil er foreslået, er Freundlich-isotermen den oftest anvendte til beskrivelse af adsorptionsprocesser. Mere udførlige oplysninger om fortolkning og betydning af adsorptionsmodeller er givet i henvisning (41)(45)(S0)(81)(S2).
Anmærkning: Det skal nævnes, at man ikke kan sammenholde værdierne af KF (Freundlich-adsorptionskoefficienten) for forskellige stoffer, medmindre de pågældende KF-værdier udtrykkes i samme enheder (83).
1.9.2.5.2.
Formålet med dette forsøg er at undersøge, om et kemisk stof adsorberes reversibelt eller irreversibelt til en jordtype. Denne oplysning er vigtig, da også desorptionsprocessen spiller en vigtig rolle for et kemisk stofs opførsel i jord på en given lokalitet. Desuden er desorptionsdata nyttige som inddata for computermodeller af et stofs udvaskning og afstrømning i opløst form. Ønskes en desorptionsundersøgelse, anbefales det at udføre nedenstående undersøgelse på hvert system, for hvilket det var muligt at bestemme Kd nøjagtigt i det foregående adsorptionskinetikforsøg.
Ligesom for adsorptionskinetikundersøgelsen kan der vælges: a) den parallelle metode og b) den sekventielle metode. Valg af metode er op til eksperimentatoren, som må tage hensyn til de forhåndenværende laboratoriefaciliteter og ressourcer.
|
a) |
Den parallelle metode: For hver jordtype, som udvælges til desorptionsundersøgelsen, fremstilles prøver med ens jord/opløsning forhold, således at antal prøver er lig det antal tidsintervaller, hvori adsorptionskinetikken ønskes undersøgt. Tidsintervallerne bør helst være de samme som i adsorptionskinetikforsøget; dog kan den samlede tid om nødvendigt forlænges, for at systemet kan nå desorptionsligevægt. I hvert forsøg (én jordtype, én opløsning), køres én blindprøve. Blindprøven består af jord og 0,01 M CaCl2-opløsning uden stofprøve og med henholdsvis vægt og volumen identisk med forsøgets. Som kontrolprøve underkastes prøvestof i 0,01 M CaCl2-opløsning (uden jord) samme testprocedure. Alle blandingerne af jord og opløsning omrystes, til adsorptionsligevægt er nået {som tidligere bestemt i trin 2). Derefter adskilles faserne ved centrifugering, og den vandige fase fjernes så fuldstændigt som muligt. Det fjernede rumfang væske erstattes af samme rumfang 0,01 M CaCl2 uden prøvestof, og de nye blandinger omrystes igen. Den vandige fase i første prøveglas opsamles så fuldstændig som muligt og måles efter f.eks. 2 h, væsken i det andet prøveglas efter 4 h, i det tredje prøveglas efter 6 h, osv., indtil desorptionsligevægt er nået. |
|
b) |
Den sekventielle metode: Efter adsorptionskinetikforsøget centrifugeres blandingen, og den vandige fase fjernes så fuldstændigt som muligt. Det fjernede rumfang væske erstattes af samme rumfang 0,01 M CaCl2 uden prøvestof. Den nye blanding omrystes, indtil desorptionsligevægt er nået. I dette tidsrum centrifugeres blandingen ved fastlagte tidsintervaller, så faserne adskilles. En lille prøve af den vandige fase analyseres straks for prøvestof; derefter fortsætter forsøget med den oprindelige blanding. Rumfanget af hver enkelt prøve skal være mindre end 1 % af det samlede rumfang. Der tilsættes samme mængde frisk fremstillet 0,01 M CaCl2-opløsning til blandingen for at bevare uændret forhold mellem jord og opløsning, og omrystningen fortsætter indtil næste tidsinterval. |
Adsorptionsprocenten beregnes for hvert tidspunkt ( ) og/eller tidsinterval (
) og/eller tidsinterval ( ) (afhængigt af, hvad undersøgelsen kræver) og afsættes mod tiden. Desorptionskoefficienten Kdes ved ligevægt beregnes ligeledes. Alle de ligninger, som skal anvendes, er givet i afsnittet »Data og rapportering« og i tillæg 5.
) (afhængigt af, hvad undersøgelsen kræver) og afsættes mod tiden. Desorptionskoefficienten Kdes ved ligevægt beregnes ligeledes. Alle de ligninger, som skal anvendes, er givet i afsnittet »Data og rapportering« og i tillæg 5.
Resultater af desorptionskinetikforsøget
Fælles afbildninger af desorptionsprocenten 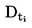 og adsorptionsprocenten
og adsorptionsprocenten  som funktion af tiden giver mulighed for at at vurdere adsorptionsprocessens reversibilitet. Hvis desorptionsligevægt nås. selv om det tager dobbelt så lang tid som at nå adsorptionsligevægt, og hvis den samlede desorption udgør over 75 % af den adsorberede mængde, anses adsorptionen for reversibel.
som funktion af tiden giver mulighed for at at vurdere adsorptionsprocessens reversibilitet. Hvis desorptionsligevægt nås. selv om det tager dobbelt så lang tid som at nå adsorptionsligevægt, og hvis den samlede desorption udgør over 75 % af den adsorberede mængde, anses adsorptionen for reversibel.
1.9.2.5.3.
Freundlich-desorptionsisotermer bestemmes for de jordtyper, som anvendes i forsøget til bestemmelse af adsorptionsisotermer. Desorptionstesten udføres som beskrevet i afsnittet »Desorptionskinetik«, blot med den forskel, at den vandige fase kun analyseres én gang, ved desorptionsligevægt. Den samlede masse desorberet prøvestof beregnes. Indholdet af prøvestof, som stadig er adsorberet til jorden ved desorptionsligevægt, afsættes som funktion af ligevægtskoncentrationen af prøvestoffet i opløsningen (se »Data og rapportering« samt tillæg 5).
2. DATA OG RAPPORTERING
Analysedata fremlægges i tabelform (se tillæg 6). Enkeltmålinger og beregnede gennemsnit angives. Der gives grafiske fremstillinger af adsorptionsisotermerne. Beregningerne foretages som nedenfor beskrevet.
I forsøget sættes vægten af 1 cm3 vandig opløsning til 1 g. Forholdet jord/opløsning kan angives i enheder w/w eller w/vol. med samme talværdi.
2.1. ADSORPTION
Adsorptionen ( ) defineres som den procentdel af det fra forsøgets begyndelse tilstedeværende stof, som adsorberes på jorden under de givne forsøgsbetingelser. Såfremt prøvestoffet er stabilt og ikke i nævneværdig grad adsorberes til beholderens væg, beregnes adsorptionsprocenten
) defineres som den procentdel af det fra forsøgets begyndelse tilstedeværende stof, som adsorberes på jorden under de givne forsøgsbetingelser. Såfremt prøvestoffet er stabilt og ikke i nævneværdig grad adsorberes til beholderens væg, beregnes adsorptionsprocenten  til hvert af tidspunkterne ti efter ligningen:
til hvert af tidspunkterne ti efter ligningen:
|
|
(3) |
hvor:
|
|
= |
adsorptionsprocent til tiden ti ( %) |
|
|
= |
masse af prøvestof adsorberet til jorden til tiden ti (μg) |
|
m0 |
= |
masse af prøvestof i prøveglasset ved prøvens begyndelse (μg). |
Udførlige oplysninger om, hvordan den procentuelle adsorption 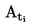 beregnes ved brug af den parallelle og sekventielle metode, findes i tillæg 5.
beregnes ved brug af den parallelle og sekventielle metode, findes i tillæg 5.
Fordelingskoefficienten Kd er forholdet mellem indholdet af stoffet i jordfasen og massekoncentrationen af stoffet i den vandige opløsning under testbetingelserne efter indtræden af ligevægt.
|
|
(4) |
hvor:
|
|
= |
indhold af stof adsorberet på jorden ved adsorptionsligevægt (μg g-1) |
|
|
= |
massekoncentration af stof i vandig fase ved adsorptionsligevægt (μg cm-3). Koncentrationen bestemmes ved analyse under hensyntagen til resultaterne af blindprøverne |
|
|
= |
masse af stof adsorberet på jorden ved adsorptionsligevægt (μg) |
|
|
= |
masse af stof i opløsningen ved adsorptionsligevægt (μg) |
|
msoil |
= |
mængde jordfase, angive; som tor masse af jord (g) |
|
V0 |
= |
begyndelsesrumfang af vandig fase i kontakt med jorden (cm3). |
Forholdet mellem Aeq og Kd er givet ved;
|
|
(5) |
hvor:
|
Aeq |
= |
adsorptionsprocent ved adsorptionsligevægt, %. |
Den normaliserede adsorptionskoefficient for organisk kulstof Koc angiver sammenhængen mellem fordelingskoefficienten Kd og jordprøvens organiske kulstofindhold:
|
|
(6) |
hvor:
|
% oc |
= |
procentdel organisk kulstof i jordprøven (g g-1). |
Koc-koefficienten angiver ved én enkelt værdi fordelingen af hovedsagelig ikke-polære organiske stoffer mellem organisk kulstof i jord eller slam og vand. Adsorptionen af sådanne kemiske stoffer er korreleret med det organiske indhold af det sorberende faste stof (7); således afhænger Koc-værdierae af de særlige karakteristika af humusfraktionerne, hvis sorptionsevne afviger betydeligt som følge af forskelle i oprindelse, tilblivelse osv.
2.1.1. Adsorptionsisotermer
Freundlich-adsorptionsisotermligningen giver sammenhængen mellem den adsorberede mængde prøvestof og koncentrationen af opløst prøvestof ved ligevægt (ligning 8).
Data behandles som angivet under »Adsorption«, og for hvert prøveglas beregnes indholdet af prøvestof adsorberet på jorden efter adsorptionstesten ) (andetsteds betegnet x/m). Det antages, at der er indtrådt ligevægt, og at
) (andetsteds betegnet x/m). Det antages, at der er indtrådt ligevægt, og at 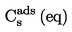 udtrykker ligevægtsværdien:
udtrykker ligevægtsværdien:
|
|
(7) |
Freundlich-adsorptionsligningen er vist i (8):
|
|
(8) |
eller, på lineær form:
|
|
(9) |
hvor:
|
|
= |
Freundlich-adsorptionskoefficient; den har kun dimensionen cm3 g-1, såfremt l/n = 1: i alle andre tilfælde indføres hældningen l/n i dimensionen af |
|
n |
= |
regressionskonstant; l/n ligger sædvanligvis i intervallet 0,7-1,0, hvilket viser, at sorptionsdata ofte er let ulineære. |
Ligning 8 og 9 afsættes, og værdierne af og l/n beregnes ved regressionsanalyse ved anvendelse af ligning 9. Korrelationskoefficienten r2 for den logaritmiske ligning beregnes ligeledes. Et eksempel på sådanne afbildninger er givet i figur 2.
og l/n beregnes ved regressionsanalyse ved anvendelse af ligning 9. Korrelationskoefficienten r2 for den logaritmiske ligning beregnes ligeledes. Et eksempel på sådanne afbildninger er givet i figur 2.

2.1.2. Massebalance
Massebalancen (MB) defineres som den procentdel af den nominelle mængde stof ved testens begyndelse, som kan genfindes analytisk efter en adsorptionstest.
Behandlingen af data vil afvige, hvis det opløste stof er fuldstændig blandbart med vand. For opløsningsmidler blandbare med vand kan man ved at behandle data som beskrevet under »Desorption« bestemme den mængde stof, som genfindes ved ekstraktion med opløsningsmiddel. Er opløsningsmidlet mindre blandbart med vand, må den genfundne mængde bestemmes.
Beregning af massebalancen MB for adsorption: leddet (mE) antages at svare til den samlede masse prøvestof ekstraheret af jorden og fra prøveglassets overflade med organisk opløsningsmiddel:
|
|
(10) |
hvor:
|
MB |
= |
massebalance ( %) |
|
mE |
= |
samlet masse prøvestof ekstraheret fra jord og prøveglassets vægge i to trin (μg) |
|
C0 |
= |
initial massekoncentration af prøveopløsning i berøring med jorden (ug cm-3) |
|
Vrec |
= |
rumfang supernatant, som er opsamlet efter indtræden af adsorptionsligevægt (cm-3). |
2.2. DESORPTION
Desorptionen (D) defineres som den procentdel af den tidligere adsorberede stofmængde, som desorberes under forsøgsbetingelserne:
|
|
(11) |
hvor:
|
|
= |
desorptionsprocent til tidspunktet ti ( %) |
|
|
= |
masse af prøvestof desorberet fra jorden til tiden ti (μg) |
|
|
= |
masse af prøvestof adsorberet til jorden ved adsorptionsligevægt (μg). |
Udførlige oplysninger om, hvordan desorptionsprocenten  beregnes ved brug af den parallelle og sekventielle metode, er givet i tillæg 5.
beregnes ved brug af den parallelle og sekventielle metode, er givet i tillæg 5.
Den tilsyneladende desorptionskoefficient (Kdes) er forholdet mellem indholdet af resterende stof i jordfasen og massekoncentrationen af desorberet stof i den vandige opløsning under testbetingelserne efter indtræden af desorptionsligevægt:
|
|
(12) |
hvor:
|
Kdes |
= |
desorptionskoefficient (cm3 g-1) |
|
|
= |
samlet masse af prøvestof desorberet fra jorden ved desorptionsligevægt (μg) |
|
VT |
= |
samlet masse af vandig fase i kontakt med jorden under desorptionskinetikforsøget (cm3). |
Vejledning tor beregning af (eq) findes i tillæg 5 under overskriften »Desorption«.
(eq) findes i tillæg 5 under overskriften »Desorption«.
Bemærkning:
Hvis den efterfølgende adsorptionstest blev udført med den parallelle metode, sættes rumfanget VT i ligning 12 lig V0.
2.2.1. Desorptionsisotermer
Freundlich-ligningen for adsorptionsisotermer udtrykker sammenhængen mellem den mængde prøvestof, som stadig er absorberet på jorden, og koncentrationen af prøvestof ved desorptionsligevægt (ligning 16).
For hvert prøveglas beregnes indholdet af prøvestof, som stadig er adsorberet til jorden ved desorptionsligevægt, på følgende måde:
|
|
(13) |
 defineres som:
defineres som:
|
|
(14) |
hvor:
|
|
= |
indhold af prøvestof, som ved desorptionsligevægt stadig er adsorberet til jorden (μg g-1) |
|
|
= |
masse af stof, som er bestemt analytisk i den vandige fase ved desorptionsligevægt (μg) |
|
|
= |
masse af prøvestof, som på grund af ufuldstændig volumenudskiftning er tilbage efter indtræden af adsorptionsligevægtt (μg) |
|
|
= |
masse af stof i opløsningen ved adsorptionsligevægt (μg) |
|
|
(15) |
|
|
= |
rumfang opløsning, som ved desorptionsligevægt er udtaget af prøveglasset med henblik på bestemmelse af prøvestof (cm3) |
|
VR |
= |
rumfang supernatant, som efter opnåelse af adsorptionsligevægt er fjernet fra prøveglasset og erstattet af samme rumfang 0,01 M CaCl2-opløsning (cm3). |
Freundlich-desorptionsligningen er vist i (16):
|
|
(16) |
eller, på lineær form:
|
|
(17) |
hvor:
|
|
= |
Freundlich-desorptionskoefficient |
|
n |
= |
regressionskonstant |
|
|
= |
= massekoncentration af stof i vandig fase ved desorptionsligevægt (μg cm-3). |
Ligningerne 16 og 17 kan afsættes grafisk, og værdien af  og l/n beregnes ved regressionsanalyse ved anvendelse af ligning 17.
og l/n beregnes ved regressionsanalyse ved anvendelse af ligning 17.
Bemærkning:
Hvis Freundlich-adsorptions- eller desorptionseksponenten l/n er lig 1, vil Freundlich-bindingskonstanten for adsorption eller desorption ( og
og ) være lig ligevægtskonstanterne (Kd og Kdes) for henholdsvis adsorption og desorption, og ved afbildning af Cs mod Caq fås en ret linje. Er eksponenterne forskellige fra 1, bliver en afbildning af Cs som funktion af Caq ulineær, og adsorptions- og desorptionskonstanterne vil ændre sig langs isotermerne.
) være lig ligevægtskonstanterne (Kd og Kdes) for henholdsvis adsorption og desorption, og ved afbildning af Cs mod Caq fås en ret linje. Er eksponenterne forskellige fra 1, bliver en afbildning af Cs som funktion af Caq ulineær, og adsorptions- og desorptionskonstanterne vil ændre sig langs isotermerne.
2.2.2. Testrapport
Testrapporten bør indeholde følgende oplysninger:
|
a) |
fuldstændig identifikation af de anvendte jordprøver, med angivelse af: |
|
b) |
stedsbestemmelse for lokaliteten (bredde og længde) |
|
c) |
prøveindsamlingsdato |
|
d) |
anvendelsesmønster (f.eks. landbrugsjord, skov osv.) |
|
e) |
dybde af prøvetagning |
|
f) |
sand-/silt-/lerindhold |
|
g) |
pH-værdi (i 0,01 M CaCl2) |
|
h) |
organisk kulstofindhold |
|
i) |
indhold af organisk stof |
|
j) |
kvælstofindhold |
|
k) |
C/N-forhold |
|
l) |
kationbytningskapacitet (mmol/kg) |
|
m) |
udtømmende oplysninger om indsamling og opbevaring af jordprøver |
|
n) |
når det er hensigtsmæssigt, alle relevante oplysninger til fortolkning af prøvestoffets adsorption/desorption |
|
o) |
henvisninger, som angiver de metoder, der er anvendt til bestemmelse af hver parameter |
|
p) |
oplysninger om prøvestoffet, hvis det er hensigtsmæssigt |
|
q) |
forsøgstemperaturen |
|
r) |
centrifugeringsbetingelser |
|
s) |
analysemetode anvendt til analyse af prøvestoffet |
|
t) |
begrundelse af enhver brug af opløsningsmiddel til fremstilling af stamopløsningen af prøvestoffet |
|
u) |
forklaring af eventuelle korrektioner foretaget i beregningerne |
|
v) |
data i henhold til formularbladet (tillæg 6) og grafiske fremstillinger |
|
w) |
alle oplysninger og bemærkninger, som kan være til hjælp ved fortolkning af testresultaterne. |
3. HENVISNINGER
|
(1) |
Kukowski H. and Brummer G., (1987). Investigations on the Adsorption and Desorption of Selected Chemicals in Soils. UBA Report 106 02 045, Part II. |
|
(2) |
Fränzle O., Kuhnt G. and Vetter L., (1987). Selection of Representative Soils in the EC-Territory. UBA Report 106 02 045, Part I. |
|
(3) |
Kuhnt G. and Muntau H. (Eds.) EURO-Soils: Identification, Collection, Treatment, Characterisation. Special Publication No 1.94.60, Joint Research Centre. European Commission, ISPRA, December 1994. |
|
(4) |
OECD Test Guidelines Programme, Final Report of the OECD Workshop on Selection of Soils/Sediments, Belgirate, Italy, 18.-20. januar 1995 (June 1995). |
|
(5) |
US Environment Protection Agency: Pesticide Assessment Guidelines, Subdivision N, Chemistry: Environmental Fate, Series 163-1, Leaching and Adsorption/Desorption Studies, Addendum 6 on Data Reporting, 540/09-88-096, Date: 1/1988. |
|
(6) |
US Environment Protection Agency: Prevention, Pesticides and Toxic Substances, OPPTS Harmonized Test Guidelines, Series 835-Fate. Transport and Transformation Test Guidelines, OPPTS No: 835.1220 Sediment and Soil Adsorption/Desorption Isotherm. EPA No: 71 2-C-96-048, April 1996. |
|
(7) |
ASTM Standards, E 1195-85, Standard Test Method for Determining a Sorption Constant (Koc) for an Organic Chemical in Soil and Sediments. |
|
(8) |
Agriculture Canada: Environmental Chemistry and Fate. Guidelines for registration of pesticides in Canada, 15 July 1987. |
|
(9) |
Netherlands Commission Registration Pesticides (1995): Application for registration of a pesticide. Section G. Behaviour of the product and its metabolites in soil, water and air. |
|
(10) |
Danish National Agency of Environmental Protection (October 1988): Criteria for registration of pesticides as especially dangerous to health or especially harmful to the environment. |
|
(11) |
BBA (1990), Guidelines for the Official Testing of Plant Protection Products, Biological Research Centre for Agriculture and Forestry, Braunschweig, Germany. |
|
(12) |
Calvet R., (1989), ’Evaluation of adsorption coefficients and the prediction of the mobilities of pesticides in soils’, in Methodological Aspects of the Study of Pesticide Behaviour in Soil (ed. P. Jamet), INRA, Paris, (Review). |
|
(13) |
Calvet R., (1980), ’Adsorption-Desorption Phenomena’ in Interactions between herbicides and the soil, (R. . Hance ed.), Academic Press, London, pp. 83-122. |
|
(14) |
Hasset J.J., and Banwart W.L., (1989), The sorption of nonpolar organics by soils and sediments' in Reactions and Movement of Organic Chemicals in Soils. Soil Science Society of America (S.S.S.A), Special Publication no. 22, pp. 31-44. |
|
(15) |
van Genuchten M.Th., Davidson J. M., and Wierenga P.J., (1974), ’An evaluation of kinetic and equilibrium equations for the prediction of pesticide movement through porous media’. Soil Sci. Soc. Am. Proc, Vol. 38(1), pp. 29-35. |
|
(16) |
McCall P.J., Laskowski D.A., Swann R.L., and Dishburger H.J., (1981), ’Measurement of sorption coefficients of organic chemicals and their use, in environmental fate analysis’, in Test Protocols for Environmental Fate and Movement of Toxicants. Proceedings of AOAC Symposium, AOAC, Washington DC. |
|
(17) |
Lambert S.M., Porter P. E., and Schieferrstein R. H., (1965), ’Movement and sorption of chemicals applied to the soil’. Weeds, 13, pp. 185-190. |
|
(18) |
Rhodes R.C, Belasco I.J., and Pease H.L., (1970) ’Determination of mobility and adsorption of agrochemicals in soils’. J.Agric.Food Chem., 18, pp. 524-528. |
|
(19) |
Russell M.H., (1995), ’Recommended approaches to assess pesticide mobility in soil’ in Environmental Behavior of Agrochemicals (ed. T.R. Roberts and P.C. Kearney), John Wiley & Sons Ltd. |
|
(20) |
Esser H.O., Hemingway R.J., Klein W., Sharp D.B., Vonk J.W. and Holland P.T., (1988), ’Recommended approach to the evaluation of the environmental behavior of pesticides’, IUPAC Reports on Pesticides (24). Pure Appl. Chem., 60, pp. 901-932. |
|
(21) |
Guth J.A., Burkhard N., and D.O. Eberle, (1976), ’Experimental models for studying the persistence of pesticides in soils’. Proc. BCPC Symposium: Persistence of Insecticides and Herbicides, pp. 137-157, BCPC, Surrey, UK. |
|
(22) |
Furminge C.G.L., and Osgerby J.M., (1967), ’Persistence of herbicides in soil’. J. Sci. Fd Agric, 18, pp. 269-273. |
|
(23) |
Burkhard N., and Guth J.A., (1981), ’Chemical hydrolysis of 2-Chloro-4,6-bis(alkyiamino)-l,3,5-triazine herbicides and their breakdown in soil under the influence of adsorption’, Pestic. Sci. 12, pp. 45-52. |
|
(24) |
Guth J.A., Gerber H.R., and Schlaepfer T., (1977), ’Effect of adsorption, movement and persistence on the biological availability of soil-applied pesticides’. Proc. Br. Crop Prot. Conf., 3, pp. 961-971. |
|
(25) |
Osgerby J.M., (1973), ’Process affecting herbicide action in soil’. Pestic. Sci., 4, pp. 247-258. |
|
(26) |
Guth J.A., (1972), ’Adsorptions- und Einwascheverhalten von Pflanzenschutzmitteln in Boden’. Schr. Reihe Ver. Wass.-Boden-Lufthyg. Berlin-Dahlem, Heft 37, pp. 143-154. |
|
(27) |
Hamaker J.W., (1975), ’The interpretation of soil leaching experiments’. in Environmental Dynamics of Pesticides (eds R. Haque and V.H. freed), pp. 135-172, Plenum Press, NY. |
|
(28) |
Helling C.S., (1971), ’Pesticide mobility in soils’. Soil Sci. Soc. Amer. Proc., 35, pp. 732-210. |
|
(29) |
Hamaker J.W., (1972), ’Diffusion and volatilization’ in Organic chemicals in the soil environment (C.A.I. Goring and J. W. Hamaker eds), Vol. I, pp. 49-143. |
|
(30) |
Burkhard N. and Guth J.A., (1981), ’Rate of volatilisation of pesticides from soil surfaces; Comparison of calculated results with those determined in a laboratory model system’. Pestic. Sci. 12, pp. 37-44. |
|
(31) |
Cohen S.Z., Creeger S.M., Carsel R.F., and Enfield C.G., (1984), ’Potential pesticide contamination of groundwater from agricultural uses’, in Treatment and Disposal of Pesticide Wastes, pp. 297-325, Acs Symp. Ser. 259, American Chemical Society, Washington, DC. |
|
(32) |
Gustafson D.J., (1989), ’Groundwater ubiquity score: a simple method for assessing pesticide leachability’. J. Environ. Toxic. Chem., 8(4), pp. 339-357. |
|
(33) |
Leistra M., and Dekkers W.A., (1976). ’Computed effects of adsorption kinetics on pesticide movement in soils’. J. of Soil Sci., 28, pp. 340-3 50. |
|
(34) |
Bromilov R.H., and Leistra M., (1980), ’Measured and simulated behavior of aldicarb and its oxydation products in fallow soils’. Pest. Sci., 11, pp. 389-395. |
|
(35) |
Green R.E., and Karickoff S.W., (1990), ’Sorption estimates for modeling’, in Pesticides in the Soil Environment: Process, Impacts and Modeling (ed. H.H. Cheng). Soil Sci. Soc. Am., Book Series no. 2, pp. 80-101. |
|
(36) |
Lambert S. M., (1967), ’Functional relationship between sorption in soil and chemical structure’. J. Agri. Food Chem., 15, pp. 572-576. |
|
(37) |
Hance R.J., (1969), ’An empirical relationship between chemical structure and the sorption of some herbicides by soils’. J. Agri. Food Chem., 17, pp. 667-668. |
|
(38) |
Briggs G.G. (1969), ’Molecular structure of herbicides and their sorption by soils’. Nature, 223, 1288. |
|
(39) |
Briggs G.G. (1981). Theoretical and experimerital relationships between soil adsorption, octanol-water partition coefficients, water solubilities, bioconcentration factors, and the parachor'. J. Agric. Food Chem., 29, pp. 1050-1059. |
|
(40) |
Sabljic A., (1984), ’Predictions of the nature and strength of soil sorption of organic polutance by molecular topology’. J. Agric. Food Chem., 32, pp. 243-246. |
|
(41) |
Bailey G. W., and White J.L., (1970), ’Factors influencing the adsorption, desorption, and movement of pesticides in soil’. Residue Rev., 32, pp. 29-92. |
|
(42) |
Bailey G.W., J.L White and Y. Rothberg., (1968), ’Adsorption of organic herbicides by montomorillonite: Role of pH and chemical character of adsorbate’. Soil Sci. Soc. Amer. Proc. 32: pp. 222-234. |
|
(43) |
Karickhoff S.W., (1981), ’Semi-empirical estimation of sorption of hydrophobic pollutants on natural sediments and soils’. Chemosphere 10, pp. 833-846. |
|
(44) |
Paya-Perez A., Riaz M. and Larsen B., (1989), ’Soil Sorption of 6 Chlorobenzenes and 20 PCB Congeners’. Environ. Toxicol. Safety 21, pp. 1-17. |
|
(45) |
Hamaker J.W., and Thompson J.M., (1972), ’Adsorption in organic chemicals’ in Organic Chemicals in the Soil Environment (Goring C.A.I. and Hamaker J.W., eds), Vol I and II, Marcel Dekker, Inc., New York, NY, 1972, pp. 49-143. |
|
(46) |
Deli J., and Warren G.F., 1971, ’Adsorption, desorption and leaching of diphenamid in soils’. Weed Sci. 19; pp. 67-69. |
|
(47) |
Chu-Huang Wu, Buehring N., Davinson J.M. and Santelmann, (1975), ’Napropamide Adsorption, desorption and Movement in soils’. Weed Science, Vol. 23, pp. 454-457. |
|
(48) |
Haues M.H.B., Stacey M., and Thompson J. M., (1968), ’Adsorption of s-triazine herbicides by soil organic preparations’ in Isotopes and Radiation in Soil Organic Studies, p.75, International. Atomic Energy Agency, Vienna. |
|
(49) |
Pionke H.B.. and Deangelis R.J., (1980), ’Methods for distributing pesticide loss in field run-off between the solution and adsorbed phase’, CREAMS, in A Field Scale Model for Chemicals, Run-off and Erosion from Agricultural Management Systems, Chapter 19, Vol. III: Supporting Documentation, USDA Conservation Research report. |
|
(50) |
ISO Standard Compendium Environment: Soil Quality — General aspects; chemical and physical methods of analysis: biological methods of analysis. First Edition (1994). |
|
(51) |
Scheffer F., and Schachtschabel, Lehrbuch der Bodenkunde, F. Enke Verlag, Stuttgart (1982), 11th edition. |
|
(52) |
Black, Evans D.D., White J.L., Ensminger L.E., and Clark F.E., eds. ’Methods of Soil Analysis’, Vol 1 and 2, American Society of Agronomy, Madison, WI, 1982. |
|
(53) |
ISO/DIS 10381-1 Soil Quality — Sampling — Part 1: Guidance on the design of sampling programmes. |
|
(54) |
ISO/DIS 10381-2 Soil Quality — Sampling — Part 2: Guidance on sampling techniques. |
|
(55) |
ISO/DIS 10381-3 Soil Quality — Sampling — Part 3: Guidance on safety of sampling. |
|
(56) |
ISO/DIS 10381-4 Soil Quality — Sampling — Part 4: Guidance on the investigation of natural and cultivated soils. |
|
(57) |
ISO/DIS 10381-5 Soil Quality — Sampling — Part 5: Guidance on the investigation of soil contamination of urban and industrial sites. |
|
(58) |
ISO 10381-6, 1993: Soil Quality — Sampling — Part 6: Guidance on the collection, handling and storage of soil for the assessment of aerobic microbial processes in the laboratory. |
|
(59) |
Green R.E., and Yamane V.K., (1970), ’Precision in pesticide adsorption measurements’. Soil Sci. Am. Proc., 34, pp. 353-354. |
|
(60) |
Grover R., and Hance R.J. (1970), ’Effect of ratio of soil to water on adsorption of linuron and atrazine’. Soil Sci., pp. 109-138. |
|
(61) |
Boesten, J.J.T.I, ’Influence of soil/liquid ratio on the experimental error of sorption coefficients in pesticide/soil system’. Pest. Sci. 1990, 30, pp. 31-41. |
|
(62) |
Boesten, J.J.T.I. ’Influence of soil/liquid ratio on the experimental error of sorption coefficients in relation to OECD guideline 106’. Proceedings of 5th international workshop on environmental behaviour of pesticides and regulatory aspects, Brussels, 26-29 April 1994. |
|
(63) |
Bastide J., Cantier J.M., et Coste C., (1980), ’Comportement de substances herbicides dans le sol en fonction de leur structure chimique’. Weed Res. 21, pp. 227-231. |
|
(64) |
Brown D.S., and Flagg E.W., (1981), ’Empirical prediction of organic pollutants sorption in natural sediments’, Environ.Qual., 10(3), pp. 382-3S6. |
|
(65) |
Chiou C.T., Porter P.E., and Schmedding D.W., (1983), ’Partition equilibria of non-ionic organic compounds between soil organic matter and water’. Environ. Sci. Technol., 17(4), pp. 227-231. |
|
(66) |
Gerstl Z., and Mingelgrin U., (1984), ’Sorption of organic substances by soils and sediments’. J. Environm. Sci. Health, Bl 9 (3), pp. 297-312. |
|
(67) |
Vowles P.D., and Mantoura R.F.C, (1987), ’Sediment-water partition coefficient and HPLC retention factors of aromatic hydrocarbons’. Chemosphere, 16(1), pp. 109-116. |
|
(68) |
Lyman W.J., Reehl W.F.and Rosenblatt D.H. (1990). Handbook of Chemical Property Estimation Methods. Environmental Behaviour of Organic Compounds. American Chemical Society, Washington DC. |
|
(69) |
Keniga E.E., and Goring, C.A. I. (1980). ’Relationship between water solubility, soil sorption, octanol-water partitioning and concentration of chemicals in the biota’ in Aquatic Toxicology (eds),G. Eaton, et al.), pp.78-115, ASTM STP 707, Philadelphia. |
|
(70) |
Chiou C.T., Peters L.J., and Freed V.H., (1979), ’A physical concept of soil-water equilibria for non-ionic organic compounds’. Science, Vol. 206, pp. 831-832. |
|
(71) |
Hassett J.J., Bamvart W. J., Wood S.G., and Means J.C, (1981), ’Sorption of/-Naphtol: implications concerning the limits of hydrophobic sorption’. Soil Sci. Soc. Am. J. 45, pp. 38-42. |
|
(72) |
Karickhoff S.W., (1981), ’Semi-empirical estimation of sorption of hydrophobic pollutants on natural sediments and soils’. Chemosphere. Vol. 10(8), pp. 833-846. |
|
(73) |
Moreale A.. van Bladel R., (1981), ’Adsorption de 13 herbicides et insecticides par le sol. Relation solubilité-reactivité’. Revue de l'Agric. 34 (4), pp. 319-322. |
|
(74) |
Muller M.. Kordel W. (1996), 'Comparison of screening methods for the determination/estimation of adsorption coefficients on soil. Chemosphere. 32(12), pp, 2493-2504. |
|
(75) |
Kordel W. Kotthoff G., Muller M. (1995), ’HPLC — screening method for the determination of the adsorption coefficient on soil — results of a ring test’. Chemosphere 30 (7), pp. 1373-1384. |
|
(76) |
Kordel W., Stutte J., Kotthoff G. (1993), ’HPLC — screening method for the determination of the adsorption coefficient on soil — comparison of different stationary phases’. Chemosphere 27 (12), pp. 2341-2352. |
|
(77) |
Hance, R.J., (1967), ’The Speed of Attamment of Sorption Equilibria in Some Systems Involving Herbicides’. Weed Research, Vol. 7, pp. 29-36. |
|
(78) |
Koskinen W.C, and Harper S.S., (1990), ’The retention processes: mechanisms’ in Pesticides in the Soil Environment: Processes, Impacts and Modelling (ed. H.H. Cheng). Soil Sci. Soc. Am. Book Series, No. 2, Madison, Wisconsin. |
|
(79) |
Cohen S.Z., Creeger S. M., Carsel R.F., and Enfield C.G. (1984), ’Potential pesticide contamination of groundwater from agricultural uses’, in Treatment and Disposal of Pesticide Wastes, pp.297-325, ACS Symp. Ser. 259, American Chemical Society, Washington, DC. |
|
(80) |
Giles C.H., (1970), ’Interpretation and use of sorption isotherms’ in Sorption and Transport Processes in Soils. S.C.I. Monograph No. 37, pp. 14-32. |
|
(81) |
Giles, C.H; McEwan J.H.; Nakhwa, S.N. and Smith, D, (1960), ’Studies in adsorption: XI. A system of classification of solution adsorption isotherms and its use in the diagnosis of adsorption mechanisms and in measurements of pesticides surface areas of soils’. J. Chem. Soc, pp. 3973-93. |
|
(82) |
Calvet R., Tercé M., and Arvien J.C, (1980), ’Adsorption des pesticides par les sols et leurs constituants: 3. Caractéristiques générales de l'adsorption’. Ann. Agron. 31: pp. 239-251. |
|
(83) |
Bedbur E., (1996). Anomalies in the Freundlich equation', Proc. COST 66 Workshop, Pesticides in soil and the environment, 13-15 May 1996, Stratford-upon-Avon, UK. |
|
(84) |
Guth, J.A., (1985), ’Adsorption/desorption’, in Joint international Symposium, Physicochemical Properties and their Role in Environmental Hazard Assessment, July 1-3, Canterbury, UK. |
|
(85) |
Soil Texture Classification (US and FAO systems): Weed Science, 33, Suppl. 1 (1985) and Soil Sci. Soc. Arner. Proc. 26:305 (1962). |
Tillæg 1
Testplan

Tillæg 2
BETYDNINGEN AF ANALYSEMETODENS NØJAGTIGHED OG KONCENTRATIONSÆNDRINGER FOR ADSORPTIONSRESULTATERNES NØJAGTIGHED
Af følgende tabel (84) ses, at når der er meget lille forskel mellem prøvestoffets begyndelsesmasse (m0 =110 μg) og ligevægtsmasse ( =100 μg) i opløsningen, vil en fejl på 5 % på ligevægtskoncentrationsmålingen føre til en fejl på 50 % på beregningen af den stofmængde, som er adsorberet på jorden (
=100 μg) i opløsningen, vil en fejl på 5 % på ligevægtskoncentrationsmålingen føre til en fejl på 50 % på beregningen af den stofmængde, som er adsorberet på jorden ( ) og på 52,4 % på beregningen af Kd.
) og på 52,4 % på beregningen af Kd.
|
Mængde jord |
mjord |
= 10 g |
|
Opløsningens rumfang |
V0 |
=100 cm3 |
|
|
FOR-L_2008142DA.01044401.notes.0187.xml.jpg (μg) |
FOR-L_2008142DA.01044401.notes.0188.xml.jpg (μg cm-3) |
R |
FOR-L_2008142DA.01044401.notes.0189.xml.jpg (μg) |
FOR-L_2008142DA.01044401.notes.0190.xml.jpg (μg g-1) |
R‡ |
Kd* |
R‡ |
|
|
FOR A = 9 % |
|||||||
|
m0= 110 μg eller C0=1,100 μg/cm3 |
100 |
1,000 |
sand værdi |
10 |
1,00 |
sand værdi |
1 |
|
|
101 |
1,010 |
1 % |
9 |
0,90 |
10 % |
0,891 |
10,9 % |
|
|
105 |
1,050 |
5 % |
5 |
0,50 |
50 % |
0,476 |
52,4 % |
|
|
109 |
1,090 |
9 % |
1 |
0,10 |
90 % |
0,092 |
90,8 % |
|
|
|
FOR A = 55 % |
|||||||
|
m0= 110 μg eller C0=1,100 μg/cm3 |
50,0 |
0,500 |
sand værdi |
60,0 |
6,00 |
sand værdi |
12,00 |
|
|
50,5 |
0,505 |
1 % |
59,5 |
5,95 |
0,8 % |
11,78 |
1,8 % |
|
|
52,5 |
0,525 |
5 % |
57,5 |
5,75 |
4,0 % |
10,95 |
8,8 % |
|
|
55,0 |
0,550 |
10 % |
55,0 |
5,50 |
8,3 % |
10,00 |
16,7 % |
|
|
|
FOR A = 99 % |
|||||||
|
m0 = 110 μg eller C0=1,100 μg/cm3 |
1,100 |
0,011 |
sand værdi |
108,9 |
10,89 |
sand værdi |
990 |
|
|
1,111 |
0,01111 |
1 % |
108,889 |
10,8889 |
0,01 % |
980 |
1,0 % |
|
|
1,155 |
0,01155 |
5 % |
108,845 |
10,8845 |
0,05 % |
942 |
4,8 % |
|
|
1,21 |
0,0121 |
10 % |
108,790 |
10,8790 |
0,10 % |
899 |
9,2 % |
|
Hvor:
|
|
= |
|
|
|
= |
masse af prøvestof i jordfasen ved ligevægt, μg |
|
|
= |
masse af prøvestof i vandfasen ved ligevægt, μg |
|
|
= |
indhold af prøvestof i jordfasen ved ligevægt, μg g-1 |
|
|
= |
massekoncentration af prøvestoffet i vandig fase ved ligevægt, μg cm-3 |
|
R |
= |
analysefejl på bestemmelse af |
|
R‡ |
= |
beregnet fejl som følge af analysefejl R. |
Tillæg 3
TEKNIKKER TIL SKØNSMÆSSIG BEREGNING AF Kd
|
1. |
Ved hjælp af teknikker til skønsmæssig beregning kan størrelsen af Kd forudsiges på grundlag af korrelationer med f.eks. værdier af Pow (12)(39)(63-68), vandopløselighedsdata (12)(19)(21)(39)(68-73) eller polaritetsdata afledt ved brug af omvendt fase HPLC (74-76). Som vist i tabel 1 og 2 beregnes Koc eller Kom og dermed indirekte Kd af ligningerne:
|
|
2. |
Princippet om disse korrelationer hviler på to forudsætninger: (1) et stofs adsorption er hovedsagelig bestemt af jordens organiske stofindhold, og (2) de indgående vekselvirkninger er hovedsagelig af ikke-polær art. For disse korrelationer gælder følgelig: (1) de gælder ikke eller kun i nogen udstrækning for polære stoffer, og (2) de gælder ikkeved meget lille organisk stofindhold i jorden (12). Desuden gælder, at skønt der er fundet tilfredsstillende korrelation mellem Pow og adsorptionen (19), kan det samme ikke siges om forholdet mellem vandopløselighed og adsorptionsgrad (19)(21); undersøgelser heraf har hidtil givet meget modstridende resultater. |
|
3. |
Nogle eksempler på korrelationen mellem adsorptionskoefficient og oktanol-vand fordelingskoefficient samt vandopløselighed er givet henholdsvis i tabel 1 og 2. Tabel 1 Eksempler på korrelation mellem adsorptionsfordelingskoefficienten og oktanol-vand fordelingskoefficienten; flere eksempler er givet i (12) og (68)
Tabel 2 Eksempler på korrelationen mellem adsorptionsfordelingskoefficient og vandopløselighed; flere eksempler er givet i (68) og (69)
|
Tillæg 4
BEREGNINGER TIL FASTLÆGGELSE AF CENTRIFUGERINGSBETINGELSER
|
1. |
Centrifugeringstiden er givet ved følgende formel, idet partiklerne forudsættes at være kugleformede:
For enkelhedens skyld er alle parametre angivet i ikke-SI-enheder (g, cm) hvor
I praksis fordobler man sædvanligvis de beregnede tider for at sikre fuldstændig separation. |
|
2. |
Ligning 1 kan forenkles yderligere, hvis opløsningens viskositet (η) og massefylde (ρaq) sættes lig viskositeten og massefylden afvand ved 25 oC, η = 8,95 × 10-3 g s-1 cm-1 og ρaq = 1,0 g cm-3. Centrifugeringstiden er da givet ved ligning 2:
|
|
3. |
Af ligning 2 ses, at to parametre er vigtige ved fastlæggelse af centrifugeringsbetingelseme, nemlig tid (t) og hastighed (rpm), når man vil udskille partikler med en given størrelse (i vort tilfælde radius 0,1 μm): (1) jordens massefylde og (2) længden af blandingen i centrifugeglasset (Rb-Rt), dvs. den distance, som en jordpartikel tilbagelægger fra overfladen af opløsningen til glassets bund; for et givet rumfang er det klart, at længden af blandingen i røret bestemmes af kvadratet på rørets radius. |
|
4. |
Figur 1 viser sammenhængen mellem centrifugeringstiden (t) og centrifugeringshastigheden (rpm) for forskellige jordmassefylder (ps (Figur la) og forskellige længder af blandingen i centrifugeglassene (Figur 2a). Af figur la ses betydningen af jordens massefylde tydeligt; for en typisk centrifugeringshastighed på 3000 rpm er centrifugeringstiden ca.240 min for jord med massefylde 1,2 g cm men kun 50 min., når massefylden er 2,0 g cm Tilsvarende ses af figur 1b, at for en typisk centrifugeringshastighed på 3000 rpm er centrifugeringstiden ca. 50 min for en længde af blandingen på 10 cm men kun 7 min for en længde på 1 cm. Det er imidlertid vigtigt at finde et optimalt forhold mellem den centrifugering, som kræver mindst mulig længde, og den. som gør det let for eksperimentatoren at adskille faserne efter centrifugering. |
|
5. |
Ved fastlæggelse af forsøgsbetingelserne med henblik på adskillelse af fast og flydende fase er det desuden vigtigt at tage hensyn til eventuel tilstedeværelse af en tredje »pseudo«-fase, kolloiderne. Disse partikler, der er under 0,2 μm, kan i væsentlig grad påvirke hele absorptionsmekanismen for et stof i en jordsuspension. Når der centrifugeres som ovenfor beskrevet, forbliver kolloiderne i den vandige fase og bliver analyseret sammen med den vandige fase. Derfor går oplysningerne om deres indflydelse tabt. Hvis det laboratorium, som forestår undersøgelsen, har faciliteter til ultracentrifugering eller ultrafiltrering, kan adsorption/desorption af et stof i jord undersøges mere tilbundsgående, herunder stoffets adsorption på kolloiderne, I så tilfælde bør der anvendes ultracentrifugering ved 60 000 rpm eller ultrafiltrering med en filterporøsitet på 100 000 Dalton til at adskille de tre faser jord, kolloider og opløsning. Den forsøgsprotokol bør desuden ændres tilsvarende, således at alle tre faser analyseres for stoffet. |
Figur 1a.
Fig, 1b.

Tillæg 5
BEREGNING AF ADSORPTION A ( %) OG DESORPTION D ( %)
Tidsplanen for proceduren er følgende:

Ved alle beregninger er forudsat, at prøvestoffet er stabilt og ikke adsorberes nævneværdigt til beholderens vægge.
ADSORPTION A (A %)
a) Den parallelle metode
Adsorptionsprocenten beregnes for hvert prøveglas (i) ved hvert tidspunkt (ti) efter ligningen:
|
|
(1) |
Leddene i denne ligning kan beregnes på følgende måde:
|
m0 = C0 · V0 (μg) |
(2) |
|
|
(3) |
hvor:
|
|
= |
adsorptionsprocent ( %) til tiden ti |
|
|
= |
masse af prøvestof adsorberet til jorden på det tidspunkt ti, da analysen udføres (μg); |
|
m0 |
= |
masse af prøvestof i prøveglasset ved prøvens begyndelse (μg) |
|
C0 |
= |
initial massekoncentration afprøveopløsning i berøring med jorden (μg cm-3) |
|
|
= |
massekoncentration af stof i vandig fase på det tidspunkt ti, da analysen udføres (μg cm-3); denne koncentration bestemmes ved analyse med hensyntagen til blindværdierne |
|
V0 |
= |
initialt rumfang af prøveopløsning i kontakt med jorden (cm3). |
Værdierne af adsorptionsprocenten  eller
eller afsættes på en kurve mod tiden, og tiden til indtræden af sorptionsligevægt bestemmes. Eksempler på sådanne kurver er givet i hhv. figur 1 og figur 2.
afsættes på en kurve mod tiden, og tiden til indtræden af sorptionsligevægt bestemmes. Eksempler på sådanne kurver er givet i hhv. figur 1 og figur 2.

b) Den sekventielle metode
I de følgende ligninger er der taget hensyn til, at adsorptionsbestemmelsen sker ved måling af prøvestoffet i små prøver af vandig fase til bestemte tidsintervaller.
|
— |
Inden for hvert tidsinterval beregnes mængden af stof adsorberet på jorden på følgende måde:
|
|
— |
Adsorptionsprocenten i hvert tidsinterval,
medens adsorptionsprocenten
Adsorptionsværdierne |
|
— |
Ved ligevægtstidspunktet teq:
|
De ovenfor anvendte parametre er defineret som følger:
|
|
= |
masse af prøvestof adsorberet til jorden henholdsvis i løbet af tidsintervallerne Δt1, Δt2,..., Δtn (μg); |
|
|
= |
masse af prøvestof målt i en prøve |
|
|
= |
masse af stof adsorberet på jorden ved adsorptionsligevægt (µg) |
|
|
= |
masse af stof i opløsningen ved adsorptionsligevægt (µg) |
|
|
= |
rumfang af den prøve, hvori prøvestoffet måles (cm3) |
|
|
= |
adsorptionsprocent svarende til et tidsinterval Δti (%) |
|
|
= |
adsorptionsprocent ved adsorptionsligevægt (%). |
DESORPTION D (%)
Tidspunktet t0 for start af desorptionskinetikforsøget regnes som det tidspunkt, hvor det maksimale opsamlede rumfang af prøvestofopløsningen (efter indtræden af adsorptionsligevægt) erstattes et tilsvarende rumfang 0,01 M CaCl2-opløsning.
a) Den parallelle metode
Til tiden ti måles massen af prøvestof i den vandige fase udtaget af prøveglas i ( ).og den desorberede masse beregnes af ligningen:
).og den desorberede masse beregnes af ligningen:
|
|
(13) |
Ved desorptionsligevægt er ti = teq og derfor  =
= 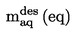 .
.
Massen af prøvestof desorberet i et tidsinterval (Δt1) er givet ved ligningen:
|
|
(14) |
Desorptionsprocenten beregnes:
til tidspunktet ti af ligningen:
|
|
(15) |
og i et tidsinterval (Δt1) af ligningen:
|
|
(16) |
hvor:
|
|
= |
desorptionsprocent til tidspunktet ti ( %); |
|
|
= |
desorprionsprocent svarende til et tidsinterval Δti ( %)masse af prøvestof desorberet til tidspunktet ti (μg) |
|
|
= |
masse af prøvestof desorberet til tidspunktet ti (µg) |
|
|
= |
masse af prøvestof desorberet af jorden i løbet af et tidsinterval Δt1 (μg) |
|
|
= |
masse af prøvestof bestemt analytisk til et tidspunkt ti i et rumfang opløsning |
|
|
= |
masse af prøvestof, som er til overs efter indtræden af adsorptionsligevægt som følge af ufuldstændig volumenudskiftning (μg) |
|
|
(17) |
|
|
= |
masse af prøvestof i opløsningen ved adsorptionsligevægt (µg) |
|
VR |
= |
rumfang supernatant, som efter opnåelse af adsorptionsligevægt er fjernet fra prøveglasset og erstattet af samme rumfang 0,01 M CaCl2-opløsning (cm3) |
|
|
= |
rumfang af opløsning udtaget af prøveglas (i) til bestemmelse af prøvestof i desorptionskinetikforsøget (cm3). |
Værdierne af desorptionen 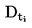 eller
eller  (afhængigt af, hvad der er nødvendigt i undersøgelsen) afsættes mod tiden, og tidsrummet til indtræden af sorptionsligevægt bestemmes.
(afhængigt af, hvad der er nødvendigt i undersøgelsen) afsættes mod tiden, og tidsrummet til indtræden af sorptionsligevægt bestemmes.
b) Den sekventielle metode
I følgende ligninger er der taget hensyn til, at adsorptionsbestemmelsen sker ved måling af prøvestoffet i små prøver  af vandig fase (den sekventielle metode i 1.9. Testens præstationer). Forudsætninger: a) det rumfang supernatant, som er fjernet fra glasset efter adsorptionskinetikforsøget er erstattet af samme rumfang 0,01 M CaCl2-opløsning (VR), og b) det samlede rumfang vandig fase i kontakt med jorden (VT) under desorptionskinetikforsøget forbliver uændret og er givet ved ligningen:
af vandig fase (den sekventielle metode i 1.9. Testens præstationer). Forudsætninger: a) det rumfang supernatant, som er fjernet fra glasset efter adsorptionskinetikforsøget er erstattet af samme rumfang 0,01 M CaCl2-opløsning (VR), og b) det samlede rumfang vandig fase i kontakt med jorden (VT) under desorptionskinetikforsøget forbliver uændret og er givet ved ligningen:
|
|
(18) |
Til tidspunktet ti:
|
— |
måles massen af prøvestof i en lille prøve
|
|
— |
ved desorptionsligevægt er ti = teq og derfor |
|
— |
kan desorptionsprocenten
|
I et tidsinterval (Δt1):
beregnes mængden af stof desorberet i for hvert tidsinterval på følgende måde:
|
— |
for det første tidsinterval Δt1 = ti-t0
|
|
— |
for det andet tidsinterval Δt1 = t2-t1
|
|
— |
for det n'te tidsinterval Δtn = tn-tn-1
|
Endelig beregnes desorptionsprocenten i hvert tidsinterval,  ved hjælp af følgende ligning:
ved hjælp af følgende ligning:
|
|
(24) |
medens desorptionsprocenten  , til tidspunktet ti er givet ved ligningen:
, til tidspunktet ti er givet ved ligningen:
|
|
(25) |
hvor de ovenfor anvendte parametre er defineret således:
|
|
= |
masse af prøvestof desorberet henholdsvis i løbet af tidsintervallet Δt1, Δt2, ..., Δtn (μg) |
||
|
|
= |
masse af prøvestof målt i en prøve henholdsvis til tidspunktet t1, t2, ..., tn (μg) |
||
|
|
= |
masse af prøvestof målt i en prøve |
||
|
VT |
= |
samlet rumfang af vandig fase i kontakt med jorden under desorptionskinetikforsøget udført med den sekventielle metode (cm3) |
||
|
|
= |
masse af prøvestof, som er til overs efter indtræden af adsorptionsligevægt som følge af ufuldstændig volumenudskiftning (μg)
|
||
|
VR |
= |
rumfang supernatant, som efter indtræden af adsorptionsligevægt er fjernet fra prøveglasset og erstattet af samme rumfang 0,01 M CaCl2-opløsnmg (cm3) |
||
|
|
= |
rumfang af prøve udtaget af prøveglas (i) til analyseformål i desorptionskinetikforsøget udført med den sekventielle metode (cm3)
|
Tillæg 6
ADSORPTION-DESORPTION I JORD: DATARAPPORTERINGSBLADE
Afprøvet stof:
Afprøvet jord:
Jordens tørstofmasseindhold (105 oC, 12 h): %
Temperatur: oC
Analysemetodens egnethed
|
Afvejet jord |
g |
|
|
Jord: tør masse |
g |
|
|
Rumfang CaCl2-opløsning |
cm3 |
|
|
Nominel koncentreret endelig opløsning |
μg cm-3 |
|
|
Analytisk bestemt koncentreret endelig opløsning |
μg cm-3 |
|
Princip i anvendt analysemetode:
Kalibrering af analysemetode:
Afprøvet stof:
Afprøvet jord:
Jordens tørstofmasseindhold (105 oC, 12 h): %
Temperatur: oC
|
Anvendt analysemetodik: |
Indirekte |
|
Parallel |
|
Sekventiel |
|
|
|
Direkte |
|
|
|
|
|
Adsorptionstest: udtagne prøver
|
|
Symbol |
Enhed |
Ekvilibreringstid |
Ekvilibreringstid |
Ekvilibreringstid |
Ekvilibreringstid |
||||
|
Prøveglas nr. |
|
|
|
|
|
|
|
|
|
|
|
Afvejet jord |
— |
g |
|
|
|
|
|
|
|
|
|
Jord: tør masse |
mSoil |
g |
|
|
|
|
|
|
|
|
|
Rumfang vand i afvejet jord (beregnet) |
VWS |
cm3 |
|
|
|
|
|
|
|
|
|
Rumfang 0,01 M CaCl2-opløsning til ekvilibrering af jord |
|
cm3 |
|
|
|
|
|
|
|
|
|
Rumfang stamopløsning |
|
cm3 |
|
|
|
|
|
|
|
|
|
Samlet rumfang vandig fase i kontakt med jord |
V0 |
cm3 |
|
|
|
|
|
|
|
|
|
Begyndelseskoncentration af prøveopløsning |
C0 |
μg cm-3 |
|
|
|
|
|
|
|
|
|
Masse af prøvestof ved testens begyndelse |
m0 |
μg |
|
|
|
|
|
|
|
|
|
Efter omrøring og centrifugering |
||||||||||
|
INDIREKTE METODE |
||||||||||
|
Parallelle metode |
||||||||||
|
Koncentration af prøvestof i vandig fase inklusive korrektion for blindværdi |
|
μg cm-3 |
|
|
|
|
|
|
|
|
|
Sekventielle metode |
||||||||||
|
Målt masse af prøvestof i prøve Va A |
|
μg |
|
|
|
|
|
|
|
|
|
DIREKTE METODE |
||||||||||
|
Masse af prøvestof adsorberet på jord |
|
μg |
|
|
|
|
|
|
|
|
|
Beregning af adsorption |
||||||||||
|
Adsorption |
|
% |
|
|
|
|
|
|
|
|
|
|
|
% |
|
|
|
|
|
|
|
|
|
Gennemsnitsværdier |
|
|
|
|
|
|
||||
|
Adsorptionskoefficient |
Kd |
cm3 g-1 |
|
|
|
|
|
|
|
|
|
Gennemsnitsværdier |
|
|
|
|
|
|
||||
|
Adsorptionskoefficient |
Koc |
cm3 g-1 |
|
|
|
|
|
|
|
|
|
Gennemsnitsværdier |
|
|
|
|
|
|
||||
Afprøvet stof:
Afprøvet jord:
Jordens tørstofmasseindhold (105 oC, 12 h): %
Temperatur: oC
Adsorptionstest: blindprøver og kontrolprøver
|
|
Symbol |
Enhed |
Blindprøve |
Blindprøve |
Kontrolprøve |
|||
|
Glas nr. |
|
|
|
|
|
|
|
|
|
Afvejet jord |
|
g |
|
|
|
|
0 |
0 |
|
Mængde vand i afvejet jord (beregnet) |
|
cm3 |
|
|
|
|
— |
— |
|
Tilsat rumfang 0,01 M CaCl2-opløsning |
|
cm3 |
|
|
|
|
|
|
|
Tilsat rumfang stamopløsning af prøvestof |
|
cm3 |
0 |
0 |
|
|
|
|
|
Samlet rumfang vandig fase (beregnet) |
|
cm3 |
|
|
|
|
— |
— |
|
Begyndelseskoncentration af prøvestof i vandig fase |
|
μg cm-3 |
|
|
|
|
|
|
|
Efter omrøring og centrifugering |
||||||||
|
Koncentration i vandig fase |
|
μg cm-3 |
|
|
|
|
|
|
Bemærkning: Tilføj flere kolonner om nødvendigt.
Afprøvet stof:
Afprøvet jord:
Jordens tørstofmasseindhold (105 oC, 12 h):
Temperatur:
Massebalance
|
|
Symbol |
Enhed |
|
|
|
|
|
Prøveglas nr. |
|
|
|
|
|
|
|
Afvejet jord |
— |
g |
|
|
|
|
|
Jord: tør masse |
msoil |
g |
|
|
|
|
|
Rumfang vand i afvejet jord (beregnet) |
VWS |
ml |
|
|
|
|
|
Rumfang 0,01 M CaCl2-opløsning til ekvilibrering af jord |
|
ml |
|
|
|
|
|
Rumfang stamopløsning |
|
cm3 |
|
|
|
|
|
Samlet rumfang vandig fase i kontakt med jord |
V0 |
cm3 |
|
|
|
|
|
Begyndelseskoncentration af prøveopløsning |
C0 |
μg cm-3 |
|
|
|
|
|
Ekvilibreringstid |
— |
h |
|
|
|
|
|
Efter omrøring og centrifugering |
||||||
|
Koncentration af prøvestof i vandig fase ved adsorptionsligevægt inklusive blindværdikorrektion |
|
μg cm-3 |
|
|
|
|
|
Ekvilibreringstid |
teq |
h |
|
|
|
|
|
Første fortynding med opløsningsmiddel |
||||||
|
Fjernet rumfang vandig fase |
vrec |
cm3 |
|
|
|
|
|
Tilsat rumfang opløsningsmiddel |
ΔV |
cm3 |
|
|
|
|
|
Første ekstraktion med opløsningsmiddel |
||||||
|
Signalstof i opløsningsmiddel |
SE1 |
var. |
|
|
|
|
|
Koncentreret prøvestof i opløsningsmiddel |
CE1 |
μg cm-3 |
|
|
|
|
|
Masse stof ekstraheret fra jord og beholdervægge |
mE1 |
μg |
|
|
|
|
|
Anden fortynding med opløsningsmiddel |
||||||
|
Fjernet rumfang opløsningsmiddel |
ΔVS |
cm3 |
|
|
|
|
|
Tilsat rumfang opløsningsmiddel |
ΔV |
cm3 |
|
|
|
|
|
Anden ekstraktion med opløsningsmiddel |
||||||
|
Signalstof i opløsningsmiddelfase |
SE2 |
var. |
|
|
|
|
|
Koncentreret prøvestof i opløsningsmiddel |
CE2 |
μg cm-3 |
|
|
|
|
|
Masse stof ekstraheret fra jord og beholdervægge |
mE2 |
μg |
|
|
|
|
|
Samlet masse prøvestof ekstraheret i to trin |
mE |
μg |
|
|
|
|
|
Massebalance |
MB |
% |
|
|
|
|
Afprøvet stof:
Afprøvet jord:
Jordens tørstofmasseindhold (105 oC, 12 h): %
Temperatur: oC
Adsorptionsisotermer
|
|
Symbol |
Enhed |
|
|
|
|
|
|
|
|
|
Prøveglas nr. |
|
|
|
|
|
|
|
|
|
|
|
Afvejet jord |
— |
g |
|
|
|
|
|
|
|
|
|
Jord: tør masse |
E |
g |
|
|
|
|
|
|
|
|
|
Rumfang vand i afvejet jord (beregnet) |
VWS |
cm3 |
|
|
|
|
|
|
|
|
|
Rumfang 0,01 M CaCl2-opløsning til ekvilibrering af jord |
|
cm3 |
|
|
|
|
|
|
|
|
|
Tilsat rumfang stamopløsning |
|
cm3 |
|
|
|
|
|
|
|
|
|
Samlet rumfang vandig fase i kontakt med jorden (beregnet) |
V0 |
cm3 |
|
|
|
|
|
|
|
|
|
Koncentration, opøsning |
C0 |
μg cm-3 |
|
|
|
|
|
|
|
|
|
Ekvilibreringstid |
— |
h |
|
|
|
|
|
|
|
|
|
Efter omrøring og centrifugering |
||||||||||
|
Stofkoncentration, vandig fase, inklusive blindkorrektion |
|
μg cm-3 |
|
|
|
|
|
|
|
|
|
Temperatur |
|
oC |
|
|
|
|
|
|
|
|
|
Adsorptionsmasse pr. enhed jord |
|
μg g-1 |
|
|
|
|
|
|
|
|
Regressionsanalyse:
størrelse af KF ads:
størrelse af l/n:
regressionskoefficient r2:
Afprøvet stof:
Afprøvet jord:
Jordens tørstofmasseindhold (105 oC, 12 h): %
Temperatur: oC
|
Anvendt analysemetodik: |
Indirekte |
|
Parallel |
|
Sekventiel |
|
Desorptionstest
|
|
Symbol |
Enhed |
Tidsinterval |
Tidsinterval |
Tidsinterval |
Tidsinterval |
|
|
Prøveglas ner. fra adsorptionstrin |
|
|
|
|
|
|
|
|
Masse af stof adsorberet til jord ved adsorptionsligevægt |
|
μg |
|
|
|
|
|
|
Fjernet rumfang vandig fase, erstattet med 0,01 M CaCl2 |
VR |
cm3 |
|
|
|
|
|
|
Samlet rumfang vandig fase i berøring med jorden |
PM |
V0 |
cm3 |
|
|
|
|
|
SM |
VT |
cm3 |
|
|
|
|
|
|
Masse af prøvestof, som er til overs efter indtræden af adsorptionsligevægt som følge af ufuldstændig volumenudskiftning |
|
μg |
|
|
|
|
|
|
Desorptionskinetik |
|||||||
|
Målt mass stof desorberet fra jorden til tiden ti |
|
μg |
|
|
|
|
|
|
Rumfang opløsning udtaget af prøveglas (i) til bestemmelse af prøvestof |
PM |
Vf i |
cm3 |
|
|
|
|
|
SM |
va D |
cm3 |
|
|
|
|
|
|
Masse stof desorberet fra jorden til tiden ti (beregnet) |
|
μg |
|
|
|
|
|
|
Masse stof desorberet fra jorden i tidsintervallet Δti (beregnet) |
|
μg |
|
|
|
|
|
|
Desorptionsprocent |
|||||||
|
Desorption til tiden ti |
Dti |
% |
|
|
|
|
|
|
Desorption til tidsintervallet Δti |
|
% |
|
|
|
|
|
|
Tilsyneladende desorptionskoefficent |
Kdes |
|
|
|
|
|
|
PM: Parallelle metode.
SM: Sekventielle metode.
C.19. BESTEMMELSE AF ADSORPTIONSKOEFFICIENTEN (Koc ) I JORD OG I KLOAKSLAM MED HPLC (HIGH PERFORMANCE LIQUID CHROMATOGRAPHY)
1. METODE
Metoden er gengivet efter OECD TG 121 (2000).
1.1. INDLEDNING
Stoffers sorptionsegenskaber i jord og kloakslam kan beskrives ved parametre, som bestemmes eksperimentelt med prøvemetode C.18. En vigtig parameter er adsorptionskoefficienten, der defineres som forholdet mellem stoffets koncentration i jord/slam og stoffets koncentration i den vandige fase ved adsorptionsligevægt. Adsorptionskoefficienten normaliseret efter jordens organiske kulstofindhold. Koc er nyttig som indikator for et kemisk stofs bindingsevne til organisk materiale i jord og kloakslam og kan bruges til at sammenligne forskellige kemiske stoffer. Denne parameter kan bestemmes ved korrelering med vandopløseligheden og fordelingskoefficienten i n-oktanol/vand (1)(2)(3)(4)(5)(6)(7).
I den her beskrevne forsøgsmetode anvendes HPLC til bestemmelse af adsorptionskoefficienten Koc i jord og i kloakslam (8). De derved beregnede værdier er mere pålidelige end dem, der bygger på QSAR-beregninger (9). Som beregningsmetode kan den ikke helt erstatte batch-ligevægtsforsøgene i prøvemetode C.18. Den beregnede Koc-værdi kan imidlertid være nyttig ved valg af passende testparametre til adsorptions-/desorptionsundersøgelser med prøvemetode C.18 gennem beregning af Kd (fordelingskoefficient) eller Kf (Freundlich-adsorptionskoefficient) efter ligning 3 (jf. 1.2).
1.2. DEFINITIONER
Kd: Fordelingskoefficienten defineres som forholdet mellem ligevægtskoncentrationerne C af et opløst prøvestof i et tofasesystem bestående af et sorptionsmiddel (jord eller kloakslam) og en vandig fase; den er dimensionsløs, når koncentrationerne i begge faser udtrykkes på w/w basis. Angives koncentrationen i den vandige fase på vægt/volumen basis, bliver enheden ml g-1. Kd kan variere med absorptionsmidlets egenskaber og kan være koncentrationsafhængig.
|
|
(1) |
hvor:
|
Csoil |
= |
ligevægtskoncentration af prøvestof i jord (μg g-1) |
|
Csiudge |
= |
ligevægtskoncentration af prøvestof i slam (μg g-1) |
|
Caq |
= |
ligevægtskoncentration af prøvestof i vandig fase (μg g-1, μg ml-1). |
Kf: Freundlich-adsorptionskoefficienten defineres som prøvestoffets koncentration i jord eller kloakslam (x/m), når ligevægtskoncentrationen Caq den vandige fase er lig én; enheden er μg g-1 adsorptionsmiddel. Størrelsen kan afhænge af adsorptionsmidlets egenskaber.
|
|
(2) |
hvor:
|
x/m |
= |
mængde prøvestof x (μg) adsorberet på en mængde adsorptionsmiddel m (g) ved ligevægt |
|
l/n |
= |
Freundlich-adsorptionsisotermens hældning |
|
Caq |
= |
ligevægtskoncentration af prøvestof; vandig fase (μg ml-1) |
Ved
Koc: Fordelingskoefficient (Kd) eller Freundlich-adsorptionskoefficient (Kf) normaliseret efter adsorptionsmidlets organiske kulstofindhold (foc); denne er navnlig for uioniserede kemiske stoffer en vigtig indikator for størrelsen af adsorptionen mellem et stof og adsorptionsmidlet og kan bruges til at sammenligne forskellige kemiske stoffer. Alt efter dimensionen af Kd og Kf kan Koc være dimensionsløs eller have enheden ml g-1eller μg g-1organisk stof.
|
|
(3) |
Forholdet mellem Koc og Kd er ikke altid lineært, således kan Koc-værdierne variere fra jordtype til jordtype, dog med langt mindre variabilitet end Kd eller Kf.
Adsorptionskoefficienten (Koc afledes af kapacitetsfaktoren (k') ved hjælp af en kalibreringskurve over log k' som funktion af logoc for de valgte referencestoffer.
|
|
(4) |
hvor:
|
tR: |
= |
HPLC-retentionstiden for prøve- og referencestof (minutter) |
|
t0: |
= |
HPLC-dødtid (minutter) (se 1.8.2). |
Pow : Oktanol-vand-fordelingskoefficienten defineres som forholdet mellem koncentrationerne af opløst stof i n-oktanol og vand; den er dimensionsløs.
|
|
(5) |
1.3. REFERENCESTOFFER
Før metoden anvendes, bør man kende strukturformel, renhed og eventuel dissociationskonstant. Det er nyttigt at kende opløselighed i vand og organiske opløsningsmidler, oktanol-vand-fordelingskoefficient og hydrolyseegenskaber.
For at korrelere de målte HPLC-retentionsdata for et prøvestof med dets adsorptionskoefficient Koc må der opstilles en kalibreringskurve over log Koc som funktion af log k'. Der kræves mindst seks referencepunkter, heraf mindst ét over og ét under den forventede værdi for prøvestoffet. Metodens nøjagtighed kan forbedres væsentligt ved at anvende referencestoffer, som er strukturmæssigt beslægtet med prøvestoffet. Foreligger sådanne data ikke, er det op til brugeren at vælge passende kalibreringsstoffer. I så fald bør der vælges et mere generelt sæt strukturmæssigt forskelligartede stoffer. De stoffer og Koc-værdier, som kan anvendes, er i tillægget angivet i tabel 1 for kloakslam og tabel 3 for jord. Valg af andre kalibreringsstoffer bør begrundes.
1.4. PRØVEMETODENS PRINCIP
Der udføres HPLC på analysekolonner pakket med en gængs cyanopropyl-faststoffase med både lipofile og polære grupper. Der anvendes en moderat polær stationær fase baseret på silicamatrix:
|
— O — Si |
— CH2 — CH2 — CH2 |
— CN |
|
silica |
ikke polært fyldstof |
polær gruppe |
Prøvemetodens princip svarer til prøvemetode A.8 (fordelingskoefficient, HPLC-metode). Når prøvestoffet føres gennem kolonnen sammen med den mobile fase, reagerer prøvestoffet med den stationære fase. Prøvestoffets fordeling mellem mobil og stationær fase bevirker, at det bliver forsinket. Den stationære fases dobbelte sammensætning med både polære og upolære bindingssteder giver mulighed for interaktion med de polære og ikke-polære grupper i et molekyle på samme måde som det er tilfældet for et organisk stof i en grundsubstans af jord eller kloakslam. Derved er det muligt at fastlægge forholdet mellem retentionstiden på søjlen og adsorptionskoefficienten på organisk materiale.
pH har væsentlig indflydelse på sorptionsegenskaberne, navnlig af polære stoffer. I landbrugsjord og i tanke i spildevandsbehandlingsanlæg ligger pH normalt mellem 5,5 og 7,5. For ioniserbare stoffer bør der udføres to forsøg med både den ioniserede og den ikke-ioniserede form i passende bufferopløsninger, dog kun når mindst 10 % af prøvestoffet er dissocieret ved pH mellem 5,5 og 7,5.
Da vurderingen alene bygger på forholdet mellem retentionen på HPLC-kolonnen og adsorptionskoefficienten, er en kvantitativ analysemetode unødvendig, og kun retentionstiden behøver bestemmes. Hvis man råder over et egnet sæt referencestoffer og kan anvende standardiserede forsøgsbetingelser, er dette en hurtig og effektiv metode til at få et skøn over adsorptionskoefficienten Koc.
1.5. PRØVENS ANVENDELIGHED
HPLC-metoden kan anvendes på kemiske stoffer (mærkede eller umærkede), til hvilke der findes et egnet detektionssystem (f.eks. spektrofotometer, strålingsdetektor), og som er tilstrækkeligt stabile inden for forsøgsvarigheden. Den kan især være nyttig for kemiske stoffer, som er vanskelige at undersøge i andre eksperimentelle systemer (f.eks. stoffer, som er flygtige, eller hvis vandopløselighed er for ringe til at give målelige koncentrationer, og stoffer med høj affinitet til inkuberingssystemernes overflade). Metoden er anvendelig til blandinger, som giver dårligt adskilte elueringsbånd. I så fald bør øvre og nedre grænse for log Koc-værdierne af stofferne i testblandingen angives.
Urenheder kan undertiden vanskeliggøre fortolkningen af HPLC-resultater, men er dog af underordnet betydning, når blot prøvestoffet kan identificeres sikkert ad analytisk vej og kan adskilles fra urenhederne.
Metoden er valideret for stofferne i tabel 1 i tillægget og er endvidere anvendt på en række andre kemiske stoffer i følgende grupper:
|
— |
aromatiske aminer (f.eks. trifluralin, 4-chloranilin, 3,5-dinitroanilin, 4-methylanilin, N-methylanilin, 1-naphthylamin) |
|
— |
aromatiske carboxylsyreestere (f.eks. benzoesyremethylester, 3,5-dinitrobenzoesyreethylester) |
|
— |
aromatiske carbonhydrider (f.eks. toluen, xylen, ethylbenzen, nitrobenzen) |
|
— |
aryloxyphenoxypropionsyreestere (f.eks. diclofopmethyl, fenoxapropethyl, fenoxaprop-P-ethyl) |
|
— |
benzimidazol- og imidazol-fungicider (f.eks. carbendazim. fuberidazol, triazoxid) |
|
— |
carboxylsyreamider (f.eks. 2-chlorbenzamid, N.N-dimethylbenzamid, 3,5-dinitrobenzamid, N-methylbenzamid, 2-nitrobenzamid, 3-nitrobenzamid) |
|
— |
chlorerede carbonhydrider (f.eks. endosulfan, DDT. hexachlorbenzen, quintozen, 1.2,3-trichlorbenzen) |
|
— |
insekticider af typen organiske fosforforbindelser (f.eks. azinphosmethyl, disulfoton, fenamiphos, isofenphos, pyrazophos. sulprofos, triazophos) |
|
— |
phenoler (f.eks. phenol, 2-nitrophenol. 4-nitrophenol, pentachlorphenol, 2.4.6-trichlorphenol. 1-naphthol) |
|
— |
derivater af phenylurinstof (f.eks. isoproturon. monolinuron. pencycuron) |
|
— |
pigmentfarvestoffer (f.eks. Acid Yellow 219. Basic Blue 41, Direct Red SI) |
|
— |
polyaromatiske carbonhydrider (f.eks. acenaphthen, naphthalen) |
|
— |
1,3,5-triazin-herbicider (f.eks. prometryn, propazin, simazin, terbutryn) |
|
— |
triazolderivater (f.eks. tebuconazol, triadimefon, tradimenol, triapenthenol). |
Metoden er ikke anvendelig til stoffer, som reagerer enten med eluenten eller den stationære fase. Den er ligeledes ikke anvendelig til stoffer, som udviser specifik interaktion med uorganiske komponenter (f.eks. kompleksdannelse med lermineraler). Metoden virker ikke nødvendigvis på overfladeaktive stoffer, uorganiske stoffer og middelstærke til stærke organiske syrer og baser. Log Koc-værdier mellem 1,5 og 5,0 kan bestemmes. Til bestemmelse af ioniserbare stoffer må anvendes en bufret mobil fase, men der må tages forholdsregler til undgåelse af udfældning af bufferkomponenter eller prøvestof.
1.6. KVALITETSKRITERIER
1.6.1. Nøjagtighed
Normalt kan adsorptionskoefficienten af et prøvestof bestemmes inden for +/- 0,5 logaritmeenhed af den tilsvarende værdi bestemt ved batch-ligevægtsmetoden (se tabel 1 i tillægget). Bedre nøjagtighed kan opnås, hvis de anvendte referencestoffer er strukturelt beslægtet med prøvestoffet.
1.6.2. Repeterbarhed
Bestemmelserne skal foretages mindst som dobbeltbestemmelser. Log Koc-værdier baseret på enkeltmålinger bør højst afvige 0,25 logaritmeenhed indbyrdes.
1.6.3. Reproducerbarhed
De hidtidige erfaringer med anvendelsen af metoden underbygger dens validitet, En undersøgelse af HPLC-metoden på 48 stoffer (hovedsagelig pesticider), for hvilke der forelå pålidelige data vedrørende Koc i jord, resulterede i en korrelationskoefficient på R = 0,95 (10)(l 1).
Der er gennemført en sammenlignende laboratorieundersøgelse med 11 deltagende laboratorier med henblik på at forbedre og validere metoden (12). Resultaterne er gengivet i tabel 2 i tillægget.
1.7. BESKRIVELSE AF PRØVEMETODEN
1.7.1. Indledende skøn over adsorptionskoefficienten
Oktanol-vand fordelingskoefficienten Pow (= Kow) og, i nogen udstrækning, vandopløseligheden kan — navnlig for uioniserede stoffer — benyttes som indikatorer for adsorptionsgraden og således bruges til indledende afgrænsning af området. Der er udgivet en række nyttige korrelationer for forskellige grupper af kemiske stoffer (1)(2)(3)(4)(5)(6)(7).
1.7.2. Apparatur
Der anvendes en væskekromatograf forsynet med en impulsfri pumpe og en egnet detektor. Det anbefales at bruge en injektionsventil med injektionssløjfe. Der anvendes gængse kemisk bundne cyanopropyl-harpikser på silicabasis (f.eks. Hypersil og Zorbax CN). En forkolonne af samme materiale kan placeres mellem injektions-system og analysekolonne. Kolonner fra forskellige leverandører kan variere betydeligt i adskillelsesevne. Vejledende skal man opnå følgende kapacitetsfaktorer k'; log k' > 0,0 for log Koc = 3,0 og log k' > 0,4 for log Koc = 2,0 ved anvendelse af en mobil fase bestående af methanol/vand 55/45 %.
1.7.3. Mobile faser
Der er afprøvet forskellige mobile faser, og følgende to anbefales:
|
— |
methanol/vand (55/45 % v/v) |
|
— |
methanol/0,01 M citratbuffer pH 6,0 (55/45 % v/v). |
Til fremstilling af elueringsvæsken anvendes methanol af HPLC-kvalitet og destilleret vand eller citratbuffer. Blandingen afgasses før brug. Elueringen skal være isokratisk (dvs. af ensartet styrke). Er methanol/vand-blandinger ikke egnede, kan andre blandinger af organiske opløsningsmidler og vand anvendes, f.eks. ethanol/vand eller acetonitril/vand. For ioniserbare stoffer anbefales, at pH stabiliseres med bufferopløsning. Der må træffes foranstaltninger til at undgå saltudfældning og tæring af kolonnen, som kan forekomme ved visse blandinger af organisk fase og buffer.
Der må ikke anvendes additiver som f.eks. ionparreagenser, da de kan påvirke sorptionsegenskaberne af den stationære fase. Sådanne ændringer af den stationære fase kan være irreversible. Af denne grund er det ubetinget nødvendigt, at eventuelle forsøg med additiver udføres på separate kolonner.
1.7.4. Opløste stoffer
Prøve- og referencestoffer opløses i den mobile fase.
1.8. UDFØRELSE AF FORSØGET
1.8.1. Prøvningsbetingelser
Temperaturen bør registreres under af målingerne. Det anbefales stærkt, at kolonnerummet er temperaturreguleret for at sikre konstante betingelser under kalibrerings- og overslagsforsøg og måling af prøvestoffet.
1.8.2. Bestemmelse af dødtid t0
Til bestemmelse af dødtiden t0 kan anvendes to forskellige metoder (se også 1.2).
1.8.2.1. Bestemmelse af dødtiden t0 ved hjælp af en homolog serie
Denne fremgangsmåde har vist sig give pålidelige og ensartede t0-værdier. Nærmere enkeltheder herom kan findes i prøvemetode A.8: Fordelingskoefficient (n-oktanol/vand), HPLC-metode.
1.8.2.2. Bestemmelse af dødtid t0 ved hjælp af inaktive stoffer, som ikke tilbageholdes af kolonnen
Teknikken bygger på, at der indsprøjtes en opløsning af formamid, urinstof eller natriumnitrat. Bestemmelserne skal udføres mindst som dobbeltbestemmelser.
1.8.3. Bestemmelse af retentionstider tR
Referencestoffer vælges som beskrevet i 1.3. Til bestemmelse af disse stoffers retentionstid kan de indsprøjtes som en blandet standard, forudsat at det er godtgjort, at retentionstiden for den enkelte referencestandard er upåvirket af tilstedeværelsen af de andre referencestandarder. Kalibrering skal ske med regelmæssige intervaller mindst to gange dagligt for at tage højde for eventuelle uventede ændringer i kolonnens præstationer. Det bedste er at udføre kalibreringsindsprøjtningerne før og efter indsprøjtning af prøvestoffet for at bekræfte, at retentionstiderne er uændrede. Prøvestofferne indsprøjtes separat i mindst mulig mængde (undgå overbelastning af kolonnen), og deres retentionstider bestemmes.
For at gøre målingerne mere pålidelige bør de udføres mindst som dobbeltbestemmelser. Log Koc-værdier afledt af enkeltmålinger bør højst afvige 0,25 logaritmeenhed fra hinanden.
1.8.4. Bedømmelse
Kapacitetsfaktorerne k' beregnes af dødtiden t0 og retentionstiderne tR for de valgte referencestoffer efter ligning 4 (se 1.2). Log k'-værdierne for referencestofferne afbildes derefter mod de tilhørende log Koc-værdier fra batchligevægtsforsøgene, som er givet i tabel 1 og 3 i tillægget. Ved hjælp af denne kurve anvendes log k'-værdien for et prøvestof derefter til beregning af dets Koc-værdi. Viser de faktiske resultater, at log Koc for prøvestoffet er uden for kalibreringsområdet, bør forsøget gentages med et andet, mere velegnet referencestof.
2. DATA OG RAPPORTERING
Rapporten skal indeholde følgende oplysninger:
|
— |
identitet og renhed af prøve- og referencestof, samt pKa-værdi hvis relevant |
|
— |
beskrivelse af apparatur og forsøgsbetingelser, f.eks. type og dimension af analysekolonne (og forkolonne), detektionsmåde, mobil fase (forholdet mellem komponenterne, samt pH), temperaturområde under målingerne |
|
— |
dødtiden og metoden anvendt til bestemmelse heraf |
|
— |
mængdeprøve- og referencestof, som indføres i kolonnen |
|
— |
retentionstiderne for de referencestoffer, der anvendes til kalibrering |
|
— |
enkeltheder vedrørende den tilnærmede regressionslinje (log k' mod log Koc) og grafisk fremstilling af regressionslinjen |
|
— |
gennemsnitlig retention og beregnet d log Koc-værdi for prøvestoffet |
|
— |
kromatogrammer. |
HENVISNINGER
|
(1) |
W.J. Lyman, W.F. Reehl, D.H. Rosenblatt (ed). (1990). Handbook of chemical property estimation methods, Chap. 4, McGraw-Hill, New York. |
|
(2) |
J. Hodson, N.A. Williams (1988). The estimation of the adsorption coefficient (Koc) for soils by HPLC. Chemosphere, 17, 167. |
|
(3) |
G.G. Briggs (1981). Theoretical and experimental relationships between soil adsorption, octanol-water partition coefficients, water solubilities, bioconcentration factors, and the parachor. J. Agric. Food Chem., 29, pp. 1050-1059. |
|
(4) |
C.T. Chiou, P.E. Porter, D.W. Schmedding (1983). Partition equilibria of nonionic organic compounds between soil organic matter and water. Environ. Sci. Technol., 1 7, pp. 227-231. |
|
(5) |
Z. Gerstl, U. Mingelgrin (1984). Sorption of organic substances by soils and sediment. J. Environm. Sci. Health, B19, pp. 297-312. |
|
(6) |
C.T. Chiou, L.J. Peters, V.H. Freed (1979). A physical concept of soil water equilibria for nonionic organic compounds, Science, 106, pp. 831-832. |
|
(7) |
S.W. Karickhoff (1981). Semi-empirical estimation of sorption of hydrophobic pollutants on natural sediments and soils, Chemosphere, 10, pp. 833-846. |
|
(8) |
W. Kordel, D. Hennecke, M. Herrmann (1997). Application of the HPLC-screening method for the determination of the adsorption coefficient on sewage sludges. Chemosphere, 35(1/2), pp. 121-128. |
|
(9) |
M. Mueller, W. Kordel (1996). Comparison of screening methods for the estimation of adsorption coefficients on soil. Chemosphere, 32(12), pp. 2493-2504. |
|
(10) |
W. Kordel, J. Stutte, G. Kotthoff (1993). HPLC-screening method for the determination of the adsorption coefficient in soil-comparison of different stationary phases, Chemosphere, 27(12), pp. 2341-2352. |
|
(11) |
B. von Oepen, W. Kordel, W. Klein (1991) Sorption of nonpolar and polar compounds to soils: Processes, measurements and experience with the applicability of the modified OECD Guideline 106, Chemosphere, 22, pp. 285-304. |
|
(12) |
W. Kordel, G. Kotthoff, J. Muller (1995). HPLC-screening method for the determination of the adsorption coefficient on soil-results of a ring test, Chemosphere, 30(7), pp. 1373-1384. |
Tillæg
Tabel 1
Sammenligning af Koc-værdier for jordarter og kloakslam, samt værdier beregnet ved HPLC-screeningsmetoden (31) (32)
|
Stof |
CAS nr. |
kloakslam |
Log Koc ved HPLC |
Δ |
Log Koc for jordtyper |
Log Koc ved HPLC |
Δ |
|
Atrazin |
1912-24-9 |
1,66 |
2,14 |
0,48 |
1,81 |
2,20 |
0,39 |
|
Linuron |
330-55-2 |
2,43 |
2,96 |
0,53 |
2,59 |
2,89 |
0,30 |
|
Fenthion |
55-38-9 |
3,75 |
3,58 |
0,17 |
3,31 |
3,40 |
0,09 |
|
Monuron |
150-68-5 |
1,46 |
2,21 |
0,75 |
1,99 |
2,26 |
0,27 |
|
Phenanthren |
85-01-8 |
4,35 |
3,72 |
0,63 |
4,09 |
3,52 |
0,57 |
|
Benzoesyrephenylester |
93-99-2 |
3,26 |
3,03 |
0,23 |
2,87 |
2,94 |
0,07 |
|
Benzamid |
55-21-0 |
1,60 |
1,00 |
0,60 |
1,26 |
1,25 |
0,01 |
|
4-Nitrobenzamid |
619-80-7 |
1,52 |
1,49 |
0,03 |
1,93 |
1,66 |
0,27 |
|
Acetanilid |
103-84-4 |
1,52 |
1,53 |
0,01 |
1,26 |
1,69 |
0,08 |
|
Anilin |
62-53-3 |
1,74 |
1,47 |
0,27 |
2,07 |
1,64 |
0,43 |
|
2,5-Dichloroanilin |
95-82-9 |
2,45 |
2,59 |
0,14 |
2,55 |
2,58 |
0,03 |
Tabel 2
Resultater af det laboratoriesammenlignende forsøg (11 deltagende laboratorier), som er gennemført med henblik på at forbedre og validere HPLC-metoden (33)
|
Stof |
CAS nr. |
Log Koc |
Koc |
Log Koc |
|
(OECD 106) |
(HPLC metode) |
|||
|
Atrazin |
1912-24-9 |
1,81 |
78 ± 16 |
1,89 |
|
Monuron |
150-68-5 |
1,99 |
100 ± 8 |
2,00 |
|
Triapenthenol |
77608-88-3 |
2,37 |
292 ± 58 |
2,47 |
|
Linuron |
330-55-2 |
2,59 |
465 ± 62 |
2,67 |
|
Fenthion |
55-38-9 |
3,31 |
2 062 + 648 |
3,31 |
Tabel 3
Anbefalede referencestoffer til HPLC-screeningsmetoden baseret på jordadsorptionsdata
|
Referencestof |
CAS nr. |
Gennemsnitlige log Koc-værdier fra batchligevægt |
Nummer på Koc-data |
Log S.D. |
Kilde |
|
Acetanilid |
103-84-4 |
1,25 |
4 |
0,48 |
|
|
Phenol |
108-95-2 |
1,32 |
4 |
0,70 |
|
|
2-Nitrobenzamid |
610-15-1 |
1,45 |
3 |
0,90 |
|
|
N.N-dimethylbenzamid |
611-74-5 |
1,52 |
2 |
0,45 |
|
|
4-Methylbenzamid |
619-55-6 |
1,78 |
3 |
1,76 |
|
|
Methylbenzoat |
93-58-3 |
1,80 |
4 |
1,08 |
|
|
Atrazin |
1912-24-9 |
1,81 |
3 |
1,08 |
|
|
Isoproturon |
34123-59-6 |
1,86 |
5 |
1,53 |
|
|
3-Nitrobenzamid |
645-09-0 |
1,95 |
3 |
1,31 |
|
|
Anilin |
62-53-3 |
2,07 |
4 |
1,73 |
|
|
3,5-Dinitrobenzamid |
121-81-3 |
2,31 |
3 |
1,27 |
|
|
Carbendazim |
10605-21-7 |
2,35 |
3 |
1,37 |
|
|
Triadimenol |
55219-65-3 |
2,40 |
3 |
1,85 |
|
|
Triazoxid |
72459-58-6 |
2,44 |
3 |
1,66 |
|
|
Triazophos |
24017-47-8 |
2,55 |
3 |
1,78 |
|
|
Linuron |
330-55-2 |
2,59 |
3 |
1,97 |
|
|
Naphthalen |
91-20-3 |
2,75 |
4 |
2,20 |
|
|
Endosulfan-diol |
2157-19-9 |
3,02 |
5 |
2,29 |
|
|
Methiocarb |
2032-65-7 |
3,10 |
4 |
2,39 |
|
|
Acid Yellow 219 |
63405-85-6 |
3,16 |
4 |
2,83 |
|
|
1,2,3-Trichlorbenzen |
87-61-6 |
3,16 |
4 |
1,40 |
|
|
γ-HCH |
58-89-9 |
3,23 |
5 |
2,94 |
|
|
Fenthion |
55-38-9 |
3,31 |
3 |
2,49 |
|
|
Direct Red 81 |
2610-11-9 |
3,43 |
4 |
2,68 |
|
|
Pyrazophos |
13457-18-6 |
3,65 |
3 |
2,70 |
|
|
α-Endosulfan |
959-98-8 |
4,09 |
5 |
3,74 |
|
|
Diclofop-methyl |
51338-27-3 |
4,20 |
3 |
3,77 |
|
|
Phenanthren |
8 5-01-8 |
4,09 |
4 |
3,83 |
|
|
Basic Blue 4] (mix) |
26850-47-5 12270-13- |
4,89 |
4 |
4,46 |
|
|
DDT |
50-29-3-2 |
5,631— |
1 |
— |
C.20. DAPHNIA MAGNA FORMERINGSTEST
1. METODE
Denne testmetode, der undersøger kemikaliers toksiske indflydelse på formeringsevnen, svarer til OECD TG 211 (1998).
1.1. INDLEDNING
Det primære mål for testen er at vurdere kemikaliers indvirkning på Dafphnia magnas formeringsevne.
1.2. DEFINITIONER OG ENHEDER
Forældregenerationen er de hundafnier, som er til stede ved testens begyndelse, og hvis formeringsevne er genstand for undersøgelsen.
Afkom er de dafnier, der er født af forældregenerationen under testen.
Laveste koncentration med observeret effekt (LOEC) er den laveste kemikaliekoncentration, der i testen har vist sig at resultere i en statistisk signifikant indflydelse (p < 0,05) på formeringen og dødelighed for forældregenerationen inden for den fastlagte eksponeringstid, sammenlignet med kontrollen. Imidlertid skal alle undersøgte koncentrationer, der ligger over LOEC, have haft en skadelig virkning, som er større end eller lig med den, som er observeret ved LOEC. Hvis ikke disse to krav opfyldes, skal der gøres fuldstændigt rede for fastlæggelsen af LOEC (og dermed NOEC).
Koncentration uden observeret effekt (NOEC) er den testkoncentration umiddelbart under LOEC, som ved sammenligning med kontrollen ikke har nogen statistisk signifikant effekt (p < 0,05) inden for en fastlagt eksponeringstid.
EC X er den koncentration af testkemikaliet opløst i vand, som resulterer i en x procent reduktion i formeringen af Daphnia magna inden for en fastlagt eksponeringstid.
Den sande formeringshastighed er det mål for vækst, der integrerer formering og den aldersbetingede dødelighed (20)(21)(22). I en »steady state« population vil den være lig nul. For populationer i vækst vil den være positiv, og for populationer, der mindskes, vil den være negativ. Det er klart, at den sidste situation er uholdbar og vil ende med udslettelse af populationen.
Den påviselige grænseværdi er den laveste koncentration, der ses at have effekt, men som ikke kan kvantiteres.
Den målelige grænseværdi er den laveste koncentration, der kan kvantiteres.
Mortalitet et dyr anses for værende dødt, når det ikke kan bevæge sig, dvs. når det ikke er i stand til at svømme, eller hvis der ikke ses bevægelser af vedhæng eller postabdomen inden for 15 sekunder efter forsigtig omrystning af undersøgelsesbeholderen. (Hvis en anden definition anvendes, skal det meddeles sammen med dets reference).
1.3. PRINCIPPET FOR TESTMETODEN
Unge hundafnier (forældregenerationen), yngre end 24 timer ved testens begyndelse, udsættes for testkemikaliet opløst i vand i en række koncentrationer. Testens varighed er 21 dage. Ved testens afslutning vurderes det totale antal efterkommere efter den stadig levende forældregeneration. Det betyder, at afkom af dafnier, der er døde i løbet af testen, ikke inkluderes i beregningerne. Forældregenerationens formeringsevne kan udtrykkes på andre måder (f.eks. antal levende efterkommere pr moderdyr fra den første dag, afkom blev observeret), men skal rapporteres sammen med det totale antal afkom frembragt pr. levende moderdyr ved testens afslutning. Formeringsevnen for de dyr, der blev eksponeret for testkemikaliet, sammenlignes med formeringsevnen i kontrollen/kontrollerne for at fastlægge den laveste koncentration med observeret effekt (LOEC) og dermed koncentrationen uden observeret effekt (NOEC). Så vidt gørligt skal data desuden analyseres ved hjælp af en regressionsmodel for at vurdere de koncentrationer, der ville forårsage en x % reduktion i formeringsevnen (dvs. EC50. EC20 eller EC10).
Forældregenerationens overlevelse og tiden indtil den første yngel skal også meddeles. Andre effekter af kemikaliet på parametre såsom vækst (dvs. længde) og eventuelt den sande formeringshastighed kan tænkes undersøgt.
1.4. INFORMATION OM TESTKEMIKALIET
Resultatet fra en akut toksicitetstest (se metode C.2, del I) udført med Daphnia magna bør være tilgængelig. Resultatet kan være nyttigt for valget af en passende række af testkoncentrationer i formeringstestene. Opløseligheden i vand og testkemikaliets damptryk bør være kendt ligesom en pålidelig analytisk metode til kvantitering af kemikaliet i testopløsningerne med angivelse af effektivitet med hensyn til genfinding og den målelige grænseværdi.
Data for testkemikaliet, som kan være nyttige for at fastslå testbetingelserne, inkluderer strukturformel, stoffets renhed, lysstabilitet, stabilitet i testforløbet, pKa, Pow. og resultater af test for spontan nedbrydning (se metode C.4)
1.5. TESTENS VALIDITET
Hvis en test skal være valid skal følgende resultat-krav opfyldes af kontrollen/kontrollerne:
|
— |
forældregenerationens mortalitet (hundafnier) må ikke overstige 20 % ved afslutning af testen |
|
— |
det gennemsnitlige antal levende afkom pr overlevende forældre-hundafnie ved testens afslutning er ≥ 60. |
1.6. BESKRIVELSE AF TESTMETODEN
1.6.1. Apparatur
Bægerglas og andet apparatur, der kommer i kontakt med testopløsningerne, skal være fremstillet af rent glas eller andet kemisk inert materiale. Karrene til undersøgelsen vil almindeligvis være af glas.
Yderligere nødvendigt apparatur kan være følgende:
|
— |
iltmålingsapparatur (med mikroelektrode eller andet passende udstyr til måling af ilt i små volumina) |
|
— |
fyldestgørende apparatur til temperaturkontrol |
|
— |
pH-meter |
|
— |
apparatur til måling af vands hårdhedsgrad |
|
— |
apparatur til måling af totalkoncentrationen af organisk kulstof (TOC) i vand eller apparatur til bestemmelse af det kemiske iltbehov (COD) |
|
— |
fyldestgørende apparatur til kontrol af lys-regime og måling af lys-intensitet. |
1.6.2. Testorganisme
Den art, der skal benyttes i testen, er Daphnia magna Straus. Andre arter af dafnier kan anvendes, under forudsætning af at de på passende måde opfylder validitetskriterierne. (Det relevante validitetskriterium for dafniearten er formeringsevnen, som den ses i kontrollerne). Hvis der anvendes andre dafniearter, skal de klart defineres og brugen af dem begrundes.
En genotypebestemmelse som identifikation af arten vil være at foretrække. Undersøgelser (1) har vist, at formeringsevnen af Klon A (som stammer fra IRCHA i Frankrig) (3) er stabil med hensyn til det relevante validitetskriterium med sit gennemsnit på ≥ 60 stk. afkom pr. forældredafnie, der overlever ved de dyrkningsbetingelser, som nærværende metode beskriver. Andre kloner er imidlertid acceptable, hvis det kan påvises, at dafniekulturen opfylder validitetskriterierne for en test.
Ved testens begyndelse skal dyrene være yngre end 24 timer og må ikke være først udrugede afkom. De skal være efterkommere af en sund population (dvs. uden tegn på stress, såsom høj mortalitet, tilstedeværelse af handyr og ephippier, forsinkelse i fremkomst at første udrugede afkom, misfarvning osv.) De dyr, der siden skal levere forsøgsdyr, skal dyrkes under betingelser (lys. temperatur, medium, fodring og antal dyr pr. volumenenhed), der svarer til dem, der anvendes i testen. Hvis dafnierne rutinemæssigt lever i et andet medium end det, der anvendes i testen, er det god praksis at inkludere en prætest-akklimatiseringsperiode på normalt tre uger (dvs. en generation) for at undgå at stresse forældregenerationen.
1.6.3. Testmedium
Det anbefales at anvende et veldefineret medium i denne test. Derved undgås brugen af additiver (f.eks. tang, udtræk af jord), som er vanskeligt at karakterisere, men derimod at forbedre muligheden for standardisering laboratorierne imellem. Elendt M4 (4) og M7 media (se tillæg 1) er fundet velegnede til dette formål. Andre medier kan imidlertid også bruges (f.eks. (5)(6)), forudsat at dafnie-kulturen opfylder testens validitetskriterier.
Hvis der anvendes medier, der indeholder udefinerede tilsætningsstoffer, skal disse klart specificeres, og der skal fremskaffes oplysninger til testrapporten om sammensætning, særlig med hensyn til kulstofindholdet, da det kan optræde som tilskud til diæten. Det anbefales at fastlægge det totale organiske kulstof- (TOC) og/eller kemiske iltkrav (COD) til det organiske additiv i stamopløsningen, og at der foretages en vurdering af det medfølgende tilskud til TOC/COD i testmediet. Det anbefales, at niveauet af TOC i mediet (før tilsætning af alger) er under 2 mg/l (7).
Det er vigtigt at gøre sig klart i forbindelse med undersøgelse af kemikalier, der indeholder metaller, at testmediets egenskaber (f.eks. hårdhed, cheleringskapacitet) kan have indflydelse på testkemikaliets toksicitet. Af den grund er et fuldt ud defineret medium ønskværdigt. For tiden er de eneste fuldt ud definerede medier, som er egnede til langtidskulturer med Daphnia magna Elendt M4 og M7. Begge indeholder EDTA som cheleringsmiddel. Work har påvist (2), at cadmiums tilsyneladende toksicitet generelt er lavere, når formeringstesten udføres med M4 og M7 end med medier uden EDTA. Af den grund er M4 og M7 ikke at anbefale, når der skal undersøges kemikalier indeholdende metaller, ligesom andre medier kendt for at have chelerende stoffer ligeledes bør undgås. Til kemikalier, der indeholder metaller, tilrådes brugen af andre medier som f.eks. hårdt ferskvand tilsat ASTM (7), som ikke indeholder EDTA, hvortil sættes ekstrakt af tang (8). Denne kombination af hårdt ferskvand tilsat ASTM med ekstrakt af tang er også velegnet til langtidskulturer og undersøgelse af Daphnia magna (2), endskønt den udøver en let chelerende effekt på grund af den organiske komponent i den tilsatte tangekstrakt.
Under hele testen bør koncentrationen af opløst ilt være over 3 mg/l. pH bør være inden for 6-9 og normalt ikke variere mere end 1,5 enheder. Hårdhed over 140 rng/l (målt som CaCO3) anbefales. Test på dette niveau og derover har vist formeringsevne i overensstemmelse med validitetskriterierne (9)(10).
1.6.4. Testopløsninger
Testopløsninger med de valgte koncentrationer fremstilles almindeligvis ved fortynding af en stamopløsning. Stamopløsninger bør almindeligvis fremstilles ved opløsning af de relevante kemikalier i testmediet.
Brugen af organiske opløsningsmidler eller dispergenser er i nogle tilfælde nødvendige til fremstilling af en passende koncentreret stamopløsning, men alle anstrengelser bør tages i anvendelse for at undgå brugen af sådanne materialer. Eksempler på passende opløsningsmidler er acetone, ethanol, methanol, dimethylformamid og triethylene glycol. Eksempler på dispergenser er Cremophor RH40, methylcellulose 0,01 % og HCO-40. Under alle omstændigheder bør test-kemikaliet i testopløsningen ikke overskride graden af opløselighed i testmediet.
Opløsningsmidler anvendes til fremstilling af en stamopløsning, som kan doseres i vand med stor nøjagtighed. Med den anbefalede koncentration af opløsningsmiddel i det endelige testmedium (dvs. 0,1 ml/l) vil de ovenfor nævnte opløsningsmidler ikke være toksiske, og de vil heller ikke øge et kemikaliums vandopløselighed.
Dispergensernes funktion er at hjælpe til med en nøjagtig dosering og dispersion. Med den anbefalede koncentration af dispergens i det endelige testmedium (dvs. 0,1 ml/l) vil de ovenfor nævnte dispergenser ikke være toksiske, og de vil heller ikke øge et kemikaliums vandopløselighed.
TESTENS DESIGN
Tilsætning til måleglassene og de påfølgende håndteringer af dem bør foretages på en randomiseret måde. Hvis dette ikke gøres, risikerer man bias, som kunne opfattes som resultatet af forskellige koncentrationer. Helt specielt kan dette ses, hvis undersøgelsesrækker håndteres systematisk efter behandling eller koncentrationer, at tidsrelaterede resultater, som f.eks. persontræthed eller anden fejlmulighed, kan medføre større effekt ved de højere koncentrationer. Yderligere, hvis testresultaterne kan tænkes påvirkede af begivenheder ved testens start eller af omgivelserne, som f.eks. karrenes placering i laboratoriet, bør man overveje at dele testen op i grupper.
1.8. FREMGANGSMÅDE
1.8.1. Omstændigheder ved ekspositionen
1.8.1.1. Varighed
Testen varer 21 dage.
1.8.1.2. Påfyldning
Moderdyrene holdes individuelt med ét dyr pr måleglas med 50-100 ml medium i hvert måleglas.
Af hensyn til målinger af koncentrationen af testkemikaliet kan større volumina være nødvendige; det er dog tilladt at foretage den kemiske analyse på en pulje sammensat fra flergangsbestemmelser. Hvis der anvendes volumina større end 100 ml, kan det være nødvendigt at øge fødemængden for at sikre tilstrækkelig fødetilgang for dafnien og overensstemmelse med validitetskriterierne. Hvis der anvendes gennemstrømningstest, kan det af tekniske grunde være nødvendigt med alternative design (f.eks. fire grupper med 10 dyr i et større testvolumen), men enhver ændring i testdesign skal meddeles.
1.8.1.3. Antal dyr
Ved semistatiske test skal der være mindst 10 dyr, hver holdt separat, for hver testkoncentration og mindst 10 dyr, hver holdt separat, i kontrolserien.
Det har ved gennemstrømningstest vist sig passende med 40 dyr opdelt i grupper med 10 dyr for hver testkoncentration (1). Man kan anvende et mindre antal testorganismer, og der anbefales da et minimum på 20 dyr pr. koncentration opdelt i dobbelt- eller flergangsbestemmelser med et tilsvarende antal dyr (f.eks. fire bestemmelser med hver fem dafnier). Det bemærkes, at ved test, hvor dyrene holdes gruppevis, vil det ikke være muligt at udtrykke formeringsevnen som det totale antal levende efterkommere frembragt for hvert levende forældredyr, dersom forældredyr dør i løbet af forsøget. I de tilfælde bør man udtrykke formeringsevnen som det totale antal levende afkom pr. forældredyr, som var i live ved testens begyndelse.
1.8.1.4. Fodring
I semistatiske forsøg bør fordring foretages daglig eller i hvert fald tre gange ugentlig (dvs. i sammenhæng med udskiftning af medium). Afvigelser fra dette (f.eks. for gennemstrømningstest) skal meddeles.
Under forsøget bør diæten for forældredyr fortrinsvis være levende algeceller af en eller flere af følgende: Chlorella sp., Selenastrum capricornutum (nu Pseudokirchneriella subcapitata (11)) og Scenedesmus subspicatus. Den tilførte diæt skal baseres på mængden af organisk kulstof (C) for hvert forældredyr. Undersøgelser (12) har vist, at for Daphnia magna er det tilstrækkeligt med fodermængder af størrelsesordenen 0,1 til 0,2 mg C/Daphnia/døgn med henblik på det ifølge validitetskriterierne ønskede antal afkom. Fodermængden kan enten være konstant gennem hele forsøgsperioden eller, om det ønskes, kan der gives en lavere mængde til at begynde med og siden lade mængden forøges for at tage hensyn til forældredyrets vækst under forløbet. I sidstnævnte tilfælde bør fodermængden i hele perioden stadig være inden for de anbefalede mængder på 0,1 til 0,2 mg C/Daphnia/døgn.
Måling af kulstofindhold er tidsrøvende, men hvis erstatningsmålinger, som f.eks. celletal af alger eller lysabsorption, anvendes for nemheds skyld for at afmåle den korrekte fødemængde, må hvert laboratorium fremstille et nomogram, som sammenholder erstatningsmålingen med kulstofindholdet i algekulturen (se tillæg 2 for rådgivning om konstruktion af nomogram). Nomogrammer skal kontrolleres mindst en gang årligt og hyppigere, hvis betingelserne for algerne er ændrede. Det er vist, at lysabsorption er en bedre erstatning end celletælling til beregning af kulstofindhold (13).
Man bør tilføre dafnierne algesuspensionen i koncentreret form, således at mindst muligt af algekulturmediet overføres til forsøgskarret. Opkoncentrering af alger kan foretages ved centrifugering efterfulgt af resuspension i destilleret vand eller deioniseret vand eller dafniekulturmedium.
1.8.1.5. Lys
16 timers lys med en intensitet, der ikke overstiger 15-20 μE m-2 s-1.
1.8.1.6. Temperatur
Testmediets temperatur skal ligge mellem 18 og 22 oC. I det enkelte forsøg bør temperaturen, om muligt, ikke variere mere end 2 oC (f.eks. 18-20, 19-21 eller 20-22 oC). Det kan være på sin plads at anvende et forsøgskar yderligere til brug for temperaturmonitoreringen.
1.8.1.7. Gennemluftning
Måleglassene må ikke gennemluftes under forsøget.
1.8.2. Testkoncentrationer
Normalt bør der være mindst fem testkoncentrationer arrangeret i en geometrisk række med en adskillelsesfaktor, som helst ikke bør overstige 3.2, og der skal udføres det passende antal af flergangsbestemmelser for hver testkoncentration (se 1.8.1.3). Begrundelse skal angives, hvis færre end fem koncentrationer undersøges. Kemikalier bør ikke testes i koncentrationer, der overstiger opløseligheden i testmediet.
Når range for koncentrationerne skal fastlægges, bør følgende tages i betragtning:
|
i) |
Hvis formålet er at opnå LOEC/NOEC, skal den laveste testkoncentration være så lav, at frugtbarheden ved denne koncentration ikke adskiller sig signifikant fra kontrollens. Hvis det ikke er tilfældet, skal testen gentages med formindsket laveste koncentration. |
|
ii) |
Hvis formålet er at opnå LOEC/NOEC, bør den højeste testkoncentration være så høj, at frugtbarheden ved denne koncentration er signifikant lavere end i kontrollen. Hvis det ikke er tilfældet, skal testen gentages med forøget højeste koncentration. |
|
iii) |
Hvis ECX for indvirkning på formeringsevner, beregnes, er det tilrådeligt, at der anvendes tilstrækkeligt mange koncentrationer til, at ECX kan defineres med passende sikkerhed. Hvis EC50 for indvirkning på formeringsevnen beregnes, er det tilrådeligt, at den højeste testkoncentration er større end den ved EC50. Ellers vil sikkerhedsgrænserne for EC50 blive meget vide, og selvom man kan beregne EC50, er det ikke muligt i tilstrækkelig grad at vurdere, om forsøget har været fyldestgørende. |
|
iv) |
Range for testkoncentrationerne bør ikke inkludere koncentrationer, der har en statistisk signifikant indflydelse på forældredyrets overlevelse, da det vil ændre undersøgelsens natur fra en simpel formeringstest til en kombineret formerings- og overlevelsestest, som stiller krav om mere komplekse statistiske analyser. |
Kendskab på forhånd til testkemikaliets toksicitet (f.eks. fra en hurtig test og/eller fra undersøgelser over range) kan hjælpe til at finde passende testkoncentrationer.
Når et opløsningsmiddel eller et dispergens er anvendt i præparationen af testopløsningen (se 1.6.4), bør slutkoncentrationen i måleglasset ikke være større end 0,1 ml/l og desuden være den samme i alle måleglassene.
1.8.3. Kontroller
Sideløbende med testserien bør udføres en testmediumkontrol og ligeledes, hvis det er relevant, en kontrolserie, som indeholder opløsningsmiddel eller dispergens i koncentrationer som dem, der anvendes i forsøget. Der skal udføres et passende antal flergangsbestemmelser (se 1.8.1.3).
Almindeligvis vil variationskoefficienten for det gennemsnitlige antal afkom pr forældredyr i kontrollen/kontrollerne i en vellykket test være ≤ 25, og dette skal meddeles fra testdesign, hvor dyrene har været holdt alene.
1.8.4. Fornyelse af medium
Hyppigheden for fornyelse af mediet afhænger af testkemikaliets stabilitet, men fornyelse bør foretages mindst tre gange om ugen. Hvis man ved fra forudgående stabilitetsundersøgelser, at testkemikaliet ikke er stabilt (dvs. uden for range 80-120 % af den nominelle koncentration eller falder under de 80 % af den målte begyndelseskoncentration) og ændrer sig i løbet af den maksimale fornyelsesperiode (dvs. tre døgn), bør man overveje at skifte mediet hyppigere eller at gå over til en gennemstrømningstest.
Når mediet skal fornyes i semistatiske test, er der fremstillet en anden række af måleglas, og forældredyret flyttes over til dem f.eks. ved hjælp af en glaspipette med en passende diameter. Mindst muligt medium bør overflyttes sammen med dafnien.
1.8.5. Observationer
Resultatet af undersøgelser gjort under forsøget skal noteres på data-ark (se eksempler i tillæg 3 og 4). Hvis der ønskes andre målinger (se 1.3 og 1.8.8), kan det være nødvendigt med andre undersøgelser.
1.8.6. Afkom
Det er bedst dagligt at fjerne og tælle afkom fra hvert forældredyr fra første dag, der ses yngel, for at hindre ynglen i at indtage den føde, der var tiltænkt forældredyret. Undersøgelsens formål kræver kun, at man tæller antal levende afkom, men tilstedeværelsen af aborterede æg eller dødt afkom bør også noteres.
1.8.7. Mortalitet
Dødelighed blandt forældredyrene bør noteres dagligt og i hvert fald samtidigt med, at afkommet tælles.
1.8.8. Andre parametre
Selvom metoden principielt er designet til at vurdere virkning på formeringsevnen, er det muligt, at andre virkninger kan kvantiteres, så der kan fortages statistiske beregninger. Målinger af vækst er særdeles ønskværdige, eftersom de giver informationer om mulige subletale virkninger, som kan tænkes at være mere nyttige end måling alene af formeringsevnen; det anbefales, at man ved forsøgets afslutning måler længden af forældredyrene (dvs. kropslængden minus den anale spina). Andre parametre kan måles eller beregnes såsom tiden til fremkomst af den første yngel (og den påfølgende yngel), antal og størrelse af yngel pr. dyr, antallet af aborteret yngel, tilstedeværelse af handyr eller ephippier, populationens egentlige formeringshastighed.
1.8.9. Hyppighed af analyser og målinger
Ilt-koncentration, temperatur, hårdhedsgrad og pH-værdier bør måles i hvert fald hver uge i nye og gamle medier, i kontroller og i opløsninger med den højeste koncentration af testkemikaliet.
Mens forsøget løber, måles koncentrationen af testkemikaliet med regelmæssige intervaller.
I semistatiske test, hvor koncentrationen af testkemikaliet forventes at forblive inden for ± 20 % af den nominelle koncentration (dvs. inden for range 80-120 % — se 1.4 og 1.8.4), anbefales det som et minimum at måle højeste og laveste testkoncentration ved fremstillingen og i forbindelse med udskiftning ved en lejlighed i den første uge af forsøget (dvs, analyser skal udføres på en prøve fra samme opløsning — frisk fremstillet og ved en fornyelse). Disse målinger bør gentages mindst ugentligt derefter.
I forbindelse med test, hvor testkemikaliet ikke forventes at holde sig inden for ± 20 % af den nominelle koncentration, er det nødvendigt at analysere alle testkoncentrationer, når de er frisk fremstillede og ved udskiftning. I de test, imidlertid, hvor testkemikaliets målte begyndelseskoncentration ikke ligger inden for ± 20 % af den nominelle koncentration, men hvor der er tilstrækkelig bevis for, at begyndelsesværdierne kan genfindes og er stabile (dvs. inden for range 80-120 % af begyndelseskoncentrationen), kan kemiske målinger reduceres i uge 2 og 3 til kun at omfatte den højeste og den laveste testkoncentration. I alle tilfælde behøver man før fornyelse kun at bestemme testkemikaliets koncentration i et af måleglassene i en flergangsbestemmelse for hver af testkoncentrationerne.
Anvendes en gennemstrømningstest, er et analyseregime svarende til de semistatiske test passende (men i dette tilfælde er måling på »gamle« opløsninger ikke mulig). I den første uge kan det være tilrådeligt at øge antallet af prøvetagninger (f.eks. tre hold målinger) for at sikre, at testkoncentrationerne holder sig stabile. I den type test bør gennemstrømningshastigheden for fortyndingsvæske og testkemikalium kontrolleres daglig.
Hvis det viser sig, at koncentrationen af det undersøgte kemikalium på tilfredsstillende måde er holdt inden for ± 20 % af den nominelle eller målte begyndelseskoncentration gennem hele testforløbet, kan resultaterne baseres på de nominelle eller målte begyndelseskoncentrationer. Hvis afvigelsen fra den nominelle eller målte begyndelseskoncentration er større end ± 20 %, skal resultaterne udtrykkes som tidsvægtet gennemsnit (se tillæg 5).
2. DATA OG RAPPORTERING
2.1. BEHANDLING AF RESULTATER
Formålet med denne test er at bestemme virkningen af testkemikaliet på det totale antal levende afkom pr, forældredyr, som er i live ved forsøgets afslutning. Det totale antal afkom pr. forældredyr skal beregnes for hver beholder (dvs. hver enkeltbestemmelse). Viser det sig ved en enkeltbestemmelse, at forældredyret dør under forsøget, eller at det er af hankøn, ekskluderes denne enkeltbestemmelse fra analysen. Analysen vil så blive baseret på et reduceret antal enkeltbestemmelser.
Vurderingen af LOEC og dermed NOEC for virkningen af kemikaliet på det totale antal afkom forudsætter en beregning af det gennemsnitlige antal afkom på tværs af enkeltbestemmelserne for hver koncentration samt den samlede residuale standarddeviation. Dette kan gøres ved hjælp af en variansanalyse (ANOVA). Gennemsnittet for hver koncentration skal så sammenholdes med kontrolgennemsnittet ved hjælp af en passende metode til multiple sammenligninger. Dunnetts eller Williams test kan være anvendelige (14) (15) (16) (17). Det er nødvendigt at kontrollere, at ANOVA-forudsætningen om varianshomogenitet holder stik. Det anbefales, at dette gøres grafisk snarere end ved en formel signifikanstest (18). Et passende alternativ er Bartletts test. Hvis den nævnte forudsætning ikke holder, bør man overveje at transformere data med henblik på sikring af homogenitet før udførelse af ANOVA eller at udføre en vægtet ANOVA. Størrelsen af den virkning, der kan påvises med ANOVA (dvs. mindste signifikante forskel), skal beregnes og meddeles.
Til vurdering af den koncentration, som medfører en 50 % reduktion af det totale antal afkom (dvs. EC50), konstrueres en passende kurve, f.eks. en logistisk kurve, som er tilpasset data ved hjælp af en statistisk metode, f.eks. de mindste kvadraters metode. Kurven skal parameteriseres, således at EC50 og standardfejlen på denne kan aflæses direkte. Dette vil lette beregningen af sikkerhedsgrænserne for EC50. Medmindre der er gode grunde ti! at foretrække andre sikkerhedsgrænser, skal tosidede 95 %-sikkerhedsgrænser meddeles. Tilpasningsproceduren skal helst tillade en vurdering af signifikansen af den manglende overensstemmelse. Man kan gøre dette grafisk, eller man kan inddele den residuale kvadratsum i to komponenter: manglende overensstemmelse og tilfældig variation, hvorefter man udfører en signifikanstest for den manglende overensstemmelse. Eftersom behandlinger, der medfører højere frugtbarhed, må formodes at udvise større varians i antallet af afkom end behandlinger, der medfører lav frugtbarhed, bør man overveje at vægte de observerede data, så de afspejler de forskellige varianser i de forskellige behandlingsgrupper (se baggrundsinformation i henvisning (18)).
Ved analysen af data fra den endelige »ringtest« (2) tilpassedes en logistisk kurve ved hjælp af følgende model, selvom andre passende modeller kan anvendes:
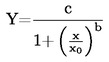
hvor:
|
Y |
= |
totale antal afkom pr. forældredyr, som var i live ved forsøgets afslutning (beregnet for hver beholder) |
|
x |
= |
substanskoncentrationen |
|
c |
= |
det forventede antal afkom, når x = 0 |
|
x0 |
= |
EC50 i populationen |
|
b |
= |
hældningen. |
Denne model vil formentlig være passende i et stort antal tilfælde, men der vil være forsøg, hvor den ikke er adækvat. Man bør vurdere modellens validitet som foreslået ovenfor. I nogle tilfælde vil det være passende at anvende en hormese-model, hvor lave koncentrationer giver en øget virkning (19).
Andre virkningskoncentrationer, såsom EC10 eller EC20 kan også estimeres. men de! kan i så fald være formålstjenligt at anvende en parameterisering af modellen, som er forskellig fra den, der anvendtes til at estimere EC50.
2.2. TESTRAPPORT
Test-rapporten skal indeholde:
2.2.1. For test-kemikaliet:
|
— |
fysiske natur og relevante fysisk-kemiske egenskaber |
|
— |
data vedrørende den kemiske identifikation inklusive renhedsgrad. |
2.2.2. For dyrearten:
|
— |
klonen (om den er blevet genotypebestemt), leverandør eller kilde (hvis kendt) og dyrkningsbetingelserne. Hvis en anden art end Daphnia magna anvendes, skal det meddeles og begrundes. |
2.2.3. Testomstændigheder:
|
— |
den anvendte test-procedure (f.eks. semistatisk eller gennemstrømning, volumen, antal dafnier pr liter ved påfyldning |
|
— |
lysperiode og lysintensitet |
|
— |
testdesign (f.eks. antal flergangsbestemmelser, antal forældredyr pr måleglas) |
|
— |
detaljer vedrørende kulturmediet |
|
— |
eventuel tilsætning af organisk materiale, inkluderende sammensætning, kilde, fremstillingsmetode, stamopløsningens TOC/COD, vurdering af resulterende TOC/COD i testmediet |
|
— |
detaljeret information angående fodring, inklusive mængde (i mg C/Dafnia/døgn) og liste (f.eks. fodertype(r)) inklusive algernes specifikke navn (arten) og, hvis kendt, herkomst og dyrkningsbetingelserne |
|
— |
måde til fremstilling af stamopløsninger og hyppighed af fornyelse (evt. opløsningsmidler eller dispergenser og deres koncentrationer skal opgives). |
2.2.4. Resultater
|
— |
resultater af ethvert forudgående studium af test-kemikaliets stabilitet |
|
— |
de nominelle testkoncentrationer og resultaterne af alle analyser til bestemmelse af koncentrationen af testkemikaliet i måleglasset (se eksempler på dataark i tillæg 4); metodens genfindingsevne og dens grænser skal også meddeles |
|
— |
vandets kvalitet i måleglasset (dvs. pH, temperatur, koncentrationen af opløst ilt og TOC og/eller COD og hårdhedsgrad, hvor dette er relevant) (se eksempler på data-ark i tillæg 3) |
|
— |
fuld fortegnelse over levende afkom efter hvert moderdyr (se eksempler på data-ark i tillæg 3) |
|
— |
antal døde moderdyr og dagen for hændelsen (se eksempler på data-ark i tillæg 3) |
|
— |
variationskoefficiemen for frugtbarhed i kontrollerne (baseret på det totale antal afkom pr forældredyr, som er i live ved testens afslutning) |
|
— |
det totale antal af levende afkom pr. forældredyr (for hver af flergangsbestemmelserne), som er i live ved testens afslutning plottet over for koncentrationen af testkemikaliet |
|
— |
laveste koncentration med observeret effekt (LOEC) med hensyn til frugtbarhed, inklusive en beskrivelse af den anvendte statistiske fremgangsmåde og en angivelse af, hvilken virkningsstørrelse der kunne måles og koncentration uden observeret effekt (NOEC) med hensyn til frugtbarhed: hvor relevant opgives ligeledes LOEC/NOEC-mortaliteten for forældredyrene |
|
— |
hvor relevant ECx med hensyn til frugtbarhed og sikkerhedsgrænser og en graf af den model, der var anvendt til beregningen, hældningen på dosis-responskurven og standardfejlen |
|
— |
andre observerede biologiske virkninger eller målinger: der skal rapporteres enhver biologisk virkning, som blev observeret eller målt (f.eks. forældredyrenes vækst) inklusive relevante begrundelser |
|
— |
forklaringer for enhver afvigelse fra testmetoden. |
HENVISNINGER
|
(1) |
OECD Test Guideline Programme, Report of the Workshop on the Daphnia magna Pilot Ring Test, Sheffield University, UK. 20-21 March 1993. |
|
(2) |
OECD Environmental Health and Safety Publications. Series on Testing and Assessment No. 6. Report of the Final Ring Test of the Daphnia magna Reproduction Test Paris. 1997. |
|
(3) |
Baird D.J,, Barber J., Bradley M.C, Soares A.M.V.M. and Calow P. (1991). A comparative study of genotype sensitivity to acute toxic stress using clones of Daphnia magna Strauss. Ecotoxicology and Environmental Safety, 21, pp. 257-265. |
|
(4) |
Elendt B.P., (1990). Selenium deficiency in Crustacea; An ultrastructural approach to antennal damage in Daphnia magna Straus. Protoplasma, 154, pp. 25-33. |
|
(5) |
EPA (1993). Methods for Measuring the Acute Toxicity of Effluents and Receiving Waters to Freshwater and Marine Organisms. (Fourth ed.). EPA/600/4-90/027F. C. I. Weber (ed), USEPA, Cincinnati, Ohio. |
|
(6) |
Vigano L, (1991) Suitability of commercially available spring waters as standard medium for culturing Daphnia magna. Bull. Environ. Contam. Toxicol, 47, pp. 775-782. |
|
(7) |
ASTM (1988). Standard Guide for Conducting Acute Toxicity Tests with Fishes, Macromvertebrates and Amphibians. E729-88a. American Society for Testing and Materials, Philadelphia P.A. 20 pp. |
|
(8) |
Baird D.J., Soares A.M.V.M., Girling A., Barber J., Bradley M.C. and Calow P. (1989). The long term maintenance of Daphnia magna Straus for use in ecotoxicological tests; problems and prospects. In: Proceedings of the 1st European Conference on Ecotoxicology. Copenhagen 1988 (H.Løkke, H. Tyle & F. Bro-Rasmussen. Eds.), pp. 144-148. |
|
(9) |
Parkhurst B.R., Forte J.L and Wright G.P. (1981). Reproducibility of a life-cycle toxicity test with Daphnia magna. Bull. Environ. Contam. and Toxicol., 26, pp. 1-8. |
|
(10) |
Cowgill U.M. and Milazzo D.P. (1990) The sensitivity of two cladocerans to water quality variables: salinity and hardness. Arch. Hydrobiol., 120(2), pp. 185-196. |
|
(11) |
Korshikov (1990). Pseudokirchneriella subcapitata Hindak, F-1990. Biologice Prace, 36, 209. |
|
(12) |
Sims I.R., Watson S. and Holmes D. (1993). Toward a standard Daphnia juvenile production test. Environmental Toxicology and Chemistry, 12, pp. 2053-2058. |
|
(13) |
Sims i. (1993). Measuring the growth of phytoplankton: the relationship between total organic carbon with three commonly used parameters of algal growth. Arch. Hydrobiol., 128, pp. 459-466. |
|
(14) |
Dunnett C.W., (1955). A multiple comparisons procedure for comparing several treatments with a control. j. Amer. Statist. Assoc, 50, pp. 1096-1121. |
|
(15) |
Dunnett C.W., (1964). New tables for multiple comparisons with a control. Biometrics, 20, pp. 4S2-491. |
|
(16) |
Williams D.A. (1971). A test for differences between treatment means when several dose levels are compared with a zero dose control. Biometrics 27, pp. 103-117. |
|
(17) |
Williams D.A. (1972). The comparison of several dose levels with a zero dose control. Biometrics, 28, pp. 510-531. |
|
(18) |
Draper N.R. and Smith H. (1981). Applied Regression Analysis, second edition, Wiley, N.Y. |
|
(19) |
Brain P. and Cousens R. (1989). An equation to describe dose responses where there is stimulation of growth at low doses. Weed Research, 29, pp. 93-96. |
|
(20) |
Wilson E.O. and Bossert, W.H. (1971). A Primer of Population Biology. Sinauer Associates Inc. Publishers. |
|
(21) |
Poole R.W. (1974). An Introduction to quantitative Ecology. McGraw-Hill Series in Population Biolog}', New York, pp. 532. |
|
(22) |
Meyer). S., Ingersoll C.G., McDonald L.L and Boyce M.S. (1986). Estimating uncertainty in population growth rates: Jackknife vs bootstrap techniques. Ecology, 67. pp. 1156-1166. |
Tillæg 1
KOMPLET FREMSTILLING AF MEDIERNE M7 OG M4
Akklimatisering til Medierne Elendt M7 og M4
Nogle laboratorier har haft problemer med at overflytte datnier direkte til M4- (1) og M7-medierne. Imidlertid har en gradvis akklimatisering givet nogen succes; man flytter dafnierne fra deres eget medium til et 30 % Elendt, siden til et 60 % Elendt og så til et 100 % Elendt. Akklimatiseringsperioden kan være på op til en måned.
FREMSTILLING
Sporelementer
Separate stamopløsninger (I) med de forskellige sporstoffer fremstilles primært i vand af passende renhedsgrad, f.eks. de-ioniseret, destilleret vand eller vand renset ved omvendt osmose. Ud fra disse forskellige stamopløsninger (I) fremstilles en sekundær stamopløsning (II), som indeholder alle sporstofferne (kombinationsopløsningen), som følger:
|
Stamopløsning I (enkelt stof) |
Antal mg sat til 1 liter vand |
Koncentrationer (refererende til M4) (fold) |
For at fremstille stamopløsning Il tilsættes nedennævnte antal ml af stamopløsning i til 1 liter vand |
|
|
M4 |
M7 |
|||
|
H3BO3 |
57 190 |
20 000 |
1,0 |
0,25 |
|
MnCl2 * 4H2O |
7 210 |
20 000 |
1,0 |
0,25 |
|
LiCl |
6 120 |
20 000 |
1,0 |
0,25 |
|
RbCl |
1 420 |
20 000 |
1,0 |
0,25 |
|
SrCl2 * 6 H20 |
3 040 |
20 000 |
1,0 |
0,25 |
|
NaBr |
320 |
20 000 |
1,0 |
0,25 |
|
Na2Mo04 * 2 H2O |
1 260 |
20 000 |
1,0 |
0,25 |
|
CuCl2 * 2 H2O |
335 |
20 000 |
1,0 |
0,25 |
|
ZnCl2 |
260 |
20 000 |
1,0 |
1,0 |
|
CoCl2 * 6 H2O |
200 |
20 000 |
1,0 |
1,0 |
|
Kl |
65 |
20 000 |
1,0 |
1,0 |
|
Na2SeO3 |
43,8 |
20 000 |
1,0 |
1,0 |
|
NH4VO3 |
11,5 |
20 000 |
1,0 |
1,0 |
|
Na2EDTA * 2 H2O |
5 000 |
2 000 |
— |
— |
|
FeSO4 * 7 H20 |
1 991 |
2 000 |
_ |
_ |
|
Na2EDTA- og FeSO4-opløsningerne fremstilles hver for sig. hældes sammen og autoklaveres straks. Dette giver: |
||||
|
21 Fe-EDTA-opløsning |
|
1 000 |
20,0 |
5,0 |
M4- og M7-medierne
M4- og M7-medierne fremstilles ved hjælp af stamopløsning II, makronæringsstofferne og vitaminerne som følger:
|
|
Antal mg sat til 1 liter vand |
Koncentrationer (relateret til medium M4) (fold) |
Antal ml af stamopløsning, der sættes til 1 liter for at fremstille mediet |
|||||
|
M4 |
M7 |
|||||||
|
Stamopløsning II samlede sporstoffer |
|
20 |
50 |
50 |
||||
|
Stamopløsning med makro-næringsstoffer (enkelte stoffer) |
||||||||
|
CaCl2 * 2 H2O |
293 800 |
1 000 |
1,0 |
1,0 |
||||
|
MgSO4* 7 H2O |
246 600 |
2 000 |
0,5 |
0,5 |
||||
|
KCl |
58 000 |
10 000 |
0,1 |
0,1 |
||||
|
NaHCO3 |
64 800 |
1 000 |
1,0 |
1,0 |
||||
|
Na2SiO3 * 9 H2O |
50 000 |
5 000 |
0,2 |
0,2 |
||||
|
NaNO3 |
2 740 |
10 000 |
0,1 |
0,1 |
||||
|
KH2PO4 |
1 430 |
10 000 |
0,1 |
0,1 |
||||
|
K2HPO4 |
1 840 |
10 000 |
0,1 |
0,1 |
||||
|
Kombineret vitaminopløsning |
— |
10 000 |
0,1 |
0,1 |
||||
|
Den kombinerede vitaminopløsning fremstilles ved at sætte de 3 vitaminer til 1 liter vand som vist nedenfor: |
||||||||
|
Thiaminhydrochlorid |
750 |
10 000 |
— |
— |
||||
|
Cyanocobalamin (B12) |
10 |
10 000 |
— |
— |
||||
|
Biotin |
7,5 |
10 000 |
— |
— |
||||
|
Den kombinerede vitaminopløsning opbevares i små mængder i frossen tilstand. Vitaminerne sættes til mediet kort før brugen.
|
||||||||
Tillæg 2
ANALYSE FOR DET TOTALE ORGANISKE KULSTOF (TOC) OG FREMSTILLING AF NOMOGRAM TIL TOC-INDHOLDET I ALGEFODERET
Det erkendes, at kulstofindholdet i algefoderet almindeligvis ikke vil blive målt direkte, men ud fra korrelationer (dvs. nomogrammer med erstatningsmålinger, såsom antallet af algeceller eller lysabsorbans).
TOC bør måles ved oxidation ved høje temperaturer snarere end ved UV eller persulphate metoder. (Se: The Instrumental Determination of Total Organic Carbon, Total Oxygen Demand and Related Determinands 1979, 1980; 49 High Holborn, London WC1V 6HB).
Med henblik på fremstilling af nomogrammer skal algerne adskilles fra dyrkningsmediet ved centrifugering med påfølgende resuspension i destilleret vand. Mål erstatningsparameteren og TOC-koncentrationen i hver prøve som en tredobbelt bestemmelse. Blindværdier måles i destilleret vand, og TOC-koncentrationen trækkes fra TOC-værdien i algeprøven.
Nomogrammet skal være lineært i hele området over kulkoncentrationer. Eksempler ses nedenfor:
NB: Værdierne fra det her viste nomogram må ikke anvendes; det er essentielt at laboratorierne selv fremstiller deres egne nomogrammer.


Tillæg 3
EKSEMPEL PÅ DATAARK TIL NOTATER OM FORNYELSE AF MEDIUM, FYSISK/KEMISK MONITORERING, FODRING, FORMERING OG MORTALITET BLANDT VOKSNE DAFNIER
|
Eksperiment nr. |
Startdato: |
Klon: |
Medium: |
Fodertype: |
Testkemikalium: |
Nominel koncentration: |
||||||||||||||||||
|
Dag |
0 |
1 |
2 |
3 |
4 |
5 |
6 |
7 |
8 |
9 |
10 |
11 |
12 |
13 |
14 |
15 |
16 |
17 |
18 |
19 |
20 |
21 |
|
|
|
Fornyelse af medium (sæt kryds) |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
pH (37) |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
ny |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
gammel |
|
|
|
O2 mg/l (37) |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
ny |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
gammel |
|
|
|
Temperatur ( oC) (37) |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
ny |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
gammel |
|
|
|
Fodring (sæt kryds) |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Antal levende afkom (38) |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Total |
|
Måleglas 1 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
2 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
3 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
4 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
5 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
6 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
7 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
8 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
9 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
10 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Total |
|
|
Kumulativ voksenmortalitet (39) |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Tillæg 4
EKSEMPEL PÅ DATAARK MED RESULTATER AF KEMISKE ANALYSER
a) Målte koncentrationer
|
Nominel koncentration |
Prøver i uge 1 |
Prøver i uge 2 |
Prøver i uge 3 |
|||
|
ny |
gammel |
ny |
gammel |
ny |
gammel |
|
|
|
|
|
|
|
|
|
b) Målte koncentrationer i procent af nominelle
|
Nominel koncentration |
Prøver i uge 1 |
Prøver i uge 2 |
Prøver i uge 3 |
|||
|
ny |
gammel |
ny |
gammel |
ny |
gammel |
|
|
|
|
|
|
|
|
|
Tillæg 5
BEREGNING AF TIDSVÆGTET GENNEMSNIT
Tidsvægtet gennemsnit
Idet koncentrationen af testkemikaliet kan falde i perioden mellem fornyelsen af medierne, er det nødvendigt at overveje, hvilken koncentration der skal vælges som repræsentant for variationen af koncentrationer, som den voksne dafnie bliver udsat for. Valget skal baseres på såvel biologiske som statistiske betragtninger. Hvis for eksempel formeringen formodes mest påvirket af den højeste koncentration, så skal den anvendes. Hvis imidlertid den akkumulerede effekt eller langtidseffekten fra det toksiske stof regnes for mere vigtigt, så er gennemsnitskoncentrationen mere relevant. I dette tilfælde er den tidsvægtede gennemsnitskoncentration det passende at anvende, eftersom den tager hensyn til variationen af hvert øjebliks koncentration i tidsforløbet.
Figur 1
Eksempel på tidsvægtet gennemsnit

Figur 1 viser som eksempel et (forenklet) forsøg over syv dage med en fornyelse af mediet på dag 0, 2 og 4.
|
|
Zigzaglinjen repræsenterer koncentrationen på ethvert tidspunkt. Faldet i koncentration formodes at følge en eksponential henfaldsproces. |
|
|
De seks angivne punkter repræsenterer de målte koncentrationer ved begyndelsen og slutningen af hver fornyelsesperiode. |
|
|
Den tykke, solide linje angiver placeringen af det tidsvægtede gennemsnit. |
Det tidsvægtede gennemsnit er beregnet, så arealet under det tidsvægtede gennemsnit har samme størrelse som arealet under koncentrationskurven. Beregningen af det ovenfor viste eksempel ses i tabel 1.
Tabel 1
Beregning af tidsvægtet gennemsnit
|
Fornyelse nr. |
Dage |
Konc0 |
Konc1 |
Ln(Konc0) |
Ln(Konc1) |
Areal |
|
1 |
2 |
10,000 |
4,493 |
2,303 |
1,503 |
13,767 |
|
2 |
2 |
11,000 |
6,037 |
2,398 |
1,798 |
16,544 |
|
3 |
3 |
10,000 |
4,066 |
2,303 |
1,403 |
19,781 |
|
Totale antal dage: 7 |
Totale areal |
50,091 |
||||
|
TW gennemsnit |
7,156 |
|||||
|
»Dage« er antallet af dage i fornyelsesperioden. »Konc0« er den målte koncentration ved begyndelsen af hver fornyelsesperiode. »Konc1« er den målte koncentration ved afslutningen af hver fornyelsesperiode. »Ln(Konc0)« er den naturlige logaritme til Konc0. »Ln(Konc1l)« er den naturlige logaritme til Konc1. »Arealet« er arealet under eksponentialkurven for hver fornyelsesperiode. Det beregnes således:
|
||||||
Det tidsvægtede gennemsnit (TW gennemsnit) er det »totale areal« divideret med de »totale antal dage«.
Tabellen vil selvfølgelig strække sig over 21 dage i forbindelse med dafnieformeringstesten.
Det er klart, at når der kun foretages målinger ved begyndelsen og slutningen af hver fornyelsesperiode, kan man ikke bekræfte, at henfaldsprocessen er eksponentiel. En anderledes kurve ville resultere i en anderledes beregning for arealet. En eksponential henfaldsproces er dog ikke usandsynlig, og den er den bedste kurve at anvende, når der ikke foreligger andre informationer.
Stor omhu må udvises, hvis en kemisk analyse ikke kan påvise test-kemikaliet ved afslutningen af en fornyelsesperiode. Medmindre man kan påvise, med hvilken hastighed testkemikaliet forsvandt fra opløsningen, er det ikke muligt at finde frem til et realistisk areal under kurven, hvilket medfører, at det er umuligt at finde frem til et rimeligt tidsvægtet gennemsnit.
C.21. JORDMIKROORGANISMER: KVÆLSTOFOMDANNELSESTEST
1. METODE
Metoden er gengivet efter OECD TG 216 (2000).
1.1. INDLEDNING
Den beskrevne metode er en laboratoriemetode til undersøgelse af langtidsvirkningerne på jordmikroorganismers kvælstofomdannende aktivitet af enkeltstående eksponering for kemiske stoffer. Metoden er hovedsagelig baseret på anbefalinger fra Plantebeskyttelsesorganisationen for Europa og Middelhavsområderne (1). Der er endvidere taget hensyn til guidelines fra den tyske Biologische Bundesanstalt (2), den amerikanske miljøstyrelse (3), SETAC (4) og Den Internationale Standardiseringsorganisation (5). På en OECD-workshop om valg af jord-/sedimenttype, som blev holdt i Belgirate, Italien, i 1995 (6), enedes man om antal og art af de jordtyper, som skal anvendes i denne test. Anbefalingerne for indsamling, håndtering og opbevaring af jordprøver er baseret på en ISO-vejledning (7) og på anbefalingerne fra workshoppen i Belgirate, Ved vurdering af prøvestoffers toksiske egenskaber kan det være nødvendigt at bestemme virkningerne på den mikrobielle aktivitet i jord, f.eks. når der kræves oplysninger om plantebeskyttelsesmidlers potentielle bivirkninger på mikrofloraen i jorden, eller når jordmikroorganismer forventes udsat for andre kemiske stoffer end plantebeskyttelsesmidler. Kvælstofomdannelsesprøven udføres for at bestemme virkningerne af sådanne kemiske stoffer på mikrofloraen i jorden. Ved test af landbrugskemikalier (f.eks. plantebeskyttelsesmidler, kunstgødning og skovbrugskemikalier) udføres både kvælstofomdannelses- og kulstofomdannelsestest. For andre kemiske stoffer end landbrugskemikalier er kvælstofomdannelsestesten tilstrækkelig. Viser sådanne kemiske stoffer sig imidlertid i kvælstofomdannelsestesten at have en EC50-værdi i samme område som gængse nitrifikationsinhibitorer (f.eks. nitrapyrin), kan en kulstofomdannelsestest udføres for at skaffe yderligere oplysninger.
Jord består af levende og ikke levende komponenter, som er til stede i komplekse og heterogene blandinger. Mikroorganismer spiller en vigtig rolle i nedbrydning og omdannelse af det organiske stof i frugtbar jord, og mange arter bidrager til flere forskellige aspekter af jordens frugtbarhed. Hvis der gennem længere tid gribes ind i disse biokemiske processer, kan det gribe ind i næringsstofcyklussen, således at jordens frugtbarhed ændrer sig. Kulstof- og kvælstofomdannelse finder sted i al frugtbar jord. Skønt det alt efter jordtypen er forskellige mikrobielle samfund, som er ansvarlige for disse processer, følger omdannelsen i det væsentlige altid samme vej.
Den her beskrevne forsøgsmetode anvendes til bestemmelse af stoffers langsigtede skadevirkning på kvælstofomdannelsesprocessen i jordens aerobe dæklag. Metoden giver desuden mulighed for at bedømme stoffernes indvirkning på den mikrobielle kulstofomdannelse i jorden. Efter nedbrydning af bindingerne mellem kulstof og kvælstof dannes nitrat. Finder man lige stor nitratproduktion i den behandlede jord og i kontroljord, er de vigtigste kulstotfnedbrydningsveje derfor højst sandsynligt intakte og funktionsdygtige. Det ved testen anvendte substrat (finmalet lucernemel) har et gunstigt kulstof-/kvælstofforhold (sædvanligvis mellem 12/1 og 16/1). Dette mindsker kulstofmanglen under testen, og hvis de mikrobielle samfund beskadiges af et kemikalie, kan de muligvis komme på fode inden for 100 dage.
De test, som metoden oprindeligt er baseret på, er hovedsagelig udviklet til stoffer, for hvilke den til jorden tilførte mængde er kendt på forhånd. Det er f.eks. tilfældet for plantebeskyttelsesmidler, hvor spredemængden i marken kendes. For landbrugskemikalier er det tilstrækkeligt at teste to doser, som er relevante for den forventede eller forudsete spredemængde. Landbrugskemikalier kan enten testes som aktivt indholdsstof eller som færdigformuleret produkt. Testen er dog ikke begrænset til landbrugskemikalier. Ved at ændre dels den tilførte mængde teststof til jorden, dels metoden til vurdering af data kan man desuden anvende testen på kemiske stoffer, hvor man ikke kender den mængde, som vil nå jorden. For andre kemiske stoffer end landbrugskemikalier bestemmes virkningen af en række koncentrationer på kvælstofomdannelsen. Data fra disse forsøg anvendes til at opstille en dosis-responskurve og beregne værdier af ECx hvor x er den definerede virkning i %.
1.2. DEFINITIONER
Kvælstofomdannelse: den fuldstændige mikrobielle nedbrydning af kvælstofholdigt organisk stof via ammonifikations- og nitrifikationprocesser til det pågældende uorganiske nitrat som slutprodukt.
EC x (effektiv koncentration): den koncentration af prøvestoffet i jord, som medfører en hæmning på x procent af kvælstofomdannelsen til nitrat.
EC 50 (effektiv mediankoncentration): den koncentration af prøvestoffet i jord, som medfører en 50 % hæmning af kvælstoffets omdannelse til nitrat.
1.3. REFERENCESTOFFER
Ingen.
1.4. FORSØGSMETODENS PRINCIP
Siet jord forbedres med finmalet plantemel og behandles enten med prøvestoffet eller lades ubehandlet (kontrol). Til test af landbrugskemikalier anbefales mindst to prøvekoncentrationer, som vælges i henhold til den højeste forventede koncentration i marken. Efter 0, 7, 14 og 28 døgns inkubation ekstraheret prøver af behandlet jord og kontroljord med et passende opløsningsmiddel, og nitratmængden i ekstrakterne bestemmes. Nitratomdannelsen i de behandlede prøver sammenholdes med omdannelsen i kontrolprøverne, og den procentuelle afvigelse af de behandlede prøver fra kontrolprøverne beregnes. Alle forsøg løber over mindst 28 døgn. Er forskellen mellem behandlet og ubehandlet jord 25 % eller derover på dag 28, fortsættes målingerne indtil højst 100 dage. Ved test af andre stoffer end landbrugskemikalier tilsættes en række koncentrationer af prøvestoffet til jordprøverne, og den dannede mængde kvælstof i behandlede prøver og kontrolprøver måles efter 28 døgns inkubation. Resultaterne af forsøg med flere koncentrationer analyseres efter en regressionsmodel, og ECx-værdierne beregnes (dvs. EC50, EC25 og/eller EC10). Jf. definitionerne.
1.5. FORSØGETS VALIDITET
Vurdering af forsøgsresultater med landbrugskemikalier bygger på nogle forholdsvis små forskelle (gennemsnitligt ± 25 %) mellem nitratkoncentrationen i kontrolprøver og i behandlet jord, hvorfor store variationer i kontrolprøverne kan give forkerte resultater. Afvigelsen mellem gentagne kontrolprøver skal derfor være mindre end ± 15 %.
1.6. BESKRIVELSE AF FORSØGSMETODEN
1.6.1 Apparatur
Prøvebeholderne skal være udført i kemisk inaktivt materiale. De skal have passende kapacitet svarende til den metode, der anvendes til inkubation af jordprøverne, dvs. inkubation af den samlede mængde eller som en række individuelle jordprøver (jf. 1.7.1.2), Der skal være draget omsorg både for at vandtabet minimeres og at gasser kan udveksles under prøven (f.eks. kan prøvebeholderne tildækkes med perforeret polyethylenfolie). Ved test af flygtige stoffer skal anvendes forseglede og gastætte beholdere. Beholderne skal have en størrelse, så deres rumfang er ca. en fjerdedel udfyldt af jordprøven.
Der anvendes standardlaboratorieudstyr, herunder følgende:
|
— |
til omrystning: mekanisk rystebord eller tilsvarende |
|
— |
centrifuge (3 000 g) eller filtreringsanordning (med nitratfrit filterpapir) |
|
— |
instrument til nitratanalyse med tilstrækkelig følsomhed og reproducerbarhed. |
1.6.2. Jordtyper og antal heraf
Der anvendes én enkelt jordtype. Følgende jordkarakteristika anbefales:
|
— |
sandindhold: mindst 50 % og ikke over 75 % |
|
— |
pH: 5,5-7,5 |
|
— |
organisk kulstofindhold: 0,5-1,5 % |
|
— |
den mikrobielle biomasse skal måles (8)(9), og dens kulstofindhold skal udgøre mindst 1 % af jordens samlede organiske kulstofindhold. |
I de fleste tilfælde vil en jord med sådanne egenskaber repræsentere den værst tænkelige situation, da prøvestoffets adsorption er minimal og dets tilgængelighed for mikrofloraen maksimal. Sædvanligvis behøver man derfor ikke udføre forsøg med andre jordtyper. Under visse omstændigheder, f.eks. når prøvestoffet hovedsagelig forventes anvendt i bestemte jordtyper som sur skovbundsjord eller når der er tale om elektrostatisk ladede kemiske stoffer, kan det være nødvendigt at anvende en yderligere jordtype.
1.6.3. Indsamling og opbevaring af jordprøver
1.6.3.1. Indsamling
Der skal foreligge detaljerede historiske oplysninger om det markareal, hvorfra prøvejorden er indsamlet. Disse oplysninger skal omfatte nøjagtig beliggenhed, plantedække, dato for behandlinger med plantebeskyttelsesmidler, behandlinger med organisk gødning og kunstgødning, tilførsler af biologisk materiale og uheldsbetingede forureningsepisoder. Til indsamling af jord skal vælges et areal, som kan anvendes gennem længere tid. Egnede er arealer med vedvarende græs, marker med årlig kornhøst (bortset fra majs) eller tæt tilsået grøngødskning. De valgte prøvetagningsarealer må ikke have været behandlet med plantebeskyttelsesmidler i mindst ét år forud for prøvetagningen. Endvidere må der ikke have været anvendt organisk gødning inden for de sidste seks måneder. Anvendelse af mineralsk gødning er kun acceptabel, når det sker i overensstemmelse med afgrødens behov, og når jordprøverne tidligst udtages tre måneder efter udbringning af gødningen. Jord, som har været behandlet med kunstgødning med kendt biocid virkning (f.eks calciumeyanamid) bør ikke anvendes.
Prøvetagning bør undgås under eller umiddelbart efter lange perioder (mere end 30 dage) med tørke eller oversvømmelse. Af pløjet jord tages prøver fra en dybde af 0 ned til 20 cm. For græsningsarealer eller andre jordtyper, hvor der ikke pløjes i længere perioder (mindst én vækstsæson), kan den maksimale prøvetagningsdybde være en smule mere end 20 cm (f.eks. ned til 25 cm).
Jordprøverne skal transporteres i sådanne beholdere og under sådanne temperaturforhold, at jordens oprindelige egenskaber med sikkerhed ikke ændrer sig nævneværdigt.
1.6.3.2. Opbevaring
Der bør fortrinsvis anvendes jord, som er frisk optaget fra marken. Er opbevaring i laboratoriet ikke til at undgå, kan jorden opbevares i mørke ved 4 + 2 oC i indtil tre måneder. Under opbevaringen skal aerobe forhold være sikret. Er jorden indsamlet fra områder, hvor den er frossen mindst tre måneder årligt, kan opbevaring i seks måneder ved minus 18 oC til minus 22 oC overvejes. Den mikrobielle biomasse af de opbevarede jordprøver måles før hvert forsøg, og biomassens kulstofindhold skal udgøre mindst 1 % af jordens samlede organiske kulstofindhold (se 1.6.2).
1.6.4. Håndtering og klargøring af jordprøver til forsøget
1.6.4.1. Præinkubering
Har jorden været opbevaret (jf. 1.6.3.2), anbefales præinkubering i et tidsrum på mellem 2 og 28 døgn. Under præinkuberingen skal jordens temperatur- og fugtindhold svare til forsøgsbetingelserne (se 1.6.4.2 og 1.7.1.3).
1.6.4.2. Fysisk-kemiske egenskaber
Jorden befries manuelt for store genstande (f.eks. sten, plantedele osv.) og sies derefter våd uden for megen udtørring til en partikelstørrelse på højst 2 mm. Jordprøvens fugtindhold justeres ved hjælp af destilleret eller demineraliseret vand til en værdi mellem 40 % og 60 % af den maksimale vandholdende evne.
1.6.4.3. Forbedring med organisk substrat
Jorden skal forbedres med et passende organisk substrat, f.eks. finmalet lucernegræs-grønmel (hovedkomponent: Medicago saliva) med etC/N-forhold mellem 12/1 og 16/1. Det anbefalede lucerne-jord-forhold er 5 g lucerne pr. kilogram jord (tør vægt).
1.6.5. Klargøring af prøvestoffet til tilføring til jorden
Prøvestoffet tilføres normalt ved hjælp af et bærestof. Bærestoffet kan være vand (til vandopløselige stoffer) eller et inaktivt fast stof som finkornet kvartssand (partikelstørrelse: 0,1-0,5 mm). Man bør undgå andre flydende bærestoffer end vand (f.eks. organiske opløsningsmidler som acetone eller chloroform), da de kan skade mikrofloraen. Anvendes sand som bærestof, kan det coates med prøvestof, som er opløst eller opslæmmet i et passende opløsningsmiddel. I så fald skal opløsningsmidlet fjernes ved fordampning før opblanding med jorden. For at få den bedst mulige fordeling af prøvestoffet i jorden anbefales et forhold på 10 g sand pr. kilogram jord (tør vægt). Kontrolprøver behandles kun med den tilsvarende mængde vand og/eller kvartssand.
Ved test af flygtige kemiske stoffer må tab under behandlingen så vidt muligt undgås, og det må tilstræbes at sikre ensartet fordeling i jorden (f.eks. bør prøvestoffet sprøjtes i jorden i flere punkter).
1.6.6. Prøvekoncentrationer
Af landbrugskemikalier skal anvendes mindst to koncentrationer. Den lave koncentration skal afspejle mindst den maksimale mængde, som under praktiske forhold forventes at nå jorden, og den høje koncentration skal være et multiplum af den lave koncentration. Ved beregning af den tilsatte prøvestofkoncentration til jorden forudsættes homogen tilsætning til en dybde af 5 cm og en bulkdensitet af jorden på 1,5. For landbrugskemikalier, som tilføres jorden direkte, og for kemiske stoffer, for hvilke det kan forudsiges, i hvilken mængde de når jorden, anbefales som prøvekoncentrationer den maksimale forventede eksponeringskoncentration (Predicted Environmental Concentration, PEC) samt fem gange denne koncentration. Stoffer, som forventes tilført jorden flere gange i løbet af én sæson, skal testes ved koncentrationer, som fås ved at gange eksponeringskoncentrationen med det maksimale forventede antal tilførsler. Den højeste testede koncentration må dog ikke svare til mere end ti gange den maksimale mængde ved én tilførsel. Afprøves andre stoffer end landbrugskemikalier, skal der anvendes mindst fem koncentrationer, som danner en kvotientrække. De afprøvede koncentrationer skal dække det område, der er nødvendigt tor at fastlægge ECx-værdierne.
1.7. UDFØRELSE AF FORSØGET
1.7.1. Eksponeringsbetingelser
1.7.1.1. Behandlingsprøver og kontrolprøver
Ved test af landbrugskemikalier deles jorden i tre portioner med samme vægt. To portioner blandes med bærestof indeholdende produktet, den tredje med bærestof uden produkt (kontrol). Det anbefales at benytte mindst tre gentagelser for både behandlet og ubehandlet jord. Ved test af andre stoffer end landbrugskemikalier deles jorden i seks portioner med samme vægt. Fem af prøverne blandes med bærestof indeholdende prøvestof, og den sjette blandes med bærestof uden prøvestof. Der anbefales tre gentagelser for både behandlings- og kontrolgruppe. Der drages omsorg for at sikre ensartet fordeling af prøvestoffet i de behandlede jordprøver. Under blandingen bør komprimering og kugledannelse af jorden undgås.
1.7.1.2 Inkubering af jordprøver
Inkubering af jordprøver kan ske på to måder: som bulkprøve af behandlet og ubehandlet jord eller som en række lige store delprøver af hver behandlet og ubehandlet jord. Ved test af flygtige prøvestoffer må forsøget dog kun udføres med en række særskilte delprøver. Når jorden inkuberes som bulkprøve, tilberedes store mængder af hver behandlet og ubehandlet jord, og under forsøget udtages efter behov delprøver til analyse. Hvor stor en mængde, som fra begyndelsen skal klargøres til hver behandlings-og kontrolprøve, afhænger af antal delprøver, antal gentagelser af analyserne og det forventede maksimale antal prøvetagningstidspunkter. Når jorden inkuberes som bulkprøve, skal den blandes grundigt før udtagning af delprøver. Når jorden inkuberes som en række særskilte jordprøver, skal hver behandlet og ubehandlet bulkjord deles i det nødvendige antal delprøver, og disse anvendes efter behov, Forventes flere end to prøvetagningstidspunkter i forsøget, tilberedes så mange delprøver, at de rækker til alle gentagelser og alle prøvetagningstidspunkter. Der inkuberes mindst tredobbelte prøver af prøvejorden under aerobe betingelser (se 1.7.1,1). Til alle forsøg skal anvendes passende beholdere med tilstrækkeligt headspace til at undgå opståen af anaerobe betingelser. Ved test af flygtige prøvestoffer udføres forsøget kun med en række særskilte delprøver.
1.7.1.3. Prøvningsbetingelser og forsøgsvarighed
Forsøget udføres i mørke ved en rumtemperatur på 20 ± 2 oC, Under forsøget holdes prøvens fugtindhold på en værdi svarende til mellem 40 % og 60 % af jordens maksimale vandholdende evne (se 1.6.4.2) med en variation på = 5 %. Efter behov kan tilsættes destilleret, demineraliseret vand.
Forsøgenes mindste varighed er 28 døgn. Ved test af landbrugskemikalier sammenholdes nitratdannelsen i behandlede prøver og kontrolprøver. Hvis forskellen mellem dem på dag 28 er over 25 %, fortsættes forsøget, indtil forskellen mellem dem er 25 % eller derunder, dog højst i 100 dage. For andre stoffer end landbrugskemikalier ophører forsøget efter 28 dage. På dag 28 bestemmes nitratmængden i behandlede prøver og kontrolprøver, og ECx-værdierne beregnes.
1.7.2. Prøvetagningsplan for analyse af jord
1.7.2.1. Prøvetagningsplan for jordprøver
Ved test af landbrugskemikalier analyseres jordprøverne for nitrat på dag 0, 7, 14 og 28. Er det nødvendigt at forlænge forsøget, fortsættes målingerne efter dag 28 med 14 dages mellemrum.
Testes andre stoffer end landbrugskemikalier, skal der anvendes mindst fem koncentrationer, og jordprøverne analyseres for nitrat ved eksponeringsperiodens start (dag 0) og slutning (dag 28). En mellemliggende måling, f.eks. dag 7, kan foretages hvis det anses for nødvendigt. Data registreret på dag 28 anvendes til bestemmelse af stoffets ECx-værdi. Om ønsket kan værdierne for kontrolprøverne på dag 0 anvendes som jordens startindhold af nitrat.
1.7.2.2. Analyse af jordprøver
Nitratmængden i hver gentagelse for behandlede prøver og kontrolprøver bestemmes ved hvert prøvetagningstidspunkt, Nitratindholdet i jorden ekstraheres ved udrystning med en passende ekstraktionsvæske, f.eks. en 0,1 M kaliumchloridopløsning. Det tilrådes at anvende en mængde KCl-opløsning på 5 ml pr. gram tør jord. For at optimere ekstraktionen bør beholderne med jord og ekstraktionsvæske ikke være mere end halvt fyldte. Blandingerne omrystes ved 150 o./min, i 60 minutter. Blandingerne centrifugeres eller filtreres, og væskefaserene analyseres for nitrat. Partikelfri væskeformige ekstrakter kan opbevares ved 20 ± 5 oC i indtil seks måneder.
2. DATA
2.1. BEHANDLING AF RESULTATER
Ved test af landbrugskemikalier skal den dannede nitratmængde i hver gentagelse af jordprøven registreres, og gennemsnitsværdien for alle gentagelser angives i tabelform. Kvælstofomdannelsen bedømmes ved egnede og almindeligt anerkendte statistiske metoder (f.eks F-test, 5 % signifikansniveau). Den dannede mængde nitrat angives som mg nitrat/kg tør jord/døgn. Nitratdannelsen ved hver behandling sammenholdes med kontrolprøvens, og den procentuelle afvigelse fra kontrolprøven beregnes.
Ved test af andre stoffer end landbrugskemikalier bestemmes den dannede mængde nitrat for hver gentagelse, og der opstilles en dosis-responskurve til bestemmelse af ECx-værdierne. Den nitratmængde (dvs. mg nitrat/kg tør jord), som er fundet i de behandlede prøver efter 28 døgn, sammenholdes med den, der er bestemt i kontrolprøverne. Af disse data beregnes for hver prøvestofkoncentration hæmningen i procent. Disse procentværdier afsættes mod koncentrationen, og ECx-værdierne beregnes. Desuden bestemmes konfidensgrænser (p = 0,95) for de beregnede ECx-værdier ved hjælp af standardmetoder (10)(l 1)(12).
Prøvestoffer med højt kvælstofindhold kan bidrage til den mængde nitrat, som dannes under forsøget. Hvis disse stoffer testes i høj koncentration (f.eks. stoffer som forventes anvendt til gentagen påførsel), skal passende kontrolprøver indgå i forsøget (dvs. jord med prøvestof, men uden plantemel). Data fra disse kontrolprøver skal indgå i beregningerne af ECx.
2.2. FORTOLKNING AF RESULTATER
Når man ved test af landbrugskemikalier finder, at forskellen mellem kvælstofdannelsen i prøver, som er behandlet med den laveste mængde (dvs. den forventede maksimale koncentration), og i kontrolprøven ikke er over 25 % på noget prøvetagningstidspunkt efter dag 28. kan produktet anses for at være uden langtidsvirkning på kvælstofomdannelsen i jord. Ved vurdering af forsøg med andre stoffer end landbrugskemikalier anvendes EC50, EC25 og/eller EC10-værdier.
3. RAPPORTERING
Forsøgsrapporten skal indeholde følgende oplysninger:
|
|
Fuldstændig identifikation af den anvendte jord, med angivelse af:
|
|
|
Prøvestof:
|
|
|
Substrat:
|
|
|
Forsøgsbetingelser:
|
|
|
Resultater:
|
4. HENVISNINGER
|
(1) |
EPPO (1994). Decision-Making Scheme for the Environmental Risk Assessment of Plant Protection Chemicals. Chapter 7: Soil Microflora. EPPO Bulletin 24: 1-16, 1994. |
|
(2) |
BBA (1990), Effects on the Activity of the Soil Microflora. BBA Guidelines for the Official Testing of Plant Protection Products, VI, 1-1 (2. udg., 1990). |
|
(3) |
EPA (1987). Soil Microbial Community Toxicity Test, EPA 40 CFR Part 797.3700. Toxic Substances Control Act Test Guidelines; Proposed rule. 28. september 1987. |
|
(4) |
SETAC-Europe (1995). Procedures for assessing the environmental fate and ecotoxicity of pesticides, Ed. M.R. Lynch, Pub. SETAC-Europe, Bruxelles. |
|
(5) |
ISO/DIS 14238 (1995). Soil Quality — Determination of Nitrogen Mineralisation and Nitrification in Soils and the Influence of Chemicals on these Processes. Technical Committee ISO/TC 190/SC 4: Soil Quality — Biological Methods. |
|
(6) |
OECD (1995). Final Report of the OECD Workshop on Selection of Soils/Sediments, Belgirate, Italien, 18.-20. januar 1995. |
|
(7) |
ISO 10381-6 (1993). Soil quality — Sampling. Guidance on the collection, handling and storage of soil for the assessment of aerobic microbial processes in the laboratory. |
|
(8) |
ISO 14240-1 (1997), Soil quality — Determination of soil microbial biomass — Part 1: Substrate-induced respiration method. |
|
(9) |
ISO 14240-2 (1997), Soil quality — Determination of soil microbial biomass — Part 2: Fumigation-extraction method. |
|
(10) |
Litchfield, J.T. and Wilcoxon F. (1949). A simplified method of evaluating dose-effect experiments. Jour, Pharmacol, and Exper. Then, 96, 99-113. |
|
(11) |
Finney, D.J. (1971), Probit Analysis. 3. udg., Cambridge, London og New York. |
|
(12) |
Finney, D.J. (1978), Statistical Methods in biological Assay. Griffin, Weycombe, UK. |
C.22. JORDMIKROORGANISMER: KULSTOFOMDANNELSESTEST
1. METODE
Metoden er gengivet efter OECD TG 217 (2000).
1.1. INDLEDNING
Den beskrevne metode er en laboratoriemetode til undersøgelse af langtidsvirkningerne på jordmikroorganismers kulstofomdannende aktivitet af enkeltstående eksponering for plantebeskyttelsesmidler og eventuelt også andre kemiske stoffer. Metoden er hovedsagelig baseret på anbefalinger fra Plantebeskyttelsesorganisationen for Europa og Middelhavsområderne (1). Der er endvidere taget hensyn til guidelines fra den tyske Biologische Bundesanstalt (2), den amerikanske miljøstyrelse (3), og SETAC (4). På en OECD-workshop om valg af jord-/sedimenttype, som blev holdt i Belgirate. Italien, i 1995 (5), enedes man om antal og art af de jordtyper, som skal anvendes i denne test, Anbefalingerne for indsamling, håndtering og opbevaring af jordprøver er baseret på en ISO-vejledning (6) og på anbefalingerne fra workshoppen i Belgirate.
Ved vurdering af prøvestoffers toksiske egenskaber kan det være nødvendigt at bestemme virkningerne på den mikrobielle aktivitet i jord, f.eks. når der kræves oplysninger om plantebeskyttelsesmidlers potentielle bivirkninger på mikrofloraen i jorden, eller når jordmikroorganismer forventes udsat for andre kemiske stoffer end plantebeskyttelsesmidler. Kulstofomdannelsestesten udføres for at bestemme virkningerne af sådanne kemiske stoffer på mikrofloraen i jorden. Ved test af landbrugskemikalier (f.eks. plantebeskyttelsesmidler, kunstgødning og skovbrugskemikalier) udføres både kulstofomdannelses- og kvælstofomdannelsestest. For andre kemiske stoffer end landbrugskemikalier er kvælstofomdannelsestesten tilstrækkelig. Viser sådanne kemiske stoffer sig imidlertid i kvælstofomdannelsestesten at have en EC50-værdi i samme område som gængse nitrifikationsinhibitorer (f.eks. nitrapyrin), kan en kulstofomdannelsestest udføres for at skaffe yderligere oplysninger.
Jord består af levende og ikke levende komponenter, som er til stede i komplekse og heterogene blandinger. Mikroorganismer spiller en vigtig rolle i nedbrydning og omdannelse af det organiske stof i frugtbar jord, og mange arter bidrager til flere forskellige aspekter af jordens frugtbarhed. Hvis der gennem længere tid gribes ind i disse biokemiske processer, kan det gribe ind i næringsstofcyklussen, således at jordens frugtbarhed ændrer sig. Kulstof- og kvælstofomdannelse finder sted i al frugtbar jord. Skønt det alt efter jordtypen er forskellige mikrobielle samfund, som er ansvarlige for disse processer, følger omdannelsen i det væsentlige altid samme vej.
Den her beskrevne forsøgsmetode anvendes til bestemmelse af stoffers langsigtede skadevirkning på kulstofomdannelsesprocessen i jordens aerobe dæklag. Metoden er følsom for ændringer i størrelse og aktivitet af de mikrobielle samfund, som er ansvarlige for kulstofomdannelsen, da den udsætter disse samfund for både kemisk stress og kulstofmangel. Der anvendes en sandet jord med lavt indhold af organisk stof. Jorden behandles med prøvestoffet og inkuberes under betingelser, som giver mulighed for et hurtigt mikrobielt stofskifte. Under disse betingelser udtømmes kilderne til umiddelbart tilgængeligt kulstof i jorden hurtigt. Derved opstår der kulstofmangel, som både forårsager celledød og inducerer dvaletilstand og/eller sporedannelse, Hvis forsøget løber over mere end 28 dage, kan man i kontrolprøver (med ubehandlet jord) følge disse reaktioner som et gradvist tab i metabolisk aktiv mikrobiel biomasse (7). Hvis biomassen i jord, som er udsat for kulstofmangel, under prøvebetingelserne påvirkes af tilstedeværelse af et kemisk stof, vil den ikke nødvendigvis vende tilbage til samme niveau som kontrolprøvens. Forstyrrelser, som på et vilkårligt tidspunkt under prøven er fremkaldt af prøvestoffet, vil således ofte vare ved indtil forsøgets slutning.
De test, som metoden oprindeligt er baseret på, er hovedsagelig udviklet til stoffer, for hvilke den til jorden tilførte mængde er kendt på forhånd. Det er f.eks. tilfældet for plantebeskyttelsesmidler, hvor spredemængden i marken kendes. For landbrugskemikalier er det tilstrækkeligt at teste to doser, som er relevante for den forventede eller forudsete spredemængde. Landbrugskemikalier kan enten testes som aktivt indholdsstof eller som færdigformuleret produkt. Testen er dog ikke begrænset til kemiske stoffer med forudsigelig miljøkoncentration. Ved at fendre dels den tilførte mængde prøvestof til jorden, dels metoden til vurdering af data kan man desuden anvende testen på kemiske stoffer, hvor man ikke kender den mængde, som vil nå jorden. For andre kemiske stoffer end landbrugskemikalier bestemmes virkningen af en række koncentrationer på kulstofomdannelsen. Data fra disse forsøg anvendes til at opstille en dosis-responskurve og beregne værdier af ECx, hvor x er den definerede virkning i %.
1.2. DEFINITIONER
Kulstofomdannelse, mikrobiel nedbrydning af organisk stof til kuldioxid som uorganisk slutprodukt.
EC x (effektiv koncentration), den koncentration af prøvestoffet i jord, som medfører en x % hæmning af kulstoffets omdannelse til kuldioxid.
EC 50 (effektiv mediankoncentration), den koncentration af prøvestoffet i jord, som medfører en 50 % hæmning af kulstoffets omdannelse til kuldioxid.
1.3. REFERENCESTOFFER
Ingen.
1.4. FORSØGSMETODENS PRINCIP
Siet jord behandles enten med prøvestoffet eller lades ubehandlet (kontrol). Til test af landbrugskemikalier anbefales mindst to prøvekoncentrationer, som vælges i henhold til den højeste forventede koncentration i marken. Efter inkubation i 0, 7, 14 og 28 døgn blandes prøver af behandlet jord og kontroljord med glucose, og den glucoseinducerede respiration måles gennem 12 timer uden afbrydelse, Respirationshastigheden angives enten som kuldioxidproduktionshastighed (mg kuldioxid/kg tør jord/h) eller som iltforbrugshastighed (mg ilt/kg jord/h). Den gennemsnitlige respirationshastighed i de behandlede jordprøver sammenholdes med kontrolprøvernes, og den procentuelle afvigelse af de behandlede prøver fra kontrolprøverne beregnes. Alle forsøg løber over mindst 28 døgn. Hvis der på dag 28 er mere end 25 % forskel mellem behandlet og ubehandlet jord, fortsættes målingerne med 14 dages intervaller indtil højst 100 dage. Ved test af andre stoffer end landbrugskemikalier tilsættes prøvestoffet i en række koncentrationer til jordprøverne, og den glucoseinducerede respirationshastighed (dvs. den gennemsnitlig kuldioxiddannelse eller det gennemsnitlige iltforbrug) måles efter 28 døgn. Resultaterne af forsøg med flere koncentrationer analyseres efter en regressionsmodel, og ECx-værdierne beregnes (dvs. EC.50, EC25 og/eller EC10). Jf. definitionerne.
1.5. FORSØGETS VALIDITET
Vurdering af forsøgsresultater med landbrugskemikalier bygger på nogle forholdsvis små forskelle (gennemsnitligt ± 25 %) mellem kuldioxidafgivelse eller iltforbrug i kontrolprøver og i behandlede jordprøver, hvorfor store variationer i kontrolprøverne kan give forkerte resultater. Afvigelsen mellem gentagne kontrolprøver skal derfor være mindre end ± 15 %.
1.6. BESKRIVELSE AF FORSØGSMETODEN
1.6.1. Apparatur
Prøvebeholderne skal være udført i kemisk inaktivt materiale. De skal have passende kapacitet svarende til den metode, der anvendes til inkubation af jordprøverne, dvs. inkubation af den samlede mængde eller som en række særskilte jordprøver (jf. 1.7.1.2). Der skal være draget omsorg både for minimering af vandtabet og udveksling af gasser under forsøget (f.eks. kan prøvebeholderne tildækkes med perforeret polyethylenfolie). Ved test af flygtige stoffer skal anvendes forseglede og gastætte beholdere. Beholderne skal have en størrelse, så jordprøven udfylder ca. en fjerdedel af deres rumfang.
Til bestemmelse af glucoseinduceret respiration skal anvendes inkubationssystemer og apparatur til måling af kuldioxidproduktion eller iltforbrug. Eksempler på sådanne systemer og apparatur findes i litteraturen (8) (9) (10) (11).
1.6.2. Jordtyper og antal heraf
Der anvendes én enkelt jordtype. Følgende jordkarakteristika anbefales:
|
— |
sandindhold: mindst 50 % og ikke over 75 % |
|
— |
pH: 5,5-7,5 |
|
— |
organisk kulstofindhold: 0,5-1,5 % |
|
— |
den mikrobielle biomasse skal måles (12)(13), og dens kulstof indhold skal udgøre mindst 1 % af jordens samlede organiske kulstofindhold, |
I de fleste tilfælde vil en jord med sådanne egenskaber repræsentere den værst tænkelige situation, da prøvestoffets adsorption er minimeret og dets tilgængelighed for mikrofloraen maksimal. Sædvanligvis behøver man derfor ikke udføre forsøg med andre jordtyper. Under visse omstændigheder, f.eks, når prøvestoffet hovedsagelig forventes anvendt i bestemte jordtyper som sur skovbundsjord eller når der er tale om elektrostatisk ladede kemiske stoffer, kan det være nødvendigt at anvende en yderligere jordtype.
1.6.3. Indsamling og opbevaring af jordprøver
1.6.3.1. Indsamling
Der skal foreligge detaljerede historiske oplysninger om det markareal, hvorfra prøvejorden er indsamlet, Disse oplysninger skal omfatte nøjagtig beliggenhed, plantedække, dato for behandlinger med plantebeskyttelsesmidler, behandlinger med organisk gødning og kunstgødning, tilførsler af biologisk materiale og uheldsbetingede forureningsepisoder. Til indsamling af jord skal vælges et areal, som kan anvendes gennem længere tid. Egnede er arealer med vedvarende græs, marker med årlig kornhøst (bortset fra majs) eller tæt tilsået grøngødskning, De valgte prøvetagningsarealer må ikke have været behandlet med plantebeskyttelsesmidler i mindst ét år forud for prøvetagningen. Endvidere må der ikke have været anvendt organisk gødning inden for de sidste seks måneder. Anvendelse af mineralsk gødning er kun acceptabel, når det sker i overensstemmelse med afgrødens behov, og når jordprøverne tidligst udtages tre måneder efter udbringning af gødningen. Jord, som har været behandlet med kunstgødning med kendt biocid virkning (f.eks calciumcyanamid) bør ikke anvendes.
Prøvetagning bør undgås under eller umiddelbart efter lange perioder (mere end 30 dage) med tørke eller oversvømmelse. Af pløjet jord tages prøver fra en dybde af 0 ned til 20 cm. For græsningsarealer eller andre jordtyper, hvor der ikke pløjes i længere perioder (mindst én vækstsæson), kan den maksimale prøvetagningsdybde være en smule mere end 20 cm (f.eks. ned til 25 cm). Jordprøverne skal transporteres i sådanne beholdere og under sådanne temperaturforhold, at jordens oprindelige egenskaber med sikkerhed ikke ændrer sig nævneværdigt,
1.6.3.2. Opbevaring
Der bør fortrinsvis anvendes jord, som er frisk optaget fra marken. Er opbevaring i laboratoriet ikke til at undgå, kan jorden opbevares i mørke ved 4 ± 2 oC i indtil tre måneder. Under opbevaringen skal aerobe forhold være sikret. Er jorden indsamlet fra områder, hvor den er frossen mindst tre måneder årligt, kan opbevaring i seks måneder ved minus 18 oC overvejes. Den mikrobielle biomasse af de opbevarede jordprøver måles før hvert forsøg, og biomassens kulstofindhold skal udgøre mindst 1 % af jordens samlede organiske kulstofindhold (se 1.6.2).
1.6.4. Håndtering og klargøring af jordprøver til forsøget
1.6.4.1. Præinkubering
Har jorden været opbevaret (jf. 1.6.4.2 og 1.7.1.3), anbefales præinkubering i et tidsrum på mellem 2 og 28 døgn. Under præinkuberingen skal jordens temperatur- og fugtindhold svare til forsøgsbetingelserne (se 1.6.4.2 og 1.7.1.3).
1.6.4.2 Fysisk-kemiske egenskaber
Jorden befries manuelt for store genstande (f.eks. sten, plantedele osv.) og sies derefter våd uden for megen udtørring til en partikelstørrelse på højst 2 mm. Jordprøvens fugtindhold justeres ved hjælp af destilleret eller demineraliseret vand til en værdi mellem 40 % og 60 % af den maksimale vandholdende evne.
1.6.5. Klargøring af prøvestoffet til tilføring til jorden
Prøvestoffet tilføres normalt ved hjælp af et bærestof. Bærestoffet kan være vand (til vandopløselige stoffer) eller et inaktivt fast stof som finkornet kvartssand (partikelstørrelse: 0,1-0,5 mm), Man bør undgå andre flydende bærestoffer end vand (f.eks. organiske opløsningsmidler som acetone eller chloroform), da de kan skade mikrofloraen. Anvendes sand som bærestof, kan det coates med prøvestof, som er opløst eller opslæmmet i et passende opløsningsmiddel. I så fald skal opløsningsmidlet fjernes ved fordampning før opblanding med jorden. For at få den bedst mulige fordeling af prøvestoffet i jorden anbefales et forhold på 10 g sand pr. kilogram jord (tør vægt). Kontrolprøver behandles kun med den tilsvarende mængde vand og/eller kvartssand.
Ved test af flygtige kemiske stoffer må tab under behandlingen undgås, og det må tilstræbes at sikre ensartet fordeling i jorden (f.eks. bør prøvestoffet sprøjtes i jorden i flere punkter).
1.6.6. Prøvekoncentrationer
Af landbrugskemikalier og andre kemiske stoffer med forudsigelig eksponeringskoncentration testes mindst to koncentrationer. Den lave koncentration skal afspejle mindst den maksimale mængde, som under praktiske forhold forventes at nå jorden, og den høje koncentration skal være et multiplum af den lave koncentration, Ved beregning af den tilsatte prøvestofkoncentration til jorden forudsættes homogen tilsætning til en dybde af 5 cm og en bulkdensitet af jorden på 1,5. For landbrugskemikalier, som tilføres jorden direkte, og for kemiske stoffer, for hvilke det kan forudsiges, i hvilken mængde de vil nå jorden, anbefales som prøvekoncentrationer den forventede eksponeringskoncentration (Predicted Environmental Concentration, PEC) samt fem gange denne koncentration. Stoffer, som forventes tilført jorden flere gange i løbet af én sæson, skal testes ved koncentrationer, som fås ved at gange eksponeringskoncentrationen med det maksimale forventede antal tilførsler, De højeste testede koncentration må dog ikke svare til mere end ti gange den maksimale mængde ved én tilførsel.
Afprøves andre stoffer end landbrugskemikalier, skal der anvendes mindst fem koncentrationer, som danner en kvotientrække, De afprøvede koncentrationer skal dække det område, der er nødvendigt for at fastlægge ECx-værdierne.
1.7. UDFØRELSE AF FORSØGET
1.7.1. Eksponeringsbetingelser
1.7.1.1. Behandlingsprøver og kontrolprøver
Ved test af landbrugskemikalier deles jorden i tre portioner med samme vægt, To portioner blandes med bærestof indeholdende produktet, den tredje med bærestof uden produkt (kontrol), Det anbefales at benytte mindst tre gentagelser for både behandlet og ubehandlet jord. Ved test af andre stoffer end landbrugskemikalier deles jorden i seks portioner med samme vægt. Fem af prøverne blandes med bærestof indeholdende prøvestof, den sjette med bærestof uden prøvestof. Der anbefales tre gentagelser for både behandlings- og kontrolgruppe. Der drages omsorg for at sikre ensartet fordeling af prøvestoffet i de behandlede jordprøver. Under blandingen bør komprimering og kugledannelse af jorden undgås.
1.7.1.2. Inkubering af jordprøver
Inkubering af jordprøver kan ske på to måder: som bulkprøve af behandlet og ubehandlet jord eller som en række lige store delprøver af hver behandlet og ubehandlet jord. Ved test af flygtige prøvestoffer må forsøget dog kun udføres med en række særskilte delprøver. Når jorden inkuberes som bulkprøve, tilberedes store mængder af hver behandlet og ubehandlet jord, og under forsøget udtages efter behov delprøver til analyse, Hvor stor en mængde, som fra begyndelsen skal klargøres til hver behandlings-og kontrolprøve, afhænger af antal delprøver, antal gentagelser af analyserne og det forventede maksimale antal prøvetagningstidspunkter. Når jorden inkuberes som bulkprøve, skal den blandes grundigt før udtagning af delprøver. Når jorden inkuberes som en række særskilte jordprøver, skal hver behandlet og ubehandlet bulkjord deles i det nødvendige antal delprøver, og disse anvendes efter behov. Forventes flere end to prøvetagningstidspunkter i forsøget, tilberedes så mange delprøver, at de rækker til alle gentagelser og alle prøvetagningstidspunkter. Der inkuberes mindst tredobbelte prøver af prøvejorden under aerobe betingelser (se 1.7.1.1). Til alle forsøg skal anvendes passende beholdere med tilstrækkeligt headspace til at undgå opståen af anaerobe betingelser, Ved test af flygtige prøvestoffer udføres forsøget kun med en række særskilte delprøver.
1.7.1.3. Prøvningsbetingelser og forsøgsvarighed
Forsøget udføres i mørke ved en rumtemperatur på 20 ± 2 oC. Under forsøget holdes prøvens fugtindhold på en værdi svarende til mellem 40 % og 60 % af jordens maksimale vandholdende evne (se 1.6.4.2) med en variation på ± 5 %. Efter behov kan tilsættes destilleret, demineraliseret vand.
Forsøgenes mindste varighed er 28 døgn, Ved test af landbrugskemikalier sammenholdes kuldioxidafgivelse eller iltforbrug i behandlede prøver og kontrolprøver. Hvis forskellen mellem dem på dag 28 er over 25 %, fortsættes forsøget, indtil forskellen mellem dem er 25 % eller derunder, dog højst i i 100 dage. For andre stoffer end landbrugskemikalier ophører forsøget efter 28 dage. På dag 28 bestemmes kuldioxidafgivelse eller iltforbrug i behandlede prøver og kontrolprøver, og ECx-værdierne beregnes.
1.7.2. Prøvetagning og analyse af jord
1.7.2.1. Prøvetagningsplan for jordprøver
Ved test af landbrugskemikalier analyseres jordprøverne for glucoseinduceret respirationshastighed på dag 0, 7, 14 og 28. Er det nødvendigt at forlænge forsøget, fortsættes målingerne efter dag 28 med 14 dages mellemrum.
Testes andre stoffer end landbrugskemikalier, skal der anvendes mindst fem koncentrationer, og jordprøverne analyseres for glucoseinduceret respirationshastighed ved eksponeringsperiodens start (dag 0) og slutning (dag 28). En mellemliggende måling, f.eks. på dag 7, kan foretages, hvis det anses for nødvendigt. Data registreret på dag 28 anvendes til bestemmelse af stoffets ECx-værdi. Om ønsket kan dag 0-værdierne for kontrolprøveme anvendes som startværdier for mængden af metabolisk aktiv biomasse i jorden (12).
1.7.2.2. Måling af glucoseinduceret respirationshastighed
Den glucoseinducerede respirationshastighed i hver gentagelse for behandlede prøver og kontrolprøver bestemmes ved hvert prøvetagningstidspunkt. Jordprøverne blandes med en tilstrækkelig mængde glucose til at udløse øjeblikkelig maksimal respiratorisk respons. De nødvendige mængde glucose til fremkaldelse af maksimal respiratorisk respons fra en given jord kan bestemmes i en indledende test, hvor der bruges en række glucosekoncentrationer (14). For sandjord med 0,5-1,5 % organisk kulstof er 2 000 mg til 4 000 mg glucose pr, kg tør jord dog sædvanligvis tilstrækkeligt. Glucosen kan finmales med rent kvartssand (10 g sand/kg tør jord) og iblandes homogent i jorden.
De glucoseforbedrede jordprøver inkuberes i et passende apparat med henblik på måling af respirationshastighed enten kontinuerligt, hver time eller hver anden time (se 1.6.1) ved 20 ± 2 oC. Den afgivne kuldioxid eller den forbrugte ilt måles i 12 timer uden afbrydelse, og målingen begynder snarest muligt, dvs. inden for en til to timer efter glucosetilsætningen. Den samlede kuldioxidproduktion eller det samlede iltforbrug i løbet af de 12 timer måles, og den gennemsnitlige respirationshastighed bestemmes.
2. DATA
2.1. BEHANDLING AF RESULTATER
Ved test af landbrugskemikalier registreres kuldioxidafgivelse eller iltforbrug i hver gentagelse af jordprøven, og gennemsnitsværdien for alle gentagelser angives i tabelform. Resultaterne bedømmes ved egnede og almindeligt anerkendte statistiske metoder (f.eks F-test, 5 % signifikansniveau). Den glucoseinducerede respirationshastighed angives i mg kuldioxid/kg tør jord/h eller mg ilt/kg tør jord/h. Den gennemsnitlige kuldioxidproduktionshastighed eller iltforbrugshastighed for hver behandling sammenholdes med kontrolprøvens, og den procentuelle afvigelse fra kontrolprøven beregnes.
Ved test af andre stoffer end landbrugskemikalier bestemmes kuldioxidproduktion eller iltforbrug for hver gentagelse, og der opstilles en dosis-responskurve til bestemmelse af ECx-værdierne. Den glucoseinducerede respirationshastighed (dvs. mg kuldioxid/kg tør jord/h eller mg ilt/kg tør jord/h), som findes i de behandlede prøver efter 28 dage, sammenholdes med kontrolprøvernes. Af disse data beregnes hæmningen i procent for hver prøvestofkoncentration. Disse procentværdier afsættes mod koncentrationen, og ECx-værdierne beregnes ved hjælp af statistiske metoder. Desuden bestemmes konfidensgrænser (p = 0,95) for de beregnede ECxværdier ved hjælp af standardmetoder (15)(16)(17).
2.2. FORTOLKNING AF RESULTATER
Når man ved test af landbrugskemikalier finder, at forskellen mellem respirationshastigheden i prøver behandlet med den laveste mængde (dvs. den forventede maksimale koncentration) og i kontrolprøve ikke er over 25 % på noget prøvetagningstidspunkt efter dag 28, kan produktet anses for at være uden langtidsvirkning på kulstofomdannelsen i jord. Ved vurdering af forsøg med andre stoffer end landbrugskemikalier anvendes EC50, EC25 og/eller EC10-værdier.
3. RAPPORTERING
FORSØGSRAPPORT
Forsøgsrapporten skal indeholde følgende oplysninger:
|
|
Fuldstændig identifikation af den anvendte jord, med angivelse af:
|
|
|
Prøvestof:
|
|
|
Forsøgsbetingelser:
|
|
|
Resultater:
|
4. HENVISNINGER
|
(1) |
EPPO (1994). Decision-Making Scheme for the Environmental Risk Assessment of Plant Protection Chemicals. Chapter 7: Soil Microflora. EPPO Bulletin 24: 1-16, 1994. |
|
(2) |
BBA (1990). Effects on the Activity of the Soil Microflora. BBA Guidelines for the Official Testing of Plant Protection Products, VI, 1-1 (2. udg., 1990). |
|
(3) |
EPA (1987). Soil Microbial Community Toxicity Test. EPA 40 CFR Part 797.3700. Toxic Substances Control Act Test Guidelines; Proposed rule, 28. september 1987. |
|
(4) |
SETAC-Europe (1995). Procedures for assessing the environmental fate and ecotoxicity of pesticides, Ed. M.R. Lynch, Pub. SETAC-Europe, Bruxelles. |
|
(5) |
OECD (1995). Final Report of the OECD Workshop on Selection of Soils/Sediments, Belgirate, Italien, 18-20. januar 1995. |
|
(6) |
ISO 10381-6 (1993). Soil quality — Sampling. Guidance on the collection, handling and storage of soil for the assessment of aerobic microbial processes in the laboratory. |
|
(7) |
Anderson. J.P.E. (1987). Handling and Storage of Soils for Pesticide Experiments, in »Pesticide Effects on Soil Microflora«. Eds. L. Somerville and M.P. Greaves, Kap. 3: 45-60. |
|
(8) |
Anderson, J.P.E. (1982). Soil Respiration, in »Methods of Soil Analysis — Part 2: Chemical and Microbiological Properties«. Agronomy Monograph No 9. Eds. A.L. Page, R.H. Miller and D.R. Keeney. 41: 831-871. |
|
(9) |
ISO 11266-1. (1993). Soil Quality — Guidance on Laboratory Tests for Biodegradation in Soil: Part 1. Aerobic Conditions. |
|
(10) |
ISO 14239 (1997E). Soil Quality — Laboratory incubation systems for measuring the mineralization of organic chemicals in soil under aerobic conditions. |
|
(11) |
Heinemeyer, O., Insam, H., Kaiser, E.A, and Walenzik, G, (1989), Soil microbial biomass and respiration measurements; an automated technique based on infrared gas analyses. Plant and Soil, 116: 77-81. |
|
(12) |
ISO 14240-1 (1997). Soil quality — Determination of soil microbial biomass —Part 1: Substrate-induced respiration method. |
|
(13) |
ISO 14240-2 (1997). Soil quality — Determination of soil microbial biomass — Part 2: Fumigation-extraction method. |
|
(14) |
Malkomes, H.-P, (1986), Einfluβ von Glukosemenge auf die Reaktion der Kurzzeit-Atmung im Boden Gegenüber Pflanzenschutzmitteln, Dargestellt am Beispiel eines Herbizide, (Influence of the Amount of Glucose Added to the Soil on the Effect of Pesticides in Short-Term Respiration, using a Herbicide as an Example). Nachrichtenbl. Deut. Pflanzenschutzd., Braunschweig, 38: 113-120. |
|
(15) |
Litchfield, J.T. and Wilcoxon, F. (1949), A simplified method of evaluating dose-effect experiments, Jour, Pharmacol, and Exper.Ther., 96, 99-113. |
|
(16) |
Finney, D.J. (1971). Probit Analysis. 3. udg., Cambridge, London og New York. |
|
(17) |
Finney D.J. (1978). Statistical Methods in biological Assay. Griffin, Weycombe, UK. |
C.23. AEROB OG ANAEROB OMDANNELSE I JORD
1. METODE
Metoden er gengivet efter OECD TG 307 (2002)
1.1. INDLEDNING
Denne forsøgsmetode er baseret på eksisterende guidelines (1)(2)(3)(4)(5)(6)(7)(8)(9). Den her beskrevne metode er til brug ved undersøgelse af kemiske stoffers aerobe og anaerobe omdannelse i jord. Ved forsøgene bestemmes i) prøvestoffets omdannelseshastighed og ii) art, dannelseshastighed og nedbrydningshastighed af de omdannelsesprodukter, som planter og jordorganismer kan blive udsat for. Sådanne undersøgelser er nødvendige for kemiske stoffer, som tilføres jorden direkte eller må forventes at nå frem til jordmiljøet. Resultaterne af sådanne laboratorieforsøg kan desuden anvendes til at opstille protokoller for prøvetagning og analyse i tilknyttede markundersøgelser.
Til vurdering af omdannelsesveje er aerobe og anaerobe forsøg med én jordtype sædvanligvis tilstrækkelige (8)(10)(11). Omdannelseshastigheden skal bestemmes i mindst tre yderligere jordtyper (8)(10).
På en OECD-workshop om valg af jord-/sedimenttype i Belgirate, Italien, i 1995 (10) enedes man specielt om antal og art af de jordtyper, som skal anvendes i dette forsøg. De testede jordtyper skal være repræsentative for de miljøforhold, hvor stoffet skal anvendes eller vil blive udledt. For eksempel skal kemiske stoffer, som kan blive udledt i subtropisk til tropisk klima, testes med Ferrasoler eller Nitosoler (FAO-systemet). Workshoppen resulterede desuden i anbefalinger for indsamling, håndtering og opbevaring af jordprøver, baseret på ISO-vejledningen (15). Anvendelse af overrislet jord (til risdyrkning) er ligeledes tilgodeset i metoden.
1.2. DEFINITIONER
Prøvestof: et vilkårligt stof, som enten er moderstof eller et relevant omdannelsesprodukt.
Omdannelsesprodukter: alle stoffer, som dannes ved biotisk eller abiotisk omdannelse af prøvestoffet, herunder CO3 og stoffer, som er til stede i bundne rester.
Bundne rester:»Bundne rester« repræsenterer de stoffer i jord, planter eller dyr, som efter ekstraktion bliver tilbage i den pågældende matrix, enten som moderstof eller som metabolitter/omdannelsesprodukter. Ekstraktionsmetoden må ikke bevirke væsentlige ændringer af selve stofferne eller af matrixens struktur. Bindingens art kan delvis klarlægges gennem matrixomdannende ekstraktionsmetoder og avancerede analyseteknikker. Indtil nu har man på denne måde identificeret kovalente bindinger, ionbindinger og sorptionsbindinger foruden indlejring. Sædvanligvis vil dannelse af bundne rester indebære væsentligt nedsat fysiologisk opløselighed (bioaccessibility) og biotilgængelighed (bioavailability) (12) (ændret efter IUPAC 1984 (13)).
Aerob omdannelse: reaktioner, som finder sted i tilstedeværelse af molekylært oxygen (14).
Anaerob omdannelse: reaktioner, som finder sted i fravær af molekylært oxygen (14).
Jord: en blanding af mineralske og organisk-kemiske bestanddele, hvoraf sidstnævnte indeholder højmolekylære forbindelser med stort kulstof- og kvælstofindhold og bebos af små organismer (hovedsagelig mikroskopiske). Jord kan håndteres i to tilstande:
|
a) |
uforstyrret, som den har udviklet sig med tiden, i karakteristiske lag af forskellige jordtyper |
|
b) |
forstyrret, således som den sædvanligvis findes i agerjord eller når der opgraves prøver til anvendelse i denne forsøgsmetode (14). |
Mineralisering: fuldstændig nedbrydning af en organisk forbindelse til CO2 og H2O under aerobe betingelser, og til CH4, CO2 og H2O under anaerobe betingelser. Når der i denne forsøgsmetode anvendes 14C-mærkede forbindelser, forstås ved mineralisering en omfattende nedbrydning, hvorunder et mærket kulstof atom oxideres under frigivelse af den pågældende mængde 14CO2 (14).
Halveringstid: t0,5 er den tid, der forløber, til et prøvestof er 50 % omdannet, når omdannelsen kan beskrives ved første ordens kinetik; den er koncentrationsuafhængig.
DT 50 (Disappearance Time 50): den tid, der forløber, til koncentrationen af prøvestoffet er aftaget med 50 %: følger omdannelsen ikke første ordens kinetik, er DT50 forskellig fra halveringstiden t0,5.
DT 75 (Disappearance Time 75): den tid, der forløber, til koncentrationen af prøvestoffet er aftaget med 75 %.
DT 90 (Disappearance Time 90): den tid, der forløber, til koncentrationen af prøvestoffet er aftaget med 90 %.
1.3. REFERENCESTOFFER
Der skal anvendes referencestoffer til karakterisering og/eller identifikation af omdannelsesprodukter med spektroskopiske og/eller kromatografiske metoder.
1.4. FORSØGSMETODENS ANVENDELIGHED
Forsøgsmetoden kan anvendes på alle kemiske stoffer (umærkede eller radioaktivt mærkede), for hvilke man råder over en tilstrækkelig nøjagtig og følsom analysemetode. Metoden er anvendelig på tungflygtige, ikke-flygtige, vandopløselige og vanduopløselige forbindelser. Metoden bør ikke anvendes på kemikalier, som er letflygtige fra jord (f.eks. rygningsmidler eller organiske opløsningsmidler) og således ikke vil forblive i jorden under de her beskrevne forsøgsbetingelser.
1.5. OPLYSNINGER OM PRØVESTOFFET
Til måling af omdannelseshastigheden kan anvendes mærket eller umærket prøvestof. Til undersøgelse af omdannelsesveje og opstilling af massebalance skal anvendes mærket stof. 14C-mærkning anbefales, men andre isotoper, f.eks. 13C, 15N, 3H og 32P kan også anvendes. Mærket skal så vidt muligt placeres i molekylets mest stabile del(e) (40). Prøvestoffets renhed skal være mindst 95 %.
Før man foretager forsøg med aerob og anaerob omdannelse i jord, skal der foreligge følgende oplysninger om prøvestoffet:
|
a) |
vandopløselighed (metode A.6) |
|
b) |
opløselighed i organiske opløsningsmidler |
|
c) |
damptryk (metode A.4) og Henrys konstant |
|
d) |
n-oktanol/vand fordelingskoefficient (metode A.8) |
|
e) |
kemisk stabilitet i mørke (hydrolyse) (metode C.7) |
|
f) |
pKa, hvis molekylet er tilbøjeligt til at protoneres eller deprotoneres (OECD Guideline 112) (16). |
Af andre nyttige oplysninger kan nævnes data vedrørende prøvestoffet toksicitet over for jordmikroorganismer (prøvemetode C.21 og C.22) (16).
Der skal foreligge analysemetoder (herunder metoder til ekstraktion og oprensning) til kvantificering og identifikation af prøvestoffet og dets omdannelsesprodukter.
1.6. METODENS PRINCIP
Jordprøverne behandles med prøvestoffet og inkuberes i mørke i biometerkolber eller i gennemstrømningssystemer under kontrollerede laboratoriebetingelser (ved konstant temperatur og jordfugtighed). Efter passende tidsintervaller ekstraheres jordprøverne og analyseres for moderstof og omdannelsesprodukter. Flygtige produkter indsamles ligeledes til analyse ved hjælp af passende adsorptionsanordninger. Ved at benytte 14C-mærket stof kan de forskellige mineraliseringshastigheder af prøvestoffet måles gennem opsamling af den udviklede 14CO2 og opstilling af en massebalance indbefattende dannelse af jordbundne rester.
1.7. KVALITETSKRITERIER
1.7.1. Genfindingsgrad
Ved ekstraktion og analyse af i det mindste dobbelte jordprøver umiddelbart efter tilsætning af prøvestoffet fås et første fingerpeg om analysemetodens repeterbarhed og om, hvor homogent prøvestoffet er blevet tilført. Genfindingsgraden for senere forsøgsstadier fås af de respektive massebalancer, Genfindingsgraden skal være mellem 90 % og 110 % for mærkede kemiske stoffer (8) og mellem 70 % og 110 % for umærkede stoffer (3).
1.7.2. Analysemetodens repeterbarhed og følsomhed
Repeterbarheden af analysemetoden (heri ikke medregnet udvindingsgraden ved den indledende ekstraktion) til kvantitativ bestemmelse af prøvestof og omdannelsesprodukter kan kontrolleres ved dobbelt analyse på den samme ekstrakt af jord, som er inkuberet tilstrækkelig længe til at dannelse af omdannelsesprodukter har fundet sted.
Analysemetodens detektionsgrænse for prøvestoffet og for omdannelsesprodukteme må ikke være over 0,01 mg-kg-1 jord (som prøvestof), og ikke over 1 % af den tilsatte mængde. Grænsen for kvantitativ bestemmelse skal ligeledes angives.
1.7.3. Nøjagtighed af omdannelsesdata
Gennem regressionsanalyse på bestemmelserne af prøvestoffets koncentration som funktion af tiden fås de nødvendige oplysninger om omdannelseskurvens pålidelighed og mulighed for at bestemme konfidensgrænser for halveringstiderne (for pseudo første ordens kinetik) eller DT50-værdier og, i givet fald, DT75- og DT90-værdier.
1.8. BESKRIVELSE AF PRØVEMETODEN
1.8.1. Udstyr og kemiske reagenser
Som inkuberingssystem anvendes statiske lukkede systemer eller egnede gennemstrømningssystemer (7)(17). Eksempler på egnede gennemstrømningsapparater til jordinkubering og biometerkolber er vist i hhv. figur 1 og 2. Hver type inkuberingssystem har sine fordele og begrænsninger (7)(17).
Der kræves standardlaboratorieudstyr, specielt følgende:
|
— |
Apparater til GLC, HPLC og TLC med passende detektionssystemer til analyse af radioaktivt mærkede eller umærkede stoffer eller til omvendt isotopfortyndingsmetode |
|
— |
Instrumenter til identifikation (f.eks. MS, GC-MS, HPLC-MS, NMR osv.) |
|
— |
Væskescintillationstæller |
|
— |
Ovn til forbrænding af radioaktivt materiale |
|
— |
Centrifuge |
|
— |
Ekstraktionsapparat (f.eks, centrifugeglas til kold ekstraktion og Soxhlet-apparat til kontinuerlig ekstraktion med recirkulation); |
|
— |
Udstyr til koncentrering af opløsninger og ekstrakter (f.eks. rotationsfordamper) |
|
— |
Vandbad |
|
— |
Mekanisk blandemaskine (f.eks, æltemaskine, rotationsmixer). |
Anvendte kemiske reagenser kan f.eks. omfatte:
|
— |
NaOH, analytisk kvalitet. 2 mol.dm-3, eller anden egnet base (f.eks. KOH, ethanolamin) |
|
— |
H2SO4, analytisk kvalitet, 0,05 mol.dm-3 |
|
— |
Ethylenglycol, analytisk kvalitet |
|
— |
Faste absorbenter som natronkalk og polyurethancylindre |
|
— |
Organiske opløsningsmidler af analytisk kvalitet, f.eks. acetone, methanol osv. |
|
— |
Scintillationsvæske. |
1.8.2. Tilsætning af prøvestof
Med henblik på tilsætning til jorden og fordeling heri kan prøvestoffet opløses i vand (demineraliseret eller destilleret) eller, når det er nødvendigt, i mindst mulige mængder acetone eller andre organiske opløsningsmidler (6), hvori prøvestoffet er tilstrækkelig opløseligt og stabilt. Det valgte opløsningsmiddel må imidlertid i den anvendte mængde ikke have væsentlig indflydelse på den mikrobielle aktivitet i jorden (jf. 1.5 og 1.9.2-1.9.3). Man bør undgå opløsningsmidler som virker hæmmende på den mikrobiologiske aktivitet, således chloroform, dichlormethan og andre halogenerede opløsningsmidler.
Prøvestoffet kan også tilsættes i fast form. f.eks. blandet i kvartssand (6) eller i en lille delprøve af prøvejorden, som er lufttørret og steriliseret. Tilsættes prøvestoffet ved hjælp af et opløsningsmiddel, skal man lade opløsningsmidlet fordampe før tilsætning af den spikede delprøve til den oprindelige usterile jordprøve.
For sædvanlige kemikalier, hvis hovedtilførselsvej til jord er gennem kloakslam/landbrug, tilsætter man først prøvestoffet til slammet, som derefter tilsættes til jordprøven. (jf. 1.9.2 og 1.9.3).
Det kan ikke anbefales at bruge formulerede produkter rutinemæssigt, For prøvestoffer med ringe opløselighed kan anvendelse af det formulerede stof dog være et hensigtsmæssigt alternativ.
1.8.3. Jordtyper
1.8.3.1. Valg af jordtype
Til fastlæggelse af omdannelsesvejen kan anvendes en repræsentativ jordtype; en sandblandet lerjord (sandy loam), siltblandet lerjord (silty loam), lerjord (loam) eller en lerblandet sandjord (loamy sand) (i henhold til FAO's og USDA's klassifikation (18)) med pH 5,5-8,0, organisk kulstofindhold 0,5-2,5 % og en mikrobiel biomasse på mindst 1 % af det totale organiske kulstofindhold anbefales (10).
Til undersøgelser af omdannelseshastigheden skal anvendes mindst tre yderligere jordtyper, som repræsenterer et udsnit af relevante jordtyper. Jordtyperne skal være forskellige hvad angår organisk kulstofindhold, pH, lerindhold og mikrobiel biomasse (10).
Alle jordtyper skal i det mindste være karakteriseret med hensyn til tekstur ( % sand, % silt, % ler) (i henhold til FAO's og USDA's klassifikation (18)), pH, kationbytterkapacitet, organisk kulstofindhold, bulkdensitet og vandretentionsegenskaber (41) og mikrobiel biomasse (kun ved aerobe undersøgelser). Til fortolkning af resultaterne kan nærmere oplysninger om jordens egenskaber være nyttige, Til bestemmelse af jordegenskaber kan anvendes de metoder, som anbefales i henvisning (19)(20)(21)(22)(23). Til bestemmelse af mikrobiel biomasse anvendes SIR-metoden (substratinduceret respiration) (25)(26) eller alternative metoder (20).
1.8.3.2. Indsamling, håndtering og opbevaring af jord
Der skal foreligge detaljerede historiske oplysninger om det markareal, hvorfra prøvejorden er indsamlet. Disse oplysninger skal omfatte nøjagtig beliggenhed, plantedække, behandling med kemiske stoffer, behandling med organisk gødning og kunstgødning, tilførsel af biologisk materiale og anden forurening. Har jorden inden for de foregående fire år været behandlet med prøvestoffet eller stoffer strukturelt beslægtet hermed, må den ikke anvendes til omdannelsesundersøgelserne (10)(15).
Jorden skal være frisk indsamlet fra marken (fra A-horisonten eller det øverste 20 cm lag) med et indhold af jordvand, som letter sigtning. For andre jordtyper end jord fra overrislede marker bør man undgå prøvetagning under eller umiddelbart efter lange perioder (> 30 dage) med tørke, frysning eller oversvømmelse (14). Prøverne skal transporteres på en måde, som bevirker mindst mulig ændring i vandindholdet og skal opbevares mørkt med så fri adgang for luften som muligt. Hertil er en løst snøret polyethylenpose sædvanligvis velegnet.
Jorden skal behandles snarest muligt efter prøveindsamlingen. Vegetation, større jordfaunadele og sten fjernes, før jorden sigtes gennem en 2 mm si, som fjerner småsten, fauna- og planterester. Omfattende tørring og formaling af jorden før sigtning bør undgås (15).
Når prøvetagning i marken er vanskelig (jorden frossen eller snedækket), kan prøven tages fra jord, som opbevares i drivhus under plantedække (f.eks. græs eller græs/kløver). Undersøgelser med frisk indsamlet jord fra marken må stærkt foretrækkes, men skal den indsamlede og behandlede jord opbevares, før forsøget påbegyndes, må det ske under passende betingelser og i begrænset tidsrum (4-2 oC i højst 3 måneder) for at bevare den mikrobielle aktivitet (42). Detaljerede anvisninger for indsamling, håndtering og opbevaring af jord til anvendelse i bioomdannelsesforsøg kan findes i (8)(10)(15)(26)(27).
Før den behandlede jord anvendes til dette forsøg, skal den præinkuberes for at kimene kan bringes til spiring og fjernes, og for at ligevægten i den mikrobielle metabolisme kan genetableres efter skiftet fra prøvetagnings- eller opbevaringsbetingelser til inkubationsbetingelser. Sædvanligvis vil en præinkubationseriode på mellem 2 og 28 døgn under tilnærmelsesvis samme temperatur- og fugtighedsbetingelser som under det faktiske forsøg være tilstrækkelig (15). Opbevarings- og præinkubationsperioden må tilsammen ikke være over tre måneder.
1.9. UDFØRELSE AF FORSØGET
1.9.1. Forsøgsbetingelser
1.9.1.1. Forsøgstemperatur
I hele forsøgsperioden skal jordprøverne inkuberes i mørke ved en konstant temperatur, som er repræsentativ for de klimabetingelser, hvorunder benyttelse eller udledning vil finde sted. En temperatur på 20 ± 2 oC anbefales for alle prøvestoffer, som kan tilføres jorden i tempereret klima. Temperaturen skal kontrolleres.
For kemikalier, som tilføres eller udledes under koldere klimaforhold (f.eks. i nordlige lande og i efterårs- og vinterperioden) bør yderligere jordprøver desuden inkuberes ved lavere temperatur (f.eks. 10 ± 2 oC).
1.9.1.2. Fugtindhold
Ved omdannelsesforsøg under aerobe forhold skal jordens fugtindhold (43) justeres til og holdes på en pF-værdi mellem 2,0 og 2,5 (3). Jordens fugtindhold angives som masseenheder vand pr. masseenhed tør jord og bør regelmæssigt kontrolleres (f.eks. med 2 ugers mellemrum) ved vejning af inkubationskolberne, og vandtabet kompenseres ved tilsætning af vand (helst sterilfiltreret ledningsvand). Ved tilsætning af vand bør vises omhu, så man undgår eller minimerer tab af prøvestof og/eller omdannelsesprodukter ved fordampning og/eller eventuel fotodegradering.
Til omdannelsesforsøg under anaerobe forhold og under betingelser svarende til overrislet mark skal jorden vandmættes ved overrisling.
1.9.1.3. Aerobe inkubationsbetingelser
I gennemstrømningssystemer opretholdes aerobe betingelser ved periodisk skylning eller ved kontinuerlig ventilation med befugtet luft. I biometerkolber opretholdes luftskiftet ved diffusion.
1.9.1.4. Sterile aerobe betingelser
For at skaffe viden om eventuel abiotisk omdannelse af prøvestoffet kan jordprøverne steriliseres (vedr. sterilisationsmetoder henvises til henvisning (16) og (29)), behandles med sterilt prøvestof (f.eks. med en opløsning tilsat gennem et sterilfilter) og beluftes med befugtet steril luft som beskrevet i 1.9.1.3. For jord fra overrislet mark skal jord og vand steriliseres, og inkubation skal finde sted som beskrevet i 1.9.1.6.
1.9.1.5. Anaerobe inkubationsbetingelser
For at etablere og opretholde anaerobe betingelser skal jorden — efter behandling med prøvestoffet og inkubation under aerobe betingelser i 30 døgn, dog højst én halveringstid eller DT50 — vandmættes (et vandlag på 1-3 cm), og inkubationssystemet skylles med en inaktiv gas (f.eks. kvælstof eller argon) (44). Forsøgsopstillingen skal gøre det muligt at måle f.eks. pH, oxygenkoncentration og redoxpotentiale og skal omfatte anordninger til indsamling af flygtige produkter. Systemer af biometertypen skal være lukkede for at undgå luftindtrængen ved diffusion.
1.9.1.6. lnkubationsbetingelser svarende til overrislet mark
For at undersøge omdannelsen i overrislede rismarker oversvømmes jorden med et vandlag på 1-5 cm, og prøvestoffet tilsættes vandfasen (9). En jorddybde på mindst 5 cm anbefales. Systemet ventileres med luft under aerobe betingelser. Den vandige fases pH, oxygenkoncentration og redoxpotentiale overvåges og registreres. Der kræves en præ-inkubationsperiode på mindst to uger, før omdannelsesundersøgelserne påbegyndes (se 1.8.3.2).
1.9.1.7. Forsøgets varighed
Undersøgelserne af omdannelseshastighed og -vej bør normalt ikke strække sig over mere end 120 dage (45) (3)(6)(8), da der herefter må forventes at indtræffe et fald i den mikrobiologiske aktivitet med tiden i et kunstigt laboratoriesystem, som er afskåret fra naturlig fornyelse. Forsøgene kan fortsætte i længere perioder (f.eks. 6 eller 12 måneder), når det er nødvendigt for at beskrive nedbrydning af prøvestof og dannelse og nedbrydning af hovedomdannelsesprodukter (8), Eventuelle længere inkubationsperioder skal begrundes i forsøgsrapporten og skal være ledsaget af målinger af biomasse under og ved slutningen af sådanne perioder.
1.9.2. Forsøgets udførelse
50 til 200 g jord (på tør vægtbasis) anbringes i hver inkubationskolbe (se, figur 1 og 2 i bilag 3), og jorden behandles med prøvestoffet efter en af de i 1.8.2 beskrevne metoder. Når der anvendes organiske opløsningsmidler til påføring af prøvestoffet, fjernes disse fra jorden ved fordampning. Jorden blandes derefter grundigt med spatel og/eller ved omrystning af kolben. Finder forsøget sted under betingelser svarende til overrislet mark, skal jord og vand blandes grundigt efter tilsætning af prøvestoffet. Små alikvoter (f.eks. 1 g) af den behandlede jord analyseres for prøvestoffet til kontrol af, at fordelingen er ensartet. En alternativ metode er beskrevet nedenfor.
Behandlingsmængden skal svare til den højeste i brugsanvisningen anbefalede spredemængde af et planteskyttelsesmiddel, idet dette iblandes homogent til en passende dybde i marken (f.eks. de øverste 10 cm (46) jord). For kemiske stoffer, der tilføres ved bladgødskning eller påføres jorden uden iblanding, vil den mest passende dybde til beregning af den mængde kemisk stof, der skal tilsættes hver kolbe, være 2,5 cm. For kemiske stoffer, som iblandes jorden, er den mest passende dybde den iblandingsdybde, som foreskrives i brugsanvisningen. Generelt gælder, at den den mængde, der skal tilføres af et kemisk stof, bestemmes på grundlag af den mest relevante tilførselsvej; er den vigtigste tilførselsvej således gennem kloakslam, skal stoffet tilsættes slammet i en koncentration, som afspejler den forventede koncentration i slammet, og den til jorden tilsatte mængde slam skal afspejle den normale mængde slam, som landbrugsjord belastes med. Er denne koncentration ikke høj nok til, at hovedomdannelsesprodukterne kan identificeres, kan inkubation af separate jordprøver med en større tilsætningsmængde være nyttig, men man bør undgå mængder så store, at de påvirker jordens mikrobielle funktioner (jf. 1.5 og 1.8.2).
Alternativt kan en større portion (dvs. 1 til 2 kg) jord behandles med prøvestoffet, blandes grundigt i en passende blandemaskine og derefter overføres til inkubationskolberne i små portioner på 50 til 200 g (f.eks. ved hjælp af prøvedelere). Små alikvoter (f.eks. 1 g) af den behandlede jordportion analyseres for prøvestoffet til kontrol af, at fordelingen er ensartet. Denne fremgangsmåde må foretrækkes, da den giver en mere ensartet fordeling af prøvestoffet i jorden.
Desuden skal der inkuberes ubehandlede jordprøver ved samme betingelser (aerobe) som prøver behandlet med prøvestof. Disse prøver anvendes til måling af biomasse under og efter afslutning af forsøgene.
Når prøvestoffet tilføres jorden opløst i et eller flere organiske opløsningsmidler, skal jordprøverne inkuberes med samme mængde opløsningsmiddel (-midler) og ved samme betingelser (aerobe) som prøver behandlet med prøvestof. Disse prøver anvendes til måling af biomasse før, under og efter afslutning af forsøgene for at kontrollere, om opløsningsmidlerne har indvirkning på den mikrobielle biomasse.
Kolberne med behandlet jord tilsluttes enten det i figur 1 beskrevne gennemstrømningssystem eller lukkes med den i figur 2 viste absorptionskolonne (se bilag 3).
1.9.3. Prøvetagning og måling
Ved de respektive tidsintervaller udtages et par inkubationskolber, og jordprøverne ekstraheres med passende opløsningsmidler af forskellig polaritet og analyseres for prøvestof og/eller omdannelsesprodukter. I en veltilrettelagt undersøgelse er der tilstrækkeligt med kolber til, at der kan bruges to kolber ved hver prøvetagning. Endvidere udtages absorptionsopløsninger eller faste absorbenter med forskellige tidsintervaller (i den første måned med 7-døgns intervaller, efter én måned med 17-døgns intervaller) under og efter inkubationen af hver jordprøve og analyseres for flygtige produkter. Derudover skal der indgå jordprøver udtaget direkte efter tilsætning (0-dages prøve) i mindst 5 forskellige prøvetagningspunkter. Tidsintervallerne vælges sådan, at mønsteret for nedbrydning af prøvestoffet og for omdannelsesprodukternes dannelse og nedbrydning kan fastlægges (f.eks. ved 0. 1, 3, 7 dage; 2, 3 uger; 1, 2, 3 måneder osv.).
Når der bruges 14C-mærket prøvestof, bliver den ikke-ekstraherbare radioaktivitet kvantificeret ved forbrænding, og der opstilles en massebalance for hvert prøvetagningsinterval.
Ved inkubation under anaerobe forhold og med overrisling af jorden kan jord- og vandfasen enten sammen analyseres for prøvestof og omdannelsesprodukter, eller faserne kan adskilles ved filtrering eller centrifugering før ekstraktion og analyse.
1.9.4. Frivillige prøver
Det kan være nyttigt at foretage yderligere forsøg under aerobe, ikke-sterile betingelser ved andre temperaturer og jordfugtigheder for at belyse temperaturens og jordfugtighedens indflydelse på omdannelseshastigheden af et prøvestof og/eller dets omdannelsesprodukter i jord.
Yderligere karakterisering af ikke-ekstraherbar radioaktivitet kan søges foretaget f.eks. med superkritisk væskeekstraktion.
2. DATA
2.1. BEHANDLING AF RESULTATER
Mængden af prøvestof, omdannelsesprodukter, flygtige stoffer (kun i %) og ikke ekstraherbart stof angives i % af den tilførte begyndelseskoncentration og, i givet fald, i mg.kg-1 jord (på tør vægtbasis) for hvert prøvetagningsinterval. For hvert prøvetagningsinterval opstilles en massebalance i procent af den tilførte begyndelseskoncentration. En grafisk fremstilling af prøvestoffets koncentration mod tiden giver mulighed for bestemmelse af dets omdannelseshalveringstid eller DT50. De vigtigste omdannelsesprodukter skal identificeres og deres koncentration ligeledes afbildes grafisk mod tiden for at vise deres dannelses- og nedbrydningshastighed. Ved »vigtigste omdannelsesprodukter« forstås dem, der på noget tidspunkt i løbet af undersøgelsen repræsenterer ≥ 10 % af den tilførte mængde.
De opsamlede flygtige produkter giver et vist fingerpeg om prøvestoffets og omdannelsesprodukterne potentielle flygtighed fra jord.
Der foretages en mere præcis bestemmelse af halveringstider eller DT50-værdier og, i givet fald. DT75- og DT90-værdier, ved beregning med passende kinetiske modeller. I rapporten angives værdierne af halveringstider og DT50 sammen med beskrivelsen af den anvendte model, reaktionsordenen og determinationskoefficienten (r2). Første ordens kinetik antages at være gældende, medmindre r2 < 0,7. Når det er hensigtsmæssigt, anvendes beregningerne også på hovedomdannelsesprodukterne. Eksempler på passende modeller er beskrevet i henvisning (31) til (35).
For kinetikforsøg ved forskellige temperaturer beskrives omdannelseshastighederne som funktion af temperaturen inden for det eksperimentelle temperaturområde med brug af Arrhenius' ligning i formen:
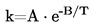 or
or  ,
,
hvor ln A og B er regressionskonstanter givet ved henholdsvis skæringspunkt og hældning af den bedst tilnærmede linje dannet ved lineær regression af ln k mod 1/T, idet k er hastighedskonstanten og T temperaturen i Kelvin. Man må være opmærksom på, at Arrchenius-ligningen kun er gyldig inden for et begrænset temperaturområde, når omdannelsen sker ved mikrobiel aktivitet.
2.2. EVALUERING OG FORTOLKNING AF RESULTATER
Skønt forsøgene finder sted i et kunstigt laboratoriesystem, kan man ved hjælp af resultaterne bestemme både prøvestoffets omdannelseshastighed og den hastighed, hvormed omdannelsesprodukterne dannes og nedbrydes under feltbetingelser (36)(37).
Ved at undersøge omdannelsesvejen for et prøvestof får man oplysninger om, hvordan det tilførte stof undergår strukturændringer i jorden gennem kemiske og mikrobielle reaktioner.
3. RAPPORTERING
FORSØGSRAPPORT
Forsøgsrapporten skal indeholde følgende:
|
|
Prøvestof:
|
|
|
Referencestoffer:
|
|
|
Prøvejordtyper:
|
|
|
Omstændigheder ved forsøget:
|
|
|
Resultater
|
4. HENVISNINGER
|
(1) |
US- Environmental Protection Agency (1982). Pesticide Assessment Guidelines. Subdivision N. Chemistry: Environmental Fate. |
|
(2) |
Agriculture Canada (1987). Environmental Chemistry and Fate. Guidelines for registration of pesticides in Canada. |
|
(3) |
European Union (EU) (1995). Commission Directive 95/36/EC of 14 July 1995 amending Council Directive 91/414/EEC concerning the placing of plant protection products on the market. Annex II, Part A and Annex III. Part A: Fate and Behaviour in the Environment. |
|
(4) |
Dutch Commission for Registration of Pesticides (1995). Application for registration of a pesticide. Section G: Behaviour of the product and its metabolites in soil, water and air. |
|
(5) |
BBA (1986). Richtlinie für die amtliche Prüfung von Pflanzenschutzmitteln, Teil IV, 4-1. Verbleib von Pflanzenschutzmitteln im Boden — Abbau, Umwandlung und Metabolismus. |
|
(6) |
ISO/DIS 11266-1 (1994). Soil Quality -Guidance on laboratory tests for biodegradation of organic chemicals in soil — Part 1: Aerobic conditions. |
|
(7) |
ISO 14239 (1997). Soil Quality — Laboratory incubation systems for measuring the mineralization of organic chemicals in soil under aerobic conditions. |
|
(8) |
SETAC (1995). Procedures for Assessing the Environmental Fate and Ecotoxicity of Pesticides. Mark R. Lynch, Ed. |
|
(9) |
MAFF — Japan 2000 — Draft Guidelines for transformation studies of pesticides in soil — Aerobic metabolism study in soil under paddy field conditions (flooded). |
|
(10) |
OECD (1995). Final Report of the OECD Workshop on Selection of Soils/Sediments. Belgirate, Italy, 18-20 January 1995. |
|
(11) |
Guth, J.A. (1980). The study of transformations. In Interactions between Herbicides and the Soil (R.J. Hance, Ed.), Academic Press, 123-157. |
|
(12) |
DFG: Pesticide Bound Residues in Soil. Wiley — VCH (1998). |
|
(13) |
T.R. Roberts: Non-extractable pesticide residue in soils and plants. Pure Appl. Chem. 56, 945-956 (IUPAC 1984). |
|
(14) |
OECD Test Guideline 304 A: Inherent Biodegradability in Soil (adopted 12 May 1981). |
|
(15) |
ISO 10381-6 (1993). Soil Quality — Sampling . Part 6: Guidance on the collection, handling and storage of soil for the assessment of aerobic microbial processes in the laboratory. |
|
(16) |
Annex V to Dir. 67/548/EEC. |
|
(17) |
Guth, J.A. (1981). Experimental approaches to studying the fate of pesticides in soil. In Progress in Pesticide Biochemistry. D.H. Hutson, T.R. Roberts, Eds. J. Wiley & Sons. Vol 1, 85-114. |
|
(18) |
Soil Texture Classification (US and FAO systems): Weed Science, 33, Suppl. 1 (1985) and Soil Sci. Soc. Amer. Proc. 26:305 (1962). |
|
(19) |
Methods of Soil Analysis (1986). Part 1, Physical and Mineralogical Methods. A. Klute, Ed.) Agronomy Series No 9, 2nd Edition. |
|
(20) |
Methods of Soil Analysis (1982). Part 2, Chemical and Microbiological Properties. A.L. Page, R.H. Miller and D.R. Kelney, Eds. Agronomy Series No 9, 2nd Edition. |
|
(21) |
ISO Standard Compendium Environment (1994). Soil Quality — General aspects; chemical and physical methods of analysis; biological methods of analysis. First Edition. |
|
(22) |
Mückenhausen, E. (1975). Die Bodenkunde und ihre geologischen, geomorphologischen, mineralogischen und petrologischen Grundlagen. DLG-Verlag, Frankfurt, Main. |
|
(23) |
Scheffer, F., Schaehtschabel, P. (1975). Lehrbuch der Bodenkunde. F. Enke Verlag, Stuttgart. |
|
(24) |
Anderson, J.P.E., Domsch, K.H. (1978) A physiological method for the quantitative measurement of microbial biomass in soils. Soil Biol. Biochem. 10, 215-221. |
|
(25) |
ISO 14240-1 and 2 (1997). Soil Quality — Determination of soil microbial biomass — Part 1: Substrate-induced respiration method. Part 2: fumigation-extraction method. |
|
(26) |
Anderson, J.P.E. (1987). Handling and storage of soils for pesticide experiments, In Pesticide Effects on Soil Microflora, L. Somerville. M.P. Greaves, Eds. Taylor & Francis, 45-60. |
|
(27) |
Kato, Yasuhiro. (1998). Mechanism of pesticide transformation in the environment; Aerobic and bio-transformation of pesticides in aqueous environment. Proceedings of the 16th Symposium on Environmental Science of Pesticide, 105-120. |
|
(28) |
Keuken O., Anderson J.P.E. (1996). Influence of storage on biochemical processes in soil. In Pesticides, Soil Microbiology and Soil Quality. 59-63 (SETAC-Europe). |
|
(29) |
Stenberg B., Johansson M,, Pell M., Sjödahl-Svensson K., Stenström J., Torstensson L. (1996), Effect of freeze and cold storage of soil on microbial activities and biomass. In Pesticides, Soil Microbiology and Soil Quality, 68-69 (SETAC-Europe). |
|
(30) |
Gennari, M., Negre, M., Ambrosoli, R, (1987). Effects of ethylene oxide on soil microbial content and some chemical characteristics. Plant and Soil 102, 197-200. |
|
(31) |
Anderson, J.P.E. (1975). Einfluss von Temperatur und Feuchte auf Verdampfung, Abbau und Festlegung von Diallat im Boden. Z. PflKrankh Pflschutz, Sonderheft VII, 141-146. |
|
(32) |
Hamaker, J.W. (1976). The application of mathematical modelling to the soil persistence and accumulation of pesticides. Proc. BCPC Symposium: Persistence of Insecticides and Herbicides, 181-199. |
|
(33) |
Goring, C.A.I., Laskowski, DA, Hamaker, J.W., Mcikle, R.W. (1975). Principles of pesticide degradation in soil. In »Environmental Dynamics of Pesticides«. R. Haque and V.H. Freed, Eds., 135-172. |
|
(34) |
Timme, G,, Frehse, H,, Laska, V, (1986). Statistical interpretation and graphic representation of the degradational behaviour of pesticide residues. II, Pflanzenschutz — Nachrichten Bayer 39, 188-204. |
|
(35) |
Timme, G., Frehse, H. (1980). Statistical interpretation and graphic representation of the degradational behaviour of pesticide residues. 1. Pflanzenschutz — Nachrichten Bayer 33, 47-60. |
|
(36) |
Gustafson D.I., Holden L.R. (1990). Non-linear pesticide dissipation in soil; a new model based on spatial variability. Environm. Sci.Technol. 24. 1032-1041. |
|
(37) |
Hurle K., Walker A. (1980). Persistence and its prediction. In Interactions between Herbicides and the Soil (R.J. Hance, Ed.), Academic Press, 83-122. |
Bilag 1
VANDTENSION, MARKKAPACITET OG VANDHOLDENDE EVNE (47)
|
Højde af vandsøjle [cm] |
pF (48) |
bar (49) |
Bemærkninger |
|
107 |
7 |
104 |
Tør jord |
|
1,6 · 104 |
4,2 |
16 |
Visnegrænse |
|
104 |
4 |
10 |
|
|
103 |
3 |
1 |
|
|
6 · 102 |
2,8 |
0,6 |
|
|
3,3 · 102 |
2,5 |
0,33 (50) |
Område for Markkapacitet (51) |
|
102 |
2 |
0,1 |
|
|
19 |
1,8 |
0,06 |
|
|
33 |
1,5 |
0,033 |
|
|
10 |
1 |
0,01 |
Vandholdende evne (tilnærmet) |
|
1 |
0 |
0,001 |
Vandmættet jord |
Vandpotentialet måles i cm vandsøjle eller i bar. På grund af sugetensionens store variationsområde angives denne simpelthen som pF-værdien, som svarer til logaritmen til værdien i cm vandsøjle.
Markkapaciteten er den mængde vand. som en naturlig jord kan tilbageholde mod tyngdekraften 2 døgn efter længerevarende regn eller efter tilstrækkelig kunstvanding. Den bestemmes i uforstyrret jord in situ i marken. Målingen er således ikke anvendelig på laboratorieprøver af forstyrret jord. Markkapaciteten bestemt i forstyrret jord kan udvise store systematiske afvigelser.
Den vandholdende evne (WHC) bestemmes i laboratoriet med uforstyrret og forstyrret jord ved at mætte en jordsøjle med vand ved kapillartransport. Den er specielt nyttig til forstyrret jord og kan være indtil 30 % større end markkapaciteten (1). Den er endvidere lettere al bestemme eksperimentelt end pålidelige værdier af markkapaciteten.
Noter
Bilag 2
FUGTINDHOLD I JORD (G VAND PR. 100 G TØR JORD) FOR FORSKELLIGE JORDTYPER FRA FORSKELLIGE LANDE
|
|
|
Fugtindhold i jord ved |
||
|
Jordtype |
Land |
|||
|
|
|
Vandholdende evne (WHC) (52) |
pF = 1,8 |
pF = 2,5 |
|
Sand |
Tyskland |
28,7 |
8,8 |
3,9 |
|
Lerblandet sand |
Tyskland |
50,4 |
17,9 |
12,1 |
|
Lerblandet sand |
Schweiz |
44,0 |
35,3 |
9,2 |
|
Siltler |
Schweiz |
72,8 |
56,6 |
28,4 |
|
Lerjord |
Brasilien |
69,7 |
38,4 |
27,3 |
|
Lerjord |
Japan |
74,4 |
57,8 |
31,4 |
|
Sandblandet lerjord |
Japan |
82,4 |
59,2 |
36,0 |
|
Siltler |
USA |
47,2 |
33,2 |
18,8 |
|
Sandblandet lerjord |
USA |
40,4 |
25,2 |
13,3 |
Bilag 3
Figur 1
Eksempel på gennemstrømningsapparat til undersøgelser af omdannelsen af kemikalier i jord (53) (54)

Figur 2
Eksempel på kolbe af biometertypen til undersøgelse af omdannelsen af kemiske stoffer i jord (55)

C.24. AEROB OG ANAEROB OMDANNELSE I AKVATISKE SEDIMENTSYSTEMER
1. METODE
Metoden er gengivet efter OECD TG 308 (2002).
1.1. INDLEDNING
Kemiske stoffer kan blive tilført lave eller dybe overfladevande gennem direkte tilledning, spredning af aerosoler, afstrømning, afløb, affaldsbortskaffelse, spildevand fra industri, husholdning eller landbrug samt atmosfærisk deposition. Den her beskrevne laboratoriemetode er bestemt til vurdering af aerob og anaerob omdannelse af organisk-kemiske stoffer i akvatiske sedimentsystemer. Den er baseret på eksisterende Guidelines (1)(2)(3)(4)(5)(6). På en OECD-workshop om valg af jord-/sedimenttype i Belgirate, Italien, i 1995 (7) enedes man specielt om antal og art af de sedimenter, som skal anvendes i dette forsøg. Workshoppen udsendte desuden anbefalinger for indsamling, håndtering og opbevaring af sedimentprøver, baseret på ISO-vejledningen (8). Sådanne undersøgelser er nødvendige for kemiske stoffer, som anvendes direkte i vandmiljøet eller som må påregnes at nå frem til vandmiljøet ad ovennævnte veje.
I naturlige akvatiske sedimentsystemer hersker der ofte aerobe forhold i den øvre vandfase. Det overfladiske sedimentlag kan enten være aerobt eller anaerobt, mens den dybere del af sedimentet sædvanligvis er anaerob. For at tage alle disse muligheder i betragtning beskrives i dette dokument både aerobe og anaerobe forsøg. Det aerobe forsøg simulerer en aerob vandsøjle over et aerobt sedimentlag, som befinder sig over en anaerob gradient. Det anaerobe forsøg simulerer et fuldstændig anaerobt vandsedimentsystem. Hvis omstændighederne gør det nødvendigt at afvige væsentligt fra disse anbefalinger, f.eks. ved at bruge intakte sedimentkerner eller sedimenter, som kan have været udsat for prøvestoffet, foreligger der andre metoder til dette formål (9).
1.2. DEFINITIONER
Der skal i alle tilfælde anvendes SI-enheder.
Prøvestof: et vilkårligt stof, hvad enten det er moderstof eller et relevant orndannelsesprodukt.
Omdannelsesprodukter: alle stoffer, som dannes ved biotisk eller abiotisk omdannelse af prøvestoffer, herunder CO2 og bundne rester.
Bundne rester:»Bundne rester« repræsenterer de stoffer i jord, planter eller dyr, som efter ekstraktion persisterer i den pågældende matrix, enten som moderstof eller som metabolitter/omdannelsesprodukter. Ekstraktionsmetoden må ikke bevirke væsentlige ændringer af selve stofferne eller af matrixens struktur. Bindingens art kan delvis klarlægges gennem matrixomdannende ekstraktionsmetoder og avancerede analyseteknikker. Indtil nu er der på denne måde identificeret kovalente bindinger, ionbindinger og sorptionsbindinger foruden indlejring. Sædvanligvis vil dannelse af bundne rester medføre væsentligt nedsat fysiologisk opløselighed (bioaccessibility) og biotilgængetighed (bioavailability) (10) (ændret efter IUPAC 1984 (11)).
Aerob omdannelse: (oxiderende): reaktioner, som finder sted i tilstedeværelse af molekylært oxygen (12).
Anaerob omdannelse: reaktioner, som finder sted i fravær af molekylært oxygen (12).
Naturligt vand: overfladevande udtaget fra damme, floder, vandløb osv.
Sediment: en blanding af mineralske og organisk-kemiske bestanddele, hvoraf sidstnævnte indeholder højmolekylære forbindelser med stort kulstof- og kvælstofindhold, Det afsættes af naturligt vand og danner grænseflade til dette vand.
Mineralisering; fuldstændig nedbrydning af en organisk forbindelse til CO2 og H2O under aerobe betingelser, og til CH4, CO2 og H2O under anaerobe betingelser. Når der i forbindelse med denne forsøgsmetode anvendes radioaktivt mærkede forbindelser, forstås ved mineralisering en vidtgående nedbrydning af molekylet, hvorunder et mærket kulstofatom oxideres eller reduceres under frigivelse af den pågældende mængde af henholdsvis 14CO2 og 14CH4.
Halveringstid: t0,5 er den tid, der forløber, til et prøvestof er 50 % omdannet, når omdannelsen kan beskrives ved første ordens kinetik; den er uafhængig af begyndelseskoncentrationen.
DT 50 (Disappearance Time 50): den tid, der forløber, til koncentrationen af prøvestoffet er aftaget med 50 %.
DT 75 (Disappearance Time 75): den tid, der forløber, til koncentrationen af prøvestoffet er aftaget med 75 %.
DT 90 (Disappearance Time 90): den tid, der forløber, til koncentrationen af prøvestoffet er aftaget med 90 %.
1.3. REFERENCESTOFFER
Til identifikation og kvantitativ bestemmelse af omdannelsesprodukter med spektroskopiske og kromatografiske metoder skal anvendes referencestoffer.
1.4. OPLYSNINGER OM PRØVESTOFFET
Til måling af omdannelseshastigheden kan anvendes umærket eller isotopmærket prøvestof, men radioaktivt mærket stof må foretrækkes. Til undersøgelse af omdannelsesveje og opstilling af massebalance skal anvendes mærket stof. 14C-mærkning anbefales, men andre isotoper, f.eks. 13C, 15N, H og "P, kan også anvendes. Mærket skal så vidt muligt placeres i molekylets mest stabile del(e) (56). Prøvestoffets kemiske og/eller radiokemiske renhed skal være mindst 95 %.
Før man udfører et forsøg, skal der foreligge følgende oplysninger om prøvestoffet:
|
a) |
vandopløselighed (metode A.6) |
|
b) |
opløselighed i organiske opløsningsmidler |
|
c) |
damptryk (metode A.4) og Henrys konstant |
|
d) |
n-oktanol/vand fordelingskoefficient (metode A.8) |
|
e) |
adsorptionskoefficient (Kd, Kf eller, i givet fald, Koc (metode C. 18) |
|
f) |
hydrolyse (metode C.7) |
|
g) |
syredissociationskonstant (pKa) (OECD Guideline 112) (13) |
|
h) |
prøvestoffets kemiske struktur og placeringen af eventuel isotopmærkning. |
Bemærkning; Den temperatur, ved hvilken disse målinger har fundet sted, skal angives.
Af andre nyttige oplysninger kan nævnes data vedrørende prøvestoffet toksicitet over for mikroorganismer, data vedrørende umiddelbar og/eller naturlig bionedbrydelighed og data vedrørende aerob og anaerob omdannelse i jord.
Der skal foreligge analysemetoder (herunder metoder til ekstraktion og oprensning) til identifikation og kvantitativ bestemmelse af prøvestoffet og dets omdannelsesprodukter i vand og i sediment (se 1.7.2).
1.5. FORSØGSMETODENS PRINCIP
I den her beskrevne forsøgsmetode anvendes et aerobt og et anaerobt akvatisk sedimentsystem (se bilag 1), som giver mulighed for:
|
i) |
måling af prøvestoffets omdannelseshastighed i et vand-sediment system |
|
ii) |
måling af prøvestoffets omdannelseshastighed i sedimentet |
|
iii) |
måling af mineraliseringshastigheden for prøvestoffet og/eller dets omdannelsesprodukter (når der anvendes 14C-mærket prøvestof) |
|
iv) |
identifikation og kvantitativ bestemmelse af omdannelsesprodufeter i vand- og sedimentfaser, herunder massebalance (når der anvendes mærket prøvestof) |
|
v) |
måling af fordelingen af prøvestoffet og dets omdannelsesprodukter mellem de to faser under inkubation i mørke (for at undgå f.eks. algevækst) ved konstant temperatur, Halveringstid. DT50, DT75 og DT90 bestemmes, når data berettiger det, men bør ikke ekstrapoleres ud over forsøgsperioden (se 1.2). |
Til både aerobe og anaerobe forsøg kræves mindst to sedimenter og de tilhørende vande (7). I visse tilfælde kan det dog være nødvendigt at anvende flere end to akvatiske sedimenter, f.eks. når det kemiske stof kan optræde i ferskvands- og/eller marint miljø.
1.6. FORSØGSMETODENS ANVENDELIGHED
Forsøgsmetoden kan anvendes på alle kemiske stoffer (umærkede eller mærkede), til hvilke man råder over en tilstrækkelig nøjagtig og følsom analysemetode. Metoden kan anvendes på tungflygtige, ikke-flygtige, vandopløselige og tungtopløselige forbindelser. Metoden bør ikke anvendes på kemiske stoffer, som er letflygtige fra vand (f.eks. rygningsmidler eller organiske opløsningsmidler) og således ikke vil forblive i vand og/eller sediment under de her beskrevne forsøgsbetingelser.
Metoden er hidtil anvendt til undersøgelse af omdannelsen af kemiske stoffer i ferske vande og sedimenter, men kan i princippet også anvendes på flodmundingssystemer/marine systemer. Den er ikke egnet til simulering af betingelserne i strømmende vand (f.eks floder) eller i det åbne hav.
1.7. KVALITETSKRITERIER
1.7.1. Genfindingsgrad
Ved ekstraktion og analyse af i det mindste dobbelte vand- og sedimentprøver umiddelbart efter tilsætning af prøvestoffet fås et første fingerpeg om analysemetodens repeterbarhed og om, hvor homogent prøvestoffet er blevet tilført. Genfindingsgraden for senere forsøgsstadier fås af de respektive massebalancer (når der bruges mærket stof). Genfindingsgraden skal være mellem 90 % og 110 % for mærkede kemiske stoffer (6) og mellem 70 % og 110 % for umærkede stoffer.
1.7.2. Analysemetodens repeterbarhed og følsomhed
Repeterbarheden af analysemetoden (heri ikke medregnet udvindingsgraden ved den indledende ekstraktion) til kvantitativ bestemmelse af prøvestof og omdannelsesprodukter kan kontrolleres ved dobbelt analyse på den samme ekstrakt af vand- eller sedimentprøverne, efter at disse er inkuberet tilstrækkelig længe til, at dannelse af omdannelsesprodukter har fundet sted.
Analysemetodens detektionsgrænse for prøvestoffet og for omdannelsesprodukterne må ikke være over 0,01 mg kg-1 i vand eller sediment (som prøvestof) og ikke over 1 % af den startmængde, der er tilsat prøvesystemet. Grænsen for kvantitativ bestemmelse skal angives.
1.7.3. Nøjagtighed af omdannelesdata
Gennem regressionsanalyse på bestemmelserne af prøvestoffets koncentration som funktion af tiden fås relevante oplysninger om omdannelseskurvens nøjagtighed samt mulighed for at fastlægge konfidensgrænser for halveringstiderne (når der gælder pseudo første ordens kinetik) eller DT50-værdier, og i givet fald DT75- og DT90-værdier.
1.8. BESKRIVELSE AF METODEN
1.8.1. Prøvesystem og apparatur
Til forsøget skal anvendes glasbeholdere (f.eks. flasker, centrifugeglas), medmindre de indledende data (således n-oktanol-vand fordelingskoefficient, sorptionsdata osv.) viser, at prøvestoffet kan adhærere til glas, i hvilket tilfælde det kan blive nødvendigt at benytte et alternativt materiale (som Teflon). Når prøvestoffet vides at adhærere til glas, kan problemet afhjælpes ved hjælp af eneller flere af følgende metoder:
|
— |
bestemmelser a fden masse prøvestof og omdannelsesprodukter, som sorberes på glas |
|
— |
vask af alt glasapparatur med opløsningsmiddel ved forsøgets afslutning |
|
— |
anvendelse af formulerede produkter (se også 1.9.2) |
|
— |
brug af øget mængde co-solvent ved tilsætning af prøvestof til systemet; anvendes co-solvent, må det ikke bevirke solvolyse af prøvestoffet. |
Eksempler på typiske prøveopstillinger, dvs. gennemstrømningssystemer og biometersystemer, er vist i hhv, bilag 2 og 3 (14). Andre anvendelige inkubationssystemer er beskrevet i henvisning (15). Forsøgsopstillingen skal give mulighed for udskiftning af luft eller kvælstof og indsamling af flygtige produkter, Forsøgsopstillingen skal være dimensioneret således, at den opfylder de krav, der stilles af forsøget (jf. 1.9.1). Udluftning kan enten ske ved let gennembobling eller ved at der ledes luft eller kvælstof hen over vandoverfladen. I sidstnævnte tilfælde kan forsigtig omrøring ovenfra være nødvendig for at opnå bedre fordeling af ilt eller kvælstof i vandet, Der må ikke anvendes CO2-fri luft, da det kan medføre stigning i vandets pH. I begge tilfælde bør forstyrrelse af sedimentet så vidt muligt undgås. Tungflygtige kemiske stoffer skal testes i et biometer-system med forsigtig omrøring af vandoverfladen. Lukkede kar med headspace af enten atmosfærisk luft eller kvælstof og indvendige glas til opsamling af flygtige produkter kan ligeledes anvendes (16). Ved den aerobe prøve kræves regelmæssig udskiftning af headspace-gassen for at kompensere for biomassens iltforbrug.
Egnede udskillere til flygtige opsamlingsprodukter kan være, men er ikke begrænset til en 1 M opløsning af kaliumhydroxid eller natriumhydroxid til udskillelse af kuldioxid (57) og ethylenglycol, ethanolamtn eller 2 % paraffin i xylen til organiske forbindelser. Flygtige stoffer, som dannes under anaerobe forhold, således methan, kan f.eks, opsamles med molekylsi, Sådanne flygtige stoffer kan f.eks, forbrændes til CO2 ved, at gassen ledes gennem et kvartsrør fyldt med CuO ved en temperatur på 900 oC, og den dannede CO2 opsamles i en absorber med alkali (17).
Der kræves laboratorieudstyr til kemisk analyse af prøvestoffet og omdannelsesprodukter (f.eks. ved gasvæskekromatografi (GLC), high performance væskekromatografi (HPLC), tyndtlagskromatografi (TLC), massespektroskopi (MS), gaskromatografi-massespektroskopi (GC-MS), væskekromatografi-massespektroskopi (LC-MS), kernemagnetisk resonans (NMR) mv.), herunder detektionssystemer for radioaktivt mærkede eller umærkede kemiske stoffer. Når radioaktivt mærket stof anvendes, skal der desuden bruges en væskescintillationstæller og en forbrændingsovn (til forbrænding af sedimentprøve før analyse af radioaktivitet).
Herudover skal der efter behov bruges øvrigt standardlaboratoneudstyr til fysisk-kemisk og biologisk bestemmelse (se tabel 1, 1.8.2.2), glasapparatur, kemikalier og reagenser.
1.8.2. Type og antal akvatiske sedimenter
Prøvetagningsstedet skal vælges i henhold til formålet med det pågældende forsøg. Ved valg af prøvetagningssted må der tages hensyn til oplysninger om eventuel tidligere tilførsel fra landbrug, industri eller husholdninger til vandene opstrøms for prøvearealet. Har sedimenterne inden for de foregående fire år været forurenet med prøvestoffet eller stoffer strukturelt beslægtet hermed, bør de ikke anvendes.
1.8.2.1. Valg af sediment
Til aerobe forsøg anvendes normalt to sedimenter (7). De to valgte sedimenter skal være forskellige med hensyn til organisk kulstofindhold og tekstur. Det ene sediment skal have højt organisk kulstofindhold (2,5-7,5 %) og fin tekstur, det andet skal have lavt organisk kulstofindhold (0,5-2,5 %) og grov tekstur. Forskellen i organisk kulstofindhold skal normalt være mindst 2 %. Ved »fin tekstur« forstås, at indholdet af [ler + silt] (58) > 50 %, og ved »grov tekstur« forstås, at indholdet af [ler + silt| < 50 %. Forskellen i indholdet af [ler + silt] i de to sedimenter bør normalt være mindst 20 %. I tilfælde, hvor et kemisk stof også kan blive ført til marine vande, skal mindst det ene vandsedimentsystem være af marin oprindelse.
Til det rent anaerobe forsøg tages to sedimentprøver (sammen med de tilhørende vande) fra det anaerobe område af overfladevandområderne (7). Der drages omsorg for, at ilten ikke får adgang til sediment- eller vandfasen under håndtering og transport.
Også andre parametre kan være vigtige ved valg af sedimenter og må tages i betragtning i de enkelte tilfælde. For eksempel vil sedimenternes pH-område være vigtig ved testning af stoffer, hvis omdannelse og/eller sorption kan være pH-afhængig. pH-afhængig sorption kan afspejle sig i prøvestoffets pKa-værdi.
1.8.2.2. Karakterisering af vand-sediment prøver
I nedenstående tabel er angivet de nøgleparametre, der skat måles og rapporteres (med henvisning til den anvendte metode) for både vand og sediment, samt på hvilket trin i forsøget disse parametre skal bestemmes. Til orientering er metoder til bestemmelse af de pågældende parametre givet i henvisning (18)(19)(20)(21).
Derudover kan der i det enkelte tilfælde være behov for måling og af andre parametre (f.eks, for ferskvand; partikler, alkalinitet, hårdhed, ledningsevne, NO3/PO4 (forhold og enkeltværdier); for sedimenter: kationbytrerkapacitet, vandholdende evne, karbonat, total kvælstof og fosfor; og for marine systemer: salinitet). Analyse af sedimenter og vand for nitrat, sulfat, biotilgængeligt jern og eventuelle andre elektronacceptorer kan desuden være nyttig til vurdering af redoxforholdene, specielt i forbindelse med anaerob omdannelse.
Måling af parametre til karakterisering af vand-sediment prøver(7)(22)(23)
|
Parameter |
Forsøgsfase |
|||||
|
mark prøvetagning |
efter håndteri ng |
start på akklimatise ring |
start på forsøg |
under forsøg |
forsøgets slutning |
|
|
Vand |
||||||
|
Oprindelse/kilde |
X |
|
|
|
|
|
|
Temperatur |
X |
|
|
|
|
|
|
pH |
X |
|
X |
X |
X |
X |
|
totalt organisk kulstof (TOC) |
|
|
X |
X |
|
X |
|
O2-koncentration* |
X |
|
X |
X |
X |
X |
|
Redoxpotentiale* |
|
|
X |
X |
X |
X |
|
Sediment |
||||||
|
Oprindelse/kilde |
X |
|
|
|
|
|
|
Lagdybde |
X |
|
|
|
|
|
|
pH |
|
X |
X |
X |
X |
X |
|
Partikelstørrelsesfordeling |
|
X |
|
|
|
|
|
totalt organisk kulstof (TOC) |
|
X |
X |
X |
|
X |
|
Mikrobiel biomasse (60) |
|
X |
|
X |
|
X |
|
Redoxpotentiale (59) |
Observation (farve/lugt) |
|
X |
X |
X |
X |
1.8.3. Indsamling, håndtering og opbevaring
1.8.3.1. Indsamling
Udtagning af prøver af sediment skal ske i overensstemmelse med udkastet til ISO-vejledningen for prøvetagning af bundsediment (8). Sedimentprøver skal tages fra hele den øverste 5 til 10 cm dybe del af sedimentlaget. Det tilhørende vand skal indsamles fra samme areal eller lokalitet og på samme tidspunkt som sedimentet. Til det anaerobe forsøg skal prøven af sedimentet og det tilhørende vand udtages og transporteres uden adgang af ilt (28) (jf. 1.8.2.1). I litteraturen beskrives nogle prøvetagningsanordninger(8)(23).
1.8.3.2. Håndtering
Sedimentet adskilles fra vandet ved filtrering og vådsigtes gennem en 2 mm sigte med overskud af vand fra indsamlingsstedet, og det overskydende vand herfra kasseres. Derefter blandes kendte mængder sedimenter og vand i det ønskede forhold (jf, 1.9.1) i inkubationsflasker og forberedes til akklimatiseringsperioden (jf. 1.8.4). Til det anaerobe forsøg skal al håndtering ske under udelukkelse af ilt (29)(30)(31)(32)(33).
1.8.3.3. Opbevaring
Brug af frisk udtagede prøver af sediment og vand må stærkt anbefales, men er opbevaring nødvendig, skal sediment og vand sigtes som beskrevet ovenfor og opbevares sammen under vand (et 6-10 cm vandlag), i mørke ved 4 ± 2 oC (61) i højst 4 uger (7)(8)(23). Prøver, som skal anvendes til aerobe forsøg, skal opbevares med fri luftadgang (f.eks i åbne beholdere), hvorimod prøver til anaerobe forsøg opbevares under udelukkelse af ilt. Under transport og opbevaring må ikke forekomme frysning af sediment og vand eller udtørring af sedimentet.
1.8.4. Klargøring af prøverne af sediment/vand til forsøget
Før prøvestoffet tilsættes, skal der hengå en akkltmatiseringsperiode, hvor hver sediment/vandprøve er anbragt i den inkubationsbeholder, som skal bruges i hovedforsøget, og akklimatiseringen skal finde sted under nøjagtig samme betingelser som ved inkubationen under forsøget (se 1.9.1). Akklimatiseringsperioden skal svare til den tid, systemet er om at nå en rimelig stabilitet, således som det ses af pH, oxygenkoncentrationen i vandet, redoxpotentiale af sediment og vand, og den makroskopiske adskillelse af faserne, Akklimatiseringen skal normalt vare mellem en og to uger og bør ikke overstige fire uger. Resultaterne af de bestemmelser, der er foretaget i denne periode, skal angives i rapporten.
1.9. UDFØRELSE AF FORSØGET
1.9.1. Forsøgsbetingelser
Forsøget udføres i inkubationsapparat (se 1.8.1) med et vand:sediment volumenforhold mellem 3:1 og 4:1 og et sedimentslag 2,5 cm (±0,5 cm). Det anbefales at bruge mindst 50 g sediment (på tørvogtsbasis) per inkubationsbeholder.
Forsøget udføres i mørke ved konstant temperatur mellem 10 og 30 oC, En temperatur på (20 ± 2) oC er passende. Når det er hensigtsmæssigt, kan man i de enkelte tilfælde overveje en yderligere lav temperatur (f.eks. 10 oC), alt efter hvilke oplysninger forsøget skal give. Inkubationstemperaturen skal overvåges og rapporteres.
1.9.2. Behandling og tilsætning af prøvestoffet
Der anvendes én prøvekoncentration af det kemiske stof (62). For plantebeskyttelsesmidler, som tilføres vandområderne direkte, anvendes som maksimal spredemængde den på mærkaten angivne maksimumsdosering, beregnet på grundlag af overfladearealet af vandet i prøvekarret. I alle andre tilfælde beregnes den anvendte koncentration på grundlag af prognoser baseret på de udledte mængder. Der skal drages omsorg for at sikre, at den tilførte mængde prøvestof er tilstrækkelig til at give mulighed for at karakterisere dets omdannelsesvej og omdannelsesprodukternes dannelse og nedbrydning. Det kan være nødvendigt at tilsætte højere doser (f.eks. 10 gange) i tilfælde, hvor stoffets koncentrationer ved forsøgets begyndelse er nær detektionsgrænsen og/eller hvis de vigtigste omdannelsesprodukter ikke let kan detekteres, når de optræder i en mængde på 10 % af den af prøvestoffet tilførte mængde. Anvendes højere testkoncentrationer, må disse dog ikke have nævneværdig ugunstig virkning på den mikrobielle aktivitet af vand-sediment systemet. For at opnå konstant prøvestofkoncentration i prøvekar af forskellige dimensioner vil det eventuelt blive fundet hensigtsmæssigt at korrigere den tilførte stofmængde på grundlag af dybden af vandsøjlen i karret i forhold til vanddybden i marken (som sættes til 100 cm, men andre dybder kan anvendes). Et eksempel på beregningsmåden er givet i bilag 4.
Ideelt skal prøvestoffet tilsættes prøvesystemets vandfase som vandig opløsning. Når det er uomgængelig nødvendigt, kan der bruges små mængder vandblandbare opløsningsmidler (som f.eks, acetone eller ethanol) til at tilføre og fordele prøvestoffet, men den tilsatte mængde må ikke udgøre over 1 % v/v eller have ugunstig virkning på den mikrobielle aktivitet af prøvesystemet. Den vandige opløsning af prøvestoffet skal fremstilles med omhu — det kan være hensigtsmæssigt at anvende en generatorsøjle og forblanding for at sikre fuldstændig homogenitet. Efter tilsætning af den vandige opløsning til prøvesystemet anbefales, at vandfasen blandes forsigtigt, så sedimentet forstyrres mindst muligt.
Det kan normalt ikke anbefales at anvende det færdigformulerede produkt, da indholdsstofferne i formuleringen kan påvirke prøvestoffets og/eller omdannelsesprodukternes fordeling mellem vand- og sedimentfase. For prøvestoffer med ringe opløselighed i vand kan anvendelse af det formulerede stof dog være et hensigtsmæssigt alternativ.
Antallet af inkubationsbeholdere afhænger af antal prøvetagningstidspunkter (se 1.9.3). Der skal indgå tilstrækkeligt mange prøvesystemer til, at der kan ofres to systemer for hvert prøvetagningstidspunkt. Anvendes der kontrolenheder med hvert akvatisk sedimentsystem, skal disse ikke behandles med prøvestof. Kontrolenhederne kan benyttes til at bestemme sedimentets mikrobielle biomasse og det totale organiske kulstofindhold i vand og sediment ved forsøgets slutning. To af kontrolenhederne (dvs. én kontrolenhed af hvert akvatisk sediment) kan anvendes til at overvåge de ønskede parametre i sediment og vand i akklimatiseringsperioden (se tabellen i 1.8.2.2). Når prøvestoffet tilføres ved hjælp af et opløsningsmiddel, skal der indgå to ekstra kontrolenheder til bestemmelse af ugunstig påvirkning af den mikrobielle aktivitet i prøvesystemet,
1.9.3. Forsøgsvarighed og prøvetagning
Forsøget bør normalt ikke strække sig over mere 100 dage (6) og skal løbe, indtil nedbrydningsvej og vand/sediment fordelingsmønster er klarlagt, eller indtil 90 % af prøvestoffet er spredt ved omdannelse og/eller fordampning. Antal prøvetagningstidspunkter skal være mindst seks (inklusive tidspunktet nul), idet en hensigtsmæssig prøvetagningsplan og forsøgets varighed fastlægges i en frivillig indledende undersøgelse (se 1.9.4), medmindre der fra tidligere undersøgelser foreligger tilstrækkelige data om prøvestoffet. For hydrofobe prøvestoffer kan ekstra prøvetagningspunkter være nødvendige i den indledende del af forsøgsperioden for at bestemme fordelingsforholdet mellem vand- og sedimentfase.
På passende prøvetagningstidspunkter udtages komplette (ens) inkubationsbeholdere til analyse. Sedimentet og det overliggende vand analyseres hver for sig (63). Overfladevandet fjernes forsigtigt med mindst mulig forstyrrelse af sedimentet. Til ekstraktion og karakterisering af prøvestof og omdannelsesprodukter skal anvendes egnede analysemetoder. Der drages omsorg for at fjerne materiale, som kan være adsorberet til inkubationsbeholderen eller til de forbindelsesslanger, der anvendes til opsamling af flygtige stoffer.
1.9.4. Frivillig forundersøgelse
Kan forsøgsvarighed og prøvetagningsfrekvens ikke skønsmæssigt fastlægges ud fra andre relevante undersøgelser af prøvestoffet, vil en frivillig forundersøgelse eventuelt blive fundet hensigtsmæssig og skal i så fald udføres under de forsøgsbetingelser, som påtænkes for den endelige undersøgelse. Hvis en sådan forundersøgelse gennemføres, skal relevante forsøgsomstændigheder og resultater for denne kortfattet rapporteres.
1.9.5. Målinger og analyse
For hvert prøvetagningstidspunkt måles og rapporteres koncentrationen af prøvestof og omdannelsesprodukter i vand og sediment (som koncentration og som procent af tilført mængde). Normalt skal omdannelsesprodukter, som på noget prøvetagningstidspunkt er detekteret som ≥10 % af den mængde radioaktivitet, der er tilført det samlede vand-sediment-system, identificeres, medmindre undladelse heraf med rimelighed kan begrundes. Også for omdannelsesprodukter med konstant stigende koncentration hen gennem forsøget må identifikation overvejes selv om deres koncentrationen ikke er over de nævnte grænser, da dette tyder på, at de er persistente. Der må ske en konkret vurdering heraf i det enkelte tilfælde, og begrundelser gives i rapporten.
Resultater fra opsamlingen af gasser/flygtige stoffer (CO2 og andet, dvs. flygtige organiske forbindelser), rapporteres for hvert prøvetagningstidspunkt. Mineraliseringshastigheder skal ligeledes rapporteres. Ikke-ekstraherbare (bundne) rester i sediment rapporteres for hvert prøvetagningspunkt.
2. DATA
2.1. BEHANDLING AF RESULTATER
For hvert prøvetagningstidspunkt beregnes en total massebalance eller genfindingsgrad (se 1.7.1) for den tilsatte radioaktivitet. Resultaterne angives i procent af tilsat mængde radioaktivitet. For hvert prøvetagningstidspunkt skal fordelingen af radioaktivitet mellem vand og sediment angives som koncentration og som procentdel.
Halveringstiden, DT50 og, i givet fald, DT75 og DT90 for prøvestoffet beregnes med tilhørende konfidensgrænser (jf. 1.7.3). Oplysninger om prøvestoffets spredning i vand og sediment kan opnås ved hjælp af passende evalueringsværktøjer. Disse kan række fra anvendelse af pseudo første ordens kinetik og empirisk kurvetilpasning med grafisk eller numerisk løsning til mere komplekse analyser, f.eks. baseret på en- eller flerkompartmentmodeller. Nærmere detaljer kan hentes i litteraturen (35)(36)(37).
Alle metoderne har stærke og svage sider og er meget forskellige i kompleksitet, Antagelse af første ordens kinetik kan være en oversimplificering af nedbrydnings- og fordelingsprocesserne, men når den er er mulig, resulterer den i et letforståeligt begreb (hastighedskonstanten eller halveringstiden), som er værdifuldt i simuleringsmodeller og beregninger af forventede koncentrationer i miljøet. Empiriske metoder eller lineær transformation kan give bedre tilpasning af kurver til data og dermed mulighed for bedre skøn over halveringstider, DT50- og, i givet fald. DT75- og DT90-værdier. De afledte konstanter har imidlertid begrænset anvendelse. Af kompartmentmodeller kan uddrages en række nyttige konstanter, som har værdi ved risikovurderinger og beskriver nedbrydningshastigheden i forskellige kompartments og det kemiske stofs fordeling. Desuden bør de anvendes ved bestemmelse af hastighedskonstanter for dannelse og nedbrydning af hovedomdannelsesprodukterne. I alle tilfælde skal valget af den anvendte metode begrundes, og eksperimentatoren skal grafisk og/eller statistisk påvise tilpasningsgraden.
3. RAPPORTERING
3.1. FORSØGSRAPPORT
Rapporten skal indeholde følgende oplysninger:
|
|
Prøvestof;
|
|
|
Referencestoffer:
|
|
|
Testsedimenter og -vande:
|
|
|
Forsøgsbetingelser:
|
|
|
Resultater:
|
4. HENVISNINGER
|
(1) |
BBA-Guidelines for the examination of plant protectors in the registration process. (1990). Part IV, Section 5-1: Degradability and fate of plant protectors in the water/sediment system. Germany. |
|
(2) |
Commission for registration of pesticides: Application for registration of a pesticide. (1991). Part G. Behaviour of the product and its metabolites in soil, water and air, Section G.2.1 (a). The Netherlands. |
|
(3) |
MAFF Pesticides Safety Directorate. (1992). Preliminary guideline for the conduct of biodegradability tests on pesticides in natural sediment/water systems. Ref No SC 9046. United Kingdom. |
|
(4) |
Agriculture Canada: Environmental chemistry and fate. (1987). Guidelines for registration of pesticides in Canada. Aquatic (Laboratory) — Anaerobic and aerobic. Canada. pp 35-37. |
|
(5) |
US-EPA: Pesticide assessment guidelines, Subdivision N. Chemistry: Environmental fate (19S2). Section 162-3, Anaerobic aquatic metabolism. |
|
(6) |
SETAC-Europe publication. (1995). Procedures for assessing the environmental fate and ecotoxicity of pesticides. Ed. Dr Mark R. Lynch. SETAC-Europe, Brussels. |
|
(7) |
OECD Test Guidelines Programme. (1995). Final Report of the OECD Workshop on Selection of Soils/sediments, Belgirate, Italy, 18-20 January 1995. |
|
(8) |
ISO/DIS 5667-12. (1994). Water quality — Sampling — Part 12: Guidance on sampling of bottom sediments. |
|
(9) |
US-EPA (1998a). Sediment/water microcosm biodegradation test. Harmonised Test Guidelines (OPPTS 835.3180). EPA 712-C-98-080. |
|
(10) |
DFG: Pesticide Bound Residues in Soil. Wiley-VCH (1998). |
|
(11) |
T.R. Roberts: Non-extractable pesticide residues in soils and plants. Pure Appl. Chem. 56, 945-956 (IUPAC 1984). |
|
(12) |
OECD Test Guideline 3O4A: Inherent Biodegradability in Soil (adopted 12 May 1981). |
|
(13) |
OECD (1993): Guidelines for Testing of Chemicals. Paris. OECD (1994-2000): Addenda 6-11 to Guidelines for the Testing of Chemicals. |
|
(14) |
Scholz, K., Fritz R., Anderson C. and Spiteller M. (1988) Degradation of pesticides in an aquatic model ecosystem. BCPC — Pests and Diseases, 3B-4, 149-158. |
|
(15) |
Guth, J.A. (1981). Experimental approaches to studying the fate of pesticides in soil. In Progress in Pesticide Biochemistry (D.H. Hutson, T.R, Roberts, Eds.), Vol. 1, 85-114, J. Wiley & Sons. |
|
(16) |
Madsen, T., Kristensen, P. (1997). Effects of bacterial inoculation and non-ionic surfactants on degradation of polycyclic aromatic hydrocarbons in soil. Environ. Toxicol. Chem. 16, 631-637. |
|
(17) |
Steber, J., Wierich, P. (1987), The anaerobic degradation of detergent range fatty alcohol ethoxylates. Studies with l4C-labelled model surfactants. Water Research 21, 661-667. |
|
(18) |
Black, C.A. (1965). Methods of Soil Analysis. Agronomy Monograph No. 9. American Society of Agronomy, Madison. |
|
(19) |
APHA (1989). Standard Methods for Examination of Water and Wastewater (17th edition). American Public Health Association, American Water Works Association and Water Pollution Control Federation, Washington D.C. |
|
(20) |
Rowell, D.L. (1994). Soil Science Methods and Applications. Longman. |
|
(21) |
Light, T.S. (1972). Standard solution for redox potential measurements. Anal. Chemistry 44, 1038-1039. |
|
(22) |
SETAC-Europe publication (1991). Guidance document on testing procedures for pesticides in freshwater mesocosms. From the Workshop »A Meeting of Experts on Guidelines for Static Field Mesocosms Tests«, 3-4 July 1991. |
|
(23) |
SETAC-Europe publication. (1993). Guidance document on sediment toxicity tests and bioassays for freshwater and marine environments. From the Workshop On Sediment Toxicity Assessment (WOSTA), 8-10 November 1993. Eds.: I.R. Hill, P. Matthiessen and F. Heimbach. |
|
(24) |
Vink, J.P.M., van der Zee, S.E.A.T.M. (1997). Pesticide biotransformation in surface waters: multivariate analyses of environmental factors at field sites. Water Research 3 1, 2858-2868. |
|
(25) |
Vink, J.P.M., Schraa, G., van der Zee, S.E.A.T.M. (1999). Nutrient effects on microbial transformation of pesticides in nitrifying waters. Environ. Toxicol, 329-338. |
|
(26) |
Anderson, T.H., Domsch, K.H. (1985). Maintenance carbon requirements of actively-metabolising microbial populations under in-situ conditions. Soil Biol. Biochem. 17, 197-203. |
|
(27) |
ISO-14240-2. (1997). Soil quality — Determination of soil microbial biomass — Part 2: Fumigation-extraction method. |
|
(28) |
Beelen, P. Van and F. Van Keulen. (1990), The Kinetics of the Degradation of Chloroform and Benzene in Anaerobic Sediment from the River Rhine. Hydrobiol. Bull. 24 (1), 13-21. |
|
(29) |
Shelton, D.R. and Tiedje, J.M. (1984). General method for determining anaerobic biodegradation potential. App. Environ. Microbiol. 47, 850-857. |
|
(30) |
Birch, R.R., Biver, C, Campagna, R., Gledhill, W.E., Pagga, U., Steber. J., Reust, H. and Bontinck, W.J. (1989). Screening of chemicals for anaerobic biodegradation. Chemosphere 19, 1527-1550. |
|
(31) |
Pagga, U. and Beimborn. D.B. (1993). Anaerobic biodegradation tests for organic compounds. Chemoshpere 27, 1499-1509. |
|
(32) |
Nuck, B.A. and Federle, T.W. (1986), A batch test for assessing the mineralisation of 14C- radiolabelled compounds under realistic anaerobic conditions. Environ. Sci. Technol. 30, 3597-3603. |
|
(33) |
US-EPA (1998b). Anaerobic biodegradability of organic chemicals. Harmonised Test Guidelines (OPPTS 835.3400). EPA 712-C-98-090. |
|
(34) |
Sijm, Haller and Schrap (1997). Influence of storage on sediment characteristics and drying sediment on sorption coefficients of organic contaminants, Bulletin Environ. Contam, Toxicol, 58, 961-968. |
|
(35) |
Timme, G., Frehse H. and Laska V. (1986) Statistical interpretation and graphic representation of the degradational behaviour of pesticide residues II, Pflanzenschutz — Nachrichten Bayer, 39, 187-203. |
|
(36) |
Timme, G., Frehse, H. (1980) Statistical interpretation and graphic representation of the degradational behaviour of pesticide residues I. Pflanzenschutz — Nachrichten Bayer, 33, 47-60. |
|
(37) |
Carlton, R.R. and Allen, R. (1994). The use of a compartment model for evaluating the fate of pesticides in sediment/water systems. Brighton Crop Protection Conference — Pest and Diseases, pp 1349-1354. |
Bilag 1
VEJLEDNING I AEROBE OG ANAEROBE TESTSYSTEMER
Aerobt testsystem
Det her beskrevne aerobe testsystem består af et aerobt vandlag (typisk iltkoncentration fra 7 til 10 mg.l-1) og et sedimentlag, som er aerobt ved overfladen og anaerobt under overfladen (typisk gennemsnitligt redoxpotentiale (Eh) i sedimentlagets anaerobe zone: - 80 til - 190 mV). Der ledes befugtet luft hen over vandoverfladen i hver inkubationsenhed for at opretholde tilstrækkelig iltkoncentration i head space.
Anaerobt testsystem
Forsøgsmetoden med det anaerobe testsystem er i hovedsagen den samme som for det aerobe system, bortset fra at der ledes befugtet kvælstof over vandoverfladen for at opretholde et nitrogenfyldt headspace. Sediment og vand anses for anaerobt, når redoxpotentialet (Eh) er under - 100 mV.
I det anaerobe forsøg omfatter bestemmelsen af mineraliseringen måling af den udviklede kuldioxid og methan.
Bilag 2
EKSEMPEL PÅ GENNEMSTRØMNINGSAPPARAT

Bilag 3
EKSEMPEL PÅ APPARAT AF BIOMETERTYPEN

Bilag 4
EKSEMPEL PÅ BEREGNING AF MÆNGDE TILSAT PRØVEBEHOLDERNE
|
Flankens indvendige diameter: |
= 8 cm |
|
Dybde af vandsøjle, eksklusive sediment: |
= 12 cm |
|
Overfladeareal: 3,142 × 42 |
= 50,3 cm2 |
|
Spredemængde: 500 g prøvestof/ha svarer til 5 μg/cm2 |
|
|
Total μg: 5 × 50,3 |
= 251,5 μg |
|
Mængden korrigeres til en dybde på 100 cm: 12 × 251,5 : 100 |
= 30,18 μg |
|
Vandsøjlens rumfang: 50,3 × 12 |
= 603 ml |
|
Koncentrationen i vand: 30,18 : 603 |
= 0,050 μg/ml eller 50 μg/l |
(1) Ifølge resultaterne af sådanne ringtest og en teknisk berigtigelse af ISO 6341 ligger EC50 — 24 h for kaliumdichromat (K2Cr2O7) mellem 0,6 mg/l og 1,7 mg/l.
(2) Vand af passende renhed, f.eks. deioniseret, destilleret eller behandlet ved omvendt osmose, fortrinsvis med ledningsevne ikke over 10 μS.cm-1.
(3) Gennemsnittet af D1 og D2 skal ikke tages, hvis der er en betydelig forskel.
(4) Gennemsnittet af D1 og D2 skal ikke tages, hvis der er en betydelig forskel.
(5) eller ved inkuberingens afslutning.
(6) Germemsnittet af D1 og D2 skal ikke tages, hvis der er en betydelig forskel.
(7) eller ved inkuberingens afslutning.
(8) Tag ikke gennemsnittet, hvis der er en betydelig forskel mellem replikaterne.
(9) Tag ikke gennemsnittet, hvis der er en betydelig forskel mellem replikaterne.
(10) Brugte opløsninger, der indeholder mercurisalte, behandles således, at der ikke spredes kviksølv: i miljøet.
(11) Mabey og Mill anbefaler brug af borat- eller acetatbuffer i stedet for fosfat (11).
(12) Sådanne oplysninger kan komme fra andre kilder, således hydrolysedata for strukturelt beslægtede forbindelser, fra litteraturen eller fra andre semikvantitative forprøver med hydrolyse af prøvestoffet i en tidligere udviklingsfase.
(13) Hvis kurven over logaritmisk transformerede data som funktion af tiden ikke peger på en lineær funktion (svarende til en førsteordens reaktion), kan hastighedskonstanten for hydrolyse af prøvestoffet ikke bestemmes af ligning [3].
(14) De i tabellerne angivne pH-værdier er beregnet af potentialemålinger ved hjælp af Sørensens standardligninger (1909). De tilsvarende pH-værdier er 0,04 enhed højere end værdierne i tabellerne.
(15) For at undgå skimmelvækst tilsættes en lille krystal thymol eller tilsvarende.
(16) Gennemsnit af tredobbeltbestemmelse.
(17) Meyer, A., Orti G. (1993) Proc, Royal Society of London. Series B., Vol. 252, p. 231.
(18) Der udtages vandprøve efter aftapning af mindst tre kammerrumfang vand.
Tallene i parentes angiver, hvor mange prøver (vand, fisk) der skal udtages, hvis der foretages supplerende prøveudtagning.
|
Anmærking |
: |
Forud for testen er k2 for en log Pow på 4,0 skønnet til 0,652 dag-1. Forsøgets samlede varighed sættes til 3 × up = 3 × 4,6 dage, dvs. 14 dage. For skøn af »up« henvises der til bilag 3. |
(19) Meyer, A., Bierman, C.H. and Orti, G. (1993). The phylogenetic position of the zebrafish (Danio rerio), a model system in developmental biology: an invitation to the comparative method. Proc. R. Soc. Lond. B. 252, 231-236.
(20) En række bestående af fem (eller flere) successive koncentrationer kan vælges fra en kolonne. Midterpunkterne mellem koncentrationerne i kolonne (x) findes i kolonne (2x + 1), De anførte værdier kan repræsentere koncentrationer udtrykt i procent pr. volumen eller vægt (mg/l eller μg/l). Værdierne kan multipliceres eller divideres med en hvilken som helst potens af 10, alt efter hvad der er aktuelt. Kolonne 1 kan eventuelt anvendes, hvis der har været betydelig tvivl om toksicitetsniveauet.
(21) OECD. Paris, 1992, Test Guideline 210, »Fish, Early-life Stage Toxicity Test«.
(22) Gældende embryoner.
(23) Gældende for larver.
(24) Mørke for embryoner og larver op til en uge efter klækning, undtagen ved inspektion. Siden svagt lys i resten af testen.
(25) I henhold til FAO og US-systemet (85).
(26) Værdierne af de respektive variable bør fortrinsvis ligge i det anførte område. Hvis det imidlertid er vanskeligt at finde passende jordmateriale, kan værdier under den anførte mindsteværdi godtages.
(27) For jordtyper med mindre end 0,3 % organisk kulstof kan sammenhængen mellem organisk kulstofindhold og adsorption være forstyrret. Det anbefales således at anvende jord med et organisk kulstofindhold på mindst 0,3 %.
(29) Kurver over koncentrationen af prøvestof i den vandige fase  som funktion af tiden kunne også anvendes til at vurdere, om der nås et ligevægtsplateau (se fig. 2 i tillæg 5).
som funktion af tiden kunne også anvendes til at vurdere, om der nås et ligevægtsplateau (se fig. 2 i tillæg 5).
(30) DT-50: den tid, der forløber, til 50 % af prøvestoffer er nedbrudt.
(31) W. Kördel, D. Hennecke, M. Herrmann (1997). Application of the HPLC-screening method for the determination of the adsorption coefficient on sewage sludges. Chemosphere, 35(1/2), pp. 121-128.
(32) W. Kördel, D. Hennecke, C. Franke (1997). Determination of the adsorption-coefficients of organic substances on sewage sludges. Chemosphere, 35 (1/2), pp. 107-119.
(33) W. Kördel, G. Kotthoff, J. Muller (1995). HPLC-screening method for the determination of the adsorption coefficient on soil-results of a ring test. Chemosphere. 30(7), pp. 1373-1384.
(34) W. Kördel, J. Müller (1994). Bestimmung des Adsorptionskoeffizienten organischer Chemikalien mit der HPLC. UBA R & D Report No 106 01 044 (1994).
(35) B.V. Oepen. W. Kördel. W. Klein (1991). Chemosphere, 22. pp, 285-304.
(36) Data stillet til rådighed af industrien.
(37) Angiv hvilket glas, der blev anvendt til undersøgelsen.
(38) Anfør aborteret afkom som AB i det relevante felt.
(39) Anfør mortalitet for ethvert voksent dyr som M i det relevante felt.
(40) Indeholder prøvestoffet f.eks. én ring, skal denne ring være mærket; indeholder prøvestoffet to eller flere ringe, kan separate forsøg være påkrævet for at undersøge, hvad der sker med de enkelte mærkede ringe, og for at skaffe anvendelige oplysninger om dannelse af omdannelsesprodukter.
(41) En jordtypes vandretentionsegenskaber kan måles som markkapacitet, vandkapacitet eller vandets sugetension (pF-værdi). Forklaringer findes i bilag 1. I prøverapporten angives, om jordtypernes vandretentionsegenskaber og bulkdensitet er bestemt i uforstyrrede markprøver eller i forstyrrede (behandlede) prøver.
(42) Nyere forskningsresultater viser, at jord fra tempererede zoner ligeledes kan opbevares ved - 20 oC i over tre måneder (28)(29) uden nævneværdigt tab i mikrobiel aktivitet.
(43) Jorden må hverken være for våd eller for tør til at opretholde tilstrækkelig luft- og næringsstoftilførsel til jordens mikroflora. Til optimal mikrobiel vækst anbefales et fugtindhold på mellem 40 og 60 % vandholdende evne (WHC) eller fra 0,1 til 0,33 bar (6). Sidstnævnte område svarer til et pF-område på 2,0-2,5. Det typiske fugtindhold i forskellige jordtyper er angivet i bilag 2.
(44) Aerobe betingelser er fremherskende i overjord og endda i den underliggende jord, som påvist i et EU-finansieret forskningsprojekt (K. Takagi et al. (1992). Microbial diversity and activity in subsoils; Methods, field site, seasonal variation in subsoil temperatures and oxygen contents. Proc. Internat. Symp. Environm. Aspects Pesticides Microbiol., 270-277, 17-21 August 1992, Sigtuna, Sverige). Anaerobe betingelser kan lejlighedsvis optræde, når jorden er oversvømmet efter kraftig nedbør eller når rismarker overrisles.
(45) Aerobe undersøgelser kan afsluttes længe inden 120 døgn, forudsat at omdannelse og mineralisering er løbet fuldstændig til ende på det pågældende tidspunkt. Afslutning af forsøget er mulig efter 120 dage eller når mindst 90 % af prøvestoffet er omdannet, men kun hvis der er dannet mindst 5 % CO2.
(46) Begyndelseskoncentrationen på arealbasis beregnes af følgende udtryk:

|
Csoil |
= |
Begyndelseskoncentrationen i jord (mg.kg-1) |
|
A |
= |
Spredemængde (kg.ha-1); 1 = tykkelse af markjordlaget (m): d = tør bulkdensitet af jorden (kg.m-3). |
Som tommelfingerregel resulterer en spredemængde på 1 kg.ha-1 i en jordkoncentration på ca. 1 mg.kg-1 i et 10 cm tykt lag (idet bulkdensiteten er sat til 1 g.cm-3).
(47) Mückenhausen, E. (1975). Die Bodenkunde und ihre geologischen, geomorphologischen, mineralogischen und petrologischen Grundlagen. DLG-Verlag, Frankfurt, Main.
(48) pF = logaritmisk angivelse af cm vandsøjle.
(49) 1 bar = 105 Pa.
(50) Svarende til et omtrentligt vandindhold på 10 % i sand, 35 % i lerjord og 45 % i ler.
(51) Markkapaciteten er ikke konstant, men varierer alt efter jordtypen mellem pF 1,5 og 2,5.
(52) Vandholdende evne.
(53) Guth, J.A. (1980). The study of transformations. In Interactions between Herbicides and the Soil (RJ. Hance, Ed.), Academic Press, 123-157.
(54) Guth, J.A. (1981). Experimental approaches to studying the fate of pesticides in soil. In Progress in Pesticide Biochemistry. D.H. Hutson, T.R. Roberts, Eds. J. Wiley & Sons. Vol 1, 85-114.
(55) Anderson, J.P.E. (1975). Einfluss von Temperatur und Feuchte auf Verdampfung, Abbau und Festlegung von Diallat im Boden. Z. PflKrankh Pflschutz, Sonderheft VII, 141-146.
(56) Indeholder prøvestoffet f.eks. én ring, skal denne ring være mærket; indeholder prøvestoffet to eller flere ringe, kan separate forsøg være nødvendige for at undersøge de enkelte mærkede ringes skæbne og for at få anvendelige oplysninger om dannelse af omdannelsesprodukter.
(57) Da sådanne alkaliske absorptionsvæsker også optager kuldioxiden i ventilationsluften og den, der er dannet ved respiration i aerobe forsøg, må de udskiftes regelmæssigt for at de ikke skal blive mættet og tabe absorptionsevnen.
(58) [Ler + silt] er den mineralfraktion af sedimentet, hvis partikelstørrelse er < 50 μm.
(59) Nyere forskning har vist, at målinger af vandets iltindhold og redoxpotentiale hverken har mekanistisk eller prædiktiv værdi med hensyn til mikrobielle populationers vækst og udvikling i overfladevande (24)(25). Bestemmelse af biokemisk iltforbrug (BOD, ved prøvetagning i marken, ved forsøgets start og slutning) og koncentrationen af mikro- og makronæringsstofferne Ca, Mg og Mn (ved forsøgets start og slutning) i vand samt måling af totalt kvælstof og totalt fosfor i sedimenter (ved prøvetagning i marken, ved forsøgets start og slutning) kan være mere velegnede som værktøjer til forståelse og vurdering aerobe biomdannelseshastigheder og -veje.
(60) Metoder baseret på mikrobiel respirationshastighed (26), rygning (27) eller kolonitælling (f.eks. bakterier, aktinomyceter, svampe og totalkolonier) til aerobe forsøg; methandannelseshastigheden ved anaerobe forsøg.
(61) Nyere undersøgelser har vist. at der ved opbevaring ved 4 oC kan indtræde et fald i sedimentets organiske kulstofindhold, muligvis med nedsat mikrobiel aktivitet til følge (34).
(62) Prøvning med endnu en koncentration kan være nyttig for stoffer, som tilføres overfladevandene ad forskellige veje med deraf følgende forskelle i koncentration, når blot den laveste koncentration kan analyseres med tilstrækkelig nøjagtighed.
(63) Hvis anaerobe omdannelsesprodukter let kan blive reoxideret, skal der opretholdes anaerobe betingelser under prøvetagning og analyse.




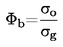
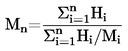

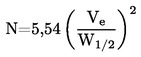
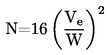

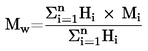
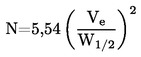


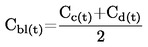



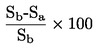






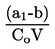






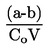



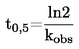
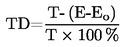






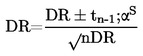








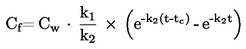




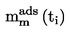
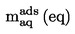

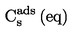
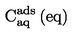






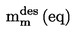





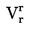




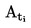

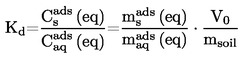
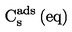








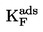
 (μg
(μg


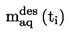

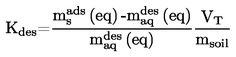




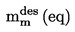

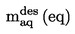







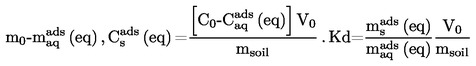

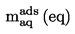








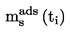





 beregnes ved følgende ligning:
beregnes ved følgende ligning:
 til tidspunktet t
til tidspunktet t
 eller
eller  (afhængigt af, hvad der er nødvendigt i undersøgelsen) afsættes som funktion af tiden, og tiden til indtræden af sorptionsligevægt bestemmes.
(afhængigt af, hvad der er nødvendigt i undersøgelsen) afsættes som funktion af tiden, og tiden til indtræden af sorptionsligevægt bestemmes.
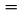

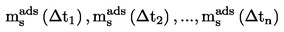
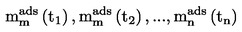




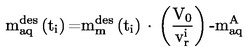

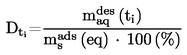


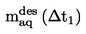

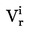 , som udtages til analysen (μg)
, som udtages til analysen (μg)
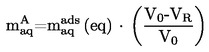



 , og den desorberede masse beregnes af ligningen:
, og den desorberede masse beregnes af ligningen:
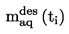 =
= 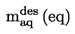 .
. beregnes af følgende ligning:
beregnes af følgende ligning:


 og
og
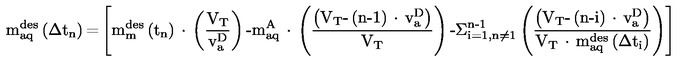 og
og






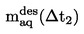

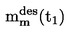

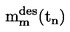
 henholdsvis til tidspunktet t
henholdsvis til tidspunktet t