

EUR-Lex Access to European Union law
This document is an excerpt from the EUR-Lex website
Document 32010D0015
2010/15/: Commission Decision of 16 December 2009 laying down guidelines for the management of the Community Rapid Information System RAPEX established under Article 12 and of the notification procedure established under Article 11 of Directive 2001/95/EC (the General Product Safety Directive) (notified under document C(2009) 9843)
2010/15/: Kommissionens afgørelse af 16. december 2009 om retningslinjer for forvaltningen af fællesskabssystemet for hurtig udveksling af oplysninger (RAPEX), jf. artikel 12 i direktiv 2001/95/EF (direktivet om produktsikkerhed i almindelighed), og notifikationsproceduren, jf. samme direktivs artikel 11 (meddelt under nummer K(2009) 9843)
2010/15/: Kommissionens afgørelse af 16. december 2009 om retningslinjer for forvaltningen af fællesskabssystemet for hurtig udveksling af oplysninger (RAPEX), jf. artikel 12 i direktiv 2001/95/EF (direktivet om produktsikkerhed i almindelighed), og notifikationsproceduren, jf. samme direktivs artikel 11 (meddelt under nummer K(2009) 9843)
OJ L 22, 26.1.2010, p. 1–64
(BG, ES, CS, DA, DE, ET, EL, EN, FR, IT, LV, LT, HU, MT, NL, PL, PT, RO, SK, SL, FI, SV)
Special edition in Croatian: Chapter 15 Volume 023 P. 3 - 66
 No longer in force, Date of end of validity: 11/11/2018; ophævet ved 32019D0417
No longer in force, Date of end of validity: 11/11/2018; ophævet ved 32019D0417
- Date of document:
- 16/12/2009
- Date of effect:
- 18/12/2009; virkningstidspunkt meddelelsesdato
- Date of notification:
- 18/12/2009
- Date of end of validity:
- 11/11/2018; ophævet ved 32019D0417
- Author:
- Europa-Kommissionen
- Form:
- Beslutning
- Addressee:
- De syvogtyve medlemsstater: Belgien, Bulgarien, Tjekkiske Republik, Danmark, Tyskland, Estland, Irland, Grækenland, Spanien, Frankrig, Italien, Cypern, Letland, Litauen, Luxembourg, Ungarn, Malta, Nederlandene, Østrig, Polen, Portugal, Rumænien, Slovenien, Slovakiet, Finland, Sverige, Det Forenede Kongerige
- Treaty:
- Traktat om Den Europæiske Unions funktionsmåde , Traktat om Den Europæiske Union
- Legal basis:
-
- 32001L0095 - A11P1L3 32001L0095 - N8PT8
- Link
- Select all documents based on this document
- Link
- Link
- Select all documents mentioning this document No data available in the table
- Modifies:
-
Relation Act Comment Subdivision concerned From To Repeal 32004D0418
No data available in the table
- Modified by:
-
Relation Act Comment Subdivision concerned From To Corrected by 32010D0015R(01) (FI) Corrected by 32010D0015R(02) Repealed by 32019D0417 12/11/2018 - Link
- EUROVOC descriptor:
- Subject matter:
- Directory code:
|
26.1.2010 |
DA |
Den Europæiske Unions Tidende |
L 22/1 |
KOMMISSIONENS AFGØRELSE
af 16. december 2009
om retningslinjer for forvaltningen af fællesskabssystemet for hurtig udveksling af oplysninger (RAPEX), jf. artikel 12 i direktiv 2001/95/EF (direktivet om produktsikkerhed i almindelighed), og notifikationsproceduren, jf. samme direktivs artikel 11
(meddelt under nummer K(2009) 9843)
(2010/15/EU)
EUROPA-KOMMISSIONEN HAR —
under henvisning til traktaten om Den Europæiske Union og traktaten om Den Europæiske Unions funktionsmåde,
under henvisning til Europa-Parlamentets og Rådets direktiv 2001/95/EF af 3. december 2001 om produktsikkerhed i almindelighed (1), særlig artikel 11, stk. 1, tredje afsnit, og bilag II, punkt 8,
efter høring af det rådgivende udvalg, der er nedsat i henhold til artikel 15 i direktiv 2001/95/EF, og
ud fra følgende betragtninger:
|
(1) |
Ved artikel 12 i direktiv 2001/95/EF er der indført et fællesskabssystem for hurtig udveksling af oplysninger (RAPEX) med det formål at muliggøre hurtig udveksling af oplysninger mellem medlemsstaterne og Kommissionen om foranstaltninger og forholdsregler, der træffes vedrørende produkter, som udgør en alvorlig risiko for forbrugernes sundhed og sikkerhed. |
|
(2) |
RAPEX er med til at forhindre og begrænse udbuddet af produkter, der udgør en alvorlig risiko for forbrugernes sundhed og sikkerhed, ligesom det letter overvågningen af, hvor effektivt og konsekvent markedsovervågning og håndhævelsesaktiviteter gennemføres i medlemsstaterne. Systemet danner udgangspunkt for klarlæggelse af behovet for fællesskabsforanstaltninger og grundlag for en ensartet håndhævelse af Fællesskabets produktsikkerhedskrav og derigennem et velfungerende indre marked. |
|
(3) |
Notifikationsproceduren, som er indført ved artikel 11 i direktiv 2001/95/EF, danner grundlag for udveksling af oplysninger mellem medlemsstaterne og Kommissionen om foranstaltninger, der træffes vedrørende produkter, som udgør en ikke-alvorlig risiko for forbrugernes sundhed og sikkerhed. |
|
(4) |
Meddelelser indgivet i henhold til artikel 11 i direktiv 2001/95/EF bidrager til at sikre et ensartet, højt forbrugersundhedsniveau og til at sikre ensartede vilkår overalt i det indre marked. |
|
(5) |
Med henblik på at lette forvaltningen af RAPEX-systemet og notifikationsproceduren som indført ved artikel 11 i direktiv 2001/95/EF bør Kommissionen udarbejde retningslinjer til regulering af forskellige aspekter af disse notifikationsprocedurer og især præcisere, hvad meddelelser skal indeholde. Retningslinjerne bør omfatte en standardmeddelelsesformular, kriterier for meddelelser vedrørende risici, hvis virkninger ikke rækker ud over eller ikke kan række ud over medlemsstatens område, samt kriterier vedrørende klassificering af meddelelserne i overensstemmelse med, hvor meget disse haster. Der bør i retningslinjerne ligeledes fastlægges praktiske rammer for aktiviteterne, herunder tidsfrister for de forskellige trin i notifikationsprocedurerne. |
|
(6) |
For at sikre en korrekt anvendelse af RAPEX og notifikationsproceduren som indført ved artikel 11 i direktiv 2001/95/EF bør der i retningslinjerne desuden fastlægges en risikovurderingsmetode og frem for alt specifikke kriterier for identificering af alvorlige risici. |
|
(7) |
Den 29. april 2004 vedtog Kommissionen beslutning 2004/418/EF om retningslinjer for forvaltningen af fællesskabssystemet for hurtig udveksling af oplysninger (RAPEX) og for meddelelser indgivet i henhold til artikel 11 i direktiv 2001/95/EF (2). Punkt 8 i bilag II til direktiv 2001/95/EF samt punkt 1.2 i retningslinjerne i bilaget til beslutning 2004/418/EF foreskriver, at retningslinjerne skal ajourføres regelmæssigt på grundlag af den seneste udvikling og opnåede erfaringer. |
|
(8) |
Fem år efter vedtagelsen af beslutning 2004/418/EF er det samlede antal meddelelser indgivet via RAPEX og notifikationsproceduren som indført ved artikel 11 i direktiv 2001/95/EF firedoblet, og antallet er stadig stigende. Markedsovervågningsmyndighederne har intensiveret deres håndhævelsesindsats (blandt andet med deltagelse i fælles markedsovervågningsprojekter), ligesom de nationale myndigheder med ansvar for kontrollen ved de ydre grænser nu deltager mere aktivt i produktsikkerhedsaktiviteter. |
|
(9) |
På baggrund af denne udvikling og for at sikre mere effektive notifikationsprocedurer i overensstemmelse med bedste praksis er det nødvendigt at ajourføre retningslinjerne. |
|
(10) |
Hovedmålsætningen med denne afgørelse er at fastlægge nye retningslinjer, der giver en klarere afgrænsning af anvendelsesområdet for RAPEX og for notifikationsproceduren som indført ved artikel 11 i direktiv 2001/95/EF, beskriver notifikationskriterierne og regulerer en række elementer i notifikations- og tilbagemeldingsprocedurerne, såsom hvilke oplysninger medlemsstaterne skal fremlægge, bestemmelser om fortrolig behandling, tilbagekaldelse af meddelelser, opfølgning på meddelelser samt organisatoriske aspekter. |
|
(11) |
Med udgangspunkt i bestemmelserne i punkt 2 i bilag II til direktiv 2001/95/EF omfatter de nye retningslinjer forbedrede retningslinjer for risikovurdering af forbrugerprodukter, hvori kriterierne for identificering af alvorlige risici præciseres. |
|
(12) |
De nye retningslinjers struktur og indhold gør det muligt at tilpasse dem, hvis og når det er nødvendigt, så der kan indarbejdes regler vedrørende den notifikationsprocedure, der er indført ved artikel 22 i Europa-Parlamentets og Rådets forordning (EF) nr. 765/2008 af 9. juli 2008 om kravene til akkreditering og markedsovervågning i forbindelse med markedsføring af produkter og om ophævelse af Rådets forordning (EØF) nr. 339/93 (3), ifølge hvilken RAPEX skal anvendes til udveksling af oplysninger og til meddelelser som led i sikkerhedsforanstaltninger, f.eks. vedrørende legetøj. |
|
(13) |
Retningslinjerne henvender sig til alle myndigheder i medlemsstaterne, der arbejder med forbrugerproduktsikkerhed og deltager i RAPEX-netværket i overensstemmelse med direktiv 2001/95/EF, herunder markedsovervågningsmyndighederne med ansvar for overvågning af, om forbrugerprodukter opfylder de relevante sikkerhedskrav, og myndighederne med ansvar for kontrollen ved de ydre grænser. Kommissionen bør bruge retningslinjerne som referencedokument i forvaltningen af RAPEX og notifikationsproceduren som indført ved artikel 11 i direktiv 2001/95/EF — |
VEDTAGET FØLGENDE AFGØRELSE:
Artikel 1
Retningslinjerne for forvaltningen af fællesskabssystemet for hurtig udveksling af oplysninger (RAPEX), jf. artikel 12 i direktiv 2001/95/EF (direktivet om produktsikkerhed i almindelighed), og notifikationsproceduren, jf. samme direktivs artikel 11, findes i bilaget til nærværende afgørelse.
Artikel 2
Beslutning 2004/418/EF ophæves.
Artikel 3
Denne afgørelse er rettet til medlemsstaterne.
Udfærdiget i Bruxelles, den 16. december 2009.
På Kommissionens vegne
Meglena KUNEVA
Medlem af Kommissionen
(1) EFT L 11 af 15.1.2002, s. 4.
(2) EUT L 151 af 30.4.2004, s. 83.
(3) EUT L 218 af 13.8.2008, s. 30.
BILAG
Retningslinjer for forvaltningen af fællesskabssystemet for hurtig udveksling af oplysninger (RAPEX), jf. artikel 12 i direktiv 2001/95/EF (direktivet om produktsikkerhed i almindelighed), og notifikationsproceduren, jf. samme direktivs artikel 11
INDHOLD
DEL I — Status og målgruppe for retningslinjerne
|
1. |
Status og mål for samt ajourføring af retningslinjerne |
|
1.1. |
Status |
|
1.2. |
Mål |
|
1.3. |
Ajourføring |
|
2. |
Hvem henvender retningslinjerne sig til |
DEL II — Fællesskabssystemet for hurtig udveksling af oplysninger (RAPEX), jf. artikel 12 i direktivet om produktsikkerhed i almindelighed
|
1. |
Indledning |
|
1.1. |
Målene med RAPEX |
|
1.2. |
Hvad består RAPEX af? |
|
2. |
RAPEX-notifikationskriterier |
|
2.1. |
Forbrugerprodukter |
|
2.1.1. |
Produkter, der er omfattet af RAPEX |
|
2.1.2. |
Produkter, der ikke er omfattet af RAPEX |
|
2.2. |
Foranstaltninger |
|
2.2.1. |
Kategorier af foranstaltninger |
|
2.2.2. |
Type foranstaltninger |
|
2.2.3. |
Obligatoriske foranstaltninger truffet af myndigheder med ansvar for kontrollen ved de ydre grænser |
|
2.2.4. |
Udelukkelse af generelt gældende obligatoriske foranstaltninger |
|
2.2.5. |
Underretningstidspunkt |
|
2.2.6. |
Meddelende myndigheder |
|
2.2.7. |
RAPEX-meddelelser vedrørende meddelelser fra virksomheder |
|
2.3. |
Alvorlig risiko |
|
2.3.1. |
Alvorlig risiko |
|
2.3.2. |
Risikovurderingsmetode |
|
2.3.3. |
Vurderingsmyndighed |
|
2.3.4. |
Risikovurdering i virksomhedsmeddelelser |
|
2.4. |
Virkninger på tværs af grænserne |
|
2.4.1. |
Internationale hændelser |
|
2.4.2. |
Lokale hændelser |
|
3. |
Meddelelser |
|
3.1. |
Meddelelsestyper |
|
3.1.1. |
RAPEX-meddelelser |
|
3.1.2. |
Meddelelser til orientering |
|
3.2. |
Meddelelsers indhold |
|
3.2.1. |
Fuldstændige oplysninger |
|
3.2.2. |
Oplysninger, der skal fremlægges |
|
3.2.3. |
Ajourføring af oplysninger |
|
3.2.4. |
Ansvaret for de formidlede oplysninger |
|
3.3. |
Fortrolighed |
|
3.3.1. |
Videregivelse af oplysninger generelt |
|
3.3.2. |
Undtagelser fra de generelle regler |
|
3.3.3. |
Anmodninger om fortrolig behandling |
|
3.3.4. |
Håndtering af meddelelser, der behandles fortroligt |
|
3.3.5. |
Tilbagetrækning af anmodninger om fortrolig behandling |
|
3.4. |
Kommissionens gennemgang af meddelelser |
|
3.4.1. |
Korrekthed |
|
3.4.2. |
Fuldstændighed |
|
3.4.3. |
Anmodninger om supplerende oplysninger |
|
3.4.4. |
Undersøgelser |
|
3.5. |
Validering og formidling af meddelelser |
|
3.5.1. |
Validering og formidling af meddelelser |
|
3.5.2. |
Validering af meddelelser om sikkerhedsaspekter, der behandles på EU-plan |
|
3.6. |
Oplysninger om farlige produkter sendt af Kommissionen |
|
3.7. |
Opfølgning på meddelelser |
|
3.7.1. |
Opfølgningen for de forskellige typer meddelelser |
|
3.7.2. |
Målene med opfølgningen |
|
3.7.3. |
Opfølgningsmetoder |
|
3.8. |
Permanent sletning af en meddelelse i RAPEX-applikationen |
|
3.8.1. |
Situationer, hvor meddelelser kan slettes |
|
3.8.2. |
Anmodende medlemsstat |
|
3.8.3. |
Indholdet af anmodningen |
|
3.8.4. |
Beslutninger om sletning af meddelelser |
|
3.9. |
Midlertidig fjernelse af en RAPEX-meddelelse fra RAPEX-hjemmesiden |
|
3.9.1. |
Situationer, hvori meddelelser kan fjernes midlertidigt |
|
3.9.2. |
Anmodende medlemsstat |
|
3.9.3. |
Indholdet af anmodningen |
|
3.9.4. |
Beslutning om at fjerne en meddelelse |
|
3.9.5. |
Fornyet offentliggørelse af en meddelelse |
|
3.10. |
Frister for indgivelse af RAPEX-meddelelser |
|
3.10.1. |
Frister |
|
3.10.2. |
Krisesituationer |
|
4. |
Tilbagemeldinger |
|
4.1. |
Underretning om opfølgende foranstaltninger |
|
4.2. |
Indholdet af tilbagemeldinger |
|
4.2.1. |
Meddelte oplysninger |
|
4.2.2. |
Fuldstændige tilbagemeldinger |
|
4.2.3. |
Ajourføring af validerede tilbagemeldinger |
|
4.2.4. |
Ansvaret for tilbagemeldinger |
|
4.3. |
Fortrolighed |
|
4.4. |
Kommissionens gennemgang af tilbagemeldinger |
|
4.4.1. |
Korrekthed og fuldstændighed |
|
4.4.2. |
Anmodninger om supplerende oplysninger |
|
4.5. |
Validering og formidling af tilbagemeldinger |
|
4.6. |
Permanent sletning af en tilbagemelding i RAPEX-applikationen |
|
4.7. |
Frister for tilbagemeldinger |
|
5. |
Forvaltningen af RAPEX-netværkene |
|
5.1. |
RAPEX-kontaktpunkter |
|
5.1.1. |
Tilrettelæggelse |
|
5.1.2. |
Opgaver |
|
5.2. |
RAPEX-netværk etableret i EU-regi og på nationalt plan |
|
5.2.1. |
Netværket af RAPEX-kontaktpunkter |
|
5.2.2. |
RAPEX-netværk etableret på nationalt plan |
|
5.3. |
RAPEX-kommunikationen — praktiske og tekniske rammer |
|
5.3.1. |
Sprog |
|
5.3.2. |
RAPEX-onlineapplikation |
|
5.3.3. |
Forvaltningen af RAPEX uden for normal arbejdstid |
DEL III — Notifikationsproceduren, jf. artikel 11 i direktivet om produktsikkerhed i almindelighed
|
1. |
Baggrund og mål |
|
2. |
Notifikationskriterier |
|
3. |
Meddelelser |
|
4. |
Tilbagemeldinger |
|
5. |
Praktiske og tekniske rammer |
DEL IV — Tillæg
|
1. |
Standardmeddelelsesformular |
|
2. |
Tilbagemeldingsformular |
|
3. |
Tidsfrister — medlemsstaterne |
|
4. |
Tidsfrister — Kommissionen |
|
5. |
Retningslinjer for risikovurdering af forbrugerprodukter |
DEL I
STATUS OG MÅLGRUPPE FOR RETNINGSLINJERNE
1. Status og mål for samt ajourføring af retningslinjerne
1.1. Status
Retningslinjerne for forvaltningen af fællesskabssystemet for hurtig udveksling af oplysninger (RAPEX) som indført ved artikel 12 i direktiv 2001/95/EF (direktivet om produktsikkerhed i almindelighed) og notifikationsproceduren som indført ved samme direktivs artikel 11 (i det følgende benævnt »retningslinjerne«) vedtages af Kommissionen (1) efter artikel 11, stk. 1, i og punkt 8 i bilag II til Europa-Parlamentets og Rådets direktiv 2001/95/EF af 3. december 2001 om produktsikkerhed i almindelighed (2) (i det følgende benævnt »DPSA«), bistået af et rådgivende udvalg, som består af medlemsstaternes repræsentanter og er nedsat i henhold til artikel 15, stk. 3, i DPSA.
I punkt 8 i bilag II til DPSA hedder det: »Efter proceduren i direktivets artikel 15, stk. 3, udarbejder og ajourfører Kommissionen regelmæssigt retningslinjer for Kommissionens og medlemsstaternes forvaltning af RAPEX.« Artikel 11, stk. 1, i DPSA foreskriver endvidere, at retningslinjerne, der udarbejdes til brug for RAPEX-notifikationsproceduren, også skal omfatte regler for diverse elementer i notifikationsproceduren som indført ved artikel 11 i DPSA. Retningslinjerne regulerer derfor anvendelsen/forvaltningen af såvel RAPEX-notifikationsproceduren, der er indført ved artikel 12 i DPSA, som den notifikationsprocedure, der er indført ved artikel 11 i DPSA.
Retningslinjerne udgør et selvstændigt dokument, som danner rammerne for den RAPEX-notifikationsprocedure, der er indført ved artikel 12 i DPSA. Denne procedure anvendes til præventive og restriktive foranstaltninger, der træffes vedrørende forbrugerprodukter, der udgør en alvorlig risiko for forbrugernes sundhed og sikkerhed. Retningslinjernes struktur og indhold gør det imidlertid muligt at tilpasse dem, hvis og når det er nødvendigt, så der kan indarbejdes regler vedrørende den notifikationsprocedure, der er indført ved artikel 22 i Europa-Parlamentets og Rådets forordning (EF) nr. 765/2008 af 9. juli 2008 om kravene til akkreditering og markedsovervågning i forbindelse med markedsføring af produkter og om ophævelse af Rådets forordning (EØF) nr. 339/93 (3).
Medlemsstaterne (4) samt ansøgerlande, tredjelande og internationale organisationer, der indrømmes adgang til RAPEX (på de vilkår, der er omhandlet i artikel 12, stk. 4, i DPSA), deltager i systemet i overensstemmelse med bestemmelserne i DPSA og retningslinjerne.
1.2. Mål
Ifølge DPSA skal der udarbejdes retningslinjer, som angiver enkle og klare kriterier og praktiske regler, med henblik på at gøre det lettere at forvalte notifikationssystemerne som indført ved artikel 11 og 12 i DPSA. Retningslinjerne har følgende formål:
|
— |
at præcisere anvendelsesområdet for de to notifikationssystemer |
|
— |
at fastsætte notifikationskriterierne for de to notifikationssystemer |
|
— |
at definere, hvad meddelelser og tilbagemeldinger, der sendes inden for rammerne af de to notifikationssystemer, skal indeholde, navnlig hvilke oplysninger der skal gives, og hvilke formularer der skal anvendes |
|
— |
at fastlægge, hvordan medlemsstater skal følge op på en modtaget meddelelse, samt hvilke oplysninger der skal gives i den forbindelse |
|
— |
at beskrive, hvordan Kommissionen skal behandle meddelelser og tilbagemeldinger |
|
— |
at fastsætte tidsfrister for de forskellige typer foranstaltninger, der træffes inden for rammerne af de to notifikationssystemer |
|
— |
at fastlægge de praktiske og tekniske rammer i henholdsvis Kommissionens regi og på medlemsstatsplan, der er nødvendige for, at notifikationssystemerne kan fungere effektivt |
|
— |
at fastlægge en risikovurderingsmetode og frem for alt kriterier for identificering af alvorlige risici. |
1.3. Ajourføring
Retningslinjerne vil regelmæssigt blive ajourført af Kommissionen efter rådgivningsproceduren på grundlag af opnåede erfaringer og den seneste udvikling på produktsikkerhedsområdet.
2. Hvem henvender retningslinjerne sig til
Retningslinjerne henvender sig til alle myndigheder i medlemsstaterne, der arbejder med forbrugerproduktsikkerhed og deltager i RAPEX-netværket, herunder markedsovervågningsmyndighederne med ansvar for overvågning af, om forbrugerprodukterne opfylder de relevante sikkerhedskrav, og myndighederne med ansvar for kontrollen ved de ydre grænser.
Kommissionen bør bruge retningslinjerne som referencedokument i forvaltningen af RAPEX-systemet som indført ved artikel 12 i DPSA og notifikationsproceduren som indført ved samme direktivs artikel 11.
DEL II
FÆLLESSKABSSYSTEMET FOR HURTIG UDVEKSLING AF OPLYSNINGER (RAPEX), JF. ARTIKEL 12 I DIREKTIVET OM PRODUKTSIKKERHED I ALMINDELIGHED
1. Indledning
1.1. Målene med RAPEX
Ved artikel 12 i DPSA er der indført et fællesskabssystem for hurtig udveksling af oplysninger (RAPEX)
RAPEX blev indført med henblik på at:
|
— |
skabe en ramme for hurtig udveksling af oplysninger mellem medlemsstaterne og Kommissionen om præventive og restriktive foranstaltninger, der træffes vedrørende forbrugerprodukter, som udgør en alvorlig risiko for forbrugernes sundhed og sikkerhed |
|
— |
orientere medlemsstaterne og Kommissionen om konklusionerne på opfølgning fra de nationale myndigheders side med hensyn til de oplysninger, der udveksles via RAPEX. |
RAPEX spiller en vigtig rolle på produktsikkerhedsområdet; systemet supplerer andre foranstaltninger, der træffes i nationalt regi og på fællesskabsplan for at sikre et højt forbrugersikkerhedsniveau i EU.
RAPEX-oplysningerne bidrager til at:
|
— |
forhindre og begrænse udbuddet af farlige produkter for forbrugerne |
|
— |
overvåge, hvor effektivt og konsekvent medlemsstaternes myndigheder gennemfører markedsovervågning og håndhævelsesaktiviteter |
|
— |
klarlægge behovet for og skabe grundlaget for indgriben på EU-plan |
|
— |
sikre en ensartet håndhævelse af EU's produktsikkerhedskrav og derigennem et velfungerende indre marked. |
1.2. Hvad består RAPEX af?
RAPEX består af en række elementer, som supplerer hinanden, og som alle er af afgørende betydning for et effektivt samarbejde. De vigtigste af dem er:
|
— |
de lovmæssige rammer for forvaltningen af systemet (dvs. DPSA og retningslinjerne) |
|
— |
onlineapplikationen (i det følgende benævnt »RAPEX-applikationen«), som gør det muligt for medlemsstaterne og Kommissionen hurtigt at udveksle oplysninger via en webbaseret platform |
|
— |
netværket af RAPEX-kontaktpunkter, som består af de enkelte RAPEX-kontaktpunkter med ansvar for forvaltningen af RAPEX i alle medlemsstaterne |
|
— |
de nationale RAPEX-netværk, der er etableret i samtlige medlemsstater og omfatter RAPEX-kontaktpunkterne og alle myndigheder, der arbejder med at garantere sikkerheden ved forbrugerprodukter |
|
— |
Kommissionens RAPEX-team i afdelingen med ansvar for DPSA, som gennemgår og validerer dokumenter, der indgives via RAPEX, og vedligeholder RAPEX-systemet og sørger for, at det fungerer korrekt |
|
— |
RAPEX-hjemmesiden (5), hvor der er adgang til resuméer af RAPEX-meddelelserne i overensstemmelse med artikel 16, stk. 1, i DPSA |
|
— |
RAPEX-udgivelser, såsom RAPEX-statistikker, RAPEX-årsrapporter og diverse pr-materiale. |
2. RAPEX-notifikationskriterier
RAPEX, som er indført ved artikel 12 i DPSA, anvendes til foranstaltninger, der forhindrer, begrænser eller indfører særlige bestemmelser for markedsføring og brug af forbrugerprodukter, der udgør en alvorlig risiko for forbrugernes sundhed og sikkerhed.
I henhold til DPSA er det obligatorisk for medlemsstaterne at deltage i RAPEX, og medlemsstaterne har således en juridisk forpligtelse til at underrette Kommissionen, når følgende fire notifikationskriterier er opfyldt:
|
— |
Produktet er et forbrugerprodukt. |
|
— |
Produktet er omfattet af foranstaltninger, der forhindrer, begrænser eller indfører særlige bestemmelser for den eventuelle markedsføring eller brug af produktet (i det følgende benævnt »præventive og restriktive foranstaltninger«). |
|
— |
Produktet udgør en alvorlig risiko for forbrugernes sundhed og sikkerhed. |
|
— |
Den alvorlige risiko har virkninger på tværs af grænserne. |
2.1. Forbrugerprodukter
2.1.1. Produkter, der er omfattet af RAPEX
I henhold til artikel 2, litra a), i DPSA er følgende produkter omfattet af RAPEX:
|
— |
»produkter, der er bestemt for forbrugerne« — produkter, der er udformet og fremstillet til og stilles til rådighed for forbrugerne |
|
— |
»migrerende produkter« — produkter, der er udformet og fremstillet til erhvervsdrivende, men som under med rimelighed forudsigelige betingelser kan forventes anvendt af forbrugere. Der er her tale om produkter fremstillet til erhvervsmæssig brug, der stilles til rådighed for forbrugerne, som kan købe og betjene dem uden at være i besiddelse af nogen særlig viden eller uddannelse, f.eks. boremaskiner, vinkelslibere eller bordsave, der er udformet og fremstillet til erhvervsdrivende, men også tilbydes på forbrugermarkedet (dvs. forbrugerne kan uden videre købe dem i butikkerne og kan selv betjene dem uden at have fået nogen specifik oplæring). |
Såvel produkter bestemt for forbrugerne som migrerende produkter kan gives til forbrugerne uden beregning, købes af forbrugerne og leveres til forbrugerne i forbindelse med en tjenesteydelse. Alle disse tre situationer er omfattet af RAPEX.
Produkter, der leveres til forbrugerne i forbindelse med en tjenesteydelse, omfatter:
|
— |
produkter, der leveres til forbrugerne, og som tages med og anvendes et andet sted end på tjenesteyderens adresse, f.eks. biler og græsslåmaskiner, der lejes eller leases i et udlejningsforetagendes lokaler, eller tatoveringsfarver og implantater (der ikke er klassificeret som medicinsk udstyr), som implanteres under forbrugerens hud af en tjenesteyder |
|
— |
produkter, der anvendes på en tjenesteyders adresse, forudsat at forbrugerne selv aktivt betjener produktet (f.eks. starter en maskine, har mulighed for at stoppe den eller påvirker maskinens virkemåde ved at ændre dens position eller intensitet under anvendelsen). Et eksempel på sådanne produkter er solarier, der anvendes i solcentre og fitnesscentre. Forbrugernes anvendelse af produktet skal være aktiv og forbundet med en væsentlig grad af kontrol. En rent passiv anvendelse, såsom en kundes brug af shampoo i forbindelse med hårvask hos en frisør eller passagerers brug af en bus, betragtes ikke som forbrugeranvendelse. |
Udstyr, som en tjenesteyder anvender eller betjener i forbindelse med levering af en tjenesteydelse, f.eks. udstyr betjent af tjenesteyderen, med hvilket forbrugeren transporteres eller rejser, falder derimod uden for anvendelsesområdet for RAPEX, og systemet kan derfor ikke benyttes til meddelelser vedrørende sådanne produkter.
2.1.2. Produkter, der ikke er omfattet af RAPEX
RAPEX omfatter ikke:
|
1) |
produkter, der ikke er omfattet af definitionen af et »produkt« i artikel 2, litra a), i DPSA:
|
|
2) |
produkter, der er omfattet af specifikke, tilsvarende notifikationssystemer indført ved anden EU-lovgivning:
|
2.2. Foranstaltninger
2.2.1. Kategorier af foranstaltninger
Alle kategorier af præventive og restriktive foranstaltninger, der træffes vedrørende markedsføring og anvendelse af forbrugerprodukter, som udgør en alvorlig risiko for forbrugernes sundhed og sikkerhed, er omfattet af RAPEX-underretningspligten. Artikel 8, stk. 1, litra b)-f), i DPSA indeholder en liste over de forskellige kategorier af foranstaltninger, der er omfattet af underretningspligten i RAPEX, herunder foranstaltninger vedrørende:
|
— |
mærkning af et produkt med passende advarsler om de risici, produktet kan være forbundet med |
|
— |
fastsættelse af betingelser, der skal være opfyldt før markedsføringen af et produkt |
|
— |
advarsler til forbrugerne om de risici, et produkt kan udgøre for bestemte personer |
|
— |
midlertidigt forbud mod, at et produkt leveres, tilbydes leveret eller udstilles |
|
— |
forbud mod markedsføring af et produkt og eventuelle ledsageforanstaltninger |
|
— |
tilbagetrækning fra markedet af et produkt |
|
— |
tilbagekaldelse af et produkt fra forbrugerne |
|
— |
destruktion af et produkt, der er trukket eller kaldt tilbage. |
Inden for RAPEX-systemet bruges begrebet »tilbagetrækning« udelukkende for foranstaltninger, der har til formål at forhindre, at et farligt produkt distribueres, udstilles og tilbydes til forbrugerne, mens »tilbagekaldelse« udelukkende bruges om foranstaltninger, der har til formål at få returneret et farligt produkt, som producenten eller distributøren allerede har stillet til rådighed for forbrugeren.
2.2.2. Type foranstaltninger
Præventive og restriktive foranstaltninger vedrørende farlige produkter kan træffes enten på initiativ af en producent eller en distributør, der har markedsført og/eller distribueret produktet (»frivillige foranstaltninger«), eller efter påbud herom fra en myndighed i en medlemsstat med beføjelser til at overvåge, at produkter opfylder de relevante sikkerhedskrav (»obligatoriske foranstaltninger«)
I RAPEX forstås følgende ved henholdsvis obligatoriske og frivillige foranstaltninger:
|
— |
Obligatoriske foranstaltninger: Foranstaltninger, som en medlemsstats myndigheder træffer eller beslutter at træffe, ofte i form af en administrativ afgørelse, som forpligter en producent eller en distributør til at træffe præventive eller restriktive foranstaltninger over for et specifikt produkt, som den pågældende har stillet til rådighed på markedet. |
|
— |
Frivillige foranstaltninger:
|
I henhold til artikel 12, stk. 1, i DPSA skal såvel obligatoriske som frivillige foranstaltninger meddeles via RAPEX.
2.2.3. Obligatoriske foranstaltninger truffet af myndigheder med ansvar for kontrollen ved de ydre grænser
Foranstaltninger, der træffes af myndigheder med ansvar for kontrollen ved de ydre grænser, og som forhindrer, at forbrugerprodukter, der udgør en alvorlig risiko for forbrugernes sundhed og sikkerhed, markedsføres i EU (f.eks. beslutninger om stop for import ved EU's grænser), skal meddeles til Kommissionen via RAPEX ligesom foranstaltninger, der træffes af markedsovervågningsmyndighederne og begrænser markedsføringen eller anvendelsen af et produkt.
2.2.4. Udelukkelse af generelt gældende obligatoriske foranstaltninger
Almindeligt gældende retsakter, der vedtages på nationalt plan for at forhindre eller begrænse markedsføring og anvendelse af en eller flere generelt definerede kategorier af forbrugerprodukter, fordi disse udgør en alvorlig risiko for forbrugernes sundhed og sikkerhed, meddeles ikke til Kommissionen via RAPEX-systemet. Alle sådanne nationale foranstaltninger, der kun gælder for generelt definerede kategorier af produkter, såsom alle produkter generelt eller alle produkter med en bestemt funktion — og ikke (kategorier af) produkter, der er identificeret nærmere i kraft af mærke, særligt udseende, producent, forhandler, modelnavn eller nummer osv. — meddeles til Kommissionen i henhold til Europa-Parlamentets og Rådets direktiv 98/34/EF af 22. juni 1998 om en informationsprocedure med hensyn til tekniske standarder og forskrifter samt forskrifter for informationssamfundets tjenester (12).
2.2.5. Underretningstidspunkt
I henhold til artikel 12, stk. 1, i DPSA skal medlemsstaterne øjeblikkelig underrette Kommissionen om præventive og restriktive foranstaltninger via RAPEX. Denne bestemmelse gælder for både obligatoriske og frivillige foranstaltninger, om end underretningstidspunktet ikke er det samme.
|
— |
Obligatoriske foranstaltninger Sådanne foranstaltninger meddeles via RAPEX, umiddelbart efter at foranstaltningerne eller beslutningen herom er truffet, selv hvis det er sandsynligt, at der vil blive indgivet en klage over foranstaltningerne på nationalt plan, hvis en sådan klage allerede behandles, eller hvis foranstaltningen er omfattet af bestemmelser om obligatorisk offentliggørelse. Denne fremgangsmåde er i overensstemmelse med RAPEX-målsætningen, dvs. at sikre en hurtig udveksling af oplysninger mellem medlemsstaterne og Kommissionen med henblik på at forhindre levering og anvendelse af produkter, der udgør en alvorlig risiko for forbrugernes sundhed eller sikkerhed. |
|
— |
Frivillige foranstaltninger Producenter og distributører har i henhold til artikel 5, stk. 3, i DPSA pligt til at underrette medlemsstatens kompetente myndigheder om alle frivillige forholdsregler og foranstaltninger, de træffer for at forhindre, at forbrugerne udsættes for risici på grund af produkter, som de har stillet til rådighed på markedet (i det følgende benævnt »virksomhedsmeddelelse«). Den myndighed i en medlemsstat, der modtager en sådan meddelelse fra en virksomhed, bruger de pågældende oplysninger som grundlag for en RAPEX-meddelelse (hvis alle RAPEX-notifikationskriterierne i artikel 12, stk. 1, er opfyldt) og sender denne umiddelbart efter at have modtaget meddelelsen fra virksomheden. Såfremt der vedtages frivillige foranstaltninger i form af en aftale mellem en producent eller en distributør og en myndighed i en medlemsstat eller på grundlag af en anbefaling fra en myndighed til en producent eller en distributør, indgives der en RAPEX-meddelelse herom, umiddelbart efter at aftalen er indgået eller anbefalingen vedtaget. |
Med henblik på en ensartet overholdelse af RAPEX-underretningspligten er der i tillæg 3 til retningslinjerne fastsat specifikke frister for indgivelse af meddelelser til Kommissionen via RAPEX (13).
2.2.6. Meddelende myndigheder
Såvel obligatoriske som frivillige foranstaltninger meddeles via RAPEX af det nationale RAPEX-kontaktpunkt, som er ansvarligt for alle oplysninger, som det pågældende land formidler via systemet (14).
2.2.7. RAPEX-meddelelser vedrørende meddelelser fra virksomheder
I henhold til artikel 5, stk. 3, i DPSA skal producenter og distributører (på samme tid) indberette oplysninger vedrørende et farligt produkt til de kompetente myndigheder i alle de medlemsstater, hvor det farlige produkt er stillet til rådighed. Bilag I til DPSA indeholder betingelserne for og nærmere oplysninger om denne type meddelelser.
I disse situationer gælder RAPEX-underretningspligten for alle medlemsstater, der modtager en virksomhedsmeddelelse. Med henblik på at forenkle anvendelsen i praksis af artikel 12, stk. 1, i DPSA og for at undgå unødvendig overlapning mellem RAPEX-meddelelser er det dog aftalt med medlemsstaterne, at en RAPEX-meddelelse kun skal indgives af den medlemsstat, hvori den meddelende producent/distributør er etableret (i det følgende benævnt »hovedmedlemsstaten«). Når Kommissionen har valideret en RAPEX-meddelelse og videreformidlet den i systemet, skal andre medlemsstater (især dem, der har modtaget den samme virksomhedsmeddelelse) reagere på den pågældende RAPEX-meddelelse.
Undlader en hovedmedlemsstat at indgive en RAPEX-meddelelse inden udløbet af de frister, der er angivet i tillæg 3 til retningslinjerne, og at underrette Kommissionen og andre medlemsstater om grunden til forsinkelsen, kan enhver anden medlemsstat, der har modtaget den samme virksomhedsmeddelelse, indgive en meddelelse via RAPEX.
2.3. Alvorlig risiko
2.3.1. Alvorlig risiko
Inden en myndighed i en medlemsstat beslutter at indgive en RAPEX-meddelelse, foretager den altid den nødvendige risikovurdering for at undersøge, hvorvidt det produkt, den påtænkte meddelelse vedrører, udgør en alvorlig risiko for forbrugernes sundhed og sikkerhed, og dermed hvorvidt et af RAPEX-notifikationskriterierne er opfyldt.
Eftersom RAPEX ikke er beregnet til udveksling af oplysninger om produkter, der udgør ikke-alvorlige risici, kan meddelelser om foranstaltninger truffet i relation til sådanne produkter ikke sendes via RAPEX i henhold til artikel 12 i DPSA.
2.3.2. Risikovurderingsmetode
I tillæg 5 til retningslinjerne beskrives den risikovurderingsmetode, medlemsstaternes myndigheder skal anvende til at vurdere, hvor stor en risiko et forbrugerprodukt udgør for forbrugernes sundhed og sikkerhed, og til at afgøre, hvorvidt en RAPEX-meddelelse er påkrævet.
2.3.3. Vurderingsmyndighed
Risikovurderingen foretages altid af en myndighed i en medlemsstat, som har gennemført de relevante undersøgelser og truffet de relevante foranstaltninger, eller som har overvåget frivillige foranstaltninger truffet af en producent eller en distributør i relation til et farligt produkt.
Inden der sendes en RAPEX-meddelelse til Kommissionen, kontrolleres risikovurderingen (skal være indeholdt i meddelelsen), der er foretaget af en myndighed i en medlemsstat, altid af RAPEX-kontaktpunktet. Eventuelle tvivlsspørgsmål afgøres af kontaktpunktet i samråd med den ansvarlige myndighed, inden en meddelelse formidles via RAPEX.
2.3.4. Risikovurdering i virksomhedsmeddelelser
Meddelelser om farlige forbrugerprodukter, der indgives af producenter og distributører til de kompetente myndigheder i medlemsstater i henhold til artikel 5, stk. 3, i DPSA, skal indeholde en detaljeret beskrivelse af risikoen. De nationale myndigheder, der modtager sådanne meddelelser, gennemgår disse og analyserer de fremlagte risikovurderinger. Hvis en myndighed i en medlemsstat på grundlag af de meddelte oplysninger og en uafhængig risikovurdering konkluderer, at det produkt, der er indgivet meddelelse om (i det følgende benævnt »det indberettede produkt«), udgør en alvorlig risiko for forbrugernes sundhed og sikkerhed, sendes en RAPEX-meddelelse om det pågældende produkt øjeblikkelig til Kommissionen (artikel 12, stk. 1, fjerde afsnit, i DPSA).
Risikovurderinger foretaget af producenter og distributører er ikke bindende for medlemsstaternes myndigheder. En myndighed i en medlemsstat vil derfor kunne nå frem til en anden konklusion vedrørende risikovurderingen end den, man har draget en virksomhedsmeddelelse.
2.4. Virkninger på tværs af grænserne
2.4.1. Internationale hændelser
I henhold til artikel 12 i DPSA indgiver en medlemsstat udelukkende en RAPEX-meddelelse, hvis den vurderer, at virkningerne af risiciene ved et farligt produkt rækker ud over eller kan række ud over dens område (i det følgende benævnt »virkninger på tværs af grænserne« eller »international hændelse«).
På baggrund af de frie varebevægelser i det indre marked, og i lyset af at produkter importeres til EU ad forskellige distributionskanaler og forbrugerne køber produkter under ophold i udlandet og via internettet, anbefales det, at de nationale myndigheder fortolker kriteriet vedrørende virkninger på tværs af grænserne forholdsvis bredt. En RAPEX-meddelelse indgives således, hvis:
|
— |
det ikke kan udelukkes, at et farligt produkt er blevet solgt til forbrugere i mere end én EU-medlemsstat, eller |
|
— |
det ikke kan udelukkes, at et farligt produkt er blevet solgt til forbrugere via internettet, eller |
|
— |
produktet har oprindelse i et tredjeland og kan formodes at være blevet importeret til EU ad flere forskellige distributionskanaler. |
2.4.2. Lokale hændelser
Foranstaltninger truffet vedrørende et produkt, der udgør en alvorlig risiko, som kun kan have virkning lokalt (en »lokal hændelse«), meddeles ikke via RAPEX. Dette gælder situationer, hvor en myndighed i en medlemsstat har grund til at formode, at et produkt ikke er og ikke vil blive stillet til rådighed (på nogen som helst måde) for forbrugerne i andre medlemsstater, dvs. foranstaltninger vedrørende et lokalt produkt, der er fremstillet og udelukkende distribueres i én medlemsstat.
Lokale hændelser skal dog meddeles til Kommissionen, men dette skal ske i henhold til artikel 11 i DPSA, og kun hvis hændelsen giver anledning til information om produktsikkerhed, der kan formodes at være af interesse for andre medlemsstater, især oplysninger om foranstaltninger, der træffes som reaktion på en ny risikotype, som ikke har været genstand for andre meddelelser, en ny risikotype, som er resultatet af en kombination af produkter, eller en ny, farlig produkttype eller –kategori (artikel 12, stk. 1, andet afsnit, i DPSA).
3. Meddelelser
3.1. Meddelelsestyper
3.1.1. RAPEX-meddelelser
Der findes to typer RAPEX-meddelelser, nemlig »artikel 12-meddelelser« og »artikel 12-meddelelser, der nødvendiggør hasteforanstaltninger«.
|
— |
Hvis alle RAPEX-notifikationskriterierne som fastsat i artikel 12 i DPSA (jf. del II, kapitel 2, i retningslinjerne) er opfyldt, klassificeres den RAPEX-meddelelse, som en medlemsstat udarbejder og sender til Kommissionen, i RAPEX-applikationen som en »artikel 12-meddelelse«. |
|
— |
Den RAPEX-meddelelse, som en medlemsstat, der indgiver en meddelelse (i det følgende benævnt »den meddelende medlemsstat«), udarbejder og sender til Kommissionen, i tilfælde af at alle RAPEX-notifikationskriterierne er opfyldt, og et produkt ydermere udgør en livstruende risiko, og/eller der er indtruffet ulykker med dødelig udgang, samt i andre tilfælde, hvor en RAPEX-meddelelse nødvendiggør hasteforanstaltninger i samtlige medlemsstater, klassificeres i RAPEX-applikationen som en »artikel 12-meddelelse, der nødvendiggør hasteforanstaltninger«. |
RAPEX-kontaktpunktet i den meddelende medlemsstat kontrollerer inden indgivelsen af enhver RAPEX-meddelelse, at denne opfylder samtlige RAPEX-notifikationskriterier, og at den skal sendes via RAPEX-applikationen som en »artikel 12-meddelelse« eller en »artikel 12-meddelelse, der nødvendiggør hasteforanstaltninger«.
3.1.2. Meddelelser til orientering
Såfremt en meddelelse ikke kan sendes gennem systemet som en RAPEX-meddelelse, kan kontaktpunktet vælge at benytte RAPEX-applikationen til at sende de pågældende oplysninger til orientering. Sådanne meddelelser klassificeres i RAPEX-applikationen som »meddelelser til orientering« og kan sendes i følgende situationer:
|
a) |
Alle RAPEX-notifikationskriterierne i artikel 12 i DPSA er opfyldt, men den pågældende meddelelse indeholder ikke alle de oplysninger (primært vedrørende produktidentifikation og distributionskanaler), der er nødvendige, for at andre medlemsstater effektivt kan følge op på meddelelsen (15). En meddelelse, for hvilken det gælder, at produktbetegnelse, mærke og billede mangler, og det indberettede produkt derfor ikke kan identificeres behørigt og ikke kan skelnes fra andre produkter af samme kategori eller type på markedet, er et eksempel på en meddelelse, der kan formidles via RAPEX-applikationen som en »meddelelse til orientering«. Vurderingen af, hvorvidt en meddelelse indeholder tilstrækkelige oplysninger til andre medlemsstater til at sikre den nødvendige opfølgning, foretages altid fra sag til sag. |
|
b) |
En medlemsstat er bekendt med, at et forbrugerprodukt, der udbydes på EU-markedet, udgør en alvorlig risiko for forbrugernes sundhed og sikkerhed, men producenten eller distributøren har endnu ikke truffet præventive og restriktive foranstaltninger, ligesom der ikke er truffet og heller ikke foreligger en beslutning om at træffe sådanne foranstaltninger fra en myndighed i en medlemsstat (artikel 12, stk. 1, fjerde afsnit, i DPSA). Såfremt oplysninger om et sådant produkt formidles via RAPEX-applikationen, inden der er truffet foranstaltninger, underretter den meddelende medlemsstat efterfølgende (hurtigst muligt og under alle omstændigheder inden for de frister, der er angivet i tillæg 3 til retningslinjerne) Kommissionen om den endelige afgørelse, der er truffet vedrørende det indberettede produktet (med især oplysninger om, hvilke præventive eller restriktive foranstaltninger der er truffet, eller en begrundelse for, at sådanne foranstaltninger ikke er truffet). |
|
c) |
En medlemsstat beslutter at meddele præventive og restriktive foranstaltninger, der træffes vedrørende et forbrugerprodukt, som udgør en alvorlig risiko for forbrugernes sundhed og sikkerhed og kun har virkning lokalt (»lokal hændelse«). Giver en meddelelse om en »lokal hændelse« anledning til information om produktsikkerhed, der kan formodes at være af interesse for andre medlemsstater, sendes den imidlertid i henhold til artikel 11 i DPSA, jf. punkt 2.4.2. |
|
d) |
En meddelelse vedrører et forbrugerprodukt, for hvilken sikkerhedsaspekterne (især spørgsmålet om, hvor stor en risiko varen udgør for forbrugernes sundhed og sikkerhed) behandles på EU-plan med det formål at sikre en fælles indfaldsvinkel til risikovurdering og/eller håndhævelse i alle medlemsstaterne (16). |
|
e) |
Det kan ikke med sikkerhed slås fast, at et eller flere af RAPEX-notifikationskriterierne er opfyldt, men den pågældende meddelelse giver anledning til information om produktsikkerhed, der kan formodes at være af interesse for andre medlemsstater. En meddelelse om et produkt, der ikke endegyldigt kan klassificeres som et forbrugerprodukt, men indeholder oplysninger om en ny type risiko for forbrugernes sundhed og sikkerhed, er et eksempel på en meddelelse, der kan formidles via RAPEX-applikationen som en »meddelelse til orientering«. |
RAPEX-kontaktpunktet angiver i forbindelse med indsendelse af en »meddelelse til orientering« tydeligt, hvad begrundelsen er for at indsende denne.
3.2. Meddelelsers indhold
3.2.1. Fuldstændige oplysninger
Meddelelser bør være så udtømmende som muligt. Standardmeddelelsesformularen findes i tillæg 1 til retningslinjerne. Alle rubrikker i meddelelsesformularen udfyldes med de nødvendige oplysninger. Foreligger de nødvendige oplysninger ikke på tidspunktet for indgivelse af en meddelelse, angives dette tydeligt i formularen af den meddelende medlemsstat med en forklaring. Så snart de manglende oplysninger foreligger, ajourfører den meddelende medlemsstat den pågældende meddelelse. Den ajourførte meddelelse gennemgås af Kommissionen, inden den valideres og videreformidles i systemet.
RAPEX-kontaktpunkterne instruerer alle nationale myndigheder, der deltager i RAPEX-netværket, i, hvilke oplysninger standardmeddelelsesformularen skal indeholde for at være behørigt udfyldt. Dette er med til at sikre, at de oplysninger, som de pågældende myndigheder giver RAPEX-kontaktpunktet, er korrekte og fuldstændige.
Medlemsstaterne overholder de fastsatte tidsfrister og forsinker ikke en RAPEX-meddelelse om et produkt, der udgør en meget alvorlig eller livstruende risiko for forbrugernes sundhed og sikkerhed, blot fordi dele af de oplysninger, der er påkrævet i henhold til retningslinjerne, endnu ikke foreligger.
Kontaktpunktet kontrollerer, inden det indgiver en meddelelse, at der ikke allerede er indgivet meddelelse om det pågældende produkt via applikationen fra en anden medlemsstat (for at undgå unødvendige dubletter). Er der allerede indgivet meddelelse om produktet, sender kontaktpunktet — frem for at udarbejde en ny meddelelse — en tilbagemelding på den allerede indsendte meddelelse og fremlægger desuden eventuelle supplerende oplysninger, der kunne være relevante for myndighederne i andre medlemsstater, såsom supplerende identifikationsnumre på køretøjer, en detaljeret liste over importører og distributører, supplerende afprøvningsrapporter osv.
3.2.2. Oplysninger, der skal fremlægges
Meddelelser, der sendes til Kommissionen via RAPEX, indeholder følgende typer oplysninger:
|
— |
Oplysninger, der gør det muligt at identificere det indberettede produkt, dvs. produktkategori, produktbetegnelse, mærke, model- og/eller typenummer, stregkode, vareparti- eller serienummer, toldkode og beskrivelse af produktet og dets emballage ledsaget af billeder af produktet, dets emballage og dets mærkning. En detaljeret og præcis produktidentifikation er en hjørnesten inden for markedsovervågning og håndhævelse, idet en sådan identifikation gør det muligt for de nationale myndigheder at identificere det indberettede produkt, at skelne det fra andre produkter af samme eller lignende type eller kategori på markedet, at finde det på markedet samt at træffe eller indgå aftale om hensigtsmæssige foranstaltninger. |
|
— |
Oplysninger om, hvor produktet stammer fra, dvs. oprindelsesland for samt navn, adresse og kontaktoplysninger, herunder telefonnummer og e-mail-adresse, på producent og eksportører. Medlemsstaterne fremlægger især alle tilgængelige oplysninger om producenter og eksportører i tredjelande, der har et tæt samarbejde med EU om produktsikkerhed. Kommissionen holder således RAPEX-kontaktpunkterne løbende informeret om den seneste udvikling på dette område. Følgende dokumenter skal vedlægges formularen, for så vidt de foreligger: kopier af ordrer, købekontrakter, fakturaer, forsendelsesdokumenter, toldangivelser osv. Detaljerede oplysninger om producenter i tredjelande gør det muligt for Kommissionen at fremme en mere effektiv håndhævelse i de pågældende lande, ligesom de bidrager til at begrænse antallet af farlige forbrugerprodukter, der eksporteres til EU. |
|
— |
Oplysninger vedrørende sikkerhedskravene til det indberettede produkt, herunder referencenumre og titler på gældende bestemmelser og standarder. |
|
— |
En risikobeskrivelse af det indberettede produkt, herunder en beskrivelse af resultaterne af laboratorietest eller visuelle undersøgelser, afprøvningsrapporter og attester, der dokumenterer, at det indberettede produkt ikke opfylder sikkerhedskravene, og en fuldstændig risikovurdering med konklusioner og oplysninger om kendte ulykker eller hændelser. |
|
— |
Oplysninger om de forskellige omsætningsled for det indberettede produkt i medlemsstaterne, og især information om bestemmelseslandene, plus oplysninger om importører samt, i det omfang de relevante data foreligger, om distributører af det indberettede produkt. |
|
— |
Oplysninger om trufne foranstaltninger, herunder navnlig deres art (obligatoriske eller frivillige), kategori (f.eks. tilbagetrækning fra markedet/tilbagekaldelse fra forbrugerne), anvendelsesområde (f.eks. landsdækkende/lokale), ikrafttrædelsesdato og varighed (f.eks. ikke-tidsbegrænsede/midlertidige). |
|
— |
Angivelse af, hvorvidt en meddelelse, dele af den og/eller tillæg hertil skal behandles fortroligt. Anmodninger om fortrolig behandling ledsages altid af en begrundelse, hvori det tydeligt er angivet, hvad anmodningen er baseret på. |
Medlemsstaterne opfordres til at indsamle og fremlægge oplysninger om omsætningsleddene for det indberettede produkt i lande uden for EU, der har et tæt samarbejde med EU om produktsikkerhed.
3.2.3. Ajourføring af oplysninger
Den meddelende medlemsstat underretter (hurtigst muligt og under alle omstændigheder inden for de frister, der er angivet i tillæg 3 til retningslinjerne) Kommissionen om nye situationer eller forhold, der gør det nødvendigt at ændre en meddelelse, som er formidlet via RAPEX-applikationen. Medlemsstaterne underretter især Kommissionen om eventuelle ændringer (f.eks. som følge af en retsafgørelse under en klageprocedure) i status for de meddelte foranstaltninger eller i risikovurderingen eller nye afgørelser vedrørende fortrolig behandling.
Kommissionen gennemgår oplysningerne fra den meddelende medlemsstat og ajourfører de pågældende oplysninger i RAPEX-applikationen og på RAPEX-hjemmesiden, i det omfang det er nødvendigt.
3.2.4. Ansvaret for de formidlede oplysninger
I punkt 10 i bilag II til DPSA hedder det: »Den medlemsstat, der foretager meddelelsen, er ansvarlig for de meddelte oplysninger«.
RAPEX-kontaktpunktet i den meddelende medlemsstat og den ansvarlige nationale myndighed sørger for, at de oplysninger, der meddeles via RAPEX, navnlig produkt- og risikobeskrivelser, er nøjagtige, så det pågældende produkt ikke på nogen måde kan forveksles med lignende produkter af samme kategori eller type på EU-markedet.
RAPEX-kontaktpunktet og den myndighed, der er involveret i notifikationsproceduren (f.eks. i kraft af at den foretager risikovurderingen af det indberettede produkt eller tilvejebringer oplysninger om distributionskanalerne), er ansvarlige for de oplysninger, der meddeles via RAPEX. RAPEX-kontaktpunktet kontrollerer og validerer alle meddelelser, som den modtager fra de ansvarlige myndigheder, inden den sender meddelelserne videre til Kommissionen.
Kommissionens aktiviteter, herunder gennemgang af meddelelserne samt validering og videreformidling af dem via RAPEX-applikationen og offentliggørelse af dem på RAPEX-hjemmesiden, indebærer ikke, at Kommissionen påtager sig noget ansvar for de formidlede oplysninger, idet dette fortsat ligger hos den meddelende medlemsstat.
3.3. Fortrolighed
3.3.1. Videregivelse af oplysninger generelt
I henhold til artikel 16, stk. 1, i DPSA har offentligheden ret til at blive holdt informeret om farlige produkter, der udgør en risiko for deres sundhed og sikkerhed. For at opfylde denne forpligtelse offentliggør Kommissionen oversigter over nye RAPEX-meddelelser (dvs. »artikel 12-meddelelser« og »artikel 12-meddelelser, der nødvendiggør hasteforanstaltninger«) på RAPEX-hjemmesiden. Det samme gør medlemsstaterne, som ligeledes — på deres respektive sprog — informerer borgerne om produkter, der udgør en alvorlig risiko for forbrugerne, og om foranstaltninger, der er truffet for at eliminere en sådan risiko. Disse oplysninger kan udbredes via internettet, som publikationer, ved hjælp af de elektroniske medier osv.
De oplysninger, offentligheden har adgang til, er et resumé af en RAPEX-meddelelse med bl.a. de oplysninger, der er nævnt i artikel 16 i DPSA, dvs. oplysninger til identifikation af produkterne samt om de pågældende risici og foranstaltninger, der er truffet for at forhindre eller begrænse disse risici. Kommissionen og medlemsstaterne giver ikke offentligheden adgang til meddelelserne i deres helhed, især ikke detaljerede risikobeskrivelser med afprøvningsrapporter og attester eller udtømmende lister over distributionskanaler, idet visse af disse oplysninger pga. deres art er fortrolige (forretningshemmeligheder) og skal beskyttes.
3.3.2. Undtagelser fra de generelle regler
I henhold til artikel 16, stk. 1, første afsnit, i DPSA skal offentligheden have adgang til oplysningerne »med forbehold af de begrænsninger, der er nødvendige af hensyn til kontrol- og undersøgelsesaktiviteterne«, mens andet afsnit foreskriver, at Kommissionen og medlemsstaterne ikke »viderebringer oplysninger […], som i behørigt begrundede tilfælde ifølge deres natur er forretningshemmeligheder, medmindre viderebringelse af oplysninger om produkters sikkerhedsmæssige egenskaber under visse omstændigheder er nødvendig for at beskytte forbrugernes sikkerhed og sundhed«.
Med disse bestemmelser i mente skal medlemsstaterne og Kommissionen ikke give offentligheden adgang til oplysninger om et farligt produkt, som der er indgivet meddelelse om via RAPEX-applikationen, hvis videregivelse af oplysningerne ville bringe beskyttelsen af retsprocedurer, overvågnings- og efterforskningsaktiviteter eller forretningshemmeligheder i fare, medmindre der er tale om information vedrørende produkters sikkerhedsmæssige egenskaber, som det på grund af særlige omstændigheder er nødvendigt at offentliggøre for at beskytte forbrugernes sundhed og sikkerhed.
3.3.3. Anmodninger om fortrolig behandling
En meddelende medlemsstat kan i sin meddelelse anmode om fortrolig behandling. I en sådan anmodning angives det tydeligt, hvilke(n) del(e) af meddelelsen der ønskes behandlet fortroligt.
Enhver anmodning om fortrolig behandling vedlægges desuden en begrundelse, hvori det tydeligt er angivet, hvad anmodningen er baseret på, jf. artikel 16, stk. 1 og 2, i DPSA.
Anmodninger om fortrolig behandling behandles af Kommissionen. Kommissionen kontrollerer, at anmodningen er fuldstændig (dvs. angiver, hvilke dele af formularen der skal behandles fortroligt, og indeholder en begrundelse) og berettiget (dvs. er i overensstemmelse med DPSA og retningslinjerne). Kommissionen træffer beslutning om anmodningens gyldighed efter samråd med det pågældende RAPEX-kontaktpunkt.
3.3.4. Håndtering af meddelelser, der behandles fortroligt
I artikel 16, stk. 2, i DPSA hedder det: »Beskyttelsen af forretningshemmeligheder er ikke til hinder for videregivelse til de kompetente myndigheder af oplysninger, der er af betydning for effektive kontrolaktiviteter og for et effektivt markedstilsyn.« Meddelelser, der er omfattet af en beslutning om delvist eller fuldt fortrolig behandling, gennemgås af Kommissionen, hvorefter de — efter at være blevet valideret og videreformidlet via RAPEX-applikationen — lægges til grund for den sædvanlige opfølgning i medlemsstaterne. Det, at en meddelelse eller dele heraf behandles fortroligt, er ikke til hinder for, at meddelelsen håndteres og videreformidles til de kompetente nationale myndigheder via RAPEX.
Den eneste væsentlige forskel med hensyn til behandlingen og opfølgningen er, at Kommissionen og medlemsstaterne ikke videregiver dele af en meddelelse, som skal holdes hemmelige for offentligheden. De pågældende dele skal fortsat behandles fortroligt og må derfor ikke offentliggøres i nogen form. Myndigheder i medlemsstaterne, der modtager fortrolige oplysninger via RAPEX, sørger ved varetagelsen af deres opgaver for, at de pågældende oplysninger beskyttes.
3.3.5. Tilbagetrækning af anmodninger om fortrolig behandling
Den meddelende medlemsstat trækker øjeblikkelig sin anmodning om fortrolig behandling tilbage, hvis myndigheden i den pågældende medlemsstat bliver bekendt med, at grundlaget for en sådan anmodning ikke længere er til stede. Ved modtagelsen af den relevante anmodning fra den meddelende medlemsstat underretter Kommissionen samtlige medlemsstater om, at en meddelelse ikke længere skal behandles fortroligt.
En RAPEX-meddelelse, der ikke længere er omfattet af en beslutning om fuldt eller delvist fortrolig behandling, offentliggøres i overensstemmelse med de »generelle regler« for RAPEX-meddelelser.
3.4. Kommissionens gennemgang af meddelelser
Kommissionen gennemgår alle meddelelser, som den modtager via RAPEX-applikationen, inden den sender dem videre til medlemsstaterne, for at sikre, at de er korrekte og fuldstændige.
3.4.1. Korrekthed
Når den skal vurdere, om en meddelelse er korrekt, kontrollerer Kommissionen især, at:
|
— |
meddelelsen opfylder alle relevante krav i DPSA og retningslinjerne |
|
— |
der ikke allerede er indgivet meddelelse om det pågældende produkt (for at undgå unødvendige dubletter) |
|
— |
meddelelsen (indsendt af RAPEX-kontaktpunktet i den meddelende medlemsstat) er klassificeret i overensstemmelse med de kriterier, der er fastsat i kapitel 3.1 i retningslinjerne |
|
— |
de meddelte oplysninger (især risikobeskrivelsen) er i overensstemmelse med gældende produktsikkerhedsbestemmelser og de relevante standarder |
|
— |
det er den rigtige notifikationsprocedure, der er valgt. |
3.4.2. Fuldstændighed
Efter at have bekræftet, at en meddelelse er korrekt, kontrollerer Kommissionen, at den også er fuldstændig. Punkt 3.2.1 og 3.2.2 i retningslinjerne anvendes som referencegrundlag. Der sættes særlig fokus på de dele af meddelelsen, der vedrører produktidentifikation, risikobeskrivelse, foranstaltninger, sporbarhed og distributionskanaler.
Eftersom Kommissionen ikke har beføjelser til at foretage en risikovurdering af det indberettede produkt og blot kontrollerer, at den indgivne meddelelse indeholder en sådan vurdering, fremlægger den meddelende medlemsstat altid en udtømmende risikobeskrivelse, der omfatter alle de elementer, der er nævnt i punkt 3.2.2 i retningslinjerne.
3.4.3. Anmodninger om supplerende oplysninger
Såfremt Kommissionen har spørgsmål vedrørende en meddelelse i forbindelse med gennemgangen heraf, kan den vælge at suspendere valideringen af meddelelsen og anmode den meddelende medlemsstat om supplerende oplysninger eller nærmere redegørelser. Den meddelende medlemsstat fremlægger den supplerende information inden udløbet af den frist, der er angivet i Kommissionens anmodning om oplysninger.
3.4.4. Undersøgelser
Kommissionen kan om nødvendigt gennemføre en undersøgelse til vurdering af sikkerheden ved et produkt. En sådan undersøgelse vil især kunne være relevant i situationer, hvor der er alvorlige tvivl vedrørende risiciene ved et produkt, som der er indgivet meddelelse om via RAPEX-applikationen. Sådanne tvivlsspørgsmål kan enten opstå i forbindelse med Kommissionens behandling af en meddelelse eller blive påpeget over for Kommissionen af en medlemsstat (f.eks. ved en tilbagemelding) eller af en tredjepart (f.eks. en producent).
Kommissionen kan som led i sådanne undersøgelser især:
|
— |
anmode enhver medlemsstat om oplysninger eller nærmere redegørelser |
|
— |
anmode om en uafhængig risikovurdering og uafhængig afprøvning (laboratorietest eller visuelle undersøgelser) af det produkt, der undersøges |
|
— |
høre de videnskabelige udvalg, Det Fælles Forskningscenter eller andre institutioner, der er specialiserede i forbrugerprodukters sikkerhed |
|
— |
indkalde til møder med det udvalg, der er nedsat i henhold til DPSA, forbrugersikkerhedsnetværket og/eller RAPEX-kontaktpunkter og høre de relevante arbejdsgrupper med henblik på at drøfte en undersøgelses forløb. |
Hvis en undersøgelse vedrører et produkt, som der er indgivet meddelelse om via RAPEX-applikationen, kan Kommissionen suspendere valideringen af en meddelelse eller, hvor en sådan meddelelse allerede er blevet valideret og formidlet via RAPEX-applikationen, midlertidigt fjerne den oversigt, der er offentliggjort på RAPEX-hjemmesiden. Når en undersøgelse er afsluttet, har Kommissionen, afhængigt af resultaterne af undersøgelsen (og om nødvendigt efter samråd med den meddelende medlemsstat), først og fremmest følgende valgmuligheder: Den kan validere den tidligere suspenderede meddelelse og formidle den via RAPEX, den kan opretholde den validerede meddelelse i RAPEX-applikationen (med eventuelle ændringer), eller den kan permanent slette meddelelsen i RAPEX-applikationen.
Kommissionen underretter medlemsstaterne om:
|
— |
enhver beslutning fra Kommissionens side om at iværksætte en undersøgelse, idet den tydeligt angiver, hvilke forhold der ligger til grund for beslutningen |
|
— |
enhver beslutning fra Kommissionens side om at afslutte en undersøgelse, idet den fremlægger sine konklusioner og angiver eventuelle ændringer i de(n) undersøgte meddelelse(r) |
|
— |
alle nye situationer eller forhold, der konstateres i løbet af en undersøgelse. |
3.5. Validering og formidling af meddelelser
3.5.1. Validering og formidling af meddelelser
Kommissionen validerer og formidler (»validering«) via RAPEX-applikationen og inden for de frister, der er angivet i tillæg 4 til retningslinjerne, alle meddelelser, der på grundlag af undersøgelsen vurderes at være korrekte og fuldstændige.
Har den meddelende medlemsstat i løbet af en undersøgelse fået en anmodning (og om nødvendigt efterfølgende en påmindelse) om supplerende oplysninger eller nærmere redegørelser, har Kommissionen følgende muligheder:
|
— |
Hvis anmodningen om supplerende oplysninger eller nærmere redegørelser er blevet efterkommet, gennemgår Kommissionen meddelelsen på ny og validerer den, om nødvendigt med en ændret klassifikation (f.eks. fra »meddelelse til orientering« til »artikel 12-meddelelse«). |
|
— |
Hvis anmodningen om supplerende oplysninger eller nærmere redegørelser ikke er blevet efterkommet inden for den fastsatte frist, eller hvis oplysningerne/redegørelserne er utilstrækkelige, træffer Kommissionen beslutning på grundlag af den meddelte information, inden den, afhængigt af omstændighederne, enten kan validere den efter at have ændret klassifikationen (f.eks. fra »artikel 12-meddelelse« til »meddelelse til orientering«) eller beslutte ikke at validere den. |
3.5.2. Validering af meddelelser om sikkerhedsaspekter, der behandles på EU-plan
Når medlemsstaterne indbyrdes har fastlagt en fælles indfaldsvinkel til risikovurdering og/eller håndhævelse, kan Kommissionen, under hensyntagen til omstændighederne og medlemsstaternes synspunkter, først og fremmest vælge at:
|
— |
beholde de pågældende meddelelser i RAPEX-applikationen eller |
|
— |
ændre klassifikationen af de meddelelser, der er lagret i RAPEX-applikationen, eller |
|
— |
fjerne meddelelser fra RAPEX-applikationen (17). |
3.6. Oplysninger om farlige produkter sendt af Kommissionen
I punkt 9 i bilag II til DPSA hedder det: »Kommissionen kan underrette de nationale kontaktpunkter om produkter, der frembyder alvorlige risici, og som er importeret til eller eksporteret fra Det Europæiske Fællesskab og Det Europæiske Økonomiske Samarbejdsområde.«
Kommissionen kan sende medlemsstaterne oplysninger om farlige nonfoodforbrugerprodukter med oprindelse i og uden for EU, som ifølge de tilgængelige oplysninger kan formodes at være i handelen på EU-markedet. Dette vedrører primært oplysninger, som Kommissionen modtager fra tredjelande, internationale organisationer, virksomheder eller andre systemer for hurtig varsling.
Kommissionen undersøger så vidt muligt, om de pågældende oplysninger er korrekte og fuldstændige, inden den videresender dem til medlemsstaterne. Kommissionen kan imidlertid kun foretage foreløbig kontrol og kan ikke påtage sig det juridiske ansvar for gyldigheden af de informationer, den formidler, idet den hverken juridisk eller teknisk set har mulighed for at gennemføre en fuldstændig risikovurdering eller træffe håndhævelsesforanstaltninger.
3.7. Opfølgning på meddelelser
3.7.1. Opfølgningen for de forskellige typer meddelelser
Medlemsstaterne påser, at der følges behørigt op på RAPEX-meddelelser (dvs. »artikel 12-meddelelser« og »artikel 12-meddelelser, der nødvendiggør hasteforanstaltninger«) og oplysninger fra Kommissionen om farlige produkter (jf. punkt 3.6) hurtigst muligt og under alle omstændigheder inden for de frister, der er angivet i tillæg 3 til retningslinjerne.
Meddelelser til orientering kræver ikke nogen særlig opfølgning. Ofte indeholder disse meddelelser ikke de oplysninger, der er nødvendige for at sikre en effektiv håndhævelse for det indberettede produkt (f.eks. er det indberettede produkt og/eller de meddelte foranstaltninger måske ikke identificeret tilstrækkelig nøjagtigt). Medlemsstaterne opfordres dog til at sikre opfølgning på sådanne meddelelser, hvis det indberettede produkt kan formodes at være blevet stillet til rådighed for forbrugerne på deres marked, og produktidentifikationen gør det muligt at træffe foranstaltninger.
3.7.2. Målene med opfølgningen
Når en medlemsstat modtager en meddelelse, gennemgår den de oplysninger, der er indeholdt i meddelelsen, og træffer de nødvendige foranstaltninger for at:
|
— |
fastslå, om produktet er markedsført på dens område |
|
— |
vurdere, hvilke præventive eller restriktive foranstaltninger der skal træffes vedrørende det indberettede produkt, der er fundet på medlemsstatens marked, under hensyntagen til de foranstaltninger, der er truffet af den meddelende medlemsstat, samt eventuelle særlige omstændigheder, der måtte tale for at træffe andre eller slet ingen foranstaltninger |
|
— |
om nødvendigt foretage supplerende risikovurdering og afprøvning af det indberettede produkt |
|
— |
indsamle eventuelle supplerende oplysninger, der måtte være relevante for andre medlemsstater (f.eks. oplysninger om distributionskanaler for det indberettede produkt i andre medlemsstater). |
3.7.3. Opfølgningsmetoder
For at sikre en effektiv opfølgning bør de nationale myndigheder basere deres indsats på bedste praksis, således at opfølgningen omfatter:
|
— |
Kontrol på markedet De nationale myndigheder gennemfører regelmæssigt kontrol (planlagt kontrol og stikprøvekontrol) på markedet for at fastslå, om forbrugerprodukter, som der er indgivet meddelelse om via RAPEX-applikationen, stilles til rådighed for forbrugerne. |
|
— |
Samarbejde med brancheorganisationer De nationale myndigheder forsyner regelmæssigt brancheorganisationerne med oversigter over de seneste meddelelser og beder disse organisationer svare på, hvorvidt nogen af de indberettede produkter er blevet fremstillet eller distribueret af deres medlemmer. De nationale myndigheder giver kun virksomhederne resuméer af meddelelser, såsom de ugentlige oversigter, der offentliggøres på RAPEX-hjemmesiden. Meddelelser i deres helhed må ikke videregives til tredjepart, idet visse oplysninger (f.eks. detaljerede oplysninger om risikobeskrivelsen eller information om distributionskanaler) ofte er fortrolige og skal beskyttes. |
|
— |
Offentliggørelse af RAPEX-oplysninger via internettet eller elektroniske og skrevne medier De nationale myndigheder advarer løbende forbrugere og virksomheder om forbrugerprodukter, som der indgives meddelelse om via RAPEX, på deres hjemmesider og/eller via andre medier. Oplysninger offentliggjort på denne måde gør det muligt for forbrugerne at kontrollere, hvorvidt de er i besiddelse af eller anvender farlige produkter, og er ofte en kilde til nyttig feedback for de pågældende myndigheder. |
De nationale myndigheder bør anvende en kombination af forskellige opfølgningsmetoder og ikke koncentrere alle deres aktiviteter om én metode.
Især en medlemsstat, hvori en producent af, repræsentant for eller importør af det indberettede produkt er etableret (»hovedmedlemsstaten«), sørger for en passende opfølgning på meddelelser, der formidles via RAPEX-applikationen. »Hovedmedlemsstaten« har ofte bedre muligheder — juridisk og teknisk set — for at indhente oplysninger om det indberettede tilfælde, som vil gøre det nemmere for andre medlemsstater at gennemføre en effektiv opfølgning.
3.8. Permanent sletning af en meddelelse i RAPEX-applikationen
Meddelelser, der formidles via RAPEX-applikationen, gemmes i systemet i ubegrænset tid. Kommissionen kan dog, i de situationer, der er beskrevet i dette kapitel, permanent slette en meddelelse i applikationen.
3.8.1. Situationer, hvor meddelelser kan slettes
|
— |
Der foreligger dokumentation for, at et eller flere af RAPEX-notifikationskriterierne (18) ikke er opfyldt, og en RAPEX-meddelelse er således ikke berettiget. Dette gælder især i tilfælde, hvor det konstateres, at den første risikovurdering ikke er gennemført på korrekt vis, og at det indberettede produkt ikke udgør en alvorlig risiko for forbrugernes sundhed og sikkerhed. Omfattet er også situationer, hvor en klage over de meddelte foranstaltninger er blevet taget til følge ved en domstolsbehandling eller andre procedurer og ikke længere er gyldige. |
|
— |
Der er ikke truffet foranstaltninger vedrørende et produkt, som der er indgivet meddelelse om via RAPEX-applikationen (til orientering) inden beslutningen om at træffe foranstaltninger eller forholdsregler (19). |
|
— |
Efter drøftelser på EU-plan beslutter medlemsstaterne i fællesskab, at det ikke er formålstjenligt at udveksle oplysninger om visse sikkerhedsaspekter, som der er indgivet meddelelse om via RAPEX-applikationen (20). |
|
— |
Der foreligger dokumentation for, at produkter, der er omfattet af en meddelelse, ikke længere markedsføres, og at samtlige enheder, som var stillet til rådighed for forbrugerne, allerede er trukket tilbage fra markedet og kaldt tilbage fra forbrugerne i samtlige medlemsstater. |
En meddelelse kan ikke begæres slettet med den begrundelse, at det indberettede produkt er blevet ændret, så det nu opfylder alle gældende sikkerhedskrav, medmindre der fremlægges dokumentation for, at alle de farlige produkter (enheder), der var stillet til rådighed for forbrugerne, er trukket og kaldt tilbage i samtlige medlemsstater, og at de ikke længere markedsføres.
3.8.2. Anmodende medlemsstat
Kommissionen kan kun slette meddelelser i RAPEX-applikationen efter anmodning herom fra den meddelende medlemsstat, idet sidstnævnte påtager sig det fulde ansvar for de oplysninger, der formidles gennem systemet. Alle medlemsstater opfordres dog til at underrette Kommissionen om alle forhold, der vil kunne begrunde, at en meddelelse slettes.
3.8.3. Indholdet af anmodningen
Enhver anmodning om sletning ledsages af en begrundelse, hvori det er angivet, hvad anmodningen er baseret på, og af al tilgængelig dokumentation til underbygning heraf. Kommissionen gennemgår hver enkelt anmodning og efterprøver især begrundelsen og dokumentationen. Kommissionen kan udbede sig supplerende oplysninger, nærmere redegørelser eller en udtalelse fra den meddelende medlemsstat og/eller andre medlemsstater, inden den træffer sin beslutning.
3.8.4. Beslutninger om sletning af meddelelser
Beslutter Kommissionen på grundlag af den meddelte begrundelse at slette en meddelelse i RAPEX-applikationen, fjerner den meddelelsen fra:
|
— |
RAPEX-applikationen (eller gør den på anden måde usynlig for alle brugere af systemet) |
|
— |
RAPEX-hjemmesiden (om nødvendigt). |
Kommissionen underretter pr. e-mail eller på anden, tilsvarende effektiv måde samtlige medlemsstater om sletningen af en meddelelse og orienterer om nødvendigt også befolkningen via offentliggørelse af et korrigendum på RAPEX-hjemmesiden.
3.9. Midlertidig fjernelse af en RAPEX-meddelelse fra RAPEX-hjemmesiden
3.9.1. Situationer, hvori meddelelser kan fjernes midlertidigt
Hvor det er berettiget, kan Kommissionen midlertidigt fjerne en RAPEX-meddelelse fra RAPEX-hjemmesiden, især hvis den meddelende medlemsstat har mistanke om, at en risikovurdering i en indgivet meddelelse ikke er gennemført på korrekt vis, og at det indberettede produkt derfor muligvis ikke udgør en alvorlig risiko for forbrugernes sundhed og sikkerhed. En meddelelse kan midlertidigt fjernes fra RAPEX-hjemmesiden, indtil tvivlsspørgsmålet vedrørende risikovurderingen af det indberettede produkt er blevet afklaret.
3.9.2. Anmodende medlemsstat
Jf. punkt 3.8.2.
3.9.3. Indholdet af anmodningen
Jf. punkt 3.8.3.
3.9.4. Beslutning om at fjerne en meddelelse
Beslutter Kommissionen på grundlag af den meddelte begrundelse at fjerne en RAPEX-meddelelse fra RAPEX-hjemmesiden, underretter den pr. e-mail eller på anden, tilsvarende effektiv måde samtlige medlemsstater herom og orienterer om nødvendigt også befolkningen via offentliggørelse af et korrigendum på RAPEX-hjemmesiden.
3.9.5. Fornyet offentliggørelse af en meddelelse
Når grundlaget for at fjerne en meddelelse fra RAPEX-hjemmesiden ikke længere er til stede, underretter den meddelende medlemsstat øjeblikkelig Kommissionen herom. Den meddeler især Kommissionen resultaterne af en eventuel ny risikovurdering, så Kommissionen på det rigtige grundlag kan beslutte, hvorvidt en meddelelse skal opretholdes i RAPEX-applikationen og offentliggøres på RAPEX-hjemmesiden på ny eller permanent slettes i RAPEX-applikationen (efter anmodning fra den meddelende medlemsstat).
Kommissionen kan vælge at offentliggøre en RAPEX-meddelelse på RAPEX-hjemmesiden på ny efter en begrundet anmodning herom fra den meddelende medlemsstat, når risikovurderingsspørgsmålet er afklaret.
Kommissionen underretter pr. e-mail eller på anden, tilsvarende effektiv måde de øvrige medlemsstater om offentliggørelsen på ny af en RAPEX-meddelelse på RAPEX-hjemmesiden og orienterer om nødvendigt også befolkningen ved at erstatte korrigendummet på RAPEX-hjemmesiden med et nyt.
3.10. Frister for indgivelse af RAPEX-meddelelser
3.10.1. Frister (21)
Medlemsstaterne underretter hurtigst muligt og under alle omstændigheder inden for de frister, der er angivet i tillæg 3 til retningslinjerne, Kommissionen om præventive og restriktive foranstaltninger, der træffes vedrørende forbrugerprodukter, som udgør en alvorlig risiko for forbrugernes sundhed og sikkerhed. Der er på nationalt plan etableret til formålet egnede ordninger for formidling af oplysninger mellem de nationale myndigheder med ansvar for produktsikkerhed og RAPEX-kontaktpunktet for at sikre, at fristerne overholdes.
De angivne frister gælder uanset eventuelle igangværende klageprocedurer eller bestemmelser om obligatorisk offentliggørelse.
3.10.2. Krisesituationer
Før enhver »artikel 12-meddelelse, der nødvendiggør hasteforanstaltninger« ringer RAPEX-kontaktpunktet til Kommissionens RAPEX-team (på teamets mobiltelefonnummer) for at sikre omgående validering og opfølgning. Denne regel er især relevant for meddelelser, der formidles i weekenden eller i ferieperioder.
4. Tilbagemeldinger
4.1. Underretning om opfølgende foranstaltninger
Medlemsstaterne meddeler Kommissionen alle foranstaltninger til opfølgning på RAPEX-meddelelser (dvs. »artikel 12-meddelelser« og »artikel 12-meddelelser, der nødvendiggør hasteforanstaltninger«) og på oplysninger fra Kommissionen om farlige produkter (jf. punkt 3.6).
Medlemsstaterne opfordres til at underrette Kommissionen om alle foranstaltninger til opfølgning på meddelelser, der formidles til orientering.
4.2. Indholdet af tilbagemeldinger
4.2.1. Meddelte oplysninger
Resultaterne af opfølgningsaktiviteter meddeles til Kommissionen i form af tilbagemeldinger på meddelelser. Med henblik på harmonisering af de oplysninger, der meddeles, og for at begrænse arbejdsbyrden til et minimum indgiver medlemsstaterne først og fremmest tilbagemeldinger i følgende situationer:
|
— |
Fund af produktet Der sendes en tilbagemelding, når nationale myndigheder finder det indberettede produkt på markedet eller ved de ydre grænser. I tilbagemeldingen gives detaljerede oplysninger om det pågældende produkt (såsom betegnelse, mærke, modelnummer, stregkode og varepartiets nummer) samt om, hvor mange enheder der er fundet i alt. Derudover gives følgende oplysninger vedrørende de foranstaltninger, der er truffet: art (obligatoriske/frivillige), kategori (f.eks. tilbagetrækning fra markedet/tilbagekaldelse fra forbrugerne), anvendelsesområde (f.eks. landsdækkende/lokale), ikrafttrædelsesdato og varighed (f.eks. ikke-tidsbegrænsede/midlertidige). Hvis det indberettede produkt er fundet på markedet, uden at der er truffet foranstaltninger, skal det i tilbagemeldingen begrundes, at der ingen foranstaltninger er truffet. Medlemsstaterne orienterer ikke (medmindre Kommissionen anmoder om det) Kommissionen om konklusionerne på opfølgningsaktiviteter, hvis det indberettede produkt ikke er fundet på markedet. |
|
— |
Afvigende risikovurdering Der sendes en tilbagemelding, hvis konklusionerne i en risikovurdering foretaget af en myndighed i den medlemsstat, der sender tilbagemeldingen (i det følgende benævnt »den reagerende medlemsstat«), afviger fra konklusionerne i en meddelelse. Denne tilbagemelding indeholder en detaljeret risikobeskrivelse (med blandt resultaterne af afprøvninger, en risikovurdering og oplysninger om kendte ulykker og hændelser), ledsaget af den relevante dokumentation (afprøvningsrapporter, attester osv.). Den reagerende medlemsstat dokumenterer desuden, at den risikovurdering, der er fremsendt med tilbagemeldingen, er foretaget for det samme produkt som det, der er indgivet meddelelse om, dvs. et produkt af samme mærke og med samme betegnelse, modelnummer, produktionsdato, oprindelsessted osv. |
|
— |
Supplerende oplysninger Der sendes en tilbagemelding, hvis nationale myndigheder indsamler yderligere oplysninger (som led i opfølgningsaktiviteter), der kan være nyttige for markedsovervågningen og håndhævelsesindsatsen i andre medlemsstater. Medlemsstaterne opfordres til at indsamle yderligere oplysninger, der kan være relevante for myndighederne såvel i andre medlemsstater som i tredjelande, der har et tæt samarbejde med EU om produktsikkerhed. Det gælder bl.a. oplysninger om produktets oprindelse (f.eks. om oprindelsesland, producent og/eller eksportører) og om omsætningsleddene (f.eks. om bestemmelseslande, importører og distributører). Det reagerende land vedlægger al tilgængelig dokumentation vedrørende tilbagemeldingen, såsom kopier af ordrer, købekontrakter, fakturaer, toldangivelser osv. |
Kontaktpunktet i den reagerende medlemsstat sørger sammen med den ansvarlige myndighed for, at alle oplysninger i tilbagemeldingen er nøjagtige og fuldstændige, og at det pågældende produkt ikke kan forveksles med lignende produkter af samme eller lignende kategori eller type på EU-markedet.
4.2.2. Fuldstændige tilbagemeldinger
Oplysningerne i tilbagemeldinger skal være så fuldstændige som muligt. Standardtilbagemeldingsformularen findes i tillæg 2 til retningslinjerne. Foreligger visse relevante oplysninger ikke på tidspunktet for afsendelse af en tilbagemelding, angiver den reagerende medlemsstat dette i tilbagemeldingsformularen. Så snart de pågældende oplysninger foreligger, ajourfører den reagerende medlemsstat tilbagemeldingen. Den ajourførte tilbagemelding gennemgås af Kommissionen, inden den valideres og videreformidles i systemet.
RAPEX-kontaktpunktet instruerer alle myndigheder i sin egen medlemsstat, der deltager i RAPEX-netværket, i, hvilke oplysninger tilbagemeldingsformularen skal indeholde for at være udfyldt korrekt. Dette vil bidrage til at sikre, at de oplysninger, som de pågældende myndigheder giver kontaktpunktet, er korrekte og fuldstændige.
4.2.3. Ajourføring af validerede tilbagemeldinger
Den reagerende medlemsstat underretter (hurtigst muligt og under alle omstændigheder inden for de frister, der er angivet i tillæg 3 til retningslinjerne) Kommissionen om alle nye situationer eller forhold, der måtte nødvendiggøre ændringer i en tilbagemelding, som er formidlet via RAPEX-applikationen. Medlemsstaterne underretter især Kommissionen om ændringer i status for de foranstaltninger, der er truffet, og i den risikovurdering, der er fremsendt med en tilbagemelding.
Kommissionen gennemgår oplysningerne fra den reagerende medlemsstat og ajourfører om nødvendigt de pågældende oplysninger.
4.2.4. Ansvaret for tilbagemeldinger
I punkt 10 i bilag II til DPSA hedder det: »Den medlemsstat, der foretager meddelelsen, er ansvarlig for de meddelte oplysninger.«
RAPEX-kontaktpunktet og den myndighed, der er involveret i tilbagemeldingsproceduren (f.eks. i kraft af at den foretager risikovurderingen eller træffer restriktive foranstaltninger), er ansvarlige for de oplysninger, der meddeles i tilbagemeldinger. RAPEX-kontaktpunktet kontrollerer og validerer alle tilbagemeldinger udarbejdet af de pågældende myndigheder, inden den sender meddelelserne videre til Kommissionen.
Kommissionens aktiviteter, herunder gennemgang og validering af tilbagemeldinger, indebærer ikke, at Kommissionen påtager sig noget ansvar for de formidlede oplysninger, idet dette fortsat ligger hos den reagerende medlemsstat.
4.3. Fortrolighed
En reagerende medlemsstat kan i tilbagemeldingen anmode om fortrolig behandling. I en sådan anmodning angives det tydeligt, hvilke(n) del(e) af tilbagemeldingen der ønskes behandlet fortroligt. Enhver anmodning om fortrolig behandling vedlægges desuden en begrundelse, hvori det tydeligt er angivet, hvad anmodningen er baseret på.
Kommissionen gennemgår anmodninger om fortrolig behandling for at efterprøve, at de er berettigede (dvs. er i overensstemmelse med DPSA og retningslinjerne) og fuldstændige (dvs. angiver, hvilke dele af formularen, der skal behandles fortroligt, og indeholder en begrundelse). Kommissionen træffer den endelige beslutning om fortrolighed efter samråd med det ansvarlige RAPEX-kontaktpunkt.
Kommissionen og medlemsstaterne behandler tilbagemeldinger med anmodning om fortrolig behandling på samme måde som andre tilbagemeldinger. Det, at en tilbagemelding eller dele heraf behandles fortroligt, er ikke til hinder for, at tilbagemeldingen formidles til de kompetente nationale myndigheder via RAPEX-systemet. Kommissionen og medlemsstaterne videregiver dog ingen dele af en tilbagemelding, som skal holdes hemmelige for offentligheden. Disse oplysninger er fortrolige og må derfor ikke offentliggøres i nogen form.
Den reagerende medlemsstat trækker øjeblikkelig sin anmodning om fortrolig behandling tilbage, hvis myndigheden i den pågældende medlemsstat bliver bekendt med, at grundlaget for en sådan anmodning ikke længere er til stede. Efter at have modtaget den relevante anmodning underretter Kommissionen samtlige medlemsstater om, at en tilbagemelding ikke længere skal behandles fortroligt.
4.4. Kommissionens gennemgang af tilbagemeldinger
4.4.1. Korrekthed og fuldstændighed
Kommissionen gennemgår alle tilbagemeldinger, som den modtager via RAPEX-applikationen, inden de valideres og sendes videre til medlemsstaterne. Ved denne kontrol fokuseres der på, om de meddelte oplysninger er korrekte og fuldstændige.
Kommissionen kontrollerer, om en tilbagemelding opfylder alle relevante krav i DPSA og i retningslinjerne, og om der er anvendt den rigtige tilbagemeldingsprocedure. Efter at have bekræftet, at en tilbagemelding er korrekt, kontrollerer Kommissionen, at den også er fuldstændig. Punkt 4.2.2 i retningslinjerne anvendes som referencegrundlag i forbindelse med denne kontrol.
Kommissionen tildeler tilbagemeldinger med risikovurderinger særlig opmærksomhed. Den efterprøver navnlig, at risikobeskrivelsen er fuldstændig, klart formuleret og veldokumenteret, og at det er godtgjort, at risikovurderingen vedrører det produkt, der er indgivet meddelelse om.
4.4.2. Anmodninger om supplerende oplysninger
Kommissionen kan, inden den validerer en tilbagemelding, anmode den reagerende medlemsstat om at fremlægge supplerende oplysninger eller nærmere redegørelser inden for en bestemt frist. Valideringen af en tilbagemelding kan gøres betinget af, at de ønskede oplysninger modtages.
Kommissionen kan bede en hvilken som helst — og frem for alt den meddelende — medlemsstat om at udtale sig om en valideret tilbagemelding. Medlemsstaten forelægger sin udtalelse for Kommissionen inden for den af sidstnævnte fastsatte frist. Den meddelende medlemsstat meddeler ydermere Kommissionen, hvorvidt der er behov for at ændre meddelelsen (f.eks. risikovurderingen) eller dennes status (f.eks. permanent sletning i systemet).
4.5. Validering og formidling af tilbagemeldinger
Alle tilbagemeldinger, der er gennemgået og fundet korrekte og fuldstændige, valideres og formidles (»validering«) af Kommissionen inden for de frister, der er angivet i tillæg 4 til retningslinjerne.
Kommissionen validerer ikke tilbagemeldinger, hvortil der er knyttet en risikovurdering, der afviger fra vurderingen i den meddelelse, tilbagemeldingen vedrører, hvis ikke risikovurderingen er fuldstændig, klart formuleret og veldokumenteret, eller hvis det ikke er godtgjort, at risikovurderingen vedrører det produkt, der er indgivet meddelelse om.
4.6. Permanent sletning af en tilbagemelding i RAPEX-applikationen
Tilbagemeldinger, der formidles via RAPEX-applikationen, gemmes i systemet lige så længe som den meddelelse, de vedrører. Kommissionen kan permanent slette en valideret tilbagemelding i RAPEX-applikationen, hvis en meddelelse, som den pågældende tilbagemelding knytter sig til, er blevet slettet i RAPEX-applikationen (jf. kapitel 3.8 i retningslinjerne). Kommissionen kan desuden slette en valideret tilbagemelding, der tydeligvis indeholder ukorrekte oplysninger, navnlig hvis:
|
— |
et produkt, der er fundet på markedet af den reagerende medlemsstat, adskiller sig fra det produkt, der er omfattet af en meddelelse |
|
— |
en klage over de foranstaltninger, der er truffet af den reagerende medlemsstat, er blevet taget til følge ved en domstolsbehandling eller andre procedurer og efterfølgende er ophævet |
|
— |
den risikovurdering, der er foretaget af den reagerende medlemsstat, bevisligt er ukorrekt eller vedrører et andet produkt end det, der er omfattet af en meddelelse. |
Punkt 3.8.2 og 3.8.3 finder anvendelse.
Når Kommissionen beslutter at slette en tilbagemelding, fjernes denne fra RAPEX-applikationen (eller gøres på anden måde usynlig for brugere af systemet).
Kommissionen underretter pr. e-mail eller på anden, tilsvarende effektiv måde samtlige medlemsstater om sletningen af en tilbagemelding.
4.7. Frister for tilbagemeldinger
Medlemsstaterne sender en tilbagemelding til Kommissionen hurtigst muligt og under alle omstændigheder inden for de frister, der er angivet i tillæg 3 til retningslinjerne.
Der er på nationalt plan etableret til formålet egnede ordninger for formidling af oplysninger mellem alle de kompetente myndigheder og RAPEX-kontaktpunktet for at sikre, at fristerne overholdes.
De angivne frister gælder uanset eventuelle igangværende klageprocedurer eller bestemmelser om obligatorisk offentliggørelse.
5. Forvaltningen af RAPEX-netværkene
5.1. RAPEX-kontaktpunkter
Hver medlemsstat etablerer ét enkelt RAPEX-kontaktpunkt, der står for forvaltningen af RAPEX-systemet på nationalt plan. De nationale myndigheder afgør, hvilken national myndighed RAPEX-kontaktpunktet skal oprettes under. Hver medlemsstat sørger desuden for, at dets nationale RAPEX-netværks struktur sikrer en effektiv informationsstrøm mellem RAPEX-kontaktpunktet og de forskellige myndigheder, der deltager i RAPEX.
5.1.1. Tilrettelæggelse
Den enkelte medlemsstat forsyner RAPEX-kontaktpunktet med de ressourcer og den information, det behøver for at kunne varetage sine arbejdsopgaver, og først og fremmest at forvalte systemet med effektive backupfunktioner/driftskontinuitet.
RAPEX-kontaktpunktet har sin egen e-mail-konto for deltagelse i RAPEX-systemet, som alle embedsmænd i det pågældende kontaktpunkt har adgang til (f.eks. rapex@ …). Arbejdsmailkonti eller private e-mail-konti for de embedsmænd, der er ansvarlige for RAPEX-kontaktpunktet, bør ikke anvendes som kontaktpunktets e-mail-konto. RAPEX-kontaktpunktet har også direkte telefon- og faxnumre, som det kan kontaktes på i og uden for normal arbejdstid.
5.1.2. Opgaver
RAPEX-kontaktpunktets vigtigste opgaver er at:
|
— |
tilrettelægge og lede arbejdet i det nationale RAPEX-netværk i overensstemmelse med retningslinjerne |
|
— |
uddanne og bistå alle myndigheder i netværket i anvendelsen af RAPEX |
|
— |
sikre, at alle RAPEX-opgaver i henhold til DPSA og retningslinjerne udføres på behørig vis, og især at alle nødvendige oplysninger (dvs. meddelelser, tilbagemeldinger, supplerende oplysninger osv.) meddeles til Kommissionen inden for de fastsatte frister |
|
— |
formidle oplysninger mellem Kommissionen og de nationale markedsovervågningsmyndigheder og myndigheder med ansvar for kontrollen ved de ydre grænser |
|
— |
kontrollere og validere alle oplysninger, der modtages fra de kompetente myndigheder, inden de sendes videre til Kommissionen via RAPEX-applikationen |
|
— |
kontrollere, om der allerede er indgivet meddelelse om et produkt, eller om oplysninger vedrørende det pågældende produkt allerede er blevet formidlet via RAPEX-applikationen, inden der indgives en meddelelse (for at undgå dubletter) |
|
— |
påtage sig ansvaret (sammen med den pågældende myndighed) for de oplysninger, der meddeles via RAPEX-applikationen |
|
— |
deltage i RAPEX-kontaktpunkternes arbejdsgruppemøder og andre arrangementer vedrørende forvaltningen af RAPEX |
|
— |
komme med forslag til forbedring af systemet |
|
— |
straks underrette Kommissionen om eventuelle tekniske problemer med RAPEX-applikationen |
|
— |
koordinere alle RAPEX-relaterede nationale aktiviteter og initiativer |
|
— |
forklare interesserede parter, hvordan RAPEX-systemet fungerer, og hvilke forpligtelser de har ifølge DPSA, navnlig virksomhedsmeddelelsesforpligtelsen i henhold til artikel 5, stk. 3. |
5.2. RAPEX-netværk etableret i EU-regi og på nationalt plan
5.2.1. Netværket af RAPEX-kontaktpunkter
Kommissionen tilrettelægger og leder arbejdet i netværket af RAPEX-kontaktpunkter. Dette netværk består af alle de RAPEX-kontaktpunkter, der er udpeget i medlemsstaterne.
Kommissionen indkalder regelmæssigt til møde i netværket af RAPEX-kontaktpunkter med henblik på at drøfte, hvordan systemet fungerer (f.eks. for at orientere om den seneste udvikling inden for RAPEX og udveksle erfaringer og knowhow), og styrke samarbejdet mellem kontaktpunkterne.
5.2.2. RAPEX-netværk etableret på nationalt plan
RAPEX-kontaktpunktet tilrettelægger og leder arbejdet i sit nationale RAPEX-netværk. Dette netværk består af:
|
— |
RAPEX-kontaktpunktet |
|
— |
markedsovervågningsmyndighederne med ansvar for overvågning af sikkerheden ved forbrugerprodukter |
|
— |
myndighederne med ansvar for kontrollen ved de ydre grænser. |
RAPEX-kontaktpunkterne opfordres til at fastsætte formelle regler for tilrettelæggelsen og forvaltningen af det nationale RAPEX-netværk, så det sikres, at alle involverede myndigheder kender deres rolle og ansvarsområder i RAPEX-systemet. Reglerne kan være bindende eller ikke-bindende, og de fastsættes i overensstemmelse med retningslinjerne.
RAPEX-kontaktpunktet afholder regelmæssigt møder i det nationale RAPEX-netværk med henblik på sammen med alle de involverede myndigheder at drøfte, hvordan RAPEX forvaltes og fungerer, og for at give uddannelseskurser. Et møde i et nationalt RAPEX-netværk kan afholdes i forbindelse med et RAPEX-seminar, hvis det organiseres af Kommissionen i den pågældende medlemsstat.
5.3. RAPEX-kommunikationen — praktiske og tekniske rammer
5.3.1. Sprog
I forbindelse med valget af sprog i meddelelser og tilbagemeldinger og i kommunikationen mellem RAPEX-kontaktpunkterne og Kommissionen skal der tages behørigt hensyn til målene med RAPEX, ligesom sprogvalget skal sikre en hurtig udveksling af oplysninger mellem medlemsstaterne og Kommissionen om forbrugerprodukter, der udgør en alvorlig risiko for forbrugernes sundhed og sikkerhed.
5.3.2. RAPEX-onlineapplikation
Kommissionen etablerer og administrerer en webbaseret applikation til kommunikationen i RAPEX-systemet. Medlemsstaterne anvender denne applikation til at udarbejde og indgive meddelelser og tilbagemeldinger via RAPEX, og Kommissionen bruger den til at validere de dokumenter, den modtager.
Kommissionen giver alle RAPEX-kontaktpunkter og kompetente nationale myndigheder samt de relevante tjenestegrene i Kommissionen adgang til applikationen. Kommissionen opretter så mange brugere som muligt i applikationen under hensyntagen til behov og tekniske begrænsninger. Kommissionen fastsætter reglerne for, hvem der skal have adgang til applikationen.
I situationer, hvor RAPEX-applikationen midlertidigt er ude af drift (af andre årsager end på grund af almindeligt, planlagt vedligeholdelsesarbejde), sender medlemsstaterne kun RAPEX-meddelelser til Kommissionen (dvs. »artikel 12- meddelelser« og »artikel 12 meddelelser, der nødvendiggør hasteforanstaltninger«). Indgivelsen af meddelelser til orientering og tilbagemeldinger indstilles, indtil RAPEX-applikationen er oppe at køre igen. Så længe applikationen er ude af drift, sendes RAPEX-meddelelser til Kommissionen pr. e-mail til adressen sanco-reis@ec.europa.eu eller til en anden, på forhånd oplyst e-mail-adresse. Hvis e-mail-overførsel ikke er mulig, sendes RAPEX-meddelelser til Kommissionen pr. fax til det på forhånd oplyste faxnummer. Det er ikke nødvendigt at sende meddelelser via en medlemsstats faste repræsentation ved EU.
5.3.3. Forvaltningen af RAPEX uden for normal arbejdstid
RAPEX-systemet er altid aktivt. Kommissionen og RAPEX-kontaktpunkterne sørger for, at de embedsmænd, der har ansvaret for forvaltningen af RAPEX, altid kan kontaktes (pr. telefon eller e-mail eller på anden, tilsvarende effektiv måde), og at de kan træffe de nødvendige foranstaltninger, også i krisesituationer og uden for normal arbejdstid, f.eks. i weekenden eller i ferieperioder.
Kommissionen giver RAPEX-kontaktpunkterne kontaktoplysninger på Kommissionens RAPEX-team, herunder navne og e-mail-adresser samt telefon- og faxnumre på embedsmænd, som de kan komme i kontakt med i og uden for normal arbejdstid.
RAPEX-kontaktpunkterne giver Kommissionen deres kontaktoplysninger, herunder navnene på de embedsmænd, der arbejder i kontaktpunktet, navn og adresse på den myndighed, kontaktpunktet er oprettet under, og e-mail-adresser samt telefon- og faxnumre på embedsmænd, som kan kontaktes i og uden for normal arbejdstid. RAPEX-kontaktpunkterne underretter straks Kommissionen om enhver ændring i kontaktoplysningerne. Kommissionen offentliggør kontaktoplysningerne på RAPEX-kontaktpunkterne på RAPEX-hjemmesiden.
DEL III
NOTIFIKATIONSPROCEDUREN, JF. ARTIKEL 11 I DIREKTIVET OM PRODUKTSIKKERHED I ALMINDELIGHED
1. Baggrund og mål
Ved artikel 11 i direktivet om produktsikkerhed i almindelighed er der indført en notifikationsprocedure med henblik på udveksling af oplysninger mellem medlemsstaterne og Kommissionen om foranstaltninger, der træffes vedrørende forbrugerprodukter, som udgør en ikke-alvorlig risiko for forbrugernes sundhed og sikkerhed.
Artikel 11-notifikationssystemet skal (trods lighedspunkter og indbyrdes forbindelser) anvendes som en uafhængig procedure, adskilt fra den notifikationsprocedure, der er indført ved artikel 12 i DPSA (»RAPEX«).
Artikel 11-notifikationsproceduren har to hovedformål:
|
— |
At bidrage til et velfungerende indre marked Det første formål med artikel 11-notifikationsproceduren er at sikre, at Kommissionen underrettes om foranstaltninger truffet af nationale myndigheder, som begrænser markedsføringen på EU-markedet af produkter, der udgør en ikke-alvorlig risiko for forbrugernes sundhed og sikkerhed. Dette formål svarer til målsætningen med den procedure for beskyttelsesforanstaltninger, der er indført ved diverse sektordirektiver, og som har til formål at sikre, at Kommissionen holdes orienteret om præventive og restriktive foranstaltninger, der træffes af nationale myndigheder, og at den kan vurdere, hvorvidt den pågældende begrænsning af den frie bevægelighed for det indberettede produkt er i overensstemmelse med EU-lovgivningen og ikke uretmæssigt krænker princippet om frie varebevægelser. Artikel 11-notifikationsproceduren supplerer proceduren for beskyttelsesforanstaltninger og sikrer, at Kommissionen holdes orienteret om præventive og restriktive foranstaltninger truffet af nationale myndigheder, som ikke er omfattet af sidstnævnte procedure. |
|
— |
At forhindre markedsføring og forbrugernes anvendelse af farlige produkter (som ikke udgør en alvorlig risiko) Det andet formål med artikel 11-notifikationsproceduren er at sikre, at medlemsstaterne hurtigt kan udveksle oplysninger om produkter, der udgør en ikke-alvorlig risiko for forbrugernes sundhed og sikkerhed, og at forhindre eller begrænse markedsføringen og anvendelsen af dem i EU. Dette svarer til formålet med RAPEX, idet RAPEX dog kun omfatter produkter, der udgør en alvorlig risiko for forbrugernes sundhed og sikkerhed. |
2. Notifikationskriterier
Artikel 11-notifikationsproceduren finder kun anvendelse på foranstaltninger, der træffes af nationale myndigheder for at begrænse markedsføringen af produkter, der udgør en ikke-alvorlig risiko for forbrugernes sundhed og sikkerhed, trække dem tilbage fra markedet eller kalde dem tilbage fra forbrugerne. Det betyder, at denne procedure ikke anvendes til meddelelser om frivillige foranstaltninger.
Hvis følgende fem kriterier er opfyldt, har medlemsstaterne en juridisk forpligtelse til at underrette Kommissionen i henhold til artikel 11 i DPSA:
|
— |
Det pågældende produkt er et forbrugerprodukt. |
|
— |
Det er genstand for restriktive foranstaltninger truffet af nationale myndigheder (obligatoriske foranstaltninger). |
|
— |
Det udgør en ikke-alvorlig risiko for forbrugernes sundhed og sikkerhed. |
|
— |
Virkningerne af risikoen rækker ud over eller kan række ud over én medlemsstats område, eller de rækker ikke eller kan ikke række ud over dens område, men foranstaltningerne giver anledning til oplysninger, der kan formodes at være af betydning for produktsikkerheden i andre medlemsstater. |
|
— |
De trufne foranstaltninger er ikke anmeldelsespligtige i henhold til nogen notifikationsprocedure indført ved EU-lovgivning (f.eks. RAPEX som indført ved artikel 12 i DPSA eller den procedure for beskyttelsesforanstaltninger, der er indført ved diverse sektordirektiver). |
Nedennævnte kapitler i retningslinjernes del II er relevante for artikel 11-notifikationsproceduren:
|
— |
Kapitel 2.1 om forbrugerprodukter (definition af et forbrugerprodukt) |
|
— |
Kapitel 2.2 om restriktive foranstaltninger (kategorier af restriktive foranstaltninger, definition af obligatoriske foranstaltninger, underretningstidspunkt og meddelende myndigheder) |
|
— |
Kapitel 2.3 om risikovurdering (risikovurderingsmetode, vurderingsmyndighed) |
|
— |
Kapitel 2.4 om virkninger på tværs af grænserne (internationale hændelser, lokale hændelser). |
3. Meddelelser
Hvis alle notifikationskriterier er opfyldt, udarbejder medlemsstaten en meddelelse og sender den til Kommissionen under anvendelse af RAPEX-applikationen. Standardmeddelelsesformularen findes i tillæg 1 til retningslinjerne.
Alle meddelelser, der sendes via RAPEX-applikationen i henhold til artikel 11 i DPSA, klassificeres i systemet som »artikel 11-meddelelser«.
RAPEX-kontaktpunktet i den meddelende medlemsstat sikrer, at enhver meddelelse opfylder alle de notifikationskriterier, der er fastsat ved artikel 11 i DPSA.
Nedennævnte kapitler i retningslinjernes del II er relevante for artikel 11-notifikationsproceduren:
|
— |
Kapitel 3.2 om indholdet af meddelelser (fuldstændighed, oplysninger, der skal fremlægges, ajourføring af oplysninger, ansvaret for de formidlede oplysninger) |
|
— |
Kapitel 3.3 om fortrolighed (videregivelse af oplysninger, undtagelser fra de almindelige regler, anmodninger om fortrolig behandling, håndtering af meddelelser, der behandles fortroligt, og tilbagetrækning af anmodninger om fortrolig behandling) |
|
— |
Kapitel 3.4 om Kommissionens gennemgang af meddelelser (korrekthed, fuldstændighed, anmodninger om supplerende oplysninger, undersøgelser) |
|
— |
Kapitel 3.5 om validering af meddelelser |
|
— |
Kapitel 3.8 om permanent sletning af en meddelelse i RAPEX-applikationen (situationer, hvor meddelelser kan slettes, anmodende medlemsstat, indholdet af anmodningen, beslutninger om sletning). |
Medlemsstaterne sender en »artikel 11-meddelelse« hurtigst muligt og under alle omstændigheder inden for de frister, der er angivet i tillæg 3 til retningslinjerne. Kapitel 3.10 om frister i retningslinjernes del II finder anvendelse.
4. Tilbagemeldinger
Medlemsstaterne opfordres til at sikre opfølgning på »artikel 11-meddelelser«, hvor det er sandsynligt, at produktidentifikation vil gøre det muligt at træffe præventive og restriktive foranstaltninger. Medlemsstaterne opfordres ligeledes til at orientere Kommissionen om konklusionerne på opfølgningsaktiviteter, der gennemføres i relation til »artikel 11-meddelelser«.
Nedennævnte kapitler i retningslinjernes del II er relevante for artikel 11-notifikationsproceduren:
|
— |
Kapitel 3.7 om opfølgningsaktiviteter (mål, opfølgningsmetoder) |
|
— |
Kapitel 4.2 om indholdet af tilbagemeldinger (meddelte oplysninger, fuldstændighed, ajourføring, ansvaret for tilbagemeldinger) |
|
— |
Kapitel 4.3 om fortrolighed |
|
— |
Kapitel 4.4 om Kommissionens gennemgang af tilbagemeldinger (korrekthed og fuldstændighed, anmodninger om supplerende oplysninger) |
|
— |
Kapitel 4.5 om validering af tilbagemeldinger |
|
— |
Kapitel 4.6 om permanent sletning af tilbagemeldinger i RAPEX-applikationen. |
5. Praktiske og tekniske rammer
»Artikel 11-meddelelser« og tilbagemeldinger herpå udarbejdes og sendes til Kommissionen af RAPEX-kontaktpunkterne under anvendelse af RAPEX-applikationen. Punkt 5.1-5.3 i retningslinjernes del II om forvaltningen af RAPEX-netværkene (etableret i EU-regi og på nationalt plan) samt om de praktiske og tekniske rammer (sprog, onlineapplikation og anvendelsen af systemet uden for normal arbejdstid) er relevante for artikel 11-notifikationsproceduren.
DEL IV
TILLÆG
1. Standardmeddelelsesformular


2. Tilbagemeldingsformular

3. Tidsfrister — medlemsstaterne
|
Notifikationsprocedure |
Opgave |
Frist |
||||||
|
Fællesskabssystemet for hurtig udveksling af oplysninger (RAPEX), jf. artikel 12 i direktivet om produktsikkerhed i almindelighed |
Meddelelser |
Indsendelse af »artikel 12-meddelelse, der nødvendiggør hasteforanstaltninger« |
3 dage efter:
|
|||||
|
Indsendelse af »artikel 12-meddelelse« |
10 dage efter:
|
|||||||
|
Bekræftelse af foranstaltninger, hvis meddelelsen er indsendt inden beslutningen om at træffe foranstaltninger |
45 dage efter indgivelse af meddelelsen |
|||||||
|
Ajourføring af en meddelelse |
5 dage efter modtagelse af oplysningerne om nye situationer eller forhold, der nødvendiggør ændringer i meddelelsen |
|||||||
|
Tilbagemeldinger |
Opfølgning på: |
»artikel 12-meddelelse, der nødvendiggør hasteforanstaltninger« |
20 dage efter modtagelse af meddelelsen |
|||||
|
»artikel 12-meddelelse« og »meddelelse sendt af Europa-Kommissionen« |
45 dage efter modtagelse af meddelelsen |
|||||||
|
Tilbagemelding på: |
»artikel 12-meddelelse, der nødvendiggør hasteforanstaltninger« |
3 dage efter:
|
||||||
|
»artikel 12-meddelelse« og »meddelelse sendt af Europa-Kommissionen« |
5 dage efter:
|
|||||||
|
Ajourføring af en tilbagemelding |
5 dage efter modtagelse af oplysninger om nye situationer eller forhold, der nødvendiggør ændringer i tilbagemeldingen |
|||||||
|
Notifikationsprocedure, jf. artikel 11 i direktivet om produktsikkerhed i almindelighed |
Meddelelser |
Indsendelse af »artikel 11-meddelelse« |
10 dage efter gennemførelsen af »obligatoriske foranstaltninger« |
|||||
|
Ajourføring af meddelelsen |
5 dage efter modtagelse af oplysninger om nye situationer eller forhold, der nødvendiggør ændringer i meddelelsen |
|||||||
4. Tidsfrister — Kommissionen
|
Notifikationsprocedure |
Opgave |
Frist |
|
|
Fællesskabssystemet for hurtig udveksling af oplysninger (RAPEX), jf. artikel 12 i direktivet om produktsikkerhed i almindelighed |
Meddelelser |
Validering af »artikel 12-meddelelse, der nødvendiggør hasteforanstaltninger« |
3 dage efter modtagelse af meddelelsen |
|
Validering af »artikel 12-meddelelse« |
5 dage efter modtagelse af meddelelsen |
||
|
Validering af »meddelelse til orientering« |
10 dage efter modtagelse af meddelelsen |
||
|
Tilbagemeldinger |
Validering af tilbagemelding på »artikel 12-meddelelse, der nødvendiggør hasteforanstaltninger« |
3 dage efter modtagelse af tilbagemeldingen |
|
|
Validering af tilbagemelding på »artikel 12-meddelelse« og »meddelelse sendt af Europa-Kommissionen« |
5 dage efter modtagelse af tilbagemeldingen |
||
|
Validering af tilbagemelding på »meddelelse til orientering« |
10 dage efter modtagelse af tilbagemeldingen |
||
|
Notifikationsprocedure, jf. artikel 11 i direktivet om produktsikkerhed i almindelighed |
Meddelelser |
Validering af »artikel 11-meddelelse« |
10 dage efter modtagelse af meddelelsen |
|
Tilbagemeldinger |
Validering af tilbagemelding på »artikel 11-meddelelse« |
10 dage efter modtagelse af tilbagemeldingen |
|
5. Retningslinjer for risikovurdering af forbrugerprodukter
INDHOLD
|
1. |
Indledning |
|
2. |
Risikovurdering — en kort gennemgang |
|
2.1. |
Risiko — en kombination af fare og sandsynlighed |
|
2.2. |
En risikovurdering i tre trin |
|
2.3. |
Gode råd |
|
3. |
Risikovurderingen trin for trin |
|
3.1. |
Produktet |
|
3.2. |
Faren ved et produkt |
|
3.3. |
Forbrugeren |
|
3.4. |
Skadesscenarie: trinnene på vejen til en personskade |
|
3.5. |
Skadens alvorlighed |
|
3.6. |
Sandsynligheden for skaden |
|
3.7. |
Bestemmelse af risikoen |
|
4. |
Fra risiko til foranstaltninger |
|
5. |
Risikovurderingsproceduren — i store træk |
|
6. |
Eksempler |
|
6.1. |
Klapstol |
|
6.2. |
Kontaktsikringer |
|
6.3. |
Følsomhedsanalyse |
Skema 1 — Forbrugere
Skema 2 — Farer, typiske skadesscenarier og typiske personskader
Skema 3 — Skadens alvorlighed
Skema 4 — Risikoniveau på grundlag af kombinationen af skadens alvorlighed og sandsynligheden for den
Anvendte begreber
1. Indledning
Man kan komme til skade i forbindelse med anvendelsen af forbrugerprodukter. For eksempel kan man brænde sig på et varmt strygejern, skære sig på en saks eller en kniv eller få hudskader af et rensemiddel. Denne type skader er ikke hyppigt forekommende, idet de fleste ved eller har adgang til instrukser i, hvordan forbrugerprodukter anvendes på forsvarlig vis. Risikoen for skader vil dog altid være til stede.
Risikoen kan vurderes på forskellige måder. Der er anvendt en række forskellige metoder til at måle risikoen ved forbrugerprodukter, såsom nomogrammetoden (22), matrixmetoden (23) og den metode, der hidtil er blevet anbefalet anvendt i EU's system for hurtig varsling (RAPEX) (24). Mens der altid har været enighed om de overordnede principper for risikovurdering, har man løbende arbejdet med at udvikle metoder til måling af risici. Dette har ført til indbyrdes afvigende resultater, som har affødt diskussioner og overvejelser om, hvad der ville være den optimale praksis.
Formålet med disse retningslinjer for risikovurdering er derfor at forbedre situationen og, inden for rammerne af direktivet om produktsikkerhed i almindelighed (25), fastlægge en gennemskuelig og praktisk anvendelig metode, som medlemsstaternes kompetente myndigheder med fordel kan anvende, når de vurderer risikoen ved nonfoodforbrugerprodukter. Disse retningslinjer er baseret på en risikovurderingsmetode, der er udviklet til andre formål (26) og tilpasset de særlige krav, der gælder for nonfoodforbrugerprodukter.
Der vil i sagens natur være behov for en vis portion uddannelse, inden disse retningslinjer kan bruges i praksis, men ekspertisen inden for risikovurdering vil i høj grad lette opgaven. Til understøttelse heraf vil de risikovurderingsansvarlige (i det følgende benævnt »risikoanalytikerne«) udveksle synspunkter, idet den ekspertise og erfaring, der er opbygget igennem årene, er uvurderlig.
Ved at opbygge en risikovurderingsmetode i små, overkommelige trin gør disse retningslinjer det lettere at fokusere på de relevante aspekter af et produkt, dets bruger(e) og dets anvendelse(r) samt helt fra starten at få afdækket spørgsmål, der måtte være uenighed om risikoanalytikerne imellem, så man undgår tidskrævende diskussioner. Retningslinjerne burde således udmønte sig i ensartede og holdbare risikovurderingsresultater baseret på håndfast dokumentation og forskning og derigennem almindelig enighed om, hvilken risiko de mange forbrugerprodukter kan være forbundet med.
En kort gennemgang af og et flow-chart over risikovurderingsproceduren i overensstemmelse med disse retningslinjer findes i afsnit 5 — Med »forbrugerprodukter« menes i disse retningslinjer udelukkende nonfoodforbrugerprodukter.
Det er ikke meningen, at disse retningslinjer skal træde i stedet for andre retningslinjer, som kan omhandle meget specifikke produkter, eller som specifikt er udstukket i lovgivningen, f.eks. inden for kemikalier, kosmetiske midler, lægemidler eller medicinsk udstyr. Det anbefales på det kraftigste at anvende disse specifikke retningslinjer, fordi de er skræddersyede, men det vil altid være op til den enkelte risikoanalytiker at afgøre, hvordan risikoen ved et produkt bedst vurderes.
Det er heller ikke tanken, at producenterne skal anvende disse retningslinjer »blot for at undgå alvorlige risici«, når de udtænker og fremstiller produkter. Forbrugerprodukter skal være sikre, og disse retningslinjer har til formål at gøre det nemmere for myndighederne at identificere alvorlige risici i tilfælde, hvor et produkt — til trods for at producenten har gjort sit bedste — ikke er sikkert.
2. Risikovurdering — en kort gennemgang
2.1. Risiko — en kombination af fare og sandsynlighed
Ved risiko forstås almindeligvis noget, der udgør en trussel mod menneskers helbred eller endog deres liv, eller som kan forårsage væsentlig materiel skade. Desuagtet løber mennesker risici i bevidsthed om de potentielle skader, fordi skaden ikke altid sker. Eksempler:
|
— |
At kravle op ad en stige er altid forbundet med muligheden for at falde ned og komme til skade. At »falde ned« er derfor »indbygget i stigen«; det er en integrerende del af det at bruge en stige og kan ikke udelukkes. At »falde ned« kaldes derfor den iboende fare ved en stige. Denne fare giver sig imidlertid ikke altid udslag i en konkret hændelse, idet mange mennesker bruger stiger uden at falde ned og komme til skade. Deraf kan man udlede, at der er en vis mulighed (eller sandsynlighed), men ikke nogen sikkerhed for, at den iboende fare giver sig udslag i en konkret hændelse. Mens farer altid er til stede, kan sandsynligheden for, at faren giver sig udslag i en konkret hændelse, begrænses til et minimum, f.eks. ved at den person, der kravler op ad stigen, udviser forsigtighed. |
|
— |
At anvende et rensemiddel, der indeholder natriumhydroxid, til at skabe gennemløb i tilstoppede afløb er altid forbundet med muligheden for meget alvorlige hudskader, hvis produktet kommer i kontakt med huden, eller endog permanent blindhed, hvis brugeren får dråber af produktet i øjet. Dette skyldes, at natriumhydroxid er et meget ætsende stof, og rensemidlet rummer derfor en iboende fare. Ved en korrekt anvendelse af rensemidlet giver faren sig imidlertid ikke altid udslag i en konkret hændelse. Korrekt anvendelse kan omfatte brug af gummihandsker og beskyttelsesbriller. Derved beskyttes hud og øjne, og sandsynligheden for skader begrænses væsentligt. |
Risiko er således kombinationen af henholdsvis alvorligheden af og sandsynligheden for de potentielle skader.
2.2. En risikovurdering i tre trin
Risikoen bestemmes i tre trin:
|
1. |
Der forudses et skadesscenarie, hvori et givent produkts iboende fare giver sig udslag i, at forbrugeren (jf. skema 1), kommer til skade. Det fastlægges, hvor alvorlige forbrugerens skader er. En målestok i forbindelse med måling af produktets iboende fare er alvorligheden af de skadelige virkninger, produktet kan have for en forbrugers sundhed. Risikoanalytikeren forudser derfor et »skadesscenarie«, som trin for trin beskriver, hvordan faren resulterer i den pågældende skade hos forbrugeren (jf. skema 2). Med skadesscenariet beskrives kort sagt den ulykke, forbrugeren har med det pågældende produkt, og alvorligheden af forbrugerens skader som følge af ulykken. Alvorligheden af en personskade kan variere afhængigt af faren ved produktet, måden, produktet anvendes på af forbrugeren, typen af forbruger, der anvender produktet, og meget andet (jf. afsnit 3). Jo alvorligere skaden er, desto alvorligere er faren, der resulterede i den, og vice versa. »Alvorligheden af skaden« er derfor et redskab til at måle faren. I disse retningslinjer opereres der med fire alvorlighedsgrader — fra skader, efter hvilke helbredstilstanden normalt er 100 % genoprettelig, til meget alvorlige skader, der fører til varig invaliditet på over godt 10 % eller endog dødsfald (jf. skema 3). |
|
2. |
Sandsynligheden for, at forbrugeren i praksis kommer til skade som følge af produktets iboende fare, bestemmes. Skadesscenariet beskriver, hvordan faren resulterer i skader hos forbrugeren, men der er kun en vis sandsynlighed for, at scenariet bliver til virkelighed. Sandsynligheden kan udtrykkes som en procentdel/brøkdel, f.eks. »> 50 %« eller »> 1/1 000« (jf. skema 4, venstre side). |
|
3. |
Risikoen beregnes ved at kombinere faren (udtrykt som skadens alvorlighed) og sandsynligheden (udtrykt som en procentdel/brøkdel). Denne kombination kan foretages ved at aflæse begge værdier i det relevante skema (jf. skema 4); i skemaet angives risikoniveauet som »alvorlig«, »høj«, »middel« og »lav« risiko. |
Når der kan forudses flere forskellige skadesscenarier, bestemmes risikoen for hvert af de pågældende scenarier, idet den højeste risiko angives som »den faktiske risiko« ved produktet. Den højeste risiko er normalt af største betydning, fordi kun foranstaltninger tilrettelagt efter den højeste risiko gør det muligt at sikre et højt beskyttelsesniveau.
På den anden side kan en identificeret risiko være lavere end den højeste risiko, men alligevel nødvendiggøre særlige risikoreducerende foranstaltninger. Det er i givet fald vigtigt også at træffe foranstaltninger mod denne risiko, således at alle risici begrænses effektivt.
Når ovenstående trin er fuldendt, er risikovurderingen i det store og hele tilendebragt.
I slutningen af afsnit 5 findes et flow-chart over opbygningen af en risikovurdering.
2.3. Gode råd
Indhent oplysninger
Som det fremgår af ovenstående eksempler, er det på hvert af de beskrevne trin i en risikovurdering nødvendigt at forudse, hvad der vil kunne ske, og hvor sandsynligt det er, at det faktisk sker. Dette skyldes, at produktet, der er genstand for vurderingen, normalt ikke vil have været årsag til en ulykke, og at risikoen derfor (endnu) ikke vil have givet sig udslag i en konkret hændelse. Tidligere erfaringer med lignende produkter vil være nyttige i den forbindelse, og det samme gælder diverse andre oplysninger om produktet, f.eks. om udformning, mekanisk stabilitet, kemisk sammensætning, virkemåde, brugsanvisning (herunder evt. risikohåndteringsvejledning), målgruppe (og forbrugertyper, som produktet ikke er bestemt for), afprøvningsrapporter, ulykkesstatistikker og EU's database over personskader (IDB) (27), samt oplysninger om klager fra forbrugerne, om forskellige forbrugeres adfærd i forbindelse med anvendelse af produktet og om tilfælde af tilbagekaldelse af produktet. Også produktkrav, der er fastsat i lovgivningen, i produktstandarder eller i tjeklister (som f.eks. i ISO 14121: Maskinsikkerhed — Risikovurdering), kan være nyttige kilder til information.
De produkter, der skal vurderes, kan imidlertid være meget specielle, og nævnte kilder vil derfor ikke nødvendigvis give de nødvendige oplysninger. De indsamlede oplysninger kan desuden være ufuldstændige, indbyrdes modstridende eller ikke 100 % pålidelige. Dette vil især kunne være tilfældet med ulykkesstatistikker, hvor kun produktkategorien er registreret. Det, at der kun er registreret få eller slet ingen ulykker, eller at der ikke har været tale om alvorlige ulykker, er ikke tilstrækkeligt grundlag for at formode, at risikoen er lav. Også produktspecifikke statistikker skal anvendes med betydelige forbehold, eftersom produktet — såvel dets udformning som dets sammensætning — kan have ændret sig med tiden. Oplysningerne skal altid vurderes med et kritisk øje.
Feedback fra andre eksperter på dette felt kan være særlig nyttig, idet de kan trække på deres erfaringer i praksis og bidrage med input, der ikke nødvendigvis springer i øjnene ved vurderingen af en produktrisiko. Kolleger på feltet kan desuden komme med gode råd vedrørende vurdering af risikoen for forskellige forbrugertyper, herunder sårbare forbrugere såsom børn (jf. tabel 1), idet sidstnævnte måske vil håndtere et produkt anderledes end andre forbrugere. De vil tillige kunne være behjælpelige med at vurdere risikoen for forskellige typer skader, som et produkt vil kunne være årsag til, samt hvorledes de pågældende skader opstår ved anvendelse af produktet. Derudover kan de bedømme, om et skadesscenarie er »helt teoretisk«, dvs. for usandsynligt, og hjælpe risikoanalytikeren til at koncentrere sig om mere realistiske antagelser.
Feedback fra erfarne kolleger kan derfor, selv om den ikke er obligatorisk, være nyttig på flere måder. En risikoanalytiker under en myndighed kan rådføre sig med kolleger under samme myndighed, under andre myndigheder, i virksomheder, i andre lande, i videnskabelige sammenslutninger og andre steder. Omvendt kan en risikoanalytiker i en virksomhed drage nytte af sine kontakter med myndigheder og andre, når et nyt eller forbedret produkt skal vurderes inden markedsføring.
Eksisterende risikovurderinger ajourføres naturligvis, hver gang der foreligger nye oplysninger.
Foretag en følsomhedsanalyse af din risikovurdering
Lykkes det ikke at indhente de nødvendige, meget specifikke oplysninger med diverse informationssøgninger og forespørgsler hos ekspertkolleger, kan en såkaldt følsomhedsanalyse være nyttig. I denne analyse antages for hver parameter i risikovurderingen henholdsvis en højere og en lavere værdi end den først valgte, hvorefter disse underkastes hele risikovurderingsproceduren. Af de risikoniveauer, man således når frem til, vil det fremgå, hvor følsomt risikoniveauet reagerer på input af lavere og højere værdier. På denne måde kan det vurderes, inden for hvilket spektrum risikoen ved produktet reelt vil befinde sig.
Kan den mest sandsynlige værdi beregnes for hver enkelt parameter, underkastes disse mest sandsynlige værdier proceduren, og det risikoniveau, man således når frem til, vil være den mest sandsynlige risiko.
I afsnit 6 er vist et eksempel på en følsomhedsanalyse.
Lad andre efterprøve din risikovurdering
Feedback fra kolleger er også nyttig i forbindelse med færdiggørelse af risikovurderingen. Kollegerne vil kunne give deres besyv med vedrørende de antagelser og beregninger, der er foretaget på de tre ovenfor beskrevne trin. De vil bidrage med deres erfaring og derved være med til at skabe en mere holdbar, velfunderet, gennemskuelig og i sidste ende mere acceptabel risikovurdering. Det anbefales derfor, at der så vidt muligt indhentes rådgivning fra kolleger på fagområdet, eventuelt inden for rammerne af en gruppedrøftelse, inden der lægges sidste hånd på en risikovurdering. Det bør med sådanne grupper, som f.eks. kan bestå af 3-5 medlemmer, tilstræbes at samle en palet af ekspertise af relevans for det produkt, der vurderes, dvs. ingeniører, kemikere, (mikro-)biologer, statistikere, produktsikkerhedschefer og andre. Gruppedrøftelser vil være til særlig stor gavn, når der er tale om et nyt produkt på markedet, som aldrig tidligere er blevet vurderet.
Risikovurderinger skal være velfunderede og realistiske. Da sådanne vurderinger nødvendiggør en række antagelser, vil forskellige risikoanalytikere imidlertid kunne nå frem til forskellige konklusioner, afhængigt af hvilke oplysninger og anden dokumentation de har kunnet finde, eller på grund af deres forskellige erfaringsgrundlag. Det er af samme grund nødvendigt for risikoanalytikere at tale sammen for at nå til fuld, eller i det mindste principiel enighed. Den trinopdelte risikovurdering, der er beskrevet i disse retningslinjer, burde dog sikre, at sådanne diskussioner bliver mere frugtbare. Hvert enkelt trin i en risikovurdering skal beskrives præcist og detaljeret. Derved bliver det muligt hurtigt at identificere eventuelle stridspunkter, og det bliver lettere at nå til enighed. Dette vil gøre risikovurderinger mere acceptable.
Dokumenter din risikovurdering
Det er vigtigt, at du dokumenterer din risikovurdering og beskriver produktet og alle de parametre, du vælger, mens du udvikler den, såsom resultaterne af afprøvninger, type(r) forbrugere, som du vælger til dit eller dine skadesscenarier, samt sandsynlighederne med de tilgrundliggende oplysninger og antagelser. Dette vil gøre det muligt for dig entydigt at dokumentere, hvordan du har beregnet risikoniveauet, ligesom det vil være nemmere for dig at ajourføre din vurdering, samtidig med at du får registreret alle ændringer.
Mange farer, mange skader — men kun én risiko
Hvor der er identificeret flere farer, flere skadesscenarier, forskellige alvorlighedsgrader for de pågældende skader eller forskellige sandsynligheder, underkastes hvert af disse elementer hele risikovurderingsproceduren med henblik på at få bestemt risikoen for hvert enkelt. Et produkt kan som følge heraf have flere forskellige risikoniveauer. Den samlede risiko ved produktet er da det højeste fastlagte risikoniveau, fordi det normalt er foranstaltninger tilrettelagt efter den højeste risiko, der mest effektivt begrænser risikoen. Kun i visse særlige tilfælde vil en risiko, der er lavere end den højeste risiko, kunne betragtes som særlig betydningsfuld, fordi den kan nødvendiggøre særlige risikohåndteringsforanstaltninger.
Som et eksempel på flere risici kan en hammer have et skrøbeligt hoved og et svagt skaft, som begge kan gå i stykker, når hammeren bruges, og resultere i, at forbrugeren kommer til skade. Hvis de relevante scenarier er forbundet med forskellige risikoniveauer, angives den højeste risiko som »den faktiske risiko« ved hammeren.
Man kan argumentere for følgende:
|
— |
Den tilsyneladende mest alvorlige fare bør tillægges den største betydning, fordi denne ville føre til de alvorligste skader. I ovenstående eksempel med hammeren kunne denne fare bestå i, at hammerhovedet gik i stykker, idet brugeren kunne få stykker af det ødelagte hoved i øjet og potentielt miste synet. Derimod ville hammerens skaft, hvis det brækkede, aldrig blive splintret i små stykker, der kunne gøre lige så stor skade på øjnene. Dette ville imidlertid være en vurdering af faren, ikke en risikovurdering. Med en risikovurdering ses der også på sandsynligheden for, at en personskade faktisk vil indtræffe. Den »mest alvorlige fare« kan således resultere i en skade, der er langt mindre sandsynlig end en mindre alvorlig fare, og derfor være forbundet med en lavere risiko. Omvendt kan et scenarie, der indebærer en mindre alvorlig personskade, være langt mere sandsynligt end et scenarie, der indebærer dødsfald, således at den mindre alvorlige personskade kan udgøre en højere risiko. |
|
— |
Den højeste sandsynlighed for, at et skadesscenarie bliver til virkelighed, bør være afgørende ved bestemmelsen af »den faktiske risiko« ved produktet. I ovenstående eksempel med hammeren indebærer det mest sandsynlige skadesscenarie, såfremt hammerskaftet er meget svagt, at skaftet brækker, og dette scenarie bør derfor være afgørende. |
I så fald ville man imidlertid ikke tage hensyn til alvorligheden af de øjenskader, der kunne blive resultatet af, at hammerens hoved gik i stykker. At se på sandsynligheden alene ville derfor ikke give et helhedsbillede.
Sammenfattende kan det konkluderes, at risiko er en afbalanceret kombination af faren og sandsynligheden for den personskade, faren kan resultere i. Risiko er hverken udtryk for faren eller for sandsynligheden, men begge disse elementer på samme tid. Ved at lade den højeste risiko være »den faktiske risiko« ved produktet opnår man den mest effektive produktsikkerhed (undtagen når der er tale om visse særlige risici, som nødvendiggør særlige risikohåndteringsforanstaltninger, jf. ovenfor).
Kan risici akkumuleres?
For snart sagt ethvert produkt kan der fastlægges flere skadesscenarier, der indebærer flere forskellige risici. For eksempel kan en vinkelsliber være forbundet med en risiko for elektrisk stød, fordi elektriske ledninger kan være for udsatte, og en risiko for brand, fordi der ved normal brug kan gå ild i værktøjet på grund af overophedning. Hvis begge disse risici betragtes som »høje«, skal de da lægges sammen, så den samlede risiko ved vinkelsliberen er »alvorlig«?
I de tilfælde, hvor et produkt er forbundet med flere risici, vil en af disse risici i sagens natur frembyde en større sandsynlighed for en konkret hændelse med deraf følgende personskader end de øvrige risici. Den samlede mulighed for en personskade er derfor større. Dette betyder imidlertid ikke nødvendigvis, at den samlede risiko også er højere:
|
— |
Den samlede sandsynlighed beregnes ikke ved blot at lægge de relevante sandsynligheder sammen. Det er nødvendigt med mere komplekse beregninger, og resultatet af disse er altid en sandsynlighed, der er lavere end summen af alle de relevante sandsynligheder. |
|
— |
Der er en forskel på en faktor 10 fra ét sandsynlighedsniveau til det næste (jf. skema 4). Dette betyder, at mange forskellige scenarier skulle gøre sig gældende på samme niveau, hvis det skulle resultere i en højere samlet sandsynlighed (og eventuelt risiko). |
|
— |
Sandsynlighedsværdier er beregninger, der ikke nødvendigvis er 100 % nøjagtige, da de ofte bevidst foretages, så de hælder til den »sikre« side, for at garantere et højt beskyttelsesniveau. Det er derfor mere hensigtsmæssigt at se på en mere nøjagtig beregning af sandsynligheden for et scenarie, der indebærer den højeste risiko, end det er at lægge løseligt anslåede sandsynligheder for alskens scenarier sammen. |
|
— |
Med lidt god vilje kunne man udvikle hundredvis af skadesscenarier. Hvis man derfor blot lagde de relevante risici sammen, ville den samlede risiko afhænge af antallet af udviklede skadesscenarier og ville derfor kunne stige »uendeligt«. Dette ville være formålsløst. |
Risici akkumuleres således ikke bare. Hvis mere end én relevant risiko gør sig gældende, vil det dog kunne være nødvendigt hurtigere at træffe foranstaltninger for at styre risiciene eller at træffe mere vidtgående foranstaltninger. For eksempel ville det med to risici kunne være nødvendigt øjeblikkeligt at fjerne et produkt fra markedet og kalde det tilbage fra forbrugerne, mens det med en enkelt risiko måske vil være tilstrækkeligt at stoppe salget af det pågældende produkt.
Risikohåndteringen afhænger af mange faktorer og ikke kun antallet af risici, som et produkt kan være forbundet med på en og samme tid. Nedenfor ser vi derfor nærmere på sammenhængen mellem risiko og risikohåndtering (afsnit 4).
Overholdelse af grænseværdier i henhold til lovgivning og standarder
Som led i markedsovervågningen testes forbrugerprodukter ofte mod grænseværdier eller krav, der er fastsat i lovgivningen og i produktsikkerhedsstandarder. Et produkt, der overholder den/det eller de relevante grænseværdier eller krav (28), antages at være sikkert for så vidt angår de sikkerhedskarakteristika, den eller de pågældende værdier eller krav dikterer. Denne antagelse er realistisk, fordi den/det eller de relevante grænseværdier eller krav fastsættes under hensyntagen til de risici, produktet er forbundet med ved den påtænkte og med rimelighed forudsigelige anvendelse. Producenterne har således behov for, at deres produkter overholder de pågældende værdier eller krav, fordi de så kun behøver at se på de risici ved deres produkter, der ikke er omfattet af den/det eller de pågældende grænseværdier eller krav.
Et eksempel på en grænseværdi i
|
— |
lovgivningen er grænsen på 5 mg benzen pr. kg i legetøj, som ikke må overskrides, jf. punkt 5 i bilag XVII til REACH-forordningen (29), som ændret ved Kommissionens forordning (EF) nr. 552/2009 (30) |
|
— |
en standard er cylinderen til smådele: små dele af et legetøj til børn under 36 måneder må ikke rummes fuldstændig i cylinderen som beskrevet i legetøjsstandarden (31). Hvis de gør, er de forbundet med en risiko. |
Et produkt antages ikke at være sikkert, hvis det ikke overholder de fastsatte grænseværdier. Dette indebærer, når der er tale om grænseværdier fastsat i
|
— |
lovgivningen, f.eks. om kosmetiske midler eller om begrænsning af markedsføring og anvendelse, at produktet ikke må stilles til rådighed på markedet |
|
— |
standarder, at producenten alligevel, med en selvstændig risikovurdering af det pågældende produkt, kan forsøge at dokumentere, at produktet er lige så sikkert, som det ville være, hvis det faktisk overholdt standardens grænseværdi. Dette vil dog kunne være forbundet med mere arbejde — og vil i visse tilfælde kunne være umuligt, f.eks. i tilfældet med ovennævnte cylinder til smådele — end simpelthen at producere produktet i overensstemmelse med standardens grænseværdi. |
Manglende overholdelse af grænseværdier indebærer ikke nødvendigvis, at det pågældende produkt er forbundet med en »alvorlig risiko« (som er det højeste risikoniveau, der opereres med i disse retningslinjer). For at sikre hensigtsmæssige risikoreducerende foranstaltninger vil en risikovurdering derfor være påkrævet for de dele af et produkt, der ikke overholder eller ikke er omfattet af lovgivning eller en standard.
Derudover er en risikovurdering påkrævet for visse produkter, såsom kosmetiske midler, selv når de overholder de i lovgivningen fastsatte grænseværdier. Med denne risikovurdering skal sikkerheden dokumenteres for hele produktet (32).
Sammenfattende kan det konkluderes, at overholdelse af grænseværdier fastsat ved lovgivning eller i standarder giver formodning om sikkerhed, men at dette ikke nødvendigvis er tilstrækkeligt.
Specifikke retningslinjer for risikovurdering i særlige tilfælde
Hvad angår kemikalier foreligger der specifikke instrukser i, hvordan en risikovurdering gennemføres (33), og de behandles af samme grund ikke nærmere i disse retningslinjer. Der gælder dog de samme principper som for »almindelige« forbrugerprodukter:
|
— |
identificering og vurdering af farer — dette svarer til at fastlægge skadens alvorlighed, som beskrevet ovenfor |
|
— |
vurdering af eksponeringen — på dette trin udtrykkes eksponeringen som den sandsynlige dosis af kemikaliet, forbrugeren kan optage ad oral vej, ved indånding eller gennem huden (på hver enkelt måde eller tilsammen) i forbindelse med anvendelse af produktet som forudset i skadesscenariet. — Dette svarer til at bestemme sandsynligheden for, at skaden faktisk vil indtræffe |
|
— |
karakterisering af risikoen — dette trin består kort beskrevet i at sammenholde den dosis af kemikaliet, forbrugeren kan formodes at ville optage (= eksponering), med det afledte nuleffektniveau (Derived No-Effect Level) (DNEL) for det pågældende kemikalie. Såfremt eksponeringen ligger tilstrækkelig langt under DNEL, eller med andre ord hvis risikokarakteriseringskvotienten (RCR) ligger klart under 1, anses risikoen for at være begrænset tilstrækkeligt. — Dette svarer til at bestemme risikoniveauet. Hvis dette er tilstrækkelig lavt, vil det ikke nødvendigvis være nødvendigt med risikohåndteringsforanstaltninger. |
Eftersom der kan være flere farer ved et kemikalie, bestemmes risikoen normalt for den »primære virkning på sundheden«, som er den sundhedsmæssige virkning (eller »endpoint« såsom akut toksicitet, irritation, sensibilisering, carcinogenicitet, mutagenicitet eller reproduktionstoksicitet), der anses for alvorligst.
Også for kosmetiske midler foreligger der særlige retningslinjer (34), ligesom der kan være udarbejdet specifikke vejledninger for andre produkter eller anvendelser.
Det anbefales på det kraftigste at anvende sådanne specifikke vejledninger, fordi disse er skræddersyet til de pågældende produkter. I tilfælde, hvor de ifølge specifikke vejledninger påkrævede oplysninger ikke foreligger eller ikke kan beregnes, kan nærværende retningslinjer dog anvendes til en foreløbig risikovurdering. Denne risikovurdering vil skulle foretages med behørig omhu og opmærksomhed for at undgå fejlfortolkninger.
3. Risikovurderingen trin for trin
I denne del beskrives det detaljeret, hvilke elementer der skal tages i betragtning, og hvilke spørgsmål der skal stilles i forbindelse med en risikovurdering.
3.1. Produktet
Produktet identificeres entydigt. Identifikationen omfatter produktbetegnelse, mærke, modelnavn, typenummer, et eventuelt produktionspartinummer, enhver attest, der ledsager produktet, en børnesikret lukning, hvis en sådan findes, navn på den ansvarlige for markedsføringen af produktet samt oprindelsesland. Et billede af produktet, emballagen og mærkepladen (hvor det er relevant) og afprøvningsrapport(er), der beskriver faren/farerne ved produktet, kan ligeledes betragtes som en del af produktbeskrivelsen.
I visse tilfælde kan faren være begrænset til en bestemt del af produktet, som kan være adskilt fra dette og også kan stilles til rådighed for forbrugerne separat. I disse tilfælde er det tilstrækkeligt at vurdere den pågældende del af produktet. Et eksempel herpå er genopladelige batterier til bærbare computere, som kan blive overophedet.
Beskrivelsen af produktet omfatter al mærkning, der kan være relevant for risikovurderingen, især advarsler. Brugsanvisninger kan ligeledes indeholde relevante oplysninger om risikoen ved produktet og om, hvordan denne begrænses mest muligt, hvilket f.eks. kan være ved at bruge personlige værnemidler eller ved at sørge for, at børn ikke anvender produktet. Et eksempel herpå er en kædesav.
Nogle produkter, f.eks. saml selv-møbler, vil desuden skulle samles af forbrugeren selv inden brug. Er monteringsvejledningen tilstrækkeligt letforståelig, så det anvendelsesklare produkt opfylder alle relevante sikkerhedskrav? Eller vil forbrugerne kunne begå fejl, når de samler produktet, som kunne skabe uforudsete risici?
En risikovurdering bør altid omfatte hele produktets levetid. Dette er særlig vigtigt ved vurderingen af risiciene ved et nyudviklet produkt. Vil farens art og omfang ændre sig med produktets alder og brug? Vil nye farer opstå, efterhånden som produktet bliver ældre, eller måske på grund af med rimelighed forudsigelig uhensigtsmæssig anvendelse? Hvad er tidshorisonten for produktfejl? Hvad er produktets levetid/holdbarhed? Hvor længe anvendes produktet i praksis af forbrugeren, inden det bliver til affald?
Yderligere overvejelser vil kunne være nødvendige, såfremt et produkt bliver ubrugeligt efter et bestemt tidsrum, selv hvis det aldrig anvendes. Eksempler herpå er elektriske varmetæpper og varmepuder. De elektriske ledninger i disse produkter er normalt tynde og bliver skrøbelige efter ti år, selv hvis produktet aldrig anvendes. Varmetrådene kan komme i kontakt med hinanden og forårsage en kortslutning, så der går ild i sengetøjet.
Endelig bør enhver risikovurdering også omfatte produktets emballage.
3.2. Faren ved et produkt
Fare er et produkts iboende potentiale til at forårsage skade på den forbruger, der anvender produktet. Faren kan antage flere forskellige former:
|
— |
mekanisk fare, f.eks. pga. skarpe kanter, som man kan skære sig på, eller meget små åbninger, som man kan få fingrene i klemme i |
|
— |
kvælningsfare (intern luftvejsblokering), f.eks. pga. smådele, der kan rive sig løs fra et legetøj og blive slugt af et barn, som bliver kvalt |
|
— |
kvælningsfare (ekstern luftvejsblokering), f.eks. pga. snorene i lukningen på en anorakhætte, som kan være årsag til strangulering |
|
— |
elektrisk fare, f.eks. pga. strømførende elektriske dele, der kan give elektriske stød |
|
— |
ophednings- eller brandfare, f.eks. en varmeblæser, som der går ild i på grund af overophedning, hvorved forbrugeren pådrager sig forbrændinger |
|
— |
termisk fare, f.eks. den varme yderside af en ovn, som kan give forbrændinger |
|
— |
kemisk fare, f.eks. et giftigt stof, der kan give en forbruger øjeblikkelig forgiftning ved indtagelse, eller et carcinogent stof, der kan forårsage kræft på lang sigt. Nogle kemikalier er kun farlige for forbrugeren, hvis denne udsættes for dem gentagne gange |
|
— |
mikrobiologisk fare, f.eks. bakteriologisk kontaminering af kosmetiske midler, som kan være årsag til hudinfektioner |
|
— |
støjfare, f.eks. legetøjsmobiltelefoner med alt for høje ringetoner, som kan give børn høreskader |
|
— |
andre farer, såsom eksplosions- eller implosionsfare og fare på grund af sonisk og ultrasonisk tryk, væsketryk eller laserstråler. |
Farer er i disse retningslinjer inddelt i grupper på grundlag af et produkts størrelse, form og overflade, potentiel, kinetisk eller elektrisk energi, ekstreme temperaturer og andre faktorer, jf. skema 2. Skemaet er kun ment som vejledning, og den enkelte risikoanalytiker bør tilpasse scenariet til det produkt, der vurderes. Det er klart, at ikke alle typer farer er relevante for alle produkter.
Skema 2 burde dog gøre det lettere for risikoanalytikere at indkredse og identificere alle potentielle farer ved forbrugerprodukter, der gøres til genstand for en vurdering. Hvis et produkt frembyder flere farer, underkastes hver enkelt af disse en særskilt risikovurdering, idet den højeste risiko angives som »den faktiske risiko« ved produktet. Risici, der nødvendiggør særlige risikohåndteringsforanstaltninger, indberettes naturligvis også, så det bliver muligt at begrænse samtlige risici.
Bemærk, at en enkelt fare kan resultere i flere forskellige skader i et og samme scenarie. For eksempel vil funktionsfejl på en motorcykels bremser kunne være årsag til en ulykke og resultere i skader på motorcyklistens hoved, hænder og ben og tillige forbrændinger, hvis benzinen antændes i forbindelse med ulykken. I dette tilfælde ville alle skaderne tilhøre samme skadesscenarie, og det ville være nødvendigt at foretage en samlet vurdering af alvorligheden af alle de pågældende skader. Nævnte skader er selvfølgelig samlet set meget alvorlige. — Der tilføjes dog ikke flere forskellige skader i forskellige scenarier.
I forbindelse med de daglige markedsovervågningsaktiviteter vil det kunne være tilstrækkeligt at vurdere risikoen for en enkelt fare. Hvis den risiko, der knytter sig til den pågældende fare, er tilstrækkeligt grundlag for risikohåndteringsforanstaltninger, kan disse træffes uden videre. Risikoanalytikeren bør dog forvisse sig om, at den identificerede risiko er den højeste eller en af de højeste risici, så det sikres, at risikohåndteringsforanstaltningerne bliver tilstrækkeligt effektive. Dette er altid tilfældet, når risikoen er alvorlig, eftersom dette er det højeste risikoniveau, der opereres med i disse retningslinjer. I tilfælde, hvor der er tale om en mindre end alvorlig risiko, vil det til gengæld kunne være nødvendigt med yderligere risikovurderinger samt eventuelt særlige risikohåndteringsforanstaltninger på et senere stadium. Sammenfattende kan det konkluderes, at antallet af nødvendige risikovurderinger vil blive begrænset til et minimum, efterhånden som der opbygges risikovurderingserfaring som led i markedsovervågningsaktiviteterne.
Identificering af farer ved hjælp af afprøvninger og standarder
Farer identificeres og måles ofte ved hjælp af afprøvninger. Sådanne afprøvninger samt beskrivelser af, hvordan de gennemføres, kan være indeholdt i europæiske eller internationale standarder. Et produkt, der overholder en »harmoniseret« europæisk standard (»EN …«), som der er offentliggjort henvisninger til i Den Europæiske Unions Tidende, antages at være sikkert (dog kun for så vidt angår de sikkerhedskarakteristika, der er omfattet af den eller de pågældende værdier eller standarder). Det kan i disse tilfælde antages, at produktet kun er forbundet med en minimal risiko, således at der testes et højt beskyttelsesniveau med hensyn til den pågældende fare.
Der kan dog være tilfælde, hvor der ikke foreligger formodning om sikkerhed, og i disse tilfælde vil der skulle foretages en særligt veldokumenteret risikovurdering og anmodes om ændring af den pågældende harmoniserede standard.
Omvendt vil det, såfremt et produkt ikke består testen, normalt kunne formodes, at der eksisterer en risiko, medmindre producenten kan dokumentere, at produktet er sikkert.
Produkter kan godt være forbundet med en risiko, selv om de ikke er årsag til personskader
Produkter kan være forbundet med en risiko, selv om de ikke er farlige, hvis de ikke er egnede til den påtænkte anvendelse. Eksempler på dette ses inden for personlige værnemidler og redningsudstyr, såsom de refleksjakker, bilister tager på efter et trafikuheld. Disse jakker har til formål at påkalde sig opmærksomheden hos modkørende bilister og andre medtrafikanter og advare dem om, at der er sket et uheld, især om natten. Hvis refleksstriberne er for små eller ikke reflekterer lyset tilstrækkelig effektivt, ses jakkerne imidlertid måske ikke og beskytter derfor ikke brugerne efter hensigten. Sådanne jakker udgør derfor en risiko, selv om de ikke er farlige i sig selv. — Et andet eksempel er en solcreme, der er mærket »høj beskyttelse« (solbeskyttelsesfaktor 30), men kun giver »lav beskyttelse« (faktor 6). Resultatet kan være alvorlig solskoldning.
3.3. Forbrugeren
Forbrugerens evner og adfærd kan have endog meget stor indflydelse på risikoniveauet i forbindelse med anvendelsen af et produkt. Det er derfor afgørende nødvendigt at danne sig et klart billede af, hvilken type forbruger skadesscenariet er relevant for.
Det kan være nødvendigt at udtænke skadesscenarier med forskellige typer forbrugere for at identificere den højeste risiko og dermed »den faktiske risiko« ved produktet. Det er f.eks. ikke tilstrækkeligt kun at se på de mest sårbare forbrugere, idet sandsynligheden for skadelige virkninger på disse forbrugere i det pågældende scenarie kan være så lille, at risikoen er lavere end i et skadesscenarie med en ikke-sårbar forbruger.
Med i betragtningerne tages også personer, der ikke selv anvender produktet, men måske opholder sig i nærheden af brugeren. For eksempel kan en kædesav være årsag til, at flyvende træsplinter rammer en anden tilstedeværende person i øjet. Tilstedeværende personer kan således være udsat for en alvorlig trussel, selv om brugeren selv håndterer risikoen ved kædesaven effektivt ved at bruge sikkerhedsudstyr og ved at iagttage alle andre risikohåndteringsforanstaltninger, producenten måtte have angivet. Af samme grund bør der, f.eks. i brugsanvisningen til kædesaven, advares om risiciene for tilstedeværende personer, ligesom det bør angives, hvordan disse risici begrænses mest muligt.
Ved udviklingen af et skadesscenarie bør der derfor tages hensyn til følgende aspekter vedrørende forbrugertyper og den måde, de anvender produktet på. Listen er ikke udtømmende, men den skulle gerne være et incitament for risikoanalytikere til at beskrive deres skadesscenarier tilstrækkelig detaljeret. Det bedes bemærket, at der med »forbruger« også menes personer, der ikke selv anvender produktet, men kan blive berørt af det, fordi de opholder sig i nærheden af dette:
|
— |
Påtænkt/ikke-påtænkt bruger: Den påtænkte bruger af et produkt kan let anvende dette, fordi brugeren følger produktanvisningerne eller allerede kender den pågældende produkttype, herunder også åbenlyse og ikke-åbenlyse farer ved produktet. Faren ved produktet giver sig derfor måske ikke udslag i en konkret hændelse, og risikoen ved produktet er dermed muligvis mindre væsentlig. Den ikke-påtænkte bruger kender måske ikke produktet og vil ikke nødvendigvis være opmærksom på de(n) relevante fare(r). Brugeren risikerer derfor at komme til skade, og forbrugerrisikoen er således højere. Risikoen kan følgelig være forskellig for henholdsvis en påtænkt og en ikke-påtænkt bruger, afhængigt af produktet og måden at anvende det på. |
|
— |
Sårbare forbrugere: Der kan sondres mellem flere kategorier af sårbare og meget sårbare forbrugere: børn (0-36 måneder, > 36 måneder til < 8 år og 8-14 år) og andre, såsom ældre mennesker (jf. skema 1). Fælles for disse er, at de har vanskeligere ved at genkende en fare, f.eks. børn, som — når de kommer i berøring med en varm overflade — først efter omkring 8 sekunder bemærker varmen (og da allerede har brændt sig), mens voksne mærker varme med det samme. Sårbare forbrugere kan også have vanskeligt ved at orientere sig om advarsler i produkters mærkning, eller de kan have særlige problemer med at anvende et produkt, de aldrig har anvendt før. De kan også opføre sig på en måde, der gør dem mere udsatte, f.eks. små børn, der kravler og putter ting i munden. Børn kan også blive tiltrukket af produkter, der appellerer til deres sanser, hvilket betyder, at sådanne produkter bliver til højrisikoprodukter i hænderne på børn. På den anden side skulle forældres eller andre voksnes opsyn normalt forhindre, at børnene uden videre udsætter sig for fare. Dertil kommer, at forbrugere, der ikke normalt er sårbare, kan blive sårbare i særlige situationer, f.eks. når anvisninger eller advarsler på et produkt er skrevet på et sprog, som forbrugeren ikke forstår. Endelig gælder det specifikt for kemikalier, at børn kan være mere påvirkelige af giftige kemikalier end den voksne gennemsnitsforbruger. Børn må derfor ikke behandles, som om de er »små voksne«. Sammenfattende kan det konkluderes, at et produkt, der normalt er sikkert for en gennemsnitsvoksen, ikke nødvendigvis er sikkert for sårbare forbrugere. Dette skal tages i betragtning, når alvorligheden af og sandsynligheden for en personskade (se nedenfor) og dermed risikoen bestemmes. |
|
— |
Påtænkt og med rimelighed forudsigelig anvendelse: Forbrugere kan anvende et produkt til andre formål end det, produktet er beregnet til, selv om produktanvisningerne — herunder eventuelle advarsler — er let forståelige. Eftersom advarsler således ikke nødvendigvis er 100 % effektive, må der i en risikovurdering også tages højde for andre anvendelser end de påtænkte. Dette aspekt er af særlig stor betydning for producenten af et produkt, fordi denne skal sikre, at produktet er sikkert under alle med rimelighed forudsigelige anvendelsesbetingelser. Det vil i nogle tilfælde være nødvendigt at basere »med rimelighed forudsigelig anvendelse« på de erfaringer, der er gjort, fordi de officielle ulykkesstatistikker og øvrige informationskilder ikke indeholder de relevante oplysninger. Det vil da kunne være svært at sondre mellem »med rimelighed forudsigelige« og »helt teoretiske« scenarier. Disse retningslinjer er imidlertid også relevante for »helt teoretiske« scenarier, selv hvor disse er forbundet med meget alvorlige personskader, idet sandsynligheden for sådanne scenarier altid vil være meget lille. Dette kan med en vis sandsynlighed forhindre, at sådanne scenarier tillægges for stor betydning ved bestemmelsen af den samlede risiko ved produktet. |
|
— |
Anvendelsens hyppighed og varighed: Nogle forbrugere vil anvende et produkt oftere end andre, ligesom nogle vil anvende det i længere tid end andre. Dette afhænger af, hvor meget produktet appellerer til den enkelte forbruger, samt af, hvor letanvendeligt det er. Daglig eller langvarig anvendelse vil kunne gøre en forbruger fuldstændig fortrolig med et produkt og dets karakteristika, herunder farerne ved det samt produktanvisninger og advarselsmærkning, hvilket vil gøre risikoen mindre væsentlig. Daglig eller langvarig anvendelse vil på den anden side kunne gøre forbrugeren for vant til produktet og føre til brugertræthed og deraf følgende letsindig tilsidesættelse af anvisninger og advarsler, hvilket vil øge risikoen. Endelig vil daglig eller langvarig brug også kunne fremskynde produktets ældning, og dele, der ikke kan holde til sådan hyppig anvendelse, vil kunne svigte og skabe en fare — og muligvis en personskade — hvilket også øger risikoen. |
|
— |
Genkendelse af farer, sikker adfærd og beskyttelsesudstyr: Nogle produkter er kendt for de farer, de frembyder, såsom sakse, knive, gør det selv-boremaskiner, kædesave, rulleskøjter, cykler, motorcykler og biler. I alle de nævnte tilfælde er faren ved produktet velkendt eller åbenbar, eller den er beskrevet i produktanvisningerne, som vil omfatte relevante risikohåndteringsforanstaltninger. Forbrugeren kan på det grundlag udvise forsigtighed eller bruge personlige værnemidler såsom handsker, hjelm eller sikkerhedssele og derved anvende produktet på en måde, der begrænser risikoen til et minimum. I andre tilfælde er faren ved produktet måske ikke så let at få øje på, hvilket f.eks. gælder kortslutning i et strygejern, ligesom advarsler i produktets mærkning kan blive overset eller misforstået, og forbrugerne vil kun sjældent kunne træffe forebyggende foranstaltninger. |
|
— |
Forbrugerens adfærd i forbindelse med en hændelse: Hvor en fare påvirker forbrugeren som en konkret hændelse, kan resultatet være personskader. Det er derfor vigtigt for en risikovurdering at overveje, hvordan forbrugeren vil reagere. Vil han stille og roligt lægge produktet fra sig og træffe forebyggende foranstaltninger og f.eks. forsøge at bekæmpe en brand, som produktet har været årsag til, eller vil han kaste det fra sig i panik? Sårbare forbrugere, især børn, vil som bekendt ikke nødvendigvis opføre sig på samme måde som andre, ikke sårbare forbrugere. |
|
— |
Forbrugerens kulturelle baggrund og den måde, produktet anvendes på i forbrugerens hjemland, kan have betydning for risikoen ved et produkt. Producenterne er i høj grad nødt til at tage hensyn til sådanne kulturelle forskelle, når de lancerer et nyt produkt på et marked. Producenternes erfaringer på dette område kan derfor være en værdifuld informationskilde for de myndigheder, der gennemfører en risikovurdering. |
3.4. Skadesscenarie: trinnene på vejen til en personskade
De fleste skadesscenarier omfatter følgende tre hovedtrin:
|
1. |
Produktet har en »defekt« eller kan i sin forudsigelige levetid være årsag til en »farlig situation«. |
|
2. |
»Defekten« eller »den farlige situation« resulterer i en ulykke. |
|
3. |
Ulykken resulterer i en personskade. |
Disse tre trin kan igen inddeles i flere trin for at anskueliggøre, hvordan faren ved produktet kan resultere i personskader osv. Disse »trin på vejen til en personskade« skal imidlertid beskrives klart og kortfattet, uden at detaljeringsgraden og antallet af trin overdrives. I takt med at erfaringsgrundlaget udbygges, vil det blive stadig lettere at indkredse betingelserne, under hvilke en given personskade vil indtræffe, samt »den korteste vej til en personskade« (eller »den kritiske vej til en personskade«).
Det nemmeste er nok at tage udgangspunkt i et scenarie, hvori forbrugeren, som produktet er tiltænkt, anvender produktet i overensstemmelse med produktanvisningerne eller, såfremt sådanne ikke foreligger, i overensstemmelse med normal håndtering og brug. Får man med denne vurdering det højeste risikoniveau, vil der normalt ikke være behov for at gennemføre flere vurderinger, og der vil kunne træffes passende risikoreducerende foranstaltninger. Tilsvarende vil det, hvor en hændelse indberettes som en specifik forbrugerklage, kunne være tilstrækkeligt med et enkelt skadesscenarie som grundlag for at drage konklusioner vedrørende de nødvendige risikoreducerende foranstaltninger.
I andre tilfælde kan man udvikle yderligere scenarier for at inkludere sårbare forbrugere, især børn (jf. skema 1), større eller mindre afvigelser fra normal brug, anvendelse under anderledes klimatiske forhold, f.eks. stærk kulde eller høj varme, ugunstige anvendelsesbetingelser såsom mangel på klart dagslys eller ordentlig belysning, anvendelse som underforstået, da produktet blev solgt (f.eks. må en lampe, der sælges i en legetøjsbutik, nødvendigvis også vurderes for risikoen ved den, når den anvendes af et barn), anvendelse i hele produktets levetid (herunder slitage) osv. Hvert enkelt scenarie underkastes hele risikovurderingsproceduren.
Hvor produktet frembyder flere farer, udvikles der skadesscenarier og dermed risikoscenarier for hver af disse. Det er dog muligt at begrænse antallet af skadesscenarier ved at foretage en sandsynlighedskontrol til efterprøvelse af, hvorvidt et skadesscenarie kan være forbundet med en foranstaltningskrævende risiko.
Af alle de scenarier, der udvikles, vil det normalt være scenariet med den højeste tilknyttede risiko (= »den faktiske risiko« ved produktet), der vil være afgørende for, hvilke risikoreducerende foranstaltninger der skal træffes, eftersom det er foranstaltninger tilrettelagt efter den højeste risiko, der begrænser risikoen mest effektivt. En undtagelse fra reglen kunne være en særlig risiko, der er lavere end den højeste risiko og udspringer af en anden fare, og som vil kunne håndteres ved hjælp af særlige foranstaltninger, idet den højeste risiko naturligvis også vil skulle være omfattet.
Som tommelfingerregel kan man sige, at skadesscenarier kan være forbundet med det højeste risikoniveau, når
|
— |
de(n) relevante personskade(r) har en af de højeste alvorlighedsgrader (niveau 4 eller 3) |
|
— |
den samlede sandsynlighed for et skadesscenarie er forholdsvis høj (mindst > 1/100). |
Skema 4 giver yderligere vejledning desangående. Dette vil kunne bidrage til at begrænse antallet af scenarier.
Beslutningen om antallet af skadesscenarier træffes naturligvis af risikoanalytikeren og vil afhænge af, hvor mange faktorer det er nødvendigt at tage hensyn til ved bestemmelsen af »den faktiske risiko« ved produktet. Det er derfor umuligt at angive et specifikt tal for, hvor mange skadesscenarier der vil være nødvendige i hvert enkelt tilfælde.
Som en hjælp til at udvikle et passende antal scenarier indeholder disse retningslinjer et skema med typiske skadesscenarier (skema 2). Disse scenarier skal tilpasses det enkelte produkt, forbrugertypen og andre relevante forhold.
3.5. Skadens alvorlighed
Den personskade, en fare kan resultere i for forbrugeren, kan have forskellige alvorlighedsgrader. Skadens alvorlighed afspejler således den virkning, faren har for forbrugeren under de betingelser, der er beskrevet i skadesscenariet.
Skadens alvorlighed kan afhænge af følgende:
|
— |
faretypen (jf. listen over farer ovenfor og i skema 2). En mekanisk fare, f.eks. skarpe kanter, kan resultere i sårskader på fingrene; disse opdages med det samme, og forbrugeren vil træffe foranstaltninger til at få helet sine sår. En kemisk fare, derimod, kan være årsag til udvikling af kræft. Dette opdager man normalt ikke umiddelbart, og sygdommen, som anses for meget alvorlig, fordi det er meget vanskeligt og nogle gange umuligt at helbrede kræft, manifesterer sig måske først efter mange år |
|
— |
hvor omfattende faren er. For eksempel vil en overflade, der er opvarmet til 50 °C, måske give lette forbrændinger, mens en overflade med en temperatur på 180 °C vil give svære forbrændinger |
|
— |
hvor længe faren påvirker forbrugeren som en konkret hændelse. Ved kortvarig kontakt med et produkt, der udgør en raspningsfare, vil forbrugeren måske kun få overfladiske hudafskrabninger, mens han risikerer at miste større hudområder ved længere tids kontakt |
|
— |
hvilken legemsdel der skades. Mens det for eksempel gør ondt at få gennemboret huden af en spids genstand, er der tale om en mere alvorlig og måske endda livstruende skade, hvis det er øjet, der gennembores |
|
— |
hvilken virkning faren har på en eller flere legemsdele. En elektrisk fare kan resultere i et elektrisk stød, der medfører bevidstløshed, og efterfølgende være årsag til en brand, som måske forårsager lungeskader hos den bevidstløse person som følge af indånding af røgen. |
|
— |
forbrugertype og -adfærd. Et produkt, der er mærket med en advarsel, kan anvendes af en voksen forbruger, uden at denne kommer til skade, fordi forbrugeren indretter sig efter, hvordan produktet skal anvendes. Børn og andre sårbare forbrugere (jf. skema 1), der ikke kan læse eller forstå advarselsmærkningen, kan derimod komme meget alvorligt til skade. |
Med henblik på at måle alvorligheden af (en) personskade(r) er det i skema 3 i disse retningslinjer vist, hvordan skader inddeles i fire kategorier afhængigt af deres reversibilitet, dvs. hvorvidt og i hvilket omfang helbredstilstanden kan genoprettes efter skaden. Denne kategorisering er udelukkende ment som en vejledning, og en risikoanalytiker bør om nødvendigt ændre kategorien, idet det relevante angives i risikovurderingen.
Omfatter risikovurderingen flere skadesscenarier, klassificeres alvorligheden af hver enkelt personskade særskilt og gøres til en del af hele risikovurderingsproceduren.
Eksempel: En forbruger bruger en hammer til at slå et søm i en væg. Hammerens hoved er for skrøbeligt (pga. forkert materialevalg) og går i stykker, og et af disse stykker rammer forbrugeren i øjet med tilstrækkelig kraft til at medføre blindhed. Skaden kategoriseres således som »øjenskader, fremmedlegeme i øje: permanent synstab (på ét øje)«, som er en niveau 3-skade i skema 3.
3.6. Sandsynligheden for skaden
»Sandsynligheden for skaden« er sandsynligheden for, at et skadesscenarie faktisk vil give sig udslag i en konkret personskade i et produkts forventede levetid.
Det er ikke let at beregne denne sandsynlighed, men hvis et scenarie beskrives som en række trin, kan hvert af disse trin tildeles en sandsynlighed, hvorefter den samlede sandsynlighed for det pågældende scenarie kan beregnes ved at gange disse delsandsynligheder med hinanden. Denne trinvise fremgangsmåde burde gøre det lettere at beregne den samlede sandsynlighed. Hvis der udvikles mere end ét scenarie, må den samlede sandsynlighed naturligvis beregnes for hver enkelt af disse scenarier.
Beskrives et skadesscenarie imidlertid som ét enkelt trin, kan sandsynligheden for scenariet også kun bestemmes i ét samlet trin. Der vil i dette tilfælde dog kun være tale om et løseligt skøn, som vil kunne blive udsat for alvorlig kritik og afstedkomme, at der sættes spørgsmålstegn ved risikovurderingen som helhed. En mere gennemskuelig tildeling af sandsynligheder i et flertrinsscenarie er derfor at foretrække, især fordi de enkelte delsandsynligheder kan baseres på ubestridelig dokumentation.
Der opereres i disse retningslinjer med otte sandsynlighedsniveauer til klassificering af den samlede sandsynlighed: fra < 1/1 000 000 til > 50 % (jf. skema 4, venstre side). Følgende eksempel med et hammerhoved, der går i stykker, da brugeren slår et søm i en væg, skal illustrere, hvordan hvert trin tildeles en sandsynlighed, og hvordan den samlede sandsynlighed klassificeres:
|
Trin 1: |
Hammerens hoved går i stykker, da brugeren forsøger at slå et søm i en væg, fordi det materiale, hammerhovedet er fremstillet i, ikke er stærkt nok. Den lave brudstyrke er konstateret med en afprøvning, og med den oplyste svaghed sættes sandsynligheden for, at hammerhovedet går i stykker inden for hammerens i øvrigt forventede levetid, til 1/10. |
|
Trin 2: |
Et stykke af hammeren rammer brugeren, da hammeren går i stykker. Sandsynligheden for, at dette sker, sættes til 1/10, idet den del af overkroppen, der er udsat for de løsrevne stykker af hammeren, formodes at dække 1/10 af den halvkreds, der kan tegnes ud fra væggen. Det er klart, at hvis brugeren stod meget tæt på væggen, ville den pågældendes krop dække en større del af halvkredsen, hvormed sandsynligheden ville være højere. |
|
Trin 3: |
Stykket rammer brugeren i hovedet. Hovedet vurderes at udgøre 1/3 af overkroppen, og sandsynligheden er derfor 1/3. |
|
Trin 4: |
Stykket rammer brugeren i øjet. Øjnene vurderes at dække ca. 1/20 af hovedets areal, og sandsynligheden er derfor 1/20. |
Ved at gange sandsynlighederne for ovennævnte trin med hinanden får man en samlet sandsynlighed for scenariet på 1/10 * 1/10 * 1/3 * 1/20 = 1/6 000. Dette svarer til > 1/10 000 (jf. skema 4, venstre side).
Når den samlede sandsynlighed er beregnet for et skadesscenarie, kontrolleres det, at tallene er realistiske. Dette kræver ganske stor erfaring, hvilket betyder, at det vil være hensigtsmæssigt at søge bistand hos personer med risikovurderingserfaring (jf. punktet »Lad andre efterprøve din risikovurdering« ovenfor). Efterhånden som der opbygges erfaring med anvendelsen af disse retningslinjer, skulle det blive nemmere at beregne sandsynlighed, ligesom opgaven vil blive lettere i takt med det stadig større antal eksempler.
Tildelingen af sandsynligheder til forskellige skadesscenarier for det samme produkt vil kunne indebære følgende:
|
— |
Når produktet anvendes af mere sårbare forbrugere i et scenarie, vil det kunne være nødvendigt at opjustere den samlede sandsynlighed, fordi mere sårbare forbrugere lettere kommer til skade. Dette gælder først og fremmest børn, fordi de normalt ikke besidder den erfaring, der er nødvendig for at kunne træffe forebyggende foranstaltninger — tværtimod (jf. også punktet »Sårbare forbrugere« under afsnit 3.3). |
|
— |
Når risikoen er åbenbar, f.eks. i kraft af advarselsmærkning, vil det kunne være nødvendigt at sætte sandsynligheden lavere, fordi brugeren vil anvende produktet med større forsigtighed for så vidt muligt at undgå at komme til skade. Dette vil ikke nødvendigvis være tilfældet for et skadesscenarie med et (lille) barn eller en anden sårbar bruger (jf. skema 1), der ikke kan læse. |
|
— |
I forbindelse med indberetning af ulykker, der passer ind i skadesscenariet, kan sandsynligheden for det pågældende scenarie øges. I tilfælde, hvor der kun er indberettet få ulykker, eller hvor man slet ikke har kendskab til ulykker, vil det kunne være nyttigt at spørge producenten af produktet, om den pågældende har kendskab til ulykker eller skadelige virkninger forårsaget af produktet. |
|
— |
Skal et forholdsvis stort antal betingelser være opfyldt, for at personskaden indtræffer, vil den samlede sandsynlighed for scenariet normalt være lavere. |
|
— |
Sandsynligheden kan øges, hvis betingelserne for, at skaden indtræffer, hurtigt vil være opfyldt. |
|
— |
Hvis resultaterne af afprøvningen af et produkt langt fra lever op til de relevante grænseværdier (fastsat i den relevante standard eller i lovgivningen), kan sandsynligheden for, at (scenariet med) personskaden indtræffer, være højere, end hvis produktet testes til at ligge tæt på grænseværdierne. |
»Sandsynligheden for skaden« er i dette tilfælde sandsynligheden for, at skadesscenariet faktisk bliver til virkelighed. Sandsynligheden er således ikke udtryk for befolkningens eksponering for et produkt generelt, beregnet ved f.eks. at konstatere, at der sælges så og så mange millioner enheder af produktet på markedet, og derefter tage i betragtning, at nogle få af disse produkter måske vil svigte. Overvejelser af denne art spiller imidlertid en rolle, når der skal træffes beslutning om de relevante risikoreducerende foranstaltninger (jf. afsnit 4).
Ulykkesstatistikker, selv produktspecifikke statistikker, skal ligeledes anvendes med forsigtighed, når de bruges til at beregne sandsynlighed. Omstændighederne ved en indberettet ulykke er måske ikke beskrevet tilstrækkelig udførligt, produktet kan have ændret sig med tiden, producenten kan være en anden osv. Dertil kommer, at mindre ulykker ikke nødvendigvis er blevet indberettet til de ansvarlige for indsamling af oplysninger til statistikkerne. Ulykkesstatistikker kan dog under alle omstændigheder kaste lys over skadesscenarier og sandsynligheden for dem.
3.7. Bestemmelse af risikoen
Når alvorligheden af og sandsynligheden for personskaden er fastlagt, om muligt med flere forskellige skadesscenarier, aflæses risikoniveauet i skema 4. Skema 4 kombinerer alvorligheden af og sandsynligheden for skaden, og den højeste risiko er »den faktiske risiko« ved produktet. Risici, der nødvendiggør særlige risikohåndteringsforanstaltninger, bør også indberettes med henblik på at sikre, at samtlige risici begrænses til et minimum.
Der opereres i disse retningslinjer med fire risikoniveauer: alvorlig, høj, middel og lav. Forskellen i risiko fra ét personskadealvorligheds- eller sandsynlighedsniveau til det næste er normalt ét niveau. Dette er i tråd med erfaringen i øvrigt, som siger, at risikoen ikke automatisk øges gradvist i takt med gradvise ændringer i inputfaktorerne. Når skadens alvorlighed stiger fra niveau 1 til niveau 2 (skema 4, højre side), går visse risikoniveauer dog to niveauer op, nemlig fra middel til alvorlig og fra lav til høj. Dette skyldes, at der i disse retningslinjer opereres med fire alvorlighedsgrader for personskader, mens den oprindelige metode (jf. indledningen) omfattede fem grader. Fire alvorlighedsgrader anses imidlertid for normalt, når der er tale om forbrugerprodukter, idet de sikrer en tilstrækkeligt holdbar alvorlighedsberegning; beregninger med fem niveauer ville blive overdrevent forfinede, eftersom det ikke er muligt at bestemme hverken skadens alvorlighed eller dens sandsynlighed helt nøjagtigt.
Ved afslutningen af risikovurderingen — hvad enten denne vedrører et enkelt skadesscenarie eller den samlede risiko ved produktet — ses der på, om risikoniveauet forekommer realistisk, ligesom de relevante usikkerhedselementer i beregningerne tages i betragtning. Dette kan omfatte efterprøvelse af, at risikoanalytikeren har lagt de bedst mulige tilgængelige oplysninger til grund for sine beregninger og antagelser. Også feedback fra kolleger og andre eksperter kan være nyttig.
Derudover kan en følsomhedsanalyse være meget værdifuld (jf. eksemplet i afsnit 6.3). Hvor meget ændrer risikoniveauet sig, når en personskades alvorlighed eller sandsynlighed går et niveau op eller ned? Ændrer risikoniveauet sig slet ikke, er det ret sandsynligt, at det er beregnet korrekt. Ændrer det sig derimod, grænser risikoniveauet muligvis til det urealistiske. I så fald er det nødvendigt at revurdere skadesscenarierne og den tildelte alvorlighedsgrad og sandsynlighed for den eller de pågældende personskader. Ved afslutningen af følsomhedsanalysen bør risikoanalytikeren føle sig forvisset om, at risikoniveauet er tilstrækkelig realistisk, og at han kan dokumentere dette og videreformidle de relevante oplysninger.
4. Fra risiko til foranstaltninger
Når risikovurderingen er tilendebragt, vil den normalt blive brugt som grundlag for at beslutte, hvorvidt der er behov for foranstaltninger til at begrænse risikoen og derigennem forhindre skadelige virkninger på en forbrugers sundhed. Selv om gennemførelse af foranstaltninger er et selvstændigt skridt i forhold til risikovurderingen, gennemgås nedenfor visse aspekter for at vise, hvordan der kunne følges op på identificerede risici.
Foranstaltninger som led i markedsovervågningen vil ofte omfatte kontakt mellem den kompetente myndighed og producenten, importøren eller distributøren. Dette kan gøre det lettere for myndigheden at afgøre, hvordan den pågældende risiko kan håndteres mest effektivt.
Er der tale om en alvorlig risiko ved et forbrugerprodukt, vil de risikoreducerende foranstaltninger kunne omfatte tilbagetrækning fra markedet eller tilbagekaldelse fra forbrugerne. Lavere risikoniveauer indebærer normalt mindre vidtgående foranstaltninger. Det vil i sådanne tilfælde kunne være tilstrækkeligt at mærke produktet med advarsler eller forbedre produktanvisningerne for at gøre produktet sikkert. Uanset risikoniveauet bør myndigheden således overveje, om der skal træffes foranstaltninger, og i givet fald hvilke.
Der er imidlertid ikke nogen automatisk sammenkædning af risikoniveau og foranstaltninger. Der kan for et produkt, der frembyder flere »mindre end alvorlige« risici og dermed ikke en alvorlig samlet risiko, være behov for hasteforanstaltninger, fordi enhver af de pågældende risici ret hurtigt kan give sig udslag i konkrete hændelser. Produktets risikomønster kan give formodning om manglende kvalitetskontrol i fremstillingsprocessen.
Det er også vigtigt at tage hensyn til, i hvilket omfang befolkningen som helhed er udsat. Hvis produktet findes på markedet i stort antal og derfor anvendes af mange forbrugere, kan selv en enkelt »mindre end alvorlig« risiko nødvendiggøre hurtig indgriben for at undgå skadelige virkninger på forbrugernes sundhed.
»Mindre end alvorlige« risici kan også nødvendiggøre foranstaltninger, hvis det pågældende produkt vil kunne være årsag til dødsulykker, selv hvis sandsynligheden for sådanne ulykker er ekstremt lav. Dette kunne være tilfældet med en lukkeanordning på drikkevareemballage, der kan løsrive sig og blive slugt af et barn, som bliver kvalt. Denne risiko vil kunne fjernes blot ved at ændre på lågets udformning, uden at der nødvendigvis vil være behov for andre foranstaltninger. Der vil muligvis endda kunne indrømmes en overgangsperiode, hvor der kan sælges ud af produktet, såfremt risikoen for en dødsulykke virkelig er ekstremt lav.
Andre potentielt risikorelevante aspekter er befolkningens opfattelse af risiko og mulige konsekvenser heraf, kulturelt og politisk følsomme spørgsmål og fremstillingen af risici i medierne. Disse aspekter kan være særlig relevante, når det drejer sig om sårbare forbrugere, især børn. Det vil være op til den eller de nationale markedsovervågningsmyndigheder at afgøre, hvilke foranstaltninger der er behov for.
Foranstaltninger til imødegåelse af en risiko kan også afhænge af selve produktet og de »minimale risici, der er forenelige med produktets anvendelse, og som under hensyntagen til et højt beskyttelsesniveau […] anses for acceptable« (35). En sådan minimal risiko kan forventes at ville være meget lavere for legetøj, hvor børn er involveret, end for en kædesav, som er kendt for at frembyde så høj en risiko, at det er nødvendigt at bruge beskyttelsesudstyr for at holde risikoen på et håndterbart niveau.
Endelig kan det være nødvendigt at træffe foranstaltninger, selv om der slet ikke er nogen risiko, blandt andet hvis et produkt ikke overholder gældende forskrifter/lovgivning (f.eks. pga. ufuldstændig mærkning).
Sammenfattende kan det konkluderes, at der ikke er nogen automatik i overgangen fra risiko til foranstaltninger. Overvågningsmyndighederne vil tage hensyn til en række faktorer, herunder de ovenfor beskrevne. Proportionalitetsprincippet skal altid indgå i beslutningsgrundlaget, og foranstaltningerne skal være effektive.
5. Risikovurderingsproceduren — i store træk
1. Beskriv produktet og faren ved det.
Beskriv produktet entydigt. Knytter faren sig til produktet som helhed eller kun en (adskillelig) del af produktet?
Frembyder produktet kun én fare? Er der flere farer? Vejledende henvises til skema 2.
Registrer, hvilke(n) standard(er) eller lovgivning produktet er omfattet af.
2. Fastlæg, hvilken type forbruger du vil lade være omfattet af dit skadesscenarie for det farlige produkt.
Lad dit første skadesscenarie omfatte den påtænkte bruger og den påtænkte anvendelse af produktet. Inddrag andre forbrugere (jf. skema 1) og anvendelser til yderligere scenarier.
3. Beskriv et skadesscenarie, hvori den eller de produktfarer, du har valgt, er årsag til (en) skade(r) eller (en) skadelig(e) virkning(er) på den valgte forbrugers sundhed.
Beskriv trinnene, der fører til den eller de pågældende personskader, klart og kortfattet og uden for mange detaljer (»den korteste vej til en personskade«/»den kritiske vej til en personskade«). Hvis dit scenarie indebærer flere samtidige skader, inkluderes de alle i det samme scenarie.
Tag ved beskrivelsen af skadesscenariet hensyn til anvendelsens hyppighed og varighed og graden af genkendelse af de(n) pågældende fare(r) blandt forbrugerne samt til, om der er tale om en sårbar forbruger (især børn), og til beskyttelsesudstyr, forbrugerens adfærd i tilfælde af en ulykke, forbrugerens kulturelle baggrund og andre faktorer, som efter din mening er vigtige for risikovurderingen.
Jf. vejledningen i afsnit 3.3 og skema 2.
4. Bestem alvorligheden af personskaden.
Bestem alvorlighedsgraden (1 til 4) af skaden på forbrugeren. Hvis forbrugeren pådrager sig flere skader i dit skadesscenarie, bestem da alvorligheden af alle disse skader tilsammen.
Jf. vejledningen i skema 3.
5. Bestem sandsynligheden for skadesscenariet.
Tildel hvert trin i dit skadesscenarie en sandsynlighed. Beregn den samlede sandsynlighed for dit skadesscenarie ved at gange sandsynlighederne med hinanden.
Jf. vejledningen i skema 4, venstre side.
6. Bestem risikoniveauet.
Kombiner alvorligheden af personskaden og den samlede sandsynlighed for skadesscenariet og aflæs risikoniveauet i skema 4.
7. Kontrollér, at risikoniveauet er realistisk.
Forekommer risikoniveauet ikke realistisk, eller er du usikker på alvorligheden af skaden/skaderne eller sandsynligheden/sandsynlighederne, rykker du disse et niveau op og ét ned og beregner risikoen igen. Med denne »følsomhedsanalyse« vil du kunne se, om risikoen ændrer sig, når du ændrer dine input.
Hvis risikoniveauet forbliver det samme, har du god grund til at have tiltro til din risikovurdering. Ændrer niveauet sig let, vil det kunne være en god idé at lade din beregning hælde til den sikre side og lade det højeste risikoniveau være »den faktiske risiko« ved forbrugerproduktet.
Du kan også diskutere, hvor realistisk risikoniveauet er, med erfarne kolleger.
8. Udarbejd flere forskellige skadesscenarier for at identificere den højeste risiko ved produktet.
Gør følgende, hvis risikoniveauet i dit første skadesscenarie er lavere end det højeste af de risikoniveauer, der opereres med i disse retningslinjer, og hvis du har en formodning om, at produktet kan udgøre en højere risiko end den identificerede:
|
— |
udvælg andre forbrugere (herunder sårbare forbrugere, især børn) |
|
— |
identificer andre anvendelser (herunder med rimelighed forudsigelige anvendelser) |
for at afgøre, i hvilket skadesscenarie produktet frembyder den højeste risiko.
Den højeste risiko er normalt »den faktiske risiko« ved produktet, der gør det muligt at træffe de mest effektive risikohåndteringsforanstaltninger. I visse særlige tilfælde kan en bestemt fare indebære en risiko, der er lavere end den højeste risiko, og nødvendiggøre særlige risikohåndteringsforanstaltninger. Dette bør indgå i overvejelserne.
Som tommelfingerregel kan man sige, at skadesscenarier kan være forbundet med det højeste risikoniveau, der opereres med i disse retningslinjer, når:
|
— |
de(n) relevante personskade(r) mindst er af alvorlighedsgrad 3 eller 4 |
|
— |
den samlede sandsynlighed for et skadesscenarie er mindst > 1/100. |
Jf. vejledningen i skema 4.
9. Dokumenter din risikovurdering og del den med andre.
Sørg for åbenhed i processen og beskriv alle de usikkerhedsmomenter, du er stødt på under udarbejdelsen af din risikovurdering.
Afsnit 6 i disse retningslinjer indeholder eksempler på risikovurderingsrapportering.
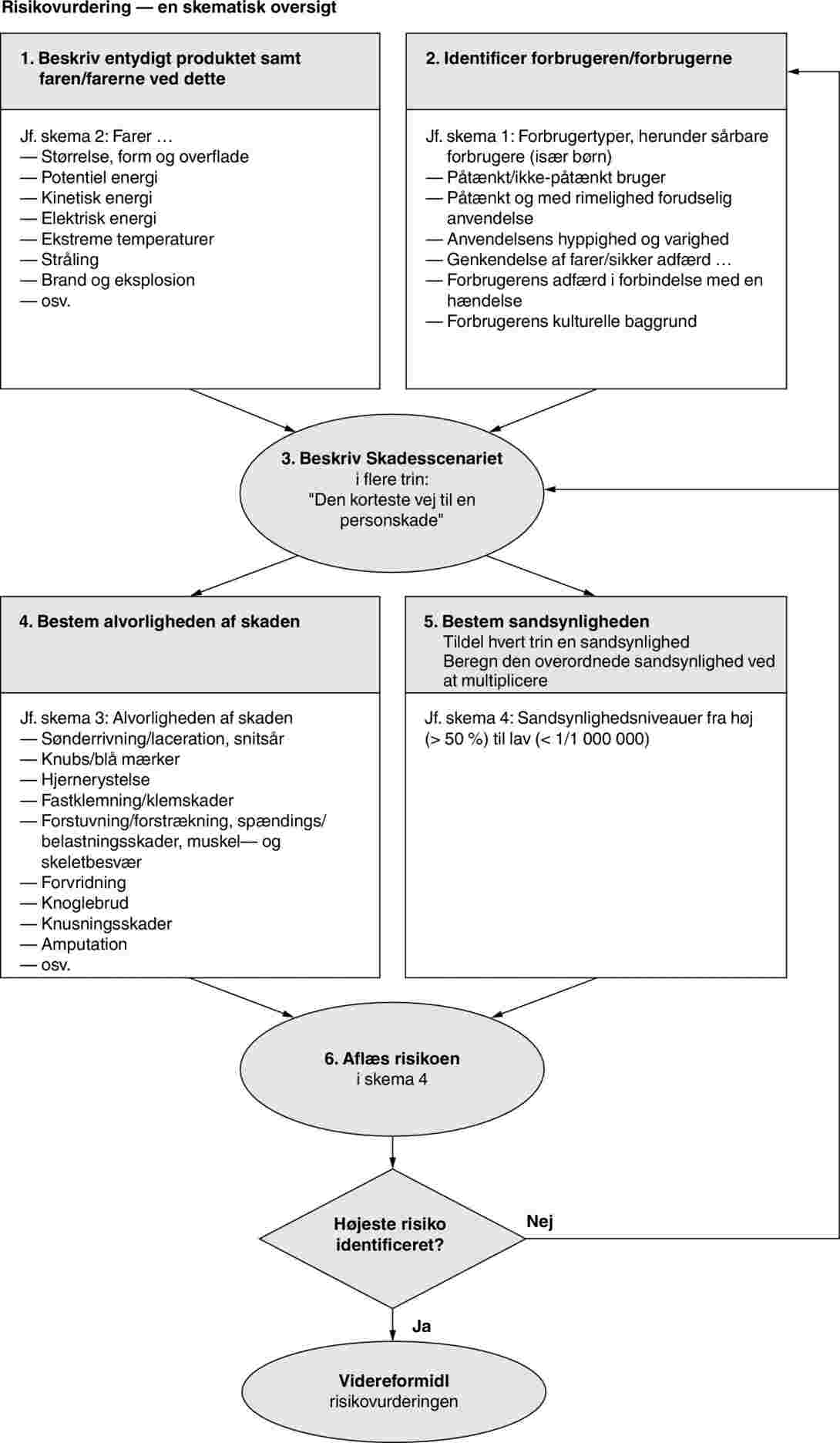
6. Eksempler
6.1. Klapstol
![]()
En klapstol er forsynet med en foldemekanisme, som er udformet på en sådan måde, at brugerens fingre kan komme i klemme mellem sædet og foldemekanismen. Dette kan resultere i brud på eller endog tab af en eller flere fingre.
Bestemmelse af risikoen/risiciene
|
Skadesscenarie |
Skadens art og placering |
Skadens alvorlighed |
Sandsynligheden for skaden |
Samlet sandsynlighed |
Risiko |
|
|
Brugeren klapper stolen ud, holder ved en fejl (fordi han er uopmærksom/distraheres) i sædet tæt ved det bageste hjørne og får en finger i klemme mellem sæde og ryg |
Mindre alvorlig klemning af finger |
1 |
Stolen klappes ud |
1 |
1/500 |
Lav risiko |
|
Der holdes i sædet ved det bageste hjørne, mens stolen klappes ud |
1/50 |
|||||
|
En finger kommer i klemme |
1/10 |
> 1/1 000 |
||||
|
Fingeren klemmes mindre alvorligt |
1 |
|||||
|
Brugeren klapper stolen ud, holder ved en fejl (fordi han er uopmærksom/distraheres) i sædet på siden af dette og får en finger i klemme mellem sæde og forbindelsesskinne |
Mindre alvorlig klemning af finger |
1 |
Stolen klappes ud |
1 |
1/500 |
Lav risiko |
|
Der holdes på siden af sædet, mens stolen klappes ud |
1/50 |
|||||
|
En finger kommer i klemme |
1/10 |
> 1/1 000 |
||||
|
Fingeren klemmes mindre alvorligt |
1 |
|||||
|
Brugeren klapper stolen ud, men foldemekanismen binder, så brugeren forsøger at presse sædet ned og holder ved en fejl (fordi han er uopmærksom/distraheres) i sædet tæt ved hjørnet og får en finger i klemme mellem sæde og ryg |
Brud på finger |
2 |
Stolen klappes ud |
1 |
1/500 000 |
Lav risiko |
|
Foldemekanismen binder |
1/1 000 |
|||||
|
Der holdes i sædet i hjørnerne, mens stolen klappes ud |
1/50 |
|||||
|
En finger kommer i klemme |
1/10 |
> 1/1 000 000 |
||||
|
Der opstår brud på fingeren |
1 |
|||||
|
Brugeren klapper stolen ud, men foldemekanismen binder, så brugeren forsøger at presse sædet ned og holder ved en fejl (fordi han er uopmærksom/distraheres) i sædet på siden af dette og får en finger i klemme mellem sæde og forbindelsesskinne |
Brud på finger |
2 |
Stolen klappes ud |
1 |
1/500 000 |
Lav risiko |
|
Foldemekanismen binder |
1/1 000 |
|||||
|
Der holdes på siden af sædet, mens stolen klappes ud |
1/50 |
|||||
|
En finger kommer i klemme |
1/10 |
> 1/1 000 000 |
||||
|
Brud på finger |
1 |
|||||
|
Brugeren sidder på stolen, ønsker at flytte denne og forsøger at løfte den ved at holde fast i stolen på sædets bageste del og får en finger i klemme mellem sæde og ryg |
Tab af finger |
3 |
Brugeren sidder på stolen |
1 |
1/6 000 |
Høj risiko |
|
Brugeren flytter stolen, mens han sidder på den |
1/2 |
|||||
|
Brugeren holder bag på stolen, mens han flytter den |
1/2 |
|||||
|
Stolen klapper delvis sammen, hvorved der opstår et mellemrum mellem ryg og sæde |
1/3 |
> 1/10 000 |
||||
|
En finger er anbragt mellem ryg og sæde |
1/5 |
|||||
|
Fingeren kommer i klemme |
1/10 |
|||||
|
Brugeren mister fingeren (eller en del af den) |
1/10 |
|||||
|
Brugeren sidder på stolen, ønsker at flytte denne og forsøger at løfte den ved at holde fast i stolen på sædets bageste del og får en finger i klemme mellem sæde og forbindelsesskinne |
Tab af finger |
3 |
Brugeren sidder på stolen |
1 |
1/6 000 |
Høj risiko |
|
Brugeren flytter stolen, mens han sidder på den |
1/2 |
|||||
|
Brugeren holder bag på stolen, mens han flytter den |
1/2 |
|||||
|
Stolen klapper delvis sammen, hvorved der opstår et mellemrum mellem ryg og sæde |
1/3 |
> 1/10 000 |
||||
|
En finger er anbragt mellem ryg og sæde |
1/5 |
|||||
|
Fingeren kommer i klemme |
1/10 |
|||||
|
Brugeren mister fingeren (eller en del af den) |
1/10 |
|||||
Den samlede risiko ved klapstolen er således en »høj risiko«.
6.2. Kontaktsikringer
![]()
Dette eksempel handler om kontaktsikringer. Dette er anordninger, som brugerne (forældre) indsætter i elektriske stikkontakter for at forhindre, at små børn kommer i kontakt med strømførende dele ved at stikke en lang genstand af metal ind i et af hullerne i stikkontakten og får et (dødbringende) elektrisk stød.
Hullerne i den specifikke kontaktsikring (som stikproppens ben stikkes igennem) er så små, at stikproppens ben kan sidde fast i dem. Dette betyder, at brugeren kan komme til at trække sikringen ud af stikkontakten, når stikket trækkes ud. Brugeren opdager måske ikke, at dette er sket.
Bestemmelse af risikoen/risiciene
|
Skadesscenarie |
Skadens art og placering |
Skadens alvorlighed |
Sandsynligheden for skaden |
Samlet sandsynlighed |
Risiko |
|
|
Sikringen trækkes ud af stikkontakten, som derefter er ubeskyttet. Et barn leger med en tynd genstand med ledningsevne, som kan stikkes ind i stikkontakten, og kommer i kontakt med højspænding med døden til følge |
Elektrisk stød med døden til følge |
4 |
Sikringen trækkes ud |
9/10 |
27/160 000 |
Alvorlig risiko |
|
Det opdages ikke, at sikringen er trukket ud |
1/10 |
|||||
|
Et barn leger med en tynd genstand med ledningsevne |
1/10 |
|||||
|
Barnet leger uden opsyn |
1/2 |
> 1/10 000 |
||||
|
Barnet stikker genstanden ind i stikkontakten |
3/10 |
|||||
|
Barnet kommer i kontakt med strøm |
1/2 |
|||||
|
Elektrisk stød med døden til følge (uden strømafbryder) |
1/4 |
|||||
|
Sikringen trækkes ud af stikkontakten, som derefter er ubeskyttet. Et barn leger med en tynd genstand med ledningsevne, som kan stikkes ind i stikkontakten, kommer i kontakt med højspænding og får elektrisk stød |
2.-gradsforbrændinger |
1 |
Sikringen trækkes ud |
9/10 |
81/160 000 |
Lav risiko |
|
Det opdages ikke, at sikringen er trukket ud |
1/10 |
|||||
|
Et barn leger med en tynd genstand med ledningsevne |
1/10 |
|||||
|
Barnet stikker genstanden ind i stikkontakten |
3/10 |
|||||
|
Barnet kommer i kontakt med strøm |
1/2 |
> 1/10 000 |
||||
|
Barnet leger uden opsyn |
1/2 |
|||||
|
Forbrændinger forårsaget af elektrisk strøm (uden strømafbryder) |
3/4 |
|||||
|
En stikkontakt er ubeskyttet. Et barn leger med en tynd genstand med ledningsevne, som kan stikkes ind i stikkontakten, kommer i kontakt med højspænding og dør af det |
Elektrisk stød med døden til følge |
4 |
Et barn leger med en tynd genstand med ledningsevne |
1/10 |
3/80 000 |
Høj risiko |
|
Barnet leger uden opsyn |
1/100 |
|||||
|
Barnet stikker genstanden ind i stikkontakten |
3/10 |
|||||
|
Barnet kommer i kontakt med strøm |
1/2 |
> 1/100 000 |
||||
|
Elektrisk stød med døden til følge (uden strømafbryder) |
1/4 |
|||||
Den samlede risiko ved kontaktsikringerne er således »alvorlig«.
6.3. Følsomhedsanalyse
De faktorer, der anvendes til beregning af risikoen for et skadesscenarie, nemlig alvorligheden af og sandsynligheden for personskaden, må ofte baseres på skøn. Dette skaber usikkerhed. Især sandsynligheden kan være vanskelig at beregne, idet det kan være vanskeligt at forudsige f.eks. forbrugeres adfærd. Foretager en person ofte eller kun lejlighedsvis en bestemt handling?
Det er derfor vigtigt at se på usikkerhedsgraden for de to faktorer og foretage en følsomhedsanalyse. Formålet med denne analyse er at fastlægge, hvor meget risikoniveauet ændrer sig, når de faktorer, man har beregnet sig frem til, ændrer sig. Eksemplet nedenfor viser kun udsvingene i sandsynligheden, idet personskadens alvorlighed normalt kan forudsiges med større sikkerhed.
En praktisk måde at foretage følsomhedsanalysen på er at gentage risikovurderingen for et bestemt scenarie, men sådan at der anvendes en anden sandsynlighed for ét eller flere trin i scenariet. For eksempel vil et stearinlys, der indeholder frø, kunne være årsag til en brand, fordi frøene kan flamme op og skabe høje flammer. Der vil kunne gå ild i møbler eller gardiner, og personer, der ikke befinder sig i rummet, vil kunne indånde giftige dampe og dø af røgforgiftning.
|
Skadesscenarie |
Skadens art og placering |
Skadens alvorlighed |
Sandsynligheden for skaden |
Resulterende sandsynlighed |
Risiko |
||||||||
|
Frø eller bønner flammer op og skaber høje flammer. Der går ild i møbler eller gardiner. Personer, der ikke befinder i rummet, indånder giftige dampe |
Forgiftning med døden til følge |
4 |
|
0,00675 > 1/1 000 |
Alvorlig |
Sandsynlighederne for de forskellige trin i scenariet er beregnet som vist i skemaet.
Den samlede sandsynlighed er 0,00675, hvilket svarer til > 1/1 000 i skema 4. Konklusionen er på dette grundlag, at der er tale om en »alvorlig risiko«. Bemærk, at den eksakte sandsynlighed ligger tættere på 1/100 end på 1/1 000, hvilket allerede giver en vis tillid til risikoniveauet, fordi vi her ligger lidt længere inde i »alvorlig risiko«-området i skema 4, end placeringen i > 1/1 000-rækken umiddelbart giver indtryk af.
Lad os antage, at vi nærer en vis usikkerhed mht. sandsynligheden på 5 % for, at personer indånder de giftige dampe. Vi kan da f.eks. sætte denne langt lavere, nemlig til 0,1 % (0,001 = én ud af tusind). Hvis vi foretager beregningen igen med denne antagelse, får vi en samlet sandsynlighed på 0,000135, hvilket svarer til > 1/10 000. Risikoen er dog stadig alvorlig. Selv hvis sandsynligheden af en eller anden grund var en faktor 10 lavere, ville risikoen stadig være høj. Det vil sige, at selv om sandsynligheden kan variere 10- til 100-fold, vil vi stadig nå frem til en alvorlig eller høj risiko (idet den i sidstnævnte tilfælde vil være ret tæt på »alvorlig«). På grundlag af denne følsomhedsanalyse kan vi således roligt vurdere risikoen til at være alvorlig.
Generelt bør risikovurdering dog baseres på de realistisk set værst tænkelige tilfælde, således at man ikke beregner hver eneste faktor overdrevent pessimistisk, men bestemt heller ikke for optimistisk.
Skema 1
Forbrugere
|
Forbrugere |
Beskrivelse |
|
Meget sårbare forbrugere |
Helt små børn: 0-36 måneder Andre: personer med svære og komplicerede handicap |
|
Sårbare forbrugere |
Små børn: børn på over 36 måneder og under 8 år Større børn: børn på 8-14 år Andre: personer med nedsat fysisk, sensorisk eller psykisk funktionsevne (f.eks. delvist handicappede personer og ældre mennesker, herunder gruppen på over 65, med delvist nedsat fysisk og psykisk funktionsevne) eller med begrænset erfaring og viden |
|
Andre forbrugere |
Forbrugere, der ikke er sårbare eller meget sårbare |
Skema 2
Farer, typiske skadesscenarier og typiske personskader
|
Faregruppe |
Fare (produktegenskaber) |
Typiske skadesscenarier |
Typiske skader |
|
Størrelse, form og overflade |
Produktet udgør en forhindring |
En person snubler over produktet og falder, eller personen støder ind i produktet |
Knubs/blå mærker; knoglebrud; hjernerystelse |
|
|
Produktet er lufttæt |
Produktet dækker en persons (typisk et barns) mund og/eller næse eller blokerer de indre luftveje |
Kvælning (ekstern luftvejsblokering) |
|
|
Produktet er eller indeholder en lille del |
En person (et barn) sluger den lille del; delen sætter sig fast i svælget og blokerer luftvejene |
Kvælning (intern luftvejsblokering) |
|
|
Mulighed for, at en lille del bides af produktet |
En person (et barn) sluger den lille del; delen sætter sig fast i spiserøret |
Blokering af spiserøret |
|
|
Skarpt hjørne eller spids genstand |
En person støder ind i skarpt hjørne eller rammes af spids genstand i bevægelse; dette resulterer i gennemboringsskader eller penetrerende skader |
Gennemboringsskader; blænding, fremmedlegeme i øjet; høreskader, fremmedlegeme i øre |
|
|
Skarpt hjørne |
En person støder ind i skarpt hjørne; dette sønderriver huden eller skærer gennem væv |
Sønderrivning/laceration, sårskader; amputation |
|
|
Glat overflade |
En person går på den glatte overflade, glider og falder |
Knubs/blå mærker; knoglebrud; hjernerystelse |
|
|
Ru overflade |
En person glider hen over/langs ru overflade; dette forårsager friktion og/eller raspning |
Hudafskrabninger |
|
|
Mellemrum eller åbning mellem dele |
En person får en legemsdel eller hele kroppen i klemme i en åbning, og fingeren, armen, halsen, hovedet, hele kroppen eller beklædningsgenstande sidder fast; personen kommer til skade på grund af vægt eller bevægelse |
Knusningsskader, knoglebrug, amputation, strangulering |
|
Potentiel energi |
Begrænset mekanisk stabilitet |
Et produkt vælter; en person oven på produktet falder fra højde, eller en person i nærheden af produktet rammes af dette; elektrisk produkt vælter og går i stykker, så strømførende dele blotlægges, eller fortsætter med at virke, så overflader i nærheden ophedes |
Knubs/blå mærker; forvridning; forstuvning/forstrækning; knoglebrud; hjernerystelse; knusningsskader; elektrisk stød; forbrændinger |
|
|
Begrænset mekanisk styrke |
Et produkt bryder sammen på grund af overvægt; en person oven på produktet falder fra højde, eller en person i nærheden af produktet rammes af dette; elektrisk produkt vælter og går i stykker, så strømførende dele blotlægges, eller fortsætter med at virke, så overflader i nærheden ophedes |
Knubs/blå mærker; forvridning; knoglebrud; hjernerystelse; knusningsskader; elektrisk stød; forbrændinger |
|
|
Brugeren befinder sig højt oppe |
En person, der befinder sig højt oppe oven på produktet, mister balancen, har ikke noget at holde fast i og falder fra højde |
Knubs/blå mærker; forvridning; knoglebrud; hjernerystelse; knusningsskader |
|
|
Elastisk element eller fjeder |
Et komprimeret elastisk element eller en spændt fjeder udløses pludseligt; en person, som befinder sig i bevægelsesbanen, rammes af produktet |
Knubs/blå mærker; forvridning; knoglebrud; hjernerystelse; knusningsskader |
|
|
Væske eller gas under tryk (eller vakuum) |
Væske eller gas under tryk frigives pludseligt, og en person i nærheden rammes, eller produktet imploderer, hvorved genstande flyver gennem luften |
Forvridning; knoglebrud; hjernerystelse; knusningsskader; sårskader (jf. under »Brand og eksplosion«) |
|
Kinetisk energi |
Produkt i bevægelse |
En person, som befinder sig i produktets bevægelsesbane, rammes af produktet eller køres over |
Knubs/blå mærker; forstuvning/forstrækning; knoglebrud; hjernerystelse; knusningsskader |
|
|
Dele, der bevæger sig mod hinanden |
En person får en legemsdel i klemme mellem de bevægelige dele, mens de bevæger sig mod hinanden; den pågældende legemsdel sidder fast og udsættes for tryk (knuses) |
Knubs/blå mærker; forvridning; knoglebrud; knusningsskader |
|
|
Dele, der bevæger sig forbi hinanden |
En person får en legemsdel i klemme mellem de bevægelige dele, mens de bevæger sig tæt forbi hinanden (saksebevægelse); den pågældende legemsdel sidder fast mellem de bevægelige dele og udsættes for tryk (knuses) |
Sønderrivning/laceration, sårskader; amputation |
|
|
Roterende dele |
En legemsdel, hår eller beklædningsgenstande på en person vikles ind i den roterende del; derved udsættes personen for et træk |
Knubs/blå mærker; knoglebrud; sønderrivning/laceration (hovedhuden); strangulering |
|
|
Roterende dele tæt på hinanden |
En legemsdel, hår eller beklædningsgenstande på en person fanges af de roterende dele; derved udsættes personen for et træk, og den pågældende legemsdel udsættes for tryk |
Knusningsskader, knoglebrug, amputation, strangulering |
|
|
Acceleration |
En person, der befinder sig på det accelererende produkt, mister balancen, har ikke noget at holde fast i og falder af i fart |
Forvridning; knoglebrud; hjernerystelse; knusningsskader |
|
|
Flyvende genstande |
En person rammes af den flyvende genstand og pådrager sig skader (afhængigt af den kraft, hvormed det sker) |
Knubs/blå mærker; forvridning; knoglebrud; hjernerystelse; knusningsskader |
|
|
Vibration |
En person, der holder produktet, mister balancen og falder, eller langvarig kontakt med det vibrerende produkt resulterer i neurologiske forstyrrelser, knogle- og ledbesvær, skader på rygsøjlen eller karforstyrrelser |
Knubs/blå mærker; forvridning; knoglebrud; knusningsskader |
|
|
Støj |
En person udsættes for støj fra produktet. Afhængigt af lydniveauet og afstanden til produktet kan resultatet være tinnitus og tab af hørelse |
Høreskader |
|
Elektrisk energi |
Højspænding/lavspænding |
En person rører ved en del af produktet, som er under højspænding; personen får et elektrisk stød og dør måske af det |
Elektrisk stød |
|
|
Varmeudvikling |
Et produkt bliver varmt; en person, som rører ved det, brænder sig måske, eller produktet udsender smeltede partikler, damp osv., der rammer en person |
Forbrændinger, skoldning |
|
|
Strømførende dele placeret for tæt på hinanden |
En lysbue eller gnister opstår mellem de strømførende dele. Der opstår måske brand og kraftig stråling |
Øjenskader; forbrændinger, skoldning |
|
Ekstreme temperaturer |
Åben ild |
En person, der befinder sig i nærheden af flammerne, bliver måske forbrændt, eventuelt efter at der er gået ild i hans tøj |
Forbrændinger, skoldning |
|
|
Varme overflader |
En person ved ikke, at overfladen er varm, og rører ved den; personen brænder sig |
Forbrændinger |
|
|
Varme væsker |
En person, der håndterer en beholder med væske, spilder noget af væsken; væsken kommer i kontakt med huden og forårsager skoldning |
Skoldning |
|
|
Varme gasser |
En person indånder varme gasser, som et produkt udsender, hvilket resulterer i lungeforbrændinger, eller dehydreres efter længere tids udsættelse for varm luft |
Forbrændinger |
|
|
Kolde overflader |
En person ved ikke, at overfladen er kold, og rører ved den; personen får forfrysninger |
Forfrysninger |
|
Stråling |
Ultraviolette stråler, laserstråler |
En persons hud eller øjne udsættes for stråler fra produktet |
Forbrændinger, skoldning; neurologiske forstyrrelser; øjenskader; hudkræft, mutation |
|
|
Kilde til kraftige elektromagnetiske felter (EMF), lavfrekvente eller højfrekvente (mikrobølger) |
En person befinder sig tæt på kilden til det elektromagnetiske felt (EMF) og kroppen (centralnervesystemet) udsættes for dette |
Neurologiske (hjerne-)skader, leukæmi (børn) |
|
Brand og eksplosion |
Brændbare stoffer |
En person befinder sig tæt på det brændbare stof; en antændingskilde antænder stoffet; derved pådrager personen sig skader |
Forbrændinger |
|
|
Eksplosive blandinger |
En person befinder sig tæt på den eksplosive blanding; en antændingskilde forårsager en eksplosion; personen rammes af trykbølgen, brændende materialer og/eller flammer |
Forbrændinger, skoldning; øjenskader, fremmedlegeme i øje; høreskader, fremmedlegeme i øre |
|
|
Antændingskilder |
Antændingskilden forårsager en brand; en person såres af flammerne eller forgiftes af gasser fra branden |
Forbrændinger; forgiftning |
|
|
Overophedning |
Et produkt bliver overophedet; brand, eksplosion |
Forbrændinger, skoldning; øjenskader, fremmedlegeme i øje; høreskader, fremmedlegeme i øre |
|
Toksicitet |
Giftigt fast stof/giftig væske |
En person nedsvælger stoffet/væsken fra et produkt, f.eks. ved at putte produktet i munden, og/eller får stoffet/væsken på huden |
Akut forgiftning; irritation, dermatitis |
|
En person får fast eller flydende stof, f.eks. opkast, i luftvejene (lungeaspiration) |
Akut lungeforgiftning (aspirationspneumoni); infektion |
||
|
|
Giftig gas, giftige dampe eller giftigt støv |
En person indånder stoffet fra et produkt og/eller får stoffet på huden |
Akut lungeforgiftning; irritation, dermatitis |
|
|
Sensibiliserende stof |
En person nedsvælger stoffet fra et produkt, f.eks. ved at putte produktet i munden, og/eller får stoffet på huden, og eller personen indånder gas, dampe eller støv |
Sensibilisering; allergisk reaktion |
|
|
Lokalirriterende eller ætsende fast stof eller væske |
En person nedsvælger stoffet/væsken fra et produkt, f.eks. ved at putte produktet i munder, og/eller får stoffet/væsken på huden eller i øjnene |
Irritation, dermatitis; ætsning af huden; øjenskader, fremmedlegeme i øje |
|
|
Lokalirriterende eller ætsende gas eller dampe |
En person indånder stoffet fra et produkt og/eller får stoffet på huden eller i øjnene |
Irritation, dermatitis; ætsning af huden; akut forgiftning eller ætsning af lunger eller øjne |
|
|
CMR-stof |
En person nedsvælger stoffet fra et produkt, f.eks. ved at putte produktet i munden, og/eller får stoffet på huden, og eller personen indånder stoffet som gas, dampe eller støv |
Kræft, mutation, reproduktionstoksicitet |
|
Mikrobiologisk kontaminering |
Mikrobiologisk kontaminering |
En person kommer i kontakt med et kontamineret produkt ved nedsvælgning, indånding eller kontakt med huden |
Infektion, lokal eller systemisk |
|
Betjeningsrelaterede farer |
Usund arbejdsstilling |
Produktets udformning er skyld i en usund arbejdsstilling hos den, der betjener produktet |
Spændings-/belastningsskader; muskel- og skeletbesvær |
|
|
Overanstrengelse |
Produktets udformning gør, at det kræver en betydelig kraftudfoldelse at betjene det |
Forstrækning eller spændings-/belastningsskader; muskel- og skeletbesvær |
|
|
Anatomisk uegnethed |
Produktets udformning er ikke tilpasset den menneskelige anatomi, hvilket gør det vanskeligt eller umuligt at betjene |
Forstrækning eller spændings-/belastningsskader |
|
|
Tilsidesættelse af behov for personlig beskyttelse |
Produktets udformning gør det vanskeligt for en person, der bærer beskyttelsesudstyr, at håndtere eller betjene produktet |
Diverse skader |
|
|
Mulighed for utilsigtet (de)aktivering |
Det er let at (de)aktivere produktet, hvilket resulterer i uønsket aktivering heraf |
Diverse skader |
|
|
Utilstrækkelig funktionsdygtighed |
Produktets udformning er skyld i, at en person fejlbetjener det, eller et produkt med en indbygget sikkerhedsfunktion yder ikke den forventede beskyttelse |
Diverse skader |
|
|
Manglende standsning |
En person ønsker at deaktivere produktet, men det fortsætter med at køre, i en situation hvor dette er uønsket |
Diverse skader |
|
|
Uventet igangsætning |
Et produkt sættes ud af drift under et strømsvigt, men begynder at køre igen på en sådan måde, at der opstår fare |
Diverse skader |
|
|
Standsning ikke mulig |
En person er i en nødsituation ikke i stand til at deaktivere produktet |
Diverse skader |
|
|
Dele, der ikke passer sammen |
En person forsøger at fastgøre en del, men er nødt til at bruge for mange kræfter på at passe den ind, og produktet går i stykker, eller delen sidder ikke godt nok fast og løsriver sig under anvendelsen |
Forstuvning/forstrækning eller spændings-/belastningsskader; sønderrivning/laceration, sårskader; knubs/blå mærker; fastklemning/klemskader |
|
|
Manglende eller ukorrekt indbygget beskyttelse |
Farlige dele er tilgængelige for brugeren |
Diverse skader |
|
|
Utilstrækkelige advarselstekster og –symboler |
Brugeren overser advarselstekster og/eller forstår ikke symboler |
Diverse skader |
|
|
Utilstrækkelige advarselssignaler |
Brugeren ser eller hører ikke advarselssignaler (optiske signaler eller lyd), hvorved betjeningen bliver farlig |
Diverse skader |
|
NB: Dette skema er udelukkende ment som vejledning, og de typiske skadesscenarier bør tilpasses den enkelte risikovurdering. Der findes specifikke retningslinjer for risikovurdering af kemikalier, kosmetiske midler og muligvis også andre produkter. Det anbefales på det kraftigste at anvende disse specifikke retningslinjer i forbindelse med vurdering af sådanne produkter. Jf. afsnit 3.2. |
|||
Skema 3
Skadens alvorlighed
Indledning
Der opereres i disse retningslinjer for risikovurdering med fire niveauer for skaders alvorlighed. Det er vigtigt at holde sig for øje, at alvorlighed nødvendigvis må vurderes fuldstændig objektivt. I denne fase er målet ikke at afgøre, hvorvidt den enkelte personskade er acceptabel, men at sammenholde alvorligheden af forskellige scenarier og fastsætte prioriteringer. En hvilken som helst skade, der nemt kunne have været undgået, vil være vanskelig at acceptere for en forbruger. Myndighederne vil imidlertid med god ret kunne vælge at bruge flere ressourcer på at undgå varige skader end på at forebygge midlertidige gener.
Med henblik på en vurdering af omfanget af konsekvenserne (akutte personskader eller andre skader på helbredet) kan der udledes objektive kriterier af på den ene side omfanget af den lægebehandling, der er nødvendig, og på den anden side konsekvenserne for den tilskadekomnes tilværelse efterfølgende. Begge disse faktorer kunne udtrykkes som en omkostning, men det kan være vanskeligt at sætte tal på omkostningerne ved konsekvenserne af skader på helbredet.
Kombinerer man disse kriterier, kan de fire niveauer defineres således:
|
1. |
Personskade eller konsekvens, som efter basal behandling (førstehjælp, normalt ikke udført af en læge) ikke i væsentlig grad hæmmer en person i dagligdagen eller er årsag til meget stærke smerter; almindeligvis er konsekvenserne 100 % genoprettelige. |
|
2. |
Personskade eller konsekvens, som kan nødvendiggøre et besøg på skadestuen, men generelt ikke kræver hospitalsindlæggelse. Den tilskadekomne kan være påvirket af skaden i en begrænset periode, men højst 6 måneder, og den pågældendes helbredstilstand genoprettes helt eller næsten helt. |
|
3. |
Personskade eller konsekvens, som normalt kræver hospitalsindlæggelse, og som vil efterlade den tilskadekomne påvirket af skaden i over 6 måneder eller medføre varigt funktionstab. |
|
4. |
Personskade eller konsekvens, som medfører eller kunne medføre døden, herunder hjernedød; konsekvenser, som påvirker forplantning eller afkom; alvorligt tab af lemmer og/eller funktionstab, som medfører en invaliditetsgrad på over godt 10 %. |
I nedenstående skema, som hverken er præskriptivt eller udtømmende, men skal ses som en rettesnor, gives eksempler på personskader på alle fire niveauer. Der kan være nationalt betingede forskelle, enten af kulturel art eller affødt af de indbyrdes forskelle på sundhedssystemer og økonomiske ordninger. Afvigelser fra den i skemaet beskrevne klassifikation vil imidlertid være til hinder for en ensartet vurdering af risici i EU; sådanne afvigelser skal derfor klart angives og begrundes i risikovurderingsrapporten.
|
Skadens art |
Skadens alvorlighed |
|||
|
1 |
2 |
3 |
4 |
|
|
Sønderrivning/laceration, sårskader |
Overfladisk |
Ydre (dyb) (> 10 cm længde på kroppen) (> 5 cm længde i ansigtet) — syningskrævende Sene eller led Senehinden eller hornhinden |
Synsnerve Halsarterie Luftrøret Indre organer |
Bronkiegren Strubehovedet Hovedpulsåren Rygmarven (nedre del) Dyb laceration af indre organer Overskåren (øvre) rygmarv Hjernen (svær læsion/funktionsforstyrrelser) |
|
Knubs/blå mærker (hudafskrabninger/blodudtrædninger, hævelser, væskeansamlinger) |
Overfladisk ≤ 25 cm2 i ansigtet ≤ 50 cm2 på kroppen |
Mere alvorlig > 25 cm2 i ansigtet > 50 cm2 på kroppen |
Luftrøret Indre organer (mindre) Hjertet Hjernen Lunge, med blod eller luft i brystet |
Hjernestammen Rygmarv, med lammelse til følge |
|
Hjernerystelse |
— |
Bevidstløshed af meget kort varighed (minutter) |
Længerevarende bevidstløshed |
Koma |
|
Fastklemning/klemskader |
Mindre alvorlig klemning |
— |
(Beskrivelse af det endelige resultat — knubs/blå mærker, knusningsskader, knoglebrud, forvridning, amputation — alt efter hvad der er relevant) |
(Samme resultat som for kvælning/strangulering) |
|
Forstuvning/forstrækning, spændings-/belastningsskader, muskel- og skeletbesvær |
Ekstremiteter Led Ryggen (ingen forvridning eller brud) |
Overbelastning af ledbånd i knæ |
Sprængning/iturivning af ledbånd eller sener Overrivning af muskler Piskesmæld |
— |
|
Forvridning |
— |
Ekstremiteter (fingre, tæer, hænder eller fødder) Albue Kæbe Løse tænder |
Ankel Håndled Skulder Hofte Knæ Ryggen |
Rygsøjlen |
|
Knoglebrud |
— |
Ekstremiteter (fingre, tæer, hænder eller fødder) Håndled Arm Ribben Brystbenet Næsen Tand Kæbe Knogler omkring øje |
Ankel Ben (lår- og underben) Hofte Lår Kraniet Ryggen (mindre kompressionsbrud) Kæbe (alvorligt) Strubehovedet Flere brud på ribben Blod eller luft i brystet |
Nakken Rygsøjlen |
|
Knusningsskader |
— |
— |
Ekstremiteter (fingre, tæer, hænder eller fødder) Albue Ankel Håndled Underarm Ben Skulder Luftrøret Strubehovedet Bækkenet |
Rygmarven Nakken (midterste/nederste del) Brystet (omfattende knusningsskader) Hjernestammen |
|
Amputation |
— |
— |
Finger/fingre Tå/tæer Hånd Fod Arm (del af) Ben Øje |
Begge ekstremiteter |
|
Stik-/gennemboringsskader |
Begrænset dybde, kun i huden |
Dybere end underhuden Bugvæggen (ingen organer berørt) |
Øje Indre organer Brystvæggen |
Hovedpulsåren Hjertet Bronkiegren Dybe skader på organer (lever, nyre, tarm osv.) |
|
Nedsvælgning/slugning |
— |
— |
Skader på indre organer (Desuden beskrives indre luftvejsblokering, hvor den slugte genstand sætter sig fast ved strubehovedet) |
Varige skader på indre organ |
|
Indre luftvejsblokering |
— |
— |
Der blokeres for iltforsyningen til hjernen — ingen varige konsekvenser |
Der blokeres for iltforsyningen til hjernen — varige konsekvenser |
|
Kvælning/strangulering |
— |
— |
Der blokeres for iltforsyningen til hjernen — ingen varige konsekvenser |
Kvælning/strangulering med døden til følge |
|
Neddykning/drukning |
— |
— |
— |
Druknedød |
|
Forbrændinger/skoldning (pga. varme, kulde eller kemisk stof) |
1.-grads — op til 100 % af legemsoverfladen 2.-grads - < 6 % af legemsoverfladen |
2.-grads – 6-15 % af legemsoverfladen |
2.-grads – 16-35 % af legemsoverfladen, eller 3.-grads — op til 35 % af legemsoverfladen Inhalationsforbrændinger |
2.-grads eller 3.-grads - > 35 % af legemsoverfladen Inhalationsforbrændinger - åndedrætshjælp nødvendig |
|
Elektrisk stød |
(Jf. under forbrændinger, idet elektrisk strøm også kan forårsage forbrændinger) |
Lokale virkninger (forbigående kramper eller muskellammelse) |
— |
Elektrisk stød med døden til følge |
|
Neurologiske forstyrrelser |
— |
— |
Epileptisk anfald |
— |
|
Øjenskader, fremmedlegeme i øje |
Forbigående, ikke-behandlingskrævende smerter i øje |
Forbigående synstab |
Delvist synstab Permanent synstab (på ét øje) |
Permanent synstab (på begge øjne) |
|
Høreskader, fremmedlegeme i øre |
Forbigående, ikke-behandlingskrævende smerter i øre |
Forbigående svækkelse af hørelsen |
Delvist tab af hørelsen Fuldstændigt tab af hørelsen (på ét øre) |
Fuldstændigt tab af hørelsen (på begge ører) |
|
Forgiftning med stoffer (nedsvælgning, indånding, kontakt med huden) |
Diarré, opkastning, lokale symptomer |
Genoprettelige skader på indre organer, såsom lever eller nyre, mindre alvorlig hæmolytisk anæmi |
Varige skader på indre organer, såsom strubehovedet, maven, lever eller nyre, hæmolytisk anæmi, genoprettelige skader på nervesystemet |
Varige skader på nervesystemet Død |
|
Irritation, dermatitis, betændelse eller ætsning forårsaget af stoffer (indånding, kontakt med huden) |
Mild lokalirritation |
Genoprettelige øjenskader Genoprettelige systemiske skader Betændelsestilstand |
Lunger, respirationsinsufficiens, kemisk lungebetændelse Varige systemiske skader Delvist synstab Ætsning |
Lunger - åndedrætshjælp nødvendig Pulsløshed |
|
Allergisk reaktion eller sensibilisering |
Mild eller lokal allergisk reaktion |
Allergisk reaktion, udbredt allergisk kontaktdermatitis |
Kraftig sensibilisering, resulterende i allergi over for flere stoffer |
Anafylaktisk reaktion, chok Død |
|
Langvarige skader pga. kontakt med stoffer eller udsættelse for stråler |
Diarré, opkastning, lokale symptomer |
Genoprettelige skader på indre organer, såsom lever eller nyre, mindre alvorlig hæmolytisk anæmi |
Skader på nervesystemet, såsom organisk psykosyndrom (OPS) eller kronisk toksisk encefalopati (»malersyndrom«). Varige skader på indre organer, såsom strubehovedet, maven, lever eller nyre, hæmolytisk anæmi, genoprettelige skader på nervesystemet |
Kræft (leukæmi) Virkninger for forplantningsevnen Virkninger for afkom Svækkelse af centralnervesystemet |
|
Mikrobiologisk infektion |
|
Genoprettelige skader |
Varige skader |
Infektion — længerevarende hospitalsindlæggelse nødvendig, antibiotikaresistente organismer Død |
Skema 4
Risikoniveau på grundlag af kombinationen af skadens alvorlighed og sandsynligheden for den
|
Sandsynligheden for skader i produktets forudsigelige levetid |
Skadens alvorlighed |
||||
|
1 |
2 |
3 |
4 |
||
|
Høj |
> 50 % |
H |
S |
S |
S |
|
|
> 1/10 |
M |
S |
S |
S |
|
> 1/100 |
M |
S |
S |
S |
|
|
> 1/1 000 |
L |
H |
S |
S |
|
|
> 1/10 000 |
L |
M |
H |
S |
|
|
> 1/100 000 |
L |
L |
M |
H |
|
|
> 1/1 000 000 |
L |
L |
L |
M |
|
|
Lav |
< 1/1 000 000 |
L |
L |
L |
L |

|
S – Alvorlig risiko |
|
H – Høj risiko |
|
M – Middel risiko |
|
L – Lav risiko |
Anvendte begreber
Fare: Farekilde, der indebærer en mulighed for at komme til skade. En målestok for faren i forbindelse med en risikovurdering er alvorligheden af den potentielle personskade.
Faren ved et produkt: Fare, der udspringer af produktets egenskaber/karakteristika.
Risiko: Afbalanceret kombination af en fare og sandsynligheden for, at en personskade vil indtræffe. Risiko er hverken udtryk for faren eller for sandsynligheden, men begge disse elementer på samme tid.
Risikovurdering: Procedure til identificering og vurdering af farer, som omfatter tre trin:
|
— |
identificering af en fares alvorlighed |
|
— |
bestemmelse af sandsynligheden for, at en forbruger vil komme til skade som følge af den pågældende fare |
|
— |
kombinering af faren og sandsynligheden. |
Risikoniveau: Grad af risiko, som kan være »alvorlig«, »høj«, »middel« eller »lav«. Når risikoniveauet (det højeste) er bestemt, er risikovurderingen tilendebragt.
Risikohåndtering: Opfølgning, som er et selvstændigt skridt i forhold til risikovurderingen og har til formål at begrænse eller eliminere en risiko.
(1) I retningslinjerne menes der med »Kommissionen« normalt det RAPEX-team, der er nedsat under Kommissionens afdeling med ansvar for direktivet om produktsikkerhed i almindelighed, og til andre relevante tjenestegrene i Kommissionen.
(2) EFT L 11 af 15.1.2002, s. 4.
(3) EUT L 218 af 13.8.2008, s. 30.
(4) I dette dokument forstås ved »medlemsstaterne« alle EU's medlemslande samt de lande, der har undertegnet aftalen om Det Europæiske Økonomiske Samarbejdsområde (EØS).
(5) www.ec.europa.eu/rapex.
(6) EFT L 31 af 1.2.2002, s. 1.
(7) EFT L 311 af 28.11.2001, s. 67.
(8) EFT L 311 af 28.11.2001, s. 1.
(9) EFT L 169 af 12.7.1993, s. 1.
(10) EFT L 331 af 7.12.1998, s. 1.
(11) EFT L 189 af 20.7.1990, s. 17.
(12) EFT L 204 af 21.7.1998, s. 37.
(13) Punkt 3.10 i retningslinjerne indeholder flere oplysninger om disse frister.
(14) Punkt 5.1 i retningslinjerne indeholder flere oplysninger om RAPEX-kontaktpunkterne og deres ansvarsområder.
(15) Punkt 3.7 indeholder flere oplysninger om opfølgning.
(16) Punkt 3.5.2 og 3.8.1 indeholder flere oplysninger om meddelelser om sikkerhedsaspekter, der behandles på EU-plan.
(17) Punkt 3.1.2, litra d), og punkt 3.8.1 indeholder flere oplysninger om meddelelser om sikkerhedsaspekter, der behandles på EU-plan.
(18) Kapitel 2 indeholder flere oplysninger om RAPEX-notifikationskriterierne.
(19) Punkt 3.1.2, litra b), indeholder flere oplysninger om meddelelser, der sendes via RAPEX-applikationen, inden der træffes foranstaltninger.
(20) Punkt 3.1.2, litra d), og punkt 3.5.2 indeholder flere oplysninger om meddelelser om sikkerhedsaspekter, der behandles på EU-plan.
(21) Alle frister i disse retningslinjer er angivet i kalenderdage.
(22) Benis, H. G. (1990): A Product Risk Assessment Nomograph (rapport udarbejdet for New Zealands Ministry of Consumer Affairs), februar 1990. Citeret i: Europa-Kommissionen (2005): Establishing a Comparative Inventory of Approaches and Methods Used by Enforcement Authorities for the Assessment of the Safety of Consumer Products Covered by Directive 2001/95/EC on General Product Safety and Identification of Best Practices (rapport udarbejdet af Risk & Policy Analysts (RPA), Loddon, Norfolk, UK).
(23) Metode, der anvendes af de belgiske myndigheder. Citeret i: Europa-Kommissionen (2005): Establishing a Comparative Inventory of Approaches and Methods Used by Enforcement Authorities for the Assessment of the Safety of Consumer Products Covered by Directive 2001/95/EC on General Product Safety and Identification of Best Practices (rapport udarbejdet af Risk & Policy Analysts (RPA), Loddon, Norfolk, UK).
(24) Kommissionens beslutning 2004/418/EF af 29. april 2004 om retningslinjer for forvaltningen af fællesskabssystemet for hurtig udveksling af oplysninger (RAPEX) og for meddelelser indgivet i henhold til artikel 11 i direktiv 2001/95/EF (EUT L 151 af 30.4.2004, s. 83).
(25) Europa-Parlamentets og Rådets direktiv 2001/95/EF af 3. december 2001 om produktsikkerhed i almindelighed (EFT L 11 af 15.1.2002, s. 4).
(26) Kinney, G. F., Wiruth, A. D. (1976): Practical risk analysis for safety management. China Lake, California: NWC Technical Publication 5865, Naval Weapons Center, California, juni 1976.
(27) https://webgate.ec.europa.eu/idbpa/.
(28) NB: Der skal altid tages hensyn til usikkerheden i forbindelse med sammenholdelse af et testresultat med en grænseværdi. Se f.eks.
|
— |
»Report on the relationship between analytical results, measurement uncertainty, recovery factors and the provisions of EU food and feed legislation […]« (http://ec.europa.eu/food/food/chemicalsafety/contaminants/report-sampling_analysis_2004_en.pdf). |
|
— |
den sammenfattende rapport »Preparation of a working document in support of the uniform interpretation of legislative standards and the laboratory quality standards prescribed under Directive 93/99/EEC« (http://ec.europa.eu/food/fs/scoop/9.1_sr_en.pdf). |
(29) Europa-Parlamentets og Rådets forordning (EF) nr. 1907/2006 af 18. december 2006 om registrering, vurdering og godkendelse af samt begrænsninger for kemikalier (REACH), om oprettelse af et europæisk kemikalieagentur og om ændring af direktiv 1999/45/EF og ophævelse af Rådets forordning (EØF) nr. 793/93 og Kommissionens forordning (EF) nr. 1488/94 samt Rådets direktiv 76/769/EØF og Kommissionens direktiv 91/155/EØF, 93/67/EØF, 93/105/EF og 2000/21/EF (EUT L 396 af 30.12.2006, s. 1).
(30) EUT L 164 af 26.6.2009, s. 7.
(31) Standard EN 71-1:2005, del 8.2 + A6:2008.
(32) Artikel 7a, stk. 1, litra d), i Rådets direktiv 76/768/EØF (EFT L 262 af 27.9.1976, s. 169).
(33) REACH-forordningen. De relevante REACH-vejledninger findes på følgende adresse: http://echa.europa.eu/.
Det Europæiske Kemikalieagentur (2008): The Guidance on Information Requirements and Chemical Safety Assessment (http://guidance.echa.europa.eu/docs/guidance_document/information_requirements_en.htm).
(34) Den Videnskabelige Komité for Forbrugsvarer (VKF): The SCCP’s Notes of Guidance for the Testing of Cosmetic Ingredients and their Safety Evaluation, 6. udgave, den 19. december 2006 (http://ec.europa.eu/enterprise/cosmetics/html/testing_guidance.htm).
(35) Dette er taget fra definitionen af et »sikkert produkt« i artikel 2, litra b), i direktiv 2001/95/EF.






