|
3.
|
Se adaugă următoarele capitole:
„B.49. TESTUL IN VITRO DE MICRONUCLEE PE CELULE DE MAMIFERE
INTRODUCERE
|
1.
|
Testul in vitro de micronuclee (MNvit) este un test de genotoxicitate pentru identificarea micronucleelor (MN) în citoplasma celulelor în interfază. Micronucleele pot avea ca origine fragmente de cromozomi acentrici (și anume, fără centrometru) sau cromozomi integrali incapabili să migreze către polii celulei în timpul etapei de anafază a divizării celulare. Testul detectează activitatea substanțelor chimice clastogene și aneugene (substanțe și amestecuri) (1) (2) în celulele care au efectuat diviziune celulară în timpul sau după expunerea la substanța de test. Această metodă de testare (MT) autorizează utilizarea de protocoale incluzând sau nu citoclasina B (citoB), un inhibitor de polimerizare a actinei. Adăugarea de citoB anterior mitozei vizate permite identificarea și analiza selectivă a frecvenței micronucleelor în celulele care au încheiat procesul de mitoză, aceste celule fiind binucleate (3) (4). Prezenta MT autorizează, de asemenea, utilizarea de protocoale fără blocarea citocinezei, cu condiția existenței posibilității de a demonstra faptul că populația celulară analizată a efectuat mitoza.
|
|
2.
|
Pe lângă faptul că permite identificarea substanțelor chimice (substanțe și amestecuri) care induc formarea de micronuclee, testul MNvit, asociat blocajului citocinezei, marcării imunochimice a kinetocorilor sau hibridizării cu sonde centrometrice/telometrice [hibridizare fluorescentă in situ (FISH)], poate de asemenea, să furnizeze informații cu privire la mecanismele care stau la originea leziunilor cromozomiale și a formării micronucleelor (5) (6) (7) (8) (9) (10) (11) (12) (13) (14) (15) (16). Aceste proceduri de marcare și de hibridizare pot fi utilizate atunci când se constată o creștere a formării de micronuclee, iar experimentatorul dorește să stabilească dacă respectiva creștere este rezultatul unor evenimente clastogenice și/sau aneugene.
|
|
3.
|
Micronucleele reprezintă leziunile transmise celulelor-fiice, în timp ce aberațiile cromozomiale observate în celulele aflate în metafază pot să nu fie transmise. Deoarece micronucleele prezente în celulele în interfază pot fi evaluate în mod relativ obiectiv, personalul laboratorului trebuie doar să determine dacă celulele au efectuat mitoza și să identifice numărul de celule care conțin un micronucleu. Prin urmare, preparatele pot fi examinate relativ rapid, iar analiza poate fi automatizată. Acest lucru permite numărarea a mii în loc de sute de celule pentru fiecare tratament, mărind astfel puterea testului. În sfârșit, dat fiind faptul că micronucleele pot să provină de la cromozomi întârziați, testul poate permite detectarea agenților de inducție aneuploidică, dificil de studiat în cadrul testelor de aberații cromozomiale clasice, de exemplu orientarea OCDE de testare nr. 473 (capitolul B.10 din prezenta anexă) (17). Cu toate acestea, testul MNvit nu permite diferențierea substanțelor chimice care induc poliploidie de cele care induc clastogenicitate fără utilizarea de tehnici speciale precum metoda FISH descrisă la puncul 2.
|
|
4.
|
Testul MNvit este o metodă in vitro care utilizează în general culturi de celule umane sau de la rozătoare. Acesta constituie un instrument de bază complet pentru investigarea in vitro a potențialului de leziuni cromozomiale datorită capacității acestuia de evidențiere a agenților aneugeni și clastogeni.
|
|
5.
|
Testul MNvit este fiabil și eficient pe o diversitate de tipuri de celule în prezența sau absența citoB. Validitatea testului MNvit este atestată de numeroase date obținute utilizându-se diferite linii celulare provenite de la rozătoare (CHO, V79, CHL/IU și L5178Y) și limfocite umane (18) (19) (20) (21) (22) (23) (24) (25) (26) (27) (28) (29) (30) (31). Acestea includ, în special, studii internaționale de validare coordonate de Société Française de Toxicologie Génétique (SFTG) (18) (19) (20) (21) (22) și rapoartele elaborate de International Workshop on Genotoxicity Testing (4) (16). În plus, datele disponibile au făcut obiectul unei noi evaluări în cadrul unui studiu de validare retrospectiv fondat pe forța probatorie a datelor, efectuat de Centrul European pentru validarea metodelor alternative (ECVAM) al Comisiei Europene, iar metoda de testare a fost declarată valabilă din punct de vedere științific de către Comitetul științific consultativ al ECVAM (ESAC) (32) (33) (34). Utilizarea unei linii celulare limfoblastoide umane TK6 (35), de celule HepG2 (36) (37) și de celule embrionare primare de hamster sirian (38) a fost descrisă, fără a fi însă utilizată în studiile de validare.
|
DEFINIȚII
|
6.
|
Definițiile utilizate sunt furnizate în apendicele 1.
|
CONSIDERAȚII INIȚIALE
|
7.
|
Testele efectuate in vitro necesită, în general, o sursă exogenă de activare metabolică, cu excepția cazului în care celulele utilizate sunt compatibile din punct de vedere metabolic cu substanțele testate. Sistemele de activare metabolică de acest tip nu reproduc integral condițiile in vivo. Ar trebui să se acorde atenție evitării condițiilor care ar conduce la rezultate pozitive false care nu reflectă mutagenitatea intrinsecă și care pot proveni de la factori precum modificările importante de pH, de osmolalitate sau din valori mari ale citotoxicității (39) (40) (41). În cazul în care substanța de test cauzează o modificare a pH-ului mediului în timpul adăugării sale, pH-ul trebuie ajustat, de preferat tamponând soluția-mamă astfel încât toate volumele să rămână identice pentru toate concentrațiile testate și pentru toate substanțele martor.
|
|
8.
|
Pentru a analiza inducția micronucleelor, este esențial ca mitoza să se fi produs atât în culturile tratate, cât și la cele netratate. Cea mai prolifică etapă în materie de date în ceea ce privește numărarea micronucleelor este în celulele care au finalizat o mitoză în timpul sau după tratamentul cu substanța de test.
|
PRINCIPIUL TESTULUI
|
9.
|
Culturile celulare de origine umană sau mamiferă sunt expuse la substanța de test în prezența și în absența unei surse exogene de activare metabolică, cu excepția cazului în care sunt utilizate celule dotate cu capacitate metabolică adecvată. Toate testele includ utilizarea concomitentă de solvenți/vehiculi (VC) și substanțe chimice martor pozitive (PC).
|
|
10.
|
În timpul sau după expunerea la substanța de test, celulele se plasează în culturi pentru o perioadă suficientă pentru a permite leziunii cromozomiale sau a fusului să conducă la formarea de micronuclee în celulele aflate în interfază. Pentru inducția aneuploidiei, substanța de test trebuie în general să fie prezentă în timpul producerii mitozei. Celulele recoltate în interfază sunt colorate și analizate pentru a detecta prezența micronucleelor. În principiu, numărarea micronucleelor trebuie realizată în celulele care au efectuat o mitoză completă în timpul expunerii la substanța de test sau în timpul perioadei de post-expunere, dacă aceasta este prevăzută în cadrul testului. În culturile care au fost tratate cu un agent de blocare a citocinezei, numărarea se efectuează doar pe celulele binucleate. În absența unui agent de blocare a citocinezei, este important să se demonstreze că celulele analizate au efectuat cel mai probabil diviziunea celulară în timpul sau după expunerea la substanța de test. Pentru toate protocoalele, este important să se demonstreze că proliferarea celulară a avut loc atât în culturile tratate, cât și în cele martor, iar gradul de citotoxicitate sau citostază indus de substanța de test este evaluat pentru toate culturile (sau culturile paralele) în care se realizează numărarea micronucleelor.
|
DESCRIEREA TESTULUI
Pregătiri
|
11.
|
În cadrul testului pot fi utilizate limfocite primare din sânge periferic uman în cultură (5) (19) (42) (43) și mai multe linii celulare provenite de la rozătoare precum CHO, V79, CHL/IU, și L5178Y (18) (19) (20) (21) (22) (25) (26) (27) (28) (30). Utilizarea de alte linii celulare sau alte tipuri de celule trebuie justificată pe baza performanței demonstrate a acestora în cadrul testului, astfel cum este indicat în secțiunea referitoare la criteriile de acceptabilitate. Deoarece frecvența obișnuită a micronucleelor va influența sensibilitatea testului, se recomandă utilizarea unor tipuri de celule cu o frecvență obișnuită de formare a nucleelor slabă și stabilă.
|
|
12.
|
Limfocitele din sângele periferic uman utilizate sunt obținute de la persoane tinere (aproximativ 18-35 de ani), sănătoase, nefumătoare, fără expuneri recente cunoscute la substanțe chimice sau radiații genotoxice. Dacă celulele provenite de la mai mulți donatori sunt puse în comun în vederea utilizării, trebuie precizat numărul de donatori. Frecvența micronucleelor crește odată cu vârsta, iar această tendință este mai marcantă la femei decât la bărbați (44), aspect care trebuie luat în considerare în momentul selectării donatorilor pentru o punere în comun a celulelor.
|
Medii și condiții de cultură
|
13.
|
Trebuie utilizate un mediu de cultură și condiții de incubare (recipiente de cultură, concentrație de CO2, temperatură și umiditate) adecvate pentru menținerea culturilor. Liniile celulare stabilite și sușele celulare trebuie verificate regulat pentru a verifica stabilitatea cariotipului și absența contaminării cu micoplasme și nu trebuie utilizate dacă au fost contaminate sau în cazul în care cariotipul s-a modificat. Durata normală a ciclului celular în condiții de cultură aplicate într-un laborator de testare trebuie cunoscută. În cazul în care se utilizează metoda de blocare a citocinezei, concentrația agentului inhibitor al citocinezei se adaptează la tipul de celulă utilizat și s-a dovedit capacitatea acestuia de a produce suficiente celule binucleate pentru a se putea proceda la numărarea acestora.
|
Pregătirea culturilor
|
14.
|
Linii celulare stabilite și sușe celulare: celulele sunt multiplicate plecând de la culturile-mamă, plasate într-un mediu de cultură la o densitate astfel încât culturile să nu acopere complet suprafața mediului, iar culturile în suspensie să nu atingă o densitate excesivă înainte de recoltarea acestora, și sunt incubate la 37 °C.
|
|
15.
|
Limfocite: sânge integral tratat cu un anticoagulant (de exemplu, heparină) sau limfocite separate sunt plasate în cultură în prezența unui mitogen, de exemplu fitohemaglutinină (PHA), anterior expunerii la substanța de test sau la citoB.
|
Activare metabolică
|
16.
|
Recurgerea la un sistem de activare metabolică exogen este necesară în cazul utilizării de celule dotate cu o capacitate metabolică endogenă inadecvată. Sistemul utilizat cel mai frecvent este o fracție postmitocondrială îmbogățită cu un cofactor (S9) preparată din ficat provenit de la rozătoare tratat cu agenți de inducție enzimatică, de exemplu Aroclor 1254 (45) (46) sau un amestec de fenobarbitonă și β-naftoflavonă (46) (47) (48) (49). Utilizarea acestui amestec nu este contrară prevederilor Convenției de la Stockholm privind poluanții organici persistenți (50) și nici Regulamentului (CE) nr. 850/2004 privind poluanții organici persistenți (66) și s-a dovedit a fi la fel de eficientă ca și Aroclor 1254 pentru inducția de oxidaze cu funcție mixtă (46) (47) (48) (49). Fracția S9 este în general utilizată cu o concentrație cuprinsă între 1-10 % (v/v) în mediul de test final. Alegerea unui sistem de activare metabolică poate depinde de clasa substanței chimice, iar în unele cazuri poate fi mai adecvată utilizarea mai multor concentrații ale fracției S9.
|
|
17.
|
Liniile celulare modificate genetic pentru a produce enzime de activare specifice, umane sau de rozătoare, pot elimina necesitatea utilizării unui sistem de activare metabolică exogen și pot fi utilizate ca celule de test. În astfel de cazuri, alegerea liniilor celulare utilizate trebuie justificată științific, de exemplu prin relevanța oxidazelor cu funcție mixtă pentru metabolismul substanței de test (51) și reactivitatea acestora la substanțele clastogene și aneugene cunoscute (a se vedea secțiunea separată referitoare la criteriile de acceptabilitate). Trebuie recunoscut faptul că substanța testată poate să nu fie metabolizată de oxidaza (oxidazele) cu funcție mixtă exprimate; în acest caz, rezultatele negative nu ar indica faptul că substanța de test nu poate induce micronuclee.
|
Prepararea substanței de test
|
18.
|
Substanțele chimice solide sunt dizolvate într-un vehicul sau în solvenți adecvați și diluate, dacă este cazul, anterior tratamentului aplicat celulelor. Substanțele chimice lichide pot fi adăugate direct sistemelor de testare și/sau diluate înaintea tratamentului. Gazele sau substanțele volatile necesită o modificare adecvată a protocoalelor standard, de exemplu utilizarea de recipiente închise ermetic (52) (53). Trebuie utilizate preparate proaspete ale substanței de test, cu excepția cazului când datele reținute demonstrează stabilitatea preparatelor în condiții de stocare.
|
Condiții de testare
Solvenți/vehicul
|
19.
|
Solventul/vehiculul nu trebuie să reacționeze cu substanța de test și nici să altereze supraviețuirea celulelor sau menținerea activității S9 la concentrația utilizată. Dacă sunt utilizați alți solvenți/vehicule decât cei bine stabiliți (de exemplu, apă, mediu de cultură celulară, dimetilsulfoxid), utilizarea acestora trebuie justificată prin date care să indice compatibilitatea acestora cu substanța de test, precum și absența genotoxicității. Se recomandă ca, în măsura în care este posibil, să se ia în considerare în primă instanță utilizarea unui solvent/vehicul apos.
|
Utilizarea de citoB ca agent de blocare a citocinezei
|
20.
|
Unul dintre cele mai importante aspecte în efectuarea testului MNvit este garantarea faptului că celulele numărate au încheiat mitoza în timpul tratamentului sau în timpul perioadei de incubare posttratament, dacă aceasta este prevăzută în cadrul metodei. CitoB este agentul folosit cel mai frecvent pentru blocarea citocinezei, deoarece inhibă asamblarea actinei împiedicând astfel separarea celulelor-fiice după mitoză, conducând la formarea de celule binucleate (5) (54) (55). Prin urmare, numărarea micronucleelor poate fi limitată la celulele care au fost supuse procesului de mitoză în timpul sau după tratament. Efectul substanței de test asupra cineticii proliferării celulare poate fi măsurat concomitent. CitoB trebuie utilizat ca agent de blocare a citocinezei atunci când sunt utilizate limfocite umane, deoarece durata ciclului celular este variabilă în funcție de culturi și donatori și pentru că nu toate limfocitele reacționează la PHA. Au fost utilizate și alte metode pentru a determina dacă celulele liniilor celulare utilizate și care fac obiectul unei numerotări au efectuat o mitoză; metodele respective sunt detaliate mai jos (a se vedea puncul 26).
|
|
21.
|
Pentru fiecare tip de celulă, laboratorul determină concentrația adecvată de citoB pentru a obține frecvența optimală de celule binucleate în culturile martor tratate cu solvent/vehicul. Concentrația adecvată de citoB este, în general, cuprinsă între 3 și 6 μg/ml.
|
Măsurarea proliferării celulare și a citotoxicității și alegerea concentrațiilor de expunere
|
22.
|
În momentul determinării celei mai mari concentrații a substanței de test, trebuie evitate concentrațiile susceptibile să producă false răspunsuri pozitive, precum cele care produc citotoxicitate excesivă, un precipitat în mediul de cultură și modificări importante de pH sau osmolalitate (39) (40) (41).
|
|
23.
|
Măsurătorile proliferării celulare se efectuează pentru a asigura că celulele tratate au efectuat o mitoză în timpul testului și că tratamentele sunt realizate la niveluri adecvate de citotoxicitate (a se vedea punctul 29). Citotoxicitatea se determină cu sau fără activare metabolică în celulele care nu necesită activare metabolică cu ajutorul creșterii relative a numărului de celule (RICC) sau dublarea relativă a populației (RPD) (a se vedea formulele din apendicele 2) cu excepția situației când se utilizează citoB, caz în care citotoxicitatea poate fi determinată cu ajutorul indicelui de replicare (IR) (a se vedea formulele din apendicele 2).
|
|
24.
|
Tratarea culturilor cu citoB și măsurarea frecvențelor relative ale celulelor mononucleate, binucleate și multinucleate în fiecare cultură furnizează o metodă precisă de cuantificare a efectului unui tratament asupra proliferării celulare și a activității citotoxice sau citostatice (5) și asigură că numai celule care s-au divizat în timpul tratamentului sau după acesta sunt luate în calcul.
|
|
25.
|
În studiile cu citoB, citostaza/citotoxicitatea poate fi cuantificată cu ajutorul indicelui de proliferare a celulelor a căror citocineză a fost blocată (CBPI) (5) (26) (56) sau poate fi derivată din indicele de replicație pornind de la cel puțin 500 de celule pentru fiecare cultură (a se vedea formulele din apendicele 2). Atunci când citoB este utilizată pentru a evalua proliferarea celulară, un CBPI sau RI trebuie determinat având la bază cel puțin 500 de celule pentru fiecare cultură. Aceste măsuri pot servi, printre altele, la estimarea citotoxicității prin compararea valorilor obținute pentru culturile tratate și cele martor. Evaluarea markerilor de citotoxicitate (de exemplu, grad de confluență, număr de celule, apoptoză, necroză, numărare de celule în metafază) pot furniza informații utile.
|
|
26.
|
În studiile fără citoB, este necesar să se demonstreze că celulele numărate dintr-o cultură au realizat diviziunea în timpul sau ulterior tratamentului cu substanța de test, în caz contrar existând riscul de producere a unor răspunsuri fals negative. Metodele care au fost utilizate pentru a garanta că celulele divizate sunt cele numărate includ incorporarea și ulterior detectarea bromodeoxiuridinei (BrdU) pentru identificarea celulelor replicate (57), formarea de clone în momentul în care celule provenite de la linii celulare permanente sunt tratate și numărate in situ pe o lamă de microscop [indice de proliferare (PI)] (25) (26) (27) (28) sau măsurarea dublării relative a populației (RPD) sau creșterea relativă a numărului de celule (RICC) sau alte metode dovedite (16) (56) (58) (59) (a se vedea formulele din anexa 2). Evaluarea altor markeri de citotoxitate sau citostază (de exemplu, grad de confluență, număr de celule, apoptoză, necroză, numărare de celule în metafază) poate furniza informații utile.
|
|
27.
|
Trebuie evaluate cel puțin trei concentrații analizabile. În acest scop, poate fi necesară efectuarea experimentului cu ajutorul unui număr mai mare de concentrații la distanță apropiată și analizarea formării micronucleelor în concentrațiile care prezintă o plajă de citotoxicități adecvate. O strategie alternativă este reprezentată de realizarea unui test de citotoxicitate preliminar pentru a restrânge spectrul testului definitiv.
|
|
28.
|
Cea mai mare concentrație trebuie să vizeze producerea unui nivel de toxicitate 55 ± 5 %. Niveluri mai mari pot induce leziuni cromozomiale ca efect secundar al citotoxicității (60). În prezența citotoxicității, concentrațiile de test selectate trebuie să acopere un interval cuprins între citotoxicitate zero sau ușoară și o citotoxicitate de 55 ± 5 %.
|
|
29.
|
Atunci când nu se observă nicio citotoxicitate sau precipitat, cea mai mare concentrație de test trebuie să corespundă celei mai mici concentrații dintre următoarele: 0,01 M, 5 mg/mL sau 5 μl/mL. Concentrațiile selectate pentru analiză sunt, în general, separate de un factor de maxim 10. Pentru substanțele de test care prezintă o curbă concentrație-răspuns caracterizată de o pantă puternică, poate fi necesară reducerea factorului de separare a concentrațiilor substanței de test astfel încât culturile aflate în plajele de toxicitate inferioare sau medii să fie, de asemenea, analizate.
|
|
30.
|
Atunci când solubilitatea este un factor de limitare, concentrația maximă, dacă aceasta nu este limitată de citotoxicitate, corespunde concentrației maxime care permite observarea unei precipitații minime în condiții de cultură, cu condiția ca aceasta să nu interfereze cu numărarea. Evaluarea precipitării trebuie să se facă prin metode precum microscopia optică, notând precipitatul persistent sau care apare în timpul perioadei de cultură (până la sfârșitul tratamentului).
|
Martori
|
31.
|
Martori concomitenți pozitivi și tratați cu solvent/vehicul, cu sau fără activare metabolică, trebuie incluși în fiecare experiment.
|
|
32.
|
Martorii pozitivi sunt necesari pentru a demonstra că celulele utilizate și protocolul de testare permit identificarea substanțelor clastogene și aneugene și pentru a dovedi capacitatea metabolică a preparatului S9. Martorii pozitivi trebuie să facă apel la inductori cunoscuți de formare de micronuclee la concentrații susceptibile să determine o creștere ușoară sau reproductibilă a frecvenței de fond și să demonstreze sensibilitatea sistemului de testare. Concentrațiile martorilor pozitivi trebuie alese astfel încât efectele să fie clare, dar să nu dezvăluie imediat identitatea lamelelor codate celui care le interpretează.
|
|
33.
|
Un clastogen care necesită activare metabolică (de exemplu, ciclofosfamidă; benzo[a]piren) trebuie utilizat pentru a demonstra atât competența metabolică, cât și abilitatea sistemului de testare de detectare a clastogenelor. Alte substanțe martor pozitive pot fi utilizate, dacă acest lucru este justificat. Deoarece unele substanțe martor pozitive care necesită activare metabolică pot fi active fără activare metabolică exogenă în anumite condiții de tratament sau la anumite linii celulare, necesitatea activării metabolice, precum și activitatea preparării S9 trebuie testată pe linia celulară selectată și la concentrațiile selectate.
|
|
34.
|
În prezent, niciun aneugen cunoscut nu necesită activare metabolică pentru a prezenta activitate genotoxică (16). Martorii pozitivi acceptați în prezent pentru activitatea aneugenică sunt, de exemplu, colchicina și vinblastina. Pot fi utilizate și alte substanțe chimice dacă acestea induc micronuclee doar sau în principal prin intermediul activității aneugene. Pentru a evita necesitatea a două substanțe martor pozitive (pentru clastogenitate și aneugenitate) fără activare metabolică, martorul pentru aneugenitate poate fi utilizat ca martor pozitiv fără S9, iar martorul pentru clastogenitate poate fi utilizat pentru a testa caracterul adecvat al sistemului de activare metabolică utilizat. Martorii pozitivi atât pentru clastogenitate, cât și pentru aneugenitate se utilizează pentru celulele care nu necesită S9. Martorii pozitivi sugerați sunt indicați în apendicele 3.
|
|
35.
|
Utilizarea de substanțe martor pozitive aparținând aceleiași clase chimice poate fi luată în considerare dacă sunt disponibile substanțe chimice adecvate. Toate substanțele martor pozitive trebuie să fie adaptate tipului de celulă în cauză și condițiilor de activare.
|
|
36.
|
Substanțe martor tratate cu solvent/vehicul trebuie incluse la momentul fiecărei recolte. În plus, substanțe martor negative netratate (fără solvent/vehicul) trebuie, de asemenea, utilizate cu excepția cazului când datele publicate sau obținute în urma testelor martor realizate anterior de către laborator demonstrează că solventul ales nu induce aciune genotoxică sau alte efecte nocive la concentrațiile utilizate.
|
PROCEDURA DE TESTARE
Schema de tratament
|
37.
|
Pentru a maximiza probabilitatea detectării unei acțiuni aneugene sau clastogene într-un stadiu specific al ciclului celular, este important ca un număr suficient de celule să fie tratate cu substanța de test în toate stadiile ciclului celular. Schema de tratament pentru liniile celulare și culturile de celule primare poate, prin urmare, să difere într-o oarecare măsură de cea pentru limfocite, care necesită o stimulare mitogenică pentru a demara ciclul celular, și care sunt detaliate la punctele 41-43 (16).
|
|
38.
|
Considerațiile teoretice și datele publicate (18) indică faptul că majoritatea substanțelor aneugene și clastogene sunt detectate în urma unui tratament de scurtă durată, de 3 până la 6 ore, în prezența și în absența preparatului S9, urmat de îndepărtarea substanței de test și o perioadă de creștere echivalentă cu 1,5-2,0 cicluri celulare (6). Eșantioanele de celule sunt prelevate după un timp echivalent cu aproximativ 1,5-2,0 ori durata unui ciclu celular normal (și anume netratat) fie după începutul tratamentului, fie la sfârșitul acestuia (a se vedea tabelul 1). Durata eșantionării sau a recuperării poate fi extinsă dacă se cunoaște sau se presupune că substanța de test afectează durata ciclului celular (de exemplu, dacă se testează nucleotide analoage).
|
|
39.
|
Din cauza citotoxicității potențiale a preparatelor S9 pentru celulele de mamifere cultivate, tratamentul de expunere prelungită echivalent cu 1,5-2,0 cicluri celulare normale este utilizat doar în absența preparatului S9. În tratamentul extins, sunt posibile diferite opțiuni pentru a permite tratarea celulelor cu substanța de test în absența sau în prezența citoB. Aceste opțiuni vizează situațiile în care pot exista preocupări referitoare la interacțiunile posibile dintre substanța de test și citoB.
|
|
40.
|
Tabelul 1 prezintă diferitele scheme de tratament propuse. Schemele de tratament general pot fi modificate în funcție de stabilitatea sau de reactivitatea substanței de test sau de caracteristicile de creștere ale celulelor utilizate. Toate tratamentele trebuie să înceapă și să se încheie în timpul etapei de creștere exponențială a celulelor. Aceste scheme sunt prezentate mai detaliat la punctele 41-47 mai jos.
Tabelul 1
Tratamentul celulelor și timpii de recoltă pentru testul MNvit
|
Limfocite, celule primare și linii celulare tratate cu citoB
|
+ S9
|
Tratare timp de 3-6 ore în prezența S9;
înlăturarea S9 și a mediului de tratare;
adăugarea unui mediu proaspăt și a citoB;
recoltarea după 1,5-2,0 cicluri celulare normale
|
|
– S9
Expunere scurtă
|
Tratare timp de 3-6 ore;
înlăturarea mediului de tratare;
adăugarea unui mediu proaspăt și a citoB;
recoltarea după 1,5-2,0 cicluri celulare normale.
|
|
– S9
Expunere prelungită
|
Opțiunea A: Tratare timp de 1,5-2 cicluri normale celulare în prezența citoB;
recoltare la sfârșitul perioadei de expunere.
Opțiunea B: Tratare timp de 1,5-2,0 cicluri celulare normale;
înlăturarea substanței de test;
adăugarea unui mediu proaspăt și a citoB;
recoltarea după 1,5-2,0 cicluri celulare normale.
|
|
Linii celulare tratate fără citoB
(schemă de tratament identică cu schemele de tratament descrise mai sus, cu excepția faptului că nu se adaugă citoB)
|
|
Limfocite, celule primare și linii celulare cu citoB
|
41.
|
În cazul limfocitelor, cea mai eficientă metodă constă în demararea expunerii la substanța de test la 44-48 de ore după stimularea cu PHA, odată dispărută sincronizarea ciclului celular (5). În timpul testului inițial, celulele sunt tratate timp de 3-6 ore cu substanța de test în absența și în prezența S9. Mediul de tratament este apoi eliminat și înlocuit cu un mediu proaspăt conținând citoB, iar celulele sunt recoltate după 1,5-2,0 cicluri celulare normale.
|
|
42.
|
Dacă rezultatele ambelor teste inițiale de tratament scurt (3-6 ore) sunt negative sau echivoce, se utilizează un alt tratament cu o expunere extinsă fără S9. Două opțiuni de tratament, ambele acceptabile, sunt disponibile. Cu toate acestea, ar putea fi mai adecvată utilizarea opțiunii A pentru limfocitele stimulate a căror creștere exponențială poate să intre în declin la 96 de ore după stimulare. De altfel, culturile de celule nu trebuie să acopere în întregime suprafața mediului înainte de eșantionarea finală în cadrul opțiunii B.
|
—
|
Opțiunea A: Celulele sunt tratate cu substanța de test timp de 1,5-2,0 cicluri celulare normale și recoltate la sfârșitul perioadei de tratament.
|
|
—
|
Opțiunea B: Celulele sunt tratate cu substanța de test timp de 1,5-2,0 cicluri celulare normale. Mediul de tratament este înlăturat și înlocuit cu un mediu proaspăt iar celulele sunt recoltate după alte 1,5 -2,0 cicluri celulare normale.
|
|
|
43.
|
Tratamentul celulelor primare și al liniilor celulare este similar cu cel aplicat limfocitelor, cu excepția faptului că stimularea acestora cu PHA timp de 44-48 de ore nu este necesară. Celulele, altele decât limfocitele, trebuie expuse astfel încât, la încheierea studiului, celulele să se afle încă în etapa de creștere exponențială.
|
Linii celulare fără citoB
|
44.
|
Celulele trebuie tratate timp de 3-6 ore în prezența și în absența S9. Mediul de tratament este înlăturat și înlocuit cu un mediu proaspăt, iar celulele sunt recoltate după 1,5-2,0 cicluri celulare normale.
|
|
45.
|
Dacă rezultatele ambelor teste inițiale de tratament scurt (3-6 ore) sunt negative sau echivoce, se utilizează un alt tratament cu o expunere extinsă (fără S9). Două opțiuni de tratament, ambele acceptabile, sunt disponibile:
|
—
|
Opțiunea A: Celulele sunt tratate cu substanța de test timp de 1,5-2,0 cicluri celulare normale și recoltate la sfârșitul perioadei de tratament.
|
|
—
|
Opțiunea B: Celulele sunt tratate cu substanța de test timp de 1,5-2,0 cicluri celulare normale. Mediul de tratament este înlăturat și înlocuit cu un mediu proaspăt, iar celulele sunt recoltate după alte 1,5-2,0 cicluri celulare normale.
|
|
|
46.
|
În culturile monostrat, pot fi prezente celule mitotice (recunoscute după forma rotundă a acestora și după faptul că se detașează de pe suprafață) la sfârșitul perioadei de tratament de 3-6 ore. Deoarece celulele mitotice se detașează ușor, acestea pot fi pierdute în momentul înlăturării mediului conținând substanța de test. Pentru a evita pierderea de celule în mitoză susceptibile să formeze micronuclee, trebuie acordată o atenție deosebită colectării cu grijă a acestora în momentul spălării culturilor și apoi reintroducerii acestora în culturi la momentul recoltei.
|
Număr de culturi
|
47.
|
Trebuie utilizate culturi duplicat pentru fiecare concentrație a substanței de test, precum și pentru culturile de vehicul/solvent și de martori negativi. În cazul în care rezultatele anterioare de laborator au demonstrat că variația între culturile duplicat este minimă, poate fi utilizată o singură cultură pentru fiecare concentrație. În cazul utilizării de culturi unice, se recomandă analizarea unui număr mai mare de concentrații.
|
Recoltarea celulelor și pregătirea lamelelor
|
48.
|
Fiecare cultură este recoltată și prelucrată separat. Prepararea celulelor poate implica tratament hipotonic, dar această etapă nu este necesară dacă celulele sunt răspândite în mod corespunzător în alt mod. Pot fi folosite diferite tehnici pentru prepararea lamelelor, cu condiția ca preparatele celulare obținute pentru numărare să fie de foarte bună calitate. Citoplasma celulară este conservată pentru a permite detectarea de eventuale nuclee și (în cadrul metodei blocării citocinezei) identificarea fiabilă a celulelor binucleate.
|
|
49.
|
Lamelele pot fi colorate prin metode diferite precum Giemsa sau coloranți fluorescenți specifici ai ADN (59). Utilizarea unui colorant specific pentru ADN [de exemplu, portocaliu de acridină (61) sau Hoechst 33258 plus pironină-Y (62)] poate elimina o parte dintre artefactele asociate utilizării unui colorant nespecific al ADN. Anticorpii kinetocori, metoda FISH cu sonde ADN pancentrometrice sau marcarea in situ prin amorsare cu ajutorul amorselor pancentrometrice, împreună cu un colorant de contrast al ADN adecvat, pot fi utilizați pentru a identifica conținutul (cromozomi/fragmente cromozomiale) micronucleelor dacă informațiile de ordin mecanic cu privire la formarea acestora prezintă interes (15)(16). Alte metode de diferențiere a efectelor clastogene și aneugene pot fi utilizate, cu condiția ca eficacitatea acestora să fi fost demonstrată.
|
Analiză
|
50.
|
Toate lamelele, inclusiv cele cu solvent/vehicul și cu substanțele martor, trebuie codate individual înainte de analiza la microscop. De asemenea, eșantioanele codate pot fi analizate cu ajutorul unui sistem automat validat de citometrie în flux sau de analiză a imaginilor.
|
|
51.
|
În culturile tratate cu citoB, frecvența micronucleelor se analizează în cel puțin 2 000 de celule binucleate pe concentrație (cel puțin 1 000 de celule binucleate pe cultură; două culturi pe concentrație). În cazul culturilor unice, trebuie analizate cel puțin 2 000 de celule binucleate pe concentrație din culturile respective. Dacă numărul de celule binucleate disponibile într-o cultură pentru fiecare concentrație este considerabil mai mic de 1 000 sau de 2 000 în cazul culturilor unice și dacă nu este detectată nicio creștere semnificativă a numărului de micronuclee, testul trebuie reluat pe un număr mai mare de celule sau la concentrații mai puțin toxice, în funcție de situație. Se recomandă precauție pentru a nu număra celulele binucleate cu formă neregulată sau ale căror nuclee prezintă o diferență de mărime considerabilă; de asemenea, celulele binucleate nu trebuie confundate cu celulele multinucleate răspândite neuniform. Celulele care conțin mai mult de două nuclee principale nu sunt analizate deoarece frecvența de referință a micronucleelor poate fi mai mare în acest tip de celule (63) (64). Numărarea celulelor mononucleate este acceptabilă dacă substanța de test interferează cu activitatea citoB.
|
|
52.
|
Pentru liniile celulare testate fără citoB, micronucleele trebuie analizate pentru cel puțin 2 000 de celule pe concentrație (cel puțin 1 000 de celule pe cultură; două culturi pe concentrație). Dacă se utilizează o singură cultură pe concentrație, sunt examinate cel puțin 2 000 de celule din cultura respectivă.
|
|
53.
|
Dacă se utilizează citoB, trebuie determinat un CBPI sau un RI pentru a evalua proliferarea celulară (a se vedea apendicele 2) utilizând cel puțin 500 de celule pe cultură. Atunci când tratamentele sunt efectuate în absența citoB, este esențial să se facă dovada că celulele analizate au făcut obiectul unei proliferări, conform punctelor 24-27.
|
Criterii de acceptabilitate
|
54.
|
Orice laborator care propune utilizarea testului MNvit descris în prezenta MT trebuie să demonstreze capacitatea sa de a detecta în mod fiabil și precis substanțele chimice a căror activitate aneugenă și clastogenă este cunoscută, cu sau fără activare metabolică, precum și a substanțelor chimice negative, utilizând substanțele chimice de referință din apendicele 3. Pentru a atesta capacitatea sa de a efectua corect prezenta MT, laboratorul trebuie să demonstreze că celulele ale căror micronuclee au fost numărate au efectuat o diviziune celulară dacă testul este realizat fără utilizare de citoB.
|
|
55.
|
Substanțele chimice din apendicele 3 sunt recomandate pentru utilizare ca substanțe chimice de referință. Pot fi incluse substanțe de substituție sau suplimentare dacă activitatea acestora este cunoscută, dacă induc formarea de micronuclee prin intermediul acelorași mecanisme de acțiune și dacă relevanța acestora este dovedită pentru substanțele de test din cadrul procedurii MNvit. Justificarea poate include un studiu de validare acoperind o mare diversitate de substanțe sau axat pe un spectru mai restrâns, limitat la clasa chimică a substanței de test sau la mecanismul de leziune studiat.
|
|
56.
|
Martorii tratați cu solvent/vehicul și culturile netratate prezintă frecvențe reproductibile de micronuclee slabe și coerente (de regulă, 5-25 micronuclee/1 000 de celule pentru tipurile de celule indicate la puncul 11). Alte tipuri de celule pot prezenta plaje de răspuns diferite, care trebuie determinate în momentul validării acestora pentru utilizare în cadrul testului MNvit. Datele provenite de la martori negativi, solvenți și martori pozitivi trebuie utilizate pentru a stabili plaje de valori de referință. Aceste valori trebuie utilizate în stabilirea caracterului adecvat al martorilor pozitivi/negativi concomitenți pentru un experiment.
|
|
57.
|
În cazul în care sunt propuse modificări minore la protocolul de test (de exemplu, utilizarea de tehnici de numărare automate și nu manuale; utilizarea unui nou tip de celulă), atunci eficacitatea modificărilor trebuie demonstrată pentru ca protocolul modificat să poată fi considerat acceptabil pentru utilizare. Demonstrarea eficienței include demonstrarea faptului că pot fi detectate mecanismele majore de ruptură cromozomială, de pierdere sau de câștig cromozomial și că pot fi obținute rezultate pozitive și negative adecvate pentru clasa de substanțe în cauză sau pentru întreg spectrul de substanțe de test.
|
DATE ȘI RAPORTARE
Tratamentul rezultatelor
|
58.
|
Dacă se utilizează tehnica blocării citocinezei, doar frecvențele celulelor binucleate prezentând micronuclee (independent de numărul de micronuclee dintr-o celulă) se utilizează pentru evaluarea inducției de micronuclee. Numărarea celulelor prezentând unul, două sau mai multe micronuclee poate oferi informații utile, dar nu este obligatorie.
|
|
59.
|
Trebuie efectuate măsurători paralele de citotoxicitate și/sau citostază pentru toate culturile martor tratate sau tratate cu solvent/vehicul (58). De asemenea, trebuie calculat CBPI sau indicele de replicație (RI) pentru toate culturile tratate și cele martor, pentru a măsura întârzierea ciclului celular în cazul utilizării metodei blocării citocinezei. În absența citoB, trebuie măsurate dublarea relativă a populației (RPD) sau creșterea relativă a numărului de celule (RICC) sau indicele de proliferare (PI) (a se vedea apendicele 2).
|
|
60.
|
Datele se indică individual pentru fiecare cultură. În plus, toate datele sunt sintetizate sub formă de tabele.
|
|
61.
|
Substanțele chimice care induc micronuclee în testul MNvit pot face aceasta deoarece induc ruptură cromozomială, pierdere cromozomială sau o combinație a celor două. Alte analize utilizând anticorpi antikinetocori, sonde centrometrice in situ, sau alte metode permit să se determine dacă mecanismul inducției de micronuclee se datorează activității clastogene și/sau aneugene.
|
Evaluarea și interpretarea rezultatelor
|
62.
|
Nu există nicio cerință referitoare la verificarea cu ajutorul unor teste suplimentare a răspunsurilor pozitive sau negative clare. Rezultatele echivoce pot fi clarificate prin analiza a încă 1 000 de celule din toate culturile pentru a păstra caracterul orb al testului. Dacă această abordare nu permite obținerea unor rezultate satisfăcătoare, trebuie efectuate teste suplimentare. În cadrul experimentelor suplimentare realizate astfel, se are în vedere modificarea parametrilor studiului pentru a lărgi sau pentru a restrânge condițiile, după caz. Parametrii de studiu care ar putea fi modificați includ spațierea dintre concentrațiile de test, calendarul privind tratamentele și recolta celulelor și/sau condițiile de activare metabolică.
|
|
63.
|
Mai multe criterii permit determinarea unui rezultat pozitiv precum o creștere legată de concentrație sau o creștere semnificativă din punct de vedere statistic a numărului de celule conținând micronuclee. Relevanța biologică a rezultatelor trebuie avută în vedere în primă instanță. Faptul că valorile observate sunt cuprinse sau nu în plaja de valori de referință poate furniza indicii utile în momentul evaluării semnificației biologice a răspunsului. Evaluarea rezultatelor testului se poate sprijini pe metode statistice adecvate (65). Cu toate acestea, rezultatele testelor statistice se evaluează în raport cu relația doză-răspuns. Reproductibilitatea și datele istorice trebuie, de asemenea, luate în considerare.
|
|
64.
|
Deși majoritatea experimentelor oferă rezultate clare pozitive sau negative, în unele cazuri setul de date nu permite conturarea unei judecăți categorice cu privire la activitatea substanței de test. Rezultatele echivoce sau discutabile pot surveni indiferent de numărul de repetări ale testului.
|
|
65.
|
Rezultatele pozitive în cadrul unui test MNvit indică faptul că substanța de test induce rupere sau ruptură cromozomială, în culturile de celule de mamifere. Rezultatele negative indică faptul că, în condițiile de testare, substanța de test nu induce rupere și/sau câștig sau pierdere cromozomială în culturile de celule de mamifere.
|
Raportul de testare
|
66.
|
Raportul de testare trebuie să includă cel puțin următoarele informații, dacă acestea sunt relevante pentru efectuarea studiului:
|
|
Substanța chimică de test:
|
—
|
date de identificare, numărul din registrul CAS și numărul CE;
|
|
—
|
starea fizică și puritate;
|
|
—
|
proprietățile fizice și chimice relevante pentru realizarea studiului;
|
|
—
|
reactivitatea substanței de test cu solventul/vehiculul sau mediul de cultură celulară;
|
|
|
|
Solvent/vehicul:
|
—
|
justificarea alegerii solventului/vehiculului;
|
|
—
|
solubilitatea și stabilitatea substanței de test în solvent/vehicul;
|
|
|
|
Celule:
|
—
|
tipul și sursa celulelor utilizate;
|
|
—
|
caracterul adecvat al tipului de celulă utilizat;
|
|
—
|
absența micoplasmei, dacă este cazul;
|
|
—
|
informații privind durata ciclului celular, timpul de dublare sau indicele de proliferare;
|
|
—
|
în cazul utilizării de limfocite, sexul, vârsta și numărul donatorilor de sânge, dacă se aplică;
|
|
—
|
în cazul utilizării de limfocite, a se preciza dacă este expus sânge total sau limfocite izolate;
|
|
—
|
numărul de pasaje, dacă se aplică;
|
|
—
|
metode de întreținere a culturilor celulare, dacă se aplică;
|
|
—
|
numărul modal de cromozomi;
|
|
—
|
durata normală a unui ciclu celular (martor negativ);
|
|
|
|
Condiții de testare:
|
—
|
identitatea substanței de blocare a citocinezei (de exemplu, citoB), dacă se utilizează, concentrația sa și durata de expunere a celulelor;
|
|
—
|
justificarea selectării concentrațiilor și a numărului de culturi, inclusiv date privind citotoxicitatea și limitele de solubilitate, dacă acestea sunt disponibile;
|
|
—
|
compoziția mediului, concentrația de CO2, dacă se aplică;
|
|
—
|
concentrațiile substanței de test;
|
|
—
|
concentrația (și/sau volumul) vehiculului și a substanței de test adăugate;
|
|
—
|
temperatura și timpul de incubație;
|
|
—
|
perioada de recoltă după tratament;
|
|
—
|
densitatea celulară la momentul însămânțării, dacă se aplică;
|
|
—
|
tipul și compoziția sistemului de activare metabolică, inclusiv criteriile de acceptabilitate;
|
|
—
|
substanțele martor pozitive și negative;
|
|
—
|
metodele de preparare a lamelelor și tehnica de colorare utilizată;
|
|
—
|
criteriile de identificare a micronucleelor;
|
|
—
|
numărul de celule analizate;
|
|
—
|
metodele de măsurare a citotoxicității;
|
|
—
|
orice alte informații suplimentare cu privire la citotoxicitate;
|
|
—
|
criteriile în funcție de care un studiu poate fi considerat ca fiind pozitiv, negativ sau echivoc;
|
|
—
|
metoda (metodele) de analiză statistică utilizate;
|
|
—
|
metode precum utilizarea de anticorpi kinetocori, pentru a determina dacă micronucleele conțin cromozomi întregi sau fragmentați, dacă se aplică;
|
|
|
|
Rezultate:
|
—
|
măsurarea citotoxicității utilizate, de exemplu CBPI sau RI în cazul metodei de blocare a citocinezei; RICC, RPD sau PI atunci când nu sunt utilizate metode de blocare a citocinezei; alte observații, dacă este cazul, de exemplu gradul de confluență celulară, apoptoză, necroză, numărarea celulelor în metafază, frecvența celulelor binucleate;
|
|
—
|
date privind pH-ul și osmolalitatea mediului de tratament, dacă acestea au fost determinate;
|
|
—
|
definiția celulelor acceptabile pentru analiză;
|
|
—
|
distribuția celulelor mono-, bi- și multinucleate dacă se utilizează o metodă de blocare a citocinezei;
|
|
—
|
numărul de celule cu micronuclee, furnizat separat pentru fiecare cultură tratată și cultură martor, precizându-se dacă acestea provin din celule binucleate sau mononucleate, după caz;
|
|
—
|
relația concentrație-răspuns, dacă este posibil;
|
|
—
|
date (concentrații și solvenți) privind substanțele martor negative (solvent/vehicul) și pozitive concomitente;
|
|
—
|
date istorice privind substanțele martor negative (solvent/vehicul) și pozitive, inclusiv intervale de valori, medii și devieri standard și intervalul de încredere (de exemplu, 95 %);
|
|
—
|
analiză statistică; valorile P, dacă este cazul.
|
|
|
BIBLIOGRAFIE
|
(1)
|
Kirsch-Volders, M. (1997), Towards a validation of the micronucleus test. Mutation Res., 392, 1-4.
|
|
(2)
|
Parry, J.M. and Sors, A. (1993), The detection and assessment of the aneugenic potential of environmental chemicals: the European Community aneuploidy project, Mutation Res., 287, 3-15.
|
|
(3)
|
Fenech, M. and Morley, A.A. (1985), Solutions to the kinetic problem in the micronucleus assay, Cytobios., 43, 233-246.
|
|
(4)
|
Kirsch-Volders, M., Sofuni, T., Aardema, M., Albertini, S., Eastmond, D., Fenech, M., Ishidate, M. Jr, Lorge, E., Norppa, H., Surralles, J., von der Hude, W. and Wakata, A. (2000), Report from the In Vitro Micronucleus Assay Working Group, Environ. Mol. Mutagen., 35, 167-172.
|
|
(5)
|
Fenech, M. (2007), Cytokinesis-block micronucleus cytome assay, Nature Protocols, 2(5), 1084-1104.
|
|
(6)
|
Fenech, M. and Morley, A.A. (1986), Cytokinesis-block micronucleus method in human lymphocytes: effect of in-vivo ageing and low dose X-irradiation, Mutation Res., 161, 193-198.
|
|
(7)
|
Eastmond, D.A. and Tucker, J.D. (1989), Identification of aneuploidy-inducing agents using cytokinesis-blocked human lymphocytes and an antikinetochore antibody, Environ. Mol. Mutagen., 13, 34-43.
|
|
(8)
|
Eastmond, D.A. and Pinkel, D. (1990), Detection of aneuploidy and aneuploidy-inducing agents in human lymphocytes using fluorescence in-situ hybridisation with chromosome-specific DNA probes, Mutation Res., 234, 9-20.
|
|
(9)
|
Miller, B.M., Zitzelsberger, H.F., Weier, H.U. and Adler, I.D. (1991), Classification of micronuclei in murine erythrocytes: immunofluorescent staining using CREST antibodies compared to in situ hybridization with biotinylated gamma satellite DNA, Mutagenesis, 6, 297-302.
|
|
(10)
|
Farooqi, Z., Darroudi, F. and Natarajan, A.T. (1993), The use of fluorescence in-situ hybridisation for the detection of aneugens in cytokinesis-blocked mouse splenocytes, Mutagenesis, 8, 329-334.
|
|
(11)
|
Migliore, L., Bocciardi, R., Macri, C. and Lo Jacono, F. (1993), Cytogenetic damage induced in human lymphocytes by four vanadium compounds and micronucleus analysis by fluorescence in situ hybridization with a centromeric probe, Mutation Res., 319, 205-213.
|
|
(12)
|
Norppa, H., Renzi, L. and Lindholm, C. (1993), Detection of whole chromosomes in micronuclei of cytokinesis-blocked human lymphocytes by antikinetochore staining and in situ hybridization, Mutagenesis, 8, 519-525.
|
|
(13)
|
Eastmond, D.A, Rupa, D.S. and Hasegawa, L.S. (1994), Detection of hyperdiploidy and chromosome breakage in interphase human lymphocytes following exposure to the benzene metabolite hydroquinone using multicolor fluorescence in situ hybridization with DNA probes, Mutation Res., 322, 9-20.
|
|
(14)
|
Marshall, R.R., Murphy, M., Kirkland, D.J. and Bentley, K.S. (1996), Fluorescence in situ hybridisation (FISH) with chromosome-specific centromeric probes: a sensitive method to detect aneuploidy, Mutation Res., 372, 233-245.
|
|
(15)
|
Zijno, P., Leopardi, F., Marcon, R. and Crebelli, R. (1996), Analysis of chromosome segregation by means of fluorescence in situ hybridization: application to cytokinesis-blocked human lymphocytes, Mutation Res., 372, 211-219.
|
|
(16)
|
Kirsch-Volders, M., Sofuni, T., Aardema, M., Albertini, S., Eastmond, D., Fenech, M., Ishidate Jr., M., Lorge, E., Norppa, H., Surrallés, J., von der Hude, W. and Wakata, A. (2003), Report from the in vitro micronucleus assay working group. Mutation Res., 540, 153-163.
|
|
(17)
|
OECD (1997), In Vitro Mammalian Chromosome Aberration Test, Test Guideline No. 473, OECD Guidelines for Testing of Chemicals, OECD, Paris. Disponibil la: [www.oecd.org/env/testguidelines]
|
|
(18)
|
Lorge, E., Thybaud, V., Aardema, M.J., Oliver, J., Wakata, A., Lorenzon G. and Marzin, D. (2006), SFTG International collaborative Study on in vitro micronucleus test. I. General conditions and overall conclusions of the study, Mutation Res., 607, 13-36.
|
|
(19)
|
Clare, G., Lorenzon, G., Akhurst, L.C., Marzin, D., van Delft, J., Montero, R., Botta, A., Bertens, A., Cinelli, S., Thybaud, V. and Lorge, E. (2006), SFTG International collaborative study on the in vitro micronucleus test. II. Using human lymphocytes, Mutation Res., 607, 37-60.
|
|
(20)
|
Aardema, M.J., Snyder, R.D., Spicer, C., Divi, K., Morita, T., Mauthe, R.J., Gibson, D.P., Soelter, S., Curry, P.T., Thybaud, V., Lorenzon, G., Marzin, D. and Lorge, E. (2006), SFTG International collaborative study on the in vitro micronucleus test, III. Using CHO cells, Mutation Res., 607, 61-87.
|
|
(21)
|
Wakata, A., Matsuoka, A., Yamakage, K., Yoshida, J., Kubo, K., Kobayashi, K., Senjyu, N., Itoh, S., Miyajima, H., Hamada, S., Nishida, S., Araki, H., Yamamura, E., Matsui, A., Thybaud, V., Lorenzon, G., Marzin, D. and Lorge, E. (2006), SFTG International collaborative study on the in vitro micronucleus test, IV. Using CHO/IU cells, Mutation Res., 607, 88-124.
|
|
(22)
|
Oliver, J., Meunier, J.-R., Awogi, T., Elhajouji, A., Ouldelhkim, M.-C., Bichet, N., Thybaud, V., Lorenzon, G., Marzin, D. and Lorge, E. (2006), SFTG International collaborative study on the in vitro micronucleus test, V. Using L5178Y cells, Mutation Res., 607, 125-152.
|
|
(23)
|
Albertini, S., Miller, B., Chetelat, A.A. and Locher, F. (1997), Detailed data on in vitro MNT and in vitro CA: industrial experience, Mutation Res., 392, 187-208.
|
|
(24)
|
Miller, B., Albertini, S., Locher, F., Thybaud, V. and Lorge, E. (1997), Comparative evaluation of the in vitro micronucleus test and the in vitro chromosome aberration test: industrial experience, Mutation Res., 392, 45-59.
|
|
(25)
|
Miller, B., Potter-Locher, F., Seelbach, A., Stopper, H., Utesch, D. and Madle, S. (1998), Evaluation of the in vitro micronucleus test as an alternative to the in vitro chromosomal aberration assay: position of the GUM Working Group on the in vitro micronucleus test. Gesellschaft fur Umwelt-Mutations-forschung, Mutation Res., 410, 81-116.
|
|
(26)
|
Kalweit, S., Utesch, U., von der Hude, W. and Madle, S. (1999), Chemically induced micronucleus formation in V79 cells – comparison of three different test approaches, Mutation Res. 439, 183-190.
|
|
(27)
|
Kersten, B., Zhang, J., Brendler Schwaab, S.Y., Kasper, P. and Müller, L. (1999), The application of the micronucleus test in Chinese hamster V79 cells to detect drug-induced photogenotoxicity, Mutation Res. 445, 55-71.
|
|
(28)
|
von der Hude, W., Kalweit, S., Engelhardt, G., McKiernan, S., Kasper, P., Slacik-Erben, R., Miltenburger, H.G., Honarvar, N., Fahrig, R., Gorlitz, B., Albertini, S., Kirchner, S., Utesch, D., Potter-Locher, F., Stopper, H. and Madle, S. (2000), In vitro micronucleus assay with Chinese hamster V79 cells – results of a collaborative study with in situ exposure to 26 chemical substances, Mutation Res., 468, 137-163.
|
|
(29)
|
Garriott, M.L., Phelps, J.B. and Hoffman, W.P. (2002), A protocol for the in vitro micronucleus test, I. Contributions to the development of a protocol suitable for regulatory submissions from an examination of 16 chemicals with different mechanisms of action and different levels of activity, Mutation Res., 517, 123-134.
|
|
(30)
|
Matsushima, T., Hayashi, M., Matsuoka, A., Ishidate, M. Jr., Miura, K.F., Shimizu, H., Suzuki, Y., Morimoto, K., Ogura, H., Mure, K., Koshi, K. and Sofuni, T. (1999), Validation study of the in vitro micronucleus test in a Chinese hamster lung cell line (CHL/IU), Mutagenesis, 14, 569-580.
|
|
(31)
|
Elhajouji, A., and Lorge, E. (2006), Special Issue: SFTG International collaborative study on in vitro micronucleus test, Mutation Res., 607, 1-152.
|
|
(32)
|
ECVAM (2006), Statement by the European Centre for the Validation of Alternative Methods (ECVAM) Scientific Advisory Committee (ESAC) on the scientific validity of the in vitro micronucleus test as an alternative to the in vitro chromosome aberration assay for genotoxicity testing. ESAC 25th meeting, 16-17 November 2006, Disponibil la: [http://ecvam.jrc.it/index.htm]
|
|
(33)
|
ESAC (2006), ECVAM Scientific Advisory Committee (ESAC) Peer Review, Retrospective Validation of the In Vitro Micronucleus Test, Summary and Conclusions of the Peer Review Panel, Disponibil la: [http://ecvam.jrc.it/index.htm]
|
|
(34)
|
Corvi, R., Albertini, S., Hartung, T., Hoffmann, S., Maurici, D., Pfuhler, S, van Benthem, J., Vanparys P. (2008), ECVAM Retrospective Validation of in vitro Micronucleus Test (MNT), Mutagenesis, 23, 271-283.
|
|
(35)
|
Zhang, L.S., Honma, M., Hayashi, M., Suzuki, T., Matsuoka, A. and Sofuni, T. (1995), A comparative study of TK6 human lymphoblastoid and L5178Y mouse lymphoma cell lines in the in vitro micronucleus test, Mutation Res., 347, 105-115.
|
|
(36)
|
Ehrlich, V., Darroudi, F., Uhl, M., Steinkellner, S., Zsivkovits, M. and Knasmeuller, S. (2002), Fumonisin B1 is genotoxic in human derived hepatoma (HepG2) cells, Mutagenesis, 17, 257-260.
|
|
(37)
|
Knasmüller, S., Mersch-Sundermann, V., Kevekordes, S., Darroudi, F., Huber, W.W., Hoelzl, C., Bichler, J. and Majer, B.J. (2004), Use of human-derived liver cell lines for the detection of environmental and dietary genotoxicants; current state of knowledge, Toxicol., 198, 315-328..
|
|
(38)
|
Gibson, D.P., Brauninger, R., Shaffi, H.S., Kerckaert, G.A., LeBoeuf, R.A., Isfort, R.J. and Aardema, M.J. (1997), Induction of micronuclei in Syrian hamster embryo cells: comparison to results in the SHE cell transformation assay for National Toxicology Program test chemicals, Mutation Res., 392, 61-70.
|
|
(39)
|
Scott, D., Galloway, S.M., Marshall, R.R., Ishidate, M. Jr., Brusick, D., Ashby, J. and Myhr, B.C. (1991), International Commission for Protection Against Environmental Mutagens and Carcinogens, Genotoxicity under extreme culture conditions. A report from ICPEMC Task Group 9, Mutation Res., 257, 147-205.
|
|
(40)
|
Morita, T., Nagaki, T., Fukuda, I. and Okumura, K. (1992), Clastogenicity of low pH to various cultured mammalian cells, Mutation Res., 268, 297-305.
|
|
(41)
|
Brusick, D. (1986), Genotoxic effects in cultured mammalian cells produced by low pH treatment conditions and increased ion concentrations, Environ. Mutagen., 8, 789-886.
|
|
(42)
|
Fenech, M. and Morley, A.A. (1985), Measurement of micronuclei in lymphocytes, Mutation Res., 147, 29-36.
|
|
(43)
|
Fenech, M. (1997), The advantages and disadvantages of cytokinesis-blood micronucleus method, Mutation Res., 392, 11-18.
|
|
(44)
|
Bonassi, S., Fenech, M., Lando, C., Lin, Y.P., Ceppi, M., Chang, W.P., Holland, N., Kirsch-Volders, M., Zeiger, E., Ban, S., Barale, R., Bigatti, M.P., Bolognesi, C., Jia, C., Di Giorgio, M., Ferguson, L.R., Fucic, A., Lima, O.G., Hrelia, P., Krishnaja, A.P., Lee, T.K., Migliore, L., Mikhalevich, L., Mirkova, E., Mosesso, P., Muller, W.U., Odagiri, Y., Scarffi, M.R., Szabova, E., Vorobtsova, I., Vral, A. and Zijno, A. (2001), HUman MicroNucleus Project: international database comparison for results with the cytokinesis-block micronucleus assay in human lymphocytes, I. Effect of laboratory protocol, scoring criteria and host factors on the frequency of micronuclei, Environ. Mol. Mutagen. 37, 31-45.
|
|
(45)
|
Maron, D.M. and Ames, B.N. (1983), Revised methods for the Salmonella mutagenicity test, Mutation Res., 113, 173-215.
|
|
(46)
|
Ong, T.-m., Mukhtar, M., Wolf, C.R. and Zeiger, E. (1980), Differential effects of cytochrome P450-inducers on promutagen activation capabilities and enzymatic activities of S-9 from rat liver, J. Environ. Pathol. Toxicol., 4, 55-65.
|
|
(47)
|
Elliott, B.M., Combes, R.D., Elcombe, C.R., Gatehouse, D.G., Gibson, G.G., Mackay, J.M. and Wolf, R.C. (1992), Alternatives to Aroclor 1254-induced S9 in in-vitro genotoxicity assays. Mutagenesis, 7, 175-177.
|
|
(48)
|
Matsushima, T., Sawamura, M., Hara, K. and Sugimura, T. (1976), A safe substitute for Polychlorinated Biphenyls as an Inducer of Metabolic Activation Systems, In: de Serres, F.J., Fouts, J. R., Bend, J.R. and Philpot, R.M. (eds), In Vitro Metabolic Activation in Mutagenesis Testing, Elsevier, North-Holland, pp. 85-88.
|
|
(49)
|
Johnson, T.E., Umbenhauer, D.R. and Galloway, S.M. (1996), Human liver S-9 metabolic activation: proficiency in cytogenetic assays and comparison with phenobarbital/beta-naphthoflavone or Aroclor 1254 induced rat S-9, Environ. Mol. Mutagen., 28, 51-59.
|
|
(50)
|
UNEP (2001), Stockholm Convention on Persistent Organic Pollutants, United Nations Environment Programme (UNEP). Disponibil la: [http://www.pops.int/]
|
|
(51)
|
Doherty, A.T., Ellard, S., Parry, E.M. and Parry, J.M. (1996), An investigation into the activation and deactivation of chlorinated hydrocarbons to genotoxins in metabolically competent human cells, Mutagenesis, 11, 247-274.
|
|
(52)
|
Krahn, D.F., Barsky, F.C. and McCooey, K.T. (1982), CHO/HGPRT Mutation Assay: Evaluation of Gases and Volatile Liquids, In: Tice, R.R., Costa, D.L. and Schaich, K.M. (eds), Genotoxic Effects of Airborne Agents. New York, Plenum, pp. 91-103.
|
|
(53)
|
Zamora, P.O., Benson, J.M., Li, A.P. and Brooks, A.L. (1983), Evaluation of an exposure system using cells grown on collagen gels for detecting highly volatile mutagens in the CHO/HGPRT mutation assay, Environ. Mutagenesis 5, 795-801.
|
|
(54)
|
Fenech, M. (1993), The cytokinesis-block micronucleus technique: a detailed description of the method and its application to genotoxicity studies in human populations, Mutation Res., 285, 35-44.
|
|
(55)
|
Phelps, J.B., Garriott, M.L., and Hoffman, W.P. (2002), A protocol for the in vitro micronucleus test. II. Contributions to the validation of a protocol suitable for regulatory submissions from an examination of 10 chemicals with different mechanisms of action and different levels of activity, Mutation Res., 521, 103-112.
|
|
(56)
|
Kirsch-Volders, M., Sofuni, T., Aardema, M., Albertini, S., Eastmond, D., Fenech, M., Ishidate, M. Jr., Kirchner, S., Lorge, E., Morita, T., Norppa, H., Surralles, J., Vanhauwaert, A. and Wakata, A. (2004), Corrigendum to «Report from the in vitro micronucleus assay working group», Mutation Res., 564, 97-100.
|
|
(57)
|
Pincu, M., Bass, D. and Norman, A. (1984), An improved micronuclear assay in lymphocytes, Mutation Res., 139, 61-65.
|
|
(58)
|
Lorge, E., Hayashi, M., Albertini, S. and Kirkland, D. (2008), Comparison of different methods for an accurate assessment of cytotoxicity in the in vitro micronucleus test. I. Theoretical aspects, Mutation Res., 655, 1-3.
|
|
(59)
|
Surralles, J., Xamena, N., Creus, A., Catalan, J., Norppa, H. and Marcos, R. (1995), Induction of micronuclei by five pyrethroid insecticides in whole-blood and isolated human lymphocyte cultures, Mutation Res., 341, 169-184.
|
|
(60)
|
Galloway, S. (2000), Cytotoxicity and chromosome aberrations in vitro: Experience in industry and the case for an upper limit on toxicity in the aberration assay, Environ. Molec. Mutagenesis 35, 191-201.
|
|
(61)
|
Hayashi, M., Sofuni, T., and Ishidate, M. Jr. (1983), An Application of Acridine Orange Fluorescent Staining to the Micronucleus Test, Mutation Res., 120, 241-247.
|
|
(62)
|
MacGregor, J. T., Wehr, C. M., and Langlois, R. G. (1983), A Simple Fluorescent Staining Procedure for Micronuclei and RNA in Erythrocytes Using Hoechst 33258 and Pyronin Y, Mutation Res., 120, 269-275.
|
|
(63)
|
Hayashi, M., Sofuni, T. and Ishidate, M. Jr. (1983), An application of acridine orange fluorescent staining to the micronucleus test, Mutation Res., 120, 241-247.
|
|
(64)
|
Fenech, M., Chang, W.P., Kirsch-Volders, M., Holland, N., Bonassi, S. and Zeiger, E. (2003), HUMN project: detailed description of the scoring criteria for the cytokinesis-block micronucleus assay using isolated human lymphocyte cultures, Mutation Res., 534, 65-75.
|
|
(65)
|
Hoffman, W.P., Garriott, M.L. and Lee, C. (2003), In vitro micronucleus test, In: Encyclopedia of Biopharmaceutical Statistics, Second edition. S. Chow (ed.), Marcel Dekker, Inc. New York, NY, pp. 463-467.
|
|
(66)
|
Regulamentul (CE) Nr. 850/2004 al Parlamentului European și al Consiliului din 29 aprilie 2004 privind poluanții organici persistenți și de modificare a Directivei 79/117/CEE, JO L 229, 30.4.2004, p. 5.
|
Apendicele 1
Definiții
Aneugen: orice substanță sau proces care, prin interacțiunile cu componentele celulei mitotice sau meiotice în timpul diviziunii celulare, conduce la apariția aneuploidiei în celule sau organisme.
Aneuploidie: orice deviere de la numărul diploid (sau haploid) normal de cromozomi, cu unul sau mai mulți cromozomi, dar nu cu un set (seturi) întreg (întregi) de cromozomi (poliploidie).
Apoptoză: moarte celulară programată caracterizată printr-o succesiune de etape conducând la dezintegrarea celulelor în particule membranare care sunt apoi eliminate prin fagocitoză sau excreție.
Proliferare celulară: creșterea numărului de celule ca rezultat al diviziunii celulare mitotice.
Centromer: regiunea ADN-ului unui cromozom unde cele două cromatide sunt legate între ele și pe care cei doi kinetocori sunt fixați unul lângă altul.
Clastogen: orice substanță sau proces care cauzează aberații cromozomiale structurale în populații de celule sau organisme.
Citocineză: procesul de diviziune celulară care survine imediat după mitoză pentru a forma două celule fiice, fiecare conținând un nucleu unic.
Indice de proliferare a celulelor a căror citocineză a fost blocată (Cytokinesis-Block Proliferation index, CBPI): proporția de celule provenite de la a doua diviziune în populația tratată în raport cu populația martor netratată (a se vedea formulele din apendicele 2).
Citostază: inhibarea creșterii celulare (a se vedea formulele din apendicele 2).
Citotoxicitate: efecte dăunătoare asupra structurii sau funcțiilor celulare antrenând în final moartea celulară.
Genotoxic: termen general cuprinzând toate tipurile de leziuni ale ADN-ului sau ale cromozomilor, inclusiv ruperi, aducții, rearanjări, mutații, aberații cromozomiale ș aneuploidie. Nu toate tipurile de efecte genotoxice au drept rezultat mutații sau leziuni cromozomiale stabile.
Celule în interfază: celule care nu se află în etapa de mitoză.
Kinetocor: structură proteică situată la nivelul centromerului unui cromozom la care se asociază fibrele fusoriale în timpul diviziunii celulare, permițând mișcarea ordonată a cromozomilor-fii către polii celulelor-fiice.
Micronuclee: nuclee mici, separate și adiționale nucleelor principale ale celulelor, formate în timpul telofazei mitozei sau meiozei pornind de la cromozomi întregi sau de la fragmente de cromozomi întârziați.
Mitoză: divizarea nucleului celular, în general, descompus în profază, prometafază, metafază, anafază și telofază.
Indice mitotic: raportul dintre numărul de celule în metafază și numărul total de celule observate într-o populație celulară; acesta oferă un indiciu cu privire la gradul de proliferare celulară a populației respective.
Mutagen: agent care produce o modificare ereditară uneia sau mai multor secvențe de perechi de bază de ADN genic sau structurii cromozomilor (aberații cromozomiale).
Nondisjuncție: lipsa separării unei perechi de cromatide, care nu reușesc prin urmare să migreze în mod corespunzător către celulele-fiice formate, fapt care determină prezența unui număr anormal de cromozomi în celulele-fiice.
Poliploidie: aberații cromozomiale numerice în celule sau organisme implicând unul sau mai multe seturi întregi de cromozomi, spre deosebire de unul sau mai mulți cromozomi izolați (aneuploidie).
Indice de proliferare (PI): metodă de măsurare a citotoxicității atunci când nu este folosită citoB (a se vedea formula din apendicele 2).
Creștere relativă a numărului de celule (Relative Increase in Cell Count, RICC): metodă de măsurare a citotoxicității atunci când nu este folosită citoB (a se vedea formula din apendicele 2).
Dublarea relativă a populației (Relative Population Doubling, RPD): metodă de măsurare a citotoxicității atunci când nu este folosită citoB (a se vedea formula din apendicele 2).
Indice de replicație (RI): proporția ciclurilor de diviziune celulară complete într-o cultură tratată în raport cu cultura martor netratată, în timpul perioadei de expunere și de recuperare (a se vedea formula din apendicele 2).
Substanță chimică de test (denumită, de asemenea, substanță de test): orice substanță sau amestec testat cu ajutorul prezentei MT.
Apendicele 2
Formule pentru evaluarea citotoxicității
|
1.
|
În cazul utilizării citoB, evaluarea citotoxicității se bazează pe indicele de proliferare a celulelor a căror citocineză a fost blocată (CBPI) sau pe indicele de replicație (RI) (16) (58). CBPI indică numărul mediu de cicluri celulare pe celulă efectuate în timpul perioadei de expunere la citoB și poate fi utilizat pentru a calcula proliferarea celulară. RI indică numărul relativ de nuclee prezente în culturile tratate comparativ cu culturile martor și poate fi utilizat pentru calcularea procentajului de citostază:
% citostază = 100 – 100{(CBPIT – 1) ÷ (CBPIC – 1)}
cu:
|
T
|
=
|
cultură tratată cu substanța chimică de test
|
|
C
|
=
|
cultură martor tratată cu vehicul
|
unde:

Astfel, un CBPI egal cu 1 (toate celulele sunt mononucleate) este echivalent cu o citostază de 100 %.
Citostază = 100 – RI
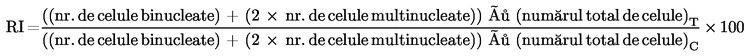
|
T
|
=
|
culturi tratate
|
|
C
|
=
|
culturi martor
|
|
|
2.
|
Astfel, un RI de 53 % înseamnă că, comparativ cu numărul de celule care au efectuat o diviziune celulară pentru a forma celule binucleate și multinucleate în cultura martor, doar 53 % din celule au efectuat o diviziune în cultura tratată, și anume o citostază de 47 %.
|
|
3.
|
În absența utilizării citoB, se recomandă evaluarea citotoxicității pe baza creșterii relative a numărului de celule (RICC) sau a dublării relative a populației (RPD) (58), deoarece ambele iau în considerare proporția de celule care au efectuat o diviziune celulară.


unde:
dublarea populației = [log (număr de celule post-tratamet ÷ numărul inițial de celule)] ÷ log 2
|
|
4.
|
Astfel, un RICC sau un RPD de 53 % indică o citotoxicitate/citostază de 47 %.
|
|
5.
|
Utilizând indicele de proliferare (PI), citotoxicitatea poate fi evaluată prin intermediul numărării clonelor compuse dintr-o celulă (cl1), 2 celule (cl2), 3 până la 4 celule (cl4) și 5 până la 8 celule (cl8)

|
|
6.
|
PI este, de asemenea, utilizat ca un parametru fiabil și util de citotoxicitate și pentru liniile celulare cultivate in situ în absența citoB (25) (26) (27) (28).
|
Apendicele 3
Substanțe chimice de referință recomandate pentru evaluarea performanței
(16)
|
Categorie
|
Substanță chimică
|
nr. CAS
|
nr. CE
|
| 1. Clastogeni activi fără activare metabolică
|
|
|
Cistozină arabinozidă
|
147-94-4
|
205-705-9
|
|
|
Mitomicină C
|
50-07-7
|
200-008-6
|
| 2. Clastogeni necesitând activare metabolică
|
|
|
Benzo(a)piren
|
50-32-8
|
200-028-5
|
|
|
Ciclofosfamidă
|
50-18-0
|
200-015-4
|
| 3. Aneugeni
|
|
|
Colchicină
|
64-86-8
|
200-598-5
|
|
|
Vinblastină
|
143-67-9
|
205-606-0
|
| 4. Substanțe negative
|
|
|
Di(2-etilhexil)ftalat
|
117-81-7
|
204-211-0
|
|
|
Acid nalidixic
|
389-08-2
|
206-864-7
|
|
|
Piren
|
129-00-0
|
204-927-3
|
|
|
Clorură de sodiu
|
7647-14-5
|
231-598-3
|
B.50. SENSIBILIZAREA DERMICĂ: TESTUL LOCAL PE GANGLIONI LIMFATICI: DA
INTRODUCERE
|
1.
|
Orientările OCDE privind testarea substanțelor chimice și metodele de testare UE bazate pe acestea sunt periodic revizuite în lumina progresului științific, modificând necesitățile de reglementare și considerațiile referitoare la bunăstarea animalelor. Prima metodă de testare (MT) (B.42) pentru determinarea sensibilizării dermice la șoarece, testul local pe ganglioni limfatici (LLNA; orientarea OCDE privind testarea nr. 429), a fost revizuită (1). Detaliile privind validarea LLNA, precum și o revizuire a lucrărilor asociate au fost publicate (2)(3)(4)(5)(6)(7)(8)(9). În cadrul metodei LLNA, marcajul radioizotopic cu timidină sau iod este utilizat pentru a măsura proliferarea limfocitelor și, prin urmare, testul are o aplicare limitată în regiunile unde achiziționarea, utilizarea sau eliminarea produselor radioactive pun probleme. LLNA: DA (dezvoltată de către Daicel Chemical Industries, Ltd.) este o variantă non-radioactivă a LLNA, care cuantifică conținutul de adenozină trifosfat (ATP) prin bioluminiscență ca indicator al proliferării celulare. Metoda de testare LLNA: DA a fost validată, revizuită și recomandată de către o echipă internațională de examinare care a recunoscut utilitatea acesteia în identificarea, între anumite limite, a substanțelor chimice care au sau nu un efect de sensibilizare a pielii (10) (11) (12) (13). Prezenta MT vizează evaluarea potențialului de sensibilizare dermică a substanțelor chimice (substanțe și amestecuri) la animale. Capitolul B.6 din prezenta anexă și orientarea OCDE privind testarea nr. 406 utilizează testele pe cobai, și anume testul de maximizare la cobai și testul Buehler (14). LLNA (capitolul B.42 din prezenta anexă; orientarea OCDE privind testarea OECD nr. 429) și cele două variante non-radioactive, LLNA: DA (capitolul B.50 din prezenta anexă; orientarea OCDE privind testarea nr. 442 A) și LLNA: BrdU-ELISA (capitolul B.51 din prezenta anexă; orientarea OCDE privind testarea nr. 442 B) sunt mai avantajoase decât testele pe cobai din B.6 și orientarea OCDE privind testarea nr. 406 (14) în termeni de reducere și de rafinare a utilizării animalelor.
|
|
2.
|
În mod similar cu LLNA, LLNA: DA studiază faza de inducție a sensibilizării dermice și furnizează date cantitative adecvate pentru evaluarea relației doză-răspuns. În plus, abilitatea de a detecta agenții de sensibilizare dermică fără a recurge la radiomarcarea ADN-ului elimină riscurile profesionale de expunere la radiații și problemele legate de gestionarea deșeurilor. În schimb, aceasta ar putea presupune o creștere a numărului de șoareci utilizați pentru detectarea agenților de sensibilizare dermică determinând, cu toate acestea, diminuarea numărului de cobai pentru testarea potențialului de sensibilizare dermică (și anume, B.6; orientarea OCDE privind testarea nr. 406) (14).
|
DEFINIȚII
|
3.
|
Definițiile utilizate sunt furnizate în apendicele 1.
|
CONSIDERAȚII INIȚIALE ȘI LIMITE
|
4.
|
Metoda LLNA: DA este o variantă a metodei LLNA vizând identificarea potențialului de sensibilizare dermică al substanțelor chimice, între anumite limite. Acest lucru nu implică în mod necesar faptul că LLNA: DA trebuie să înlocuiască sistematic LLNA sau testele pe cobai (și anume, B.6; orientarea OCDE privind testarea nr. 406) (14), ci mai curând, că testul reprezintă un instrument de o calitate similară putând fi folosit ca alternativă și ale cărui rezultate pozitive sau negative nu mai necesită, în general, o confirmare suplimentară (10) (11). Anterior derulării studiului, laboratorul de testare trebuie să colecteze toate informațiile disponibile cu privire la substanța de test. Astfel de informații includ identitatea și structura chimică a substanței de test; proprietățile sale fizico-chimice; rezultatele tuturor celorlalte teste de toxicitate in vitro sau in vivo pentru substanța de test și datele toxicologice privind substanțele chimice asociate din punct de vedere structural. Aceste informații trebuie analizate pentru a determina dacă metoda LLNA: DA este adecvată pentru substanța de test [având în vedere incompatibilitatea anumitor tipuri de substanțe chimice cu LLNA: DA (a se vedea puncul (5)] și pentru a contribui la alegerea dozelor adecvate.
|
|
5.
|
LLNA: DA este o metodă in vivo și, prin urmare, nu elimină utilizarea animalelor pentru evaluarea sensibilizării alergice de contact. Cu toate acestea, comparativ cu testele pe cobai (B.6; orientarea OCDE privind testarea nr. 406), aceasta are potențialul de a reduce numărul de animale utilizate în acest scop (14). Mai mult, LLNA: DA oferă o rafinare importantă (reducerea durerii și a suferinței) a modului în care sunt utilizate animalele pentru testarea sensibilizării alergice de contact, deoarece, spre deosebire de B.6 și orientarea OCDE privind testarea nr. 406, LLNA: DA nu se bazează pe declanșarea reacțiilor de hipersensibilitate dermică prin manifestarea declanșării. În ciuda avantajelor metodei LLNA: DA comparativ cu B.6 și orientarea OCDE privind testarea nr. 406 (14), există anumite limite care ar putea impune utilizarea metodei B.6 sau a orientării OCDE privind testarea nr. 406 [de exemplu, testarea anumitor metale, rezultate fals pozitive cu anumite substanțe iritante pentru piele (precum anumite substanțe tensioactive) (6) (1 și capitolul B.42 din prezenta anexă), solubilitatea substanței de test]. În plus, anumite substanțe sau clase de substanțe chimice conținând grupele funcționale demonstrate ca acționând ca posibili factori de confuzie (16) ar putea impune utilizarea testelor pe cobai [și anume B.6; testarea OCDE privind testarea nr. 406 (14)]. Limitele identificate pentru LLNA (1 și capitolul B.42 din prezenta anexă) se recomandă ca fiind aplicabile și în cazul LLNA: DA (10). În plus, utilizarea LLNA: DA ar putea fi inadecvată în cazul testării substanțelor care modifică nivelurile ATP (de exemplu, substanțele care acționează ca inhibitori ai ATP) sau al celor care perturbă măsurarea exactă a ATP intracelular (de exemplu, prezența de enzime care degradează ATP, prezența de ATP extracelular în ganglionul limfatic). În afara acestor limite identificate, LLNA: DA se aplică oricăror substanțe de test care nu prezintă proprietăți susceptibile să afecteze acuratețea testului LLNA: DA. Mai mult, trebuie luată în considerare posibilitatea obținerii de rezultate pozitive ambigue pentru care valorile indicelui de stimulare (IS) sunt cuprinse între 1,8 și 2,5 (a se vedea punctele 31-32). Acest fapt se bazează pe baza de date de validare de 44 de substanțe prezentând un IS ≥ 1,8 (a se vedea punctul (6) pentru care LLNA: DA a identificat corect toți cei 32 de agenți de sensibilizare LLNA, dar a identificat incorect trei dintre cei 12 agenți de non-sensibilizare LLNA prezentând un IS cuprins între 1,8 și 2,5 (și anume rezultate pozitive de limită) (10). Cu toate acestea, deoarece același set de date a fost utilizat pentru stabilirea valorilor IS și pentru calcularea proprietăților estimative ale testului, rezultatele indicate ar putea supraestima proprietățile estimative reale.
|
PRINCIPIUL METODEI DE TESTARE
|
6.
|
Principiul fundamental care stă la baza LLNA: DA este acela că agenții de sensibilizare induc o proliferare a limfocitelor în ganglionii limfatici care drenează porțiunea unde se aplică substanța de test. Proliferarea este proporțională cu doza aplicată și cu puterea alergenului și oferă o modalitate simplă de a obține măsurători cantitative ale sensibilizării. Proliferarea este măsurată prin compararea proliferării medii din fiecare grup de test cu proliferarea medie din grupul martor tratat cu vehicul (VC). Se calculează raportul dintre proliferarea medie din fiecare grup tratat și proliferarea medie din grupul VC, denumit IS, iar acesta trebuie să aibă o valoare ≥ 1,8 pentru a se putea evalua în continuare substanța de test ca și potențial agent de sensibilizare dermică. Procedurile descrise aici se bazează pe cuantificarea conținutului de ATP prin bioluminiscență (factor cunoscut ca fiind în corelație cu numărul de celule vii) (17) pentru a indica creșterea numărului de celule proliferate în ganglionii limfatici auriculari de drenare (18) (19). Această tehnică prin bioluminiscență utilizează enzima luciferază pentru a cataliza formarea luminii pornind de la ATP și luciferină conform reacției următoare:
ATP + luciferină + O
2
 oxiluciferină + AMP + PPi
+ CO
2 + lumină
oxiluciferină + AMP + PPi
+ CO
2 + lumină
Intensitatea luminii emise urmărește o traiectorie liniară în funcție de concentrația ATP și se măsoară cu ajutorul unui luminometru. Testul luciferină-luciferază constituie o metodă preciză de cuantificare a ATP și este utilizat în numeroase aplicații (20).
|
DESCRIEREA TESTULUI
Selectarea speciilor de animale
|
7.
|
Specia selectată pentru acest test este șoarecele. Studiile de validare a LLNA: DA au fost efectuate utilizându-se exclusiv animale din rasa CBA/J, considerată, prin urmare, rasa preferată (12) (13). Se folosesc femele adulte tinere, nulipare și care nu sunt însărcinate. La începutul studiului, animalele trebuie să aibă între 8-12 săptămâni, iar diferențele de greutate dintre acestea trebuie să fie minime și să nu depășească 20 % din greutatea medie. Se pot utiliza și alte rase și masculi dacă se generează suficiente date pentru a demonstra că nu există diferențe semnificative în răspunsul la LLNA: DA specifice rasei și/sau sexului.
|
Condiții de adăpostire și de hrănire
|
8.
|
Șoarecii trebuie adăpostiți în grupuri (21), cu excepția cazului în care sunt furnizate justificări științifice adecvate pentru adăpostirea acestora în incinte individuale. Temperatura sălii în care se află animalele de experiență trebuie să fie de 22 °C (± 3 °C). Deși umiditatea relativă trebuie să fie de cel puțin 30 % și este preferabil să nu depășească 70 % decât în timpul operațiunilor de curățare a sălii, obiectivul de umiditate este ca aceasta să fie de 50-60 %. Iluminatul trebuie să fie artificial, alternând 12 ore de lumină cu 12 ore de întuneric. Pentru alimentație, se poate utiliza hrană convențională de laborator, cu furnizarea unei cantități nelimitate de apă potabilă.
|
Pregătirea animalelor
|
9.
|
Animalele se selectează aleatoriu, se marchează pentru a putea fi identificate individual (dar nu se folosește niciun tip de crotalie) și se țin în cuști timp de cel puțin 5 zile înainte de a se începe administrarea dozei astfel încât să se aclimatizeze la condițiile de laborator. Toate animalele sunt examinate înainte de începerea tratamentului, pentru a se stabili că nu prezintă leziuni observabile la nivel dermic.
|
Pregătirea dozelor
|
10.
|
Substanțele chimice solide trebuie dizolvate sau suspendate în solvenți/vehicule și diluate, dacă este cazul, anterior aplicării acestora pe urechea șoarecilor. Substanțele chimice lichide pot fi aplicate pure sau diluate anterior dozării. Substanțele chimice insolubile precum cele observate în general la dispozitivele medicale ar trebui să fie supuse unui proces de extracție amplificată într-un solvent adecvat pentru a indica toți constituenții care pot fi extrași pentru testare anterior aplicării pe urechea șoarecilor. Substanțele de test ar trebui preparate zilnic, cu excepția cazului în care datele privind stabilitatea demonstrează faptul că acestea pot fi stocate.
|
Verificarea fiabilității
|
11.
|
Substanțele martor pozitive (PC) contribuie la demonstrarea performanței adecvate a testului răspunzând în mod adecvat și reproductibil la o substanță de sensibilizare de test pentru care magnitudinea răspunsului este bine cunoscută. Este recomandată includerea unei PC concomitente deoarece aceasta demonstrează competența laboratorului de a realiza corect fiecare test și permite evaluarea comparabilității și reproductibilității intra-laborator și inter-laborator. O serie de autorități de reglementare solicită, de asemenea, existența unei PC pentru fiecare studiu, utilizatorii sunt astfel încurajați să consulte autoritățile relevante înainte de realizarea testului LLNA: DA. Prin urmare, utilizarea sistematică a unei PC concomitente este încurajată pentru a evita necesitatea efectuării unor teste suplimentare pe animale, fapt ce este câteodată impus prin utilizarea unei PC testate periodic (a se vedea punctul 12). PC trebuie să reacționeze pozitiv la LLNA: DA la un nivel de expunere estimat să determine o creștere a IS ≥ 1,8 față de grupul martor negativ (NC). Doza de PC trebuie aleasă astfel încât să nu cauzeze iritare dermică excesivă sau toxicitate sistemică, iar inducerea răspunsului să fie reproductibilă, dar nu excesivă (de exemplu, un IS > 10 ar fi excesiv). Substanțele preferate pentru PC sunt aldehida hexil-cinamică 25 % (nr. CAS 101-86-0) și eugenol 25 % (nr. CAS 97-53-0,) în acetonă: ulei de măsline (4:1, v/v). Pot exista situații în care, cu o justificare adecvată, pot fi utilizate alte PC care îndeplinesc criteriile de mai sus.
|
|
12.
|
În timp ce includerea unui grup PC concomitent este recomandată, pot exista situații în care testarea periodică (la intervale ≤ 6 luni) a PC poate fi adecvată pentru laboratoarele care efectuează LLNA: DA în mod regulat (efectuează LLNA: DA cel puțin o dată pe lună) și care dețin o bază de date stabilită cu PC care demonstrează capacitatea laboratorului de a obține rezultate reproductibile și exacte cu PC. Performanța adecvată în ceea ce privește LLNA: DA poate fi demonstrată cu succes prin generarea de rezultate pozitive consecvente în cazul PC în cel puțin 10 teste independente efectuate pe o perioadă de timp rezonabilă (mai mică de un an).
|
|
13.
|
Un grup PC concomitent trebuie inclus întotdeauna în cazul în care se aduce o modificare procedurală la LLNA: DA (modificarea personalului instruit, modificarea materialelor și/sau a reactivilor utilizați în metoda de testare, modificarea echipamentului utilizat în metoda de testare, modificarea sursei animalelor de test), iar astfel de modificări trebuie documentate în rapoarte de laborator. Trebuie avut în vedere impactul modificărilor asupra caracterului adecvat al bazei de date istorice constituite anterior în ceea ce privește determinarea necesității de a constitui o nouă bază de date istorice pentru a documenta consecvența rezultatelor referitoare la PC.
|
|
14.
|
Experimentatorii trebuie să fie conștienți de faptul că decizia de a derula un studiu PC în mod periodic și nu concomitent are consecințe asupra acurateții și acceptabilității rezultatelor negative obținute în urma finalizării unui test fără PC concomitent, în intervalul dintre fiecare test periodic cu PC. De exemplu, dacă se obține un rezultat fals negativ la un test periodic al PC, rezultatele negative ale substanței de test obținute în intervalul dintre ultimul test acceptabil periodic al PC și testul PC periodic inacceptabil pot fi puse sub semnul întrebării. Implicațiile rezultatelor trebuie atent analizate în momentul luării deciziei de a include teste concomitente ale PC sau de a efectua doar teste periodice ale PC. Trebuie avută în vedere, de asemenea, utilizarea unui număr mai mic de animale în grupul PC concomitent în cazul în care acest lucru este justificat științific și dacă laboratorul demonstrează, pe baza datelor istorice specifice laboratorului, că poate fi utilizat un număr mai mic de șoareci (22).
|
|
15.
|
Deși substanța utilizată ca martor pozitiv trebuie testată împreună cu un vehicul despre care se știe că va provoca un răspuns constant (de exemplu, acetonă: ulei de măsline; 4:1, v/v), este posibil să existe unele situații prevăzute de lege în care este necesară, de asemenea, testarea cu un vehicul non-standard (preparat cu relevanță clinică/chimică) (23). În cazul în care substanța utilizată ca PC concomitent este testată într-un vehicul diferit față de substanța de test, atunci trebuie inclusă un VC separat pentru PC concomitent.
|
|
16.
|
În cazurile în care se evaluează substanțe de test aparținând unei clase chimice speciale sau care dau rezultate situate într-un anumit interval, substanțele etalon pot, de asemenea, să fie utile pentru a demonstra că metoda de testare funcționează corespunzător pentru detectarea potențialului de sensibilizare dermică al acestor tipuri de substanțe de test. Substanțele etalon adecvate trebuie să prezinte următoarele proprietăți:
|
—
|
similaritate structurală și funcțională cu clasa chimică a substanței de test;
|
|
—
|
caracteristici fizico-chimice cunoscute;
|
|
—
|
date justificative obținute cu ajutorul LLNA: DA;
|
|
—
|
date justificative obținute cu ajutorul altor modele animale și/sau de la om.
|
|
PROCEDURA DE TESTARE
Număr de animale și doze
|
17.
|
Fiecare grup tratat trebuie să fie format din minimum patru animale și se folosesc cel puțin trei concentrații ale substanței de test, plus un grup martor negativ tratat numai cu vehiculul utilizat împreună cu substanța de test și un martor pozitiv (paralel sau recent, în funcție de politica laboratorului în ceea ce privește punctele 11-15). Trebuie avută în vedere testarea de doze multiple pe martorul pozitiv, în special dacă testarea grupului martor pozitiv se face pe bază intermitentă. Animalele din grupurile martor trebuie manipulate și tratate în mod identic cu animalele din grupurile tratate, cu excepția faptului că nu li se administrează tratamentul cu substanța de test.
|
|
18.
|
Doza și vehiculul se aleg pe baza recomandărilor oferite în referințele (2) și (24). Dozele consecutive se aleg în mod normal dintr-o gamă adecvată de concentrații de 100 %, 50 %, 25 %, 10 %, 5 %, 2,5 %, 1 %, 0,5 % etc. Alegerea seriilor de concentrații utilizate trebuie însoțită de o justificare științifică adecvată. Eventualele date existente privind toxicitatea (de exemplu, toxicitatea acută și iritația dermică) și informații privind structura și caracteristicile fizico-chimice ale substanței de test de interes (și/sau a substanțelor asociate din punct de vedere structural) trebuie avute în vedere la alegerea celor trei concentrații consecutive astfel încât cea mai mare concentrație să maximizeze expunerea, evitând în același timp toxicitatea sistemică și/sau iritația dermică locală excesivă (24) (25). În lipsa unor astfel de informații, ar putea fi necesară o metodă pre-screening de testare (a se vedea punctele 21-24).
|
|
19.
|
Vehiculul nu trebuie să interfereze cu rezultatul testării sau să-l influențeze și trebuie selectat pe baza maximizării solubilității în vederea obținerii concentrației maxime posibile, producând în același timp o soluție/suspensie adecvată pentru aplicarea substanței de test. Vehiculele recomandate sunt acetona: ulei de măsline (4:1, v/v), N,N-dimetilformamidă, metil etil cetonă, propilen glicol și dimetil sulfoxid (6), dar se pot utiliza și altele dacă sunt furnizate justificări științifice suficiente. În anumite situații este posibil să fie necesară utilizarea unui solvent cu relevanță clinică sau a unui preparat comercial sub forma căruia este comercializată substanța de test ca martor suplimentar. Trebuie acordată o atenție specială pentru ca substanțele de test hidrofile să fie încorporate într-un sistem vehicul care să umezească pielea și să nu curgă imediat, prin încorporarea unor dizolvanți adecvați (de exemplu, 1 % Pluronic® L92). Prin urmare, vehiculele formate numai din apă trebuie evitate.
|
|
20.
|
Prelucrarea ganglionilor limfatici de la șoareci individuali permite evaluarea variabilității inter-indivizi și o comparare statistică a măsurătorilor privind substanța de test și a celor privind grupul martor tratat cu vehicul (a se vedea puncul 33). În plus, evaluarea posibilității de a reduce numărul de șoareci în grupul PC este realizabilă dacă se colectează date individuale referitoare la animale (22). Mai mult, o serie de autorități de reglementare solicită colectarea de date individuale referitoare la animale. Colectarea regulată de date specifice fiecărui animal contribuie la bunăstarea animalelor prin evitarea testelor duplicat care ar fi necesare dacă rezultatele obținute în alt mod (de exemplu, cu date colective pentru grupuri de animale) ar trebui ulterior înaintate unor autorități de reglementare care prevăd alte cerințe (de exemplu, furnizarea de date individuale).
|
Testarea preliminară
|
21.
|
În absența informațiilor necesare pentru determinarea celei mai mari doze pentru a fi testată (a se vedea punctul 18), trebuie efectuat un test pre-screening pentru a stabili doza adecvată de test în cadrul LLNA: DA. Scopul testării preliminare este de a furniza orientare în selectarea nivelului maxim al dozei care poate fi utilizat în cadrul studiului principal LLNA: DA, în cazul în care nu sunt disponibile informații privind concentrația care induce toxicitate sistemică (a se vedea punctul 24) și/sau iritație dermică locală excesivă (a se vedea punctul 23). Doza maximă testată trebuie să fie de 100 % din substanța de test pentru lichide sau concentrația maximă posibilă pentru substanțele solide sau suspensii.
|
|
22.
|
Testarea preliminară se derulează în condiții identice cu cele necesare studiului principal LLNA: DA, cu excepția cazului în care nu există o evaluare a proliferării ganglionilor limfatici și pot fi utilizate mai puține animale în grupul testat. Se recomandă una sau două animale pe grup testat. Toți șoarecii sunt observați zilnic pentru identificarea semnelor clinice de toxicitate sistemică sau de iritație locală la locul aplicării. Greutatea corporală este consemnată anterior testării preliminare și anterior finalizării acesteia (ziua 8). Ambele urechi ale fiecărui șoarece sunt observate pentru depistarea eritemelor și se punctează folosind tabelul 1 (25). Măsurătorile privind grosimea urechii sunt efectuate cu ajutorul unui instrument de măsurare a grosimii (de exemplu, micrometru digital sau instrumentul de măsurare a grosimii Peacock Dial) în ziua 1 (pre-doză), ziua 3 (după aproximativ 48 de ore de la administrarea primei doze), ziua 7 (24 de ore înainte de încheiere) și ziua 8. În plus, în ziua 8, grosimea urechii poate fi măsurată pornind de la un eșantion de ureche, prelevat după eutanasierea animalelor. Iritația dermică locală excesivă este indicată printr-un punctaj acordat eritemului de ≥ 3 și/sau o creștere a grosimii urechii de cel puțin 25 % în oricare zi a măsurării (26) (27). Cea mai mare doză selectată pentru studiul principal LLNA: DA este doza imediat inferioară din seria de concentrații pre-screening (a se vedea punctul 18) care nu induce toxicitate sistemică și/sau iritație dermică locală excesivă.
Tabelul 1
Punctaje acordate eritemelor
|
Observație
|
Punctaj
|
|
Fără eritem
|
0
|
|
Eritem foarte ușor (abia perceptibil)
|
1
|
|
Eritem bine definit
|
2
|
|
Eritem moderat spre grav
|
3
|
|
Eritem grav (roșu violaceu) cu formare de escare care împiedică punctarea eritemului
|
4
|
|
|
23.
|
Pe lângă o creștere cu 25 % a grosimii urechii (26) (27), o creștere semnificativă din punct de vedere statistic a grosimii urechii la șoarecii tratați comparativ cu șoarecii martor a fost, de asemenea, utilizată pentru a identifica substanțele iritante din cadrul LLNA (28) (29) (30) (31) (32) (33) (34). Cu toate acestea, deși pot surveni creșteri semnificative din punct de vedere statistic atunci când grosimea urechii este sub 25 %, acestea nu au fost asociate în mod specific cu iritația excesivă (30) (31) (32) (33) (34).
|
|
24.
|
Următoarele observații clinice pot indica toxicitate sistemică (35) atunci când sunt utilizate ca parte a unei evaluări integrate și, prin urmare, pot indica doza maximă utilizată în studiul LLNA: DA principal: modificări ale funcției sistemului nervos (de exemplu, piloerecție, ataxie, tremor și convulsii); modificări ale comportamentului (de exemplu, agresivitate, modificări ale activității de curățare, modificare semnificativă a nivelului de activitate); tulburări respiratorii (de exemplu, modificări ale frecvenței și intensității respirației precum dispnee, respirație greoaie și zgomotoasă) și modificări legate de consumul de hrană și apă. În plus, semnele de letargie și/sau lipsa reacției și orice semne clinice indicative ale unei dureri sau suferințe mai mari sau persistente sau o scădere a greutății corporale cu mai mult de 5 % din ziua 1 până în ziua 8, precum și mortalitatea trebuie incluse în evaluare. Animalele muribunde sau cele care manifestă dureri evidente sau prezintă semne de suferință gravă și prelungită sunt eutanasiate (36).
|
Schema de tratament a studiului principal
|
25.
|
Pentru acest test, schema de tratament este următoarea:
— Ziua 1: Se identifică și se înregistrează greutatea fiecărui animal în parte, precum și toate observațiile clinice. Se aplică o soluție apoasă de lauril sulfat de sodiu (SLS) 1 % în regiunea dorsală a fiecărei urechi cu ajutorul unei pensule muiate în soluția SLS, astfel încât să se acopere întreaga suprafață dorsală a fiecărei urechi cu patru până la cinci aplicări. După o oră de la aplicarea tratamentului cu SLS, se aplică 25 μL de substanță de test diluată în mod adecvat, doar din vehicul sau din PC (concomitent sau recent, în funcție de politica laboratorului cu privire la punctele 11-15), în regiunea dorsală a fiecărei urechi.
— Zilele 2, 3 și 7: Se repetă tratamentul prealabil cu soluție apoasă de SLS 1 % apoi se aplică substanța de test urmând aceeași procedură ca în ziua 1.
— Zilele 4, 5 și 6: Nu se aplică tratament.
— Ziua 8: Se înregistrează greutatea fiecărui animal și toate observațiile clinice. După aproximativ 24-30 de ore de la începutul aplicării (în ziua 7), animalele se eutanasiază. Se excizează ganglionii limfatici auriculari de drenare de la fiecare ureche de la toate animalele și se prelucrează separat în soluție tamponată cu fosfat (PBS) pentru fiecare animal. Detalii și diagrame privind identificarea și disecția ganglionilor limfatici pot fi găsite în referință (22). Pentru a monitoriza în continuare răspunsul dermic local în studiul principal, pot fi incluși în protocolul de studiu parametri adiționali precum punctarea eritemului din zona urechii sau măsurări ale grosimii urechii (obținute fie cu ajutorul unui instrument de măsurare a grosimii, fie prin determinări ale greutății eșantioanelor de urechi ulterior necropsiei).
|
Prepararea suspensiilor celulare
|
26.
|
Pentru fiecare șoarece, se prepară o suspensie de celule izolate provenind din ganglionii limfatici (LNC) excizați bilateral, prin plasarea ganglionilor între două lamele din sticlă pe care se exersează o presiune ușoară pentru a strivi eșantioanele. După ce s-a verificat faptul că țesutul s-a răspândit adecvat, se separă cele două lamele. Se suspendă țesutul de pe ambele lamele în PBS, înclinând fiecare lamelă deasupra cutiei Petri apoi se clătește cu PBS răzuind concomitent țesutul de pe lamelă cu o rașchetă pentru celule. Ganglionii limfatici de la animalele din NC fiind mici, este important ca aceștia să fie tratați cu grijă pentru a se evita orice efecte artificiale induse valorilor IS. Volumul total de PBS utilizat pentru clătirea celor două lamele este de 1 mL. Se omogenizează ușor suspensia de LNC în cutia Petri cu ajutorul rașchetei pentru celule. Se prelevează apoi o alicotă de 20 μL de suspensie LNC cu o micropipetă, având grijă să nu se desprindă membrana vizibilă cu ochiul liber, apoi aceasta se amestecă cu 1,98 mL de PBS pentru a se obține un eșantion de 2 mL. Un al doilea eșantion de 2 mL se prepară respectând aceeași procedură, astfel încât să fie preparate 2 eșantioane pentru fiecare animal.
|
Determinarea proliferării celulare (măsurarea conținutului de ATP din limfocite)
|
27.
|
Creșterile de conținut ATP din ganglionii limfatici se măsoară prin metoda luciferină/luciferază cu un set de măsurare a ATP care măsoară bioluminiscența, exprimată în unități relative de luminiscență (RLU). Timpul dintre eutanasierea animalelor și cuantificarea conținutului de ATP pentru fiecare individ trebuie să fie menținut constant și să nu depășească aproximativ 30 de minute deoarece se consideră că ATP se diminuează treptat de la momentul eutanasierii (12). Astfel, seria de proceduri de la excizarea nodulilor limfatici auriculari până la măsurarea conținutului de ATP trebuie să fie realizată în 20 de minute, conform unui program pre-stabilit valabil pentru toate animalele. Luminiscența datorată conținutului de ATP trebuie măsurată în toate eșantioanele de 2 mL, și anume un total de două măsurători ale ATP pentru fiecare animal. Se determină apoi media rezultatelor, iar aceasta este utilizată în calculele următoare (a se vedea puncul 30).
|
OBSERVAȚII
Observații clinice
|
28.
|
Fiecare șoarece se examinează atent cel puțin o dată pe zi pentru identificarea oricăror semne clinice, fie de iritații la locul aplicării, fie de toxicitate sistemică. Toate observațiile se consemnează sistematic în înregistrările individuale pentru fiecare șoarece. Planurile de monitorizare trebuie să includă criterii pentru identificarea promptă a șoarecilor care prezintă toxicitate sistemică, iritație dermică locală excesivă sau coroziunea pielii în vederea eutanasierii (36).
|
Greutatea corporală
|
29.
|
Astfel cum se arată la punctul 25, greutatea corporală a fiecărui animal se măsoară la începutul testului și în momentul prevăzut pentru eutanasiere.
|
CALCULAREA REZULTATELOR
|
30.
|
Rezultatele pentru fiecare grup tratat se exprimă printr-un indice de stimulare (IS) mediu. IS se calculează împărțind media RLU/șoarece în fiecare grup tratat cu substanța de test și grupul PC la media RLU/șoarece în grupul martor tratat cu solvent/vehicul (VC). IS mediu pentru VC este, prin urmare, egal cu 1.
|
|
31.
|
Un rezultat este considerat pozitiv atunci când IS ≥ 1,8 (10). Cu toate acestea, intensitatea relației doză-răspuns, semnificația statistică și coerența răspunsurilor obținute cu solvent/vehicul și PC pot fi, de asemenea, utilizate în declararea unui rezultat limită (și anume, cu valori ale IS cuprinse între 1,8 și 2,5) ca fiind pozitiv (2) (3) (37).
|
|
32.
|
În cazul unui răspuns pozitiv ambiguu cu valori ale IS cuprinse între 1,8 și 2,5, experimentatorii pot examina informații adiționale precum relația doză-răspuns, manifestările de toxicitate sistemică sau iritație excesivă și, dacă este cazul, semnificația statistică împreună cu valorile IS pentru a confirma că astfel de rezultate sunt pozitive (10). Diferitele proprietăți ale substanței de test trebuie, de asemenea, luate în considerare, inclusiv o eventuală analogie structurală cu agenți de sensibilizare dermică cunoscuți, declanșarea unei iritații dermice excesive la șoarece și natura relației doză-răspuns observată. Aceste considerații, precum și altele, sunt examinate în detaliu într-un alt document (4).
|
|
33.
|
Colectarea de date pentru fiecare șoarece permite o analiză statistică a existenței și a intensității relației doză-răspuns pe baza datelor. Orice evaluare statistică poate include o evaluare a relației doză-răspuns, precum și comparări ale grupurilor testate adaptate corespunzător (de exemplu, comparări pe perechi de grupuri de doză cu grupul martor concomitent tratat cu solvent/vehicul). Analizele statistice pot include, de exemplu, o regresie liniară sau testul William pentru evaluarea funcției doză-răspuns, precum și testul Dunnett pentru comparațiile pe perechi. Pentru alegerea metodei adecvate de analiză statistică, experimentatorul trebuie să aibă în vedere posibilele inegalități la nivelul varianțelor și alte probleme conexe care ar putea impune o transformare a datelor sau o analiză statistică non-parametrică. În orice caz, experimentatorul ar putea fi nevoit să calculeze indicii de stimulare și să efectueze analizele statistice cu sau fără anumite puncte de date (denumite uneori «valori aberante»).
|
DATE ȘI RAPORTARE
Date
|
34.
|
Datele se sistematizează sub formă de tabele indicând valorile RLU pentru fiecare animal, media RLU/animal pentru fiecare grup, marja de eroare asociată (de exemplu, deviația standard, eroarea standard asociată mediei) și indicele de stimulare mediu pentru fiecare grup tratat în raport cu grupul martor concomitent tratat cu solvent/vehicul.
|
Raportul de testare
|
35.
|
Raportul de testare trebuie să includă următoarele informații:
|
|
Substanțele chimice de test și cele martor:
|
—
|
date de identificare (de exemplu, numărul CAS și numărul CE, dacă se cunosc; sursa; puritatea; impuritățile cunoscute; numărul lotului);
|
|
—
|
starea fizică și proprietățile fizice și chimice (de exemplu, volatilitate, stabilitate, solubilitate);
|
|
—
|
în cazul amestecurilor, compoziția și procentul relativ al fiecărui constituent;
|
|
|
|
Solvent/vehicul:
|
—
|
date de identificare (puritate; concentrație, dacă este cazul; volumul utilizat);
|
|
—
|
justificarea alegerii vehiculului;
|
|
|
|
Animalele de experiență:
|
—
|
proveniența șoarecilor CBA;
|
|
—
|
statutul microbiologic al animalelor, dacă se cunoaște;
|
|
—
|
numărul și vârsta animalelor;
|
|
—
|
sursa animalelor, condițiile de adăpostire, regimul alimentar etc.;
|
|
|
|
Condiții de testare:
|
—
|
sursa, numărul lotului, datele furnizate de producător cu privire la asigurarea calității/controlului de calitate pentru setul ATP;
|
|
—
|
detalii privind prepararea și aplicarea substanței de testat;
|
|
—
|
justificarea dozei alese (inclusiv rezultatele testării preliminare, dacă este cazul);
|
|
—
|
concentrațiile vehiculului și ale substanței de test utilizate și cantitatea totală de substanță aplicată;
|
|
—
|
detalii privind calitatea hranei și a apei (inclusiv tipul/sursa regimului alimentar, sursele de apă);
|
|
—
|
detalii privind schemele de tratament și de eșantionare;
|
|
—
|
metodele de măsurare a toxicității;
|
|
—
|
criteriile pentru stabilirea studiilor ca fiind pozitive sau negative;
|
|
—
|
detalii privind oricare abateri de la protocol și explicații referitoare la modul în care abaterea afectează conceptul studiului și rezultatele;
|
|
|
|
Verificarea fiabilității:
|
—
|
un rezumat al rezultatelor obținute la ultima verificare a fiabilității, inclusiv informații privind substanța de test, concentrația și vehiculul utilizat;
|
|
—
|
rezultatele martorilor specifici laboratorului de testare pentru pozitiv concomitent și/sau istoric, precum și pentru martorul negativ concomitent;
|
|
—
|
în cazul în care nu a fost inclus un grup PC concomitent, datele și raportul de laborator pentru cel mai recent grup PC periodic și un raport detaliind datele istorice privind PC specifice laboratorului, astfel încât să se justifice de ce nu a fost realizat un grup PC concomitent;
|
|
|
|
Rezultate:
|
—
|
greutatea fiecărui animal la începutul testului și în momentul prevăzut pentru eutanasiere, precum și media și marja de eroare asociată (de exemplu, deviația standard, eroarea standard asociată mediei) pentru fiecare grup tratat;
|
|
—
|
momentul declanșării și semnele de toxicitate, inclusiv iritația dermică la locul administrării, dacă este cazul, pentru fiecare animal;
|
|
—
|
ora eutanasierii animalului și ora măsurării conținutului de ATP pentru fiecare animal;
|
|
—
|
un tabel cu valorile RLU și IS pe șoarece pentru fiecare grup tratat;
|
|
—
|
media și marja de eroare asociată (de exemplu, deviația standard, eroarea standard asociată mediei) a RLU/șoarece pentru fiecare grup tratat și rezultatele analizei valorilor aberante în cadrul fiecărui grup;
|
|
—
|
indicii de stimulare obținuți și determinarea adecvată a variabilității ținând cont de variațiile dintre animale atât în grupurile tratate cu substanța de test, cât și în grupurile martor;
|
|
—
|
analize statistice, dacă este cazul;
|
|
|
|
Discutarea rezultatelor:
|
—
|
un scurt comentariu privind rezultatele, analiza relației doză-răspuns și analize statistice, dacă este cazul, și o concluzie care să indice dacă substanța de test trebuie considerată agent de sensibilizare dermică.
|
|
|
BIBLIOGRAFIE
|
(1)
|
OECD (2010), Skin Sensitisation: Local Lymph Node Assay, Test Guideline No. 429, Guidelines for the Testing of Chemicals, OECD, Paris. Disponibil la: [http://www.oecd.org/env/testguidelines]
|
|
(2)
|
Chamberlain, M. and Basketter, D.A. (1996), The local lymph node assay: status of validation. Food Chem, Toxicol., 34, 999-1002.
|
|
(3)
|
Basketter, D.A., Gerberick, G.F., Kimber, I. and Loveless, S.E. (1996), The local lymph node assay: A viable alternative to currently accepted skin sensitisation tests. Food Chem, Toxicol., 34, 985-997.
|
|
(4)
|
Basketter, D.A., Gerberick, G.F. and Kimber, I. (1998), Strategies for identifying false positive responses in predictive sensitisation tests. Food Chem. Toxicol., 36, 327-333.
|
|
(5)
|
Van Och, F.M.M., Slob, W., De Jong, W.H., Vandebriel, R.J. and Van Loveren, H. (2000), A quantitative method for assessing the sensitising potency of low molecular weight chemicals using a local lymph node assay: employment of a regression method that includes determination of uncertainty margins. Toxicol., 146, 49-59.
|
|
(6)
|
ICCVAM (1999), The murine local lymph node Assay: A test method for assessing the allergic contact dermatitis potential of chemicals/compounds: The results of an independent peer review evaluation coordinated by the Interagency Coordinating Committee on the Validation of Alternative Methods (ICCVAM) and the National Toxicology Program Center for the Evaluation of Alternative Toxicological Methods (NICETAM). NIH Publication No: 99-4494. Research Triangle Park, N.C. Disponibil la: [http://iccvam.niehs.nih.gov/docs/immunotox_docs/llna/llnarep.pdf]
|
|
(7)
|
Dean, J.H., Twerdok, L.E., Tice, R.R., Sailstad, D.M., Hattan, D.G., Stokes, W.S. (2001), ICCVAM evaluation of the murine local lymph node assay: II. Conclusions and recommendations of an independent scientific peer review panel. Reg. Toxicol. Pharmacol. 34, 258-273.
|
|
(8)
|
Haneke, K.E., Tice, R.R., Carson, B.L., Margolin, B.H., Stokes, W.S. (2001), ICCVAM evaluation of the murine local lymph node assay: III. Data analyses completed by the national toxicology program interagency center for the evaluation of alternative toxicological methods. Reg. Toxicol. Pharmacol. 34, 274-286.
|
|
(9)
|
Sailstad, D.M., Hattan, D., Hill, R.N., Stokes, W.S. (2001), ICCVAM evaluation of the murine local lymph node assay: I. The ICCVAM review process. Reg. Toxicol. Pharmacol. 34, 249-257.
|
|
(10)
|
ICCVAM (2010), ICCVAM Test Method Evaluation Report. Nonradioactive local lymph node assay: modified by Daicel Chemical Industries, Ltd., based on ATP content test method protocol (LLNA: DA). NIH Publication No. 10-7551A/B. Research Triangle Park, NC: National Institute of Environmental Health Sciences. Disponibil la: [http://iccvam.niehs.nih.gov/methods/immunotox/llna-DA/TMER.htm]
|
|
(11)
|
ICCVAM (2009), Independent Scientific Peer Review Panel Report: Updated validation status of new versions and applications of the murine local lymph node assay: a test method for assessing the allergic contact dermatitis potential of chemicals and products. Research Triangle Park, NC: National Institute of Environmental Health Sciences. Disponibil la: [http://iccvam.niehs.nih.gov/docs/immunotox_docs/LLNAPRPRept2009.pdf].
|
|
(12)
|
Idehara, K., Yamagishi, G., Yamashita, K. and Ito, M. (2008), Characterization and evaluation of a modified local lymph node assay using ATP content as a non-radio isotopic endpoint. J. Pharmacol. Toxicol. Meth., 58, 1-10.
|
|
(13)
|
Omori, T., Idehara, K., Kojima, H., Sozu, T., Arima, K., Goto, H., Hanada, T., Ikarashi, Y., Inoda, T., Kanazawa, Y., Kosaka, T., Maki, E., Morimoto, T., Shinoda, S., Shinoda, N., Takeyoshi, M., Tanaka, M., Uratani, M., Usami, M., Yamanaka, A., Yoneda, T., Yoshimura, I. and Yuasa, A. (2008), Interlaboratory validation of the modified murine local lymph node assay based on adenosine triphosphate measurement. J. Pharmacol. Toxicol. Meth., 58, 11-26.
|
|
(14)
|
OECD (1992), Skin Sensitisation, Test Guideline No. 406, Guidelines for Testing of Chemicals, OECD, Paris. Disponibil la: [http://www.oecd.org/env/testguidelines]
|
|
(15)
|
Kreiling, R., Hollnagel, H.M., Hareng, L., Eigler, L., Lee, M.S., Griem, P., Dreessen, B., Kleber, M., Albrecht, A., Garcia, C. and Wendel, A. (2008), Comparison of the skin sensitising potential of unsaturated compounds as assessed by the murine local lymph node assay (LLNA) and the guinea pig maximization test (GPMT). Food Chem. Toxicol., 46, 1896-1904.
|
|
(16)
|
Basketter, D., Ball, N., Cagen, S., Carrillo, J.C., Certa, H., Eigler, D., Garcia, C., Esch, H., Graham, C., Haux, C., Kreiling, R. and Mehling, A. (2009), Application of a weight of evidence approach to assessing discordant sensitisation datasets: implications for REACH. Reg. Toxicol. Pharmacol., 55, 90-96.
|
|
(17)
|
Crouch, S.P., Kozlowski, R., Slater, K.J. and Fletcher J. (1993), The use of ATP bioluminescence as a measure of cell proliferation and cytotoxicity. J. Immunol. Meth., 160, 81-88.
|
|
(18)
|
Ishizaka, A., Tono-oka, T. and Matsumoto, S. (1984), Evaluation of the proliferative response of lymphocytes by measurement of intracellular ATP. J. Immunol. Meth., 72, 127-132.
|
|
(19)
|
Dexter, S.J., Cámara, M., Davies, M. and Shakesheff, K.M. (2003), Development of a bioluminescent ATP assay to quantify mammalian and bacterial cell number from a mixed population. Biomat., 24, 27-34.
|
|
(20)
|
Lundin A. (2000), Use of firefly luciferase in ATP-related assays of biomass, enzymes, and metabolites. Meth. Enzymol., 305, 346-370.
|
|
(21)
|
ILAR (1996), Institute of Laboratory Animal Research (ILAR) Guide for the Care and Use of Laboratory Animals. 7th ed. Washington, DC: National Academies Press.
|
|
(22)
|
ICCVAM (2009), Recommended Performance Standards: Murine Local Lymph Node Assay, NIH Publication Number 09-7357, Research Triangle Park, NC: National Institute of Environmental Health Science. Disponibil la: [http://iccvam.niehs.nih.gov/docs/immunotox_docs/llna-ps/LLNAPerfStds.pdf]
|
|
(23)
|
McGarry, H.F. (2007), The murine local lymph node assay: regulatory and potency considerations under REACH. Toxicol., 238, 71-89.
|
|
(24)
|
Kimber, I., Dearman, R.J., Scholes E.W. and Basketter, D.A. (1994), The local lymph node assay: developments and applications. Toxicol., 93, 13-31.
|
|
(25)
|
OECD (2002), Acute Dermal Irritation/Corrosion, Test Guideline No. 404, Guidelines for Testing of Chemicals, OECD, Paris. Disponibil la: [http://www.oecd.org/env/testguidelines]
|
|
(26)
|
Reeder, M.K., Broomhead, Y.L., DiDonato, L. and DeGeorge, G.L. (2007), Use of an enhanced local lymph node assay to correctly classify irritants and false positive substances. Toxicologist, 96, 235.
|
|
(27)
|
ICCVAM (2009), Nonradioactive Murine Local Lymph Node Assay: Flow Cytometry Test Method Protocol (LLNA: BrdU-FC) Revised Draft Background Review Document. Research Triangle Park, NC: National Institute of Environmental Health Sciences. Disponibil la: [http://iccvam.niehs.nih.gov/methods/immunotox/fcLLNA/BRDcomplete.pdf].
|
|
(28)
|
Hayes, B.B., Gerber, P.C., Griffey, S.S. and Meade, B.J. (1998), Contact hypersensitivity to dicyclohexylcarbodiimide and diisopropylcarbodiimide in female B6C3F1 mice. Drug. Chem. Toxicol., 21, 195-206.
|
|
(29)
|
Homey, B., von Schilling, C., Blumel, J., Schuppe, H.C., Ruzicka, T., Ahr, H.J., Lehmann, P. and Vohr, V.W. (1998), An integrated model for the differentiation of chemical-induced allergic and irritant skin reactions. Toxicol. Appl. Pharmacol., 153, 83-94.
|
|
(30)
|
Woolhiser, M.R., Hayes, B.B. and Meade, B.J. (1998), A combined murine local lymph node and irritancy assay to predict sensitisation and irritancy potential of chemicals. Toxicol. Meth., 8, 245-256.
|
|
(31)
|
Hayes, B.B. and Meade, B.J. (1999), Contact sensitivity to selected acrylate compounds in B6C3F1 mice: relative potency, cross reactivity, and comparison of test methods. Drug Chem. Toxicol., 22, 491-506.
|
|
(32)
|
Ehling, G., Hecht, M., Heusener, A., Huesler, J., Gamer, A.O., van Loveren, H., Maurer, T., Riecke, K., Ullmann, L., Ulrich, P., Vandebriel, R. and Vohr, H.W. (2005), A European inter-laboratory validation of alternative endpoints of the murine local lymph node assay: first round. Toxicol., 212, 60-68.
|
|
(33)
|
Vohr, H.W. and Ahr, H.J. (2005), The local lymph node assay being too sensitive? Arch. Toxicol., 79, 721-728.
|
|
(34)
|
Patterson, R.M., Noga, E. and Germolec, D. (2007), Lack of evidence for contact sensitisation by Pfiesteria extract. Environ. Health Perspect., 115, 1023-1028.
|
|
(35)
|
ICCVAM (2009), Report on the ICCVAM-NICEATM/ECVAM/JaCVAM Scientific Workshop on Acute Chemical Safety Testing: Advancing In Vitro Approaches and Humane Endpoints for Systemic Toxicity Evaluations. Research Triangle Park, NC: National Institute of Environmental Health Sciences. Disponibil la: [http://iccvam.niehs.nih.gov/methods/acutetox/Tox_workshop.htm]
|
|
(36)
|
OECD (2000), Guidance Document on the Recognition, Assessment and Use of Clinical Signs as Humane Endpoints for Experimental Animals Used in Safety Evaluation, Environmental Health and Safety Monograph Series on Testing and Assessment No. 19, ENV/JM/MONO(2000)7, OECD, Paris. Disponibil la: [http://www.oecd.org/env/testguidelines]
|
|
(37)
|
Kimber, I., Hilton, J., Dearman, R.J., Gerberick, G.F., Ryan, C.A., Basketter, D.A., Lea, L., House, R.V., Ladies, G.S., Loveless, S.E. and Hastings, K.L. (1998), Assessment of the skin sensitisation potential of topical medicaments using the local lymph node assay: An interlaboratory exercise. J. Toxicol. Environ. Health, 53 563-79.
|
|
(38)
|
OECD (2005), Guidance Document on the Validation and International Acceptance of New or Updated Test Methods for Hazard Assessment, Environment, Health and Safety Monograph Series on Testing and Assessment No. 34, ENV/JM/MONO (2005)14, OECD, Paris. Disponibil la: [http://www.oecd.org/env/testguidelines]
|
Apendicele 1
DEFINIȚII
Acuratețe: Gradul de apropiere dintre rezultatele metodei de testare și valorile de referință acceptate. Aceasta constituie una dintre caracteristicile de performanță ale metodei de testare și unul dintre aspectele relevanței acesteia. Termenul este adesea utilizat în paralel cu termenul sinonim «concordanță» pentru a desemna proporția de rezultate corecte ale unei metode de testare (38).
Substanță etalon: O substanță de sensibilizare sau de non-sensibilizare utilizată ca standard pentru comparația cu o substanță de test. O substanță etalon ar trebui să aibă următoarele proprietăți: (i) sursă (surse) compatibilă (compatibile) și sigure; (ii) asemănări structurale și funcționale cu clasa de substanțe care este testată; (iii) caracteristici fizico-chimice cunoscute; (iv) date justificative privind efecte cunoscute; și (v) influență cunoscută în intervalul de răspuns dorit.
Fals negativ: O substanță de test identificată incorect ca negativă sau inactivă printr-o metodă de testare, aceasta fiind de fapt pozitivă sau activă.
Fals pozitiv: O substanță de test identificată incorect ca pozitivă sau activă în urma unui test, aceasta fiind de fapt negativă sau inactivă.
Pericol: Potențialul de efect negativ asupra mediului sau sănătății. Efectul negativ se manifestă doar atunci când există o expunere la un nivel suficient.
Reproductibilitatea inter-laborator: Un indicator al măsurii în care diferite laboratoare calificate, utilizând același protocol și testând aceleași substanțe de test, pot produce rezultate similare din punct de vedere calitativ și cantitativ. Reproductibilitatea inter-laborator este determinată în timpul proceselor de validare prealabilă și de validare și indică măsura în care un test poate fi transferat cu succes inter-laborator, denumită, de asemenea, reproductibilitate între laboratoare (38).
Reproductibilitatea intra-laborator: O determinare a măsurii în care persoane calificate din cadrul aceluiași laborator pot repeta cu succes rezultatele la momente diferite, utilizând un protocol specific. Aceasta este denumită, de asemenea, reproductibilitate în cadrul laboratorului (38).
Valoare aberantă: O valoare aberantă este o observație considerabil diferită de alte valori în cadrul unui eșantion aleatoriu dintr-o populație.
Asigurarea calității: Un proces de gestionare prin care aderarea la standardele și cerințele de testare în laborator, procedurile de păstrare a înregistrărilor și acuratețea transferului de date sunt evaluate de către persoane independente de cele care au efectuat testarea.
Fiabilitate: Măsura în care o metodă de testare poate fi reprodusă în timp, în același laborator sau în laboratoare diferite, folosind același protocol. Aceasta se evaluează prin calcularea reproductibilității intra-laborator sau inter-laborator (38).
Sensibilizare dermică: Un proces imunologic care rezultă atunci când un individ susceptibil este expus topic la un alergen chimic favorizant care provoacă un răspuns cutanat imun ce poate conduce la dezvoltarea unei sensibilizări de contact.
Indice de stimulare (IS): O valoare calculată pentru a evalua potențialul de sensibilizare dermică al unei substanțe de test, și anume raportul dintre proliferarea înregistrată la grupurile tratate și cea înregistrată la grupul martor concomitent tratat cu vehicul.
Substanța de test (denumită, de asemenea, substanță chimică de test): Orice substanță sau amestec testat prin intermediul prezentei metode de testare.
B.51. SENSIBILIZARE DERMICĂ: TESTUL LOCAL PE GANGLIONI LIMFATICI: BrdU-ELISA
INTRODUCERE
|
1.
|
Orientările OCDE privind testarea substanțelor chimice și metodele de testare UE bazate pe acestea sunt periodic revizuite în lumina progresului științific, modificând necesitățile de reglementare și considerațiile referitoare la bunăstarea animalelor. Prima metodă de testare (MT) (B.42) pentru determinarea sensibilizării dermice la șoarece, testul local pe ganglioni limfatici (LLNA; orientarea OCDE privind testarea nr. 429), a fost revizuită (1 și capitolul B.42 din prezenta anexă). Detaliile privind validarea LLNA, precum și o revizuire a lucrărilor asociate au fost publicate (2) (3) (4) (5) (6) (7) (8) (9). În cadrul metodei LLNA, marcajul radioizotopic cu timidină sau iod este utilizat pentru a măsura proliferarea limfocitelor, prin urmare, testul are o aplicare limitată în regiunile unde achiziționarea, utilizarea sau eliminarea produselor radioactive pun probleme. LLNA: BrdU-ELISA (Enzyme-Linked Immunosorbent Assay – testul de imunoabsorbție prin enzimă legată) este o variantă non-radioactivă a MT LLNA, care utilizează 5-bromo-2-dezoxiuridina (BrdU) (nr. CAS 59-14-3) non-radiomarcată într-un test de tip ELISA pentru a măsura proliferarea limfocitelor. Metoda de testare LLNA: BrdU-ELISA a fost validată, revizuită și recomandată de către o echipă internațională de examinare care a recunoscut utilitatea acesteia în identificarea, între anumite limite, a substanțelor chimice care au sau nu un efect de sensibilizare a pielii (10) (11) (12). Prezenta MT vizează evaluarea potențialului de sensibilizare dermică al substanțelor chimice (substanțe și amestecuri) la animale. Capitolul B.6 din prezenta anexă și orientarea OCDE privind testarea nr. 406 utilizează testele pe cobai, și anume testul de maximizare la cobai și testul Buehler (14). LLNA (capitolul B.42 din prezenta anexă; orientarea OCDE privind testarea OECD nr. 429) și cele două variante non-radioactive, LLNA: BrdU-ELISA (capitolul B.51 din prezenta anexă; orientarea OCDE privind testarea nr. 442 B) și LLNA: DA (capitolul B.50 din prezenta anexă; orientarea OCDE privind testarea nr. 442 A), sunt mai avantajoase decât testele pe cobai din B.6 și orientarea OCDE privind testarea nr. 406 (13) în termeni de reducere și de rafinare a utilizării animalelor.
|
|
2.
|
În mod similar cu LLNA, metoda LLNA: BrdU-ELISA studiază faza de inducție a sensibilizării dermice și furnizează date cantitative adecvate pentru evaluarea relației doză-răspuns. În plus, abilitatea de a detecta agenții de sensibilizare dermică fără a recurge la radiomarcarea ADN-ului elimină riscurile profesionale de expunere la radiații și problemele legate de gestionarea deșeurilor. În schimb, aceasta ar putea presupune o creștere a numărului de șoareci utilizați pentru detectarea agenților de sensibilizare dermică determinând, cu toate acestea, diminuarea numărului de cobai pentru testarea potențialului de sensibilizare dermică (și anume, B.6; orientarea OCDE privind testarea nr. 406) (13).
|
DEFINIȚII
|
3.
|
Definițiile utilizate sunt furnizate în apendicele 1.
|
CONSIDERAȚII INIȚIALE ȘI LIMITE
|
4.
|
Metoda LLNA: BrdU-ELISA este o variantă a metodei LLNA vizând identificarea potențialului de sensibilizare dermică al substanțelor chimice, între anumite limite. Aceasta nu implică în mod necesar faptul că LLNA: BrdU-ELISA trebuie să înlocuiască sistematic LLNA sau testele pe cobai (și anume, B.6; orientarea OCDE privind testarea nr. 406) (13), ci, mai curând, că testul reprezintă un instrument de o calitate similară care poate fi utilizat ca alternativă și ale cărui rezultate pozitive sau negative nu mai necesită, în general, o confirmare suplimentară (10) (11). Anterior derulării studiului, laboratorul de testare trebuie să colecteze toate informațiile disponibile cu privire la substanța de test. Astfel de informații includ identitatea și structura chimică a substanței de test; proprietățile sale fizico-chimice; rezultatelor tuturor celorlalte teste de toxicitate in vitro sau in vivo pentru substanța de test și datele toxicologice privind substanțele chimice asociate din punct de vedere structural. Aceste informații trebuie analizate pentru a determina dacă metoda LLNA: BrdU-ELISA este adecvată pentru substanța de test [având în vedere incompatibilitatea anumitor tipuri de substanțe chimice cu LLNA: BrdU-ELISA (a se vedea puncul 5) și pentru a contribui la alegerea dozelor adecvate.
|
|
5.
|
Metoda LLNA: BrdU-ELISA este o metodă in vivo și, prin urmare, nu elimină utilizarea animalelor pentru evaluarea sensibilizării alergice de contact. Cu toate acestea, comparativ cu testele pe cobai (B.6; orientarea OCDE privind testarea nr. 406), aceasta are potențialul de a reduce numărul de animale utilizate în acest scop (13). Mai mult, LLNA: BrdU-ELISA oferă o rafinare importantă a modului în care sunt utilizate animalele pentru testarea sensibilizării alergice de contact, deoarece, spre deosebire de B.6 și orientarea OCDE privind testarea nr. 406, LLNA: BrdU-ELISA nu se bazează pe declanșarea reacțiilor de hipersensibilitate dermică prin manifestarea declanșării. În plus, LLNA: BrdU-ELISA nu necesită utilizarea unui adjuvant ca în cazul testului de maximizare pe cobai (capitolul B.6 din prezenta anexă, 13). Astfel, LLNA: BrdU-ELISA reduce suferința animalelor. În ciuda avantajelor LLNA: BrdU-ELISA comparativ cu B.6 și cu orientarea OCDE privind testarea nr. 406 (13), există anumite limite care ar putea impune utilizarea metodei B.6 sau a orientării OCDE privind testarea nr. 406 [de exemplu, testarea anumitor metale, rezultate fals pozitive cu anumite substanțe iritante pentru piele (precum anumite substanțe tensioactive) (6) (1 și capitolul B.42 din prezenta anexă), solubilitatea substanței de test]. În plus, anumite substanțe sau clase de substanțe chimice conținând grupele funcționale demonstrate ca reprezentând posibili factori de confuzie (15) ar putea impune utilizarea testelor pe cobai [de exemplu, B.6; orientarea OCDE privind testarea nr. 406 (13)]. Limitele identificate pentru LLNA (1 și capitolul B.42 din prezenta anexă) se recomandă ca fiind aplicabile și în cazul LLNA: BrdU-ELISA (10). În afara acestor limite identificate, LLNA: BrdU-ELISA se aplică oricăror substanțe de test care nu prezintă proprietăți susceptibile să afecteze acuratețea testului LLNA: BrdU-ELISA. Mai mult, trebuie luată în considerare posibilitatea obținerii de rezultate pozitive limită pentru care valorile indicelui de stimulare (IS) sunt cuprinse între 1,6 și 1,9 (a se vedea punctele 31-32). Acest fapt se bazează pe baza de date de validare alcătuită din 43 de substanțe prezentând un IS ≥ 1,6 (a se vedea puncul 6) pentru care LLNA: BrdU-ELISA a identificat corect toți cei 32 de agenți de sensibilizare LLNA, dar a identificat incorect doi dintre cei 11 agenți de non-sensibilizare LLNA prezentând un IS cu valori cuprinse între 1,6 și 1,9 (și anume rezultate pozitive de limită) (10). Cu toate acestea, deoarece același set de date a fost utilizat pentru stabilirea valorilor IS și pentru calcularea proprietăților estimative ale testului, rezultatele indicate ar putea supraestima proprietățile estimative reale.
|
PRINCIPIUL METODEI DE TESTARE
|
6.
|
Principiul fundamental care stă la baza LLNA: BrdU-ELISA este acela că agenții de sensibilizare induc o proliferare a limfocitelor în ganglionii limfatici care drenează porțiunea unde se aplică substanța de test. Proliferarea este proporțională cu doza aplicată și cu puterea alergenului și oferă o modalitate simplă de a obține măsurători cantitative ale sensibilizării. Proliferarea este măsurată prin compararea proliferării medii din fiecare grup de test cu proliferarea medie din grupul martor tratat cu vehicul (VC). Se calculează raportul dintre proliferarea medie din fiecare grup tratat și proliferarea medie din grupul VC, denumit IS, care trebuie să aibă o valoare ≥ 1,6 pentru a se putea evalua în continuare substanța de test ca potențial agent de sensibilizare dermică. Procedurile descrise aici se bazează pe măsurarea conținutului de BrdU pentru a indica creșterea numărului de celule în proces de proliferare în ganglionii limfatici auriculari de drenare. BrdU este un analog al timidinei care se încorporează în același mod în ADN-ul celulelor în proces de proliferare. Încorporarea BrdU se măsoară cu ELISA, care utilizează un anticorp specific pentru BrdU marcat, de asemenea, cu peroxidază. Peroxidaza reacționează apoi cu un substrat adăugat special pentru a produce un compus colorat dozat cu un sistem de redare a plăcilor de microtitrare la o absorbanță specifică.
|
DESCRIEREA TESTULUI
Selectarea speciilor de animale
|
7.
|
Specia selectată pentru acest test este șoarecele. Studiile de validare a LLNA: BrdU-ELISA au fost efectuate utilizându-se exclusiv animale din rasa CBA/JN, considerată, prin urmare, rasa preferată (10) (12). Se folosesc femele adulte tinere, nulipare și care nu sunt însărcinate. La începutul studiului, animalele trebuie să aibă între 8-12 săptămâni, iar diferențele de greutate dintre acestea trebuie să fie minime și să nu depășească 20 % din greutatea medie. Se pot utiliza și alte rase și masculi dacă se generează suficiente date pentru a demonstra că nu există diferențe semnificative în răspunsul la LLNA: BrdU-ELISA specifice rasei și/sau sexului.
|
Condiții de adăpostire și de hrănire
|
8.
|
Șoarecii trebuie adăpostiți în grupuri (16), cu excepția cazului în care sunt furnizate justificări științifice adecvate pentru adăpostirea acestora în incinte individuale. Temperatura sălii în care se află animalele de experiență trebuie să fie de 22 °C ± 3 °C. Deși umiditatea relativă trebuie să fie de cel puțin 30 % și este preferabil să nu depășească 70 % decât în timpul operațiunilor de curățare a sălii, obiectivul de umiditate este ca aceasta să fie de 50-60 %. Iluminatul trebuie să fie artificial, alternând 12 ore de lumină cu 12 ore de întuneric. Pentru alimentație, se poate utiliza hrană convențională de laborator, cu furnizarea unei cantități nelimitate de apă potabilă.
|
Pregătirea animalelor
|
9.
|
Animalele se selectează aleatoriu, se marchează pentru a putea fi identificate individual (dar nu se folosește niciun tip de crotalie) și se țin în cuști timp de cel puțin 5 zile înainte de a se începe administrarea dozei astfel încât să se aclimatizeze la condițiile de laborator. Toate animalele sunt examinate înainte de începerea tratamentului, pentru a se stabili că nu prezintă leziuni observabile la nivel dermic.
|
Pregătirea dozelor
|
10.
|
Substanțele chimice solide trebuie dizolvate sau suspendate în solvenți/vehicule și diluate, dacă este cazul, anterior aplicării acestora pe urechea șoarecilor. Substanțele chimice lichide pot fi aplicate pure sau diluate anterior dozării. Substanțele chimice insolubile precum cele observate în general la dispozitivele medicale ar trebui să fie supuse unui proces de extracție amplificată într-un solvent adecvat pentru a indica toți constituenții care pot fi extrași pentru testare anterior aplicării pe urechea șoarecilor. Substanțele de test ar trebui preparate zilnic, cu excepția cazului în care datele privind stabilitatea demonstrează faptul că acestea pot fi stocate.
|
Verificarea fiabilității
|
11.
|
Substanțele martor pozitive (PC) contribuie la demonstrarea performanței adecvate a testului răspunzând în mod adecvat și reproductibil la o substanță de sensibilizare de test pentru care magnitudinea răspunsului este bine cunoscută. Este recomandată includerea unei PC concomitente deoarece aceasta demonstrează competența laboratorului de a realiza corect fiecare test și permite evaluarea comparabilității și reproductibilității intra-laborator și inter-laborator. Anumite autorități solicită, de asemenea, existența unei PC pentru fiecare studiu; prin urmare, utilizatorii sunt încurajați să consulte autoritățile relevante înainte de realizarea testului LLNA: BrdU-ELISA. Prin urmare, utilizarea sistematică a unei PC concomitente este încurajată pentru a evita necesitatea efectuării unor teste suplimentare pe animale, fapt care este câteodată impus prin utilizarea unei PC testate periodic (a se vedea puncul 12). PC trebuie să reacționeze pozitiv la LLNA: BrdU-ELISA la un nivel de expunere estimat să determine o creștere a IS ≥ 1,6 față de grupul martor negativ (NC). Doza de PC trebuie aleasă astfel încât să nu cauzeze iritare dermică excesivă sau toxicitate sistemică, iar inducerea răspunsului să fie reproductibilă, dar nu excesivă (de exemplu, un SI > 14 ar fi excesiv). Substanțele preferate pentru PC sunt aldehida hexil-cinamică 25 % (nr. CAS 101-86-0) și eugenol 25 % (nr. CAS 97-53-0) în acetonă: ulei de măsline (4:1, v/v). Pot exista situații în care, cu o justificare adecvată, este posibil să fie utilizate alte PC care îndeplinesc criteriile de mai sus.
|
|
12.
|
În timp ce includerea unui grup PC concomitent este recomandată, pot exista situații în care testarea periodică (la intervale ≤ 6 luni) a PC poate fi adecvată pentru laboratoarele care efectuează LLNA: BrdU-ELISA în mod regulat (efectuează LLNA: BrdU-ELISA cel puțin o dată pe lună) și care dețin o bază de date stabilită cu PC care demonstrează capacitatea laboratorului de a obține rezultate reproductibile și exacte cu PC. Performanța adecvată în ceea ce privește LLNA: BrdU-ELISA poate fi demonstrată cu succes prin generarea de rezultate pozitive consecvente în cazul PC în cel puțin 10 teste independente efectuate pe o perioadă de timp rezonabilă (mai mică de un an).
|
|
13.
|
Un PC concomitent trebuie inclus întotdeauna în cazul în care se aduce o modificare procedurală la LLNA: BrdU-ELISA (modificarea personalului instruit, modificarea materialelor și/sau a reactivilor utilizați în metoda de testare, modificarea echipamentului utilizat în metoda de testare, modificarea sursei animalelor de test), iar astfel de modificări trebuie documentate în rapoarte de laborator. Trebuie avut în vedere impactul modificărilor asupra caracterului adecvat al bazei de date istorice constituite anterior în determinarea necesității de a constitui o nouă bază de date istorice pentru a documenta consecvența rezultatelor referitoare la PC.
|
|
14.
|
Experimentatorii trebuie să fie conștienți de faptul că decizia de a derula un studiu PC în mod periodic și nu concomitent are consecințe asupra acurateții și acceptabilității rezultatelor negative obținute în urma finalizării unui test fără PC concomitent, în intervalul dintre fiecare test periodic cu PC. De exemplu, dacă se obține un rezultat fals negativ la un test periodic al PC, rezultatele negative ale substanței de test obținute în intervalul dintre ultimul test acceptabil periodic al PC și testul PC periodic inacceptabil pot fi puse sub semnul întrebării. Implicațiile rezultatelor trebuie atent analizate în momentul luării deciziei de a include teste concomitente ale PC sau de a efectua doar teste periodice ale PC. Trebuie avută în vedere, de asemenea, utilizarea unui număr mai mic de animale în grupul PC concomitent în cazul în care acest lucru este justificat științific și dacă laboratorul demonstrează, pe baza datelor istorice specifice laboratorului, că poate fi utilizat un număr mai mic de șoareci (17).
|
|
15.
|
Deși substanța utilizată ca martor pozitiv trebuie testată împreună cu un vehicul despre care se știe că va provoca un răspuns constant (de exemplu, acetonă: ulei de măsline; 4:1, v/v), este posibil să existe unele situații prevăzute de lege în care este necesară și testarea cu un vehicul non-standard (preparat cu relevanță clinică/chimică) (18). În cazul în care substanța utilizată ca PC concomitent este testată într-un vehicul diferit față de substanța de test, atunci trebuie inclus un VC separat pentru PC concomitent.
|
|
16.
|
În cazurile în care se evaluează substanțe de test aparținând unei clase chimice speciale sau care dau rezultate situate într-un anumit interval, substanțele etalon pot, de asemenea, să fie utile pentru a demonstra că metoda de testare funcționează corespunzător pentru detectarea potențialului de sensibilizare dermică al acestor tipuri de substanțe de test. Substanțele etalon adecvate trebuie să prezinte următoarele proprietăți:
|
—
|
similaritate structurală și funcțională cu clasa chimică a substanței de test;
|
|
—
|
caracteristici fizico-chimice cunoscute;
|
|
—
|
date justificative provenind de la LLNA: BrdU-ELISA;
|
|
—
|
date justificative provenind de la alte modele animale și/sau de la om.
|
|
PROCEDURA DE TESTARE
Număr de animale și doze
|
17.
|
Fiecare grup tratat trebuie să fie format din minimum patru animale și se folosesc cel puțin trei concentrații ale substanței de test, plus un grup martor negativ tratat numai cu vehiculul utilizat împreună cu substanța de test și un martor pozitiv (paralel sau recent, în funcție de politica laboratorului în ceea ce privește punctele 11-15). Trebuie avută în vedere testarea de doze multiple pe martorul pozitiv, în special dacă testarea grupului martor pozitiv se face pe bază intermitentă. Animalele din grupurile martor trebuie manipulate și tratate în mod identic cu animalele din grupurile tratate, cu excepția faptului că nu li se administrează tratamentul cu substanța de test.
|
|
18.
|
Doza și vehiculul se aleg pe baza recomandărilor oferite în referințele 2 și 19. Dozele consecutive se aleg în mod normal dintr-o gamă adecvată de concentrații de 100 %, 50 %, 25 %, 10 %, 5 %, 2.5 %, 1 %, 0,5 % etc. Alegerea seriilor de concentrații utilizate trebuie însoțită de o justificare științifică adecvată. Eventualele date existente privind toxicitatea (de exemplu, toxicitatea acută și iritația dermică) și informații privind structura și caracteristicile fizico-chimice ale substanței de test în cauză (și/sau a substanțelor asociate din punct de vedere structural) trebuie avute în vedere la alegerea celor trei concentrații consecutive, astfel încât cea mai mare concentrație să maximizeze expunerea, evitând în același timp toxicitatea sistemică și/sau iritația dermică locală excesivă (19) (20 și capitolul B.4 din prezenta anexă). În lipsa unor astfel de informații, ar putea fi necesară o metodă preliminară de testare (a se vedea punctele 21-24).
|
|
19.
|
Vehiculul nu trebuie să interfereze cu rezultatul testării sau să-l influențeze și trebuie selectat pe baza maximizării solubilității în vederea obținerii celei mai mari concentrații posibile, producând în același timp o soluție/suspensie adecvată pentru aplicarea substanței de test. Vehiculele recomandate sunt acetona: ulei de măsline (4:1, v/v), N,N-dimetilformamidă, metil etil cetonă, propilen glicol și dimetil sulfoxid (6), dar se pot utiliza și altele dacă sunt furnizate justificări științifice suficiente. În anumite situații este posibil să fie necesară utilizarea unui solvent cu relevanță clinică sau a unui preparat comercial sub forma căruia este comercializată substanța de test ca martor suplimentar. Trebuie acordată o atenție specială pentru ca substanțele de test hidrofile să fie încorporate într-un sistem vehicul care să umezească pielea și să nu curgă imediat, prin încorporarea unor dizolvanți adecvați (de exemplu, 1 % Pluronic® L92). Prin urmare, vehiculele formate numai din apă trebuie evitate.
|
|
20.
|
Prelucrarea ganglionilor limfatici de la șoareci individuali permite evaluarea variabilității inter-animale și o comparare statistică a diferențelor dintre substanța de test și măsurătorile pe grupul martor tratat cu vehicul (a se vedea puncul 33). În plus, evaluarea posibilității de a reduce numărul de șoareci în grupul PC este realizabilă dacă se colectează date individuale referitoare la animale (17). Mai mult, anumite autorități de reglementare solicită colectarea de date individuale referitoare la animale. Colectarea regulată de date specifice fiecărui animal contribuie la bunăstarea animalelor prin evitarea testelor duplicat care ar fi necesare dacă rezultatele obținute în alt mod (de exemplu, cu date colective pentru grupuri de animale) ar trebui ulterior înaintate unor autorități de reglementare cu alte cerințe (de exemplu, furnizarea de date individuale).
|
Testarea preliminară
|
21.
|
În absența informațiilor necesare pentru determinarea celei mai mari doze pentru a fi testată (a se vedea punctul 18), trebuie efectuat un test pre-screening pentru a stabili doza adecvată de test în cadrul LLNA: BrdU-ELISA. Scopul testării preliminare este de a furniza orientare în selectarea nivelului maxim al dozei care poate fi utilizat în cadrul studiului principal LLNA: BrdU-ELISA, în cazul în care nu sunt disponibile informații privind concentrația care induce toxicitate sistemică (a se vedea punctul 24) și/sau iritație dermică locală excesivă (a se vedea punctul 23). Doza maximă testată trebuie să fie de 100 % din substanța de test pentru lichide sau concentrația maximă posibilă pentru substanțele solide sau suspensii.
|
|
22.
|
Testarea preliminară se derulează în condiții identice cu cele necesare studiului principal LLNA: BrdU-ELISA, cu excepția cazului în care nu există o evaluare a proliferării ganglionilor limfatici și pot fi utilizate mai puține animale în grupul testat. Se recomandă unul sau două animale pe grup testat. Toți șoarecii sunt observați zilnic pentru identificarea semnelor clinice de toxicitate sistemică sau de iritație locală la locul aplicării. Greutatea corporală este consemnată anterior testării preliminare și anterior finalizării acesteia (ziua 6). Ambele urechi ale fiecărui șoarece sunt observate pentru depistarea eritemelor, iar rezultatul se notează în tabelul 1 (20 și capitolul B.4 din prezenta anexă). Măsurătorile privind grosimea urechii sunt efectuate cu ajutorul unui instrument de măsurare a grosimii (de exemplu, micrometru digital sau instrumentul de măsurare a grosimii Peacock Dial) în ziua 1 (pre-doză), ziua 3 (după aproximativ 48 de ore de la administrarea primei doze) și în ziua 6. În plus, în ziua 6, grosimea urechii poate fi măsurată pornind de la un eșantion de ureche, prelevat după eutanasierea animalelor. Iritația dermică locală excesivă este indicată printr-un punctaj acordat eritemului de ≥ 3 și/sau o creștere a grosimii urechii de cel puțin 25 % în oricare zi a măsurării (21) (22). Cea mai mare doză selectată pentru studiul principal LLNA: BrdU-ELISA este doza imediat inferioară din seria de concentrații (a se vedea punctul 18) care nu induce toxicitate sistemică și/sau iritație dermică locală excesivă.
Tabelul 1
Punctaje acordate eritemelor
|
Observație
|
Punctaj
|
|
Fără eritem
|
0
|
|
Eritem foarte ușor (abia perceptibil)
|
1
|
|
Eritem bine definit
|
2
|
|
Eritem moderat spre grav
|
3
|
|
Eritem grav (roșu violaceu) cu formare de escare care împiedică punctarea eritemului
|
4
|
|
|
23.
|
Pe lângă o creștere cu 25 % a grosimii urechii (21) (22), o creștere semnificativă din punct de vedere statistic a grosimii urechii la șoarecii tratați comparativ cu șoarecii martor a fost, de asemenea, utilizată pentru a identifica substanțele iritante din cadrul LLNA (22) (23) (24) (25) (26) (27) (28). Cu toate acestea, deși pot surveni creșteri semnificative din punct de vedere statistic atunci când grosimea urechii este sub 25 %, acestea nu au fost asociate în mod specific cu iritația excesivă (25) (26) (27) (28) (29).
|
|
24.
|
Următoarele observații clinice pot indica toxicitate sistemică (30) atunci când sunt utilizate ca parte a unei evaluări integrate și, prin urmare, pot indica doza maximă utilizată în studiul LLNA: BrdU-ELISA principal: modificări ale funcției sistemului nervos (de exemplu, piloerecție, ataxie, tremor și convulsii); modificări ale comportamentului (de exemplu, agresivitate, modificări ale activității de curățare, modificare semnificativă a nivelului de activitate); tulburări respiratorii (de exemplu, modificări ale frecvenței și intensității respirației precum dispnee, respirație greoaie și zgomotoasă) și modificări legate de consumul de hrană și apă. În plus, semnele de letargie și/sau lipsa reacției și orice semne clinice indicative ale unei dureri sau suferințe mai mari sau persistente sau o scădere a greutății corporale cu mai mult de 5 % din ziua 1 până în ziua 6, precum și mortalitatea trebuie incluse în evaluare. Animalele muribunde sau cele care manifestă dureri evidente sau prezintă semne de suferință gravă și prelungită sunt eutanasiate (31).
|
Schema de tratament a studiului principal
|
25.
|
Pentru acest test, schema de tratament este următoarea:
— Ziua 1: Se identifică și se înregistrează greutatea fiecărui animal în parte, precum și toate observațiile clinice. Se aplică 25 μL de substanță de test diluată în mod adecvat, doar din vehicul sau din PC (concomitent sau recent, în funcție de politica laboratorului cu privire la punctele 11-15), în regiunea dorsală a fiecărei urechi.
— Zilele 2 și 3: Se repetă procedura de aplicare efectuată în ziua 1.
— Ziua 4: Nu se aplică tratament.
— Ziua 5: Se injectează intraperitoneal 0,5 ml (5 mg/șoarece) de soluție BrdU (10 mg/ml).
— Ziua 6: Se înregistrează greutatea fiecărui animal și toate observațiile clinice. După aproximativ 24 de ore (24 h) de la injecția cu BrdU, animalele se eutanasiază. Se excizează ganglionii limfatici auriculari de drenare de la fiecare ureche de la toate animalele și se prelucrează separat în soluție tamponată cu fosfat (PBS) pentru fiecare animal. Detalii și diagrame privind identificarea și disecția ganglionilor limfatici pot fi consultate în referința (17). Pentru a monitoriza în continuare răspunsul dermic local în studiul principal, pot fi incluși în protocolul de studiu parametri adiționali precum punctarea eritemului din zona urechii sau măsurări ale grosimii urechii (obținute fie cu ajutorul unui instrument de măsurare a grosimii, fie prin determinări ale greutății eșantioanelor de urechi ulterior necropsiei).
|
Prepararea suspensiilor celulare
|
26.
|
Pentru fiecare șoarece, se prepară o suspensie de celule izolate provenind din ganglionii limfatici (LNC) excizați bilateral printr-o separare mecanică ușoară realizată cu ajutorul unei site din oțel inoxidabil cu ochiuri de 200 μm sau o altă tehnică acceptabilă de generare a unei singure suspensii celulare (de exemplu, utilizarea unui mojar din plastic de unică folosință pentru a strivi ganglionii limfatici înainte de a-i trece printr-o sită din nylon cu ochiuri de 70 μm). Procedura pentru prepararea suspensiei LNC constituie o etapă crucială în cadrul testului și, prin urmare, trebuie stăpânită în prealabil de către fiecare operator. În plus, ganglionii limfatici prelevați de la animalele din grupul NC sunt mici și, prin urmare, este importantă manipularea cu grijă a acestora pentru a evita orice perturbare a valorilor IS. În orice caz, volumul suspensiei de LNC trebuie să corespundă unui volum optimizat prestabilit (aproximativ 15 ml). Volumul optimizat se bazează pe atingerea unei absorbanțe medii a grupului NC situată între 0,1-0,2.
|
Determinarea proliferării celulare (măsurarea conținutului de BrdU din ADN-ul limfocitelor)
|
27.
|
BrdU se măsoară cu ajutorul unui set de măsurare ELISA disponibil în comerț (de exemplu Roche Applied Science, Mannheim, Germania, nr. catalog 11 647 229 001). Pe scurt, se adaugă 100 μl de suspensie de LNC în compartimentele unei microplăci cu baza plată, în triplicate. După fixarea și denaturarea LNC, se adaugă anticorpul anti-BrdU în fiecare compartiment și se lasă să reacționeze. Ulterior, anticorpul anti-BrdU se îndepărtează prin spălare, apoi se adaugă soluția de substrat și se lasă pentru a permite sintetizarea cromogenului. Se măsoară absorbanța la 370 nm cu o lungime de undă de referință fixată la 492 nm. În toate cazurile, condițiile de testare trebuie optimizate (a se vedea punctul 26).
|
OBSERVAȚII
Observații clinice
|
28.
|
Fiecare șoarece se examinează atent cel puțin o dată pe zi pentru identificarea oricăror semne clinice, fie de iritații la locul aplicării, fie de toxicitate sistemică. Toate observațiile se consemnează sistematic în înregistrările individuale pentru fiecare șoarece. Planurile de monitorizare trebuie să includă criterii pentru identificarea promptă a șoarecilor care prezintă toxicitate sistemică, iritație dermică locală excesivă sau coroziunea pielii în vederea eutanasierii (31).
|
Greutatea corporală
|
29.
|
Astfel cum se arată la punctul 25, greutatea corporală a fiecărui animal se măsoară la începutul testului și în momentul prevăzut pentru eutanasiere.
|
CALCULAREA REZULTATELOR
|
30.
|
Rezultatele pentru fiecare grup tratat se exprimă printr-un IS mediu. IS se calculează împărțind indicele de marcare BrdU mediu al fiecărui grup tratat cu substanța de test și PC la indicele de marcare BrdU mediu al grupului martor tratat cu solvent/vehicul (VC). IS mediu pentru VC este, prin urmare, egal cu 1.
Indicele de marcare BrdU se calculează după cum urmează:
indice de marcare BrdU = (ABSem – ABS blankem) – (ABSref – ABS blankref)
unde em = lungimea undei de emisie; și ref = lungimea undei de referință.
|
|
31.
|
Un rezultat este considerat pozitiv atunci când IS ≥ 1,6 (10). Cu toate acestea, intensitatea relației doză-răspuns, semnificația statistică și coerența răspunsurilor obținute cu solvent/vehicul și PC pot fi, de asemenea, utilizate în declararea unui rezultat limită (și anume cu valori ale IS cuprinse între 1,6 și 1,9) ca fiind pozitiv (3) (6) (32).
|
|
32.
|
În cazul unui răspuns pozitiv ambiguu cu valori ale IS cuprinse între 1,6 și 1,9, experimentatorii pot examina informații adiționale precum relația doză-răspuns, manifestările de toxicitate sistemică sau iritație excesivă și, dacă este cazul, semnificația statistică împreună cu valorile IS pentru a confirma că astfel de rezultate sunt pozitive (10). Trebuie luate în considerare diferitele proprietăți ale substanței de test, inclusiv o eventuală analogie structurală cu agenți de sensibilizare dermică cunoscuți, declanșarea unei iritații dermice excesive la șoarece și natura relației doză-răspuns observată. Aceste considerații, precum și altele, sunt examinate în detaliu în alt document (4).
|
|
33.
|
Colectarea de date pentru fiecare șoarece permite o analiză statistică a existenței și a intensității relației doză-răspuns pe baza datelor. Orice evaluare statistică poate include o evaluare a relației doză-răspuns, precum și comparații între grupurile testate adaptate corespunzător (de exemplu, comparații pe perechi de grupuri de doză cu grupul martor concomitent tratat cu solvent/vehicul). Analizele statistice pot include, de exemplu, o regresie liniară sau testul William pentru evaluarea funcției doză-răspuns, precum și testul Dunnett pentru comparațiile pe perechi. Pentru alegerea metodei adecvate de analiză statistică, experimentatorul trebuie să aibă în vedere posibilele inegalități la nivelul varianțelor și alte probleme conexe care ar putea impune o transformare a datelor sau o analiză statistică non-parametrică. În orice caz, experimentatorul ar putea fi nevoit să calculeze indicii de stimulare și să efectueze analizele statistice cu sau fără anumite puncte de date (denumite câteodată «valori aberante»).
|
DATE ȘI RAPORTARE
Date
|
34.
|
Datele se sistematizează sub formă de tabele, indicând indicii de marcare BrdU pentru fiecare animal, media indicilor de marcare BrdU/animal pentru fiecare grup, marja de eroare asociată (de exemplu, deviația standard, eroarea standard asociată mediei) și IS mediu pentru fiecare grup tratat în raport cu grupul martor concomitent tratat cu solvent/vehicul.
|
Raportul de testare
|
35.
|
Raportul de testare trebuie să includă următoarele informații:
|
|
Substanțele chimice de test și cele martor:
|
—
|
date de identificare (de exemplu, numărul CAS și CE, dacă se cunosc; sursa; puritatea; impuritățile cunoscute; numărul lotului);
|
|
—
|
starea fizică și proprietățile fizice și chimice (de exemplu, volatilitate, stabilitate, solubilitate);
|
|
—
|
în cazul amestecurilor, compoziția și procentul relativ al fiecărui constituent;
|
|
|
|
Solvent/vehicul:
|
—
|
date de identificare (puritate; concentrație, dacă este cazul; volumul utilizat);
|
|
—
|
justificarea alegerii vehiculului;
|
|
|
|
Animalele de experiență:
|
—
|
proveniența șoarecilor de rasa CBA;
|
|
—
|
statutul microbiologic al animalelor, dacă se cunoaște;
|
|
—
|
numărul și vârsta animalelor;
|
|
—
|
sursa animalelor, condițiile de adăpostire, regimul alimentar etc.;
|
|
|
|
Condiții de testare:
|
—
|
sursa, numărul lotului, datele furnizate de producător cu privire la asigurarea calității/controlului de calitate (sensibilitatea și specificitatea anticorpilor, limitele de detecție) pentru setul ELISA;
|
|
—
|
detalii privind prepararea și aplicarea substanței de testat;
|
|
—
|
justificarea dozei alese (inclusiv rezultatele testării preliminare, dacă este cazul);
|
|
—
|
concentrațiile vehiculului și ale substanței de test utilizate și cantitatea totală de substanță aplicată;
|
|
—
|
detalii privind calitatea hranei și a apei (inclusiv tipul/sursa regimului alimentar, sursele de apă);
|
|
—
|
detalii privind schemele de tratament și de eșantionare;
|
|
—
|
metodele de măsurare a toxicității;
|
|
—
|
criteriile pentru stabilirea studiilor ca fiind pozitive sau negative;
|
|
—
|
detalii privind oricare abateri de protocol și explicații referitoare la modul în care acestea afectează conceptul studiului și rezultatele;
|
|
|
|
Verificarea fiabilității:
|
—
|
un rezumat al rezultatelor obținute la ultima verificare a fiabilității, inclusiv informații privind substanța de test, concentrația și vehiculul utilizat;
|
|
—
|
rezultatele martorilor specifici laboratorului de testare pentru martorul pozitiv concomitent și/sau istoric, precum și pentru martorul negativ concomitent (solvent/vehicul);
|
|
—
|
în cazul în care nu a fost inclus un grup PC concomitent, datele și raportul de laborator pentru cel mai recent grup PC periodic și un raport detaliind datele istorice privind PC specifice laboratorului astfel încât să se justifice de ce nu a fost realizat un grup PC concomitent;
|
|
|
|
Rezultate:
|
—
|
greutatea fiecărui animal la începutul testului și în momentul prevăzut pentru eutanasiere, precum și media și marja de eroare asociată (de exemplu, deviația standard, eroarea standard asociată mediei) pentru fiecare grup tratat;
|
|
—
|
momentul declanșării și semnele de toxicitate, inclusiv iritația dermică la locul administrării, dacă este cazul, pentru fiecare animal;
|
|
—
|
un tabel al indicilor de marcare BrdU și cu valorile IS pentru fiecare șoarece și pentru fiecare grup tratat;
|
|
—
|
media și marja de eroare asociată (de exemplu, deviația standard, eroarea standard asociată mediei) a indicilor de marcare BrdU/șoarece pentru fiecare grup tratat și rezultatele analizei valorilor aberante în cadrul fiecărui grup tratat;
|
|
—
|
indicii de stimulare obținuți și determinarea adecvată a variabilității ținând cont de variațiile dintre animale atât în grupurile tratate cu substanța de test, cât și în grupurile martor;
|
|
—
|
analize statistice, dacă este cazul;
|
|
|
|
Discutarea rezultatelor:
|
—
|
un scurt comentariu privind rezultatele, analiza relației doză-răspuns și analize statistice, dacă este cazul, precum și o concluzie care să indice dacă substanța de test trebuie considerată agent de sensibilizare dermică.
|
|
|
BIBLIOGRAFIE
|
(1)
|
OECD (2010), Skin Sensitisation: Local Lymph Node Assay, Test Guideline No. 429, Guidelines for the Testing of Chemicals, OECD, Paris. Disponibil la: [http://www.oecd.org/env/testguidelines]
|
|
(2)
|
Chamberlain, M. and Basketter, D.A. (1996), The local lymph node assay: status of validation. Food Chem. Toxicol., 34, 999-1002.
|
|
(3)
|
Basketter, D.A., Gerberick, G.F., Kimber, I. and Loveless, S.E. (1996), The local lymph node assay: A viable alternative to currently accepted skin sensitisation tests. Food Chem. Toxicol., 34, 985-997.
|
|
(4)
|
Basketter, D.A., Gerberick, G.F. and Kimber, I. (1998), Strategies for identifying false positive responses in predictive sensitisation tests. Food Chem. Toxicol., 36, 327-33.
|
|
(5)
|
Van Och, F.M.M., Slob, W., De Jong, W.H., Vandebriel, R.J. and Van Loveren, H. (2000), A quantitative method for assessing the sensitising potency of low molecular weight chemicals using a local lymph node assay: employment of a regression method that includes determination of uncertainty margins. Toxicol., 146, 49-59.
|
|
(6)
|
ICCVAM (1999), The murine local lymph node Assay: A test method for assessing the allergic contact dermatitis potential of chemicals/compounds: The results of an independent peer review evaluation coordinated by the Interagency Coordinating Committee on the Validation of Alternative Methods (ICCVAM) and the National Toxicology Program Center for the Evaluation of Alternative Toxicological Methods (NICETAM). NIH Publication No: 99-4494. Research Triangle Park, N.C. Disponibil la: [http://iccvam.niehs.nih.gov/docs/immunotox_docs/llna/llnarep.pdf]
|
|
(7)
|
Dean, J.H., Twerdok, L.E., Tice, R.R., Sailstad, D.M., Hattan, D.G., Stokes, W.S. (2001), ICCVAM evaluation of the murine local lymph node assay: II. Conclusions and recommendations of an independent scientific peer review panel. Reg. Toxicol. Pharmacol., 34(3), 258-273.
|
|
(8)
|
Haneke, K.E., Tice, R.R., Carson, B.L., Margolin, B.H., Stokes, W.S. (2001), ICCVAM evaluation of the murine local lymph node assay: III. Data analyses completed by the national toxicology program interagency center for the evaluation of alternative toxicological methods. Reg. Toxicol. Pharmacol., 34(3), 274-286.
|
|
(9)
|
Sailstad, D.M., Hattan, D., Hill, R.N., Stokes, W.S. (2001), ICCVAM evaluation of the murine local lymph node assay: I. The ICCVAM review process. Reg. Toxicol. Pharmacol., 34(3), 249-257.
|
|
(10)
|
ICCVAM (2010), ICCVAM Test Method Evaluation Report. Nonradioactive local lymph node assay: BrdU-ELISA Test Method Protocol (LLNA: BrdU-ELISA). NIH Publication No. 10-7552A/B. Research Triangle Park, NC: National Institute of Environmental Health Sciences. Disponibil la: [http://iccvam.niehs.nih.gov/methods/immunotox/llna-ELISA/TMER.htm]
|
|
(11)
|
ICCVAM (2009), Independent Scientific Peer Review Panel Report: Updated validation status of new versions and applications of the murine local lymph node assay: a test method for assessing the allergic contact dermatitis potential of chemicals and products. Research Triangle Park, NC: National Institute of Environmental Health Sciences. Disponibil la: [http://iccvam.niehs.nih.gov/docs/immunotox_docs/LLNAPRPRept2009.pdf]
|
|
(12)
|
Takeyoshi, M., Iida, K., Shiraishi, K. and Hoshuyama, S. (2005), Novel approach for classifying chemicals according to skin sensitising potency by non-radioisotopic modification of the local lymph node assay. J. Appl. Toxicol., 25, 129-134.
|
|
(13)
|
OECD (1992), Skin Sensitisation, Test Guideline No. 406, Guidelines for Testing of Chemicals, OECD, Paris. Disponibil la: [http://www.oecd.org/env/testguidelines]
|
|
(14)
|
Kreiling, R., Hollnagel, H.M., Hareng, L., Eigler, L., Lee, M.S., Griem, P., Dreessen, B., Kleber, M., Albrecht, A., Garcia, C. and Wendel, A. (2008), Comparison of the skin sensitising potential of unsaturated compounds as assessed by the murine local lymph node assay (LLNA) and the guinea pig maximization test (GPMT). Food Chem. Toxicol., 46, 1896-1904.
|
|
(15)
|
Basketter, D., Ball, N., Cagen, S., Carrilo, J.C., Certa, H., Eigler, D., Garcia, C., Esch, H., Graham, C., Haux, C., Kreiling, R. and Mehling, A. (2009), Application of a weight of evidence approach to assessing discordant sensitisation datasets: implications for REACH. Reg. Toxicol. Pharmacol., 55, 90-96.
|
|
(16)
|
ILAR (1996), Institute of Laboratory Animal Research (ILAR) Guide for the Care and Use of Laboratory Animals. 7th ed. Washington, DC: National Academies Press.
|
|
(17)
|
ICCVAM (2009), Recommended Performance Standards: Murine Local Lymph Node Assay. NIH Publication Number 09-7357. Research Triangle Park, NC: National Institute of Environmental Health Sciences. Disponibil la:[http://iccvam.niehs.nih.gov/docs/immunotox_docs/llna-ps/LLNAPerfStds.pdf]
|
|
(18)
|
McGarry, H.F. (2007), The murine local lymph node assay: regulatory and potency considerations under REACH. Toxicol., 238, 71-89.
|
|
(19)
|
Kimber, I., Dearman, R.J., Scholes E.W. and Basketter, D.A. (1994), The local lymph node assay: developments and applications. Toxicol., 93, 13-31.
|
|
(20)
|
OECD (2002), Acute Dermal Irritation/Corrosion, Test Guideline No. 404, Guidelines for Testing of Chemicals, OECD, Paris. Disponibil la: [http://www.oecd.org/env/testguidelines]
|
|
(21)
|
Reeder, M.K., Broomhead, Y.L., DiDonato, L. and DeGeorge, G.L. (2007), Use of an enhanced local lymph node assay to correctly classify irritants and false positive substances. Toxicologist, 96, 235.
|
|
(22)
|
ICCVAM (2009), Nonradioactive Murine Local Lymph Node Assay: Flow Cytometry Test Method Protocol (LLNA: BrdU-FC) Revised Draft Background Review Document. Research Triangle Park, NC: National Institute of Environmental Health Sciences. Disponibil la: [http://iccvam.niehs.nih.gov/methods/immunotox/fcLLNA/BRDcomplete.pdf].
|
|
(23)
|
Hayes, B.B., Gerber, P.C., Griffey, S.S. and Meade, B.J. (1998), Contact hypersensitivity to dicyclohexylcarbodiimide and diisopropylcarbodiimide in female B6C3F1 mice. Drug Chem. Toxicol., 21, 195-206.
|
|
(24)
|
Homey, B., von Schilling, C., Blumel, J., Schuppe, H.C., Ruzicka, T., Ahr, H.J., Lehmann, P. and Vohr, V.W. (1998), An integrated model for the differentiation of chemical-induced allergic and irritant skin reactions. Toxicol. Appl. Pharmacol., 153, 83-94.
|
|
(25)
|
Woolhiser, M.R., Hayes, B.B. and Meade, B.J. (1998), A combined murine local lymph node and irritancy assay to predict sensitisation and irritancy potential of chemicals. Toxicol. Meth., 8, 245-256.
|
|
(26)
|
Hayes, B.B. and Meade, B.J. (1999), Contact sensitivity to selected acrylate compounds in B6C3F1 mice: relative potency, cross reactivity, and comparison of test methods. Drug. Chem. Toxicol., 22, 491-506.
|
|
(27)
|
Ehling, G., Hecht, M., Heusener, A., Huesler, J., Gamer, A.O., van Loveren, H., Maurer, T., Riecke, K., Ullmann, L., Ulrich, P., Vandebriel, R. and Vohr, H.W. (2005), A European inter- laboratory validation of alternative endpoints of the murine local lymph node assay: first round. Toxicol., 212, 60-68.
|
|
(28)
|
Vohr, H.W. and Ahr, H.J. (2005), The local lymph node assay being too sensitive? Arch. Toxicol., 79, 721-728.
|
|
(29)
|
Patterson, R.M., Noga, E. and Germolec D. (2007), Lack of evidence for contact sensitisation by Pfiesteria extract. Environ. Health Perspect., 115, 1023-1028.
|
|
(30)
|
ICCVAM (2009), Report on the ICCVAM-NICEATM/ECVAM/JaCVAM Scientific Workshop on Acute Chemical Safety Testing: Advancing In Vitro Approaches and Humane Endpoints for Systemic Toxicity Evaluations. Research Triangle Park, NC: National Institute of Environmental Health Sciences. Disponibil la: [http://iccvam.niehs.nih.gov/methods/acutetox/Tox_workshop.htm].
|
|
(31)
|
OECD (2000), Guidance Document on the Recognition, Assessment and Use of Clinical Signs as Humane Endpoints for Experimental Animals Used in Safety Evaluation, Environmental Health and Safety Monograph Series on Testing and Assessment No. 19, ENV/JM/MONO(2000)7, OECD, Paris. Disponibil la: [http://www.oecd.org/env/testguidelines]
|
|
(32)
|
Kimber, I., Hilton, J., Dearman, R.J., Gerberick, G.F., Ryan, C.A., Basketter, D.A., Lea, L., House, R.V., Ladies, G.S., Loveless, S.E. and Hastings, K.L. (1998), Assessment of the skin sensitisation potential of topical medicaments using the local lymph node assay: An interlaboratory exercise. J. Toxicol. Environ.l Health, 53, 563-79.
|
|
(33)
|
OECD (2005), Guidance Document on the Validation and International Acceptance of New or Updated Test Methods for Hazard Assessment, Environment, Health and Safety Monograph Series on Testing and Assessment No. 34, ENV/JM/MONO(2005)14, OECD, Paris. Disponibil la: [http://www.oecd.org/env/testguidelines]
|
Apendicele 1
DEFINIȚII
Acuratețe: Gradul de apropiere dintre rezultatele metodei de testare și valorile de referință acceptate. Aceasta constituie una dintre caracteristicile de performanță ale metodei de testare și unul dintre aspectele relevanței acesteia. Termenul este adesea utilizat în paralel cu termenul sinonim «concordanță», pentru a desemna proporția de rezultate corecte ale unei metode de testare (33).
Substanță etalon: O substanță de sensibilizare sau de non-sensibilizare utilizată ca standard pentru comparația cu o substanță de test. O substanță etalon ar trebui să aibă următoarele proprietăți: (i) sursă (surse) compatibilă (compatibile) și sigure; (ii) asemănări structurale și funcționale cu clasa de substanțe care este testată; (iii) caracteristici fizico-chimice cunoscute; (iv) date justificative privind efecte cunoscute; și (v) influență cunoscută în intervalul de răspuns dorit.
Fals negativ: O substanță de test identificată incorect ca negativă sau inactivă printr-o metodă de testare, ea fiind de fapt pozitivă sau activă (33).
Fals pozitiv: O substanță de test identificată incorect ca pozitivă sau activă în urma unui test, ea fiind de fapt negativă sau inactivă (33).
Pericol: Potențialul de efect negativ ecologic sau pentru sănătate. Efectul negativ se manifestă doar atunci când există o expunere la un nivel suficient.
Reproductibilitatea inter-laborator: Un indicator al măsurii în care diferite laboratoare calificate, utilizând același protocol și testând aceleași substanțe de test, pot produce rezultate similare din punct de vedere calitativ și cantitativ. Reproductibilitatea inter-laborator este determinată în timpul proceselor de pre-validare și de validare și indică măsura în care un test poate fi transferat cu succes inter-laborator, denumită de asemenea, reproductibilitate inter-laborator (33).
Reproductibilitatea intra-laborator: O determinare a măsurii în care persoane calificate din cadrul aceluiași laborator pot repeta cu succes rezultatele la momente diferite, utilizând un protocol specific. Aceasta este denumită, de asemenea, reproductibilitate în cadrul laboratorului (33).
Valoare aberantă: O valoare aberantă este o observație considerabil diferită de alte valori în cadrul unui eșantion aleatoriu dintr-o populație.
Asigurarea calității: Un proces de management prin care sunt evaluate aderarea la standardele și cerințele de testare în laborator, procedurile de păstrare a înregistrărilor și acuratețea transferului de date de către persoane independente de cele care au efectuat testarea.
Fiabilitate: Măsura în care o metodă de testare poate fi reprodusă în timp, în același laborator sau în laboratoare diferite, folosind același protocol. Aceasta se evaluează prin calcularea reproductibilității intra-laborator sau inter-laborator (33).
Sensibilizare dermică: Un proces imunologic care rezultă atunci când un individ susceptibil este expus topic la un alergen chimic favorizant care provoacă un răspuns cutanat imun care poate conduce la dezvoltarea unei sensibilizări de contact.
Indice de stimulare (IS): O valoare calculată pentru a evalua potențialul de sensibilizare dermică al unei substanțe de test, și anume raportul dintre proliferarea înregistrată la grupurile tratate și cea înregistrată la grupul martor concomitent tratat cu vehicul.
Substanța de test (denumită, de asemenea, substanță chimică de test): Orice substanță sau amestec testat utilizându-se prezenta metodă de testare.
” |


 In force
In force





