

EUR-Lex Access to European Union law
This document is an excerpt from the EUR-Lex website
Document 32008R0273
Commission Regulation (EC) No 273/2008 of 5 March 2008 laying down detailed rules for the application of Council Regulation (EC) No 1255/1999 as regards methods for the analysis and quality evaluation of milk and milk products
Kommissionens forordning (EF) nr. 273/2008 af 5. marts 2008 om gennemførelsesbestemmelser til Rådets forordning (EF) nr. 1255/1999 for så vidt angår metoder til analyse og kvalitetsvurdering af mælk og mejeriprodukter
Kommissionens forordning (EF) nr. 273/2008 af 5. marts 2008 om gennemførelsesbestemmelser til Rådets forordning (EF) nr. 1255/1999 for så vidt angår metoder til analyse og kvalitetsvurdering af mælk og mejeriprodukter
OJ L 88, 29.3.2008, p. 1–115
(BG, ES, CS, DA, DE, ET, EL, EN, FR, IT, LV, LT, HU, MT, NL, PL, PT, RO, SK, SL, FI, SV)
Special edition in Croatian: Chapter 03 Volume 010 P. 139 - 253
 No longer in force, Date of end of validity: 06/02/2018; ophævet ved 32018R0150
No longer in force, Date of end of validity: 06/02/2018; ophævet ved 32018R0150
|
29.3.2008 |
DA |
Den Europæiske Unions Tidende |
L 88/1 |
KOMMISSIONENS FORORDNING (EF) Nr. 273/2008
af 5. marts 2008
om gennemførelsesbestemmelser til Rådets forordning (EF) nr. 1255/1999 for så vidt angår metoder til analyse og kvalitetsvurdering af mælk og mejeriprodukter
KOMMISSIONEN FOR DE EUROPÆISKE FÆLLESSKABER HAR –
under henvisning til traktaten om oprettelse af Det Europæiske Fællesskab,
under henvisning til Rådets forordning (EF) nr. 1255/1999 af 17. maj 1999 om den fælles markedsordning for mælk og mejeriprodukter (1), særlig artikel 10 og 15, artikel 26, stk. 3, artikel 29, stk. 1, og artikel 31, stk. 4, og
ud fra følgende betragtninger:
|
(1) |
Ved Kommissionens forordning (EF) nr. 213/2001 (2) er der fastsat gennemførelsesbestemmelser til Rådets forordning (EF) nr. 1255/1999 for så vidt angår metoder til analyse og kvalitetsvurdering af mælk og mejeriprodukter. I lyset af den tekniske udvikling inden for analysemetoder er der behov for at foretage yderligere væsentlige ændringer. Af hensyn til klarhed og effektivitet og på grund af antallet af ændringer og deres tekniske karakter bør forordning (EF) nr. 213/2001 ophæves og erstattes af en ny forordning. |
|
(2) |
De krav til mælks og mejeriprodukters sammensætning og kvalitet, som er fastsat i forbindelse med ordningerne i forordning (EF) nr. 1255/1999, skal verificeres for at sikre, at disse krav opfyldes nøje. |
|
(3) |
Referencemetoderne for sådanne verifikationer er ofte metoder, der er offentliggjort af internationale organisationer såsom Den Europæiske Standardiseringsorganisation (CEN), Det Internationale Mejeriforbund (IDF), Den Internationale Standardiseringsorganisation (ISO) og Association of Official Analytical Chemists (AOAC International), og som regelmæssigt ajourføres af disse organisationer. I nogle tilfælde er der fastsat en EF-referencemetode; i andre tilfælde er ingen referencemetode specificeret i EF-reglerne. For at sikre en ensartet anvendelse af referencemetoder bør der opstilles en liste over referencemetoder, og det bør fastsættes, at Kommissionen skal tilpasse listen, når det er nødvendigt. |
|
(4) |
Anvendelsen af rutinemetoder bør ikke udelukkes. Minimumsbetingelserne for anvendelse af dem bør derfor angives. |
|
(5) |
For at sikre en ensartet praksis for vurdering af analyseresultaterne bør der endvidere fastsættes fælles metoder; det forholder sig på samme måde med organoleptisk bedømmelse af de pågældende produkter og omprøvning af resultater, der bestrides. |
|
(6) |
For visse analyser findes der for tiden ikke internationalt accepterede referencemetoder, der er valideret, og der foreligger derfor ingen oplysninger om variationen af analyseresultater fra laboratorium til laboratorium. Der bør derfor fastsættes metoder på EF-plan, der er valideret efter internationalt fastsatte regler, og som bør anvendes som referencemetoder. |
|
(7) |
Ved Kommissionens forordning (EF) nr. 1898/2005 (3) er der fastsat gennemførelsesbestemmelser til Rådets forordning (EF) nr. 1255/1999 for så vidt angår foranstaltninger til afsætning af fløde, smør og koncentreret smør på EF's marked, og den foreskriver under visse omstændigheder tilsætning af røbestof til fløde, smør og koncentreret smør for at sikre, at disse produkters endelige anvendelse er korrekt. For at ordningen kan fungere korrekt, er det vigtigt at tilsætte røbestoffer. For at sikre lige behandling af de erhvervsdrivende, der deltager i ordningen, bør der så vidt muligt fastsættes fælles metoder for bestemmelse af nogle af disse røbestoffer. |
|
(8) |
Der kan ydes en støtte til privat oplagring af ost af fåremælk efter artikel 9 i forordning (EF) nr. 1255/1999. Der kan ydes en særlig restitution for de samme produkter efter artikel 31 i samme forordning. Der kan importeres ost fremstillet af fåre-, gede- eller bøffelmælk eller af blandinger af fåre-, gede- og bøffelmælk til EF fra bestemte tredjelande ifølge præferenceordninger. På grund af ovennævnte bestemmelser er det nødvendigt ved passende kontrol at efterprøve, at der ikke er iblandet komælk i de pågældende produkter. Der bør derfor fastsættes en EF-referencemetode til påvisning af komælk, uden at dette berører anvendelsen af rutinemetoder, hvis disse opfylder bestemte kriterier. |
|
(9) |
Efter Kommissionens forordning (EØF) nr. 2921/90 af 10. oktober 1990 om ydelse af støtte til skummetmælk med henblik på fremstilling af kasein og kaseinater (4) skal det påvises, at der ikke findes coliforme bakterier i de pågældende produkter. Den internationalt anerkendte referencemetode, der er accepteret på internationalt plan til påvisning af coliforme bakterier i mælk og mejeriprodukter, er ISO 4831. Der er fastsat en EF-referencemetode til påvisning af coliforme bakterier baseret på ovennævnte standard. |
|
(10) |
I Rådets forordning (EØF) nr. 2658/87 af 23. juli 1987 om told- og statistiknomenklaturen og den fælles toldtarif (5) differentieres toldsatserne for foderblandinger henhørende under toldposition 2309 ud fra indholdet af mejeriprodukter. For at sikre en ensartet anvendelse af de pågældende bestemmelser bør der fastlægges en metode til bestemmelse af lactoseindholdet, der er obligatorisk for alle medlemsstater, og i denne forbindelse bør der vælges en generelt anerkendt metode. |
|
(11) |
I forordning (EF) nr. 1255/1999 er det fastsat, at visse krav til kvalitet skal opfyldes, for så vidt angår smør og skummetmælkspulver til intervention eller skummetmælkspulver til dyrefoder. Der bør derfor fastsættes referencemetoder til efterprøvning af, om disse krav er opfyldt. |
|
(12) |
Nogle metoder indføres for første gang ved denne forordning. Der bør fastsættes tilstrækkelig tid fra forordningens ikrafttrædelse til, at laboratorierne på korrekt vis kan indføre og anvende disse nye metoder. Når en referencemetode, der henvises til i bilag I, ændres og offentliggøres af den organisation, der udvikler standarderne, bør laboratorierne have en frist på seks måneder til at opdatere deres analyseprocedurer, så de bringes i overensstemmelse med den nye standard. |
|
(13) |
Foranstaltningerne i denne forordning er i overensstemmelse med udtalelse fra Forvaltningskomitéen for Mælk og Mejeriprodukter - |
UDSTEDT FØLGENDE FORORDNING:
KAPITEL I
ALMINDELIGE BESTEMMELSER
Artikel 1
Formål og anvendelsesområde
1. I denne forordning fastsættes visse referencemetoder til kemisk, fysisk og mikrobiologisk analyse og til organoleptisk bedømmelse af mælk og mejeriprodukter i forbindelse med ordningerne i den fælles markedsordning for mælk og mejeriprodukter, der er fastsat i forordning (EF) nr. 1255/1999, samt reglerne for anvendelsen af metoderne.
2. Listen over referencemetoder for de i stk. 1 omhandlede analyser er anført i bilag I.
3. Kommissionen ajourfører listen efter proceduren i artikel 42 i forordning (EF) nr. 1255/1999.
Artikel 2
Rutinemetoder
Der kan anvendes rutinemetoder til de analyser, der er fastsat i EF-reglerne, hvis de er korrekt kalibreret og regelmæssigt kontrolleres i forhold til referencemetoden. Resultaterne sammenlignes under hensyntagen til konstante systematiske fejl, repeterbarhed og reproducerbarhed.
I tilfælde af tvistemål er det resultat, der opnås med referencemetoden, afgørende.
Medlemsstaterne underretter Kommissionen om anvendelsen af rutinemetoder til de analyser, der er omhandlet i artikel 1.
KAPITEL II
ANALYSEMETODER
Artikel 3
Vurdering af, om en sending overholder en forskriftsmæssig grænse
Bortset fra analysen af røbestoffer anvendes bilag II til at fastslå, at forskriftsmæssige krav til sammensætning er overholdt.
Artikel 4
Organoleptisk bedømmelse
1. Med hensyn til mælk og mejeriprodukter, bortset fra smør til offentlig oplagring, benytter medlemsstaterne som referencemetode for organoleptisk bedømmelse enten IDF-standard 99C:1997 eller andre lignende metoder, som de meddeler til Kommissionen.
Metoderne i bilag III anvendes til at efterprøve dommernes arbejde og pålideligheden af resultaterne af organoleptiske bedømmelser.
2. Med hensyn til smør til offentlig oplagring anvendes metoderne i bilag III til at efterprøve dommernes arbejde og pålideligheden af resultaterne af organoleptiske bedømmelser.
Den metode, der er fastsat i bilag IV, anvendes som referencemetode for organoleptisk bedømmelse.
Artikel 5
Røbestoffer
1. Den i bilag V fastsatte analysemetode benyttes som referencemetode til bestemmelse af indholdet af triglycerid af enantiksyre i smør, smørolie og fløde.
2. Den i bilag VI fastsatte analysemetode benyttes som referencemetode til bestemmelse af vanillin i koncentreret smør, smør og fløde.
3. Den i bilag VII fastsatte analysemetode benyttes som referencemetode til bestemmelse af mængden af ethylester af β-apo-8'-karotinsyre i koncentreret smør og smør.
4. Den i bilag VIII fastsatte analysemetode benyttes som referencemetode til bestemmelse af indholdet af β-sitosterol eller stigmasterol i smør og koncentreret smør.
5. Tilsætningen af røbestof til koncentreret smør, smør eller fløde anses for at være i overensstemmelse med de relevante EF-regler, hvis de opnåede resultater svarer til specifikationerne i punkt 10 og 11 i bilag V og punkt 8 i bilag VI, VII og VIII.
Artikel 6
Påvisning af kasein af komælk
1. Den i bilag IX fastsatte referenceanalysemetode benyttes for at sikre, at ost udelukkende fremstillet af fåre-, gede- eller bøffelmælk eller af blandinger af fåre-, gede- og bøffelmælk ikke indeholder kasein af komælk.
Kasein af komælk anses for at være til stede, hvis indholdet af kasein af komælk i den analyserede prøve er lig med eller overstiger indholdet i referenceprøven med 1 % komælk, jf. bilag IX.
2. Der kan anvendes rutinemetoder til påvisning af kasein af komælk i de i stk. 1 omhandlede oste på følgende betingelser:
|
a) |
Påvisningsgrænsen må højst være 0,5 %. |
|
b) |
Der må ikke forekomme falsk positive resultater. |
|
c) |
Kasein af komælk skal være påviseligt med den fornødne følsomhed selv efter lange modningsperioder, som det kan være tilfældet under almindelige handelsforhold. |
Hvis et af ovenstående krav ikke er opfyldt, anvendes de referencemetoder, der er fastsat i bilag IX.
Artikel 7
Påvisning af coliforme bakterier
Coliforme bakterier i smør, skummetmælkspulver, kasein og kaseinater påvises i overensstemmelse med den referencemetode, der er fastsat i bilag X.
Artikel 8
Bestemmelse af lactoseindhold
Indholdet af lactose i produkter henhørende under position 2309 bestemmes i overensstemmelse med den referencemetode, der er fastsat i bilag XI.
Artikel 9
Påvisning af løbevalle
1. Løbevalle i skummetmælkspulver til offentlig oplagring påvises i overensstemmelse med den referencemetode, der er fastsat i bilag XII.
2. Løbevalle i skummetmælkspulver og blandinger til dyrefoder påvises i overensstemmelse med den referencemetode, der er fastsat i bilag XII. Hvis der påvises løbevalle, bør bilag XIII finde anvendelse.
Artikel 10
Påvisning af kærnemælk
Kærnemælk i skummetmælkspulver påvises i overensstemmelse med den referencemetode, der er fastsat i bilag XIV.
Artikel 11
Påvisning af restkoncentrationer af antimikrobielle stoffer
Restkoncentrationer af antimikrobielle stoffer i skummetmælkspulver påvises i overensstemmelse med den referencemetode, der er fastsat i bilag XV.
Artikel 12
Bestemmelse af indholdet af skummetmælkspulver
Indholdet af skummetmælkspulver i foderblandinger bestemmes i overensstemmelse med den referencemetode, der er fastsat i bilag XVI.
Artikel 13
Påvisning af stivelse
Stivelse i skummetmælkspulver, denatureret mælkepulver og foderblandinger påvises i overensstemmelse med den referencemetode, der er fastsat i bilag XVII.
Artikel 14
Bestemmelse af vandindholdet i tørret fløde
Vandindholdet i tørret fløde bestemmes i overensstemmelse med den referencemetode, der fastsat i bilag XVIII.
Artikel 15
Bestemmelse af vandindholdet i syrnet kærnemælkspulver
Vandindholdet i syrnet kærnemælkspulver bestemmes i overensstemmelse med den referencemetode, der er fastsat i bilag XIX.
Artikel 16
Bestemmelse af mælkefedts renhed
Mælkefedts renhed bestemmes i overensstemmelse med den referencemetode, der er fastsat i bilag XX.
KAPITEL III
ALMINDELIGE OG AFSLUTTENDE BESTEMMELSER
Artikel 17
Kvalitetssikring
Analyser foretages i laboratorier, der råder over et system til analysekvalitetssikring, herunder interne kvalitetskontrolprocedurer. Laboratorier, der ikke er akkrediterede, deltager i præstationsprøvningsordninger mindst en gang om året, og deres resultater må højst afvige 2σR (referencemetodens standardafvigelse for reproducerbarhed) fra konsensusværdien. Der skal foreligge en detaljeret beskrivelse af de anvendte systemer i laboratoriet.
Laboratorier, der er akkrediterede i overensstemmelse med artikel 12 i Europa-Parlamentets og Rådets forordning (EF) nr. 882/2004 af 29. april 2004 om offentlig kontrol med henblik på verifikation af, at foderstof- og fødevarelovgivningen samt dyresundheds- og dyrevelfærdsbestemmelserne overholdes (6), er fritaget fra kravet om at deltage i præstationsprøvninger.
Artikel 18
Prøveudtagning og bestridelse af analyseresultater
1. Prøveudtagning foretages i overensstemmelse med den relevante forskrift for det pågældende produkt. Hvis der ikke er fastsat bestemmelser om prøveudtagning, anvendes bestemmelserne i ISO 707 | IDF 50, Mælk og mælkeprodukter. Vejledning om prøveudtagning.
2. Laboratorierapporter om analyseresultaterne skal indeholde tilstrækkelige oplysninger til at muliggøre en evaluering af resultaterne efter bilag II og XXI.
3. Med henblik på de analyser, der er fastsat i EF-reglerne, skal der udtages dobbelte prøver.
4. Den i bilag XXI beskrevne procedure anvendes i tilfælde, hvor resultaterne af en analyse ikke accepteres af den erhvervsdrivende.
5. Inden fem arbejdsdage efter prøveudtagningen skal prøveudtagningen om muligt gentages, hvis producenten godtgør, at prøveudtagningsproceduren ikke var korrekt. Er en ny prøveudtagning ikke mulig, accepteres sendingen.
Artikel 19
Overgangsperiode
Senest 12 måneder efter denne forordnings ikrafttrædelse foretages en vurdering af, om bilag II er overholdt. Medlemsstaterne underretter om nødvendigt straks Kommissionen, hvis der opstår større problemer i denne periode med den statistiske kontrolprocedure.
Artikel 20
Ophævelse
Forordning (EF) nr. 213/2001 ophæves.
Henvisninger til den ophævede forordning betragtes som henvisninger til nærværende forordning og læses i overensstemmelse med sammenligningstabellen i bilag XXII.
Artikel 21
Ikrafttrædelse
Denne forordning træder i kraft på tredjedagen efter offentliggørelsen i Den Europæiske Unions Tidende.
Den anvendes fra den 31. marts 2008.
Denne forordning er bindende i alle enkeltheder og gælder umiddelbart i hver medlemsstat.
Udfærdiget i Bruxelles, den 5. marts 2008.
På Kommissionens vegne
Mariann FISCHER BOEL
Medlem af Kommissionen
(1) EFT L 160 af 26.6.1999, s. 48. Senest ændret ved forordning (EF) nr. 1152/2007 (EUT L 258 af 4.10.2007, s. 3). Med virkning fra den 1. juli 2008 erstattes forordning (EF) nr 1255/1999 af forordning (EF) nr. 1234/2007 (EUT L 299 af 16.11.2007, s. 1).
(2) EFT L 37 af 7.2.2001, s. 1.
(3) EUT L 308 af 25.11.2005, s. 1. Senest ændret ved forordning (EF) nr. 1546/2007 (EUT L 337 af 21.12.2007, s. 68).
(4) EFT L 279 af 11.10.1990, s. 22. Senest ændret ved forordning (EF) nr. 1487/2006 (EUT L 278 af 10.10.2006, s. 8).
(5) EFT L 256 af 7.9.1987, s. 1. Senest ændret ved forordning (EF) nr. 733/2007 (EUT L 169 af 29.6.2007, s. 1).
(6) EUT L 165 af 30.4.2004, s. 1.
BILAG I
(Artikel 1)
LISTE OVER REFERENCEMETODER
Indeks min. = minimum, maks. = maksimum, bilag = bilag til den nævnte forordning, FT = fedtfri tørstof, PI = peroxidindeks, U = udseende, S = smag, K = konsistens, SK = samlet kimindhold, ITB = indhold af termofile bakterier, MS = medlemsstat, IDF = Det Internationale Mejeriforbund, ISO = Den Internationale Standardiseringsorganisation, IUPAC = International Union of Pure and Applied Chemistry, ADPI = American Dairy Products Institute, SKM = sødet koncentreret mælk, IMF = inddampet mælk eller fløde.
DEL A
|
Kommissionens forordning |
Produkt |
Parameter |
Grænse (1) |
Referencemetode |
Bemærkning |
||||||
|
Forordning (EF) nr. 2771/1999 — Offentlig oplagring |
Usaltet smør |
Fedtstof |
Min. 82 % m/m |
ISO 17189:2003|IDF 194:2003 |
|
||||||
|
|
|
Vand |
Maks. 16 % m/m |
ISO 3727-1:2001|IDF 80-1:2001 |
|
||||||
|
|
|
FT |
Maks. 2 % m/m |
ISO 3727-2:2001|IDF 80-2:2001 |
|
||||||
|
|
|
Fedtsyrer |
1,2 mmol/100 g fedtstof |
ISO 1740:2004|IDF 6:2004 |
|
||||||
|
|
|
PI (maks.) |
0,3 mækv. ilt/1 000 g fedtstof |
ISO 3976:2006|IDF 74:2006 |
Bemærkning 1 |
||||||
|
|
|
Coliforme bakterier |
Ikke påviselige i 1 g |
Bilag X |
Bemærkning 3 |
||||||
|
|
|
Andre fedtstoffer end mælkefedt |
Ikke påviselige ved triglyceridanalyse |
Bilag XX |
|
||||||
|
|
|
Sterolrøbestoffer |
Ikke påviselige, β-sitosterol ≤ 40 mg/kg |
Bilag VIII |
|
||||||
|
|
|
Andre røbestoffer |
|
|
|
||||||
|
|
|
|
Ikke påviseligt |
Bilag VI |
|
||||||
|
|
|
|
≤ 6 mg/kg |
Bilag VII |
|
||||||
|
|
|
|
Ikke påviselige |
Bilag V |
|
||||||
|
|
|
Organoleptiske egenskaber |
Mindst 4 point af 5 for U, S og K |
Bilag IV |
|
||||||
|
|
|
Vandfordeling |
Mindst 4 point |
ISO 7586:1985 — IDF 112A:1989 |
|
||||||
|
Forordning (EF) nr. 2771/1999 Privat oplagring |
Usaltet smør |
Fedtstof |
Min. 82 % m/m |
ISO 17189:2003|IDF 194:2003 |
|
||||||
|
|
|
Vand |
Maks. 16 % m/m |
ISO 3727-1:2001|IDF 80-1:2001 |
|
||||||
|
|
|
FT |
Maks. 2 % m/m |
ISO 3727-2:2001|IDF 80-2:2001 |
|
||||||
|
Forordning (EF) nr. 2771/1999 Privat oplagring |
Saltet smør |
Fedtstof |
Min. 80 % m/m |
ISO 17189:2003|IDF 194:2003 |
|
||||||
|
|
|
Vand |
Maks. 16 % m/m |
ISO 3727-1:2001|IDF 80-1:2001 |
|
||||||
|
|
|
FT (ekskl. salt) |
Maks. 2 % m/m |
ISO 3727-2:2001|IDF 80-2:2001 |
|
||||||
|
|
|
Salt |
Maks. 2 % m/m |
ISO 15648:2004|IDF 179:2004 |
|
||||||
|
Forordning (EF) nr. 1898/2005 kapitel II |
Usaltet smør |
Fedtstof |
Min. 82 % m/m |
ISO 17189:2003|IDF 194:2003 |
|
||||||
|
|
|
Andre fedtstoffer end mælkefedt |
|
Bilag XX |
|
||||||
|
|
|
Vand |
Maks. 16 % m/m |
ISO 3727-1 2001|IDF 80-1:2001 |
|
||||||
|
|
|
FT |
Maks. 2 % m/m |
ISO 3727-2:2001|IDF 80-2:2001 |
|
||||||
|
|
|
Røbestoffer: |
|
|
|
||||||
|
|
|
|
Se bilag VIII |
Bilag VIII |
|
||||||
|
|
|
|
Se bilag VI |
Bilag VI |
|
||||||
|
|
|
|
Se bilag VII |
Bilag VII |
|
||||||
|
|
|
|
Se bilag V |
Bilag V |
|
||||||
|
Forordning (EF) nr. 1898/2005 kapitel II |
Saltet smør |
Fedtstof |
Min. 80 % m/m |
ISO 17189:2003|IDF 194:2003 |
|
||||||
|
|
|
Andre fedtstoffer end mælkefedt |
|
Bilag XX |
|
||||||
|
|
|
Vand |
Maks. 16 % m/m |
ISO 3727-1:2001|IDF 80-1:2001 |
|
||||||
|
|
|
FT (ekskl. salt) |
Maks. 2 % m/m |
ISO 3727-2:2001|IDF 80-2:2001 |
|
||||||
|
|
|
Salt |
Maks. 2 % m/m |
ISO 15648:2004|IDF 179:2004 |
|
||||||
|
|
|
Røbestoffer: |
|
|
|
||||||
|
|
|
|
Se bilag VIII |
Bilag VIII |
|
||||||
|
|
|
|
Se bilag VI |
Bilag VI |
|
||||||
|
|
|
|
Se bilag VII |
Bilag VII |
|
||||||
|
|
|
|
Se bilag V |
Bilag V |
|
||||||
|
Forordning (EF) nr. 1898/2005 kapitel II |
Koncentreret smør |
Fedtstof |
Min. 99,8 % m/m |
IDF 24:1964 |
|
||||||
|
|
|
Vand og FT |
Maks. 0,2 % m/m |
ISO 5536:2002|IDF 23:2002 (vandindhold) IDF 24:1964 (SNF) |
|
||||||
|
|
|
Fedtsyrer |
1,2 mmol/100 g fedtstof |
ISO 1740:2004|IDF 6:2004 |
|
||||||
|
|
|
PI (maks.) |
0,5 mækv. ilt/1 000 g fedtstof |
ISO 3976:2006|IDF 74:2006 |
Bemærkning 1 |
||||||
|
|
|
Andre fedtstoffer end mælkefedt |
Ingen |
Bilag XX |
|
||||||
|
Smag |
Ren |
||||||||||
|
Lugt |
Ingen fremmed lugt |
||||||||||
|
Andet |
Ingen neutraliseringsmidler, antioxidanter og konserveringsmidler |
||||||||||
|
|
|
Røbestoffer: |
|
|
|
||||||
|
Se bilag VIII |
Bilag VIII |
|||||||||
|
Se bilag VI |
Bilag VI |
|||||||||
|
Se bilag VII |
Bilag VII |
|||||||||
|
Se bilag V |
Bilag V |
|||||||||
|
Forordning (EF) nr. 1898/2005 kapitel II |
Fløde |
Fedtstof |
Min. 35 % m/m |
ISO 2450:1999 — IDF 16 C:1987 |
|
||||||
|
|
|
Andre fedtstoffer end mælkefedt |
|
Bilag XX |
|
||||||
|
|
|
Røbestoffer: |
|
|
|
||||||
|
Se bilag VIII |
|
Bemærkning 2 |
||||||||
|
|
|
|
Se bilag VI |
Bilag VI |
|
||||||
|
|
|
|
Se bilag VII |
|
Bemærkning 2 |
||||||
|
|
|
|
Se bilag V |
Bilag V |
|
||||||
|
Forordning (EF) nr. 1898/2005 kapitel III |
Koncentreret smør |
Fedtstof |
Min. 96 % m/m |
|
Bemærkning 2 |
||||||
|
|
|
Andre fedtstoffer end mælkefedt |
|
Bilag XX |
|
||||||
|
|
|
FT |
Maks. 2 % m/m |
|
Bemærkning 2 |
||||||
|
|
|
Røbestoffer: |
|
|
|
||||||
|
15 g/100 kg koncentreret smør: |
Bilag VIII |
|||||||||
|
|
|
|
17 g/100 kg koncentreret smør: |
Bilag VIII |
|
||||||
|
|
|
|
10,34 kg/100 t koncentreret smør: |
Bilag V |
|
||||||
|
|
|
|
|
|
Bemærkning 2 |
||||||
|
|
|
|
|
|
Bemærkning 2 |
||||||
|
|
|
Lecithin (E 322) |
Maks. 0,5 % m/m |
|
Bemærkning 2 |
||||||
|
|
|
NaC1 |
Maks. 0,75 % m/m |
ISO 15648:2004|IDF 179:2004 |
|
||||||
|
|
|
Fedtsyrer |
1,2 mmol/100 g fedtstof |
ISO 1740:2004|IDF 6:2004 |
|
||||||
|
|
|
PI (maks.) |
Maks. 0,5 mækv. ilt/1 000 g fedtstof |
ISO 3976:2006|IDF 74:2006 |
Bemærkning 1 |
||||||
|
|
|
Smag |
Ren |
|
|
||||||
|
|
|
Lugt |
Ingen fremmed lugt |
|
|
||||||
|
|
|
Andet |
Ingen neutraliseringsmidler, antioxidanter og konserveringsmidler |
|
|
||||||
|
Forordning (EF) nr. 1898/2005 kapitel IV |
Usaltet smør |
Fedtstof |
Min. 82 % m/m |
ISO 17189:2003|IDF 194:2003 |
|
||||||
|
|
|
Vand |
Maks. 16 % m/m |
ISO 3727-1:2001|IDF 80-1:2001 |
|
||||||
|
|
|
FT |
Maks. 2 % m/m |
ISO 3727-2:2001|IDF 80-2:2001 |
|
||||||
|
Forordning (EF) nr. 1898/2005 kapitel IV |
Saltet smør |
Fedtstof |
Min. 80 % m/m |
ISO 17189:2003|IDF 194:2003 |
|
||||||
|
|
|
Vand |
Maks. 16 % m/m |
ISO 3727-1:2001|IDF 80-1:2001 |
|
||||||
|
|
|
FT (ekskl. salt) |
Maks. 2 % m/m |
ISO 3727-2:2001|IDF 80-2:2001 |
|
||||||
|
|
|
Salt |
Maks. 2 % m/m |
ISO 15648:2004|IDF 179:2004 |
|
||||||
|
Artikel 9 og afsnit II i forordning (EF) nr. 1255/1999 |
Ost af fåre- og/eller gedemælk |
Komælk |
< 1 % m/m |
Bilag IX |
|
||||||
|
Forordning (EØF) nr. 2921/90 |
Bilag I — Syrekasein |
Vand |
Maks. 12,00 % m/m |
ISO 5550:2006|IDF 78:2006 |
|
||||||
|
|
|
Fedtstof |
Maks. 1,75 % m/m |
ISO 5543:2004|IDF127:2004 |
|
||||||
|
|
|
Frie syrer |
Maks. 0,30 ml 0,1N NaOH-opløsning/g |
ISO 5547:1978 — IDF 91:1979 |
|
||||||
|
Forordning (EØF) nr. 2921/90 |
Bilag I — Løbekasein |
Vand |
Maks. 12,00 % m/m |
ISO 5550:2006|IDF 78:2006 |
|
||||||
|
|
|
Fedtstof |
Maks. 1,00 % m/m |
ISO 5543:2004|IDF 127:2004 |
|
||||||
|
|
|
Aske |
Min. 7,50 % m/m |
ISO 5545:1978 — IDF 90:1979 |
|
||||||
|
Forordning (EØF) nr. 2921/90 |
Bilag I — Kaseinat |
Vand |
Maks. 6,00 % m/m |
ISO 5550:2006|IDF 78:2006 |
|
||||||
|
|
|
Mælkeprotein |
Min. 88,00 % m/m |
ISO 5549:1978 — IDF 92:1979 |
|
||||||
|
|
|
Fedtstoffer og aske |
Maks. 6,00 % m/m |
ISO 5543:2004|IDF 127:2004 |
|
||||||
|
|
|
Aske (fixed ash) |
|
ISO 5544:1978 — IDF 89:1979 |
|
||||||
|
|
|
Aske |
|
ISO 5545:1978 — IDF 90:1979 |
|
||||||
|
Forordning (EØF) nr. 2921/90 |
Bilag II — Syrekasein |
Vand |
Maks. 10,00 % m/m |
ISO 5550:2006|IDF 78:2006 |
|
||||||
|
|
|
Fedtstof |
Maks. 1,50 % m/m |
ISO 5543:2004|IDF 127:2004 |
|
||||||
|
|
|
Frie syrer |
Maks. 0,20 ml 0,1 N NaOH-opløsning/g |
ISO 5547:1978 — IDF 91:1979 |
|
||||||
|
|
|
SK (maks.) |
30 000/g |
ISO 4833:2003 |
Bemærkning 3 |
||||||
|
|
|
Coliforme bakterier |
Ingen i 0,1 g |
Bilag X |
Bemærkning 3 |
||||||
|
|
|
ITB (maks.) |
5 000/g |
ISO 4833:2003 |
Bemærkning 3 og 4 |
||||||
|
Forordning (EØF) nr. 2921/90 |
Bilag II — Løbekasein |
Vand |
Maks. 8,00 % m/m |
ISO 5550:2006|IDF 78:2006 |
|
||||||
|
|
|
Fedtstof |
Maks. 1,00 % m/m |
ISO 5543:2004|IDF 127:2004 |
|
||||||
|
|
|
Aske |
Min. 7,50 % m/m |
ISO 5545:1978 — IDF 90:1979 |
|
||||||
|
|
|
SK (maks.) |
30 000/g |
ISO 4833:2003 |
Bemærkning 3 |
||||||
|
|
|
Coliforme bakterier |
Ingen i 0,1 g |
Bilag X |
Bemærkning 3 |
||||||
|
|
|
ITB (maks.) |
5 000/g |
ISO 4833:2003 |
Bemærkning 3 og 4 |
||||||
|
Forordning (EØF) nr. 2921/90 |
Bilag II — Kaseinat |
Vand |
Maks. 6,00 % m/m |
ISO 5550:2006|IDF 78:2006 |
|
||||||
|
|
|
Mælkeprotein |
Min. 88,00 % m/m |
ISO 5549:1978 — IDF 92:1979 |
|
||||||
|
|
|
Fedtstoffer og aske |
Maks. 6,00 % m/m |
ISO 5543:2004|IDF 127:2004 ISO 5544:1978 — IDF 89:1979 eller ISO 5545:1978 — IDF 90:1979 |
|
||||||
|
|
|
SK (maks.) |
30 000/g |
ISO 4833:2003 |
Bemærkning 3 |
||||||
|
|
|
Coliforme bakterier |
Ingen i 0,1 g |
Bilag X |
Bemærkning 3 |
||||||
|
|
|
ITB (maks.) |
5 000/g |
ISO 4833:2003 |
Bemærkning 3 og 4 |
||||||
|
Forordning (EØF) nr. 2921/90 |
Bilag III — Kaseinat |
Vand |
Maks. 6,00 % m/m |
ISO 5550:2006|IDF 78:2006 |
|
||||||
|
|
|
Mælkeprotein |
Min. 85,00 % m/m |
ISO 5549:1978 — IDF 92:1979 |
|
||||||
|
|
|
Fedtstof |
Maks. 1,50 % m/m |
ISO 5543:2004|IDF 127:2004 |
|
||||||
|
|
|
Lactose |
Maks. 1,00 % m/m |
ISO 5548:2004|IDF 106:2004 |
|
||||||
|
|
|
Aske |
Maks. 6,50 % m/m |
ISO 5544:1978 — IDF 89:1979 eller ISO 5545:1978 — IDF 90:1979 |
|
||||||
|
|
|
SK (maks.) |
30 000/g |
ISO 4833:2003 |
Bemærkning 3 |
||||||
|
|
|
Coliforme bakterier |
Ingen i 0,1 g |
Bilag X |
Bemærkning 3 |
||||||
|
|
|
ITB (maks.) |
5 000/g |
ISO 4833:2003 |
Bemærkning 3 og 4 |
||||||
|
Forordning (EF) nr. 2799/1999 |
Foderblandinger og skummetmælkspulver (til dyrefoder) |
Vand (syrnet kærnemælkspulver) |
Maks. 5 % m/m |
Bilag XIX |
|
||||||
|
|
|
Protein |
31,4 % (min.) i det fedtfrie tørstof |
ISO 8968-1|2|3:2001|IDF 20-1|2|3:2001 |
|
||||||
|
|
|
Vand (skummetmælkspulver) |
Maks. 5 % m/m |
ISO 5537:2004|IDF 26:2004 |
|
||||||
|
|
|
Fedtstof (skummetmælkspulver) |
Maks. 11 % m/m |
ISO 1736:2000 — IDF 9C:1987 |
|
||||||
|
|
|
Løbevalle (skummetmælkspulver) |
Ingen |
Bilag XIII |
Bemærkning 6 |
||||||
|
|
|
Stivelse (skummetmælkspulver) |
Ingen |
Bilag XVII |
|
||||||
|
|
|
Vand (blanding) |
Maks. 5 % m/m i det fedtfrie tørstof |
ISO 5537:2004|IDF 26:2004 |
|
||||||
|
|
|
Fedtstof (blanding) |
|
Kommissionens direktiv 84/4/EØF (EFT L 15 af 18.1.1984, s. 29) |
|
||||||
|
|
|
Løbevalle (blanding) |
Ingen |
Bilag XIII |
|
||||||
|
|
|
Indhold af skummetmælkspulver (i det endelige produkt) |
Min. 50 % m/m |
Bilag XVI |
|
||||||
|
|
|
Fedtstof (i det endelige produkt) |
Min. 2,5 % m/m eller 5 % m/m |
Kommissionens direktiv 84/4/EØF (EFT L 15 af 18.1.1984, s. 29) |
Bemærkning 7 |
||||||
|
|
|
Stivelse (i det endelige produkt) |
Min. 2 % m/m |
Bilag XVII |
Bemærkning 8 |
||||||
|
|
|
Kobber (i det endelige produkt) |
25 ppm |
Kommissionens direktiv 78/633/EØF (EFT L 206 af 26.7.1987, s. 43) |
|
||||||
|
Forordning (EF) nr. 214/2001 |
Skummetmælkspulver (spraymetoden) |
Fedtstof |
Maks. 1,0 % m/m |
ISO 1736:2000 — IDF 9C:1987 |
|
||||||
|
|
|
Protein |
31,4 % (2)(min.) i det fedtfrie tørstof |
ISO 8968-1/2:2001|IDF 20-1/2:2001 |
|
||||||
|
|
|
Vand |
Maks. 3,5 % m/m |
ISO 5537:2004|IDF 26:2004 |
|
||||||
|
|
|
Syreindhold |
Maks., 19,5 ml, 0,1N NaOH, 10 g fedtfrit tørstof |
ISO 6091:1980 — IDF 86:1981 |
|
||||||
|
|
|
Lactater |
Maks. 150 mg/100 g fedtfrit tørstof |
ISO 8069:2005|IDF 69:2005 |
|
||||||
|
|
|
Fosfatase |
Negativ |
ISO 11816-1:2006|IDF 155-1:2006 |
|
||||||
|
|
|
Uopløselighed |
Maks. 0,5 ml ved 24 °C |
ISO 8156:2005|IDF 129:2005 |
|
||||||
|
|
|
Brændte partikler |
Filter A eller B (15,0 mg) |
ADPI (1990) |
|
||||||
|
|
|
SK |
40 000/g |
ISO 4833:2003 |
Bemærkning 3 |
||||||
|
|
|
Coliforme bakterier |
Negativ/0,1 g |
Bilag X |
Bemærkning 3 |
||||||
|
|
|
Kærnemælk |
Negativ |
Bilag XIV |
|
||||||
|
|
|
Løbevalle |
Negativ |
Bilag XII |
|
||||||
|
|
|
Sur valle |
Negativ |
|
Bemærkning 2 |
||||||
|
|
|
Antimikrobielle stoffer |
|
Bilag XV |
|
DEL B
Referencemetoderne i del B anvendes ved analyse af produkter, der er omfattet af en af forordningerne i første kolonne.
|
Kommissionens forordning |
Produkt |
KN-kode |
Parameter |
Grænse |
Referencemetode |
Bemærkning |
|
Forordning (EF) nr. 2658/87 Forordning (EF) nr. 2535/2001 Forordning (EF) nr. 1282/2006 |
Mælk og fløde, ikke koncentreret og ikke tilsat sukker eller andre sødemidler |
0401 |
Fedtstof (≤ 6 % m/m) |
Grænserne svarer til de grænser, der er specificeret i beskrivelsen af KN-koden for det særlige produkt, og som eventuelt er præciseret i Kommissionens forordning (EØF) nr. 3846/87 (EFT L 366 af 24.12.1987, s. 1), del 9 i nomenklaturen over eksportrestitutioner, eller i forordning (EF) nr. 2535/2001 (EFT L 341 af 22.12.2001, s. 29) |
ISO 1211:2001 — IDF 1D:1996 |
|
|
|
|
|
Fedtstof (> 6 % m/m) |
|
ISO 2450:1999 — IDF 16C:1987 |
|
|
|
Mælk og fløde, koncentreret eller tilsat sukker eller andre sødemidler |
0402 |
Fedtstof (flydende form) |
|
ISO 1737:1999 — IDF 13C:1987 |
|
|
|
|
|
Fedtstof (fast form) |
|
ISO 1736:2000 — IDF 9C:1987 |
|
|
|
|
|
Protein |
|
ISO 8968-1|2|3:2001|IDF20-1|2|3:2001 |
|
|
|
|
|
Saccharose (normalt indhold) |
|
ISO 2911:2004| IDF 35:2004 |
|
|
|
|
|
Saccharose (ringe indhold) |
|
|
Bemærkning 2 |
|
|
|
|
Tørstof (SKM) |
|
ISO 6734:1989 — IDF 15B:1991 |
|
|
|
|
|
Tørstof (IMF) |
|
ISO 6731:1989 — IDF 21B:1987 |
|
|
|
|
|
Vand (mælkepulver) |
|
ISO 5537:2004/IDF 26:2004 |
|
|
|
|
|
Vand (flødepulver) |
|
Bilag XVIII |
|
|
|
Kærnemælk, fermenteret eller syrnet mælk og fløde, også koncentreret, tilsat sukker eller andre sødemidler |
0403 |
Fedtstof |
|
ISO 1211:2001 — IDF 1D:1996 ISO 1736:2000 — IDF 9C:1987 ISO 2450:1999 — IDF 16 C:1987 ISO 7208:1999 — IDF 22B:1987 ISO 8262-3:2005 — IDF 124-3:2005 |
|
|
|
|
|
Protein |
|
ISO 8968-1|2|3:2001|IDF 20-1|2|3:2001 |
|
|
|
|
|
Saccharose (normalt indhold) |
|
ISO 2911:2004|IDF 35:2004 |
|
|
|
|
|
Saccharose (ringe indhold) |
|
|
Bemærkning 2 |
|
|
|
|
Vand (syrnet kærnemælkspulver) |
|
Bilag XIX |
|
|
|
|
|
Vand (usyrnet kærnemælkspulver) |
|
ISO 5537:2004|IDF26:2004 |
|
|
|
|
|
Tørstof (andre produkter) |
|
Metoder, der er godkendt af den kompetente myndighed |
|
|
|
Valle, også koncentreret eller tilsat sukker eller andre sødemidler; varer bestående af naturlige mælkebestanddele |
0404 |
Fedtstof |
|
ISO 1736:2000 — IDF 9C:1987 ISO 2450:1999 — IDF 16C:1987 ISO 7208:1999 — IDF 22B:1987 |
|
|
|
|
|
Protein |
|
ISO 8968-1|2|3:2001|IDF 20-1|2|3:2001 |
|
|
|
|
|
Saccharose (normalt indhold) |
|
ISO 2911:2004|IDF 35:2004 |
|
|
|
|
|
Saccharose (ringe indhold) |
|
|
Bemærkning 2 |
|
|
|
0404 90 |
Protein |
|
ISO 8968 1/2 2001|IDF 20-1/2:2001 |
|
|
|
|
|
Vand |
|
IDF 21B:1987 |
|
|
|
|
|
Tørstof |
|
ISO 6734:1989 — IDF 15B:1991 |
|
|
|
|
|
(Koncentrerede produkter) |
|
ISO 6731:1989 — IDF 21B:1987 |
|
|
|
Smør og andre mælkefedtstoffer; smørbare mælkefedtprodukter |
0405 |
Fedtstof (hvis ≤ 85 % m/m) |
|
ISO 17189:2003|IDF 194:2003 |
|
|
|
|
Smør |
Vand |
|
ISO 3727-1:2001|IDF 80-1:2001 |
|
|
|
|
|
FT |
|
ISO 3727-2:2001|IDF 80-2:2001 |
|
|
|
|
|
NaCl |
|
ISO 15648:2004|IDF 179:2004 |
|
|
|
|
|
Fedtstof (hvis > 99 % m/m) |
|
IDF 24:1964 |
|
|
|
Smørolie |
|
Vand (hvis fedtstof < 99 % m/m) |
|
ISO 5536:2002|IDF 23:2002 |
|
|
|
Ost og ostemasse |
0406 |
Fedtstof |
|
ISO 1735:2004|IDF 5:2004 |
|
|
|
|
|
Tørstof |
|
ISO 5534:2004|IDF 4:2004 |
|
|
|
|
|
Tørstof (ricotta) |
|
ISO 2920:2004|IDF 58:2004 |
|
|
|
|
|
NaCl |
|
ISO 5943:2006|IDF 88:2006 |
|
|
|
|
|
Lactose |
|
ISO 5765-1/2:2002|IDF 79-1/2:2002 |
|
|
Forordning (EF) nr. 2658/87 |
Foderblandinger |
2309 |
Lactose |
|
Bilag XI |
|
Bemærkninger til listen over EU-referencemetoder.
Bemærkning 1: Isolering af mælkefedt som beskrevet i ISO 1740:1991 (beskyttelse mod lys).
Bemærkning 2: Der er ikke fastsat nogen referencemetode. Metoder, der er godkendt af den kompetente myndighed.
Bemærkning 3: Prøven skal forberedes ifølge ISO 8261:2001|IDF 122:2001.
Bemærkning 4: Inkubering i 48 timer ved 55 °C. Der skal træffes forholdsregler mod udtørring af dyrkningsmediet.
Bemærkning 5: % m/m FT = % m/m tørstof — % m/m fedtstof.
Bemærkning 6: Kommissionens direktiv 84/4/EØF.
Bemærkning 7: Kommissionens forordning (EF) nr. 2799/1999 (EFT L 340 af 31.12.1999, s. 3-27).
Bemærkning 8: Kommissionens direktiv 78/633/EØF.
(1) Jf. dog kravene i særforordningen.
(2) Mindsteindhold af protein bliver 34 % pr. 1. september 2009.
BILAG II
(Artikel 3)
VURDERING AF, OM EN SENDING OVERHOLDER DEN FORSKRIFTSMÆSSIGE GRÆNSE
1. PRINCIP
Når der i de relevante bestemmelser er fastsat detaljerede prøveudtagningsprocedurer, følges sådanne procedurer. I alle andre tilfælde anvendes der en prøve bestående af mindst 3 prøveenheder udtaget tilfældigt af den sending, der forevises til kontrol. En blandingsprøve kan forberedes. Resultatet sammenlignes med de forskriftsmæssige grænser ved beregning af et konfidensinterval på 95 % som to gange standardafvigelsen, hvor den relevante standardafvigelse afhænger af, 1) om metoden er valideret ved internationalt samarbejde med værdier for σr og σR, eller 2) om der ved intern validering er beregnet en intern reproducerbarhed. Konfidensintervallet er da det samme som resultatets måleusikkerhed.
2. METODE VALIDERET VED INTERNATIONALT SAMARBEJDE
I dette tilfælde er standardafvigelsen for repeterbarhed σr og standardafvigelsen for reproducerbarhed σR blevet fastlagt, og laboratoriet kan dokumentere overholdelse af karakteristika for den validerede metodes ydeevne.
Det aritmetiske gennemsnit  af de n gentagne målinger beregnes.
af de n gentagne målinger beregnes.
Den ekspanderede måleusikkerhed (k = 2) af  som
som

Hvis det endelige måleresultat x beregnes med en formel af typen x = y1 + y2, x = y1 – y2, x = y1. y2 oder x = y1 / y2, følges de sædvanlige procedurer til kombinering af standardafvigelser i sådanne tilfælde.
Sendingen anses for ikke at overholde den øvre forskriftsmæssige grænse UL, hvis

ellers anses den for at overholde UL.
Sendingen anses for ikke at overholde den nedre forskriftsmæssige grænse LL, hvis

ellers anses den for at overholde LL.
3. INTERN VALIDERING MED BEREGNING AF DEN INTERNE STANDARDAFVIGELSE FOR REPRODUCERBARHED
Hvis der anvendes metoder, der ikke er angivet i denne forordning, og der ikke er fastlagt præcisionsmålinger, skal der foretages en intern validering. Den interne standardafvigelse for repeterbarhed sir og den interne standardafvigelse for reproducerbarhed siR skal anvendes i stedet for henholdsvis σr og σ R i formlen til beregning af den ekspanderede usikkerhed U.
Afgørelser træffes i henhold til punkt 1. Hvis sendingen imidlertid bedømmes til ikke at overholde den forskriftsmæssige grænse, gentages målingerne ved anvendelse af den metode, der er angivet i denne forordning, og der træffes afgørelse i henhold til punkt 1.
BILAG III
(Artikel 4)
EVALUERING AF DOMMERE OG PÅLIDELIGHEDEN AF RESULTATER AF ORGANOLEPTISKE BEDØMMELSER
Følgende procedurer benyttes, hvis der gøres brug af karaktergivningsmetoder (IDF — standard 99C:1997).
A) BESTEMMELSE AF »REPETERBARHEDSINDEKS«
Der analyseres mindst ti prøver som blinde dobbeltanalyser af en dommer inden for en periode på 12 måneder. Dette vil sædvanligvis finde sted i flere omgange. Resultaterne af de forskellige produkters karakteristika evalueres ved anvendelse af følgende formel:

hvor:
|
w1 |
: |
repeterbarhedsindeks |
|
xi1 |
: |
point for første evaluering af prøve xi |
|
xi2 |
: |
point for anden evaluering af prøve xi |
|
n |
: |
antal prøver |
De prøver, der skal evalueres, skal afspejle et bredt kvalitetsudsnit. wI må ikke overstige 1,5 (fempunktsskalaer).
B) BESTEMMELSER AF »AFVIGELSESINDEKSET«
Dette indeks skal anvendes til at tjekke, om en dommer anvender samme skala for kvalitetsvurdering som en gruppe erfarne dommere. De point, som dommeren har givet, sammenlignes med gennemsnittet af de point, som dommerpanelet har givet.
Følgende formel anvendes for evalueringen af resultaterne:

hvor:
|
xi1; xi2 |
: |
se litra A) |
|
|
: |
dommerpanelets gennemsnitspoint for henholdsvis første og anden evaluering af prøven xi |
|
n |
: |
antal prøver (mindst 10 pr. tolvmånedersperiode). |


De prøver, der skal evalueres, skal afspejle et bredt kvalitetsudsnit. DI må ikke overstige 1,5 (fempunktsskalaer).
Medlemsstaterne giver meddelelse om alle vanskeligheder, der er opstået under anvendelsen af denne metode.
Hvis det konstateres, at en dommer overskrider grænsen på 1,5 point for afvigelsesindekset eller repeterbarhedsindekset, gennemfører den officielle myndigheds særligt sagkyndige atter et eller flere randomiserede præstationstjek på prøver, som vedkommende har klassificeret i de seneste uger, eller den/de særligt sagkyndige gennemfører et eller flere check, hvor de ledsager den pågældende dommer. Det er nødvendigt med nøje tilsyn for at afgøre, om dommerne fortsat skal varetage deres hverv. De konstaterede forhold bør dokumenteres, og dokumentationen bør opbevares til brug ved opfølgende indsats.
C) SAMMENLIGNING AF DE OPNÅEDE RESULTATER I FORSKELLIGE REGIONER AF EN MEDLEMSSTAT OG I FORSKELLIGE MEDLEMSSTATER
Hvis det er relevant, foretages der mindst én gang om året en test, som giver mulighed for at sammenligne resultaterne af de vurderinger, som dommere fra forskellige regioner har foretaget. Konstateres der væsentlige forskelle, træffes de nødvendige foranstaltninger for at finde årsagerne hertil og nå frem til sammenlignelige resultater.
Medlemsstaterne kan tilrettelægge test, som giver mulighed for at sammenligne de resultater, der er opnået af deres egne dommere og af dommere fra nabomedlemsstater. I tilfælde af væsentlige forskelle skal der foretages en tilbundsgående undersøgelse for at nå frem til sammenlignelige resultater.
Medlemsstaterne underretter Kommissionen om resultaterne af disse sammenligninger.
BILAG IV
(Artikel 4)
ORGANOLEPTISK BEDØMMELSE AF SMØR
1. FORMÅL
Formålet med denne metode til organoleptisk bedømmelse af smør er at give en ensartet metode, der kan benyttes i samtlige medlemsstater.
Nærmere detaljer findes i den aktuelle udgave af IDF International Standard for Milk and Milk Products, IDF 99 — Parts 1,2,3 on Sensory Evaluation.
2. DEFINITIONER
Ved »organoleptisk bedømmelse« (vurdering) forstås undersøgelse af et produkts egenskaber ved hjælp af sanseorganerne.
Ved »panel« forstås en gruppe udvalgte dommere, der under vurderingen arbejder uden indbyrdes kontakt og uden at påvirke hinanden.
Ved »dommer« forstås en person, der er udvalgt på grund af vedkommendes evne til at foretage en organoleptisk test. Denne type dommer har ikke nødvendigvis stor erfaring.
Ved »særligt sagkyndig dommer« forstås en person, der har stor sensorisk følsomhed og erfaring med sensorisk metodologi, og som er i stand til at foretage konsistente og pålidelige organoleptiske bedømmelser af diverse produkter. Denne type dommer skal have en god langtidshukommelse hvad angår sanseindtryk.
Ved »karaktergivning« forstås et panels organoleptiske bedømmelse efter en talskala. Der skal benyttes en nomenklatur for fejl.
Ved »klassificering« forstås en kvalitetssortering, der sker på grundlag af karaktergivningen.
Ved »kontroldokumenter« forstås dokumenter, hvori noteres de enkelte point for hver egenskab samt produktets endelige klasse (samme dokument kan også anvendes til angivelse af kemisk sammensætning).
3. PRØVELOKALE
Nærmere detaljer findes i ISO 8589 og ISO/DIS 22935-2 | IDF 99-2 afsnit 7.
Der skal træffes forholdsregler, så dommerne i prøvelokalet ikke bliver påvirket af udefra kommende faktorer.
Prøvelokalet skal være frit for fremmede lugte og let at rengøre. Væggene skal have en lys farve og må ikke være reflekterende.
Prøvelokalet og dets belysning skal være således, at de produktegenskaber, der skal bedømmes, ikke påvirkes.
Lokalet skal være udstyret med passende termostatisk styring, så smørret kan holde en konstant temperatur. Smørret bør ved klassificeringen have en temperatur på 12 °C (±2 °C).
4. UDVÆLGELSE AF DOMMERE
En dommer skal være velkendt med smørprodukter og kunne foretage organoleptisk klassificering. Vedkommendes egnethed bør overvåges regelmæssigt (mindst en gang om året) af den kompetente myndighed.
4.1. Nærmere detaljer findes i ISO/DIS 22935-1 | IDF 99-1 afsnit 4 (hvervning) og afsnit 5.1 vedrørende generelle krav og screeningtest, der kan benyttes, inden en ny dommer anvendes officielt.
Løbende efteruddannelse er af afgørende betydning, og der bør regelmæssigt afholdes fælles kurser. Nærmere oplysninger om uddannelse af paneler findes i ISO 8586-1.
4.2. Grunduddannelsen bør omfatte følgende:
|
— |
generel teori om og praktisk betydning af organoleptisk bedømmelse |
|
— |
metoder, skalaer og beskrivelse af sanseindtryk |
|
— |
påvisning og genkendelse af organoleptiske egenskaber og specifikke sensoriske termer |
|
— |
grundlæggende viden om smørfremstilling |
|
— |
validerede referencer og prøver, der kan hjælpe dommeren til at identificere specifikke smagsindtryk og smagsintensitet i produktet. |
5. KRAV TIL PANELET
Antallet af dommere i panelet bør være ulige og mindst tre personer. Flertallet af dem skal være medarbejdere ved den kompetente myndighed eller autoriserede personer, der ikke er ansat i mejeribruget.
Panellederen har ansvaret for hele proceduren og kan deltage i panelet.
Flere faktorer skal tages i betragtning inden vurderingen, for at dommerne kan opnå en optimal dømmekraft:
|
— |
Dommerne må ikke lide af sygdomme, der kan påvirke deres dømmekraft. Hvis sådanne sygdomme opstår, skal en anden person indlemmes i panelet. |
|
— |
Dommerne skal overholde tiden for deltagelse i bedømmelsen og afsætte tilstrækkelig tid til bedømmelsen. |
|
— |
Dommerne skal undgå at bruge stærkt duftende produkter såsom parfume, aftershave og deodorant og bør undlade at spise stærkt aromatiserede (krydrede) fødevarer osv. |
|
— |
Dommerne må hverken ryge, spise eller drikke andet end vand i den sidste halve time inden bedømmelsen. |
6. PRÆSTATIONER
Alle dommere bør regelmæssigt deltage i paneler for organoleptisk bedømmelse for at bevare deres kompetence. Hyppigheden afhænger af, hvor meget smør der produceres, men vedkommende bør om muligt deltage i mindst et panel pr. måned.
Erfarne dommere bør ligeledes deltage i flere paneler om året, om muligt mindst en gang pr. kvartal.
7. UDTAGNING OG FORBEREDELSE AF PRØVER
Det er afgørende, at prøvernes identitet er skjult under bedømmelsen, så enhver form for eventuel partiskhed undgås. Prøverne bør forsynes med en kode.
Dette skal ske inden bedømmelsen. Der bør fastsættes krav til smørrets temperatur under transporten til prøvelokalet (6 °C ± 2 °C).
Når den organoleptiske bedømmelse foretages på et kølehus, udtages prøven med en smørstikker. Hvis den organoleptiske bedømmelse finder sted på et andet sted end i kølehuset, skal der udtages en prøve på mindst 500g. Under vurderingen bør smørret have en temperatur på 12 °C (±2 °C) (NB: i ISO/DIS 22935-2 | IDF 99-2 er smørrets temperatur ved bedømmelsen fastsat til 14 °C ±2 °C). Store afvigelser bør for enhver pris undgås.
8. VURDERING AF VÆRDIEN AF DE ENKELTE EGENSKABER
8.1. Den organoleptiske bedømmelse skal omfatte følgende egenskaber: udseende, konsistens, lugt og smag.
Udseende omfatter farve, synlig renhed, fravær af fysisk kontaminering, fravær af mugvækst og ensartet vandfordeling. Vandfordelingen afprøves efter IDF-standard 112A/1989.
Konsistens omfatter formstabilitet, tekstur og hårdhed. Smørbarhed kan kontrolleres med fysiske metoder, hvis en enkeltmedlemsstat ønsker det for at imødekomme forbrugerkrav. Kommissionen kan på et senere tidspunkt beslutte at harmonisere metoderne.
Formstabilitet drejer sig om produktets sammenhængsevne ved forbrug. Formstabilitet forbindes normalt med hårdhed og smørbarhed, og den bør være ensartet i hele produktet. Den hænger nøje sammen med teksturen og udtrykker produktets evne til at bære sin egen vægt. Den konstateres ved modstanden under gennemskæring og kan måles mekanisk og ved fornemmelsen i munden og mellem fingrene.
Smag er de egenskaber, der opfattes i munden, primært via tungens smagsløg.
Lugt er de egenskaber, der opfattes af næsen og lugtesansen.
En signifikant afvigelse fra den anbefalede temperatur forhindrer pålidelig vurdering af konsistens, lugt og smag. Temperaturen er af altafgørende betydning.
Klassificeringen af smør skal udskydes, hvis temperaturen ligger uden for det anbefalede interval.
8.2. Egenskaberne skal bedømmes organoleptisk hver for sig. Der gives karakterer efter tabel 1.
8.3. Det kan være ønskværdigt, at dommerne, inden de begynder bedømmelsen, sammen giver karakterer for en eller flere referenceprøver med hensyn til udseende, konsistens samt lugt og smag, så der bliver vurderet ensartet.
8.4. Karaktererne for, at produktet kan godkendes, er som følger:
Ved karaktergivningen følges afsnit 7 — Nomenclature, and description of criteria applicable to points.
|
|
Maksimum |
Krævet |
|
Udseende |
5 |
4 |
|
Konsistens |
5 |
4 |
|
Smag/lugt |
5 |
4 |
|
— |
Hvis den krævede karakter ikke opnås, skal fejlen beskrives. |
|
— |
De karakterer, den enkelte dommer giver for hver egenskab, skal optegnes i kontroldokumentet. |
|
— |
Produktet godkendes eller afvises på grundlag af en flertalsbeslutning. |
|
— |
Der bør ikke ofte forekomme tilfælde (ikke mere end ét pr. 20 prøver), hvor de enkelte dommeres karakter for hver egenskab giver en forskel på over et point. Hvis det sker, må panellederen kontrollere, om panelet er kompetent. |
9. TILSYN
Lederen af et panel, som skal være en medarbejder ved den kompetente myndighed og kan være medlem af panelet, skal være generelt ansvarlig for hele proceduren. Vedkommende skal optegne de enkelte dommeres karakter for hver egenskab i kontroldokumentet og attestere, om produktet er godkendt eller afvist.
10. NOMENKLATUR
Jf. vedlagte tabel 2.
11. HENVISNINGER
FIL-IDF 99C:1997 Sensory evaluation of dairy products by scoring — Reference method
ISO/DIS 22935 | IDF 99 International Standard for Milk and Milk Products — Sensory analysis — Parts 1-3
ISO 8586-1 Sensorisk analyse — Generel vejledning i udvælgelse, træning og overvågning af assessorer — del 1
ISO 8589 Sensory analysis — General guidance for the design of test rooms
FIL-IDF 112A:1989 Butter — Determination of water dispersion value.
Tabel 1
Bedømmelsesskema for smør
|
Udseende |
Konsistens |
Smag og lugt |
||||||
|
Point |
Nr. (1) |
Bemærkninger |
Point (kvalitetsklasse) |
Nr. (1) |
Bemærkninger |
Point (kvalitetsklasse) |
Nr. (1) |
Bemærkninger |
|
5 |
|
Meget tilfredsstillende idealtype højeste kvalitet (ensartet tør) |
5 |
|
Meget tilfredsstillende idealtype højeste kvalitet (godt smørbar) |
5 |
|
Meget tilfredsstillende idealtype højeste kvalitet (absolut ren, fineste lugt) |
|
4 |
|
Tilfredsstillende (2) ingen åbenlyse fejl |
4 |
17 18 |
Tilfredsstillende (2) hård blød |
4 |
|
Tilfredsstillende (2) ingen åbenlyse fejl |
|
3 |
1 2 3 4 5 6 7 8 |
Temmelig tilfredsstillende (mindre fejl) synlige vanddråber ikke ensartet farve, tofarvet stribet skjoldet, marmoreret plettet olieudskillelse for stærk farve poret (åben tekstur) |
3 |
14 15 16 17 18 |
Temmelig tilfredsstillende (mindre fejl) skør, smuldrende dejagtig, fedtet klæbrig hård blød |
3 |
21 22 25 27 33 34 35 |
Temmelig tilfredsstillende (mindre fejl) uren fremmed smag syrlig kogt smag, sveden smag fodersmag grov, bitter oversaltet |
|
2 |
1 3 4 5 6 10 11 12 |
Utilfredsstillende (åbenlyse fejl) synlige vanddråber stribet skjoldet, marmoreret plettet olieudskillelse fremmede bestanddele muggen uopløst salt |
2 |
14 15 16 17 18 |
Utilfredsstillende (åbenlyse fejl) skør, smuldrende dejagtig, fedtet klæbrig hård blød |
2 |
21 22 23 25 32 33 34 35 36 38 |
Utilfredsstillende (åbenlyse fejl) uren fremmed smag gammel smag syrlig oxideret smag, metallisk smag fodersmag grov, bitter oversaltet muggen, fad, rådden kemikaliesmag |
|
1 |
1 3 4 5 6 7 9 10 11 12 |
Særdeles utilfredsstillende (udtalte fejl) synlige vanddråber stribet skjoldet, marmoreret plettet olieudskillelse for stærk farve kornet fremmede bestanddele muggen uopløst salt |
1 |
14 15 16 17 18 |
Særdeles utilfredsstillende (udtalte fejl) skør, smuldrende dejagtig, fedtet klæbrig hård blød |
1 |
22 24 25 26 28 29 30 31 32 34 35 36 37 38 |
Særdeles utilfredsstillende (udtalte fejl) fremmed smag osteagtig, syrlig ostesmag syrlig gærsmag muggen smag harsk olieagtig, trannet talgagtig oxideret smag, metallisk smag grov, bitter oversaltet muggen, fad, rådden maltagtig kemikaliesmag |
Tabel 2
Nomenklatur over fejl ved smør
|
||
|
||
|
||
|
||
|
||
|
||
|
||
|
||
|
||
|
||
|
||
|
||
|
||
|
||
|
||
|
||
|
||
|
||
|
||
|
||
|
||
|
||
|
||
|
||
|
||
|
||
|
||
|
||
|
||
|
||
|
||
|
||
|
||
|
||
|
||
|
||
|
||
|
||
|
||
|
||
|
(1) Tabel 2.
(2) De fejl, der er nævnt under »tilfredsstillende«, må kun bestå i meget små afvigelser fra idealtypen.
(3) Denne benævnelse bør bruges så sjældent som muligt og kun, når fejlen ikke kan beskrives mere præcist.
BILAG V
(Artikel 5)
BESTEMMELSE AF INDHOLDET AF TRIGLYCERID AF ENANTIKSYRE I SMØR, SMØROLIE OG FLØDE VED GASKROMATOGRAFISK ANALYSE AF TRIGLYCERIDER
1. FORMÅL
Denne metode anvendes til bestemmelse af indholdet af triglycerid af enantiksyre i smør, smørolie og fløde.
2. TERMER OG DEFINITIONER
Indhold af enantiksyre: indholdet af triglycerid af enantiksyre bestemt ved den i denne metode angivne procedure.
Bemærkning: Indholdet af enantiksyre udtrykkes i kg pr. ton produkt for smørolie og smør, og det udtrykkes i kg pr. ton mælkefedt for fløde.
3. PRINCIP
Mælkefedt ekstraheres fra de forskellige produkter ifølge ISO 14156 | IDF 172:2001. Den kvantitative bestemmelse af indholdet af triglycerid af enantiksyre i det ekstraherede fedt foretages ved hjælp af gaskromatografi med kapillarkolonne (GC). Resultatet for prøven vurderes ved reference til triglycerid af capronsyre som intern standard.
Bemærkning: Tributyrin er også en tilfredsstillende intern standard.
4. REAGENSER
Der må kun anvendes reagenser af anerkendt analysekvalitet.
4.1. n-Hexan
4.2. Standardtriglycerid af capronsyre, mindst 99 % rent.
4.3. Standardtriglycerid af enantiksyre, mindst 99 % rent.
4.4. Vandfri natriumsulfat (Na2SO4).
5. APPARATUR
Sædvanligt laboratorieudstyr, især følgende:
5.1. Analysevægt, nøjagtighed 1 mg
5.2. Målekolber, 10 ml og 20 ml
5.3. Centrifugeglas, 30 ml
5.4. Rotationsinddamper
5.5. Ovn, som kan holde en temperatur på 50 °C ±5 °C
5.6. Filtrerpapir, medium porøsitet, diameter ca. 15 cm
Gaskromatografiudstyr
5.7.1. Gaskromatograf udstyret med split/splitless-injektor eller direkte injektion på kolonnen og en flammeionisationsdetektor (FID)
GC-kolonne med en stationær fase, der med godt resultat er blevet anvendt til triglyceridseparation (100 % dimethylpolysiloxan eller 5 % phenyl-95 % methylpolysiloxan). Den stationære fase, kolonnehøjden (4-15 m), den indre diameter (0,22-0,50 mm) og filmtykkelse (0,12 μm eller derover) vælges under hensyntagen til laboratoriets erfaring og det anvendte injektionssystem. Under alle omstændigheder skal den valgte kolonne frembringe såvel en komplet separation mellem opløsningsmidlets top og triglycerid af capronsyre som en resolution til basislinjen mellem toppene for triglycerid af capronsyre og enantiksyre. Nedenfor er anført eksempler på betingelserne.
5.7.2.1. Eksempel på betingelser ved anvendelse af en split-injektor
|
— |
Bæregas: helium |
|
— |
Tryk ved kolonnetop: 100 kPa |
|
— |
Kolonne: 12 m lang kolonne i kvartsglas, 0,5 mm indre diameter, 0,1 μm filmtykkelse |
|
— |
Stationær fase: 100 % dimethylpolysiloxan eller 5 % phenyl-95 % dimethylpolysiloxan (f.eks. HT5) |
|
— |
Kolonnetemperatur: initialtemperatur på 130 °C, opretholdt i 1 min., øget med 20 °C/min. til 260 °C og dernæst øget med 30 °C/min. til 360 °C; opretholdes i 10 min. ved 360 °C |
|
— |
Detektortemperatur: 370 °C |
|
— |
Injektortemperatur: 350 °C |
|
— |
Splitforhold: 1:30 |
|
— |
Indsprøjtet mængde af prøven: 1 μl. |
5.7.2.2. Eksempel på betingelser ved anvendelse af injektor på kolonnen
|
— |
Bæregas: hydrogen (konstant flow-system) |
|
— |
Tryk ved kolonnetop: 89 kPa |
|
— |
Kolonne: 4 m lang kolonne i kvartsglas, 0,32 mm indre diameter, 0,25 μm filmtykkelse |
|
— |
Stationær fase: 5 % phenyl, 95 % dimethylpolysiloxan |
|
— |
Kolonnetemperatur: initialtemperatur på 60 °C, opretholdt i 2 min., øget med 35 °C/min. til 340 °C, opretholdt ved denne temperatur i 5 min. |
|
— |
Detektortemperatur: 350 °C |
|
— |
Indsprøjtet mængde af prøven: 1 μl. |
5.8. Injektionssprøjte, 5 μl.
6. PRØVEUDTAGNING
Det er vigtigt, at laboratoriet modtager en prøve, der reelt er repræsentativ, og som ikke er skadet eller forandret under transport eller oplagring.
Prøveudtagning indgår ikke i den metode, der er angivet i dette bilag. En anbefalet metode til prøveudtagning findes i IDF: standard 50C: 1995 eller ISO 707-1997- Mælk og mælkeprodukter. Vejledning om prøveudtagning.
7. FREMGANGSMÅDE
7.1. Forberedelse af klargjort prøve og analyseprøve
Fremgangsmåden følger ISO 14156 | IDF 172:2001.
7.1.1. Smørolie og smør
7.1.1.1. Der smeltes 50-100 g af den klargjorte prøve i ovnen (5.5).
7.1.1.2. 0,5-1,0 g vandfri natriumsulfat (5.4) placeres i et foldet filtrerpapir.
7.1.1.3. Fedtet filtreres gennem filtrerpapiret, der indeholder vandfri natriumsulfat, og filtratet opsamles i et bægerglas, der hele tiden findes i ovnen (5.5). Når det smeltede smør dekanteres over på filtrerpapiret, skal der udvises omhu, for at der ikke følger valle med.
7.1.2. Fløde
7.1.2.1. Den klargjorte prøve opvarmes til en temperatur på 20 °C ±2 °C.
7.1.2.2. Prøven blandes eller røres grundigt.
7.1.2.3. En passende mængde af den klargjorte prøve fortyndes, så der fås 100 ml analyseprøve med en massefraktion af fedt på ca. 4 %.
7.1.2.4. For at ekstrahere fedtet fra fløden følges samme fremgangsmåde som for rå mælk og homogeniseret mælk (se ISO 14156 | IDF 172:2001, § 8.3).
7.1.2.5. 1 g af det ekstraherede fedt afvejes med 1 mg nøjagtighed i en 10 ml målekolbe (5.2). Derefter tilsættes 1 ml opløsning 7.2.2. Der fyldes n-hexan (4.1) i op til 10 ml, hvorefter der homogeniseres.
7.1.2.6. 1 ml af opløsning 7.1.1.2 fyldes i en 10 ml målekolbe (5.2), og der fortyndes op til 10 ml med n-hexan (4.1).
7.2. Forberedelse af kalibreringsstandarder
7.2.1. 100 mg triglycerid af enantiksyre (4.3) opløses i 10 ml n-hexan (4.1).
7.2.2. 100 mg triglycerid af capronsyre (4.2) opløses i 10 ml n-hexan (4.1).
7.2.3. 1 ml af opløsning 7.2.2 fyldes i en 10 ml målekolbe (5.2). Der fyldes n-hexan (4.1) i op til 10 ml.
7.2.4. 1 ml af opløsning 7.2.1 og 1 ml af opløsning 7.2.2 fyldes i en 10 ml målekolbe (5.2). Der fyldes n-hexan (4.1) i op til 10 ml.
7.2.5. 1 ml af opløsning 7.2.4 fyldes i en 10 ml målekolbe (5.2), og der fyldes n-hexan (4.1) i op til 10 ml.
7.3. Kromatografisk bestemmelse
7.3.1. Der indsprøjtes 1 μl standardopløsning 7.2.5 to gange.
7.3.2. Der indsprøjtes 1 μl af hver prøveopløsning.
Bemærkning: Hvis der benyttes direkte injektion på kolonnen, bør både standardopløsningen og prøveopløsningerne fortyndes mere.
7.3.3. Fremgangsmåden 7.3.1 gentages for hver tredje prøve, for at prøverne ligger tæt på dobbeltanalyser af standardopløsningen. Resultaterne baseres på gennemsnittet af responsfaktorerne fra standardkromatogrammerne.
8. BEREGNING AF RESULTATER
For hvert kromatogram integreres toparealet for triglyceriderne af enantiksyre og capronsyre.
Instrukserne følges for hver sekvens, dvs. for et sæt af prøver med standardopløsninger før og efter (bracketing), idet den standardopløsning, der indsprøjtes to gange umiddelbart før, kaldes STD1, og den standardopløsning, der indsprøjtes to gange umiddelbart efter, kaldes STD2.
8.1. Kalibrering
8.1.1. Der foretages en beregning af responsfaktoren for hver dobbeltanalyse af STD1, Rf1(a) og Rf1(b).
Rf1 (a) eller (b) = (topareal for triglycerid af capronsyre/topareal for triglycerid af enantiksyre) × 100
Den gennemsnitlige responsfaktor Rf1 beregnes således:
Rf1 = (Rf1(a) + Rf1(b)) / 2
8.1.2. Den gennemsnitlige responsfaktor STD2, Rf2 beregnes på lignende måde.
8.1.3. Den gennemsnitlige responsfaktor Rf beregnes således:
Rf = (Rf1 + Rf2) /2
8.2. Klargjorte prøver
For hvert prøvekromatogram, der ligger mellem STD1 og STD2, beregnes enantiksyreindholdet, C (kg/ton):
C = (topareal for triglycerid af enantiksyre × Rf × 100)/(topareal for triglycerid af capronsyre × Wt × 1 000)
hvor:
|
— |
Wt = fedtstofvægt (g) |
|
— |
100 = prøvens fortyndingsvolumen |
|
— |
1 000 = omregningsfaktor (for μg/g til kg/ton). |
For smørprøver tages smørrets fedtindhold i betragtning, og der beregnes et korrigeret indhold, Csmør (kg/ton smør)
Csmør = Cfedt × F
hvor F er smørrets fedtindhold.
9. PRÆCISION
Nærmere oplysninger om en sammenlignende laboratorieprøvning vedrørende smør i overensstemmelse med ISO 5725-1 og ISO 5725-2 om præcisionsmetode findes i punkt 12.
Værdierne for repeterbarheds- og reproducerbarhedsgrænser udtrykkes for en 95 % sandsynlighedsgrænse og må ikke anvendes til andre koncentrationsintervaller eller matrixer end de angivne.
9.1. Repeterbarhed
De absolutte forskelle mellem to enkeltresultater, der er opnået med den samme metode på identisk prøvemateriale, på det samme laboratorium, udført af den samme person med det samme udstyr og med et kort tidsinterval, bliver i højst 5 % af tilfældene større end 0,35 kg/t.
9.2. Reproducerbarhed
De absolutte forskelle mellem to enkeltresultater, der er opnået med den samme metode på identisk prøvemateriale på forskellige laboratorier udført af forskellige personer med forskelligt udstyr, bliver i højst 5 % af tilfældene større end 0,66 kg/t.
10. TOLERANCERGRÆNSER: NEDRE GRÆNSER (VED UTILSTRÆKKELIGE MÆNGDER)
10.1. Der udtages tre prøver af produktet med røbestof for at kontrollere, om produktet er tilsat røbestof på korrekt måde.
10.2. Smør og koncentreret smør
10.2.1. Der iblandes 11 kg mindst 95 % ren triglycerid af enantiksyre pr. t smør, dvs. 10,45 kg/t.
10.2.2. Resultaterne af analysen af de tre prøver af produktet anvendes til at kontrollere mængden og homogeniteten af det iblandede røbestof, og det laveste af disse resultater sammenholdes med følgende grænseværdier:
|
— |
9,51 kg/t (95 % af den mindste iblanding for 95 % ren triglycerid af enantiksyre, enkeltbestemmelse) |
|
— |
6,89 kg/t (70 % af den mindste iblanding for 95 % ren triglycerid af enantiksyre, enkeltbestemmelse) |
|
— |
Koncentrationen af røbestoffet i den prøve, der giver det laveste resultat, benyttes sammen med interpolering mellem 9,51 kg/t og 6,89 kg/t. |
10.3. Fløde
10.3.1. Der iblandes 10 kg mindst 95 % ren triglycerid af enantiksyre pr. t mælkefedt, dvs. 9,50 kg/t mælkefedt med røbestof.
10.3.2. Resultaterne af analysen af de tre prøver af produktet anvendes til at kontrollere mængden og homogeniteten af det iblandede røbestof, og det laveste af disse resultater sammenholdes med følgende grænseværdier:
|
— |
8,60 kg/t (95 % af den mindste iblanding for 95 % ren triglycerid af enantiksyre, enkeltbestemmelse) |
|
— |
6,23 kg/t (70 % af den mindste iblanding for 95 % ren triglycerid af enantiksyre, enkeltbestemmelse) |
|
— |
Koncentrationen af røbestoffet i den prøve, der giver det laveste resultat, benyttes sammen med interpolering mellem 8,60 kg/t og 6,23 kg/t. |
11. TOLERANCEGRÆNSER: ØVRE GRÆNSER (VED EN MÆNGDE, DER ER MERE END 20 % FOR STOR)
11.1. Der udtages tre prøver af produktet med røbestof for at kontrollere, om produktet er tilsat røbestof på korrekt måde.
11.2. Smør og koncentreret smør
11.2.1. Resultaterne af analysen af de tre prøver af produktet anvendes til at kontrollere mængden og homogeniteten af det iblandede røbestof, og gennemsnittet af disse resultater sammenholdes med følgende grænseværdier:
|
— |
Den øvre grænse er 12,96 kg/t. |
11.3. Fløde
11.3.1. Resultaterne af analysen af de tre prøver af produktet anvendes til at kontrollere mængden og homogeniteten af det iblandede røbestof, og gennemsnittet af disse resultater sammenholdes med følgende grænseværdier:
|
— |
Den øvre grænse er 11,82 kg/t. |
12. SUPPLERENDE OPLYSNINGER: STATISTISK ANALYSE AF RESULTATER AF BESTEMMELSEN AF TRIENANTOAT I SMØRFEDT VED TRIGLYCERIDANALYSE
Der er gennemført fire fællesafprøvninger for at bestemme trienantoatindholdet i smør med røbestof.
Ni laboratorier deltog i den første ringtest, og der forelå ingen specifikationer om, hvilke analysemetoder der skulle anvendes.
Ti laboratorier deltog i den anden ringtest, og der blev anvendt fire forskellige metoder:
|
— |
kvantitativ bestemmelse af methylheptanoat ved anvendelse af n-nonan eller methylnonanoat som intern standard |
|
— |
kvantitativ bestemmelse af trienantoat ved anvendelse af tricaproat som intern standard |
|
— |
kvantitativ bestemmelse af methylheptanoat ved anvendelse af en kalibreringsprøve/-blanding |
|
— |
kvantitativ bestemmelse af methylheptanoat ved anvendelse af en kalibreringsblanding. |
Hvis methylestere af fedtsyrer (FAME) blev analyseret, blev der desuden anvendt to forskellige methyleringsprocedurer (De Francesco og Christopherson & Glass).
På grundlag af resultaterne blev der udvalgt to metoder til den tredje ringtest:
|
— |
kvantitativ bestemmelse af methylheptanoat ved anvendelse af n-nonan eller methylnonanoat som intern standard |
|
— |
kvantitativ bestemmelse af trienantoat ved anvendelse af tricaproat som intern standard. |
Resultaterne fra 7 laboratorier viste, at FAME-metoden havde en større variabilitet, og det blev derfor besluttet kun at bestemme trienantoat som triglycerid ved anvendelse af proceduren med kvantitativ bestemmelse af trienantoat ved anvendelse af tricaproat som intern standard. Endvidere skal triglyceridanalysen gennemføres med kapillarkolonne.
I den fjerde ringtest blev der rundsendt fire prøver (A, B, C, D), og der blev fremlagt resultater fra ni laboratorier (tabel 1-2).
To laboratorier (DE og EU) analyserede prøverne ved anvendelse af FAME-metoden.
På grund af det begrænsede antal laboratorier er der foretaget en statistisk beregning både af det komplette datasæt (tabel 1-2), inkl. FAME-resultaterne, og af dataene fra triglyceridanalysen.
Outliertest:
|
— |
Prøve A: Dixon, Cochran og Grubbs-test på niveau 1 og 5 % viste én afvigende værdi (outlier) for et laboratorium. |
|
— |
Prøve B: Grubbs-test på niveau 5 % viste én afvigende værdi (outlier) for et laboratorium. |
|
— |
Prøve C: Dixon og Grubbs-test på niveau 1 og 5 % viste én afvigende værdi (outlier) for et laboratorium. |
|
— |
Prøve D: Dixon og Grubbs-test på niveau 1 og 5 % viste én afvigende værdi (outlier) for et laboratorium. |
Den afvigende værdi er blevet udelukket fra beregningen.
Det bør bemærkes, at resultaterne fremkommet ved FAME-metoden aldrig blev betragtet som afvigende ved de anvendte test.
Præcisionsparametre
Tabel 1 og 2 viser resultaterne fra alle laboratorierne og præcisionsparametrene, der er beregnet ud fra et acceptabelt antal (8) laboratorier, men som desværre ikke hidrører fra den samme analysemetode.
Tabel 3 og 4 viser kun de resultater, der hidrører fra triglyceridmetoden og de tilsvarende præcisionsparametre. En accept af disse parametre forudsætter, at det lave antal laboratorier (6) accepteres.
Figur 3 og 4 viser tendensen for Sr og SR beregnet for de 4 prøver ud fra de to nævnte datasæt.
Tabel 5 viser værdierne for Sr og SR sammen med de tilsvarende puljede værdier og de samlede parametre for r og R.
Endelig er den kritiske forskel for en 95 % sandsynlighedsgrænse blevet beregnet.
Tabel 1
Statistiske resultater af triglycerid- + FAME*-metoderne
|
Prøve A |
|
R1 |
R2 |
GNSN |
Antal laboratorier efter eliminering af laboratorier med afvigende resultater |
8 |
|
RENNES |
FR1 |
11,0 |
11,1 |
11,1 |
Antal afvigende resultater |
1 |
|
RIKILT |
NL |
11,2 |
11,2 |
11,2 |
Afvigende resultater |
DК |
|
ZPLA |
DE* |
11,6 |
11,8 |
11,7 |
Gennemsnitsværdi |
11,3 |
|
ADAS |
GB |
11,4 |
11,2 |
11,3 |
Korrekt værdi |
11,0 |
|
CNEVA |
FR2 |
11,4 |
11,4 |
11,4 |
Standardafvigelse for repeterbarhed (Sr) |
0,09 |
|
LODI |
IT |
11,1 |
11,3 |
11,2 |
Relativ standardafvigelse for repeterbarhed (RSDr%) |
0,80 |
|
ËÈLA |
FI |
11,3 |
11,2 |
11,3 |
Repeterbarhed r (95 %) |
0,26 |
|
ISPRA |
UE* |
11,0 |
11,0 |
11,0 |
Relativ repeterbarhed r % |
2,24 |
|
D.V.F.A. |
DK |
13,3 |
11,8 |
12,6 |
Standardafvigelse for reproducerbarhed (SR) |
0,23 |
|
|
|
|
|
|
Relativ standardafvigelse for reproducerbarhed (RSDR%) |
2,04 |
|
|
|
|
|
|
Reproducerbarhed R (95 %) |
0,84 |
|
|
|
|
|
|
Relativ reproducerbarhed R % |
5,71 |
|
Prøve B |
|
R1 |
R2 |
GNSN |
Antal laboratorier efter eliminering af laboratorier med afvigende resultater |
8 |
|
RENNES |
FR1 |
12,7 |
12,8 |
12,8 |
Antal afvigende resultater |
1 |
|
RIKILT |
NL |
13,5 |
13,3 |
13,4 |
Afvigende resultater |
DK |
|
ZPLA |
DE* |
14,0 |
13,8 |
13,9 |
Gennemsnitsværdi |
13,4 |
|
ADAS |
GB |
13,4 |
13,5 |
13,5 |
Korrekt værdi |
13,5 |
|
CNEVA |
FR2 |
13,3 |
13,4 |
13,4 |
Standardafvigelse for repeterbarhed (Sr) |
0,14 |
|
LODI |
IT |
13,9 |
13,5 |
13,7 |
Relativ standardafvigelse for repeterbarhed (RSDr%) |
1,04 |
|
EELA |
FI |
13,4 |
13,2 |
13,3 |
Repeterbarhed r (95 %) |
0,40 |
|
ISPRA |
UE* |
13,2 |
13,3 |
13,3 |
Relativ repeterbarhed r % |
2,91 |
|
D.V.F.A. |
DK |
14,1 |
14,8 |
14,5 |
Standardafvigelse for reproducerbarhed (SR) |
0,35 |
|
|
|
|
|
|
Relativ standardafvigelse for reproducerbarhed (RSDR%) |
2,61 |
|
|
|
|
|
|
Reproducerbarhed R (95 %) |
0,99 |
|
|
|
|
|
|
Relativ reproducerbarhed R % |
7,31 |
Tabel 2
Statistiske resultater af triglycerid- + FAME*-metoderne
|
Prøve C |
|
R1 |
R2 |
GNSN |
Antal laboratorier efter eliminering af laboratorier med afvigende resultater |
8 |
|
RENNES |
FR1 |
8,9 |
9,2 |
9,1 |
Antal afvigende resultater |
1 |
|
RIKILT |
NL |
9,2 |
9,3 |
9,3 |
Afvigende resultater |
DK |
|
ZPLA |
DE* |
9,2 |
9,4 |
9,3 |
Gennemsnitsværdi |
9,3 |
|
ADAS |
GB |
9,5 |
9,3 |
9,4 |
Korrekt værdi |
9,3 |
|
CNEVA |
FR2 |
9,4 |
9,4 |
9,4 |
Standardafvigelse for repeterbarhed (Sr) |
0,14 |
|
LODI |
IT |
9,2 |
9,5 |
9,4 |
Relativ standardafvigelse for repeterbarhed (RSDr%) |
1,50 |
|
EELA |
FI |
9,4 |
9,6 |
9,5 |
Repeterbarhed r (95 %) |
0,40 |
|
ISPRA |
UE* |
9,4 |
9,3 |
9,4 |
Relativ repeterbarhed r % |
4,20 |
|
D.V.F.A. |
DK |
10,7 |
10,9 |
10,8 |
Standardafvigelse for reproducerbarhed (SR) |
0,17 |
|
|
|
|
|
|
Relativ standardafvigelse for reproducerbarhed (RSDR%) |
1,82 |
|
|
|
|
|
|
Reproducerbarhed R (95 %) |
0,47 |
|
|
|
|
|
|
Relativ reproducerbarhed R % |
5,10 |
|
Prøve D |
|
R1 |
R2 |
GNSN |
Antal laboratorier efter eliminering af laboratorier med afvigende resultater |
8 |
|
RENNES |
R1 |
1,6 |
1,6 |
1,6 |
Antal afvigende resultater |
1 |
|
RIKILT |
NL |
2,1 |
2,1 |
2,1 |
Afvigende resultater |
DK |
|
ZPLA |
DE* |
2,3 |
2,3 |
2,3 |
Gennemsnitsværdi |
2,1 |
|
ADAS |
GB |
2,1 |
2,2 |
2,2 |
Korrekt værdi |
2,1 |
|
CNEVA |
FR2 |
2,1 |
2,1 |
2,1 |
Standardafvigelse for repeterbarhed (Sr) |
0,08 |
|
LODI |
IT |
2,2 |
1,9 |
2,1 |
Relativ standardafvigelse for repeterbarhed (RSDr%) |
3,81 |
|
EELA |
FI |
2,3 |
2,3 |
2,3 |
Repeterbarhed r (95 %) |
0,22 |
|
ISPRA |
UE* |
2,3 |
2,3 |
2,3 |
Relativ repeterbarhed r % |
10,67 |
|
D.V.F.A. |
DK |
3,4 |
2,9 |
3,2 |
Standardafvigelse for reproducerbarhed (SR) |
0,24 |
|
|
|
|
|
|
Relativ standardafvigelse for reproducerbarhed (RSDR%) |
11,43 |
|
|
|
|
|
|
Reproducerbarhed R (95 %) |
0,67 |
|
|
|
|
|
|
Relativ reproducerbarhed R % |
32,00 |
Tabel 3
Statistiske resultater af triglycerid- + FAME*-metoderne
|
Prøve A |
|
R1 |
R2 |
GNSN |
Antal laboratorier efter eliminering af laboratorier med afvigende resultater |
6 |
|
RENNES |
FR1 |
11,0 |
11,1 |
11,1 |
Antal afvigende resultater |
1 |
|
RIKILT |
NL |
11,2 |
11,2 |
11,2 |
Afvigende resultater |
DК |
|
ADAS |
GB |
11,4 |
11,2 |
11,3 |
Gennemsnitsværdi |
11,2 |
|
CNEVA |
FR2 |
11,4 |
11,4 |
11,4 |
Korrekt værdi |
11,0 |
|
LODI |
IT |
11,1 |
11,3 |
11,2 |
Standardafvigelse for repeterbarhed (Sr) |
0,09 |
|
ËÈLA |
FI |
11,3 |
11,2 |
11,3 |
Relativ standardafvigelse for repeterbarhed (RSDr%) |
0,80 |
|
D.V.F.A. |
DK |
13,3 |
11,8 |
12,6 |
Repeterbarhed r (95 %) |
0,25 |
|
|
|
|
|
|
Relativ repeterbarhed r % |
2,24 |
|
|
|
|
|
|
Standardafvigelse for reproducerbarhed (SR) |
0,13 |
|
|
|
|
|
|
Relativ standardafvigelse for reproducerbarhed (RSDR%) |
1,16 |
|
|
|
|
|
|
Reproducerbarhed R (95 %) |
0,36 |
|
|
|
|
|
|
Relativ reproducerbarhed R % |
3,25 |
|
Prøve B |
|
R1 |
R2 |
GNSN |
Antal laboratorier efter eliminering af laboratorier med afvigende resultater |
6 |
|
RENNES |
FR1 |
12,7 |
12,8 |
12,8 |
Antal afvigende resultater |
1 |
|
RIKILT |
NL |
13,5 |
13,3 |
13,4 |
Afvigende resultater |
DК |
|
ADAS |
GB |
13,4 |
13,5 |
13,5 |
Gennemsnitsværdi |
13,3 |
|
CNEVA |
FR2 |
13,3 |
13,4 |
13,4 |
Korrekt værdi |
13,5 |
|
LODI |
IT |
13,9 |
13,5 |
13,7 |
Standardafvigelse for repeterbarhed (Sr) |
0,15 |
|
EELA |
FI |
13,4 |
13,2 |
13,3 |
Relativ standardafvigelse for repeterbarhed (RSDr%) |
1,13 |
|
D.V.F.A. |
DK |
14,1 |
14,8 |
14,5 |
Repeterbarhed r (95 %) |
0,42 |
|
|
|
|
|
|
Relativ repeterbarhed r % |
3,16 |
|
|
|
|
|
|
Standardafvigelse for reproducerbarhed (SR) |
0,33 |
|
|
|
|
|
|
Relativ standardafvigelse for reproducerbarhed (RSDR%) |
2,48 |
|
|
|
|
|
|
Reproducerbarhed R (95 %) |
0,93 |
|
|
|
|
|
|
Relativ reproducerbarhed R % |
6,94 |
Tabel 4
Statistiske resultater af triglyceridmetoden
|
Prøve C |
|
R1 |
R2 |
GNSN |
Antal laboratorier efter eliminering af laboratorier med afvigende resultater |
6 |
|
RENNES |
FR1 |
8,9 |
9,2 |
9,1 |
Antal afvigende resultater |
1 |
|
RIKILT |
NL |
9,2 |
9,3 |
9,3 |
Afvigende resultater |
DК |
|
ADAS |
GB |
9,5 |
9,3 |
9,4 |
Gennemsnitsværdi |
9,3 |
|
CNEVA |
FR2 |
9,4 |
9,4 |
9,4 |
Korrekt værdi |
9,3 |
|
LODI |
IT |
9,2 |
9,5 |
9,4 |
Standardafvigelse for repeterbarhed (Sr) |
0,15 |
|
ËÈLA |
FI |
9,4 |
9,6 |
9,5 |
Relativ standardafvigelse for repeterbarhed (RSDr%) |
1,61 |
|
D.V.F.A. |
DK |
10,7 |
10,9 |
10,8 |
Repeterbarhed r (95 %) |
0,42 |
|
|
|
|
|
|
Relativ repeterbarhed r % |
4,51 |
|
|
|
|
|
|
Standardafvigelse for reproducerbarhed (SR) |
0,19 |
|
|
|
|
|
|
Relativ standardafvigelse for reproducerbarhed (RSDR%) |
2,04 |
|
|
|
|
|
|
Reproducerbarhed R (95 %) |
0,53 |
|
|
|
|
|
|
Relativ reproducerbarhed R % |
5,71 |
|
Prøve D |
|
R1 |
R2 |
GNSN |
Antal laboratorier efter eliminering af laboratorier med afvigende resultater |
6 |
|
RENNES |
FR1 |
1,6 |
1,6 |
1,6 |
Antal afvigende resultater |
1 |
|
RIKILT |
NL |
2,1 |
2,1 |
2,1 |
Afvigende resultater |
DK |
|
|
|
|
|
|
Gennemsnitsværdi |
2,1 |
|
ADAS |
GB |
2,1 |
2,2 |
2,2 |
Korrekt værdi |
2,1 |
|
CNEVA |
FR2 |
2,1 |
2,1 |
2,1 |
Standardafvigelse for repeterbarhed (Sr) |
0,09 |
|
LODI |
IT |
2,2 |
1,9 |
2,1 |
Relativ standardafvigelse for repeterbarhed (RSDr%) |
4,29 |
|
EELA |
FI |
2,3 |
2,3 |
2,3 |
Repeterbarhed r (95 %) |
0,26 |
|
D.V.F.A. |
DK |
3,4 |
2,9 |
3,2 |
Relativ repeterbarhed r % |
12,01 |
|
|
|
|
|
|
Standardafvigelse for reproducerbarhed (SR) |
0,25 |
|
|
|
|
|
|
Relativ standardafvigelse for reproducerbarhed (RSDR%) |
11,90 |
|
|
|
|
|
|
Reproducerbarhed R (95 %) |
0,69 |
|
|
|
|
|
|
Relativ reproducerbarhed R % |
33,32 |
Tabel 5
Repeterbarhed og reproducerbarhed (med FAME)
|
|
Antal laboratorier |
Afvigende resultat |
Repeterbarhed Sr (95 %) |
Reproducerbarhed SR (95 %) |
||
|
Prøve A |
8 |
1 |
0,09 |
0,23 |
||
|
Prøve B |
8 |
1 |
0,14 |
0,35 |
||
|
Prøve C |
8 |
1 |
0,14 |
0,17 |
||
|
Prøve D |
8 |
1 |
0,08 |
0,24 |
||
|
Puljet værdi |
|
|
0,116 |
0,256 |
||
|
|
|
|
R |
R |
||
|
Puljet værdi * 2,8 |
|
|
0,324 |
0,716 |
||
|
CrD95 =0,40 Minimumsrenhed angivet for trienantoat = 95 % Minimumsgrænse angivet for trienantoat i smørfedt = 11 kg/t Med et konventionelt konfidensinterval på 95 % må gennemsnittet af de to resultater ikke være lavere end:
|
||||||
Repeterbarhed og reproducerbarhed (uden FAME)
|
|
Antal laboratorier |
Afvigende resultat |
Repeterbarhed Sr (95 %) |
Reproducerbarhed SR (95 %) |
||
|
Prøve A |
6 |
1 |
0,09 |
0,13 |
||
|
Prøve B |
6 |
1 |
0,15 |
0,33 |
||
|
Prøve C |
6 |
1 |
0,15 |
0,19 |
||
|
Prøve D |
6 |
1 |
0,09 |
0,25 |
||
|
Puljet værdi |
|
|
0,124 |
0,237 |
||
|
|
|
|
r |
R |
||
|
Puljet værdi * 2,8 |
|
|
0,347 |
0,663 |
||
|
CrD95 = 0,36 Minimumsrenhed angivet for trienantoat = 95 % Minimumsgrænse angivet for trienantoat i smørfedt = 11 kg/t Med et konventionelt konfidensinterval på 95 % må gennemsnittet af de to resultater ikke være lavere end:
|
||||||
Figur 1 (1)
Forsøgsresultater: Prøve A

Forsøgsresultater: Prøve B
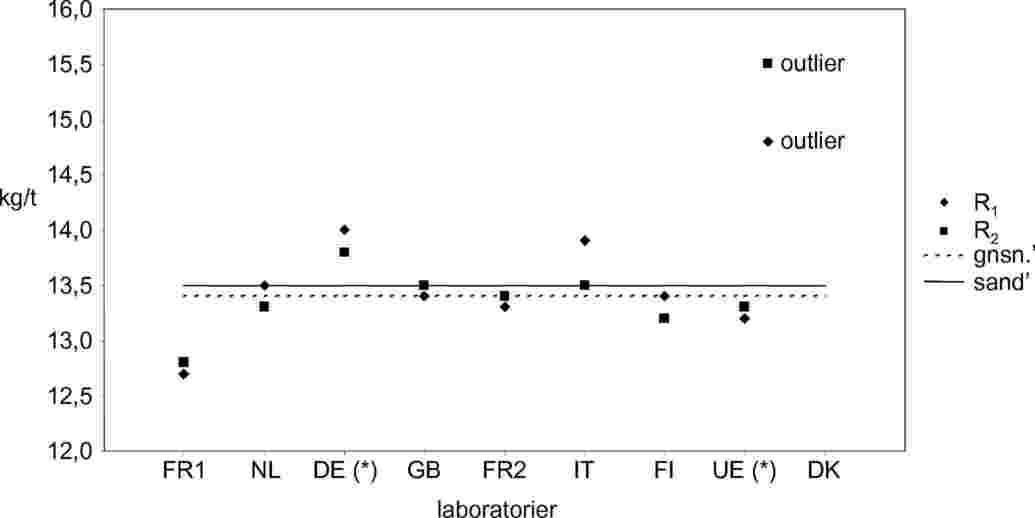
Forsøgsresultater: Prøve C
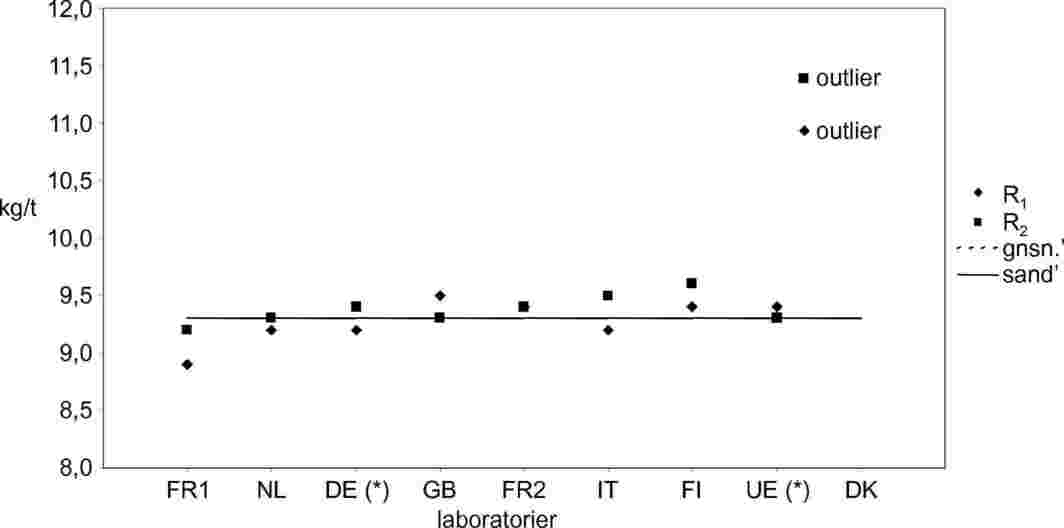
Forsøgsresultater: Prøve D

Figur 2
Standardafvigelse for repeterbarhed og reproducerbarhed på forskellige niveauer (TG+FAME)

Figur 3
Standardafvigelse for repeterbarhed og reproducerbarhed på forskellige niveauer (TG)

Figur 4
Eksempel ved direkte injektion på kolonnen

(1) = FAME-metode
BILAG VI
(Artikel 5)
BESTEMMELSE AF INDHOLDET AF VANILLIN I KONCENTRERET SMØR, SMØR OG FLØDE VED HØJTRYKSVÆSKEKROMATOGRAFI
1. FORMÅL OG ANVENDELSESOMRÅDE
Kvantitativ bestemmelse af vanillin i koncentreret smør, smør og fløde.
2. PRINCIP
Ekstraktion af en kendt prøvemængde ved hjælp af en blanding af isopropanol/ethanol/acetonitril (1:1:2). Udskillelse af størsteparten af fedtstoffet ved afkøling (temperatur mellem –15 og –20 °C) efterfulgt af centrifugering.
Efter fortynding med vand bestemmes vanillinindholdet ved højtryksvæskekromatografi (HPLC).
3. APPARATUR
Sædvanligt laboratorieapparatur, herunder især følgende:
3.1. Fryser med en arbejdstemperatur mellem –15 °C og –20 °C
3.2. Injektionssprøjter (engangssprøjter) 2 ml
3.3. Membranfiltre (porestørrelse: 0,45 μm), modstandsdygtige over for en opløsning med 5 % ekstraktionsmiddel (4.4)
3.4. Væskekromatografisk system bestående af pumpe (flow 1,0 ml/min.), injektionsenhed (20 μl injektion, automatisk eller manuel), UV-detektor (306 nm, fuldt udslag 0,01 absorptionsenhed), skriver eller integrator samt kolonnetermostat indstillet til 25 °C
3.5. Analysekolonne (250 mm x 4,6 mm indvendig diameter) pakket med LiChrospher RP 18 (Merck, 5 μm) eller tilsvarende
3.6. Forkolonne (ca. 20 mm x 3 mm indvendig diameter) pakket tørt med LiChrospher RP 18 (5-10 μm) eller tilsvarende
3.7. Centrifuge, som arbejder ved 2 000 omdrejninger pr. minut.
4. REAGENSER
Alle reagenser skal være af anerkendt analysekvalitet.
4.1. Isopropanol
4.2. Ethanol, 96 % (v/v)
4.3. Acetonitril
4.4. Ekstraktionsopløsning
Isopropanol (4.1), ethanol (4.2) og acetonitril (4.3) blandes i forholdet 1:1:2 (v/v)
Vanillin (4-hydroxy-3-methoxybenzaldehyd) ≥ 98 %
4.5.1. Vanillinstamopløsning (= 500 μg/ml)
Ca. 50 mg (»CM« mg) vanillin (4.5) afvejes med en nøjagtighed på 0,1 mg i en 100 ml målekolbe; der tilsættes 25 ml ekstraktionsmiddel (4.4) og fyldes op med vand.
4.5.2. Vanillinstandardopløsning (= 10 μg/ml)
Der afpipetteres 5 ml vanillinstamopløsning (4.5.1) i en 250 ml målekolbe, hvorefter der fyldes op med vand.
4.5.3. Methanol, HPLC-kvalitet
4.5.4. Iseddike
4.5.5. Vand, HPLC-kvalitet
4.5.6. Mobil fase til HPLC
300 ml methanol (4.5.3), ca. 500 ml vand (4.5.5) og 20,0 ml iseddike (4.5.4) blandes i en 1 000 ml målekolbe, og der fyldes op med vand (4.5.5). Blandingen filtreres gennem et filter (0,45 μm) (3.3).
5. FREMGANGSMÅDE
5.1. Forberedelse af prøven
5.1.1. Smør
Prøven opvarmes, indtil den begynder at smelte. Lokal opvarmning til ca. 30 °C skal undgås. Smørret må under ingen omstændigheder skille i to faser. Når prøven er blevet tilstrækkeligt blød, homogeniseres den ved rystning. Der omrøres i 15 sek., inden der udtages en prøve. Der afvejes med en nøjagtighed på 1 mg ca. 5 g (»SM« g) smør i en 100 ml målekolbe.
5.1.2. Koncentreret smør
Umiddelbart inden prøveudtagningen anbringes beholderen med koncentreret smør i en ovn ved 40-50 °C, indtil smørret er fuldstændig smeltet. Prøven blandes ved forsigtig omdrejning eller omrøring. For voldsom bevægelse med dannelse af luftbobler skal undgås. Der afvejes med en nøjagtighed på 1 mg ca. 4g (»SM« g) koncentreret smør i en 100 ml målekolbe.
5.1.3. Fløde
Prøven opvarmes i vandbad eller i inkubator ved en temperatur på 35-40 °C. Fedtstoffet fordeles ensartet ved forsigtig omdrejning og om nødvendigt ved omrøring. Prøven afkøles hurtigt til 20 ± 2 °C. Prøven skal fremtræde ensartet, ellers gentages proceduren. Der afvejes med en nøjagtighed på 1 mg ca. 10g (»SM« g) fløde i en 100 ml målekolbe.
5.2. Fremstilling af prøveopløsning
Der tilsættes ca. 75 ml ekstraktionsopløsning (4.4) til analyseprøven (5.1.1, 5.1.2 eller 5.1.3), og der omrøres eller omrystes kraftigt i ca. 15 min. og fyldes op med ekstraktionsopløsning (4.4). Ca. 10 ml af dette ekstrakt overføres til et reagensglas med prop. Glasset stilles i fryseren (3.1) i ca. 30 min. Det afkølede ekstrakt centrifugeres i 5 minutter ved ca. 2 000 omdrejninger pr. minut, og der dekanteres straks. Den dekanterede opløsning henstår, til den har nået stuetemperatur. Der afpipetteres 5 ml heraf i en 100 ml målekolbe, og der fyldes op med vand. En delprøve heraf filtreres på membranfilter (3.3) ved anvendelse af en injektionssprøjte (3.2). Filtratet er klart til HPLC-analyse.
5.3. Kalibrering
Der afpipetteres 5 ml vanillinstandardopløsning (4.5.2) i en 100 ml målekolbe. Der tilsættes 5 ml ekstraktionsopløsning (4.4) og fyldes op med vand til mærket. Denne opløsning indeholder 0,5 μg vanillin pr. ml.
5.4. Bestemmelse ved HPLC
Kromatografisystemet stabiliseres i ca. 30 min. Standardopløsningen (5.3) indsprøjtes. Dette gentages, indtil forskellen i topareal eller tophøjde mellem to på hinanden følgende indsprøjtninger er mindre end 2 %. Under de beskrevne forhold har vanillin en retentionstid på ca. 9 min. Der foretages en dobbeltanalyse af standardopløsningen (5.3) med indsprøjtning af 20 μl. Der indsprøjtes 20 μl prøveopløsning (5.2). Arealet eller højden af den fremkomne vanillintop bestemmes. Efter 10 indsprøjtninger af prøveopløsninger (5.2) gentages dobbeltanalysen af standardopløsningen (5.3).
6. BEREGNING AF RESULTATER
Det gennemsnitlige topareal (eller tophøjde) (»AC«) for de vanillintoppe, der er fremkommet ved de to dobbeltanalyser før og efter hver batch af prøveopløsninger (4 toparealer eller tophøjder), beregnes.
Responsfaktoren (R) beregnes således:

hvor CM er massen af vanillin i mg (4.5.1).
Indholdet (i mg/kg) af vanillin (C) i analyseprøven beregnes ved følgende formel:

hvor:
|
AS |
= |
toparealet eller tophøjden af vanillintoppen fra analyseprøven |
|
SM |
= |
massen af analyseprøven i g (5.1.1, 5.1.2 eller 5.1.3). |
Bemærkning: Når fløden analyseres for vanillin, skal koncentrationen af røbestoffet udtrykkes som mg røbestof/kg mælkefedt. Dette gøres ved, at man multiplicerer C med 100/f, hvor f er flødens fedtindhold i procent (m/m).
|
20 |
= |
en faktor, hvor der tages hensyn til fortyndingerne af standard- og analyseprøven |
|
0,96 |
= |
korrektionsfaktoren for fedtindholdet ved første fortynding af analyseprøven |
Bemærkning: I stedet for toparealer kan der benyttes tophøjder (se punkt 8.3).
7. METODENS NØJAGTIGHED
7.1. Repeterbarhed (r)
Forskellen mellem resultaterne af to bestemmelser af det samme prøvemateriale, der foretages umiddelbart efter hinanden af den samme person med det samme udstyr, må ikke overstige 16 mg/kg.
7.2. Reproducerbarhed (R)
Forskellen mellem resultaterne af to bestemmelser af det samme prøvemateriale, der foretages i forskellige laboratorier med forskelligt udstyr, må ikke overstige 27 mg/kg.
8. TOLERANCEGRÆNSER
8.1. Der skal tages tre prøver fra produktet med røbestof for at kontrollere homogeniteten.
Røbestof fra vanille eller syntetisk vanillin
8.2.1. Iblandingen af 4-hydroxy-3-methoxybenzaldehyd er 250g pr. t koncentreret smør eller smør. For fløde med røbestof er iblandingen 250g pr. t mælkefedt.
8.2.2. Resultaterne af analysen af de tre prøver af produktet anvendes til at kontrollere mængden og homogeniteten af det iblandede røbestof, og det laveste af disse resultater sammenholdes med følgende grænseværdier:
|
— |
220,8 mg/kg (95 % af den mindste iblanding) |
|
— |
158,3 mg/kg (70 % af den mindste iblanding). |
Koncentrationen af røbestoffet i den prøve, der giver det laveste resultat, benyttes sammen med interpolering mellem 220,8 mg/kg og 158,3 mg/kg.
Røbestof kun fra vanilleplantens frugt eller komplette ekstrakter heraf
8.3.1. Iblandingen af 4-hydroxy-3-methoxybenzaldehyd er 100g pr. t koncentreret smør eller smør. For fløde med røbestof er iblandingen 100g pr. t mælkefedt.
8.3.2. Resultaterne af analysen af de tre prøver af produktet anvendes til at kontrollere mængden og homogeniteten af det iblandede røbestof, og det laveste af disse resultater sammenholdes med følgende grænseværdier:
|
— |
78,3 mg/kg (95 % af den mindste iblanding) |
|
— |
53,3 mg/kg (70 % af den mindste iblanding). |
Koncentrationen af røbestoffet i den prøve, der giver det laveste resultat, benyttes sammen med interpolering mellem 78,3 mg/kg og 53,3 mg/kg.
9. BEMÆRKNINGER
9.1. Genfindingen af vanillin, der er tilsat i en mængde på 250 mg pr. kg smørolie, varierer fra 97,0 % til 103,8 %. I gennemsnit er der fundet 99,9 % med en standardafvigelse på 2,7 %.
9.2. Standardopløsningen indeholder 5 % ekstraktionsopløsning for at kompensere for topforbredning, som følge af at prøveopløsningerne indeholder 5 % ekstraktionsopløsning. Derved kan der ved den kvantitative bestemmelse benyttes tophøjder.
9.3. Analysen bygger på en lineær kalibreringskurve, der går gennem nulpunktet.
9.4. Med passende fortyndinger af standardopløsningen (4.5.2) bør lineariteten kontrolleres, første gang analysen udføres, og derefter regelmæssigt og efter udskiftninger eller reparationer på HPLC-udstyret. Vanillin kan nedbrydes til vanillinsyre, divanillin eller andre bestanddele som følge af naturligt forekommende enzymer i upasteuriseret fløde eller produkter heraf.
BILAG VII
(Artikel 5)
BESTEMMELSE AF ETHYLESTER AF β-APO-8'-CAROTENSYRE I KONCENTRERET SMØR OG SMØR VED SPEKTROMETRI
1. FORMÅL OG ANVENDELSESOMRÅDE
Det er en metode til kvantitativ bestemmelse af ethylester af β-apo-8'-carotensyre (apocarotenester) i koncentreret smør og smør. Apocarotenester er summen af alle stoffer, der er til stede i et ekstrakt af prøver fremstillet på de i metoden beskrevne betingelser, og som absorberer lys ved 440 nm.
2. PRINCIP
Smørfedt opløses i petroleumsether (light petroleum), og absorbansen måles ved 440 nm. Indholdet af apocarotenester bestemmes ved reference til en ekstern standard.
3. APPARATUR
3.1. Målepipetter på 0,25, 0,50, 0,75 og 1,0 ml
3.2. Spektrofotometer, der er egnet til brug ved 440 nm (og 447-449 nm), og som er udstyret med kuvetter med en lysvej på 1 cm
3.3. Målekolber, 20 ml og 100 ml
3.4. Analysevægt med en følsomhed på 0,1 mg, der kan veje med en nøjagtighed på 1 mg, og som kan aflæses med en nøjagtighed på 0,1 mg
3.5. Ovn, 45 °C ± 1 °C
3.6. Hurtigfiltrerende askefri filtre.
4. REAGENSER
Alle reagenser skal være af anerkendt analysekvalitet.
Opslæmning af apokarotenester (ca. 20 %)
4.1.1. Opslæmningens indhold bestemmes som følger:
Opslæmningen opvarmes til mellem 45 °C og 50 °C og homogeniseres i den uåbnede originalbeholder. Ca. 400 mg afvejes nøjagtigt ned i en 100 ml målekolbe, opløses i 20 ml chloroform (4.4) og målekolben fyldes op til mærket med cyclohexan (4.5). 5 ml af denne opløsning fortyndes til 100 ml med cyclohexan (opløsning A). 5 ml af opløsning A fortyndes til 100 ml med cyclohexan. Absorbansen måles ved 447-449 nm (maksimum måles med cyclohexan som reference i kuvetter med 1 cm lysvej).
Indhold af apocarotenester P (%) = (Abs max × 40 000) / (Msusp × 2 550) eller efter formlen: (Absmax / 2 550) × (100 / 5) × (100 / 5) × (100 / Msusp)
|
Absmax |
= |
måleopløsningens absorbans ved maksimum |
|
Msusp |
= |
opslæmningens masse (g) |
|
2 550 |
= |
referenceværdi Abs (1 %, 1 cm) |
|
P |
= |
opslæmningens renhed (indhold) (%) |
Bemærkning: En opslæmning af apocarotenester er følsom for luft, varme og lys. Den kan opbevares i ca. 12 måneder i uåbnet originalbeholder (forseglet under kvælstof) på et køligt sted. Efter åbning skal indholdet anvendes inden for kort tid.
4.1.2. Standardopløsning af apocarotenester, ca. 0,2 mg/ml
0,100 g ± 1 mg opslæmning af apocarotenester (4.1.1) afvejes (Wg), opløses i petroleumsether (4.2) og overføres kvantitativt til en 100 ml målekolbe, hvorefter der fyldes op til mærket med petroleumsether.
Denne opløsning indeholder (W × P) / 10 mg/ml apocarotenester.
Bemærkning: Opløsningen skal opbevares på et køligt sted i mørke. Ubrugt opløsning kasseres efter en måneds forløb.
4.2. Petroleumsether (kogepunktsinterval 40-60 °C)
4.3. Natriumsulfat, vandfrit, granuleret og forinden tørret ved 102 °C i to timer
4.4. Chloroform
4.5. Cyclohexan.
5. FREMGANGSMÅDE
5.1. Forberedelse af prøven
5.1.1. Koncentreret smør
Prøven smeltes i ovn ved ca. 45 °C.
5.1.2. Smør
Prøven smeltes i ovn ved ca. 45 °C. En repræsentativ portion filtreres gennem et filter, der indeholder ca. 10 g vandfrit natriumsulfat (4.3), afskærmet fra stærk naturlig eller kunstig belysning, bibeholdt ved en temperatur på 45 °C. En passende mængde smørfedt opsamles.
5.2. Bestemmelse
Ca. 1 g koncentreret smør (eller ekstraheret smørfedt (5.1.2)) (M) afvejes med 1 mg nøjagtighed. Det overføres kvantitativt til en 20 ml målekolbe (V), der fyldes op til mærket med petroleumsether (4.2), og blandes grundigt.
En delmængde overføres til en 1 cm kuvette, og absorbansen måles ved 440 nm med petroleumsether som reference. Koncentrationen af apocarotenester i opløsningen fås ud fra den fremkomne standardkurve (C μ/ml).
5.3. Kalibrering
0, 0,25, 0,5, 0,75 og 1,0 ml standardopløsning af apocarotenester (4.1.2) pipetteres i fem 100 ml målekolber. Der fyldes op til mærket med petroleumsether (4.2) og blandes.
Opløsningernes koncentrationer går fra 0 til 2 μg/ml og angives nøjagtigt ved reference til koncentrationen af standardopløsningen (4.1.2) (W. × P) / 10 mg/ml. Absorbansen måles ved 440 nm med petroleumsether (4.2) som reference.
Absorbanserne afbildes på y-aksen mod koncentrationen af apocarotenester ad x-aksen. Standardkurvens ligning beregnes.
6. BEREGNING AF RESULTATER
6.1. Indholdet af apocarotenester udtrykt i mg/kg produkt angives ved:
koncentreret smør: (C × V)/M
smør: 0,82 (C × V)M
hvor:
|
C |
= |
indhold af apocarotenester i μg/ml aflæst på kalibreringskurven (5.3) |
|
V |
= |
volumen (ml) af prøveopløsningen (5.2) |
|
M |
= |
massen (g) af analyseprøven (5.2) |
|
0,82 |
= |
en korrektionsfaktor for smørfedtindholdet i smør. |
7. METODENS NØJAGTIGHED
7.1. Repeterbarhed
7.1.1. Analyse af smør
Forskellen mellem resultaterne af to bestemmelser af det samme prøvemateriale, der foretages umiddelbart efter hinanden af den samme person med det samme udstyr, må ikke overstige 1,4 mg/kg.
7.1.2. Analyse af koncentreret smør
Forskellen mellem resultaterne af to bestemmelser af det samme prøvemateriale, der foretages umiddelbart efter hinanden af den samme person med det samme udstyr, må ikke overstige 1,6 mg/kg.
7.2. Reproducerbarhed
7.2.1. Analyse af smør
Forskellen mellem resultaterne af to bestemmelser af det samme prøvemateriale, der foretages i forskellige laboratorier med forskelligt udstyr, må ikke overstige 4,7 mg/kg.
7.3. Analyse af koncentreret smør
Forskellen mellem resultaterne af to bestemmelser af det samme prøvemateriale, der foretages i forskellige laboratorier med forskelligt udstyr, må ikke overstige 5,3 mg/kg.
7.4. Kilde for præcisionsdata
Præcisionsdataene blev fastlagt på grundlag af et forsøg i 1995, der omfattede elleve laboratorier og tolv prøver af smør med røbestof (seks blinde dobbeltprøver) og tolv prøver af koncentreret smør med røbestof (seks blinde dobbeltprøver).
8. TOLERANCEGRÆNSER
8.1. Der udtages tre prøver af produktet med røbestof for at kontrollere, om produktet er tilsat røbestof på korrekt måde.
8.2. Smør
8.2.1. Under hensyntagen til baggrundsabsorbansen er iblandingen for smør 22 mg/kg.
8.2.2. Resultaterne af analysen af de tre prøver af produktet anvendes til at kontrollere mængden og homogeniteten af det iblandede røbestof, og det laveste af disse resultater sammenholdes med følgende grænseværdier:
|
— |
17,7 mg/kg (95 % af den mindste iblanding) |
|
— |
12,2 mg/kg (70 % af den mindste iblanding). |
Koncentrationen af røbestoffet i den prøve, der giver det laveste resultat, benyttes sammen med interpolering mellem 17,7 mg/kg og 12,2 mg/kg.
8.3. Koncentreret smør
8.3.1. Under hensyntagen til baggrundsabsorbansen er iblandingen for koncentreret smør 24 mg/kg.
Resultaterne af analysen af de tre prøver af produktet anvendes til at kontrollere mængden og homogeniteten af det iblandede røbestof, og det laveste af disse resultater sammenholdes med følgende grænseværdier:
|
— |
19,2 mg/kg (95 % af den mindste iblanding) |
|
— |
13,2 mg/kg (70 % af den mindste iblanding). |
Koncentrationen af røbestoffet i den prøve, der giver det laveste resultat, benyttes sammen med interpolering mellem 19,2 mg/kg og 13,2 mg/kg.
BILAG VIII
(Artikel 5)
BESTEMMELSE AF SITOSTEROL OG STIGMASTEROL I KONCENTRERET SMØR OG SMØR VED GASKROMATOGRAFI I KAPILLARKOLONNE
1. FORMÅL OG ANVENDELSESOMRÅDE
Metoden består i en procedure til kvantitativ bestemmelse af sitosterol og stigmasterol i koncentreret smør og smør. Ved sitosterol forstås summen af β-sitosterol og 22-dihydro-β-sitosterol, da de andre sitosteroler anses for at være uden større betydning.
2. PRINCIP
Det koncentrerede smør og smørret forsæbes med kaliumhydroxid i en ethanolopløsning og uforsæbelige bestanddele ekstraheres med diethylether.
Sterolerne omdannes til trimethylsilylethere og analyseres ved gaskromatografi i en kapillarkolonne med betulin som intern standard.
3. APPARATUR
3.1. En 150 ml kolbe til forsæbning med tilbagesvaler med glasslib
3.2. 500 ml skilletragte
3.3. 250 ml kolber
3.4. Trykudligningstragte, 250 ml eller tilsvarende, til opsamling af fordampet diethylether
3.5. Glaskolonne, 350 mm × 20 mm, med sintret glasskive
3.6. Vandbad eller varmekappe
3.7. Prøveglas, 2 ml
Gaskromatograf (med splitningssystem), som egner sig til brug med kapillarkolonne, og som består af:
3.8.1. en termostatstyret kolonneovn, som kan opretholde den ønskede temperatur inden for ±1 °C
3.8.2. injektionsblok med temperaturindstilling
3.8.3. flammeioniseringsdetektor og konverter/forstærker
3.8.4. skriver med integrator, som kan anvendes sammen med konverteren/forstærkeren (3.8.3)
3.9. Kapillarkolonne i kvartsglas, coated med BP1 eller tilsvarende (eller anden kolonne med mindst tilsvarende resolution) i en ensartet lagtykkelse på 0,25 μm. Kolonnen skal kunne adskille trimethylsilylderivater af lanosterol og sitosterol. En BP1-kolonne på 12 m med 0,2 mm indre diameter er velegnet
3.10. Mikrosprøjte på 1 μl med hærdet nål til gaskromatografi.
4. REAGENSER
Alle reagenser skal være af anerkendt analysekvalitet. Der anvendes destilleret vand eller vand af mindst tilsvarende renhed.
4.1. Ethanol, mindst 95 %.
4.2. Kaliumhydroxid, 60 %; 600 g kaliumhydroxid (minimum 85 %) opløses i vand og fortyndes til 1 liter med vand.
Betulin, mindst 99 % renhed
Betulinopløsninger i diethylether (4.4)
4.3.1.1. Betulinopløsningen til bestemmelse af sitosterol skal være af en koncentration på 1,0 mg/ml.
4.3.1.2. Betulinopløsningen til bestemmelse af stigmasterol skal være af en koncentration på 0,4 mg/ml.
4.4. Analytisk ren diethylether (fri for peroxider og rester)
4.5. Natriumsulfat, vandfrit, granuleret og forinden tørret ved 102 °C i to timer.
4.6. Silyleringsreagens, f.eks. TRI-SIL (kan fås fra Pierce Chemical Co., katalognummer 49001) eller tilsvarende (NB: TRI-SIL er brandfarligt og giftigt, ætsende og kan være kræftfremkaldende. Laboratoriepersonalet skal have kendskab til sikkerhedsbestemmelserne for TRI-SIL og handle derefter)
4.7. Lanosterol
Sitosterol af en kendt renhedsgrad på ikke under 90 % (P)
Bemærkning 1: Renhedsgraden af standardmaterialer, som anvendes til kalibrering, fastsættes efter standardiseringsmetoden. Det forudsættes, at samtlige steroler, som findes i prøven, kommer til udtryk på kromatogrammet, at toppenes samlede areal repræsenterer 100 % af sterolindholdet og at sterolerne fremkalder samme detektorrespons. Systemets linearitet skal kontrolleres for de aktuelle koncentrationsområder.
4.8.1. Sitosterolstandardopløsning: Der fremstilles en opløsning af sitosterol (4.8) i diethylether (4.4), der indeholder ca. 0,5 mg/ml, (med en nøjagtighed på 0,001 mg/ml) (W1)
Stigmasterol af en kendt renhedsgrad på ikke under 90 % (P)
4.9.1. Stigmasterolstandardopløsning: Der fremstilles en opløsning af stigmasterol (4.9) i diethylether (4.4), der indeholder ca. 0,2 mg/ml, (med en nøjagtighed på 0,001 mg/ml (W1).
4.10. Blanding til kontrol af resolution. Der fremstilles en opløsning af 0,05 mg lanosterol (4,7) og 0,5 mg sitosterol (4.8) pr. ml diethylether (4.4).
5. METODE
5.1. Fremstilling af standardopløsninger til kromatografi
Den interne standardopløsning (4.3.1) tilsættes til den pågældende sterolstandardopløsning samtidig med tilsætning til den forsæbede prøve (se 5.2.2).
5.1.1. Standardkromatografiopløsning for sitosterol: Der overføres 1 ml sitosterolstandardopløsning (4.8.1) til hvert af to prøveglas (3.7), og diethyletheren afdampes med kvælstof. Der tilsættes 1 ml intern standardopløsning (4.3.1.1), og diethyletheren afdampes med kvælstof.
5.1.2. Standardkromatografiopløsning for sitosterol: Der overføres 1 ml sitosterolstandardopløsning (4.9.1) til hvert af to prøveglas (3.7), og diethyletheren afdampes med kvælstof. Der tilsættes 1 ml intern standardopløsning (4.3.1.2), og diethyletheren afdampes med kvælstof.
5.2. Tilberedning af uforsæbelige bestanddele
5.2.1. Smørprøven smeltes ved opvarmning til højst 35 °C. Den blandes omhyggeligt ved omrøring.
Der afvejes 1 g smør (W2) eller koncentreret smør (W2), med 1 mg nøjagtighed, i en 150 ml kolbe (3.1). Der tilsættes 50 ml ethanol (4.1) og 10 ml kaliumhydroxidopløsning (4.2). Tilbagesvaleren påsættes, og kolben opvarmes til ca. 75 °C i 30 minutter. Svaleren tages af, og kolben afkøles til rumtemperatur.
5.2.2. Der tilsættes 1,0 ml intern standardopløsning (4.3.1.1) til kolben, hvis indholdet af sitosterol skal bestemmes, eller 4.3.1.2, hvis indholdet af stigmasterol skal bestemmes. Opløsningen blandes omhyggeligt. Kolbens indhold overføres kvantitativt til en 500 ml skilletragt (3.2). Kolben skylles med 50 ml vand og dernæst med 250 ml diethylether (4.4). Skilletragten rystes kraftigt i to minutter, hvorefter man lader faserne adskilles. Det nedre vandige lag tappes af, og etherlaget vaskes ved at ryste kolben med fire hold vand a 100 ml.
Bemærkning 2:: For at undgå emulsionsdannelse skal de første to skylninger med vand foretages forsigtigt (kolben vendes ti gange). Ved tredje skylning kan der rystes kraftigt i 30 sekunder. En eventuel emulsion kan brydes ved at tilsætte 5 til 10 ml ethanol. Hvis der tilsættes ethanol, skal der foretages yderligere to grundige skylninger med vand.
5.2.3. Det klare, sæbefrie etherlag ledes igennem en glaskolonne (3.5), som indeholder 30 g vandfri natriumsulfat (4,5). Etheren opsamles i en 250 ml kolbe (3.3). Der tilsættes en kogesten, og etheren afdampes til næsten tørhed i vandbad eller varmekappe, mens overskydende opløsningsmiddel omhyggeligt opsamles.
Bemærkning 3: Hvis prøveekstrakter inddampes til fuldstændig tørhed ved for høj temperatur, kan der ske tab af sterol.
5.3. Fremstilling af trimethylsilylethere
5.3.1. Den resterende etheropløsning i kolben hældes i et 2 ml prøveglas (3.7) med 2 ml diethylether, og etheren afdampes med kvælstof. Kolben skylles med yderligere to hold diethylether a 2 ml, og indholdet hældes hver gang over i prøveglasset, og etheren afdampes med kvælstof hver gang.
5.3.2. Prøven silyleres ved tilsætning af 1 ml TRI-SIL (4.6). Prøveglasset lukkes og rystes kraftigt for at opløse prøven. Hvis prøven stadig ikke er fuldstændigt opløst, opvarmes den til 65-70 °C. Prøven henstår i mindst fem minutter, inden den indsprøjtes i gaskromatografen. Standardopløsningerne silyleres på samme måde som prøverne. Opløsningen til kontrol af resolution (4.10) silyleres på samme måde.
Bemærkning 4: Silylering skal foretages i vandfrit miljø. Ved ufuldstændig silylering ses en top tæt ved betulintoppen.
Ethanol kan give interferens ved silylering. Dette kan skyldes utilstrækkelig skylning ved ekstraktionen. Hvis dette problem forekommer, kan der foretages en femte skylning med kraftig rystning i 30 sekunder ved ekstraktionen.
5.4. Gaskromatografisk analyse
5.4.1. Valg af kromatografibetingelser
Gaskromatografen indstilles som angivet i betjeningsvejledningen.
De vejledende kromatografibetingelser er som følger:
|
— |
kolonnetemperatur: 265 °C |
|
— |
injektorblokkens temperatur: 265 °C |
|
— |
detektorens temperatur: 300 °C |
|
— |
bæregassens gennemstrømningshastighed: 0,6 ml/min. |
|
— |
hydrogentryk: 84 kPa |
|
— |
lufttryk: 155 kPa |
|
— |
splitningssystemet indstilles til mellem 10:1 og 50:1 og optimeres i henhold til betjeningsvejledningen, hvorefter detektorresponsens linearitet kontrolleres inden for det aktuelle koncentrationsområde. Bemærkning 5: Det er særlig vigtigt, at injektionsblokkens belægning rengøres regelmæssigt. |
|
— |
indsprøjtet mængde: 1 μl trimethylsilyletheropløsning. |
Systemet skal være i ligevægt og basislinjen stabil, inden analysen påbegyndes.
Disse betingelser kan fraviges, hvis særlige forhold ved kolonnen og gaskromatografen gør det nødvendigt for at opnå kromatogrammer, som opfylder følgende krav:
|
— |
Sitosteroltoppen skal være tydeligt adskilt fra lanosteroltoppen. I figur 1 ses, hvordan et kromatogram af en silyleret blanding til kontrol af resolutionen (4.10) typisk skal se ud. |
|
— |
Den relative retentionstid for følgende steroler skal være ca.:
|
|
— |
Retentionstiden for betulin skal være ca. 24 minutter. |
5.4.2. Analyseprocedure
Der indsprøjtes 1 μl silyleret standardopløsning (stigmasterol eller sitosterol), og integratorens kalibreringsparametre indstilles.
Herefter indsprøjtes yderligere 1 μl silyleret standardopløsning for at bestemme responsfaktorerne for betulin.
Der indsprøjtes 1 μl silyleret prøveopløsning, og toppene aflæses. Hver kromatografisk serie skal begyndes med og efterfølges af indsprøjtning af standardopløsninger.
Som hovedregel kan der indsprøjtes seks prøver i hver serie.
Bemærkning 6: Integration af stigmasteroltoppen skal også omfatte en evt. hale som afgrænset af punkt 1, 2 og 3 i figur 2b.
Integration af sitosteroltoppen skal også omfatte toppen for 22-dihydro-β-sitosterol (stigmastanol), som eluerer umiddelbart efter sitosterol (figur 3b), når den samlede mængde sitosterol bestemmes.
6. BEREGNING AF RESULTATER
6.1. Arealet af steroltoppe og betulintoppe i begge referencekromatogrammer omkring en serie bestemmes, og R1 beregnes således:
R1 = (gennemsnitsarealet af steroltoppen i standardopløsningen)/(gennemsnitsarealet af betulintoppen i standardopløsningen)
Arealet af steroltoppen (stigmasterol og sitosterol) og betulintoppen i prøven bestemmes, og R2 beregnes således:
R2 = (arealet af steroltoppen i prøven)/(arealet af betulintoppen i prøven)
|
W1 |
= |
sterolindhold (i mg) i 1 ml standardopløsning (4.8.1 eller 4.9.1) |
|
W2 |
= |
prøvens vægt i g (5.2.1) |
|
P |
= |
renhedsgrad af sterolstandarden (4.8 eller 4.9) |
Sterolindhold i prøven (mg/kg) = ((R 2) / (R 1)) × ((W 1) / (W 2)) × P × 10
7. METODENS PRÆCISION
7.1. Smør
7.1.1. Repeterbarhed
7.1.1.1. Stigmasterol
Forskellen mellem resultaterne af to bestemmelser af det samme prøvemateriale, der foretages umiddelbart efter hinanden af den samme person med det samme udstyr, må ikke overstige 19,3 mg/kg.
7.1.1.2. Sitosterol
Forskellen mellem resultaterne af to bestemmelser af det samme prøvemateriale, der foretages umiddelbart efter hinanden af den samme person med det samme udstyr, må ikke overstige 23,0 mg/kg.
7.1.2. Reproducerbarhed
7.1.2.1. Stigmasterol
Forskellen mellem resultaterne af to bestemmelser af det samme prøvemateriale, der foretages i forskellige laboratorier med forskelligt udstyr, må ikke overstige 31,9 mg/kg.
7.1.2.2. Sitosterol
Forskellen mellem resultaterne af to bestemmelser af det samme prøvemateriale, der foretages i forskellige laboratorier med forskelligt udstyr, må ikke være større end 8,7 % af gennemsnittet af resultaterne.
7.1.3. Kilde for præcisionsdata
Præcisionsdataene er fastlagt på grundlag af et eksperiment, som i 1992 blev foretaget med otte laboratorier og seks prøver (blindbestemmelse med tre dobbeltprøver) for stigmasterol samt seks prøver (blindbestemmelse med tre dobbeltprøver) for sitosterol.
7.2. Koncentreret smør
7.2.1. Repeterbarhed
7.2.1.1. Stigmasterol
Forskellen mellem resultaterne af to bestemmelser af det samme prøvemateriale, der foretages umiddelbart efter hinanden af den samme person med det samme udstyr, må ikke overstige 10,2 mg/kg.
7.2.1.2. Sitosterol
Forskellen mellem resultaterne af to bestemmelser af det samme prøvemateriale, der foretages umiddelbart efter hinanden af den samme person med det samme udstyr, må ikke være større end 3,6 % af gennemsnittet af resultaterne.
7.2.2. Reproducerbarhed
7.2.2.1. Stigmasterol
Forskellen mellem resultaterne af to bestemmelser af det samme prøvemateriale, der foretages i forskellige laboratorier med forskelligt udstyr, må ikke overstige 25,3 mg/kg.
7.2.2.2. Sitosterol
Forskellen mellem resultaterne af to bestemmelser af det samme prøvemateriale, der foretages i forskellige laboratorier med forskelligt udstyr, må ikke være større end 8,9 % af gennemsnittet af resultaterne.
7.2.3. Kilde for præcisionsdata
Præcisionsdataene er fastlagt på grundlag af et eksperiment, som i 1991 blev foretaget med ni laboratorier og seks prøver (blindbestemmelse med tre dobbeltprøver) for stigmasterol samt seks prøver (blindbestemmelse med tre dobbeltprøver) for sitosterol.
8. TOLERANCEGRÆNSER
8.1. Der udtages tre prøver af produktet med røbestof for at kontrollere, om produktet er tilsat røbestof på korrekt måde.
8.2. Smør
8.2.1. Stigmasterol
8.2.1.1. Der iblandes 150 g mindst 95 % ren stigmasterol pr. t smør, dvs. 142,5 mg/kg, eller 170 g mindst 85 % ren stigmasterol pr. t smør, dvs. 144,5 mg/kg.
8.2.1.2. Resultaterne af analysen af de tre prøver af produktet anvendes til at kontrollere mængden og homogeniteten af det iblandede røbestof, og det laveste af disse resultater sammenholdes med følgende grænseværdier:
|
— |
115,8 mg/kg (95 % af den mindste iblanding for 95 % ren stigmasterol) |
|
— |
117,7 mg/kg (95 % af den mindste iblanding for 85 % ren stigmasterol) |
|
— |
80,1 mg/kg (70 % af den mindste iblanding for 95 % ren stigmasterol) |
|
— |
81,5 mg/kg (70 % af den mindste iblanding for 85 % ren stigmasterol). |
Koncentrationen af røbestoffet i den prøve, der giver det laveste resultat, benyttes sammen med interpolering mellem 115,8 mg/kg og 80,1 mg/kg eller mellem 117,7 mg/kg og 81,5 mg/kg.
8.2.2. Sitosterol
8.2.2.1. Der iblandes 600 g mindst 90 % ren sitosterol pr. t smør, dvs. 540 mg/kg.
8.2.2.2. Resultaterne af analysen af de tre prøver af produktet anvendes til at kontrollere mængden og homogeniteten af det iblandede røbestof, og det laveste af disse resultater sammenholdes med følgende grænseværdier:
|
— |
482,6 mg/kg (95 % af den mindste iblanding for 90 % ren sitosterol) |
|
— |
347,6 mg/kg (70 % af den mindste iblanding for 90 % ren sitosterol). |
Koncentrationen af røbestoffet i den prøve, der giver det laveste resultat, benyttes sammen med interpolering mellem 482,6 mg/kg og 347,6 mg/kg.
8.3. Koncentreret smør
8.3.1. Stigmasterol
8.3.1.1. Der iblandes 150 g mindst 95 % ren stigmasterol pr. t koncentreret smør, dvs. 142,5 mg/kg eller 170 g mindst 85 % ren stigmasterol pr. t koncentreret smør, dvs. 144,5 mg/kg.
8.3.1.2. Resultaterne af analysen af de tre prøver af produktet anvendes til at kontrollere mængden og homogeniteten af det iblandede røbestof, og det laveste af disse resultater sammenholdes med følgende grænseværdier:
|
— |
118,5 mg/kg (95 % af den mindste iblanding for 95 % ren stigmasterol) |
|
— |
120,4 mg/kg (95 % af den mindste iblanding for 85 % ren stigmasterol) |
|
— |
82,9 mg/kg (70 % af den mindste iblanding for 95 % ren stigmasterol) |
|
— |
84,3 mg/kg (70 % af den mindste iblanding for 85 % ren stigmasterol). |
Koncentrationen af røbestoffet i den prøve, der giver det laveste resultat, benyttes sammen med interpolering mellem 118,5 mg/kg og 82,9 mg/kg eller mellem 120,4 mg/kg og 84,3 mg/kg.
8.3.2. Sitosterol
8.3.2.1. Der iblandes 600 g mindst 90 % ren sitosterol pr. t koncentreret smør, dvs. 540 mg/kg.
8.3.2.2. Resultaterne af analysen af de tre prøver af produktet anvendes til at kontrollere mængden og homogeniteten af det iblandede røbestof, og det laveste af disse resultater sammenholdes med følgende grænseværdier:
|
— |
480,9 mg/kg (95 % af den mindste iblanding for 90 % ren sitosterol) |
|
— |
345,9 mg/kg (70 % af den mindste iblanding for 90 % ren sitosterol). |
Koncentrationen af røbestoffet i den prøve, der giver det laveste resultat, benyttes sammen med interpolering mellem 480,9 mg/kg og 345,9 mg/kg.
Figur 1
Kromatogram af opløsning til kontrol af adskillelsesevne
Det er bedst, hvis toppene er fuldstændig adskilt, dvs. At toppen for lanosterol går helt ned til basislinjen, før sitosteroltoppen begynder. En vis overlapning kan dog accepteres.

|
Figur 2a |
Figur 2b |
|
Stigmasterolstandardopløsning |
Smørprøve denatureret med stigmasterol |
|
|
|
|
Bemœrk: Integration af stigmasteroltoppen skal også omfatte en eventuel hale som afgrænset af punkt 1, 2 og 3. |
|
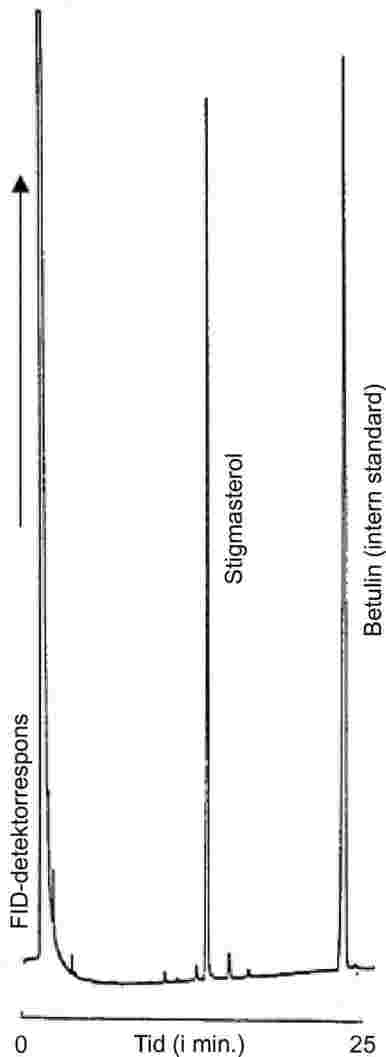

|
Figur 3a |
Figur 3b |
|
β-sitosterolstandardopløsning |
Smørprøve denatureret med β-sitosterol |
|
|
|
|
Bemœrk: β-sitosterol indeholder ofte et fremmedstof (stigmastanol), som eluerer umiddelbart efter β-sitosterol. Det samlede indhold af β-sitosterol findes ved at lægge disse to toppes arealer sammen. |
|


BILAG IX
(Artikel 6)
REFERENCEMETODE TIL PÅVISNING AF KOMÆLK OG KOMÆLKSKASEINAT I OST FREMSTILLET AF FÅRE-, GEDE- ELLER BØFFELMÆLK ELLER AF BLANDINGER AF FÅRE-, GEDE- OG BØFFELMÆLK
1. FORMÅL
Påvisning af komælk og komælkskaseinat i ost fremstillet af fåre-, gede- eller bøffelmælk eller af blandinger af fåre-, gede- og bøffelmælk ved isoelektrisk fokusering af γ-kaseiner efter plasminolyse.
2. ANVENDELSESOMRÅDE
Metoden er egnet til følsom og specifik påvisning af rå og varmebehandlet komælk og komælkskaseinat i frisk og modnet ost fremstillet af fåre-, gede- eller bøffelmælk eller af blandinger af fåre-, gede- og bøffelmælk. Den er ikke egnet til påvisning af, om mælk og ost er forfalsket med varmebehandlede kovalleproteinkoncentrater.
3. PRINCIP
3.1. Isolering af kaseiner fra ost og referenceopløsninger
3.2. Opløsning af isolerede kaseiner, som derefter underkastes plasminspaltning (EC.3.4.21.7).
3.3. Isoelektrisk fokusering af de plasminbehandlede kaseiner i tilstedeværelse af urea og farvning af proteinerne.
3.4. Vurdering af de farvede γ3- og γ2-kaseinmønstre (tegn på komælk) ved sammenligning af mønstret fra prøven med dem, der fremkom i samme gel fra referenceopløsninger med 0 % og 1 % komælk.
4. REAGENSER
Medmindre andet er angivet, skal der anvendes kemikalier af analysekvalitet. Vandet skal være dobbeltdestilleret eller af tilsvarende renhed.
Bemærkning: Nedenstående detaljer gælder for laboratoriefremstillede polyacrylamidgeler, der indeholder urea, af størrelsen 265 × 125 × 0,25 mm. Hvis der bruges gel af en anden størrelse eller type, skal separationsbetingelserne måske justeres.
Isoelektrisk fokusering
4.1. Reagenser til fremstilling af polyacrylamider, der indeholder urea
4.1.1. Stamopløsning
Dissolve:
,85 g acrylamid
0,15 g N, N'-metylen-bis-acrylamid (BIS)
48,05 g urea
15,00 g glycerol (87 % w/w)
opløses i vand, og der fyldes op til 100 ml. Opbevares i brun glasflaske i køleskab.
Bemærkning: En forblandet acrylamid/BIS-opløsning, der fås i handelen, kan anvendes frem for de angivne vægte af neurotoksiske acrylamider. Hvis en sådan opløsning indeholder 30 % w/v acrylamid og 0,8 % w/v BIS, skal der bruges et volumen på 16,2 ml i formuleringen i stedet for de angivne vægte. Stamopløsningens holdbarhed er højst 10 døgn. Hvis dens ledningsevne er over 5 μS, afioniseres den ved omrøring med 2 g Amberlite MB-3 i 30 minutter og filtreres derefter gennem en 0,45 μm membran.
4.1.2. Gelopløsning
Der laves en gelopløsning ved blanding af additiver og amfolytter med stamopløsningen (4.1.1).
9,0 ml stamopløsning
24 mg β-alanin
500 μl amfolyt pH 3,5-9,5 (1)
250 μl amfolyt pH 5-7 (1)
250 μl amfolyt pH 6-8 (1).
Gelopløsningen blandes og afgasses i 2-3 minutter i et ultralydsbad eller i vakuum.
Bemærkning: Gelopløsningen laves straks før ophældningen (6.2).
4.1.3. Katalysatoropløsninger
4.1.3.1. N, N, N'N'-tetrametylendiamin (TEMED)
4.1.3.2. 40 % w/v ammoniumpersulfat (PER)
800 mg PER opløses i vand, og der fyldes op til 2 ml.
Bemærkning: Der skal altid bruges frisklavet PER-opløsning.
4.2. Kontaktvæske
Petroleum eller flydende paraffin.
4.3. Anodeopløsning
5,77 g fosforsyre (85 % w/w) opløses i vand og fortyndes til 100 ml med vand.
4.4. Katodeopløsning
2,00 natriumhydroxid opløses i vand og fortyndes til 100 ml med vand.
Forberedelse af prøven
4.5. Reagenser til proteinisolering
4.5.1. Fortyndet eddikesyre (25,0 ml iseddike fortyndet til 100 ml ved vand).
4.5.2. Dichlormethan
4.5.3. Acetone
4.6. Proteinopløsende buffer
Dissolve:
5,75 g glycerol (87 % w/w)
24,03 g urea
250 mg dithiothreitol
opløses i vand, og der fyldes op til 50 ml.
Bemærkning: Opbevares i køleskab — holdbarhed 1 uge.
4.7. Reagenser til plasminspaltning af kaseiner
4.7.1. Ammoniumcarbonatbuffer
En 0,2 mol/l ammoniumhydrogencarbonatopløsning (1,58 g/100 ml vand) med 0,05 mol/l ethylendiamintetra-eddikesyre (EDTA, 1,46 g/100 ml) titreres med en 0,2 mol/l ammoniumcarbonatopløsning (1,92 g/100 ml vand) med 0,05 mol/l EDTA til pH 8.
4.7.2. Bovin plasmin (EC. 3.4.21.7), aktivitet mindst 5 U/ml
4.7.3. ε-Aminocapronsyre til enzymhæmning
2,624 g ε-aminocapronsyre (6-amino-n-heksanonsyre) opløses i 100 ml 40 % (v/v) ethanol.
4.8. Standardopløsninger
4.8.1. Godkendte referenceopløsninger af en blanding af løbebehandlet skummet fåre- og gedemælk indeholdende 0 % og 1 % komælk kan fås hos Kommissionen, Instituttet for Referencemålinger og -Materialer, B-2440 Geel, Belgien.
4.8.2. Fremstilling af midlertidige laboratoriestandardopløsninger af løbebehandlet bøffelmælk indeholdende 0 % og 1 % komælk.
Skummetmælken fremstilles ved centrifugering af bøffelmælk eller rå tankkomælk ved 37 °C ved 2 500 g i 20 minutter. Efter hurtig afkøling af glas og indhold til 6-8 °C fjernes det øverste fedtlag fuldstændigt. Til fremstilling af 1 %-standardopløsningen tilsættes 5,00 ml skummet komælk til 495 ml skummet bøffelmælk i et 1 l bæger. pH-værdien justeres til 6,4 ved tilsætning af fortyndet mælkesyre (10 % w/v). Temperaturen justeres til 35 °C, og 100 μl kalveløbe tilsættes (løbeaktivitet: 1:10 000, ca. 3 000 U/ml). Der omrøres i et minut, hvorefter opløsningen henstår tildækket med alufolie ved 35 °C i en time, så ostemassen kan dannes. Når ostemassen er dannet, frysetørres al den løbebehandlede sødmælk uden forudgående homogenisering eller afdræning af vallen. Efter frysetørringen formales produktet, så der fremkommer et homogent pulver. Til fremstilling af 0 %-standardopløsningen følges samme metode med brug af skummet bøffelmælk. Standardopløsningerne skal opbevares ved –20 °C.
Bemærkning: Det kan tilrådes at kontrollere bøffelmælkens renhed ved isoelektrisk fokusering af de plasminbehandlede kaseinater, inden standardopløsningerne fremstilles.
Reagenser til proteinfarvning
4.9. Fikservæske
150 g trichloreddikesyre opløses i vand, og der fyldes op til 1 000 ml.
4.10. Affarvningsopløsning
500 ml methanol og 200 ml iseddike fortyndes til 2 000 ml med destilleret vand.
Bemærkning: Affarvningsopløsningen frisklaves hver dag. Den kan fremstilles ved at blande lige store mængder stamopløsninger af 50 % (v/v) methanol og 20 % (v/v) iseddike.
4.11. Farvningsopløsninger
4.11.1. Farvningsopløsning (stamopløsning 1)
3,0 g Coomassie Brilliant Blue G-250 (C.I. 42655) opløses i 1 000 ml 90 % (v/v) methanol ved hjælp af en magnetisk omrører (ca. 45 minutter) og filtreres gennem to middelhastigheds-foldefiltre.
4.11.2. Farvningsopløsning (stamopløsning 2)
5,0 g kobbersulfatpentahydrat opløses i 1 000 ml 20 % (v/v) eddikesyre.
4.11.3. Farvningsopløsning (arbejdsopløsning)
125 ml af hver af stamopløsningerne (4.11.1, 4.11.2) sammenblandes umiddelbart før farvningen.
Bemærkning: Farvningsopløsningen bør laves samme dag, den skal bruges.
5. APPARATUR
5.1. Glasplader (265 × 125 × 4 mm), gummirulle (bredde 15 cm) og vandret flade
5.2. Gelbærefolie (265 × 125 mm)
5.3. Dækfolie (280 × 125 mm). Der anbringes en strimmel klæbebånd (280 × 6 × 0,25 mm) på hver langside (jf. figur 1)
5.4. Elektrofokuseringskammer med køleplade (f.eks. 265 × 125 mm) og egnet spændingskilde (≥ 2,5 kV) eller automatisk elektroforeseapparat
5.5. Cirkulationskryostat, termostatstyret ved 12 ± 0,5 °C
5.6. Centrifuge, justerbar til 3 000 g
5.7. Elektrodestrimler (længde ≥ 265 mm)
5.8. Plastdråbetæller til anode- og katodeopløsninger
5.9. Analyseprøveapplikatorer (10 × 5 mm, viskose- eller lavproteinabsorberingsfilterpapir)
5.10. Saks, skalpel og pincetter af rustfrit stål
5.11. Farvnings- og affarvningsskåle (f.eks. 280 × 150 mm instrumentbakker) af rustfrit stål eller glas
5.12. Justerbar stanghomogenisator (10 mm diameter), skala 8 000-20 000 rpm
5.13. Magnetomrører
5.14. Ultralydsbad
5.15. Filmsvejser
5.16. 25 μl mikropipetter
5.17. Vakuumkoncentrator eller frysetørrer
5.18. Termostatstyret vandbad, der kan indstilles til 35 og 40 ± 1 °C, med rysteaggregat
5.19. Densitometerudstyr med aflæsning ved λ = 634 nm.
6. FREMGANGSMÅDE
6.1. Forberedelse af prøven
6.1.1. Isolering af kaseiner
En mængde svarende til 5 g tør ostemasse eller standardopløsninger afvejes i et 100 ml centrifugeglas. Der tilsættes 60 ml destilleret vand og homogeniseres med en stanghomogenisator (8 000-10 000 rpm). pH-værdien justeres til 4,6 med fortyndet eddikesyre (4.5.1), og der centrifugeres (5 minutter — 3 000 g). Fedtet og vallen afhældes, restindholdet homogeniseres i 40 ml destilleret vand (pH-værdien justeret til 4,5 med fortyndet eddikesyre (4.5.1)) ved 20 000 rpm. Der tilsættes 20 ml dichlormethan (4.5.2), homogeniseres igen og centrifugeres (5 minutter — 3 000 g). Kaseinlaget, der flyder mellem den vandige og den organiske fase (jf. figur 2), fjernes med en spatel, og begge faser afhældes. Kaseinet homogeniseres igen i 40 ml destilleret vand (som ovenfor) og 20 ml dichlormethan (4.5.2), og der centrifugeres. Dette gentages, indtil begge ekstraktionsfaser er farveløse (2-3 gange). Proteinresterne homogeniseres med 50 ml acetone (4.5.3) og filtreres gennem et middelhastigheds-foldefilterpapir. Resterne vaskes på filtret med to særskilte portioner acetone på hver 25 ml og henstår til tørring i luft eller en kvælstofstrøm, hvorefter de finknuses i en morter.
Bemærkning: Tørre kaseinisolater bør opbevares ved –20 °C.
6.1.2. Plasminspaltning af β-kaseiner for at intensivere γ-kaseiner
25 mg isoleret kasein (6.1.1) opslæmmes i 0,5 ml ammoniumcarbonatbuffer (4.7.1) og homogeniseres i 20 minutter, f.eks. ved ultralydsbehandling. Der opvarmes til 40 °C og tilsættes 10 μl plasmin (4.7.2), blandes og inkuberes i en time ved 40 °C under stadig omrystning. Til hæmning af enzymer tilsættes 20 μl ε-aminocapronsyre (4.7.3) og derefter yderligere 200 mg fast urea og 2 mg dithiothreitol.
Bemærkning: For at få større symmetri i de fokuserede kaseinbånd kan det tilrådes at frysetørre opløsningen efter tilsætningen af ε-aminocapronsyre og derefter opløse resterne i 0,5 ml ureabuffer (4.6).
6.2. Fremstilling af polyacrylamidgeler, der indeholder urea
Gelbærefolien (5.2) rulles med et par dråber vand ud på en glasplade (5.1), og eventuelt overskydende vand fjernes med papirhåndklæde- eller serviet. Dækfolien (5.3) udrulles med afstandsstykker (0,25 mm) på en anden glasplade på samme måde. Pladen anbringes plant på en vandret flade.
10 μl TEMED (4.1.3.1) tilsættes til den tilberedte, afgassede gelopløsning (4.1.2), og efter omrystning 10 μl PER-opløsning (4.1.3.2). Opløsningen blandes grundigt og hældes straks derefter jævnt ud på midten af dækfolien. Den ene side af gelbærefolien (med bæresiden nedad) anbringes på dækfoliepladen og sænkes langsomt, således at der danner sig en gelfilm mellem folierne, som spreder sig regelmæssigt uden bobledannelse (jf. figur 3). Gelbærepladen sænkes forsigtigt helt ned ved hjælp af en tynd spatel, og der lægges endnu tre plader oven på den, der skal tjene som vægte. Når polymiseringen er fuldstændig (ca. 60 minutter), overføres den polymeriserede gel på gelbærefolien sammen med dækfolien, ved at glaspladerne vippes. Bærefoliens bagside rengøres omhyggeligt for at fjerne gelrester og urea. Gelsandwichen svejses til et filmglas og opbevares i køleskab (højst 6 uger).
Bemærkning: Dækfolien med afstandsstykkerne kan genbruges. Polyacrylamiden kan skæres i mindre størrelser, hvad der kan anbefales, når der kun er få prøver, eller hvis der anvendes et automatisk elektroforeseapparat (2 geler — størrelse 4,5 × 5 cm).
6.3. Isoelektrisk fokusering
Køletermostaten indstilles til 12 °C. Gelbærefoliens bagside aftørres med petroleum, hvorefter et par dråber petroleum (4.2) dryppes på midten af køleblokken. Derpå rulles gelsandwichen (med bæresiden nedad) ud på den, men det skal undgås, at der danner sig bobler. Eventuel overskydende petroleum tørres af, og dækfolien fjernes. Elektrodestrimlerne gennemvædes med elektrodeopløsningerne (4.3 og 4.4), tilskæres i gelens længde og anbringes de givne steder (elektrodeafstand 9,5 cm).
Fokuseringen foretages på nedenstående betingelser:
6.3.1. Gelstørrelse 265 × 125 × 0,25 mm
|
Trin |
Tid (min.) |
Spænding (V) |
Strøm (mA) |
Effekt (W) |
Volttimer (Vh) |
||
|
30 |
maksimum 2 500 |
maksimum 15 |
konstant 4 |
ca. 300 |
||
|
60 |
maksimum 2 500 |
maksimum 15 |
konstant 4 |
ca. 1 000 |
||
|
60 |
maksimum 2 500 |
maksimum 5 |
maksimum 20 |
ca. 3 000 |
||
|
40 |
maksimum 2 500 |
maksimum 6 |
maksimum 20 |
ca. 3 000 |
|||
|
30 |
maksimum 2 500 |
maksimum 7 |
maksimum 25 |
ca. 3 000 |
Bemærkning: Hvis gelens tykkelse eller bredde ændres, skal strøm og effekt justeres herefter (f.eks. fordobles elstrøm og effekt, hvis der benyttes en 265 × 125 × 0,5 mm gel).
6.3.2. Eksempel på et spændingsprogram for et automatisk elektroforeseapparat (2 geler på 5,0 × 4,5 cm); elektroder anbringes uden strimler direkte på gelen:
|
Trin |
Spænding |
Strøm |
Effekt |
Temp. |
Volttimer |
||
|
1 000 V |
10,0 mA |
3,5 W |
8 °C |
85 Vh |
||
|
250 V |
5,0 mA |
2,5 W |
8 °C |
30 Vh |
||
|
1 200 V |
10,0 mA |
3,5 W |
8 °C |
80 Vh |
||
|
1 500 V |
5,0 mA |
7,0 W |
8 °C |
570 Vh |
Prøveapplikatoren anbringes på andet trin ved 0 Vh.
Prøveapplikatoren fjernes på andet trin ved 30 Vh.
6.4. Proteinfarvning
6.4.1. Proteinfiksering
Elektrodestrimlerne aftages, så snart der er slukket for strømmen, og gelen kommes straks i en farvnings- eller affarvningsskål fyldt med 200 ml fikservæske (4.9). Den henstår i 15 minutter under stadig omrystning.
6.4.2. Vask og farvning af gelpladen
Fikservæsken drænes grundigt, og gelpladen vaskes i 30 sekunder to gange med 100 ml affarvningsopløsning (4.10). Affarvningsopløsningen afhældes, og skålen fyldes med 250 ml farvningsopløsning (4.11.3). Man lader farven udvikle sig i 45 minutter under forsigtig omrystning.
6.4.3. Affarvning af gelpladen
Farvningsopløsningen afhældes, og gelpladen vaskes med 100 ml affarvningsopløsning (4.10) to gange, hvorefter der omrystes med 200 ml affarvningsopløsning i 15 minutter. Affarvningstrinnet gentages mindst 2-3 gange, indtil baggrunden er klar og ufarvet. Derefter skylles gelpladen med destilleret vand (to gange 2 minutter) og tørres i luft (2-3 timer) eller med en hårtørrer (10-15 minutter).
Bemærkning 1: Fiksering, vask, farvning og affarvning foretages ved 20 °C. Der må ikke benyttes højere temperaturer.
Bemærkning 2: Hvis man foretrækker mere følsom sølvfarvning (f.eks. Silver Staining Kit, Protein, Pharmacia Biotech, Code No. 17-1150-01), skal plasminbehandlede kaseinprøver fortyndes til 5 mg/ml.
7. VURDERING
Vurderingen foretages ved sammenligning af proteinmønstrene i den ukendte prøve med referenceprøver på samme gel. Påvisning af komælk i ost fremstillet af fåre-, gede- eller bøffelmælk eller af blandinger af fåre-, gede- og bøffelmælk sker via de γ3- og γ2-kaseiner, hvis isoelektriske punkter ligger mellem pH 6,5 og pH 7,5 (jf. figur 4a, 4b og 5). Påvisningsgrænsen er under 0,5.
7.1. Visuel vurdering
Med henblik på visuel vurdering af indholdet af komælk kan det tilrådes at justere koncentrationerne af prøve- og standardkoncentrationerne, så der fremkommer samme intensitet af fåre-, gede- og/eller bøffel-γ2- og γ3-kaseiner (jf. »γ2 E, G, B« og »γ3 E, G, B« i figur 4a, 4b og 5). Derefter kan indholdet af komælk (mindre end, lig med eller højere end 1 %) i den ukendte prøve bedømmes direkte ved at sammenligne med intensiteten af de bovine γ3- og γ2-kaseiner (jf. »γ3 C« og »γ2 C« i figur 4a, 4b og 5) med 0 %- og 1 %-standardopløsningen (får, ged) eller midlertidige laboratoriestandardopløsninger (bøffel) analyseret på samme gel.
7.2. Densitometrisk vurdering
Om muligt benyttes densitometri (5.19) til bestemmelse af forholdet mellem toparealerne for ko-, fåre-, gede- og/eller bøffel- γ2- og γ3-kaseiner i figur 5. Denne værdi sammenlignes med forholdet mellem toparealerne for γ2- og γ3-kaseiner i 1 %-standardopløsningen (får, ged) eller midlertidige laboratoriestandardopløsninger (bøffel) analyseret på samme gel.
Bemærkning: Metoden fungerer tilfredsstillende, hvis der er et klart, positivt tegn på ko-γ2- og γ3-kaseiner i 1 %-standardopløsningen, men ikke i 0 %-standardopløsningen. Hvis det ikke er tilfældet, forbedres metoden ved, at den følges ganske nøje i alle detaljer.
En prøve betragtes som positiv, hvis ko-γ2- og γ3-kaseinerne eller de tilsvarende toparealforhold er lig med eller højere end niveauet for 1 %-standardopløsningen.
8. LITTERATURHENVISNINGER
|
1. |
Addeo F., Moio L., Chianese L., Stingo C., Resmini P., Berner I, Krause I., Di Luccia A., Bocca A.: Use of plasmin to increase the sensitivity of the detection of bovine milk in ovine and/or caprine cheese by gel isoelectric focusing of γ2-caseins. Milchwissenschaft 45, 708-711 (1990). |
|
2. |
Addeo F., Nicolai M.A., Chianese L., Moio L., Spagna Musso S., Bocca A., Del Giovine L.: A control method to detect bovine milk in ewe and water buffalo cheese using immunoblotting. Milchwissenschaft 50, 83-85 (1995). |
|
3. |
Krause I., Berner I, Klostermeyer H.: Sensitive detection of cow milk in ewe and goat milk and cheese by carrier ampholyte — and carrier ampholyte/immobilized pH gradient — isoelectric focusing of γ-caseins using plasmin as signal amplifier. in: Electrophoresis-Forum 89 (B. J. Radola, ed.) pp 389-393, Bode-Verlag, München (1989). |
|
4. |
Krause Ι., Belitz H.-D., Kaiser K.-P.: Nachweis von Kuhmilch in Schaf and Ziegenmilch bzw. -käse durch isoelektrische Fokussierung in harnstoffhaltigen Polyacrylamidgelen. Z. Lebensm. Unters. Forsch. 174, 195-199 (1982). |
|
5. |
Radola B.J.: Ultrathin-layer isoelectric focusing in 50-100 μm polyacrylamide gels on silanised glass plates or polyester films. Electrophoresis 1, 43-56 (1980). |
Figur 1
Skematisk tegning af dækfolie

Figur 2
Kaseinlaget, der flyder mellem den vandige og den organiske fase efter centrifugering
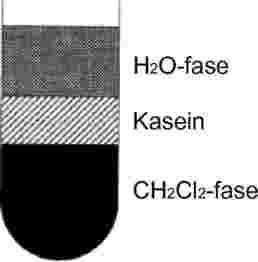
Figur 3
Viftemetode til støbning af ultratynde polyacrylamidgeler

a = afstandsbånd (0,25 mm); b = dækfolie (5.3); c og e = glasplader (5.1); d = gelopløsning (4.1.2); f = gelbærefolie (5.2)
Figur 4a
Isoelektrisk fokusering af plasminbehandlede kaseinater fra ost fremstillet af fåre- eller gedemælk med forskellige mængder komælk.

% CM = procent komælk; C = ko; E = får; G = ged
Den øverste halvdel af IEF-gelen ses.
Figur 4b
Isoelektrisk fokusering af plasminbehandlede kaseinater fra ost fremstillet af blandinger af fåre-, gede- og bøffelmælk med forskellige mængder komælk.

% CM = procent komælk; 1 + = prøve indeholdende 1 % komælk og tilsat rent komælkskasein midt på sporet. C = ko; E = får; G = ged; B = bøffel
IEF-gelens totale separationsafstand ses.
Figur 5
Overlejring af densitogrammer af standardopløsninger (STD) og osteprøver fra en blanding af fåre- og gedemælk efter isoelektrisk fokusering.

a og b = standardopløsninger med 0 og 1 % komælk; c til g = osteprøver med 0, 1, 2, 3 og 7 % komælk; C = ko; E = får; G = ged.
Den øverste halvdel af IEF-gelen er scannet ved λ = 634 nm.
(1) Produkterne Ampholine® pH 3,5-9,5 (Pharmacia) og Resolyte® pH 5-7 og pH 6-8 (BDH, Merck) har vist sig særligt velegnede til at opnå den ønskede separation af γ-kaseiner.
(2) Prøveapplikation: Efter præfokusering (første trin), afpipetteres 18 μl af prøven og standardopløsningerne over på prøveapplikatorerne (10 × 5 mm), der anbringes med en indbyrdes afstand på 1 mm og 5 mm fra anoden og trykkes let ned. Fokuseringen foretages på ovenstående betingelser, og prøveapplikatorerne borttages forsigtigt efter de 60 minutters prøvefokusering.
BILAG X
(Artikel 7)
REFERENCEMETODE TIL PÅVISNING AF COLIFORME BAKTERIER I SMØR, SKUMMETMÆLKSPULVER OG KASEIN/KASEINAT
1. FORBEREDELSE AF PRØVER
ISO-standard 8261
2. FREMGANGSMÅDE
ISO-standard 4831
Prøver svarende til 1 g smør eller 0,1 g skummetmælkspulver eller kasein/kaseinat podes på dyrkningssubstratet.
Der podes tre reagensglas pr. prøve.
3. RESULTATER
Hvis de 3 reagensglas giver 3 negative resultater, er kravet »opfyldt«.
Hvis de 3 reagensglas giver 2 eller 3 positive resultater, er kravet »ikke opfyldt«.
Hvis de 3 reagensglas giver 2 negative resultater, gentages analysen to gange (med to reagensglas).
|
— |
Hvis begge resultater er negative, er kravet »opfyldt«. |
|
— |
Hvis mindst 1 resultat er positivt, er kravet »ikke opfyldt«. |
BILAG XI
(Artikel 8)
BESTEMMELSE AF LACTOSE I FODERBLANDINGER
1. FORMÅL OG ANVENDELSESOMRÅDE
Bestemmelse af lactose i foderblandinger.
2. HENVISNING
Indholdet af lactose defineres som masseprocenten som bestemt ved den beskrevne fremgangsmåde.
3. DEFINITION
Indholdet af vandfri lactose udtrykkes som g pr. 100 g.
4. PRINCIP
Foderblandingen rekonstitueres med vand. Der tilsættes en Biggs-opløsning til en fortyndet afvejet delprøve, så foderblandingens fedt- og proteinkomponentfraktioner udfældes. Prøven filtreres (eller centrifugeres) og filtratet (eller supernatanten) injiceres på en kationbytnings-HPLC-kolonne på blyform ved anvendelse af vand af HPLC-renhed som mobil fase. Den eluerede lactose påvises med et differentialrefraktometer (1).
5. REAGENSER
5.1. Generelt
Der anvendes udelukkende reagenser af anerkendt analysekvalitet, medmindre andet er foreskrevet, og afgasset vand af HPLC-renhed.
5.2. Lactose
D-Lactosemonohydrat ((C12H22)O11. H2O) kan optage ekstra fugtighed. Inden anvendelsen måles det reelle vandindhold med Karl Fisher-metoden, eller den ekstra fugtighed fjernes ved at anbringe lactose i en ovn ved 105 °C i 8 timer (lactosen mister ikke sit krystalvand ved denne behandling).
5.3. Koncentreret Biggs/Szijarto-opløsning (2)
9,10 g zinkacetatdihydrat (Zn(CH3COO)2.2H2O) og 5,46 g phosphorwolframsyremonohydrat (H3[P(W3O10)4.xH2O]) opløses i ca. 70 ml med vand af HPLC-renhed (6.8) i en 100 ml målekolbe.
Der tilsættes 5,81 ml iseddike (CH3COOH). Der fortyndes til 100 ml med vand af HPLC-renhed (6.8) og blandes. Opløsningen kan opbevares ved stuetemperatur i 1 år.
5.4. Fortyndet Biggs/Szijarto-opløsning
25 ml koncentreret Biggs/Szijarto-opløsning (5.3) fortyndes med vand til 500 ml i en målekolbe. Opløsningen kan opbevares ved stuetemperatur i 1 måned.
5.5. Fremstilling af vand af HPLC-renhed
Ultrarent vand (6.8) filtreres ved hjælp af vakuumfiltrering (6.9). For at forbedre pumpeevnen og for at opnå en stabil basislinje afgasses den mobile fase dagligt ved anvendelse af en af de disponible metoder, f.eks. gennemstrømning med helium, sonikering, vakuum eller inline-afgasning.
Bemærkning: For at forlænge kolonnens levetid er det afgørende, at elueringsvæskens kuldioxidindhold er så lavt som muligt, og at genoptagelse hindres.
6. APPARATUR
Sædvanligt laboratorieudstyr, herunder især følgende:
6.1. HPLC-ionbytterharpikskolonne
Kolonnepakning: 8 % tværbundet polystyren-divinylbenzen-copolymer forsynet med kationbyttergrupper på blyform.
Kolonnestørrelse: Længde 300 mm, indvendig diameter ca. 8 mm.
Anden diameterstørrelse er mulig, forudsat at gennemstrømningshastigheden tilpasses.
6.2. Forkolonne
Forkolonnen er en kombination af en separat kationbytter (H+) og en anionbytter (CO3-), der hver især er pakket i kolonner på ca. 30 mm × 4,6 mm (L × ID) (f.eks. mikroforkolonner i en mikroforkolonneholder) og serieforbundne eller forbundet i form af en mixed bed, der består af AG 50W-X4, — 400 mesh (H+) og AG3-X4A, 200-400 mesh (OH-) i forholdet 35:65 (m/m) manuelt pakket i en kolonne på ca. 20 × 9 mm (L x ID).
6.3. Kolonneovn
Ovn, som kan holde en konstant temperatur på 85 °C ± 1 °C.
6.4. HPLC-pumpe
Pumpe, der kan generere en konstant gennemstrømningshastighed (< 0,5 % fluktuationer) ved 0,2-1,0 m/min.
6.5. HPLC-injektor
Autosampler, der kan injicere 25 µL med en repeterbarhed på < 0,5 %.
Alternativt kan en manuel anordning anvendes (samme krav som til autosampleren).
6.6. HPLC-detektor
Meget følsom RI-detektor (RI = brydningsindeks) med støj på < 5,10-9 RI-enheder.
6.7. Integrator
Software eller en dedikeret integrator til opsamling og behandling af data og til at generere toparealer og tophøjder, som kan konverteres til lactosekoncentrationer.
6.8. Vandrensningsenhed
System, der kan fremstille ultrarent vand (type 1) med en modstandsevne på > 14 MΩ/cm.
6.9. Enhed til filtrering af opløsningsmiddel
System, der kan filtrere vandet gennem et membranfilter med porestørrelse 0,45 μm.
Bemærkning: Mange vandrensningsenheder (6.8) har en indbygget 0,45 eller 0,2 µm filtrering. Ekstra filtrering kan undlades, hvis vandet anvendes direkte.
6.10. Analysevægt
Vægten skal kunne aflæses med en nøjagtighed på 0,1 mg.
6.11. Vandbad
Vandbad, som kan holde en temperatur på 40 °C ±0,5 °C.
6.12. Centrifuge
Centrifugen skal kunne yde mindst 3 000 g for eppendorfrør eller lignende eller større typer prøveglas.
6.13. Målekolbe 50 ml
Rumfang 50 ml, klasse A
Bemærkning: Der kan anvendes andre størrelser kolber under hensyntagen til rumfangsfaktoren.
6.14. Målekolbe 100 ml
Rumfang 100 ml, klasse A
6.15. Målepipette
Målepipette 10 ml
Bemærkning: Alternativt kan der anvendes en håndpipette 5 ml ved at tilsætte 2 gange 5 ml reagens (5.3).
7. PRØVEUDTAGNING
Det er vigtigt, at laboratoriet modtager en prøve, der er udtaget i overensstemmelse med ISO 707/IDF 50 (3), som reelt er repræsentativ, og som ikke er blevet skadet under transport eller opbevaring.
8. FREMSTILLING AF LACTOSESTANDARDOPLØSNING
8.1. Standardopløsning 1
Ca. 50 mg omhyggeligt afvejet (med en nøjagtighed på 0,1 mg) lactosemonohydrat (5.2) opløses i en 100 ml målekolbe (6.14), der fyldes op med vand til mærket.
8.2. Standardopløsning 2
Ca. 100 mg omhyggeligt afvejet (med en nøjagtighed på 0,1 mg) lactosemonohydrat (5.2) opløses i en 100 ml målekolbe (6.14), der fyldes op med vand til mærket.
Bemærkning: Standardopløsningerne kan opbevares ved ca. 5 °C i maks. 1 uge.
9. FORBEREDELSE AF PRØVEN
9.1. Rekonstituering af prøven
Ca. 5 g af pulveret afvejes i en 50 ml kolbe (6.13), og vægten noteres med en nøjagtighed på 1 mg (W1(11)). Der tilsættes 50 ml vand, og vægtforøgelsen (W2 (11)) noteres med en nøjagtighed på 0,01 g. Den lukkede kolbe placeres i vandbad (6.11) i 30 min. og vendes nogle gange i løbet af dette tidsrum. Der afkøles derefter til stuetemperatur.
9.2. Behandling af prøven
Ca. 1 g af denne opløsning placeres i en 50 ml målekolbe (6.13), vægten noteres med en nøjagtighed på 1 mg (W3 (11)), og der tilsættes 20 ml vand og derefter 10 ml fortyndet Biggs/Szijarto-reagens (5.4), og der fyldes op med vand til mærket. Flasken vendes forsigtigt 5 gange i de første 30 min.
Efter 1 time udtages en delprøve, som centrifugeres (6.12) ved 3 000 g i 10 min. (større g-værdi kan anvendes i tilsvarende kortere tid). En delprøve af supernatanten anvendes til HPLC-analyse.
10. HPLC-BESTEMMELSE
10.1. Indledende forberedelse af HPLC
10.1.1. Installering af kolonne og forkolonne
Forkolonnen (6.2) installeres uden for kolonneovnen (6.3) og kolonnen (6.1) i ovnen.
Bemærkning: Hvis ovnen ikke indeholder rør til foropvarmning af elueringsvæsken, er det nødvendigt, at elueringsvæsken passerer gennem ca. 15 cm rør af rustfrit stål i ovnen, inden den kommer ind i kolonnen (det er absolut nødvendigt, at elueringsvæsken er opvarmet, inden den kommer ind i kolonnen, ellers vil der forekomme topforbredning).
10.1.2. Detektor og indledende gennemstrømningshastighed
For at få en stabil basislinje, tændes detektoren (6.6) mindst 24 timer inden analysestart. Detektorens interne temperatur indstilles til 35 °C. Gennemstrømningshastigheden indstilles til 0,2 ml/min. (6.4) i mindst 20 min., og kolonneovnen (6.3) indstilles til stuetemperatur.
10.1.3. Kolonneovn og endelig gennemstrømningshastighed
Kolonneovnen (6.3) indstilles til 85 °C. Når den temperatur er nået, øges gennemstrømningshastigheden efter 30 min. gradvis fra 0,2 ml/min. til 0,6 ml/min. (6.4). Systemet skal bringes i ligevægt med den pågældende gennemstrømningshastighed og ved 85 °C i 2 timer, eller indtil der opnås en stabil basislinje.
10.1.4. Integration
Parametrene (6.7) for dataopsamling og integration vælges med omhu, f.eks. datasignaleringshastighed, følsomhed, tidskonstant, topbredde og tærskelværdi.
Lactoses retentionstid er ca. 11 min.
Bemærkning: Mange edb-programmer til dataopsamling (6.7) giver en nem måling af antallet af teoretiske bunde. Antallet af teoretiske bunde i standardopløsning 1 (8.1) måles regelmæssigt, og kolonnen (6.1) udskiftes, når bundantallet er 25 % lavere end begyndelsestallet for en ny kolonne.
10.1.5. Test af forkolonne
Forkolonnens (6.2) evne til at eliminere salte fra prøven kontrolleres regelmæssigt (mindst en gang pr. sekvens) ved at injicere 25 µl 0,05 % natriumchloridopløsning. Når der viser sig toppe, bør forkolonnen udskiftes.
10.2. Kørsel med standardopløsninger
I begyndelsen af hver analyserække injiceres 25 µl (6.5) standardopløsning 1 (8.1) og derefter standardopløsning 2 (8.2). Dette gentages for hver 10.-20. prøve og ligeledes som afslutning på sekvensen.
10.3. Kørsel med prøver
Der injiceres 25 µl af supernatanten (9.2) fra prøven.
11. BEREGNING OG ANGIVELSE AF RESULTATER
11.1. Kalibrering
Sædvanligvis anvendes tophøjder til at beregne resultaterne, men hvis signalet indeholder for meget støj, kan toparealer anvendes (kvantitativ bestemmelse ved hjælp af tophøjde påvirkes ikke i så høj grad af bestanddele, der forekommer i lavere koncentrationer, og som er delvis, men utilstrækkeligt adskilt fra lactosetoppen).
Softwaren (6.7) bør beregne en lineær kalibreringskurve, som skal gå gennem nulpunktet. Kurven kontrolleres for eventuel ikke-linearitet (åbenbar ikke-linearitet skyldes sandsynligvis en fejl ved fremstillingen af standardopløsningerne 1 (8.1) eller 2 (8.2), dårlig integration eller, mindre sandsynligt, dårligt fungerende injektor).
Som input anvendes de beregnede lactosekoncentrationer i mg/ml af standardopløsningerne 1 (8.1) eller 2 (8.2) som vandfri lactose.
Kalibreringslinjens hældning (RF) defineres af areal/koncentration i mg/ml.
11.2. Prøver
Resultatet af analysen udtrykkes i g/100 g og beregnes ved hjælp af softwaren (6.7) eller med følgende formel:
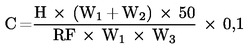
hvor:
|
C |
: |
lactosekoncentration i g/100 g pulver |
|
H |
: |
prøvens tophøjde for lactose |
|
RF |
: |
responsfaktor (eller hældning) for kalibreringen udtrykt i mV/mg/ml |
|
W1 |
: |
pulverprøvens vægt (g) (9.1) |
|
W2 |
: |
vægten (g) af det vand, der er tilsat pulverprøven (9.1) |
|
W3 |
: |
den rekonstituerede pulveropløsnings vægt (g) (9.2) |
|
50 |
: |
den anvendte målekolbes rumfang (9.2) |
|
0,1 |
: |
omregning af resultatet til g/100 g |
12. PRÆCISION
De værdier, der er fremkommet ved denne sammenlignende laboratorieprøvning, gælder ikke nødvendigvis for andre koncentrationsintervaller og matrixer end de angivne. Repeterbarheds- og reproducerbarhedsværdierne afledes fra en sammenlignende laboratorieprøvning, der gennemføres i overensstemmelse med ISO 5725 (4).
12.1. Repeterbarhed
De absolutte forskelle mellem to enkeltresultater, der er opnået med den samme metode på identisk prøvemateriale, på det samme laboratorium, udført af den samme person med det samme udstyr og med kort tidsinterval, bliver i højst 5 % af tilfældene større end xxx (bestemmes ved en ringtest) (5).
12.2. Reproducerbarhed
Den absolutte forskel mellem to enkeltresultater, der er opnået med den samme metode på identisk testmateriale på forskellige laboratorier udført af forskellige personer med forskelligt udstyr, bliver i højst 5 % af tilfældene større end 0,5 g/100 g bestemmes ved en ringtest).
13. LITTERATURHENVISNINGER
(1) J. Koops en C. Olieman, Netherlands Milk and Dairy Journal, 39 (1985) 89-106.
(2) D.A. Biggs en L. Szijarto, Journal of Dairy Science, 46 (1963) 1196.
(3) ISO 707 (IDF 50), Mælk og mælkeprodukter. Vejledning om prøveudtagning.
(4) ISO 5725-1, Nøjagtighed (korrekthed og præcision) af målemetoder og resultater. Del 1: Generelle principper og definitioner.
(5) ISO 5725-2, Nøjagtighed (korrekthed og præcision) af målemetoder og resultater. Del 2: Grundlæggende metode til bestemmelse af repeterbarhed og reproducerbarhed for en standardiseret målemetode.
BILAG XII
(Artikel 9)
PÅVISNING AF LØBEVALLE I SKUMMETMÆLKSPULVER TIL OFFENTLIG OPLAGRING VED HJÆLP AF HØJTRYKSVÆSKEKROMATOGRAFISK BESTEMMELSE AF KASEINMAKROPEPTIDINDHOLDET (HPLC-METODE)
1. FORMÅL OG ANVENDELSESOMRÅDE
Med denne metode kan det ved bestemmelse af kaseinmakropeptider påvises, om der er løbevalle til stede i skummetmælkspulver.
2. HENVISNING
International standard ISO 707 — Mælk og mælkeprodukter. Vejledning om prøveudtagning, i overensstemmelse med retningslinjerne i bilag I, punkt 2, litra c), sidste afsnit.
3. DEFINITION
Indholdet af løbevalletørstof defineres som indholdet udtrykt i vægtprocent ved bestemmelse af indholdet af kaseinmakropeptider ved den beskrevne fremgangsmåde.
4. PRINCIP
|
— |
Opløsning af skummetmælkspulveret, fjernelse af fedtstoffer og proteiner ved hjælp af trichloreddikesyre efterfulgt af centrifugering eller filtrering |
|
— |
Bestemmelse af mængden af kaseinmakropeptider (CMP) i supernatanten ved HPLC |
|
— |
Vurdering af resultatet i forhold til standardprøver af skummetmælkspulver, som ikke indeholder eller er tilsat en kendt mængde vallepulver. |
5. REAGENSER
Alle reagenser skal være af anerkendt analysekvalitet. Der anvendes destilleret vand eller vand af mindst tilsvarende renhed.
5.1. Opløsning af trichloreddikesyre
240 g trichloreddikesyre (CCl3COOH) opløses i vand og fortyndes til 1 000 ml. Opløsningen skal forblive klar og farveløs.
5.2. Elueringsvæske med pH 6,0
1,74 g kaliumhydrogenphosphat (K2HPO4), 12,37 g kaliumdihydrogenphosphat (KH2PO4) og 21,41 g natriumsulfat (Na2SO4) opløses i ca. 700 ml vand. Om nødvendigt indstilles pH til 6,0 ved hjælp af fortyndet phosphorsyre eller en kaliumhydroxidopløsning.
Der fyldes op til 1 000 ml med vand og blandes.
Bemærkning: Elueringsvæskens sammensætning kan opdateres for at efterkomme standarderne eller anbefalinger fra fabrikanten af kolonnepakningsmaterialet.
Før brugen filtreres elueringsvæsken gennem et membranfilter med porestørrelse 0,45 μm.
5.3. Opløsning til skylning af kolonnerne
En volumendel acetonitril (CH3CN) blandes med ni volumendele vand. Før brugen filtreres opløsningen gennem et membranfilter med porestørrelse 0,45 μm.
Bemærkning: Der kan til skylning anvendes andre opløsninger, der har baktericid virkning, og som ikke ændrer kolonnernes adskillelsesevne.
5.4. Standardprøver
5.4.1. Skummetmælkspulver, der opfylder kravene i denne forordning, benævnt [0].
5.4.2. Samme pulver tilsat 5 % (m/m) løbevallepulver med normal sammensætning, benævnt [5].
6. APPARATUR
6.1. Analysevægt
6.2. Centrifuge, der kan præstere en centrifugalkraft på 2 200 g, med tilhørende lukkede centrifugeglas på ca. 50 ml.
6.3. Mekanisk rysteapparat
6.4. Magnetomrører
6.5. Glastragte med diameter på ca. 7 cm
6.6. Filtrerpapir, mellemfint, med diameter på ca. 12,5 cm
6.7. Filtreringsudstyr af glas med tilhørende membranfiltre med porestørrelse 0,45 μm
6.8. Målepipetter på 10 ml (ISO 648, klasse A, eller ISO/R 835) eller et dispenseringssystem, som muliggør gennemløb af 10,0 ml på 2 min.
6.9. Dispenseringssystem, som muliggør gennemløb af 20,0 ml vand ved ca. 50 °C
6.10. Termostatstyret vandbad indstillet på 25 ± 0,5 °C
HPLC-udstyr bestående af:
6.11.1. Pumpe
6.11.2. Manuel eller automatisk injektionsenhed på 15-30 μl
6.11.3. To serieforbundne kolonner, TSK 2 000 SW (længde 30 cm, indvendig diameter 0,75 cm) eller kolonner af tilsvarende effektivitet (f.eks. single TSK 2 000-SWxl, single Agilent Technologies Zorbax GF 250), og en forkolonne (3 cm × 0,3 cm) pakket med I 125 eller et lige så effektivt materiale
6.11.4. Termostatstyret kolonneovn indstillet på 35 ± 1 °C
6.11.5. UV-detektor med variabel bølgelængde, hvormed der ved 205 nm kan måles med en følsomhed på 0,008 A
6.11.6. Integrator, der kan integrere over de enkelte toppe
Bemærkning: Der kan arbejdes med kolonner ved stuetemperatur, men adskillelsesevnen bliver i så fald lidt mindre. Temperatursvingningerne gennem en analyserække må her ikke overstige ± 5 °C.
7. PRØVEUDTAGNING
7.1. Prøveudtagningen sker efter metoden i den internationale standard ISO 707. Medlemsstaterne kan dog anvende en anden metode til prøveudtagning, hvis den følger principperne i ovennævnte standard.
7.2. Prøven opbevares under sådanne forhold, at der hverken kan ske ødelæggelse eller ændring af sammensætningen.
8. FREMGANGSMÅDE
8.1. Forberedelse af prøven
Pulveret overføres til en beholder, der er cirka dobbelt så stor som pulverets volumen, og som har et lufttæt låg. Beholderen lukkes straks. Mælkepulveret blandes omhyggeligt, ved at beholderen vendes flere gange rundt.
8.2. Analyseprøve
I et centrifugeglas (6.2) eller en tilproppet kolbe (50 ml) afvejes 2,000 ± 0,001 g analyseprøve.
8.3. Fjernelse af fedtstoffer og proteiner
8.3.1. Der tilsættes 20 ml 50 °C varmt vand til analyseprøven. Pulveret opløses ved omrøring i 5 min. med et rysteapparat (6.3). Reagensglasset anbringes i vandbadet (6.10), og det afventes, at der indtræder en ligevægt ved 25 °C.
8.3.2. I løbet af 2 min. tilsættes 10,0 ml trichloreddikesyreopløsning (5.1) på 25 °C under kraftig omrøring med magnetomrøreren (6.4). Glasset anbringes i vandbadet (6.10), hvori det henstår i 60 minutter.
8.3.3. Der centrifugeres (6.2) ved 2 200 g i 10 min. eller filtreres gennem papirfilter (6.6), idet de første 5 ml filtrat bortkastes.
8.4. Kromatografisk bestemmelse
8.4.1. Af supernatanten eller filtratet (8.3.3) indsprøjtes en nøjagtigt afmålt mængde på mellem 15 og 30 μl i HPLC-apparatet (6.11) under en gennemstrømningshastighed på 1,0 ml elueringsvæske (5.2) pr. minut.
Bemærkninger: 1. Den kan anvendes en anden gennemstrømningshastighed afhængigt af de anvendte kolonners indvendige diameter eller kolonnefabrikantens anvisninger.
Bemærkninger: 2. Elueringsvæsken (5.2) holdes på 85 °C under hele kromatograferingen for at holde den luftfri og fri for bakterievækst. Andre foranstaltninger med tilsvarende virkning er acceptable.
Bemærkninger: 3. Hver gang kromatograferingen afbrydes, skylles kolonnen med vand. Den må aldrig efterlades med elueringsvæske (5.2) i.
Skal kolonnerne stå ubenyttede hen i mere end 24 timer, skylles de først med vand og dernæst med opløsning (5.3) i mindst 3 timer med en gennemstrømningshastighed på 0,2 ml pr. minut.
8.4.2. Resultaterne af kromatograferingen af analyseprøven [E] foreligger i form af et kromatogram, hvor hver top identificeres ved sin retentionstid, RT, nemlig:
|
Top II: |
kromatogrammets anden top, hvis retentionstid er ca. 12,5 min. |
|
Top III: |
kromatogrammets tredje top, der svarer til CMP, og hvis retentionstid er 15,5 min. |
Valg af kolonne(r) kan påvirke de forskellige toppes retentionstid betydeligt.
Integratoren (6.11.6) beregner automatisk arealet A af hver top, nemlig:
|
AII: |
arealet af top II |
|
AIII: |
arealet af top III |
For at afsløre eventuelle afvigelser enten som følge af, at apparaturet eller kolonnerne ikke har fungeret tilfredsstillende, eller på grund af den analyserede prøves oprindelse eller art må alle kromatogrammer bedømmes visuelt, før en kvantitativ tolkning påbegyndes.
I tvivlstilfælde gentages analysen.
8.5. Kalibrering
8.5.1. Med standardprøverne (5.4) følges den i punkt 8.2 til 8.4.2 beskrevne fremgangsmåde nøje.
Der benyttes frisk fremstillede opløsninger, da CMP nedbrydes i 8 % trichloreddikesyreopløsning. Indholdet heraf falder med ca. 0,2 % pr. time ved 30 °C.
8.5.2. Før kromatografering af prøverne konditioneres kolonnerne ved gentagen indsprøjtning af opløsningen (8.5.1) af standardprøven (5.4.2), indtil den top, der svarer til CMP, har konstant areal og retentionstid.
8.5.3. Kalibreringsfaktorerne R bestemmes ved at indsprøjte filtrat (8.5.1) i samme mængde som prøverne.
9. ANGIVELSE AF RESULTATER
9.1. Formler og beregning
9.1.1. Beregning af responsfaktorerne R
|
Top II: |
RII = 100/(AII[0]) |
hvor:
|
RII |
= |
responsfaktoren for top II |
|
AII [0] |
= |
det areal af top II for standardprøven [0], som er fremkommet under 8.5.3 |
|
Top III: |
RIII = W/(AIII[5] – AIII[0]) |
hvor:
|
RIII |
= |
responsfaktoren for top III |
|
AIII [0] og AIII [5] |
= |
de arealer af top III for henholdsvis standardprøve [0] og [5], som er fremkommet under 8.5.3 |
|
W |
= |
indholdet af valle i standardprøven [5], nemlig 5 |
9.1.2. Beregning af de relative toparealer for prøven [E]
SII[E] = RII × AII[E]
SIII[E] = RIII × AIII[E]
SIV[E] = RIV × AIV[E]
hvor:
|
SII [E], SIII [E], SIV [E] |
= |
de relative arealer af henholdsvis top II, III og IV for prøven [E] |
|
AII [E], AIII [E] |
= |
de arealer af henholdsvis top II og III for prøven [E], som er fremkommet under 8.4.2 |
|
RII, RIII |
= |
de under 9.1.1 beregnede responsfaktorer |
9.1.3. Beregning af de relative retentionstider for top III for prøven [E] RRTIII[E] = (RTIII[E])/(RTIII[5])
hvor:
|
RRTIII [E] |
= |
den relative retentionstid for top III for prøven [E] |
|
RTIII [E] |
= |
retentionstiden for top III for prøven [E], som er fremkommet under 8.4.2 |
|
RTIII [5] |
= |
retentionstiden for top III for standardprøven [5], som er fremkommet under 8.5.3 |
9.1.4. Det er vist eksperimentelt, at den relative retentionstid for top III, RRTIII [E], er ligefrem proportional med vallepulverindholdet op til 10 %.
|
— |
ved et indhold på > 5 % er RRTIII [E] < 1,000 |
|
— |
ved et indhold på ≤ 5 % er RRTIII [E] ≥ 1,000. |
Den tilladte usikkerhed på RRTIII er ±0,002.
Normalt afviger RRTIII [0]-værdien kun lidt fra 1,034. Alt efter kolonnernes tilstand kan denne værdi nærme sig, men skal altid være større end 1,000.
9.2. Beregning af det procentvise indhold af løbevallepulver i prøven
W = SIII[E] – [1, 3 + (SIII[0] – 0, 9)]
hvor:
|
W |
= |
indholdet af løbevalle i prøven [E], i % m/m |
|
SIII [E] |
= |
det relative areal af top III for analyseprøven [E], som er fremkommet under 9.1.2 |
|
1,3 |
= |
det relative middelareal af top III udtrykt i g løbevalle pr. 100 g bestemt i uforfalsket skummetmælkspulver af forskellig oprindelse. Denne værdi er bestemt eksperimentelt |
|
SIII [0] |
= |
det relative areal af top III, som er lig med RIII × AIII [0]. Disse størrelser er bestemt under henholdsvis 9.1.1 og 8.5.3 |
|
(SIII [0] – 0,9) |
= |
korrektionen af det relative middelareal 1,3, når SIII [0] ikke er 0,9. Det relative middelareal af top III for standardprøven [0] er bestemt eksperimentelt til 0,9. |
9.3. Metodens nøjagtighed
9.3.1. Repeterbarhed
Forskellen mellem resultaterne af to bestemmelser, der er udført samtidig eller med kort tids mellemrum af samme person, med samme apparatur og på samme prøve, må ikke overstige 0,2 % m/m.
9.3.2. Reproducerbarhed
Forskellen mellem to resultater, som to forskellige laboratorier er nået frem til med samme prøve, må ikke overstige 0,4 % m/m.
9.4. Fortolkning
9.4.1. Der er ikke valle til stede, hvis det relative areal af top III, SIII [E], udtrykt i gram løbevalle pr. 100 gram produkt er ≤ 2,0 + (SIII[0] – 0,9), hvor
|
2,0 |
= |
er den højest tilladte værdi for det relative areal af top III; der er her taget hensyn til det relative areal af top III, nemlig 1,3, usikkerheden som følge af variationer i skummetmælkspulverets sammensætning og metodens reproducerbarhed (9.3.2) |
|
(SIII [0] – 0,9) |
= |
er korrektionen, når arealet SIII [0] afviger fra 0,9 (jf. punkt 9.2) |
9.4.2. Hvis det relative areal af top III, SIII [E] er > 2,0 + (SIII[0] – 0,9) og det relative areal af top II, SII [E] samtidig er ≤ 160, beregnes indholdet af løbevalle som angivet i punkt 9.2.
Hvis det relative areal af top III, SIII [E] er > 2,0 + (SIII[0] – 0,9) og det relative areal af top II, SII [E] samtidig er ≤ 160, bestemmes det samlede proteinindhold (P %); herefter undersøges figur 1 og 2.
9.4.3.1. De tal, der fremkommer efter analyse af prøver af uforfalsket skummetmælkspulver med et højt samlet proteinindhold, er anført i figur 1 og 2.
Den fuldt optrukne linje svarer til regressionslinjen, for hvilken koefficienterne beregnes ved hjælp af de mindste kvadraters metode.
Med den stiplede linje fastsættes den øvre grænse for det relative areal af top III med en sandsynlighed for ikke at blive overskredet i 90 % af tilfældene.
Ligningerne for de stiplede linjer i figur 1 og 2 er henholdsvis lig med:
|
SIII = 0,376 P % – 10,7 |
(figur 1) |
|
SIII = 0,0123 SII [E] + 0,93 |
(figur 2), |
hvor
|
SIII |
= |
er det relative areal af top III beregnet enten på grundlag af det samlede proteinindhold eller på grundlag af det relative areal af top SII [E] |
|
P % |
= |
er det samlede proteinindhold udtrykt som vægtprocent |
|
SII [E] |
= |
er det i punkt 9.1.2 beregnede relative areal af prøven |
Disse ligninger svarer til det i punkt 9.2 anførte tal 1,3.
Forskellen (T1 og T2) mellem det fundne relative areal SIII [E] og det relative areal SIII fremgår af følgende ligninger: T1 = SIII[E] – [(0,376 P % – 10,7) + (SIII[0] – 0,9)]T2 = SIII[E] – [(0,0123 SII[E] + 0,93) + (SIII[0] – 0,9)]
|
Hvis T1 og/eller T2 |
er mindre end eller lig med nul, kan et indhold af løbevalle ikke påvises. |
|
Hvis T1 og T2 |
er større end nul, kan det sluttes, at der er løbevalle til stede. |
Løbevalleindholdet beregnes ved hjælp af følgende formel: W = T2+0,91
hvor:
0,91 svarer til den lodrette afstand mellem den fuldt optrukne linje og den stiplede linje.
Skummetmælkspulver

Skummetmælkspulver

BILAG XIII
(Artikel 9)
BESTEMMELSE AF LØBEVALLETØRSTOF I SKUMMETMÆLKSPULVER OG BLANDINGER I HENHOLD TIL FORORDNING (EF) Nr. 2799/1999
1. FORMÅL: PÅVISNING AF TILSÆTNING AF LØBEVALLETØRSTOF TIL FØLGENDE PRODUKTER:
|
a) |
Skummetmælkspulver som defineret i artikel 2 i forordning (EF) nr. 2799/1999 |
|
b) |
Blandinger som defineret i artikel 4 i forordning (EF) nr. 2799/1999. |
2. REFERENCER: INTERNATIONAL STANDARD ISO 707
3. DEFINITION
Indholdet af løbevalletørstof defineres som indholdet udtrykt i vægtprocent ved bestemmelse af indholdet af kaseinmakropeptider ved den beskrevne fremgangsmåde.
4. PRINCIP
Indholdet af kaseinomakropeptider bestemmes i overensstemmelse med bilag XII. Prøver, der giver et positivt resultat, analyseres for kaseinmakropeptid A ved højtryksvæskekromatografi med omvendt fase (HPLC-metoden). Alternativt analyseres prøver direkte ved HPCL-procedure med omvendt fase. Vurdering af resultatet i forhold til standardprøver af skummetmælkspulver med og uden et kendt procentvist indhold af vallepulver. Resultater over 1 % (m/m) er tegn på, at der er løbevalletørstof til stede.
5. REAGENSER
Alle reagenser skal være af anerkendt analysekvalitet. Der anvendes destilleret vand eller vand af mindst tilsvarende renhed. Acetonitril skal være af spektroskopisk kvalitet eller HPLC-kvalitet.
De reagenser, der er nødvendige for metoden, er beskrevet i bilag XII til nærværende forordning.
Reagenser til HPLC med omvendt fase.
5.1. Opløsning af trichloreddikesyre
240 g trichloreddikesyre (CCl3COOH) opløses i vand og fortyndes til 1 000 ml. Opløsningen skal forblive klar og farveløs.
5.2. Elueringsvæske A og B
Elueringsvæske A: 150 ml acetonitril (CH3CN), 20 ml isopropanol (CH3CHOHCH3) og 1,00 ml trifluoreddikesyre (TFA, CF3COOH) blandes i en 1 000 ml målekolbe. Derefter fyldes op til 1 000 ml med vand.
Elueringsvæske B: 550 ml acetonitril, 20 ml isopropanol og 1,00 ml TFA blandes i en 1 000 ml målekolbe. Derefter fyldes op til 1 000 ml med vand. Før brugen filtreres elueringsvæsken gennem et membranfilter med porestørrelse 0,45 μm.
5.3. Opbevaring af kolonnen
Efter analyserne skylles kolonnen med elueringsvæske B (under anvendelse af en gradient), og derefter skylles den med acetonitril (under anvendelse af en gradient i 30 minutter). Kolonnen opbevares i acetonitril.
5.4. Standardprøver
5.4.1. Skummetmælkspulver, der opfylder kravene til offentlig oplagring, benævnt [0].
5.4.2. Samme pulver tilsat 5 % (m/m) løbevallepulver med normal sammensætning, benævnt [5].
5.4.3. Samme pulver tilsat 50 % (m/m) løbevallepulver med normal sammensætning, benævnt (1).
6. APPARATUR
Det apparatur, der er nødvendigt for metoden, er beskrevet i bilag XII til nærværende forordning.
6.1. Analysevægt
6.2. Centrifuge, der kan præstere en centrifugalkraft på 2 200 g, med tilhørende lukkede centrifugeglas på ca. 50 ml.
6.3. Mekanisk rysteapparat
6.4. Magnetomrører
6.5. Glastragte med diameter på ca. 7 cm
6.6. Filtrerpapir, mellemfint, med diameter på ca. 12,5 cm
6.7. Filtreringsudstyr af glas med tilhørende membranfiltre med porestørrelse 0,45 μm
6.8. Målepipetter på 10 ml (ISO 648, klasse A, eller ISO/R 835) eller et dispenseringssystem, som muliggør gennemløb af 10,0 ml på 2 min.
6.9. Dispenseringssystem, som muliggør gennemløb af 20,0 ml vand ved ca. 50 °C
6.10. Termostatstyret vandbad indstillet på 25 ± 0,5 °C
HPLC-udstyr bestående af:
6.11.1. Binært gradientpumpesystem
6.11.2. Manuel eller automatisk injektionsenhed på 100 μl
6.11.3. En Agilent Technologies Zorbax 300 SB-C3-kolonne (længde: 25 cm, indvendig diameter: 0,46 cm) eller en tilsvarende bredporet siliciumbaseret kolonne med omvendt fase
6.11.4. Termostatstyret kolonneovn indstillet på 35 ± 1 °C
6.11.5. UV-detektor med variabel bølgelængde, hvormed der ved 210 nm kan måles med en følsomhed på 0,02 Å (om nødvendigt kan en større bølgelængde på op til 220 nm benyttes).
6.11.6. Integrator, der kan integrere til en fælles basislinje eller over de enkelte toppe
Bemærkning: Der kan arbejdes med kolonnen ved stuetemperatur, hvis stuetemperaturen ikke svinger med mere end 1 °C. I modsat fald vil der være for stor variation i retentionstiden for CMPA.
7. PRØVEUDTAGNING
7.1. Prøveudtagningen sker efter metoden i den internationale standard ISO 707. Medlemsstaterne kan dog anvende en anden metode til prøveudtagning, hvis den følger principperne i ovennævnte standard.
7.2. Prøven opbevares under sådanne forhold, at der hverken kan ske ødelæggelse eller ændring af sammensætningen.
8. FREMGANGSMÅDE
8.1. Forberedelse af prøven
Pulveret overføres til en beholder, der er cirka dobbelt så stor som pulverets volumen, og som har et lufttæt låg. Beholderen lukkes straks. Mælkepulveret blandes omhyggeligt ved, at beholderen vendes flere gange rundt.
8.2. Analyseprøve
I et centrifugeglas (6.2) eller en tilproppet kolbe (50 ml) afvejes 2,00 ± 0,001 g analyseprøve.
Bemærkning: Når det drejer sig om blandinger, afvejes en sådan mængde af analyseprøven, at den affedtede prøve svarer til 2,00 g.
8.3. Fjernelse af fedtstoffer og proteiner
8.3.1. Der tilsættes 20 ml 50 °C varmt vand til analyseprøven. Pulveret opløses ved omrøring med et rysteapparat (6.3) i 5 minutter eller i 30 minutter, såfremt det drejer sig om syrnet kærnemælk. Reagensglasset anbringes i vandbadet (6.10), og det afventes, at der indtræder en ligevægt ved 25 °C.
8.3.2. I løbet af 2 min. tilsættes 10,0 ml trichloreddikesyreopløsning (5.1) på 25 °C under kraftig omrøring med magnetomrøreren (6.4). Glasset anbringes i vandbadet (6.10), hvori det henstår i 60 minutter.
8.3.3. Der centrifugeres (6.2) ved 2 200 g i 10 min. eller filtreres gennem papirfilter (6.6), idet de første 5 ml filtrat bortkastes.
8.4. Kromatografisk bestemmelse
8.4.1. Der foretages en HPLC-analyse som beskrevet i bilag XII. Hvis der opnås et negativt resultat, indeholder den analyserede prøve ikke løbevalle i påviselige mængder. Hvis der opnås et positivt resultat, skal den nedenfor beskrevne HPLC-metode med omvendt fase anvendes. Alternativt kan HPLC-metoden med omvendt fase anvendes direkte. Tilstedeværelse af syrnet kærnemælkspulver kan give anledning til falsk positive resultater ved anvendelse af den metode, der er beskrevet i bilag XII. HPLC-metoden med omvendt fase udelukker denne mulighed.
8.4.2. Før HPLC-analysen med omvendt fase gennemføres, må gradientbetingelserne optimeres. En retentionstid på 26 minutter ±2 minutter for CMPA er optimal for gradientsystemer med et dødvolumen på ca. 6 ml (volumenet fra det punkt, hvor opløsningerne blandes, til det punkt, hvor injektionssløjfen slutter). For gradientsystemer med et mindre dødvolumen (f.eks. 2 ml) bruges 22 minutter som optimal retentionstid.
Der anvendes opløsninger af standardprøverne (5.4) uden og med 50 % løbevalle.
Af supernatanten eller filtratet (8.3.3) indsprøjtes 100 μl i HPLC-apparatet under anvendelse af de kontrolgradientvilkår, der fremgår af tabel 1.
Tabel 1
Kontrolgradientvilkår for optimering af kromatografien
|
Tid (min.) |
Gennemstrømning (ml/min.) |
% A |
% B |
Kurve |
|
Start |
1,0 |
90 |
10 |
* |
|
27 |
1,0 |
60 |
40 |
lineær |
|
32 |
1,0 |
10 |
90 |
lineær |
|
37 |
1,0 |
10 |
90 |
lineær |
|
42 |
1,0 |
90 |
10 |
lineær |
Sammenligning af de to kromatogrammer vil vise beliggenheden af toppen af CMPΑ.
Ved anvendelse af nedenstående formel kan den oprindelige sammensætning af den opløsning, der skal bruges til den normale gradient (se 8.4.3), beregnes: % B = 10 – 2,5 + (13,5 + (RTcmpA – 26) / 6) × 30 / 27 % B = 7,5 + (13,5 + (RTcmpA – 26) / 6) × 1,11
hvor:
|
RTcmpA |
: |
retentionstiden for CMPΑ ved anvendelse af kontrolgradienten |
|
10 |
: |
den oprindelige % B af kontrolgradienten |
|
2,5 |
: |
% B midtvejs minus % B ved begyndelsen ved anvendelse af den normale gradient |
|
13,5 |
: |
midtvejspunkt for kontrolgradienten |
|
26 |
: |
den krævede retentionstid for CMPΑ |
|
6 |
: |
hældningskoefficient for kontrolgradienten og den normale gradient |
|
30 |
: |
% B på begyndelsestidspunktet minus % B efter 27 minutter ved anvendelse af kontrolgradienten |
|
27 |
: |
varighed for kontrolgradienten. |
8.4.3. Der udtages opløsninger af analyseprøverne.
Af supernatanten eller filtratet (8.3.3) indsprøjtes en nøjagtigt afmålt mængde på 100 μl i HPLC-apparatet under en gennemstrømningshastighed på 1,0 ml elueringsvæske (5.2) pr. minut.
Sammensætningen af elueringsvæsken ved analysens begyndelse opnås fra 8.4.2. Den er normalt tæt på A:B = 76:24 (5.2). Umiddelbart efter indsprøjtningen startes en lineær gradient, således at B bliver 5 % højere efter 27 minutter. Derefter startes en lineær gradient, hvorved elueringsvæskens indhold af B kommer op på 90 % i løbet af 5 minutter. Denne sammensætning opretholdes i 5 minutter, hvorefter sammensætningen i løbet af 5 minutter ændres ved hjælp af en lineær gradient til den oprindelige sammensætning. Afhængigt af pumpesystemets indre volumen kan den næste indsprøjtning foretages 15 minutter efter, at de oprindelige betingelser er nået.
Bemærkninger 1. Retentionstiden for CMPA bør være 26 minutter ± 2 minutter. Dette kan opnås ved at ændre begyndelses- og slutbetingelserne for den første gradient. Imidlertid skal forskellen i % B for begyndelses- og slutbetingelserne for den første gradient fortsat være 5 % B.
Bemærkninger 2. Elueringsvæskerne må afgasses tilstrækkeligt og skal forblive afgassede. Dette er vigtigt for gradientpumpesystemets tilfredsstillende virkning. Standardafvigelsen for retentionstiden for CMPA-toppen skal være på under 0,1 min. (n = 10).
Bemærkninger 3. Efter hver femte prøve skal referenceprøven (5) indsprøjtes og benyttes til beregning af en ny responsfaktor R (9.1.1).
8.4.4. Resultaterne af kromatograferingen af analyseprøven [E] foreligger i form af et kromatogram, hvor CMPA-toppen identificeres ved sin retentionstid på ca. 26 minutter.
Integratoren (6.11.6) beregner automatisk CMPA-toppens højde H: Basislinjens beliggenhed skal kontrolleres for hvert kromatogram. Analysen eller integrationen skal gentages, hvis basislinjens beliggenhed ikke er korrekt.
Bemærkning: Hvis CMPA-toppen er tilstrækkelig adskilt fra andre toppe, bør der anvendes dal-til-dal integration, ellers anvendes linjer vinkelret på en fælles basislinje, hvis startpunkt skal ligge tæt på CMPA-toppen (dvs. ikke ved t = 0 min!). For standardprøven og prøverne anvendes samme integrationstype, og ved anvendelse af fælles basislinje kontrolleres det, at den er den samme for prøverne og standardprøven.
For at afsløre eventuelle afvigelser enten som følge af, at apparaturet eller kolonnen ikke har fungeret tilfredsstillende, eller på grund af den analyserede prøves oprindelse eller art, skal alle kromatogrammer bedømmes visuelt før en kvantitativ tolkning påbegyndes. I tvivlstilfælde gentages analysen.
8.5. Kalibrering
8.5.1. Med standardprøverne (5.4.1 og 5.4.2) følges den i punkt 8.2 til 8.4.4 beskrevne fremgangsmåde nøje. Der benyttes frisk fremstillede opløsninger, da CMP nedbrydes i 8 % trichloreddikesyreopløsning ved stuetemperatur. Ved 4 °C forbliver opløsningen stabil i 24 timer. Skal der foretages en lang række analyser, er det ønskeligt at anvende en kølet prøvebakke i den automatiske injektor.
Bemærkning: 8.4.2 kan udelades, hvis % B på begyndelsesbetingelserne er kendt fra tidligere analyser.
Kromatogrammet for referenceprøven [5] skal svare til figur 1. På denne figur er der inden CMPA-toppen to små toppe. Det er vigtigt at opnå en lignende separation.
8.5.2. Før den kromatografiske bestemmelse af prøverne indsprøjtes 100 μl af standardprøven uden løbevalle [0] (5.4.1).
Kromatogrammet må ikke indeholde nogen top ved retentionstiden for CMPA-toppen.
8.5.3. Responsfaktorerne R bestemmes ved at indsprøjte filtrat (8.5.1) i samme mængde som prøverne.
9. ANGIVELSE AF RESULTATER
9.1. Formler og beregning
9.1.1. Beregning af responsfaktoren R:
CMPA-toppen: R = W/H
hvor:
|
R |
= |
responsfaktoren for CMPA-toppen |
|
H |
= |
højden af CMPA-toppen |
|
W |
= |
mængden af valle i standardprøven [5]. |
9.2. Beregning af det procentvise indhold af løbevallepulver i prøven
W(E) = R × H(E)
hvor:
|
W(E) |
= |
indholdet af løbevalle i prøven [E], i % m/m |
|
R |
= |
responsfaktoren for CMPA-toppen (9.1.1) |
|
H(E) |
= |
højden af CMPA-toppen af prøven [E]. |
Hvis W(E) er større end 1 % og forskellen mellem retentionstiden og retentionstiden for standardprøven [5] er mindre end 0,2 min., er der løbevalletørstof til stede.
9.3. Metodens nøjagtighed
9.3.1. Repeterbarhed
Forskellen mellem resultaterne af to bestemmelser, der er udført samtidig eller med kort tids mellemrum af samme person, med samme apparatur og på samme prøve, må ikke overstige 0,2 % m/m.
9.3.2. Reproducerbarhed
Ikke fastslået.
9.3.3. Linearitet
For mellem 0 og 16 % løbevalle skal der opnås en lineær sammenhæng med en korrelationskoefficient > 0,99.
9.4. Fortolkning
Grænsen på 1 % er fastsat i overensstemmelse med bestemmelserne i forordning (EØF) nr. 214/2001, bilag XIX, punkt 9.2 og 9.4.1, som omfatter usikkerheden i forbindelse med reproducerbarhed.
Figur 1
Ni –4,6 standard

(1) Løbevallepulver med normal sammensætning og skummetmælkspulver med tilsætning kan erhverves fra Nizo, Kernhemseweg 2, PO Box 20, NL-6710 BA Ede, Nederlandene. Pulver, der giver samme resultater som Nizo-pulveret, kan dog også bruges.
BILAG XIV
(Artikel 10)
SKUMMETMÆLKSPULVER: KVANTITATIV BESTEMMELSE AF PHOSPHATIDYLSERIN- OG PHOSPHATIDYLETHANOLAMININDHOLD
Metode: HPLC-metode i omvendt fase
1. FORMÅL OG ANVENDELSESOMRÅDE
Denne metode er en procedure for kvantitativ bestemmelse af phosphatidylserin (PS) og phosphatidylethanolamin (PE) i skummetmælkspulver (SMP) og egner sig til påvisning af kærnemælksklumper i SMP.
2. DEFINITION
Indhold af PS + PE: massefordelingen af stof bestemt ved anvendelse af denne procedure. Resultatet udtrykkes som milligram phosphatidylethanolamindipalmitoyl (PEDP) pr. 100 g pulver.
3. PRINCIP
Ekstraktion af aminophospholipider fra rekonstitueret mælkepulver ved hjælp af methanol. Bestemmelse af PS og PE som o-phthaldialdehyd (OPA) derivater ved HPLC i omvendt fase og ved fluorescenspåvisning. Kvantificering af PS- og PE-indholdet i analyseprøven ved sammenholdelse med en standardprøve indeholdende en kendt mængde PEDP.
4. REAGENSER
Alle reagenser skal være af anerkendt analysekvalitet. Der anvendes destilleret vand eller vand af mindst tilsvarende renhed, medmindre andet er angivet.
4.1. Standardmateriale: PEDP, mindst 99 % rent
Bemærkning: Standardmateriale skal opbevares ved –18 °C.
4.2. Reagenser til forberedelse af standardprøve og analyseprøve
4.2.1. Methanol af HPLC-renhed
4.2.2. Chloroform af HPLC-renhed
4.2.3. Tryptamin-monohydrochlorid
4.3. Reagenser til o-phthaldialdehyd-derivatisering
4.3.1. Natriumhydroxid, 12 M opløsning i vand
4.3.2. Borsyre, 0,4 M opløsning i vand, indstillet til pH 10,0 med natriumhydroxid (4.3.1)
4.3.3. 2-mercaptoethanol
4.3.4. o-phthaldialdehyd (OPA)
4.4. HPLC-elueringsvæsker
4.4.1. Elueringsvæsker fremstilles ved anvendelse af reagenser af HPLC-renhed.
4.4.2. Vand af HPLC-renhed
4.4.3. Methanol af fluorimetri-testet renhed
4.4.4. Tetrahydrofuran
4.4.5. Natriumdihydrogenphosphat
4.4.6. Natriumacetat
4.4.7. Eddikesyre.
5. APPARATUR
5.1. Analysevægt, der kan veje med en nøjagtighed på 1 mg, og som kan aflæses med en nøjagtighed på 0,1 mg
5.2. Bægerglas på 25 og 100 ml
5.3. Pipetter på 1 og 10 ml
5.4. Magnetomrører
5.5. Målepipetter på 0,2, 0,5 og 5 ml
5.6. Målekolber på 10, 50 og 100 ml
5.7. Sprøjter på 20 og 100 μl
5.8. Ultralydsbad
5.9. Centrifuge, som arbejder ved 27 000 × g
5.10. Prøveglas på ca. 5 ml
5.11. Måleglas på 25 ml
5.12. pH-meter med en nøjagtighed på 0,1 pH-enhed
HPLC-udstyr
5.13.1. Gradient-pumpesystem, i stand til at arbejde ved 1,0 ml/min. ved 200 bar
5.13.2. Autosampler med mulighed for derivatisering
5.13.3. Kolonneopvarmningsenhed, som kan holde kolonnens temperatur på 30 °C ± 1 °C
5.13.4. Fluorescensdetektor, der kan fungere på en exciteringsbølgelængde på 330 nm og en emissionsbølgelængde på 440 nm.
5.13.5. Integrator eller databehandlingsprogrammel, som er i stand til at måle toparealer
5.13.6. En Lichrosphere — 100 kolonne (250 × 4,6 mm) eller en tilsvarende kolonne pakket med octadecylisan (C 18) med en partikelstørrelse på 5 μm.
6. PRØVEUDTAGNING
Prøveudtagning skal ske i overensstemmelse med ISO standard 707.
7. FREMGANGSMÅDE
7.1. Forberedelse af den interne standardopløsning
7.1.1. 30,0 ± 0,1 mg tryptamin-monohydrochlorid (4.2.3) afvejes i en 100 ml målekolbe (5.6) og der fyldes op til mærket med methanol (4.2.1).
7.1.2. Med en pipette (5.3) overføres 1 ml af denne opløsning til en 10 ml målekolbe (5.6), og der fyldes op til mærket med methanol (4.2.1) for at opnå en 0,15 mM tryptaminkoncentration.
7.2. Forberedelse af analyseopløsningen
7.2.1. 1,000 ± 0,001 g af SMP-prøven i et 25 ml bægerglas (5.2). Der tilsættes 10 ml destilleret vand ved 40 °C ± 1 °C med pipette (5.3) og omrøres med magnetomrører (5.4) i 30 minutter for at opløse alle klumper.
7.2.2. 0,2 ml af den rekonstituerede mælk pipetteres (5.5) over i en 10 ml målekolbe (5.6), hvorefter der med en sprøjte (5.7) tilsættes 100 μl 0,15 mM tryptaminopløsning (7.1) og fyldes op til mærket med methanol (4.2.1). Der blandes omhyggeligt ved at vende kolben på hovedet flere gange; anbringelse i ultralydsbad (5.8) i 15 min.
7.2.3. Derefter centrifugeres (5.9) ved 27 000 g × g i 10 min., og supernatanten indsamles i et prøveglas (5.10).
Bemærkning: Analyseopløsningen skal opbevares ved 4 °C, til HPLC-analysen er gennemført.
7.3. Forberedelse af den eksterne standardopløsning
7.3.1. 55,4 mg PEDP (4.1) afvejes i en 50 ml målekolbe (5.6), og der tilsættes ca. 25 ml chloroform (4.2.2) ved anvendelse af et måleglas (5.11). Den tilproppede kolbe opvarmes til 50 °C ± 1 °C, og der blandes omhyggeligt, indtil PEDP opløses. Derefter afkøles kolben til 20 °C, og der fyldes op til mærket med methanol (4.2.1) og blandes ved at vende kolben på hovedet flere gange.
7.3.2. 1 ml af denne opløsning pipetteres (5.3) over i en 100 ml målekolbe (5.6), og der fyldes op til mærket med methanol (4.2.1). 1 ml af denne opløsning pipetteres (5.3) over i en 10 ml målekolbe (5.6), hvorefter der tilsættes 100 μl (5.7) 0,15 mM tryptaminopløsning (7.1) og fyldes op til mærket med methanol (4.2.1). Der blandes ved at vende kolben på hovedet flere gange.
Bemærkning: Standardanalyseopløsningen skal opbevares ved 4 °C indtil HPLC-analysen.
7.4. Forberedelse af derivateringsreagenset
25,0 ± 0,1 mg OPA (4.3.4) afvejes i en 10 ml målekolbe (5.6), hvorefter der tilsættes 0,5 ml (5.5) methanol (4.2.1) og blandes omhyggeligt for at opløse OPA. Der fyldes op til mærket med borsyreopløsning (4.3.2) og tilsættes 20 μl 2-mercaptoethanol (4.3.3) med en sprøjte (5.7).
Bemærkning: Derivateringsreagenset skal opbevares ved 4 °C i et brunt prøveglas. Det er holdbart i en uge.
7.5. Bestemmelse ved HPLC
7.5.1. Elueringsvæsker (4.4)
Væske A: 0,3 mM natriumdihydrogenphosphat- og 3 mM natriumacetatopløsning (indstillet til pH 6,5 ±0,1 med eddikesyre), methanol: tetrahydrofuran = 558:440:2 (v/v/v)
Væske B: methanol
7.5.2. Foreslået elueringsgradient:
|
Tid (min.) |
Væske A (%) |
Væske B (%) |
Gennemstrømningshastighed (ml/min.) |
|
Start |
40 |
60 |
0 |
|
0,1 |
40 |
60 |
0,1 |
|
5,0 |
40 |
60 |
0,1 |
|
6,0 |
40 |
60 |
1,0 |
|
6,5 |
40 |
60 |
1,0 |
|
9,0 |
36 |
64 |
1,0 |
|
10,0 |
20 |
80 |
1,0 |
|
11,5 |
16 |
84 |
1,0 |
|
12,0 |
16 |
84 |
1,0 |
|
16,0 |
10 |
90 |
1,0 |
|
19,0 |
0 |
100 |
1,0 |
|
20,0 |
0 |
100 |
1,0 |
|
21,0 |
40 |
60 |
1,0 |
|
29,0 |
40 |
60 |
1,0 |
|
30,0 |
40 |
60 |
0 |
Bemærkning: Elueringsgradienten vil kunne skulle ændres lidt for at opnå den opløsning, der er vist i figur 1.
Kolonnetemperatur: 30 °C.
7.5.3. Injektionsvolumen: 50 μl derivateringsreagens og 50 μl prøveopløsning.
7.5.4. Kolonneligevægt
Ved daglig opstart af systemet gennemskylles kolonnen med 100 % væske B i 15 min., hvorefter der indstilles A: 40:60 og bringes i ligevægt ved 1 ml/min. i 15 min. Der foretages en blindprøve ved injektion af methanol (4.2.1).
Bemærkning: Forud for opbevaring over længere tid gennemskylles kolonnen med methanol: chloroform = 80: 20 (v/v) i 30 min.
7.5.5. Indholdet af PS + PE i analyseprøven bestemmes.
7.5.6. Rækken af kromatografianalyser gennemføres under overholdelse af et konstant tidsforhold mellem de enkelte analyserunder for at opnå konstante retentionstider. Den eksterne standardopløsning (7.3) indsprøjtes for hver 5.-10. analyseopløsning for at beregne responsfaktoren.
Bemærkning: Kolonnen skal renses ved gennemskylning med 100 % væske B (7.5.1) i mindst 30 min. for hver 20.-25. analyserunde.
7.6. Integrationsmåde
7.6.1. PEDP-top
PEDP elueres som en enkelt top. Toparealet bestemmes ved dal-til-dal-integration.
7.6.2. Tryptamin-top
Tryptamin elueres som en enkelt top (figur 1). Toparealet bestemmes ved dal-til-dal-integration.
7.6.3. Grupper af PS- og PE-toppe
Under de beskrevne betingelser (figur 1) elueres PS som to delvis uopløste hovedtoppe efter en mindre top. PE elueres som 3 delvis uopløste hovedtoppe. Hele arealet for hver samling af toppe bestemmes ved at sætte basislinjen som rapporteret i figur 1.
8. BEREGNING OG ANGIVELSE AF RESULTATER
Indholdet af PS og PE i analyseprøven beregnes således: C = 55,36 × ((A2)/(A1)) × ((T1)/(T2))
hvor:
|
C |
= |
indhold af PS eller PE (mg/100 g pulver) i analyseprøven |
|
A1 |
= |
topareal af PEDP i standardprøveopløsningen (7.3) |
|
A2 |
= |
topareal af PS eller PE i analyseopløsningen (7.2) |
|
T1 |
= |
topareal af tryptamin i standardprøveopløsningen (7.3) |
|
T2 |
= |
topareal af tryptamin i analyseprøveopløsningen (7.2). |
9. METODENS NØJAGTIGHED
Bemærkning: Værdierne for repeterbarhed blev beregnet efter den internationale IDF-standard (1). Den foreløbige reproducerbarhedsgrænse er beregnet efter standardens bilag III, punkt b).
9.1. Repeterbarhed
Den relative standardafvigelse for repeterbarheden, som er et udtryk for variationen i uafhængige analyseresultater, der er opnået af samme person under anvendelse af samme apparatur på samme betingelser på samme analyseprøve inden for et kort tidsinterval, må ikke overstige 2 %. Hvis der opnås to bestemmelser under disse betingelser, bør den relative forskel mellem de to resultater ikke overstige 6 % af det aritmetiske gennemsnit af resultaterne.
9.2. Reproducerbarhed
Hvis to bestemmelser foretages af laboranter i forskellige laboratorier under anvendelse af forskelligt apparatur på forskellige betingelser for analyse og samme analyseprøve, må den relative forskel mellem de to resultater ikke overstige 11 % af det aritmetiske gennemsnit af resultaterne.
10. LITTERATURHENVISNINGER
10.1. Resmini P., Pellegrino L., Hogenboom J.A., Sadini V., Rampilli M., »Detection of buttermilk solids in skimmilk powder by HPLC quantification of aminophospholipids.« Sci. Tecn. Latt.-Cas., 39,395 (1988).
Figur 1
HPLC-mønster for OPA-derivater af phosphatidylserin (PS) og phosphatidylethanolamin (PE) i methanolekstrakt af rekonstitueret skummetmælkspulver. Integrationsmåde for toppene af PS, PE og tryptamin (intern standard) rapporteres.
(1) International IDF-standard 135B/1991. Milk and Milk products. Precision characteristics of analytical methods. Outline of collaborative study procedure.
BILAG XV
(Artikel 11)
PÅVISNING AF RESTKONCENTRATIONER AF ANTIMIKROBIELLE STOFFER I SKUMMETMÆLKSPULVER
Der skal anvendes en mikrobeinhibitor-screeningtest, hvor Geobacillus stearothermophilus var. calidolactis ATCC 10149 (identisk med stamme C953) anvendes som test-mikroorganisme, og som er tilstrækkelig følsom til at påvise 4 μg benzylpenicillin pr. kg mælk og 100 μg sulfadimidin pr. kg mælk. Analysesæt findes i handelen og kan anvendes, hvis de har den tilstrækkelige følsomhed for benzylpenicillin og sulfadimidin.
Til testen anvendes rekonstitueret skummetmælkspulver (1 g pulver +9 ml destilleret vand). Testen gennemføres som beskrevet i ISO/TS 26844:2006 Milk and milk products — Determination of antimicrobial residues — Tube diffusion test IDF — Bulletin nr. 258/1991, sektion 1, kapitel 2, eller efter anvisningerne fra fabrikanten i prøvesættet (1).
Positive resultater fortolkes således:
|
1. |
Forekomst af β-lactamer kan bekræftes ved at gentage analyse efter tilsætning af penicillinase til testsystemet (2): Negativt resultat: Det inhibiterende stof er et β-lactam-antibiotikum. Fortsat positivt resultat: Det inhibiterende stof kan ikke identificeres ved denne procedure. Der fortsættes med punkt 2. |
|
2. |
Forekomst af sulfonamider kan bekræftes ved at gentage analyse efter tilsætning af ρ-aminobenzosyre til testsystemet: Negativt resultat: Det inhibiterende stof er et sulfonamid. Fortsat positivt resultat: Det inhibiterende stof kan ikke identificeres ved denne procedure. Der fortsættes med punkt 3. |
|
3. |
Forekomst af en kombination af et β-lactam og et sulfonamid kan bekræftes ved at gentage analyse efter tilsætning af pencillinase + ρ-aminobenzosyre til testsystemet: Negativt resultat: De inhibiterende stoffer er et β-lactam antibiotikum og et sulfonamid. Positivt resultat: Det inhibiterende stof kan ikke identificeres ved denne procedure. |
(1) Vigtig note: Falsk-positive resultater kan forekomme, når skummetmælkspulver analyseres. Det er derfor vigtigt at verificere, at det anvendte testsystem ikke giver falsk-positive resultater.
(2) Visse β-lactamer er mindre følsomme over for β-lactamase. I sådanne tilfælde anbefales en supplerende forbehandling af prøven (1 ml analyseprøve med 0,3 ml penasekoncentrat ved 37 °C i 2 timer).
BILAG XVI
(Artikel 12)
KVANTITATIV BESTEMMELSE AF SKUMMETMÆLKSPULVER I FODERBLANDINGER VED ENZYMATISK KOAGULATION AF PARA-KASEIN
1. FORMÅL
Kvantitativ bestemmelse af skummetmælkspulver i foderblandinger ved koagulering af para-kaseinet ved hjælp af løbe-enzymer.
2. ANVENDELSESOMRÅDE
Denne metode anvendes ved analyse af foderblandinger, som indeholder mindst 10 % skummetmælkspulver. Tilstedeværelsen af store mængder kærnemælkspulver og/eller visse ikke-mælkeproteiner kan forårsage interferens.
3. PRINCIP
3.1. Kaseinet i foderblandingen opløses ved ekstraktion med en natriumcitratopløsning.
3.2. Calciumionkoncentrationen justeres til det niveau, der er nødvendigt for udfældningen af para-kasein, ved tilsætning af løbe.
3.3. Kvælstofindholdet i det udfældede para-kasein bestemmes ved hjælp af Kjeldahl-metoden, som er beskrevet i ISO-standard 8968-2:2001/IDF 20-2:2001, og resultatet beregnes på basis af et mindsteinhold på 27,5 % kasein i skummetmælkspulver (jf. 8.1).
4. REAGENSER
Der anvendes reagenser af analysekvalitet. Vandet skal enten være destilleret eller af tilsvarende renhed. Alle anvendte reagenser og opløsninger skal være fri for kvælstof, med undtagelse af løbe-enzymet (4.5).
4.1. Trinatriumcitrat 2-hydrat (1 % vægt/vol.-opløsning)
4.2. Calciumchlorid (ca. 5M-opløsning).
75 g CaCl2 · 2 H2O opløses i 100 ml destilleret vand ved rystning (NB: en eksotermisk reaktion er mulig). Opløsningen henstår til næste dag, hvor den filtreres. Opløsningen opbevares i køleskab.
4.3. 0,1 N natriumhydroxid
4.4. 0,1 N saltsyre
4.5. Flydende kalveløbe (styrke ca. 100 IMCU/ml, jf. ISO-standard 11815/IDF 157). Opbevares i køleskab ved 4-6 °C.
4.6. Reagenser til kvantitativ bestemmelse af kvælstofindholdet som anført for Kjedahl-metoden i ISO 8968-2:2001/IDF 20-2:2001.
5. APPARATUR
Sædvanligt laboratorieudstyr og især:
5.1. Morter eller mølle
5.2. Analysevægt, der kan veje med en nøjagtighed på 1 mg, og som kan aflæses med en nøjagtighed på 0,1 mg
5.3. Bordcentrifuge (500 g eller 2 000 til 3 000 omdr./min.), udstyret med centrifugerør på 50 ml og 2 000 g.
5.4. Magnetomrører med magnetpinde på 10 til 15 mm
5.5. 150-200 ml bægerglas
5.6. 250 ml til 500 ml kolber
5.7. Glastragte med en diameter på 60 til 80 mm
5.8. Hurtigfiltrerende askefri rundfiltre med en diameter på 150 mm (Whatman nr. 41 eller lignende)
5.9. Pipetter af forskellig størrelse
5.10. Termostatreguleret vandbad indstillet på 37 °C ± 1° C
5.11. pH-meter med en nøjagtighed på 0,1 pH-enhed
5.12. Termometre med en nøjagtighed på ±1 °C.
6. FREMGANGSMÅDE
6.1. Forberedelse af prøven
10 til 20 g af prøven formales i morter eller mølle for at opnå en homogen blanding.
6.2. Opløsning af mælkepulveret og udskillelse af det uopløselige bundfald
6.2.1. 1,000 ± 0,002 g af den homogeniserede foderblanding (6.1) afvejes direkte i et 50 ml-centrifugerør. Der tilsættes 30 ml trinatriumcitratopløsning (4.1), som i forvejen er opvarmet til 45 °C ± 2 °C og blandes i mindst 5 min. med magnetomrører eller ved kraftig manuel rystning.
6.2.2. Der centrifugereres 500 g ved 2 000 til 3 000 omdr./min i 10 min, hvorefter den klare, vandige opløsning dekanteres over i et 150 til 200 ml bægerglas. Undgå at overføre uopløselige partikler fra bunden af centrifugerøret.
6.2.3. Der foretages yderligere to ekstraktioner på bundfaldet efter samme fremgangsmåde, og hver gang dekanteres der oven i den første.
6.2.4. Dersom der viser sig et lag af fedtstof på overfladen af den opsamlede dekanterede opløsning, anbringes opløsningen i køleskab, indtil fedtet er størknet, og det fjernes derefter ved hjælp af en spatel.
6.3. Koagulering af kaseinet ved hjælp af løbe-enzymer
6.3.1. 2 ml calciumchlorid (4.2) tilsættes dråbevis under omrøring til den samlede opløsning (ca. 100 ml). pH-værdien justeres til 6,4-6,5 ved tilsætning af opløsninger af natriumhydroxid (4,3) eller saltsyre (4.4). Opløsningen sættes i et termostatreguleret vandbad ved 37 °C ± 1 °C i 15 til 20 min. for at opnå saltbalance. Dette viser sig ved, at opløsningen bliver lettere uklar.
6.3.2. Væsken overføres til et centrifugeglas, og bundfaldet fjernes ved centrifugering ved 2 000 g i 10 minutter. Supernatanten overføres til et andet centrifugeglas, uden at bundfaldet vaskes.
6.3.3. Supernatanten opvarmes atter til 37 °C ± 1 °C. Under omrøring tilsættes der dråbevis 0,5 ml kalveløbeopløsning (4.5) til ekstraktet. Koaguleringen indtræder i løbet af 2 minutter.
6.3.4. Prøven anbringes igen i vandbadet og henstår ved 37 °C ± 1 °C i 15 minutter. Prøven fjernes fra vandbadet, og koaglet skilles ved omrøring. Der centrifugeres ved 2 000 g i 10 minutter. Supernatanten filtreres gennem et passende papirfilter (5.8), som gemmes. Bundfaldet i centrifugeglasset vaskes med 50 ml ca. 35 °C varmt vand under omrøring.
Der centrifugeres igen ved 2 000 g i 10 minutter. Supernatanten filtreres igennem det samme filter, som er benyttet ovenfor.
6.4. Bestemmelse af kaseinkvælstof
6.4.1. Efter vaskning overføres bundfaldet kvantitativt til det i punkt 6.3.4 omhandlede filter med destilleret vand. Det tørrede filterpapir anbringes i en Kjeldahl-kolbe. Kvælstofindholdet bestemmes ved Kjeldahl-metoden, som beskrevet i ISO-standard 8968-2:2001/IDF 20-2:2001.
7. BLINDPRØVE
7.1. Der foretages regelmæssigt en blindbestemmelse ved mineralisering ifølge Kjeldahl-metoden, som beskrevet i ISO-standard 8968-2:2001/IDF 20-2:2001, på et askefrit filter (5.8), der er fugtet med en blanding af 90 ml natriumcitratopløsning (4.1), 2 ml mættet calciumchloridopløsning (4.2), 0,5 ml flydende løbe (4.5) og derefter vasket med 3 × 15 ml destilleret vand.
7.2. Forbruget af syre ved titreringen af blindprøven fratrækkes den mængde (4.4), der er anvendt ved titrering af prøven.
8. ANGIVELSE AF RESULTATER
8.1. Procenten af skummetmælkspulver i foderblandinger beregnes ved hjælp af følgende formel:

hvor
N er det fundne indhold af para-kasein-kvælstof i blandingen udtrykt i procent
27,5 er en faktor til omregning af den fundne kaseinmængde til skummetmælkspulver
2,81 og 0,908 er korrektionsfaktorer fundet ved regressionsanalyse.
9. METODENS NØJAGTIGHED
9.1. Repeterbarhed
I mindst 95 % af de undersøgte tilfælde har den samme prøve analyseret af samme person i samme laboratorium ved to bestemmelser ikke givet en større afvigelse end 2,3 g skummetmælkspulver pr. 100 g foderblanding.
9.2. Reproducerbarhed
I mindst 95 % af de undersøgte tilfælde har den samme prøve analyseret af to forskellige laboratorier ikke givet en større afvigelse end 6,5 g skummetmælkspulver pr. 100 g foderblanding.
10. BEMÆRKNINGER
10.1. Tilstedeværelse af en stor procentdel af visse ikke-mælkeproteiner og specielt sojaprotein kan, for så vidt de er blevet opvarmet med mælkepulveret, medføre for høje resultater, fordi de udfældes sammen med para-kasein.
10.2. Tilstedeværelse af kærnemælkspulver kan undertiden medføre for lave resultater, da bestemmelsen kun omfatter den fedtfrie del af prøven. Tilstedeværelse af sur kærnemælk kan give meget lave resultater på grund af en ufuldstændig opløsning i citrat-koncentrationen.
10.3. Tilstedeværelse af mere end 0,5 % lecithin kan ligeledes medføre for lave resultater.
10.4. Foderblandinger med indhold af skummetmælkspulver, der har været opvarmet til høje temperaturer (high heat), kan give for høje resultater på grund af, at visse mælkeserumproteiner udfældes sammen med para-kaseinet.
BILAG XVII
(Artikel 13)
PÅVISNING AF STIVELSE I SKUMMETMÆLKSPULVER, DENATURERET MÆLKEPULVER OG FODERBLANDINGER
1. FORMÅL
Denne metode anvendes til påvisning af stivelse, der bruges som røbestof i denatureret mælkepulver.
Metodens påvisningsgrænse er ca. 0,05 g stivelse pr. 100 g prøve.
2. PRINCIP
Der benyttes samme reaktion som ved jodometri:
|
— |
kolloidernes binding af fri jod i vandig opløsning |
|
— |
stivelsesmicellernes absorption og farvedannelse. |
3. REAGENSER
3.1. Jodopløsning
|
— |
Jod: 1,0 g |
|
— |
Kaliumjodid: 2,0 g |
|
— |
Destilleret vand: 100 ml. |
|
— |
1,0 g jod og 2,0 g kaliumjodid opløses i vand i en 100 ml målekolbe. Der fortyndes til 100 ml med vand og blandes. |
4. APPARATUR
4.1. Analysevægt
4.2. Kogende vandbad
4.3. Reagensglas, 25 mm × 200 mm.
5. FREMGANGSMÅDE
1,0 g af prøven afvejes med en nøjagtighed på 0,1 g og hældes på reagensglas (4,3).
Der tilsættes 20 ml destilleret vand, og glasset rystes, så vandet fordeler sig i prøven.
Glasset anbringes i kogende vandbad (4,2) og henstår i fem minutter.
Glasset tages op af vandbadet og afkøles ved rumtemperatur.
Der tilsættes 0,5 ml jodopløsning (3,1), glasset rystes, og farven iagttages.
6. ANGIVELSE AF RESULTATER
Hvis indholdet farves blåt, viser det, at prøven indeholder naturlig stivelse.
Hvis prøven indeholder modificeret stivelse, må den ikke farves blå.
7. BEMÆRKNINGER
Farven, farvestyrken og stivelsens udseende under mikroskop varierer afhængigt af den naturlige stivelses oprindelse (f.eks. majs eller kartoffel), og hvilken type modificeret stivelse der findes i prøven.
Indeholder prøven modificeret stivelse, farves den violet, rød eller brun, alt efter i hvilken grad den naturlige stivelses krystallinske struktur er modificeret.
BILAG XVIII
(Artikel 14)
BESTEMMELSE AF VANDINDHOLDET I TØRRET FLØDE
1. FORMÅL
I dette bilag angives en metode til bestemmelse af vandindholdet i tørret fløde.
2. TERMER OG DEFINITIONER
I dette bilag forstås ved:
»vandindhold« massetabet bestemt ved den i dette bilag angivne procedure.
Det angives i masseprocent.
3. PRINCIP
En analyseprøve tørres ved 102 ± 2 °C til en konstant masse, og prøven vejes for at bestemme massetabet.
4. APPARATUR
Sædvanligt laboratorieudstyr, herunder især følgende:
4.1. Analysevægt, der kan veje med en nøjagtighed på 1 mg, og som kan aflæses med en nøjagtighed på 0,1 mg
4.2. Tørreovn, velventileret, termostatstyret, som kan holde en temperatur på 102 ± 2 °C i hele ovnrummet
4.3. Ekssikkator med frisktørret silicagel med indikator for vandindhold eller et andet effektivt tørremiddel
4.4. Fladbundede skåle, dybde ca. 25 mm, diameter ca. 50 mm, af passende materiale (f.eks. glas, rustfrit stål, nikkel eller aluminium) med tætsluttende låg, der er lette af tage af
4.5. Flasker med tætsluttende propper til blanding af laboratorieprøverne.
5. PRØVEUDTAGNING
Det er vigtigt, at laboratoriet modtager en prøve, som reelt er repræsentativ, og som ikke er blevet skadet eller forandret under transport eller opbevaring.
Prøveudtagning beskrives ikke i den metode, der er angivet i dette bilag. En anbefalet prøveudtagningsmetode findes i ISO 707|IDF 50.
Prøven opbevares, så der hverken kan ske ødelæggelse eller ændring af sammensætningen.
6. FORBEREDELSE AF PRØVEN
Prøven blandes grundigt ved gentagne gange at ryste og vende beholderen (om nødvendigt efter at have placeret alle prøver i en lufttæt beholder, der er tilstrækkelig stor til, at blandingen kan foretages).
Hvis dette ikke resulterer i komplet homogenitet, udtages analyseprøverne (til 2 enkeltbestemmelser) fra en forberedt prøve to steder, der ligger så langt fra hinanden som muligt.
7. FREMGANGSMÅDE
7.1. Klargøring af skålen
7.1.1. En skål uden låg på og dens låg (punkt 4.4) opvarmes i tørreovnen (4.2) ved 102 ± 2 °C i mindst 1 time.
7.1.2. Låget lægges på skålen, som placeres i ekssikkatoren (4.3), afkøles til temperaturen i vejerummet og vejes med 0,1 mg nøjagtighed til nærmeste mg.
7.2. Analyseprøve
Ca. 1-3g af den klargjorte prøve (6) placeres i skålen, der dækkes med låget og vejes med 0,1 mg nøjagtighed til nærmeste mg.
7.3. Bestemmelse
7.3.1. Låget tages af skålen, som placeres sammen med låget i tørreovnen (4.2) ved 102 ± 2 °C i 2 timer.
7.3.2. Låget lægges igen på skålen, som placeres i ekssikkatoren, afkøles til temperaturen i vejerummet og vejes med 0,1 mg nøjagtighed til nærmeste mg.
7.3.3. Låget tages af skålen, som opvarmes igen sammen med låget i tørreovnen i 1 time. Proceduren i 7.3.2 gentages.
7.3.4. Opvarmningen og vejningen gentages, indtil massen falder med 1 mg eller derunder, eller den stiger mellem to på hinanden følgende vejninger.
Til beregningen anvendes den laveste registrerede masse.
8. BEREGNING OG ANGIVELSE AF RESULTATER
8.1. Beregning
Vandindholdet udtrykt i g pr. 100g svarer til:

hvor:
m0 er massen i g af skålen og låget (7.1.2)
m1 er massen i g af skålen, låget og analyseprøven inden tørring (7.2)
m2 er massen i g af skålen, låget og analyseprøven efter tørring (7.3.4).
Resultatet angives med 2 decimaler.
9. PRÆCISION
Bemærkning: Repeterbarheds- og reproducerbarhedsværdierne blev afledt fra en sammenlignende laboratorieprøvning (se Steiger, G. Bulletin of IDF No 285/1993, s. 21-28), der blev gennemført i overensstemmelse med IDF-standard 135B:1991. Milk and milk products — Precision characteristics of analytical methods — Outline of collaborative study procedure.
9.1. Repeterbarhed
Den absolutte forskel mellem to uafhængige enkeltresultater, der er opnået med den samme metode på identisk prøvemateriale, på det samme laboratorium, udført af den samme person med det samme udstyr og med et kort tidsinterval, bliver i højst 5 % af tilfældene større end 0,20g vand pr. 100g produkt.
9.2. Reproducerbarhed
Den absolutte forskel mellem to uafhængige enkeltresultater, der er opnået med den samme metode på identisk testmateriale på forskellige laboratorier udført af forskellige personer med forskelligt udstyr, bliver i højst 5 % af tilfældene større end 0,40g vand pr. 100g produkt.
10. PRØVERAPPORT
Prøverapporten skal indeholde følgende oplysninger:
|
— |
alle de oplysninger, der er nødvendige for fuldstændig identifikation af prøven |
|
— |
den prøveudtagningsmetode, der er anvendt (hvis den kendes) |
|
— |
den prøvemetode, der er anvendt med henvisning til dette bilag |
|
— |
alle prøvebetingelser, der ikke er specificeret i dette bilag, eller som anses for valgfrie, samt en detaljeret beskrivelse af alle forhold, der kan have indvirket på resultatet. |
Opnået resultat, og, hvis repeterbarheden er blevet kontrolleret, det endelige registrerede resultat.
BILAG XIX
(Artikel 15)
BESTEMMELSE AF VANDINDHOLDET I SYRNET KÆRNEMÆLKSPULVER
1. FORMÅL
Bestemmelse af vandindholdet i syrnet kærnemælkspulver, som oprindeligt er bestemt til foder.
2. PRINCIP
Prøven tørres under vakuum. Massetabet bestemmes ved vejning.
3. APPARATUR
3.1. Analysevægt, der kan veje med en nøjagtighed på 1 mg, og som kan aflæses med en nøjagtighed på 0,1 mg
3.2. Skåle af korrosionsbestandigt metal eller glas med lufttætte låg; nyttearealet skal være stort nok til, at prøven kan fordeles med ca. 0,3 g/cm2.
3.3. Elektrisk opvarmet justerbart vakuumtørreskab med oliepumpe, som er forsynet med en anordning til tilførsel af tørret varmluft gennem et tårn med f.eks. calciumoxid eller calciumsulfat (med fugtighedsindikator).
3.4. Ekssikkator med et effektivt tørremiddel.
3.5. Ventileret tørreovn med termostatregulering (ved 102 ± 2 °C).
4. FREMGANGSMÅDE
En skål (3.2) med låg opvarmes i tørreovnen (3.5) i mindst 1 time. Låget lægges på skålen, som straks flyttes over i en ekssikkator (3.4), afkøles til rumtemperatur og vejes med 0,1 mg nøjagtighed til nærmeste mg.
Låget tages af skålen, og ca. 5g af prøven placeres i skålen, som vejes med 0,1 mg nøjagtighed til nærmeste mg. Skålen stilles med låg ind i vakuumtørreskabet (3.3), som på forhånd er opvarmet til 83 °C. For at undgå at temperaturen i tørreskabet falder for meget, stilles skålen ind så hurtigt som muligt.
Trykket indstilles til 100 torr (13,3 kPa), og prøven tørres til konstant vægt (ca. 4 timer) ved dette tryk under tilførsel af varm, tør luft.
Tørringstiden regnes fra det tidspunkt, hvor temperaturen i tørreskabet atter er nået op på 83 °C. Efter tørringstiden er udløbet, bringes vakuumtørreskabet forsigtigt op på atmosfærisk tryk. Efter åbning af vakuumtørreskabet lukkes skålen straks med låget, tages ud af skabet, henstilles til afkøling i 30 til 45 minutter i ekssikkatoren (3.4) og vejes med 0,1 mg nøjagtighed til nærmeste mg. Der tørres i yderligere 30 minutter i vakuumtørreskabet (3.3) ved 83 °C, og derefter vejes på ny. Opvarmningen og vejningen gentages, indtil massen af skålen med låg falder med 1 mg eller derunder, eller den stiger mellem to på hinanden følgende vejninger. Til beregningen anvendes den laveste registrerede masse.
5. BEREGNING
% vandindhold = (m1 – m2) / (m1 – m0) × 100 %
hvor
|
m0 |
er massen af skålen og låget |
|
m1 |
er massen af skålen, låget og analyseprøven inden tørring |
|
m2 |
er massen af skålen, låget og analyseprøven efter tørring. |
Resultatet registreres med en nøjagtighed på 0,1 g pr. 100 g.
6. PRÆCISION
6.1. Repeterbarhed
Den absolutte forskel mellem to uafhængige enkeltresultater, der er opnået med den samme metode på identisk prøvemateriale, på det samme laboratorium, udført af den samme person med det samme udstyr og med et kort tidsinterval, bliver i højst 5 % af tilfældene større end 0,4 g vand pr. 100 g kærnemælkspulver.
6.2. Reproducerbarhed
Den absolutte forskel mellem to uafhængige enkeltresultater, der er opnået med den samme metode på identisk testmateriale på forskellige laboratorier udført af forskellige personer med forskelligt udstyr, bliver i højst 5 % af tilfældene større end 0,6 g vand pr. 100 g syrnet kærnemælkspulver.
6.3. Kilde for præcisionsdata
Præcisionsdataene blev bestemt ud fra et forsøg, der blev udført i 1995, og som omfattede otte laboratorier og 12 prøver (seks blinde dobbeltanalyser).
BILAG XX
(Artikel 16)
REFERENCEMETODE TIL BESTEMMELSE AF MÆLKEFEDTS RENHED VED GASKROMATOGRAFISK ANALYSE AF TRIGLYCERIDER — REVISION 2
1. FORMÅL OG ANVENDELSESOMRÅDE
Denne standard fastsætter en referencemetode til bestemmelse af mælkefedts renhed ved gaskromatografisk analyse af triglycerider. Såvel vegetabilske fedtstoffer som animalske fedtstoffer, f.eks. oksetalg og svinefedt, kan påvises.
Ved brug af veldefinerede triglyceridformler bestemmes mælkefedts renhed. Generelt anvendes metoden til tankkomælk eller produkter heraf, uanset fodrings-, avls- eller laktationsforholdene. Kun fodring med ekstraordinært stort indhold af rene vegetabilske olier, f.eks. rapsolie, kan forårsage falsk positive resultater. Mejeriprodukter fra enkeltkøer kan ligeledes forårsage et falsk positivt resultat.
Metoden anvendes i særdeleshed til fedt ekstraheret fra mejeriprodukter, der angiveligt indeholder rent mælkefedt med uændret sammensætning, f.eks. smør, fløde, mælk og mælkepulver. Teknologisk behandling af mælkefedt, f.eks. fjernelse af cholesterol eller fraktionering, kan forårsage et falsk positivt resultat. Det gælder også mælkefedt fra skummetmælk eller kærnemælk. Metoden kan ikke altid anvendes til fedt ekstraheret fra ost, fordi modningsprocessen kan påvirke fedtsammensætningen så meget, at der fås et falsk positivt resultat.
Bemærkning 1: Smørsyre (n-butansyre) (C4) forekommer udelukkende i mælkefedt og gør det muligt at foretage kvantitative skøn over lave til middelstore mængder af mælkefedt i vegetabilske og animalske fedtstoffer. Da massefraktionen af C4 varierer meget fra 3,1 % til 3,8 %, er det imidlertid vanskeligt at tilvejebringe kvalitative og kvantitative oplysninger om fremmede fedtstoffer ved massefraktioner af rent mælkefedt på op til 20 % [1].
Bemærkning 2: Kvantitative resultater kan i praksis ikke afledes af vegetabilske fedtstoffers sterolindhold, da det er forskelligt alt efter produktions- og forarbejdningsbetingelser. Desuden er den kvalitative bestemmelse af fremmede fedtstoffer ved hjælp af steroler tvetydig.
2. DEFINITION
Ved »mælkefedts renhed« forstås fravær af vegetabilske og animalske fedtstoffer bestemt ved den i denne standard angivne procedure.
Bemærkning: Renheden bestemmes ved anvendelse af S-værdier, som beregnes på grundlag af triglyceridsammensætningen. Massefraktionerne af triglycerid udtrykkes i procent.
3. PRINCIP
Fedt ekstraheret fra mælk eller mejeriprodukter analyseres ved gaskromatografi med en pakket kolonne eller en kort kapillarkolonne for at bestemme triglyceriderne (TG), adskilt pr. kulstofnummer. S-værdierne beregnes ved at indsætte massefraktionen udtrykt i % af fedtmolekyler af forskellig størrelse (C24 til C54, kun lige kulstofnumre) i egnede TG-formler. Hvis S-værdierne overskrider de grænser, der er fastsat for rent mælkefedt, er der påvist forekomst af fremmed fedtstof.
Bemærkning 1: Det er blevet påvist at såvel pakkede kolonner som kapillarkolonner er egnede og ligestillede [2-4].
Bemærkning 2: S-værdien er en sum af TG-massefraktioner, der hver ganges med fastlagte faktorer.
4. REAGENSER
Alle reagenser skal være af anerkendt analysekvalitet.
4.1. Bæregas: kvælstof eller, alternativt, helium eller brint, alle med en renhedsgrad på 99,995 % eller derover.
Standardiseret fedt til standardisering af standardmælkefedt, jf. punkt 7.3.3
4.2.1. Standardtriglycerider, mættede; egnede produkter kan fås i handelen
4.2.2. Standardcholesterol
4.3. Methanol(CH3OH), vandfri
4.4. n-Hexan (CH3(CH2)4CH3)
4.5. n-Heptan (CH3(CH2)5CH3)
4.6. Andre gasser: brint, med en renhedsgrad på 99,995 % eller derover, uden organiske urenheder (CnHm < 1 µl/l); syntetisk luft, uden organiske urenheder (CnHm < 1 µl/l)
4.7. Vandfri natriumsulfat (Na2SO4).
5. APPARATUR
Sædvanligt laboratorieudstyr, herunder især følgende:
5.1. Højtemperaturgaskromatograf
Højtemperaturgaskromatografen skal være egnet til temperaturer på mindst 400 °C og være forsynet med en flammeioniseringsdetektor (FID). Septa, der anvendes i injektoren, skal kunne modstå høje temperaturer og vise meget lille blødning. Ved gaskromatografi med kapillarkolonner anvendes direkte injektion på kolonnen. Der anvendes altid grafitferruler til at tilslutte kolonnen og injektor- og/eller detektorindsatser (hvor det er relevant).
5.2. Kromatografikolonne
5.2.1. Pakket kolonne
Der anvendes en glaskolonne med en indvendig diameter på 2 mm og en længde på 500 mm, som er pakket med en stationær fase af 3 % OV-1 på 125 µm til 150 µm (100 til 120 mesh) Gas ChromQ (1). Forberedelse, silanisering, pakning og konditionering af kolonnen er beskrevet i tillæg A.
Alternativt kan der anvendes en kapillarkolonne (5.2.2).
5.2.2. Kapillarkolonne
Der anvendes en kort kapillarkolonne, f.eks. med en længde på 5 m, med en ikke-polær stationær fase, der kan modstå temperaturer på 400 °C eller derover (2). Kolonnen konditioneres ved at foretage 20 analyser af en mælkefedtopløsning (7.2) på 2-3 dage med de indstillinger, der er angivet i punkt 7.3.4.2. Derefter skal responsfaktorerne (7.3.3) ligge tæt på 1 og under 1,20.
Bemærkning: Kolonner med forskellige dimensioner og forskellige ikke-polære, højtemperaturresistente faser kan anvendes, når blot deres ydeevne er i overensstemmelse med denne standard. Se også punkt 7.3.4.2.
5.3. 1-3 ml Extrelut-kolonne med silicagel, anvendes udelukkende til ekstraktion af mælkefedt efter punkt 7.1.3
5.4. Grafitferruler, der kan modstå temperaturer på 400 °C eller derover; anvendes til at tilslutte gaskromatografkolonnen og injektor- og/eller detektorindsatser
5.5. Vandbad, som kan holde en temperatur på 50 °C ± 2 °C
5.6. Ovn, som kan fungere ved 50 °C ± 2 °C og 100 °C ± 2 °C
5.7. Mikroliterpipette
5.8. Målepipette på 5 ml
5.9. 50 ml rundbundet kolbe
5.10. Erlenmayerkolbe, nominelt rumfang 250 ml
5.11. Tragt
5.12. Finporet filtrerpapir
5.13. Rotationsfordamper
5.14. Prøveglas, nominelt rumfang 1 ml, der kan lukkes med polytetrafluoroethylen-foret hætte eller skruelåg af aluminium
5.15. Injektionssprøjte; sprøjtens stempel må ikke trænge ind i kanylespidsen (for pakket kolonne)
Bemærkning: Med sådanne sprøjter opnås en bedre repeterbarhed for resultaterne.
5.16. Analysevægt, der kan veje med en nøjagtighed på 1 mg, og som kan aflæses med en nøjagtighed på 0,1 mg
6. PRØVEUDTAGNING
Der skal sendes en repræsentativ prøve til laboratoriet. Prøven må ikke være blevet skadet eller forandret under transport eller opbevaring.
Prøveudtagning beskrives ikke i den metode, der er angivet i dette bilag. En anbefalet prøveudtagningsmetode findes i ISO 707|IDF 50 [5].
7. FREMGANGSMÅDE
7.1. Forberedelse af prøver
En af følgende tre metoder til mælkefedtekstraktion benyttes til forberedelse af prøver.
7.1.1. Isolering fra smør eller smørolie
50-100 g af prøven smeltes ved 50 °C i vandbad (5.5) eller ovn (5.6). 0,5-1,0 g natriumsulfat (4.7) anbringes i et foldet filtrerpapir (5.12). En 250 ml erlenmeyerkolbe (5.10) og en tragt (5.11) med filterindsats forvarmes i ovnen (5.6) til 50 °C. Den smeltede prøves fedtlag filtreres, mens kolben, tragten og filteret, der er forvarmet, holdes inde ovnen. Der sørges omhyggeligt for, at der ikke overføres valle.
Kun hvis der kun er en begrænset prøvemængde til rådighed, kan der anvendes en mindre prøve, og fremgangsmåden bør tilpasses tilsvarende. Håndtering af en mindre analyseprøve indebærer dog en større risiko for, at man får en ikke-repræsentativ prøve.
Bemærkning 1: Smør kan fremstilles af fløde ved hjælp af kærning og grundig skylning af de udkærnede smørkorn.
Bemærkning 2: Det mælkefedt, der fremkommer ved anvendelse af fremgangsmåden i punkt 7.1.1, vil være næsten helt fri for fosfolipider.
7.1.2. Ekstraktion efter Röse–Gottlieb-metoden
Fedtfraktionen ekstraheres fra prøven ved hjælp af den gravimetriske metode, der er beskrevet i en af standarderne ISO 1211 IDF 001D, ISO 2450IDF 016C eller ISO 7328IDF 116A.
Bemærkning: Hvis der forekommer fosfolipider i mælkefedtet, fremkommer der en cholesteroltop, der øges med ca. 0,1 %. TG-sammensætningen standardiseret til 100 % inkl. cholesterol bliver derved kun påvirket i uvæsentligt omfang.
7.1.3. Ekstraktion fra mælk ved brug af silicagelkolonner
Med en mikroliterpipette (5.7) tilsættes 0,7 ml prøve lunet til 20 °C til en 1-3 ml Extrelut-kolonne (5.3). Man lader den fordele sig ensartet på silicagelen i ca. 5 min.
Til denaturering af proteinlipidkomplekserne tilsættes der med målepipette (5.8) 1,5 ml methanol (4.3) til Extrelut-kolonnen. Derefter ekstraheres fedtfraktionen fra prøven med 20 ml n-hexan (4.4). n-Hexanen tilsættes langsomt i små mængder. Opløsningsmidlet samles i en 50 ml rundbundet kolbe (5.9), som er tørret til en konstant, kendt masse, der er vejet med 0,1 mg nøjagtighed til nærmeste mg.
Efter ekstraktion henstår kolonnen til afdrypning, indtil den er tom. Opløsningsmidlerne afdampes fra eluatet på en rotationsfordamper (5.13) ved en vandbadstemperatur på 40-50 °C. Når opløsningsmidlerne er afdampet, tørres den rundbundede kolbe, der vejes med 0,1 mg nøjagtighed til nærmeste mg. Fedtmængden bestemmes ved at trække den tørrede, tomme rundbundede kolbes masse fra den fremkomne masse.
Bemærkning: Fedtekstraktioner ifølge Gerber-, Wibull-Berntrop- eller Schmid-Bondszinsky-Ratzlaff-metoderne eller ved isolering af mælkefedt ved brug af detergenter (BDI-metode) er ikke egnet til TG-analyse, da store mængder partielle glycerider eller fosfolipider kan medgå i fedtfasen ved disse metoder. Derfor er anvendelsen af dette bilag begrænset for visse produkter, bl.a. ost.
7.2. Fremstilling af prøveopløsningen
Til gaskromatografi med pakket kolonne fremstilles der en 5 %-opløsning (volumenfraktion) af fedtstoffet (tilvejebragt efter punkt 7.1) i n-hexan (4.4) eller n-heptan (4.5). Afhængigt af kolonnedimensionerne anvendes en koncentration på 1 % (0,53 mm, stor indvendig diameter) eller lavere til direkte injektion på en kapillarkolonne.
På grundlag af den anvendte kolonne og fedtmassen fremkommet efter punkt 7.1.3 bestemmes, hvor meget opløsningsmiddel (4.4 eller 4.5) der skal tilsættes til prøvematerialet i kolben ved vejning med 0,1 mg nøjagtighed til nærmeste mg. Resten opløses fuldstændigt.
Ca. 1 ml af prøveopløsningen overføres til et prøveglas (5.14).
7.3. Kromatografisk triglyceridbestemmelse
7.3.1. Basislinjens drift
For at minimere basislinjens stigning skal kolonnen konditioneres som angivet i punkt 5.2.2 (kapillarkolonne) eller 5.2.1 (pakket kolonne).
Bemærkning: På grund af den høje kolonnetemperatur er TG-analysen særligt modtagelig for en basislinjestigning for de høje kulstofnumre.
7.3.2. Injektionsteknik
7.3.2.1. Pakket kolonne
For at undgå fraktionering benyttes der »hot injection«, hvorved der opnås bedre kvantitativ bestemmelse af de højtkogende TG-komponenter. Kanylen fyldes med luft ved at trække fedtopløsningen op i sprøjten. Kanylen indsættes i injektoren. Kanylen opvarmes inden injektionen i ca. 3 sek. Derefter injiceres sprøjtens indhold meget hurtigt.
7.3.2.2. Kapillarkolonne
Når der anvendes kold, direkte injektion på kolonnen (7.3.4.2), indsættes sprøjtens kanyle, og der injiceres straks. Kanylen skal blive i injektionsporten i et tidsrum, så der undgås bred haledannelse for opløsningsmidlets top.
Bemærkning: Det optimale tidsrum er typisk 3 sek.
7.3.3. Kalibrering
7.3.3.1. Generelt
Til kalibrering af prøverne gennemføres 2-3 analyser af standardiseret mælkefedt ved begyndelsen af hver dag. Den seneste analyse af det standardiserede mælkefedt anvendes til at bestemme TG's og cholesterols responsfaktorer RFsi (massefraktion/arealfraktion), og responsfaktorerne anvendes til de efterfølgende prøver (jf. 9.1):
![]() (1)
(1)
hvor
|
w si |
er massefraktionen udtrykt i % af hver TG eller cholesterol i det standardiserede mælkefedt |
|
A si |
er den numeriske værdi af toparealet for hver TG eller cholesterol i det standardiserede mælkefedt. |
Enten 7.3.3.2 eller 7.3.3.3 anvendes til at frembringe standardiseret mælkefedt med en kendt TG-sammensætning.
7.3.3.2. Standardiseret mælkefedt, der fås i handelen
Responsfaktoren for prøvens enkeltbestanddele bestemmes bedst ved at anvende standardiseret mælkefedt med en certificeret TG-sammensætning.
Bemærkning: En egnet standard er CRM 519 (vandfrit mælkefedt), der sælges af Institute for Reference Materials and Measurements (IRMM), Geel, Belgien (3).
7.3.3.3. Laboratoriefremstillet standardiseret mælkefedt
Der forberedes ca. 1 g blanding af de standardiserede fedtstoffer (jf. 4.2), der mindst indeholder de mættede TG'er C24, C30, C36, C42, C48 og C54 samt cholesterol og desuden helst C50 og C52, og som vejes med 0,1 mg nøjagtighed til nærmeste mg, med henblik på at opnå en relativ TG-sammensætning, der svarer til mælkefedt.
Der foretages gentagne analyser af en opløsning af blandingen af de standardiserede fedtstoffer i n-hexan (4.4) eller n-heptan (4.5) efter 7.3.4. I samme sekvens analyseres gentagne gange mælkefedt med en gennemsnitlig sammensætning.
TG-responsfaktorerne bestemmes ud fra blandingen af de standardiserede fedtstoffer. Mellemliggende responsfaktorer for TG'er, der ikke forekommer i blandingen, kan findes ved matematisk interpolering. Responsfaktorerne for mælkefedtet anvendes til at fremstille en standardiseret sammensætning. Det således fremstillede standardiserede mælkefedt har en holdbarhed på flere år, hvis det opbevares i kvælstof ved en temperatur på maks. –18 °C.
7.3.4. Kromatografiske betingelser
Bemærkning: Anvendelse af pakkede kolonner eller kapillarkolonner resulterer generelt i en resolution, der svarer til figur 1. Adskillelse af TG'er med lige numre observeres normalt ikke og skal undgås.
7.3.4.1. Pakket kolonne
|
a) |
Temperaturprogram: Ovnen indstilles til 210 °C (initialtemperatur). Denne temperatur holdes i 1 min. Derefter øges temperaturen gradvis med 6 °C/min. til 350 °C. Denne temperatur (sluttemperatur) holdes i 5 min. |
|
b) |
Detektor- og injektortemperatur: Begge indstilles til 370 °C. |
|
c) |
Bæregas: Der anvendes kvælstof ved en konstant gennemstrømningshastighed på 40 ml/min. Det nøjagtige bæregasflow reguleres således, at C54 elueres ved 341 °C. |
|
d) |
Analysevarighed: 29,3 min. |
|
e) |
Injektionsvolumen: Der injiceres 0,5 µl af en 5 %-prøveopløsning (volumenfraktion). |
Hvis der ikke gennemføres TG-analyser, holdes ovnens initialtemperatur som angivet i litra a), detektor- og injektortemperatur som angivet i litra b) og bæregasflowet som angivet i litra c) på et konstant niveau, også natten over og i weekender og på helligdage. Dette sikrer, at kolonnen fungerer optimalt.
7.3.4.2. Kapillarkolonne
|
a) |
Temperaturprogram: Ovnen indstilles til 80 °C (initialtemperatur). Denne temperatur holdes i 0,5 min. Derefter øges temperaturen gradvis med 50 °C/min. til 190 °C og derefter med 6 °C/min. til 350 °C. Denne temperatur (sluttemperatur) holdes i 5 min. |
|
b) |
Detektortemperatur: Indstilles til 370 °C. |
|
c) |
Bæregas: Der anvendes kvælstof ved en konstant gennemstrømningshastighed på 3 ml/min. |
|
d) |
Analysevarighed: 34,4 min. |
|
e) |
Injektionsvolumen: Der injiceres 0,5 µl af en 1 %-prøveopløsning (volumenfraktion). |
Indstillingerne opretholdes også ved standby for at sikre den bedst ydeevne (jf. 7.3.4.1).
De analyseindstillinger, der er angivet i 7.3.4.2, er egnede til anvendelse med en kolonne med stor indvendig diameter (0,53 mm), jf. 5.2.2. Der kan gælde andre betingelser, hvis der anvendes en anden kolonnestørrelse eller fase.
8. INTEGRATION, EVALUERING OG KONTROL AF ANALYSEPRÆSTATION
Kromatogrammets toppe evalueres med et integrationssystem, hvormed basislinjen kan plottes og reintegreres. Figur 1 viser et korrekt integreret kromatogram, men figur 2 viser en sporadisk fejl på basislinjen, der ender efter C54, og som påvirker procentdelene for alle TG'er. Ikke desto mindre udelukkes toppe, der eluerer efter C54 fra evalueringen.
TG'er med ulige acyl-C-nummer (2n + 1) kombineres med det forudgående TG med lige nummer (2n). Det lave C56-indhold tages ikke i betragtning. De resterende TG'ers arealandel, inkl. cholesterol, multipliceres med de tilsvarende responsfaktorer for det standardiserede mælkefedt (seneste kalibrering), og summen sættes til 100 %, jf. 9.1.
Figur 1
Eksempel på et triglyceridkromatogram af mælkefedt med korrekt indstillet basislinje

Figur 2
Eksempel på et triglyceridkromatogram af mælkefedt med ukorrekt indstillet basislinje

For at kontrollere målebetingelserne sammenlignes med variationskoefficienterne (VK), udtrykt i %, for de forskellige TG'er i tabel 1 beregnet ud fra 19 på hinanden følgende analyser af den samme mælkefedtprøve.
Hvis VK'erne ligger betragteligt højere end de værdier, der fremgår af tabel 1, er de kromatografiske betingelser ikke egnede.
Bemærkning: Værdierne i tabel 1 er ikke obligatoriske og er blot en vejledning for kvalitetskontrollen.
Hvis der imidlertid accepteres højere VK-værdier, skal grænserne for repeterbarhed og reproducerbarhed i punkt 10 alligevel overholdes.
Tabel 1
Variationskoefficienter for triglyceridindholdet (19 på hinanden følgende analyser)
|
Triglycerid |
VK % |
|
C24 |
10,00 |
|
C26 |
2,69 |
|
C28 |
3,03 |
|
C30 |
1,76 |
|
C32 |
1,03 |
|
C34 |
0,79 |
|
C36 |
0,25 |
|
C38 |
0,42 |
|
C40 |
0,20 |
|
C42 |
0,26 |
|
C44 |
0,34 |
|
C46 |
0,37 |
|
C48 |
0,53 |
|
C50 |
0,38 |
|
C52 |
0,54 |
|
C54 |
0,60 |
9. BEREGNING OG ANGIVELSE AF RESULTATER
9.1. Triglyceridsammensætning
9.1.1. Beregning
Massefraktionen, wi , beregnes for hvert TG (for i = C24, C26, C28, C30, C32, C34, C36, C38, C40, C42, C44, C46, C48, C50, C52 og C54) og cholesterol udtrykt i % af prøvens samlede TG-indhold ved hjælp af følgende ligning:
 (2)
(2)
hvor
|
Ai |
er den numeriske værdi af toparealet for hver TG i prøven |
|
RF si |
er responsfaktoren for hver TG bestemt ved kalibrering (7.3.3). |
9.1.2. Angivelse af resultater
Resultatet angives med 2 decimaler.
9.2. S-værdier
9.2.1. Beregning
9.2.1.1. S-værdierne, udtrykt i %, beregnes ved at indsætte den beregnede wi (9.1.1) for de relevante TG-andele i ligning 3-7. Alle ligningerne anvendes, uanset hvilken type fremmed fedtstof man formoder forekommer.
9.2.1.2. Sojaolie, solsikkeolie, olivenolie, rapsolie, hørfrøolie, hvedekimolie, majskimolie, bomuldsfrøolie og fiskeolie
S = 2,098 3 · w C30 + 0,728 8 · w C34 + 0,692 7 · w C36 + 0,635 3 · w C38 + 3,745 2 · w C40 – 1,292 9 · w C42 + 1,354 4 · w C44 + 1,701 3 · w C46 + 2,528 3 · w C50 (3)
9.2.1.3. Kokosfedt og palmekernefedt
S = 3,745 3 · w C32 + 1,113 4 · w C36 + 1,364 8 · w C38 + 2,154 4 · w C42 + 0,427 3 · w C44 + 0,580 9 · w C46 + 1,292 6 · w C48 + 1,030 6 · w C50 + 0,995 3 · w C52 + 1,239 6 · w C54 (4)
9.2.1.4. Palmeolie og oksetalg
S = 3,664 4 · w C28 + 5,229 7 · w C30 – 12,507 3 · w C32 + 4,428 5 · w C34 – 0,201 0 · w C36 + 1,279 1 · w C38 + 6,743 3 · w C40 – 4,271 4 · w C42 +6,373 9 · w C46 (5)
9.2.1.5. Svinefedt
S = 6,512 5 · w C26 + 1,205 2 · w C32 + 1,733 6 · w C34 + 1,755 7 · w C36 + 2,232 5 · w C42 + 2,800 6 · w C46 + 2,543 2 · w C52 + 0,989 2 · w C54 (6)
9.2.1.6. Alle
S = – 2,757 5 · w C26 + 6,407 7 · w C28 + 5,543 7 · w C30 – 15,324 7 · w C32 + 6,260 0 · w C34 + 8,010 8 · w C40 – 5,033 6 · w C42 + 0,635 6 · w C44 + 6,017 1 · w C46 (7)
9.2.2. Angivelse af resultater
Resultatet angives med 2 decimaler.
9.3. Påvisning af fremmede fedtstoffer
De fem S-værdier fremkommet efter 9.2.1 sammenlignes med de tilsvarende S-grænser i tabel 2.
Prøven betragtes som rent mælkefedt, hvis alle fem S-værdier ligger under grænserne i tabel 2. Hvis en eller flere af S-værdierne ligger over den pågældende grænse, anses prøven for at indeholde et fremmed fedtstof.
Skønt de specifikke ligninger 3-6 er mere følsomme over for visse fremmede fedtstoffer end ligning 7 (alle) (jf. tabel B.1), kan der ikke på grundlag af et positivt resultat ved kun én af ligningerne 3-6 drages konklusioner om typen af fremmed fedtstof.
I tillæg B beskrives en procedure til beregning af indholdet af vegetabilske eller animalske fedtstoffer i det forfalskede mælkefedt. Proceduren er ikke valideret og er udelukkende ment som information.
Tabel 2
S-grænser for rent mælkefedt
|
Fremmed fedtstof |
Ligning |
S-grænser (4) |
|
Sojaolie, solsikkeolie, olivenolie, rapsolie, hørfrøolie, hvedekimolie, majskimolie, bomuldsfrøolie og fiskeolie |
(3) |
98,05-101,95 |
|
Kokosfedt og palmekernefedt |
(4) |
99,42-100,58 |
|
Palmeolie og oksetalg |
(5) |
95,90-104,10 |
|
Svinefedt |
(6) |
97,96-102,04 |
|
Alle |
(7) |
95,68-104,32 |
10. PRÆCISION
10.1. Sammenlignende laboratorieprøvning
Repeterbarheds- og reproducerbarhedsværdierne blev bestemt på grundlag af ligning 3-7 ved anvendelse af rent mælkefedt og må ikke anvendes på andre matrixer end de anførte.
10.2. Repeterbarhed
Den absolutte forskel mellem to enkeltresultater, der er opnået med den samme metode på identisk prøvemateriale, på det samme laboratorium, udført af den samme person med det samme udstyr og med et kort tidsinterval, overskrider i højst 5 % af tilfældene de grænser, der er anført i tabel 3.
Tabel 3
Repeterbarhedsgrænser, r, for ligning 3-7
|
Fremmed fedtstof |
Ligning |
r % |
|
Sojaolie, solsikkeolie, olivenolie, rapsolie, hørfrøolie, hvedekimolie, majskimolie, bomuldsfrøolie og fiskeolie |
(3) |
0,67 |
|
Kokosfedt og palmekernefedt |
(4) |
0,12 |
|
Palmeolie og oksetalg |
(5) |
1,20 |
|
Svinefedt |
(6) |
0,58 |
|
Alle |
(7) |
1,49 |
10.3. Reproducerbarhed
Den absolutte forskel mellem to enkeltresultater, der er opnået med den samme metode på identisk prøvemateriale på forskellige laboratorier udført af forskellige personer med forskelligt udstyr, overskrider i højst 5 % af tilfældene de grænser, der er anført i tabel 4.
Tabel 4
Reproducerbarhedsgrænser, R, for ligning 3-7
|
Fremmed fedtstof |
Ligning |
R % |
|
Sojaolie, solsikkeolie, olivenolie, rapsolie, hørfrøolie, hvedekimolie, majskimolie, bomuldsfrøolie og fiskeolie |
(3) |
1,08 |
|
Kokosfedt og palmekernefedt |
(4) |
0,40 |
|
Palmeolie og oksetalg |
(5) |
1,81 |
|
Svinefedt |
(6) |
0,60 |
|
Alle |
(7) |
2,07 |
11. MÅLEUSIKKERHED
Med repeterbarhedsgrænsen (r) og reproducerbarhedsgrænsen (R) kan den ekspanderede usikkerhed for en S-værdi beregnes.
Ved at medregne den ekspanderede usikkerhed (baseret på dobbeltanalyser) i S-værdierne i tabel 2 fås udvidede S-værdier, som er anført i tabel 5.
Tabel 5
Udvidede S-grænser for rent mælkefedt, inkl. den ekspanderede usikkerhed
|
Fremmed fedtstof |
Ligning |
Udvidede S-grænser |
|
Sojaolie, solsikkeolie, olivenolie, rapsolie, hørfrøolie, hvedekimolie, majskimolie, bomuldsfrøolie og fiskeolie |
(3) |
97,36-102,64 |
|
Kokosfedt og palmekernefedt |
(4) |
99,14-100,86 |
|
Palmeolie og oksetalg |
(5) |
94,77-105,23 |
|
Svinefedt |
(6) |
97,65-102,35 |
|
Alle |
(7) |
94,42-105,58 |
12. TESTRAPPORT
Testrapporten skal indeholde følgende oplysninger:
|
— |
alle de oplysninger, der er nødvendige for fuldstændig identifikation af prøven |
|
— |
den prøveudtagningsmetode, der er anvendt (hvis den kendes) |
|
— |
den prøvemetode, der er anvendt med henvisning til dette bilag |
|
— |
alle prøvebetingelser, der ikke er specificeret i dette bilag, eller som anses for valgfrie, samt en detaljeret beskrivelse af alle forhold, der kan have indvirket på resultatet |
|
— |
opnået resultat, og, hvis repeterbarheden er blevet kontrolleret, det endelige registrerede resultat. |
(1) Eksempel på et egnet produkt, der kan fås i handelen. Oplysningen gives som en hjælp til brugere af dette bilag, men den udgør ikke en officiel godkendelse af produktet.
(2) CP-Ultimetal SimDist (5 m × 0,53 mm × 0,17 µm) er et eksempel på et egnet produkt, der kan fås i handelen. Oplysningen gives som en hjælp til brugere af dette bilag, men den udgør ikke en officiel godkendelse af produktet.
(3) Eksempel på et egnet produkt, der kan fås i handelen. Oplysningen gives som en hjælp til brugere af dette bilag, men den udgør ikke en officiel godkendelse af produktet.
(4) Beregnet på et konfidensniveau på 99 %, så tilsat fremmed fedtstof kun angives, hvis påvisningsgrænserne for den relevante ligning er overskredet (jf. tabel B.1).
TILLÆG A
(normativt)
KLARGØRING AF PAKKET KOLONNE
A.1 REAGENSER OG APPARATUR
A.1.1 Toluen (C6H5CH3)
A.1.2 Opløsning af dimethyldichlorsilan [Si(CH3)2Cl2]
50 ml dimethyldichlorsilan opløses i 283 ml toluen (A.1.1)
A.1.3 Kakaosmøropløsning, med en massefraktion på 5 % kakaosmør, i n-hexan (4.4) eller n-heptan (4.5)
A.1.4 Stationær fase, 3 % OV-1 på 125/150 µm (100-120 mesh) GasChromQ (1)
Bemærkning: Partikelangivelsen er omregnet til μm ifølge BS 410 (alle dele) [6]
A.1.5 Glaskolonne med en indvendig diameter på 2 mm og en længde på 500 mm, U-formet
Apparatur til fyldning af den pakkede kolonne
A.1.6.1 Kolonne til påfyldning, med påskruede endestykker og et mærke, der viser, hvortil den krævede mængde stationære fase kan påfyldes
A.1.6.2 Finmasket si (maskestørrelse på ca. 100 μm) med skruelåg, der er egnet til at forsegle glaskolonnen som i fig. A.3
A.1.6.3 Silaniseret glasuld, deaktiveret
A.1.6.4 Vibrator til hjælp til ensartet fordeling af den stationære fase ved påfyldningen
A.1.6.5 Udstyr til silanisering af kolonnens glasoverflade
A.1.6.6 Woulff-flaske
A.1.6.7 Vandsugepumpe
A.2 SILANISERING (DEAKTIVERING AF GLASOVERFLADEN)
Efter at Woulff-flasken (A.1.6.6) er forbundet med vandsugepumpen (A.1.6.7), dyppes slange 2 (jf. figur A.1) ned i dimethyldichlorsilanopløsningen (A.1.2). Kolonnen (A.1.5) fyldes med opløsningen ved at lukke hanen. Hanen åbnes igen, og derefter fjernes de to slanger. Kolonnen fastgøres på et stativ og fyldes helt op med dimethyldichlorsilanopløsning (A.1.2) ved hjælp af en pipette.
Figur A.1
Silaniseringsapparatur
![]()
Signaturforklaring
|
1 |
slange 1 |
|
2 |
slange 2 |
|
3 |
vandsugepumpe |
|
4 |
hane |
|
5 |
glaskolonne |
|
6 |
dimethyldichlorsilan og toluen |
Kolonnen henstår i 20-30 min. Derefter udskiftes Woulff-flasken med en sugekolbe. Kolonnen tømmes ved at tilslutte den til vandsugepumpen (A.1.6.7) (jf. figur A.2). Den tømte kolonne gennemskylles successivt med 75 ml toluen (A.1.1) og 50 ml methanol (4.3) ved at dyppe slange 2 i opløsningsmidlet. Den skyllede kolonne tørres i ovnen (5.6) ved 100 °C i ca. 30 min.
Figur A.2
Skylleapparatur
![]()
Signaturforklaring
|
1 |
slange 1 |
|
2 |
slange 2 |
|
3 |
vandsugepumpe |
|
4 |
sugekolbe |
|
5 |
glaskolonne |
|
6 |
skyllemiddel |
A.3 PÅFYLDNING
Til kolonnepåfyldning anvendes apparaturet i figur A.3. Den stationære fase (A.1.4) fyldes i påfyldningskolonnen (A.1.6.1) op til mærket. Den nedre del af den glaskolonne, der skal fyldes, forsegles med en ca. 1 cm lang tot silaniseret, komprimeret glasuld (A.1.6.3). Kolonnens ende lukkes med den finmaskede si (A.1.6.2).
Figur A.3
Påfyldning af glaskolonnen

Signaturforklaring
|
1 |
kvælstofindtag |
|
2 |
påfyldningskolonne, der fyldes med OV-1 op til mærket |
|
3 |
glaskolonne, der skal fyldes |
|
4 |
skruelåg med filter, som glasfiberen og den stationære fase presses imod |
Kolonnen fyldes med den stationære fase under tryk (300 kPa og et kvælstofflow). For at få en ensartet, sammenhængende og fast pakning føres en vibrator op og ned i glaskolonnen under fyldningen. Efter fyldningen presses en massiv tot silaniseret glasuld (A.1.6.3) ind i den anden ende af den pakkede kolonne. De ender, der rager frem, afskæres. Glasuldstotten presses nogle få millimeter ind i kolonnen med en spatel.
A.4 KONDITIONERING
Under trin a) til c) må kolonnens bageste ende ikke forbindes med detektoren for at undgå forurening. Den fyldte kolonne (A.3) konditioneres således:
|
a) |
Kolonnen skylles med kvælstof i 15 min. med en gennemstrømningshastighed på 40 ml/min og ovnen indstillet på 50 °C. |
|
b) |
Kolonnen opvarmes med 1 °C/min. indtil 355 °C med et kvælstofflow på 10 ml/min. |
|
c) |
Kolonnen henstår i 12-15 timer ved 355 °C. |
|
d) |
Der injiceres to gange 1 μl kakaosmøropløsning (A.1.3) ved anvendelse af samme temperaturprogram som for den pakkede kolonne, jf. 7.3.4.1. Bemærkning: Kakaosmør består næsten udelukkende af højtkogende C50 til C54 TG'er, hvilket reducerer indsatsen i forbindelse med kolonnekonditionering for så vidt angår de respektive responsfaktorer. |
|
e) |
Der injiceres 20 gange 0,5 µl mælkefedtopløsning efter 7.2 på 2-3 dage med de indstillinger, der er angivet i punkt 7.3.4.1.
|
(1) Eksempel på et egnet produkt, der kan fås i handelen. Oplysningen gives som en hjælp til brugere af dette bilag, men den udgør ikke en officiel ISO- eller IDF-godkendelse af produktet.
TILLÆG B
(vejledende)
KVANTITATIV BESTEMMELSE AF INDHOLDET AF FREMMEDE FEDTSTOFFER
B.1 GENERELT
I tabel B.1 angives påvisningsgrænserne for de forskellige fremmede fedtstoffer beregnet med en konfidens på 99 %. Den midterste kolonne viser påvisningsgrænserne for de bedste specifikke ligninger 3-6.
Påvisningsgrænserne for ligning 7 (totalligningen), der er angivet i kolonne til højre, er noget højere. I princippet behøves kun ligning 7 til kvantitativ bestemmelse af fremmede fedtstoffer.
Der kan med samtlige ligninger påvises blandinger af forskellige fremmede fedtstoffer. Variationen af TG-sammensætningen mellem forskellige enkeltprøver af en enkelt type af fremmede fedtstoffer har ingen væsentlig indflydelse på påvisningsgrænserne.
Når såvel de specifikke ligninger som totalligningen anvendes, gælder de specifikke ligningers påvisningsgrænser. S-værdien for totalligningen er dog i visse tilfælde (B.2) nødvendig for en kvantitativ bestemmelse.
Tabel B.1
99 % påvisningsgrænser for fremmed fedtstof tilsat til mælkefedt i %
|
Fremmed fedtstof |
Specifik ligning % |
Totalligning % |
|
Sojabønneolie |
2,1 |
4,4 |
|
Solsikkeolie |
2,3 |
4,8 |
|
Olivenolie |
2,4 |
4,7 |
|
Kokosolie |
3,5 |
4,3 |
|
Palmeolie |
4,4 |
4,7 |
|
Palmekernefedt |
4,6 |
5,9 |
|
Rapsolie |
2,0 |
4,4 |
|
Hørfrøolie |
2,0 |
4,0 |
|
Hvedekimolie |
2,7 |
6,4 |
|
Majskimolie |
2,2 |
4,5 |
|
Bomuldsfrøolie |
3,3 |
4,4 |
|
Svinefedt |
2,7 |
4,7 |
|
Oksetalg |
5,2 |
5,4 |
|
Hærdet fiskeolie |
5,4 |
6,1 |
B.2 BEREGNING
Der foretages kun en kvantitativ bestemmelse af fremmede fedtstoffer, hvis mindst en af S-værdierne (tabel 2 eller tabel 5) er overskredet. For at få kvantitative oplysninger beregnes massefraktionen, w f, af fremmede fedtstoffer eller af blanding af fremmede fedtstoffer, udtrykt i %, i prøven ved hjælp af følgende formel:

hvor
|
S |
er resultatet ved indsættelse af TG-data for mælkefedt, som der er tilsat et fremmed fedtstof eller en blanding af fremmede fedtstoffer, i en af ligningerne 3-7 |
|
S f |
er en konstant, der afhænger af, hvilken type fremmed fedtstof der er tilsat. |
Hvis typen af fremmed fedtstof, der er tilsat til mælkefedt, ikke kendes, anvendes en generel S f-værdi på 7,46 (tabel B.2). S-værdien fremkommet ved ligning 7 anvendes altid, også selv om dens S-grænser ikke er overskredet, men en anden lignings grænser er overskredet.
For kendte fremmede fedtstoffer indsættes deres specifikke S f-værdier (tabel B.2) i ligning B.1. Den relevante ligning vedrørende fremmede fedtstoffer vælges blandt ligningerne 3-6 for at beregne S.
Tabel B.2
Sf-værdier af forskellige fremmede fedtstoffer
|
Fremmed fedtstof |
Sf |
|
Ukendt |
7,46 |
|
Sojabønneolie |
8,18 |
|
Solsikkeolie |
9,43 |
|
Olivenolie |
12,75 |
|
Kokosolie |
118,13 |
|
Palmeolie |
7,55 |
|
Palmekerneolie |
112,32 |
|
Rapsolie |
3,30 |
|
Hørfrøolie |
4,44 |
|
Hvedekimolie |
27,45 |
|
Majskimolie |
9,29 |
|
Bomuldsfrøolie |
41,18 |
|
Svinefedt |
177,55 |
|
Oksetalg |
17,56 |
|
Fiskeolie |
64,12 |
B.3 ANGIVELSE AF RESULTATER
Resultatet angives med 2 decimaler.
Litteraturhenvisninger
|
1. |
Molkentin, J., Precht, D. Representative determination of the butyric acid content in European milk fats. Milchwissenschaft, 52, 1987, pp. 82-85 |
|
2. |
Precht, D., Molkentin, J. Quantitative triglyceride analysis using short capillary columns. Chrompack News, 4, 1993, pp. 16-17 |
|
3. |
Molkentin, J., Precht, D. Comparison of packed and capillary columns for quantitative gas chromatography of triglycerides in milk fat. Chromatographia, 39, 1994, pp. 265-270 |
|
4. |
Molkentin, J., Precht, D. Equivalence of packed and capillary GC columns with respect to suitability for foreign fat detection in butter using the triglyceride formula method. Chromatographia, 52, 2000, pp. 791-797 |
|
5. |
ISO 707IDF 50, Mælk og mælkeprodukter. Vejledning om prøveudtagning |
|
6. |
BS 410:1988, Test sieves — Technical requirements and testing |
|
7. |
Precht, D. Control of milk fat purity by gas chromatographic triglyceride analysis. Kieler Milchwirtsch. Forschungsber., 43, 1991, pp. 219-242 |
|
8. |
Precht, D. Detection of adulterated milk fat by fatty acid and triglyceride analyses. Fat Sci. Technol., 93, 1991, pp. 538-544 |
|
9. |
DIN 10336:1994, Nachweis und Bestimmung von Fremdfetten in Milchfett anhand einer gaschromatographischen Triglyceridanalyse [Påvisning og bestemmelse af fremmede fedtstoffer i mælkefedt ved gaskromatografisk triglyceridanalyse] |
|
10. |
Kommissionen for De Europæiske Fællesskaber: Consideration of results from the first, second, third, fourth, fifth and sixth EEC collaborative trial: Determination of triglycerides in milk fat; dok. nr. VI/2644/91, VI/8.11.91, VI/1919/92, VI 3842/92. VI/5317/92, VI/4604/93 |
|
11. |
Molkentin, J. Detection of foreign fat in milk fat from different continents by triacylglycerol analysis. Eur. J. Lipid Sci. Technol., 109, 2007, pp. 505-510 |
BILAG XXI
(Artikel 18)
PROCEDURE, SOM SKAL FØLGES, NÅR RESULTATERNE AF EN ANALYSE BESTRIDES (KEMISK ANALYSE)
1. Der foretages en yderligere analyse på et andet laboratorium, der er godkendt af den kompetente myndighed, ved anvendelse af den relevante metode efter anmodning fra den erhvervsdrivende, forudsat at forseglede dobbeltprøver af produktet er disponible og har været opbevaret på passende måde hos den kompetente myndighed. Anmodningen skal indgives inden syv arbejdsdage efter meddelelsen af resultaterne af den første analyse. Analysen foretages senest 21 arbejdsdage efter modtagelsen af anmodningen. Den kompetente myndighed sender efter anmodning fra den erhvervsdrivende og på dennes bekostning disse prøver til et andet laboratorium. Dette laboratorium skal være autoriseret til at udføre officielle analyser og skal have en dokumenteret kompetence for de pågældende relevante analyser.
2. De ekspanderede usikkerheder (k = 2) af gennemsnittet  af de
af de  gange foretagne målinger i laboratorium 1 af gennemsnittet
gange foretagne målinger i laboratorium 1 af gennemsnittet 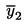 af de
af de  gange foretagne målinger i laboratorium 2 er henholdsvis
gange foretagne målinger i laboratorium 2 er henholdsvis
3.  og
og 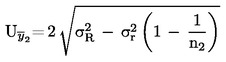 , hvor
, hvor  er standardafvigelsen for repeterbarhed, og
er standardafvigelsen for repeterbarhed, og ![]() er standardafvigelsen for reproducerbarhed ved den relevante metode. Hvis det endelige måleresultat y beregnes med en formel af typen
er standardafvigelsen for reproducerbarhed ved den relevante metode. Hvis det endelige måleresultat y beregnes med en formel af typen  ,
,  ,
,  eller
eller 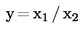 , følges de sædvanlige procedurer til kombinering af standardafvigelser i sådanne tilfælde for at finde usikkerheden.
, følges de sædvanlige procedurer til kombinering af standardafvigelser i sådanne tilfælde for at finde usikkerheden.
4. For at afprøve, om de to laboratoriers resultater er i overensstemmelse metodens standardafvigelse for reproducerbarhed ![]() , beregnes differensens ekspanderede usikkerhed
, beregnes differensens ekspanderede usikkerhed  :
:
5.  . Hvis den absolutte værdi af differencens mellem laboratoriernes gennemsnit
. Hvis den absolutte værdi af differencens mellem laboratoriernes gennemsnit 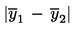 ikke er større en usikkerheden
ikke er større en usikkerheden  ,
,
![]() ,
,
er de to laboratoriers resultater i overensstemmelse med standardafvigelsen for reproducerbarhed  , og det aritmetiske gennemsnit af de to laboratoriers gennemsnit
, og det aritmetiske gennemsnit af de to laboratoriers gennemsnit
 ,
,
anføres som det endelige resultat. Dets ekspanderede usikkerhed er
 .
.
Sendingen afvises for ikke at overholde den øvre forskriftsmæssige grænse UL, hvis
 ;
;
ellers anses den for at overholde UL.
Sendingen afvises for ikke at overholde den nedre forskriftsmæssige grænse LL, hvis
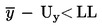 ;
;
ellers anses den for at overholde LL.
Hvis den absolutte værdi af differensen mellem laboratoriernes gennemsnit 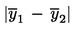 er større end usikkerheden
er større end usikkerheden  ,
,
![]() ,
,
er de to laboratoriers resultater ikke i overensstemmelse med standardafvigelsen for reproducerbarhed.
I så fald afvises sendingen for ikke at overholde forskrifterne, hvis den anden analyse bekræfter den første. Ellers accepteres sendingen.
Den kompetente myndighed meddeler den erhvervsdrivende de endelige resultater hurtigst muligt. Omkostningerne ved den anden analyse påhviler den erhvervsdrivende, hvis sendingen afvises.
BILAG XXII
SAMMENLIGNINGSTABEL
|
Forordning (EF) nr. 213/2001 |
Nærværende forordning |
|
Artikel 1 |
Artikel 1 |
|
Artikel 2 |
Artikel 1 |
|
Artikel 3 |
Artikel 2 |
|
— |
Artikel 3 |
|
Artikel 4 |
— |
|
Artikel 5 |
— |
|
Artikel 6 |
Artikel 4 |
|
Artikel 7 |
Artikel 18 |
|
Artikel 8 |
— |
|
Artikel 9 |
Artikel 5 |
|
Artikel 10 |
Artikel 6 |
|
Artikel 11 |
Artikel 7 |
|
Artikel 12 |
Artikel 8 |
|
Artikel 13 |
Artikel 9 |
|
Artikel 14 |
Artikel 10 |
|
Artikel 15 |
Artikel 11 |
|
Artikel 16 |
Artikel 12 |
|
Artikel 17 |
Artikel 13 |
|
— |
Artikel 14 |
|
Artikel 18 |
Artikel 15 |
|
Artikel 19 |
Artikel 16 |
|
|
Artikel 17 |
|
|
Artikel 19 |
|
Artikel 20 |
— |
|
Artikel 21 |
— |
|
Artikel 22 |
Artikel 20 |
|
Artikel 23 |
Artikel 21 |