PŘÍLOHA
Příloha nařízení (ES) č. 440/2008 se mění takto:
|
1) |
V části A se doplňuje tato kapitola: „A.25 DISOCIAČNÍ KONSTANTY VE VODĚ (TITRAČNÍ METODA – SPEKTROFOTOMETRICKÁ METODA – KONDUKTOMETRICKÁ METODA) ÚVOD Tato zkušební metoda je rovnocenná Pokynu OECD pro zkoušení (TG) č. 112 (1981). Předpoklady
Pomocné informace
Použití metody
Podklady pro metodu Tato zkušební metoda je založena na metodách uvedených v odkazech v oddíle „Literatura“ a na předběžném návrhu dokumentu Návod pro oznamování před výrobou, EPA, 18. srpna 1978. METODA – ÚVOD, ÚČEL, ROZSAH, RELEVANTNOST, POUŽITÍ A OMEZENÍ ZKOUŠKY Disociace látky ve vodě má význam pro posuzování jejího dopadu na životní prostředí. Závisí na ní forma látky, což určuje její chování a přepravu. Může ovlivňovat absorpci chemické látky do půdy a sedimentů a adsorpci do biologických buněk. Definice a jednotky Disociace je reverzibilní rozdělení molekuly na dvě nebo více chemických látek, které mohou mít charakter iontů. Tento proces obecně vyjadřuje rovnice RX ⇌ R ++ X – a koncentrační rovnovážná konstanta určující reakci je
Například v konkrétním případě, kde R je vodík (látkou je kyselina), je tato konstanta
nebo
Referenční látky Následující referenční látky není nutné používat vždy při zkoušení nové chemické látky. Jsou uvedeny především proto, aby bylo možné čas od času provést kalibraci metody a aby bylo možné porovnat výsledky v případě, že se použije jiná metoda.
Bylo by užitečné mít látku s několika hodnotami pK, jak je uvedeno níže v oddíle „Princip zkušební metody“. Takovou látkou by mohla být:
Princip zkušební metody Popisovaný chemický proces je celkově jen mírně závislý na teplotě v teplotním rozpětí, které je běžné v životním prostředí. Ke stanovení disociační konstanty je nutné změřit koncentrace disociované a nedisociované formy chemické látky. Příslušnou konstantu lze stanovit na základě znalosti stechiometrie disociační reakce uvedené výše v oddíle „Definice a jednotky“. V konkrétním případě popsaném v této zkušební metodě se látka chová jako kyselina nebo jako zásada a stanovení se nejpohodlněji provede určením relativních koncentrací iontové a neiontové formy látky a pH roztoku. Vztah mezi těmito pojmy popisuje rovnice pro výpočet pKa v oddíle „Definice a jednotky“ výše. Některé látky vykazují více než jednu disociační konstantu a lze odvodit obdobné rovnice. Některé z metod zde popsaných jsou vhodné také pro nekyselé/bazické disociace. Kritéria kvality Opakovatelnost Stanovení disociační konstanty by se mělo provést opakovaně (minimálně tři stanovení) s tolerancí ± 0,1 log. POPIS ZKUŠEBNÍCH POSTUPŮ Existují dvě základní metody pro stanovení pKa. Jedna zahrnuje titraci známého množství látky se standardní kyselinou nebo případně zásadou; druhá zahrnuje stanovení relativní koncentrace ionizované a neionizované formy a její závislosti na pH. Příprava Metody založené na těchto zásadách lze rozdělit na titrační, spektrofotometrické a konduktometrické. Zkušební roztoky Při titrační metodě a konduktometrické metodě by chemická látka měla být rozpuštěna v destilované vodě. Při spektrofotometrické a jiných metodách se používají pufrované roztoky. Koncentrace zkoušené chemické látky by neměla přesahovat 0,01 M nebo polovinu saturační koncentrace, podle toho, která z hodnot je nižší, a pro přípravu roztoku by měla být použita nejčistší dostupná forma látky. Je-li látka jen mírně rozpustná, může být před přípravou výše uvedených koncentrací rozpuštěna v malém množství rozpouštědla mísitelného s vodou. Roztoky by měly být za využití Tyndallova jevu zkontrolovány na přítomnost emulzí, zejména pokud byl použit kosolvent pro zvýšení rozpustnosti. Použijí-li se pufrované roztoky, neměla by koncentrace pufru přesáhnout 0,05 M. Podmínky zkoušky Teplota Teplota by měla být regulována s přesností nejméně ± 1°C. Stanovení by se mělo provádět při 20 °C. Existuje-li podezření na významnou závislost na teplotě, mělo by být stanovení prováděno alespoň při dvou jiných teplotách. Teplotní intervaly by v tomto případě měly být 10 °C a regulace teploty ± 0,1 °C. Analýzy Metoda bude určována povahou látky, která se testuje. Musí být dostatečně citlivá, aby umožňovala stanovení různých druhů chemických látek při každé koncentraci zkušebního roztoku. Provedení zkoušky Titrační metoda Zkušební roztok se testuje titrací se standardním roztokem zásady nebo případně kyseliny, přičemž po každém přidání titračního roztoku se změří pH. Před dosažením bodu ekvivalence by měl být roztok přidán v rostoucích objemech alespoň desetkrát. Je-li rovnováhy dosaženo dostatečně rychle, může se použít záznamový potenciometr. Pro tuto metodu musí být přesně známo celkové množství látky a její koncentrace. Je nutné vyloučit oxid uhličitý. Podrobnosti postupu, preventivní opatření a výpočet jsou uvedeny ve standardních zkouškách, např. v položkách (1), (2), (3) a (4) seznamu literatury. Spektrofotometrická metoda Vlnová délka se zjistí, pokud ionizovaná a neionizovaná forma látky má znatelně rozdílné absorpční koeficienty. Absorpční spektrum UV/VIS se zjistí z roztoků o konstantní koncentraci za takové hodnoty pH, kde je látka v podstatě neionizovaná, kde je plně ionizovaná a při několika mezilehlých hodnotách pH. Toho lze dosáhnout buď přidáním podílů koncentrované kyseliny (zásady) do poměrně velkého objemu roztoku látky v mnohosložkovém pufru, nejprve při vysoké (nízké) hodnotě pH (viz položka 5 seznamu literatury), anebo přidáním stejných objemů zásobního roztoku látky např. ve vodě nebo v methanolu ke konstantním objemům různých pufrovaných roztoků, jež pokrývají žádoucí rozpětí hodnot pH. Z hodnot pH a hodnot absorbance při zvolené vlnové délce se vypočítá dostatečný počet hodnot pKa za použití alespoň 5 hodnot pH, kdy je látka ionizována z nejméně 10 % a z méně než 90 %. Další podrobnosti o zkoušce a metoda výpočtu jsou uvedeny v odborné literatuře (1). Konduktometrická metoda Pomocí elektrického článku o nízké, známé článkové konstantě se změří vodivost přibližně 0,1 M roztoku látky ve vodivostní vodě. Změří se rovněž vodivost několika přesně připravených zředění tohoto roztoku. Koncentrace se pokaždé sníží na polovinu a celá série by měla pokrývat koncentrace nejméně v rozsahu jednoho řádu. Mezní vodivost při nekonečném zředění se zjistí provedením obdobného pokusu se sodnou solí a extrapolací. Míru disociace pak lze vypočítat z vodivosti každého roztoku pomocí Onsagerovy rovnice, a disociační konstantu je tedy možné vypočítat pomocí Ostwaldova zřeďovacího zákona jako K = α2C/(1 – α), kde C je koncentrace v molech na litr a α je disociovaná frakce. Je nutné vyloučit CO2. Další podrobnosti o zkoušce a metoda výpočtu jsou uvedeny v normách a odborné literatuře (1), (6) a (7). ÚDAJE A PŘEDKLÁDÁNÍ ZPRÁV Zpracování výsledků Titrační metoda Vypočítá se pKa pro 10 měřených bodů na titrační křivce. Vypočítá se střední hodnota těchto hodnot pKa a směrodatná odchylka. Měl by být přiložen graf závislosti hodnot pH na objemu standardní zásady nebo kyseliny spolu s vyjádřením v tabulce. Spektrofotometrická metoda Hodnoty absorbance a pH v každém spektru se uvedou v tabulce. Z mezilehlých údajů ve spektrech se vypočítá alespoň pět hodnot pKa a vypočte se rovněž střední hodnota těchto výsledků a směrodatná odchylka. Konduktometrická metoda Vypočítá se ekvivalentní vodivost Λ pro každou koncentraci kyseliny a pro každou koncentraci směsi jednoho ekvivalentu kyseliny plus 0,98 ekvivalentu hydroxidu sodného bez uhličitanů. Přebytek kyseliny má zabránit přebytku OH– v důsledku hydrolýzy. Hodnota 1/Λ se vynese do grafu v závislosti na √C a hodnotu Λo soli lze stanovit extrapolací na nulovou koncentraci. Hodnotu Λo kyseliny lze vypočítat pomocí hodnot H+ a Na+ uvedených v odborné literatuře. Hodnotu pKa je možné vypočítat z α = Λi /Λo a Ka = α2C/(1 – α) pro každou koncentraci. Přesnější hodnoty Ka lze získat provedením úprav na mobilitu a aktivitu. Měla by se vypočítat střední hodnota a směrodatné odchylky z hodnot pKa. Závěrečná zpráva Předložit by se měly všechny naměřené údaje a vypočítané hodnoty pKa spolu s metodou výpočtu (nejlépe ve formě tabulky, jak se doporučuje v položce seznamu literatury (1)) a stejně tak i výše popsané statistické parametry. U titračních metod by měly být uvedeny podrobnosti o standardizaci titračních roztoků. U spektrofotometrické metody by měla být předložena všechna spektra. U konduktometrické metody by měly být uvedeny podrobnosti týkající se stanovení článkové konstanty. Měly by se uvést informace o použitém postupu, analytických metodách a povaze všech použitých pufrů. Měla by se uvést zkušební teplota (teploty). LITERATURA
|



|
2) |
V části B se kapitola B.5 nahrazuje tímto: „B.5 AKUTNÍ DRÁŽDIVÉ/LEPTAVÉ ÚČINKY NA OČI ÚVOD Tato metoda je rovnocenná metodě Pokynu OECD pro zkoušení (TG) 405 (2012). Pokyny OECD pro zkoušení chemických látek jsou pravidelně přezkoumávány s cílem zajistit, aby odrážely nejlepší dostupné vědecké poznatky. Při předchozích přezkumech těchto pokynů pro zkoušení byla zvýšená pozornost věnována možným zlepšením, jichž lze dosáhnout vyhodnocováním veškerých existujících informací o zkoušené chemické látce s cílem vyhnout se nadbytečným zkouškám na laboratorních zvířatech a zohlednit tak zásadu dobrého zacházení se zvířaty. Pokyn pro zkoušení (TG) 405 (přijatý v roce 1981 a aktualizovaný v letech 1987, 2002 a 2012) obsahuje doporučení, aby byla před provedením popsané zkoušky akutních dráždivých/leptavých účinků na oči in vivo provedena analýza závažnosti důkazů (1) u existujících relevantních údajů. Pokud nejsou dostatečné údaje k dispozici, doporučuje se získat je pomocí metody postupného zkoušení (2) (3). Strategie zkoušení zahrnuje provedení validovaných a uznaných zkoušek in vitro a je popsána v doplňku této zkušební metody. Pro účely nařízení (ES) č. 1907/2006 o registraci, hodnocení, povolování a omezování chemických látek (REACH) (2) je v příslušném pokynu agentury ECHA uvedena rovněž integrovaná strategie zkoušení (21). Zkoušky na zvířatech by se měly provádět pouze v případě, kdy bylo po posouzení dostupných alternativních metod zjištěno, že tyto zkoušky jsou nezbytné a že použití takových zkoušek je přiměřené. V době přípravy této aktualizované zkušební metody existují případy, kdy použití této zkušební metody je stále nezbytné nebo je podle některých legislativních přístupů vyžadováno. Poslední aktualizace se zaměřila hlavně na použití analgetik a anestetik, aniž by to ovlivnilo základní koncept a strukturu TG 405. Výbor ICCVAM (3) a nezávislá mezinárodní vědecká komise pro odbornou revizi posoudily užitečnost a omezení běžného používání topických anestetik, systémových analgetik a humánních opatření při zkouškách bezpečnosti chemických látek z hlediska jejich dráždivých účinků na oči in vivo (12). Přezkum dospěl k závěru, že použití topických anestetik a systémových analgetik by mohlo z větší části nebo zcela zamezit bolestem a utrpení, aniž by ovlivnilo výsledek zkoušky, a bylo doporučeno, aby tyto látky byly vždy používány. Tato zkušební metoda bere tento posudek v úvahu. Při testování akutních dráždivých a leptavých účinků na oči by se topická anestetika, systémová analgetika a humánní opatření měly používat rutinně. Výjimky z jejich použití by měly být zdůvodněny. Vylepšení popsaná v této metodě podstatně sníží bolest a utrpení zvířat nebo jim zamezí ve většině zkušebních situací, kdy je testování bezpečnosti pro oči in vivo stále nezbytné. Vyvážené preventivní zmírňování bolesti by mělo zahrnovat i) běžné podávání topických anestetik (např. proparacain nebo tetracain) a systémových analgetik (např. buprenorfin) před expozicí, ii) rutinní plán systémové analgezie (např. buprenorfin a meloxicam) v postexpoziční době, iii) plánované prohlídky, sledování a zaznamenávání klinických příznaků bolesti a/nebo utrpení u zvířat a iv) plánované prohlídky, sledování a zaznamenávání povahy, závažnosti a progrese veškerých poškození očí. Další podrobnosti jsou uvedeny v níže popsaných aktualizovaných postupech. Po podání zkoušené chemické látky by další topická anestetika ani analgetika neměla být podávána, aby nedošlo k ovlivnění studie. Analgetika s protizánětlivým působením (např. meloxicam) by neměla být aplikována lokálně a použité dávky by neměly systémově ovlivňovat účinky na oči. Definice jsou uvedeny v dodatku této zkušební metody. VÝCHOZÍ ÚVAHY V zájmu spolehlivého vědeckého přístupu a dobrého zacházení se zvířaty by se ke zkouškám in vivo nemělo přistupovat, dokud nebyly všechny dostupné údaje významné pro možné leptavé/dráždivé účinky chemické látky na oči vyhodnoceny na základě analýzy závažnosti důkazů. Takové údaje zahrnují důkazy získané z existujících studií na lidech a/nebo laboratorních zvířatech, důkazy leptavých/dráždivých účinků na oči jedné nebo více strukturně příbuzných látek nebo směsí těchto látek, údaje dokládající vysokou kyselost nebo zásaditost chemické látky (4) (5) a výsledky validovaných a uznaných zkoušek leptavých účinků na kůži a leptavých a dráždivých účinků na oči in vitro nebo ex vivo (6) (13) (14) (15) (16) (17). Tyto studie mohly být vypracovány před analýzou závažnosti důkazů nebo na základě zjištění této analýzy. U některých chemických látek může taková analýza naznačovat nutnost provedení studií možných leptavých/dráždivých účinků chemické látky na oči in vivo. Ve všech takových případech by před zvážením použití oční zkoušky in vivo měla být přednostně nejprve provedena studie leptavých účinků chemické látky na kůži in vitro a/nebo in vivo a vyhodnocena v souladu se strategií postupného zkoušení uvedenou v rámci zkušební metody B.4 (7), nebo v souladu se strategií integrovaného zkoušení, která je popsána v pokynech agentury ECHA (21). Strategie postupného zkoušení, která zahrnuje provedení validovaných zkoušek leptavých/dráždivých účinků na oči in vitro a ex vivo, je popsána v doplňku této zkušební metody a pro účely nařízení REACH v pokynech agentury ECHA (21). Doporučuje se, aby se podle této strategie zkoušení postupovalo před prováděním zkoušek in vivo. U nových chemických látek se doporučuje metoda zkoušení po krocích, díky které lze získat vědecky spolehlivé údaje o leptavých/dráždivých účincích chemické látky. U existujících chemických látek, o jejichž leptavých/dráždivých účincích na kůži a oči není k dispozici dostatek údajů, lze tuto strategii využít pro doplnění chybějících informací. Použití jiné strategie nebo postupu zkoušení nebo rozhodnutí nepoužít metodu zkoušení po krocích musí být odůvodněno. PRINCIP ZKOUŠKY IN VIVO Po předchozím ošetření systémovým analgetikem a zavedení vhodného topického anestetika se chemická látka, která má být zkoušena, nanese v jedné dávce na jedno oko pokusného zvířete, přičemž neexponované oko slouží jako kontrola. V předem určených intervalech se na základě lézí spojivky, rohovky a duhovky vyhodnotí stupeň dráždivých/leptavých účinků na oči. Popíší se také jiné účinky na oko a nežádoucí systémové účinky, aby bylo možno provést úplné posouzení účinků. Doba studie by měla být dostatečně dlouhá, aby bylo možné zhodnotit vratnost nebo nevratnost účinků. Zvířata, která v jakékoli fázi zkoušky vykazují příznaky značného utrpení a/nebo bolesti nebo léze odpovídající humánním ukazatelům popsaným v této zkušební metodě (viz odstavec 26), se humánně utratí a chemická látka se odpovídajícím způsobem vyhodnotí. Kritéria rozhodování o humánním usmrcení umírajících a značně trpících zvířat lze nalézt v pokynech OECD (8). PŘÍPRAVA NA ZKOUŠKU IN VIVO Výběr druhů Upřednostňovaným laboratorním zvířetem je albinotický králík; používají se zdravá mladá dospělá zvířata. Použití jiných kmenů nebo druhů je třeba zdůvodnit. Příprava zvířat 24 hodin před zkouškou se u každého z předběžně vybraných pokusných zvířat provede vyšetření obou očí. Zvířata, u kterých se zjistí podráždění očí, oční defekt nebo poškození rohovky, se nepoužijí. Podmínky chovu a krmení Zvířata by měla být chována samostatně. Teplota v místnosti pro pokusná zvířata by měla být u králíků 20 °C (± 3 °C). Ačkoli by relativní vlhkost vzduchu měla být minimálně 30 % a pokud možno by kromě doby úklidu místnosti neměla přesáhnout 70 %, cílem by měla být hodnota 50–60 %. Osvětlení by mělo být umělé a mělo by se střídat 12 hodin světla a 12 hodin tmy. Je třeba se vyvarovat nadměrné intenzity světla. Ke krmení lze použít konvenční laboratorní stravu s neomezeným přísunem pitné vody. POSTUP ZKOUŠKY Použití topických anestetik a systémových analgetik K zamezení nebo minimalizaci bolesti a utrpení při postupech zkoušení bezpečnosti pro oči se doporučují následující postupy. Lze je nahradit alternativními postupy, u nichž bylo zjištěno, že stejně dobře nebo lépe zamezují nebo zbavují bolesti a utrpení.
Aplikace zkoušené chemické látky Zkoušená chemická látka se aplikuje každému zvířeti do spojivkového vaku jednoho oka tak, že se spodní víčko lehce odchlípne od oční bulvy. Víčka se pak asi na jednu sekundu lehce přidrží u sebe, aby nedošlo ke ztrátě látky. Druhé oko, na které se látka neaplikuje, slouží jako kontrola. Výplach Oči pokusných zvířat se do 24 hodin po instilaci zkoušené chemické látky nevyplachují, s výjimkou pevných látek (viz odstavec 18) a okamžitých leptavých nebo dráždivých účinků. Po 24 hodinách je možné v případě potřeby oči vypláchnout. Použití satelitní skupiny zvířat k vyšetření vlivu výplachu očí se nedoporučuje, pokud to není vědecky odůvodněno. Je-li satelitní skupina nezbytná, použijí se dva králíci. Podmínky výplachu se podrobně zdokumentují, např. doba výplachu, složení a teplota promývacího roztoku, trvání, objem a rychlost aplikace. Úroveň dávek 1) Zkoušení kapalin Při zkoušení kapalin se použije dávka 0,1 ml. K instilaci chemické látky přímo do oka by se neměly používat aerosolové rozstřikovače s čerpadlem. Před instilací 0,1 ml do oka by se měla tekutina z rozstřikovače vypudit a shromáždit do nádobky. 2) Zkoušení pevných látek Při zkoušení pevných látek, past a chemických látek obsahujících částice se použije objem 0,1 ml nebo hmotnost nejvýše 100 mg. Zkoušená chemická látka se rozdrtí na jemný prášek. Objem pevného materiálu se stanoví až po opatrném zhutnění, např. poklepáním odměrnou nádobkou. Pokud nebyla zkoušená pevná chemická látka odstraněna z oka pokusného zvířete pomocí fyziologických mechanismů do doby prvního pozorování po 1 hodině po aplikaci, lze oko vypláchnout fyziologickým roztokem nebo destilovanou vodou. 3) Zkoušení aerosolů Před instilací látek do oka se doporučuje zachytit látky obsažené v rozstřikovačích nebo aerosolech do nádobky. Jedinou výjimkou jsou chemické látky obsažené v aerosolové nádobce pod tlakem, které z důvodu odpařování zachytit nelze. V takových případech se oko podrží otevřené a chemická látka se do oka aplikuje přímým vstříknutím trvajícím jednu sekundu ze vzdálenosti 10 cm. Vzdálenost se může lišit podle tlaku v rozprašovači a jeho obsahu. Je třeba dbát na to, aby nedošlo k poškození oka tlakem z rozprašovače. Případně bude nutné vyhodnotit možnost „mechanického“ poškození oka v důsledku rozprašovacího tlaku. Odhad dávky z aerosolu lze provést simulací zkoušky takto: látka se nastříká na odvažovací papír otvorem o velikosti králičího oka, který se nachází bezprostředně před papírem. Na základě zvýšení hmotnosti papíru se odhadne množství látky, která se vstříkne do oka. U těkavých chemických látek lze dávku odhadnout pomocí zvážení odběrové nádobky před odstraněním zkoušené chemické látky a po jejím odstranění. Počáteční zkouška (zkouška dráždivých/leptavých účinků na oči in vivo na jednom zvířeti) Důrazně se doporučuje, aby se zkouška in vivo provedla zpočátku na jednom zvířeti (viz doplněk této zkušební metody: „Strategie postupného zkoušení dráždivých a leptavých účinků na oči“). Na základě pozorování by mělo být možné určit závažnost a vratnost předtím, než se přejde k potvrzující zkoušce na druhém zvířeti. Pokud výsledky této zkoušky naznačují, že chemická látka má leptavé nebo vysoce dráždivé účinky na oči při použití popsaného postupu, další zkoušení dráždivých účinků na oči se neprovádí. Potvrzující zkouška (zkouška dráždivých účinků na oči in vivo na dalších zvířatech) Pokud během počáteční zkoušky není pozorován leptavý nebo silně dráždivý účinek, měla by se dráždivá nebo negativní reakce potvrdit na maximálně dalších dvou zvířatech. Pokud je během počáteční zkoušky pozorován dráždivý účinek, doporučuje se, doporučuje se provést potvrzující zkoušku metodou postupného zkoušení vždy na jednom zvířeti spíše než exponováním dalších dvou zvířat současně. Pokud se u druhého zvířete projeví leptavé nebo silně dráždivé účinky, ve zkoušce se nepokračuje. Pokud výsledky u druhého zvířete jsou dostačující a umožňují stanovit kategorii nebezpečnosti, žádné další zkoušení se neprovádí. Doba pozorování Doba pozorování by měla být dostatečně dlouhá, aby bylo možné plně zhodnotit míru a vratnost pozorovaných účinků. Zkouška by však měla být ukončena vždy, pokud zvíře vykazuje příznaky značné bolesti nebo utrpení (8). Za účelem určení vratnosti účinků by se zvířata měla obvykle pozorovat 21 dnů po podání zkoušené chemické látky. Pokud je vratnost pozorována před uplynutím 21 dnů, zkouška se v tomto okamžiku ukončí. Klinická pozorování a hodnocení očních reakcí Oči by měly být komplexně zhodnoceny na přítomnost či nepřítomnost očních lézí hodinu po aplikaci zkoušené chemické látky a poté alespoň jednou denně. Zvířata by měla být pozorována v prvních třech dnech několikrát denně, aby se zajistilo, že rozhodnutí o utracení budou přijata včas. U zkušebních zvířat by se měly alespoň dvakrát denně po celou dobu studie pravidelně hodnotit na klinické příznaky bolesti a/nebo utrpení (např. opakované škrábání nebo tření oka, nadměrné mrkání, nadměrné slzení) (9) (10) (11) v minimálně šestihodinových nebo v případě potřeby i delších intervalech mezi jednotlivými pozorováními. Je nezbytné i) náležitě posoudit, zda jsou u zvířat patrné známky bolesti a utrpení, aby bylo možno přijmout podložená rozhodnutí, zda je nutné zvýšit dávkování analgetik, a ii) posoudit, zda jsou u zvířat patrné stanovené humánní ukazatele, aby bylo možné přijmout informované rozhodnutí, zda je nutné je humánně utratit, či nikoli, a zajistit, že tato rozhodnutí budou přijata včas. Běžně by se mělo používat zbarvení fluoresceinem a v případě potřeby (např. při posuzování hloubky poškození v případě zvředovatění rohovky) se jako pomůcka při detekci a měření poškození oka a při hodnocení, zda bylo dosaženo stanovených konečných ukazatelů pro humánní utracení, použije ruční štěrbinová lampa. Pro ilustraci lze pořídit digitální fotografie pozorovaných lézí a vést trvalý záznam o rozsahu poškození očí. Jakmile se získají konečné informace, nemělo by zkoušení na zvířatech probíhat déle, než je nezbytné. Zvířata se známkami značných bolesti nebo utrpení se neprodleně humánně utratí a chemická látka se odpovídajícím způsobem vyhodnotí. Humánně se utratí ta zvířata, u nichž došlo po instilaci k těmto očním lézím (popis stupňů lézí viz tabulka 1): perforace rohovky nebo výrazné zvředovatění rohovky včetně stafylomu; krev v přední komoře oční; zákal rohovky stupně 4; absence reakce na osvit (reakce duhovky stupně 2) přetrvávající 72 hodin; zvředovatění spojivkové membrány; nekróza spojivek nebo slzné žlázy; nebo odlupování nekrotické hmoty. Tento postup je dán nevratností takových lézí. Dále se doporučuje použít následující oční léze jako humánní koncové ukazatele pro ukončení studií před uplynutím plánované 21denní doby pozorování. Tyto léze jsou považovány za předpověď závažných dráždivých nebo leptavých poškození a poškození, u nichž se neočekává plná vratnost před ukončením 21denní doby pozorování: značná hloubka poškození (např. zvředovatění rohovky zasahující dále než do povrchových vrstev stromatu), poškození limbu > 50 % (doložené vyblednutím spojivkové tkáně) a silná infekce očí (výtok hnisu). Za užitečné kritérium, jež může ovlivnit klinické rozhodnutí o předčasném ukončení studie, lze také považovat kombinaci vaskularizace povrchu rohovky (tj. pannus, povrchový zánět rohovky), oblastí zbarvených fluoresceinem, které se na základě každodenního posuzování v čase nezmenšují, a/nebo nedostatečné reepitelizace 5 dní po aplikaci zkoušené chemické látky. Jednotlivě však tyto nálezy nejsou dostatečným důvodem k ukončení studie. Jakmile jsou zjištěny závažné účinky na oči, je třeba se obrátit na praktického nebo kvalifikovaného laboratorního veterinárního lékaře nebo osobu odborně způsobilou k identifikaci klinických lézí, aby se provedlo klinické vyšetření, jež stanoví, zda je nutné v důsledku kombinace těchto účinků studii předčasně ukončit. Stupně očních reakcí (spojivek, rohovky a duhovky) by měly být zjišťovány a zaznamenávány po 1, 24, 48 a 72 hodinách od aplikace zkoušené chemické látky (tabulka 1). Zvířata, u nichž oční léze nevzniknou, nelze utratit dříve než 3 dny po instilaci. Zvířata s očními lézemi, které nejsou závažné, se pozorují, dokud léze nevymizí, nebo po 21 dnů, po jejichž uplynutí se studie ukončí. Pozorování, jejichž cílem je stanovit stav lézí a jejich vratnost či nevratnost, by měla být prováděna a zaznamenávána minimálně po 1 hodině, 24 hodinách, 48 hodinách, 72 hodinách, 7 dnech, 14 dnech a 21 dnech. V nezbytných případech by se pozorování mělo provádět častěji s cílem určit, zda by pokusná zvířata měla být z humánních důvodů utracena nebo kvůli negativním výsledkům ze studie vyjmuta. Při každém vyšetření by měly být zaznamenány stupně očních lézí (tabulka 1). Zaznamenány by měly být také jakékoli další oční léze (např. zánět, zbarvení, změny v přední komoře oční) nebo nepříznivé systémové účinky. Vyšetřování reakcí lze usnadnit použitím binokulární lupy, ruční štěrbinové lampy, očního mikroskopu nebo jiných vhodných zařízení. Po zaznamenání pozorování po 24 hodin mohou být oči zvířat dále vyšetřeny fluoresceinem. Hodnocení reakcí očí je nevyhnutelně subjektivní. S cílem podporovat harmonizaci hodnocení reakcí očí a napomoci zkušebním laboratořím a těm, kdo provádějí a interpretují pozorování, musí být zaměstnanci provádějící pozorování odpovídajícím způsobem kvalifikováni pro práci s používaným systémem vyhodnocování. ÚDAJE A PŘEDKLÁDÁNÍ ZPRÁV Vyhodnocení výsledků Stupně dráždivých účinků na oči se zhodnotí ve spojení s povahou a stupněm závažnosti lézí a jejich vratností nebo nevratností. Jednotlivé stupně nepředstavují absolutní měřítko dráždivých vlastností chemické látky, neboť se hodnotí také jiné účinky zkoušené chemické látky. Jednotlivé stupně by spíše měly být považovány za referenční hodnoty, které jsou smysluplné, pouze pokud se opírají o úplný popis a hodnocení veškerých pozorování. Závěrečná zpráva Závěrečná zpráva by měla obsahovat tyto informace:
Interpretace výsledků Extrapolace výsledků studií dráždivých účinků na oči laboratorních zvířat na člověka je platná jen v omezené míře. Albinotický králík je ve většině případů na dráždivé a leptavé látky citlivější než člověk. Je třeba dbát na to, aby byla při interpretaci údajů vyloučena dráždivost pramenící ze sekundární infekce. LITERATURA
Tabulka 1 Stupnice očních lézí
Dodatek DEFINICE Kyselá/alkalická rezerva : U acidních přípravků je to množství (v g) hydroxidu sodného/100 g přípravku, které je nutné k dosažení konkrétní hodnoty pH. U alkalických přípravků je to množství (v g) hydroxidu sodného odpovídající g kyseliny sírové/100 g přípravku, které je nutné k dosažení konkrétní hodnoty pH (Young et al. (1988)). Chemická látka : Chemická substance nebo směs. Nedráždivé látky : Látky, jež nejsou klasifikovány jako látky s dráždivými účinky na oči kategorie I, II, nebo III EPA; nebo jako látky s dráždivými účinky na oči kategorie 1, 2, 2A nebo 2B GHS; nebo jako kategorie 1 nebo 2 EU (17) (18) (19). Látka s leptavými účinky na oči : a) Chemická látka, která způsobuje nevratné poškození oční tkáně; b) chemické látky, jež jsou klasifikovány jako látky s dráždivými účinky na oči kategorie 1 GHS nebo látky s dráždivými účinky na oči kategorie I EPA nebo kategorie 1 EU (17) (18) (19). Látka s dráždivými účinky na oči : a) Chemická látka, která vyvolává vratnou změnu v oku; b) chemické látky, jež jsou klasifikovány jako látky s dráždivými účinky na oči kategorie II nebo III EPA; nebo látky s dráždivými účinky na oči kategorie 2, 2A nebo 2B GHS; nebo kategorie 2 EU (17) (18) (19). Látka se silně dráždivými účinky na oči : a) Chemická látka, která způsobuje poškození oční tkáně, které nezmizí do 21 dnů od aplikace, nebo způsobuje závažné fyzické zhoršení vidění; b) chemické látky, jež jsou klasifikovány jako látky s dráždivými účinky na oči kategorie 1 GHS nebo látky s dráždivými účinky na oči kategorie I EPA nebo kategorie 1 EU (17) (18) (19). Zkoušená chemická látka : Jakákoli chemická substance nebo směs zkoušená pomocí této zkušební metody. Postupný přístup : Strategie postupného zkoušení, kde se všechny existující informace o zkoušené chemické látce přezkoumávají ve stanoveném pořadí s použitím hodnocení závažnosti důkazů na každém stupni, aby se zjistilo, zda jsou k dispozici dostačující informace pro rozhodnutí o klasifikaci nebezpečnosti, než se postoupí do dalšího stupně. Pokud lze potenciál dráždivých účinků zkoušené látky určit na základě existujících informací, další zkoušení není nutné. Jestliže potenciál dráždivých účinků zkoušené látky nelze určit na základě existujících informací, použije se postupný přístup zkoušení na zvířatech po krocích, dokud se nestanoví jednoznačná klasifikace. Závažnost důkazů (postup) : Silné a slabé stránky souboru informací se použijí jako základ pro vyvození závěrů, které z jednotlivých údajů nemusí být zjevné. DOPLNĚK ZKUŠEBNÍ METODY B.5 (4) STRATEGIE POSTUPNÉHO ZKOUŠENÍ DRÁŽDIVÝCH A LEPTAVÝCH ÚČINKŮ NA OČI Obecné informace V zájmu spolehlivého vědeckého přístupu a dobrého zacházení se zvířaty je důležité vyhýbat se zbytečnému používání zvířat a minimalizovat jakékoli zkoušky, které u zvířat pravděpodobně vyvolají závažné reakce. Veškeré informace týkající se potenciálních dráždivých/leptavých účinků chemické látky na oči by se měly zhodnotit před zvažováním zkoušení in vivo. Může již existovat dostatek důkazů pro klasifikaci zkoušené chemické látky podle jejích potenciálních dráždivých nebo leptavých účinků na oči, aniž by bylo nutné provádět zkoušky na laboratorních zvířatech. Proto využití analýzy závažnosti důkazů a strategie postupného zkoušení sníží potřebu provádět zkoušení in vivo, zejména pokud chemická látka pravděpodobně vyvolá závažné reakce. Doporučuje se, aby se stávající informace týkající se dráždivých a leptavých účinků chemické látky na oči zhodnotily pomocí analýzy závažnosti důkazů, na jejímž základě se rozhodne o tom, zda by se k lepší charakteristice tohoto potenciálu měly provést další studie, jiné než oční studie in vivo. Jsou-li nutné další studie, doporučuje se využít strategie postupného zkoušení a jejím prostřednictvím získat relevantní experimentální údaje. U látek, které doposud nebyly zkoušeny, by se měla strategie postupného zkoušení využít k získání údajů, které jsou nezbytné k vyhodnocení jejich leptavých nebo dráždivých účinků na oči. Původní strategie zkoušení popsaná v tomto doplňku byla vypracována na semináři OECD (1). Následně byla tato strategie potvrzena a rozpracována v Harmonizovaném integrovaném systému klasifikace nebezpečnosti chemických látek pro lidské zdraví a jejich vlivu na životní prostředí, který schválilo 28. společné zasedání Výboru pro chemické látky a Pracovní skupiny pro chemické látky v listopadu 1998 (2) a aktualizovala skupina odborníků OECD v roce 2011. Ačkoliv tato strategie zkoušení není nedílnou součástí zkušební metody B.5, vyjadřuje doporučovaný postup stanovení dráždivých/leptavých účinků na oči. Tento postup představuje nejlepší praktický i etický standard pro zkoušení dráždivých/leptavých účinků na oči in vivo. Zkušební metoda obsahuje pokyny pro provádění zkoušky in vivo a shrnuje faktory, kterým je nutno před zvažováním takové zkoušky věnovat pozornost. Strategie postupného zkoušení představuje přístup posuzování stávajících údajů o dráždivých a leptavých účincích chemických látek na oči a odstupňovaný přístup k získávání relevantních údajů o chemických látkách, u kterých je nutné provést další studie, nebo u nichž žádné studie nebyly doposud provedeny. Zkušební strategie zahrnuje provedení validovaných a uznaných zkoušek in vitro a ex vivo a poté, za specifických okolností, provedení studie zkušební metodou B.4 (3) (4). Popis strategie zkoušení po krocích Před zahájením zkoušek v rámci strategie postupného zkoušení (viz obrázek) se vyhodnotí veškeré dostupné informace, aby se mohlo rozhodnout o nutnosti provést zkoušku na očích in vivo. Ačkoliv lze podstatné informace získat z vyhodnocení jednotlivých parametrů (např. extrémní hodnota pH), stávající informace by se měly hodnotit jako celek. Všechny relevantní údaje o účincích zkoušené chemické látky a jejích strukturních obdob se vyhodnotí na základě rozhodnutí založeného na posouzení závažnosti důkazů a toto rozhodnutí se zdůvodní. Zvláštní důraz by měl být kladen na již existující údaje o účincích chemické látky na člověka a na zvířata a dále na výsledky zkoušení in vitro nebo ex vivo. Studie žíravých chemických látek in vivo by se měly provádět co nejméně. Mezi faktory uvedené ve strategii zkoušení patří:
STRATEGIE ZKOUŠENÍ A HODNOCENÍ DRÁŽDIVÝCH/LEPTAVÝCH ÚČINKŮ NA OČI
LITERATURA
|
|
3) |
V části B se kapitola B.10 nahrazuje tímto: „B.10 ZKOUŠKA NA CHROMOZÓMOVÉ ABERACE U SAVCŮ IN VITRO ÚVOD Tato zkušební metoda je rovnocenná pokynu OECD pro zkoušení (TG) č. 473 (2016). Je součástí série zkušebních metod v oblasti genetické toxikologie. Byl vypracován dokument OECD, který obsahuje stručné informace o zkoušení v oblasti genetické toxikologie a přehled posledních změn, které byly v těchto pokynech ke zkoušení provedeny (1). Zkouška na chromozómové aberace u savců in vitro má identifikovat chemické látky, které způsobují strukturní chromozómové aberace v kultivovaných buňkách savců (2) (3) (4). Rozlišují se dva typy strukturních aberací: chromozómové a chromatidové. Při zkouškách na chromozómové aberace in vitro by mohla vzniknout polyploidie (včetně endoreduplikace). Aneugenní látky mohou vyvolat polyploidii, avšak z polyploidie samotné nevyplývá aneugenický potenciál a může prostě ukazovat na narušení buněčného cyklu nebo na cytotoxicitu (5). Tato zkouška není určena k měření aneuploidie. K detekci aneuploidie by bylo možné doporučit zkoušku na přítomnost mikrojader in vitro (6). Ke zkoušce na chromozómové aberace in vitro mohou být použity stabilizované buněčné linie nebo primární buněčné kultury lidského původu nebo pocházející z hlodavců. Použité buňky by měly být vybrány na základě schopnosti růstu v kultuře, stálosti karyotypu (včetně počtu chromozómů) a spontánní četnosti výskytu chromozómových aberací (7). Údaje, které jsou v současné době k dispozici, neumožňují předkládat jednoznačná doporučení, avšak naznačují, že při hodnocení chemické nebezpečnosti je důležité zvážit stav p53, genetickou stálost (stálost karyotypu), reparační schopnost DNA a původ buněk (lidské versus pocházející z hlodavců) vybraných ke zkoušení. Uživatelům této zkušební metody se proto doporučuje, aby při detekování indukce chromozómových aberací zvážili vliv těchto i jiných buněčných charakteristik na vlastnosti buněčné linie. Použité definice jsou uvedeny v dodatku 1. VÝCHOZÍ ÚVAHY A OMEZENÍ Zkoušky prováděné in vitro zpravidla vyžadují použití exogenního zdroje metabolické aktivace, pokud nejsou buňky metabolicky kompetentní s ohledem na zkoušené chemické látky. Systém exogenní metabolické aktivace zcela nenapodobuje podmínky in vivo. Je třeba se zcela vyvarovat podmínek, které by vedly k falešným pozitivním výsledkům, tj. k poškození chromozómů, jež není způsobeno přímou interakcí mezi zkoušenou chemickou látkou a chromozómy; takové podmínky zahrnují změny pH nebo osmolality (8) (9) (10), interakce se složkami média (11) (12) nebo nadměrné úrovně cytotoxicity (13) (14) (15) (16). Tato zkouška se používá k odhalování chromozómových aberací, jež mohou být výsledkem klastogenního působení. Analýza indukce chromozómových aberací by se měla provádět pomocí buněk v metafázi. Je proto důležité, že buňky by měly dosáhnout mitózy jak v exponovaných, tak v neovlivněných kulturách. Při zkoušení vyráběných nanomateriálů mohou být potřebné určité úpravy metody, které však v této zkušební metodě nejsou popsány. Před použitím této zkušební metody na směs za účelem získání údajů pro zamýšlené použití v právních předpisech by se mělo zvážit, zda tato metoda může pro tento účel poskytnout přiměřené výsledky, a pokud ano, proč je tomu tak. Takové úvahy nejsou nutné, pokud existuje právní požadavek na zkoušení dané směsi. PRINCIP ZKOUŠKY Buněčné kultury lidského a savčího původu jsou vystaveny zkoušené chemické látce za přítomnosti exogenního zdroje metabolické aktivace i bez něho, pokud ovšem nejsou použity buňky s dostatečnou schopností metabolismu (viz odstavec 13). Ve vhodných předem stanovených intervalech po začátku expozice buněčných kultur zkoušené chemické látce se přidá látka zastavující metafázi (např. colcemid nebo kolchicin), buňky se shromáždí, obarví a u buněk v metafázi je mikroskopicky analyzována přítomnost chromozómových a chromatidových aberací. POPIS METODY Příprava Buňky Mohou být použity různé buněčné linie (např. vaječník křečka čínského (CHO), plíce křečka čínského V79, plíce křečka čínského (CHL/IU, TK6) nebo primární buněčné kultury včetně lymfocytů periferní krve člověka nebo jiných savců (7). Volba buněčných linií by měla být odborně zdůvodněna. Použijí-li se primární buňky, mělo by se vzhledem k ochraně pokusných zvířat, tam, kde je to možné, zvážit použití primárních buněk lidského původu. V takovém případě se odběr vzorků provede v souladu s etickými zásadami a předpisy platnými pro člověka. Lymfocyty periferní krve člověka by měly být získány od mladých (přibližně ve věku 18–35 let) nekuřáků, o nichž není známo, že by trpěli nemocemi nebo že by byli nedávno vystaveni genotoxickým činitelům (např. chemickým látkám, ionizujícímu záření) na úrovních, které by mohly zvýšit spontánní výskyt chromozómových aberací. Tím by se mělo zajistit, že spontánní výskyt chromozómových aberací bude nízký a stálý. Spontánní výskyt chromozómových aberací se s věkem zvyšuje a tento trend je více patrný u žen než u mužů (17) (18). Pokud se společně používají buňky pocházející od více než jednoho dárce, měl by být počet dárců uveden. Je nezbytné prokázat, že buňky se dělily od počátku ošetření zkoušenou chemickou látkou až do odběru buněk k analýze. Buněčné kultury jsou udržovány ve fázi exponenciálního růstu buněk (buněčné linie) nebo jsou stimulovány k dělení (primární kultury lymfocytů), aby bylo možné exponovat buňky v různých stadiích buněčného cyklu, protože citlivost stadií na zkoušené chemické látky nemusí být známa. Primární buňky, které k dělení potřebují stimulaci mitogenními látkami, během expozice zkoušené chemické látce již zpravidla nejsou dále synchronizovány (např. lidské lymfocyty po 48hodinové mitogenní stimulaci). Použití synchronizovaných buněk v průběhu expozice se nedoporučuje, avšak lze je akceptovat v odůvodněných případech. Média a kultivační podmínky Pro udržování kultur by mělo být použito vhodné kultivační médium a inkubační podmínky (kultivační nádoby, případně zvlhčená atmosféra s 5 % CO2, inkubační teplota 37 °C). U buněčných linií by se měla pravidelně kontrolovat stabilita modální hodnoty počtu chromozómů a mělo by se zjišťovat, zda nejsou kontaminovány mykoplasmaty (7) (19). Pokud jsou kultury kontaminované nebo se modální hodnota počtu chromozómů změnila, neměly by se dotyčné kultury používat. Pro buněčné linie nebo primární kultury použité ve zkušební laboratoři by měla být stanovena normální délka buněčného cyklu, která by měla být v souladu s buněčnými charakteristikami uvedenými v literatuře (20). Příprava kultur Buněčné linie: buňky se pomnoží z kmenových kultur, nasadí se do kultivačního média v takové hustotě, aby u buněk v suspenzích nebo v monovrstvách pokračoval exponenciální růst až do sběru buněk (např. mělo by se zamezit konfluenci u buněk rostoucích v monovrstvách). Lymfocyty: krev ošetřená antikoagulantem (např. heparinem) nebo oddělené lymfocyty se kultivují (např. po dobu 48 hodin u lidských lymfocytů) za přítomnosti mitogenu [např. fytohemaglutinin (PHA) u lidských lymfocytů], aby se indukovalo dělení buněk před expozicí zkoušené chemické látce. Metabolická aktivace Při použití buněk, které mají nedostatečnou endogenní metabolickou schopnost, by se měly použít exogenní metabolizující systémy. Nejčastěji používaným systémem, který je standardně doporučován, pokud není zdůvodněno použití jiného systému, je postmitochondriální frakce (S9) doplněná kofaktory. Tato frakce se připravuje z jater hlodavců (zpravidla potkanů) exponovaných látkami indukujícími enzymy, jako je Aroclor 1254 (21) (22) (23) nebo kombinace fenobarbitalu a β-naftoflavonu (24) (25) (26) (27) (28) (29). Posledně jmenovaná kombinace není v rozporu se Stockholmskou úmluvou o perzistentních organických znečišťujících látkách (30) a bylo prokázáno, že je stejně účinná jako Aroclor 1254 pro indukování oxidáz se smíšenou funkcí (24) (25) (26) (28). Frakce S9 je v konečném testovacím médiu obvykle používána v koncentracích od 1 % do 2 % obj., ale koncentraci je možné zvýšit až na 10 %. V průběhu aplikace by se neměly používat látky, jež snižují mitotický index, zejména látky, které tvoří komplexy s vápníkem (31). Volbu typu a koncentrace systému exogenní metabolické aktivace nebo metabolického induktoru, který se použije, je třeba provádět s ohledem na typ zkoušených chemických látek Příprava zkoušené chemické látky Pevné zkoušené chemické látky by měly být před aplikací na buňky rozpuštěny ve vhodných rozpouštědlech a popřípadě zředěny (viz odstavec 23). Kapalné zkoušené látky mohou být přidány přímo k testovacímu systému a/nebo mohou být před aplikací zředěny. Plynné a těkavé zkoušené chemické látky by se měly zkoušet za použití vhodně upravených standardních protokolů, například prostřednictvím ošetření buněk v neprodyšně uzavřených kultivačních nádobách (32) (33) (34). Zkoušená chemická látka by měla být připravena bezprostředně před aplikací, pokud údaje o stálosti neprokazují možnost skladování. Zkušební podmínky Rozpouštědla Rozpouštědlo by mělo být zvoleno tak, aby optimalizovalo rozpustnost zkoušené chemické látky, aniž by nepříznivě ovlivnilo provádění zkoušky, např. měnilo růst buněk, ovlivňovalo integritu zkoušené chemické látky, reagovalo s kultivačními nádobami, narušovalo systém metabolické aktivace. Doporučuje se pokud možno nejdříve zvážit použití vodného rozpouštědla (nebo kultivačního média). Dobře zavedenými rozpouštědly jsou například voda nebo dimethylsulfoxid. Podíl organických rozpouštědel by zpravidla neměl být větší než 1 % obj. a podíl vodných rozpouštědel (fyziologického roztoku nebo vody) v konečném expozičním médiu by neměl být větší než 10 % obj. Použijí-li se jiná než dobře zavedená rozpouštědla (např. ethanol nebo aceton), mělo by být jejich použití podloženo údaji o jejich kompatibilitě se zkoušenou chemickou látkou a zkušebním systémem a o tom, že nejsou v použité koncentraci genotoxické. Pokud takové podpůrné údaje neexistují, je důležité zařadit neexponované kontroly (viz dodatek 1) prokazující, že zvolené rozpouštědlo nevyvolává žádné škodlivé nebo klastogenní účinky. Měření buněčné proliferace a cytotoxicity a volba expozičních koncentrací Při stanovení nejvyšší koncentrace zkoušené chemické látky by neměly být použity koncentrace, které jsou schopny vyvolat falešnou pozitivní odpověď, jako např. koncentrace, jež vyvolávají nadměrnou cytotoxicitu (viz odstavec 22), srážení kultivačního média (viz odstavec 23) nebo zřetelné změny pH nebo osmolality (viz odstavec 5). Pokud zkoušená chemická látka v době jejího přidání způsobuje zřetelnou změnu hodnoty pH média, pH může být upraveno pufrováním konečného expozičního média tak, aby nedošlo k falešným pozitivním výsledkům a aby byly zachovány vhodné kultivační podmínky. Provádí se měření buněčné proliferace, aby bylo zajištěno, že dostatečný počet ošetřených buněk prodělal během zkoušky mitózu a že expozice je prováděna při vhodných úrovních cytotoxicity (viz odstavce 18 a 22). Cytotoxicita by měla být stanovena v hlavním experimentu s metabolickou aktivací a bez ní za použití vhodných indikátorů buněčné smrti a růstu. Hodnocení cytotoxicity při předběžné zkoušce může být užitečné a může napomoci k lepšímu určení koncentrací, jež mají být použity při hlavní zkoušce, avšak předběžná zkouška není povinná. Pokud se provede, nemůže nahradit měření cytotoxicity při hlavním experimentu. Vhodnými metodami pro posouzení cytotoxicity v cytogenetických zkouškách jsou relativní zdvojnásobení populace (RPD) nebo relativní nárůst počtu buněk (RICC) (13) (15) (35) (36) (55) (příslušné vzorce viz dodatek 2). V případě dlouhodobé expozice a dobách odběru delších než 1,5násobek normální délky buněčného cyklu po zahájení zkoušky (tj. celkově delších než trojnásobek délky buněčného cyklu) by použití RPD mohlo vést k podhodnocení cytotoxicity (37). Za těchto okolností měření RICC může být lepším ukazatelem nebo v případě použití RPD by hodnocení cytotoxicity po 1,5násobku normální délky buněčného cyklu mohlo být užitečné. U lymfocytů v primárních kulturách je mírou cytotoxických/cytostatických účinků mitotický index (MI), avšak ten je ovlivněn tím, po jaké době od expozice se měří, použitým mitogenem a možným narušením buněčného cyklu. MI je však přijatelný, protože jiné metody stanovení cytotoxicity mohou být obtížné a nepraktické a nemusí platit pro cílovou populaci lymfocytů rostoucích v reakci na stimulaci pomocí PHA. Ačkoli doporučenými parametry cytotoxicity jsou pro buněčné linie RICC a RPD a pro primární kulturu lymfocytů MI, mohly by další užitečné informace poskytnout i jiné ukazatele (např. integrita buněk, apoptóza, nekróza, buněčný cyklus). Měly by se vyhodnotit nejméně tři zkušební koncentrace (nepočítaje v to kontroly s rozpouštědlem a pozitivní kontroly), které splňují kritéria přijatelnosti (vhodná cytotoxicita, počet buněk atd.). Bez ohledu na typy buněk (buněčné linie nebo primární kultury lymfocytů) mohou být pro každou zkušební koncentraci použity buď jednotlivé exponované kultury nebo replikáty.I když je doporučováno použití duplicitních kultur, jednu kulturu lze rovněž akceptovat za předpokladu, že v této jediné kultuře je hodnocen stejný celkový počet buněk jako v duplicitních kulturách. Použití jedné kultury je opodstatněné zejména v případě, že jsou posuzovány více než 3 koncentrace (viz odstavec 31). Výsledky získané z duplicitních nezávislých kultur při dané koncentraci lze pro účely analýzy údajů sloučit (38). U zkoušených látek, jež vykazují malou cytotoxicitu nebo nevykazují žádnou cytotoxicitu, budou obvykle vhodné přibližně dvojnásobné až trojnásobné intervaly koncentrací. V případě cytotoxicity by měly zvolené zkušební koncentrace pokrývat rozpětí od koncentrace vyvolávající cytotoxicitu, jak je popsáno v odstavci 22, po koncentrace, při nichž dochází ke střední a nízké cytotoxicitě nebo nedochází k žádné cytotoxicitě. Mnohé zkoušené chemické látky vykazují strmé křivky závislosti odezvy na koncentraci, a aby bylo možné získat údaje při nízké a střední cytotoxicitě nebo podrobně studovat závislost účinku na dávce, může být nezbytné zvolit menší rozdíly mezi jednotlivými koncentracemi a/nebo více než tři koncentrace (ať už pro jednu kulturu, nebo replikáty), zejména v případě kdy je požadován opakovaný experiment (viz odstavec 47). Je-li maximální koncentrace odvozena od cytotoxicity, měla by být nejvyšší koncentrace zvolena tak, aby se dosáhlo 55 ± 5 % cytotoxicity za použití doporučených parametrů cytotoxicity (tj. snížení RICC a RPD u buněčných linií a snížení MI u primárních kultur lymfocytů na 45 ± 5 % u souběžné negativní kontroly). Je třeba dbát na to, aby byly interpretovány pouze ty pozitivní výsledky, které jsou vykazovány při vyšší hranici tohoto rozmezí cytotoxicity 55 ± 5 % (13). V případě špatně rozpustných zkoušených chemických látek, které nejsou cytotoxické při koncentracích nižších, než je jejich rozpustnost, by nejvyšší analyzovaná koncentrace měla na konci expozice zkoušené chemické látce vyvolat zákal nebo sraženinu viditelnou pouhým okem nebo pomocí inverzního mikroskopu. Dokonce i v případech, kdy k výskytu cytotoxicity dochází při koncentracích vyšších, než je rozpustnost, se doporučuje testovat pouze při jedné koncentraci vyvolávající zákal nebo viditelné sraženiny, neboť sraženina může vést k falešným účinkům. Při koncentraci, kdy vzniká sraženina, je třeba dbát na to, aby sraženina nemohla ovlivňovat provádění zkoušky (např. barvení nebo hodnocení). Užitečné může být stanovit rozpustnost v kultivačním médiu před pokusem. Není-li pozorována sraženina nebo omezení cytotoxicity, měla by nejvyšší zkušební koncentrace odpovídat 10 mM, 2 mg/ml nebo 2 μl/ml, podle toho, která z uvedených hodnot je nejnižší (39) (40) (41). Pokud u zkoušené chemické látky není stanoveno její složení, např. u látek s neznámým nebo proměnlivým složením, komplexních reakčních produktů nebo biologického materiálu (UVCB) (42), přírodního extraktu atd., může v případě nízké cytotoxicity být horní koncentrace vyšší (např. 5 mg/ml), aby se zvýšila koncentrace každé ze složek. Je však třeba poznamenat, že u humánních léčivých přípravků se tyto požadavky mohou lišit (43). Kontroly Pro každý časový interval sběru buněk by měly být použity souběžné negativní kontroly (viz odstavec 15) skládající se ze samotného rozpouštědla v kultivačním médiu a zpracované stejným způsobem jako exponované kultury. Souběžné pozitivní kontroly jsou nutné pro prokázání schopnosti laboratoře identifikovat klastogeny za daných experimentálních podmínek a pro prokázání účinnosti exogenního systému metabolické aktivace, všude kde je to možné. Příklady pozitivních kontrol jsou uvedeny v tabulce 1 níže. V odůvodněných případech mohou být použity jiné pozitivní kontrolní chemické látky. Jelikož zkoušky na buňkách savců in vitro za účelem stanovení genetické toxicity jsou dostatečně standardizované, může se použití pozitivních kontrol omezit na experimenty s kontrolními klastogeny vyžadujícími metabolickou aktivaci. Pokud je provedena souběžně se zkouškou bez aktivace se stejnou dobou trvání expozice, tato jediná odpověď v pozitivní kontrole prokáže činnost metabolického aktivačního systému a citlivost testovacího systému. Dlouhodobá expozice (bez S9) by však měla mít svou vlastní pozitivní kontrolu, protože doba trvání expozice bude odlišná než při zkoušce za použití metabolické aktivace. Každá pozitivní kontrola by měla být použita při jedné nebo více koncentracích, u nichž se očekává, že poskytnou reprodukovatelný a detekovatelný nárůst oproti pozadí, čímž se prokáže citlivost testovacího systému (tj. aby byl účinek zřetelný, ale aby při odečtu nevyšla ihned najevo identita kódovaného preparátu), a reakce by neměla být narušena cytotoxicitou přesahující limitní hodnoty stanovené ve zkušební metodě. Tabulka 1. Referenční chemické látky doporučené pro posouzení způsobilosti laboratoře a pro výběr pozitivních kontrol.
POSTUP Expozice zkoušené chemické látce Proliferující buňky se vystaví zkoušené chemické látce jak za přítomnosti metabolického aktivačního systému, tak bez něho. Doba sběru buněk Pro důkladné hodnocení, které by bylo zapotřebí pro vyvození závěru o negativním výsledku, by měly být splněny všechny tři následující zkušební podmínky za použití krátkodobé expozice s metabolickou aktivací a bez ní a dlouhodobé expozice bez metabolické aktivace (viz odstavce 43, 44 a 45):
V případě, že kterékoli z výše uvedených experimentálních uspořádání vedlo k pozitivní odpovědi, nebude nutné zkoumat kterýkoli z ostatních režimů aplikace. Příprava preparátů pro analýzu chromozómů Do buněčné kultury se obvykle 1–3 hodiny před ukončení kultivace přidá colcemid nebo kolchicin. Pro přípravu preparátů pro analýzu chromozómů se každá buněčná kultura zpracovává zvlášť. Příprava preparátů pro analýzu chromozómů zahrnuje hypotonizaci buněk, fixaci a obarvení buněk. Na konci doby expozice v trvání 3–6 hodin se v monovrstvách mohou vyskytovat mitotické buňky (které se vyznačují okrouhlým tvarem a odchlipováním od povrchu). Jelikož lze tyto mitotické buňky snadno oddělit, mohou být při odstranění média obsahujícího zkoušenou chemickou látku ztraceny. Pokud v době sběru buněk existují důkazy podstatného nárůstu počtu mitotických buněk v porovnání s kontrolami, což pravděpodobně ukazuje na zastavení mitózy, pak by buňky měly být odebrány odstředěním a přidány zpět do kultur, aby se zabránilo ztrátě buněk, u nichž probíhá mitóza a mohou zde vznikat chromozómové aberace. Analýza Všechny preparáty, včetně pozitivních a negativních kontrol, by měly být před mikroskopickou analýzou chromozómových aberací nezávisle kódovány. Poněvadž při fixaci u částu buněk v metafázi často dochází ke ztrátě chromozómů, měly by tudíž vyšetřované buňky obsahovat centromery v počtu rovném modální hodnotě +/- 2. Aby bylo možno vyhodnotit zkoušenou chemickou látku jako jasně negativní, u každé koncentrace a kontroly by mělo být hodnoceno alespoň 300 dobře rozprostřených metafází. Jsou-li využívány k replikám, mělo by být těchto 300 buněk rovnoměrně rozděleno mezi tyto kultury. Pokud je pro každou koncentraci použita jediná kultura (viz odstavec 21), mělo by být hodnoceno alespoň 300 dobře rozprostřených metafází v této jedné kultuře. Hodnocení 300 buněk má tu výhodu, že se zvyšuje statistická síla zkoušky a navíc budou zřídka pozorovány nulové hodnoty (předpokládá se, že pouze u 5 % buněk) (44). Počet hodnocených metafází lze snížit, je-li pozorován velký počet chromozómových aberací a zkoušená chemická látka je považována za jasně pozitivní. Měly by být hodnoceny buňky se strukturními chromozómovými aberacemi včetně gapů a s vyloučením gapů. Zlomy a gapy jsou definovány v Dodatku 1 podle (45) (46). Chromatidové a chromozómové aberace by měly být zaznamenávány odděleně a klasifikovány podle podtypů (zlomy, výměny). Postupy používané v laboratoři by měly zajistit, aby analýzu chromozómových aberací prováděli dobře odborně připravení hodnotitelé a aby tato analýza byla případně podrobena odborné revizi. Ačkoli je účelem zkoušky detekovat strukturní chromozómové aberace, je důležité zaznamenat četnost výskytu polyploidií a endoreduplikací, jsou-li tyto jevy pozorovány (viz odstavec 2). Způsobilost laboratoře Aby laboratoř prokázala dostatečnou zkušenost s testem před rutinním použitím, měla by provést sérii experimentů s referenčními pozitivními chemickými látkami, jež působí prostřednictvím různých mechanismů, a s různými negativními kontrolami (s použitím různých rozpouštědel/vehikul). Odpovědi těchto pozitivních a negativních kontrol by měly být v souladu s odbornou literaturou. Toto se nepoužije na laboratoře, které tuto zkušenost mají, tj. které mají k dispozici databázi historických údajů, jak je definována v odstavci 37. U vybraných pozitivních chemických látek (viz tabulka 1 v odstavci 26) by měla být testována krátká a dlouhá expozice za nepřítomnosti metabolické aktivace a rovněž krátká expozice za přítomnosti metabolické aktivace, aby byla prokázána způsobilost pro detekci klastogenních chemických látek a stanovení účinnosti systému metabolické aktivace. Rozsah koncentrací by u vybraných chemických látek měl být zvolen tak, aby poskytl opakovatelná zvýšení účinku v jednotlivých koncentracích ve srovnání s úrovní pozadí, aby se tak prokázala citlivost a dynamické rozpětí testovacího systému. Historické kontrolní údaje Laboratoř by měla stanovit:
Při prvním získávání údajů o historické distribuci negativních kontrol by měly být výsledky provedených negativních kontrol v souladu s publikovanými kontrolními údaji, pokud existují. S tím, jak je do distribuce kontrol přidáváno více údajů z experimentů, měly by aktuální negativní kontroly v ideálním případě spadat do 95 % rozmezí této distribuce (44) (47). Historická databáze negativních kontrol dané laboratoře by měla být vytvořena na základě nejméně 10 experimentů, nejlépe však by měla zahrnovat nejméně 20 experimentů provedených za srovnatelných zkušebních podmínek. V laboratořích by měly být uplatňovány metody řízení jakosti, jako např. kontrolní diagramy (např.C-charts nebo X-bar charts (48)), z nichž je patrné, jak proměnlivé jsou jejich údaje o pozitivních a negativních kontrolách a že metodika je v dané laboratoři „pod kontrolou“ (44). Další doporučení, jak získat a používat historické údaje (tj. kritéria pro zařazení údajů do historické databáze a jejich vyřazení a kritéria přijatelnosti pro daný experiment), lze najít v literatuře (47). Jakékoli změny zkušebního protokolu by měly být zvažovány z hlediska jejich souladu se stávajícími historickými kontrolními databázemi laboratoře. Jakýkoli větší změna by měla vést k zavedení nové historické kontrolní databáze. Údaje o negativních kontrolách by měly zahrnovat výskyt buněk s chromozómovými aberacemi z jedné kultury nebo ze souhrnu replikovaných kultur, jak je popsáno v odstavci 21. Výsledky aktuálních negativních kontrol by v ideálním případě měly spadat do 95 % rozmezí distribuce v historické databázi negativních kontrol laboratoře (44) (47). Pokud jsou údaje z aktuálních negativních kontrol mimo 95 % rozmezí, může být přijatelné je zahrnout do dosavadní distribuce kontrol, pokud tyto údaje nejsou mimořádně odlehlé a pokud existuje důkaz o tom, že testovací systém je „pod kontrolou“ (viz odstavec 37), a důkaz o tom, že nedošlo k technické nebo lidské chybě. ÚDAJE A PŘEDKLÁDÁNÍ ZPRÁV Vyjádření výsledků Měl by se vyhodnotit procentuální podíl buněk se strukturními chromozómovými aberacemi. Chromatidové a chromozómové aberace klasifikované podle podtypů (zlomy, výměny) by měly být uvedeny odděleně spolu s jejich počty a četností v experimentálních a kontrolních kulturách. Gapy se zaznamenávají a uvádějí odděleně, ale nezahrnují se do celkové četnosti výskytu aberací. Zaznamená se procentuální podíl polyploidie a/nebo endoreduplikovaných buněk, pokud jsou pozorovány. Měly by být zaznamenány výsledky souběžného stanovení cytotoxicity u všech exponovaných kultur a negativních a pozitivních kontrolních kultur v hlavních experimentech pro hodnocení aberací. Měly by být uvedeny údaje pro jednotlivé kultury. Všechny údaje by měly být shrnuty ve formě tabulek. Kritéria přijatelnosti Přijetí zkoušky je založeno na následujících kritériích:
Hodnocení a interpretace výsledků Za předpokladu, že všechna kritéria přijatelnosti byla splněna, lze zkoušenou chemickou látku považovat za jasně pozitivní, pokud v některé ze zkoumaných experimentálních uspořádání (viz odstavec 28):
Jsou-li splněna všechna tato kritéria, má se za to, že zkoušená chemická látka je schopna vyvolávat chromozómové aberace v kultivovaných savčích buňkách v tomto testovacím systému. Doporučení ohledně nejvhodnějších statistických metod lze najít v literatuře (49) (50) (51). Za předpokladu, že byla splněna všechna kritéria přijatelnosti, lze zkoušenou chemickou látku považovat za jasně negativní, pokud za všech zkoumaných experimentálních uspořádání (viz odstavec 28):
Pak se má za to, že zkoušená chemická látka není schopna vyvolávat chromozómové aberace v kultivovaných savčích buňkách v tomto testovacím systému. Ověření jasně pozitivní či jasně negativní odpovědi se nepožaduje. V případě, že odpověď není ani jasně negativní, ani jasně pozitivní, jak je popsáno výše, nebo s cílem napomoci při stanovení biologického významu výsledku, by měly být údaje vyhodnoceny na základě odborného posouzení a/nebo dalším šetřením. Užitečné by mohlo být hodnocení většího počtu buněk (je-li vhodné) nebo provedení opakovaného experimentu, případně s použitím upravených zkušebních podmínek (např. rozsah koncentrací, jiné podmínky metabolické aktivace (např. koncentrace S9 nebo původu S9)). V ojedinělých případech dokonce ani po dalším šetření nebude možné na základě daného souboru údajů učinit závěr, zda jsou výsledky pozitivní či negativní, a proto bude učiněn závěr, že odpověď na zkoumanou chemickou látku je neurčitá. Nárůst počtu polyploidních buněk může znamenat, že zkoušená chemická látka má schopnost potlačit mitotické procesy a indukovat numerické chromozómové aberace (52). Nárůst počtu buněk s endoreduplikovanými chromozómy může znamenat, že zkoušené chemické látky mají schopnost potlačit progresi buněčného cyklu (53) (54) (viz odstavec 2). Výskyt polyploidních buněk a buněk s endoreduplikovanými chromozómy by se proto měl zaznamenat odděleně. Závěrečná zpráva Závěrečná zpráva by měla obsahovat tyto informace:
LITERATURA
Dodatek 1 DEFINICE Aneuploidie : jakákoli odchylka od normálního diploidního (nebo haploidního) počtu chromozómů o jeden nebo více chromozómů, avšak nikoli o celou sadu (nebo více sad) chromozómů (polyploidie). Apoptóza : programovaná buněčná smrt, která se vyznačuje řadou kroků vedoucích k rozpadu buněk na membránově vázané části, které jsou poté odstraněny fagocytózou nebo rozkladem. Buněčná proliferace : zvyšování počtu buněk v důsledku jejich mitotického dělení. Chemická látka : chemická substance nebo směs. Chromatidový zlom : přerušení jedné chromatidy, při kterém dojde k jasnému odchýlení jedné z chromatid. Chromatidový gap : oblast bez zabarvení (achromatická léze) jedné chromatidy, ve které existuje minimální přerušení chromatidy. Chromatidová aberace : strukturní poškození chromozómu v podobě zlomu jedné chromatidy nebo zlomu a opětného spojení chromatid. Chromozómová aberace : strukturní poškození chromozómu v podobě zlomu nebo zlomu a spojení obou chromatid v tomtéž místě. Klastogen : jakákoli chemická látka, která způsobuje strukturní chromozómové aberace v populacích buněk nebo eukaryotických organismů. Koncentrace : odkazuje na konečné koncentrace zkoušené chemické látky v kultivačním médiu. Cytotoxicita : u zkoušek prováděných podle této zkušební metody s použitím buněčných linií se cytotoxicita vyjadřuje jako snížení relativního zdvojnásobení populace (RPD) nebo relativního nárůstu počtu buněk (RICC) u exponovaných buněk v porovnání s negativní kontrolou (viz odstavec 17 a dodatek 2). U zkoušek prováděných podle této zkušební metody s použitím primárních kultur lymfocytů se cytotoxicita vyjadřuje jako snížení mitotického indexu (MI) exponovaných buněk v porovnání s negativní kontrolou (viz odstavec 18 a dodatek 2). Endoreduplikace : proces, při kterém v jádře po S-fázi replikace DNA nedojde k mitóze, nýbrž následuje další S-fáze. Výsledkem jsou chromozómy se 4, 8, 16 … chromatidami. Genotoxický : obecný termín zahrnující všechny typy poškození DNA nebo chromozómů včetně jejich rozlámání, delecí, aduktů, modifikací a propojení nukleotidů, přestavby, genových mutací, chromozómových aberací a aneuploidie. Ne všechny druhy genotoxických účinků způsobují mutace nebo stálé poškození chromozómů. Mitotický index (MI) : podíl buněk, které se nacházejí v metafázi, z celkového počtu buněk v populaci; udává stupeň proliferace této populace. Mitóza : proces dělení buněčného jádra, který se obvykle člení na profázi, prometafázi, metafázi, anafázi a telofázi. Mutagenní : produkující dědičné změny sekvence (sekvencí) párů bází DNA v genech nebo struktury chromozómů (chromozómové aberace). Numerická aberace : odchylka počtu chromozómů od normálního počtu obvyklého u použitého typu buněk. Polyploidie : numerické chromozómové aberace v buňkách nebo organismech, které se týkají celé sady (celých sad) chromozómů, na rozdíl od jednoho chromozómu nebo jednotlivých chromozómů (aneuploidie). Stav p53 : protein p53 se podílí na regulaci buněčného cyklu, apoptózy a reparace DNA. Buňky, jež mají nedostatek funkčního proteinu p53, nejsou schopny zastavovat buněčný cyklus nebo odstraňovat poškozené buňky prostřednictvím apoptózy nebo jiných mechanismů (např. indukcí reparace DNA) souvisejících s funkcemi p53 v reakci na poškození DNA, by teoreticky měly být náchylnější ke genovým mutacím nebo chromozómovým aberacím. Relativní nárůst počtu buněk (RICC) : zvýšení počtu buněk v chemicky ošetřených kulturách oproti zvýšení v neexponovaných kulturách, poměr vyjádřený v procentech. Relativní zdvojnásobení populace (RPD) : zvýšení počtu pozorovaných zdvojnásobení populace v chemicky ošetřených kulturách oproti zvýšení v neexponovaných kulturách, poměr vyjádřený v procentech. S9 jaterní frakce : supernatant homogenátu jater po odstředění při 9 000 g, tj. surový jaterní extrakt. S9 směs : směs S9 jaterní frakce a kofaktorů nezbytných pro metabolickou aktivitu enzymů. Kontrola s rozpouštědlem : obecný termín k označení kontrolních kultur, ke kterým se přidává pouze rozpouštědlo používané k rozpuštění zkoušené chemické látky. Strukturní aberace : mikroskopicky pozorovatelné změny struktury chromozómů při buněčném dělení ve stadiu metafáze; jeví se jako delece a fragmenty, intrachromozómální nebo interchromozómální změny. Zkoušená chemická látka : jakákoli chemická látka nebo směs zkoušená pomocí této zkušební metody. Neexponované kontroly : kultury, které se neexponují (tj. ani chemické látce, ani rozpouštědlu), ale zpracovávají se souběžně stejným způsobem jako kultury, k nimž se se přidává zkoušená chemická látka. Dodatek 2 VZORCE PRO HODNOCENÍ CYTOTOXICITY Mitotický index (MI):
Doporučuje se použít relativní nárůst počtu buněk (RICC = Relative Increase in Cell Counts) nebo relativní zdvojnásobení populace (RPD = Relative Population Doubling), jelikož oba tyto ukazatele zohledňují poměr buněčné populace, která se rozdělila.
kde: Zdvojnásobení populace = [log (počet buněk po expozici ÷ počáteční počet buněk)] ÷ log 2. Například hodnota 53 % RICC nebo RPD označuje 47 % cytotoxicity/cytostáze a 55 % cytotoxicity/cytostáze měřené pomocí MI znamená, že skutečný MI je 45 % kontroly. V každém případě by měl být změřen počet buněk před expozicí a počet buněk v exponované kultuře a v negativní kontrolní kultuře. V minulosti byl jako parametr cytotoxicity užíván RCC (tj. počet buněk v exponovaných kulturách / počet buněk v kontrolních kulturách), nyní se již nedoporučuje, protože může cytotoxicitu podhodnotit. V negativních kontrolních kulturách by mělo být zdvojnásobení populace slučitelné s požadavkem na odběr buněk po takové době od aplikace, která odpovídá 1,5násobku normální délky buněčného cyklu, a mitotický index by měl být natolik vyšší, aby se získal dostatečný počet buněk v mitóze a aby bylo možné spolehlivě vypočítat snížení o 50 %. |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||



|
4) |
V části B se kapitola B.11 nahrazuje tímto: „B.11 Zkouška na chromozómové aberace v buňkách kostní dřeně savců ÚVOD Tato zkušební metoda je rovnocenná Pokynu OECD pro zkoušení (TG) č. 475 (2016). Je součástí série zkušebních metod v oblasti genetické toxikologie. Byl vypracován dokument OECD, který obsahuje stručné informace o zkoušení v oblasti genetické toxikologie a přehled posledních změn, které byly v těchto pokynech ke zkoušení provedeny (1). Zkouška na chromozómové aberace v buňkách kostní dřeně savců in vivo je zvláště významná k posouzení genotoxicity, neboť zde probíhá aktivní metabolismus in vivo, farmakokinetika a procesy reparace DNA, které přispívají k účinku testované látky, jakkoli mohou být různé u různých druhů. Zkouška in vivo je rovněž užitečná pro další výzkum genotoxicity zjištěné v systémech in vitro. Zkouška na chromozómové aberace u savců in vivo je používána pro detekci strukturních chromozómových aberací indukovaných zkoušenou chemickou látkou v buňkách kostní dřeně savců, obvykle hlodavců (2) (3) (4) (5). Rozlišují se dva typy strukturních aberací – chromozómové a chromatidové. U většiny genotoxických chemických látek jsou indukované aberace chromatidového typu, avšak chromozómové aberace se rovněž vyskytují. Poškození chromozómů a související jevy jsou příčinou mnoha geneticky podmíněných chorob u člověka a existuje mnoho důkazů o tom, že pokud tyto léze a související jevy způsobují změny v onkogenech a v tumor-supresorových genech, mají podíl na indukci rakoviny u člověka a v experimentálních systémech. Při zkouškách na chromozómové aberace in vivo mohou vznikat polyploidie (včetně endoreduplikací). Z nárůstu polyploidie samotné však nevyplývá aneugenický potenciál a může prostě ukazovat na narušení buněčného cyklu nebo na cytotoxicitu. Tato zkouška není určena k detekci aneuploidie. Pro detekci aneuploidie in vivo a in vitro lze doporučit mikronukleus test v savčích erytrocytech in vivo (kapitola B.12 této přílohy) nebo zkoušku in vitro na přítomnost mikrojader v buňkách savců (kapitola B.49 této přílohy). Definice použitých termínů jsou uvedeny v dodatku 1. VÝCHOZÍ ÚVAHY Při této zkoušce jsou běžně používáni hlodavci, avšak v některých případech, je-li to vědecky zdůvodněno, mohou být vhodné i jiné druhy. Cílovou tkání je v této zkoušce kostní dřeň, poněvadž je vysoce vaskularizovanou tkání a obsahuje populaci buněk s rychlým cyklem, které se snadno izolují a zpracovávají. Vědecké zdůvodnění použití jiných druhů než potkanů a myší by mělo být uvedeno ve zprávě. Jsou-li použity jiné druhy než hlodavci, doporučuje se detekci chromozómových aberací v buňkách kostní dřeně začlenit do jiné vhodné zkoušky toxicity. Jestliže existuje důkaz o tom, že se zkoušená chemická látka (chemické látky) nebo reaktivní metabolit (metabolity) nedostanou do cílové tkáně, není vhodné tuto zkoušku použít. Před použitím této zkušební metody na směs za účelem získání údajů pro zamýšlené použití v právních předpisech by se mělo zvážit, zda tato metoda může pro tento účel poskytnout přiměřené výsledky, a pokud ano, proč je tomu tak. Takové úvahy nejsou nutné, pokud existuje právní požadavek na zkoušení dané směsi. PRINCIP ZKUŠEBNÍ METODY Zvířata se vhodným způsobem vystaví zkoušené chemické látce a ve vhodném okamžiku po expozici se humánně utratí. Před utracením se zvířatům podá látka zastavující metafázi (např. kolchicin nebo colcemid). Z buněk kostní dřeně se poté připraví preparáty chromozómů, obarví se a analyzují se chromozómové aberace buněk v metafázi. OVĚŘENÍ ZPŮSOBILOSTI LABORATOŘE Experimenty pro ověření způsobilosti Aby bylo možné před zahájením rutinního zkoušení zjistit, zda laboratoř má s danou zkouškou dostatečné zkušenosti, měla by prokázat schopnost reprodukovat očekávané výsledky z publikovaných údajů (např. (6)), pokud jde o četnost chromozómových aberací, nejméně se dvěma pozitivními kontrolními chemickými látkami (včetně slabých reakcí indukovaných nízkými dávkami u pozitivních kontrol), např. látkami uvedenými v tabulce 1, a s kontrolami s kompatibilním vehikulem/rozpouštědlem (viz odstavec 22). Při těchto experimentech by se měly používat dávky, jež poskytují reprodukovatelné nárůsty v závislosti na dávce, a měly by prokázat citlivost a dynamické rozpětí zkušebního systému ve sledované tkáni (kostní dřeň) za použití hodnotící metody, jež má být v laboratoři uplatňována. Tento požadavek neplatí pro laboratoře, které tuto zkušenost mají, tj. které mají dostupnou databázi historických údajů, jak je definována v odstavcích 10–14. Historické kontrolní údaje V průběhu šetření způsobilosti by laboratoř měla stanovit:
Při prvním získávání údajů o dosavadní distribuci negativních kontrol by měly být výsledky provedených negativních kontrol v souladu s publikovanými kontrolními údaji, pokud existují. S tím, jak je do distribuce kontrol přidáváno více údajů z experimentů, měly by souběžné negativní kontroly v ideálním případě spadat do 95 % rozmezí této distribuce. Historická databáze negativních kontrol laboratoře by měla být statisticky robustní, aby byla zajištěna schopnost laboratoře posuzovat distribuci svých údajů o negativních kontrolách. V literatuře se doporučuje, že nezbytným minimem může být 10 experimentů, nejlépe však by databáze měla zahrnovat nejméně 20 experimentů provedených za srovnatelných zkušebních podmínek. V laboratořích by měly být uplatňovány metody řízení jakosti, jako např. kontrolní diagramy (např.C-charts nebo X-bar charts (7)), z nichž je patrné, jak proměnlivé jsou jejich údaje a že metodika je v dané laboratoři „pod kontrolou“. Další doporučení, jak získat a používat dosavadní údaje (tj. kritéria pro zařazení údajů do historické databáze a jejich vyřazení a kritéria přijatelnosti pro daný experiment), lze najít v literatuře (8). Pokud laboratoř neprovedla dostatečný počet experimentů, aby mohla během ověření šetření způsobilosti (popsaného v odstavci 9) stanovit statisticky robustní distribuci negativních kontrol (viz odstavec 11), je přijatelné, aby distribuce byla stanovena během prvních rutinních zkoušek. Uplatňování tohoto přístupu by mělo vycházet z doporučení uvedených v literatuře (8) a výsledky negativních kontrol získané při těchto experimentech by měly zůstat v souladu s publikovanými údaji o negativních kontrolách. Jakékoli změny zkušebního protokolu by měly být zvažovány z hlediska jejich dopadu na výsledné údaje tak, aby zůstaly v souladu se stávající historickou kontrolní databází laboratoře. Pouze případy větších rozdílů ve zkušebním protokolu by měly vést k zavedení nové historické kontrolní databáze, přičemž její odlišnost od dřívější distribuce se stanoví na základě odborného posouzení (viz odstavec 11). V průběhu opětovného stanovení nemusí být k provedení aktuální zkoušky nutná úplná databáze negativních kontrol, pokud laboratoř prokáže, že hodnoty jejích souběžných negativních kontrol zůstávají v souladu s její předchozí databází nebo s odpovídajícími publikovanými údaji. Údaje o negativních kontrolách by měly zahrnovat výskyt strukturních chromozómových aberací (s vyloučením gapů) u každého zvířete. Výsledky souběžných negativních kontrol by v ideálním případě měly být v 95 % rozmezí distribuce negativních kontrol v historické databázi laboratoře. Pokud jsou údaje ze souběžných negativních kontrol mimo 95 % rozmezí, může být přijatelné je zařadit mezi dosavadní distribuci kontrol, pokud tyto údaje nejsou mimořádně odlehlé a pokud existuje důkaz o tom, že testovací systém je „pod kontrolou“ (viz odstavec 11), a neexistuje důkaz o tom, že by došlo k technické nebo lidské chybě. POPIS METODY Příprava Výběr druhu zvířat Měly by být použity běžně používané laboratorní kmeny zdravých mladých pohlavně dospělých zvířat. Běžně je používán potkan, ale vhodná může být také myš. Použit může být i jakýkoli jiný vhodný druh savce, je-li ve zprávě poskytnuto vědecké zdůvodnění. Podmínky chovu a krmení zvířat Teplota v místnosti pro zvířata by u hlodavců měla být 22 °C (± 3 °C). Relativní vlhkost vzduchu by v ideálním případě měla být 50–60 %, ale minimálně 40 % a neměla by pokud možno přesáhnout 70 % kromě doby úklidu místnosti. Osvětlení by mělo být umělé a mělo by se střídat 12 hodin světla a 12 hodin tmy. Ke krmení lze použít konvenční laboratorní krmivo s neomezeným přísunem pitné vody. Výběr potravy může být ovlivněn nutností zajistit dostatečné mísení se zkoušenou látkou, je-li látka podávána touto cestou. Hlodavci by měli být chováni v malých skupinách (nejvýše po pěti v každé kleci) téhož pohlaví a se stejnou aplikací, pokud se nepředpokládá agresivní chování, přednostně v klecích s pevnou podlahou s vhodným obohacením životního prostředí. Zvířata mohou být umístěna v klecích individuálně, pouze je-li to vědecky odůvodněné. Příprava zvířat Obvykle se používají zdravá mladá dospělá zvířata (u hlodavců ideálně ve věku 6–10 týdnů na počátku expozice, ale přijatelná jsou i zvířata poněkud starší), která se náhodně přiřadí do kontrolních a experimentálních skupin. Jednotlivá zvířata se jednoznačně identifikují humánní, co nejméně invazivní metodou (např. kroužkováním, označením štítkem, pomocí mikročipu nebo biometrické identifikace, avšak nikoli nastřihnutím ucha nebo prstu dolní končetiny) a nechají se v laboratorních podmínkách alespoň pět dní aklimatizovat. Klece by měly být uspořádány tak, aby byl vliv umístění klecí minimalizován. Mělo by se zabránit vzájemné kontaminaci pozitivní kontroly a zkoušené chemické látky. V okamžiku zahájení studie by měla být odchylka v hmotnosti zvířat minimální a neměla by překročit ± 20 % střední hodnoty hmotnosti zvířat daného pohlaví. Příprava dávek Pevné zkoušené chemické látky by se měly rozpustit nebo suspendovat ve vhodných rozpouštědlech nebo vehikulech nebo by měly být přimíchány do potravy nebo pitné vody před podáním dávky zvířatům. Kapalné zkoušené chemické látky mohou být podávány přímo nebo mohou být před podáním zředěny. Pro účely expozice inhalací lze zkoušené chemické látky v závislosti na jejich fyzikálně-chemických vlastnostech podávat jako plyn, páru nebo pevný/kapalný aerosol. Měly by být použity čerstvě připravené zkoušené chemické látky, pokud údaje o stálosti neprokazují možnost skladování a nestanoví vhodné podmínky pro skladování. Rozpouštědlo/vehikulum Rozpouštědlo/vehikulum by nemělo mít při použitých hladinách dávek toxické účinky a mělo by být vyloučeno podezření, že reaguje se zkoušenou chemickou látkou. Jsou-li použita jiná než známá rozpouštědla/vehikula, mělo by být jejich zařazení podloženo referenčními údaji o jejich kompatibilitě. Doporučuje se pokud možno nejprve zvážit použití vodných rozpouštědel/vehikul. Jako příklad běžně používaných kompatibilních rozpouštědel/vehikul lze uvést vodu, fyziologický roztok, roztok methylcelulosy, roztok sodné soli karboxymethylcelulosy, olivový olej a kukuřičný olej. Neexistují-li historické nebo publikované kontrolní údaje prokazující, že zvolené atypické rozpouštědlo/vehikulum nevyvolává žádné strukturní aberace nebo jiné zhoubné účinky, měla by být provedena počáteční studie za účelem stanovení přijatelnosti kontroly s tímto rozpouštědlem/vehikulem. Kontroly Pozitivní kontroly V každé zkoušce by měla být obvykle zařazena jedna skupina zvířat vystavených pozitivní kontrolní chemické látce. Od toho lze upustit, jestliže zkušební laboratoř prokázala způsobilost k provádění zkoušky a stanovila historické rozmezí pozitivních kontrol. Není-li zařazena souběžná pozitivní kontrolní skupina, měly by být v každém experimentu zařazeny hodnotící kontroly (fixované a neobarvené preparáty). Tyto hodnotící kontroly lze získat tak, že se do hodnocení studie zařadí vhodné referenční vzorky, které byly získány a uchovány ze samostatného experimentu s pozitivními kontrolami prováděného pravidelně (např. každých 6–18 měsíců) v laboratoři, kde se provádí daná zkouška, například během testování způsobilosti a poté pravidelně podle potřeby. Chemické látky pro pozitivní kontrolu by měly spolehlivě vyvolat pozorovatelné zvýšení výskytu buněk se strukturními chromozómovými aberacemi nad spontánní úroveň. Dávky pozitivní kontroly by měly být zvoleny tak, aby byly účinky zřetelné, ale aby hodnotitel okamžitě nezjistil identitu kódovaného preparátu. Je přijatelné, aby látka v pozitivní kontrole byla podávána jiným způsobem než zkoušená chemická látka, podle odlišného harmonogramu expozice, a aby se odběr vzorků prováděl pouze v jediném okamžiku. Pro pozitivní kontrolu navíc může být vzato v úvahu použití chemických látek ze stejné chemické třídy, je-li to vhodné. Příklady pozitivních kontrolních chemických látek jsou uvedeny v tabulce 1. Tabulka 1. Příklady pozitivních kontrolních chemických látek
Negativní kontroly V každém expozičním intervalu by měla být zařazena souběžná negativní kontrolní skupina zvířat, které jinak podstupují stejný proces jako exponované skupiny až na to, že nejsou vystavena zkoušené chemické látce. Používá-li se při podávání zkoušené chemické látky rozpouštědlo/vehikulum, měla by toto rozpouštědlo/vehikulum dostávat i kontrolní skupina. Pokud však dosavadní údaje o negativních kontrolách v různých intervalech expozice v dané zkušební laboratoři prokazují konzistentní údaje o variabilitě zvířat a četnosti buněk se strukturními aberacemi, může být pro negativní kontrolu nezbytný pouze jeden interval expozice. Je-li u negativních kontrol použit jediný odběr buněk, měl by se provést v prvním (nejkratším) intervalu expozice použitém ve studii. POSTUP Počet a pohlaví zvířat Obecně je známo, že účinky zjištěné v mikronukleus testu jsou podobné u samců a samic (9) a předpokládá se, že tomu tak bude i u strukturních chromozómových aberací; většinu studií proto je možné provést s kterýmkoli z obou pohlaví. Existence údajů prokazujících významné rozdíly mezi samci a samicemi (např. rozdíly v systémové toxicitě, metabolismu, biologické dostupnosti, toxicita pro kostní dřeň atd. získané např. ve studii ke stanovení rozsahu) by byla důvodem pro použití zvířat obojího pohlaví. V tomto případě může být vhodné provést studii s oběma pohlavími, např. jako součást studie toxicity po opakovaných dávkách. V případě použití obou pohlaví by mohlo být vhodné použít faktoriálního uspořádání experimentu. Podrobnosti ohledně toho, jak analyzovat údaje získané při tomto uspořádání experimentu, jsou uvedeny v dodatku 2. Velikost skupin při zahájení studie by měla být stanovena tak, aby poskytla minimálně pět analyzovatelných zvířat jednoho pohlaví, nebo každého pohlaví, jsou-li použita obě, z každé skupiny. Je-li expozice člověka chemickým látkám specifická pro určité pohlaví, jako je tomu například u některých léčivých přípravků, měla by být zkouška provedena se zvířaty odpovídajícího pohlaví. Určitým vodítkem pro maximální typické požadavky na počet zvířat může být, že studie s buňkami kostní dřeně při dvou odběrech se třemi skupinami exponovanými dávce jednou souběžnou negativní kontrolní skupinou plus jednou pozitivní kontrolní skupinou (každá skupina složena z pěti zvířat jednoho pohlaví) by vyžadovala 45 zvířat. Dávkování Jestliže se provádí předběžná studie pro stanovení rozsahu dávek, poněvadž nejsou k dispozici vhodné údaje, jež by mohly pomoci při stanovení dávek, pak by měla být provedena ve stejné laboratoři, se stejným druhem, kmenem, pohlavím a za stejného režimu expozice, které se použijí v hlavní studii (10). Cílem studie by mělo být zjistit maximální tolerovanou dávku, která je definována jako nejvyšší dávka, která bude po dobu trvání zkoušky tolerována bez příznaků toxicity omezující studii (např. vyvolání poklesu tělesné hmotnosti nebo cytotoxicity hematopoetického systému, avšak nikoli smrti nebo příznaků bolesti, utrpení nebo strádání, kdy je nezbytné zvířata humánně utratit (11)). Nejvyšší dávku lze rovněž definovat jako dávku, která vyvolává určité známky toxicity pro kostní dřeň. Výjimku z kritérií pro stanovení dávek mohou představovat chemické látky, jež vykazují saturaci toxikokinetických vlastností nebo indukují detoxifikační procesy, které mohou vést ke snížení expozice po dlouhodobé aplikaci a jež by měly být hodnoceny v každém jednotlivém případě. Aby bylo možné získat informace o dávce a účinku, měla by úplná studie zahrnovat negativní kontrolní skupinu a minimálně tři úrovně dávek, které jsou zpravidla odstupňovány faktorem 2, avšak nepřevyšujícím 4. Pokud ve studii ke stanovení rozsahu dávek nebo na základě stávajících údajů zkoušená chemická látka nevyvolává toxicitu, měla by nejvyšší dávka při jednorázovém podávání odpovídat 2 000 mg/kg tělesné hmotnosti. Pokud však zkoušená chemická látka způsobuje toxicitu, měla by být největší podávanou dávkou maximální tolerovaná dávka a úrovně dávek by měly přednostně pokrývat rozmezí od maximální dávky až po dávku vyvolávající malou toxicitu nebo nevyvolávající žádnou toxicitu. Je-li toxicita pro cílovou tkáň (kostní dřeň) pozorována na všech testovaných úrovních dávek, doporučuje se další studie s netoxickými dávkami. Studie, jejichž cílem je úplnější charakteristika kvantitativních informací o dávce a účinku, mohou vyžadovat zařazení doplňkových skupin zvířat exponovaných dalším dávkám. Pro některé typy zkoušených chemických látek (např. humánních léčivých přípravků), na něž se vztahují specifické požadavky, se tyto limity mohou lišit. Limitní zkouška Pokud experimenty pro určení rozsahu dávek nebo stávající údaje získané u příbuzných kmenů zvířat naznačují, že aplikace limitní dávky (viz níže) nevyvolá pozorovatelné toxické účinky (ani pokles proliferace kostní dřeně nebo jiné příznaky cytotoxicity cílové tkáně), a není-li na základě studií genotoxicity in vitro nebo na základě údajů o látkách, které mají příbuznou strukturu, očekávána genotoxicita, nepovažuje se úplná studie se třemi úrovněmi dávek za nutnou, za předpokladu, že bylo prokázáno, že zkoušená chemická látka (látky) dosahuje (dosahují) cílové tkáně (kostní dřeně). V takových případech může být postačující jediná úroveň expozice v limitní dávce. Při delší než 14denní aplikaci je limitní dávka 1 000 mg/kg tělesné hmotnosti/den. Při 14denní a kratší aplikaci je limitní dávka 2 000 mg/kg tělesné hmotnosti/den. Podávání dávek Při plánování zkoušky by měl být zvážen předpokládaný způsob expozice člověka. Proto lze zvolit odůvodněné způsoby expozice např. stravou, v pitné vodě, lokálně, subkutánně, nitrožilně, orálně (pomocí žaludeční sondy), inhalací, intratracheálně nebo implantací. Způsob podávání by měl být v každém případě zvolen tak, aby zajistil odpovídající expozici cílové tkáně (tkání). Intraperitoneální injekce se obecně nedoporučují, protože se nejedná o předpokládaný způsob expozice člověka, a mohla by být použita pouze s konkrétním vědeckým zdůvodněním. Je-li zkoušená chemická látka přimíchána do potravy nebo pitné vody, zejména v případě jednorázové dávky, mělo by se dbát na to, aby prodleva mezi příjmem krmiva a vody a odběrem vzorku byla dostatečná, aby umožnila detekci účinků (viz odstavce 33–34). Maximální objem kapaliny, který může být najednou podán žaludeční sondou nebo injekčně, závisí na velikosti pokusného zvířete. Toto množství by obvykle nemělo překročit 1 ml/100 g tělesné hmotnosti, s výjimkou vodných roztoků, kde lze použít maximálně 2 ml/100 g tělesné hmotnosti. Použití objemů větších než tento by mělo být odůvodněno. Až na dráždivé a žíravé zkoušené chemické látky, které obvykle při vyšších koncentracích vykazují zesílené účinky, by měla být variabilita zkušebního objemu minimalizována nastavením koncentrace zajišťující podávání konstantního objemu v závislosti na tělesné hmotnosti při všech úrovních dávky. Plán expozice Zkoušené chemické látky jsou obvykle podávány jednorázově, ale mohou být podávány také ve dvou dávkách (tzn. dvě nebo více dávek v týž den v rozmezí ne více než 2–3 hodiny), aby bylo usnadněno podávání velkých objemů. Za těchto okolností, nebo je-li zkoušená chemická látka podávána inhalací, by měl být harmonogram odběru buněk stanoven na základě doby podání poslední dávky nebo ukončení expozice. Pro tuto zkoušku je k dispozici málo údajů o vhodnosti protokolu s opakovanými dávkami. Avšak za situace, kdy je žádoucí integrovat tuto zkoušku se zkouškou toxicity po opakovaných dávkách, by se mělo dbát na to, aby nedošlo ke ztrátě mitotických buněk s poškozenými chromozómy, což může nastat u toxických dávek. Taková integrace je přijatelná, pokud nejvyšší dávka je větší než limitní dávka nebo je stejná (viz odstavec 29) a pokud je exponované skupině podávána limitní dávka po dobu trvání expozice. Je-li žádoucí integrace s jinými studiemi, měl by být za vhodnou zkoušku na chromozómových aberacích in vivo považován mikronukleus test (zkušební metoda B.12). Vzorky kostní dřeně by měly být odebrány ve dvou různých intervalech od jednorázové aplikace. U hlodavců by se odběr měl provádět po takové době od aplikace, která odpovídá 1,5násobku normální délky buněčného cyklu (jenž trvá obvykle 12–18 hodin od expozice). Poněvadž doba nezbytná pro příjem a metabolismus zkoušené chemické látky (látek) a rovněž pro účinky na kinetiku buněčného cyklu může mít vliv na optimální časový interval pro detekci chromozómových aberací, doporučuje se provést další odběr po 24 hodinách od prvního odběru. V prvním intervalu expozice by měly být aplikovány všechny exponované skupiny a proveden odběr buněk. V dalším intervalu (intervalech) expozice je potřebné podat pouze nejvyšší dávku. Je-li na základě vědeckého odůvodnění aplikace rozložena do více než jednoho dne, měl by být odběr zpravidla proveden po takové době od poslední aplikace, která odpovídá 1,5násobku normální délky buněčného cyklu. Po expozici a před odběrem vzorků se zvířatům intraperitoneálně podá vhodná dávka látky zastavující metafázi (např. colcemidu nebo kolchicinu) a po vhodné době se u nich odeberou vzorky. U myši je tato doba do odebrání vzorku přibližně 3–5 hodin a u potkana je to 2–5 hodin. Z kostní dřeně se odeberou buňky, zhotoví se nátěry, ty se fixují a obarví a buňky se analyzují se na chromozómové aberace (12). Pozorování Všeobecné klinické pozorování by se mělo provádět nejméně jednou denně, přednostně ve stejnou dobu (stejné doby) a s uvážením doby očekávaného maximálního účinku po podání látky. Nejméně dvakrát denně po dobu podávání dávek by měla být provedena prohlídka všech zvířat za účelem zjištění morbidity a mortality. Všechna zvířata by měla být zvážena na počátku studie, nejméně jednou týdně během studií s opakovanou dávkou a při utracení. Při studiích s alespoň týdenním trváním by se alespoň jednou týdně měla změřit spotřeba krmiva. Pokud je zkoušená chemická látka podávána v pitné vodě, měla by se spotřeba vody měřit při každé výměně vody a alespoň jednou týdně. Zvířata, která vykazují neletální indikátory nadměrné toxicity, by měla být humánně utracena před uplynutím doby zkoušky (11). Expozice cílových tkání Ve vhodnou dobu (vhodné doby) by měl být odebrán vzorek krve, aby mohlo být provedeno vyšetření hladiny zkoušené chemické látky v plazmě za účelem prokázání, že došlo k expozici kostní dřeně, pokud je to požadováno a pokud neexistují jiné údaje o expozici (viz odstavec 44). Příprava kostní dřeně a chromozómů Buňky kostní dřeně se získávají z femuru nebo tibie zvířat ihned po humánním utracení, hypotonizují se a fixují. Buňky v metafázi se poté zavedenými metodami nanesou na podložní sklíčka a obarví se (viz (3) (12)). Analýza Všechny preparáty, včetně preparátů pozitivních a negativních kontrol, by měly být před analýzou označeny samostatným kódem určeným náhodným výběrem tak, aby hodnotiteli nebyly známy zkušební podmínky. Jako měřítko cytotoxicity by měl být u všech exponovaných zvířat (včetně pozitivních kontrol), neexponovaných zvířat pro negativní kontrolu a zvířat pro negativní kontrolu s vehikulem/rozpouštědlem stanoven mitotický index, a to alespoň u 1 000 buněk na jedno zvíře. U každého zvířete by mělo být analyzováno alespoň 200 metafází na strukturní chromozómové aberace včetně gapů a s vyloučením gapů (6), Pokud však z historické databáze negativních kontrol vyplývá, že střední četnost spontánního výskytu strukturních chromozómových aberací v dané laboratoři je < 1 %, mělo by se zvážit vyhodnocení dalších buněk. Chromatidové a chromozómové aberace by měly být zaznamenávány odděleně a klasifikovány podle podtypů (zlomy, výměny). Postupy používané v laboratoři by měly zajistit, aby analýzu chromozómových aberací prováděli dobře odborně připravení hodnotitelé a a aby tato analýza případně byla podrobena odborné revizi. Vzhledem k tomu, že při přípravě preparátů často dochází k poškození části buněk v metafázi s následnou ztrátou chromozómů, měly by vyšetřované buňky obsahovat centromery v počtu nejméně 2n ± 2, kde n je haploidní počet chromozómů pro daný druh. ÚDAJE A PŘEDKLÁDÁNÍ ZPRÁV Zpracování výsledků Údaje pro jednotlivá zvířata by měly být zpracovány ve formě tabulky. U každého zvířete by měl být uveden mitotický index, počet hodnocených buněk v metafázi, počet aberací na každou buňku v metafázi a procentuální podíl buněk se strukturními chromozomovými aberacemi. Pro exponované a kontrolní skupiny by měly být uvedeny různé typy strukturních chromozómových aberací s jejich počtem a četností. Gapy, jakož i polyploidní buňky a buňky s endoreplikovanými chromozómy se zaznamenávají odděleně. Četnost výskytu gapů se zaznamenává, ale do analýzy celkové četnosti strukturních aberací se nezahrnuje. Neexistuje-li důkaz o rozdílu v odpovědi mezi pohlavími, mohou být pro účely statistické analýzy tyto údaje sloučeny. Uvedeny by měly být i údaje o toxicitě u zvířat a klinické příznaky. Kritéria přijatelnosti Přijatelnost zkoušky určují tato kritéria:
Hodnocení a interpretace výsledků Za předpokladu, že všechna kritéria přijatelnosti byla splněna, lze zkoušenou chemickou látku považovat za jasně pozitivní, pokud:
Je-li v určitém konkrétním čase odběru zkoumána pouze nejvyšší dávka, je zkoušená chemická látka považována za jasně pozitivní, pokud existuje statisticky významný nárůst v porovnání se souběžnou negativní kontrolou a výsledky jsou mimo distribuci dosavadních údajů o negativních kontrolách (např. 95 % rozmezí dle Poissonova rozdělení). Doporučení ohledně vhodných statistických metod lze najít v literatuře (13). Při provádění analýzy dávky a odezvy by měly být analyzovány alespoň tři skupiny exponované různé dávce. Při statistických testech by mělo být zvíře použito jako experimentální jednotka. Pozitivní výsledky zkoušky na chromozómové aberace znamenají, že zkoušená látka indukuje strukturální chromozómové aberace v kostní dřeni druhu zvířat použitého k testování. Za předpokladu, že všechna kritéria přijatelnosti byla splněna, je zkoušená chemická látka považována za jasně negativní, pokud ve všech zkoumaných zkušebních podmínkách:
Doporučení ohledně nejvhodnějších statistických metod lze najít v literatuře (13). Důkazy o expozici kostní dřeně zkoušené chemické látce mohou zahrnovat pokles mitotického indexu nebo měření hladiny zkoušené chemické látky (látek) v plazmě nebo v krvi. V případě nitrožilního podání není důkazů o expozici zapotřebí. Alternativně mohou být k prokázání expozice kostní dřeně použity údaje o absorpci, distribuci, metabolismu a exkreci (ADME) získané při nezávislé studii za použití stejného způsobu expozice a se stejným druhem. Negativní výsledky znamenají, že zkoušená chemická látka za podmínek zkoušky neindukuje strukturní chromozómové aberace v kostní dřeni druhu zvířat použitého k testování. Ověření jasně pozitivní či jasně negativní odpovědi se nepožaduje. V případech, kdy odpověď není jasně negativní nebo pozitivní, a s cílem napomoci při stanovení biologického významu výsledku (např. slabý nebo okrajový nárůst), by měly být údaje vyhodnoceny na základě odborného posouzení a/nebo dalším vyšetřením u provedených experimentů. V některých případech by mohlo být užitečné analyzovat více buněk nebo provést opakovaný experiment za použití upravených zkušebních podmínek. V ojedinělých případech dokonce ani po dalším šetření nebude možné na základě daných údajů učinit závěr, zda zkoušená chemická látka přináší pozitivní či negativní výsledky, a proto bude učiněn závěr, že výsledek studie je neurčitý. Četnost polyploidie a endoreduplikovaných metafází v celkovém počtu metafází by se měla zaznamenat odděleně. Nárůst počtu polyploidních buněk / endoreduplikovaných buněk může znamenat, že zkoušená chemická látka má schopnost potlačit mitotické procesy nebo progresi buněčného cyklu (viz odstavec 3). Závěrečná zpráva Závěrečná zpráva by měla obsahovat tyto informace: Souhrn
LITERATURA
Dodatek 1 DEFINICE Aneuploidie : jakákoli odchylka od normálního diploidního (nebo haploidního) počtu chromozómů o jeden nebo více chromozómů, avšak nikoli o násobky celé sady (nebo více sad) chromozómů (viz polyploidie). Centromera : oblast (oblasti) chromozómu, k níž (k nimž) se během dělení buněk připojí dělící vřeténko umožňující uspořádaný pohyb dceřiných chromozómů k pólům dceřiných buněk. Chemická látka : chemická substance nebo směs. Chromatidová aberace : strukturní poškození chromozómu v podobě zlomu jedné chromatidy nebo zlomu a opětného spojení chromatid. Chromozómová aberace : strukturní poškození chromozómu v podobě zlomu nebo zlomu a spojení obou chromatid v tomtéž místě. Endoreduplikace : proces, při kterém v jádře po S-fázi replikace DNA nedojde k mitóze, nýbrž následuje další S-fáze. Výsledkem jsou chromozómy se 4, 8, 16 … chromatidami. Gap : achromatická léze menší než šířka jedné chromatidy a s minimálním přerušením chromatid. Mitotický index : podíl počtu buněk, které se nacházejí v mitóze, z celkového počtu buněk v populaci, který je mírou proliferace této populace. Numerická aberace : odchylka počtu chromozómů od normálního počtu obvyklého u použitého druhu zvířat (aneuploidie). Polyploidie : numerická chromozómová aberace zahrnující odchylku počtu celých sad chromozómů, na rozdíl od odchylky počtu v části sady chromozómů (viz aneuploidie). Strukturní chromozómové aberace : mikroskopicky pozorovatelné změny struktury chromozómů při buněčném dělení ve stadiu metafáze; jeví se jako delece a fragmenty, intrachromozómální nebo interchromozómální změny. Zkoušená chemická látka : jakákoli chemická substance nebo směs zkoušená pomocí této zkušební metody. Dodatek 2 FAKTORIÁLNÍ POKUS KE ZJIŠTĚNÍ ROZDÍLŮ MEZI POHLAVÍMI VE ZKOUŠCE NA CHROMOZÓMOVÉ ABERACE IN VIVO Faktoriální pokus a jeho analýza Při tomto pokusu se na každé úrovni koncentrace testuje minimálně 5 samců a 5 samic a výsledkem je uspořádání s použitím minimálně 40 zvířat (20 samců a 20 samic plus příslušné pozitivní kontroly). Tento pokus, který je jedním z nejjednodušších faktoriálních pokusů, je rovnocenný dvoufaktorové analýze rozptylu, přičemž hlavními prvky jsou úroveň koncentrace a pohlaví. Údaje je možné analyzovat pomocí mnoha standardních statistických softwarových balíčků, např. SPSS, SAS, STATA, Genstat a také pomocí programu R. Analýza dělí variabilitu v souboru údajů na variabilitu mezi pohlavími, variabilitu mezi úrovněmi koncentrace a variabilitu interakce mezi pohlavími a koncentracemi. Každý z těchto znaků se testuje oproti odhadu variability u replicitních zvířat ve skupinách zvířat stejného pohlaví, jimž je podávána stejná koncentrace látky. Veškeré podrobnosti základní metodiky jsou dostupné v mnoha standardních učebnicích statistiky (viz odkazy) a v „nápovědě“ statistických programů. Analýza se provádí zkoumáním znaku interakce pohlaví x koncentrace v tabulce ANOVA (5). Neexistuje-li významný znak interakce, kombinované hodnoty za obě pohlaví nebo za všechny úrovně koncentrace poskytují validní statistické testy variability mezi úrovněmi založené na zkoumání kumulovaného znaku vnitroskupinové variability pomocí analýzy ANOVA. Analýza pokračuje rozdělením odhadu variability mezi koncentracemi na kontrasty, což zajišťuje test na lineární a kvadratické kontrasty odpovědí na všech úrovních koncentrace. Pokud existuje významná interakce pohlaví x koncentrace, lze tento znak také rozdělit na lineární a kvadratický člen kontrastů interakce pohlaví. Pomocí těchto znaků se testuje, zda jsou odpovědi na koncentraci u obou pohlaví paralelní, nebo zda je odpověď u každého pohlaví odlišná. Odhad kumulované vnitroskupinové variability může být použit k párovému testování rozdílu mezi středními hodnotami. Tato srovnání by mohla být provedena mezi středními hodnotami u obou pohlaví a mezi středními hodnotami u různých úrovní koncentrace, např. za účelem porovnání s úrovněmi negativní skupiny. V případech, kdy existuje významná interakce, lze provádět srovnání mezi středními hodnotami při různých koncentracích v rámci jednotlivých pohlaví nebo mezi středními hodnotami u pohlaví při stejné koncentraci. Odkazy Existuje množství učebnic statistiky, které se zabývají teorií, uspořádáním, metodikou, analýzou a interpretací faktoriálních pokusů od nejjednodušších dvoufaktorových analýz až po složitější formy používané v metodice experimentálního uspořádání. Následující seznam není vyčerpávající. V některých pracích jsou uvedeny vypracované příklady srovnatelných uspořádání, v některých případech i s kódem pro provádění analýz pomocí různých počítačových programů.
|
|
5) |
V části B se kapitola B.12 nahrazuje tímto: „B.12 Mikronukleus test v savčích erytrocytech ÚVOD Tato zkušební metoda je rovnocenná Pokynu OECD pro zkoušení (TG) č. 474 (2016). Je součástí série zkušebních metod v oblasti genetické toxikologie. Byl vypracován dokument OECD, který obsahuje stručné informace o zkoušení v oblasti genetické toxikologie a přehled posledních změn, které byly v těchto pokynech ke zkoušení provedeny (1). Mikronukleus test v savčích erytrocytech in vivo je zvláště významný k posouzení genotoxicity, neboť zde probíhá aktivní metabolismus in vivo, farmakokinetika a procesy reparace DNA, které přispívají k účinku testované látky, jakkoli mohou být různé u různých druhů. Zkouška in vivo je rovněž užitečná pro další výzkum genotoxicity zjištěné v systémech in vitro. Mikronukleus test v savčích erytrocytech in vivo je používán k detekci poškození chromozómů nebo mitotického aparátu erytroblastů, které je indukováno zkoušenou chemickou látkou. V tomto testu se hodnotí tvorba mikrojader v erytrocytech odebraných z kostní dřeně nebo z buněk periferní krve, obvykle hlodavců. Účelem mikronukleus testu v savčích erytrocytech je identifikovat chemické látky, které způsobují cytogenetické poškození, jehož výsledkem je tvorba mikrojader obsahujících nereplikující se (lagging) chromozómové fragmenty nebo celé chromozómy. Když se erytroblast kostní dřeně mění na nezralý erytrocyt (někdy též označovaný jako polychromatický erytrocyt nebo retikulocyt), hlavní jádro je vypuzeno a mikrojádra, která při tom mohou vzniknout, zůstávají v cytoplasmě. Zviditelnění nebo detekce mikrojader jsou v těchto buňkách usnadněny tím, že neobsahují hlavní jádro. Nárůst výskytu nezralých erytrocytů s mikrojádry v exponovaných zvířatech je známkou indukovaných strukturních nebo numerických chromozómových aberací. Nově vytvořené erytrocyty s mikrojádry se zjišťují a kvantifikují obarvením a poté následuje buď vizuální hodnocení pomocí mikroskopu, nebo automatizovaná analýza. Počítání dostačujícího počtu nezralých erytrocytů v periferní krvi nebo kostní dřeni dospělých zvířat se značně usnadní použitím automatizovaného hodnotícího systému. Tyto systémy jsou přijatelnými alternativami manuálního hodnocení (2). Z komparativních studií vyplývá, že tyto metody, při nichž se používají vhodné kalibrační standardy, mohou zajistit lepší mezilaboratorní a vnitrolaboratorní reprodukovatelnost a větší citlivost než manuální hodnocení pomocí mikroskopu (3) (4). Automatizované systémy pro měření četnosti erytrocytů s mikrojádry zahrnují kromě jiného průtokové cytometry (5), platformy pro analýzu obrazu (6)(7) a laserové skenovací cytometry (8). Ačkoli se to v rámci tohoto testu obvykle neprovádí, lze odlišit chromozómové fragmenty od celých chromozómů podle řady kritérií. Tato kritéria zahrnují identifikaci přítomnosti nebo nepřítomnosti kinetochoru nebo centromerické DNA, jež jsou obojí charakteristické pro neporušené chromozómy. Nepřítomnost kinetochoru nebo centromerické DNA naznačuje, že mikrojádro obsahuje pouze fragmenty chromozómů, zatímco jejich přítomnost ukazuje na ztrátu chromozómů. Definice použitých termínů jsou uvedeny v dodatku 1. VÝCHOZÍ ÚVAHY Cílovou tkání pro genetická poškození v tomto testu je kostní dřeň mladých dospělých hlodavců, protože v této tkáni vznikají erytrocyty. Detekce mikrojader v nezralých erytrocytech v periferní krvi je možná i u jiných druhů savců, u nichž byla prokázána dostatečná citlivost při detekci chemické látky, které v těchto buňkách způsobují strukturní nebo numerické chromozómové aberace (indukcí mikrojader v nezralých erytrocytech), a je-li uvedeno vědecké zdůvodnění. Hlavním konečným ukazatelem je četnost výskytu nezralých erytrocytů s mikrojádry. Četnost výskytu zralých erytrocytů, jež obsahují mikrojádra v periferní krvi, může být rovněž použita jako koncový ukazatel u druhů bez silné slezinné selekce proti buňkám s mikrojádry a jsou-li zvířata trvale exponována po dobu, jež přesahuje délku života erytrocytu u použitého druhu (např. 4 týdny nebo více u myši). Jestliže existuje důkaz o tom, že se zkoušená chemická látka (chemické látky) nebo reaktivní metabolit (metabolity) nedostanou do cílové tkáně, není vhodné tuto zkoušku použít. Před použitím této zkušební metody na směs za účelem získání údajů pro zamýšlené použití v právních předpisech by se mělo zvážit, zda tato metoda může pro tento účel poskytnout přiměřené výsledky, a pokud ano, proč je tomu tak. Takové úvahy nejsou nutné, pokud existuje právní požadavek na zkoušení dané směsi. PRINCIP ZKUŠEBNÍ METODY Zvířata se vhodným způsobem vystaví zkoušené chemické látce. Použije-li se kostní dřeň, zvířata se ve vhodném okamžiku po expozici humánně utratí, odebere se kostní dřeň, připraví se preparát a obarví se (9) (10) (11) (12) (13) (14) (15). Při použití periferní krve se krev ve vhodném okamžiku (vhodných okamžicích) po expozici odebere, připraví se preparát a obarví se (12) (16) (17) (18). Při akutní expozici je důležité zvolit takové časy odběru kostní dřeně nebo krve, kdy je možné detekovat indukci nezralých erytrocytů s mikrojádry související s expozicí. V případě odběru periferní krve musí též uplynout dostatek času, aby se tyto jevy mohly objevit v cirkulující krvi. Preparáty se analyzují na přítomnost mikrojader, a to vizualizací pomocí mikroskopu, analýzou obrazu, průtokovou cytometrií nebo laserovou skenovací cytometrií. OVĚŘENÍ ZPŮSOBILOSTI LABORATOŘE Experimenty pro ověření způsobilosti Aby bylo možné před zahájením rutinního zkoušení zjistit, zda laboratoř má s danou zkouškou dostatečné zkušenosti, měla by prokázat schopnost reprodukovat očekávané výsledky z publikovaných údajů (17) (19) (20) (21) (22), pokud jde o četnost výskytu mikrojader, nejméně se dvěma pozitivními kontrolními chemickými látkami (včetně slabých odpovědí indukovaných nízkými dávkami u pozitivních kontrol), např. látkami uvedenými v tabulce 1, a s kontrolami s kompatibilním vehikulem/rozpouštědlem (viz odstavec 26). Při těchto experimentech by se měly používat dávky, jež poskytují reprodukovatelné nárůsty v závislosti na dávce, a měly by prokázat citlivost a dynamické rozpětí zkušebního systému ve sledované tkáni (kostní dřeni nebo periferní krvi) za použití hodnotící metody, jež má být v laboratoři uplatňována. Tento požadavek neplatí pro laboratoře, které tuto zkušenost mají, tj. které mají k dispozici historickou databázi, jak je definována v odstavci 14–18. Historické kontrolní údaje V průběhu šetření způsobilosti by laboratoř měla stanovit:
Při prvním získávání údajů o dosavadní distribuci negativních kontrol by měly být výsledky provedených negativních kontrol v souladu s publikovanými kontrolními údaji, pokud existují. S tím, jak je do distribuce kontrol přidáváno více údajů z experimentů, měly by souběžné negativní kontroly v ideálním případě spadat do 95 % rozmezí této distribuce. Historická databáze negativních kontrol laboratoře by měla být statisticky robustní, aby byla zajištěna schopnost laboratoře posuzovat distribuci svých údajů o negativních kontrolách. V literatuře se doporučuje, že nezbytným minimem může být 10 experimentů, nejlépe však by databáze měla zahrnovat nejméně 20 experimentů provedených za srovnatelných zkušebních podmínek. V laboratořích by měly být uplatňovány metody řízení jakosti, jako např. kontrolní diagramy (např.C-charts nebo X-bar charts (23)), z nichž je patrné, jak proměnlivé jsou jejich údaje a že metodika je v dané laboratoři „pod kontrolou“. Další doporučení, jak získat a používat dosavadní údaje (tj. kritéria pro zařazení údajů do historické databáze a jejich vyřazení a kritéria přijatelnosti pro daný experiment), lze najít v literatuře (24). Pokud laboratoř neprovedla dostatečný počet experimentů, aby mohla během šetření způsobilosti (popsaného v odstavci 13) stanovit statisticky robustní distribuci negativních kontrol (viz odstavec 15), je přijatelné, aby distribuce byla stanovena během prvních rutinních zkoušek. Uplatňování tohoto přístupu by mělo vycházet z doporučení uvedených v literatuře (24) a výsledky negativních kontrol získané při těchto experimentech by měly zůstat v souladu s publikovanými údaji o negativních kontrolách. Jakékoli změny zkušebního protokolu by měly být zvažovány z hlediska jejich dopadu na výsledné údaje tak, aby zůstaly v souladu se stávající historickou kontrolní databází laboratoře. Pouze případy větších rozdílů ve zkušebním protokolu by měly vést k zavedení nové historické kontrolní databáze, přičemž její odlišnost od dřívější distribuce se stanoví na základě odborného posouzení (viz odstavec 15). V průběhu opětovného stanovení nemusí být k provedení aktuální zkoušky nutná úplná databáze negativních kontrol, pokud laboratoř prokáže, že hodnoty jejích souběžných negativních kontrol zůstávají v souladu s její předchozí databází nebo s odpovídajícími publikovanými údaji. Údaje o negativních kontrolách by měly zahrnovat výskyt nezralých erytrocytů s mikrojádry u každého zvířete. Výsledky souběžných negativních kontrol by v ideálním případě měly být v 95 % rozmezí distribuce v historické databázi negativních kontrol laboratoře. Pokud jsou údaje ze souběžných negativních kontrol mimo 95 % rozmezí, může být přijatelné je zařadit mezi dosavadní distribuci kontrol, pokud tyto údaje nejsou mimořádně odlehlé a pokud existuje důkaz o tom, že testovací systém je „pod kontrolou“ (viz odstavec 15), a neexistuje důkaz o tom, že by došlo k technické nebo lidské chybě. POPIS METODY Příprava Výběr druhu zvířat Měly by být použity běžně používané laboratorní kmeny zdravých mladých pohlavně dospělých zvířat. Použit lze myši, potkany nebo jiné vhodné druhy savců. Při použití periferní krve se musí stanovit, že detekci indukovaných mikrojader u zvoleného druhu neohrozí odstraňování buněk s mikrojádry z krevního oběhu ve slezině. Toto bylo jasně prokázáno u periferní krve myši a potkana (2). Vědecké zdůvodnění použití jiných druhů než potkanů a myší by mělo být uvedeno ve zprávě. Jsou-li použity jiné druhy než hlodavci, doporučuje se měření indukovaných mikrojader začlenit do jiné vhodné zkoušky toxicity. Podmínky chovu a krmení zvířat Teplota v místnosti pro zvířata by u hlodavců měla být 22 °C (± 3 °C). Relativní vlhkost vzduchu by v ideálním případě měla být 50–60 %, ale minimálně 40 % a neměla by pokud možno přesáhnout 70 % kromě doby úklidu místnosti. Osvětlení by mělo být umělé a mělo by se střídat 12 hodin světla a 12 hodin tmy. Ke krmení lze použít konvenční laboratorní krmivo s neomezeným přísunem pitné vody. Výběr potravy může být ovlivněn nutností zajistit dostatečné mísení se zkoušenou látkou, je-li látka podávána touto cestou. Hlodavci by měli být chováni v malých skupinách (nejvýše po pěti v každé kleci) téhož pohlaví a se stejnou aplikací, pokud se nepředpokládá agresivní chování, přednostně v klecích s pevnou podlahou s vhodným obohacením životního prostředí. Zvířata mohou být umístěna v klecích individuálně, pouze je-li to vědecky odůvodněné. Příprava zvířat Obvykle se používají zdravá mladá dospělá zvířata (u hlodavců ideálně ve věku 6–10 týdnů na počátku expozice, ale přijatelná jsou i zvířata poněkud starší), která se náhodně přiřadí do kontrolních a experimentálních skupin. Jednotlivá zvířata se jednoznačně identifikují humánní, co nejméně invazivní metodou (např. kroužkováním, označením štítkem, pomocí mikročipu nebo biometrické identifikace, avšak nikoli nastřihnutím ucha nebo prstu dolní končetiny) a nechají se v laboratorních podmínkách alespoň pět dní aklimatizovat. Klece by měly být uspořádány tak, aby byl vliv umístění klecí minimalizován. Mělo by se zabránit vzájemné kontaminaci pozitivní kontroly a zkoušené chemické látky. V okamžiku zahájení studie by měla být odchylka v hmotnosti zvířat minimální a neměla by překročit ± 20 % střední hodnoty hmotnosti zvířat daného pohlaví. Příprava dávek Pevné zkoušené chemické látky by se měly rozpustit nebo suspendovat ve vhodných rozpouštědlech nebo vehikulech nebo by měly být přimíchány do potravy nebo pitné vody před podáním dávky zvířatům. Kapalné zkoušené chemické látky mohou být podávány přímo nebo mohou být před podáním zředěny. Pro účely expozice inhalací lze zkoušené chemické látky v závislosti na jejich fyzikálně-chemických vlastnostech podávat jako plyn, páru nebo pevný/kapalný aerosol. Měly by být použity čerstvě připravené zkoušené chemické látky, pokud údaje o stálosti neprokazují možnost skladování a nestanoví vhodné podmínky pro skladování. Zkušební podmínky Rozpouštědlo/vehikulum Rozpouštědlo/vehikulum by nemělo mít při použitých úrovních dávek toxické účinky a nemělo by být schopno chemické reakce se zkoušenými chemickými látkami. Jsou-li použita jiná než známá rozpouštědla/vehikula, mělo by být jejich zařazení podloženo referenčními údaji o jejich kompatibilitě. Doporučuje se pokud možno nejprve zvážit použití vodných rozpouštědel/vehikul. Jako příklad běžně používaných kompatibilních rozpouštědel/vehikul lze uvést vodu, fyziologický roztok, roztok methylcelulosy, roztok sodné soli karboxymethylcelulosy, olivový olej a kukuřičný olej. Neexistují-li historické nebo publikované kontrolní údaje prokazující, že zvolené atypické rozpouštědlo/vehikulum neindukuje mikrojádra nebo jiné zhoubné účinky, měla by být provedena počáteční studie za účelem stanovení přijatelnosti kontroly s tímto rozpouštědlem/vehikulem. Kontroly Pozitivní kontroly V každé zkoušce by měla být obvykle zařazena jedna skupina zvířat vystavených pozitivní kontrolní chemické látce. Od toho lze upustit, jestliže zkušební laboratoř prokázala způsobilost k provádění zkoušky a stanovila historické rozmezí pozitivních kontrol. Není-li zařazena souběžná pozitivní kontrolní skupina, měly by být v každém experimentu zařazeny hodnotící kontroly (fixované a nezbarvené preparáty nebo vzorky buněčné suspenze podle toho, co je vhodné pro danou hodnotící metodu). Tyto hodnotící kontroly lze získat tak, že se do hodnocení studie zařadí vhodné referenční vzorky, které byly získány a uchovány ze samostatného experimentu s pozitivními kontrolami prováděného pravidelně (např. každých 6–18 měsíců), například během testování způsobilosti a poté pravidelně podle potřeby. Chemické látky pro pozitivní kontrolu by měly spolehlivě vyvolat pozorovatelné zvýšení výskytu buněk s mikrojádry nad spontánní úroveň. Při použití manuálního hodnocení za použití mikroskopie by dávky u pozitivní kontroly měly být zvoleny tak, aby byl účinek zřetelný, ale aby hodnotitel okamžitě nezjistil identitu kódovaného vzorku. Je přijatelné, aby látka v pozitivní kontrole byla podávána jiným způsobem než zkoušená chemická látka, podle odlišného harmonogramu expozice, a aby se odběr vzorků prováděl pouze v jediném okamžiku. Pro pozitivní kontrolu navíc může být vzato v úvahu použití chemických látek ze stejné chemické třídy, je-li to vhodné. Příklady pozitivních kontrolních chemických látek jsou uvedeny v tabulce 1. Tabulka 1. Příklady chemických látek pro pozitivní kontroly. Chemické látky a registrační číslo CAS (CASRN) ethyl-methansulfonát [CASRN 62-50-0] methyl-methansulfonát [CASRN 66-27-3] 1-ethyl-1-nitrosomočovina [CASRN 759-73-9] mitomycin C [CASRN 50-07-7] cyklofosfamid (cyklofosfamid monohydrát) [CASRN 50-18-0 (CASRN 6055-19-2)] 2,4,6-tris(aziridin-1-yl)- 1,3,5-triazin [CASRN 51-18-3] kolchicin [CASRN 64-86-8] nebo vinblastin [CASRN 865-21-4] – jako aneugeny Negativní kontroly V každém expozičním intervalu by měla být zařazena souběžná negativní kontrolní skupina zvířat, která jinak podstupují stejný proces jako exponované skupiny až na to, že nejsou vystavena zkoušené chemické látce. Používá-li se při podávání zkoušené chemické látky rozpouštědlo/vehikulum, měla by toto rozpouštědlo/vehikulum dostávat i kontrolní skupina. Pokud však dosavadní údaje o negativních kontrolách v různých intervalech expozice v dané zkušební laboratoři prokazují konzistentní údaje o variabilitě zvířat a četnosti buněk s mikrojádry, může být pro negativní kontrolu nezbytný pouze jeden odběr. Je-li u negativních kontrol použit jediný odběr, měl by se provést v prvním (nejkratším) intervalu expozice použitém ve studii. Při použití periferní krve je u krátkodobých studií přijatelný vzorek odebraný před expozicí namísto souběžné negativní kontroly, pokud výsledné údaje jsou konzistentní s historickou kontrolní databází dané zkušební laboratoře. Bylo prokázáno, že u potkana vzorek s malými objemy odebraný před expozicí (např. méně než 100 μl/den) má minimální dopad na četnost výskytu mikrojader na pozadí (25). POSTUP Počet a pohlaví zvířat Indukce mikrojader je v zásadě u samic a samců podobná, a většinu studií je proto možné provést s kterýmkoli z obou pohlaví (26). Existence údajů prokazujících významné rozdíly mezi samci a samicemi (např. rozdíly v systémové toxicitě, metabolismu, biologické dostupnosti, toxicita pro kostní dřeň atd. získané např. ve studii ke stanovení rozsahu dávek) by byla důvodem pro použití zvířat obojího pohlaví. V tomto případě může být vhodné provést studii s oběma pohlavími, např. jako součást studie toxicity po opakovaných dávkách. V případě použití obou pohlaví by mohlo být vhodné použít faktoriálního uspořádání experimentu. Podrobnosti ohledně toho, jak analyzovat údaje získané při tomto uspořádání experimentu, jsou uvedeny v dodatku 2. Velikost skupin při zahájení studie by měla být stanovena tak, aby poskytla minimálně pět analyzovatelných zvířat jednoho pohlaví, nebo každého pohlaví, jsou-li použita obě, z každé skupiny. Je-li expozice člověka chemickým látkám specifická pro určité pohlaví, jako je tomu například u některých léčivých přípravků, měla by být zkouška provedena se zvířaty odpovídajícího pohlaví. Určitým vodítkem pro maximální typické požadavky na počet zvířat může být, že studie s kostní dření prováděné podle parametrů uvedených v odstavci 37 se třemi exponovanými skupinami a souběžnými negativními a kontrolními skupinami (každá skupina složena z pěti zvířat jednoho pohlaví) by vyžadovala 25 až 35 zvířat. Dávkování Provádí-li se studie pro zjištění rozsahu dávek, poněvadž nejsou k dispozici vhodné údaje, jež by mohly pomoci při stanovení dávek, měla by být provedena ve stejné laboratoři, se stejným druhem, kmenem, pohlavím a za stejného režimu expozice, které se použijí v hlavní studii (27). Cílem studie by mělo být zjistit maximální tolerovanou dávku, která je definována jako nejvyšší dávka, která bude po dobu trvání zkoušky tolerována bez příznaků toxicity omezující studii (např. vyvolání poklesu tělesné hmotnosti nebo cytotoxicity hematopoetického systému, avšak nikoli smrti nebo příznaků bolesti, utrpení nebo strádání, kdy je nezbytné zvířata humánně utratit (28)). Nejvyšší dávka může být také definována jako dávka vyvolávající v kostní dřeni toxicitu (např. snížení podílu nezralých erytrocytů z celkového počtu erytrocytů v kostní dřeni nebo v periferní krvi o více než 50 %, avšak nejméně na 20 % kontrolní hodnoty). Při analýze CD71 pozitivních buněk v periferním krevním oběhu (tj. pomocí průtokové cytometrie) tato velmi mladá frakce nezralých erytrocytů odpovídá na toxické podněty rychleji než větší kohorta nezralých erytrocytů s pozitivní RNA. Vyšší zjevná toxicita proto může být zřejmá při uspořádání s akutní expozicí, kdy se zkoumá frakce CD71 pozitivních nezralých erytrocytů v porovnání s těmi uspořádáními, jež identifikují nezralé erytrocyty na základě obsahu RNA. A proto, jestliže se při experimentech využívá pěti nebo méně dnů expozice, může být nejvyšší dávka chemické látky způsobující toxicitu definována jako dávka, která způsobuje statisticky významné snížení podílu nezralých CD71 pozitivních erytrocytů z celkového množství erytrocytů, avšak nikoli na úroveň menší než 5 % kontrolní hodnoty (29). Výjimku z kritérií pro stanovení dávek mohou představovat chemické látky, jež vykazují saturaci toxikokinetických vlastností nebo indukují detoxifikační procesy, které mohou vést ke snížení expozice po dlouhodobé aplikaci, a jež by měly být hodnoceny v každém jednotlivém případě. Aby bylo možné získat informace o dávce a účinku, měla by úplná studie zahrnovat negativní kontrolní skupinu a minimálně tři úrovně dávek, které jsou zpravidla odstupňovány faktorem 2, avšak nepřevyšujícím 4. Pokud zkoušená chemická látka ve studii ke stanovení rozsahu dávek nebo na základě existujících údajů nevyvolává toxicitu, měla by být nejvyšší dávka 1 000 mg/kg tělesné hmotnosti/den při době podávání 14 dní nebo delší nebo 2 000 mg/kg tělesné hmotnosti/den při dobách podávání kratších než 14 dní. Pokud však zkoušená chemická látka způsobuje toxicitu, měla by být největší podávanou dávkou maximální tolerovaná dávka a úrovně dávek by měly přednostně pokrývat rozmezí od maximální dávky až po dávku vyvolávající malou toxicitu nebo nevyvolávající žádnou toxicitu. Je-li toxicita pro cílovou tkáň (kostní dřeň) pozorována na všech testovaných úrovních dávek, doporučuje se další studie s netoxickými dávkami. Studie, jejichž cílem je úplnější charakteristika kvantitativních informací o dávce a odezvě, mohou vyžadovat zařazení doplňkových skupin zvířat exponovaných dalším dávkám. Pro některé typy zkoušených chemických látek (např. humánních léčivých přípravků), na něž se vztahují specifické požadavky, se tyto limity mohou lišit. Limitní zkouška Pokud experimenty pro určení rozsahu dávek nebo existující údaje získané u příbuzných kmenů zvířat naznačují, že aplikace limitní dávky (viz níže) nevyvolá pozorovatelné toxické účinky (ani pokles proliferace kostní dřeně nebo jiné příznaky cytotoxicity cílové tkáně), a není-li na základě studií genotoxicity in vitro nebo na základě údajů o látkách, které mají příbuznou strukturu, očekávána genotoxicita, nepovažuje se úplná studie se třemi úrovněmi dávky za nutnou, za předpokladu, že bylo prokázáno, že zkoušená chemická látka (látky) dosahuje (dosahují) cílové tkáně (kostní dřeně). V takových případech může být postačující jediná úroveň expozice v limitní dávce. Pro 14denní a delší aplikaci je limitní dávka 1 000 mg/kg tělesné hmotnosti/den. Pro kratší než 14denní aplikaci je limitní dávka 2 000 mg/kg tělesné hmotnosti/den. Podávání dávek Při plánování zkoušky by měl být zvážen předpokládaný způsob expozice člověka. Proto lze zvolit odůvodněné způsoby expozice např. potravou, v pitné vodě, lokálně, subkutánně, nitrožilně, orálně (pomocí žaludeční sondy), inhalací, intratracheálně nebo implantací. Způsob podávání by měl být v každém případě zvolen tak, aby zajistil odpovídající expozici cílové tkáně (tkání). Intraperitoneální injekce se obecně nedoporučují, protože se nejedná o předpokládaný způsob expozice člověka, a mohla by být použita pouze s konkrétním vědeckým zdůvodněním. Je-li zkoušená chemická látka přimíchána do potravy nebo pitné vody, zejména v případě jednorázové dávky, mělo by se dbát na to, že prodleva mezi příjmem krmiva a vody a odběrem vzorku by měla být dostatečná, aby umožnila detekci účinků (viz odstavec 37). Maximální objem kapaliny, který může být najednou podán žaludeční sondou nebo injekčně, závisí na velikosti pokusného zvířete. Toto množství by obvykle nemělo překročit 1 ml/100 g tělesné hmotnosti, s výjimkou vodných roztoků, kde lze použít maximálně 2 ml/100 g tělesné hmotnosti. Použití objemů větších než tento by mělo být odůvodněno. Až na dráždivé a žíravé zkoušené chemické látky, které obvykle při vyšších koncentracích vykazují zesílené účinky, by měla být variabilita zkušebního objemu minimalizována nastavením koncentrace zajišťující podávání konstantního objemu v závislosti na tělesné hmotnosti při všech úrovních dávky. Plán expozice Přednostně se provádějí 2 nebo více aplikací v intervalu 24 hodin, zejména při začlenění tohoto testu do jiných studií toxicity. Alternativně lze podat jedinou dávku, pokud je to vědecky odůvodněno (např. chemické látky, o nichž je známo, že blokují buněčný cyklus). Zkoušené chemické látky mohou být podávány také ve dvou dávkách, tzn. dvě nebo více dávek v týž den v rozmezí ne více než 2–3 hodiny, aby bylo usnadněno podávání velkých objemů. Za těchto okolností, nebo je-li zkoušená chemická látka podávána inhalací, by měl být harmonogram odběru buněk stanoven na základě doby podání poslední dávky nebo ukončení expozice. Zkouška může být prováděna na myších nebo potkanech jedním z těchto způsobů:
Popřípadě mohou být použity jiné režimy podávání dávek nebo odběru vzorků, je-li to vědecky zdůvodněno a k usnadnění integrace s jinými zkouškami toxicity. Pozorování Všeobecné klinické pozorování by se mělo provádět nejméně jednou denně, přednostně ve stejnou dobu (stejné doby) a s uvážením doby očekávaného maximálního účinku po podání látky. Nejméně dvakrát denně po dobu podávání dávek by měla být provedena prohlídka všech zvířat za účelem zjištění morbidity a mortality. Všechna zvířata by měla být zvážena na počátku studie, nejméně jednou týdně během studií s opakovanou dávkou a při utracení. Při studiích s alespoň týdenním trváním by se alespoň jednou týdně měla změřit spotřeba krmiva. Pokud je zkoušená chemická látka podávána v pitné vodě, měla by se spotřeba vody měřit při každé výměně vody a alespoň jednou týdně. Zvířata, která vykazují neletální indikátory nadměrné toxicity, by měla být humánně utracena před uplynutím doby zkoušky (28). Za určitých okolností by mohla být sledována tělesná teplota zvířete, protože hypertermie a hypotermie indukovaná expozicí může způsobovat falešné výsledky (32) (33) (34). Expozice cílových tkání Krevní vzorek by měl být odebrán ve vhodnou dobu (vhodné doby), aby mohlo být provedeno vyšetření hladiny zkoušené chemické látky v plazmě za účelem prokázání, že došlo k expozici kostní dřeně, pokud je to požadováno a pokud neexistují jiné údaje o expozici (viz odstavec 48). Příprava kostní dřeně nebo krve Buňky kostní dřeně se obvykle získávají z femuru nebo tibie zvířat ihned po jejich humánním usmrcení. Buňky se odeberou a zavedenými metodami se preparují a obarví. Malé objemy periferní krve lze podle příslušných standardů dobrého zacházení se zvířaty získat buď postupem, který umožňuje přežití zkušebního zvířete, např. odběrem z ocasní žíly nebo jiné vhodné krevní cévy, anebo srdeční punkcí nebo odběrem vzorku z velké cévy při utracení zvířete. U erytrocytů získaných z kostní dřeně nebo z periferní krve je možné, v závislosti na metodě analýzy, buňky ihned supravitálně obarvit (16) (17) (18), připravit preparáty roztěrem a poté je obarvit pro mikroskopii nebo vhodně fixovat a obarvit pro analýzu pomocí průtokové cytometrie. Použitím barviva specifického pro DNA [např. akridinová oranž (35) nebo Hoechst 33258 a pyronin-Y (36)] se lze vyhnout některým artefaktům spojeným s použitím barviva nespecifického pro DNA. Tato výhoda nebrání použití konvenčních barviv (např. Giemsa pro analýzu pod mikroskopem). Přídavné systémy [např. celulosová kolona k odstranění buněk obsahujících jádra (37) (38)] lze použít za předpokladu, že se prokázalo, že jsou kompatibilní se systémy pro preparaci vzorků v laboratoři. Jsou-li tyto metody použitelné, lze k identifikaci povahy mikrojader (chromozóm/chromozómový fragment) využít protilátky proti kinetochorům (39), hybridizační metodu FISH využívající pan-centromerické DNA-sondy (40), případně značení in situ pomocí primerů se specifickou pan-centromerickou vazbou spolu s vhodným kontrastním barvením DNA (41), aby bylo možné určit, zda mechanismus indukce mikrojader je důsledkem klastogenního a/nebo aneugenního působení. K rozlišení mezi klastogeny a aneugeny lze použít i jiné metody, pokud se ukázaly jako účinné. Analýza (manuální a automatizovaná) Všechny preparáty nebo vzorky k analýze, včetně preparátů nebo vzorků pozitivní a negativní kontroly, by měly být před kterýmkoli typem analýzy označeny samostatným kódem a určeny náhodným výběrem tak, aby hodnotiteli používajícímu ruční postupy nebyly známy podmínky expozice; takové kódování není nutné při použití automatizovaných systémů hodnocení, které nejsou závislé na vizuální kontrole a nemohou být ovlivněny neobjektivností obsluhy. Pro každé zvíře se stanoví podíl nezralých erytrocytů z celkového (nezralé + zralé) množství erytrocytů, přičemž se v případě kostní dřeně použije celkově alespoň 500 erytrocytů a v případě periferní krve alespoň 2 000 erytrocytů (42). U každého zvířete by se mělo vyšetřit alespoň 4 000 nezralých erytrocytů na výskyt mikrojader v nezralých erytrocytech (43). Pokud z historické databáze negativních kontrol vyplývá, že střední četnost výskytu nezralých erytrocytů s mikrojádry v dané laboratoři je < 0,1 %, mělo by se zvážit vyhodnocení dalších buněk. Při analýze vzorků by podíl počtu nezralých erytrocytů z celkového počtu všech hodnocených erxtrocytů u exponovaných zvířat neměl být menší než 20 % podílu v kontrolách s vehikulem/rozpouštědlem při hodnocení pod mikroskopem a ne méně než přibližně 5 % podílu nezralých erytrocytů z celkového množství erytrocytů v kontrolách s vehikulem/rozpouštědlem při hodnocení CD71 + nezralých erytrocytů cytometrickými metodami (viz odstavec 31) (29). Například ve zkoušce s kostní dření, která je hodnocena mikroskopicky, pokud by podíl nezralých erytrocytů v kostní dřeni u kontrol byl 50 %, horní limit toxicity by představoval 10 % nezralých erytrocytů. Poněvadž slezina potkanů erytrocyty s mikrojádry zachycuje a ničí, v zájmu zachování vysoké citlivosti zkoušky při analýze periferní krve potkana se upřednostňuje omezit analýzu nezralých erytrocytů s mikrojádry na nejmladší frakci. Při použití automatizovaných analytických metod lze tyto nejnezralejší erytrocyty identifikovat na základě jejich vysokého obsahu RNA nebo vysoké hladiny transferinových receptorů (CD71+) vyjádřené na jejich povrchu (31). Z přímého srovnání různých metod barvení však vyplynulo, že uspokojivé výsledky lze získat různými metodami, včetně konvenčního zbarvení akridinovou oranží (3)(4). ÚDAJE A PŘEDKLÁDÁNÍ ZPRÁV Zpracování výsledků Údaje pro jednotlivá zvířata by měly být zpracovány ve formě tabulky. Pro každé analyzované zvíře by měl být uveden počet vyšetřených nezralých erytrocytů, počet nezralých erytrocytů s mikrojádry a podíl nezralých erytrocytů z celkového množství erytrocytů. Jestliže jsou myši exponovány nepřetržitě čtyři týdny nebo déle, měly by být také uvedeny údaje o počtu a podílu zralých erytrocytů s mikrojádry, byly-li shromažďovány. Uvedeny by měly být i údaje o toxicitě u zvířat a klinické příznaky. Kritéria přijatelnosti Přijatelnost zkoušky určují tato kritéria:
Hodnocení a interpretace výsledků Za předpokladu, že všechna kritéria přijatelnosti byla splněna, lze zkoušenou chemickou látku považovat za jasně pozitivní, pokud:
Je-li v určitém konkrétním čase odběru zkoumána pouze nejvyšší dávka, je zkoušená chemická látka považována za jasně pozitivní, pokud existuje statisticky významný nárůst v porovnání se souběžnou negativní kontrolou a výsledky jsou mimo distribuci dosavadních údajů o negativních kontrolách (např. mimo 95 % rozmezí dle Poissonova rozdělení). Doporučení ohledně nejvhodnějších statistických metod lze najít v literatuře (44) (45) (46) (47). Při provádění analýzy dávky a účinku by měly být analyzovány alespoň tři skupiny exponované různé dávce. Při statistických testech by mělo být zvíře použito jako experimentální jednotka. Pozitivní výsledky mikronukleus testu znamenají, že zkoušená látka indukuje mikrojádra, jež jsou důsledkem chromozómového poškození nebo poškození mitotického aparátu erytroblastu testovaného druhu. V případě, že cílem zkoušky bylo detekovat centromery v mikrojádrech, pak indukce mikrojader obsahujících centromery (centromerickou DNA nebo kinetochory, jež ukazují na ztrátu celých chromozómů) zkoušenou chemickou látkou je důkazem, že zkoušená chemická látka je aneugenní. Za předpokladu, že všechna kritéria přijatelnosti byla splněna, je zkoušená chemická látka považována za jasně negativní, pokud ve všech zkoumaných zkušebních podmínkách:
Doporučení ohledně nejvhodnějších statistických metod lze najít v literatuře (44) (45) (46) (47). Důkazy o expozici kostní dřeně zkoušené chemické látce mohou zahrnovat pokles poměru nezralých a zralých erytrocytů nebo měření hladiny zkoušené chemické látky v plazmě nebo krvi. V případě nitrožilního podání není důkazů o expozici zapotřebí. Alternativně mohou být k prokázání expozice kostní dřeně použity údaje o absorpci, distribuci, metabolismu a exkreci (ADME) získané při nezávislé studii za použití stejného způsobu expozice a se stejným druhem. Negativní výsledky znamenají, že zkoušená chemická látka za podmínek zkoušky neprodukuje mikrojádra v nezralých erytrocytech druhu zvířat použitého k testování. Ověření jasně pozitivní či jasně negativní odpovědi se nepožaduje. V případech, kdy odpověď není jasně negativní nebo pozitivní, a s cílem napomoci při stanovení biologického významu výsledku (např. slabý nebo okrajový nárůst), by měly být údaje vyhodnoceny na základě odborného posouzení a/nebo dalším vyšetřením u provedených experimentů. V některých případech by mohlo být užitečné analyzovat více buněk nebo provést opakovaný experiment za použití upravených zkušebních podmínek. V ojedinělých případech dokonce ani po dalším šetření nebude možné na základě daných údajů učinit závěr, zda zkoušená chemická látka přináší pozitivní či negativní výsledky, a proto bude učiněn závěr, že výsledek studie je neurčitý. Závěrečná zpráva Závěrečná zpráva by měla obsahovat tyto informace:
LITERATURA
Dodatek 1 DEFINICE Centromera : oblast (oblasti) chromozómu, k níž (k nimž) se během dělení buněk připojí dělící vřeténko umožňující uspořádaný pohyb dceřiných chromozómů k pólům dceřiných buněk. Chemická látka : chemická substance nebo směs. Erytroblast : rané stadium vývoje erytrocytu, bezprostředně předcházející nezralému erytrocytu, kdy buňka ještě obsahuje jádro. Kinetochor : proteinová struktura tvořící centromeru eukaryotických buněk, jež během mitózy a meiózy spojuje s polymery mikrotubulů z mitotického vřeténka a při dělení buněk působí tak, že odtahuje sesterské chromatidy od sebe. Mikrojádra : malá jádra existující odděleně od hlavních buněčných jader a vedle nich, vytvářená během telofáze mitózy (meiózy) nereplikujícími se (lagging) chromozómovými fragmenty nebo celými chromozómy. Normochromatický nebo zralý erytrocyt : plně zralý erytrocyt, který ztratil reziduální RNA, jež zůstává po vypuzení jádra a/nebo který ztratil jiné buněčné markery s krátkou životností, jež zpravidla mizí po vypuzení jádra následujícím po konečném dělení erytroblastu. Polychromatický nebo nezralý erytrocyt : nově vzniklý erytrocyt v přechodném stadiu vývoje, který se obarví modrou a červenou složkou klasických krevních barviv jako např. Wright-Giemsa v důsledku přítomnosti reziduální RNA v nově vzniklé buňce. Takové nově vzniklé buňky jsou přibližně stejné jako retikulocyty, které se vizualizují pomocí vitálního barviva, jež způsobí, že se reziduální RNA shlukne do retikula. K identifikaci nově vzniklých červených krvinek se nyní často používají i jiné metody včetně monochromatického barvení RNA fluorescenčními barvivy nebo označení povrchových markerů s krátkou životností, jako je CD71, fluorescenčními protilátkami. Jak polychromatické erytrocyty, tak retikulocyty i CD71 pozitivní erytrocyty jsou nezralé erytrocyty, ačkoli každý z nich má poněkud odlišné věkové rozložení. Retikulocyt : nově vzniklý erytrocyt obarvený vitálním barvivem, jež způsobuje, že se reziduální RNA shlukne do charakteristického retikula. Retikulocyty a polychromatické erytrocyty mají podobné věkové rozložení buněk. Zkoušená chemická látka : jakákoli chemická substance nebo směs zkoušená pomocí této zkušební metody. Dodatek 2 FAKTORIÁLNÍ POKUS KE ZJIŠTĚNÍ ROZDÍLŮ MEZI POHLAVÍMI V TESTU MIKROJADER IN VIVO Faktoriální pokus a jeho analýza Při tomto pokusu se na každé úrovni koncentrace testuje minimálně 5 samců a 5 samic a výsledkem je uspořádání s použitím minimálně 40 zvířat (20 samců a 20 samic plus příslušné pozitivní kontroly). Tento pokus, který je jedním z nejjednodušších faktoriálních pokusů, je rovnocenný dvoucestné analýze rozptylu, přičemž hlavními prvky jsou úroveň koncentrace a pohlaví. Údaje je možné analyzovat pomocí mnoha standardních statistických softwarových balíčků, např. SPSS, SAS, STATA, Genstat a také pomocí programu R. Analýza dělí variabilitu v souboru údajů na variabilitu mezi pohlavími, variabilitu mezi úrovněmi koncentrace a variabilitu interakce mezi pohlavími a koncentracemi. Každý z těchto znaků se testuje oproti odhadu variability u replicitních zvířat ve skupinách zvířat stejného pohlaví, jimž je podávána stejná koncentrace látky. Veškeré podrobnosti základní metodiky jsou dostupné v mnoha standardních učebnicích statistiky (viz odkazy) a v „nápovědě“ statistických programů. Analýza se provádí zkoumáním znaku interakce pohlaví x koncentrace v tabulce ANOVA (6). Neexistuje-li významný znak interakce, kombinované hodnoty za obě pohlaví nebo za všechny úrovně koncentrace poskytují validní statistické testy variability mezi úrovněmi založené na zkoumání kumulovaného znaku vnitroskupinové variability pomocí analýzy ANOVA. Analýza pokračuje rozdělením odhadu variability mezi koncentracemi na kontrasty, což zajišťuje test na lineární a kvadratické kontrasty odpovědí na všech úrovních koncentrace. Pokud existuje významná interakce pohlaví x koncentrace, lze tento znak také rozdělit na lineární a kvadratický člen kontrastů interakce pohlaví. Pomocí těchto znaků se testuje, zda jsou odpovědi na koncentraci u obou pohlaví paralelní, nebo zda je odpověď u každého pohlaví odlišná. Odhad kumulované vnitroskupinové variability může být použit k párovému testování rozdílu mezi středními hodnotami. Tato srovnání by mohla být provedena mezi středními hodnotami u obou pohlaví a mezi středními hodnotami u různých úrovní koncentrace, např. za účelem porovnání s úrovněmi negativní skupiny. V případech, kdy existuje významná interakce, lze provádět srovnání mezi středními hodnotami při různých koncentracích v rámci jednotlivých pohlaví nebo mezi středními hodnotami u pohlaví při stejné koncentraci. Odkazy Existuje množství učebnic statistiky, které se zabývají teorií, uspořádáním, metodikou, analýzou a interpretací faktoriálních pokusů od nejjednodušších dvoufaktorových analýz až po složitější formy používané v metodice „experimentálního uspořádání“. Následující seznam není vyčerpávající. V některých pracích jsou uvedeny vypracované příklady srovnatelných uspořádání, v některých případech i s kódem pro provádění analýz pomocí různých počítačových programů.
|
|
6) |
V části B se zrušuje kapitola B.15. |
|
7) |
V části B se zrušuje kapitola B.16. |
|
8) |
V části B se zrušuje kapitola B.18. |
|
9) |
V části B se zrušuje kapitola B.19. |
|
10) |
V části B se zrušuje kapitola B.20. |
|
11) |
V části B se zrušuje kapitola B.24. |
|
12) |
V části B se kapitola B.47 nahrazuje tímto: „B.47 Zkušební metoda využívající zákal a propustnost rohovky skotu pro identifikaci i) chemických látek vyvolávajících vážné poškození očí a ii) chemických látek, které není nutné klasifikovat jako látky mající dráždivé účinky na oči nebo vyvolávající vážné poškození očí ÚVOD Tato metoda je rovnocenná Pokynu OECD pro zkoušení (TG) č. 437 (2013). Zkušební metodu pro zákal a propustnost rohovky u skotu (BCOP – Bovine Corneal Opacity and Permeability) hodnotil Koordinační výbor mezi různými organizacemi pro ověřování platnosti alternativních metod (ICCVAM) spolu s Evropským střediskem pro validaci alternativních metod (ECVAM) a Japonským střediskem pro validaci alternativních metod (JaCVAM) v letech 2006 a 2010 (1) (2). Při prvním hodnocení byla zkušební metoda BCOP hodnocena z hlediska její užitečnosti pro identifikaci chemických látek (látek a směsí) vyvolávajících závažné poškození očí (1). Při druhém hodnocení byla zkušební metoda BCOP hodnocena z hlediska její užitečnosti pro identifikaci chemických látek (látek a směsí), které nejsou klasifikovány jako látky mající dráždivé účinky na oči nebo vyvolávající vážné poškození očí (2). Databáze pro validaci BCOP obsahovala celkem 113 látek a 100 směsí (2) (3). Na základě těchto hodnocení a jejich odborné revize se dospělo k závěru, že tato zkušební metoda může správně identifikovat chemické látky (látky i směsi) vyvolávající vážné poškození očí (kategorie 1) a také látky, které není nutné klasifikovat jako látky mající dráždivé účinky na oči nebo vyvolávající vážné poškození očí, jak je definuje Globálně harmonizovaný systém klasifikace a označování chemických látek (GHS) Organizace spojených národů (OSN) (4) a nařízení (ES) č. 1272/2008 o klasifikaci, označování a balení látek a směsí (CLP) (7), a byla proto potvrzena jako vědecky platná pro oba výše uvedené účely. Vážným poškozením očí se rozumí vyvolání poškození oční tkáně nebo zhoršení vidění po aplikaci zkoušené chemické látky na povrch oka, které není plně vratné do 21 dnů po aplikaci. Zkoušené chemické látky vyvolávající vážné poškození očí jsou klasifikovány v kategorii 1 GHS OSN. Chemické látky, které nejsou klasifikovány jako látky mající dráždivé účinky na oči nebo vyvolávající vážné poškození očí, jsou definovány jako chemické látky, které nesplňují požadavky pro klasifikaci v kategorii 1 nebo 2 (2A nebo 2B) GHS OSN, tj. označují se jako chemické látky bez kategorie GHS OSN. V této zkušební metodě je uvedeno doporučené použití i to, jaká jsou omezení zkušební metody BCOP na základě jejích hodnocení. Hlavní rozdíly mezi původní verzí Pokynu OECD pro zkoušení z roku 2009 a aktualizovanou verzí z roku 2013 se mimo jiné týkají: použití zkušení metody BCOP k identifikaci chemických látek, které není nutné klasifikovat podle systému GHS OSN (odstavce 2 a 7); vyjasnění týkajících se použitelnosti zkušební metody BCOP pro testování alkoholů, ketonů a pevných látek (odstavce 6 a 7) a látek a směsí (odstavec 8); vyjasnění toho, jak by měla být testována smáčedla a směsi smáčedel (odstavec 28); aktualizace a vyjasnění týkajících se pozitivních kontrol (odstavce 39 a 40); aktualizace kritérií rozhodování zkušební metody BCOP (odstavec 47); aktualizace kritérií přijatelnosti studie (odstavec 48); aktualizace prvků protokolu o zkoušce (odstavec 49); aktualizace dodatku 1 týkající se definic; vložení dodatku 2 o prediktivní schopnosti zkušební metody BCOP v různých klasifikačních systémech; aktualizace dodatku 3 týkající se seznamu chemických látek pro prokázání způsobilosti; a aktualizace dodatku 4 týkající se držáku rohovky pro zkoušku BCOP (odstavec 1) a opacitometru (odstavce 2 a 3). V současnosti se obecně uznává, že jediná zkouška dráždivých účinků na oči in vitro nemůže v dohledné budoucnosti nahradit Draizeovu oční zkoušku in vivo, pokud jde o předpověď celé škály dráždivosti různých tříd chemických látek. Draizeovu oční zkoušku (5) však možná bude možné nahradit strategickou kombinací několika alternativních zkušebních metod v rámci strategie (odstupňovaného) zkoušení. Postup „shora dolů“ (5) má být použit, pokud se na základě existujících informací očekává, že chemická látka má vysokou schopnost vyvolávat dráždivé účinky, zatímco postup „zdola nahoru“ (5) se má použít, jestliže se na základě existujících informací předpokládá, že chemická látka nevyvolává tak intenzivní dráždivé účinky, aby vyžadovaly její klasifikaci. Zkušební metoda BCOP je zkušební metoda in vitro, kterou lze za určitých okolností a s některými omezeními použít pro klasifikaci nebezpečnosti chemických látek pro oči a pro označování těchto chemických látek. I když se BCOP nepovažuje za úplnou náhradu za oční test in vivo na králících, doporučuje se tuto zkušební metodu použít jako počáteční krok v rámci strategie zkoušení, jako je např. postup „shora dolů“, jejž navrhuje Scott et al. (5) pro identifikaci chemických látek vyvolávajících vážné poškození očí, tj. chemických látek, které je třeba klasifikovat v kategorii 1 GHS OSN bez dalšího zkoušení (4). Zkušební metodu BCOP se rovněž doporučuje použít k identifikaci chemických látek, které není nutné klasifikovat jako látky mající dráždivé účinky na oči nebo vyvolávající vážné poškození očí, jak jsou definovány v systému GHS OSN (látky bez kategorie GHS OSN) (4), a to v rámci strategie zkoušení, jako je např. postup „zdola nahoru“ (5). U chemické látky, o níž se na základě výsledků zkušební metody BCOP nepředpokládá, že vyvolává vážné poškození očí nebo naopak nemůže být klasifikována jako látka mající dráždivé účinky na oči / vyvolávající vážné poškození očí (látka bez kategorie), by však pro stanovení konečné klasifikace bylo nutné další zkoušení (in vitro a/nebo in vivo). Účelem této zkušební metody je popsat postupy používané k hodnocení potenciálních nebezpečných účinků zkoušené chemické látky na oči měřené její schopností způsobit zákal a zvýšit propustnost v izolované rohovce skotu. Toxické účinky na rohovku se měří: i) sníženou propustností světla (zákal) a ii) zvýšenou propustností barviva fluoresceinu sodného (propustnost). Hodnocení zákalu a propustnosti rohovky po expozici zkoušené chemické látce se kombinují ve výsledné skóre podráždění in vitro (IVIS – In Vitro Irritancy Score), které se používá pro klasifikaci úrovně dráždivých účinků zkoušené chemické látky. Definice jsou uvedeny v dodatku 1. VÝCHOZÍ ÚVAHY A OMEZENÍ Tato zkušební metoda je založena na protokolu zkušební metody BCOP vypracovaném výborem ICCVAM (6) (7), který byl původně vyvinut na základě informací získaných z protokolu Institutu pro vědu in vitro (IIVS) a INVITTOX protokolu č. 124 (8). Protokol INVITTOX byl použit pro studii o předběžném ověřování platnosti sponzorovanou Evropským společenstvím a vypracovanou v letech 1997–1998. Oba tyto protokoly vycházely z metodiky zkušební metody BCOP poprvé publikované Gautheronem et al. (9). Zkušební metodu BCOP je možné použít k identifikaci chemických látek vyvolávajících vážné poškození očí, jak je definováno v systému GHS OSN, tj. chemických látek, které je třeba klasifikovat v kategorii 1 GHS OSN (4). Je-li zkušební metoda BCOP použita pro tento účel, má celkovou přesnost 79 % (150/191), míru falešné pozitivity 25 % (32/126) a míru falešné negativity 14 % (9/65) ve srovnání s údaji ze zkušební metody in vivo na očích králíků klasifikovanými podle klasifikačního systému GHS OSN (3) (viz dodatek 2, tabulka 1). Pokud se z databáze vyloučí zkoušené chemické látky v určitých chemických (tj. alkoholy, ketony) nebo fyzikálních třídách (tj. pevné látky), má zkušební metoda BCOP v klasifikačním systému GHS OSN celkovou přesnost 85 % (111/131), míru falešné pozitivity 20 % (16/81) a míru falešné negativity 8 % (4/50) (3). Potenciální nedostatky zkušební metody BCOP, je-li použita k identifikaci chemických látek vyvolávajících vážné poškození očí (kategorie 1 GHS OSN), vyplývají z vysoké míry falešné pozitivity pro alkoholy a ketony a z vysoké míry falešné negativity pro pevné látky sledované v databázi ověřování platnosti (1) (2) (3). Jelikož však ne všechny alkoholy a ketony jsou zkušební metodou BCOP nepřiměřeně určeny a některé jsou správně určeny jako látky v kategorii 1 GHS OSN, nejsou tyto dvě organické funkční skupiny považovány za skupiny nacházející se mimo oblasti použitelnosti této zkušební metody. Je na uživateli této zkušební metody, aby rozhodl, zda případnou nepřiměřenou předpověď u alkoholu nebo ketonu lze akceptovat, nebo zda by mělo být provedeno další zkoušení s použitím přístupu založeného na posuzování závažnosti důkazů. Pokud jde o míru falešné negativity u pevných látek, je třeba poznamenat, že v Draizeově zkoušce dráždivých účinků na oči in vivo mohou pevné látky vést k různým a extrémním podmínkám expozice, což může v důsledku vyústit v irelevantní předpovědi jejich skutečné schopnosti vyvolávat dráždivé účinky (10). Nutno též podotknout, že míra falešné negativity zjištěná ve validační databázi ICCVAM (2) (3) v rámci identifikace chemických látek vyvolávajících vážné poškození očí (kategorie 1 GHS OSN) v žádném případě nevedla k hodnotě IVIS ≤ 3, což je kritérium používané k identifikaci zkoušené chemické látky jako látky bez kategorie GHS OSN. Míry falešné negativity BCOP v této souvislosti navíc nejsou kriticky důležité, protože všechny zkoušené chemické látky, které indukují hodnotu 3 < IVIS ≤ 55, by byly následně zkoušeny dalšími náležitě validovanými zkouškami in vitro nebo, jako poslední možnost, na králících, v závislosti na požadavcích právních předpisů, s využitím strategie postupného zkoušení v rámci přístupu hodnocení závažnosti důkazů. Vzhledem k tomu, že některé pevné chemické látky zkušební metoda BCOP správně určuje jako látky v kategorii 1 GHS OSN, ani toto skupenství není považováno za nacházející se mimo oblast použitelnosti této zkušební metody. Zkoušející by mohli zvážit použití této zkušební metody na všechny druhy chemických látek, přičemž hodnota IVIS > 55 by měla být považována za ukazatel reakce indukující vážné poškození očí, které je třeba klasifikovat v kategorii 1 GHS OSN bez dalšího zkoušení. Jak již však bylo uvedeno, kladné výsledky získané u alkoholů a ketonů by se měly z důvodu možné nadhodnocené předpovědi interpretovat opatrně. Zkušební metodu BCOP lze též použít k identifikaci chemických látek, které není nutné klasifikovat jako látky mající dráždivé účinky na oči nebo vyvolávající vážné poškození očí podle klasifikačního systému GHS OSN (4). Je-li zkušební metoda BCOP použita pro tento účel, má celkovou přesnost 69 % (135/196), míru falešné pozitivity 69 % (61/89) a míru falešné negativity 0 % (0/107) ve srovnání s údaji ze zkušební metody in vivo na očích králíků klasifikovanými podle klasifikačního systému GHS OSN (3) (viz dodatek 2, tabulka 2). Zjištěná míra falešné pozitivity (chemické látky bez kategorie GHS OSN ve zkouškách in vivo indukují hodnotu IVIS > 3, viz odstavec 47) je značně vysoká, avšak v této souvislosti nikoli kriticky důležitá, protože všechny zkoušené chemické látky, které indukují hodnotu 3 < IVIS ≤ 55, by byly následně zkoušeny jinými náležitě validovanými zkouškami in vitro nebo, jako poslední možnost, zkouškou na králících, v závislosti na požadavcích právních předpisů, s využitím strategie postupného zkoušení v rámci přístupu posuzování váhy důkazů. Zkušební metoda BCOP nevykazuje žádné specifické nedostatky v případě zkoušení alkoholů, ketonů a pevných látek, pokud je jejím účelem identifikovat chemické látky, které není nutné klasifikovat jako látky mající dráždivé účinky na oči nebo vyvolávající vážné poškození očí (tj. látky bez kategorie GHS OSN) (3). Zkoušející by mohli zvážit použití této zkušební metody na všechny druhy chemických látek, přičemž negativní výsledek (hodnota IVIS ≤ 3) by měl být uznán jako ukazatel toho, že látku není nutné klasifikovat (tj. látky bez kategorie GHS OSN). Jelikož zkušební metodou BCOP lze správně identifikovat pouze 31 % chemických látek, které není nutné klasifikovat jako látky mající dráždivé účinky na oči nebo vyvolávající vážné poškození očí, neměla by být tato zkušební metoda první volbou pro zahájení postupu „zdola nahoru“ (5), pokud jsou k dispozici jiné validované a uznané metody in vitro s podobně vysokou citlivostí, ale s vyšší specifičností. Databáze pro validaci BCOP obsahovala celkem 113 látek a 100 směsí (2) (3). Zkušební metoda BCOP je proto považována za použitelnou jak ke zkoušení látek, tak ke zkoušení směsí. Zkušební metoda BCOP se nedoporučuje pro identifikaci zkoušených chemických látek, které by měly být klasifikovány jako látky mající dráždivé účinky na oči (kategorie 2 nebo kategorie 2A GHS OSN), nebo zkoušených chemických látek, které by měly být klasifikovány jako látky mající mírně dráždivé účinky na oči (kategorie 2B GHS OSN), vzhledem k značnému počtu chemických látek kategorie 1 GHS OSN podhodnoceně klasifikovaných v kategorii 2, 2A nebo 2B GHS OSN a látek bez kategorie GHS OSN nadhodnoceně klasifikovaných v kategorii 2, 2A nebo 2B GHS OSN (2) (3). Pro tento účel může být nezbytné další zkoušení s použitím jiné vhodné metody. V případě všech postupů týkajících se očí skotu a rohovek skotu by se měly dodržovat právní předpisy a postupy pro zacházení s materiály získanými ze zvířat, které kromě jiného zahrnují tkáně a mimobuněčné tekutiny. Doporučují se univerzální laboratorní preventivní opatření (11). Zkušební metoda nebere v úvahu poškození spojivky a duhovky, ale zabývá se účinky na rohovku, které jsou důležitým faktorem pro stanovení klasifikace in vivo při posuzování klasifikace GHS OSN. Reverzibilitu lézí rohovky nelze zkušební metodou BCOP hodnotit samu o sobě. Na základě studií prováděných na očích králíků bylo navrženo, aby se posouzení počáteční hloubky poškození rohovky použilo k identifikaci některých typů nevratných účinků (12). Jsou však nezbytné další vědecké poznatky pro pochopení toho, jakým způsobem dochází k nevratným účinkům, které nejsou spojeny s počáteční vysokou úrovní poškození. A konečně je třeba uvést, že zkušební metoda BCOP neumožňuje posouzení možnosti systémové toxicity spojené s expozicí očí. Tato zkušební metoda se bude pravidelně aktualizovat, tak jak budou zohledňovány nové informace a údaje. Bude-li například zapotřebí komplexněji charakterizovat poškození rohovky, může být potenciálně užitečná histopatologie. Jak je uvedeno v Dokumentu OECD (GD) č. 160 (13), uživatelům se doporučuje, aby uchovali rohovku a připravili vzorky pro histopatologii, které lze použít pro vytvoření databáze a kritérií rozhodování, jež mohou dále zvýšit přesnost této zkušební metody. Každá laboratoř, která provádí tuto zkušební metodu poprvé, by měla použít referenční chemické látky uvedené v dodatku 3. Laboratoř může použít tyto chemické látky k prokázání své odborné způsobilosti k provádění zkušební metody BCOP před předložením výsledků získaných zkušební metodou BCOP pro regulační účely klasifikace nebezpečnosti. PODSTATA ZKOUŠKY Zkušební metoda BCOP je organotypický model, který zajišťuje krátkodobé udržování běžné fyziologické a biochemické funkce rohovky skotu in vitro. V této zkušební metodě se poškození zkoušenou chemickou látkou posuzuje kvantitativním měřením změn v zákalu a propustnosti rohovky za pomoci opacitometru (přístroje na měření průhlednosti) a spektrofotometru, který pracuje ve viditelné oblasti spektra. Obě měření se používají k výpočtu skóre podráždění in vitro (IVIS) za účelem přidělení klasifikační kategorie nebezpečnosti z hlediska podráždění in vitro, která slouží pro předpovídání potenciálu očního podráždění zkoušenou chemickou látkou in vivo (viz kritéria rozhodování v odstavci 48). Zkušební metoda BCOP používá izolované rohovky z očí čerstvě poráženého skotu. Zákal rohovky se měří kvantitativně jako množství průchodu světla rohovkou. Propustnost se měří kvantitativně jako množství barviva fluoresceinu sodného, které prochází celou tloušťkou rohovky a zjišťuje se v mediu zadní komory. Zkoušené chemické látky se aplikují na epiteliální povrch rohovky přidáním do přední komory držáku rohovky. V dodatku 4 je uveden popis a schématický nákres držáku rohovky používaný ve zkušební metodě BCOP. Držáky rohovky lze komerčně získat z různých zdrojů, nebo je lze zkonstruovat. Zdroj a věk očí skotu a výběr druhů zvířat Skot posílaný na jatka se zpravidla poráží buď pro lidskou spotřebu, nebo pro jiná komerční použití. Jako zdroj rohovek pro použití ve zkušební metodě BCOP se používají pouze zdravá zvířata, vyhovující požadavkům pro vstup do lidského potravinového řetězce. Jelikož skot má široký rozsah hmotnosti v závislosti na chovu, věku a pohlaví, neexistuje žádné doporučení týkající se hmotnosti zvířat v době porážky. Mohou se vyskytnout rozdíly ve velikosti rohovky, když se používají oči od zvířat různého věku. Rohovky o vodorovném průměru > 30,5 mm a s hodnotami základní tloušťky rohovky (CCT) ≥ 1100 μm se obvykle získávají ze skotu staršího osmi let, zatímco rohovky o vodorovném průměru < 28,5 mm a CCT < 900 μm se obvykle získávají ze skotu mladšího pěti let (14). Z těchto důvodů se zpravidla nepoužívají oči ze skotu staršího 60 měsíců. Oči ze skotu mladšího 12 měsíců se tradičně nepoužívají, protože oči se ještě vyvíjejí a tloušťka a průměr rohovky jsou mnohem menší, než jsou zaznamenány u očí dospělého skotu. Používání rohovek z mladých zvířat (tj. 6 až 12 měsíců starých) je však přípustné, jelikož mají určité výhody, např. zvýšená dostupnost, úzký věkový rozsah a nižší nebezpečnost související s potenciální expozicí zaměstnanců bovinní spongiformní encefalopatii (15). Protože by bylo účelné další hodnocení vlivu velikosti nebo tloušťky rohovky na reakci na leptavé a dráždivé chemické látky, uživatelům se doporučuje oznamovat odhadovaný věk a/nebo hmotnost zvířat, které poskytují rohovky používané ve studii. Získávání a doprava očí do laboratoře Oči shromažďují zaměstnanci jatek. Aby se minimalizovaly mechanické a jiné druhy poškození očí, měly by se oči odstranit co nejdříve po úmrtí a okamžitě po odstranění i během dopravy by měly být chlazeny. Aby se zamezilo expozici očí potenciálně dráždivým chemickým látkám, zaměstnanci jatek by neměli při oplachování hlavy zvířete používat čisticí prostředky. Oči by měly být ve vhodně velké nádobě úplně ponořeny do vychlazeného Hanksova vyváženého solného roztoku (HBSS) a dopraveny do laboratoře tak, aby se minimalizovalo poškození a/nebo bakteriální znečištění. Jelikož oči se získávají během procesu porážení, mohou být vystaveny krvi a jiným biologickým materiálům, včetně baktérií a jiných mikroorganismů. Proto je důležité zajistit, aby riziko znečištění bylo minimalizováno (např. držením nádoby, která obsahuje oči, v mokrém ledu, přidáním antibiotik do Hanksova vyváženého solného roztoku (HBSS) použitého k uchování očí během dopravy [např. penicilín se 100 m.j./ml a streptomycin se 100 μg/ml]). Časový interval mezi získáním očí a použitím rohovky ve zkušební metodě BCOP by měl být co nejkratší (získání a použití se uskutečňuje zpravidla tentýž den) a měl by být prokázán, aby se nenarušily výsledky analýzy. Tyto výsledky jsou založeny na kritériích výběru očí a také na reakcích na pozitivní a negativní kontroly. Všechny oči použité v analýze by měly být ze stejné skupiny očí získaných v určitý den. Kritéria výběru očí používaných ve zkušební metodě BCOP Jakmile se oči dostanou do laboratoře, jsou pečlivě vyšetřeny z hlediska vad, včetně zvýšeného zákalu, škrábanců a neovaskularizace. Smějí se používat pouze rohovky z očí, které tyto vady nemají. Kvalita každé rohovky se také hodnotí v pozdějších krocích analýzy. Rohovky, které mají zákal větší než sedm zákalových jednotek nebo zákal odpovídající pro použitý opacitometr a držáky rohovky i po počáteční jednohodinové době vyrovnání, se vyřadí (POZNÁMKA: opacitometr by měl být kalibrován podle norem zákalu používaných k určení zákalových jednotek, viz dodatek 4). Každá zkušební skupina (zkoušená chemická látka, paralelní negativní a pozitivní kontroly) pozůstává nejméně ze třech očí. V případě rohovek pro negativní kontrolu by se ve zkušební metodě BCOP měly používat tři rohovky. Protože všechny rohovky jsou vyříznuty z celé oční koule a vloženy do držáku rohovky, existuje při zpracování možnost vzniku artefaktů, které pak mohou mít vliv na hodnoty zákalu a propustnosti rohovky (včetně negativní kontroly). Kromě toho se hodnoty zákalu a propustnosti rohovek z negativních kontrol používají ke korekci hodnot zákalu a propustnosti rohovek získaných u pozitivních kontrol a u rohovek ovlivněných zkoušenou látkou ve výpočtech IVIS. POSTUP Příprava očí Rohovky, které nemají vady, se vyřezávají s 2 až 3mm okrajem očního bělma, který zůstává a pomáhá v následném ošetření, přičemž se musí dávat pozor, aby se zamezilo poškození epitelu a endotelu rohovky. Oddělené rohovky se zasazují do zvlášť k tomu určených držáků rohovek složených z předních a zadních částí, které které jsou ve styku s epiteliální nebo endoteliální stranou rohovky. Obě komory se úplně zaplní předehřátým Eaglovým minimálním médiem (EMEM = Eagle's Minimum Essential Medium) bez fenolové červeně (nejdříve zadní komora), při tom je třeba zajistit, aby se nevytvořily žádné bubliny. Zařízení se potom temperuje na teplotu 32 ± 1 °C nejméně po dobu jedné hodiny, aby se rohovky mohly teplotně vyrovnat s médiem a dosáhnout v co největším rozsahu běžné metabolické aktivity (přibližná teplota povrchu rohovky in vivo je 32 °C). Po vyrovnání teplot se čerstvé předehřáté médium EMEM bez fenolové červeně přidá do obou komor a pro každou rohovku se změří základní hodnoty zákalu. Všechny rohovky, které vykazují makroskopické poškození tkání (např. poškrábání, pigmentace, neovaskularizace) nebo zákal větší než sedm jednotek zákalu nebo větší než odpovídající hodnota pro použitý opacitometr a držáky rohovky, se vyřadí. Minimálně tři rohovky se vyberou jako rohovky pro negativní kontrolu (nebo pro kontrolu s rozpouštědlem). Zbylé rohovky se potom rozdělí do zkušebních skupin a do skupin pro pozitivní kontrolu. Jelikož tepelná kapacita vody je ve srovnání se vzduchem vyšší, voda poskytuje stabilnější teplotní podmínky pro inkubaci. Proto se doporučuje používání vodní lázně pro udržování držáku rohovky a jeho obsahu při teplotě 32 ± 1 °C. Vzduchové inkubátory však lze také použít za předpokladu, že budou přijata opatření k zachování tepelné stability (např. předehřátím držáků a médií). Aplikace zkoušené chemické látky Používají se dva různé zkušební protokoly, jeden pro tekutiny a smáčedla (pevná nebo tekutá) a jeden pro pevné látky, které nejsou smáčedla. Tekutiny se zkoušejí neředěné. Obdobně jako tekutiny se zpravidla zkoušejí polotuhé, krémové a voskovité látky. Čistá smáčedla se zkoušejí při koncentraci 10 % (m/V) v 0,9 % roztoku chloridu sodného, destilované vody nebo jiného rozpouštědla, u něhož se prokázalo, že nemá nepříznivé účinky na zkušební systém. U alternativních koncentrací roztoku by mělo být předloženo příslušné zdůvodnění. Směsi obsahující smáčedla by se měly zkoušet nezředěné nebo zředěné na vhodnou koncentraci v závislosti na příslušném scénáři expozice in vivo. Pro zkoušenou koncentraci by mělo být předloženo příslušné zdůvodnění. Rohovky jsou vystaveny tekutinám a smáčedlům po dobu 10 minut. Použití jiných dob expozice by mělo být doprovázeno odpovídajícím vědeckým zdůvodněním. Definice smáčedla a směsi obsahující smáčedla jsou uvedeny v dodatku 1. Pevné látky, které nejsou smáčedla, se zpravidla zkoušejí jako roztoky nebo suspenze při koncentraci 20 % m/V v 0,9 % roztoku chloridu sodného, destilované vody nebo jiného rozpouštědla, u něhož se prokázalo, že nemá nepříznivé účinky na zkušební systém. Za určitých okolností a s náležitým vědeckým zdůvodněním se pevné látky mohou zkoušet také čisté s přímou aplikací na povrch rohovky a s použitím metody otevřených komor (viz odstavec 32). Rohovky jsou vystaveny pevným látkám po dobu čtyř hodin, ale podobně jako u tekutých látek a smáčedel lze s odpovídajícím vědeckým zdůvodněním použít alternativní doby expozice. Je možné použít různé metody zkoušení v závislosti na fyzické povaze a chemických vlastnostech zkoušené chemické látky (např. pevné látky, tekutiny, viskózní látky versus neviskózní tekutiny). Kritický faktorem je nutnost zajistit, že zkoušená chemická látka náležitě pokrývá epiteliální povrch a že je příslušně odstraněna během oplachování. Metoda uzavřených komor se obvykle používá pro neviskózní až mírně viskózní tekuté zkoušené chemické látky, zatímco metoda otevřených komor se zpravidla používá pro poloviskózní a viskózní tekuté zkoušené chemické látky a pro čisté pevné látky. V metodě uzavřených komor se dostatečné množství zkoušené chemické látky (750 μL) k pokrytí epiteliální strany rohovky zavádí do přední komory přes dávkovací otvory na horním povrchu komory a otvory jsou potom během expozice uzavřeny komorovými zátkami. Je důležité zajistit, aby každá rohovka byla vystavena zkoušené chemické látce po příslušnou dobu. V metodě otevřených komor se před zkoušením odstraní z přední komory zajišťovací kroužek okénka a skleněné okénko. Kontrolní nebo zkoušená chemická látka (750 μl nebo dostatečné množství zkoušené chemické látky, aby úplně pokryla rohovku) se aplikuje přímo na epiteliální povrch rohovky pomocí mikropipety. Pokud je zkoušenou látku obtížné pipetovat, lze použít objemovou tlakovou pipetu (positive displacement pipette), což pomůže v dávkování. Špička objemové pipety se zasune do dávkovací špičky injekční stříkačky tak, aby látku bylo možné zavést pod tlakem do špičky vytěsňování. Píst injekční stříkačky se stlačí současně s tím, jak se píst pipety vytahuje nahoru. Pokud se ve špičce pipety objeví vzduchové bubliny, zkoušená chemická látka se odstraní (vytlačí) a postup se opakuje, dokud špička není naplněná bez vzduchových bublin. V případě potřeby lze použít běžnou injekční stříkačku (bez jehly), protože umožňuje měření přesného objemu zkoušené chemické látky a snadnější aplikaci na epiteliální povrch rohovky. Po dávkování se skleněné okénko umístí znovu na přední komoru, aby se obnovil uzavřený systém. Inkubace po expozici Po době expozice se zkoušená chemická látka, chemická látka pro negativní kontrolu nebo chemická látka pro pozitivní kontrolu odstraní z přední komory a epitel se nejméně třikrát opláchne (nebo se oplachuje tak dlouho, dokud nepřestanou být viditelné stopy zkoušené chemické látky) médiem EMEM (obsahující fenolovou červeň). Na oplachování se používá médium obsahující fenolovou červeň, protože změnou barvy fenolové červeně lze sledovat, jak účinné bylo oplachování kyselých nebo alkalických zkušebních chemických látek. Rohovky se proplachují více než třikrát, pokud je fenolová červeň ještě zbarvená (do žluta nebo do nachova), nebo pokud je zkoušená chemická látka stále viditelná. Jakmile již v médiu není zkoušená chemická látka, rohovky se naposledy opláchnou médiem EMEM (bez fenolové červeně). EMEM (bez fenolové červeně) se používá na konečný oplach proto, aby se před měřením zákalu zajistilo odstranění fenolové červeně z přední komory. Přední komora se pak naplní čerstvým EMEM bez fenolové červeně. U tekutin nebo smáčedel se rohovky po oplachu inkubují po dobu dalších dvou hodin při teplotě 32 ± 1 °C. Delší doba po expozici by byla za určitých okolností účelná a mohla by se zvážit případ od případu. Rohovky zkoušené s pevnými látkami se důkladně propláchnou na konci doby čtyřhodinové expozice, ale nevyžadují si další inkubaci. Zákal a propustnost každé rohovky se v případě tekutých látek a smáčedel zaznamená na konci doby inkubace po expozici a v případě pevných látek, které nejsou smáčedla, na konci doby čtyřhodinové expozice. Každá rohovka se sleduje rovněž vizuálně a případná pozorování se zaznamenají (např. odlupování tkáně, zbytky zkoušené chemické látky, nejednotné struktury rohovky). Tato pozorování by mohla být důležitá, protože mohou souviset se změnami v údajích opacitometru. Chemické látky pro kontrolu Do každého pokusu se zařazují souběžné negativní kontroly nebo kontroly s rozpouštědlem/vehikulem a pozitivní kontroly. Když se zkouší tekutá látka při 100 % koncentraci, souběžná negativní kontrola (např. 0,9 % roztok chloridu sodného nebo destilovaná voda) se zahrne do zkušební metody BCOP, takže jemožné zjistit nespecifické změny ve zkušebním systému a zajistit základní úroveň účinku pro konečné analýzy. Tím se rovněž zajistí, že podmínky analýzy nebudou mít za následek nevhodnou dráždivou reakci. Když se zkouší rozředěná tekutina, smáčedlo nebo pevná látka, skupina pro souběžnou kontrolu s rozpouštědlem/vehikulem se zahrne do zkušební metody BCOP tak, aby bylo možné zjistit nespecifické změny ve zkušebním systému a zajistit základnu pro koncové body analýzy. Lze použít pouze rozpouštědlo/vehikulum, u kterého se prokázalo, že nemá nepříznivé účinky na zkušební systém. Do každého pokusu se zařadí chemická látka, o níž se ví, že vyvolává pozitivní reakce, jako souběžná pozitivní kontrola, aby se ověřila integrita zkušebního systému a jeho správné fungování. Aby bylo možné vyhodnotit proměnlivost reakce na pozitivní kontrolu v čase, rozsah dráždivé reakce by neměl být nadměrný. Příklady pozitivních kontrol pro kapalné zkoušené chemické látky jsou 100 % ethanol nebo 100 % dimethylformamid. Příkladem pevných zkoušených látek pro pozitivní kontrolu je 20 % (m/V) imidazol v 0,9 % roztoku chloridu sodného. Srovnávací chemické látky jsou účelné pro hodnocení potenciálu podráždění očí neznámými chemickými látkami nebo specifickými třídami chemických látek či produktů nebo pro hodnocení potenciálu relativního podráždění látkou s dráždivými účinky na oči ve specifickém rozmezí dráždivých reakcí. Měřené koncové účinky Zákal se určuje množstvím průchodu světla rohovkou. Zákal rohovky se kvantitativně měří za pomoci opacitometru a výsledkem jsou hodnoty zákalu měřené v kontinuálním rozsahu. Propustnost se určuje množstvím barviva fluoresceinu sodného, které proniká všemi vrstvami buněk rohovky (tj. epitelem na vnějším povrchu rohovky přes endotel na vnitřním povrchu rohovky). Jeden ml roztoku fluoresceinu sodného (4 nebo 5 mg/ml, když se příslušně zkoušejí tekuté látky a smáčedla nebo pevné látky, které nejsou smáčedla) se přidává do přední komory držáku rohovky, která je spojena s epiteliální stranou rohovky, zatímco zadní komora, která je spojena s endoteliální stranou rohovky, se naplní čerstvým médiem EMEM. Držák se potom ve vodorovné poloze inkubuje po dobu 90 ± 5 min. na teplotu 32 ± 1 °C. Množství fluoresceinu sodného, který přechází do zadní komory, se kvantitativně měří za pomoci spektrofotometrie UV/VIS. Spektrofotometrická měření hodnocená ve 490 nm se zaznamenají jako optická hustota (OD490) nebo hodnoty absorbance, které se měří v kontinuálním rozsahu. Hodnoty propustnosti fluoresceinu se stanoví s použitím hodnot OD490 na základě spektrofotometru viditelného světla s použitím běžné optické délky 1 cm. Alternativně se může použít analyzátor destiček s 96 jamkami, pokud i) lze stanovit rozsah linearity měření analyzátoru destiček pro určení hodnot fluoresceinu OD490; a ii) v 96jamkové destičce je použit správný objem vzorků, jehož výsledkem jsou hodnoty OD490 odpovídající běžné optické délce 1 cm (to by si mohlo vyžadovat úplně plnou jamku [obvykle 360μL]). ÚDAJE A PŘEDKLÁDÁNÍ ZPRÁV Hodnocení údajů Jakmile se hodnoty zákalu a střední hodnoty propustnosti (OD490) upraví na zákal pozadí a hodnoty propustnosti OD490pro negativní kontrolu, střední hodnoty zákalu a propustnosti OD490pro každou zkušební skupinu by se měly spojit do empiricky odvozeného vzorce, aby se vypočítal stav podráždění in vitro (IVIS) pro každou zkušební skupinu takto: IVIS = střední hodnota zákalu + (15 × střední hodnota propustnosti OD490). Sina et al. uvádějí, že tento vzorec byl odvozen během laboratorních a mezilaboratorních studií. Údaje vytvořené pro řady 36 sloučenin v multilaboratorní studii byly předmětem vícerozměrné analýzy s cílem určit nejvhodnější rovnováhu mezi údaji in vivo a in vitro. Tuto analýzu provedli vědci ve dvou samostatných firmách a odvodili téměř totožné rovnice. Hodnoty zákalu a propustnosti by se měly rovněž posuzovat samostatně, aby se určilo, zda zkoušená chemická látka způsobuje poleptání nebo silné podráždění pomocí pouze jednoho nebo dvou koncových účinků (viz kritéria rozhodování). Kritéria rozhodování Dále jsou uvedeny hraniční hodnoty IVIS pro identifikaci zkoušených chemických látek vyvolávajících vážné poškození očí (kategorie 1 GHS OSN) a zkoušených chemických látek, které není nutné klasifikovat jako látky mající dráždivé účinky na oči nebo vyvolávající vážné poškození očí (bez kategorie GHS OSN):
Kritéria přijatelnosti studie Zkouška se považuje za přijatelnou, pokud pozitivní kontrola vykazuje IVIS spadající do obvyklých odchylek historické střední hodnoty, kterou je třeba aktualizovat nejméně každé tři měsíce nebo pokaždé, když se přijatelná zkouška vykonává v laboratořích, kde se zkoušky nedělají často (tj. méně než jednou měsíčně). Výsledkem reakce na negativní kontrolu nebo na kontrolu s rozpouštědlem/vehikulem by měly být hodnoty zákalu a propustnosti, které jsou nižší než stanovené horní meze pro hodnoty zákalu pozadí a prostupnosti u rohovek skotu exponované příslušnou negativní kontrolou nebo kontrolou s rozpouštědlem/vehikulem. U zkoušené chemické látky s jednoznačnou výslednou klasifikací by mělo stačit jedno provedení zkoušky za použití alespoň tří rohovek. Avšak v případě hraničních výsledků při prvním provedení zkoušky by se mělo zvážit druhé provedení zkoušky (ale není nezbytně nutné) a v případě nesouhlasných výsledků střední hodnoty IVIS mezi prvními dvěma provedeními zkoušky i třetí provedení zkoušky. V této souvislosti se výsledek prvního provedení zkoušky považuje za hraniční, pokud předpovědi na základě tří rohovek byly nesouhlasné tak, že:
Závěrečná zpráva Závěrečná zpráva by měla zahrnovat následující informace, pokud jsou relevantní pro provedení studie:
LITERATURA
Dodatek 1 DEFINICE Přesnost : Přesnost shody mezi výsledky zkušební metody a přijatými referenčními hodnotami. Je to měření výsledku zkušební metody a jednoho aspektu „důležitosti“. Termín se často používá namísto „souladu“, kterým se rozumí podíl správných výsledků zkušební metody. Srovnávací chemická látka : Látka použitá jako norma pro srovnání se zkoušenou látkou. Srovnávací látka by měla mít tyto vlastnosti: i) nesporný a spolehlivý zdroj (nesporné a spolehlivé zdroje); ii) strukturální a funkční podobnost s třídou zkoušených látek; iii) známé fyzikální/chemické vlastnosti; iv) podpůrné údaje o známých účincích a v) známá síla v rozsahu žádoucí reakce. Přístup „zdola nahoru“ : Přístup zkoušení po krocích používaný u chemických látek, u nichž se předpokládá, že je není nutné klasifikovat jako látky mající dráždivé účinky na oči nebo vyvolávající vážné poškození očí, který začíná odlišením chemických látek, u nichž není nutná klasifikace (negativní výsledek), od ostatních chemických látek (pozitivní výsledek). Chemická látka : Látka nebo směs. Rohovka : Průhledná přední část oční bulvy, která zahrnuje duhovku a zornici a propouští světlo dovnitř oka. Zákal rohovky : Měření rozsahu zákalu rohovky po expozici zkoušené chemické látce. Zvýšený zákal rohovky je příznačný pro poškození rohovky. Zákal lze hodnotit subjektivně, jak se to dělá ve Draizeově oční zkoušce na králících, nebo objektivně s přístrojem jako je „opacitometr“. Propustnost rohovky : Kvantitativní měření poškození epitelu rohovky určením množství barviva fluoresceinu sodného, který prochází všemi vrstvami buněk rohovky. Podráždění očí : vyvolání změn na oku po aplikaci zkoušené chemické látky na přední plochu oka, které jsou plně vratné do 21 dní od aplikace. Tento pojem je zaměnitelný s výrazy „vratné účinky na oko“ a „kategorie 2 GHS OSN“ (4). Míra falešné negativity : Podíl všech pozitivních chemických látek falešně zjištěných zkušební metodou jako negativní. Je to jeden z ukazatelů výsledku zkušební metody. Míra falešné pozitivity : Podíl všech negativních látek falešně zjištěných zkušební metodou jako pozitivní. Je to jeden z ukazatelů výsledku zkušební metody. Nebezpečnost : Základní charakteristika látky nebo situace, která má potenciál vyvolat nepříznivé účinky, jsou-li organismus, systém nebo (sub)populace vystaveny této látce. Skóre podráždění in vitro (IVIS) : Empiricky odvozený vzorec používaný ve zkušební metodě BCOP, přičemž střední hodnoty zákalu a propustnosti se pro každou zkušební skupinu spojí do jedné hodnoty in vitro pro každou zkušební skupinu. IVIS = střední hodnota zákalu + (15 × střední hodnota propustnosti). Nevratné účinky na oči : Viz „vážné poškození očí“. Směs : Směs nebo roztok tvořený dvěma nebo více látkami, které v něm nereagují (4). Negativní kontrola : Neovlivněná replika, která obsahuje všechny složky zkušebního systému. Tento vzorek se zpracovává se vzorky zkoušené chemické látky a jinými kontrolními vzorky, aby se zjistilo, zda rozpouštědlo vzájemně reaguje se zkušebním systémem. Neklasifikované : Chemické látky, které nejsou klasifikované jako látky mající dráždivé účinky na oči (kategorie 2, 2A nebo 2B GHS OSN) nebo jako látky vyvolávající vážné poškození očí (kategorie 1 GHS OSN). Tento pojem je zaměnitelný s pojmem „bez kategorie GHS OSN“. Opacitometr : Přístroj používaný k měření „zákalu rohovky“ kvantitativním hodnocením světelné propustnosti přes rohovku. Typický přístroj má dvě části, každá s vlastním zdrojem světla a fotobuňkou. Jedna část se používá pro zkoušenou rohovku a druhá pro kalibraci a vynulování přístroje. Světlo z halogenové lampy se posílá přes kontrolní část (prázdná komora bez okének nebo tekutiny) do fotobuňky a vyrovnané se světlem se posílá do fotobuňky přes zkušební část, ve které se nachází komora obsahující rohovku. Rozdíl ve světelné propustnosti z fotobuněk se porovnává a numerická hodnota zákalu se zobrazí na digitální obrazovce. Pozitivní kontrola : Replika obsahující všechny složky zkušebního systému a zkoušená s chemickou látkou, o níž je známo, že způsobuje pozitivní reakci. Aby se zajistilo, že proměnlivost v reakci na pozitivní kontrolu bude možné časem vyhodnotit, rozsah pozitivní reakce by neměl být nadměrný. Vratné účinky na oči : Viz „dráždivé účinky na oči“. Spolehlivost : Měření rozsahu, v jakém může být zkušební metoda časem reprodukovatelná v laboratořích a mezi laboratořemi, když se provádí s použitím stejného protokolu. Hodnotí se to výpočtem laboratorní a mezilaboratorní reprodukovatelnosti a laboratorní opakovatelnosti. Vážné poškození očí : Tvorba poškození tkání v oku nebo závažné fyzikální slábnutí vidění po aplikaci zkoušené látky na přední plochu oka, které není plně vratné do 21 dní od aplikace. Tento pojem je zaměnitelný s výrazy „nevratné účinky na oči“ a „kategorie 1 GHS OSN“ (4). Kontrola s rozpouštědlem/vehikulem : Neošetřený vzorek obsahující všechny složky zkušebního systému, včetně rozpouštědla nebo vehikula, který se zpracovává se vzorky exponované zkoušené chemické látky a jinými kontrolními vzorky, aby se zjistila základní reakce vzorků zkoušených se zkoušenou chemickou látkou rozpuštěnou ve stejném rozpouštědle nebo vehikulu. Pokud se vzorek zkouší souběžnou negativní kontrolou, ukazuje také, zda rozpouštědlo nebo vehikul vzájemně reaguje se zkušebním systémem. Látka : Chemické prvky nebo jejich sloučeniny v přírodním stavu nebo získané výrobním procesem včetně všech přídatných látek nutných k uchování stability výrobku a všech nečistot vznikajících v použitém procesu, ale s vyloučením všech rozpouštědel, která je možno oddělit bez ovlivnění stability látky nebo změny jejího složení (4). Smáčedlo : Nazývané též povrchově aktivní látka, je látka, jako např. detergent, která je schopna snížit povrchové napětí kapaliny a umožnit jí vytvářet pěnu nebo pronikat do pevných látek; je též známa jako surfaktant. Směs obsahující smáčedla : V kontextu této zkušební metody je to směs obsahující jedno nebo více smáčedel v konečné koncentraci > 5 %. Přístup „shora dolů“ : Přístup zkoušení po krocích používaný u chemických látek, u nichž se předpokládá, že způsobují vážné poškození očí, který začíná odlišením chemických látek vyvolávajících vážné poškození očí (pozitivní výsledek) od jiných chemických látek (negativní výsledek). Zkoušená chemická látka : Jakákoli látka nebo směs zkoušená touto zkušební metodou. Strategie postupného zkoušení : Strategie postupného zkoušení, kde se všechny existující informace o zkoušené chemické látce přezkoumávají ve stanoveném pořadí s použitím postupu váhy důkazů na každém stupni, aby se zjistilo, zda je k dispozici dost informací pro rozhodnutí o klasifikaci nebezpečnosti, než se postoupí do dalšího stupně. Pokud potenciál dráždivých účinků zkoušené chemické látky lze určit na základě existujících informací, další zkoušení není nutné. Jestliže potenciál dráždivých účinků zkoušené látky nelze určit na základě existujících informací, použije se stupňovitý postupný postup zkoušení na zvířatech po krocích, dokud se nestanoví jednoznačná klasifikace. Globálně harmonizovaný systém klasifikace a označování chemických látek Organizace spojených národů (GHC OSN) : Systém navrhující klasifikaci látek a směsí podle standardizovaných typů a úrovní fyzikálních, zdravotních a environmentálních nebezpečí a řešící příslušné komunikační prvky, jako jsou piktogramy, signální slova, výroky o nebezpečí, výroky o bezpečnostních opatřeních a bezpečnostní datové listy, které obsahují informace o jejich nežádoucích účincích s ohledem na ochranu lidí (včetně zaměstnavatelů, dělníků, přepravců, spotřebitelů a osob reagujících na havárie) a životního prostředí (4). Kategorie 1 GHS OSN : Viz „vážné poškození očí“. Kategorie 2 GHS OSN : Viz „dráždivé účinky na oči“. Bez kategorie GHS OSN : Chemické látky, které nesplňují požadavky na klasifikaci v kategorii 1 nebo 2 (2A nebo 2B) GHS OSN. Tento pojem je zaměnitelný s výrazem „neklasifikované“. Validovaná zkušební metoda : Zkušební metoda, u níž byly provedeny validační studie k určení její relevantnosti (včetně přesnosti) a spolehlivosti pro konkrétní účel. Je nutné poznamenat, že validovaná zkušební metoda nemusí poskytnout postačující výsledek z hlediska přesnosti a spolehlivosti, aby byla shledána přijatelnou pro navržený účel. Závažnost důkazů : Postup zvažování silných a slabých stránek různých informací v dosahování a podpoře závěru týkajícího se potenciálu nebezpečnosti látky. Dodatek 2 PREDIKTIVNÍ SCHOPNOST ZKUŠEBNÍ METODY BCOP Tabulka 1 Prediktivní schopnost BCOP při identifikaci chemických látek vyvolávajících vážné poškození očí [GHS OSN / CLP EU: kat. 1 versus jiné než kat. 1 (kat. 2 + bez kat.); EPA USA: kat. I versus jiné než kat I (kat. II + kat. III + kat. IV)]
Tabulka 2 Prediktivní schopnost BCOP při identifikaci chemických látek, které není nutné klasifikovat jako látky mající dráždivé účinky na oči nebo vyvolávající vážné poškození očí [GHS OSN / CLP EU:. bez kat. versus jiné než bez kat. (kat. 1 + kat. 2); EPA USA: kat. IV versus jiné než kat. IV (kat. I + kat. II + kat. III)]
Dodatek 3 CHEMICKÉ LÁTKY PRO PROKÁZÁNÍ ZPŮSOBILOSTI PRO ZKUŠEBNÍ METODU BCOP Dříve než začnou laboratoře rutinně používat tuto zkušební metodu, měly by prokázat svou odbornou způsobilost správným určením klasifikace nebezpečnosti pro oči u 13 chemických látek doporučených v tabulce 1. Tyto chemické látky byly vybrány tak, aby představovaly rozsah reakcí na nebezpečnost pro oči na základě výsledků oční zkoušky in vivo na králících (TG 405) (17) a klasifikačního systému GHS OSN (tj. kategorie 1, 2A, 2B, nebo neklasifikované) (4). Dalšími kritérii výběru bylo to, zda látka je komerčně dostupná, zda jsou k dispozici kvalitní referenční údaje in vivo a zda jsou kvalitní údaje in vitro získané ze zkušební metody BCOP. Referenční údaje jsou dostupné v publikacích Streamlined Summary Document (Racionalizovaný souhrnný dokument) (3) a v Background Review Document (Základní dokument o přezkumu) ICCVAM pro zkušební metodu BCOP (2) (18). Tabulka 1 Doporučené chemické látky pro prokázání způsobilosti k provádění zkušební metody BCOP
Dodatek 4 DRŽÁK ROHOVEK BCOP Držáky rohovek pro BCOP se vyrábějí z inertního materiálu (např. polypropylenu). Držáky se skládají ze dvou polovin (přední a zadní komora) a mají dvě podobné cylindrické vnitřní komory. Každá komora je zkonstruována tak, že pojme objem přibližně 5 ml a končí skleněným okénkem, přes které se zaznamenávají měření zákalu. Každá vnitřní komora má průměr 1,7 cm a hloubku 2,2 cm (11). Použije se těsnící kroužek umístěný na zadní komoře, aby se zamezilo úniku látky. Rohovky jsou umístěny na endoteliální straně pod těsnícím kroužkem zadních komor a přední komory se nacházejí na epiteliální straně rohovek. Komory se udržují na místě za pomoci třech šroubů z nerezové oceli umístěných na vnějších okrajích komory. Na konci každé komory se nachází skleněné okénko, které lze odstranit, aby byl snadný přístup k rohovce. Jeden těsnící kroužek je také umístěn mezi skleněným okénkem a komorou, aby se zamezilo úniku látky. Dva otvory v horní části každé komory umožňují zavedení a odstranění média a zkoušených chemických látek. Po dobu zkoušení a inkubace jsou uzavřeny gumovými uzávěry. Průchod světla přes držáky rohovky se potenciálně může měnit, neboť účinky běžného opotřebení nebo nahromadění reziduí určitých chemických látek ve vyvrtaných otvorech nebo na skleněném okénku vnitřní komory mohou ovlivnit rozptyl nebo odrazivost světla. Důsledkem by mohla být zvýšení nebo snížení základního průchodu světla (a tedy snížení nebo zvýšení naměřených údajů zákalu) přes držáky rohovky a ta se mohou projevit jako znatelné změny původně očekávaných naměřených údajů zákalu rohovky v jednotlivých komorách (tj. původní hodnoty zákalu rohovky v konkrétních držácích rohovky se mohou běžně lišit o více než 2 nebo 3 jednotky zákalu od očekávaných základních hodnot). Každá laboratoř by měla zvážit zavedení programu pro hodnocení změn průchodu světla přes držáky rohovky v závislosti na povaze zkoušených chemismů a na frekvenci použití komor. Ke stanovení základních hodnot je možné držáky rohovky před běžným použitím zkontrolovat změřením základních hodnot zákalu (nebo průchodu světla) komor naplněných kompletním médiem bez rohovek. Držáky rohovky se pak pravidelně kontrolují na změny průchodu světla během doby použití. Každá laboratoř může stanovit frekvenci kontrol držáků rohovky na základě zkoušených chemických látek, frekvenci použití a pozorování změn základních hodnot zákalu rohovky. Jsou-li pozorovány znatelné změny průchodu světla přes držáky rohovky, je nutno zvážit přiměřené postupy čištění a/nebo leštění vnitřního povrchu držáku rohovky nebo jejich výměnu. Držák rohovky: viz připojený nákres. 
Dodatek 5 OPACITOMETR Opacitometr je přístroj pro měření světelné propustnosti. Například u zařízení OP-KIT od výrobce Electro Design (Riom, Francie), které se používá při hodnocení v rámci zkušební metody BCOP, se světlo z halogenové lampy posílá přes kontrolní část (prázdná komora bez okének nebo tekutiny) do fotobuňky a vyrovnané se světlem se posílá do fotobuňky přes zkušební část, ve které se nachází komora obsahující rohovku. Rozdíl ve světelné propustnosti z fotobuněk se porovnává a numerická hodnota zákalu se zobrazí na digitální obrazovce. Tím se určí zákalové jednotky. Lze použít jiné typy opacitometrů s odlišnou konstrukcí (např. nevyžadující paralelní měření kontrolní a zkušební přihrádky), je-li prokázáno, že poskytnou podobné výsledky jako u validovaného zařízení. Opacitometr by měl poskytnout lineární odezvu za pomoci řady údajů o zákalu, které zahrnují mezní hodnoty používané pro různé klasifikace popsané v modelu předpovědí (tj. až do mezní hodnoty, která určuje leptavost/silnou dráždivost). Aby se zajistily lineární a přesné údaje až do 75–80 zákalových jednotek, je nutné opacitometr kalibrovat s použitím řady kalibračních přístrojů. Kalibrátory se vloží do kalibrační komory (komora pro rohovku určená k uložení kalibrátorů) a odečítá se údaj na opacitometru. Kalibrační komora je určena k držení kalibrátorů přibližně ve stejné vzdálenosti mezi světlem a fotobuňkou, v jaké se umístí rohovky během měření zákalu. Referenční hodnoty a nastavený počáteční bod závisí na typu použitého zařízení. Lineárnost údajů opacitometru by měla být zajištěna vhodnými postupy (specifickými podle typu přístroje). Například u zařízení OP-KIT od výrobce Electro Design (Riom, Francie) se opacitometr nejprve kalibruje na zákal 0 zákalových jednotek s použitím kalibrační komory bez kalibračního materiálu. Potom se do kalibrační komory umístí jeden po druhém tři různé kalibrátory a měří se zákaly. Výsledkem kalibrátorů 1, 2 a 3 by měly být údaje o zákalu rovnající se příslušně nastaveným hodnotám 75, 150, a 225 zákalových jednotek, ± 5 %. |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
13) |
V části B se kapitola B.48 nahrazuje tímto: „B.48 Zkušební metoda odděleného kuřecího oka pro zjišťování i) chemických látek vyvolávajících vážné poškození očí a ii) chemických látek, které není nutné klasifikovat jako látky mající dráždivé účinky na oči nebo vyvolávající vážné poškození očí ÚVOD Tato metoda je rovnocenná Pokynu OECD pro zkoušení (TG) č. 438 (2013). Zkušební metodu odděleného kuřecího oka (ICE – Isolated Chicken Eye) hodnotil Koordinační výbor mezi různými organizacemi pro ověřování platnosti alternativních metod (ICCVAM) spolu s Evropským střediskem pro validaci alternativních metod (ECVAM) a Japonským střediskem pro validaci alternativních metod (JaCVAM) v letech 2006 a 2010 (1) (2) (3). Při prvním hodnocení byla zkušební metoda ICE schválena jako vědecky platná zkušební metoda, kterou lze použít jako screeningovou zkoušku pro zjišťování chemických látek (látek a směsí) vyvolávajících vážné poškození očí (kategorie 1), jak ho definuje Globálně harmonizovaný systém klasifikace a označování chemických látek (GHS) Organizace spojených národů (OSN) (1) (2) (4) a nařízení (ES) č. 1272/2008 o klasifikaci, označování a balení látek a směsí (nařízení CLP) (12). Při druhém hodnocení byla zkušební metoda ICE hodnocena z hlediska jejího použití jako screeningové zkoušky pro identifikaci chemických látek, které nejsou klasifikovány jako látky mající dráždivé účinky na oči nebo vyvolávající vážné poškození očí, jak je definuje GHS OSN (3) (4). Na základě výsledků validační studie a doporučení komise odborné revize bylo ponecháno původní doporučení na používání zkušební metody ICE pro klasifikaci chemických látek vyvolávajících vážné poškození očí (kategorie 1 GHS OSN) a dostupná databáze zůstala od původní validace, kterou provedl výbor ICCVAM, beze změn. V té době nebyla předložena žádná doporučení na rozšíření oblasti použitelnosti zkušební metody ICE také na další kategorie. Bylo provedeno nové hodnocení souboru údajů in vitro a in vivo použitého při validační studii, se zaměřením na hodnocení užitečnosti zkušební metody ICE pro zjišťování chemických látek, které není nutné klasifikovat jako látky vyvolávající podráždění očí nebo vážné poškození očí (5). Toto nové hodnocení dospělo k závěru, že zkušební metodu ICE lze též použít pro zjišťování chemických látek, které není nutné klasifikovat jako látky mající dráždivé účinky na oči nebo vyvolávající vážné poškození očí, jak je definuje GHS OSN (4) (5). V této zkušební metodě jsou uvedena doporučená použití a omezení zkušební metody ICE na základě těchto hodnocení. Hlavními rozdíly mezi původní verzí z roku 2009 a aktualizovanou verzí Pokynů OECD ke zkoušení z roku 2013 jsou mimo jiné použití zkušební metody ICE pro zjišťování chemických látek, které není nutné klasifikovat podle klasifikačního systému GHS OSN, aktualizace prvků protokolu o zkoušce, aktualizace dodatku 1 týkající se definic a aktualizace dodatku 2 týkající se chemických látek pro prokázání způsobilosti. V současnosti se obecně uznává, že jediná zkouška dráždivých účinků na oči in vitro nemůže v dohledné budoucnosti nahradit Draizeovu oční zkoušku in vivo, pokud jde o předpověď celé škály dráždivosti různých tříd chemických látek. Draizeovu oční zkoušku (6) však možná bude možné nahradit strategickou kombinací několika alternativních zkušebních metod v rámci strategie (odstupňovaného) zkoušení. Postup „shora dolů“ (7) má být použit, pokud se na základě existujících informací očekává, že chemická látka má vysokou schopnost vyvolávat dráždivé účinky, zatímco postup „zdola nahoru“ (7) se má použít, jestliže se na základě existujících informací předpokládá, že chemická látka nevyvolává tak intenzivní dráždivé účinky, aby vyžadovaly její klasifikaci. Zkušební metoda ICE je zkušební metoda in vitro, kterou lze za určitých okolností a s některými omezeními, jež jsou popsány v odstavcích 8 až 10, použít pro klasifikaci nebezpečnosti chemických látek pro oči a pro označování chemických látek. I když se zkušební metoda ICE nepovažuje za úplnou náhradu za oční test in vivo na králících, doporučuje se tuto zkušební metodu použít jako počáteční krok v rámci strategie zkoušení, jako je např. postup „shora dolů“, jejž navrhuje Scott et al. (7) pro identifikaci chemických látek vyvolávajících vážné poškození očí, tj. chemických látek, které je třeba klasifikovat v kategorii 1 GHS OSN bez dalšího zkoušení (4). Zkušební metodu ICE se rovněž doporučuje použít pro zjišťování chemických látek, které není nutné klasifikovat jako látky mající dráždivé účinky na oči nebo vyvolávající vážné poškození očí, jak je definuje systém GHS OSN (látky bez kategorie) (4), a lze ji tedy použít jako počáteční krok v rámci strategie zkoušení postupem „zdola nahoru“ (7). U chemické látky, o níž se na základě použití zkušení metody ICE nepředpokládá, že vyvolává vážné poškození očí nebo že by měla být klasifikována jako látka mající dráždivé účinky na oči / vyvolávající vážné poškození očí, by však pro stanovení konečné klasifikace bylo nutné další zkoušení (in vitro a/nebo in vivo). Použití zkušební metody ICE v rámci postupu „zdola nahoru“ podle jiných klasifikačních systémů než GHS OSN by mělo být předem konzultováno s příslušnými regulačními orgány. Účelem této zkušební metody je popsat postupy používané k hodnocení potenciálních nebezpečných účinků zkoušené chemické látky na oči měřené její schopností způsobit či nezpůsobit toxicitu v izolovaném kuřecím oku. Toxické účinky na rohovku se měří i) kvalitativním hodnocením zákalu, ii) kvalitativním hodnocením poškození epitelu na základě aplikace fluoresceinu do oka (zadržování fluoresceinu), iii) kvantitativním měřením zvýšené tloušťky (otok) a iv) kvalitativním hodnocením makroskopického morfologického poškození povrchu. Hodnocení zákalu a otoku rohovky a poškození po expozici zkoušené chemické látce se posuzuje samostatně a potom se spojí za účelem odvození klasifikace dráždivých účinků na oči. Definice jsou uvedeny v dodatku 1. VÝCHOZÍ ÚVAHY A OMEZENÍ Tato zkušební metoda je založena na protokolu doporučeném v pokynu OECD č. 160 (8) zpracovaném v návaznosti na mezinárodní studii ICCVAM o ověřování platnosti (1) (3) (9) s přispěním Evropského střediska pro ověřování platnosti alternativních metod, Japonského střediska pro validaci alternativních metod a oddělení toxikologie a aplikované farmakologie TNO Quality of Life (Nizozemsko). Protokol vychází z informací získaných z uveřejněných protokolů a také z aktuálního protokolu, který používá TNO (10) (11) (12) (13) (14). Při validaci této zkušební metody byla testována široká škála chemických látek a empirická databáze validační studie obsahuje celkem 152 chemických látek, včetně 72 látek a 80 směsí (5). Zkušební metodu lze použít pro pevné látky, kapaliny, emulze a gely. Kapaliny mohou být vodné či nevodné, pevné látky mohou být ve vodě rozpustné či nerozpustné. Plyny a aerosoly dosud nebyly ve validační studii hodnoceny. Zkušební metodu ICE je možné použít k identifikaci chemických látek vyvolávajících vážné poškození očí, tj. chemických látek, které je třeba klasifikovat v kategorii 1 GHS OSN (4). Při použití pro tento účel zjištěná omezení zkušební metody ICE vyplývají z vysoké míry falešné pozitivity pro alkoholy a z vysoké míry falešné negativity pro pevné látky a smáčedla (1) (3) (9). Míra falešné pozitivity však v této souvislosti (chemické látky kategorie 1 GHS OSN identifikované jako látky, které nepatří do kategorie 1) není kriticky důležitá, neboť všechny zkoušené chemické látky, jež byly shledány negativními, by byly následně zkoušeny jinou náležitě validovanou zkouškou (zkouškami) in vitro nebo, jako poslední možnost, zkouškou na králících, v závislosti na požadavcích právních předpisů, s využitím strategie postupného zkoušení v rámci přístupu založeného na posuzování závažnosti důkazů. Je třeba poznamenat, že v Draizeově zkoušce dráždivých účinků na oči in vivo mohou pevné látky vést k různým a extrémním podmínkám expozice, což může v důsledku vyústit v irelevantní předpovědi jejich skutečné schopnosti vyvolávat dráždivé účinky (15). Zkoušející by mohli zvážit použití této zkušební metody na všechny druhy chemických látek, přičemž pozitivní výsledek by měl být považován za ukazatel vážného poškození očí, tj. klasifikace v kategorii 1 GHS OSN, bez dalšího zkoušení. Kladné výsledky získané u alkoholů by se však měly interpretovat opatrně z důvodu rizika nepřiměřené předpovědi. Je-li zkušební metoda ICE použita pro zjišťování chemických látek vyvolávajících vážné poškození očí (kategorie 1 GHS OSN), má celkovou přesnost 86 % (120/140), míru falešné pozitivity 6 % (7/113) a míru falešné negativity 48 % (13/27) ve srovnání s údaji ze zkušební metody in vivo na očích králíků klasifikovanými podle klasifikačního systému GHS OSN (4) (5). Zkušební metodu ICE lze též použít k identifikaci chemických látek, které není nutné klasifikovat jako látky mající dráždivé účinky na oči nebo vyvolávající vážné poškození očí podle klasifikačního systému GHS OSN (4). Použití zkušební metody ICE v rámci postupu „zdola nahoru“ podle jiných klasifikačních systémů by mělo být předem konzultováno s příslušnými regulačními orgány. Tuto zkušební metodu lze použít pro všechny druhy chemických látek, přičemž negativní výsledek by mohl být uznán pro neklasifikování chemické látky jako látky mající dráždivé účinky na oči a vyvolávající vážné poškození očí. Na základě jednoho výsledku z validační databáze však u barviv s účinkem proti usazeninám obsahujících organická rozpouštědla může dojít k předpovědi podhodnocující jejich potenciál dráždivých účinků (5). Je-li zkušební metoda ICE použita pro zjišťování chemických látek, které není nutné klasifikovat jako látky vyvolávající dráždivé účinky na oči a vážné poškození očí, má celkovou přesnost 82 % (125/152), míru falešné pozitivity 33 % (26/79) a míru falešné negativity 1 % (1/73) ve srovnání s údaji ze zkušební metody in vivo na očích králíků klasifikovanými podle systému GHS OSN (4) (5). Pokud se z databáze vyloučí zkoušené chemické látky v určitých chemických třídách (tj. barviva s účinkem proti usazeninám obsahující organická rozpouštědla), je přesnost zkušební metody ICE v klasifikačním systému GHS OSN 83 % (123/149), míra falešné pozitivity 33 % (26/78) a míra falešné negativity 0 % (0/71) (4) (5). Zkušební metoda ICE se nedoporučuje pro identifikaci zkoušených chemických látek, které by měly být klasifikovány jako látky mající dráždivé účinky na oči (kategorie 2 nebo kategorie 2A GHS OSN), nebo zkoušených chemických látek, které by měly být klasifikovány jako látky mající mírně dráždivé účinky na oči (kategorie 2B GHS OSN), vzhledem k značnému počtu chemických látek kategorie 1 GHS OSN podhodnoceně klasifikovaných v kategorii 2, 2A nebo 2B GHS OSN a látek bez kategorie GHS OSN nadhodnoceně klasifikovaných v kategorii 2, 2A nebo 2B GHS OSN. Pro tento účel může být nezbytné další zkoušení s použitím jiné vhodné metody. Veškeré postupy u kuřecích očí by měly dodržovat právní předpisy a postupy pro zacházení s lidskými materiály nebo materiály získanými ze zvířat, které kromě jiného zahrnují tkáně a mimobuněčné tekutiny. Doporučují se univerzální laboratorní preventivní opatření (16). Zkušební metoda ICE nebere v úvahu poškození spojivky a duhovky, které se hodnotí ve zkušební metodě dráždivosti pro oči u králíků, ale zabývá se účinky na rohovku, které jsou důležitým faktorem pro stanovení klasifikace in vivo při posuzování klasifikace GHS OSN. Ačkoli reverzibilitu lézí rohovky nelze ve zkušební metodě ICE hodnotit samu o sobě, na základě studií o očích králíků bylo přesto navrženo, že by se posouzení počáteční hloubky poškození rohovky mohlo použít k určení některých druhů nevratných účinků (17). Jsou zejména nezbytné další vědecké poznatky pro pochopení toho, jakým způsobem dochází k nevratným účinkům, které nejsou spojeny s počáteční vysokou úrovní poškození. A konečně je třeba uvést, že zkušební metoda ICE neumožňuje posouzení potenciálu systémové toxicity spojené s expozicí očí. Tato zkušební metoda se bude pravidelně aktualizovat, tak jak budou zohledňovány nové informace a údaje. Bude-li například zapotřebí komplexněji charakterizovat poškození rohovky, může být potenciálně užitečná histopatologie. Pro vyhodnocení této možnosti se uživatelům doporučuje, aby uchovali oči a připravili vzorky pro histopatologii, které lze použít pro vytvoření databáze a kritérií rozhodování, jež mohou dále zvýšit přesnost této zkušební metody. Organizace OECD vypracovala pokyny k použití zkušebních metod pro hodnocení oční toxicity in vitro, jež zahrnují podrobné postupy při shromažďování histopatologických vzorků a informace, kam mají být vzorky a/nebo histopatologické údaje zasílány (8). Každá laboratoř, která zavádí tuto analýzu, by měla použít chemické látky pro zkoušení způsobilosti uvedené v dodatku 2. Laboratoř může použít tyto chemické látky k prokázání své odborné způsobilosti v používání zkušební metody ICE před předložením údajů ICE pro regulační účely klasifikace nebezpečí. PODSTATA ZKOUŠKY Zkušební metoda ICE je organotypický model, který zajišťuje krátkodobé uchování kuřecího oka in vitro. V této zkušební metodě se poškození zkoušenou chemickou látkou posuzuje určením otoku a zákalu rohovky a zadržování fluoresceinu. Zatímco dva poslední parametry se týkají kvalitativního hodnocení, analýza otoku rohovky zajišťuje kvantitativní hodnocení. Každé měření se buď převádí na kvantitativní hodnotu používanou k výpočtu celkového ukazatele podráždění, nebo je přiřazeno kvalitativní kategorizaci, která se používá ke stanovení klasifikace nebezpečnosti pro oči in vitro buď v kategorii 1 GHS OSN, nebo jako látky neklasifikované v systému GHS OSN. Každý z těchto výsledků potom může být použit k předpovědi schopnosti zkoušené látky vyvolávat vážné poškození očí in vivo nebo k předpovědi, že se nevyžaduje klasifikace nebezpečnosti zkoušené chemické látky pro oči (viz kritéria rozhodování). Není však možné stanovit klasifikaci chemických látek, u nichž nebylo s použitím zkušební metody ICE předpovězeno, že vyvolávají vážné poškození očí nebo že je není nutné klasifikovat. Zdroj a věk kuřecích očí Tradičně se pro tuto analýzu používají kuřecí oči získané z jatek, kde se kuřata zabíjejí pro lidskou spotřebu, čímž se eliminuje potřeba laboratorních zvířat. Používají se pouze oči zdravých zvířat, které se považují za vhodné pro vstup do lidského potravinového řetězce. Ačkoli nebyla zpracována kontrolní studie pro hodnocení optimálního věku kuřat, věk a hmotnost kuřat tradičně používaných v této zkušební metodě odpovídá mladým kuřatům tradičně zpracovávaným na jatkách pro drůbež (tj. přibližně 7 týdnů staré, o váze 1,5–2,5 kg). Shromažďování a doprava očí do laboratoře Hlavy by se měly odstranit ihned po usmrcení kuřat obvykle elektrickým šokem a naříznutím krku za účelem zbavení krve. Místní zdroj kuřat blízko laboratoře by měl být umístěn tak, aby hlavy kuřat mohly být dopraveny z jatek do laboratoře dostatečně rychle s cílem minimalizovat poškození a/nebo bakteriální znečištění. Časový interval mezi shromážděním kuřecích hlav, odstraněním očí a jejich umístěním do superfúzní komory by měl být co nejkratší (zpravidla do dvou hodin), aby se zajistilo splnění kritérií přijatelnosti zkoušky. Všechny oči použité v analýze by měly být ze stejné skupiny očí shromážděných v určitý den. Protože oči se oddělují v laboratoři, neporušené hlavy se dopravují z jatek za okolní teploty (obyčejně mezi 18 °C a 25 °C) v umělohmotných krabicích, které jsou navlhčeny ubrousky namočenými v izotonickém fyziologickém roztoku. Kritéria výběru a počet očí používaných v ICE Oči, které po enukleaci mají vysoké základní zbarvení fluoresceinem (tj. > 0,5) nebo vysokou hodnotu zákalu rohovky (tj. > 0,5), se vyřadí. Každá zkušební skupina a souběžná pozitivní kontrola pozůstává nejméně ze třech očí. Skupina pro negativní kontrolu nebo pro kontrolu s rozpouštědlem (používá-li se jiné rozpouštědlo než fyziologický roztok) pozůstává nejméně z jednoho oka. V případě pevných látek, které vedou k výsledku, kdy látku není nutné klasifikovat v systému GHS OSN, se doporučuje druhé provedení zkoušky se třema očima, která negativní výsledek potvrdí nebo vyvrátí. POSTUP Příprava očí Oční víčka se opatrně vyříznou a přitom se dává pozor, aby se nepoškodila rohovka. Neporušenost rohovky se rychle posoudí kápnutím 2 % (objemové hmotnosti) fluoresceinu sodného aplikovaného na povrch rohovky po dobu několika vteřin, který se potom opláchne izotonickým fyziologickým roztokem. Oči ošetřené fluoresceinem se pak zkoumají pod mikroskopem se štěrbinovou lampou, aby se zajistilo, že se rohovka nepoškodí (tj. hodnoty zadržení fluoresceinu a zákalu rohovky ≤ 0,5). Není-li oko poškozeno, vyřízne se z lebky a přitom se dává pozor, aby se nepoškodila rohovka. Oční bulbus se vyjme z očnice tak, že se chirurgickou pinzetou pevně uchopí mžurka a oční sval se ustřihne zahnutými tupě zakončenými nůžkami. Je důležité zamezit poškození rohovky nadměrným tlakem (tj. kompresní artefakty). Když je oko vyjmuto z očnice, viditelná část očního nervu by se měla ponechat. Jakmile se oko vyjme z očnice, položí se na absorpční podložku a mžurka a jiné pojivové tkáně se ustřihnou. Odstraněné oko se nasadí na držák z nezeravějící oceli s rohovkou ve svislé poloze. Držák se potom přenese do komory superfúzního aparátu (18). Držáky by se měly v tomto speciálním zkušebním přístroji postavit tak, aby se kapky izotonického fyziologického roztoku dostávaly k celé rohovce (3–4 kapky za minutu nebo 0,1 až 0,15 ml/min). Teplota komor přístroje by měla být řízena na 32 ± 1,5 °C. V dodatku 3 je schématický nákres typického superfúzního aparátu a očních držáků, které je možné obchodně získat nebo zkonstruovat. Přístroje mohou být upraveny, aby splňovaly potřeby jednotlivé laboratoře (např. umístění různého počtu očí). Když se oči vloží do superfúzního aparátu, znovu se prozkoumají pod mikroskopem se štěrbinovou lampou, aby se zjistilo, zda nebyly poškozeny během vyřezávání. Tloušťka rohovky by se v této době měla také měřit na vrcholu rohovky s použitím zařízení pro měření hloubky na mikroskopu se štěrbinovou lampou. Oči s i) hodnotou zadržení fluoresceinu > 0.5; (i) zákalem rohovky > 0.5 nebo iii) jakýmikoli dalšími znaky poškození by se měly vyměnit. V případě očí nevyřazených na základě jakéhokoli z těchto kritérií se musí vyřadit jednotlivé oči s tloušťkou rohovky, která se o víc než 10 % odchyluje od střední hodnoty pro všechny oči. Uživatelé by si měli být vědomi toho, že mikroskopy se štěrbinovou lampou by mohly ukazovat různou tloušťku rohovky, je-li rozdílné nastavení šířky štěrbiny. Šířka štěrbiny by se měla nastavit na 0,095 mm. Poté, co byly oči prozkoumány a schváleny, inkubují se po dobu přibližně 45 až 60 minut, aby se uvedly do rovnováhy se zkušebním systémem před dávkováním. Po době vyrovnání se pro tloušťku rohovky a zákal zaznamená nulové referenční měření, které slouží jako základ (tj. čas = 0). Skóre fluoresceinu stanovené při pitvě je základní hodnotou pro měření tohoto koncového účinku. Aplikace zkoušené chemické látky Ihned po nulovém referenčním měření se oči (v držáku) vyjmou ze superfúzního aparátu, postaví se do vodorovné polohy a na rohovku se aplikuje zkoušená chemická látka. Tekuté zkoušené chemické látky se zpravidla zkoušejí neředěné, mohou se však ředit, pokud se to považuje za nutné (např. jako součást návrhu studie). Preferovaným roupouštědlem pro ředěné chemické látky je fyziologický roztok. Za řízených podmínek lze však také použít alternativní rozpuštědla, ale vhodnost jiných rozpouštědel než fyziologického roztoku by se měla prokázat. Tekuté zkoušené chemické látky se aplikují na rohovku tak, že celý povrch rohovky se rovnoměrně pokryje zkoušenou chemickou látkou; obvyklý objem je 0,03 ml. Podle možnosti by se pevné zkoušené chemické látky měly co nejjemněji rozetřít pomocí třecí misky a těrky nebo srovnatelným drticím prostředkem. Prášek se aplikuje na rohovku tak, že se celý povrch rohovky rovnoměrně pokryje zkoušenou chemickou látkou; obvyklé množství je 0,03 g. Zkoušená chemická látka (tekutá nebo pevná) se aplikuje po dobu 10 vteřin a pak se z oka opláchne izotonickým fyziologickým roztokem (přibližně 20 ml) při okolní teplotě. Oko (v držáku) se potom v původní svislé poloze vrátí do superfúzního aparátu. V případě potřeby lze použít další opláchnutí po desetivteřinové aplikaci a v následných časových intervalech (např. po zjištění reziduí zkoušené chemické látky na rohovce). Množství fyziologického roztoku dodatečně použitého pro opláchnutí obecně není kriticky důležité, ale pozorování přilnutí chemické látky k rohovce je důležité. Chemické látky pro kontrolu Do každého pokusu by měly být zařazeny souběžné negativní kontroly nebo kontroly s rozpouštědlem/vehikulem a pozitivní kontroly. Když se zkoušejí 100 % tekuté látky nebo pevné látky, fyziologický roztok se ve zkušební metodě ICE používá pro souběžnou negativní kontrolu, aby se zjistily nespecifické změny ve zkušebním systému a aby se zajistilo, že podmínky analýzy nebudou mít za následek nevhodnou dráždivou reakci. Když se zkoušejí zředěné tekuté látky, do zkušební metody se zařadí skupina pro souběžnou kontrolu s rozpouštědlem/vehikulem, aby se zjistily nespecifické změny ve zkušebním systému a aby se zajistilo, že podmínky analýzy nebudou mít za následek nevhodnou dráždivou reakci. Jak je uvedeno v bodě 31, lze použít pouze rozpouštědlo/vehikulum, u kterého se prokázalo, že nemá nepříznivé účinky na zkušební systém. Do každého pokusu se zařadí známá látka s dráždivými účinky na oči jako souběžná pozitivní kontrola, aby se ověřilo, že je vyvolána odpovídající reakce. Protože analýza ICE se v této zkušební metodě používá ke zjišťování leptavých nebo silných dráždivých látek, pozitivní kontrola by měla být takovou referenční chemickou látkou, která v této zkušební metodě vyvolá silnou reakci. Aby se však zajistilo, že proměnlivost v reakci na pozitivní kontrolu bude možné časem vyhodnotit, rozsah silné reakce by neměl být nadměrný. Měl by se nashromáždit dostatek údajů pro pozitivní kontrolu in vitro, aby bylo možné vypočítat statisticky definovaný, přijatelný rozsah pozitivní kontroly. Nejsou-li údaje o zkušební metodě ICE z minulosti pro určitou pozitivní kontrolu k dispozici, bude nutné uskutečnit studie za účelem získání těchto informací. Příklady tekutých zkoušených chemických látek pro pozitivní kontroly jsou 10 % kyselina octová nebo 5 % benzalkonium chlorid, zatímco příklady pevných zkoušených chemických látek pro pozitivní kontroly jsou hydroxid sodný nebo imidazol. Srovnávací chemické látky jsou účelné pro hodnocení potenciálu podráždění očí neznámými chemickými látkami nebo specifickými třídami chemických látek či produktů nebo pro hodnocení potenciálu relativního podráždění látkou s dráždivými účinky na oči ve specifickém rozmezí dráždivých reakcí. Měřené koncové účinky Zkoušené rohovky se hodnotí před expozicí a po 30, 75, 120, 180 a 240 minutách (± 5 minut) po oplachu po ošetření. Tyto časové body zajišťují odpovídající počet měření během čtyřhodinové doby zkoušení a přitom ponechávají dostatek času mezi měřeními, aby potřebná pozorování byla uskutečněna pro všechny oči. Hodnocené koncové účinky jsou zákal rohovky, zduření rohovky, zadržení fluoresceinu v rohovce a morfologické účinky (např. tvorba jamek nebo uvolnění epitelu). Všechny koncové účinky kromě zadržování fluoresceinu (který se určuje pouze před expozicí a 30 minut po expozici zkoušené látce) se určují v každém výše uvedeném časovém intervalu. Doporučují se fotografické snímky, aby se zdokumentoval zákal rohovky, zadržování fluoresceinu, morfologické účinky a histopatologie, pokud se provádí. Po poslední prohlídce po 4 hodinách se uživatelům doporučuje uložit oči do vhodného fixačního média (např. neutrálního pufrovaného formalínu) pro případné histopatologické zkoumání (podrobněji viz odstavec 14 a odkaz (8)). Otok rohovky se určuje měřením tloušťky rohovky, které se provádí optickým pachymetrem pod mikroskopem se štěrbinovou lampou. Vyjadřuje se jako procento a vypočítává se z měření tloušťky rohovky podle následujícího vzorce:
Střední procento otoku rohovky se u všech zkoušených očí vypočítává pro všechny časové body pozorování. Na základě nejvyšší střední hodnoty otoku rohovky pozorované v jakémkoli časovém bodu se potom každé zkoušené chemické látce přidělí celková hodnota kategorie (viz odstavec 51). Zákal rohovky se vyhodnotí s použitím plochy rohovky pro hodnocení, která je nejvíce zakalená, jak je uvedeno v tabulce 1. Střední hodnota zákalu rohovky se u všech zkoušených očí vypočítává pro všechny časové body pozorování. Na základě nejvyšší střední hodnoty zákalu rohovky pozorované v jakémkoli časovém bodě se potom každé zkoušené chemické látce přidělí celková hodnota kategorie (viz odstavec Tabulka 1. Hodnoty zákalu rohovky.
Zadržení fluoresceinu se hodnotí pouze při měření v časovém intervalu 30 minut, jak je uvedeno v tabulce 2. Střední hodnota zadržování fluoresceinu se poté u všech zkoušených očí vypočítává pro časový interval 30minutového pozorování a používá pro celkovou hodnotu kategorie přidělenou každé zkoušené chemické látce (viz odstavec 51). Tabulka 2. Hodnoty zadržování fluoresceinu.
Morfologické účinky zahrnují „jizvy“ na epiteliálních buňkách rohovky, „uvolňování“ epitelu, „zdrsnění“ povrchu rohovky a „nalepení“ zkoušené látky na rohovku. Tato zjištění se mohou ve své závažnosti měnit a mohou se vyskytovat souběžně. Klasifikace těchto zjištění je subjektivní podle interpretace výzkumníka. ÚDAJE A PŘEDKLÁDÁNÍ ZPRÁV Hodnocení údajů Výsledky ze zákalu a otoku rohovky a zadržování fluoresceinu by se měly hodnotit zvlášť, aby se pro každý koncový účinek vytvořila třída ICE. Třídy ICE pro každý koncový účinek se pak spojí za účelem vytvoření klasifikace dráždivých účinků pro každou zkoušenou chemickou látku. Kritéria rozhodování Poté, co byl každý koncový účinek vyhodnocen, lze na základě předem určeného rozmezí přiřadit třídy ICE. Interpretace otoku (tabulka 3) a zákalu rohovky (tabulka 4) a zadržování fluoresceinu (tabulka 5) s použitím čtyř tříd ICE se uskutečňuje podle níže uvedených měřítek. Je důležité poznamenat, že hodnoty otoku rohovky uvedené v tabulce 3 platí pouze v případě, že se tloušťka měří mikroskopem se štěrbinovou lampou (například Haag-Streit BP900) se zařízením pro měření hloubky č. 1 a při nastavení šířky štěrbiny 9 Tabulka 3. Klasifikační kritéria ICE pro otok rohovky.
Tabulka 4. Klasifikační kritéria ICE pro zákal.
Tabulka 5. Klasifikační kritéria ICE pro střední zadržování fluoresceinu.
Klasifikace in vitro zkoušené chemické látky se hodnotí pomocí klasifikace GHS, která odpovídá kombinaci kategorií získaných pro otok rohovky, zákal rohovky a zadržování fluoresceinu, jak je popsáno v tabulce 6. Tabulka 6. Celkové klasifikace in vitro.
Kritéria přijatelnosti studie Zkouška se považuje za přijatelnou, pokud vede ke zjištění, že souběžné negativní kontroly nebo kontroly s vehikulem/rozpouštědlem odpovídají chemickým látkám neklasifikovaným podle systému GHS a souběžné pozitivní kontroly odpovídají chemickým látkám klasifikovaným v kategorii 1 systému GHS. Závěrečná zpráva Závěrečná zpráva by měla zahrnovat následující informace, pokud jsou relevantní pro provedení studie:
LITERATURA
Dodatek 1 DEFINICE Přesnost : Přesnost shody mezi výsledky zkušební metody a přijatými referenčními hodnotami. Je to měření výsledku zkušební metody a jednoho aspektu „důležitosti“. Termín se často používá namísto „souladu“, kterým se rozumí podíl správných výsledků zkušební metody. Srovnávací chemická látka : Látka použitá jako norma pro srovnání se zkoušenou látkou. Srovnávací látka by měla mít tyto vlastnosti: i) nesporný a spolehlivý zdroj (nesporné a spolehlivé zdroje); ii) strukturální a funkční podobnost s třídou zkoušených látek; iii) známé fyzikální/chemické vlastnosti; iv) podpůrné údaje o známých účincích a v) známá síla v rozsahu žádoucí reakce. Přístup „zdola nahoru“ : Přístup zkoušení po krocích používaný u chemických látek, u nichž se předpokládá, že je není nutné klasifikovat jako látky mající dráždivé účinky na oči nebo vyvolávající vážné poškození očí, který začíná odlišením chemických látek, u nichž není nutná klasifikace (negativní výsledek), od ostatních chemických látek (pozitivní výsledek). Chemická látka : Látka nebo směs. Rohovka : Průhledná přední část oční bulvy, která zahrnuje duhovku a zornici a propouští světlo dovnitř oka. Zákal rohovky : Měření rozsahu zákalu rohovky po expozici zkoušené chemické látce. Zvýšený zákal rohovky je příznačný pro poškození rohovky. Otok rohovky : Objektivní měření rozsahu zduření rohovky ve zkoušce ICE po expozici zkoušené látce. Vyjadřuje se procentuálně a vypočítává se z měření základní tloušťky rohovky (před dávkováním) a tloušťky zaznamenávané v pravidelných intervalech po vystavení zkoušené chemické látce ve zkoušce ICE. Stupeň otoku rohovky je příznačný pro poškození rohovky. Dráždivé účinky na oči : Vyvolání změn na oku po aplikaci zkoušené chemické látky na přední plochu oka, které jsou plně vratné do 21 dní od aplikace. Tento pojem je zaměnitelný s výrazy „vratné účinky na oči“ a „kategorie 2 GHS OSN“ (4). Míra falešné negativity : Podíl všech pozitivních chemických látek falešně zjištěných zkušební metodou jako negativní. Je to jeden z ukazatelů výsledku zkušební metody. Míra falešné pozitivity : Podíl všech negativních látek falešně zjištěných zkušební metodou jako pozitivní. Je to jeden z ukazatelů výsledku zkušební metody. Zadržování fluoresceinu : Subjektivní měření rozsahu fluoresceinu sodného ve zkoušce ICE, který je zadržován buňkami epitelu v rohovce po expozici zkoušené látce. Stupeň zadržování fluoresceinu je příznačný pro poškození epitelu rohovky. Nebezpečnost : Základní charakteristika látky nebo situace, která má potenciál vyvolat nepříznivé účinky, jsou-li organismus, systém nebo (sub)populace vystaveny této látce. Nevratné účinky na oči : viz „vážné poškození očí“ a „kategorie 1 GMS OSN“. Směs : Směs nebo roztok tvořený dvěma nebo více látkami, které v něm nereagují (4). Negativní kontrola : Nezkoušená replika, která obsahuje všechny složky zkušebního systému. Tento vzorek se zpracovává se vzorky zkoušené chemické látky a jinými kontrolními vzorky, aby se zjistilo, zda rozpouštědlo vzájemně reaguje se zkušebním systémem. Neklasifikované : Látky, které nejsou klasifikovány jako látky mající dráždivé účinky na oči (kategorie 2 GHS OSN) nebo jako látky vyvolávající vážné poškození očí (kategorie 1 GHS OSN). Tento pojem je zaměnitelný s pojmem „bez kategorie GHS OSN“. Pozitivní kontrola : Replika obsahující všechny složky zkušebního systému a zkoušená s chemickou látkou, o níž je známo, že způsobuje pozitivní reakci. Aby se zajistilo, že proměnlivost v reakci na pozitivní kontrolu bude možné časem vyhodnotit, rozsah silné reakce by neměl být nadměrný. Spolehlivost : Měření rozsahu, v jakém může být zkušební metoda časem reprodukovatelná v laboratořích a mezi laboratořemi, když se provádí s použitím stejného protokolu. Hodnotí se to výpočtem laboratorní a mezilaboratorní reprodukovatelnosti a laboratorní opakovatelnosti. Vratné účinky na oči : viz „dráždivé účinky na oči“ a „kategorie 2 GMS OSN“. Vážné poškození očí : Tvorba poškození tkání v oku nebo závažné fyzikální slábnutí vidění po aplikaci zkoušené látky na přední plochu oka, které není plně vratné do 21 dní od aplikace. Tento pojem je zaměnitelný s výrazy „nevratné účinky na oči“ a „kategorie 1 GHS OSN“ (4). Mikroskop se štěrbinovou lampou : Přístroj používaný k přímému vyšetření oka se zvětšením pod binokulárním mikroskopem a vytvořením stereoskopického, přímého obrazu. Ve zkušební metodě ICE se tento přístroj používá k prohlížení předních struktur kuřecího oka a také k objektivnímu měření tloušťky rohovky s přídavným zařízením na měření hloubky. Kontrola s rozpouštědlem/vehikulem : Neošetřený vzorek obsahující všechny složky zkušebního systému, včetně rozpouštědla nebo vehikula, který se zpracovává se vzorky exponovaných zkoušených chemických látek a s jinými kontrolními vzorky, aby se zjistila základní reakce vzorků zkoušených se zkoušenou chemickou látkou rozpuštěnou ve stejném rozpouštědle nebo vehikulu. Pokud se vzorek zkouší souběžnou negativní kontrolou, ukazuje také, zda rozpouštědlo nebo vehikul vzájemně reaguje se zkušebním systémem. Látka : Chemické prvky nebo jejich sloučeniny v přírodním stavu nebo získané výrobním procesem včetně všech přídatných látek nutných k uchování stability výrobku a všech nečistot vznikajících v použitém procesu, ale s vyloučením všech rozpouštědel, která je možno oddělit bez ovlivnění stability látky nebo změny jejího složení (4). Smáčedlo : Nazývané též povrchově aktivní látka, je látka, jako např. detergent, která je schopna snížit povrchové napětí kapaliny a umožnit jí vytvářet pěnu nebo pronikat do pevných látek; je též známa jako sufraktant. Přístup „shora dolů“ : Přístup zkoušení po krocích používaný u chemických látek, u nichž se předpokládá, že způsobují vážné poškození očí, který začíná odlišením chemických látek vyvolávajících vážné poškození očí (pozitivní výsledek) od jiných chemických látek (negativní výsledek). Zkoušená chemická látka : Jakákoli látka nebo směs zkoušená pomocí této zkušební metody. Strategie postupného zkoušení : Strategie postupného zkoušení, kde se všechny existující informace o zkoušené chemické látce přezkoumávají ve stanoveném pořadí s použitím postupu váhy důkazů na každém stupni, aby se zjistilo, zda je k dispozici dost informací pro rozhodnutí o klasifikaci nebezpečnosti, než se postoupí do dalšího stupně. Pokud potenciál dráždivých účinků zkoušené chemické látky lze určit na základě existujících informací, další zkoušení není nutné. Jestliže potenciál dráždivých účinků zkoušené látky nelze určit na základě existujících informací, použije se stupňovitý postupný postup zkoušení na zvířatech po krocích, dokud se nestanoví jednoznačná klasifikace. Globálně harmonizovaný systém klasifikace a označování chemických látek Organizace spojených národů (GHC OSN) : Systém navrhující klasifikaci látek a směsí podle standardizovaných typů a úrovní fyzikálních, zdravotních a environmentálních nebezpečí a řešící příslušné komunikační prvky, jako jsou piktogramy, signální slova, výroky o nebezpečí, výroky o bezpečnostních opatřeních a bezpečnostní datové listy, které obsahují informace o jejich nežádoucích účincích s ohledem na ochranu lidí (včetně zaměstnavatelů, dělníků, přepravců, spotřebitelů a osob reagujících na havárie) a životního prostředí (4). Kategorie 1 GHS OSN : Viz „vážné poškození očí“ a/nebo „nevratné účinky na oči“. Kategorie 2 GHS OSN : Viz „dráždivé účinky na oči“ a/nebo „vratné účinky na oči“. Bez kategorie GHS OSN : Látky, které nesplňují požadavky na klasifikaci v kategorii 1 nebo 2 (2A nebo 2B) GHS OSN. Tento pojem je zaměnitelný s výrazem „neklasifikované“. Validovaná zkušební metoda : Zkušební metoda, u níž byly provedeny validační studie k určení její relevantnosti (včetně přesnosti) a spolehlivosti pro konkrétní účel. Je nutné poznamenat, že validovaná zkušební metoda nemusí poskytnout postačující výsledek z hlediska přesnosti a spolehlivosti, aby byla shledána přijatelnou pro navržený účel. Závažnost důkazů : Postup zvažování silných a slabých stránek různých informací v dosahování a podpoře závěru týkajícího se potenciálu nebezpečnosti látky. Dodatek 2 CHEMICKÉ LÁTKY PRO PROKÁZÁNÍ ZPŮSOBILOSTI PRO ZKUŠEBNÍ METODU ICE Dříve než začnou laboratoře rutinně používat tuto zkušební metodu, která dodržuje tuto zkušební metodu, měly by prokázat svou odbornou způsobilost správným určením klasifikace nebezpečnosti pro oči u 13 chemických látek doporučených v tabulce 1. Tyto chemické látky byly vybrány tak, aby představovaly rozsah reakcí na nebezpečnost pro oči na základě výsledků oční zkoušky in vivo na králících (TG 405) a klasifikačního systému GHS OSN (tj. kategorie GHS OSN 1, 2A, 2B, nebo neklasifikované) (4) (6). Dalšími kritérii výběru bylo to, zda je látka komerčně dostupná, zda jsou k dispozici kvalitní referenční údaje in vivo a zda jsou kvalitní údaje získané ze zkušební metody ICE in vitro. Referenční údaje jsou dostupné v publikacích SSD (Summary Document Supporting OECD Test Guideline 438) (5) a v Background Review Documents (Základní dokumenty o přezkumu) ICCVAM pro zkušební metodu ICE (9). Tabulka 1 Doporučené chemické látky k prokázání odborné způsobilosti pro ICE
Dodatek 3 SCHÉMATICKÝ NÁKRES SUPERFÚZNÍHO APARÁTU A DRŽÁKŮ OČÍ ICE (Další generické popisy superfúzního aparátu a držáků očí viz Burton a kol. (18)) 
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||

 , která se rovná 0,095 mm. Uživatelé by si měli být vědomi, že mikroskopy se štěrbinovou lampou mohou poskytovat různé naměřené hodnoty tloušťky rohovky, je-li nastavena odlišná šířka štěrbiny.
, která se rovná 0,095 mm. Uživatelé by si měli být vědomi, že mikroskopy se štěrbinovou lampou mohou poskytovat různé naměřené hodnoty tloušťky rohovky, je-li nastavena odlišná šířka štěrbiny.|
14) |
V části B se kapitola B.49 nahrazuje tímto: „B.49 Mikronukleus test v savčích buňkách in vitro ÚVOD Tato zkušební metoda je rovnocenná Pokynu OECD pro zkoušení (TG) č. 487 (2016). Je součástí série zkušebních metod v oblasti genetické toxikologie. Byl vypracován dokument OECD, který obsahuje stručné informace o zkoušení v oblasti genetické toxikologie a přehled posledních změn, které byly v těchto pokynech ke zkoušení provedeny (1). Mikronukleus test v savčích buňkách in vitro (MNvit) je zkouška genotoxicity pro zjišťování přítomnosti mikrojader v cytoplazmě interfázních buněk. Mikrojádra mohou pocházet z acentrických fragmentů chromozomů (tzn. fragmentů, jimž chybí centromera) nebo z celých chromozomů, které nejsou při buněčném dělení schopny se ve stádiu anafáze rozejít k pólům. Zkouška MNvit je tedy metoda in vitro, která poskytuje komplexní základ pro zkoumání in vitro schopnosti látek poškozovat chromozomy, protože je možné zjistit aneugeny a rovněž klastogeny v buňkách, které prodělaly buněčné dělení během expozice nebo po expozici zkoušené látce, (2) (3) (podrobněji viz odstavec 13). Mikrojádra představují poškození přenesené na dceřiné buňky, zatímco chromozomové aberace zjištěné v metafázních buňkách se přenášet nemusejí. V obou případech tyto změny nemusí být slučitelné s přežitím buněk. Tato zkušební metoda umožňuje používat protokoly s inhibitorem polymerace aktinu, cytochalasinem B (cytoB), i bez něho. Přidání cytoB před mitózou vede ke vzniku buněk, které jsou dvoujaderné, a proto umožňuje identifikaci a analýzu mikrojader pouze v buňkách, které prodělaly jednu mitózu (4) (5). Tato zkušební metoda rovněž umožňuje použití protokolů bez blokování cytokineze, pokud existují důkazy, že analyzovaná populace buněk prodělala mitózu. Kromě použití zkoušky MNvit pro identifikaci chemických látek, které vyvolávají vznik mikrojader, lze další informace o mechanismech poškození chromozomů a tvorbě mikrojader získat rovněž použitím imunochemického značení kinetochorů nebo hybridizace za použití centromerických/telomerických sond (fluorescenční hybridizace in situ – FISH) (6) (7) (8) (9) (10) (11) (12) (13) (14) (15) (16) (17). Postupy značení a hybridizace lze použít, pokud dojde ke zvýšené tvorbě mikrojader a zkoušející chce určit, zda je toto zvýšení výsledkem klastogenního a/nebo aneugenního působení. Jelikož mikrojádra v interfázních buňkách lze posuzovat poměrně objektivně, laboratorní personál musí pouze zjistit počet dvoujaderných buněk v případě, je-li použit cytoB, a výskyt buněk s mikrojádry v každém případě. Díky tomu lze preparáty vyhodnotit poměrně rychle a analýzu lze automatizovat. Takto lze při každé aplikaci vyhodnotit tisíce namísto stovek buněk, čímž se zvyšuje výkonnost této metody. A konečně, jelikož mikrojádra mohou vznikat z opožděných chromozomů, je možno zjišťovat látky, které vyvolávají aneuploidii a jež lze obtížně studovat za použití běžných zkoušek na chromozomové aberace, např. podle kapitoly B.10 této přílohy (18). Zkouška MNvit popsaná v této zkušební metodě však neumožňuje odlišit chemické látky vyvolávající změny počtu chromozomů a/nebo polyploidii od látek vyvolávajících klastogenicitu bez použití speciálních postupů, jako je například metoda FISH uvedená v odstavci 4. Zkouška MNvit je spolehlivá a lze ji provádět u široké škály různých typů buněk, za přítomnosti nebo nepřítomnosti cytoB. Existuje množství údajů, jež potvrzují platnost zkoušky MNvit za použití různých druhů buněk (kultur buněčných linií nebo primárních buněčných kultur) (19) (20) (21) (22) (23) (24) (25) (26) (27) (28) (29) (30) (31) (32) (33) (34) (35) (36). Patří mezi ně zejména mezinárodní validační studie koordinované společností Société Française de Toxicologie Génétique (SFTG) (19) (20) (21) (22) (23) a zprávy z Mezinárodního semináře o zkoušení genotoxicity (5) (17). Dostupné údaje byly rovněž přehodnoceny v retrospektivní validační studii v rámci přístupu založeného na hodnocení závažnosti důkazů, provedené Evropským střediskem pro validaci alternativních metod (ECVAM) Evropské komise (EK), a zkušební metoda byla potvrzena jako vědecky platná vědeckým poradním výborem ECVAM (ESAC) (37) (38) (39). Ke zkoušce MNvit na buňkách savců mohou být použity kultury buněčných linií nebo primární buněčné kultury lidského původu nebo pocházející z hlodavců. Jelikož četnost mikrojader v rámci přirozeného pozadí ovlivní citlivost testu, doporučuje se používat druhy buněk se stabilní a definovanou četností tvorby mikrojader v rámci přirozeného pozadí. Použité buňky se vybírají na základě jejich schopnosti dobrého růstu v kultuře, stálosti karyotypu (včetně počtu chromozomů) a spontánní četnosti výskytu mikrojader (40). Údaje, které jsou v současné době k dispozici, neumožňují předkládat jednoznačná doporučení, avšak naznačují, že při hodnocení chemické nebezpečnosti je důležité zvážit stav p53, genetickou stálost (stálost karyotypu), reparační schopnost DNA a původ buněk (lidské versus pocházející z hlodavců) vybraných ke zkoušení. Uživatelům této zkušební metody se proto doporučuje, aby při detekování indukce mikrojader zvážili vliv těchto i jiných buněčných charakteristik na vlastnosti buněčné linie. Použité definice jsou uvedeny v dodatku 1. VÝCHOZÍ ÚVAHY A OMEZENÍ Zkoušky prováděné in vitro zpravidla vyžadují použití exogenního zdroje metabolické aktivace, pokud nejsou buňky metabolicky kompetentní s ohledem na zkoušené chemické látky. Systém exogenní metabolické aktivace zcela nenapodobuje podmínky in vivo. Je nutno zabránit vzniku podmínek, které by mohly vést k uměle pozitivním výsledkům, jež neodrážejí genotoxicitu zkoušených chemických látek. Takové podmínky zahrnují změny pH (41) (42) (43) nebo osmolality, interakce s kultivačním médiem (44) (45) nebo nadměrné úrovně cytotoxicity (viz odstavec 29). Pro analýzu indukce vzniku mikrojader je nezbytné, aby proběhla mitóza v exponovaných i neexponovaných kulturách. Nejinformativnějším stadiem pro hodnocení výskytu mikrojader jsou buňky, které prodělaly jednu mitózu během expozice nebo po expozici zkoušené chemické látce. Při zkoušení vyráběných nanomateriálů je nutné v této zkušební metodě provést určité úpravy, avšak ty v této zkušební metodě nejsou popsány. Před použitím této zkušební metody na směs za účelem získání údajů pro zamýšlené použití v právních předpisech by se mělo zvážit, zda tato metoda může pro tento účel poskytnout přiměřené výsledky, a pokud ano, proč je tomu tak. Takové úvahy nejsou nutné, pokud existuje právní požadavek na zkoušení dané směsi. PODSTATA ZKOUŠKY Buněčné kultury lidského a savčího původu jsou vystaveny zkoušené chemické látce za přítomnosti exogenního zdroje metabolické aktivace i bez něho, pokud ovšem nejsou použity buňky s dostatečnou schopností metabolismu (viz odstavec 19). Během expozice nebo po expozici zkoušené chemické látce se buňky kultivují dostatečně dlouho, aby poškození chromozomů nebo jiné účinky na buněčný cyklus / dělení buněk vedlo v interfázových buňkách ke vzniku mikrojader. Pro vyvolání aneuploidie by měla být zkoušená chemická látka obvykle přítomna při mitóze. Získané a obarvené interfázní buňky se analyzují na přítomnost mikrojader. V ideálním případě by měla být přítomnost mikrojader zjištěna pouze v těch buňkách, které prodělaly mitózu během expozice zkoušené chemické látce nebo během postexpoziční doby, pokud se používá. V kulturách, které byly ošetřeny blokátorem cytokineze, se toho snadno dosáhne tím, že se zaznamenají pouze dvoujaderné buňky. V případě nepřítomnosti blokátoru cytokineze je nutno prokázat, že analyzované buňky pravděpodobně, na základě nárůstu buněčné expozice, prodělaly buněčné dělení během expozice nebo po expozici zkoušené látce. U všech protokolů je důležité prokázat, že došlo k proliferaci buněk jak v kontrolních, tak v exponovaných kulturách, a také je třeba ve všech kulturách, u nichž se hodnotí výskyt mikrojader, posoudit míru cytotoxicity nebo cytostáze vyvolané zkoušenou chemickou látkou. POPIS METODY Buňky Lze použít kultivované primární lidské nebo savčí lymfocyty z periferní krve (7) (20) (46) (47) a celou řadu buněčných linií pocházejících z hlodavců, např. buňky CHO, V79, CHL/IU a L5178Y, nebo lidské buněčné linie, např. TK6 (19) (20) (21) (22) (23) (26) (27) (28) (29) (31) (33) (34) (35) (36) (viz odstavec 6). Pro testování mikrojader byly použity i jiné buněčné linie, např. HT29 (48), Caco-2 (49), HepaRG (50) (51), HepG2 (52) (53), A549 a primární buňky z embryí syrského křečka (54), avšak dosud nebyly dostatečně validovány. Použití těchto buněčných linií a druhů by proto mělo být odůvodněno na základě jejich prokázané funkčnosti ve zkoušce, jak je popsáno v oddíle „Kritéria přijatelnosti“. Bylo zaznamenáno, že cytoB může potenciálně ovlivnit růst buněk L5178Y, a proto se nedoporučuje jej s touto buněčnou linií používat (23). Použijí-li se primární buňky, mělo by se z důvodu dobrého zacházení se zvířaty tam, kde je to možné, zvážit použití buněk lidského původu a odběr vzorků se provede v souladu s etickými zásadami a předpisy platnými pro člověka. Lidské lymfocyty z periferní krve by měly být získány od mladých (přibližně ve věku 18–35 let) nekuřáků, o nichž není známo, že by trpěli nemocemi nebo že by byli nedávno vystaveni genotoxickým látkám (např. chemickým látkám, ionizujícímu záření) na úrovních, které by mohly zvýšit výskyt buněk s mikrojádry na pozadí. Tím by se zajistilo, že výskyt buněk s mikrojádry na pozadí bude nízký a stálý. Výskyt buněk s mikrojádry na pozadí se s věkem zvyšuje a tento trend je více patrný u žen než u mužů (55). Pokud se společně používají buňky pocházející od více než jednoho dárce, měl by být počet dárců uveden. Je nezbytné prokázat, že buňky se dělily od počátku ovlivnění zkoušenou chemickou látkou až do odběru buněčných vzorků. Buněčné kultury jsou udržovány ve fázi exponenciálního růstu (buněčné linie) nebo jsou stimulovány k dělení (primární kultury lymfocytů), aby bylo možné exponovat buňky v různých stadiích buněčného cyklu, protože citlivost stadií buněk na zkoušené chemické látky nemusí být známa. Primární buňky, které k dělení potřebují stimulaci mitogenními látkami, během expozice zkoušené chemické látce již zpravidla nejsou synchronizovány (např. lidské lymfocyty po 48hodinové mitogenní stimulaci). Použití synchronizovaných buněk v průběhu expozice zkoušení chemické látce se nedoporučuje, avšak lze je akceptovat v odůvodněných případech. Média a podmínky kultivace Pro udržování kultur by se měla používat vhodná kultivační média a inkubační podmínky (kultivační nádoby, případně zvlhčená atmosféra s 5 % CO2, teplota 37 °C). U buněčných linií by se měla pravidelně kontrolovat stabilita modální hodnoty počtu chromozomů a mělo by se zjišťovat, zda nejsou kontaminovány mykoplasmaty. Pokud jsou buňky kontaminované nebo se modální hodnota počtu chromozomů změnila, neměly by se takové kultury používat. Pro buněčné linie nebo primární kultury použité ve zkušební laboratoři by měla být stanovena normální délka buněčného cyklu, která by měla být v souladu s buněčnými charakteristikami uvedenými v literatuře. Příprava kultur Buněčné linie: buňky se pomnoží z kmenových kultur, nasadí se do kultivačního média v takové hustotě, aby u buněk v suspenzích nebo v monovrstvách pokračoval exponenciální růst až do sběru buněk (např. mělo by se zamezit konfluenci u buněk rostoucích v monovrstvách). Lymfocyty: plná krev ošetřená antikoagulantem (např. heparinem) nebo oddělené lymfocyty se před expozicí zkoušené chemické látce a cytoB kultivují (např. 48 hodin v případě lidských lymfocytů) za přítomnosti mitogenu (např. fytohemaglutininu (PHA) v případě lidských lymfocytů), aby se vyvolalo buněčné dělení. Metabolická aktivace Při použití buněk s nedostatečnou endogenní metabolickou schopností by se měly použít exogenní metabolizující systémy. Nejčastěji používaným systémem, který je standardně doporučován, pokud není zdůvodněno použití jiného systému, je kofaktorem dotovaná postmitochondriální frakce (S9) připravená z jater hlodavců (zpravidla potkanů) zpracovaná činidlem indukujícím enzymy, jako je Aroclor 1254 (56) (57), nebo směsí fenobarbitalu a b-naftoflavonu (58) (59) (60). Použití uvedené směsi není v rozporu se Stockholmskou úmluvou o perzistentních organických znečišťujících látkách (61) a bylo prokázáno, že je stejně účinná jako Aroclor 1254 pro indukování oxidáz se smíšenou funkcí (58) (59) (60). Frakce S9 je v konečném testovacím médiu obvykle používána v koncentracích od 1 % do 2 % obj., avšak koncentraci je možné zvýšit do 10 %. V průběhu aplikace by se neměly používat produkty, jež snižují mitotický index, zejména komplexotvorná činidla pro vápník (62). Volbu typu a koncentrace systému exogenní metabolické aktivace nebo metabolického induktoru, který se použije, může ovlivnit třída zkoušených chemických látek. Příprava zkoušené chemické látky Pevné zkoušené chemické látky by se před aplikací na buňky měly připravit ve vhodných rozpouštědlech a popřípadě zředit. Kapalné látky lze přidat přímo do zkušebních systémů a/nebo je lze před použitím k ošetření zkušebních systémů zředit. Plynné a těkavé zkoušené chemické látky by se měly zkoušet za použití vhodně upravených standardních protokolů, například prostřednictvím ošetření buněk v neprodyšně uzavřených nádobách (63) (64) (65). Měly by se používat čerstvě připravené zkoušené látky, pokud údaje o stálosti neprokazují možnost skladování. Zkušební podmínky Rozpouštědla Rozpouštědlo by mělo být zvoleno tak, aby optimalizovalo rozpustnost zkoušené chemické látky, aniž by nepříznivě ovlivnilo provádění zkoušky, např. měnilo růst buněk, ovlivňovalo integritu zkoušené chemické látky, reagovalo s kultivačními nádobami, narušovalo systém metabolické aktivace. Doporučuje se pokud možno nejprve zvážit použití vodného rozpouštědla (nebo kultivačního média). Dobře zavedenými rozpouštědly jsou voda nebo dimethylsulfoxid (DMSO). Podíl organických rozpouštědel by obvykle neměl být větší než 1 % obj. Je-li cytoB rozpuštěn v DMSO, celkové množství organického rozpouštědla použitého na zkoušenou chemickou látku a na cytoB by nemělo přesahovat 1 % obj., v opačném případě by se měly použít neexponované kontroly, aby se zajistilo, že podíl organického rozpouštědla nebude mít žádný nepříznivý účinek. Podíl vodných rozpouštědel (fyziologického roztoku nebo vody) v konečném expozičním médiu by neměl být větší než 10 % obj. Jsou-li použita jiná než osvědčená rozpouštědla (např. ethanol nebo aceton), mělo by být jejich použití podloženo údaji o jejich kompatibilitě se zkoušenou chemickou látkou a zkušebním systémem a o tom, že nejsou v použité koncentraci genotoxické. Pokud takové podpůrné údaje neexistují, je důležité zařadit neexponované kontroly (viz dodatek 1) a také kontroly s rozpouštědlem, aby se prokázalo, že zvolené rozpouštědlo nevyvolává žádné škodlivé účinky ani účinky na chromozomy (např. aneuploidii nebo klastogenicitu). Používání cytoB jako blokátoru cytokineze Jedním z nejdůležitějších hledisek při provádění zkoušky MNvit je zajistit, aby hodnocené buňky prodělaly mitózu během expozice zkoušené látce nebo během postexpoziční inkubační doby, pokud se používá. Proto by se hodnocení výskytu mikrojader mělo omezit pouze na buňky, které prodělaly mitózu během ošetření zkoušenou látkou nebo po něm. CytoB je látka, která se nejčastěji používá k blokování cytokineze, protože inhibuje sestavení aktinu, a tím zabraňuje oddělení dceřiných buněk po mitóze, což vede ke vzniku dvoujaderných buněk (6) (66) (67). Při použití cytoB lze zároveň měřit účinek zkoušené chemické látky na kinetiku buněčné prolifereace. CytoB by se měl používat jako blokátor cytokineze při použití lidských lymfocytů, protože délky trvání buněčného cyklu budou u různých dárců proměnlivé a protože ne všechny lymfocyty budou reagovat na stimulaci PHA. Pro jiné druhy buněk není použití cytoB povinné, pokud je možné stanovit, že prodělaly dělení, jak je popsáno v odstavci 27. CytoB se mimoto obvykle nepoužívá v případech, kdy se výskyt mikrojader ve vzorcích hodnotí pomocí metod průtokové cytometrie. V laboratoři by se měla stanovit vhodná koncentrace cytoB pro každý druh buněk, aby se dosáhlo optimální četnosti dvoujaderných buněk v kontrolních kulturách s rozpouštědlem, a mělo by se prokázat, že tato koncentrace poskytuje dostatečné množství dvoujaderných buněk pro hodnocení. Vhodná koncentrace cytoB obvykle činí 3 až 6 μg/ml (19). Měření buněčné proliferace a cytotoxicity a volba expozičních koncentrací Při určování nejvyšší koncentrace zkušební chemické látky je nutno se vyhnout koncentracím, které mají schopnost produkovat arteficielní pozitivní odezvy, například koncentracím, jež vyvolávají velmi vysokou cytotoxicitu (viz odstavec 29), vysrážení v kultivačním médiu (viz odstavec 30) a výrazné změny pH nebo osmolality (viz odstavec 9). Pokud zkoušená chemická látka v době jejího přidání způsobuje zřetelnou změnu hodnoty pH média, bylo by možné pH upravit pufrováním konečného expozičního média tak, aby nedošlo k falešným pozitivním výsledkům a aby byly zachovány vhodné kultivační podmínky. Měření buněčné proliferace se provádí proto, aby bylo zajištěno, že dostatek ošetřených buněk prodělal během zkoušky mitózu a že se jednotlivé expozice provádějí při vhodných úrovních cytotoxicity (viz odstavec 29). Cytotoxicita by měla být stanovena v hlavním experimentu s metabolickou aktivací a bez metabolické aktivace za použití vhodné indikace zániku a růstu buněk (viz odstavce 26 a 27). Hodnocení cytotoxicity při počáteční předběžné zkoušce může být užitečné a může napomoci lepšímu stanovení koncentrací, jež mají být použity při hlavní zkoušce, avšak předběžná zkouška není povinná. Pokud se provede, neměla by nahrazovat měření cytotoxicity v hlavním experimentu. Ošetření kultur cytoB a měření relativních četností jednojaderných, dvoujaderných a vícejaderných buněk v kultuře představuje přesnou metodu kvantifikace účinku na buněčnou proliferaci a cytotoxického nebo cytostatického působení látky použité k ovlivnění kultury (6) a zajišťuje, že se mikroskopicky hodnotí pouze buňky, které prodělaly buněčné dělení během expozice nebo po ní. K odhadu cytotoxického a cytostatického působení za expozice porovnáním hodnot u exponovaných a kontrolních kultur se doporučuje použít proliferační index při blokování cytokineze (CBPI) (6) (27) (68) nebo replikační index (RI) získaný z nejméně 500 buněk v každé kultuře (příslušné vzorce viz dodatek 2). Užitečné informace může poskytnout i hodnocení ostatních ukazatelů cytotoxicity (např. integrity buněk, apoptózy, nekrózy, počítání metafází, buněčný cyklus), avšak tyto ukazatele by neměly být používány namísto CBPI nebo RI. Ve studiích bez použití cytoB je nutno prokázat, že buňky v kultuře se rozdělily, takže podstatná část z hodnocených buněk prodělala buněčné dělení během expozice zkoušené chemické látce nebo po ní, jinak mohou vznikat falešně negativní odpovědi. K odhadu cytotoxického a cytostatického působení expozice se doporučuje použít měření relativního zdvojnásobení populace (RPD) nebo relativního nárůstu počtu buněk (RICC) (17) (68) (69) (70) (71) (příslušné vzorce viz dodatek 2). Při prodloužení časů odběru vzorků (např. expozice po dobu odpovídající 1,5–2násobku normální délky buněčného cyklu a odebrání po další době odpovídající 1,5–2násobku normální délky buněčného cyklu, což celkově vede k časům odběru delším než doba odpovídající 3–4násobku normální délky buněčného cyklu, jak je popsáno v oddílech 38 a 39) by při použití měření RPD mohlo vést k podhodnocenému odhadu cytotoxicity (71). Za těchto okolností by mohlo být lepším měřítkem RICC nebo by bylo užitečné provést odhad na základě hodnocení cytotoxicity po době odpovídající 1,5–2násobku normální délky buněčného cyklu. Užitečné doplňující informace může poskytnout i hodnocení ostatních ukazatelů cytotoxicity nebo cytostáze (např. integrity buněk, apoptózy, nekrózy, počítání metafází, proliferačního indexu (PI), buněčného cyklu, nukleoplasmatických můstků nebo jaderných pupenů), avšak tyto ukazatele by neměly být používány namísto RPD nebo RICC. Měly by se vyhodnotit nejméně tři zkušební koncentrace (nepočítaje v to kontroly s rozpouštědlem a pozitivní kontroly), které splňují kritéria přijatelnosti (vhodná cytotoxicita, počet buněk atd.). Bez ohledu na typy buněk (buněčné linie nebo primární kultury lymfocytů) lze pro každou zkušební koncentraci použít repliky nebo jednu exponovanou kulturu. Lze doporučit použití duplicitních kultur, avšak jednu kulturu lze rovněž akceptovat za předpokladu, že v této jediné kultuře nebo v duplicitních kulturách je hodnocen stejný celkový počet buněk. Použití jedné kultury je opodstatněné zejména v případě, že jsou posuzovány více než 3 koncentrace (viz odstavce 44–45). Výsledky získané z nezávislých kultur použitých k opakování při dané koncentraci lze pro účely analýzy údajů sloučit. U zkoušených látek, jež vykazují malou cytotoxicitu nebo nevykazují žádnou cytotoxicitu, budou obvykle vhodné přibližně dvojnásobné až trojnásobné intervaly koncentrací. V případě cytotoxicity by měly zvolené zkušební koncentrace pokrývat rozpětí od koncentrace vyvolávající cytotoxicitu, jak je popsáno v odstavci 29, po koncentrace, při nichž dochází ke střední a nízké cytotoxicitě nebo nedochází k žádné cytotoxicitě. Mnohé zkoušené chemické látky vykazují strmé křivky závislosti odezvy na koncentraci, a aby bylo možné získat údaje při nízké a střední cytotoxicitě nebo podrobně studovat závislost odezvy na dávce, může být nezbytné zvolit menší rozdíly mezi jednotlivými koncentracemi a/nebo více než tři koncentrace (pro jednu kulturu nebo repliky), zejména v situacích, kdy jsou nutné opakované experimenty (viz odstavec 60). Je-li maximální koncentrace odvozena od cytotoxicity, měla by být nejvyšší koncentrace zvolena tak, aby se dosáhlo 55 ± 5 % cytotoxicity za použití doporučených parametrů cytotoxicity (tj. snížení RICC a RPD u buněčných linií, není-li použit cytoB, a snížení CBPI nebo RI, je-li použit cytoB, na 45 ± 5 % u souběžné negativní kontroly) (72). Je třeba dbát na to, aby byly interpretovány pouze ty pozitivní výsledky, které jsou vykazovány při vyšší hranici tohoto rozmezí cytotoxicity 55 ± 5 % (71). V případě špatně rozpustných zkoušených chemických látek, které nejsou cytotoxické při koncentracích nižších, než je jejich rozpustnost, by nejvyšší analyzovaná koncentrace měla na konci expozice zkoušené chemické látce vyvolat zákal nebo sraženinu viditelnou pouhým okem nebo pomocí inverzního mikroskopu. Dokonce i v případech, kdy k výskytu cytotoxicity dochází při koncentracích vyšších, než je rozpustnost, se doporučuje testovat pouze při jedné koncentraci indukující zákal nebo viditelné sraženiny, neboť sraženina může vést k falešným účinkům. Při koncentraci, kdy vzniká sraženina, je třeba dbát na to, aby sraženina nemohla ovlivňovat provádění zkoušky (např. barvení nebo hodnocení). Užitečné může být stanovit rozpustnost v kultivačním médiu před pokusem. Není-li pozorována sraženina nebo cytotoxicita omezující hodnocení, měla by největší zkušební koncentrace odpovídat 10 mM, 2 mg/ml nebo 2 μl/ml, podle toho, která z uvedených hodnot je nejnižší (73) (74) (75). Pokud u zkoušené chemické látky není stanoveno její složení, např. u látek s neznámým nebo proměnlivým složením, komplexních reakčních produktů nebo biologického materiálu (UVCB) (76), přírodního extraktu atd., bude možná muset být při nedostatečné cytotoxicitě nejvyšší koncentrace vyšší (např. 5 mg/ml), aby se zvýšila koncentrace každé ze složek. Je však třeba poznamenat, že u humánních léčivých přípravků se tyto požadavky mohou lišit (93). Kontroly V okamžiku každého odběru by měly být použity souběžné negativní kontroly (viz odstavec 21) skládající se ze samotného rozpouštědla v kultivačním médiu a zpracované stejným způsobem jako exponované kultury. K prokázání schopnosti laboratoře stanovit klastogeny a aneugeny za podmínek použitého zkušebního protokolu a účinnosti exogenního systému metabolické aktivace mohou být (případně) nutné souběžné pozitivní kontroly. Příklady pozitivních kontrol jsou uvedeny v tabulce 1 níže. V odůvodněných případech mohou být k pozitivní kontrole použity jiné chemické látky. V současné době nejsou známy žádné aneugenní látky, které by ke svému genotoxickému působení vyžadovaly metabolickou aktivaci (17). Jelikož zkoušky genetické toxicity savčích buněk in vitro při krátkodobé expozici prováděné souběžně s metabolickou aktivací a bez metabolické aktivace s použitím stejné doby trvání expozice jsou již dostatečně standardizovány, je možné použití pozitivních kontrol omezit na testování klastogenu vyžadujícího metabolickou aktivaci. V tomto případě klastogenní reakce jediné pozitivní kontroly prokáže činnost systému metabolické aktivace i citlivost zkušebního systému. Dlouhodobá expozice (bez S9) by však měla mít svou vlastní pozitivní kontrolu a doba trvání expozice bude odlišná než při zkoušce za použití metabolické aktivace. Je-li pro jedinou pozitivní kontrolu při krátkodobé expozici s metabolickou aktivací a bez metabolické aktivace zvolen klastogen, měl by být při dlouhodobé expozici bez metabolické aktivace zvolen aneugen. Pro pozitivní kontroly pro klastogenicitu i pro aneugenicitu by měly být použity metabolicky kompetentní buňky, které nevyžadují S9. Každá pozitivní kontrola by měla být použita při jedné nebo více koncentracích, u nichž se očekává, že poskytnou reprodukovatelný a detekovatelný nárůst oproti pozadí, čímž se prokáže citlivost testovacího systému (tj. aby byl účinek zřetelný, ale aby při odečtu nevyšla ihned najevo identita kódovaného preparátu), a reakce by neměla být narušena cytotoxicitou přesahující limitní hodnoty stanovené ve zkušební metodě. Tabulka 1. Referenční chemické látky doporučené pro posouzení způsobilosti laboratoře a pro výběr pozitivních kontrol
POSTUP Harmonogram aplikace Aby se maximalizovala pravděpodobnost detekce aneugenní nebo klastogenní látky působící v určitém stadiu buněčného cyklu, je důležité, aby byl zkoušené chemické látce vystaven dostatečný počet buněk ve všech fázích jejich buněčného cyklu. Všechny expozice by měly začít a skončit v době exponenciálního růstu buněk a v době odběru vzorků by měl růst buněk pokračovat. Harmonogram aplikace pro buněčné linie a primární buněčné kultury se proto může poněkud lišit od harmonogramu aplikace u lymfocytů, které k zahájení svého buněčného cyklu potřebují mitogenní stimulaci (17). U lymfocytů je nejúčinnějším postupem začít s aplikací zkoušené chemické látky 44–48 hodin po stimulaci PHA, kdy se buňky budou dělit asynchronicky (6). Publikované údaje (19) ukazují, že většina aneugenních a klastogenních látek bude detekována za použití krátkodobé expozice v trvání 3 až 6 hodin za přítomnosti i nepřítomnosti S9, po níž následuje odstranění zkoušené látky a odběr v době, která odpovídá 1,5–2násobku normální délky buněčného cyklu od zahájení expozice (7). Pro důkladné hodnocení, které by bylo zapotřebí pro vyvození závěru o negativním výsledku, by měly být splněny všechny tři následující zkušební podmínky za použití krátkodobé expozice s metabolickou aktivací a bez ní a dlouhodobé expozice bez metabolické aktivace (viz odstavce 56, 57 a 58):
V případě, že kterákoli z výše uvedených zkušebních podmínek vede k pozitivní odpovědi, je možné, že nebude nutné zkoumat kterýkoli z ostatních režimů aplikace. Pokud je známo nebo existuje podezření, že zkoušená chemická látka ovlivňuje délku buněčného cyklu (např. při zkoušení analogů nukleosidů), a to zejména u p53 kompetentních buněk (35) (36) (77), lze intervaly odběru vzorků nebo obnovy kultur prodloužit o další dobu odpovídající až 1,5–2násobku normální délky buněčného cyklu (tj. celkem na 3–4násobek délky buněčného cyklu po zahájení krátkodobé a dlouhodobé expozice). Tyto možnosti jsou určeny pro situace, které se týkají možných interakcí mezi zkoušenou chemickou látkou a cytoB. Při použití prodloužených intervalů odběru vzorků (tj. celkově na dobu odpovídající 3–4násobku délky buněčného cyklu kultur) je třeba dbát na to, aby se buňky v této době stále aktivně dělily. Například u lymfocytů může exponenciální růst v čase 96 hodin od stimulace klesat a buněčné kultury v monovrstvách mohou splývat. Navrhované harmonogramy expozice buněk jsou uvedeny v tabulce 2. Tyto obecné harmonogramy expozice lze upravit (což by mělo být odůvodněno) v závislosti na stabilitě a reaktivitě zkoušené chemické látky nebo zvláštních růstových charakteristikách použitých buněk. Tabulka 2. Doby expozic a odběru buněk v rámci zkoušky MNvit
Na konci doby expozice v trvání 3–6 hodin se v kulturách v monovrstvách mohou vyskytovat mitotické buňky (které se vyznačují okrouhlým tvarem a odchlipováním od povrchu). Jelikož lze tyto mitotické buňky snadno oddělit, mohou být při odstranění média obsahujícího zkoušenou chemickou látku ztraceny. Pokud v době sklizně existují důkazy podstatného nárůstu počtu mitotických buněk v porovnání s kontrolami, což pravděpodobně ukazuje na zastavení mitózy, pak by buňky měly být odebrány odstředěním a přidány zpět do kultury, aby se zabránilo ztrátě buněk, u nichž probíhá mitóza a tedy může dojít k indukci mikrojader / chromozómových aberací. Odběr buněk a příprava preparátů Buňky z každé kultury by se měly odebírat a zpracovávat zvlášť. Příprava buněk může zahrnovat hypotonizaci, ale tento krok není nutný, pokud se dosáhne odpovídajícího rozprostření buněk jiným způsobem. Při přípravě preparátů lze použít různé postupy za předpokladu, že zajistí získání vysoce kvalitních buněčných preparátů pro vyhodnocení. Měly by být zachovány buňky s neporušenou buněčnou membránou a neporušenou cytoplazmou, což umožni detekci mikrojader a (při použití metody s blokováním cytokineze) spolehlivou identifikaci dvoujaderných buněk. Preparáty lze barvit pomocí různých metod, jako je Giemsa nebo fluorescenčními barvivy se specifickou vazbou na DNA. Použitím vhodných fluorescenčních barviv (jako je např. akridinová oranž (78) nebo Hoechst 33258 a pyronin-Y (79)) se lze vyhnout některým artefaktům spojeným s použitím barviva bez specifické vazby na DNA. Je-li zapotřebí získat mechanistické informace o tvorbě mikrojader, lze k identifikaci jejich obsahu použít protilátky proti kinetochorům, hybridizační metodu FISH využívající pan-centrometrické DNA-sondy nebo značení in situ pomocí primerů se specifickou pan-centrometrickou vazbou spolu s vhodným kontrastním barvením DNA (celé chromozomy se zbarví, zatímco acentrické fragmenty chromozomů nikoli) (16) (17). K rozlišení mezi klastogeny a aneugeny lze použít i jiné metody, pokud se ukázaly jako účinné a byly validovány. Například u některých buněčných linií by užitečné informace mohlo poskytnout také měření jader sub-2N jako hypodiploidních jevů pomocí takových postupů, jako je analýza obrazu, laserová skenovací cytometrie nebo průtoková cytometrie (80) (81) (82). Také morfologická pozorování jader by mohla odhalit známky možné aneuploidie. Rovněž zkouška chromozomových aberací v metafázi, provedená nejlépe se stejným druhem buněk a podle protokolu se srovnatelnou citlivostí, by mohla být užitečná pro stanovení toho, zda jsou mikrojádra důsledkem poškození chromozomů (vzhledem k tomu, že ztráta chromozomů by při zkoušce chromozomových aberací nebyla zjištěna). Analýza Všechny preparáty včetně preparátů z kontrol s rozpouštědlem a z neexponovaných (jsou-li použity) a pozitivních kontrol by před mikroskopickou analýzou četnosti výskytu mikrojader měly být označeny samostatným kódem. Při použití automatizovaného systému hodnocení, jako je např. průtoková cytometrie, laserová skenovací cytometrie nebo analýza obrazu, by měly být použity vhodné techniky, aby se kontrolovalo, zda nedochází k jakémukoli zkreslení či posunu ve výsledcích. Ať už se pro vyčíslení mikrojader používá automatizovaná platforma, či nikoli, měly by být souběžně posouzeny hodnoty CBPI, RI, RPD nebo RICC. V případě kultur ošetřených pomocí cytoB by se měla analyzovat četnost výskytu mikrojader nejméně u 2 000 dvoujaderných buněk pro každou koncentraci a z každé kontroly (83), které se rovnoměrně rozdělí mezi replikami, jsou-li použity. V případě použití jediné kultury na každou dávku (viz odstavec 28) by se mělo vyhodnotit nejméně 2 000 dvoujaderných buněk na každou kulturu v rámci této jediné kultury (83). Pokud je k dispozici k vyhodnocení pro každou koncentraci podstatně méně než 1 000 dvoujaderných buněk z každé kultury (u duplicitních kultur), nebo než 2 000 (u jedné kultury) a není-li zjištěn výrazný nárůst výskytu mikrojader, musí se zkouška zopakovat za použití většího počtu buněk nebo při méně cytotoxických koncentracích (podle toho, co z těchto dvou možností připadá v úvahu). Je třeba dbát na to, aby se nezapočítávaly dvoujaderné buňky, které mají nepravidelný tvar nebo jejichž dvě jádra se svou velikostí navzájem značně liší. Dále, dvoujaderné buňky by neměly být zaměňovány za špatně rozprostřené mnoho jaderné buňky. Buňky obsahující více než dvě hlavní jádra by se neměly analyzovat na přítomnost mikrojader, jelikož základní četnost mikrojader může být v těchto buňkách vyšší (84). Započtení jednojaderných buněk je přijatelné, pokud se prokáže, že zkoušená chemická látka narušuje působení cytoB. V takových případech by mohla být užitečná opakovaná zkouška bez cytoB. Započtení jednojaderných buněk spolu s hodnocením dvoujaderných buněk by mohlo poskytnout užitečné informace (85) (86), ale není povinné. V případě buněčných linií testovaných bez cytoB by se měla analyzovat četnost výskytu mikrojader nejméně u 2 000 dvoujaderných buněk pro každou zkušební koncentraci a z každé kontroly (83), které se rovnoměrně rozdělí mezi replikami, jsou-li použity. Pokud je na každou koncentraci použita jediná kultura (viz odstavec 28), mělo by být hodnoceno nejméně 2 000 buněk na kulturu v rámci této jediné kultury. Pokud je k dispozici k vyhodnocení pro každou koncentraci podstatně méně než 1 000 buněk z každé kultury (u duplicitních kultur), nebo než 2 000 (u jedné kultury) a není-li zjištěn výrazný nárůst výskytu mikrojader, musí se zkouška zopakovat za použití většího počtu buněk nebo při méně cytotoxických koncentracích (podle toho, co z těchto dvou možností připadá v úvahu). Při použití cytoB by se měla určit hodnota CBPI nebo RI k posouzení buněčné proliferace (viz dodatek 2) za použití nejméně 500 buněk z každé kultury. Provádí-li se expozice bez přítomnosti cytoB, je nezbytné prokázat, že buňky v kultuře se rozdělily, jak je vysvětleno v odstavcích 24 až 28. Způsobilost laboratoře Aby bylo možné před zahájením rutinního zkoušení zjistit, zda laboratoř má s danou zkouškou dostatečné zkušenosti, měla by provést sérii experimentů s referenčními pozitivními chemickými látkami, jež působí prostřednictvím různých mechanismů (alespoň s jednou látkou s metabolickou aktivací a jednou bez metabolické aktivace a jednou působící prostřednictvím aneugenního mechanismu, které se vyberou ze seznamu chemických látek uvedeného v tabulce 1), a s různými negativními kontrolami (včetně kontrol s neexponovanými kulturami a kontrol s různými rozpouštědly/vehikuly). Odpovědi těchto pozitivních a negativních kontrol by měly být v souladu s odbornou literaturou. Tento požadavek neplatí pro laboratoře, které tuto zkušenost mají, tj. které mají k dispozici historickou databázi historických údajů, jak je definována v odstavcích 49 až 52. Měl by být zkoumán výběr z chemických látek pro pozitivní kontroly (viz tabulka 1) s krátkou a dlouhou expozicí za nepřítomnosti metabolické aktivace a rovněž s krátkou expozicí za přítomnosti metabolické aktivace, aby byla prokázána způsobilost pro detekci klastogenních a aneugenních chemických látek, stanovení účinnosti systému metabolické aktivace a prokázání vhodnosti postupů hodnocení (mikroskopická vizuální analýza, průtoková cytometrie, laserová skenovací cytometrie nebo analýza obrazu). Rozsah koncentrací by u vybraných chemických látek měl být zvolen tak, aby poskytl opakovatelná zvýšení účinku v závislosti na dávce nad úroveň pozadí, aby se tak prokázala citlivost a dynamické rozpětí testovacího systému. Historické kontrolní údaje Laboratoř by měla stanovit:
Při prvním získávání údajů o historické distribuci negativních kontrol by měly být výsledky souběžných negativních kontrol v souladu s publikovanými negativními kontrolními údaji, pokud existují. S tím, jak je do distribuce kontrol přidáváno více údajů z experimentů, měly by být souběžné negativní kontroly v ideálním případě uvnitř 95 % kontrolních limitů této distribuce (87) (88). Nejprve by měla být vybudována historická databáze negativních kontrol dané laboratoře na základě nejméně 10 experimentů, nejlépe však by měla zahrnovat nejméně 20 experimentů provedených za srovnatelných zkušebních podmínek. V laboratořích by měly být uplatňovány metody řízení jakosti, jako např. regulační diagramy (např. takzvané C-charts nebo X-bar charts (88)), z nichž je patrné, jak proměnlivé jsou jejich údaje o pozitivních a negativních kontrolách a že metodika je v dané laboratoři „pod kontrolou“ (83). Další doporučení, jak získat a používat historické údaje (tj. kritéria pro zařazení údajů do historické databáze a jejich vyřazení a kritéria přijatelnosti pro daný experiment), lze najít v literatuře (87). Jakékoli změny zkušebního protokolu by měly být zvažovány z hlediska jejich souladu se stávajícími historickými kontrolními databázemi laboratoře. Jakákoliv větší změna by měla vést k zavedení nové historické kontrolní databáze. Údaje o negativních kontrolách by měly zahrnovat výskyt buněk s mikrojádry z jedné kultury nebo ze souhrnu replikovaných kultur, jak je popsáno v odstavci 28. Výsledky souběžných negativních kontrol by v ideálním případě měly spadat do 95 % rozmezí distribuce v historické databázi negativních kontrol laboratoře (87) (88). Pokud jsou údaje ze souběžných negativních kontrol mimo 95 % rozmezí může být přijatelné je zahrnout do dosavadní distribuce kontrol, pokud tyto údaje nejsou mimořádně odlehlé a pokud existují důkazy o tom, že testovací systém je „pod kontrolou“ (viz odstavec 50), a důkazy o tom, že nedošlo k technické nebo lidské chybě. ÚDAJE A PŘEDKLÁDÁNÍ ZPRÁV Vyjádření výsledků Pokud se používá technika blokování cytokineze, slouží k hodnocení indukce vzniku mikrojader jen četnost dvoujaderných buněk s mikrojádry (nezávisle na počtu mikrojader v každé buňce). Samostatně lze podat zprávu o počítání počtu buněk s jedním, dvěma nebo více mikrojádry, které by mohlo poskytnout užitečné informace, ale není povinné. Souběžně by se mělo provádět měření cytotoxicity a/nebo cytostáze u všech ošetřených kultur a kultur v negativních a pozitivních kontrolách (16). Při použití metody s blokováním cytokineze by se měly vypočítat hodnoty CBPI nebo RI u všech ošetřených a kontrolních kultur jako ukazatele zpoždění buněčného cyklu. V případě nepřítomnosti cytoB by se měly použít hodnoty RPD nebo RICC (viz dodatek 2). Měly by se uvést údaje za jednotlivé kultury. Kromě toho se všechny údaje shrnou do tabulky. Kritéria přijatelnosti Přijetí zkoušky je založeno na následujících kritériích:
Hodnocení a interpretace výsledků Za předpokladu, že všechna kritéria přijatelnosti byla splněna, lze zkoušenou chemickou látku považovat za jasně pozitivní, pokud v některém ze zkoumaných experimentálních uspořádání (viz odstavce 36–39):
Jsou-li všechna tato kritéria splněna, má se za to, že zkoušená chemická látka je schopna vyvolávat poškození chromozomů a/nebo zvýšení či snížení jejich počtu v tomto zkušebním systému. Doporučení ohledně nejvhodnějších statistických metod lze též najít v literatuře (90) (91) (92). Za předpokladu, že byla splněna všechna kritéria přijatelnosti, se zkoušená chemická látka považuje za jasně negativní, pokud za všech zkoumaných zkušebních podmínek (viz odstavce 36–39):
Pak se má za to, že zkoušená chemická látka není schopna vyvolávat poškození chromozomů a/nebo zvýšení či snížení počtu chromozomů v tomto zkušebním systému. Doporučení ohledně nejvhodnějších statistických metod lze též najít v literatuře (90) (91) (92). Ověření jasně pozitivní či jasně negativní odpovědi se nepožaduje. V případě, že odpověď není ani jasně negativní, ani jasně pozitivní, jak je popsáno výše, nebo s cílem napomoci při stanovení biologického významu výsledku by měly být údaje vyhodnoceny na základě odborného posouzení a/nebo dalším šetřením. Užitečné by mohlo být hodnocení dalších buněk (je-li vhodné) nebo provedení opakovaného experimentu, případně za použití upravených zkušebních podmínek (např. rozsah koncentrací, jiné podmínky metabolické aktivace (např. koncentrace S9 nebo původ S9)). V ojedinělých případech dokonce ani po dalším šetření nebude možné na základě daného souboru údajů dospět k závěru o pozitivitě či negativitě výsledku, a bude proto učiněn závěr, že výsledek je neurčitý. Chemické látky, které vyvolávají vznik mikrojader při zkoušce MNvit, mají tento účinek proto, že vyvolávají poškození chromozomů, ztrátu chromozomů nebo kombinaci obou těchto dějů. Při určování, zda je mechanismus vyvolání tvorby mikrojader důsledkem klastogenního a/nebo aneugenního působení, lze uplatnit další analýzy za použití protilátek proti kinetochorům, sond in situ se specifickou vazbou na centromery nebo jiných metod. Závěrečná zpráva Závěrečná zpráva by měla obsahovat následující informace:
LITERATURA
Dodatek 1 DEFINICE Aneugen : jakákoli chemická látka nebo proces, který v interakci se složkami mechanismu mitotického a meiotického cyklu buněčného dělení vede k aneuploidii v buňkách nebo organismech. Aneuploidie : jakákoli odchylka od normálního diploidního (nebo haploidního) počtu chromozomů o jeden nebo více chromozomů, avšak nikoli o celou sadu (nebo více sad) chromozomů (polyploidie). Apoptóza : programovaná buněčná smrt, která se vyznačuje řadou kroků vedoucích k rozpadu buněk na membránově vázané části, které jsou poté odstraněny fagocytózou nebo rozkladem. Buněčná proliferace : zvyšování počtu buněk v důsledku jejich mitotického dělení. Centromera : oblast DNA v chromozomu, kde se stýkají obě chromatidy a k níž jsou bok po boku připojeny oba kinetochory. Chemická látka : chemická substance nebo směs. Koncentrace : odkazuje na konečné koncentrace zkoušené chemické látky v kultivačním médiu. Klastogen : jakákoli chemická látka nebo děj, který způsobuje strukturní chromozomové aberace v populacích buněk nebo eukaryotických organismů. Cytokineze : proces rozdělení buňky ihned po mitóze, čímž vzniknou dvě dceřiné buňky, z nichž každá obsahuje jedno jádro. Proliferační index při blokování cytokineze (CBPI) : podíl počtu buněk z druhého dělení v ošetřené populaci k jejich počtu v neošetřené kontrole (příslušný vzorec viz dodatek 2). Cytostáze : inhibice růstu buněk (příslušný vzorec viz dodatek 2). Cytotoxicita : u zkoušek prováděných podle této zkušební metody za přítomnosti cytochalasinu B se cytotoxicita vyjadřuje jako snížení proliferačního indexu při blokování cytokineze (CBPI) nebo replikačního indexu (RI) exponovaných buněk v porovnání s negativní kontrolou (viz odstavec 26 a dodatek 2). U zkoušek prováděných podle této zkušební metody prováděných za nepřítomnosti cytochalasinu B se cytotoxicita vyjadřuje jako snížení relativního zdvojnásobení populace (RPD) nebo relativního nárůstu počtu buněk (RICC) exponovaných buněk v porovnání s negativní kontrolou (viz odstavec 27 a dodatek 2). Genotoxický : obecný termín zahrnující všechny typy poškození DNA nebo chromozomů včetně jejich rozlámání, delecí, aduktů, modifikací a propojení nukleotidů, přestavby, genových mutací, chromozomových aberací a aneuploidie. Ne všechny druhy genotoxických účinků způsobují mutace nebo stálé poškození chromozomů. Interfázní buňky : buňky, které se nenacházejí ve stádiu mitózy. Kinetochor : struktura obsahující proteiny, jež se vytváří u centromery chromozomu, na kterou se při dělení buněk napojují vlákna dělicího vřeténka, což umožňuje správný pohyb dceřiných chromozomů k pólům dceřiných buněk. Mikrojádra : malá jádra existující odděleně od hlavních jader a vedle nich, vytvářená během telofáze mitózy (meiózy) nereplikujícími se (lagging) chromozomovými fragmenty nebo celými chromozomy. Mitóza : proces dělení buněčného jádra, který se obvykle člení na profázi, prometafázi, metafázi, anafázi a telofázi. Mitotický index : poměr buněk v metafázi vydělený celkovým počtem buněk zjištěných v populaci buněk; udává stupeň buněčné proliferace této populace. Mutagenní : produkující dědičné změny sekvence (sekvencí) párů bází DNA v genech nebo struktury chromozomů (chromozomové aberace). Nondisjunkce : děj, při kterém se párové chromatidy nerozloučí a správně nerozdělí do vznikajících dceřiných buněk, což vede ke vzniku dceřiných buněk s abnormálními počty chromozomů. Stav p53 : protein p53 se podílí na regulaci buněčného cyklu, apoptóze a reparaci DNA. Buňky, jež mají nedostatek funkčního proteinu p53, nejsou schopny zastavovat buněčný cyklus nebo odstraňovat poškozené buňky prostřednictvím apoptózy nebo jiných mechanismů (např. indukcí reparace DNA) souvisejících s funkcemi p53 v reakci na poškození DNA, by teoreticky měly být náchylnější ke genové mutaci nebo chromozómovým aberacím. Polyploidie : numerické chromozomální aberace v buňkách nebo organismech, které se týkají celé sady (celých sad) chromozomů, na rozdíl od jednoho chromozomu nebo jednotlivých chromozomů (aneuploidie). Proliferační index (PI) : metoda pro měření cytotoxicity bez použití cytoB (příslušný vzorec viz dodatek 2). Relativní nárůst počtu buněk (RICC) : metoda pro měření cytotoxicity bez použití cytoB (příslušný vzorec viz dodatek 2). Relativní zdvojnásobení populace (RPD) : metoda pro měření cytotoxicity bez použití cytoB (příslušný vzorec viz dodatek 2). Replikační index (RI) : odíl počtu dokončených cyklů buněčného dělení v ošetřené kultuře v porovnání s jejich počtem v neošetřené kontrole během expoziční doby a obnovy kultury (příslušný vzorec viz dodatek 2). S9 jaterní frakce : supernatant homogenátu jater po odstředění při 9 000 g, tj. surový jaterní extrakt. Směs S9 : směs S9 jaterní frakce a kofaktorů nezbytných pro metabolickou aktivaci enzymů. Kontrola s rozpouštědlem : obecný termín k označení kontrolních kultur, ke kterým se přidává pouze rozpouštědlo používané k rozpuštění zkoušené chemické látky. Zkoušená chemická látka : jakákoli chemická substance nebo směs, která se zkouší za použití této zkušební metody. Neexponovaná/neošetřená kontrola : kultury, které se neexponují (tj. ani chemické látce, ani rozpouštědlu), ale zpracovávají se souběžně stejným způsobem jako kultury, k nimž se se přidává zkoušená chemická látka. Dodatek 2 VZORCE PRO HODNOCENÍ CYTOTOXICITY Při použití cytoB by mělo být hodnocení cytotoxicity založeno na proliferačním indexu při blokování cytokineze (CBPI) nebo na replikačním indexu (RI) (17) (69). Hodnota CBPI udává průměrný počet jader na buňku a lze ji použít k výpočtu proliferace buněk. Hodnota RI udává relativní počet buněčných cyklů na buňku v době jejího vystavení cytochalasinu B v ošetřených kulturách ve srovnání s kontrolními kulturami a lze ji použít k výpočtu procenta cytostáze: % cytostáze = 100–100{(CBPIT – 1) ÷ (CBPIC – 1)} přičemž:
kde:
Hodnota CBPI = 1 (všechny buňky jsou jednojaderné) tedy odpovídá 100 % cytostázi. Cytostáze = 100 – RI
Hodnota RI = 53 % tedy znamená, že v porovnání s počtem buněk, z nichž v kontrolní kultuře vznikly rozdělením dvoujaderné a vícejaderné buňky, se v ošetřené kultuře rozdělilo pouze 53 % tohoto počtu, což znamená 47 % cytostázi. Když se cytoB nepoužije , doporučuje se hodnotit cytotoxicitu na základě relativního nárůstu počtu buněk (RICC) nebo relativního zdvojnásobení populace (RPD) (69), jelikož oba tyto ukazatele zohledňují poměr buněčné populace, která se rozdělila.
kde: Zdvojnásobení populace = [log (počet buněk po ošetření ÷ počáteční počet buněk)] ÷ log 2 Hodnota RICC nebo RPD ve výši 53 % tedy znamená 47 % cytotoxicitu/cytostázi. Za použití indexu proliferace (PI) lze posoudit cytotoxicitu spočítáním klonů tvořených 1 buňkou (cl1), 2 buňkami (cl2), 3 až 4 buňkami (cl4) a 5 až 8 buňkami (cl8).
Hodnota PI se používá jako cenný a spolehlivý parametr cytotoxicity i pro buněčné linie kultivované in vitro bez přítomnosti cytoB (35) (36) (37) (38) a lze ji považovat za užitečný doplňkový parametr. V každém případě by počet buněk před expozicí měl být v exponované kultuře a v negativní kontrolní kultuře stejný. V minulosti byl jako parametr cytotoxicity užíván RCC (tj. počet buněk v exponovaných kulturách / počet buněk v kontrolních kulturách), nyní se již nedoporučuje, protože může cytotoxicitu podhodnotit. Při použití automatizovaných systémů hodnocení, například průtokové cytometrie, laserové skenovací cytometrie nebo analýzy obrazu, lze počet buněk ve vzorci nahradit počtem jader. V negativních kontrolních kulturách by zdvojnásobení populace nebo replikační index měly být slučitelné s požadavkem na odběr buněčných vzorků po expozici v čase odpovídajícím přibližně 1,5–2násobku normální délky buněčného cyklu. |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||




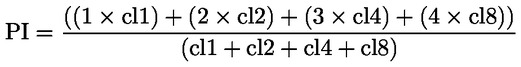
|
15) |
V části B se doplňují tyto kapitoly: „B.59 Senzibilizace kůže in chemico: zkouška přímé reaktivity peptidů (DPRA) ÚVOD Tato zkušební metoda je rovnocenná Pokynu OECD pro zkoušení (TG) č. 442C (2015). Jako látka senzibilizující kůži se označuje látka, která po styku s kůží vyvolává alergickou odpověď, jak ji definuje globálně harmonizovaný systém klasifikace a označování chemických látek Organizace spojených národů (GHS OSN) (1) a nařízení Evropské unie (ES) č. 1272/2008 o klasifikaci, označování a balení látek a směsí (CLP) (18). Tato zkušební metoda popisuje postup in chemico (zkoušku přímé reaktivity peptidů – DPRA), který se má podpůrně použít k rozlišování mezi látkami senzibilizujícími kůži a látkami nesenzibilizujícími kůži v souladu s GHS OSN a CLP. Existuje všeobecná shoda, pokud jde o klíčové biologické procesy, jež zapříčiňují senzibilizaci kůže. Dosavadní znalosti o chemických a biologických mechanismech souvisejících se senzibilizací kůže byly shrnuty ve formě Dráhy nežádoucího účinku (AOP) (2) počínaje molekulární iniciační událostí přes průběžné události až po nepříznivý účinek, a sice alergickou kontaktní dermatitidu u lidí nebo kontaktní přecitlivělost u hlodavců. V rámci AOP pro senzibilizaci kůže je molekulární iniciační událostí kovalentní vazba elektrofilních látek na nukleofilní centra v kožních proteinech. Hodnocení senzibilizace kůže bylo obvykle prováděno na pokusných zvířatech. Klasické metody založené na morčatech, Magnussonova-Klingmannova maximalizační zkouška na morčatech (GPMT) a Bühlerova zkouška (zkušební metoda B.6 (3)), studují indukční i provokační fázi senzibilizace kůže. Uznána byla i zkouška na myších, a sice zkouška s vyšetřením lokálních lymfatických uzlin (LLNA, zkušební metoda B.42 (4)) a její dvě neradioaktivní modifikace, LLNA: DA (zkušební metoda B.50 (5)) a LLNA: BrdU-ELISA (zkušební metoda B.51 (6)), které všechny posuzují výlučně odpověď na indukci; neboť ve srovnání se zkouškami na morčatech jsou vhodnější z hlediska dobrého zacházení se zvířaty a objektivního měření indukční fáze senzibilizace kůže. V poslední době jsou za vědecky platné metody pro hodnocení nebezpečnosti chemických látek pro senzibilizaci kůže považovány mechanisticky založené zkušební metody in chemico a in vitro. Protože však každá z těchto nyní dostupných zkušebních metod bez použití zvířat mechanisticky pokrývá AOP jen v omezeném rozsahu, bude pro úplné nahrazení nyní používaných zkoušek na zvířatech zapotřebí kombinace metod bez použití zvířat (in silico, in chemico, in vitro) v rámci integrovaného přístupu ke zkouškám a posuzování (IATA) (2) (7). Zkouška DPRA je navržena ke zkoumání molekulární iniciační události AOP pro senzibilizaci kůže, konkrétně reaktivity proteinů, pomocí kvantifikace reaktivity zkoušených chemických látek vůči modelovým syntetickým peptidům obsahujícím lysin nebo cystein (8). Na základě procentuálních hodnot úbytku cysteinových a lysinových peptidů se pak látka zařadí do jedné ze čtyř tříd reaktivity, což pomáhá rozlišovat mezi látkami senzibilizujícími kůži a látkami nesenzibilizujícími kůži (9). Zkušební metoda DPRA byla validována ve validační studii provedené pod vedením Referenční laboratoře Evropské unie pro alternativy ke zkouškám na zvířatech (EURL ECVAM), následně byla podrobena nezávislé odborné revizi, kterou provedl vědecký poradní výbor (ESAC) EURL ECVAM, a uznána za vědecky platnou (10) pro použití v rámci integrovaných přístupů IATA na podporu rozlišení mezi látkami senzibilizujícími a nesenzibilizujícími kůži pro účely klasifikace a označování nebezpečnosti. Příklady využití údajů DPRA v kombinaci s jinými informacemi jsou uvedeny v odborné literatuře (11) (12) (13) (14). Definice jsou uvedeny v dodatku 1. VÝCHOZÍ ÚVAHY, POUŽITELNOST A OMEZENÍ Korelace mezi reaktivitou proteinů a potenciálem senzibilizace kůže byla spolehlivě zjištěna (15) (16) (17). Ačkoli se jedná o molekulární iniciační události AOP pro senzibilizace kůže, vazba na proteiny představuje pouze jeden klíčový jev, takže informace o reaktivitě proteinů získané zkušebními i nezkušebními metodami nemusí samy o sobě stačit k vyvození závěru o neexistenci potenciálu chemických látek způsobovat senzibilizaci kůže. Údaje získané pomocí této zkušební metody by proto měly být zvažovány v kontextu integrovaných přístupů, jako je např. IATA, v kombinaci s jinými doplňujícími informacemi např. pocházejícími ze zkoušek in vitro zkoumajících jiné klíčové události v rámci metody AOP pro stanovení senzibilizace kůže, jakož i z nezkušebních metod včetně údajů odvozených z analogických chemických látek. Tuto zkušební metodu lze v kombinaci s jinými doplňujícími informacemi v rámci přístupu IATA použít na podporu rozlišení mezi látkami senzibilizujícími kůži (tj. kategorie 1 GHS OSN /CLP) a látkami nesenzibilizujícími kůži. Tuto zkušební metodu nelze používat samu o sobě ani k zařazení látek senzibilizujících kůži do podkategorií 1A a 1B, jak je definuje systém GHS OSN / CLP, ani k predikci účinnosti těchto látek při rozhodování v rámci posuzování bezpečnosti. V závislosti na regulačním rámci však kladný výsledek získaný zkušební metodou DPRA může být použit sám o sobě ke klasifikaci chemické látky v kategorii 1 GHS OSN / CLP. U zkušební metody DPRA se prokázalo, že ji lze přenést do laboratoří, které mají zkušenosti s analýzou pomocí vysokoúčinné kapalinové chromatografie (HPLC). Míra reprodukovatelnosti předpovědí, které lze od této zkušební metody očekávat, činí řádově 85 % v rámci laboratoří a 80 % mezi laboratořemi (10). Výsledky validační studie (18) a zveřejněných studií (19) celkově ukazují, že přesnost zkušební metody DPRA při odlišení senzibilizujících látek (tj. kategorie 1 GHS OSN / CLP) od nesenzibilizujících látek činí 80 % (N=157) při citlivosti 80 % (88/109) a specifičnosti 77 % (37/48) ve srovnání s výsledky zkušební metody LLNA. V případě DPRA je pravděpodobnější, že povede k příliš nízké předpovědi u chemických látek vykazujících nízkou až střední potenci způsobovat senzibilizaci kůže (tj. podkategorie 1B GHS OSN / CLP), než u chemických látek vykazujících vysokou potenci způsobovat senzibilizaci kůže (tj. podkategorie 1A GHS OSN / CLP (18) (19). Zde uvedené hodnoty přesnosti DPRA jako samostatné zkušební metody jsou však pouze orientační, protože tato zkušební metoda by měla být zvažována v kombinaci s jinými zdroji informací v rámci přístupu IATA a v souladu s ustanoveními odstavce 9 výše. Při hodnocení metod posuzování senzibilizace kůže bez použití zvířat by se dále mělo vést v patrnosti, že ani zkouška LLNA, ani jiné zkoušky na zvířatech nemusí plně odrážet situaci existující u druhu, který je předmětem zájmu, tj. u lidí. Na základě celkových dostupných údajů bylo prokázáno, že zkušební metodu DPRA lze použít v případě zkoušených chemických látek, jež obsahují různé organické funkční skupiny, mají různé reakční mechanismy, různou potenci způsobovat senzibilizaci kůže (jak bylo stanoveno ve studiích in vivo) a různé fyzikálně-chemické vlastnosti (8) (9) (10) (19). Celkově vzato z těchto informací vyplývá, že zkušební metoda DPRA je užitečná a může přispět ke zjištění nebezpečí senzibilizace kůže. Termín „zkoušená chemická látka“ se v této zkušební metodě používá k označení toho, co se zkouší, a nesouvisí s použitelností zkušební metody DPRA ke zkoušení látek a/nebo směsí. Tuto zkušební metodu nelze použít k testování sloučenin kovů, protože je o nich známo, že s proteiny reagují prostřednictvím jiných mechanismů, než je kovalentní vazba. Zkoušená chemická látka by měla být rozpustná ve vhodném rozpouštědle při konečné koncentraci 100 mM (viz odstavec 18). Chemické látky, které nejsou při této koncentraci rozpustné, je nicméně přesto možné testovat při koncentracích nižších, ve kterých jsou rozpustné. V takovém případě by přesto mohl být pozitivní výsledek použit na podporu identifikace zkoušené chemické látky jako látky senzibilizující kůži, avšak z negativního výsledku by se neměl vyvozovat závěr, že není dostatečné reaktivní. V současnosti jsou k dispozici jen omezené informace o použitelnosti zkušební metody DPRA ke zkoušení směsí se známým složením (18) (19). DPRA je nicméně považována za technicky použitelnou k testování vícesložkových látek a směsí se známým složením (viz odstavec 10). Před použitím této zkušební metody ke zkoušení směsi za účelem získání údajů pro zamýšlené použití v právních předpisech by mělo být zváženo, zda tato metoda může pro tento účel poskytnout přiměřené výsledky, a pokud ano, proč je tomu tak. Takové posouzení není nutné, pokud existuje právní požadavek na zkoušení dané směsi. Vzhledem k definovanému molárnímu poměru zkoušené chemické látky k peptidu není možné stávající predikční model použít ke zkoušení komplexních směsí s neznámým složením nebo látek s neznámým nebo proměnlivým složením, komplexních reakčních produktů nebo biologického materiálu (tj. látek UVCB). Pro tento účel bude nutné vyvinout nový predikční model založený na gravimetrické metodě. V případech, kdy mohou být předloženy důkazy prokazující nepoužitelnost této zkušební metody ke zkoušení jiných specifických kategorií chemických látek, neměla by se tato zkušební metoda na tyto specifické kategorie chemických látek používat. Tato zkušební metoda je metodou in chemico, která nezahrnuje metabolický systém. Pomocí této zkušební metody není možné detekovat chemické látky, které pro prokázání svého potenciálu senzibilizace kůže potřebují enzymatickou bioaktivaci (tj. prohapteny). V literatuře se uvádí, že v některých případech byly za pomocí této zkušební metody správně detekovány chemické látky, které se stávají senzibilizujícími látkami po abiotické přeměně (tj. prehapteny) (18). Vzhledem k výše uvedenému by se měly negativní výsledky získané touto zkušební metodou interpretovat v kontextu stanovených omezení a ve spojení s jinými zdroji informací v rámci přístupu IATA. Při zkoušení chemických látek, které se na peptidy kovalentně neváží, ale podporují jejich oxidaci (dimerizaci cysteinů), by mohlo potenciálně dojít k příliš vysokému odhadu úbytku peptidů, což může vést k falešným pozitivním předpovědím a/nebo k zařazení do vyšší třídy reaktivity (viz odstavce 29 a 30). Jak bylo popsáno výše, zkušební metoda DPRA pomáhá rozlišovat mezi látkami senzibilizujícími kůži a látkami nesenzibilujícími kůži. Může však také potenciálně přispět k hodnocení senzibilizační potence (11), je-li používána v rámci integrovaných přístupů, například IATA. Pro určení toho, jak mohou výsledky DPRA potenciálně přispět k posuzování potence, však bude nutný další výzkum, přednostně založený na údajích o účincích na člověka. PODSTATA ZKOUŠKY DPRA je metoda in chemico, která kvantifikuje zbytkovou koncentraci peptidů obsahujících cystein nebo lysin po 24hodinové inkubaci se zkoušenou chemickou látkou při teplotě 25 ± 2,5 °C. K usnadnění detekce obsahují syntetické peptidy fenylalanin. Relativní koncentrace peptidů se měří pomocí vysokoúčinné kapalinové chromatografie (HPLC) s gradientovou elucí a UV detekcí při vlnové délce 220 nm. Poté se vypočítají procentuální hodnoty úbytku cysteinových a lysinových peptidů a použijí se v predikčním modelu (viz odstavec 29), což umožní zařadit zkoušenou chemickou látku do jedné ze čtyř tříd reaktivity, které se podpůrně používají k rozlišení mezi senzibilizujícími a nesenzibilizujícími látkami. Před rutinním používáním popsané metody, která se řídí touto zkušební metodou, by měly laboratoře prokázat odbornou způsobilost použitím deseti látek pro prokazování způsobilosti, které jsou uvedeny v dodatku 2. POSTUP Tato zkušební metoda je založena na protokolu DPRA DB-ALM č. 154 (20), což je protokol použitý při validační studii provedené za koordinace EURL ECVAM. Při provádění a používání této metody v laboratoři se doporučuje použít tento protokol. Dále je uveden popis hlavních složek a postupů zkušební metody DPRA. Použije-li se jiné nastavení vysokoúčinné kapalinové chromatografie (HPLC), měla by být prokázána jeho rovnocennost validovanému nastavení, které je popsáno v protokolu DB-ALM (např. testováním vhodných látek uvedených v dodatku 2). Příprava peptidů obsahujících cystein nebo lysin Zásobní roztoky cysteinu (Ac-RFAACAA-COOH) a lysinu (Ac-RFAAKAA-COOH) obsahující syntetické peptidy o čistotě vyšší než 85 % a přednostně v rozmezí 90–95 % by se měly připravovat čerstvé bezprostředně před jejich inkubací se zkoušenou chemickou látkou. Konečná koncentrace cysteinových peptidů ve fosfátovém pufru s pH 7,5 by měla být 0,667 mM a konečná koncentrace lysinových peptidů v pufru octanu amonného s pH 10,2 by měla být 0,667 mM. Série provedení HPLC by se měla nastavit tak, aby trvání analýzy HPLC bylo kratší než 30 hodin. Při nastavení HPLC použitém ve validační studii a popsaném v této zkušební metodě je možné v rámci jednoho provedení HPLC zpracovat až 26 analytických vzorků (jež zahrnují vzorky se zkoušenou chemickou látkou, pozitivní kontrolu a přiměřený počet kontrol s rozpouštědlem na základě počtu jednotlivých rozpouštědel použitých ve zkoušce, přičemž každý z nich se testuje třikrát). U všech replik analyzovaných v rámci jednoho provedení by měly být použity totožné zásobní roztoky cysteinových a lysinových peptidů. U jednotlivých šarží peptidů se před použitím doporučuje ověřit správnou rozpustnost. Příprava zkoušené chemické látky Před provedením zkoušky by měla být posouzena rozpustnost zkoušené chemické látky ve vhodném rozpouštědle za dodržení postupu rozpouštění popsaného v protokolu DPRA DB-ALM (20). Ve vhodném rozpouštědle se zkoušená chemická látka rozpustí beze zbytku. Jelikož při zkoušce DPRA se zkoušená chemická látka inkubuje ve velkém přebytku s cysteinovými nebo lysinovými peptidy, stačí k ověření toho, že se zkoušená chemická látka (a všechny její složky v případě zkoušení vícesložkové látky nebo směsi) rozpustila, vizuální kontrola tvorby čirého roztoku. Vhodnými rozpouštědly jsou acetonitril, voda, směs voda/acetonitril v poměru 1:1, izopropanol, aceton nebo směs aceton/acetonitril v poměru 1:1. Lze použít i jiná rozpouštědla, pokud nemají vliv na stabilitu peptidů, sledovanou pomocí referenční kontroly C (tj. vzorky sestávající pouze z peptidů rozpuštěných ve vhodném rozpouštědle; viz dodatek 3). Poslední možností, není-li zkoušená chemická látka rozpustná v žádném z těchto rozpouštědel, je pokusit se ji rozpustit ve 300 μl DMSO a výsledný roztok zředit přidáním 2 700 μl acetonitrilu, a pokud zkoušená chemická látka není v této směsi rozpustná, pokusit se rozpustit stejné množství zkoušené chemické látky v 1 500 μl DMSO a výsledný roztok zředit přidáním 1 500 μl acetonitrilu. Zkoušená chemická látka by měla být předem odměřena do skleněných zkumavek a bezprostředně před zkoušením rozpuštěna ve vhodném rozpouštědle k získání 100 mM roztoku. U směsí a vícesložkových látek se známým složením by se měla stanovit jediná čistota součtem podílů jejich složek (s výjimkou vody) a měla by se stanovit celková molekulová hmotnost na základě jednotlivých molekulových hmotností každé složky směsi (s výjimkou vody) a jejich jednotlivých podílů. Výsledná čistota a molekulová hmotnost by poté měly být použity k výpočtu hmotnosti zkoušené chemické látky nezbytné pro přípravu 100 mM roztoku. U polymerů, u kterých není možné stanovit dominantní molekulovou hmotnost, lze pro přípravu 100 mM roztoku posoudit molekulovou hmotnost monomeru (nebo zdánlivou molekulovou hmotnost různých monomerů tvořících polymer). Při zkoušení směsí, vícesložkových látek nebo polymerů se známým složením by však mělo být zváženo také zkoušení chemické látky v neředěném stavu. V případě kapalin by měla být zkoušena čistá chemická látka jako taková bez předchozího ředění prostřednictvím inkubace s cysteinovými peptidy v molárním poměru 1:10 a s lysinovými peptidy v molárním poměru 1:50. V případě pevných látek by měla být zkoušená chemická látka rozpuštěna v maximální rozpustné koncentraci ve stejném rozpouštědle, které bylo použito na přípravu roztoku se zdánlivou molekulovou hmotností 100 mM. Poté by měla zbýt zkoušena jako taková bez dalšího ředění tak, že se inkubuje s cysteinovými peptidy v molárním poměru 1:10 a s lysinovými peptidy v molárním poměru 1:50. Souhlasné výsledky (reaktivita nebo nereaktivita) mezi roztokem se zdánlivou molekulovou hmotností 100 mM a roztokem čisté chemické látky by měly umožnit vyvodit na základě tohoto výsledku spolehlivý závěr. Příprava pozitivní kontroly, referenčních kontrol a kontrol koelucí Jako pozitivní kontrola by měl být použit cinnamaldehyd (č. CAS 104-55-2, potravinářské čistoty ≥95 %) v roztoku o koncentraci 100 mM v acetonitrilu. Lze použít i jiné vhodné pozitivní kontroly přednostně poskytující hodnoty úbytku středního rozsahu, pokud jsou k dispozici dosavadní údaje umožňující odvodit srovnatelná kritéria přijatelnosti daného provedení zkoušky. Do série provedení HPLC by navíc měly být zařazeny referenční kontroly (tj. vzorky obsahující pouze peptidy rozpuštěné ve vhodném rozpouštědle), které se používají k ověření vhodnosti systému HPLC před analýzou (referenční kontroly A), k ověření stability referenčních kontrol v časovém průběhu (referenční kontroly B) a k ověření toho, že rozpouštědlo použité k rozpuštění zkoušené chemické látky nemá vliv na procentuální úbytek peptidů (referenční kontroly C) (viz dodatek 3). Pro každou chemickou látku se použije vhodná referenční kontrola k výpočtu procentuálního úbytku peptidů vyvolaného touto chemickou látkou (viz odstavec 26). Do série provedení by rovněž měla být pro každou zkoušenou chemickou látku, která se analyzuje, zařazena kontrola koeluce sestávající pouze ze zkoušené chemické látky, s cílem detekovat případnou koeluci zkoušené chemické látky s lysinovými nebo cysteinovými peptidy. Inkubace zkoušené chemické látky s roztoky cysteinových a lysinových peptidů Roztoky cysteinových a lysinových peptidů by se měly inkubovat ve skleněných zkumavkách s automatickým dávkovačem se zkoušenou chemickou látkou v poměru 1:10 pro cysteinové a 1:50 pro lysinové peptidy. Je-li ihned po přidání roztoku zkoušené chemické látky do roztoku peptidů zpozorována sraženina v důsledku nízké rozpustnosti zkoušené chemické látky ve vodě, nelze si v tomto případě být jistý, jaké množství zkoušené chemické látky zůstalo v roztoku a bude moci reagovat s peptidy. Proto by v takovém případě pozitivní výsledek mohl být i přesto použit, ale negativní výsledek je nejistý a měl by být interpretován s velkou opatrností (viz též ustanovení odstavce 11 o zkoušení chemických látek, které nejsou rozpustné až do koncentrace 100 mM). Před provedením analýzy HPLS by reakční roztok měl být ponechán v temnu při teplotě 25 ± 2,5 °C po dobu 24 ± 2 hodin. Každá zkoušená chemická látka by se měla analyzovat třikrát s každým z obou druhů peptidů. Před analýzou HPLC se musí vzorky vizuálně zkontrolovat. Je-li zjištěna sraženina nebo fázová separace, lze vzorky odstředit při nízké rychlosti (100–400xg), aby se sraženina z preventivních důvodů vytlačila ke dnu zkumavky, neboť velké množství sraženiny může ucpat trubice nebo kolony HPLC. Je-li sraženina nebo fázová separace zjištěna po inkubační době, může v případě negativního výsledku dojít k podhodnocenému odhadu a závěr o nedostatečné reaktivitě nelze učinit s dostatečnou jistotou. Příprava kalibrační křivky standardů pro HPLC Kalibrační křivka standardů by měla být stanovena pro cysteinové i pro lysinové peptidy. Standardy peptidů by měly být připraveny ve 20 % nebo 25 % pufračním roztoku acetonitrilu za použití fosfátového pufru (pH 7,5) pro cysteinové peptidy a pufru octanu sodného (pH 10,2) pro lysinové peptidy. S použitím standardu sériového ředění pro zásobní roztok s peptidy (0,667 mM) by se mělo připravit 6 kalibračních roztoků pokrývajících rozsah od 0,534 do 0,0167 mM. Do kalibrační křivky standardů by měla být rovněž zanesena slepá zkouška s ředicím pufrem. Vhodné kalibrační křivky by měly mít r2 > 0,99. Příprava a analýza HPLC Před provedením analýzy by se měla ověřit vhodnost systému HPLC. Úbytek peptidů se sleduje pomocí HPLC s připojeným UV detektorem (fotodiodový detektor nebo absorpční detektor s fixní vlnovou délkou se signálem 220 nm). Do systému HPLC se nainstaluje vhodná kolona. Nastavení HPLC popsané ve validační studii používá přednostně kolonu Zorbax SB-C-18 o rozměrech 2,1 mm × 100 mm × 3,5 micronu. S touto kolonou HPLC na reverzní fázi by se celý systém měl nechat před provedením analýzy nejméně po dobu 2 hodin ustálit při teplotě 30 °C s 50 % fází A (0,1 % obj. roztok kyseliny trifluoroctové ve vodě) a 50 % fází B (0,085 % obj. roztok kyseliny trifluoroctové v acetonitrilu). Analýza HPLC by se měla provádět za průtoku 0,35 ml/min a s lineárním gradientem od 10 % do 25 % acetonitrilu za 10 minut, s následným prudkým zvýšením na 90 % acetonitrilu za účelem odstranění jiných materiálů. Injekcí by měly být nastříknuty stejné objemy každého standardu, vzorku a kontroly. Kolona by se mezi injekcemi měla znovu nechat ustálit za původních podmínek po dobu 7 minut. Použije-li se jiná kolona HPLC na reverzní fázi, bude možná nutné upravit výše popsané nastavení parametrů, aby se zajistila přiměřená eluce a integrace cysteinových a lysinových peptidů, včetně vstřikovaného objemu, který se může lišit (obvykle v rozmezí od 3 do 10 μl) podle použitého systému. Důležité je, že při použití jiného nastavení HPLC by měla být prokázána jeho rovnocennost s validovaným nastavením, které je popsáno výše (např. testováním vhodných látek uvedených v dodatku 2). Absorbance se měří při 220 nm. Je-li použit fotodiodový detektor, měla by se rovněž zaznamenávat absorbance při 258 nm. Je třeba poznamenat, že některé dodávky acetonitrilu by mohly mít negativní dopad na stabilitu peptidů, a toto se musí posoudit při použití nové šarže acetonitrilu. Jako ukazatel koeluce lze použít poměr plochy píků při 220 nm k ploše píků při 258 nm. Poměr v rozmezí 90 % < střední hodnota (19) poměru plochy u kontrolních vzorků < 100 % u každého vzorku by znamenal přesvědčivou známku toho, že ke koeluci nedošlo. Mohou se vyskytnout zkoušené chemické látky, které by mohly podporovat oxidaci cysteinových peptidů. Pík dimerizovaného cysteinového peptidu je možné sledovat vizuálně. Pokud se zdá, že došlo k dimerizaci, mělo by to být zaznamenáno, neboť může dojít k příliš vysokému odhadu procentuálního úbytku peptidů, což vede k falešným pozitivním předpovědím a/nebo k zařazení do vyšší třídy reaktivity (viz odstavce 29 a 30) Analýzu cysteinových a lysinových peptidů pomocí metody HPLC lze provádět souběžně (jsou-li k dispozici dva systémy HPLC), nebo v různých dnech. Provádí-li se analýza v různých dnech, pak by se všechny roztoky zkušebních chemických látek měly čerstvě připravit pro obě analýzy v každý příslušný den. Analýza by měla být načasována tak, aby se zajistilo, že vstříknutí prvního vzorku začne po uplynutí 22 až 26 hodin od okamžiku, kdy byla zkoušená chemická látka smíchána s roztokem peptidu. Série provedení HPLC by se měla nastavit tak, aby trvání analýzy HPLC bylo kratší než 30 hodin. Při nastavení HPLC, které bylo použito ve validační studii a je popsáno v této zkušební metodě, lze v jedné sérii provedení HPLC zpracovat až 26 analytických vzorků (viz též odstavec 17). Příklad série provedení analýzy HPLC je uveden v dodatku 3. ÚDAJE A PŘEDKLÁDÁNÍ ZPRÁV Vyhodnocení údajů Koncentrace cysteinových nebo lysinových peptidů se stanoví v každém vzorku fotometricky při 220 nm změřením plochy píku (plocha pod křivkou) vhodných píků a výpočtem koncentrace peptidů pomocí lineární kalibrační křivky odvozené od standardů. Procentuální úbytek peptidů se stanoví v každém vzorku změřením plochy píku, která se vydělí střední hodnotou plochy píku příslušných referenčních kontrol C (viz dodatek 3) podle níže uvedeného vzorce.
Kritéria přijatelnosti Aby se provedení mohlo považovat za platné, měla by být splněna tato kritéria:
Není-li jedno nebo více těchto kritérií splněno, mělo by se provedení opakovat. Aby se výsledky zkoušení zkoušené chemické látky mohly považovat za platné, měla by být splněna tato kritéria:
Predikční model Pro každou zkoušenou chemickou látku se vypočítá střední hodnota procentuálního úbytku cysteinu a procentuálního úbytku lysinu. Záporný úbytek se při výpočtu střední hodnoty považuje za „0“. S použitím predikčního modelu cystein 1:10 / lysin 1:50, jak je uvedeno v tabulce 1, by rozlišení mezi látkami senzibilizujícímu kůži a látkami nesenzibilizujícími kůži v rámci přístupu IATA mělo umožnit použití prahové hodnoty průměrného úbytku peptidů ve výši 6,38 %. Uplatnění tohoto predikčního modelu pro zařazení zkoušené chemické látky do příslušné třídy reaktivity (tj. nízká, střední a vysoká reaktivita) se snad může osvědčit jako užitečný zdroj informací pro posouzení potence v rámci přístupu IATA. Tabulka 1 Predikční model cystein 1:10 / lysin 1:50 (20)
Mohly by se vyskytnout případy, kdy zkoušená chemická látka (látka nebo jedna nebo několik složek vícesložkové látky nebo směsi) při 220 nm značně absorbuje a má stejný retenční čas jako peptid (koeluce). Koeluci lze vyřešit mírnou úpravou nastavení HPLC, aby se více oddělil eluční čas zkoušené chemické látky a eluční čas peptidu. Při použití jiného nastavení HPLC ve snaze vyřešit koeluci by měla být prokázána jeho rovnocennost s validovaným nastavením (např. testováním vhodných látek uvedených v dodatku 2). Dojde-li ke koeluci, pík peptidu nelze integrovat a výpočet procentuálního úbytku peptidu není možný. Jestliže dojde ke koeluci takových zkoušených chemických látek s cysteinovými i lysinovými peptidy, měla by být analýza označena za „neprůkaznou“. V případech, kdy dojde ke koeluci pouze s lysinovým peptidem, lze použít predikční model cystein 1:10 uvedený v tabulce Tabulka 2 Predikční model cystein 1:10 (22)
Mohly by se vyskytnout jiné případy, kdy se retenční čas zkoušené chemické látky a retenční čas některého z peptidů zcela nepřekrývá. V takových případech lze hodnoty procentuálního úbytku peptidu odhadnout a použít je v predikčním modelu cystein 1:10 / lysin 1:50, avšak zařazení zkoušené chemické látky do třídy reaktivity nelze provést s přesností. Jediná analýza HPLC pro hodnocení cysteinových i lysinových peptidů by měla být pro zkoušenou chemickou látku dostačující, pokud je výsledek jednoznačný. Avšak v případě výsledků přibližujících se prahové hodnotě použité pro rozlišení mezi pozitivními a negativními výsledky (tj. hraničních výsledků) může být nezbytné další zkoušení. V situacích, kdy se střední hodnota procentuálního úbytku nachází v rozmezí 3 % až 10 % v predikčním modelu cystein 1:10 / lysin 1:50 nebo kdy se procentuální úbytek cysteinu nachází v rozmezí 9 % až 17 % v predikčním modelu cystein 1:10, by mělo být zváženo druhé provedení a v případě nesouhlasných výsledků prvního a druhého provedení by mělo být zváženo třetí provedení. Závěrečná zpráva Závěrečná zpráva by měla obsahovat následující informace:
LITERATURA
Dodatek 1 DEFINICE Přesnost : Přesnost shody mezi výsledky zkušební metody a přijatými referenčními hodnotami. Je to měření výsledku zkušební metody a jednoho aspektu „důležitosti“. Tento výraz se často používá namísto výrazu „soulad“ a rozumí se jím podíl správných výsledků zkušební metody (21). AOP (Dráha nežádoucího účinku) : Sled událostí od chemické struktury cílové chemické látky nebo skupiny podobných chemických látek přes molekulární iniciační událost po účinek in vivo, který je předmětem zájmu (2). Kalibrační křivka : Vztah mezi hodnotou experimentální odpovědi a analytickou koncentrací (rovněž nazývaná standardní křivka) známé látky. Chemická látka : Látka nebo směs. Variační koeficient : Míra variability, která se vypočítává pro skupinu údajů z replik tak, že se směrodatná odchylka vydělí průměrem. Lze ho vynásobit 100 za účelem vyjádření v procentech. Nebezpečnost : Základní charakteristika látky nebo situace, která má potenciál vyvolat nepříznivé účinky, jsou-li organismus, systém nebo (sub)populace vystaveny této látce. IATA (integrovaný přístup ke zkoušení a posuzování) : Strukturovaný přístup používaný pro zjišťování nebezpečnosti (potenciál), charakterizaci rizik (potence) a/nebo posouzení bezpečnosti (potenciál/potence a expozice) chemické látky nebo skupiny chemických látek, jenž strategicky integruje a váží všechny důležité údaje s cílem poskytnout informace pro přijetí regulačního rozhodnutí, které se týká potenciální nebezpečnosti a/nebo rizika a/nebo potřeby dalšího cíleného, a tudíž minimálního zkoušení. Molekulární iniciační událost : Narušení biologického systému na molekulární úrovni vyvolané chemickou látkou, jež bylo identifikováno jako počáteční událost v mechanismu nepříznivých účinků. Směs : Směs nebo roztok tvořený dvěma nebo více látkami, které v něm nereagují (1). Jednosložková látka : Látka definovaná svým kvantitativním složením, v níž je přítomna jedna hlavní složka v míře alespoň 80 % hmot. Vícesložková látka : Látka definovaná svým kvantitativním složením, v níž je přítomna více než jedna hlavní složka v koncentraci ≥ 10 % hmot. a < 80 % hmot. Vícesložková látka je výsledkem výrobního procesu. Rozdíl mezi směsí a vícesložkovou látkou spočívá v tom, že směs se získá smísením dvou nebo více látek bez chemické reakce. Vícesložková látka je výsledkem chemické reakce. Pozitivní kontrola : Replika obsahující všechny složky zkušebního systému s přidáním látky, o níž je známo, že způsobuje pozitivní reakci. Aby se zajistilo, že proměnlivost v reakci na pozitivní kontrolu bude možné časem vyhodnotit, rozsah pozitivní reakce by neměl být nadměrný. Referenční kontrola : Neexponovaný vzorek obsahující všechny složky zkušebního systému, včetně rozpouštědla nebo vehikula, který se zpracovává s exponovanými zkoušenými chemickými látkami a jinými kontrolními vzorky, aby se zjistila základní reakce vzorků zkoušených se zkoušenou chemickou látkou rozpuštěnou ve stejném rozpouštědle nebo vehikulu. Když se použije souběžně s negativní kontrolou, ukazuje také, zda rozpouštědlo nebo vehikul vzájemně reaguje se zkušebním systémem. Relevantnost : Popis vztahu zkoušky k účinku, který je předmětem zájmu, a také toho, zda je zkouška smysluplná a použitelná pro konkrétní účel. Vyjadřuje, do jaké míry zkouška správně měří nebo predikuje biologický účinek, který je předmětem zájmu. Relevantnost zahrnuje také posouzení přesnosti (shody) zkušební metody (21). Spolehlivost : Měření rozsahu, v jakém může být zkušební metoda časem reprodukovatelná v laboratořích a mezi laboratořemi, když se provádí s použitím stejného protokolu. Hodnotí se to výpočtem laboratorní a mezilaboratorní reprodukovatelnosti a laboratorní opakovatelnosti (21). Reprodukovatelnost : Shoda mezi výsledky získanými při zkoušení téže chemické látky za použití stejného zkušebního protokolu (viz spolehlivost) (21). Citlivost : Podíl všech pozitivních/aktivních chemických látek, které jsou na základě zkušební metody správně zatříděny. Je měřítkem přesnosti zkušební metody, která poskytuje kategorické výsledky, a je důležitým prvkem při hodnocení relevantnosti zkušební metody (21). Specifičnost : Podíl všech negativních/neaktivních chemických látek, které jsou na základě zkušební metody správně zatříděny. Je měřítkem přesnosti zkušební metody, která poskytuje kategorické výsledky, a je důležitým prvkem při hodnocení relevantnosti zkušební metody (21). Látka : Chemické prvky nebo jejich sloučeniny v přírodním stavu nebo získané výrobním procesem včetně všech přídatných látek nutných k uchování stability výrobku a všech nečistot vznikajících v použitém procesu, ale s vyloučením všech rozpouštědel, která je možno oddělit bez ovlivnění stability látky nebo změny jejího složení (1). Vhodnost systému : Určení výkonnosti (např. citlivosti) přístrojů pomocí analýzy referenčního standardu před zavedením analytické sady (22). Zkoušená chemická látka : Termín „zkoušená chemická látka“ se používá k označení toho, co se podrobuje zkoušení. Globálně harmonizovaný systém klasifikace a označování chemických látek Organizace spojených národů (GHS OSN) : Systém navrhující klasifikaci látek a směsí podle standardizovaných typů a úrovní fyzikálních, zdravotních a environmentálních nebezpečí a řešící příslušné komunikační prvky, jako jsou piktogramy, signální slova, výroky o nebezpečí, výroky o bezpečnostních opatřeních a bezpečnostní datové listy, které obsahují informace o jejich nežádoucích účincích s ohledem na ochranu lidí (včetně zaměstnavatelů, dělníků, přepravců, spotřebitelů a osob reagujících na havárie) a životního prostředí (1). UVCB : Látky s neznámým nebo proměnlivým složením, komplexní reakční produkty nebo biologický materiál. Validovaná zkušební metoda : Zkušební metoda, u níž se má za to, že je dostatečně relevantní a spolehlivá pro určitý účel, a která je založena na vědecky spolehlivých zásadách. Zkušební metoda není nikdy validovaná v absolutním smyslu, ale pouze pro určitý konkrétní účel (21). Dodatek 2 LÁTKY PRO PROKÁZÁNÍ ZPŮSOBILOSTI Senzibilizace kůže in chemico: zkouška přímé reaktivity peptidů Dříve než začnou laboratoře běžně používat tuto zkušební metodu, měly by prokázat odbornou způsobilost správným získáním předpokládané předpovědi DPRA u 10 vhodných látek doporučených v tabulce 1 a získáním hodnot úbytku cysteinu a lysinu, které spadají do příslušného referenčního rozpětí, u 8 z 10 vhodných látek pro každý peptid. Tyto vhodné látky byly vybrány tak, aby představovaly rozpětí účinků z hlediska nebezpečí senzibilizace kůže. Dalšími kritérii výběru bylo, aby byly komerčně dostupné, aby k nim byly k dispozici vysoce kvalitní referenční údaje získané in vivo a vysoce kvalitní údaje získané zkouškou DPRA in vitro a aby byly použity ve validační studii provedené za koordinace EURL ECVAM k prokázání úspěšného provedení této zkušební metody v laboratořích, které se na studii podílely. Tabulka 1 Doporučené látky k prokázání odborné způsobilosti k provádění zkoušky přímé reaktivity peptidů
Dodatek 3 PŘÍKLADY ANALYTICKÉ SÉRIE
B.60 Senzibilizace kůže in vitro: zkušební metoda s vyšetřením luciferázy ARE-Nrf2 ÚVOD Tato zkušební metoda je rovnocenná Pokynu OECD pro zkoušení (TG) č. 442D (2015). Jako látka senzibilizující kůži se označuje látka, která po styku s kůží vyvolává alergickou odpověď, jak ji definuje globálně harmonizovaný systém klasifikace a označování chemických látek Organizace spojených národů (GHS OSN) (1) a nařízení Evropské unie (ES) č. 1272/2008 o klasifikaci, označování a balení látek a směsí (CLP) (27). Tato zkušební metoda popisuje postup in vitro (zkoušku luciferázy ARE-Nrf2), který má být podpůrně použit k rozlišování mezi látkami senzibilizujícími kůži a látkami nesenzibilizujícími kůži v souladu s GHS OSN a s nařízením CLP. Existuje všeobecná shoda, pokud jde o klíčové biologické procesy, jež zapříčiňují senzibilizaci kůže. Dosavadní znalosti o chemických a biologických mechanismech spojených se senzibilizací kůže byly shrnuty ve formě Dráhy nežádoucích účinků (AOP) (2), která se odvíjí od molekulární iniciační události přes průběžné události až k výslednému nepříznivému účinku na zdraví, tj. alergickou kontaktní dermatitidu u lidí nebo kontaktní přecitlivělost u hlodavců (2) (3). Molekulární iniciační událostí je kovalentní navázání elektrofilních látek na nukleofilní centra v kožních proteinech. Druhá klíčová událost v této metodě AOP se odehrává v keratinocytech a zahrnuje zánětlivé odpovědi, jakož i expresi genů spojenou se specifickými dráhami buněčné signalizace, např. dráhami závislými na antioxidačním/elektrofilním responsním elementu (ARE). Třetí klíčovou událostí je aktivace dendritických buněk, typicky hodnocená podle exprese specifických buněčných povrchových markerů, chemokinů a cytokinů. Čtvrtou klíčovou událostí je proliferace T buněk, která se nepřímo hodnotí ve zkoušce s vyšetřením lokálních lymfatických uzlin na myších (4). Hodnocení senzibilizace kůže bylo obvykle prováděno na pokusných zvířatech. Klasické metody založené na morčatech, Magnussonova-Klingmannova maximalizační zkouška na morčatech (GPMT) a Bühlerova zkouška (zkušební metoda B.6 (5)), studují indukční i provokační fázi senzibilizace kůže. Uznána byla i zkouška na myších, a sice zkouška vyšetřením lokálních místních uzlin (LLNA, zkušební metoda B.42 (4)) a její dvě neradioaktivní modifikace, LLNA: DA (zkušební metoda B.50 (6)) a LLNA: BrdU-ELISA (zkušební metoda B.51 (7)), které všechny posuzují výlučně odpověď na indukci; neboť oproti zkouškám na morčatech poskytují výhody z hlediska dobrého zacházení se zvířaty, i pokud jde o objektivní měření indukční fáze senzibilizace kůže. V poslední době jsou za vědecky platné metody pro hodnocení nebezpečnosti chemických látek pro senzibilizaci kůže považovány mechanisticky založené zkušební metody in chemico a in vitro. Protože však každá z těchto nyní dostupných zkušebních metod bez použití zvířat mechanisticky pokrývá AOP jen v omezeném rozsahu, bude pro úplné nahrazení nyní používaných zkoušek na zvířatech zapotřebí kombinace metod bez použití zvířat (in silico, in chemico, in vitro) v rámci integrovaného přístupu ke zkouškám a posuzování (IATA) (2) (3). Tato zkušební metoda (zkouška luciferázy ARE-Nrf2) se se navrhuje ke zkoumání druhé klíčové události, jak je vysvětleno v odstavci 2. V literatuře se uvádí, že látky senzibilizující kůži indukují geny, které jsou regulovány antioxidačním responsním elementem (ARE) (8) (9). Slabé elektrofilní látky, jako jsou látky senzibilizující kůži, mohou působit na senzorový protein Keap1 (Kelch-like ECH-associated protein 1) například kovalentní modifikací jeho zbytkového cysteinu, což vede k jeho disociaci od trankripčního faktoru Nrf2 (nuclear factor-erythroid 2-related factor 2). Disociovaný Nrf2 může pak aktivovat geny závislé na ARE, např. ty, které kódují detoxifi kační enzymy fáze II (8) (10) (11). V současnosti je k dispozici jediná zkouška, která vyhovuje zde popsané zkušební metodě in vitro s vyšetřením luciferázy ARE-Nrf2, a to zkouška KeratinoSensTM, u které již byly dokončeny validační studie (9) (12) (13) s následnou nezávislou odbornou revizí, kterou provedla Referenční laboratoř Evropské unie pro alternativy ke zkouškám na zvířatech (EURL ECVAM) (14). Zkouška KeratinoSensTM byla uznána za vědecky platnou při použití v rámci přístupu IATA na podporu rozlišování mezi látkami senzibilizujícími kůži a látkami nesenzibilizujícími kůži za účelem klasifikace nebezpečnosti a označování (14). Laboratoře, které chtějí tuto zkušební metodu provádět, mohou získat rekombinantní buněčnou linii použitou ve zkoušce KeratinoSensTM po uzavření licenční dohody se subjektem, který tuto zkušební metodu vyvinul (15). Definice jsou uvedeny v dodatku 1. VÝCHOZÍ ÚVAHY, POUŽITELNOST A OMEZENÍ Jelikož se aktivace dráhy Keap1-Nrf2-ARE vztahuje pouze ke druhé klíčové události AOP pro senzibilizaci kůže, není pravděpodobné, že informace získané touto zkušební metodou na základě aktivace této dráhy, jsou-li použity samy o sobě, by byly dostatečné pro vyvození závěru o potenciálu chemických látek senzibilizovat kůži. Údaje získané pomocí této zkušební metody by proto měly posuzovány v kontextu integrovaných přístupů, např. IATA, v kombinaci s jinými doplňujícími informacemi získanými např. ze zkoušek in vitro, které zkoumají jiné klíčové události AOP pro senzibilizaci kůže, a také nezkušebními metodami včetně údajů odvozených z analogických chemických látek. Příklady využití údajů zkušební metody s vyšetřením luciferázy ARE-Nrf2 v kombinaci s jinými informacemi jsou uvedeny v literatuře (13) (16) (17) (18) (19). Tuto zkušební metodu je možné v rámci IATA podpůrně použít k rozlišování mezi látkami senzibilizujícími kůži (tj. kategorie 1 GHS OSN / CLP) a látkami nesenzibilizujícími kůži. Tuto zkušební metodu nelze používat samu o sobě, a to ani k zařazení látek senzibilizujících kůži do podkategorií 1A a 1B, jak je definuje systém GHS OSN / CLP, ani k predikci účinnosti těchto látek při rozhodování v rámci posuzování bezpečnosti. V závislosti na regulačním rámci však pozitivní výsledek může být sám o sobě použit ke klasifikaci chemické látky v kategorii 1 GHS OSN / CLP. Na základě souboru údajů z validační studie a z interního laboratorního testování použitého pro nezávislou odbornou revizi této zkušební metody bylo prokázáno, že zkoušku KeratinoSensTM lze přenést do laboratoří, které mají zkušenosti s kultivací buněk. Míra reprodukovatelnosti předpovědí, které lze od této zkušební metody očekávat, činí řádově 85 % v rámci laboratoří a 80 % mezi laboratořemi (14). Přesnost (77 % – 155/201), citlivost (78 % – 71/91) a specifičnost (76 % – 84/110) zkoušky KeratinoSensTM pro rozlišení mezi látkami senzibilizujícími kůži (tj. kategorie 1 UN GHS/CLP) a látkami nesenzibilizujícími kůži ve srovnání s výsledky zkoušky LLNA byly vypočteny na základě všech údajů, které byly předloženy EURL ECVAM za účelem vyhodnocení a odborné revize této zkušební metody. Tyto údaje jsou obdobné jako nedávno zveřejněné údaje založené na výsledcích interního testování asi 145 látek (přesnost 77 %, citlivost 79 %, specifičnost 72 %) (13). V případě zkoušky KeratinoSensTM je pravděpodobnější, že povede k příliš nízké předpovědi u chemických látek vykazujících nízkou až střední potenci způsobovat senzibilizaci kůže (tj. podkategorie 1B GHS OSN / CLP), než u chemických látek vykazujících vysokou potenci způsobovat senzibilizaci kůže (tj. podkategorie 1A GHS OSN / CLP) (13) (14). Celkově vzato tyto informace naznačují, že zkouška KeratinoSensTM může být užitečná a může přispět k zjištění nebezpečí senzibilizace kůže. Zde uvedené hodnoty přesnosti zkoušky KeratinoSensTM jako samostatné zkušební metody jsou však pouze orientační, neboť tato zkušební metoda by měla být posuzována v kombinaci s jinými zdroji informací v rámci IATA a v souladu s ustanoveními odstavce 9 výše. Při hodnocení metod posuzování senzibilizace kůže bez použití zvířat by se dále mělo vést v patrnosti, že ani LLNA a ani jiné zkoušky na zvířatech nemusí plně odrážet situaci existující u druhu, který je předmětem zájmu, tj. u lidí. Termín „zkoušená chemická látka“ se v této zkušební metodě používá k označení toho, co se zkouší, a netýká se použitelnosti zkušební metody luciferázy ARE-Nrf2 ke zkoušení látek a/nebo směsí. Na základě nyní dostupných údajů bylo prokázáno, že zkoušku KeratinoSensTM lze použít ke zkoušení chemických látek, které obsahují různé organické funkční skupiny, mají různé reakční mechanismy, různou potenci způsobovat senzibilizaci kůže (jak byla stanovena ve studiích in vivo) a různé fyzikálně-chemické vlastnosti (9) (12) (13) (14). Zkoušeny byly hlavně jednosložkové látky, ačkoli určité omezené množství údajů existuje i o zkoušení směsí (20). Zkušební metoda je nicméně technicky použitelná i na zkoušení vícesložkových látek a směsí. Před použitím této zkušební metody ke zkoušení směsi za účelem získání údajů pro zamýšlené použití v právních předpisech by však mělo být zváženo, zda tato metoda může pro tento účel poskytnout přiměřené výsledky, a pokud ano, proč je tomu tak. Takové úvahy nejsou nutné, pokud existuje právní požadavek na zkoušení dané směsi. Při zkoušení vícesložkových látek nebo směsí by se mimoto měly uvážit možné rušivé vlivy cytotoxických složek na pozorované odpovědi. Zkušební metodu lze použít ke zkoušení rozpustných chemických látek nebo látek, jež tvoří stabilní disperzi (tj. koloid nebo suspenzi, ve které se zkoušená chemická látka neusazuje ani se neodděluje od rozpouštědla do různých fází) buď ve vodě, nebo v DMSO (včetně všech složek zkoušených chemických látek v případě zkoušení vícesložkové látky nebo směsi). Zkoušené chemické látky, které nesplňují tyto podmínky při nejvyšší požadované konečné koncentraci 2 000 μM (viz odstavec 22), lze přesto zkoušet při nižších koncentracích. V takovém případě by výsledky splňující kritéria pro pozitivitu popsaná v odstavci 39 i přesto mohly být použity na podporu identifikace zkoušené chemické látky jako látky senzibilizující kůži, ale negativní výsledek získaný při koncentracích < 1 000 μM by měl být považován za nepřesvědčivý (viz predikční model v odstavci 39). Obecně byly úspěšně zkoušeny látky s hodnotou LogP do 5, zatímco na extrémně hydrofobní látky s hodnotou LogP vyšší než 7 se podle současných poznatků použitelnost této zkušení metody nevztahuje (14). O látkách, u nichž hodnota LogP leží mezi 5 a 7, jsou k dispozici jen omezené informace. Negativní výsledky by se měly interpretovat obezřetně, protože látky s výlučnou reaktivitou vůči reziduím lysinů může tato zkušební metoda detekovat jako negativní. Z důvodu omezené metabolické schopnosti použité buněčné linie (21) a vzhledem k experimentálním podmínkám mohou prohapteny (tj. chemické látky vyžadující enzymatickou aktivaci, například prostřednictvím enzymů P450) a prehapteny (tj. chemické látky aktivované autooxidací) zejména při nízké míře oxidace rovněž vykazovat negativní výsledky. Na druhé straně zkoušené chemické látky, které nepůsobí jako senzibilizující látka, ale jsou nicméně chemickými stresory, mohou vést k falešně pozitivním výsledkům (14). Ne vždy je též možné spolehlivě posuzovat vysoce cytotoxické zkoušené chemické látky. Rovněž tak zkoušené chemické látky, jež ovlivňují enzym luciferázu, mohou zkreslit aktivitu luciferázy ve zkouškách založených na zkoušení buněk, neboť způsobují buď očividnou inhibici, nebo zvýšenou luminiscenci (22). Uvádí se například, že koncentrace fytoestrogenu vyšší než 1 μM ovlivnila luminiscenční signály v jiných zkouškách reporterových genů založených na luciferáze v důsledku nadměrné aktivace reporterového genu luciferázy (23). V důsledku toho je nutné pečlivě zkoumat exprese luciferázy získané při vysoké koncentraci fytoestrogenů nebo podobných chemických látek, u nichž existuje podezření, že vyvolávají nadměrnou aktivaci reporterového genu luciferázy podobně jako fytoestrogeny (23). V případech, kdy mohou být předloženy důkazy prokazující nepoužitelnost této zkušební metody na jiné specifické kategorie chemických látek, neměla by se tato zkušební metoda ke zkoušení těchto specifických kategorií používat. Kromě podpory rozlišování mezi látkami senzibilizujícími kůži a látkami nesenzibilizujícími kůži zkouška KeratinoSensTM rovněž poskytuje informace o vztahu koncentrace a účinku, které při použití v rámci integrovaných přístupů jako IATA mohou potenciálně přispět k hodnocení senzibilizační potence (19). Ke stanovení toho, jak mohou výsledky zkoušky KeratinoSensTM přispět k hodnocení potence (24) a k zařazování senzibilizujících látek do podkategorií GHS OSN / CLP, však bude zapotřebí další výzkum, přednostně založeny na spolehlivých údajích o účincích na člověka. PODSTATA ZKOUŠKY Zkušební metoda s vyšetřením luciferázy ARE-Nrf2 používá permanentní adherentní buněčné linie odvozené z lidských keratinocytů HaCaT stabilně transfektované volitelným plasmidem. Buněčná linie obsahuje gen luciferázy pod transkripční kontrolou konstitutivního promotoru sloučeného s elementem ARE z genu, o kterém je známo, že je up-regulován kontaktními senzibilizátory (25) (26). Signál luciferázy odráží aktivaci látek senzibilizujících endogenní Mrf2 závislé geny, a závislost signálu luciferázy na Nrf2 v rekombinantní buněčné linii byla prokázána (27). To umožňuje kvantitativní měření (pomocí lumuniscenční detekce) indukce genu luciferázy za použití dobře známých substrátů pro luciferázu produkujících světlo, jako ukazatele aktivity transkripčního faktoru Nrf2 v buňkách po expozici elektrofilním látkám. Zkoušené chemické látky se při zkoušce KeratinoSens™ považují za pozitivní, pokud vyvolávají statisticky významnou indukci aktivity luciferázy nad stanovenou prahovou hodnotou (tj. > 1,5násobný nebo 50 % růst) při menší než stanovené koncentraci, která významně neovlivňuje životaschopnost buněk (tj. nižší než 1 000 μM a při koncentraci, při které je životaschopnost buněk vyšší než 70 % (9) (12)). Za tímto účelem se stanoví maximální násobek indukce aktivity luciferázy oproti (negativní) kontrole s rozpouštědlem (Imax). Jelikož jsou buňky vystaveny řadě koncentrací zkoušených chemických látek, měly by se dále z křivky dávka-účinek interpolovat koncentrace potřebné pro vyvolání statisticky významné aktivity luciferázy nad prahovou hodnotou (tj. hodnota EC1,5) (výpočty viz odstavec 32). Měla by být rovněž prováděna paralelní měření cytotoxicity, aby bylo možné posoudit, zda úrovně indukce aktivity luciferázy nastávají při sub-cytotoxických koncentracích. Před rutinním použitím zkoušky se stanovením luciferázy ARE-Nrf2 by měly laboratoře prokázat odbornou způsobilost použitím deseti k tomu určených látek, které jsou uvedeny v dodatku 2. Jsou k dispozici technické parametry metody (Performance Standards) (28), které usnadní validaci nových nebo pozměněných zkušebních metod luciferázy ARE-Nrf2 in vitro obdobných zkoušce KeratinoSens™ a umožní včas provést změnu této zkušební metody, aby do ní mohly být zahrnuty. Vzájemné uznávání údajů bude podle postupu dohodnutého v rámci OECD zaručeno pouze u zkušebních metod validovaných podle těchto výkonnostních standardů, pokud tyto zkušební metody byly podrobeny revizi a pokud je OECD zahrnula do odpovídajícího pokynu ke zkoušení. POSTUP Jedinou zkouškou, kterou popsaná zkušební metoda v současnosti zahrnuje, je vědecky platná zkouška KeratinoSensTM (9) (12) (13) (14). Jsou k dispozici standardní pracovní postupy pro zkoušku KeratinoSensTM a měly by být používány pro provádění a využívání této zkušební metody v laboratoři (15). Laboratoře, jež chtějí tuto zkušební metodu provádět, mohou získat rekombinantní buněčnou linii použitou ve zkoušce KeratinoSensTM po uzavření licenční dohody se subjektem, který tuto zkušební metodu vyvinul. V následujících odstavcích je uveden popis hlavních složek a postupů zkušební metody se stanovením luciferázy ARE-Nrf2. Příprava kultur keratinocytů Měla by být použita transgenní buněčná linie, která má stabilní inzerci reporterového genu luciferázy řízeného elementem ARE (např. buněčná linie KeratinoSens™). Buňky se po obdržení pomnoží (např. 2 až 4 pasáže) a skladují se zmrazené jako homogenní kmen. Buňky z tohoto původního kmene se mohou pomnožit až do maximálního počtu pasáží (tj. 25 v případě KeratinoSensTM) a použijí se pro rutinní zkoušení s použitím vhodného udržovacího média (v případě KeratinoSensTM se jedná o DMEM (Dulbeccova modifikace Eagleova média, obsahující sérum a geneticin). Buňky by pro zkoušení měly být z 80 % až 90 % konfluentní a je třeba dbát na to, aby nikdy nevyrostly do úplné konfluence. Jeden den před zkoušením se buňky odeberou a rozdělí na destičky o 96 jamkách (10 000 buněk na každou jamku v případě KeratinoSensTM). Je třeba se vyvarovat sedimentace buněk během nasazování, aby se zajistila distribuce homogenního počtu buněk do jamek. Pokud by se tak nestalo, může z tohoto kroku vzejít vysoká variabilita mezi jednotlivými jamkami. V každém opakování se použijí tři repliky pro měření aktivity luciferázy a jedna souběžná replika se použije pro zkoušku životaschopnosti buněk. Příprava zkoušené chemické látky a kontrolních látek Zkoušená chemická látka a kontrolní látky se připraví v den zkoušení. Při zkoušce KeratinoSensTM se zkoušené chemické látky rozpustí v dimethylsulfoxidu (DMSO) na žádoucí konečnou koncentraci (např. 200 mM). Roztoky v DMSO lze považovat za samosterilizující, takže sterilní filtrace není nutná. Chemická látka, která není rozpustná v DMSO, se rozpustí v sterilní vodě nebo v kultivačním médiu a roztoky se sterilizují např. filtrací. V případě zkoušené chemické látky, která nemá definovanou molekulovou hmotnost (MW) se připraví zásobní roztok v standardní koncentraci (40 mg/ml nebo 4 % (m/V)) používané ve zkoušce KeratinoSensTM. V případě použití jiných rozpouštědel než DMSO, vody nebo kultivačního média by mělo být uvedeno dostatečné vědecké zdůvodnění. Na základě zásobních roztoků zkoušené chemické látky v DMSO se provádí sériové ředění pomocí DMSO k získání 12 základních koncentrací chemické látky, která má být zkoušena (od 0,098 do 200 mM ve zkoušce KeratinoSensTM). U chemických látek nerozpustných v DMSO se k získání základních koncentrací provádí ředění sterilní vodou nebo sterilním kultivačním médiem. Nezávisle na použitém rozpouštědle se poté základní koncentrace dále naředí v poměru 1:25 v kultivačním médiu obsahujícím sérum, a nakonec se použije k expozici buněk po dalším zředění ředicím faktorem 4, takže konečné koncentrace zkoušené chemické látky při zkoušce KeratinoSensTM jsou v rozsahu od 0,98 do 2 000 μM. V odůvodněných případech lze použít jiné koncentrace (např. v případě cytotoxicity nebo špatné rozpustnosti). Pro negativní kontrolu (s rozpouštědlem) se ve zkoušce KeratinoSensTM používá DMSO (č. CAS 67-68-5, v ≥ 99 % čistotě), pro tuto kontrolu se připraví šest jamek na každé destičce. Provede se stejné zředění, jak je popsáno u základních koncentrací v odstavci 22, takže konečná koncentrace negativní kontroly (s rozpouštědlem) je 1 %, což neovlivní životaschopnost buněk a odpovídá to stejné koncentraci DMSO, která je použita u zkoušené chemické látky a v pozitivní kontrole. U zkoušené chemické látky nerozpustné v DMSO, u které se ředění prováděla ve vodě, musí být úroveň DMSO ve všech jamkách konečného zkušebního roztoku upravena na 1 %, tj. na stejnou úroveň, která byla použita u ostatních zkoušených chemických látek a u kontrolních látek. Pozitivní kontrolou, která se používá v případě zkoušky KeratinoSensTM je cinnamaldehyd (č. CAS 14371-10-9, o čistotě ≥ 98 %), pro který se připraví řada základních koncentrací v rozsahu od 0,4 do 6,4 mM v DMSO (ze zásobního roztoku 6,4 mM) a rozpustí se tak, jak je uvedeno u základních koncentrací v odstavci 22, takže konečná koncentrace pozitivní kontroly je v rozsahu 4 až 64 μM. Lze použít jiné vhodné pozitivní kontroly, nejlépe poskytující hodnoty EC1,5 ve střední části rozsahu, pokud jsou k dispozici historické údaje k odvození srovnatelných kritérií přijatelnosti provedení. Aplikace zkoušené chemické látky a kontrolních látek U každé zkoušené chemické látky a pozitivní kontrolní látky je pro předpověď účinku (pozitivní nebo negativní) postačující jeden experiment, který sestává nejméně ze dvou nezávislých opakování, každé s třemi replikami (tj. n = 6). V případě neshodných výsledků mezi dvěma nezávislými opakováními by se mělo provést třetí opakování se třemi replikami (tj. n=9). Každé nezávislé opakování se provádí v různý den s čerstvým zásobním roztokem zkoušených chemických látek a s nezávisle sklizenými buňkami. Buňky však mohou pocházet ze stejné pasáže. Po nasazení buněk, jak je popsáno v odstavci 20, se buňky kultivují 24 hodin na mikrotitračních destičkách s 96 jamkami. Médium se poté odstraní a nahradí čerstvým kultivačním médiem (v případě zkoušky KeratinoSensTM je to 150 μl kultivačního média obsahujícího sérum, ale bez geneticinu, do něhož se přidá 50 μl 25násobně rozředěné zkoušené chemické látky a kontrolních látek. Nejméně jedna jamka na každé destičce by se měla ponechat prázdná (bez buněk a bez expozice), aby bylo možné posoudit hodnoty pozadí. Exponované destičky se poté ve zkoušce KeratinoSensTM inkubují asi 48 hodin při teplotě 37 ± 1 °C za přítomnosti 5 % CO2. Mělo by se dbát na to, aby nedošlo k vypařování těkavých zkoušených chemických látek a ke vzájemné kontaminaci zkoušenou chemickou látkou mezi jamkami, např. tak, že se destičky před inkubací se zkoušenými chemickými látkami přikryjí fólií. Měření aktivity luciferázy K zajištění odpovídajících údajů o luminiscenci jsou kriticky důležité tři faktory:
Před vlastním zkoušením by měl být proveden kontrolní experiment, jak je popsáno v dodatku 3, aby se zajistilo, že tyto tři body budou splněny. Po 48hodinové době expozice zkoušené chemické látce a kontrolním látkám ve zkoušce KeratinoSensTM se buňky opláchnou fosfátovým pufrem a do každé jamky se přidá na 20 minut za pokojové teploty příslušný lýzový pufr pro získání údajů o luminiscenci. Destičky s buněčným lyzátem se poté vloží do luminometru pro získání údajů, což je ve zkoušce KeratinoSensTM naprogramováno tak, že se i) přidá substrát pro luciferázu do každé jamky (tj. 50 μl), ii) počká 1 sekundu a iii) integruje aktivita luciferázy po 2 sekundy. V případě, že se použije jiné nastavení, např. v závislosti na použitém modelu luminometru, mělo by to být odůvodněno. Je možné rovněž použít luminiscenční substrát za předpokladu, že byl úspěšně splněn pokus na kontrolu kvality uvedený v dodatku 3. Posouzení cytotoxicity Při zkoušce životaschopnosti buněk KeratinoSensTM se médium po 48hodinové expoziční době nahradí čerstvým médiem obsahujícím MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-difenyltetrazolium-bromid, thiazolylová modř č. CAS 298-93-1) a buňky se inkubují 4 hodiny při teplotě 37 °C za přítomnosti 5 % CO2. Médium s MTT se poté odstraní a buňky se přes noc lýzují (např. přidáním 10 % roztoku SDS do každé jamky). Po protřepání se změří absorpce při 600 nm pomocí fotometru. ÚDAJE A PŘEDKLÁDÁNÍ ZPRÁV Vyhodnocení údajů Ve zkoušce KeratinoSensTM se vypočítávají tyto parametry:
Rovnice 1:
kde
EC1,5 se vypočítá lineární interpolací podle rovnice2 a celková hodnota EC1,5 se vypočítá jako geometrický průměr z jednotlivých opakování. Rovnice 2:
kde
Životaschopnost se vypočítá podle rovnice 3: Rovnice 3:
kde
IC50 a IC30 se vypočítají lineární interpolací podle rovnice 4 a celkové hodnoty IC50 a IC30 se vypočítají jako geometrický průměr z jednotlivých opakování. Rovnice 4:
kde
U každé koncentrace vykazující větší než 1,5násobnou indukci aktivity luciferázy se vypočte statistická významnost (např. pomocí dvouvýběrového Studentova t-testu), přičemž se hodnoty luminiscence u tří replicitních vzorků porovnají s hodnotami luminiscence zjištěné v jamkách (negativní) kontroly s rozpouštědlem, což umožní stanovit, zda je indukce aktivity luciferázy statisticky významná (p < 0,05). Nejnižší koncentrace s větší než 1,5násobnou indukcí aktivity luciferázy je hodnotou určující hodnotu EC1,5. V každém případě se zkontroluje, zda je tato hodnota nižší než hodnota IC30, což nasvědčuje tomu, že při koncentraci určující hodnotu EC1,5 je menší než 30 % snížení životaschopnosti buněk. Doporučuje se tyto údaje zkontrolovat vizuálně s použitím grafů. Není-li zjištěna jasná křivka dávka-účinek nebo pokud je získaná křivka dávka-účinek dvoufázová (tj. protíná prahovou hodnotu 1,5 dvakrát), měl by být experiment opakován, aby se ověřilo, zda je to specifické pro danou chemickou látku nebo zda je to důsledkem experimentálního artefaktu. V případě, že dvoufázová odezva je reprodukovatelná v nezávislém experimentu, měla by být zaznamenána nižší hodnota EC1,5 (koncentrace, kdy je prahová hodnota 1,5 protnuta poprvé). V ojedinělých případech, kdy je zjištěna statisticky nevýznamná indukce nad 1,5násobnou hodnotou, po níž následuje vyšší koncentrace se statisticky významnou indukcí, se výsledky získané z tohoto opakování považují za platné a pozitivní pouze v případě, že při koncentraci, jež není cytotoxická, byla získána statisticky významná indukce vyšší než prahová hodnota 1,5). A konečně, u chemických látek, jež vyvolávají 1,5násobnou nebo vyšší indukci při nejnižší koncentraci 0,98 μM, se na základě vizuální kontroly křivky dávka-účinek stanoví hodnota EC1,5 < 0,98. Kritéria přijatelnosti Při použití zkoušky KeratinoSensTM by měla být splněna tato kritéria přijatelnosti: Zaprvé, indukce aktivity luciferázy získaná s pozitivní kontrolou, cinnamaldehydem, by měla být statisticky významná nad prahovou hodnotou 1,5 (tj. s použitím T-testu) alespoň při jedné ze zkoušených koncentrací (od 4 do 64 μM). Zadruhé, hodnota EC1,5 by měla ležet mezi dvěma směrodatnými odchylkami od historické střední hodnoty zkušebního zařízení (např. mezi 7 μM a 30 μM na základě souboru údajů použitého pro validaci), která by měla být pravidelně aktualizována. Dále, průměrná indukce ve třech replikách s cinnamaldehydem při koncentraci 64 μM by měla být mezi 2 a 8. Není-li toto posledně uvedené kritérium splněno, měla by se pečlivě zkontrolovat křivka dávka-účinek cinnamaldehydu a zkoušky lze přijmout pouze v případě, že existuje jednoznačný vztah dávka-účinek s rostoucí indukcí aktivity luciferázy při rostoucích koncentracích v pozitivní kontrole. Průměrný variační koeficient u naměřených údajů luminiscence v negativní kontrole (s rozpouštědlem) s DMSO by pak měl být pod hodnotou 20 % při každém opakování, které se skládá z 6 jamek testovaných třikrát. Je-li variabilita vyšší, výsledky se nepoužijí. Interpretace výsledků a predikční model Předpověď získaná zkouškou KeratinoSensTM se považuje za pozitivní, jsou-li splněny všechny 4 následující podmínky ve 2 ze 2 opakování nebo v týchž 2 ze 3 opakování, jinak se předpověď získaná zkouškou KeratinoSensTM považuje za negativní (schéma č. 1):
Pokud v daném opakování jsou všechny tři první podmínky splněny, avšak nelze zjistit jasný vztah dávka-účinek při indukci luciferázy, pak by výsledek tohoto opakování měl být považován za nepřesvědčivý a může být nutné další zkoušení (schéma č. 1). Rovněž tak negativní výsledek získaný s koncentracemi < 1 000 μM (nebo < 200 μg/ml v případě zkoušených chemických látek bez definované molekulové hmotnosti) by také měl být považován za nepřesvědčivý (viz odstavec 11). Schéma č. 1 Predikční model používaný při zkoušce KeratinoSensTM. Předpověď KeratinoSensTM by měla být hodnocena v rámci přístupu IATA a v souladu s ustanoveními odstavců 9 a 11.
V ojedinělých případech zkoušené chemické látky, jež indukují aktivitu luciferázy na úrovni velmi blízké cytotoxickým úrovním, mohou být při některých opakováních pozitivní na necytotoxických úrovních (tj. při koncentraci určující hodnotu EC1,5 menší než (<) koncentrace IC30), a při jiných opakováních mohou být pozitivní pouze na cytotoxických úrovních (tj. při koncentraci určující hodnotu EC1,5 větší než (>) koncentrace IC30). Takové zkoušené chemické látky musí být přezkoušeny s užší analýzou vztahu dávka-účinek a za použití nižšího ředicího faktoru (např. zředění 1:1,33 nebo √2 (= 1,41) mezi jamkami), aby bylo možné stanovit, zda došlo k indukci na cytotoxických úrovních, či nikoli (9). Závěrečná zpráva Závěrečná zpráva by měla obsahovat tyto informace:
LITERATURA
Dodatek 1 DEFINICE Přesnost : Přesnost shody mezi výsledky zkušební metody a přijatými referenčními hodnotami. Je to měření výsledku zkušební metody a jednoho aspektu „důležitosti“. Tento výraz se často používá namísto výrazu „soulad“ a rozumí se jím podíl správných výsledků zkušební metody (29). AOP (mechanismy nežádoucích účinků) : Sled událostí od chemické struktury cílové chemické látky nebo skupiny podobných chemických látek přes molekulární iniciační událost po účinek in vivo, který je předmětem zájmu (2). ARE : Antioxidační responzivní element (též nazývaný EpRE, elektrofilní responzivní element), je responzivní element, který se nachází v oblasti protisměrných promotorů mnohých cytoprotektivních genů a genů kódujících enzymy II. fáze. Je-li aktivován prostřednictvím transkripčního faktoru Nfr2, zprostředkovává transkripční indukci těchto genů. Chemická látka : Chemická substance nebo směs. Variační koeficient : Míra variability, která se vypočítává pro skupinu údajů z replik tak, že se směrodatná odchylka vydělí průměrem. Lze ho vynásobit 100 za účelem vyjádření v procentech. EC1,5 : Interpolovaná koncentrace pro získání 1,5násobné indukce luciferázy. IC30 : Koncentrace vyvolávající snížení životaschopnosti buněk o 30 %. IC50 : Koncentrace vyvolávající snížení životaschopnosti buněk o 50 %. Nebezpečnost : Základní charakteristika látky nebo situace, která má potenciál vyvolat nepříznivé účinky, jsou-li organismus, systém nebo (sub)populace vystaveny této látce. IATA (integrovaný přístup ke zkoušení a posuzování) : Strukturovaný přístup používaný pro zjišťování nebezpečnosti (potenciál), charakterizaci rizik (potence) a/nebo posouzení bezpečnosti (potenciál/potence a expozice) chemické látky nebo skupiny chemických látek, jenž strategicky integruje a váží všechny důležité údaje s cílem poskytnout informace pro přijetí regulačního rozhodnutí, které se týká potenciální nebezpečnosti a/nebo rizika a/nebo potřeby dalšího cíleného, a tudíž minimálního zkoušení. Imax : Faktor maximální indukce aktivity luciferázy ve srovnání s (negativní) kontrolou s rozpouštědlem naměřený při jakékoli koncentraci zkoušené chemické látky. Keap1 : (Kelch-like ECH-associated protein 1) je senzorový protein, který dokáže regulovat aktivitu transkripčního faktoru Nrf2. Za neindukovaných podmínek senzorový protein Keap1 směruje transkripční faktor Nrf2 na ubikvitinylaci a proteolytickou degradaci v proteazomu. Kovalentní modifikace reaktivních reziduí cysteinu v Keap 1 prostřednictvím malých molekul může vést k disociaci Nrf2 z Keap1 (8) (10) (11). Směs : Směs nebo roztok tvořený dvěma nebo více látkami, které v něm nereagují (1). Jednosložková látka : Látka definovaná svým kvantitativním složením, v níž je přítomna jedna hlavní složka v míře alespoň 80 % hmot. Vícesložková látka : Látka definovaná svým kvantitativním složením, v níž je přítomna více než jedna hlavní složka v koncentraci ≥ 10 % hmot. a < 80 % hmot. Vícesložková látka je výsledkem výrobního procesu. Rozdíl mezi směsí a vícesložkovou látkou spočívá v tom, že směs se získá smísením dvou nebo více látek bez chemické reakce. Vícesložková látka je výsledkem chemické reakce. Nrf2 : Nuclear factor (erythroid-derived 2)-like 2, je transkripční faktor zapojený do mechanismu antioxidační odpovědi. Není-li Nrf2 ubikvitinylován, hromadí se se v cytoplasmě a přesouvá se do jádra, kde se spojuje s ARE v oblasti protisměrných promotorů mnohých cytoprotektivních genů a vyvolává jejich transkripci (8) (10) (11). Pozitivní kontrola : Replika obsahující všechny složky zkušebního systému a látku, o níž je známo, že způsobuje pozitivní reakci. Aby se zajistilo, že proměnlivost v reakci na pozitivní kontrolu bude možné časem vyhodnotit, rozsah pozitivní reakce by neměl být nadměrný. Relevantnost : Popis vztahu zkoušky k účinku, který je předmětem zájmu, a také toho, zda je zkouška smysluplná a použitelná pro konkrétní účel. Vyjadřuje, do jaké míry zkouška správně měří nebo predikuje biologický účinek, který je předmětem zájmu. Relevantnost zahrnuje také posouzení přesnosti (shody) zkušební metody (29). Spolehlivost : Měření rozsahu, v jakém může být zkušební metoda časem reprodukovatelná v laboratořích a mezi laboratořemi, když se provádí s použitím stejného protokolu. Hodnotí se to výpočtem laboratorní a mezilaboratorní reprodukovatelnosti a laboratorní opakovatelnosti. Hodnotí se na základě výpočtu laboratorní a mezilaboratorní reprodukovatelnosti a laboratorní opakovatelnosti (29). Reprodukovatelnost : Shoda mezi výsledky získanými při zkoušení téže chemické látky za použití stejného zkušebního protokolu (viz spolehlivost) (29). Citlivost : Podíl všech pozitivních/aktivních chemických látek, které jsou na základě zkušební metody správně zatříděny. Je měřítkem přesnosti zkušební metody, která poskytuje kategorické výsledky, a je důležitým prvkem při hodnocení relevantnosti zkušební metody (29). Kontrola s rozpouštědlem/vehikulem : Replika obsahující všechny složky zkušebního systému s výjimkou zkoušené chemické látky, ale včetně rozpouštědla, které se používá. Používá se ke stanovení účinku na pozadí u vzorků exponovaných zkoušené chemické látce, která je rozpuštěna ve stejném rozpouštědle. Specifičnost : Podíl všech negativních/neaktivních chemických látek, které jsou na základě zkušební metody správně zatříděny. Je měřítkem přesnosti zkušební metody, která poskytuje kategorické výsledky, a je důležitým prvkem při hodnocení relevantnosti zkušební metody (29). Látka : Chemické prvky nebo jejich sloučeniny v přírodním stavu nebo získané výrobním procesem včetně všech přídatných látek nutných k uchování stability výrobku a všech nečistot vznikajících v použitém procesu, ale s vyloučením všech rozpouštědel, která je možno oddělit bez ovlivnění stability látky nebo změny jejího složení (1). Zkoušená chemická látka : Termín „zkoušená chemická látka“ se používá k označení toho, co se podrobuje zkoušení. Globálně harmonizovaný systém klasifikace a označování chemických látek Organizace spojených národů (GHS OSN) : Systém navrhující klasifikaci látek a směsí podle standardizovaných typů a úrovní fyzikálních, zdravotních a environmentálních nebezpečí a řešící příslušné komunikační prvky, jako jsou piktogramy, signální slova, výroky o nebezpečí, výroky o bezpečnostních opatřeních a bezpečnostní datové listy, které obsahují informace o jejich nežádoucích účincích s ohledem na ochranu lidí (včetně zaměstnavatelů, dělníků, přepravců, spotřebitelů a osob reagujících na havárie) a životního prostředí (1). UVCB : Látky s neznámým nebo proměnlivým složením, komplexní reakční produkty nebo biologický materiál. Validovaná zkušební metoda : Zkušební metoda, u níž se má za to, že je dostatečně relevantní a spolehlivá pro určitý účel, a která je založena na vědecky spolehlivých zásadách. Zkušební metoda není nikdy validovaná v absolutním smyslu, ale pouze pro určitý konkrétní účel (29). Dodatek 2 LÁTKY PRO PROKÁZÁNÍ ZPŮSOBILOSTI Senzibilizace kůže in vitro: zkušební metoda se stanovením luciferázy ARE-Nrf2 Dříve než začnou laboratoře běžně používat tuto zkušební metodu, měly by prokázat odbornou způsobilost správným získáním předpokládané předpovědi KeratinoSensTM u 10 vhodných látek doporučených v tabulce 1 a získáním hodnot EC1,5 a IC50, které spadají do příslušného referenčního rozpětí, u nejméně 8 z 10 vhodných látek. Tyto látky pro prokázání způsobilosti byly vybrány tak, aby představovaly rozpětí účinků na nebezpečí senzibilizace kůže. Jinými kritérii výběru bylo, aby byly komerčně dostupné, aby k nim byly k dispozici vysoce kvalitní referenční údaje získané in vivo a vysoce kvalitní údaje získané in vitro zkouškou KeratinoSensTM. Tabulka 1 Doporučené látky pro prokázání odborné způsobilosti k provádění zkoušky KeratinoSens™.
Dodatek 3 KONTROLA KVALITY MĚŘENÍ LUMINISCENCE Základní experiment k zajištění optimálních měření luminiscence při zkoušce KeratinoSensTM Pro zajištění spolehlivých výsledků při měření luminometrem jsou kriticky důležité tyto tři parametry:
Před zkoušením se doporučuje zajistit odpovídající měření luminiscence pomocí testování kontrolní destičky s níže uvedeným uspořádáním (trojitá analýza).: Uspořádání destičky při prvním přípravném pokusu
EGDMA= ethylenglykol-dimethakrylát (č. CAS: 97-90-5), chemická látka vyvolávající silnou indukci CA= cinnamaldehyd (č. CAS: 104-55-2), pozitivní referenční látka Analýza kontroly kvality by měla prokázat:
B.61 Zkušební metoda průniku fluoresceinu ke zjišťování látek s leptavými a silně dráždivými účinky na oči ÚVOD Tato zkušební metoda odpovídá Pokynu OECD pro zkoušení (TG) č. 460 (2012). Zkušební metoda průniku fluoresceinu (Fluorescein Leakage – FL) je zkušební metoda in vitro, kterou lze používat za určitých podmínek a se specifickými omezeními ke klasifikaci chemických látek (látek a směsí) jako látek s leptavými účinky na oči a silně dráždivých látek, jak je definuje Globálně harmonizovaný systém klasifikace a označování chemických látek (GHS) Organizace spojených národů (OSN), nařízení (ES) č. 1272/2008 o klasifikaci, označování a balení látek a směsí (CLP) (31) (kategorie 1) a Agentura pro ochranu životního prostředí (EPA) USA (kategorie I) (1) (2). Pro účely této zkušební metody jsou látky se silně dráždivými účinky na oči definovány jako chemické látky, které způsobují poškození oční tkáně po podání zkoušené chemické látky, které není vratné do 21 dní, nebo způsobují vážné fyzické zhoršení vidění, a látky s leptavými účinky na oči jsou chemické látky, které způsobují nevratné poškození oční tkáně. Tyto látky jsou klasifikovány v kategorii 1 GHS OSN, v kategorii 1 CLP EU nebo v kategorii I EPA USA. Zkušební metoda FL není považována za dostatečnou pro úplné nahrazení oční zkoušky in vivo na králících, avšak zkoušku FL se doporučuje používat v rámci strategie odstupňovaného zkoušení pro účely klasifikace a označování v právních předpisech. Zkušební metodu FL se tak doporučuje použít jako úvodní krok v rámci přístupu „shora dolů“ ke zjišťování látek s leptavými / silně dráždivými účinky na oči, a zvláště pak ke zkoušení určitých druhů chemických látek (tj. látek a směsí rozpustných ve vodě) (3) (4). V současnosti se všeobecně uznává, že jediná zkouška dráždivých účinků na oči in vitro nemůže v dohledné budoucnosti nahradit oční zkoušku in vivo (zkušební metoda B.5, OECD (5)), pokud jde o předpověď v celé škále dráždivosti různých tříd chemických látek. Oční zkoušku in vivo však možná bude možné nahradit strategickou kombinací několika alternativních zkušebních metod v rámci strategie (odstupňovaného) zkoušení. Postup „shora dolů“ je určen pro případy, kdy se na základě existujících informací předpokládá, že chemická látka má vysokou schopnost vyvolat dráždivé účinky. Na základě predikčního modelu, který je podrobně popsán v odstavci 35, může zkušební metoda FL identifikovat chemické látky v omezené oblasti použitelnosti jako látky s leptavými / silně dráždivými účinky na oči (kategorie 1 GHS OSN; kategorie 1 CLP EU; kategorie I EPA USA) bez jakéhokoli dalšího zkoušení. Totéž se předpokládá u směsí, ačkoli směsi nebyly při validaci použity. Při použití strategie postupného zkoušení uvedené ve zkušební metodě B.5 (5) lze zkušební metodu FL použít ke stanovení dráždivých/leptavých účinků chemických látek na oči. Chemickou látku, o níž se na základě použití metody FL nepředpokládá, že je látkou s leptavými / silně dráždivými účinky na oči, by však bylo třeba zkoušet jednou nebo několika dalšími zkušebními metodami (in vitro a/nebo in vivo), které jsou schopny přesně identifikovat i) chemické látky, které ve zkoušce FL in vitro ke zjištění látek s leptavými / silně dráždivými účinky na oč (kategorie 1 GHS OSN; kategorie 1 CLP EU; kategorie I EPA USA) vykazují falešné negativní výsledky; ii) chemické látky, které nejsou klasifikovány jako látky s leptavými/dráždivými účinky na oči (bez kategorie GHS EOSN; bez kategorie CLP EU; kategorie IV EPA USA); a/nebo iii) chemické látky se středně silnými / slabými dráždivými účinky na oči (kategorie 2A a 2B GHS OSN; kategorie 2 CLP EU; kategorie II a III EPA USA). Účelem této zkušební metody je popsat postupy používané k hodnocení potenciálních leptavých nebo silně dráždivých účinků zkoušené chemické látky na oči vyjádřených jako její schopnost poškozovat nepropustné konfluentní monovrstvy buněk epitelu. Integrita transepiteliální propustnosti je důležitou funkcí epitelu, např. epitelu ve spojivkách a v rohovce. Transepiteliální propustnost je řízena různými těsnými spoji. Bylo prokázáno, že zvýšení propustnosti epitelu rohovky in vivo koreluje s mírou zánětu a poškození povrchu, jež jsou zaznamenávány s postupujícím podrážděním oka. Při zkušební metodě FL se toxické účinky po krátké expozici zkoušené chemické látce měří pomocí zvýšení propustnosti fluorosceinu sodného přes epiteliální monovrstvu Madin-Darbyho psích ledvinových (MDCK, Madin-Darby Canine Kidney) buňek kultivovaných na propustných vložkách. Výše úniku fluoresceinu, k němuž dojde, je přímo úměrná poškození těsných spojů, desmosomálních spojů a buněčných membrán, jež způsobila chemická látka, a lze ji použít k odhadu potenciálu oční toxicity zkoušené chemické látky. Schéma buněk MDCK kultivovaných na membráně vložky pro zkušební metodu FL je uvedeno v dodatku 1. Definice jsou uvedeny v dodatku 2. VÝCHOZÍ ÚVAHY A OMEZENÍ Tato zkušební metoda je založena INVITTOX protokolu č. 71 (6), který byl hodnocen v mezinárodní validační studii Evropským střediskem pro validaci alternativních metod (ECVAM) ve spolupráci s Koordinačním výborem mezi různými organizacemi pro ověřování platnosti alternativních metod (ICCVAM) USA a s Japonským střediskem pro validaci alternativních metod (JaCVAM). Zkušební metoda FL se nedoporučuje ke zjišťování chemických látek, které by měly být klasifikovány jako látky se slabými / středně silnými dráždivými účinky, nebo chemických látek (látek a směsi), které by neměly být klasifikovány jako látky s dráždivými účinky na oči (tj. kategorie 2A/2B nebo bez kategorie GHS OSN; kategorie 2 nebo bez kategorie CLP EU; kategorie II/III/IV EPA USA), jak prokázala validační studie (3) (7). Zkušební metodu lze používat pouze ke zkoušení chemických látek (látek a směsí) rozpustných ve vodě. Zkušební metoda FL obvykle přesně předpovídá potenciál chemických látek mít silně dráždivé účinky na oči, pokud jsou tyto chemické látky rozpustné ve vodě a/nebo pokud není toxický účinek ovlivněn zředěním (7). Pro určení chemické látky jako rozpustné ve vodě by za zkušebních podmínek měla být rozpustná ve sterilním Hanksově vyváženém solném roztoku (HBSS) při koncentrací ≥ 250 mg/ml (jedna dávka nad mezní hodnotou 100 mg/ml) s obsahem vápníku (v koncentraci 1,0–1,8 mM), bez fenolové červeně. Je-li však zkoušená chemická látka rozpustná při koncentraci nižší než 100 mg/ml, ale při této koncentraci již vyvolává 20 % indukci FL (což znamená, že FL20 < 100 mg/ml), lze ji přesto klasifikovat v kategorii 1 GHS nebo kategorii I EPA. Omezení zjištěná u této zkušební metody vylučují z oblasti její použitelnosti silné kyseliny a zásady, buněčná fixativa a vysoce těkavé chemické látky. Tyto chemické látky mají mechanismy, které zkušební metoda FL neměří, např. vysokou koagulaci, saponifikaci nebo specifické reaktivní chemismy. Jiná zjištěná omezení této metody vyplývají z výsledků její prediktivní schopnosti, pokud jde o zbarvené a viskózní chemické látky. Poukazuje se na to, že oba tyto druhy chemických látek je obtížné z monovrstvy odstranit po krátké době expozice a že kvalitu předpovědí této zkušební metody by mohlo zlepšit, pokud by bylo použito více oplachovacích kroků. Pevné chemické látky rozpuštěné v kapalině mohou vést k tvorbě sraženin a může být obtížné stanovit konečnou koncentraci. Jsou-li látky v rámci těchto chemických a fyzikálních tříd vyřazeny z databáze, přesnost zkušební metody FL v klasifikačních systémech EU, EPA a GHS je podstatně vyšší (7). Vzhledem k účelu této zkušební metody (tj. zjišťovat pouze látky s leptavými / silně dráždivými účinky na oči) není míra falešné negativity (viz odstavec 13) kriticky důležitá, protože takové chemické látky by byly následně zkoušeny jinými náležitě validovanými zkouškami in vitro nebo zkouškou na králících, v závislosti na požadavcích právních předpisů, s využitím strategie postupného zkoušení v rámci přístupu založeného na posuzování váhy důkazů (5) (viz též odstavce 3 a 4). Další zjištěná omezení zkušební metody FL vyplývají z míry falešné negativity a falešné pozitivity. Při použití zkušební metody FL jako počátečního kroku v rámci přístupu „shora dolů“ ke zjištění ve vodě rozpustných látek a směsí s leptavými / silně dráždivými účinky na oči (kategorie 1 GHS OSN; kategorie 1 CLP EU; kategorie I EPA USA), se míra falešné pozitivity zkušební metody FL pohybovala od 7 % (7/103; GHS OSN a CLP EU) do 9 % (9/99; EPA USA) a míra falešné negativity se pohybovala od 54 % (15/28; EPA USA) do 56 % (27/48; GHS OSN a CLP EU) ve srovnání s výsledky získanými in vivo. Skupiny chemických látek, které ve zkušební metodě FL vykazují falešně pozitivní nebo falešně negativní výsledky, zde nejsou definovány. Určitá technická omezení jsou specifická pro kulturu buněk MDCK. Těsné spoje, které blokují průchod barviva fluoresceinu sodného monovsrstvou, jsou s rostoucím počtem buněčných pasáží stále více narušovány. Neúplná tvorba těsných spojů vede ke zvýšenému úniku fluoresceinu u neexponovaných kontrol. Proto je důležité stanovit maximální přípustný průnik v neexponovaných kontrolách (viz odstavec 38: 0 % průnik). Stejně jako u všech zkoušek in vitro se buňky mohou v průběhu času transformovat, a je tedy nezbytné, aby byla pro tyto zkoušky stanovena rozpětí pro počet pasáží. Stávající oblast použitelnosti zkušební metody by bylo možné v některých případech rozšířit, avšak až po analýze rozšířeného souboru údajů o zkoumaných zkoušených chemických látkách, přednostně získaného prostřednictvím jejich zkoušení (3). Tato zkušební metoda se bude podle potřeby aktualizovat, tak jak budou zohledňovány nové informace a údaje. Každá laboratoř, která poprvé provádí tuto analýzu, by měla použít referenční chemické látky uvedené v dodatku 3. Laboratoře mohou použít tyto chemické látky k prokázání své odborné způsobilosti k provádění zkušební metody FL před předložením výsledků získaných zkušební metodou FL pro regulační účely klasifikace nebezpečnosti. PODSTATA ZKOUŠKY Zkušební metoda FL je zkouška in vitro založená na cytotoxicitě a funkci buněk, která se provádí na konfluentní monovrstvě tubulárních epiteliálních buněk MDCK CB997, které se kultivují na polopropustných vložkách a modelují neproliferující stav epitelu rohovky in vivo. Buněčná linie MDCK je dobře zavedená a tvoří těsné spoje a desmosomální spoje podobné těm, které se nacházejí na apikální straně epitelu spojivek a rohovky. Těsné a desmosomální spoje in vivo zabraňují pronikání rozpuštěných látek a cizích materiálů do epitelu rohovky. Ztráta transepiteliální nepropustnosti v důsledku poškození těsných spojů a desmosomálních spojů je jedním z časných jevů při podrážděních oka vyvolaných chemickou látkou. Zkoušená chemická látka se nanese na konfluentní vrstvu buněk kultivovaných na apikální straně vložky. K vyjádření obvyklé rychlosti clearance při expozici člověka se běžně používá krátká expozice v délce 1 minuty. Výhodou krátké expoziční doby je, že látky a směsi založené na vodě je možné zkoušet v čistém stavu, pokud se dají po ukončení expozice snadno odstranit. To umožňuje přímější srovnání výsledků s účinky chemických látek na lidi. Zkoušená chemická látka se poté odstraní a na apikální stranu monovrstvy se na dobu 30 minut nanese netoxické, vysoce fluorescenční barvivo fluorescein sodný. Poškození těsných spojů způsobené zkoušenou chemickou látkou se určuje množstvím fluoresceinu, které během stanoveného časového úseku unikne přes vrstvu buněk. Množství barviva fluoresceinu sodného, jež pronikne monovrstvou a membránou vložky do stanoveného objemu roztoku v jamce (do které barvivo fluorescein sodný uniká), se určí spektrofluorimetrickým měřením koncentrace fluoresceinu v jamce. Velikost úniku fluoresceinu (FL) se vypočítá podle údajů o intenzitě fluorescence naměřených ve dvou kontrolách: ve slepé kontrole a v kontrole s maximálním únikem. Únik vyjádřený v procentech, a tedy míra poškození těsných spojů, se vyjádří v poměru k těmto kontrolám, a to u každé ze stanovených koncentrací zkoušené chemické látky. Poté se vypočítá hodnota FL20 (tj. koncentrace, která způsobuje 20 % únik fluoresceinu v poměru k hodnotě zaznamenané v neexponované konfluentní monovrstvě a ve vložkách bez buněk). Hodnota FL20 (v mg/ml) se použije v predikčním modelu ke zjišťování látek s leptavými účinky na oči a silně dráždivých látek (viz odstavec 35). Důležitou součástí profilu toxicity zkoušené chemické látky je zotavení, které se rovněž posuzuje zkouškou dráždivých účinků na oči in vivo. Z předběžných analýz vyplývá, že údaje o zotavení (až do 72 hodin po expozici chemické látce) by potenciálně mohly zvýšit prediktivní schopnost INVITTOX protokolu č. 71, avšak je zapotřebí další hodnocení opírající se o doplňující údaje, přednostně získané dalším zkoušením (6). Tato zkušební metoda se bude podle potřeby aktualizovat, tak jak budou zohledňovány nové informace a údaje. POSTUP Příprava monovrstvy buněk Monovrstva buněk MDCK CB997 se připraví použitím sub-konfluentních buněk, které se kultivují v baňkách s buněčnou kulturou v roztoku DMEM/Nutrient Mix F12 (1x koncentrát s L-glutaminem, 15 mM HEPES, vápníkem (v koncentraci 1,0–1,8 mM) a10 % tepelně dezaktivovaným FCS/FBS). Důležité je, aby všechna média/roztoky používané v průběhu zkoušky FL obsahovaly vápník v koncentraci mezi 1,8 mM (200 mg/l) a 1,0 mM (111 mg/l), což zajistí tvorbu a pevnost těsných spojů. Mělo by se kontrolovat rozpětí počtu buněčných pasáží, aby se zajistila rovnoměrná a reprodukovatelná tvorba těsných spojů. Počet buněčných pasáží by nejlépe měl být v rozpětí 3 až 30 od rozmrazení, protože buňky v tomto rozpětí pasáží mají podobnou funkčnost, což napomáhá reprodukovatelnosti zkoušky. Před provedením zkušební metody FL se buňky vyjmou z baňky trypsinizací, odstředí se a přiměřené množství buněk se nasadí do vložek umístěných v destičkách s 24 jamkami (viz dodatek 1). K násadě buněk by se měly použít vložky o průměru 12 mm s membránou vyrobenou ze smíšených esterů celulosy, o tloušťce 80 až 150 μm a velikosti pórů 0,45 μm. Ve validační studii byly použity vložky Millicell-HA o průměru 12 mm. Vlastnosti vložky a druh membrány jsou důležité, neboť mohou ovlivnit růst buněk a vazbu na chemickou látku. Některé druhy chemických látek se mohou vázat na membránu vložky Millicell-HA, což by mohlo ovlivnit interpretaci výsledků. Jsou-li použity jiné membrány, měla by být prokázána jejich rovnocennost pomocí vhodných chemických látek (viz dodatek 3). Vazba chemické látky na membránu vložky je obvyklejší u chemických látek vytvářející kationty, jako je např. benzalkoniumchlorid, které jsou přitahovány k membráně s nábojem (7). Vazba chemické látky na membránu vložky může prodloužit dobu expozice chemickou látkou, což vede k nadhodnocení toxického potenciálu chemické látky, avšak může to rovněž fyzicky snížit únik fluoresceinu přes vložku v důsledku vazby barviva na chemickou látku tvořící kationty vázanou na membránu vložky, což vede k podhodnocení toxického potenciálu dané chemické látky. Tento aspekt lze snadno sledovat tak, že se samotná membrána vystaví nejvyšší koncentraci chemické látky, která se zkouší, a poté se přidá barvivo fluorescein sodný v normální koncentraci na standardní dobu (kontrola bez buněk). Vznikne-li vazba barviva fluoresceinu sodného, membrána vložky se po opláchnutí zkušebního materiálu zbarví žlutě. Pro interpretaci účinku chemické látky na buňky je tudíž důležité znát vazební vlastnosti zkoušené chemické látky. Nasazením buněk do vložek by měla vzniknout konfluentní monovrstva v době expozice chemické látce. Do každé vložky by mělo být vloženo 1,6 × 105 buněk (400 μl buněčné suspenze o hustotě 4 × 105 buněk/ml). Za těchto podmínek se po 96 hodinách obvykle vytvoří konfluentní jednovrstvá kultura. Před aplikací látek by se vložky měly vizuálně zkontrolovat a zaznamenat viditelná poškození, aby se zajistilo, která poškození zaznamenaná při vizuální kontrole, která je popsána v odstavci 30, byla způsobena manipulací. Buněčné kultury MDCK by měly být udržovány v inkubátorech ve zvlhčeném ovzduší, při 5 % ± 1 % CO2 a teplotě 37 ± 1 °C. Buňky by neměly být kontaminovány bakteriemi, viry, mykoplasmaty ani houbovými mikroorganismy. Aplikace zkoušených a kontrolních chemických látek Pro každé provedení pokusu by měl být připraven čerstvý zásobní roztok zkoušené chemické látky a použit do 30 minut od přípravy. Zkoušená chemická látka by se měla připravit v roztoku HBSS s obsahem vápníku (v koncentraci 1,0 až 1,8 mM), bez fenolové červeně, aby se zabránilo vazbě na proteiny v séru. Před zkoušením by se měla posoudit rozpustnost chemické látky v roztoku HBSS při koncentraci 250 mg/ml. Jestliže chemická látka při této koncentraci vytváří stabilní suspenzi nebo emulzi (tj. zachovává jednotnost a neusazuje se ani se nedělí na více než jednu fázi) po dobu 30 minut, může být HBSS přesto použito jako rozpouštědlo. Pokud se však zjistí, že chemická látka je za této koncentrace v HBSS nerozpustná, mělo by se zvážit použití jiných zkušebních metod než FL. Použití lehkého minerálního oleje jako rozpouštědla v případech, kdy se zjistí, že chemická látka není v HBSS rozpustná, by mělo být zvažováno s obezřetností, neboť není k dispozici dostatek údajů umožňujících učinit závěr o funkčnosti zkoušky FL za takových podmínek. Všechny chemické látky, které mají být zkoušeny, se připraví v roztoku HBSS s obsahem vápníku (v koncentraci 1,0 až 1,8 mM), bez fenolové červeně, pocházejícím ze zásobního roztoku, v pěti pevně stanovených koncentracích zředěných na bázi hmotnosti na jednotku objemu: 1, 25, 100, 250 mg/ml a čistý nebo nasycený roztok. Při zkoušení pevné chemické látky by měla být zařazena velmi vysoká koncentrace 750 mg/ml. Chemickou látku v této koncentraci možná bude nutné na buňky aplikovat pomocí objemové pipety. Pokud se zjistí toxicita při koncentraci mezi 25 a 100 mg/ml, měly by se následující koncentrace zkoušet dvakrát: 1, 25, 50, 75, 100 mg/ml. Hodnota FL20 by se měla odvodit z těchto koncentrací, za předpokladu, že byla splněna kritéria přijatelnosti zkoušky. Zkoušené chemické látky se aplikují na konfluentní buněčné monovrstvy po odstranění média pro kultivaci buněk a po dvojím opláchnutí sterilním, teplým (37 °C) roztokem HBSS s obsahem vápníku (v koncentraci 1,0 až 1,8 mM), bez fenolové červeně. Předtím byly vizuálně zkontrolovány filtry, zda nevykazují dříve vzniklá poškození, která by mohla být chybně přičtena potenciální neslučitelnosti se zkoušenými chemickými látkami. Měly by být použity nejméně tři repliky pro každou koncentraci zkoušené chemické látky a pro kontroly v každém provedení zkoušky. Po expozici o délce 1 minuty za pokojové teploty by měla být zkoušená chemická látka pečlivě odstraněna odsátím, monovrstva by se měla dvakrát opláchnout sterilním teplým (37 °C) roztokem HBSS s obsahem vápníku (v koncentraci 1,0 až 1,8 mM), bez fenolové červeně, a ihned by se měl změřit průnik fluoresceinu. Souběžné negativní a pozitivní kontroly je nutné používat pro každé provedení zkoušky, aby se prokázalo, že integrita monovrstev (v negativní kontrole) a citlivost buněk (v pozitivní kontrole) je v rámci definovaného dosavadního rozpětí přijatelnosti. Doporučenou chemickou látkou pro pozitivní kontrolu je Brij 35 (č. CAS 9002-92-0) v koncentraci 100 mg/ml. Tato koncentrace by měla vykázat přibližně 30 % průnik fluoresceinu (přijatelné rozpětí je 20 až 40 % průnik fluoresceinu, tj. poškození vrstvy buněk). Doporučenou chemickou látkou pro negativní kontrolu je roztok HBSS s obsahem vápníku (v koncentraci 1,0 až 1,8 mM), bez fenolové červeně (neexponovaná, slepá zkouška) V každém provedení zkoušky by měla být zařazena také kontrola s maximálním průnikem umožňující výpočet hodnot FL20. Maximální průnik se stanoví pomocí kontrolní vložky bez buněk. Stanovení propustnosti fluoresceinu Ihned po odstranění zkoušené a kontrolní chemické látky se do vložek (např. typu Milicell-HA) přidá 400μl roztoku fluoresceinu sodného o koncentraci 0,1 mg/ml (0,01 % (m/V) v HBSS s obsahem vápníku (v koncentraci 1,0 až 1,8 mM], bez fenolové červeně). Kultury se udržují 30 minut při pokojové teplotě. Na konci inkubace s fluoresceinem se z každé jamky opatrně vyjmou vložky. Provede se vizuální kontrola každého filtru a zaznamená se každé poškození, ke kterému mohlo dojít při manipulaci. V roztoku, který zbyl v jamkách po odstranění vložek, se vyčíslí množství fluoresceinu, který pronikl přes monovrstvu a podložku. Měření se provádějí spektrofluorimetrem při excitační vlnové délce 485 mm a emisní vlnové délce 530 mm. Citlivost spektrofluorimetru by měla být nastavena tak, aby byl co nejvyšší numerický rozdíl mezi maximálním FL (vložka bez buněk) a minimálním FL (vložka s konfluentní monovrstvou ošetřená negativní kontrolní látkou). Kvůli rozdílnosti použitého spektrofluorimetru se doporučuje použít takovou citlivost, která poskytne intenzitu fluorescence > 4 000 v kontrole s maximálním průnikem fluoresceinu. Maximální hodnota FL by neměla být větší než 9 999. Maximální intenzita průniku fluorosceinu by se měla nacházet v lineárním rozpětí použitého spektrofluorimetru. Interpretace výsledků a predikční model Velikost průniku fluoresceinu je úměrná poškození těsných spojů vyvolanému chemickou látkou. Procentuální hodnota průniku fluoresceinu pro každou zkoušenou koncentraci chemické látky se vypočítá z hodnot FL získaných pro zkoušenou chemickou látku ve srovnání s hodnotami FL získanými z negativní kontroly (údaje naměřené z konfluentní monovrstvy ošetřené negativní kontrolní látkou) a z kontroly s maximálním únikem (údaje naměřené z množství fluoresceinu, který pronikl přes vložku bez buněk). Střední hodnota intenzity fluorescence při maximálním průniku = x Střední hodnota intenzity fluorescence při 0 % průniku (v negativní kontrole) = y Střední hodnota 100 % průniku se získá odečtením střední hodnoty 0 % průniku od střední hodnoty maximálního průniku, tj. x – y = z Procentuální hodnota průniku pro každou pevně stanovenou dávku se získá odečtením 0 % průniku od střední hodnoty intenzity fluorescence získané měřeními tří replik (m) a vydělením této hodnoty 100 % průnikem tj. % FL = [(m-y) / z] × 100 %, kde:
Pro výpočet koncentrace chemické látky vyvolávající 20 % FL se použije tato rovnice: FLD = [(A-B) / (C-B)] × (MC – MB) + MB kde:
Níže je uvedena mezní hodnota FL20 pro předpověď chemických látek jako látek s leptavými / silně dráždivými účinky na oči:
Zkušební metoda FL se doporučuje pouze ke zjišťování ve vodě rozpustných látek s leptavými a silně dráždivými účinky na oči (kategorie 1 GHS OSN, kategorie 1 CLP EU, kategorie I EPA USA) (viz odstavce 1 a 10). Aby bylo možné identifikovat chemické látky (látky a směsi) rozpustné ve vodě (3) (6) (7) jako „látky vyvolávající závažné poškození očí“ (kategorie 1 GHS OSN / CLP EU) nebo jako „látky s leptavými nebo silně dráždivými účinky na oči“ (kategorie 1 EPA USA), měla by chemická látka indukovat hodnotu FL20 ≤ 100 mg/ml. Přijatelnost výsledků Střední hodnota při maximálním průniku fluorosceinu (x) by měla být vyšší než 4 000 (viz odstavec 31), střední hodnota při 0 % průniku (y) by měla být rovna nebo menší než 300 a střední hodnota při 100 % průniku (z) by se měla nacházet mezi 3 700 a 6 000. Zkouška se považuje za přijatelnou, pokud pozitivní kontrola vyvolala 20 % až 40 % poškození vrstvy buněk (vyjádřeno v % průniku fluoresceinu). ÚDAJE A PŘEDKLÁDÁNÍ ZPRÁV Údaje U každého provedení se uvedou ve formě tabulky údaje o jednotlivých replicitních jamkách (např. hodnoty intenzity fluorescence a vypočtené procentuální údaje o průniku fluoresceinu pro každou zkoušenou chemickou látku včetně její klasifikace). Dále by měly být uvedeny střední hodnoty ± směrodatné odchylky u jednotlivých měření replik při každém provedení zkoušky. Závěrečná zpráva Závěrečná zpráva by měla obsahovat tyto informace:
LITERATURA
Dodatek 1 SCHÉMA BUNĚK MDCK KULTIVOVANÝCH NA MEMBRÁNĚ VLOŽKY PRO ZKUŠEBNÍ METODU FL Konfluentní vrstva buněk MDCK se kultivuje na polopropustné membráně vložky. Vložky jsou umístěny do jamek destiček s 24 jamkami. 
Schéma převzato z: Wilkinson, P.J. (2006), Development of an in vitro model to investigate repeat ocular exposure, Ph.D. Thesis, University of Nottingham, UK. Dodatek 2 DEFINICE Přesnost : Přesnost shody mezi výsledky zkušební metody a přijatými referenčními hodnotami. Je to měření výsledku zkušební metody a jednoho aspektu „důležitosti“. Termín se často používá namísto „souladu“, kterým se rozumí podíl správných výsledků zkušební metody. Chemická látka : Látka nebo směs. Kategorie I EPA : Chemické látky, jež vyvolávají poleptání (nevratné poškození oční tkáně) nebo zasažení rohovky či dráždění přetrvávající déle než 21 dní (2). CLP EU (nařízení (ES) č. 1272/2008 o klasifikaci, označování a balení látek a směsí) : Toto nařízení provádí v Evropské unii (EU) systém GHS OSN pro klasifikaci chemických látek (látek a směsí). Míra falešné negativity : Podíl všech pozitivních chemických látek falešně zjištěných zkušební metodou jako negativní. Je to jeden z ukazatelů výsledku zkušební metody. Míra falešné pozitivity : Podíl všech negativních látek falešně zjištěných zkušební metodou jako pozitivní. Je to jeden z ukazatelů výsledku zkušební metody. FL20 : Hodnota, kterou lze odhadnout stanovením koncentrace, při které zkoušená chemická látka způsobuje 20 % únik fluoresceinu skrz vrstvu buněk. Průnik fluoresceinu : Množství fluoresceinu, které pronikne vrstvou buněk, určené spektrofluorimetrickým měřením. GHS (Globálně harmonizovaný systém klasifikace a označování chemických látek zavedený Organizací spojených národů (OSN)) : Systém navrhující klasifikaci látek a směsí podle standardizovaných typů a úrovní fyzikálních, zdravotních a environmentálních nebezpečí a řešící příslušné komunikační prvky, jako jsou piktogramy, signální slova, výroky o nebezpečí, výroky o bezpečnostních opatřeních a bezpečnostní datové listy, které obsahují informace o jejich nežádoucích účincích s ohledem na ochranu lidí (včetně zaměstnavatelů, dělníků, přepravců, spotřebitelů a osob reagujících na havárie) a životního prostředí. Kategorie 1 GHS : Tvorba poškození tkání v oku nebo závažné fyzikální slábnutí vidění po aplikaci zkoušené látky na přední plochu oka, které není plně vratné do 21 dní od aplikace. Nebezpečnost : Základní charakteristika látky nebo situace, která má potenciál vyvolat nepříznivé účinky, jsou-li organismus, systém nebo (sub)populace vystaveny této látce. Směs : V kontextu GHS OSN se používá k označení směsi nebo roztoku složeného ze dvou nebo více látek, které v něm nereagují. Negativní kontrola : Neovlivněná replika, která obsahuje všechny složky zkušebního systému. Tento vzorek se zpracovává se vzorky zkoušené chemické látky a jinými kontrolními vzorky, aby se zjistilo, zda rozpouštědlo vzájemně reaguje se zkušebním systémem. Neklasifikovány : Chemické látky, které nejsou klasifikovány jako látky s dráždivými účinky na oči kategorie 1, 2A nebo 2B GHS OSN, kategorie 1 nebo 2 CLP EU, kategorie I, II nebo III EPA USA. Látka s leptavými účinky na oči : a) chemická látka, která způsobuje nevratné poškození tkání očí; b) chemické látky, které jsou klasifikovány jako látky s dráždivými účinky na oči kategorie 1 GHS OSN, kategorie 1 CLP EU nebo kategorie I EPA USA. Látka s dráždivými účinky na oči : a) chemická látka, která způsobuje vratnou změnu v oku po aplikaci na přední plochu oka; b) chemické látky, které jsou klasifikovány jako látky s dráždivými účinky na oči kategorie 2A nebo 2B GHS OSN, kategorie 2 CLP EU, nebo kategorie II nebo III EPA USA. Látka se silně dráždivými účinky na oči : a) chemická látka, která způsobuje poškození tkání v oku po aplikaci na přední plochu oka, které není vratné do 21 dnů od aplikace, nebo způsobuje vážné fyzické zhoršení vidění; b) chemické látky, které jsou klasifikovány jako látky s dráždivými účinky na oči kategorie 1 GHS OSN, kategorie 1 CLP EU nebo kategorie I EPA USA. Pozitivní kontrola : Replika obsahující všechny složky zkušebního systému a zkoušená s chemickou látkou, o níž je známo, že způsobuje pozitivní reakci. Aby se zajistilo, že proměnlivost v reakci na pozitivní kontrolu bude možné časem vyhodnotit, rozsah pozitivní reakce by neměl být extrémní. Vhodné chemické látky : Podsoubor ze seznamu referenčních chemických látek, které může nezkušená laboratoř použít k prokázání způsobilosti k provádění validované referenční zkušební metody. Relevantnost : Popis vztahu zkoušky k účinku, který je předmětem zájmu, a také toho, zda je zkouška smysluplná a použitelná pro konkrétní účel. Vyjadřuje, do jaké míry zkouška správně měří nebo predikuje biologický účinek, který je předmětem zájmu. Relevantnost zahrnuje také posouzení přesnosti (shody) zkušební metody (8). Spolehlivost : Měření rozsahu, v jakém může být zkušební metoda časem reprodukovatelná v laboratořích a mezi laboratořemi, když se provádí s použitím stejného protokolu. Hodnotí se to výpočtem laboratorní a mezilaboratorní reprodukovatelnosti a laboratorní opakovatelnosti. Hodnotí se výpočtem laboratorní a mezilaboratorní reprodukovatelnosti a laboratorní opakovatelnosti. Náhradní zkouška : Zkouška, která je koncipována tak, aby nahradila běžně používanou a uznávanou zkoušku, která slouží k určení nebezpečnosti a/nebo posouzení rizika a u níž je zjištěno, že poskytuje stejnou nebo lepší úroveň ochrany zdraví lidí nebo zvířat či případně ochrany životního prostředí ve srovnání s uznávanou zkouškou ve všech možných zkušebních situacích a pro všechny zkoušené chemické látky. Citlivost : Podíl všech pozitivních/aktivních chemických látek, které jsou na základě zkoušky správně zatříděny. Je to měřítko přesnosti zkušební metody, která dává kategorické výsledky, a je důležitým prvkem při hodnocení relevance zkušební metody (8). Vážné poškození očí : Je to tvorba poškození tkání v oku nebo závažné fyzikální slábnutí vidění po aplikaci zkoušené chemické látky na přední plochu oka, které není plně vratné do 21 dní od aplikace. Kontrola s rozpouštědlem/vehikulem : Neošetřený vzorek obsahující všechny složky zkušebního systému, včetně rozpouštědla nebo vehikula, který se zpracovává s exponovanými zkoušenými chemickými látkami a jinými kontrolními vzorky, aby se zjistila základní reakce vzorků zkoušených se zkoušenou chemickou látkou rozpuštěnou ve stejném rozpouštědle nebo vehikulu. Pokud se vzorek zkouší souběžnou negativní kontrolou, ukazuje také, zda rozpouštědlo nebo vehikul vzájemně reaguje se zkušebním systémem. Specifičnost : Podíl všech negativních/neaktivních chemických látek, které jsou na základě zkušební metody správně zatříděny. Je to měřítko přesnosti zkušební metody, která dává kategorické výsledky, a je důležitým prvkem při hodnocení relevance zkušební metody. Látka : V kontextu GHS OSN jsou to chemické prvky a jejich sloučeniny v přírodním stavu nebo získané výrobním procesem, včetně všech přídatných látek nezbytných k uchování stability výrobků a veškerých nečistot vznikajících v daném procesu, avšak s výjimkou všech rozpouštědel, která lze oddělit bez ovlivnění stability látky nebo změny jejího složení. Zkoušená chemická látka : Jakákoli látka nebo směs zkoušená touto zkušební metodou. Strategie postupného zkoušení : Strategie postupného zkoušení, kde se všechny existující informace o zkoušené chemické látce přezkoumávají ve stanoveném pořadí s použitím postupu váhy důkazů na každém stupni, aby se zjistilo, zda je k dispozici dost informací pro rozhodnutí o klasifikaci nebezpečnosti, než se postoupí do dalšího stupně. Pokud potenciál dráždivých účinků zkoušené chemické látky lze určit na základě existujících informací, další zkoušení není nutné. Jestliže potenciál dráždivých účinků zkoušené látky nelze určit na základě existujících informací, použije se stupňovitý postupný postup zkoušení na zvířatech po krocích, dokud se nestanoví jednoznačná klasifikace. Validovaná zkušební metoda : Zkušební metoda, u níž byly provedeny validační studie k určení její relevantnosti (včetně přesnosti) a spolehlivosti pro konkrétní účel. Je nutné poznamenat, že validovaná zkušební metoda nemusí poskytnout postačující výsledek z hlediska přesnosti a spolehlivosti, aby byla shledána přijatelnou pro navržený účel (8). Závažnost důkazů : Postup zvažování silných a slabých stránek různých informací v dosahování a podpoře závěru týkajícího se potenciálu nebezpečnosti látky. Dodatek 3 CHEMICKÉ LÁTKY PRO PROKÁZÁNÍ ZPŮSOBILOSTI PRO ZKUŠEBNÍ METODU FL Dříve než začnou laboratoře rutinně používat tuto zkušební metodu, měly by prokázat svou odbornou způsobilost správným určením klasifikace leptavých účinků na oči u 8 chemických látek doporučených v tabulce 1. Tyto chemické látky byly vybrány tak, aby představovaly rozpětí reakcí na lokální podráždění/poleptání očí, a to na základě výsledků oční zkoušky in vivo na králících (TG OECD 405, zkušební metoda B.5 (5)) (tj. kategorie 1, 2A, 2B, nebo bez klasifikace podle GHS OSN). Vzhledem k validované užitečnosti zkoušky FL (tj. zjišťovat pouze látky vyvolávající leptavé / silně dráždivé účinky na oči) však existují pouze dva výsledky zkoušky pro účely klasifikace (látka mající leptavé / silně dráždivé účinky na oči nebo látka nemající leptavé / silně dráždivé účinky na oči) k prokázání způsobilosti. Jinými kritérii výběru bylo, aby chemické látky byly komerčně dostupné, aby k nim byly k dispozici vysoce kvalitní referenční údaje in vivo a aby existovaly vysoce kvalitní údaje získané zkušební metodou FL. Z tohoto důvodu byly vybrány osvědčené chemické látky ze „Základního dokumentu o revizi zkoušky úniku fluoresceinu jako alternativní metody ke zkoušení dráždivých účinků na oči“ (8), který byl použit pro zpětnou validaci zkušební metody FL. Tabulka 1 Doporučené chemické látky pro prokázání odborné způsobilosti ke zkoušce FL
B.62 Alkalický kometový test in vivo na savcích ÚVOD Tato zkušební metoda je rovnocenná Pokynu OECD pro zkoušení (TG) č. 489 (2016). Alkalický kometový test in vivo (jednobuněčná gelová elektroforéza) (dále označovaný pouze jako kometový test) se používá k detekci přerušení vláken DNA v buňkách nebo jádrech izolovaných z mnoha tkání zvířete, obvykle hlodavce, které byly vystaveny potenciálně genotoxickému materiálu (materiálům). Tento kometový text byl podroben revizi a různé skupiny odborníků zveřejnily svá doporučení (1) (2) (3) (4) (5) (6) (7) (8) (9) (10). Tato zkušební metoda je součástí série zkušebních metod genetické toxikologie. Byl vypracován dokument OECD, který obsahuje stručné informace o zkoušení v oblasti genetické toxikologie a přehled posledních změn, které byly v těchto pokynech ke zkoušení provedeny (11). Účelem kometového testu je zjišťovat chemické látky, které způsobují poškození DNA. V zásaditém prostředí (> pH 13) může kometový test odhalit jednoduché a dvojné zlomy vláken, které jsou například důsledkem přímých interakcí s DNA, existence oblastí DNA, které jsou nestabilní vůči zásadám, nebo jsou důsledkem přechodných zlomů vláken DNA vyvolaných vyštěpovací reparací DNA. Tyto zlomy je možné opravit, což znamená, že následky nejsou trvalé, nebo, mohou být pro buňku smrtící, anebo mohou vést k mutaci, která představuje trvalou životaschopnou změnu. Mohou rovněž vést k poškození chromozomů, které je mimo jiné spojeno s mnoha nemocemi u lidí, včetně rakoviny. Formální validační experiment kometového testu in vivo na hlodavcích byl proveden v letech 2006 až 2012 za koordinace Japonského střediska pro validaci alternativních metod (JaCVAM) a ve spolupráci s Evropským střediskem pro validaci alternativních metod (ECVAM), Meziagenturním koordinačním výborem pro validaci alternativních metod (ICCVAM) a Meziagenturním střediskem pro hodnocení alternativních toxikologických metod (NICEATM) národního toxikologického programu (NTP) (12). Tato zkušební metoda obsahuje rovněž doporučené použití a omezení kometového testu a je založena na závěrečném protokolu (12), který byl použit ve validačním experimentu, a na dalších příslušných zveřejněných i nezveřejněných (chráněných laboratorních) údajích. Definice nejdůležitějších termínů jsou uvedeny v dodatku 1. Je třeba konstatovat, že při tomto testu lze použít řadu různých platforem (mikroskopické preparáty, gelové skvrny, 96jamkové destičky atd.). Z praktických důvodů se v celé zbývající části tohoto dokumentu používá výraz „preparát“, avšak zahrnuje i všechny ostatní platformy. VÝCHOZÍ ÚVAHY A OMEZENÍ Kometový test je metodou měření zlomů vláken DNA v eukaryotických buňkách. Samostatné buňky/jádra ponořené do agarózy se nechají na podložním sklíčku s detergentem a při vysoké koncentraci soli lyzovat. Tímto krokem lýzy se vyluhují buněčné a jaderné membrány a umožní se uvolnění svinutých smyček DNA, které se zpravidla označují jako nukleoidy a fragmenty DNA. Elektroforéza při vysokém pH vede ke vzniku struktur podobajícím se kometám, které při použití vhodných fluorescenčních barviv lze pozorovat prostřednictvím fluorescenční mikroskopie; fragmenty DNA podle své velikosti putují od „hlavy“ k „ohonu“ a intenzita „ohonu komety“ v poměru k celkové intenzitě (hlava plus ohon) vyjadřuje množství přerušení DNA (13) (14) (15). Alkalický kometový test in vivo je zvláště vhodný k posouzení genotoxické nebezpečnosti proto, že odezvy při tomto testu jsou závislé na absorpci, distribuci, metabolismu a exkreci (ADME) in vivo a rovněž na procesech reparace DNA. Tyto jevy mohou u různých druhů zvířat, různých tkání a při různých druzích poškození DNA probíhat různě. Aby byly splněny požadavky šetrného zacházení se zvířaty, zejména snížení používání zvířat (zásady 3R nahrazení (replacement), snižování (reduction), zmírňování (refinement)), může být tento test také začleněn do jiných toxikologických studií, např. do studií toxicity po opakovaných dávkách (10) (16) (17), nebo lze jeho cílový ukazatel kombinovat s jinými cílovými ukazateli genotoxicity, např. zkoušky savčích erytrocytárních mikrojader in vivo (18) (19) (20). Kometový test se nejčastěji provádí na hlodavcích, ačkoli byl prováděn i na jiných druzích savců a na jiných zvířatech než na savcích. Použití jiných druhů než hlodavců by v každém jednotlivém případě mělo být vědecky a eticky zdůvodněno a důrazně se doporučuje provádět kometový test na jiných druzích než na hlodavcích pouze v rámci jiných studií toxicity, a nikoli jako samostatný test. Výběr způsobu expozice a tkáně (tkání), jež mají být zkoušeny, by měl vycházet ze všech dostupných/existujících poznatků o zkoušené chemické látce, např. zamýšleného/předpokládaného způsobu expozice lidí, metabolismu a distribuce, možných účinků v místě kontaktu, strukturálních rizik, jiných údajů o genotoxicitě a toxicitě a z účelu studie. Proto, je-li to vhodné, je možné genotoxický potenciál zkoušených chemických látek zkoušet na cílové tkáni (tkáních), na nichž se zkouší karcinogenní a/nebo jiné toxické účinky. Tento test je rovněž považován za užitečný pro další výzkum genotoxicity zjištěné způsobem in vitro. Je vhodné provést kometový test in vivo na tkáni, která je předmětem zájmu, jestliže lze důvodně předpokládat, že tato tkáň bude dostatečně exponována. Zkouška byla nejobsáhleji validována na somatických tkáních samice potkana v kolaborativních studiích, jako byl experiment střediska JaCVAM (12) a zkouška popsaná v publikaci Rothfuss et al., 2010 (10). V mezinárodním validačním experimentu střediska JaCVAM byla použita játra a žaludek. Játra proto, že jsou nejaktivnějším orgánem v metabolismu chemických látek a často též cílovým orgánem karcinogenity. Žaludek proto, že je obvykle prvním místem kontaktu s chemickými látkami po expozici orální cestou, ačkoli by za tkáně kontaktu měly být považovány i jiné oblasti trávicí soustavy jako dvanáctník a lačník, které z hlediska lidského organismu mohou být považovány za relevantnější než žláznatý žaludek hlodavců. Je třeba dbát na to, aby tyto tkáně nebyly vystaveny nadměrně vysokým koncentracím zkoušené chemické látky (21). Tuto metodu lze v zásadě použít ke zkoušení jakékoli tkáně, ze které lze odebrat jednobuněčné/jednojaderné suspenze. Chráněné údaje některých laboratoří prokazují její úspěšné použití na mnoha různých tkáních a existuje množství publikací, z nichž vyplývá použitelnost této metody pro zkoušení jiných orgánů nebo tkání než jater a žaludku, např. lačníku (22), ledvin (23) (24), kůže (25) (26) nebo močového měchýře (27) (28), plic a buněk bronchoalveolární laváže (což je důležité při inhalačních studiích chemických látek) (29) (30), a prováděly se i zkoušky s více orgány současně (31) (32). Předmětem zájmu mohou být genotoxické účinky na zárodečné buňky, avšak je třeba poznamenat, že standardní alkalický kometový test, jak je popsán v této zkušební metodě, není považován za vhodný pro měření zlomů vláken DNA ve zralých zárodečných buňkách. Při průzkumu literatury o použití kometového testu k výzkumu genotoxicity zárodečných buněk byly totiž zaznamenány vysoké a proměnlivé úrovně poškození buněk pozadí (33), a proto se má za to, že předtím, než bude možné do této zkušební metody začlenit kometový test na zralých zárodečných buňkách (např. spermatu), musí se provést změny v protokolu a hlubší standardizační a validační studie. Doporučený režim expozice popsaný v této zkušební metodě navíc není optimální a pro analýzu zlomů vláken DNA ve zralých spermiích, pro jeho použitelnost by byla nezbytná delší expozice nebo delší interval odběru vzorků. V literatuře jsou popsány genotoxické účinky měřené kometovým testem v testikulárních buňkách v různých fázích jejich diferenciace (34) (35). Je však třeba poznamenat, že gonády obsahují směs somatických a zárodečných buněk. Z tohoto důvodu pozitivní výsledky v celých gonádách (varlatech) nemusí nutně odrážet poškození zárodečných buněk; nicméně nasvědčují tomu, že zkoušená chemická látka (látky) a/nebo její metabolity dospěly až do gonád. Za standardních zkušebních podmínek kometového testu nelze spolehlivě zjistit překřížení vláken DNA. Za určitých upravených zkušebních podmínek by bylo možné zjistit překřížení DNA-DNA a DNA-protein a jiné bazické změny, jako např. zoxidované báze (23) (36) (37) (38) (39). K tomu, aby se daly nezbytné změny protokolu náležitě charakterizovat, byl by zapotřebí další výzkum. Zjišťování činitelů způsobujících překřížení vláken tak není prvořadým úkolem testu, jak je zde popsán. Test není vhodný, a to ani s úpravami, ke zjišťování aneugenů. Za současného stavu poznání jsou s kometovým testem in vivo spojena i některá další omezení (viz dodatek 3). Lze předpokládat, že tato zkušební metoda bude v budoucnu přezkoumána a v případě potřeby revidována vzhledem k získaným zkušenostem. Před použitím této zkušební metody ke zkoušení směsi s cílem získat údaje pro zamýšlené regulační účely by mělo být zváženo, zda tato metoda může pro tento účel poskytnout přiměřené výsledky, a pokud ano, proč je tomu tak. Takové úvahy nejsou nutné, pokud existuje právní požadavek na zkoušení dané směsi. PODSTATA METODY Zvířata se vhodným způsobem exponují zkoušené chemické látce. Podrobný popis dávkování a odběru vzorků je uveden v odstavcích 36 až 40. Ve zvoleném čase (časech) odběru vzorků se tkáně, které jsou předmětem zájmu, vyříznou a připraví se z nich jednobuněčná/jednojaderná suspenze (bude-li to považováno za užitečné, např. u jater, lze povést perfuzi in situ) a uloží se do měkkého agaru tak, aby se na sklíčkách znehybnily. Buňky/jádra se ošetří lýzovým pufrem, aby se odstranila buněčná a/nebo jaderná membrána, a exponuje se silné zásadě, např. s hodnotou pH ≥13, aby se umožnilo rozvinutí šroubovice DNA a odloučení uvolněných smyček a fragmentů DNA. Jaderná DNA v agaru se poté podrobí elektroforéze. Normální, nefragmentované molekuly DNA zůstanou v pozici, ve které byla jaderná DNA v agaru, zatímco veškerá fragmentovaná DNA a uvolněné smyčky DNA migrovaly směrem k anodě. Po elektroforéze se DNA zviditelní pomocí vhodného fluorescenčního barviva. Preparáty by se měly analyzovat za pomoci mikroskopu a plně automatizovaného nebo poloautomatizovaného analytického systému. Rozsah, v němž DNA migruje během elektroforézy, a vzdálenost, na niž migruje, odráží množství a velikost fragmentů DNA. Kometový test má několik cílových ukazatelů. Pro posouzení poškození DNA bylo doporučeno stanovit obsah DNA v ohonu (procentuální zastoupení DNA v ohonu ( % tail DNA) nebo procentuální intenzita ohonu ( % tail intensity)) (12) (40) (41) (42). Po analýze dostatečného počtu jader se údaje analyzují vhodnými metodami a vyhodnotí se výsledek testu. Je třeba poznamenat, že byly zkoumány změny různých kroků metodiky, včetně přípravy vzorků, podmínek elektroforézy, parametrů vizuální analýzy (např. intenzita barviva, intenzita světla žárovky mikroskopu a použití filtrů u mikroskopu a dynamika kamery) a podmínek okolního prostředí (např. osvětlení pozadí), které mohou migraci DNA ovlivnit (43) (44) (45) (46). OVĚŘENÍ ODBORNÉ ZPŮSOBILOSTI LABORATOŘE Každá laboratoř by měla dosáhnout způsobilosti k provádění pokusů tím, že prokáže schopnost získat dostatečně kvalitní suspenze s jedinou buňkou nebo jádrem pro každou cílovou tkáň (tkáně) každého druhu, který se použije. Kvalita preparátů bude hodnocena především podle procent DNA v ohonu u zvířat ošetřených vehikulem, která patří do reprodukovatelného nízkého rozpětí. Dosavadní údaje naznačují, že střední hodnota procentuálního zastoupení DNA v ohonu ve skupině (na základě střední hodnoty mediánů – podrobněji o těchto termínech viz odstavec 57) u jater potkanů by přednostně neměla být vyšší než 6 %, což by se shodovalo s hodnotami získanými ve validačním experimentu JaCVAM (12) a jinými zveřejněnými i chráněnými údaji. V současnosti neexistuje dostatek údajů, aby bylo možné předložit doporučení týkající se optimálních nebo přijatelných rozpětí pro jiné tkáně. To nebrání použití jiných tkání, je-li to odůvodněno. V závěrečné zprávě by měl být uveden náležitý přehled účinnosti kometového testu v těchto tkáních v poměru k údajům z publikované literatury nebo chráněným údajům. Za prvé je žádoucí, aby nízké rozpětí procentuálního zastoupení DNA v ohonu v kontrolách poskytovalo dostatečně dynamické rozpětí pro zjištění pozitivního účinku. Za druhé by každá laboratoř měla být schopna reprodukovat očekávané odezvy na přímé mutageny a promutageny s různými mechanismy působení, jak se doporučuje v tabulce 1 (odstavec 29). Pozitivní látky lze vybrat z validačního experimentu JaCVAM (12) nebo případně s odůvodněním i z jiných zveřejněných údajů (viz odstavec 9), které prokazují jasné pozitivní odezvy ve tkáních, které jsou předmětem zájmu. Měla by být rovněž prokázána schopnost detekovat slabé účinky známých mutagenů, např. EMS, při nízkých dávkách, například určením vztahu dávka-účinek s odpovídajícím počtem dávek a intervaly jejich podávání. Nejprve by se úsilí mělo zaměřit na dosažení způsobilosti ke zkoušení nejčastěji používaných tkání, tj. jater hlodavců, kde lze provést srovnání s existujícími údaji a očekávanými výsledky. Zároveň by měly být shromažďovány údaje vztahující se k jiným tkáním, např. žaludku/dvanáctníku/lačníku, krvi atd. Laboratoř musí prokázat způsobilost ke zkoušení každé jednotlivé tkáně každého druhu, který plánuje zkoušet, a bude muset prokázat, že v této tkáni lze získat přijatelnou pozitivní odezvu na známý mutagen (např. EMS). Měly by být shromažďovány údaje o kontrolách s vehikulem / negativních kontrolách, aby bylo možné prokázat reprodukovatelnost údajů o negativních odezvách a prokázat, že byly řádně kontrolovány technické aspekty testu, nebo aby bylo možné navrhnout stanovení nových historických rozpětí kontrol (viz odstavec 22). Je třeba poznamenat, že mnohé tkáně je možné odebírat při pitvě a zpracovat pro kometovou analýzu, avšak laboratoř musí být způsobilá odebírat vzorky více tkání od jediného zvířete, čímž se zajistí, že jakékoli potenciální léze DNA nebudou opomenuty a že analýza komet nebude zpochybněna. Zásadně důležitý může být časový interval od utracení do odstranění tkání pro zpracování (viz odstavec 44). Při dosahování způsobilosti k této zkoušce musí být brán ohled na dobré zacházení se zvířaty, proto při zvyšování způsobilosti v různých aspektech testu lze použít tkáně ze zvířat použitých v jiných zkouškách. Nemusí být rovněž nezbytné v různých fázích zavádění nové zkušební metody v laboratoři provádět celou zkoušku a při rozvíjení potřebných dovedností lze použít méně zvířat nebo nižší zkušební koncentrace. Historické kontrolní údaje Během šetření způsobilosti by laboratoř měla vytvořit databázi historických údajů, aby mohla stanovit rozsah a distribuci pozitivních a negativních kontrol pro příslušné tkáně a druhy zvířat. Doporučení, jak shromáždit a používat historické údaje (tj. kritéria pro zařazení údajů do databáze a jejich vyřazení a kritéria přijatelnosti pro daný experiment), lze najít v literatuře (47). Různé tkáně a různé druhy, jakož i různá vehikula a způsoby podávání mohou vést k různým hodnotám procentuálního zastoupení DNA v ohonu u negativních kontrol. Je proto důležité stanovit rozsahy negativních kontrol pro každou tkáň a pro každý druh. V laboratořích by měly být uplatňovány metody řízení jakosti, např. regulační diagramy (např. diagramy typu C nebo χ s pruhem (48)), z nichž je patrné, jak proměnlivé jsou jejich údaje a že metodika je v dané laboratoři „pod kontrolou“. Aby bylo možné detekovat slabé účinky, bude také možná zapotřebí optimalizovat výběr vhodných látek pro pozitivní kontrolu, rozsahy dávkování a zkušební podmínky (např. podmínky elektroforézy) (viz odstavec 17). Jakékoli změny zkušebního protokolu by měly být zvažovány z hlediska jejich souladu se stávající databází kontrolních údajů laboratoře. Jakýkoli větší rozdíl ve zkušebním protokolu by měl vést k zavedení nové historické kontrolní databáze. POPIS METODY Příprava Výběr zvířecích druhů Obvykle se používají běžné laboratorní kmeny zdravých, mladých a pohlavně zralých zvířat (ve věku 6 až 10 týdnů na počátku expozice, i když přijatelné je i použití poněkud starších zvířat). Při výběru druhu hlodavců by se mělo vycházet z i) druhů používaných v jiných studiích toxicity (aby bylo možné porovnat údaje a provádět integrované studie), ii) druhů, u kterých se ve zkoušce karcinogenity (při zkoumání mechanismu karcinogeneze) rozvinuly nádory nebo iii) druhů, jejichž metabolismus nejvíce odpovídá lidskému, jsou-li takové známy. Při této zkoušce jsou rutinně používáni hlodavci. Lze však použít i jiné druhy, je-li to eticky a vědecky odůvodněno. Podmínky chovu a krmení zvířat Teplota v místnosti s pokusnými zvířaty by u hlodavců v ideálním případě měla být 22 °C (± 3 °C). Relativní vlhkost vzduchu by v ideálním případě měla mít hodnotu 50 až 60 %, minimálně však 30 %, a neměla by pokud možno přesáhnout 70 % kromě doby úklidu místnosti. Osvětlení by mělo být umělé a mělo by se střídat 12 hodin světla a 12 hodin tmy. Ke krmení lze použít konvenční laboratorní krmivo s neomezeným přísunem pitné vody. Výběr druhu potravy může být ovlivněn její dostatečnou mísitelností se zkoušenou chemickou látkou, je-li látka podávána tímto způsobem. Hlodavci by měli být umístěni v klecích v malých skupinkách (obvykle maximálně pět zvířat) stejného pohlaví, pokud se neočekává agresivní chování. Zvířata mohou být umístěna v klecích individuálně, pouze je-li to vědecky odůvodněné. Měly by být pokud možno použity pevné podlahy, protože pletivové podlahy mohou způsobit vážná poranění (49). Musí být zajištěno přiměřené obohacení životního prostředí. Příprava zvířat Zvířata se náhodným výběrem rozdělí do kontrolních a experimentálních skupin. Zvířata se jednoznačně identifikují a před začátkem expozice se nechají v laboratorních podmínkách alespoň pět dní aklimatizovat. Musí být použita co nejméně invazivní metoda jednoznačné identifikace zvířat. Vhodnými metodami jsou například kroužkování, označení štítkem, pomocí mikročipu nebo biometrické identifikace. Zkracování posledních článků končetin a vystříhávání tkání z uší není při těchto zkouškách vědecky odůvodněné. Klece by měly být uspořádány tak, aby byl případný vliv jejich polohy minimalizován. V okamžiku zahájení studie by měla být odchylka v hmotnosti zvířat minimální a neměla by překročit ± 20 %. Příprava dávek Pevné zkoušené chemické látky by se před podáním dávky zvířatům měly rozpustit nebo suspendovat ve vhodných vehikulech nebo přimíchat do potravy nebo pitné vody. Kapalné zkoušené chemické látky mohou být podávány přímo nebo se před podáním zředí. Pro účely expozice inhalací lze zkoušené chemické látky v závislosti na jejich fyzikálně-chemických vlastnostech podávat v podobě plynu, páry nebo pevného/kapalného aerosolu (50) (51). Používat by se měla čerstvě připravená zkoušená chemická látka, pokud údaje o stabilitě neprokazují možnost skladování a nestanoví vhodné podmínky pro skladování. Zkušební podmínky Vehikulum Vehikulum by nemělo mít při použitých objemech dávek toxické účinky a mělo by být vyloučeno podezření, že reaguje se zkoušenou chemickou látkou. Jsou-li použita jiná než známá vehikula, mělo by být jejich zařazení podloženo referenčními údaji o jejich kompatibilitě, pokud jde o pokusná zvířata, způsob podávání a cílový ukazatel. Doporučuje se nejprve zvážit použití vodného rozpouštědla/vehikula. Je třeba poznamenat, že některá (zejména viskózní) vehikula mohou vyvolat zánět a zvýšit úroveň zlomů vláken DNA v pozadí v místě kontaktu, zejména při vícenásobném podávání. Kontroly Pozitivní kontroly V současnosti by měla být běžně do každé zkoušky zařazena skupina nejméně 3 analyzovatelných zvířat jednoho pohlaví nebo každého pohlaví, jsou-li použita obě (viz odstavec 32), exponovaných pozitivní kontrolní látce. V budoucnosti bude možná možné prokázat odpovídající způsobilost a potřebu pozitivních kontrol snížit. Odebírají-li se vzorky vícekrát (např. při protokolu s jediným podáním dávky), je nezbytné zařadit pozitivní kontroly pouze při jednom odběru, avšak mělo by být zajištěno vyvážené uspořádání (viz odstavec 48). V souběžných pozitivních kontrolách není nezbytné podávat látky stejným způsobem jako zkoušenou chemickou látku, ovšem při měření účinků v místě kontaktu je důležité použít stejný způsob podávání. O pozitivních kontrolních látkách by mělo být známo, že indukují zlomy vláken DNA ve všech tkáních, které jsou předmětem zájmu u zkoušené chemické látky, a vhodnou látkou pro pozitivní kontrolu je pravděpodobně EMS, neboť vyvolala zlomy vláken DNA ve všech tkáních, které byly dosud zkoumány. Dávky pozitivních kontrolních látek by měly být vybrány tak, aby vyvolaly střední účinky, které nejlépe dokládají dobré provedení a citlivost zkoušky, a mohly by být založeny na křivkách dávka-účinek vytvořených laboratoří během prokazování způsobilosti. U zvířat souběžné pozitivní kontroly by procentuální zastoupení DNA v ohonu mělo odpovídat rozpětí, které bylo předem laboratoří stanoveno pro každou jednotlivou tkáň a každý odběr vzorků u daného druhu (viz odstavec 16). Příklady pozitivních kontrolních látek a některé z jejich cílových tkání (u hlodavců) jsou uvedeny v tabulce 1. Je možné vybrat jiné látky než látky uvedené v tabulce 1, je-li to vědecky odůvodněno. Tabulka 1 Příklady pozitivních kontrolních látek a některé z jejich cílových tkání Látky a jejich číslo CAS ethylmethansulfonát (CAS RN 62-50-0) pro veškeré tkáně 1-ethyl 1-nitrosomočovina (CAS RN 759-73-9) pro játra a žaludek, dvanáctník nebo lačník methylmethansulfonát (CAS RN 66-27-3) pro játra, žaludek, dvanáctník nebo lačník, plíce a buňky bronchoalveolární laváže (BAL), ledviny, močový měchýř, plíce, varlata a kostní dřeň / krev N-methyl-N′-nitro-N-nitrosoguanidin (CAS RN: 70-25-7) pro žaludek, dvanáctník nebo lačník 1,2-dimethylhydrazin-dihydrochlorid (CAS RN 306-37-6) pro játra a střeva 1-methyl-1-nitrosomočovina (CAS RN 684-93-5) pro játra, kostní dřeň, krev, ledviny, žaludek, dvanáctník a mozek Negativní kontroly Do každé zkoušky by pro každý odběr a pro každou tkáň měla být zařazena skupina zvířat pro negativní kontrolu ošetřených pouze vehikulem, s kterou se jinak zachází stejně jako s exponovanými skupinami. U zvířat negativní kontroly by procentuální zastoupení DNA v ohonu mělo být uvnitř rozpětí pro pozadí, které laboratoř předem stanoví pro každou jednotlivou tkáň a každý odběr vzorků u daného druhu (viz odstavec 16). Neexistují-li dosavadní nebo publikované údaje o kontrolách prokazující, že zvolené vehikulum, počet podávaných dávek nebo způsob jejich podávání vyvolává zhoubné nebo genotoxické účinky, měly by se před provedením úplné studie provést počáteční studie ke stanovení přijatelnosti kontroly s vehikulem. POSTUP Počet a pohlaví zvířat Existuje málo údajů týkajících se zvířat samičího pohlaví, na jejichž základě by bylo možné provést srovnání mezi oběma pohlavími v souvislosti s kometovým testem, avšak obecně jsou jiné genotoxické odezvy in vivo u zvířat samčího a samičího pohlaví podobné, a proto většinu studií lze provést se zvířaty kteréhokoli pohlaví. Použití zvířat obou pohlaví podpoří údaje prokazující významné rozdíly mezi samci a samicemi (např. rozdíly v systémové toxicitě, metabolismu, biologické dostupnosti atd. zjištěné např. ve studii ke stanovení rozsahu dávek pro studie toxicity po opakovaných dávkách). V tom případě může být vhodné provést studii s oběma pohlavími, např. jako součást studie toxicity po opakovaných dávkách. V případě použití obou pohlaví by mohlo být vhodné použít faktoriální pokus. Podrobnosti ohledně toho, jak pomocí tohoto pokusu analyzovat údaje, jsou uvedeny v dodatku 2. Velikost skupin při zahájení studie (a během stanovení způsobilosti) by měla být stanovena tak, aby poskytla minimálně pět analyzovatelných zvířat jednoho pohlaví nebo každého pohlaví, jsou-li použita obě, z každé skupiny (méně ve skupině souběžné pozitivní kontroly – viz odstavec 29). Je-li expozice člověka chemickým látkám specifická pro určité pohlaví, jako je tomu například u některých farmaceutických látek, měla by být zkouška provedena se zvířaty odpovídajícího pohlaví. Určitým vodítkem pro maximální typické požadavky na počet zvířat může být, že studie provedená podle parametrů uvedených v odstavci 33 se třemi exponovanými skupinami a souběžnými negativními a pozitivními kontrolami (každá skupina složená z pěti zvířat jednoho pohlaví) by vyžadovala 25 až 35 zvířat. PLÁN EXPOZICE Zvířata by měla být každodenně exponována po dobu 2 nebo více dnů (tj. dvě nebo více expozic v přibližně 24hodinových intervalech), a vzorky by měly být odebírány každých 2 až 6 hodin (nebo v čase Tmax) od první expozice. Vzorky ze studií s prodlouženým režimem podávání (např. každodenní podávání po dobu 28 dnů) jsou přijatelné. Byla prokázána úspěšná kombinace kometového testu a testu erytrocytárních mikrojader (10) (19). Je však třeba pečlivě zvážit logistiku spojenou s odběrem vzorků tkání pro kometovou analýzu spolu s požadavky na odběr tkáňových vzorků pro jiné druhy toxikologických hodnocení. Ve většině případů není vhodný odběr vzorků 24 hodin po poslední dávce, který je typický pro studium obecné toxicity. Použití jiných harmonogramů expozice a odběru vzorků by mělo být odůvodněno (viz dodatek 3). Mohla by být například použita jediná expozice s více odběry vzorků, avšak je třeba poznamenat, že studie s jediným podáním dávky bude vyžadovat více zvířat, jelikož je zapotřebí více odběrů, avšak někdy může být upřednostněna, např. jestliže zkoušená chemická látka po opakovaném podání vyvolává nadměrnou úroveň toxicity. Ať je zkouška provedena jakýmkoli způsobem, je přijatelná, pokud zkoušená chemická látka vyvolá pozitivní odezvu nebo, v případě negativní studie, pokud jsou podány přímé či nepřímé důkazy o expozici cílové tkáně (tkání) nebo o toxicitě pro cílovou tkáň (tkáně) nebo o tom, že bylo dosaženo limitní dávky (viz odstavec 36). Zkoušené chemické látky mohou být podávány také ve dvou dávkách, tj. dvě expozice v týž den v intervalu nanejvýš 2 až 3 hodin, aby se usnadnilo podávání velkých objemů. Za těchto podmínek by doby odběru vzorků měly být stanoveny na základě doby posledního podání dávky (viz odstavec 40). Dávkování Provádí-li se studie ke stanovení rozsahu dávek, poněvadž nejsou k dispozici vhodné údaje z jiných relevantních studií, které by mohly pomoci při stanovení dávek, měla by podle současného přístupu k provádění studií ke stanovení rozsahu dávek být provedena ve stejné laboratoři, se stejným druhem, kmenem, pohlavím a za stejného režimu expozice, které se použijí v hlavní studii. Cílem této studie by mělo být zjistit maximální tolerovanou dávku (MTD), která je definována jako dávka vyvolávající slabé toxické účinky po dobu trvání zkoušky (například zjevné klinické příznaky jako nenormální chování nebo reakce, menší pokles tělesné hmotnosti nebo cytotoxicita cílové tkáně), nikoli však smrt nebo důkazy o bolesti, utrpení nebo strádání vyžadující utracení. U netoxické chemické látky při době podávání 14 dnů nebo delší činí maximální (limitní) dávka 1 000 mg/kg tělesné hmotnosti na den. Při kratší než 14denní aplikaci je maximální (limitní) dávka 2 000 mg/kg tělesné hmotnosti na den. Pro některé druhy zkoušených chemických látek (např. farmaceutické látky používané v humánní medicíně), na něž se vztahují specifické požadavky, se tyto limity mohou lišit. Výjimku z kritérií pro stanovení dávek mohou představovat chemické látky, jež vykazují saturaci toxikokinetických vlastností nebo indukují detoxifikační procesy, jež mohou vést ke snížení expozice po dlouhodobém podávání; tyto látky by měly být hodnoceny v každém jednotlivém případě. V akutní a subakutní verzi kometového textu by kromě maximální dávky (MTD, maximální únosné dávky, maximální expozice nebo limitní dávky) měl být pro každý odběr vzorků vybrána klesající řada alespoň dvou dalších vhodně odstupňovaných úrovní (pokud možno v intervalu menším než 10), aby se prokázala odezva v závislosti na dávce. Použité úrovně dávek by však pokud možno měly pokrývat rozsah od maximální dávky až po dávku vyvolávající malou toxicitu nebo nevyvolávající žádnou toxicitu. Je-li toxicita pro cílovou tkáň (kostní dřeň) pozorována na všech testovaných úrovních dávek, doporučuje se další studie s netoxickými dávkami (viz odstavce 54–55). Studie, jejichž cílem je úplnější výzkum tvaru křivky dávka-účinek, mohou vyžadovat zařazení doplňkové exponované skupiny (skupin). Podávání dávek Při plánování zkoušky by měl být zvážen předpokládaný způsob expozice člověka. Proto lze oprávněně zvolit způsoby expozice např. stravou, v pitné vodě, lokálně, subkutánně, nitrožilně, orálně (pomocí žaludeční sondy), inhalací, intratracheálně nebo implantací. Způsob podávání by měl být v každém případě zvolen tak, aby zajistil odpovídající expozici cílové tkáně (tkání). Intraperitoneální injekce se obecně nedoporučuje, protože se nejedná o typický odpovídající způsob expozice člověka, a měla by být použita pouze s konkrétním zdůvodněním (např. u některých pozitivních kontrolních látek, pro výzkumné účely nebo u některých léčiv, která se podávají intraperitoneální cestou). Maximální objem kapaliny, který lze najednou podán žaludeční sondou nebo injekčně, závisí na velikosti pokusného zvířete. Tento objem by neměl překročit 1 ml na 100 g tělesné hmotnosti, s výjimkou vodných roztoků, kde lze použít 2 ml na 100g tělesné hmotnosti. Použití větších objemů, než jsou tyto (pokud je povolují právní předpisy o šetrném zacházení se zvířaty), je třeba odůvodnit. Různých úrovní dávky by mělo být pokud možno dosaženo úpravou koncentrace aplikační formy, aby se zajistil konstantní objem v poměru k tělesné hmotnosti na všech úrovních dávek. Doba odběru vzorků Doba odběru vzorků je kritická proměnná, neboť je určována dobou, která je nezbytná k dosažení maximální koncentrace zkoušených chemických látek v cílové tkáni a k vyvolání zlomů vláken DNA, avšak dříve, než byly tyto zlomy odstraněny, napraveny nebo než vedly k zániku buňky. Trvání některých lézí, jež vedou k zlomům vláken DNA detekovaným při kometovém testu, může být, alespoň u některých chemických látek zkoušených in vitro, velmi krátké (52) (53). Existuje-li podezření na výskyt takových přechodných lézí DNA, měla by být přijata opatření ke zmírnění jejich ztráty tím, že se vzorky tkání odeberou včas, možná dříve než ve standardních dobách, které jsou uvedeny níže. Optimální doba (doby) odběru vzorků mohou záviset na chemické látce nebo na způsobu jejího podávání, což vede například k rychlé expozici tkání při nitrožilním podávání nebo při expozici inhalací. Proto by doby odběru vzorků měly být pokud možno stanoveny na základě kinetických údajů (např. čas (Tmax), ve kterém je dosažena maximální koncentrace v plazmě nebo ve tkáni (Cmax), nebo v rovnovážném stavu v případě podávání ve více dávkách). Pokud kinetické údaje neexistují, vhodným kompromisem pro měření genotoxicity je odebírat vzorky 2 až 6 hodin po poslední expozici v případě dvou nebo více expozic nebo po 2 až 6 hodinách a po 16 až 24 hodinách po jediném podání dávky; mělo by se však dbát na to, aby se pitva všech zvířat provedla ve stejném intervalu po poslední (nebo jediné) dávce. Při výběru vhodných dob odběru vzorků mohou být též použity informace o vzniku toxických účinků v cílových orgánech (jsou-li takové informace k dispozici). Pozorování Všeobecné klinické pozorování zdravotního stavu pokusných zvířat by se mělo provádět a zaznamenávat nejméně jednou denně, nejlépe ve stejnou dobu (stejné doby) každého dne a s uvážením doby očekávaného maxima účinku po podání dávky (54). Nejméně dvakrát denně by měla být provedena prohlídka všech zvířat za účelem zjištění morbidity a mortality. Při déletrvajících studiích by všechna zvířata měla být nejméně jednou týdně zvážena, a rovněž při ukončení zkoušky. Spotřeba potravy by se měla měřit při každé výměně krmiva a alespoň jednou týdně. Pokud je zkoušená chemická látka podávána v pitné vodě, měla by se spotřeba vody měřit při každé výměně vody a alespoň jednou týdně. Zvířata, která vykazují neletální indikátory nadměrné toxicity, by měla být utracena před uplynutím doby zkoušky a pro kometovou analýzu se zpravidla nepoužívají. Odběr tkání Jelikož vyvolání zlomů vláken DNA (komet) je možné zkoumat prakticky v jakékoli tkáni, mělo by být jasně uvedeno zdůvodnění tkáně (tkání), které se budou odebírat, a mělo by vycházet z důvodu provedení této studie a z veškerých existujících údajů o absorpci, distribuci, metabolismu a exkreci (ADME), o genotoxicitě, karcinogenitě nebo jiné toxicitě zkoušených chemických látek, které jsou předmětem výzkumu. Důležitými faktory, které je nutno zvážit, jsou mimo jiné způsob podávání dávek (založený na pravděpodobném způsobu (způsobech) expozice u lidí), predikovaná distribuce a absorpce tkání, vliv metabolismu a možný mechanismus působení zkoušených chemických látek. Nejčastěji zkoumanou tkání jsou játra, o kterých také existuje nejvíce údajů. Pokud některé podkladové informace chybějí a pokud předmětem zájmu nejsou některé konkrétní tkáně, byl by odběr vzorků z jater oprávněný, neboť především v nich se odehrává xenobiotický metabolismus a jsou často vystaveny působení jak mateřské látky (látek), tak metabolitu (metabolitů). V některých případech může být nejvhodnější zkoumání místa přímého kontaktu (např. žláznatého žaludku nebo dvanáctníku/lačníku u chemických látek podávaných orálně nebo do plic v případě inhalovaných chemických látek). Další nebo alternativní tkáně by měly být vybírány na základě konkrétních důvodů, proč se zkouška provádí, ale může být užitečné zkoumat více tkání stejných zvířat za předpokladu, že laboratoř prokázala způsobilost ke zkoušení těchto tkání a k manipulaci s více tkáněmi současně. Příprava vzorků U procesů popsaných v následujících odstavcích (44–49) je důležité, že všechny roztoky nebo stabilní suspenze by měly být používány před skončením jejich použitelnosti nebo, pokud je to možné, měly být čerstvě připraveny. V následujících odstavcích jsou rovněž doby nezbytné k i) odstranění každé tkáně po provedení pitvy, ii) zpracování každé tkáně do buněčných/jaderných suspenzí a iii) zpracování suspenze a přípravě preparátů považovány za kritické proměnné (viz definice, dodatek 1) a při zavádění metody a při prokazování způsobilosti by měly být stanoveny přijatelné časové intervaly pro každý z těchto kroků. Ve vhodnou dobu (doby) po poslední expozici zkoušené chemické látce se zvířata utratí v souladu s platnými právními předpisy o šetrném zacházení se zvířaty a se zásadami 3R- nahrazování, snižování a zmírňování. Vybraná tkáň (tkáně) se vyjme, oddělí a část se odebere pro kometový test, podle standardních postupů by se kousek této tkáně měl odříznout a uložit do formaldehydového roztoku nebo do vhodné fixační látky pro případnou histopatologickou analýzu (viz odstavec 55). Tkáň určená pro kometový test se vloží do rozmělňovacího pufru, dostatečně se propláchne studeným rozmělňovacím pufrem, aby se odstranila zbytková krev a až do zpracování se uchovává se v ledovém rozmělňovacím pufru. Může se rovněž provést perfuze (např. jater, ledvin) in situ. Existuje celá řada metod izolace buněk/jader, které jsou popsány v literatuře. Tyto metody zahrnují rozřezání tkání, např. jater nebo ledvin, na malé kousky, seškrabání povrchu sliznice v případě gastrointestinálního traktu, homogenizaci a enzymatickou digesci. V průběhu validačního experimentu JaCVAM byly zkoumány pouze izolované buňky, a proto se při zavádění metody, aby se při prokazování způsobilosti bylo možné odvolat na údaje z experimentu JaCVAM, upřednostňují izolované buňky. Bylo však prokázáno, že ať byly použity izolované buňky nebo jádra, nebyl ve výsledcích zkoušky podstatný rozdíl (8). Srovnatelné výsledky daly také různé metody izolace buněk/jader (např. homogenizace, krájení, enzymatická digesce a filtrace na sítu) (55). Mohou tedy být použity buď izolované buňky, nebo izolovaná jádra. Laboratoř by měla důkladně vyhodnotit metody izolace jediné buňky / jediného jádra specifické pro každou tkáň a ověřit je. Jak bylo uvedeno v odstavci 40, některé léze, jež vedou k zlomům vláken DNA detekovaným při kometovém testu, mohou mít jen velmi krátké trvání (52) (53). Proto, ať je na přípravu jednobuněčných/jednojaderných suspenzí použita kterákoli metoda, je důležité, aby tkáně byly zpracovány co nejdříve po utracení zvířat a uchovávány v podmínkách, které snižují ústup lézí (např. uchováváním tkáně při nízké teplotě). Buněčné suspenze by měly být uchovávány na ledu, dokud nejsou připraveny k použití, aby tak mohla být prokázána minimální variabilita mezi vzorky a přiměřeným účinkem u pozitivních a negativních kontrol. PŘÍPRAVA PREPARÁTŮ Příprava preparátů by se měla provést co nejdříve (v ideálním případě do jedné hodiny) po přípravě izolovaných buněk/jader, ale teplota a časový interval mezi smrtí zvířete a přípravou preparátů by měly být přísně řízeny a ověřovány v podmínkách laboratoře. Objem buněčné suspenze přidaný do agarózy s nízkým bodem tání (obvykle 0,5 až 1,0 %) na přípravu preparátů by neměl snížit procentuální podíl agarózy s nízkým bodem tání na méně než 0,45 %. Optimální hustota buněk bude stanovena systémem analýzy obrazu, který se používá pro hodnocení komet. Lýza Podmínky lýzy jsou rovněž kritickou proměnnou a mohou ovlivňovat zlomy vláken u některých specifických modifikací DNA (určité alkylace DNA a bazické adukty). Proto se doporučuje, aby podmínky lýzy byly pro všechny preparáty v experimentu co nejvíce konstantní. Jakmile jsou preparáty připraveny, měly by se ponořit alespoň na hodinu (nebo přes noc) do chlazeného lyzovacího roztoku o teplotě asi 2 až 8 °C za tlumeného osvětlení, např. pomocí žlutého světla (nebo chráněné před světlem), což zabrání vystavení bílému světlu, jež může obsahovat UV složky. Po této inkubační době by preparáty před krokem alkalického rozvinutí měly být opláchnuty, aby se odstranil zbytkový detergent a soli. To lze provést pomocí přečištěné vody, neutralizačního pufru nebo fosfátového pufru. Použít lze také elektroforézní pufr. To by zachovalo zásadité prostředí v elektroforézní komoře. Rozvinutí a elektroforéza Preparáty by měly být náhodným výběrem umístěny na platformu zařízení pro ponorkovou elektroforézu obsahující dostatečné množství elektroforézního roztoku, aby povrch preparátů byl zcela pokryt (hloubka pokrytí by měla být konzistentní při jednotlivých provedeních). U jiného druhu zařízení pro elektroforézu při kometovém testu, tj. zařízení s aktivním chlazením, cirkulací a vysokokapacitním zdrojem energie, bude vyšší vrstva roztoku na povrchu mít za následek silnější elektrický proud při udržování konstantního napětí. Preparáty by měly být vkládány do elektroforézní nádoby ve vyváženém uspořádání, aby se zmírnily účinky jakýchkoli trendů nebo okrajové účinky uvnitř nádoby a minimalizovala se variabilita mezi jednotlivými sadami, tj. při každém provedení elektroforézy by měl být stejný počet preparátů z každého zvířete zařazeného do studie a měly by být zařazeny vzorky z různých exponovaných skupin, negativních a pozitivních kontrol. Preparáty by měly být ponechány alespoň 20 minut v klidu, aby se mohla rozvinout DNA, a poté podrobeny elektroforéze za řízených podmínek, které umožní dosáhnout co nejvyšší citlivosti a dynamického rozpětí zkoušky (tj. povedou k přijatelným úrovním procent DNA v ohonu u negativních a pozitivních kontrol, jež maximalizují citlivost). Míra migrace DNA je přímo úměrná délce trvání elektroforézy a také potenciálu (ve V/cm). Na základě experimentu JaCVAM by to mohlo být 0,7 V/cm za dobu nejméně 20 minut. Délka trvání elektroforézy je považována za kritickou proměnnou a době elektroforézy by měla být stanovena tak, aby se optimalizovalo dynamické rozpětí. Delší doba elektroforézy (např. 30 nebo 40 minut pro dosažení co největší citlivosti) u známých mutagenů obvykle vedou k silnějším pozitivním odezvám. Delší doby elektroforézy však mohou také vést k nadměrné migraci u kontrolních vzorků. Při každém experimentu by mělo být udržováno konstantní napětí a proměnlivost ostatních parametrů by měla být v úzkém vymezeném rozpětí, např. v experimentu JaCVAM hodnota 0,7 V/cm poskytla počáteční proud 300 mA. Hloubka pufru by měla být upravena tak, aby bylo dosaženo požadovaných podmínek a ty byly zachovány po celou dobu pokusu. Hodnota proudu na začátku a na konci doby elektroforézy by měla být zaznamenána. Optimální podmínky by měly být určeny při počátečním prokázání způsobilosti v příslušné laboratoři pro každou testovanou tkáň. Teplota elektroforézního roztoku během rozvinutí a elektroforézy by měla být udržována na nízké hodnotě, obvykle 2 až 10 °C (10). Teplota elektroforézního roztoku na počátku rozvinutí, na začátku elektroforézy a na konci elektroforézy by se měla zaznamenat. Po ukončení elektroforézy by preparáty měly být ponořeny/propláchnuty v neutralizačním pufru nejméně po dobu 5 minut. Gely lze obarvit a hodnotit v „čerstvém“ stavu (např. během 1 až 2 dnů) nebo je lze dehydratovat pro pozdější hodnocení (např. během 1 až 2 týdnů po obarvení) (56). Tyto podmínky by však měly být při prokázání způsobilosti ověřeny a měly by být získány historické údaje, které se uchovávají zvlášť pro každou z těchto podmínek. Ve druhém z uvedených případů by preparáty měly být dehydrovány ponořením do absolutního ethanolu alespoň na 5 minut, nechají se usušit na vzduchu a poté se uskladní buď při pokojové teplotě, nebo v nádobě v chladničce, až do hodnocení. Metody měření Komety by se měly hodnotit kvantitativně pomocí automatizovaného nebo poloautomatizovaného systému analýzy obrazu. Preparáty se obarví vhodným fluorescenčním barvivem, např. SYBR Gold, Green 1, propidiumiodidem nebo ethidiumbromidem a změří se při vhodném zvětšení (např. 200x) mikroskopem vybaveným epifluorescencí a vhodnými detektory nebo digitálním fotoaparátem (např. CCD). Buňky lze rozdělit do tří kategorií, jak je popsáno v atlasu vyobrazení komet (57), a sice hodnotitelné, nehodnotitelné a tzv. „hedgehogs“ („ježci“, bližší objasnění viz odstavec 56). Aby nedošlo k falešným výsledkům, k získání procentuálního zastoupení DNA v ohonu by měly být hodnoceny pouze hodnotitelné buňky (s jasně vyjádřenou hlavou a ohonem, bez rušivých vlivů sousedních buněk). Četnost výskytu nehodnotitelných buněk není nutné uvádět. Četnost výskytu „hedgehogs“ by měla být stanovena na základě vizuálního hodnocení (neboť nepřítomnost jasně vyjádřené hlavy bude znamenat, že je nelze snadno odhalit analýzou obrazu) alespoň 150 buněk v každém vzorku (bližší objasnění viz odstavec 56) a zdokumentována zvlášť. Všechny preparáty k analýze, včetně preparátů pozitivních a negativních kontrol, by měly být označeny samostatným kódem a hodnoceny „naslepo“ tak, aby hodnotiteli nebyly známy zkušební podmínky. Z každého vzorku (na každou tkáň z každého zvířete) by mělo být analyzováno alespoň 150 buněk (s vyloučením „hedgehogs“ – viz odstavec 56), Hodnocení 150 buněk z každého zvířete při nejméně 5 zvířatech na každou dávku (v souběžné pozitivní kontrole méně – viz odstavec 29) poskytuje podle analýzy Smitha et al., 2008 (5) odpovídající statistickou váhu. Jsou-li použity preparáty, mohlo by to znamenat hodnocení 2 nebo 3 preparátů z každého vzorku, pokud jsou použity skupiny s pěti zvířaty. Mělo by být pozorováno několik oblastí preparátu o hustotě zajišťující, že nedojde k překrývání ohonů. Oblasti na okraji preparátů by se hodnotit neměly. Zlomy vláken DNA by se v kometovém testu měly měřit podle nezávislých cílových ukazatelů, jako je např. procentuální zastoupení DNA v ohonu, délka ohonu a moment ohonu („tail moment“, definovaný jako součin délky ohonu a procentuálního zastoupení DNA v ohonu). Lze provést všechna tři měření, pokud je použit vhodný softwarový systém pro analýzu obrazu. Pro hodnocení a interpretaci výsledků se však doporučuje použít procentuální zastoupení DNA v ohonu (též známé jako procento intenzity ohonu) (12) (40) (41) (42), které se stanoví jako intenzita fragmentů DNA v ohonu vyjádřená v procentech z celkové intenzity buňky (13). Poškození tkání a cytotoxicita Pozitivní nálezy v kometovém testu nemusejí být pouze důsledkem genotoxicity, ale k nárůstům migrace DNA může vést také toxicita cílové tkáně (12) (41). A naopak u známých genotoxinů je často zjištěna nízká nebo střední cytotoxocita (12), z čehož plyne, že pouze kometovým testem není možné rozlišit migraci DNA vyvolanou genotoxicitou od migrace DNA způsobené cytotoxicitou. Pokud jsou zjištěny nárůsty migrace DNA, doporučuje se nicméně provést vyšetření jednoho nebo více ukazatelů cytotoxicity, což může napomoci při interpretaci těchto nálezů. Nárůsty migrace DNA za přítomnosti jasných příznaků cytotoxicity by měly být interpretovány s obezřetností. Byla navržena celá řada měřítek cytotoxicity, přičemž za důležité měřítko toxicity tkání jsou považovány histopatologické změny. S nárůsty migrace DNA byla spojována taková zjištění jako zánět, infiltrace buněk, apoptotické nebo nekrotické změny, avšak jak prokázal validační experiment JaCVAM (12), není k dispozici žádný konečný seznam histopatologických změn, které jsou vždy spojeny se zvýšenou migrací DNA. Užitečné informace o poškození tkání mohou poskytnout i změny v klinických biochemických ukazatelích (např. AST, ALT) a posoudit lze i další ukazatele, jako je aktivace kaspázy, barvivo TUNEL, barvivo Annexin V atd. O použití posledně jmenovaných ukazatelů ve studiích in vivo však existují jen omezené publikované údaje a některé mohou být méně spolehlivé než jiné. „Hedgehogs“ (nebo též „clouds“ či „ghost cells“) jsou buňky, které pod mikroskopem tvoří obraz sestávající z malé nebo chybějící hlavy a z velkých roztroušených ohonů a jsou považovány za těžce poškozené buňky, ačkoli etiologie „hedgehogs“ je nejistá (viz dodatek 3). V důsledku jejich vzhledu jsou měření procentuálního zastoupení DNA v ohonu pomocí obrazové analýzy nespolehlivá, a proto se by buňky „hedgehogs“ měly hodnotit zvlášť. Výskyt „hedgehogs“ by měl být zaznamenán a uveden ve zprávě a jakýkoli významný nárůst, o němž se má za to, že je vyvolán zkoušenou chemickou látkou, by měl být pečlivě zkoumán a interpretován. Při takovém posuzování může pomoci znalost potenciálního mechanismu působení zkoušené chemické látky. ÚDAJE A PŘEDKLÁDÁNÍ ZPRÁV Zpracování výsledků Experimentální jednotkou je zvíře, a proto by měly být předloženy jak údaje o jednotlivých zvířatech, tak souhrnné výsledky ve formě tabulky. Vzhledem k hierarchické povaze údajů se doporučuje určit mediánovou hodnota procentuálního zastoupení DNA v ohonu pro každý preparát a vypočítat střední hodnotu těchto mediánových hodnot pro každé zvíře (12). Ze středních hodnot pro jednotlivá zvířata se pak určí skupinová střední hodnota. Všechny tyto hodnoty by měly být v závěrečné zprávě uvedeny. Možné jsou i jiné přístupy (viz odstavec 53), pokud jsou vědecky a statisticky odůvodněny. Statistickou analýzu lze provést různými způsoby (58) (59) (60) (61). Při výběru statistické metody, která má být použita, by měla být zvážena nutnost transformace (např. log nebo druhá odmocnina) údajů a/nebo přičtení malého čísla (např. 0,001) ke všem (i k nenulovým) hodnotám, aby se zmírnily účinky nulových buněčných hodnot, jak bylo popsáno ve výše uvedených odkazech. Podrobnosti k analýze interakcí expozice/pohlaví, jsou-li použita obě pohlaví, a k následné analýze údajů v případě, že jsou zjištěny nebo nejsou zjištěny rozdíly, jsou uvedeny v dodatku 2. Uvedeny by měly být i údaje o toxicitě a klinické příznaky. Kritéria přijatelnosti Přijetí zkoušky je založeno na následujících kritériích:
Hodnocení a interpretace výsledků Za předpokladu, že všechna kritéria přijatelnosti byla splněna, lze zkoušenou chemickou látku považovat za jasně pozitivní, pokud:
Jsou-li splněna všechna tato kritéria, má se za to, že zkoušená chemická látka je schopna vyvolávat zlomy vláken DNA v tkáních zkoušených v tomto zkušebním systému. Je-li splněno pouze jedno nebo dvě z těchto kritérií, viz odstavec 62. Za předpokladu, že všechna kritéria přijatelnosti byla splněna, lze zkoušenou chemickou látku považovat za jasně negativní, pokud:
Zkoušená chemická látka je pak považována za látku nevyvolávající poškození vláken DNA v tkáních zkoumaných v tomto zkušebním systému. Ověření jasně pozitivního či jasně negativního účinku není nutné. V případě, že účinek není jasně negativní ani jasně pozitivní (tj. ne všechna kritéria uvedená v odstavcích 59 nebo 60 jsou splněna) a aby se napomohlo stanovení biologického významu výsledku, měly by být údaje vyhodnoceny na základě odborného posudku a/nebo by měl být provedeno další šetření, pokud je to vědecky odůvodněné. Užitečné by mohlo být hodnocení dalších buněk (je-li vhodné) nebo provedení opakovaného experimentu, případně za optimalizovaných zkušebních podmínek (např. intervaly koncentrací, jiné způsoby podávání dávek, jiné doby odběru vzorků nebo jiné tkáně). Ve vzácných případech ani po dalším šetření nebude možné na základě daného souboru údajů dospět k závěru o pozitivitě či negativitě výsledku, a bude proto učiněn závěr, že výsledek je neurčitý. Ke zhodnocení biologického významu pozitivního nebo neurčitého výsledku jsou nutné informace o cytotoxicitě na cílovou tkáň (viz odstavce 54–55). V případě, že jsou pozitivní nebo neurčitá zjištění pozorována pouze za přítomnosti zjevných důkazů cytotoxicity, měla by být studie vyhodnocena jako neurčitá z hlediska genotoxicity, pokud neexistuje dostatek informací podporujících jednoznačný závěr. V případě negativního výsledku studie, kdy existují známky toxicity při všech zkoušených dávkách, lze doporučit další studii při netoxických dávkách. Závěrečná zpráva Závěrečná zpráva by měla obsahovat tyto informace:
LITERATURA
Dodatek 1 DEFINICE Alkalická jednobuněčná gelová elektroforéza : citlivá metoda detekce primárního poškození DNA na úrovni jednotlivé buňky/jádra. Chemická látka : chemická substance nebo směs. Kometa : tvar, který nabývají nukleoidy poté, co jsou vystaveny jednomu elektroforéznímu poli, vzhledem k podobnosti kometám: hlavou je jádro a ohon tvoří DNA migrující z jádra ven v elektrickém poli. Kritická proměnná / kritický parametr : je to proměnná protokolu, jejíž malá změna může mít velký dopad na závěry zkoušky. Kritické proměnné mohou být u různých tkání různé. Kritické proměnné by neměly být měněny, zejména v průběhu zkoušky, aniž by se zvážilo, jak může změna změnit odezvu ve zkoušce vyjádřenou například velikostí a variabilitou v pozitivních a negativních kontrolách. V závěrečné zprávě by měly být uvedeny změny kritických proměnných provedené během zkoušky nebo ve srovnání se standardním protokolem dané laboratoře a každá změna by měla být odůvodněna. Intenzita ohonu nebo procentuální zastoupení DNA v ohonu : odpovídá intenzitě ohonu komety v poměru k celkové intenzitě (hlavy plus ohonu). Zohledňuje množství zlomů DNA vyjádřené v procentech. Zkoušená chemická látka : Jakákoli chemická substance nebo směs zkoušená touto zkušební metodou. UVCB : látky s neznámým nebo proměnlivým složením, komplexní reakční produkty nebo biologický materiál. Dodatek 2 FAKTORIÁLNÍ POKUS KE ZJIŠTĚNÍ ROZDÍLŮ MEZI POHLAVÍMI V KOMETOVÉM TESTU IN VIVO Faktoriální pokus a jeho analýza Při tomto pokusu se na každé úrovni koncentrace testuje minimálně 5 samců a 5 samic a výsledkem je uspořádání s použitím minimálně 40 zvířat (20 samců a 20 samic plus příslušné pozitivní kontroly). Tento pokus, který je jedním z nejjednodušších faktoriálních pokusů, je rovnocenný dvoucestné analýze rozptylu, přičemž hlavními efekty jsou úroveň koncentrace a pohlaví. Údaje je možné analyzovat pomocí mnoha standardních statistických počítačových programů, např. SPSS, SAS, STATA, Genstat a také pomocí programu R. Analýza dělí variabilitu v souboru údajů na variabilitu mezi pohlavími, variabilitu mezi úrovněmi koncentrace a variabilitu interakce mezi pohlavími a koncentracemi. Každý z těchto znaků se testuje ve srovnání s odhadem variability mezi paralelními zvířaty v rámci skupin zvířat stejného pohlaví, kterým je podávána stejná koncentrace látky. Veškeré podrobnosti příslušné metodiky jsou dostupné v mnoha běžných učebnicích statistiky (viz odkazy) a v příručkách ke statistickým programům. Analýza se provádí zkoumáním znaku interakce pohlaví x koncentrace v tabulce ANOVA (35). Neexistuje-li význačný znak interakce, kombinované hodnoty za obě pohlaví nebo za všechny úrovně koncentrace poskytují validní statistické testy variability mezi úrovněmi založené na zkoumání kumulovaného znaku vnitroskupinové variability pomocí analýzy ANOVA. Analýza pokračuje rozdělením odhadu variability mezi koncentracemi na kontrasty, což vede k testování lineárních a kvadratických kontrastů odezev na všech úrovních koncentrace. Pokud existuje významná interakce pohlaví x koncentrace, lze tento znak také rozdělit na kontrasty interakce lineární x pohlaví a kvadratický x pohlaví. Pomocí těchto členů se testuje, zda jsou odpovědi na koncentraci u obou pohlaví paralelní, nebo zda je odpověď u každého pohlaví odlišná. Odhad kumulované vnitroskupinové variability může být použit k párovému testování rozdílu mezi středními hodnotami. Tato srovnání by mohla být provedena mezi středními hodnotami u obou pohlaví a mezi středními hodnotami u různých úrovní koncentrace, např. za účelem porovnání s úrovněmi negativní skupiny. V případech, kdy existuje významná interakce, lze provádět srovnání mezi středními hodnotami při různých koncentracích v rámci jednotlivých pohlaví nebo mezi středními hodnotami každého pohlaví při stejné koncentraci. Odkazy Existuje množství učebnic statistiky, které se zabývají teorií, uspořádáním, metodikou, analýzou a interpretací faktoriálních pokusů od nejjednodušších dvoufaktorových analýz až po složitější formy používané v metodice experimentálního uspořádání. Následující seznam není úplný. V některých pracích jsou uvedeny vypracované příklady srovnatelných uspořádání, v některých případech i s kódem pro provádění analýz pomocí různých počítačových programů.
Dodatek 3 OMEZENÍ ZKOUŠKY V SOUČASNOSTI Za současného stavu poznání jsou s kometovým testem in vivo spojena i některá omezení. Očekává se, že tato omezení budou redukována nebo úžeji definována, jakmile bude k dispozici více zkušeností s použitím zkoušky, které umožní řešit příslušné bezpečnostní otázky v právních předpisech.
Odkazy
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||



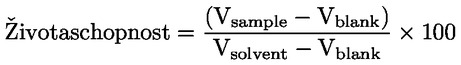


|
16) |
V části C se kapitola C.13 nahrazuje tímto: „C.13 Bioakumulace v rybách: expozice ve vodním prostředí a v potravě ÚVOD Tato zkušební metoda je rovnocenná Pokynu OECD pro zkoušení (TG) č. 305 (2012). Revize této zkušební metody sleduje dva hlavní cíle. Za prvé má začlenit zkoušku bioakumulace v potravě (36) vhodnou pro stanovení bioakumulačního potenciálu látek s velmi nízkou rozpustností ve vodě. Za druhé má vytvořit zkušební metodu, ve které se ve vhodných případech z důvodu dobrých životních podmínek zvířat používá méně ryb a která je nákladově efektivnější. V letech po přijetí konsolidované zkušební metody C.13 (1) bylo zkoušeno mnoho látek a laboratoře i regulační orgány získaly značné zkušenosti. To vedlo k přesvědčení, že při splnění určitých kritérií (srov. odstavec 88) je možné složitost zkoušky snížit a že lze uplatnit postupný přístup. Zkušenosti také ukázaly, že biologické faktory, např. růst ryb a obsah lipidů v nich, mohou mít značný dopad na výsledky a že je zapotřebí je brát v úvahu. Bylo rovněž zjištěno, že zkoušení látek velmi málo rozpustných ve vodě nemusí být technicky proveditelné. U látek s velmi nízkou rozpustností ve vodě může mít expozice touto cestou omezený význam ve srovnání s expozicí v potravě. To vedlo k vývoji zkušební metody, ve které jsou ryby exponovány prostřednictvím potravy (srov. odstavce 7 až 14 a odstavec 97 a následující). V roce 2010 byla provedena validace (mezilaboratorní porovnávací zkouška) zkoušky expozice v potravě. Hlavní změny jsou tyto:
Před provedením zkoušky bioakumulace je třeba získat tyto informace o zkoušené látce:
Nezávisle na zvolené metodě expozice nebo schématu odběru vzorků je v této zkušební metodě popsán postup pro charakterizaci potenciálu látek bioakumulovat se v rybách. Ačkoli jsou průtokové zkušební režimy preferovány, připouštějí se i semistatické režimy, jsou-li splněna kritéria validace (srov. odstavce 24 a 113). V případě expozice v potravě není průtokový systém nezbytný pro udržení koncentrace zkoušené látky ve vodním prostředí, ale pomůže udržet přiměřené koncentrace rozpuštěného kyslíku a zajistit čistou vodu a také eliminovat vlivy např. výměšků. Nezávisle na zvolené zkušební metodě jsou ve zkušební metodě dostatečně popsány podrobnosti pro provedení zkoušky, přičemž je poskytnuta dostatečná volnost pro přizpůsobení experimentálního uspořádání podmínkám v jednotlivých laboratořích a různým vlastnostem zkoušených látek. Zkouška expozice ve vodním prostředí je nejvhodnější pro stabilní organické látky s hodnotou log K OW od 1,5 do 6,0 (13), ale může být také použita na silně hydrofobní látky (s hodnotou log K OW > 6,0), pokud lze prokázat stabilní koncentraci plně rozpuštěné zkoušené látky ve vodě. Jestliže nelze prokázat stabilní koncentraci zkoušené látky ve vodě, studie ve vodním prostředí by nebyla vhodná a bylo by nutné pro zkoušení látky na rybách použít postup expozice v potravě (i když interpretace a využití výsledků zkoušky s potravou může záviset na regulačním rámci). Předběžné odhady biokoncentračního faktoru (BCF, někdy označovaného jako K B) pro organické látky s hodnotou log K OW až asi 9,0 lze získat pomocí rovnice Binteina et al. (14). Předběžně odhadnutý biokoncentrační faktor může být pro silně hydrofobní látky vyšší než hodnota biokoncentračního faktoru v rovnovážném stavu (BCFSS), která je očekávána z laboratorních experimentů, zejména pokud je pro předběžný odhad použit jednoduchý lineární model. Mezi parametry, které charakterizují bioakumulační potenciál, patří rychlostní konstanta příjmu (k 1), rychlostní konstanty úbytku včetně rychlostní konstanty vylučování (k 2), biokoncentrační faktor v rovnovážném stavu (BCFSS), kinetický biokoncentrační faktor (BCFK) a potravní bioobohacovací faktor (BMF) (38). Radioizotopově značené zkoušené látky mohou usnadnit analýzu vody, vzorků potravy a ryb a mohou být použity v případě, že by měla být provedena identifikace a kvantifikace metabolitů. Měří-li se pouze celkový obsah radioaktivního zbytku (např. po spálení nebo solubilizaci tkáně), je hodnota BCF nebo BMF založena na součtu mateřské látky, všech zadržených metabolitů a také asimilovaného uhlíku. Hodnoty BCF nebo BMF založené na celkovém obsahu radioaktivního zbytku tedy nemohou být přímo srovnatelné s hodnotou BCF nebo BMF získanou specifickou chemickou analýzou pouze mateřské látky. V izotopových studiích mohou být pro stanovení BCF nebo BMF mateřské látky před analýzou zařazeny separační postupy, např. TLC, HPLC nebo GC (39). Jsou-li použity separační metody a pokud má být hodnota BCF nebo BMF založena na koncentraci mateřské látky v rybách, a nikoli na celkovém obsahu radioizotopově značeného zbytku, měla by se provést identifikace a kvantifikace mateřské látky a příslušných metabolitů (40) (srov. odstavec 65). Je možné rovněž kombinovat studii metabolismu ryb nebo distribuce in vivo se studií bioakumulace analýzou a identifikací zbytku v tkáních. Možnost metabolismu lze předpovědět vhodnými prostředky (např. souborem nástrojů QSAR OECD (15) a patentovanými programy QSAR). Rozhodnutí, zda provézt zkoušku expozice ve vodním prostředí nebo v potravě a v jakém uspořádání by mělo vycházet z faktorů uvedených v odstavci 3, které se zváží spolu s příslušným regulačním rámcem. Například pro látky, které mají vysokou hodnotu K OW, ale přesto vykazují značnou rozpustnost ve vodě s ohledem na citlivost dostupných analytických metod, by měla být v první řadě zvážena zkouška expozice ve vodním prostředí. Je však možné, že informace o rozpustnosti těchto hydrofobních druhů látek ve vodě nejsou konečné, takže před přijetím rozhodnutí, kterou zkušební metodu použít, by měla být zkoumána možnost přípravy stabilních, měřitelných koncentrací ve vodním prostředí (stabilní emulze nejsou přípustné) použitelných pro studii expozice ve vodním prostředí. Na základě „hraničních“ kritérií rozpustnosti ve vodě a rozdělovacího koeficientu oktanol/voda není možné vydat přesný normativní pokyn, jaké metody mají být použity, protože zřetelný vliv na použitelnost metody mají z výše uvedených důvodů také jiné faktory (analytické metody, rozklad, adsorpce atd.). Hodnota log K OW vyšší než 5 a rozpustnost vody nižší než ~0,01 až 0,1 mg/l vymezují okruh látek, jejichž zkoušení prostřednictvím expozice ve vodním prostředí může být stále obtížnější. Měly by se zvážit i jiné faktory, které mohou ovlivnit výběr zkoušky, včetně schopnosti látky adsorbovat na zkušebních nádržích, přístrojích a vybavení, její stability ve vodném roztoku v porovnání s její stabilitou v rybí potravě (17) (18) atd. Informace o takových praktických aspektech mohou být dostupné z jiných dokončených studií ve vodním prostředí. Další informace týkající se hodnocení aspektů provádění bioakumulačních studií jsou k dispozici v literatuře (např. (19)). U látek, kde rozpustnost nebo zachování koncentrace ve vodním prostředí, jakož i analýza této koncentrace (koncentrací) nepředstavuje omezení pro uskutečnění metody expozice ve vodním prostředí, se pro stanovení bioakumulačního potenciálu látky upřednostňuje tato metoda. V každém případě by se mělo ověřit, že zkušební koncentrace, které mají být použity ve vodním prostředí, jsou v rozmezí rozpustnosti ve zkušebním médiu ve vodním prostředí. Pro zachování stabilních koncentrací rozpuštěné zkoušené látky lze použít různé metody, jako např. použití zásobních roztoků nebo pasivních dávkovacích systémů (např. sloupcové eluční metody), pokud lze prokazatelně udržet stabilní koncentrace a pokud zkušební média se nezmění z hodnot doporučených v odstavci 27. U silně hydrofobních látek (log K OW > 5 a rozpustnost menší než ~ 0,01 až 0,1 mg/l) může být zkoušení prostřednictvím expozice ve vodním prostředí stále obtížnější. Důvodem pro omezení může být skutečnost, že není možné zachovat koncentraci ve vodním prostředí v míře, která je považována za dostatečně konstantní (např. v důsledku sorpce na skle expozičních nádob nebo rychlého příjmu rybami), nebo že koncentrace ve vodním prostředí, které mají být použity, jsou tak nízké, že jsou na úrovni analyticky stanovené meze kvantifikace nebo jsou ještě nižší (41). Pro tyto vysoce hydrofobní látky se doporučuje zkouška expozice v potravě, pokud je tato zkouška v souladu s příslušným regulačním rámcem a s potřebami posouzení rizik. U povrchově aktivních látek by se mělo zvážit, zda zkouška biokoncentrace ve vodním prostředí je vzhledem k vlastnostem látky proveditelná; v opačném případě je patrně vhodnější studie expozice v potravě. Povrchově aktivní látky jsou činidla, která působí na povrchu, což snižuje mezifázové napětí mezi dvěma kapalinami. Jejich amfifilní povaha (tj. obsahují jak hydrofilní, tak hydrofobní složku) způsobuje, že se akumulují na povrchu, např. na rozhraní voda-vzduch, na rozhraní voda-potrava a na skleněných stěnách, což narušuje stanovení jejich koncentrace ve vodním prostředí. Zkouškou expozice v potravě je možné u komplexních směsí se složkami s různými mezemi rozpustnosti ve vodě obejít některé z aspektů expozice, neboť srovnatelná expozice u všech složek směsi je pravděpodobnější než při metodě vodního prostředí (srov. (20)) Nutno poznamenat, že postup expozice v potravě poskytuje potravní bioobohacovací faktor (BMF), a nikoli biokoncentrační faktor (BCF) (42). K dispozici jsou metody, jimiž lze odhadnout kinetický biokoncentrační faktor (BCFK) z údajů získaných při studii potravy (jak se pojednává v dodatku 8), avšak tyto metody je třeba používat s obezřetností. Obecně tyto metody předpokládají kinetiku prvního řádu a jsou použitelné pouze na některé skupiny sloučenin. Použitelnost těchto metod pro povrchově aktivní látky je nepravděpodobná (viz odstavec 12). Minimalizované nastavení zkoušky expozice ve vodním prostředí s menším počtem odběrů vzorků s cílem snížit počet ryb a/nebo zdrojů (srov. odstavec 83 a dále), by mělo být použito pouze pro látky, při jejichž příjmu a vylučování lze přibližně očekávat kinetiku prvního řádu (tj. obecně nedisociované organické látky, srov. odstavec 88). C.13 – I Zkouška biokoncentrace v rybách s expozicí ve vodním prostředí PODSTATA ZKOUŠKY Zkouška má dvě fáze: fázi expozice (příjem) a poexpoziční fázi (vylučování). Během fáze příjmu je skupina ryb stejného druhu vystavena jedné nebo více zvoleným koncentracím zkoušené látky v závislosti na vlastnostech zkoušené látky (srov. odstavec 49). Poté jsou přemístěny do média, které zkoušenou látku neobsahuje, aby začala fáze vylučování. Fáze vylučování je vždy nezbytná, není-li příjem látky během fáze příjmu nevýznamný. Koncentrace zkoušené látky v rybách nebo na nich (nebo v jejich specifikovaných tkáních) se sleduje v průběhu obou fází zkoušky. Vedle zkušební skupiny se za stejných podmínek, s výjimkou přítomnosti zkoušené látky, udržuje kontrolní skupina ryb, aby mohly být na odpovídající kontrolní skupině porovnány možné nepříznivé účinky pozorované při biokoncentračních zkouškách a aby byly získány koncentrace zkoušené látky v pozadí (43). Při zkoušce expozice ve vodním prostředí trvá fáze příjmu obvykle 28 dnů. Její délku lze v případě potřeby prodloužit (srov. odstavec 18) nebo zkrátit, je-li prokázáno, že je rovnovážného stavu dosaženo dříve (viz dodatek 1, definice a jednotky). Délku fáze příjmu a dobu potřebnou k ustavení rovnovážného stavu lze předpovědět pomocí rovnice v dodatku 5. Fáze vylučování poté začne, když ryby již nejsou exponovány zkušební látce, po jejich přemístění do čisté nádrže se stejným médiem, ale bez zkoušené látky. Je-li to možné, vypočítá se biokoncentrační faktor přednostně jednak jako poměr koncentrace v rybách (Cf) a koncentrace ve vodě (Cw) v rovnovážném stavu (BCFSS; viz dodatek 1, definice) a jednak jako kinetický biokoncentrační faktor (BCFK, viz dodatek 1, definice), který se odhadne jako poměr rychlostních konstant příjmu (k1 ) a vylučování (k2 ) za předpokladu kinetiky prvního řádu (44). Nedojde-li k rovnovážnému stavu do 28 dnů, pak se buď vypočítá BCF s použitím kinetického přístupu (viz odstavec 38), nebo je možné fázi příjmu prodloužit. Pokud by to mělo vést k tomu, že k dosažení rovnovážného stavu bude zapotřebí neprakticky dlouhá fáze příjmu (srov. odstavce 37 a 38, dodatek 5), upřednostní se kinetický přístup. Alternativně by u vysoce hydrofobních látek měla být zváženo provedení studie s expozicí v potravě (45), pokud zkouška expozice v potravě je v souladu s příslušným regulačním rámcem. Rychlostní konstanta příjmu, rychlostní konstanta (nebo konstanty, jsou-li použity složitější modely) vylučování (úbytku), biokoncentrační faktor (v rovnovážném stavu a/nebo kinetický), a je-li to možné, také intervaly spolehlivosti každého z těchto parametrů se vypočítají z modelu, který nejlépe popisuje naměřené koncentrace zkoušené látky v rybách a ve vodě (srov. dodatek 5). Růst hmotnosti ryb během zkoušky povede k poklesu koncentrace zkoušené látky v rostoucích rybách (takzvané zředění růstem) a kinetický BCF bude tedy podhodnocen, pokud nebude upraven o růst (srov. odstavce 72 a 73). Hodnota BCF je založena na celkové koncentraci v rybách (tj. vypočítává se z celkové živé hmotnosti ryby). Pro zvláštní účely však mohou být použity specifikované tkáně nebo orgány (např. svalovina, játra), je-li ryba dostatečně velká nebo je-li možné ji rozdělit na jedlý (vykostěný) podíl a nejedlý podíl (vnitřnosti). Vzhledem k tomu, že u mnoha organických látek existuje zřetelný vztah mezi schopností biokoncentrace a hydrofobií, existuje také odpovídající vztah mezi obsahem tuku v zkušebních rybách a pozorovanou biokoncentrací takových látek. Aby se tedy snížilo kolísání výsledků pro tyto látky s vysokou lipofilností (tj. s log K OW > 3), měla by biokoncentrace kromě přímého odvození ze studie být také vyjádřena v normalizované podobě, a to pro ryby s 5 % obsahem tuku (na základě hmotnosti celého těla). To je nezbytné pro získání základu, na kterém bude možné vzájemně porovnávat výsledky pro jednotlivé látky a/nebo zkušební druhy. Hodnota 5 % obsahu tuku se široce používá, neboť představuje průměrný obsah tuku v rybách běžně používaných v této zkušební metodě (21). INFORMACE O ZKOUŠENÉ LÁTCE Kromě vlastností zkoušené látky uvedených v úvodu (odstavec 3) jsou dalšími požadovanými informacemi toxicita pro druh ryby, který má být při zkoušce použit, nejlépe asymptotická hodnota LC50 (tj. časově nezávislá) a/nebo toxicita odhadnutá při dlouhodobých zkouškách na rybách (např. zkušební metody C.47 (22), C.15 (23), C.14 (24)). K dispozici musí být vhodná analytická metoda o známé přesnosti a citlivosti pro kvantitativní stanovení látky ve zkušebních roztocích a v biologickém materiálu a dále podrobnosti o přípravě a uchovávání vzorků. Měly by být také známy analytické meze pro stanovení obsahu zkoušené látky jak ve vodě, tak v rybích tkáních. Je-li použita radioizotopově značená zkoušená látka, měla by mít nejvyšší čistotu (např. nejlépe > 98 %) a měla by být známa aktivita nečistot vyjádřená v procentech. VALIDITA ZKOUŠKY Aby byla zkouška platná, musí být splněny tyto podmínky:
REFERENČNÍ LÁTKY Použití referenčních látek o známém biokoncentračním potenciálu a nízkém metabolismu by mohlo být užitečné pro kontrolu experimentálního postupu, je-li požadována (např. když laboratoř nemá předchozí zkušenosti se zkouškou nebo když se zkušební podmínky změnily). POPIS METODY Přístroje a pomůcky U všech částí zařízení je třeba se vyhýbat použití materiálů, které se mohou rozpouštět, sorbovat nebo vyluhovat a mají nepříznivý účinek na ryby. Lze použít standardní pravoúhlé nebo válcové nádrže vyrobené z inertního materiálu a mající vhodný objem s ohledem na rychlost nasazování (srov. odstavec 43). Použití měkkých trubek z plastu by mělo být minimalizováno. Měly by být použity trubky z polytetrafluoretylenu (teflon), z korozivzdorné oceli a/nebo ze skla. Zkušenosti ukázaly, že pro zkoušené látky s vysokými absorpčními koeficienty, jako jsou syntetické pyrethroidy, je nutné použít silanizované sklo. V těchto případech musí být zařízení po použití zlikvidováno. Upřednostňuje se vystavit zkušební systémy koncentracím zkoušené látky, která má být ve studii použita, tak dlouho, kolik je zapotřebí k prokázání udržení stabilních expozičních koncentrací, dříve než jsou zavedeny zkušební organismy. Voda Ke zkoušce se zpravidla používá přírodní voda, která by měla být získána z nekontaminovaného zdroje stálé kvality. Pro zaručení jednotné kvality po celou dobu však může být vhodnější rekonstituovaná voda (tj. demineralizovaná voda, do které byly přidány specifické živiny ve známých množstvích). Ředicí voda, což je voda použitá ke smísení se zkoušenou látkou před vstupem do zkušební nádrže (viz odstavec 30), by měla mít takovou kvalitu, která umožní přežití zvoleného druhu ryby po dobu aklimatizace a v průběhu fází zkoušky, aniž by vykazoval abnormální vzhled nebo chování. V ideálním případě by mělo být prokázáno, že zkušební druh může v ředicí vodě přežít, růst a rozmnožovat se (např. zkouškou s laboratorní kulturou nebo zkouškou toxicity během životního cyklu). Ředicí voda by měla být charakterizována alespoň hodnotou pH, tvrdostí, celkovým obsahem pevných látek, celkovým obsahem organického uhlíku (TOC (47)) a podle možnosti také obsahem amoniaku a dusičnanů, alkalitou a v případě mořských druhů salinitou. Všechny parametry, které jsou důležité pro optimální prospívání ryb, nejsou známy; v dodatku 2 jsou přesto uvedeny doporučené maximální koncentrace pro řadu parametrů sladké a mořské vody. Ředicí voda by měla mít po celou dobu zkoušky stejnou kvalitu. Hodnota pH by na začátku zkoušky měla být v rozmezí 6,0 až 8,5, avšak během zkoušky by se pH nemělo lišit o více než ±0,5. Pro ujištění, že ředicí voda nebude mít přílišný vliv na výsledky zkoušky (například tvorbou komplexů se zkoušenou látkou) nebo nepříznivý vliv na stav obsádky ryb, by měly být odebírány v pravidelných intervalech její vzorky pro analýzu, alespoň na začátku a na konci zkoušky. Je-li kvalita vody relativně konstantní, mělo by být stanovení těžkých kovů (např. Cu, Pb, Zn, Hg, Cd a Ni), hlavních aniontů a kationtů (např. Ca2+, Mg2+, Na+, K+, Cl–, a SO4 2–), pesticidů (např. celkový obsah organofosforových a organochlorových pesticidů), celkový obsah organického uhlíku a suspendovaných látek provedeno např. každé tři měsíce. Je-li prokázáno, že kvalita ředicí vody je konstantní po dobu alespoň jednoho roku, mohou být stanovení méně častá a intervaly lze prodloužit (např. na šest měsíců). Celkový přirozený obsah částic a rovněž obsah organického uhlíku (TOC) v ředicí vodě by měl být co nejnižší, aby nedošlo k adsorpci zkoušené látky na organických látkách, což může snížit její biologickou dostupnost, a může tak vést k podhodnocení BCF. Maximální přijatelná hodnota je 5 mg/l pro částice (sušina zachycená filtrem 0,45 μm) a 2 mg/l pro celkový organický uhlík (srov. dodatek 2). Je-li to nezbytné, měla by být ředicí voda před použitím filtrována. Příspěvek k obsahu organického uhlíku ve zkušební vodě od zkušebních ryb (exkrety) a ze zbytků potravy by měl být udržován na co nejmenší úrovni (srov. odstavec 46). Zkušební roztoky Zásobní roztok zkoušené látky se připraví ve vhodné koncentraci. Zásobní roztok by měl být připraven nejlépe jednoduchým mechanickým mícháním nebo protřepáním zkoušené látky s ředicí vodou. Vhodnou alternativou může být v některých případech použití dávkovacího systému s desorpcí tuhé fáze. Použití rozpouštědel nebo dispergátorů (solubilizačních činidel) se zpravidla nedoporučuje (srov. (25)); použití těchto látek však může být přijatelné pro vytvoření zásobního roztoku o vhodné koncentraci, ale je třeba vyvinout maximální úsilí, aby se takové látky používaly v co nejmenší míře a neměla by být překročena jejich kritická koncentrace micel (je-li to relevantní). Rozpouštědly, která mohou být použita, jsou aceton, ethanol, methanol, dimethylformamid a triethylenglykol; dispergátory, které byly použity, jsou Tween 80, 0,01 % methylcelulosa a HCO-40. Koncentrace rozpouštědla v konečném zkušebním médiu by měla být stejná jako při všech expozicích (tj. bez ohledu na koncentraci zkoušené látky) a neměla by být vyšší než příslušné prahové hodnoty toxicity stanovené pro rozpouštědlo za zkušebních podmínek. Maximální koncentrace je 100 mg/l (nebo 0,1 ml/l). Není pravděpodobné, že koncentrace rozpouštědla 100 mg/l významně změní maximální koncentraci rozpuštěné zkoušené látky, které lze v médiu dosáhnout (25). Příspěvek rozpouštědla (společně s příspěvkem zkoušené látky) k celkovému obsahu organického uhlíku ve zkušební vodě by měl být znám. V průběhu zkoušky by neměla koncentrace organického uhlíku ve zkušební nádrži překročit koncentraci organického uhlíku pocházejícího ze zkoušené látky a z rozpouštědla nebo solubilizačního činidla (48), je-li použito, o více než 10 mg/l (± 20 %). Obsah organických látek může mít významný účinek na množství volně rozpuštěné zkoušené látky během průtokových zkoušek na rybách, zejména u vysoce lipofilních látek. Důležité informace o poměru mezi vázanými a volně rozpuštěnými sloučeninami, přičemž se předpokládá, že volně rozpustné sloučeniny představují biologicky dostupnou frakci, může poskytnout mikroextrakce tuhé fáze (srov. odstavec 60). Koncentrace zkoušené látky by měla být nižší než je mez rozpustnosti zkoušené látky ve zkušebních médiích, nehledě na použití rozpouštědla nebo solubilizačního činidla. Snadno biologicky odbouratelná rozpouštědla je třeba používat rozvážně, neboť mohou způsobit problémy s růstem bakterií v průtokových zkouškách. Není-li možné připravit zásobní roztok bez použití solubilizačního činidla, měla by se zvážit vhodnost studie expozice ve vodním prostředí v porovnání se studií expozice v potravě. Při průtokových zkouškách je pro zajištění koncentrací zkoušené látky ve zkušebních nádržích nezbytný systém, který nepřetržitě dávkuje a ředí zásobní roztok zkoušené látky (např. měřicí čerpadlo, proporcionální ředění, saturační systém) nebo dávkovací systém s desorpcí tuhé fáze. Přijatelná je výměna nejlépe pěti objemů v každé zkušební nádrži za den. Upřednostňuje se průtokový režim, není-li to však možné (např. jsou-li zkušební organismy nepříznivě ovlivňovány), může být použita semistatická technika za předpokladu, že jsou splněna kritéria validity (srov. odstavec 24). Rychlosti průtoku zásobního roztoku a ředicí vody by měly být kontrolovány jak 48 hodin před zkouškou, tak poté alespoň jednou denně během zkoušky. Do této kontroly se zahrne stanovení rychlosti průtoku v každé zkušební nádrži a zajistí se, aby se rychlost průtoku neměnila o více než 20 % v rámci jedné nádrže nebo mezi nádržemi. Výběr druhů Důležitými kritérii pro výběr druhu jsou jeho dostupnost, možnost získat jej ve vyhovující velikosti a jeho bezproblémový chov v laboratoři. Dalšími kritérii pro výběr druhu ryb jsou jeho rekreační, komerční a ekologický význam a rovněž srovnatelná citlivost, úspěšné dřívější použití atd. Doporučené druhy jsou uvedeny v dodatku 3. Mohou být použity i jiné druhy, avšak zkušební postup musí být upraven, aby byly vytvořeny vhodné zkušební podmínky. V takovém případě by měly být uvedeny důvody pro výběr druhu a experimentální metoda. Obecně platí, že použití menších druhů ryb obecně zkrátí dobu potřebnou k dosažení rovnovážného stavu, avšak může být zapotřebí většího počtu ryb (vzorků), aby bylo možné dostatečně analyzovat obsah lipidů a koncentrace zkoušené látky v rybách. Navíc je možné, že rozdíly v rychlosti dýchání a metabolismu mezi mladými a staršími rybami mohou narušit porovnání výsledků mezi různými zkouškami a zkušebními druhy. Nutno poznamenat, že druhy ryb zkoušené během (nedospělého) životního stadia s rychlým růstem mohou zkomplikovat interpretaci údajů. Chov ryb (příslušný expozici ve vodním prostředí a v potravě) Obsádka ryb by se měla nechat aklimatizovat alespoň dva týdny (srov. odstavec 28) při zkušební teplotě a krmí se odpovídající potravou (srov. odstavec 45). Voda i potrava by měly být stejného druhu jako ty, které se použijí během zkoušky. Po 48 hodinách aklimatizace se zaznamená mortalita a použijí se tato kritéria:
Ryby použité pro zkoušku by neměly vykazovat pozorovatelné příznaky nemoci a abnormality. Všechny nemocné ryby se vymění. Dva týdny před zkouškou nebo v průběhu zkoušky nesmí být léčeny u ryb žádné nemoci. PROVEDENÍ ZKOUŠKY Předběžná zkouška V zájmu optimalizace zkušebních podmínek konečné zkoušky nebo stanovení, zda je nutné provést úplnou zkoušku, může být užitečné provést předběžný experiment, např. výběr koncentrace (koncentrací) zkoušené látky, délka fáze příjmu a fáze vylučování. Uspořádání předběžné zkoušky by mělo být takové, aby se získaly nezbytné informace. Lze zvážit, zda by k odvození hodnoty BCF postačila minimalizovaná zkouška nebo zda je nutná úplná studie (o minimalizované zkoušce srov. odstavce 83 až 95). Podmínky expozice Délka fáze příjmu Předpověď délky fáze příjmu lze získat z praktických zkušeností (např. z dřívější studie nebo ze studie akumulace strukturně podobné látky) nebo z určitých empirických vztahů využívajících znalosti buď rozpustnosti ve vodě, nebo rozdělovacího koeficientu n-oktanol/voda pro zkoušenou látku (za předpokladu, že se příjem řídí kinetikou prvního řádu, srov. dodatek 5). Fáze příjmu trvá 28 dnů, není-li prokázáno, že je rovnovážného stavu dosaženo dříve (viz dodatek 1, definice a jednotky). Rovnovážného stavu je dosaženo tehdy, je-li při grafickém znázornění křivka časové závislosti koncentrace zkoušené látky v rybách (C f ) rovnoběžná s časovou osou a tři po sobě jdoucí analýzy C f ve vzorcích odebraných v intervalech alespoň dvou dnů se mezi sebou neliší více než o ± 20 % a nedojde-li v době mezi první a poslední následnou analýzou k výraznému nárůstu hodnoty C f . Analyzují-li se spojené vzorky, jsou nezbytné alespoň čtyři po sobě jdoucí analýzy. V případě zkoušených látek, jejichž příjem probíhá pomalu, jsou vhodnější sedmidenní intervaly. Nedosáhne-li se rovnovážného stavu do 28 dnů, pak se buď vypočítá hodnota BCF pouze pomocí kinetické metody, která není závislá na dosažení rovnovážného stavu, anebo může být fáze příjmu prodloužena do jeho dosažení, přičemž se provádějí další měření, nebo na 60 dnů, podle toho, co nastane dříve. Koncentrace zkoušené látky v rybách na konci fáze příjmu také musí být dostatečně vysoká, aby byl zajištěn spolehlivý odhad hodnoty k 2 z fáze vylučování. Není-li zjištěn výrazný příjem po 28 dnech, lze zkoušku zastavit. Délka fáze vylučování U látek splňujících kinetiku prvního řádu je doba odpovídající polovině délky fáze příjmu obvykle dostatečná, aby se přiměřeně snížil (např. o 95 %) obsah látky v organismu (pro vysvětlení odhadu – srov. dodatek 5). Jestliže doba nezbytná pro dosažení 95 % úbytku je neprakticky dlouhá, překračuje například dvakrát běžnou délku fáze příjmu (tj. více než 56 dnů), může být použita kratší doba (např. dokud není koncentrace zkoušené látky menší než 10 % koncentrace v rovnovážném stavu). V případě látek se složitějším charakterem příjmu a vylučování, než jaký představuje model ryby s jedním kompartmentem řídícím se kinetikou prvního řádu, však mohou být nezbytné delší fáze vylučování. Je-li zaznamenán a/nebo se předpokládá takový složitější charakter, doporučuje se obrátit se o radu k biostatistikovi a/nebo farmakokinetikovi a zajistit vhodné nastavení zkoušky. Pokud se fáze vylučování prodlouží, může být počet ryb pro odběr vzorků omezený a výsledky mohou být ovlivněny rozdíly v růstu mezi rybami. Délka fáze však bude určena také dobou, po kterou zůstává koncentrace zkoušené látky v rybách nad analyticky stanovenou mezí kvantifikace. Počty zkušebních ryb Počet ryb na jednu zkušební koncentraci se zvolí tak, aby byly pro každý odběr vzorků k dispozici nejméně čtyři ryby. Ryby by měly být slučovány pouze v případě, že analýza jediné ryby je neproveditelná. Je-li cílem větší přesnost při stanovení křivky (a odvozených parametrů) nebo jsou-li požadovány studie metabolismu (např. za účelem rozlišení mezi metabolity a mateřskou látkou při použití radioizotopově značených zkoušených látek), bude nezbytný větší počet ryb pro každý odběr vzorků. Obsah lipidů by měl být stanoven na stejném biologickém materiálu, jaký je použit ke stanovení koncentrace zkoušené látky. Pokud by to nebylo proveditelné, mohou být zapotřebí další ryby (srov. odstavce 56 a 57). Použijí-li se dospělé (tj. pohlavně zralé) ryby, neměly by být ve stavu tření nebo vytřené nedávno před zkouškou nebo v jejím průběhu. Mělo by se též uvést, zda byli v experimentu použiti samci, nebo samice, nebo obě pohlaví. Jsou-li použita obě pohlaví, mělo by být před započetím expozice prokázáno, že rozdíl v růstu a v obsahu lipidů mezi oběma pohlavími není významný; zejména pokud se předpokládá, že bude nezbytné sloučení rybích samců a samic, aby byly zajištěny zaznamenatelné koncentrace látky a/nebo zaznamenatelný obsah lipidů. V každé zkoušce se vyberou ryby podobné hmotnosti, tak aby hmotnost nejmenší z nich nebyla nižší než dvě třetiny hmotnosti největší ryby. Všechny by měly být stejného stáří a měly by pocházet ze stejného zdroje. Vzhledem k tomu, že hmotnost a stáří ryby mohou mít významný vliv na hodnoty BCF (12), musí být tyto podrobnosti přesně zaznamenány. Doporučuje se zvážit krátce před zahájením zkoušky dílčí vzorek obsádky s cílem odhadnout střední hodnotu hmotnosti (srov. odstavec 61). Nasazování Měl by se zvolit vysoký poměr množství vody k množství ryb, aby se minimalizovalo snížení koncentrace zkoušené sloučeniny ve vodě způsobené přidáním ryb na začátku zkoušky, a také proto, aby nedošlo k poklesu koncentrace rozpuštěného kyslíku. Je důležité, aby rychlost nasazování byla přiměřená použitému druhu. V každém případě se obvykle doporučuje rychlost nasazování 0,1 až 1,0 g ryb (živá hmotnost) na litr vody za den. Vysoká rychlost nasazování může být zvolena, je-li prokázáno, že požadovaná koncentrace zkoušené látky může být udržována v mezích ± 20 % a že koncentrace rozpuštěného kyslíku neklesne pod 60 % nasycení (srov. odstavec 24). Při volbě vhodného režimu nasazování má být přihlédnuto k obvyklému přirozenému prostředí ryb. Například ryby žijící u dna mohou vyžadovat při stejném objemu vody větší plochu dna v akváriu než pelagické druhy ryb. Krmení Během aklimatizace a po dobu zkoušky se ryby krmí vhodnou potravou se známým obsahem tuku a celkových bílkovin podávanou v množství dostatečném pro udržení zdravého stavu a pro udržení tělesné hmotnosti (určitý růst je přípustný). Ryby se krmí denně v průběhu aklimatizace a po dobu zkoušky stanoveným množstvím podle použitého druhu ryb, zkušebních podmínek a kalorické hodnoty potravy (např. u pstruha duhového přibližně 1 až 2 % tělesné hmotnosti za den). Množství potravy by mělo být zvoleno tak, aby se zamezilo rychlému růstu a velkému nárůstu obsahu lipidů. Aby bylo možno zachovat stejné množství potravy, měla by být případně, například jednou týdně, nově přepočítána velikost krmných dávek. Hmotnost ryb v každé nádrži může být pro tento výpočet odhadnuta z hmotnosti ryb naposledy odebraných z dotyčné nádrže. Ryby, které zůstaly v nádrži, se neváží. Nezkrmená potrava a exkrety by se měly denně odstraňovat ze zkušebních nádrží odsátím krátce po krmení (30 minut až 1 hodina). Nádrže by se měly udržovat v celém průběhu zkoušky v co nejvyšší čistotě, aby byla koncentrace organických látek co nejnižší (srov. odstavec 29), neboť přítomnost organického uhlíku může snižovat biologickou dostupnost zkoušené látky (12). Poněvadž mnoho krmiv pochází z rybí moučky, mělo by se zajistit, že krmivo nebude ovlivňovat výsledky zkoušky ani vyvolávat nepříznivé účinky, např. kontaminací pesticidy nebo stopami pesticidů, těžkými kovy a/nebo přímo zkušební látkou. Světlo a teplota Doporučuje se fotoperioda 12 až 16 hodin a teplota (± 2 °C) by měla odpovídat zkušebnímu druhu (srov. dodatek 3). Druh a charakteristiky osvětlení by měly být známy. Měla by být věnována pozornost možné fototransformaci zkoušené látky za světelných podmínek studie. Mělo by být použito vhodné osvětlení, aby ryby nebyly vystaveny fotoproduktům nevyskytujícím se v přírodě. V některých případech může být vhodné použít filtr pro odfiltrování UV záření s vlnovou délkou kratší než 290 nm. Zkušební koncentrace Zkouška byla původně určena pro nepolární organické látky. U látek tohoto druhu se očekává, že bude dostatečná expozice ryb jediné koncentraci, jelikož se neočekávají žádné účinky koncentrace, ačkoli příslušný regulační rámec může vyžadovat použití dvou koncentrací. Jsou-li zkoušeny látky, které nepatří do této oblasti, nebo pokud jsou známy jiné známky možné závislosti na koncentraci, měla by být zkouška provedena s dvěma nebo více koncentracemi. Je-li zkoušena pouze jedna koncentrace, mělo by se uvést odůvodnění použití jediné koncentrace (srov. odstavec 79). Zkoušená koncentrace by také měla být co nejnižší, nakolik je to praktické nebo technicky možné (tj. nikoli blízká mezi rozpustnosti), V některých případech lze předpokládat, že biokoncentrace látky je závislá na koncentraci ve vodě (např. u kovů, kde příjem u ryb je možné alespoň částečně regulovat). V takovém případě je nezbytné, aby byly zkoušeny nejméně dvě, ale nejlépe více koncentrací (srov. odstavec 49), které jsou běžné v životním prostředí. Také v případě látek, u kterých zkoušené koncentrace z praktických důvodů musí být blízké mezi rozpustnosti, se doporučuje zkoušet nejméně dvě koncentrace, protože to může poskytnout informace o spolehlivosti zkušebních koncentrací. Zvolené zkušební koncentrace by měly zahrnovat koncentraci reálnou v životním prostředí i koncentraci, která odpovídá účelu konkrétního posouzení. Koncentrace zkoušené látky by se měla (měly) vybrat tak, aby byly nižší než úroveň jejích chronických účinků nebo než 1 % její asymptotické hodnoty akutní LC50, v rozpětí běžném pro životní prostředí a aby byla nejméně o jeden řád vyšší než je mez kvantifikace této látky ve vodě při použité analytické metodě. Nejvyšší přípustnou zkušební koncentraci lze také stanovit dělením akutní 96hodinové LC50 příslušným poměrem akutní/chronické letální dávky (např. u některých látek může být koeficient přibližně tři, ale u několika je vyšší než 100). Použije-li se druhá koncentrace, měla by být navýšena faktorem 10. Není-li to možné z důvodu kritéria toxicity (které limituje vyšší zkušební koncentraci) a z důvodu meze analytické metody (která limituje nižší zkušební koncentraci), může být použit nižší faktor než 10 nebo by mělo být zváženo použití zkoušené látky (nejvyšší čistoty, např. nejlépe > 98 %) značené radioizotopově. Je třeba dát pozor, aby žádná z použitých koncentrací nebyla vyšší než mez rozpustnosti zkoušené látky ve zkušebním médiu. Kontroly Kromě zkušebních sérií by měl být proveden kontrolní experiment s ředicí vodou, která popřípadě obsahuje rozpouštědlo (srov. odstavce 30 a 31). Četnost měření kvality vody V průběhu zkoušky by mělo být v každé zkušební a kontrolní nádrži měřeno množství rozpuštěného kyslíku, TOC, pH a teplota. Celková tvrdost a popřípadě salinita by měly být měřeny v kontrolních experimentech a v jedné nádrži. Jsou-li zkoušeny dvě nebo více koncentrací, měří se tyto parametry při vyšší (nebo nejvyšší) koncentraci. Množství rozpuštěného kyslíku a popřípadě salinita by měly být měřeny alespoň třikrát – na začátku, přibližně uprostřed a na konci fáze příjmu – a jednou týdně ve fázi vylučování. Hodnota TOC by měla být měřena na začátku zkoušky (24 hodin a 48 hodin před započetím fáze příjmu) před nasazením ryb a alespoň jednou týdně jak v průběhu fáze příjmu, tak v průběhu fáze vylučování. Teplota by měla být měřena a zaznamenávána denně, pH na začátku a na konci každé fáze a tvrdost vody jednou v průběhu každé zkoušky. Teplota by měla být měřena nejlépe nepřetržitě alespoň v jedné nádrži. Odběr vzorků a analýza ryb a vody Harmonogram odebírání vzorků ryb a vody Voda ze zkušebních nádrží by se pro stanovení koncentrace zkoušené látky měla odebírat před nasazením ryb a během fáze příjmu i během fáze vylučování. Vzorky vody by se měly odebírat před krmením, ve stejnou dobu jako vzorky ryb. Častější odběr vzorků může být užitečný pro zajištění stabilních koncentrací po nasazení ryb. Během fáze příjmu by se měly stanovovat koncentrace zkoušené látky, aby se ověřilo, že jsou v souladu s kritérii validity (odstavec 24). Pokud analýzy vzorků vody na počátku fáze vylučování ukážou, že zkoušená látka není detekována, lze to použít jako odůvodnění, že se ve zkušební vodě v kontrolním experimentu nebude provádět stanovení zkoušené látky po zbytek fáze vylučování. Ryby by se ke stanovení zkoušené látky měly odebírat alespoň pětkrát během fáze příjmu a alespoň čtyřikrát během fáze vylučování. Vzhledem k tomu, že v mnoha případech bude obtížné s tímto počtem vzorků vypočítat rozumně přesný odhad hodnoty BCF, zejména jde-li o jinou než jednoduchou kinetiku prvního řádu, může být účelné odebírat vzorky v obou fázích častěji (srov. dodatek 4). Obsah lipidů by se měl stanovovat na stejném biologickém materiálu, jako je použit ke stanovení koncentrace zkoušené látky, nejméně na začátku a na konci fáze příjmu a na konci fáze vylučování. Pokud by to nebylo uskutečnitelné, měly by být při každém z týchž tří odběrů nezávisle odebrány alespoň tři ryby pro stanovení obsahu lipidů. Na začátku experimentu by měl být odpovídajícím způsobem upraven počet ryb v každé nádrži (49). Alternativně, pokud u kontrolních ryb (tj. u ryb z kmenové populace) nejsou zjištěna žádná významná množství zkoušené látky, je možné u těchto ryb analyzovat pouze obsah lipidů a analýzu zkoušené látky v exponované skupině (skupinách) (a související hodnoty rychlostní konstanty příjmu, rychlostní konstanty vylučování a BCF) lze upravit o změny podle obsahu lipidů u kontrolní skupiny během zkoušky (50). Na mrtvých nebo nemocných rybách se analýza chemické látky ani obsahu lipidů neprovádí. Příklad přijatelného časového rozvrhu odběru vzorků je uveden v příloze 4. Jiné plány lze snadno odvodit pomocí předpokládané hodnoty K OW, s níž se vypočítá expoziční doba pro 95 % příjem (výpočty viz dodatek 5). V odběru vzorků by se mělo pokračovat během fáze příjmu do dosažení rovnovážného stavu (srov. dodatek 1, definice a jednotky) nebo se fáze příjmu ukončí jinak (po 28 nebo 60 dnech, viz odstavce 37 a 38). Před začátkem fáze vylučování by se ryby měly přemístit do čistých nádrží. Odběr vzorků a příprava vzorků Vzorky vody pro analýzy by se měly odebírat například odsáváním potrubím z inertního materiálu ze středu zkušební nádrže. Biologicky nedostupná frakce zkoušené látky se nedá často oddělit od biologicky dostupné frakce ani filtrací, ani odstředěním. Použije-li se separační metoda, mělo by se v závěrečné zprávě vždy uvést odůvodnění nebo validace této separační metody vzhledem k obtížím s biologickou dostupností. Zejména v případě vysoce hydrofobních látek (tj. látek s hodnotou log K OW > 5) (12) (26), kdy by mohlo dojít k adsorpci na filtrovou matrici nebo na odstřeďovací nádoby, by vzorky neměly být takto zpracovávány. Namísto toho je třeba přijmout opatření pro to, aby byly nádrže udržovány co nejčistší (srov. odstavec 46), a obsah organického uhlíku by měl být pravidelně monitorován jak během fáze příjmu, tak během fáze vylučování.(srov. odstavec 53). Aby se zamezilo možným problémům se sníženou biologickou dostupností, mohou být v případě špatně rozpustných a vysoce hydrofobních látek použity pro odběr vzorků metody mikroextrakce pevné fáze. Ryba určená pro odběr vzorků by se měla okamžitě usmrtit nejvhodnější humánní metodou (při měření celých ryb by neměly být prováděny žádné další postupy než omytí vodou (srov. odstavec 28) a důkladné osušení). Ryba se zváží a změří se celková délka (51). U každého jedince by naměřená hmotnost a délka měly být vztaženy ke koncentrací analyzované látky (a případně k obsahu lipidů), například tak, že se každé rybě ve vzorku přidělí jedinečný identifikační kód. Ryby a voda se pokud možno analyzují ihned po odběru s cílem předejít degradaci nebo jiným ztrátám a vypočítat přibližné rychlostní konstanty příjmu a vylučování ještě v průběhu zkoušky. Okamžitá analýza rovněž zabrání opožděnému stanovení plató (rovnovážného stavu) při jeho dosažení. Neprovádí-li se analýza okamžitě, měly by se vzorky uchovat vhodným způsobem. Před zahájením studie je třeba zjistit informace o řádné metodě uchovávání s ohledem na danou zkoušenou látku – například hluboké zmrazení, udržování při 4 °C, vyluhování atd. Délka uchovávání by měla být zvolena tak, aby se zajistilo, že látka během uchovávání nedegraduje. Kvalita analytické metody Vzhledem k tomu, že celý postup je určen hlavně správností, přesností a citlivostí analytické metody použité pro analýzu zkoušené látky, je třeba experimentálně kontrolovat, že správnost, přesnost a reprodukovatelnost analýzy látky a rovněž výtěžek zkoušené látky jak z vody, tak ze vzorků ryb jsou pro danou analytickou metodu dostatečné. To by mělo být součástí předběžných zkoušek. Kontroluje se také, zda se zkoušená látka nevyskytuje v použité ředicí vodě. Hodnoty koncentrace zkoušené látky ve vodě a v rybách získané ve zkoušce se podle potřeby korigují na výtěžky a hodnoty pozadí v kontrolních experimentech. Vzorky ryb a vody by se měly zpracovávat tak, aby se minimalizovala kontaminace a ztráty (např. v důsledku adsorpce odběrným zařízením). Analýza vzorků ryb Je-li ve zkoušce použit materiál značený radioizotopy, je možné provést analýzu na celkovou aktivitu (tj. pro mateřskou látku i s metabolity) nebo lze provést separaci, tak aby mohla být mateřská látka analyzována samostatně. Má-li být hodnota BCF založena na mateřské látce, měly by být charakterizovány hlavní metabolity, a to minimálně na konci fáze příjmu (srov. odstavec 6). Hlavní metabolity jsou ty metabolity, které představují ≥ 10 % celkového množství reziduí v tkáních ryb, metabolity, které představují ≥ 5 % ve dvou po sobě jsoucích odběrech vzorků, metabolity, které vykazují rostoucí úroveň v průběhu celé fáze příjmu a metabolity se známými toxikologickými obavami. Je-li hodnota BCF pro celé ryby vyjádřená celkovou aktivitou reziduí ≥ 500, může být účelné – a pro určité kategorie látek, např. pesticidy, se to důrazně doporučuje – identifikovat a kvantifikovat hlavní metabolity Některé regulační orgány mohou kvantifikaci takových metabolitů vyžadovat. Jsou-li produkty rozkladu představující ≥ 10 % celkových radioizotopově značených reziduí v rybách identifikovány a kvantifikovány, doporučuje se také identifikovat a kvantifikovat produkty rozkladu ve zkušební vodě. Pokud by to nebylo proveditelné, je třeba to ve zprávě odůvodnit. Koncentrace zkoušené látky by měla být obvykle stanovena pro každou jednotlivou zváženou rybu. Není-li to možné, mohou být vzorky při každém odběru sdružovány, avšak sdružování omezuje statistické postupy, které lze na údaje aplikovat, takže by měl být do zkoušky nasazen dostatečný počet ryb vyhovující požadovanému sdružování, statistickému postupu a statistické váze. Jako úvod k příslušným postupům sdružování je možno použít odkazy (27) a (28) v seznamu literatury. Hodnota BCF by kromě hodnoty odvozené ze studie (srov. odstavec 21) měla být vyjádřena jako hodnota normalizovaná na ryby s 5 % obsahem lipidů (na základě živé hmotnosti), pokud nelze prokázat, že zkoušená látka se neakumuluje hlavně v lipidech. Je-li to možné, měl by být obsah lipidů v rybách stanoven při každém odběru a pokud možno v extraktu připraveném pro analýzu zkoušené látky, neboť lipidy musí být často před chromatografickou analýzou odstraněny. Analýza zkoušené látky však často vyžaduje zvláštní extrakční postupy, které by mohly být v rozporu se zkušebními metodami ke stanovení lipidů. V tomto případě (dokud nebudou k dispozici vhodné nedestruktivní instrumentální metody) se doporučuje pro stanovení obsahu lipidů v rybách použít jinou strategii (srov. odstavec 56). Pro stanovení obsahu lipidů by měly být použity vhodné metody (20). Jako standardní metodu lze doporučit extrakční metodu za pomoci chloroformu/methanolu (29), ale jako alternativní metoda se doporučuje Smedesova metoda (31). Tato metoda se vyznačuje srovnatelnou účinností extrakce, vysokou přesností, použitím méně toxických organických rozpouštědel a snadným provedením. Lze použít i jiné metody, jejichž přesnost je srovnatelná s doporučenými metodami, je-li to náležitě odůvodněno. Je důležité použitou metodu podrobně popsat. Měření růstu ryb Na začátku zkoušky je třeba jednotlivě zvážit pět až deset ryb z kmenové populace a změřit jejich celkovou délku. Mohou to být tytéž ryby, které se použijí na analýzu lipidů (srov. odstavec 56). Před provedením analýzy chemické látky nebo lipidů by měla být stanovena hmotnost a délka ryb použitých pro každý odběr vzorků jak ze zkušební skupiny, tak z kontrolního experimentu. Měření těchto ryb určených pro odběr vzorků lze použít k odhadu hmotnosti a délky ryb, které zůstávají ve zkušební a kontrolní nádrži (srov. odstavec 45). ÚDAJE A PŘEDKLÁDÁNÍ ZPRÁV Zpracování výsledků Křivku příjmu zkoušené látky je třeba sestrojit vynesením koncentrace látky v/na rybách (nebo ve specifikovaných tkáních) v průběhu fáze příjmu proti času v lineárním měřítku. Dosáhla-li křivka plató, tj. pokud začíná být rovnoběžná s časovou osou, vypočítá se hodnota BCF v rovnovážném stavu (BCFSS) z poměru:
Vývoj hodnoty C f může být ovlivněn růstem ryb (srov. odstavce 72 a 73). Střední hodnotu zkušební koncentrace (Cw ) ovlivňuje kolísání v čase. Lze očekávat, že časově vážená průměrná koncentrace je pro studie bioakumulace relevantnější a přesnější, i když je kolísání v rámci příslušného rozpětí platnosti (srov. odstavec 24). Časově váženou průměrnou koncentraci ve vodě lze vypočítat podle oddílu 1 dodatku 5. Kinetický biokoncentrační faktor (BCFK) by se měl stanovit jako poměr k 1/k 2, dvou kinetických rychlostních konstant prvního řádu. Rychlostní konstanty k 1 a k 2 a hodnotu BCFK je možno odvodit tak, že se křivkou současně prokládají údaje jak o fázi příjmu, tak o fázi vylučování. Alternativně lze hodnotu k 1 a k 2 stanovit postupně (popis a porovnání těchto metod – viz dodatek 5). Rychlostní konstantu vylučování (k 2) bude možná nutné upravit o zředění růstem (srov. odstavce 72 a 73). Není-li křivka příjmu a/nebo křivka vylučování zjevně křivkou prvního řádu, měly by být použity složitější modely (viz odkazy v příloze 5) a měl by být konzultován biostatistik a/nebo farmakokinetik. Údaje o hmotnosti/délce ryb V tabulce se uvedou živé hmotnosti a celkové délky jednotlivých ryb ve všech intervalech odběru vzorků zvlášť pro zkušební a pro kontrolní skupinu během fáze příjmu (včetně kmenové populace na začátku příjmu) a během fáze vylučování. U každého jedince by naměřená hmotnost a délka měly být vztaženy ke koncentraci analyzované chemické látky, např. tak, že se každé rybě ve vzorku přidělí jedinečný identifikační kód. Preferovaným měřítkem růstu pro účely úpravy hodnot kinetického BCF o zředění růstem je hmotnost (o metodě používané k úpravě údajů o zředění růstem viz odstavec 73 a dodatek 5). Úprava o zředění růstem a normalizace lipidů Růst ryb během fáze vylučování může snížit naměřené koncentrace chemické látky v rybách s tím důsledkem, že celková rychlostní konstanta vylučování (k 2) je větší než ta, která by vyplynula pouze z procesů odstranění (např. dýchání, metabolismus, vyplavením). Kinetické biokoncentrační faktory by měly být upraveny o zředění růstem. Také hodnota BCFSS bude ovlivněna růstem, ale pro úpravu BCFSS o růst není k dispozici žádný schválený postup. V případech výrazného růstu by měla být odvozena také hodnota BCFK upravená o růst (BCFKg), neboť může být relevantnějším měřítkem biokoncentračního faktoru. Obsah lipidů v zkušebních rybách (který je úzce spojen s bioakumulací hydrofobních látek) může v praxi výrazně kolísat, takže je nezbytná normalizace na stanovený obsah lipidů v rybách (5 % hmot.), aby bylo možné smysluplně vyjádřit jak kinetické biokoncentrační faktory, tak biokoncentrační faktory v rovnovážném stavu, pokud není možné namítnout, že zkoušená látka se neakumuluje především v lipidech (např. některé perfluorované látky se mohou vázat na proteiny). Rovnice a příklady těchto výpočtů lze najít v dodatku 5. Aby bylo možno upravit kinetický BCF o zředění růstem, je třeba rychlostní konstantu vylučování upravit o růst. Tato rychlostní konstanta vylučování upravená o růst (k 2g) se vypočítá tak, že se od celkové rychlostní konstanty vylučování (k 2) odečte rychlostní konstanta růstu (k g, získaná z naměřených údajů o hmotnosti). Biokoncentrační faktor upravený o růst se pak vypočítá dělením rychlostní konstanty příjmu (k 1) rychlostní konstantou vylučování upravenou o růst (k 2g) (srov. dodatek 5). V některých případech lze tento přístup zpochybnit. Například u velmi pomalu se vylučujících látek zkoušených na rychle rostoucích rybách může být odvozená hodnota k 2g velmi malá a chyba ve dvou rychlostních konstantách, které se používají pro její odvození, se tak stává kritickou a v některých případech může být odhad hodnoty k g větší než hodnota k 2. Alternativní přístup, který obchází nutnost úpravy o zředění růstem, zahrnuje použití údajů o hmotnosti vylučování zkoušené látky na každou rybu (základem je celá ryba), a nikoli obvyklých údajů o hmotnosti zkoušené látky na jednotku hmotnosti ryby (koncentraci). Toho lze snadno dosáhnout, neboť zkoušky podle této metody by měly spojovat naměřené koncentrace v tkáních s hmotností jednotlivých ryb. Jednoduchý postup, jak to provést, je uveden v dodatku 5. Upozorňujeme, že hodnota k 2 by přesto měla být ve zprávě uvedena, i když se použije tento alternativní přístup. Měl by rovněž být uveden kinetický biokoncentrační faktor a biokoncentrační faktor v rovnovážném stavu ve vztahu k standardnímu 5 % hmot. obsahu lipidů v rybách, pokud není možné namítnout, že zkoušená látka se neakumuluje především v lipidech. Údaje o koncentraci v rybách, nebo hodnoty BCF, se normalizují podle poměru mezi 5 % obsahem lipidů a skutečnou (individuální) střední hodnotou obsahu lipidů (v % živé hmotnosti) (srov. dodatek 5). Jestliže byly analýzy chemické látky a lipidů prováděny na týchž rybách, měly by být normalizované údaje o lipidech jednotlivých ryb použity pro výpočet BCF s normalizovanými lipidy. Alternativně, je-li růst kontrolních a exponovaných ryb podobný, může být pro úpravu o lipidy použít pouze obsah lipidů v rybách z kontrolního experimentu (srov. odstavec 56). Metoda pro výpočet BCF s normalizovanými lipidy je popsána v dodatku 5. Interpretace výsledků Pokud se naměřené koncentrace zkoušených roztoků pohybují na úrovních blízkých mezi detekce analytické metody, je nutno výsledky interpretovat s opatrností. Aby se vyloučily toxické účinky, průměrný růst ve zkušební i v kontrolní skupině by se v zásadě neměl výrazně lišit. Rychlostní konstanty růstu nebo křivky růstu v obou skupinách by se měly porovnávat vhodným postupem (52). Jasně vymezené křivky příjmu a úbytku jsou ukazatelem dobré kvality údajů o biokoncentraci. Pokud jde o rychlostní konstanty, aby je bylo možno považovat za spolehlivé, měl by výsledek testu dobré shody pro model bioakumulace být v souladu s rozdělením χ5 (tj. nízké procento chyb měření (32)) (srov. dodatek 5). Je-li použito více než jedna zkušební koncentrace, rozdíl konstant příjmu/vylučování mezi těmito zkušebními koncentracemi by měl být nižší než 20 % (53). Pokud bude vyšší, měla by být indikována závislost na koncentraci. Pozorované významné rozdíly v rychlostních konstantách příjmu/vylučování mezi použitými zkušebními koncentracemi by měly být zaznamenány a mělo by být uvedeno jejich možné vysvětlení. 95 % interval spolehlivosti hodnot BCF u dobře navržených studií se obecně blíží ± 20 % odvozené hodnoty BCF. Jsou-li zkoušeny dvě nebo více koncentrací, výsledky obou nebo všech koncentrací se použijí k ověření, zda jsou výsledky konzistentní, a k prokázání, zda existuje závislost na koncentraci. Zkouší-li se pouze jedna koncentrace s cílem snížit použití zvířat a/nebo zdrojů, mělo by být uvedeno odůvodnění použití pouze jedné koncentrace. Výsledná hodnota BCFSS je pochybná, je-li BCFK je výrazně větší než BCFSS, jelikož to může ukazovat na to, že nebylo dosaženo rovnovážného stavu nebo že nebylo vzato v úvahu zředění růstem a ztrátové procesy. V případech, kdy je BCFSS o mnoho vyšší než BCFK, mělo by se zkontrolovat, zda při odvození rychlostních konstant příjmu a vylučování nedošlo k chybám a odvození by se mělo přehodnotit. Odhad hodnoty BCFK by mohlo zlepšit použití odlišný postupu vynášení údajů na křivku (srov. dodatek 5). Závěrečná zpráva Kromě informací o zkoušené látce uvedených v odstavci 3 závěrečná zpráva obsahuje tyto informace:
C.13 – II Minimalizovaná zkouška na rybách s expozicí ve vodním prostředí ÚVOD Rostoucí zkušenosti, které při provádění a interpretaci úplné zkoušky získaly laboratoře a regulační orgány, ukazují, že – s určitými výjimkami – pro odhad rychlostních konstant příjmu a vylučování platí kinetika prvního řádu. Rychlostní konstanty příjmu a vylučování tak lze odhadnout při minimu odběrů a odvodit hodnotu kinetického BCF. Původním účelem zkoumání alternativního uspořádání studií BCF bylo vyvinout jednodušší zkoušku, kterou by bylo možné použít jako krok průběžného testování za účelem zamítnutí nebo potvrzení odhadů BCF založených na K OW a QSAR, a vyhnout se tak nutnosti provádět úplnou studii mnoha látek a minimalizovat náklady a použití zvířat tím, že se sníží odběry vzorků a počet prováděných analytických sérií. Při dodržení hlavního uspořádání podle předchozí zkušební metody, aby bylo možné výsledky zkoušky začlenit do existujících údajů BCF a usnadnila se interpretace zkoušek a údajů, bylo cílem poskytnout dostatečně správné a přesné odhady BCF pro přijímání rozhodnutí při posuzování rizik. Platí mnohé stejné úvahy jako v úplné zkoušce, např. kritéria validity (srov. odstavec 24) a zastavení zkoušky, pokud je na konci fáze příjmu zjištěn nevýznamný příjem (srov. odstavce 16 a 38). Látky, způsobilé pro minimalizované uspořádání zkoušky, by měly patřit do celkové oblasti látek, pro které byla tato zkušební metoda vyvinuta, tj. nepolární organické látky (srov. odstavec 49). Pokud se vyskytne jakýkoli ukazatel, že látka, která má být zkoušena, by se mohla chovat jinak (např. jasná odchylka od kinetiky prvního řádu), měla by se pro účely právních předpisů provést úplná zkouška. Minimalizovaná zkouška obvykle netrvá kratší dobu než standardní test ke stanovení BCF, ale provádí se při ní méně odběrů ryb. V případě rychle vylučovaných látek však je možné vylučovací fázi zkrátit, aby nedošlo k tomu, že koncentrace v rybách klesnou před ukončením zkoušky na hodnotu nižší, než je mez detekce/kvantifikace. Minimalizovanou zkoušku expozice ryb s jedinou koncentrací lze použít ke stanovení, zda je nutná úplná zkouška a jsou-li výsledné údaje používané pro výpočet rychlostních konstant a BCF robustní (srov. odstavec 93), lze od úplné zkoušky upustit, pokud je výsledná hodnota BCF vzdálená od předmětných hodnot stanovených v právních předpisech. V některých případech může být výhodné provést zkoušku v minimalizovaném uspořádání s více než jednou zkušební koncentrací jako předběžný test ke stanovení toho, zda odhady BCF určité látky jsou závislé na koncentraci. Pokud odhady BCF z minimalizované zkoušky nasvědčují závislosti na koncentraci, bude nezbytné provést úplnou zkoušku. Jestliže na základě takové minimalizované zkoušky jsou odhady BCF na koncentraci nezávislé, avšak výsledky nejsou považovány za konečné, mohla by se jakákoli následná úplná zkouška provést při jediné koncentraci, což sníží použití zvířat v porovnání s úplnou zkouškou se dvěma (nebo více) koncentracemi. Látky potenciálně vhodné pro minimalizovanou zkoušku by měly:
HARMONOGRAM ODBĚRU VZORKŮ PRO STUDIE S MINIMALIZOVANÝM USPOŘÁDÁNÍM Odběr vzorků ryb Odběr vzorků ryb je omezen na čtyři odběry:
Odběr vzorků vody Při minimalizovaném uspořádání se vzorky vody odebírají jako při úplné studii (srov. odstavec 54) nebo alespoň pětkrát ve stejných intervalech ve fázi příjmu a každý týden ve fázi vylučování. Změny v uspořádání S ohledem na vlastnosti zkoušené látky, platné předpovědi QSAR a konkrétní účel studie je možné zvážit některé změny v uspořádání studie:
Výpočty Argumentem, který svědčí pro použití tohoto přístupu, je skutečnost, že biokoncentrační faktor lze v úplné zkoušce stanovit buď jako biokoncentrační faktor v rovnovážném stavu (BCFSS) tak, že se vypočítá poměr koncentrace zkoušené látky v rybích tkáních a koncentrace zkoušené látky ve vodě, nebo se vypočítá kinetický biokoncentrační faktor (BCFK) jako poměr rychlostní konstanty příjmu k 1 a rychlostní konstanty vylučování k 2. BCFK je platný i v případě, kdy nebylo dosaženo rovnovážné koncentrace látky během fáze příjmu, za předpokladu, že příjem i vylučování probíhají přibližně v souladu s kinetickými procesy prvního řádu. Ke stanovení rychlostních konstant příjmu a vylučování jsou jako absolutní minimum požadovány dva odběry vzorků, jeden na konci fáze příjmu (tj. na začátku fáze vylučování) a druhý na konci (nebo po uplynutí podstatné části) fáze vylučování. Pro kontrolu kinetiky příjmu a vylučování se doporučuje zařadit mezi oba výše uvedené odběry vzorků ještě třetí odběr (57). Výpočty viz dodatky 5 a 6. Interpretace výsledků K posouzení validity a informační hodnoty zkoušky ověřte, že fáze vylučování je delší než poločas. Rovněž hodnota BCFKm (kinetický BCF odvozený z minimalizované zkoušky) by se měl porovnat s hodnotou BCFSS z minimalizované zkoušky (což je BCFSS vypočtený na konci fáze příjmu, za předpokladu, že bylo dosaženo rovnovážného stavu. To lze pouze předpokládat, neboť počet odběrů vzorků není dostatečný, aby to bylo možné prokázat). Je-li BCFKm < BCFSS z minimalizované zkoušky, pak by upřednostněnou hodnotou měl být BCFSS z minimalizované zkoušky. Je-li BCFKm menší než 70 % BCFSS z minimalizované zkoušky, nejsou výsledky validní a měla by se provést úplná zkouška. Pokud minimalizovaná zkouška vede k BCFKm poblíž jakékoli hodnoty, která je předmětem právní úpravy, měla by se provést úplná zkouška. Je-li výsledek vzdálený od jakékoli hodnoty, která je předmětem právní úpravy, nemusí být úplná zkouška nezbytná nebo lze provést úplnou zkoušku s jedinou koncentrací, pokud to vyžaduje platný právní rámec. Jestliže se po minimalizované zkoušce s jednou koncentraci zjistí, že úplná zkouška je nezbytná, lze tuto úplnou zkoušku provést při druhé koncentraci. Jsou-li výsledky shodné, lze od další úplné zkoušky při jiné koncentraci upustit, neboť se nepředpokládá, že biokoncentrace látky je závislá na koncentraci. Pokud byla provedena minimalizovaná zkouška při dvou koncentracích a pokud výsledky nevykazují závislost na výsledku, lze provést úplnou zkoušku pouze s jednou koncentrací (srov. odstavec 87). Závěrečná zpráva Závěrečná zpráva o minimalizované zkoušce by měla obsahovat všechny informace požadované v případě úplné zkoušky (srov. odstavec 81), s výjimkou těch, které není možné získat (např. křivka vyjadřující dobu potřebnou k dosažení rovnovážného stavu a biokoncentrační faktor v rovnovážném stavu; namísto uvedeného faktoru by měl být uveden BCFss z minimalizované zkoušky). Měla by rovněž obsahovat zdůvodnění, proč byla použita minimalizovaná zkouška, a výslednou hodnotu BCFKm. C.13 – III: Zkouška bioakumulace v rybách s expozicí v potravě ÚVOD Metoda popsaná v tomto oddíle by se měla používat pro látky, u kterých není možné použít metodiku expozice ve vodním prostředí (například protože není možné udržet stabilní, měřitelnou koncentraci této látky ve vodě nebo není možné za 60 dnů expozice dosáhnout jejího dostatečného obsahu v těle; viz předchozí oddíly věnované metodě expozice ve vodním prostředí). Je však třeba si uvědomit, že koncovým bodem této zkoušky bude potravní bioobohacovací faktor (BMF), a nikoli bioakumulační faktor (BCF) (58). V květnu 2001 byla na konferenci SETAC Europe, která se konala v Madridu, předložena nová metoda pro zkoušení bioakumulace organických látek špatně rozpustných ve vodě. Tato práce navazovala na různé studie bioakumulace uvedené v literatuře, používající metodu dávkování zahrnující obohacená krmiva (např. (36)). Na počátku roku 2004 byl na zasedání pracovní skupiny EU PBT předložen návrh protokolu (38) určeného k měření bioakumulačního potenciálu organických látek špatně rozpustných ve vodě, u kterých nebylo možné použít standardní metodu bioakumulace s expozicí ve vodě, spolu s podpůrným podkladovým dokumentem (39). Dalším odůvodněním ve prospěch metody bylo, že k potenciální expozici takovým špatně rozpustným látkám (tj. s hodnotou log K OW >5) v životním prostředí může dojít hlavně prostřednictvím potravy (viz (40) (41) (42) (43) (44)). Z tohoto důvodu se v některých zveřejněných nařízeních o chemických látkách odkazuje na zkoušky expozice v potravě (59). Je však třeba si uvědomit, že zde popsaná metoda se pečlivě vyhýbá expozici ve vodné fázi a že hodnotu BMF z této zkušební metody tedy nelze přímo srovnávat s hodnotou BMF ze studie za reálných podmínek (ve které je možné kombinovat expozici ve vodě i potravě). Tento oddíl této zkušební metody je založen na tomto protokolu (38) a představuje novou metodu, která se v předchozí verzi zkušební metody C.13 neobjevila. Tato alternativní zkouška umožňuje za řízených laboratorních podmínek přímo zkoumat způsob expozice v potravě. Potenciální výzkumníci se odkazují na odstavce 1 až 14 této zkušební metody, kde najdou informace ohledně toho, kdy je možné zkoušku expozice v potravě upřednostnit před zkouškou expozice ve vodním prostředí. Informace o různých aspektech látek jsou dostupné a měly by být před provedením zkoušky zváženy. Lze zvážit použití radioizotopově značených zkoušených látek za stejných okolností jako při metodě expozice ve vodním prostředí (srov. odstavce 6 a 65). Metodu expozice v potravě lze použít ke zkoušení více než jedné látky v rámci jediné zkoušky, pokud jsou splněna určitá kritéria; tato kritéria jsou dále rozvedena v odstavci 112. Kvůli jednoduchosti zde uvedená metodika popisuje zkoušku, ve které je použita pouze jedna zkoušená látka. Zkouška s expozicí v potravě se v mnohém podobá metodě expozice ve vodním prostředí, vyjma způsobu expozice. Mnohé zde popsané aspekty této metody se proto překrývají s metodou expozice ve vodním prostředí, která byla popsána v předchozím oddíle. Tam, kde je to možné, se odkazuje na odstavce v předchozím oddíle, ale v zájmu srozumitelnosti a pochopení je určitá míra zdvojení nevyhnutelná. PODSTATA ZKOUŠKY Lze použít průtokové nebo semistatické podmínky (srov. odstavec 4); průtokové podmínky se doporučují pro omezení potenciální expozice zkoušené látce prostřednictvím vody v důsledku jakékoli desorpce z obohacené potravy nebo výměšků. Zkouška se skládá ze dvou fází: příjmu (s krmivem obohaceným zkoušenou látkou) a vylučování (s čistým, neupraveným krmivem) (srov. odstavec 16). Ve fázi příjmu se „exponované“ skupině denně podává stanovená dávka komerční rybí potravy o známém složení, obohacené zkoušenou látkou. V ideálním případě by ryby měly požít všechnu nabízenou potravu (srov. odstavec 141). Rybám je poté během fáze vylučování podávána čistá, neupravená obchodní rybí potrava. Stejně jako při metodě expozice ve vodním prostředí může být podle potřeby použito více než jedna zkušební skupina s různými koncentracemi přidané zkoušené látky, ale pro většinu vysoce hydrofobních organických zkoušených látek je jedna zkušební skupina dostatečná (srov. odstavce 49 a 107). Jsou-li použity semistatické podmínky, měly by se ryby na konci fáze příjmu přemístit do nového média a/nebo do nové zkušební nádrže (pro případ, že médium a/nebo přístroje a pomůcky použité ve fázi příjmu byly kontaminovány zkoušenou látkou prostřednictvím vyplavování). Koncentrace zkoušené látky v rybách se měří v obou fázích zkoušky. Vedle skupiny ryb, kterým se podává obohacená potrava (zkušební skupina) je držena kontrolní skupina ryb za stejných podmínek a krmena stejně až na to, že dávky komerční rybí potravy nejsou obohaceny zkoušenou látkou. Tato kontrolní skupina umožňuje u neexponovaných ryb kvantifikovat hodnoty zkoušené látky v pozadí a slouží ke srovnání s jakýmikoli nepříznivými účinky spojenými s expozicí zjištěnými v zkušební skupině (skupinách) (60). Umožňuje rovněž porovnání rychlostních konstant růstu mezi skupinami jako kontrolu, že byla požita podobná množství nabízené potravy (při vysvětlení různých rychlostních konstant růstu by měly být vzaty v úvahu také potenciální rozdíly v chutnosti jednotlivých krmiv; srov. odstavec 138). Je důležité, aby během fáze příjmu i vylučování byla zkušební i kontrolní skupině podávána nutričně rovnocenná potrava. Na základě zkušeností subjektů, které tuto metodu vyvinuly (38) (39), je fáze příjmu, která trvá 7 až 14 dnů, obvykle dostatečná. Toto rozmezí by mělo minimalizovat náklady na provedení zkoušky, a přitom by u většiny látek mělo přesto zaručovat dostatečnou expozici. V některých případech však je možné fázi příjmu prodloužit (srov. odstavec 127). Během fáze příjmu koncentrace látky v rybách nemusí dosáhnout rovnovážného stavu, takže zpracování údajů a výsledky této metody jsou obvykle založeny na kinetické analýze reziduí ve tkáních. (Poznámka: Zde je možné použít rovnice pro odhadnutí doby potřebné k dosažení rovnovážného stavu stejně jako u zkoušky expozice ve vodním prostředí – viz dodatek 5). Fáze vylučování začíná, když je rybám poprvé podána neobohacená dávka, a obvykle trvá až 28 dnů nebo do doby, kdy zkoušenou látku již není možné v celých rybách kvantifikovat, podle toho, co nastane dříve. Fázi vylučování je možné zkrátit nebo ji prodloužit na více než 28 dnů, v závislosti na tom, jak se s postupem času mění naměřené koncentrace chemické látky a velikost ryb. Tato metoda umožňuje stanovit pro zkušební látku v rybách látkově specifický poločas (t1/2, z rychlostní konstanty vylučování k2), účinnost vstřebání (absorpce ve střevech; a), kinetický potravní bioobohacovací faktor (BMFK), o růst upravený kinetický potravní bioobohacovací faktor (BMFKg) a o lipidy upravený (61) kinetický potravní bioobohacovací faktor (BMFKL) (a/nebo o růst a lipidy upravený kinetický potravní bioobohacovací faktor (BMFKgL). Stejně jako v metodě expozice ve vodním prostředí, nárůst hmotnosti ryb během zkoušky povede k ředění zkoušené látky v rostoucích rybách, a hodnota (kinetického) BCF bude tedy podhodnocena, nebude-li upravena o růst (srov. odstavce 162 a 163). Jestliže se odhadem stanoví, že ve fázi příjmu bylo dosaženo rovnovážného stavu, je možné rovněž vypočítat orientační hodnotu BMF v rovnovážném stavu. Jsou k dispozici postupy, jež umožňují odhadnout hodnotu kinetického bioobohacovacího faktoru (BCFK) z údajů získaných ve studii potravy (např. (44) (45) (46) (47) (48)). O silných a slabých stránkách těchto postupů se pojednává v dodatku 8. Zkouška byla vyvinuta především pro zkoumání špatně rozpustných nepolárních organických látek, jejichž příjem a vylučování u ryb se přibližně řídí kinetikou prvního řádu. V případě, že se zkouší látka, jejíž příjem a vylučování se ani přibližně neřídí kinetikou prvního řádu, měly by být použity složitější modely (viz odkazy v dodatku 5) a měl by být konzultován biostatistik a/nebo farmakokinetik. Hodnota BMF se obvykle stanovuje pomocí analýzy zkoušené látky v celých rybách (na základě živé hmotnosti). Je-li to důležité pro účely studie, lze odebrat specifické tkáně (např. svalovinu, játra), pokud se ryba rozdělí na jedlý a nejedlý podíl (srov. odstavec 21). Lze též provést vyjmutí a samostatnou analýzu gastrointestinálního traktu ke stanovení příspěvku ke koncentracím v celých rybách při odběrech vzorků na konci fáze příjmu a u začátku fáze vylučování anebo v rámci metody hmotnostní bilance. Měl by se měřit obsah lipidů v celých rybách ve vzorku, aby mohly být koncentrace upraveny o lipidy, přičemž je třeba vzít v úvahu obsah lipidů v potravě i v rybách (srov. odstavce 56 a 57 a dodatek 7). Je nutné měřit a zaznamenávat hmotnost jedinců ve vzorku, a to ve spojitosti s koncentrací analyzované chemické látky u daného jedince (např. lze ji udávat s použitím jedinečného identifikačního kódu pro každou rybu ve vzorku), za účelem vypočítání růstu, ke kterému může dojít během zkoušky. Měřit by se pokud možno měla také celková délka ryb (62). Údaje o hmotnosti jsou též nezbytné pro odhadování hodnoty BCF s použitím údajů o vylučování ze zkoušky s expozicí v potravě. INFORMACE O ZKOUŠENÉ LÁTCE Měly by být k dispozici informace o zkoušené látce uvedené v odstavcích 3 a 22. Analytická metoda pro zkoumání koncentrací zkoušené látky ve vodě obvykle není nezbytná; požadovány jsou metody s dostatečnou citlivostí pro měření koncentrací v rybí potravě a rybích tkáních. Metodu je možné použít ke zkoušení více než jedné látky v rámci jediné zkoušky. Zkoušené látky by však měly být navzájem slučitelné, aby vzájemně nereagovaly nebo neměnily svou chemickou identitu po přidání do rybí potravy. Cílem je, aby se naměřené výsledky pro každou společně zkoušenou látku výrazně nelišily od výsledků, které by se získaly, kdyby byly provedeny samostatné zkoušky každé ze zkoušených látek. Předběžnou analytickou studií by mělo být stanoveno, že z vícenásobně obohacené potravy a ze vzorku rybí tkáně lze vytěžit každou z těchto látek s i) vysokým výtěžkem (např. > 85 % nominální hodnoty) a ii) nezbytnou citlivostí pro zkoušení. Celková dávka společně zkoušených látek by měla být nižší než kombinovaná koncentrace, která by mohla vyvolat toxické účinky (srov. odstavec 51). Uspořádání zkoušky by mělo také brát v úvahu možné nepříznivé účinky na ryby a potenciální účinky interakce (např. metabolické účinky) spojené se zkoušením více látek současně. Současného zkoušení ionizovatelných látky je třeba se vyvarovat. Pokud jde o expozici, je tato metoda vhodná i pro složité směsi (srov. odstavec 13, ačkoli budou platit stejná omezení při analýze jako u kterékoli jiné metody). VALIDITA ZKOUŠKY Aby byla zkouška platná, musí být splněny tyto podmínky (srov. odstavec 24):
REFERENČNÍ LÁTKY Pokud laboratoř dříve tuto zkoušku neprováděla nebo došlo k podstatným změnám (např. změna rybího kmene nebo dodavatele, jiný druh ryb, významná změna velikosti ryb, rybí potravy nebo metody jejího obohacení atd.), doporučuje se provedení studie odborné způsobilosti s referenční látkou. Referenční látka se používá především ke stanovení, zda je postup obohacení potravy odpovídající a zda zajistí maximální homogenitu a biologickou dostupnost zkoušených látek. Příkladem, který byl použit v případě nepolárních hydrofobních látek, je hexachlorbenzen (HCB), ale vzhledem k nebezpečnosti HCB by mělo být zváženo použití jiných látek, pro které existují spolehlivé údaje o příjmu a bioobohacení (63). Pokud se referenční látka použije, měly by být v závěrečné zprávě o ní uvedeny základní informace včetně názvu, čistoty, čísla CAS, struktury, údajů o toxicitě (pokud jsou k dispozici), stejně jako u zkoušených látek (srov. odstavce 3 a 22). POPIS METODY Přístroje a pomůcky Materiál, přístroje a pomůcky by měly být používány tak, jak je popsáno v metodě expozice ve vodním prostředí (srov. odstavec 26). Měl by být použit průtokový zkušební systém nebo statický systém s obnovováním média, který dodává do zkušebních nádrží dostatečný objem ředicí vody. Rychlost průtoku se zaznamená. Voda Zkušební voda by měla být používána tak, jak je popsáno v metodě expozice ve vodním prostředí (srov. odstavce 27 až 29). Zkušební médium by mělo mít charakteristiky, jak bylo popsáno, a mělo by mít v průběhu zkoušky stejnou kvalitu. Přirozený obsah částic a celkový organický uhlík by před začátkem zkoušky měl být co nejnižší (≤ 5 mg/l u částic; ≤ 2 mg/l u celkového organického uhlíku). Hodnotu TOC je nutné měřit pouze před zkouškou v rámci charakterizace zkušební vody (srov. odstavec 53). Potrava Doporučuje se použít komerčně dostupnou rybí potravu (plovoucí a/nebo pomalu klesající vločkové krmivo), u které je udán alespoň obsah proteinů a tuku. Krmivo by mělo obsahovat vločky o stejné velikosti, aby se zvýšila účinnost expozice potravy, tj. ryby požijí více potravy, než kdyby požily větší kousky a menší nechaly bez povšimnutí. Vločky by měly mít vhodnou velikost odpovídající velikosti ryb na začátku zkoušky (např. lze použít vločky o průměru asi 0,6 až 0,85 mm pro ryby o celkové délce 3 až 7 cm a o průměru 0,85 až 1,2 mm pro ryby o celkové délce od 6 do 12 cm). Velikost vloček je možné upravit podle růstu ryb na začátku fáze vylučování. Příklad vhodného složení potravy, dodávané komerčně, je uveden v dodatku 7. Při vývoji této metody byla používána zkušební potrava s celkovým obsahem tuku 15 až 20 % hmot. Rybí potrava s tak vysokou koncentrací lipidů nemusí být v některých regionech dostupná. V takových případech by bylo možné provést studie s nižší koncentrací lipidů v potravě, a je-li to nezbytné, vhodně upravit (na základě předběžného zkoušení) množství potravy, aby se ryby udržely v dobrém zdravotním stavu. Celkový obsah lipidů ve výživě zkušební skupiny a kontrolní skupiny je nutné změřit a zaznamenat před zahájením zkoušky a na konci fáze příjmu. V závěrečné zprávě by měly být předloženy podrobné údaje o analýze živin, vlhkosti, vlákniny a popela a pokud možno reziduí minerálů a pesticidů (např. „standardních“ hlavních znečišťujících látek) poskytnuté dodavatelem komerčního krmiva. Při obohacování krmiva zkoušenou látkou by se mělo všemožně dbát na to, aby se zajistila homogenita celého množství zkušební potravy. Koncentrace zkušební látky v potravě pro zkušební skupinu by měla být zvolena s ohledem na citlivost analytické metody, toxicitu zkoušené látky (hodnota NOEL, pokud je známa) a příslušné fyzikálně-chemické údaje. Je-li použita referenční látka, měla by být přednostně zavedena v koncentraci odpovídající asi 10 % koncentrace zkoušené látky (nebo v každém případě v co možná nejnižší koncentraci), podrobena analýze citlivosti (např. u hexachlorbenzenu bylo zjištěno, že jeho přijatelná koncentrace v potravě činí 1 až 100 μg/g; vice informací o účinnosti asimilace HCB viz (47)). Obohacení rybí potravy zkoušenou látkou lze provést několika způsoby v závislosti na jejích fyzikálních charakteristikách a na její rozpustnosti (podrobněji o metodách obohacení viz dodatek 7):
Někdy, např. u méně hydrofobních zkoušených látek, kde je pravděpodobnější, že budou z potravy desorbovat, může být nezbytné přelít připravené krmivové vločky malým množstvím kukuřičného oleje nebo rybího tuku (viz odstavec 142). V takových případech je třeba podobně upravit i potravu pro kontrolní skupinu a pro měření lipidů použít konečnou připravenou potravu. Je-li použita referenční látka, výsledky by měly být srovnatelné s údaji ze studií popsaných v literatuře, které byly provedeny za podobných podmínek a při srovnatelném množství potravy (srov. odstavec 45), a parametry referenční látky by měly splňovat příslušná kritéria v odstavci 113 (třetí, čtvrtá a pátá odrážka). Je-li jako vehikulum pro zkoušenou látku použit olej nebo nosné rozpouštědlo, mělo by se odpovídající množství stejného vehikula (bez zkoušené látky) smísit s krmivem pro kontrolní skupinu, aby byla zachována rovnocennost s obohacenou potravou. Je důležité, aby během fáze příjmu i vylučování byla zkušební i kontrolní skupině podávána nutričně rovnocenná potrava. Obohacená potrava by se měla uchovávat za podmínek, kdy zůstane zachována stabilita zkoušené látky v krmné směsi (např. v chladničce) a tyto podmínky se uvedou ve zprávě. Výběr druhu ryby Použity mohou být druhy ryb stanovené pro expozici ve vodním prostředí (srov. odstavec 32 a dodatek 3). Ve studiích potravní bioakumulace s organickými látkami před zveřejněním této zkušební metody byli běžně používáni pstruh duhový (Oncorhynchus mykiss), kapr obecný (Cyprinus carpio) a jeleček velkohlavý (Pimephales promelas). Zkušební druh by měl mít takové chování při krmení, které vede k rychlému požití podané dávky krmiva, aby se zajistilo, že působení jakéhokoli faktoru ovlivňujícího koncentraci zkoušené látky v potravě (např. vyplavování do vody a možnost expozice ve vodním prostředí) bude omezeno na minimum. Měly by být použity ryby v rámci velikostního/hmotnostního rozpětí (srov. dodatek 3). Ryby by neměly být tak malé, aby to znesnadnilo analýzy na bázi jednotlivých ryb. Druhy testované ve stadiu života s rychlým růstem mohou zkomplikovat interpretaci údajů a vysoká tempa růstu mohou ovlivnit výpočet účinnosti vstřebání (64). Chov ryb Aklimatizace a kritéria přijatelnosti mortality a nemocí jsou stejné jako pro metodu expozice ve vodním prostředí před provedením zkoušky (srov. odstavce 33–35). PROVEDENÍ ZKOUŠKY Práce předcházející studii a zkouška ke stanovení rozsahu Před studií je nezbytná analytická práce za účelem prokázání výtěžku látky z obohacené potravy / obohacené rybí tkáně. Zkouška ke stanovení rozsahu za účelem výběru vhodné koncentrace v potravě není vždy nezbytná. Pro účely prokázání, že nebyly zjištěny žádné nepříznivé účinky, a hodnocení chutnosti obohacené potravy, citlivosti analytické metody ve vztahu k rybám a potravě a výběru vhodného množství potravy a vhodných intervalů odběru vzorků během fáze vylučování atd. mohou být podniknuty předběžné pokusy s krmením, ale nejsou povinné. Předběžná studie může být cenná pro odhadnutí počtu ryb potřebného pro odběry vzorků během fráze vylučování. Může vést k výraznému snížení použitého počtu ryb, zvláště u zkoušených látek, které jsou zvlášť náchylné k metabolismu. Podmínky expozice Délka fáze příjmu Obvykle postačuje fáze příjmu v délce 7 až 14 dnů, během které je jedné skupině ryb denně podávána kontrolní potrava a druhé skupině ryb zkušební potrava v pevně stanovených dávkách v závislosti na zkušebním druhu a na zkušebních podmínkách, např. v případě pstruha duhového je to 1 až 2 % tělesné hmotnosti (živé hmotnosti). Množství potravy by mělo být zvoleno tak, aby se zamezilo rychlému růstu a velkému nárůstu obsahu lipidů. Fázi příjmu je možné v případě potřeby prodloužit na základě praktických zkušeností z předchozích studií nebo na základě znalostí o příjmu/vylučování zkoušené (nebo analogické) látky rybami. Začátek zkoušky je definován jako okamžik prvního podání obohacené potravy. Experimentální den trvá od okamžiku podání krmiva do okamžiku krátce (např. jednu hodinu) před příštím krmením. První experimentální den příjmu trvá od doby prvního podání obohacené potravy a končí krátce před druhým podáním obohacené potravy. V praxi fáze příjmu končí krátce (např. jednu hodinu) před prvním podáním potravy neobohacené zkoušenou látkou, neboť ryby budou v mezidobí 24 hodin pokračovat v trávení obohacené potravy a v absorbování zkoušené látky. Je důležité zajistit, aby bylo dosaženo dostatečně vysoké (netoxické) množství zkoušené látky v těle vzhledem k analytické metodě, takže je možné měřit její pokles alespoň o jeden řád během fáze vylučování. Ve zvláštních případech lze použít prodlouženou fázi příjmu (až do 28 dnů) s dodatečným odběrem vzorků, aby se získaly informace o kinetice příjmu. Během fáze příjmu nemusí koncentrace v rybách dosáhnout rovnovážného stavu. Zde, stejně jako při zkoušce expozice ve vodním prostředí, je možné použít rovnice pro odhadování doby potřebné k dosažení rovnovážného stavu, jako indikaci, jak dlouho pravděpodobně potrvá, než budou dosaženy hodnotitelné koncentrace v rybách (srov. dodatek 5). V některých případech může být známo, že v důsledku slabé analytické citlivosti nebo nízké účinnosti asimilace nebude příjem látky rybami po dobu 7 až 14 dnů postačovat k tomu, aby použitá koncentrace v potravě dosáhla dostatečně vysoké koncentrace v rybách, a aby tak bylo možné analyzovat pokles alespoň o jeden řád během fáze vylučování. V takových případech může být výhodné prodloužit původní fázi příjmu na více než 14 dnů nebo, zejména u látek s vysokou metabolizací, by měla být zvážena vyšší koncentrace v potravě. Je však třeba dávat pozor, aby množství v těle během fáze příjmu bylo udržováno na úrovni nižší, než je (odhadovaná) stálá koncentrace bez účinků (NOEC) na rybí tkáně (srov. odstavec 138). Délka fáze vylučování Vylučování typicky trvá až 28 dnů a začíná, jakmile je rybám v zkušební skupině po fázi příjmu podávána čistá, neexponovaná potrava. Vylučování začíná s prvním podáním „neobohacené“ potravy, a nikoli bezprostředně po posledním podání „obohacené“ potravy, neboť ryby v mezidobí 24 hodin pokračují v trávení potravy a absorpci zkoušené látky, jak je uvedeno v odstavci 126. První vzorek ve fázi vylučování se tedy odebírá krátce před druhým podáním neobohacené potravy. Tato fáze vylučování je určena k zachycení látek s potenciálním poločasem až 14 dnů, což je v souladu s poločasem bioakumulujících látek (65), takže 28 dnů představuje dva poločasy takových látek. V případě látek s velmi vysokou bioakumulací může být výhodné fázi vylučování prodloužit (pokud to naznačí předběžné zkoušení). Je-li látka vylučována velmi pomalu, takže nemusí být možné stanovit přesný poločas ve fázi vylučování, může tato informace přesto postačovat pro účely hodnocení jako příznak vysoké úrovně bioakumulace. A naopak, je-li látka vylučována tak rychle, že není možné spolehlivě odvodit koncentraci v čase nula (koncentrace na konci fáze příjmu / začátku fáze vylučování, C 0,d) ani hodnotu k 2, lze učinit konzervativní odhad hodnoty k 2 (srov. dodatek 7). Pokud před uplynutím celé 28denní doby analýzy ryb odebraných v dřívějších intervalech (např. 7denním nebo 14denním) ukážou, že látka byla vylučována v menším množství, než jsou meze kvantifikace, pak následný odběr vzorků může být přerušen a zkouška se ukončí. V nemnohých případech se může stát, že na konci fáze příjmu (nebo při odebrání druhého vzorku ve fázi vylučování) nedojde k žádnému měřitelnému příjmu zkoušené látky. Pokud lze prokázat, že i) byla splněna kritéria platnosti v odstavci 113 a ii) nedostatečný příjem není důsledkem některých jiných nedostatků zkoušky (např. nedostatečná délka fáze příjmu, nesprávný postup obohacení potravy, který vedl ke špatné biologické dostupnosti, nedostatečná citlivost analytické metody, ryby nepojídající potravu atd.), bylo by možné studii ukončit, aniž by bylo nutné ji opakovat s delší fází příjmu. Pokud předběžná práce naznačila, že k tomu může dojít, lze doporučit, je-li to možné, analýzu výměšků na přítomnost nestrávené zkušební látky v rámci metody hmotnostní bilance. Počty zkušebních ryb Podobně jako ve zkoušce expozice ve vodním prostředí by měly být vybrány ryby podobné hmotnosti a délky, tak aby hmotnost nejmenší z nich nebyla nižší než dvě třetiny hmotnosti největší ryby (srov. odstavec 42). Celkový počet ryb pro studii by se měl zvolit na základě harmonogramu odběru vzorků (nejméně jeden vzorek na konci fáze příjmu a čtyři až šest vzorků během fáze vylučování, ale v závislosti na délce fází), s ohledem na citlivost analytické metody, na koncentraci, která bude pravděpodobně dosažena na konci fáze příjmu (na základě předchozích znalostí) a na délku fáze vylučování (pokud předchozí znalosti umožňují ji odhadnout). Při každém odběru by mělo být odebráno pět až deset ryb, přičemž před analýzou chemické látky nebo analýzou lipidů se změří parametry růstu (hmotnost a celková délka). Kvůli vlastní variabilitě velikosti, rychlosti růstu a fyziologie ryb a pravděpodobnému různému množství podávané potravy, které požije každá z ryb, by v každém intervalu mělo být odebráno alespoň pět ryb ze zkušební skupiny a pět z kontrolní skupiny, aby bylo možné náležitě stanovit průměrnou koncentraci a její kolísání. Kolísání koncentrace u použitých ryb pravděpodobně přispěje k celkové neřízené variabilitě ve zkoušce více, než je variabilita vlastní použitým analytickým metodám, a v některých případech tak opravňuje k použití až deseti ryb na každý odběr. Jestliže však na začátku vylučování nejsou u ryb kontrolní skupiny měřitelné koncentrace zkoušené látky v pozadí, může být dostatečná chemická analýza dvou nebo tří ryb z kontrolní skupiny pouze při konečném odběru vzorků, pokud jsou zbývající ryby z kontrolní skupiny při stejných odběrech přesto odebírány pro stanovení hmotnosti a celkové délky (takže pro stanovení růstu se odebere stejný počet ryb z exponované a kontrolní skupiny). Ryby by měly být uskladněny, jednotlivě zváženy (dokonce, i když se následně ukáže jako nezbytné výsledky získané z vzorků zkombinovat) a změřena jejich celková délka. Při standardní zkoušce například s 28denním vylučováním zahrnující pět odběrů ve fázi vylučování to znamená celkem 59–120 ryb z exponované a 50–110 ryb z kontrolní skupiny, za předpokladu, že metoda analýzy dané látky umožňuje provést analýzu obsahu lipidů na týchž rybách. Pokud analýzu lipidů není možné provést na stejném vzorku jako analýzu chemické látky a pokud není proveditelné ani použití ryb z kontrolní skupiny pouze k analýze lipidů (srov. odstavec 56), bylo by zapotřebí dalších 15 ryb (tři z kmenové populace na začátku zkoušky, po třech z kontrolní a zkušební skupiny na začátku vylučování a po třech z kontrolní a zkušební skupiny na konci pokusu). Příklad harmonogramu odběru vzorků s počty ryb lze najít v dodatku 4. Nasazování Měl by být použit podobně vysoký poměr množství vody k množství ryb, jako při metodě expozice ve vodním prostředí (srov. odstavce 43 a 44). Ačkoli vyšší rychlost nasazování množství ryb na množství vody nemá při této zkoušce vliv na zkušební koncentrace, pro zachování odpovídajících koncentrací rozpuštěného kyslíku a pro minimalizaci stresu u zkušebních organismů se doporučuje rychlost nasazování 0,1 až 1,0 g ryb (živé hmotnosti) na litr vody za den. Zkušební potrava a krmení Během aklimatizace by rybám měla být podávána vhodná potrava, jak je popsáno výše (odstavec 117). Jestliže se zkouška provádí za průtokových podmínek, měl by být přítok vody na dobu krmení pozastaven. Potrava pro zkušební skupinu by se během zkoušky měla řídit tím, co je popsáno výše (odstavce 116 až 121). Při zohlednění faktorů specifických pro danou látku, analytické citlivosti, předpokládané koncentrace v potravě za podmínek obvyklých v životním prostředí a úrovní chronické toxicity / obsahu látky v těle by výběr cílové obohacující koncentrace měl brát v úvahu chutnost potravy (aby ryby krmivo neodmítaly). Ve zprávě by měla být zdokumentována nominální koncentrace zkoušené látky při obohacení. Na základě zkušeností poskytuje koncentrace při obohacení v rozmezí 1 až 1 000 μg/g praktické pracovní rozpětí pro zkoušené látky, které nevykazují specifický mechanismus toxicity. Pokud například působí prostřednictvím nespecifického mechanismu, neměla by být úroveň reziduí v tkáních vyšší než 5 μmol/g lipidů, neboť je pravděpodobné, že rezidua nad touto úrovní budou vyvolávat chronické účinky (19) (48) (50) (66). U jiných látek by se mělo dbát na to, aby nedošlo k nepříznivým účinkům vyvolaným akumulovanou expozicí (srov. odstavec 127). To platí zejména v případě, že se současně zkouší více než jedna látka (srov. odstavec 112). Vhodné množství zkoušené látky lze dostat do rybí potravy jedním ze tří způsobů, jak je popsáno v odstavci 119 a v dodatku 7. Metody a postupy obohacování potravy by měly být zdokumentovány ve zprávě. Rybám v kontrolní skupině se podává neupravená potrava obsahující ekvivalentní množství neobohaceného olejového vehikula, pokud bylo použito v obohacené potravě ve fázi příjmu, nebo ošetřená „čistým“ rozpouštědlem, pokud bylo pro přípravu potravy pro zkušební skupinu použito nosné rozpouštědlo. Upravená a neupravená potrava by měla být měřena analyticky nejméně třikrát pro stanovení koncentrace zkoušené látky před začátkem fáze příjmu a na jejím konci. Po expozici v obohacené potravě (ve fázi příjmu) se rybám (v obou skupinách) podává neupravená potrava (ve fázi vylučování). Rybám se podávají pevně stanovené dávky (závislé na druhu, např. v případě pstruha duhového přibližně 1 až 2 % živé hmotnosti těla za den). Množství potravy by mělo být zvoleno tak, aby se zamezilo rychlému růstu a velkému nárůstu obsahu lipidů. Přesné množství potravy stanovené během experimentu je třeba zaznamenat. Počáteční krmení by mělo vycházet z naplánovaných stanovení hmotnosti u kmenové populace těsně před zahájením zkoušky. Množství krmiva by mělo být upraveno na základě živé hmotnosti ryb určených pro vzorek při každém odběru vzorků, aby bylo možné zohlednit růst během experimentu. Hmotnost a délku těla ryb ve zkušební a kontrolní nádrži je možné odhadnout z hmotností a celkových délek ryb odebraných při každém odběru; ryby zůstávající ve zkušební a kontrolní nádrži se neváží ani neměří. Je důležité zachovat stejné množství potravy v průběhu celého experimentu. Krmení by se mělo sledovat, aby bylo zajištěno, že ryby očividně požijí všechnu potravu, která jim byla podána, a že tak ve výpočtech budou použity odpovídající hodnoty míry pojídání potravy. Při výběru množství potravy by mělo být přihlédnuto k předběžným experimentům s krmením, což zajistí, že bude spotřebována všechna potrava podaná při krmení, které se provádí jednou denně. Pokud potrava pravidelně zůstává nepožita, lze doporučit rozdělení dávky a část podat v rámci jednoho krmení zařazeného v každém experimentálním dnu navíc (např. nahradit jednorázové denní krmení krmením, při kterém se podá polovina tohoto množství dvakrát denně). Pokud je nezbytné použít tento postup, ke druhému krmení by mělo dojít ve stanovenou dobu a mělo by se načasovat tak, aby proběhla co možná nejdelší doba před odběrem vzorků ryb (např. doba druhého krmení se stanoví v první polovině experimentálního dne). Ačkoli ryby většinou pojídají potravu rychle, je důležité zajistit, aby látka zůstala v potravě absorbována. Je třeba dbát na to, aby se zamezilo rozpuštění látky z potravy ve vodě, čímž by byly ryby vystaveny koncentracím zkoušené látky ve vodním prostředí, a nejen prostřednictvím potravy. Toho lze dosáhnout odstraněním veškeré nepožité potravy (a výměšků) ze zkušební a kontrolní nádrže do jedné hodiny, ale nejlépe do 30 minut od krmení. Kromě toho lze použít systém, kdy je voda soustavně čištěna přes filtr z aktivního uhlíku s cílem absorbovat jakékoli „rozpuštěné“ znečišťující látky. Rychlému odplavení částic potravy a rozpuštěných látek mohou napomoci průtokové systémy (67). V některých případech může pomoci zmírnit tento problém poněkud upravený způsob přípravy obohacené potravy (viz odstavec 119). Světlo a teplota Stejně jako při metodě expozice ve vodním prostředí (srov. odstavec 48) se doporučuje fotoperioda 12 až 16 hodin a teplota vhodná pro použitý zkušební druh (± 2 °C) (srov. dodatek 3). Druh a charakteristiky osvětlení by měly být známy a zdokumentovány. Kontroly Měla by se použít jedna kontrolní skupina, ve které je rybám podávána stejná potrava jako rybám ve zkušební skupině, ale bez přítomnosti zkoušené látky v krmivu. Pokud byl k obohacení krmiva ve zkušební skupině použit olej nebo nosné rozpouštědlo, mělo by být krmivo pro kontrolní skupinu ošetřeno přesně stejným způsobem, avšak bez zkoušené látky, takže dávky ve zkušební skupině a v kontrolní skupině jsou rovnocenné (srov. odstavce 121 a 139). Četnost měření kvality vody Podmínky popsané v metodě expozice ve vodním prostředí platí i zde, s tou výjimkou, že TOC je třeba měřit pouze před zkouškou v rámci charakterizace zkušební vody (srov. odstavec 53). Odběr vzorků a analýza ryb a potravy Analýza vzorků potravy Vzorky zkušební a kontrolní potravy by měly být analyzovány nejméně třikrát pro stanovení zkoušené látky a obsahu lipidů alespoň před začátkem fáze příjmu a na jejím konci. Metody analýzy a postupy pro zajištění homogenity potravy je třeba uvést v závěrečné zprávě. Vzorky by měly být analyzovány na přítomnost zkoušené látky pomocí zavedené a validované metody. V rámci přípravy studie by měla být stanovena mez kvantifikace, procento výtěžku, interference a analytická variabilita v zamýšlené vzorkovací matici. Pokud se zkouší radioizotopově značený materiál, měly by být vzaty v úvahu podobné okolnosti jako při metodě expozice ve vodním prostředí, přičemž namísto analýzy vody se provádí analýza potravy (srov. odstavec 65). Analýza ryb Při každém odběru vzorku ryb bude odebráno po 5 až 10 jedincích z exponované a kontrolní skupiny (v některých případech lze počet kontrolních ryb snížit; viz odstavec 134). Odběry by se měly uskutečnit ve stejnou dobu každý experimentální den (ve vztahu k době krmení) a měly by být načasovány tak, aby se pravděpodobnost toho, že během fáze příjmu a v rané části fáze vylučování zůstane ve střevech potrava, minimalizovala, a zabránilo se tak nevěrohodným příspěvkům k celkovým koncentracím zkoušené látky (tj. ryby určené pro vzorek by měly být vyjmuty na konci experimentálního dne, přičemž je třeba mít na paměti, že experimentální den začíná v době krmení a končí v době příštího krmení, přibližně po 24 hodinách. Vylučování začíná od prvního podání neobohacené potravy; srov. odstavec 128). První vzorek ve fázi vylučování (odebraný krátce před druhým krmením neobohacenou potravou) je důležitý, neboť extrapolace o jeden den nazpět od tohoto měření se používá k odhadu koncentrace v čase nula (C 0,d, koncentrace v rybách na konci příjmu / začátku vylučování). Volitelně je možné vyjmout z ryb gastrointestinální trakt a analyzovat ho samostatně na konci příjmu a v den 1 a 3 vylučování. Při každém odběru by ryby měly být vyjmuty z obou zkušebních nádrží a mělo by s nimi být zacházeno stejným způsobem, jak je popsáno v metodě expozice ve vodním prostředí (srov. odstavce 61–63). Koncentrace zkoušené látky v celých rybách (živé hmotnosti) se měří alespoň na konci fáze příjmu a během fáze vylučování v kontrolní i ve zkušební skupině. Během fáze vylučování se doporučuje provést čtyři až šest odběrů (např. 1., 3., 7., 14. a 28. den). Volitelně lze zařadit dodatečný odběr po 1 až 3denním příjmu za účelem odhadu účinnosti asimilace z lineární fáze příjmu u ryb, který bude stále blízko začátku doby expozice. Existují dvě hlavní odchylky od harmonogramu: i) jestliže se použije prodloužená fáze příjmu pro účely zkoumání kinetiky příjmu, dojde k dodatečným odběrům vzorků během fáze příjmu, a bude tedy nutné zařadit dodatečné ryby (srov. odstavec 126; ii) jestliže byla studie na konci fáze příjmu ukončena z důvodu neexistence měřitelného příjmu (srov. odstavec 131). Jednotlivé ryby, které se odeberou ve vzorku, by měly být zváženy (a změřena jejich celková délka), aby bylo možné stanovit rychlostní konstanty růstu. Koncentrace látky ve specifické rybí tkáni (jedlé a nejedlé podíly) lze rovněž měřit na konci fáze příjmu a ve vybraných dobách fáze vylučování. Pokud se zkouší radioizotopově značený materiál, měly by být vzaty v úvahu podobné okolnosti jako při metodě expozice ve vodním prostředí, přičemž namísto analýzy vody se provádí analýza potravy (srov. odstavec 65). Při pravidelném používání referenční látky (srov. odstavec 25) se upřednostňuje měřit koncentrace ve zkušební skupině na konci fáze příjmu a ve všech dobách fáze vylučování stanovených pro danou zkoušenou látku (na celých rybách); v kontrolní skupině je nutné analyzovat koncentrace pouze na konci fáze příjmu (na celých rybách). Za určitých okolností (například pokud metody analýzy přítomnosti zkoušené látky a referenční látky nejsou slučitelné, takže by pro dodržení harmonogramu odběru vzorků bylo nutné použít dodatečné ryby, lze použít jiný přístup s cílem minimalizovat požadovaný počet dodatečných ryb, jak je uvedeno dále. Koncentrace referenční látky se měří pouze během fáze vylučování, a to v den 1, 3 a ve dvou dalších odběrech zvolených tak, aby bylo možné provést spolehlivé odhady koncentrace v čase nula (C 0,d) a hodnoty k 2 referenční látky. Obsah lipidů u jednotlivých ryb by měl být stanoven při každém odběru nebo alespoň na začátku a na konci fáze příjmu a na konci fáze vylučování (srov. odstavce 56 a 67). V závislosti na analytické metodě (srov. odstavec 67 a dodatek 4) lze možná použít stejné ryby ke stanovení obsahu lipidů i koncentrací zkoušené látky. To je upřednostňováno z důvodu minimalizace počtu ryb. Pokud by to však nebylo možné, lze použít stejný postup, který je popsán v metodě expozice ve vodním prostředí (možnosti alternativního měření lipidů viz odstavec 56). Metodu, která byla použita ke kvantifikaci obsahu lipidů, je třeba zdokumentovat v závěrečné zprávě. Kvalita analytické metody Měly by být prováděny experimentální kontroly s cílem zajistit specifičnost, přesnost a reprodukovatelnost analytické metody pro danou látku a rovněž výtěžky zkoušené látky z potravy a z ryb. Měření růstu ryb Na začátku zkoušky je třeba zvážit vzorek ryb z kmenové populace (a změřit jejich celkovou délku). Tyto ryby by měly být odebrány krátce před prvním podáním obohaceného krmiva a přiděleny k experimentálnímu dnu 0. Počet ryb v tomto vzorku by měl být přinejmenším stejný jako počet ryb ve vzorcích během zkoušky. Některé z nich mohou být tytéž ryby, které jsou použity pro analýzu lipidů před začátkem fáze příjmu (srov. odstavec 153). Při každém odběru vzorků se ryby nejprve zváží a změří se jejich délka. U každého jedince by naměřená hmotnost (a délka) měla být vztažena ke koncentraci analyzované chemické látky (a případně k obsahu lipidů), například tak, že se každé rybě ve vzorku přidělí jedinečný identifikační kód. Měření těchto ryb ve vzorku lze použít k odhadu hmotnosti (a délky) ryb, které zůstávají ve zkušební a kontrolní nádrži. Experimentální hodnocení Každý den se má sledovat případná mortalita a výsledky se zaznamenají. Měla by být prováděna další pozorování, zda nedochází k nepříznivým účinkům, například k abnormálnímu chování nebo pigmentaci, a výsledky se zaznamenají. Ryby jsou považovány za mrtvé, pokud u nich nedochází k dýchacím pohybům a nelze u nich detekovat žádnou reakci na slabý mechanický podnět. Veškeré mrtvé nebo zjevně umírající ryby musí být odstraněny. ÚDAJE A PŘEDKLÁDÁNÍ ZPRÁV Zpracování výsledků Výsledky zkoušky se použijí k odvození rychlostní konstanty vylučování (k 2) jako funkce celkové živé hmotnosti ryb. Vypočítá se rychlostní konstanta růstu, k g, založená na střední hodnotě nárůstu hmotnosti ryb a případně se použije k získání rychlostní konstanty vylučování upravené o růst, k 2g. Ve zprávě by rovněž měla být uvedena účinnost asimilace (a; absorpce ze střev), kinetický bioobohacovací faktor (BMFK) (v případě potřeby upravený o růst, BMFKg), jeho hodnota upravená o lipidy (BMFKL nebo BMFKgL, je-li upravená o zředění růstem) a množství potravy. Je-li možné provést odhad doby potřebné k dosažení rovnovážného stavu (např. 95 % rovnovážného stavu neboli t 95 = 3,0/k 2), lze rovněž uvést odhad BMF v rovnovážném stavu (BMFSS) (srov. odstavce 105 a 106 a dodatek 5), pokud hodnota t 95 naznačuje, že mohlo být dosaženo podmínek rovnovážného stavu. U této hodnoty BMFSS by měla být použita stejná úprava o lipidy jako u kineticky odvozeného BMF (BMFK), čímž se získá hodnota upravená o lipidy, BMFSSL (je nutné upozornit na to, že neexistuje žádný schválený postup pro úpravu BMF v rovnovážném stavu o ředění růstem). Vzorce a příklady výpočtů jsou uvedeny v dodatku 7. Jsou k dispozici postupy, jež umožňují odhadnout hodnotu kinetického biokoncentračního faktoru (BCFK) z údajů získaných ve studii potravy. O tom se pojednává v dodatku 8. Údaje o hmotnosti/délce ryb Živé hmotnosti a délky jednotlivých ryb ve všech časových úsecích se uvedou v tabulkách zvlášť za zkušební a za kontrolní skupinu ve všech dnech odběru vzorků během fáze příjmu (kmenová populace na začátku příjmu; kontrolní skupina a zkušební skupina na konci příjmu, a provádí-li se raná fáze (např. ve dnech 1–3 příjmu)), a během vylučování (např. ve dnech 1, 2, 4, 7, 14, 28 za kontrolní a za zkušební skupinu). Pro účely úpravy o zředění růstem je upřednostňovaným měřítkem hmotnost. Metoda (metody), které se používají k úpravě údajů o zředění růstem, jsou uvedeny dále (odstavce 162 a 163). Údaje o koncentraci zkoušené látky v rybách Měření reziduí zkoušené látky v jednotlivých rybách (nebo ve sdružených vzorcích ryb, pokud měření jednotlivých ryb není možné), vyjádřených jako koncentrace v hmotnostních procentech z živé hmotnosti, se uvedou v tabulkách za zkušební a za kontrolní ryby při jednotlivých odběrech vzorků. Pokud byla prováděna analýza lipidů na každé rybě ve vzorku, lze odvodit jednotlivé koncentrace upravené o lipidy, vyjádřené jako koncentrace lipidů (v hmotnostních procentech lipidů z živé hmotnosti) a uvést ve formě tabulky.
Rychlost vylučování a bioobohacovací faktor Aby bylo možné z údajů vypočítat bioobohacovací faktor, je třeba nejprve získat hodnotu účinnosti asimilace (absorpce zkoušené látky ve střevech, α). K tomu by měla být použita rovnice A7.1 z dodatku 7, pro kterou je nutné znát odvozenou koncentraci v rybách v čase nula fáze vylučování (C 0,d), (celkovou) rychlostní konstantu vylučování (k 2), koncentraci v potravě (C food), rychlostní konstantu pojídání potravy (I) a délku fáze příjmu (t). Směrnice a úsek lineárního vztahu mezi ln (koncentrací) a fází vylučování se uvedou jako celková rychlostní konstanta vylučování (k 2 = směrnice) a koncentrace v čase nula (C 0,d = eintercept), jak je uvedeno výše. Odvozené hodnoty by měly být zkontrolovány, zda jsou biologicky možné (např. účinnost asimilace jako zlomek není větší než 1). Hodnota (I) se vypočítá vydělením hmotnosti potravy hmotností ryb, kterým byla podána potrava každý den (je-li podáváno množství odpovídající 2 % tělesné hmotnosti, bude hodnota (I) 0,02). Množství potravy použité ve výpočtu však možná bude nutné upravit o růst ryb (to lze provést tak, že se známá rychlostní konstanta růstu použije k odhadu hmotnosti ryb při každém odběru vzorků během fáze příjmu; viz dodatek 7). V případech, kdy hodnoty k 2 a C 0,d není možné odvodit, protože například koncentrace klesla pod mez detekce pro druhý vzorek ve fázi vylučování, lze provést konzervativní odhad k 2 a „horní odhad“ BMFk (srov. dodatek 7). Jakmile je získána účinnost asimilace (α), je možné vypočítat bioobohacovací faktor tak, že se α vynásobí rychlostní konstantou pojídání potravy (I) a výsledek se dělí (celkovou) rychlostní konstantou vylučování (k 2). Bioobohacovací faktor upravený o růst se vypočítá stejně, ale s použitím rychlostní konstanty vylučování upravené o růst (k 2g; viz odstavce 162 a 163). Alternativní odhad účinnosti asimilace lze odvodit, jestliže byla provedena analýza tkáně z ryb odebraných v rané, lineární fázi fáze příjmu; srov. odstavec 151 a dodatek 7. Tato hodnota představuje nezávislý odhad účinnosti asimilace pro v podstatě neexponovaný organismus (tj. ryby jsou blízko začátku fáze příjmu). Účinnost asimilace odhadnutá z údajů o vylučování se obvykle používá k odvození BMF. Úprava o lipidy a úprava o zředění růstem Růst ryb během fáze vylučování může snížit naměřené koncentrace chemické látky v rybách s tím důsledkem, že celková rychlostní konstanta vylučování, k2 , je větší než ta, která by vyplynula pouze z procesů odstranění (např. metabolismus, vyplavení). Obsah lipidů v zkušebních rybách (který je úzce spojen s bioakumulací hydrofobních látek) a obsah lipidů v potravě může v praxi dosti kolísat, takže je nezbytná jejich úprava, aby bylo možné odvodit bioobohacovací faktory smysluplným způsobem. Bioobohacovací faktor by měl být upraven o zředění růstem (stejně jako se upravuje kinetický BCF v metodě expozice ve vodním prostředí) a upraven o obsah lipidů v potravě v porovnání s obsahem lipidů v rybách (faktor úpravy o lipidy). Rovnice a příklady těchto výpočtů lze najít v dodatku 5 a dodatku 7. Aby bylo možné provést úpravu o ředění růstem, je třeba vypočítat rychlostní konstantu vylučování upravenou o růst (k 2g) (rovnice viz dodatek 5). Tato rychlostní konstanta vylučování upravená o růst (k 2g) se poté použije k výpočtu bioobohacovacího faktoru upraveného o růst, jak je uvedeno v odstavci 73. V některých případech není tento postup možný. Alternativní přístup, který obchází nutnost úpravy o zředění růstem, zahrnuje použití údajů o hmotnosti vylučování zkoušené látky na každou rybu (základem je celá ryba), a nikoli obvyklých údajů o hmotnosti zkoušené látky na jednotku hmotnosti ryby (koncentraci). Toho lze snadno dosáhnout, neboť zkoušky podle této metody by měly vztahovat zaznamenané koncentrace v tkáních k hmotnosti jednotlivých ryb. Jednoduchý postup, jak to provést, je uveden v dodatku 5. Upozorňujeme, že hodnota k 2 by přesto měla být odhadnuta a uvedena ve zprávě, i když se použije tento alternativní přístup. Aby bylo možné provést úpravu o obsah lipidů v potravě a v rybách, jestliže analýza lipidů nebyla provedena u všech ryb ve vzorku, odvodí se střední hodnota frakcí lipidů (v procentech hmotnosti) v rybách a v potravě (68). Faktor úpravy o lipidy (Lc ) se poté vypočítá tak, že se střední hodnota frakce lipidů v rybách dělí střední hodnotou frakce lipidů v potravě. Bioobohacovací faktor, upravený o růst či nikoli, se vydělí faktorem úpravy o lipidy a vypočítá se bioobohacovací faktor upravený o lipidy. Jestliže byla analýza chemické látky a analýza lipidů provedena na týchž rybách při každém odběru vzorků, pak údaje o tkáních jednotlivých ryb upravené o lipidy mohou být přímo použity k výpočtu BMF upraveného o lipidy (srov. (37)). Grafickým znázorněním údajů o koncentracích upravených o lipidy se získá hodnota C 0,d na základě lipidů a hodnota k 2. Poté může proběhnout matematická analýza za pomoci stejných rovnic jako v dodatku 7, avšak účinnost asimilace (a) se vypočítá s použitím rychlostní konstanty pojídání potravy s normalizovanými lipidy (I lipid) a koncentrace v potravě na základě lipidů (C food-lipid). Parametry upravené o lipidy se poté podobně použijí k výpočtu BMF (povšimněte si, že pro výpočet hodnoty BMFKgL upravené o růst, upravené o lipidy by se také úprava rychlostní konstantou růstu měla použít na frakci lipidů, a nikoli na živou hmotnost ryb). Interpretace výsledků Aby se vyloučily toxické účinky, průměrný růst ve zkušební i v kontrolní skupině by se v zásadě neměl výrazně lišit. Rychlostní konstanty růstu nebo křivky růstu v obou skupinách by se měly porovnávat vhodným postupem (69). Závěrečná zpráva Po ukončení studie se vypracuje závěrečná zpráva obsahující informace o zkoušené látce, zkušebním druhu a zkušebních podmínkách, které jsou uvedeny v odstavci 81 (stejně jako u metody expozice ve vodním prostředí). Navíc jsou požadovány tyto informace:
LITERATURA
Dodatek 1 DEFINICE A JEDNOTKY
LITERATURA
Dodatek 2 NĚKTERÉ CHEMICKÉ CHARAKTERISTIKY PŘIJATELNÉ ŘEDICÍ VODY
Dodatek 3 DRUHY RYB DOPORUČENÉ PRO ZKOUŠKU
Různé estuarinní nebo mořské druhy byly používány méně často, například:
Sladkovodní ryby uvedené v tabulce se snadno chovají a/nebo jsou dobře dostupné po celý rok, zatímco dostupnost mořských nebo estuarinních ryb je částečně omezena na určité země. Lze je množit a chovat buď v rybích farmách, nebo v laboratoři za kontrolovaných zdravotních a parazitologických podmínek tak, aby pokusné ryby byly zdravé a byly známého původu. Tyto ryby jsou dostupné v mnoha částech světa. LITERATURA
Dodatek 4 HARMONOGRAMY ODBĚRU VZORKŮ PRO ZKOUŠKY EXPOZICE VE VODNÍM PROSTŘEDÍ A V POTRAVĚ 1. Teoretický příklad harmonogramu odběru vzorků pro úplnou zkoušku biokoncentrace látky s log KOW = 4 s expozicí ve vodním prostředí.
2. Teoretický příklad harmonogramu odběru vzorků pro zkoušku bioakumulace látky v potravě po 10denní fázi příjmu a 42denní fázi vylučování.
Poznámka k načasování fází a odběrů vzorků: Fáze příjmu začíná prvním podáním obohacené potravy. Experimentální den trvá od jednoho krmení do doby krátce před příštím krmením o 24 hodin později. První odběr vzorků (č. 1 v tabulce) by se měl uskutečnit krátce před prvním krmením. Odběr vzorků během studie by v ideálním případě měl být proveden krátce před krmením následujícího dne (tj. asi 23 hodin po krmení v den odběru vzorků). Fáze příjmu končí krátce před prvním podáním neobohacené potravy, kdy začíná fáze vylučování (ryby v zkušební skupině budou v mezidobí 24 hodin po posledním podání obohacené potravy pravděpodobně ještě trávit obohacené krmivo). To znamená, že vzorek na konci fáze příjmu by měl být odebrán krátce před prvním podáním neobohacené potravy a první vzorek ve fázi vylučování by měl být odebrán asi 23 hodin po prvním podání neobohaceného krmiva. Dodatek 5 VŠEOBECNÉ VÝPOČTY
1. ÚVOD Obecný model bioakumulace v rybách ve vodním prostředí lze popsat z hlediska procesů příjmu a úbytku, nehledě na příjem potravy. Diferenciální rovnice (dCf/dt) popisující rychlost změny koncentrace v rybách (mg/kg/den) je dána rovnicí (1):
kde
U bioakumulujících látek lze očekávat, že nejpodstatnější hodnotou zkušební koncentrace ve vodě (Cw) v rámci přípustného rozpětí kolísání (srov. odstavec 24) je časově vážená průměrná hodnota (TWA). TWA koncentrace ve vodě se doporučuje vypočítat postupem uvedeným v dodatku 6 zkušební metody C.20 (2). Je třeba poznamenat, že pokud se očekává exponenciální rozpad mezi obnoveními, např. při semistatickém uspořádání zkoušky, je vhodná logaritmická transformace koncentrace ve vodě. Při průtokovém systému logaritmická transformace zkušebních koncentrací možná nebude nutná. Jsou-li odvozeny TWA koncentrace ve vodě, měly by být uvedeny ve zprávě a použity v následných výpočtech. Při standardní zkoušce BCF na rybách je možné příjem a vylučování popsat jako dva procesy s kinetickou prvního řádu.
V rovnovážném stavu, předpokládáme-li, že růst a metabolismus jsou zanedbatelné (tj. hodnoty kg a km není možné odlišit od nuly), se rychlost příjmu rovná rychlosti vylučování, a kombinací rovnice A5.2 a rovnice A5.3 tedy získáme tento vztah:
kde
Poměr k1/k2 se označuje jako kinetický BCF (BCFK) a měl by se rovnat BCF v rovnovážném stavu (BCFSS) získanému z poměru koncentrace v rybách v rovnovážném stavu ke koncentraci ve vodě v rovnovážném stavu, avšak může dojít k odchylkám, pokud rovnovážný stav nebyl jasně stanoven nebo pokud byly u kinetického BCF provedeny úpravy o růst. Jelikož však k1 a k2 jsou konstanty, k odvození hodnoty BCFK není nutné dosáhnout rovnovážného stavu. Na základě těchto prvních rovnic prvního řádu jsou v tomto dodatku 5 uvedeny všeobecné výpočty nezbytné pro bioakumulační metody expozice jak ve vodním prostředí, tak v potravě. Oddíly 5, 6 a 8 se ovšem týkají pouze metody expozice ve vodním prostředí, ale jsou zde uvedeny, neboť se jedná o „všeobecné“ postupy. Použití metod postupného (oddíly 4 a 5) a současného (oddíl 6) stanovení umožňuje výpočet konstant příjmu a vylučování, které se používají k odvození kinetických BCF. Metoda postupného stanovení hodnoty k2 (oddíl 4) je důležitá pro zkoušku expozice v potravě, neboť je nutná jak pro výpočet účinnosti asimilace, tak pro výpočet BMF. Výpočty, které jsou specifické pro studii potravy, jsou podrobně popsány v dodatku 7. 2. PŘEDPOVĚĎ DÉLKY FÁZE PŘÍJMU Před provedením zkoušky lze získat odhad hodnoty k2 a odtud lze získat dobu nezbytnou pro dosažení rovnovážného stavu z empirických vztahů mezi k2 a rozdělovacím koeficientem n-oktanol/voda (KOW) nebo mezi k1 a BCF. Je však třeba si uvědomit, že rovnice v tomto oddíle platí pouze v případě, že se příjem a vylučování řídí kinetikou prvního řádu. Pokud tomu tak zjevně není a je-li žádoucí mít předpovědi fáze příjmu, doporučuje se konzultovat biostatistika a/nebo farmakokinetika. Odhad hodnoty k2 (jednotka/den) lze získat několikerým způsobem. Mohly by být například nejprve použity tyto empirické vztahy (87):
nebo
W = střední hodnota hmotnosti exponovaných ryb (v gramech živé hmotnosti) na konci fáze příjmu / začátku fáze vylučování (88) Jiné související vztahy viz (6). Při odhadu hodnoty k2 může být výhodné použít složitější modely, například pokud je pravděpodobné, že může dojít k výraznému metabolismu (7) (8). Čím je však model složitější, tím obezřetněji je třeba přistupovat k interpretaci předpovědí. Například, přítomnost nitroskupin by mohla indikovat rychlý metabolismus, ale není tomu tak vždy. Uživatel by proto při plánování studie měl zvážit výsledky prediktivní metody s ohledem na chemickou strukturu a na jakékoli jiné důležité informace (například předběžné studie). Dobu potřebnou pro dosažení určitého procenta rovnovážného stavu lze s použitím odhadu k2 získat z obecné kinetické rovnice popisující příjem a vylučování (s kinetikou prvního řádu), za předpokladu, že růst a metabolismus jsou zanedbatelné. Dojde-li během studie k výraznému růstu, nebudou níže popsané odhady spolehlivé. V takových případech je lepší použít hodnotu upravenou o růst, k2 g, jak je popsáno dále (viz oddíl 7 tohoto dodatku).
nebo, je-li Cw konstantní:
Jestliže se blíží dosažení rovnovážného stavu (t → ∞), lze rovnici A5.10 zjednodušit (srov. (9) (10)) a získáme:
nebo
BCF × Cw se pak blíží koncentraci v rybách v rovnovážném stavu (Cf-SS). [Poznámka: stejný postup lze použít při odhadování BMF v rovnovážném stavu v rámci zkoušky expozice v potravě. V tomto případě se hodnota BCF ve shora uvedené rovnici nahradí BMF a hodnota Cw se nahradí Cfood, koncentrací v potravě.] Rovnice A5.10 může být přepsána na rovnici:
nebo
Pomocí rovnice A5.14 je možné předpovědět dobu potřebnou pro dosažení určitého procenta rovnovážného stavu, když hodnota k2 je předem odhadnuta za použití rovnice A5.5 nebo rovnice A5.6. Je pravidlem, že statisticky optimální délka fáze příjmu pro získání statisticky přijatelných údajů (BCFK) je doba potřebná k tomu, aby křivka sestrojená vynesením logaritmu koncentrace zkoušené látky v rybách proti času v lineárním měřítku dosáhla alespoň 50 % rovnovážného stavu (tj. 0,69/k2 ), ale ne více než 95 % rovnovážného stavu (tj. 3,0/k2 ) (11). V případě, že akumulace překročí hodnotu 95 % rovnovážného stavu, je proveditelný výpočet BCFSS. Doba potřebná pro dosažení 80 % rovnovážného stavu činí (za použití rovnice A5.14):
nebo
Podobně doba potřebná pro dosažení 95 % rovnovážného stavu je:
Například délka fáze příjmu (tj. doba potřebná pro dosažení určitého procenta rovnovážného stavu, např. t80 nebo t95) u látky s hodnotou log KOW = 4 by byla (při použití rovnice A5.5, rovnice A5.16 a rovnice A5.17): logk 2 = 1,47 – 0,414 4 k2 = 0,652 den– 1
nebo Alternativně výraz:
může být eventuálně použit pro výpočet doby potřebné k dosažení efektivního rovnovážného stavu (teSS) (12). V případě zkoušené látky s hodnotou log KOW = 4 to vede k výsledku: teSS = 6,54 · 10 – 3 · 104 + 55,31 = 121 hours 3. PŘEDPOVĚĎ DÉLKY FÁZE VYLUČOVÁNÍ Předpověď doby potřebné ke snížení obsahu určitého procenta původní koncentrace v těle lze rovněž získat z obecné rovnice vyjadřující příjem a vylučování (za předpokladu kinetiky prvního řádu, viz rovnice A5.9 (1) (13). Ve fázi vylučování se má za to, že hodnota Cw (nebo Cfood v případě studie potravy) je nulová. Rovnice může pak být zjednodušena na rovnici:
nebo
kde Cf,0 je koncentrace na počátku fáze vylučování. 50 % vyloučení bude dosaženo v čase t50:
nebo
Podobně 95 % vyloučení bude dosaženo v čase:
Je-li pro první fázi zvoleno dosažení 80 % příjmu (1,6/ k2 ) a pro fázi vylučování je zvoleno dosažení 95 % úbytku (3,0/ k2 ), pak je délka fáze vylučování přibližně dvojnásobkem délky fáze příjmu. Nelze ztrácet ze zřetele, že odhady jsou založeny na předpokladu, že procesy příjmu a vylučování se budou řídit kinetikou prvního řádu. Nejedná-li se zjevně o kinetiku prvního řádu, nejsou tyto odhady platné. 4. METODA POSTUPNÉHO STANOVENÍ: STANOVENÍ RYCHLOSTNÍ KONSTANTY VYLUČOVÁNÍ (ÚBYTKU) K2 U většiny údajů týkajících se biokoncentrace se předpokládá, že jsou „dostatečně“ dobře popsány jednoduchým dvoukompartmentovým nebo dvouparametrovým modelem, jak je patrné z přímky, která je aproximací bodů pro koncentrace v rybách (na logaritmické škále) během fáze vylučování.
Je třeba si povšimnout, že odchylky od přímky mohou znamenat složitější model vylučování, než je kinetika prvního řádu. Pro analýzu typů vylučování, které se odchylují od kinetiky prvního řádu, mohou být použity grafické metody. Pro výpočet k2 pro více odběrů vzorků se provede lineární regrese ln (koncentrace) v čase. Směrnice regresní přímky je odhadovaná hodnota rychlostní konstanty vylučování k2 (89). Z úseku lze snadno vypočítat průměrnou koncentraci v rybách na začátku fáze vylučování (C0,d; která se rovná průměrné koncentraci v rybách na konci fáze příjmu) (včetně rozpětí chyby) (89):
Pro výpočet hodnoty k2 , jsou-li k dispozici pouze dva odběry vzorků (jako při minimalizovaném uspořádání), nahraďte průměrné koncentrace v této rovnici
kde ln (Cf1) a ln (Cf2) jsou přirozené logaritmy koncentrací v čase t1 resp. v čase t2 , přičemž t2 a t1 jsou časy, kdy byly tyto dva vzorky odebrány, vzhledem k začátku vylučování (90). 5. METODA POSTUPNÉHO STANOVENÍ: STANOVENÍ RYCHLOSTNÍ KONSTANTY PŘÍJMU K1 (POUZE PRO METODU EXPOZICE VE VODNÍM PROSTŘEDÍ) K nalezení hodnoty k1 založené na souboru po sobě jdoucích údajů o koncentracích ve fázi příjmu použijte počítačový program pro vyjádření tohoto modelu:
kde hodnota k2 je dána předchozím výpočtem, Cf(t) je koncentrace v rybách v čase t a Cw(t) koncentrace ve vodě v čase t. Pro výpočet hodnoty k1 , jsou-li k dispozici pouze dva odběry vzorků (jako při minimalizovaném uspořádání), se použije tento vzorec:
kde k2 je dána předchozím výpočtem, Cf je koncentrace v rybách na začátku fáze vylučování a Cw je průměrná koncentrace ve vodě během fáze příjmu (91). Posouzení dobré shody je možné provést vizuální kontrolou směrnic k1 a k2 vynesených do grafu proti naměřeným údajům při odběrech. Jestliže se ukáže, že metodou postupného stanovení byl získán špatný odhad hodnoty k1 , měla by se pro výpočet k1 a k2 použít metoda současného stanovení (viz následující oddíl 6). Výsledné směrnice by opět měly být při vizuální kontrole dobré shody porovnány s naměřenými údaji vynesenými do grafu. Je-li shoda stále špatná, může to znamenat, že se nejedná o kinetiku prvního řádu a měly by se použít jiné, složitější modely. 6. METODA SOUČASNÉHO STANOVENÍ PRO VÝPOČET RYCHLOSTNÍCH KONSTANT PŘÍJMU A VYLUČOVÁNÍ (ÚBYTKU) (POUZE PRO METODU EXPOZICE VE VODNÍM PROSTŘEDÍ) Pomocí počítačových programů lze nalézt hodnoty k1 a k2 založené na souboru po sobě jdoucích údajů o koncentracích a na modelu:
kde
Tímto postupem se přímo získají směrodatné chyby pro odhady hodnot k1 a k12 . Když se k1 / k2 v rovnici A5.25 a v rovnici A5.26 nahradí hodnotou BCF (srov. rovnice A5.4), je také možné odhadnout směrodatnou chybu a 95 % interval spolehlivosti BCF. To je užitečné zvláště při porovnávání různých odhadů v důsledku transformace dat. Závislou proměnnou (koncentrace v rybách) je možné proložit křivkou s ln transformací nebo bez ní a lze vyhodnotit výslednou nejistotu BCF. Jelikož mezi oběma parametry k1 a k2 , jsou-li odhadovány současně, existuje silná korelace, lze doporučit nejprve vypočítat k2 pouze z údajů o vylučování (viz výše); hodnotu k2 je ve většině případů možné s poměrně vysokou přesností odhadnout z křivky vylučování. Hodnotu k1 lze poté vypočítat z údajů o příjmu pomocí nelineární regrese (92). Při postupném stanovení se doporučuje použít stejnou transformaci dat. Posouzení dobré shody lze provést vizuální kontrolou výsledných směrnic vynesených do grafu proti naměřeným údajům při odběrech. Jestliže se ukáže, že touto metodou byl získán špatný odhad hodnoty k1 , měl by se pro výpočet k1 a k2 použít postup současného stanovení. Proložený model by opět měl být vizuální kontrolou dobré shody porovnán s naměřenými údaji vynesenými do grafu a výsledné odhady parametrů k1 a k k2 a výsledný BCF a jejich směrodatné chyby a/nebo intervaly nejistoty by měly být porovnány z hlediska různých druhů shody. Je-li shoda malá, může to znamenat, že se nejedná o kinetiku prvního řádu a měly by se použít jiné, složitější modely. Jednou z nejběžnějších komplikací je růst ryb během zkoušky. 7. ÚPRAVA KINETICKÉHO BCF A BMF O ZŘEDĚNÍ RŮSTEM V tomto oddíle se popisuje standardní postup úpravy o růst ryb během zkoušky (tzv. „ředění růstem“), která je platná pouze tehdy, jedná-li se o kinetiku prvního řádu. Pokud existují příznaky, že se nejedná o kinetiku prvního řádu, doporučuje se obrátit se konzultovat biostatistika, aby úprava o ředění růstem byla provedena správně, nebo použít postup založený na celkové hmotnosti, který je popsán níže. V některých případech tato metoda úpravy o ředění růstem není dostatečně přesná nebo nefunguje (například u velmi pomalu vylučovaných látek zkoušených na rychle rostoucích rybách může být odvozená rychlostní konstanta vylučování upravená ředění růstem, k2g , velmi nízká, a chyba ve dvou rychlostních konstantách používaných k jejímu odvození se tak stává zásadně důležitou, a v některých případech mohou být odhadované hodnoty kg větší než k2 ). V takových případech lze použít alternativní postup (tj. hmotnostní přístup), který funguje i tehdy, pokud nebyla dodržena kinetika prvního řádu, a při kterém není nutné tuto úpravu provádět. Tento postup je nastíněn na konci tohoto oddílu. Postup úpravy o růst odečtením rychlostní konstanty růstu Při standardní metodě jsou všechny údaje o individuální hmotnosti a délce převáděny na přirozené logaritmy a ln (hmotnost) nebo ln (1/hmotnost) se vynese do grafu proti času (den), zvlášť pro exponovanou a pro kontrolní skupinu. Stejný postup se provádí zvlášť pro údaje z fáze příjmu a z fáze vylučování. Při úpravě o ředění růstem je obecně vhodnější pro odvození rychlostní konstanty růstu (kg ) použít údaje o hmotnosti z celé studie, ale statisticky významné rozdíly mezi rychlostními konstantami růstu odvozenými pro fázi příjmu a pro fázi vylučování mohou znamenat, že by měla být použita rychlostní konstanta vylučovací fáze. Celkové rychlosti růstu pro exponovanou a kontrolní skupinu získané ze studií ve vodním prostředí lze použít pro kontrolu jakýchkoli účinků spojených s expozicí. Běžnými statistickými postupy se pomocí metody nejmenších čtverců vypočítá lineární závislost ln (hmotnost ryb) na čase (den)a ln (1/hmotnost) na čase (den) pro každou skupinu (exponované a kontrolní skupiny a jednotlivé údaje, nikoli denní střední hodnoty) za celou dobu studie a za fáze příjmu a vylučování. Vypočítají se odchylky ve směrnicích přímek a použijí se pro hodnocení statistické významnosti (p = 0,05) rozdílů směrnic (rychlostní konstanty růstu) s použitím Studentova t-testu (nebo analýzy ANOVA, pokud je zkoušena více než jedna koncentrace). Pro účely úpravy o růst se zpravidla upřednostňují údaje o hmotnosti. Údaje o délce, které se zpracovávají stejným způsobem, mohou být užitečné pro porovnání účinků spojených s expozicí mezi kontrolní a zkušební skupinou. Pokud analýza údajů o hmotnosti nevykáže statisticky významný rozdíl, lze údaje z exponované a kontrolní skupiny sloučit a vypočítat celkovou rychlostní konstantu růstu ryb (kg ) ve studii jako celkovou směrnici lineární korelace. Jsou-li zjištěny statisticky významné rozdíly, uvedou se rychlostní konstanty růstu pro každou skupinu ryb a/nebo pro každou fázi studie zvlášť. Rychlostní konstanta pro každou zkušební skupinu by poté měla být použita pro účely úpravy o ředění růstem za tuto skupinu. Pokud byly zjištěny statistické rozdíly mezi rychlostními konstantami ve fázi příjmu a ve fázi vylučování, měly by být použity odvozené rychlostní konstanty ve fázi vylučování. Vypočítanou rychlostní konstantu růstu (kg , vyjádřenou jako den-1) lze odečíst od celkové rychlostní konstanty vylučování (k2 ) a získá se rychlostní konstanta vylučování upravená o růst k2g :
Rychlostní konstanta příjmu se vydělí rychlostní konstantou vylučování upravenou o růst a získá se kinetický kinetický BCF upravený o růst označovaný jako BCFKg (nebo BMFKg).
Rychlostní konstanta růstu odvozená pro studii potravy se použije v rovnici A7.5 k výpočtu BMFKg upraveného o růst (srov. dodatek 7). Metoda úpravy o růst založená na hmotnosti Alternativu k výše uvedené „metodě odečtení rychlostní konstanty růstu“, při které není nutné provádět úpravu o růst, je možné použít následujícím způsobem. Zásada spočívá v tom, že se použijí údaje o vylučování na základě hmotnosti celých ryb, a nikoli na základě koncentrace. Koncentrace v tkáních ve fázi vylučování (hmotnost zkoušené látky/jednotková hmotnost ryb) se převede na hmotnost zkoušené látky/ryba: koncentrace a hmotnosti jednotlivých ryb se porovnají ve formě tabulky (např. pomocí počítačové tabulky) a každá koncentrace se vynásobí celkovou hmotností ryb při daném měření, čímž se získá soubor hmotnost zkoušené látky/ryba pro všechny vzorky z fáze vylučování. Výsledné údaje přirozeného logaritmu hmotnosti látky se vynesou do grafu proti délce experimentu (fáze vylučování), jak by se to provedlo obvykle. Pro metodu expozice ve vodním prostředí se běžným způsobem odvodí rychlostní konstanta vylučování (viz oddíly 4 a 6; za povšimnutí stojí, že „normální“ hodnota k2 by měla být použita v rovnicích vynesení křivky pro hodnotu k1 ), a odvodí se rychlostní konstantu růstu z výše uvedených údajů. Jelikož výsledná hodnota rychlostní konstanty vylučování je nezávislá na růstu, neboť byla odvozena na základě hmotnosti na celou rybu, měla by být označena jako k2g , a nikoli jako k2 . 8. NORMALIZACE LIPIDŮ NA 5 % OBSAH LIPIDŮ (POUZE PRO METODU EXPOZICE VE VODNÍM PROSTŘEDÍ) Měly by rovněž být uvedeny výsledky stanovení BCF (kinetického a v rovnovážném stavu) ve vztahu k standardnímu obsahu lipidů ve výši 5 % živé hmotnosti, pokud není možné namítnout, že zkoušená látka se neakumuluje především v lipidech (např. některé perfluorované látky se mohou vázat na proteiny). Údaje o koncentraci v rybách, neboli BCF, je nutné převést na 5 % obsah lipidů vztažený k živé hmotnosti. Pokud byly k měření koncentrací látky a obsahu lipidů při všech odběrech použity tytéž ryby, musí se každá naměřená koncentrace v jednotlivých rybách upravit o obsah lipidů dané ryby.
kde
Pokud analýza lipidů nebyla provedena u všech ryb ve vzorku, použije se k normalizaci BCF střední hodnota obsahu lipidů. Pro úpravu BCF v rovnovážném stavu by měla být použita střední hodnota zaznamenaná na konci fáze příjmu ve zkušební skupině. Pokud jde o normalizaci kinetického BCF, existují některé případy, kdy se vyžaduje jiný postup, například pokud se obsah lipidů během fáze příjmu nebo vylučování zřetelně změnil. Každopádně by se však mělo rutinně používat množství potravy, které minimalizuje dramatické změny obsahu lipidů.
kde
LITERATURA
Dodatek 6 ODDÍL ROVNIC PRO ZKOUŠKU EXPOZICE VE VODNÍM PROSTŘEDÍ: MINIMALIZOVANÉ USPOŘÁDÁNÍ ZKOUŠKY Argumentem, který svědčí pro použití tohoto přístupu, je skutečnost, že biokoncentrační faktor lze v úplné zkoušce stanovit buď jako biokoncentrační faktor v rovnovážném stavu (BCFSS) tak, že se vypočítá poměr koncentrace zkoušené látky v rybích tkáních a koncentrace zkoušené látky ve vodě, nebo se vypočítá kinetický biokoncentrační faktor (BCFK) jako poměr rychlostní konstanty příjmu k1 a rychlostní konstanty vylučování k2 . BCFK je platný i v případě, kdy nebylo dosaženo rovnovážné koncentrace látky během fáze příjmu, za předpokladu, že příjem i vylučování probíhají přibližně v souladu s kinetickými procesy prvního řádu. Je-li měření koncentrace látky v tkáních (Cf1) prováděno v době, kdy expozice končí (t1 ), a koncentrace v tkáni (Cf2) se změří znovu po uplynutí doby (t2 ), lze odhadnout rychlostní konstantu vylučování (k2 ) pomocí rovnice A5.22 z dodatku 5. Rychlostní konstantu příjmu, k1 , je pak možné stanovit algebraicky pomocí rovnice A5.23 z dodatku 5 (kde Cf je rovno Cf1 a t je rovno t1 ) (1). Kinetický biokoncentrační faktor pro minimalizované uspořádání (označený jako BCFKm pro odlišení od kinetických biokoncentračních faktorů stanovených jinými metodami) tedy je:
Koncentrace nebo výsledky by měly být upraveny o ředění růstem a normalizovány na 5 % obsah lipidů v rybách, jak je popsáno v dodatku 5. Hodnota BCFSS při minimalizovaném uspořádání je rovna BCF vypočtenému na konci fáze příjmu za předpokladu, že bylo dosaženo rovnovážného stavu. To však lze pouze předpokládat, neboť počet odběrů vzorků není dostatečný, aby bylo možno rovnost prokázat.
kde Cf-minSS = koncentrace v rybách v předpokládaném rovnovážném stavu na konci fáze příjmu (mg/kg živé hmotnosti) Cw-minSS = koncentrace ve vodě v předpokládaném rovnovážném stavu na konci fáze příjmu (mg/l). LITERATURA
Dodatek 7 ODDÍL ROVNIC PRO ZKOUŠKU EXPOZICE V POTRAVĚ
1. PŘÍKLAD POMĚRU SLOŽEK VE VHODNÉ KOMERČNÍ RYBÍ POTRAVĚ
2. PŘÍKLADY ZPŮSOBŮ OBOHACOVÁNÍ POTRAVY Všeobecně Potrava pro kontrolní skupinu by se měla připravovat přesně stejným způsobem jako obohacená potrava, ale bez zkoušené látky. S cílem zkontrolovat koncentraci obohacené potravy by měly být za pomoci vhodné extrakční metody extrahovány tři vzorky dávkované potravy a v těchto extraktech změřena koncentrace nebo radioaktivita zkoušené látky. Měly by být prokázány vysoké analytické výtěžky (> 85 %) s nízkým kolísáním mezi vzorky (tři vzorky koncentrací látky odebrané na začátku zkoušky by se neměly lišit o více než ± 15 % od střední hodnoty). Během zkoušky potravy by se měly odebrat k analýze tři vzorky potravy v den 0 a na konci fáze příjmu za účelem stanovení obsahu zkoušené látky v potravě. Příprava rybí potravy s kapalnou zkoušenou látkou (v čistém stavu) Stanoví se cílová, nominální zkušební koncentrace v exponované rybí potravě, např. 500 μg zkoušené látky/g potravy. Přiměřené množství (podle molární hmotnosti nebo podle specifické radioaktivity) čisté chemické látky se přidá do známé hmotnosti rybí potravy ve skleněné nádobě nebo v baňce rotační odparky. Hmotnost rybí potravy by měla být dostatečná s ohledem na délku fáze příjmu (přičemž se vezme v úvahu potřeba zvýšit množství při každém krmení v důsledku růstu ryb). Rybí krmivo/zkoušená látka by se měly přes noc promísit pomalým převracením (např. pomocí RRM nebo rotací, použije-li se baňka rotační odparky). Obohacená potrava by se až do použití měla uchovávat za podmínek, kdy zůstane zachována stabilita zkoušené látky v krmném mixu (např. v chladničce). Příprava rybí potravy s kukuřičným olejem nebo s rybím tukem jako vehikulem Pevné zkoušené látky by se měly rozmělnit v hmoždíři na jemný prášek. Kapalné zkoušené látky lze přidat přímo do kukuřičného oleje nebo rybího tuku. Zkoušená látka se rozpustí ve známém množství kukuřičného oleje nebo rybího tuku (např. 5 až 15 ml). Dávkovaný olej se kvantitativně přenese do rotační odparky vhodné velikosti. Baňka použitá pro přípravu dávkovaného oleje by se měla propláchnout dvěma malými díly oleje a ty se přidají do baňky rotační odparky, aby se zajistilo, že bude přenesena veškerá rozpuštěná zkoušená látka. Aby se zajistilo úplné rozpuštění/disperze v oleji (nebo pokud se ve studii používá více než jedna zkoušená látka), přidá se mikromíchačka, baňka se uzavře zátkou a směs se nechá přes noc rychle míchat. Přiměřené množství rybího krmiva (obvykle ve formě vloček) pro zkoušku se přidá do baňky a obsah baňky se homogenně míchá stálým otáčením skleněné baňky alespoň po domu 30 minut, ale nejlépe přes noc. Obohacená potrava se poté vhodně uskladní (např. v chladničce), aby se zajistila stabilita zkoušené látky v potravě až do použití. Příprava rybí potravy s organickým rozpouštědlem Vhodné množství zkoušené látky (podle molární hmotnosti nebo podle specifické radioaktivity) dostatečné pro dosažení cílové dávky se rozpustí ve vhodném organickém rozpouštědle (např. cyklohexan nebo aceton; 10 až 40 ml, ale v případě potřeby i větší objem, podle množství potravy, které se má obohatit). Poměrný díl nebo všechen tento roztok (přidávaný po částech) se smíchá s přiměřeným množstvím rybí potravy dostatečným k tomu, aby bylo ve zkoušce dosaženo požadované nominální úrovně dávky. Potravu a zkoušenou látka je možné smísit v nádobě mixéru z nerezové oceli a čerstvě připravené dávky rybí potravy dva dny ponechat v nádobě v laboratorních podmínkách (za občasného míchání), aby se umožnilo odpaření přebytečného rozpouštědla, nebo smísit v baňce rotační odparky se stálou rotací. Přebytečné rozpouštědlo lze v případě potřeby „odfouknout“ proudem vzduchu nebo dusíku. Je třeba dát pozor, aby zkoušená látka při odstraňování rozpouštědla nekrystalizovala. Obohacená potrava by se až do použití měla uchovávat za podmínek, kdy zůstane zachována stabilita zkoušené látky v krmném mixu (např. v chladničce). 3. VÝPOČET ÚČINNOSTI ASIMILACE A BIOOBOHACOVACÍHO FAKTORU Aby bylo možné vypočítat účinnost asimilace, měla by se nejprve odhadnout celková rychlostní konstanta vylučování podle oddílu 4 dodatku 5 („metodou postupného stanovení“, tj. standardní lineární regresí) s použitím střední hodnoty koncentrace vzorků z fáze vylučování. Rychlostní konstanta pojídání potravy, I, a délka fáze příjmu, t, jsou známými parametry studie. Cfood, střední hodnota naměřené koncentrace zkoušené látky v potravě, je měřená proměnná ve studii. C0,d, koncentrace zkoušené látky v rybách na konci fáze příjmu, je obvykle odvozena z úseku grafického znázornění závislosti hodnoty ln (koncentrace) na dnu vylučování. Účinnost asimilace látky (a, absorpce zkoušené látky ve střevech) se vypočítá jako:
kde: C0,d = odvozená koncentrace v rybách v čase nula fáze vylučování (mg/kg) k2 = celková (neupravená o růst) rychlostní konstanta vylučování (den-1) vypočítaná podle rovnic v oddílu 3 dodatku 5 I = rychlostní konstanta pojídání potravy (g potravy/g ryby/den) Cfood = koncentrace v potravě (mg/kg potravy) t= délka krmení (dny) Avšak má-li být získána přesná účinnost asimilace, a, bude možná nutné rychlostní konstantu pojídání potravy, I, která se používá ve výpočtu, upravit o růst ryb. Při zkoušce, kdy ryby během fáze příjmu (ve které se neprovádí žádná úprava množství krmiva, aby bylo zachováno stanovené množství potravy) vykazují výrazný růst, bude s postupem fáze příjmu efektivní množství potravy nižší než původně stanovené, což vede k vyšší „skutečné“ účinnosti asimilace. (Pro výpočet celkového BMF to ovšem není důležité, neboť členy I v rovnici A7.1 a v rovnici A7.4 se ve skutečnosti vyruší). Střední hodnotu rychlostní konstanty pojídání potravy upravenou o zředění růstem, Ig, lze odvodit několika způsoby, ale přímým a přesným způsobem je použít známou rychlostní konstantu růstu (kg ) k odhadu hmotností zkušebních ryb při odběrech vzorků během fáze příjmu, tj.:
kde Wf(t)= střední hodnota hmotnosti ryb v den t fáze příjmu Wf,0 = střední hodnota hmotnosti ryb na začátku experimentu Tímto způsobem lze odhadnout (alespoň) střední hodnotu hmotnosti ryb v poslední den expozice (Wf,end-of-uptake). Jelikož množství potravy bylo stanoveno na základě Wf,0, je možné pomocí těchto dvou hodnot hmotnosti vypočítat efektivní množství potravy pro každý den fáze příjmu. Rychlostní konstantu pojídání potravy upravenou o růst, Ig (g potravy/g ryby/den), která se v případech rychlého růstu během fáze příjmu použije místo I, je pak možné vypočítat jako
Jakmile byla získána účinnost asimilace, lze vypočítat BMF tak, že účinnost vynásobíme rychlostní konstantou pojídání potravy I (nebo Ig , používá-li se k výpočtu α) a výsledek dělíme celkovou rychlostní konstantou vylučování k2 :
Také bioobohacovací faktor upravený o růst by se měl vypočítat stejným způsobem, s použitím rychlostní konstanty vylučování upravené o růst (odvozené podle dodatku 5 oddílu 7). A opět, pokud byla k výpočtu α použita hodnota Ig, měla by být použita i zde namísto I:
kde: α = účinnost asimilace (absorpce zkoušené látky ve střevech) k2 = celková (neupravená o růst) rychlostní konstanta vylučování (den-1) vypočítaná podle rovnic v oddílu 3 dodatku 5 k2g = rychlostní konstanta vylučování upravená o růst (jednotka/den) I = rychlostní konstanta pojídání potravy (g potravy/g ryby/den) Poločas upravený o růst (t1/2 ) se vypočítá takto:
Účinnost asimilace potravy lze odhadnout také v případě, že byla stanovena rezidua v tkáních během lineární fáze příjmu (mezi dny 1 a 3). V tomto případě lze účinnost asimilace (α) látky stanovit takto:
kde Cfish(t)= koncentrace zkoušené látky v rybách v čase t (mg/kg živé hmotnosti). 4. ÚPRAVA O LIPIDY Jestliže byl obsah lipidů měřen u týchž ryb, na kterých byla prováděna analýza chemické látky při všech odběrech, pak by měly jednotlivé koncentrace být upraveny na základě lipidů a hodnoty ln (koncentrace upravená o lipidy) zaneseny do grafu v závislosti na vylučování (vyjádřené ve dnech), čímž se získají hodnoty C0,d a k2. Účinnost asimilace (rovnice A7.1) pak lze vypočítat na základě lipidů s použitím hodnoty Cfood na základě lipidů (tj. Cfood se vynásobí střední hodnotou frakce lipidů v potravě). Následným výpočtem podle rovnice A7.4 a rovnice A7.5 se přímo získá hodnota BMF upravená o lipidy (a upravená o zředění růstem). Jinak se střední hodnotu frakce lipidů (v hmotnostním vyjádření) v rybách a v potravě odvodí pro exponovanou a kontrolní skupinu (u potravy a ryb kontrolní skupiny je to obvykle z údajů naměřených na začátku a na konci expozice; u ryb ze zkušební skupiny je to obvykle pouze z údajů naměřených na konci expozice). V některých studiích se může obsah lipidů znatelně zvýšit; v takových případech je vhodnější použít střední hodnotu koncentrace lipidů v rybách vypočítanou z naměřených hodnot na konci fáze expozice a na konci fáze vylučování. Obecně by údaje získané od zkušební skupiny měly být používány pouze k odvození obou frakcí lipidů. Faktor úpravy o lipidy (Lc) se vypočítá jako:
kde Lfish je střední hodnota frakce lipidů v rybách a Lfood je střední hodnota frakce lipidů v potravě. Faktor úpravy o lipidy se používá k výpočtu bioobohacovacího faktoru upraveného o lipidy (BMFL):
5. HODNOCENÍ ROZDÍLŮ MEZI NAMĚŘENOU KONCENTRACÍ V ČASE NULA (C0,m) A ODVOZENOU KONCENTRACÍ V ČASE NULA (C0,d) Měla by se porovnat naměřená koncentrace v čase nula (C0,m) a odvozená koncentrace v čase nula (C0,d). Pokud jsou si velmi blízké, podporuje to model prvního řádu, který se používá k odvození parametrů vylučování. V některých studiích může být mezi odvozenou hodnotou v čase nula C0,d a střední hodnotou naměřené koncentrace v čase nula zřetelný rozdíl. C0,m (viz poslední odrážku odstavce 159 této zkušební metody). Je-li C0,d mnohem nižší než C0,m (C0,d << C0,m), může tento rozdíl nasvědčovat přítomnosti nestrávené obohacené potravy ve střevech. To lze experimentálně zjistit provedením samostatné analýzy vyříznutých střev, pokud byly na konci fáze příjmu odebrány dodatečné vzorky (celých ryb) a uskladněny. Pokud však statisticky validní test odlehlých hodnot lineární regrese fáze vylučování ukáže, že výsledky z prvního odběru vzorků ve fázi vylučování jsou nepřiměřené vysoké, může být vhodné provést lineární regresi za účelem odvození hodnoty k2 , ale vypustit první koncentraci ve fázi vylučování. V takových případech, pokud se nejistota v lineární regresi značně sníží a pokud je jasné, že kinetika vylučování byla přibližně kinetikou prvního řádu, může být vhodné použít výsledné hodnoty C0,d a k2 ve výpočtu účinnosti asimilace. Ve zprávě by to mělo být podrobně zdůvodněno. Je také možné, že ve fázi vylučování působila jiná kinetika než kinetika prvního řádu. Je-li to pravděpodobné (tj. pokud se jeví, že přirozený logaritmus transformovaných dat tvoří křivku v porovnání s přímkou na grafu lineární regrese), pak není pravděpodobné, že výpočty hodnot k2 a C0,d jsou platné a měl by být konzultován biostatistik. Je-li C0,d mnohem vyšší než naměřená hodnota (C0,d >> C0,m), může to znamenat, že látka byla vyloučena velmi rychle (tj. odběry se přiblížily mezi kvantifikace analytické metody ve velmi rané části fáze vylučování, viz oddíl 6 níže; že při vylučování došlo k odchylce od kinetiky prvního řádu; že v lineární regresi pro odvození k2 a C0,d došlo k chybě; nebo že při některých odběrech vzorků se vyskytl problém s naměřenými koncentracemi ve studii. V takových případech by měl být zkontrolován graf lineární regrese z hlediska vzorků na hranici nebo poblíž meze kvantifikace, z hlediska odlehlých hodnot a očividného zakřivení (jež naznačují, že se nejedná o kinetiku prvního řádu), a toto by mělo být zaznamenáno ve zprávě. Jakékoli následné přehodnocení lineární regrese s cílem zlepšit odhadované hodnoty by mělo být popsáno a odůvodněno. Jsou-li zjištěny zřetelné odchylky od kinetiky prvního řádu, pak není pravděpodobné, že výpočty k2 a C0,d jsou platné, a měl by být konzultován biostatistik. 6. POKYNY PRO VELMI RYCHLE SE VYLUČUJÍCÍ ZKOUŠENÉ LÁTKY Jak bylo již poukázáno v odstavci 129 zkušební metody, některé látky se mohou vylučovat tak rychle, že není možné spolehlivě odvodit koncentraci v čase nula, C0,d, a hodnotu k2 , protože ve vzorcích odebraných ve velmi rané části fáze vylučování (tj. počínaje druhým odběrem ve fázi vylučování) nelze látku již prakticky změřit (koncentrace uvedené při mezi kvantifikace). Tato situace byla zjištěna při mezilaboratorní porovnávací zkoušce provedené na podporu této zkušební metody s benzo[a]pyrenem a byla zdokumentována ve validační zprávě této metody. V takových případech není možné spolehlivě provést lineární regresi a je pravděpodobné, že z ní vyplyne nereálně vysoký odhad C0,d, který povede k účinnosti asimilace zjevně mnohem vyšší než 1. V těchto případech je možné vypočítat konzervativní odhad k2 a „horní odhad“ BMF. S použitím těch odběrů ve fázi vylučování, kdy byla měřena koncentrace, až do první „nedetekované“ koncentrace (koncentrace stanovené na mezi kvantifikace) včetně, se lineární regresí získá odhad hodnoty k2 (pomocí přirozeného logaritmu transformovaných údajů o koncentraci proti času). V případech tohoto druhu je pravděpodobné, že zahrnují pouze dva odběry vzorků (např. dny odběru 1 a 2 fáze vylučování), a k2 pak lze odhadnout pomocí rovnice A5.22 v dodatku 5. Tento odhad k2 může být použit k odhadnutí účinnosti asimilace podle rovnice A7.1, přičemž v případech, kdy odhadovaná hodnota C0,d by zjevně byla mnohem vyšší, než jaké by mohlo být dosaženo při zkoušce, se hodnota C0,d v rovnici se nahradí naměřenou koncentrací v čase nula (C0,m). Pokud C0,m nebylo možné změřit, měla by být použita mez detekce v rybí tkáni. Jestliže se takto v některých případech získá hodnota α > 1, má se za to, že účinnost asimilace je „v nejhorším případě“ 1. Maximální BMFK pak lze odhadnout pomocí rovnice A7.4 a měl by být označen jako hodnota „mnohem menší než“ (<<). Například při studii prováděné s množstvím krmiva 3 % a poločasem vyloučení kratším než 3 dny a s α, které je „v nejhorším případě“ 1, bude hodnota BMFK pravděpodobně nižší než asi 0,13. Vzhledem k účelu tohoto odhadu a k tomu, že hodnoty budou ve své povaze konzervativní, není nezbytné je upravovat o zředění růstem nebo o obsah lipidů v rybách a v potravě. Dodatek 8 POSTUPY PRO ODHAD PŘEDBĚŽNÝCH BCF Z ÚDAJŮ ZÍSKANÝCH VE STUDII EXPOZICE V POTRAVĚ Studie potravy je do této zkušební metody zahrnuta pro zkoušení bioakumulace látek, které v praxi není možné zkoušet metodou expozice ve vodním prostředí. Metodou expozice ve vodním prostředí se získá biokoncentrační faktor, zatímco metoda expozice v potravě vede přímo k informacím o potenciálu bioobohacení prostřednictvím krmení. Informace o biokoncentraci ve vodním prostředí jsou požadovány při mnohých režimech chemické bezpečnosti (např. při posuzování rizik a v rámci globálně harmonizovaného systému klasifikace). Existuje tudíž potřeba používat údaje získané ve zkoušce potravy k odhadu biokoncentračního faktoru, který je srovnatelný s výsledkem zkoušek prováděných podle metody expozice ve vodním prostředí (94). V tomto oddíle se zkoumají postupy, kterými je možné se přitom řídit, přičemž se však uznávají nedostatky, se kterými jsou tyto odhady nutně spojeny. Studie potravy měří vylučování s cílem získat rychlostní konstantu vylučování, k2. Je-li možné odhadnout rychlostní konstantu příjmu na základě dostupných údajů v situaci, kdy byly ryby exponovány zkoušené látce prostřednictvím vody, pak by bylo možné odhadnout kinetický BSF. Odhad rychlostní konstanty příjmu pro expozici ve vodě závisí na množství předpokladů, z nichž všechny budou přispívat k nejistotě odhadu. Tento přístup k odhadování BCF předpokládá, že celková rychlost vylučování (včetně přispívajících faktorů jako distribuce v těle a jednotlivé procesy vylučování) je nezávislá na způsobu expozice, který se použije k získání obsahu zkoušené látky v těle. Hlavní předpoklady, na kterých je založen tento postup odhadování, lze shrnout takto: vylučování, jež následuje po příjmu potravy, je stejné jako vylučování po expozici dané látce ve vodním prostředí, příjem z vody by se řídil kinetikou prvního řádu, podle metody použité pro odhad příjmu:
databáze („školicí sada“) použitá při vývoji metody odhadování příjmu je reprezentativní pro látku, která se posuzuje. V některých veřejně dostupných publikacích jsou odvozeny rovnice, které příjem ryb z vody prostřednictvím žaber vztahují k rozdělovacímu koeficientu n-oktanol/voda látky, k hmotnosti ryb (1) (2) (3) (4), k objemu a/nebo obsahu lipidů, k propustnosti membrány / difusi (5) (6), k objemu provzdušňování ryb (7) a vycházejí z přístupu prchavosti / hmotnostní bilance (8) (9) (10). Podrobné praktické hodnocení těchto metod podávají Crookes & Brooke (11). V této souvislosti je užitečná také Barberova publikace (12) zaměřená na modelování bioakumulace prostřednictvím příjmu potravy, neboť obsahuje příspěvky z modelů rychlosti příjmu žábrami. Tomuto aspektu byl věnován také jeden z oddílů podkladového dokumentu k protokolu zkoušky potravy z roku 2004 (13). Většina z těchto modelů zřejmě byla odvozena s použitím omezených databází. U modelů, pro něž jsou k dispozici podrobnosti o databázi použité pro vybudování těchto modelů, se zdá, že použité druhy látek mají často podobnou strukturu nebo třídu (z hlediska funkčnosti, např. chlorované organické látky). To jen zvyšuje nejistotu, zda je možné použít nějaký model k předpovídání rychlostní konstanty příjmu různých druhů látek, spolu s dalšími problémy specifickými pro tuto zkoušku, jako je druh zvířat, teplota atd. Z přezkumu metod, které jsou k dispozici (11), vyplynulo, že žádná z těchto metod není „korektnější“ než ostatní. Proto by mělo být podáno jednoznačné odůvodnění použitého modelu. Tam, kde je k dispozici několik metod, jejichž použití lze odůvodnit, bylo by vhodné předložit několik odhadů hodnoty k1 (a tedy BCF) nebo rozpětí hodnot k1 (a BCF) podle několika metod odhadu příjmu. Vhledem k rozdílným druhům modelů a souborů dat použitých při jejich vývoji by však nebylo vhodné stanovovat střední hodnotu z odhadů odvozených různými způsoby. Někteří výzkumníci tvrdí, že odhady BCF tohoto druhu je nezbytné upravit o biologickou dostupnost, aby bylo možné zohlednit adsorpci látky k rozpuštěnému organickému uhlíku (DOC) za podmínek expozice ve vodním prostředí, a uvést tak odhad do souladu s výsledky studií expozice ve vodním prostředí (např. (13) (14)). Tato úprava však nemusí být vhodná vzhledem k nízkým úrovním DOC požadovaným ve studii expozice ve vodním prostředí pro vypracování odhadu „pro nejhorší případ“ (tj. poměr biologicky dostupné látky a látky měřené v roztoku). U vysoce hydrofobních látek může být příjem na žábrách omezený mírou pasivní difuse poblíž povrchu žaber; v tom případě je možné, že úprava zohledňuje spíše tento účinek, než ten, pro který byla určena. Doporučuje se zaměřit se na metody, jež vyžadují vstupy se snadno dostupnými údaji o látkách, které se zkoušejí podle zde uvedené studie potravy (tj. log KOW, hmotnost ryb). Použít lze i jiné metody, které vyžadují složitější vstupy, avšak kde budou možná nutná další měření při zkoušce nebo důkladné znalosti o dané zkoušené látce nebo o druhu ryb, které nemusí být široce dostupné. Výběr modelu může být navíc ovlivněn úrovní validace a oblastí použitelnosti (přehled různých metod a jejich srovnání, viz (11)). Je třeba mít na paměti, že výsledný odhad k1 a odhadnutá hodnota BCF jsou nejisté a pro získání celkového obrazu o bioakumulačním potenciálu látky je, stejně jako odvozený BMF a parametry látky (např. velikost molekul), možná bude nutné přezkoumat analýzou průkaznosti výsledků. Interpretace a použití těchto parametrů může záviset na regulačním rámci. LITERATURA
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||








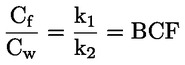






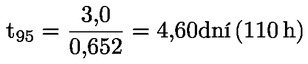








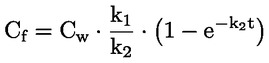

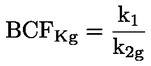


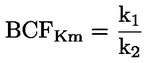
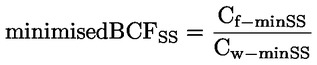
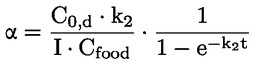






|
17) |
V části C se kapitola C.20 nahrazuje tímto: „C.20 Zkouška toxicity pro reprodukci u Daphnia magna ÚVOD Tato zkušební metoda je rovnocenná Pokynu OECD pro zkoušení (TG) č. 211 (2012). Pokyny OECD pro zkoušení se pravidelně přezkoumávají a aktualizují s ohledem na vědecký pokrok. Pokyn č. 211 ke zkoušce toxicity pro reprodukci vychází z Pokynu pro zkoušení č. 202, část II, Zkouška toxicity pro reprodukci u rodu Daphnia (1984). Všeobecně se uznávalo, že údaje ze zkoušek prováděných podle Pokynu pro zkoušení č. 202 mohou být proměnlivé. Vzhledem k tomu bylo vyvinuto značné úsilí ke zjištění příčin této proměnlivosti s cílem získat lepší zkušební metodu. Pokyn pro zkoušení č. 211 je založen na výsledku těchto výzkumných činností, mezilaboratorních porovnávacích zkoušek a validačních studií provedených v letech 1992 (1), 1994 (2) a 2008 (3). Hlavní rozdíly mezi původní verzí (TG 202, 1984) a druhou verzí (TG 211, 1998) Pokynu pro zkoušku toxicity pro reprodukci byly tyto:
Použité definice jsou uvedeny v dodatku 1. PODSTATA ZKOUŠKY Hlavním cílem zkoušky je posoudit účinky chemických látek na reprodukční schopnost druhu Daphnia magna. Za tímto účelem se mladé samičí dafnie (rodičovské organismy) na začátku zkoušky mladší než 24 hodin exponují zkoušené látce přidané do vody v určitém rozsahu koncentrací. Zkouška trvá 21 dní. Na konci zkoušky se odhadne celkový počet vylíhnutých živých potomků. Reprodukční schopnost rodičovských organismů lze vyjádřit jiným způsobem (např. jako počet živých potomků pocházejících z jednoho rodičovského organismu za den, a to od prvního dne, kdy bylo potomstvo pozorováno), který se však uvede doplňkově vedle celkového počtu vylíhnutých živých potomků na konci zkoušky. Vzhledem ke zvláštnímu uspořádání semistatické zkoušky ve srovnání s jinými metodami zkoušení toxicity pro reprodukci bezobratlých je také možné spočítat živé potomky pocházející z každého jednotlivého rodičovského organismu. To na rozdíl od jiných metod zkoušení toxicity pro reprodukci bezobratlých umožňuje v případě, že rodičovský organismus v době zkoušky náhodně a/nebo bezděčně uhyne, vyloučit jeho potomstvo z hodnocení údajů. A tedy, dojde-li k mortalitě rodičovských organismů v exponovaných paralelních skupinách, mělo by být posouzeno, zda tato mortalita odpovídá vztahu koncentrace a účinku, či nikoli, např. zda existuje významný pokles účinku v závislosti na koncentraci zkoušené chemické látky s kladnou směrnicí (k tomu lze použít statistický test, například Cochranovu-Armitageovu zkoušku trendu). Jestliže mortalita neodpovídá vztahu koncentrace a účinku, měly by být tyto paralelní skupiny s mortalitou rodičovských organismů z analýzy výsledků zkoušky vyloučeny. Pokud mortalita odpovídá vztahu koncentrace a účinku, měla by být mortalita rodičovských organismů označena za účinek zkoušené chemické látky a paralelní skupiny by neměly být z analýzy vyloučeny. Uhyne-li rodičovský organismus během zkoušky, tj. náhodně v důsledku nesprávné manipulace nebo nehody nebo bezděčně z nevysvětlitelného důvodu nesouvisejícího s účinkem zkoušené chemické látky, nebo se ukáže, že šlo o samčí organismus, je paralelní skupina z analýzy vyloučena (podrobněji viz odstavec 51). Toxický účinek zkoušené chemické látky na reprodukční schopnost se vyjadřuje jako LCx a ECx, přičemž údaje se zanášejí do vhodného modelu nelineární regresí s cílem odhadnout koncentraci, která by způsobila mortalitu nebo snížení reprodukční schopnosti ve výši x %, nebo alternativně jako hodnota NOEC/LOEC (4). Zkoušené koncentrace by měly přednostně zahrnovat nejnižší z použitých koncentrací s pozorovatelnými účinky (např. EC10), což znamená, že tato hodnota je vypočtena interpolací, a nikoli extrapolací. Musí být rovněž uvedeno přežití rodičovských organismů a okamžik vylíhnutí prvních potomků. Zkoumány mohou být i jiné účinky na parametry, jako je růst (např. délka), a možná i vnitřní míra růstu populace (viz odstavec 44). INFORMACE O ZKOUŠENÉ LÁTCE Výsledky zkoušky akutní toxicity (viz kapitola C.2 této přílohy: Zkouška akutní imobilizace dafnií (Daphnia sp.)) provedené u Daphnia magna mohou být užitečné pro volbu vhodného rozsahu zkušebních koncentrací ve zkouškách toxicity pro reprodukci. Měly by být známy rozpustnost zkoušené chemické látky ve vodě a tlak jejích par a měla by být k dispozici spolehlivá analytická metoda kvantitativního stanovení chemické látky ve zkušebních roztocích, a to s doloženým výtěžkem a mezí stanovitelnosti. Užitečnými informacemi o zkoušené chemické látce pro účely stanovení zkušebních podmínek mohou být strukturní vzorec, čistota chemické látky, stabilita na světle, stabilita v podmínkách zkoušky pKa, Pow a výsledky zkoušky snadné biologické rozložitelnosti (viz kapitoly C.4 (Stanovení „snadné“ biologické rozložitelnosti) a C.29 (hodnocení snadné biologické rozložitelnosti – uvolňování CO2 v těsně uzavřených lahvičkách) této přílohy). VALIDITA ZKOUŠKY Má-li být zkouška platná, musí být u kontrolní skupiny (kontrolních skupin) splněna tato kritéria reprodukční schopnosti:
Poznámka: Stejné kritérium validity (20 %) lze použít pro náhodnou a bezděčnou mortalitu rodičovských organismů v kontrolních skupinách a také pro každou ze zkušebních koncentrací. POPIS METODY Přístroje a pomůcky Zkušební nádoby nebo jiné pomůcky a přístroje, které přijdou do styku se zkušebními roztoky, by měly být skleněné nebo z jiného chemicky inertního materiálu. Zkušebními nádobami jsou obvykle skleněné kádinky. Kromě toho bude nezbytné některé nebo veškeré toto zařízení:
Zkušební organismy Ke zkoušce se použije druh Daphnia magna Straus (95). Klon by měl být identifikován přednostně uvedením genotypu. Výzkum (1) ukázal, že reprodukční schopnost klonu A (který pochází z institutu IRCHA ve Francii) (5) stabilně splňuje kritérium validity, jímž je 60 potomků na přeživší rodičovský organismus při kultivaci za podmínek popsaných v této metodě. Jiné klony jsou však přijatelné, splňuje-li kultura dafnií prokazatelně kritéria validity pro tuto zkoušku. Na začátku zkoušky by měly být dafnie nejvýše 24 hodin staré a neměly by pocházet z první generace potomků. Měly by pocházet ze zdravé kultury (tj. neměly by vykazovat známky stresu, např. zvýšenou mortalitu, přítomnost jedinců samčího pohlaví a efipií, zpoždění produkce prvních potomků, přítomnost jinak zbarvených jedinců atd.). Organismy z kmenové kultury by měly být chovány v podmínkách podobných těm, které mají být použity při zkoušce (osvětlení, teplota, médium, krmení a počet dafnií na jednotku objemu). Liší-li se kultivační médium pro dafnie, které má být použito při zkoušce, od média používaného k rutinní kultivaci dafnií, doporučuje se zařadit před zkouškou obvykle třítýdenní aklimatizační období (tj. v délce odpovídající jedné generaci), aby nedošlo u rodičovských organismů ke stresu. Zkušební médium Doporučuje se, aby bylo v této zkoušce použito přesně definované médium. Tím se lze vyhnout použití přísad (např. mořských řas, extraktu z půdy), které je obtížné charakterizovat, a zvýšit tak vyhlídky mezilaboratorní standardizace. Elendtova média M4 (6) a M7 (viz odstavec 2) byla shledána pro tento účel jako vyhovující. Jiná média (např. (7, 8)) jsou však přijatelná, splňuje-li reprodukční schopnost kultury dafnií prokazatelně kritéria validity pro tuto zkoušku. Použije-li se médium, které obsahuje nestanovené přísady, měly by být tyto přísady jasně specifikovány a v závěrečné zprávě se uvedou informace o složení, a to zejména o obsahu organického uhlíku, neboť může přispívat k dodávané potravě. Doporučuje se, aby byl stanoven celkový organický uhlík (TOC) a/nebo chemická spotřeba kyslíku (COD) výchozího preparátu organické přísady a aby byl odhadnut výsledný příspěvek k TOC nebo COD ve zkušebním médiu. Dále se doporučuje, aby byly úrovně TOC v médiu (tj. před přidáním řas) nižší než 2 mg/l (9). Při zkoušení chemických látek obsahujících kovy je důležité si uvědomit, že vlastnosti zkušebního média (např. tvrdost, chelatační schopnosti) mohou mít vliv na toxicitu chemické látky. Z tohoto důvodu je žádoucí, aby bylo médium přesně definováno. V současnosti jsou však jedinými přesně definovanými médii, o nichž je známo, že jsou vhodná pro dlouhodobou kultivaci kultury druhu Daphnia magna, Elendtova média M4 a M7. Obě média obsahují chelatační činidlo EDTA. Práce (2) ukázaly, že provádí-li se zkouška v médiích M4 a M7, je „zdánlivá toxicita“ kadmia zpravidla nižší než v médiu, které neobsahuje EDTA. Média M4 a M7 se tedy nedoporučují pro zkoušení chemických látek obsahujících kovy a neměla by se používat ani jiná média obsahující činidla známá svou chelatační schopností. V případě chemických látek obsahujících kovy se doporučuje používat jiné médium, například rekonstituovanou tvrdou sladkou vodu podle ASTM (9), která neobsahuje EDTA. Tato kombinace rekonstituované tvrdé sladké vody podle ASTM a extraktu z řas (10) je vhodná pro dlouhodobou kultivaci druhu Daphnia magna (2). Na začátku zkoušky a v jejím průběhu by měla být koncentrace rozpuštěného kyslíku vyšší než 3 mg/l. Hodnota pH by měla být v rozmezí 6 až 9 a zpravidla by se neměla v žádné zkoušce měnit o více než 1,5. Doporučuje se tvrdost nad 140 mg/l (jako CaCO3). Ve zkouškách při této a vyšší tvrdosti reprodukční schopnost prokazatelně vyhovovala kritériím validity (11, 12). Zkušební roztoky Zkušební roztoky o zvolených koncentracích se obvykle připraví ředěním zásobního roztoku. Zásobní roztoky se připraví, pokud možno bez použití rozpouštědel nebo dispergátorů, nejlépe mechanickým mícháním nebo protřepáváním zkoušené chemické látky ve zkušebním médiu, např. protřepáváním, mícháním, pomocí ultrazvuku nebo jinými vhodnými metodami. Upřednostňuje se exponovat zkušební systémy koncentracím zkoušené chemické látky, která má být ve studii použita, tak dlouho, jak je zapotřebí k prokázání udržení stabilních zkušebních koncentrací, dříve než jsou zavedeny zkušební organismy. Je-li zkoušená chemická látka obtížně rozpustná ve vodě, měl by se použít postup popsaný v Pokynu OECD pro manipulaci se složitými látkami (13). Rozpouštědla nebo dispergátory by se používat neměly, ale v některých případech mohou být pro vytvoření zásobního roztoku o vhodné koncentraci nezbytné. Kromě zkušebních koncentrací by měla být nasazena kontrolní skupina v ředicí vodě s odpovídajícím počtem paralelních skupin a, je-li to nevyhnutelné, kontrolní skupina s rozpouštědlem s odpovídajícím počtem paralelních skupin. Při zkoušce by měla být užita pouze rozpouštědla nebo dispergátory, u nichž bylo šetřením prokázáno, že nemají žádné významné nebo mají jen minimální účinky na proměnné odezvy. Příklady vhodných rozpouštědel (např. aceton, ethanol, methanol, dimethylformamid a triethylenglykol) a dispergátorů (např. Cremophor RH40, 0,01 % methylcelulosa a HCO-40) jsou uvedeny v (13). Použije-li se rozpouštědlo nebo dispergátor, neměla by jeho konečná koncentrace být větší než 0,1 ml/l (13) a ve všech zkušebních nádobách s výjimkou kontrolní skupiny s ředicí vodou by měla být stejná koncentrace. Mělo by však být vynaloženo maximální úsilí k tomu, aby bylo rozpouštědlo udržováno v co nejnižší koncentraci. POSTUP Podmínky expozice Délka expozice Zkouška trvá 21 dní. Nasazování Rodičovské organismy se umístí po jednom do zkušebních nádob s 50 až 100 ml média (u druhu Daphnia magna; menší objemy lze použít zejména pro menší dafnie, např. druh Ceriodaphnia dubia) v každé nádobě, pokud není pro zkoušení nezbytné průtokové uspořádání zkoušky. V některých případech může být nezbytné použít větší objemy, aby bylo možné splnit požadavky analytické metody použité pro stanovení koncentrace zkoušené chemické látky, třebaže je sdružování paralelních vzorků k chemické analýze rovněž přípustné. Jsou-li použity objemy větší než 100 ml, může být nezbytné zvýšit dávky podávané dafniím, aby se zajistila odpovídající dostupnost potravy a splnila kritéria validity. Zkušební organismy U semistatických zkoušek se nasadí jednotlivě alespoň 10 organismů pro každou zkušební koncentraci a alespoň 10 organismů jednotlivě do kontrolních skupin. U průtokových zkoušek se ukázalo vhodným rozdělit pro každou koncentraci 40 organismů do čtyř skupin po 10 organismech (1). Může být použit menší počet zkušebních organismů, přičemž se doporučuje na jednu koncentraci rozdělit minimálně 20 organismů do dvou nebo více paralelních skupin o stejném počtu organismů (např. čtyři paralelní skupiny po pěti dafniích). Je třeba poznamenat, že u zkoušek, při nichž se organismy nasazují jako skupiny, nebude možné ze statistické analýzy vyloučit žádné potomky, pokud dojde po zahájení reprodukce k bezděčné/náhodné mortalitě rodičovských organismů, a v takovém případě se proto reprodukční schopnost vyjádří jako celkový počet živých potomků pocházejících z jednoho rodičovského organismu přítomného na začátku zkoušky. Expozice se rozvrhne pro jednotlivé zkušební nádoby a veškerá další manipulace se zkušebními nádobami se provádí náhodně. Nedodržení této zásady může vést k jednostrannému vychýlení, které může být považováno za účinek koncentrace. Je-li například s experimentálními jednotkami manipulováno v pořadí varianty nebo v pořadí koncentrací, mohl nějaký časově podmíněný efekt, např. únava obsluhy nebo jiné pochybení, vést k vyšším účinkům při vyšších koncentracích. Mohou-li být výsledky ovlivněny počátečními podmínkami zkoušky nebo okolními podmínkami, jako je např. umístění v laboratoři, mělo by být dále zváženo blokové uspořádání zkoušky. Krmení U semistatických zkoušek se dafnie krmí přednostně denně, nejméně však třikrát týdně (tj. s výměnou média). Mělo by být vzato v úvahu možné zředění zkušební koncentrace v důsledku přidání potravy, čemuž by mělo být v co největší míře zabráněno suspensí řas v náležité koncentraci. Odchylky od této praxe (např. u průtokových zkoušek) se uvedou ve zprávě. Potrava rodičovských organismů během zkoušky by měla sestávat přednostně z živých jednobuněčných řas jednoho nebo více těchto druhů: Chlorella sp., Pseudokirchneriella subcapitata (dříve Selenastrum capricornutum) a Desmodesmus subspicatus (dříve Scenedesmus subspicatus). Dodávaná potrava by měla vycházet z množství organického uhlíku (C) dodávaného na jeden rodičovský organismus. Výzkum (14) ukázal, že u druhu Daphnia magna je množství potravy obsahující od 0,1 do 0,2 mg C na dafnii za den dostatečné na to, aby bylo dosaženo požadovaného živého potomstva nezbytného pro splnění kritérií validity. Potrava může být dodávána buď v konstantních dávkách po celou dobu zkoušky, nebo podle požadavku v nižších dávkách na začátku zkoušky a poté ve zvyšujících se dávkách s ohledem na růst rodičovských organismů. V takovém případě by mělo množství potravy po celou dobu obsahovat doporučené množství 0,1 až 0,2 mg C na dafnii za den. Použije-li se k určení požadovaného množství potravy zástupný ukazatel, jako je počet jednobuněčných řas nebo absorbance ve viditelném světle (z praktických důvodů, neboť měření obsahu uhlíku je časově náročné), měla by každá laboratoř vytvořit vlastní nomogram vztahu zástupného ukazatele a obsahu uhlíku v kultuře řas (viz dodatek 3 s návodem na vytvoření nomogramu). Nomogramy by měly být ověřovány jednou ročně a častěji v případě, že se změní podmínky kultivace řas. Absorbance ve viditelném světle byla shledána lepším zástupným ukazatelem celkového obsahu uhlíku než počet buněk (15). Dafniím by měla být dodávána koncentrovaná suspense řas, aby se minimalizoval objem kultivačního média řas uváděný do zkušební nádoby. Zkoncentrování řas lze dosáhnout centrifugací a následnou resuspenzí v kultivačním médiu dafnií. Osvětlení 16 hodin světla při intenzitě nepřekračující 15 až 20 μE · m– 2 · s– 1 naměřené na hladině vody v nádobě. Měří-li se intenzita osvětlení luxmetry, ekvivalentní rozpětí 1 000 až 1 500 lx pro chladné bílé světlo odpovídá přibližně doporučené světelné intenzitě 15–20 μE·m– 2 · s– 1). Teplota Teplota zkušebního média by měla být v rozmezí 18–22 °C. V žádné zkoušce by však teplota neměla v těchto mezích kolísat o více než 2 °C (např. 18 až 20, 19 až 21 nebo 20 až 22 °C). Pro účely sledování teploty může být vhodné použít další zkušební nádobu. Provzdušňování Zkušební nádoby by neměly být během zkoušky provzdušňovány. Uspořádání zkoušky Zkouška ke stanovení rozsahu Je-li to nezbytné, provede se zkouška ke stanovení rozsahu například s pěti koncentracemi zkoušené chemické látky a s dvěma paralelními skupinami pro každou expozici a s kontrolní skupinou. Při rozhodování o rozsahu koncentrací, které ve zkoušce ke stanovení rozsahu mají být použity, mohou být užitečné i další informace o akutní toxicitě pro druh Daphnia magna a/nebo pro jiné vodní organismy, získané při zkouškách s obdobnými chemickými látkami nebo z literatury. Zkoušky pro stanovení rozsahu trvají 21 dní nebo tak dlouho, aby bylo možné spolehlivě předpovědět úrovně účinků. Na konci zkoušky se vyhodnotí reprodukce dafnií. Počet rodičovských organismů a výskyt potomstva by měl být zaznamenán. Hlavní zkouška Zpravidla se použije alespoň pět zkušebních koncentrací zahrnujících účinnou koncentraci (např. ECx a tvořících geometrickou řadu s faktorem nepřekračujícím 3,2. Měl by se použít přiměřený počet paralelních zkušebních skupin v rámci každé zkušební koncentrace (viz odstavce 24–25). Použití méně než pěti koncentrací by mělo být zdůvodněno. Chemické látky by neměly být zkoušeny při koncentracích vyšších, než je jejich rozpustnost ve zkušebním médiu. Před provedením experimentu se doporučuje posoudit statistickou sílu uspořádání zkoušek a použití vhodných statistických metod (4). Při volbě rozsahu koncentrací by měly být zohledněny tyto zásady:
Nejsou-li při nejvyšší koncentraci v rámci zkoušky ke stanovení rozsahu (např. při 10 mg/l) zjištěny žádné účinky nebo pokud je zkoušená chemická látka na základě nezaznamenané toxicity pro jiné organismy a/nebo na základě nízkého/žádného příjmu je s vysokou pravděpodobností málo toxická nebo netoxická, je možné provést zkoušku toxicity pro reprodukci jako limitní zkoušku s použitím zkušební koncentrace např. 10 mg/l a kontrolní skupiny. K exponované i ke kontrolní skupině by mělo být použito deset paralelních skupin. Pokud by bylo nutné provést limitní zkoušku v průtokovém systému, bude stačit méně paralelních skupin. Limitní zkouška umožní prokázat, že při mezní koncentraci není žádný statisticky významný účinek, ale jsou-li účinky zaznamenány, bude zpravidla nutné provést úplnou zkoušku. Kontroly Kromě zkušební série se nasadí jedna série kontrolních skupin pro kontrolu zkušebního média a podle potřeby jedna série kontrolních skupin obsahujících rozpouštědlo nebo dispergátor. Použije-li se rozpouštědlo nebo dispergátor, měla by být jejich koncentrace stejná jako koncentrace v nádobách se zkoušenou chemickou látkou. Použije se vhodný počet paralelních skupin (viz odstavce 23–24). V dobře vedených zkouškách by měl být variační koeficient středního počtu živých potomků připadajících na rodičovský organismus v kontrolní skupině (kontrolních skupinách) zpravidla ≤ 25 % a měl být uveden v návrzích zkoušek s oddělenými jedinci. Obnovování zkušebního média Četnost obnovování média bude záviset na stabilitě zkoušené chemické látky, médium se však obnovuje alespoň třikrát týdně. Není-li podle předběžných zkoušek stability (viz odstavec 7) koncentrace zkoušené látky po maximální dobu mezi obnoveními média (tj. po tři dny) stálá (tj. nepohybuje-li se v intervalu 80–120 % nominální koncentrace nebo klesá-li pod 80 % naměřené počáteční koncentrace), mělo by být zváženo častější obnovování média nebo průtoková zkouška. Při obnovování média v semistatických zkouškách se připraví druhá série zkušebních nádob a rodičovské organismy se do nich přenesou např. skleněnou pipetou o vhodném průměru. Objem média přeneseného s dafniemi by měl být co nejmenší. Pozorování Výsledky pozorování prováděných během zkoušky se zaznamenávají do přehledu údajů (viz příklady v dodatcích 4 a 5). Jsou-li požadována další měření (viz odstavec 44), může být nezbytné provádět další pozorování. Potomstvo Potomstvo pocházející z každého rodičovského organismu se od jeho prvního výskytu nejlépe denně vybírá a počítá, aby nespotřebovávalo potravu určenou pro rodičovský organismus. Pro účely této zkušební metody je třeba počítat pouze živé potomstvo, avšak přítomnost nevylíhnutých vajíček nebo mrtvého potomstva se zaznamená. Mortalita Mortalita rodičovských organismů se zaznamenává přednostně denně, nebo by měla být stanovována alespoň při počítání potomstva. Další parametry Třebaže je tato zkušební metoda navržena hlavně k posouzení účinků na reprodukční schopnost, mohou však být i jiné účinky kvantifikovány v míře dostatečné pro statistickou analýzu. Může být zaznamenána reprodukční schopnost na jeden přeživší rodičovský organismus, tj. počet žijících potomků vylíhnutých během zkoušky na jeden přeživší rodičovský organismus To je možné porovnat s hlavní proměnnou odezvy (reprodukční schopnost na jeden rodičovský organismus na začátku zkoušky, který během zkoušky bezděčně nebo náhodně neuhynul). Dojde-li k mortalitě rodičovských organismů v exponovaných paralelních skupinách, mělo by být posouzeno, zda tato mortalita odpovídá vztahu koncentrace a odezvy, či nikoli, např. zda existuje významný pokles odezvy v závislosti na koncentraci zkoušené chemické látky s pozitivní směrnicí (k tomu lze použít statistický test, například Cochranovu-Armitageovu zkoušku trendu). Jestliže mortalita neodpovídá vztahu koncentrace a odezvy, měly by být tyto paralelní skupiny s mortalitou rodičovských organismů z analýzy výsledků zkoušky vyloučeny. Odpovídá-li mortalita vztahu koncentrace a odezvy, měla by být mortalita rodičovských organismů označena za účinek zkoušené chemické látky a paralelní skupiny by neměly být z analýzy výsledků zkoušky vyloučeny. Zvláště měření růstu jsou žádoucí, neboť poskytují informace o možných subletálních účincích, které mohou být užitečné vedle samotných měření reprodukce; doporučuje se na konci zkoušky změřit délku rodičovských organismů (tj. délku těla bez zadečkového hrotu). Mezi další parametry, které lze měřit nebo vypočítávat, patří doba do vylíhnutí prvních plůdků (a následně dalších potomků), počet a velikost potomků na jeden organismus, počet nevylíhnutých plůdků, přítomnost novorozenců samčího pohlaví (OECD, 2008) nebo efipií a možná i vnitřní míra růstu populace (viz dodatek 1 s definicemi a dodatek 7 ke zjištění pohlaví novorozenců). Četnost analytických stanovení a měření Koncentrace kyslíku, teplota, tvrdost a pH se měří alespoň jednou týdně před obnovením média a po něm, v kontrolních nádobách a u nejvyšší zkušební koncentrace chemické látky. Během zkoušky se koncentrace zkoušené chemické látky stanovují v pravidelných intervalech. U semistatických zkoušek (s obnovováním média), u nichž se má koncentrace zkoušené chemické látky pohybovat v rozmezí ±20 % nominálních hodnot (tj. od 80 do 120 % – viz odstavce 6, 7 a 39), se doporučuje analyzovat alespoň nejvyšší a nejnižší zkušební koncentrace ihned po jejich přípravě a při obnovování, a to jednou během prvního týdne zkoušky (tj. analýzy se provedou na vzorku z téhož roztoku – při jeho přípravě a při obnovování). Tato stanovení se poté opakují alespoň v týdenních intervalech. U zkoušek, u nichž se nepředpokládá, že se bude koncentrace zkoušené chemické látky pohybovat v rozmezí ± 20 % nominální hodnoty, je nezbytné analyzovat všechny zkušební koncentrace, a to ihned po jejich přípravě a při obnovování. U zkoušek, u nichž není naměřená počáteční koncentrace zkoušené chemické látky v rozsahu ± 20 % nominální koncentrace, avšak u nichž lze dostatečně prokázat, že počáteční koncentrace jsou reprodukovatelné a stálé (tj. od 80 do 120 % počátečních koncentrací), by mohla být stanovení ve 2. a 3. týdnu zkoušky omezena na nejvyšší a nejnižší zkušební koncentrace. Ve všech případech je stanovení koncentrací zkoušené chemické látky před obnovením třeba provést pro každou koncentraci pouze u jedné paralelní nádoby. U průtokových zkoušek je vhodný podobný vzorkovací režim, jaký je popsán u semistatických zkoušek (v tomto případě se však neprovádí měření „starých“ roztoků). Doporučuje se však zvýšit počet odběrů v prvním týdnu (např. tři sady měření) pro ujištění, že jsou zkušební koncentrace stálé. V těchto typech zkoušky se průtok ředicí vody a zkoušené chemické látky kontroluje denně. Lze-li prokázat, že byla po celou dobu zkoušky koncentrace zkoušené látky uspokojivě udržována v rozmezí ± 20 % nominální hodnoty nebo naměřené počáteční koncentrace, mohou být výsledky založeny na nominálních nebo naměřených počátečních hodnotách. Je-li odchylka od nominální nebo naměřené počáteční koncentrace větší než ± 20 %, vyjádří se výsledky jako časově vážené střední hodnoty (pro pokyny k výpočtu viz dodatek 6). ÚDAJE A PŘEDKLÁDÁNÍ ZPRÁV Zpracování výsledků Účelem této zkoušky je stanovit účinky zkoušené chemické látky na reprodukční schopnost. Celkový počet potomků na rodičovský organismus se vypočte pro každou zkušební nádobu (tj. pro každou paralelní nádobu). Reprodukci je také možné vypočítat na základě produkce živých potomků přeživším rodičovským organismem. Ekologicky nejvýznamnější proměnnou účinku však je celkový počet živých potomků pocházejících z jednoho rodičovského organismu, který neuhyne náhodně (96) nebo bezděčně (97) během zkoušky. Jestliže rodičovský organismus během zkoušky náhodně nebo bezděčně uhyne nebo se zjistí, že jde o samečka, vyloučí se nádoba z experimentu. Analýza se poté provede se zmenšeným počtem paralelních nádob. Dojde-li k mortalitě rodičovských organismů v exponovaných paralelních skupinách, mělo by být posouzeno, zda tato mortalita odpovídá vztahu koncentrace a účinku, či nikoli, např. zda existuje významný pokles účinku v závislosti na koncentraci zkoušené chemické látky se sklonem k pozitivnímu účinku (k tomu lze použít statistický test, například Cochranovu-Armitageovu zkoušku trendu). Jestliže mortalita neodpovídá vztahu koncentrace a účinku, měly by být tyto paralelní skupiny s mortalitou rodičovských organismů z analýzy výsledků zkoušky vyloučeny. Odpovídá-li mortalita vztahu koncentrace a účinku, měla by být mortalita rodičovských organismů označena za účinek zkoušené chemické látky a paralelní skupiny by neměly být z analýzy výsledků zkoušky vyloučeny. Souhrnně vyjádřeno, jsou-li pro vyjádření účinků použity LOEC a NOEC nebo ECx, doporučuje se vypočítat účinek na reprodukci s použitím obou proměnných odezvy uvedených výše, tj.
a poté jako konečný výsledek použít nejnižší hodnotu NOEC a LOEC nebo ECx vypočítanou pomocí jednoho z těchto dvou proměnných účinků. Před provedením statistické analýzy, např. postupů ANOVA, porovnání zkušební skupiny s kontrolními skupinami pomocí Studentova t-testu, Dunnettova testu, Williamsova testu nebo Jonckheerova-Terpstrova testu sestupného trendu se doporučuje zvážit transformaci údajů, je-li to nezbytné pro splnění požadavků daného statistického testu. Jako neparametrické alternativy lze zvážit Dunnův nebo Mannův-Whitneyův test. Pro střední hodnoty jednotlivých koncentrací se vypočtou 95 % intervaly spolehlivosti. Počet přeživších rodičovských organismů v neexponovaných kontrolních skupinách je kritériem validity zkoušky a je třeba jej zdokumentovat. V závěrečné zprávě by měly být zdokumentovány také všechny ostatní škodlivé účinky, např. nenormální chování a významné toxikologické nálezy. ECx Hodnoty ECx, včetně s nimi souvisejících nižších a vyšších hladin intervalu spolehlivosti, se vypočítají vhodnou statistickou metodou (např. pomocí logistické neboli Weibullovy funkce, zkrácenou Spearmanovou-Karberovou metodou nebo prostou interpolací). Pro výpočet EC10, EC50 nebo kterékoli jiné hodnoty EC se úplný soubor údajů podrobí regresní analýze. NOEC/LOEC Jestliže se při statistické analýze očekává stanovení NOEC/LOEC, je třeba použít vhodné statistické metody podle dokumentu OECD č. 54 – Současné přístupy ve statistické analýze údajů o ekotoxicitě: Pokyny pro aplikaci (4). Nepříznivé účinky zkoušené chemické látky v porovnání s kontrolou se zpravidla zkoumají jednostranným testováním hypotéz při p ≤ 0,05. Normalitu rozdělení a homogenitu rozptylu lze prověřit vhodným statistickým testem, např. Shapirovým-Wilkovým testem, případně Leveneovým testem (p ≤ 0,05). Je možné provést jednocestnou analýzu ANOVA a následné mnohonásobné srovnávací testy. Pro výpočet, zda existují významné rozdíly (p ≤ 0,05) mezi kontrolními skupinami a různými koncentracemi zkoušené chemické látky, je možné použít mnohonásobné srovnávání (např. Dunnettův test) nebo testy sestupného trendu (např. Williamsův test nebo Jonckheerův-Terpstrův test sestupného trendu) (výběr doporučeného testu se provede podle dokumentu OECD č. 54 (4)). Nebo lze k určení NOEC a LOEC použít neparametrické metody (např. Bonferroniho U-test podle Holma nebo Jonckheerovu-Terpstrovu zkoušku trendu). Limitní zkouška Byla-li provedena limitní zkouška (porovnání kontrolní skupiny a pouze jedné exponované skupiny) a jsou-li splněny podmínky pro parametrické zkušební postupy (normalita rozdělení, homogenita rozptylu), je možné hodnotit spojité proměnné pomocí Studentova testu (t-testu). Pokud tyto požadavky nejsou splněny, lze použít t-test nestejných rozptylů (např. Welchův test) nebo neparametrický test, např. Mannův-Whitneyův U-test. Pro určení významných rozdílů mezi kontrolními skupinami (kontrolní a kontrolní s rozpouštědlem nebo dispergátorem) je možné každou kontrolní skupinu podrobit opakovaným zkouškám, jak bylo popsáno u limitní zkoušky. Pokud tyto zkoušky neodhalí významné rozdíly, údaje ze všech opakování s kontrolou a kontrolou s rozpouštědlem je možné sloučit. V opačném případě by všechny expozice měly být porovnány s kontrolou s rozpouštědlem. Závěrečná zpráva Závěrečná zpráva obsahuje následující informace:
LITERATURA
Dodatek 1 DEFINICE Pro účely této zkušební metody se použijí tyto definice: Náhodná mortalita : mortalita, která nesouvisí s chemickou látkou a je vyvolána nahodilými okolnostmi (tj. známou příčinou). Chemická látka : chemická substance nebo směs. ECx : koncentrace zkoušené chemické látky rozpuštěné ve vodě, která pro danou expoziční dobu vede k x % snížení reprodukce dafnií. Bezděčná mortalita : mortalita, která nesouvisí s chemickou látkou a nemá známou příčinu. Vnitřní míra růstu populace : míra růstu populace, v níž je zahrnuta reprodukční schopnost a mortalita související s věkem (1) (2) (3). V rovnovážném stavu bude nulový. Pro rostoucí populaci bude kladná a pro snižující se populaci bude záporná. Je zřejmé, že při záporném růstu nelze druh zachovat a nakonec dojde k jeho vyhynutí. Mez detekovatelnosti : nejnižší koncentrace, v níž lze látku detekovat, nikoli však stanovit. Mez stanovitelnosti : nejnižší koncentrace, v níž lze látku kvantitativně naměřit. Nejnižší koncentrace s pozorovanými účinky (lowest observed effect concentration, LOEC) : nejnižší zkušební koncentrace, při níž je pro danou expoziční dobu pozorován statisticky významný účinek chemické látky na reprodukci a na mortalitu rodičovských organismů (při p < 0,05) ve srovnání s kontrolou. Všechny zkušební koncentrace vyšší než LOEC by měly mít škodlivé účinky stejné nebo větší, než jsou účinky pozorované při LOEC. Nelze-li tyto dvě podmínky splnit, mělo by být podrobně vysvětleno, jak byla LOEC (a tedy i NOEC) zvolena. Mortalita : organismus se zaznamená jako mrtvý, je-li nepohyblivý, tj. není-li schopen plavat nebo není-li u končetin nebo zadečku pozorován pohyb do 15 sekund po lehkém zatřepání zkušební nádobou. (Použije-li se jiná definice, měla by být uvedena společně s odkazem na literaturu.) Koncentrace bez pozorovaných účinků (no observed effect concentration, NOEC) : zkušební koncentrace bezprostředně nižší než LOEC, která při srovnání s kontrolní skupinou nemá pro danou expoziční dobu statisticky významný účinek (při p < 0,05). Potomstvo : mladé dafnie pocházející z rodičovských organismů v průběhu zkoušky. Rodičovské organismy : dafnie samičího pohlaví, které jsou přítomné na začátku zkoušky, a jejich reprodukční schopnost je předmětem studie Reprodukční schopnost : počet živých potomků pocházejících z rodičovských organismů za dobu zkoušky. Zkoušená chemická látka : jakákoli chemická substance nebo směs zkoušená touto zkušební metodou. LITERATURA
Dodatek 2 PŘÍPRAVA PŘESNĚ DEFINOVANÝCH ELENDTOVÝCH MÉDIÍ M7 A M4 Aklimatizace na Elendtova média M7 a M4 Některé laboratoře narazily na obtíže při přímém přenosu dafnií do médií M4 (1) a M7. Určitého úspěchu však bylo dosaženo postupnou aklimatizací, tj. s přenesením z vlastního média do 30 % Elendtova média, poté do 60 % Elendtova média a nakonec do 100 % Elendtova média. Délky dob aklimatizace mohou být až jeden měsíc. Příprava Stopové prvky Ve vodě vhodné čistoty, např. v deionizované nebo destilované vodě nebo ve vodě přečištěné reverzní osmózou, se nejprve připraví samostatné zásobní roztoky (I) jednotlivých stopových prvků. Z těchto oddělených zásobních roztoků (I) se připraví jeden zásobní roztok (II) obsahující všechny stopové prvky (směsný roztok), tj.:
Média M4 a M7 Média M4 a M7 se připraví ze zásobního roztoku II, makroživin a vitaminů tímto způsobem:
Kombinovaný vitaminový zásobní roztok se uskladňuje zmrazený v malých stejnoměrných dílech. Vitaminy se do média přidávají krátce před použitím.
Dodatek 3 ANALÝZA CELKOVÉHO ORGANICKÉHO UHLÍKU (TOC) A VYTVOŘENÍ NOMOGRAMU PRO OBSAH TOC V KRMIVU NA BÁZI ŘAS Je známo, že obsah uhlíku v krmivu na bázi řas se obvykle neměří přímo, ale stanoví se z korelací (tj. z nomogramů) se zástupnými ukazateli, jako jsou počet buněk řas nebo absorbance ve viditelném světle. TOC by měl být stanoven spíše vysokoteplotní oxidací než pomocí UV nebo persíranovou metodou. (Návod viz: The Instrumental Determination of Total Organic Carbon, Total Oxygen Demand and Related Determinands 1979, HMSO 1980; 49 High Holborn, London WC1V 6HB.) Pro účely vytvoření nomogramu se řasy oddělí od růstového média odstředěním a poté se resuspendují v destilované vodě. Zástupný ukazatel a koncentrace TOC se měří v každém vzorku třikrát. Provede se analýza slepého pokusu s destilovanou vodou a jeho koncentrace TOC se odečte od koncentrace TOC ve vzorku s řasami. Nomogram by měl být lineární v požadovaném rozsahu koncentrací uhlíku. Příklady jsou uvedeny níže.

Chlorella vulgaris var. viridis (CCAP 211/12). Regresní křivka vztahu koncentrace v mg suché hmotnosti na litr a množství uhlíku v mg C na litr. Údaje z koncentrovaných suspenzí buněk semikontinuálně dávkově kultivovaných buněk, resuspendovaných v destilované vodě. osa x: mg C na litr koncentrovaného krmiva na bázi řas osa y: mg suché hmotnosti koncentrovaného krmiva na bázi řas na litr Korelační koeficient: -0,980 
Chlorella vulgaris var. viridis (CCAP 211/12). Regresní křivka vztahu počtu buněk a množství uhlíku v mg C na litr. Údaje z koncentrovaných suspenzí buněk semikontinuálně dávkově kultivovaných buněk, resuspendovaných v destilované vodě. osa x: mg C na litr koncentrovaného krmiva na bázi řas osa y: počet buněk koncentrovaného krmiva na bázi řas na litr (x 108) Korelační koeficient: -0,926 
Chlorella vulgaris var. viridis (CCAP 211/12). Regresní křivka vztahu absorbance a množství uhlíku v mg C na litr (optická dráha 1 cm). Údaje z koncentrovaných suspenzí buněk semikontinuálně dávkově kultivovaných buněk, resuspendovaných v destilované vodě. osa x: mg C na litr koncentrovaného krmiva na bázi řas osa y: absorbance koncentrovaného krmiva na bázi řas zředěného v poměru 1/10 při 440 nm Korelační koeficient: – 0,998 Dodatek 4 PŘÍKLAD FORMULÁŘE PŘEHLEDU ÚDAJŮ PRO ZAZNAMENÁNÍ OBNOVY MÉDIA, SLEDOVANÝCH FYZIKÁLNĚ-CHEMICKÝCH ÚDAJŮ, KRMENÍ, REPRODUKČNÍ SCHOPNOSTI DAFNIÍ A MORTALITY RODIČOVSKÝCH ORGANISMŮ
Dodatek 5 PŘÍKLAD FORMULÁŘE PŘEHLEDU ÚDAJŮ PRO ZAZNAMENÁNÍ VÝSLEDKŮ CHEMICKÉ ANALÝZY a) Naměřené koncentrace
b) Naměřené koncentrace vyjádřené v procentech nominální koncentrace
Dodatek 6 VÝPOČET ČASOVĚ VÁŽENÉ STŘEDNÍ HODNOTY Časově vážená střední hodnota Vzhledem k tomu, že mezi výměnami média může koncentrace zkoušené chemické látky klesat, je nezbytné zvážit, jaká koncentrace bude považována za reprezentativní pro rozsah koncentrací, jimž byly rodičovské organismy dafnií vystaveny. Výběr by měl být založen na biologických i statistických úvahách. Vychází-li se například z toho, že reprodukce je ovlivňována hlavně maximální koncentrací, jíž byl organismus vystaven, měla by být použita maximální koncentrace. Považuje-li se však za významnější akumulované nebo dlouhodobé působení toxické chemické látky, má větší opodstatnění průměrná koncentrace. V takovém případě je vhodným průměrem časově vážená střední koncentrace, neboť zohledňuje časové změny okamžité koncentrace. Obrázek 1 Příklad časově vážené střední hodnoty 
Na obrázku 1 je znázorněn příklad (zjednodušené) zkoušky trvající sedm dní s obnovou média v den 0, 2 a 4.
Časově vážená střední hodnota je vypočtena tak, aby se plocha pod čarou pro časově váženou střední hodnotu rovnala ploše pod čarou průběhu koncentrace. Výpočet pro výše uvedený příklad je ilustrován v tabulce 1. Tabulka 1 Výpočet časově vážené střední hodnoty
Plocha je plocha pod exponenciální křivkou pro každé období mezi obnoveními média. Vypočte se z rovnice:
Časově vážená střední hodnota (ČV střední hodnota) se vypočte jako podíl celkové plochy a celkového počtu dnů. Pro zkoušku toxicity pro reprodukci na dafniích se tabulka pochopitelně rozšíří tak, aby pokrývala 21 dnů. Je zřejmé, že provádí-li se měření pouze na začátku a na konci období mezi obnoveními média, není možné potvrdit, že je proces rozkladu skutečně exponenciální Jiná křivka by vedla k jinému způsobu výpočtu „plochy“. Exponenciální rozklad však není nepřijatelný a jemu odpovídající křivka je pravděpodobně optimální, chybí-li jiné informace. Je však nezbytné postupovat opatrně, není-li v médiu na konci období mezi jeho obnoveními zjištěno žádné množství zkoušené chemické látky. Není-li možné odhadnout, jak rychle chemická látka z roztoku zmizela, není možné získat realistickou plochu pod křivkou, a není tedy možné získat rozumnou časově váženou střední hodnotu. Dodatek 7 POKYNY KE ZJIŠTĚNÍ POHLAVÍ NOVOROZENCŮ K produkci novorozenců samčího pohlaví může dojít za měnících se podmínek životního prostředí, např. zkracující se fotoperiody, teploty, klesající koncentrace potravy a rostoucí hustoty populace (Hobaek and Larson, 1990; Kleiven et al., 1992). Produkce samečků je také známou reakcí na činitele regulující růst určitých druhů hmyzu (Oda et al., 2005). Za podmínek, kdy chemické stresory vyvolávají pokles reprodukce potomstva u partenogenetických samiček, lze očekávat zvýšený počet samečků (IECD, 2008). Na základě dostupných informací není možné předpovědět, jaký poměr pohlaví nebo který koncový ukazatel reprodukční schopnosti bude citlivější; existují však známky toho (viz validační zpráva, část 1), že zmíněné zvýšení počtu samečků by mohlo být méně citlivé než snížení potomstva. Poněvadž hlavním účelem této zkušební metody je posoudit počet vylíhnutých potomků, je pozorování výskytu samečků jen volitelné. Je-li tento volitelný ukazatel ve studii hodnocen, mělo by být použito další kritérium validity zkoušky, a to, že počet samečků v kontrolních skupinách není větší než 5 %. Nejpraktičtějším a nejjednodušším způsobem, jak rozlišit pohlaví u dafnií, je použít jejich fenotypové charakteristiky, neboť samečkové a samičky jsou geneticky identičtí a jejich pohlaví je určováno životním prostředím. Samečci a samičky se liší délkou a morfologií prvních tykadel, které jsou u samečků delší než u samiček (obrázek 1). Tento rozdíl je patrný ihned po narození, ačkoli s růstem se vyvíjejí i jiné druhotné pohlavní znaky (viz např. obrázek 2, Olmstead and LeBlanc, 2000). Za účelem pozorování morfologického pohlaví by novorozenci pocházející od každého zkušebního organismu měli být pipetou přeneseni do Petriho misky se zkušebním médiem. Médium je udržováno na minimu s cílem omezit pohyb organismů. Pozorování prvních tykadel je možné provádět pod stereomikroskopem (× 10–60). Obrázek 1 Sameček (vlevo) a samička (vpravo) D. magna ve stáří 24 hodin. Samečky lze od samiček odlišit podle délky a morfologie prvních tykadel, jak je vyznačeno v kroužcích (Tatarazako et al., 2004). 
ODKAZY Hobaek A and Larson P. 1990. Sex determination in Daphnia magna. Ecology 71: 2255-2268. Kleiven O.T., Larsson P., Hobaek A. 1992. Sexual reproduction in Daphnia magna requires three stimuli. Oikos 65, 197-206. Oda S., Tatarazako N, Watanabe H., Morita M., and Iguchi T. 2005. Production of male neonates in Daphnia magna (Cladocera, Crustacea) exposed to juvenile hormones and their analogs. Chemosphere 61:1168-1174. OECD, 2008. Validation report for an enhancement of OECD TG 211 Daphnia magna reproduction test. OECD Series on Testing and Assessment, Number 88. Organisation for Economic Co-operation and Development, Paris. Olmstead, A.W., LeBlanc, G.A., 2000. Effects of endocrine-active chemicals on the development characteristics of Daphnia magna. Environmental Toxicology and Chemistry 19:2107-2113. Tatarazako, N., Oda, S., Abe, R., Morita M. and Iguchi T., 2004. Development of a screening method for endocrine disruptors in crustaceans using Daphnia magna (Cladocera, Crustacea). Environmental Science 17, 439-449. |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||

|
(18) |
V části C, kapitole C.29 se odstavec 66 nahrazuje tímto:
|
|
19) |
V části C se doplňují nové kapitoly, které znějí: „C.47 Zkouška toxicity na rybách v raném stadiu života ÚVOD
PODSTATA ZKOUŠKY
INFORMACE O ZKOUŠENÉ CHEMICKÉ LÁTCE
PLATNOST ZKOUŠKY
POPIS METODY Zkušební komory
Výběr druhu
Chov matečných ryb
Chov oplodněných jiker, embryí a plůdků
Voda
Zkušební roztoky
POSTUP Podmínky expozice Doba trvání zkoušky
Nasazování
Světlo a teplota
Krmení
Zkušební koncentrace
Kontroly
Četnost analytických stanovení a měření
Pozorování 26. Stadia vývoje embrya : embryonální stadium na začátku expozice zkoušené chemické látce by mělo být identifikováno co nejpřesněji. Lze to provést za použití reprezentativního vzorku vhodně uchovávaných a očištěných jiker. 27. Líhnutí a přežívání : Líhnutí a přežívání se pozoruje alespoň jednou denně a zaznamenávají se počty. Zjistí-li se na jikrách mykóza v raném stadiu vývoje embryí (např. v den 1 nebo 2 zkoušky), tyto jikry se spočítají a odstraní. Uhynulá embrya, plůdky a nedospělé ryby se odstraní ihned po nalezení, neboť se mohou rychle rozkládat a činností ostatních ryb mohou být porušeny. Při odstraňování uhynulých jedinců se postupuje s mimořádnou opatrností, aby se fyzicky nepoškodily sousední jikry/plůdky. Příznaky úmrtí se liší podle druhu a životního stadia. Například:
28. Neobvyklý vzhled : počet plůdků nebo nedospělých ryb vykazujících neobvyklý tělesný vzhled se zaznamenává v přiměřených intervalech závisejících na trvání zkoušky a na povaze popisované abnormality. Je třeba si uvědomit, že neobvyklé plůdky a nedospělé ryby se vyskytují přirozeně a u některých druhů jich může být v kontrole (kontrolách) řádově několik procent. Jsou-li deformace a související neobvyklé chování považovány za tak závažné, že daný organismus je vystaven značnému utrpení a nevratným změnám, lze je ze zkoušky vyřadit. Taková zvířata je třeba usmrtit a pro účely následné analýzy údajů by s nimi mělo být zacházeno jako s uhynulými. U většiny druhů doporučených pro tuto zkušební metodu byl zdokumentován normální vývoj embryí (9) (10) (11) (12). 29. Neobvyklé chování : abnormality, např. hyperventilace, nekoordinované plavání, netypická nečinnost a netypické chování při krmení, se zaznamenávají v přiměřených intervalech závisejících na trvání zkoušky (např. jednou denně u teplovodních druhů). Tyto účinky, přestože je obtížné je kvantifikovat, mohou – jsou-li pozorovány – pomoci při interpretaci údajů o úhynu. 30. Hmotnost : na konci zkoušky se zváží všechny přežívající ryby alespoň v jednom opakování (zaznamená se počet ryb v opakování a střední hmotnost jedné ryby): upřednostňuje se živá hmotnost (po osušení do sucha), avšak uvedeny mohou být také údaje o suché hmotnosti (13). 31. Délka : na konci zkoušky se změří individuální délky. Doporučuje se použít celkovou délku, avšak došlo-li k uhnití ocasní ploutve nebo k erozi ploutví, lze použít standardní délku. Pro všechny ryby v dané zkoušce by měla být použita stejná metoda. Individuální délku lze měřit např. posuvným měřítkem, digitálním fotoaparátem nebo kalibrovaným okulárním mikrometrem. Typické minimální délky jsou uvedeny v dodatku 2. ÚDAJE A PŘEDKLÁDÁNÍ ZPRÁV Zpracování výsledků
Závěrečná zpráva
LITERATURA
Dodatek 1 DEFINICE
Dodatek 2 ZKUŠEBNÍ PODMÍNKY, DÉLKA ZKOUŠKY A KRITÉRIA PŘEŽITÍ PRO DOPORUČENÉ DRUHY
Dodatek 3 POKYNY PRO KRMENÍ A CHOV MATEČNÝCH A ZKUŠEBNÍCH RYB DOPORUČENÝCH DRUHŮ
Dodatek 4 NĚKTERÉ CHEMICKÉ CHARAKTERISTIKY DOPORUČENÉ ŘEDICÍ VODY
Dodatek 5 STATISTICKÉ POSTUPY KE STANOVENÍ NOEC Obecné informace Jednotkou analýzy je nádrž (opakování). Při průběžných měřeních, např. velikosti, je tak třeba vypočítat střední nebo mediánové hodnoty opakování a tyto hodnoty opakování jsou údaje pro analýzu. Měla by být doložena průkaznost použité zkoušky, přednostně na základě odpovídající databáze dosavadních údajů každé laboratoře. Pro každý ukazatel by měla být uvedena velikost účinku, který lze pomocí statistického testu, který má být použit, určit s průkazností 75–80 %. Databáze dostupné v době vývoje této zkušební metody stanoví, jaké průkaznosti zkoušky je možné dosáhnout při doporučených statistických postupech. Každá jednotlivá laboratoř by měla prokázat svou schopnost splnit tento požadavek na průkaznost buď provedením vlastní statistické analýzy, nebo prokázáním toho, že variační koeficient (CV) pro každou odezvu nepřekračuje 90. percentil variačních koeficientů použitých při vývoji pokynu pro zkoušení. Tyto hodnoty CV jsou uvedeny v tabulce 1. Jsou-li k dispozici pouze střední nebo mediánové hodnoty opakování, není třeba CV v rámci opakování brát v úvahu. Tabulka 1 90. percentil CV pro vybrané sladkovodní druhy
Téměř při všech statistických testech, které se používají k hodnocení laboratorních toxikologických studií, jsou předmětem zájmu srovnání mezi zkušebními skupinami a kontrolami. Z tohoto důvodu není vhodné vyžadovat provedení ANOVA F-testu významnosti před použitím Dunnettova nebo Williamsova testu nebo provedení Kruskalova-Wallisova testu významnosti před použitím Jonckheerova-Terpstrova, Mannova-Whitneyova nebo Dunnova testu (Hochberg a Tamhane 1987, Hsu 1996, Dunnett 1955, 1964, Williams 1971, 1972, 1975, 1977, Robertson et al., 1988, Jonckheere 1954, Dunn 1964). Dunnettův test zahrnuje korekci multiplikace a jeho míra falešné pozitivity a míra falešné negativity je nepříznivě ovlivňována použitím F-testu pro snížení rozptylu. Podobně Williamsův a Jonckheerův-Terpstrův test sestupného trendu používající při každém kroku hladinu významnosti 0,05 zajišťuje celkovou 5 % míru falešné pozitivity a tato míra a síla testu jsou nepříznivě ovlivňovány použitím F-testu nebo Kruskalova-Wallisova testu pro snížení rozptylu. U Mannova-Whitneyova a Dunnova testu musí být provedena korekce multiplikace a doporučuje se použít Bonferroniho-Holmovu korekci. Důkladný rozbor většiny doporučení ohledně testování hypotéz a ověřování předpokladů, z nichž tyto zkoušky vycházejí, je podán v dokumentu OECD (2006), který také obsahuje obsáhlý seznam literatury. Nakládání s kontrolami při použití rozpouštědla Je-li použito rozpouštědlo, měla by být zařazena jak kontrola s ředicí vodou, tak kontrola s rozpouštědlem. Tyto dvě kontroly by měly být porovnávány pro každou odezvu, a nejsou-li mezi kontrolami shledány významné rozdíly, pro účely statistické analýzy by měly být jejich údaje sloučeny. V opačném případě by měla být pro stanovení NOEC nebo odhad ECx použita kontrola s rozpouštědlem, a kontrola s vodou se nepoužije. Viz omezení v kritériích platnosti (odstavec 7). Pro stanovení délky, hmotnosti, podílu uhynulých jiker při líhnutí nebo uhynulých plůdků nebo abnormálních plůdků a pro určení prvního nebo posledního dne líhnutí nebo vyplavání by měl být použit T-test nebo Mannův-Whitneyův test, kterým se porovná kontrola s ředicí vodou a kontrola s rozpouštědlem při hladině významnosti 0,05, přičemž všechny zkušební skupiny se neberou v úvahu. Výsledky těchto testů je třeba uvést ve zprávě. Měření velikosti (délka a hmotnost) Individuální hodnoty délky a hmotnosti ryb lze popsat pomocí normálního rozdělení nebo logaritmicko-normálního rozdělení. V každém případě střední hodnoty opakování vykazují sklon k normální distribuci na základě centrální limitní věty, jak potvrzují údaje z více než 100 studií toxicity v raném stadiu života provedených na třech druzích sladkovodních ryb. Alternativně, pokud údaje nebo databáze dosavadních údajů nasvědčují logaritmicko-normálnímu rozdělení hodnot individuální velikostí ryb, lze vypočítat logaritmus střední hodnoty z individuálních hodnot ryb v opakování, a údaje pro analýzu pak budou hodnotami inverzní funkce k logaritmům středních hodnot daného opakování. Údaje by měly být hodnoceny z hlediska jejich souladu s normálním rozdělením a homogenitou rozptylu. Pro tento účel by měly být použity reziduální hodnoty získané z modelu ANOVA s koncentrací jako jedinou vysvětlující klasifikační proměnnou. Lze je vizuálně stanovit z bodových grafů a histogramů nebo z lodyhových grafů. Alternativně je možné použít formální test, např. Shapirův-Wilkův nebo Andersonův-Darlingův test. Soulad s homogenitou rozptylu lze posoudit na základě vizuálního zkoumání stejného bodového grafu nebo formálně na základě Levenova testu. Pouze parametrické testy (např. Williamsův, Dunnettův) je nutné hodnotit z hlediska normality nebo homogenity rozptylu. Pozornost by měla být věnována možným odlehlým hodnotám a jejich vlivu na analýzu. Použít lze Tukeyův test odlehlých hodnot a vizuální kontrolu stejných grafů reziduálních hodnot, které jsou popsány výše. Je třeba připomenout, že pozorování se týkají celých opakování, takže vypuštění odlehlé hodnoty z analýzy by se mělo provést pouze po pečlivé úvaze. Statistickými testy, které využívají charakteristik experimentálního uspořádání a biologického očekávání, jsou testy sestupného trendu, např. Williamsův a Jonckheerův-Terpstrův test. Tyto testy předpokládají monotónní vztah koncentrace-odezva a údaje by měly být posuzovány z hlediska souladu s těmito předpoklady. Toto posouzení lze provést vizuálně z bodového grafu středních hodnot opakování proti zkušební koncentraci. Bude užitečné na tento bodový graf přenést po částech lineární graf spojující střední hodnoty koncentrací vážené velikostí vzorku opakování. Velká odchylka tohoto po částech lineárního grafu od monotónnosti by ukazovala na to, že je možná nutné použít jiný než trendový test. Alternativně lze použít formální testy. Jednoduchý formální test spočívá v porovnání lineárních a kvadratických kontrastů středních hodnot koncentrací. Je-li kvadratický kontrast signifikantní a lineární kontrast není signifikantní, ukazuje to na možný problém s monotónností, která by měla být podrobena dalšímu hodnocení na základě grafů. Tam, kde může být problematická normalita nebo homogenita rozptylu, lze tyto kontrasty vytvořit z transformovaných údajů seřazených do pořadí. Lze použít alternativní postupy, např. Bartholomewův test monotónnosti, ale ty zvyšují složitost. Obrázek 2 Vývojový diagram měření velikosti (délky a hmotnosti) pro účely stanovení NOEC 
Nejsou-li údaje v souladu s požadavky pro tyto testy, hodnota NOEC se stanoví sestupnou aplikací Williamsova nebo Jonckheerova-Terpstrova testu. Podrobnosti o těchto postupech jsou uvedeny v dokumentu OECD (2006). V případě údajů, které nejsou v souladu s požadavky pro test sestupného trendu, lze použít Dunnettův test nebo Tamhaneho-Dunnettův test, přičemž v obou těchto testech je začleněna korekce multiplikace. Tyto testy předpokládají normalitu a, v případě Dunnettova testu, homogenitu rozptylu. Nejsou-li výše uvedené podmínky splněny, je možné použít Dunnův neparametrický test. Podrobnosti o všech těchto testech obsahuje dokument OECD (2006). Přehled o tom, jak nalézt test, který potřebujete, podává obrázek 2. Přežití jiker při líhnutí a plůdků Údaje představují podíly jiker, které se vylíhnou, nebo plůdků, které přežijí v jednotlivých opakováních. Tyto podíly by měly být hodnoceny z hlediska extra-binomické variability, která je při takových měřeních obvyklá, ale není univerzální. Schéma na obrázku 3 obsahuje pokyny pro výběr vhodného testu; podrobný popis je uveden v textu. Obvykle se používají dva testy. Je to Taronův C(α) test (Tarone, 1979) a chí-kvadrát testy, přičemž každý z nich se použije na každou zkušební koncentraci zvlášť. Vyskytne-li se alespoň v jedné zkušební koncentraci extra-binomická variabilita, měly by být použity metody, které to zohledňují. Vzorec 1 Taronův C(α) test (Tarone 1979)
kde je střední hodnota podílu dané koncentrace, m je počet nádrží (opakování), nj je počet subjektů v opakování j, a xj je počet subjektů s odezvou v tomto opakování, např. nevylíhlých nebo mrtvých. Tento test se použije na každou koncentraci zvlášť. Lze jej považovat za upravený chí-kvadrát test, avšak omezené simulace síly provedené Taronem ukázaly, že má větší sílu než chí-kvadrát test. Obrázek 3 Schéma hodnocení úhynu jiker při líhnutí a úhynu plůdků při stanovení NOEC 
Neexistuje-li významný důkaz o extra-binomické variabilitě, lze použít sestupný Cochranův-Armitageův test. Tento test nepřihlíží k opakováním, takže pokud takový důkaz existuje, Raova-Scottova korekce ke Cochranovu-Armitageovu testu (RSCA) bere v úvahu opakování, velikost opakování a extra-binomickou variabilitu a doporučuje se ji použít. Mezi alternativní testy patří sestupný Williamsův a Jonckheerův-Terpstrův test a Dunnettův test, jak jsou popsány v oddíle týkajícím se měření velikosti. Tyto testy jsou platné bez ohledu na to, zda existuje extra-binomická variabilita, avšak mají poněkud menší sílu (Agresti 2002, Morgan 1992, Rao a Scott 1992, 1999, Fung et al. 1994, 1996). První nebo poslední den líhnutí nebo vyplavání Odezva je vyjádřena celým číslem, udává den zkoušky, ve kterém je zpozorováno uvedené zjištění v dané nádrži (opakování). Rozpětí hodnot je zpravidla velmi omezené a často se vyskytuje vysoký podíl propojených hodnot, např. tentýž první den líhnutí je zpozorován ve všech kontrolních opakováních a možná při jedné nebo dvou nejnižších zkušebních koncentracích. Parametrické testy, např. Williamsův a Dunnettův test, nejsou pro takové údaje vhodné. Pokud neexistují důkazy o výrazné nemotónnosti, je pro detekci účinků zkoušené chemické látky velmi silný sestupný Jonckheerův-Terpstrův test. Jinak lze použít Dunnův test. Abnormality plůdků Odezvou je počet plůdků, u nichž je shledáno, že jsou nějakým způsobem abnormální. Tato odezva se většinou vyskytuje málo a je spojena se stejnými problémy, jako při stanovení prvního dne líhnutí, a rovněž tak je někdy za odezvu na koncentraci pokládána chybně. Pokud údaje alespoň přibližně sledují monotónní křivku odezvy na koncentraci, je pro detekci účinků silný sestupný Jonckheerův-Terpstrův test. Jinak lze použít Dunnův test. ODKAZY Agresti, A. (2002); Categorical Data Analysis, second edition, Wiley, Hoboken. Dunnett C. W. (1955); A multiple comparison procedure for comparing several treatments with a control, J. American Statistical Association 50, 1096-1121. Dunn O. J. (1964 ); Multiple Comparisons Using Rank Sums, Technometrics 6, 241-252. Dunnett C. W. (1964); New tables for multiple comparisons with a control, Biometrics 20, 482-491. Fung, K.Y., D. Krewski, J.N.K. Rao, A.J. Scott (1994); Tests for Trend in Developmental Toxicity Experiments with Correlated Binary Data, Risk Analysis 14, 639-648. Fung, K.Y, D. Krewski, R.T. Smythe (1996); A comparison of tests for trend with historical controls in carcinogen bioassay, Canadian Journal of Statistics 24, 431-454. Hochberg, Y. a A. C. Tamhane (1987); Multiple Comparison Procedures, Wiley, New York. Hsu, J.C. (1996); Multiple Comparisons: Theory and Methods; Chapman and Hall/CRC Press, Boca Raton. Jonckheere A. R. (1954); A distribution-free k-sample test against ordered alternatives, Biometrika 41, 133. Morgan, B.J.T. (1992); Analysis of Quantal Response Data, Chapman and Hall, London. OECD (2006). Current approaches in the statistical analysis of ecotoxicity data: A guidance to application. Series on Testing and Assessment, No. 54. Organisation for Economic Co-operation and Development, OECD, Paris.. Rao J.N.K. a Scott A.J. (1992) – A simple method for the analysis of clustered binary data, Biometrics 48, 577-585. Rao J.N.K. a Scott A.J. (1999) – A simple method for analyzing overdispersion in clustered Poisson data, Statistics in Medicine 18, 1373-1385. Robertson, T., Wright F.T. a Dykstra R.L. (1988); Order restricted statistical inference, Wiley. Tarone, R.E. (1979); Testing the goodness of fit of the Binomial distribution, Biometrika 66, 585-590. Williams D.A. (1971); A test for differences between treatment means when several dose levels are compared with a zero dose control, Biometrics 27, 103-117. Williams D.A. (1972); The comparison of several dose levels with a zero dose control, Biometrics 28, 519-531. Williams D. A. (1975); The Analysis of Binary Responses from Toxicological Experiments Involving Reproduction and Teratotlogy, Biometrics 31, 949-952. Williams D.A. (1977); Some inference procedures for monotonically ordered normal means, Biometrika 64, 9-14. Dodatek 6 STATISTICKÉ POSTUPY PRO REGRESNÍ ODHADY Obecné informace K proložení modelu se používají pozorování střední hodnoty opakování (délka a šířka) nebo podíly v opakováních (úhyn jiker při líhnutí a úhyn plůdků) (OECD 2006). Obecně se doporučuje použít váženou regresi, přičemž se váží velikostí vzorků opakování. Možné jsou i jiné způsoby vážení, například vážení předpovězenou střední hodnotou odezvy nebo kombinací této hodnoty a velikosti vzorku opakování. Vážení převrácenou hodnotou variability vzorků v rámci koncentrací se nedoporučuje (Bunke et al., 1999, Seber a Wild, 2003, Motulsky a Christopoulos 2004, Huet et al. 2003). Při jakékoli transformaci odezev před analýzou by měla být zachována nezávislost pozorování a hodnota ECx a její intervaly spolehlivosti by měly být vyjádřeny v původních jednotkách měření, a nikoli v transformovaných jednotkách. Například 20 % změna logaritmu délky se nerovná 20 % změně délky (Lyles et al. 2008, Draper a Smith 1999). Přehled kroků pro odhad ECx podává schéma na obrázku 4. Podrobnosti jsou popsány v textu pod ním. Obrázek 4 Schéma odhadování střední hodnoty délky, hmotnosti nebo podílu uhynulých jiker při líhnutí nebo plůdků pro účely stanovení ECx, podrobněji viz text 
Úvahy týkající se úhynu jiker při líhnutí a úhynu plůdků Pro posouzení úhynu vylíhnutých jiker a plůdků je obecně nejlépe proložit klesající model, pokud se neproloží probitový model, jak je popsáno níže. To znamená, že by se měl modelovat podíl nevylíhnutých jiker nebo uhynulých plůdků. Důvodem pro tento postup je, že ECx se vztahuje ke koncentraci, při které dojde ke změně rovnající se x % střední hodnoty odezvy kontroly. Je-li v kontrole 5 % nevylíhnutých jiker a modeluje se neúspěšné vylíhnutí, pak hodnota EC20 představuje koncentraci, při které se změna rovná 20 % z těchto 5 % neúspěšných vylíhnutí v kontrole, a to znamená změnu o 0,2 × 0,05 = 0,01 neboli o 1 procentní bod na výsledných 6 % neúspěšného vylíhnutí. Tak malou změnu není možné smysluplně odhadnout z údajů, které jsou k dispozici, a není biologicky důležitá. Naproti tomu, modeluje-li se podíl vylíhlých jiker, jejich podíl v kontrole by v tomto příkladu byl 95 % a 20 % snížení střední hodnoty kontroly by znamenalo změnu o 0,95 × 0,2 = 0,18, čili z 95 % úspěšného vylíhnutí na 77 % (= 95 – 18) úspěšného vylíhnutí a tuto koncentraci vyvolávající účinek je možné odhadnout a lze předpokládat, že bude předmětem většího zájmu. Při měřeních velikosti takový problém nevzniká, ačkoli nepříznivé účinky na velikost zpravidla znamenají snížení velikosti. Modely pro stanovení velikosti (délky nebo hmotnosti) a úspěšného líhnutí jiker nebo přežití plůdků Kromě Brainova-Cousensova hormetického modelu jsou všechny tyto modely popsány a doporučeny v dokumentu OECD (2006). O položkách, které jsou označeny OECD 2 až 5, se pojednává také v rámci experimentů ekotoxocity v publikaci Slob (2002). Existuje samozřejmě mnoho jiných modelů, které mohou být užitečné. Bunke et al. (1999) podává výčet četných modelů, které zde nejsou uvedeny, a existuje velké množství odkazů na jiné modely. V experimentech na ekotoxicitu jsou jako obzvláště vhodné doporučovány níže uvedené modely, které se široce používají. S pěti zkušebními koncentracemi a kontrolou
S šesti nebo více zkušebními koncentracemi a kontrolou
Existují-li viditelné známky hormeze (což při stanovení úspěšného líhnutí z jiker nebo přežití plůdků není pravděpodobné, ale někdy bývá zjištěna při pozorování velikosti),
Alternativní modely pro neúspěšné líhnutí z jiker a úhyn plůdků
Míra shody jediného modelu
Porovnání modelů
Kvalita odhadu ECx Interval spolehlivosti (CI) pro ECx by neměl být příliš široký. Pro rozhodnutí toho, jak široký má být interval spolehlivosti ECx, aby ECx byla stále užitečná, je nutný statistický posudek. Simulace vhodnosti regresního modelu pro údaje o líhnutí z jiker a o velikosti ukazují, že přibližně 75 % intervalů spolehlivosti pro ECx (x = 10, 20 nebo 30) nepokrývá více než dvě zkušební koncentrace. Z toho vyplývá obecný pokyn ohledně toho, co je přijatelné, a praktický pokyn ohledně toho, co je dosažitelné. Mnozí autoři tvrdí, že je nutné uvádět intervaly spolehlivosti pro všechny parametry modelu a že široké intervaly spolehlivosti pro parametry modelu ukazují na modely, které jsou nepřijatelné (Ott a Longnecker 2008, Alvord a Rossio 1993, Motulsky a Christopoulos 2004, Lyles et al. 2008, Seber a Wild 2003, Bunke et al. 1999, Environment Canada 2005). Interval spolehlivosti pro ECx (nebo pro kterýkoli jiný parametr modelu) by neměl obsahovat nulu (Motulsky a Christopoulos 2004). Toto je regrese ekvivalentní minimálnímu významnému rozdílu, na který často odkazují metody testování hypotéz (např. Wang et al. 2000). Odpovídá také intervalu spolehlivosti pro střední hodnoty odezev, neboť LOEC neobsahuje střední hodnotu kontroly. Bylo by možné pochybovat, zda jsou tyto parametrické odhady vědecky přijatelné. Jestliže např. interval spolehlivosti pro y0 je ± 20 %, není žádný odhad EC10 věrohodný. Předpovídá-li model 20 % účinek při koncentraci C a maximální pozorovaný účinek při C a nižších koncentracích je 10 %, pak hodnota EC20 není věrohodná (Motulsky a Christopoulos 2004, Wang et al. 2000, Environment Canada 2005). Stanovení ECx by nemělo vyžadovat extrapolaci mimo rozmezí pozitivních koncentrací (Draper a Smith 1999, OECD 2006). Například obecným pokynem by mohlo být, aby hodnota ECx nebyla více než cca 25 % pod nejnižší zkoušenou koncentrací nebo nad nejvyšší zkoušenou koncentrací. ODKAZY Alvord, W.G., Rossio, J.L. (1993); Determining confidence limits for drug potency in immunoassay, Journal of Immunological Methods 157, 155-163. Brain P. a Cousens R. (1989); An equation to describe dose responses where there is stimulation of growth at low doses. Weed res. 29: 93-96. Bunke, O., Droge, B. a Polzehl, J. (1999). Model selection, transformations and variance estimation in nonlinear regression. Statistics 33, 197-240. Collett, D. (2002); Modelling Binary Data, second edition, Chapman and Hall, London. Collett, D. (2003); Modelling Survival Data in Medical Research, second edition, Chapman a Hall, London. Draper, N.R. a Smith, H. (1999); Applied Regression Analysis, third edition. New York: John Wiley & Sons. Environment Canada (2005); Guidance Document on Statistical Methods for Environmental Toxicity Tests, Report EPS 1/RM/46 Huet, S., A. Bouvier, M.-A. Poursat, E. Jolivet (2003); Statistical Tools for Nonlinear Regression: A Practical Guide with S-PLUS and R Examples, Springer Series in Statistics, New York. Lyles, R. H., C. Poindexter, A. Evans, M. Brown, a C.R. Cooper (2008); Nonlinear Model-Based Estimates of IC50 for Studies Involving Continuous Therapeutic Dose-Response Data, Contemp Clin Trials. 2008 November; 29(6): 878–886. Morgan, B.J.T. (1992); Analysis of Quantal Response Data, Chapman and Hall, London. Motulsky, H., A. Christopoulos (2004); Fitting Models to Biological Data Using Linear and Nonlinear Regression: A Practical Guide to Curve Fitting, Oxford University Press, USA. O'Hara Hines, R. J. a J. F. Lawless (1993); Modelling Overdispersion in Toxicological Mortality Data Grouped over Time, Biometrics Vol. 49, s. 107-121 OECD (2006); Current approaches in the statistical analysis of ecotoxicity data: A guidance to application. Series Testing and Assessment, No. 54, Organisation for Economic Co-operation and Development, OECD, Paris. Ott, R.L., M.T. Longnecker, An Introduction to Statistical Methods and Data Analysis, sixth edition, 2008, Brooks-Cole, Belmont, CA Ratkowsky, D.A. (1993); Principles of nonlinear regression, Journal of Industrial Microbiology 12, 195-199. Seber, G.A.F., C.J. Wild, Nonlinear Regression, Wiley, 2003 Slob W. (2002); Dose-response modelling of continuous endpoints. Toxicol. Sci., 66, 298-312 Wang, Q., D.L. Denton, a R. Shukla (2000); Applications and Statistical Properties Of Minimum Significant Difference-Based Criterion Testing In a Toxicity Testing Program, Environmental Toxicology and Chemistry, Vol. 19, s. 113–117, 2000. C.48 Krátkodobá zkouška působení na reprodukci ryb ÚVOD
VÝCHOZÍ ÚVAHY A OMEZENÍ
PODSTATA ZKOUŠKY
KRITÉRIA PŘIJATELNOSTI ZKOUŠKY
POPIS METODY Přístroje a pomůcky
Voda
Zkušební roztoky
Chov ryb
Předexpozice a výběr ryb
USPOŘÁDÁNÍ ZKOUŠKY
Výběr zkušebních koncentrací
POSTUP Výběr a vážení zkušebních ryb
Podmínky expozice Doba trvání
Krmení
Světlo a teplota
Četnost analytických stanovení a měření
Pozorování
Přežití
Chování a vzhled
Plodnost
Humánní usmrcení ryb
Pozorování druhotných pohlavních znaků
Vitelogenin (VTG)
Hodnocení histopatologie gonád
ÚDAJE A PŘEDKLÁDÁNÍ ZPRÁV Hodnocení reakcí biologických markerů pomocí analýzy rozptylu (ANOVA)
Zpráva o výsledcích zkoušky
POKYNY K INTERPRETACI A PŘIJETÍ VÝSLEDKŮ
LITERATURA
Dodatek 1 ZKRATKY A DEFINICE Chemická látka : látka nebo směs. CV : variační koeficient. ELISA : enzymová imunosorpční analýza. Osa HPG : hypothalamo-hypofýzo-gonadální osa. Velikost násady : živá hmotnost ryb na jednotku objemu vody. MTC : maximální tolerovaná koncentrace, představuje přibližně 10 % mediánu letální koncentrace (LC50). Hustota obsádky : počet ryb na jednotku objemu vody. Zkoušená chemická látka : jakákoli látka nebo směs zkoušená touto zkušební metodou. VTC : vitelogenin je fosfolipoglykoprotein, prekurzor proteinů vaječného žloutku, který se obvykle vyskytuje u sexuálně aktivních samiček všech vejcorodých druhů. Dodatek 2 EXPERIMENTÁLNÍ PODMÍNKY PRO SCREENINGOVOU ZKOUŠKU ENDOKRINNÍHO PŮSOBENÍ NA RYBÁCH
Dodatek 3 NĚKTERÉ CHEMICKÉ CHARAKTERISTIKY DOPORUČENÉ ŘEDICÍ VODY
Dodatek 4A TŘECÍ PODKLAD PRO DANIO PRUHOVANÉ Vytírací rošt : celoskleněná miska na nástroje, např. o rozměrech (d × š × v) 22 × 15 × 5,5 cm, pokrytá snímatelnou drátěnou mřížkou z nerezové oceli (šířka otvorů 2 mm). Mřížka by měla pokrývat misku na nástroje v místech pod úrovní okraje. 
Na mřížku se připevní třecí podklad. Měl by vytvořit strukturu, do které budou ryby vplouvat. Vhodné jsou například umělé akvarijní rostliny vyrobené ze zeleného plastového materiálu (poznámka: je třeba vzít v potaz možnou adsorpci zkoušené chemické látky na plastový materiál). Plastový materiál se dostatečně dlouho proplachuje v dostatečném objemu horké vody, aby se zajistilo, že se do zkušební vody nedostanou žádné chemické látky. Při použití skleněných materiálů je třeba zajistit, aby se ryby při prudkých pohybech neporanily a netísnily. Vzdálenost mezi roštem a skleněnými tabulemi by měla být alespoň 3 cm, aby se zajistilo, že ke tření nebude docházet mimo rošt. Jikry vytřené do roštu propadávají mřížkou a 45–60 minut od začátku osvětlení je lze odebírat. Průsvitné jikry nejsou přilnavé a lze je snadno spočítat pomocí bočního světla. Při použití pěti samiček v každé nádrži se počet jiker do 20 za den považuje za nízký, do 100 za den za středně vysoký a více než 100 jiker za den za vysoký. Třecí rošt se odstraní, odeberou se jikry a třecí rošt se poté opět vsadí do zkušební nádrže, a to co nejpozději večer nebo velmi časně zrána. Doba do opětovného vsazení by neměla překročit jednu hodinu, protože jinak může pod vlivem třecího podkladu dojít k individuálnímu páření a tření v neobvyklé době. Pokud situace vyžaduje pozdější vsazení třecího roštu, mělo by se tak stát alespoň 9 hodin po začátku osvětlení. V tuto pozdní denní dobu již tření vyvoláno nebývá. Dodatek 4B TŘECÍ PODKLAD PRO JELEČKA VELKOHLAVÉHO Do jedné zkušební komory se umístí dvě nebo tři kombinované třecí roury a plastové/keramické/skleněné rošty nebo rošty z nerezové oceli (např. šedý žlábek polokruhového tvaru o délce 80 mm uložený na podložce se zvednutými okraji o délce 130 mm) (viz obrázek). Bylo prokázáno, že pro třecí podklad jsou vhodné starší roury z PVC nebo keramiky (Thorpe et al, 2007). Roury se doporučuje obrousit, aby se zlepšila přilnavost. Rošt by rovněž měl být odstíněn, aby se rybám zabránilo v přístupu k vypadlým jikrám, pokud u použitého třecího podkladu nebylo prokázáno, že na něm jikry dobře ulpívají. 
Účelem základny je pojmout veškeré jikry, které neulpěly na povrchu roury a které by tak spadly na dno nádrže (nebo jikry, které jsou kladeny přímo na plochou plastovou základnu). Všechny třecí podklady by se před použitím měly nejméně 12 hodin proplachovat v ředicí vodě. Thorpe KL, Benstead R, Hutchinson TH, Tyler CR, 2007. An optimised experimental test procedure for measuring chemical effects on reproduction in the fathead minnow, Pimephales promelas. Aquatic Toxicology, 81, s. 90–98. Dodatek 5A POSUZOVÁNÍ DRUHOTNÝCH POHLAVNÍCH ZNAKŮ U JELEČKA VELKOHLAVÉHO ZA ÚČELEM ZJIŠTĚNÍ URČITÝCH CHEMICKÝCH LÁTEK S ENDOKRINNÍM PŮSOBENÍM Přehled Mezi potenciálně důležité charakteristiky tělesného vzhledu dospělých jelečků velkohlavých při zkouškách endokrinních disruptorů patří zbarvení těla (tj. světlé/tmavé), forma zbarvení (tj. přítomnost či nepřítomnost svislých pruhů), tvar těla (tj. tvar hlavy a hrudní oblasti, rozšíření břicha) a druhotné pohlavní znaky specifické pro tento druh (tj. počet a velikost pářicích hrbolků, velikost dorzálního polštářku a kladélka). Pářicí hrbolky se nacházejí na hlavě (dorzální polštářek) reprodukčně aktivních jelečků velkohlavých a obvykle jsou uspořádány symetricky po obou stranách (Jensen et al., 2001). U samiček v kontrolních skupinách a u nedospělých samečků a samiček nebyl vývoj hrbolků zaznamenán (Jensen et al., 2001). Samečci mohou mít až osm jednotlivých hrbolků kolem očí a mezi nosními dírkami. Nejvíce hrbolků největší velikosti se nachází ve dvou souběžných řadách bezprostředně pod nosními dírkami a nad tlamou. Ryby často mají také skupiny hrbolků pod dolní čelistí. Ty, které jsou nejblíže k tlamě, se zpravidla vyskytují v jednom páru, zatímco více ventrálně se mohou nacházet i po čtyřech. Skutečný počet hrbolků je zřídka větší než 30 (v rozmezí 18–28; Jensen et al., 2001). Nejvíce hrbolků má podobu jednotlivých, zaoblených struktur, jejichž výška se přibližně rovná jejich poloměru. Většina reprodukčně aktivních samečků má rovněž alespoň některé hrbolky zvětšené a vystouplé do té míry, že je nelze odlišit jako jednotlivé struktury. Některé druhy chemických látek narušujících činnost endokrinních žláz mohou způsobit abnormální výskyt určitých druhotných pohlavních znaků u opačného pohlaví. Například agonisté androgenního receptoru, jako je 17-alfa-methyltestosteron nebo 17-beta-trenbolon, mohou způsobit, že se pářicí hrbolky vyvinou u samiček jelečka velkohlavého (Smith, 1974; Ankley et al., 2001; 2003), a agonisté estrogenního receptoru mohou způsobit snížení počtu nebo velikosti pářicích hrbolků u samečků (Miles-Richardson et al., 1999; Harries et al., 2000). Následuje popis, jak charakterizovat pářicí hrbolky u jelečka velkohlavého, založený na postupech používaných v laboratoři americké Agentury na ochranu životního prostředí (EPA) v Duluthu ve státě Minnesota. Specifické produkty a/nebo vybavení lze nahradit srovnatelnými materiály, které jsou k dispozici. Prohlídka se nejlépe provádí pomocí lupy s osvětlením nebo pomocí preparačního mikroskopu s osvětlením a s objektivem 3X. Ryby se prohlížejí z dorzálního směru a anteriorní stranou dopředu (hlavou k pozorovateli).
Počítání hrbolků a jejich klasifikace Bylo zjištěno šest specifických oblastí pro posuzování přítomnosti a vývoje hrbolků u dospělých jelečků velkohlavých. Ke zmapování umístění a množství přítomných hrbolků byla vytvořena šablona (viz konec tohoto dodatku). U každého organismu se zaznamená počet hrbolků a jejich velikost pak lze kvantitativně seřadit takto: 0 – nevyskytují se, 1 – přítomné, 2 – zvětšené a 3 – výrazně zvětšené (obrázek 1). Stupeň 0 – žádné hrbolky se nevyskytují. Stupněm 1 (přítomné) se označí všechny hrbolky s jedním vrcholem, jejichž výška se přibližně rovná jejich poloměru. Stupeň 2 (zvětšené) se projevuje tkání v hvězdicovitém tvaru, který má obvykle velkou radiální základnu s rýhami nebo brázdami směřujícími od středu. Horní část hrbolku je často spíše rozeklaná, ale může být i poněkud zaoblená. Při stupni 3 (výrazně zvětšené) jsou hrbolky obvykle poměrně velké a zaoblené, s málo rozlišenou strukturou. Někdy se hrbolky mohou vyskytovat společně a vytvářet jednolitou strukturu v určité oblasti nebo v několika oblastech najednou (B, C a D, jak jsou popsány níže). Zbarvení a tvar jsou podobné jako u stupně 2, ale někdy je obtížné je navzájem rozlišit. Použití tohoto klasifikačního systému zpravidla vede k celkovému hodnocení hrbolků nižšímu než 50 u normálních samečků v kontrolních skupinách, kteří mají 18–20 hrbolků (Jensen et al., 2001). Obrázek 1 
Skutečný počet hrbolků u některých ryb může být v konkrétní oblasti klasifikace větší, než je počet políček v šabloně. Nastane-li tento případ, lze vpravo nebo vlevo od daného políčka vepsat další klasifikační čísla. Šablona proto nemusí být symetrická. Další způsob zmapování hrbolků, které jsou párové nebo spojené vertikálně podél horizontální roviny tlamy, lze provést tak, že se do jednoho políčka uvedou klasifikační body dvou hrbolků. Oblasti mapování:
ODKAZY
Dodatek 5B POSUZOVÁNÍ DRUHOTNÝCH POHLAVNÍCH ZNAKŮ U HALANČÍKA JAPONSKÉHO ZA ÚČELEM ZJIŠTĚNÍ URČITÝCH CHEMICKÝCH LÁTEK S ENDOKRINNÍM PŮSOBENÍM Následuje popis měření bradavkovitých výběžků (*10), které jsou druhotným pohlavním znakem u halančíka japonského (Oryzias latipes).
Měření
Obrázek 1. Schéma znázorňující rozdíl mezi pohlavími v tvaru a velikosti řitní ploutve. A – sameček, B – samička. Oka, T. B., 1931. On the processes on the fin rays of the male of Oryzias latipes and other sex characters of this fish. J. Fac. Sci., Tokyo Univ., IV, 2, s. 209-218. 
Obrázek 2. A – výběžky na článcích paprsků řitní ploutve. J. P. – článek, A. S. – osová vzdálenost, P. – výběžek. B – distální konec ploutvového paprsku. Na hrotu paprsku se nacházejí actinotrichia (Act.). Oka, T. B., 1931. On the processes on the fin rays of the male of Oryzias latipes and other sex characters of this fish. J. Fac. Sci., Tokyo Univ., IV, 2, s. 209-218. 
Obrázek 3. Fotografie rybího těla, na které je znázorněno místo řezu při fixaci gonád ve fixačním roztoku, nejedná-li se o 10 % neutrální pufrovaný formalín. V takovém případě se zbývající část těla pomocí břitvy odřízne mezi anteriorní oblastí řitní ploutve a řitním otvorem (červená čára), hlavová část rybího těla se uloží do fixačního roztoku pro gonády a ocasní část se uloží do 10 % neutrálního pufrovaného formalínu. 
Dodatek 6 DOPORUČENÉ POSTUPY PRO ODBĚR VZORKŮ ZA ÚČELEM ANALÝZY VITELOGENINU Je třeba dbát na to, aby nedošlo ke vzájemné kontaminaci mezi vzorky VTG samečků a samiček. Postup 1A: Jeleček velkohlavý, odběr krve z ocasní žíly/tepny Po anestetizaci se skalpelem částečně oddělí ocasní stopka a heparinizovanou mikrohematokritovou kapilárou se odebere krev z ocasní žíly/tepny. Po odebrání krve se rychle izoluje plazma, a to odstředěním po dobu 3 minut při zrychlení 15 000 g (nebo alternativně po dobu 10 minut při zrychlení 15 000g a při teplotě 4 °C). Je-li to požadováno, lze po odstředění stanovit procento hematokritu. Část plazmy se poté přenese z mikrohematokritové kapiláry a uloží se do odstředivkové zkumavky s 0,13 jednotkami aprotininu (inhibitor proteázy) při teplotě – 80 °C do doby, než bude možné provést stanovení VTG. Podle velikosti jelečka velkohlavého (závisející na pohlaví) se objem plazmy, kterou lze odebrat, zpravidla nachází v rozmezí od 5 do 60 mikrolitrů z každé ryby (Jensen et al., 2001). Postup 1B: Jeleček velkohlavý, odběr krve ze srdce Alternativně lze krev odebrat také srdeční punkcí pomocí heparinizované injekční stříkačky (1 000 jednotek heparinu na ml). Krev se přenese do Eppendorfových zkumavek (uložených na ledu) a poté odstředí (po dobu 5 minut při zrychlení 7 000g a pokojové teplotě). Plazma se přenese do čistých Eppendorfových zkumavek (v poměrných částech, pokud je to s ohledem na objem plazmy proveditelné), urychleně se zmrazí při teplotě – 80 °C a takto se ponechá až do analýzy (Panter et al., 1998). Postup 2A: Halančík japonský, vyříznutí jater Vyjmutí zkušební ryby ze zkušební komory
Vyříznutí jater
Po odnětí jater je mrtvé tělo ryby k dispozici pro histologii gonád a měření druhotných pohlavních znaků. Vzorek Pokud se vzorky jater odebrané u zkušebních ryb krátce po vyříznutí nepoužijí pro předběžnou úpravu, skladují se při teplotě ≤ – 70 °C. Obrázek 1. Nůžkami se vede řez těsně před prsními ploutvemi. 
Obrázek 2. Pomocí nůžek se rozstřihne střední linie břicha až k bodu, který se nachází přibližně 2 mm kraniálně od řitního otvoru. 
Obrázek 3. Pinzetou se rozevřou břišní stěny, aby se odkryla játra a další vnitřní orgány. (Alternativně lze břišní stěny laterálně přišpendlit.) Šipka označuje játra. 
Obrázek 4. Játra se pomocí pinzety vypreparují a vyříznou. 
Obrázek 5. Pinzetou se jemně odtáhnou střeva. 
Obrázek 6. Oba konce střev a veškeré střevní okruží se oddělí nůžkami. 
Obrázek 7 (samička). U samiček je postup stejný. 
Obrázek 8. Dokončený postup. 
Postup 2B: Halančík japonský (Oryzias latipes), předúprava jater pro analýzu vitelogeninu Ze soupravy pro analýzu ELISA se vezme lahev s homogenizačním pufrem a ochladí se v ledové tříšti (teplota roztoku ≤ 4 °C). Je-li použit homogenizační pufr ze systému ELISA EnBio, roztok se rozmrazí při pokojové teplotě a poté se lahev ochladí v ledové tříšti. Objem pufru pro homogenizaci jater se vypočítá na základě jejich hmotnosti (na mg hmotnosti jater se přidá 50 μl homogenizačního pufru). Například je-li hmotnost jater 4,5 mg, objem pufru pro homogenizaci jater je 225 μl. Připraví se seznam objemů homogenizačního pufru pro všechna játra. Příprava jater pro předúpravu
Provedení předúpravy 1) Přidání homogenizačního pufru Kontrolou seznamu se zjistí, jaký objem homogenizačního pufru má být přidán pro daný konkrétní vzorek jater, a mikropipeta (rozmezí objemů 100–1 000 μl) se nastaví na příslušný objem. Na mikropipetu se nasadí čistý hrot. Z lahve s činidlem se nabere homogenizační pufr a přidá se do zkumavky o objemu 1,5 ml obsahující játra. Podle výše uvedeného postupu se homogenizační pufr přidá do všech mikrozkumavek o objemu 1,5 ml obsahujících játra. Hrot mikropipety není třeba měnit za nový. Je-li hrot kontaminován nebo existuje-li podezření, že je kontaminován, musí se vyměnit. 2) Homogenizace jater
3) Odstředění suspendovaného homogenátu jater
4) Odběr supernatantu
Skladování vzorku Mikrozkumavky o objemu 0,5 ml obsahující supernatant homogenátu jater se skladují při teplotě ≤ – 70 °C do doby, než jsou použity pro analýzu ELISA. Postup 3A: Danio pruhované, odběr krve z ocasní žíly/tepny Ihned po anestetizaci se příčně oddělí ocasní stopka a z ocasní tepny/žíly se odebere krev pomocí heparinizované mikrohematokritové kapiláry. Objemy krve se nacházejí v rozmezí od 5 do 15 mikrolitrů v závislosti od velikosti ryby. Do mikrokapiláry se přidá stejný objem pufru s aprotininem (6 mikrogramů aprotininu na ml fosfátového pufru (PBS)) a plazma se od krve oddělí odstředěním (po dobu 5 minut při zrychlení 600 g). Plazma se shromáždí do zkušebních zkumavek a uskladní se při teplotě – 20 °C do doby, než se analyzuje na VTG nebo na jiné proteiny, jež jsou předmětem zájmu. Postup 3B: Danio pruhované, odběr krve srdeční punkcí Aby nedošlo ke koagulaci krve a rozložení proteinu, vzorky se odebírají ve fosfátovém pufru (PBS) obsahujícím heparin (1 000 jednotek/ml) a inhibitor proteázy aprotinin (2 TIU/ml). Jako složky pro přípravu tohoto pufru se doporučuje použít heparin, amonnou sůl a lyofilizovaný aprotinin. Odběr krve se doporučuje provést injekční stříkačkou (o objemu 1ml) s fixovanou tenkou jehlou (např. značky Braun Omnikan-F). Injekční stříkačka by se měla předem naplnit pufrem (přibližně 100 mikrolitrů) tak, aby malé objemy krve z každé ryby byly plně eluovány. Vzorky krve se odeberou srdeční punkcí. Ryba se nejprve anestetizuje roztokem MS-222 (v koncentraci 100 mg/l). Správný režim anestezie umožní uživateli rozeznat srdeční tep dania pruhovaného. Při provádění srdeční punkce se píst injekční stříkačky udržuje pod slabým tlakem. Množství krve, která lze odebrat, se nacházejí v rozmezí 20–40 mikrolitrů. Po srdeční punkci se směs krve/pufru vloží do zkušební zkumavky. Plazma se oddělí od krve odstředěním (po dobu 20 minut při 5 000g) a uskladní se při teplotě – 80 °C do doby, než bude použita pro analýzu. Postup 3C: Standardní operační postup: danio pruhované, homogenizace hlavy a ocasu
Dodatek 7 VZORKY OBOHACENÉ VITELOGENINEM A REFERENČNÍ STANDARD PRO OPAKOVATELNOST Každý den provádění zkoušek na VTG se analyzuje obohacený vzorek vytvořený pomocí referenčního standardu pro opakovatelnost. Pro přípravu referenčního standardu pro opakovatelnost se použije VTG z jiné šarže, než jaká byla použita pro přípravu kalibračních standardů pro zkoušku, která se nyní provádí. Obohacený vzorek se připraví tak, že se známé množství standardu pro opakovatelnost přidá do vzorku plazmy samečků z kontrolní skupiny. Vzorek se obohatí tak, aby se dosáhlo koncentrace VTG 10krát až 100krát vyšší než očekávaná koncentrace vitelogeninu u samečků ryb z kontrolní skupiny. Vzorek plazmy samečků z kontrolní skupiny, který se obohacuje, může pocházet z jedné ryby, nebo to může být směsný vzorek z několika ryb. Dílčí vzorek neobohacené plazmy samečků z kontrolní skupiny se analyzuje alespoň ve dvou duplikátních jamkách. Rovněž obohacený vzorek se analyzuje alespoň ve dvou duplikátních jamkách. Za účelem stanovení očekávané koncentrace se střední hodnota množství vitelogeninu z obou neobohacených vzorků plazmy samečků z kontroly přičte k vypočtenému množství VTG přidaného za účelem obohacení vzorků. Poměr mezi touto očekávanou koncentrací a naměřenou koncentrací se zaznamená spolu s výsledky všech souborů zkoušek provedených v týž den. Dodatek 8 SCHÉMA ROZHODOVÁNÍ PRO STATISTICKOU ANALÝZU 
C.49 Zkouška akutní toxicity na rybích embryích (FET) ÚVOD
PODSTATA ZKOUŠKY
VÝCHOZÍ ÚVAHY
VALIDITA ZKOUŠKY
POPIS METODY
Přístroje a pomůcky
Zkušební komory
Voda a zkušební podmínky
Zkušební roztoky
Chov matečných ryb
Zkoušení odborné způsobilosti
Produkce jiker
Diferenciace jiker
POSTUP Podmínky expozice
Zkušební koncentrace
Kontroly
Začátek expozice a doba trvání zkoušky
Distribuce jiker na 24jamkových destičkách
Pozorování
Analytická měření
LIMITNÍ ZKOUŠKA
ÚDAJE A PŘEDKLÁDÁNÍ ZPRÁV Zpracování výsledků
Závěrečná zpráva
LITERATURA
Dodatek 1 DEFINICE Hlavní ukazatel : způsobuje účinek na úrovni populace. Blastula : buněčný útvar obklopující animální pól, který zakrývá určitou část žloutku. Chemická látka : látka nebo směs. Epibolie : masivní proliferace převážně epidermálních buněk ve fázi gastrulace embrya a jejich pohyb od dorzální k ventrální straně, v důsledku kterého jsou vrstvy endodermálních buněk internalizovány během procesu podobnému invaginaci (vchlípení) a žloutek je vstřebán do embrya. Průtoková metoda : zkouška, při níž zkušební roztoky plynule protékají zkušebním systémem po celou dobu trvání expozice. Vnitřní kontrola destičky : vnitřní kontrola sestávající z naplnění 4 jamek ve 24jamkové destičce ředicí vodou má za cíl zjistit potenciální kontaminaci destiček způsobenou výrobcem nebo výzkumníkem během zkušebního postupu a jakýkoli účinek na destičku, který může ovlivnit výsledek zkoušky (např. teplotní gradient). IUPAC : International Union of Pure and Applied Chemistry (Mezinárodní unie pro čistou a užitou chemii). Chovná voda : voda, ve které se provádí chov dospělých ryb. Medián letální koncentrace (LC50) : koncentrace zkoušené chemické látky, která usmrtí přibližně 50 % zkušebních organismů během celé zkoušky. Semistatická (obnovovací) zkouška : zkouška, při níž se zkušební roztoky pravidelně obnovují po stanovených časových úsecích (např. každých 24 hodin). SMILES : Simplified Molecular Input Line Entry Specification (zjednodušená molekulární specifikace pro vstupní řádky). Somit : somity jsou bloky mezodermu ve vyvíjejícím se embryu obratlovců rozmístěné po stranách neurální trubice, z nichž později vzniká škára (dermatomy), kosterní svaly (myotomy) a obratle (sklerotomy). Statická metoda : zkouška, při níž se zkušební roztoky během celé zkoušky nemění. Zkoušená chemická látka : jakákoli látka nebo směs zkoušená pomocí této zkušební metody. UVCB : látky s neznámým nebo proměnlivým složením, komplexní reakční produkty nebo biologický materiál. Dodatek 2 CHOV, MNOŽENÍ A TYPICKÉ PODMÍNKY PRO ZKOUŠKY AKUTNÍ TOXICITY NA EMBRYÍCH DANIA PRUHOVANÉHO
Dodatek 3 NORMÁLNÍ VÝVOJ DANIA PRUHOVANÉHO PŘI TEPLOTĚ 26 C 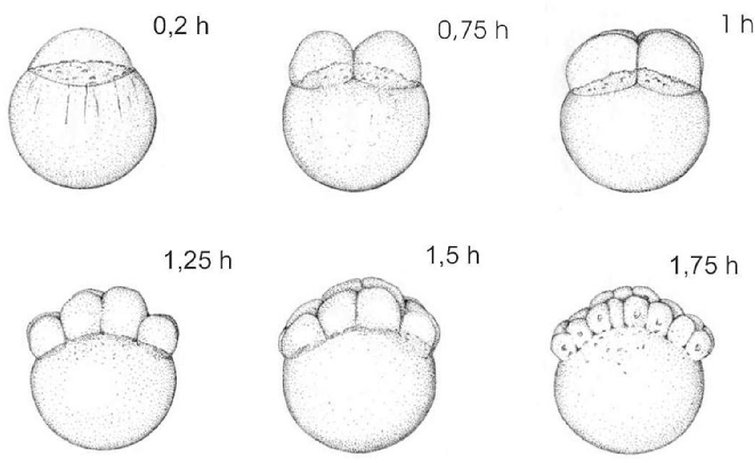


Dodatek 4 Obrázek 1 Rozvržení 24jamkových destiček 
Obrázek 2 Schéma postupu při zkoušce akutní toxicity na embryích dania pruhovaného (zleva doprava): tvorba jiker, sběr jiker, předexpoziční doba bezprostředně po oplodnění ve skleněných nádobách, výběr oplodněných jiker s inverzním nebo binokulárním mikroskopem a rozdělení oplodněných jiker do připravených 24jamkových destiček s příslušnými zkušebními koncentracemi / kontrolami, n = požadovaný počet jiker pro každou zkušební koncentraci / kontrolu (zde 20), hpf = hodin po oplodnění. 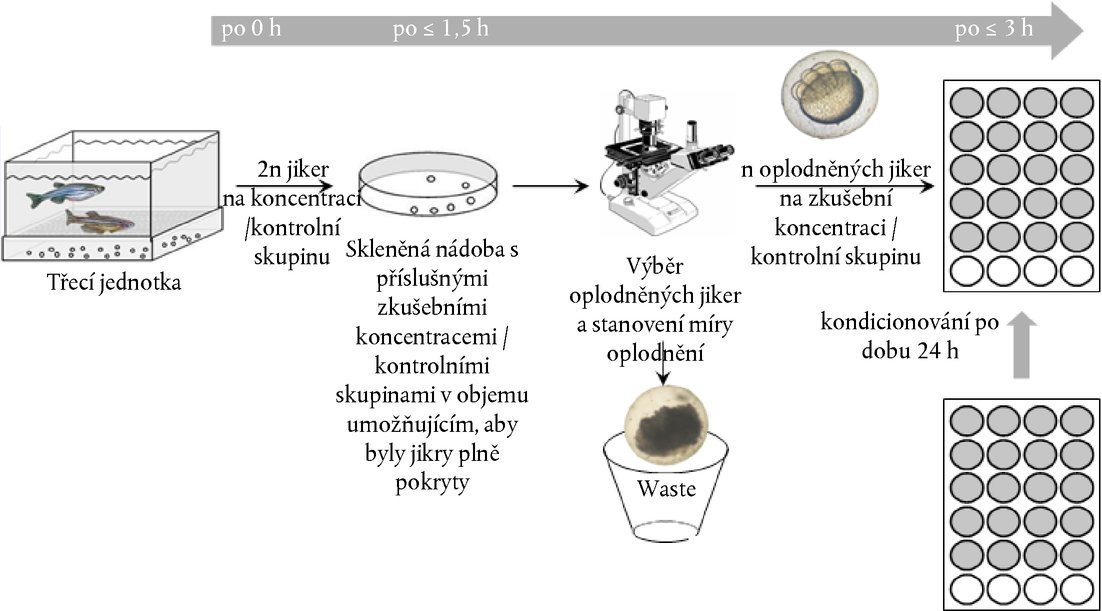
Dodatek 5 ATLAS UKAZATELŮ LETALITY PRO ZKOUŠKU AKUTNÍ TOXICITY NA EMBRYÍCH DANIA PRUHOVANÉHO Následující hlavní ukazatele naznačují akutní toxicitu a následně uhynutí embryí: koagulace embrya, neoddělení ocasu, nepřítomnost tvorby somitů a nepřítomnost srdečního tepu. K ilustraci těchto ukazatelů byly vybrány následující mikroskopické snímky. Obrázek 1 Koagulace embrya: 
Při osvětlení v jasném poli jsou na koagulovaných embryích dania pruhovaného viditelné různé neprůhledné útvary. Obrázek 2 Nepřítomnost tvorby somitů: 
Třebaže je embryo dania pruhovaného ve věku 24 h na snímku a) opožděné ve vývoji přibližně o 10 h, vykazuje dobře vyvinuté somity (→), zatímco embryo na snímku b) nevykazuje žádné známky tvorby somitů (→). Ačkoli embryo dania pruhovaného ve věku 48 h na snímku c) vykazuje výrazný otok žloutkového váčku (*), zároveň je zřetelně patrná tvorba somitů (→), zatímco embryo dania pruhovaného ve věku 96 h vyobrazené na snímku d) nevykazuje žádné známky tvorby somitů (→). Na embryu vyobrazeném na snímku d) si povšimněte také zakřivení páteře (skoliózy) a otoku perikardu (*). Obrázek 3 Neoddělení ocasního pupenu, 
Obrázek 4 Nepřítomnost srdečního tepu 
Nepřítomnost srdečního tepu je z povahy věci obtížné prokázat z mikroskopického snímku. Na nepřítomnost srdečního tepu ukazuje to, že nedochází ke stahům srdce (dvojitá šipka). Nepohyblivost krevních buněk např. v břišní aortě (→ na vloženém snímku) není ukazatelem nepřítomnosti srdečního tepu. Povšimněte si také neexistence tvorby somitů v tomto embryu (*, spíše homogenní než členitý vzhled svalových tkání). Pozorování pro zaznamenání nepřítomnosti srdečního tepu by mělo trvat nejméně jednu minutu při minimálně 80 násobném zvětšení. C.50 Zkouška toxicity na Myriophyllum spicatum bez sedimentu ÚVOD
PODSTATA ZKOUŠKY
INFORMACE O ZKOUŠENÉ CHEMICKÉ LÁTCE
VALIDITA ZKOUŠKY
REFERENČNÍ CHEMICKÁ LÁTKA
POPIS METODY Přístroje a pomůcky
Zkušební organismy
Kultivace
Zkušební médium
Zkušební roztoky
Zkušební a kontrolní skupiny
Expozice
Zkušební podmínky
Doba trvání
Měření a analytická stanovení
Četnost měření a analytická stanovení
Limitní zkouška
ÚDAJE A PŘEDKLÁDÁNÍ ZPRÁV Proměnné odezvy
Průměrná specifická růstová rychlost
Výtěžek
Doba zdvojnásobení
Vynášení křivek závislosti koncentrace-odezva
Odhad ECx
Statistické postupy
Předkládání zpráv
LITERATURA
Dodatek 1 DEFINICE
Dodatek 2 MODIFIKOVANÉ ANDREWSOVO MÉDIUM PRO ZÁSOBNÍ KULTURY A PŘEDKULTURY Andrewsovo médium potřebné pro zásobní kultury a předkultury se připraví z pěti samostatně připravených zásobních živných roztoků s přidáním 3 % sacharózy. Tabulka 1 Složení Andrewsova živného roztoku: (norma ASTM E 1913-04)
Zásobní roztoky lze držet v chladničce po dobu 6 měsíců (při 5 až 10 °C). Pouze zásobní roztok č. 5 má omezenou životnost (dva měsíce). Tabulka 2 Příprava zásobního roztoku 3.1 na přípravu zásobního roztoku 3
Připravený zásobní roztok 3.1 (tabulka 2) se hluboce zmrazí v poměrných dílech přibližně po 11 ml (nejméně při – 18 °C). Hluboce zmrazené části mají životnost pět let. Pro přípravu zásobního roztoku 3 se zásobní roztok 3.1 rozmrazí, 10 ml z něho se odlije do odměrné baňky o objemu 1 l a dolije se velmi čistá voda až po značku 1 l. Pro získání Andrewsova modifikovaného média se přibližně 2 500 ml velmi čisté vody nalije do odměrné baňky o objemu 5 l. Po přidání 50 ml každého zkušebního roztoku se odměrná baňka z 90 % naplní velmi čistou vodou a pH se upraví na 5,8. Poté se přidá 150 g rozpuštěné sacharózy (3 % na každých 5 l); poté se do odměrné baňky dolije velmi čistá voda až po značku 5 l. Nakonec se živným roztokem naplní Schottovy baňky o objemu 1 l a autoklávují se při 121 °C po dobu 20 minut. Takto získaný živný roztok lze udržovat sterilní v chladničce (při 5 až 10 °C) po dobu tří měsíců. Modifikované Andrewsovo médium pro zkoušku toxicity bez sedimentu Z pěti zásobních živných roztoků již popsaných v tabulkách 1 a 2 se připraví desetinásobně koncentrované modifikované Andrewsovo médium, které je požadováno pro získání živných roztoků, přidáním 30 % sacharózy. Provede se to tak, že přibližně 100 ml velmi čisté vody se nalije do odměrné baňky o objemu 1 l. Po přidání 100 ml každého ze zásobních roztoků se pH nastaví na 5,8. Poté se přidá 30 % rozpuštěná sacharóza (300 g na 1 000 ml); poté se do odměrné baňky dolije velmi čistá voda až po značku 1 l. Nakonec se živným roztokem naplní Schottovy baňky o objemu 0,5 l a autoklávují se při 121 °C po dobu 20 minut. Takto získaný desetinásobně koncentrovaný modifikovaný živný roztok lze udržovat sterilní v chladničce (při 5 až 10 °C) po dobu tří měsíců. Dodatek 3 UCHOVÁVÁNÍ ZÁSOBNÍCH KULTUR V tomto dodatku 3 se popisuje zásobní kultura Myriophyllum spicatum L (103), vodní dvouděložné rostliny rostoucí pod hladinou, která je druhem stolístku z čeledi Haloragaceae. Od června do srpna jeho nenápadné růžově bílé květy vyrůstají nad vodní hladinu. Rostliny jsou zakořeněny v zemi soustavou pevných oddenků a lze se s nimi setkat po celé severní polokouli v eutrofních, avšak neznečištěných stojatých vodách s větším obsahem vápníku a s bahnitým substrátem. Myriophyllum spicatum upřednostňuje sladkou vodu, ale vyskytuje se i v brakických vodách. Pro zásobní kulturu bez sedimentu v laboratorních podmínkách jsou zapotřebí sterilní rostliny. Sterilní rostliny jsou dostupné z ekotoxikologické laboratoře německého Umweltbundesamt (Spolkový úřad pro životní prostředí, Německo). Alternativně lze zkušební organismy připravit z nesterilních rostlin v souladu s normou ASTM E 1913-04. Následuje postup pro kultivaci Myriophyllum sibiricum odebraných v terénu – výtah ze standardních pokynů ASTM: „Začínáte-li od nesterilních rostlin sbíraných v terénu, odeberte na podzim turiony M. sibiricum. Vložte turiony do akvária o objemu 20 l obsahujícího 5 cm sterilního sedimentu, který je pokryt křemenným pískem nebo například Turface®, a 18 l reagenční vody. Akvárium provzdušněte a udržujte při teplotě 15 oC a ozářenosti 200 až 300 μmol m– 2 s– 1 po dobu 16 hodin za den. Rostlinnou kulturu v akváriu lze udržovat jako zásobní zdroj rostlin v případě, že sterilní rostlinné kultury jsou zničeny mechanickým poškozením v růstové komoře, kontaminací nebo z jiného důvodu. Rostliny pěstované v akváriu nejsou sterilní a sterilní kultury není možné udržovat ve vsádkovém kultivačním systému. Za účelem sterilizace kultury se rostliny vyjmou z akvária a oplachují pod tekoucí deionizovanou vodou asi po dobu 0,5 h. V aseptických podmínkách v komoře s laminárním prouděním vzduchu se rostliny dezinfikují po dobu necelých 20 minut (dokud většina rostlinných tkání nevybledne a pouze rostoucí vrcholek je ještě zelený) v 3 % (m/V) roztoku chlornanu sodného obsahujícím 0,01 % vhodného smáčedla. Dezinfekční přípravek a rostlinný materiál se protřepou. Segmenty s několika kolínky se přenesou do sterilních kultivačních zkumavek obsahujících 45 ml sterilizovaného modifikovaného Andrewsova média a uzavřou se plochými uzávěry pro kultivační zkumavky. Do každé zkušební komory se vloží pouze jeden segment rostliny. K zajištění uzávěru kultivační nádoby se použije laboratorní těsnicí fólie. Po založení sterilní kultury by se segmenty rostlin obsahující několik kolínek vždy po deseti až dvanácti dnech měly přemístit do nových zkušebních komor s čerstvým kapalným živným médiem. Jak bylo prokázáno kultivací v miskách za použití agaru, rostliny musí být sterilní a zůstat sterilní po dobu osmi týdnů předtím, než lze zahájit zkoušení.“ Jelikož modifikované Andrewsovo médium obsahuje sacharózu (která stimuluje růst hub a bakterií), musí se veškerá manipulace s materiálem a roztoky a kultivace provádět za sterilních podmínek. Veškeré kapaliny a vybavení se před použitím sterilizují. Sterilizace se provádí horkým vzduchem (210 °C) po dobu 4 hodin nebo v autoklávu po dobu 20 minut při teplotě 121 °C. Všechny baňky, misky, mísy atd. a jiné vybavení navíc těsně před použitím projdou úpravou plamenem na sterilním laboratorním stole. Zásobní kultury je možné uchovávat při sníženém osvětlení a teplotě (50 μE m– 2 s– 1, 20 ± 2 °C) po delší dobu, bez potřeby je znovu zakládat. Růstové médium pro Myriophyllum může být stejné jako médium použité pro zkoušení, ale pro zásobní kultury je možné použít jiná média bohatá na živiny. Segmenty rostlin se axenicky (jednodruhově) rozdělí do několika Erlenmeyerových baněk o objemu 500 ml a/nebo Fernbachových baněk o objemu 2 000 ml, do každé z nich se nalije přibližně 450, respektive 1 000 ml modifikovaného Andrewsova média. Baňky se poté axenicky uzavřou celulózovou zátkou. Dále je zcela nezbytné důkladné ošetření náčiní plamenem na sterilním laboratorním stole těsné před použitím. V závislosti na počtu a velikosti se rostliny přibližně každé tři týdny mají přemístit do čerstvého živného roztoku. Pro tuto obnovenou kulturu lze použít vrcholky, jakož i segmenty střední části stonků. Počet a velikost přenesených rostlin (nebo segmentů rostlin) závisí na tom, kolik rostlin je potřeba. Je například možné přemístit pět segmentů výhonku do jedné Fernbachovy baňky a tři segmenty výhonku do jedné Erlenmeyerovy baňky, každý o délce 5 cm. Veškeré zakořeněné, kvetoucí, mrtvé nebo jinak zjevně odlišné části se vyřadí. Obrázek 1 Řezání rostlin pro zásobní kulturu a předkulturu po třech týdnech kultivace. 
Kultivace rostlin se má provádět v Erlenmeyerových baňkách o objemu 500 ml a Fernbachových baňkách o objemu 2 000 l v chladicím inkubátoru při teplotě 20 ± 2 °C za nepřetržitého světla přibližně 100 až 150 μE m– 2 s– 1 nebo 6 000 až 9 000 luxů (vydávaného komorovým osvětlením barevné teploty „teplé bílé světlo“). Obrázek 2 Kultivace rostlin v chladicím inkubátoru s komorovým osvětlením. 
Je třeba používat chemicky čisté (umyté kyselinou) a sterilní skleněné kultivační nádoby a dodržovat aseptické podmínky manipulace. V případě kontaminace zásobní kultury, např. řasami, houbami nebo bakteriemi, je třeba připravit novou kulturu nebo by pro obnovení dané kultury měla být použita zásobní kultura z jiné laboratoře. Dodatek 4 UDRŽOVÁNÍ PŘEDKULTURY A PŘÍPRAVA ZKUŠEBNÍCH ORGANISMŮ PRO ZKOUŠENÍ Pro získání předkultury se výhonky ze zásobní kultury nařežou na segmenty, každý s dvěma přesleny; segmenty se vloží do Fernbachových baněk naplněných modifikovaným Andrewsovým médiem (s 3 % sacharózy). Každá baňka může obsahovat až 50 segmentů výhonků. Je však třeba dbát na to, aby byly segmenty vitální a neměly žádné kořeny ani boční větve nebo jejich pupeny (viz obrázek 1 v dodatku 3). Organismy v předkultuře se kultivují po 14 až 21 dnů za sterilních podmínek v komoře za podmínek prostředí se střídáním fází 16 hodin světla / 8 hodin tmy. Intenzita světla se zvolí v rozmezí 100 až 150 μE m– 2 s– 1. Teplota ve zkušebních nádobách by měla být 23 ± 2 °C. Jelikož modifikované Andrewsovo médium obsahuje sacharózu (která stimuluje růst řas, hub a bakterií), musí se zkušební roztoky připravovat a kultivovat za sterilních podmínek. Veškeré kapaliny a vybavení se před použitím sterilizují. Sterilizace se provádí horkým vzduchem (210 °C) po dobu 4 hodin nebo v autoklávu po dobu 20 minut při teplotě 121 °C. Všechny baňky, misky, mísy atd. a jiné vybavení navíc těsně před použitím projdou úpravou plamenem na sterilním laboratorním stole. Výhonky se axenicky odstraní z baněk předkuktury, přičemž se vybere materiál, který je co nejvíce homogenní. Pro každé zkoušení je zapotřebí nejméně 60 zkušebních organismů (zkoušení s osmi koncentracemi zkoušené chemické látky). Pro zkoušení se z předkultur odeberou čerstvé boční větve, zkrátí se na 2,5 cm od báze (měřeno pravítkem) a přenesou se do kádinky se sterilním modifikovaným Andrewsovým roztokem. Tyto čerstvé boční větve lze použít na zkoušku toxicity na Myriophyllum spicatum bez sedimentu. Obrázek 2 Řezání rostlin z předkultury na zkoušku toxicity na Myriophyllum spicatum bez sedimentu. 
C.51 Zkouška toxicity na Myriophyllum spicatum v systému voda-sediment ÚVOD
PODSTATA ZKOUŠKY
INFORMACE O ZKOUŠENÉ CHEMICKÉ LÁTCE
VALIDITA ZKOUŠKY
REFERENČNÍ CHEMICKÁ LÁTKA
POPIS METODY Zkušební zařízení
Zkušební organismy
Kultivace zkušebního organismu
Sedimenty
Zkušební médium
Uspořádání experimentu
Koncentrace zkoušené chemické látky a kontrolní skupiny
Limitní zkouška
Zkušební roztoky
POSTUP ZKOUŠKY
Zaváděcí fáze
Výběr jednotného rostlinného materiálu
Expozice prostřednictvím vodní fáze
Expozice prostřednictvím sedimentu
Udržování úrovně vody po dobu trvání zkoušky
Zkušební podmínky
Doba trvání zkoušky
Měření a analytická stanovení
Četnost měření a analytická stanovení
Analytická měření zkoušené chemické látky
VYHODNOCENÍ ÚDAJŮ
Proměnné odezvy
Průměrná specifická růstová rychlost
Výtěžek
Vynášení křivek závislosti koncentrace-odezva
Odhad ECx
Statistické postupy
PŘEDKLÁDÁNÍ ZPRÁV
LITERATURA
Dodatek 1 SLOŽENÍ SMARTOVA A BARKOVA MÉDIA
Dodatek 2 DEFINICE
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||




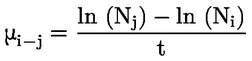

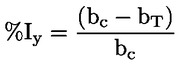

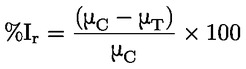

(1) Pro teplotu 20 °C není hodnota k dispozici, ale lze předpokládat, že variabilita výsledků měření je vyšší než závislost na teplotě, již lze očekávat.
(2) Nařízení Evropského parlamentu a Rady (ES) č. 1907/2006 ze dne 18. prosince 2006 o registraci, hodnocení, povolování a omezování chemických látek, o zřízení Evropské agentury pro chemické látky, o změně směrnice 1999/45/ES a o zrušení nařízení Rady (EHS) č. 793/93, nařízení Komise (ES) č. 1488/94, směrnice Rady 76/769/EHS a směrnic Komise 91/155/EHS, 93/67/EHS, 93/105/ES a 2000/21/ES. Úř. věst. L 304, 22.11.2007, s. 1.
(3) Interagency Coordinating Committee on the Validation of Alternative Methods (Meziagenturní koordinační výbor pro validaci alternativních metod), USA.
(*1) Zaznamená se oblast zákalu rohovky.
(4) K použití integrované strategie zkoušení dráždivých účinků na oči podle nařízení REACH viz též pokyny vydané agenturou ECHA Guidance on information requirements and chemical safety assessment, Chapter R.7a: Endpoint specific guidance (Pokyny ohledně požadavků na informace a pro posouzení chemické bezpečnosti, kapitola R.7a: Pokyny pro jednotlivé ukazatele) http://echa.europa.eu/documents/10162/13632/information_requirements_r7a_en.pdf
(5) Statistikové, kteří používají metodu modelování, např. pomocí obecných lineárních modelů (GLM), mohou k této analýze přistupovat odlišným, byť srovnatelným způsobem, ale nebudou nutně odvozovat tradiční tabulku ANOVA, což se datuje zpět k algoritmickým postupům statistických výpočtů vyvinutým před érou počítačů.
(6) Statistikové, kteří používají metodu modelování, např. pomocí obecných lineárních modelů (GLM), mohou k této analýze přistupovat různým, byť srovnatelným způsobem, ale nebudou nutně odvozovat tradiční tabulku ANOVA, což se datuje zpět k algoritmickým postupům k výpočtu statistik vyvinutým před érou počítačů.
(7) Nařízení Evropského parlamentu a Rady (ES) č. 1272/2008 ze dne 16. prosince 2008 o klasifikaci, označování a balení látek a směsí, o změně a zrušení směrnic 67/548/EHS a 1999/45/ES a o změně nařízení (ES) č. 1907/2006, Úř. věst. L 353, 31.12.2008, s. 1.
(8) Chemické třídy byly přiděleny každé zkoušené chemické látce s použitím standardního klasifikačníhoschématu na základě klasifikačního systému Národní knihovny názvů léků a léčivých látek (MeSH)(dostupný na: http://www.nlm.nih.gov/mesh).
(9) Založena na oční zkoušce in vivo na králících (OECD TG 405) (17) a s použitím GHS OSN (4).
(10) Klasifikace jako 2A nebo 2B závisí na interpretaci kritéria GHS OSN pro rozlišování mezi těmito dvěmakategoriemi, tj. 1 ze 3 versus 2 ze 3 zvířat s účinky v den 7 nezbytnými pro stanovení klasifikace v kategorii2A Studie in vivo byla provedena na 3 zvířatech. Všechny sledované vlastnosti kromě zčervenání spojivek ujednoho zvířete se ke dni 7 nebo dříve vrátily zpět na nulovou hodnotu. Toto jedno zvíře, které se ke dni 7zcela neuzdravilo, mělo hodnotu zčervenání spojivek 1 (v den 7) a plně se uzdravilo v den 10.
(11) Poskytnuté rozměry vycházejí z držáku rohovek, který se používá u krav ve věku od 12 do 60 měsíců. V případě, že se používají zvířata ve věku od 6 do 12 měsíců, držák by musel být zkonstruován tak, aby každá komora měla objem 4 ml a aby každá vnitřní komora měla průměr 1,5 cm a hloubku 2,2 cm. U každého nově zkonstruovaného držáku rohovek je velmi důležité, aby poměr exponované plochy povrchu rohovky k objemu zadní komory byl stejný jako poměr v tradičním držáku rohovek. Je to nutné k zajištění, že hodnoty propustnosti budou stanoveny správně pro výpočet IVIS za pomoci navrženého vzorce.
(12) Nařízení Evropského parlamentu a Rady (ES) č. 1272/2008 ze dne 16. prosince 2008 o klasifikaci, označování a balení látek a směsí, o změně a zrušení směrnic 67/548/EHS a 1999/45/ES a o změně nařízení (ES) č. 1907/2006, Úř. věst. L 353, 31.12.2008, s. 1.
(*2) Nejvyšší střední hodnota zjištěná v kterémkoli časovém bodě.
(*3) Maximální střední hodnota zjištěná v kterémkoli časovém bodě (na základě hodnot zákalu uvedených v tabulce 1).
(*4) Na základě hodnot uvedených v tabulce 2.
(*5) Výskyt těchto kombinací je méně pravděpodobný.
(13) Chemické třídy byly přiděleny každé zkoušené chemické látce s použitím standardního klasifikačního schématu na základě klasifikačního systému Národní knihovny názvů léků a léčivých látek (MeSH) (dostupný na: http://www.nlm.nih.gov/mesh).
(14) Založena na oční zkoušce in vivo na králících (Pokyn OECD (TG) č. 405) a s použitím GHS OSN (4) (6).
(15) Na základě výsledků ICE uvedených v tabulce 6.
(16) Kombinace jiných hodnot ICE, než jsou hodnoty uvedené v tabulce 6 pro identifikaci chemických látek bez kategorie GHS a chemických látek kategorie 1 GHS (viz tabulka 6).
(17) Klasifikace v kategorii 2A nebo 2B závisí na interpretaci kritéria GHS OSN pro rozlišování mezi těmito dvěma kategoriemi, tj. 1 ze 3 versus 2 ze 3 zvířat s účinky v den 7 nezbytnými pro stanovení klasifikace v kategorii 2A. Studie in vivo byla provedena na 3 zvířatech. Všechny sledované vlastnosti kromě zčervenání spojivek u jednoho zvířete se ke dni 7 nebo dříve vrátily zpět na nulovou hodnotu. Toto jedno zvíře, které se ke dni 7 zcela neuzdravilo, mělo hodnotu zčervenání spojivek 1 (v den 7) a plně se uzdravilo v den 10.
(18) Nařízení Evropského parlamentu a Rady (ES) č. 1272/2008 ze dne 16. prosince 2008 o klasifikaci, označování a balení látek a směsí, o změně a zrušení směrnic 67/548/EHS a 1999/45/ES a o změně nařízení (ES) č. 1907/2006, Úř. věst. L 353, 31.12.2008, s. 1.
(19) Střední hodnotou se v celém tomto dokumentu rozumí aritmetický průměr.
(20) Tyto údaje vyjadřují statisticky odvozené prahové hodnoty a nesouvisí s přesností měření.
(21) Předpověď DPRA by měla být zvažována v rámci přístupu IATA a v souladu s ustanoveními odstavců 9 a12.
(22) Tyto údaje vyjadřují statisticky odvozené prahové hodnoty a nesouvisí s přesností měření.
(23) Předpověď DPRA by měla být zvažována v rámci přístupu IATA a v souladu s ustanoveními odstavců 9 a 12.
(24) Předpovědi nebezpečnosti (a potence) in vivo vycházejí z údajů ze zkoušek LLNA (19). Potence in vivo je odvozena s použitím kritérií navržených střediskem ECETOC (23).
(25) Předpověď DPRA by měla být zvažována v rámci přístupu IATA a v souladu s ustanoveními odstavců 9 a 11.
(26) Rozpětí stanovená na základě alespoň 10 hodnot úbytku získaných 6 nezávislými laboratořemi.
(27) Nařízení Evropského parlamentu a Rady (ES) č. 1272/2008 ze dne 16. prosince 2008 o klasifikaci, označování a balení látek a směsí, o změně a zrušení směrnic 67/548/EHS a 1999/45/ES a o změně nařízení (ES) č. 1907/2006, Úř. věst. L 353, 31.12.2008, s. 1.
(28) Předpovědi nebezpečnosti (a potence) in vivo vycházejí z údajů ze zkoušek LLNA (13). Potence in vivo je odvozena použitím kritérií střediskem ECETOC (24).
(29) Předpověď KeratinoSens™ by měla být hodnocena v rámci přístupu IATA a v souladu s ustanoveními odstavců 9 a 11 této zkušební metody.
(30) Na základě dosavadních zaznamenaných hodnot (12).
(31) Nařízení Evropského parlamentu a Rady (ES) č. 1272/2008 ze dne 16. prosince 2008 o klasifikaci, označování a balení látek a směsí, o změně a zrušení směrnic 67/548/EHS a 1999/45/ES a o změně nařízení (ES) č. 1907/2006, Úř. věst. L 353, 31.12.2008, s. 1.
(32) Chemické třídy byly přiděleny každé zkoušené chemické látce s použitím standardního klasifikačního schématu na základě klasifikačního systému Národní knihovny názvů léků a léčivých látek (MeSH) (dostupný na: http://www.nlm.nih.gov/mesh).
(33) Založena na výsledcích oční zkoušky in vivo na králících (Pokyn OECD pro zkoušení č. 405, zkušební metoda B.5) a s použitím klasifikace podle GHS OSN a CLP EU.
(34) Na základě výsledků získaných zkušební metodou FL (INVITTOX protokolu č. 71(6)).
(35) Statistikové, kteří používají metodu modelování, např. s použitím obecných lineárních modelů (GLM), mohou k této analýze přistupovat různým, byť srovnatelným způsobem, ale nebudou nutně odvozovat tradiční tabulku ANOVA, která se datuje zpět k algoritmickým metodám výpočtu statistických údajů vyvinutým před érou počítačů.
(36) Definice a jednotky viz dodatek 1.
(37) Někdy se označuje jako P OW; stanovuje se metodou třepací láhve popsanou ve zkušební metodě A.8 (4), metodou vysokoúčinné kapalinové chromatografie (HPLC) popsanou ve zkušební metodě A.24 (5) a metodou pomalého míchání popsanou ve zkušební metodě A.23 (6). Ke stanovení hodnoty log KOW.se někdy používá metoda generátorových kolon. K dispozici je jen omezený počet studií, které využívají tuto metodu, a to především pro chlorované bifenyly a dibenzodioxiny (např. Li and Doucette, 1993) (3). V případě látek, které by mohly ionizovat, by se log K OW měl vztahovat na nedisociovanou formu.
(38) Definice a jednotky viz dodatek 1.
(39) TLC: chromatografie na tenké vrstvě; HPLC: vysokotlaká kapalinová chromatografie; GC: plynová chromatografie
(40) V některých regulačních rámcích může být analýza metabolitů povinná, jsou-li splněny určité podmínky (srov. odstavec 65).
(41) Obecně by měřené koncentrace ve vodě během fáze příjmu měly být alespoň o jeden řád větší než mez kvantifikace, aby ve vylučovací fázi bylo možné měřit více než jeden poločas obsahu látky v organismu.
(42) Definice a jednotky viz dodatek 1.
(43) U většiny zkoušených látek by v ideálním případě neměly být ve vodě kontrolní skupiny zaznamenány žádné stopy zkoušené látky. Koncentrace v pozadí by se měly týkat pouze materiálů vyskytujících se v přírodě (např. některých kovů) a látek, které jsou všudypřítomné v životním prostředí.
(44) Nejedná-li se zjevně o kinetiku prvního řádu, měly by být použity složitější modely (viz odkazy v dodatku 5) a měl by být konzultován biostatistik.
(45) Ve zkoušce biokoncentrace může být příjem kvůli nízké rozpustnosti ve vodě omezen na nízké expoziční koncentrace, zatímco ve zkoušce expozice v potravě lze dosáhnout mnohem vyšších expozičních koncentrací.
(46) U vícesložkových látek, UVCB a směsí by pro stanovení vhodných expozičních koncentrací měla být posouzena rozpustnost každé významné složky ve vodě.
(47) TOC zahrnuje organický uhlík z částic a rozpuštěný organický uhlík, tj. TOC = POC + DOC.
(48) Ačkoli se to obecně nedoporučuje, je-li rozpouštědlo nebo solubilizační činidlo použito, měl by být pro zhodnocení koncentrace organického uhlíku ve zkušebních nádržích organický uhlík pocházející z tohoto činidla připočten k organickému uhlíku ze zkoušené látky.
(49) Není-li obsah lipidů analyzován u stejných ryb jako zkoušená látka, měly by mít ryby alespoň podobnou hmotnost a (je-li to podstatné) měly by být stejného pohlaví.
(50) Tato alternativa je platná pouze v případě, že ryby jsou ve všech zkušebních skupinách chovány v podobném počtu, ryby se odebírají stejným způsobem a jsou stejně krmeny. To zajistí, aby růst ryb ve všech zkušebních skupinách byl podobný, pokud je zkoušená koncentrace nižší než rozpětí toxicity. Je-li růst podobný, lze očekávat, že také obsah lipidů bude podobný. Odlišný růst v kontrole by nasvědčoval účinku látky a studii by zneplatnil.
(51) Kromě hmotnosti by se měla zaznamenat celková délka, protože porovnání míry nárůstu délky během zkoušky je dobrým ukazatelem toho, zda došlo k nepříznivému účinku.
(52) Je možné provést t-test rychlostní konstanty růstu za účelem, aby se prozkoumalo, zda se liší růst mezi kontrolními a zkušebními skupinami, F-test v případě analýzy rozptylu. V případě potřeby lze použít F-test nebo test poměru pravděpodobnosti jako pomůcku při výběru vhodného růstového modelu (monografie OECD 54 (32)).
(53) Tyto procentuální hodnoty předpokládají, že analytické metody jsou spolehlivé a že poločas je < 14 dnů. Pokud jsou analytické metody méně spolehlivé nebo pokud se poločas (výrazně) prodloužil, budou tato čísla větší.
CL: interval spolehlivosti (pokud je možné ho odhadnout).
SD: směrodatná odchylka (pokud je možné ji odhadnout).
(56) Minimalizovaná zkouška může být ve skutečnosti použita k prokázání rychlého metabolismu, pokud je známo, že rychlý metabolismus je pravděpodobný.
(57) Jsou-li měřeny pouze dva odběry vzorků, lze odhad intervalů spolehlivosti BCFKm provést s použitím pomocí metod postupného výpočtu (bootstrap). Pokud jsou rovněž k dispozici údaje z mezilehlých odběrů, lze intervaly spolehlivosti BCFKm vypočítat stejně jako v úplné zkoušce.
(58) Definice a jednotky viz dodatek 1.
(59) Pro účely nařízení (ES) č. 1907/2006 o registraci, hodnocení, povolování a omezování chemických látek (REACH) (Úř. věst. L 396, 30.12.2006, s. 1) se o této otázce pojednává v „Pokynech ohledně požadavků na informace a posuzování chemické bezpečnosti“, kapitoly R.7c, R.7.10.3.1; R.7.10.4.1 a obrázek R7.10–2.
(60) U většiny zkoušených látek by v ideálním případě neměly být ve vodě kontrolní skupiny žádné nálezy. Koncentrace v pozadí by se měly týkat pouze materiálů vyskytujících se v přírodě (např. některých kovů) a látek, které jsou všudypřítomné v životním prostředí.
(61) Jelikož je BMF definován jako poměr koncentrace látky v organismu a koncentrace látky v potravě podávané tomuto organismu v rovnovážném stavu, berou se v úvahu lipidy provedením úpravy o obsah lipidů v organismu a v potravě, a tento postup se tudíž přesněji označuje jako „úprava“. Tento postup se liší od „normalizace“ na stanovený obsah lipidů v organismu, která se provádí při zkoušce bioakumulace s expozicí ve vodním prostředí.
(62) Celková délka by se měla zaznamenávat i v průběhu zkoušky, neboť je to dobrý ukazatel toho, zda nenastal nějaký nepříznivý účinek.
(63) HCB je uveden v přílohách A a C Stockholmské úmluvy a v přílohách I a III nařízení (ES) č. 850/2004 o perzistentních organických znečišťujících látkách (Úř. věst. L 158, 30.4.2004, s. 7).
(64) V důsledku rychlého růstu během fáze příjmu se sníží skutečné množství potravy pod úroveň původně stanovenou na začátku expozice.
(65) Ve studii expozice ve vodním prostředí by 14denní poločas odpovídal hodnotě BCF cca 10 000 l/kg při použití ryb o hmotnosti 1 g s odpovídající rychlostní konstantou příjmu asi 500 l/kg/den (podle rovnice Sijma et al (46)).
(66) Poněvadž skutečnou vnitřní koncentraci je možné stanovit teprve po provedení zkoušky, je zapotřebí odhad předpokládané vnitřní koncentrace (založený např. na očekávané hodnotě BMF a na koncentraci v potravě; viz rovnice A5.8 v dodatku 5).
(67) Přítomnosti zkoušené látky ve zkušebním médiu v důsledku vyměšování ryb nebo vyplachování z potravy možná nebude možné zcela zabránit. Jednou z možností je změřit koncentraci látky ve vodě na konci fáze příjmu, zejména pokud je použito semistatické uspořádání, což pomůže stanovit, zda došlo k expozici ve vodním prostředí, či nikoli.
(68) Tento postup je specifický pro studii potravy na rozdíl od postupu, kterým se řídí zkouška expozice ve vodním prostředí, a proto byl použit výraz „úprava“, a nikoli „normalizace“, aby se předešlo zmatení pojmů – viz též poznámku pod čarou v odstavci 106.
(69) Je možné provést t-test rychlostní konstanty růstu za účelem zkoumání, zda se liší růst mezi kontrolní a zkušební skupinou, nebo F-test v případě analýzy rozptylu. V případě potřeby lze použít F-test nebo test poměru pravděpodobnosti jako pomůcku při výběru vhodného růstového modelu (monografie OECD 54 (32)).
(70) Ve volné přírodě k největší expozici ve vodním prostředí pravděpodobně dochází prostřednictvím pojídání velmi hydrofobních látek, takže odhadnutý BCF není striktně reprezentativní pro vyjádření bioakumulačního potenciálu takové látky.
CL: interval spolehlivosti (pokud je možné ho odhadnout).
SD: směrodatná odchylka (pokud je možné ji odhadnout).
(73) Meyer et al. (1)
(74) Je třeba poznamenat, že v samotné zkoušce se upřednostňuje hmotnost jako měřítko změn rychlostních konstant velikosti a růstu. Uznává se však, že praktičtější je poměřovat ryby délkou, pokud mají být na začátku pokusu vybírány vizuálně (tj. z kmenové populace).
(75) Toto délkové rozpětí je uvedeno v Metodách zkoušení nových chemických látek atd., které vycházejí z japonského zákona o kontrole chemických látek.
(76) Hodnoty v závorkách jsou počty vzorků (vody, ryb), které mají být odebrány, je-li prováděn dodatečný odběr.
(77) Předběžný odhad k2 pro log KOW = 4,0 je 0,652/den. Celková délka experimentu je stanovena na 3 × tSS = 3 × 4,6 dnů, tj. 14 dnů. Odhad hodnoty tSS viz dodatek 5.
(78) Odběr vzorku vody po dodání alespoň tří „objemů nádrže“.
(79) Tyto ryby se odebírají z kmenové populace.
(80) Je-li nutná větší přesnost nebo studie metabolismu, jež vyžadují více ryb, měly by se tyto ryby (odebírat zejména na konci fáze příjmu a fáze vylučování (srov. odstavec 40).
(81) Pro analýzu obsahu lipidů mohou být zapotřebí přinejmenším další 3 ryby, není-li možné použít tytéž ryby odebrané za účelem měření koncentrací látky na začátku zkoušky, na konci fáze příjmu a na konci fáze vylučování. Poznámka: V mnoha případech by mělo být možné použít pouze 3 ryby z kontrolní skupiny (srov. odstavec 56).
(82) Po 3 vzorcích potravy z kontrolní a exponovaní skupiny, na kterých se analyzují koncentrace zkoušené látky a obsah lipidů.
(83) Ryby se odebírají z kmenové populace co nejblíže k zahájení studie; alespoň 3 ryby z kmenové populace by na začátku zkoušky měly být odebrány za účelem analýzy obsahu lipidů.
(84) (Nepovinný) odběr vzorků v rané fázi příjmu poskytne údaje k výpočtu asimilace zkoušené látky podávané v potravě, které lze porovnat s účinností asimilace vypočtenou z údajů o fázi vylučování.
(85) 5 ryb navíc může být odebráno k analýze jednotlivých tkání.
(86) Pro analýzu obsahu lipidů mohou být zapotřebí přinejmenším další 3 ryby, není-li možné použít tytéž ryby odebrané za účelem měření koncentrací látky na začátku zkoušky, na konci fáze příjmu a na konci fáze vylučování. Poznámka: V mnoha případech by mělo být možné použít pouze 3 kontrolní ryby (srov. odstavce 56 a 153).
(87) Jako u každého empirického vztahu i zde by se mělo ověřit, zda zkoušená látka spadá do oblasti použitelnosti daného vztahu.
(88) Hmotnost ryb na konci fáze příjmu lze odhadnout z údajů v dřívějších studiích nebo na základě znalosti pravděpodobného růstu daného zkušebního druhu z typické hmotnosti při zahájení zkoušky během typické délky fáze příjmu (např. 28 dnů).
(89) Ve většině programů, které umožňují provádět lineární regresi, jsou uváděny také směrodatné chyby a interval spolehlivosti (CI) odhadů, např. v programu Microsoft Excel s použitím souboru nástrojů Data Analysis.
(90) Na rozdíl od metody lineární regrese se pomocí tohoto vzorce nezíská směrodatná chyba hodnoty k2 .
(91) Na rozdíl od postupu linearizace se pomocí této metody obvykle nezíská směrodatná chyba nebo interval spolehlivosti odhadované hodnoty k1 .
(92) Je třeba si uvědomit, že nejistota při odhadu hodnoty k2 není v bioakumulačním modelu používána správně, je-li tento model při stanovení k1 v rámci metody postupného stanovení v podstatě považován za konstantní. Výsledná nejistota BCF při uplatnění metod současného a postupného stanovení bude proto různá.
(93) V některých regionech může být k dostání pouze rybí potrava s koncentrací lipidů, která je mnohem menší než tato horní mez. V takových případech by bylo možné provést studie s nižší koncentrací lipidů v potravě, tak jak je dodávána, a vhodně upravit množství potravy, aby se ryby udržely v dobrém zdravotním stavu. Lipidy v potravě by se neměly uměle zvyšovat přidáním nadbytečného oleje.
(94) Ve volné přírodě k největší expozici ve vodním prostředí pravděpodobně dochází prostřednictvím pojídání velmi hydrofobních látek, takže odhadnutý BCF není striktně reprezentativní pro vyjádření bioakumulačního potenciálu takové látky.
(95) Jiné dafnie mohou být použity, pokud splňují příslušná kritéria validity (kritérium validity týkající se reprodukční schopnosti kontrolních skupin by mělo být relevantní pro všechny druhy). Použijí-li se jiné druhy dafnií, musí být jasně identifikovány a jejich použití musí být zdůvodněno.
(96) Náhodná mortalita: mortalita, která nesouvisí s chemickou látkou a je vyvolaná nahodilými okolnostmi (tj. známou příčinou).
(97) Bezděčná mortalita: mortalita, která nesouvisí s chemickou látkou a nemá známou příčinu.
(*6) Uvede se, ve které nádobě bylo měření provedeno.
(*7) Do příslušné buňky se zaznamenají nevylíhnuté plůdky jako „AB“ (aborted broods).
(*8) Do příslušné buňky se zaznamená mortalita rodičovských organismů jako „M“.
(98) Typická minimální střední celková délka není kritériem validity, ale odchylky pod uvedenou hodnotou by měly být pečlivě zkoumány z hlediska citlivosti zkoušky. Minimální střední celková délka je odvozena od souboru údajů, které jsou v současnosti dostupné.
(99) Konkrétní zkušební kmen pstruha duhového může vyžadovat použití jiných teplot. Matečné ryby musí být udržovány při stejné teplotě, jako je teplota používaná pro jikry. Po získání jiker od komerčního chovatele je nezbytná krátká adaptace na zkušební teplotu (např. 1 až 2 hodiny) po přejímce.
(100) Tma pro plůdky do jednoho týdne po vylíhnutí kromě doby, kdy jsou kontrolovány, poté se po dobu zkoušky vystaví osvětlení (fotoperioda 12–16 hodin) (4).
(101) Světelný režim by při jakýchkoli stanovených zkušebních podmínkách měl být konstantní.
(102) Při jakékoli dané zkoušce musí být tato hodnota dodržena s přesností na ± 20/00.
(*9) Potrava by měla být podávána do sytosti. Zbytky potravy a výkaly se, je-li to nutné, odstraní, aby nedošlo k hromadění odpadu.
|
FBS |
zmrazené žábronožky solné; dospělé Artemia sp |
|
BSN |
larvy (nauplia) žábronožky solné; čerstvě vylíhnuté |
|
BSN48 |
larvy (nauplia) žábronožky solné; 48 hodin staré |
(1) plůdky se žloutkovým váčkem krmení nevyžadují
(2) filtrované ze smíšené kultury
(3) granule z fermentačního procesu
(*10) Bradavkovité výběžky se obvykle objevují pouze u dospělých samečků a nacházejí se na paprscích ploutví, a to od druhého do sedmého nebo osmého paprsku od posteriorního konce řitní ploutve (obrázek 1 a 2). Na prvním ploutevním paprsku od posteriorního konce řitní ploutve se výběžky vyskytují jen zřídka. Uvedené standardní operační postupy zahrnují měření výběžků na prvním ploutevním paprsku (pořadové číslo ploutevního paprsku v tomto standardním operačním postupu označuje pořadí od posteriorního konce řitní ploutve).
(*11) Homogenizační pufr:
|
— |
(50 mM Tris-HCl pH 7,4; 1 % koktejl inhibitoru proteázy (Sigma)): 12 ml Tris-HCl pH 7,4 + 120 μl koktejlu inhibitoru proteázy. |
|
— |
TRIS: TRIS-ULTRA PURE (ICN), např. od výrobce Bie & Berntsen, Dánsko. |
|
— |
Koktejl inhibitoru proteázy: od výrobce Sigma (pro tkáně savců), produktové číslo P 8340. |
POZNÁMKA: Homogenizační pufr by měl být použit ve stejný den, kdy byl vyroben. Během použití se má uchovávat na ledu.
(103) Carl von Linné (* 23. května 1707 v Råshultu u Älmhultu; † 10. ledna 1778 v Uppsale).
(*12) Demineralizovaná (tj. destilovaná nebo deionizovaná) voda.