

EUR-Lex Access to European Union law
This document is an excerpt from the EUR-Lex website
Document 32003R2003
Regulation (EC) No 2003/2003 of the European Parliament and of the Council of 13 October 2003 relating to fertilisers (Text with EEA relevance)
Регламент (ЕО) № 2003/2003 на Европейския парламент и на Съвета от 13 октомври 2003 година относно тороветеТекст от значение за ЕИП.
Регламент (ЕО) № 2003/2003 на Европейския парламент и на Съвета от 13 октомври 2003 година относно тороветеТекст от значение за ЕИП.
OJ L 304, 21.11.2003, p. 1–194
(ES, DA, DE, EL, EN, FR, IT, NL, PT, FI, SV)
Special edition in Czech: Chapter 13 Volume 032 P. 467 - 660
Special edition in Estonian: Chapter 13 Volume 032 P. 467 - 660
Special edition in Latvian: Chapter 13 Volume 032 P. 467 - 660
Special edition in Lithuanian: Chapter 13 Volume 032 P. 467 - 660
Special edition in Hungarian Chapter 13 Volume 032 P. 467 - 660
Special edition in Maltese: Chapter 13 Volume 032 P. 467 - 660
Special edition in Polish: Chapter 13 Volume 032 P. 467 - 660
Special edition in Slovak: Chapter 13 Volume 032 P. 467 - 660
Special edition in Slovene: Chapter 13 Volume 032 P. 467 - 660
Special edition in Bulgarian: Chapter 13 Volume 043 P. 3 - 196
Special edition in Romanian: Chapter 13 Volume 043 P. 3 - 196
Special edition in Croatian: Chapter 13 Volume 011 P. 76 - 269
 No longer in force, Date of end of validity: 15/07/2022; отменен от 32019R1009
No longer in force, Date of end of validity: 15/07/2022; отменен от 32019R1009
- Date of document:
- 13/10/2003
- Date of effect:
- 11/12/2003; влизане в сила дата на публикуване + 20 виж член 38
- Date of effect:
- 11/06/2005; влизане в сила виж член 38
- Date of end of validity:
- 15/07/2022; отменен от 32019R1009
- Author:
- Европейски парламент, Съвет на Европейския съюз
- Responsible body:
- Directorate-General for Internal Market, Industry, Entrepreneurship and SMEs
- Form:
- Регламент
- Additional information:
- COD 2001/0212, отнася се за ЕИП, разширяване на ЕИП от 22005D0008
- Authentic language:
- испански, датски, немски, гръцки, английски, френски, италиански, нидерландски, португалски, фински, шведски, исландски, норвежки
- Procedure number:
- Link
- European Parliament - Legislative observatory
- Treaty:
- Договор за създаване на Европейската общност
- Legal basis:
- Proposal:
- Link
- Select all documents based on this document
- Link
- Link
- Select all documents mentioning this document
- Internal procedures based on this legislative basic act
-
- Draft regulation Amending Regulation (EC) No 2003/2003 relating to fertilisers for the purposes of adapting Annex I;
No data available in the table
- Modifies:
-
Relation Act Comment Subdivision concerned From To Repeal 31976L0116 Repeal 31977L0535 Repeal 31980L0876 Repeal 31987L0094 Implicit repeal 31989L0284 11/12/2003 Implicit repeal 31989L0530 частично отменяне член 8 11/12/2003 Implicit repeal 31997L0063 частично отменяне член 1 11/12/2003 Implicit repeal 31997L0063 частично отменяне член 2 11/12/2003 Implicit repeal 31997L0063 частично отменяне член 3 11/12/2003
No data available in the table
- Modified by:
-
Relation Act Comment Subdivision concerned From To Corrected by 32003R2003R(01) (PL) Corrected by 32003R2003R(02) (DA, ES, IT, LT, NL, SL) Corrected by 32003R2003R(03) (BG) Corrected by 32003R2003R(04) (SV) Corrected by 32003R2003R(05) (EL) Modified by 32004R0885 поправка приложение 1 01/05/2004 Modified by 32004R2076 поправка приложение 1 24/12/2004 Derogated in 32006D0347 31/12/2007 Derogated in 32006D0348 Derogated in 32006D0349 Modified by 32006R1791 изпълнение приложение 1 01/01/2007 Modified by 32007R0162 поправка приложение 4 12/03/2007 Modified by 32007R0162 поправка приложение 1 12/03/2007 Modified by 32008R1107 поправка приложение 4 28/11/2008 Modified by 32008R1107 поправка приложение 1 28/11/2008 Modified by 32009R0219 поправка член 31 20/04/2009 Modified by 32009R0219 заместване член 29.4 20/04/2009 Modified by 32009R0219 заместване член 32 20/04/2009 Modified by 32009R1020 поправка приложение 5 18/11/2009 Modified by 32009R1020 поправка приложение 4 18/11/2009 Modified by 32009R1020 поправка приложение 1 18/11/2009 Modified by 32009R1020 поправка приложение 3 18/11/2009 Modified by 32011R0137 поправка приложение I 09/03/2011 Modified by 32011R0137 поправка приложение IV 09/03/2011 Modified by 32012R0223 поправка приложение IV 04/04/2012 Modified by 32012R0223 поправка приложение I 04/04/2012 Modified by 32013R0463 поправка приложение IV 07/06/2013 Modified by 32013R0463 поправка приложение I 07/06/2013 Modified by 32013R0463 поправка приложение II 07/06/2013 Modified by 32014R1257 изпълнение приложение IV SECTION B 15/12/2014 Modified by 32014R1257 поправка приложение I 15/12/2014 Modified by 32016R1618 добавка приложение I SECTION E.3.2 TABL текст 29/09/2016 Modified by 32016R1618 заместване приложение IV SECTION B текст 29/09/2016 Modified by 32016R1618 заместване приложение I SECTION F.2 TABL текст 29/09/2016 Modified by 32016R1618 добавка приложение IV SECTION B текст 29/09/2016 Modified by 32016R1618 добавка приложение I SECTION E.3.1 TABL текст 29/09/2016 Repealed by 32019R1009 16/07/2022 Modified by 32019R1102 заместване приложение IV раздел B текст 18/07/2019 Modified by 32019R1102 заместване приложение I таблица F.1 текст 18/07/2019 Modified by 32019R1102 добавка приложение IV раздел B текст 18/07/2019 Modified by 32020R1666 добавка приложение I таблица D точка 2.3 01/12/2020 Modified by 32021R0862 добавка приложение I раздел C.1 таблица точка 8 20/06/2021 - Subsequent related instruments:
-
- Amendment proposed by 52004PC0148(02)
- Amendment proposed by 52007PC0824
- Amendment proposed by 52016PC0157
- Instruments cited:
- Link
- EUROVOC descriptor:
- Subject matter:
- Directory code:
|
13/ 43 |
BG |
Официален вестник на Европейския съюз |
3 |
32003R2003
|
L 304/1 |
ОФИЦИАЛЕН ВЕСТНИК НА ЕВРОПЕЙСКИЯ СЪЮЗ |
РЕГЛАМЕНТ (ЕО) № 2003/2003 НА ЕВРОПЕЙСКИЯ ПАРЛАМЕНТ И НА СЪВЕТА
от 13 октомври 2003 година
относно торовете
(текст от значение за ЕИП)
ЕВРОПЕЙСКИЯТ ПАРЛАМЕНТ И СЪВЕТЪТ НА ЕВРОПЕЙСКИЯ СЪЮЗ,
като взеха предвид Договора за създаване на Европейската общност, и по-специално член 95 от него,
като взеха предвид предложението на Комисията (1),
като взеха предвид становището на Европейския икономически и социален комитет (2),
в съответствие с процедурата, установена в член 251 от Договора (3),
като имат предвид, че:
|
(1) |
Директива 76/116/ЕИО на Съвета от 18 декември 1975 г. относно сближаване на законодателствата на държавите-членки относно торовете (4), Директива 80/876/ЕИО на Съвета от 15 юли 1989 г. относно сближаването на законодателствата на държавите-членки, свързани с торове от еднокомпонентен амониев нитрат с високо азотно съдържание (5), Директива 87/94/ЕИО на Комисията относно сближаване на законодателствата на държавите-членки, свързани с процедурите за контрол на характеристиките, границите и устойчивостта на детонация на торове от прост амониев нитрат с високо азотно съдържание (6) и Директива 77/535/ЕИО на Комисията от 22 юни 1977 г. относно сближаване на законодателствата на държавите-членки, свързани с методите за вземане на проби и анализ за торове (7), бяха съществено изменени няколко пъти. В съответствие със съобщението на Комисията до Европейския парламент и Съвета „По-опростено законодателство за вътрешния пазар“ (SLIM) и „План за действие за единния пазар“, тези директиви следва, в интерес на яснотата, да се отменят и да се заместят с единствен правен инструмент. |
|
(2) |
Съдържанието на законодателството на Общността относно торовете е с изразено технически характер. Поради това регламентът е най-подходящият правен инструмент, тъй като налага пряко на производителите точни изисквания за прилагането му по едно и също време и по един и същ начин в цялата Общност. |
|
(3) |
Във всяка държава-членка торовете трябва да се отличават с определени технически характеристики, установени със задължителни разпоредби. Тези разпоредби, които засягат по-специално състава и дефинициите на видовете торове, обозначаването на тези видове, тяхната идентификация и техните опаковки, се различават в различните държави-членки. Тези различия затрудняват търговията в рамките на Общността и поради това следва да бъдат хармонизирани. |
|
(4) |
Тъй като целта на предложеното действие, а именно, да се гарантира вътрешния пазар на торове, не може да бъде постигната в достатъчна степен, ако няма общи технически критерии, и поради това може поради мащаба на действието да бъде постигната по-добре на ниво на Общността, Общността може да приеме мерки в съответствие с принципа на субсидиарност, както е постановен в член 5 от Договора. В съответствие с принципа на пропорционалност, установен в цитирания член, съдържанието на настоящия регламент не надхвърля необходимото, за да се постигне тази цел. |
|
(5) |
Необходимо е да се определят на ниво на Общността обозначаването, дефинирането и състава на някои торове (ЕО торове). |
|
(6) |
Следва също така да бъдат постановени правила на Общността относно идентификацията, проследяемостта и етикетирането на ЕО торовете и относно затварянето на опаковките. |
|
(7) |
Следва да бъде създадена процедура на ниво на Общността, която да бъде следвана, когато държава-членка счита за необходимо да ограничи пускането на пазара на ЕО торове. |
|
(8) |
Производството на торове подлежи в различна степен на колебания поради производствените технологии или основните материали. Вземането на проби и процедурите на анализ могат също така да варират. Поради това е необходимо да се определят допустими отклонения за декларираното съдържание на хранителни елементи. В интерес на селскостопанския потребител е препоръчително тези допустими отклонения да се поддържат в тесни граници. |
|
(9) |
Официалният контрол по спазването при ЕО торовете на изискванията на настоящия регламент относно качеството и състава следва да бъдат извършвани от лаборатории, които са одобрени от държавите-членки и са съобщени на Комисията. |
|
(10) |
Амониевият нитрат е основна съставка на разнообразни продукти, някои от които са предназначени за използване като торове, а други като експлозиви. Необходимо е, като се има предвид особената природа на амониево-нитратните торове с високо съдържание на азот и логически следващите изисквания относно обществената безопасност, здравето и безопасността на работниците, да се установят допълнителни правила на Общността за ЕО торовете от този вид. |
|
(11) |
Някои от тези продукти биха могли да бъдат опасни и биха могли в някои случаи да бъдат използвани за цели, различни от тези, за които са били предназначени. Това може силно да застраши сигурността на лицата и собствеността. Поради това производителите следва да бъдат задължени да предприемат подходящи стъпки за избягване на такова използване и по-специално да осигурят проследяемостта на такива торове. |
|
(12) |
В интерес на обществената безопасност е особено важно да се определят на ниво на Общността характеристиките и свойствата, които различават амониево-нитратните ЕО торове с високо съдържание на азот от различните видове амониев нитрат, използван при производството на продукти, използвани като експлозиви. |
|
(13) |
Амониево-нитратните ЕО торове с високо съдържание на азот следва да отговарят на някои характеристики, за да се осигури тяхната безвредност. Производителите следва да осигурят всички амониево-нитратни торове с високо съдържание на азот да преминават през изпитания за устойчивост на детонация преди пускането на тези торове на пазара. |
|
(14) |
Необходимо е да се установят правила за методите на затворените термични цикли, дори ако тези методи могат да не симулират всички условия, които възникват при транспортиране и съхраняване. |
|
(15) |
Торовете могат да бъдат замърсени с вещества, които могат потенциално да представляват заплаха за здравето на човека и животните и за околната среда. В допълнение към становището на Научния комитет по токсичността, екотоксичността и околната среда (SCTEE), Комисията възнамерява да се заеме с проблема на неумишлено съдържание на кадмий в минерални торове и, когато е целесъобразно, ще изготви предложение за регламент, който тя възнамерява да представи на Европейския парламент и на Съвета. Когато е целесъобразно, подобен преглед ще бъде предприет за други замърсители. |
|
(16) |
Целесъобразно е да се създаде процедура, която да се спазва от всички производители или техните представители, които искат да включат нов вид тор в приложение I, за да използва маркировката „ЕО тор“. |
|
(17) |
Необходимите мерки за прилагането на настоящия регламент следва да бъдат одобрени в съответствие с Решение 1999/468/ЕО на Съвета от 28 юни 1999 г. относно установяването на процедурите за упражняване на изпълнителните правомощия, предоставени на Комисията (8). |
|
(18) |
Държавите-членки следва да налагат санкции по отношение на нарушения на разпоредбите на настоящия регламент. Те могат да предвиждат производител, който нарушава член 27, да може да бъде глобяван със сума, еквивалентна на десетократната пазарна стойност на изпратената стока, която не отговаря на изискванията. |
|
(19) |
Директиви 76/116/ЕИО, 77/535/ЕИО, 80/876/ЕИО и 87/94/ЕИО следва да бъдат отменени, |
ПРИЕХА НАСТОЯЩИЯ РЕГЛАМЕНТ:
ДЯЛ I
ОБЩИ РАЗПОРЕДБИ
ГЛАВА I
Обхват и дефиниции
Член 1
Обхват
Настоящият регламент се прилага за продукти, които се пускат на пазара като торове, обозначени „ЕО торове“.
Член 2
Дефиниции
По смисъла на настоящия регламент се прилагат следните дефиниции:
|
а) |
„Тор“ означава материал, чиято основна функция е да осигурява хранителни елементи за растенията. |
|
б) |
„Първичен хранителен елемент“ означава само елементите азот, фосфор и калий. |
|
в) |
„Вторичен хранителен елемент“ означава елементите калций, магнезий, натрий и сяра. |
|
г) |
„Хранителен микроелемент“ означава елементите бор, кобалт, мед, желязо, манган, молибден и цинк, които са извънредно важни за растежа на растенията, в количества, които са малки в сравнение с тези на основните и вторичните хранителни елементи. |
|
д) |
„Неорганичен тор“ означава тор, в който декларираните хранителни елементи са под формата на минерали, получени чрез екстракция или чрез физически и/или химически процеси. Калциевият цианамид, карбамидът и неговите кондензационни и свързани продукти и торовете, които съдържат хелатирани или комплексообразувани хранителни микроелементи, могат при съгласие да бъдат класифицирани като неорганични торове. |
|
е) |
„Хелатиран хранителен микроелемент“ означава хранителен микроелемент, който се държи от една от молекулите, изброени в раздел Д.3.1 от приложение I. |
|
ж) |
„Комплексно образуван хранителен микроелемент“ означава хранителен микроелемент, който се държи от една от молекулите, изброени в раздел Д.3.2 от приложение I. |
|
з) |
„Тип торове“ означава торове с общ вид обозначение, както е посочено в приложение I. |
|
и) |
„Еднокомпонентен тор“ означава азотен, фосфорен или калиев тор, който има подлежащо на деклариране съдържание на само един от основните хранителни елементи. |
|
й) |
„Съставен тор“ означава тор, който има подлежащо на деклариране съдържание на най-малко два основни хранителни елементи и е получен химически начин или чрез смесване или чрез комбинация на двата начина. |
|
к) |
„Комплексен тор“ означава съставен тор, получен чрез химическа реакция, чрез разтваряне или в неговото твърдо състояние чрез гранулиране, който има подлежащо на деклариране съдържание на най-малко два основни хранителни елемента. В твърдо състояние всяка гранула съдържа всички хранителни елементи в техния деклариран състав. |
|
л) |
„Смесен тор“ означава тор, получен чрез сухо смесване на няколко торове, без химическа реакция. |
|
м) |
„Листен тор“ означава тор, който е подходящ за прилагане върху листа и хранителният елемент се поема от листата на посева. |
|
н) |
„Течен тор“ означава тор в суспензия или разтвор. |
|
о) |
„Разтвор на тор“ означава течен тор, който няма твърди частици. |
|
п) |
„Суспензия на тор“ означава тор в две фази, в който твърдите частици се подържат в суспензия в течната фаза. |
|
р) |
„Деклариране“ означава обявяване на количеството на хранителните елементи, включително техните форми и разтворимост, гарантирани в рамките на специфицираните допустими отклонения. |
|
с) |
„Декларирано съдържание“ означава съдържанието на елемент или негов окис, което в съответствие със законодателството на Общността, е дадено на етикет на ЕО тор или в съответен придружаващ документ. |
|
т) |
„Допустимо отклонение“ означава разрешено отклонение на измерената стойност на съдържанието на хранителен елемент от декларираната стойност. |
|
у) |
„Европейски стандарт“ означава стандарти на CEN (Европейски комитет по стандартизация), които са били официално признати от Общността, и позоваванията на които са били публикувани в Официален вестник на Европейските общности |
|
ф) |
„Опаковка“ означава съд, който може да се запечата, използван за съхраняване, защита, манипулиране и разпространяване на торове, който съдържа не повече от 1 000 kg. |
|
х) |
„Насипно състояние“ означава тор, който не е пакетиран, както е предвидено в настоящия регламент. |
|
ц) |
„Пускане на пазара“ означава доставка на тор, срещу заплащане или безплатно, или съхраняване за целите на доставка. Внасянето на тор на митническата територия на Европейската общност се счита за пускане на пазара. |
|
ч) |
„Производител“ означава физическо или юридическо лице, което отговаря за пускане на пазара на тор; по-специално производител, вносител, опаковчик, който работи за своя сметка, или всяко лице, което променя характеристиките на тор, се счита за производител. Независимо от това дистрибутор, който не променя характеристиките на тора, не се счита за производител. |
ГЛАВА II
Пускане на пазара
Член 3
ЕО тор
Тор, който принадлежи към вида торове, изброени в приложение I, и отговаря на условията, установени в настоящия регламент, може да бъде обозначен като ЕО тор.
Обозначението „ЕО тор“ не се използва за тор, който не отговаря на изискванията на настоящия регламент.
Член 4
Установяване в рамките на Общността
Производителят е установен в рамките на Общността и е отговорен за съответствието на „ЕО тор“ с разпоредбите на настоящия регламент.
Член 5
Свободно обръщение
1. Без да се засяга член 15 и останалото законодателство на Общността, държавите-членки няма да забраняват, ограничават или възпрепятстват пускането на пазара на торове, маркирани като „ЕО тор“, които отговарят на разпоредбите на настоящия регламент, на основание на състав, идентификация, етикетиране и пакетиране и други разпоредби, съдържащи се в настоящия регламент.
2. Торове, които са маркирани като „ЕО тор“ в съответствие с настоящия регламент, са в свободно обращение в рамките на Общността.
Член 6
Задължителни заявления
1. За да бъдат удовлетворени изискванията на член 9, държавите-членки могат да предвидят посочването на съдържанията на азот, фосфор и калций на торове, които се пускат на техния пазар, да бъде изразено по следния начин:
|
а) |
азот само в елементарна форма (N); и или |
|
б) |
фосфор и калий само в елементарна форма (Р, К); или |
|
в) |
фосфор и калий само под формата на оксиди (Р2О5, К2О); или |
|
г) |
фосфор и калий само в двете елементарна и оксидна форма едновременно. |
Когато е избрана възможността да се предвиди съдържанието на фосфор и калий да се изразява под формата на елементи, всички позовавания на оксидната форма в приложенията се разбират като съдържание в елементарна форма и цифровите стойности се преобразуват, като се използват следните коефициенти:
|
а) |
фосфор (Р) = фосфорен пентаоксид (Р2О5) × 0,436; |
|
б) |
калий (К) = калиев оксид (К2О) × 0,830. |
2. Държавите-членки могат да разпоредят да се изрази съдържанието на калций, магнезий, натрий и сяра на вторични хранителни торове, и когато са изпълнени условията на член 17, на основните хранителни торове, пуснати на техните пазари:
|
а) |
под формата на оксиди (CaO, MgO, Na2O, SO3); или |
|
б) |
под елементарна форма (Ca, Mg, Na, S); или |
|
в) |
в двете форми. |
За да се преобразува съдържанието на калциевия оксид, магнезиевия оксид, натриевия оксид и серния триоксид в съдържание на калций, магнезий, натрий и сяра, се използват следните коефициенти:
|
а) |
калций (Са) = калциев оксид (СаО) × 0,715; |
|
б) |
магнезий (Mg) = магнезиев оксид (MgO) × 0,603; |
|
в) |
натрий (Na) = натриев оксид (Na2O) × 0,742; |
|
г) |
сяра (S) = серен триоксид (SO3) × 0,400. |
За изчисленото съдържание на елемент или окис, обявените цифри се закръглят до най-близкия разред след десетичната запетая.
3. Държавите-членки не възпрепятстват пускането на пазара на „ЕО тор“, етикетиран в двете форми, посочени в параграфи 1 и 2.
4. Съдържанието на един или повече от хранителните микроелементи бор, кобалт, мед, желязо, манган, молибден или цинк в ЕО торове, спадащи към видовете торове, изброени в раздели А, Б, В и Г от приложение I, се декларират, когато са изпълнени следните условия:
|
а) |
хранителните микроелементи са добавени най-малко в минималните количества, определени в раздел Д.2.2 и Д.2.3 от приложение I, |
|
б) |
ЕО торът продължава да отговаря на изискванията на раздели А, Б, В и Г от приложение I. |
5. Когато хранителните микроелементи са нормални съставки на суровини, предназначени да доставят основни (N, P, K) и вторични (Ca, Mg, Na, S) хранителни елементи, те могат да се декларират при условие че тези хранителни микроелементи присъстват най-малко в минималните количества, определени в раздели Д.2.2 и Д.2.3 от приложение I.
6. Съдържанието на хранителни микроелементи се декларира по следния начин:
|
а) |
за торове, спадащи към видове торове, изброени в раздел Д.1 от приложение I, в съответствие с изискванията, постановени в колона 6 от този раздел; |
|
б) |
за смеси на торове, посочени в буква а), съдържащи поне два различни хранителни микроелемента и отговарящи на изискванията на раздел Д.2.1 от приложение I и за торове, спадащи към видовете торове, изброени в раздели А, Б, В и Г от приложение I, като се посочва:
|
Когато хранителният микроелемент е изцяло водоразтворим, се декларира само водоразтворимото съдържание.
Когато хранителен микроелемент е химически свързан с органична молекула, съдържанието на хранителния микроелемент, съдържащ се в тора, се декларира непосредствено след водоразтворимото съдържание като процент от масата на продукта, последван от един от термините „хелатиран с“ или „комплексно образуван с“, с наименованието на органичната молекула, както е изложено в раздел Д.3 от приложение I. Наименованието на органичната молекула може да се замени с нейните инициали.
Член 7
Идентификация
1. Производителят снабдява ЕО торовете с идентификационните маркировки, изброени в член 9.
2. Ако торовете са пакетирани, тези идентификационни маркировки се поставят върху пакетите или прикрепените етикети. Ако торовете са в насипно състояние, тези маркировки се вписват в придружаващите документи.
Член 8
Проследяемост
Без да се засяга член 26, параграф 3, производителят, за да осигури проследяемостта на ЕО торовете, поддържа документация за произхода на торовете. Тази документация се предоставя на разположение за инспекция от държавите-членки за периода, през който торът се доставя на пазара и за допълнителен срок от 2 години, след като производителят е спрял да го доставя.
Член 9
Маркировки
1. Без да се засягат други правила на Общността, пакетите, етикетите и придружаващите документи, посочени в член 7, са снабдени със следните маркировки:
|
а) |
Задължителна идентификация
|
|
б) |
Идентификации, които не са задължителни
|
Указанията, предвидени в буква б), не трябва да противоречат на указанията, посочени в буква а), и трябва да бъдат ясно отделени от тях.
2. Всички маркировки, посочени в параграф 1, трябва да бъдат ясно отделени от всяка друга информация на пакетите, етикетите и придружаващите документи.
3. Течните торове могат да се пускат на пазара, само ако производителят осигурява подходящи допълнителни указания, които обхващат по-специално, температура на съхранение и предотвратяването на инциденти при съхраняването.
4. Подробни правила за прилагането на настоящия член се приемат съгласно процедурата, посочена в член 32, параграф 2.
Член 10
Етикетиране
1. Етикетите или маркировките, отпечатани на опаковката и съдържащи данните, посочени в член 9, трябва да бъдат поставени на видно място. Етикети трябва да се прикачат към опаковката или към използваната система за неговото затваряне. Ако тази система се състои от пломба, тази пломба трябва да съдържа името или марката на опаковчика.
2. Маркировките, посочени в параграф 1, трябва да бъдат и трябва да остават незаличими и ясно четливи.
3. При торове в насипно състояние, посочени във второто изречение на член 7, параграф 2, копие от документите, съдържащи идентифициращите маркировки, трябва да придружава стоките и да бъде достъпно за целите на инспекцията.
Член 11
Езици
Етикетът, маркировките на опаковката и придружаващите документи трябва да са най-малкото на националния език или езици на държавата-членка, в която ЕО торът се търгува.
Член 12
Опаковане
При пакетираните ЕО торове опаковката трябва да бъде затворена по такъв начин или с такова устройство, че при отваряне устройството, затварящата пломба или самата опаковка да се поврежда непоправимо. Могат да се използват вентилни торби.
Член 13
Допустими отклонения
1. Съдържанието на хранителни елементи в ЕО торовете отговаря на допустимите отклонения, съдържащи се в приложение II, които са предназначени да позволяват отклонения при производство, вземане на проби и анализ.
2. Производителят не трябва систематично да се възползва от допустимите отклонения, дадени в приложение II.
3. Не се разрешават допустими отклонения по отношение на минималните и максималните съдържания, посочени в приложение I.
Член 14
Изисквания към тора
Определен вид тор може да бъде включен в приложение I, само ако:
|
а) |
осигурява хранителни елементи по ефективен начин; |
|
б) |
предвидени са необходимите методи за вземане на проби, извършване на анализ и ако се изисква, методи за изпитване. |
|
в) |
при нормални условия на използване не въздейства неблагоприятно върху здравето на човека, животните или растенията, или върху околната среда. |
Член 15
Защитна клауза
1. Когато държава-членка има основателни причини да счита, че специфичен ЕО тор, въпреки че удовлетворява изискванията на настоящия регламент, представлява опасност за безопасността или здравето на хора, животни или растения или застрашава околната среда, тя може временно да забрани пускането на пазара на този тор на своята територия или да го направи предмет на специални условия. Тя незабавно информира останалите държави-членки и Комисията за това, като посочва причините за това нейно решение.
2. Комисията приема решение по въпроса в рамките на 90 дни от получаването на информацията в съответствие с процедурата, посочена в член 32, параграф 2.
3. Разпоредбите на настоящия регламент не изключват вземане на мерки от Комисията или от държавата-членка, които са оправдани от гледна точка на обществената безопасност, да забраняват, ограничават или възпрепятстват пускането на пазара на ЕО торове.
ДЯЛ II
РАЗПОРЕДБИ ЗА СПЕЦИФИЧНИ ТИПОВЕ ТОРОВЕ
ГЛАВА I
Неорганични торове с основни хранителни елементи
Член 16
Обхват
Настоящата глава се прилага за неорганични торове с основни хранителни елементи, твърди или течни, еднокомпонентни или сложни, включително за тези, които съдържат второстепенни хранителни елементи и/или микроелементи с минимално хранително съдържание, определено в раздели А, Б, В, Д.2.2 и Д.2.3 от приложение I.
Член 17
Деклариране на второстепенни хранителни елементи в торове с основни хранителни елементи
Съдържанието на калций, магнезий, натрий и сяра може да се декларира като съдържание на второстепенни хранителни елементи на ЕО торове, спадащи към видовете торове, изброени в раздели А, Б и В от приложение I, при условие че тези елементи присъстват най-малко в следните минимални количества:
|
а) |
2 % калциев оксид (СаО), т.е. 1,4 % Са; |
|
б) |
2 % магнезиев оксид (MgO), т.е. 1,2 % Mg; |
|
в) |
3 % натриев оксид (Na2O), т.е. 2,2 % Na; |
|
г) |
5 % серен триоксид (SO3), т.е. 2 % S. |
В този случай допълнителното маркиране, определено в член 19, параграф 2, точка ii) се добавя към обозначението на вида.
Член 18
Калций, магнезий, натрий и сяра
1. Декларирането на съдържанието на магнезий, натрий и сяра на торовете, изброени в раздели А, Б и В от приложение I, се изразява по един от следните начини:
|
а) |
общото съдържание, изразено като процент от масата на тора; |
|
б) |
общото съдържание и съдържанието, разтворимо във вода, изразено като процент от масата на тора, когато разтворимото съдържание е най-малкото една четвърт от общото съдържание; |
|
в) |
когато елементът е напълно разтворим във вода, се декларира само разтворимото съдържание като процент от масата. |
2. Освен ако не е определено друго в приложение I, декларирането на съдържанието на калций се прави, само ако той е разтворим във вода, и се изразява като процент от масата на тора.
Член 19
Идентификация
1. В допълнение към задължителните идентифициращи маркировки, посочени в член 9, параграф 1, буква а), се посочват маркировките, определени в параграфи 2, 3, 4, 5 и 6 от настоящия член.
2. Следното се посочва след обозначението на вида съставни торове:
|
i) |
Химическите символи на декларираните второстепенни хранителни елементи в скоби и след символите на основните хранителни елементи. |
|
ii) |
Числа, които показват съдържанието на основните хранителни елементи. Декларираното съдържание на вторичен хранителен елемент се посочва в скоби след съдържанието на основния хранителен елемент. |
3. Обозначението на вида на тора се следва само от цифри, които показват съдържанието на основни и второстепенни хранителни елементи.
4. Когато се декларират хранителни микроелементи, се използват думите „с хранителни микроелементи“ или думата „с“, последвана от наименованието или наименованията и химическите символи на присъстващите хранителни микроелементи.
5. Декларираното съдържание на основни хранителни елементи и второстепенни хранителни елементи се дава като процент от масата като цяло число или при необходимост, когато съществува подходящ метод за анализ, до един знак след десетичната запетая.
При торове, които съдържат повече от един деклариран хранителен елемент, редът за основните хранителни елементи е следният: N, P2O5 и/или P, K2O и/или K, и за второстепенните хранителни елементи: CaO и/или Ca, MgO и/или Mg, Na2O и/или Na, SO3 и/или S.
Декларираното съдържание на хранителни микроелементи указва наименованието и символа на всеки хранителен микроелемент, като се посочва процентът от масата, както е определено в раздели Д.2.2 и Д.2.3 от приложение I и в съответствие с разтворимостта.
6. Формите и разтворимостта на хранителните елементи също се изразява като процент от масата на тора, освен когато приложение I изрично предвижда това съдържание да се изрази по друг начин.
Броят на разредите след десетичната запетия е един, освен при хранителни микроелементи, при които той е съгласно определеното в раздели Д.2.2 и Д.2.3 от приложение I.
ГЛАВА II
Неорганични торове с второстепенни хранителни елементи
Член 20
Обхват
Настоящата глава се прилага за неорганични торове с второстепенни хранителни елементи, твърди или течни, еднокомпонентни или сложни, включително тези, които съдържат микроелементи, с минимално хранително съдържание, определено в раздели Г, Д.2.2 и Д.2.3 от приложение I.
Член 21
Идентификация
1. В допълнение към задължителните идентифициращи маркировки, посочени в член 9, параграф 1, буква а), се посочват маркировките, установени в параграфи 2, 3, 4 и 5 от настоящия член.
2. Когато се декларират хранителни микроелементи, се използват думите „с хранителни микроелементи“ или думата „с“, последвана от наименованието или наименованията и химическите символи на съдържащите се хранителни микроелементи.
3. Декларираното съдържание на второстепенни хранителни елементи се дава като процент от масата, като цяло число или при необходимост, когато съществува подходящ метод за анализ, до един знак след десетичната запетая.
При торове, които съдържат повече от един деклариран хранителен елемент, редът за второстепенните хранителни елементи е следният:
|
|
CaO и/или Ca, MgO и/или Mg, Na2O и/или Na, SO3 и/или S. |
Декларираното съдържание на хранителни микроелементи съдържа наименованието и символа на всеки хранителен микроелемент, като се посочва процентът от масата, както е определено в раздели Д.2.2 и Д.2.3 от приложение I и съгласно разтворимостта.
4. Формите и разтворимостта на хранителните елементи също се изразява като процент от масата на тора, освен когато приложение I изрично предвижда това съдържание да се изрази по друг начин.
Броят на разредите след десетичната запетия е един, освен при хранителни микроелементи, когато това е специфицирано в раздели Д.2.2 и Д.2.3 от приложение I.
5. Освен ако не е определено друго в приложение I, съдържанието на калций се декларира, само ако той е разтворим във вода и се изразява като процент от масата на тора.
ГЛАВА III
Неорганични торове с хранителни микроелементи
Член 22
Обхват
Настоящата глава се прилага за неорганични торове с хранителни микроелементи, твърди или течни, с минимално съдържание на азот, постановено в раздели Д.1 и Д.2.1 от приложение I.
Член 23
Идентификация
1. В допълнение към задължителните идентифициращи маркировки, посочени в чл. 9, параграф 1, буква а), трябва се посочват маркировките, установени в параграфи 2, 3, 4 и 5 от настоящия член.
2. Когато торът съдържа повече от един хранителен микроелемент, се посочва обозначението на вида „смес от хранителни микроелементи“, последвано от наименованията и химическите символи на присъстващите хранителни микроелементи.
3. За торове, които съдържат само един хранителен микроелемент (раздел Д.1 от приложение I), декларираното съдържание на хранителен микроелемент се дава като процент от масата, като цяло число, или при необходимост, до един знак след десетичната запетая.
4. Формите и разтворимостта на хранителните микроелементи се изразява като процент от масата на тора, освен когато приложение I изрично предвижда това съдържание да се изрази по друг начин.
Броят на разредите след десетичната запетия при хранителните микроелементи е както това е определено в раздел Д.2.1 от приложение I.
5. Под задължителните декларации или незадължителните декларации на етикета и в придружаващите документи по отношение на продуктите в раздели Д.1 и Д.2.1 от приложение I се изписва следното:
„Да се използва само при призната нужда. Да не се превишават подходящите норми на дозата.“
Член 24
Опаковане
ЕО торовете, обхванати от разпоредбите на настоящата глава, се опаковат.
ГЛАВА IV
Амониево-нитратни торове с високо съдържание на азот
Член 25
Обхват
За целите на настоящата глава амониево-нитратни торове с високо съдържание на азот, еднокомпонентни и сложни, са продукти на базата на амониев нитрат, произведени за използване като торове и съдържащи повече от 28 % от масата на азот по отношение на амониевия нитрат.
Този вид торове може да съдържа някои неорганични или инертни вещества.
Веществата, използвани за производството на този вид торове, не трябва да увеличават чувствителността към топлина или тенденцията за детониране.
Член 26
Мерки и контрол на безопасността
1. Производителят осигурява еднокомпонентните амониево-нитратни торове с високо съдържание на азот да отговарят на разпоредбите на раздел 1 от приложение III.
2. Проверката, анализът и изпитването за официален контрол на еднокомпонентни амониево-нитратни торове с високо съдържание на азот, предвидено в настоящата глава, се извършва в съответствие с методите, описани в раздел 3 от приложение III.
3. За да се осигури възможност за проследяване на амониево-нитратни ЕО торове с високо съдържание на азот, пуснати на пазара, производителят поддържа документация за наименованията и адресите на обектите и на операторите на обектите, на които са били произведени торът и неговите основни компоненти. Тази документация се предоставя на разположение за инспектиране от държавите-членки, докато продуктът се доставя на пазара и за допълнителен срок от 2 години, след като производителят спре да го доставя.
Член 27
Изпитване на устойчивост на детонация
Без да се засягат мерките, посочени в член 26, производителят осигурява всеки вид амониево-нитратен ЕО тор с високо съдържание на азот, пуснат на пазара, да е преминал през изпитването на устойчивост на детонация, описано в раздели 2, 3 (метод 1, точка 3) и 4 на приложение III към настоящия регламент. Това изпитване се извършва от някои от одобрените лаборатории, посочени в член 30, параграф 1 или член 33, параграф 1. Производителите представят резултатите от изпитванията на компетентния орган на съответната държава-членка най-малко 5 дни преди пускането на тора на пазара или най-малко 5 дни преди пристигането на тора на границата на Европейската общност при внос. След това производителят продължава да гарантира, че доставките на тора, пуснат на пазара, са в състояние да преминат изпитването, посочено по-горе.
Член 28
Опаковане
Амониево-нитратни торове с високо съдържание на азот се предоставят на крайния потребител само в пакетирана форма.
ДЯЛ III
ОЦЕНКА НА СЪОТВЕТСТВИЕ НА ТОРОВЕ
Член 29
Контрол
1. Държавите-членки могат да подлагат торове, маркирани като „ЕО тор“, на официални мерки за контрол за целите на проверката за спазването на настоящия регламент.
Държавите-членки могат да налагат такси, които не превишават разходите за изпитания, необходими за такива мерки за контрол, но това не задължава производителите да повтарят изпитанията или да плащат за повторни изпитания, когато първото изпитване, извършено от лаборатория, която отговаря на условията на член 30 и изпитването е показало съответствие с изискванията на дадения тор.
2. Държавите-членки осигуряват вземането на проби и извършването на анализ за официален контрол на ЕО торове, спадащи към видовете, изброени в приложение I, да се извършват в съответствие с приложения III и IV.
3. Спазването на настоящия регламент по отношение на съответствието на видове торове и спазването на декларираното съдържание на хранителни елементи и/или декларираното съдържание, изразено като форми и разтворимост на тези хранителни вещества, може да се проверява при официални инспекции само с помощта на методи за вземане на проби и анализ, установени в съответствие с приложения III и IV и вземащи предвид допустимите отклонения, определени в приложение II.
4. Адаптирането и осъвременяването на методите за измерване, вземане на проби и анализ следват процедурата, посочена в член 32, параграф 2, и при възможност следва да използват европейските стандарти. Същата процедура се прилага за приемането на правила за прилагане, необходими за определяне на мерките за контрол, предвидени в този член и в членове 8, 26 и 27 от настоящия регламент. Тези правила трябва по-специално да отговорят на въпроса за честотата, с която е необходимо да се повтарят тези изпитания, както и мерките, които се разработват за осигуряване на това, торът, пуснат на пазара, да е идентичен с изпитания тор.
Член 30
Лаборатории
1. Държавите-членки нотифицират Комисията за списъка на одобрените лаборатории на техните територии, които са компетентни да предоставят необходимите услуги за проверка на спазването по отношение на ЕО торовете на изискванията на настоящия регламент. Тези лаборатории трябва да отговарят на стандартите, посочени в раздел Б от приложение V. Това нотифициране се извършва до 11 юни 2004 г. и след това във връзка с всяко следващо изменение.
2. Комисията публикува списъка на одобрените лаборатории в Официален вестник на Европейския съюз.
3. Когато една държава-членка има основателна причина да счита, че одобрена лаборатория не отговаря на стандартите, посочени в параграф 1, тя повдига въпроса в комитета, посочен в член 32. Ако комитетът се съгласи, че лабораторията не отговаря на стандартите, Комисията отстранява името ѝ от списъка, посочен в параграф 2.
4. Комисията приема решение по въпроса в рамките 90 дни от получаването на информацията в съответствие с процедурата, посочена в член 32, параграф 2.
5. Комисията публикува изменения списък в Официален вестник на Европейския съюз.
ДЯЛ IV
ЗАКЛЮЧИТЕЛНИ РАЗПОРЕДБИ
ГЛАВА I
Изменение на приложенията
Член 31
Нови ЕО торове
1. Включването на нов вид тор в приложение I към настоящия регламент се приема в съответствие с процедурата, посочена в член 32, параграф 2.
2. Производител или негов представител, които желаят да предложат нов вид тор за включване в приложение I и от които се изисква да съставят техническо досие за тази цел, осъществяват това, като вземат предвид техническата документация, посочена в раздел А от приложение V.
3. Измененията, необходими за адаптиране на приложенията към техническия напредък, се приемат в съответствие с процедурата, посочена в член 32, параграф 2.
Член 32
Процедура на комитета
1. Комисията се подпомага от комитет.
2. Когато се прави позоваване на настоящия параграф, се прилагат членове 5 и 7 от Решение 1999/468/ЕО, като се вземат предвид разпоредбите на член 8 от него.
Срокът, постановен в член 5, параграф 6 от Решение 1999/468/ЕО, се установява на три месеца.
3. Комитетът приема свой процедурен правилник.
ГЛАВА II
Преходни разпоредби
Член 33
Компетентни лаборатории
1. Без да се засягат разпоредбите на член 30, параграф 1, държавите-членки могат през преходния период до 11 декември 2007 г. да продължат да прилагат своите национални разпоредби за упълномощаване на компетентните лаборатории за предоставяне на необходимите услуги за проверка на спазването по отношение на ЕО торовете на изискванията на настоящия регламент.
2. Държавите-членки нотифицират списъка на тези лаборатории на Комисията, като дават подробна информация за схемата на тяхното упълномощаване. Тази нотификация се извършва до 11 юни 2004 г. и при всяко последващо изменение.
Член 34
Опаковане и етикетиране
Независимо от член 35, параграф 1, маркировките, опаковките, етикетите и придружаващите документи на ЕО торове, предвидени в предходни директиви, могат да продължат да се използват до 11 юни 2005 г.
ГЛАВА III
Заключителни разпоредби
Член 35
Отменени директиви
1. Директиви 76/116/ЕИО, 77/535/ЕИО, 80/876/ЕИО и 87/94/ЕИО се отменят.
2. Позоваването на отменените директиви се тълкува като позоваване на настоящия регламент. По-специално, дерогациите от член 7 от Директива 76/116/ЕИО, които са предоставени от Комисията по член 95, параграф 6 от Договора, се тълкуват като дерогации от член 5 от настоящия регламент и продължават да са в действие, независимо от влизането в сила на настоящия регламент. До приемането на санкции по член 36 държавите-членки могат да продължат да прилагат санкции за нарушения на националните правила, като прилагат директивите, посочени в параграф 1.
Член 36
Санкции
Държавите-членки установяват правила за санкции, приложими за нарушения на разпоредбите на настоящия регламент, и приемат всички необходими мерки за тяхното прилагане. Предвидените санкции трябва да бъдат ефективни, съответни и възпиращи.
Член 37
Национални разпоредби
Държавите-членки нотифицират Комисията до 11 юни 2005 г. за всички национални разпоредби, приети в съответствие с член 6, параграф 1, член 6, параграф 2, член 29, параграф 1 и член 36 от настоящия регламент и я нотифицират незабавно за всички последващи изменения, които ги засягат.
Член 38
Влизане в сила
Настоящият регламент влиза в сила на двадесетия ден след публикуването му в Официален вестник на Европейския съюз с изключение на член 8 и член 26, параграф 3, които влизат в сила на 11 юни 2005 г.
Настоящият регламент е задължителен в своята цялост и се прилага пряко във всички държави-членки.
Съставено в Люксембург на 13 октомври 2003 година.
За Европейския парламент
Председател
P. COX
За Съвета
Председател
G. ALEMANNO
(1) ОВ C 51 Е, 26.2.2002 г., стр. 1 и ОВ C 227 Е, 24.9.2002, стр. 503.
(2) ОВ C 80, 3.4.2002 г., стр. 6.
(3) Становище на Европейския парламент от 10 април 2002 г. (ОВ C 127 Е, 29.5.2002 г., стр. 160.), Обща позиция на Съвета от 14 април 2003 г. (ОВ C 153 Е, 1.7.2003 г., стр. 56) и Решение на Европейския парламент от 2 септември 2003 г. (все още непубликувано в Официален вестник).
(4) ОВ L 24, 30.1.1976 г., стр. 21. Директива, последно изменена с Директива 98/97/ЕО на Европейския парламент и на Съвета (ОВ L 18, 23.1.1999 г., стр. 60).
(5) ОВ L 250, 23.9.1980 г., стр. 7. Директива, изменена с Директива 97/63/ЕО на Европейския парламент и на Съвета (ОВ L 335, 6.12.1997 г., стр. 15).
(6) ОВ L 38, 7.2.1987 г., стр. 1. Директива, изменена с Директива 88/126/ЕИО (ОВ L 63, 9.3.1988 г., стр. 12).
(7) ОВ L 213, 22.8.1977 г., стр. 1. Директива, последно изменена с Директива 95/8/ЕО (ОВ L 86, 20.4.1995 г., стр. 41).
(8) ОВ L 184, 17.7.1999 г., стр. 23.
СЪДЪРЖАНИЕ
ПРИЛОЖЕНИЕ I — Списък на видовете ЕО торове…
А. Неорганични еднокомпонентни торове с основни хранителни елементи …
А.1. Азотни торове …
А.2. Фосфатни торове …
А.3. Калиеви торове…
Б. Неорганични сложни торове с основни хранителни елементи …
Б.1. NPK торове …
Б.2. NP торове …
Б.3. NK торове …
Б.4. PK торове …
В. Неорганични течни торове …
В.1. Еднокомпонентни течни торове …
В.2. Сложни течни торове …
Г. Неорганични торове с второстепенни хранителни елементи …
Д. Неорганични торове с хранителни микроелементи …
Д.1. Торове, съдържащи само един хранителен микроелемент …
Д.1.1. Бор…
Д.1.2. Кобалт …
Д.1.3. Мед …
Д.1.4. Желязо …
Д.1.5. Манган …
Д.1.6. Молибден …
Д.1.7. Цинкv …
Д.2. Минимално съдържание на хранителен микроелемент, процентно съдържание на елементите в тора …
Д.3. Списък на разрешени хелатиращи и комплексообразуващи агенти за хранителни микроелементи…
ПРИЛОЖЕНИЕ II — Допустими отклонения…
1. Неорганични еднокомпонентни торове с основни хранителни елементи — абсолютна стойност в проценти от масата, изразена като N, P2O5, K2O, MgO, CL …
2. Неорганични сложни торове с основни хранителни елементи …
3. Второстепенни хранителни елементи в торове …
4. Хранителни микроелементи в торове …
ПРИЛОЖЕНИЕ III — Технически разпоредби за амониево-нитратни торове с високо съдържание на азот…
1. Характеристики и гранични стойности за еднокомпонентни амониево-нитратни торове с високо съдържание на азот …
2. Описание на изпитване на детонация на амониево-нитратни торове с високо съдържание на азот…
3. Методи за проверка на спазването на граничните стойности, определени в приложения III-1 и III-2…
4. Определяне на устойчивост на детонация …
ПРИЛОЖЕНИЕ IV — Методи за вземане на проби и анализ…
А. Метод за вземане на проби за контрол на торове …
1. Цел и обхват …
2. Служители, които вземат проби …
3. Дефиниции …
4. Апаратура …
5. Количествени изисквания …
6. Указания за вземане, приготвяне и опаковане на пробите…
7. Опаковане на окончателни проби …
8. Отчет за вземане на проби …
9. Местоназначение на пробите …
Б. Методи за анализ на торове …
Общи наблюдения …
Общи разпоредби, свързани с анализа на торовете …
Метод 1 — Приготвяне на проби за анализ …
Методи 2 — Азот …
Метод 2.1 — Определяне на амонячен азот …
Методи 2.2 — Определяне на нитратен и амонячен азот …
Метод 2.2.1 — Определяне на нитратен и амонячен азот по метода на Улш …
Метод 2.2.2 — Определяне на азотен и амонячен азот по метода на Арнд…
Метод 2.2.3 — Определяне на нитратен и амонячен азот по метода на Деварда …
Метод 2.3 — Определяне на общ азот …
Метод 2.3.1 — Определяне на общ азот в калциев цианамид без нитрати …
Метод 2.3.2 — Определяне на общ азот в калциев цианамид, съдържащ нитрати …
Метод 2.3.3 — Определяне на общ азот в карбамид …
Метод 2.4 — Определяне на цианамиден азот …
Метод 2.5 — Спектрофотометрично определяне на биурет в карбамид …
Методи 2.6 — Определяне на различни форми на азот в една и съща проба …
Метод 2.6.1 — Определяне на различни форми на азот в една и съща проба, съдържаща азот като нитратен, амонячен, карбамиден и цианамиден азот …
Метод 2.6.2 — Определяне на различни форми на азот в една и съща проба, съдържаща азот само като нитратен, амонячен, карбамиден …
Метод 3 — Фосфор …
Методи 3.1 — Екстракции …
Метод 3.1.1 — Екстракция на фосфор, разтворим в минерални киселини …
Метод 3.1.2 — Екстракция на фосфор, разтворим в 2 % мравчена киселина …(20 g на литър) …
Метод 3.1.3 — Екстракция на фосфор, разтворим в 2 % лимонена киселина …(20 g на литър) …
Метод 3.1.4 — Екстракция на фосфор, разтворим в неутрален амониев цитрат …
Метод 3.1.5 - Екстракция с алкален амониев цитрат …
Метод 3.1.5.1 — Екстракция на разтворим фосфор по Петерман при 65 °С …
Метод 3.1.5.2 — Екстракция на разтворим фосфор по Петерман при температура на околната среда…
Метод 3.1.5.3 — Екстракция на разтворим фосфор в алкален амониев цитрат на Жули …
Метод 3.1.6 — Екстракция на водоразтворим фосфор …
Метод 3.2 — Определяне на извлечен фосфор (гравиметричен метод с използване на хинолин фосфомолибден) …
Метод 4 — Калий …
Метод 4.1 — Определяне на съдържанието на водоразтворим калийv …
Метод 5 — …
Метод 6 — Хлор 116 …
Метод 6.1. — Определяне на хлориди при отсъствието на органичен материал …
Методи 7 — Степен на смилане …
Метод 7.1 — Определяне на степен на смилане (суха процедура) …
Метод 7.2 — Определяне на степен на смилане на меки естествени фосфати …
Метод 8 — Вторични хранителни елементи …
Метод 8.1 — Екстракция на общ калций, общ магнезий, общ натрий и обща сяра под формата на сулфати …
Метод 8.2 — Екстракция на обща сяра, присъстваща в различни форми …
Метод 8.3 — Екстракция на водоразтворими калций, магнезий, натрий и сяра (под формата на сулфати) …
Метод 8.4 — Екстракция на водоразтворима сяра, когато сярата е в различни форми …
Метод 8.5 — Екстракция на и определяне на елементарна сяра …
Метод 8.6 — Манганометрично определяне на извлечен калций след утаяване под формата на оксалат …
Метод 8.7 — Определяне на магнезий с атомна абсорбционна спектрометрия …
Метод 8.8 — Комплексометрично определяне на магнезий …
Метод 8.9 — Определяне на сулфати …
Метод 8.10 — Определяне на извлечен натрий …
Методи 9 — Хранителни микроелементи при концентрация, по-малка или равна на 10 % …
Метод 9.1 — Екстракция общо на хранителни микроелементи …
Метод 9.2 — Екстракция на водоразтворими хранителни микроелементи …
Метод 9.3 — Отстраняване на органични съединения от екстракти на торове …
Метод 9.4 — Определяне на хранителни микроелементи в екстракти на торове с атомна абсорбционна спектрометрия (обща процедура) …
Метод 9.5 — Определяне на бор в екстракти на торове с спектрометрия с азометин-Н…
Метод 9.6 — Определяне на кобалт в екстракти на торове с атомна абсорбционна спектрометрия …
Метод 9.7 — Определяне на мед в екстракти на торове с атомна абсорбционна спектрометрия …
Метод 9.8 — Определяне на желязо в екстракти на торове с атомна абсорбционна спектрометрия …
Метод 9.9 — Определяне на манган в екстракти на торове с атомна абсорбционна спектрометрия …
Метод 9.10 — Определяне на молибден в екстракти на торове със спектрометрия с комплекс с амониев тиоцианит …
Метод 9.11 — Определяне на цинк в екстракти на торове с атомна абсорбционна спектрометрия …
Методи 10 — Хранителни микроелементи с концентрация, по-голяма от 10 %…
Метод 10.1 — Екстракция общо на хранителни микроелементи …
Метод 10.2 — Екстракция на водоразтворими хранителни микроелементи …
Метод 10.3 — Отстраняване на органични съединения от екстракти на торове …
Метод 10.4 — Определяне на хранителни микроелементи в екстракти на торове с атомна абсорбционна спектрометрия (обща процедура) …
Метод 10.5 — Определяне на бор в екстракти на торове с помощта на ацадаметрично титруване …
Метод 10.6 — Определяне на кобалт в екстракти на торове с гравиметричен метод с 1-нитрозо-2-нафтол …
Метод 10.7 — Определяне на мед в екстракти на торове с титриметричен метод …
Метод 10.8 — Определяне на желязо в екстракти на торове с атомна абсорбционна спектрометрия…
Метод 10.9 — Определяне на манган в екстракти на торове чрез титруване …
Метод 10.10 — Определяне на молибден в екстракти на торове с гравиметричен метод с 8-хидрохинолин …
Метод 10.11 — Определяне на цинк в екстракти на торове с атомна абсорбционна спектрометрия…
ПРИЛОЖЕНИЕ V…
А. Списък на документите, с които трябва да се консултират производителите или техните представители, за да съставят техническо досие за нов вид тор за добавяне към приложение I от настоящия регламент …
Б. Стандарти за акредитиране за лаборатории, които са компетентни и упълномощени да предоставят необходимата услуга за проверка на спазването по отношение на ЕО торовете на изискванията на настоящия регламент и неговите приложения …
ПРИЛОЖЕНИЕ I
СПИСЪК НА ВИДОВЕТЕ ЕО ТОРОВЕ
А. Неорганични еднокомпонентни торове с основни хранителни елементи
А.1. Азотни торове
|
№ |
Означение на вида |
Данни за метода на получаване и основни съставки |
Минимално съдържание на хранителни елементи (проценти от масата) Данни за изразяване на хранителни елементи Други изисквания |
Други данни за означението на вида |
Съдържание на хранителните елементи за обявяване; Форми и разтворимости на хранителните елементи; Други критерии |
||||||||
|
1 |
2 |
3 |
4 |
5 |
6 |
||||||||
|
1а) |
Калциев нитрат |
Продукт, получен по химичен път, съдържащ калциев нитрат като основна съставка. Възможно е да съдържа амониев нитрат |
15 % N азот, изразен като общ азот или нитратен азот и амонячен азот. Максимално съдържание на амонячен азот: 1,5 % N |
|
Общ азот Допълнителни незадължителни данни:
|
||||||||
|
1б) |
Калциево-магнезиев нитрат (нитрат на варовик и магнезий) |
Продукт, получен по химичен път, съдържащ калциев нитрат и магнезиев нитрат като основни съставки |
13 % N азот, изразен като нитратен азот. Минимално съдържание на магнезий във форма на водоразтворими соли, изразени като магнезиев оксид: 5 % MgO |
|
Нитратен азот Водоразтворим магнезиев оксид |
||||||||
|
1в) |
Магнезиев нитрат |
Продукт, получен по химичен път, съдържащ като основна съставка магнезиев нитрат хексахидрат |
10 % N азот, изразен като нитратен азот. |
Когато се пуска на пазара във форма на кристали, може да се добави забележка „в кристална форма“ |
Нитратен азот Водоразтворим магнезиев оксид |
||||||||
|
14 % MgO Магнезий, изразен като водоразтворим магнезиев оксид |
|||||||||||||
|
2а) |
Натриев нитрат |
Продукт, получен по химичен път, съдържащ натриев нитрат като основна своя съставка |
15 % N азот, изразен като нитратен азот |
|
Нитратен азот |
||||||||
|
2б) |
Чилска селитра |
Продукт, получен от чилска селитра, съдържащ натриев нитрат като основна съставка |
15 % N азот, изразен като нитратен азот |
|
Нитратен азот |
||||||||
|
3а) |
Калциев цианамид |
Продукт, получен по химичен път, съдържащ калциев цианамид като основна своя съставка, калциев оксид и възможно малки количества амониеви соли и карбамид |
18 % N азот, изразен като общ азот, като най-малко 75 % от обявявания азот е във форма на цианамид |
|
Общ азот |
||||||||
|
3 б) |
Нитрифициран калциев цианамид |
Продукт, получен по химичен път, съдържащ калциев цианамид като основна своя съставка, калциев оксид и възможно малки количества амониеви соли и карбамид, плюс добавен нитрат |
18 % N азот, изразен като общ азот, най-малко 75 % от обявявания не-нитратен азот да бъде свързан във форма на цианамид. Съдържание на нитратен азот:
|
|
Общ азот Нитратен азот |
||||||||
|
4 |
Амониев сулфат |
Продукт, получен по химичен път, съдържащ амониев сулфат като основна съставка |
20 % N Азот, изразен като амонячен азот |
|
Амонячен азот |
||||||||
|
5 |
Амониев нитрат или калциево-амониев нитрат |
Продукт, получен по химичен път, съдържащ амониев сулфат като основна съставка, който може да съдържа пълнители, като смлян варовик, калциев сулфат, смлян доломит, магнезиев сулфат, кизерит |
20 % N азот, изразен като нитратен азот и амонячен азот, всяка от тези две форми на азот е около половината от азотното съдържание. Виж приложения III.1 и III.2 към настоящия регламент, ако се изисква. |
Означението „калциево-амониев нитрат“ е запазено изключително за тор, съдържащ само калциев карбонат (например варовик) и/или магнезиев карбонат и калциев карбонат (например, доломит) в допълнение към амониевия нитрат. Минималното съдържание на тези карбонати трябва да е 20 %, а тяхната чистота — най-малко 90 % |
Общ азот Нитратен азот Амонячен азот |
||||||||
|
6 |
Амониев сулфат-нитрат |
Продукт, получен по химичен път, съдържащ като основна съставка амониев нитрат и амониев сулфат |
25 % N азот, изразен като амонячен азот и нитратен азот. Минимално съдържание на нитратен азот: 5 % |
|
Общ азот Нитратен азот Амонячен азот |
||||||||
|
7 |
Магнезиев сулфонитрат |
Продукт, получен по химичен път, съдържащ като основна съставка амониев нитрат, амониев сулфат и магнезиев сулфат |
19 % N азот, изразен като амонячен азот и нитратен азот. Минимално съдържание на нитратен азот: 6 % N |
|
Общ азот Амонячен азот |
||||||||
|
5 % MgO Магнезий под форма на водоразтворими соли, изразени като общ магнезиев оксид |
|
Нитратен азот Водоразтворим магнезиев оксид |
|||||||||||
|
8 |
Магнезиево-амониев нитрат |
Продукт, получен по химичен път, съдържащ амониеви нитрати и магнезиеви соли (доломит магнезиев карбонат и/или магнезиев сулфат) като основна своя съставка |
19 % N азот, изразен като амонячен азот и нитратен азот. Минимално съдържание на нитратен азот: 6 % N |
|
Общ азот Амонячен азот Нитратен азот |
||||||||
|
5 % MgO Магнезий, изразени като общ магнезиев оксид |
|
Общ магнезиев оксид и възможно водоразтворим магнезиев оксид |
|||||||||||
|
9 |
Карбамид |
Продукт, получен по химичен път, съдържащ карбонил диамид като основна своя съставка |
44 % N Общ карбамид азот (включително биурет). Максимално съдържание на биурет: 1,2 % |
|
Общ азот, изразен като карбамид азот |
||||||||
|
10 |
Кротонилиден дикарбамид |
Продукт, получен при реакцията на карбамид с кротоналдехид Мономерно химическо съединение |
28 % N азот, изразен като общ азот Най-малко 25 % N от кротонилиден дикарбамид Максимално съдържание на карбамид азот: 3 % |
|
Общ азот Карбамид азот, когато той поне 1 % от масата Азот от кротонилиден дикарбамид |
||||||||
|
11 |
Изобутилиден дикарбамид |
Продукт, получен при реакция на карбамид с изобутиралдехид Мономерно химическо съединение |
28 % N азот, изразен като общ азот. Най малко 25 % N от изобутилиден дикарбамид Максимално съдържание на карбамид азот: 3 % |
|
Общ азот Карбамид азот, когато той поне 1 % от масата Азот от изобутилиден дикарбамид |
||||||||
|
12 |
Карбамиден формалдехид |
Продукт, получен при реакцията на карбамид с формалдехид и съдържащ като основна своя съставка молекули на карбамид формалдехид Полимерно химично съединение |
36 % N азот, изразен като общ азот Най-малко 3/5 от обявеното съдържание на азот трябва да бъде разтворимо в гореща вода Най-малко 31 % N от карбамид формалдехид Максимално съдържание на амиден азот: 3 %. |
|
Общ азот Карбамид азот, когато той поне 1 % от масата Азот от формалдехиден карбамид, която е разтворима в студена вода Азот от формалдехиден карбамид, която е разтворима в гореща вода |
||||||||
|
13 |
Азотен тор, съдържащ кротонилиден дикарбамид |
Продукт, получен по химичен път, съдържащ кротонилиден дикарбамид и еднокомпонентен азотен тор [Списък А-1, с изключение на продукти 3а), 3б) и 5] |
18 % N азот, изразен като общ азот. Най-малко 3 % в амонячна и/или азотна и/или карбамидна форма Най-малко 1/3 от обявеното съдържание на общ азот трябва да е получено от кротонилиден дикарбамид Максимално съдържание на биурет:
|
|
Общ азот За всяка форма, в която са в количество най-малко 1 %:
Азот от кротонилиден дикарбамид |
||||||||
|
14 |
Азотен тор, съдържащ изобутилиден дикарбамид |
Продукт, получен по химичен път, съдържащ изобутилиден дикарбамид и еднокомпонентен азотен тор [Списък А-1, с изключение на продукти 3а), 3б) и 5] |
18 % N азот, изразен като общ азот. Най-малко 3 % в амонячна и/или нитратна и/или амидна форма Най-малко 1/3 от обявеното съдържание на общ азот трябва да е получено от изобутилиден дикарбамид Максимално съдържание на биурет:
|
|
Общ азот За всяка форма, в която са в количество най-малко 1 %:
Азот от изобутилиден дикарбамид |
||||||||
|
15 |
Азотен тор съдържащ карбамид формалдехид |
Продукт, получен по химичен път, съдържащ карбамид формалдехид и еднокомпонентен азотен тор [Списък А-1, с изключение на продукти 3а), 3б) и 5] |
18 % N азот, изразен като общ азот. Най-малко 3 % в амонячна и/или азотна и/или амидна форма Най-малко 1/3 от обявеното съдържание на азот трябва да е получено от карбамид формалдехид. Азотът от карбамид формалдехида трябва да съдържа най-малко 3/5 азот, който е разтворим в гореща вода. Максимално съдържание на биурет:
|
|
Общ азот За всяка форма, в която са в количество най-малко 1 %:
Азот от карбамид формалдехид Азот от карбамид формалдехид, разтворим в студена вода Азот от карбамид формалдехид, разтворим в гореща вода |
||||||||
|
16 |
Амониев сулфат с инхибитор на нитификацията (дициандиамид) |
Продукт, получен по химичен път, съдържащ амониев сулфат и дициандиамид |
20 % N азот, изразен като общ азот Максимално съдържание на амонячен азот: 18 % Максимално съдържание на азот от дициандиамид: 1,5 % |
|
Общ азот Амонячен азот Азот от дициандиамид Техническа информация (1) |
||||||||
|
17 |
Амониев сулфонитрат с инхибитор на нитрификацията (дициандиамид) |
Продукт, получен по химичен път, съдържащ амониев сулфонитрат и дициандиамид |
24 % N азот, изразен като общ азот. Минимално съдържание на амонячен азот: 3 % Минимално съдържание на азот от дициандиамид: 1,5 % |
|
Общ азот Амонячен азот Азот от дициандиамид Техническа информация (1) |
||||||||
|
18 |
Карбамид-амониев сулфат |
Продукт, получен по химичен път от карбамид и амониев сулфат |
30 % N азот, изразен като амонячен азот и амиден азот Минимално съдържание на амонячен азот: 4 % Минимално съдържание на сяра, изразено като серен триоксид: 12 % Максимално съдържание на биурет: 0,9 % |
|
Общ азот Амонячен азот Карбамид азот Водоразтворим серен триоксид |
А. 2 Фосфатни торове
Когато е предписан критерий за големина на частиците за основните съставни материали на торове, продавани във форма на гранули (торове 1, 3, 4, 5, 6 и 7), тя се установява с подходящ аналитичен метод.
|
№ |
Обозначение на вида |
Данни за метода на получаване и основни съставки |
Минимално съдържание на хранителни елементи (процент от масата) Данни за изразяване на хранителни елементи Други изисквания |
Други данни за обозначението на вида |
Съдържание на хранителни елементи за обявяване Форми и разтворимости на хранителните елементи други критерии |
||||||||
|
1 |
2 |
3 |
4 |
5 |
6 |
||||||||
|
1 |
Основна шлака:
|
Продукт, получен при топене на желязо при третиране на фосфатни стопилки и съдържащ силикофосфати като негови основни съставки |
12 % Р2О5 Фосфор, изразен като фосфорен пентаоксид, разтворим в минерални киселини, поне 75 % от обявеното съдържание на фосфорния пентаоксид да е разтворимо в 2 % лимонена киселина или Р2О5 Фосфор, изразен като фосфорен пентаоксид, разтворим в 2 % лимонена киселина Размер на частиците:
|
|
Общ фосфорен пентаоксид (разтворим в минерални киселини) 75 % от който (да се посочи като% от масата) е разтворим в 2 % лимонена киселина (за търговия във Франция, Италия, Испания, Португалия и Гърция) Общ фосфорен пентаоксид (разтворим в минерални киселини) и фосфорен пентаоксид, разтворим в 2 % лимонена киселина (за търговия в Обединеното кралство) Фосфорен пентаоксид, разтворим в 2 % лимонена киселина (за търговия в Германия, Белгия, Дания, Ирландия, Люксембург, Нидерландия и Австрия) |
||||||||
|
2а) |
Обикновен суперфосфат |
Продукт, получен при реакцията на смлян минерален фосфат със сярна киселина и съдържащ калциев фосфат като основна съставка, както и калциев сулфат |
16 % Р2О5 Фосфор, изразен като Р2О5 разтворим в неутрален амониев цитрат. Най-малко 93 % от обявеното съдържание на Р2О5 да е водоразтворимо Проба за изпитване: 1 g |
|
Фосфорен пентаоксид, разтворим в неутрален амониев цитрат Водоразтворим фосфорен пентаоксид |
||||||||
|
2б) |
Концентриран суперфосфат |
Продукт, получен при реакцията на смлян минерален фосфат със сярна киселина и фосфорна киселина и съдържащ монокалциев фосфат като основна съставка, както и калциев сулфат |
25 % P2O5 Фосфор, изразен като P2O5 разтворим в неутрален амониев цитрат. Най-малко 93 % от обявеното съдържание на P2O5 да е водоразтворимо Проба за изпитване: 1 g |
|
Фосфорен пентаоксид, разтворим в неутрален амониев цитрат Водоразтворим фосфорен пентаоксид |
||||||||
|
2в) |
Троен суперфосфат |
Продукт, получен при реакцията на смлян минерален фосфат с фосфорна киселина и съдържащ монокалциев фосфат като основна съставка |
38 % P2O5 Фосфор, изразен като P2O5 разтворим в неутрален амониев цитрат, най-малко 93 % от обявеното съдържание на P2O5 да е водоразтворимо Проба за изпитване: 3 g |
|
Фосфорен пентаоксид, разтворим в неутрален амониев цитрат Водоразтворим фосфорен пентаоксид |
||||||||
|
3 |
Частично разтворим фосфорит |
Продукт, получен при частично разтваряне на смлян фосфорит със сярна киселина или фосфорна киселина и съдържащ монокалциев фосфат, трикалциев фосфат и калциев сулфат |
20 % P2O5 Фосфор, изразен като P2O5 разтворим в минерални киселини, най-малко 40 % от обявеното съдържание на P2O5 да е водоразтворимо. Размер на частиците:
|
|
Общ фосфорен пентаоксид (разтворим в минерални киселини) Водоразтворим фосфорен пентаоксид |
||||||||
|
4 |
Дикалциев фосфат |
Продукт, получен при утаяване на разтворена фосфорна киселина от минерални фосфати или кости, и съдържащ дикалциев фосфат дихидрат като основна съставка |
38 % P2O5 Фосфор, изразен като P2O5, разтворим в алкален амониев цитрат (Петерман). Размер на частиците:
|
|
Фосфорен пентаоксид, разтворим в алкален амониев цитрат |
||||||||
|
5 |
Калциниран фосфат |
Продукт, получен при топлинна обработка на смлян фосфорит с алкални съединения и салицилова киселина, и съдържащ основен калциев фосфат и калциев силикат, като основни съставки |
25 % P2O5 Фосфор, изразен като P2O5, разтворим в алкален амониев цитрат (Петерман) Размер на частиците:
|
|
Фосфорен пентаоксид, разтворим в алкален амониев цитрат |
||||||||
|
6 |
Калциев алумофосфат |
Продукт, получен във аморфна форма чрез топлинна обработка и смилане, съдържащ алуминиеви и калциеви фосфати като основни съставки |
30 % P2O5 Фосфор, изразен като P2O5 разтворим в минерални киселини, най-малко 75 % от обявеното съдържание на P2O5 да е разтворимо в разтворим алуминиев цитрат (Жули) Размер на частиците:
|
|
Общ фосфорен пентаоксид (разтворим в минерални киселини) Фосфорен пентаоксид, разтворим в алкален амониев цитрат |
||||||||
|
7 |
Мек смлян фосфорит |
Продукт, получен чрез смилане на меки минерални фосфати и съдържащ трикалциев фосфат и калциев карбонат като основни съставки |
25 % P2O5 Фосфор, изразен като P2O5 разтворим в неорганични киселини. Най-малко 55 % от обявеното съдържание на P2O5 да е разтворимо в 2 % мравчена киселина Размер на частиците:
|
|
Общ фосфорен пентаоксид (разтворим в минерални киселини) Фосфорен пентаоксид, разтворим в 2 % мравчена киселина Процент от масата но материала, който минава през сито с отвори 0,063 mm |
А.3 Калиеви торове
|
№ |
Обозначение на вида |
Данни за метода на получаване и основни съставки |
Минимално съдържание на хранителни елементи (% от масата) изразяване на хранителни елементи; други изисквания |
Други данни за обозначението на вида |
Хранително съдържание за обявяване Форми и разтворимости на хранителните елементи други критерии |
|
1 |
2 |
3 |
4 |
5 |
6 |
|
1 |
Каинит |
Продукт, получен от необработени калиеви соли |
10 % К2О Калий, изразен като водоразтворим К2О |
Могат да бъдат добавяни обичайни търговски наименования |
Водоразтворим калиев оксид Водоразтворим магнезиев оксид |
|
5 % MgO Магнезий във форма на водоразтворими соли, изразен като MgO |
|||||
|
2 |
Обогатена каинитова сол |
Продукт, получен от необработени калиеви соли, обогатени чрез смесване с калиев хлорид |
18 % К2О Калий, изразен като водоразтворим К2О |
Могат да бъдат добавяни обичайни търговски наименования |
Водоразтворим калиев оксид Възможно споменаване на съдържането на Водоразтворим магнезиев оксид, когато е по-високо от 5 % магнезиев оксид |
|
3 |
Калиев хлорид |
Продукт, получен от необработени калиеви соли и съдържащ калиев хлорид като основна съставка |
37 % К2О Калий, изразен като водоразтворим К2О |
Могат да бъдат добавяни обичайни търговски наименования |
Водоразтворим калиев оксид |
|
4 |
Калиев хлорид, съдържащ магнезиеви соли |
Продукт, получен от необработени калиеви соли, с добавени магнезиеви соли и съдържащ калиев хлорид като негова основна съставка |
37 % К2О Калий, изразен като водоразтворим К2О |
|
Водоразтворим калиев оксид Водоразтворим магнезиев оксид |
|
5 % MgO Магнезий във форма на водоразтворим соли, изразен като MgO |
|
||||
|
5 |
Калиев сулфат |
Продукт, получен по химичен път от калиеви соли и съдържащ калиев сулфат като основна съставка |
47 % К2О Калий, изразен като водоразтворим К2О Максимално съдържание на хлорид: 3 % Cl |
|
Водоразтворим калиев оксид Възможно споменаване на съдържанието хлориди |
|
6 |
Калиев сулфат, съдържащ магнезиева сол |
Продукт, получен по химичен път от калиеви соли, възможно с добавка на магнезиеви соли, и съдържащ калиев сулфат и магнезиев сулфат като негова основна съставка |
22 % К2О Калий изразен като водоразтворим К2О |
Могат да бъдат добавяни обичайни търговски наименования |
Водоразтворим калиев оксид Водоразтворим магнезиев оксид Възможно споменаване на съдържанието хлориди |
|
8 % MgO Магнезий във форма на водоразтворим соли, изразен като MgO Максимално съдържание на хлорид: 3 % Cl |
|||||
|
7 |
Кизерит с калиев сулфат |
Продукт, получен от кизерит с добавка на калиев сулфат |
8 % MgO Магнезий изразен като водоразтворим MgO |
Могат да бъдат добавяни обичайни търговски наименования |
Водоразтворим калиев оксид Водоразтворим магнезиев оксид Възможно споменаване на съдържанието хлориди |
|
6 % К2О Калий изразен като водоразтворим К2О Общо MgO + K2O: 20 % Максимално съдържание на хлорид: 3 % Cl |
Б. Неорганични сложни торове с основни хранителни елементи
Б.1. NPK торове
|
Б.1.1. |
Обозначение на вида: |
NPK торове |
|||
|
Данни за метода на получаване: |
Продукт, получен по химичен начин или чрез смесване, без добавка на органични хранителни вещества от животински или растителен произход. |
||||
|
Минимално съдържание на хранителни елементи (процент от масата) |
|
|
Форми, разтворимости и съдържание на хранителни елементи, обявени в колони 4, 5 и 6 Размер на частиците |
Данни за идентификация на торовете Други изисквания |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
N |
P2O5 |
K2O |
N |
P2O5 |
K2O |
||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
1 |
2 |
3 |
4 |
5 |
6 |
||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|
Водоразтворим К2О |
|
Съдържанието на Р2О5 разтворимо само в минерални киселини не трябва да превишава 2 %. Пробата за изпитване на разтворимости (2) и (3) е 1 g.
Този тор трябва да се продава под обозначението „NPK тор, съдържащ мек смлян фосфорит“ или „NPK тор, съдържащ частично разтворен фосфорит“. За този вид 2а) Пробата за изпитване на разтворимост (3) е 3 g. |
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
Размери на частиците на основните фосфатни съставки
|
|
Обявява се в съответствие с разтворимости (1) и (7), след приспадане на разтворимостта във вода: Този вид тор трябва да съдържа:
Този вид тор трябва да се търгува под обозначението „NPK тор, съдържащ калциев алумофосфат“.
Обявяването на разтворимостта на Р2О5 трябва да бъде дадена в съответствие с със следните разтворимости:
|
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
Б.1.2. |
Обозначение на вида: |
NPK тор, съдържащ кротонилиден дикарбамид или изобутилиден дикарбамид или карбамид формалдехид (както е подходящо) |
|||||||||
|
Данни за метода на получаване: |
Продукт, получен по химичен път, без добавка на органични хранителни елементи от животински или растителен произход и съдържащ кротонилидинен дикарбамид или изобутилиден дикарбамид или карбамид формалдехид. |
||||||||||
|
Минимално съдържание на хранителни елементи (% от масата) |
|
|
Форми, разтворимости и съдържание на хранителни елементи, обявено в колони 4, 5 и 6; Размер на частиците |
Данни за идентификация на торовете; Други изисквания |
||||||||||||||||||||||||||||||||||||||||||||
|
N |
P2O5 |
K2O |
N |
P2O5 |
K2O |
||||||||||||||||||||||||||||||||||||||||
|
1 |
2 |
3 |
4 |
5 |
6 |
||||||||||||||||||||||||||||||||||||||||
|
|
Водоразтворим К2О |
|
1. NPK тор, несъдържащ томасова шлака, калцинирани фосфати, алуминиево-калциеви фосфати, частично разтворен фосфорит и мек смлян фосфат, трябва да се обявява в съответствие с разтворимости (1), (2) или (3):
Съдържанието на Р2О5 разтворимо само в минерални киселини не трябва да превишава 2 %. За този вид 1 пробата за изпитване за определяне на разтворимости (2) и (3) трябва да бъде 1 g. |
|
||||||||||||||||||||||||||||||||||||||||
Б.2. NP торове
|
Б.2.1. |
Обозначение на вида: |
NP торове |
|||
|
Данни за метода на получаване: |
Продукт, получен по химичен път или чрез смесване, без добавка на органични хранителни елементи от животински или растителен произход. |
||||
|
Минимално съдържание на хранителни елементи (проценти от масата) |
|
|
Форми, разтворимост и хранително съдържание, обявено в колони 4, 5 и 6; Размер на частиците |
Данни за идентификация на торовете Други изисквания |
||||||||||||||||||||||||||||||||||||||||||||||||||
|
N |
P2O5 |
K2O |
N |
P2O5 |
K2O |
||||||||||||||||||||||||||||||||||||||||||||||
|
1 |
2 |
3 |
4 |
5 |
6 |
||||||||||||||||||||||||||||||||||||||||||||||
|
|
|
|
Съдържанието на Р2О5, разтворим само в неорганични киселини не трябва да превишава 2 %. Проба за изпитване на разтворимости (2) и (3) е 1 g.
Той се обявява в съответствие с разтворимости (1), (3) и (4). Този вид тор трябва да съдържа:
Този тор трябва да се продава под обозначението „NP тор, съдържащ алуминиево-калциев фосфат“. За този вид 2а), пробата за изпитване на разтворимости (3) е 3 g. |
|
||||||||||||||||||||||||||||||||||||||||||||||
|
Размери на частиците на основните фосфатни съставки
|
|
Той се обявява в съответствие с разтворимости (1) и (7), след приспадане на разтворимостта във вода. Този вид тор трябва да съдържа:
Този тор се пуска на пазара с обозначение „NP тор, съдържащ калциев алумофосфат“.
Обявяването на разтворимостта на Р2О5 трябва да бъде в съответствие със следните разтворимости:
|
|
||||||||||||||||||||||||||||||||||||||||||||||||
|
Б.2.2. |
Обозначение на вида: |
NP торове, съдържащи кротонилиден дикарбамид или изобутилиден дикарбамид или карбамид формалдехид (както е подходящо) |
|||||||
|
Данни за метода на получаване: |
Продукт, получен по химически начин, без добавка на органични хранителни елементи от животински или растителен произход и съдържащи кротонилиден дикарбамид или изобутилиден дикарбамид или карбамид формалдехид. |
||||||||
|
Минимално съдържание на хранителни елементи (% от масата) |
|
|
Форми, разтворимости и съдържание на хранителни елементи, обявено в колони 4, 5 и 6; Размер на частиците |
Данни за идентификация на торовете; Други изисквания |
||||||||||||||||||||||||||||||||||||||
|
N |
P2O5 |
K2O |
N |
P2O5 |
K2O |
||||||||||||||||||||||||||||||||||
|
1 |
2 |
3 |
4 |
5 |
6 |
||||||||||||||||||||||||||||||||||
|
|
|
|
NP тор, без томасова шлака, калцинирани фосфати, калциеви алумофосфати, частично разтворен фосфорит и мек смлян фосфат, трябва да се обявява в съответствие с разтворимости (1), (2) или (3):
Съдържанието на Р2О5, разтворим само в неорганични киселини не трябва да превишава 2 %. Пробата за изпитване на разтворимости (2) и (3) е 1 g. |
|
||||||||||||||||||||||||||||||||||
Б.3. NК торове
|
Б.3.1. |
Обозначение на вида: |
NK тор |
|||
|
Данни за метода на получаване: |
Продукт, получен по химичен път или чрез смесване, без добавка на органични хранителни вещества от животински или растителен произход. |
||||
|
Минимално съдържание на хранителни елементи (% от масата) |
|
|
Форми, разтворимости и съдържание на хранителни елементи, обявено в колони 4, 5 и 6; Размер на частиците |
Данни за идентификация на торовете; Други изисквания |
||||||||||||||||||||||||
|
N |
P2O5 |
K2O |
N |
P2O5 |
K2O |
||||||||||||||||||||
|
1 |
2 |
3 |
4 |
5 |
6 |
||||||||||||||||||||
|
|
Водоразтворим К2О |
|
|
|
||||||||||||||||||||
|
Б.3.2. |
Обозначение на вида: |
NK тор, съдържащ кротонилиден дикарбамид или изобутилиден дикарбамид или карбамид формалдехид (както е подходящо) |
|||||||
|
Данни за метода на получаване: |
Продукт, получен по химичен път без добавка на органични хранителни вещества от животински или растителен произход и съдържащ кротонилиден дикарбамид или изобутилиден дикарбамид или карбамид формалдехид (както е подходящо). |
||||||||
|
Минимално съдържание на хранителни елементи (% от масата) |
|
|
Форми, разтворимости и съдържание на хранителни елементи, обявено в колони 4,5 и 6; Размер на частиците |
Данни за идентификация на торовете Други изисквания |
||||||||||||||||||||||||||||||||||
|
N |
P2O5 |
K2O |
N |
P2O5 |
K2O |
||||||||||||||||||||||||||||||
|
1 |
2 |
3 |
4 |
5 |
6 |
||||||||||||||||||||||||||||||
|
|
Водоразтворим К2О |
|
|
|
||||||||||||||||||||||||||||||
Б.4. PK торове
|
Обозначение на вида: |
PK тор |
||||
|
Данни за метода на получаване: |
Продукт, получен по химичен път или чрез смесване, без добавка на органични хранителни вещества от животински или растителен произход. |
||||
|
Минимално съдържание на хранителни елементи (% от масата) |
|
|
Форми, разтворимости и съдържание на хранителни елементи, обявено в колони 4,5 и 6; Размер на частиците |
Данни за идентификация на торовете Други изисквания |
||||||||||||||||||||||||||||||||||||||||||
|
N |
P2O5 |
K2O |
N |
P2O5 |
K2O |
||||||||||||||||||||||||||||||||||||||
|
1 |
2 |
3 |
4 |
5 |
6 |
||||||||||||||||||||||||||||||||||||||
|
|
|
Водоразтворим К2О |
|
Съдържанието на Р2О5, разтворим само в неорганични киселини не трябва да превишава 2 %. Проба за изпитване на разтворимости (2) и (3) е 1 g.
Той се обявява в съответствие с разтворимости (1), (3) и (4): Този вид тор трябва да съдържа:
Този тор трябва да се продава под означението „PK тор съдържащ мек смлян фосфорит“ или „PK тор съдържащ частично разтворен фосфорит“. За този вид 2(а) пробата за изпитване за определяне на разтворимост (3) е 3 g. |
|
||||||||||||||||||||||||||||||||||||||
|
Размери на частиците на основните фосфатни съставки
|
|
Той се обявява в съответствие с разтворимост (1) и (7), след приспадане на разтворимостта във вода. Този вид тор трябва да съдържа:
Този вид тор трябва да се търгува под означение „PK тор, съдържащ калциев алумофосфат“.
Обявяването на разтворимостта на Р2О5 трябва да е в съответствие със следните разтворимости:
|
|
||||||||||||||||||||||||||||||||||||||||
В. Неорганични течни торове
В.1. Еднокомпонентни течни торове
|
№ |
Обозначение на вида |
Данни за метода на получаване и основни съставки |
Минимално съдържание на хранителни елементи (% от масата) изразяване на хранителни елементи; други изисквания |
Други данни за означението на вида |
Хранително съдържание за обявяване Форми и разтворимости на хранителните елементи други критерии |
||||||||||
|
1 |
2 |
3 |
4 |
5 |
6 |
||||||||||
|
1 |
Разтвор на азотен тор |
Продукт, получен по химичен път и чрез разтваряне във вода, във форма, устойчива при атмосферно налягане, без добавка на органични хранителни вещества от животински или растителен произход. |
15 % N Азот, изразен като общ азот, или, ако има само една форма, нитратен азот или амонячен азот или амиден азот. Максимално съдържание на биурет:
|
|
Общ азот, за всяка форма, която са в количество не по-малко от 1 %, нитратен азот, амонячен азот и/или карбамид азот. Ако съдържанието на биурет е по-малко от 0,2 %, може да се добави „с ниско съдържание на биурет“. |
||||||||||
|
2 |
Разтвор на карбамид и амониев нитрат |
Продукт, получен по химичен път и чрез разтваряне във вода, съдържащ амониев нитрат и карбамид |
26 % N азот, изразен като общ азот, когато амидният азот е около ½ от азота. Максимално съдържание на биурет: 0,5 % |
|
Общ азот Нитратен азот, амонячен азот и карбамид азот Ако съдържанието на биурет е по-малко от 0,2 %, може да се добави „с ниско съдържание на биурет“. |
||||||||||
|
3 |
Разтвор на калциев нитрат |
Продукт, получен чрез разтваряне на калциев нитрат във вода |
8 % N азот, изразен като нитратна форма с максимум 1 % амонячен азот. Калций, изразен като водоразтворим СаО |
След вида се поставя, ако е подходящо, едно от следните указания:
|
Общ азот Водоразтворим калциев оксид за целите, посочени в колона 5 Незадължителен:
|
||||||||||
|
4 |
Разтвор на магнезиев нитрат |
Продукт, получен по химичен път и чрез разтваряне на магнезиев нитрат във вода |
6 % N азот, изразен като нитратен азот. |
|
Нитратен азот Водоразтворим магнезиев оксид |
||||||||||
|
9 % MgO Магнезий изразен като водоразтворим магнезиев оксид. Минимално рН: 4 |
|
||||||||||||||
|
5 |
Суспензия на калциев нитрат |
Продукт, получен чрез суспендиране на калциев нитрат във вода |
8 % N Азот, изразен като общ азот или нитратен и амонячен азот Максимално съдържание на амонячен азот е 1,0 % |
След вида се поставя по подходящ начин едно от следните указания:
|
Общ азот Нитратен азот Водоразтворим калциев оксид, предназначен за използване при условията, посочени в колона 5 |
||||||||||
|
14 % СаО Калцият, изразен като водоразтворим СаО |
|||||||||||||||
|
6 |
Разтвор на азотен тор с карбамид формалдехид |
Продукт, получен по химичен път или чрез разтваряне във вода на карбамид формалдехид и азотен тор от списък А.1 на настоящия регламент, с изключение на видовете 3, а), 3, б) и 5 |
18 % N, изразен като общ азот Най-малко една трета от обявеното съдържание на общ азот трябва да бъде от карбамид формалдехид Максимално съдържание на биурет: (амиден N + N от карбамид формалдехид) × 0,026 |
|
Общ азот За всяка форма в количество най-малко 1 %:
Азот от карбамид формалдехид |
||||||||||
|
7 |
Суспензия от азотен тор с карбамид формалдехид |
Продукт, получен по химичен път или чрез разтваряне във вода на карбамид формалдехид и азотен тор от списък А.1 на настоящия регламент, с изключение на видовете 3, а), 3, б) и 5 |
18 % N, азот, изразен като общ азот Най-малко една трета от обявеното съдържание на азот трябва да произлиза от карбамид формалдехид, от което най-малко три пети трябва да са разтворими в гореща вода Максимално съдържание на биурет: (карбамид N + N от карбамид формалдехид) × 0,026 |
|
Общ азот За всяка форма възлизаща на най-малко 1 %:
Азот от карбамид формалдехид Азот от карбамид формалдехид, който е разтворим в студена вода Азот от карбамид формалдехид, който е разтворим само в гореща вода |
В.2. Сложни течни торове
|
В.2.1. |
Обозначение на вида: |
Разтвор на NPK торове |
|||||
|
Данни за метода на получаване: |
Продукт, получен по химичен път и чрез разтваряне във вода, във форма устойчива при атмосферно налягане, без добавка на органични хранителни вещества от животински или растителен произход. |
||||||
|
Минимално съдържание на хранителни елементи (проценти от масата): |
|
|
Форми, разтворимост и съдържание на хранителни елементи, обявено в колони 4, 5 и 6 Размер на частиците |
Данни за идентификация на торовете; други изисквания |
||||||||||||||||||||||||
|
N |
P2O5 |
K2O |
N |
P2O5 |
K2O |
||||||||||||||||||||
|
1 |
2 |
3 |
4 |
5 |
6 |
||||||||||||||||||||
|
Водоразтворим P2O5 |
Водоразтворим К2О |
|
Водоразтворим P2O5 |
|
||||||||||||||||||||
|
В.2.2. |
Обозначение на вида: |
NPK тор — суспензия |
|||||
|
Данни за метода на получаване: |
Продукт в течна форма, чийто хранителни елементи са получени от субстанции, както суспендирани във вода или разтворени, без добавки на органични хранителни вещества от животински или растителен произход. |
||||||
|
Минимално съдържание на хранителни елементи (% от масата): |
|
|
Форми, разтворимости и съдържание на хранителни елементи, обявено в колони 4, 5 и 6; Размер на частиците |
Данни за идентификация на торовете; Други изисквания |
||||||||||||||||||||||||||||||||||
|
N |
P2O5 |
K2O |
N |
P2O5 |
K2O |
||||||||||||||||||||||||||||||
|
1 |
2 |
3 |
4 |
5 |
6 |
||||||||||||||||||||||||||||||
|
|
Водоразтворим К2О |
|
Торовете не трябва да съдържат томасова шлака, калциеви алумофосфати, калциниран фосфат, частично разтворен фосфорит и мек смлян фосфат
|
|
||||||||||||||||||||||||||||||
|
В.2.3. |
Обозначение на вида: |
Разтвор на NP торове |
|||||
|
Данни за метода на получаване: |
Продукт, получен по химичен път и чрез разтваряне във вода, във форма устойчива на атмосферно налягане, без добавка на органични хранителни вещества от животински или растителен произход. |
||||||
|
Минимално съдържание на хранителни елементи (% от масата): |
|
|
Форми, разтворимости и съдържание на хранителни елементи, обявено в колони 4, 5 и 6; Размер на частиците |
Данни за идентификация на торовете; Други изисквания |
||||||||||||||||||
|
N |
P2O5 |
K2O |
N |
P2O5 |
K2O |
||||||||||||||
|
1 |
2 |
3 |
4 |
5 |
6 |
||||||||||||||
|
(1) Водоразтворим P2O5 |
|
|
Водоразтворим Р2О5 |
|
||||||||||||||
|
В.2.4. |
Обозначение на вида: |
NP тор — суспензия |
|||||
|
Данни за метода на получаване: |
Продукт в течна форма, чиито хранителни елементи са получени от субстанции, както суспендирани във така и разтворени, без добавка на органични хранителни вещества от животински или растителен произход. |
||||||
|
Минимално съдържание на хранителни елементи (проценти от масата): |
|
|
Форми, разтворимост и съдържание на хранителни елементи, обявено в колони 4, 5 и 6 Размер на частиците |
Данни за идентификация на торовете; Други изисквания |
||||||||||||||||||||||||||||
|
N |
P2O5 |
K2O |
N |
P2O5 |
K2O |
||||||||||||||||||||||||
|
1 |
2 |
3 |
4 |
5 |
6 |
||||||||||||||||||||||||
|
|
|
|
Торовете могат да не съдържат томасова шлака, калциеви алумофосфати, калцинирани фосфати, частично разтворен фосфорит и мек смлян фосфат |
|
||||||||||||||||||||||||
|
В.2.5. |
Обозначение на вида: |
NK тор — разтвор |
|||||
|
Данни за метода на получаване: |
Продукт, получен по химичен път и чрез разтваряне във вода във форма устойчива на атмосферно налягане, без добавка на органични хранителни елементи от животински или растителен произход. |
||||||
|
Минимално съдържание на хранителни елементи (% от масата): |
|
|
Форми, разтворимост и съдържание на хранителни елементи, обявено в колони 4, 5 и 6; Размер на частиците |
Данни за идентификация на торовете; Други изисквания |
||||||||||||||||||||||||
|
N |
P2O5 |
K2O |
N |
P2O5 |
K2O |
||||||||||||||||||||
|
1 |
2 |
3 |
4 |
5 |
6 |
||||||||||||||||||||
|
|
Водоразтворим К2О |
|
|
|
||||||||||||||||||||
|
В.2.6. |
Обозначение на вида: |
NK тор — суспензия |
|||||
|
Данни за метода на получаване: |
Продукт в течна форма, чиито хранителни елементи са получени от субстанции, както суспендирани във вода така и разтворени, без добавки на органични хранителни елементи от животински или растителен произход. |
||||||
|
Минимално съдържание на хранителни елементи (% от масата): |
|
|
Форми, разтворимост и съдържание на хранителни елементи, обявено в колони 4, 5 и 6; Размер на частиците |
Данни за идентификация на торовете; Други изисквания |
||||||||||||||||||||||||
|
N |
P2O5 |
K2O |
N |
P2O5 |
K2O |
||||||||||||||||||||
|
1 |
2 |
3 |
4 |
5 |
6 |
||||||||||||||||||||
|
|
Водоразтворим К2О |
|
|
|
||||||||||||||||||||
|
В.2.7. |
Обозначение на вида: |
PK тор — разтвор |
|||
|
Данни за метода на получаване: |
Продукт, получен по химичен път и чрез разтваряне във вода, без добавка на органични хранителни вещества от животински или растителен произход. |
||||
|
Минимално съдържание на хранителни елементи (% от масата): |
|
|
Форми, разтворимост и съдържание на хранителни елементи, обявено в колони 4, 5 и 6; Размер на частиците |
Данни за идентификация на торовете; Други изисквания |
||||||||||
|
N |
P2O5 |
K2O |
N |
P2O5 |
K2O |
||||||
|
1 |
2 |
3 |
4 |
5 |
6 |
||||||
|
|
Водоразтворим P2O5 |
Водоразтворим К2О |
|
Водоразтворим P2O5 |
|
||||||
|
В.2.8. |
Обозначение на вида: |
PK тор — суспензия |
|||
|
Данни за метода на получаване: |
Продукт в течна форма, чиито хранителни елементи са получени от субстанции, както суспендирани във вода така и разтворени без добавки на органични хранителни вещества от животински или растителен произход. |
||||
|
Минимално съдържание на хранителни елементи (проценти от масата) и други изисквания: |
|
|
Форми, разтворимост и съдържание на хранителни елементи, обявено в колони 4, 5 и 6; Размер на частиците |
Данни за идентификация на торовете; Други изисквания |
||||||||||||||||||||
|
N |
P2O5 |
K2O |
N |
P2O5 |
K2O |
||||||||||||||||
|
1 |
2 |
3 |
4 |
5 |
6 |
||||||||||||||||
|
|
|
Водоразтворим К2О |
|
Торовете не трябва да съдържат томасова шлака, калциев алумофосфат, калцинирани фосфати, частично разтворим фосфат или природни фосфати |
|
||||||||||||||||
Г. Неорганични торове с второстепенни хранителни елементи
|
№ |
Обозначение на вида |
Данни за метода на получаване и основни съставки |
Минимално съдържание на хранителни елементи (% от масата) Данни за изразяване на хранителни елементи; Други изисквания |
Други данни за означението на вида |
Съдържание на хранителни елементи; Форми и разтворимост на хранителните елементи; други критерии |
||||
|
1 |
2 |
3 |
4 |
5 |
6 |
||||
|
1 |
Калциев сулфат |
Продукт от естествен или промишлен произход, съдържащ калциев сулфат с различна степен на хидратиране |
25 % СаО 35 % SO3 Калций и сяра, изразени като общо съдържание на СаО + SO3 Степен на смилане:
|
Могат да се добавят обичайни търговски наименования |
Общ серен триоксид Незадължително: общ СаО |
||||
|
2 |
Разтвор на калциев хлорид |
Разтвор на калциев хлорид с промишлен произход |
12 % СаО Калция изразен като водоразтворим СаО |
|
Калциев оксид Незадължително: За листно подхранване |
||||
|
3 |
Елементарна сяра |
Сравнително пречистен естествен или промишлен продукт |
98 % S (245 % SO3) Сярата изразена като SO3 |
|
Общ серен триоксид |
||||
|
4 |
Кизерит |
Продукт, с минерален произход, съдържащ като основна съставка магнезиев сулфат |
24 % MgO 45 % SO3 Магнезият и сярата се изразяват като водоразтворим магнезиев оксид и серен триоксид |
Могат да се добавят обичайните търговски наименования |
Водоразтворим магнезиев оксид Незадължително: водоразтворим серен триоксид |
||||
|
5 |
Магнезиев сулфат |
Продукт, съдържащ като основна съставка магнезиев сулфат хептахидрат |
15 % MgO 28 % SO3 Магнезият и сярата се изразяват като водоразтворими магнезиев оксид и серен триоксид |
Могат да се добавят обичайни търговски наименования |
Водоразтворим магнезиев оксид Незадължително: водоразтворим серен триоксид |
||||
|
5.1 |
Разтвор на магнезиев сулфат |
Продукт, получен чрез разтваряне във вода на магнезиев сулфат с промишлен произход |
5 % MgO 10 % SO3 Магнезият и сярата се изразяват като водоразтворими магнезиев оксид и серен анхидрид |
Могат да се добавят обичайни търговски наименования |
Водоразтворим магнезиев оксид Незадължително: водоразтворим серен анхидрид |
||||
|
5.2 |
Магнезиев хидроксид |
Продукт, получен по химичен път и съдържащ като основна съставка магнезиев хидроксид |
60 % MgO Размер на частици: най-малко 99 % да преминават през сито с размер на отворите 0,063 mm |
|
Общ магнезиев оксид |
||||
|
5.3 |
Суспензия на магнезиев хидроксид |
Продукт, получен чрез суспендиране на тор от вида 5.2 |
24 % MgO |
|
Общ магнезиев оксид |
||||
|
6 |
Разтвор на магнезиев хлорид |
Продукт, получен чрез разтваряне на магнезиев хлорид с промишлен произход |
13 % MgO Магнезий се изразява като магнезиев оксид Максимално съдържание на калций: 3 % СаО |
|
Магнезиев оксид |
Д. Неорганични торове с микроелементи
Обяснителна бележка: Следните бележки са приложими за цялата част Д.
Бележка 1: Хелатообразуващ елемент може да бъзе обозначен с помощта на своите инициали, както е изложено в Д.3.
Бележка 2: Ако продуктът не оставя твърда утайка след разтваряне във вода, той може да бъде описан като „за разтваряне“.
Бележка 3: Когато определен микроелемент е в хелатирана форма, трябва да се заяви диапазона на рН, който гарантира приемлива стабилност на хелатираната фракция.
Д.1. Торове, съдържащи само един микроелемент
Д.1.1. Бор
|
№ |
Обозначение на вида |
Данни за метода на получаване и основни съставки |
Минимално съдържание на хранителни елементи (% от масата), изразяване на хранителни елементи; Други изисквания |
Други данни за означението на вида |
Съдържание на хранителни елементи за обявяване; Форми и разтворимости на хранителните елементи; други критерии |
|
1 |
2 |
3 |
4 |
5 |
6 |
|
1а |
Борна киселина |
Продукт, получен чрез взаимодействие на киселина с борат |
14 % водоразтворим Б |
Може да се добавят обичайните търговски наименования |
Водоразтворим бор (Б) |
|
1б |
Натриев борат |
Продукт, получен по химичен път, съдържащ като основна съставка натриев борат |
10 % водоразтворим Б |
Може да се добавят обичайните търговски наименования |
Водоразтворим бор (Б) |
|
1в |
Калциев борат |
Продукт, получен от колеманит или пандермит, съдържащ като свой основен компонент калциеви борати |
7 % общ Б Размер на частиците: най-малко 98 % да преминават през сито с размер на отворите 0,063 mm. |
Може да се добавят обичайните търговски наименования |
Общ бор (Б) |
|
1г |
Боров етанол амин |
Продукт, получен при реакция на борна киселина с етанол амин |
8 % водоразтворим В |
|
Водоразтворим бор (В) |
|
1д |
Разтвор на тор, съдържащ бор |
Продукт, получен при разтваряне на видове 1а и/или 1б и/или 1г |
2 % водоразтворим В |
Обозначението трябва да включва наименованието на бор съдържащите съставки |
Водоразтворим бор (В) |
|
1е |
Тор, съдържащ бор в суспензия |
Продукт, получен чрез суспендиране на видове 1а и/или 1б и/или 1г във вода |
2 % водоразтворим В |
Обозначението трябва да включва наименованието на бор съдържащите съставки |
Водоразтворим бор (В) |
Д.1.2. Кобалт
|
№ |
Обозначение на вида |
Данни за метода на получаване и основни съставки |
Минимално съдържание на хранителни елементи (% от масата); изразяване на хранителни елементи; други изисквания |
Други данни за означението на вида |
Съдържание на хранителни елементи за обявяване Форми и разтворимости на хранителните елементи други критерии |
||||
|
1 |
2 |
3 |
4 |
5 |
6 |
||||
|
2а |
Кобалтова сол |
Продукт, получен по химичен път и съдържащ неорганична кобалтова сол като основна съставка |
19 % водоразтворим Со |
Обозначението трябва да включва наименованието на неорганичния анион |
Водоразтворим кобалт (Со) |
||||
|
2б |
Кобалтов хелат |
Водоразтворим продукт, получен чрез химично свързване на кобалт с хелатиращ агент |
2 % водоразтворим Со, най-малко 8/10 от обявяваната стойност да е хелатирана |
Наименование на хелатиращия агент |
Водоразтворим кобалт (Со) Хелатиран кобалт (Со) |
||||
|
2в |
Разтвор на тор, съдържащ кобалт |
Продукт, получен при разтваряне във вода на видове 2а и/или 2б |
2 % водоразтворим Со |
Означението трябва да включва:
|
Водоразтворим кобалт (Со) Хелатиран кобалт (Со), ако присъства |
Д.1.3. Мед
|
№ |
Обозначение на вида |
Данни за метода на получаване и основни съставки |
Минимално съдържание на хранителни елементи (% от масата); изразяване на хранителни елементи; други изисквания |
Други данни за означението на вида |
Съдържание на хранителни елементи за обявяване Форми и разтворимости на хранителните елементи други критерии |
||||
|
1 |
2 |
3 |
4 |
5 |
6 |
||||
|
3а |
Медна сол |
Продукт, получен по химичен път, съдържащ неорганична сол като основна съставка |
20 % водоразтворима Cu |
Означението трябва да включва наименованието на неорганичния анион |
Водоразтворима мед (Cu) |
||||
|
3б |
Меден оксид |
Продукт, получен по химичен път съдържащ меден оксид като основна съставка |
70 % обща Cu Размер на частиците: Най-малко 98 % да преминават през сито с размер 0,063 mm. |
|
Обща мед (Cu) |
||||
|
3в |
Меден хидроксид |
Продукт, получен по химичен път, съдържащ меден хидроксид като основна съставка |
45 % общ Cu Размер на частиците: най-малко 98 % да преминават през сито с размер 0,063 mm |
|
Обща мед (Cu) |
||||
|
3г |
Меден хелат |
Водоразтворим продукт, получен при химично свързване на мед с хелатиращ агент |
9 % водоразтворима Cu, най-малко 8/10 от обявяваната стойност, на който е била хелатирана |
Наименование на хелатиращия агент |
Водоразтворима мед (Cu) Хелатирана мед(Cu) |
||||
|
3д |
Тор, съдържащ мед |
Продукт, получен чрез смесване на видове 3а и/или 3б и/или 3в и/или 3г и пълнител, който не е хранителна съставка, и не е токсичен |
5 % обща Cu |
Означението трябва да включва:
|
Обща мед (Cu) Водноразтворима мед (Cu), са в количество най-малко ¼ от общата мед Хелатирана мед (Cu), ако присъства |
||||
|
3е |
Разтвор на тор, съдържащ мед |
Продукт, получен чрез разтваряне на видове 3а и/или един от вид 3г във вода |
3 % водоразтворима Cu |
Означението трябва да включва:
|
Водоразтворима мед (Cu) Хелатирана мед (Cu), ако присъства |
||||
|
3ж |
Меден оксихлорид |
Продукт, получен по химичен път, съдържащ меден оксихлорид [Cu2Cl(OH)3] като основна съставка |
50 % обща Cu Размер на частиците: най-малко 98 % да преминават през сито с размер 0,063 mm |
|
Обща мед (Cu) |
||||
|
3з |
Суспензия на меден оксихлорид |
Продукт, получен чрез суспендиране на вид 3ж |
17 % обща Cu |
|
Обща мед (Cu) |
Д.1.4. Желязо
|
№ |
Обозначение на вида |
Данни за метода на получаване и основни съставки |
Минимално съдържание на хранителни елементи (процент от масата) Данни за изразяване на хранителни елементи Други изисквания |
Други данни за означението на вида |
Съдържание на хранителни елементи за обявяване Форми и разтворимости на хранителните елементи други критерии |
||||||
|
1 |
2 |
3 |
4 |
5 |
6 |
||||||
|
4а |
Желязна сол |
Продукт, получен по химичен път, съдържащ желязна сол като основна съставка |
12 % водоразтворимо желязо |
Означението трябва да включва наименованието на неорганичния анион |
Водоразтворимо желязо (Fe) |
||||||
|
4б |
Железен хелат |
Водоразтворим продукт, получен при химично свързване на желязо с хелатиращ агент, посочен в списъка на част Д.3 от приложение I. |
5 % водоразтворимо желязо, най-малко 80 % от обявяваното съдържание да е хелатирано |
Наименование на хелатиращия агент |
|
||||||
|
4в |
Разтвор на тор, съдържащ желязо |
Продукт, получен при разтваряне на видове 4а и/или един от вид 4б във вода |
2 % водоразтворимо желязо |
Означението трябва да включва:
|
Водоразтворимо желязо (Fe) Хелатирано желязо (Fe), ако присъства |
Д.1.5. Манган
|
№ |
Обозначение на вида |
Данни за метода на получаване и основни съставки |
Минимално съдържание на хранителни елементи (% от масата); изразяване на хранителни елементи; други изисквания |
Други данни за означението на вида |
Съдържание на хранителни елементи; форми и разтворимости на хранителните елементи; други критерии |
||||
|
1 |
2 |
3 |
4 |
5 |
6 |
||||
|
5а |
Манганова сол |
Продукт, получен по химичен път, съдържащ неорганична сол на мангана (Mn) като основна съставка |
17 % водоразтворим Mn |
Означението трябва да включва наименованието на свързания анион |
Водоразтворим манган (Mn) |
||||
|
5б |
Манганов хелат |
Водоразтворим продукт, получен чрез химично свързване на манган с хелатиращ агент |
5 % водоразтворим Mn, най-малко 8/10 от обявяваното съдържание да е хелатирано |
Наименование на хелатиращия агент |
Водоразтворим кобалт (Mn) Хелатиран кобалт (Mn) |
||||
|
5в |
Манганов оксид |
Продукт, получен по химичен път, съдържащ манганови оксиди като основни съставки |
40 % общ Mn Размер на частиците: най-малко 80 % да преминават през сито с отвори 0,063 mm |
|
Общ манган (Mn) |
||||
|
5г |
Тор, съдържащ манган |
Продукт, получен при смесване на видове 5а и 5в |
17 % общ Mn |
Означението трябва да включва наименованието на манган съдържащите съставки |
Общ манган (Mn) Водоразтворим манган (Mn), ако е най-малко ¼ от общия манган |
||||
|
5д |
Разтвор на тор, съдържащ манган |
Продукт, получен чрез разтваряне на видове 5а и/или 5б във вода |
3 % водоразтворим Mn |
Означението трябва да съдържа:
|
Водоразтворим манган (Mn) Хелатиран манган (Mn), ако присъства |
Д.1.6. Молибден
|
№ |
Обозначение на вида |
Данни за метода на получаване и основни съставки |
Минимално съдържание на хранителни елементи (процент от масата) Данни за изразяване на хранителни елементи Други изисквания |
Други данни за означението на вида |
Съдържание на хранителни елементи за обявяване Форми и разтворимости на хранителните елементи други критерии |
|
1 |
2 |
3 |
4 |
5 |
6 |
|
6а |
Натриев молибдат |
Продукт, получен по химичен път, съдържащ натриев молибдат като основна съставка |
35 % водоразтворим Мо |
|
Водоразтворим молибден (Мо) |
|
6б |
Амониев молибдат |
Продукт, получен по химичен път, съдържащ амониев молибдат като основна съставка |
50 % водоразтворим Мо |
|
Водоразтворим молибден (Мо) |
|
6в |
Тор, съдържащ молибден |
Продукт, получен при смесване на видове 6а и 6б |
35 % водоразтворим Мо |
Означението трябва да съдържа наименованията на молибденовите компоненти |
Водоразтворим молибден (Мо) |
|
6г |
Разтвор на тор, съдържащ молибден |
Продукт, получен чрез разтваряне на видове 6а и/или 6б във вода |
3 % водоразтворим Мо |
Означението трябва да съдържа наименованията на молибденовите компоненти |
Водоразтворим молибден (Мо) |
Д.1.7. Цинк
|
№ |
Обозначение на вида |
Данни за метода на получаване и основни съставки |
Минимално съдържание на хранителни елементи (процент от масата) Данни за изразяване на хранителни елементи Други изисквания |
Други данни за означението на вида |
Хранително съдържание за обявяване Форми и разтворимости на хранителните елементи други критерии |
||||
|
1 |
2 |
3 |
4 |
5 |
6 |
||||
|
7а |
Цинкова сол |
Продукт, получен по химичен път, съдържащ неорганична сол на цинк като основна съставка |
15 % водоразтворим Zn |
Означението трябва да включва наименованието на неорганичния анион |
Водоразтворим цинк (Zn) |
||||
|
7б |
Цинков хелат |
Водоразтворим продукт, получен чрез химично свързване на цинк с хелатиращ агент |
5 % водоразтворим Zn, най-малко 8/10 от обявеното съдържание да е хелатирано |
Наименование на хелатиращия агент |
Водоразтворим цинк (Zn) Хелатиран цинк (Zn) |
||||
|
7в |
Цинков оксид |
Продукт, получен по химичен път, съдържащ цинков оксид като основна съставка |
70 % общ Zn Размер на частиците: най-малко 80 % преминават през сито с размер 0,063 mm |
|
Общ цинк (Zn) |
||||
|
7г |
Тор, съдържащ цинк |
Продукт, получен при смесване на видове 7а и 7в |
30 % общ Zn |
Наименованието трябва да включва наименованието на цинковите компоненти |
Общ цинк (Zn) Водоразтворим цинк (Zn), ако е най-малко ¼ от общия цинк |
||||
|
7д |
Разтвор на тор, съдържащ цинк |
Продукт, получен чрез разтваряне на видове 7а и/или 7б във вода |
3 % водоразтворим Zn |
Означението трябва да включва:
|
Водоразтворим цинк (Zn) Хелатиран цинк (Zn), ако присъства |
Д.2. Минимално съдържание на микроелементи, процент от масата на торовете
Д.2.1. Твърди или течни смеси на микроелементи
|
|
Когато микроелементът присъства под следната форма |
|
|
изцяло в неорганична форма |
като хелат или комплексно съединение |
|
|
За хранителен микроелемент: |
||
|
Бор (В) |
0,2 |
0,2 |
|
Кобалт (Со) |
0,02 |
0,02 |
|
Мед (Cu) |
0,5 |
0,1 |
|
Желязо (Fe) |
2,0 |
0,3 |
|
Манган (Mn) |
0,5 |
0,1 |
|
Молибден (Мо) |
0,02 |
— |
|
Цинк (Zn) |
0,5 |
0,1 |
Минимално общо съдържание на микроелементи в твърда смес: 5 % от масата на торовете.
Минимално общо съдържание на микроелементи в течна смес: 2 % от масата на торовете.
Д.2.2. ЕО торове, съдържащи основни и/или второстепенни хранителни елементи с микроелементи за почвено торене
|
Микроелемент |
За посеви и тревни култури |
За градинарски цели |
|
Бор (В) |
0,01 |
0,01 |
|
Кобалт (Со) |
0,002 |
— |
|
Мед (Cu) |
0,01 |
0,002 |
|
Желязо (Fe) |
0,5 |
0,02 |
|
Манган (Mn) |
0,1 |
0,01 |
|
Молибден (Мо) |
0,001 |
0,001 |
|
Цинк (Zn) |
0,01 |
0,002 |
Д.2.3. ЕО торове, съдържащи основни и/или второстепенни хранителни елементи с микроелементи за листно подхранване
|
Бор (В) |
0,010 |
|
Кобалт (Со) |
0,002 |
|
Мед (Cu) |
0,002 |
|
Желязо (Fe) |
0,020 |
|
Манган (Mn) |
0,010 |
|
Молибден (Мо) |
0,001 |
|
Цинк (Zn) |
0,002 |
Д.3. Списък на разрешени хелатообразуващи и комплексообразуващи елементи
Следните продукти са разрешени при условие че отговарят на изискванията на Директива 67/548/ЕИО (2), съответно изменена.
Д.3.1. Хелатиращи агенти (3)
Натриева, калиева или амониева киселина или соли на:
|
Етилен диамин тетраоцетна киселина |
EDTA |
C10H16O8N2 |
|
Диетилен триамин пентаоцетна киселина |
DTPA |
C14H23O10N3 |
|
[o,o]: етилендиамин-ди (о-хидроксфенил оцетна) киселина |
EDDHA |
C18H20O6N2 |
|
[о,р]: етилен диамин-N-(о-хидроксифенил оцетна) киселина |
EDDHA |
C18H20O6N2 |
|
хидрокси-2 етилетилен диамин триоцетна киселина |
HEEDTA |
C10H18O7N2 |
|
[о,о]: етилен диамин-ди (о-хидрокси-о-метилфенил оцетна киселина |
EDDHMA |
C20H24O6N2 |
|
[о,р]: етилен диамин-ди (о-хидрокси-р-метил фенил оцетна киселина |
EDDHMA |
C20H24O6N2 |
|
[р,о]: етилен диамин-ди (р-хидрокси-о-метил фенил оцетна киселина |
EDDHMA |
C20H24O6N2 |
|
[2,4]: етилендиамин ди-(2-хидрокси-4-метил фенил оцетна киселина |
EDDCHA |
C20H20O10N2 |
|
[2,5]: етилендиамин ди-(2-хидрокси-5-метил фенил оцетна киселина |
EDDCHA |
C20H20O10N2 |
|
[5,2]: етилендиамин ди-(5-хидрокси-2-метил фенил оцетна киселина |
EDDCHA |
C20H20O10N2 |
Д.3.2. Комплексообразуващи агенти:
Списъкът трябва да се изготви.
(1) Към всяка опаковка или насипна доставка трябва да се прилага възможно най-пълна техническа информация от лицето, отговорно за пускането на продукта на пазара. Тази информация трябва да дава възможност на потребителя да определя нормите и времето за торене в зависимост от отглежданата култура.
(2) ОВ L 196, 16.8.1967 г., стр. 1.
(3) Хелатиращите агенти трябва да идентифицират и определят количествено съгласно Европейски стандарт EN 13368, част 1 и част 2, доколкото този стандарт обхваща горните агенти.
ПРИЛОЖЕНИЕ II
ДОПУСТИМИ ОТКЛОНЕНИЯ
Допустимите отклонения, дадени в настоящото приложение, са отрицателни стойности в процент от масата.
Допустимите отклонения, разрешени по отношение на обявяваните съдържания на хранителни елементи в различните видове ЕО торове, са както следва:
1. Абсолютни стойности на неорганични еднокомпонентни торове с основни хранителни елементи в проценти от масата, представени като N, P2O5, K2O, MgO, Cl
1.1. Азотни торове
|
Калциев нитрат |
0,4 |
||
|
Калциево-магнезиев нитрат |
0,4 |
||
|
Натриев нитрат |
0,4 |
||
|
Чилска селитра |
0,4 |
||
|
Калциев цианамид |
1,0 |
||
|
Нитрифициран калциев цианамид |
1,0 |
||
|
Амониев сулфат |
0,3 |
||
|
Амониев нитрат или калциево-амониев нитрат: |
|||
|
0,8 |
||
|
0,6 |
||
|
Амониев сулфат-нитрат |
0,8 |
||
|
Магнезиев сулфонитрат |
0,8 |
||
|
Магнезиево-амониев нитрат |
0,8 |
||
|
Карбамид |
0,4 |
||
|
Суспензия на калциев нитрат |
0,4 |
||
|
Разтвор на азотен тор с карбамид формалдехид |
0,4 |
||
|
Суспензия на азотен тор с карбамид |
0,4 |
||
|
Карбамид-амониев сулфат |
0,5 |
||
|
Разтвор на азотен тор |
0,6 |
||
|
Разтвор на карбамид и амониев нитрат |
0,6 |
||
1.2. Фосфорни торове
|
Томасова шлака: |
|||
|
0,0 |
||
|
1,0 |
||
Други фосфорни торове
|
Р2О5 разтворим в: |
(№ на тора в приложение I) |
|
||
|
(3, 6, 7) |
0,8 |
||
|
(7) |
0,8 |
||
|
(2а, 2б, 2в) |
0,8 |
||
|
4, 5, 6) |
0,8 |
||
|
(2а, 2б, 3) |
0,9 |
||
|
(2в) |
1,3 |
1.3. Калиеви торове
|
Каинит |
1,5 |
||
|
Обогатена каинитова сол |
1,0 |
||
|
Калиев хлорид: |
|||
|
1,0 |
||
|
0,5 |
||
|
Калиев хлорид, съдържащ магнезиева сол |
1,5 |
||
|
Калиев сулфат |
0,5 |
||
|
Калиев сулфат, съдържащ магнезиева сол |
1,5 |
||
1.4. Други компоненти
|
хлорид |
0,2 |
2. Неорганични сложни торове с основни хранителни елементи
2.1. Хранителни елементи
|
N |
1,1 |
|
P2O5 |
1,1 |
|
K2O |
1,1 |
2.2. Общо отрицателно отклонение от обявяваната стойност
|
двойни торове |
1,5 |
|
тройни торове |
1,9 |
3. Второстепенни хранителни елементи в торове
Допустимите отклонения, позволени по отношение на обявеното съдържание на калций, магнезий, натрий и сяра, трябва да бъдат една четвърт от обявяваните съдържания на тези хранителни елементи до максимум 0,9 % в абсолютно отношение на СаО, MgO, Na2O и SO3, т.е. 0,64 за Ca, 0,55 за Mg, 0,67 за Na и 0,36 за S.
4. Хранителни микроелементи в торовете
Допустимото отклонение, разрешено по отношение на обявеното съдържание на хранителните микроелементи, е:
|
— |
0,4 % в абсолютни стойности за съдържание повече от 2 %, |
|
— |
една пета от обявяваната стойност за съдържание не повече 2 %. |
Допустимото отклонение, разрешено по отношение на обявената стойност за различните форми на азот или обявявана разтворимост на фосфорен пентаоксид е една пета от цялото съдържание на въпросния хранителен елемент с не повече от 2 % от масата, при условие че цялото съдържание на този хранителен елемент остава в рамките на границите, определени в приложение I и допустимите отклонения, определени по-горе.
ПРИЛОЖЕНИЕ III
ТЕХНИЧЕСКИ ИЗИСКВАНИЯ ЗА АМОНИЕВО-НИТРАТНИ ТОРОВЕ С ВИСОКО СЪДЪРЖАНИЕ НА АЗОТ
1. Характеристики и пределно допустими стойности за еднокомпонентни амониево-нитратни торове с високо съдържание на азот
1.1. Порьозност (маслопоглъщаемост)
Маслопоглъщаемостта на тор, който трябва да е преминал през два термични цикъла с температура в диапазона от 25 до 50 °С и да отговаря на условията на част 2 на раздел 3 от настоящото приложение, не трябва да надвишава 4 % по маса.
1.2. Горими съставки
Процентът от масата на горимите съставки, изразени като въглерод, не трябва да превишава 0,2 % за торове, които имат съдържание на азот най-малко 31,5 % по ма са и не трябва да превишават 0,4 за торове със съдържание на азот най-малко 28 %, но по-малко от 31,5 %.
1.3. рН
Разтвор на 10 g тор в 100 ml вода трябва да има рН най-малко 4,5.
1.4. Размер на частиците
Не повече от 5 % от масата на тора трябва да преминава през сито с размер на отворите 1 mm и не повече от 3 mm от масата на тора трябва да преминават през сито с размер на отворите 0,5 mm.
1.5. Хлор
Максималното съдържание на хлор е определено на 0,02 % по маса.
1.6. Тежки метали
Тежки метали не следва да се добавят умишлено и всички следи, които са случайни за производствения процес, не трябва да превишават границите, фиксирани от Комитета.
Съдържанието на мед не трябва да бъде по-високо от 10 mg/kg.
Не са определени граници за други тежки метали.
2. Описание на изпитването за устойчивост на детонация на амониево-нитратни торове с високо съдържание на азот
Изпитването трябва да се извършва с представителна проба тор. Преди да се изпита за устойчивост на детонация, цялата маса на пробата трябва да бъде подложена на пет термични цикъла, отговарящи на условията на част 3 от раздел 3 на настоящото приложение.
Торът трябва да бъде подложен на изпитване на детонация в хоризонтална стоманена тръба при следните условия:
|
— |
безшевна стоманена тръба, |
|
— |
дължина на тръбата: най-малко 1 000 mm, |
|
— |
номинален външен диаметър: най-малко 114 mm, |
|
— |
номинална дебелина на стената най-малко 5 mm, |
|
— |
възпламенител: типът и масата на избрания възпламенител трябва да бъдат такива, че да максимално да се увеличи детонационното налягане, приложено на пробата, за да се определи неговата чувствителност на предаването на детонация, |
|
— |
температура на изпитване: 15—25 °С, |
|
— |
оловни цилиндри за индикиране на детонации: диаметър 50 mm и височина 100 mm, |
|
— |
поставени на интервали 150 mm и поддържащи тръбата в хоризонтално положение. Изпитването трябва да се извърши два пъти. Изпитването се счита за завършено, ако при двете изпитвания един или повече от поддържащите оловни цилиндри е разрушен по-малко от 5 %. |
3. Методи за проверка на съответствието на границите, определени в приложения III-1 и III-2
Метод 1
Методи за прилагане на термични цикли
1. Обект и област на приложение
Настоящият документ определя процедурите за прилагането на термични цикли преди извършването на изпитване на маслопоглъщаемост за амониево-нитратни торове с високо съдържание на азот и изпитване на устойчивост за еднокомпонентни и сложни амониево-нитратни торове с високо съдържание на азот.
Счита се, че методите на затворените термични цикли, както са описани в този раздел, симулират в достатъчна степен условията, предвидени в обхвата на прилагането на дял II, глава IV. Независимо от това тези методи могат да не симулират в достатъчна степен всички условия, възникнали при транспортиране и съхранение.
2. Термични цикли, посочени в приложение III, точка 1
2.1. Област на приложение
Настоящата процедура е за осъществяване на термични цикли преди определяне на маслопоглъщаемостта на торове.
2.2. Принцип и определения
В ерленмайерова колба се нагрява пробата от температура на околната среда до 50 °С и се поддържа тази температура в продължение на два часа (фаза при 50 °С). След това се охлажда пробата до достигане на температура 25 °С и се поддържа тази температура в продължение на два часа (фаза при 25 °С) Двете последователни фази при 50 °С и 25 °С образуват един термичен цикъл. След подлагане на два термични цикли, пробата за изпитване се държи при температура 20 ± 3 °С за определяне на стойността на задържане на масла.
2.3. Апаратура
Обичайна лабораторна апаратура, в частност:
|
— |
водни бани, термостатирани на 25 (±1) и 50 (±1) °С съответно, |
|
— |
ерленмайерови колби с обем 150 ml. |
2.4. Процедура
Поставя се всяка проба от 70 (±5) g в ерленмайерова колба, която след това се затваря със запушалка.
На всеки два часа колбата се премества от банята 50 °С в банята 25 °С и обратно.
Поддържа се постоянна температура на водата във всяка баня и в движение чрез бързо разбъркване, за да се осигури водното ниво да е над нивото на пробата. Запушалката се предпазва от кондензация с калпаче от порест каучук.
3. Термични цикли, които се използват за приложение III, точка 2
3.1. Област на приложение
Тази процедура се отнася за осъществяване на термични цикли преди извършване на тест за устойчивост на детонации.
3.2. Принцип и определение
Пробата се нагрява в херметична камера от температура на околната среда до 5 °С и тази температура се поддържа в продължение на един час (фаза при 50 °С). След това пробата се охлажда до достигане на температура 25 °С и се поддържа тази температура в продължение на един час (фаза при 25 °С). Двете последователни фази при 50 °С и 25 °С образуват един термичен цикъл. След подлагане на два термични цикъла, пробата за изпитване се поддържа при температура 20 ± 3 °С до изпитването на детонации.
3.3. Апаратура
|
— |
водна баня, термостатирана в температурния диапазон 20 до 51 °С с минимална скорост на нагряване и охлаждане 10 °С/h или две водни бани, една термостатирана на температура 20 °С, а другата на 51 °С. Водата във ваните се разбърква постоянно. Обемът на ваните следва да бъде достатъчно голям, за да гарантира свободната циркулация на водата. |
|
— |
камера от неръждаема стомана, изцяло херметизирана и снабдена с термодвойка в центъра. Външната ширина на кутията е 45 (±2) mm и дебелината на стената е 1,5 mm (виж фигура 1). Височината и дължината на камерата могат да се изберат така че да подхождат на размерите на водната баня, например дължина 600 mm, височина 400 mm. |
3.4. Процедура
Поставя се количество тор, достатъчно за единична детонация в камерата и се затваря капакът. Поставя се кутията във водната баня. Нагрява сее водата до 51 °С и се измерва температурата в центъра на пробата. Един час след като температурата в центъра достигне 50 °С водата се охлажда. Един час след като температурата в центъра достигне 25 °С водата се нагрява, за да започне вторият цикъл. При две бани камерата се прехвърля в другата баня след всеки период на нагряване/охлаждане.
Фигура 1

Метод 2
Определяне на маслопоглъщаемостта
1. Обект и област на приложение
Настоящият документ дефинира процедурата за определяне на маслопоглъщаемостта на еднокомпонентни амониево-нитратни торове с високо съдържание на азот.
Този метод е приложим за гранулирани торове, които не съдържат маслоразтворими съставки.
2. Дефиниция
Маслопоглъщаемост на тор: количеството масло, задържано от тора, установено при определени работни условия и изразено като процент от масата.
3. Принцип
Пълно потапяне на пробата за изпитване в газьол за определен период, последвано от оттичане на излишъка при определени условия. Измерване на увеличението на масата на изпитваната проба.
4. Реактив
Газьол
Вискозитет: макс. 5 mPas при 40 °С
Плътност: 0,8 до 0,85 g/ml при 20 °С
Съдържание на сяра: ≤ 1,0 % (m/m)
Пепел: ≤ 0,1 (m/m)
5. Апаратура
Обичайната лабораторна апаратура и:
|
5.1. |
Везни с точност 0,01 g. |
|
5.2. |
Бехерови чаши с обем 500 ml. |
|
5.3. |
Фуния от пластмасов материал, за предпочитане с цилиндрична форма в горния край, диаметър около 200 mm |
|
5.4. |
Лабораторно сито, отвори 0,5 mm, съответстващо на фунията (5.3). Бележка:Размерът на фунията и ситото е такъв, че да осигури натрупване само на няколко слоя гранули да лежат един върху друг и маслото да може лесно да се отцежда. |
|
5.5. |
Филтърна хартия, клас за бързо филтриране, нагъната, мека, маса 150 g/m2. |
|
5.6. |
Абсорбираща тъкан (лабораторно качество). |
6. Процедура
6.1. Извършват се две индивидуални определяния в бърза последователност на отделни порции от една и съща проба за изпитване.
6.2. Отстраняват се частиците, по-малки от 0,5 mm, като се използва ситото (5.4). Претегля се с точност до 0,01 g приблизително 50 g от пробата в бехеровата чаша (5.2). Добавя се достатъчно газьол (раздел 4), за да покрие напълно парчетата и се разбърква внимателно, за да се осигури пълно омокряне на повърхностите на всички частици. Покрива се бехеровата чаша с часовниково стъкло и се оставя да престои един час при 25 (±2) °С.
6.3. Филтрира се цялото съдържание на бехеровата чаша през фунията (5.3) с лабораторното сито (5.4). Оставя се частта, задържана върху ситото, да престои един час, така че да може да се оттече излишният газьол.
6.4. Поставят се два листа филтърна хартия (5.5) (около 500 × 500 mm) един върху друг на гладка повърхност. Сгъват се четирите ръба на двете филтърни хартии нагоре до ширина около 40 mm, за да се предотврати изпадането на гранулите. Поставят се два пласта абсорбираща тъкан (5.6) в центъра на филтърната хартия. Изсипва се цялото съдържание на ситото (5.4) върху абсорбиращите тъкани и гранулите се разпределят равномерно с мека плоска четка. След две минути се повдига едната страна на тъканите, за да се прехвърлят парчетата върху филтърните хартии отдолу и се разпределят равномерно върху тях с четката. Поставя се друг лист филтърна хартия, с ръбове обърнати нагоре по подобен начин, върху пробата и гранулите се търкалят между филтърните хартии с кръгови движения, като се оказва лек натиск. След всеки осем кръгови движения се прави пауза, за да се повдигнат противоположните ръбове на филтърните хартии и да се върнат в центъра гранулите, които са се търкулнали по периферията. Спазва се следната процедура: правят се четири пълни кръгови движения, отначало по посока на часовниковата стрелка, след това обратно. След това се частиците се търкулват обратно в центъра, както е описано по-горе. Тази процедура трябва да се извърши три пъти (24 кръгови движения, два пъти повдигане на ръбовете). Внимателно се вмъква нов лист филтърна хартия между долния и горния лист и се оставят парчетата да се изтъркалят върху новия лист чрез повдигане на ръбовете на горния лист. Покриват се парчетата с нов лист филтърна хартия и се повтаря процедурата, както е описана по-горе. Веднага след търкалянето парчетата се изсипват в тарирана тарелка и се претеглят отново с точност до 0,01 g, за да се определи масата на задържаното количество газьол.
6.5. Повтаряне на процедурата на прехвърляне и повторно измерване
Ако се установи, че количеството газьол, задържаното в порцията, е по-голямо от 2 g, пробата се поставя върху нов комплект филтърни хартии и процедурата на търкаляне се повтаря, като се повдигат ъглите в съответствие с раздел 6.4 (два пъти осем кръгови движения, едно повдигане). След това пробата отново се претегля.
7. Изразяване на резултатите
7.1. Метод на изчисление и формула
Задържането на масло от всяко определяне (6.1), изразено като процент от масата на пресятата проба за изпитване, се дава с уравнението:

където:
|
|
m1 е масата, в грамове, на пресятата част от пробата за изпитване (6.2), |
|
|
m2 е масата, в грамове, на пробата за изпитване съгласно раздел 6.4 или 6.5 съответно като резултат на последното измерване. |
Като резултат се взема средноаритметичното от двете отделни определяния.
Метод 3
Определяне на горими съставки
1. Обект и област на приложение
Настоящият документ дефинира процедурата за определяне на горимото съдържание на едносъставни амониево-нитратни торове с високо съдържание на азот.
2. Принцип
Въглеродният диоксид, произвеждан от неорганични пълнители, се отстранява предварително с киселина. Органичните съединения се окисляват с помощта на смес на хромна и сярна киселина. Образуваният въглероден диоксид се абсорбира в разтвор на бариев хидроксид. Утайката се разтваря в разтвор на солна киселина и се измерва чрез остатъчно титруване с разтвор на натриев хидроксид.
3. Реактиви
3.1. Хромов (VI) триоксид Cr2O3 аналитично качество;
3.2. Сярна киселина, 60 % обемно: наливат се 360 ml вода в бехерова чаша един литър и внимателно се добавят 640 ml сярна киселина (плътност при 20 °С = 1,83 g/ml);
3.3. Сребърен нитрат: 0,1 mol/l разтвор;
3.4. Бариев хидроксид
Претеглят се 15 g бариев хидроксид [Ba(OH)2.8H2O] и се разтварят напълно в гореща вода. Оставя се да се охлади и се прехвърля в колба един литър. Допълва се до отметката и се смесва. Филтрира се през нагъната филтърна хартия.
3.5. Солна киселина: 0,1 mol/l стандартен разтвор;
3.6. Натриев хидроксид: 0,1 mol/l стандартен разтвор;
3.7. Бромфенолово синьо: разтвор на 0,4 g във вода;
3.8. Фенолфталеин: разтвор на два g на литър в 60 % обемни етанол;
3.9. Натронкалк: размер на частиците около 1,0 до 1,5 mm;
3.10. Деминерализирана вода, прясно прекипяна, за отстраняване на въглеродния диоксид
4. Апаратура
4.1. Обичайно лабораторно оборудване, по-специално:
|
— |
филтърен тигел с плоча от синтеровано стъкло и обем 15 ml; диаметър на плочата: 20 mm; обща височина 50 mm; порьозност 4 (диаметър на порите от 5 до 15 mm), |
|
— |
бехерова чаша 600 ml. |
4.2. Подаване на азот под налягане.
4.3. Апарат, изработен от следните части и сглобен, ако е възможно, с помощта на сферични шлифове (виж фигура 2).
|
4.3.1. |
Абсорбционна колонка (А), дълга около 200 ml и с диаметър 30 mm пълна с натронкалк (3.9), задържан в тръбата със стъклена вата. |
|
4.3.2. |
Реакционна колба (В) със странично разклонение и кръгло дъно. |
|
4.3.3. |
Фракционна колона на Вигрьо, дълга около 150 mm (С′). |
|
4.3.4. |
Кондензатор с две повърхности (С), дълъг 200 mm. |
|
4.3.5. |
Бутилка на Дрексел (D), действаща като уловител на излишната киселина при дестилацията. |
|
4.3.6. |
Ледена баня (Е) за охлаждане на бутилката на Дрексел |
|
4.3.7. |
Два абсорбционни съда (F1) и (F2), 32 до 35 mm в диаметър, газовият разпределител, но които се състои от 10 mm диск от синтеровано стъкло с ниска порьозност. |
|
4.3.8. |
Вакуумпомпа и устройство (G), регулиращо засмукването и представляващо Т-образна стъклена тръбичка, свързана с апаратурата, като свободният край е съединен с фина капилярна тръбичка чрез къс каучуков маркуч и щипка. Внимание:Използването на разтвор от кипяща хромна киселина в апаратурата, намираща се под вакуум, е опасна операция и изисква подходящи предпазни мерки. |
5. Процедура
5.1. Проба за изпитване
Претеглят се около 10 g амониев нитрат с точност до 0,001 g.
5.2. Отстраняване на карбонатите
Пробата за изпитване се поставя в реакционната колба (В). Добавят се 100 ml сярна киселина (3.2). Гранулите се разтварят за около 10 минути при температура на околната среда. Сглобява се апаратурата, както е показано на диаграмата: свързва се единият край на абсорбционната тръба (А) към източника на азот (4.2) посредством обратен клапан, съдържащи еквивалент на налягане 5 до 6 mm живак, и другия край към захранващата тръбичка, която влиза в реакционната колба. Поставя се фракционна колона на Вигрьо (С′) и кондензаторът (С) на място с подаване на охлаждаща вода. Азотът се нагласява така че потокът му да е умерен, разтворът се нагрява до кипене и се задържа при тази температура в течение на 2 минути. В края на този период не трябва да се наблюдава никакво отделяне на мехури. Ако се виждат мехурчета, нагряването се продължава още 30 минути. Разтворът се оставя да се охлади в продължение най-малко на 20 минути, като азотът продължава да се пропуска през него.
Монтирането на апаратурата се довършва, както е посочено на диаграмата, като се свързва тръбата на кондензатора към бутилката на Дрексел (D) и бутилката към абсорбционните съдове F1 и F2. Азотът трябва да продължи да се пропуска през разтвора през време на монтирането на апаратурата. Във всеки един от абсорбционните съдове (F1 и F2) се добавят бързо 50 ml разтвор от бариев хидроксид (3.4).
Пропуска се поток от азотни мехурчета в продължение на 10 минути. Разтворът в абсорберите трябва да остане бистър. Ако това не се случи, процесът за отстраняване на карбонатите трябва да се повтори.
5.3. Окисляване и абсорбция
След изваждане на тръбата, подаваща азот, през страничния разтвор на реакционната колба (В) се вкарват 20 g хромен триоксид (3.1) и 6 ml разтвор на сребърен нитрат (3.3). Апаратурата се свързва с вакуумпомпата и падането на азот се регулира, така че през абсорберите със синтеровано стъкло (F1, F2) да преминава постоянен поток от газови мехурчета.
Реакционната колба (В) се нагрява, докато течността закипи, и кипенето се поддържа в продължение на един час и половина (1). Може да е необходимо да се настрои вентила за регулиране на вакуума (G), за да се контролира азотният поток, тъй като е възможно утаеният по време на изпитването бариев карбонат да блокира дисковете от синтеровано стъкло. Операцията е сполучлива, когато разтворът на бариевия хидроксид в абсорбера F2 остане бистър. В противен случай изпитанието се повтаря. Спира се нагряването и се разглобява апаратурата. Измиват се разпределителите (3.10) отвътре и отвън, за да се отстрани бариевият хидроксид, като промивните води се събират в съответния абсорбер. Поставят се разпределителите един след друг в бехерова чаша 600 ml, която ще бъде използвана след това при определянето.
Бързо се филтрира под вакуум най-напред съдържанието на абсорбер F2 и след това на абсорбер F1, като се използва тигелът от синтеровано стъкло. Събира се утайката чрез измиване на абсорберите с вода (3.10) и се измива тигелът с 50 ml от същата вода. Поставя се тигелът в бехеровата чаша 600 ml и се добавя около 100 ml вряла вода (3.10) Вкарват се 50 ml вряла вода в двата абсорбера и се пропуска азот през разпределителите в продължение на 5 минути. Водата се събира с тази от бехеровата чаша. Операцията се повтаря още веднъж, за да се осигури разпределителите да са старателно измити.
5.4. Измерване на карбонатите с органичен произход
Добавят се пет капки фенолфталеин (3.8) към съдържанието на бехеровата чаша. Разтворът става червен. Титрува се солна киселина (3.5) капка по капка, докато розовият цвят изчезне. Разтворът в тигела се разбърква добре, за да се провери дали розовият цвят няма да се появи отново. Добавят се пет капки бромфенолово синьо (3.7) и се титрува със солна киселина (3.5), докато разтворът стане жълт. Добавят се още 10 ml солна киселина.
Разтворът се нагрява до кипене и се оставя да кипи още най-много 1 минута. Внимателно се проверява да не остане утайка в течността.
Оставя се да се охлади и се титрува остатъчно с разтвор на натриев хидроксид (3.6).
6. Празна проба
Празната проба се провежда по същата процедура, като се използват същите количества от всички реактиви.
7. Изразяване на резултатите
Съдържанието на горими съставки (С), изразено като въглерод в проценти от масата на пробата, се дава с формулата:
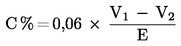
където:
|
Е |
= |
масата в грамове на порцията за изпитване, |
|
V1 |
= |
общ обем в ml на 0,1 mol/l солна киселина, добавен след промяната на цвета на фенолфталеина, |
|
V2 |
= |
обем в ml на 0,1 mol/l разтвор на натриев хидроксид, използван за обратно титруване. |
Фигура 2

Метод 4
Определяне на стойността на рН
1. Обект и област на приложение
Настоящият документ дефинира процедурата за измерване стойността на рН на разтвор на еднокомпонентен амониево-нитратен тор с високо съдържание на азот.
2. Принцип
Измерване на рН на разтвор на амониев нитрат с помощта на рН метър
3. Реактиви
Дестилирана или деминарилизирана вода, без въглероден диоксид.
3.1. Буферен разтвор, рН 6,88 при 20 °С
Разтварят се 3,40 ± 0,01 g калиев дихидроген ортофосфат (КН2РО4) в приблизително 400 ml вода. След това се разтварят 3,55 ± 0,01 g динатриев хидроген ортофосфат (Na2HPO4) в приблизително 400 ml вода. Прехвърлят се двата разтвора без загуба в градуирана колба от 1 000 ml, долива се до отметката и се смесва. Този разтвор се съхранява в херметично затворен съд.
3.2. Буферен разтвор, рН 4,00 при 20 °С
Разтворете 10,21 ± 1 g калиев хидроген фталат (КНС8О4Н4) във вода, прехвърля се без загуба в градуирана колба от 1 000 ml и се смесва.
Този разтвор се съхранява в херметично затворен съд.
3.3. Могат да се използват стандартни рН разтвори, които са на разположение на пазара.
4. Апаратура
рН-метър, оборудван със стъклен и каломелов електрод или еквивалентни на тях, с чувствителност 0,05 рН единици.
5. Процедура
5.1. Калибриране на рН-метъра
Калибрира се рН-метъра (4) при температура 20 (±1) °С, като се използват буферни разтвори (3.1), (3.2) или (3.3). Пропуска се бавен поток азот върху повърхността на разтвора и се поддържа по време на изпитването.
5.2. Определение
Добавят се 100,0 ml вода върху 10 (± 0,01) g проба в бехерова чаша 250 ml. Неразтворените частици се отстраняват чрез филтруване, декантиране или центрофугиране на течността. Измерва се стойността на рН на бистрия разтвор при температура 20 (±1) °С според същата процедура, както при калибрирането на рН-метъра.
6. Представяне на резултатите
Резултатите се изразяват в рН единици, с точност до 0,1 единици, и се посочва температурата.
Метод 5
Определяне на размера на частиците
1. Обект и област на приложение
Този метод дефинира процедурата за определяне на еднокомпонентен амониево-нитратен тор с високо съдържание на азот.
2. Принцип
Пробата за изпитване се пресява през комплект от 3 сита ръчно или с механични средства. Масата, задържана на всяко едно сито, се отчита и се изчислява процентът на материала, преминаващ през съответните сита.
3. Апаратура
3.1. Телени изпитателни сита с диаметър 200 mm с отвори 2,0 mm, 1,0 mm и 0,5 mm съответно. Един капак и един събирателен съд за тези сита.
3.2. Везни с точност 0,1 g.
3.3. Механично вибрационно сито (ако е налично), в състояние да осигури вертикално и хоризонтално движение на пробата за изпитване.
4. Процедура
4.1. Пробата се разделя на представителни порции от около 100 g.
4.2. Една от тези проби се претегля с точност до 0,1 g.
4.3. Комплектът сита се подрежда във възходящ ред: сборник, 0,5 mm, 1 mm, 2 mm, като претеглената проба за изпитване се поставя на горното сито. Поставя се плътно капакът върху комплекта сита.
4.4. Разклаща се ръчно или с машина, като се придава както вертикално, така и хоризонтално движение и ако се работи ръчно, се почуква от време на време. Този процес продължава в течение на 10 минути или докато количеството, преминаващо през всяко сито за 1 минута, е по-малко от 0,1 g.
4.5. Ситата се отделят от комплекта едно по едно и се събира задържаният материал, като се изчетква внимателно обратната страна с мека четка, ако е необходимо.
4.6. Претегля се задържаният материал на всяко сито и този, събран в дъното, с точност до 0,1 g.
5. Оценка на резултатите
5.1. Масата на отделните фракции се изчислява като процент от общата маса на фракциите (а не от началното количество на пробата).
Изчислява се процентът в сборника (т.е. < 0,5 mm): А %
Изчислява се процентът, задържан в ситото 0,5 mm: в %
Изчислява се процентът, преминаващ през сито 1,0 mm, т.е. (А + В) %
Сумата от масите на фракциите трябва да бъде до 2 % от първоначално взетата маса.
5.2. Трябва да се проведат най-малко два отделни анализа, като отделните резултати за А не трябва да се различават с повече от 1,0 % абсолютни и за В — с повече от 1,5 % абсолютни. Ако това не е така, изпитването се повтаря.
6. Изразяване на резултатите
Взема се средноаритметичната стойност, получени за А, от една страна, и за А + В, от друга.
Метод 6
Определяне на съдържанието хлор (като хлориден йон)
1. Обект и област на приложение
Този метод дефинира процедурата за определяне на съдържанието на хлор (като хлориден йон) в еднокомпонентен амониево-нитратни тор с високо съдържание на азот.
2. Принцип
Хлоридните йони във воден разтвор се определят чрез потенциометрично титруване със сребърен нитрат в кисела среда.
3. Реактиви
Дестилирана или деминерализирана вода, несъдържаща хлорни йони.
3.1. Ацетон AR.
3.2. Концентрирана азотна киселина (плътност при 20 °С = 1,40 g/ml).
3.3. Сребърен нитрат 0,1 mol/l стандартен разтвор. Този разтвор се съхранява в кафява стъклена бутилка.
3.4. Сребърен нитрат 0,004 mol/l стандартен разтвор — този разтвор се приготвя по време на употреба.
3.5. Калиев хлорид 0,1 mol/l стандартен сравнителен разтвор. 3,7276 g калиев хлорид с аналитично качество, предварително изсушен в продължение на 1 час в пещ при 130 °С и охладен в ексикатор до стайна температура, се претегля с точност до 0,1 mg. Разтваря се в малко количество вода, разтворът се прехвърля без загуба в мерителна колба от 500 ml, разрежда се до марката и се хомогенизира.
3.6. Калиев хлорид, 0,004 mol/l стандартен сравнителен разтвор — този разтвор се приготвя по време на употреба.
4. Апаратура
4.1. Потенциометър със сребърен индикаторен електрод и каломелов сравнителен електрод, чувствителност 2 mV, с обхват от – 500 до + 500 mV.
4.2. Мост, съдържащ наситен разтвор на калиев нитрат, свързан към каломеловия електрод (4.1), с поставени в краищата му порьозни запушалки.
4.3. Магнитна бъркалка с разбъркващ елемент с тефлоново покритие.
4.4. Микробюрета с фино заострен връх, градуирана с деления 0,01 ml.
5. Процедура
5.1. Стандартизиране на разтвора от сребърен нитрат
Вземат се 5,00 ml и 10,00 ml от стандартния референтен разтвор на калиевия хлорид (3.6) и се поставят в две ниски бехерови чаши с удобен обем (например 250 ml). Извършва се следното титруване на съдържанието на всяка бехерова чаша:
Добавят се 5 ml разтвор на азотната киселина (3.2), 120 ml ацетон (3.1) и достатъчно вода, за да се постигне общ обем около 150 ml. Поставя се пръчката на магнитната бъркалка (4.3) в бехеровата чаша и бъркалката се включва. Потапя се сребърният електрод (4.1) и свободният край на моста (4.2) в разтвора. Свързват се електродите на потенциометъра (4.1) и след проверка на нулата на апарата, се отбелязва стойността на началния потенциал.
Титрува се, като се използва микробюретата (4.4), добавят се първоначално 4 или 9 ml съответно от разтвора на сребърния нитрат, съответстващ и на използвания стандартен референтен разтвор на калиевия хлорид. Продължава се добавянето на порции от 0,1 ml за 0,004 mol/l разтвори и на порции от 0,05 ml за 0,1 mol/l разтвори. След всяко добавяне се изчаква стабилизиране на потенциала.
Добавените обеми се записват заедно със съответните стойности на потенциала в първите две колони на таблицата.
В третата колона на таблицата се записват последователните нараствания (Δ1Е) на потенциала Е. В четвъртата колона се записват разликите (Δ2Е), положителни или отрицателни, между нарастванията на потенциала (Δ1Е). Краят на титруването съответства на добавянето на порция (V1) 0,1 или 0,05 ml от разтвора на сребърен нитрат, което съответства на максималната стойност на Δ1Е.
За да се изчисли точният обем (Veq) на разтвора на сребърен нитрат в края на реакцията, се използва формулата:

където
|
|
V0 е общия обем, в ml, на разтвора на сребърния нитрат, непосредствено под обема, който дава максималното нарастване Δ1Е, |
|
|
V1 е обема, в ml, на последната порция на добавения разтвор на сребърен нитрат (0,1 или 0,05 ml), |
|
|
b е последната положителна стойност на Δ2Е, |
|
|
В е сборът на абсолютните стойности на последните положителни стойности на Δ2Е и първите отрицателни стойности на Δ2Е (виж примера в таблица 1). |
5.2. Празна проба
Провежда се празна проба и тя се взема предвид при изчисляване на крайния резултат.
Резултатът V4 от празната проба на реактивите се дава в ml с формулата:

където:
|
|
V2 е стойността в ml на точния обем (Veq) на разтвора на сребърен нитрат, отговарящ на титруването на 10 ml от използвания стандартен сравнителен разтвор на калиев хлорид. |
|
|
V3 е стойността в ml, на точния обем (Veq) на разтвора на сребърния нитрат за титруване на 5 ml от използвания стандартен сравнителен разтвор на калиев хлорид. |
5.3. Изпитване за проверка
Празната проба може да служи едновременно като проверка за задоволителното функциониране на апаратурата и за точното прилагане на процедурата на изпитване.
5.4. Определяне
Взема се част от пробата в границите от 10 до 20 g и се претегля с точност до 0,01 g. Прехвърля се количествено в бехерова чаша 250 ml. Добавят се 20 ml вода, 5 ml разтвор на азотна киселина (3.2), 120 ml ацетон (3.1) и достатъчно вода, за да се доведе общият обем около 150 ml.
Разбъркващият елемент на магнитната бъркалка (4.3) се поставя в чашата, чашата се поставя върху магнитната бъркалка и тя се пуска в действие. В разтвора се потапят сребърният електрод (4.1) и свободният край на моста (4.2). Електродите се съединяват с потенциометъра (4.1) и след проверка на нулата на апарата, се отчита стойността на началния потенциал.
Титрува се с разтворът на сребърния нитрат чрез добавка от микробюрета (4.4) на порции от 0,1 ml. След всяко добавяне се изчаква стабилизирането на потенциала.
Титруването продължава, както е дадено в 5.1, като се започва от четвъртия параграф: „Записват се добавените обеми и съответните стойности на потенциала в първите две колони на таблицата.…“.
6. Изразяване на резултатите
Резултатите от анализа се изразяват като процент хлор, съдържащ се в пробата, представена за анализ. Съдържанието на хлор (Cl) в проценти се изчислява по формулата:

където:
|
|
Т е концентрацията използвания разтвор на сребърен нитрат в mol/l, |
|
|
V4 е резултатът в ml от празната проба (5.2), |
|
|
V5 е стойността в ml на Veq, съответстваща на определянето (5.4), |
|
|
m е масата в грамове на пробата за изпитване. |
Таблица 1: Пример
|
Обем на разтвора на сребърен нитрат V (ml) |
Потенциал Е (mV) |
Δ1Е |
Δ2Е |
|
4,80 |
176 |
|
|
|
4,90 |
211 |
35 |
+37 |
|
5,00 |
283 |
72 |
–49 |
|
5,10 |
306 |
23 |
–10 |
|
5,20 |
319 |
13 |
|
|
|
|||

Метод 7
Определяне на съдържанието на мед
1. Обект и област на приложение
Този метод дефинира процедурата за определяне на съдържанието мед в еднокомпонентни амониево нитратни торове с високо съдържание на азот.
2. Принцип
Пробата се разтваря в разредена солна киселина и съдържанието на мед се определя чрез атомно абсорбционна спектрофотометрия.
3. Реактиви
3.1. Солна киселина (плътност при 20 °С = 1,18 g/ml).
3.2. Солна киселина, разтвор 6 mol/l.
3.3. Солна киселина, разтвор 0,5 mol/l.
3.4. Амониев нитрат.
3.5. Водороден пероксид, 30 % (w/v)
3.6. Разтвор на мед (2) (разреден): претегля се с точност до 0,001 g 1 g чиста мед, разтваря се в 25 ml 6 mol/l разтвор на солна киселина (3.2), добавят се на порции 5 ml водороден пероксид (3.5) и се разрежда до един литър с вода. 1 ml от този разтвор съдържа 1 000 μg мед (Cu)
3.6.1. Меден разтвор (разреден): разреждат се 10 ml от изходния разтвор (3.6) до 100 ml с вода, а след това 10 ml от получения разтвор се разреждат до 100 ml с вода. 1 ml от последното разреждане съдържа 10 μg мед (Cu).
Този разтвор се приготвя непосредствено преди употреба.
4. Апаратура
Атомно абсорбционен спектрометър с лампа за определяне на мед (324,8 nm).
5. Процедура
5.1. Приготвяне на разтвора за анализ
Претегля се с точност до 0,001 g 25 g от пробата, поставят се в бехерова чаша от 400 ml, добавят се внимателно 20 ml солна киселина (3.1) (може да се наблюдава бурна реакция поради образуването на въглероден диоксид). Добавя се още солна киселина, ако е необходимо. Когато бурното отделяне на газове спре, пробата се изпарява до сухост на парна баня, като се разбърква от време на време със стъклена пръчка. Добавят се 15 ml 6 mol/l разтвор на солна киселина (3.2) и 120 ml вода. Разбърква се със стъклена пръчка, която трябва да се остави в бехеровата чаша, и се покрива бехеровата чаша с часовниково стъкло. Разтворът се нагрява до кипене внимателно, до приключване на разтварянето и след това се охлажда.
Прехвърля се разтворът количествено в градуирана колба от 250 ml чрез промиване на бехеровата чаша с 5 ml 6 mol/l солна киселина (3.2) и два пъти с 5 ml вряща вода, долива се до отметката 0,5 mol/l солна киселина (3.3) и се смесва внимателно.
Филтрира се през филтърна хартия, несъдържаща мед (3), като се изхвърлят първите 50 ml.
5.2. Разтвор на празна проба
Приготвя се разтвор на празна проба, в който не е включена само пробата и се взема предвид при изчисляването на крайните резултати.
5.3. Определяне
5.3.1. Приготвяне на разтвори от пробата и празна проба
Разрежда се разтворът на пробата (5.1) и разтворът на празната проба (5.2) с 0,5 mol/l разтвор на солна киселина до концентрация на медта в рамките на оптималния измервателен обхват на спектрофотометъра. Обикновено не е необходимо разреждане.
5.3.2 Приготвяне на калибровъчни разтвори
Чрез разреждане на изходния разтвор (3.6.1) с 0,5 mol/l разтвор на солна киселина (3.3) се приготвят вай-малко пет стандартни разтвори, съответстващи на оптималния измервателен обхват на спектрофотометъра (0 до 5,0 mg/l мед). Преди да се допълнят до отметката, към всеки разтвор се добавя амониев нитрат (3.4), за да се постигне концентрация 100 mg на ml.
5.4. Измерване
Спектрофотометърът (4) се настройва на дължина на вълната 324,8 nm. Използва се окисляващ въздушно-ацетиленов пламък. Впръскват се последователно, по три пъти, калибровъчните разтвори (5.3.2), разтворът на пробата и разтворът на празната проба (5.3.1), като устройството се промива старателно с дестилирана вода след всяко впръскване. Начертава се калибровъчната крива, като средните стойности на абсорбцията за всеки използван стандартен разтвор се нанасят на ординатната ос и съответните концентрации на мед в μg/ml на абсцисната ос.
Определя се концентрацията на мед в крайния разтвор на пробата и разтвора на празната проба се отчита по стандартната крива.
6. Изразяване на резултатите
Изчислява се съдържанието на мед в пробата, като се взема предвид масата на пробата за изпитване, извършените разреждания по време на анализа и стойността на празната проба. Изразете резултата като mg Cu/kg.
4. Определяне устойчивостта на детонация
4.1. Обект и област на приложение
Този метод дефинира процедурата на работа при определяне устойчивостта към детонация на амониево-нитратни торове с високо съдържание на азот.
4.2. Принцип
Пробата за изпитване се затваря в стоманена тръба и се подлага на детонационен удар от експлозивен междинен иницииращ заряд. Разпространението на детонацията се определя от степента на деформация при свиване на оловните цилиндри, върху които тръбата лежи хоризонтално по време на изпитването.
4.3. Материали
4.3.1. Пластичен експлозив, съдържащ 83 до 86 % пентрит
Плътност: 1 500 до 1 600 kg/m3
Скорост на детонация: 7 300 до 7 700 m/sec
Маса: 500 (±1) g
4.3.2. Седем дължини на гъвкав детонационен шнур с неметално покритие (кожух)
Тегло на пълнеж: 11 до 13 g/m
Дължина на всеки шнур: 400 (±2) mm
4.3.3. Пресована гранула от вторичен експлозив с жлеб за поставяне на детонатор.
Експлозив: хексоген/восък 95/5 или тетрил, или подобен вторичен експлозив със или без добавен графит.
Плътност: 1 500 до 1 600 kg/m3
Диаметър: 19 до 21 mm
Височина: 19 до 23 mm
Централен жлеб за поставяне на детонатора: с диаметър 7 до 7,3 mm и с дълбочина 12 mm.
4.3.4. Безшевна стоманена тръба, както е специфицирана в ISO 65 — 1981 — „Тежки серии“, с номинални размери DN 100 (4)
Външен диаметър: 113,1 до 115,0 mm
Дебелина на стената: 5,0 до 6,5 mm
Дължина: 1 005 (±2) mm
4.3.5. Дънна плоча
|
Материал |
: |
стомана с добри заваряващи свойства |
|
Размери |
: |
160 × 160 mm |
|
Дебелина |
: |
5 до 6 mm |
4.3.6. Шест оловни цилиндъра
|
Диаметър |
: |
50 (±1) mm |
|
Височина |
: |
100 до 101 mm |
|
Материал |
: |
рафинирано (пречистено) олово, чистота най-малко 99,5 %. |
4.3.7. Стоманен блок
Дължина: най-малко 1 000 mm
Ширина: най-малко 150 mm
Височина: най-малко 150 mm
Маса: най-малко 300 kg, ако няма твърда основа на стоманения блок.
4.3.8. Пластмасов или картонен цилиндър за заряда на възпламенителя
Дебелина на стената: 1,5 до 2,5 mm
Диаметър: 92 до 96 mm
Височина: 64 до 67 mm.
4.3.9. Детонатор (електрически или неелектрически) с инициираща сила 8 до 10
4.3.10. Дървен диск
|
Диаметър |
: |
92 до 96 mm. Диаметърът трябва да пасва с вътрешния диаметър на пластмасовия или картонен цилиндър (4.3.8) |
|
Дебелина |
: |
20 mm |
4.3.11. Дървен прът със същите размери като детонатора (4.3.9)
4.3.12. Шивашки карфици (максимална дължина 20 mm)
4.4. Процедура
4.4.1. Приготвяне на заряда на възпламенителя за поставяне в стоманена тръба
Има два метода за иницииране на експлозива в заряда на възпламенителя, в зависимост от наличността на оборудване.
4.4.1.1. Седемточково едновременно иницииране
Зарядът на възпламенителя, приготвен за използване, е показан на фигура 1.
4.4.1.1.1. Пробиват се отвори в дървения диск (4.3.10), успоредни на оста на диска, през центъра и през шест точки, симетрично разпределени по концентричен кръг с диаметър 55 mm. Диаметърът на отворите трябва да бъде 6 до 7 mm (виж сечение А—В на фигура 1), в зависимост от диаметъра на използвания детонационен шнур (4.3.2).
4.4.1.1.2. Отрязват се седем дължини от гъвкавия детонационен шнур (4.3.2), всяка една дълга по 400 mm, като се избягват загуби на експлозив в краищата чрез гладко срязване и незабавно уплътняване на отрязаните краища с лепенка. Всяка една от седемте дължини се поставя (напъхва) през седемте отвора на дървения диск (4.3.10), докато краищата им се подадат няколко сантиметра от другата страна на диска. След това се вмъква малка шивашка топлийка (4.3.12) наклонено на текстилния кожух на всяка дължина шнур 5 до 6 mm от техните краища и се поставя лепенка около външната страна на отрязъците шнур във форма на лента, широка 2 cm, непосредствено до карфицата. Накрая се изтегля по-дълга част от шнура, за да може да допре карфицата до дървения диск.
4.4.1.1.3. Оформя се пластичният експлозив (4.3.1) като цилиндър с диаметър 92 до 96 mm, в зависимост от диаметъра на цилиндъра (4.3.8). Поставя се цилиндърът изправен на равна повърхност и се вкарва оформеният експлозив. След това се поставя дървения диск (4), носещ седемте отрязъка детонаторен шнур отгоре на цилиндъра и се натиска надолу върху експлозива. Регулира се височината на цилиндъра (64 до 67 mm), така че неговият горен ръб да не излиза извън нивото на дървото. Накрая се фиксира цилиндърът към дървения диск с телбод или малки гвоздеи по цялата обиколка.
4.4.1.1.4. Свободните краища на седемте отрязъка на детонационен шнур се групират около обиколката на дървения прът (4.3.11), така че техните краища да бъдат наравно с равнината, перпендикулярна на пръчката. Те се задържат като сноп около пръчката с помощта на лепенка (5).
4.4.1.2. Централно иницииране с пресована гранула
Зарядът на възпламенителя, готов за използване, е показан на фигура 2.
4.4.1.2.1. Приготвяне на пресована гранула
10 g вторичен експлозив (4.3.3) се поставят, вземайки необходимите предпазни мерки, в матрица с вътрешен диаметър от 19 до 21 mm и се пресоват до точните размери и плътност.
(Съотношението диаметър: височина трябва да бъде грубо 1:1).
В центъра на дъното на матрицата се намира издатина с височина 12 mm и диаметър 7,0 до 7,3 mm (в зависимост от диаметъра на използвания детонатор), който образува в пресованата гранула цилиндрично гнездо за последващо вмъкване на детонатора.
4.4.1.2.2. Приготвяне на заряда за възпламенителя
Пластичният експлозив (4.3.1) се поставя в цилиндъра (4.3.8), стоящ изправен на гладка повърхност, след това се притиска надолу с дървена щанца, за да се придаде цилиндрична форма на експлозива с централен улей (жлеб). Вкарва се пресованата гранула в този жлеб. Покрива се цилиндрично оформеният експлозив, съдържащ пресованата гранула, с дървен диск (4.3.10) с централен отвор с диаметър 7,0 до 7,3 mm за вкарване на детонатора. Дървеният диск и цилиндърът се свързват по диагонал с лепяща лента (лепенка). Осигурява се съосност на отвора, пробит в диска, и жлеба в пресованата гранула чрез вкарване на дървена пръчка (4.3.11).
4.4.2. Приготвяне на стоманените тръби за детонационни изпитвания
В единия край на стоманената тръба (4.3.4) се пробиват два диаметрално противоположни отвора с диаметър 4 mm перпендикулярно през страничните страни на разстояние 4 mm от края.
Заварява се челно дънната плоча (4.3.5) на противоположния край на тръбата, като ъгълът между дънната плоча и стената на тръбата е пълни деветдесет градуса по цялата обиколка на тръбата.
4.4.3. Пълнене и зареждане на стоманената тръба
Виж фигури 1 и 2.
4.4.3.1. Пробата за изпитване, стоманената тръба и зарядът на възпламенителя трябва да бъдат кондиционирани до температура 20 (±5) °С. От 16 до 18 kg от пробата за изпитване са необходими за две изпитвания на детонация.
4.4.3.2. Поставя се тръбата изправена с нейното квадратно дънна плоча върху твърда повърхност, за предпочитане бетон. Напълва се тръбата до около една трета от нейната височина с пробата за изпитване и се пуска от 10 cm вертикално на пода пет пъти, за да се уплътнят частиците или гранулите, колкото е възможно по-плътно в тръбата. За да се ускори уплътняването, тръбата се подлага на вибриране чрез удряне на страничните стени със 750 до 1 000 g чук между отделните пускания общо 10 пъти.
Този метод за зареждане се повтаря с още една порция от пробата за изпитване. Накрая трябва да се направи допълнително добавка по такъв начин, че след уплътняване чрез вдигане и пускане на тръбата 10 пъти и общо 20 ритмични удари с чука, зарядът да запълни тръбата до разстояние 70 mm от нейния отвор.
Височината на запълване с проба за изпитване трябва да бъде регулирана, така че зарядът на възпламенителя (4.4.1.1 или 4.4.1.2), който трябва да се вкара по-късно, да бъде в непосредствен контакт с пробата по цялата ѝ повърхност.
4.4.3.3. Вкарва се зарядът на възпламенителя в тръбата, така че да е в контакт с пробата. Горната повърхност на дървения диск трябва да бъде 6 mm под края на тръбата. Осигурява се сигурен плътен контакт между експлозива и пробата за изпитване чрез добавка и отстраняване на малки количества от пробата. Както е показано на фигури 1 и 2, през отворите близо до края на тръбата трябва да се вкарат шплентове и техните краища да се разтворят срещу тръбата.
4.4.4. Разполагане на стоманената тръба и оловните цилиндри (виж фигура 3)
4.4.4.1. Основите на оловните цилиндри (4.3.6) се номерират от 1 до 6. Правят се шест отметки на 150 mm една от друга по централната линия на стоманения блок (4.3.7), лежащ на хоризонтална основа, с първа отметка най-малкото 75 mm от ръба на блока. Поставя се изправен оловен блок на всяка от тези отметки, с основата на всеки блок центрирана върху съответната отметка.
4.4.4.2. Поставя се стоманената тръба, приготвена съгласно 4.4.3 хоризонтално върху оловните цилиндри, така че оста на тръбата да е успоредна на централната линия на стоманения блок и завареният край на тръбата се издава на 50 mm след оловен цилиндър № 6. За да се предотврати претъркалянето на тръбата, се вкарват малки дървени клинове между горната част на оловните цилиндри и стената на тръбата (по един от всяка страна) или се поставя дървена кръстачка между тръбата и стоманения блок.
Бележка: Трябва да се провери дали тръбата е в контакт с всичките шест оловни цилиндри. Леко изкривяване на повърхността на тръбата може да се компенсира чрез завъртане на тръбата около нейната надлъжна ос. Ако някой от оловните цилиндри е твърде висок, той се почуква цилиндър внимателно с чук до достигане на изискваната височина.
4.4.5. Подготовка за детонация
4.4.5.1. Апаратурата съгласно 4.4.4 се поставя в бункер или друга подходящо подготвена подземна площадка (например, мина или тунел). Трябва да се осигури температурата на стоманената тръба да се поддържа 20 (±5) °С преди детонацията.
Бележка: Ако няма на разположение такива площадки за взривяване, работата може, ако е необходимо, да се извърши в облицована с бетон шахта покрита отгоре с дървени греди. Детонацията може да причини изхвърляне на стоманени фрагменти с висока кинетична енергия. Поради това взривяването трябва да се извършва на подходящо разстояние от жилища или оживени пътища.
4.4.5.2. Ако се използва заряд на възпламенител със седемточково иницииране, трябва да се гарантира, че детонационните шнурове са изпънати, както е дадено в бележката под черта на 4.4.1.1.4 и са подредени, доколкото е възможно, по-хоризонтално.
4.4.5.3. Накрая се отстранява дървения прът и се заменя с детонатора. Взривяването не се извършва, докато зоната за безопасност не е евакуирана и персоналът за изпитването не се е прикрил.
4.4.5.4. Експлозивът се детонира.
4.4.6. Оставя се достатъчно време димовете (газообразни и понякога токсични разпадни продукти, като нитрозни газове) да се разсеят. След това се събират оловните цилиндри и се измерва тяхната височина с измервателен пергел на Верние.
За всеки от маркираните оловни цилиндри се записва степента на сплескване, изразена като процент от оригиналната височина от 100 mm. Ако цилиндрите са смачкани наклонено, се записват най-високата и най-ниската стойност и се изчислява средноаритметичната стойност.
4.4.7. Ако е необходимо, може да се използва проба за непрекъснато измерване на скоростта. Пробата трябва да се вкара по дължината на оста на тръбата или по дължината на нейната странична страна.
4.4.8. За всяка проба трябва да се проведат по две детонационни изпитвания.
4.5. Протокол от изпитването
В отчета за изпитването за всяко детонационно изпитване трябва да бъдат дадени стойностите на следните параметри:
|
— |
фактически измерените стойности за външния диаметър на стоманената тръба и за дебелината на стената, |
|
— |
твърдостта на стоманената тръба по Бринел, |
|
— |
температурите на тръбата и на пробата малко преди възпламеняването, |
|
— |
плътност (kg/м3) на пробата в стоманената тръба, |
|
— |
височината на всеки оловен цилиндър след възпламеняване, отбелязвайки номера на съответния цилиндър, |
|
— |
използвания метод на иницииране на заряда на възпламенителя. |
4.5.1. Оценка на резултатите от изпитванията
Ако при всяко възпламеняване сплескването поне на един оловен цилиндър не надминава 5 %, изпитването се счита за приключено и пробата съответства на изискванията на приложение III.2.
Фигура 1

Фигура 2

Фигура 3
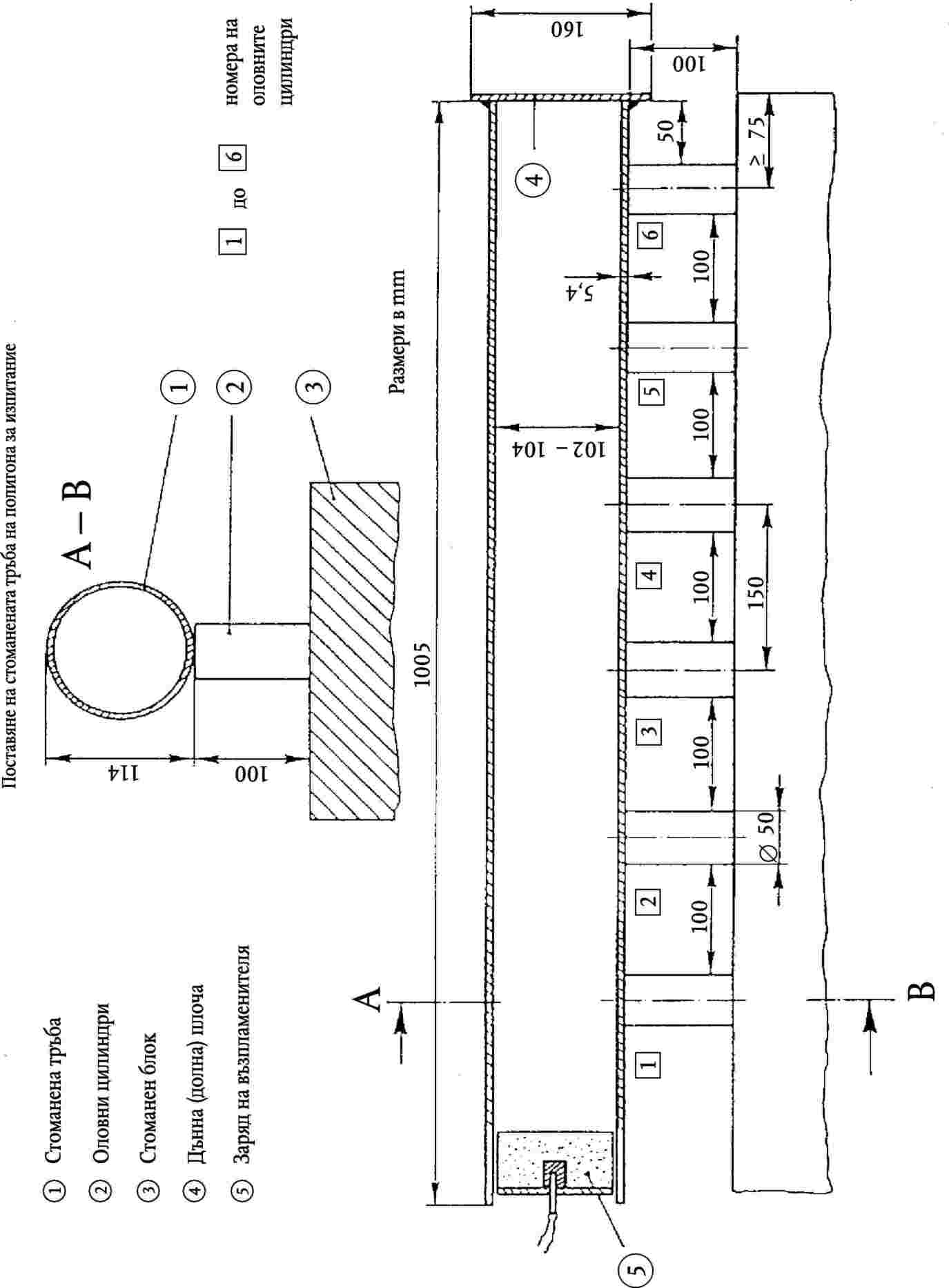
(1) Времето за реакция от един час и половина е достатъчно при повечето органични вещества в присъствието на катализатор сребърен нитрат.
(2) Може да използва стандартен разтвор на мед от търговската мрежа.
(3) Whatman 541 или еквивалентна на нея.
(4) Диаметърът на диска трябва винаги да съответства на вътрешния диаметър на цилиндъра.
NB Когато шестте периферни отрязъка шнур са стегнати след сглобяване, централният шнур трябва да остане леко хлабав.
ПРИЛОЖЕНИЕ IV
МЕТОДИ ЗА ВЗЕМАНЕ НА ПРОБИ И АНАЛИЗ
А. МЕТОД ЗА ВЗЕМАНЕ НА ПРОБИ ЗА КОНТРОЛ НА ТОРОВЕ
ВЪВЕДЕНИЕ
Правилното вземане на проби е трудна операция, която изисква най-голямо внимание. Поради това трябва да се наблегне на необходимостта от получаването на достатъчно представителна проба.
Методът за вземане на проба, описан по-долу, трябва да се спазва стриктно и точно от специалисти с опит в конвенционалната процедура за вземане на проби.
1. Цел и обхват
Пробите, предназначени за контрол на торове по отношение н а качество и състава, трябва да се вземат съгласно методите, описани по-долу. Получените по този начин проби се считат за представителни за изследваните партиди.
2. Служители, които вземат проби
Пробите се вземат от служители специалисти, упълномощени за тази цел от държавите-членки.
3. Дефиниции
Изследвана партида: Количество продукт, представляващо единица и имащо характеристики, за които се приема, че са еднородни.
Единична проба: Количество, взето от една точка на изследваната партида.
Обединена проба: Съвкупността от постепенно нарастващи проби, взети от една и съща изследвана партида.
Съкратена проба: Представителна част от обединената проба, получена от нея чрез съкращаване.
Крайна проба: Представителна част от съкратената проба.
4. Апаратура
4.1. Апаратурата за вземане на проби трябва да бъде направена от материали, които не могат да влияят на характеристиките на продуктите, от които се вземат проби. Тази апаратура може да бъде официално одобрена от държавите-членки.
4.2. Препоръчвана апаратура за вземане на проби от твърди торове
4.2.1. Ръчно вземане на проби
|
4.2.1.1. |
Плоскодънна лопата с вертикални страни |
|
4.2.1.2. |
Сонда за вземане на проби с дълъг прорез или отделения. Размерите на сондата за вземане на проби трябва да съответстват на характеристиките на изследваната партида (дълбочина на контейнер, размери на чувалите и т.н.) и частиците на тора. |
4.2.2. Механично вземане на проби
Може да се използва механичен инструмент за вземане на проби от торове в поток.
4.2.3. Делител
За вземане на постепенно нарастващи проби и за подготовката на съкратената и крайна проба се използва инструмент, проектиран да разделяне на пробата на равни части.
4.3. Препоръчван прибор за вземане на проби от течни торове
4.3.1. Ръчно вземане на проби
Отворена тръба, сонда, бутилка или друго подходящо приспособление за вземане на случайни проби от изследвана та партида.
4.3.2. Механично вземане на проби
Може да се използва механичен прибор за вземане на проби от течни торове в поток.
5. Количество на пробите
5.1. Изследвана партида
Размерът на изследваната партида трябва да дава възможност за вземане на проба от всеки от съставните ѝ части.
5.2. Единични проби
5.2.1. Насипни твърди торове или течни торове в контейнери над 100 kg.
|
5.2.1.1. |
Изследвани партиди, ненадвишаващи 2,5 тона:
|
|
5.2.1.2. |
Изследвани партиди, от 2,5 тона до 80 тона:
|

|
5.2.1.3. |
Изследвани партиди, надвишаващи 80 тона:
|
5.2.2. Опаковани твърди торове или течни торове в контейнери (= опаковки, всеки от които не надвишава 100 kg)
|
5.2.2.1. |
Опаковки по-тежки от 1 kg
|

|
5.2.2.2. |
Опаковки, ненадвишаващи 1 kg:
|
5.3. Обединена проба
Изисква се една обединена проба за изследвана партида. Общата маса на единичните проби, съставляващи обединената проба, не трябва да бъде по-малка от следното:
|
5.3.1. |
Насипни твърди торове или течни торове в контейнери над 100 kg: 4 kg. |
|
5.3.2. |
Опаковани твърди торове или течни торове в контейнери (= опаковки, всеки от които не надвишава 100 kg)
|
|
5.3.3. |
Проба за изпитване на амониево-нитратен тор съгласно приложение III.2: 75 kg |
5.4. Крайни проби
Обединената проба дава крайната проба чрез съкращаване, когато е необходимо. Изисква се анализ на поне една крайна проба. Масата на пробата за анализ не трябва да под 500 g.
5.4.1. Твърди и течни торове
5.4.2. Проба за изпитване на амониево-нитратен тор
Обединената проба дава крайната проба чрез съкращаване, когато е необходимо.
5.4.2.1. Минимална маса на крайната проба за изпитване съгласно приложение III.1: 1 kg
5.4.2.2. Минимална маса на крайната проба за изпитване съгласно приложение III.2: 25 kg
6. Инструкции за вземане, приготвяне и опаковане на пробите
6.1. Общи положения
Пробите трябва да се вземат и приготвят колкото е възможно най-бързо, като се вземат необходимите предпазни мерки за осигуряване те да останат представителни за изпробвания тор. Инструментите, повърхностите и съдовете, предназначени за получаване на проби, трябва да са чисти и сухи.
При течните торове, ако е възможно, изследваната партида трябва да се разбърка преди вземането на проба.
6.2. Единични проби
Единичните проби трябва да се вземат произволно от цялата изследвана партида и трябва да бъдат с приблизително еднакво количество.
6.2.1. Насипни твърди торове или течни торове в контейнери, надвишаващи 100 kg
Трябва да се направи въображаемо разделяне на изследваната партида на няколко приблизително равни части. Броят на частите, съответстващ на броя на единичните проби, изисквани в съответствие с точка 5.2, трябва да се избере произволно и да се вземе поне една проба от всяка от тези части. Когато не е възможно да се спазят изискванията на точка 5.1 при вземане на проби от насипни торове или течни торове в контейнери, надвишаващи 100 kg, вземането на проби трябва да се извърши при преместването на изследваната партида (товарене или разтоварване). В този случай пробите се вземат от произволно избрани въображаеми части, както е дефинирано по-горе при преместването им.
6.2.2. Опаковани твърди торове или течни торове в контейнери (= опаковки) всеки от които не надвишава 100 kg
След като са избрани изискваният брой опаковки за вземане на проба, както е посочено в точка 5.2, част от съдържанието на всяка опаковка се отстранява. Когато е необходимо, пробите се вземат след изпразване на опаковките поотделно.
6.3. Приготвяне на обединена проба
Единичните проби се смесват, за да се получи обединена проба.
6.4. Приготвяне на крайната проба
Материалът на обединената проба се хомогенизира внимателно (3).
Ако е необходимо, обединената проба трябва да бъде съкратена до най-малко 2 kg (съкратена проба) чрез използване на механичен разделител или чрез квартуване.
Приготвят се поне три крайни проби с приблизително еднакво количество и отговарящи на количествените изисквания на точка 5.4. Всяка проба се слага в подходящ херметичен контейнер. Трябва да се вземат всички необходими предпазни мерки, за да се избегне промяна на характеристиките на пробата.
За изпитанията по приложение III, раздели 1 и 2, крайните проби трябва да се съхраняват при температура между 0 °С и 25 °С.
7. Опаковане на крайните образци
Контейнерите се претеглят и етикетират (общият етикет трябва да бъде включен в пломбата) по такъв начин, че те не могат да бъдат отворени, без да се повреди пломбата.
8. Отчет за вземане на проба
Отчет трябва да се съхранява за всяко вземане на проба. Той трябва да позволява всяка изследвана партида да бъде идентифицирана недвусмислено.
9. Местоназначение на пробите
За всяка изследвана партида поне една крайна проба се изпраща, колкото е възможно по-бързо, на одобрена аналитична лаборатория или изпитателна институция заедно с информацията, необходима за анализа или изпитването.
Б. МЕТОДИ ЗА АНАЛИЗ НА ТОРОВЕ
(Виж съдържанието стр. 2.)
Общи наблюдения
Лабораторно оборудване
В описанията на методите общото лабораторно оборудване не е дефинирано прецизно, освен размерът на колбите и пипетите. При всички случаи лабораторната апаратура трябва да бъде добре почистена, особено когато се определят малки количества елементи.
Контролни изпитвания
Преди анализа е необходимо да се осигури цялата апаратура да функционира добре и аналитичната технология да се прилага правилно с използване на подходящи химически съединения с известен състав (например амониев сулфат, моно калциев фосфат и т.н.). Независимо от това, резултатите от анализираните торове могат да показват погрешен химически състав, ако аналитичният метод не се следва строго. От друга страна, някои определяния са емпирични и са свързани с продукти със сложен химичен състав. Препоръчва се, когато са на разположение, лабораториите да използват стандартни референтни торове с точно определен състав.
Общи разпоредби, свързани с методите за химически анализ на торове
1. Реактиви
Освен ако не е посочено друго в метода за анализ, всички реактиви трябва да бъдат с аналитична чистота. Когато се анализират хранителни микроелементи, чистотата на реактивите трябва да се проверява с помощта на празна проба. В зависимост от получения резултат, може да е необходимо извършването на допълнително пречистване.
2. Вода
Когато в описанието на операциите разтваряне, разреждане, изплакване или промиване, описани в методите за анализ, не се посочва какъв да бъде разтворителят или разредителят, се подразбира използването на вода. Нормално е водата да бъде деминализирана или дестилирана. В тези специфични случаи, както са посочени в метода за анализ, тази вода ще трябва да бъде подложена на специфични пречистващи процеси.
3. Лабораторно оборудване
С оглед на оборудването, обичайно използвано в контролните лаборатории, апаратурата, описана в методите за анализ, включва специални инструменти и апарати или такива, които са предмет на специфични изисквания. Това оборудване трябва да бъде идеално чисто, особено когато се определят малки количества. Лабораторията трябва да осигури точността на всички използвани мерителни стъклени съдове, като ги съобрази със съответните метрологични стандарти.
Метод 1
Подготовка на пробата за анализ
1. Обхват
Този метод дефинира процедурата за приготвяне на пробата за анализ, взета от крайната проба.
2. Принцип
Приготвянето на крайна проба, получена в лабораторията, представлява серия от операции, обичайно включващи пресяване, смилане и смесване, извършени така че:
|
— |
от една страна, най-малкото претеглено количество според методите за анализ е представително за лабораторната проба, |
|
— |
от друга страна, размерът на частиците на тора да не се е променил от приготвянето до такава степен, че неговата разтворимост в различните извличащи реактиви да е съществено повлияна. |
3. Апаратура
Разделител на проби (по избор).
Сита с отвори 0,2 и 0,5 mm.
Колби от 250 ml със запушалки.
Порцеланов пестик и хаван или лабораторна мелница.
4. Избор на третиране, което ще се използва
Предварителна забележка
Ако продуктът е подходящ, трябва да се съхранява само представителна част от крайната проба.
4.1. Крайни проби, които не трябва да се смилат
Калциев нитрат, калциево-магнезиев нитрат, натриев нитрат, калиев нитрат, калциев цианамид, калциево-азотен цианамид, амониев сулфат, амониеви нитрати с над 30 % азот, карбамид, основна шлака, природен фосфат направен частично разтворим, утаен дехидриран дикалциев фосфат, калциниран фосфат, калциев алумофосфат, мек смлян фосфорит.
4.2. Крайни проби които трябва да се разделят и час от които трябва да се смелят
Това са продукти, по отношение на които някои определяния се извършват без предварително смилане (например степента на смилането), а други определяния се извършват след смилане. Те включват всички сложни торове, съдържащи фосфатни съставки: основна шлака, калциев алумофосфат, калцинират фосфат, мек смлян фосфорит и естествен фосфат, направен частично разтворим. В тази връзка, крайната проба се разделя на две части, които да са колкото е възможно по-идентични с използване на разделител на проби или чрез квартуване.
4.3. Крайни проби по отношение на които всички определяния се извършват в смляно състояние
Необходимо е да се смели само представителна част от крайната проба. Това са всички останали торове в списъка, които не са дадени в точки 4.1 и 4.2.
5. Метод
Частта от крайната проба, посочена в 4.2 и 4.3, се пресява бързо през сито с отвори 0,5 mm. Остатъка се смила грубо така че да се получи продукт, в който има минимум фини частици, и след това се пресява. Смилането трябва да се извършва при такива условия, че веществото да не е забележимо нагрято. Операцията се повтаря толкова пъти, колкото е необходимо, докато върху ситото няма остатък. Това трябва да се извърши, колкото е възможно по-бързо, за да се предотврати приемането или загубата на съставки (вода, амоняк). Целият смлян и пресят продукт се поставя в чиста колба със запушалка.
Преди да се извърши претегляне за анализ, цялата проба трябва да се размеси добре.
6. Специални случаи
|
а) |
Торове, представляващи смес от няколко категории кристали; В този случай често се получава разделяне. Следователно безусловно важно е пробата да се начупи и пресее през сито с отвори 0,200 mm. Например: смеси от амониев фосфат и калиев фосфат. При тези продукти се препоръчва смилането на цялото количество крайна проба. |
|
б) |
Остатък, който е труден за смилане и не съдържа торови вещества; Претеглете остатъка и го вземете под внимание при изчисляване на крайния резултат. |
|
в) |
Продукти, които се разлагат при загряване; Смилането трябва да се извършва по такъв начин, да се избегне всякакво загряване. В този случай е за предпочитане да се използва пестик и хаван за смилането. Например: сложни торове, съдържащи калциев цианамид и карбамид. |
|
г) |
Продукти които са необичайно влажни или при смилането са добили пастообразна форма; За осигуряване на хомогенност трябва да се избере сито, което има най-малките отвори, позволяващи раздробяване на бучките ръчно или с пестика. Това може да се наложи при смеси, някои от съставките на които съдържат кристализационна вода. |
Методи 2
Азот
Метод 2.1
Определяне на амонячен азот
1. Обхват
Този метод дефинира процедурата за определяне на амонячен азот.
2. Област на приложение
Всички азотни торове, включително сложните торове, в които азотът се среща изключително във формата на амониеви соли или във формата на амониеви соли заедно с нитрати.
Не се прилага за торове, които съдържат карбамид, цианамид и други органични азотни съединения.
3. Принцип
Отделяне на амоняк с помощта на излишък от натриев хидроксид; поглъщане на образувания амоняка в даден обем стандартна сярна киселина и титруване на излишъка от киселина с помощта на стандартен разтвор на натриев и калиев хидроксид.
4. Реактиви
Дестилирана или деминерализирана вода, несъдържаща въглероден диоксид и всякаква азотни съединения.
4.1. Разредена солна киселина: един обем HCl (d20 = 1,18 g/ml) плюс един обем вода
|
за вариант а. |
||
|
|||
|
за вариант б (виж бележка 2) |
||
|
|||
|
за вариант в (виж бележка 2) |
||
|
4.8. Натриев хидроксид, приблизително 30 % NaOH (d20 = 1,33 g/ml), несъдържащ амоняк.
4.9. Индикаторни разтвори
4.9.1. Смесен индикатор
Разтвор А: Разтваря се 1 g метилово червено в 37 ml разтвор на натриев хидроксид 0,1 mol/l и се долива вода до един литър.
Разтвор Б: Разтваря се 1 g метиленово синьо във вода и се долива до един литър.
Смесва се един обем от А с два обема от Б.
Този индикатор е виолетов в кисел разтвор, сив в неутрален разтвор и зелен в алкален разтвор. Използват се 0,5 ml (10 капки) от него.
4.9.2. Индикаторен разтвор метилово червено
Разтваря се 0,1 g метилово червено в 50 ml от 95 %-ен етанол. Допълва се до 100 ml с вода и се филтрира, ако е необходимо. Този индикатор може да се използва (четири до пет капки) вместо предходния.
4.10. Пемза на гранули, промита в солна киселина и накалена
4.11. Амониев сулфат за анализ
5. Апаратура
5.1. Дестилационен апарат, състоящ се от облодънна колба с достатъчна вместимост, свързана с хладник с помощта на капкоуловител.
Забележка 1
Различните видове оборудване, одобрено и препоръчвано за това определяне, е показано с всички характеристики на конструкцията на фигури 1, 2, 3 и 4.
5.2. Пипети от 10, 20, 25, 100 и 200 ml
5.3. Мерителна колба от 500 ml
5.4. Ротационна клатачна машина (35 до 40 оборота в минута)
6. Подготовка на пробата
Виж метод 1.
7. Метод за анализ
7.1. Приготвяне на разтвора
Извършва се изпитване на разтворимост на пробата във вода при стайна температура и в пропорция 2 % (W/V). Претегля се с точност до 0,001 g съгласно индикациите в таблица 1, количество от 5, 7 или 10 g от приготвената проба и се слага в мерителна колба от 500 ml. В зависимост от получения резултат от изпитването на разтворимост се продължава, както следва:
|
а) |
Продукти напълно разтворими във вода; В колбата се добавя необходимото количество вода за разтваряне на пробата. Разклаща се и когато напълно се разтвори, се допълва до обема и се хомогенизира много добре. |
|
б) |
Продукти, които не са напълно разтворими във вода; В колбата се добавят 50 ml вода и след това 20 ml солна киселина (4.1). Разклаща се. Оставя се в покой, докато спре отделянето на въглероден диоксид. Добавят се 400 ml вода и се клати половин час на ротационната клатачна машина (5.4). Обемът се допълва с вода, смесва се и се филтрира през сух филтър в сух приемен съд. |
7.2. Анализ на разтвора
В съответствие с избрания вариант в приемната колба се поставя отмерено количество стандартна сярна киселина, както е посочено в таблица 1. Добавя се подходящо количество от избрания индикаторен разтвор (4.9.1 или 4.9.2) и ако е необходимо, вода, за да се получи минимум 50 ml обем. Краят на наставката на хладника трябва да е под повърхността на разтвора.
Пренася се с прецизна пипета, съгласно подробностите, дадени в таблицата, аликвотна част (4) от бистрия разтвор в дестилационната колба на апарата. Добавя се вода, за да се получи общ обем около 350 ml и няколко зърна пемза, за регулиране на кипенето.
Сглобява се дестилационният апарат. Към съдържани ето на дестилационната колба се добавят 10 ml разтвор на концентриран натриев хидроксид (4.8) за разтваряне на изследваната проба. Бавно затоплете колбата, за да избегнете бурното завиране. Когато кипенето започне, се дестилира със скорост около 100 ml за 10 до 15 минути. Общият обем на дестилата трябва да бъде около 250 ml (5). Когато вече няма амоняк за отделяне, приемната колба се снижава, така че наставката на хладника да е над повърхността на течността.
Полученият дестилат се проверява с подходящ реактив, за да е сигурно, че цялото количество амоняк се е дестилирало докрай. Наставката на хладника се промива с малко вода и излишната киселина се титрува със стандартния разтвор на натриев или калиев хидроксид, посочен за възприетия вариант (виж бележка 2).
Бележка 2:
Стандартни разтвори с различна концентрация могат да се използват за обратно титруване при условие че обемите, използвани за титруване, не превишават, доколкото това е възможно, 40 до 45 ml.
7.3. Празна проба
Празна проба се прави при същите условия и се използва при изчисляване на крайния резултат.
7.4. Контролно изпитване
Преди извършване на анализа се проверява дали апаратурата работи добре и дали се прилага правилното методът чрез използване на аликвотна част прясно приготвен разтвор на амониев сулфат (4.11), съдържащ максималното количество азот според предписанията за избрания вариант.
8. Изразяване на резултатите
Резултатът от анализа се изразява като процент на амонячния азот в тора, получен при анализа.
9. Приложения
Както е дадено в забележка 1 в точка 5.1 „Апаратура“, фигури 1, 2, 3 и 4 се отнасят за конструкционните характеристики на различни видове оборудване, използвано в настоящия документ.
Таблица 1
Определяне на амонячния азот и на амонячния и нитратен азот в торове
Таблица за претегляне, разреждане и изчисляване, които трябва да се извършат за вариантите а, б и в на метода
Вариант а
Приблизително максимално количество за дестилиране: 50 mg.
Сярна киселина 0,1 mol/l за поставяне в приемната колба: 50 ml.
Обратно титруване с NaOH или KOH 0,1 mol/l.
|
Деклариран (% N) |
Количество за претегляне (g) |
Разреждане (ml) |
Разтвор на пробата за дестилация (ml) |
Изразяване на резултата (6) (% N = (5 — А) F) |
|
0—5 |
10 |
500 |
50 |
(50 — А) × 0,14 |
|
5—10 |
10 |
500 |
25 |
(50 — А) × 0,28 |
|
10—15 |
7 |
500 |
25 |
(50 — А) × 0,40 |
|
15—20 |
5 |
500 |
25 |
(50 — А) × 0,56 |
|
20—40 |
7 |
500 |
10 |
(50 — А) × 1,00 |
Вариант б
Приблизително максимално количество за дестилиране: 100 mg.
Сярна киселина 0,2 mol/l, която се поставя в приемната колба: 50 ml.
Обратно титруване с NaOH или KOH 0,2 mol/l.
|
Деклариран (% N) |
Количество за претегляне (g) |
Разреждане (ml) |
Разтвор на пробата за дестилиране (ml) |
Изразяване на резултата (7) (% N = (5 — А) F) |
|
0—5 |
10 |
500 |
100 |
(50 — А) × 0,14 |
|
5—10 |
10 |
500 |
50 |
(50 — А) × 0,28 |
|
10—15 |
7 |
500 |
50 |
(50 — А) × 0,40 |
|
15—20 |
5 |
500 |
50 |
(50 — А) × 0,56 |
|
20—40 |
7 |
500 |
20 |
(50 — А) × 1,00 |
Вариант в
Приблизително максимално количество за дестилиране: 200 mg.
Сярна киселина 0,5 mol/l, която се поставя в приемната колба: 35 ml.
Обратно титруване с NaOH или KOH 0,5 mol/l.
|
Деклариран (% N) |
Количество за претегляне (g) |
Разреждане (ml) |
Разтвор на пробата за дестилиране (ml) |
Изразяване на резултата (8) (% N = (5 — А) F) |
|
0—5 |
10 |
500 |
200 |
(35 — А) × 0,175 |
|
5—10 |
10 |
500 |
100 |
(35 — А) × 0,350 |
|
10—15 |
7 |
500 |
100 |
(35 — А) × 0,500 |
|
15—20 |
5 |
500 |
100 |
(35 — А) × 0,700 |
|
20—40 |
7 |
500 |
50 |
(35 — А) × 1,400 |
Фигура 1

Фигура 2

Фигура 3

Фигура 4
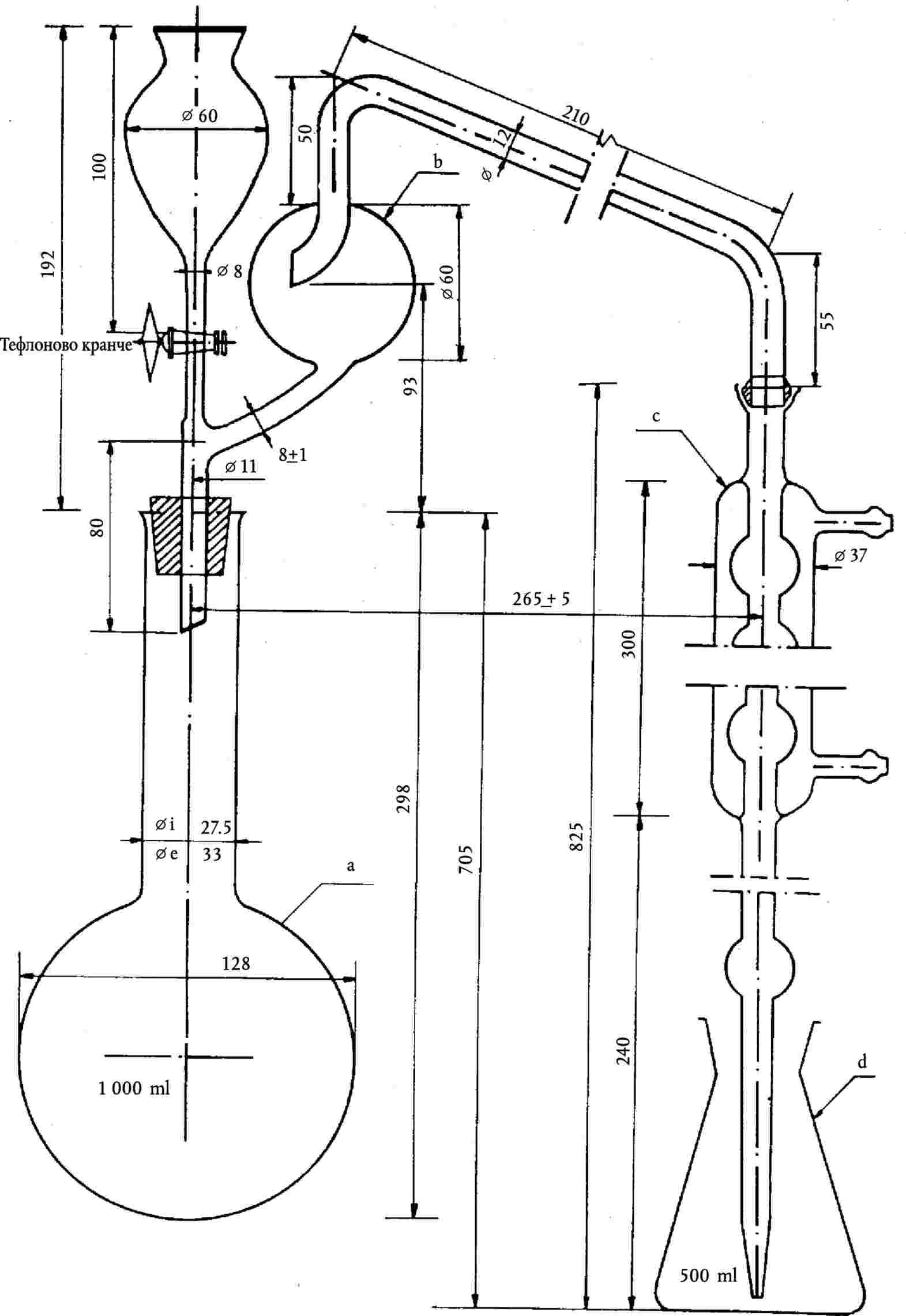
Ключ към фигури 1, 2, 3 и 4
Фигура 1
|
а) |
Облодънна колба с дълго гърло с обем 1 000 ml; |
|
б) |
Дестилационна тръба с пръ капкоуловител, свързана към хладник чрез сферичен шлиф (№ 18) (сферичният шлиф към хладника може да се замести от подходяща каучукова връзка); |
|
в) |
Фуния с тефлоново кранче за добавяне на натриев хидроксид (по същия начин кранчето може да се замести с каучукова връзка с щипка); |
|
г) |
Хладник с шест сфери и сферичен шлиф (№ 18) на входа, а на изхода е свързан към стъклена наставка с къса каучукова връзка (когато връзката към дестилационната тръба е каучукова, сферичният шлиф може да се замени с подходяща каучукова втулка); |
|
д) |
Колба от 500 ml, в която се събира дестилатът. |
Оборудването е направено от борсиликатно стъкло.
Фигура 2
|
а) |
Облодънна колба с късо гърло, с обем 1 000 ml и със сферичен шлиф (№ 35); |
|
б) |
Дестилационна тръба с капкоуловител, оборудвана със сферичен шлиф (№ 35) на входа и сферичен шлиф (№ 18) а отстрани свързана към фуния с тефлоново кранче за добавяне на натриев хидроксид; |
|
в) |
Хладник с шест сфери и сферичен шлиф (№ 18) на входа, а на изхода е свързан към стъклена наставка с къса каучукова връзка; |
|
г) |
Колба от 500 ml, в която се събира дестилатът. |
Оборудването е направено от борсиликатно стъкло.
Фигура 3
|
а) |
Облодънна колба с дълго гърло, с обем 750 или 1 000 ml и с разширена част на отвора; |
|
б) |
Дестилационна тръба с капкоуловител и сферичен шлиф (№ 18) на изхода; |
|
в) |
Тръбно коляно със сферичен шлиф (№ 18) на входа и и конусовиден шлиф на изхода (връзката към дестилационната тръба може да се осъществи с каучукова връзка вместо сферичен шлиф); |
|
г) |
Хладник с шест сфери, свързан на изхода към стъклена наставка с къса каучукова връзка; |
|
д) |
Колба от 500 ml, в която се събира дестилатът. |
Оборудването е направено от борсиликатно стъкло.
Фигура 4
|
а) |
Облодънна колба с дълго гърло, с обем 1 000 ml и с разширен отвор; |
|
б) |
Дестилационна тръба с капкоуловител и сферичен шлиф (№ 18), на изхода, свързана отстрани към фуния с тефлоново кранче за добавяне на натриев хидроксид (подходяща каучукова запушалка би могла да се използва вместо сферичния шлиф, а кранчето може да се замени с каучукова връзка с подходяща щипка); |
|
в) |
Хладник с шест сфери и сферичен шлиф (№ 18) на входа, а на изхода свързан към стъклена наставка с каучукова връзка (когато връзката към дестилационната тръба е каучукова, сферичната връзка би могла да се замени с подходяща каучукова втулка); |
|
г) |
Колба от 500 ml, в която се събира дестилатът. |
Оборудването е направено от борсиликатно стъкло.
Методи 2.2
Определяне на нитратен и амонячен азот
Метод 2.2.1
Определяне на нитратен и амонячен азот съгласно Улш
1. Обхват
Настоящият документ дефинира процедурата за определяне на нитратния и амонячния азот с редукция съгласно Улш.
2 Област на приложение
Всички азотни торове, включително сложните торове, в които азотът се намира изключително в нитратна форма или амонячна и нитратна форма.
3. Принцип
Редуциране на нитрати и нитрити до амоняк с помощта на желязо на прах в кисела среда и отделяне на така образувания амоняк с добавяне на излишък на натриев хидроксид; дестилиране на амоняка и поглъщане на амоняка в определен обем на разтвор на стандартна сярна киселина. Титруване на излишната сярна киселина с помощта на стандартен разтвор на натриев или калиев хидроксид.
4. Реактиви
Дестилирана или деминерализирана вода, несъдържаща въглероден диоксид и азотни съединения.
4.1. Разредена солна киселина: един обем HCl (d20 = 1,18 g/ml) плюс един обем вода.
4.2. Сярна киселина: 0,1 mol/l.
4.3. Разтвор на натриев или калиев хидроксид, несъдържащ карбонати: 0,1 mol/l.
4.4. Разтвор на сярна киселина, приблизително 30 % H2SO4 (w/v), несъдържащ амоняк.
4.5. Желязо на прах, редуцирано във водород (предписаното количество желязо трябва да може да редуцира поне 0,05 g нитратен азот).
4.6. Натриев хидроксид, приблизително 30 % NaOH (d20 = 1,33 g/ml) несъдържащ амоняк.
4.7. Индикаторни разтвори
4.7.1. Смесен индикатор
Разтвор А: Разтваря се 1 g метилово червено в 37 ml разтвор на натриев хидроксид 0,1 mol/l и се допълва до един литър с вода.
Разтвор Б: Разтваря се 1 g метиленово синьо във вода и се допълва до един литър.
Смесва се един обем от А с два обема от разтвор Б.
Този индикатор е виолетов в кисел разтвор, сив в неутрален разтвор и зелен в алкален разтвор. Използва се 0,5 ml (10 капки).
4.7.2. Индикаторен разтвор на метилово червено
Разтваря се 0,1 g метилово червено в 50 ml от 95 % етанол. Долива се до 100 ml с вода и се филтрува, ако е необходимо.
Този индикатор може да се използва (четири до пет капки) вместо предходния.
4.8. Пемза на гранули, промити в солна киселина и накалени
4.9. Натриев нитрат за анализ
5. Апаратура
Виж метод 2.1 „Определяне на амонячен азот“.
6. Приготвяне на пробата
Виж метод 2.1 „Определяне на амонячен азот“.
7. Метод за анализ
7.1. Приготвяне на разтвора
Виж метод 2.1 „Определяне на амонячен азот“.
7.2. Процедура
В приемната колба се поставя точно отмерено количество от 50 ml стандартна сярна киселина, както е посочено в таблица 1 на метод 2.1. (вариант а), и се добавя подходящо количество от избрания индикаторен разтвор (4.7.1 или 4.7.2). Краят на наставката на хладника трябва да е под повърхността на стандартната киселина в приемната колба.
С прецизна пипета се прехвърля аликвотна част от бистрия разтвор, както е посочено в таблица 1 на метод 2.1 (вариант а), и се поставя в дестилиращата колба на апарата. Добавят се 350 ml вода, 20 ml от 30 % разтвор на сярна киселина (4.4), разбърква се и се добавят 5 g редуцирано желязо (4.5). Промива се гърлото на колбата с няколко милилитра вода и в нея се поставя малка фуния с дълга опашка. Загрява се в кипяща водна баня около час и след това опашката на фунията се промива с няколко милилитра вода.
Като се внимава да не се причини загуба на амоняк, към съдържанието на дестилационната колба се добавят 50 ml концентриран разтвор на натриев хидроксид (4.6) или, в случаите, когато за разтворяне на пробата се използва 20 ml солна киселина (1 + 1) (4.1), 60 ml концентриран разтвор на натриев хидроксид (4.6). Дестилационният апарат се сглобява. Амонякът се дестилира по процедурата, дадена в метод 2.1.
7.3. Изпитване на празна проба
Празна проба се прави при същите условия и се използва при изчисляване на крайния резултат.
7.4. Контролно изпитване
Преди извършване на анализа, се проверява дали апартурата работи правилно и дали се прилага правилното методът, като се вземе аликвотна част прясно приготвен разтвор на натриев нитрат (4.9), съдържащ от 0,045 до 0,050 g азот.
8. Изразяване на резултатите
Резултатът от анализа се изразява като процент нитратен азот или комбинация от амонячен и нитратен азот, съдържащ в тора, получен при анализа.
Метод 2.2.2
Определяне на нитратен и амонячен азот по метода на Арнд
1. Обхват
Настоящият документ дефинира процедурата за определяне на нитратния и амонячния азот с редукция по метода Арнд (модифицирана за всеки от вариантите а, б и в).
2. Област на приложение
Виж метод 2.2.1.
3. Принцип
Редукция на нитрати и нитрити до амоняк в неутрален воден разтвор с метална сплав, състояща се от 60 % мед и 40 % магнезий (сплав на Арнд) в присъствието на магнезиев хлорид (МgСl2).
Дестилиране на амоняка и определяне на поглъщането му в определен обем стандартна сярна киселина. Титруване на излишната сярна киселина с помощта на стандартен разтвор на натриев или калиев хидроксид.
4. Реактиви
Дестилирана или деминерализирана вода, без въглероден диоксид и азотни съединения.
4.1. Разредена солна киселина: един обем HCl (d20 = 1,18 g/ml) плюс един обем вода
|
за вариант а. |
||
|
|||
|
за вариант б (вж. бележка 2, метод 2.1) |
||
|
|||
|
за вариант в (вж. бележка 2, метод 2.1) |
||
|
4.8. Разтвор на натриев хидроксид, приблизително 2 mol/l.
4.9. Сплав на Арнд за анализ: стрита на на прах така че да минава през сито с отвори по малки от 1 mm2.
4.10. 20 %-ен разтвор на магнезиев хлорид
Разтварят се 200 g магнезиев хлорид (MgCl2. 6H2O) в приблизително 600 до 700 ml вода в плоскодънна колба от 1 литър. За да се избегне образуването на пяна, се добавят 15 g магнезиев сулфат (MgSO4.7 H2O).
След разтварянето се добавят 2 g магнезиев оксид и няколко гранули пемза и суспензията се концентрира до 200 ml чрез кипене, като по този начин се отстранява всякакъв остатък от амоняк от реактивите. Охлажда се, обемът се допълва до един литър и се филтрира.
4.11. Индикаторни разтвори
4.11.1. Смесен индикатор
Разтвор А: Разтваря се 1 g метилово червено в 37 ml разтвор на натриев хидроксид 0,1 mol/l и се долива вода до един литър.
Разтвор Б: Разтваря се 1 g метиленово синьо във вода и се долива до един литър.
Смесва се един обем от разтвор А с два обема от разтвор Б.
Този индикатор е виолетов в кисел разтвор, сив в неутрален разтвор и зелен в алкален разтвор. Използват се 0,5 ml (10 капки).
4.11.2. Индикаторен разтвор метилово червено
Разтворят се 0,1 g метилово червено в 50 ml от 95 % етанол. Допълва се до 100 ml с вода и, ако е необходимо, се филтрира. Този индикатор може да се използва (четири до пет капки) вместо предходния.
4.11.3. Индикаторен разтвор конгочервено
Разтворят се 3 g конгочервено в един литър топла вода и се филтрира, ако е необходимо, след охлаждане. Този индикатор може да се използва вместо двата, описани по-горе, при неутрализацията на киселите екстракти преди дестилиране като се използват 0,5 ml на 100 ml течност за неутрализиране.
4.12. Противоударни гранули от пемза, промита със солна киселина и накалена.
4.13. Натриев нитрат за анализ.
5. Апаратура
Виж метод 2.1 „Определяне на амонячен азот“.
6. Приготвяне на пробата
Виж метод 1.
7. Метод за анализ
7.1. Приготвяне на разтвора за анализ
Виж метод 2.1 „Определяне на амонячен азот“.
7.2. Анализ на разтвора
Съгласно избрания вариант се поставя в събирателната колба точно измерено количество стандартна сярна киселина, както е посочено в таблица 1 на метод 2.1. Добавя се подходящо количество от избрания индикаторен разтвор (4.11.1 или 4.11.2) и накрая достатъчно вода, за да се получи обем от най-малко 50 ml. Краят на разширителната тръба на хладника трябва да бъде под повърхността на разтвора.
Като се използва прецизна пипета, се взема аликвотна част от бистлия разтвор съгласно таблица 1. Поставя се в дестилиращата колба.
Добавя се достатъчно вода, за да се получи общ обем 350 ml (виж бележка 1), 10 g сплав на Арнд (4.9), 50 ml разтвор на магнезиев хлорид (4.10) и няколко парченца пемза (4.12). Бързо се свързва колбата към дестилатора. Нагрява се внимателно в продължение на 30 минути. След това се увеличава нагряването, за да се дестилира амоняка. Продължава дестилацията за около час. След като мине това време, остатъкът в колбата трябва да има сиропена консистенция. Когато дестилирането свърши, количеството излишна киселина в събирателната колба се титрува съгласно процедурата в метод 2.1.
Бележка 1:
Когато разтворът на пробата е кисел (добавяне на 20 ml НСl (4.1) за разтваряне на пробата), взетата за анализ аликвотната част се неутрализира по следния начин: към дестилационната колба, съдържаща взетата аликвотна част, се добавят около 250 ml вода, необходимото количество от един от индикаторите (4.11.1, 4.11.2 или 4.11.3) и се разклаща внимателно.
Неутрализира се с 2 mol/l разтвор на натриев хидроксид (4.8) и се подкиселява с капка солна киселина (4.1) След това се процедира, както е посочено в 7.2 (втори ред).
7.3. Празна проба
Извършва се изпитване на празна проба при същите условия и се използва при изчисляване на крайния резултат.
7.4. Контролно изпитване
Преди извършване на анализа се проверява дали апаратурата работи правилно и дали се прилага правилно методът с използване на аликвотна част на прясно приготвен разтвор на амониев сулфат (4.13), съдържащ от 0,050 до 0,150 g нитратен азот в зависимост от избрания вариант.
8. Изразяване на резултатите
Виж метод 2.2.1.
Метод 2.2.3
Определяне на нитратен и амонячен азот съгласно Деварда
1. Обхват
Настоящият документ дефинира процедурата за определяне на нитратния и амонячния азот с редукция по метода Деварда (модифицирана за всеки от вариантите а), б) и в).
2. Област на приложение
Виж метод 2.2.1.
3. Принцип
Редуциране на нитрати и нитрити до амоняк в силно алкален разтвор с помощта на метална сплав, съставена от 45 % Al, 5 % Zn и 50 % Cu (сплав на Деварда). Дестилиране на амоняка и определяне на добива на амоняк в известен обем стандартен разтвор на сярна киселина. Титруване на излишната сярна киселина с помощта на стандартен разтвор на натриев или калиев хидроксид.
4. Реактиви
Дестилирана или деминерализирана вода, без въглероден диоксид и всички азотни съединения.
4.1. Разредена солна киселина: един обем HCl (d = 1,18 g/ml) плюс един обем вода
|
за вариант а. |
||
|
|||
|
за вариант б (виж бележка 2,метод 2.1). |
||
|
|||
|
за вариант в. (виж бележка 2, метод 2.1). |
||
|
4.8. Сплав на Деварда за анализ
Стрива се на прах така че 90 до 100 % да минава през сито с отвори, по-малки от 0,25 mm2, 50 до 75 % да минава през сито с отвори 0,075 mm2.
Препоръчва се използване на готов продукт в опаковки, съдържащи максимум 100 g.
4.9. Разтвор на натриев хидроксид, приблизително 30 % NaOH (d20 = 1,33 g/ml), несъдържащ амоняк.
4.10. Индикаторни разтвори
4.10.1. Смесен индикатор
Разтвор А: Разтваря се 1 g метилово червено в 37 ml разтвор на натриев хидроксид 0,1 mol/l и се допълва до един литър с вода.
Разтвор Б: Разтваря се 1 g метиленово синьо във вода и се долива до един литър.
Смесва се един обем от А с два обема от Б.
Този индикатор е виолетов в кисел разтвор, сив в неутрален разтвор и зелен в алкален разтвор. Използват се 0,5 ml (10 капки).
4.10.2. Индикаторен разтвор метилово червено
Разтваря се 0,1 g метилово червено в 50 ml от 95 % етанол. Долива се до 100 ml с вода и при необходимост се филтрира.
Този индикатор може да се използва (четири до пет капки) вместо предходния.
4.11. Етанол, 95 до 96 %.
4.12. Натриев нитрат за анализ.
5. Апаратура
Виж метод 2.1
5.1. Дестилатор, състоящ се от облодънна колба с подходящ обем, свързана към хладник с дестилираща тръба с капкоуловител, оборудван допълнително с уловител на мехури в приемната колба за предотвратяване загуба на амоняк.
Типът на одобрения апарат за това определяне е показан с всички конструкционни характеристики на фигура 5.
5.2. Пипети от 10, 20, 25, 50, 100 и 200 ml.
5.3. Мерителна колба от 500 ml.
5.4. Ротационна клатачна машина (35 до 40 оборота в минута).
6. Приготвяне на пробата
Виж метод 1.
7. Процедура
7.1. Приготвяне на разтвора за анализ
Виж метод 2.1 „Определяне на амонячен азот“.
7.2. Анализ на разтвора
Количеството нитратен азот в аликвотната част на разтвора не трябва да превишава максималното количество, посочено в таблица 1.
Съгласно избрания вариант в приемната колба се поставя точно измерено количество стандартна сярна киселина, както е посочено в таблица 1. Добавя се подходящо количество от избрания индикаторен разтвор (4.10.1 или 4.10.2) и накрая достатъчно вода, за да се получи обем от 50 ml. Краят на разширителната тръба на хладника трябва да бъде под повърхността на разтвора.
С прецизна пипета се взема аликвотна част от бистрия разтвор съгласно таблица 1 на метод 2.1. Поставя се в дестилационна колба.
Добавя се достатъчно вода, за да се получи общ обем 250 до 300 ml, 5 ml етанол (4.11) и 4 g сплав на Деварда (4.8) (виж бележка 2).
Като се внимава да се избегне загубата на амоняк, в колбата се добавят около 30 ml 30 % разтвор на натриев хидроксид (4.9) и накрая, ако пробите са кисело разтворими, допълнително количество, достатъчно да неутрализира количеството солна киселина (4.1), което присъства във взетата за анализа аликвотната част. Свързва се дестилационната колба към дестилатора, като се осигурява херметичност на връзката. Внимателно се разклаща колбата, за да се смеси съдържанието.
Нагрява се внимателно, така че отделянето на водород да се намали чувствително в продължение на около половин час и течността да закипи. Дестилацията продължава, като се увеличава нагряването, така че най-малко 200 ml течност да се дестилират за около 30 минути (дестилацията не трябва да продължава повече от 45 минути).
Когато дестилирането приключи, се демонтира приемната колба от апарата, внимателно се измиват разширителната тръба и уловителят на мехури, като се събира отмитото в колбата за титруване. Излишната киселина се титрува съгласно процедурата в метод 2.1.
Забележка 2
В присъствието на калциеви соли като калциев нитрат и калциев амониев нитрат е необходимо да се добави преди дестилирането за всеки грам от пробата в аликвота 0,700 g натриев фосфат (Na2HPO4. 2H2O), за да се предотврати образуването на Ca(OH)2.
7.3. Празна проба
Празната проба се прави при същите условия и се използва при изчисляване на крайния резултат.
7.4. Контролно изпитване
Преди извършване на анализа, трябва да се провери дали апаратурата работи добре и дали се прилага правилно методът, като се вземе аликвотна част от прясно приготвен разтвор на натриев нитрат (4.12), съдържащ от 0,050 до 0,150 g нитратен азот в зависимост от избрания вариант.
8. Изразяване на резултатите
Виж метод 2.2.1.
Фигура 5

Метод 2.3
Определяне на общ азот
Метод 2.3.1
Определяне на общия азот в калциев цианамид без нитрати
1. Обхват
Настоящият документ дефинира процедурата за определяне на общия азот в калциев цианамид, несъдържащ нитрати.
2. Област на приложение
Изключително за калциев цианамид (несъдържащ нитрати).
3. Принцип
След келдалово разлагане образуваният амонячен азот се замества от натриев хидроксид, който се събира и определя в стандартен разтвор на сярна киселина.
4. Реактиви
Дестилирана или деминерализирана вода, несъдържаща въглероден диоксид и всички азотни съединения.
4.1. Разредена сярна киселина (d20 = 1,54 g/ml): един обем сярна киселина (d20 = 1,84 g/ml) плюс един обем вода.
4.2. Калиев сулфат за анализ.
4.3. Меден оксид (CuO): 0,3 до 0,4 g за всяко определяне или еквивалентно количество меден сулфат пентахидрат (CuSO4.5H2O), от 0,95 до 1,25 g за всяко определение.
4.4. Разтвор на натриев хидроксид, приблизително 30 % NaOH (d20 = 1,33 g/ml), несъдържащ амоняк
|
за вариант а (виж метод 2.1). |
||
|
|||
|
за вариант б (виж бележка 2, метод 2.1). |
||
|
|||
|
за вариант в (виж бележка 2, метод 2.1). |
||
|
4.11. Индикаторни разтвори
4.11.1. Смесен индикатор
Разтвор А: Разтваря се 1 g метилово червено в 37 ml разтвор на натриев хидроксид 0,1 mol/l и се допълва до един литър с вода.
Разтвор Б: Разтваря се 1 g метиленово синьо във вода и се долива до един литър.
Смесва се един обем от А с два обема от Б.
Този индикатор е виолетов в кисел разтвор, сив в неутрален разтвор и зелен в алкален разтвор. Използва се 0,5 ml (10 капки).
4.11.2. Индикаторен разтвор метилово червено
Разтваря се 0,1 g метилово червено в 50 ml от 95 % етанол. Долива се до 100 ml с вода и се филтрира, ако е необходимо. Този индикатор може да се използва (четири до пет капки) вместо предходния.
4.12. Пемза на гранули, промита със солна киселина и накалена.
4.13. Калиев тиоцианат за анализ.
5. Апаратура
5.1. Дестилатор, виж метод 2.1 „Определяне на амонячен азот“.
5.2. Келдалова колба с дълго гърло с подходящ обем.
5.3. Пипети от 50, 100 и 200 ml.
5.4. Мерителна колба от 250 ml.
6. Приготвяне на пробата
Виж метод 1.
7. Процедура
7.1. Приготвяне на разтвора за анализ
Претегля се с точност до 0,001 g 1 g от пробата и се поставя в Келдаловата колба. Добавят се 50 ml разредена сярна киселина (4.1), 10 до 15 g калиев сулфат (4.2) и предписания катализатор (4.3). Загрява се бавно, за да се отдели водата, кипи се слабо в продължение на два часа, оставя се да се охлади и се разрежда със 100 до 150 ml вода. Охлажда се отново, прехвърля се количествено суспензията в мерителна колба от 250 ml, долива се до марката с вода, разклаща се и се филтрира в суха колба.
7.2. Анализ на разтвора
С пипета се прехвърля, съгласно избрания вариант (виж метод 2.1), 50, 100 или 200 ml от така получения разтвор и амонякът се дестилира, както е описано в метод 2.1, като се добавя достатъчен разтвор на натриев хидроксид (4.4), за да се осигури значителен излишък.
7.3. Празна проба
Извършва се изпитване на празна проба при същите условия и се използва при изчисляване на крайния резултат.
7.4. Контролно изпитване
Преди извършване на анализа се проверява дали апаратурата работи добре и дали се прилага правилно методът, като се използва аликвотна част на стандартен разтвор на калиев тиоцианат (4.13), съответстваща на концентрацията на азот в пробата.
8. Изразяване на резултатите
Резултатите се изразяват като процент азот (N), съдържащ се в тора, получен при анализа.
Вариант а: % N = (50 — А) × 0,7
Вариант б: % N = (50 — А) × 0,7
Вариант в: % N = (35 — А) × 0,875
Метод 2.3.2
Определяне на общия азот в калциев цианамид съдържащ нитрати
1. Обхват
Настоящият документ дефинира процедурата за определяне на общия азот в калциев цианамид.
2. Област на приложение
Изключително за калциев цианамид, съдържащ нитрати.
3. Принцип
Методът на Келдал не може да се приложи пряко за калциеви цианамиди, съдържащи нитрати. Поради тази причина нитратният азот се редуцира до амоняк с желязо на прах и калаен хлорид преди келдаловото разлагане.
4. Реактиви
Дестилирана или деминерализирана вода, несъдържаща въглероден диоксид и всички азотни съединения.
4.1. Сярна киселина (d20 = 1,84 g/ml).
4.2. Желязо на прах редуцирано във водород.
4.3. Калиев фосфат, фино пулверизиран, за анализ
|
за вариант а (виж метод 2.1). |
||
|
|||
|
за вариант б (виж бележка 2, метод 2.1). |
||
|
|||
|
за вариант в (виж бележка 2, метод 2.1) |
||
|
4.10. Индикаторни разтвори
4.10.1. Смесен индикатор
Разтвор А: Разтваря се 1 g метилово червено в 37 ml разтвор на натриев хидроксид 0,1 mol/l и се долива до един литър с вода.
Разтвор Б: Разтваря се 1 g метиленово синьо във вода и се долива до един литър.
Смесва се един обем от разтвор А с два обема от разтвор Б.
Този индикатор е виолетов в кисел разтвор, сив в неутрален разтвор и зелен в алкален разтвор. Използва се 0,5 ml (10 капки).
4.10.2. Индикаторен разтвор метилово червено
Разтваря се 0,1 g метилово червено в 50 ml от 95 % етанол. Долива се до 100 ml с вода и се филтрира, ако е необходимо. Този индикатор може да се използва (четири до пет капки) вместо предходния.
4.11. Разтвор на калаен хлорид
Разтваря се 120 g SnCl2. 2H2O в 400 ml концентрирана солна киселина (d20 = 1,18 g/ml) и се долива до един литър с вода. Разтворът трябва да бъде напълно бистър и да се приготви непосредствено преди употреба. Много е важно да се провери редукционната способност на калаения хлорид.
Забележка
Разтварят се 0,5 g SnCl2. 2H2O в концентрирана солна киселина (d20 = 1,18 g/ml) и се долива до 50 ml с вода. След това се добавят 5 g сегнетова сол (калиево-натриев тартарат) и достатъчно количество натриев бикарбонат за анализ, за да покаже разтворът алкална реакция при тест с лакмусова хартия.
Титрува се с 0,1 mol/l йоден разтвор в присъствието на разтвор на скорбяла като индикатор.
1 ml йоден разтвор 0,1 mol/l съответства на 0,01128 g SnCl2.2H2O.
Най-малко 80 % от общия калай в разтвора, приготвен по този начин, трябва да е в двувалентна форма. За титруване трябва да се използват най-малко 35 ml от йоден разтвор 0,1 mol/l.
4.12. Разтвор на натриев хидроксид, приблизително 30 % NaOH (d20 = 1,33 g/ml), несъдържаща амоняк
4.13. Стандартен нитратно-амонячен разтвор
Претеглят се 2,5 g калиев нитрат за анализ и 10,16 g амониев сулфат за анализ и се поставят в мерителна колба от 250 ml. Разтварят се във вода и се долива до 250 ml. 1 ml от този разтвор съдържа 0,01 g азот.
4.14. Пемза на гранули, промита със солна киселина и накалена.
5. Апаратура
Виж метод 2.3.1.
6. Приготвяне на пробата
Виж метод 1.
7. Процедура
7.1. Приготвяне на разтвора за анализ
Претегля се 1 g от пробата с точност до 0,001 g и се поставя в келдаловата колба. Добавят се 0,5 g желязо на прах (4.2) и 50 ml калаен хлорид (4.11), разбърква се и се оставя за половин час. През това време се разбърква отново след 10 и 20 минути. След това се добавят 10 g калиев сулфат (4.3) и 30 ml сярна киселина (4.1). Довежда се до кипене и процесът продължава един час след появата на бели пари. Оставя се да се охлади и се разрежда с 100 до 150 ml вода. Суспензията се пренася количествено в мерителна колба от 250 ml, охлажда се и се долива до марката с вода, разбърква се и се филтрира през сух филтър в сух съд. Вместо след това суспензията да се прекара през сифон, за да приложи вариант а, б или в, използвани в метод 2.1, амонячният азот в този разтвор може също така да се дестилира пряко след добавяне на достатъчно натриев хидроксид, за да се осигури голям излишък (4.12).
7.2. Анализ на разтвора
С пипета се пренасят, съгласно избрания вариант а, б или в (виж метод 2.1), 50, 100 или 200 ml от така получения разтвор. Амонякът се дестилира по процедурата, описана в метод 2.1, като в дестилационната колба се добавя достатъчно разтвор на натриев хидроксид (4.12), за да се осигури значителен излишък.
7.3. Празна проба
Извършва се изпитване на празна проба при същите условия и то се използва при изчисляването на крайния резултат.
7.4. Контролно изпитване
Преди извършване на анализа се проверява дали апаратурата работи добре и дали се прилага правилно методът със стандартен разтвор (4.13), съдържащ количества от амонячен и нитратен азот в количества, съответстващи на количествата цианамид и нитратен азот, съдържащи се в нитратния калциев цианамид.
За тази цел се поставя 20 ml стандартен разтвор (4.13) в келдаловата колба.
Анализът се извършва съгласно метода, описан в 7.1 и 7.2.
8. Изразяване на резултатите
Резултатите се изразяват като процент азот (N), съдържащ се в тора, получен при анализа.
Вариант а: % N = (50 — А) × 0,7
Вариант б: % N = (50 — А) × 0,7
Вариант в: % N = (35 — А) × 0,875
Метод 2.3.3
Определяне на общия азот в карбамид
1. Обхват
Настоящият документ дефинира процедурата за определяне на общия азот в карбамид.
2. Област на приложение
Настоящият метод се прилага изключително за карбамидни торове, несъдържащи нитрати.
3. Принцип
Карбамидът се превръща количествено в амоняк чрез кипене в присъствието на сярна киселина. Полученият по този начин амоняк се дестилира от алкална среда и дестилатът се събира с излишък на стандартна сярна киселина. Излишната киселина се титрува с помощта на стандартен алкален разтвор.
4. Реактиви
Дестилирана или деминерализирана вода, несъдържаща въглероден диоксид и всички азотни съединения.
4.1. Сярна киселина (d20 = 1,84 g/ml).
4.2. Разтвор на натриев хидроксид, приблизително 30 % NaOH (d20 = 1,33 g/ml), несъдържащ амоняк.
|
за вариант а (виж метод 2.1). |
||
|
|||
|
за вариант б (виж бележка 2, метод 2.1). |
||
|
|||
|
за вариант в (виж бележка 2, метод 2.1). |
||
|
4.9. Индикаторни разтвори
4.9.1. Смесен индикатор
Разтвор А: Разтваря се 1 g метилово червено в 37 ml разтвор на натриев хидроксид 0,1 mol/l и се допълва до един литър с вода.
Разтвор Б: Разтваря се 1 g метиленово синьо във вода и се долива до един литър.
Смесва се един обем от А с два обема от Б.
Този индикатор е виолетов в кисел разтвор, сив в неутрален разтвор и зелен в алкален разтвор. Използват се 0,5 ml (10 капки).
4.9.2. Индикаторен разтвор метилово червено
Разтваря се 0,1 g метилово червено в 50 ml от 95 % етанол. Долива се до 100 ml с вода и се филтрира, ако е необходимо. Този индикатор може да се използва (четири до пет капки) вместо предходния.
4.10. Пемза на гранули, промита със солна киселина и накалена.
4.11. Карбамид за анализ.
5. Апаратура
5.1. Дестилационен апарат, виж метод 2.1 „Определяне на амонячен азот“.
5.2. Мерителна колба от 500 ml.
5.3. Пипети от 25, 50, и 100 ml.
6. Приготвяне на пробата
Виж метод 1.
7. Процедура
7.1 Приготвяне на разтвора за анализ
Претеглят се 2,5 g с точност до 0,001 g от приготвената проба, поставят се в келдаловата колба от 300 ml и се овлажнява с 20 ml вода. При разбъркване се добавят 20 ml концентрирана сярна киселина (4.1) и няколко стъклени перли, за да се предотврати кипенето с енергично отделяне на пара. За да се избегне образуването на пръски, в гърлото на колбата се поставя стъклена фуния с дълга опашка. Отначало се загрява бавно, след това температурата се увеличава, докато се появят бели пари (30 до 40 минути).
Охлажда се и се разрежда със 100 до 150 ml вода. Прехвърля се количествено в мерителна колба от 500 ml, като се изхвърля утайката. Оставя се да се охлади при стайна температура, долива се до обема вода, разбърква се и ако е необходимо, се филтрира през сух филтър в сух приемник.
7.2. Анализ на разтвора
С прецизна пипета се прехвърля 25, 50 или 100 ml от така получения разтвор в дестилационната колба съгласно избрания вариант (виж метод 2.1). Амонякът се дестилира, както е описано в метод 2.1, като се добавя достатъчно NaOH (d20 = 1,33 g/ml), (4.2) в дестилационната колба, за да се осигури значителен излишък.
7.3. Празна проба
Извършва се изпитване на празна проба при същите условия и то се използва при изчисляване на крайния резултат.
7.4. Контролно изпитване
Преди извършване на анализа се проверява дали апаратурата работи добре и дали се прилага правилно методът, като се използва аликвотна част на прясно приготвен разтвор на карбамид (4.11).
8. Изразяване на резултатите
Резултатите се изразяват като процент азот (N), съдържащ се в тора, получен при анализа.
Вариант а: % N = (50 — А) × 1,12
Вариант б: % N = (50 — А) × 1,12
Вариант в: % N = (35 — А) × 1,40
Метод 2.4
Определяне цианамиден азот
1. Обхват
Настоящият документ дефинира процедурата за определяне на цианамиден азот.
2. Област на приложение
Калциев цианамид и смеси от калциеви цианамид и нитратни.
3. Принцип
Цианамидният азот се утаява като сребърен комплекс и се определя по метода на Келдал.
4. Реактиви
Дестилирана или деминерализирана вода, несъдържаща въглероден диоксид и всички азотни съединения.
4.1. Ледена оцетна киселина.
4.2. Амонячен разтвор, съдържащ 10 % амонячен газ от масата (d20 = 0,96 g/ml).
4.3. Амонячен сребърен разтвор по Толенс
Смесва се 500 ml 10 % разтвор на сребърен нитрат (AgNO3) във вода с 500 ml 10 % амоняк (4.2).
Да не се излага ненужно на светлина, топлина или въздух. Разтворът нормално трае години. Докато разтворът е бистър, реактивът е с добро качество.
4.4. Концентрирана сярна киселина (d20 = 1,84 g/ml).
4.5. Калиев сулфат за анализ.
4.6. Меден оксид (CuO): 0,3 до 0,4 g за всяко определяне или еквивалентно количество меден сулфат пентахидрат (CuSO4.5H2O) от 0,95 до 1,25 g за всяко определяне.
4.7. Разтвор на натриев хидроксид, приблизително 30 % NaOH (d20 = 1,33 g/ml), несъдържащ амоняк.
4.8. Сярна киселина: 0,1 mol/l.
4.9. Разтвор на натриев или калиев хидроксид, несъдържащ карбонати: 0,1 mol/l.
4.10. Индикаторни разтвори.
4.10.1. Смесен индикатор.
Разтвор А: Разтваря се 1 g метилово червено в 37 ml разтвор на натриев хидроксид 0,1 mol/l и се допълва до един литър с вода.
Разтвор Б: Разтваря се 1 g метиленово синьо във вода и се долива до един литър.
Смесва се един обем от разтвор А с два обема от разтвор Б.
Този индикатор е виолетов в кисел разтвор, сив в неутрален разтвор и зелен в алкален разтвор. Използва се 0,5 ml (10 капки).
4.10.2. Индикаторен разтвор метилово червено.
Разтваря се 0,1 g метилово червено в 50 ml от 95 % етанол. Долива се до 100 ml с вода и се филтрира, ако е необходимо. Този индикатор може да се използва (четири до пет капки) вместо предходния.
4.11. Пемза на гранули, промита със солна киселина и накалена.
4.12. Калиев тиоцианид за анализ.
5. Апаратура
5.1. Дестилатор, виж метод 2.1 „Определяне на амонячен азот“.
5.2. Мерителна колба от 500 ml (например Стохман).
5.3. Келдалова колба с дълго гърло и подходящ обем (300 до 500 ml).
5.4. Пипета от 50 ml.
5.5. Ротационна клатачна машина (35 до 40 оборота в минута).
6. Приготвяне на пробата
Виж метод 1.
7. Процедура
7.1. Предпазни мерки
Когато се използва разтвор на амонячно сребро, трябва да се носят предпазни очила. Веднага след като се образува тънка мембрана на повърхността на течността, може да възникне експлозия и затова е извънредно важно да се работи с най-голяма предпазливост.
7.2. Приготвяне на разтвора за анализ
Претеглят се 2,5 g от пробата с точност до 0,001 g и се поставят в малък стъклен хаван. Пробата се смила три пъти с вода, като водата се излива след всяко смилане в мерителна колба на Стохман от 500 ml (5.2). Прехвърля се количествено пробата в мерителната колба на Стохман от 500 ml, като хаванът, пестикът и фунията се промиват с вода. Долива се вода до около 400 ml. Добавят се 15 ml оцетна киселина (4.1). Разклаща се на ротационната клатачна машина (5.5) в продължение на два часа.
Долива се до 500 ml вода, хомогенизира се и се филтрира.
Анализът трябва да де извърши колкото е възможно по-бързо.
7.3. Анализ на разтвора
Прехвърлят се 50 ml от филтрата в бехерова чаша 250 ml.
Добавя се амонячен разтвор (4.2), докато не стане леко алкален и след това се добавят 30 ml топъл амонячен сребърен нитрат (4.3), за да се утаи жълтият сребърен комплекс на цианамида.
Оставя се да престои една нощ, след което се филтрира, а утайката се промива със студена вода, докато се освободи от амоняка.
Поставя се филтърът и утайката, все още влажни, в келдалова колба, добавят се 10 до 15 g калиев сулфат (4.5) и катализатора (4.6) в предписаната пропорция, след това 50 ml вода и 25 ml концентрирана сярна киселина (4.4).
Колбата се загрява бавно, като се разклаща внимателно, докато съдържанието започне да кипи. Увеличава се топлината, поддържа се кипенето, докато съдържанието стане безцветно или бледо зелено.
Кипенето продължава един час, след това се оставя да изстине.
Прехвърля се течността количествено от келдаловата колба в дестилационната колба, добавят се няколко гранули пемза (4.11) и се долива вода до общ обем приблизително 350 ml. Разбърква се и се охлажда.
Амонякът се дестилира съгласно метод 2.1, вариант а, като се добавя достатъчно разтвор на NaOH (4.7), за да се осигури значителен излишък.
7.4. Празна проба
Извършва се изпитване на празна проба при същите условия и се използва при изчисляване на крайния резултат.
7.5. Контролно изпитване
Преди извършване на анализа се проверява дали апаратурата работи добре и дали се прилага правилното методът, като се използва аликвотна част на стандартен разтвор на калиев тиоцианит (4.12), съответстващ на 0,05 g азот.
8. Изразяване на резултатите
Резултатът се изразява като процент цианамиден азот, съдържащ се в тора, получен при анализа.

Метод 2.5
Спектрофотометрично определяне на биурет в карбамид
1. Обхват
Настоящият документ дефинира процедурата за определяне на биурет в карбамид.
2. Област на приложение
Методът се прилага изключително за карбамид.
3. Принцип
В алкална среда, в присъствието на калиево-натриев тартарат, биуретът и двувалентната мед образуват виолетово медно съединение. Оптичната плътност на разтвора се измерва при дължина на вълната около 546 nm (нанометра).
4. Реактиви
Дестилирана или деминерализирана вода, несъдържаща въглероден диоксид и амоняк. Качеството на тази вода е особено важно за това определяне.
4.1. Метанол.
4.2. Разтвор на сярна киселина, около 0,1 mol/l.
4.3. Разтвор на натриев хидроксид, около 0,1 mol/l.
4.4. Алкален разтвор на калиево-натриев тартарат.
В мерителна еднолитрова колба се разтварят 40 g натриев хидроксид в 500 ml вода и се оставя да изстине. Добавят се 50 g калиево-натриев тартарат (NaKC4H4O6. H2O). Долива се до марката. Оставя се да престои 24 часа преди употреба.
4.5. Разтвор на меден сулфат.
В мерителна еднолитрова колба се разтварят 15 g меден сулфат пентахидрат (CuSO4. 5H2O) в 500 ml вода. Долива се до марката.
4.6. Прясно приготвен стандартен разтвор на биурет.
В мерителна колба от 250 ml се разтваря 0,250 g чист биурет (9) във вода. Долива се до 250 ml. В 1 ml от този разтвор съдържа 0,001 g биурет.
4.7. Индикаторен разтвор.
В мерителна колба от 100 ml се разтваря 0,1 g метилово червено в 50 ml от 95 % етанол. Долива се до 100 ml с вода. Филтрира се, ако останат неразтворени частици.
5. Апаратура
5.1. Спектрометър или фотометър с филтри с чувствителност и точност, които позволяват да се репродуцират мерки по-малки от 0,5 % Т (10).
5.2. Мерителни колби от 100, 250 и 1 000 ml.
5.3. Градуирани пипети от 2, 5, 10, 25 и 50 ml или бюрета от 25 ml градуирана на деления 0,05 ml.
5.4. Бехарева чаша от 250 ml.
6. Приготвяне на пробата
Виж метод 1.
7. Процедура
7.1. Приготвяне на калибровъчната крива
Прехвърлят се 0, 2, 5, 10, 25 и 50 ml от аликвота на стандартен разтвор на биурет (4.6) в серия от седем мерителни колби 100 ml. Допълва се до около 50 ml с вода, добавя се една капка индикатор (4.7) и се неутрализира, ако е необходимо, със сярна киселина 0,1 mol/l (4.2). При разбъркване се добавят 20 ml от алкалния тартаратен разтвор (4.4), след това 20 ml разтвор на меден сулфат (4.5).
Бележка:
Тези разтвори (4.4 и 4.5) трябва да се дозират с две прецизни бюрети или още по-добре с пипети.
Долива се до 100 ml с дестилирана вода, разбърква се и се оставя да престои 15 минути при 30(± 2) °С.
С „0“ стандартен разтвор на биурет ката референция се измерва оптичната плътност на всеки разтвор при дължина на вълната около 546 nm, като се използва клетки с подходяща дължина на пътя.
Начертава се калибровъчната крива, като се използват оптичните плътности като ординати и съответните количества биурет (в милиграми) като абсциси.
7.2. Приготвяне на разтвора за анализ
Претеглят се 10 g от приготвената проба с точност до 0,001 g. Разтварят се в около 150 ml вода в мерителна колба от 250 ml и се долива до марката. Филтрира се, ако е необходимо.
Забележка 1:
Ако пробата за анализ съдържа повече от 0,015 g амонячен азот, тя се разтваря в бехерова чаша 250 ml в 50 ml метанол (4.1). Редуцира се чрез изпарение до обем от около 25 ml. Прехвърля се количествено в мерителна колба от 250 ml. Долива се до марката с вода. Филтрира се, ако е необходимо, през сух филтър в сух съд.
Забележка 2:
Отстраняване на опалесценция: ако има някакво колоидно вещество, могат да възникнат трудности при филтрирането. Разтворът, предназначен за анализ, в този случай се приготвя, както следва: разтваря се пробата за анализ в 150 ml вода, добавят се 2 ml солна киселина 1 mol/l и разтворът се филтрира през два плоски много фини филтри в мерителна колба от 250 ml. Филтрите се промиват с вода и се долива до марката. Процесът продължава съгласно метода, описан в 7.3 „Определяне“.
7.3. Определяне
В съответствие с очакваното съдържание на биурет се прехвърлят 25 до 50 ml от разтвора, посочен в 7.2, с пипета, поставя се това количество в мерителна колба от 100 ml и се неутрализира, ако е необходимо, с 0,1 mol/l реактив (4.2 или 4.3), както се изисква, с използване на метилово червено като индикатор, и се добавя със същата точност, като тази, използвана при начертаването на калибровъчната крива, 20 ml от алкалния разтвор на калиево-натриевия тартарат (4.4) и 20 ml от медния разтвор (4.5). Долива се до обема, разбърква се старателно и се оставя да престои 15 минути при 30(± 2) °С.
След това се извършват фотометричните измервания и се изчислява количеството биурет в карбамида.
8. Изразяване на резултатите
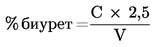
където
|
|
„С“ е масата, в милиграми, на биурета, отчетена от калибровъчната крива, |
|
|
„V“ е обемът на аликвотната част. |
9. Приложение
Ако „Jo“ е интензитетът на снопа монохроматичните лъчи (на определена дължина на вълната), преди да мине през прозрачно тяло, а „J“ е интензитетът след преминаването то:
|
— коефициент на пропускане: |
|
|
— непрозрачност: |
|
|
— оптична плътност: |
|
|
— оптична плътност на единица оптичен цикъл: |
|
|
— коефициент на специфична оптична плътност: |
|


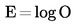


където
s= дебелина на слоя в сантиметри,
c= концентрацията в милиграми на литър,
k= специфичен коефициент за всяко вещество в закона на Ламбер-Беър.
Методи 2.6
Определяне на различни форми на азота в една и съща проба
Метод 2.6.1
Определяне на различни форми на азота в една и съща проба в торове съдържащи нитратен, амонячен, карбамиден и цианамиден азот
1. Обхват
Настоящият документ дефинира процедурата за определяне на всяка една форма в присъствието на всяка друга форма.
2. Област на приложение
Всички торове, предвидени в приложение I, които съдържат азот в различни форми.
3. Принцип
3.1. Общ разтворим и неразтворим азот
Съгласно списъка на стандартните торове (приложение I), това определяне е приложимо за продукти, които съдържат калциев цианамид.
|
3.1.1. |
При отсъствие на нитрати изпитваната проба се минерализира чрез директно келдалово разлагане. |
|
3.1.2. |
При наличие на нитрати изпитваната проба се минерализира чрез пряко келдалово разлагане след редуциране с помощта на желязо на прах и калаен хлорид. |
В двата случая амонякът се определя съгласно метод 2.1.
Бележка:
Ако анализът показва съдържание на неразтворим азот по-голямо от 0,5 %, заключението е, че торът съдържа други форми на неразтворим азот, които не са включени в списъка на приложение I.
3.2. Форми на разтворим азот
Следното се определя от различни аликвотни части, взети от един и същи разтвор на пробата:
|
3.2.1. |
Общ разтворим азот:
|
|
3.2.2. |
Общ разтворим азот с изключение на нитратен азот чрез келдалово разлагане след елиминиране в кисела среда на нитратния азот с железен сулфат, като амоняка се определя, както е описано в метод 2.1. |
|
3.2.3. |
Нитратен азот с разлика.
|
|
3.2.4. |
Амонячен азот:
|
|
3.2.5. |
Амиден азот:
|
|
3.2.6. |
Цианамиден азот, чрез утаяване като сребърно съединение, като азотът в утайката се преценява по метода на Келдал. |
4. Реактиви
Дестилирана или деминерализирана вода.
4.1. Калиев сулфат за анализ.
4.2. Желязо на прах, редуцирано с водород (предписаното количество желязо трябва да може да редуцира най-малко 50 mg нитратен азот).
4.3. Калиев тиоцианит за анализ.
4.4. Калиев нитрат за анализ.
4.5. Амониев сулфат за анализ.
4.6. Карбамид за анализ.
4.7. Разредена сярна киселина 1:1 по обем: един обем сярна киселина (d20 = 1,84 g/ml) в един обем вода.
4.8. Стандартен разтвор на сярна киселина: 0,2 mol/l.
4.9. Концентриран разтвор на натриев хидроксид. Воден разтвор при приблизително 30 % (w/v) на NaOH, несъдържащ амоняк.
4.10. Стандартен разтвор на натриев или калиев хидроксид: 0,2 mol/l, несъдържащ карбонати
4.11. Калаено-хлориден разтвор
Разтварят се 120 g SnCl2.2H2O в 400 ml концентрирана солна киселина (d20 = 1,18 g/ml) и се долива до един литър с вода. Разтворът трябва да е идеално бистър и да е приготвен непосредствено преди употреба.
Бележка:
От първостепенна важност е да се провери редукционната способност на калаения хлорид. Разрежда се 0,5 g SnCl2. 2H2O в концентрирана солна киселина (d20 = 1,18 g/ml) и се долива до 50 ml с вода. След това се добавят 5 g сегнетова сол (калиево-натриев тартарат) и достатъчно количество натриев бикарбонат за анализ, за да покаже разтворът алкална реакция при тест с лакмусова хартия.
Титрува се с 0,1 mol/l йоден разтвор в присъствието на разтвор на скорбяла като индикатор.
1 ml йоден разтвор 0,1 mol/l съответства на 0,01128 g SnCl2.2H2O.
Най-малко 80 % от общия калай в разтвора, приготвен по този начин, трябва да бъде в двувалентна форма. За титруване трябва да се използват най-малко 35 ml йоден разтвор 0,1 mol/l.
4.12. Сярна киселина (d20 = 1,84 g/ml).
4.13. Разредена солна киселина: един обем солна киселина (d20 = 1,18 g/ml) плюс един обем вода.
4.14. Оцетна киселина: 96 до 100 %.
4.15. Разтвор на сярна киселина, съдържащ около 30 % H2SO4 (w/v).
4.16. Железен сулфат: кристален, FeSO4 = 7H2O.
4.17. Стандартен разтвор на сярна киселина: 0,1 mol/l.
4.18. Октилов алкохол.
4.19. Наситен разтвор на калиев карбонат.
4.20. Стандартен разтвор на натриев или калиев хидроксид: 0,1 mol/l (несъдържащ карбонати).
4.21. Наситен разтвор на бариев хидроксид.
4.22. Разтвор на натриев карбонат: при 10 % (w/v).
4.23. Солна киселина: 2 mol/l.
4.24. Стандартен разтвор на солна киселина: 0,1 mol/l.
4.25. Разтвор на уреаза
Прави се суспензия на 0,5 g активна уреаза в 100 ml дестилирана вода, като се използва солна киселина 0,1 mol/l (4.24), регулира се рН на 5,4, измерено с рН-метър.
4.26. Ксантхидрол
Разтвор при 5 % етанол или метанол (4.31) (не се използват продукти, които дават високо съдържание на неразтворимо вещество). Разтворът може да се съхранява три месеца в добре затворена бутилка на неосветено място.
4.27. Меден окис (CuO): 0,3 до 0,4 g за всяко определяне или еквивалентно количество меден сулфат пентахидрат (CuSO4. 5H2O) от 0,95 до 1,25 g за всяко определяне.
4.28. Пемза на гранули, промита със солна киселина и накалена.
4.29 Индикаторни разтвори.
4.29.1. Разтвор А: Разтваря се 1 g метилово червено в 37 ml разтвор на натриев хидроксид 0,1 mol/l и се допълва до един литър с вода.
Разтвор Б: Разтваря се 1 g метиленово синьо във вода и се долива до един литър.
Смесва се един обем от А с два обема от Б.
Този индикатор е виолетов в кисел разтвор, сив в неутрален разтвор и зелен в алкален разтвор. Използват се 0,5 ml (10 капки) от този индикаторен разтвор.
4.29.2. Индикаторен разтвор метилово червено.
Разтваря се 0,1 g метилово червено в 50 ml от 95 % етанол. Долива се до 100 ml с вода и се филтрира, ако е необходимо. Този индикатор може да се използва (четири до пет капки) вместо предходния.
4.30. Индикаторни хартии.
Лакмус бромтимолово синьо (или други хартии, чувствителни на рН от 6 до 8).
4.31. Етанол или метанол: разтвор 95 %.
5. Апаратура
5.1. Дестилатор.
Виж метод 2.1.
5.2. Апарати за определянето на амонячен азот по аналитичен метод 7.2.5.3 (виж фигура 6).
Апаратурата се състои от приемник със специална форма с шлифовано стъклено гърло, със странично гърло, свързваща тръба с капкоуловител и перпендикулярна тръба за вкарване на въздух. Тръбите могат да бъдат свързани към приемника с помощта на проста перфорирана каучукова втулка. Важно е да се даде подходяща форма на края на тръбите, които вкарват въздух, тъй като газовите мехури трябва да се разпределят идеално в целите разтвори, които се съдържат в приемника и абсорбера. Най-доброто решение се състои от малки гъбообразни части с външен диаметър 20 mm и шест отвора от 1 mm по периферията.
5.3. Апарат за оценка на амиден азот съгласно технологията на уреаза (7.2.6.1)
Тя се състои от ерленмайерова колба от 300 ml с разделяща фуния и малък абсорбер (виж фигура 7).
5.4. Ротационна клатачна машина (35 до 40 оборота в минута).
5.5. рН-метър.
5.6. Пригодена пеш
5.7. Стъклария:
пипети от 2, 5, 10, 20, 25, 50 и 100 ml,
Келдалови колби с дълго гърло от 300 и 500 ml,
мерителни колби от 100, 250, 500 и 1 000 ml,
тигели от синтеровано стъкло, диаметър на порите от 5 до 15 μ,
хавани.
6. Приготвяне на пробата
Виж метод 1.
7. Технологии за анализ
7.1. Общ разтворим и неразтворим азот.
7.1.1. При отсъствие на нитрати
7.1.1.1. Разлагане
Претегля се с точност до 0,001 g количество от пробата, съдържащо най-много 100 mg азот. Поставя се в колбата на дестилатора (5.1). Добавя се 10 до 15 g калиев сулфат (4.1), катализатора (4.27) и няколко гранули пемза (4.28). Добавят се 50 ml разредена сярна киселина (4.7) и се смесва внимателно. Отначало се загрява, като внимателно се разбърква от време на време, докато вече не се образуват пари. След това се нагрява, така че течността да кипи постоянно. Поддържа се кипенето в продължение на един час след като разтвора стане бистър, като се предотвратява полепването на органични вещества по стените на колбата. Оставя се да се охлади. Внимателно се добавят около 350 ml вода, като се смесва. Разтварянето трябва да е колкото е възможно по-пълно. Оставя се да се охлади и се свързва колбата към дестилатора (5.1).
7.1.1.2. Дестилиране на амоняк
Прехвърля се с прецизна пипета в приемника на апарата 50 ml стандартен разтвор на сярна киселина 0,2 mol/l (4.8). Добавя се индикатор (4.29.1 или 4.29.2). Накрайникът на хладника трябва да бъде най-малко 1 cm под нивото на разтвора.
Като се вземат необходимите предпазни мерки, за да се избегне загуба на амоняк, внимателно се добавя към дестилационната колба достатъчно количество от концентрирания разтвор на натриев хидроксид (4.9), за да стане течността силно алкална (120 ml по правило са достатъчни). Проверява се чрез добавяне на няколко капки фенолфталеин. Накрая на дестилацията разтворът в колбата трябва да бъде все още ясно алкален). Регулира се нагряването на колбата така че да се дестилират 150 ml за половин час. Тества се с индикаторна хартия (4.30) дали дестилирането е приключило. Ако не е, дестилират се допълнително 50 ml и тестът се повторя, докато допълнителният дестилат реагира неутрално на индикаторната хартия (4.30). След това приемникът се сваля, дестилират се няколко милиметра повече и се промива накрайникът на хладника. Титрува се излишъкът от киселина със стандартен разтвор на калиев или натриев хидроксид 0,2 mol/l (4.10), докато индикаторът промени своя цвят.
7.1.1.3. Празна проба
Извършва се изпитване на празна проба при същите условия и то се използва при изчисляване на крайния резултат.
7.1.1.4 Изразяване на резултата

където
|
а |
= |
ml стандартен разтвор на натриев или калиев хидроксид 0,2 mol/l, използван за изпитване на празната проба, извършено чрез прехвърля се с пипетане в приемника на апарата (5.1) на 50 ml стандартен разтвор на сярна киселина 0,2 mol/l (4.8), |
|
А |
= |
ml стандартен разтвор на натриев или калиев хидроксид 0,2 mol/l, използван за анализа, |
|
М |
= |
маса на изпитваната проба, в грамове. |
7.1.2 При наличието на нитрат
7.1.2.2.1. Проба за изпитване
Претегля се с точност до 0,001 g количество от пробата, съдържаща не повече от 40 mg нитратен азот.
7.1.2.2. Редуциране на нитрат
Смесва се пробата за изпитване в малък хаван с 50 ml вода. Прехвърля се с минимално количество дестилирана вода в келдалова колба от 500 ml. Добавят се 5 g редуцирано желязо (4.2) и 50 ml разтвор на калаен хлорид (4.11). Разклаща се и се оставя да престои в продължение на половин час. През времето на престоя се разбърква отново след 10 и 20 минути.
7.1.2.3. Келдалово разлагане
Добавят се 30 ml сярна киселина (4.12), 5 g калиев сулфат (4.1), предписаното количество катализатор (4.27) и няколко гранули пемза (4.28). Нагрява се внимателно леко наклонената колба. Топлината се увеличава бавно и разтворът се разклаща често, за да се поддържа сместа суспензирана: течността потъмнява и след това се избистря с образуването на жълто-зелена безводна суспензия на железен сулфат. Нагряването продължава един час след получаване на бистър разтвор, като се поддържа точката на бавно кипене. Оставя се да се охлади. Внимателно се смива съдържанието на колбата с малко вода и се добавя малко по малко 100 ml вода. Смесва се и се прехвърля съдържанието на колбата в мерителна колба от 500 ml. Допълва се до марката с вода. Хомогенизира се. Филтрира се през сух филтър в сух събирателен съд.
7.1.2.4. Анализ на разтвора
Прехвърля се с пипета в колбата на дестилатора (5.1) аликвотна част, съдържаща най-много 100 mg азот. Разрежда се до около 350 ml с дестилирана вода, добавят се няколко гранули пемза (4.28), свързва се колбата към дестилатора и определянето продължава, както е описано в 7.1.1.2.
7.1.2.5. Празна проба
Виж 7.1.1.3.
7.1.2.6 Изразяване на резултата
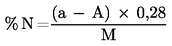
където
|
а |
= |
ml стандартен разтвор на натриев или калиев хидроксид 0,2 mol/l, използван за изпитване на празната проба, извършено чрез прехвърля се с пипетане в приемника на апарата (5.1) на 50 ml стандартен разтвор на сярна киселина 0,2 mol/l (4.8), |
|
А |
= |
ml стандартен разтвор на натриев или калиев хидроксид 0,2 mol/l, използван за анализа, |
|
М |
= |
маса на изпитваната проба, в грамове, присъстваща в аликвотната част, взета в 7.1.2.4. |
7.2. Форми на разтворим азот
7.2.1. Приготвяне на разтвора за анализиране
Претеглят се 10 g от пробата с точност до 1 mg и се поставят в мерителна колба от 500 ml.
7.2.1.1. В случай на торове, които не съдържат цианамиден азот
Добавят се в колбата 50 ml вода и след това 20 ml разредена солна киселина (4.13). Разклаща се и се оставя да престои, докато образуването на въглероден диоксид престане. След това се добавят 400 ml вода и се разклаща в продължение на половин час на ротационна клатачна машина (5.4). Долива се до обема с вода, смесва се и се филтрира през сух филтър в сух събирателен съд.
7.2.1.2. В случай на торове, които съдържат цианамиден азот.
Добавят се в колбата 400 ml и няколко капки метилово червено (4.29.2). Ако е необходимо, разтворът се подкиселява с използване на оцетна киселина (4.14). Добавят се 15 ml оцетна киселина (4.14). Разклаща се на ротационната клатачна машина в продължение на два часа (5.4). Ако е необходимо, разтворът се подкиселява отново по време на работа, като се използва оцетна киселина (4.14). Долива се до обема с вода, смесва се, филтрира се незабавно през сух филтър в сух приемник и незабавно се определя цианамидният азот.
И в двата случая се определят различните разтворими форми на азот в същия ден, когато е направен разтворът, като се започне с цианамидния азот и карбамидния азот, ако те присъстват.
7.2.2. Общ разтворим азот
7.2.2.1. При отсъствие на нитрат
Прехвърля се с пипета се в келдалова колба от 300 ml аликвотна част на филтрат (7.2.1.1. или 7.2.1.2), съдържащ най-много100 mg азот. Добавят се 15 ml концентрирана сярна киселина (4.12), 0,4 g меден оксид или 1,25 g меден сулфат (4.27) и няколко гранули пемза (4.28). Първо се загрява внимателно, за да започне изваряването и след това при по-високи температури, докато течността стане безцветна или слабо зеленикава и ясно се виждат бели пари. След охлаждане количествено се прехвърля разтворът в дестилационната колба, разрежда се до около 500 ml с вода и се добавят няколко гранули пемза (4.28). Свързва се колбата към дестилатора (5.1) и определянето продължава, както е описано в 7.1.1.2.
7.2.2.2 При наличието на нитрат
Прехвърля се с прецизна пипета в ерленмайрова колба от 500 ml аликвотна част от филтрата (7.2.1.1 или 7.2.1.2), съдържаща не повече от 40 mg нитратен азот. На този етап на анализа общото количество на азота не е важно. Добавя се 10 ml сярна киселина на 30 % (4.15), 5 g редуцирано желязо (4.2) и веднага се покрива ерленмайеровата колба с наблюдателно стъкло. Нагрява се внимателно, докато реакцията е стабилна, но не бурна. В този момент нагряването се прекратява и се оставя колбата да престои в продължение на най-малко три часа при температура на околната среда. С вода количествено се прехвърля течността в мерителна колба от 250 ml, като неразтвореното желязо се оставя в другия съд и се допълва до марката с вода. Смесва се старателно и се прехвърля с прецизна пипета в келдалова колба от 300 ml аликвотна част, съдържаща не повече от 100 mg азот. Добавят се 15 ml концентрирана сярна киселина (4.12), 0,4 g меден оксид или 1,25 g меден сулфат (4.27) и няколко гранули пемза (4.28). Първо се загрява внимателно, за да започне изваряването, и след това при по-високи температури, докато течността стане безцветна или слабо зеленикава и се образуват бели пари. След охлаждане разтворът се прехвърля количествено в дестилационната колба, разрежда се до около 500 ml с вода и се добавят няколко гранули пемза (4.28). Свързва се колбата към дестилатора (5.1) и определянето продължава, както е описано в 7.1.1.2.
7.2.2.3. Празна проба
Виж 7.1.1.3.
7.2.2.4. Изразяване на резултата

където
|
а |
= |
ml стандартен разтвор на натриев или калиев хидроксид 0,2 mol/l, използван за изпитване на празната проба, извършено чрез прехвърля се с пипетане в приемника на апарата (5.1) на 50 ml стандартен разтвор на сярна киселина 0,2 mol/l (4.8), |
|
А |
= |
ml стандартен разтвор на натриев или калиев хидроксид 0,2 mol/l, използван за анализа, |
|
М |
= |
маса на изпитваната проба, в грамове, присъстваща в аликвотната част, взета в 7.2.2.1 или 7.2.2.2. |
4.2.3. Общ разтворим азот с изключение на нитратен азот
Прехвърля се с прецизна пипета в келдалова колба от 300 ml аликвотна част на филтрата (7.2.1.1 или 7.2.1.2), съдържаща не повече от 50 mg азот за определяне. Разрежда се до 100 ml с вода, се добавя 5 g железен сулфат (4.16), 20 ml концентрирана сярна киселина (4.12) и няколко гранули пемза (4.28). Първо се загрява внимателно, за да започне изваряването и след това при по-високи температури, докато се появят бели пари. Изваряването продължава 15 минути. Спира се нагряването, добавя се меден оксид (4.27) като катализатор и се поддържа такава температура, че да се отделят бели пари в продължение допълнително на 10 до 15 минути. След охлаждане количествено се прехвърля разтворът в Келдаловата колба на апарата (5.1). Разрежда се до около 500 ml с вода и се добавят няколко гранули пемза (4.28). Свързва се колбата към дестилатора и определянето продължава, както е описано в 7.1.1.2.
7.2.3.1. Празна проба
Виж 7.1.1.3.
7.2.3.2. Изразяване на резултата

където
|
а |
= |
ml стандартен разтвор на натриев или калиев хидроксид 0,2 mol/l, използван за изпитване на празната проба, извършено чрез прехвърля се с пипетане в приемника на апарата (5.1) на 50 ml стандартен разтвор на сярна киселина 0,2 mol/l (4.8), |
|
А |
= |
ml стандартен разтвор на натриев или калиев хидроксид 0,2 mol/l, използван за анализа, |
|
М |
= |
маса на изпитваната проба, в грамове, присъстваща в аликвотната част. |
7.2.4. Нитратен азот
7.2.4.1. При отсъствие на калциев цианамид
Получава се от разликата между резултатите, получени в 7.2.2.4 и 7.2.3.2, и/или резултата, получен в 7.2.2.4, и сумата на резултатите, получени в (7.2.5.2 или 7.2.5.5) и (7.2.6.3 или 7.2.6.5 или 7.2.6.6).
7.2.4.2. При наличие на калциев цианамид
Получава се от разликата между резултатите, получени в 7.2.2.4 и 7.2.3.2, и между резултатите, получени в 7.2.2.4, и сумата на резултатите, получени в (7.2.5.5), (7.2.6.3 или 7.2.6.5 или 7.2.6.6) и (7.2.7).
7.2.5. Амонячен азот
7.2.5.1. Само при наличие на амонячен азот и амонячен плюс нитратен азот.
Прехвърля се с прецизна пипета в колбата на дестилатора (5.1) аликвотна част от филтрата (7.2.1.1), съдържаща не повече от 100 mg амонячен азот. Добавя се вода, за да се получи общ обем от около 350 ml, и се добавят няколко гранули пемза (4.28) за улесняване на кипенето. Свързва се колбата към дестилатора, добавят се 20 ml разтвор на натриев хидроксид (4.9) и се дестилира, както е описано в 7.1.1.2.
7.2.5.2. Изразяване на резултата
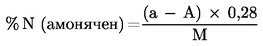
където
|
а |
= |
ml стандартен разтвор на натриев или калиев хидроксид 0,2 mol/l, използван за изпитване на празната проба, извършено чрез прехвърля се с пипетане в приемника на апарата (5.1) на 50 ml стандартен разтвор на сярна киселина 0,2 mol/l (4.8), |
|
А |
= |
ml стандартен разтвор на натриев или калиев хидроксид 0,2 mol/l, използван за анализа, |
|
М |
= |
маса на изпитваната проба, в грамове, присъстваща в аликвотната част. |
7.2.5.3. При наличие на карбамиден и/или цианамиден азот
Прехвърля се с прецизна пипета в сухата колба на апарата (5.2) аликвотна проба от филтрата (7.2.1.1 или 7.2.1.2), съдържаща не повече от 20 mg амонячен азот. След това се сглобява апаратът. Прехвърля се с пипетат се в ерленмайерова колба от 300 ml 50 ml стандартен разтвор на сярна киселина 0,1 mol/l (4.17) и достатъчно вода, за да бъде нивото на течността приблизително 5 cm над отвора на доставящата тръба. Добавя се през страничното гърло на колбата дестилирана вода така че обемът да стане около 50 ml. Смесва се. За да се избегне образуването на пяна при аерацията, се добавят няколко капки октилов алкохол (4.18). След това разтворът се прави алкален, като се използва 50 ml наситен разтвор на калиев карбонат (4.19) и веднага започва изключването на амоняка, който по този начин се освобождава от студената суспензия. Силният поток въздух, необходим за това (поток от приблизително три литра в минута), се пречиства преди това чрез преминаване през измиващи колби, съдържащи разредена сярна киселина и разреден натриев хидроксид. Вместо използване на въздух под налягане е възможно да се работи във вакуум (водна помпа) при условие че входящата тръба е свързана достатъчно херметично към събирателния съд, използван за възстановяване на амоняка. Елиминирането на амоняка по правило завършва след три часа. Независимо от това, добре е да се провери това чрез смяна на приемната колба. Когато операцията завърши, отделя се колбата от апарата, измива се накрайникът на тръбата и страните на колбата с малко дестилирана вода. Титрува се излишъкът на киселината със стандартен разтвор на натриев хидроксид 0,1 mol/l (4.20), докато индикаторът стане сив (4.29.1).
7.2.5.4. Празна проба
Виж 7.1.1.3.
7.2.5.5. Изразяване на резултата

където
|
а |
= |
ml стандартен разтвор на натриев или калиев хидроксид 0,1 mol/l, използван за изпитване на празната проба, извършено чрез прехвърля се с пипетане в ерленмайеровата колба от 300 ml на апарата (5.2) на 50 ml стандартен разтвор на сярна киселина 0,1 mol/l (4.17), |
|
А |
= |
ml стандартен разтвор на натриев или калиев хидроксид 0,1 mol/l, използван за анализа, |
|
М |
= |
маса на изпитваната проба, в грамове, присъстваща в аликвотната част, взета за анализа. |
7.2.6. Карбамиден азот
7.2.6.1. Уреазен метод
Прехвърля се с прецизна пипета в мерителна колба от 500 ml алоквотна част на филтрата (7.2.1.1 или 7.2.1.2), съдържаща не повече от 250 mg карбамиден азот. За да се утаят фосфатите, се добавя известно количество наситен разтвор на бариев хидроксид (4.21), докато не се получи допълнително утаяване. След това се елиминира излишъкът от бариеви йони (и всички разтворени калциеви йони) с помощта на разтвор на 10 % натриев карбонат (4.22).
Оставя се да се утаи и се проверява дали е станало пълно отделяне на утайката. Долива се до марката, смесва се и се филтрира през нагънат филтър. Прехвърля се с пипетат се 50 ml от филтрата в ерленмайеровата колба от 300 ml на апарата (5.3). Подкиселява се филтратът със солна киселина 2 mol/l (4.23) до постигане на рН 3,0, измерено с рН метър (5.5). След това рН се повишава до 5,4 с натриев хидроксид 0,1 mol/l (4.20).
За да се избегне загубата на амоняк при разлагането с уреаза, затваря се ерленмайеровата колба със запушалка, снабдена с разделяща фуния и малък уловител на мехури, съдържаща точно 2 ml стандартен разтвор на солна киселина 0,1 mol/l (4.24). Добавят се през разделящата фуния 20 ml разтвор на уреаза (4.25) и се оставя да престои в продължение на един час при 20 до 25 °С. След това се прехвърля се с пипета 25 ml стандартен разтвор на солна киселина 0,1 mol/l (4.24) в разделящата фуния, оставя се да премине в разтвора и след това се измива с малко вода. По същия начин количествено се прехвърля съдържанието на защитния събирателен съд в разтвора, съдържащ се в ерленмайеровата колба. Титрува се излишъкът на киселината със стандартен разтвор на натриев хидроксид 0,1 mol/l (4.20) до получаването на рН 5,4, измерено с рН метър.
7.2.6.2. Празна проба
Виж 7.1.1.3.
7.2.6.3. Изразяване на резултата

където
|
а |
= |
ml стандартен разтвор на натриев или калиев хидроксид 0,1 mol/l, използван за изпитване на празната проба, извършено чрез прехвърля се с пипетане в приемника на апарата (5.1) на 50 ml стандартен разтвор на сярна киселина 0,2 mol/l (4.8), |
|
А |
= |
ml стандартен разтвор на натриев или калиев хидроксид 0,1 mol/l, използван за анализа, |
|
М |
= |
маса на изпитваната проба, в грамове, присъстваща в аликвотната част, взета за анализа. |
Забележки:
|
1. |
След утаяването с разтворите на бариев хидроксид и натриев карбонат се долива до марката, филтрира се и се неутрализира колкото е възможно по-бързо. |
|
2. |
Изпитване с титруване може също така да се извърши с индикатора (4.29.2), но тогава крайната точка се наблюдава по-трудно. |
7.2.6.4. Гравиметричен метод с ксантхидрол
Прехвърля се с прецизна пипета в бехерова чаша 250 ml аликвотна част от филтрата (7.2.1.1 или 7.2.1.2), съдържаща не повече от 20 mg карбамид. Добавят се 40 ml оцетна киселина (4.14). Разбърква се със стъклена пръчка в продължение на една минута, оставя се утайката да се утаи за пет минути. Филтрира се през плоска подложка в бехерова чаша 100 ml, измива се с няколко милилитра оцетна киселина (4.14). След това към филтрата се добавят капка по капка 10 ml ксантхидрол (4.26), като се разбърква непрекъснато със стъклена пръчка. Оставя се да се утаи, докато се появява утайка, разбърква се отново в продължение една или две минути. Оставя се да престои един час и половина. Филтрира се през стъклен филтриращ тигел, който е бил преди това подсушен и претеглен, като се натиска леко надолу. Измива се три пъти с 5 ml етанол (4.31), без да се прави опит да се елиминира цялата оцетна киселина. Поставя се в пещ и се оставя при температура 130 °С в продължение на един час (да не се надвишават 145 °С). Оставя се да се охлади в сушилня и се претегля.
7.2.6.5. Изразяване на резултата

където
м1= маса на получената утайка, в грамове,
М2= маса на пробата, изразена в грамове, присъстваща в аликвотната част, взета за оценката.
Коригира се заради празната проба. Биуретът може по принцип да бъде измерен с карбамидния азот без голяма грешка, тъй като неговото съдържание остава малко в абсолютната стойност при съставните торове.
7.2.6.6. Метод на разликата
Карбамиден азот може още да бъде изчислен съгласно следната таблица:
|
Случай |
Нитратен N |
Амонячен N |
Цианамиден N |
Карбамиден N |
|
1 |
Отсъствие |
Наличие |
Наличие |
(7.2.2.4) — (7.2.5.5 + 7.2.7) |
|
2 |
Наличие |
Наличие |
Наличие |
(7.2.3.2) — (7.2.5.5 + 7.2.7) |
|
3 |
Отсъствие |
Наличие |
Отсъствие |
(7.2.2.4) — (7.2.5.5) |
|
4 |
Наличие |
Наличие |
Отсъствие |
(7.2.3.2) —(7.2.5.5) |
7.2.7. Цианамиден азот
Взема се една аликвотна част от филтрата (7.2.1.2), съдържаща 10 до 30 mg цианамиден азот и се поставя в бехерова чаша 250 ml. Анализът продължава съгласно метод 2.4.
8. Проверка на резултатите
8.1. В някои случаи може да се открие разлика между общия азот, пряко измерен от пробата (7.1) и общия разтворим азот (7.2.2). Независимо от това, разликата не трябва да бъде по-голяма от 0,5 %. Ако не е така, торът съдържа форми на неразтворим азот, които не се съдържат в списъка в приложение 1.
8.2. Преди всеки анализ се проверява дали апаратурата работи добре и дали се прилага правилно методът със стандартен разтвор, включващ различни форми на азот, в пропорции, подобни на тези на пробата за изпитване. Този стандартен разтвор се приготвя от стандартни разтвори на калиев тиоцианит (4.3), калиев нитрат (4.4), амониев сулфат (4.5) и карбамид (4.6).
Фигура 6
Апаратура за оценка на амонячен азот (7.2.5.3)
Фигура 7
Апаратура за оценка на амонячен азот (7.2.6.1)

Метод 2.6.2
Определяне на различни форми на азот в торове, съдържащи азот само под формата на нитратен, амонячен и карбамиден азот
1. Цел
Целта на настоящия документ е да специфицира опростен метод за определяне на различни форми на азот в торове, съдържащи азот само като нитратен, амонячен и карбамиден азот.
2. Област на приложение
Настоящият метод може да се използва за всички торове, посочени в приложение I, които съдържат изключително нитратен, амонячен или карбамиден азот.
3. Принцип
Правят се следните определяния на различни части на един и същи разтвор на пробата:
|
3.1. |
Общ разтворим азот:
|
|
3.2. |
Общ разтворим азот с изключение на нитратен азот, чрез Келдалово разлагане след елиминиране в кисела среда на нитратния азот с помощта на железен сулфат, като амонякът се определя, както е описано в метод 2.1; |
|
3.3. |
Нитратен азот, с разликата между 3.1.2 и 3.2 или между общия разтворим азот (3.1.2) и сумата на амонячния и карбамиден азот (3.4 + 3.5); |
|
3.4. |
Амонячен азот, чрез студено дестилиране след слаба алкализация; амонякът се получава в разтвор на сярна киселина и се определя, както в метод 2.1; |
|
3.5. |
Карбамиден азот, или:
|
4. Реактиви
Дестилирана или деминерализирана вода.
4.1. Калиев сулфат за анализ
4.2. Желязо за анализ, редуцирано с водород (специфицираното количество желязо трябва да може да редуцира най-малко 50 mg нитратен азот)
4.3. Калиев нитрат за анализ
4.4. Амониев сулфат за анализ
4.5. Карбамид за анализ
4.6. Разтвор на сярна киселина: 0,2 mol/l
4.7. Концентриран разтвор на натриев хидроксид: воден разтвор при приблизително 30 % (w/v) на NaOH, несъдържащ амоняк
4.8. Разтвор на натриев или калиев хидроксид: 0,2 mol/l, несъдържащ амоняк
4.9. Сярна киселина: плътност (d20 = 1,84 g/ml)
4.10. Разредена солна киселина: един обем солна киселина (d20 = 1,18 g/ml) плюс един обем вода
4.11. Оцетна киселина: 96 до 100 %
4.12. Разтвор на сярна киселина, съдържащ около 30 % H2SO4 (w/v), несъдържащ амоняк
4.13. Железен сулфат: кристален, FeSO4.7H2O
4.14. Титруван разтвор на сярна киселина: 0,1 mol/l
4.15. Октилов алкохол
4.16. Наситен разтвор на калиев карбонат
4.17. Натриев или калиев хидроксид: 0,1 mol/l
4.18. Наситен разтвор на бариев хидроксид
4.19. Разтвор на натриев карбонат: при 10 % (w/v)
4.20. Солна киселина: 2 mol/l
4.21. Разтвор на солна киселина: 0,1 mol/l
4.22. Разтвор на уреаза
Прави се суспензия на 0,5 g активна уреаза в 100 ml дестилирана вода, като се използва солна киселина 0,1 mol/l (4.21). Регулира се рН на 5,4, измерено с рН метър.
4.23. Ксантхидрол
5 % разтвор в етанол или метанол (4.28) (не се използват продукти с висока пропорция на неразтворимо вещество). Разтворът може да се съхранява три месеца в добре затворена бутилка на неосветено място.
4.24. Катализатор
Меден окис (CuO): 0,3 до 0,4 g за определяне или еквивалентно количество меден сулфат пентахидрат (CuSO4.5H2O) от 0,95 до 1,25 g за всяко определяне.
4.25. Пемза на гранули, промита със солна киселина и накалена
4.26. Индикаторни разтвори
4.26.1. Смесен индикатор
Разтвор А: Разтваря се 1 g метилово червено в 37 ml разтвор на натриев хидроксид 0,1 mol/l и се допълва до един литър с вода.
Разтвор Б: Разтваря се 1 g метиленово синьо във вода и се долива до един литър.
Смесва се един обем от разтвор А с два обема от разтвор Б.
Този индикатор е виолетов в кисел разтвор, сив в неутрален разтвор и зелен в алкален разтвор. Използват се 0,5 ml (10 капки) от този индикатор.
4.26.2. Индикаторен разтвор метилово червено
Разтварят се 0,1 g метилово червено в 50 ml от 95 % етанол. Долива се до 100 ml с вода и се филтрира, ако е необходимо. Четири до пет капки от този индикатор може да се използват вместо предходния.
4.27. Индикаторни хартии
Лакмус бромтимолово синьо (или други хартии, чувствителни на рН от 6 до 8)
4.28. Етанол или метанол: 95 % (w/v)
5. Апаратура
5.1. Дестилатор
Виж метод 2.1.
5.2. Апарат за оценка на амонячния азот (7.5.1)
Виж метод 2.6.1 и фигура 6.
5.3. Апарат за оценка на карбамидния азот с технологията на уреаза (7.6.1)
Виж метод 2.6.1 и фигура 7.
5.4. Ротационна клатачна машина (35 до 40 оборота в минута)
5.5. рН метър
5.6. Стъклария:
прецизни пипети 2, 5, 10, 20, 25, 50 и 100 ml,
Келдалови колби с дълго гърло 300 и 500 ml,
мерителни колби 100, 250, 500 и 1 000 ml,
тигел от синтеровано стъкло, диаметър на порите 5 до 15 μ,
хаван.
6. Приготвяне на пробата
Виж метод 1.
7. Методи
7.1. Приготвяне на разтвор за анализ
Претегля се 10 g от пробата с точност до 1 mg и се прехвърля в мерителна колба от 500 ml. Добавят се 50 ml вода и след това 20 ml разредена солна киселина (4.10). Разклаща се. Оставя се да престои, докато освобождаването на целия въглероден диоксид завърши. Добавят се 400 ml вода. Разклаща се в продължение на половин час (5.4). Долива се до обема с вода, хомогенизира се и се филтрира през сух филтър в сух съд.
7.2. Общ азот
7.2.1. При отсъствие на нитрати
Прехвърля се с пипета се в келдалова колба от 300 ml част от филтрат (7.1), съдържащ максимум 100 mg азот. Добавят се 15 ml концентрирана сярна киселина (4.9), 0,4 g меден оксид или 1,25 g меден сулфат (4.24) и няколко стъклени перли за контролиране на кипенето. Първо се загрява внимателно, за да започне реакцията и след това по-силно, докато течността стане безцветна или слабо зеленикава и ясно се виждат бели пари. След охлаждане количествено се прехвърля разтворът в дестилационната колба, разрежда се до около 500 ml с вода и се добавят няколко гранули пемза (4.25). Свързва се колбата към дестилатора (5.1) и определянето продължава, както е описано в 7.1.1.2, метод 2.6.1.
7.2.2. При наличие на нитрати
Прехвърля се с пипета в ерленмайрова колба от 500 ml част от филтрата (7.1), съдържаща не повече от 40 mg нитратен азот. На този етап на анализа общото количество на азота не е важно. Добавят се 10 ml сярна киселина на 30 % (4.12), 5 g редуцирано желязо (4.2) и веднага се покрива ерленмайеровата колба с наблюдателно стъкло. Нагрява се внимателно, докато реакцията е стабилна, но не бурна. В този момент нагряването се прекратява и се оставя колбата да престои в продължение на най-малко три часа при температура на околната среда. Прехвърля се течността в мерителна колба от 250 ml, като се игнорира неразтвореното желязо. Допълва се до марката с вода. Хомогенизира се старателно. Прехвърля се с пипета в келдалова колба от 300 ml част, съдържаща максимум 100 mg азот. Добавят се 15 ml концентрирана сярна киселина (4.9), 0,4 g меден оксид или 1,25 g меден сулфат (4.24) и няколко стъклени перли за контролиране на кипенето. Първо се загрява внимателно, за да започне реакцията и след това по-силно, докато течността стане безцветна или слабо зеленикава и ясно се виждат бели пари. След охлаждане количествено се прехвърля разтворът в дестилационната колба, разрежда се до около 500 ml с вода и се добавят няколко гранули пемза (4.25). Свързва се колбата към дестилатора (5.1) и определянето продължава, както е описано в 7.1.1.2, метод 2.6.1.
7.2.3. Празна проба
Извършва се изпитване на празна проба при същите условия и се то се посочва при изчисляване на крайния резултат.
7.2.4. Изразяване на резултата

където
|
а |
= |
ml титруван разтвор на натриев или калиев хидроксид 0,2 mol/l (4.8), използван за изпитване на празната проба, извършено чрез поставяне в приемника на апарата на 50 ml титруван разтвор на сярна киселина 0,2 mol/l (4.6), |
|
А |
= |
ml титруван разтвор на натриев или калиев хидроксид 0,2 mol/l (4.8), използван за анализа, |
|
М |
= |
маса на изпитваната проба, в грамове, присъстваща в аликвотната част, взет в 7.2.1 или 7.2.2. |
7.3. Общ азот с изключение на нитратен азот
7.3.1. Анализ
Прехвърля се с пипета в келдалова колба от 300 ml аликвотна част на филтрата (7.1), съдържаща не повече от 50 mg азот за определяне. Разрежда се до 100 ml с вода, се добавят 5 g железен сулфат (4.13), 20 ml концентрирана сярна киселина (4.9) и няколко стъклени перли за контролиране на кипенето. Първо се загрява слабо, след това по-силно, докато се появят бели пари. Реакцията продължава 15 минути. Спира се нагряването, добавя се меден оксид 0,4 g меден оксид или 1,25 g меден сулфат (4.24) като катализатор. Подновява се нагряването и се поддържа отделянето на бели пари в продължение на 10 до 15 минути. След охлаждане съдържанието на келдаловата колба се прехвърля количествено в дестилационната колба (5.1). Разрежда се до около 500 ml с вода и се добавят няколко гранули пемза (4.25). Свързва се колбата към дестилатора и определянето продължава, както е описано в 7.1.1.2, метод 2.6.1.
7.3.2. Празна проба
Виж 7.2.3.
7.3.3. Изразяване на резултата

където
|
а |
= |
ml титруван разтвор на натриев или калиев хидроксид 0,2 mol/l (4.8), използван за изпитване на празната проба, извършено чрез поставяне в приемника на апарата на 50 ml титруван разтвор на сярна киселина 0,2 mol/l (4.6), |
|
А |
= |
ml титруван разтвор на натриев или калиев хидроксид 0,2 mol/l, използван за анализа, |
|
М |
= |
маса на изпитваната проба, в грамове, присъстваща в аликвотната част, използван при определянето. |
7.4. Нитратен азот
Получава се от разликата между:
|
|
7.2.4. – (7.5.3 + 7.6.3) или |
|
|
7.2.4. – (7.5.3 + 7.6.5) или |
|
|
7.2.4. – (7.5.3 + 7.6.6) |
7.5 Амонячен азот
7.5.1. Анализ
Прехвърля се с пипета в сухата колба на апарата (5.2) част от филтрата (7.1), съдържаща максимум 20 mg амонячен азот. Свързва се към апарата. Прехвърля се с пипета се в ерленмайерова колба от 300 ml 50 ml титруван разтвор на сярна киселина 0,1 mol/l (4.14) и достатъчно вода, за да бъде нивото на течността приблизително 5 cm над отвора на доставящата тръба. Добавя се през страничното гърло на колбата дестилирана вода така че обемът да стане около 50 ml. Смесва се. За да се избегне образуването на пяна при аерацията, се добавят няколко капки октилов алкохол (4.15). Добавят се 50 ml наситен разтвор на калиев карбонат (4.16) и веднага се започва изключване на амоняка, който се освобождава по този начин от студената суспензия. Силният поток въздух, необходим за целта (поток от приблизително три литра в минута), се пречиства преди това чрез преминаване през измиващи колби, съдържащи разредена сярна киселина и разреден натриев хидроксид. Вместо с въздух под налягане може да се работи във вакуум (водна смукателна помпа) при условие че връзките между апаратите са херметични.
Елиминирането на амоняка по принцип завършва след три часа.
Независимо от това, желателно е да се провери това чрез смяна на ерленмайеровата колба. Когато операцията приключи, ерленмайлеровата колба се отделя от апарата, измива се накрайникът на входната тръба и страните на колбата с малко дестилирана вода и се титрува излишъкът на киселината със стандартен разтвор на натриев хидроксид 0,1 mol/l (4.17).
7.5.2. Празна проба
Виж 7.2.3.
7.5.3.N Изразяване на резултата

където
|
а |
= |
ml титруван разтвор на натриев или калиев хидроксид 0,1 mol/l (4.17), използван за изпитване на празната проба, извършено чрез поставяне в ерленмайеровата колба от 300 ml на апарата (5.2) на 50 ml титруван разтвор на сярна киселина 0,1 mol/l (4.14), |
|
А |
= |
ml титруван разтвор на натриев или калиев хидроксид 0,1 mol/l (4.17), използван за анализа, |
|
М |
= |
маса на изпитваната проба, в грамове, присъстваща в аликвотната част, използван за анализа. |
7.6. Карбамиден азот
7.6.1. Метод с уреаза
Прехвърля се с пипета в мерителна колба от 500 ml част от филтрата (7.1), съдържаща не повече от 250 mg карбамиден азот. За да се утаят фосфатите, се добавя подходящо количество наситен разтвор на бариев хидроксид (4.18), докато не се предизвиква допълнително утаяване. Елиминира се излишъкът от бариеви йони (и всички разтворени калциеви йони) с помощта на разтвор на натриев карбонат 10 % (4.19). Оставя се да се утаи и се проверява дали е станало пълно отделяне на утайката. Долива се до марката, хомогенизира се и се филтрира през нагънат филтър. Прехвърля се с пипета се 50 ml от филтрата в ерленмайеровата колба от 300 ml на апарата (5.3). Подкиселява се филтратът със солна киселина 2 mol/l (4.20) до постигане на рН 3,0, измерен с рН метър. След това рН се повишава до 5,4 с натриев хидроксид 0,1 mol/l (4.17). За да се избегне загубата на амоняк при хидролизата с уреаза, ерленмайеровата колба се затваря със запушалка, снабдена с разделяща фуния и малък уловител на мехури, съдържаща точно 2 ml стандартен разтвор на солна киселина 0,1 mol/l (4.21). Добавя се през разделящата фуния 20 ml разтвор на уреаза (4.22). Оставя се да престои в продължение на един час при 20 до 25 °С. Прехвърля се с пипета 25 ml стандартен разтвор на солна киселина 0,1 mol/l (4.21) в разделящата фуния, оставя се да премине в разтвора и след това се измива с малко вода. По същия начин количествено се прехвърля съдържанието на защитния съд в разтвора, съдържащ се в ерленмайеровата колба. Титрува се излишъкът на киселината със стандартен разтвор на натриев хидроксид 0,1 mol/l (4.17) до получаването на рН 5,4, измерено с рН метър.
Забележки:
|
1. |
След утаяването с разтворите на бариев хидроксид и натриев карбонат се долива до марката, филтрира се и се неутрализира колкото е възможно по-бързо. |
|
2. |
Титруването също така може да се определи с използване на индикатора (4.26), въпреки че промяната на цвета се наблюдава по-трудно. |
7.6.2. Празна проба
Виж 7.2.3.
7.6.3. Изразяване на резултата

където
|
а |
= |
ml титруван разтвор на натриев или калиев хидроксид 0,1 mol/l (4.17), използван за изпитване на празната проба, извършено при точно същите условия като при анализа, |
|
А |
= |
ml титруван разтвор на натриев или калиев хидроксид 0,1 mol/l (4.17), използван за анализа, |
|
М |
= |
маса на пробата, изразена в грамове, присъстваща в аликвотната част, използвана за анализа. |
7.6.4. Гравиметричен метод с ксантодрол
Прехвърля се с пипета се в бехерова чаша 100 ml част от филтрата (7.1), съдържаща не повече от 20 mg карбамид. Добавят се 40 ml оцетна киселина (4.11). Разбърква се със стъклена пръчка в продължение на една минута. Оставя се утайката да се отдели за пет минути. Филтрира се и се измива с няколко милилитра оцетна киселина (4.11). Добавя се към филтрата капка по капка 10 ml ксантхидрол (4.23), като се разбърква непрекъснато със стъклена пръчка. Оставя се да престои, докато се появи утайка, разбърква се отново в продължение една или две минути. Оставя се да престои един час и половина. Филтрира се през стъклен филтриращ тигел, който е бил преди това подсушен и претеглен с леко натискане надолу. Измива се три пъти с 5 ml етанол (4.28), без да се прави опит да се елиминира цялата оцетна киселина. Поставя се в пещ и се поддържа температура 130 °С в продължение на един час (да не се превишава 145 °С). Оставя се да се охлади в сушилня и се претегля.
7.6.5. Изразяване на резултата

карбамиден
където= m = маса на получената утайка, в грамове,
М= маса на пробата, в грамове, присъстваща в аликвотната част, използвана при определянето.
Коригира се заради празната проба. Биуретът може по принцип да бъде измерен с карбамидния азот без някаква голяма грешка, тъй като неговото ниво остава малко в абсолютната стойност при съставните торове.
7.6.6. Метод на разликата
Карбамидният азот може да се изчисли, както е посочено следната таблица:
|
Случай |
Нитратен азот |
Амонячен азот |
Карбамиден азот |
|
1 |
Липса |
Наличие |
(7.2.4) — (7.5.3) |
|
2 |
Наличие |
Наличие |
(7.3.3) — (7.5.3) |
8. Проверка на резултатите
Преди всеки анализ се проверява функционирането на апаратурата и дали се прилагат правилно методите със стандартен разтвор, съдържащ различни форми на азот, в пропорции, подобни на тези на пробата. Този стандартен разтвор се приготвя от стандартни разтвори на калиев нитрат (4.3), амониев сулфат (4.4) и карбамид (4.5).
Методи 3
Фосфор
Методи 3.1
Екстракции
Метод 3.1.1
Екстракция на фосфор разтворим в неорганични (минерални) киселини
1. Обхват
Настоящият документ описва процедурата са определяне на фосфор, разтворим в неорганични киселини.
2. Област на приложение
Приложим изключително за фосфатните торове, изброени в приложение I.
3. Принцип
Екстракция на фосфора в тора със смес на азотна киселина и сярна киселина.
4. Реактиви
Дестилирана или деминерализирана вода.
4.1. Сярна киселина (d20 = 1,84 g/ml)
4.2. Азотна киселина (d20 = 1,40 g/ml)
5. Оборудване
Стандартно лабораторно оборудване.
5.1. Келдалова колба с обем най-малко 500 ml или облодънна колба 250 ml със стъклена тръба, образуваща обратен хладник.
5.2. Мерителна колба от 500 ml.
6. Приготвяне на пробата
Виж метод 1.
7. Процедура
7.1. Проба
Претеглят се 2,5 g от приготвената проба с точност до 0,001 g и се поставят в келдалова колба.
7.2. Екстракция
Добавят се 15 ml вода, за да се суспензира веществото. Добавя се 20 ml азотна киселина (4.2) и внимателно се добавя 30 ml сярна киселина (4.1).
Когато първоначалната бурна реакция спре, съдържанието се довежда бавно до кипене и се вари в продължение на 30 минути. Оставя се да се охлади и след това внимателно се добавя със смесване около 150 ml вода. Изваряването продължава 15 минути.
Охлажда се напълно и се прехвърля течността количествено в мерителна колба от 500 ml. Допълва се до обема, смесва се и се филтрира през сух нагънат филтър, без фосфати, като първата част от филтрата се изхвърля.
7.3. Определяне
Определянето на фосфора ще се извърши по метод 3.2 на аликвотната част на разтвора, получен по този начин.
Метод 3.1.2
Екстракция на фосфор разтворим в 2 % мравчена киселина (20 g на литър)
1. Обхват
Настоящият документ дефинира процедурата за определяне на фосфор разтворим в 2 % мравчена киселина (20 g на литър).
2. Област на приложение
Изключително меки естествени фосфати.
3. Принцип
За се направи разлика между твърди естествени фосфати и меки естествени фосфати, фосфорът, разтворим в мравчена киселина, се извлича при специфични условия.
4. Реактиви
4.1. Мравчена киселина, 2 % (20 g на литър)
Бележка
Долива се 82 ml мравчена киселина (концентрация 98 до 100 %; d20 = 1,22 g/ml) до 5 литра с дестилирана вода.
5. Апаратура
Стандартно лабораторно оборудване
5.1. Мерителна колба от 500 ml (например Стохман)
5.2. Ротационна клатачна машина (35 до 40 оборота в минута)
6. Приготвяне на пробата
Виж метод 1.
7. Процедура
7.1. Проба
Претеглят се 5 g от приготвената проба с точност до 0,001 g и се поставят в суха мерителна колба на Стохман от 500 ml с широко гърло (5.1)
7.2. Екстракция
Като непрекъснато се върти колбата с ръка, се добавя 2 % мравчена киселина при 20 (±1) °С (4.1), докато тя е приблизително 1 cm под марката и се долива до обема. Затваря се колбата с гумена запушалка и се разклаща в продължение на 30 минути при 20(± 1) °С на ротационна клатачна машина (5.2).
Филтрира се разтворът през сух нагънат филтър, без фосфати, сух стъклен приемник. Изхвърля се първата част от филтрата.
7.3. Определяне
Определя се фосфорът по метод 3.2 в аликвотната част на напълно бистър филтрат.
Метод 3.1.3
Екстракция на фосфор разтворим в 2 % лимонена киселина (20 g на литър)
1. Обхват
Настоящият документ дефинира процедурата за определяне на фосфор разтворим в 2 % лимонена киселина (20 g на литър).
2. Област на приложение
Приложим само за видове основна шлака (виж приложение I.А).
3. Принцип
Екстракция на фосфор от тора с разтвор на 2 % лимонена киселина (20 g на литър) при дадени условия.
4. Реактиви
Дестилирана или деминерализирана вода.
4.1. Разтвор на 2 % лимонена киселина (20 g/l), приготвена от кристализирала лимонена киселина (C6H8O7.H2O)
Бележка
Проверява се концентрацията на този разтвор на лимонена киселина чрез титруване със стандартен разтвор 0,1 mol/l на натриев хидроксид, като се използва фенолфталеин като индикатор.
Ако концентрацията е правилна, трябва да бъдат използвани 28,55 ml стандартен разтвор.
5. Апаратура
5.1. Ротационна клатачна машина (35 до 40 оборота в минута)
6. Приготвяне на пробата
Анализът се извършва на продукта, получен след внимателно смесване на оригиналната проба, за да се осигури нейната хомогенност. Виж метод 1.
7. Процедура
7.1. Проба
Претеглят се g 5 g от приготвената проба с точност до 0,001 и се поставят в суха колба с достатъчно широко гърло, с обем 600 ml, позволяваща доброто разклащане на течността.
7.2. Екстракция
Добавя се 500 (±1) ml разтвор на лимонена киселина при 20(± 1) °С. При добавянето на първите милилитри реактив се разклаща силно с ръка, за да се спре образуването на буци и да се предотврати полепването на веществото по стените. Затваря се колбата с гумена запушалка и се разклаща на ротационната клатачна машина (5.1) точно 30 минути при температура при 20(± 2) °С.
Филтрира се незабавно през сух нагънат филтър, без фосфати, в сух стъклен събирателен съд и се изхвърлят първите 20 ml от филтрата. Филтрирането продължава до получаването на достатъчно количество филтрат за извършване на определянето на фосфора.
7.3. Определяне
Определянето на фосфорния екстракт ще се извършва съгласно метод 3.2 на аликвотна част на разтвора, получен по този начин.
Метод 3.1.4
Екстракция на фосфор разтворим в неутрален амониев цитрат
1. Обхват
Настоящият документ дефинира процедурата за определяне на фосфор, който е разтворим в неутрален амониев цитрат.
2. Област на приложение
Всички торове по отношение на които разтворимостта в неутрален амониев цитрат е постановена (виж приложение I).
3. Принцип
Екстракция на фосфор при температура 65 °С с използване на неутрален разтвор на амониев цитрат (рН 7,0) при специфични условия.
4. Реактиви
Дестилирана или деминерализирана вода
4.1. Неутрален разтвор на амониев цитрат (рН 7,0)
Този разтвор трябва да съдържа на литър 185 g кристализирала лимонена киселина и трябва да има специфично тегло 1,09 при 20 °С и рН 7,0.
Реактивът се приготвя, както следва:
|
|
Разтварят се 370 g кристализирала лимонена киселина (C6H8O7.H2O) в около 1,5 литра вода и се прави приблизително неутрален разтвор чрез добавяне на 345 ml разтвор на амониев хидроксид (28 до 29 % NH3). Ако концентрацията на NH3 е по-ниска от 28 %, се добавя съответно по-голямо количество разтвор на амониев хидроксид и се разрежда лимонената киселина в съответно по-малки количества вода. |
|
|
Охлажда се и се прави неутрален като се поддържат електродите на рН-метъра, потопени в разтвора. Добавя се амоняк, при 28 до 29 % NH3, капка по капка, като се разбърква непрекъснато (с механическа бъркалка) до получаване на точно рН 7,0 при температура 20 °С. В този момент обемът се допълва до два литра и рН се тества отново. Реактивът се съхранява в затворен съд и редовно се проверява рН. |
5. Апаратура
5.1. Бехерова чаша 2 литра
5.2. рН метър
5.3. Ерленмайерова колба от 200 или 250 ml
5.4. Мерителни колби от 500 ml и 2 000 ml
5.5. Водна баня, която може да се настрои термостатично на 65 °С, оборудвана с подходяща бъркалка (виж фигура 8).
6. Приготвяне на пробата
Виж метод 1.
7. Процедура
7.1. Проба
Прехвърля се 1 до 3 g от тора за анализиране (виж приложение I, части А и Б от регламента) в ерленмайерова колба от 200 или 250 ml, съдържаща 100 ml разтвор на амониев цитрат предварително нагрят до 65 °С.
7.2. Анализ на разтвора
Запушва се ерленмайеровата колба и се разклаща, за да се суспензира торът, без да се образуват буци. Маха се запушалката за момент, за да се балансира налягането и отново се затваря колбата. Поставя се колбата във водна баня, настроена да поддържа съдържанието на колбата на точно 65 °С и се свързва с бъркалката (виж фигура 8). По време на бъркането нивото на суспензията в колбата трябва да остава постоянно под нивото на водата във водната баня (11). Механичното разбъркване се регулира, за да се осигури пълна суспензия.
След разбъркване точно един час, отстранява се ерленмайеровата колба от водната баня.
Охлажда се незабавно под течаща вода до температурата на околната среда и незабавно, количествено се прехвърля съдържанието на ерленмайеровата колба в мерителна колба 500 ml със струя вода (измиване на бутилка). Долива се до обема с вода. Смесва се старателно. Филтрира се през сух нагънат филтър (средна скорост без фосфати) в сух съд, изхвърляйки първата част на филтрата (около 50 ml).
След това ще се събере около 100 ml бистър филтрат.
7.3. Определяне
Определя се фосфорът на екстракта, получен по този начин съгласно метод 3.2.
Фигура 8

Методи 3.1.5
Екстракция с алкален амониев цитрат
Метод 3.1.5.1
Екстракция на разтворим фосфор съгласно Petermann при 65 °С
1. Обхват
Настоящият документ дефинира процедурата за определяне на фосфор в алкален амониев цитрат.
2. Област на приложение
Изключително обезводнен фосфат с отделена утайка (CaHPO4.2H2O).
3. Принцип
Екстракция на фосфор при температура 65 °С с алкален разтвор на амониев цитрат (Petermann) при специфицирани условия.
4. Реактиви
Дестилирана вода или деминерализирана вода със същите характеристики като дестилираната вода.
4.1. Разтвор на Petermann.
4.2. Характеристики
Лимонена киселина (C6H8O7.H2O): 173 g на литър.
Амоняк: 42 g на литър амонячен азот.
рН между 9,4 и 9,7.
Приготвяне от диамониев цитрат
Разтваря се 931 g диамониев цитрат (молекулна маса 226,19) в около 3 500 ml вода петлитрова стандартна колба. Поставя се в баня с течаща вода, смесва се и се охлажда и се добавя на малки количества амоняк. Например, за d20 = 906 g/ml, съответстващо на ниво 20,81 % по маса на амонячен азот, е необходимо да се използва 502 ml амониев разтвор. Регулира се температурата на 20 °С, прехвърля се в петлитрова стандартна колба, допълва до марката с дестилирана вода и се смесва.
Препарат от лимонена киселина и амоняк
Разредете 865 g монохидрат на лимонена киселина в около 2 500 ml дестилирана вода в съд с обем около пет литра. Поставете съда в студена вана и добавете малки количества, като разклащате продължително, разтвор на амоняк като използвате фуния, чиято дръжка е потопена в разтвор на лимонена киселина. Например, за d20 = 906 g/ml, която отговаря на ниво 20,81 % от масата на амониевия азот, необходимо е да се добавят 1 114 ml амониев разтвор. Нагласете температурата до 20 °C, прехвърлете в петлитрова стандартна колба, допълнете до указаната маркировка с дестилирана вода и разбъркайте.
Проверява се съдържанието на амонячен азот, както следва:
Прехвърля се 25 ml от разтвора в стандартна колба от 250 ml и се допълва до обема с дестилирана вода. Смесва се. Определя се съдържанието на амоняк на 25 ml от този разтвор, като се следва метод 2.1. Ако разтворът е правилен, трябва да се използват 15 ml 0,5 N mol/l H2SO4.
Ако силата на амонячния азот е по-голяма от 42 g на литър, NH3 може да бъде изключен с поток инертен газ или чрез умерено нагряване за довеждане обратно рН до 9,7. Извършва се второ определяне.
Ако силата на амонячния азот е по-малка от 42 g на литър, ще бъде необходимо да се добави маса М от амонячния разтвор:

или обем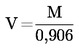 при температура 20 °С
при температура 20 °С
Ако V е по-малко от 25 ml, добавете го директно в петлитрова колба с обем V × 0,173 g лимонена киселина на прах.
Ако V е по-малък от 25 ml, ще бъде удобно да се направи нов литър реактив по следния начин.
Претегля се 173 g лимонена киселина. Разтваря се в 500 ml вода. Вземайки посочените предпазни мерки, се добавя не повече от 225 + V × 1 206 ml амониев разтвор, който беше използван за приготвянето на пет литра реактив. Долива се до обем с вода. Смесва се.
Този литър се смесва с предварително приготвените 4 975 ml.
5. Апаратура
5.1. Водна баня, която може да поддържа температура 65(± 1) °С
5.2. Мерителна колба от 500 ml (например: Stohmann)
6. Приготвяне на пробата
Виж метод 1.
7. Процедура
7.1. Проба
Претегля се 1 g от приготвената проба с точност до 0,001 g и се прехвърля в мерителна колба 500 ml (5.2).
7.2 Екстракция
Добавя се 200 ml алкален разтвор на амониев цитрат (4.1). Запушва колбата и се разклаща колбата силно с ръка, за да се избегне образуването на буци и да се предотврати залепването по стените.
Поставя се колбата във водна баня, настроена на 65 °С, и се разклаща на всеки пет минути през първия половин час. След всяко разклащане се повдига запушалката, за да се изравни налягането. Нивото на водата във водната баня трябва да бъде над нивото на разтвора в колбата. Оставя се колбата във водната баня още един час при 65 °С и се разклаща на всеки 10 минути. Отстранява се колбата, охлажда се до температура над 20 °С, долива се до обем 500 ml с вода. Смесва се и филтрира се през суха гофрирана филтърна хартия, без фосфати, като се изхвърля първата част на филтрата.
7.3 Определяне
Определянето на извлечения фосфор ще се извършва по метод 3.2 на аликвотна част на разтвора, получен по този начин.
Метод 3.1.5.2
Екстракция на разтворим фосфор съгласно Petermann при температура на околната среда
1. Обхват
Настоящият документ дефинира процедурата за определяне на фосфор, разтворим в студен алкален амониев цитрат.
2. Област на приложение
Изключително за дезинтегрирани фосфати.
3. Принцип
Екстракция на фосфор при температура около 20 °С с алкален разтвор на амониев цитрат (разтвор на Petermann) при специфични условия.
4. Реактив
Виж метод 3.1.5.1.
5. Апаратура
5.1. Стандартно лабораторно оборудване и мерителна колба от 250 ml (например Стохман)
5.2. Ротационна клатачна машина (35 до 40 оборота в минута)
6. Приготвяне на пробата
Виж метод 1.
7. Процедура
7.1. Проба
Претегля се 2,5 g от приготвената проба с точност до 0,001 g и се поставя в мерителна колба 250 ml (5.1).
7.2. Екстракция
Добавя се малко от разтвора на Petermann при 20 °С, разклаща се силно, за да спре образуването на буци и да се предотврати залепването на веществото по стените на колбата. Долива се до марката с разтвор на Petermann и се затваря колбата с гумена запушалка.
Разклаща се в продължение на два часа на ротационната клатачна машина (5.2). Филтрира се незабавно след това през сух нагънат филтър, без фосфати, в сух съд, като се изхвърля първата част на филтрата.
7.3. Определяне
Определянето на фосфора ще се извърши по метод 3.2 на аликвотна част но получения по този начин разтвор.
Метод 3.1.5.3
Екстракция на фосфор разтворим в алкален амониев цитрат на Жули
1. Обхват
Настоящият документ дефинира процедурата за определяне на фосфор, разтворим в алкален амониев цитрат на Жули.
2. Област на приложение
Всички концентрирани и съставни фосфатни торове, в които фосфата се появява в алуминиево-калциева форма.
3. Принцип
Екстракция чрез силно разклащане с алкален разтвор на амониев цитрат с дефинирана спецификация (и, когато е подходящо в присъствието на оксин) при около 20 °С.
4. Реактиви
Дестилирана или деминерализирана вода.
4.1. Алкален разтвор на амониев цитрат на Жули
Този разтвор съдържа 400 g лимонена киселина и 153 g NH3 на литър. Неговото свободно съдържание на амоняк е приблизително 55 g на литър. Може да бъде приготвен по някой от методите, описани по-долу.
|
4.1.1. |
В мерителна колба един литър се разтваря 400 g лимонена киселина (C6H8O7.H2O) в приблизително 600 ml амоняк (d20 = 0,925 g/ml, т.е. 200 g NH3 на литър). След това се добавя лимонена киселина в количества от 50 до 80 g, като се поддържа температурата под 50 °С. Долива се до обем един литър с амоняк. |
|
4.1.2. |
В еднолитрова градуирана колба, се разтварят 432 g двуосновен амониев џитрат (C6H14H2O7) в 300 ml вода. Добавят се 440 ml амоний (d20 = 0,923 g/ml). Обемът се допълва с вода докато достигне един литър. |
Бележка:
Проверка на общото съдържание на амоняк.
Взема се проба 10 ml на цитратния разтвор и се поставя в колба от 250 ml. Долива се до обема дестилирана вода. Определя се съдържанието на амонячен азот в 25 ml на този разтвор съгласно метод 2.1.
1 ml H2SO4 0,5 mol/l = 0,008516 g NH3
При тези условия реактивът се счита коректен, когато количеството милилитри при титруване се окаже между 17,7 и 18 ml.
Ако това не е така, се добавя 4,25 ml амоняк (d20 = 0,925 g/l) на 0,1 ml под 18 ml, посочени по-горе.
4.2. 8-хидроксикинолин (оксин) на прах
5. Апаратура
5.1. Стандартно лабораторно оборудване и малък хаван от стъкло или порцелан с пестик.
5.2. Мерителни колби от 500 ml
5.3. Мерителна колба от 1 000 ml
5.4. Ротационна клатачна машина (35 до 40 оборота в минута)
6. Приготвяне на пробата
Виж метод 1.
7. Процедура
7.1. Проба
Претегля се 1 g от приготвената проба с точност до 0,0005 g и се поставя в малък хаван. Добавят се 10 капки цитрат (4.1), за да се навлажни и се разчупва внимателно с пестика.
7.2. Екстракция
Добавят се 20 ml амониев цитрат (4.1) и се смесва до паста. Оставя се да се утаи за около една минута.
Декантира се течността в мерителна колба от 500 ml, като се прецеждат частиците, които са могли да избегнат предишната влажна дезинтеграция. Добавят се 20 ml от цитратния разтвор (4.1) към остатъка, раздробява се както по-горе, и се декантира течността в мерителна колба. Повтаря се процесът четири пъти, така че накрая на петия път целият продукт да може да се излее в колбата. Общото количество цитрат, използвано за тези процеси, трябва да бъде приблизително 100 ml.
Измива се пестикът и хаванът над мерителната колба с 40 ml дестилирана вода.
Запушената колба се разклаща в продължение на три часа на ротационната клатачна машина (5.4).
Оставете колбата изправена за 15—16 часа, разклатете я при същите условия за три часа. Температурата по време на целия процес трябва да бъде 20(± 2) °С.
Долива се до марката на мерителната колба дестилирана вода. Филтрира се през сух филтър, изхвърля се първата част на филтрата и бистрият филтрат се събира в суха колба.
7.3. Определяне
Оценката на извлечения фосфор ще се извърши по метод 3.2 на аликвотна част но получения по този начин разтвор.
8. Приложение
Използването на оксин прави възможно използването на този метод за торове, съдържащи магнезий. Това използване се препоръчва, когато отношението на съдържанието на магнезий и фосфорен анхидрид е по-високо от 0,03 (Mg/P2O5 > 0,03). Ако е така, се добавя 3 g оксин към навлажнената проба за анализиране. Още повече, че използването на оксин при отсъствие на магнезий не е вероятно да повлияе на последващото определяне. При известно отсъствие на магнезий, обаче, е възможно да не се използва оксин.
Метод 3.1.6
Екстракция на водоразтворим фосфор
1. Обхват
Настоящият документ дефинира процедурата за определяне на фосфор, разтворим във вода.
2. Област на приложение
Всички торове, включително съставни, в които се определя водоразтворим фосфор.
3. Принцип
Екстракция чрез разклащане при специфични условия
4. Реактиви
Дестилирана или деминерализирана вода.
5. Апаратура
5.1. Мерителна колба от 500 ml (например Стохман)
5.2. Ротационна клатачна машина (35 до 40 оборота в минута)
6. Приготвяне на пробата
Виж метод 1.
7. Процедура
7.1. Проба
Претеглят се 5 g от приготвената проба с точност до 0,001 g и се поставят в мерителна колба от 500 ml (5.1).
7.2. Екстракция
Добавят се в колбата 450 ml вода, температурата на която трябва да бъде между 20 и 25 °С.
Разклаща се на ротационната клатачна машина(5.2) в продължение на 30 минути.
След това се долива до марката вода, смесва се старателно чрез разклащане и се филтрира през сух нагънат филтър, без фосфати, в сух съд.
7.3. Определяне
Оценката на извлечения фосфор ще се извърши по метод 3.2 на аликвотна част на получения по този начин разтвор.
Метод 3.2
Определяне на извлечен фосфор
(Гравиметричен метод с използване на хинолов фосфомолибдат)
1. Обхват
Настоящият документ дефинира процедурата за определяне на фосфор в екстракти от торове.
2. Област на приложение
Методът е приложим за всички екстракти на торове (12) за определянето на различни форми на фосфор.
3. Принцип
След възможна хидролиза на различни форми на фосфор, различни от ортофосфати, ортофосфатираните йони се утаяват в кисела среда под формата на хинолов фосфомолибдат.
След филтриране и измиване утайката се изсушава при 250 °С и се претегля.
При условията, посочени по-горе, веществата, които е вероятно да бъдат намерени в разтвора (органични и неорганични киселини, амониеви йони, разтворими силикати и т.н.), не оказват въздействие, ако реактив на базата молибден или амониев молибден се използва при утаяването.
4. Реактиви
Дестилирана или деминерализирана вода.
4.1. Концентрирана азотна киселина (d20 = 1,40 g/ml)
4.2. Приготвяне на реактива
4.2.1. Приготвяне на реактива на базата на натриев молибдат
Разтвор А: Разтваря се 70 g дехидрат на натриев молибдат в 100 ml дестилирана вода.
Разтвор Б: Разтваря се 60 g монохидрат на лимонен киселина в 100 ml дестилирана вода и се добавя 85 g контрирана азотна киселина (4.1).
Разтвор В: Разбърква се разтвор А в разтвор Б, за да се получи разтвор В.
Разтвор Г: Към 50 ml дестилирана вода се добавя 35 g концентрирана азотна киселина (4.1), след това 5 ml прясно дестилиран хинолин. Добавя се този разтвор към разтвор В, смесва се старателно и се оставя да престои една нощ на тъмно. След това се долива до 500 ml с дестилирана вода, смесва се отново и се филтрира през синтерована стъклена фуния (5.6).
4.2.2. Приготвяне на реактив на базата на амониев молибдат
Разтвор А: В 300 ml дестилирана вода се разтваря 100 g амониев молибдат, като се загрява слабо и се разбърква от време на време.
Разтвор Б: Разтваря се 120 g монохидрат на лимонената киселина в 200 ml дестилирана вода и се добавят 170 g концентрирана азотна киселина (4.1).
Разтвор В: Добавят се 10 ml прясно дестилиран хинолин към 70 ml концентрирана азотна киселина (4.1).
Разтвор Г: Излива се бавно, като се разбърква добре, разтвор А в разтвор Б. След старателно размесване се добавя разтвор В към тази смес и се допълва до един литър. Оставя се да престои два дни на тъмно място и се филтрира през синтерованата стъклена фуния (5.6).
Реактивите 4.2.1. и 4.2.2 могат да бъдат използвани по един и същ начин. И двата трябва да се съхраняват на тъмно в полиетиленови бутилки.
5. Апаратура
5.1. Стандартно лабораторно оборудване и ерленмайерова колба от 500 ml с широко гърло.
5.2. Градуирани пипети от 10, 25 и 50 ml
5.3. Филтър-тигел с порьозност 5 до 20 μm
5.4. Бухнерова колба
5.5. Пещ за сушене регулирана на 250 (+10) °С
5.6. Синтерована стъклена фуния съд с порьозност 5 до 20 μm
6. Процедура
6.1. Третиране на разтвора
С пипета се взема аликвотна част от екстракта на тора (виж таблица 2), съдържаща около 0,01 g Р2О5, и се поставя в ерленмайерова колба от 500 ml. Добавя се 15 ml концентрирана азотна киселина (13) (4.1) и разредете с вода докато достигне количество от 100 ml.
Таблица 2
Определяне на аликвотните части на фосфорни разтвори
|
%P2O5 в тора |
%Р в тора |
Проба за анализ (g) |
Разреждане (на ml) |
Проба (ml) |
Разреждане (на ml) |
Проба за утаяване (ml) |
Коефициент на превръщане на хинолов фосфомолибдат (F) в %P2O5 |
Коефициент на превръщане на хинолов фосфомолибдат (F′) в %Р |
|
5-10 |
2.2-4-4 |
1 |
500 |
— |
— |
50 |
32,074 |
13,984 |
|
5 |
500 |
— |
— |
10 |
32,074 |
13,984 |
||
|
10-25 |
4.4-11.0 |
1 |
500 |
— |
— |
25 |
64,148 |
27,968 |
|
5 |
500 |
50 |
500 |
50 |
64,148 |
27,968 |
||
|
+25 |
+11 |
1 |
500 |
— |
— |
10 |
160,370 |
69,921 |
|
5 |
500 |
50 |
500 |
25 |
128,296 |
55,937 |
6.2. Хидролиза
Ако се очаква присъствие в разтвора на метафосфати, пирофосфати или полифосфати, хидролизата се извършва, както следва.
Довежда се съдържанието на ерленмайеровата колба до кипене бавно и се поддържа тази температура до завършването на хидролизата (това обикновено отнема един час). Трябва да се внимава да се избегнат загуби от изплискване и излишно изпаряване, което може да намали първоначалния обем с повече от половината, чрез монтиране на обратен хладник. След хидролизата се долива вода до първоначалния обем.
6.3. Претегляне на тигела
Изсушава се филтър-тигела (5.3) в продължение най-малко на 15 минути в пещ за сушене, настроена на 250(± 10 °С). След охлаждане в сушилен шкаф се претегля.
6.4 Отделяне на утайката
Киселинният разтвор, който се съдържа в ерленмайеровата колба се нагрява, докато започне да кипи. След това започва разделянето на утайката на хинолов фосфомолибдат чрез добавяне на 40 ml утаяващ реактив (реактив 4.2.1 или 4.2.2) (14) капка по капка, като се разбърква постоянно. Поставя се ерленмайеровата колба в парна баня, оставя се там в продължение на 15 минути, като от време навреме се разклаща. Разтворът може да бъде филтриран веднага или след охлаждането му.
6.5. Филтриране и измиване
Филтрира се разтвора под вакуум чрез декантиране. Измива се утайката в ерленмайеровата колба с 30 ml вода. Разтворът се декантира и се филтрира. Процесът се повтаря пет пъти. Остатъкът от утайката се прехвърля количествено в тигела, като се измива с вода. Измива се четири пъти с 20 ml вода, като се оставя водата да се изтече от събирателния съд преди всяко добавяне. Утайката се изсушава старателно.
6.6 Изсушаване и претегляне
Избърсва се външната част на тигела с филтърна хартия. Поставя се този тигел в пещ за сушене и се държи там, докато неговата маса остане постоянна при температура 250 °С (5.5) (обикновено 15 минути). Оставя се да се охлади в сушилен шкаф при температура на околната среда и бързо се претегля.
6.7. Празна проба
За всяка серия определяния се извършва изпитване на празна проба, като се използват само реактиви и разтвори в пропорциите, използвани при екстракцията (цитратен разтвор и т.н.), които се отчитат при изчисляването на крайния резултат.
6.8. Проверка
Извършва се определянето, като се използва алоквотна част на разтвор на калиев двуводороден фосфат, съдържащ 0,01 g Р2О5.
7. Изразяване на резултата
Ако се използват пробите за анализиране и разрежданията, показани в таблица 2, се прилагат следните формули:
|
|
или |

|
|
|

където
|
А |
= |
маса (в грамове, на хиноловия фосфомолибдат, |
|
а |
= |
маса в грамове, на хиноловия фосфомолибдат, получена при изпитване на празната проба, |
|
F и F′ |
= |
коефициенти, дадени в последните две колони на таблица 2. |
При проби за анализ и разреждания, които се различават от тези в таблица 2, се прилагат следните формули:
|
|
или |

|
|
|

където
|
f и f′ |
= |
коефициенти на конверсия на хинолов фосфомолибдат в Р2О5 = 0,032074, (f) или в Р = 0,013987 (f′), |
|
D |
= |
коефициент на разреждане, |
|
M |
= |
маса, в грамове, на анализираната проба. |
Метод 4
Калий
Метод 4.1
Определяне на съдържанието на водоразтворим калий
1. Обхват
Настоящият документ дефинира процедурата за определяне на калий, разтворим във вода.
2. Област на приложение
Всички калиеви торове в списъците на приложение I.
3. Принцип
Калият в анализираната проба се разтваря във вода. След елиминиране или фиксиране на веществата, които могат да повлияят на количественото определяне, калият се утаява в леко алкална среда под формата на калиев тетрафенилборат.
4. Реактиви
Дестилирана или деминерализирана вода.
4.1. Формалдехид
Бистър разтвор на формалдехид при 25 до 35 %.
4.2. Калиев хлорид за анализ
4.3. Разтвор на натриев хидроксид: 10 mol/l
Трябва да се внимава да се използва натриев хидроксид, несъдържащ калий.
4.4. Индикаторен разтвор
Разтваря се 0,5 g фенолфталеин в 90 % етанол и се долива до обем 100 ml.
4.5. EDTA разтвор
Разтваря се 4 g безводна двунатриева сол на етилендиаминтетраоцетна киселина във вода в мерителна колба от 100 ml. Допълва се до обема и се смесва.
Реактивът се съхранява в пластмасов съд.
4.6. STPB разтвор
Разтваря се 32,5 g натриев тетрафенилборат в 480 ml вода и 2 ml разтвор на натриев хидроксид (4.3) и 20 ml разтвор на магнезиев хлорид (100 g MgCl2. 6H2O на литър).
Разбърква се в продължение на 15 минути и се филтрира през фин филтър без пепел.
Този реактив се съхранява в пластмасов съд.
4.7. Течност за измиване
Разтворете 20 ml от разтвора STPB (4.6) с вода докато достигне количество 1 000 ml.
4.8. Бромна вода
Наситен бромен разтвор с вода.
5. Апаратура
5.1. Мерителни колби от 1 000 ml
5.2. Бехерова чаша от 250 ml
5.3. Филтър-тигел с порьозност 5 до 20 μ
5.4. Пещ регулирана на 120(± 10) °С
5.5. Сушилен шкаф
6. Приготвяне на пробата
Виж метод 1.
При калиеви соли пробата трябва да се смели достатъчно фино, за да се получи представителна проба за анализ. За тези продукти трябва да се използва метод 1, точка 6, буква а).
7. Процедура
7.1. Проба
Претегля се 10 g от приготвената проба с точност до 0,001 g (5 g за калиеви соли, съдържащи повече от 50 % калиев оксид). Поставя се тази проба за изпитване в бехеров съд от 600 ml с приблизително 400 ml вода.
Довежда се до кипене и се оставя да кипи 30 минути. Охлажда се, прехвърля се количествено в мерителна колба от 1 000 ml, долива се до обема, смесва се и се филтрира в сух приемник. Изхвърлят се първите 50 ml от филтрата (виж бележката по процедурата към 7.6).
7.2. Приготвяне на аликвотната част за отделяне на утайката
Прехвърля се с пипета аликвотна част от филтрата, съдържаща 25 до 50 mg калий (виж таблица 3) и се поставя в бехеров съд 250 ml. Ако се изисква, се долива до 50 ml с вода.
За да се отстранят всички взаимни влияния, се добавя 10 ml EDTA разтвор (4.5), няколко капки разтвор на фенолфталеин (4.4) и чрез разбърква се добавя, капка по капка, разтвор на натриев хидроксид (4.3), докато стане червен. След това накрая се добавя още няколко капки натриев хидроксид, за да се осигури излишък (обикновено 1 ml натриев хидроксид е достатъчен да неутрализира пробата и осигури излишък).
За да се елиминират по-голямата част от амоняка (виж бележката по процедурата към точка 7.6, буква б), загрява се слабо в продължение на 15 минути.
Ако е необходимо, се добавя вода, за да се постигне обем 60 ml.
Довежда се разтвора до кипене, отстранява се бехеровата чаша от топлината и се добавя 10 ml формалдехид (4.1). Добавя се няколко капки фенолфталеин и ако е необходимо, още натриев хидроксид, докато се появи отчетлив червен цвят. Покрива се бехеровата чаша с наблюдателно стъкло и се поставя в парна баня за 15 минути.
7.3. Претегляне на тигела
Изсушава се филтър-тигела (виж точка 5 „Апаратура“) до постоянна маса (около 15 минути) в пещ при 120 °С (5.4).
Оставя се тигелът да се охлади в сушилен шкаф и след това се претегля.
7.4. Отделяне на утайката
Отстранява се бехеровата чаша от парната баня, разбърква се капка по капка 10 ml STPB разтвор (4.6). Това добавяне отнема около две минути. Изчаква се поне 10 минути преди филтриране.
7.5. Филтриране и измиване
Филтрира се под вакуум в претегления тигел, измива се бехеровата чаша с течността за измиване (4.7), измива се утайката три пъти с течността за измиване (60 ml общо течност за измиване) и два пъти с 5 до 10 ml вода.
Измива се старателно утайката.
7.6. Изсушаване и претегляне
Избърсва се външната част на тигела с филтърна хартия. Поставя се тигела с неговото съдържание в пещ за сушене в продължение на един час и половина при температура 120 °С. Оставя се да се охлади в сушилен шкаф при температура на околната среда и бързо се претегля.
Бележка по процедурата
|
а) |
Ако филтратът е тъмен на цвят, прехвърля се с пипета аликвотна част, съдържаща най-много 100 mg К2О и се поставя в мерителна колба от 100 ml, се добавя бромова вода и се довежда до кипене, за да се елиминира целият излишен бром. След охлаждане се долива до обема, филтрира се и се определя количествено калият в аликвотната част на филтрата. |
|
б) |
Когато присъства малко или никакъв азот, не е нужно да кипи 15 минути. |
7.7. Аликвотни части, които трябва да се вземат като проби, и коефициенти за конверсия
Таблица 3
За метод 4
|
% К2O в тора |
% К в тора |
Проба за анализ (g) |
Проба на разтвор на екстракта за разреждане (ml) |
Разреждане (на ml) |
Аликвотна част за вземане от пробата за утаяване (ml) |
Коефициент на превръщане (F) |
Коефициент на превръщане F′ |
|
5—10 |
4,2—8,3 |
10 |
— |
— |
50 |
26,280 |
21,812 |
|
10—20 |
8,3—16,6 |
10 |
— |
— |
25 |
52,560 |
43,624 |
|
20—50 |
16,6—41,5 |
10 |
или — |
|
10 |
131,400 |
109,060 |
|
|
|
или 50 |
250 |
50 |
131,400 |
109,060 |
|
|
над 50 |
над 41,5 |
5 |
или — |
— |
10 |
262,800 |
218,120 |
|
|
|
или 50 |
250 |
50 |
262,800 |
218,120 |
7.8. Празна проба
За всяка серия определяния се извършва изпитване на празна проба, като се използват само реактиви и разтвори в пропорциите, използвани при анализа определянето, които се отчитат при изчисляването на крайния резултат.
7.9. Контролно изпитване
За да се извърши контрол на метода за анализ, се извършва определяне на алоквотна част на воден разтвор на калиев хлорид, съдържащ най-много 40 mg К2О.
8. Изразяване на резултата
Ако се използват пробите за анализиране и разрежданията, показани в таблица 3, се прилагат следните формули:
|
|
или |

|
|
|
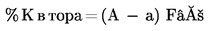
където
|
А |
= |
маса, в грамове, на утайката от пробата, |
|
а |
= |
маса в грамове, на утайката от празната проба, |
|
F и F′ |
= |
коефициенти (виж таблица 3). |
При проби и разреждания, които се различават от тези в таблица 3, използват се следните формули:
|
|
или |

|
|
|
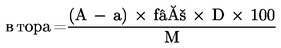
където
|
f |
= |
коефициент на конверсия, КТРВ в К2O = 0,1314, |
|
f′ |
= |
коефициенти на конверсия, КТРВ в К = 0,109, |
|
D |
= |
коефициент на разреждане, |
|
M |
= |
маса, в грамове, на анализираната проба. |
Метод 5
Няма позиция
Метод 6
Хлор
Метод 6.1
Определяне на хлориди при отсъствие на органичен материал
1. Обхват
Настоящият документ дефинира процедурата за определяне на хлор при отсъствие на органичен материал.
2. Област на приложение
Всички торове, които не съдържат органичен материал.
3. Принцип
Утайката на хлоридите, разтворени във вода, се разделят в кисела среда с излишък на стандартен разтвор на сребърен нитрат. Излишъкът се титрува с разтвор на амониев тиоцианит в присъствието на железен амониев сулфат (метод на Фолхард).
4. Реактиви
Дестилирана или деминерализирана вода, несъдържаща хлориди.
4.1. Нитробензин или диетилов етер
4.2. Азотна киселина: 10 mol/l
4.3. Индикаторен разтвор
Разтваря се 40 g железен амониев сулфат Fe2(SO4)3.(NH4)2SO4.24H2O във вода и се долива до един литър.
4.4. Стандартен разтвор на сребърен нитрат: 0,1 mol/l
Приготвяне
Тъй като тази сол е хигроскопична и не може да се души без риск от разлагане, препоръчва се да се претеглят приблизително 9 g, да се разтворят във вода и да се долее до един литър. Регулира се на концентрация 0,1 mol/l с титруване на AgNO3 0,1 mol/l.
5. Апаратура
5.1. Ротационна клатачна машина (35 до 40 оборота в минута)
5.2. Бюрети
5.3. Мерителна колба от 500 ml
5.4. Конична (ерленмайерова) колба от 250 ml
6. Приготвяне на пробата
Виж метод 1.
7. Процедура
7.1. Проба и приготвяне на разтвора
Поставят се 5 g от пробата, претеглени с точност до 0,001 g, в мерителна колба от 500 ml и се добавя 450 ml вода. Смесва се в продължение на половин час на клатачна машина (5.1). Долива се до 500 ml с дестилирана вода. Смесва се и се филтрира в бехерова чаша.
7.2 Определяне
Взема се аликвотна част на филтрата, съдържаща не повече от 0,150 g хлор. Например, 25 ml (0,25 g), 50 ml (0,5 g) или 100 ml (1 g). Ако пробата е по-малка от 50 ml, е необходимо да се допълни обемът до 50 ml с дестилирана вода.
Добавя се 5 ml азотна киселина 10 mol/l (4.2), 20 ml индикаторен разтвор (4.3) и две капки стандартен разтвор на амониев тиоцианат (проба от последния реактив се взема с бюрета, регулирана на нула за тази цел).
С бюрета след това се добавя стандартен разтвор на сребърен нитрат (4.4), докато има излишък от 2 до 5 ml. Добавя се 5 ml нитробензин или 5 ml диетилов етер (4.1), за да се агломерира утайката. Титрува се излишният сребърен нитрат с амониев тиоцианат 0,1 mol/l (4.5), докато се появи червено-кафяв цвят, който остава след леко разклащане на колбата.
Бележка
Нитробензинът или диетиловият етер (но преди всичко нитробензинът) предотвратяват реагирането на сребърния нитрат с йоните на тиоцианата. Така се получава ясна смяна на цвета.
7.3. Празна проба
Извършва се изпитване на празна проба (изпускайки пробата) при същите условия и това се отчита при изчисляването на окончателния резултат.
7.4. Контролно изпитване
Преди извършване на оценяването, проверява се точността на метода, като се използва аликвотна част на прясно приготвен разтвор на калиев хлорид, който съдържа известно количество от порядъка на 100 mg хлор.
8. Изразяване на резултата
Резултатът се изразява като процент на хлорида, съдържащ се в пробата, както е била получена за анализ.
Изчислява се процента на хлора (Cl) с формулата:

където
|
VZ |
= |
брой милилитри сребърен нитрат 0,1 mol/l, |
|
Vcz |
= |
брой милилитри сребърен нитрат 0,1 mol/l, използван при изпитването на празната проба |
|
Va |
= |
брой милилитри амониев тиоцианат 0,1 mol/l, |
|
Vca |
= |
брой милилитри амониев тиоцианат 0,1 mol/l, използван при изпитването на празната проба, |
|
M |
= |
маса, в грамове, на взетата проба (7.2). |
Методи 7
Степен на смилане
Метод 7.1
Определяне на степен на смилане
(суха процедура)
1. Обхват
Настоящият документ дефинира процедурата за определяне на степента на смилане.
2. Област на приложение
Всички ЕО тип торове, в чиито изисквания са дадени степен на смилане с използване на сита 0,630 и 0,160 mm.
3. Принцип
Чрез механично отсяване се определят количествата на продукта с размери на гранулата по-голям от 0,630 mm и тези с размер на гранулата между 0,160 и 0,630 mm и се изчислява процента на степента на смилане.
4. Апаратура
4.1. Механична клатачна машина на сито
4.2. Сита с отвори 0,160 и 0,630 mm съответно стандартен обхват (20 cm диаметър и 5 cm височина).
5. Процедура
Претеглят се 50 g от субстанцията с точност 0,05 g. Сглобяват се двете сита и събиращия съд на клатачната машина (4.1), като ситото с по-големи отвори се поставя отгоре. Поставя се анализираната проба отгоре. Пресява се 10 минути и се отстранява частта, събрана отдолу. Пуска се апаратът отново и след една минута се проверява дали количеството, събрано на дъното през това време, не е повече 250 mg. Повтаря се процесът (за една минута всеки път, докато събраното количество е по-малко от 250 mg. Претегля се останалият материал на двете сита отделно.
6. Изразяване на резултата
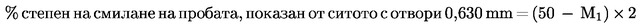

където
М1= маса, в грамове на остатъка върху ситото с отвори 0,630 mm,
М2= маса, в грамове на остатъка върху ситото с отвори 0,160 mm,
Отделеното от ситото с отвори 0,630 е вече елиминирано.
Резултатите от тези изчисления се закръгляват до най-близкото цяло число.
Метод 7.2
Определяне на степента на смилане на меки естествени фосфати
1. Обхват
Настоящият метод е за определяне степента на смилане на меки естествени фосфати.
2. Област на приложение
Меки естествени фосфати.
3. Принцип
При проби с фини частици може да се случи агломерация, която да направи пресяването трудно. Поради тази причина нормално се използва мокро пресяване.
4. Реактиви
Разтвор на натриев хексаметафосфат: 1 %.
5. Апаратура
5.1. Сита с отвори 0,063 и 0,125 mm съответно стандартен обхват (20 cm диаметър и 5 cm височина); събиращи съдове
5.2. Стъклена фуния с диаметър 20 cm, монтирана на стойка
5.3. Бехерови чаши 250 ml
5.4. Пещ за сушене
6. Метод за анализ
6.1. Вземане на проба
Претегля се 50 g от субстанцията с точност до 0,05 g. Измиват се двете страни на ситото и се поставят ситата с отвори 0,125 mm над ситото с отвори 0,063 mm.
6.2. Процедура
Поставя се анализираната проба върху горното сито. Пресява се под малка струя студена вода (може да се използва вода от канализацията), докато водата е практическа чиста, когато преминава през ситото. Трябва да се внимава дебитът на водата да е такъв, че долното сито никога да не се пълни с вода.
Когато остатъкът на горното сито изглежда, че остава повече или по-малко постоянен, ситото се маха и се поставя междувременно в събиращ съд.
Мокрото пресяване продължава през по-долното сито няколко минути, докато водата, преминаваща през него, стане почти бистра.
Заменя се ситото 0,125 mm над ситото 0,063 mm. Прехвърля се натрупаното от събиращия съд върху ситото и се пресява отново под малка струя вода, докато тази вода стани почти бистра още веднъж.
Остатъците се прехвърлят количествено в различни бехерови чаши с помощта на фунията. Суспензира се всеки остатък чрез напълване бехеровите чаши с вода. Оставя се ги да престоят няколко минути, декантира се колкото е възможно повече вода.
Поставя се бехеровите чаши в пещта за сушене при 150 °С за два часа.
Оставят се да се охладят, остатъците се отделят и се претеглят.
7. Изразяване на резултатите
Резултатите от тези изчисления се закръгляват до най-близкото цяло число.


където
М1= маса, в грамове на остатъка върху ситото с отвори 0,125 mm,
М2= маса, в грамове на остатъка върху ситото с отвори 0,063 mm.
8. Забележки
Ако се наблюдава присъствието на буци след пресяването, анализът трябва да се извърши отново по следния начин.
|
|
Изсипват се бавно 50 g от пробата в колба един литър, съдържаща 500 ml разтвор на натриев хексаметафосфат, бъркайки постоянно. Запушва се колбата и се разклаща силно с ръка, за да се разбият буците. Прехвърля се цялата суспензия върху горното сито и се измива старателно колбата. Анализът продължава, както е описано в 6.2. |
Методи 8
Вторични хранителни елементи
Метод 8.1
Екстракция на общ калций, общ магнезий, общ натрий и общ фосфор под формата на сулфати
1. Обхват
Настоящият документ дефинира процедурата за извличане на общ калций, общ магнезий, общ натрий и общ фосфор под формата на сулфати, така че едно и също извличане да може да се използва за определянето на всеки изискван хранителен елемент.
2. Област на приложение
Настоящият метод се прилага за ЕО торове, за които в настоящия регламент се изисква деклариране на общ калций, общ магнезий, общ натрий и общ фосфор под формата на сулфати.
3. Принцип
Разтваряне чрез кипене в разредена солна киселина.
4. Реактиви
4.1. Разредена солна киселина
Един обем солна киселина (d20 = 1,18 g/ml) плюс един обем вода.
5. Апаратура
Електрически котлон с регулируема температура.
6. Приготвяне на пробата
Виж метод 1.
7. Процедура
7.1 Проба за изпитване
Калций, магнезий, натрий и сяра под формата на сулфати се извличат от проба за изпитване с тегло пет грама, претеглени с точност до 1 милиграм.
Независимо от това, когато торът съдържа повече от 15 % сяра (S) т.е. 37,5 % SO3 и повече от 18,8 % калций (Са) т.е. 26,3 % СаО, извличането на калций и сяра се извършва на проба за изпитване един грам, претеглен с точност до един милиграм. Екстрактът трябва да бъде напълно прозрачен. Поставя се пробата за изпитване в бехерова чаша 600 ml.
7.2. Приготвяне на разтвора
Прибавя се приблизително 400 ml вода и като се внимава, когато пробата съдържа значително количество карбонати, 50 ml разредена солна киселина (4.1) на малки количества. Довежда се до кипене, което се поддържа 30 минути. Оставя се да се охлади, като се разбърква от време на време. Декантира се количествено в мерителна колба от 500 ml. Долива се до обема вода, смесва се. Прекарва се през сух филтър в сух съд, като се изхвърля първата част. Екстрактът трябва да бъде напълно прозрачен. Запушва се, ако филтратът няма да се използва незабавно.
Метод 8.2
Екстракция на обща сяра присъстващи под различни форми
1. Обхват
Настоящият документ дефинира процедурата за извличане на обща сяра, съдържаща се в торове в елементарна форма и/или други химически комбинации.
2. Област на приложение
Настоящият метод се прилага за ЕО торове, за които в настоящия регламент се изисква деклариране на общ фосфор под различни форми (елементарна, тиосулфат, сулфит, сулфат).
3. Принцип
Елементарният фосфор се превръща в алкална среда в полисулфиди и тиосулфати. Те, заедно с всички сулфити, които могат да присъстват, след това се окисляват с водороден прекис. Различните форми на сяра по този начин се превръщат в сулфат, който се определя чрез отделяне на утайката на бариев сулфат (метод 8.9).
4. Реактиви
4.1. Разредена солна киселина
Един обем солна киселина (d20 = 1,18 g/ml) плюс един обем вода.
4.2. Разтвор на натриев диоксид, NaOH, 30 % минимум (d20 = 1,33 g/ml)
4.3. Разтвор на водороден прекис, 30 % w/w
4.4. Воден разтвор на бариев хлорид BaCl2.2H2O, 122 грама на литър
5. Апаратура
Електрически котлон с регулируема температура.
6. Приготвяне на пробата
Виж метод 1.
7. Процедура
7.1. Проба за изпитване
Претегля се с точност до един милиграм количество от тора, съдържащо между 80 и 350 милиграма сяра (S) или между 200 и 875 милиграма SO3.
Като правило (когато S < 15 %), претеглят се 2,5 грама. Поставя се пробата за изпитване в бехерова чаша 400 ml.
7.2. Окисляване
Добавят се 20 милилитра разтвор на натриев хидроксид (4.2) и 20 милилитра вода. Покрива се с наблюдателно стъкло. Оставя се да кипи пет минути на котлон (5.1). Сваля се от котлона. Като се използва струя гореща вода, се събира сярата, полепнала по стените на бехеровата чаша, и се вари в продължение на 20 минути. Оставя се да се охлади.
Добавят се 2 милилитра инкременти на водороден прекис (4.3), докато не се появи реакция. Ще бъде необходим от шест до осем ml водороден прекис. Оставя се окисляването да продължи един час, след това се довежда до кипене в продължение на половин час. Оставя се да се охлади.
7.3. Приготвяне на разтвора за анализ
Добавят се приблизително 50 ml вода и 50 ml разтвор на солна киселина (4.1).
|
— |
Ако нивото на сярата (S) е по-ниско от 5 %:
|
|
— |
Ако нивото на сярата (S) е по-високо от 5 %:
|
Метод 8.3
Екстракция на калций, магнезий, натрий и сяра, разтворими във вода
(под формата на сулфати)
1. Обхват
Настоящият документ дефинира процедурата за екстракция на калций, магнезий, натрий и сяра разтворими във вода (под формата на сулфати), така че един и същ екстракт да може да бъде използван за всеки изискван хранителен елемент.
2. Област на приложение
Този метод се прилага само за торове, за които в приложение I е предвидено деклариране на калций, магнезий, натрий и сяра, разтворими във вода.
3. Принцип
Хранителните елементи се разтварят в кипяща вода.
4. Реактиви
Дестилирана или деминерализирана вода с еквивалентно качество.
5. Апаратура
Електрически котлон с регулируема температура.
6. Приготвяне на пробата
Виж метод 1.
7. Процедура
7.1. Проба за изпитване
|
а) |
Когато торовете не съдържат сяра или когато те съдържат, в същото време, не повече от 3 % сяра (S) т.е. 7,5 % SO3 и не повече от 4 % калций (Са) т.е. 5,6 % СаО, се претеглят пет грама тор с точност до един милиграм. |
|
б) |
Когато торовете съдържат повече от 3 % сяра (S) и повече от 4 % калций (Са), се претегля един грам тор с точност до един милиграм. |
Поставя се пробата за изпитване в бехерова чаша 600 ml.
7.2. Приготвяне на разтвора
Добавят се приблизително 400 ml вода и се вари в продължение на 30 минути. Оставя се да се охлади, като се разбърква от време на време, и се декантира количествено в мерителна колба от 500 ml. Допълва се до обема вода и се смесва.
Филтрира се през сух филтър в сух съд. Изхвърля се първоначалните части на филтрата. Филтратът трябва да бъде напълни прозрачен.
Слага се запушалка, ако разтворът няма да се използва незабавно.
Метод 8.4
Екстракция на водоразтворима сяра, когато сярата е под различни форми
1. Обхват
Настоящият документ дефинира процедурата за извличане на разтворима във вода сяра, съдържаща се в торове под различна форма.
2. Област на приложение
Настоящият метод се прилага за торове, за които е предвидено деклариране в приложение I на разтворим във вода серен триоксид.
3. Принцип
Сярата се разтваря в студена вода и се превръща в сулфат чрез окисляване с водороден прекис в алкална среда.
4. Реактиви
4.1. Разредена солна киселина
Един обем солна киселина (d20 = 1,18 g/ml) плюс един обем вода.
4.2. Разтвор на натриев диоксид, NaOH, 30 % минимум (d20 = 1,33 g/ml)
4.3. Разтвор на водороден прекис, 30 % w/w
5. Апаратура
5.1. Мерителна колба Стохман от 500 ml
5.2. Ротационна клатачна машина, 30 до 40 оборота в минута.
5.3. Електрически котлон с регулируема температура.
6. Приготвяне на пробата
Виж метод 1.
7. Процедура
7.1. Проба за изпитване
|
а) |
Когато торове не съдържат сяра или когато те съдържат, в същото време, не повече от 3 % сяра (S) т.е. 7,5 % SO3 и не повече от 4 % калций (Са) т.е. 5,6 % СаО, се претеглят пет грама тор с точност до един милиграм. |
|
б) |
Когато торове съдържат повече от 3 % сяра (S) и повече от 4 % калций (Са), се претегля един грам тор с точност до един милиграм. |
Поставя се пробата за изпитване в колба от 500 ml (5.1).
7.2. Приготвяне на разтвора
Добавя се приблизително 400 ml вода. Запушва се. Разклаща се (5.2) в продължение на 30 минути. Добавя се до обема вода и се смесва. Филтрира се през сух филтър в сух съд. Слага се запушалка, ако разтворът няма да се използва незабавно.
7.3. Окисляване на аликвотна част за анализиране
Взема се аликвотна част от разтвора на екстракта, не превишаваща 50 милилитра, и ако е възможно, съдържаща между 20 и 100 милиграма сяра.
Долива се до обем 50 ml вода. Добавя се три милилитра разтвор на натриев хидроксид (4.2) и два милилитра разтвор на водороден прекис (4.3). Покрива се с наблюдателно стъкло и се вари на слаб огън един час на котлона (5.3). Продължава се добавянето на инкременти от един милилитър разтвор на водороден прекис, докато реакцията продължава (максимално количество пет милилитра).
След това се оставя да се охлади. Маха се наблюдателното стъкло и се измива долната част в бехерова чаша. Добавя се приблизително 20 ml разредена солна киселина (4.1). Долива се до приблизително 300 милилитра с вода.
Определя се съдържанието на сулфати в целия окислен разтвор в съответствие с метод 8.9.
Метод 8.5
Екстракция и определяне на елементарна сяра
Предупреждение
Този метод за анализ включва използването на въглероден дисулфид (CS2). Специални мерки за безопасност трябва да се вземат, по специално по отношение на:
|
— |
съхраняването на CS2, |
|
— |
защитно оборудване за персонала, |
|
— |
професионална хигиена, |
|
— |
предотвратяване на пожари и експлозии, |
|
— |
изхвърляне на реактива. |
Този метод изисква персонал с високи умения и подходящо оборудвана лаборатория.
1. Обхват
Настоящото дефинира процедурата за извличане на елементарна сяра, съдържаща се в торове.
2. Област на приложение
Настоящият метод се прилага за EO торове, за които е предвидено деклариране в приложение I на сяра в елементарна форма.
3. Принцип
След отстраняване на разтворимите съединения, елементарната сяра се извлича с използване на въглероден дисулфид, последвано от гравиметрично определяне на извлечената сяра.
4. Реактиви
Въглероден дисулфид
5. Апаратура
5.1. Екстракционна колба от 100 ml с шлифована стъклена запушалка
5.2. Сокслетов апарат с подходящи филтърни елементи
5.3. Вакуумен ротационен изпарител
5.4. Електрически котлон, подпомаган от вентилатор, настроен на 90(± 2) °С
5.5. Порцеланови блюда на Петри с диаметър от пет до седем сантиметра, не превишаващи височина от пет сантиметра.
5.6. Електрически котлон с регулируема температура
6. Приготвяне на пробата
Виж метод 1.
7. Процедура
7.1. Проба за изпитване
Претеглят се пет грама от пробата с точност до един милиграм и се поставят в екстракционната гилза на Сокслетов апарат (5.2).
7.2. Отделяне на сярата
Промийте старателно съдържанията с топла вода, за да премахнете всички разтворими съединения. Изсушете в печка на 90 °C за най-малко един час. Поставете филтъра в апарата Soxhlet (5.2).
Поставя се няколко стъклени перли в колбата на апарата (5.1) и се претегля (Р0). След това се добавя 50 милилитра въглероден дисулфид (4.1).
Свързва се апаратът и се оставя елементарната сяра да бъде извлечена за шест часа. Изключва се нагряването и след охлаждане се откача колбата. Свързва се колбата към ротационния изпарител (5.3) и се изпарява, докато съдържанието на колбата се втвърди в гъбеста маса.
Изсушава се колбата в пещта при 90 °С (5.4) (по принцип е необходим един час) докато се получи постоянна маса (Р1).
7.3. Определяне на чистотата на елементарната сяра
Някои елементи могат да са били извлечени с въглеродния дисулфид заедно с елементарната сяра. Чистотата на елементарната сяра се определя, както следва:
|
|
Хомогенизира се съдържанието на колбата, колкото може по-старателно, и се отстраняват два до три грама. Претегля се с точност до 1 милиграм (n). Поставя се в блюдо на Петри (5.5). Претегля се блюдото и съдържанието заедно (Р2). Поставя се на котлона (5.6), който е температура, непревишаваща 220 °С, така че да не причини изгаряне на сярата. Продължава се сублимирането три до четири часа, докато се получи постоянна маса (Р3). |
Бележка:
За някои торове може да не е необходимо да се определя, колко е чиста сярата. В този случай се пропуска стъпка 7.2.
8. Изразяване на резултати
Процентното съдържание на елементарна сяра (S) на тора е, както следва:
|
|
|

|
|
|

|
|
|

където
|
m |
= |
масата на пробата за изпитване на тора в грамове, |
|
P0 |
= |
масата на колбата на Сикслет в грамове, |
|
P1 |
= |
масата на колбата на Сокслет и нечистата сяра след изсушаване, |
|
n |
= |
масата на нечистата сяра за пречистване м грамове, |
|
P2 |
= |
масата на блюдото на Петри, |
|
P3 |
= |
масата на блюдото на Петри след сублимирането на сярата в грамове. |
Метод 8.6
Манганиметрично определяне на извлечен калций след утаяване под формата на оксалат
1. Обхват
Настоящият документ дефинира процедурата за извличане на калций в екстракти на торове.
2. Област на приложение
Настоящият метод се прилага за ЕО торове, за които в приложение I е предвидено деклариране на общ и/или водоразтворим калций.
3. Принцип
Утаяване на калций, съдържащ се в аликвот на извлечения разтвор под формата на оксалат, който се определя чрез титруване с използване на калиев перманганат.
4. Реактиви
4.1. Разредена солна киселина
Един обем солна киселина (d20 = 1,18 g/ml) в един обем вода.
4.2. 1:10 разредена сярна киселина
Един обем сярна киселина (d20 = 1,84 g/ml) в десет обема вода.
4.3. 1:1 разреден амонячен разтвор
Един обем амоняк (d20 = 0,88 g/ml) в един обем вода.
4.4. Наситен разтвор на амонячен оксалат [(NH4)2C2O4H2O] при температура на околната среда (приблизително 40 грама на литър)
4.5. Разтвор на лимонена киселина, 30 % (m/v)
4.6. Разтвор на амонячен хлорид, 5 % (m/v)
4.7. Разтвор на бромотимол син в етанол, при 95 %, 0,1 % (m/v)
4.8. Разтвор на бромкрезолово зелено в етанол, при 95 %, 0,04 % (m/v)
4.9. Стандартен разтвор на калиев перманганат, 0,02 mol/l
5. Апаратура
5.1. Филтърен тигел с порьозност 5 до 20 μ от синтеровано стъкло
5.2. Гореща водна баня
6. Приготвяне на аликвота за анализиране
Взема се с пипета аликвотна част от разтвора на екстракта, получен по метод 8.1 или 8.3, съдържаща между 15 и 50 mg Са (= 21 до 70 милиграма СаО). Обемът на този аликвот трябва да бъде v2. Излива се в бехерова чаша от 400 ml. Ако е необходимо, се неутрализира (обръщане на индикатор (4.7) от зелен на син) с няколко капки разтвор на амоняк (4.3).
Добавя се един милилитър разтвор на лимонена киселина (4.5) и пет милилитра разтвор на амонячен хлорид (4.6).
7. Утаяване на калиевия оксалат
Добавя се приблизително 100 ml вода. Довежда се до кипене, добавят се осем до десет капки индикаторен разтвор (4.8) и, бавно, 50 милилитра горещ разтвор на амониев оксилат (4.4). Ако се образува утайка, тя се разтваря чрез добавяне на няколко капки солна киселина (4.1). Неутрализира се много бавно с амонячен разтвор (4.3), като се бърка постоянно до рН от 4,4 до 4,6 (превръщане на индикатор (4.8) от зелен в син). Поставя се бехеровата чаша в кипяща гореща водна баня (5.2) за приблизително 30 минути.
Отстранява се бехеровата чаша от банята, оставя се да престои един час и се филтрира в тигела (5.1).
8. Титруване на оксалатната утайка
Измива се бехеровата чаша и тигела, докато излишният амониев оксалат се отстрани напълно (това може да се провери с отсъствието на хлорид в измиващата вода). Поставя се тигелът в бехеровата чаша от 400 ml и се разтваря утайката с 50 ml гореща сярна киселина (4.2). Добавя се вода в бехеровата чаша, за да се получи обем приблизително 100 ml. Довежда се до температура от 70 до 80 °С и се титрува капка по капка с разтвор на перманганата (4.9), докато розовия цвят се задържи минута. Този обем е n.
9. Изразяване на резултатите
Съдържанието на калций (Са) в тора е, както следва:
|
|
|
 ,
,където
|
n |
= |
брой милилитри използван перманганат, |
|
m |
= |
масата на пробата за изпитване в грамове, |
|
v2 |
= |
обем на аликвота в милилитри, |
|
v1 |
= |
обем на разтвора на екстракта в милилитри |
|
t |
= |
концентрация на разтвора на перманганата в mol/l. |
CaO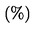 = Ca
= Ca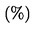 × 1,400
× 1,400
Метод 8.7
Определяне на магнезий чрез атомна абсорбционна спектрометрия
1. Обхват
Настоящият документ описва процедура за определяне на магнезий в екстракти на торове.
2. Област на приложение
Процедурата се прилага за образци на ЕО торове, получени по методи 8.1 и 8.3, за които се изисква деклариране на общ и/или разтворим във вода магнезий, с изключение на следните торове, изброени в приложение I.Г, свързани с вторични хранителни елементи:
|
— |
вид 4 (кизерит), |
|
— |
вид 5 (магнезиев сулфат) и вид 5.1 (разтвор на магнезиев сулфат), |
|
— |
и с изключение на следните торове, изброени в приложение I, част А, точка 3, свързани с калиеви торове: |
|
— |
вид 7 (кизерит с калиев фосфат), |
|
— |
за който се прилага метод 8.8. |
Методът, изложен по-долу, се прилага за всички екстракти на торове, съдържащи елементи в количества, които могат да повлияят на комплексометричното определяне на магнезия.
3. Принцип
След подходящо разреждане на екстракта магнезият се определя чрез атомна абсорбционна спектрометрия.
4. Реактиви
4.1. Солна киселина, 1 mol/l
4.2. Солна киселина, 0,5 mol/l
4.3. Стандартен разтвор на магнезий1 1,00 милиграма в милилитър.
|
4.3.1. |
Разтварят се 1,013 грама магнезиев сулфат (MgSO4.7H2O) в 0,5 mol/l разтвор на солна киселина (4.2). |
|
4.3.2. |
Претеглят се 1,658 грама магнезиев оксид (MgO), калциниран преди това за отстраняване на всички следи от карбонати. Поставя се в бехерова чаша със 100 ml вода и 120 ml 1 mol/l солна киселина (4.1). Когато се разтвори, количествено се декантира в мерителна колба от 1 000 ml. Долива се до обема с вода и се смесва. или |
|
4.3.3. |
Търговски стандартен разтвор Лабораторията отговаря за изпитването на такива разтвори. |
4.4. Разтвор на стронциев хлорид
Разтваря се 75 g стронциев хлорид (SrCl2 6H2O) в разтвор на солна киселина (4.2) и се долива до 500 милилитра със същия разтвор на киселината.
5. Апаратура
Спектрометър, подходящ за атомна абсорбция, с магнезиева лампа, настроена на 285,2 nm.
Пламък въздух-ацетилен.
6. Приготвяне на пробата
Виж методи 8.1 и 8.3.
7. Процедура
7.1. Ако торът има декларирано съдържание на магнезий (Mg) повече от 6 % (т.е. 10 % като MgO), се взема 25 милилитра (V1) от разтвора на екстракта. Прехвърля се в мерителна колба от 100 ml, долива се до обема с вода и се смесва. Коефициентът на разреждане е D1 = 100/V1.
7.2. Вземат се с пипета 10 милилитра от разтвора на екстракта (6) или разтвора (7.1). Прехвърля се в мерителна колба от 200 ml. Допълва се до обема с 0,5 mol/l разтвор на солна киселина (4.2) и се смесва. Коефициентът на разреждане е 200/10.
7.3. Разрежда се този разтвор (7.2) с 0,5 mol/l солна киселина (4.2), така че да се получи концентрация в оптималното работно поле на спектрометъра (5.1). V2 е обемът на пробата в 100 ml. Коефициентът на разреждане е D2 = 100/V2.
Окончателния разтвор трябва да съдържа10 %(v/v) разтвор на стронциев хлорид (4.4).
7.4. Приготвяне на разтвор на празна проба
Приготвя се разтвор на празна проба, като се повтаря цялата процедура от екстракцията (метод 8.1 или 8.3); пропуска се само пробата за изпитване на тора.
7.5. Приготвяне на калибровъчни разтвори
Чрез разреждане на стандартния разтвор(4.3) с 0,55 mol/l солна киселина се приготвят най-малкото пет калибровъчни разтвора с увеличаваща се концентрация в рамките на оптималния измервателния диапазон на апарата (5.1).
Тези разтвори трябва да съдържат 10 % v/v на разтвор на стронциев хлорид (4.4).
7.6. Измерване
Настройва се спектрометърът на (5.1) на дължината на вълната 285,2 nm.
Впръскват се последователно калибровъчните разтвори (7.5), разтворът на пробата (7.3) и разтворът на празната проба (7.4), като се измива инструментът с разтвор, който ще се измерва след това. Операцията се повтаря три пъти. Начертава се калибровъчната крива, като се използват средните аритметични абсорбции на всяко калибриране (7.5) за ордината, а съответните концентрации на магнезия в μg/ml за абсциси. Определя се концентрацията на магнезия в пробата (7.3) Xs и празната проба (7.4) Xb с позоваване на калибровъчната крива.
8. Изразяване на резултатите
Изчислява се количеството на магнезия (Mg) или магнезиевия оксид (MgO) в пробата с позоваване на калибровъчната крива и се вземе предвид празната проба.
Процентът на магнезия (Mg) в тора е равен на:
|
|
|
 ,
,където
|
|
Хs = концентрацията на анализирания разтвор, записан на калибровъчната крива, в μg/ml. |
|
|
Хb = концентрацията на разтвора на празната проба, записан на калибровъчната крива, в μg/ml. |
|
|
D1 = коефициент на разреждането, когато разтворът се разрежда (7.1). |
|
— |
Той е равен на четири, ако са взети 25 ml. |
|
— |
Той е равен на едно, когато разтворът не е разтворен. |
|
— |
D2 = коефициентът на разреждането е 7.3. |
|
— |
М = масата на пробата за изпитване по време на екстракцията. |
|
— |
MgO |
 = Mg
= Mg /0,6
/0,6Метод 8.8
Определяне на магнезий чрез комплексометрия
1. Обхват
Настоящият документ дефинира процедурата за извличане на магнезий в екстракти на торове.
2. Област на приложение
Настоящият метод се прилага за ЕС торове, за които в е предвидено деклариране на общ и/или водоразтворим магнезий:
|
— |
торове, изброени в приложение I: еднокомпонентни азотни торове, вид 1b + 1c (калциево-магнезиев нитрат), вид 7 (магнезиев сулфонитрат), вид 8 (азотни торове с магнезий) и еднокомпонентни калиеви торове, вид 2 (обогатен каинит), вид 4 (калиев хлорид, съдържащ магнезий), вид 6 (калиев сулфат, съдържащ магнезиева сол), |
|
— |
торове, изброени в приложение I.Г, свързани с вторични хранителни елементи. |
3. Принцип
Магнезия се разтваря с методи 8.1 и/или 8.3. Първо титруване: с EDTA Ca и Mg в присъствието на Eriochrome черен-Т. Второ титруване: с EDTA Са в присъствието на калсеин или калкон въглеродна киселина. Определяне на магнезий с разлика.
4. Реактиви
4.1. Стандартен 0,05 mol/l разтвор на магнезий:
|
4.1.1. |
Разтварят се 1, 232 грама магнезиев сулфат (MgSO4 7H2О) в 0,5 mol/l разтвор на солна киселина (4.11) и се долива до 100 ml със същата киселина; или |
|
4.1.2. |
Претеглят се 2,016 грама магнезиев оксид, предварително калциниран с цел отстраняване на всички следи от карбонизация. Поставя се в бехерова чаша със 100 милилитра вода. |
Чрез разбъркване се добавят приблизително 120 милилитра солна киселина приблизително 1 mol/l (4.12).
След разтваряне количествено се прехвърля в мерителна колба от 1 000 ml. Долива се до обема и се смесва.
Един милилитър от този разтвор трябва да съдържа 1,216 милиграма Mg (= 2,016 милиграма MgО).
Лабораторията отговаря за изпитване на силата на този стандартен разтвор.
4.2. 0,05 моларен разтвор на EDTA
Претеглят се 18,61 грама дихидратна двунатриева сол на етилендиаминтетраоцетна (C10H14N2O8.2H2O)., поставят се в бехерова чаша от 1 000 милилитра и се разтварят в 600 до 800 милилитра вода. Допълва се до обема и се смесва. Проверява се този разтвор със стандартния разтвор (4.1) като се взема проба от 20 милилитра от него и се титрува съгласно процедурата за анализ, описана в (7.2)
Един милилитър от EDTA разтвора трябва да съответства на 1,216 милиграма Mg (= 2,016 милиграма MgО) и на 2,004 милиграма Са (= 2,804 милиграма СаО) (виж забележки 10.1 и 10.6).
4.3. 0,05 моларен стандартен разтвор на калций
Претеглят се 5,004 грама сух калциев карбонат. Поставят се го в бехерова чаша със 100 милилитра вода. Постоянно се разбърква в 120 милилитра солна киселина приблизително 1 mol/l (4.12).
Довежда се до кипене, за да се изкара въглеродният диоксид, охлажда се, прехвърля се количествено в мерителна колба от един литър, допълва се до обема вода и се смесва. Проверява се този разтвор спрямо EDTA разтвор (4.2), като се следва процедурата за анализ (7.3). Един милилитър от този разтвор трябва да съдържа 2,004 милиграма Са (= 2,804 милиграма СаО) и трябва да съответства на 1 милилитър 0,05 моларен EDTA разтвор (4.2).
4.4. Калсеинен индикатор
Внимателно се смесва в хаван 1 g калсеин с 100 g натриев хлорид. Използват се 10 милиграма от тази смес. Индикаторът се променя от зелен на оранжев. Титруване трябва да се извършва, докато се получи оранжев цвят без зелени отсенки.
4.5. Индикатор калкон въглеродна киселина
Разтваря се 400 милиграма калкон въглеродна киселина в 100 милилитра метанол. Този разтвор може да се съхранява само приблизително четири седмици. Индикаторът се променя от червен на син. Титруването трябва да се извършва докато се получи син цвят без червени отсенки.
4.6. Индикатор ериохром черен-Т
Разтваря се 300 милиграма ериохром черен-Т в 25 милилитра пропанол-1 и 15 милилитра триетаноламин. Този разтвор може да се съхранява само приблизително четири седмици. Индикаторът се променя от червен на син. Титруването трябва да се извършва докато се получи син цвят без червени отсенки. Той си променя цвета само когато присъства магнезий. Ако е необходимо, се добавя един милилитър стандартен разтвор (4.1).
Когато присъстват и калций, и магнезий, EDTA първо формира комплекс с калция и след това с магнезия. В този случай два елемента се определят едновременно.
4.7. Разтвор на калиев цианид
2 % воден разтвор на KCN. (Не се прехвърля с пипета с уста и виж 10.7).
4.8. Разтвор на калиев хидроксид и калиев цианид
Разтваря се 280 грама КОН и 66 грама KCN във вода, долива се до един литър и се смесва.
4.9. Буферен разтвор рН 10,5
В мерителна колба от 500 ml се разтваря 33 грама амониев хлорид в 200 милилитра вода, се добавя 250 ml амоняк (d20 = 0,91 g/ml) и се долива до обема с вода и се смесва. Тества се рН на разтвора редовно.
4.10. Разредена солна киселина: един обем солна киселина (d20 = 1,18 g/ml) плюс един обем вода
4.11. Разтвор на солна киселина приблизително 0,5 mol/l
4.12. Разтвор на солна киселина приблизително 1 mol/l
4.13. 5 mol/l разтвор на натриев хидроксид
5. Апаратура
5.1. Магнитна или механична бъркалка
5.2. рН-метър
6. Контролно изпитване
Извършва се определяне на аликвотни части на разтвори (4.1) и 4.3), така че отношението Са/Mg е приблизително равно на отношението на разтвора за анализ. В тази връзка се вземат (а) милилитра стандартен разтвор на Мg (4.3) и (b – a) милилитра стандартен разтвор (4.1), като (а) и (b) са количеството милилитри от EDTA разтвора, използвани при двете титрувания, извършени на разтвора за анализиране. Тази процедура е коректна, само ако разтворите на EDTA, калция и магнезия са точно еквивалентни. Ако не е така, е необходимо да се извършат корекции.
7. Приготвяне на разтвора за анализ
Виж методи 8.1 и 8.3.
8. Определяне
8.1. Аликвотни проби за вземане
Аликвотната част ще съдържа, доколкото е възможно, между 9 и 18 милиграма магнезий (= 15 до 30 милиграма МgO).
8.2. Титруване в присъствието на ериохром черен-Т
Прехвърля се с пипета се аликвотна част (8.1) на разтвора за анализиране в бехерова чаша от 400 ml. Неутрализира се излишната киселина с 5 mol/l разтвор на натриев хидроксид (4.12) с използване на рН метър. Разрежда се с вода до приблизително 100 милилитра. Добавят се 5 милилитра буферен разтвор (4.9). Измерената рН трябва да бъде 10,5 ± 0,1. Добавя се 2 милилитра разтвор на калиев цианид (4.7) и три капки индикатор ериохром черен-Т (4.6). Титрува се с EDTA разтвор (4.2). Разбърка се бавно с бъркалката (5.1) (виж 10.2, 10.3 и 10.4). „b“ е количеството милилитра 0,05 mol/l EDTA разтвор.
8.3. Титруване в присъствието на калсеин или калкон въглеродна киселина
Прехвърля се с пипета аликвотна част на разтвора за анализиране, равна на взетата при горното титруване, и се поставя в бехерова чаша от 400 ml. Неутрализира се излишната киселина с 5 mol/l разтвор на натриев хидроксид (4.13) с използване на рН метър. Разрежда се с вода до приблизително 100 милилитра. Добавя се 10 милилитра разтвор КОН/ KCN (4.8) и индикатора (4.4 или 4.5). Титрува се с EDTA разтвор (4.2), като се разбърква бавно с бъркалката (5.1) (виж 10.2, 10.3 и 10.4). „а“ е количеството милилитра 0,05 mol/l EDTA разтвор.
9. Изразяване на резултатите
За ЕО торовете, за които е приложим метода, (5 грама тор в 500 милилитра екстракт) процентното съдържание на тора е:
|
|
|
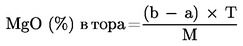
|
|
|

където
|
a |
= |
количество милилитри 0,05 mol/l EDTA разтвор, използвани за титруването в присъствието на калсеин или калкон въглеродна киселина, |
|
b |
= |
количество милилитри 0,05 mol/l EDTA разтвор, използвани за титруването в присъствието на ериохром черен-Т, |
|
M |
= |
маса на пробата, присъстваща в взетата аликвотна част (в грамове), |
|
T |
= |
0,2016 × mol/l на EDTA разтвор/0,05 (виж 4.2). |
|
T′ |
= |
0,1216 × mol/l на EDTA разтвор/0,05 (виж 4.2). |
10. Забележки
10.1. Стохиометричното отношение EDTA/метал при комлексометричния анализ е винаги 1:1, независимо от валенцията на метала и това, че EDTA е четиривалентен. EDTA разтворът за титруване и стандартните разтвори ще бъдат моларни, а не нормални.
10.2. Комплексометричните индикатори са често чувствителни на въздуха. Разтворът може да загуби цвят при титруването. В този случай трябва да се добавят една или две капки индикатор. Това е вярно особено при ериохром черен-Т и калкон въглеродна киселина.
10.3. Комплексите за индикатор на метал са често относително стабилни и може да отнеме известно време цветът да се промени. Затова последните капки EDTA трябва да се добавят бавно и се добавя капка 0,05 mol/l разтвор на магнезий (4.1) или калций (4.3), за да се осигури, че промяната на цвета все още не е станала. Това е особено вярно при комплекса ериохром-магнезий.
10.4. Смяната на цвета на индикатора трябва да се наблюдава не вертикално, а хоризонтално в целия разтвор и бехеровата чаша трябва да бъде разположена срещу бял фон в добре осветено положение. Смяната на цвета на индикатора може също така да се наблюдава лесно като бехеровата чаша се постави върху грапаво стъкло, осветено умерено отдолу (лампа 25 вата).
10.5. Този анализ изисква известен опит. Задачата ще включва, inter alia, наблюдаване на промените на цвета на стандартните разтвори 4.1 и 4.3. Препоръчва се определянията да се извършват от един и същ лаборант.
10.6. Ако се използва EDTA разтвор с гарантирана сила (Titrisol, Normex, например), това може да опрости контрола на еквивалентността на стандартните разтвори 4.1, 4.2 и 4.3.
10.7. Разтворите, съдържащи калиев цианид, не трябва да се изливат в мивката, докато цианида не се превърне в безвредно съединение, например, чрез окисляване със натриев хипохлорид след алкализация.
Метод 8.9
Определяне на сулфати
1. Обхват
Настоящият документ дефинира процедурата за определяне на сяра, присъстваща в екстракти на торове под формата на сулфати.
2. Област на приложение
Този метод се прилага за определяне на сулфати, присъстващи в екстракциите, извършени с методи 8.1, 8.2, 8.3 и 8.4.
3. Принцип
Гравиметрично определяне на бариев сулфат.
4. Реактиви
4.1. Разредена солна киселина
Един обем солна киселина (d20 = 1,18 g/ml) и един обем вода.
4.2. Разтвор на бариев хлорид BaCl22Н2O: 122 грама на литър
4.3. Разтвор на сребърен нитрат: 5 грама на литър
5. Апаратура
5.1. Порцеланови тигели
5.2. Гореща водна баня
5.3. Пещ за сушене, настроена на 105(± 1) °С
5.4. Електрическа пещ, настроена на 800(± 50) °С
6. Процедура
6.1. Вземане на проба от разтвора
Прехвърля се с пипета се аликвотна част на един от разтвори от екстракциите, посочени в 2, който съдържа между 20 и 100 милиграма S или 50 до 250 милиграма SO3.
Поставя се този аликвот в бехерова чаша с подходящ обем. Добавя се 20 милилитра разредена солна киселина (4.1). Долива се до около 300 ml вода.
6.2. Приготвяне на утайката
Довежда се разтвора до кипене. Добавя се, капка по капка, около 20 милилитра разтвор на бариев хлорид (4.2), като разтворът се разбърква силно. Кипва се в продължение на няколко минути.
Поставя се бехеровата чаша, покрита с наблюдателно стъкло, в гореща водна баня (5.2) за един час. След това се оставя гореща (около 60 °С), докато течността, плаваща отгоре, е бистра. Декантира се бистрият разтвор през филтър за бавно филтриране без пепел. Измива се утайката няколко пъти с гореща вода. Продължава се отмиването на утайката върху филтъра, докато филтратът се освободи от хлориди. Това може да се провери с използване на разтвор на сребърен нитрат (4.3).
6.3. Опепеляване и претегляне на утайката
Поставя се филтърната хартия и утайката в порцеланов тигел (5.1), предварително претеглен с точност до 0,1 милиграм. Изсушава се в пещта (5.3) и се опепелява при приблизително 800 °С за половин час (5.4). Оставя се да се охлади в сушилен шкаф и се претегля с точност до 0,1 милиграм.
7. Изразяване на резултатите
Един милиграм бариев сулфат съответства на 0,137 милиграма S или на 0,343 милиграма SO3.
Процентното съдържание на сяра в тора е, както следва:
|
|
|

|
|
|

където
w= масата на утайката на бариевия сулфат в милиграми,
v1= обем на разтвора на екстракта в милилитри,
v2= аликвотен обем в милилитри,
m= маса на пробата за изпитване в грамове.
Метод 8.10
Определяне на извлечен натрий
1. Обхват
Настоящият документ дефинира процедурата за определяне на натрия в екстракти на торове.
2. Област на приложение
Този метод се прилага за ЕО торове, за които в приложение I се предвижда деклариране на натрия.
3. Принцип
След подходящо разреждане на екстракта, получен посредством метод 8.1 и/или 8.3, съдържанието на натрий на разтвора се определя с помощта на спектрометрия на емисията на пламък.
4. Реактиви
4.1. Разредена солна киселина
Един обем солна киселина (d20 = 1,18 g/ml) плюс един обем вода.
4.2. Алуминиев нитрат Al(NO3)3.9H2O
4.3. Цезиев хлорид, CsCl
4.4. Безводен натриев хлорид, NaCl
4.5. Разтвор на цезиев хлорид и алуминиев нитрат
Разтваря се във вода 50 грама цезиев хлорид (4.3) и 250 грама алуминиев нитрат (4.2) в мерителна колба от 1 000 милилитра. Долива се до обема вода и се смесва.
4.6. Стандартен разтвор на натрий един милиграм/милилитър Na
Разтваря се във вода 2,542 грама натриев хлорид (4.4) в мерителна колба от 1 000 милилитра. Добавя се 10 милилитра солна киселина (4.1). Долива се до обема вода и се смесва.
5. Апаратура
Спектрометър, оборудван за емисия на пламък, настроен на 589,3 nm.
6. Калибровъчни разтвори
6.1. Поставя се 10 милилитра стандартен разтвор (4.6) в мерителна колба от 250 милилитра. Долива се до обема и се смесва. Концентрация на разтвора: 40 μg/ml натрий.
6.2. Поставя се 0, 5, 10, 15, 20, 25 милилитра от междинния разтвор (6.1) в мерителни колби от 100 милилитра. Добавя се 10 милилитра от разтвор (4.5). Долива се до обема и се смесва. Концентрация на разтворите: 0, 2, 4,6, 8, 10 μg/ml натрий.
7. Приготвяне на разтвори за измерване
В зависимост от очакваното съдържание на натрий на разтвора на екстракта, получен по метод 8.1 или 8.3 (пет грама тор в 500 милилитра) се извършва разреждане в съответствие със следната таблица:
|
Na2O FOR-L_2003304BG.01007301.notes.0053.xml.jpg |
Na (%) |
Междинно разреждане |
Окончателно разреждане |
Степен на разреждане |
||
|
Проба (ml) (v2) |
Разреждане до ml (v3) |
Проба (ml) (v4) |
Разреждане до ml |
|||
|
3—5 |
2,2—3,7 |
10 |
50 |
10 |
100 |
50 |
|
5—10 |
3,7—7,4 |
10 |
100 |
10 |
100 |
100 |
|
10—20 |
7,4—15 |
10 |
100 |
5 |
100 |
200 |
|
20—38 |
15—28 |
5 |
100 |
5 |
100 |
400 |
Извършва се междинното разреждане с вода. За окончателното разреждане се добавят 10 милилитра от разтвор (4.5) в мерителна колба от 100 милилитра.
За пробата за изпитване един грам обемът на окончателното разреждане (v4) се умножава по пет.
8. Определяне
Приготвя се спектрометърът (5.1) за измерване при 589,3 nm. Калибрира се инструментът чрез измерване на реакцията на калибровъчните разтвори (6.2). След това се регулира чувствителността на инструмента, за да се използва цялата негова скала, когато се използва най-концентрираният калибровъчен разтвор. Измерва се реакцията на разтвора на пробата за анализ (7). Тази операция се повтаря три пъти.
9. Изчисляване на резултатите
Начертава се калибровъчната крива, като средната реакция за всеки калибровъчен разтвор се поставя по ординатата, а съответстващите концентрации, изразени в μg на милилитър — по абсцисата. От нея се определя концентрацията на натрий на разтвора за изпитване. Резултатите се изразяват като процент от пробата.
Процентното съдържание на натрий в тора е, както следва:
|
|
|
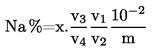
|
|
|
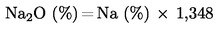
където
|
x |
= |
концентрация на разтвора, поставен в спектрометъра, м μg/ml, |
|
v1 |
= |
обем на разтвора на екстракта в милилитри, |
|
v2 |
= |
обем на аликвота при междинното разреждане в милилитри, |
|
v3 |
= |
обем на междинното разреждане в милилитри, |
|
v4 |
= |
обем аликвота в ml на окончателното разреждане (до 100 милилитри), |
|
m |
= |
маса на пробата за изпитване в грамове. |
Методи 9
Хранителни микроелементи при концентрация по-малка или равна на 10 %
Метод 9.1
Екстракция на общо хранителни микроелементи
1. Обхват
Този метод дефинира процедурата за извличане на следните хранителни микроелементи: общ бор, общ кобалт, обща мед, общо желязо, общ манган, общ молибден и общ цинк. Целта е да се извършат минимален брой извличания, като се използва, когато е възможно, един и същи екстракт за определяне на общото ниво на всяко от хранителните микроелементи, изброени по-горе.
2. Област на приложение
Тази процедура се отнася за ЕС торове, покрити от приложение I.Д, съдържащи един или повече от следните хранителни микроелементи: бор, кобалт, мед, о желязо, манган, молибден и цинк. Тя е приложима за всеки хранителен микроелемент с декларираното съдържание по-малко или равно на 10 %.
3. Принцип
Разтваряне в кипяща разредена солна киселина.
Бележка:
Извличането е емпирично и може да не е количествено в зависимост от продукта или други съставки на тора. По специално, при някои манганови оксиди, извлеченото количество може да бъде съществено по-малко от общото количество манган, което продукта съдържа. Производителят на тора е отговорен да осигури декларираното количество действително да съответства на количеството, извлечено при присъщите условия на метода.
4. Реактиви
4.1. Разтвор на разредена солна киселина (HCl), около 6 mol/l
Смесва се 1 обем солна киселина (d20 = 1,18 g/ml) с един обем вода.
4.2. Концентриран амониев разтвор (NH4OH, d20 = 0,9 g/mol)
5. Апаратура
Електрически котлон с променлив температурен контрол.
Бележка:
Когато се определя съдържанието на бор на екстракта, не се използва борсиликатна стъклария. Тъй като методът включва кипене, тефлонът и кварцът са за предпочитане. Стъкларията се изплаква старателно, ако е била измита с миещи препарати, съдържащи борати.
6. Приготвяне на пробата
Виж метод 1.
7. Процедура
7.1. Проба за изпитване
Взема се количество от тора, тежащо между 2 и 10 g, в зависимост от декларираното съдържание на елемента в продукта. Следващата таблица трябва да се използва за получаване на окончателния разтвор, който след подходящо разреждане ще бъде в рамките на измервателния диапазон за всеки метод. Пробите трябва да бъдат претеглени с точност до 1 mg.
|
Декларирано съдържание на хранителен микроелемент в тора (%) |
< 0,01 |
0,01 < 5 |
≥ 5—10 |
|
Маса на пробата за изпитване (g) |
10 |
5 |
2 |
|
Маса на елемента в пробата (mg) |
1 |
0,5—250 |
100—200 |
|
Обем на екстракта V (ml) |
250 |
500 |
500 |
|
Концентрация на елемента в екстракта (mg/l) |
4 |
1—500 |
20—400 |
Поставя се пробата в бехерова чаша от 250 ml.
7.2. Приготвяне на разтвора
Ако е необходимо, пробата се овлажнява с малко вода, добавят се 10 ml от разредената солна киселина (4.1) на грам тор внимателно, на малки количества. След това се добавят около 50 ml вода. Покрива се бехеровата чаша с наблюдателно стъкло и се смесва. Довежда се до кипене на котлона и се вари 30 минути. Оставя се да се охлади, като се разбърква от време на време. Прехвърля се количествено в мерителна колба от 250 или 500 ml (виж таблицата). Долива се до обема с вода и се смесва старателно. Филтрира се през сух филтър в сух съд. Изхвърля се първата порция. Екстрактът трябва да бъде идеално бистър.
Препоръчва се определянето да се извърши незабавно на аликвотна част на бистрия филтрат. Ако не, съдът трябва да се запуши.
Забележка:
Екстракти, в които трябва да се определи съдържанието на бор: регулира се рН между 4 и 6 с концентриран амоняк (4.2).
8. Определяне
Определянето на всеки хранителен микроелемент се извършва на аликвотните части, посочени в метода за всеки индивидуален хранителен елемент.
Ако е необходимо, се отстраняват органичните хелатообразуващи или комплексообразуващи субстанции от аликвотната част на екстракта, като се използва метод 9.3. При определяне с атомна абсорбционна спектрометрия такова отстраняване може да не е необходимо.
Метод 9.2
Екстракция на хранителни микроелементи, разтворими във вода
1. Обхват
Този метод дефинира процедурата за извличане на форми, разтворими във вода, на следните хранителни микроелементи: бор, кобалт, мед, желязо, манган, молибден и цинк. Целта е да се извършат минимален брой извличания, като се използва, когато е възможно, един и същи екстракт за определяне на общото ниво на всяко от хранителните микроелементи, изброени по-горе.
2. Област на приложение
Тази процедура се отнася за ЕС торове, покрити от приложение I, съдържащи един или повече от следните хранителни микроелементи: бор, кобалт, мед, о желязо, манган, молибден и цинк. Тя е приложима за всеки хранителен микроелемент, на който декларираното съдържание е по-малко или равно на 10 %.
3. Принцип
Хранителните микроелементи се извличат чрез разклащане на тор във вода при 20(± 2) °С.
Бележка
Извличането е емпирично и може да е или не количествено.
4. Реактиви
4.1. Разтвор на разредена солна киселина, около 6 mol/l
Смесва се 1 обем солна киселина (d20 = 1,18 g/ml) с един обем вода.
5. Апаратура
5.1. Ротационна клатачна машина, настроен на около 35 до 40 rpm.
5.2. рН метър.
Бележка:
Когато се определя съдържанието на бор на екстракта, не се използва борсиликатна стъклария. Тъй като методът включва кипене, тефлонът и кварцът са за предпочитане. Измива се стъкларията старателно, ако е била измита с измиващи препарати, съдържащи борати.
6. Приготвяне на пробата
Виж метод 1.
7. Процедура
7.1. Проба за изпитване
Взема се количество от тора, тежащо между 2 и 10 g, в зависимост от декларираното съдържание на елемента в продукта. Следващата таблица трябва да се използва за получаване на окончателния разтвор, който след подходящо разреждане ще бъде в рамките на измервателния диапазон за всеки метод. Пробите трябва да бъдат претеглени с точност до 1 mg.
|
Декларирано съдържание на хранителен микроелемент в тора (%) |
< 0,01 |
0,01 < 5 |
≥ 5—10 |
|
Маса на пробата за изпитване (g) |
10 |
5 |
2 |
|
Маса на елемента в пробата (mg) |
1 |
0,5—250 |
100—200 |
|
Обем на екстракта V (ml) |
250 |
500 |
500 |
|
Концентрация на елемента в екстракта (mg/l) |
4 |
1—500 |
200—400 |
Поставя се пробата в бехерова чаша от 250 или 500 ml (съгласно таблицата).
7.2. Приготвяне на разтвора
Добавя се около 200 ml вода в колба от 250 ml или 400 ml вода в колба от 500 ml.
Запушва се добре колбата. Разклаща се силно с ръка, за да се разпръсне пробата, след това колбата се поставя на клатачната машина и се разклаща 30 минути.
Долива се до обема вода и се смесва старателно.
7.3. Приготвяне на разтвора за изпитване
Филтрира се незабавно в чиста суха колба. Запушва се колбата. Извършва се определянето незабавно след филтрирането.
Бележка:
Ако филтратът постепенно става замъглен, прави се друго извличане в колба с обем Vе, като се следват 7.1 и 7.2. Филтрира се в калибрирана колба с обем W, която е била предварително подсушена и са били сложени 5,00 ml разредена солна киселина (4.1). Филтрирането се спира в момента, когато се достигне калибровъчната отметка. Смесва се старателно.
При тези условия стойността V в изразяването на резултата е:
|
|
|

Разреждането в изразяването на резултатите зависи от тази стойност V.
8. Определяне
Определянето на всеки хранителен микроелемент се извършва на аликвотните части, посочени в метода за всеки индивидуален хранителен елемент.
Ако е необходимо, се отстраняват органичните хелатообразуващи или коплексообразуващи субстанции от аликвотната част на екстракта, като се използва метод 9.3. При определяне с атомна абсорбционна спектрометрия такова отстраняване може да не е необходимо.
Метод 9.3
Отстраняване на органични съединения от екстракти на торове
1. Обхват
Този метод дефинира процедурата за отстраняване на органични съединения от екстракти на торове.
2. Област на приложение
Тази процедура е приложима за анализиране на проби на торове, извлечени по методи 9.1 и 9.2, за които с приложение I.Д към настоящия регламент се изисква деклариране на общ и/или водоразтворим елемент.
Бележка:
Присъствието на малки количества органични вещества обикновено не влияе на определянето с помощта на атомна абсорбционна спектрометрия.
3. Принцип
Органичните съединения в аликвотна част на екстракта се окисляват с водороден прекис.
4. Реактиви
4.1. Разтвор на разредена солна киселина (HCl), около 0,5 mol/l
Смесва се 1 обем солна киселина (d20 = 1,18 g/ml) с 20 обема вода.
4.2. Разтвор на водороден прекис (30 % Н2О2, d20 = 1,11 g/ml), без хранителни микроелементи
5. Апаратура
Електрически котлон с променлив температурен контрол
6. Процедура
Вземат се 25 ml от разтвора на екстракта, получен по метод 9.1 или метод 9.2, и се поставя в бехерова чаша от 100 ml. При метод 9.2 се добавят 5 ml разтвор на разредена солна киселина (4.1). След това се добавя 5 ml разтвор на водороден прекис (4.2). Покрива се с наблюдателно стъкло. Оставя се да се окисли при стайна температура за около един час, след това бавно се довежда до кипене и се вари в продължение на половин час. Ако е необходимо, се добавят допълнителни 5 ml водороден прекис към разтвора след като се охлади. След това се кипва, за да се отстрани излишният водороден прекис. Оставя се да се охлади и се прехвърля количествено в мерителна колба от 50 ml и се допълва до обема. При необходимост се филтрира.
Трябва да се взема предвид това разреждане, когато се вземат аликвотни части и се изчислява процента на хранителния микроелемент в продукта.
Метод 9.4
Определяне на хранителни микроелементи в екстракти на торове чрез атомна абсорбционна спектрометрия
(обща процедура)
1. Обхват
Настоящият метод дефинира общата процедура за определяне на нивата на някои хранителни микроелементи чрез атомна абсорбционна спектрометрия.
2. Област на приложение
Настоящият метод се прилага за анализиране на проби на торове, извлечени по методи 9.1 и 9.2, за които с приложение I.Д към настоящия регламент се изисква деклариране на общ и/или водоразтворим елемент.
Адаптирането на тази процедура за различните хранителни микроелементи е описано подробно в методите, дефинирани специфично за всеки елемент.
Бележка:
В повечето случаи наличието на малки количества органично вещество няма да влияе на определянията чрез атомна абсорбционна спектрометрия.
3. Принцип
След като екстрактът е бил третиран, ако е необходимо, за намаляване или елиминиране на влияещи химически видове, то той се разрежда, така че неговата концентрация да е в оптималния диапазон на спектрометъра на дължина на вълната, подходяща за определяния хранителен микроелемент.
4. Реактиви
4.1. Разреден разтвор на солна киселина (HCl), около 6 mol/l:
Смесва се един обем солна киселина (d20 = 1,18 g/ml) с един обем вода.
4.2. Разреден разтвор на солна киселина (HCl), около 0,5 mol/l:
Смесва се един обем солна киселина (d20 = 1,18 g/ml) с 20 обема вода.
4.3. Разтвори на лантанови соли (10 g лантан на литър)
Този реактив се използва за определяне на кобалт, желязо, манган и цинк. Той може да бъде приготвен:
|
а) |
с лантанов оксид, разтворен в солна киселина (4.1). Поставя се 11,73 g лантанов оксид (La2O3) в 150 ml вода в мерителна колба от един литър и се добавят 120 ml 6 mol/l солна киселина (4.1). Оставя се да се разтвори и след това се долива до 1 литър с вода и се смесва старателно. Този разтвор е приблизително 0,5 mol/l в солна киселина. |
|
б) |
или с разтвори на лантанов хлорид, сулфат или нитрат. Разтваря се 26,7 g лантанов хлорид хептахидрат (LaCl3 7H2O) или 31,2 g лантанов нитратен хексахидрат [La(NO3)3 6H2O], или 26,2 g лантанов сулфатен нонахидрат [La2 (SO4)3 9H2O] в 150 ml вода и след това се добавя 85 ml 6 mol/l солна киселина (4.1). Оставя се да се разтвори и след това се долива до един литър с вода. Смесва се старателно. Този разтвор е приблизително 0,5 mol/l в солна киселина. |
4.4. Калибровъчни разтвори
За тяхното приготвяне виж индивидуалните методи за определяне на всеки хранителен микроелемент.
5. Апаратура
Атомен абсорбционен спектрометър, снабден с източници, излъчващи радиационна характеристика на определяния хранителен микроелемент.
Лаборантът трябва да следва указанията на производителя и да бъде запознат с материала. Апаратът трябва да позволява фонова корекция, така че да може да бъде използван, когато е необходимо (Со и Zn). Използваните газове са въздух и ацетилен.
6. Приготвяне на разтвора за анализ
6.1. Приготвяне на разтвор на екстракти на определяните микроелементи
Виж методи 9.1 и/или 9.2 и, ако е подходящо, 9.3.
6.2. Третиране на разтвора за изпитване
Разрежда се аликвотна част на екстракта, получен по метод 9.1, 9.2 или 9.3, с вода и/или солна киселина (4.1) или (4.2), така че да се получи окончателен разтвор за измерване с концентрация на определяния елемент, която е подходяща за използвания диапазон на калибриране (7.2) и концентрация на солната киселина най-малко 0,5 mol/l и не повече от 2,5 mol/l. Тази операция може да изисква едно или повече последователни разреждания.
Взема се аликвотна част от окончателния разтвор, получен чрез разреждането на екстракта, като (а) е неговия обем в ml, и се излива в мерителна колба от 100 ml. При определяне съдържанието на кобалт, желязо, манган или цинк се добавя 10 ml разтвор на лантанова сол (4.3). Долива се до обема с 0,5 mol/l разтвор на солна киселина (4.2) и се смесва старателно. Това е окончателния разтвор за измерване. D е коефициентът на разреждане.
7. Процедура
7.1. Приготвяне на разтвор на празна проба
Приготвя се разтвор на празна проба чрез повтаряне на цялата процедура от етапа на извличане, като се пропуска само пробата за изпитване на тора.
7.2. Приготвяне на калибровъчни разтвори
От работния калибровъчен разтвор, приготвен с използване на метода, даден за всеки индивидуален хранителен микроелемент, в измервателни колби от 100 ml се приготвя серия от най-малко пет калибровъчни разтвора с увеличаваща се концентрация в рамките на оптималния измерителен диапазон на спектрометъра. Ако е необходимо, концентрацията на солната киселина се регулира, за да е колкото е възможно по-близо до тази на разредения разтвор за изпитване (6.2). За определяне на кобалт, желязо, манган или цинк се добавя 10 ml от същия разтвор на лантанова сол, като използвания в 6.2. Долива се до обема с 0,5 mol/l разтвор на солна киселина (4.2) и се смесва старателно.
7.3. Определяне
Приготвя се спектрометърът (5) за определянето и се регулира на дължината на вълната, дадена в метода за съответния индивидуален хранителен микроелемент.
Впръскват се три пъти последователно калибровъчните разтвори (7.2), разтворът за изпитване (6.2) и разтворът на празната проба (7.1), като се отбелязва всеки резултат и се измива инструментът с дестилирана вода между индивидуалните впръсквания.
Построява се калибровъчната крива чрез построяване на средните показания на спектрометъра за всеки калибровъчен разтвор (7.2) по ординатата и съответната концентрация на елемента, изразена в μg/ml по абсцисата.
От тази крива се определят концентрациите на съответния хранителен микроелемент в разтвора за изпитване хs (6.2) и разтвора на празната проба xb (7.1), като тези концентрации се изразяват в μg на ml.
8. Изразяване на резултатите
Процентът на хранителния микроелемент (Е) в тора е равно на:
|
|
|
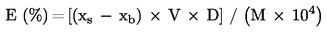
или ако се използва метод 9.3:
|
|
|

където
|
|
Е е количеството на определяния хранителен микроелемент, изразено като процент от тора; |
|
|
хs е концентрацията на разтвора за изпитване (6.2) в μg/ml; |
|
|
xb е концентрацията на разтвора на празната проба (7.1) в μg/ml; |
|
|
V е обемът на екстракта, получен метод 9.1. или 9.2, в ml; |
|
|
D е коефициент, съответстващ на разреждането, извършено в 6.2; |
|
|
М е масата на пробата за изпитване, взета в съответствие метод 9.1 или 9.2, в грамове. |
Изчисляване на коефициента на разреждане D:
|
|
Ако (а1), (а2), (а3),…, (аi) и (а) са аликвоните части и (v1), (v2), (v3),….., (vi) и (100) са обемите в ml, съответстващи на съответните разреждания, коефициентът на разреждане D ще бъде равен на:
|
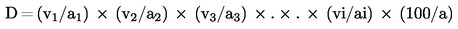
Метод 9.5
Определяне на бор в екстракти на торове с помощта на спектрометрия с азометан-Н
1. Обхват
Този метод описва процедурата за определяне на бор в екстракти на торове.
2. Област на приложение
Настоящият метод се прилага за анализиране на проби на торове, извлечени по методи 9.1 и 9.2, за които с приложение I към настоящия регламент се изисква деклариране на общ и/или разтворим във вода бор.
3. Принцип
В разтвор на иземетин-Н борените йони формират жълт комплекс, чиято концентрация се определя с молекулярна абсорбционна спектрометрия при 410 nm. Интерфериращите йони са маскирани с EDTA.
4. Реактиви
4.1. EDTA буферен разтвор
Поставя се в мерителна колба от 500 ml, съдържаща 300 ml вода,:
|
— |
75 g амониев ацетат (NH4OOCCH3); |
|
— |
10 g двунатриева сол на етилен диамин тетралимонена киселина (Na2EDTA); |
|
— |
40 ml оцетна киселина (CH3COO, d20 = 1,05 g/ml). |
Долива се до обема вода и се смесва старателно. рН на разтвора, проверен с помощта на стъклен електрод, трябва да бъде 4,8 ± 0,1.
4.2. Разтвор на азометин-Н
Поставя се в мерителна колба от 200 ml
|
— |
10 ml буферен разтвор (4.1); |
|
— |
400 mg азометин-Н (C17H12NNAO8S2); |
|
— |
2 g абсорбираща киселина (C6H8O6); |
|
— |
Долива се до обема и се смесва старателно. Не се приготвят големи количества от този реактив, тъй като е стабилен само няколко дена. |
4.3. Борени калибровъчни разтвори
4.3.1. Борен основен разтвор (100 μg/ml)
Разтваря се 0,5719 g борна киселина (H2BO3) във вода в мерителна колба от 1 000 ml. Долива се до обема вода и се смесва старателно. Прехвърля се в пластмасова бутилка за съхраняване в хладилник.
4.3.2. Борен работен разтвор (10 μg/ml)
Поставя се 50 ml от основния разтвор (4.3.1) в измерителна колба от 500 ml. Долива се до обема вода и се смесва старателно.
5. Апаратура
Спектрометър, подходящ за молекулярна абсорбция с клетки 10 mm оптичен път и настроен на дължина на вълната 410 nm.
6. Приготвяне на разтвора за анализ
6.1. Приготвяне на боровия разтвор
Виж методи 9.1 и/или, ако е подходящо, 9.3.
6.2. Приготвяне на пробата за изпитване
Разрежда се аликвотна част на екстракта (6.1), за да се получи концентрация на бора, както е специфицирана в 7.2. Може да са необходими две последователни разреждания. D е коефициентът на разреждане.
6.3. Приготвяне на коригиращ разтвор
Ако разтворът за изпитване (6.2) е оцветен, се приготвя съответстващ коригиращ разтвор чрез поставяне в пластмасова колба от 5 ml от разтвора за изпитване (6.2), 5 ml EDTA буферен разтвор (4.1) и 5 ml вода и се смесва старателно.
7. Процедура
7.1. Приготвяне на разтвор на празна проба
Приготвя се разтвор на празна проба, като се повтаря цялата процедура от етапа на извличане; пропуска само пробата за изпитване на тора.
7.2. Приготвяне на калибровъчни разтвори
Прехвърлят се 0, 5, 10, 15, 20 и 25 ml от работния калибровъчен разтвор (4.3.3) в серия мерителни колби 100 ml. Долива се до 100 ml вода и се смесва старателно. Тези разтвори съдържат между 0 и 2,5 μg/ml бор.
7.3. Проявяване на цвят
Прехвърлят се 5 ml от калиброващите разтвори (7.2), разтворите за изпитване (6.2) и разтвора на празната проба в серия пластмасови колби. Добавя се 5 ml EDTA буферен разтвор (4.1). Добавя се 5 ml от разтвор на азометин-Н (4.2).
Смесва се старателно и се оставя да се прояви цвят на тъмно 2 1/2 до 3 часа.
7.4. Определяне
Измерва се абсорбцията на разтворите, получени при 7.3, и, ако е подходящо, на коригиращия разтвор (6.3) спрямо вода при дължина на вълната 410 nm. Преди всяко отчитане клетките се измиват с вода.
8. Изразяване на резултатите
Начертава се калиброваща крива на концентрацията калиброващите разтвори (7.2) по абсцисата и абсорбцията, показана от спектрометъра (7.4) по ординатата.
От калибровъчната крива се отчита концентрацията на бора в празната проба (7.1), концентрацията та бора в разтвора за изпитване (6.2) и, ако разтворът за изпитване е оцветен, коригираната концентрация на разтвора за изпитване. За да се изчисли последната, се изважда абсорбцията на коригиращия разтвор (6.3) от абсорбцията на разтвора за изпитване (6.2) и се определя коригираната концентрация на разтвора за изпитване. Отбелязва се концентрацията на разтвора за изпитване (6.2), с и без корекция, X (xs) и на празната проба (xb).
Процентът на бора в тора се дава с:
|
|
|

или ако се използва метод 9.3:
|
|
|

където
|
|
В е количеството на бор, изразено като процент от тора; |
|
|
хs е концентрацията на разтвора за изпитване (6.2); |
|
|
xb е концентрацията на разтвора на празната проба (7.1); |
|
|
V е обемът на екстракта, получен метод 9.1. или 9.2; |
|
|
D е коефициент, съответстващ на разреждането, извършено в 6.2; |
|
|
М е масата на пробата за изпитване, взета в съответствие метод 9.1 или 9.2. |
Изчисляване на коефициента на разреждане D: Ако (а1) и (а2) са аликвотните части и (v1) и (v2) са обемите в ml, съответстващи на съответните разреждания, коефициентът на разреждане D ще бъде равен на:
|
|
|

Метод 9.6
Определяне на кобалт в екстракти на тор чрез атомна абсорбционна спектрометрия
1. Обхват
Настоящият метод описва процедура за определяне на кобалт в екстракти на торове.
2. Област на приложение
Процедурата е приложима за анализиране на проби на торове, извлечени по методи 9.1 и 9.2, за които с приложение I.Д към настоящия регламент се изисква деклариране на общ и/или водоразтворим кобалт.
3. Принцип
След подходящо третиране и разреждане на екстракта съдържанието на кобалт се определя чрез атомна абсорбционна спектрометрия.
4. Реактиви
4.1. Разреден разтвор на солна киселина, около 6 mol/l:
Виж метод 9.4 (4.1).
4.2 Разреден разтвор на солна киселина, около 0,5 mol/l:
Виж метод 9.4 (4.2).
4.3. Разтвори на лантанови соли (10 g лантан на литър)
Виж метод 9.4 (4.3).
4.4. Калибровъчни разтвори на кобалт
4.4.1. Основен разтвор на кобалт (1 000 μg/ml)
В бехерова чаша от 250 ml се претегля 1 g кобалт с точност до 0,1 mg, добавя се 25 ml 6 mol/l солна киселина (4.1) и се нагрява на котлон до пълното разтваряне на кобалта. Когато се охлади, количествено се прехвърля в мерителна колба от 1 000 ml. Долива се до обема с вода и се смесва старателно.
4.4.2. Работен разтвор на кобалт (100 μg/ml)
Поставя се 10 ml основен разтвор (4.4.1) в мерителна колба от 100 ml. Долива се до обема с 0,5 mol/l разтвор на солна киселина (4.2) и се смесва старателно.
5. Апаратура
Атомен абсорбционен спектрометър: виж метод 9.4(5). Инструментът трябва да бъде снабден с източник на лъчи, характерни за кобалта (240,7 nm). Спектрометърът трябва да позволява извършването на фонова корекция.
6. Приготвяне на разтвора за анализ
6.1. Разтвор на екстракт на кобалт
Виж методи 9.1 и/или 9.2 и, ако е подходящо, 9.3.
6.2. Приготвяне на разтвора за изпитване
Виж метод 9.4 (6.2). Разтворът за изпитване трябва да съдържа 10 % (v/v) разтвор на лантанова сол (4.3).
7. Процедурdddа
7.1 Приготвяне на разтвор на празна проба
Виж метод 9.4 (7.1). Разтворът за празната проба трябва да съдържа 10 % (v/v) разтвор на лантанова сол, използван в 6.2.
7.2. Приготвяне на калибровъчни разтвори
Виж метод 9.4. (7.2.).
За оптимален диапазон на определяне от 0 до 5 μg/ml кобалт се поставя 0, 0,5, 1, 2, 3, 4 и 5 ml съответно от работния разтвор (4.4.2) в серия мерителни колби 100 ml. Ако е необходимо, се регулира концентрацията на солна киселина да бъде колкото е възможно по-близо до тази на разтвора за изпитване. Добавя се във всяка колба 10 ml разтвор на лантанова сол, използвана в 6.2. Долива се до 100 ml с 0,5 mol/l разтвор на солна киселина (4.2) и се смесва старателно. Тези разтвори съдържат съответно 0, 0,5, 11, 2, 3, 4 и 5 μg/ml кобалт.
7.3. Определяне
Виж метод 9.4 (7.3). Приготвя се спектрометърът (5) за измерване на дължината на вълната 240,7 nm.
8. Изразяване на резултатите
Виж метод 9.4 (8).
Процентът на кобалта се да с:
|
|
|

или ако се използва метод 9.3:
|
|
|

където
|
|
Со е количеството на кобалта, изразено като процент от тора; |
|
|
хs е концентрацията на разтвора за изпитване (6.2); |
|
|
xb е концентрацията на разтвора на празната проба (7.1); |
|
|
V е обемът на екстракта, получен метод 9.1. или 9.2; |
|
|
D е коефициент, съответстващ на разреждането, извършено в 6.2; |
|
|
М е масата на пробата за изпитване, взета в съответствие метод 9.1 или 9.2. |
Изчисляване на коефициента на разреждане D: Ако (а1), (а2), (а3,…, (аi) и (а) са аликвотните части и (v1), (v2), (v3),…, (v1), (v2), (v3),…, (vi) и (100) са обемите в ml, съответстващи на съответните разреждания, коефициентът на разреждане D ще бъде равен на:
|
|
|
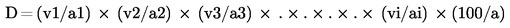
Метод 9.7
Определяне на мед в екстракти на тор чрез атомна абсорбционна спектрометрия
1. Обхват
Настоящият метод описва процедура за определяне на мед в екстракти на торове.
2. Област на приложение
Процедурата е приложима за анализиране на проби на торове, извлечени по методи 9.1 и 9.2, за които с приложение I.Д към настоящия регламент се изисква деклариране на общ и/или водоразтворима мед.
3. Принцип
След подходящо третиране и разреждане на екстракта съдържанието на мед се определя чрез атомна абсорбционна спектрометрия.
4. Реактиви
4.1. Разреден разтвор на солна киселина, около 6 mol/l:
Виж метод 9.4 (4.1).
4.2. Разреден разтвор на солна киселина, около 0,5 mol/l:
Виж метод 9.4 (4.2).
4.3. Разтвор на водороден прекис (30 % H2O2, d20= 1,11 g/ml) без хранителни микроелементи
4.4. Калибровъчни разтвори на мед
4.4.1. Основен разтвор на мед (1 000 μg/ml)
В бехерова чаша 250 ml се претегля 1 g мед с точност до 0,1 mg, добавя се 25 ml 6 mol/l солна киселина (4.1) и 5 ml разтвор на водороден прекис (4.3). Нагрява на котлон до пълното разтваряне на меда. Когато се охлади, се прехвърля количествено в мерителна колба от 1 000 ml. Долива се до обема с вода и се смесва старателно.
4.4.2 Работен разтвор на мед (100 μg/ml)
Поставя се 10 ml основен разтвор (4.4.1) в мерителна колба от 200 ml. Долива се до обема с 0,5 mol/l разтвор на солна киселина (4.2) и се смесва старателно.
5. Апаратура
Атомен абсорбционен спектрометър: виж метод 9.4 (5). Инструментът трябва да бъде снабден с източник на лъчи, характеристични за меда (324,8 nm).
6. Приготвяне на разтвора за анализ
6.1. Разтвор на екстракт на мед
Виж методи 9.1 и/или 9.2 и, ако е подходящо, 9.3.
6.2. Приготвяне на разтвора за изпитване
Виж метод 9.4 (6.2).
7. Процедура
7.1. Приготвяне на разтвор на празна проба
Виж метод 9.4 (7.1).
7.2. Приготвяне на калибровъчни разтвори
Виж метод 9.4 (7.2).
За оптимален диапазон на определяне от 0 до 5 μg/ml мед се поставя 0, 0,5, 1, 2, 3, 4 и 5 ml съответно от работния разтвор (4.4.2) в серия мерителни колби 100 ml. Ако е необходимо, се регулира концентрацията на солна киселина, така че да е колкото е възможно по-близо до тази на разтвор за изпитване (6.2). Долива се до 100 ml с 0,5 mol/l разтвор на солна киселина (4.2) и се смесва старателно. Тези разтвори съдържат съответно 0, 0,5, 1, 2, 3, 4 и 5 μg/ml мед.
7.3 Определяне
Виж метод 9.4 (7.3). Приготвя се спектрометърът (5) за измерване на дължината на вълната 324.8 nm.
8 Изразяване на резултатите
Виж метод 9.4 (8).
Процентът на медта се дава с:
|
|
|

или ако се използва метод 9.3:
|
|
|
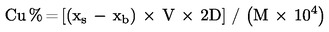
където
|
|
Сu е количеството на медта, изразено като процент от тора; |
|
|
хs е концентрацията в μg/ml на разтвора за изпитване (6.2); |
|
|
xb е концентрацията в μg/ml на разтвора на празната проба (7.1); |
|
|
V е обемът в ml на екстракта, получен по метод 9.1. или 9.2; |
|
|
D е коефициент, съответстващ на разреждането, извършено в 6.2; |
|
|
М е масата на пробата за изпитване, взета в съответствие метод 9.1 или 9.2, в грамове. |
Изчисляване на коефициента на разреждане D: Ако (а1), (а2), (а3),…, (аi) и (а) са аликвотните части и (v1), (v2), (v3),….., (vi) и (100) са обемите в ml, съответстващи на съответните разреждания, коефициентът на разреждане D ще бъде равен на:
|
|
|
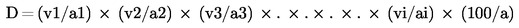
Метод 9.8
Определяне на желязо в екстракти на тор чрез атомна абсорбционна спектрометрия
1. Обхват
Настоящият метод описва процедура за определяне на желязо в екстракти на торове.
2. Област на приложение
Процедурата е приложима за анализиране на проби на торове, извлечени по методи 9.1 и 9.2, за които с приложение I.Д към настоящия регламент се изисква деклариране на общ и/или водоразтворимо желязо.
3. Принцип
След подходящо третиране и разреждане на екстракта съдържанието на желязо се определя чрез атомна абсорбционна спектрометрия.
4. Реактиви
4.1. Разреден разтвор на солна киселина, около 6 mol/l:
Виж метод 9.4 (4.1).
4.2. Разреден разтвор на солна киселина, около 0,5 mol/l:
Виж метод 9.4 (4.2).
4.3. Разтвор на водороден прекис (30 % H2O2, d20= 1,11 g/ml), без хранителни микроелементи
4.4. Разтвори на лантанови соли (10 g лантан на литър)
Виж метод 9.4 (4.3).
4.5. Калибровъчни разтвори на желязо
4.5.1. Основен разтвор на желязо (1 000 μg/ml)
В бехерова чаша от 500 ml се претегля 1 g желязо с точност до 0,1 mg , добавя се 200 ml 6 mol/l солна киселина (4.1) и 15 ml разтвор на водороден прекис (4.3) и се нагрява на котлон до пълното разтваряне на желязото. Когато се охлади, се прехвърля количествено в мерителна колба от 1 000 ml. Долива се до обема с вода и се смесва старателно.
4.5.2. Работен разтвор на желязо (100 μg/ml)
Поставя се 20 ml основен разтвор (4.5.1) в мерителна колба от 200 ml. Долива се до обема с 0,5 mol/l разтвор на солна киселина (4.2) и се смесва старателно.
5. Апаратура
Атомен абсорбционен спектрометър: виж метод 9.4 (5). Инструментът трябва да бъде снабден с източници на лъчи, характеристични за желязото (248,3 nm).
6. Приготвяне на разтвора за анализ
6.1. Разтвор на екстракт на желязо
Виж методи 9.1 и/или 9.2 и, ако е подходящо, 9.3.
6.2. Приготвяне на разтвора за изпитване
Виж метод 9.4 (6.2). Разтворът за изпитване трябва да съдържа 10 % (v/v) разтвор на лантанова сол.
7. Процедура
7.1. Приготвяне на разтвор на празна проба
Виж метод 9.4 (7.1). Разтворът за празната проба трябва да съдържа 10 % (v/v) разтвор на лантанова сол, използван в 6.2.
7.2. Приготвяне на калибровъчни разтвори
Виж метод 9.4 (7.2).
За оптимален диапазон на определяне от 0 до 10 μg/ml желязо се поставя 0, 2, 4, 6, 8 и 10 ml съответно от работния разтвор (4.5.2) в серия мерителни колби от 100 ml. Ако е необходимо, се регулира концентрацията на солна киселина, така че да е колкото е възможно по-близо до тази на разтвора за изпитване. Добавя се във всяка колба 10 ml разтвор на лантанова сол, използвана в 6.2. Долива се с 0,5 mol/l разтвор на солна киселина (4.2) и се смесва старателно. Тези разтвори съдържат съответно 0, 2, 4, 6, 8 и 10 μg/ml желязо.
7.3. Определяне
Виж метод 9.4 (7.3). Приготвя се спектрометърът (5) за измерване на дължината на вълната 248,3 nm.
8. Изразяване на резултатите
Виж метод 9.4 (8).
Процентът на желязото се дава с:
|
|
|
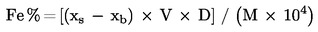
или ако се използва метод 9.3:
|
|
|
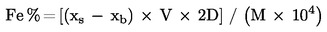
където
|
|
Fe е количеството на желязото, изразено като процент от тора; |
|
|
хs е концентрацията на разтвора за изпитване (6.2) в μg/ml; |
|
|
xb е концентрацията на разтвора на празната проба (7.1) в μg/ml; |
|
|
V е обемът на екстракта, получен метод 9.1. или 9.2, в ml; |
|
|
D е коефициент, съответстващ на разреждането, извършено в 6.2; |
|
|
М е масата на пробата за изпитване, взета в съответствие метод 9.1 или 9.2. |
Изчисляване на коефициента на разреждане D: Ако (а1), (а2), (а3),…, (аi) и (а) са аликвотните части и (v1), (v2), (v3),…, (vi) и (100) са обемите в ml, съответстващи на съответните разреждания, коефициентът на разреждане D ще бъде равен на:
|
|
|

Метод 9.9
Определяне на манган в екстракти на тор чрез атомна абсорбционна спектрометрия
1. Обхват
Настоящият метод описва процедура за определяне на манган в екстракти на торове.
2. Област на приложение
Процедурата е приложима за анализиране на проби на торове, извлечени по методи 9.1 и 9.2, за които с приложение I.Д към настоящия регламент се изисква деклариране на общ и/или водоразтворим манган.
3. Принцип
След подходящо третиране и разреждане на екстракта съдържанието на манган се определя чрез атомна абсорбционна спектрометрия.
4. Реактиви
4.1. Разреден разтвор на солна киселина, около 6 mol/l:
Виж метод 9.4 (4.1).
4.2. Разреден разтвор на солна киселина, около 0,5 mol/l:
Виж метод 9.4 (4.2).
4.3. Разтвори на лантанови соли (10 g лантан на литър)
Виж метод 9.4 (4.3).
4.4. Калибровъчни разтвори на манган
4.4.1. Основен разтвор на манган (1 000 μg/ml)
В бехерова чаша от 250 ml се претегля 1 g манган с точност до 0,1 mg , добавя се 25 ml 6 mol/l солна киселина (4.1) и се нагрява на котлон до пълното разтваряне на мангана. Когато се охлади, се прехвърля количествено в мерителна колба от 1 000 ml. Долива се до обема с вода и се смесва старателно.
4.4.2. Работен разтвор на манган (100 μg/ml)
Разрежда се 20 ml основен разтвор (4.4.1) в 0,5 mol/l разтвор на солна киселина (4.2) в мерителна колба от 200 ml. Долива се до обема с 0,5 mol/l разтвор на солна киселина (4.2) и се смесва старателно.
5. Апаратура
Атомен абсорбционен спектрометър: виж метод 9.4 (5). Инструментът трябва да бъде снабден с източник на лъчи, характеристични за мангана (279,6 nm).
6. Приготвяне на разтвора за анализ
6.1. Разтвор на екстракт на манган
Виж методи 9.1 и/или 9.2 и, ако е подходящо, 9.3.
6.2. Приготвяне на разтвора за изпитване
Виж метод 9.4 (6.2). Разтворът за изпитване трябва да съдържа 10 % (v/v) разтвор на лантанова сол (4.3).
7. Процедура
7.1. Приготвяне на разтвор на празна проба
Виж метод 9.4 (7.1). Разтворът за празната проба трябва да съдържа 10 % (v/v) разтвор на лантанова сол, използван в 6.2.
7.2. Приготвяне на калибровъчни разтвори
Виж метод 9.4 (7.2).
За оптимален диапазон на определяне от 0 до 5 μg/ml манган се поставя 0, 0,5, 1, 2, 3, 4 и 5 ml съответно от работния разтвор (4.4.2) в серия мерителни колби от 100 ml. Ако е необходимо, се регулира концентрацията на солна киселина, така че да е колкото е възможно по-близо до тази на разтвора за изпитване. Добавя се във всяка колба 10 ml разтвор на лантанова сол, използвана в 6.2. Долива се до 100 ml с 0,5 mol/l разтвор на солна киселина (4.2) и се смесва старателно. Тези разтвори съдържат съответно 0, 0,5, 1, 2, 3, 4 и 5 μg/ml манган.
7.3. Определяне
Виж метод 9.4 (7.3). Приготвя се спектрометърът (5) за измерване на дължината на вълната 279,6 nm.
8. Изразяване на резултатите
Виж метод 9.4 (8).
Процентът на мангана в тора е, както следва:
|
|
|
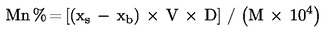
или ако се използва метод 9.3:
|
|
|
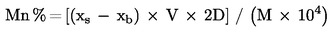
където
|
|
Мn е количеството на мангана, изразено като процент от тора; |
|
|
хs е концентрацията на разтвора за изпитване (6.2) в μg/ml; |
|
|
xb е концентрацията на разтвора на празната проба (7.1) в μg/ml ; |
|
|
V е обемът на екстракта, получен метод 9.1. или 9.2, в ml; |
|
|
D е коефициент, съответстващ на разреждането, извършено в 6.2; |
|
|
М е масата на пробата за изпитване, взета в съответствие метод 9.1 или 9.2. |
Изчисляване на коефициента на разреждане D: ако (а1), (а2), (а3,…, (аi) и (а) са аликвотните части и (v1), (v2), (v3),…, (vi) и (100) са обемите в ml, съответстващи на съответните разреждания, коефициентът на разреждане D ще бъде равен на:
|
|
|

Метод 9.10
Определяне съдържанието на молибден в торови екстракти чрез спектрометрия на комплекс с амиониев тиоцианат
1. Обхват
Настоящият метод описва процедура за определяне съдържанието на молибден в торови екстракти.
2. Област на приложение
Тази процедура се прилага към екстракти на проби на торове, получени чрез методи 9.1 и 9.2, за които в приложение I.Д към настоящия регламент съществува изискване за обявяване съдържанието на молибден.
3. Принцип
Молибденът (V) образува комплекс [MoO(SCN)5] — в кисела среда с SCN-йони.
Комплексът се екстрахира с помощта на n-бутил ацетат. Йоните, влизащи в реакция, като тези на желязото, остават във водната фаза. Жълто-оранжевият цвят се определя от молекулярната абсорбционна спектрометрия на 470 nm.
4. Реактиви
4.1. Разреден разтвор на солна киселина (HCl), приблизително 6 mol/l:
Виж метод 9.4 (4.1).
4.2. Меден разтвор (70 mg/l) в солна киселина 1,5 mol/l:
Разтварят се 275 mg меден сулфат (CuSO4 5H2O), измерени с точност до 0,1 mg в 250 ml разтвор на солна киселина 6 mol/l (4.1) в мерителна колба от 1 000 ml. Допълва се с вода до обема и се хомогенизира.
4.3. Разтвор на аскорбинова киселина (50 g/l):
Разтварят се 50 g аскорбинова киселина (C6H8O6) в мерителна колба от 1 000 ml. Допълва се с вода до обема, хомогенизира се и се държи в хладилник.
4.4. n-бутилов ацетат
4.5. Разтвор на амониев тиоцианат, 0,2 mol/l
В мерителна колба от 1 000 ml във вода се разтварят 15,224 g NH4SCN. Допълва се с вода до обема, хомогенизира се и се съхранява в бутилка с тъмен цвят.
4.6. Калаен разтвор (50 g/l) в солна киселина 2 mol/l:
Този разтвор трябва да бъде идеално прозрачен и приготвен непосредствено преди употреба. Трябва да се използва много чист калаен хлорид, в противен случай разтворът няма да бъде прозрачен.
За да се приготвят 100 ml от разтвора, се разтварят 5 g (SnCl22H2O) в 35 ml разтвор на HCl 6 mol/l (4.1). Добавят се 10 ml меден разтвор (4.2). Допълва се с вода до обема и се хомогенизира.
4.7. Калибровъчни разтвори на молибден
4.7.1. Основен молибденов разтвор (500 μg/ml)
Разтварят се 0,920 g амониев молибдат [(NH4)6Mo7O24 4H2O)], претеглени с точност до 0,1 mg в солна киселина 6 mol/l (4.1) в мерителна колба от 1 000 ml. Допълва се до обема с този разтвор и се хомогенизира.
4.7.2. Молибденов разтвор за непосредствена употреба (25 μg/ml)
Поставят се 25 ml от основния разтвор (4.7.1) в мерителна колба от 500 ml. Допълва се от обема със солна киселина 6 mol/l (4.1) и се хомогенизира.
4.7.3. Работен молибденов разтвор (2,5 μg/ml)
Поставят се 10 ml от разтвора за непосредствена употреба (4.7.2) в мерителна колба от 100 ml. Допълва се от обема със солна киселина 6 mol/l (4.1) и се хомогенизира.
5. Апаратура
5.1. Спектрометър, пригоден за молекулярна абсорбция с кювета с 20 mm оптична пътека, настроен за дължина на вълната 470 nm.
5.2. 200 или 250 ml разделителни фунии.
6. Подготовка на разтвора за анализ
6.1. Разтвор от молибденов екстракт
Виж методи 9.1 и/или 9.2 и, ако е подходящо, 9.3.
6.2. Приготвяне на разтвора
Разрежда се аликвотна част от екстракта (6.1) със солна киселина 6 mol/l (4.1), така че да се получи съответната концентрация на молибден. Факторът на разреждане е D.
Взема се аликвотна част а) от разтвора на екстракта, съдържаща 1 до 12 μg молибден и се поставя в разделителната фуния (5.2). Допълва се до 50 ml със солна киселина 6 mol/l (4.1).
7. Процедура
7.1. Празна проба
Приготвя се празна проба чрез повтаряне на цялата процедура от етапа на екстракцията, като се пропуска само пробата от тора.
7.2. Приготвяне на серия от калибровъчни разтвори:
Приготвя се серия от най-малко шест калибровъчни разтвора с нарастваща концентрация в съответствие с оптималния диапазон на чувствителност на спектрометъра.
За диапазона 0—12,5 μg молибден поставете съответно 0, 1, 2, 3, 4 и 5 ml работен разтвор (4.7.3) в разделителните фунии (5.2). Допълнете до 50 ml със солна киселина 6 mol/l (4.1). Фуниите съдържат съответно 0, 2,5, 5, 7,5, 10 и 12,5 μg молибден.
7.3. Отделяне на комплекса
Към всяка разделителна фуния (6.2, 7.1 и 7.2) се добавят в следния ред:
|
— |
10 ml меден разтвор (4.2), |
|
— |
20 ml разтвор на аскорбинова киселина (4.3); |
хомогенизира се и се изчаква две до три минути. След това се добавят:
|
— |
10 ml n-бутилов ацетат (4.4), като се използва прецизна пипета, |
|
— |
20 ml разтвор на тиоцианат (4.5). |
Разклаща се в продължение на една минута, за да се екстрахира комплексът в органична фаза; оставя се да се утаи; след отделянето на двете фази, се отстранява цялата водната фаза и се изхвърля; след това органичната фаза се промива с:
|
— |
10 ml разтвор на калаен хлорид (4.6). |
Разклаща се в продължение на една минута. Оставя се да се утаи и се отстранява цялата водната фаза. Прехвърля се органичната фаза в епруветка; така ще могат да бъдат събирани капките вода в суспензията.
7.4. Определяне
Измерва се оптичната плътност на разтворите, получени по 7.3 при дължина на вълната от 470 nm, като се използва 0 μg/ml калибровъчен молибденов разтвор (7.2) като референция.
8. Изразяване на резултатите
Начертава се калибровъчната крива, като по абсцисата се нанасят съответните маси молибден в калибровъчните разтвори (7.2), изразени в μg, и съответните стойности на оптичната плътност (7.4), отчетени от спектрометъра — по ординатата.
От тази крива се определя масата на молибдена в разтвора за изпитване (6.2) и празната проба (7.1). Тези маси се обозначават съответно като (xs) и (xb).
Процентното съдържание на молибден в тора се изчислява, както следва:
|
|
|
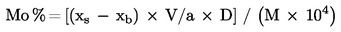
или ако се използва метод 9.3:
|
|
|
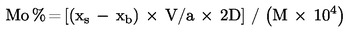
където:
|
|
Мо е количеството молибден, изразено като процент в тора; |
|
|
а е обемът в ml на аликвотата, взета от последния разреден разтвор (6.2); |
|
|
xs е масата на Мо в μg в разтвора за изпитване (6.2); |
|
|
xb е масата на Мо в μg в празната проба (7.1), обемът на която съответства на обем (а) на аликвотата в разтвора за изпитване (6.2); |
|
|
V е обемът в ml на разтвора от екстракта съгласно метод 9.1 или 9.2; |
|
|
D е факторът на разреждане по 6.2; |
|
|
М е масата в грамове на анализираната проба в съответствие с метод 9.1 и 9.2. |
Изчисляването на фактора на разреждане D: ако (a1) и (a2) са аликвотните части и (v1) и (v2) са обемите, съответстващи на съответните им разреждания, факторът на разреждане ще бъде равен на:
|
|
|

Метод 9.11
Определяне на цинк в екстракти на тор чрез атомна абсорбционна спектрометрия
1. Обхват
Настоящият метод процедура за определяне на цинк в екстракти на торове.
2. Област на приложение
Процедурата е приложима за анализиране на проби на торове, извлечени по методи 9.1 и 9.2, за които с приложение I.Д към настоящия регламент се изисква деклариране на общ и/или водоразтворим цинк.
3. Принцип
След подходящо третиране и разреждане на екстракта съдържанието на цинк се определя чрез атомна абсорбционна спектрометрия.
4. Реактиви
4.1. Разреден разтвор на солна киселина, около 6 mol/l:
Виж метод 9.4 (4.1).
4.2. Разреден разтвор на солна киселина, около 0,5 mol/l:
Виж метод 9.4 (4.2).
4.3. Разтвори на лантанови соли (10 g лантан на литър)
Виж метод 9.4 (4.3).
4.4. Калибровъчни разтвори на цинк
4.4.1. Основен разтвор на цинк (1 000 μg/ml)
В мерителна колба от 1 000 ml се разтваря 1 g цинк на прах, претеглен с точност до 0,1 mg, в 25 ml 6 mol/l солна киселина (4.1). Когато се разтвори напълно,се долива до обема с вода и се смесва старателно.
4.4.2. Работен разтвор на цинк (100 μg/ml)
Разрежда се 20 ml основен разтвор (4.4.1) в 0,5 mol/l разтвор на солна киселина (4.2) в мерителна колба от 200 ml. Долива се до обема с 0,5 mol/l разтвор на солна киселина (4.2) и се смесва старателно.
5. Апаратура
Атомен абсорбционен спектрометър: виж метод 9.4 (5). Инструментът трябва да бъде снабден с източници на лъчи, характеристични за цинка (213,8 nm). Спектрометърът трябва да позволява коригиране на фона.
6. Приготвяне на разтвора за анализ
6.1. Разтвор на екстракт на цинк
Виж методи 9.1 и/или 9.2 и, ако е подходящо, 9.3.
6.2. Приготвяне на разтвора за изпитване
Виж метод 9.4 (6.2). Разтворът за изпитване трябва да съдържа 10 % (v/v) разтвор на лантанова сол (4.3).
7. Процедура
7.1. Приготвяне на разтвор на празна проба
Виж метод 9.4 (7.1). Разтворът за празната проба трябва да съдържа 10 % (v/v) разтвор на лантанова сол, използван в 6.2.
7.2. Приготвяне на калибровъчни разтвори
Виж метод 9.4 (7.2).
За оптимален диапазон на определяне от 0 до 5 μg/ml цинк се поставя 0, 0,5, 1, 2, 3, 4 и 5 ml съответно от работния разтвор (4.4.2) в серия мерителни колби 100 ml. Ако е необходимо, се регулира концентрацията на солна киселина, така че да е колкото е възможно по-близо до тази на разтвора за изпитване. Добавя се във всяка колба 10 ml разтвор на лантанова сол, използвана в 6.2. Долива се до 100 ml с 0,5 mol/l разтвор на солна киселина (4.2) и се смесва старателно. Тези разтвори съдържат съответно 0, 0,5, 1, 2, 3, 4 и 5 μg/ml цинк.
7.3. Определяне
Виж метод 9.4 (7.3). Приготвя се спектрометърът (5) за измерване на дължината на вълната 213,8 nm.
8. Изразяване на резултатите
Виж метод 9.4 (8).
Процентът на цинка в тора се изчислява, както следва:
|
|
|

Ако се използва метод 9.3:
|
|
|

където
|
|
Zn е количеството на цинка, изразено като процент от тора; |
|
|
хs е концентрацията на разтвора за изпитване (6.2) в μg/ml ; |
|
|
xb е концентрацията на разтвора на празната проба (7.1) в μg/ml ; |
|
|
V е обемът на екстракта, получен метод 9.1. или 9.2, в ml; |
|
|
D е коефициент, съответстващ на разреждането, извършено в 6.2; |
|
|
М е масата на пробата за изпитване, взета в съответствие метод 9.1 или 9.2. |
Изчисляване на коефициента на разреждане D: Ако (а1), (а2), (а3,…, (аi) и (а) са аликвотните части и (v1), (v2), (v3),…, (vi) и (100) са обемите в ml, съответстващи на съответните разреждания, коефициентът на разреждане D ще бъде равен на:
|
|
|

Методи 10
Хранителни микроелементи при концентрация по-големи от 10 %
Метод 10.1
Екстракция на общото съдържание на хранителни микроелементи
1. Обхват
Този метод дефинира процедурата за извличане на следните хранителни микроелементи: общ бор, общ кобалт, обща мед, общо желязо, общ манган, общ молибден и общ цинк. Целта е да се извършат минимален брой извличания, като се използва, когато е възможно, един и същи екстракт за определяне на общото ниво на всеки от хранителните микроелементи, изброени по-горе.
2. Област на приложение
Тази процедура се отнася за торове в Общността, покрити от приложение I.Д, съдържащи един или повече от следните хранителни микроелементи: бор, кобалт, мед, желязо, манган, молибден и цинк. Тя е приложима за всеки хранителен микроелемент, на който декларираното съдържание е по-голямо от 10 %.
3. Принцип
Разтваряне в кипяща разредена солна киселина.
Бележка
Извличането е емпирично и може да не е количествено в зависимост от продукта или други съставки на тора. По специално, при някои манганови оксиди, извлеченото количество може да бъде съществено по-малко от общото количество манган, което продукта съдържа. Производителят на тора е отговорен да осигури декларираното количество действително да съответства на количеството, извлечено при присъщите условия на метода.
4. Реактиви
4.1. Разтвор на разредена солна киселина (HCl), около 6 mol/l
Смесва се 1 обем солна киселина (d20 = 1,18 g/ml) с един обем вода.
4.2. Концентриран амониев разтвор (NH4OH, d20 = 0,9 g/mol)
5. Апаратура
5.1. Електрически котлон с променлив температурен контрол.
5.2. pH метър
Бележка
Когато се определя съдържанието на бор на екстракта, не се използва борсиликатна стъклария. Тъй като методът включва кипене, тефлона и кварца са за предпочитане. Измива се стъкларията старателно, ако е била измита с измиващи препарати, съдържащи борати.
6. Приготвяне на пробата
Виж метод 1.
7. Процедура
7.1. Проба за изпитване
Взема се количество от тора, тежащо между 1 и 2 g, в зависимост от декларираното съдържание на елемента в продукта. Следващата таблица трябва да се използва за получаване на окончателния разтвор, който след подходящо разреждане ще бъде в рамките на измервателния диапазон за всеки метод. Пробите трябва да бъдат претеглени с точност до 1 mg.
|
Декларирано съдържание на хранителен микроелемент в тора (%) |
> 10 < 25 |
≥ 25 |
|
Маса на пробата за изпитване (g) |
2 |
1 |
|
Маса на елемента в пробата (mg) |
> 200 < 500 |
≥ 250 |
|
Обем на екстракта V (ml) |
500 |
500 |
|
Концентрация на елемента в екстракта (mg/l) |
> 400 < 1 000 |
≥ 500 |
Поставя се пробата в бехерова чаша от 250 ml.
7.2. Приготвяне на разтвора
Ако е необходимо, пробата се овлажнява с малко вода, внимателно, на малки количества се добавят 10 ml от разредената солна киселина (4.1) на грам тор. След това се добавя около 50 ml вода. Покрива се бехеровата чаша с наблюдателно стъкло и се смесва. Довежда се до кипене на котлона и се вари 30 минути. Оставя се да се охлади, като се разбърква от време на време. Прехвърля се количествено в мерителна колба от 500 ml. Долива се до обема с вода и се смесва старателно. Филтрира се през сух филтър в сух съд. Първата порция се изхвърля. Екстрактът трябва да бъде идеално бистър.
Препоръчва се определянето да се извърши незабавно на аликвотна част на бистрия филтрат. Ако не, съдът трябва да се запуши.
Бележка
Екстракти, в които трябва да се определи съдържанието на бор: регулира се рН между 4 и 6 с концентриран амоняк (4.2).
8. Определяне
Определянето на всяко хранителен микроелемент се извършва на аликвотните части, посочени в метода за всеки индивидуален хранителен микроелемент.
Методите 10.5, 10.6, 10.7, 10.9 и 10.10 не могат да се използват за определяне на елементи, присъстващи в хелатена или комплексирана форма. В такъв случай трябва да се използва Метод 10.3 преди определянето.
При определяне с ААС (методи 10.8, 10.11) такова третиране не е необходимо.
Метод 10.2
Екстракция на хранителни микроелементи, разтворими във вода
1. Обхват
Този метод дефинира процедурата за извличане на форми, разтворими във вода, на следните хранителни микроелементи: бор, кобалт, мед, желязо, манган, молибден и цинк. Целта е да се извършат минимален брой извличания, като се използва, когато е възможно, един и същи екстракт за определяне на общото ниво на всеки от хранителните микроелементи, изброени по-горе.
2. Област на приложение
Тази процедура се отнася за торовете в Общността, покрити от приложение I.Д, съдържащи един или повече от следните хранителни микроелементи: бор, кобалт, мед, о желязо, манган, молибден и цинк. Тя е приложима за всеки хранителен микроелемент, на което декларираното съдържание е по-голямо от 10 %.
3. Принцип
Хранителните микроелементи се извличат чрез разклащане на тор във вода при 20(± 2) °С.
Бележка
Извличането е емпирично и може да е или не количествено.
4. Реактиви
4.1. Разтвор на разредена солна киселина (HCl), около 6 mol/l
Смесва се 1 обем солна киселина (d20 = 1,18 g/ml) с един обем вода.
5. Апаратура
5.1. Ротационна клатачна машина, настроен на около 35 до 40 rpm.
Бележка
Когато се определя съдържанието на бор на екстракта, не се използва борсиликатна стъклария. Тъй като методът включва кипене, тефлона и кварца са за предпочитане. Измива се стъкларията старателно, ако е била измита с измиващи препарати, съдържащи борати.
6. Приготвяне на пробата
Виж метод 1.
7. Процедура
7.1. Проба за изпитване
Взема се количество от тора, тежащо между 1 и 2 g, в зависимост от декларираното съдържание на елемента в продукта. Следващата таблица трябва да се използва за получаване на окончателния разтвор, който след подходящо разреждане ще бъде в рамките на измервателния диапазон за всеки метод. Пробите трябва да бъдат претеглени с точност до 1 mg.
|
Декларирано съдържание на хранителен микроелемент в тора (%) |
> 10 < 25 |
≥ 25 |
|
Маса на пробата за изпитване (g) |
2 |
1 |
|
Маса на елемента в пробата (mg) |
> 200 < 500 |
≥ 250 |
|
Обем на екстракта V (ml) |
500 |
500 |
|
Концентрация на елемента в екстракта (mg/l) |
> 400 < 1 000 |
≥ 500 |
Поставя се пробата в бехерова чаша от 500 ml.
7.2. Приготвяне на разтвора
Добавят се около 400 ml вода.
Колбата се запушва добре. Разклаща се силно с ръка, за да се разпръсне пробата, след това се поставя колбата на клатачната машина и се разклаща 30 минути.
Долива се до обема вода и се смесва старателно.
7.3. Приготвяне на разтвора за изпитване
Филтрира се незабавно в чиста суха колба. Запушва се колбата. Извършва се определянето незабавно след филтрирането.
Бележка
Ако филтратът постепенно става замъглен, прави се друго извличане в колба с обем Vе, като се следват 7.1 и 7.2. Филтрира се калибрирана колба с обем W, която е била предварително подсушена и са били сложени 5 ml разредена солна киселина (4.1). Филтрирането се спира в момента, когато се достигне калибровъчната отметка. Смесва се старателно.
При тези условия стойността V в изразяването на резултата е:
|
|
|

Разреждането в изразяването на резултатите зависи от тази стойност V.
8. Определяне
Определянето на всеки хранителен микроелемент се извършва на аликвотните части, посочени в метода за всеки индивидуален хранителен микроелемент.
Методите 10.5, 10.6, 10.7, 10.9 и 10.10 не могат да се използват за определяне на елементи, присъстващи в хелатена или комплексирана форма. В такъв случай трябва да се използва Метод 10.3 преди определянето.
При определяне с ААС (методи 10.8 10.11) такова третиране не е необходимо.
Метод 10.3
Отстраняване на органични съединения от екстракти на торове
1. Обхват
Този метод дефинира процедурата за отстраняване на органични съединения от екстракти на торове.
2. Област на приложение
Тази процедура е приложима за анализиране на проби на торове, извлечени по методи 10.1 и 10.2, за които с приложение I.Д към настоящия регламент се изисква деклариране на общ и/или разтворим във вода елемент.
Бележка
Присъствието на малки количества органични вещества обикновено не влияе на определянето с помощта на атомна абсорбционна спектрометрия.
3. Принцип
Органичните съединения в аликвотна част на екстракта се окисляват с водороден прекис.
4. Реактиви
4.1. Разтвор на разредена солна киселина (HCl), около 0,5 mol/l
Смесва се 1 обем солна киселина (d20 = 1,18 g/ml) с 20 обема вода.
4.2. Разтвор на водороден прекис (30 % Н2О2, d20 = 1,11 g/ml), без хранителни микроелементи
5. Апаратура
Електрически котлон с променлив температурен контрол.
6. Процедура
Взема се 25 ml от разтвора на екстракта, получен по метод 10.1 или метод 10.2, и се поставя в бехерова чаша от 100 ml. При метод 10.2 се добавя 5 ml разтвор на разредена солна киселина (4.1). След това се добавя 5 ml разтвор на водороден прекис (4.2). Покрива се с наблюдателно стъкло. Оставя се да се окисли при стайна температура за около един час, след това бавно се довежда до кипене и се вари в продължение на половин час. Ако е необходимо, се добавя допълнителни 5 ml водороден прекис към разтвора след като се охлади. След това се кипва, за да се отстрани излишният водороден прекис. Оставя се да се охлади, прехвърля се количествено в измервателна колба 50 ml и се допълва до обема. При необходимост се филтрира.
Следва да се взема предвид това разреждане, когато се вземат аликвотни части и се изчислява процентът на хранителния микроелемент в продукта.
Метод 10.4
Определяне на хранителни микроелементи в екстракти на торове чрез атомна абсорбционна спектрометрия
(обща процедура)
1. Обхват
Настоящият метод дефинира общата процедура за определяне на нивата на желязо и цинк в екстракти на торове чрез атомна абсорбционна спектрометрия.
2. Област на приложение
Настоящият метод се прилага за анализиране на проби на торове, извлечени по методи 10.1 и 10.2, за които с приложение I.Д към настоящия регламент се изисква деклариране на общи и/или разтворими във вода желязо и цинк.
Адаптирането на тази процедура за различните микрохранителни елементи са описани подробно в методите, дефинирани специфично за всеки елемент.
Бележка:
В повечето случаи наличието на малки количества органично вещество няма да влияе на определянията чрез атомна абсорбционна спектрометрия.
3. Принцип
След като екстрактът е бил третиран, когато е необходимо, за намаляване или елиминиране на влияещи химически видове, екстракта се разрежда така че неговата концентрация да е в оптималния диапазон на спектрометъра на дължина на вълната, подходяща за определяния хранителен микроелемент.
4. Реактиви
4.1. Разреден разтвор на солна киселина (HCl), около 6 mol/l:
Смесва се един обем солна киселина (d20 = 1,18 g/ml) с един обем вода.
4.2. Разреден разтвор на солна киселина (HCl), около 0,5 mol/l:
Смесва се един обем солна киселина (d20 = 1,18 g/ml) с 20 обема вода.
4.3. Разтвори на лантанови соли (10 g лантан на литър)
Този реактив се използва за определяне на желязо и цинк. Той може да бъде приготвен:
|
а) |
с лантанов оксид, разтворен в солна киселина (4.1). Поставя се 11,73 g лантанов оксид (La2O3) в 150 ml вода в мерителна колба от един литър и се добавя 120 ml 6 mol/l солна киселина (4.1). Оставя се да се разтвори и след това се долива до 1 литър с вода и се смесва старателно. Този разтвор е приблизително 0,5 mol/l в солна киселина; или |
|
б) |
с разтвори на лантанов хлорид, сулфат или нитрат. Разтваря се 26,7 g лантанов хлорид хептахидрат (LaCl3 7H2O) или 31,2 g лантанов нитратен хексахидрат [La(NO3)3 6H2O], или 26,2 g лантанов сулфатен нонахидрат [La2 (SO4)3 9H2O] в 150 ml вода и след това се добавя 85 ml 6 mol/l солна киселина (4.1). Оставя се да се разтвори и след това се долива до един литър с вода. Смесва се старателно. Този разтвор е приблизително 0,5 mol/l в солна киселина. |
4.4. Калибровъчни разтвори
За тяхното приготвяне виж индивидуалните методи за определяне на всеки хранителен микроелемент.
5. Апаратура
Атомен абсорбционен спектрометър, снабден с източници, излъчващи радиационна характеристика на определяния хранителен микроелемент.
Лаборантът трябва да следва указанията на производителя и да бъде запознат с материала. Апаратът трябва да позволява фонова корекция, така че да може използван, когато е необходимо (например Zn). Използваните газове са въздух и ацетилен.
6. Приготвяне на разтвора за анализ
6.1. Приготвяне на разтвори на екстракта съдържащ елементи за определяне
Виж методи 10.1 и/или 10.2 и, ако е подходящо, 10.3.
6.2. Третиране на разтвора за изпитване
Разрежда се аликвотна част на екстракта, получен по метод 10.1, 10.2 или 10.3, с вода и/или солна киселина (4.1) или (4.2), така че да се получи окончателен разтвор за измерване, концентрация на определяния елемент, която е подходяща за използвания диапазон на калибриране (7.2) и концентрация на солната киселина най-малко 0,5 mol/l и не повече от 2,5 mol/l. Тази операция може да изисква едно или повече последователни разреждания.
Окончателният разтвор трябва да се получи чрез поставяне на аликвотна част на разредения екстракт в мерителна колба от 100 ml. (а) е неговия обем в ml. Добавя се 10 ml разтвор на лантанова сол (4.3). Долива се до обема с 0,5 mol/l разтвор на солна киселина (4.2) и се смесва старателно. D е коефициентът на разреждане.
7. Процедура
7.1. Приготвяне на разтвор на празна проба
Приготвя се разтвор на празна проба чрез повтаряне на цялата процедура от етапа на извличане, изпускайки само пробата за изпитване на тора.
7.2. Приготвяне на калибровъчни разтвори
От работния калибровъчен разтвор, приготвен с използване на метода, даден за всеки индивидуален хранителен микроелемент, в мерителни колби от 100 ml се приготвя серия от най-малко пет калибровъчни разтвора с увеличаваща се концентрация в рамките на оптималния измервателен диапазон на спектрометъра. Ако е необходимо, концентрацията на солната киселина се регулира, така че да е колкото е възможно по-близо до тази на разредения разтвор за изпитване (6.2). За определяне на желязо или цинк се добавя 10 ml от същия разтвор на лантанова сол, като използвания в 6.2. Допълва се до обема с 0,5 mol/l разтвор на солна киселина (4.2) и се смесва старателно.
7.3. Определяне
Приготвя се спектрометърът (5) за определянето и се регулира на дължината на вълната, дадена в метода за съответния индивидуален микроелемент.
Впръскват се три пъти последователно калибровъчните разтвори (7.2), разтворът за изпитване (6.2) и разтворът на празната проба (7.1), като се отбелязва всеки резултат и инструментът се измива с дестилирана вода между индивидуалните впръсквания.
Построява се калибровъчната крива чрез построяване на средните показания на спектрометъра за всеки калибровъчен разтвор (7.2) по ординатата и съответната концентрация на елемента, изразена в μg/ml по абсцисата.
От тази крива се определят концентрациите на съответния хранителен микроелемент в разтвора за изпитване хs (6.2) и разтвора на празната проба xb (7.1), като тези концентрации се изразяват в μg на ml.
8. Изразяване на резултатите
Процентът на хранителния микроелемент (Е) в тора е равен на:
|
|
|

или ако се използва метод 10.3:
|
|
|

където
|
|
Е е количеството на определяния хранителен микроелемент, изразен като процент от тора; |
|
|
хs е концентрацията на разтвора за изпитване (6.2) в μg/ml; |
|
|
xb е концентрацията на разтвора на празната проба (7.1) в μg/ml; |
|
|
V е обема на екстракта, получен метод 10.1. или 10.2, в ml; |
|
|
D е коефициент, съответстващ на разреждането, извършено в 6.2; |
|
|
М е масата на пробата за изпитване, взета в съответствие метод 10.1 или 10.2, в грамове. |
Изчисляване на коефициента на разреждане D:
|
|
Ако (а1), (а2), (а3),…, (аi) и (а) са аликвоните части и (v1), (v2), (v3),…, (vi) и (100) са обемите в ml, съответстващи на съответните разреждания, коефициентът на разреждане D ще бъде равен на:
|
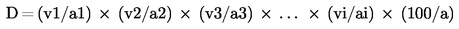
Метод 10.5
Определяне на бор в екстракти на торове с помощта на ацидиметрично титруване
1. Обхват
Този метод дефинира процедурата за определяне на съдържанието на бор в екстракти на торове.
2. Област на приложение
Настоящият метод се прилага за анализиране на проби на торове, извлечени по методи 10.1 и 10.2, за които с приложение I.Д към настоящия регламент се изисква деклариране на общ и/или водоразтворим бор.
3. Принцип
Манито-борен комплекс се образува чрез долната реакция на борат с манитол.
|
|
|
Комплексът се третира с разтвор на натриев хидроксид до рН 6,3.
4. Реактиви
4.1. Разтвор на индикатор метилово червено
Разтваря се 0,1 g метилово червено (C15H15N3O2) в 50 ml етанол (95 % в мерителна колба от 100 ml). Долива се до обема от 100 ml с вода. Смесва се старателно.
4.2. Разреден разтвор на солна киселина , около 0,5 mol
Смесва се един обем солна киселина HCl (d20 = 1,18 g/ml) с 20 обема вода.
4.3. Разтвор на натриев хидроксид, около 0,5 mol/l
Трябва да бъде без въглероден диоксид. Разтваря се 20 g натриев диоксид (NaOH) на гранули в едва мерителна колба от един литър, съдържаща около 800 ml кипяща вода. Когато разтворът се охлади, долива се до 1 000 ml с вряща вода и се смесва добре.
4.4. Стандартен разтвор на натриев хидроксид, около 0,025 mol/l
Трябва да бъде без въглероден диоксид. Разрежда се 0,5 mol/l разтвор на натриев хидроксид (4.3) 20 пъти с гореща вода и се смесва добре. Стойността на разтвора, изразена като бор (В), трябва да се определи (виж параграф 9).
4.5. Борен калибровъчен разтвор (100 μg/ml В)
Разтваря се 0,5719 g борна киселина (Н3ВО3), претеглени с точност до 0,1 mg в мерителна колба от 1 000 ml. Долива се до обема с вода и се смесва добре. Прехвърля се в пластмасова бутилка за съхраняване в хладилник.
4.6. D-манитол (C6H14O6) на прах
4.7. Натриев хлорид (NaCl)
5. Апаратура
5.1. рН метър със стъклен електрод
5.2. Магнитна бъркалка
5.3. Бехерова чаша от 400 ml с тефлонова пръчка
6. Приготвяне на разтвора за анализ
6.1. Приготвяне на борения разтвор
Виж методи 10.1, 10.2 и, когато е подходящо, 10.3.
7. Процедура
7.1. Изпитване
Поставя се в бехерова чаша от 400 ml (5.3) аликвот (а) на екстракта (6.1), съдържащ 2 до 4 mg В. Добавя се 150 ml вода.
Добавят се няколко капки разтвор на индикатора метилово червено (4.1).
При екстракция с метод 10.2 се подкиселява чрез добавяне на 0,5 mol/l солна киселина (4.2) до точката на промяна на индикаторния разтвор. След това се добавя още 0,5 ml 0,5 mol/l солна киселина (4.2).
След добавяне на 3 g натриев хлорид (4.7) се довежда до кипене, за да се изкара въглеродният диоксид. Оставя се да се охлади. Поставя се бехеровата чаша на магнитната бъркалка (5.2) и се вмъкват електродите на предварително калибрирания рН-метър (5.1).
Регулира се рН на точно 6,3 — първо с 0,5 mol/l разтвор на натриев хидроксид (4.3), след това с разтвор 0,025 mol/l (4.4).
Добавят се 20 g D-манитол (4.6), разтваря се напълно и се смесва старателно. Титрува се с 0,025 mol/l разтвор на натриев хидроксид (4.4) до рН 6,3 (поне 1 минута стабилност). Х1 е изискваният обем.
8. Разтвор на празна проба
Приготвя се разтвор на празна проба, като се повтаря цялата процедура от етапа на приготвяне на разтвора; пропуска се само торът. Х0 трябва да бъде изисквания обем.
9. Стойност на бор (В) на разтвора на натриев хидроксид (4.4)
Прехвърлят се с пипета се 20 ml (2,0 mg В) от калибровъчния разтвор в бехерова чаша от 400 ml и се добавят няколко капки индикаторен разтвор метилово червено (4.1). Добавят се 3 g натриев хлорид (4.7) и разтвор на солна киселина (4.2) до точката на промяна на цвета на индикаторния разтвор (4.1).
Долива се до обем около 150 ml и постепенно се довежда до кипене, така че да се елиминира въглеродният диоксид. Оставя се да се охлади. Поставя се бехеровата чаша на магнитната бъркалка (5.2) и се вкарват предварително калибрираните електроди на рН метъра (5.1). Регулира се рН точно на 6,3, първо с 0,5 mol/l разтвор на натриев хидроксид (4.3), след това с 0,025 mol/l разтвор (4.4).
Добавя се 20 g D-манитол (4.6), разтваря се напълно и се смесва старателно. Титрува се с 0,025 mol/l разтвор на натриев хидроксид (4.4) до рН 6,3 (поне 1 минута стабилност). V1 е изискваният обем.
Приготвя се празна проба по същия начин, като се добавят 20 ml за калибровъчния разтвор. Необходимото количество е V0.
Стойността на бора (F) в mg/ml на стандартния разтвор на NaOH (4.4) е както следва:
|
|
|

1 ml от точно 0,025 mol/l разтвор на натриев хидроксид съответства на 0,27025 mg В.
10. Изразяване на резултатите
Процентът на бор в тора се дава с:
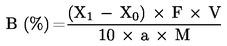
където
|
|
В (%) е процент на бора в тора; |
|
|
Х1 е обема, в ml на 0,025 mol/l разтвор на натриeв хидроксид (4.4), необходим ма разтвора за изпитване; |
|
|
Х0 е обема, в ml на 0,025 mol/l разтвор на натриeв хидроксид (4.4), необходим ма разтвора на празната проба; |
|
|
F е стойността на бора (В), в mg/ml, на 0,025 mol/l разтвор на натриeв хидроксид (4.4); |
|
|
V е обемът в ml на разтвора на екстракта, получен в съответствие с метод 10.1 или 10.2; |
|
|
а е обема, ml на аликвота (7.1), взета от разтвора на екстракта (6.1); |
|
|
М е масата, в грамове, на пробата за изпитване, взета в съответствие с метод 10.1 или 10.2. |
Метод 10.6
Определяне на кобалт в екстракти на торове по гравиметричен метод с 1-нитрозо-2- нафтол
1. Обхват
Настоящият метод дефинира процедура за определяне на кобалт в екстракти на торове.
2. Област на приложение
Тази процедура е приложима за екстракти от проби на торове, получени с метод 10.1 или метод 10.2, за които с приложение I.Д към настоящия регламент се изисква деклариране на съдържанието на кобалт.
3. Принцип
Кобалт III се съединява с 1-нитрозо-2- нафтол, за да даде червена утайка Co(C10H6ONO)3. 2H2O. След като кобалтът, присъстващ в екстракта, е бил доведен до състояние кобалт III, кобалтът се утаява в среда на оцетна киселина с разтвор на 1-нитрозо-2- нафтол. След филтриране утайката се измива и изсушава до постоянна маса и след това се претегля като Co(C10H6ONO)3. 2H2O.
4. Реактиви
4.1. Разтвор на водороден прекис, (H2O2, d20 = 1,11 g/ml) 30 %
4.2. Разтвор на натриев хидроксид, около 2 mol/l
Разтваря се 8 g натриев хидроксид на гранули в 100 ml вода.
4.3. Разреден разтвор на солна киселина HCl, около 6 mol/l
Смесва се един обем солна киселина (d20 = 1,18 g/ml) с един обем вода.
4.4. Оцетна киселина (99,7 % CH3CO2H) (d20 = 1,05 g/ml)
4.5. Разтвор на оцетна киселина (1:2), около 6 mol/l
Смесва се един обем оцетна киселина (4.4) с 2 обема вода.
4.6. Разтвор на 1-нитрозо-2-нафтол в 100 ml оцетна киселина (4.4). Добавя се 100 ml хладка вода. Филтрира се веднага. Получения разтвор трябва да бъде използван незабавно.
5. Апаратура
5.1. Филтърен тигел Р 16/ISO 4793, порьозност 4, обем 30 до 50 ml
5.2. Пещ за сушене при 130(± 2) °С
6. Приготвяне на разтвора за анализ
6.1. Приготвяне на кобалтовия разтвор
Виж методи 10.1 или 10.2.
6.2. Приготвяне на разтвор за анализиране
Поставя се аликвот на екстракта, съдържащ не повече от 20 mg Со в бехерова чаша от 400 ml. Ако екстрактът е получен съгласно метод 10.2, подкисилен с пет капки солна киселина (4.3). Добавя се около 10 ml разтвор на водороден прекис (4.1). Оставя се оксидантът да действа в студено състояние 15 минути, след това се долива до около 100 ml с вода. Покрива се бехеровата чаша с наблюдателно стъкло. Довежда се разтвора да точка на кипене и се оставя да ври около 10 минути. Охлажда се. Прави се го алкален с разтвор на натриев хидроксид (4.2) капка по капка, докато започне да се утаява черна кобалтова утайка.
7. Процедура
Добавя се 10 ml оцетна киселина (4.4) и се долива до около 200 ml с вода. Нагрява се до завиране. С помощта на бюрета, се добавя 20 ml разтвор на 1-нитрозо-2-нафтол капка по капка, като се бърка постоянно. Завършва се със силно разбъркване, така че утайката да коагулира.
Филтрира се през предварително претеглен филтърен тигел (5.1), като се внимава да не се задръсти тигелът. Като се има това предвид, се осигурява течността да остава над утайката през целия процес на филтриране.
Измива се бехеровата чаша с разредена оцетна киселина (4.5), за да се отстрани цялата утайка. Измива се утайката върху филтъра с разредена оцетна киселина (4.5) и след това три пъти с гореща вода.
Изсушава се в пещ за сушене (5.2) при 130(± 2) °С, докато се получи постоянна маса.
8. Изразяване на резултатите
1 mg утайка на Co(C10H6ONO)3.2H2O съответства на 0,096381 mg Со.
Процентът на кобалт (Со) в тора се дава с:
 ,
,
където
|
|
Х е масата в mg на утайката; |
|
|
V е обемът в ml на разтвора на екстракта, получен в съответствие с метод 10.1 или метод 10.2; |
|
|
а е обемът в ml на аликвота, взет от последното разреждане; |
|
|
D е коефициентът на разреждане на този аликвот; |
|
|
М е масата в g на пробата за изпитване. |
Метод 10.7
Определяне на мед в екстракти на торове с титриметрични методи
1. Обхват
Настоящият метод дефинира процедурата за определяне на мед в екстракти на торове.
2. Област на приложение
Тази процедура е приложима за екстракти от проби на торове, получени с метод 10.1 или метод 10.2, за които с приложение I.Д към настоящия регламент се изисква деклариране на съдържанието на мед.
3. Принцип
Медните йони се редуцират в кисела среда с калиев йодид:
|
|
|
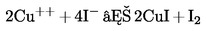
Освободеният по този начин йод се титрува със стандартен разтвор на натриев тиосулфат в присъствието на скорбяла като индикатор в съответствие с:
|
|
|

4. Реактиви
4.1. Азотна киселина (HNO3, d20 = 1,40 g/ml
4.2. Карбамид [(NH2)2 C = 0]
4.3. Разтвор на амониев бифлуорид (NH4HF2), 10 % w/v
Разтворът се съхранява в пластмасов съд.
4.4. Разтвор на амониев хидроксид (1 + 1)
Смесва се един обем амоняк (NH4OH, d20 = 0.9 g/ml) с 1 обем вода.
4.5. Стандартен разтвор на натриев тиосулфат
Разтваря се 7,812 g натриев тиосулфат пентахидрат (Na2S2O35H2O) с вода в мерителна колба от 1 литър. Този разтвор трябва да се приготви, така че 1 ml = 2 mg мед. За стабилизиране се прибавя няколко капки хлороформ. Разтворът трябва да се съхранява в стъклен съд и да бъде защитен от пряка светлина.
4.6. Калиев йодид (KI)
4.7. Разтвор на калиев тиоцианат (KSCN) (25 % w/v)
Разтворът се съхранява в пластмасов съд.
4.8. Разтвор на нишесте (около 0,5 %)
Поставя се 2,5 g нишесте в бехерова чаша от 600 ml. Добавя се около 500 ml вода. Кипва се, като се разбърква. Охлажда се до температурата на околната среда. Разтворът има кратък срок на съхранение. Този срок може да бъде удължен чрез добавяне на около 10 mg живачен йодид.
5. Приготвяне на разтвора за анализиране
Приготвяне на медения разтвор
Виж методи 10.1 и 10.2.
6. Процедура
6.1. Приготвяне на разтвор за титруване
Поставя се аликвотна част на разтвора, съдържаща не по-малко от 20—40 mg мед в ерленмайерова колба от 500 ml.
Изкарва се целият излишен кислород, като се кипва за кратко. Долива се до обем около 100 ml вода. Добавят се 5 ml азотна киселина (4.1), довежда се до кипене и се оставя да ври около половин минута.
Отстранява се ерленмайеровата колба от нагряващия апарат, добавя се около 3 g карбамид (4.2) и отново се вари за около половин минута.
Отстранява се апаратът от загряващия апарат и се добавя 200 ml студена вода. Когато е необходимо, се охлажда съдържанието на ерленмайеровата колба до температурата на околната среда.
Постепенно се добавя разтвор на амониев хидроксид (4.4), докато разтворът стане син. След това се добавя 1 ml в излишък.
Добавят се 50 ml разтвор на амониев бифлуорид (4.3) и се смесва.
Добавят се 10 g калиев йодид (4.6) и се разтваря.
6.2. Титруване на разтвора
Поставя се ерленмайеровата колба на магнитна бъркалка. Поставя се пръчката в ерленмайерова колба и бъркалката се настройва на желаната скорост.
С помощта на бюрета се добавя стандартен разтвор на натриев тиосулфат (4.5), докато кафявият цвят на йода, освободен от разтвора, стане по-малко интензивен.
Добавят се 10 ml разтвор на скорбяла (4.8).
Продължава се титруването с разтвор на натриев тиосулфат (4.5) и се титрува, докато виолетовият цвят напълно изчезне.
Добавете 20 ml разтвор на калиев тиоцианат (4.7) и продължете титруването докато напълно изчезне виолетово-синия цвят.
Отбелязва се обемът за приложения тиосулфатен разтвор.
7. Изразяване на резултатите
1 ml стандартен разтвор на натриев тиосулфат (4.5) съответства на 2 mg мед.
Процентът мед в тора се дава с:

където
|
|
Х е обемът в ml на използвания разтвор натриев тиосулфат; |
|
|
V е обемът в ml на разтвора на екстракта, получен в съответствие с метод 10.1или метод 10.2; |
|
|
а е обемът в ml на аликвота, взет от последното разреждане; |
|
|
М е масата в g на пробата за изпитване, третирана в съответствие с метод 10.1 или метод 10.2. |
Метод 10.8
Определяне на желязо в екстракти на тор чрез атомна абсорбционна спектрометрия (ААС)
1. Обхват
Настоящият метод описва процедура за определяне на желязо в екстракти на торове.
2. Област на приложение
Процедурата е приложима за екстракти от проби на торове, получени по методи 10.1 и 10.2, за които с приложение I.Д към настоящия регламент се изисква деклариране на общо и/или водоразтворимо желязо.
3. Принцип
След подходящо третиране и разреждане на екстракта съдържанието на желязо се определя чрез атомна абсорбционна спектрометрия.
4. Реактиви
4.1. Разтвор на солна киселина, около 6 mol/l:
Виж метод 10.4 (4.1).
4.2. Разтвор на солна киселина, около 0,5 mol/l:
Виж метод 10.4 (4.2).
4.3. Разтвор на водороден прекис (30 % H2O2, d20 = 1,11 g/ml), несъдържащ хранителни микроелементи
4.4. Разтвори на лантанови соли (10 g лантан на литър)
Виж метод 10.4 (4.3).
4.5. Калибровъчни разтвори на желязо
4.5.1. Основен разтвор на желязо (1 000 μg/ml)
В бехерова чаша 500 ml се претегля с точност до 0,1 mg 1 g желязо, добавя се 200 ml 6 mol/l солна киселина (4.1) и 15 ml разтвор на водороден прекис (4.3) и се нагрява на котлон до пълното разтваряне на желязото. Когато се охлади, прехвърля се количествено в мерителна колба от 1 000 ml. Долива се до обема с вода и се смесва старателно.
4.5.2. Работен разтвор на желязо (100 μg/ml)
Поставя се 20 ml основен разтвор (4.5.1) в мерителна колба от 200 ml. Долива се до обема с 0,5 mol/l разтвор на солна киселина (4.2) и се смесва старателно.
5. Апаратура
Атомен абсорбционен спектрометър: виж метод 10.4 (5). Инструментът трябва да бъде снабден с източници на лъчи, характеристични за желязото (248,3 nm).
6. Приготвяне на разтвора за анализ
6.1. Разтвор на екстракт на желязо
Виж методи 10.1 и/или 10.2 и, ако е подходящо, 10.3.
6.2. Приготвяне на разтвора за изпитване
Виж метод 10.4 (6.2). Разтворът за изпитване трябва да съдържа 10 % (v/v) разтвор на лантанова сол.
7. Процедура
7.1. Приготвяне на разтвор на празна проба
Виж метод 10.4 (7.1). Разтворът за празната проба трябва да съдържа 10 % (v/v) разтвор на лантанова сол, използван в 6.2.
7.2. Приготвяне на калибровъчни разтвори
Виж метод 10.4 (7.2).
За оптимален диапазон на определяне от 0 до 10 μg/ml желязо се поставя 0, 2, 4, 6, 8 и 10 ml съответно от работния разтвор (4.5.2) в серия мерителни колби 100 ml. Ако е необходимо, се регулира концентрацията на солна киселина, така че да е колкото е възможно по-близо до тази на разтвор за изпитване. Добавя се във всяка колба 10 ml разтвор на лантанова сол, използвана в 6.2. Долива се с 0,5 mol/l разтвор на солна киселина (4.2) и се смесва старателно. Тези разтвори съдържат съответно 0, 2, 4, 6, 8 и 10 μg/ml желязо.
7.3. Определяне
Виж метод 10.4 (7.3). Приготвя се спектрометърът (5) за измерване на дължината на вълната 248,3 nm.
8. Изразяване на резултатите
Виж метод 10.4 (8).
Процентът на желязото в тора се дава с:
|
|
|

Ако се използва метод 10.3:
|
|
|

където
|
|
Fe е количеството на желязото, изразено като процент от тора; |
|
|
хs е концентрацията на разтвора за изпитване (6.2) в μg/ml; |
|
|
xb е концентрацията на разтвора на празната проба (7.1) в μg/ml; |
|
|
V е обемът на екстракта, получен по метод 10.1. или 10.2, в ml; |
|
|
D е коефициент, съответстващ на разреждането, извършено в 6.2; |
|
|
М е масата на пробата за изпитване, взета в съответствие метод 10.1 или 10.2. |
Изчисляване на коефициента на разреждане D: ако (а1), (а2), (а3,…, (аi) и (а) са аликвотните части и (v1), (v2), (v3),…, (vi) и (100) са обемите в ml, съответстващи на съответните разреждания, коефициентът на разреждане D ще бъде равен на:
|
|
|
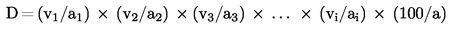
Метод 10.9
Определяне на манган в екстракти на торове чрез титруване
1. Обхват
Този метод описва процедурата за определяне на манган в екстракти на торове.
2. Област на прилагане
Тази процедура се прилага към екстракти от проби на торове, получени по методи 10.1 и 10.2, за които се изисква декларация за магнезий по приложение I.Д към настоящия регламент.
3. Принцип
Ако в екстракта присъстват хлорни йони, те се изкарват от чрез варене на екстракта със сярна киселина. Манганът се окислява с натриев бисмут в среда на азотна киселина. Образуваният перманганат се редуцира с излишък на железен сулфат. Този излишък се титрува с разтвор на калиев перманганат.
4. Реактиви
4.1. Концентрирана сярна киселина (H2SO4, d20 = 1,84 g/ml)
4.2. Сярна киселина, около 9 mol/l
Внимателно се смесва 1 обем концентрирана сярна киселина (4.1) с един обем вода.
4.3. Азотна киселина, 6 mol/l
Смесва се 3 обема азотна киселина (HNO3, d20 = 1,40 g/ml) с четири обема вода.
4.4. Азотна киселина, 0,3 mol/l
Смесва се 1 обем азотна киселина 6 mol/l с 19 обема вода.
4.5. Натриев бисмутат (NaBiO3) (85 %).
4.6. Кизелгур
4.7. Ортофосфорна киселина, 15 mol/l (H3PO4, d20 = 1,71 g/ml)
4.8. Разтвор на железен сулфат, 0,15 mol/l
Разтваря се 41,6 g на хептахидрат на железен сулфат (FeSO4.7H2O) в мерителна колба от 1 литър.
Добавя се 25 ml концентрирана сярна киселина (4.1) и 25 ml фосфорна киселина (4.7). Долива се до 1 000 ml. Смесва се.
4.9. Разтвор на калиев перманганат, 0,020 mol/l
Претегля се 3,160 g калиев перманганат (KМnO4) с точност до 0,1 mg. Разтваря се и се долива до 1 000 ml с вода.
4.10. Разтвор на сребърен нитрат, 0,1 mol/l
Разтваря се 1,7 g сребърен нитрат(AgNO3) във вода и се долива до 100 ml.
5. Апаратура
5.1. Филтърен тигел P16/ISO 4793, порьозност 4, обем 50 ml, монтиран на филтруваща колба от 500 ml.
5.2. Магнитна бъркалка
6. Приготвяне на разтвора за анализиране
6.1. Разтвор на манганов екстракт
Виж методи 10.1 и 10.2. Ако не е известно дали присъстват хлорни йони, се извършва тестване на разтвора с една капка разтвор на сребърен нитрат (4.10).
6.2. При отсъствие на хлорни йони, се поставя аликвота на екстракта, съдържащ от 10 до 20 mg манган, във висока бехерова чаша от 400 ml. Довежда се обемът до около 25 ml чрез изпаряване или чрез добавяне на вода. Добавя се 2 ml концентрирана сярна киселина (4.1).
6.3. Ако присъстват хлорни йони, е необходимо те да бъдат отстранени, както следва:
|
|
Поставя се аликвот на екстракта, съдържащ от 10 до 20 mg манган във висока бехерова чаша от 400 ml. Добавя се 5 ml от 9 mol/l сярна киселина (4.2). Под смукателен чадър за дим се довежда се до кипене на котлон и се оставя да ври, докато се освобождават изобилни бели пари. Продължава се, докато обемът намалее до около 2 ml (тънък слой сиропоподобна течност на дъното на бехеровата чаша). Оставя се да се охлади до температурата на околната среда. |
|
|
Внимателно се добавя 25 ml вода и отново се тества за наличието на хлориди с една капка разтвор на сребърен нитрат (4.10). Ако все още остават хлориди, се повторя операцията чрез добавяне на 5 ml от 9 mol/l сярна киселина (4.2). |
7. Процедура
Добавя се 25 ml от 6 mol/l азотна киселина (4.3) и 2,5 g натриев бисмутат (4.5) в бехерова чаша от 400 ml, съдържаща разтвора за изпитване. Разбърква се силно три минути с магнитната бъркалка (5.2).
Добавя се 50 ml от 0,3 mol/l азотна киселина (4.4) и се разбърква отново. Филтрира се в празен съд през тигел (5.1), дъното на който е покрит с кизелгур (4.6). Измива се тигелът няколко пъти с 0,3 mol/l азотна киселина (4.4), докато се получи безцветен филтрат.
Прехвърлят се филтратът и измиващият разтвор в бехерова чаша от 500 ml. Смесва се и се добавя 25 ml от 0,15 mol/l разтвор на железен сулфат (4.8). Ако филтратът стане жълт след добавянето на железния сулфат, се добавя 3 ml от 15 mol/l ортофосфорна киселина (4.7).
Титрува се с бюрета излишният железен сулфат с 0,02 mol/l разтвор на калиев перманганат (4.9), докато сместа стане розова и цветът остане стабилен за една минута. Извършва се изпитване на празна проба при същите условия, като се пропуска само пробата.
Бележка:
Окисленият разтвор не трябва да влиза в контакт с гума.
8. Изразяване на резултатите
1 ml от 0,02 mol/l разтвор на калиевия перманганат съответства на 1,099 mg манган (Mn).
Процентът на мангана в тора се дава с:

където
|
|
xb е обемът в ml на перманганата, използван за празната проба; |
|
|
хs е обемът в ml на перманганата, използван за пробата за изпитване; |
|
|
V е обемът на екстракта, получен по метод 10.1. или 10.2, в ml; |
|
|
а е обемът в ml на разтвора на екстракта в съответствие с метод 10.1. и 10.2; |
|
|
М е масата в g на пробата за изпитване. |
Метод 10.10
Определяне на молибден в екстракти на торове с гравиметричен метод с 8-хидроксихинолин
1. Обхват
Настоящият документ дефинира процедура за определяне на молибден в екстракти на торове.
2. Област на приложение
Тази процедура е приложима за екстракти от проби на торове, получени с метод 10.1 и метод 10.2, за които с приложение I.Д към настоящия регламент се изисква деклариране на съдържанието на молибден.
3. Принцип
Нивото на молибден се определя чрез утаяване като молибденов оксинат при специфични условия.
4. Реактиви
4.1. Разтвор на сярна киселина, приблизително 1 mol/l
Внимателно се изливат 55 ml сярна киселина (H2SO4, d20 = 1,84 g/ml) в мерителна колба от 1 литър, съдържаща 800 ml вода. След охлаждане се долива до един литър. Смесва се.
4.2. Разреден амонячен разтвор (1:3)
Смесва се 1 обем концентриран разтвор на амоняк (NH4OH, d20 = 0,9 g/ml) с 3 обема вода.
4.3. Разреден разтвор на оцетна киселина (1:3)
Смесва се 1 обем концентрирана оцетна киселина (99,7 % CH3COOH, d20 = 1,049 g/ml) с 3 обема вода.
4.4. Разтвор на двунатриева сол в етилен диамен тетраоцетна киселина (EDTA)
Разтваря се 5 g от Na2EDTA във вода в мерителна колба от 100 ml. Долива се до марката и се смесва.
4.5. Буферен разтвор
В мерителна колба от 100 ml се разтваря 15 ml концентрирана оцетна киселина и 30 g амонячен ацетат във вода. Долива се до 100 ml.
4.6. Разтвор на 7- хидроксихинолин
В мерителна колба от 100 ml се разтваря 3 g 8-хидроксихинолин в 5 ml концентрирана оцетна киселина. Добавя се 80 ml вода. Добавя се амонячния разтвор (4.2) капка по капка, докато разтворът стане мътен, и след това се добавя оцетна киселина (4.3) — докато разтворът отново стане бистър.
Долива се до 100 ml вода.
5. Апаратура
5.1. Филтърен тигел Р 16/ISO 4793, порьозност 4, обем 30 ml
5.2. рН метър със стъклен електрод
5.3. Пещ за сушене при 130 до 135 °С
6. Приготвяне на разтвора за анализ
6.1. Приготвяне на молибденовия разтвор. Виж метод 10.1 и метод 10.2.
7. Процедура
7.1. Приготвяне на пробата за изпитване
Поставя се аликвотна част, съдържаща от 25 до 100 mg Мо в бехерова чаша от 250 ml. Долива се до 50 ml с вода.
Регулира се разтвора на рН 5 чрез добавяне на разтвор на сярна киселина (4.1) капка по капка. Добавя се 15 ml EDTA разтвор (4.4) и след това 5 ml от буферния разтвор (4.5). Долива се до около 80 ml с вода.
7.2. Получаване и измиване на утайката
Получаване на утайката
Загрява се леко разтворът. Като се бърка постоянно, се добавя разтворът на оксина (4.6). Утаяването продължава, докато спре образуването на отлагане. Добавя се още реактив, докато разтворът, плаващ отгоре, стане слабо жълт. Количество от 20 ml обикновено трябва да бъде достатъчно. Утайката продължава да се нагрява слабо в продължение на две до три минути.
Филтриране и измиване
Филтрира се през филтърен тигел (5.1). Измива се няколко пъти с 20 ml гореща вода. Измиващата вода трябва постепенно да става безцветна, което показва, че повече не присъства оксин.
7.3. Претегляне на утайката
Изсушава се утайката при 130 до 135 °С до постоянна маса (най-малко един час).
Оставя се да се охлади в сушилен шкаф и след това се претегля.
8. Изразяване на резултатите
1 mg молибденов оксинат MoO2(C9H6ON) съответства на 0,2305 mg Мо.
Процентът на молибдена в тора се дава с:

където
|
|
Х е масата в mg на утайката молибденовия оксинат; |
|
|
V е обемът в ml на разтвора на екстракта в съответствие с методи 10.1 или 10.2; |
|
|
а е обемът в ml на аликвота, взет от последното разреждане; |
|
|
D е коефициентът на разреждане на този аликвот; |
|
|
М е масата в g на пробата за изпитване. |
Метод 10.11
Определяне на цинк в екстракти на тор чрез атомна абсорбционна спектрометрия (ААС)
1. Обхват
Настоящият метод описва процедура за определяне на цинк в екстракти на торове.
2. Област на приложение
Процедурата е приложима за анализиране на проби на торове, извлечени по методи 10.1 и 10.2, за които с приложение I.Д към настоящия регламент се изисква деклариране на цинк.
3. Принцип
След подходящо третиране и разреждане на екстракта нивото на цинк се определя чрез атомна абсорбционна спектрометрия.
4. Реактиви
4.1. Разреден разтвор на солна киселина, около 6 mol/l:
Виж метод 10.4 (4.1).
4.2. Разреден разтвор на солна киселина, около 0,5 mol/l:
Виж метод 10.4 (4.2).
4.3 Разтвори на лантанови соли (10 g лантан на литър)
Виж метод 10.4 (4.3).
4.4 Калибровъчни разтвори на цинк
4.4.1. Основен разтвор на цинк (1 000 μg/ml)
В мерителна колба от 1 000 ml се разтваря 1 g цинк на прах, претеглен с точност до 0,1 mg, в 25 ml 6 mol/l солна киселина (4.1). Когато се разтвори напълно, се долива до обема с вода и се смесва старателно.
4.4.2. Работен разтвор на цинк (100 μg/ml)
Разрежда се 20 ml основен разтвор (4.4.1) в 0,5 mol/l разтвор на солна киселина (4.2) в мерителна колба от 200 ml. Долива се до обема с 0,5 mol/l разтвор на солна киселина и се смесва старателно.
5. Апаратура
Атомен абсорбционен спектрометър.
Виж метод 10.4 (5). Инструментът трябва да бъде снабден с източник на лъчи, характеристични за цинка (213,8 nm). Спектрометърът трябва да позволява коригиране на фона.
6. Приготвяне на разтвора за анализ
6.1. Разтвор на екстракт на цинк
Виж методи 10.1 и/или 10.2.
6.2. Приготвяне на разтвора за изпитване
Виж метод 10.4 (6.2). Разтворът за изпитване трябва да съдържа 10 % (v/v) разтвор на лантанова сол (4.3).
7. Процедура
7.1. Приготвяне на разтвор на празна проба
Виж метод 10.4 (7.1). Разтворът за празната проба трябва да съдържа 10 % (v/v) разтвор на лантанова сол, използван в 6.2.
7.2. Приготвяне на калибровъчни разтвори
Виж метод 10.4 (7.2). За оптимален диапазон на определяне от 0 до 5 μg/ml цинк се поставя 0, 0,5, 1, 2, 3, 4 и 5 ml съответно от работния разтвор (4.4.2) в серия мерителни колби от 100 ml. Ако е необходимо, се регулира концентрацията на солна киселина, така че да е колкото е възможно по-близо до тази на разтвор за изпитване. Добавя се във всяка колба 10 ml разтвор на лантанова сол, използвана в 6.2. Долива се до 100 ml с 0,5 mol/l разтвор на солна киселина (4.2) и се смесва старателно.
Тези разтвори съдържат съответно 0, 0,5, 1, 2, 3, 4 и 5 μg/ml цинк.
7.3. Определяне
Виж метод 10.4 (7.3). Приготвя се спектрометърът (5) за измерване на дължината на вълната 213,8 nm.
8. Изразяване на резултатите
Виж метод 10.4 (8).
Процентът на цинка в тора се дава с:
|
|
|
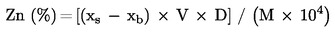
Ако се използва метод 9.3:
|
|
|

където
|
|
Zn е количеството на цинка, изразено като процент от тора; |
|
|
хs е концентрацията на разтвора за изпитване в μg/ml; |
|
|
xb е концентрацията на разтвора на празната проба в μg/ml; |
|
|
V е обемът на екстракта, получен метод 10.1. или 10.2, в ml; |
|
|
D е коефициент, съответстващ на разреждането, извършено в 6.2; |
|
|
М е масата на пробата за изпитване, взета в съответствие метод 10.1 или 10.2. |
Изчисляване на коефициента на разреждане D:
|
|
ако (а1), (а2), (а3,…, (аi) и (а) са аликвотните части и (v1), (v2), (v3),…, (vi) и (100) са обемите в ml, съответстващи на съответните разреждания, коефициентът на разреждане D ще бъде равен на:
|

(1) Когато полученото число е дроб, тя се закръгля до най-близкото цяло число.
(2) За опаковки, чието съдържание не надвишава 1 kg, единичната проба е съдържанието на един оригинален пакет.
(3) Всички бучки се раздробяват (ако е необходимо чрез отделянето им от пробата и връщане в пробата след това).
(4) Количеството на амонячен азот, съдържащ се в аликвотната част, взета съгласно таблица 1, ще бъде приблизително:
|
— |
0,05 g за вариант а, |
|
— |
0,10 g за вариант б, |
|
— |
0,20 g за вариант в. |
(5) Хладникът трябва да бъде регулиран така че да се осигури непрекъснат поток на кондензат. Дестилирането трябва да приключи за 30 до 40 минути.
(6) За целите на формулата за изчисляване на резултата:
|
— |
= |
50 или 35 |
= |
милиметри стандартен разтвор на сярна киселина, който се поставя в приемната колба, |
|
— |
= |
А |
= |
милиметри натриев или калиев хидроксид, използван за обратно титруване, |
|
— |
= |
F |
= |
коефициент включващ претегленото количество, разреждането, аликвотната част на разтвора на колбата за дестилиране и обемния еквивалент. |
(7) а) За целите на формулата за изчисляване на резултата:
|
— |
= |
50 или 35 |
= |
милиметри стандартен разтвор на сярна киселина за поставяне в събирателната колба, |
|
— |
= |
А |
= |
милиметри натриев или калиев хидроксид, използван за обратно титруване, |
|
— |
= |
F |
= |
коефициент включващ претегленото количество, разреждането, аликвотната част на разтвора на колбата за дестилиране и обемния еквивалент. |
(8) За целите на формулата за изчисляване на резултата:
|
— |
= |
50 или 35 |
= |
милиметри стандартен разтвор на сярна киселина за поставяне в събирателната колба, |
|
— |
= |
А |
= |
милиметри натриев или калиев хидроксид, използван за обратно титруване, |
|
— |
= |
F |
= |
коефициент включващ претегленото количество, разреждането, аликвотната част на разтвора на колбата за дестилиране и обемния еквивалент. |
(9) Биуретът може да бъде пречистен предварително чрез промиване в амонячен разтвор (10 %), след което с ацетон и се изсуши във вакуум.
(10) Виж точка 9 „Приложение“.
(11) Ако няма на разположение механична бъркалка, колбата може да се разклаща ръчно на всеки пет минути.
(12) Фосфор, разтворим в неорганични киселини, фосфор, разтворим във вода, фосфор, разтворим в разтвори на амониев цитрат, фосфор, разтворим в 2 % лимонена киселина, и фосфор, разтворим в 2 % мравчена киселина.
(13) 21 ml, когато разтворът за утаяване съдържа повече от 15 ml цитратен разтвор (неутрален цитрат, алкален цитрат на Петерман или Жоли.
(14) За утаяване на фосфатни разтвори, които съдържат повече от 15 ml цитратен разтвор (неутрален, Петерман или Жоли), които са били подкиселени с 21 ml концентрирана азотна киселина (вж. бележка под линия към 6.1), се използват 80 ml утаяващ реактив.
ПРИЛОЖЕНИЕ V
А. СПИСЪК НА ДОКУМЕНТИТЕ, С КОИТО ТРЯБВА ДА СЕ КОНСУЛТИРАТ ПРОИЗВОДИТЕЛИТЕ ИЛИ ТЕХНИТЕ ПРЕДСТАВИТЕЛИ, ЗА ДА СЪСТАВЯТ ТЕХНИЧЕСКО ДОСИЕ ЗА НОВ ТИП ТОР, КОЙТО ДА БЪДЕ ДОБАВЕН В ПРИЛОЖЕНИЕ І КЪМ НАСТОЯЩИЯ РЕГЛАМЕНТ
1. Насока за съставяне на техническо досие за кандидатстване за определяне на торове за „ЕО тор“.
Официален вестник на Европейските общности, С 138, 20.5.1994 г., стр. 4.
2. Директива 91/155/ЕИО на Комисията от 5 март 1991 г. относно дефиниране и определяне на подробни правила за системата за специфична информация, свързана с опасни препарати при прилагане на член 10 от Директива 88/379/ЕИО.
Официален вестник на Европейските общности, L 76/35, 22.3.1991 г., стр. 35.
3. Директива 93/112/ЕО на Комисията от 10 декември 1993 г. за изменение и на Директива 91/155/ЕИО на Комисията относно дефиниране и определяне на подробни правила за системата за специфична информация, свързана с опасни препарати при прилагане на член 10 от Директива 88/379/ЕИО.
Официален вестник на Европейските общности, L 314,16.12.1993 г., стр. 38.
Б. СТАНДАРТИ ЗА АКРЕДИТИРАНЕ НА ЛАБОРАТОРИИ, КОМПЕТЕНТНИ ДА ПРЕДОСТАВЯТ НЕОБХОДИМАТА УСЛУГА ЗА ПРОВЕРКА НА СЪОТВЕТСТВИЕТО НА ЕО ТОРОВЕ С ИЗИСКВАНИЯТА НА НАСТОЯЩИЯ РЕГЛАМЕНТ И НЕГОВИТЕ ПРИЛОЖЕНИЯ
1. Стандарт, приложим на нивото на лабораториите:
|
|
EN ISO/EIС 17025, Общи изисквания за компетенция на лаборатории за изпитване и калибриране. |
2. Стандарт, приложим на ниво акредитиращи органи:
|
|
EN 45003, Система за акредитиране на лаборатории за изпитване и калибриране, общи изисквания за работа и признаване. |






