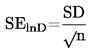ISSN 1830-3617
doi:10.3000/18303617.L_2009.220.bul
Официален вестник
на Европейския съюз
L 220

Издание на български език
Законодателство
Година 52
24 август 2009 г.
|
ISSN 1830-3617 doi:10.3000/18303617.L_2009.220.bul |
||
|
Официален вестник на Европейския съюз |
L 220 |
|
|
|
||
|
Издание на български език |
Законодателство |
Година 52 |
|
Съдържание |
|
I Актове, приети по силата на Договорите за ЕО/Евратом, чието публикуване е задължително |
Страница |
|
|
|
РЕГЛАМЕНТИ |
|
|
|
* |
Регламент (ЕО) № 761/2009 на Комисията от 23 юли 2009 година за изменение с цел адаптиране към техническия прогрес на Регламент (ЕО) № 440/2008 за определяне на методи за изпитване в съответствие с Регламент (ЕО) № 1907/2006 на Европейския парламент и на Съвета относно регистрацията, оценката, разрешаването и ограничаването на химикали (REACH) ( 1 ) |
|
|
|
|
|
(1) текст от значение за ЕИП |
|
BG |
Актовете, чиито заглавия се отпечатват с нормален шрифт, са актове по текущо управление на селскостопанската политика и имат кратък срок на действие. Заглавията на всички останали актове се отпечатват с удебелен шрифт и се предшестват от звезда. |
I Актове, приети по силата на Договорите за ЕО/Евратом, чието публикуване е задължително
РЕГЛАМЕНТИ
|
24.8.2009 |
BG |
Официален вестник на Европейския съюз |
L 220/1 |
РЕГЛАМЕНТ (ЕО) № 761/2009 НА КОМИСИЯТА
от 23 юли 2009 година
за изменение с цел адаптиране към техническия прогрес на Регламент (ЕО) № 440/2008 за определяне на методи за изпитване в съответствие с Регламент (ЕО) № 1907/2006 на Европейския парламент и на Съвета относно регистрацията, оценката, разрешаването и ограничаването на химикали (REACH)
(текст от значение за ЕИП)
КОМИСИЯТА НА ЕВРОПЕЙСКИТЕ ОБЩНОСТИ,
като взе предвид Договора за създаване на Европейската общност,
като взе предвид Регламент (EО) № 1907/2006 на Европейския парламент и на Съвета от 18 декември 2006 г. относно регистрацията, оценката, разрешаването и ограничаването на химикали (REACH), за създаване на Европейска агенция по химикали, за изменение на Директива 1999/45/EО и за отмяна на Регламент (EИО) № 793/93 на Съвета и на Регламент (EО) № 1488/94 на Комисията, както и на Директива 76/769/EИО на Съвета и Директиви 91/155/EИО, 93/67/EИО, 93/105/EО и 2000/21/EО на Комисията (1), и по-специално член 13, параграф 3 от него,
като има предвид, че:
|
(1) |
Регламент (ЕО) № 440/2008 на Комисията (2) съдържа методите за изпитване с цел определяне на физико-химическите свойства, токсичността и екотоксичността на вещества, които се използват за целите на Регламент (ЕО) № 1907/2006. |
|
(2) |
Необходимо е да се актуализира Регламент (ЕО) № 440/2008, за да се включат в него промени в някои от методите за изпитване, както и множество нови методи за изпитване, приети от ОИСР. Заинтересованите страни са консултирани по това предложение. Посочените изменения адаптират въпросните методи към научно-техническия прогрес. |
|
(3) |
Разпоредбите относно парното налягане следва да бъдат преразгледани във връзка с включването на новия ефузионен метод. |
|
(4) |
Необходимо е да бъде включен нов метод за измерване на претегления по дължина средногеометричен диаметър на влакна (length weighted geometric mean diameter of fibres). |
|
(5) |
Подходящо е да се актуализира Регламент (ЕО) № 440/2008, за да се включи приоритетно нов метод за изпитване in vitro за кожно дразнене, с цел да се намали до минимум броят на използваните за опитни цели животни в съответствие с Директива 86/609/ЕИО на Съвета от 24 ноември 1986 г. за сближаване на законовите, подзаконовите и административните разпоредби на държавите-членки относно защитата на животните, използвани за опитни и други научни цели (3). Въпреки че един проектометод за изпитване in vitro за кожно дразнене все още е в процес на обсъждане в рамките на ОИСР, е уместно в разглеждания изключителен случай методът В 46 да бъде включен в посочения регламент. Методът В 46 следва да бъде актуализиран възможно най-скоро след като бъде постигнато споразумение в рамките на ОИСР или ако се появи допълнителна информация, обосноваваща такова преразглеждане. |
|
(6) |
Разпоредбите във връзка с изпитването чрез инхибиране на водорасли трябва да бъдат преразгледани, за да се включат в тях допълнителни видове и да се изпълнят изискванията по отношение на оценката на риска и класифицирането на химикали. |
|
(7) |
Необходимо е да се добави нов метод за измерване на аеробната минерализация на повърхностни води чрез симулативно изпитване за биоразграждане и нов метод за оценка на токсичността по отношение на рода Lemna посредством изпитване чрез инхибиране на растежа. |
|
(8) |
Регламент (ЕО) № 440/2008 следва да бъде съответно изменен. |
|
(9) |
Мерките, предвидени в настоящия регламент, са в съответствие със становището на комитета, създаден съгласно член 133 от Регламент (ЕО) № 1907/2006, |
ПРИЕ НАСТОЯЩИЯ РЕГЛАМЕНТ:
Член 1
Приложението към Регламент (ЕО) № 440/2008 се изменя, както следва:
|
1. |
Част А се изменя, както следва:
|
|
2. |
Част Б се изменя, както следва: Добавя се глава Б.46, представена в приложение III към настоящия регламент. |
|
3. |
Част В се изменя, както следва:
|
Член 2
Настоящият регламент влиза в сила на третия ден след публикуването му в Официален вестник на Европейския съюз.
Настоящият регламент е задължителен в своята цялост и се прилага пряко във всички държави-членки.
Съставено в Брюксел на 23 юли 2009 година.
За Комисията
Stavros DIMAS
Член на Комисията
(1) ОВ L 396, 30.12.2006 г., стр. 1.
(2) ОВ L 142, 31.5.2008 г., стр. 1.
(3) ОВ L 358, 18.12.1986 г., стр. 1.
ПРИЛОЖЕНИЕ I
|
A.4. |
НАЛЯГАНЕ НА ПАРИТЕ |
1. МЕТОД
Този метод е еквивалентен на OECD TG 104 (2004).
1.1. ВЪВЕДЕНИЕ
Този преразгледан вариант на метод А.4 (1) включва един допълнителен метод; ефузионен метод: изотермична термогравиметрия, предназначена за вещества с много ниски налягания на парите (до 10–10 Pa). С оглед на необходимостта от методика, по-специално по отношение на определянето на налягането на парите при вещества с ниско налягане на парите, се оценяват още веднъж други методики за този метод по отношение на други интервали на приложимост.
В условията на термодинамично равновесие налягането на парите на чистото вещество зависи само от температурата. Основните принципи са описани в (2) и (3).
Единствена методика за измерване, приложима за целия обхват на налягания на парите от под 10–10 до 105 Pa, не съществува. Този метод включва осем метода за измерване на налягането на парите, които могат да се прилагат в различни обхвати от парни налягания. В таблица 1 различните методи са сравнени по отношение на обхватите на прилагането им и измерването. Методите могат да бъдат прилагани за съединения, които не се разлагат при условията на изследването. В случаите, в които експерименталните методи не могат да бъдат приложени по технически причини, налягането на парите може да бъде оценено, като препоръчителният метод за оценка е даден в допълнението.
1.2. ОПРЕДЕЛЕНИЯ И МЕРНИ ЕДИНИЦИ
Налягане на парите на вещество е налягането на наситените пари над твърдо или течно вещество.
Трябва да се използва единицата за налягане по SI, която е паскал (Ра). По-долу са дадени други мерни единици, които са използвани в миналото, заедно с техните коефициенти на преобразуване:
|
1 Torr |
= |
1 mm Hg |
= |
1,333 × 102 Pa |
|
1 атмосфера |
= |
1,013 × 105 Pa |
|
|
|
1 bar |
= |
105 Pa |
|
|
Eдиницата за температура по SI е Келвин (K). Преобразуването на градуси по Целзий в келвини се извършва по формулата.
T = t + 273,15
където Т е келвиновата температура или абсолютната температура в келвини, а t е температурата, изразена в градуси по Целзий.
Таблица 1
|
Метод на измерване |
Вещества |
Оценка повторяемост |
Оценка възпроизводимост |
Препоръчителен обхват |
|
|
твърди |
течни |
||||
|
Динамичен метод |
Нискотопими |
Да |
до 25 % от 1 до 5 % |
до 25 % от 1 до 5 % |
от 103 Pa до 2 × 103 Pa от 2 × 103 Pa до 105 Pa |
|
Статичен метод |
Да |
Да |
от 5 до 10 % |
от 5 до 10 % |
от 10 Pa до 105 Pa от 10–2 Pa до 105 Pa (1) |
|
Метод на изотенископа |
Да |
Да |
от 5 до 10 % |
от 5 до 10 % |
от 102 Pa до 105 Pa |
|
Ефузионен метод: везна за измерване на налягането на парите |
Да |
Да |
от 5 до 20 % |
до 50 % |
от 10–3 до 1 Pa |
|
Ефузионен метод: Кнудсенова клетка |
Да |
Да |
от 10 до 30 % |
— |
от 10–10 до 1 P |
|
Ефузионен метод: изотермична термогравиметрия |
Да |
Да |
от 5 до 30 % |
до 50 % |
от 10–10 до 1 Pa |
|
Метод с насищане на газ |
Да |
Да |
от 10 до 30 % |
до 50 % |
от 10–10 до 103 Pa |
|
Метод с въртящ се ротор |
Да |
Да |
от 10 до 20 % |
— |
от 10–4 до 0,5 Pa |
1.3. ПРИНЦИП НА ИЗСЛЕДВАНЕТО
В общия случай налягането на парите се определя при различни температури. В ограничен температурен интервал логаритъмът от налягането на парите на дадено чисто вещество е линейна функция на реципрочната стойност на абсолютната температура в съответствие с уравнението на Клапейрон—Клаузиус:
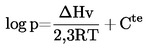
където:
|
p |
= |
налягането на парите в паскали |
|
ΔHv |
= |
скритата топлина на изпарение в J mol–1 |
|
R |
= |
универсалната газова константа, 8,314 J mol–1 K–1 |
|
T |
= |
температурата в K |
1.4. ВЕЩЕСТВА ЗА СРАВНЕНИЕ
Не е необходимо да се използват вещества за сравнение. Преди всичко те служат за периодична проверка на действието на метода, както и да позволят сравнение на резултатите, получени по различни методи.
1.5. ОПИСАНИЕ НА МЕТОДА
1.5.1. Динамичен метод (метод на Котрел)
1.5.1.1. Принцип
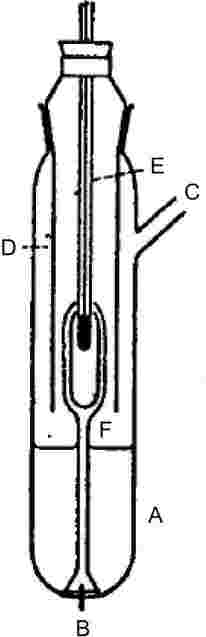
Налягането на парите се определя, като се измери температурата на кипене на веществото при няколко определени налягания, грубо между 103 и 105 Ра. Този метод се препоръчва и за определяне на температурата на кипене. Той е подходящ за тази цел до 600 K. На дълбочина от 3 до 4 cm температурите на кипене на течностите са приблизително с 0,1 °C по-високи отколкото на повърхността поради хидростатичното налягане на стълба течност. При метода на Котрел (4) термометърът е поставен в парите над повърхността на течността и кипящата течност се принуждава непрекъснато да се самоизпомпва над резервоара на термометъра. Резервоарът е покрит с тънък слой течност, който при атмосферно налягане е в равновесие с парите. Така термометърът отчита действителната температура на кипене без грешки, дължащи се на прегряване или на хидростатичното налягане. Използваната първоначално от Котрел помпа e показана на фигура 1. Кипящата течност се съдържа в тръбата А. Равномерното кипене е улеснено от платинената тел В, захваната към дъното. Страничната тръба С води към кондензатор, а обвивката D пречи на студения кондензат да достига термометъра Е. Когато течността А кипи, през двата клона на помпата F над резервоара на термометъра преминават мехурчета и течност.
|
Фигура 1
|
Фигура 2
|
Помпа на Котрел (4)
|
A: |
Термодвойка |
|
B: |
Вакуумен буферен обем |
|
C: |
Манометър |
|
D: |
Вакуум |
|
E: |
Точка на измерване |
|
F: |
Нагревател около 150 W |
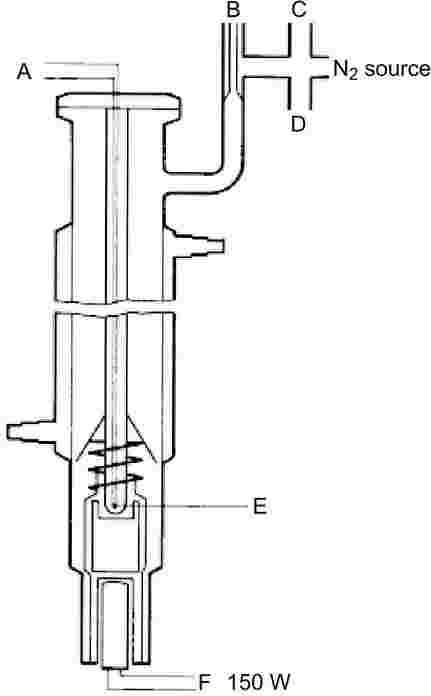
1.5.1.2. Апаратура
На фигура 2 e показан много точен уред, използващ принципа на Котрел. Той се състои от тръба със зона на кипене в долната част, охладител в средната част и изпускателен канал и фланец в горната част. Помпата на Котрел е разположена в зоната на кипене, която се нагрява с електрически елемент. Температурата се измерва с термодвойка в защитна обвивка или със съпротивителен термометър, който се вкарва през фланеца най-горе. Изпускателният канал е свързан към системата за регулиране на налягането. Последната се състои от вакуумпомпа, буферен обем, маностат за подаване на азот за регулиране на налягането и манометър.
1.5.1.3. Методика
Веществото се поставя в зоната на кипене. Проблеми могат да възникнат с веществата, които не са в прахообразно състояние, но в някои случаи те могат да се решат, като се нагрее охлаждащият кожух. Апаратът се затваря плътно с фланеца и веществото се дегазира. С този метод не могат да се измерват вещества, които образуват пяна.
След това се задава най-ниското желано налягане и се включва нагряването. В същото време температурният датчик се свързва със записващо устройство.
Равновесието е достигнато, когато при постоянно налягане се отчита една и съща температура на кипене. Трябва да се внимава много кипенето да не става на тласъци. Освен това в охладителя трябва да се извършва пълна кондензация на парите. Когато се определя налягането на парите на нискотопими твърди вещества, трябва да се вземат мерки да не се блокира кондензаторът.
След като се регистрира тази равновесна точка, се задава по-високо налягане. Процесът продължава по този начин до достигане на налягане от 105 Ра (приблизително от 5 до 10 измервателни точки общо). За проверка равновесните точки трябва да се повторят и при намаляване на налягането.
1.5.2. Статичен метод
1.5.2.1. Принцип
При статичния метод (5) налягането на парите при термодинамично равновесие се определя за дадена температура. Този метод е подходящ за вещества и многокомпонентни течности, както и за твърди тела в обхвата от 10 до 105 Pa и ако се внимава, също и в обхвата от 10–1 до 10 Pa.
1.5.2.2. Апаратура
Оборудването се състои от термостатирана вана (точност ±0,2 K), контейнер за пробата, свързан към вакуумна линия, манометър и система за регулиране на налягането. Камерата за проби (фигура 3a) е свързана към вакуумната линия през клапан и диференциален манометър (U-образна тръба, съдържаща подходяща манометрична течност), който служи за нулев индикатор. В зависимост от обхвата на налягането и химичните процеси с веществото в U-образната тръба могат да бъдат използвани живак, силикони и фталати. По екологични съображения обаче, ако е възможно, употребата на живак трябва да се избягва. Изпитваното вещество не бива да се разтваря забележимо или да реагира с течността в U-образната тръба. Вместо U-образна тръба може да се използва манометър (фигура 3б). За обхвата от нормално налягане до 102 Ра в манометъра може да се използва живак, докато силиконовите течности и фталатите са подходящи под 102 Ра до 10 Ра. Съществуват други манометри, които могат да се използват под 102 Pa, а под 10–1 Pa могат да бъдат използвани дори капацитивни манометри с нагряваща се мембрана. Температурата се измерва върху външната стена на съда, съдържащ пробата, или вътре в самия съд.
1.5.2.3. Методика
При използване на апарата в съответствие с фигура 3а U-образната тръба се напълва с избраната течност, която трябва да се дегазира при повишена температура, преди да започне отчитането. Изпитваното вещество се поставя в апарата и се дегазира при понижена температура. В случая на многокомпонентна проба температурата трябва да бъде достатъчно ниска, за да се гарантира, че съставът на материала не се променя. Равновесие може да се установи по-бързо чрез разбъркване. Пробата може да се охлажда с течен азот или сух лед, но трябва да се внимава да се избегне кондензацията на въздуха или на помпената течност. Клапанът над съда с пробата се отваря и в продължение на няколко минути се извършва засмукване, за да се отстрани въздухът. Ако е необходимо, операцията по дегазирането трябва да се повтори няколко пъти.
|
Фигура 3a
|
Фигура 3б
|
Когато пробата се нагрява при затворен клапан, налягането на парите се увеличава. Това променя равновесието на течността в U-образната тръба. За да се компенсира тази промяна, в апарата през клапан се подава азот или въздух, докато диференциалният индикатор на налягане се върне в нулево положение. Необходимото за целта налягане може да се отчете от манометъра или от измервателен уред с по-голяма точност. Това налягане съответства на налягането на парите на веществото при дадената температура на измерване. При използване на апарата от фигура 3б налягането на парите се отчита директно.
Налягането на парите се определя през подходящи малки интервали (приблизително от 5 до 10 измервателни точки общо) до достигане на желаната максимална температура.
За проверка отчитането при ниски температури трябва да се повтори. Ако стойностите, получени при това повторно отчитане, не съвпадат с кривата, получена при повишаване на температурата, това може да се дължи на една от следните причини:
|
i) |
пробата все още съдържа въздух (например при високовискозни материали) или нискокипящи вещества, които се отделят при нагряването; |
|
ii) |
веществото претърпява химична реакция в изследвания температурен интервал (например разграждане, полимеризация). |
1.5.3. Метод на изотенископа
1.5.3.1. Принцип
Изотенископът (6) се основава на принципа на статичния метод. Методът включва поставяне на проба в колба, поддържана при постоянна температура и свързана към манометър или вакуумпомпа. По-летливите примеси на веществото се отстраняват чрез дегазиране при намалена температура. Налягането на парите на пробата при избрани температури се уравновесява от известно по стойност налягане на инертен газ. Изотенископът е разработен за измерване на налягане на пари на определени течни въглеводороди, но може да се използва и за изследване на твърди вещества. Методът обикновено не е подходящ за многокомпонентни системи. Резултатите включват само малки грешки при проби, съдържащи нелетливи примеси. Препоръчителният обхват е от 102 до 105 Pa.
1.5.3.2. Апаратура
Пример за измервателен уред е показан на фигура 4. Можете да намерите пълно описание в ASTM D 2879—86 (6).
1.5.3.3. Методика
Когато се изпитват течности, самото вещество служи за флуид в диференциалния манометър. В изотенископа се поставя количество течност, достатъчно да запълни резервоара и късия клон на манометъра. Изотенископът се прикачва към система за вакуум и се евакуира, след което се напълва с азот. Евакуирането и прочистването на системата се повтарят два пъти, за да се отстрани остатъчният кислород. Така напълненият изотенископ се поставя в хоризонтално положение, така че пробата да се разстели в тънък слой в резервоара за пробата и в манометъра. Налягането на системата се намалява до 133 Ра и пробата внимателно се нагрява до прага на закипяване (отстраняване на разтворените газове). След това изотенископът се поставя в такова положение, че пробата да се върне в резервоара и да изпълни късия клон на манометъра. Налягането се поддържа на 133 Pa. Заостреният край на резервоара за пробата се нагрява на малък пламък, докато отделената от пробата пара се разшири достатъчно, за да измести част от пробата в горната част на резервоара и в клона на манометъра в посока към манометричната зона, като по този начин се създава изпълнено с пари и свободно от азот пространство. След това изотенископът се поставя в термостатирана вана и налягането на азота се регулира, докато се изравни с това на пробата. При равновесие налягането на азота е равно на налягането на парите на веществото.
Фигура 4

При твърди вещества в зависимост от обхвата от налягания и температури се използват манометрични течности, като силиконови флуиди или фталати. Дегазираната течност на манометъра се помества в едно разширение върху дългото рамо на изотенископа. След това твърдото вещество, което ще бъде изследвано, се поставя в резервоара и се дегазира при повишена температура. После изотенископът се накланя така, че течността в манометъра да навлезе в U-образната тръба.
1.5.4. Ефузионен метод: везна за измерване на налягането на парите (7)
1.5.4.1. Принцип
Проба от изпитваното вещество се загрява в малка пещ и се поставя в евакуиран стъклен звънец. Пещта е покрита с капак, в който има малки отвори с известни диаметри. Парите на веществото, които се пропускат през един от отворите, се насочват към блюдо на везна с голяма чувствителност, която също е затворена в евакуирания стъклен звънец. При някои постановки блюдото на везната се намира в хладилна камера, осигуряваща разсейване на топлината навън чрез топлопроводност, като блюдото се охлажда чрез лъчист топлообмен, така че изпусканите пари кондензират върху него. Въздействието от струята на парите създава усилие върху везните. Налягането на парите може да се намери по два начина: пряко от силата върху блюдото, а също и от скоростта на изпаряване по формулата на Херц—Кнудсен (2):
където:
|
G |
= |
скоростта на изпаряване (kg s–1 m–2) |
|
M |
= |
моларната маса (g mol–1) |
|
T |
= |
температурата (K) |
|
R |
= |
универсалната газова константа (J mol–1 K–1) |
|
P |
= |
налягането на парите (Pa) |
Препоръчителният обхват е от 10–3 до 1 Pa.
1.5.4.2. Апаратура
Основният принцип на апаратурата е показан на фигура 5.
Фигура 5

|
А: |
Опорна плоча |
F: |
Хладилна камера и охлаждаща шина |
|
B: |
Измервателен уред от магнитоелектричната система |
G: |
Изпарителна пещ |
|
C: |
Стъклен звънец |
H: |
Дюаров съд с течен азот |
|
D: |
Везна с блюдо |
I: |
Измерване на температурата на пробата |
|
E: |
Устройство за измерване на подналягане |
J: |
Изследвано вещество |
1.5.5. Ефузионен метод: Кнудсенова клетка
1.5.5.1. Принцип
Методът се основава на измерване на масата от изпитваното вещество, която изтича под формата на пари за единица време от Кнудсенова клетка (8) през микроотверстие в условията на свръхвакуум. Масата на изпуснатите пари може да се получи или като се определи загубата на маса от клетката, или чрез кондензация на парите при ниска температура и определяне на количеството на изпареното вещество, като се използва хроматографски анализ. Налягането на парите се изчислява чрез прилагане на зависимостта на Херц—Кнудсен (вж. точка 1.5.4.1) с корекционни коефициенти, които зависят от параметрите на апаратурата (9). Препоръчителният обхват е от 10 –10 до 1 Pa (10)(11)(12)(13)(14).
1.5.5.2. Апаратура
Основният принцип на апаратурата е показан на фигура 6.
Фигура 6
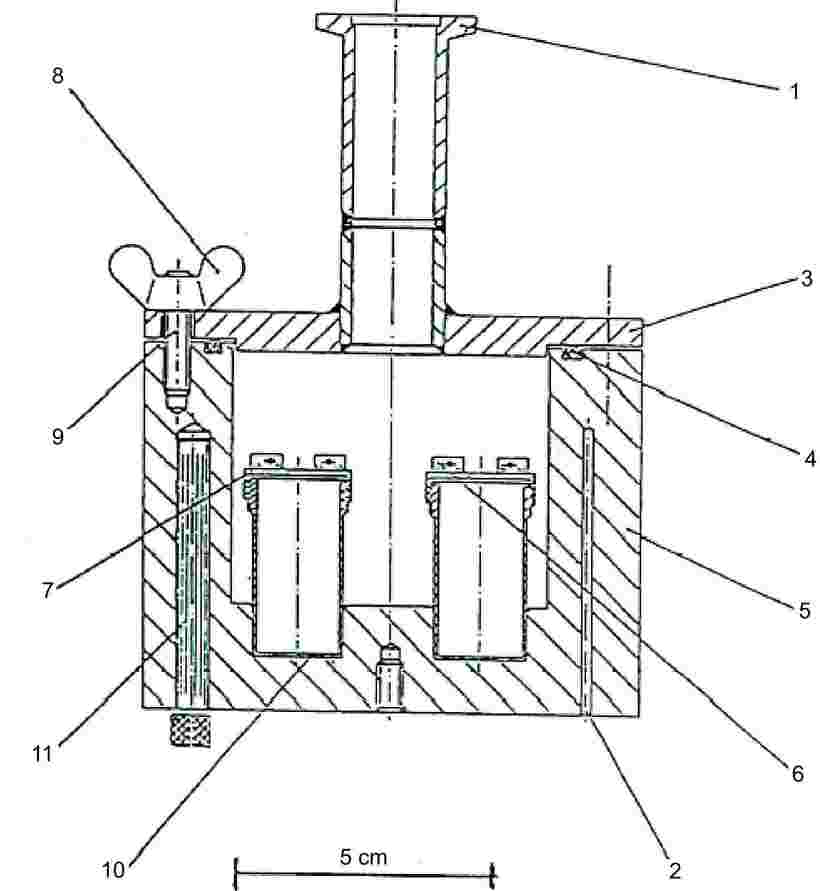
|
1: |
Връзка към вакуума |
7: |
Капак с резба |
|
2: |
Гнезда за платинов съпротивителен термометър или за следене и регулиране на температурата |
8: |
Крилчати гайки |
|
3: |
Капак за вакуумния резервоар |
9: |
Болтове |
|
4: |
О-пръстен |
10: |
Ефузионни клетки от неръждаема стомана |
|
5: |
Алуминиев вакуумен резервоар |
11: |
Нагревателен елемент |
|
6: |
Устройство за монтиране и изваждане на ефузионните клетки |
|
|
1.5.6. Ефузионен метод: изотермична термогравиметрия
1.5.6.1. Принцип
Този метод се основава на определянето на повишени скорости на изпаряване на изследваното вещество при повишени температури и атмосферно налягане чрез използване на термогравиметрия (10)(15)(16)(17)(18)(19)(20). Скоростите на изпаряване vT се определят чрез излагане на избраното съединение на бавен поток от инертен газ и следене на намаляването на теглото при определени фиксирани температури T (в келвини) през съответни интервали от време. Наляганията на парите pT се изчисляват въз основа на стойностите vT, като се използва линейната зависимост между логаритъма на налягането на парите и логаритъма на скоростта на изпаряване. Ако е необходимо, може да се извърши екстраполиране за температури 20 и 25 °C чрез регресионен анализ на log pT като функция на 1/T. Този метод е подходящ за вещества с ниски налягания на парите като 10–10 Pa (10–12 mbar) и с чистота възможно най-близка до 100 %, за да се избегне неточно тълкуване на измерените намалявания на теглото.
1.5.6.2. Апаратура
Основният принцип на опитната постановка е показан на фигура 7.
Фигура 7

Плочата с пробата, висяща на микровезна в камера с регулиране на температурата, се обдухва със струя от сух газ азот, който отнася изпарените молекули от изследваното вещество. След напускане на камерата газовата струя се пречиства в сорбционен модул.
1.5.6.3. Методика
Изследваното вещество се нанася върху повърхността на матирана стъклена плоча под формата на хомогенен слой. В случая на твърди материали плочата се намокря равномерно с разтвор на веществото в подходящ разтворител и се изсушава в инертна атмосфера. За измерването плочата с покритието се окачва в термогравиметричния анализатор, след което теглото непрекъснато се измерва във функция от времето.
Скоростта на изпаряване vT при определена температура се изчислява въз основа на намаляването на теглото Δm на плочата с пробата по формулата:

където F е площта на покритието от изследваните вещества — обикновено площта на плочата с пробата, а t е времето за намаляване на теглото с Δm.
Налягането на парите pT се изчислява въз основа на зависимостта му от скоростта на изпаряване vT:
Log pT = C + D log vT
където C и D са константи, специфични за използваната опитна постановка, които зависят от диаметъра на измервателната камера и от дебита на газа. Тези константи трябва да бъдат определени еднократно чрез измерване на набор от съединения с известно налягане на парите и регресионен анализ на log pT като функция на log vT (11)(21)(22).
Зависимостта между налягането на парите pТ и температурата в градуси по Келвин се дава от:
Log pT = A + B 1/T
където A и B са константи, които се получават чрез регресионен анализ на log pT като функция на 1/T. По тази формула налягането на парите може да бъде изчислено за всяка друга температура чрез екстраполация.
1.5.7. Метод с насищане на газ (23)
1.5.7.1. Принцип
При стайна температура и известен дебит инертен газ се пропуска през или над проба от изследваното вещество достатъчно бавно, за да се осигури насищане. Постигането на насищане в газовата фаза е от критично значение. Пренасяното вещество се улавя в общия случай чрез сорбент и се определя количеството му. Алтернатива на улавянето на парите и последващия им анализ са аналитичните методи с непрекъснат характер (като газовата хроматография), които могат да се използват за определяне на количеството на пренасяното вещество. Налягането на парите се изчислява, като се приема, че е в сила законът за идеалния газ и че общото налягане на газова смес е равно на сумата от наляганията на участващите в сместа газове. Парциалното налягане на изследваното вещество, т.е. налягането на парите, се изчислява въз основа на известния общ газов обем и теглото на пренасяното вещество.
Методиката с насищане на газ е приложима за твърди и течни вещества. Тя може да се използва за налягания на парите до минимум 10–10 Pa (10)(11)(12)(13)(14). Методът е най-надежден за налягания на парите под 103 Pa. Над 103 Pa за наляганията на парите по принцип се определят завишени стойности, вероятно поради образуването на аерозол. Тъй като измерванията на налягането на парите се извършват при стайна температура, не е необходимо екстраполиране на данни от високи температури и екстраполацията от високи температури, която често води до сериозни грешки, е избегната.
1.5.7.2. Апаратура
Методиката изисква използване на камера с постоянна температура. Скицата на фигура 3 представлява камера, съдържаща три прободържателя за твърди проби и три прободържателя за течни проби, които позволяват трикратен анализ на твърда или течна проба. Температурата се регулира с точност ±0,5 °C или по-висока.
Фигура 8

В общия случай като инертен газ носител се използва азот, но в някои случаи може да е необходимо да се използва друг газ (24). Газът носител трябва да бъде сух. Газовият поток се разделя на 6 потока, управлявани с иглени клапани (диаметър на отвора приблизително 0,79 mm), и навлиза в камерата през медна тръба с вътрешен диаметър 3,8 mm. След изравняване на температурата газът преминава през пробата и уловителя със сорбента и излиза от камерата.
Твърдите проби се зареждат в стъклена тръба с вътрешен диаметър 5 mm между запушалки от стъклена вата (вж. фигура 9). На фигура 10 е показан прободържател за течна проба и система от сорбенти. Най-възпроизводимият метод за измерване на налягането на парите на течност е да се разпредели течността върху стъклени гранули или върху инертен сорбент, като например кварц, и да се запълни прободържателят с тези гранули. Като алтернативен вариант газът носител може да бъде принуден да преминава през груба фрита и да барботира през стълб от течното вещество, обект на изследването.
|
Фигура 9
|
Фигура 10
|
Системата от сорбенти съдържа предна и спомагателна сорбентна част. При много ниски налягания на парите сорбентът задържа много малки количества и адсорбцията върху стъклената вата и стъклената тръба между пробата и сорбента може да се окаже сериозен проблем.
Охлажданите със сух лед (CO2) уловители са друг ефикасен начин за натрупване на изпареното вещество. Те не пораждат обратно налягане в колоната за насищане и също така е лесно отстраняването на уловеното вещество с определяне на количеството му.
1.5.7.3. Методика
Дебитът на изходящия газ носител се измерва при стайна температура. Дебитът се проверява често по време на опита, за да се гарантира, че общият обем на газа носител не е с точно определена стойност. Предпочита се непрекъснато следене с масов дебитомер. За насищането на газовата фаза може да е необходимо значително време на контакт и съответно доста ниски дебити на газа (25).
Накрая на опита предната и спомагателната част на сорбента се анализират поотделно. Съединението се десорбира от всяка от частите чрез добавяне на разтворител. Получените разтвори се подлагат на количествен анализ, за да се определи теглото, десорбирано от всяка от частите. Изборът на метода за анализ (както и изборът на сорбента и разтворителя за десорбцията) се определя от природата на изследваното вещество. Ефективността на десорбцията се определя, като се инжектира предварително известно количество от пробата върху сорбента, десорбира се и се анализира полученото количество. Важно е ефективността на десорбцията да се проверява при или близо до концентрацията на пробата при условията на опита.
За да се гарантира че газът носител е наситен с изследваното вещество, се използват три различни дебита на газа. Ако изчисленото налягане на парите не показва зависимост от дебита, се приема, че газът е наситен.
Налягането на парите се изчислява по формулата:

където:
|
p |
= |
налягане на парите (Pa) |
|
W |
= |
масата на изпареното вещество, обект на изследването (g) |
|
V |
= |
обема на наситения газ (m3) |
|
R |
= |
универсалната газова константа, 8,314 (J mol–1 K–1) |
|
T |
= |
температурата (K) |
|
M |
= |
моларната маса на изследваното вещество (g mol–1) |
Измерените обеми трябва да се коригират според разликите в температурата и налягането между дебитомера и колоната за насищане.
1.5.8. Въртящ се ротор
1.5.8.1. Принцип
Този метод използва измервателен уред за вискозитет с въртящ се ротор, в който измервателният елемент е малко стоманено топче, което е окачено в магнитно поле и се привежда във въртеливо движение чрез въртящо поле (26)(27)(28). Наличието на измервателни намотки позволява да се измерва скоростта му на въртене. Когато топчето достигне дадена скорост на въртене (обикновено около 400 оборота в секунда), задвижването му се прекратява и скоростта му започва да намалява поради триенето с газовете. Намаляването на скоростта на въртене се измерва като функция на времето. Налягането на парите се получава от забавянето на стоманеното топче, което зависи от налягането. Препоръчителният обхват е от 10–4 до 0,5 Pa.
1.5.8.2. Апаратура
На фигура 11 е показан схематичен чертеж на опитната постановка. Измервателната глава е поставена в термостатиран съд с регулиране на температурата с точност до 0,1 °С. Контейнерът с пробата е поставен в отделен съд, също с регулиране на температурата с точност до 0,1 °С. Всички останали части на постановката се поддържат на по-висока температура, за да се предотврати кондензиране. Целият апарат е свързан към система с висок вакуум.
Фигура 11

2. РЕЗУЛТАТИ И ПРОТОКОЛИРАНЕ
2.1. РЕЗУЛТАТИ
При всеки от описаните методи налягането на парите трябва да се определи поне за две температури. За да се провери линейността на кривата на налягането на парите, е за предпочитане да се направят три или повече измервания в интервала 0÷50 °С. В случая на ефузионния метод (кнудсенова клетка и изотермична термогравиметрия) и на метода с насищане на газ вместо 0÷50 °С се препоръчва температурен интервал на измерване 120÷150 °С.
2.2. ПРОТОКОЛ ОТ ИЗПИТВАНЕТО
Протоколът от изпитването трябва да съдържа следната информация:
|
— |
използван метод, |
|
— |
точна спецификация на веществото (вид и примеси) и предварителни етапи на пречистване, ако има такива, |
|
— |
най-малко две стойности на налягането на парите и температурата, а за предпочитане три или повече стойности, в интервала 0 ÷ 50 °С (или 120 ÷ 150 °С), |
|
— |
ако за избрания метод е технически възможно, поне една от температурите следва да бъде по-малка или равна на 25 °С, |
|
— |
всички изходни данни, |
|
— |
кривата на зависимостта на log p от 1/Т, |
|
— |
оценка за налягането на парите при 20 или 25 °С. |
Ако се наблюдава преход (промяна на агрегатното състояние, разграждане), трябва да се отбележи следната информация:
|
— |
естество на промяната, |
|
— |
температура, при която се извършва промяната при атмосферното налягане, |
|
— |
налягане на парите при 10 и 20 °С под температурата на прехода и 10 и 20 °С над тази температура (освен ако преходът не е от твърдо към газообразно състояние). |
Цялата информация и забележките, свързани с интерпретацията на резултатите, трябва да се включат в протокола, особено когато става дума за примеси или за агрегатното състояние на веществото.
3. ЛИТЕРАТУРА
|
(1) |
Официален вестник на Европейските общности L 383 А, 26—47 (1992 г.). |
|
(2) |
Ambrose, D. (1975). Experimental Thermodynamics, Vol. II, Le Neindre, B., and Vodar, B., Eds., Butterworths, London. |
|
(3) |
Weissberger R., ed. (1959). Technique of Organic Chemistry, Physical Methods of Organic Chemistry, 3rd ed., Vol. I, Part I. Chapter IX, Interscience Publ., New York. |
|
(4) |
Glasstone, S. (1946). Textbook of Physical Chemistry, 2nd ed., Van Nostrand Company, New York. |
|
(5) |
NF T 20—048 AFNOR (September 1985). Chemical products for industrial use — Determination of vapour pressure of solids and liquids within a range from 10–1 to 105 Pa — Static method. |
|
(6) |
ASTM D 2879—86, Standard test method for vapour pressure — temperature relationship and initial decomposition temperature of liquids by isoteniscope. |
|
(7) |
NF T 20—047 AFNOR (September 1985). Chemical products for industrial use –Determination of vapour pressure of solids and liquids within range from 10–3 to 1 Pa — Vapour pressure balance method. |
|
(8) |
Knudsen, M. (1909). Ann. Phys. Lpz., 29, 1979; (1911), 34, 593. |
|
(9) |
Ambrose, D., Lawrenson, I.J., Sprake, C.H.S. (1975). J. Chem. Thermodynamics 7, 1173. |
|
(10) |
Schmuckler, M.E., Barefoot, A.C., Kleier, D.A., Cobranchi, D.P. (2000), Vapor pressures of sulfonylurea herbicides; Pest Management Science 56, 521—532. |
|
(11) |
Tomlin, C.D.S. (ed.), The Pesticide Manual, Twelfth Edition (2000) |
|
(12) |
Friedrich, K., Stammbach, K., Gas chromatographic determination of small vapour pressures determination of the vapour pressures of some triazine herbicides. J. Chromatog. 16 (1964), 22—28 |
|
(13) |
Grayson, B.T., Fosbraey, L.A., Pesticide Science 16 (1982), 269—278. |
|
(14) |
Rordorf, B.F., Prediction of vapor pressures, boiling points and enthalpies of fusion for twenty-nine halogenated dibenzo-p-dioxins, Thermochimia Acta 112 Issue 1 (1987), 117—122. |
|
(15) |
Gückel, W., Synnatschke, G., Ritttig, R., A Method for Determining the Volatility of Active Ingredients Used in Plant Protection; Pesticide Science 4 (1973) 137—147. |
|
(16) |
Gückel, W., Synnatschke, G., Ritttig, R., A Method for Determining the Volatility of Active Ingredients Used in Plant Protection II. Application to Formulated Products; Pesticide Science 5 (1974) 393—400. |
|
(17) |
Gückel, W., Kaestel, R., Lewerenz, J., Synnatschke, G., A Method for Determining the Volatility of Active Ingredients Used in Plant Protection. Part III: The Temperature Relationship between Vapour Pressure and Evaporation Rate; Pesticide Science 13 (1982) 161—168. |
|
(18) |
Gückel, W., Kaestel, R., Kroehl, T., Parg, A., Methods for Determining the Vapour Pressure of Active Ingredients Used in Crop Protection. Part IV: An Improved Thermogravimetric Determination Based on Evaporation Rate; Pesticide Science 45 (1995) 27—31. |
|
(19) |
Kroehl, T., Kaestel, R., Koenig, W., Ziegler, H., Koehle, H., Parg, A., Methods for Determining the Vapour Pressure of Active Ingredients Used in Crop Protection. Part V: Thermogravimetry Combined with Solid Phase MicroExtraction (SPME); Pesticide Science, 53 (1998) 300—310. |
|
(20) |
Tesconi, M., Yalkowsky, S.H., A Novel Thermogravimetric Method for Estimating the Saturated Vapor Pressure of Low-Volatility Compounds; Journal of Pharmaceutical Science 87(12) (1998) 1512—20. |
|
(21) |
Lide, D.R. (ed.), CRC Handbook of Chemistry and Physics, 81th ed.(2000), Vapour Pressure in the Range -25 °C to 150 °C. |
|
(22) |
Meister, R.T. (ed.), Farm Chemicals Handbook, Vol. 88 (2002) |
|
(23) |
40 CFR, 796. (1993). pp 148—153, Office of the Federal Register, Washington DC |
|
(24) |
Rordorf B.F. (1985). Thermochimica Acta 85, 435. |
|
(25) |
Westcott et al. (1981). Environ. Sci. Technol. 15, 1375. |
|
(26) |
Messer G., Röhl, P., Grosse G., and Jitschin W. (1987). J. Vac. Sci. Technol. (A), 5(4), 2440. |
|
(27) |
Comsa G., Fremerey J.K., and Lindenau, B. (1980). J. Vac. Sci. Technol. 17(2), 642. |
|
(28) |
Fremerey, J.K. (1985). J. Vac. Sci. Technol. (A), 3(3), 1715. |
(1) При използване на капацитивен манометър.
Допълнение
Метод за оценка
ВЪВЕДЕНИЕ
Стойностите по оценка на налягането на парите могат да се използват:
|
— |
за да се прецени кой от експерименталните методи е подходящ, |
|
— |
за да се осигури приблизителна оценка или пределна стойност в случаите, в които експерименталният метод не може да бъде приложен по технически причини. |
МЕТОД ЗА ОЦЕНКА
Налягането на парите на течности и твърди вещества може да се изчисли, като се използва преобразуваната зависимост на Уотсън (а). Опитно е необходимо да се определи само нормалната температура на кипене. Методът е приложим в целия обхват на наляганията от 105 до 10–5 Ра.
Подробна информация за метода е дадена в „Наръчника на методите за оценка на химичните свойства“ (б). Вижте също OECD Environmental Monograph No.67 (c).
МЕТОДИКА НА ИЗЧИСЛЯВАНЕ
Налягането на парите се изчислява, както следва:

където:
|
T |
= |
температурата, за която се прави изчислението |
|
Tb |
= |
нормалната температура на кипене |
|
PVP |
= |
налягането на парите при температура Т |
|
ΔHVb |
= |
топлината на изпарение |
|
ΔZb |
= |
коефициентът на свиваемост (оценен на 0,97) |
|
m |
= |
опитно определен коефициент, зависещ от агрегатното състояние при температурата, за която се прави изчислението |
Освен това,

където KF е опитно определен коефициент, отчитащ полярността на веществото. В препратка (б) са дадени коефициентите KF за няколко типа съединения.
Доста често могат да се намерят данни, в които е дадена температурата на кипене при понижено налягане. В такъв случай налягането на парите се изчислява, както следва:
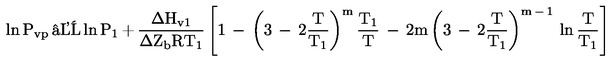
където T1 е температурата на кипене при пониженото налягане Р1.
ПРОТОКОЛ
Когато се използва метод за оценка, в протокола трябва да се включи пълна документация за изчисленията.
ЛИТЕРАТУРА
|
(а) |
Watson, K.M. (1943). Ind. Eng. Chem, 35, 398. |
|
(б) |
Lyman, W.J., Reehl, W.F., Rosenblatt, D.H. (1982). Handbook of Chemical Property Estimation Methods, McGraw-Hill. |
|
(в) |
OECD Environmental Monograph No.67. Application of Structure-Activity Relationships to the Estimation of Properties Important in Exposure Assessment (1993). |
ПРИЛОЖЕНИЕ II
|
A.22. |
ПРЕТЕГЛЕНА СПРЯМО ДЪЛЖИНАТА СРЕДНОГЕОМЕТРИЧНА СТОЙНОСТ НА ДИАМЕТЪРА НА ВЛАКНА |
1. МЕТОД
1.1. ВЪВЕДЕНИЕ
Настоящият метод описва процедура за измерване на претеглената спрямо дължината средногеометрична стойност на диаметъра (LWGMD) на насипни изкуствени минерални влакна (MMMF). Тъй като LWGMD на всички влакна попада с 95 % вероятност в 95-процентния доверителен интервал (LWGMD ± две стандартни грешки) на пробата, определената стойност (стойност от изпитването) ще бъде долната 95-процентна доверителна граница на пробата (т.е. LWGMD — 2 стандартни грешки). Методът е въз основа на актуализация (юни 1994 г.) на проект на HSE за процедура за отрасъла, съгласуван на среща между Европейската асоциация на отрасъла на керамичните влакна (ECFIA) и HSE в Честър на 26.9.1993 г. и е разработен за и от втори междулабораторен опит (1, 2). Настоящият метод за измерване може да се използва за охарактеризиране на диаметъра на влакната на насипни материали или продукти, които съдържат MMMF, включително и огнеупорни керамични влакна (RCF), изкуствени стъкловлакна (MMVF), кристални и поликристални влакна.
Претеглянето спрямо дължината е начин за компенсация на ефекта от разпределяне на диаметъра при късане на дълги влакна по време на вземане на проби или работа с материала. Геометричните статистически данни (средногеометрична стойност) се използват за измерване на разпределенията на големините на диаметрите на MMMF, тъй като тези диаметри обикновено имат разпределения на големината, които са приблизително логнормални разпределения.
Измерването на дължината и на диаметъра е както трудоемък, така и изискващ време процес, но ако бъдат измерени само онези влакна, които допират една безкрайно тънка линия от зрителното поле на сканиращ електронен микроскоп (СЕМ), тогава вероятността да бъде избрано определено влакно е пропорционална на неговата дължина. Тъй като то отчита дължината при изчисленията за претегляне спрямо дължината, единственото необходимо измерване е на диаметъра, и LWGMD–2СГ може да се изчисли, както е описано.
1.2. ОПРЕДЕЛЕНИЯ
Частица: Обект със съотношение на дължината към ширината, по-малко от 3:1.
Влакно: Обект със съотношение на дължината към ширината (стройност) най-малко 3:1.
1.3. ОБХВАТ И ОГРАНИЧЕНИЯ
Методът е предназначен да разглежда разпределения на диаметъра, когато медианният диаметър е от 0,5 μm до 6 μm. По-големи диаметри могат да се измерват с по-малко увеличение на СЕМ, но в такъв случай методът ще бъде все по-ограничителен за по-тесни разпределения на влакната, като при медианни диаметри под 0,5 μm се препоръчва измерване с трансмисионен електронен микроскоп (TEM).
1.4. ПРИНЦИП НА МЕТОДА НА ИЗПИТВАНЕ
Чрез ядково вземане на проба се вземат определен брой представителни проби от плъст от влакна или от влакна в насипно състояние. Дължината на така получените влакна се намалява чрез раздробяване и една представителна подпроба се разпръсква във вода. Вземат се аликвотни части от пробата и се прецеждат през поликарбонатен филтър с диаметър на порите 0,2 μm и се подготвят за изследване със сканиращ електронен микроскоп. Диаметрите на влакната се измерват с увеличение на екрана × 10 000 или по-голямо (1), като се използва метод на подбор в обсега на линия, който дава безпристрастна оценка на медианния диаметър. Изчислява се долният доверителен интервал от 95 % (въз основа на едностранно изпитване), за да се направи оценка за най-нискатa стойност на средногеометричния диаметър на влакното на материала.
1.5. ОПИСАНИЕ НА МЕТОДА НА ИЗПИТВАНЕ
1.5.1. Безопасност/предпазни мерки
Личното експониране на намиращи се във въздуха влакна следва да бъде сведено до минимум и при боравене със сухите влакна следва да се използва смукателен шкаф или защитна камера с ръкавици. Следва да се провежда периодичен мониторинг на личното експониране, за да се определи ефективността на методите за контрол. При боравене с MMMF следва да се използват ръкавици за еднократна употреба, за да се намали дразненето на кожата и да се избегне кръстосано замърсяване.
1.5.2. Апаратура/оборудване
|
— |
Преса и форми (способни да постигнат 10 MPa). |
|
— |
Поликарбонатни капилярни филтри (25 mm диаметър) с размер на порите 0,2 μm. |
|
— |
Мембранен филтър от целулозен естер с размер на порите 5 μm като подсигуряващ филтър. |
|
— |
Стъклен прибор за филтриране (или филтриращи системи за еднократна употреба), който да побира филтри с диаметър 25 mm (напр. стъкления комплект за микроанализи на Millipore, тип № XX10 025 00). |
|
— |
Прясно дестилирана вода, филтрирана през филтър с размер на порите 0,2 μm, за да се отстранят микроорганизмите. |
|
— |
Апарат за метализиране чрез запрашване със златна или златна/паладиева мишена. |
|
— |
Сканиращ електронен микроскоп с разделителна способност, достигаща 10 nm и работещ с увеличение × 10 000. |
|
— |
Разни: шпатули, скалпел тип 24, пинсети, тръби за СЕМ, въглеродно лепило или въглеродна залепваща лента, колоиден разтвор на сребро. |
|
— |
Ултразвукова сонда или настолна ултразвукова вана. |
|
— |
Прибор за вземане на ядкова проба или лабораторна замба за вземането на ядкови проби от плъст от MMMF. |
1.5.3. Процедура на изпитване
1.5.3.1. Взимане на проби
За плъст и кече се използва 25-милиметрова ядкова сонда или лабораторна замба, за да се вземат проби от напречното сечение. Те следва да бъдат разположени равномерно по широчината при малка дължина на плъстта или ако са налични големи дължини от плъстта, се вземат проби от произволно избрани зони. Същото оборудване може да се използва за вземане на случайни проби от свободна маса влакна. По възможност се вземат шест проби, които да отразят пространствените промени в насипния материал.
Шестте ядкови проби се раздробяват в 50-милиметрова форма при 10 MPa. Материалът се размесва с шпатула и се пресова отново при 10 МРа, след което се изважда от формата и се поставя в запечатана стъклена бутилка.
1.5.3.2. Подготовка на пробата
Ако е необходимо, свързващото органично вещество може да се отстрани, като влакната се поставят за около един час в пещ при 450 °С.
Оформете пробата с конус и след това я разделете на четири части (това следва да се извърши в смукателен шкаф).
С помощта на шпатула добавете малко количество (< 0,5 g) от пробата в 100 ml прясно дестилирана вода, която е била филтрирана през 0,2 μm мембранен филтър (могат да се използват алтернативни източници на свръхчиста вода, ако е доказано, че удовлетворяват изискванията). Разбърква се изцяло с помощта на ултразвукова сонда при мощност 100 W и регулирана така, че да се образува кавитация. (Ако не разполагате със сонда, използвайте следния метод — многократно разклатете и обърнете обратно в продължение на 30 секунди; в настолна ултразвукова вана за 5 минути; след това многократно разклатете и обърнете обратно за още 30 секунди).
Непосредствено след разбъркването на влакната вземете няколко аликвотни части (напр. три аликвотни части от 3, 6 и 10 ml) с пипета с широк отвор (2—5 ml вместимост).
Филтрирайте вакуумно всяка взета аликвотна част през поликарбонатен филтър 0,2 μm с допълнителен подсигуряващ филтър МЕС с пори 5 µm с помощта на 25-милиметрова стъклена фуния за филтриране с цилиндричен резервоар. Приблизително 5 ml от филтрираната дестилирана вода следва да се постави във фунията, а аликвотната част бавно се добавя във водата с помощта на пипета, чийто връх се държи под повърхността на течността. След тази процедура резервоарът и пипетата трябва да бъдат добре промити, тъй като фините влакна обикновено се разполагат в по-голяма степен по повърхността.
Внимателно отстранете филтъра и го отделете от допълнителния филтър, преди да го сложите в съд за изсушаване.
Със скалпел тип 24 отрежете с възвратно-постъпателно движение четвърт сектор или половината от филтъра с филтрираните отложения. Внимателно прикрепете отрязаната част към предметната плочка на СЕМ, като използвате залепваща въглеродна лепенка или въглеродно лепило. За подобряване на електрическия контакт по краищата на филтъра и предметната плочка следва да се нанесе колоидно сребро най-малко на три места. Когато лепилото/колоидното сребро изсъхне, чрез метализиране чрез запрашване покрийте с приблизително 50 nm златен или златен/паладиев слой повърхността на отложенията.
1.5.3.3. Калибриране и работа със СЕМ
1.5.3.3.1. Калибриране
Калибрирането на СЕМ следва да се проверява поне веднъж седмично (най-добре веднъж дневно) чрез използване на сертифицирана калибрираща решетка. Калибрирането се проверява спрямо сертифициран стандарт и ако измерената стойност (СЕМ) не е в рамките на ±2 % от сертифицираната стойност, тогава калибрирането на СЕМ трябва да се регулира и да се провери отново.
СЕМ следва да може да измери най-малко минимален видим диаметър от 0,2 µm, като се използва реална матрица образец при увеличение x 2 000.
1.5.3.3.2. Работа
Със СЕМ следва да се работи с увеличение 10 000 (2) при условия, които осигуряват добра резолюция и задоволителен образ при бавна скорост на сканиране, например 5 секунди на кадър. Въпреки че работните изисквания на различните СЕМ може да варират, обикновено за постигане на най-добра видимост и резолюция при материали със сравнително ниско атомно тегло следва да се използва ускорително напрежение 5—10 keV с малка големина на петното и късо работно разстояние. Тъй като се използва линейно трасиране, следва да се използва наклон от 0o, за да се сведе до минимум повторното фокусиране или ако СЕМ е с евцентрично стъпало, следва да се използва евцентричното работно разстояние. Ако материалът не съдържа малки (по диаметър) влакна и диаметрите на влакната са големи (> 5 µm), може да се използва по-малко увеличение.
1.5.3.4. Измерване
1.5.3.4.1. Изследване с ниско увеличение за оценка на пробата
Първоначално пробата се изследва с ниско увеличение, за да се определи дали има сплъстяване на големи влакна и да се оцени гъстотата на влакната. При наличие на прекомерно сплъстяване се препоръчва да се приготви нова проба.
За статистическата точност е необходимо да се измери минимален брой влакна и високата им гъстота може да изглежда за предпочитане, тъй като изследването на празни полета отнема време и не допринася за анализа. Обаче ако филтърът е претоварен, става трудно да се измерят всички измерими влакна и тъй като малките влакна може да са закрити от по-големи, могат да бъдат пропуснати.
Отклонение към надценяване на LWGMD може да възникне при гъстота на влакната над 150 влакна на 1 милиметър линейно трасиране. От друга страна, ниските концентрации на влакна биха увеличили времето за анализ и често е по-рентабилно да се изготви проба с гъстота на влакната, близка до оптималната, отколкото да се продължава броенето при филтри с ниска концентрация. Оптималната гъстота на влакната би трябвало да даде средно около едно или две преброими влакна за зрително поле при увеличение 5 000. Независимо от това оптималната гъстота ще зависи от размера (диаметъра) на влакната, така че е необходимо операторът да използва известна експертна преценка, за да реши дали плътността на влакното е близка до оптималната или не.
1.5.3.4.2. Претегляне спрямо дължината на диаметрите на влакната
Отчитат се единствено онези влакна, които допират (или пресичат) една (безкрайно) тънка линия, очертана на екрана на СЕМ. Поради тази причина се начертава хоризонтална (или вертикална) линия през центъра на екрана.
Като алтернативен метод се поставя точка в центъра на екрана и пробата се сканира без прекъсване в една посока през филтъра. Диаметърът на всяко влакно със стройност, по-голяма от 3:1, което допира или пресича тази точка, се измерва и записва.
1.5.3.4.3. Измерване на влакната
Препоръчва се да се измерят минимум 300 влакна. Всяко влакно се измерва само един път в точката на пресичане с линията или в точката, очертана върху образа (или близо до точката на пресичане, ако ръбовете на влакното са неясни). Ако попаднете на влакна с различни напречни сечения, следва да се използва измерване, представящо средния диаметър на влакното. Трябва да се внимава при определяне на ръбовете и измерване на най-късото разстояние между ръбовете на влакното. Измерването може да се извърши в момента или впоследствие на базата на съхранени образи или снимки. Препоръчва се използването на полуавтоматични системи за измерване на образи, които прехвърлят данните директно в електронна таблица, тъй като пестят време, отстраняват грешки при вписването и изчисленията могат да бъдат автоматизирани.
Краищата на дълги влакна следва да се проверяват при ниско увеличение, за да се гарантира, че те не завиват обратно в полето на измерване и че са измерени само веднъж.
2. ДАННИ
2.1. ОБРАБОТВАНЕ НА РЕЗУЛТАТИТЕ
Диаметрите на влакната обикновено не са с нормално разпределение. Въпреки това чрез логаритмично преобразуване е възможно да се получи разпределение, което се приближава до нормалното.
Изчислете средноаритметичната стойност (среден InD) и стандартното отклонение (SDInD) на логаритъма с основа е на стойностите (lnD) на n диаметри на влакна (D).
|
|
(1) |
|
|
(2) |
Стандартното отклонение се дели на квадратния корен на броя измервания (n), за да се получи стандартната грешка (SElnD).
|
|
(3) |
Извадете два пъти стандартната грешка от средната стойност и изчислете експоненциалната на тази стойност (средната стойност минус две стандартни грешки), за да получите средногеометричната стойност минус две стандартни геометрични грешки.
|
|
(4) |
3. ДОКЛАДВАНЕ
ПРОТОКОЛ ОТ ИЗПИТВАНЕТО
Протоколът от изпитването следва да включва най-малко следната информация.
|
— |
Стойността на LWGMD-2SE. |
|
— |
Всички отклонения и особено онези, които биха могли да повлияят на прецизността и точността на резултатите, с подходящи обосновки. |
4. ИЗПОЛЗВАНА ЛИТЕРАТУРА
|
1. |
B. Tylee SOP MF 240. Health and Safety Executive. Февруари 1999 г. |
|
2. |
G. Burdett and G. Revell. Development of a standard method to measure the length-weigthed geometric mean fibre diameter: Results of the Second inter-laboratory exchange. IR/L/MF/94/07. Project R42.75 HPD. Health and Safety Executive. Research and Laboratory Services Division. 1994 г. |
(1) Тази увеличителна стойност е указана за влакна от 3 µm, за влакна от 6 µm може да е по-подходящо увеличение × 5 000.
(2) За влакна от 3 μm вж. предходната бележка.
ПРИЛОЖЕНИЕ III
|
Б.46. |
IN VITRO КОЖНО ДРАЗНЕНЕ: ИЗПИТВАНЕ ВЪРХУ МОДЕЛ НА РЕКОНСТРУИРАН ЧОВЕШКИ ЕПИДЕРМИС |
1. МЕТОД
1.1. ВЪВЕДЕНИЕ
Кожно дразнене означава предизвикване на обратимо увреждане на кожата вследствие на прилагането на изпитвано вещество за период от време до 4 часа [както е определено в Глобалната хармонизирана система за класифициране и етикетиране на химически вещества (GHS) на Организацията на обединените нации (ООН)](1). Този метод за изпитване обхваща in vitro процедура, която в зависимост от изискванията за информация може да позволи определяне на дразнещия ефект на химични вещества върху кожата като напълно самостоятелно алтернативно заместващо изпитване в рамките на стратегия за изпитване, при подход, основан на преценка на тежестта на доказателствата (2).
Оценката на кожното дразнене обикновено включва използването на лабораторни животни (вж. метод B.4)(3). Във връзка със съображенията за хуманно отношение към животните метод B.4 позволява определянето на корозивността за/дразненето на кожата с прилагането на стратегия на етапно изпитване, използвайки валидирани in vitro и ex vivo методи, като по този начин се избягват болката и страданието на животните. За свързаната с корозивността част на стратегията за етапно изпитване от метод B.4 е полезно използването на три валидирани метода за in vitro изпитване (указания за изпитване) — B.40, B.40bis и TG 435 (4, 5, 6).
Настоящият метод на изпитване се основава на модели на реконструиран човешки епидермис, които поради цялостното си устройство (използването на човешки епидермални кератиноцити като източник на клетки, представителна тъканна и клетъчна структура) силно наподобяват биохимичните и физиологичните свойства на горните слоеве на човешката кожа, т.е. на епидермиса. Процедурата, описана в настоящия метод на изпитване, позволява определянето на опасността от дразнещите вещества в съответствие с категория 2 по GHS на ООН. Този метод на изпитване включва също набор от стандарти за ефективност за оценката на подобни и модифицирани методи на изпитване, основани на реконструиран човешки епидермис (7).
Изследвания, завършени преди валидирането на метода, както и за оптимизирането и за валидирането му, са проведени за два метода за in vitro изпитване (8, 9, 10, 11, 12, 13, 14, 15, 16, 17) — предлаганите на пазара EpiSkin™ и EpiDerm™, използващи модели на реконструиран човешки епидермис. Тези посочени в библиографията изследвания се основават на оценка на критериите за класифициране с R 38. Някои аспекти, свързани с преизчисляването за целите на GHS, са разгледани в изследването, посочено под № 25 от библиографията. Методи с ефективност, еквивалентна на EpiSkin™ (валидиран референтен метод 1), се препоръчват като напълно самостоятелен заместващ изпитвателен метод на in vivo изпитването върху зайци за класифициране на дразнещи вещества от категория 2 по GHS. Методи с ефективност, еквивалентна на EpiDerm™ (валидиран референтен метод 2), се препоръчват само като изпитвателен метод за скрининг или като част от стратегия за етапно изпитване в подход, основан на преценка на тежестта на доказателствата, за класифицирането на дразнещи вещества от категория 2 по GHS. Преди даден in vitro метод, предложен за изпитване на кожно дразнене върху модел на реконструиран човешки епидермис, да може да се използва за регулаторни цели, следва да се определят неговата надеждност, приложимост (точност) и ограничения за предложената употреба, за да се гарантира, че методът е сравним с валидирания референтен метод 1 в съответствие със стандартите за ефективност, определени в настоящия метод за изпитване (допълнение).
Два други метода за in vitro изпитване върху реконструиран човешки епидермис са валидирани в съответствие с изискванията на настоящия метод за изпитване и показват сходни резултати като валидирания референтен метод 1 (18). Това са модифицираният метод за изпитване EpiDerm™ (модифициран референтен метод 2) и методът за изпитване SkinEthic RHE™ („аз също“ метод 1).
1.2. ОПРЕДЕЛЕНИЯ
В настоящия метод за изпитване се прилагат следните определения:
Точност: Степента на близост на съвпадение на резултатите от метода за изпитване и приетите референтни стойности. Това е мярка за възможностите на метода и един от аспектите на неговата приложимост. Терминът често се използва взаимозаменяемо с термина „съответствие“, за да се обозначи относителният дял на верните резултати при даден метод за изпитване.
Химично вещество за контрол на партидите: Вещество, предизвикващо определена величина на отговор в средната част на диапазона на жизнеспособността на клетките от дадена тъкан.
Клетъчна жизнеспособност: Параметър, измерващ общата активност на клетъчната популация, например като способност на клетъчните митохондриални дехидрогенази да намаляват постъпването на виталния оцветител MTT ([3 — (4,5-диметилтиазол-2-ил)-2,5-дифенилтетразолиев бромид, Тиазолил синьо) в клетките, което, в зависимост от измерената крайна точка и използваната процедура на изпитване, корелира с общия брой и/или с виталността на живите клетки.
ET50 : Времето на експозиция, необходимо за намаляване на жизнеспособността на клетките с 50 % след прилагане на маркиращо вещество при определена фиксирана концентрация, вж. също IC50.
Честота на фалшивонегативните резултати: Делът от всички вещества, даващи положителна реакция, която е погрешно определена от метода за изпитване като отрицателна. Това е един от показателите за възможностите на метода за изпитване.
Честота на фалшивопозитивните резултати: Делът от всички неактивни вещества, даващи отрицателна реакция, която е определена погрешно като положителна. Това е един от показателите за възможностите на метода за изпитване.
Неопределена доза: Количеството от изпитваното вещество, приложено върху кожата, което надхвърля количеството, необходимо за пълно и равномерно покриване на повърхността на кожата.
GHS (Globally Harmonized System of Classification and Labelling of Chemicals, Глобална хармонизирана система за класифициране и етикетиране на химикали): Система за класифициране на вещества и смеси съгласно стандартизирани видове и нива на физични, здравни и екологични опасности заедно със съответните съобщителни елементи, като пиктограми, сигнални думи, предупреждения за опасност, препоръки за безопасност и информационни листове за безопасност, с цел да предава информация за вредните им въздействия с оглед предпазването на хората (включително работодатели, работници, превозвачи, потребители и лица, които оказват помощ при спешни ситуации) и околната среда (1), въведена в ЕС посредством Регламент (ЕО) № 1272/2008.
IC50 : Концентрацията на маркиращото вещество, при която жизнеспособността на тъканите се намалява с 50 % (IC50) след фиксирано време на експозиция, вж. също ET50.
Стандарти за ефективност: Стандарти въз основа на валидиран референтен метод, които предоставят основата за оценка на сравнимостта на предложен метод за изпитване, който е сходен механистично и функционално на референтния. Включени са: I) съществени елементи на метода за изпитване; II) минимален списък от референтни вещества, избрани измежду веществата, използвани за демонстрация на приемливата ефективност на валидирания референтен метод; и III) сравнимите нива за сравнение на точност и надеждност въз основа на получените за валидирания референтен метод, които предложеният метод за изпитване трябва да демонстрира, когато се оценява чрез използване на минималния списък с референтни вещества.
Надеждност: Мярка за степента, в която метод за изпитване може да се извърши възпроизводимо в една и съща лаборатория и между различни лаборатории в течение на времето, когато се използва една и съща процедура. Определя се, като се изчислява вътрешно- и междулабораторната възпроизводимост.
Чувствителност: Относителният дял от всички положителни/активни вещества, които са класифицирани правилно чрез изпитването. Това е мярка за точността на метод за изпитване, която предоставя категорийни резултати и е важен фактор при оценката на приложимостта на метода за изпитване.
Специфичност: Относителният дял от всички негативни/неактивни вещества, които са класифицирани правилно чрез изпитването. Това е мярка за точността на метода за изпитване, която предоставя категорийни резултати и е важен фактор при оценката на приложимостта на метода за изпитване.
Кожно дразнене: Предизвикването на обратимо увреждане на кожата вследствие на прилагането на изпитвано вещество за период от време до 4 часа. Кожното дразнене е локално възникваща, неимуногенна реакция, която се появява скоро след третирането (24). Главната му характеристика е обратимият процес, който включва възпалителни реакции и повечето от клиничните признаци на дразнене (еритем, оток, сърбеж и болка), свързани с възпалителен процес.
1.3. ОБХВАТ И ОГРАНИЧЕНИЯ
Ограничение на изпитванията върху реконструиран човешки епидермис, обхванати от настоящия метод за изпитване, е, че те класифицират вещества като дразнители на кожата само в съответствие с категория 2 от GHS на ООН. Тъй като те не позволяват класифициране на веществата в незадължителната категория 3, както е определена в GHS на ООН, всички останали вещества няма да бъдат класифицирани (без категория). В зависимост от регулаторните нужди и възможното бъдещо включване на нови крайни точки, усъвършенстване или разработване на нови изпитвания „аз също“, може да се наложи настоящият метод за изпитване да бъде преразглеждан.
Настоящият метод за изпитване позволява да се идентифицират опасностите от дразнещи еднокомпонентни вещества (19), но не предоставя адекватна информация за корозивността им за кожата. Газовете и аерозолите не могат да се изпитват, а смесите все още не са оценени с валидизационно проучване.
1.4. ПРИНЦИП НА ИЗПИТВАНЕТО
Изпитваното вещество се прилага локално на триизмерен модел на реконструиран човешки епидермис, състоящ се от нормални, получени от човек епидермални кераноцити, които са култивирани да образуват многослоен силно диференциран модел на човешкия епидермис. Той се състои от базален, спинозен и гранулозен слой и от многослоен рогов слой, съдържащ междуклетъчни ламелни липидни слоеве, подредени по начин, аналогичен на този, който се наблюдават in vivo.
Принципът на изпитването върху модел на реконструиран човешки епидермис се основава на предпоставката, че дразнещите вещества са способни да проникнат в роговия слой чрез дифузия и са цитотоксични за клетките в по-долните слоеве. Жизнеспособността на клетката се измерва чрез дехидрогеназно преобразуване на виталния оцветител MTT [3 — (4,5-диметилтиазол-2-ил)-2,5-дифенилтетразолиев бромид, Тиазолил синьо; EINECS номер: 206—069—5, CAS номер: 298—93—1], в синя формазанова сол, която се измерва количествено след екстракция от тъканите (20). Дразнещите вещества се определят по способността им да намаляват жизнеспособността на клетките под определени прагови нива (т.е. ≤ 50 % за дразнители от категория 2 по GHS на ООН). Веществата, при които жизнеспособността на клетките е над определеното прагово ниво, няма да бъдат класифицирани (т.е. > 50 %, без категория).
Моделираните системи на реконструиран човешки епидермис може да се използват за изпитване на твърди вещества, течности, полутвърди вещества и восъци. Течностите могат да бъдат или да не бъдат водни разтвори; твърдите вещества може да са разтворими или неразтворими във вода. В случаите, когато това е възможно, твърдите вещества следва да се изпитват под формата на фин прах. Тъй като във валидирането на системите за изпитване върху модели на реконструиран човешки епидермис бяха включени 58 внимателно подбрани вещества, представляващи широк спектър класове химически вещества, очаква се методите да са общоприложими за различните класове химически вещества (16). Във валидирането са включени 13 дразнителя от категория 2 по СХС. Следва да се отбележи, че във валидирането не бяха включени неразяждащи киселини, основи, соли и други неорганични вещества, а някои известни класове органични дразнители, като например водородни пероксиди, феноли и повърхностноактивни вещества или не бяха включени, или бяха включени само частично.
1.5. ДОКАЗВАНЕ НА ГОДНОСТ
Преди рутинната употреба на валидиран метод, който се придържа към настоящия метод за изпитване, лабораториите може да пожелаят да демонстрират техническа годност, като използват десетте вещества, препоръчани в таблица 1. Съгласно настоящия метод за изпитване класифицирането в незадължителната категория 3 по GHS на ООН се счита за класифициране като това без категория. За демонстриране на сравнима надеждност и точност преди използването за регулаторни изпитвания на нови сходни („аз също“) методи, разработени съгласно настоящия метод за изпитване и структурно и функционално сходни на валидирани референтни методи, както и на модификации на валидирани методи, следва да се приложат стандартите за ефективност, описани в допълнението към настоящия метод за изпитване.
Таблица 1
Веществата за оценка на годност, които са подгрупа на референтните вещества, посочени в допълнението
|
Вещество |
CAS номер |
Резултат in vivo |
Агрегатно състояние |
Категория по GHS |
|
1-нафтилоцетна киселина |
86—87—3 |
0 |
Твърдо |
Без категория |
|
изопропанoл |
67—63—0 |
0,3 |
Течно |
Без категория |
|
метилов стеарат |
112—61—8 |
1 |
Твърдо |
Без категория |
|
хептил бутират |
5870—93—9 |
1,7 |
Течно |
Незадължителна категория 3 |
|
хексил салицилат |
6259—76—3 |
2 |
Течно |
Незадължителна категория 3 |
|
цикламен алдехид |
103—95—7 |
2,3 |
Течно |
Категория 2 |
|
1-бромхексан |
111—25—1 |
2,7 |
Течно |
Категория 2 |
|
бутил метакрилат |
97—88—1 |
3 |
Течно |
Категория 2 |
|
1-метил-3-фенил-1-пиперазин |
5271—27—2 |
3,3 |
Твърдо |
Категория 2 |
|
хептанал |
111—71—7 |
4 |
Течно |
Категория 2 |
1.6. ОПИСАНИЕ НА МЕТОДА
По-долу са описани елементите и процедурите на изпитване за оценка на кожно дразнене върху модел на реконструиран човешки епидермис. Моделът на реконструиран човешки епидермис може да се изгради, изготви или закупи (например EpiSkin™, EpiDerm™ и SkinEthic RHE™). Стандартни протоколи за метода на изпитване за EpiSkin™, EpiDerm™ и SkinEthic RHE™ могат да се открият на интернет адрес [http://ecvam.jrc.ec.europa.eu] (21, 22, 23). Изпитването следва да се провежда съгласно следните изисквания:
1.6.1. Елементи на модела на реконструиран човешки епидермис
1.6.1.1. Общи условия за модела
За изграждането на епитела следва да се използват нормални човешки кератиноцити. Под функционалния рогов слой трябва да има множество слоеве от жизнеспособни епителни клетки (базален слой, stratum spinosum, stratum granulosum). Роговият слой трябва да е многослоен и да съдържа основния липиден профил, за да създаде функционална преграда, която е достатъчно здрава да устои на бързото проникване на цитотоксични маркиращи вещества, например натриев додецил сулфат (SDS) или Triton X-100. Бариерната функция може да бъде оценена или чрез определяне на концентрацията, при която маркиращо вещество намалява жизнеспособността на тъканите с 50 % (IC50) след фиксирано време на експозиция, или чрез определяне на времето на експозиция, необходимо да се намали жизнеспособността с 50 % (ET50) след прилагане на маркиращото вещество с определена фиксирана концентрация. Задържащите свойства на модела следва да предотвратяват преминаването на материал около роговия слой към жизнеспособната тъкан, което би довело до неадекватно моделиране на експозицията на кожата. Моделът на кожа следва да не е заразен с бактерии, вируси, микоплазма или гъбички.
1.6.1.2. Изисквания за функционалния модел
1.6.1.2.1. Жизнеспособност
За предпочитане е определянето на величината на жизнеспособността да се извърши чрез оцветяване с MTT (20). Оптичната плътност (ОП) на екстахирания (разтворен) оцветител от тъканта, третирана с отрицателната контрола (ОК), следва да бъде най-малко 20 пъти по-голяма от ОП на разтворителя, който се използва за екстракцията. Следва да се документира, че тъканта, третирана с ОК, е стабилна в култура (дава сходни измервания на жизнеспособността) за периода на продължителност на експозиция при изпитването.
1.6.1.2.2. Бариерна функция
Бариерната функция на роговия слой и липидният му състав следва да бъдат достатъчни, за да устоят на бързото проникване на цитотоксични маркиращи вещества, например SDS или Triton X-100, оценено с IC50 или ET50.
1.6.1.2.3. Морфология
Персонал с необходимата квалификация следва да проведе хистологично изследване на реконструираната кожа/реконструирания епидермис, за да се демонстрира, че структурата е подобна на човешка кожа/човешки епидермис (включително многослойния stratum corneum).
1.6.1.2.4. Възпроизводимост
Резултатите, получени с метода при използването на определен модел, трябва да демонстрират възпроизводимост в течение на времето, за предпочитане чрез подходящо химично вещество за контрол на партидите (вещество, предизвикващо определена величина на отговор (вж. допълнението).
1.6.1.2.5. Контрол на качеството (КК) на модела
Всяка използвана партида от модели на епидермис следва да отговаря на определени критерии за продукция за пускане в обращение, сред които най-значими са тези за жизнеспособност (точка 1.6.1.2.1) и за бариерна функция (точка 1.6.1.2.2). Доставчикът на модела на кожа (или изследователят, ако се използва собствен модел) трябва да определи приемлив интервал (горна и долна граница) за IC50 или за ET50. След получаването на тъканите преградните им свойства следва да бъдат проверени от лабораторията. За надеждно предвиждане на дразнещия ефект могат да се приемат само резултати, получени с отговарящи на изискванията тъкани. По-долу като пример са показани интервалите на приемливост на валидираните референтни методи.
Таблица 2
Примери за критерии на контрол на качеството на партида за пускане в обращение
|
|
Долна граница на приемане |
Средна стойност на приемливия интервал |
Горна граница на приемане |
|
Валидиран референтен метод 1 (третиране за 18 часа с SDS) |
IC50 = 1,0 mg/ml |
IC50 = 2,32 mg/ml |
IC50 = 3,0 mg/ml |
|
Валидиран референтен метод 2 (1 % Triton X100) |
ET50 = 4,8 h |
ET50 = 6,7 h |
ET50 = 8,7 h |
1.6.1.3. Прилагане на изпитваните и контролните вещества
За всяко третиране и за контролите следва да се използват достатъчен брой тъканни репликати (най-малко три репликата на изпитване). За течни, както и за твърди вещества, за равномерното покриване на кожната повърхност следва да се прилага достатъчно количество от изпитваното вещество, като същевременно се избягва прилагането на неопределена доза (вж. 1.2 Определения), т.е. следва да се използват най-малко 25 μL/cm2 или 25 mg/cm2. Преди прилагането на твърдите вещества повърхността на епидермиса следва да се овлажни с дейонизирана или дестилирана вода, за да се осигури добър контакт с кожата. При всяка възможност твърдите вещества следва да се изпитват под формата на фин прах. В края на периода на експозиция изпитваното вещество трябва внимателно да се измие от повърхността на кожата с буферен воден разтвор или 0,9 % NaCl. В зависимост от използвания модел на реконструиран човешки епидермис времето на експозиция може да варира от 15 до 60 минути, а температурата на инкубация между 20 и 37 °C. За подробности вж. стандартните работни процедури за трите метода (21, 22, 23).
За да се демонстрира, че жизнеспособността (ОК), преградната функция и получената в резултат тъканна чувствителност (ПК) на тъканите са в границите на определен предходно установен интервал на приемане, при всяко изследване трябва да се използват едновременни отрицателни (ОК) и положителни контроли (ПК). Като вещество за положителна контрола е предложен 5 % воден разтвор на SDS. Като вещества за отрицателни контроли са предложени вода или физиологичен разтвор с фосфатен буфер (PBS).
1.6.1.4. Измерване на жизнеспособността на клетките
Най-важният елемент на процедурата за изпитване е измерванията на жизнеспособността да не се провеждат веднага след експозицията на изпитваните вещества, а след достатъчно дълъг период на инкубация на промитите в прясна среда тъкани след третирането им. Този период позволява както възстановяването от леките дразнещи въздействия, така и появата на ясно изразени цитотоксични ефекти. По време на фазата на оптимизиране на изпитването (9, 10, 11, 12, 13) беше установено, че оптималният период на инкубация след третирането е 42 часа и поради това той беше използван при валидирането на референтните методи на изпитване.
Преобразуващият опит с MTT е утвърден количествен метод, който следва да се използва за измерване на клетъчната жизнеспособност. Той е съвместим с използване в триизмерни тъканни модели. Кожната проба се поставя в разтвор на MTT с подходяща концентрация (т.е. 0,3—1 mg/mL) за 3 часа. След това утаеният син формазанов продукт се извлича от тъканта с помощта на разтворител (например изопропанол, киселинен изопропанол), а концентрацията на формазан се измерва, като се определи ОП при 570 nm и ширина на филтърната дължина на вълната най-много ±30 nm.
Оптичните свойства на изпитваното вещество или химичното му въздействие върху MTT могат да повлияят на изпитването, като доведат до погрешна оценка на жизнеспособността (защото изпитваното вещество може да предотврати, да предизвика или да промени оцветяването). Това може да се случи, ако определено изпитвано вещество не е отстранено напълно от кожата чрез промиването или когато проникне през епидермиса. Ако изпитваното вещество, което действа пряко върху MTT, е естествено оцветено или се оцветява по време на третиране на тъканта, следва да се използват допълнителни контроли, за да се открие и коригира въздействието на изпитваното вещество върху техниката за измерване на жизнеспособността. За подробно описание как да се установява пряка редукция на MTT, следва да се ползва протоколът за валидираните референтни методи методи (21, 22, 23). Неспецифичният цвят (НСЦ), дължащ се на тази намеса, не трябва да превишава 30 % от ОК (за корекции). Ако НСЦ > 30 %, изпитваното вещество се счита за несъвместимо с изпитването.
1.6.1.5. Критерии за приемливост на опита
За всеки опит, използващ валидни партиди (вж. точка 1.6.1.2.5), тъканите, третирани с ОК, следва да показват оптична плътност, отразяваща състоянието на тъканите, които са преминали през всички етапи по транспортиране и приемане, както и целият процес по протокола за оценка на дразнещия ефект. Стойностите на ОП на контролите не трябва да са по-ниски от предходно установените долни граници. По подобен начин тъканите, третирани с ПК, т.е. 5 % воден разтвор на SDS, следва да демонстрират запазената чувствителност на тъканите и способността им да реагират на дразнещо вещество при условията на всеки отделен опит (например жизнеспособност ≤ 40 % за валидирания референтен метод 1 и ≤ 20 % за валидирания референтен метод 2). Следва да се определят подходящи критерии за оценка на вариабилността между тъканните репликати (например ако се използват стандартните отклонения, те трябва да са ≤ 18 %).
2. ДАННИ
2.1. ДАННИ
За всяко третиране следва да се представят във вид на таблица данни от всяко измерване на отделните репликати на пробите (например данни за стойностите на ОП и изчислената процентна клетъчната жизнеспособност за всяко изпитвано вещество, включително класифициране), включително данни от повторни опити, ако е целесъобразно. В допълнение следва да се докладват средните аритметични стойности ± стандартното отклонение за всяка серия опити. За всяко изпитвано вещество следва да се докладват наблюдаваните взаимодействия с реагента MTT, като трябва да се посочат и оцветените изпитвани вещества.
2.2. ТЪЛКУВАНЕ НА РЕЗУЛТАТИТЕ
Получените стойности за ОП за всяка изпитвана проба могат да се използват за изчисляването на процентната жизнеспособност в сравнение с тази на ОК, която е определена за 100 %. Граничната стойност за процента на клетъчната жизнеспособност, която да разграничава дразнещите от некласифицираните изпитвани вещества, и статистическата(ите) процедура(и), използвана(и) за оценка на резултатите и определяне на дразнещите вещества, трябва да бъдат ясно определени и документирани, както и доказано целесъобразни. Граничните стойности за прогнозиране на дразнене, свързани с валидираните референтни методи, са показани по-долу:
Изпитваното вещество се счита за дразнещо за кожата в съответствие с категория 2 по СХС на ООН:
|
i) |
ако жизнеспособността на тъканта след експозиция и инкубация след третирането е по-малка или равна (≤) на 50 %. |
Изпитваното вещество не се класифицира като дразнещо за кожата:
|
ii) |
ако жизнеспособността на тъканта след експозиция и инкубация след третирането е по-голяма (>) от 50 %. |
3. ДОКЛАДВАНЕ
3.1. ПРОТОКОЛ ОТ ИЗПИТВАНЕТО
Протоколът от изпитването включва следната информация:
Изпитвани и контролни вещества:
|
— |
химично(и) наименование(я) по IUPAC или по CAS и номер по CAS, ако е известен, |
|
— |
чистота и състав на веществото (в тегловни проценти), |
|
— |
физико-химични свойства, които имат отношение към провеждането на изследването (напр. агрегатно състояние, стабилност и летливост, pH, разтворимост във вода, ако е известна), |
|
— |
третиране на изпитваните/контролните вещества преди изпитването, ако има такова (например загряване, смилане), |
|
— |
условия за съхранение. |
Обосновка на използваните модел на кожа и използван протокол на изпитването.
Условия на изпитване:
|
— |
използвана клетъчна система, |
|
— |
информация за калибрирането на измервателния уред и за ширината на филтрираната дължина на вълната, използвани за измерване на клетъчната жизнеспособност (например спектрофотометър), |
|
— |
пълна придружаваща информация за конкретния използван модел на кожа, включително качествените му показатели; това следва да включва, но не се ограничава до:
|
|
— |
подробности за използваната процедура на изпитване, |
|
— |
използвани дози при изпитването, продължителност на експозицията и инкубационен период след третирането, |
|
— |
описание на всякакви изменения на процедурата на изпитване, |
|
— |
препратка към предходно установените данни за модела; това следва да включва, но не се ограничава до:
|
|
— |
описание на използваните критериите за оценка, включително обосновката за избора на граничната(ите) точка(и) за прогнозния модел. |
Резултати:
|
— |
таблично представяне на данните от отделните проби; |
|
— |
описание на други наблюдавани ефекти. |
Обсъждане на резултатите.
Заключения.
4. БИБЛИОГРАФИЯ
|
1. |
Организация на обединените нации (ООН) (2007 г.). Световна хармонизирана система за класификация и етикетиране на химически вещества (СХС), второ преработено издание, ООН Ню Йорк и Женева, 2007 г. Достъпно на: http://www.unece.org/trans/danger/publi/ghs/ghs_rev02/02files_e.html. |
|
2. |
REACH: Ръководство за изискванията за информация и за оценка на химическата безопасност. Достъпно на: http://guidance.echa.europa.eu/docs/guidance_document/information_requirements_en.htm?time=1232447649. |
|
3. |
Метод за изпитване B.4. ОСТРА ТОКСИЧНОСТ; ДРАЗНЕНЕ/КОРОЗИВНОСТ ЗА КОЖАТА. |
|
4. |
Метод за изпитване B.40. IN VITRO КОРОЗИВНОСТ ЗА КОЖАТА: ТРАНСКУТАННО ИЗПИТВАНЕ TER НА ЕЛЕКТРИЧЕСКОТО СЪПРОТИВЛЕНИЕ. |
|
5. |
Метод за изпитване B.40 BIS. IN VITRO КОРОЗИВНОСТ ЗА КОЖАТА: ИЗПИТВАНЕ ВЪРХУ МОДЕЛ НА ЧОВЕШКА КОЖА. |
|
6. |
OECD (2006 г.). Test Guideline 435. OECD Guideline for the Testing of Chemicals. In Vitro Membrane Barrier Test Method. Приет на 19 юли 2006 г. Достъпен на: http://www.oecd.org/document/22/0,2340,en_2649_34377_1916054_1_1_1_1,00.html. |
|
7. |
ECVAM (2009) Performance Standards for applying human skin models to in vitro skin irritation. Достъпен в „Download Study Documents“ на: http://ecvam.jrc.ec.europa.eu. |
|
8. |
Fentem, J.H., Briggs, D., Chesné, C., Elliot, G.R., Harbell, J.W., Heylings, J.R., Portes, P., Roguet, R., van de Sandt, J.J.M. & Botham, P. (2001). A prevalidation study on in vitro tests for acute skin irritation. Results and evaluation by the Management Team. Toxicology in Vitro 15, 57—93 стр. |
|
9. |
Portes, P., Grandidier, M.H., Cohen, C. & Roguet, R.(2002 г.). Refinement of the EPISKIN protocol for the assessment of acute skin irritation of chemicals: follow-up to the ECVAM prevalidation study. Toxicology in Vitro 16, 765—770 стр. |
|
10. |
Kandárová, H., Liebsch, M., Genschow, E., Gerner, I., Traue, D., Slawik, B. & Spielmann, H. (2004). Optimisation of the EpiDerm test protocol for the upcoming ECVAM validation study on in vitro skin irritation tests. ALTEX 21, 107—114 стр. |
|
11. |
Kandárová, H., Liebsch, M., Gerner, I., Schmidt, E., Genschow, E., Traue, D. & Spielmann H. (2005) The EpiDerm Test Protocol fort the Upcoming ECVAM Validation Study on In Vitro Skin Irritation Tests — An Assessment of the Performance of the Optimised Test. ATLA 33, 351—367 стр. |
|
12. |
Cotovio, J., Grandidier, M.—H., Portes, P., Roguet, R. & G. Rubinsteen. (2005). The In Vitro Acute Skin Irritation of Chemicals: Optimisation of the EPISKIN Prediction Model within the Framework of the ECVAM Validation Process. ATLA 33, 329—249 стр. |
|
13. |
Zuang, V., Balls, M., Botham, P.A., Coquette, A., Corsini, E., Curren, R.D., Elliot, G.R., Fentem, J.H., Heylings, J.R., Liebsch, M., Medina, J., Roguet, R., van De Sandt, J.J.M., Wiemann, C. & Worth, A.(2002). Follow-up to the ECVAM prevalidation study on in vitro tests for acute skin irritation. Доклад 2 на работната група по кожно дразнене на ECVAM. ATLA 30,109—129 стр. |
|
14. |
Spielmann, H., Hoffmann, S., Liebsch, M., Botham, P., Fentem, J., Eskes, C., Roguet, R., Cotovió, J., Cole, T., Worth, A., Heylings, J., Jones, P., Robles, C., Kandárová, H., Gamer, A., Remmele, M., Curren, R., Raabe, H., Cockshott, A., Gerner, I. and Zuang, V. (2007) The ECVAM International Validation Study on In Vitro Tests for Acute Skin Irritation: Report on the Validity of the EPISKIN and EpiDerm Assays and on the Skin Integrity Function Test. ATLA 35, 559—601 стр. |
|
15. |
Hoffmann, S. (2006). ECVAM Skin Irritation Validation Study Phase II: Analysis of the Primary Endpoint MTT and the Secondary Endpoint IL1-α. 135 стр. + приложения. Достъпен в „Download Study Documents“ на: http://ecvam.jrc.ec.europa.eu. |
|
16. |
Eskes, C., Cole, T., Hoffmann, S., Worth, A., Cockshott, A., Gerner, I. & Zuang. V (2007) ECVAM International Validation Study on In Vitro Tests for Acute Skin Irritation: Selection of Test Chemicals. ATLA 35, 603—619 стр. |
|
17. |
J. Cotovio, M.—H. Grandidier, D. Lelièvre, R. Roguet, E. Tinois-Tessonneaud, J. Leclaire (2007). In vitro acute skin irritancy of chemicals using the validated EPISKIN model in a tiered strategy — Results and performances with 184 cosmetic ingredients, AATEX, Special Issue-proceedings from WC6. Vol.14, 351—358 стр. |
|
18. |
Декларация на ESAC относно актуализираните EpiDerm и подобни на SkinEthic опити. |
|
19. |
ЕО (2006 г.). Регламент (EО) № 1907/2006 на Европейския парламент и на Съвета от 18 декември 2006 г. относно регистрацията, оценката, разрешаването и ограничаването на химични вещества и препарати (REACH), за създаване на Европейска агенция по химикали, за изменение на Директива 1999/45/EО и за отмяна на Регламент (EИО) № 793/93 на Съвета и Регламент (EО) № 1488/94 на Комисията, както и Директива 76/769/EИО на Съвета и Директиви 91/155/EИО, 93/67/EИО, 93/105/EО и 2000/21/EО на Комисията. Официален вестник на Европейския съюз L 396/1 от 30.12.2006 г. Служба за публикации, Люксембург. |
|
20. |
Mosman, T. (1983) Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. Journal of Immunological Methods 65, 55 — 63 стр. |
|
21. |
EpiSkin™ SOP, Version 1.6 (January 2005). Validation of the EpiSkin Skin Irritation Test — 42 Hours Assay For The Prediction of Acute Skin Irritation of Chemicals. Достъпен в „Download Study Documents“ на: http://ecvam.jrc.ec.europa.eu. |
|
22. |
EpiDerm™ SOP, Version 5.0 (October 2004). Draft Standard Operating Procedure. In Vitro Skin Irritation Test: Human Skin Model. Model: EpiDerm™-200. Достъпен в „Download Study Documents“ на: http://ecvam.jrc.ec.europa.eu. |
|
23. |
SkinEthic RHE™ SOP. Ще е достъпен в „Download Study Documents“ на: http://ecvam.jrc.ec.europa.eu. |
|
24. |
Harvell, J.D., Lamminstausta, K, Maibach H.I. (1995) Irritant contact dermatitis IN: Guin J.D. Practical Contact Dermatitis Mc Graw-Hill New York, 7 — 18 стр. |
|
25. |
Griesinger C, Barroso J & Zuang V: ECVAM background document on the recent adaptations of the ECVAM performance standards for in vitro skin irritation testing in the context of the drafting process of an EU test method and an OECD draft test guideline. Ispra, November 13, 2008. |
Допълнение
Оценка на функционалните характеристики на предложени in vitro модели на реконструиран човешки епидермис за кожно дразнене
ВЪВЕДЕНИЕ
Предложените за използване по настоящия метод за изпитване процедури следва да бъдат оценени, за да се определят надеждността и точността им, като се използват вещества, които са представителни за пълната скала на резултатите за дразнене на Draize. Когато оценката се извършва с използване на 20 препоръчителни референтни вещества (таблица 2), предложените процедури трябва да имат стойности на надеждност и точност, сравними с тези на валидирания референтен метод 1 (таблица 3) (1). Стандартните стойности на надеждност и точност, които трябва да бъдат достигнати, са представени в раздели II и III по-долу. Включени са некласифицирани и класифицирани (категория 2 по GHS на ООН) вещества, представящи съответните класове химически вещества, така че стойностите на надеждност и ефективност (чувствителност, специфичност, относителен дял на фалшиво негативни и фалшиво позитивни резултати, точност) за предложения метод за изпитване да могат да бъдат сравнени с тези на валидирания референтен метод 1. Надеждността на метода за изпитване, както и неговата способност правилно да идентифицира дразнещи вещества от категория 2 по GHS на ООН, следва да бъдат определени, преди използването му за изпитване на нови вещества.
СТАНДАРТИ ЗА ЕФЕКТИВНОСТ
Стандартите за ефективност включват следните три елемента: I) съществени елементи на метода за изпитване; II) референтни вещества; и III) определени стойности на точност и надеждност (2). Настоящите стандарти за ефективност се основават на стандартите за ефективност, определени след приключването на валидирането на изпитването за кожно дразнене ECVAM (3).
I) Съществени елементи на метода за изпитване
Общи условия за модела
За изграждането на епитела следва да се използват нормални човешки кератиноцити. Под функционалния рогов слой трябва да има множество слоеве от жизнеспособни епителни клетки (базален слой, stratum spinosum, stratum granulosum). Роговият слой трябва да е многослоен и да съдържа основния липиден профил, за да създаде функционална преграда, която е достатъчно здрава да устои на бързото проникване на цитотоксични маркиращи вещества, например натриев додецил сулфат (SDS) или Triton X-100. Бариерната функция може да бъде оценена или чрез определяне на концентрацията, при която маркиращо вещество намалява жизнеспособността на тъканите с 50 % (IC50) след фиксирано време на експозиция, или чрез определяне на времето на експозиция, необходимо да се намали жизнеспособността с 50 % (ET50) след прилагане на маркиращото вещество с определена фиксирана концентрация. Задържащите свойства на модела следва да предотвратяват преминаването на материал около роговия слой към жизнеспособната тъкан, което би довело до неадекватно моделиране на експозицията на кожата. Моделът на кожа следва да не е заразен с бактерии, вируси, микоплазма или гъбички.
Функционални изисквания за модела
Жизнеспособност
За предпочитане е определянето на величината на жизнеспособността да се извърши чрез оцветяване с МТТ (4). Оптичната плътност (ОП) на извлечения (разтворен) оцветител от тъканта, третирана с отрицателната контрола (ОК), следва да бъде най-малко 20 пъти по-голяма от ОП на извличащия разтворител. Следва да се документира, че тъканта, третирана с ОК, е стабилна в култура (дава сходни измервания на жизнеспособността) за периода на продължителност на експозиция при изпитването.
Бариерна функция
Бариерната функция на роговия слой и липидният му състав следва да бъдат достатъчни, за да устоят на бързото проникване на цитотоксични маркиращи вещества, например SDS или Triton X-100, оценено с IC50 или ET50.
Морфология
Персонал с необходимата квалификация следва да проведе хистологично изследване на реконструираната кожа/реконструирания епидермис, за да се демонстрира структура, подобна на човешка кожа/човешки епидермис (включително многослойния stratum corneum).
Възпроизводимост
Резултатите, получени с метода при използването на определен модел, трябва да демонстрират възпроизводимост в течение на времето, за предпочитане чрез подходящо химично вещество за контрол на партидите (вещество, предизвикващо определена величина на отговор) (вж. определенията в раздел 1.2).
Контрол на качеството (КК) на модела
Всяка използвана партида от модели на епидермис следва да отговаря на определени критерии за продукция за пускане в обращение, сред които най-значими са тези за жизнеспособност и за бариерна функция. Доставчикът на модела на кожа (или изследователят, ако се използва собствен модел) трябва да определи приемлив интервал (горна и долна граница) за IC50 или за ET50. След получаването на тъканите преградните им свойства следва да бъдат проверени от лабораторията. За надеждно предвиждане на дразнещия ефект могат да се приемат само резултати, получени с отговарящи на изискванията тъкани. По-долу като пример са показани интервалите на приемливост на валидираните референтни методи.
Таблица 1
Примери за критерии на контрол на качеството на партида за пускане в обращение
|
|
Долна граница на приемане |
Средна стойност на приемливия интервал |
Горна граница на приемане |
|
Валидиран референтен метод 1 (третиране 18 часа с SDS) |
IC50 = 1,0 mg/ml |
IC50 = 2,32 mg/ml |
IC50 = 3,0 mg/ml |
|
Валидиран референтен метод 2 (1 % Triton X100) |
ET50 = 4,8 h |
ET50 = 6,7 h |
ET50 = 8,7 h |
II) Референтни вещества
Референтните вещества се използват, за да се определи дали надеждността и точността на предложен нов in vitro метод за изпитване върху реконструиран човешки епидермис, за който е доказано, че е достатъчно структурно и функционално сходен на валидираните референтни методи, и който представлява незначителна промяна на валидиран референтен метод, показват резултати, сравними с тези на валидиран референтен метод 1 (1). Изброените в таблица 2 двадесет вещества включват вещества, представящи различни представляващи интерес класове химически вещества, както и вещества от категория 2 по GHS на ООН. Веществата, включени в списъка, обхващат 10 вещества от категория 2 по GHS на ООН, 3 вещества от незадължителна категория 3 по GHS на ООН и 7 вещества „без категория“. Съгласно настоящия метод за изпитване класифицирането в незадължителната категория 3 по GHS на ООН се счита за класифициране като това без категория. Тези референтни вещества представляват минималния брой вещества, които следва да бъдат използвани за оценка на надеждността и точността на предложен метод за изпитване върху реконструиран човешки епидермис за кожно дразнене. Ако дадено вещество от списъка не е на разположение, биха могли да бъдат използвани други вещества, за които са налице адекватни in vivo референтни данни. По желание към минималния списък на референтните вещества могат да бъдат прибавени допълнителни вещества, представящи други класове вещества и за които са налице адекватни in vivo референтни данни, с цел допълнителна оценка на точността на предложения метод за изпитване.
Таблица 2
Референтни вещества за определяне на стойностите на точност и надеждност за модели на реконструиран човешки епидермис за кожно дразнене
|
Вещество (1) |
CAS № |
EINECS № |
Агрегатно състояние |
Резултат in vivo |
GHS in vitro категория |
GHS in vivo категория |
|
1-бромо-4-хлорбутан |
6940-78-9 |
230-089-3 |
Течно |
0 |
Категория 2 |
Без категория |
|
Диетилфталат |
84-66-2 |
201-550-6 |
Течно |
0 |
Без категория |
Без категория |
|
Нафтилоцетна киселина |
86-87-3 |
201-705-8 |
Твърдо |
0 |
Без категория |
Без категория |
|
Алил феноксиацетат |
7493-74-5 |
231-335-2 |
Течно |
0,3 |
Без категория |
Без категория |
|
Изопропанол |
67-63-0 |
200-661-7 |
Течно |
0,3 |
Без категория |
Без категория |
|
4-метил-тио-бензалдехид |
3446-89-7 |
222-365-7 |
Течно |
1 |
Категория 2 |
Без категория |
|
Метилов стеарат |
112-61-8 |
203-990-4 |
Твърдо |
1 |
Без категория |
Без категория |
|
Хептил бутират |
5870-93-9 |
227-526-5 |
Течно |
1,7 |
Без категория |
Незадължителна категория 3 |
|
Хексил салицилат |
6259-76-3 |
228-408-6 |
Течно |
2 |
Без категория |
Незадължителна категория 3 |
|
Три-изобутил-фосфат |
126-71-6 |
204-798-3 |
Течно |
2 |
Категория 2 |
Незадължителна категория 3 |
|
1-деканол |
112-30-1 |
203-956-9 |
Течно |
2,3 |
Категория 2 |
Категория 2 |
|
Цикламен алдехид |
103-95-7 |
203-161-7 |
Течно |
2,3 |
Категория 2 |
Категория 2 |
|
1-бромхексан |
111-25-1 |
203-850-2 |
Течно |
2,7 |
Категория 2 |
Категория 2 |
|
2-хлорометил-3,5-диметил-4-метоксипиридин хидрохлорид |
86604-75-3 |
434-680-9 |
Твърдо |
2,7 |
Категория 2 |
Категория 2 |
|
а-терпинеол |
98-55-5 |
202-680-6 |
Течно |
2,7 |
Категория 2 |
Категория 2 |
|
Ди-н-пропил дисулфид |
629-19-6 |
211-079-8 |
Течно |
3 |
Без категория |
Категория 2 |
|
Бутил метакрилат |
97-88-1 |
202-615-1 |
Течно |
3 |
Категория 2 |
Категория 2 |
|
Бензенетиол, 5 - (1,1-диметилетил)-2-метил |
7340-90-1 |
438-520-9 |
Течно |
3,3 |
Категория 2 |
Категория 2 |
|
1-метил-3-фенил-1-пиперазин |
5271-27-2 |
431-180-2 |
Твърдо |
3,3 |
Категория 2 |
Категория 2 |
|
Хептанал |
111-71-7 |
203-898-4 |
Течно |
4 |
Категория 2 |
Категория 2 |
Веществата, изброени в таблица 2, са разпределени така, че да бъдат представителни за 58-те вещества, използвани за международното изследване за валидиране на изпитването за кожно дразнене ECVAM (1). Техният избор е въз основа на следните критерии:
|
— |
веществата се предлагат на пазара |
|
— |
те са представителни за пълната скала на резултатите за дразнене на Draize (от не-дразнител до силен дразнител) |
|
— |
те имат ясно определена химична структура |
|
— |
те са представителни за възпроизводимостта и капацитета на предвиждане на утвърдения метод, определени в международното изследване за валидиране на ECVAM |
|
— |
те са представителни за функционалността на химическите вещества, използвани в процеса на валидиране |
|
— |
те нямат изключително токсичен профил (например канцерогенен или токсичен по отношение на репродуктивната система), нито пък разходите за изваждане от употреба са прекалено високи. |
III) Определени стойности на точност и надеждност
Стойностите за ефективността (чувствителност, специфичност, относителен дял на фалшивонегативни и фалшивопозитивни резултати, точност) за предложения метод за изпитване трябва да бъдат сравними с тези на утвърдения референтен метод 1 (таблица 3), т.е. чувствителността трябва да бъде равна или по-голяма (≥) от 80 %, специфичността трябва да бъде равна или по-голяма (≥) от 70 % и точността трябва да бъде равна или по-голяма (≥) от 75 %. Изчисляването на стойностите за ефективността трябва да се извърши, като се използват всички класификации, получени за 20-те вещества в различните участващи лаборатории. Класификацията на всяко вещество във всяка лаборатория трябва да бъде получена, като се използва средната стойност на жизнеспособност по време на три отделно проведени изпитвания (проведени най-малко три валидни изпитвания).
Таблица 3
Стойности за ефективността на валидирания референтен метод 1 (2)
|
Метод за изпитване |
Брой вещества |
Чувствителност |
Специфичност |
Фалшивонегативни резултати |
Фалшивопозитивни резултати |
Точност |
|
Валидиран референтен метод 1 (3) |
58 |
87,2 % (4) |
71,1 % (5) |
12,8 % |
29,9 % |
74,7 % |
|
Валидиран референтен метод 1 (3) |
20 |
90 % |
73,3 % |
10 % |
26,7 % |
81,7 % |
Надеждността на предложения метод за изпитване трябва да бъдат сравнима с тази на утвърдените референтни методи.
Вътрешнолабораторна възпроизводимост
Оценката на вътрешнолабораторната вариабилност трябва да показва съвпадение на класификациите (категория 2/без категория), получени при различни независимо проведени изпитвания на 20-те еталонни вещества в рамките на една лаборатория, равно или по-голямо (≥) от 90 %.
Междулабораторна възпроизводимост
Оценката на междулабораторната възпроизводимост не е от съществено значение, ако предложеният метод за изпитване се използва само в една лаборатория. За методи, които подлежат на трансфериране между лаборатории, оценката на вътрешнолабораторната вариабилност трябва да показва съвпадение на класификациите (категория 2/без категория), получени при различни независимо проведени изпитвания на 20-те референтни вещества, препоръчително в рамките на най-малко три лаборатории, равно или по-голямо (≥) от 80 %.
БИБЛИОГРАФИЯ
|
1. |
Spielmann, H., Hoffmann, S., Liebsch, M., Botham, P., Fentem, J., Eskes, C., Roguet, R., Cotovió, J., Cole, T., Worth, A., Heylings, J., Jones, P., Robles, C., Kandárová, H., Gamer, A., Remmele, M., Curren, R., Raabe, H., Cockshott, A., Gerner, I. and Zuang, V. (2007) The ECVAM International Validation Study on In Vitro Tests for Acute Skin Irritation: Report on the Validity of the EPISKIN and EpiDerm Assays and on the Skin Integrity Function Test. ATLA 35, 559—601 стр. |
|
2. |
OECD (2005) Guidance Document No. 34 on the validation and international acceptance of new or updated test methods for hazard assessment. OECD, Paris. |
|
3. |
ECVAM (2007) Performance Standards for applying human skin models to in vitro skin irritation. Достъпен в „Download Study Documents“ на: http://ecvam.jrc.ec.europa.eu. Консултиран на 27.10.2008 г. |
|
4. |
Mosman, T. (1983) Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. Journal of Immunological Methods 65, 55—63 стр. |
|
5. |
Eskes, C., Cole, T., Hoffmann, S., Worth, A., Cockshott, A., Gerner, I. & Zuang. V (2007) ECVAM International Validation Study on In Vitro Tests for Acute Skin Irritation: Selection of Test Chemicals. ATLA 35, 603—619 стр. |
(1) 20-те референтни вещества са представителна извадка от 58-те вещества, които бяха използвани първоначално за валидирането на референтен метод 1 (EpiSkin™). Налице е също пълен списък на изпитваните вещества и критериите за подбора им (5).
(2) Таблица 3 предоставя стойностите за ефективността на валидирания референтен метод 1 по отношение на неговата годност правилно да идентифицира дразнещи вещества (категория 2 по GHS на ООН) и некласифицирани вещества (без категория, включително незадължителна категория 3) съответно за 58-те и 20-те еталонни вещества (таблица 2).
(3) EpiSkin™
(4) Въз основа на 13 дразнителя от категория 2 по GHS.
(5) Въз основа на 45 дразнителя от категория 3 по GHS или химически вещества без категория по GHS.
ПРИЛОЖЕНИЕ IV
|
C.3. |
СЛАДКОВОДНИ ВОДОРАСЛИ И ЦИАНОБАКТЕРИИ, ИЗПИТВАНЕ ЗА ПОТИСКАНЕ НА РАСТЕЖА |
1. МЕТОД
Този метод е еквивалентен на ОИСР TG 201 (2006) (1).
1.1. УВОД
Методите за изпитване периодично се преразглеждат и актуализират в контекста на научния напредък. Методът на изпитване C.3 трябваше да бъде преразгледан, за да се включат допълнителни видове и за да отговори на изискванията за оценяване на риска и класифициране на химикали. Преразглеждането беше извършено въз основа на обширен практически опит, научен напредък в сферата на изследванията за токсичност при водораслите и широкообхватна регулаторна употреба, която е налице след първоначалното приемане.
1.2. ОПРЕДЕЛЕНИЯ
За целите на настоящия метод на изпитване се използват следните определения и съкращения:
Биомаса: сухото тегло от жива материя, присъстваща в дадена популация, изразено посредством определен обем; например mg изпитван разтвор от водорасли/литър. Обикновено „биомаса“ се определя като маса, но в настоящото изпитване думата се използва за маса за обем. Също така в настоящото изпитване сурогатите на биомаса, например преброяването на клетки, флуоресценция и т.н., се измерват по принцип и следователно употребата на термина „биомаса“ се отнася и до измервания на тези сурогати.
Коефициент на вариация: безразмерна мярка на неравномерността на параметър, определена като съотношението между стандартното отклонение и средната стойност. Това също така може да бъде изразено като процентна стойност. Средният коефициент на вариация на средната специфична скорост на растеж в повторните проби от контролни култури се изчислява, както следва:
|
1. |
Изчисляване на % CV на средната специфична скорост на растеж от дневните скорости на растеж/скоростите на растеж по сектори за съответната повторна проба. |
|
2. |
Изчисляване на средната стойност на всички стойности, изчислени в точка 1, за да се получи средният коефициент на вариация на дневната специфична скорост на растеж/специфична скорост на растеж по сектори в повторните проби от контролни култури. |
ECx: концентрацията на изпитваното вещество, разтворено в изпитваната среда, което води до x % (например 50 %) намаляване на растежа на изпитвания организъм в рамките на даден период на експозиция (който трябва да бъде посочен изрично, ако се отклонява от пълната или нормалната продължителност на изпитването). За да се обозначи недвусмислено стойността на EC, получена от скоростта на растеж или от добива, се използват съответно символите „ErC“ и „EyC“.
Среда на растеж: напълно синтетичната културална среда, в която растат изпитваните водорасли, когато са изложени на изпитваното вещество. Изпитването вещество обикновено ще бъде разтваряно в средата за изпитване.
Скорост на растеж (средна специфична скорост на растеж): логаритмичното нарастване на биомасата по време на периода на експозиция.
Най-ниска концентрация на наблюдавано въздействие (LOEC) е най-ниската изпитвана концентрация, при която за веществото се наблюдава статистически значимо понижаващо въздействие върху растежа (при p < 0,05) в сравнение с контролната проба, в рамките на дадено време на експозиция. Въпреки това всички изпитвани концентрации, по-големи от LOEC, трябва да имат вредно въздействие, равно или по-голямо от наблюдаваното при LOEC. Когато тези две условия не могат да бъдат изпълнени, трябва да се даде пълно обяснение за това, как е избрана LOEC (и следователно NOEC).
Концентрация без наблюдавано въздействие (NOEC): изпитваната концентрация непосредствено под LOEC.
Променлива на реакция: променлива за оценяване на токсичността, получена от измерваните параметри, описващи биомаса чрез различни методи на изчисление. За този метод скоростите на растеж и добив са променливи на реакция, получени от прякото измерване на биомаса или който и да е от посочените сурогати.
Специфична скорост на растеж: променлива на реакция, определена като частното на разликата на естествените логаритми на параметър на наблюдение (биомаса в настоящия метод за изпитване) и съответния период от време.
Добив: стойността на измерваната променлива в края на периода на експозиция минус стойността на измерваната променлива в началото на периода на експозиция, която изразява увеличаването на биомасата по време на изпитването.
1.3. ПРИЛОЖИМОСТ НА ИЗПИТВАНЕТО
Настоящият метод за изпитване е най-лесно приложим към водоразтворими вещества, за които е вероятно при условията на изпитването да останат във водата. За изпитването на вещества, които са летливи, силно адсорбиращи, оцветени, със слаба водоразтворимост или вещества, които могат да повлияят на наличността на хранителни вещества или минерали в средата на изпитване, може да са нужни определени изменения на описаната процедура (например затворена система, предварителна обработка на съдовете за изпитване). Указания за някои подходящи изменения са дадени в (2), (3) и (4).
1.4. ПРИНЦИП НА ИЗПИТВАНЕТО
Целта на настоящото изпитване е да се определят въздействията на дадено вещество върху растежа на сладководните микроводорасли и/или цианобактерии. Експоненциално растящите изпитвани организми са изложени на действието на изпитваното веществото в култури на партиди, обикновено за период от 72 часа. Въпреки относително кратката продължителност на изпитването могат да бъдат оценени въздействията върху няколко поколения.
Реакцията на системата е намаляване на растежа в серии култури от водорасли (изпитвани единици), изложени на действието на различни концентрации на изпитваното вещество. Реакцията се оценява като функция на концентрацията на експозиция в сравнение със средния растеж на повторни, неизложени на въздействие контролни култури. За пълното представяне на реакцията на системата на токсични въздействия (оптимална чувствителност) на културите се дава възможност за неограничен експоненциален растеж в условия на достатъчно хранителни вещества и непрекъсната светлина в течение на достатъчен период от време, за да се измери намаляването на специфичната скорост на растеж.
Растежът и потискането на растежа се определят количествено чрез измервания на биомасата от водорасли като функция на времето. Биомасата на водорасли се определя като сухо вещество за единица обем, например mg водорасли на литър изпитван разтвор. Въпреки това сухото вещество се измерва трудно и следователно се използват сурогатни параметри. От тези сурогати най-често се използва преброяване на клетките. Други сурогатни параметри включват обем на клетката, флуоресценция, оптична плътност и т.н. Трябва да е известен факторът на преобразуване между измервания сурогатен параметър и биомасата.
Крайната цел на изпитването е потискането на растежа, изразено като логаритмично нарастване в биомасата (средната специфична скорост на растеж) по време на периода на експозиция. От средните специфични скорости на растеж, регистрирани в поредица от изпитвани разтвори, се определя концентрацията, при която се осъществява потискане на скоростта на растежа до определените x % (например 50 %) и се изразява като ErCx (например ErC50).
За прилагането на настоящия метод съгласно регулаторната рамка на ЕС изчисляването на резултатите следва да се базира върху средната специфична скорост на растеж поради причините, описани в раздел 2.2 по-долу. Допълнителната променлива на реакция, използвана в настоящия метод на изпитване, е добивът, който може да е нужен за изпълнението на специфични регулаторни изисквания в някои държави. Той се определя като биомасата в края на периода на експозиция минус биомасата в началото на периода на експозиция. От добива, регистриран в редица от изпитвани разтвори, се изчислява концентрацията, при която се осъществява потискане на добива до определените x % (например 50 %) и се изразява като EyCx (например EyC50).
В допълнение най-ниската концентрация на наблюдавано въздействие (LOEC) и липсата на концентрация на наблюдавано въздействие (NOEC) могат да бъдат определени по статистически начин.
1.5. ИНФОРМАЦИЯ ЗА ИЗПИТВАНОТО ВЕЩЕСТВО
Информацията за изпитваното вещество, която може да се използва при установяването на условията за изпитване, включва структурната формула, чистотата, устойчивостта на светлина, устойчивостта при условията на изпитването, свойствата на поглъщане на светлината, pKa и резултатите от изследванията на трансформация, включително биоразграждането във вода.
Разтворимостта във вода, коефициентът за разделяне октанол/вода (Pow) и парното налягане на изпитваното вещество трябва да са известни и да е на разположение валидиран метод за количествено измерване на веществото в изпитваните разтвори с отчетена ефективност на възстановяването и граница на откриването.
1.6. РЕФЕРЕНТНО ВЕЩЕСТВО
Референтното(ите) вещество(а), като например 3,5-дихлорфенол, използван в международното кръгово изпитване (4), може(гат) да бъде(ат) изпитвано(и) като средство за проверка на процедурата за изпитване. Калиевият дихромат също може да бъде използван като референтно вещество за зелените водорасли. Желателно е референтното вещество да се изпитва най-малко два пъти годишно.
1.7. ВАЛИДНОСТ НА ИЗПИТВАНЕТО
За да е валидно изпитването, трябва да бъдат изпълнени следните критерии за резултати:
|
— |
Биомасата в контролните култури би трябвало да се е увеличила експоненциално с фактор най-малко 16 в рамките на 72-часовия период на изпитване. Това съответства на специфична скорост на растеж от 0,92 ден–1. За най-често използваните видове скоростта на растеж обикновено е значително по-висока (вижте допълнение 1). Този критерий може да не бъде изпълнен, когато се използват видове, които растат по-бавно от тези, изброени в допълнение 1. В този случай периодът на изпитване се удължава, за да се получи поне 16-кратен растеж на контролните култури, като същевременно растежът трябва да бъде експоненциален през целия период на изпитване. Периодът на изпитване може да бъде намален на поне 48 часа, за да се поддържа неограничен експоненциален растеж по време на изпитването, при условие че се постига минималният фактор за умножение 16. |
|
— |
Средният коефициент на вариация за специфични скорости на растеж по сектори (дни 0—1, 1—2 и 2—3 за 72-часови изпитвания) в контролните култури (вижте раздел 1.2, „коефициент на вариация“) не трябва да превишава 35 %. Вижте втората алинея от раздел 2.2.1 за изчисляване на специфичната скорост на растеж по сектори. Този критерий се прилага за средната стойност на коефициентите на вариация, изчислени за повторните контролни култури. |
|
— |
Коефициентът на вариация на средните специфични скорости на растеж по време на целия период на изпитване в повторни контролни култури не трябва да превишава 7 % в изпитванията с Pseudokirchneriella subcapitata и Desmodesmus subspicatus. За други, по-рядко изпитвани видове, стойността не бива да превишава 10 %. |
1.8. ОПИСАНИЕ НА МЕТОДА
1.8.1. Апаратура
Съдовете за изпитване и другите апарати, които ще бъдат в контакт с изпитваните разтвори, трябва да бъдат направени изцяло от стъкло или друг химически инертен материал. Предметите трябва да бъдат добре почистени, за да се гарантира, че няма органични или неорганични замърсители, които да повлияят на растежа на водораслите или върху състава на изпитваните разтвори.
Съдовете за изпитване обикновено са стъклени колби с размери, които позволяват достатъчен обем на културата за измервания по време на изпитването и достатъчен пренос на маса от CO2 от атмосферата (вижте втората алинея от раздел 1.8.9). Отбележете, че обемът на течността трябва да бъде достатъчен за аналитични изчисления (вижте петата алинея от раздел 1.8.11).
В допълнение ще се изискват някои или всички от следните уреди:
|
— |
Апаратура за култивиране: препоръчва се шкаф или камера, в който/която избраната инкубационна температура да може да бъде поддържана при ±2 °C. |
|
— |
Уреди за измерване на светлината: важно е да се отбележи, че методът на измерване на интензитета на светлината, и по-специално видът на рецептора (колектора), ще окаже въздействие върху измерената стойност. Препоръчително е измерванията да бъдат направени с използването на сферичен (4 π) рецептор (който реагира на пряка и отразена светлина от всички ъгли над и под равнината на измерване), или 2 π рецептор (който реагира на светлината от всички ъгли над равнината на измерване). |
|
— |
Апарат за определяне на биомасата от водорасли. Преброяването на клетки, което е най-често използваният сурогатен параметър за биомаса от водорасли, може да бъде направено с използването на електронен брояч на частици, микроскоп с броячна камера или поточен цитометър. Другите заместители на биомасата могат да бъдат измерени с използването на поточен цитометър, флуориметър, спектрофотометър или колориметър. Полезно е да се изчисли коефициент на превръщане, свързващ броя на клетките със сухото тегло. За да се осигурят полезни измервания при ниски концентрации на биомаса при използването на спектрофотометър, може да е необходимо да се използват кювети с дължина на светлинния път поне 4 cm. |
1.8.2. Изпитвани организми
Могат да се използват няколко вида неприкрепени микроводорасли и цианобактерии. Щамовете, изброени в допълнение 1, са демонстрирани като подходящи при използването на процедурата на изпитване, определена в настоящия метод на изпитване.
Ако се използват други видове, следва да се докладва щамът и/или произходът. Трябва да бъде потвърдено, че експоненциалният растеж на избраните изпитвани водорасли може да бъде поддържан по време на периода на изпитване при преобладаващите условия.
1.8.3. Среда на растеж
Препоръчват се две алтернативни среди на растеж, средите на ОИСР и на AAP. Съставът на тези среди е показан в допълнение 2. Отбележете, че първоначалната стойност на pH и буферната способност (регулираща увеличаването на pH) на двете среди са различни. Следователно резултатите от изпитванията могат да бъдат различни в зависимост от използваната среда, особено когато се изпитват йонизиращи вещества.
Може да е необходимо изменение на средата на растеж за определени цели, например когато се изпитват метали или хелатни агенти или за изпитване при различни стойности на pH. Използването на изменена среда трябва да бъде подробно описано и обосновано (3)(4).
1.8.4. Начална концентрация на биомасата
Началната биомаса в изпитваните култури трябва да бъде еднаква във всички изпитвани култури и достатъчно ниска, за да позволява експоненциален растеж през целия инкубационен период без риск от изчерпване на хранителните вещества. Началната биомаса не трябва да превишава 0,5 mg/l като сухо тегло. Препоръчват се следните начални концентрации на клетки:
|
Pseudokirchneriella subcapitata |
5 x 103-104 |
клетки/ml |
|
Desmodesmus subspicatus |
2-5 x 103 |
клетки/ml |
|
Navicula pelliculosa |
104 |
клетки/ml |
|
Anabaena flos-aquae |
104 |
клетки/ml |
|
Synechococcus leopoliensis |
5 x 104-105 |
клетки/ml |
1.8.5. Концентрации на изпитваното вещество
Диапазонът на концентрация, в който е възможно да настъпят въздействия, може да бъде определен на базата на резултати от изпитвания за откриване на диапазон. За крайното определящо изпитване трябва да се изберат поне пет концентрации, подредени в геометрична поредица с фактор, който не превишава 3,2. За изпитвани вещества, показващи полегата крива на реагиране на концентрация, може да бъде обоснован и по-висок фактор. Поредицата от концентрации трябва по възможност да обхваща диапазона, при който се постига 5—75 % потискане на скоростта на растеж на водораслите.
1.8.6. Повторни проби и контролни проби
Планът на изпитването следва да включва три повторни проби от всяка една изпитвана концентрация. Ако не се изисква определяне на NOEC, планът на изпитването може да бъде изменен, за да се увеличи броят на концентрациите и да се намали броят на повторните проби за всяка концентрация. Броят на контролните повторни проби трябва да бъде поне три и в най-добрия случай следва да бъде два пъти повече от броя на повторните проби, използвани за всяка една изпитвана концентрация.
Може да бъде приготвен отделен набор от изпитвани разтвори за аналитично определяне на концентрациите на изпитваното вещество (вижте четвърта и шеста алинея от раздел 1.8.11).
Когато се използва разтворител, за да се направи разтворимо изпитваното вещество, в плана на изпитването трябва да бъдат включени допълнителни контролни проби, съдържащи разтворителя в същата концентрация, каквато е използвана в изпитваните култури.
1.8.7. Подготвяне на инокулант
За да се адаптират изпитваните водорасли към условията за изпитване и да се гарантира, че водораслите са във фаза на експоненциален растеж, когато се използват за инокулация на изпитваните разтвори, се приготвя инокулант в изпитваната среда 2—4 дни преди започване на изпитването. Биомасата от водорасли следва да бъде приспособена, за да се осигури преобладаване на експоненциалния растеж в инокуланта до започване на изпитването. Инокулантът се инкубира при същите условия като изпитваните култури. Измерете увеличаването на биомасата в инокуланта, за да гарантирате, че растежът е в рамките на нормалния диапазон за изпитвания щам при условията за култивиране. В допълнение 3 е описан пример за процедурата на култивиране на водорасли. За да се избегне синхронното делене на клетките по време на изпитването, би могло да се наложи втори стадий в размножаването на инокуланта.
1.8.8. Приготвяне на изпитваните разтвори
Всички разтвори за изпитване трябва да съдържат еднакви концентрации на средата на растеж и началната биомаса на изпитваните водорасли. Изпитваните разтвори на избраните концентрации обикновено се приготвят чрез смесване на стандартен разтвор на изпитваното вещество със средата на растеж и инокуланта. Стандартните разтвори обикновено се приготвят чрез разтваряне на веществото в изпитваната среда.
Разтворители, като например ацетон, терт-бутилов алкохол и диметилформамид, могат да се използват като носители за добавяне на вещества със слаба водоразтворимост към изпитваната среда (2)(3). Концентрацията на разтворителя не трябва да превишава 100 µl/l и към всички култури (включително и контролните) в изпитваните серии следва да се добави същата концентрация на разтворителя.
1.8.9. Инкубация
Запушете съдовете за изпитване с въздухопропускливи запушалки. Съдовете се разклащат и поставят в апаратурата за култивиране. По време на изпитването е необходимо водораслите да се държат в суспензия и да се улесни преносът на CO2. За тази цел следва да се осъществява постоянно разклащане или разбъркване. Културите следва да бъдат поддържани при температура в диапазона от 21 до 24 °C, регулирана с отклонение от ±2 °C. За видовете, различни от посочените в допълнение 1, например тропическите видове, може да са подходящи по-високи температури, при условие че могат да бъдат изпълнени критериите за валидност. Препоръчва се колбите да бъдат поставяни на случаен принцип и местата им в инкубатора да бъдат сменяни всеки ден.
pH на контролната среда не би трябвало да се увеличава с повече от 1,5 единици по време на изпитването. За метали и смеси, които частично се йонизират при стойности на pH около стойността на pH при изпитването, може да е нужно ограничаване на отклонението на pH, за да се получат възпроизводими и добре дефинирани резултати. Отклонение < 0,5 pH единици е технически осъществимо и може да бъде постигнато с осигуряване на адекватен процент на пренос на маса от CO2 от заобикалящия въздух към изпитвания разтвор, например чрез увеличаване на честотата на разклащане. Друга възможност е да се намали потребността от CO2 чрез намаляване на първоначалната биомаса или продължителността на изпитването.
Повърхността, където се инкубират културите, следва да получава непрекъсната хомогенна флуоресцентна светлина, например от вида „студена бяла светлина“ или „дневна светлина“. Щамовете от водорасли и цианобактерии се различават в изискванията си за светлина. Интензитетът на светлината следва да бъде избран така, че да подхожда на използвания изпитван организъм. За препоръчваните видове зелени водорасли интензитетът на светлината на нивото на изпитваните разтвори се избира в диапазона на 60—120 µE·m–2·s–1, когато се измерва в диапазона на фотосинтетичната ефективна дължина на вълната от 400—700 nm с използването на подходящ рецептор. Някои видове, по-специално Anabaena flos-aquae, растат добре при ниски интензитети на светлина и могат да бъдат увредени при високи интензитет. За такива видове следва да се избере среден интензитет на светлината в диапазона 40—60 µE·m–2·s–1. (За уреди за измерване на светлината, калибрирани в луксове, еквивалентният диапазон от 4 440—8 880 lux за студена бяла светлина съответства приблизително на препоръчвания интензитет на светлината от 60—120 µE·m–2·s–1). Интензитетът на светлината не се отклонява с повече от ±15 % от средния интензитет на светлината върху инкубационната площ.
1.8.10. Продължителност на изпитването
Продължителността на изпитването обикновено е 72 часа. Въпреки това може да се използва по-кратка или по-дълга продължителност на изпитванията, при условие че са спазени всички критерии за валидност в раздел 1.7.
1.8.11. Измервания и аналитични определения
Биомасата от водорасли във всяка колба се определя най-малко веднъж дневно по време на изпитването. Ако измерванията се правят на малки обеми, взети от изпитвания разтвора с пипета, те не бива да се връщат обратно.
Измерването на биомасата се извършва чрез ръчно преброяване на клетките с микроскоп или електронен брояч на частици (чрез преброяване на клетки и/или обем на биомаса). Могат да се използват алтернативни техники, например поточна цитометрия, in vitro или in vivo хлорофилна флуоресценция (6)(7) или оптична плътност, при условие че може да бъде демонстрирана задоволителна корелация с биомасата в диапазона на биомасата, която се явява по време на изпитването.
Стойността на pH на разтворите се измерва в началото и в края на изпитването.
При условие че е налице аналитична процедура за определяне на изпитваното вещество в използвания диапазон на концентрацията, изпитваните разтвори следва да бъдат анализирани, за да се потвърдят първоначалните концентрации и поддържането на концентрациите на експозиция по време на изпитването.
Анализът на концентрацията на изпитваното вещество в началото и края на изпитването при ниска и висока изпитвана концентрация, както и на концентрация около очакваната EC50 може да е достатъчен в случаите, при които има вероятност концентрациите на експозиция да варират с по-малко от 20 % от номиналните стойности по време на изпитването. Препоръчва се анализ на всички концентрации в началото и в края на изпитването, ако вероятността концентрациите да останат в рамките на 80—120 % от номиналните е малка. За летливи, нестабилни или силно адсорбиращи изпитвани вещества се препоръчва допълнително вземане на проби за анализ на 24-часови интервали по време на периода на експозиция, за да се определи по-добре загубата на изпитвано вещество. За тези вещества ще са необходими допълнителни повторни проби. Във всички случаи определянето на концентрациите на изпитваното вещество трябва да бъде извършвано само в един съд с повторна проба при всяка една изпитвана концентрация (или в съдържанието на съдовете, групирани от повторна проба).
Изпитваната среда, приготвена специално за анализ на концентрации на експозиция по време на изпитването, следва да бъде третирана по същия начин както тези, използвани за изпитването, т.е. те трябва да бъдат инокулирани с водорасли и инкубирани при еднакви условия. Ако се изисква анализ на концентрацията на разтвореното изпитвано вещество, може да е нужно да се отделят водораслите от средата. За предпочитане е отделянето да се прави чрез центрофугиране при ниска сила на притегляне, достатъчна за утаяване на водораслите.
Ако има доказателство, че концентрацията на веществото, което се изпитва, е била задоволително поддържана в рамките на ±20 % от номиналната или измерваната първоначална концентрация през цялото време на изпитването, анализът на резултатите може да бъде базиран на номиналните или измерените първоначални стойности. Ако отклонението от номиналната или измерената първоначална концентрация е по-голямо от ±20 %, анализът на резултатите следва да се основава на средната геометрична концентрация по време на експозиция или на модели, описващи намаляването на концентрацията на изпитваното вещество (3)(8).
Изпитването за потискане на растежа на водораслите е по-динамична система за изпитване от повечето други краткосрочни изпитвания за водна токсичност. Като следствие може да е трудно да се определят действителните концентрации на експозиции, особено за адсорбиращи вещества, изпитвани при ниски концентрации. В такива случаи изчезването на веществото от разтвор чрез адсорбция към увеличаващата се биомаса от водорасли не означава, че то е изгубено от системата за изпитване. Когато се анализира резултатът на изпитването, следва да се провери дали намаляването на концентрацията на изпитваното вещество в процеса на изпитване се придружава от намаляване в потискането на растежа. Ако това е така, може да се обмисли прилагането на подходящ модел, който описва намаляването на концентрацията на изпитваното вещество (8). В противен случай може да е подходящо анализът да се базира на резултатите от първоначалните (номинални или измерени) концентрации.
1.8.12. Други наблюдения
Следва да бъдат извършени наблюдения с микроскоп, за да се потвърди нормалният и здрав външен вид на инокуланта, както и да се види дали има промени във външния вид на водораслите (които могат да бъдат причинени от експозицията на въздействието на изпитваното вещество) в края на изпитването.
1.8.13. Гранично изпитване
При определени условия, например когато предварителното изпитване показва, че изпитваното вещество няма токсични въздействия при концентрации до 100 mg·l–1 или до неговата граница на разтворимост в изпитваната среда (в зависимост от това кое е по-ниско), може да бъде осъществено гранично изпитване, включващо сравнение на реакциите в контролна група и една от третираните групи (100 mg·l–1 или концентрация, равна на границата на разтворимост). Настоятелно се препоръчва това да бъде подкрепено от анализ на концентрацията на експозиция. Всички предишни описани условия на изпитване и критерии за валидност се прилагат към граничното изпитване, с изключение на това, че броят на третираните повторни проби трябва да бъде най-малко шест. Променливите на реакция в контролната и третираната група могат да бъдат анализирани с използването на статистически тест, за да се сравнят средните стойности, например t-тест на Student. Ако променливостта на двете групи не е еднаква, трябва да бъде извършен t-тест, предназначен за различни видове променливост.
1.8.14. Изменение за силно оцветените вещества
Ирадиацията (интензитетът на светлината) трябва да бъде в най-високия край на диапазона, предписан в настоящия метод за изпитване: 120 µE m–2 s–1 или по-висока.
Светлинният път трябва да бъде скъсен посредством намаляване на обема на изпитваните разтвори (в диапазона 5—25 ml).
Трябва да се приложи достатъчно раздвижване (например посредством умерено разклащане), за да се получи висока честота на експозиция на водораслите на висока ирадиация на повърхността на културата.
2. ДАННИ
2.1. ПОСТРОЯВАНЕ НА КРИВИ НА РАСТЕЖА
Биомасата в съдовете за изпитване може да се изрази в единици на сурогатния параметър, използван за измерване (например брой клетки, флуоресценция).
Табулирайте изчислената концентрация на биомасата в изпитваните култури и контролните проби заедно с концентрациите на изпитвания материал и времената на измерване, записани с разделителна способност най-малко час, за да начертаете графики на кривите на растежа. На този първи етап може да са полезни както логаритмични, така и линейни скали, но логаритмичните скали са задължителни и в общия случай дават по-добро представяне на вариациите в модела на растежа по време на периода за изпитване. Отбележете, че експоненциалният растеж дава права линия, когато се начертае в логаритмична скала, и че ъгълът на наклона на линията (наклон) показва специфичната скорост на растеж.
Като използвате графиките, проверете дали контролните култури растат експоненциално с очакваната скорост през цялото изпитване. Разгледайте внимателно всички точки с данни и вида на графиките и проверете необработените данни и процедурите за евентуални грешки. По-специално проверете точките с данни, за които изглежда, че се отклоняват поради системна грешка. Ако е очевидно, че могат да се установят и/или предположат процедурни грешки с голяма вероятност, специфичната точка с данни се маркира като отклонение и не се включва в последващия статистически анализ. (Нулева концентрация на водорасли в един от двата или трите съда с повторни проби може да е индикация, че съдът не е инокулиран правилно или не е бил почистен добре). Причините за отхвърлянето на точка с данни като отклонение трябва да се формулират ясно в протокола от изпитването. Приемливите причини са само (редки) грешки в процедурата, а не просто недостатъчно добра точност. Статистическите процедури за идентифициране на отклонения имат ограничено използване за този тип проблем и не могат да заменят експертната преценка. Препоръчително е точките с отклонения (маркирани като такива) да се запазят между точките с данни, показани в последващите графични или таблични представяния на данни.
2.2. ПРОМЕНЛИВИ НА РЕАКЦИЯ
Целта на изпитването е да се определят въздействията на изпитваното вещество върху растежа на водораслите. Настоящият метод на изпитване описва две променливи на реакция, тъй като държавите-членки имат различни предпочитания и регулаторни нужди. За да се приемат резултатите от изпитването във всички държави-членки, въздействията следва да се оценяват, като се използват и двете променливи на реакция a) и б), описани по-долу.
|
a) |
Средна специфична скорост на растеж: тази променлива на реакция се изчислява на базата на логаритмичното нарастване на биомасата през периода на изпитването, изразено за ден. |
|
б) |
Добив: тази променлива на реакция представлява биомасата в края на изпитването минус началната биомаса. |
За прилагането на настоящия метод съгласно регулаторната рамка на ЕС изчисляването на резултатите следва да се базира върху средната специфична скорост на растеж поради причините, описани по-долу. Би трябвало да се отбележи, че стойностите на токсичността, изчислени с използването на тези две променливи на реакция, не са сравними и тази разлика трябва да се разпознава, когато се използват резултатите от изпитването. Стойностите на ECx, базирани на средната специфична скорост на растеж (ErCx), в общия случай ще са по-високи от резултатите, базирани на добива (EyCx), ако се придържате към условията за изпитване на настоящия метод за изпитване поради математическата база на съответните подходи. Това не бива да се тълкува като разлика в чувствителността между двете променливи на реакция, просто двете стойности са математически различни. Понятието за средна специфична скорост на растеж се основава на общия модел на експоненциален растеж на водораслите в неограничени култури, където токсичността се оценява на базата на въздействията върху скоростта на растежа, без да зависи от абсолютното ниво на специфичната скорост на растеж на контролната проба, от наклона на кривата на реакция на концентрация или от продължителността на изпитването. Противоположно на това резултатите, базирани на добива, зависят от всички тези други променливи. EyCx зависи от специфичната скорост на растеж на вида водорасли, използвани във всяко от изпитванията, и от максималната специфична скорост на растеж, която може да варира между видовете и дори между различните щамове на водорасли. Тази променлива на реакция не трябва да се използва за сравняване на чувствителността към токсични вещества между видовете водорасли или дори между различните щамове. Докато използването на средната специфична скорост на растеж за оценка на токсичността се предпочита от научна гледна точка, оценките на токсичността, базирани на добива, също са включени в настоящия метод на изпитване, така че да бъдат удовлетворени действащите регулаторни изисквания в някои държави.
2.2.1. Средна скорост на растеж
Средната специфична скорост на растеж за определен период се изчислява като логаритмичното нарастване на биомасата от уравнението за всеки отделен съд с контролни проби и третирани проби:
 (ден–1)
(ден–1)
където:
|
µ i—j |
е средната специфичната скорост на растеж за време от i до j; |
|
X i |
е биомасата в момента i; |
|
X j |
е биомасата в момента j. |
Изчислете средната стойност за скоростта на растеж заедно с оценки на променливостта за всяка група третирани проби и контролна група.
Изчислете средната специфична скорост на растеж в течение на целия период на изпитването (обикновено 0—3 дни) с използване на първоначално инокулираната биомаса като начална стойност, а не измерената начална стойност, защото по този начин обикновено се получава по-голяма точност. Ако оборудването, което се използва за измерване на биомасата, позволява достатъчно точно определяне на малката биомаса на инокулума (например поточен цитометър), тогава може да се използва концентрацията на началната биомаса. Освен това оценете скоростта на растеж по сектори, изчислена като специфичните скорости на растеж за всеки ден по време на изпитването (дни 0—1, 1—2 и 2—3), и проверете дали скоростта на растеж на контролната проба остава постоянна (вижте критериите за валидност, раздел 1.7). Значително по-ниската специфична скорост на растеж в първия ден в сравнение с общата средна специфична скорост на растеж може да е показател за латентна фаза. Докато латентната фаза може да се сведе до минимум и практически да се елиминира в контролните култури чрез правилното размножаване на предварителна култура, латентната фаза в експонираната култура може да е индикатор за възстановяване след първоначален токсичен стрес или намалена експозиция поради загуба на изпитвано вещество (включително сорбция върху биомасата на водораслите) след първоначалната експозиция. От тук следва, че скоростта на растеж по сектори може да се оцени, за да се направи оценка на въздействията на изпитваното вещество, които възникват при процеса на експозиция. Наличието на съществени разлики между скоростта на растеж по сектори и средната скорост на растеж показва, че има отклонение от постоянния експоненциален растеж и има основание за внимателна проверка на кривите на растежа.
Процентното потискане на скоростта на растежа за всяка третирана повторна проба се изчислява от уравнението:

където:
|
% Ir: |
процентно потискане на средната специфична скорост на растеж; |
|
µC: |
средна стойност за средната специфична скорост на растеж (µ) в контролната група; |
|
µT: |
средна специфична скорост на растеж за третираната повторна проба. |
Когато за приготвянето на изпитваните разтвори се използват разтворители, за изчисляването на процентното потискане следва да се използват контролните проби на разтворителите, а не контролните проби без разтворители.
2.2.2. Добив
Добивът се изчислява като биомасата в края на изпитването минус началната биомаса за всеки отделен съд с контролни проби и с третирани проби. За всяка изпитвана концентрация и контролна проба следва да се изчисли средната стойност на добива заедно с оценки на променливостта. Процентното потискане на добива (%Iy) може да се изчисли за всяка третирана повторна проба, както следва:

където:
|
% Iy: |
процентно потискане на добива; |
|
YC: |
средна стойност за добива в контролната група; |
|
YT: |
стойност на добива в третираната повторна проба. |
2.3. ПОСТРОЯВАНЕ НА КРИВАТА НА РЕАКЦИЯ НА КОНЦЕНТРАЦИЯ
Нанесете процента на потискане като функция на логаритъма на концентрацията на изпитваното вещество и внимателно проверете графиката, като не отчитате точките с данни, които са изолирани като отклонения в първата фаза. Прекарайте гладка линия през точките с данни на око или чрез компютърна интерполация, за да получите първо впечатление от съотношението между концентрацията и реакцията, а след това продължете, като приложите по-подробен метод, за предпочитане някой компютризиран статистически метод. В зависимост от предполагаемото използване на данните, качеството (точност) и количеството на данните, както и наличието на инструменти за анализ на данни, може да се реши (и понякога е добре обосновано) на този етап анализът на данните да се прекрати и просто да се отчетат ключовите величини EC50 и EC10 (и/или EC20) от нанесената на око крива (вижте също раздела по-долу относно стимулираните въздействия). Обоснованите причини да не се използва статистически метод може да включват:
|
— |
данните не са подходящи за компютризирани методи, чрез които да се дадат по-надеждни резултати от тези, които могат да се получат чрез експертна оценка — в такива ситуации някои компютърни програми може дори да не дадат надеждно решение (повторенията може да не съвпадат и др.), |
|
— |
реакциите на стимулиран растеж не могат да се обработят адекватно с използване на наличните компютърни програми (вижте по-долу). |
2.4. СТАТИСТИЧЕСКИ ПРОЦЕДУРИ
Целта е да се получи количествено съотношение на реакция на концентрация чрез регресионен анализ. Може да се използва тегловна линейна регресия след извършване на преобразуване на данните от ответната реакция в линейни — например в единици пробит или логит или Weibull (9), но предпочитаните техники са процедурите на нелинейна регресия, които по-добре обработват неизбежните неравномерности в данните и отклоненията от гладките разпределения. При приближаването на нулево или пълно потискане подобни неравномерности може да бъдат увеличени от преобразуването, като по този начин пречат на анализа (9). Следва да се отбележи, че стандартните методи на анализ с помощта на преобразувания на пробит, логит или Weibull са предназначени за използване с дискретни данни (например смъртност или оцеляване) и трябва да бъдат изменени, за да се приспособят към данните за растежа или биомасата. Специфичните процедури за определяне на стойностите на ECx от непрекъснати данни могат да се намерят в (10)(11) и (12). Използването на нелинеен регресионен анализ е допълнително описано в допълнение 4.
За анализа на всяка от променливите на реакция използвайте съотношението на реакция на концентрация за да изчислите точковите оценки за стойностите на ECx. Когато е възможно, следва да се определят 95 % доверителни граници за всяка прогноза. Степента на съответствие на данните от реакцията и регресионния модел следва да се оцени графично или статистически. Регресионният анализ следва да се извърши като се използват реакциите на отделните повторни проби, а не средните стойности на група от третирани проби. Ако обаче налагането на нелинейна крива е трудно или несполучливо поради твърде голямото разсейване в данните, проблемът може да се заобиколи чрез извършване на регресия върху средните стойности на група като практичен начин за намаляване на влиянието на точки, за които има съмнения, че се отклоняват твърде много. Използването на тази възможност следва да се отбележи в протокола от изпитването като отклонение от нормалната процедура, тъй като нанасянето на отделните повторни проби на кривата не е дало добър резултат.
Оценките на EC50 и доверителните граници могат да се получат и чрез използване на линейна интерполация с оценка на извадковата грешка (13), ако наличните регресионни модели/методи не са подходящи за данните.
За оценяването на LOEC и следователно на NOEC и за въздействието на изпитваното вещество върху скоростта на растеж е необходимо да се сравнят средните стойности на третирана проба като се използват техниките за анализ на дисперсията (ANOVA). След това средната стойност за всяка концентрация трябва да се сравни със средната стойност на контролна проба с помощта на подходящ метод за многократно сравняване или метод за тестване на тенденция. Тестът на Dunnett или William може да е удобен (14)(15)(16)(17)(18). Необходимо е да се оцени дали е в сила предположението в ANOVA за хомогенност на променливостта. Тази оценка може да се извърши графично или чрез формален тест (18). Подходящи са тестовете на Levene или Bartlett. Невярното предположение за хомогенност на променливостта понякога може да се коригира чрез логаритмично преобразуване на данните. Ако хетерогенността на променливостта е много голяма и не може да се коригира чрез преобразувания, следва да се обмисли анализ с методи като низходящите тестове на Jonkheere за тенденции. Допълнителни указания относно определянето на NOEC могат да се намерят в (12).
Последните научни разработки доведоха до препоръката концепцията за NOEC да бъде премахната и заменена с точкови оценки на ECx, базирани на регресия. Не е установена подходящата стойност на x за това изпитване на водорасли. Изглежда, че диапазонът от 10 до 20 % е подходящ (в зависимост от избраната променлива на реакция) и е по-добре да бъдат отчетени и двете стойности EC10 и EC20.
2.5. СТИМУЛИРАНЕ НА РАСТЕЖА
Понякога се наблюдава стимулиране на растежа (отрицателно потискане) при ниски концентрации. Това може да е резултат от хормезис („токсична стимулация“) или от добавянето на стимулиращи растежа фактори с изпитвания материал към използваната минимална среда. Отбележете, че добавянето на неорганични хранителни вещества не би трябвало да има пряко въздействие, защото изпитваната среда следва да поддържа излишък от хранителни вещества през цялото време на изпитването. Обикновено стимулацията с ниска доза може да се пренебрегне при изчисляването на EC50, освен ако не е много крайна. При все това, ако е крайна или ако трябва да се изчисли стойността на ECx за ниско x, може да са необходими специални процедури. Ако е възможно, следва да се избягва заличаване на реакциите при стимулиране от анализа на данните, а ако наличният софтуер за нанасяне на криви не може да приеме незначителна стимулация, би могло да се използва линейна интерполация с оценка на извадковата грешка. Ако стимулацията е крайна, може да се обмисли използване на хормезен модел (19).
2.6. НЕТОКСИЧНО ПОТИСКАНЕ НА РАСТЕЖА
Поглъщащите светлина изпитвани материали могат да предизвикат намаляване на скоростта на растежа, тъй като засенчването намалява количеството налична светлина. Такива въздействия от физическо естество следва да бъдат отделени от токсичните въздействия чрез изменение на условията на изпитването, а първите следва да се отчитат отделно. Насоки могат да се намерят в (2) и (3).
3. ОТЧИТАНЕ
3.1. ПРОТОКОЛ ОТ ИЗПИТВАНЕТО
Протоколът от изпитването трябва да включва следното:
Изпитвано вещество:
|
— |
физическо естество и съответни физикохимични свойства, включително граница на водоразтворимост, |
|
— |
данни за химичната идентификация, включително чистота. |
Изпитвани видове:
|
— |
щам, доставчик или източник и използваните условия за културата. |
Условия на изпитването:
|
— |
дата на началото на изпитването и неговата продължителност, |
|
— |
описание на схемата на изпитването: съдове за изпитването, обеми на културите, плътност на биомасата в началото на изпитването, |
|
— |
състав на средата, |
|
— |
изпитвани концентрации и повторни проби (например брой на повторните проби, брой на изпитваните концентрации и използваната геометрична прогресия), |
|
— |
описание на подготвянето на изпитваните разтвори, включително използването на разтворители и др., |
|
— |
апаратура за култивиране, |
|
— |
интензитет и качество на светлината (източник, хомогенност), |
|
— |
температура, |
|
— |
изпитвани концентрации: номинални тествани концентрации и всички резултати от анализи за определяне на концентрацията на изпитваното вещество в съдовете за изпитване. Следва да се отчете ефективността за възстановяване на метода и границата на количествената оценка в тестовата матрица, |
|
— |
всички отклонения от настоящия метод на изпитване, |
|
— |
метод за определянето на биомасата и доказателство за корелация между измерения параметър и сухото тегло. |
Резултати:
|
— |
стойности на pH в началото и края на изпитването при всички третирани проби, |
|
— |
биомасата за всяка колба при всяка точка на измерване и метод за измерване на биомасата, |
|
— |
криви на растеж (графика на биомасата в зависимост от времето), |
|
— |
изчислените променливи на реакция за всяка третирана повторна проба със средни стойности и коефициент на вариациите за повторните проби, |
|
— |
графично представяне на съотношението концентрация/ефект, |
|
— |
оценки на токсичността за променливите на реакция, например EC50, EC10, EC20 и свързаните доверителни интервали. Ако са изчислени — LOEC и NOEC, както и статистическия метод за тяхното определяне, |
|
— |
ако е използван ANOVA, мащабът на въздействието, което може да се открие (например най-малката значима разлика), |
|
— |
всяка стимулация на растеж, открита в която и да е третирана проба, |
|
— |
всички други наблюдавани ефекти, например морфологични изменения във водораслите, |
|
— |
обсъждане на резултатите, включително всяко влияние върху резултата от изпитването, появило се поради отклонения от настоящия метод на изпитване. |
4. ЛИТЕРАТУРА
|
(1) |
OECD TG 201 (2006) Freshwater Alga and Cyanobacteria, Growth Inhibition Test |
|
(2) |
ISO 1998: Water quality — Guidance for algal growth inhibition tests with poorly soluble materials, volatile compounds, metals and waster water. ISO/DIS 14442 |
|
(3) |
OECD 2000: Guidance Document on Aquatic Toxicity Testing of Difficult Substances and mixtures. Environmental Health and Safety Publications. Series on Testing and Assessment, no. 23. |
|
(4) |
ISO 1998: Water quality — Sampling — Part 16: General Guidance for Biotesting. ISO 5667—16. |
|
(5) |
ISO 1993: Water quality — Algal growth inhibition test. ISO 8692 |
|
(6) |
Mayer, P., Cuhel, R. and Nyholm, N. (1997). A simple in vitro fluorescence method for biomass measurements in algal growth inhibition tests. Water Research 31: 2525—2531. |
|
(7) |
Slovacey, R.E. and Hanna, P.J. In vivo fluorescence determinations of phytoplancton chlorophyll, Limnology & Oceanography 22,5 (1977), pp.919—925 |
|
(8) |
Simpson, S.L., Roland, M.G.E., Stauber, J.L. and Batley, G.E. (2003) Effect of declining toxicant concentrations on algal bioassay endpoints. Environ. Toxicol. Chem 22, 2073—2079. |
|
(9) |
Christensen, E.R., Nyholm, N. (1984): Ecotoxicological Assays with Algae: Weibull Dose-Response Curves. Env. Sci. Technol. 19, 713—718. |
|
(10) |
Nyholm, N. Sørensen, P.S., Kusk, K.O. and Christensen, E.R. (1992): Statistical treatment of data from microbial toxicity tests. Environ. Toxicol. Chem. 11, 157—167. |
|
(11) |
Bruce, R.D., and Versteeg, D.J. (1992). A statistical procedure for modelling continuous toxicity data. Env. Toxicol. Chem. 11:1485—1494. |
|
(12) |
OECD. (2004). Guidance Document on Statistical Analysis of Ecotoxicity Data. |
|
(13) |
Norberg-King T.J. (1988) An interpolation estimate for chronic toxicity: The ICp approach. National Effluent Toxicity Assessment Center Technical Report 05—88. USEPA, Duluth, MN. |
|
(14) |
Dunnett, C.W. (1955) A multiple comparisons procedure for comparing several treatments with a control. J. Amer. Statist. Assoc. 50: 1096—1121 |
|
(15) |
Dunnett, C.W. (1964) New tables for multiple comparisons with a control. Biometrics 20: 482—491. |
|
(16) |
Williams, D.A. (1971) A test for differences between treatment means when several dose levels are compared with a zero dose control. Biometrics 27: 103—117. |
|
(17) |
Williams, D.A. (1972) The comparison of several dose levels with a zero dose control. Biometrics 28: 510—531. |
|
(18) |
Draper, N.R. and Smith, H. (1981). Applied Regression Analysis, second edition. Wiley, New York. |
|
(19) |
Brain P. and Cousens R. (1989). An equation to describe dose-responses where there is stimulation of growth at low doses. Weed Research, 29, 93—96. |
Допълнение 1
Щамове, за които е демонстрирана пригодност за изпитването
Зелени водорасли
|
— |
Pseudokirchneriella subcapitata (известни преди като Selenastrum capricornutum), ATCC 22662, CCAP 278/4, 61.81 SAG |
|
— |
Desmodesmus subspicatus (известни преди като Scenedesmus subspicatus) 86.81 SAG |
Диатомеи
|
— |
Navicula pelliculosa, UTEX 664 |
Цианобактерии
|
— |
Anabaena flos-aquae, UTEX 1444, ATCC 29413, CCAP 1403/13A |
|
— |
Synechococcus leopoliensis, UTEX 625, CCAP 1405/1 |
Източници на щамове
Препоръчаните щамове са налични в едновидови водораслови култури от следните колекции (по азбучен ред):
|
|
|
|
|
|
|
|
|
|
|
|
Външен вид и характеристики на препоръчваните видове
|
|
P. subcapitata |
D. subspicatus |
N. pelliculosa |
A. flos-aquae |
S. leopoliensis |
|
Външен вид |
Извити, усукани едноклетъчни |
Овални, повечето едноклетъчни |
Пръчици |
Вериги от овални клетки |
Пръчици |
|
Размери (L × W) µm |
8—14 × 2—3 |
7—15 × 3—12 |
7,1 × 3,7 |
4,5 × 3 |
6 × 1 |
|
Клетъчен обем (µm3/клетка) |
40—60 (1) |
60—80 (1) |
40—50 (1) |
30—40 (1) |
2,5 (2) |
|
Сухо тегло на клетката (mg/клетка) |
2—3 × 10–8 |
3—4 × 10–8 |
3—4 × 10–8 |
1—2 × 10–8 |
2—3 × 10–9 |
|
Скорост на растеж (3) (ден–1) |
1,5—1,7 |
1,2—1,5 |
1,4 |
1,1—1,4 |
2,0—2,4 |
Специфични препоръки за култивиране и работа с препоръчаните видове за изпитване
Pseudokirchneriella subcapitata и Desmodesmus subspicatus
Тези зелени водорасли като цяло се поддържат лесно в различни среди за култури. Информация относно подходящите среди е на разположение от колекциите с култури. Клетките обикновено са единични и измерването на плътността на клетките може лесно да се извърши с помощта на електронен брояч на частици или микроскоп.
Anabaena flos-aquae
Може да се използват различни среди за растеж за запазване на изходната култура. Особено важно е да се избягва възможността партидна култура да прескочи логаритмичната фаза на растеж при обновяване; възстановяването на този етап е трудно.
Anabaena flos-aquae развива агрегати от свързани помежду си вериги от клетки. Размерите на тези агрегати може да варират в зависимост от условията за култивиране. Може да e необходимо тези агрегати да се прекъснат при преброяване с микроскоп или електронен брояч на частици за определяне на биомасата.
Може да се използва обработване с ултразвук на подпробите за прекъсване на веригите, за да се намалят разминаванията при преброяването. По-дългата обработка с ултразвук от необходимата за накъсването на веригите за да станат те с по-малка дължина, може да разруши клетките. Интензивността на обработката с ултразвук и нейната продължителност трябва да са идентични за всяка третирана проба.
Пребройте достатъчно полета върху хемоцитометъра (най-малко 400 клетки), за компенсиране на разминаванията. Това ще подобри надеждността на определянето на плътността с микроскоп.
Може да се използва електронен брояч на частици за определяне на общия обем на клетките на Anabaena след прекъсването на клетъчните вериги чрез внимателна обработка с ултразвук. Енергията на ултразвука при обработката трябва да се регулира, за да се избегне разрушаването на клетките.
Използвайте вихров миксер или подобен подходящ метод, за да сте сигурни, че суспензията на водорасли, използвана за инокулиране на съдовете за изпитване, е добре разбъркана и хомогенна.
Съдовете за изпитване следва да се поставят върху въртящ се кръгово или възвратно-постъпателен шутел-апарат при около 150 оборота в минута. Алтернативно може да се използва непрекъснато разбъркване за намаляване на тенденцията при Anabaena да образува бучки. Ако се получи образуване на бучки, трябва да се внимава да се вземат представителни проби за измервания на биомасата. Може да е необходимо енергично разбъркване за раздробяване на бучките от водорасли преди вземането на проби.
Synechococcus leopoliensis
Може да се използват различни среди за отглеждане при съхраняването на изходната култура. Информация относно подходящите среди е налична от колекциите с култури.
Synechococcus leopoliensis расте като единични пръчковидни клетки. Клетките са много малки, което усложнява използването на броенето с микроскоп за измервания на биомасата. От полза са електронни броячи на частици, оборудвани за броене на частици до размери от приблизително 1 µm. Флуоресцентните измервания in vitro също са приложими.
Navicula pelliculosa
Може да се използват различни среди за отглеждане при съхраняването на изходната култура. Информация относно подходящите среди е налична от колекциите с култури. Отбележете, че в средата е нужен силикат.
Navicula pelliculosa може да образува агрегати при определени условия за отглеждане. Поради производството на липиди, клетките на водораслите понякога проявяват тенденция за натрупване в повърхностен слой. При такива обстоятелства трябва да се предприемат специални мерки, когато се вземат подпроби за определяне на биомасата, за да се вземат представителни проби. Може да е нужно енергично разбъркване, например с използване на вихров миксер.
(1) Измерено с електронен брояч на частици.
(2) Изчислено от размерите.
(3) Най-често наблюдавана скорост на растеж в среда на ОИСР при интензитет на светлината приблизително 70 µE·m–2·s–1 и 21 °C.
Допълнение 2
Среда на растеж
Може да се използва една от следните две среди на растеж:
Среда ОИСР: оригиналната среда на ОИСР TG 201, също съгласно ISO 8692
US. EPA среда на AAP, също съгласно ASTM.
При приготвянето на тези среди следва да се използват реактив или химикал с високоаналитични качества и дейонизирана вода.
Състав на средата на AAP (US. EPA) и на средата на ОИСР TG 201
|
Компонент |
EPA |
ОИСР |
||
|
|
mg/l |
mM |
mg/l |
mM |
|
NaHCO3 |
15,0 |
0,179 |
50,0 |
0,595 |
|
NaNO3 |
25,5 |
0,300 |
|
|
|
NH4Cl |
|
|
15,0 |
0,280 |
|
MgCl2·6(H2O) |
12,16 |
0,0598 |
12,0 |
0,0590 |
|
CaCl2·2(H2O) |
4,41 |
0,0300 |
18,0 |
0,122 |
|
MgSO4·7(H2O) |
14,6 |
0,0592 |
15,0 |
0,0609 |
|
K2HPO4 |
1,044 |
0,00599 |
|
|
|
KH2PO4 |
|
|
1,60 |
0,00919 |
|
FeCl3·6(H2O) |
0,160 |
0,000591 |
0,0640 |
0,000237 |
|
Na2EDTA·2(H2O) |
0,300 |
0,000806 |
0,100 |
0,000269 (1) |
|
H3BO3 |
0,186 |
0,00300 |
0,185 |
0,00299 |
|
MnCl2·4(H2O) |
0,415 |
0,00201 |
0,415 |
0,00210 |
|
ZnCl2 |
0,00327 |
0,000024 |
0,00300 |
0,0000220 |
|
CoCl2·6(H2O) |
0,00143 |
0,000006 |
0,00150 |
0,00000630 |
|
Na2MoO4·2(H2O) |
0,00726 |
0,000030 |
0,00700 |
0,0000289 |
|
CuCl2·2(H2O) |
0,000012 |
0,00000007 |
0,00001 |
0,00000006 |
|
pH |
7,5 |
8,1 |
||
При изпитването с диатомеята Navicula pelliculosa и към двете среди трябва да се добави Na2SiO3·9H2О за получаване на концентрация от 1,4 mg Si/l.
Стойността на pH се получава при равновесие между карбонатната система на средата и парциалното налягане на CO2 в атмосферния въздух. Приблизителното съотношение между pH при 25 °C и моларната концентрация на бикарбонатната система е:
pHeq = 11,30 + log [HCO3]
С 15 mg NaHCO3/l, pHeq = 7,5 (среда на US. EPA), а с 50 mg NaHCO3/l, pHeq = 8,1 (среда на ОИСР).
Елементен състав на изпитваните среди
|
Елемент |
EPA |
ОИСР |
|
|
mg/l |
mg/l |
|
C |
2,144 |
7,148 |
|
N |
4,202 |
3,927 |
|
P |
0,186 |
0,285 |
|
K |
0,469 |
0,459 |
|
Na |
11,044 |
13,704 |
|
Ca |
1,202 |
4,905 |
|
Mg |
2,909 |
2,913 |
|
Fe |
0,033 |
0,017 |
|
Mn |
0,115 |
0,115 |
Приготвяне на средата на ОИСР
|
Хранително вещество |
Концентрация в изходния разтвор |
|
Изходен разтвор 1: макронутриенти |
|
|
NH4Cl MgCl2·6H2O CaCl2·2H2O MgSO4·7H2O KH2PO4 |
1,5 g·l–1 1,2 g·l–1 1,8 g·l–1 1,5 g·l–1 0,16 g·l–1 |
|
Изходен разтвор 2: желязо |
|
|
FeCl3·6H2O Na2EDTA·2H2O |
64 mg·l–1 100 mg·l–1 |
|
Изходен разтвор 3: микроелементи |
|
|
H3BO3 MnCl2·4H2O ZnCl2 CoCl2·6H2O CuCl2·2H2O Na2MoO4·2H2O |
185 mg·l–1 415 mg·l–1 3 mg·l–1 1,5 mg·l–1 0,01 mg·l–1 7 mg·l–1 |
|
Изходен разтвор 4: бикарбонат |
|
|
NaHCO3 |
50 g·l–1 |
|
Na2SiO3·9H20 |
|
Стерилизирайте изходните разтвори с мембранно филтриране (среден диаметър на порите 0,2 µm) или чрез обработка в автоклав (120 °C, 15 минути). Съхранявайте разтворите на тъмно при 4 °C.
Не обработвайте изходни разтвори 2 и 4 в автоклав, а ги стерилизирайте с мембранно филтриране.
Пригответе средата на растеж чрез добавяне на съответния обем изходен разтвор от 1 до 4 към вода:
Добавете до 500 ml стерилизирана вода:
|
— |
10 ml от изходен разтвор 1 |
|
— |
1 ml от изходен разтвор 2 |
|
— |
1 ml от изходен разтвор 3 |
|
— |
1 ml от изходен разтвор 4 |
Допълнете до 1 000 ml със стерилизирана вода
Оставете достатъчно време за постигане на равновесие на средата с атмосферния CO2, при необходимост чрез барботиране със стерилен филтриран въздух в течение на няколко часа.
Приготвяне на средата на AAP
|
A1.1. |
Добавете 1 mL от всеки изходен разтвор в A1.2.1 — A1.2.7 към приблизително 900 mL дейонизирана или дестилирана вода и след това разредете до 1 L. |
|
A1.2. |
Изходните разтвори с макронутриенти се правят чрез разтварянето на следните съставки в 500 mL дейонизирана или дестилирана вода. Реактивите A1.2.1, A1.2.2, A1.2.3 и A1.2.4 могат да се комбинират в един изходен разтвор. |
|
A1.2.1. |
NaNO3 -12,750 g. |
|
A1.2.2. |
MgCL2·6H2O -6,082 g. |
|
A1.2.3. |
CaCl2·2H2O -2,205 g. |
|
A1.2.4. |
Изходен разтвор с микронутриенти — (вижте A1.3). |
|
A1.2.5. |
MgSO4·7H2O -7,350 g. |
|
A1.2.6. |
K2HPO4 -0,522 g. |
|
A1.2.7. |
NaHCO3 -7,500 g. |
|
A1.2.8. |
Na2SiO3·9H2O — вижте забележка A1.1. Забележка A1.1 — Използвайте само видове диатомеи за тестване. Може да се добавят директно (202,4 mg) или във вид на изходен разтвор за получаването на крайна концентрация в средата от 20 mg/L Si. |
|
A1.3. |
Изходният разтвор с микронутриенти се прави чрез разтваряне на следните съставки в 500 mL дейонизирана или дестилирана вода: |
|
A1.3.1. |
H3BO3 -92,760 mg. |
|
A1.3.2. |
MnCl2·4H2O -207,690 mg. |
|
A1.3.3. |
ZnCl2 -1,635 mg. |
|
A1.3.4. |
FeCl3·6H2O -79,880 mg. |
|
A1.3.5. |
CoCl2·6H2O -0,714 mg. |
|
A1.3.6. |
Na2MoO4·2H2O -3,630 mg. |
|
A1.3.7. |
CuCl2·2H2O -0,006 mg. |
|
A1.3.8. |
Na2EDTA·2H2O -150,000 mg. [Динатриев (етилендинитрило) тетраацетат]. |
|
A1.3.9. |
Na2SeO4·5H2O -0,005 mg Вижте забележка A1.2. Забележка A1.2 — Използвайте само в среда за изходни култури на вида диатомея. |
|
A1.4. |
Регулирайте pH до 7,5 ± 0,1 с 0,1 N или 1,0 N NaOH или HCl. |
|
A1.5. |
Филтрирайте средите в стерилен контейнер през 0,22-μm мембранен филтър, ако ще се използва брояч на частици или през 0,45-μm филтър, ако няма да използвате брояч на частици. |
|
A1.6. |
Съхранявайте средата на тъмно при приблизително 4 °C до влизане в употреба. |
(1) Моларното отношение на ЕДТА към желязото с малко надхвърля единица. Това предотвратява утаяването на желязото и същевременно свежда до минимум образуването на комплекси от йони на тежки метали.
Допълнение 3
Пример на процедура за култивиране на водорасли
Общи наблюдения
Целта на култивирането въз основа на следната процедура е да се получат култури на водорасли за изпитвания на токсичност.
Трябва да се използват подходящи методи, за да се гарантира, че културите на водорасли не са заразени с бактерии. Желателни са култури с отсъствие на чужди организми, но трябва да бъдат въведени и използвани едновидови водораслови култури.
Всички операции трябва да се извършват при стерилни условия, за да се избегне замърсяването с бактерии и други водорасли.
Оборудване и материали
Вижте в метод на изпитване: апаратура.
Процедури за получаване на култури на водорасли
Приготвяне на хранителни разтвори (среди):
Всички хранителни соли на средата се приготвят като концентрирани изходни разтвори и се съхраняват на тъмно и хладно. Тези разтвори се стерилизират чрез филтриране или обработка в автоклав.
Средата се приготвя чрез добавяне на правилното количество от изходния разтвор към стерилна дестилирана вода, като се внимава да не възникнат инфекции. За твърда среда се добавя 0,8 процента агар.
Изходна култура:
Изходните култури са малки култури на водорасли, които периодично се прехвърлят в прясна среда, за да играят ролята на първоначален материал за изпитване. Ако културите не се използват периодично, те се разполагат на ивици в наклонени тръбички с агар. Те се прехвърлят в прясна среда най-малко веднъж на всеки два месеца.
Изходните култури се отглеждат в конични колби, съдържащи подходящата среда (обем около 100 ml). Когато водораслите се инкубират при 20 °C при непрекъснато осветяване е необходимо преместване веднъж седмично.
При преместването известно количество от „старата“ култура се прехвърля със стерилни пипети в колба с прясна среда, така че при бързорастящите видове началната концентрация е около 100 пъти по-малка, отколкото при старата култура.
Скоростта на растеж на вида може да се определи от кривата на растежа. Ако тя е известна, е възможно да се оцени плътността, при която културата следва да се прехвърли в нова среда. Това трябва да се направи преди културата да достигне фазата на смърт.
Предкултура:
Предкултурата е предназначена да осигури известно количество водорасли, подходящи за инокулацията на култури за изпитване. Предкултурата се инкубира при условията на изпитването и се използва докато все още расте експоненциално, обикновено след инкубационен период от 2 до 4 дни. Когато културите на водорасли съдържат деформирани или анормални клетки, те трябва да се изхвърлят.
Допълнение 4
Анализ на данните чрез нелинейна регресия
Общи съображения
Реакцията при изпитването на водорасли и други микробиални изпитвания на растеж — растеж на биомаса — по своя характер е непрекъсната или метрична променлива — скоростта на процеса, ако се използва скоростта на растеж и нейният интеграл във времето, ако е избрана биомасата. И двете се отнасят към съответната средна реакция на неекспонирани повторни контролни проби, които дават максимална реакция при наложените условия — със светлината и температурата като основни определящи фактори при изпитването на водорасли. Системата е разпределена или хомогенна и биомасата може да се разглежда като постоянен параметър, без да се вземат предвид отделните клетки. Разпределението на променливостта на типа реакция за такава система е свързано единствено с експериментални фактори (типично описвани чрез нормален логаритъм или нормално разпределение на грешки). Това е противоположно на типичните реакции на биологичния тест с количествени данни, за които допустимото отклонение (обикновено с биномно разпределение) на отделните организми често се приема за доминиращата компонента на променливостта. Реакциите на контролните проби тук са нулево или фоново ниво.
В неусложнените ситуации нормализираната или относителната реакция, r, намалява монотонно от 1 (нулево потискане) до 0 (100-процентно потискане). Отбележете, че за всички реакции е налице съответстваща грешка и че очевидно отрицателни потискания могат да се изчисляват като резултат от случайна грешка.
Регресионен анализ
Модели
Регресионният анализ има за цел да опише количествено кривата концентрация—реакция във формата на математическа регресионна функция Y = f (C) или по-често F (Z), където Z = log C. Използването на обратната функционална зависимост C = f–1 (Y) позволява да се изчислят числата за ECx, включително EC50, EC10 и EC20, както и техните 95 % доверителни граници. Доказано е, че няколко прости математически функционални форми успешно описват съотношението концентрация— реакция, получено при изпитванията за потискане растежа на водорасли. Функциите включват например логистичното уравнение, несиметричното уравнение на Weibull и функцията на разпределението на нормалния логаритъм, всички от които са сигмоидални криви, асимптотично клонящи към единица за C → 0 и към нула за C → безкрайност.
Използването на модели с непрекъсната прагова функция (например моделът на Koyman „за потискане на растежа на популация“ — Kooijman et al., 1996 г.) е предложено наскоро или се приема като алтернатива на асимптотичните модели. Този модел предполага липсата на ефекти при концентрации под определен праг, EC0+, който се оценява чрез екстраполация на съотношението концентрация— реакция до пресичането с оста на концентрацията с помощта на проста непрекъсната функция, която не е разграничима в началната точка.
Обърнете внимание, че анализът може да бъде просто минимизиране на суми от остатъчни квадрати (като се приема постоянна променливост) или претеглените квадрати, ако хетерогенността на променливостта е компенсирана
Процедура
Процедурата може да се опише както следва: изберете подходящо функционално уравнение, Y = f (C) и го съгласувайте с данните чрез нелинейна регресия. За предпочитане е да използвате измерванията от всяка отделна колба, а не средните стойности на повторните проби, за да извлечете колкото е възможно повече информация от данните. От друга страна, ако променливостта е голяма, практическият опит предполага, че средните стойности на повторните проби могат да доведат до по-устойчива математическа оценка, по-слабо повлияна от произволните грешки в данните, отколкото при запазването на всяка отделна точка с данни.
Постройте описващата крива и измерените данни и проверете дали налагането на кривата е подходящо. Анализът на остатъчни вещества може да е особено полезен инструмент за тази цел. Ако избраното функционално съотношение за налагане на реакцията на концентрация не описва добре цялата крива или съществена част от нея, като например реакцията при ниски концентрации, изберете друга възможност за налагане на кривата — например несиметрична крива като функцията на Weibull вместо симетрична. Отрицателните потискания могат да представляват проблем, например с функцията на разпределение на нормалния логаритъм, което също изисква алтернативна функция на регресия. Не се препоръчва да се обозначава с нула или малка положителна стойност на такива отрицателни стойности, защото това изкривява разпределението на грешките. Може да е подходящо да правите отделни налагания на частите от кривата като например частта на слабо потискане, за да оцените стойностите за EClow x. Изчислете от описващото уравнение (чрез „обратно изчисление“, C = f–1 (Y), оценките на характерните точки на ECx и отчетете като минимум оценките за EC50 и една или две за EClow x. Опитът от практическите изпитвания е показал, че точността на изпитването на водорасли обикновено позволява достатъчно точна оценка при 10 % ниво на потискане, ако точките с данни са достатъчно — освен ако не възникне стимулиране при ниски концентрации като фактор за смесване на ефектите. Точността на оценката за EC20 често е значително по-добра от тази на EC10, защото EC20 обикновено е разположена на приблизително линейната част на централната крива на реакция на концентрация. Понякога може да е трудно да се тълкува EC10 поради стимулирането на растежа. Така че докато EC10 обикновено се получава с достатъчна точност, препоръчително е да се отчита винаги и EC20.
Тегловни фактори
Експерименталната променливост в общия случай не е постоянна и типично включва пропорционална компонента, следователно има благоприятни условия за рутинно извършване на тегловна регресия. При такъв анализ обикновено се приема, че тегловните фактори са обратнопропорционални на променливостта:
Wi = 1/Var(ri)
Много програми за регресия имат възможност за анализ на тегловна регресия с тегловни фактори, изброени в таблица. За удобство, тегловните фактори следва да се нормализират, като се умножат с n/Σ wi (n е броят на точките с данни), така че тяхната сума да е равна на единица.
Нормализиране на реакциите
Нормализирането чрез средните стойности на реакциите от контролните проби създава някои принципни проблеми и води до получаване на твърде усложнена структура на променливост. Когато реакциите се разделят на средната стойност на реакцията на контролната проба, за да се получи процентът на потискане, се внася допълнителна грешка, предизвикана от грешката в средната стойност на контролната проба. Освен ако тази грешка не е пренебрежимо малка, тегловните фактори за регресията и доверителните граници трябва да бъдат коригирани от ковариацията с контролната проба (17). Трябва да се има предвид, че високата точност на изчислената средна стойност за реакцията на контролната проба е важна при свеждането до минимум на цялостната променливост за относителната реакция. Тази променливост е следната:
индексът i се отнася до нивото i на концентрацията, а индексът 0 — за контролните проби)
Yi = относителна реакция = ri/r0 = 1 — I = f (Ci)
с променливост:
Var (Yi) = Var (ri/r0) ≈ (∂ Yi / ∂ ri)2·Var (ri) + (∂ Yi/ ∂ r0)2·Var (r0)
и тъй като
(∂ Yi / ∂ ri) = 1/r0 и (∂ Yi / ∂ r0) = ri/r0 2
с нормално разпределени данни и mi и m0 повторни проби:
Var (ri) = σ2/mi
общата променливост на относителната реакция Yi следователно става:
Var (Yi) = σ2/(r0 2 mi) + ri 2·σ2/r0 4 m0
Грешката върху средната стойност на контролна проба е обратнопропорционална на корен квадратен от осреднените стойности на повторните контролни проби и в някои случаи може да се обоснове включването на предишни статистически данни, като по този начин в голяма степен се намалява грешката. Алтернативната процедура е данните да не се нормализират и да се налагат абсолютните стойности на реакциите, включително данните от реакциите на контролните проби, но като се въведе стойността на реакцията на контролната проба като допълнителен параметър, който се налага чрез нелинейна регресия. При използване на обикновеното уравнение за регресия с два параметъра този метод изисква налагане на три параметъра и следователно се нуждае от повече точки с данни от нелинейната регресия върху данни, които се нормализират с помощта на предварително зададена реакция от контролна проба.
Обратни доверителни интервали
Изчисляването на доверителните интервали за нелинейна регресия чрез обратна оценка е доста сложно и не е налична стандартна възможност в обикновените пакети от статистически компютърни програми. Приблизителните доверителни граници могат да бъдат получени със стандартните програми за нелинейна регресия с нова параметризация (Bruce and Versteeg, 1992), което включва повторно съставяне на математическото уравнение с желаните оценки на точките, например EC10 и EC50 като параметри, които ще се оценяват. (Нека функцията е I = f (α, β, концентрация) и като използваме дефиниционните съотношения f (α, β, EC10) = 0,1 и f (α, β, EC50) = 0,5 да заместим f (α, β, концентрация) с еквивалентна функция g (EC10, EC50, концентрация).
По-директно изчисляване (Andersen et al, 1998) се извършва чрез запазване на оригиналното уравнение и използване на разлагането на Taylor към средните стойности на ri и r0.
Напоследък станаха популярни методите „boot strap“. Тези методи използват измерените данни и често вземане на повторни извадки, насочвано чрез генератор на случайни числа, за оценка на разпределението на емпиричната променливост.
Литература
Kooijman, S.A.L.M.; Hanstveit, A.O.; Nyholm, N. (1996): No-effect concentrations in algal growth inhibition tests. Water Research, 30, 1625—1632.
Bruce, R.D. and Versteeg, D.J.(1992) A Statistical Procedure for Modelling Continuous Ecotoxicity Data. Env. Toxicol. Chem.11, 1485—1494
Andersen, J.S., Holst, H., Spliid, H., Andersen, H., Baun, A. & Nyholm, N. (1998): Continuous ecotoxicological data evaluated relative to a control response. Journal of Agricultural, Biological and Environmental Statistics, 3, 405—420.
ПРИЛОЖЕНИЕ V
|
C.25. |
АЕРОБНА МИНЕРАЛИЗАЦИЯ В ПОВЪРХНОСТНИ ВОДИ — СИМУЛАЦИОННО ИЗПИТВАНЕ ЗА БИОРАЗГРАЖДАНЕ |
1. МЕТОД
Този метод е еквивалентен на ОИСР TG 309 (2004)
1.1. ВЪВЕДЕНИЕ
Целта на това изпитване е измерването на продължителността на биоразграждането на изпитвано вещество при ниска концентрация в аеробна природна вода и количествена оценка на наблюденията под формата на кинетични уравнения. Това симулационно изпитване представлява лабораторно серийно изпитване в колба, което има за цел определяне степента на биоразграждане на органични вещества чрез изследване на проби от природни повърхностни води (прясна, полусолена или морска). То се базира на ISO/DIS 14592—1 (1), като включва и елементи от методите на изпитване C.23 и C.24 (2)(3). По избор, при голяма продължителност на времето за изпитванията серийният способ се заменя с полунепрекъсваем, с цел да се предотврати влошаването на микросистемата за изпитване. Основната цел на симулационното изпитване е определянето на минерализацията на изпитваното вещество в повърхностна вода, а минерализацията служи като основа за изразяването на кинетиката на разграждането. При все това, изпитването има и една второстепенна незадължителна цел — да се получи информация за първичното разграждане и за формирането на основните продукти от трансформацията. Идентифицирането на продуктите на трансформацията и, по възможност, определяне на концентрациите им са особено важни в случая на вещества, които се минерализират много бавно (напр. с период на полуразпад за общ остатъчен 14C, надхвърлящ 60 дни). Поради аналитични ограничения, за идентифициране и количествена оценка на основните продукти на трансформацията, обикновено би следвало да се използват по-високи концентрации на изпитваното вещество (напр. > 100 μg/l).
При това изпитване ниска концентрация означава концентрация (напр. по-малка от 1 μg/l до 100 μg/l), която е достатъчно ниска, за да гарантира, че кинетиката на биоразграждането, получена при изпитването, отразява тази, която се очаква в околната среда. Сравнено с общата маса на биоразградими въглеродни субстрати, налични в природната вода, използвана в изпитването, изпитваното вещество с ниска концентрация ще послужи като вторичен субстрат. Това предполага, че очакваната кинетика на биоразграждане е от първи порядък („ненарастваща“ кинетика), както и че изпитваното вещество може да бъде разградено чрез „кометаболизъм“. Кинетиката от първи порядък означава, че скоростта на разграждане (mg/L/ден) е пропорционална на концентрацията на субстрата, която намалява с времето. При същинската кинетика от първи порядък специфичната константа на скоростта на разграждане — k не зависи от времето и концентрацията, т.е. k не се изменя осезаемо по време на експеримента и не се влияе от добавената концентрация между отделните експерименти. По дефиниция специфичната константа на скоростта на разграждане е равна на относителната промяна на скоростта във времето: k = (1/C) · (dC/dt). Макар и при зададените условия обикновено се очаква кинетика от първи порядък, възможно е наличието на определени обстоятелства, при които да е по-подходяща друг вид кинетика. Отклонения от кинетика от първи порядък могат да се наблюдават, например ако проявата на масов трансфер, какъвто е скоростта на дифузия, вместо да ограничи скоростта на биологична реакция, ограничава скоростта на биотрансформация. Въпреки това, данните почти винаги могат да бъдат описани чрез кинетика от псевдо-първи порядък, приемайки зависима от концентрацията константа на скоростта.
Информация за биоразграждането на изпитваното вещество при по-високи концентрации (напр. от стандартни скрининг изпитвания), както и информация за абиотичната биоразградимост, продуктите на трансформацията и съответните физико-химични свойства, следва да бъде налична преди започване на изпитването, за да се подпомогне планирането на експеримента и интерпретирането на резултатите. Използването на маркирани с 14C изпитвани вещества и определянето на фазовото разпределение на 14C в края на изпитването позволяват установяването на пълната биоразградимост. При използване на немаркирани изпитвани вещества пълната биоразградимост може да бъде установена единствено при изпитване на по-високи концентрации и ако са известни всички основни продукти на трансформацията.
1.2. ОПРЕДЕЛЕНИЯ
Първично биоразграждане: Структурната промяна (трансформация) на химичното вещество под влияние на микроорганизми, в резултат на което настъпва загуба на химичната природа.
Функционално биоразграждане: Структурната промяна (трансформация) на химично вещество под влияние на микроорганизми, в резултат на което настъпва загуба на определено свойство.
Пълно аеробно биоразграждане: Разпадането на дадено химично вещество под действието на микроорганизми в присъствието на кислород до въглероден диоксид, вода и минерални соли на други налични елементи (минерализация) и производството на нова биомаса и органични продукти на микробиалния биосинтез.
Минерализация: Разлагането на дадено химично вещество или органична материя под действието на микроорганизми и при наличието на кислород до въглероден диоксид, вода и минерални соли на някакви други налични елементи.
Латентен период: Периодът от началото на изпитването до момента на адаптация на разлагащите се микроорганизми и на осезаемо увеличаване на степента на биоразграждане на дадено химично вещество или органична материя (напр. 10 % от максималното теоретично биоразграждане или по-нисък процент, в зависимост от точността на способа за измерване).
Максимална степен на биоразграждане: Степента на биоразграждане на дадено химично вещество или органична материя по време на изпитване, измерена в проценти, над която не се наблюдава допълнително биоразграждане в рамките на изпитването.
Първичен субстрат: Съвкупност от естествен въглерод и енергийни източници, осигуряващи растеж и поддържане на микробиалната биомаса.
Вторичен субстрат: Субстратна съставка, налична в толкова ниска концентрация, че при нейното биоразграждане на съответните микроорганизми се доставят незначителни количества въглерод и енергия, в сравнение с въглерода и енергията, получени при разграждането на основните субстратни съставки (първични субстрати).
Скоростна константа на разграждане: Скоростна константа от първи порядък или псевдо-първи порядък k (d–1), показва скоростта на процесите на разграждане. При серийните експерименти k се изчислява от началната част на кривата на разграждане, получена след края на латентната фаза.
Период на полуразпад, t1/2 (d): Термин, използван за характеризиране на скоростта на реакция от първи порядък. Представлява интервала от време, съответстващ на двукратно намаляване на концентрацията. Полуразпадът и скоростната константа на разграждане са свързани чрез уравнението t1/2 = ln2/k.
Период на полуразграждане, DT50 (d): термин, използван за количествено определяне на резултата от изпитванията за биоразграждане. Представлява интервала от време, включително и латентната фаза, който е необходим за достигането на стойност на биоразграждане от 50 %.
Граница на откриваемост (LOD) и граница на количествено определяне (LOQ): Границата на откриваемост (LOD) представлява концентрацията на дадено вещество, под която видът на веществото не може да бъде разграничен от аналитични продукти. Границата на количествено определяне (LOQ) представлява концентрацията на дадено вещество, под която концентрацията не може да бъде установена с приемлива точност.
Разтворен органичен въглерод (DOC): Онази част от органичния въглерод във водна проба, която не може да бъде отделена чрез определеното фазово разделяне, например чрез центрофугиране при 40 000 ms–2 в продължение на 15 минути или чрез мембранно филтриране, използвайки мембрани с отвори с диаметър 0,2 μm — 0,45 μm.
Активност на общия органичен 14C (TOA): Общата активност на органичен 14C, свързана с органичния въглерод.
Активност на разтворения органичен 14C (DOA): Общата активност на 14C, свързана с разтворения органичен въглерод.
Активност на органичен 14C под формата на твърди частици (POA): общата активност на 14C, свързана с органичния въглерод под формата на твърди частици.
1.3. ПРИЛОЖИМОСТ НА ИЗПИТВАНЕТО
Симулационното изпитване е приложимо за нелетливи или слабо летливи органични вещества, изпитвани при ниски концентрации. При използването на отворени колби в контакт с атмосферата (напр. със запушалки от вата), веществата с константи по закона на Хенри под около 1 Pa·m3/mol (приблиз. 10–5 atm·m3/mol) могат да се считат за практически нелетливи. При използването на затворени колби за парофазов анализ е възможно изпитването на слабо летливи вещества (с константа по закона на Хенри < 100 Pa·m3/mol или < 10–3 atm·m3/mol) без загуби от системата за изпитване. Загуба на вещества, маркирани с 14C, може да възникне, ако не се вземат съответните предпазни мерки, при отстраняването на въглероден диоксид. В такива ситуации може да се наложи улавянето на въглеродния диоксид във вътрешен абсорбер с основа или използването на външна абсорбционна система за въглероден диоксид (директно определяне на 14CO2; вж. 3). За определянето на кинетиката на биоразграждането концентрациите на изпитваното вещество трябва да бъдат под степента на неговата водоразтворимост. Въпреки това, следва да се отбележи, че данните от научни източници за водоразтворимостта могат да са значително по-високи от разтворимостта на изпитваното вещество в природни води. По избор може да бъде установена разтворимостта на особено слабо водоразтворими изпитвани вещества, като се използват изпитваните природни води.
Методът може да бъде използван за симулиране на биоразграждане в повърхностни води без наличие на груби частици (пелагично изпитване) или в мътна повърхностна вода, която примерно може да се намира в близост до воден/седиментен обект (изпитване със суспендирани утайки).
1.4. ПРИНЦИП НА ИЗПИТВАНЕТО
Изпитването се провежда на серии чрез инкубиране на изпитваното вещество или само с повърхностна вода (пелагично изпитване), или с повърхностна вода с наличие на суспендирани неразтворени вещества/утайки със сухо вещество от 0,01 до 1 g/l (изпитване със суспендирани утайки) с цел да се симулира воден обект със суспендирани неразтворени вещества или ресуспендирани утайки. За повечето повърхностни води е типично концентрацията на суспендираните неразтворени вещества/утайки да попада в долната част на този интервал. Колбите за изпитване се инкубират на тъмно при температура като на околната среда, при аеробни условия и с разклащане. За определянето на кинетиката на разграждане е необходимо използването на най-малко две различни концентрации на изпитваното вещество. Концентрациите трябва да се различават помежду си от 5 до 10 пъти и трябва да отразяват очаквания диапазон на концентрации в околната среда. Максималната концентрация на изпитваното вещество не трябва да надвишава 100 µg/L, но максимални концентрации на изпитване под 10 µg/L или по-ниски са предпочитани с оглед обезпечаване на кинетика на биоразграждане от първи порядък. Най-ниската концентрация не трябва да превишава 10 µg/L, но се предпочитат минимални концентрации на изпитване от 1—2 μg/L или по-ниски от 1 μg/L. Обикновено задоволителен анализ на подобни ниски концентрации може да бъде постигнат чрез използването на предлагани в търговската мрежа вещества, маркирани с 14C. Поради аналитични ограничения често е невъзможно да се измери концентрацията на вещество с изискваната точност, ако изпитваното вещество се използва в концентрация ≤ 100 µg/L (вж. втора алинея от раздел 1.7.2). По-високи концентрации на изпитваното вещество (> 100 µg/L и понякога > 1 mg/L) могат да се използват за идентифициране и количествено определяне на основните продукти на трансформация или при липсата на специфичен метод за анализ с ниска граница на откриваемост. При изпитване на вещества при високи концентрации може да се окаже невъзможно използването на резултатите за изчисляване на константата на разграждане от първи порядък и периода на полуразпад, тъй като разграждането вероятно няма да протича по кинетичен модел от първи порядък.
Процесът на разграждане се наблюдава през определени интервали от време, измервайки или остатъчния 14C, или остатъчната концентрация на изпитваното вещество, при използването на специфичен химичен анализ. Маркирането на най-стабилната част от молекулата с 14C осигурява определянето на общата минерализация, докато маркирането с 14C на по-нестабилната част от молекулата, както и използването на специфичен анализ позволява оценката единствено на първичното биоразграждане. Въпреки това, най-стабилната част не включва непременно съответната функционална група на молекулата (която може да е свързана със специфични свойства като токсичност, биоакумулиране и др.). В такъв случай по-подходящо би било използването на маркирано с 14C изпитвано вещество във функционалната му част с цел да се проследи елиминирането на специфичното свойство.
1.5. ИНФОРМАЦИЯ ЗА ИЗПИТВАНОТО ВЕЩЕСТВО
За това изпитване могат да бъдат използвани както радиомаркирани, така и немаркирани изпитвани вещества. Препоръчва се методът за маркировка с 14C, като обикновено маркировката трябва да става в най-стабилната(ите) част(и) на молекулата (вж. също раздел 1.4). При вещества, които съдържат повече от един ароматен пръстен, е за предпочитане един или два въглеродни атома във всеки пръстен да бъдат маркирани с 14C. В допълнение към това, за предпочитане е един или повече въглеродни атома от двете страни на лесно разпадащи се връзки да са маркирани с 14C. Химичната и/или радиохимичната чистота на изпитваното вещество трябва да бъдат > 95 %. За радиомаркирани вещества е за предпочитане специфична активност от приблизително 50 μCi/mg (1,85 MBq) или повече с цел да се улесни измерването на 14C при изпитвания, провеждани при ниски първоначални концентрации. Трябва да е налична следната информация за изпитваното вещество:
|
— |
разтворимост във вода [метод A.6]; |
|
— |
разтворимост в органичен(ни) разтворител(и) (вещества, използвани с разтворители, или вещества с ниска водоразтворимост); |
|
— |
дисоциационна константа (pKa), ако веществото е склонно към протониране или депротониране [ОИСР TG 112] (4); |
|
— |
парен натиск [метод A.4] и константа по закона на Хенри; |
|
— |
химична стабилност във вода и на тъмно (хидролиза) [метод C.7]. |
Когато вещества с ниска водоразтворимост се изпитват в морска вода, полезно би било да се разполага с информация за константата на обезсоляване (или константата „Setschenow“) Ks, която се определя от следното уравнение: log (S/S') = Ks Cm, където S и S' са разтворимостите на веществото съответно в прясна вода и в морска вода, а Cm е моларната концентрация на солта.
Ако изпитването се провежда като „изпитване със суспендирани утайки“ трябва да се разполага и със следната информация:
|
— |
коефициент на разпределение n-октанол/вода [метод A.8]; |
|
— |
коефициент на адсорбция [метод C.18]. |
Друга полезна информация включва:
|
— |
концентрация в околната среда, ако е известна или изчислена; |
|
— |
токсичност на изпитваното вещество спрямо микроорганизми [метод C.11]; |
|
— |
пряка и/или присъща биоразградимост [методи C.4 A—F, C.12, C.9, ОИСР TG 302 (4)]; |
|
— |
аеробна или анаеробна биоразградимост в почва и изследвания на преобразуването утайка/вода [методи C.23, C.24]. |
1.6. РЕФЕРЕНТНО ВЕЩЕСТВО
Като референтно вещество трябва да бъде използвано такова, което обикновено се разгражда лесно при аеробни условия (напр. анилин или натриев бензоат). Обикновено очакваният период на разграждане на анилина и на натриевия бензоат е под 2 седмици. Предназначението на референтното вещество е да осигури микробиална активност на водата за изпитване е в рамките на определени граници; т.е. наличието на активна микробиална популация във водата.
1.7. КРИТЕРИИ ЗА КАЧЕСТВО
1.7.1. Възстановяване
Веднага след добавянето на изпитваното вещество всяка първоначална изпитвана концентрация трябва да бъде проверявана чрез измерване на активността на 14C или в случая с немаркирани вещества — чрез химични анализи, използвайки най-малко две проби. По този начин се добива информация за приложимостта и повторяемостта на аналитичния метод, както и за хомогенността в разпределянето на изпитваното вещество. Обикновено за последващите анализи на данните се използва измерената първоначална активност 14C или концентрацията на изпитваното вещество, а не номиналната концентрация, тъй като по този начин се компенсират загубите в резултат на грешки при сорбцията и дозирането. При маркирани с 14C изпитвани вещества степента на възстановяване в края на експеримента се определя от тегловния баланс (вж. последната алинея от раздел 1.8.9.4). В идеалния случай маркираният като радиоактивен тегловен баланс трябва да е в диапазона от 90 % до 110 %, докато аналитичната точност би следвало да осигури първоначално възстановяване от порядъка на 70—110 % по отношение на немаркирани изпитвани вещества. Тези диапазони трябва да бъдат разбирани като целеви и не трябва да се използват като критерии за приемане на изпитването. По избор аналитичната точност може да бъде определена за изпитваното вещество при по-ниска концентрация от първоначалната, както и за основните продукти на трансформацията.
1.7.2. Повторяемост и чувствителност на аналитичния метод
Повторяемостта на аналитичния метод (включително и ефикасността на първоначалната екстракция) за количествено определяне на изпитваното вещество, както и продуктите на трансформацията, ако е уместно, трябва да се проверят чрез пет анализа на репликати от отделните пробовземания от повърхностните води.
Границата на откриваемост (LOD) при аналитичния метод по отношение на изпитваното вещество и на продуктите на трансформацията трябва да бъде най-малко 1 % от първоначалното количество, използвано в системата за изпитване, ако е възможно. Границата на количествено определяне (LOQ) трябва да е равна на или по-малка от 10 % от използваната концентрация. Химичните анализи на редица органични вещества и техните продукти на трансформация често изискват изпитваното вещество да се прилага в относително висока концентрация, т.е. > 100 μg/L.
1.8. ОПИСАНИЕ НА МЕТОДА НА ИЗПИТВАНЕ
1.8.1. Оборудване
Изпитването може да се извърши в конусовидни или цилиндрични колби с подходяща вместимост (напр. 0,5 или 1,0 литра), затворени със силиконови или гумени запушалки, както и в серумни колби, с непропускащи CO2 капачки (напр. с мембрана от бутиленов каучук). Друг вариант е извършване на изпитването при използването на няколко колби и изследване на целите колби, най-малко по две идентични, при всеки интервал на вземане на проби (вж. последната алинея от раздел 1.8.9.1). При нелетливи изпитвани вещества, които не са радиомаркирани, не се изисква използването на газонепропускащи тапи или капачки; за целта са подходящи тапи от памук, които предпазват от замърсители във въздуха (вж. втора алинея от раздел 1.8.9.1). Слабо летливите вещества трябва да бъдат изпитвани в система от биометричен тип при леко разбъркване на повърхността на водата. С цел по-сигурно предпазване от бактериално замърсяване, по желание стъклените съдове могат да бъдат стерилизирани преди употреба чрез загряване или автоклавиране. В допълнение към това се използва следното стандартно лабораторно оборудване:
|
— |
вибрационен плот или магнитни бъркалки за продължително разбъркване на колбите за изпитване; |
|
— |
центрофуга; |
|
— |
pH-метър; |
|
— |
турбидиметър за измерване на мътност; |
|
— |
пещ или микровълнова пещ за определяне на сухото вещество; |
|
— |
апаратура за мембранно филтриране; |
|
— |
автоклав или пещ за топлинна стерилизация на стъкленици; |
|
— |
уреди за обработка на вещества, маркирани с 14C; |
|
— |
оборудване за количествено определяне на 14C активността в проби от улавящи CO2 разтвори и, ако е необходимо, в проби от утайки; |
|
— |
аналитично оборудване за определянето на изпитваното (и на референтното) вещество, в случай че се използва специфичен химичен анализ (напр. газов хроматограф, течен хроматограф с високо налягане). |
1.8.2. Изходни разтвори на изпитвани вещества
За приготвяне на изходни разтвори на изпитвани и референтни вещества се използва дейонизирана вода (вж. първа алинея от раздел 1.8.7). Дейонизираната вода не трябва да съдържа вещества, които са токсични за микроорганизми, а разтвореният органичен въглерод (DOC) не трябва да надвишава 1 mg/L (5).
1.8.3. Вземане и транспортиране на проби от повърхностна вода
Мястото за вземане на проба от повърхностна вода трябва да бъдат избирани в съответствие с целта на изпитването за всеки отделен случай. При избора на места за вземане на проби трябва да се вземе под внимание информация за евентуални предишни земеделски, промишлени или битови въздействия. Ако е известно, че дадена водна среда е била замърсена с изпитваното вещество или с негови структурни аналози в рамките на последните четири години, тя не трябва да се използва за вземане на проби, освен ако изследователят няма за изрична цел проучването на скоростите на разграждане в замърсени в миналото обекти. На мястото на вземане на пробата трябва да се измери показателят pH и температурата на водата. Нещо повече, дълбочината на вземане на пробата и видът на водната проба (напр. цвят и мътност) трябва да бъдат отчетени (вж. раздел 3). Концентрацията на кислород и/или редоксипотенциалът във вода и в повърхностния седиментен слой трябва да бъдат измерени с цел демонстриране на аеробните условия, освен ако те не се подразбират от външния вид и наличната информация за обекта. Повърхностната вода трябва да бъде транспортирана в основно почистен контейнер. По време на транспортирането температурата на пробата не трябва да надвишава значително температурата, при която се провежда изпитването. Ако времето за транспортиране на водните проби надвиши 2 до 3 часа, се препоръчва охлаждане до 4 °C. Водната проба не трябва да бъде замръзнала.
1.8.4. Съхранение и подготовка на повърхностната вода
За предпочитане е изпитването да започне в рамките на един ден след вземане на пробата. Съхранението на водата, ако то е необходимо, трябва да бъде сведено до минимум и при всички случаи не трябва да надвишава 4 седмици. Водната проба трябва да бъде съхранявана при температура от 4 °C и аерация до момента на нейното използване. Преди употреба грубите частици трябва да бъдат отстранени, например чрез филтрация през найлонов филтър с размер на отворите около 100 μm или през груба филтърна хартия, или пък чрез утаяване.
1.8.5. Подготовка на вода заедно с утайка (по избор)
При извършване на изпитването със суспендирани утайки към колбите, съдържащи природна вода (филтрирана според указанията на раздел 1.8.4 с цел отстраняване на грубите частици), се добавя повърхностна утайка с цел получаване на суспензия; концентрацията на суспендираните неразтворени вещества трябва да бъде между 0,01 и 1 g/L. Повърхностната утайка трябва да произлиза от същия място, от което е взета водната проба. В зависимост от особеностите на водната среда, повърхностната утайка може да се характеризира или с високо съдържание на въглерод (2,5 — 7,5 %) и с фина текстура, или с ниско съдържание на въглерод (0,5 — 2,5 %) и с груба текстура (2). Повърхностната утайка може да се подготви по следния начин: извлечете няколко утаечни ядки, използвайки тръбичка от прозрачна пластмаса, изрежете горните аеробни слоеве (от повърхността до дълбочина от около максимум 5 mm) веднага след вземането на пробите и ги съберете заедно. Формираната по този начин проба от утайка трябва да бъде транспортирана в контейнер с голямо свободно въздушно пространство с цел осигуряване на аеробни условия за утайката (охладете до 4 °C, ако транспортирането ще продължи повече от 2–3 часа). Пробата от утайката трябва да бъде суспендирана във водата за изпитване в съотношение 1:10 и съхранявана при температура от 4 °C и аерира до момента на използване. Съхраняването на утайката, ако е необходимо, трябва да бъде сведено до минимум и при всички случаи не трябва да надвишава 4 седмици.
1.8.6. Полунепрекъсната процедура (по избор)
Може да бъде необходима продължителната инкубация (няколко месеца), ако има дълъг латентен период преди да е възможно измерването на значително разграждане на изпитваното вещество. Ако тази информация е налична от предходни изпитвания на веществото, изпитването може да бъде инициирано чрез използването на полунепрекъснат способ, който позволява периодичното подновяване на част от водата за изпитване или суспензията (вж. допълнение 2). Алтернативно, нормалното серийно изпитване може да бъде премине в полунепрекъснато, ако в рамките на приблизително 60 дни на изпитване чрез сериен способ не е настъпило никакво разграждане на изпитваното вещество (вж. втора алинея от раздел 1.8.8.3).
1.8.7. Добавяне на изпитваното (или на референтното) вещество
За вещества с висока водоразтворимост (> 1 mg/L) и ниска летливост (с константи по закона на Хенри < 1 Pa·m3/mol или < 10–5 atm·m3/mol) може да бъде приготвен изходен разтвор в дейонизирана вода (вж. раздел 1.8.2); подходящото количество от изходния разтвор се добавя в стъклените съдове за изпитване с цел постигане на желаната концентрация. Обемът на даден добавен изходен разтвор трябва да бъде сведен до функционалния минимум (< 10 % от крайния обем на течността, ако е възможно). Друга възможна процедура е разтварянето на изпитваното вещество в по-голям обем от водата за изпитване, което може да се счете за алтернатива на използването на органични разтворители.
Ако е неизбежно, изходните разтвори на нелетливите вещества със слаба водоразтворимост трябва да се приготвят чрез използването на летлив органичен разтворител, но количеството разтворител, добавено към системата за изпитване, не трябва да превишава 1 % v/v и не трябва да оказва неблагоприятен ефект върху микробиалната активност. Разтворителят не трябва да влияе върху стабилността на изпитваното вещество във вода. Разтворителят трябва да бъде в изключително малко количество, за да не увеличи чувствително концентрацията на DOC във водата за изпитване или в суспензията. Това трябва да се провери чрез специфичен за съответното вещество анализ, или, по възможност, чрез анализ на DOC (5). Трябва да се обезпечи ограничаването на количеството на прехвърления разтворител до абсолютно необходимото, както и да се осигури разтварянето на количеството изпитвано вещество в крайния обем вода за изпитване. Други техники за въвеждане на изпитваното вещество в стъклените съдове за изпитване могат да бъдат използвани, както е указано в (6) и (7). Когато даден органичен разтворител се използва за прилагане на изпитваното вещество, контролните проби на разтворителя, съдържащи вода за изпитване (без никакви добавки) и водата за изпитване с добавено към нея референтно вещество трябва да бъдат обработени по същия начин като използваните стъклени съдове за изпитване, допълнени с изпитвано вещество в среда от разтворител. Целта на контролните проби на разтворителите е изследването на евентуалните неблагоприятни ефекти върху микробиалната популация, причинени от разтворителя, за които свидетелства разграждането на референтното вещество.
1.8.8. Условия на изпитване
1.8.8.1. Температура на изпитване
Инкубацията трябва да се извърши на тъмно (за предпочитане) или на слаба светлина при регулирана температура (± 2 °C), която може да бъде температура на средата или стандартна температура от 20—25 °C. Температура на средата може да бъде или реалната температура на пробата към момента на вземането ѝ, или средната околна температура на мястото на вземане на пробата.
1.8.8.2. Размесване
Трябва да се осигури размесване чрез непрекъснато разклащане или разбъркване с цел поддържане на частиците и на микроорганизмите в състояние на суспензия. Размесването улеснява също трансфера на кислорода от въздуха над течността, за да се осигури поддържането на подходящи аеробни условия. Поставете колбите върху вибрационен плот (с приблизително 100 rpm) или използвайте магнитно разбъркване. Размесването не трябва да се прекъсва. Въпреки това, разклащането или разбъркването трябва да се извършват възможно най-деликатно, като същевременно с това се поддържа хомогенна суспензия.
1.8.8.3. Продължителност на изпитването
Нормалната продължителност на изпитването не трябва да надвишава 60 дни, освен ако не се използва полунепрекъсната процедура с периодично подновяване на изпитваната суспензия (вж. раздел 1.8.6 и допълнение 2). При все това периодът на изпитване за серийното изпитване може да бъде удължен до максимум 90 дни, ако разграждането на изпитваното вещество е започнало в рамките на първите 60 дни. Разграждането се наблюдава през съответните интервали от време, като се определя остатъчната активност 14C или отделения 14CO2 (вж. раздел 1.8.9.4) и/или чрез химичен анализ (раздел 1.8.9.5). Инкубационният период трябва да бъде достатъчно дълъг, за да позволи оценяването на процеса на разграждане. За предпочитане е степента на разграждане да надвишава 50 %; за бавно разграждащи се вещества степента на разграждане трябва да бъде достатъчна (обикновено над 20 % разграждане), за да позволи определянето на скоростната константа на процеса на разграждане.
Трябва да се провеждат периодични измервания на pH и концентрацията на кислород в системата за изпитване, освен ако предходният опит от подобни изпитвания в проби от вода и утайка, взети от същото място, не правят ненужно извършването на такива измервания. При определени условия метаболизмът на първичните субстрати при много по-високи концентрации във водата или утайката могат да доведат евентуално до значителна промяна на експерименталните условия по време на изпитването под въздействието на достатъчно отделен CO2 и достатъчно намаляване на кислорода.
1.8.9. Процедура
1.8.9.1. Подготовка на колби за пелагични изпитвания
Прехвърлете подходящо количество вода за изпитване в колбите за изпитване, запълвайки до около една трета от обема на колбите, но не по-малко от около 100 ml. При използването на няколко колби (за да е възможно обработването на цяла колба във всяко време на вземане на проби), подходящият обем вода за изпитване отново е около 100 ml, тъй като малките обеми проби могат да повлияят върху продължителността на латентния период. Изпитваното вещество се добавя от изходен разтвор, както е описано в раздели 1.8.2 и 1.8.7. Най-малко две различни концентрации на изпитваното вещество, с разлика помежду си от 5 до 10 пъти, трябва да бъдат използвани с цел определянето на кинетиката на разграждане и изчисляването на скоростната константа на разграждане. И двете избрани концентрации трябва да са по-малки от 100 µg/L и за предпочитане е да бъдат в диапазона < 1—10 µg/L.
Затворете колбите със запушалки или капаци, които не пропускат въздух и CO2. За немаркирани с 14C нелетливи изпитвани вещества са подходящи тапи от памук, които предпазват от замърсявания от въздуха (вж. раздел 1.8.1), при условие че е известно, че основните продукти на разграждане са нелетливи, както и при индиректно определяне на CO2 (вж. допълнение 3).
Инкубирайте колбите при избраната температура (вж. раздел 1.8.8.1). Вземете проби за химичен анализ или измервания на 14C в началото на изпитването (т.е. преди да започне биоразграждането; вж. раздел 1.7.1), а след това през определени интервали от време в процеса на изпитването. Вземането на проби може да се извърши чрез отделяне на подпроби (напр. аликвотни части от 5 ml) от всяка репликата или чрез вземане на цели колби при всяко отделно вземане на проба. Минерализацията на изпитваното вещество може да бъде определена директно или индиректно (вж. допълнение 3). Обикновено по време на фазата на разграждане са необходими минимум пет пробни точки (т.е. след края на латентния период), за да се определи с достатъчна точност скоростната константа, освен ако не съществуват основания да се смята, че три пробни точки са достатъчни за бързо разграждащите се вещества. За бавно разграждащи се вещества могат лесно да бъдат направени повече измервания по време на фазата на разграждане и в този случай трябва да се използват повече измервателни точки за определянето на k. Не може да бъде установен фиксиран времеви график за вземане на проби, тъй като скоростта на биоразграждане варира; въпреки това, в случай че процесът на разграждане е бавен, се препоръчва ежеседмично вземане на проби. Ако изпитваното вещество е бързо разградимо, вземането на проби трябва да се извършва веднъж на ден по време на първите три дни, след което на всеки втори или трети ден. При определени обстоятелства, като например при много бързо хидролизиращи вещества, може да се наложи ежечасно вземане на проби. Препоръчва се да се извърши предварително проучване преди началото на изпитването, за да се определят подходящите интервали за вземане на проби. Ако е необходимо наличието на проби за последващ специфичен анализ, препоръчително е да се вземат повече проби, след което да се изберат онези, които ще бъдат анализирани в края на експеримента, като се следва обратна последователност, т.е. последните проби се анализират първи (вж. втора алинея от раздел 1.8.9.5 за насоки относно стабилността на пробите по време на съхраняването).
1.8.9.2. Брой колби и проби
Трябва да разполагате с достатъчен брой колби за изпитване, за да осигурите:
|
— |
колби за изпитване; най-малко по две колби за всяка концентрация от изпитваното вещество (за предпочитане е най-малко 3) или няколко колби за изпитване за всяка концентрация, в случай че се вземат цели колби при всяко вземане на проби (обозначение FT); |
|
— |
колби за изпитване за изчисляване на тегловния баланс; най-малко по две колби за всяка концентрация от изпитваното вещество (обозначение FM); |
|
— |
празна проба, без изпитвано вещество; най-малко една празна колба за изпитване, съдържаща само вода за изпитване (обозначение FB); |
|
— |
референтен контрол; две колби с референтното вещество (напр. анилин или натриев бензоат, при 10 µg/l) (обозначение FC). Целта на референтния контрол е да се потвърди минимална микробиална активност. Ако е удобно, може да бъде използвано радиомаркирано референтно вещество, също и когато разграждането на изпитваното вещество се контролира чрез химични анализи; |
|
— |
стерилен контрол; една или две колби, съдържащи стерилизирана вода за изпитване за изследване на възможното абиотично разграждане или друго небиологично отстраняване на изпитваното веществото (обозначение FS). Биологичната активност може да бъде прекъсната с автоклавна обработка (121 °C; 20 мин.) на водата за изпитване или чрез добавяне на токсично вещество (напр. натриев азид (NaN3) 10—20 g/l, живачен хлорид (HgCl2) 100 mg/l или формалин 100 mg/l), както и чрез облъчване с гама лъчи. Ако се използва HgCl2, той трябва да бъде изхвърлен като токсичен отпадък. При вода, към която е добавено голямо количество утайка, не е лесно да се постигнат стерилни условия; в този случай се препоръчва повторна автоклавна обработка (напр. трикратна). Трябва да се вземе под внимание, че сорбционните характеристики на утайката могат да бъдат променени от автоклавната обработка; |
|
— |
контрол на разтворителя, със съдържание на вода за изпитване и вода за изпитване с примес на референтно вещество; паралелен анализ в колби, обработвани с еднакво количество разтворител и при използването на същата процедура като при прилагането на изпитваното вещество. Целта е да се изследват възможните неблагоприятни ефекти на разтворителя, определяйки разграждането на референтното вещество. |
При планирането на изпитването изследователят трябва да вземе под внимание относителната важност на увеличената репликатност при експеримента спрямо увеличен брой интервали на вземане на проби. Точният брой на необходимите колби ще зависи от метода, използван за измерване на разграждането (вж. трета алинея от раздел 1.8.9.1; раздел 1.8.9.4 и допълнение 3).
Две подпроби (напр. аликвотни части от 5 ml) трябва да бъдат взети от всяка колба за изпитване за всеки интервал на вземане на проба. Ако се използват няколко колби с цел вземане на цели колби, за всеки интервал на вземане на проба трябва да се използват минимум две колби (вж. първа алинея от раздел 1.8.9.1).
1.8.9.3. Подготовка на колби за изпитване със суспендирана утайка [по избор]
Добавете в изпитвателните съдове за изпитване необходимите количества вода и утайка за изпитване, ако е необходимо (вж. раздел 1.8.5). Подготовката на колбите за изпитване със суспендирана утайка е същата като при пелагичното изпитване (вж. раздели 1.8.9.1 и 1.8.9.2). За предпочитане е използването на серумни бутилки или на колби с подобна форма. Поставете затворените колби в хоризонтално положение върху вибрационен плот. Както се подразбира, отворените колби за немаркирани с 14C нелетливи вещества трябва да бъдат поставени в изправено положение; в този случай е препоръчително магнитно разбъркване и използването на магнитни бъркалки със стъклено покритие. Ако е необходимо, аерирайте бутилките за поддържане на подходящи аеробни условия.
1.8.9.4. Радиохимични измервания
Отделеният 14CO2 се измерва директно и индиректно (вж. допълнение 3). 14CO2 се определя индиректно чрез разликата между първоначалната активност 14C във вода за изпитване или в суспензия и общата остатъчна активност в съответното време на вземане на пробата, измерена след подкиселяването на пробата до pH 2—3 и отстраняването на CO2. По този начин неорганичният въглерод се отстранява и измерената остатъчната активност се обуславя от органичен материал. Индиректното определяне на 14CO2 не трябва да се използва, ако в хода на трансформация на изпитваното вещество се формират основни летливи продукти на трансформация (вж. допълнение 3). По възможност, отделянето на 14CO2 трябва да се измерва директно (вж. допълнение 3) при всеки интервал на вземане на проба в най-малко една колба за изпитване; тази процедура позволява проверката както на тегловния баланс, така и на процеса на биоразграждане, но е приложима единствено за изпитвания, провеждани със затворени колби.
Ако отделеният 14CO2 се измерва директно по време на изпитването, трябва да си осигурите повече колби за тази цел в началото на изпитването. Директното измерване на 14CO2 се препоръчва, ако по време на трансформацията на изпитваното вещество се формират основни летливи продукти на трансформация. При всяка точка на измерване допълнителните колби за изпитване се подкиселяват до pH 2—3 и 14CO2 се събира във вътрешен или външен абсорбер (вж. допълнение 3).
По избор концентрациите на маркирани с 14C изпитвани вещества и основните продукти на трансформация могат да се определят чрез използването на радиохроматография (напр. тънкослойна хроматография, RAD-TLC) или високоефективна течна хроматография HPLC с радиохимична детекция.
По избор може да се определи фазовото разпределение на остатъчната радиоактивност (вж. допълнение 1) и остатъчното изпитвано вещество и продуктите на трансформация.
В края на изпитването трябва да се определи тегловният баланс чрез директно измерване на 14CO2, използвайки отделни колби за изпитване, от които не са вземани никакви проби по време на изпитването (вж. допълнение 3).
1.8.9.5. Специфичен химичен анализ
При наличие на чувствителен специфичен аналитичен метод първоначалното биоразграждане може да бъде оценено чрез измерване на общата остатъчна концентрация на изпитваното вещество, вместо използването на техники за радиомаркиране. Ако се използва радиомаркирано вещество (за измерване на общата минерализация), могат да бъдат провеждани паралелно специфични химични анализи с цел осигуряване на полезна допълнителна информация и проверка на процедурата. Специфични химични анализи могат да се провеждат и за измерване на продуктите на трансформация, получени по време на разграждането на изпитваното вещество, като това се препоръчва по отношение на вещества, които се минерализират с периоди на полуразпад над 60 дни. Концентрацията на изпитваното вещество и продуктите на трансформация при всеки интервал на вземане на проби трябва да бъдат измервани и отчитани (като концентрация и като процент от използваното количество). Обикновено ще бъдат идентифицирани продукти на трансформация ≥ 10 % от използваната концентрация за всеки интервал на вземане на проба, освен ако няма сериозни основания за противното. Продуктите на трансформация, за които концентрациите постоянно се увеличават по време на изследването, също трябва да бъдат взети под внимание при идентифицирането, дори и техните концентрации да не превишават горните граници, тъй като това би могло да очертава някаква тенденция. Ако се допуска възможността за бърза абиотична трансформация на изпитваното вещество (напр. хидролиза), трябва да се предвидят анализи на продуктите на трансформация при контрол на стерилността. Нуждата от количествено определяне и идентифициране на продуктите на трансформация трябва да се преценява за всеки отделен случай, като съответните обосновки се посочват в протокола. Техниките за екстракция с органичен разтворител трябва да се прилагат в съответствие с инструкциите, описани в съответната аналитична процедура.
В случай че анализът се извършва в рамките на 24 часа (за предпочитане), всички проби трябва да се съхраняват при температура от 2 до 4 °C и в условия на херметизация. При по-продължителен период на съхранение пробите трябва да са замразени до температура под –18 °C или третирани с химични консерванти. Подкиселяването не е препоръчителен метод за съхраняване на пробите, тъй като подкиселените проби могат да са нестабилни. Ако пробите не се анализират в рамките на 24 часа и подлежат на по-продължителен период на съхраняване, трябва да се проведе изследване на устойчивостта в условията на съхранение, за да се установи стабилността на въпросните химични вещества при съхранение при температура под –18 °C или при обработка с химични консерванти. Ако аналитичният метод изисква или екстракция с разтворител, или екстракция в твърда фаза, екстракцията трябва да се извърши веднага след вземането на пробата или след запазването на замразената проба за максимален период от 24 часа.
В зависимост от чувствителността на аналитичния метод може да се наложи използването на по-големи обеми проби от посочените в раздел 1.8.1. Изпитването може да бъде извършено лесно с изпитвани обеми от един литър в колби с вместимост от 2—3 литра, което би позволило вземането на проби от около 100 ml.
2. ДАННИ И ДОКЛАДВАНЕ
2.1. ОБРАБОТКА НА РЕЗУЛТАТИТЕ
2.1.1. Графично изобразяване на данните
Интервалите на вземане на пробите трябва да се закръглят на цели часове (освен ако веществото не се разгражда значително за минути до часове), но не и на цели дни. Изобразявайте данните за остатъчната активност на изпитваното вещество (за маркирани с 14C вещества) или за остатъчната концентрация (за немаркирани вещества), спрямо времето както в линейна, така и в полулогаритмична графика (вж. фигури 1a, 1б). Ако се е извършило разграждане, сравнете резултатите от колби FT с тези от колби FS. Ако резултатите от колбите с изпитваното вещество (FT) и стерилните колби (FS) се различават с по-малко от 10 %, може да се предположи, че наблюдаваното разграждане е предимно абиотично. Ако разграждането в колби FS е по-ниско, данните могат да бъдат използвани за коригиране на получените от колби FT (чрез изваждане) с цел установяване на степента на биоразграждане. Когато по избор се провеждат анализи на основни продукти на трансформация, трябва да бъдат представени графики на тяхното формиране и спад в допълнение към графичното изобразяване на намаляването на изпитваното вещество.
Установете продължителността на латентния период tL от кривата на разграждането (полулогаритмична графика) чрез екстраполация на нейната линейна част до нулево разграждане или алтернативно чрез определяне на времето за разграждане в размер на около 10 % (вж. фигури 1a и 1б). От полулогаритмичната графика установете скоростната константа от първи порядък k и нейната стандартна грешка чрез линейна регресия на ln (остатъчна активност 14C или концентрация на изпитваното вещество) спрямо времето. Специално при измерванията 14C използвайте единствено данни, принадлежащи към началната линейна част на кривата след приключилата латентна фаза и отдайте предпочитание на избора на известно количество представителни данни, вместо на голям обем несигурни данни. Несигурността в случая се определя от грешки, обусловени от препоръчаното директно използване на измерени остатъчни 14C активности (вж. по-долу). В някои случаи би било уместно изчисляването на две различни скоростни константи, ако разграждането се развива по двуфазен модел. За тази цел се определят две различни фази на кривата на разграждане. Изчисленията на скоростната константа k и на периода на полуразпад t½ = ln2/k трябва да се извършват за всяка отделна репликатна колба, когато се вземат подпроби от една и съща колба, или чрез използване на средните стойности, когато в рамките на един интервал на вземане на проби се вземат цели колби (вж. последната алинея от раздел 1.8.9.2). Когато се използва първата от посочените процедури, скоростната константа и периодът на полуразпад трябва да се отчитат за всяка отделна репликатна колба като средна стойност със стандартна грешка. В случай че са били използвани високи концентрации на изпитваното вещество, кривата на разграждане може значително да се отклонява от права линия (полулогаритмична графика) и кинетиката от първи порядък може да не е валидна в този случай. В този смисъл определянето на периода на полуразпад е безпредметно. Въпреки това, за ограничен обем данни може да се приложи кинетика от първи порядък и да се установи периодът на полуразграждане DT50 (време за достигане на разграждане в размер на 50 %). Следва обаче да се има предвид, че продължителността на разграждане извън избрания обем данни не може да бъде прогнозирана на базата на показателя DT50, който единствено описва избраните данни. Предлага се широк набор от аналитични инструменти, улесняващи статистическите изчисления и апроксимацията на кривите, а използването на такъв софтуер е препоръчително.
В случай на извършване на специфични химични анализи, установете скоростните константи и периодите за първоначалното полуразграждане, както по-горе за общата минерализация. Ако първоначалното разграждане е пределният процес, понякога могат да се използват базови точки от целия ход на разграждането. Това се дължи на факта, че измерванията са директни, за разлика от измерванията на активността 14C.
Ако се използват маркирани с 14C вещества, тегловният баланс трябва да се изрази като процент от използваната първоначална концентрация, най-малкото в края на изпитването.
2.1.2. Остатъчна активност
Когато маркираната с 14C част от дадено органично вещество бъде биоразградена, основната част от 14C преминава в 14CO2, докато друга се използва за нарастване на биомасата и/или за синтез на извънклетъчни метаболити. По тази причина пълното „максимално“ биоразграждане на дадено вещество не води до 100-процентово преминаване на въглерода в неговия състав в 14CO2. Вграденият в продукти от биосинтеза 14C впоследствие бавно се отделя под формата на 14CO2 в резултат на „вторична минерализация“. По тези причини графичните изображения на остатъчната активност на органичен 14C (измерена след отстраняването на CO2) или на произведения 14CO2 спрямо времето ще покаже „снижаване“ след приключване на процеса на биоразграждане. Това усложнява кинетичното интерпретиране на данните и за целта само началната част на кривата (след края на латентния период и преди достигането на около 50 % от процеса на разграждане) трябва обикновено да бъде използвана за установяването на скоростната константа на разграждане. Ако изпитваното вещество бъде разградено, общата остатъчна активност на органичния 14C винаги е по-висока от активността 14C, свързана с остатъчното непроменено изпитвано вещество. Ако изпитваното вещество се разгражда чрез реакция от първи порядък и една постоянна фракция α се минерализира в CO2, началният наклон на кривата на концентрация на 14C (общ органичен 14C спрямо времето) ще бъде α пъти наклона на съответната крива на концентрацията на изпитваното вещество (или по-точно, частта от изпитваното вещество, маркирана с 14C). Използвайки некоригирани измервания на общата активност на органичен 14C, изчислената скоростна константа на разграждане ще бъде стабилна. Процедурите за установяване на концентрациите на изпитваното вещество от измерените радиохимични активности на базата на различни опростяващи допускания са описани в литературата (1)(8)(9)(10). Подобни процедури са най-лесно приложими за бързо разграждащи се вещества.
2.2. ИНТЕРПРЕТИРАНЕ НА РЕЗУЛТАТИТЕ
Ако бъде установено, че k зависи от добавената концентрация (т.е. ако изчислената k е приблизително една и съща при различни концентрации на изпитваното вещество), може да се предположи, че скоростната константа от първи порядък е представителна за използваните условия на изпитване, т.е. изпитваното вещество, водната проба и температурата на изпитване. Чрез експертна оценка трябва да се определи до каква степен резултатите могат да бъдат обобщени или екстраполирани към други системи. Ако се използва висока концентрация на изпитваното вещество, поради което процесът на разграждане не се развива по кинетичен модел от първи порядък, данните не могат да бъдат използвани за директно определяне на скоростната константа от първи порядък или на съответния период на полуразграждане. Въпреки това, данните, получени от изпитването при използването на висока концентрация на изпитваното вещество, могат все още да послужат за пресмятането на степента на общата минерализация и/или откриваемост и количествено определяне на продуктите на трансформацията.
Ако са известни скоростите на други процеси на преобразуване, различни от биоразграждането (напр. хидролиза или изпарение), те могат да бъдат приспаднати от нетната скорост на превръщане, констатирана по време на изпитването, за да се определи една приблизителна скорост на биоразграждане. Данни за хидролизата могат да бъдат получени например от стерилните контроли или от паралелното изпитване при използване на по-висока концентрация на изпитваното вещество.
Директното и индиректно определяне на 14CO2 (раздел 1.8.9.4 и допълнение 3) могат да бъдат използвани единствено за измерване на степента на минерализация на изпитваното вещество до CO2. За анализирането на концентрациите на маркирани с 14C изпитвани вещества и на формирането на основни продукти на трансформацията (вж. трета алинея от раздел 1.8.9.4) могат да бъдат използвани радиохроматография (RAD-TLC) или HPLC. Директното определяне на периода на полуразграждане е възможно единствено при липсата на всякакви основни продукти на трансформация (определени като ≥ 10 % от използваното количество изпитвано вещество). В случай че се наблюдава наличие на основни продукти на трансформация, отговарящи на горната дефиниция, трябва да се извърши подробна оценка на данните. Тя може да включва повторно изпитване и/или идентификация на продуктите на трансформация (вж. първа алинея от раздел 1.8.9.5), освен ако пътят на продуктите на трансформация не може да бъде достоверно описан на базата на предходния опит (напр. информация за пътя на разграждане). Тъй като съотношението на въглерода в изпитваното вещество, преобразуван в CO2, варира (до голяма степен в зависимост от концентрацията на изпитваното вещество и на други налични субстрати, от условията на изпитване и от микробиалната среда), това изпитване не позволява директно определяне на пълното биоразграждане, както при изпитването за отмиране на DOC; но резултатът е сходен с този, получен при респирометрично изпитване. По тази причина степента на минерализация ще бъде по-малка или равна на минималната степен на пълното биоразграждане. За да се получи по-пълна представа за пълното биоразграждане (минерализация и включване в биомаса), в края на изпитването следва да се извърши анализ на фазовото разпределение на 14C (вж. допълнение 1). Намиращият се в групата на частиците 14C ще се състои от 14C, включен в бактериална биомаса, и 14C, сорбиран от органични частици.
2.3. ВАЛИДНОСТ НА ИЗПИТВАНЕТО
Ако референтното вещество не се е разградило в рамките на очаквания интервал от време (по отношение на анилина и натриевия бензоат този период обикновено е под две седмици), валидността на изпитването се подлага под съмнение и трябва да бъде проверена допълнително или изпитването трябва да се повтори с нова водна проба. При кръгово изпитване по ISO на метода, в което участваха седем лаборатории от различни точки на Европа, отчетените скоростни константи на разграждане по отношение на анилина варират от 0,3 до 1,7 ден–1 (средно 0,8 d–1) при температура 20 oC и стандартна грешка ±0,4 d–1 (t½ = 0,9 дни). Установените латентни периоди варират от 1 до 7 дни. В изследваните води е установено наличие на бактериална биомаса, съответстваща на 103—104 колонообразуващи единици (CFU)/ml. Скоростите на разграждане в богатите на хранителни вещества средноевропейски води са по-високи в северните олиготрофни води, което би могло да се дължи на различния трофичен статус или на предходен контакт с химични вещества.
Общото възстановяване (тегловен баланс) в края на експеримента трябва да е между 90 % и 110 % за радиомаркираните вещества, докато първоначалното възстановяване в началото на експеримента трябва да е между 70 % и 110 % за немаркираните вещества. При все това, посочените диапазони трябва да бъдат възприемани като целеви и не трябва да се използват като критерии за приемане на изпитването.
3. ПРОТОКОЛ ОТ ИЗПИТВАНЕТО
Видът на изследването (пелагично изпитване или изпитване със суспендирани утайки) трябва да бъде ясно посочен в протокола от изпитването, който трябва също така да съдържа следната задължителна информация:
Изпитвано вещество и референтно вещество/вещества:
|
— |
тривиални наименования, химични наименования (препоръчително е използването на наименованията по IUPAC и/или CAS), CAS номера, структурни формули (показващи разположението на 14C в случай на използване на радиомаркирани вещества) и съответните физикохимични свойства на изпитваното вещество и на референтното вещество (вж. раздели 1.5 и 1.6); |
|
— |
химични наименования, CAS номера, структурни формули (показващи разположението на 14C в случай на използване на радиомаркирани вещества) и съответните физикохимични свойства на вещества, използвани като стандарти за идентифициране и количествено определяне на продуктите на трансформация; |
|
— |
чистота (примеси) на изпитваните вещества и на референтни вещества; |
|
— |
радиохимична чистота на маркираните реактиви и специфична активност (където е уместно). |
Повърхностни води:
Трябва да бъде предоставена следната задължителна информация за взетата водна проба:
|
— |
местоположение и описание на мястото на вземане на пробата, включително (ако е възможно) история на замърсяване; |
|
— |
дата и час на вземане на пробата; |
|
— |
хранителни вещества (общ N, амониеви радикали, нитрити, нитрати, общ P, разтворен ортофосфат); |
|
— |
дълбочина на вземане на пробата; |
|
— |
външен вид на пробата (напр. цвят и мътност); |
|
— |
DOC и TOC; |
|
— |
BOD; |
|
— |
температура и pH на мястото и към момента на вземане на пробата; |
|
— |
окислително-редукционен потенциал (задължително само ако аеробните условия не са очевидни); |
|
— |
соленост или проводимост (в случай на морска вода и солена вода); |
|
— |
суспендирани неразтворени вещества (при мътност на пробата); |
|
— |
всякаква друга подходяща информация за мястото на вземане на пробата към момента на вземането ѝ (напр. актуална или предходна информация за водните количества на реките или на морските течения, основни зауствания в близките околности и видове зауствания, климатични условия преди момента на вземане на пробата); |
и по избор:
|
— |
микробиална биомаса (напр. директно отчитане на акридин оранж или колонообразуващи единици); |
|
— |
неорганичен въглерод; |
|
— |
концентрация на хлорофил-a като специфичен показател за биомаса от водорасли. |
В допълнение към това в случай на извършване на изпитване със суспендирани утайки трябва да бъде представена следната информация за утайките:
|
— |
дълбочина на вземане на проби от утайки; |
|
— |
външен вид на утайките (оцветени, мътни, тинести или песъчливи); |
|
— |
текстура (напр. процент едър пясък, фин пясък, тиня и глина) |
|
— |
сухо тегло на суспендираните неразтворени вещества в g/l, концентрация на TOC или загуба на тегло при горене като показател за измерване на съдържанието на органично вещество; |
|
— |
pH |
|
— |
окислително-редукционен потенциал (задължително само ако аеробните условия не са очевидни). |
Условия на изпитване:
|
— |
период между вземането на пробата и използването ѝ за лабораторното изпитване, съхранение и предварителна обработка на пробата, дати на извършване на изследванията; |
|
— |
количество на използваното изпитвано вещество, изпитвана концентрация и референтно вещество; |
|
— |
метод за прилагане на изпитваното вещество, включително и употребата на всякакви разтворители; |
|
— |
обем на използваната повърхностна вода и на утайките (в случай на употребата на такива) и обем на взетата за анализ проба за всеки отделен интервал; |
|
— |
описание на използваната система на изпитване. |
Ако изпитването не се провежда на тъмно, трябва да се предостави информация за условията на „слабо осветление“:
|
— |
информация за метода/методите, използвани за установяване на стерилен контрол (напр. температура, време и брой автоклавни обработки); |
|
— |
температура на инкубиране; |
|
— |
информация за аналитичните техники и методи, използвани за радиохимични измервания и за проверка на тегловния баланс и за измервания на фазовото разпределение (ако се провеждат такива). |
|
— |
брой на репликатите. |
Резултати:
|
— |
проценти на възстановяване (вж. раздел 1.7.1); |
|
— |
повторяемост и чувствителност на използваните аналитични методи, включително границата на откриваемост (LOD) и границата на количествено определяне (LOQ) (вж. раздел 1.7.2); |
|
— |
всички измерени данни (включително моментите на вземане на пробите) и изчислени величини в таблична форма и криви на разграждане; за всяка изпитвана концентрация и за всяка репликатна колба трябва да се отчете коефициентът на линейна корелация за наклона на логаритмичната графика, установеният латентен период и скоростната константа от първи или псевдо първи порядък (по възможност), както и съответният период на полуразграждане (или периодът на полуразпад t50); |
|
— |
отчитане на съответните величини като средни стойности на наблюдаваните резултати при отделните репликати, напр. дължина на латентния период, скоростна константа на разграждане и период на полуразпад (или t50); |
|
— |
категоризиране на системата или като приспособена, или като неприспособена, съдейки по вида на кривата на разграждане и по евентуалното влияние на изпитваната концентрация; |
|
— |
резултати от окончателната проверка на тегловния баланс и резултати от измерванията на фазовото разпределение (при наличие на такива); |
|
— |
дял на минерализиран 14C и при използване на специфични анализи, пълна степен на първоначално разграждане; |
|
— |
идентифициране, моларна концентрация и процент на използваните продукти и основните продукти на трансформация (вж. първа алинея от раздел 1.8.9.5), където е уместно; |
|
— |
предложен път на трансформация, където е уместно; |
|
— |
обсъждане на резултатите. |
4. ЛИТЕРАТУРА
|
1. |
OECD TG 309 (2004) Aerobic Mineralisation in surface water — Simulation Biodegradation Test |
|
2. |
ISO/DIS 14592—1 (1999) Качество на водите — оценка на аеробната биоразградимост на органични съединения при ниски концентрации — част 1: Серийно изпитване в колби за разклащане с повърхностна вода или суспензии повърхностна вода/утайка. |
|
3. |
Метод за изпитване C.23. Аеробна и анаеробна трансформация в почва. |
|
4. |
Метод за изпитване C.24. Аеробна и анаеробна трансформация във водни утайки. |
|
5. |
ОИСР (1993). Насоки за изпитването на химикали. ОИСР, Париж. |
|
6. |
ISO 8245 (1999). Качество на водите — насоки за определянето на общия ограничен въглерод (TOC) и на разтворения органичен въглерод (DOC). |
|
7. |
ISO 10634 (1995). Качество на водите — насоки за подготовката и обработването на лошо водоразтворими органични вещества за последваща оценка на биоразградимостта им във водна среда. |
|
8. |
ОИСР проект (2000). Документ с насоки за изпитване за токсичност във водна среда на трудни вещества и смеси. Публикации за здравословността и безопасността на околната среда. Поредица относно изпитването и оценката. № 22 (за публикуване през лятото на 2000 г.). |
|
9. |
Simkins, S. and Alexander, M. (1984). Models for mineralization kinetics with the variables of substrate concentration and population density. Appl. Environ. Microbiol.47, 394—401. |
|
10. |
Ingerslev, F. and N. Nyholm. (2000). Shake-flask test for determination of biodegradation rates of 14C-labeled chemicals at low concentrations in surface water systems. Ecotoxicol. Environ. Saf. 45, 274—283. |
|
11. |
ISO/CD 14592—1 (1999). Доклад от кръгово изпитване: Качество на водите — оценка на аеробната биоразградимост на органични съединения при ниски концентрации част 1 — доклад от кръговото изпитване от 1998/1999 г. Серийно изпитване в колби за разклащане с повърхностна вода или суспензии вода/утайка. |
Допълнение 1
Фазово разпределение на 14C
С цел да се провери процедурата, рутинните измервания на остатъчната обща активност на органичен 14C (TOA) трябва да бъдат допълнени с измервания на тегловния баланс, включващи директно определяне на отделения 14CO2 след улавянето му в абсорбер (вж. допълнение 3). Само по себе си, действителното получаване на 14CO2 представлява недвусмислено доказателство за биоразграждане в противовес на абиотичното разграждане или други механизми на преобразуване, като изпарение и сорбция. Допълнителна полезна информация, характеризираща модела на биоразграждане, може да бъде получена от измерванията на разпределението на TOA между разтвореното състояние (активност на разтворения органичен 14C, DOA) и твърдото състояние (активност на органичния 14C под формата на твърди частици, POA) след отделяне на твърдото вещество чрез мембранен филтър или центрофуга. POA се състои от изпитваното вещество, абсорбирано или адсорбирано от микробиална биомаса и от други частици, в допълнение към въглерода от веществото на изпитването, използван за синтез на нов клетъчен материал и по този начин включен във фракцията на биомасата от частици. Образуването на разтворен органичен материал 14C може да се определи като DOA в края на процеса на биоразграждане („платото“ на кривата на разграждане спрямо времето).
Установете фазовото разпределение на остатъчния 14C в избрани проби, като филтрирате пробите през мембранни филтри с диаметри на отворите от 0,22 μm до 0,45 μm и изработени от материал, който не адсорбира значителни количества от изпитваното вещество (за целта биха могли да се използват поликарбонатни филтри). Ако сорбцията на изпитваното вещество от филтъра е прекалено голяма, за да не бъде взета под внимание (това трябва да се провери преди началото на експеримента), може да се използва високоскоростна центрофуга (2 000 g; 10 мин.), вместо филтриране.
Продължете филтрирането или центрофугирането на нефилтрираните проби, както е описано в допълнение 3. Разтворете мембранните филтри в подходящ сцинтилационен флуид и отчетете резултатите по нормалната процедура, обикновено като се използва само метода на определяне на съотношение с външен еталон за коригиране на потискането, или се използва оксидатор на проби. В случай че е използвана центрофуга, суспендирайте отново пелетата, формирана от фракцията на частиците, в 1—2 ml дестилирана вода и прехвърлете в сцинтилационна стъкленица. След това промийте двукратно с 1 ml дестилирана вода и прехвърлете водата от промиването в стъкленицата. При необходимост суспензията може да бъде внесена в гел с цел извършване на течно сцинтилационно броене.
Допълнение 2
Полунепрекъсната процедура
Може да се изисква удължено инкубиране до няколко месеца с цел постигане на достатъчно разграждане на устойчивите вещества. Обикновено продължителността на изпитването не надвишава 60 дни, освен ако характеристиките на оригиналната водна проба не се поддържат чрез подновяване на изпитваната суспензия. Въпреки това, периодът на изпитване може да бъде удължен до максимум 90 дни, без да се подновява изпитваната суспензия, ако разграждането на изпитваното вещество е започнало в рамките на първите 60 дни.
В хода на продължително инкубиране разнородността на микробиалната среда може да намалее поради различни механизми на преобразуване, както и поради евентуално изчерпване на основни хранителни вещества и първични въглеродни субстрати във водната проба. По тази причина се препоръчва използването на полунепрекъснато изпитване с цел достоверното определяне на скоростта на разграждане при слабо разграждащи се вещества. Тестът трябва да бъде стартиран с използването на полунепрекъсната процедура, ако, на базата на предходен опит, се очаква, че за постигането на 20 % разграждане на веществото е необходим инкубационен период от три месеца. Алтернативно, нормалното серийно изпитване може да бъде променено на полунепрекъснато, ако за срок от приблизително 60 дни на изпитване по сериен метод не е постигнато никакво разграждане на изпитваното вещество. Когато бъде отчетена значителна степен на разграждане (напр. > 20 %), полунепрекъсната процедура може да бъде преустановена и изпитването да продължи като сериен експеримент.
При полунепрекъснато изпитване на всеки две седмици около една трета от обема на изпитваната суспензия се заменя с прясно взета вода, като изпитваното вещество се добавя към първоначалната концентрация. Аналогично, в случай на извършване на незадължителното изпитване със суспендирани утайки към подновяващата вода към първоначалната концентрация се добавя утайка (между 0,01 и 1 g/l). При извършване на изпитването със суспендирани неразтворени вещества от утайки е важно да се поддържа напълно суспендирана система и по време на подновяването на водата, както и периодът на престой да бъде идентичен за неразтворените вещества и за водата, тъй като в противен случай желаното наподобяване на хомогенна водна система без отделни фази може да бъде нарушено. Поради тези причини при използване на полунепрекъсната процедура се предпочита първоначалната концентрация на суспендирани утайки да бъде в по-ниската част на определения диапазон.
Препоръчаното добавяне на изпитвано вещество означава, че първоначалната концентрация на изпитваното вещество не е превишена от частичното подновяване на изпитваната суспензия, следователно приспособяването, което често се наблюдава при високи концентрации на изпитвано вещество, се предотвратява. Поради факта, че процедурата обхваща както повторна инокулация, така и компенсация на изчерпаните хранителни вещества и първични субстрати, първоначалното микробиално разнообразие се възстановява и продължителността на извършване на изпитването може да бъде удължена практически до безкрайност. Когато се използва полунепрекъсната процедура, е важно да се отбележи, че остатъчната концентрация на изпитваното вещество трябва да бъде коригирана по отношение на количествата изпитвано вещество, които се добавят и отстраняват при всяко едно подновяване. Концентрацията на общото изпитвано вещество и тази на разтвореното вещество могат да бъдат използвани взаимозаменяемо по отношение на съединения, които имат незначителна абсорбционна способност. Абсорбцията е незначителна (< 5 %) при определени условия (0,1—1 g неразтворени вещества/l) за вещества с log Kow < 3 (приложимо за неутрални, липофилни съединения). Това се илюстрира от следното примерно изчисление: 0,1 g/l неразтворени вещества съответства приблизително на 10 mg въглерод на литър (въглеродна фракция, fC = 0,01). Като се приеме, че;
Log Kow (от изпитваното вещество) = 3
Koc = 0,42 x Kow
Коефициент на разпределение, Kd = fC x Koc,
след което разтворената част от общата концентрация (C-вода (Cw)/C-общо (Ct) е:
Cw/Ct = 1/(1 + Kd x SS) = 1(1 + Koc x fC x SS) = 1/(1 + 0,42 x 103 x 0,01 x 0,1 x 10–3) = 0,999
Допълнение 3
Определяне на 14CO2
Индиректно определяне на 14CO2
При рутинните измервания индиректният метод обикновено е най-бърз и най-прецизен, в случай че изпитваното вещество е нелетливо и не се преобразува в летлив продукт на трансформация. Просто прехвърлете нефилтрираните проби (напр. количество от 5 ml) в сцинтилационните стъкленици. Първоначалната нормална активност в пробите е 5 000—10 000 dpm (80— 170 Bq), а минималната първоначална активност е около 1 000 dpm. CO2 следва да бъде отстранен след подкиселяване до pH 2—3 с 1—2 капки концентрирана H3PO4 или HCl. Отстраняването на CO2 следва да се извърши чрез пропускане на въздух за около половин—един час. Друга възможност е стъклените съдове да бъдат разклатени силно за около 1—2 часа (например върху вибрационен плот за микроплата) или да бъдат подложени на по-леко разклащане в продължение на една нощ. Ефикасността на способа за отстраняване на CO2 трябва да бъде проверена (чрез удължаване на периода на аерация или на разклащане). След това трябва да бъде добавена сцинтилационна течност, подходяща за отчитане на водни проби, като пробата бъде хомогенизирана чрез вихров миксер, а радиоактивността, да бъде определена посредством течно сцинтилационно броене, като се извади фоновата активност, установена в празните проби за изпитване (FB). Освен ако водата за изпитване не е силно оцветена или не съдържа висока концентрация на частици, пробите обикновено ще покажат еднородно потискане и ще бъдат достатъчни за извършването на корекции на потискането при използването на външен еталон. Ако водата за изпитване е силно оцветена, може да се наложи корекция на потискането чрез добавяне на вътрешен еталон. Ако концентрацията на частиците е висока, може да е невъзможно получаването на хомогенен разтвор или гел, или пък разликите в потискането между пробите може да са големи. В този случай може да бъде използван описаният по-долу метод по отношение на изпитвани суспензии. Ако изпитването се провежда като изпитване със суспендирани утайки, измерването на 14CO2 може да се извърши индиректно чрез вземането на хомогенна проба от водата за изпитване/суспензия в размер на 10 ml и разделянето на фазите чрез центрофугиране при подходяща скорост (напр. 40 000 m/s2 в продължение 15 мин.). След това водната фаза трябва да бъде обработена както е описано по-горе. Активността на 14C под формата на твърди частици (POA) трябва да се определи чрез повторно суспендиране на утайката в малко количество дестилирана вода, прехвърляне в сцинтилационни стъкленици и добавяне на сцинтилационна течност с цел образуване на гел (за целта се предлагат специални сцинтилационни течности). В зависимост от характера на частиците (напр. тяхното съдържание на органични вещества), може да се приложи изгниване на пробата в продължение на една нощ с помощта на тъканен солюбилизатор и след това се извърши хомогенизиране чрез вихров миксер преди добавянето на сцинтилационна течност. Алтернативно, POA може да се определи чрез горене при излишък на кислород чрез използването на оксидатор на проби. При отчитането трябва винаги да се вземат под внимание вътрешните стандарти и е възможно да се наложи извършването на корекции на потискането, като в допълнение се използва вътрешен стандарт за всяка отделна проба.
Директно определяне на 14CO2
Ако отделеният 14CO2 се измерва директно, това трябва да бъде извършвано чрез осигуряване на повече колби в началото на изпитването, зареждане на колбите за изпитване във всяка точка на измерване, чрез подкиселяване до pH 2—3 и събиране на 14CO2 във вътрешен (поставен във всяка колба в началото на изпитването) или външен абсорбер. Като абсорбционно средство може да бъде използвана или основа (напр. 1 N разтвор на NaOH, или кубче NaOH), етаноламин или базирано на етаноламин вещество, както и предлагани в търговската мрежа абсорбанти. При директното измерване на 14CO2 колбите трябва да бъдат затворени, например с прегради от бутилов каучук.
Фигура 1а
Пример за аритметично графично изобразяване (остатъчна активност спрямо времето)

Фигура 1б
Пример за полулогаритмично графично изобразяване (ln остатъчна активност спрямо времето)

ПРИЛОЖЕНИЕ VI
|
C.26. |
ИЗПИТВАНЕ ПОТИСКАНЕТО НА РАСТЕЖА НА ВИДА LEMNA |
1. МЕТОД
Настоящият метод е еквивалентен на ОИСР TG 221 (2006) (1). Съществува широко съгласие между институциите на ЕС, че изпитването Lemna е подходяща алтернатива на изпитването с водорасли за силно оцветени вещества (2)(3).
1.1. ВЪВЕДЕНИЕ
Настоящият метод за изпитване е предназначен за оценяване на токсичността на вещества по отношение на сладководни растения от вида Lemna (водна леща). Той се базира на съществуващите насоки (4)(5)(6)(7)(8)(9), но включва и модификации на тези методи с цел да бъдат отразени най-скорошните научни изследвания и обсъждането на редица ключови въпроси. Предложеният метод е валидиран чрез международно кръгово изпитване (10).
Настоящият метод за изпитване описва изпитване за токсичност с използване на Lemna gibba и Lemna minor, като и двата вида са били пространно изследвани и са предмет на посочените по-горе стандарти. Таксономията на вида Lemna е трудна, тъй като е усложнена от съществуването на широк кръг от фенотипове. Въпреки че за Lemna може да възникне генетична изменчивост като реакция на токсични вещества, към момента няма достатъчно данни относно този източник на изменчивост, за да бъде препоръчан специфичен клонинг, който да бъде използван с настоящия метод за изпитване. Следва да се отбележи, че изпитването не е проведено при пълно отсъствие на чужди организми, но по време на процедурата за изпитване се предприемат поетапни стъпки за поддържане на минимално замърсяване с други организми.
Описани са подробности от изпитването с обновяване (полустатично или проточно) на изпитвания разтвор и без обновяване (статично) на изпитвания разтвор. В зависимост от целите на изпитването и регулаторните изисквания се препоръчва да се обмисли прилагането на полустатичния метод и на проточния метод, например за вещества, които бързо се губят от разтвора в резултат на изпаряване, фоторазграждане, утаяване или биоразграждане. Допълнителни насоки са дадени в (11).
1.2. ОПРЕДЕЛЕНИЯ
За целите на настоящия метод на изпитване са използвани следните определения и съкращения:
Биомаса: сухото тегло на живите организми, съдържащи се в една популация. В настоящото изпитване заместителите на биомасата, като количеството листовидни тела или площта на листовидните тела обикновено са измерени и поради това използването на термина „биомаса“ се отнася също и за измерванията на заместителите.
Хлороза: пожълтяване на тъканта на листовидните тела.
Клонинг: организъм или клетка, възникнал от единствен индивид чрез безполово възпроизвеждане. Следователно, индивидите от един и същи клонинг са генетично идентични.
Колония: означава съвкупност от майчини и дъщерни листовидни тела (обикновено от 2 до 4), закрепени един към друг. Понякога се посочва като растение.
ECx : концентрацията на изпитваното вещество, разтворено в средата на изпитване, която води до потискане на растежа от x % (например 50 %) на Lemna в рамките на определения период на експозиция (да бъде изрично отбелязано, ако има отклонение от пълната или нормалната продължителност на изпитването). С оглед на еднозначно обозначаване на стойността на EC, изведена от скоростта на растежа или от добива, символът „ErC“ се използва за скоростта на растежа, а „EyC“ се използва за добива, следван от използваната измервана променлива, например ErC (брой листовидни тела).
Проточно изпитване: изпитване, при което изпитваните разтвори се подменят постоянно.
Листовидно тяло: отделна/единична „листообразна“ структура на растението водна леща. Това е най-малката единица, т.е. годен за възпроизводство индивид.
Издутост: означава листовидни тела, чийто външен вид е изпъкнал или набъбнал.
Растеж: нарастване на измерваната променлива, например на броя на листовидните тела, сухото тегло, мокрото тегло или площта на листовидните тела през периода на изпитване.
Скорост на растеж (средна специфична скорост на растеж): логаритмичното нарастване на биомасата през периода на експозиция.
Най-ниска концентрация на наблюдавано въздействие (LOEC): най-ниската изпитвана концентрация, при която за веществото се наблюдава статистически значимо понижаващо въздействие върху растежа (при p < 0,05) в сравнение с контролната проба, в рамките на дадено време на експозиция. При все това, всички изпитвани концентрации над LOEC трябва да имат вредно въздействие, равно или по-голямо от наблюдаваното при LOEC. Когато тези две условия не могат да бъдат удовлетворени, трябва да се даде пълно обяснение за това как е била избрана LOEC (и следователно NOEC).
Измервани променливи: всички видове променливи, които се измерват с цел да се изрази крайната точка на изпитването с помощта на една или няколко различни променливи на реакцията. При настоящия метод измерваните променливи са броят на листовидните тела, площта на листовидните тела, свежото тегло и сухото тегло.
Монокултура: култура от един вид растение.
Некроза: мъртва (т.е. бледа или просмукана с вода) листна тъкан.
Концентрация без наблюдавано въздействие (NOEC): изпитваната концентрация, непосредствено под LOEC.
Фенотип: наблюдаемите характеристики на един организъм, определени от взаимодействието на неговите гени и неговата околна среда.
Променливи на реакцията: променливите за оценяване на токсичността, изведени от произволни измервани променливи, описващи биомасата, с помощта на различни методи за изчисление. При настоящия метод, скоростите на растежа и на добива са променливи на реакцията, изведени от измервани променливи като брой листовидни тела, площ на листовидните тела, свежо тегло или сухо тегло.
Полустатично (обновяемо) изпитване: изпитване, при което изпитваният разтвор периодично се подменя на определени интервали по време на изпитването.
Статично изпитване: метод за изпитване без обновяване на изпитвания разтвор по време на изпитването.
Крайна точка на изпитване: описва общия показател, който ще бъде изменен от химикала за изпитване по отношение на контролната проба като цел на изпитването. При настоящия метод крайната точка на изпитването е потискането на растежа, което може да се изрази чрез различни променливи на реакцията на базата на една или повече измервани променливи.
Среда на изпитване: напълно синтетичната среда на растеж, върху която растат изпитваните растения, когато са подложени на въздействието на веществото за изпитване. Веществото за изпитване обикновено се разтваря в средата на изпитване.
Добив: стойността на измервана променлива, чрез която се изразява биомасата в края на периода на експозиция минус измерваната променлива в началото на периода на експозиция.
1.3. ПРИНЦИП НА ИЗПИТВАНЕТО
Дадена е възможност експоненциално растящите растителни култури от вида Lemna да растат като монокултури при различни концентрации на изпитваното вещество за период от седем дни. Целта на изпитването е да се измерят количествено свързаните с веществото въздействия върху вегетативния растеж през този период, на базата на оценки на избрани измервани променливи. Броят на листовидните тела е главната измервана променлива. Най-малко още една измервана променлива (обща площ на листовидните тела, сухо тегло или свежо тегло) също се измерва, тъй като някои вещества могат да окажат много по-голямо въздействие върху други измервани променливи, отколкото върху броя на листовидните тела. За количествена оценка на свързани с веществата въздействия, растежът в изпитваните разтвори се сравнява с този на контролните проби и се определят концентрациите, при които се осъществява потискане на растежа до определените x % (например 50 %) и се изразяват като ECx (например EC50)
Крайната точка на изпитването е потискането на растежа, изразено като логаритмично нарастване на измерваната променлива (средна специфична скорост на растеж) през периода на експозицията. От средните специфични скорости на растеж, регистрирани в серия от изпитвани разтвори, се определя концентрацията, при която се осъществява потискане на скоростта на растежа до определените x % (например 50 %) и се изразява като ErCx (например ErC50).
Допълнителната променлива на растеж, използвана в настоящия метод на изпитване, е добивът, който може да е необходим, за да бъдат изпълнени специфични регулаторни изисквания в някои държави. Той се определя като измерваните променливи в края на периода на експозиция минус измерваните променливи в началото на периода на експозиция. От добива, регистриран в серия от разтвори на изпитване се изчислява концентрацията, при която се осъществява потискане на добива до определените x % (например 50 %) и се изразява като EyCx (например EyC50).
В допълнение концентрацията с най-ниско наблюдавано въздействие (LOEC) и концентрацията с ненаблюдавано въздействие (NOEC) могат да бъдат определени статистически.
1.4. ИНФОРМАЦИЯ ОТНОСНО ИЗПИТВАНОТО ВЕЩЕСТВО
Следва да е възможно използването на аналитичен метод с достатъчна чувствителност за количествено измерване на веществото в средата на изпитване.
Информацията относно изпитваното вещество, която може да бъде полезна при установяване на условията на изпитване, включва структурната формула, чистотата, водоразтворимостта, стабилността във вода и на светлина, pKa, Kow, парното налягане и биоразградимостта. Разтворимостта във вода и парното налягане може да се използват за изчисляване на константата по закона на Хенри, която ще покаже дали са възможни значителни загуби на изпитваното вещество през периода на изпитването. Това ще помогне да се разбере дали следва да се предприемат определени стъпки за контролиране на тези загуби. Когато информацията за разтворимостта и стабилността на изпитваното вещество е несигурна, се препоръчва те да бъдат оценени в условията на изпитване, т.е. среда на растеж, температура, режим на осветеност, които ще бъдат използвани при изпитването.
Когато контролът върху pH на средата на изпитването е особено важен, например при изпитване на метали или вещества, които са нестабилни при хидролизата, се препоръчва добавянето на буферно вещество към хранителната среда (вж. първата алинея от раздел 1.7.4). Допълнителни насоки относно изпитвани вещества, чиито физикохимични свойства затрудняват тяхното изпитване, са дадени в (11).
1.5. РЕФЕРЕНТНО ВЕЩЕСТВО
Референтното вещество (референтните вещества), като например 3,5-дихлорфенол, използвано в международното кръгово изпитване (10), може да бъде изпитвано като средство за проверка на процедурата за изпитване. Желателно е референтното вещество да се изпитва най-малко два пъти годишно или, когато изпитването се извършва по-рядко, да се извършва едновременно с определянето на токсичността на изпитваното вещество.
1.6. ВАЛИДНОСТ НА ИЗПИТВАНЕТО
За да е валидно изпитването, времето за удвояване на листовидното тяло в контролната проба трябва да бъде по-малко от 2,5 дни (60 часа), съответстващо на седемкратно увеличение за седем дни и средна специфична скорост на растеж от 0,275 d–1. Използването на средата и на условията на изпитване, описани в настоящия метод за изпитване, позволяват постигането на този критерий с помощта на статичен режим на изпитване (8). Очаква се също, че този критерий е достижим и в условия на полустатично и проточно изпитване. Изчисляването на времето за удвояване е показано в раздел 2.1.
1.7. ОПИСАНИЕ НА МЕТОДА
1.7.1. Апаратура
Цялото оборудване в контакт със средата на изпитване следва да е направено от стъкло или друг химически инертен материал. Стъклените съдове, използвани за отглеждане на култури и за изпитване, трябва да бъдат почистени от химични замърсители, които биха могли да проникнат в средата на изпитване, както и да бъдат стерилни. Съдовете за изпитването трябва да са достатъчно широки, така че листовидното тяло от различни колонии в контролните съдове да расте без припокриване в края на изпитването. Няма значение дали корените докосват дъното на съдовете за изпитване, но се препоръчва минимална дълбочина от 20 mm и минимален обем от 100 ml за всеки съд за изпитване. Изборът на съда за изпитването не е от решаващо значение, при условие че тези изисквания са изпълнени. Доказано е, че подходящи за целта са стъклени лабораторни чаши, блюда кристализатори или стъклени блюда на Петри с нужните размери. Съдовете за изпитването трябва да бъдат покрити с оглед минимизиране на изпарението и на случайното замърсяване, като същевременно се даде възможност за циркулиране на въздуха. Подходящите съдове за провеждане на изпитването и особено капаците трябва да предотвратяват засенчването или промените в спектралните характеристики на светлината.
Културите и съдовете за изпитването трябва да не се съхраняват заедно. Това се постига най-добре с използването на изолирани от въздействията на околната среда, камери, инкубатори или помещения за растеж. Необходимо е осветлението и температурата да могат да се контролират и да се поддържат на постоянно ниво (вж. раздел 1.7.8).
1.7.2. Изпитван организъм
Организмът, използван за настоящото изпитване, е Lemna gibba или Lemna minor. Кратки описания на видовете водна леща, използвани за изпитване за токсичност, са дадени в допълнение 1. Растителният материал може да бъде получен от колекция от култури, от друга лаборатория или от полето. Ако са събрани от полето, растенията следва да се съхраняват в култура в същата среда, която се използва за изпитването, в течение на минимум осем седмици преди да се използват. Полевите обекти за събиране на началните култури трябва да са без явни източници на замърсяване. Ако са получени от друга лаборатория или от колекция от култури, те следва да се съхраняват по същия начин в течение на минимум три седмици. За източника на растителен материал, вида и клонинга (ако е известен), използвани в изпитването, трябва винаги да се докладва.
Трябва да бъдат използвани монокултури, които нямат видимо замърсяване с други организми като водорасли и протозоа. Здравите растения от L. minor трябва да се състоят от колонии, включващи между два и пет листовидни тела, докато здравите колонии на L. gibba могат да съдържат до седем листовидни тела.
Качеството и еднородността на използваните за изпитването растения оказват значително влияние върху резултата от изпитването и поради това растенията трябва да бъдат грижливо подбрани. Трябва да се използват млади, бързорастящи растения без видими повреди и обезцветяване (хлороза). Признак за култури с добро качество е големият обхват на колонии, съставени от поне две листовидни тела. Голям брой отделни листовидни тела е показателен за екологичен стрес, например ограничаване на хранителните вещества, като растителен материал от такива култури не трябва да се използва за изпитване.
1.7.3. Култивиране
За намаляване на честотата за поддръжка на културата (например когато не са планирани изпитвания с Lemna за известен период), културите трябва да се съхраняват при намалено осветление и температура (4—10 °C). Подробности за култивирането са дадени в допълнение 2. Очевидни знаци за замърсяване с водорасли или други организми изискват стерилизация на повърхността на подпроба от листообразните тела на Lemna, последвана от прехвърляне в прясна среда (вж. допълнение 2). В такъв случай останалата замърсена култура трябва да се изхвърли.
Най-малко седем дни преди изпитването достатъчно колонии се прехвърлят по стерилен начин в прясна стерилна среда и се култивират в течение на 7—10 дни при условията на изпитването.
1.7.4. Среда на изпитване
Препоръчват се различни среди за Lemna minor и за Lemna gibba, както е описано по-долу. Трябва внимателно да се прецени включването на pH буфер в средата на изпитване (MOPS (4-морфолинпропанова сулфонна киселина, № по CAS: 1132—61—2; № по EINECS: 214—478—5) в средата за L. minor и NaHCO3 в средата за L. gibba), когато има предположения, че буферът може да реагира с изпитваното вещество и да повлияе на изразяването на неговата токсичност. Средата на Steinberg (12) също е приемлива, при условие че критериите за валидност са изпълнени.
За култивиране и изпитване с L. minor се препоръчва модификация на средата на растеж за Lemna по шведския стандарт (SIS). Съставът на тази среда е даден в допълнение 3.
Средата на растеж 20X — AAP, както е описана в допълнение 3, се препоръчва за култивиране и изпитване с L. gibba.
Средата на Steinberg, както е описана в допълнение 3, също е подходяща за L. minor, но би могла да се използва и за L. gibba, при условие че са изпълнени критериите за валидност.
1.7.5. Разтвори за изпитване
Разтворите за изпитване обикновено се приготвят чрез разреждане на изходните разтвори. Изходният разтвор на изпитваното вещество обикновено се приготвя чрез разтваряне на веществото в среда на растеж.
Най-високата изпитвана концентрация на изпитваното вещество обикновено не трябва да надвишава разтворимостта във вода на веществото при условията за провеждане на изпитването. Следва да се отбележи обаче, че видът Lemna плава на повърхността и може да бъде изложен на въздействието на вещества, които се събират на границата между водата и въздуха (например, слабо разтворими във вода или хидрофобни вещества или повърхностно активни вещества). При такива обстоятелства експозицията ще произтича от материал, различен от този в разтвора, и изпитваните концентрации могат, в зависимост от характеристиките на изпитваното вещество, да надвишат разтворимостта във вода. За изпитвани вещества с ниска водоразтворимост може да е необходимо да се приготви концентриран изходен разтвор или емулсия на веществото, като се използва органичен разтворител или дисперсант, за да се улесни добавянето на точни количества от изпитваното вещество към средата на изпитване и да се подпомогне неговото диспергиране и разтваряне. Трябва да се положат всички усилия, за да се избегне използването на такива материали. Не трябва да има фитотоксичност в резултат на използването на спомагателни разтворители и дисперсанти. Например обичайно използваните разтворители, които не предизвикват фитотоксичност при концентрации до 100 μl·l–1, включват ацетон и диметил формамид. Ако се използва разтворител или дисперсант, трябва да бъде отчетена неговата крайна концентрация и тя да се поддържа на минимално ниво (≤ 100 μl·l–1), а всички третирани и контролни проби трябва да съдържат еднаква концентрация на разтворителя или дисперсанта. Допълнителни насоки относно използването на дисперсанти са дадени в (11).
1.7.6. Изпитвана и контролна групи
Предварителното познаване на токсичността на изпитваното вещество по отношение на Lemna, например от изпитване за определяне на диапазон, ще помогне при избирането на подходящи изпитвани концентрации. В окончателното изпитване за токсичност обикновено би следвало да има най-малко пет изпитвани концентрации, подредени в геометрична прогресия. За предпочитане е коефициентът на разделяне между изпитваните концентрации да не надхвърля 3,2, но могат да се използват и по-големи стойности, при които кривата „концентрация—реакция“ е полегата. Трябва да се представи обосновка, ако са използвани по-малко от пет концентрации. Най-малко три идентични проби трябва да се използват за всяка от изпитваните концентрации.
При задаването на диапазона на изпитваните концентрации (за изпитване за определяне на диапазон и/или за окончателно изпитване за токсичност), трябва да се има пред вид следното:
|
— |
за определяне на ECx изпитваните концентрации трябва да обхващат стойността на ECx, за да се гарантира подходящо доверително ниво. Например, ако се оценява EC50, най-голямата изпитвана концентрация трябва да е по-голяма от стойността на EC50. Ако стойността на EC50 попада извън диапазона на изпитваните концентрации, присъединените доверителни интервали ще бъдат големи и правилната оценка за статистическото съответствие на модела може да не е възможна. |
|
— |
ако целта е да се оцени LOEC/NOEC, най-ниската изпитвана концентрация трябва да е достатъчно ниска, така че растежът да не бъде значително по-малък от този на контролната проба. В допълнение, най-високата изпитвана концентрация трябва да е достатъчно висока, така че растежът да бъде значително по-нисък от този в контролната проба. Ако това не е така, изпитването ще трябва да се повтори с използване на различен диапазон за концентрациите (освен ако най-високата концентрация е на границата на разтворимостта или максимално изискваната пределна концентрация, например 100 mg·l–1). |
Всяко изпитване трябва да включва контролни проби, състоящи се от същата хранителна среда, същия брой листовидни тела и колонии, същите условия на околната среда и процедури както при съдовете за изпитване, но без изпитваното вещество. Ако се използва спомагателен разтвор или дисперсант, трябва да бъде включена допълнително контролна третирана проба, като разтворителят/дисперсантът трябва да присъстват в същата концентрация като тази в съдовете с изпитваното вещество. Броят на съдовете за повторни контролни проби (и съдовете с разтворител, ако е приложимо) трябва да е най-малко равен, а в идеалния случай да е два пъти по-голям от броя на съдовете, използвани за всяка изпитвана концентрация.
Ако не се изисква определяне на NOEC, планът за изпитването може да бъде променен, като се увеличи броят на концентрациите и се намали броят на повторните проби за всяка от концентрациите. Но все пак броят на повторните контролни проби трябва да е най-малко три.
1.7.7. Експозиция
Колониите, състоящи се от 2 до 4 видими листовидни тела, се прехвърлят от инокулираната култура и се прехвърлят на принципа на случайността в съдовете за изпитване при стерилни условия. Всеки съд за изпитване трябва да съдържа общо от 9 до 12 листовидни тела. Броят на листовидните тела и колонии трябва да е еднакъв във всеки съд за изпитване. Придобитият опит за настоящия метод и данните от кръговото изпитване показват, че използването на три повторни проби от една третирана проба, като всяка повторна проба съдържа в началото от 9 до 12 листовидни тела, е достатъчно за откриването на различията в растежа приблизително от 4 до 7 % на потискането, изчислено чрез скоростта на растежа (10 до15 %, изчислени чрез добива) между третираните проби (10).
За минимизиране влиянието на пространствените различия в интензитета на светлината и температурата се изисква схема за случаен подбор за разполагане на съдовете за изпитване в инкубатора. При извършване на наблюдения (или при по-честа промяна на разполагането) също се изискват блокова схема или промяна на разполагането чрез случаен подбор.
Ако предварителното изпитване за стабилност показва, че концентрацията на изпитваното вещество не може да бъде поддържана (т.е. измерената концентрация пада под 80 % от първоначално измерената концентрация) по време на изпитването (7 дни), се препоръчва полустатичен режим на изпитване. В този случай колониите трябва да бъдат експонирани в прясно приготвени разтвори за изпитване и за контролни проби най-малко два пъти по време на изпитването (например, ден 3 и 5). Честотата на експониране в прясна среда ще зависи от стабилността на изпитваното вещество; може да е необходима по-голяма честота, за да се поддържа почти постоянна концентрация за силно нестабилни или летливи вещества. При някои обстоятелства може да се изисква процедура с протичане (11)(13).
Сценарият на експозиция чрез листно подхранване на растенията (пръскане) не е включен в настоящия метод за изпитване, вместо това вж. (14).
1.7.8. Инкубационни условия
Трябва постоянно да се използва топло или студено бяло флуоресцентно осветление, за да се осигури интензитет на светлината, избрана в диапазона 85—135 μE·m–2·s–1, когато се измерва във фотосинтетично активно излъчване (400—700 nm) в точки, чието разстояние от източника на светлина е еднакво с това на листовидни тела на Lemna (еквивалентно на 6 500—10 000 lux). Разликите в интензитета на светлината и избрания за площта на изпитването интензитет на светлината не трябва да нахвърлят ± 15 %. Методът за засичане и измерване на светлината, особено видът на сензора, ще оказват влияние върху измерваната стойност. Сферичните сензори (които реагират на светлина, излъчвана от всички ъгли над и под равнината на измерване) и „косинусовите“ сензори (които реагират на светлината, излъчвана от всички ъгли над равнината на измерване), са за предпочитане пред еднопосочните сензори и ще отчитат по-прецизни показания за описания тук вид многоточков източник на светлина.
Температурата в съдовете за изпитване трябва да бъде 24 ± 2 °C. Стойността на pH на контролната среда не трябва да се повишава с повече от 1,5 единици по време на изпитването. Въпреки това, отклонение с повече от 1,5 единици не прави изпитването невалидно, при условие че може да се покаже, че критериите за валидност са изпълнени. Необходимо е да се обърне допълнително внимание на отклонението в стойността на pH в някои специални случаи, например когато се изпитват нестабилни вещества или метали. Вж. (11) за допълнителни насоки.
1.7.9. Продължителност
Изпитването се прекратява 7 дни след прехвърлянето на растенията в съдовете за изпитване.
1.7.10. Измервания и аналитични определения
В началото на изпитването се преброяват и записват листовидните тела в съдовете за изпитване, като се внимава да бъде гарантирано отчитането на подаващите се, ясно видими листовидни тела. Броят на листовидните тела, които изглеждат нормални или анормални, следва да се определи в началото на изпитването и най-малко веднъж на всеки 3 дни по време на експозицията (т.е. в най-малко 2 случая по време на 7-дневния период), както и при завършване на изпитването. Трябва да бъдат отбелязани промените в развитието на растението, например в размера на листовидните тела, външния вид, показания за некроза, хлороза или набъбналост, разпадане на колонията или загуба на способността за задържане върху повърхността на водата, както и дължината и външния вид на корените. Трябва да се отбележат и значими характеристики на средата на изпитване (например наличието на неразтворен материал, растеж на водорасли в съда за изпитване).
В допълнение към определянето на броя на листовидните тела по време на изпитването, трябва да се оценят и въздействията на изпитваното вещество върху една (или повече) от следните измервани променливи:
|
i) |
обща площ на листовидните тела, |
|
ii) |
сухо тегло, |
|
iii) |
свежо тегло. |
Общата площ на листовидните тела има предимство, че може да се определи за всеки изпитван и контролен съд в началото, по време и в края на изпитването. Сухото или свежото тегло трябва да се определят в началото на изпитването от проба на инокулативната култура, която е образец на това, което се използва за начало на изпитването, както и в края на изпитването с растителен материал от всеки съд за изпитване и всеки контролен съд. Ако не се измерва площта на листовидните тела, за предпочитане е сухото пред свежото тегло.
Общата площ на листовидните тела, сухото тегло и свежото тегло могат да бъдат определени, както следва:
|
i) |
Обща площ на листовидните тела: Общата площ на листовидните тела на всички колонии може да се определи чрез анализ на изображенията. Силуетът на съда за изпитване и растенията могат да се уловят с помощта на видеокамера (т.е. чрез поставянето на съда върху светлопроницаема кутия) и полученото изображение да се дигитализира. Чрез калибриране с плоски форми на известна площ след това може да се определи общата площ на листовидните тела в съда за изпитване. Трябва да се вземат мерки за изключване на интерференцията, предизвикана от ръба на съда за изпитване. Алтернативен, но по-трудоемък подход е да се фотографират съдовете за изпитване и растенията, да се изреже полученият силует на колониите и да се определи тяхната площ с помощта на анализатор на площта на листата или с милиметрова хартия. И други техники (например съотношението по тегло на хартията между площта на силуета на колониите и единицата площ) може също да са подходящи. |
|
ii) |
Сухо тегло: Всички колонии се събират от всеки съд за изпитване и се изплакват с дестилирана или дейонизирана вода. Те се попиват за отстраняване на излишната вода и след това се изсушават при 60 °C до постоянно тегло. Всички части на корени трябва също да се включат. Сухото тегло трябва да бъде изразено с точност най-малко 0,1 mg. |
|
iii) |
Свежо тегло: Всички колонии се прехвърлят в тръби от предварително претеглен полистирол (или друг инертен материал) с малки (1 mm) дупки в заоблените долни части. След това тръбите се центрофугират с 3 000 rpm в течение на 10 минути при стайна температура. Тръбите, които сега съдържат изсушените колонии, се измерват отново и свежото тегло се пресмята чрез изваждането на теглото на празната тръба. |
1.7.10.1. Честота на измерванията и аналитичните определения
Ако се използва схема за статично изпитване, pH на всяка третирана проба трябва да се измери в началото и в края на изпитването. Ако се използва схема за полустатично изпитване, pH трябва да се измерва във всяка партида от „свеж“ разтвор за изпитване преди всяко подновяване, както и в съответните „изчерпани“ разтвори.
Интензитетът на светлината трябва да се измерва във вегетационната камера, в инкубатора или помещението в точки, които са на разстояние от източника на светлина, което е еднакво с това на листовидните тела на Lemna. Измерванията трябва да се извършват най-малко веднъж по време на изпитването. Температурата на средата в заместващия съд, който се държи при същите условия във вегетационната камера, инкубатора или помещението, трябва да се записва най-малко веднъж дневно.
По време на изпитването концентрациите на изпитваното вещество се определят през подходящи интервали. При статични изпитвания минималното изискване е концентрациите да се определят в началото и края на изпитването.
При полустатични изпитвания, при които концентрацията на изпитваното вещество не се очаква да остане в рамките на ± 20 % от номиналната концентрация, е необходимо да се анализират всички прясно приготвени изпитвани разтвори и същите разтвори при всяко обновяване (вж. третата алинея от раздел 1.7.7). Обаче при тези изпитвания, при които измерената начална концентрация на изпитваното вещество не е в рамките на ± 20 % от номиналната, но за които може да се предоставят достатъчно доказателства, показващи че началните концентрации са повторяеми и стабилни (т.е. в диапазона 80—120 % от началната концентрация), химичните определения могат да се извършат само за най-високата и най-ниската изпитвана концентрация. Във всички случаи определянето на концентрациите на изпитваното вещество преди обновяването е необходимо да се извършва само за един повторен съд за всяка изпитвана концентрация (или съдържанието на съдовете, обединени в пул от повторени проби).
Ако се използва проточно изпитване, подходящ е режим за взимане на проби, подобен на описания при полустатичните изпитвания, включително анализа в началото, по средата и в края на изпитването, но измерванията на „изчерпаните“ разтвори в този случай не са подходящи. В този тип на изпитване скоростта на потока на разреждащото и изпитваното вещество или изходният разтвор на изпитваното вещество трябва да се проверяват ежедневно.
Ако има доказателство, че концентрацията на веществото, което се изпитва, се поддържа задоволително в рамките на ± 20 % от номиналната или измерената начална концентрация през цялото време на изпитването, анализът на резултатите може да се базира на номиналните или измерените начални стойности. Ако отклонението от номиналната или измерената начална концентрация е по-голямо от ± 20 %, анализът на резултатите трябва да се базира на средногеометричната стойност на концентрацията по време на експозицията или на модели, които описват намаляването на концентрацията на изпитваното вещество (11).
1.7.11. Гранично изпитване
При някои обстоятелства, например когато едно предварително изпитване е показало, че изпитваното вещество няма токсично въздействие при концентрации до 100 mg·l–1, или до неговата граница на разтворимост в средата на изпитване (която от двете стойности е по-ниска), може да се предприеме гранично изпитване, включващо сравнение на реакциите в контролна група и една група с третирани проби (100 mg·l–1 или концентрация, равна на границата на разтворимост). Настоятелно се препоръчва това да бъде подкрепено с анализ на концентрацията на експозиция. Всички предходни условия на изпитване и критерии за валидност се прилагат и за гранично изпитване, с изключение на това, че броят на третираните повторни проби трябва да се удвои. Растежът в контролната и третираната група може да се анализира чрез използване на статистическо изпитване за сравняване на средните стойности, например t-тест на Student.
2. ДАННИ И ОТЧИТАНЕ
2.1. ВРЕМЕ НА УДВОЯВАНЕ
За определяне на времето за удвояване (Td) на броя на листовидните тела и придържането към този критерий за валидност при изследването (раздел 1.6) се използва следната формула с данните, получени от контролните съдове:
Td = ln 2/μ
където μ е средната специфична скорост на растеж, определена според описанието в първа и втора алинея от раздел 2.2.1.
2.2. ПРОМЕНЛИВИ НА РЕАКЦИЯТА
Целта на изпитването е да се определят въздействията на изпитваното вещество върху вегетативния растеж на Lemna. Настоящият метод за изпитване описва две променливи на реакцията, тъй като държавите-членки имат различни предпочитания и регулаторни нужди. За да се приемат резултатите от изпитването във всички държави-членки, въздействията следва да се оценяват с използването и на двете променливи a) и б) на реакцията, описани по-долу.
|
a) |
Средна специфична скорост на растеж: тази променлива на реакцията се изчислява на базата на промените в логаритмите на броя на листовидните тела и допълнително — на базата на промените в логаритмите на друг измерван параметър (обща площ на листовидните тела, сухо тегло или свежо тегло) с течение на времето (изразено в дни) в контролните проби и във всяка група с третирани проби. Понякога тя се нарича относителна скорост на растеж (15). |
|
б) |
Добив: тази променлива на реакцията се изчислява на базата на промените в броя на листовидните тела и допълнително — на базата на промените в логаритмите на друг измерван параметър (обща площ на листовидните тела, сухо тегло или свежо тегло) в контролните проби и във всяка група с третирани проби до края на изпитването. |
Следва да се отбележи, че стойностите на токсичността, изчислени с помощта на тези две променливи на реакцията, не са сравними и тази разлика трябва да се разпознава, когато се използват резултатите от изпитването. Стойностите на ECx, базирани на средната специфична скорост на растеж (ErCx), по правило ще бъдат по-високи от резултатите, базирани на добива (EyCx), ако се придържате към условията за изпитване на настоящия метод за изпитване, което се дължи на математическата основа на съответните подходи. Това не трябва да се интерпретира като разлика между чувствителността на двете променливи на растеж, тъй като стойностите просто са различни чисто математически. Концепцията за средната специфична скорост на растеж се основава на общия експоненциален модел за растежа на водната леща в неограничени култури, където токсичността се оценява на базата на въздействието върху скоростта на растежа, без да зависи от абсолютното ниво на специфичната скорост на растеж на контролната проба, от наклона на кривата на концентрация-реакция или продължителността на изпитването. Противоположно на това, резултатите, базирани на променливата на реакцията за добива, зависят от всички тези други променливи. EyCx зависи от специфичната скорост на растеж на вида водна леща, използван във всяко изпитване, както и от максималната специфична скорост на растеж, която може да варира между видовете и дори различните клонинги. Тази променлива на реакцията не следва да се използва за сравняване на чувствителността към токсични вещества между видовете водна леща и дори различните клонинги. Докато използването на средната специфична скорост на растеж за оценяване на токсичността е за предпочитане от научна гледна точка, оценките за токсичността, базирани на добива, които също са включени в настоящия метод за изпитване, удовлетворяват действащите регулаторни изисквания в някои държави.
Оценките за токсичността трябва да са базирани на броя на листовидните тела и на още една допълнителна измервана променлива (обща площ на листовидните тела, сухо тегло или свежо тегло), тъй като някои вещества могат да въздействат на други измервани променливи много повече, отколкото броят на листовидните тела. Това въздействие не може да бъде открито само чрез изчисляване на броя на листовидните тела.
Броят листовидни тела, както и всяка друга записана измервана променлива, т.е. общата площ на листовидните тела, сухото тегло или свежото тегло, се табулират заедно с концентрациите на изпитваното вещество за всеки измерван случай. Последващият анализ на данни, например оценяването на LOEC, NOEC или ECx, трябва да се основава на стойностите за отделните повторни проби, а не на изчислените средни стойности за група третирани проби.
2.2.1. Средна специфична скорост на растеж
Средната специфична скорост на растеж за определен период се изчислява като логаритмичното нарастване в променливите за растеж — брой на листовидните тела и още една измервана стойност (обща площ на листовидните тела, сухо тегло или свежо тегло) — като се използва формулата, дадена по-долу, за всяка повторна проба на контролната проба и третираните проби:

където:
|
— |
μi—j: |
средна специфична скорост на растеж за времето от i до j |
|
— |
Ni: |
измервана променлива в съда за изпитване или в контролния съд в момента от време i |
|
— |
Nj: |
измервана променлива в съда за изпитване или в контролния съд в момента от време j |
|
— |
t: |
времеви интервал от i до j |
За всяка група от третирани проби и контролна група да се изчисли средната стойност за скоростта на растеж заедно с оценки на променливостта.
Средната специфична скорост на растеж трябва да се изчисли за целия период на изпитване (времето „i“ в горната формула е началото на изпитването, а времето „j“ е краят на изпитването). За всяка изпитвана и контролна концентрация да се изчисли средната стойност на скоростта за растеж заедно с оценки на променливостта. Допълнително трябва да се оцени скоростта на растеж сектор по сектор с цел да се оценят въздействията на изпитваното вещество, които възникват в периода на експозиция (например чрез проверка на логаритмично преобразуваните криви на растежа). Значителните различия между скоростта на растеж по сектори и средната скорост на растеж показват отклонение от постоянния експоненциален растеж и че има основание за внимателна проверка на кривите на растежа. В този случай консервативният подход ще бъде да се сравнят специфичните скорости на растежа на третираните култури в течение на времевия интервал на максимално потискане с тези за контролните проби за същия времеви интервал.
Процентът на потискане на скоростта на растежа (Ir) тогава може да се изчисли за всяка изпитвана концентрация (група от третирани проби) съгласно следната формула:

където:
|
— |
% Ir: |
процентното потискане на средната специфична скорост на растеж |
|
— |
μC: |
средната стойност за μ в контролната проба |
|
— |
μT: |
средната стойност за μ в групата от третирани проби |
2.2.2. Добив
Въздействията върху добива се определят на базата на две измервани променливи, броя на листовидните тела и още една измервана променлива (обща площ на листовидните тела, сухо тегло или свежо тегло), които присъстват във всеки съд за изпитване в началото и в края на изпитването. За сухото тегло или свежото тегло началната биомаса се определя на базата на проба от листовидни тела, взета от същата проба, използвана за инокулацията на съдовете за изпитване (вж. втората алинея от раздел 1.7.3). За всяка изпитвана концентрация и контролна концентрация да се изчисли средната стойност на добива заедно с оценки на променливостта. Средното процентно потискане на добива (% Iy) може да се изчисли за всяка група от третирани проби, както следва:

където:
|
— |
% Iy: |
процентното намаление на добива |
|
— |
bC: |
крайната биомаса минус началната биомаса за контролната група |
|
— |
bT: |
крайната биомаса минус началната биомаса в групата от третирани проби |
2.2.3. Построяване на кривите „концентрация—реакция“
Трябва да се построят кривите концентрация—реакция, които дават съотношението на средното процентно потискане на променливата на реакцията (Ir или Iy, изчислени както е показано в последната алинея от раздел 2.2.1 или в раздел 2.2.2) и логаритъма на концентрацията на изпитваното вещество.
2.2.4. Оценяване на ECx
Оценките на ECx (например EC50) следва да се базират както на средната специфична скорост на растеж (ErCx), така и на добива (EyCx), като всяка от тях от своя страна се основава на броя на листовидните тела и една допълнителна измервана променлива (обща площ на листовидните тела, сухо тегло или свежо тегло). Това е така, тъй като има изпитвани вещества, който оказват различно въздействие върху броя на листовидните тела и други измервани променливи. Следователно исканите параметри за токсичност са четирите стойности на ECx за всяко изчислено ниво x на потискане на растежа: ErCx (брой на листовидните тела); ErCx (обща площ на листовидните тела, сухо тегло или свежо тегло); EyCx (брой на листовидните тела); и EyCx (обща площ на листовидните тела, сухо тегло или свежо тегло).
2.3. СТАТИСТИЧЕСКИ ПРОЦЕДУРИ
Целта е да се получи количествено съотношение концентрация—реакция чрез регресивен анализ. Може да се използва тегловна линейна регресия след извършване на линеаризираща трансформация на данните от ответната реакция — например в единици пробит или логит или Weibull (16), но предпочитаните техники са тези на нелинейната регресия, които по-добре обработват неизбежните неравномерности и отклонения от гладките разпределения. При приближаване на нулево или пълно потискане подобни неравномерности може да бъдат увеличени от трансформацията, като по този начин пречат на анализа (16). Трябва да се отбележи, че стандартните методи за анализ с помощта на преобразования на пробит, логит или Weibull са предназначени за използване за количествени данни (например смъртност или оцеляване) и трябва да се модифицират, за да се приспособят към данните за скоростта на растежа или добива. Специфични процедури за определяне на стойностите на ECx от непрекъснати данни могат да се намерят в (17), (18) и (19).
За анализа на всяка от променливите на реакцията да се използва съотношението концентрация—реакция, за да се изчислят точковите оценки на стойностите на ECx. По възможност трябва да се определят 95 % доверителни граници за всяка от оценките. Адекватността на данните от реакцията спрямо регресивния модел може да се оцени графично или статистически. Регресивният анализ трябва да се извърши, като се използват реакциите на отделните повторни проби, а не средните стойности на група от третирани проби.
Оценките на EC50 и доверителните граници могат да се получат също и чрез използване на линейна интерполация с оценка на извадковата грешка (20), ако наличните регресивни модели/методи са неподходящи за данните.
За оценяване на LOEC и следователно на NOEC е необходимо да се сравнят средните стойности на третирана проба, като се използва техниките за анализ на променливостта (ANOVA). Средната стойност за всяка концентрация след това трябва да се сравни със средната стойност на контролна проба с помощта на съответния метод за многократно сравняване или на изпитване за тенденция. Могат да се използват изпитвания на Dunnett или на William (21)(22)(23)(24). Необходимо е да се оцени дали предположението ANOVA за хомогенност на променливостта се запазва. Тази оценка може да се извърши графично или чрез изпитване за формална значимост (25). Подходящи са изпитванията на Levene или на Bartlett. Невъзможността за удовлетворяване на предположението за хомогенност на променливостта понякога може да се коригира чрез логаритмична трансформация на данните. Ако хетерогенността е много голяма, следва да се обмисли анализ с методи като низходящите изпитвания на Jonkheere за тенденцията. Допълнителни насоки относно определянето на NOEC могат да се намерят в (19).
Най-скорошните научни разработки водят до препоръка да бъде изоставена концепцията за NOEC и тя да бъде заменена с регресия, базирана на точкови оценки на ECx. Не е установена подходяща стойност на x за това изпитване на Lemna. Но изглежда, че диапазонът от 10 до 20 % е подходящ (в зависимост от избраната променлива на реакцията) и за предпочитане е да бъде отчетена както стойността EC10, така и EC20.
3. ОТЧИТАНЕ
3.1. ПРОТОКОЛ ОТ ИЗПИТВАНЕТО
Протоколът от изпитването трябва да включва следното:
Изпитвано вещество:
|
— |
физическа природа и физикохимични свойства, включително граница на водоразтворимост; |
|
— |
данни за химичната идентификация (например CAS номер), включително чистота. |
Изпитвани видове:
|
— |
научно наименование, клонинг (ако е известен) и източник. |
Условия на изпитване:
|
— |
използвана процедура на изпитване (статична, полустатична или проточна); |
|
— |
дата на началото на изпитването и неговата продължителност; |
|
— |
среда на изпитване; |
|
— |
описание на експерименталната постановка: съдове за изпитване и капаци, обем на разтворите, брой колонии и листовидни тела за един съд за изпитване в началото на изпитването; |
|
— |
изпитвани концентрации (съответно номинална и измерена) и брой на повторните проби за една концентрация; |
|
— |
методи за приготвяне на изходните и изпитваните разтвори, включително използването на всякакви разтворители или дисперсанти; |
|
— |
температура по време на изпитването; |
|
— |
източник на светлина, интензитет на светлината и хомогенност; |
|
— |
стойности на pH за изпитваните и контролните среди; |
|
— |
концентрации на изпитваното вещество и метод за анализ със съответните данни за оценка на качеството (изследвания за достоверност, стандартни отклонения или доверителни граници на анализите); |
|
— |
методи за определяне на броя на листовидните тела и други измервани променливи, например сухо тегло, свежо тегло или площ на листовидните тела; |
|
— |
всички отклонения от настоящия метод за изпитване. |
Резултати:
|
— |
необработени данни: брой листовидни тела и други измервани променливи във всеки съд за изпитване и контролен съд при всяко наблюдение и извършен анализ; |
|
— |
средни стойности и стандартни отклонения за всяка измервана стойност; |
|
— |
криви на растежа за всяка концентрация (препоръчително с логаритмично преобразувана измервана стойност, вж. втората алинея от раздел 2.2.1); |
|
— |
време на удвояване/скорост на растеж в контролната проба на базата на броя листовидни тела; |
|
— |
изчислените зависими променливи за всяка третирана повторна проба със средните стойности и коефициент на променливост за повторните проби; |
|
— |
графично представяне на съотношението концентрация— въздействие; |
|
— |
оценки на крайните точки на токсичност за променливите на реакцията, например EC50, EC10, EC20, и съответните доверителни интервали. Ако са изчислени, LOEC и/или NOEC и статистическите методи, използвани за тяхното определяне; |
|
— |
ако е използван ANOVA, размерът на въздействието, което може да се открие (например най-малката значима разлика); |
|
— |
всякакво стимулиране на растежа, открито в някоя третирана проба; |
|
— |
всякакви визуални знаци за фитотоксичност, както и наблюдения на изпитваните разтвори; |
|
— |
обсъждане на резултатите, включително всякакво влияние върху резултата от изпитването, вследствие на отклонения от настоящия метод за изпитване. |
4. ЛИТЕРАТУРА
|
(1) |
OECD TG 221 (2006) Lemna Sp. Growth Inhibition Test |
|
(2) |
The use of Lemna studies for coloured substances is detailed in Section 13.5.3 of the EU Manual of Decisions dated July 2006, at http://ecb.jrc.ec.europa.eu/new-chemicals |
|
(3) |
Guidance on information requirements and chemical safety assessment — Chapter R.7b: Endpoint specific guidance; Table 7.8.3 Summary of difficult substance testing issues, available at http://guidance.echa.europa.eu/docs/guidance_document/information_requirements_en.htm?time=1234958685#A |
|
(4) |
ASTM International. (2003). Standard Guide for Conducting Static Toxicity Test With Lemna gibba G3. E 1415—91 (Reapproved 1998). pp. 733—742. In, Annual Book of ASTM Standards, Vol. 11.05 Biological Effects and Environmental Fate; Biotechnology; Pesticides, ASTM, West Conshohocken, PA. |
|
(5) |
USEPA — United States Environmental Protection Agency. (1996). OPPTS 850.4400 Aquatic Plant Toxicity Test Using Lemna spp., „Public draft“. EPA 712-C-96—156. 8pp. |
|
(6) |
AFNOR — Association Française de Normalisation. (1996). XP T 90—337: Détermination de l’inhibition de la croissance de Lemna minor. 10pp. |
|
(7) |
SSI — Swedish Standards Institute. (1995). Water quality — Determination of growth inhibition (7-d) Lemna minor, duckweed. SS 02 82 13. 15pp. (in Swedish). |
|
(8) |
Environment Canada. (1999). Biological Test Method: Test for Measuring the Inhibition of Growth Using the Freshwater Macrophyte, Lemna minor. EPS 1/RM/37—120 pp |
|
(9) |
Environment Canada. (1993) Proposed Guidelines for Registration of Chemical Pesticides: Non-Target Plant Testing and Evaluation. Canadian Wildlife Service, Technical Report Series No. 145. |
|
(10) |
Sims I., Whitehouse P., and Lacey R. (1999) The ОИСР Lemna Growth Inhibition Test. Development and Ring-testing of draft ОИСР Test Guideline. R&D Technical Report EMA 003. WRc plc — Environment Agency. |
|
(11) |
OECD (2000). Guidance Document on Aquatic Toxicity Testing of Difficult Substances and Mixtures. OECD Environmental Health and Safety Publications, Series on Testing and Assessment No.23. |
|
(12) |
ISO DIS 20079. Water Quality — Determination of the Toxic Effect of Water Constituents and Waste Water to Duckweed (Lemna minor) — Duckweed Growth Inhibition Test. |
|
(13) |
Walbridge C. T. (1977). A flow-through testing procedure with duckweed (Lemna minor L.). Environmental Research Laboratory — Duluth, Minnesota 55804. US EPA Report No. EPA-600/3—77 108. September 1977. |
|
(14) |
Lockhart W. L., Billeck B. N. and Baron C. L. (1989). Bioassays with a floating plant (Lemna minor) for effects of sprayed and dissolved glyphosate. Hydrobiologia, 118/119, 353—359. |
|
(15) |
Huebert, D.B. and Shay J.M. (1993) Considerations in the assessment of toxicity using duckweeds. Environmental Toxicology and Chemistry, 12, 481—483. |
|
(16) |
Christensen, E.R., Nyholm, N. (1984): Ecotoxicological Assays with Algae: Weibull Dose-Response Curves. Env. Sci. Technol. 19, 713—718. |
|
(17) |
Nyholm, N. Sørensen, P.S., Kusk, K.O. and Christensen, E.R. (1992): Statistical treatment of data from microbial toxicity tests. Environ. Toxicol. Chem. 11, 157—167. |
|
(18) |
Bruce R.D. and Versteeg D.J. (1992) A statistical procedure for modelling continuous toxicity data. Environmental Toxicology and Chemistry, 11, 1485—1494. |
|
(19) |
OECD. (2004). Guidance Document on Statistical Analysis of Ecotoxicity Data. |
|
(20) |
Norberg-King T.J. (1988) An interpolation estimate for chronic toxicity: The ICp approach. National Effluent Toxicity Assessment Center Technical Report 05—88. USEPA, Duluth, MN. |
|
(21) |
Dunnett, C.W. (1955) A multiple comparisons procedure for comparing several treatments with a control. J. Amer. Statist. Assoc., 50, 1096—1121. |
|
(22) |
Dunnett, C.W. (1964) New tables for multiple comparisons with a control. Biometrics, 20, 482—491. |
|
(23) |
Williams, D.A. (1971) A test for differences between treatment means when several dose levels are compared with a zero dose control. Biometrics, 27: 103—117. |
|
(24) |
Williams, D.A. (1972) The comparison of several dose levels with a zero dose control. Biometrics, 28: 510—531. |
|
(25) |
Brain P. and Cousens R. (1989). An equation to describe dose-responses where there is stimulation of growth at low doses. Weed Research, 29, 93—96. |
Допълнение 1
Описание на вида Lemna
Водното растение Lemna, обикновено наричано водна леща, принадлежи на семейството Lemnaceae, което има няколко вида в четири рода, разпространени в целия свят. Техният различен външен вид и таксономия са описани изчерпателно в (1)(2). Lemna gibba и L. minor са представители на вида в умерените области и обикновено се използват за изпитвания за токсичност. И двата вида имат плаващо или потопено във водата дисковидно стъбло (листовидно тяло), а много тънък корен излиза от центъра на долната повърхност на всяко листовидно тяло. Видът Lemna рядко дава цвят и растенията се размножават чрез вегетативно създаване на нови листовидни тела (3). В сравнение с по-възрастните растения младите са по-бледи, имат по-къси корени и се състоят от две до три листовидни тела с различни размери. Малкият размер на Lemna, опростената структура, безполовото размножаване и краткото време за създаване на поколение правят растението много подходящо за лабораторни изпитвания (4)(5).
Поради възможно междувидово вариране по отношение на чувствителност, валидни са сравнения на чувствителността единствено в рамките на един вид.
Примери за видове Lemna, които са били използвани за изпитване: Референтни видове
Lemna aequinoctialis: Eklund, B. (1996). The use of the red alga Ceramium strictum and the duckweed Lemna aequinoctialis in aquatic ecotoxicological bioassays. Licentiate in Philosophy Thesis 1996:2. Dep. of Systems Ecology, Stockholm University.
Lemna major: Clark, N. A. (1925). The rate of reproduction of Lemna major as a function of intensity and duration of light. J. phys. Chem., 29: 935—941.
Lemna minor: United States Environmental Protection Agency (USEPA). (1996). OPPTS 850.4400 Aquatic Plant Toxicity Test Using Lemna spp., „Public draft“. EPA 712-C-96—156. 8pp.
Association Française de Normalisation (AFNOR). (1996). XP T 90—337: Détermination de l’inhibition de la croissance de Lemna minor. 10pp.
Swedish Standards Institute (SIS). (1995). Water quality — Determination of growth inhibition (7-d) Lemna minor, duckweed. SS 02 82 13. 15pp. (in Swedish).
Lemna gibba: ASTM International. (2003). Standard Guide for Conducting Static Toxicity Test With Lemna gibba G3. E 1415—91 (Reapproved 1998). pp. 733—742.
United States Environmental Protection Agency (USEPA). (1996). OPPTS 850.4400 Aquatic Plant Toxicity Test Using Lemna spp., „Public draft“. EPA 712-C-96—156. 8pp.
Lemna paucicostata: Nasu, Y., Kugimoto, M. (1981). Lemna (duckweed) as an indicator of water pollution. I. The sensitivity of Lemna paucicostata to heavy metals. Arch. Environ. Contam. Toxicol., 10:1959—1969.
Lemna perpusilla: Clark, J. R. et al. (1981). Accumulation and depuration of metals by duckweed (Lemna perpusilla). Ecotoxicol. Environ. Saf., 5:87—96.
Lemna trisulca: Huebert, D. B., Shay, J. M. (1993). Considerations in the assessment of toxicity using duckweeds. Environ. Toxicol. and Chem., 12:481—483.
Lemna valdiviana: Hutchinson, T.C., Czyrska, H. (1975). Heavy metal toxicity and synergism to floating aquatic weeds. Verh.-Int. Ver. Limnol., 19:2102—2111.
Източници на видове Lemna
|
University of Toronto Culture Collection of Algae and Cyanobacteria |
|
Department of Botany, University of Toronto |
|
Toronto, Ontario, Canada, M5S 3 B2 |
|
Tel: +1–416–978–3641 |
|
Fax:+1–416–978–5878 |
|
e-mail: jacreman@botany.utoronto.ca |
|
http://www.botany.utoronto.ca/utcc |
|
North Carolina State University |
|
Forestry Dept |
|
Duckweed Culture Collection |
|
Campus Box 8002 |
|
Raleigh, NC 27695–8002 |
|
United States |
|
phone 001 (919) 515–7572 |
|
astomp@unity.ncsu.edu |
|
Institute of Applied Environmental Research (ITM) Stockholm University |
|
SE-106 91 Stockholm |
|
SWEDEN |
|
Tel: +46 8 674 7240 |
|
Fax +46 8 674 7636 |
|
Federal Environmental Agency (UBA) |
|
FG III 3.4 |
|
Schichauweg 58 |
|
12307 Berlin |
|
Germany |
|
e-mail: lemna@uba.de |
|
http://www.umweltbundesamt.de/contact.htm |
Литература
|
(1) |
Hillman, W.S. (1961). The Lemnaceae or duckweeds: A review of the descriptive and experimental literature. The Botanical Review, 27:221—287. |
|
(2) |
Landolt, E. (1986). Biosystematic investigations in the family of duckweed (Lemnaceae). Vol. 2. Geobotanischen Inst. ETH, Stiftung Rubel, Zürich, Switzerland. |
|
(3) |
Björndahl, G. (1982). Growth performance, nutrient uptake and human utilization of duckweeds (Lemnaceae family). ISBN 82—991150—0—0. The Agricultural Research Council of Norway, University of Oslo. |
|
(4) |
Wang, W. (1986). Toxicity tests of aquatic pollutants by using common duckweed. Environmental Pollution, Ser B, 11:1—14. |
|
(5) |
Wang, W. (1990). Literature review on duckweed toxicity testing. Environmental Research, 52:7—22. |
Допълнение 2
Поддържане на изходната култура
Изходните култури могат да бъдат поддържани при по-ниски температури (4—10 °C) за продължителни периоди без да има нужда да бъдат възстановявани. Средата на растеж на Lemna може да е същата като тази, използвана за изпитването, но за изходните култури може да се използва друга богата хранителна среда.
Периодично няколко млади, светлозелени растения се преместват с помощта на стерилна техника в нови съдове за култури, които съдържат прясна среда. При предложените тук по-студени условия субкултивирането може да се провежда на интервали до три месеца.
Трябва да се използват химически чисти (промити с киселина) и стерилни стъклени съдове за културите, както и да се прилага асептична техника за манипулиране. При замърсяване на изходната култура, например с водорасли и гъби, е необходимо да се вземат мерки за премахване на замърсяващите организми. При замърсяване с водорасли и много други замърсяващи организми това може да се постигне с повърхностна стерилизация. Взима се проба със замърсен растителен материал и корените се отрязват. След това материалът се разклаща енергично и после се потапя в 0,5 % (v/v) разтвор на натриев хипохлорит за период между 30 секунди и 5 минути. След това растителният материал се изплаква със стерилна вода и се прехвърля на няколко партиди в съдове за култури, съдържащи прясна среда на растеж. В резултат на тази обработка, много от листовидните тела ще умрат, особено ако се използват по-дълги периоди на експозиция, но някои от преживелите обикновено не са замърсени. Те биха могли след това да се използват за инокулиране на нови култури.
Допълнение 3
Среди
За L. minor и L. gibba се препоръчват различни среди на растеж. За L. minor се препоръчва среда, модифицирана съгласно шведски стандарт (SIS), докато за L. gibba се препоръчва среда 20X AAP. Съставните елементи на двете среди са дадени по-долу. При приготвянето на тези среди трябва да се използват реактиви или химикали с аналитично качество и дейонизирана вода.
Среда на растеж за Lemna съгласно шведски стандарт (SIS)
|
— |
Изходните разтвори I—V се стерилизират чрез обработка в автоклав (120 °C, 15 минути) или чрез мембранно филтриране (размер на порите приблизително 0,2 μm). |
|
— |
Изходният разтвор VI (и по избор VII) се стерилизират само чрез мембранно филтриране; те не трябва да се обработват в автоклав. |
|
— |
Стерилните изходни разтвори трябва да се съхраняват на тъмно и хладно. Изходните разтвори I—V трябва да се изхвърлят след шест месеца, докато изходният разтвор VI (и по избор VII) има срок на съхранение един месец.
|
||||||||||||||||||||||||||||||||||||||||||||||||||
|
— |
За приготвянето на един литър среда съгласно SIS към 900 ml дейонизирана вода се добавят следните съставки:
Забележка: Може да е необходим допълнителен изходен разтвор VII (буфер MOPS) за определени изпитвани вещества (вж. последната алинея от раздел 1.4). |
|
— |
Стойността на pH се регулира на 6,5 ± 0,2 с 0,1 или 1 mol HCl или NaOH, а обемът се регулира до един литър с дейонизирана вода. |
Среда на растеж 20X AAP
Изходните разтвори се приготвят в стерилна дестилирана или дейонизирана вода.
Стерилните изходни разтвори трябва да се съхраняват на студено и тъмно. При тези условия изходните разтвори ще имат срок на съхранение най-малко 6—8 седмици.
Пет хранителни изходни разтвора (A1, A2, A3, B и C) се приготвят за средата 20X — AAP с помощта на химикали с аналитично качество. 20 ml от всеки хранителен изходен разтвор се добавят към приблизително 850 ml дейонизирана вода за изготвяне на средата на растеж. Стойността на pH се регулира до 7,5 ± 0,1 с 0,1 или 1 mol HCl или NaOH, а обемът се регулира до един литър с дейонизирана вода. След това средата се филтрира през мембранен филтър с 0,2 μm (приблизително) в стерилен контейнер.
Средата на растеж, предназначена за изпитването, трябва да се приготви 1—2 дни преди да се използва, за да се даде възможност на pH да се стабилизира. Стойността на pH в средата на растеж трябва да се провери преди да се използва и при необходимост да се регулира отново чрез добавяне на 0,1 или 1 M NaOH или HCl, както е описано по-горе.
|
№ на изходен разтвор |
Вещество |
Концентрация в изходния разтвор (g·l–1) (1) |
Концентрация в приготвената среда (mg·l–1) (1) |
Приготвена среда |
|
|
Елемент |
Концентрация (mg·l–1) (1) |
||||
|
A1 |
NaNO3 MgCl2·6H2O CaCl2·2H2O |
26 12 4,4 |
510 240 90 |
Na; N Mg Ca |
190; 84 58,08 24,04 |
|
A2 |
MgSO4·7H2O |
15 |
290 |
S |
38,22 |
|
A3 |
K2HPO4·3H2O |
1,4 |
30 |
K; P |
9,4;3,7 |
|
B |
H3BO3 MnCl2·4H2O FeCl3·6H2O Na2EDTA·2H2O ZnCl2 CoCl2·6H2O Na2MoO4·2H2O CuCl2·2H2O |
0,19 0,42 0,16 0,30 3,3 mg·l–1 1,4 mg·l–1 7,3 mg·l–1 0,012 mg·l–1 |
3,7 8,3 3,2 6,0 66 μg·l–1 29 μg·l–1 145 μg·l–1 0,24 μg·l–1 |
B Mn Fe — Zn Co Mo Cu |
0,65 2,3 0,66 — 31 μg·l–1 7,1 μg·l–1 58 μg·l–1 0,080 μg·l–1 |
|
C |
NaHCO3 |
15 |
300 |
Na; C |
220; 43 |
Бележка под линия: Теоретично подходящата крайна концентрация на бикарбонат (при която ще се избегне съществено регулиране на стойността на pH) е 15 mg/l, а не 300 mg/l. В исторически план обаче ползването на средата 20X-AAP, включително кръговото изпитване за настоящия метод, се базира на 300 mg/l. (I. Sims, P. Whitehouse и R. Lacey. (1999) The OECD Lemna Growth Inhibition Test. Development and Ring testing of draft OECD Test Guideline. R&D Technical Report EMA 003. WRc plc — Environment Agency.
Среда на Steinberg (съгласно ISO 20079)
Концентрации и изходни разтвори
|
— |
Модифицираната среда на Steinberg се използва в ISO 20079 само за Lemna minor (само ако там се разрешава Lemna minor), но изпитванията са показали, че могат да се постигнат добри резултати също и с Lemna gibba. |
|
— |
При приготвяне на средата трябва да се използват реактиви или химикали с аналитично качество и дейонизирана вода. |
|
— |
Хранителната среда да се приготвя от изходни разтвори или 10-кратно концентрирана среда, която позволява максимална концентрация на средата без утаяване. |
Таблица 1
pH-стабилизирана среда на Steinberg (модифицирана съгласно Altenburger)
|
Вещество |
Хранителна среда |
||
|
Макроелементи |
Моларна маса |
mg/l |
mmol/l |
|
KNO3 |
101,12 |
350,00 |
3,46 |
|
Ca(NO3)2 · 4H2O |
236,15 |
295,00 |
1,25 |
|
KH2PO4 |
136,09 |
90,00 |
0,66 |
|
K2HPO4 |
174,18 |
12,60 |
0,072 |
|
MgSO4 · 7H2O |
246,37 |
100,00 |
0,41 |
|
Микроелементи |
Моларна маса |
µg/l |
µmol/l |
|
H3BO3 |
61,83 |
120,00 |
1,94 |
|
ZnSO4 · 7H2O |
287,43 |
180,00 |
0,63 |
|
Na2MoO4 · 2H2O |
241,92 |
44,00 |
0,18 |
|
MnCl2 · 4H2O |
197,84 |
180,00 |
0,91 |
|
FeCl3 · 6H2O |
270,21 |
760,00 |
2,81 |
|
Двунатриев дихидрат на EDTA |
372,24 |
1 500,00 |
4,03 |
Таблица 2
Изходни разтвори (макроелементи)
|
g/l |
||
|
Изходен разтвор 1: |
|||
|
KNO3 |
17,50 |
||
|
KH2PO4 |
4,5 |
||
|
K2HPO4 |
0,63 |
||
|
Изходен разтвор 2: |
|||
|
MgSO4 · 7H2O |
5,00 |
||
|
Изходен разтвор 3: |
|||
|
Ca(NO3)2 · 4H2O |
14,75 |
||
Таблица 3
Изходни разтвори (микроелементи)
|
mg/l |
||
|
Изходен разтвор 4: |
|||
|
H3BO3 |
120,0 |
||
|
Изходен разтвор 5: |
|||
|
ZnSO4 · 7H2O |
180,0 |
||
|
Изходен разтвор 6: |
|||
|
Na2MoO4 · 2H2O |
44,0 |
||
|
Изходен разтвор 7: |
|||
|
MnCl2 · 4H2O |
180,0 |
||
|
Изходен разтвор 8: |
|||
|
FeCl3 · 6H2O |
760,00 |
||
|
Двунатриев дихидрат на EDTA |
1 500,00 |
||
|
— |
Изходните разтвори 2 и 3 и отделно разтворите от 4 до 7 могат да бъдат обединени (като се вземе под внимание нужната концентрация). |
|
— |
За по-дълъг срок на съхранение изходните разтвори да се обработват в автоклав при 121 °C в течение на 20 минути или алтернативно да се извърши стерилна филтрация (0,2 µm). За изходен разтвор 8 настоятелно се препоръчва стерилната филтрация (0,2 µm). |
Приготвяне на крайната концентрация на средата на Steinberg (модифицирана)
|
— |
Добавете 20 ml от изходните разтвори 1, 2 и 3 (вж. таблица 2) към около 900 ml дейонизирана вода, за да се избегне утаяването. |
|
— |
Добавете 1,0 ml от изходни разтвори 4, 5, 6, 7 и 8 (вж. таблица 3). |
|
— |
Стойността на pH трябва да е 5,5 ± 0,2 (регулирайте чрез добавяне на минимално количество разтвор на NaOH или HCl) |
|
— |
Регулирайте с вода до 1 000 ml. |
|
— |
Ако изходните разтвори са стерилизирани и е използвана подходяща вода, не е необходима допълнителна стерилизация. Ако стерилизацията се извършва с крайната среда, изходен разтвор 8 трябва да се добави след обработка в автоклав (при 121 °C в продължение на 20 минути). |
Приготвяне на 10-кратно концентрирана среда на Steinberg (модифицирана) за междинно съхраняване
|
— |
Добавете 20 ml от изходните разтвори 1, 2 и 3 (вж. таблица 2) към около 900 ml дейонизирана вода, за да се избегне утаяването. |
|
— |
Добавете 1,0 ml от изходните разтвори 4, 5, 6, 7 и 8 (вж. таблица 3). Регулирайте с вода до 100 ml. |
|
— |
Ако изходните разтвори са стерилизирани и е използвана подходяща вода, не е необходима допълнителна стерилизация. Ако стерилизацията е извършена с крайната среда, изходен разтвор 8 трябва да се добави след обработка в автоклав (при 121 °C в продължение на 20 минути). |
|
— |
Стойността на pH на средата (крайна концентрация) трябва да е 5,5 ± 0,2. |
(1) Освен ако не е посочено.